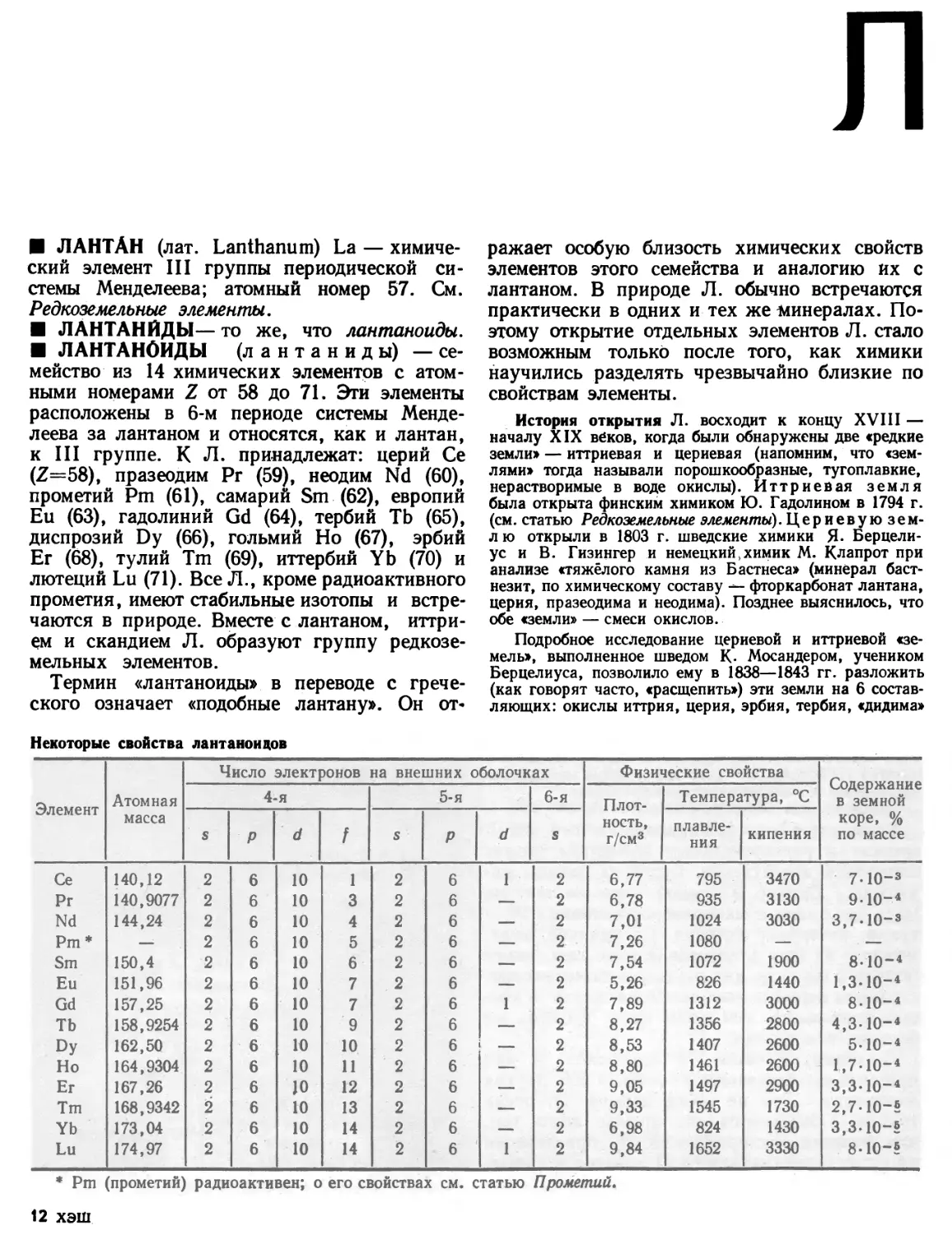
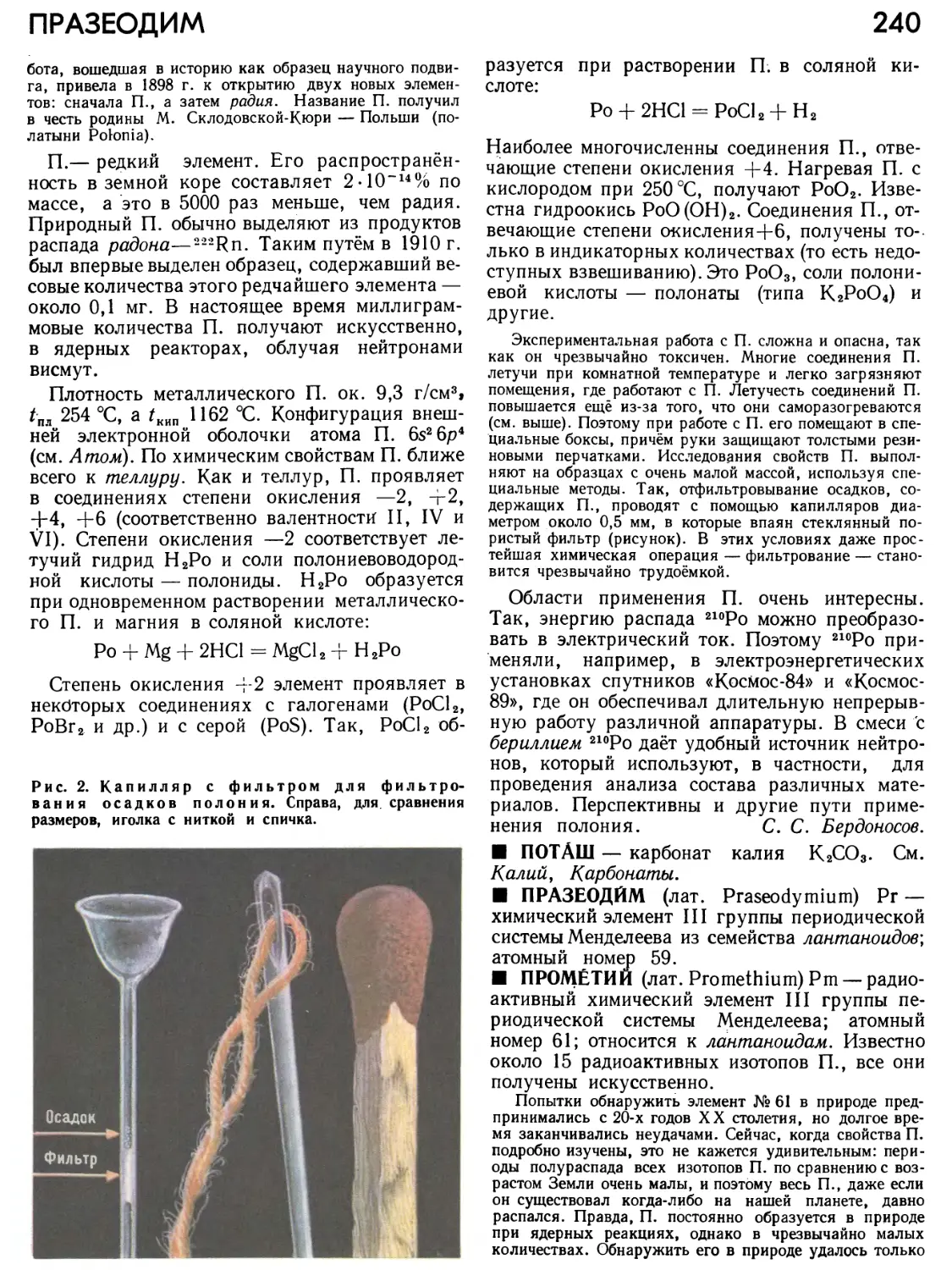
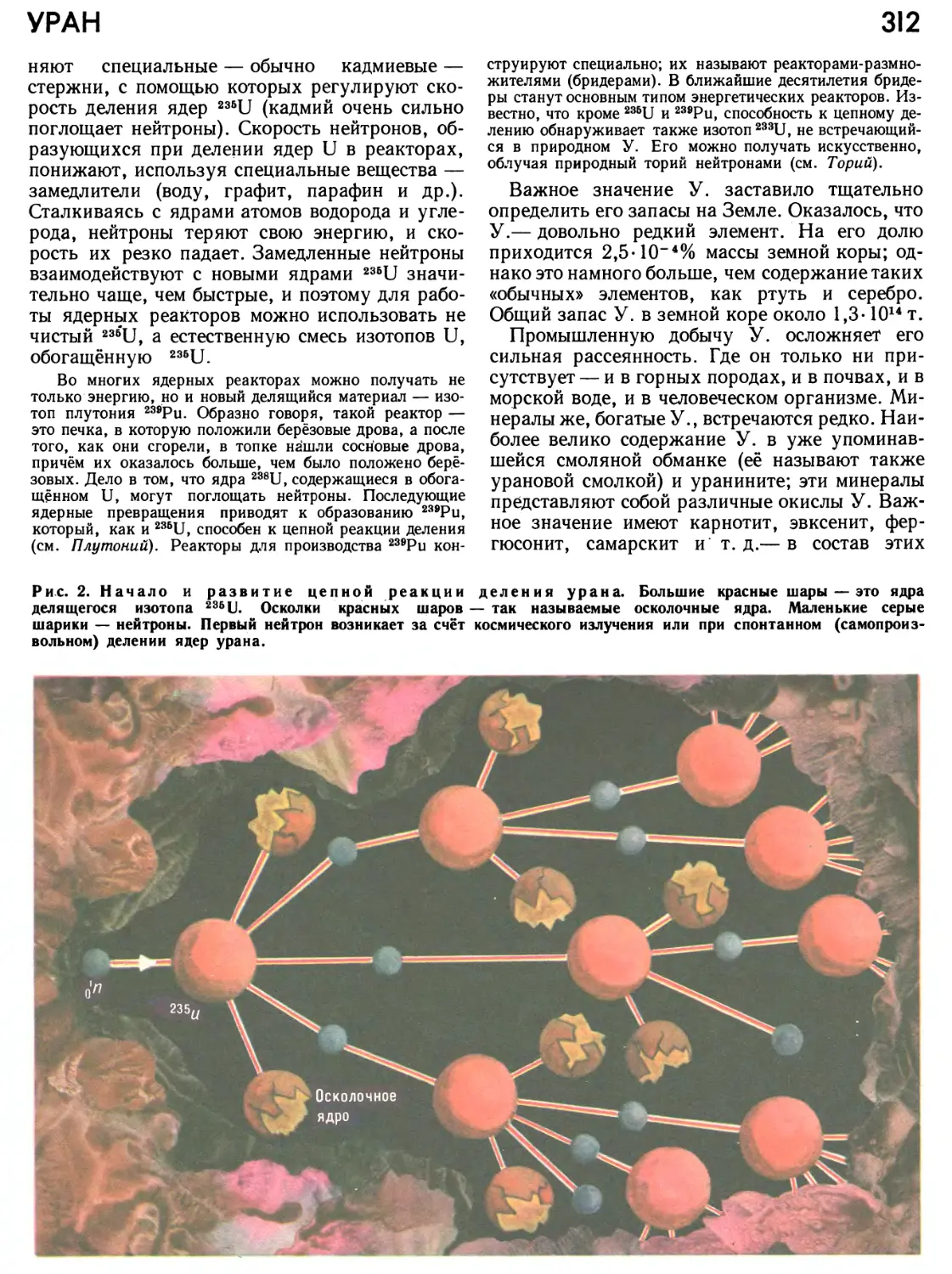
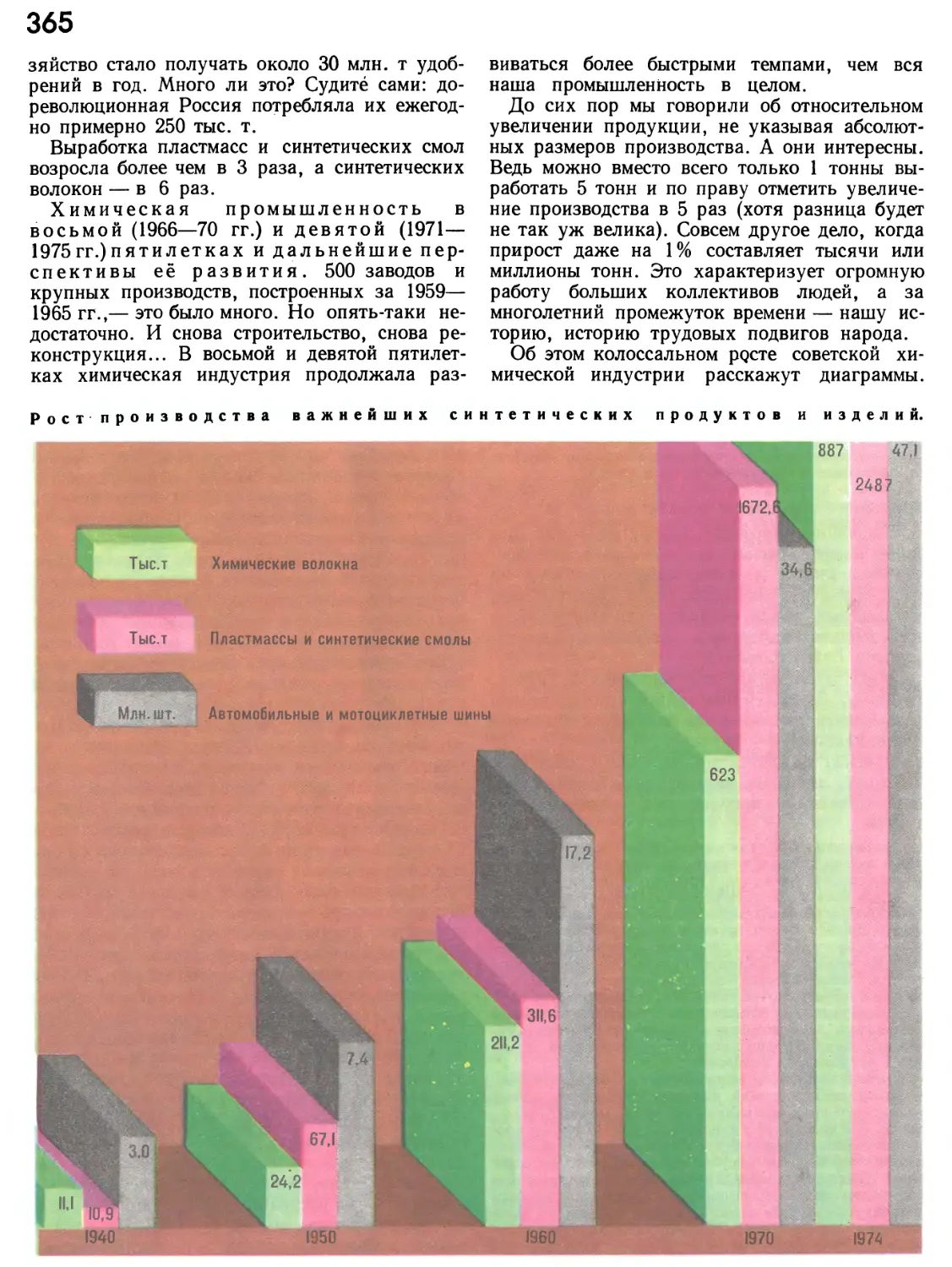
/





























































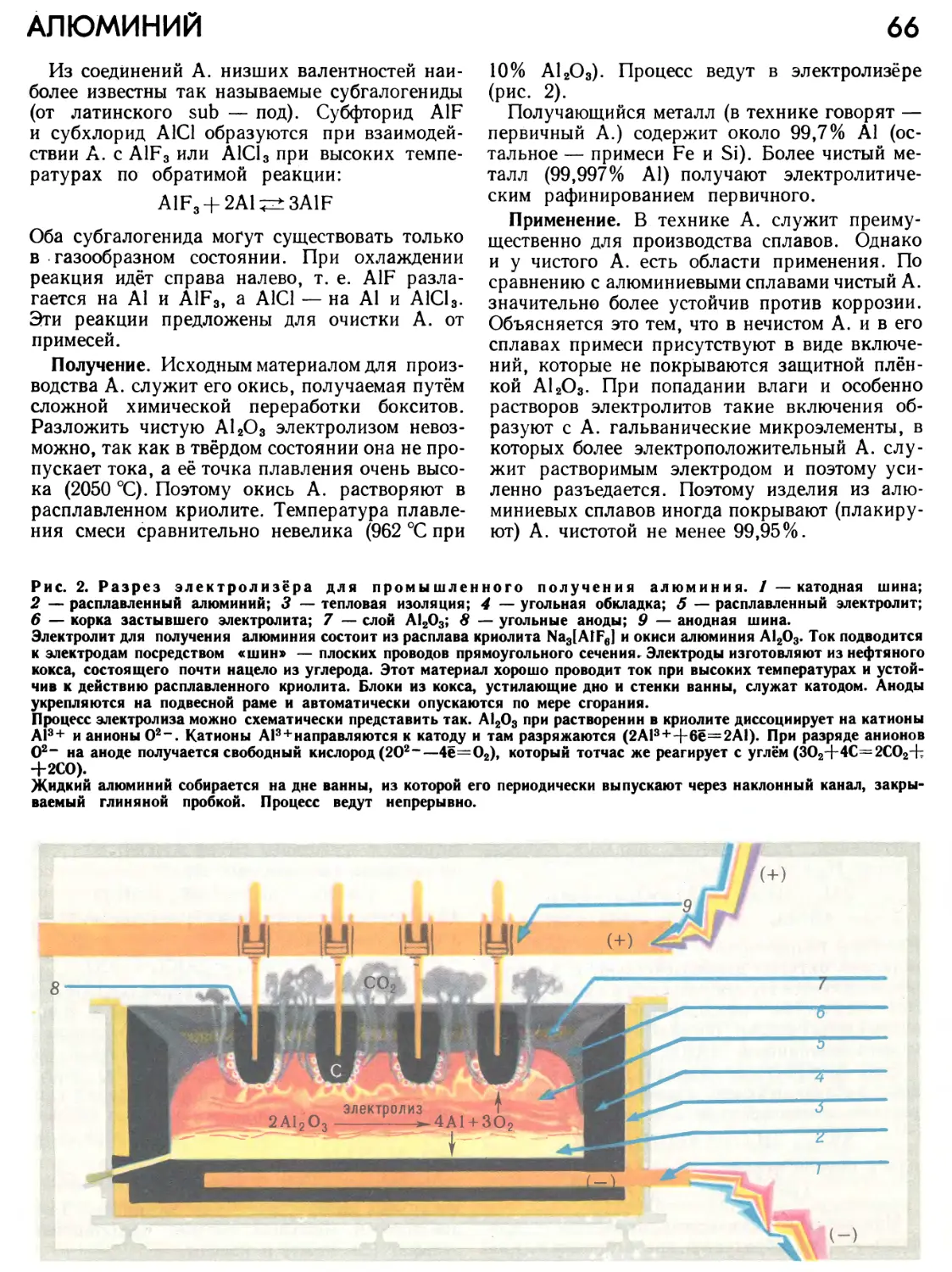
























































































































































































































































































































Автор: Прокофьев М.А.
Теги: химия неорганическая химия академия наук ссср химический справочник серия энциклопедия школьника
Год: 1975




Текст
ЭНЦИКЛОПЕДИЯ ШКОЛЬНИКА
под общей редакцией члена-корреспондента АН СССР М. А. ПРОКОФЬЕВА
НАУЧНО-РЕДАКЦИОННЫЙ СОВЕТ ИЗДАТЕЛЬСТВА
А. М. ПРОХОРОВ (председатель), И. В. АБАШИДЗЕ, П. А. АЗИМОВ, А. П. АЛЕКСАНДРОВ, В. А. АМБАРЦУМЯН, И. И. АРТОБОЛЕВСКИЙ, А. В. АРЦИХОВСКИЙ, М. С. АСИМОВ, М. П. БАЖАН, Н. В. БАРАНОВ, Н. Н. БОГОЛЮБОВ, П. У. БРОВКА, Ю. В. БРОМЛЕЙ, Б. Э. БЫХОВСКИЙ, В. X. ВАСИЛЕНКО, А. П. ВИНОГРАДОВ, В. В. ВОЛЬСКИЙ, Б. М. ВУЛ, Б. Г. ГАФУРОВ, С. Р. ГЕРШБЕРГ, В. П. ГЛУШКО, В. М. ГЛУШКОВ, Г. Н. ГОЛИКОВ, Я. С. ГРОСУЛ, А. А. ГУСЕВ (заместитель председателя), В. П. ЕЛЮТИН, В. С. ЕМЕЛЬЯНОВ, Е. М. ЖУКОВ, А. А. ИМШЕНЕЦКИЙ, Н. Н. ИНОЗЕМЦЕВ, М. И. КАБАЧНИК, С. В. КАЛЕСНИК, Г. А. КАРАВАЕВ, К. К. КАРАКЕЕВ, М. К. КАРАТАЕВ, Б. М. КЕДРОВ, Г. В. КЕЛДЫШ, В. А. КИРИЛЛИН, И, Л. КНУНЯНЦ, С. М. КОВАЛЕВ (первый заместитель председателя), Ф. В. КОНСТАНТИНОВ, В. Н. КУДРЯВЦЕВ, М. И. КУЗНЕЦОВ (заместитель председателя), Б. В. КУКАРКИН, В. Г. КУЛИКОВ, И. А. КУТУЗОВ, П. П. ЛОБАНОВ, Г. М. ЛОЗА, Ю. Е. МАКСАРЕВ, П. А. МАРКОВ, А. И. МАР-КУШЕВИЧ, Ю, Ю. МАТУЛИС, Г. И. НААН, Г. Д. ОБИЧКИН, Б. Е. ПАТОН, Я. В. ПЕЙВЕ, В. М. ПОЛЕВОЙ, М. А. ПРОКОФЬЕВ, Ю. В. ПРОХОРОВ, РАСУЛ РЗА, Н. Ф. РОСТОВЦЕВ, А. М. РУМЯНЦЕВ, Б. А. РЫБАКОВ, В. П. САМСОН, М. И. СЛАДКОВСКИЙ, В. И. СМИРНОВ, А. А. СОЛДАТОВ, Д. Н. СОЛОВЬЕВ (заместитель председателя), В. Г. СОЛОДОВНИКОВ, В. Н. СТАРОВСКИЙ, В. Н. СТОЛЕТОВ, Б. И. СТУКАЛИН, А. А. СУРКОВ, М. Л. ТЕРЕНТЬЕВ, С. А. ТОКАРЕВ, В. А. ТРАПЕЗНИКОВ, А. Т. ТУМАНОВ, Е. К. ФЕДОРОВ, М. Б. ХРАПЧЕНКО, Е. И. ЧАЗОВ, В. Н. ЧЕРНИГОВСКИЙ, Я. Е. ШМУШКИС, С. И. ЮТКЕВИЧ.
МОСКВА, 1973
НЕОРГАНИЧЕСКАЯ ХИМИЯ
Главный редактор академик И. П. АЛИМАРИН
РЕДАКЦИОННАЯ КОЛЛЕГИЯ
В. К. БЕЛЬСКИЙ, В. Л. ВАСИЛЕВСКИЙ, А. А. ГУСЕВ, Н. П. МОСТОВЕНКО (зам. главного редактора), С. А. ПОГОДИН
ИЗДАТЕЛЬСТВО «СОВЕТСКАЯ ЭНЦИКЛОПЕДИЯ»
54 (03)
Н 52
Научные консультанты — С. С. БЕР ДОНОСОВ, Д. С. ДАНИН, П. М. ЗОРКИЙ, А. И. ПЕРЕЛЬМАН, В. С. ТИТОВ
Редакторы — А. А. ЖУРКОВА, Л. Д. КОВБА, Н. П. МОСТОВЕНКО, В. А. СОЛОМЕННИКОВА
Научно-контрольная редакция — Н. П. ПРЕОБРАЖЕНСКАЯ, Я. Е. ШМУШКИС Литературная редакция — В. В. МАЙКОВА
Художественно-техническая редакция — И. А. ВЕТРОВА, И. Н. САХАРОВА, Г. В. СМИРНОВА
Корректоры — М. В. АКИМОВА, Н. Е. ЗМЕЕВА, И. Н. ЛУКЬЯНОВА, О. Н. ПАВЛОВА, Л. В. ПИТАЛЁВА, А. Ф. ПРОШКО, М. М. ШИНКАРЁВА Том комплектовала Н. А. ФЁДОРОВА Указатель составил А. Б. ДМИТРИЕВ Обложка — художник Б. Е. МАРКОВ
В работе над книгой принимали участие: Л. М. КАЧАЛОВА, Т. И. ПАВЛОВА (производственный отдел), Е. А. ВОЛКОВСКАЯ, И. С. ГЕРЦФЕЛЬД (проверка фактов), Н. М. ПРИЛЕПОВА (редакция словника), В. М. ПЕТЮШЕНКО (редакция искусства), Г. В. СОБОЛЕВСКИЙ (редакция иллюстраций).
Художники —Н. А. ДОБРОХОТОВА, Т. А. ДОБРОХОТОВА
60602—006
007(01)—75
БЗ—1 —21 —75
© ИЗДАТЕЛЬСТВО «СОВЕТСКАЯ ЭНЦИКЛОПЕДИЯ»
О химии
ВВЕДЕНИЕ
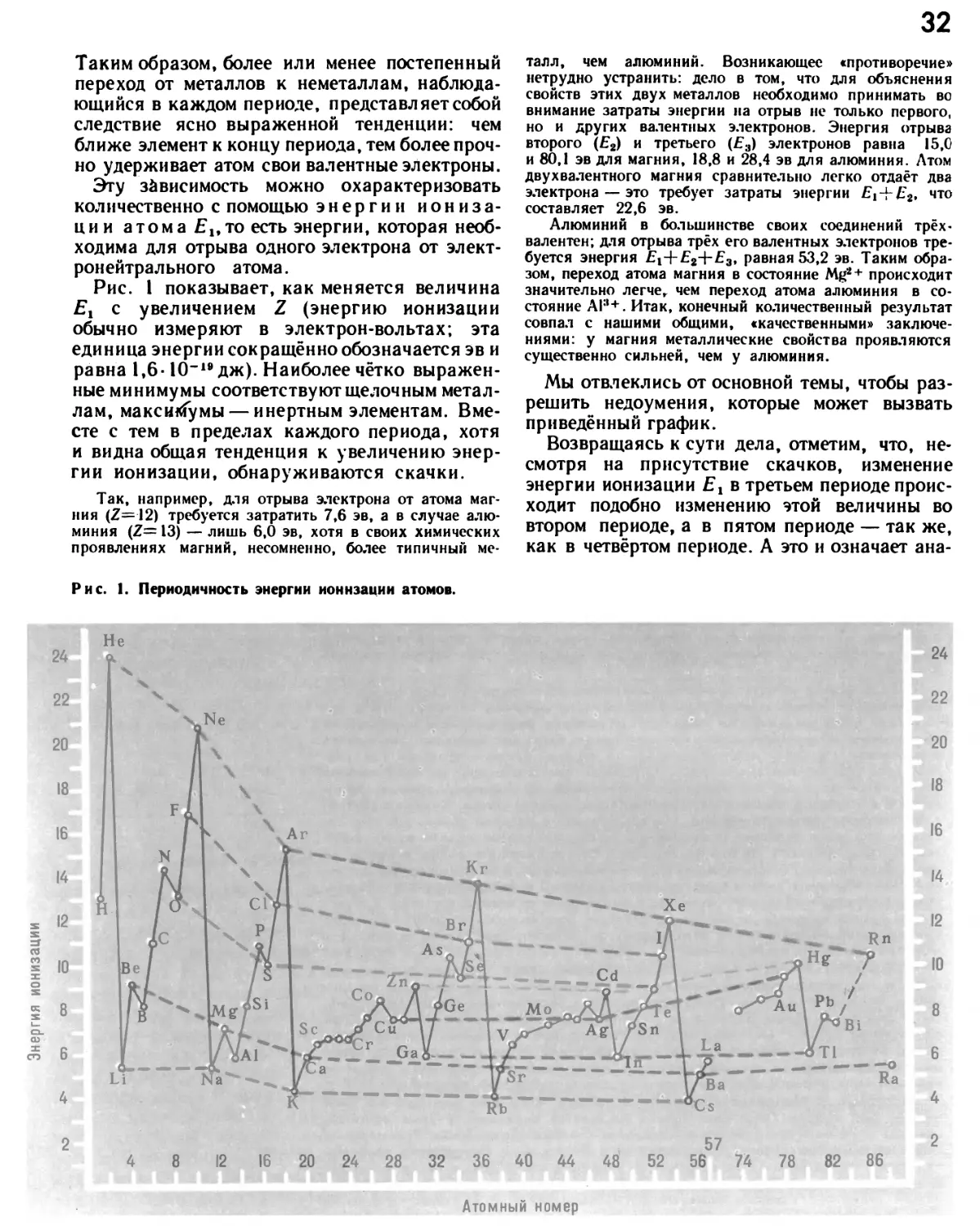
Наш земной шар — это гигантский универсальный химический комбинат, действующий вот уже миллиарды лет. Глубины Земли и её твёрдая кора, океаны и атмосфера содержат в своей первооснове химические элементы. Из них построены окружающий нас мир и мы сами. Недаром химические элементы иногда сравнивают с буквами алфавита, из ограниченного числа которых составлены все слова. Сегодня известно более 100 химических элементов; из них 89 обнаружены в природе, а остальные получены искусственно. Взаимодействие существующих в природе элементов и обеспечивает непрерывную, многосложную, неисчерпаемую работу удивительного химического комбината «Земля». И столь же непрерывно Солнце снабжает этот комбинат своей энергией. Разнообразные химические взаимодействия элементов, прежде всего четырёх из них (углерода, водорода, азота, кислорода), привели к возникновению простейших органических соединений. Постепенно усложняясь, такие соединения положили начало жизни на Земле. Всё более мощно разрастались в океане и на суше растения. Этой исполинской зелёной фабрике, дающей атмосфере кислород, обязан своим существованием животный мир. И, разумеется,— человечество.
Многие века человек использовал для своих нужд то, что ему давала сама природа: строил жилища из дерева, делал орудия труда из камня и посуду из глины, одевался в шкуры животных, силой ветра приводил в движение парусные корабли и мельничные жернова. Издревле люди, наблюдая явления природы, стремились разгадать её тайны. Обугливая дерево, обжигая глину, выплавляя металлы из руд, выделяя лекарственные вещества из растений, наши далёкие предки, сами того не подозревая, знакомились с химическими процессами. Можно считать поэтому, что химия — наука о веществах и их превращениях — одна из древнейших сфер познавательной деятельности человека.
Первые практические сведения по химии появились, как полагают, в Древнем Египте. И происхождение самого слова «химия» учёные чаще всего связывают с наименованием Древнего Египта «Хем», что означало «тёмный» или «чёрный» (по-видимому, по цвету почвы в долине Нила). Другие же исследователи связывают слово «химия» с древнегреческим «хемёйя» — искусство выплавки металлов. Ведь металлургия на протяжении веков была и остаётся по сей день обширнейшей областью практических приложений химии. Из известных в наши дни химических элементов свыше 80 — металлы и лишь около 20 — неметаллы.
В древности и в средние века металлов насчитывали только семь: золото, серебро, медь, олово, свинец, железо, ртуть. Из неметаллов раньше других стали известны углерод и сера. В средние века развитие химии— её называли тогда арабским словом «алхиМия» — определялось прежде всего поисками так называемого «философского камня»: алхимики надеялись с его помощью превращать любой металл в золото. Ко
нечно же, надежды эти были тщетными. Но кропотливые поиски не пропали даром: алхимики усовершенствовали саму технику лабораторного эксперимента и сделали немало серьёзных практических открытий. Они впервые получили в свободном состоянии ещё несколько химических элементов: мышьяк, сурьму, фосфор.
Ни в Древнем мире, ни во времена средневековья ещё не существовало научных представлений о составе и строении разных веществ. Не было и ясного понимания того, что же следует называть химическим* элементом. Четырьмя «элементами» Вселенной долго считались огонь, воздух, вода и земля, а всё в окружающем мире представлялось их сочетанием. Впервые в 1661 г. английский химик Роберт Бойль сформулировал понятие о химическом элементе как о веществе, не разложимом на более простые части. В 1748 г. Михаил Васильевич Ломоносов в России и независимо от него в 1774 г. Антуан Лавуазье во Франции открыли закон сохранения массы веществ в химических реакциях. К концу XVIII в. список известных химических элементов расширился уже до 32, и в числе новооткрытых появились кислород, азот, хлор. Был установлен состав воды и воздуха. Было получено много прежде неведомых химических соединений. А главное: в химии, остававшейся до той поры наукой качественной и описательной, утвердился, наконец, количественный подход к изучению всех процессов. И естественно, что вскоре дошла очередь до решения вопроса о строении вещества.
К представлению о прерывистом строении вещества — об атомарной природе всех тел — пришли ещё великие натурфилософы древности Левкипп, Демокрит, Лукреций Кар. Однако воззрения их были только догадками — хоть и гениальными, но не обоснованными точным естественнонаучным экспериментом. В начале XIX в. английский учёный Джон Дальтон возродил учение древних атомистов на новой основе, на обобщении строгих опытных данных. Каждому химическому элементу у Дальтона соответствовал определённый вид атомов, и количественной характеристикой элемента служил его атомный вес. Открытые в тот период законы постоянства состава и кратных отношений, вместе с ранее открытым законом сохранения массы, получили теоретическое объяснение и стали мощным стимулом для дальнейших количественных исследований.
В конце XVIII — начале XIX вв. усиленное внимание учёных начали привлекать вещества растительного и животного происхождения. Их систематическое изучение привело к появлению в первой половине XIX в. новой ветви химии, получившей название органической. Её предметом стали, за очень немногими исключениями, соединения углерода. (Особенности углерода таковы, что число его соединений составляет около 3 миллионов, в то время как соединений всех остальных элементов насчитывается лишь около 60 тысяч.)
Вторая половина XIX в. ознаменовалась в истории химии ещё и тем, что в этот период, особенно с конца 70-х гг., в самостоятельную отрасль начала оформляться новая дисциплина — физическая химия. Исторически её предназначение заключалось в том, чтобы установить закономерности химических явлений на базе общих принципов физики.
С развитием атомистических воззрений были сделаны первые попытки систематизации элементов — прежде всего по их атомным весам. Они завершились в 1869 г. великим открытием Дмитрия Ивановича Менделеева: появился периодический закон химических элементов. Он внёс ясность и порядок в хаос неисчислимых сведений, накопленных химией за многовековую историю. (К тому времени были известны уже 63 элемента и огромное множество их соединений.) Теперь стало возможным теоретическое предсказание ещё не открытых элементов и прогнозирование их свойств. С закона Менделеева начался современный этап развития химии. Но самый этот закон, в свою очередь, нуждался в физическом обосновании.
В начале XX в. физика раскрыла сложную структуру атома. Учение о строении атома и стало ключом к пониманию периодической системы
элементов. А значит — и ключом ко всем областям науки о веществе и его превращениях.
Химия обрела в атомной теории крепкий физический фундамент. Вместе с тем на протяжении нашего века химия научилась использовать для своих целей разнообразный арсенал точных физических методов исследования вещества. Это проникновение физики в химию обогатило и физику новыми научными направлениями, новыми подходами, новыми материалами. Такой синтез двух великих наук был закономерен. Развился целый комплекс наук, пограничных между физикой и химией, таких как химическая физика, химическая термодинамика, электрохимия, радиационная химия...
Для общего процесса стремительного взлёта естествознания и технических наук в XX в. это взаимопроникновение и взаимообогащение наук о природе — характерная черта. И везде, где химия соприкасалась с другими отраслями природоведения, возникли «точки роста» новых наук. Изучение химических процессов в живых организмах привело к появлению биохимии. Естественная связь химии с сельским хозяйством дала агрохимию. Контакты химии с геологией породили геохимию. Проникновение в космос ознаменовалось рождением космохимии и т. д. Словом, на вечнозелёном древе химии разрослось множество новых могучих и плодоносящих ветвей. Богатые урожаи всё новых плодов — удивительных и неожиданных — собирает с этого ветвистого древа техника.
Изучение отдельных важных для техники классов веществ тоже выделило новые самостоятельные «химии» — химию полимеров, химию полупроводников, нефтехимию...
Надёжные стимулы развитию химии практика давала всегда. Уже в середине XVIII в. великий Ломоносов имел право сказать: «Широко распростирает химия руки свои в дела человеческие... куда ни посмотрим, куда ни оглянемся — везде обращаются перед очами нашими успехи ее прилежания». В наши дни эти слова ещё более справедливы...
Никогда ещё за всю свою многовековую историю «выход» химии в жизнь и практическую деятельность людей не был столь разносторонним. Наш век можно назвать веком химии с неменьшим основанием, чем веком атома или веком кибернетики. В сущности почти всё материальное содержание человеческой цивилизации состоит сегодня так или иначе из продуктов химии, начиная с малого и кончая большим. В самом деле... Мы носим одежду и обувь, сшитые из синтетических тканей и искусственной кожи, а окрашены они химическими красителями. Мебель и посуда, бумага и лекарства делаются с помощью химии. Без неё не было бы ни фотографии, ни кино, ни телевидения. Но химия не только изменила и украсила наш быт. Она создала материалы, из которых конструируют самолёты и автомобили, электронно-вычислительные машины и синхрофазотроны, протезы суставов и кровеносных сосудов. Химии принадлежит почётное место и среди наук, подготовивших возможность межпланетных путешествий: она дала топливо и конструкционные материалы ракетам и космическим кораблям. Недаром вещества и материалы, которые рождаются в результате чудесных химических превращений, иногда называют «второй природой».
Этой «второй природе» ещё расти и расти! Перед человечеством стоит немало трудных — глобальных — проблем; обеспечение всех людей высококалорийной пищей, хорошей одеждой и удобным жильём, сохранение окружающей природной среды и рациональное использование всех природных ресурсов... Само собой разумеется, что для их разрешения необходимо прежде всего более справедливое, более совершенное социальное устройство общества на всём земном шаре, подобное тому, которое создано в Советском Союзе и других социалистических странах.
Материальное же осуществление подобных программ уже сегодня требует всё новых источников энергии, всё новых материалов, всё новых знаний. Многое ещё будет открыто и изучено химией. Вместе с физикой
и биологией, вместе с электроникой и кибернетикой, вместе со всеми другими науками химии предстоит ещё внести огромный вклад в будущее человечества.
Об энциклопедиях Мы живём в век всё более бурного накопления новых знаний. В трудном восхождении к вершинам науки на исследователей обрушивается лавина, ими же самими вызванная. Число книг и журналов растёт с поистине сказочной быстротой. К началу XIX в. во всём мире выходило около 100 научных журналов, к 1850 г.— около 1000, к 1900 г.— 10 000, а в 1964 г.—100 000... Как же отыскивать нужные сведения в этом половодье информации? Создаётся парадоксальное положение: согласно крылатому выражению, сегодня «легче открыть новый факт или вывести новую формулу, чем удостовериться в том, что они еще не были открыты или выведены раньше». Вот почему всевозможные энциклопедии служат «спасательными кругами» в океане информации.
Слово «энциклопедия» по происхождению греческое. В буквальном переводе оно означает: «круг знаний». В энциклопедиях концентрируются наиболее достоверные и существенные сведения, добытые человечеством за всю предшествующую историю цивилизации. На 10—20 лет — это миг в истории человечества, хотя и большой период в жизни одного поколения (!),— энциклопедии фиксируют достигнутый уровень знаний, помогают узнать, что уже открыто и изучено. В наши дни энциклопедиями систематически пользуются и учёные, и политические деятели, и писатели, и рабочие, и студенты... Заглядывают в энциклопедии и школьники — порою по необходимости (для подготовки к экзамену, сочинению, докладу), порою просто из любознательности. Но такому читателю, конечно, далеко не всё доступно в обычных энциклопедиях. И не только потому, что ещё не пройдены «азы» науки. Труден бывает и самый стиль изложения предмета в подобных изданиях; насыщенный информацией и предельно конспективный, он рождён потребностью сказать очень многое очень малым числом слов.
Если энциклопедия содержит знания по всем отраслям науки и практики, её называют универсальной; если в одной какой-либо области (химия или физика, музыка или театр и т. д.) — отраслевой, или специальной. Самым известным примером универсальной энциклопедии прошлого служит французская «Энциклопедия, или Толковый словарь наук, искусств и ремесел» — крупнейший памятник просветительной философии XVIII в. В нашей стране универсальные энциклопедии издавались неоднократно — таков, например, Энциклопедический словарь Брокгауза и Эфрона (82 основных тома и 4 дополнительных, 1890—1907 гг.). Таковы и три издания Большой советской энциклопедии (третье завершится в 1978 г.), и её младшие соратники на поприще идейно-философского воспитания и научного просвещения — Малая советская энциклопедия и Энциклопедический словарь. Такова хорошо знакомая нашему юному читателю Детская энциклопедия. А среди специальных энциклопедий есть и Краткая химическая в пяти томах (1961—1967 гг.).
Различным бывает не только содержание научно-справочных изданий, но и план их построения. В Большой и Малой советских энциклопедиях, в Краткой химической энциклопедии и в подавляющем большинстве других отраслевых термины (статьи) расположены по алфавиту — от «А» до «Я». Это позволяет выстроить в единый ряд огромное множество понятий-терминов (в 3-м издании БСЭ их около 100 тысяч). Это даёт и наиболее простой и экономный способ отыскать любую информацию. В других энциклопедиях, как в Детской, материал расположен не по алфавиту, а в соответствии с классификацией широких областей научного знания. «Земля», «Мир небесных тел. Числа и фигуры», «Вещество и энергия», «Растения и животные», «Техника и производство» — так называются отдельные тома Детской энциклопедии. И в каждом её томе статьи расположены тоже не по алфавиту, а по принципу собирательной систематизации. Для информационно-справочных целей такой способ расположения статей менее удобен. Однако он даёт более целостную картину
Об этой книге
какой-либо отрасли науки или области человеческой деятельности. Для очень юного читателя, постигающего целое в самых общих его чертах, такое построение педагогически оправдано.
Книга, которую вы сейчас берёте в руки, это — специальная, отраслевая Энциклопедия. Но не совсем обычная. В чём же её своеобразие?
Химия нашего времени, как вы только что убедились, необъятная область человеческого знания. Наша книга посвящена неорганической химии. Вот какое определение даётся сегодня этой дисциплине: «Неорганическая химия — наука о химических элементах и образуемых ими простых и сложных веществах (кроме соединений углерода, составляющих, за немногими исключениями, предмет органической химии)».
Все термины в этой книге, содержащие основной массив информации по неорганической химии, расположены, как и в обычных энциклопедиях, в алфавитном порядке — от «<Азота» до «Эрбия». Однако три большие статьи — «Атом», «Закон Менделеева» и «Химия служит человеку» — идут вне алфавита. Обобщающие и пространные, они служат фундаментом всей книги. Статьи «Атом» и «Закон Менделеева» открывают издание. И прежде чем начать странствия по алфавиту, стоит внимательно ознакомиться с этими ключевыми статьями. Они приводят читателя из глубин истории химии в её сегодняшний день. А статья «Химия служит человеку» завершает эту Энциклопедию, служит окном из мира химии в широкий мир созидательной человеческой деятельности.
Что же представляет собой основная — алфавитная — часть Энциклопедии «Неорганическая химия»? В ней описаны все известные на сегодня химические элементы — как в самостоятельных статьях («Бром», «Железо», «Технеций»...), так и в групповых («Актиноиды», «Галогены», «Платиновые металлы»...). Отдельные статьи посвящены и наиболее важным соединениям элементов («Аммиак», «Вода», «Серная кислота», «Углерода двуокись»...). Важнейшие технические продукты и материалы тоже описаны самостоятельно («Бетон», «Минеральные удобрения», «Стекло»...). Сведения о многих других веществах заключены в статьях о самих элементах. Вы найдёте по алфавиту и объединяющие статьи («Металлы», «Неметаллы», «Карбонаты», «Нитраты»...). Об элементах в природе, об их распространении на Земле, об источниках сырья, которыми располагает человечество, рассказано во многих других статьях книги («Атмосфера», «Биосфера», «Земная кора», «Минералы»...).
Статьи, сходные по теме (например, об отдельных химических элементах), написаны по одному плану. И это тоже один из важнейших энциклопедических принципов. Ведь только при таком однотипном — унифицированном — изложении нетрудно быстро найти нужную справку.
В книге вы найдёте и биографии ряда выдающихся учёных. Биографии (и портреты учёных) мы поместили не в алфавитном порядке, а внутри наиболее подходящих по теме статей. (Так, биография английского физика и химика Дж. Дальтона дана в статье «Атом», советского геохимика А. Е. Ферсмана — в статье «Минералы», советского металлурга И. П. Бардина— в статье «Чугун».) Мы, конечно же, не могли исчерпать список всех, кто оставил заметный след в истории неорганической химии.
При отборе материала для этой Энциклопедии мы стремились отразить на её страницах те общие идеи, веяния, направления, которые отличают сегодняшнюю химию в целом, равно и её неорганическую ветвь. Среди направлений её нынешнего развития всего важнее, пожалуй, три.
...Углубление связи химии с физикой — совместно они дают единый ключ к познанию вещества.
...Расширение влияния химий на ход современной научно-технической революции.
...Повышение роли химии в решении общенародной задачи — охраны природы.
О читателе
Все особенности в построении и содержании этой специальной Энциклопедии определяются тем, что она предназначена школьникам. Вооружить подрастающее поколение современными знаниями — эта нелёгкая задача решается в нашей стране одновременно разными путями. Один из них — создание учебников и пособий, всё более совершенных и в научном отношении, и по методам изложения. Другой — новая форма обучения — факультативные (необязательные) занятия, которые проводятся в старших классах по программам, выходящим за традиционные школьные рамки. Это расширяет кругозор подростков и помогает им прийти к сознательному выбору будущей специальности. Факультативные занятия получают всё более широкое распространение. Ими руководят не только учителя, но и энтузиасты из университетов, институтов, лабораторий, конструкторских бюро... Для таких занятий тоже нужны книги — монографии, научно-популярные издания, справочники. В новую жизнь сегодняшней школы должна внести свой вклад и наша Энциклопедия. Со временем к ней присоединятся и два других тома — один по физической химии, другой по органической и биологической химии. Ещё шире: этой книгой начинается издание общей серии — «Энциклопедия школьника»,— посвящённой разнообразным дисциплинам.
Итак, книга «Неорганическая химия» адресована тем подрастающим естествоиспытателям, для которых химия уже в школе стала любимым предметом. Иными словами, наш предполагаемый молодой читатель это тот, кто уже сделал выбор своей будущей специальности. Ею станет химия! Мы надеемся, что Энциклопедия принесёт пользу педагогам. И несомненно, что она будет настольной книгой на занятиях всевозможных химических кружков. Мы хотим, чтобы она стала верным спутником всех, кто пленён древней и вечно юной наукой — химией, проникающей в тайны природы и служащей благу человеческого общества.
В статьях химической энциклопедии школьника мы старались сочетать научную строгость и насыщенность информацией с доступным, образным и увлекательным изложением. Конечно, маленькие статьи чисто справочного характера написаны лаконично и сухо. Однако в сравнительно обширных статьях мы уходили от лаконизма и сухости обычных энциклопедий, дав в одном случае интересный пример, в другом— нешаблонное раскрытие темы, в третьем — сосредоточивая обильные сведения в иллюстрациях. Мы стремились к тому, чтобы статьи читались легко и привлекали внимание к главному, даря читателю радость познания на пути к самостоятельным открытиям. Так нам хотелось. К этому мы стремились. А как получилось на деле? Советы и замечания читателей помогут нам в следующих книгах школьной серии решить поставленные задачи точнее и совершеннее.
В создании этой Энциклопедии участвовали известные учёные. Ответственный редактор всего издания — Михаил Алексеевич Прокофьев, член-корреспондент Академии наук СССР, министр просвещения СССР. Он первым в нашей стране начал исследования по синтетической химии нуклеотидов, его работы способствуют развитию молекулярной биологии, генетики, медицины.
Главный редактор книги «Неорганическая химия»—Иван Павлович Алимарин, академик, крупнейший специалист в области современной аналитической химии. Пользуясь разработанными им методами, удаётся определять в веществах ничтожно малые примеси. Особо же чистые вещества необходимы для полупроводниковой, лазерной и других новейших областей техники.
Среди членов редколлегии — Сергей Александрович Погодин, профессор, заслуженный деятель науки и техники. Он автор основополагающей монографии по физико-химическому анализу, многих трудов по изучению металлических сплавов и по истории химии.
Над статьями Энциклопедии работал большой коллектив авторов — по преимуществу молодых учёных, сотрудников и питомцев Московского государственного университета имени М. В. Ломоносова.
ATOM
«Если бы в результате какой-то мировой катастрофы все накопленные знания оказались утерянными и к грядущим поколениям перешла только одна фраза, то какое утверждение принесло бы наибольшую информацию? Я считаю, что это—атомная гипотеза: все тела состоят из атомов—маленьких телец, которые находятся в беспрерывном движении, притягиваются на небольшом расстоянии, но отталкиваются, если одно из них плотнее прижать к другому».
Эти слова принадлежат современному американскому физику Р. Фейнману. Трудно с большей яркостью выразить колоссальную роль, которую играет в естествознании учение об атоме. Только слово «гипотеза» здесь неуместно: речь идёт о факте, который сегодня не вызывает ни малейших сомнений. Но эта уверенность — результат многовекового пути.
Рис. 1. Страницы из истории атома. а — Для древних греков вопрос о существовании атомов был проблемой чисто философской, умозрительной. Возьмём небольшую порцию вещества — капельку воды, песчинку или кристаллик поваренной соли — и разделим её пополам. Половинку снова разделим на равные части, половинку половинки — опять пополам и т. д. Наступит ли такой момент, когда очередную половинку нельзя будет никаким образом ни разбить, ни распилить даже в принципе? Демокрит утверждал, что такой момент наступит, и вот эту последнюю крупинку материи, которую уже никак нельзя раздробить, он назвал «атомос» — неделимый. Оппоненты Демокрита считали, что неделимых частиц материи нет и процесс деления можно продолжать до бесконечности.
б — Английский физик Дж. Томсон установил (1897 г.), что в состав атома входят электроны. Он полагал, что эти отрицательные частицы вкраплены в облако, имеющее положительный заряд. «Нечто вроде пудинга с изюмом», — так однажды высказался Томсон о строении атома.
в — В начале XX в. изучение радиоактивности показало, что одни химические элементы могут превращаться в другие. Так, при распаде радия образуются радон и гелий (альфа-частица). Эти открытия положили конец мифу о неразложимости атома. Оказалось, что «неделимый» всё-таки делится.
г — Эрнест Резерфорд и Нильс Бор (их портреты помещены на стр. 21 и 33) считали, что электроны в атоме движутся вокруг ядра по круговым или эллиптическим орбитам — как планеты в солнечной системе. Эта модель строения атома сыграла исключительно важную роль в развитии естествознания, но и она оказалась неточной.
12
БИОГРАФИЯ АТОМА
Учение об А. уходит своими корнями в глубокую древность. Около 2400 лет назад в трудах греческого философа Демокрита появилось слово «атомос», что значит «неделимый». Так он назвал мельчайшие частицы вещества, которые не могут дробиться.
Ломоносов, Лавуазье, Дальтон, Авогадро, Берцелиус, Канниццаро, Менделеев — вот имена прославленных учёных, каждый из которых вписал яркую страницу в биографию А. В результате их работ обнаружилась тесная связь между понятиями А. и химического элемента. Стала известной масса А., оказалось возможным оценить их размеры.
Новый этап развития учения об А. начинается с открытия явления радиоактивности. В 1896 г. французский химик А. Беккерель обнаружил самопроизвольное испускание ураном какого-то нового вида излучения. В 1898 г. также во Франции супруги М. и П. Кюри выделили из урановых руд два ещё более мощных излучателя — новые радиоактивные элементы полоний и радий. А в первые годы нашего века английские физики Э. Резерфорд и Ф. Сод-ди показали, что радиоактивность приводит к превращению одних элементов в другие. И ещё одно открытие, не менее выдающееся, было сделано на рубеже двух столетий: в 1897 г. английский учёный Дж. Томсон установил, что в состав А., представлявшегося дотоле мельчайшей частицей вещества, входят ещё более мелкие частицы —электроны. Эти великие открытия произвели переворот в естествознании — они положили конец мифу о неразложимости А.
1911 год ... Может быть, это самая важная веха в биографии А. В один из зимних дней этого года Эрнест Резерфорд, входя в лабораторию, торжественно объявил: «Теперь я знаю, как выглядит атом!». Этим восклицанием было ознаменовано рождение современного представления об А. как о системе, состоящей из очень небольшого по сравнению с размерами А. положительного ядра, в окрестности которого движутся отрицательно заряженные электроны.
С этого времени учение об А. развивается в двух хотя и связанных между собой, но всё-таки существенно разных направлениях. Одно из них — это углублённое исследование атомного ядра. Продолжая цепь своих блестящих исследований, неутомимый Резерфорд в 1919 г. провёл первую ядерную реакцию: бомбардируя А. азота ядрами гелия (а-частицами), он получил кислород. Годом позже Резерфорд ввёл понятие о протоне (ядре водорода) как о по-
Рис. 2. Половину кратчайшего расстояния между ядрами одноимённых атомов — в молекуле или'в кристалле простого вещества — принимают за «атомный радиус». При таком подходе атомы уподобляются касающимся друг друга шарам. На рисунке изображено расположение атомов в кристалле железа (кубическая объёмно-центрированная кристаллическая решётка).
ложительно заряженной элементарной частице, входящей в состав всех атомных ядер. Тогда же, в 1920 г., он предсказал существование нейтрона.
1932 год ознаменовался открытием этой нейтральной частицы, имеющей почти такую же массу, как протон. Нейтроны были экспериментально обнаружены англичанином Дж. Чэдвиком, а теоретические работы немецкого физика В. Гейзенберга, советского физика Д. Д. Иваненко и итальянского физика Э.Майорана показали, что нейтроны наряду с протонами формируют атомные ядра и определяют массу А.
Но в нашем рассказе об А. речь пойдёт главным образом о другом направлении исследований. Здесь в центре внимания оказывается не ядро А., а его электроны, образующие электронные оболочки. Эта сторона вопроса представляет первостепенный интерес для химии: именно строением электронных оболочек А. обусловлены свойства химических веществ, особенности их поведения при химических реакциях. Поэтому представления о строении электронных оболочек А. и историю формирования этих представлений мы должны рассмотреть особенно подробно.
Но прежде всего дадим общую характеристику А., которая послужит для нас стартовой площадкой.
13
МАССА И РАЗМЕРЫ АТОМОВ.
ЗАРЯД АТОМНОГО ЯДРА
Итак, ядра А. состоят из протонов и нейтронов, имеющих почти одинаковую массу, которая приблизительно равна одной углеродной единице (у. е.), то есть 1,67-10~24 г. Масса электрона гораздо меньше, чем масса протона или нейтрона; она составляет около 1/1840 у. е. Поэтому, естественно, масса А. в целом определяется практически полностью массой его ядра, а массовое число, представляющее собой суммарное число протонов и нейтронов в ядре, можно считать приблизительно равным массе А., выраженной в у.е. Эта величина меняется в широких пределах — от 1 у.е. у водорода до 250—270 у.е. у элементов, завершающих на сегодняшний день периодическую систему Менделеева.
Напротив, размеры А. определяются не ядром, а электронами, образующими его электронные оболочки. Однако электронные оболочки не имеют строго очерченных границ (подробнее этот вопрос рассмотрен ниже), и поэтому, говоря о «размерах» А., следует помнить, что такой подход достаточно условен. Размеры А. определяют по кратчайшим межъядерным расстояниям в молекулах или кристаллах простых веществ. Половина такого расстояния между ядрами одинаковых А. принимается за атомный радиус. При таком подходе А. уподобляются касающимся друг друга шарам (рис. 2).
Приведём атомные радиусы для некоторых элементов; величины радиусов принято выражать в ангстремах (1 А=10”8 см).
Элемент Атомный радиус Элемент Атомный радиус Элемент Атомный радиус
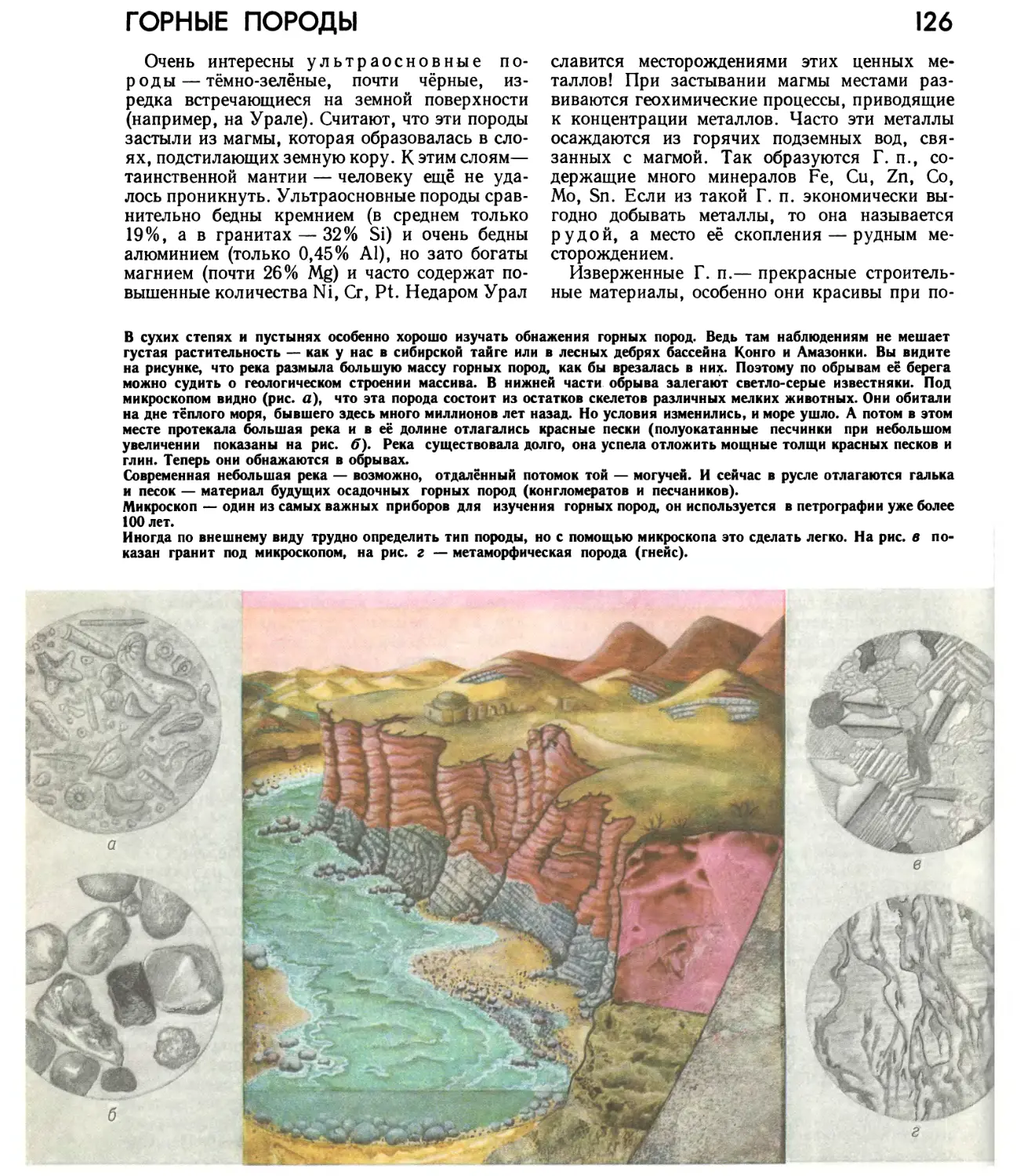
— Н 0,37
Li 1,55 Be 1.13 F 0,64
Na 1,89 Mg 1,60 С1 0,99
К 2,36 Са 1,97 Вг 1.14
Rb 2,48 Sr 2,15 I 1,33
Cs 2,68 Ba 2,21 — —
Из таблицы видно, что атомные радиусы — это величины порядка 10-8 см. В то же время радиусы ядер близки к 10”12 см. Следовательно, атомное ядро приблизительно в 10 000 раз меньше, чем А. в целом.
Важнейшей характеристикой А., обусловливающей его индивидуальность и принадлежность к данному элементу, является заряд ядра Z. Поскольку нейтроны — незаряженные частицы, величины Z определяются числом входящих в ядро протонов. Заряд протона численно равен заряду электрона, и поэтому Z удобно выражать в электронных единицах (1 электронная единица, то есть заряд электрона, равна 1,6-10"19 кулона). Выраженный таким образом заряд ядра представляет собой целое положительное число, которое обычно приводят без указания размерности. Величина Z совпадает с порядковым номером
элемента в периодической системе Менделеева (с атомным номером).
При одинаковом заряде ядра массовые числа А. могут различаться. Это значит, что в атомном ядре при одном и том же числе протонов может быть разное число нейтронов. Такие разновидности А. с одинаковыми Z, но неодинаковыми массовыми числами называются изотопами. При одном и том же заряде атомных ядер различие массы ядер изотопов на несколько у.е. не сказывается на строении электронных оболочек. Вместе с тем, как мы уже отмечали, химические и многие физические свойства А. зависят именно от строения электронных оболочек; поэтому эти свойства одинаковы или очень близки для всех изотопов данного элемента. Наибольшие различия в свойствах наблюдаются для трёх изотопов водорода: «лёгкого» водорода — протия, дейтерия и трития, массовые числа которых, соответственно, равны 1, 2 и 3.
В нейтральном А. число электронов равно заряду ядра Z. Но А. могут терять электроны, приобретая положительный заряд и превращаясь в катионы. Для отрыва от А. одного или нескольких электронов необходима затрата энергии, называемая энергией ионизации. Так, например, для превращения А. натрия в катион Na+ требуется 5,1 электрон-вольт (эв), то есть 8,2-10"19 джоуля (дж), а для превращения А. магния в катион Mg2+, идущего с отрывом двух электронов, 22,6 эв (36,2-10"19 дж).
Наряду с этим некоторые нейтральные А. могут присоединять электроны, превращаясь в отрицательно заряженные анионы. Такой процесс происходит с выделением энергии, величина которой характеризует сродство к электрону. Например, присоединение электрона к F с превращением его в ион F" сопровождается выделением 3,5эв(5,6- 10"19дж).
АТОМНЫЕ СПЕКТРЫ
И ПЛАНЕТАРНАЯ МОДЕЛЬ АТОМА
При прохождении электрического тока через разреженный аргон в разрядной трубке появляется красивое сине-голубое свечение. Неоновая разрядная трубка даёт красный цвет, а криптоновая — белый. Эти эффекты, которые знакомы каждому, кто видел световые рекламы, обусловлены атомными спектрами испускания. Такой спектр мы можем наблюдать и при нагревании вещества в пламени газовой горелки. Например, соли натрия окрашивают пламя в жёлтый цвет, причём возникает излучение со строго определённой длиной волны.
Белый свет содержит, как известно, цветные лучи, которым соответствуют всевозможные длины волн в диапазоне от 7,6-10~6 до4-10"6 см.
14
Рис. 3. Атомы каждого элемента обладают характеристическим спектром испускания. Мы приводим здесь примеры таких спектров.
Но если пропустить белый свет через раскалённые пары натрия, то окажется, что жёлтые лучи с той самой длиной волны, которая появилась в спектре испускания натриевых солей, почти полностью исчезли. Так возникает атомный спектр поглощения.
Говоря точнее, атомным спектром называется совокупность световых волн, испускаемых или поглощаемых А. данного элемента. Характерная особенность атомных спектров состоит в том, что они содержат лишь некоторые избранные длины волн X и, соответственно, частоты v (длина волны света и его частота связаны соотношением где с — скорость света). Иными словами, такой спектр оказывается линейчатым — он представляет собой дискретный набор линий, специфичный для каждого элемента (рис. 3). Этим обстоятельством широко пользуются в химическом анализе.
Ещё в середине XIX в. были хорошо изучены атомные спектры многих элементов. В 1868 г. в спектре Солнца была обнаружена линия, не соответствовавшая ни одному из ранее известных элементов; так был открыт гелий, позже обнаруженный на Земле. Однако происхождение линейчатых атомных спектров, причины их возникновения ещё долгое время оставались совершенно неясными.
Важной предпосылкой, которая впоследствии позволила объяснить спектры и создать теорию строения А., явилась фундаментальная физическая концепция, родившаяся в первые годы XX столетия. Оказалось, что испускание света происходит отдельными порциями —
квантами. Только таким образом можно было истолковать результаты, полученные немецким учёным М. Планком при исследовании излучения так называемого абсолютно чёрного тела. В физике появилась новая универсальная постоянная — постоянная Планка (h~ =6,62* 10“27 эрг-сек), с помощью которой выражается квант энергии (E=/iv).
Квантовый характер световой энергии с особенной чёткостью продемонстрировал в 1905 г. великий физик А. Эйнштейн. После его работ стало ясно, что, подобно тому, как вещество состоит из мельчайших частиц — атомов, так и световой луч состоит из фотонов, поведение которых во многом сходно с поведением частиц. Природа света оказалась двойственной: он представляет собой одновременно волну (это было известно издавна) и поток фотонов (это показал Эйнштейн).
Погружаясь в мир атомов, мы ещё столкнёмся с этой двойственностью (волна — частица): окажется, что электрон — типичная частица! — обладает в то же время свойствами волны (см. стр. 21). Именно волновые свойства электрона лежат в основе современного представления об А. Но физикам, работавшим в начале XX столетия, был известен лишь вывод Эйнштейна: свет — типичная волна! — представляет собой поток частиц — фотонов.
Это был очень важный вывод, и он позволил датскому учёному Н. Бору создать в 1913 г. во многом верную, а потому и поныне сохранившую определённое значение теорию строения А.
Джон ДАЛЬТОН. Впервые научно обосновал представления древних атомистов.
Английский физик и химик Джон Дальтон родился 6 сентября 1766 г. в Иглсфилде. Сын бедного ткача, он не имел достаточных средств получить образование в каком-либо из учебных заведений. Но жажда знаний была у юноши необычайной, он поглощал сотни книг по разным научным дисциплинам и в конце концов стал весьма эрудированным учёным. С 1793 по 1800 гг. Дальтон преподавал математику и естественные науки в Манчестерском колледже, затем давал частные уроки и читал публичные лекции.
Основные исследования Дальтона связаны с изучением физических свойств газов. Почти одновременно с Ж. Л. Гей-Люссаком он открыл (в 1802 г.) закон теплового расширение газов, а в 1803 г. установил законы давления газовых смесей (законы Дальтона).
Его занимала проблема состава газообразных химических соединений. Изучая метан и этилен, учёный обнаружил, что в этих соединениях количества углерода, приходящиеся на одно и то же количество водорода, находятся в отношении 1 : 2. Такие же простые кратные отношения он нашёл для окислов азота, для окиси и двуокиси углерода. На основе своих наблюдений Дальтон сформулировал закон кратных отношений, хорошо известный вам из учебника.
Этот закон лёг в основу химической атомистики — первой общей теории состава химических соединений — и впервые позволил дать представление об относительных атомных весах элементов. Так натурфилософское учение древних атомистов впервые получило научное обоснование. Для определения атомных весов Дальтон принял за единицу вес атома водорода. Учёный впервые употребил для обозначения химических элементов специальные символы: например символ Q обозначал водород, Q) — азот, •—углерод, Q —кислород и т. д. Сочетанием этих символов Дальтон изображал атомный состав соединений: например, —окись углерода, О®О— двуокись углерода. Эти знаки, не удобные для набора, были впоследствии заменены химическими знаками Берцелиуса. Сущность своей атомной теории Дальтон изложил в книге «Новая система химической философии».
На лекциях Дальтон пользовался как наглядным пособием моделями атомов в виде деревянных кубиков, окрашенных в различные цвета. Это дало повод его слушателям определять атомы, как «...разноцветные деревяшки, выдуманные доктором Дальтоном». Кстати сказать, Дальтон страдал недостатком зрения — «слепотой» по отношению к некоторым цветам спектра. Эту слепоту он описал в 1794 г., и с тех пор она известна под названием дальтонизма.
В 1822 г. Джон Дальтон был избран членом Лондонского королевского общества, а в 1830 г.— иностранным членом Парижской академии наук. Эти высокие отличия он получил за свои фундаментальные открытия. Умер он 27 июля 1844 г. в Манчестере. Его химическая атомистика нашла развитие в трудах многих химиков первой половины XIX в., в особенности же Я. Берцелиуса.
В модели А., предложенной Резерфордом, атомное ядро уподоблялось Солнцу, электроны— планетам, каждая из которых движется по круговой или эллиптической орбите, и весь А. в целом оказывался разительно похожим на солнечную систему. Бор существенно дополнил планетарную модель, предположив, что электроны в А. могут двигаться не по любым, а лишь по некоторым «разрешённым» орбитам и что электрон, находясь на такой орбите, сохраняет постоянный запас энергии.
По теории Бора, электрон в А. ведёт себя аналогично шарику, находящемуся на одной из ступенек лестницы (рис. 4). Во-первых, энергия шарика — здесь речь идёт о потенциальной энергии — выражается формулой V5n=mgnH, где Н — высота одной ступеньки, а п — номер ступеньки. Таким образом, допустимые значения энергии образуют дискретный (прерывный) ряд. Во-вторых, переход шарика с одной ступеньки на другую при движении вверх будет сопровождаться затратой, а при движении вниз — освобождением неко
торой порции энергии. Например, для переноса шарика с первой ступеньки на третью ему нужно сообщить энергию U3—и±=2 mgH. Наконец, в-третьих, чем ниже находится шарик, тем устойчивей его положение. Чтобы убедиться в этом, представим, что лестница начинает шататься — землетрясение! Колебания настолько велики, что шарик, упавший вниз, может снова оказаться заброшенным на одну из верхних ступенек. Но чем ниже данная ступенька, тем больше вероятность обнаружить на ней шарик по окончании землетрясения.
А теперь рассмотрим на фоне этой аналогии результаты, к которым приводит теория Бора. Сосредоточим пока своё внимание на А. водорода. Бор установил, что в этом простейшем из А. единственный электрон может приобретать лишь такие значения полной энергии, которые отвечают формуле Еп= — 2^2^-- , где т — масса электрона, е — его заряд, h — постоянная Планка. Допустимые значения энер
16
гии — энергетические уровни — обратно пропорциональны п2, но они отрицательны, и поэтому с увеличением п энергия увеличивается. Натуральное число п, представляющее собой номер энергетического уровня, называется главным квантовым числом. Последовательные энергетические уровни иногда обозначают буквами Д’, £, Л4, JV ....
Состояние А., при котором его энергия минимальна, называется основным; прочие со-
Рис. 4. Электрон в атоме подобен шарику, находящемуся на одной из ступенек лестницы. Поток белого света, пронизывающий на рисунке эту условную лестницу, содержит лучи с различными частотами — им соответствуют разные цвета. Зелёный луч отдаёт строго определённую порцию энергии (квант) электрону, который при этом перебрасывается вверх. При переходе электрона с верхней ступеньки на нижнюю излучается квант определённой частоты; в той ситуации, которая символически изображена на рисунке, при этом возникает оранжевый луч.
стояния носят название возбуждённых. Изменение состояния А. водорода сопровождается скачкообразным изменением энергии его электрона, а следовательно, и А. в целом.
Если, например, электрон перешёл с уровня энергии Еп на более низкий уровень Ek> то А. потерял энергию, рацную &Е=Еп—Ek. Эта энергия испускается в^ окружающее А. пространство в виде фотона, то естьЪ виде «порции» излучения, которое характеризуется частотой v. Энергия фотона равна hv — это и есть квант. Таким образом &Е=Еп—Ek=hv, от-куда вытекает, что v= . Следовательно, частота определяется номерами уровней п и k\ поэтому будем обозначать её vnk. Итак,
При переходе электрона с более низкого на более высокий уровень эта формула остаётся в силе, но фотон, несущий квант энергии hvnk, в таком случае не испускается, а наоборот поглощается.
Такое поведение электрона в А. водорода позволяет объяснить его атомный спектр. При облучении водорода белым светом, содержащим лучи с различными частотами, А. переходят в возбуждённые состояния; при этом поглощаются кванты энергии, но не любые, а лишь те, которые равны разности между энергиями каких-либо уровней. Так возникает линейчатый спектр поглощения. Возникновение спектра испускания обусловлено переходом возбуждённых А. в состояния с более низкой энергией; при этом излучаются кванты со специфическим набором частот. В спектре водорода, изображённом на рис. 3, видна оранжевая линия, которая соответствует переходу электрона с третьего уровня на второй, то есть частоте v32, а также зелёная и синяя линии, соответствующие частотам v42 и у52. Линии, которые отвечают другим возможным переходам, оказываются вне диапазона видимых лучей (в частности, частоты переходов в основное состояние лежат в ультрафиолетовой области спектра).
Как уже говорилось выше, состояние электрона, характеризующееся определённой энергией, Бор связывал с движением по одной из «разрешённых» орбит. Для А. водорода эти орбиты считались круговыми и были вычислены их радиусы. Однако последующие исследования показали несостоятельность такой точки зрения. Выяснилось, что движение электрона в А. нельзя связать с какой-либо геометрически фиксированной орбитой. Но в этом и нет нужды! Мы видели, что для объяснения атомного спектра водорода вполне достаточно представления о дискретных энергетических уровнях —
17
никакие орбиты в этом объяснении не фигурируют.
Мы ещё вернёмся к этому вопросу, а пока посмотрим, что дала теория Бора и её дальнейшее развитие для установления строения А. более сложных, чем А. водорода,— для многоэлектронных А.
КВАНТОВЫЕ ЧИСЛА
И МНОГОЭЛЕКТРОННЫЕ АТОМЫ
Детальный анализ свойств А. показывает, что для описания состояния электронов одного главного квантового числа п недостаточно. Необходимо добавить ещё три характеристики состояния, ещё три квантовых числа, обозначаемых символами /, mL и tns.
Число I (орбитальное квантовое число) приобретает значения от 0 до п—1. Например, если состояние электрона характеризуется значением лг 2, то I равно 0 или 1. Состояния с /=0, 1,2,3, 4 принято обозначать, соответственно, буквами s, р, d, /, g.
Число (магнитное квантовое
число) позволяет продолжить классификацию электронных состояний.Если орбитальное квантовое число равно/, то mz=0, ±1, ±2, ..., ±/. Таким образом, для s-состояния /nz=0. В случае p-состояний возможны три варианта: = = — 1, 0, 1; в случае d-состояний — пять вариантов: /пг =—2, —1, 0,1, 2 и т.д.
Наконец, число ms (спиновое квантовое число) для электрона, характеризующегося любым набором значений чисел п, / и /по равно л 1 х 1 либо либо — -у .
Конкретный смысл орбитального; магнитного и спинового квантовых чисел, к сожалению, трудно объяснить в рамках элементарных физических представлений. Упрощая очень сложную картину, которая в действительности не поддаётся наглядной трактовке, здесь можно воспользоваться сопоставлениями с некоторыми величинами, используемыми в классической механике. Орбитальное и магнитное квантовые числа I и mL можно связать с так называемым моментом количества движения электрона. Если считать, что электрон движется по круговой орбите со скоростью v, то этот момент М численно равен произведению количества движения электрона mv на его расстояние от ядра г и представляет собой вектор, направленный перпендикулярно вектору mv и вектору г. Как и энергия электрона, его момент количества движения приобретает лишь некоторые определённые, допустимые значения. Квантовое число I входит в формулу, определяющую возможные численные значения М, а через числоmt выражаются допустимые значения проекции вектора Л4 на некоторое направление. Два значения спинового квантового числа ms можно ассоциировать с двумя разными направлениями вращения электрона вокруг собственной оси.
Итак, каждое электронное состояние характеризуется определённым сочетанием значений четырёх квантовых чисел, и эту четвёрку
можно рассматривать как совокупность некоторых индексов, которые присвоены данному состоянию. Общая картина различных состояний электрона в Ал для трёх первых значений главного квантового числа приведена в таблице.
Электронная оболочка Орбитальная характеристика состояния Квантовые числа п 1 mt ms
1 (К) S 1 0 0 ± >/2
S 2 0 0 ± уг
2 (£) Р 2 1 —1 ±У2 2 1 0 ± >/2 2 1 1 ±'i
S 3 0 0 ±yt
3 (М) р 3 1 -1 ±?2 3 1 0 ±'2 3 1 1 ± </2
d 3 2 — 2 ± уг 3 2-1 ±уг 3 2 0 ± */2 3 2 1 ± у2 3 2 2 ± >/2
Каждая строка последней графы соответствует двум допустимым состояниям, отличающимся знаком спинового квантового числа. Таблицу нетрудно продолжить, переходя к большим значениям числа п,— закон её построения достаточно прост.
Приведенную таблицу удобно представить как систему клеточек, каждая из которых соответствует двум возможным состояниям электрона, отличающимся лишь знаком спинового квантового числа:
Для А. водорода эта система клеточек изображает множество возможных состояний единственного электрона; применительно к более сложным А. она может служить «обоймой», заполняя которую мы найдём строение многоэлектронного А. Для простоты будем пока говорить только об основных состояниях многоэлектронных А., то есть о состояниях с минимальной энергией.
Итак, нам предстоит распределить по данной системе клеточек электроны, входящие в состав сложного А. Руководящей идеей здесь послужит важный принцип, открытый в 1925 г. швейцарским учёным В. Паули: в А. не может быть двух электронов, находящихся в одинаковых
2 хэш
18
состояниях, то есть каждый электрон характеризуется своим собственным, индивидуальным набором четырёх квантовых чисел: п, /, mt и ms. Таким образом, в каждой клеточке нашей таблицы может разместиться пара электронов, для которых ms=±:1/2.
Электроны, которые находятся в состояниях с одинаковым значением числа м, называют принадлежащими к одной электронной оболочке*. Подобно энергетическим уровням в А. водорода, оболочки иногда обозначают буквами К, £, Л!, . Из принципа Паули выте-
кает, что каждая оболочка может содержать не более 2м2 электронов.
В первую очередь заполняются оболочки с наименьшими значениями п — им соответствует меньшая энергия электронов, а состояние с минимальной энергией наиболее устойчиво. Впрочем, как мы увидим дальше, электронные оболочки достаточно сложных по строению А. перекрываются (здесь имеется в виду перекрывание на энергетической шкале, а вовсе не в чисто геометрическом, смысле); поэтому заполнение оболочки с номером п+1 может начаться и тогда, когда оболочка с номером п ещё не заполнена.
Схематическое изображение распределения электронов по оболочкам для первых 36 элементов периодической системы Менделеева показано на рис. 5. Здесь обнаруживается ряд существенных обстоятельств, с которыми мы познакомимся, постепенно переходя от простых по строению А. к всё более и более сложным.
Оба электрона А. гелия принадлежат первой оболочке. Они отличаются лишь знаком спинового квантового числа ms. Про такие электроны говорят, что они спарены; их спины (от английского spin — вращение) антипараллель-ны, и они изображаются на схеме противоположно направленными стрелками.
Первая оболочка может вместить лишь два электрона: при п=\ существует только два возможных набора квантовых чисел. И поэтому
* К сожалению, в учебной и научно-популярной литературе нет единства в употреблении терминов, используемых при описании строения А. Так, совокупность электронов с одинаковым значением главного Квантового числа и часто называют «электронным слоем» или «энергетическим уровнем». Последний термин неудачен, поскольку, как будет видно из последующего изложения, при одинаковом значении и, но разном значении I электроны обладают различной энергией. Чтобы отразить последнее обстоятельство, часто вводят понятия «подуровня» или «подоболочки» для обозначения совокупности электродов с одинаковыми значениями п и I. Мы, однако, предпочитаем этими понятиями не пользоваться, сохраняя термин «подуровень» для описания расщепления энергетических уровней А. под влиянием внешнего поля (см. стр. 20).
в трёхэлектронном А. лития начинается заполнение второй оболочки, которая уже способна вместить восемь электронов. Заполнение этой оболочки продолжается в А. последующих шести элементов и завершается в А. неона (Z= 10) — последнего элемента 2-го периода.
Чтобы понять строение А. элементов, составляющих 2-й период системы Менделеева, необходимо сделать два важных дополнения к сказанному:
1. В многоэлектронных А., в отличие от А. воДорода, энергия электрона зависит не только от главного квантового числа м,но и от орбитального квантового числа Z; это следствие взаимного отталкивания электронов. В результате электроны, находящиеся в s- и р-состояниях, имеют разную энергию. Таким образом, вторая оболочка расщепляется на два энергетических уровня: более низкий 2s и более высокий 2р. Поэтому в А. бериллия присутствуют два спаренных электрона в s-состоянии и лишь в А. бора появляется электрон, находящийся в p-состоянии. Аналогично, третья оболочка состоит из трёх уровней: 3s, Зр и 3d, четвёртая — из уровней 4s, 4р, 4d, 4/, и т.д.
2. Электроны, находящиеся на одном энергетическом уровне (например, на 2р), стремятся иметь одинаковое значение спинового квантового числа ms, то есть для электронов с одинаковой энергией более выгодно состояние с параллельными спинами. Только тогда, когда исчерпаны все возможные варианты значения магнитного квантового числа mt (энергетический уровень заполнен наполовину), электроны начинают спариваться. Это важное правило, во многом определяющее магнитные свойства А. и их способность к образованию химических связей, установил в 1927 г. немецкий учёный Ф. Хунд.
Строение А. для элементов 3-го периода не обнаруживает каких-либо новых особенностей— здесь заполнение третьей оболочки идёт точно так же, как заполнение второй оболочки у элементов 2-го периода.
В строении элементов 4-го периода существенно сказывается то, что энергия 45-уровня несколько ниже, чем энергия Sd-уровня. Поэтому 19-й электрон калия (Z= 19) и 20-й электрон кальция (Z —20) находятся в s-состоянии, и лишь со скандия (Z=21) начинается заполнение Зй-уровня.
Далее показано, как выглядит последовательность энергетических уровней в многоэлектронных А.
ls<2s<2p<3s<3p <4s«3d <4р <5s«4d<5p<
<6s«5d«4f <6p<7s»6d«5f...
19
Отсюда видно, что четвёртая электронная оболочка «наползает» на третью, а пятая — на четвёртую; шестая же оболочка перекрывается не только с пятой, но и с четвёртой. Эти перекрывания определяют сложный характер заполнения оболочек атомов с большими порядковыми номерами (о заполнении электронных оболочек рассказано также в статье Закон Менделеева).
Понятно, что в образовании химических связей главную роль играют электроны внешней, так называемой валентной оболочки. Эти электроны наименее прочно связаны с ядром и, следовательно, наиболее подвижны. Вместе с тем строение внешних оболочек элементов, входящих в одну и ту же группу или подгруппу периодической системы, обычно вполне аналогично. Это и обусловливает периодичность свойств химических элементов. Таким образом, именно в особенностях строения А. находит объяснение периодический закон — основной закон химии.
Описанная система энергетических уровней А. напоминает этажерку весьма причудливой формы со строго фиксированными размерами полочек (рис. 6). Заполнение полочек начинается обязательно снизу. Если же А. вступает в химическое взаимодействие, то в первую очередь затрагиваются электроны верхних полочек.
«Электронная этажерка» на рис. 6 даёт наглядное представление о размещении электронов по энергетическим уровням в А. никеля. Эта этажерка соответствует той строке из схемы на рис. 5, которая относится к никелю. Эту строку можно записать и в сокращённой форме: ls22s22p63s23p63d84s2. Пользуясь уже знакомыми нам принципами строения электронных оболочек А., нетрудно расшифровать такую запись —"говорят, что она изображает электронную конфигурацию А. никеля. Часто в подобные записи электронных конфигураций включают только высшие энергетические уровни, которыми в основном определяются химические свойства элементов: в таком варианте записи конфигурация А. никеля будет выглядеть как 3d84s2.
Говоря о строении многоэлектронных А., мы всё время имели в виду А., находящиеся в основных состояниях, то есть в состояниях с минимально возможной энергией. Поглощая соответствующие кванты, А. могут переходить в возбуждённые состояния. Например, для А. гелия наряду с нормальной электронной конфигурацией 1s2 возможны конфигурации 1 $2$, ls2p — с возбуждением одного электрона, конфигурации 2s2, 2s2p — с возбуждением двух электронов. Изменением электронных конфигураций обусловлено возникновение линейчатых спектров многоэлектронных А.
А. углерода из основного состояния ls22s22p2 сравнительно легко переходит в возбуждённое состояние ls22s2p:\ в котором все четыре электрона валентной оболочки неспарены. Поскольку внешние неспаренные электроны способны спариваться с такими же электронами соседних атомов, это даёт возможность А. углерода образовать четыре ковалентные связи. Такая особенность углерода, непосредственно обусловленная строением его А., играет определяющую роль в органической химии.
Рис. 5. Схема, изображающая распределение электронов по энергетическим уровням для атомов первых 36 элементов периодической системы.
N II II I It
О II fl fit I
F II fl II II I
Ne fl II II fl II
II период
Na II fl IIIIII Mg II II IIIIII Al II It IIII II Si II II II ИII P II II IIIIII S II II IIIIII Cl II II .11IIII Ar II II IIIIII
К II If II II II Ca II II IIIIII Sc II II IIIIII Ti II II IIIIII V II fl II II fl Cr II II fl II fl Мп II fl II fl II Fe II II IIIIII Co II II IIIIII Ni II II IIIIII Си II II II fl II Zn II II IIIIII Ga fl II IIIIII Ge II fl IIIIII As II II IIIIII Se II -Il II II 1,1 Br II II fl II-II Кг II II IIIIII
II II II II I
Il II II II I I
II II II II I I I
fl II II II I I I I I
II II II II I I I I I
II IIIIIIIII I I I II IIIIII II II I I I II IIIIIIIIIIIII I II .11IIIIIIII II II fl II fl |l II II II II II fl II fl II II II II II fl II fl ll-ll II II II fl IIII fl fl IIII IIIIIIIIII II II IIII IIIIII |l II fl IIII II II fill IIII II II fl fl II fl IIII fl
2*
20
Теперь, когда читателю должна быть ясна картина строения электронных оболочек А., особо отметим, что в этой статье речь идёт главным образом о свободном, изолированном А. Если А. попадает в электрическое или магнитное поле, в его электронной структуре происходят изменения, энергетические уровни сдвигаются и расщепляются на подуровни. Последнее означает, что электроны, энергия которых в изолированном А. одинакова, могут иметь различную энергию в А., находящемся в электрическом или. магнитном поле. Такие изменения в строении А. возникают и тогда,
Рис. 6. В виде такой «этажерки» можно представить расположение электронов на энергетических уровнях атома никеля (1 s22s22pG3s23p63d84s2).
когда он оказывается в поле, создаваемом соседними А. Это обстоятельство играет важную роль при образовании химических связей.
ОСНОВЫ СОВРЕМЕННОЙ ТЕОРИИ
СТРОЕНИЯ АТОМА
Название этого раздела может вызвать недоумение. Как? Неужели всё то, что было сказано выше о строении А.,— лишь историческая справка, и только теперь начнётся рассказ об истинном положении дела? Неужели, разобравшись (может быть, не без труда) в сложной картине электронных состояний, мы так и не добрались до сути?
Поспешим успокоить читателя. Излагая бо-ровскую концепцию А., мы в основном говорили о тех её положениях, которые следует признать правильными с позиций современной науки. Однако эти положения необходимо вписать в рамки более общих, более последовательных представлений. При этом мы устраним ряд неточностей и противоречий.
Планетарная модель Резерфорда — Бора сыграла исключительно важную роль в науке и позволила объяснить многие явления. До сих пор она оказывается удобным средством описания свойств А., и в учебниках мы видим изображения миниатюрных «планетных систем», соответствующих тому или иному А. Действительно, эта модель представляется такой наглядной, законченной, логичной! К сожалению, логичность планетарной модели только кажущаяся ...
Вот как отзывались крупнейшие учёные о теории Бора вскоре после её создания:
«Если это правильно, то это означает конец физики как науки» (А. Эйнштейн, 1913 г.).
«Физика теперь снова зашла в тупик, во всяком случае для меня она слишком трудна, и я предпочел бы быть комиком в кино или кем-нибудь вроде этого и не слышать ничего о физике!» (В. Паули, 1925 г.).
И это говорят учёные, которые так много сделали для формирования взглядов на строение А.! Даже у самого Бора тогдашнее состояние учения об А. вызывало, как он выразился, «чувство грусти и безнадёжности».
В чём же противоречивость планетарной модели?
В соответствии с классической электродинамикой заряженный электрон, вращаясь вокруг ядра, должен непрерывно излучать энергию, то есть попросту терять её. И если это так, то электрон, притягиваемый ядром, неминуемо упадёт на него. Но А. существуют очень долгое время без всяких видимых изменений.
Гипотеза Бора в том и состояла, что, двигаясь по «разрешённой» орбите, электрон сохраняет свой энергетический запас неизменным, то есть не подчиняется классической теории. Но
Эрнест РЕЗЕРФОРД — основоположник современного учения о строении атома.
Английский физик Эрнест Резерфорд родился 30 августа 1871 г. в Новой Зеландии (деревушка Брайт-уотер) в семье мелкого фермера. Он был четвёртым из двенадцати детей и образование, вплоть до высшего, получал благодаря стипендиям. Когда в 1895 г. двадцатичетырёхлетний бакалавр Резерфорд по окончании Новозеландского университета приплыл из-за океана в Англию, получив право стать докторантом в знаменитой Кавендишской лаборатории, один кембриджский физик сказал: «Мы заполучили дикого кролика из страны Антиподов, и он роет глубоко!» Время показало, что этот «кролик» — атлетического сложения и бессонной работоспособности — сумел проникнуть в строение материи глубже всех своих современников. Он оставил около 350 научных работ. Не только физика, но и химия обязаны ему эпохальными открытиями. Недаром он, физик, стал лауреатом Нобелевской премии по химии (1908 г.).
Резерфорд обнаружил неоднородность радиоактивных излучений и установил существование альфа- и бета-лучей (1898 г.). Он построил вместе со своим учеником Ф. Содди теорию радиоактивного распада атомов и доказал, что этот распад — естественное превращение элементов (1901—1902 гг.). Он исследовал природу альфа-лучей и определил, что это — ионы гелия (1908 г.).
Резерфорд открыл в атоме массивную положительно заряженную сердце-вину— ядро—и построил планетарную модель атома (1909—1911 гг.). Он провёл в лаборатории первые ядерные реакции и показал возможность искусственного превращения элементов (1919 г.). Он дал имя «протон» ядру водорода и предсказал существование нейтрона (1920 г.).
Необыкновенная сила интуиции и научная отвага отличали творчество Резерфорда. А его редкостный дар учителя, щедрость мысли, требовательность и великодушие помогли ему создать мировую школу атомной и ядерной физики. К этой школе принадлежали и выдающиеся экспериментаторы, и великие теоретики. Среди его учеников и многолетних сподвижников были Нильс Бор (Дания) и Пётр Леонидович Капица (СССР).
«Вы счастливый человек, Резерфорд,— говорили ему,— вы всегда на гребне волны!» А он по праву отвечал: «Да! Но я-то и поднимаю эту волну, не так ли?..». Он вдохновлял своими идеями и своим примером целые поколения исследователей микромира — физиков и химиков всех стран. Президент Лондонского королевского общества Эрнест Резерфорд был и членом многочисленных иностранных академий, в том числе Академии наук СССР.
«Это великая штука — жизнь, я не хотел бы променять ее ни на что...»,— сказал он однажды. Он умер 19 октября 1937 г. и его прах был удостоен погребения в Вестминстерском аббатстве рядом с могилами Ньютона и Дарвина.
в то же время это движение рассматривалось в обычном механическом смысле, в частности радиусы орбит вычислялись с привлечением законов классической механики. Знаменитый английский физик У. Брэгг говорил по этому поводу, что, согласно теории Бора, физики должны были по понедельникам, средам и пятницам пользоваться старыми законами, а по вторникам, четвергам и субботам — новыми.
Почему электрон не может менять свою энергию постепенно, с постепенным изменением своей траектории? Что происходит с электроном в момент скачка, когда он находится между «разрешёнными» орбитами? И откуда, собственно говоря, берутся эти «разрешённые» орбиты, по которым электрон движется, не теряя энергии?
Планетарная модель не давала и не могла дать ответов на эти вопросы. Для химии наиболее существенно было то, что теория Бора не могла исчерпывающе объяснить возникновение химической связи, в частности связи между одинаковыми или близкими по свойствам А.
Но наука двигалась вперёд, и её развитие позволило устранить противоречия, о которых мы говорили. И сегодня планетарная модель выглядит лишь как далёкая предшественница современных представлений о строении А.
Поведение тел, которые мы наблюдаем в окружающем нас мире, описывается известными законами классической механики. Но для А. рамки этих законов оказываются слишком тесными. Здесь приходится использовать более общий подход, находящий отражение в квантовой (волновой) механике.
Один из наиболее существенных принципов квантовой механики состоит в том, что всякая движущаяся частица, в частности электрон, обладает свойствами волны. Задержимся здесь, чтобы оценить всю колоссальную значимость и необычность этого факта.
В классической механике нашими верными помощниками служат наглядные модели. Например, говоря о том, что волна — это распространение колебания, мы можем себе представить поверхность озера в ветреный день или струну, изогнувшуюся в виде синусоиды
22
(рис. 7). Но как наглядно представить себе, что движущийся электрон — это волна? Ответ на этот вопрос не слишком утешителен. В квантовой механике мы вынуждены отказаться от привычных наглядных представлений о движении тел.
Рис. 7. В каждый момент колеблющаяся струна имеет некоторую определённую форму. Эта форма может быть различной в зависимости от числа точек, остающихся неподвижными в процессе колебаний. Синусоиды, изображённые на рис. а, б, в, представляют собой как бы моментальные фотографии разных случаев колебания струны. Такая фотография окажется чёткой, если время съёмки — экспозиция, очень мало. При увеличении экспозиции изображение струны размоется и превратится в вытянутое облачко (г). Функции, описывающие колебания струны, подобны волновым функциям, описывающим поведение электрона в атоме.
Однако наличие волновых свойств у электрона — не подлежащий сомнению факт. Ещё в 1923 г. французский учёный Л. де-Бройль предположил, что всякую частицу, имеющую массу т и скорость и, можно уподобить волне, длина которой определяется формулой =hltnv. И в 1927 г. эта гипотеза получила блестящее подтверждение — была обнаружена дифракция электронов на кристаллических решётках. Дифракция — явление типично волновое; оно состоит в отклонении от закона прямолинейного распространения волн, когда на их пути встречается препятствие, соизмеримое с длиной волны. Следовательно, в этом опыте совершенно определённо была установлена волновая природа электрона. Более того, экспериментально измеренная длина волны оказалась в точном соответствии с формулой деБройля.
Для электрона массой 9,1-10“2Я г, движущегося в А. со скоростью около 10я см/сек, А — величина порядка 10~7—10“ 8 см; она близка к собственным размерам А., и именно поэтому волновые свойства электрона существенно отражаются на свойствах А. Как мы увидим дальше, последнее можно интерпретировать следующим образом: электрон в А. как бы «расплывается», превращаясь в «облако», не имеющее определённых границ.
Строго говоря, соотношение де-Бройля остаётся в силе и для тел, с которыми имеет дело классическая механика. Однако длины волн, с которыми ассоциируются эти тела в своём движении, так малы, что волновыми свойствами здесь можно пренебречь без всякого ущерба. Например, для пули массой 10 г, летящей со скоростью 660 м/сек, Х= 10“32 см. Эта величина исключительно мала по сравнению с размерами пули, что позволяет полностью пренебречь её волновыми свойствами. Если бы мы захотели изобразить облако, соответствующее движению пули, подобно тому, как это делается для электрона, то вид этого «облака» совпал бы в точности с траекторией полёта.
Здесь, однако, важно подчеркнуть, что квантовая механика в принципе применима к движению любых тел и, если мы не используем её, описывая процессы, подобные полёту пули, то только потому, что в этом нет никакой необходимости — тут отлично работает классическая механика, которая, таким образом, предстаёт как частный случай механики квантовой. Напротив, движение электронов в А. удаётся удовлетворительно описать только с использованием квантовомеханического подхода.
Движение тел и частиц как в классической, так и в квантовой механике описывается с помощью математических уравнений. И вот оказывается, что уравнение, которое на математическом языке передаёт поведение электрона в А.— дискретный характер энергетических уровней, волновые свойства электрона,— очень похоже на уравнение, описывающее волны в колеблющейся струне. В каждый момент колеблющаяся струна имеет некоторую опре
23
делённую форму. Эта форма может быть различной (рис. 7) в зависимости от числа точек, остающихся неподвижными в процессе колебаний; в физике такие точки называются узлами. Синусоиды, изображённые на рисунке, представляют собой как бы моментальные фотографии разных случаев колебания струны. Такая фотография окажется чёткой, лишь если время съёмки — экспозиция — очень мало. При увеличении экспозиции изображение струны размоется и превратится в вытянутое облачко.
Обозначим через и^х), и2(х), и3(х) ... функции, описывающие форму струны при разном числе узлов; х — это расстояние от начала струны до некоторой точки, а и(х) — это отклонение этой точки от положения равновесия в данный момент времени. Такие функции как раз и являются решениями уравнения, описывающего распространение колебаний, то есть волны в струне. Иными словами, неизвестное и(х), входящее в уравнение, приобретает значения цх(х), и2(х), и3(х) ...
В нашем изложении нет нужды выписывать это достаточно сложное уравнение (подобные уравнения, называемые дифференциальными, изучают в высшей математике). Нам важно установить лишь сущность аналогии А. и колеблющейся струны, и мы уже близки к нашей цели.
Если задать в А. некоторую систему осей координат, совместив её начало с атомным ядром, то поведение электрона можно описать с помощью волновых функций ф1(х, у, z), ф2(х, У, ?), Фз(х, у, z) ... Они получаются как решения волнового уравнения, содержащего ф(х, у, z). Это уравнение, как мы уже говорили, подобно уравнению колеблющейся струны, и роли функций ф(х, у, г) и и(х) в этих двух сходных уравнениях аналогичны.
К сожалению, физический смысл функции 4* (х, у, г) (короче, ф) разъяснить совсем не просто. Здесь-то и сказывается невозможность прибегнуть к наглядной модели. Лучший способ, отвечающий этой задаче, заключается в переходе к величине |ф|2 (знак модуля необходим потому, что волновая функция ф может приобретать и «мнимые» значения, выражаемые с помощью корней из отрицательных чисел). Эта величина представляет собой вероятность нахождения электрона в очень малом объёме в непосредственной близости от точки с координатами х, у, z. Поскольку вероятность нахождения электрона различна в разных точках пространства, говорят об электронной плотности, которая равна е |ф|2 и, следовательно, неодинакова в разных местах окружающего ядро электронного облака; сгу-
Рис. 8. Поскольку вероятность нахождения электрона в разных точках околоядерного пространства неодинакова, говорят о пропорциональной ей «электронной плотности», различной в разных местах окружающего ядро «электронндго облака». Сгустки облака соответствуют областям с максимальной вероятностью нахождения электрона, а разрежения — областям, где эта вероятность меньше. На рисунке показан вид облака для s-электрона.
стки этого облака соответствуют областям с максимальной вероятностью нахождения электрона, а разрежения — областям, где эта вероятность меньше (рис. 8). Таким образом, электрон в А. «расплывается», как колеблющаяся струна на фотографии, полученной при сколько-нибудь длительной съёмке. Разным решениям волнового уравнения — фь ф2, фз • • • соответствуют различные допустимые состояния электрона, то есть электронные облака разной формы (рис. 9).
Волновое уравнение выражает основной закон квантовой механики. Как и всякий фундаментальный закон, его нельзя вывести из более общих принципов. Волновое уравнение было открыто в 1926 г. австрийским учёным Э. Шредингером, а в анализе его физического смысла выдающаяся роль принадлежит работам немецкого учёного М. Борна.
Математическая запись уравнения Шредингера для электрона в А. имеет следующий вид:
Здесь Е — полная энергия электрона в A., U — его потенциальная энергия. В уравнение входит функция ?2(Ф). которая, как и функция ф, в конечном счёте зависит от координат электрона. Конкретное выражение этой функции можно записать только с помощью высшей математики, но это не так важно для нашего рассказа.
Теперь мы подошли к самому главному. Математический анализ уравнения Шредингера показывает, что оно имеет решения далеко не при всех значениях полной энергии Е, а лишь при некоторых избранных, отделённых друг от друга интервалами. Отсюда вытекает уже знакомый нам из теории Бора вывод: допустимые значения энергии электрона в А. образуют дискретный ряд. В случае А. водорода получается уже известная нам формула:
Р __ 2л2те1
Но не значит ли это, что подход с позиций волновой механики не даёт ничего нового по сравнению с представлениями Бора?
Давайте разберёмся. Принципиальное различие между теорией Бора и волновой механикой Шредингера состоит в том, что в первой дискретность энергетических уровней электрона постулируется, во второй — выводится из волновых свойств электрона, из двойственности: волна — частица. При этом дискретность допустимых значений энергии электрона в А. оказывается прямым следствием волновых свойств электрона.
Кроме того, в ходе проведенных рассуждений наше представление о поведении электронов в А. обогатилось очень важными, пусть менее наглядными, чем в теории Бора, но зато более правильными образами. В планетарной модели каждое устойчивое состояние электрона связано с геометрически фиксированной орбитой, а в соответствии с квантовой механикой устойчивое состояние характеризуется лишь некоторым видом электронного облака. Мы не можем проследить все этапы движения электрона и должны говорить лишь о вероятности обнаружения электрона в той или иной точке. Мысленно накладывая друг на друга всевозможные положения электрона, которые он занимает в разные моменты времени, мы приходим к образу электронного облака — электроны как бы «размазаны» в пространстве, что и приводит к неопределённости границ электронных оболочек.
Многие «роковые» вопросы, в которых безнадёжно путалась планетарная теория, в квантовой механике решаются автоматически. Так, например, вопрос о том, что происходит с электроном в процессе скачка, когда он находится в пространстве между «разрешёнными» орбитами, отпадает сам собой. Это удачно иллюстрирует рис. 10, который вместе с подписью мы позаимствовали из журнала «Химия и жизнь» (1969 г., № 1).
Неприменимость классического понятия траектории к случаю электрона, движущегося в А., отражена в п р и н-ципе неопределённости, сформулированном не
мецким физиком В. Гейзенбергом в 1927 г. Этот принцип выражается формулой:
Дх •
h
2ят
Здесь х — координата электрона, — составляющая его скорости, параллельная данной оси координат, Дх — неопределённость координаты, а Дух— неопределённость составляющей скорости. Произведение этих двух неопределённостей должно быть не меньше некоторой постоянной величины. Это значит, что чем точнее известно положение электрона в пространстве, тем более грубо мы можем оценить его скорость, и наоборот, при повышении точности измерения скорости от нас ускользнёт местоположение электрона.
4f т-1
4f т =0
Рис. 9. Электронное облако атома водорода в различных состояниях (рисунок сделан по снимкам, полученным при фотографировании специальных движущихся механических моделей).
2s т=0 Зр т = 1
3s т=0 5d т-0 5d т=1 6f т = 1
25
Советский учёный академик Л. Д. Ландау назвал открытие принципа неопределённости одним из величайших триумфов человеческого ума. «Этот принцип,— говорил Ландау,— противоречит всему тому, во что мы привыкли верить на основании своих ощущений, к чему мы привыкли с раннего детства. Мы привыкли к большим масштабам — атома же никто из нас не видел своими глазами. Поэтому мы не можем ощутить своим внутренним чутьём, как происходит движение в атоме, и тем не менее изучить это движение научными методами оказывается возможным. Открытие принципа неопределённости показало, что человек в процессе познания природы может оторваться от своего воображения, он может открыть и осознать то, что ему не под силу представить. В этом — величайшая заслуга принципа неопределённости».
Мы уже отметили, что для А. водорода решение уравнения Шредингера позволяет найти допустимые значения энергии электрона. Другой существенный результат состоит в том, что одному и тому же значению энергии электрона могут соответствовать разные волновые функции ф, то есть разные распределения плотности электронного облака. Иными словами, при одной и той же энергии электрон может находиться в разных состояниях; число таких состояний равно и2, а с учётом спина 2п2.
Картину возможных электронных состояний, каждое из которых характеризуется четвёркой квантовых чисел, мы уже рассматривали; она отражена в таблице, приведенной на стр. 17. Квантовомеханический подход позволяет добавить к этой картине конкретное изображение электронных облаков, соответствующих различным электронным состояниям (рис. 9). Вид облака определяется квантовыми числами и, Z и /Пр Как видно из рисунка, с увеличением квантовых чисел очертания электронных облаков становятся всё более причудливыми. Так, например, при переходе от 1=0 к 1=1 (то есть из s-состояния в p-состояние) облако теряет сферическую симметрию — вытягивается, разбиваясь на отдельные сгустки.
Итак, в случае простейшего из атомов — А. водорода — квантовая механика достигла существенных успехов и сумела объяснить в прекрасном согласии с экспериментальными данными все основные особенности поведения единственного электрона. Правда, картина оказалась достаточно сложной. Намного ли она усложнится, если в А. появятся два, три, четыре или даже несколько десятков электронов?
На этот вопрос можно дать два, казалось бы, взаимно исключающих друг друга ответа: и да, и нет! Да, если подойти к вопросу совершенно строго, и нет, если использовать некоторые существенно упрощающие дело приближения. Разберёмся в этом подробней.
В многоэлектронном А. электроны отнюдь не безразличны по отношению друг к другу.
Они испытывают взаимное отталкивание, и принимая во внимание сложный характер по ведения каждого электрона, учесть это оттал кивание совсем не просто. В итоге даже ДЛ5 двухэлектронного А. гелия (не говоря уже о( А., содержащих десятки электронов) не удаёт
Рис. 10. Наивная картина, которая возникает при по пытке «наглядно* изобразить переход электрона с орбить Бора л=3 на орбиту л=2. Однако понятие «траекторш электрона в атоме» ничему реальному не соответствуе* и должно быть отброшено, поскольку оно мешает понят! истинные законы движения электрона в атоме. Как ж* представить себе атом?
Вот один из возможных вариантов ответа на этот вопрос наиболее правильное и при этом наиболее наглядно* представление об атоме даёт система уравнений Шредин гера — конечно, для тех, кто сумеет сквозь неё «уви деть» атом.
26
ся получить точного решения уравнения Шредингера, а это значит, что мы не можем математически строго вывести из фундаментальных законов квантовой механики картину строения сложного А.
Выход из возникающего здесь тупика квантовая механика находит с помощью приближённых методов. В основе этих методов лежат приближения двух типов — математические и физические.
В прикладной, вычислительной математике применение приближённых методов — не редкость. Действительно, нет необходимости в строгом решении, если можно получить приближённый результат с любой требуемой степенью точности. Например, результат извлечения квадратного корня из числа 2 (то есть решение уравнения х2=2) нельзя представить в виде конечного числа. Но это нас нисколько не смущает, потому что не представляет труда вычислить значение У~2 с любым числом знаков после запятой.
Для А. гелия приближённые математические методы решения уравнения Шредингера (разумеется, гораздо более сложные, чем те приёмы, которые используются при извлечении квадратного корня) позволяют найти энергетические уровни с точностью, которая превышает точность экспериментальных спектроскопических измерений. Однако с увеличением числа электронов в А., даже при использовании самых совершенных электронных счётных машин, реально достижимая точность расчётов быстро падает. Вычислительные трудности оказываются настолько велики, что практически нет надежды получить таким путём достаточно точное решение уравнения Шредингера для А., содержащего хотя бы десяток электронов.
Гораздо более эффективны в подобных случаях физические приближения. Их сущность заключается в том, что расчёт или теоретический анализ проводят, пренебрегая одним’ или несколькими второстепенными физическими факторами. Хорошо известным примером такого приближения являются законы идеальных газов, которые не учитывают взаимного притяжения молекул. Недостаток физических приближений заключается в том, что очень часто они не позволяют достоверно определить величину ошибки, которая может возникать в результате использования данного приближения.
Основная трудность, которая возникает при анализе строения многоэлектронного А., состоит в том, что, строго говоря, мы обязаны рассматривать такой А. как единую систему, все части которой неразрывно связаны друг с другом. Это значит, что нельзя говорить об изменении состояния одного из электронов, считая состояния остальных электронов неизменными.
Строго говоря, нельзя... Ну а если всё-таки попробовать? Предприняв такую попытку, мы придём к так называемому одноэлектронному приближению; при таком подходе каждый электрон выглядит движущимся как бы в усреднённом поле остальных электронов. Сопоставление результатов, которые удаётся получить при использовании одноэлектронного приближения, с разнообразными опытными данными показывает, что это приближение достаточно хорошо передаёт общую, качественную картину строения многоэлектронного А.
Принцип Паули в том виде, как мы его сформулировали, уже содержит в себе основную идею одноэлектронного приближения — независимое рассмотрение состояния каждого отдельного электрона. Специфичность наборов квантовых чисел, описывающих электронное состояние, означает, что совокупность возможных состояний, найденная при анализе уравнения Шредингера для А. водорода, цожет послужить основой для построения системы энергетических уровней любого многоэлектронного А. Таким образом, описанные выше картины строения многоэлектронных А. также вытекают из основных законов квантовой механики (если считать, что принцип Паули является одним из них), правда, лишь при использовании одноэлектронного приближения.
В заключение коснёмся одного частного, но принципиально важного вопроса. Хорошо известна закономерность, выполняющаяся в каждой отдельной группе периодической системы: чем больше атомный номер элемента, тем слабее электроны внешней оболочки связаны с ядром. Поэтому активность щелочных металлов, связанная с потерей валентного электрона, возрастает от лития к цезию, а активность галогенов, обусловленная присоединением электрона, понижается от фтора к иоду. Часто говорят, что с увеличением числа электронных оболочек внешние электроны менее прочно связаны с ядром, потому что находятся дальше от него.
Правильно ли такое объяснение, если в действительности электрон не имеет фиксированной орбиты? Можно ли в таком случае говорить о расстоянии электрона от ядра?
Рис. 11. Мир атома поражает. Здесь всё выглядит необычно: исчезает граница между частицей и волной, электроны теряют свои траектории, а энергия дробится. Наглядная планетарная модель в свете строгой последовательной теории выглядит лишь как своего рода «проекция» картины, в действительности гораздо более сложной. В квантовой меха* нике, которая лежит в основе современного учения об атоме, мы вынуждены отказаться от привычных представлений о движении тел. Это и вдохновило художника на такое образное, фантастическое изображение атома. Нужно, однако, подчеркнуть, что здесь аллегория вовсе не равнозначна мистике, которой нет места в точной науке. «Мы привыкли к большим масштабам — атома же никто из нас не видел своими глазами. Поэтому мы не можем ощутить своим внутренним чутьём, как происходит движение в атоме, и тем не менее изучать это движение научными методами оказывается возможным... Человек в процессе познания природы может оторваться от своего воображения, он может открыть и осознать то, что ему не под силу представить»,— так говорил выдающийся советский учёный Л. Д. Ландау.
27
Ответ на эти вопросы состоит в следующем. Мы не знаем, где именно находится электрон в данный момент времени, но мы вполне можем говорить о вероятности нахождения электрона в том или ином месте околоядерного пространства. И вероятность того, что валентные электроны цезия или иода окажутся дальше от ядра, чем валентные электроны лития или фтора, гораздо больше, чем вероятность противоположной ситуации. Иными словами, электронное облако имеет максимальную плотность на таком расстоянии от ядра, которое тем больше, чем ниже находится элемент в соответствующей группе периодической системы. Поэтому геометрический подход вполне правомерен; однако важно помнить, что при этом подразумевается наиболее вероятное положение электрона.
Мир атома поражает. Здесь всё выглядит необычно: исчезает граница между частицей и волной, электроны теряют свои траектории, а энергия дробится...
Но этот мир существует — это доказано бесспорно. Однако его трудно понять, а поэтому в него трудно поверить, и иногда даже причастные к химии люди стараются отодвинуться подальше от новых представлений. Нельзя ли обойтись без всего этого? Нельзя ли
28
ограничиться простой и наглядной планетарной моделью?
Нельзя. Если мы хотим правильно судить о процессах, происходящих в А., о поведении А. при их взаимодействии, мы не имеем права выдавать за окончательный результат то, что на уровне достижений сегодняшнего дня следует считать лишь грубым приближением. Квантовая механика А. необходима химику — без нее он не в состоянии правильно описывать химические свойства, объяснять те тонкие эффекты, которые позволяет наблюдать современная экспериментальная техника. Она необходима и биологу, так как сегодня биология достигла «молекулярного уровня», то есть при изучении биологических процессов учитываются свойства и роль отдельных молекул, а эти свойства в конечном итоге определяются свойствами А. Не будет преувеличением сказать, что современное учение об А. оказывает существенное влияние на все разделы естествознания. И не только учёный, а каждый образованный человек должен знать о том, что наряду с миром, где планеты, спутники, самолёты, автомобили, люди, мячи и пылинки движутся по конкретным, наблюдаемым путям, существует «мир утраченных траекторий» — мир А.
/7. М. Зоркий.
«...Здравый человеческий рассудок, весьма почтенный спутник в четырёх стенах своего домашнего обихода, переживает самые удивительные приключения, лишь только он отважится выйти на широкий простор исследования».
Ф. Энгельс
(Маркс К. и Энгельс Ф., Соч., 2 изд., т. 20, с. 21)
ЗАКОН МЕНДЕЛЕЕВА
Металлы и силикаты, окислы и углеводороды, вода и белки... Как сильно различаются они по составу, свойствам, строению! Поистине удивительно многообразие веществ, из которых состоит окружающий нас мир. А если принять во внимание и химические соединения, которые не существуют в природе, но получены учёными в лабораториях, в списки уже известных веществ придётся включить миллионы наименований. И эти списки непрерывно расширяются...
В этом безбрежном море было бы невозможно ориентироваться, если бы не было в руках учёных и инженеров надёжнейшего компаса. Все вещества образованы лишь из нескольких десятков химических элементов, а сами элементы неукоснительно подчиняются единому закону. Этот важный закон природы — периодический закон,— открытый в 1869 году великим русским химиком Д. И. Менделеевым, служит одним из краеугольных камней фундамента, на котором зиждется химическая наука.
СУЩНОСТЬ ПЕРИОДИЧЕСКОГО ЗАКОНА
Современная формулировка периодического закона такова: свойства химических элементов (в образуемых ими простых и сложных веществах) находятся в периодической зависимости от величины заряда их атомных ядер.
Заряд атомного ядра Z (атомный номер) равен числу содержащихся в ядре протонов (см. Атом) и поэтому всегда представляет собой целую положительную величину (в единицах заряда протона). Именно эта величина определяет индивидуальность атома, его принадлежность к данному химическому элементу.
В соответствии с периодическим законом в ряду элементов, расположенных по возрастанию Z, обнаруживаются периоды. Каждый период, за исключением первого, начинается щелочным металлом и заканчивается инертным элементом:
Z I 2 3 ... 10 11 ... 18 19 ...
1-й период открывается не щелочным металлом, а водородом; впрочем, водород по некоторым свойствам подобен щелочным металлам (см. Водород).
1-й период содержит лишь два элемента, 2-й и 3-й периоды — по 8 элементов, 4-й и 5-й — по 18, 6-й — 32.
В 7-м периоде к настоящему времени известно 19 элементов (с атомными номерами от 87 по 105)*. Таким образом, ёмкости периодов неодинаковы: 1-й, 2-й и 3-й периоды принято называть малыми, все последующие — большими. В каждом периоде, начиная со второго, наблюдается закономерное изменение свойств элементов: постепенно ослабевают свойства, характерные для металлов, и, соответственно, усиливаются свойства, характерные для неметаллов.
О различиях между металлами и неметаллами сказано в статьях Металлы и Неметаллы. Различия эти касаются, конечно, как химических, так и физических свойств веществ. Но, приступая к изложению периодического закона, мы пойдём тем же путём, каким шёл сам Менделеев: проследим, как зависят от атомного номера именно химические свойства элементов (и свойства образуемых ими простых и сложных веществ).
Каковы же главные различия в химических свойствах металлов и неметаллов? Атомы металлов сравнительно легко отдают внешние (валентные) электроны; окислы и гидроокиси металлов имеют обычно основной характер; для металлов характерна способность замещать водород в кислотах с образованием солей. Наиболее активные металлы вытесняют водород даже из воды, образуя основания. Атомы неметаллов труднее отдают электроны, но легче присоединяют их, чем атомы металлов; окислы и гидроокиси неметаллов имеют кислотный характер и т.д.
Элемент
Н Не водо- ге-
роя лин
период
Li Ne ли- неон тий
2-й период
Na Аг нат- арго рий
3-й
К калий
Тй’’'
период период
* В июле 1974 г. советские физики, работающие в Объединённом институте ядерных исследований (г. Дубна) под руководством академика Г. Н. Флёрова, сообщили о синтезе 106-го элемента.
30
Закономерное изменение свойств элементов (от металлов к неметаллам) легко проследить на примере второго периода:
Z 3 4 5 6 7 8 91 0
эле- Li Be В С N О F Ne
мент ли- берил- бор угле- азот кисло- фтор неон тий лий род род
Период начинается с активного щелочного металла — лития, окисел которого Li2O и гидроокись LiOH обладают ярко выраженными основными свойствами. Следующий элемент — бериллий — также металл, но по химическим свойствам гораздо менее типичный. Это проявляется, например, в том, что гидроокись бериллия Ве(ОН)2 амфотерна; наряду с солями бериллия типа BeSO4, где Be выступает в роли катиона, существуют так называемые бериллаты типа К2ВеО2, в которых Be входит в состав аниона. Бор по химическим свойствам скорее неметалл; его окисел В2О3 и гидроокись Н3ВО3 проявляют по преимуществу кислотные свойства. Далее следуют типичные неметаллы — углерод и азот; их окислы СО2 и N2O5— ангидриды кислот (слабой угольной Н2СО3 и сильной азотной HNO3).B конце периода располагаются самые активные неметаллы — кислород и фтор. Период завершается инертным элементом неоном, практически не вступающим в химические реакции.
Такая же картина изменения свойств наблюдается и в 3-м периоде. А если элементы 2-го и 3-го периодов подписать друг под другом:
2-й Li литий Be бериллий В бор С угл е-род N азот 0 кислород F фтор N неон
3-й Na Mg А1 Si Р S С1 Аг
натрий магний алюминий кремний фос-фор .сера хлор аргон
то окажется, что элементы, записанные в ниж-
ней строчке, в своём химическом поведении во многом повторяют элементы, расположенные над ними. Так, например, натрий, как и литий, принадлежит к числу щелочных металлов; соединения элементов с серой — сульфиды — во многих отношениях подобны соединениям с кислородом —окис лам; хлор, как и фтор, обладает свойствами, характерными для галогенов; наконец, аргон, подобно неону, инертен (см. Инертные газы). В своих соединениях элементы 3-го периода обычно проявляют ту же степень окисления (валентность), что и их аналоги из 2-го периода. Это приводит к сходному виду формул важнейших химических соединений элементов-аналогов (например, LiCl и NaCl, BeSO4 и MgSO4, В2О3 и А12О3, СН4 и SiH4, HNO3 и НРО3, Н20 и H2S, BaF2 и ВаС12).
В 4-м периоде картина изменения свойств усложняется. Это связано прежде всего с увеличением числа элементов, входящих в период (см. таблицу 1). Однако восемь элементов — два начинающие период, калий и кальций (Z= 19 и 20), и завершающие его шесть, от галлия до криптона (Z от 31 до 36),— по-прежнему аналогичны соответствующим элементам 2-го и 3-го периодов. В этой восьмёрке наблюдается то же изменение свойств — от активного щелочного металла калия к типичному неметаллу (галогену) брому и, наконец, к инертному газу криптону. Существенно новое в 4-м периоде то, что в него внедряются десять элементов, нарушающих монотонное изменение свойств. Это так называемые переходные металлы — от скандия до цинка (Z от 21 до 30).
Совершенно такую же структуру имеет 5-й период: здесь присутствуют восемь элементов, подобных элементам 2-го и 3-го периодов (металлические свойства этих элементов ослабляются с увеличением Z), и десять переходных металлов, сходных по своим свойствам с соответствующими металлами из 4-го периода.
Удлинение 4-го и 5-го периодов за счёт переходных металлов приводит к тому, что в этих периодах наблюдается как бы остановка в ослаблении металлических свойств. Это проявляется, например, в том, что окислы и гидроокиси переходных металлов чаще всего имеют приблизительно одинаковый и по преимуществу основной характер.
Ещё более длительная «остановка» наступает в 6-м периоде. Это связано с появлением 14 лантаноидов (Z от 58 до 71), сходных по химическим свойствам элементов-металлов. Здесь ряд переходных металлов существенно удлиняется, он включает 24 элемента. В этом периоде металлические свойства уменьшаются ещё медленнее, чем в двух предыдущих.
В 7-м периоде пока известна лишь часть в принципе возможных элементов. Структура этого периода, по всей вероятности, должна быть подобна той, которая наблюдается в 6-м периоде. В роли аналогов лантаноидов здесь выступают актиноиды (Z от 90 до 103). За ними следуют курчатовий (Z=104), подобный по своим свойствам гафнию, и элемент с Z=105 (для него предложено название нильсборий), по-видимому, подобный танталу.
Итак, характерной чертой всех периодов оказывается ослабление металлических свойств элементов с увеличением заряда ядра атома. Обратившись к электронному строению атомов, можно констатировать, что «металличность» определяется, в первую очередь, прочностью связи электронов внешней оболочки с ядром.
Дмитрий Иванович МЕНДЕЛЕЕВ. Открыл периодический закон химических элементов.
Дмитрий Иванович Менделеев родился 8 февраля 1834 г. в Тобольске. Умер 2 февраля 1907 г. в Петербурге. Основные события жизни Менделеева, разностороннего учёного, педагога и общественного деятеля, вам известны из учебника химии для 7—8-го классов. И здесь мы лишь дополним то, что вы уже знаете. В Главном педагогическом институте в Петербурге Менделеев получил прекрасную подготовку не только по химии у профессора А. А. Воскресенского, но и по математике и физике у академиков М. В. Остроградского и Э. X. Ленца. Превосходное владение методами математики и физики позволило Менделееву применять их к решению проблем химии. В этом — большое его превосходство над своими современниками-химиками.
Уже на первых порах научной деятельности Менделеев изучает соотношение между составом, свойствами и формами химических соединений. Этим вопросам посвящены его диссертации об изоморфизме и удельных объёмах (1856 г.). Названные работы явились началом подготовки учёного к открытию периодического закона, которое датируется 1 марта 1869 г. (по старому стилю 17 февраля). Это открытие Менделеева было не плодом какого-то внезапного «озарения», но результатом долгого и упорного труда. Приступив к чтению курса неорганической химии (или, как предпочитал говорить он сам, «общей химии») и не найдя ни одного пособия, которое мог бы рекомендовать студентам, Менделеев начал писать свои знаменитые «Основы химии». По его словам, «тут много самостоятельного..., а главное — периодичность элементов, найденная именно при обработке «Основ химии». При жизни учёного «Основы химии» издавались 8 раз (последний — в 1906 г.), а затем в СССР переиздавались ещё 5 раз с дополнениями (13-е издание вышло в 1947 г.). На «Основах химии» учились многие поколения отечественных химиков. Этот классический труд был опубликован в переводах на английский, французский и немецкий языки. Нет ни одного другого руководства, которое оказало бы такое влияние на развитие и преподавание неорганической химии конца XIX — начала XX вв.
Своё величайшее открытие — периодический закон химических элементов — Менделеев сделал в возрасте 35 лет. До конца 1871 г. почти всё своё время он посвятил развитию закона и разработке периодической системы элементов. За короткий срок Менделеев сумел навести строгий порядок в мире химических элементов и впервые блестяще осуществил научный прогноз в химии.
Создав учение о периодичности, учёный намного опередил своё время. Периодическая система вскоре стала необходимым рабочим инструментом в химических исследованиях. Но лишь спустя полвека она получила физическое объяснение. «Опережать время» — этот девиз был лейтмотивом творчества Менделеева. Не сразу перечислишь все те отрасли науки и практики, где учёный высказал оригинальные идеи. Огромное теоретическое и практическое значение он придавал исследованию так называемых «неопределённых соединений», к которым относил растворы, сплавы, стёкла, шлаки. Химическая теория растворов, развитая учёным, составила эпоху в истории учения об этих системах. Исследуя свойства разреженных газов, Менделеев сделал вклад в познание газообразного состояния веществ. Он прозорливо видел в нефти не одно лишь топливо («Топить можно и ассигнациями!»),’а неисчерпаемый источник разнообразных химических продуктов. Первым высказал идею о подземной газификации каменного угля. Разработал план освоения Арктики и предложил проект мощного ледокола. Бесстрашно поднявшись на воздушном шаре для изучения верхних слоёв атмосферы, он наметил тем самым новые пути метеорологических исследований.
«Ограниченный рост промышленности совершенно непригоден нашему краю и неприличен нашему народу... Идя по-маленьку мы никогда не догоним соседей, а надо не только догнать, но и перегнать»,— писал Менделеев. Он и словом, и делом боролся за развитие производительных сил России, за расширение горной, металлургической, химической и нефтяной промышленности, за химизацию сельского хозяйства. Он стоял за доступ всех слоёв народа к образованию, за создание новых и новых учебных заведений. Грандиозные замыслы учёного сбылись только после Октябрьской революции 1917 г.
Заслуги Д. И. Менделеева получили всеобщее признание уже при его жизни. Он был избран почётным членом множества академий, учёных обществ, научных учреждений и учебных заведений разных стран мира. Его научный титул насчитывает свыше 130 названий. А в 1964 г. имя Менделеева занесено на Доску почёта науки Бриджпортского университета (США) в числе имён величайших учёных мира. В его честь — а это действительно великая честь! — назван элемент с атомным номером 101—менделевий. В 25 томах Сочинений Д. И. Менделеева (изданы в 1937—1954 гг.) читатель найдёт множество глубоких мыслей, не устаревших и в наши дни.
32
Таким образом, более или менее постепенный переход от металлов к неметаллам, наблюдающийся в каждом периоде, представляет собой следствие ясно выраженной тенденции: чем ближе элемент к концу периода, тем более прочно удерживает атом свои валентные электроны.
Эту зависимость можно охарактеризовать количественно с помощью энергии ионизации атома Е1,то есть энергии, которая необходима для отрыва одного электрона от электронейтрал ьного атома.
Рис. 1 показывает, как меняется величина Ех с увеличением Z (энергию ионизации обычно измеряют в электрон-вольтах; эта единица энергии сокращённо обозначается эв и равна 1,6-10“19 дж). Наиболее чётко выраженные минимумы соответствуют щелочным металлам, максимумы — инертным элементам. Вместе с тем в пределах каждого периода, хотя и видна общая тенденция к увеличению энергии ионизации, обнаруживаются скачки.
Так, например, для отрыва электрона от атома магния (Z=12) требуется затратить 7,6 эв, а в случае алюминия (Z= 13) — лишь 6,0 эв, хотя в своих химических проявлениях магний, несомненно, более типичный ме
талл, чем алюминий. Возникающее «противоречие» нетрудно устранить: дело в том, что для объяснения свойств этих двух металлов необходимо принимать во внимание затраты энергии на отрыв не только первого, но и других валентных электронов. Энергия отрыва второго (Е2) и третьего (Е3) электронов равна 15,0 и 80,1 эв для магния, 18,8 и 28,4 эв для алюминия. Атом двухвалентного магния сравнительно легко отдаёт два электрона — это требует затраты энергии Ei~\~E2t что составляет 22,6 эв.
Алюминий в большинстве своих соединений трёхвалентен; для отрыва трёх его валентных электронов требуется энергия Е1+£24"£з» равная 53,2 эв. Таким образом, переход атома магния в состояние Mg2+ происходит значительно легчег чем переход атома алюминия в состояние А1:,+ . Итак, конечный количественный результат совпал с нашими общими, «качественными» заключениями: у магния металлические свойства проявляются существенно сильней, чем у алюминия.
Мы отвлеклись от основной темы, чтобы разрешить недоумения, которые может вызвать приведённый график.
Возвращаясь к сути дела, отметим, что, несмотря на присутствие скачков, изменение энергии ионизации Ех в третьем периоде происходит подобно изменению этой величины во втором периоде, а в пятом периоде — так же, как в четвёртом периоде. А это и означает ана-
Энергия ионизации
Рис. 1. Периодичность энергии ионизации атомов.
2 57 2
4 8 12 16 20 24 28 32 36 40 44 48 52 56 74 78 82 86
Атомный номер
Нильс БОР — один из основоположников квантовой физики.
Датский физик Нильс Бор родился 7 октября 1885 г. в Копенгагене. В 1909 г. окончил Копенгагенский университет и с 1911 г. работал в Англии — сначала в Кембридже у Дж. Дж. Томсона (в Кавендишской лаборатории), а затем в Манчестере у Э. Резерфорда. С 1920 г. и до конца жизни руководил созданным им Институтом теоретической физики в Копенгагене, который носит теперь его имя.
«Занятно быть знакомой с гением...»,— так писала в 1904 г. о Боре-студенте его однокурсница по Копенгагенскому университету. Глубина мышления молодого датского физика-теоретика проявилась через девять лет, когда после встречи с Резерфордом он создал квантовую теорию планетарной модели атома (1913 г.).
По классическим законам такой атом был невозможен: вращаясь вокруг ядра, электроны должны были излучать свет и, теряя энергию, падали бы на ядро. Бор выдвинул два постулата: по первому — электроны на орбитах не излучали и атом пребывал в устойчивом состоянии, по второму — излучение происходило целыми порциями электромагнитной энергии при скачкообразном переходе электронов с орбиты на орбиту. Сразу получили объяснение прерывистые — линейчатые — атомные спектры. И открылся путь для физического обоснования периодической повторяемости химических свойств элементов в системе Д. И. Менделеева. Бор завершил это обоснование в 1921 г.
Однако таинственными оставались сами постулаты Бора. Противоречившие классической физике, почему они оказались истинными? Поиски ответа на этот вопрос привели к созданию новой — квантовой — механики микромира (1925— 1927 гг.). Это обессмертило имена В. Гейзенберга, Э. Шредингера, П. Дирака, В. Паули, М. Борна... «Установлением этой механики мы обязаны прежде всего изобретательности и остроумию младшего поколения физиков»,— с неизменной своей скромностью говорил Бор. Но подлинным вождём квантовой революции всегда оставался он — глава знаменитой «копенгагенской школы», через которую прошёл целый интернационал выдающихся теоретиков из многих стран Европы, Америки и Азии. Среди учеников Бора был наш прославленный академик Л. Д. Ландау.
...В 1939 г. Бор вместе с Дж. Уилером построил теорию деления ядер урана под действием нейтронов. Гитлеровская оккупация заставила его бежать из Дании. Последние годы второй мировой войны он провёл в США, работая в Лос-Аламосской атомной лаборатории. Но ещё до создания атомной бомбы он стал деятельным противником ядерного оружия.
Бор был лауреатом Нобелевской премии (1922 г.), президентом Датского королевского научного общества, членом многих других академий и научных обществ мира. Он трижды приезжал в нашу страну, а в 1929 г. был избран иностранным членом Академии наук СССР.
До конца своих дней Нильс Бор, так же как и Альберт Эйнштейн, оставался олицетворением человечности, душевной чистоты и высокого благородства. Он умер в Копенгагене 18 ноября 1962 г.
логию в свойствах элементов, занимающих аналогичные места в периодах, то есть иллюстрирует сущность периодического закона Менделеева.
СИСТЕМА ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Итак, закон Менделеева позволяет выстроить химические элементы в ряд, в котором каждый из них получает как бы специально отведённое для него место. Периодичность изменения свойств, отчётливо проявляющаяся в таком ряду, позволяет предвидеть химическое поведение элемента, основываясь на свойствах его соседей по периоду и его аналогов из других периодов. Всё это означает, что в свете периодического закона оказывается возможным рассматривать химические элементы в их взаимной связи, представить совокупность элементов в виде единой системы — периодической системы элементов. Эта система находит наглядное отображение в форме таблицы.
За 100 с лишним лет, прошедших со времени открытия периодического закона, было предложено свыше 400 вариантов графического изображения периодической системы, как плоскостных (таблицы, графики), так и объёмных (элементы размещались на поверхности конуса, цилиндра и т.п.). Но, хотя некоторые из них имеют отдельные преимущества при анализе тех или иных свойств элементов, наиболее универсальными, удобными и наглядными, а соответственно, и наиболее употребительными остаются лишь два варианта:
1) «длинная» форма таблицы, к которой мы уже обращались (таблица 1);
2) «короткая» форма (таблица 2).
Дадим сначала общую характеристику периодической системы, опираясь на «длинную» таблицу, а затем опишем особенности «короткой» формы.
3 хэш
la
ПЕРИОДИЧЕСКАЯ СИСТЕМА
4
5
6
з Li
6,941
ЛИТИЙ
и Na
22,98977
НАТРИЙ
19 К
39,098
КАЛИЙ
37 Rb
85,4678
5s’
РУБИДИЙ
55 CS 132,9054 6s1
ЦЕЗИЙ
87 Ff
7s1
ФРАНЦИЙ
___Па
4 Be
9,01218
БЕРИЛЛИЙ
12 Mg
24,305
3s2 МАГНИЙ
20 Ca
40,08
КАЛЬЦИЙ
38 Sr
87,62
5s2
СТРОНЦИЙ
56 Ba
137,34
6s2 БАРИЙ
88 Ra
226,0254
РАДИЙ
Атомный номер
Атомная масса
Название
92 lb -238,029 5f3 6tf1 7s2 — УРАН
Символ
Электронная конфигурация (высшие энергетические уровни)
Атомные массы приведены по углеродной шкале в соответствии с Международной таблицей 1973 г. В квадратных скобках приведены массовые числа наиболее устойчивых изотопов. Названия и символы элементов, приведенные в круглых скобках, не являются общепринятыми.
Шб
IV6
V6
VI6
VII6
VIII б -
21 Sc 44,9559 3d14s2 СКАНДИЙ
88,9059
ИТТРИЙ
57 La
138,9055
5dJ 6s2 ЛАНТАН
< **
89 AC
6d' 7s2
АКТИНИЙ
22 Ti
47,90
3d2 4s2
ТИТАН
23 V 50,9414 3d3 4s2 ВАНАДИЙ
24 Cr 51,996 3d5 4sl
ХРОМ
25 МП
‘ 54,9380
МАРГАНЕЦ
26 Fe
55,847
3d6 4s2 ЖЕЛЕЗО
27 Co 58,9332 3d7 4s2 КОБАЛЬТ
40 Zr
91,22
41 Nb
92,9064
2 5s2
ЦИРКОНИЙ
4d45s’
НИОБИЙ
42 Mo
95,94 4rf55s' МОЛИБДЕН
43 Тс
98,9062
4d5 5s2
ТЕХНЕЦИЙ
44 Rll
101,07
45 Rh
102,9055
РУТЕНИЙ
РОДИЙ
72 Hf
178,49
5d26s2
ГАФНИЙ
73 Ta 180,9479 5d3 6s2 ТАНТАЛ
74 W
183, SS 5d4 6s2 ВОЛЬФРАМ
75 Re 186,207 5d56s2 РЕНИЙ
76 Os
190,2
5d6 6s2
ОСМИЙ
192,22
5d76s2
ИРИДИЙ
104 Ku
[261]
КУРЧАТОВИЙ
* ЛАНТАНОИДЫ
** АКТИНОИДЫ
105 (Ns)
(НИЛЬСБОРИЙ)
58 Се 59 Pr 60 Nd 6i Pm 62 Sm
140,12 140,9077 144,24 [145] 150,4
4Z* 5rf' 6s2 4f36s2 4/4 6s2 4Z56s2 4fs 6s2
ЦЕРИЙ ПРАЗЕОДИМ НЕОДИМ ПРОМЕТИЙ САМАРИЙ
90 Th 91 Pa 92 U 93 Np 94 Pu1
232,0381 231,0359 238,029 237,0482 [244]
6d27s2 5r26d' 7s2 5/3 6d' 7s2 5/46d’7s2 5f6 7s2
ТОРИЙ ПРОТАКТИНИЙ УРАН НЕПТУНИЙ ПЛУТОНИЙ
Таблица 1
Vila Villa
ЭЛЕМЕНТОВ Д. И. МЕНДЕЛЕЕВА
Ша
IVa
Va
Па
1,0079
к1
ВОДОРОД
2 He
4,00260 .2
ГЕЛИЙ
16
Пб
28 Ni
58,70
НИКЕЛЬ
29 Cu 63,546 3d10 4s1 МЕДЬ
зо Zn
65,38
3dl04s2 ЦИНК
46 Pd
106,4
4d105s°
ПАЛЛАДИЙ
47 Ag 107,868 4d,05sl
СЕРЕБРО
48 Cd
112,40
4 d19 5 s2
КАДМИЙ
78 Pt
195,09
5d96s1
ПЛАТИНА
79 Au 196,9665 5d*° 6s'
ЗОЛОТО
80 Hg
200,59
5d'°6s2
РТУТЬ
s-элементы
БОР
5 В
10,81
12,011
УГЛЕРОД
13 А1 26,98154
АЛЮМИНИЙ
31 Ga
69,72
ГАЛЛИЙ
49 |П
114,82
ИНДИЙ
81 Т1
204,37
ТАЛЛИЙ
7 N
14,0067
АЗОТ
8 О
15,9994
2s2 2p4
КИСЛОРОД
18,99840
ФТОР
14 Si
28,086
КРЕМНИЙ
32 Ge
72,59
ГЕРМАНИЙ
82 Pb
207,2
15 Р
30,97376
ФОСФОР •
16 s
32,06
3s2 3p4
СЕРА
17 Cl
35,453
ХЛОР
34 Se
78,96
35 Вг
79,904
БРОМ
ю Ne
20,179
НЕОН
18 Ar
39,948
АРГОН
36 Кг
83,80
КРИПТОН
СЕЛЕН
СВИНЕЦ
зз As 74,9216
4s2 4p3 МЫШЬЯК
50 Stl
118,69
5s25p2 ОЛОВО
52 Те 127,60
5s2 5p4 ТЕЛЛУР
СУРЬМА
ВИСМУТ
83 Bi
208,9804
53 1 126,9045
84 Po [209]
6s2 6p4 ПОЛОНИЙ
85 At [210]
6s2 6p® АСТАТ
р-элементы
51 Sb
121,75
54 Хе
131,30
иод
КСЕНОН
86 Rn
[222]
6s26p6 РАДОН
d-элементы
f-элементы
63 Eu 151,96 4f 7 6s2 ЕВРОПИЙ 64 Gd 157,25 kt15d’ 6s2 ГАДОЛИНИЙ 65 Tb 158,9254 4f96s2 ТЕРБИЙ 66 Dy 162,50 4Z’° 6s2 ДИСПРОЗИЙ 67 Ho 164,9304 4f"6s2 ГОЛЬМИЙ 68 Er 167,26 4f12 6s2 ЭРБИЙ 69 Tm 168,9342 4f13 6s2 ТУЛИЙ 70 Yb 173,04 4fM6s2 ИТТЕРБИЙ 71 Lu 174,97 4f145dI 6s2 ЛЮТЕЦИЙ
95 Am [243] 5f7 7s2 АМЕРИЦИЙ 96 Cm [247] 5Z76d* 7s2 КЮРИЙ 97 Bk [247] 5f86d’ 7s2 БЕРКЛИЙ 98 Cf [251] 5/'° 7s2 КАЛИФОРНИЙ 99 Es [254] 57n 7s2 ЭЙНШТЕЙНИЙ loo Fm [257] 5 f12 7s2 ФЕРМИЙ 101 Md [258] 57B 7s 2 МЕНДЕЛЕВИЙ 102 (No) [255] 5 f14 7s2 (НОБЕЛИЙ) юз (Lr) [256] 57й fid1 7s2 (ЛОУРЕНСИЙ)
3
36
В предыдущем разделе, проводя сопоставление 2-го и 3-го периодов, мы уже отметили важный факт, который и есть прямое следствие периодического закона: если выписать элементы, входящие в последовательные периоды, в виде горизонтальных рядов, то в полученной таблице обнаруживаются вертикальные столбцы — подгруппы элементов, аналогичных по своим свойствам. Столбцы, открываемые элементами 1-го и 2-го периодов, называются главными подгруппами. Переходные металлы 4-го периода служат началом столбцов, называемых побочными подгруппами.
Таким образом, в периодической системе элементы подразделяются, с одной стороны, на семь периодов, а с другой стороны,— на шестнадцать подгрупп, среди которых восемь главных (1а — Villa) и восемь побочных (16 — VIII6).
Уже сам факт принадлежности элемента к главной или побочной подгруппе характеризует определённым образом многие его свойства. Для большинства элементов, входящих в главные подгруппы, типично существование солеобразующих окислов состава К20дг или КОдг/г (здесь А — номер подгруппы, в которую входит данный элемент R; первая формула соответствует нечётным значениям А, вторая — чётным). Примерами могут служить основные окислы Li2O, СаО, амфотерный А12О3, кислотные С02, N2O5, S03, С12О7. Степень окисления элементов в таких окислах достигает своего максимального значения и равна + V, то есть номеру подгруппы.
Те из элементов главных подгрупп, которые обладают преимущественно неметаллическими свойствами (элементы IVa — VIIa-подгрупп), образуют гидриды состава RH8_iV, где N по-прежнему номер подгруппы. Примерами являются СН4, NH3, Н20 и HF. В этом случае степень окисления равна N—8; здесь она отрицательна.
В каждой из главных подгрупп наблюдается закономерное усиление металлических свойств с увеличением атомного номера элемента (то есть сверху вниз). Сильнее всего металлические свойства выражены у нижнего элемента 1а-подгруппы — франция, а неметаллические— у верхнего элемента VIIa-подгруппы — фтора.
Иначе ведут себя элементы побочных подгрупп: все они — более или менее типичные металлы. Усиление металлических свойств при переходе по подгруппе сверху вниз выражено здесь далеко не так отчётливо. Максимальное положительное значение степени окисления для многих элементов побочных подгрупп, как и для элементов главных подгрупп, равно N. В качестве примеров приведём окислы цинка, скандия, титана, ванадия, хрома, марганца: ZnO, Sc2O3, TiO2, V2O5, СгО3, Mn2O7. Вместе с тем часто наблюдаются отклонения от этого
37 Rb
38 Sr
39 Y
40 Zr
41 Nb
42 Mo 43-
44 Ru
45 Rh
46 Pd
47 Ag
48 Cd
49 In
50 Sn
51 Sb
52 Те
53 I
54 Xe
Рис. 2. Лестничная форма периодической системы элементов по Н. Бору (1921 г.).
79 Au
80 Hg
81 T1 82Pb
83 Bi
84 Po
85-
86 Rn
87-
55 Cs
88 Ra
56 Ba
57 La 89 Ac 90 Th 91 Pd 92 U n 1 1 1 1 1 1 1 1 1 J
58Ce 59 Pr 60 Nd 61 - 62 Sm 63 Eu 64Gd 65Tb 66 Dy 67Ho 68Er 69 Tu 170 Yb 71 Lu 72- 73 Ta 74 W 75 — 760s 77 Ir 78 Pt
118 —
37
правила. Так, для многих элементов VII16-подгруппы соединения со степенью окисления 8 неизвестны; гораздо легче образуются соединения со значительно более низкими степенями окисления (например, FeO, Fe2O3). В соединениях золота и серебра, то есть элементов, входящих в 16-подгруппу, часто встречается степень окисления +3 (например, КАиС14, KAgF4).
Важными особенностями обладают VII16- и II 16-подгруппы. Подгруппа VII16, в отличие от прочих побочных подгрупп, содержит не три, а девять элементов, составляющих три столбца; основанием для объединения этих девяти элементов в одну подгруппу является близость их химических свойств. Подгруппа II16 включает 32 элемента (почти треть от общего числа элементов, известных на сегодня!), в том числе семейства лантаноидов и актиноидов. Лантаноиды, в таблице вынесенные в отдельную строку, вместе с лантаном (как и актиноиды вместе с актинием) обнаруживают ещё большее сходство в химических свойствах, чем элементы VII 16-подгруппы.
Вынесение лантаноидов и актиноидов за пределы общей последовательности элементов носит довольно искусственный характер; это недостаток того варианта таблицы, который мы до сих пор рассматривали. Существует, однако, и другой вариант «длинной» таблицы, где все элементы естественно вписываются в общую «иерархию» (рис. 2). Эта таблица (иногда её называют «лестничной») имеет и ещё одно преимущество: в ней удаётся наглядно отобразить встречающуюся иногда неоднозначность подразделения элементов на подгруппы.
Пример такой неоднозначности даёт водород. В «лестничной» таблице (предложенной в 1895 г. датским химиком Ю. Томсеном и разработанной в 1921 г. датским физиком Н. Бором) элементы, аналогичные по свойствам, то есть относящиеся к одной подгруппе, соединены между собой прямыми линиями. От родорода идут сразу две линии — к литию и фтору. Действительно, водород проявляет сходство и со щелочными металлами и с галогенами. Как и элементы la-подгруппы, в соединениях он обычно имеет степень окисления +1, образуя, например НС1, Н2О, H2SO4, формально подобные LiCl, Li2O, Li2SO4. С другой стороны, водород образует гидриды, в которых его степень окисления равна —1; существует аналогия между, например, LiH, СаН2, А1Н3 и LiF, CaF2, A1F3. Кроме того, подобно элементам VПа-подгруппы, водород в свободном состоянии — это газ с двухатомными молекулами. Учитывая описанную двойственность водорода, в обычных таблицах его часто помещают и Иа- и в VIIa-подгруппу (в одном из этих двух случаев — в скобках). Таблица Томсена — Бора позволяет отразить это обстоятельство более естественным путём.
Теперь перейдём к «короткой» форме периодической системы (таблица 2). Здесь а- и б-подгруппы, имеющие одинаковые номера, объединены в группы; в каждой группе элементы главной подгруппы составляют один столбец, элементы побочной подгруппы — другой. Эта особенность «короткой» формы таблицы имеет свои преимущества. Важнейшее из них со
стоит в том, что элементы, которые проявляют одинаковую высшую положительную степень окисления, равную номеру группы, как правило, оказываются вместе. Тут есть, однако, и недостаток: если в левой части таблицы элементы а- и б-подгрупп, входящие в одну группу, достаточно близки по своим свойствам, то в правой её части в одну группу попадают столь различные элементы, как хром и кислород, марганец и фтор, металлы VII 16-подгруппы и инертные элементы VIIIa-подгруппы.
Иногда между сторонниками «длинной» и «короткой» форм таблицы возникают жаркие споры. Но вряд ли для таких споров есть серьёзные основания. Каждый из двух описанных способов изображения периодической системы имеет свои достоинства и недостатки, каждый может оказаться более удобным при анализе того или иного конкретного вопроса. К тому же, между ними нет принципиального различия.
ПЕРИОДИЧЕСКАЯ СИСТЕМА И СТРОЕНИЕ АТОМА
Описывая периодическую систему, устанавливая аналогии между элементами разных периодов, мы умышленно касались главным образом химических свойств элементов, то есть шли тем же путём, что и сам Д. И. Менделеев. Однако дальнейшее развитие науки показало, что химические свойства элементов обусловлены строением их атомов, а точнее — конфигурацией внешних электронных оболочек, и что периодичность в изменении свойств — это лишь следствие периодичности, которая обнаруживается в строении атомов. Таким образом была установлена физическая сущность основного закона химии.
Рассмотрим этот вопрос подробнее. Исследования строения атомов показали, что электроны, число которых в атоме равно атомному номеру Z, распределены по электронным оболочкам. Оболочка представляет собой совокупность электронов с одинаковым значением главного квантового числа, которое совпадает с номером оболочки п (см. Атом). Оболочка с наибольшим п называется внешней (валентной).
Каждая оболочка, в свою очередь, разбивается на энергетические уровни; их принято обозначать s, р, d, f — в порядке увеличения энергии. Число уровней, которые содержит данная оболочка, равно её номеру п. Таким образом, первая оболочка содержит лишь один уровень, называемый 1s, вторая — два уровня: 2s и 2р, третья— три уровня: 3s, Зр, 3d, и т. д. Энергия электрона в атоме определяется номером оболочки и уровнем, на котором находится электрон в этой оболочке.
38
«У крепители периодического закона».
Так Д. И. Менделеев назвал учёных, чьи работы и открытия способствовали подтверждению периодического закона. В 1886 г. Менделеев разместил на толстом листе картона фотокарточки Л. Ф. Нильсона (слева), П. Э. Лекок де Буабодрана (в середине сверху), Б. Браунера (в середине снизу) и К. Винклера (справа) с их дарственными надписями. Подлинник хранится в Музее Д. И. Менделеева при Ленинградском государственном университете.
Ларс Фредерик НИЛЬСОН (родился 27 мая 1840 г. в Шёнберга — умер 14 мая 1899 г. близ Стокгольма) — шведский химик. Был профессором университета в Упсале, а затем (с 1882 г.) — Сельскохозяйственной академии в Стокгольме.
В 1879 г. Нильсон открыл химический элемент скандий, по свойствам оказавшийся тождественным с экабором, предсказанным в 1870 г. Менделеевым. Разумеется, одного этого открытия было достаточно, чтобы Нильсон встал в почётный ряд «укрепителен». Но было и ещё одно — серьёзное и беспристрастное участие в споре о валентности бериллия.
В 1878 и 1880 гг. Нильсон и С. О. Петерсон опубликовали совместные работы, в которых доказывали, что бериллий имеет атомный вес около 13,5 и трёхвалентен,* основанием послужили измерения теплоёмкости нечистого бериллия. Вывод шведских учёных противоречил периодическому закону, согласно которому бериллий как элемент II группы должен быть двухвалентным и иметь атомный вес около 9,3 (кстати сказать, ещё в 1842 г. это показал русский химик И. В. Авдеев). Но в 1884 г. те же учёные, определив плотность пара хлорида бериллия, нашли, что это двухлористый бериллий. «Мы должны отказаться от ранее защищавшегося нами мнения, что бериллий — трёхвалентный элемент. Одновременно мы признаём правильность периодического закона и в этом важном случае»,— такими словами заключили они свою новую работу. В «Основах химии» Менделеев писал: «Нильсон и Петерсон — одни из главных защитников трехатомности бериллия... доставили опытные доказательства в пользу двухатомности бериллия и, громко высказав это, показали, что в науке истина, даже при разноречиях, одинаково дорога всем».
Поль Эмиль ЛЕКОК ДЕ БУАБОДРАН (родился 18 апреля 1838 г. в г. Коньяке — умер 28 мая 1912 г. в Париже) — французский химик. Будучи богатым виноделом, Лекок де Буабодран изучил химию самостоятельно. Всю жизнь он работал в собственной лаборатории, где занимался спектральным анализом минералов.
При исследовании пиринейской цинковой обманки Лекок де Буабодран в 1875 г. открыл в ней новый элемент, который назвал галлием. Узнав об этом,
39
Менделеев написал Парижской академии наук, что галлий — это предсказанный им в 1870 г. экаалюминий. Однако, утверждал Менделеев, плотность галлия должна быть не 4,7 г/см3, как нашёл Лекок, а 5,9—6,0 г/см3. Получив это письмо, французский учёный, дотоле ничего не знавший о периодическом законе, повторил измерение плотности на тщательно очищенном галлии. Оказалось, что она действительно равна 5,96 г/см3. В 1879 Лекок де Буабодран открыл самарий, в 1886 г.— диспрозий (см. статью «Лантаноиды»). В 1878 г. Парижская академия наук избрала его своим членом-корреспондентом.
Богуслав БРАУНЕР (родился 8 мая 1855 г. в Праге — умер 15 февраля 1935 г,, там же) — чехословацкий химик. По окончании Пражской технической школы работал в 1878—1879 гг. у немецкого химика Р. Бунзена, а затем был ассистентом, доцентом и профессором (с 1897 г.) в Праге.
Познакомившись с трудом Д. И. Менделеева о периодическом законе, Браунер стал его убеждённым сторонником. Очень часто судьба больших научных открытий зависит от тех «мелких подробностей», которые либо подтверждают, либо ставят под сомнение открытие. Из статьи о Нильсоне вы уже знаете о том, что такой «подробностью» в начале жизни периодической системы оказалось положение в ней бериллия. В статьях 1878 и 1887 гг. Браунер выступал с обоснованными возражениями против выводов Нильсона и Петерсона о трёхвалентности бериллия. Он утверждал, что атомный вес бериллия равен 9,1 и формула окиси бериллия вполне соответствует положению этого элемента во II группе периодической системы.
Браунер был крупным специалистом по химии редкоземельных элементов. В 1882 г., изучая их спектры поглощения, учёный пришёл к выводу, что дидим— это смесь двух элементов. И действительно, в 1885 г. австрийский химик К. Ауэр выделил их и назвал празеодимом и неодимом. В 1900 г. Браунер предложил поместить лантаноиды в особой «интерпериодической» подгруппе IV группы системы Менделеева, начиная от церия и кончая ещё не известным элементом с атомным весом около 180. Эта мысль оказалась правильной, за исключением того, что церий и остальные лантаноиды вошли в III группу. По просьбе Менделеева Браунер написал большую статью «Элементы редких земель» для 7-го издания «Основ химии».
Клеменс Александр ВИНКЛЕР (родился 26 декабря 1838 г. во Фрейберге — умер 8 октября 1904 г. в Дрездене) — немецкий химик. Несколько поколений его предков работали на горно-металлургических предприятиях в окрестностях Фрейберга. Отец учёного Курт Винклер, ученик шведского химика И. Я. Берцелиуса, заведовал фабрикой, производившей смальту из фрейбергских кобальтовых руд. При фабрике была хорошая лаборатория, где Клеменс и получил первое знакомство с химией. В 1873 г. Винклер занял кафедру аналитической и технической химии Горной академии.
В 1885 г. в одной из шахт близ Фрейберга был найден новый минерал аргиродит, и Винклер получил его для количественного анализа. И вот химика постигла неудача — сумма найденных процентных содержаний элементов получилась не около 100%, а на 7—8% меньше. Такой «дефицит» редко случается даже у начинающих студентов, и учёный заподозрил, что «недостача» вызвана присутствием неизвестного элемента. Опыты, поиски... и, наконец, Винклер получил сероватый порошок нового элемента, названного им германием. В 1886 г. Винклер опубликовал свои исследования. Сперва он полагал, что открыл экакадмий (элемент II группы между кадмием и ртутью). Но тут же и сам Менделеев и другие химики указали, что все свойства германия совпадают с предсказанными в 1870 г. Менделеевым свойствами элемента IV группы экасилиция.
Об открытии германия Винклер писал: «Вряд ли может существовать более яркое доказательство справедливости учения о периодической системе элементов...; оно составляет, конечно, более, чем простое подтверждение смелой теории, оно знаменует собой выдающееся расширение химического поля зрения, гигантский шаг в области познания».
Винклер был большим мастером химического анализа. Он предложил ряд способов количественного определения металлов в рудах и заводских продуктах. Им создана оригинальная методика химического анализа промышленных газов. Интересно, что уже в 1883 г. учёный поднял в печати вопрос о влиянии массового сжигания каменного угля на состав атмосферы. Винклер был и требовательным профессором — считал, что верные анализы можно сделать, только соблюдая величайшие чистоту и порядок в лаборатории и даже в одежде. Он не позволял студентам надевать халаты или фартуки. «Пусть так наряжаются органики,— говорил профессор,— но аналитик должен уметь работать во фраке».
Периоды
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ Д. И. МЕНДЕЛЕЕВА
з Li
6,941
2s1
ЛИТИЙ_________
и Na
22,98977
3s1
НАТРИЙ
4
19 К
39,098
4s1
КАЛИЙ
3fts'
29 Си
63,546
МЕДЬ
5
37 Rb
85,4678
РУБИДИЙ
4cf10 5s*
47 Ag
107,868
СЕРЕБРО
6
55 C S
132,9054
6s1
ЦЕЗИЙ__________
79 Au
196,9665
5dra6s'
ЗОЛОТО
87 Fr
[223]
ФРАНЦИЙ
a 11 6
4 Be
9,01218
2s2
БЕРИЛЛИЙ_________
12 Mg
24,305
3s2
МАГНИЙ_________
20 Ca
40,08
4s2
КАЛЬЦИЙ________
зо Zn
in 65,38
3d°4j2
__________ЦИНК
38 Sr
87,62 5s2
СТРОНЦИЙ_________
48 Cd
112,40
4d10 5s2
__________КАДМИЙ
56 Ba
137,34
6?
БАРИЙ_________
80 Hg
200,59
5cf10 6s2
__________РТУТЬ
88 Ra
226,0254
7s2
РАДИЙ
5 В
10,81
2s22p'
БОР_________
13 А1
26,98154 3s2 Зр1
АЛЮМИНИЙ__________
21 Sc
44,9559
3d 4s2
_________СКАНДИЙ
3iGa
69,72
4s2 4р!
ГАЛЛИЙ
39 Y
88,9059
4<? 5s2
ИТТРИЙ
49 In
114,82
5s2 бр1
ИНДИЙ_________
57 La*
138,9055
Sof1 6s2
ЛАНТАН
81 TI
204,37
6s2 6pl
ТАЛЛИЙ
89 AC**
, [227]
7s2
АКТИНИЙ
УГЛЕРОД
14 Si
28,086
3s2 Зр2
КРЕМНИЙ_________
22 Ti
47,90
2 4s2
ТИТАН
32 Ge
72,59
4s24р2 ГЕРМАНИЙ
40 Zr
91,22
ЦИРКОНИЙ
50 Sn
118,69
ОЛОВО
5s2 5р2
5d2 6s2
72 Hf
178,49
ГАФНИЙ
82 Pb
207,2
6s2 6p2
СВИНЕЦ
6d27?
104 KU
[261]
КУРЧАТОВИЙ
a V б |
7N I
14,0067
2s2 J
АЗОТ______ I
15 Р I
30,97376 о | з?2зл
ФОСФОР_______J
23 V I
50,9414
3d34s2
_________ВАНАДИЙ j
зз As 1
74,9216
4s2 4J
МЫШЬЯК j
4iNb1
92,9064 |
4d45s'
_________НИОБИЙ I
51 Sb
121,75
5s2 5Д
СУРЬМА
5cf3 6s2
73 Ta
180,9479
ТАНТАЛ
83 Bi
208,9804
6s2 6/
ВИСМУТ_______
105 (NJ
(НИЛЬСБОРЯ
* Л A H T __________________—
58 Се 140,12 4A*Sd' 6s2 ЦЕРИЙ 59 Pr 140,9077 4/> 6s2 ПРАЗЕОДИМ 60 Nd 144,24 4f46s2 НЕОДИМ 6i Pm [145] к , 4f5 6s2 ПРОМЕТИЙ 62 Sm 150,4 6 „ 4f66s2 САМАРИЙ 63 Eu 151,96 4/76s2 ЕВРОПИЙ 64 Gd I 157,25 7 1 । 4f7 5d 6.-* ГАДОЛИНИЙ
**А Н Т И
90 Th 232,0381 6d2 7s2 ТОРИЙ 91 Ра 231,0359 5r26d'7s2 ПРОТАКТИНИЙ 92 U 238,029 5f3 6d> 7s2 УРАН 93 Np 237,0482 5f46d'7s2 НЕПТУНИЙ 94 Pu [244] 5f67s2 ПЛУТОНИЙ 95 Am [243] 7 , 5f7 7s2 АМЕРИЦИЙ 96 Cm I [247] 1 5f? 6d' 71 КЮРИЙ |
Атомные массы приведены по углеродной шкале в соответствии с Международной таблицей 1973 Названия и символы элементов, приведенные
Таблица 2
о VI
cl
80
15,9994
КИСЛОРОД
32,06
СЕРА
3d5 4s1
51,996
ХРОМ
34 Se
78,96
4s2 4р4
СЕЛЕН
d5 5s-1
42 Mo
95,94
МОЛИБДЕН
127,60
5s25о4
ТЕЛЛУР
74 W
183,85
I 5d46s2
ВОЛЬФРАМ
84 Ро
[209] , L
6s2 6р
ПОЛОНИЙ
а УП
1И
1,0079
ВОДОРОД
Is1
18,99840
2s2 2р6
ФТОР_________
17 С1
35’453 3s2Зр5
ХЛОР_________
25 МП
54,9380
3d54s2
________МАРГАНЕЦ
35 Br
79,904
БРОМ
98,9062
ТЕХНЕЦИЙ
126,9045
5s2 5р5
5d56s2
ИОД
75 Re
186,207
РЕНИЙ
85 At
[210]
6s2 6р5
АСТАТ
s-элементы
(/—элементы
а
2 Не
4,00260
ГЕЛИЙ
Is2
ю Ne
20,179
2s22p6
НЕОН
18 Аг
39,948
АРГОН
3s2 Зр6
26 Fe
55,847
ЖЕЛЕЗО
36 Кг
83,80 .
4s2 4р6
КРИПТОН
4d75s
44 Ru
101,07
РУТЕНИЙ
54 Xe
131,30
КСЕНОН
3d74s2
4d8 5s1
5s2 5p6
76OS
190,2
5d66s2
ОСМИЙ
86 Rn
6s2бр6
РАДОН
р-элементы
/-элементы
5d76s2
VIII
27 СО
58,9332
КОБАЛЬТ
45 Rh
102,9055
РОДИЙ
192,22
ИРИДИЙ
Атомный .
номер -Атомная масса
28 Ni
58,70
НИКЕЛЬ
46 Pd
106,4
4<?° 5s°
ПАЛЛАДИЙ
5d9 6s’
78 Pt
195,09
ПЛАТИНА
Символ
238,029
5f36d'7s2
Название —г У Р АН
-Электронная -1 конфигурация
(высшие энергетические уровни)
н О и д ы
65ТЬ 158,9254 4/9 6s2 ТЕРБИЙ 66 Dy 162,50 4/ ° 6s2 ДИСПРОЗИЙ 67 Но 164,9304 и 4/n 6s2 ГОЛЬМИЙ 68 Ег 167,26 4/ 2 6s2 ЭРБИЙ бэТт 168,9342 4/'3 6s2 ТУЛИЙ 70 Yb 173,04 4PI46s2 ИТТЕРБИЙ 71 Lu 174,97 4/г,45</1 6s2 ЛЮТЕЦИЙ
н о и д ы
97 Bk [247] 578 6d 7s2 БЕРКЛИЙ 98 Cf [251] 10 2 5Л10 7s2 КАЛИФОРНИЙ 99 Es [254] 5p"7s2 ЭЙНШТЕЙНИЙ loo Fm [257] 5/12 7s2 ФЕРМИЙ ioi Md [258] 5 A13 7s2 МЕНДЕЛЕВИЙ 102 (No) 12551 (НОБЕЛИЙ) юз (Lr) [256] 57м 6</'7s2 (ЛОУРЕНСИЙ)
В квадратных скобках приведены массовые числа наиболее устойчивых изотопов, в круглых скобках, не являются общепринятыми.
42
Число электронов, которые могут располагаться на том или ином уровне (то есть иметь энергию, соответствующую этому уровню), различно для разных уровней. На s-уровне любой электронной оболочки может находиться один электрон или два, но не более. Число электронов на р-уровне может достигать шести, на d-уровне их может быть десять, а на /-уровне — не более четырнадцати. В результате и ёмкость оболочек оказывается неодинаковой: первая вмещает лишь два электрона, вторая — 8, третья — 18 и т. д.
В атомах, находящихся в наиболее устойчивых (основных) состояниях, электроны стремятся занять уровни с минимальной энергией, и лишь когда такие уровни заполнены, электроны оказываются на более высоких уровнях. По своей энергии уровни образуют следующий ряд:
ls<2s<2p<3s<3p<4s^3d<4p<5s« ~4d<5p ...
Это и определяет размещение электронов в атоме по уровням и по оболочкам.
Конкретное распределение электронов по энергетическим уровням данного атома называют электронной конфигурацией. Например, для атома калия она такова:
ls22s22p63s23p64s1.
Это означает, что в атоме калия электроны распределены по четырём оболочкам. Первая оболочка содержит два электрона на s-уровне (1s2); вторая — два электрона на s-уровне и шесть на р-уровне (2s22p6); третья построена, как и вторая (3s23p6); к четвёртой оболочке атома калия относится один электрон, расположенный на s-уровне (4s1). На рис. 5 в статье Атом показана электронная конфигурация для элементов первых четырёх периодов.
Химические свойства элементов в основном зависят от электронов высших энергетических уровней; поэтому в записи электронной конфигурации часто указывают только их (например, для калия 4s1). Так и сделано в таблицах 1 и 2.
Сопоставление строения электронных оболочек атомов с положением элементов в периодической системе позволяет установить ряд важных закономерностей.
Появление первого электрона, относящегося к очередной внешней оболочке, совпадает с началом нового периода (щелочные металлы). Литий (конфигурация 2s1) открывает второй период, натрий (3s1) — третий, калий (4s1) — четвёртый и т.д. Таким образом, номер периода равен общему числу электронных оболочек у атомов данного элемента.
У элементов главных подгрупп число электронов, относящихся к внешней оболочке, равно номеру группы AL Для щелочных металлов (1а-подгруппа) конфигурация имеет вид ns1, где п — номер периода; для щелочноземельных металлов (Па-подгруппа) — ns2, а для элементов прочих главных подгрупп nsbipN-2 (исключение — гелий, атом которого в основном состоянии не имеет р-электронов). Это позволяет понять, почему максимальное значение положительной степени окисления для элементов 1а—VIIa-подгрупп обычно равно V. Вместе с тем, целиком заполненная единственная оболочка атома гелия и восьмёрки электронов (электронные октеты), составляющие внешние оболочки прочих инертных элементов, обладают особой устойчивостью, и поэтому элементы VIПа-подгруппы не достигают высоких степеней окисления и вообще не склонны к образованию химических соединений (хотя такие соединения известны).
Электронная конфигурация элементов побочных подгрупп (переходных металлов) определяется более сложными законами. Здесь при увеличении атомного номера очередной электрон чаще всего попадает на одну из внутренних недостроенных оболочек. Правда, для элементов 16- и 116-подгрупп электронная конфигурация внешних оболочек такая же, как для элементов соответствующих главных подгрупп, то есть ns1 и ns2. Однако в химических проявлениях этих элементов существенную роль играют и электроны заполненных d-уровней предшествующей (п—1)-й оболочки. Эти электроны имеют примерно такую же энергию, как и электроны уровней ns, и они часто также принимают участие в образовании химических связей. Поэтому здесь электронную конфигурацию атомов обычно записывают в виде (n— IJd^ns1 и (n—l)d10ns2; например, для элементов 4-го периода (п=4) меди и цинка конфигурация такова: Cu&ms1, Zn 3d104s2.
Электронная конфигурация элементов побочных II16— VI 16-подгрупп (кроме лантаноидов и актиноидов) чаще всего имеет вид (п—l)d^“2ns2; например, для марганца (элемента 4-го периода подгруппы VI16) 3d64s2. От этого правила отклоняются только хром, молибден и ниобий (см. таблицу 1). На двух высших энергетических уровнях металлов VII 16-подгруппы [уровни (п—l)d и ns] располагается 8, 9 или 10 электронов соответственно для трёх последовательных столбцов. Чаще всего два из них — это s-электроны, остальные — d-электроны (но и здесь есть исключения).
В рядах лантаноидов и актиноидов с увеличением заряда ядра происходит заполнение /-уровня. Их электронные конфигурации также можно было бы изобразить в виде общих формул; однако это и слишком громоздко, и число исключений здесь значительно больше. Конфигурации высших энергетических уровней этих элементов даны в таблицах 1 и 2.
Важно отметить, что, поскольку с увеличением числа электронов разность между энергиями высших уровней становится всё меньше и меньше, однозначное определение электронной конфигурации делается всё более затруднительным. Так, у одного из . актиноидов — протактиния — энергии состояний 5/26d17s2, 6d37s2, 5/16d27s2, 5/36d°7s2 настолько близки, что очень трудно установить, какая из этих записей лучше передаёт истинную картину; по-видимому, эти конфигурации достаточно свободно переходят друг в друга.
У всех элементов побочных подгрупп, несмотря на различие их электронных конфигураций, имеется одна общая особенность: их внешние оболочки содержат не более двух s-электронов и не содержат р-электронов.
Источником сведений об электронных конфигурациях служат главным образом атомные спектры (см. Атом). Современное состояние
43
теории, к сожалению, далеко не всегда позволяет дать чёткое обоснование наблюдающегося распределения электронов по энергетическим уровням. Тем не менее можно констатировать, что общая картина электронных конфигураций обнаруживает важную тенденцию: строение внешних оболочек элементов, относящихся к одной подгруппе, то есть близких по свойствам, чаще всего однотипно (или сходно). Кроме того, последовательный переход от одной подгруппы к другой сопровождается закономерным изменением электронной конфигураций.
Как уже было отмечено, у элементов главных подгрупп, кроме инертных элементов, число электронов на внешней оболочке обычно равно максимальной положительной степени окисления. В каждом периоде число валентных электронов растёт, и с этим, естественно, можно связать ослабление металлических свойств. Таким образом, закономерности, обнаруживающиеся в строении атомов, объясняют периодический закон.
Здесь необходимо, однако, сделать важную оговорку. Всё сказанное выше об электронных конфигурациях относится к изолированным, то есть к свободным, атомам. При образовании тех или иных веществ, при химических взаимодействиях, электроны в атомах перестраиваются, причём в разных случаях по-разному. Более того, благодаря обобществлению электронов при образовании связей между атомами сама индивидуальность атома в молекуле оказывается достаточно условной — электронное облако одного атома непрерывно переходит в электронное облако другого атома. В результате атомы изолированные и связанные имеют существенные различия. И при современном уровне химической теории проследить за тем, как происходит переход от свободного атома к атому в веществе, отнюдь не просто.
Между тем в периодическом законе сопоставляются свойства не свободных, а взаимодействующих атомов. Поэтому, говоря о законе Менделеева, с одной стороны, и о строении свободных атомов, с другой, важно понимать, что здесь имеется в виду не «математически точная» взаимосвязь, а только тенденция: аналогия в строении изолированных атомов приводит к подобию в их химическом поведении.
ЗНАЧЕНИЕ И ИСТОРИЯ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
Д. И. Менделеев открыл носящий его имя закон в 1869 г. (первоначальный вариант периодической системы помечен 17 февраля
ОТ1ЫТЪ СИСТЕМЫ ЭЛЕМЕНТОВЪ
ОСНОВАННОЙ НА ИХЪ АТОМНОМЪ ВЬСЪ И ХИМНЧЕСКОМЪ СХОДСТВА
Tl=50 V =51
Сг = 52
Мп -55
Ее — 56
Ni=Co=59
Н-1 Си-63,4
Ве= 9,4 Mg=24 Zn = 65,2
В=11 At = 27 Л ?=68 С = 12 Si =28 ? = 70
N=14 Р = 31 AS = 75 0 = 16 S=32 Se=79,4
F = 19 Cl =35 Br =80 Li = 7 Na = 23 K=39Rb=85,4
Ca=4O Sr =87,6 ?=45 Ce=92
2r= 90 Nb = 94 Mo= 96 Rh = 104,4 Ru -104,4
PI = 106,6
Ag = 108 Cd= 112 Ur- 116 Sn = H8 Sb- 122
Те = 128?
I =127
Cs = 133 Ba = i 37
? = 180.
Ta = 182 W = 186.
Pl = 197,4 lr = 198
Os = 199.
Hg-200
Au -197?
Bl =210?
Tl = 204
Pb = 207
?Er=56 La-94 ?Yt=60 Di = 95
?ln = 75^Th= 118?
Д. Мендельевь
Рис. 3. «Опыт системы элементов, основанной на их атомном весе и химическом сходстве». Таблица опубликована Д. И. Менделеевым отдельным изданием 17 февраля (1 марта) 1869 г.
1869 г. по старому стилю, то есть 1 марта 1869 г. по новому).
Пожалуй, о революционной роли, которую сыграл закон Менделеева в химической науке, лучше всего сказал сам автор этого открытия. «До периодического закона,— писал Д. И. Менделеев,— элементы представляли лишь отрывочные случайные явления природы; не было повода ждать каких-нибудь новых, а вновь находимые были полной неожиданной новинкой. Периодическая законность первая дала возможность видеть неоткрытые ещё элементы в такой дали, до которой невооружённое этой закономерностью зрение до сих пор не достигало».
Открытие периодического закона позволило Менделееву дать блестящий образец научного предвидения. В 1870 г. он предсказал существование трёх ещё неизвестных тогда элементов, которые назвал экасилицием, экаалюминием и экабором,— они должны были заполнить пустые клетки в периодической таблице. Более того, основываясь на свойствах соседей этих гипотетических элементов по периодической
44
системе, Менделеев сумел правильно предсказать и важнейшие свойства новых элементов (из этого соседства вытекали и принятые Менделеевым условные названия). Вскоре француз Лекок де Буабодран открыл галлий (1875 г.), швед Л. Ф. Нильсон —скандий (1879 г.), а немец К. Винклер — германий (1886 г.). Свойства этих элементов так поразительно совпадали со свойствами, предсказанными Менделеевым для экаалюминия, экабора и экасйли-ция, что сомнению в справедливости периодического закона уже не оставалось места.
Ко времени построения Менделеевым первого варианта периодической системы химикам были известны 63 элемента. За прошедшие после этого сто с лишним лет список обнаруженных в природе и искусственно полученных элементов обогатился ещё 42 названиями. В июле 1974 г. советские физики сообщили о синтезе ещё одного нового элемента, с атомным номером 106.
Все новые элементы нашли своё «законное» место в периодической системе. Правда, это потребовало значительного преобразования таблицы. Оказалось необходимым, например, ввести группу инертных элементов, семейства лантаноидов и актиноидов. И всё же современная периодическая таблица в своих основных чертах весьма близка к таблице, предложенной Менделеевым.
В 1869 г., когда Менделеев открыл свой закон, какие бы то ни было сведения о внутреннем строении атомов отсутствовали. Поэтому при систематизации элементов Менделеев, естественно, не мог исходить из заряда атомного ядра. Он расположил элементы по возрастанию атомных масс (во времена Менделеева и вплоть до последних лет в химической литературе эти величины было принято называть атомными весами). В формулировке Менделеева периодический закон звучал так: «...Если все элементы расположить в порядке по величине их атомного веса, то получится периодическое повторение свойств».
Последнее верно в той мере, в какой последовательность средних атомных масс, определяемых соотношением количеств изотопов, согласуется с последовательностью зарядов ядер. По большей части такое согласие действительно наблюдается, что и позволило Менделееву открыть периодический закон. Но есть и расхождения. * Так, средняя атомная масса теллура (Z=52) равна 127,6 углеродных единиц, а иода (Z=53)— 126,9 углеродных единиц. Однако, глубоко убеждённый в справедливости периодического закона, Менделеев в первом же варианте таблицы правильно объединил иод с прочими галогенами, а теллур — с химически подобными ему кислородом, серой и селеном, хотя это не соответствовало последовательности атомных масс.
Открытию Менделеева предшествовали многочисленные попытки классификации элементов, начавшиеся ещё в конце XVIII в. (тогда уже было известно около 30 элементов). На этом раннем этапе наибольшую роль сыграли работы великого французского химика А. Лавуазье, который в 1789 г. разделил элементы (простые вещества) на металлы и неметаллы.
В первой половине XIX в. с развитием идей атомистики появились первые опыты систематизации элемен
тов по атомным массам. Так, немецкие химики И. Де’ берейнер и М. Петтенкофер указали, что некоторые сходные по свойствам элементы (например, Li, Na, К и Mg, Са, Sr, Ва) образуют ряды, в которых атомные массы возрастают по закону арифметической прогрессии.
В 1864 г. немецкий химик Л. Мейер составил таблицу, в которой по атомным массам сопоставлялись группы сходных химических элементов. Позже (в 1870 г., то есть уже после опубликования работы Менделеева) он дал удачную иллюстрацию периодического закона с помощью кривой, отражавшей зависимость атомных объёмов от атомных масс. В том же 1864 г. англичанин У. Одлинг поместил в своём учебнике химии таблицу, где известные тогда элементы были расположены с учётом атомных масс; сходные по свойствам элементы оказывались в горизонтальных строках, вертикальные столбцы были очень близки к рядам, которые двумя годами позже в работе английского химика Дж. Ньюлендса получили название «октав». Ньюленде, располагая элементы в порядке увеличения атомных масс, констатировал, что свойства каждого восьмого элемента повторяют свойства первого, подобно чередованию нот в музыкальной гамме. Однако Ньюленде не считался с возможностью существования ещё не открытых элементов и поэтому у него оказывались формально родственными такие разные по свойствам элементы, как хлор и кобальт, бром и палладий, и т.д.
Таблицы Мейера, Одл ин га и Ньюлендса в некоторых своих чертах похожи на таблицу Менделеева. Но ни один из этих учёных не решился на основании подмеченной периодичности предсказать новые химические элементы. Ни один из них не сумел в полном объёме охватить совокупность физических и химических свойств, обнаруживающих всю глубину периодического закона. Для них периодичность была лишь удобным способом классификации — они не увидели в ней фундаментального закона природы.
Это оказалось под силу только Менделееву. И сегодня его приоритет в открытии периодического закона признан научной общественностью всего мира. Когда в 1955 г. американский учёный Г. Сиборг и его сотрудники путём ядерной реакции получили элемент № 101, они назвали его менделевием «... в признание ведущей роли великого русского химика Дмитрия Менделеева, который был первым, кто использовал периодическую систему элементов для предсказания химических свойств еще не открытых элементов ...».
Создание периодической системы тотчас же поставило перед учёными новую задачу: найти физическое обоснование вновь обнаруженного закона. На это указывал и сам Менделеев, который писал о периодическом законе, как о «новой тайне природы, еще не поддающейся рациональной концепции». Такой рациональной концепцией оказалось учение о строении атома.
В 1913 г. английский физик Г. Мозли разработал метод экспериментального определения величин зарядов ядер по рентгеновским спектрам элементов и ввёл термин «атомный номер».
45
Опытным путём он установил, что заряды ядер изменяются в соответствии с порядковыми номерами элементов в периодической таблице. Это позволило объяснить некоторые остававшиеся непонятными факты, например то, что теллур предшествует в ряду элементов иоду, хотя и имеет большую атомную массу.
Но самой крупной вехой в физическом осмыслении периодического закона явилась теория великого датского учёного Нильса Бора, который в 1921 г. высказал мысль о том, что периодичность свойств атомов определяется периодическим строением их электронных оболочек.
Подобно тому как Менделеев предсказал факт существования и свойства ряда новых элементов, Бор сумел предсказать электронную конфигурацию и спектр тогда ещё неизвестного элемента с Z=72 — гафния. Кстати, существование гафния (элемента с атомной массой, близкой к 180) предвидел и Менделеев.
В настоящее время мы выводим электронные конфигурации атомов, периодичность свойств элементов на основании квантовой механики — из уравнения Шредингера и принципа Паули (см. Атом). Квантовая механика позволила подвести под периодическую систему физическую базу. Но и периодическая система, в свою очередь, немало способствовала созданию и развитию квантовой механики.
Значение периодического закона (как и всякого фундаментального закона природы) огромно. В длинном ли, в коротком ли варианте периодическая система неизменно занимает почётное место на стене каждой химической лаборатории, на рабочем столе каждого химика — без использования этого универсального аппарата невозможно ни изучение основ химии, ни исследование её актуальных проблем. И недаром в нашей Энциклопедии статью о каждом химическом элементе мы начинаем с изображения клетки — места элемента в системе Менделеева.
Многочисленные применения периодического закона в науке и технике практически невозможно перечислить. Стремительное развитие науки, которым ознаменовалось двадцатое столетие, расширило сферу использования этого закона, вывело его далеко за пределы чистой химии. И какой бы области естествознания вы ни посвятили себя в дальнейшем — исследованию Земли, Океана или Космоса, изучению тончайших процессов, происходящих в живых организмах, или созданию новых материалов, которых ещё не знает сегодняшняя техника,—периодический закон будет служить вам путеводной звездой в многотрудном деле познания и преобразования мира.
П. М. Зоркий.
*Мощь и сила науки —во множестве фактов, цель —в обобще-нии этого множества и возведении их к началам,; Собрание фактов и гипотез —ещё не наука; оно есть только преддверие её, но преддверие, мимо которого нельзя прямо войти в святилище науки. На этих преддвериях надпись — наблюдение, предположение и опыт'».
Д. И. Менделеев (Соч., т. 15, 1949, с. 138)
КАК ПОЛЬЗОВАТЬСЯ ЭНЦИКЛОПЕДИЕЙ.
Книга посвящена неорганической химии.
Все известные на сегодня химические элементы описаны как в отдельных статьях («Алюминий», «Иод», «Уран» ...), так и в групповых («Лантаноиды», «Щелочные металлы» ...). Важнейшие неорганические соединения, технические продукты и материалы также описаны в самостоятельных статьях («Азотная кислота», «Алюминиевые сплавы», «Цемент»...). Сведения о многих и многих других веществах, которым не посвящено отдельных статей, вы найдёте, во-первых, при описании самих элементов, а во-вторых, в объединяющих статьях (например, об окислах азота в статье «Азот», о нитрате калия в статьях «Минеральные удобрения», «Нитраты», «Селитры»). В таких случаях вашим поискам поможет указатель, данный в конце книги.
Все эти и подобные им статьи, составляющие основной массив информации по неорганической химии, помещены в алфавитном порядке. Вне алфавита идут три фундаментальные статьи — «Атом» и «Закон Менделеева» (вступительные) и «Химия служит человеку» (заключительная). Биографии выдающихся учёных тоже помещены не в алфавитном порядке, а внутри наиболее подходящих по теме статей (например, биографии Д. И. Менделеева и Н. Бора в статье «Закон Менделеева»).
Заглавное слово, то есть название каждой отдельной статьи, даётся жирными прописными буквами с ударением над гласной буквой (например, БИОСФЕРА, НОМЕНКЛАТУРА НЕОРГАНИЧЕСКИХ СОЕДИНЕНИИ, ЭРБИИ). В дальнейшем тексте статьи её заглавие обозначается только начальной буквой или начальными буквами (в перечисленных примерах — «Б.», «Н. н. с.», «Э.»). Если после названия статьи даётся в разрядку другое слово (или несколько слоъ), то это означает, что наряду с первым, основным термином существуют и его общеупотребительные синонимы [например, АЛЮМИНИЯ ОКИСЬ, оке ид алюминия (III), глинозём, ...].
Выделение какого-либо слова косым шрифтом—курсивом (например, хлор в статье «Галогены») — означает, что в книге есть отдельная статья («Хлор») и что к ней следует обратиться за дополнительными сведениями. Такая система взаимных ссылок помогает установить связи между отдельными понятиями, разбросанными в Энциклопедии по воле алфавита. Система ссылок способствует и наиболее экономному изложению каких-либо общих вопросов. Так, читая статью «Железо», раздел «О магнетизме железа», вы познакомитесь с самыми общими представлениями о магнетизме вообще; в статьях же о других ферромагнетиках («Кобальт», «Никель», «Лантаноиды») эти общие положения не повторены, а заменены ссылкой (см. Железо).
Статьи, сходные по теме (например, статьи о химических элементах) написаны по одному плану. Наиболее крупные из них («Азот», «Вода», «Золото» ...) разбиты на части, каждая из которых имеет подзаголовок.
Во многих случаях (а для химических элементов — во всех без исключения) указано происхождение слова (этимология). В словах латинского происхождения этимология даётся в латинской транскрипции, в словах греческого и арабского — в русской.
АЗОТ (лат. Nitrogenium) N — химический элемент V группы периодической системы Менделеева; атомный номер 7, атомная масса 14,0067. Свободный А.— газ без цвета и запаха—состоит из двухатомных молекул N2. Природный элемент — смесь двух стабильных изотопов: 14N (99,635%) и 16N (0,365%).
Историческая справка. А.— преобладающая составная часть атмосферы. Земли. В атмосфере он и был открыт почти одновременно несколькими исследователями в конце XVIII в. Это был период, когда исследование газов — и, конечно, прежде всего воздуха — приобрело широкий размах и составило даже новое направление в химии, названное пневматической химией (от греч. слова «пнёума» — дуновение, ветер).
В 1772 г. английский учёный Д. Резерфорд провёл интереснейшие эксперименты. Он сжигал на
воздухе под стеклянным колоколом «углистые вещества» и образующуюся двуокись углерода СО2 связывал раствором едкой щёлочи. Резерфорд нашёл,- что остающаяся после этого часть воздуха — это газ, который уже не поддерживает ни горения, ни дыхания. Тот же самый газ остаётся после сжигания в таких же условиях серы или фосфора. Подобные наблюдения сделали и другие учёные: в Швеции — К- Шееле, в Англии — Г. Кавендиш и Дж. Пристли, во Франции — А. Лавуазье.
Новый газ назвали «мефитическим» (удушливым) воздухом. По мере изучения А. предлагались и другие названия, отражающие иные его свойства: «нитроген» (по-русски «селитротвор» ;— творящий, рождающий селитру), «алкалиген» (рождающий щёлочь — аммиак, который тогда называли «летучей щёлочью»).
Знаменитый Лавуазье и другие создатели первой химической номенклатуры (1787 г.) предложили термин азот (от греч. «а» — отрицательная приставка и «зоэ» — жизнь) — безжизненный; этот термин сохраняется во французском и русском языках. По этому же признаку построено и немецкое- название А.— Stickstoff (удушливое вещество). Название Nitrogenium в основе своей сохранилось в английском, итальянском и испанском языках.
Но не прошло и полувека, как выяснилось, что «безжизненный» А.— это один из главных элементов жизни. В 1836—40 гг. французский учёный Ж. Буссенго, один из основателей молодой тогда науки агрохимии, экспериментально доказал: без участия А. невозможно развитие растений. Получают же растения А. в виде его
соединений из почвы. С тех пор внесение на поля азотных удобрений стало одним из главных приёмов повышения плодородия почвы. С селитрой связывалось столько надежд землепашцев! Но ограниченность её природных месторождений ставила эти надежды под угрозу. Со временем была выдвинута и решена важная проблема: связывание свободного А. воздуха и производство его соединений в огромных масштабах.
Свою немалую роль в быстром решении этой проблемы сыграло и совершенствование военной техники. Ведь селитра — обязательная составная часть чёрного пороха, известного в Европе ещё с XIIГ в. И почти все современные взрывчатые вещества, несравненно более мощные, чем чёрный порох, это тоже соединения А.
Азот в природе. А.— один из самых распространённых элементов. В космосе он занимает четвёртое место — вслед за водородом, гелием и кислородом. Он — главная составляющая атмосферы Земли: 78,09% по объёму или 75,6% по массе (в общей сложности около 4-1016 т). Среднее содержание А. в земной коре, конечно, гораздо меньше: 1,9-10“3% (по массе). Но всё-таки каждая тонна горных пород заключает в себе около 10 л свободного А. Свободный А., наряду с аммиаком NH3 и хлоридом аммония NH4C1, присутствует в вулканических газах. Органические соединения А. находятся в нефти и угле. Живые организмы содержат около 0,3% А. в виде соединений.
Присутствие в почвах связанного А.— непременное условие плодородия земли. Получая из почвы минеральные соли, содержащие А., растения используют его для биосинтеза белков, нуклеиновых кислот, витаминов, хлорофилла — важнейших для жизни веществ. Животные, питаясь растительной пищей, вначале расщепляют растительные белки, чтобы затем из их структурных фрагментов — аминокислот — построить свои белковые структуры, свои живые ткани.
Из природных источников А. промышленное значение имеют лишь свободный А. атмосферы и месторождения двух его минералов — чилийской (NaNO3) и индийской (KNO3) селитр.
Круговорот А. в природе (см. рисунок)— сложный процесс, в котором решающую роль играют живые организмы. Особые бактерии, живущие в почве, связывают свободный А. воздуха, превращая его в минеральные соли
АЗОТ
48
Круговорот азота в природе. Весь воздушный океан, на дне которого мы живём,— это изобилие свободного азота N2 (78,09% по объёму в атмосфере). Однако и соединения азота встречаются всюду — в воздухе, воде, почве. На рисунке на фоне голубого неба и зелёных растении выделяются оранжевые пятна. Разумеется, таких пятен в природе нет. Художник условно изобразил оранжевым цветом соединения азота. Такой азот называют связанным, или фиксированным. Оранжевыми стрелками показан путь соединений азота, а белыми — движение свободного азота.
Рядом с грозовым фиолетовым облаком вы видите символическое — оранжевое (/). Здесь как бы сконцентрированы окислы азота, образующиеся в атмосфере при грозовых разрядах и действии ультрафиолетовых лучей. Сюда же при извержении вулканов поступает аммиак NH3 (2). Оранжевые стрелки (3 и 4), протянувшиеся от облака к почве и воде, показывают путь, который проходят эти соединения вместе с атмосферными осадками. Итак, почва и вода частично обогащаются связанным азотом, идущим прямо из атмосферы. Но главный источник связанного азота в почве — биохимические процессы.
Слева из общей картины «вырезано окно» в микромир почвы. Заглянем в него через гигантское увеличительное стекло. Неутомимо трудятся здесь азотфиксирующие бактерии (5). Они переводят в соединения азот воздуха, проникающего через взрыхлённую почву. Связанный азот в виде ионов NH * н NO“^ идёт на питание растений, всасывается через корни (6). Растения же, в свою очередь, служат единственным источником азотного питания для животных. В живых организмах происходят химические превращения, в результате которых азот оказывается в составе сложных органических соединений.
После отмирания растений и животных азотсодержащие органические соединения возвращаются в почву, где работают и другие микроорганизмы — денитрифицирующие бактерии. Они разлагают азотистые соединения и возвращают в атмосферу свободный азот. Посмотрите, как оранжевый цвет стрелки, идущей вверх, переходит в белый (7, £). Часть же соединений азота «запасается» в природе на сравнительно долгое время, отлагаясь в составе торфяников и углей (9).
А вот оранжевый островок в воде (/О). Здесь связывают азот некоторые водоросли (например, синезелёные). Сюда же, в Мировой океан, ручьи и реки переносят из почвы нитраты и другие растворимые соединения азота (//). И тут, как и в почве, действуют денитрификаторы, возвращающие свободный N2 в атмосферу (/2).
В центре рисунка вы видите город. Чтобы повысить урожай полей, люди вносят в почву миллионы тонн азотных удобрений; на больших площадях выращивают азотфиксирующие бобовые культуры (оранжевая стрелка протянулась из громады домов на поля).
Итак, в каждой из сфер Земли — атмосфере, гидросфере, литосфере — присутствует и свободный, и связанный азот. Равновесие между ними поддерживается процессами, о которых мы рассказали. Все они вместе и составляют круговорот азота в природе.
49
АЗОТ
и доставляя таким образом почве около 25 кг связанного А. в год на каждом гектаре. Клубеньковые же бактерии, живущие в земле не изолированно, а в симбиозе с бобовыми растениями, накапливают на каждом гектаре даже 150—400 кг связанного А. в год. Активно связывают А. воздуха и микроскопические синие водоросли, обитающие во влажной почве или в водной среде. Кроме того, А. попадает в почву в виде органических соединений после отмирания растений и животных. Бактерии разлагают органические вещества и превращают А. в аммиак и нитраты (в последнем случае говорят: происходит нитрификация). Но есть в почве и другие бактерии, действующие в обратном направлении: из ценных почвенных нитратов они высвобождают N2. Это денитрификация. В результате свободный А. возвращается в атмосферу.
Физические и химические свойства. Говоря о свойствах А., следует различать химический элемент N и простое вещество N2.
Подробно о различии понятий «химический элемент» и «простое вещество» рассказано в статье Элементы химические (ведь это различие относится не только кА., но и к водороду, кислороду, углероду и всем вообще элементам).
И всё же мы обращаем на это внимание читателя в первой (по алфавиту) статье Энциклопедии. Рассказать о химических свойствах элемента — А., значит выявить его возможные валентности и степени окисления в соединениях с другими элементами, возможность существования и устойчивость тех или иных соединений, характер химических связей атома N с атомами других элементов. При этом не столь уж важно, получены ли эти соединения прямым путём (например, NH3 — при взаимодействии N2 и Н2) или только косвенным (например, NC13—действием С12 на NH3). Для характеристики элемента А. важно, например, что существуют галогениды, в которых А. трёхвалентен и образует ковалентные связи с галогенами. Описывая химическую индивидуальность А., мы должны объяснить (исходя из знания электронной структуры атома N), почему невозможно существование галогенидов типа NX5... (см. стр. 50).
Изучая простое вещество А., говорят прежде всего о свойствах газа, состоящего из молекул N2 (при обычных условиях), и об условиях превращения его в жидкость и твёрдое тело. Охарактеризовать химию простого вещества А., значит рассказать о том, в какие реакции и при каких условиях он вступает. Например, когда мы говорим, что свободный А. не способен реагировать с галогенами, речь идёт о свойствах простого вещества N2. Итак, сначала о свойствах свободного А.— простого вещества.
Свободный А.— газ, несколько легче воздуха (плотность 1,25 г/л при 0 °C и давлении 760 мм ртутного столба). Он плохо растворяется в воде (23,3 г в 1 м3 воды). При нормальном давлении и температуре —195,8°С А. сжижается, а при —210 °C затвердевает. Плотность его в жидком состоянии 0,81 г/см3. Молекула свободного А., как мы уже знаем, состоит из двух атомов: N2. По прочности такая молекула
4 хэш
почти не имеет себе равных: чтобы разорвать её на отдельные атомы, нужно затратить очень большую энергию, 945 кдж/моль (225 ккал/моль). Даже при 3300°С только одна молекула N2 из тысячи распадается на атомы. Поэтому-то свободный А. так инертен в обычных условиях: для того чтобы он вступил в реакцию с другими простыми или сложными веществами, необходима предварительная активация молекул. Она достигается нагреванием, облучением, действием катализатора или другими способами.
Из всех металлов свободный А. реагирует в обычных условиях только с литием, давая нитрид Li3N; с натрием, магнием, кальцием, титаном подобная реакция идёт лишь при нагревании. С большинством других — менее активных — металлов А. не взаимодействует вообще. Из свободных неметаллов А. соединяется лишь с кислородом, водородом, углеродом, бором. Успешно же такие реакции идут только при весьма жёстких условиях. Так, с водородом А. взаимодействует с заметной скоростью при нагревании, повышении давления и в присутствии катализатора (см. Аммиак). Для окисления А. кислородом нужна электрическая дуга, причём не более 5% А. вступает в реакцию, образуя окись азота NO. Этот процесс требует больших затрат электроэнергии, поэтому он не получил широкого распространения в промышленности. В природе же такой процесс происходит повсеместно. Это легко понять: взаимодействие А. с кислородом воздуха при грозовых, разрядах подобно реакции в электрической дуге:
N2 + O2 = 2NO
Окись азота затем легко превращается в двуокись NO2:
2NO + О2 = 2NO2
Растворение же NO2 в дождевой воде в присутствии кислорода воздуха даёт азотную кислоту:
4NO2 + О2 + 2Н2О = 4HNO3
Подсчитано, что таким путём почва получает от 4 до 10 кг связанного А. на гектар в год.
Пропуская N2 через раскалённый кокс, удаётся'синтезировать соединение А. с углеродом — циан (точнее, дициан) N=C—feN. Когда такую реакцию проводят в присутствии щёлочи, получают цианиды, например:
2КОН + 4С + N2 - 2KCN + 2СО + Н2
С карбидом кальция, нагретым до 1000 °C, А. даёт Ca=N—C=N (цианамид кальция):
СаС2 + N2 = CaCN2 + С
Эта реакция легла в основу одного из первых методов промышленного связывания А. воз
АЗОТ
50
духа: цианамид служил исходным веществом для получения аммиака. Однако цианамидный способ сопряжён с огромным расходом энергии. Он не нашёл широкого применения и не выдержал конкуренции с прямым синтезом аммиака из А. и водорода.
Вот и все (точнее, почти все) химические превращения свободного А., которые удавалось осуществить до последнего времени. Такая его инертность доставляла и доставляет много хлопот химикам и технологам. Но нетрудно представить себе, как изменилась бы природа, не будь атмосферный А. столь инертен: Землю залили бы потоки азотной кислоты, в воздухе не осталось бы кислорода... И ещё — если бы кислород воздуха не был «разбавлен» инертным А., всё живое было бы обречено на гибель: длительное вдыхание чистого кислорода при атмосферном давлении не менее губительно, чем отсутствие этого живительного газа...
Не так давно были получены новые данные, заставляющие пересмотреть обычное представление о труднопреодолимой инертности молекулы А. Советские химики М. Е. Вольпин и В. Б. Шур обнаружили, что N2 легко реагирует с комплексными соединениями многих переходных металлов — титана, ванадия, хрома, молибдена, железа ... Реакции протекают при комнатной температуре (и даже при отрицательных температурах!), да к тому же при атмосферном давлении. Таким образом, открыты новые возможности введения А. в реакции, которые во многом напоминают процессы усвоения свободного А. микроорганизмами. Успех дальнейших работ в этом направлении означал бы подлинную революцию в химии А.
Теперь обратимся к химическим свойствам элемента А. Способность какого-либо элемента давать соединения с другими элементами, тип связей атомов друг с другом, прочность этих связей, возможные степени окисления — все эти характеристики связаны со структурой внешней электронной оболочки атома. Для А. эта структура такова: 2s22p3. Иными словами, внешняя электронная оболочка атома А. содержит 5 электронов. Поскольку радиус атома А. невелик (всего две электронные оболочки!), способность ядра в атоме А. удерживать, притягивать электроны наружной оболочки велика. Как велика? Эту способность часто оценивают величиной электроотрицательности элемента, для расчёта которой есть специальные методы. Оказывается, по своей электроотрицательности (равной 3,0) А. уступает только кислороду и фтору (электроотрицательности 3,5 и 4,0 соответственно). Поэтому А.— типичный неметалл, образующий с другими неметаллами ковалентные химические связи.
Строению наружной электронной оболочки отвечают возможные степени окисления, начиная от +5 и кончая —3. Стабильность наружной электронной оболочки достигается (как
и для других элементов второго периода) при образовании восьмёрки электронов («октета»). Учитывая, что в атоме А. есть три непарных электрона (см. Атом), понятно, почему молекула свободного А. двухатомна:
• • :n- + -n
Ведь в результате «обобществления» этих непарных электронов каждый атом А. в молекуле N2 завершает электронный октет.
Подобным же образом, обозначая точками наружные электроны атомов А. и крестиками — электроны атомов галогенов (X), легко представить себе, как достигается завершение электронных оболочек каждого из атомов в молекулах галогенидов NX3:
Поскольку «непарных» электронов в атоме А. только 3, ясно, почему невозможно существование галогенидов типа NX5.
Из производных А. наибольший интерес представляют его соединения с водородом и кислородом.
Водородные соединения. Важнейший их представитель — аммиак (NH3) — описан в отдельной статье. Осторожно окисляя аммиак гипохлоритом натрия в специальных условиях, получают H2N—NH2 (гидразин):
2NH3 + NaClO = N2H4 + NaCl + H2O
Гидразин — бесцветная жидкость, смешивающаяся с водой в любых отношениях. Подобно аммиаку, гидразин — слабое основание. С кислотами он образует соли гидразо-ния (названиедано по сходству с аммонием):
H2N—NH2 + НС1 = [H2N—NH3]+C1-
H2N—NH2 + 2HC1 = C1-[H3N—NH312+C1-
Степень окисления А. в гидразине —2; это сильный восстановитель, способный гореть на воздухе:
N2H4 + O2 = N2 + 2H2O
В гидроксил амине NH2OH степень окисления А. —1. В кислой среде это вещество — окислитель:
-1 +2 +3 -3
NH 2ОН + 2FeCl2+ЗНС1 - 2FeCl3 + NH4C1 + Н2О
В щелочной среде гидроксиламин — восстановитель:
-10 0-1
2NH2OH + I2 + 2КОН = N2 + 2KI + 4Н2О
51
АЗОТ
При взаимодействии гидразина с азотистой кислотой образуется азотистоводородная кислота HN3 или Н—N = N=N:
N2H4 + HNO2 = HN3 + 2H2O
Она устойчива только в разбавленных водных растворах, а в чистом виде или в концентрированных растворах взрывается от удара или при нагревании, распадаясь на аммиак и свободный А.:
3HN3 = NH3 + 4N2
Соли этой кислоты и тяжёлых металлов,, например азид свинца Pb(N3)2, взрываются от удара и потому используются в военной технике для изготовления капсюлей детонаторов. Азиды щелочных металлов при нагревании разлагаются без взрыва:
2NaN3-2Na + 3N2
Кислородные соединения. Известны окислы А., отвечающие всем степеням окисления от + 1 до -Их N2O — закись азота и NO — окись азота (оба — бесцветные газы), N2O3—азотистый ангидрид (синяя жидкость), NO2—двуокись азота (бурый газ), N2O5— азотный ангидрид (бесцветные прозрачные кристаллы). По современной номенклатуре неорганических соединений их называют соответственно оксид азота (I), оксид азота (II), и т.д. О существовании этих пяти окислов А. известно давно. Но их, структура, прежде казавшаяся вполне обычной, на самом деле далеко не проста, а химическое поведение каждого из этих окислов можно объяснить лишь на основе новейших представлений. Вот почему этого пока «не проходят» в средней школе.
Рассмотрим оксид азота (I) — закись N2O. Кажется вполне логичным представить себе связи атомов в этой молекуле так: N—О—N. Чёрточка здесь, как обычно, изображает связь между атомами, осуществляемую общей электронной парой. Поскольку число электронов на внешней оболочке у атома N равно 5 (обозначаем их точками), у атома О — 6 (обозначаем их крестиками), то электронная структура представилась бы так: N—О—N
: n г о: N : • XX •
Однако, приняв эту «структурную» формулу, мы вынуждены нарушить правило октета: атом N окружён не восемью, а лишь шестью электронами. Более того, в атоме N есть три непарных электрона, а по нашей структуре в образовании связи принимает участие лишь один неспаренный электрон. Почему? Это необоснованное допущение! Попробуем устранить «несправедливость».
Рассмотрим другой вариант структуры:
N = N
Кажется, теперь всё в порядке:
Правило октета не нарушено, все три непарных электрона каждого атома А. «работают», образуя электронные пары. Окислительное число А. равно +1, кислорода —2 ... И тем не менее физики скажут, что описанная нами модель неправильна. Ведь по данным рентгеноструктурного анализа в молекуле N2O все три атома выстроены в линию, а не по вершинам треугольника. Как же быть? Возможная последовательность атомов остаётся одна: NNO. Следовательно, лишь один атом А. связан с кислородом.
Легко видеть, что N2O содержит «неравноценные» атомы N. Крайний из них имеет окислительное число, равное нулю (ведь он связан с себе подобным атомом), а центральный атом имеет окислительное число +2 (ведь он связан с атомом кислорода, имеющим окислительное число —2). Ну, а «в среднем», каждый атом N формально имеет окислительное число +1: 5i2_= + l.
Электронная структура N2O может быть записана так:
То, что здесь уже сказано о структуре N2O, выглядит достаточно сложно (а ведь сказано ещё не всё!). Ещё сложнее структуры других окислов А. Современными методами установлено, что в твёрдом азотистом ангидриде N2O3 атомы N непосредственно связаны между собой (O2N — NO) и имеют разные окислительные числа (+4 для одного и +2 для другого атома). А твёрдый азотный ангидрид N2O5, оказывается, построен из (NO2) + и (NO3)_, связанных между собой ионной связью.
Интересные особенности поведения окиси NO и двуокиси NO2 связаны с тем, что и в той, и в другой молекуле есть неспаренный электрон. Поэтому для этих соединений (особенно для NO2) характерна димеризация — образование из двух молекул одной. Так, при охлаждении до комнатной температуры бурый газ NO2 конденсируется в красную жидкость, которая при дальнейшем охлаждении становится жёлтой, а при —10°С образует бесцветные кристаллы. Интенсивность окраски NO2 пропорциональна числу недимеризованных молекул. При полной димеризации окраска исчезает. Описанный процесс — это не просто физическое превращение газа в жидкость и далее в твёрдое тело, а химическая реакция, приводящая к спариванию электронов двух молекул NO2
O2N-+ NO2 O2N:NO2
По ряду причин, которые сложно объяснить, для окиси азота NO димеризация менее характерна и происходит при оч$нь низких температурах. Спаривание электронов возможно и при взаимодействии NO и NO2 между собой с образованием N2O3
охлаждение
ON + -NO2 ; • ~^ON:NOo нагревание
Легко видеть, что в N2O3 один из атомов N имеет окислительное число +2, а другой +4.
Существует и другая возможность избежать невыгодного состояния с неспаренным электроном в NO и NO2— отдать это5? неспаренный электрон. Так возникают соли типа (NO)+C1O7 и (NO2)+C1O7, в состав которых входят ионные остатки (NO)+ (нитрозоний) или (NO2)+ (нитроний).
4*
АЗОТИСТАЯ КИСЛОТА
52
Окисли N2O и NO являются несолеобразующими и не реагируют ни с водой, ни со щелочами. Окислу N2O3 соответствует слабая и неустойчивая азотистая кислота HNO2, образующая соли — нитриты. Самые же важные кислородные соединения А.— это азотная кислота, соответствующая его высшей степени окисления (+5), а также её соли — нитраты. Особое значение для живой природы имеют содержащие А. органические соединения.
Получение и применение. Огромные количества — больше 50 млн. т А. в год (1975 г.) — даёт промышленности разделение воздуха (ом. Воздух). В лаборатории А. получают нагреванием концентрированного раствора нитрита аммония:
NH4NO2 = N2 + 2H2O
На практике обычно нагревают смесь растворов сульфата аммония и нитрита натрия (в этом случае А. выделяется более равномерно):
(NH4)2SO4 + 2NaNO2 = 2N2 + Na2SO4 + 4H2O
Приготовленный таким образом А. бывает загрязнён закисью А. Исключительно чистый А. выделяется при термическом разложении азида натрия NaN3
Свыше 3/4 промышленного А. используют для синтеза аммиака, который служит исходным веществом в производстве азотной кислоты, удобрений, красителей, лекарственных препаратов, взрывчатых веществ. Свободный газообразный А. применяется и как инертная среда при получении синтетических волокон, чистых металлов и сплавов. Жидкий А. нужен для создания низких температур в криогенной технике — технике глубокого холода.
В заключение скажем, что из природного А. в промышленных масштабах выделяют его «тяжёлый» изотоп 15N. Он необходим в научных исследованиях, потому что может служить «меченым атомом».
А. обычно хранят под давлением в стальных баллонах, окрашенных в чёрный цвет и имеющих жёлтую надпись «Азот».
В. Л. Василевский.
АЗОТИСТАЯ КИСЛОТА HNO2— слабая одноосновная кислота, соответствующая степени окисления азота +3. В свободном состоянии неизвестна. Существует при низких температурах в разбавленных водных растворах. Уже при комнатной температуре разлагается:
3HNO2 = HNO3 + 2NO + Н2О
Водный раствор А. к. можно получить действием кислот на нитриты'.
NaNO2 + НС1 = NaCl + HNO2
Эту реакцию обычно применяют, когда необходимо участие А. к. в химическом процессе: она действует в момент выделения.
Взаимодействие нитрита бария с серной кислотой даёт раствор А. к., не содержащий посторонних ионов (сульфат бария практически нерастворим и выпадает в осадок):
Ba(NO2)2 + H2SO4 = BaSO4 + 2HNO2
А. к. можно получить также при взаимодействии азотистого ангидрида с водой (реакция обратима):
H,O + N2O3^2HNO2
Растворение в воде двуокиси азота NO2 даёт А. к. наряду с азотной кислотой:
2NO2 + Н2О = HNO3 + HNO2
А. к. может быть как окислителем, так и восстановителем, превращаясь соответственно в окись азота NO или в азотную кислоту HNO3. Соли А. к.— нитриты — вполне устойчивы в обычных условиях.
АЗОТИСТОКИСЛЫЕ СОЛИ—то же, что нитриты.
АЗОТИСТЫЙ АНГИДРИД, трехокись азота, оксид азота (III), N2O3—ангидрид азотистой кислоты. В обычных условиях нестабилен и распадается по уравнению (реакция обратима):
N2O3^±NO + NO2
При . пониженной температуре — синяя жидкость, кипящая при 3,5 °C с разложением и замерзающая при —102°С. Взаимодействие его с водой (0°С) даёт азотистую кислоту:
N2O3 + Н2О^2 2HNO2
С растворами щелочей образует нитриты1. N2O3 + 2NaOH = 2NaNO. + Н2О
Получается действием 50%-ной HNO3 на трёх-окись мышьяка:
2HNO3+As2O3+2H2O = 2H3AsO4+NO2+NO Смесь NO+NO2 при охлаждении даёт N2O3. См. также Азот.
АЗОТНАЯ КИСЛОТА HNO3 — сильная одноосновная кислота, соответствующая степени окисления азота +5. В свободном состоянии— бесцветная жидкость с резким удушливым запахом. В природе А. к. встречается редко: в небольших количествах её можно найти лишь в дождевой воде и воздухе, но только сразу же после грозы. А. к.— один из важнейших продуктов химической промышленности.
Физические и химические свойства. Плотность безводной А. к. 1,52 г/см3. При 84 °C А. к. кипит (с разложением), а при—41,15 °C замерзает, образуя бесцветные кристаллы. А. к. смешивается с водой в любых отношениях.
53
АЗОТНАЯ КИСЛОТА
Обычно смеси (растворы) жидкостей разделяют перегонкой: компонент с более низкой температурой кипения отгоняется первым. Однако получить этим способом чистую А. к. из её водных растворов нельзя.
Дело в том, что при кипении разбавленной А. к. сначала испаряется вода, с небольшим количеством HNO3. Раствор концентрируется. Но когда содержание А. к. возрастёт до 69%, такой раствор кипит при 121,8 °C уже как одно целое: в пар переходят совместно и HNO3 и Н2О(31%). Это — нераздельно кипящая, или азеотропная, смесь (от «а» — частица отрицания,; греч. «зёо» — киплю и «тропе» — изменение). Растворы А. к. более высокой концентрации (до 98,7%) можно получить перегонкой смеси с серной кислотой, а 100%-ную А. к.— вымораживанием раствора.
Чистая А. к. устойчива лишь ниже температуры плавления; расплавившись, она частично разлагается с выделением азотного ангидрида: 2HNO37ZiN2O5 + H2O
Равновесие этого обратимого процесса соответствует примерно 98% HNO3. А. к. остаётся бесцветной лишь в темноте. Под действием света она разлагается с выделением бурого газа — двуокиси азота NO2. Он-то и окрашивает А. к. в жёлтый цвет:
4HNO3 = 4NO2 + 2Н2О + О2
При пропускании паров А. к. через нагретую стеклянную трубку образуется смесь NO2 и кислорода. При более сильном нагревании разложение идёт глубже — до закиси азота или свободного азота:
2HNO3 = N2O + Н2О + 20 2
4HNO3 = 2N2 + 2Н2О + 5О2
Будучи очень сильным окислителем, А. к. в реакциях с металлами проявляет необычные для других кислот свойства. Лишь часть её идёт на образование соответствующей соли — нитрата, другая же часть выступает как окислитель, восстанавливаясь — в зависимости от природы металла и концентрации кислоты — +4 + 2 0 —3
до NO2, NO, N2 или NH3. Не подвержены действию А. к. лишь такие металлы, как золото, платина, родий, иридий и тантал.
Концентрированная А. к. при взаимодействии с металлами, или неметаллами обычно восстанавливается до NO2:
Си + 4HNO3 = Cu(NO3)2 + 2NO2 + 2Н2О
Неметаллы при этом окисляются до кислот с высшей степенью окисления:
О +5 +5 + 4
Р + 5HNO3 = НРО3 + 5NO2 + 2Н2О
S + 6HNO3 = H2SO4 + 6NO2 + 2Н2О
Интересно, что концентрированную А. к. можно хранить и перевозить в алюминиевых
Cu + 4HNO3 (конц.) = Cu(NO3)2+2NO2+2H2O
2NO + O2(B03fl.) = 2RO2
Рис. 1. Получение окислов азота. В пробирку а налита концентрированная азотная кислота и брошен кусочек медной проволоки. Реакция между ними приводит к выделению двуокиси азота N02— бурого цвета. По отводной трубке газ поступает в стакан с водой. Здесь по реакции N02 с Н20 образуются HNO3 и NO. Окись азота — бесцветный газ, собирается в пробирке б. Если пробирку б вынуть и перевернуть, над ней появится бурый дымок: NO на воздухе окисляется в N02.
Уравнения реакций см. на рисунке.
или стальных цистернах. Не странно ли это? Дело в том, что железо, алюминий и хром при контакте с холодной концентрированной А. к. образуют нерастворимые в ней окислы и гидроокиси, например:
2А1 + 6HNO3 = А12О3 + 6NO2 4- ЗН2О
И именно благодаря образованию на поверхности металла защитного слоя окисла дальнейшее воздействие А. к. на металл прекращается. Этот процесс называют пассивированием металлов.
Разбавленная А. к. действует на металлы иначе, чем А. к. концентрированная. По мере разбавления окислительные свойства А. к. ослабляются, а кислотные усиливаются. Продуктами реакции металлов с разбавленной А. к. могут быть или окись NO (в большинстве случаев):
3Cu + 8HNO3 = 3Cu(NO3)2 + 2NO + 4Н2О
или закись N2O:
4Zn + 10HNO3 = 4Zn(NO3)2 + N2O + 5H2O
или же аммиак NH3.~ В момент выделения аммиак реагирует с избытком А. к. и образует нитрат аммония:
4Mg +10HNO3=4Mg(NO3)2+NH4NO3+3H2O
АЗОТНАЯ КИСЛОТА
54
Таким образом, при взаимодействии металлов с А. к. водород не вытесняется. Строго говоря, если в реакции участвуют такие активные металлы, как Са или Mg, в первый момент водород, по-видимому, всё-таки образуется, но тут же реагирует с избытком А. к., окисляясь до воды:
3H + HNO3= NO + 2H2O
Концентрированная А. к. окисляет НС1: HNO3 + ЗНС1 = NOC1 + С12 + 2Н2О
Смесь 1 объёма А. к. с 3 объёмами соляной кислоты издавна называют царской водкой. Это ещё более сильный окислитель, чем А. к. Против неё не может устоять даже золото — «царь металлов»!
Многие органические вещества (бумага, солома, хлопчатобумажные волокна, скипидар) при взаимодействии с А. к. окисляются настолько быстро и энергично, что иногда загораются.
Получение и применение. Ежегодно А. к. производят десятками млн. т. Среди других минеральных кислот она по потреблению уступает только серной кислоте.
Самый давний промышленный способ получения А. к. сводится к действию на селитру избытка серной кислоты при нагревании до 150 СС:
NaNO3 + H2SO4 = HNO3 + NaHSO4
Образующаяся А. к. испаряется. Пары её конденсируют и собирают в специальных аппаратах. В России первые прописи по производству «крепкой водки» — так называли тогда А. к.— составил ещё Пётр I. Сегодня этот способ промышленного значения не имеет, так как при-, родные запасы селитры крайне ограничены.
В начале XX в. были разработаны другие методы производства А. к. Исторически первым из них был способ окисления атмосферного азота при высокой температуре с последующей переработкой образующейся окиси азота в А. к. Сырьё здесь — воздух. Аппаратурное оформление процесса несложно, но он требует большого расхода электроэнергии. Поэтому вскоре, едва только удалось осуществить промышленный синтез аммиака из азота и водорода и затем разработать методы окисления аммиака до окислов азота, описанный способ получения А. к. почти повсеместно был оставлен.
Реакцию окисления NH3 кислородом воздуха химики знали уже в 1800 г. Однако впервые в значительных количествах А. к. этим методом стали получать только с 1907 г. Выдающуюся роль в разработке промышленного производства А. к. сыграл замечательный русский инженер-химик И. И. Андреев. Независимо от других исследователей он тщательно изучил процесс окисления
Рис. 2. Схема производства азотной кислоты. Подогретый и сжатый до 7 атм воздух смешивают с аммиаком в смесителе (/). Затем аммиачно-воздушную смесь подают в контактный аппарат (2). Там происходит окисление аммиака NH3 до окиси азота NO кислородом воздуха при 875—900 °C на катализаторе, выполненном в виде сетки. Далее нитрозные газы (смесь NO, N^ О2 и паров Н2О) направляются в так называемый котёл-утилизатор (3). Он назван так потому, что в нём утилизируется часть теплоты реакции — она превращает воду в пар, используемый затем на нужды предприятия. Далее нитрозные газы проходят холодильник (4), где окончательно охлаждаются. Отсюда они поступают в абсорбционную колонну (5). Сверху сюда подаётся вода. В колонне происходят реакции:
2NO + О2 = 2NO2; 3NO2 + Н2О = 2HNO3 + NO
Образовавшаяся азотная кислота выводится снизу. Газы, отходящие из абсорбционной колонны, находятся под давлением около 5 атм. В современных установках их заставляют <работать> — не пропадать же энергии! Её используют для сжатия воздуха, необходимого в производстве азотной кислоты. Отходы производства — газовую смесь — очищают, чтобы не загрязнять воздух вредной окисью азота. С этой целью к газовой смеси добавляют небольшое количество аммиака и направляют её в реактор, где на катализаторе образуются вода и свободный азот:
6NO + 4NH3=5N2 + 6Н2О
55
АЗОТНЫЙ АНГИДРИД
аммиака и сконструировал для этой цели аппаратуру из надёжных материалов. В 1916 г. в Юзовке (ныне г. Донецк) под его руководством была пущена первая в России промышленная установка для производства А. к. окислением NH3 и амиачной селитры. По уровню техники этот завод превосходил подобные предприятия за рубежом, а стоимость его была почти в шесть раз ниже.
Процесс современного получения А. к. в промышленности состоит из двух основных стадий: первая стадия — окисление аммиака NH3 до окиси азота NO, вторая — окисление NO до двуокиси азота NO2 и растворение последней в воде.
Для проведения первой стадии газообразный аммиак и воздух прежде всего тщательно очищают от пыли, затем подогревают и смешивают в определённой пропорции (10—12% NH3). Потом эту смесь направляют в аппараты для окисления аммиака. Реакция идёт при температуре 875—900 °C:
4NH3 + 5O2= 4NO + 6H2O
Одно из важных условий процесса — активность катализатора — сплава платины и родия (95% Pt, 5% Rh) или платины, родия и палладия (93% Pt, 3% Rh, 4% Pd).
Чем выше температура в аппарате, тем полнее превращение NH3 в NO. Однако на практике её не поднимают выше 900 °C, так как при нагревании увеличиваются потери дорогостоящего катализатора. Обычно в промышленных аппаратах степень превращения NH3 составляет от 93 до 98%.
Окисление аммиака сопровождается выделением большого количества теплоты. Тепло же, как сказано выше, необходимо для предварительного подогрева газовой смеси. Поэтому оказывается возможным вести процесс без внешнего подогрева (такие процессы называются автотермичными).
Вторая технологическая стадия — окисление NO кислородом до двуокиси NO2, которая при взаимодействии с водой образует А. к.:
2NO + О2 = 2NO2
3NO2 + Н2О = 2HNO3 + NO
Выделившаяся при этом окись NO вновь окисляется кислородом до NO2.
Наиболее медленная из приведенных реакций — окисление NO в NO2. Скорость её заметно увеличивается, если проводить процесс при низких температурах (10—40 °C) и повышенных давлениях (5—10 атм).
В промышленных установках не удаётся полностью перевести все окислы азота в А. к. Часть их, правда небольшая (от 0,15 до 0,5%), направляется в специальные аппараты, где окислы разлагаются до свободного азота.В устаревших установках непрореагировавшие окислы
азота выбрасываются в атмосферу. Видели ли вы шлейфы рыжего дыма над азотно-туковыми заводами? Это печально известные «лисьи хвосты»... Окислы азота — одно из вреднейших загрязнений атмосферы. Поэтому энергичные меры по охране природы в СССР ведут к тому, что «лисьи хвосты» один за другим исчезают с нашего неба.
Способы производства А. к. различаются главным образом по применяемому давлению (атмосферное, повышенное и «комбинированное»). При комбинированном — первую стадию, окисление аммиака, проводят под атмосферным давлением, а вторую, окисление NO и поглощение NO2 водой,— под повышенным. Наиболее целесообразными считаются установки с повышенным давлением (рис. 2).
Большая часть А. к., получаемой в промышленности, содержит до 60% HNO3. Более концентрированную А. к. выделяют или перегонкой её водных растворов с серной кислотой, или взаимодействием четырёхокиси азота N2O4, кислорода и воды под давлением 50 атм и при температуре 70—85 °C.
Главный потребитель А. к.— химическая промышленность. Больше всего А. к. расходуют для производства азотных и комбинированных удобрений (см. Минеральные удобрения). А. к. идёт для получения нитратов, красителей, синтетических и искусственных волокон, смол, пластических масс... Всего не перечислить. Действием А. к. на целлюлозу получают бездымный порох — пироксилин. В цветной металлургии А. к. применяют для травления металлов, для отделения золота от серебра, и т.д. В ракетной технике — в качестве окислителя. Е. Р. Шендерей.
АЗОТНОКИСЛЫЕ СОЛИ — то же, что нитраты.
АЗОТНЫЙ АНГИДРИД, пятиокись азота, оксид азота (V), N2O5—ангидрид азотной кислоты. Бесцветные прозрачные кристаллы, плавящиеся при 30 °C. Очень неустойчив, медленно разлагается при комнатной температуре и быстро при кипении (45 °C) — по реакции:
2N2O5 = 4NO2 + О2
С водой даёт азотную кислоту:
N2O5 + Н2О = 2HNO3
Может быть получен действием фосфорного ангидрида Р2О5 на азотную кислоту:
2HNO3 + Р2О5 = 2НРО3 + N2O5
Сильный окислитель. См. также Азот.
АКТИНИДЫ — то же, что актиноиды.
АКТИНИИ
56
АКТЙНИЙ (лат. Actinium) Ас — радиоактивный химический элемент III группы периодической системы Менделеева; атомный номер 89. Серебристо-белый металл. Впервые был обнаружен в 1899 г. французским учёным А. Дебьерном в урановой руде («смоляной обманке»). Название А. получил (от греческого «актйс», род. падеж «актй-нис»—луч) благодаря своей радиоактивности. В природе встречаются 3 изотопа А.—
226Ас, 227Ас, 228Ас. Наибольшим периодом полураспада (Ti/2=21,8 года) обладает 227Ас, который может быть получен в миллиграммовых количествах. Время жизни остальных изотопов значительно короче.
Распад ядер 227Ас своеобразен. Во-первых, в отличие от большинства других радиоактивных изотопов, он происходит не по одному пути, а по двум. В 988 случаях распада из 1000 ядра 227Ас испускают 0--частицы (электроны е) и превращаются в ядра изотопа тория:
227 * __227 “
8вАс ► 90Th-f~e
В остальных 12 случаях они испускают а-частицы (ядра гелия гНе) и переходят в ядра изотопа франция:
227 д __223г? । 4т т_
89Ас ► 87сг4~2Не
Во-вторых, «актиниевые» 0--частицы обладают столь малой энергией, что почти полностью поглощаются в самих препаратах А. Поэтому их долгое время не удавалось обнаружить. Возникло даже подозрение, что радиоактивный переход |97Ас в 9o7Th противоречит одному из фундаментальных законов природы — закону сохранения заряда. В самом деле: заряд ядра возрастал на 1 (от Z=89 у Ас до Z=90 у Th), но это возрастание словно бы ничем не компенсировалось. ’Уловить 0_-частицы А. удалось лишь в 1935 г., после того как были созданы очень чувствительные приборы. И тогда-то всё стало на свои места: заряд ядра при распаде увеличивается на +1, но одновременно испускается электрон с зарядом —1, а следовательно, суммарное изменение заряда равно 0.
А.— редкий элемент. В земной коре его содержится всего лишь около 6-10-10 % по массе. Свойства металлического А. изучены слабо. Так, известно, что А. плавится при температуре около 1050°C, кипит приблизительно при 3300 СС, а надёжных экспериментальных определений плотности А. до сих пор ещё нет. Как и другие сильно радиоактивные элементы (например, полоний и радий), А. в темноте излучает голубоватое свечение. Во влажном воздухе металлический А. быстро покрывается тонкой плёнкой окиси, и она предохраняет металл от дальнейшего окисления.
В соответствии с положением А. в периодической системе, его атом имеет три внешних
(валентных) электрона (конфигурация 6dl7s2, см. Атом). А., как и его ближайший аналог лантан, в соединениях проявляет степень окисления ИЗ (валентность III). А. вообще похож на лантан по своим химическим свойствам, но его реакционная способность несколько выше. А. даёт такие же нерастворимые соединения, как и лантан: гидроокись, фосфат, карбонат и др. Гидроокись Ас(ОН)3 имеет более основной характер, чем La(OH)3. Почти все соединения А. бесцветны.
Практическое значение А. невелико. В смеси с бериллием 227Ас иногда используют для приготовления источников нейтронов, столь необходимых и физикам, и химикам, и геологам для различных исследовательских целей.
С. С. Бердоносое.
АКТИНОИДЫ — семейство из 14 химических элементов с атомными номерами Z от 90 до 103. Эти элементы расположены в 7-м периоде системы Менделеева за актинием и относятся, как и актиний, к III группе. Вот перечень А.: торий Th (Z=90), протактиний Ра (91), уран U (92), нептуний Np (93), плутоний Ри (94), америций Ат (95), кюрий Ст (96),берклий Вк (97), калифорний Cf (98), эйнштейний Es (99), фермий Fm (100), менделевий Md (101), элемент 102 (нобелий No) и элемент 103 (лоуренсий Lr); названия двух последних элементов не общеприняты и в таблице Менделеева их помещают в круглых скобках.
Все А. радиоактивны — стабильных изотопов у них нет. Только у двух А.— Th и U — есть природные изотопы с такими большими периодами полураспада (Ъ/2), что они сравнимы с возрастом Земли. Поэтому уран и торий в заметных количествах содержатся в земной коре. Оттого-то и открыты они были сравнительно давно (уран в 1789 г., а торий в 1828 г.), обычными химическими методами. Ещё один А., встречающийся в природе,— Ра, был открыт в 1913 г. как член радиоактивного ряда урана-235 (о радиоактивных рядах см. статьи Радий, Уран).
Остальных А. в природе практически уже нет. Вероятно, они существовали на нашей планете во время её формирования, но из-за высокой скорости радиоактивного распада все их изотопы давным-давно распались и полностью исчезли, превратившись в недрах Земли в другие элементы. Второе рождение этих элементов началось в 40-х гг. XX в., когда физики научились получать их искусственным путём.
По мере продвижения от U к элементу № 103 периоды полураспада даже наиболее устойчивых изотопов отдельных А. быстро уменьшу-
57
ются. Так, Ti/2 изотопа 238U равен 4,56-109 лет, а для изотопа 256103 эта константа составляет всего около 45 сек. Поэтому количества отдельных А., которые физикам удаётся накопить с помощью ядерных реакций, быстро уменьшаются по мере увеличения Z элемента.
Масса накопленного до сих пор Ри измеряется тоннами, а общее количество Ст, имеющееся во всём мире, вряд ли превышает несколько килограммов. При работе же с Es редкостью является уже образец, содержащий около 10-6 г этого элемента. Наконец, в случае Md и ещё более тяжёлых А. речь идёт только об отдельных атомах элемента. Такие количества вещества нельзя ни увидеть, ни взвесить. И разве не удивительно, что учёные все-таки нашли способы изучать свойства даже этих, самых короткоживущих элементов!
Искусственное получение А.— заслуга современной ядерной физики.
А., расположенные близко к урану, можно синтезировать, облучая ядра 238U нейтронами. Нейтроны очень удобны для ядерных реакций: они не имеют заряда и поэтому легко проникают в глубь ядер. При слиянии нейтрона и ядра 238U возникает быстро распадающийся изотоп урана 239U. Как и большинство других изотопов, ядра которых «перегружены» нейтронами, он р~-радио-активен. А когда испускаются р_-частицы — электроны, то положительный заряд ядра возрастает. В результате из 929U образуется 9339Np.
Мощный поток нейтронов позволяет ядру 239U до распада захватить не один, а несколько нейтронов. Следует быстрый р--распад сверхперегруженного нейтронами ядра. Теперь уж он сопровождается испусканием не одной, а двух или трёх р_-частиц и увеличением ядерного заряда на две-три единицы. Становится возможным рождение таких А., как Ри и Ат. Чудовищные по мощности потоки нейтронов, возникающие в момент взрыва водородной бомбы, иногда способны вызвать образование сверхтяжёлых ядер урана с массовым числом 250 и выше! Мгновенный распад таких крайне неустойчивых ядер связан с испусканием шести — восьми р~-частиц. И он приводит к возникновению таких элементов, как Es и Fm.
Разумеется, для синтеза новых А. можно облучать нейтронами не только ядра 238U, но и многие изотопы трансурановых элементов [так называют (от лат. trans — «за») элементы, следующие в периодической системе за ураном]. Однако в этом случае дело существенно осложняется просто недостатком сырья для ядерных реакций: трудно получить заметные количества изотопов исходных элементов — ведь их тоже надо создавать искусственно.
Для синтеза А. с Z^ 101 в качестве ядерных снарядов стараются использовать не нейтроны, а многозарядные ионы. Сливаясь с исходным ядром, они сразу сильно увеличивают его заряд. Снарядами служат такие ионы, как «оголённые» ядра гелия (а-частицы), а также ядра бора, кислорода, неона и других элементов. Трудность проведения реакции с многозарядными ионами легко понять; облучаемое ядро своим положительным зарядом сильно отталкивает падающую на него частицу-снаряд, тоже положительно заряженную. Это отталкивание надо преодолеть. И потому ионы разгоняют до сверхвысоких скоростей, сообщая им огромную кинетическую энергию.
АКТИНОИДЫ
Таким методом советские физики получили, например, изотоп элемента № 103:
°i?Am -|- г|О - ?50e3103 + 5jn
Название А. в переводе с греческого означает «подобные актинию». Оно дано А. по аналогии с лантаноидами — семейством 14 элементов, относящихся тоже к III группе, но расположенных в 6-м периоде за лантаном.
Мысль о существовании семейства А. впервые высказал в 1942—44 гг. американский химик Г. Сиборг. Сначала это была только теоретическая догадка. Она понадобилась затем, чтобы вывести химию радиоактивных элементов из неожиданно возникшего тупика. Это интересная и поучительная история...
К тому времени уже были открыты первые трансурановые элементы — Np и Ри. Их свойства оказались очень похожими на свойства урана. Так, у всех трёх элементов в растворе устойчивыми были степени окисления 4-4, 4-5 и 4-6. Поэтому, когда на повестку дня встал вопрос об открытии элементов с ещё большими Z, ожидалось, что они тоже окажутся аналогами U. И вот, после того как физики осуществили ядерные реакции по синтезу элементов с Z=95 и Z=96, ничтожные массы полученных продуктов перевели в раствор и химики начали искать там новые элементы — аналоги урана — по заранее заданной «урановой программе». Однако опыты оказались неудачными. Складывалось впечатление, что новых элементов в растворах просто нет. А между тем физики не сомневались, что ядерные реакции проведены правильно. В чём же могла заключаться причина неудач? Может быть, программа химиков была ошибочной?
Тогда-то Сиборг и пришёл к идее, что в 7-м периоде элементы с Z от 90 до 103, включая сам уран, являются аналогами актиния и образуют семейство А. Из гипотезы Сиборга следовала совсем иная поисковая программа: так как для актиния характерна степень окисления 4-3, то, вероятно, и для актиноидов в растворах нужно ожидать той же степени окисления. Другими словами, новые элементы стоит поискать в форме трёхзарядных катионов. Повторные опыты сразу же оказались успешными. Используя новую методику, удалось обнаружить Am (Z=95) и Cm (Z=96). Дальнейшие исследования показали, что все элементы, расположенные между Th и элементом 103, также обнаруживают черты, общие с актинием и другими А.
Итак, в 7-м периоде 14 элементов, похожих на актиний, образуют семейство со сходными химическими свойствами, как бы повторяя за-
АКТИНОИДЫ
58
90 Th 232,0381 6с/2 7s2 ТОРИЙ 91 Ра 231,0359 5f2 б#1 7s2 ПРОТАКТИНИЙ 92 U 238,029 5f3 6t? 7s2 УРАН 93 Np 237,0482 5Z46dl 7s2 НЕПТУНИЙ 94 Pu [244] 5/6 7s2 ПЛУТОНИЙ 95 Am [243] 5f7 7s2 АМЕРИЦИЙ 96 Cm [247] 5/7 6cf1 7s2 КЮРИЙ
кономерность, проявившуюся уже в 6-м периоде, где тоже 14 элементов образуют семейство лантаноидов. Замечательно, что между элементами этих семейств много общего. Чем же объясняется и то, что А. похожи друг на друга, и то, что А. и лантаноиды схожи между собой?
Ясно, что всё дело в строении внешних электронных оболочек атомов обоих семейств. Как известно, именно эти оболочки «заведуют» химией элементов.
В атомах А. семь оболочек. Число электронов на последней (7-й) и предпоследней (6-й) приблизительно одинаково у всех А.— поэтому и химические свойства этих элементов весьма схожи. Но ведь от элемента к элементу заряд ядра возрастает на единицу, и на единицу увеличивается число электронов в атоме. Куда же «деваются» эти, всё новые и новые, электроны, если самые внешние оболочки — 7-я и 6-я — у А. почти не изменяют своей структуры? А дело вот в чём.
Учёные установили, что каждая электронная оболочка содержит несколько уровней. (Число уровней зависит от номера оболочки, см. статью Атом.) Оказывается, что расположенная относительно близко к ядру 5-я оболочка атомов А. имеет пять уровней, и в одном из них — так называемом /-уровне — есть 14 вакантных мест. Всё новые и новые электроны — от Th до элемента 103 — постепенно заполняют этот уровень 5-й оболочки, не нарушая структуры самых наружных уровней. Естественно, что семейство А. насчитывает именно 14 элементов и все они химически очень похожи.
У лантаноидов число оболочек 6. Электронами у них постепенно заполняются тоже не наружные — 5-я и 6-я — оболочки, а более близкая к ядру 4-я. У неё тоже есть /-уровень, на котором может расположиться 14 электронов. Вот и получается, что у элементов обоих семейств строение трёх внешних электронных оболочек (у актиноидов — 7-й, 6-й и 5-й, а у лантаноидов — 6-й, 5-й и 4-й) оказывается сходным. Это и обусловливает близость их химических свойств.
Более точно электронные конфигурации трёх внешних оболочек атомов А. показаны в таблице (подробнее см. Атом, Закон Менделеева) .
Однако, кроме общих черт, в свойствах А. и лантаноидов наблюдаются и существенные различия. Так, А. часто образуют соединения в более высоких степенях окисления, чем +3 (это видно из таблицы). Для лантаноидов это не характерно. Причина ясна: электроны, расположенные на /-уровне 5-й оболочки в атомах А., притягиваются к ядрам, конечно, значительно слабее, чем электроны /-уровня 4-й оболочки в атомах лантаноидов. Слабее оттого, что электроны на 5/-уровне находятся дальше от ядер, чем 4 /-электроны, и к тому же
Электронные конфигурации и возможные степени окисления актиноидов в растворе.
Элемент Число электронов на трёх внешних оболочках Степени окисления (все положительны) *
5-я 6-я 7-я
S Р d f S Р d S
Th 2 6 10 0 2 6 2 2 3, 4
Pa 2 6 10 2 2 6 1 2 3, 4, 5
U 2 6 10 3 2 6 1 2 3, 4, 5, 6
Np 2 6 10 4 2 6 1 2 3, 4, 5,6,7
Pu 2 6 10 6 2 6 0 2 3, 4, 5,6,7
Am 2 6 10 7 2 6 0 2 2, 3, 4,5,6
Cm 2 6 10 7 2 6 1 2 3, 4
Bk 2 6 10 8 2 6 1 2 3, 4
Cf 2 6 10 10 2 6 0 2 2, 3, 4
Es 2 6 10 11 2 6 0 2 2, 3
Fm 2 6 10 12 2 6 0 2 2, 3
Md 2 6 10 13 2 6 0 2 1, 2, 3
№ 102 (No) 2 6 10 14 2 6 0 2 2, 3
№ 103 (Lr) 2 6 10 14 2 6 1 2 3
* Наиболее типичные выделены жирным шрифтом; для твёрдых соединений возможны иные значения.
притяжение 5 /-электронов экранировано большим числом электронных оболочек. Поэтому 5/-электроны А. подвижней и могут принимать участие в образовании химических связей. В результате первые члены семейства А., Th, Ра и U проявляют много общего с элементами соответственно IV, V и VI групп и образуют различные соединения с высокими степенями окисления: +4, +5 и +6. По ряду причин степень окисления +3 становится характерной только у америция и следующих за ним А.
А теперь коротко рассмотрим отдельные А. (сведения о тории, протактинии, уране и плутонии см. в статьях, посвящённых этим элементам).
Нептуний (лат. Neptunium) Np, Z = 93.
Первый изотоп элемента 239Np был получен в 1940 г. американскими учёными Э. М. Макмилланом и Ф. X. Эй-блсоном, когда они облучали уран нейтронами. Назван по планете Нептун, подобно тому, как уран — предшественник Np в периодической системе — получил своё название от планеты Уран.
Наиболее долгоживущим изотопом является 237Np (период полураспада Ti/3=2,14 • 106 лет,
59
АКТИНОИДЫ
97 Вк [247] 98 Cf [251] 99 Es [254] юо Fm [257] Ю1 Md [258] 102 (No) [255] юз (Lr) [256]
5/8 бс/1 7s2 б/10 7s2 5/" Is2 5f12 7s2 5f13 7s2 5fl47s2 57l46d' 7s2
БЕРКЛИЙ КАЛИФОРНИЙ ЭЙНШТЕЙНИЙ ФЕРМИЙ МЕНДЕЛЕВИЙ (НОБЕЛИЙ) (ЛОУРЕНСИЙ)
атомная масса 237,0482). В результате ряда радиоактивных превращений из 237Np образует* ся стабильный изотоп висмута 209Bi. Цепочку радиоактивных превращений, .приводящую к образованию 209Bi из 237Np, называют радиоактивным рядом нептуния. Хотя периоды полураспада всех изотопов нептуния значительно меньше возраста Земли, ничтожные количества двух изотопов этого элемента — 237Np и 239Np— были найдены в урановых рудах. Что же это значит? Да только то, что эти изотопы непрерывно образуются в процессе ядерных реакций урана с нейтронами (прилетающими, в частности, из космоса).
Элементарный нептуний — серебристо-белый ковкий металл с /пл 640°С; плотность металла 20 г/см3. По химическим свойствам Np ближе всего к U и Ри. Нептуний способен давать химические соединения со степенями окисления от +2 до +7 (валентности равны соответственно от II до VII). В твёрдых соединениях Np наиболее устойчива степень окисления +4. Прокаливание на воздухе соединений Np различных степеней окисления приводит к образованию NpO2.
В растворах существуют ионы нептуния пяти форм: Np3+, Np4+, NpO2+ (нептуноил-ион), NpO2+ (нептунил-ион) и NpOf“. Наиболее устойчив ион Np<X, соответствующий пятивалентному нептунию. Все ионы нептуния имеют характерную окраску: Np3+— голубую или фиолетовую, Np4+— жёлто-зелёную, NpO^— голубовато-зелёную, NpO|+— розовую или красную, NpOjJ-— зелёную (в щелочной среде) или коричневую (в кислой среде).
Амфотерной гидроокиси шестивалентного нептуния NpO2(OH)2 соответствуют не только соли, содержащие нептунил-ион NpO|+. Образуются и нептунаты, где нептуний находится в составе анионных групп. Например, известен малорастворимый в воде динептунат аммония (NH4)2Np2O7.
Весовые количества изотопа 237Np образуются как побочный продукт при производстве Ри в ядерных реакторах. О работе реактора см. статью Уран.
Америций (лат. Americium) Am, Z=95.
Впервые получен в 1944—45 гг. Г. Сиборгом, Р. Джеймсом, Л. Морганом и А. Гиорсо в результате облучения нейтронами изотопа плутония 239Ри. Название дацо от слова «Америка» — по аналогии с европием; послед-
ний занимает в семействе лантаноидов такое же место после лантана (шестое), что и америций в семействе актиноидов после актиния.
Наиболее устойчив изотоп 243Ат (7\/я = = 7950 лет). В свободном состоянии америций— серебристый металл; плотность равна 13,67г/см3, /пл 1173 °C, а /кип2607 °C.
Кюрий (лат. Curium) Cm, Z=96.
Искусственно синтезирован в 1944 г. Г. Сиборгом, Р. Джеймсом и А. Гиорсо при облучении 239Ри а-части-цами. Назван в честь основателей науки о радиоактивности Пьера и Марии Кюри.
Наибольший период полураспада (7\/я = = 1,64-107 лет) имеет изотоп 247Ст. Элементарный кюрий — блестящий серебристый металл, плотность около 19 г/см3, /пл 1340 °C. Изотоп 244Ст используется в «атомных батарейках». Они дают электрический ток различным приборам, устанавливаемым, например, на космических аппаратах.
Берклий (лат. Berkelium) Bk, Z=97.
Синтезирован в декабре 1949 г. С. Томпсоном, А. Гиорсо и Г. Сиборгом путём облучения а-частицами америция 241Агп. Название дано в честь научного центра Беркли (штат Калифорния, США), где были проведены многочисленные работы по синтезу и исследованию многих А.
Наиболее устойчив изотоп 247Вк (7\/я= 1380 лет). Вк в количествах, доступных для взвешивания (около 4-10"6 г), был выделен в 1958 г.
Калифорний (лат. Californium) Cf, Z=98.
Открыт в 1950 г. С. Томпсоном, А. Гиорсо, К- Стритом и Г. Сиборгом при облучении а-частицами 242Сш. Назван по месту открытия (штат Калифорния, США).
Наиболее устойчив изотоп 261Cf (7\/я около 800 лет). Первые твёрдые соединения калифорния — Cf2O3 и СЮС1 синтезированы в 1958 г.
Эйнштейний (лат. Einsteinium) Es, Z=99.
Впервые выделен из продуктов термоядерного взрыва в 1952 г. Назван в честь Альберта Эйнштейна, создателя теории относительности.
Самым долгоживущим изотопом является 264Es (7\/я=276 дней).
Фермий (лат. Fermium) Fm, Z= 100.
Обнаружен впервые в 1952 г. одновременно с эйнштейнием среди продуктов термоядерного взрыва. Назван в честь замечательного итальянского физика Энрико Ферми, который в 1942 г. осуществил первую цепную реакцию деления урана в построенном по его проекту ядерном реакторе.
Все изотопы Fm распадаются очень быстро. Наиболее устойчив 267Fm (7\/я=80 дней).
АЛЮМИНИЕВЫЕ СПЛАВЫ
Менделевий (лат. Mendelevium) Md, Z= 101.
Первые атомы этого элемента были синтезированы в 1955 г. группой учёных во главе с А. Гиорсо и Г. Сиборгом. Задача, стоявшая перед исследователями, была необычайно трудной, так как в ходе опытов ожидалось образование только считанных атомов нового элемента. Остроумно задуманные эксперименты привели к успеху.
На тонкую золотую фольгу наносили примерно 1 миллиард атомов 263Es и бомбардировали её разогнанными до высоких скоростей а-частицами. Если бы образующиеся ядра нового элемента оставались на фольге, то отделить их от Es было бы, пожалуй, гораздо сложнее, чем найти иголку в огромном стоге сена. Атомов 101-го элемента могло появиться лишь несколько, и они просто «затерялись» бы среди миллиарда атомов Es из-за близости свойств актиноидов. Исследователям пришла в голову хорошая конструктивная идея. Энергия падающих на фольгу а-частиц огромна. Их столкновение с ядрами 263Es могло приводить к созданию ядер элемента 101 в тех случаях, когда оно происходило «лоб в лоб»:
2»?Es + £Не = 22’Md + Jn
Но в этих случаях образовавшиеся ядра 266Md, приобретя в свой черёд большую энергию, должны были пробивать насквозь тонкую золотую фольгу. Их-то и следовало ловить. Для.этого использовали второй золотой листок, расположенный на небольшом удалении от первого. На этот листок-ловушку, кроме ядер Md, конечно, попадало и небольшое число ядер Es, тоже выбитых а-частицами с облучаемой мишени. После окончания облучения листок-ловушку быстро растворяли в царской водке. Золото отделяли химическим путём, а оставшийся раствор подвергали разделению по специально разработанной методике.
В .ходе таких опытов удалось обнаружить 17 атомов Md. Всего семнадцать! И тем не менее учёные сумели определить период полураспада” 266Md (около 1 часа), распознать некоторые его химические свойства и точно указать расположение нового элемента в таблице Менделеева. Постоянно руководствуясь в своих исканиях периодической системой, американские исследователи дали название новому элементу в честь её гениального создателя.
Были найдены и другие изотопы Md. Самым долгоживущим из них является 268Md (Ti/3 = =54 дня).
Советские химики установили, что Md, кроме степени окисления 4-3, может проявлять в растворах и степени 4-1 и 4-2.
60
Элемент № 102.
Первое сообщение о получении этого элемента сделала в июле 1957 г. объединённая американо-англошведская группа, работавшая в Стокгольме. Тогда же было предложено назвать его нобелием (лат. Nobelium, символ No). Однако последующие исследования, выполненные в нашей стране и в США, показали, что результаты стокгольмской группы невоспроизводимы. Это означало, что сделанное ею сообщение об открытии нового элемента ошибочно. Надёжные сведения о свойствах изотопов элемента № 102 с массовыми числами 251—256 впервые были получены в 1963—67 гг. группой советских физиков, работавших под руководством академика Г. Н. Флерова. Наши учёные, по праву первооткрывателей 102-го элемента, предложили для него название «жолиотий» в честь выдающегося французского физика и химика Жолио Кюри. Но пока (1975 г.) вопрос о названии элемента № 102 окончательно не решён, и поэтому его символ в периодической системе заключён в скобки.
Изотоп 266No, указанный в цветной таблице, считался самым устойчивым. Но уже получен более устойчивый 269No (Ti/S около 1,5 часа).
Элемент № 103.
Первые опыты по синтезу этого элемента облучением калифорния ядрами бора были выполнены в 1961 г. группой американских учёных во главе с А. Гиорсо. Они предложили назвать полученный элемент лоуренсием (лат. Lawrencium, символ Lr) в честь Эрнеста Лоуренса — изобретателя циклотрона (ускорителя, позволяющего разгонять заряженные частицы — «ядерные снаряды»—до высоких скоростей). Однако результаты этой работы в дальнейшем не подтвердились и возник вопрос о переименовании элемента № 103. Поэтому символ 103-го элемента в периодической системе (как и символ 102-го элемента) даётся в скобках.
Первые надёжные сведения об изотопе с массовым числом 256 получили советские физики Объединённого института ядерных исследований в Дубне. Установлено, что период полураспада этого изотопа составляет всего около 45 сек. Несмотря на высокую скорость распада, группе химиков, работавших в Дубне под руководством чехословацкого учёного И. Звары, удалось провести исследование ряда химических свойств нового элемента. Было найдено, что он очень похож на другие А. и резко отличается по своим свойствам от следующего за ним в таблице Менделеева трансуранового элемента курчатовия. Это подтвердило, что элемент № 103—последний в семействе актиноидов.
С. С. Бердоносов. АЛМАЗ — одна из аллотропных модификаций углерода.
АЛЮМИНИЕВЫЕ КВАСЦЫ
K2so4-A12(SO4)3-24H2O, или KAI (SO4)2-12Н2О,—см. в статьях Алюминий, Квасцы.
АЛЮМИНИЕВЫЕ СПЛАВЫ — сплавы на основе алюминия с добавками других элементов. А. с. обладают многими ценнёйшими свойствами: они легки (в 3 раза легче стали), прочны, хорошо обрабатываются, достаточно устой
61
АЛЮМИНИЕВЫЕ СПЛАВЫ
чивы к действию атмосферы и воды. А кроме того, изделиям из них нетрудно придать красивый декоративный вид. Прочность чистого А1 невелика, примерно 60—80 Мн/м2 (то есть 6— 8 кгс/мм2). Но уже получены сплавы алюминия, прочность которых в 10 раз выше: 700— 750 Мн/м2, как у среднелегированной стали. А. с. позволяют создавать относительно лёгкие, но долговечные конструкции.
Широкое промышленное использование А. с. началось после первой мировой войны 1914— 1918 гг. в связи с развитием авиации. Недаром целая группа таких сплавов носит название «авиали» (сплавы А1—Mg—Si). Если в названиях одних А. с. отражается их область применения (авиали), то в названиях других подчёркивается их состав (магналий — сплав А1 с Mg; силумин — сплав Al с Si).
Различают А. с. деформируемые — их можно прокатывать, ковать, штамповать (как дур-алюмин) — и литейные — их в расплавленном состоянии заливают в формы (как силумин).
Дуралюмин (сплав А1—Си—Mg с небольшими добавками Мп, Si) был впервые получен в 1909 г. немецким исследователем А. Вильмом. И тогда же совершенно случайно был обнаружен процесс, ведущий к повышению прочности этого сплава,— старение. Обычно со словом «старение» связываются мало приятные явления... Но старение дуралюмина привело к возрастанию его прочности вдвое, а длилось оно всего несколько суток: образец нагрели до 500 °C, быстро охладили погружением в воду и оставили при комнатной температуре. Если бы измерение его прочности произвели сразу, как это полагалось сделать по программе исследования, чудодейственное явление не было бы обнаружено!..
Через 5 суток измерение прочности показало, что новый сплав, «состарившись», стал в 7 раз прочнее чистого алюминия.
Механизм старения А. с. ещё не вполне ясен. Обратимся к более простому примеру, чем дуралюмин,— двойному сплаву алюминия с медью (А1—Си). Этот сплав (как и дуралюмин) при 500 °C представляет собой твёрдый раствор, где А1 — растворитель, Си — растворённый металл. Этот твёрдый раствор имеет ту же кристаллическую решётку, что и основной металл,— кубическую гранецентрированную. Только некоторые атомы А1 замещены в узлах решётки атомами Си. (Это возможно всегда, когда радиусы атомов разных металлов близки.)
Термин твёрдый раствор звучит странно. Ведь все привыкли называть растворами только жидкости, состав которых можно изменять без нарушения их однородности в любых отношениях или в известных пределах.
При охлаждении горячего насыщенного водного раствора соли (например, квасцов или медного купороса) из жидкости выпадают кристаллы растворённого вещества. Это — хорошо известный случай. Но представим себе, что жидкий раствор затвердел, однако из него ничего не выделилось; он как был, так и остался однородным. Это и есть твёрдый раствор. Подобно мольеровскому мещанину во дворянстве, который не знал, что он всю жизнь говорил прозой, мы, сами того не подозревая, пользуемся твёрдыми растворами и буквально окружены ими на каждом шагу. Утром мы пьём чай или кофе из стеклянного стакана, помешивая в нём ложкой из нержавеющей стали. Но стекло — это твёрдый раствор, состоящий из силикатов натрия и кальция, а нержавеющая сталь — твёрдый раствор хрома и никеля в железе. Выходя из дома, мы берёмся за дверную ручку из латуни— твёрдого раствора цинка в меди. А в трамвае или троллейбусе мы платим за проезд монетой из алюминиевой бронзы — твёрдого раствора алюминия в меди. Больше того, известные советские учёные-металлурги Н. С. Курнаков и С. А. Погодин справедливо утверждают: «Целые эпохи истории материальной культуры ... принято называть по тем металлическим твёрдым растворам, пользование которыми было преобладающим. „Золотой век“ античных поэтов отвечает применению твёрдых растворов золота с медью и серебром, бронзовая эпоха — уменью обращаться- с твёрдыми растворами олова в меди. Век железа и стали назван так по твёрдым растворам углерода и других элементов в железе. В настоящее время мы постепенно вступаем в алюминиевый век» (написано в 1940 г.).
Но вернёмся к нашему примеру.
При медленном понижении температуры из однородного твёрдого раствора меди в алюми-
Рис. 1. Лёгкие серебристые алюминиевые сплавы ... И в химической промышленности, и в авиастроении, и в электротехнике, и в сельском хозяйстве нужны трубы из алюминия и алюминиевых сплавов.
АЛЮМИНИЕВЫЕ СПЛАВЫ
62
Рис. 2. В СССР идёт огромное строительство. Из алюминиевых сплавов можно изготовить и стенные панели, и крыши, и оконные рамы ... и даже построить мост. На снимке — автодорожный мост у г. Рузы.
нии выпадает химическое соединение СиА12. (Такие взаимные соединения металлов называются интерметаллическими соединениями, или металл идами.) Это явление можно уподобить кристаллизации гидратов CuSO4-5H2O или KAI (SO4)2-12Н2О из водных растворов CuSO4 и KAI (SO4)2. При закалке — быстром охлаждении до комнатной температу-
рке. 3. Алюминиевые сплавы прекрасно зарекомендовали себя в судостроении. Из них изготовлены, например, корпуса судов на подводных крыльях.
ры — кристаллы СиА12 не успевают выделиться и сохраняется пересыщенный твёрдый раствор. Однако он неустойчив, и со временем из него всё же выпадают мельчайшие кристаллы СиА12.
Теперь стоит привлечь к объяснению старения сплавов совсем не химический образ — новую колоду игральных карт. Как легко сдвигаются с места в такой колоде совершенно гладкие карты; их поверхности — это плоскости скольжения. Если же карты проткнуть иглами, колода перестанет рассыпаться. В кристаллической решётке сплава тоже есть плоскости скольжения. Выпавшие в процессе старения из твёрдого раствора кристаллы СиА12 равномерно распределяются по всему объёму сплава и «протыкают» его, как иглы колоду карт. Сдвиги по плоскостям скольжения прекращаются. А это и значит, что сплав упрочнился. Заметим, однако, что единого мнения о механизме упрочняющего старения алюминиевых сплавов, среди учёных нет.
Термическая обработка — временное воздействие нагревания или охлаждения — самый распространённый в современной технике способ изменения свойств металлов и сплавов.
Открыв по счастливой случайности процесс старения, учёные сразу повели поиски новых химических соединений, способных упрочнить алюминий. Были созданы самые прочные в обычных условиях А. с., такие, как А1—Zn—Mg и Al—Zn—Mg—Си. И были найдены самые жаропрочные А. с.— сохраняющие свои свойства при высоких температурах, такие, как А1—Си— —Мп. Добавка тех или иных элементов к алюминию для создания нового сплава всегда определяется тем комплексом свойств, какой желательно получить. Наиболее лёгкие А. с.— это А1—Li—Mg и Al—Be—Mg, а коррозионностойкие, декоративные и электротехнические — это сплавы А1—Mg—Si (уже знакомые нам авиали).
Удалось создать материалы и совершенно нового типа, объединённые общим названием «САП» (спечёный алюминиевый порошок). В структуре САПа совмещены А1 и его прочный и тугоплавкий окисел А12О3. Готовят его так: жидкий алюминий распыляют в атмосфере азота, содержащего нужное количество кислорода. Каждая маленькая частица А1 при этом покрывается плёнкой А12О3. Полученный порошок прессуют в брикеты и спекают. Тогда плёнка окисла, окружающая каждую частицу, разрушается, и в тот же момент частицы чистого металла свариваются. Окисел же (его вес составляет до 20% веса САПа) образует сплошной «защитный» каркас по всей массе материала. Это придаёт САПу особенно большую жаропрочность. Если лучшие А. с. хорошо выдержи
63
АЛЮМИНИЙ
вают температуры не выше 250—300 °C, то САП может служить при 400—500 °C.
Около 100 лет назад Н. Г. Чернышевский, увидев алюминий, сказал, что этому металлу суждено великое будущее, что алюминий — металл социализма. И оказался прав: на наших глазах лёгкие серебристые алюминиевые сплавы входят мощным потоком в новую технику. Вот авиали! Они применяются и в судостроении, и для облицовки и перекрытий зданий, и для изготовления металлических обоев, корпусов часовых механизмов, ювелирных украшений. Это замечательный материал и для оконных рам; такие рамы не имеют швов, они легки, изящны, гигиеничны и геометрически точны. С применением авиалей построен Дворец спорта в Киеве, Дворец пионеров и универмаг «Москва» в Москве. В Кремлёвском Дворце съездов прекрасная панорама создаётся сочетанием золотых и чёрных покрытий на сплаве системы А1—Mg—Si. В нашей стране идёт огромное строительство. И в будущем для стенных панелей, подвесных потолков, крыш, оконных рам и других строительных деталей потребуются ещё сотни и сотни тысяч тонн сплавов алюминия. А сплавы А1—Mg—Мп, хорошо зарекомендовавшие себя в судостроении! Из них изготовлены, например, корпуса «Ракет», «Метеоров»—судов на подводных крыльях. Список примеров может быть далеко продолжен. И если вначале производство А. с. стимулировалось главным образом развитием авиации, то теперь — по широте применения в народном хозяйстве — они прочно заняли второе место после стали и чугуна, оставив позади давно уже известные сплавы меди.
АЛЮМЙНИЙ (лат. Aluminium) Al — химический элемент III группы периодической системы Менделеева, атомный номер 13, атомная масса 26,98154. Лёгкий серебристо-белый металл. Элемент состоит из одного устойчивого изотопа 27А1.
Историческая справка.
Соединения А. были известны с древнейших времён. Так, уже в V в. до н.э. в Египте добывались квасцы — сульфат А. и калия KA1(SO4)2* 12Н2О, которые применяли как вяжущее и дубящее средство. Древние римляне обозначали квасцы словом al шлеп
(род. падеж aluminis) .откуда и произошло название «алюминий». В 1754 г. немецкий химик А. С. Маргграф выделил из квасцов бесцветную «землю» (так назывались в то время порошкообразные, неплавкие, нерастворимые в воде вещества), которую назвал квасцовой. Позднее она получила название «глинозём», так как была обнаружена в глине. Уже в 1789 г. А. Л. Лавуазье выска
26,98154 и
А1
3s23p’
АЛЮМИНИЙ
зал предположение, что глинозём — окисел ещё неизвестного металла.
Но получить металл удалось только в 1825 г. датскому физику Г. X. Эрстеду. Действуя амальгамой калия на хлорид А1С13, он приготовил амальгаму А.; после отгонки ртути остался А. в виде серого порошка.
В 1854 г. французский химик А. Э. Сент-Клер Девиль предложил более удобный способ получения А.— нагреванием натрия с двойным хлоридом натрия и А.:
Na3[AlCl6] + 3Na = 6NaCl + Al
Уже в 1855 г. на Всемирной выставке в Париже демонстрировались слиточки А. (общим весом около 1 кг), выплавленного по способу Девиля. Но вследствие высокой стоимости и низких механических свойств А. применялся только для изготовления украшений и столовой посуды.
В 1886 г., почти одновременно и независимо друг от друга, Ч.М. Холл в США и П. Э. Эру во Франции открыли способ выплавки А. электролизом криолитоглинозёмных расплавов. (Интересное совпадение: оба изобретателя родились в 1863 г., а умерли в 1914 г.)
Уже в конце XIX — начале XX вв. электрохимическое производство А. возникло во Франции, США, Швейцарии, Германии и других странах. Важно отметить, что производство А. электролизом смогло стать реальностью только после изобретения динамомашины (так называли тогда генератор постоянного тока) и внедрения её в промышленность. Ведь до этого единственным и крайне дорогим источником электроэнергии были гальванические элементы. Расширению спроса на А. способствовало развитие авто- и авиастроения, для которых требовались прочные и лёгкие конструкционные материалы. Ими явились алюминиевые сплавы.
Как это ни удивительно, но производство А. электролизом, созданное методом «проб и ошибок», долгое время не имело научной основы. Её дал П. П. Федотьев с сотрудниками (в работах, продолжавшихся свыше 20 лет, с 1910 по 1932 г.) в Петербургском (Ленинградском) политехническом институте.
В России вопрос об организации собственного производства А. возник уже во время русско-японской войны 1904—1905 гг. Однако он на долгие годы безнадёжно утонул в дебрях министерских канцелярий. Считалось, что месторождений бокситов в стране нет, а электрическая энергия стоила очень дорого. В действительности же поисками бокситов никто серьёзно не занимался, а о строительстве гидроэлектростанций даже и не помышляли. Поэтому царское правительство предпочитало ввозить А., его сплавы и изделия из них из-за границы.
Во время войны 1914—1918 гг. Россия оказалась вынужденной покупать у стран Антанты А., необходимый для военной техники. Только в 1916 г. в районе Тихвина было открыто первое отечественное месторождение бокситов. Но производство А. смогло возникнуть в нашей стране только после Октябрьской революции.
. Решающее значение для создания советского производства А. имел разработанный в 1920 г. по указаниям В. И. Ленина план электрификации нашей страны. Без больших количеств дешёвой электроэнергии производство А. совершенно невозможно. Первая крупная гидроэлектростанция сооружена в 1926 г. на реке Волхове. 14 мая 1932 г. Волховский алюминиевый комбинат дал первый промышленный А. (Образец его вы видите на фотографии, рис. 1.) Вскоре (1933 г.) состоялся пуск Днепровского алюминиевого завода, работающего на энергии Днепрогэса имени В. И. Ленина. С течением времени удалось значительно расширить сырьевую базу советского производства А. (см. ниже). Наша алюминиевая промышленность, начатая, как принято говорить, «с нуля», заняла одно из первых мест в мире и полностью обеспечивает потребность страны в этом металле.
АЛЮМИНИИ
64
Рис. 1. Образец первого советского промышленного алюми ния. Выплавлен 14 мая 1932 г. на Волховском алюминиевом комбинате (ныне Волховский алюминиевый завод им. С. М. Кирова). Слиточек изображён в 3/4 натуральной величины. Из коллекции профессора С. А. Погодина.
Распространённость в природе. Земная кора содержит 8,05% А. по массе. По распространению в природе он занимает третье место среди всех элементов (после О и Si) и первое среди металлов. Вследствие своей высокой химической активности А. встречается только в виде соединений. Известны сотни содержащих А. минералов. Из них наиболее распространены алюмосиликаты (см. Силикаты)—составная часть изверженных горных пород — гранита, базальта, порфира, сиенита и др. Из алюмосиликатов назовём хотя бы полевые шпаты: калиевый (ортоклаз) К[AlSi3O8], натриевый (альбит) Na [ AlSi 3О8], кальциевый (анортит)
Са [ Al 2Si 2О8]. При их разрушении под действием воды и атмосферы образуется каолин — главная составная часть глин. С процессами выветривания горных пород, содержащих алюмосиликаты, связано также образование бокситов. Они состоят из смеси гидроокисей А. А1(ОН)3 и А1О (ОН), окиси железа Fe2O3 и примесей (SiO2, TiO2 и др.). Бокситы—главное сырьё для производства А. Их крупные месторождения находятся у нас на Урале, в Сибири.
В СССР разработаны и методы получения А. из новых видов сырья: алунита (квасцового камня) примерного состава KAl3ISO4]2(OH)e и в особенности нефелина NalAlSiOJ. Большие залежи этого минерала были обнаружены в 1920-х годах на Кольском полуострове экспедицией под руководством академика А. Е. Ферсмана. А в 1957 г. за промышленный способ переработки Кольского нефелина на глинозём, соду и цемент группе учёных и инженеров была присуждена Ленинская премия. По их способу вот уже много лет вырабатываются огромные количества названных продуктов.
Физические свойства. А.— серебристо-белый лёгкий металл. Плотность 2,699 г/см3, /пл 660,24 °C, /кип около 2500 °C. Металл очень пластичен (легко протягивается в проволоку и про
катывается в фольгу толщиной около 0,005 мм); обладает невысокими твёрдостью и прочностью. А.— прекрасный проводник электричества (его электропроводность составляет около 65% от электропроводности меди). Поверхность А. всегда покрыта плотным, тончайшим слоем А12О3 (см. ниже). Эта плёнка прозрачна. Поэтому покрытый ею А. отражает около 90% падающих на него световых лучей.
Химические свойства. Внешняя электронная оболочка атома А. содержит три электрона (конфигурация 3s23px, см. Атом). Поэтому для А. обычна степень окисления +3 (валентность III). В некоторых соединениях он имеет степени окисления 4-2 и 4-1.
По положению А. в ряду напряжений (см. Металлы) следовало бы ожидать, что он должен окисляться гораздо легче, чем железо. В действительности же наблюдается нечто совсем обратное. Обратите внимание на изделия из А., применяемые в домашнем быту: они не изменяются на воздухе и в воде. Химическая стойкость А. объясняется тем, что Окисная плёнка на его поверхности защищает металл от воздействия воздуха, воды, некоторых кислот.
Плёнку окиси на А. можно нарастить искусственно — электролизом. Для этого изделие из А., погружённое в электролит, делают анодом. Выделяющийся на аноде кислород в виде иона О2” окисляет А. Такой процесс (анодирование) позволяет получать на поверхности А. плёнки толщиной до 500 микрон. Это самый распространённый способ защиты А. и его сплавов от коррозии. На окисную плёнку, полученную таким путём, очень удобно наносить краски и лаки, придающие изделиям красивый внешний вид.
Весьма существенно, что если удалить каким-либо острым предметом естественную плёнку А12О3, она тут же возобновляется на воздухе. Но если поставить А. в условия, при которых сплошная окисная плёнка образоваться не сможет, истинная химическая активность А. станет очевидной. Так, если порошкообразный А. поджечь, то он сгорает с ослепительной вспышкой. Убедиться в активности А. можно и на другом, более сложном, но очень интересном опыте.
Погрузим полоску листового А. в 10%-ный раствор NaOH. Он удалит плёнку А12О3. Когда начнётся выделение водорода, вынем полоску, быстро отмоем щёлочь водой и потрём поверхность металла ваткой, смоченной в 1%-ном растворе сулемы HgCl2. При этом А. вытесняет Hg, которая образует с ним рыхлую амальгаму. На полоске амальгамированного А. при действии влаги воздуха прямо на глазах вырастает толстый налёт гидроокиси А., а если опустить полоску в воду, выделяется Н2.
А. непосредственно соединяется с галогенами, образуя галогениды типа А1Х3 (X—это F, С1, Вг, I). При сильном нагревании А. взаимодейст-
65
АЛЮМИНИИ
Чарльз Мартин ХОЛЛ — один из основателей современного способа производства алюминия.
Американский изобретатель Чарльз Мартин Холл родился 6 декабря 1863 г. в деревне Томпсон, штат Огайо. Не имея специального химического образования, Холл случайно заинтересовался работами А. Э. Сент-Клер Девиля и решил найти более дешёвый способ производства алюминия. В своей небольшой домашней лаборатории после многих неудачных опытов он провёл электролиз раствора глинозёма в расплавленном криолите. Однако электролиз в глиняном тигле не дал желаемого результата, так как происходило восстановление кремния из материала тигля. И только тогда, когда Холл выложил внутреннюю поверхность тигля углём, успех оказался полным — на дне собралось несколько небольших блестящих шариков алюминия. Это произошло 23 февраля 1886 г. А в 1888 г. в Питсбурге под руководством Холла был организован небольшой завод, выплавлявший алюминий по новому способу. Холл умер 27 декабря 1914 г. в городе Дейтона, штат Флорида.
На фотографии — статуя Холла, отлитая из алюминия. Установлена в Оберлинском колледже, где Холл учился.
вует с серой, углеродом, азотом. При этом получаются соответственно сульфид A12S3, карбид А14С3 и нитрид A1N. Вода разлагает эти соединения с образованием соответственно H2S, СН4, NH3 и А1(ОН)3.
Соляная кислота всех концентраций легко растворяет А.:
2А1 + 6НС1 = 2А1С13 - - ЗН2
Серная и азотная кислоты очень высоких концентраций (H2SO4 — свыше 90%, HNO3 — свыше 68%) на холоду на А. не действуют. При нагревании А. реагирует с ними, образуя с H2SO4 сульфат A12(SO4)3 и SO2, а с HNO3 — нитрат Al (NO3)3 и окислы азота. В тех же кислотах, разбавленных водой, А., особенно при нагревании, растворяется, выделяя соответственно Н2 и окись азота:
2А1 - 3H2SO 4= A12(SO4)3 -3 Н2
А1 4- 4HNO3 = Al (NO3)3 4- NO + 2H2O
Растворы гидроокисей и карбонатов щелочных металлов активно взаимодействуют с А., причём выделяется Н2 и образуются алюминаты. Эти соединения рассматриваются как соли очень слабых кислот, ортоалюминиевой Н3А1О3 и метаалюминиевой НАЮ2.
Соли А. в растворе гидролизованы и показывают кислую реакцию. Примером служит обратимое взаимодействие с водой хлорида А.:
А1С13 4- ЗН2ОА1(ОН)3 + ЗНС1 или в ионной форме:
А13+ +ЗОН-z±Al(OH)3
Интересна в химическом отношении гидроокись А. Она образуется при действии гидро
окисей и карбонатов щелочных металлов на растворы алюминиевых солей. А1(ОН)3 выпадает в виде белого студенистого осадка:
А1С1 з - 3NaOH = Al (ОН)3 + 3NaCl 2А1С1 з + 3Na2CO3 4- ЗН2О = 2А1 (ОН)3 4-4- 6NaCI 4- ЗСО2
Гидроокись А. амфотерна (греческое «амфоте-рбс» — и тот и другой) — она растворяется и в кислотах и в щелочах (но не вводном растворе NH3). По отношению к сильным основаниям А1 (ОН)3 ведёт себя как слабая кислота, образующая соли — алюминаты.
Из галогенидов А. практически очень важны фторид, хлорид и двойной фторид натрия и А. — криолит. В промышленности A1F3 приготовляют действием HF на А1 (ОН)3 или NaF на растворы алюминиевых солей:
А1 (ОН)3 + 3HF = A1F3 + ЗН2О
AICI з получают, пропуская С12 над смесью А12О3 с углём при 1000°С:
А12О3 4- ЗС 4- ЗС12 = 2А1С13 4- ЗСО Криолит—комплексное соединение Na3[A1FJ— играет важную роль в производстве А. В виде минерала криолит встречается в основном в Гренландии. Аборигены издавна считали его льдом (греческое «крйос» — холод, лёд), спрессованным настолько, что он уже потерял способность таять. Обычно же криолит готовят искусственно, прибавляя NaF к раствору сульфата A12(SO4)3:
Al2 (SO4)3 4- 12NaF = 2Na3[A1FJ 4- 3Na2SO4
Из других распространённых соединений 3-валентного А. большой интерес представляют сульфат (см. ниже) и квасцы.
5 хэш
АЛЮМИНИЙ
66
Из соединений А. низших валентностей наиболее известны так называемые субгалогениды (от латинского sub — под). Субфторид A1F и субхлорид А1С1 образуются при взаимодействии А. с A1F3 или А1С1 з при высоких температурах по обратимой реакции:
A1F3 + 2A1t-± 3A1F
Оба субгалогенида могут существовать только в газообразном состоянии. При охлаждении реакция идёт справа налево, т. е. A1F разлагается на А1 и A1F3, а А1С1 — на А1 и А1С13. Эти реакции предложены для очистки А. от примесей.
Получение. Исходным материалом для производства А. служит его окись, получаемая путём сложной химической переработки бокситов. Разложить чистую А12О3 электролизом невозможно, так как в твёрдом состоянии она не пропускает тока, а её точка плавления очень высока (2050 °C). Поэтому окись А. растворяют в расплавленном криолите. Температура плавления смеси сравнительно невелика (962 °C при
10% А12О3). Процесс ведут в электролизёре (рис. 2).
Получающийся металл (в технике говорят — первичный А.) содержит около 99,7% А1 (остальное — примеси Fe и Si). Более чистый металл (99,997% А1) получают электролитическим рафинированием первичного.
Применение. В технике А. служит преимущественно для производства сплавов. Однако и у чистого А. есть области применения. По сравнению с алюминиевыми сплавами чистый А. значительно более устойчив против коррозии. Объясняется это тем, что в нечистом А. и в его сплавах примеси присутствуют в виде включений, которые не покрываются защитной плёнкой А12О3. При попадании влаги и особенно растворов электролитов такие включения образуют с А. гальванические микроэлементы, в которых более электроположительный А. служит растворимым электродом и поэтому усиленно разъедается. Поэтому изделия из алюминиевых сплавов иногда покрывают (плакируют) А. чистотой не менее 99,95%.
Рис. 2. Разрез электролизёра для промышленного получения алюминия. / —катодная шина; 2 — расплавленный алюминий; 3 — тепловая изоляция; 4 — угольная обкладка; 5 — расплавленный электролит; 6 — корка застывшего электролита; 7 — слой Д12О3; 8 — угольные аноды; 9 — анодная шина.
Электролит для получения алюминия состоит из расплава криолита Na3[AIFe] и окиси алюминия Д12О3. Ток подводится к электродам посредством «шин» — плоских проводов прямоугольного сечения. Электроды изготовляют из нефтяного кокса, состоящего почти нацело из углерода. Этот материал хорошо проводит ток при высоких температурах и устойчив к действию расплавленного криолита. Блоки из кокса, устилающие дно и стенки ванны, служат катодом. Аноды укрепляются на подвесной раме и автоматически опускаются по мере сгорания.
Процесс электролиза можно схематически представить так. Д12О3 при растворении в криолите диссоциирует на катионы А13+ и анионы О2-. Катионы А13+направляются к катоду и там разряжаются (2А13++бё=2А1). При разряде анионов О2- на аноде получается свободный кислород (2О2~—4ё=О2), который тотчас же реагирует с углём (ЗО2+4С=2СО2-Ь +2СО).
Жидкий алюминий собирается на дне ванны, из которой его периодически выпускают через наклонный канал, закрываемый глиняной пробкой. Процесс ведут непрерывно.
67
АЛЮМИНИЯ ОКИСЬ
Павел Павлович ФЕДОТЬЕВ. Дал производству алюминия строгую научную основу.
Советский химик-технолог Павел Павлович Федотьев родился 21 июня 1864 г. в Благовещенске на Амуре. По окончании Петербургского технологического института (1888 г.) работал инженером на химических заводах. С 1904 г. по день смерти — профессор Петербургского (Ленинградского) политехнического института, где воспитал большую школу химиков — инженеров и исследователей.
Ещё будучи заводским работником, Федотьев недоумевал, почему важнейшие технологические процессы, созданные в XIX в. чисто эмпирическим путём, «посредством проб и ошибок», всё ещё лишены строгого научного обоснования. Одним из производств, в которые Федотьев внёс свою лепту как учёный, было получение соды аммиачным способом. Другим — производство алюминия электролизом. К исследованиям в области электрометаллургии алюминия Федотьев приступил с 1910 г. Совместно с сотрудниками он определил зависимость температур плавления криолито-глинозёмных смесей от их состава, изучил влияние различных добавок и других факторов на выход алюминия, иными словами —дал производству строгую научную основу.
Весной 1929 г. в Ленинграде на заводе «Красный выборжец», на полуза-водской установке под руководством Федотьева был получен первый советский алюминий; энергию доставила Волховская ГЭС. А в 1932 г. Волховский алюминиевый комбинат начал промышленное производство этого металла.
Член-корреспондент Академии наук СССР П. П. Федотьев деятельно участвовал в создании советской химической промышленности. Он умер 20 марта 1934 г. в Ленинграде.
Тончайший порошок А. применяется как составная часть серебристых красок, а также как горючее для вспышек при фотосъёмках. Тонкий листовой А. (алюминиевая фольга) служит превосходной обёрткой для конфет, шоколада, сыра, заменившей остродефицитное олово. Благодаря низкому удельному электросопротивлению чистый А.— хороший материал для проводов (при равной электропроводности алюминиевый провод примерно вдвое легче медного). Но механическая прочность А. невелика. Поэтому на линиях электропередач нередко прокладывают стале-алюминиевые кабели. В таких кабелях сердечник из оцинкованных стальных проволок обвит проволоками из А.
Высокая отражательная способность А. используется для изготовления зеркал: тонкий слой А. (порядка не более 0,12 микрон) наносят на поверхность стекла катодным распылением или (чаще) испарением в вакууме. Такие зеркала успешно заменяют серебряные.
При высокой температуре А. восстанавливает окислы многих металлов и неметаллов. Поэтому его применяют для получения тугоплавких металлов из окислов. При этих процессах, называемых алюминотермией (от греч. «термос»— горячий), в короткое время выделяется такое количество тепла, что реакция идёт без внешнего подогрева и все продукты получаются в жидком состоянии. Этим способом получают многие тугоплавкие металлы, например хром и марганец:
Сг20зН-2А1 = А120з + 2Сг
• ЗМп3О4 + 8А1 = 9Мп + 4А12О3 а также некоторые ферросплавы.
Применяют в технике и соли А. Так, А1С13 служит катализатором во многих органических синтезах. Сульфатом A. A12(SO4)3- 18Н2О осветляют мутную воду на водопроводных станциях. Хлопья А1 (ОН)3, образующиеся при гидролизе по обратимой реакции:
A12(SO4)3 + 6Н2О 2А1(ОН)3 + 3H2SO4 увлекают взвешенные в воде мелкие частицы, опускаются с ними на дно резервуара, и вода становится прозрачной. Сульфат А. применяется также при крашении тканей, дублении кожи, в производстве бумаги.
Алюминиевая промышленность—одна из наиболее энергоёмких отраслей народного хозяйства. На получение 1 т А. расходуется около 18 000 квт-ч электроэнергии. Не случайно, что крупные алюминиевые заводы всегда вырастают там, где мощные ГЭС дают дешёвую электроэнергию. В Сибири на базе крупных ГЭС сооружены и продолжают расширяться гиганты алюминиевой промышленности—Иркутский, Красноярский, Братский. С. А. Погодин. АЛЮМИНИЯ ОКИСЬ, оксид алюминия (III), глинозём, А12О3 — соединение алюминия с кислородом. Бесцветные кристаллы, плотность 3,96 г/см3, /пл 2050 °C, /кип свыше 3000 °C. А. о. нерастворима в воде, обладает амфотерными свойствами—взаимодействует как с кислотами, так и со щелочами (см. Алюминий). Из гидратированных форм А. о. известны А1(ОН)3 и АЮ(ОН). Гидроокись А1 (ОН)3 выпадает в виде объёмистого белого прозрачного «студня» (геля) при осторожном добавлении щёлочи к растворам солей алюминия.
АМАЛЬГАМЫ
68
А12О3 встречается в природе в виде весьма твёрдого минерала корунда. Из него делают резцы для скоростного резания стали и других металлов. Загрязнённый кварцем и окисью железа природный корунд, известный под названием наждака, издавна используют для полировки металлов и изготовления точильных камней. Красивые драгоценные камни — красный рубин, синий сапфир, зелёный восточный смарагд, фиолетовый восточный аметист — это корунд, содержащий окислы других металлов, которые и придают ему разную окраску.
Кристаллы корунда можно вырастить в лабораторных и промышленных условиях. Если расплавить А. о. в электродуге таким образом, чтобы капли падали на остриё глиняного конуса, то, охлаждаясь, они образуют большие монокристаллы, почти не отличимые от природных. А добавляя в расплав те или иные окислы, можно получить многие из перечисленных драгоценных камней. Такие камни издавна известны ювелирам и часовщикам. А сейчас их потребовала самая новая техника.
Так, рубин (А12О3 с небольшой примесью ионов Сг3+) первоначально служил материалом для часовых подшипников. Но самая современная область применения рубина — квантовая электроника. Первым оптическим квантовым генератором, созданным в 1960 г., был рубиновый лазер.
Что же такое лазер? Это источник очень мощного узкого монохроматического пучка света — фантастический «гиперболоид инженера Гарина», когда-то придуманный А. Н. Толстым, а в наши дни ставший явью. Лазерный луч может дать чрезвычайно сильный концентрированный нагрев, способный прожечь отверстие не только в бруске металла, но и в
На фотографии: в фокусе лазерного пучка в воздухе образуется световая вспышка — лазерная искра.
алмазе. Лазерное излучение передаёт информацию на огромные расстояния (наши луноходы имели лазерный канал связи с Землёй), а кроме того — информацию, в тысячи раз большую, чем обычные радиоволны.
Устройство лазера — область физики, а не химии. Скажем только, что, хотя со временем появилось много других лазерных материалов (водные растворы органических красителей, полупроводниковые соединения, СО2, смесь Не и Ne и др.), рубин остаётся среди них одним из лучших.
А. о. нашла ещё одно, несколько неожиданное применение. Дегидратацией (обезвоживанием) при 200—400 °C студнеобразного геля А1 (ОН)3 получают так называемую активную А. о. Она обладает очень большой внутренней поверхностью. Это и дало повод назвать её активной. Такая А. о. способна поглощать газы, пары и растворённые вещества. Поэтому её применяют как адсорбент в хроматографии — при разделении, очистке и анализе различных соединений.
АМАЛЬГАМЫ — сплавы ртути с другими металлами. Как известно, ртуть — жидкость. Сплавы же её с различными металлами могут быть при комнатной температуре и жидкими, и тестообразными, и твёрдыми. В одних случаях — это химические соединения, иногда очень прочные, плавящиеся при высоких температурах,— LiHg (600 °C), NaHg2 (355 °C), MgHg (630°C). В других — это жидкие и твёрдые растворы, образованные металлом и ртутью, например А. свинца и олова (о твёрдых растворах см. статью Алюминиевые сплавы). При соприкосновении с каким-либо металлом ртуть сначала смачивает его поверхность, а затем диффундирует внутрь. (Поэтому на воздухе, содержащем пары ртути, белеет золото.) Этот процесс, называемый амальгамацией, некогда широко применялся в металлургии. В виде А. металлы отделяли от пустой породы, смачивая ртутью руду. Таким образом ещё две тысячи лет назад добывали золото. Амальгамацией в металлургии благородных металлов пользуются и сегодня.
Химикам важно, что А. натрия — хороший восстановитель. Амальгамированный натрий вступает в те же реакции, что и свободный, но — и это-то всего существеннее — ведёт себя гораздо спокойнее.
А. можно получать электролизом — нужный металл выделяется под действием электрического тока из раствора своей соли и осаждается на катоде, которым служит ртуть. Кстати, именно подобным способом — электролизом смоченных водой «земель» неизвестного в то время состава — химики впервые получили в 1808 г.
69
АММИАК
А. кальция, стронция, бария, магния. Так были открыты эти элементы.
АМЕРЙЦИЙ (лат. Americium) Am — радиоактивный химический элемент с атомным номером 95, относится к актиноидам.
АММИАК NH3 — простейшее химическое соединение азота с водородом. В обычных условиях бесцветный газ с резким запахом и едким вкусом.
А.— один из важнейших продуктов химической промышленности. Синтез его из азота воздуха и водорода — основной метод получения так называемого связанного азота. В природе А. образуется при разложении азотсодержащих органических веществ. Термин «аммиак» происходит от греческих слов «хальс ам-мониакон» (аммонова соль); так назывался хлорид аммония NH4C1, который в древности добывали в оазисе Аммониум в Ливийской пустыне. По-арабски хлорид аммония назывался «ну-шадйр», откуда произошло русское «нашатырь» (в быту же нередко неправильно называют нашатырём нашатырный спирт — водный раствор аммиака).
Физические свойства и строение молекулы. А. примерно в 1,7 раза легче воздуха — плотность его при обычных условиях 0,771 г/л. При нормальном давлении и охлаждении до —33,35 °C А. превращается в бесцветную жидкость, затвердевающую при —77,7 °C. При давлении ок. 9 атм А. сжижается уже при комнатной температуре. А. хорошо растворим в воде (700 объёмов NH3 в 1 объёме Н2О при 20 °C). Растворяется он и в спиртах, бензоле, ацетоне.
Молекула NH3 имеет форму пирамиды. В её основании лежит треугольник из атомов водорода, а в вершине находится атом азота (рис. 1, о).
Обозначим условно электроны, принадлежавшие до образования молекулы А. атомам водорода, крестиками, а внешние электроны атома азота — точками. (Условно — потому что все электроны неразличимы!)
Рис. 1. Строение молекулы аммиака (а) и нона аммония (б*).
Тогда электронная формула молекулы А. будет выглядеть так:
н h:n: • X н
Азот более электроотрицателен, чем водород (см: Неметаллы и Металлы). Поэтому при образовании связи N—Н общие электронные пары «сдвигаются» к атому азота. Каждая связь N—Н становится полярной. Потому и молекула NH3 в целом полярна. Из электронной формулы видно и другое: у атома азота остаётся свободная (неподелённая) пара электронов. Это дополнительно увеличивает полярность молекулы NH3 и обусловливает многие свойства А. С полярностью молекул связана сравнительная лёгкость превращения газообразного А. в жидкость. Ведь конденсация газов определяется взаимным притяжением отдельных молекул, а у полярных молекул есть противоположно заряженные полюсы, и потому они притягиваются друг к другу сильнее, чем неполярные. По этой же причине А. имеет высокую теплоту испарения (23,37 кдж/моль, или 5,581 ккал/моль). Наконец, благодаря полярности молекул NH3 жидкий А. является, подобно воде, хорошим растворителем ионных соединений: электрическое поле его* молекул как бы расталкивает ионы в таких соединениях, помогая им разойтись.
Жидкий А. и по другим свойствам похож на воду. Он имеет высокую диэлектрическую проницаемость, равную 25,4 (у воды 81,0). Молекулы в жидком А. сильно ассоциированы, как и молекулы воды, главным образом за счёт образования водородных связей (см. об этом в статье Вода).
Интересное свойство молекул А.— их способность к структурной инверсии, т. е. к «выворачиванию наизнанку»: атом N всё время перемещается таким образом, что оказывается попеременно то над плоскостью основания,'то под ней. С такой инверсией связано электромагнитное излучение строго определённой частоты в радиодиапазоне СВЧ (сверхвысоких частот). На молекулах NH3 советские физики академики Н. Г. Басов и А. М. Прохоров в 1955 г. создали новый вид генератора излучения — так называемый мазер. Позднее (в 1960 г.) появились и первые лазеры (об этом см. в статье Алюминия окись).
Аммиачный мазер позволил создать аппаратуру для очень точного измерения времени. Благодаря таким сверхточным «молекулярным часам» установлено, что продолжительность земных суток ежегодно возрастает в среднем на 0,00043 сек.
Химические свойства. А.— весьма реакционноспособное соединение. Благодаря неподелён-ной электронной паре у атома N для NH3 особенно характерны и легко осуществимы реакции присоединения. Партнёрами А. в таких реакциях могут быть молекулы или ионы, спо-
АММИАК
70
Свежий газ
Оборотный газ
Рис. 2. Схема синтеза аммиака. Синтез аммиака проводят в специальных аппаратах, называемых колоннами синтеза. Азото-водородная смесь проходит колонну синтеза (/) и холодильник (конденсатор) (2), в котором образовавшийся аммиак NH3 охлаждается и превращается в жидкость (конденсируется). Отделение жидкого аммиака от газовой смеси происходит в сепараторе (3). Жидкий аммиак удаляется из системы, а непрореагировавшие газы поступают в циркуляционный компрессор (4), который подаёт их снова в колонну синтеза (/). В современных установках теплота, выделяющаяся при синтезе NH3, используется для нагревания смеси N2-|-H2 в колонне (/).
Жидким аммиак i
собные предоставить для электронной пары азо
та место на своих незаполненных электронных оболочках. Типичный пример — образование иона аммония (см. Аммония ион) путём присоединения иона водорода Н+ к молекуле NH3
Н
I. н—n: ч- н =
I н
или, в другой записи:
Н
X •
h:n: + н+ =
X • н
н
Н—N—Н
Н
н
н: n : н X • н
В ионе аммония все связи ковалентны и неразличимы (равноценны). Такой механизм образования химической ковалентной связи называют донорно-акцепторным, а связь — координационной. Атом азота, имеющий свободную электронную пару,— донор; ион водорода Н+, предоставляющий место этой паре на свободной электронной оболочке,— акцептор (см. Атом). Со многими солями А. образует продукты присоединения — аммиакаты (например, CuSO4-5NH3, CuSO4-4NH3-H2O). Они подобны гидратам.
По поводу растворения А. в воде существовало мнение, что это — химическое взаимодействие по схеме:
NH3 Н2О NH4OH NH4+ -J- ОН"
Однако термодинамические (то есть энергетические) расчёты показали, что на первой стадии процесса образуется вовсе не гидроокись аммония NH4OH, а гидрат аммиака NH3*H2O:
NH3 4- Н2О NH3.H2O NHt + ОН-
Оттого, что ионы NH4 и ОН- в растворах А. образуются не из NH4OH, а из NH3«H2O, таких ионов получается мало: в разбавленном водном растворе — при концентрации 1 моль/л — каждые 1000 молекул NH3 дают только 42 пары ионов NHJ и ОН-. Такие растворы проявляют слабую щелочную реакцию.
Раствор с концентрацией около 10% А. иногда называют нашатырным спиртом. Насыщенный при комнатной температуре раствор А. в воде содержит его около 25%.
При взаимодействии А. с кислотами образуются соли аммония — кислые и средние:
NH3 + H2SO4 = (NH4)HSO4 2NH3 4- H2SO4 = (NH4)2SO4
Поскольку азот в молекуле NH3 имеет окислительное число —3, А. является восстановителем. При нагревании NH3 с сильными окислителями — хлором, бромом, перекисью водорода, а также с окислами некоторых металлов образуется свободный азот:
2NH3 4-3Cl2-N2 + 6НС1
2NH3 + ЗСиО = N2 + ЗСи + ЗН2О
В кислороде А. горит:
4NH3 + ЗО2- 2N2 + 6H2O
71
АММИАК
На воздухе же А. горит, только если смесь воздуха с А. содержит 16—25% NH3.
Когда окисление А. кислородом ведут в присутствии катализатора (сплав платины с родием или др.), то образуется окись азота:
4NH3 + 5О2 = 4NO + 6Н2О
На этой реакции с последующим окислением NO в двуокись азота NO2 основаны промышленные методы получения азотной кислоты.
Для А. характерны и реакции замещения. Так, щелочные и щелочноземельные металлы при взаимодействии с А. могут давать в зависимости от условий либо нитриды (Na3N — когда замещены все атомы Н), либо амиды (NaNH2—когда замещён один атом Н). При действии избытка хлора на NH3 или насыщенный раствор NH4C1 образуется хлористый азот NC13 — очень непрочный продукт замещения водорода в А.:
NH3 + 3C12 = 3HC1 + NC13
NH4C1 + ЗС1 2 = 4НС1 + NC13
Взаимодействие А. с двуокисью углерода при температуре 150—190°С и давлении 100— 200 атм даёт очень ценный химический продукт — мочевину:
2NH3 + СО2 = (NH2)2CO + Н2О Десятки млн. т мочевины применяют как удобрение в сельском хозяйстве и как полупродукт во многих химических производствах.
Получение и применение. В лаборатории легко получить небольшие количества А., действуя щелочами или гашёной известью на растворы солей аммония, например:
NH4C1 + NaOH - NaCl + NH3 + H2O Газообразный А. освобождают от влаги, пропуская через стеклянную трубку или колонку, заполненные кусочками щёлочи.
В промышленности А. получают прямым синтезом из азота и водорода. Процесс был разработан в 1904—07 гг. немецким учёным Ф. Габером и осуществлён в 1913 г. в Германии инженером К. Бошем. Реакция
N2 + 3H2t=±2NH3 происходит только в присутствии катализаторов. Она обратима и протекает с выделением теплоты и уменьшением объёма. Поэтому наи^ более благоприятные условия образования А., с точки зрения химического равновесия,— возможно более низкая температура и возможно более высокое давление.
Однако скорость этой реакции при низких температурах мала. Поэтому были найдены такие оптимальные условия: температура 450— 550 °C, давление около 300 атм. Катализатором служит губчатое железо, содержащее так называемые промоторы — добавки окиси калия,
окиси алюминия и др. (эти добавки помогают улучшить свойства катализатора). В указанных условиях содержание аммиака на выходе из контактного аппарата составляет всего 14— 20%. Для полного превращения азото-водородной смеси в аммиак её несколько раз возвращают в контактный аппарат.
Производство А. состоит из 3 стадий: получение водорода и азота, очистка их от вредных примесей, синтез А. Источником азота служит воздух. Что касается водорода* то сейчас наиболее целесообразным признано его получение методом конверсии природного газа (см. Водород). В основе способа лежат реакции:
сн4 + н2о = со + зн2
СО + Н2О-СО2 + Н2
Схема синтеза А. изображена на рис. 2, колонна синтеза—на рис. 3.
Рис. 3. Колонна синтеза аммиака. «Человек — мера всех вещей», — сказал древнегреческий философ Протагор. Эта мысль поможет вам при взгляде на фотографию получить представление о размерах колонн для синтеза аммиака.
АММОНИЯ ИОН
72
Главный потребитель А.— производство азотной кислоты и азотных удобрений. Удобрением может служить А. или аммиачная вода, но наиболее широко применяют как азотное удобрение нитрат аммония NH4NO3 и карбамид, то есть мочевину (NH2)2CO. Во всём мире в 1970 г. на получение азотной кислоты и удобрений было израсходовано почти 45 млн. т А., что составляет примерно 4/5 всего его производства. А. используется и для получения соды, синильной кислоты, многих органических соединений.
Е. Р. Шендерей, В. Л. Василевский.
АММОНИЯ ион — сложный одновалентный катион NH4. (Об образовании А. и. из молекул аммиака NH3 и ионов водорода Н+ см. Аммиак.) А. и. представляет собой правильный тетраэдр. В центре находится атом азота, а в вершинах — атомы водорода (рис. 1, б на стр. 69). Размеры А. и. близки к размерам иона калия и его свойства весьма напоминают свойства ионов щелочных металлов. Так, все соли аммония, подобно солям щелочных металлов, растворимы в воде и образуют сходные кристаллы.
Соли аммония сильно гидролизованы в водных растворах. При нагревании они разлагаются:
NH4C1 = NH3 + НС1
Такое (термическое) разложение хлорида аммония — нашатыря — позволяет использовать эту соль для очистки металлических поверхностей при пайке. Окисные плёнки на металлах снимаются нашатырём в результате-образовании летучих хлоридов при нагревании:
СиО 4- 2НС1 =СиС12 + Н2О
ZnO + 2НС1 -ZnCl2 + Н2О
Термическая устойчивость солей аммония тем выше, чем сильнее кислота, образующая эти соли. Так, сульфат аммония гораздо устойчивее при нагревании, чем карбонат. Термическое разложение карбоната, нитрата и нитрита аммония протекает так:
(NH4)2CO3 - 2NH3 + СО2 + Н2О
NH4NO3 = N2O + 2H2O
NH4NO2 = N2 + 2Н2О
Сульфат, фосфаты и нитрат аммония — хорошие удобрения (см. также Селитры, Нитраты). АРГОН (лат. Argonum) Аг—химический элемент VIII группы периодической системы Менделеева; атомный номер 18, атомная масса 39,948; относится к инертным газам. Входит в состав атмосферы Земли (0,93% по объёму). Атмосферный А. состоит из 3 стабильных изотопов: 36Аг (0,337%), 38Аг (0,063%) и 40Аг (99,60%).
Преобладание 40Аг в естественной смеси изотопов этого элемента — причина одной из известных «аномалий» в периодической системе Менделеева: атомная масса А. несколько больше (на 0,846), чем следующего за ним в таблице калия. Эта
аномалия оставалась необъяснимой до 1913 г., когда стало ясно, что элементы должны располагаться в таблице в порядке возрастания не атомной массы, а заряда атомного ядра. Но почему 40Аг составляет более 99% природного элемента? Такое преобладание одного изотопа связано с тем, что он непрерывно образуется при радиоактивном распаде 40К. Этот изотоп калия распадается крайне медленно — период полураспада 1,3 млрд. лет. К тому же в общей смеси изотопов калия на его долю, приходится менее 0,012%, и в 1 т калия образуется за год только 3100 атомов А. Однако калий — один из наиболее распространённых элементов, и за долгую историю Земли — несколько миллиардов лет — в атмосфере успело накопиться так много А., что 1 м3 воздуха содержит 9,3 л А. Это в несколько раз больше, чем количество всех остальных инертных газов, вместе взятых.
К открытию А. привело маленькое превышение — всего на 0,0016 г/л (0,13%) — плотности азота воздуха по сравнению с плотностью азота, полученного при разложении NH4NO2. Это превышение обнаружил в 1892 г. знаменитый английский физик Дж. Рэлей. Он не счёл такую малость «ошибкой опыта», В 1894 г. Рэлей и не менее знаменитый английский химик У. Рамзай нашли в азоте воздуха примесь более тяжёлого газа. Новый газ не вступал ни в какие реакции. За ярко выраженную химическую инертность он получил название «аргон» (греч. «аргос» — недеятельный).
А. получают в процессе разделения воздуха при глубоком охлаждении. Больше 80% промышленного А. расходуется на создание защитной атмосферы при плавке, резке и сварке активных металлов. В такой инертной атмосфере получают сверхчистые материалы для полупроводниковых приборов и для реакторо- и ракетостроения.
В атмосфере А. легко возбуждается электрический разряд, обладающий сине-голубым свечением. Поэтому аргоновые трубки используют для световых реклам. А. широко применяют и для наполнения электрических ламп накаливания. Это неслучайно. Как лучше всего сделать такую лампу? Если наполнить баллон воздухом, раскалённая вольфрамовая нить мгновенно сгорит из-за окисления вольфрама кислородом воздуха. В безвоздушном стеклянном
73
АТМОСФЕРА
баллоне она быстро разрушится — испаряющиеся атомы W, покидая нить, не будут встречать препятствий со стороны атомов других веществ. В баллоне, наполненном азотом, испарение несколько затормозится из-за столкновений атомов W с молекулами N2. Но молекулы азота легки и для тяжёлых атомов W не представляют серьёзной преграды. Преимущества А. перед воздухом и азотом несомненны: его атомы и химически инертны, и тяжелее молекул азота — они способны более надёжно «сторожить» вольфрамовую нить. Серьёзный соперник А.— криптон, инертный газ с ещё большей атомной массой. Соперником может быть и инертный газ ксенон, но он очень дорог. О физических и химических свойствах А. см. Инертные газы.
С. С. Бердоносов. АРСЕНАТЫ — соли мышьяковой кислоты, например K3AsO4 (см. Мышьяк). По ряду свойств А. похожи на фосфаты. Очень ядовиты. Применяются в сельском хозяйстве для борьбы с насекомыми-вредителями.
АРСЕН ЙДЫ — соединения мышьяка с металлами. При взаимодействии с кислотами А. щелочных, щелочноземельных и некоторых других металлов выделяют мышьяковистый водород:
Zn3As2 + 6НС1 = 3ZnCl2 + 2AsH3 А. металлов III группы периодической системы (Al, Ga, In) интересны как полупроводниковые материалы.
АРСЕН ЙТЫ — соли не выделенной в свободном состоянии мышьяковистой кислоты, например NaAsO2 (см. Мышьяк). А. очень ядовиты. Подобно арсенатам применяются в сельском хозяйстве как инсектициды (от лат. in-sectum — насекомое и caedo—убиваю). Одно из очень давно известных средств для истребления насекомых-вредителей — парижская зелень — это смешанная медная соль мышьяковистой и уксусной кислот: Си(СН8СОО)2-ЗСи(AsO2)2.
АРСЙН — то же, что мышьяковистый водород AsH8.
АСТАТ (лат. Astatium) At —радиоактивный химический элемент VII группы периодической системы Менделеева; атомный номер 85; отно-
сится к галогенам. Все. изотопы А. радиоактивны и имеют небольшие периоды полураспада 71/,. Наиболее долгоживущие из них — это искусственно полученные 210At (71 /,=8,3 часа) и 211At (7,2 часа). Из изотопов А., входящих в состав естест
венных радиоактивных рядов и поэтому встречающихся в природе, выделен химическим путём 219At (7i/2=0,9 мин).
Существование тяжёлого аналога иода впервые предсказал в 1870 г. Д. И. Менделеев, назвавший предполагаемый элемент экаиодом. Несмотря на упорные поиски, его долго не могли найти в природе. В 1940 г., после того как были открыты пути искусственного синтеза элементов, Д. Корсон, К- Мак-Кензи и Э. Сегре (Калифорния, США) обнаружили, что при облучении висмута а-частицами (ядрами гелия 2Не) протекает ядер-ная реакция, приводящая к образованию элемента с атомным номером 85:
2?lBi + 42He = 281jAt + 210n
(где Jn — нейтрон).
Название А. получил от греч. слова «астатос» (неустойчивый) — у него нет долгоживущих изотопов. Существование четырёх природных изотопов А. удалось установить лишь в 1943—46 гг.
Содержание А. в природе ничтожно. По оценке геохимиков, поверхностный слой земной коры толщиной 1,6 км содержит всего 0,07 г А.— почти в 400 раз меньше, чем одного из редчайших элементов—франция. Неудивительно, что свойства А. приходится изучать на его микроколичествах: так, концентрация А. в растворах обычно составляет 10“11—10“16 моля на литр.
Внешняя электронная оболочка атома А. содержит 7 электронов и имеет конфигурацию 6$26рб (см. Атом). Если бы А. удалось получить в весомых количествах, мы, вероятно, узнали бы много интересного. А. занимает пограничное место между металлами и неметаллами. В его свойствах должны сочетаться черты и тех и других. Исследования, проведенные с микроколичествами элемента, подтверждают этот вывод. А. похож и на типичный неметалл — иод, и на металл — полоний. Подобно иоду А. проявляет степени окисления —1, +1 и +5 (валентности I и V). Подобно иоду А. летуч, но его летучесть заметно ниже, чем других галогенов. Однако, в отличие от иода, А. из растворов можно осадить сероводородом, а цинк вытесняет А. из его сернокислых растворов. Эти свойства характеризуют элемент как металл. Двойственная природа А. делает его одним из наиболее интересных с научной точки зрения элементов периодической системы Менделеева.
С. С. Бердоносов. АТМОСФЕРА — газовая оболочка планет и звёзд. А. Земли принято считать ту окружающую её газовую область, которая вращается с планетой как единое целое. Термин «А.» произошёл от греческого «атмбс» — пар, испарение и «сфайра»—шар.
А. Земли в современную геологическую эпоху состоит главным образом из азота N2 (около 78,09% по объёму) и кислорода О2 (20,95%). Довольно много в А. аргона Аг (0,93%), тогда
АТМОСФЕРА
74
как на долю других инертных газов приходятся лишь тысячные и миллионные доли процента. Содержание азота, кислорода и аргона в нижних слоях А. постоянно. Но есть в А. и такие газы, концентрация которых меняется от места к месту и даже зависит от времени года, времени суток и от многих других факторов. Непостоянно содержание в А. двуокиси углерода СО2 (в среднем 0,03% по объёму). Особенно же изменчива концентрация водяного пара — от тысячных долей процента в полярных областях Земли до 3—4% в тропиках. Поэтому все сведения о составе всегда относят к «сухой» А.
В А. много и других примесей с непостоянной концентрацией: метан СН4, двуокись серы SO2, окислы азота, водород Н2, озон О3 и т. д. Среднее их содержание в А. ничтожно. Однако в отдельных местах концентрация, например, SO2 становится опасно большой. Прежде это были лишь окрестности вулканов. Сегодня с ними, к сожалению, можно сравнить районы особенно бурной хозяйственной деятельности человека ... В А. всегда есть пыль и мелкие частицы дыма (см. также Воздух).
Общая масса А. Земли составляет около 5,15-1018 кг. Разделив эту величину на поверхность земного шара (5,1 «1018 см2), легко убедиться, что масса воздуха над каждым квадратным сантиметром земной поверхности приблизительно равна 1 кг. Поэтому среднее давление А. (около 1 кг/см2) послужило в своё время основой для выбора единицы давления, названной атмосферой (атм). В настоящее время в Интернациональной системе единиц (СИ) принята иная единица давления (кн/м2). Атмосферное давление на уровне моря, условно принятое за нормальное, равно 101,3 кн/м2 (760 мм ртутного столба). С высотой давление А. закономерно уменьшается. Так, на высоте 10 км оно составляет 210 мм рт. ст. При таком давлении вода закипает уже при температуре человеческого тела.
Посмотрите на рисунок «Атмосфера Земли». Главным критерием для выделения отдельных слоёв А. служит температура. Уже давно установлено, что с высотой температура воздуха снижается (примерно на 0,6° на каждые 100 м) и на высоте около 10 км она равна уже —55 —60 °C. В этом, самом нижнем слое А. происходят горизонтальные, вертикальные и круговые (турбулентные) перемещения воздуха. Отсюда и название слоя — тропосфера (от греч. «тропбс» — поворот).
Когда шары-зонды дали учёным возможность измерять температуру на высотах более 10 км, выяснилось неожиданное: начиная с 10—12 км, температура перестаёт понижаться. Родился термин тропопауза. Возникло предположение, что в этих условиях происходят лишь горизонтальные движения воздушных масс — так сказать, движения послойные. И часть А., расположенную над тропосферой, назвали стратосферой (от лат. stratus — слой). В дальнейшем предположение не подтвердилось: и на этой высоте воздух перемещается не только горизонтально, послойно. Однако термин «стратосфера» прочно вощёл в науку.
Сейчас к тропосфере относят слой А. в 10— 18 км и к стратосфере — примерно до 50 км над уровнем моря. В пределах стратосферы, начиная с высоты около 20—25 км, температура постепенно увеличивается, достигая 0°С у границы этого слоя А.
Остальные слои А.— мезосфера (от греч. «месос» — промежуточный), термосфера (от греч. «терме» — тепло) — тоже выделены в соответствии с изменениями температуры (вплоть до 700°C на высоте 800—1000 км). Самую внешнюю часть А. называют экзосферой (от греч. «эк», «экс» — приставка, означающая отделение, удаление).
Границы отдельных слоёв А. непостоянны, условны. Их положение зависит и от времени суток, и от географической широты местности (у экватора А. толще, чем у полюсов). Нет и резкой границы, за которой начинается «безвоздушное пространство».
Около 80% массы А. сосредоточено в тропосфере, почти 20% — в стратосфере и лишь 0,5% — во всех остальных слоях А.
Температура — это мера кинетической энергии движущихся частиц. В верхних слоях А. частицы движутся с огромными скоростями, что и отвечает высоким температурам (до 700 °C). Но поскольку газы на больших высотах крайне разрежены, число столкновений частиц с каким-нибудь телом (например, спутником) так незначительно, что их кинетическая энергия, переданная телу и превратившаяся в тепло, ничтожно мала. Так что и при температуре 700 °C в термосфере не жарко (если, конечно, защититься от палящих лучей Солнца).
Описывая структуру А., мы шли от поверхности Земли вверх. Теперь совершим обратный путь вместе с приходящим к Земле солнечным излучением, вместе с космическими лучами и с достигающей границ А. космической пылью.
Электромагнитное излучение Солнца, состоящее из радиоволн, из инфракрасных, световых (видимых глазом), ультрафиолетовых и рентгеновских лучей, ежеминутно приносит к границе А. энергию, составляющую около 8,38 дж/см2 (2 кал/см2). Это главный источник энергии физических, химических и биологических процессов на Земле.
Рентгеновское излучение — самое энергичное. И одновременно — губительное для жизни. До поверхности Земли оно не доходит, а поглощается уже в верхних слоях А., вызывая ионизацию атомов и молекул. Так в верхней А. образуется протяжённый электропроводящий слой — ионосфера. От него отражается в межпланетное пространство большая часть радиоволн, излучаемых Солнцем. Отражение коротких радиоволн земных радиостанций от ионосферы делает возможной дальнюю радиосвязь. Однако в ионосфере есть «окна» — области длин волн, для которых ионосфера «прозрачна». Эти окна используются для космической радиосвязи.
75
АТМОСФЕРА
Космическая пыль, сгорающая в А., также пополняет её ионами. От Солнца и из космического пространства в А. идёт поток заряженных частиц высоких энергий (протоны Jp, а-частицы и др.). Это — космические лучи. Их энергия так велика, что вызывает не только изменения в электронных оболочках атомов и молекул (ионизацию), но и разрушает ядра атомов, приводит к ядерным реакциям.
Как возник, например, в А. радиоактивный изотоп водорода тритий (его около 2 кг во всей А.)? Он образуется по реакции между ядрами атомов азота и нейтронами:
“N + хоп = ?Т + 32Не
Нейтроны же, в свою очередь, возникают в А. из других атомных ядер, «разбитых» действием космических лучей.
При взаимодействии ядер азота с нейтронами образуется и радиоактивный изотоп углерода:
14\т I 1П 14^ । 1 + ОП = «О + 1Р
Выше 200 км над поверхностью Земли электромагнитное излучение Солнца вызывает диссоциацию молекул азота N2 на атомы, а на меньшей высоте — диссоциацию кислорода О2. Необходимая для этого энергия «вычитается» из солнечного излучения. Атомарный кислород, взаимодействуя с недиссоциировавшими молекулами О2, даёт озон О3. Он-то и поглощает самую энергичную, самую губительную для всего живого часть ультрафиолетового излучения.
Вспомните, что основной критерий деления А. на слои — её температура. Поглощение энергии Солнца в верхних слоях А. и стратосфере вызывает нагревание А. В тропосфере вода и углекислый газ поглощают большую часть инфракрасного излучения Солнца, так что из
Рис. 1. Атмосфера Земли. Цветовая гамма рисунка условно передаёт слоистое строение атмосферы. Между слоями нет резкой границы, один плавно переходит в Другой.
Чтобы уделить достаточно места каждому слою — независимо от его протяжённости, рисунок поделён на две части и для каждой из них выбран свой масштаб. Нижние слои атмосферы — тропосфера, стратосфера — простираются всего лишь на десятки километров, и для них целесообразен более крупный масштаб (колонка справа). На сотни и тысячи километров уходят ввысь термосфера и экзосфера (колонка слева). Числа у каждой колонки обозначают высоту над уровнем моря (в км).
Наш рисунок густо заселён. Здесь и посланники Земли— самолёт (/), радиозонд (2), спутник (4), ракета (5), и гость из космоса — горящий метеорит (3).
Белые вертикальные линии условно обозначают ультрафиолетовые лучи. Неслучайно эти линии на рисунке обрываются в стратосфере. Самым жёстким ультрафиолетовым лучам преграждает дорогу озоновый экран — слой наибольшей концентрации озона. Верхние слои атмосферы, от 100 до 1000 км, могут давать свечение (люминесценцию). Такое сияние ночного неба временами наблюдается в полярных областях Земли. На рисунке полярное сияние изображено белыми пятнами слева, на высоте около 100 км (6).
3
АТМОСФЕРА
76
спектра электромагнитного излучения в А. «отсекается» как коротковолновая часть (рентгеновское и значительная доля ультрафиолетового излучения), так и длинноволновая часть (значительная доля инфракрасного и радиоизлучения). Для видимой части спектра — для всех цветов радуги! — А. прозрачна.
Значит ли это, что видимый свет полностью доходит до поверхности Земли? Нет! В А. находится в среднем около 12 тыс. км3 воды (в 10 раз больше, чем во всех реках мира!). Это — и невидимые глазу отдельные молекулы Н2О, и редкий утренний туман, и плотные облака, состоящие из капелек воды, и падающие на землю снежинки ... Но белые облака и свежий снег способны отражать до 90% падающего на них света. Облака же покрывают около половины поверхности планеты. Поэтому значительная часть пришедшего к Земле видимого света отражается и тут же отправляется в обратный путь, за пределы Земли. И ещё часть света рассеивается в А.
Пусть и физикам и лирикам будет утешением то, что благодаря способности Земли отражать и рассеивать свет наша планета выглядит из космоса красивой голубой «звездой». А неосвещённая Солнцем часть диска нашей вечной спутницы — Луны — в отражённом Землёй свете имеет красивый пепельно-серый цвет, а не тёмный, как осеннее ночное небо. Поэт Роберт Рождественский сказал о Земле:
«Она проплывает в густом голубом ореоле, прошитая Солнцем.
Продутая ветром.
Пропахшая морем ...»
Голубой цвет неба вот уже тысячелетия вдохновляет поэтов. Но долгое время он был загадкой и для учёных. В наше время и голубизна неба, и багряное зарево восходов и закатов Солнца вполне убедительно объяснены физиками. Более того, голубизна неба дала в руки физиков один из способов определения важнейшей константы— числа Авогадро.
И снова вернёмся на Землю. Дошедшая до поверхности Земли энергия электромагнитного излучения Солнца нагревает и сушу, и океан. Она расходуется на многочисленные процессы, составляющие жизнь планеты. И опять-таки в виде «обратного» — теперь уже не светового, а теплового (инфракрасного) — излучения покидает поверхность Земли. Выходит, Земля, отдав уже часть дошедшего до неё света, теперь отдаст и тепло? К счастью, нет, всего тепла Земля не отдаёт!
В чём секрет обычных парников, где выращивают, например, ранние овощи? В их рамы вставлено стекло (или полиэтиленовая плёнка), пропускающее внутрь парника много света. Этот свет частично расходуется растениями на фотосинтез, а частично переходит в тепло, превращающееся затем в обратное невидимое инфракрасное излучение. В парнике излучает всё — и почва, и стены, и сами растения... Но парник закрыт, тепло из него не выходит, температура внутри повышается.
Такую же роль «закрытого парника» играют в А. СО2 и пары Н2О — они не выпускают в
космос инфракрасное излучение поверхности Земли. Не случайно это явление называют парниковым э ффект ом. Естественно, Н2О и СО2 вбирают в себя и «прямое» инфракрасное излучение, пришедшее от Солнца. В итоге средняя годовая температура у поверхности Земли сохраняется +14,8°С.
В А. Земли происходят сложнейшие процессы формирования погоды и климата. Нагревание поверхности вызывает вертикальные перемещения больших масс воздуха. Неравномерность же нагревания на различных участках Земли (в частности, на суше и океане) создаёт разность давлений и порождает горизонтальные и круговые движения воздуха — ветры. Общая кинетическая энергия, связанная с движением А., грандиозно велика: она сравнима с энергией взрыва десятков тысяч водородных бомб. Подсчитано, что на образование небольшой стайки белых кучевых облаков расходуется за 3—4 часа столько энергии, сколько за то же время могут выработать 20 ГЭС, равных по мощности Волжской.
А., масса которой не превышает одной миллионной массы всей планеты, играет выдающуюся роль в судьбе других сфер Земли (геосфер) .
Первоначальная А. Земли возникла одновременно с гидросферой около 4,5 млрд, лет назад при образовании мантии — слоя, «подстилающего» твёрдую наружную оболочку Земли (земную кору). Тогда, как предполагают, состав А. был близок к составу нынешних вул-
Р и с. 2. Солнечная корона. В момент полнога солнечного затмения из-за тёмного края Луны видны огромные огненные языки, показанные на фотографии. Эти языки — протуберанцы — состоят из сильно раскалённого солнечного вещества, пары которого поднимаются на сотни тысяч километров над поверхностью Солнца. В 1868 г. при изучении спектра протуберанцев обнаружилось, что, кроме водорода, азота и других ранее известных элементов, в состав протуберанцев входит и какой-то элемент, ещё не известный на нашей планете, дающий жёлтую линию спектра. Так наблюдения астрономов положили начало исследованиям, которые в конце концов привели к открытию гелия и на Земле.
АТМОСФЕРА
4,5-3,5 млрд, лет назад 2 — 15 млрд, лет назад
Рис. 3. Эволюция атмосферы. Человеку, жизнь которого измеряется несколькими десятилетиями, трудно представить себе «геологические часы», на циферблате которых — миллиарды лет ...
Первичная атмосфера Земли резко отличалась от современной. 3,5—4,5 млрд, лет назад она по составу напоминала вулканические газы (Н2, N2, H2S, Н2О, СО2, СН4, SO2, NH4C1, НС1, инертные газы). Она была много плотнее нынешней, а огромные тучи пепла и пыли закрывали Солнце настолько, что у поверхности Земли было темно даже днём (рисунок слева). Формирование атмосферы шло параллельно с образованием гидросферы, которая поглотила растворимые в воде газы, оставив в атмосфере N2, СН4, инертные газы, часть СО2 и пары воды.
Не менее чем 1,5—2 млрд, лет назад появилась жизнь. Возникновение растительности (на среднем рис. в кружке микроскопическая водоросль) привело к изменению состава атмосферы. Начался новый этап ее истории — обогащение кислородом. Шли тысячелетия. Всё больше зеленела планета. Росло содержание кислорода в атмосфере (представление об этом даёт пунктир в верхней части рисунка). Голубым цветом — цветом жизни и чистого неба — передан на рисунке этот процесс, особенно бурный в последние 350 млн. лет.
350 млн.лет назад
панических газов (Н2, N2, Cl2, H2S, Н2О, СО2, СН4, SO2, NH4C1, НС1, инертные газы и др.). При охлаждении поверхности Земли пары воды конденсировались. Образовавшийся океан поглотил газы, растворимые в воде. В А. остались в основном азот, водород, метан, инертные газы, частично СО2 и пары воды. Наиболее лёгкие из них — водород и гелий — Земля не смогла удержать и большая их часть рассеялась в межпланетном пространстве. Эта же участь постигла и другие, кроме гелия, лёгкие инертные газы. Кажущееся исключение представляет в этом смысле аргон. Однако высокое содержание аргона в А. связано с постоянным возобновлением его «запаса» в результате радиоактивного распада изотопа калия 40К.
Кислорода в древней А. не было или почти не было. Некоторое количество его могло образоваться при фотохимическом разложении воды или углекислого газа. Многие учёные считают, что под действием солнечного излучения и электрических разрядов в первоначальной А. образовались органические вещества — первая ступень к возникновению жизни.
Зарождение жизни на нашей планете сыграло решающую роль в истории А.: появившиеся
растения в процессе фотосинтеза выделяли кислород, который коренным образом изменил состав А. Изменился и весь ход геологической истории Земли (см. Биосфера). Представление об эволюции атмосферы даёт рис. 2.
Бурной развитие науки и техники поставило перед человечеством серьёзную задачу: борьбу с загрязнением окружающей среды. И, конечно,— с загрязнением А. За одно только десятилетие (1960—1970 гг.) объём поступивших в А. вредных газов и пыли удвоился. Загрязнения исчисляются сотнями миллионов и даже миллиардами тонн в год. Сегодня это, как принято говорить, глобальная проблема: она касается всего человечества.
250 млн. автомобилей, бегающих по дорогам планеты, ежегодно выбрасывают в А. 200 млн. т угарного газа СО и 50 млн. т паров бензина (не говоря уже о СО2). Каждая тонна сожжённого угля забирает из А. количество кислорода, равное годовой потребности в нём десяти человек, а взамен отдаёт двуокись углерода СО2, содержание которой в А. неуклонно растёт. По мнению некоторых учёных, этот рост может привести к повышению температуры нашей планеты (напомним, что СО2 не пропускает за пределы А. теплового излучения Земли). Повышение же температуры может вызвать таяние ледников, затопление части суши и другие нарушения естественного равновесия в природных процессах. Однако другие учёные
АТМОСФЕРА
78
считают, что подобные катастрофы нам не угрожают, потому что избыток СО2 будет поглощён Мировым океаном и пойдёт на фотосинтез в морских растениях.
В нашей стране принимаются действенные меры против загрязнения окружающей среды. В постановлении Верховного Совета СССР (сентябрь 1972 г.) — одном из многочисленных законодательных актов, направленных на решение задачи охраны природы, говорится:
«Считать одной из важнейших государственных задач неустанную заботу об охране природы и лучшем использовании природных ресурсов, строгое соблюдение законодательства об охране земли, её недр, лесов и вод, животного и растительного мира, атмосферного воздуха, имея в виду, что научно-технический прогресс должен сочетаться с бережным отношением к природе и её ресурсам, способствовать созданию наиболее благоприятных условий для жизни и здоровья, для работы и отдыха трудящихся».
В СССР установлены предельно допустимые нормы содержания в А. 120 вредных веществ (в США — только для четырёх). В Москве воздух гораздо чище, чем в столице любой капиталистической страны. В Москве, благодаря выводу за её пределы и переоборудованию более 300 промышленных предприятий, концентрация одного только сернистого газа SO2 в воздухе уменьши
лась вчетверо. За последние годы значительно чище стал воздух в Ленинграде и Горьком, Донецке и Магнитогорске, во многих других промышленных городах.
Советский Союз делает всё, для того чтобы в деле охраны природы установить надёжное международное сотрудничество.
Решение этой проблемы — настоятельная необходимость. «... Либо люди сделают так, чтобы в атмосфере было меньше вредных газов, либо эти вредные газы сделают так, что на Земле станет меньше людей...» —сказал один учёный.
Данные об А. других планет Солнечной Системы, за исключением Венеры, получены косвенным путём и недостаточно полны. А. Венеры, как показали прямые измерения советских межпланетных станций «Венера-4»— «Венера-7», состоит в основном из СО2 с небольшой примесью воды (около 11 мг/л), кислорода (не свыше 0,1%) и азота (менее 2%). Температура А. у поверхности Венеры близка к 400 °C и давление — к 100 атм (в этих условиях вода, если она и есть на поверхности Венеры, ещё не кипит!). А. Марса по массе в 5 раз меньше, чем А. Земли. Давление А. там, как полагают, равно всего 65 мм рт. ст. В состав А. Марса, по-видимому, входят азот, двуокись углерода, аргон и очень небольшое количество паров воды. Кислорода в А. Марса совсем мало (в 1000 раз меньше, чем в земной А.). А. планет-гигантов— Сатурна и Юпитера — содержит водород, метан, аммиак и гелий. А. Солнца на 99,93% состоит из водорода и гелия. В. Л. Василевский.
«Мы, космонавты, по характеру нашей профессии, может быть, раньше, чем кто-либо, сталкиваемся с химией во всех её чудодейственных проявлениях. Возьмите, к примеру, топливо, которое двигает наши ракеты, сплавы и металлы, из которых они сделаны, возьмите скафандры, всю особую космическую продукцию — тысячи и тысячи больших и малых вещей, окружающих человека в его пути в космос. Всюду вы встретитесь с химией,,,».
Ю. А. Гагарин
(Гагарин Ю. А., «Комсомольская правда», 1964, 16 января, с. 2)
БАРИЙ (лат. Baryum) Ba — химический элемент II группы периодической системы Менделеева; атомный номер 56, атомная масса 137,34; относится к подгруппе щелочноземельных металлов. В природе встречаются 7 стабильных изотопов Б., среди которых преобладает 138Ва (71,66% в смеси изотопов).
Содержание Б. в земной коре 6,5 х X 10“2 % по массе (это почти в 30 раз меньше, чем магния, но в 10 раз больше, чем
цинка). В свободном виде из-за своей исключительной химической активности Б. в природе не встречается. Минералы его немногочисленны; практически важны барит BaSO4 и более редкий — витерит ВаСО3.
В 1602 г. один болонский сапожник заметил, что после прокаливания с органическими веществами минерал, названный впоследствии тяжёлым шпатом или баритом, фосфоресцирует («болонские светящиеся камни»). Лишь в 1774 г. шведский химик К- Шееле открыл, что в состав этого минерала входит окисел нового элемента (новая «земля»). А в 1808 г. английский химик Г. Дэви получил металлический Б. (в виде амальгамы) электролизом его расплавленных солей. Новый элемент был назван барием — по-гречески «барйс» означает тяжёлый; а между тем Б. — не так уж тяжёл. Он почти в полтора раза легче барита — соли, которой он обязан своим названием.
Б.— серебристо-белый сравнительно мягкий металл (мягче цинка, но твёрже свинца); его плотность 3,76 г/см3, /пл 710 °C, /кип 1640 °C.
Внешняя оболочка атома Б. содержит два электрона (конфигурации 6s2, см. Атом). Б. образует ионные соединения, в которых его степень окисления равна +2 (валентность II). Валентные электроны Б. расположены далеко от ядра и отщепляются довольно легко, чем и объясняется большая химическая активность Б. В этом отношении он более близок к щелочным металлам, чем к своим аналогам по подгруппе. На воздухе Б. быстро окисляется с об
разованием окиси и перекиси. При нагревании или резком ударе легко воспламеняется и сгорает, давая яркое жёлто-зелёное пламя. Весьма бурно разлагает воду:
Ва + 2Н2О = Ва (ОН)2 + Н2
Непосредственно соединяется с галогенами, образуя галогениды:
Ва + С12 = ВаС12
Активно взаимодействует со многими другими элементами. Хранят Б. в керосине, как и щелочные металлы.
Окись ВаО — белые кристаллы; на воздухе ВаО поглощает углекислый газ, превращаясь в карбонат ВаСО3. Если ВаО нагреть на воздухе до 500 °C, то образуется перекись ВаО2. Окись Б. при действии воды образует гидроокись (едкий барит):
ВаО + Н2О=Ва (ОН)2
Последняя хорошо растворима в воде, в отличие от гидроокисей магния и кальция. Её насыщенный водный раствор — баритовая вода — на воздухе поглощает углекислый газ и мутнеет от выпадения карбоната:
Ва (ОН)2 + СО2 = ВаСО3 + Н2О
Из обычно применяемых солей Б. хорошо растворимы хлорид ВаС12 и другие галогениды, нитрат Ba(NO3)2, сульфид BaS, хлорат Ва(С1О3)2; трудно растворимы сульфат BaSO4, карбонат ВаСО3 и хромат ВаСгО4. Образованием осадка BaSO4 пользуются в аналитической химии для качественного и количественного определения иона SO4", а также иона Ва2+:
Ва (NO3)2+Na2SO4 = BaSO4+2NaNO3
Есть и другой простой способ обнаружить Б. в его соединениях: если внести на кончике шпателя немного вещества, содержащего Б., в пламя горелки, оно окрасится в желтовато-зелёный цвет. Все растворимые соли Б. сильно ядовиты.
Основным сырьём для получения Б. и его соединений служит барит, который восстанавливают углём в пламенных печах:
BaSO4 + 4С = BaS + 4СО
БЕРИЛЛИИ
80
Образующийся растворимый BaS перерабатывают на другие соли Б. Металлический Б. получают восстановлением ВаО алюминием при 1200 °C в вакууме:
4ВаО + 2А1 = ЗВа + Ва(А1О2)2
Возгонкой в вакууме металл легко очистить до содержания примесей менее 1—10“4%. Вполне вероятно, что сверхчистый Б. обнаружит какие-то новые свойства, ценные для техники (как это случилось, например, с титаном и многими другими металлами).
А пока что применение самого металла невелико.
Обращение с чистым Б. затруднительно из-за его высокой химической активности. С другой же стороны, именно химическая активность делает Б. ценным для металлургии. В незначительных количествах его добавляют при выплавке меди и свинца для очистки этих металлов от серы и газов. Б. и его сплавы с магнием и алюминием применяют для поглощения газов в технике высокого вакуума (как геттеры) — в частности для поглощения азота (3Ba+N2=Ba3N2). Сплавы Б. со свинцом применяются в типографском деле, а из сплавов Б. с никелем изготовляют электроды запальных свечей двигателей внутреннего сгорания. Небольшие количества Б. добавляют в подшипниковые сплавы на основе РЬ.
Гораздо чаще, чем с самим Б., встречаются на практике с его соединениями. Сульфат BaSO4 стоек к действию тепла и химически активных газов, содержащихся в атмосфере индустриальных городов. Поэтому его применяют как компонент белых красок. Окрашенные же соли Б.— хромат ВаСгО4 (жёлтый) и манганат ВаМпО4 (зелёный) входят в состав цветных красок. Нитрат Ba (NO3)2 придаёт зелёный свет сигнальным ракетам и бенгальским огням. Карбонат ВаСО3 используют в производстве стекла, эмалей и глазурей. Платиноцианидом Ba [Pt (CN)4] (комплексное соединение) покрывают экраны при работе с рентгеновским и радиоактивным излучением (в кристаллах этой соли под действием излучений возбуждается яркая жёлто-зелёная флуоресценция). Многие слышали про «бариевую кашу», которой угощают пациента перед просвечиванием желудка; это — сульфат BaSO4 (хорошо поглощающий рентгеновские лучи), смешанный с водой.
Но не все применения Б. так обыденны. Есть, например, в химической технологии очень специальная, тонкая область — извлечение радия из урановых руд. В этих процессах BaSO4 и ВаС12 служат так называемыми носителями; выпадая из раствора, они захватывают и соединения радия. И таким образом концентрируют и выделяют этот очень дорогой и редкий элемент. А возможно это потому, что Ra и Ва — элементы-аналоги, близкие по химическим свойствам и ионным радиусам.
Ю. И. Романъков.
9,01218 Д
Бе
БЕРИЛЛИЙ
БЕЛ Й ЛЬНА Я ЙЗВЕСТЬ (хлорная известь)— см. в статьях Хлор, Известь.
БЕРЙЛЛИЙ (лат. Beryllium) Be — химический элемент II группы периодической системы Менделеева; атомный номер 4, атомная масса 9,01218. Светло-серый металл. Элемент имеет природный стабильный изотоп 9Ве.
Некоторые драгоценные камни — минералы Б. (аквамарин, изумруд, александрит и др.) использовались в ювелирном деле ещё до н.э. Сам элемент был открыт в виде «берилловой земли» (ВеО) в 1798 г. французским химиком Л. Вокленом, когда он пытал
ся установить общность химического состава минералов берилла и изумруда. Поскольку растворимые соли Б. сладкого вкуса, его вначале ‘ называли «глюциний» (от греч. «глюкйс»—сладкий) или «глиций». Металлический Б. был получен в 1828 г. Ф. Вёлером в Германии и независимо от него во Франции А. Бюсси по окислительно-восстановительной реакции:
ВеС12 + 2К = Be + 2КС1 Как и многие металлы, открытые в XVII — XIX вв., Б. свыше 100 лет оставался лабораторной редкостью. Применение Б. началось лишь в 40-х гг. нашего столетия, хотя его ценные качества как компонента сплавов были обнаружены уже в 1926 г., а замечательные ядерные свойства — в 1932 г.
О Б. часто говорят как о редком элементе, но в природе он распространён больше, чем, например, олово (содержание Б. в земной коре 3,8-10“4% по массе). Из минералов Б. наибольшее промышленное значение для получения металла и его соединений имеет берилл — минерал из класса силикатов Al2Be3[SieO18]. Переработка минералов Б. очень трудоёмка. В результате переработки получают двойной фторид NajBeFJ. Его расплавляют и подвергают электролизу. Б. в виде чешуек осаждается на катоде. Процесс требует больших затрат электроэнергии.
Б. плавится при 1284 °C, кипит при 2450 °C. Металл сочетает исключительно важные для техники свойства. Он очень лёгок (плотность Б. 1,848 г/см3, а плотность алюминия 2,70 г/см3), весьма тугоплавок и твёрд. Прочность же Б. такова, что пруток из него сечением 1 мм2 выдерживает груз, равный массе взрослого человека,— около 60 кг, в то время как такой же пруток из алюминия выдерживает всего лишь 11,4 кг. Кроме того, Б. обладает высокой прочностью при повышенных температурах. Так, дуралюмин (см. Алюминиевые сплавы) при нагревании до 400 °C становится в 5 раз менее прочным, чем до нагрева, а Б. только в 2 раза.
Однако для применения Б. как конструкционного материала всё ещё есть препятствия. Не вполне очищен
81
БЕТОН
ный металл хрупок. Кроме того, изготовлять изделия из него очень непросто. Традиционные методы обработки металлов — плавка и литье — оказались малопригодными для Б. из-за образования в процессе кристаллизации зёрен,, трещин и раковин. Поэтому даже очень крупные заготовки из Б. (массой до 5 т) в промышленности получают только методами порошковой металлургии (см. об этом в статьях Вольфрам, Молибден, Платиновые металлы).
Б. возглавляет главную подгруппу II группы периодической системы Менделеева. Так как внешняя электронная оболочка его атома содержит два электрона (конфигурация 2s2, см. Атом), Б. сравнительно легко отдаёт их, проявляя в соединениях степень окисления +2 (валентность II). В химическом отношении Б. сходен как с магнием, так и с алюминием. При комнатной температуре на воздухе Б. покрывается тонкой окисной плёнкой, предохраняющей его от дальнейшего окисления. С водой почти не реагирует, так как образующаяся в первый момент плёнка малорастворимой гидроокиси Ве(ОН)2 также пассивирует металл. Активно взаимодействует с НС1 и H2SO4, а при нагревании и с HNO3. Выше 800 °C Б. сгорает на воздухе с образованием ВеО. При высокой температуре он соединяется также с хлором, серой, углеродом, азотом и другими неметаллами, образуя соответственно ВеС12, BeS, Ве2С, Be3N2 (с водородом непосредственно не реагирует).
Окись Б. плавится только при 2530°C. В воде она почти нерастворима. Порошок непрокалённой ВеО хорошо растворим как в кислотах, так и в растворах щелочей:
ВеО + H2SO4 = BeSO4 + Н2О ВеО + 2КОН = К2ВеО2 + Н2О Прокалённая ВеО, напротив, химически очень стойка. Гидроокись Ве(ОН)2, так же как и А1(ОН)3, имеет амфотерный характер и диссоциирует по схеме: Ве2+4-2ОН-
Ве(ОН)2 <-> Н2ВеО2 qz±
2Н + + ВеО?" Соли с анионом ВеОг”” называются бериллатами. Свойства Ве(ОН)2 как основания выражены гораздо отчётливее кислотных. При добавлении щёлочи равновесие сдвигается в сторону образования бериллат-ионов ВеО2“.
Расцвет бериллиевой промышленности тесно связан с ядерной техникой, самолёто- и ракетостроением. Ещё в 1932 г. английский физик Дж. Чэдвик установил, что при бомбардировке а-частицами Б. испускает новые элементарные частицы:
^Ве + J2C + Jn
Так был открыт нейтрон (обозначается in или п) — элементарная частица, входящая в состав атомного ядра и не несущая заряда (лат. neuter — ни тот, ни другой).
Нейтрон — «главный снаряд» ядерной энергетики. Под действием нейтронов (или, как говорят физики, при бомбардировке нейтронами) происходит деление ядер
урана и других тяжёлых радиоактивных элементов. Делящиеся ядра, в свою очередь, испускают нейтроны — так развивается цепная ядерная реакция — могучий источник атомной энергии.
До появления ядерных реакторов смеси Ra— Be были наиболее популярными лабораторными источниками нейтронов (радий поставлял необходимые для ядерной реакции а-частицы). Но Б. оказался ещё и прекрасным замедлителем и отражателем нейтронов. Ведь ядерную реакцию необходимо регулировать! (См. об этом, например, в статьях Бор, Гафний, Уран.) И теперь в различных ядерных реакторах (в том числе и в малогабаритных) Б. выполняет свою почётную «антинейтронную работу».
Особое свойство Б.— его высокая проницаемость для рентгеновских лучей (в 17 раз выше, чем у алюминия); поэтому во всём мире Б. применяется для изготовления «окошек» в рентгеновских трубках. Благодаря высокой температуре плавления, прочности, высокой теплопроводности и малой плотности Б. находит всё возрастающее применение в самолёто- и ракетостроении. Уже сейчас Б. применяется для изготовления тормозных дисков самолётов, панелей солнечных батарей, обшивки днищ космических кораблей. Б. М. Булычев.
БЕРКЛИЙ (лат. Berkelium) Bk — радиоактивный химический элемент с атомным номером 97; относится к актиноидам.
БЕРТОЛЛЁТОВА СОЛЬ — хлорат калия КС1О3. См. Калий, Хлораты.
БЕТОН — искусственный каменный материал. Б.— самый важный и универсальный в наши дни строительный материал. Из него
Рис. 1. Гостиница «Тарасова Гора» в городе Каневе (УССР). Построена в 1961 г. «Железобетон принёс с собой новый масштаб строительства, новый внешний и внутренний облик зданий» (стр. 82).
6 хэш
БЕТОН
82
сооружают жилые дома и школы, мосты и плотины, покрытия дорог и аэродромов. Это материал не только прочный и огнестойкий, но и защищающий от радиоактивного излучения. Из чего же состоит Б. и как его приготовляют? Б. получают из смеси вяжущего вещества и воды с различными заполнителями. Такая смесь со временем твердеет, приобретая свойства камня.
В зависимости от плотности заполнителей различают четыре вида Б.: особо тяжёлый, тяжёлый, лёгкий и особо лёгкий. В особо тяжёлом и тяжёлом Б. заполнителями служат железные руды, металлы, плотные каменные породы (обычно гранит и известняк). Из этого Б. возводят крупные сооружения, в которых от Б. требуются особо высокая прочность и долговечность. Лёгкий Б. получают путём смешения воды и цемента с доменными шлаками, пемзой или вулканическим туфом. Его используют при кладке стен и перекрытий. В особо лёгком Б. (пенобетон, газобетон) почти 90% объёма занимают поры — пузырьки воздуха. Этот Б., хотя и менее прочен, но имеет гораздо лучшие тепло- и звукоизоляционные свойства. На его производство идёт значительно меньше цемента.
В перечисленных строительных Б. связующим веществом служит цемент (так называемые цементные Б.). В дорожном же строительстве наиболее распространены асфальтовые Б.— смесь битумов (отходы от переработки нефти, каменного угля) с заполнителями. Кроме того, изготовляются особые виды Б.— жаропрочный, кислотостойкий и другие с применением специальных заполнителей и вяжущих веществ. В качестве связующих большое значение приобретают полимеры. Полы из химически стойких «полимербетонов» удобны в цехах и лабораториях, где можно пролить кислоту или щёлочь. Такие полы, благодаря их высокой пластичности, придаваемой полимерными связующими, хорошо выдерживают механические колебания, вызываемые работающими машинами.
Рис. 2. Филиал Центрального музея В. И. Ленина в Ташкенте. Построен в 1970 г. «Дома из железобетона выглядят зрительно лёгкими, как будто пронизаны светом и воздухом» (стр. 82, справа).
Бетонную смесь готовят либо на бетонных заводах, либо в передвижных бетоносмесительных установках. Хорошо перемешанную массу укладывают в форму или опалубку, где она твердеет. Чтобы увеличить прочность Б. и снизить его пористость, бетонную массу уплотняют, применяя различные вибрационные устройства. Работу вибратора, уплотняющего бетонную смесь, можно увидеть на многих строительных площадках.
Б. хорошо сопротивляется сжатию, но значительно хуже (в 8—15 раз) растяжению и изгибу. Этот недостаток частично устраним, если в формы, где происходит затвердевание Б., предварительно уложить арматуру из стальной проволоки или прутьев. Так получают железобетон— самый важный и универсальный строительный материал.
Когда впервые появился железобетон? Его открытие вряд ли можно приурочить к какой-либо дате или приписать одному человеку. Глинобитные постройки со стенами, армированными прутьями, были известны издавна. Секрет упрочнения каменных построек металлическими стержнями русские зодчие знали уже в XV в. Но это был лишь «прообраз» будущего железобетона. Патент же на железобетон взял в 1867 г. садовник Монье во Франции. (Монье изготовлял цветочные кадки из проволочной сетки, покрытой цементным раствором.)
Применение железобетонных конструкций началось в XX в. и оказало огромное влияние на мировую архитектуру. Железобетон принёс с собой новый масштаб строительства, новый внешний и внутренний облик многих зданий. Если в каменном или кирпичном строении перекрытие опирается на массивные толстые стены, прорезанные небольшими окнами, то в здании с железобетонными конструкциями оно может покоиться на тонких и редко расставленных опорах. Такие дома выглядят зрительно лёгкими, как будто пронизанными светом и воздухом. Железобетона производится сейчас очень много. Только в Советском Союзе в 1973 году выпущено 102 млн. м3 сборного железобетона.
Ценные свойства Б., его стойкость к действию атмосферных осадков и к колебаниям температуры зависят от многих факторов и в первую очередь — от качества цемента, качества и количества воды, заполнителей, от степени измельчения исходных компонентов, от плотности получаемой бетонной смеси. Далеко не всякая вода годится для получения Б. Так, вода сильно минерализованная, то есть содержащая большое количество растворённых солей, обычно существенно сокращает срок службы бетонных изделий. Наиболее же пригодной оказывается питьевая вода. Качество железобетона определяется ещё и маркой стали, из которой изготовляется арматура, а также числом, толщиной и взаимным расположением арматурных стержней. Совершенствование сортов Б.— серьёзная
83
БИОСФЕРА
Рис. 3. Панорама Братской ГЭС.
«Бетон — самый важный и универсальный в наши дни строительный материал. Из него сооружают жилые дома и школы, мосты и плотины, покрытия дорог и аэродромов» (стр. 81—82).
народнохозяйственная проблема, которой занимаются многие учёные и технологи.
Важный вклад в улучшение технологии Б. и особенно в раскрытие существа процессов твердения цемента и Б. внёс советский учёный, крупный специалист по коллоидной химии, академик П. А. Ребиндер (см. Цемент).
3. А. Старикова.
БИКАРБОНАТЫ — кислые соли угольной кислоты, например Са(НСО3)2. Современное название — гидрокарбонаты. См. также Карбонаты.
БИОСФЕРА — оболочка Земли, в которой развивается жизнь во всех её формах (животные, растения, микроорганизмы). Название происходит от греч. «биос» — жизнь и «сфайра» — шар (в переносном значении — область, среда). К Б. относится и Мировой океан, где даже на глубинах более 10 км живут рыбы и другие животные. И нижняя часть атмосферы (так называемая тропосфера), где летают птицы. И поверхность суши с её лесами, степями, тундрами и прочими ландшафтами. Наконец, жизнь проникает и в твёрдую земную кору (литосферу) на глубину в несколько километров, где в подземных водах обитают различные микроорганизмы. Только температура более 100 °C определяет предел их деятельности, что ограничивает Б. снизу. Таким образом, в Б. входит почти вся
гидросфера, нижняя часть атмосферы и верхние слои земной коры.
По составу Б. очень неоднородна, в неё, как видно из рис. 1, входят и твёрдые, и жидкие, и газообразные сферы. Правомерно ли объединять их в единое целое, так ли уж велика роль живых организмов в геологической истории Земли? Ведь каждый отдельный организм мал и по сравнению с неорганическими факторами природы (теплом и холодом, ливнями и засухой, ветрами и вулканическими извержениями) роль его ничтожна. Казалось бы, удел организмов — лишь приспосабливаться к окружающей среде. Так думало большинство учёных в XIX и начале XX вв. Подобные взгляды, естественно, не способствовали развитию учения о Б., хотя само понятие о такой оболочке нашей планеты возникло уже в конце XIX в. в работах известного австрийского геолога Э. Зюсса.
Для развития учения о Б. необходим был новый подход к явлениям жизни. Его нашёл в 20-х гг. нашего столетия советский учёный академик В. И. Вернадский. Учёный не отрицал, что каждый отдельно взятый организм ничтожно мал и вынужден приспосабливаться к условиям окружающей среды. Ну а если иметь в виду деятельность всей совокупности организмов, рассмотреть их как единое целое, которое
6*
БИОСФЕРА
84
6СО2 + 6Н2О + 2,83 кдж
Вернадский назвал живым веществом? Оказывается, общая масса живого вещества на Земле не так уж мала, она превышает 1012 т. Это в основном растения — все те леса, луга, морские водоросли, которые делают нашу Землю «зелёной планетой». Гораздо меньше на Земле животных; «зоомасса» в степях, например, менее 1 % от массы растений, а в тайге — менее 0,01%.
Живое вещество образуется из простых неорганических соединений окружающей природы— из углекислого газа, воды, минеральных солей. Самый важный процесс этого типа — фотосинтез, протекающий в зелёных растениях. Схема этого процесса такова:
хлорофилл ------------► солнечный свет
—* СвН12Ов+6О2
Но это лишь общая схема процесса, его начало и конец. А каковы же его ступени, его детальный механизм? В наши дни фотосинтез изучают научные институты во многих странах мира, в том числе и в СССР. Полностью тайна фотосинтеза ещё не раскрыта, но многое уже стало ясным. Оказалось, что в зелёном листе происходит разложение воды. Кислород выделяется в окружающую среду. Водород же вовлекается в сложную систему реакций с участием углекислого газа, который поступает из атмосферы. Так синтезируются органические соединения— простейшие сахара (например, глюкоза СвН12Ов). На дальнейших ступенях фотосинтеза из простейших сахаров образуются и более сложные органические соединения.
Как известно, многие реакции в лаборатории идут лишь при нагревании, в присутствии катализаторов. А в живой природе? На первой ступени фотосинтеза растения используют энергию солнечного света, а роль катализатора играет зелёный пигмент хлорофилл. Продукты фотосинтеза — и органические соединения, и кислород — поглотили солнечную энергию, как бы «зарядились» ею. Эта энергия может выделяться в форме теплоты или совершать работу. Кто же не знает, что сахар повышает работоспособность — недаром его дают в туристических походах против усталости. Высвободившийся из Н2О под действием солнечной энергии кислород тоже приобрёл способность совершать химическую работу например окислять металлы.
Почти сплошным ковром покрывают растения земную поверхность, и непрерывно повсюду уже очень давно идёт процесс фотосинтеза — об этом говорят находки ископаемых растений в древних осадочных породах. Зелёные расте
ния практически очистили нашу атмосферу от двуокиси углерода (сейчас в ней только 0,03% СО2) и насытили её свободным кислородом. Имеется много доказательств того, что в первичной атмосфере Земли до появления зелёных растений не было свободного кислорода, а был в основном углекислый газ. Об этом косвенно свидетельствуют и данные советских космических станций об атмосфере Венеры, где жизнь земного типа отсутствует: венерианская атмосфера состоит из СО2 и практически не содержит свободного О2. Нет кислорода и на Юпитере, почти нет на Марсе. Таким образом, воздух, которым мы дышим, создан живым веществом, точнее, зелёными растениями. Интересно, что, по современным представлениям, первые организмы, возникшие около 3 млрд, лет назад, ещё не обладали способностью к фотосинтезу. Зелёные же растения появились лишь 2 млрд, лет назад, а значительное количество О2 в атмосфере накопилось, по мнению некоторых учёных, ещё позже— 1,2 млрд, лет назад.
Свободный кислород — могучий деятель на земной поверхности, «геохимический диктатор». С его появлением многие процессы на Земле стали протекать по-другому.
Так, он перевёл огромные количества серы из со--2 +в
стояния S в S и заставил её энергично мигрировать — перемещаться на Земле. В ранние геологические эпохи сера в земных ландшафтах и морях существовала в виде сульфидов или сероводорода. Это нетрудно доказать. Ведь в изверженных горных породах она до сих пор находится в основном в виде пирита FeS2 и других нерастворимых в воде сульфидов. Когда в атмосфере появился кислород, он начал окислять сульфиды, и на Земле появились сульфаты. Сера стала мигрировать на земной поверхности в виде растворимых сульфатов; в лагунах же в результате испарения отлагались пласты сульфатов мощностью в десятки метров. Напомним хотя бы о Кара-Богаз-Голе—заливе на восточном берегу Каспия, где при температуре ниже 5,5 °C отлага ется ценное сырьё — мирабилит Na2SO4« ЮН2О. Или о пластах гипса CaSO4-2H2O, распространённых по всему земному шару.
Железо и марганец в «докислородную эпоху», напротив, сравнительно энергично мигрировали в водах, так как находились в виде легко растворимых солей двухвалентных Fe и Мп. Моря и реки той далёкой эпохи содержали повышенные количества железа и марганца. Накопленный в атмосфере свободный кислород стал
Рис. 1. Биосфера Земли. Рисунок передаёт всё разнообразие земной биосферы. Конечно, для этого пришлось отказаться от привычных масштабов, и рядом оказались ландшафты, не всегда соседствующие в природе. На правой стороне рисунка показан воображаемый материк, простирающийся от полярных льдов на севере до жарких и влажных тропических лесов на юге. Между ними вы видите несколько ландшафтных зон: сначала идёт холодная бедная растительностью тундра, потом зона хвойных лесов (тайга) с её полноводными реками, болотами. С юга к ней примыкает полоса широколиственных лесов. Здесь уже теплее, жизнь богаче, чем в суровой тайге. Ещё южнее, там где климат становится суше, располагаются степи, а за ними и пустыни. Большую часть года они имеют желтовато-бурый оттенок — это цвет почв, выгоревших трав и кустарников. На рисунке такие зоны показаны жёлтым пятном. И, наконец, около экватора, где снова влажно и сыро, пышно зеленеет растительность.
Слева показана часть Мирового океана; здесь жизнь распространена до самых больших глубин. На переднем плане земная кора в разрезе, нижняя граница биосферы показана белым пунктиром. Если бы мы посмотрели в микроскоп на каплю подземной воды, взятой с большой глубины, то увидели бы различные бактерии. Это изображено на рисунке в кружке. Верхняя граница биосферы тоже отмечена белым пунктиром, она располагается в тропосфере.
85
БИОСФЕРА
БИОСФЕРА
окислять их, осаждая практически нерастворимые соединения трёхвалентного Fe и четырёхвалентного Мп. Некоторые геологи считают, что с этим связано массовое образование осадочных железных руд в древних океанах. Так, возможно, образовались железные руды Кривого Рога на Украине и Курской магнитной аномалии в РСФСР.
На истории многих и многих других элементов можно показать, как геохимический диктатор О2 изменил их поведение на Земле.
Но свободный кислород — только один из продуктов фотосинтеза, другие же его продукты — различные органические вещества. Углеводы из зелёных листьев попадают в ветви, корни и другие органы растений, где происходит синтез новых органических соединений. Животные, поедая растения, синтезируют в своём теле специфические для их организмов вещества. Остатки растений и животных служат пищей для микроорганизмов, создающих в свою очередь собственные органические соединения. Так и возникло всё разнообразие природных органических химических соединений — сотни тысяч углеводов, жиров, белков и других.
Но какова геологическая роль органических веществ на Земле? После смерти организмов их остатки, разлагаясь, частично «минерализуются», то есть снова превращаются в СО2, Н2О и минеральные соли. Часть же их захороняется в земной коре в форме торфа, углей, нефти и битумов. Местами эти горючие ископаемые образуют огромные залежи, как, например, у нас в Донбассе, Кузбассе, в нефтяных районах Азербайджана, Западной Сибири или Мангышлака. Ещё больше органических веществ рассеяно в Б. и встречается в небольших количествах повсеместно. Это — гумус почв, растворимые органические соединения в реках и озёрах (поэтому вода их местами похожа по цвету на крепкий чай). Это — примеси в известняках, глинах, песчаниках и других осадочных породах. *
Органические вещества — не инертные массы в земной коре. Везде, где есть вода, они служат пищей для микроорганизмов, окисляющих их до СО2, Н2О и т. д.
Для дыхания микробам необходим кислород, и они в первую очередь поглощают весь свободный О2, растворённый в воде; когда же он израсходуется, начинают отнимать кислород у неорганических соединений — сульфатов, нитратов, гидроокисей железа, марганца и др. Так, в болотах, илах, на дне морей и озёр, в глубоких водоносных горизонтах исчезает свободный кислород. Там сульфаты снова переходят в сульфиды, железо из трёхвалентного — в двухвалентное и т. д. Эти восстановительные процессы играют в Б., пожалуй, не меньшую геологическую роль, чем окислительные.
Обратимся снова к примеру серы. Везде, где в Б. есть органические вещества и сульфаты и нет свободного кислорода, развивается деятельность особых бактерий, разлагающих органические соединения и восстанавливающих сульфаты.
86
Их работу условно можно записать в виде реакции: C6H12Oe + 3Na2SO4 =
= 3Na2CO3 + ЗСО2 -j- 3H2S ЗН2О + энергия
Как видим, бактерии окислили сахар и добыли таким образом энергию для своей жизнедеятельности. Кислород для окисления они отняли у сульфатов, восстановив + в -2
серу из состояния S до S. В результате в воде появился сероводород — газ с характерным и неприятным запахом тухлых яиц. Сероводород, соединяясь с железом, сначала образует FeS, а в дальнейшем FeS2 (минерал пирит). Так в знаменитых одесских лиманах бактерии, живущие в илах, восстановили сульфаты и обогатили илы сероводородом и чёрным сульфидом железа, превратив их в «целебные грязи», которыми лечат болезни суставов. Такие грязи образуются и на дне других озёр и лиманов. Вблизи их созданы грязевые курорты (Пятигорск на Кавказе, Молла-Кора в Туркмении и т.д.). Л вот в Чёрном море эти микробы, пожалуй, принесли не пользу, а вред — они выработали так много H2S, что черноморская вода глубже 200 м стала безжизненной. Там развилась «зона сероводородного заражения», в которой не могут существовать рыбы и другие животные.
В земных глубинах нередко встречаются друг с другом потоки вод разного состава. Так, проникающие с поверхности «кислородные воды» (они содержат свободный растворённый кислород О2) соприкасаются с сероводородными водами (насыщенными H2S), поднимающимися из глубин. В кислородных водах легко мигрируют катионы многих металлов. При встрече их с сероводородными водами образуются нерастворимые сульфиды железа, меди, цинка, свинца, молибдена, нерастворимые минералы урана и других металлов. Так в прошлые геологические эпохи в горизонтах подземных вод и на дне морей (где в илах тоже много сероводорода) образовались руды металлов, которые ныне эксплуатируются во многих районах земного шара.
Итак, за время геологической истории живое вещество с помощью фотосинтеза разделило физико-химическую среду Земли на две противоположные: резко окислительную со свободным кислородом на земной поверхности и резко восстановительную в болотах, илах, глубоких подземных водах (рис. 2). Фотосинтез и минерализация — эти противоположные процессы образования и разрушения органических веществ — образуют единый биологический круговорот атомов в природе. (Кстати, минерализация идёт и в самих растениях, при дыхании которых органические вещества окисляются до СО2 и Н2О. Но гораздо энергичнее минерализуют органические соединения животные и особенно — как мы показали выше — микроорганизмы.)
При минерализации выделяется энергия, поглощённая при фотосинтезе. Часть её выделяется в виде теплоты, а часть совершает работу. Когда природные воды обогащаются органическими кислотами, СО2 и другими продуктами
87
БИОСФЕРА
Владимир Иванович ВЕРНАДСКИЙ — основоположник геохимии и биогеохимии.
Советский учёный Владимир Иванович Вернадский родился 12 марта 1863 г. в Петербурге в прогрессивно настроенной семье профессора-экономиста. В 1881—1885 гг. он учился в Петербургском университете, где в то время читали лекции Менделеев, Сеченов, Меншуткин, Докучаев... А во внеучебное время лучшая часть студенчества группировалась вокруг научно-литературного общества, секретарём которого был А. И. Ульянов. К этому обществу принадлежал и Вернадский. Начиная с 1890 г. в течение 21 года Вернадский преподаёт минералогию в Московском университете. Преподаёт по-новому — читает свой собственный курс, отличающийся широтой и новизной взглядов. Вокруг передового учёного группируется талантливая молодёжь, создаётся минералогическая школа. Он покинул университет в 1911 г. в числе 124 профессоров и преподавателей, протестующих против реакционной политики царизма в области просвещения. С этого времени его деятельность связана с Академией наук, действительным членом которой он был избран в 1908 г.
До В. И. Вернадского минералогия была по преимуществу наукой описательной. Изучение химического состава и внешних свойств минералов — их кристаллической формы, цвета, вида излома, блеска, твёрдости — было главным её делом. Вернадский превратил минералогию из науки описательной, статической в динамическую. Для него минерал — это не раз и навсегда застывший камень, а продукт многообразных процессов, идущих в земной коре. Ещё в 1891 г. он определил минералогию как химию земной коры. А позднее (1910—1925 гг.) создал основы геохимии — «науки об истории земных атомов». Он задумывается и над тем, какими же источниками энергии направляются геохимические процессы, то есть процессы перемещения, концентрации и рассеяния элементов — атомов в природе. Один из этих источников — Солнце, непрерывно снабжающее Землю своим теплом. Другой же мощный источник Вернадский видит в радиоактивном превращении элементов, сопровождающемся выделением огромного количества тепла. Его увлекает проблема радиоактивности, он организует первые в России экспедиции для изучения радиоактивных минералов — на Урал и в Среднюю Азию. В 1922 г. Вернадский, вопреки мнению многих авторитетов, предсказал грядущее практическое использование атомной энергии. Он предвидел здесь и опасность... «Сумеет ли человек воспользоваться этой силой, направить ее на добро, а не на самоуничтожение»,— так современно звучат его слова, сказанные ещё в 1922 г.
В последние 28 лет жизни учёный главное внимание уделял химической деятельности живых организмов в земной коре, создав ещё одну новую науку — биогеохимию. Его идеи в этой области намного опередили время. Полное признание они получили в наши дни, когда учение о биосфере легло в основу мероприятий по охране природы и борьбе с загрязнениями окружающей среды.
Он не был кабинетным учёным. В каких только экспедициях и научных экскурсиях ни побывал он в своей жизни. Только за пять лет, с 1885 г. по 1890 г., не говоря уже о работе в России, он посетил горнопромышленные области Германии, Швейцарии, Франции, Италии, Англии, Норвегии...
Вернадский был патриотом-борцом. Как и Менделеев, он ратовал за всестороннее изучение и использование полезных ископаемых, таящихся в недрах России.
Во время первой мировой войны группа выдающихся учёных во главе с Вернадским внесла в Академию наук предложение об организации постоянной Комиссии по изучению естественных производительных сил России (сокращённо— КЕПС). После Октябрьской революции 1917 г. эта комиссия, по указанию В. И. Ленина, начала огромную работу по изысканию всех видов сырья и топлива в стране и по размещению промышленности — приближению её к источникам сырья. Вернадский, возглавивший КЕПС, участвовал и в организации многих научных учреждений нашей страны, в частности Радиевого института, директором которого он был с 1922 г. по 1939 г. Сейчас его имя носит Институт геохимии и аналитической химии Академии наук СССР.
Среди учеников Вернадского имена не только крупнейших минералогов и геохимиков СССР, но и учёных других стран. Он был членом многих иностранных научных обществ и академий. Им написаны «Очерки геохимии», «Биосфера», «История минералов земной коры» и другие книги. Изданы его «Избранные сочинения» в 5 томах. За свои научные труды Вернадский в 1943 г. удостоен Государственной премии СССР.
В. И. Вернадский умер 6 января 1945 г. в Москве.
БИОСФЕРА
88
Зона окисления Зона восстановления
Рис. 2. Окислительная и восстановительная среда в биосфере. 1 —уголь; 2 —нефть; 3 — газовые залежи; 4 — микроорганизмы.
На левом рисунке изображён разрез через биосферу на материке до глубины 800 м. На земной поверхности вы видите зелёные холмы и равнины, где протекает фотосинтез. Над ними — кислородная атмосфера — продукт фотосинтеза современных и былых растений. Свободный кислород проникает в земную кору на глубину в десятки, сотни, а в горах и тысячи метров, формируя зону окисления. В водах здесь растворён кислород; горные породы часто имеют красную, бурую, жёлтую окраску, зависящую от минералов окисного (трёхвалентного) железа. На земной поверхности встречаются и очаги восстановления — например, в болотах, где микроорганизмы расходуют растворённый в воде кислород на окисление растительных остатков. Болото с чёрным торфом и восстановительной средой показано в низине.
В глубоких частях биосферы господствует восстановительная среда. Туда не проникает свободный кислород, так как он расходуется бактериями в верхней части, показанной красным цветом. Нижняя зона — зона восстановления— закрашена тёмными тонами. На больших глубинах в подземных водах также есть жизнь — она представлена микроорганизмами, которые могут существовать без свободного кислорода (4). Отнимая кислород от минеральных соединений — сульфатов, нитратов, гидроокисей, они окисляют органические вещества. Здесь накапливается много метана СН4, двуокиси углерода СО2 и других газов.
Итак, в верхней части биосферы располагается зона окисления (с очагами восстановления), на глубине — зона восстановления. Это вертикальная окислительно-восстановительная зональность.
На земной поверхности нередко наблюдается и горизонтальная окислительно-восстановительная зональность (правый рисунок): окислительная среда на возвышенностях и восстановительная — в соседнем болоте.
разложения растительных остатков, они становятся агрессивными — совершают «химическую работу». Они легко разрушают горные породы, выветривают их, превращая в почвы, кору выветривания и т. д. Таким образом, с энергетической точки зрения, биологический круговорот атомов переводит энергию солнечных лучей в химическую энергию, за счёт которой в Б. выполняется работа (разрушение горных пород и т. д.). Прав был Вернадский, когда говорил, что на земной поверхности самой мощной химической силой являются живые организмы, взятые в целом, то есть живое вещество. Это положение великого учёного было предложено именовать законом Вернадского.
Итак, миллиарды лет назад на Земле появился мощный геохимический фактор — живое вещество, преобразовавшее природу. Возникло особое планетное явление, особая оболочка —
биосфера — очень сложная саморегулирующаяся система с многочисленными связями между её частями. В последние тысячелетия произошёл новый скачок в развитии Земли как планеты — приобрела особое значение геохимическая деятельность человека. Ныне человеческое общество превратилось в могучую геохимическую силу, преобразующую природу. Эта деятельность, несущая огромные блага людям, может иметь и вредные последствия. Достаточно нарушить взаимосвязи составляющих Б. частей, и в ней могут начаться процессы, вредные для человечества.
Например, танкеры, перевозящие нефть, нередко сливают за борт её остатки. В результате тонкая плёнка нефти покрывает поверхность океана и затрудняет проникновение кислорода к микроскопическим водорослям. Они начинают задыхаться, фотосинтез ослабевает. А ведь
89
БОР
фотосинтез даёт нам кислород! Не менее важна и проблема отравления, связанная с применением ядохимикатов — соединений свинца, ртути и других элементов. Их применение в сельском хозяйстве для борьбы с вредителями позволяет сохранить урожай. Но в ряде случаев оно оказалось вредным для человека, животных и растений. Установлено, например, что в речной рыбе из 20 штатов США повышено количество ртути. Напомним и о загрязнении воздуха в крупных городах, и о гибели рыбы в реках, и о многих других нарушениях в Б., вызванных хозяйственной деятельностью человека. (Подробно вы можете прочитать об этом в статьях Атмосфера, Гидросфера.)
Вот почему охрана природы — одна из важнейших проблем, стоящих перед человечеством. Этому вопросу уже в первые годы Советской власти уделял большое внимание В. И. Ленин. В наши дни из-за грандиозного развития промышленности проблема становится всё более актуальной. В СССР приняты законы об охране природы и делается многое, чтобы биосфера и нашей страны и планеты в целом была хорошим домом для человечества. А. И. Перельман. БИСУЛЬФАТЫ — кислые соли серной кислоты, например NaHSO4. Современное название — гидросульфиты. См. также Сульфаты. БИСУЛЬФИТЫ — кислые соли сернистой кислоты, например KHSO3. Современное название гидросульфиты. См. также Сульфиты. БИХРОМАТЫ (двухромовокислые соли) — соли двухромовой кислоты, например К2СГ2О7. Современное название кислоты Н2Сг2О7 и её солей — дихромовая и дихроматы. БЛАГОРОДНЫЕ ГАЗЫ — то же, что инертные газы.
БЛАГОРОДНЫЕ МЕТАЛЛЫ —золото, серебро, а также платина и её спутники (рутений, родий, палладий, осмий, иридий), получившие своё название, главным образом, за высокую химическую стойкость и красивый внешний вид. См. Золото, Серебро, Платиновые металлы. БОР (лат. Вогит) В — химический элемент III группы периодической системы Менделеева; атомный номер 5, атомная масса 10,81. Твёрдое
кристаллическое вещество обычно серо-вато-чёрного или коричневого цвета (из-за примесей); в чистом виде бесцветен. Элемент состоит из двух изотопов: 10В (19%) и г1В (81%).
Б. довольно широко распространён в
природе (1,2-10-3% по массе в составе земной коры), то есть почти так же, как свинец. Встречается он только в виде соединений с кислородом. Таковы борная кислота (минерал сассолин) Н3ВО3, бура (тинкал) Na2B4O7-ЮН2О, кернит Na2B4O7-4H2O, ашарит MgHBO3 и другие бораты. Особенно богаты борной кислотой некоторые горячие источники и озёра Центральной Италии (Тоскана). Там на участке около 20 км2 из глубин земли поднимаются нагретые до 190 °C пары, содержащие борную кислоту. Главные месторождения боратов находятся в ГДР (Стасфурт), СССР (Казахстан), США (Калифорния).
Одно из наиболее давно известных соединений Б.— бура упоминается в алхимических рукописях (араб, «борак» или лат. borax). Отсюда и произошло название самого элемента, впервые полученного в 1808 г. французскими химиками Ж. Гей-Люссаком и Л. Тенаром. Более ста лет химики не умели выделить Б. в достаточно чистом виде. Лишь в 1909 г. был получен Б. чистотой 99%, 3 в наше время — чистотой 99,999%. Интерес к элементу резко возрос в 60-х гг.: он оказался необходимым в новейших отраслях науки и техники.
Чистый кристаллический Б. имеет плотность 2,3 г/см3, /пл 2075 °C, /кип 3860 °C. Б.—полупроводник; в обычных условиях он плохо проводит электрический ток, но при нагревании его электропроводность увеличивается (это и характерно для полупроводников).
В соответствии с положением Б. в периодической системе, его атом имеет три внешних электрона (конфигурация 2s22p1). Для Б. типична валентность III (степень окисления +3). Вообще же строение соединений Б.— один из самых сложных вопросов неорганической химии.
При обычной температуре Б. довольно инертен и взаимодействует из всех химических элементов лишь с фтором, образуя газообразный фторид BF3. С нагреванием его активность возрастает, так что он непосредственно соединяется со всеми галогенами, углеродом, азотом, фосфором. При 700 °C Б. горит на воздухе, образуя борный ангидрид В2О3. С водородом Б. не взаимодействует, бороводороды получают косвенным путём. При нагревании Б. соединяется с металлами, образуя бориды. При высокой температуре разлагает водяной пар:
2В + ЗН2О = В2О3 + ЗН2
Концентрированные азотная и серная кислоты окисляют Б. до борной кислоты:
В + 3HNO3 = Н3ВО3 + 3NO2
Другие кислоты на Б. не действуют. Концентрированные растворы щелочей медленно растворяют Б., взятый в виде порошка (но не в виде монолитных кристаллов):
2В + 2NaOH + 2Н2О = 2NaBO2 + ЗН2
NaBO2— это метаборат натрия (см. Бораты).
БОР
90
Многие соединения Б. обнаруживают большое своеобразие, привлекающее исследователей в течение вот уже полутора столетий. Атом Б., соединяясь с галогенами, образует три ковалентные связи и оказывается окружённым шестью электронами (см. ниже). Их только шесть, тогда как устойчивая внешняя электронная оболочка обычно включает в себя электронную восьмёрку. Вот почему нейтральная молекула того или иного галогенида Б. легко присоединяет нейтральную молекулу другого вещества, имеющую «лишнюю» электронную пару (например, молекулу аммиака NH3)
и :f:
н: n : + в: f : —н: n : в : f : •• •••• •••••• н :f: н :f:
Подобным образом образуется и другое соединение
:F: ’Л*’
h:f: + b:f:—h:f:b:f:
Это довольно сильная фтороборная кислота, для которой известны устойчивые соли, например K[BF4] и NHJBFJ. В таких комплексных соединениях атом Б. окружён четырьмя другими атомами, то есть Б. имеет координационное число 4. Помимо NH3*BF3,‘ известны и другие соединения, в которых Б. связан с азотом.
Особенно интересен нитрид BN — белый кристаллический порошок, удивительно напоминающий по структуре графит и с такой же, как у графита, плотностью. BN устойчив к любым химическим реагентам, кроме фтора и фтористого водорода. Известно, что графит можно использовать как «смазку», уменьшающую трение в движущихся частях машин. Нитрид Б. в этом отношении превзошёл графит. Однако сходство BN с графитом этим не ограничивается.
Как только учёным удалось приготовить из графита синтетические алмазы (см. Углерод), они решили попробовать, не произойдёт ли подобная перестройка структуры у нитрида Б. Учёных ждал полный успех. При давлении более 60 000 атм и температуре около 1350 °C графитоподобная кристаллическая структура BN превратилась в алмазоподобную. И свойства изменились настолько разительно, что полученному веществу дали новое имя — боразон. По плотности и твёрдости боразон равен алмазу, но гораздо менее хрупок и может «работать» даже при 2000 °C (в то время, как алмаз уже при 900 °C попросту сгорает на воздухе). Химическая стойкость боразона намного больше, чем «обычного» нитрида бора.
Причина необыкновенного сходства BN с углеродом (и алмазом, и графитом) такова: атом В совместно с атомом N «подражают» двум связанным между собой атомам С: у них в общем пользовании одинаковое число электронов:
В N С С
3 + 5 = 4 + 4
Строение кристаллов нитрида бора, а — гексагональный нитрид бора, б — боразон.
Этот рисунок имеет смысл сопоставить с рисунками, изображающими строение графита и алмаза (см. Углерод) и строение серого селена (см. Селен).
Обычная модификация BN по своей структуре напоминает графит — здесь также присутствуют плоские слои, состоящие из шестичленных колец. В этой структуре, как и в структуре селена, можно выделить гексагональную элементарную ячейку (на рисунке она не показана), что определяет название данной модификации. Под действием высокого давления и температуры кристаллическая структура BN перестраивается — вещество переходит в другую модификацию, называемую боразоном. В строении боразона и алмаза есть глубокая аналогия. Чтобы убедиться в этом, будем считать, что все атомы в кристаллах боразона одинаковы — тогда его кристаллическая структура окажется вполне подобной структуре алмаза.
91
и расстояния между каждой парой атомов тоже одинаковы. Кристаллические структуры двух модификаций BN изображены на рис. (а и б).
Б. соперничает с алмазом ещё в одном своём соединении — карбиде В4С (на самом деле его структура сложнее и формула В12С3). Это—чёрные блестящие кристаллы, плавящиеся только при 2350 °C. По твёрдости карбид Б. подобен алмазу и очень стоек химйчески. Понятно поэтому, что и боразон, и карбид Б. несут трудную и почётную службу при обработке твёрдых сплавов, а карбид Б. и сам входит в состав некоторых из них.
В промышленности Б. получают, разлагая природные минералы — бораты — кислотами. Выделившуюся борную кислоту превращают прокаливанием в' борный ангидрид, который затем восстанавливают металлами — Mg, Na, К или А1 при высокой температуре:
В2О3 + 3Mg = 3MgO + 2 В
Однако из-за склонности Б. взаимодействовать с металлами он получается загрязнённым. Для приготовления чистого Б. разлагают бромид ВВг3 (в парах) на раскалённой танталовой или вольфрамовой нити:
2ВВг3 = 2В + ЗВг2 (аналогично тому, как получают в чистом виде титан, цирконий и др.).
Ежегодное потребление Б. и его соединений составляет сотни тысяч тонн. Есть и многие другие, ещё не названные нами области его применения. Б. вводят в состав некоторых сплавов для повышения их прочности; поверхностное насыщение стали бором (борирование) увеличивает её твёрдость; соединения Б.— нитрид BN, фосфид ВР и др.— полупроводниковые материалы.
Ядро атома изотопа 10В способно поглощать медленные нейтроны, вызывающие цепную реакцию деления урана в атомных реакторах. Поэтому одно из самых современных применений Б.— изготовление регулирующих стержней, позволяющих поддерживать нужный режим работы реактора или останавливать его в случае необходимости.
Ядерные свойства изотопа 10В нашли и ещё одно применение. Поскольку нейтроны не несут электрического заряда, эти частицы трудно регистрировать счётчиками. На помощь пришёл Б. Нейтроны, проникающие в счётчик, заполненный газообразным BF3, вызывают ядерную реакцию:
Юр । 1п __ ?т ; । 4„
5 0 “Г Оп — 3LI 20С
Число образующихся при этом а-частиц в точности равно числу попавших в цель (то есть в ядра атомов В) нейтронов. А уж регистрировать заряженные а-частицы несложно. О нейтронах см. Бериллий, Уран; подробнее же и о нейтронах, и о ядерных реакциях вы узнаете из учебников физики.
БОРНАЯ КИСЛОТА
Б. относится к числу элементов, которые (в чрезвычайно малых количествах) необходимы для жизнедеятельности животных и растений (так называемый микроэлемент). Поэтому соединения Б.— буру и борную кислоту — вносят в почву как микроудобрения.
В. Л. Василевский. БОРАНЫ — соединения бора с водородом, например В2Нб; то же, что бороводороды. БОРАТЫ — соли борной кислоты. Такие соли большей частью отвечают не самой борной кислоте Н3ВО3, а полиборным кислотам иВ2О3-тН2О, не существующим в свободном состоянии. Например, тетраборной кислоте Н2В4О7, или 2В2О3-Н2О, отвечает соль Na2В4О7. Её кристаллогидрат — бура Na2B4O7- 10Н2О — является важнейшим из Б. В воде растворимы лишь Б. щелочных металлов. При их добавлении к «жёсткой» воде (содержащей ионы Са2+ и Mg2+) в осадок выпадают Б. кальция и магния, и таким образом жёсткость воды уменьшается. С этим связано применение Б. в составе стиральных порошков. Природные Б.— сырьё для получения бора и его соединений.
БОРИДЫ — соединения бора с другими, более электроположительными элементами, то есть главным образом с металлами. Состав Б. (как и карбидов, нитридов и т. п.) может не отвечать обычным правилам валентности — таковы, например, MgB2, ZrB2, HfB2. Б.— кристаллические вещества, как правило, очень твёрдые и тугоплавкие (так, Б. гафния HfB2 плавится при 3250 °C). Эти свойства в сочетании с чрезвычайной устойчивостью многих Б. к действию кислот сделали их важными компонентами сверхтвёрдых жаростойких сплавов. При обработке стальных деталей бором или карбидом бора на их поверхности образуется тонкий слой Б., что значительно повышает прочность таких деталей. Б. бария, лантана и церия служат материалом катодов электронных ламп, так как при нагревании легко испускают электроны (это явление называется термоэлектронной эмиссией).
БОРНАЯ КИСЛОТА (ортоборная кислота) Н3ВО3 — слабая трёхосновная кислота. Безводная Б. к.— бесцветные кристаллы. Растворима в воде (около 5 г в 100 г Н2О при 20 °C) и в спирте. Встречается в природе в виде минерала сассолина (см. Бор). При нагревании выше 100 °C Н3ВО3 отщепляет воду и образует метаборную кислоту НВО2, а затем — борный ангидрид В2О3. В промышленности Б. к. применяется для производства специального стекла и эмалей. Б. к. обладает антисептическими свойствами и широко используется в медицине. В лабораторной практике из Б. к. и её солей — боратов готовят буферные растворы. Такие
БОРОВОДОРОДЫ
растворы поддерживают определённую концентрацию ионов Н+ и ОН“ среды, что необходимо во многих химических производствах. Взаимодействием Б. к. со спиртами получают летучие эфиры Б. к., которые при поджигании горят ярко-зелёным пламенем. В аналитической химии это используют для обнаружения и идентификации Б. к.
БОРОВОДОРОДЫ (гидриды бора, бораны) — соединения бора с водородом. Известны Б., содержащие от 2 до 20 атомов бора в молекуле (В2Нв, В4Н10, В5Н9, В10Н14, В12Н12 и др.). В зависимости от числа атомов бора в молекуле Б. бывают либо газами (В2Н6, В4Н10), либо жидкостями (В5Н9), или кристаллическими веществами (В10Н14, Bi2H12). Все Б. очень ядовиты и имеют отвратительный запах.
«Архитектура» молекул Б. очень сложна: в молекулах, содержащих 4 и более атомов бора, атомы В и Н образуют тетрагональные пирамиды, октаэдры (восьмигранники), икосаэдры (двадцатигранники). Ни у одного, даже простого, Б. молекула не имеет плоской структуры.
Вопрос о строении Б. и о химических связях между атомами в их молекулах — один из самых трудных и интересных вопросов химии.
В самом деле: всё, что известно науке о свойствах Б., убеждает, что все связи в их молекулах имеют ковалентный характер. Уместно напомнить здесь в качестве примера, как образуются ковалентные связи атома углерода с другими атомами углерода или водорода (пример этот уместен ещё и потому, что формула В2Н6 внешне аналогична формуле этана С2Н6)
н НН
*• • * «X
н:с:н н;с:с:н
• X • X • X
Н НН
метан этан
(на схеме точка обозначает валентный электрон, принадлежащий атому углерода, а крестик — электрон атома водорода).
В молекуле метана СН4 каждый атом водорода отдаёт в общее пользование атому углерода один электрон. В свою очередь, атом углерода «обобществляет» с каждым атомом водорода по одному своему валентному электрону, так что образуются 4 электронные пары, каждая из которых связывает оба атома (Си Н). То же касается электронной структуры молекулы этана, только одна из общих электронных пар принадлежит совместно двум атомам углерода. Итак, мы привели пример привычной, наиболее распространённой в мире молекул, схемы образования ковалентной связи: два атома (два «центра») и связывающая их общая электронная пара.
Вернёмся теперь к соединениям бора с водородом. Приведенной здесь «привычной» схеме завязывания ковалентной связи отвеча- н ло бы образование простейшего Б.— •* ВН3 (в соответствии с тремя валентны- х.* ми электронами атома В). Н
Однако в действительности такой Б., бо р и н, не существует, а происходит его димеризация с
Структуры бороводородов.а — диборан В2Нв. Оба атома бора и два атома водорода образуют ромб в плоскости рисунка. Остальные атомы водорода лежат в перпендикулярной плоскости. Сплошными линиями показаны «обычные» ковалентные связи, тонкой пунктирной линией — «трёхцентровые» связи; б — тетраборан В4Н10. Атомы бора и водорода образуют сложную пространственную структуру. Четыре «трёхцентровые» связи показаны тонким пунктиром.
образованием диборана В2Н6. Этот экспериментальный факт никак не укладывается в типичную схему образования ковалентной связи. Ведь у атома бора в ВН3 нет свободных валентных электронов!
Изучение структуры молекулы В2Нв специальными физическими методами позволило установить, что оба атома бора и два атома водорода (из шести) находятся в вершинах ромба (рис. а). Остальные четыре атома водорода (по два у каждого атома бора) расположены в плоскости, перпендикулярной плоскости ромба. Эти четыре атома водорода образуют с бором «обычные» ковалентные связи В—Н с общей электронной парой у обоих атомов («центров») — бора и водорода (на рисунке такие «обычные» связи обозначены сплошными линиями). -Простая арифметика: н^'эти «обычные» связи израсходованы 4 электронные пары. Оставшиеся две электронные пары связывают — каждая! — не два атома, как в «обычной» схеме, а сразу три:
В------н-------В
Поэтому такую связь, в отличие от «обычной», двухцентровой ковалентной связи, называют трёхцентровой, подчёркивая тем самым, что общая электронная пара принадлежит не двум, а трём атомам совместно.
Нелегко представить себе в виде какого-то конкретного зрительного образа такую «нетипичную» связь. Несколько примитивно можно считать, что «мостиковый» атом водорода (точнее, его ядро) как бы «утоплен» в электронное облако, охватывающее оба атома бора. По своей прочности такая трёхцентровая связь слабее обычной, и «мостиковые» атомы водорода ведут себя в химических реакциях иначе, чем остальные 4 атома водорода. Ещё сложнее связи в других Б., где возможны и ещё более непривычные многоцентровые ковалентные связи (на рис. б изображена структура тетраборана В4Н10).
93
БРОМ
По свойствам Б. напоминают гидриды кремния (кремневодороды). Прямым взаимодействием бора с водородом синтезировать Б. нельзя, поэтому приходится их получать косвенными путями, например: <
2ВС13 + 6Н2 = B2He + 6НС1
Выше 700 °C все Б. разлагаются на бор и водород. При обычной температуре Б. устойчивы лишь в отсутствии воздуха и влаги. Вода разлагает Б. с выделением водорода и образованием борной кислоты:
В2Нв + 6Н2О = 2Н3ВОз + 6Н2
Кислород воздуха легко окисляет Б., так что газообразные и жидкие Б. самовоспламеняются уже при температуре ниже 100 °C:
В2Нв + ЗО2 = В2О3 + ЗН2О
При горении Б. выделяется очень много теплоты, поэтому изучается их применение как ракетного топлива. Разложением Б. пользуются для получения очень чистого кристаллического бора. В. Л. Василевский*
БРОМ (лат. Bromum) Вг — химический элемент VII группы периодической системы Менделеева; атомный номер 35, атомная масса 79,904; относится к семейству галогенов.
Свободный Б.— тяжёлая тёмно-бурая жидкость. В природе элемент представлен двумя устойчивыми изотопами — 79Вг (50,54%) и 81Вг (49,46%).
В 1825 г. французский химик А. Балар впервые выделил Б., действуя хлором на водную вытяжку из золы морских водорос
лей. Никому не известный молодой учёный стал сразу знаменитым. Некоторые видные химики иронизировали: «Не Балар открыл бром, а бром открыл Балара». Название Б. получил за резкий неприятный запах своих паров (по-гречески «бромос»—зловоние).
Хотя Б. и хорошо известен, он не принадлежит к числу широко распространённых в природе элементов. По содержанию в земной коре он занимает всего лишь 50-е место среди прочих элементов (2,1-10“4% по массе). Из-за высокой химической активности Б. встречается только в виде соединений—бромидов натрия, калия, магния (NaBr, КВг, MgBr2). Соединения Б. не образуют и больших скоплений, а лишь сопутствуют соответствующим хлоридам. Большая часть солей Б. находится в природе не в твёрдом виде, а в растворённом состоянии в морской воде, в нефтяных и буровых водах, в некоторых соляных озёрах (см. Гидросфера). В нашей стране
Б. добывают из соляных озёр Крыма, из вод Кара-Богаз-Гола и других источников.
Среди более чем 100 известных сейчас элементов лишь два при комнатной температуре на
ходятся в жидком состоянии — металл ртуть и неметалл Б. Это тяжёлая (плотность 3,1 г/см3) тёмно-бурая жидкость, которая всегда «дымит»: если открыть склянку с Б., то сейчас же начнут выделяться бурые пары. Кипит Б. при 58,78 °C, затвердевает при — 7,2 °C, образуя кра
сивые красные игольчатые кристаллы с металлическим блеском. Б. слабо растворим в воде— примерно 3,6 г Б. в 100 г воды при комнатной температуре. (Этот жёлто-бурый раствор называют бромной водой.) Несравненно лучше
растворим Б. в органических растворителях:
спирте, эфире, хлор
Ki ОС
рме, бензоле, чем пользу
ются для извлечения его из водных растворов.
Можно проделать такой опыт: в цилиндр с бромной водой добавить немного не смешивающегося с ней органического растворителя, например бензола. Как более лёгкий, бензол образует тонкий слой над водой. Если цилиндр закрыть пробкой и затем несколько раз хорошо встряхнуть, после чего дать жидкости отстояться, то весь Б. из воды перейдёт в органический растворитель. Столб воды в. цилиндре окажется прозрачным, а слой бензола — ярко окрашенным (рис.). Такой процесс извлечения вещества называется экстракцией и широко используется и в промышленности, и в лаборатории.
Экстракция брома бензолом. Бром растворяется в органической жидкости — бензоле С6Нв гораздо лучше, чем в воде. В делительную воронку с бромной водой добавим немного бензола (I), затем смесь взболтаем сильным встряхиванием (II) и дадим жидкостям расслоиться. Более лёгкий бензол соберётся вверху, над водой, а бром почти целиком перейдёт в бензольный слой (III). В результате слой бензола приобретёт интенсивную тёмнокрасную окраску. Извлечение вещества из водного раствора органической жидкостью относится к процессам, называемым экстракцией.
БРОМ
94
У атома Б. семь внешних (валентных) электронов (конфигурация 4s24p6, см. Атом). Для Б., как и для других галогенов, наиболее характерны реакции, в которых атом Вг присоединяет электрон, образуя ион Вг“. Поэтому свободный Б.— довольно сильный окислитель. В соединениях Б. может проявлять степени окисления —1, +1, +3 и +5 (валентности Г, III и V). По химическим свойствам Б. очень похож на хлор. Но в отличие от хлора и иода, соединения со степенью окисления +7 для Б. неизвестны. Б. непосредственно реагирует с различными элементами (металлами и неметаллами):
2А1 + ЗВг2 = 2А1Вг3
2Р + ЗВг2 = 2РВг3
Первая реакция проходит очень эффектно, с выделением большого количества тепла и света. Активно окисляет Б. и сложные вещества:
H2S + Br2 = S+2HBr
(можно сделать опыт: жёлто-бурая бромная вода при пропускании сероводорода обесцвечивается и становится мутной от выделившейся серы). Как и другие галогены, Б. непосредственно не взаимодействует с кислородом, углеродом и азотом, хотя его соединения с этими элементами известны. С водородом Б. реагирует лишь при нагревании, давая бромистый водород:
Вг2 + Н2 = 2НВг
Для Б. характерно образование соединений с другими галогенами — межгалогенных соединений, например BrF, BrF3, BrF5.
Как и все галогены, кроме фтора, при реакции с водой Б. даёт смесь двух кислот: бромистоводородной НВг (см. Бромистый водород) и бромноватистой:
Вг2 + Н2От±НВг + НВгО Равновесие этой обратимой реакции сильно сдвинуто влево.
Бромноватистая кислота очень слабая и существует только в растворе. Даже под действием рассеянного света она быстро разлагается с выделением активного атомарного кислорода:
НВгО = НВг + О
Все соединения Вг (4-1) — и бромноватистая кислота, и её соли гипобромиты, и BrF (газ красного цвета), и Вг2О (бурый газ) —неустойчивы.
Из соединений Вг (4-3) известен лишь трёхфтористый бром BrF3— светло-жёлтая жидкость. Водой Вгг3 разлагается:
3BrF3 4- 6Н2О = 2НВгО3 4- 9HF 4- НВг
Из всех соединений, где Б. проявляет положительные степени окисления, наиболее устойчивы производные Вг (4-5). При взаимодействии Б. со щелочами при температуре выше комнатной образуются броматы — соли бромноватой кислоты:
ЗВг2 4- 6КОН = 5КВг 4- КВгО3 4- ЗН2О
Сама бромноватая кислота существует только в растворе, но её соли вполне устойчивы при обычных условиях. Они разлагаются лишь при нагревании, причём состав продуктов разложения зависит от природы металла, образующего соли. Так, броматы К, Na, Hg разлагаются с образованием бромида и выделением кислорода:
2КВгО3 = 2КВг 4- ЗО2
Броматы Mg, Al, Zn разлагаются с образованием окисла, свободных брома и кислорода:
2Mg(BrO3)2 = 2MgO 4~ 2Вг2 4~ 5О2
В основе получения Б. как в промышленности, так и в лаборатории лежит реакция окисления бромид-иона:
- о Вг"—1е= Вг
(аналогичная реакция лежит в основе методов получения и других галогенов). Добывают Б. из озёрного и морского рассола (так называемой рапы), из буровых вод или из отходов, оставшихся после производства хлористого калия. В такой раствор (или рассол) пропускают хлор. Существует закономерность: в ряду F—С1—Вг—I каждый предыдущий вытесняет все последующие из их солей — галогенидов (подробнее см. Галогены). В нашем случае происходит такая реакция:
CL + 2КВг = Вг2 + 2КС1 или £ Л
CL + 2Вг- = Вг. + 2С1-Выделяющийся Б. отгоняют из раствора с водяным паром или с воздухом, затем очищают различными методами.
Б. чрезвычайно ядовит. Жидкий Б. вызывает сильные, трудно заживающие ожоги, а его пары раздражающе действуют на слизистую оболочку. А когда врач-невропатолог говорит больному: «Пропишу-ка я Вам бром для успокоения», то имеются в виду бромиды щелочных металлов, а не сам Б. И в таких случаях употреблять название «Б.», с точки зрения химика, неправильно.
Б. сейчас широко используется во многих областях химической промышленности. Он служит исходным веществом для получения различных бромистых солей и органических соединений. Из бромсодержащих органических соединений наиболее интересны те, которые проявляют физиологическую активность. Одни из них служат здоровью человека; это лекарства — бромурал, бромоформ, бромалин и т. д. Действие других противоположно: так, бромацетофенон и бромбензол являются сильными лакриматорами — слезоточивыми веществами. (Полиция капиталистических стран пользуется ими для разгона демонстраций.) Большое количество Б. идёт на получение дибромэтана С2Н4Вг2 и бромистого этила С2НвВг, добавляемых к бензинам для повышения их антидетона-ционных свойств.
Дело в том, что обычный антидетонатор тетраэтилсвинец (ТЭС) при сгорании образует РЬО, которая отлагается на стенках цилиндров двигателей внутреннего сгорания. При добавлении в топливо дибромэтана или
95
бромистого этилена («бромистых выносителей», как называют их в технике) образуется РЬВг2, который легко удаляется.
Самая же интересная область применения соединений Б.— фотография. Об этом см. в статье Серебро. В. К. Бельский.
БРОМАТЫ — соли бромноватой кислоты НВгО3, например КВгО3. См. Бром.
БРОМЙДЫ — соединения брома с другими, более электроположительными элементами. Б. металлов — соли бромистоводородной кислоты (см. Бромистый водород) — по большей части хорошо растворимы в воде. Мало растворимы РЬВг2 и HgBr2, практически не растворимы AgBr, Hg2Br2 и Си2Вг2. Поэтому присутствие в растворе иона Вг- можно обнаружить добавлением раствора AgNO3:
AgNO3 + KBr - AgBr + KNO3
Выпадающий в осадок AgBr—жёлтого цвета. Эта реакция используется в аналитической химии для «открытия» в растворах ионов Ag+ и Вг-.
Но бромид серебра интересен нетолько химикам-аналитикам. У него есть способность разлагаться на элементы под действием света:
2 AgBr = 2Ag + Вг2
И это делает его почти незаменимым в производстве кинофотоматериалов.
Вспомните хотя бы названия сортов фотобумаги, знакомые каждому любителю: Унибром, Фотобром, Бромпортрет, Новобром, Конта-бром и др. О фотографическом процессе см. в статье Серебро.
Б. щелочных металлов применяет в медицине в качестве успокаивающих средств (см. также Бром) и в аналитической химии как восстановители.
БРОМИСТОВОДОРОДНАЯ КИСЛОТА —раствор бромистого водорода Н Вг в воде.
БРОМИСТЫЙ ВОДОРОД НВг — соединение брома с водородом. Бесцветный газ с резким неприятным запахом. Превращается в жидкость при охлаждении до —68,7 °C. Синтезируют Б. в. из элементов при 200—300 °C:
Н2+ Вг2 = 2НВг
Температуру можно понизить добавлением катализаторов — активного угля или платинированного асбеста. Б. в. нельзя получить в чистом виде реакцией бромида металла с концентрированной серной кислотой:
NaBr + H2SO4 = NaHSO4 + НВг
Дело в том, что ион Вг“ окисляется значительно
БУРА
легче, чем ион О", и серная кислота успевает окислить часть выделяющегося Б. в.:
2НВг + H2SO4 = Вг2 + SO2 + 2Н2О
Самый же простой способ получения Б. в. в лаборатории — взаимодействие бромида фосфора РВг3 (это бесцветная жидкость) с водой:
РВг3 + ЗН2О = Н3РО3 + ЗНВг
Б. в. исключительно хорошо растворяется в воде: при 10°С 1 объём Н2О поглощает около 350 объёмов НВг. Раствор НВг в воде называется бромистоводородной кислотой. Это бесцветная (иногда желтоватая из-за следов растворённого брома) жидкость с запахом Б. в. Бромистоводородная кислота — одна из самых сильных.' Её соли называются бромидами.
БРОМНАЯ ВОДА — раствор брома в воде. БРОМНОВАТАЯ КИСЛОТА — соединение состава НВгО3, производное Вг (+5). Существует только в водных растворах. Образует соли — броматы. См. Бром.
БРОМНОВАТИСТАЯ КИСЛОТА — соединение состава НВгО, производное Вг (+1). Существует только в водных растворах. Образует соли — гипобромиты. См. Бром. БУРА Na2B4O7- 10Н2О —соль не выделенной в свободном состоянии тетраборной кислоты Н2В4О7. Важнейшее природное соединение бора, известное как минерал тин кал. Бесцветные кристаллы, растворимые в воде (3,9 г безводной соли в 100 г воды при 30 °C). Водный раствор Б. (как соли слабой кислоты) имеет щелочную реакцию. При сплавлении Б. с соединениями некоторых металлов образуются окрашенные метабораты, например зелёный Сг(ВО2)3, синий Со(ВО2)2 и др.:
Na2B4O7 + СоО = 2NaBO2 + Со (ВО2)2
В аналитической практике по образованию таких цветных стекловидных шариков — «перлов буры» судят о присутствии в исследуемом веществе того или иного металла; получают перлы в пламени газовой горелки на петельке из тонкой платиновой проволоки. Способностью расплавленной Б. растворять окислы металлов пользуются для очистки поверхностей при сварке, резке и пайке металлов.
Б.— это и распространённое дезинфицирующее средство, и консервант в пищевой промышленности, и удобрение в сельском хозяйстве.
50,9414 23
V
3d34s2
ВАНАДИЙ
ВАНАДИЙ (лат. Vanadium) V — химический элемент V группы периодической системы Менделеева; атомный номер 23, атомная масса 50,9414. Металл серо-стального цвета. Природный В. состоит из двух изотопов: 61V (99,75%) и 60V (0,25%); второй из них слабо радиоактивен (период полураспада 7\/2 = 1014 лет).
В. был открыт в 1801 г. мексиканским минералогом А. М. дель Рио в бурой свинцовой руде и назван эритронием (от греч. «эритрбс»—красный) из-за красного цвета его солей. Впоследствии, однако, сам же первооткрыватель принял эритроний за разновидность хрома. В 1830 г. шведский химик Н. Сеф-стрём обнаружил в железной руде новый элемент. Название «ванадий» было
дано элементу благодаря красивому цвету его солей, в честь древнескандинавской богини красоты Вана-дис. Через год немецкий химик Ф. Вёлер доказал тождественность В. и эритрония. Вёлер сам был очень близок к открытию В., но потерпел неудачу. Как истинный учёный, он был объективен и самокритичен. В письме к другу Вёлер писал: «Я был настоящим ослом, проглядев новый элемент в бурой свинцовой руде, и прав был Берцелиус, когда он не без иронии смеялся над тем, как неудачно и слабо, без упорства, стучался я в дом богини Ванадис». Только в 1869 г. английский химик Г. Роско получил порошок металлического В., восстановив VC12 водородом. В промышленных масштабах В. добывается с начала XX в.
В земной коре содержится 9-10"3% В. по массе, то есть несколько больше, чем меди, хрома или никеля. Это довольно распространённый, но рассеянный в породах и минералах элемент. Важным промышленным источником В. (в частности, в СССР) служат титаномагнетитовые железные руды (содержание В. достигает 1%) и осадочные железные руды (до 0,1% В.). Интересно, что один из самых богатых ванадием минералов (до 29% В.) — патронит (сульфид В. переменного состава), чрезвычайно редок. Он обнаружен лишь в одном месте — в Перу, на высоте 4700 м над уровнем моря.
Чистый В. по цвету похож на сталь, он обладает высокой твёрдостью и большой прочно
стью. В. пластичен, поддаётся обработке давлением. Плотность В. 6,11 г/см3, /пл 1900 °C, /кип 3400 °C. Вода, кислоты (за исключением фтористоводородной и азотной, а также царской водки) и щёлочи на В. не действуют. Однако при нагревании на воздухе металл интенсивно поглощает кислород и становится хрупким. При 700 °C он окисляется с образованием высшего окисла V2O5. Этот окисел не образует на поверхности металла защитной плёнкиг в отличие от окислов алюминия и многих других металлов. Он необычен в том смысле, что плавится при гораздо более низкой температуре, чем сам В.,— ниже примерно на 1200°С. При 700 °C V2O5, плавясь, стекает с поверхности металла, так что последний может окислиться полностью.
В соответствии с положением В. в периодической системе Менделеева его атом имеет пять валентных электронов (конфигурация 3d34s2, см. Атом). Соединения В. отвечают степеням окисления +2, +3, +4 и +5, то есть валентностям II, III, IV и V (последняя наиболее характерна). Им соответствуют окислы VO и V2O3 (чёрного цвета, имеют основной характер), VO2 (тёмно-синего цвета, амфотерный) и V2O6 (оранжевого цвета, кислотный).
Пятиокись V2O6— наиболее важное соединение В. Она несколько растворима в воде. Лакмусовая бумага в этом растворе краснеет, что говорит о присутствии в нём кислот В., которые не удалось, однако, выделить в чистом виде. Соли их — ванадаты, образуются при растворении V2O6 в щелочах:
V2O6 + 2NaOH = 2NaVO3 + Н2О -
(обычно состав ванадатов и соответствующих им не выделенных в свободном состоянии кислот более сложен).
Извлечение В. из руд — сложный технологический процесс; он сводится обычно к получению пятиокиси V2O5, из которой металл восстанавливают действием кальция при высокой температуре:
V2O5 + 5Са = 2V + 5СаО
В большинстве случаев целью выплавки является не чистый В., а очень важный сплав его с железом — феррованадий (содержит от
97
ВИСМУТ
35 до 70% V); в этом случае ведут восстановление смеси V2O5+Fe2O3 (вспомним, что основным ванадиевым сырьём служат некоторые железные руды). В виде феррованадия В. вводят в сталь и чугун, на свойства которых он оказывает исключительно благотворное влияние. Инструменты из стали, содержащей всего лишь 1 % V, работают вдвое эффективнее обычных. Поэтому из многочисленных сортов легированных сталей более половины содержит присадку В. Чугун же, легированный ванадием, служит вдвое дольше обычного. В железоуглеродистых сплавах, к которым относятся сталь и чугун, В. образует с углеродом карбиды — они-то и придают сплавам особую устойчивость к толчкам, ударам и вибрации. Именно такая сталь нужна для изготовления авиационных и автомобильных моторов. «Автомобильный король» Генри Форд как-то обмолвился: «Если бы не было ванадия, то не было бы и моего автомобиля».
Новым потребителем В. выступает быстро развивающаяся промышленность титановых сплавов; отдельные сплавы титана содержат до 13% V. В авиационной, ракетной промышленности и других областях техники нашли применение ниобиевые, хромовые и танталовые сплавы, содержащие присадки В. Сплавы же на основе самого В. сохраняют прочность до 1320°С, и им прочат большое будущее в космонавтике. Всего же на получение чёрных и цветных сплавов расходуется около 95% добываемого В.
Пятиокись V2O5 — отличный катализатор, заменяющий дорогую платину в производстве серной кислоты. Она же катализирует многие процессы органического синтеза. Соединения В. применяются в текстильной и лакокрасочной промышленности, в производстве стекла и глазурей. Ю. И. Романьков.
ВЕСЕЛЯЩИЙ ГАЗ — закись азота N2O. Бесцветный газ со слабым приятным запахом. Вдыхание смеси воздуха с N2O вызывает состояние, напоминающее опьянение. В медицине N2O служит для общей анестезии (обезболивания) при хирургических операциях. См. также Азот.
ВЙСМУТ (лат. Bismuthum) Bi — химический элемент V группы периодической системы Менделеева; атомный номер 83, атомная масса 208,9804. Металл серебристо-белого цвета с розоватым оттенком. Природный В. состоит из одного изотопа 209Bi. Этот изотоп радиоактивен, но с таким огромным периодом полураспада (Ti/2=2-1017 лет), что обычно его относят к стабильным.
В. был известен ещё в XV в., но долгое время его считали разновидностью олова, свинца или сурьмы.
Самостоятельным металлом В. был признан в середине XVIII в. Относительно происхождения слова «висмут» существуют разные толкования.
Изучение этимологии (происхождения тех или иных слов) — удивительно интересное занятие. Работая над этой книгой, редакторы устроили целый «научный диспут» по поводу слова «висмут» и даже написали шуточные стихи, подкреплённые, однако, самыми серьёзными ссылками на литературные источники. Вот некоторые строки:
Профессор Руска упорно твердит, Что «висмут» — искажённое арабское «ithmid».* Профессор Липман это отрицает И слово «висмут» немецким считает, Производя его от старинного «wis mat», То есть «белый металл». И учёный рад.
Но спорна весьма этимология эта: Ведь висмут красноват, а не белого цвета. А профессор доктор, Леопольд Гмелин Пишет: «Как я совершенно уверен. Висмут был назван так оттого, Что в округе Визен добывали его».
Однако лучше всех сказал Пьер Ларусс: «Etym. inconnue».** Я ничуть не боюсь Рекомендовать усердно этимологию эту На радость БСЭ и всему свету.
208,9804 33
Bi
6s26p3
ВИСМУТ
Содержание В. в земной коре составляет 9-10-7% по массе (всего лишь 71-е место среди других элементов). Встречается он и как самородный металл, и (главным образом) в виде многочисленных соединений. Основные минералы — висмутовый блеск (висмутин) Bi2S3 висмутовая охра (бисмит) ВцОзИдр. Около 90% всего добываемого в мире В. извлекают не из собственных висмутовых руд, а попутно, при добыче свинца, меди и других металлов. Самые богатые месторождения В. находятся в Боливии,на о. Тасмания и в Перу. Добуча В. в нашей стране была начата после Великой Октябрьской революции.
В.— очень легкоплавкий металл — /пл 271,3 °C; /кип 1560 °C; плотность 9,80 г/см3. В отличие от большинства металлов, В. очень хрупок; проволоку из него можно получить только продавливанием через узкое отверстие.
В. стоит особняком среди прочих металлов и по Другим физическим свойствам. При плавлении объём большинства металлов увеличивается, а при затвердевании — уменьшается. У В. же объём при плавлении уменьшается (как, кстати, и у льда). Аномалия в обоих случаях объяс
* Итмид — одно из названий сурьмяного блеска (антимонита).
** «Этимология неизвестна» или «Происхождение названия „В.\не установлено» — такую сухую справку вы найдёте в разных справочниках — от Энциклопедического словаря Ларусса XIX в. до 3-го издания Большой советской энциклопедий.
7 хэш
ВОДА
няется особенностями кристаллических структур В. и льда (см. Вода). Электрическое сопротивление В. после расплавления уменьшается более чем вдвое, а с понижением температуры резко возрастает (если понизить температуру от 0°С до —180°С, то сопротивление увеличится в 60 раз). В магнитном поле электросопротивление В. также возрастает, чем пользуются для измерения напряжённости поля. Прибор для этой цели называется висмутовой спиралью.
У атома В. пять внешних (валентных) электронов (конфигурация 6s26p3, см. Атом). По строению электронной оболочки В. аналогичен своим соседям по группе—азоту, фосфору, мышьяку и сурьме. Это обусловливает сходство некоторых химических свойств пяти перечисленных элементов. Но В. замыкает V группу снизу, и, следовательно, у него сильнее, чем у всех его аналогов, выражены металлические свойства. Наиболее характерная его степень окисления -:-3; известны и соединения со степенями окисления—3 и от — 1 до --5 (соответственно валентности I—V). Окисел Bi2O3—типичный основной окисел. А у гидроокиси Bi(OH)3 кислотная функция практически не выражена (в отличие от соответствующих азотистой, фосфористой и мышьяковистой кислот).
В сухом воздухе металл устойчив, а во влажном он покрывается с поверхности тончайшим слоем окиси. При нагревании на воздухе выше температуры плавления В. сгорает с образованием окисла Bi2O3. В ряду напряжений В., подобно меди, стоит после водорода и поэтому не вытесняет Н2 из разбавленных кислот. С соляной и разбавленной серной кислотами В. не взаимодействует. Однако разбавленная азотная кислота реагирует с ним с выделением окиси азота:
Bi - 4HNO3 = Bi (NO3)3 NO + 2H2O Непосредственно реагирует В. с галогенами и с серой.
Соединения В. со степенью окисления —5 обладают сильно выраженными окислительными свойствами. Так, висмутаты щелочных металлов (соли не выделенной в свободном состоянии висмутовой кислоты HBiO3) могут в + 2 +7
кислой среде окислять Мп до Мп, то есть являются более сильными окислителями, чем хорошо известная «марганцовка» — перманганат калия:
10KBiO3 -г 4MnSO4 -г 14H2SO4 =
= 4КМпО4 4- 3K2SO4 + 5Bi2(SO4)3 + 14Н2О
В мировом масштабе добыча В. невелика, она лишь немногим превышает добычу золота. Так как висмутовые руды, как правило, очень бедны, то они сначала подвергаются обогащению. Из окисленных руд В. извлекают
98
восстановлением углём под слоем легкоплавкого флюса:
Bi2O3 -ЗС = 2Bi - ЗСО
Области применения В. и его соединений чрезвычайно обширны. В.— прекрасный теплоноситель в энергетических ядерных реакторах.
Всем известны обычные теплоносители, служащие для обогрева (или для охлаждения) помещений или промышленных аппаратов: вода, водяной пар, топочные газы. В ядерных реакторах, где температуры гораздо выше, применяются теплоносители особого рода — расплавленные металлы или их смеси. У В. как у ядерного теплоносителя удачное сочетание свойств. Он плавится при сравнительно низкой температуре, а кипит при высокой (интервал жидкого состояния составляет около 1300°С), хорошо проводит тепло и очень стоек химически. В отличие от другого металла — теплоносителя натрия, висмутовый теплоноситель безопасен в пожарном отношении.
Но самое главное заключается в том, что В. не тормозит цепной ядерной реакции, идущей в реакторе под действием нейтронов (ядра атомов В. плохо захватывают тепловые нейтроны или, выражаясь языком ядерной физики, обладают малым сечением захвата). О цепных ядерных реакциях, идущих под действием нейтронов, см. в статье Уран.
Основная область применения В.— приготовление многочисленных легкоплавких сплавов, используемых в противопожарной аппаратуре. В автоматических огнетушителях ставятся плавкие предохранители, сделанные из сплава В. с другими металлами. Как только воздух нагреется до температуры плавления сплава (а она может быть всего лишь 47 °C), предохранительная проволочка замкнётся, контакт расплавится и.... (а дальше всё зависит от устройства помещения — либо зазвенят предупредительные звонки и загорятся сигнальные лампы... Либо, если в помещении нет людей, из труб, спрятанных в стенах, хлынут струи воды). Такой висмутовый сигнализатор предупредит нас об опасном перегреве какой-нибудь части машины или аппарата.
Известный с 1860 г. сплав Б. Вуда (50% Bi, 25% Pb, 12,5% Sn и 12,5% Cd) имеет /пл всего 68 °C. Ясно, что ложкой, изготовленной из такого сплава, горячий чай в стакане уже не размешать. Сплав В. со свинцом и ртутью плавится уже при трении и применяется для изготовления металлических карандашей.
Соединения В. применяются в медицине; так, соли В. и некоторых органических кислот служат против кожных заболеваний (дерматол, ксероформ и др.). Основной нитрат В. ещё в XVI в. применялся в театрах в качестве грима (так называемые испанские белила).
В. К. Бельский. ВОДА — окисел водорода, содержащий 88,6% кислорода и 11,4% водорода, что отвечает простейшей формуле Н2О. Эта формула знакома всем — даже людям, знающим о химии
ВОДА
99
только понаслышке. Чего уж проще — простота эта чуть ли не вошла в поговорку! Но так ли она проста, вездесущая и всем знакомая вода? Нет, это далеко не так.
Физические свойства и строение воды. Многие физические константы жидкой В. (например, плотность, теплоёмкость) приняты как эталон, образец. Её температуры плавления и кипения долгое время служили точками отсчёта стоградусной шкалы температур (О °C и 100 °C). Значит ли это, что свойства В. обычны, «Образцовы»? Нет, совсем наоборот! Трудно найти в природе другое вещество, физические свойства которого были бы так необычны, своеобразны, аномальны. Давайте забудем всё, что нам известно о В., и попробуем «открыть» её для себя заново.
Поставим перед собой вопрос: каковы должны быть температуры плавления и кипения В.? Можно ли ставить такой вопрос? А почему бы и нет, ведь периодическая система Менделеева даёт возможность представить себе свойства какого-либо соединения, зная свойства аналогичных соединений элементов той же группы. Аналоги кислорода — это сера, селен и теллур. Аналоги Н2О — 3to^H2S, H2Se и Н2Те. Если построить графики их свойств, идя в периодической системе снизу вверх, то получится картина, отражающая закономерное изменение температур кипения и плавления гидридов — Н2Те, H2Se и H2S (рис. 1). Если продолжить получившиеся линии, то окажется, что при сохранении той же закономерности В. должна быть газом, кипеть при—80 °C
Рис. 1. Температуры кипения и плавления (замерзай и я) воды и водородных соединений теллура, селена, серы. В ряду Н2Те—H2Se—H2S эти физические величины линейно уменьшаются. Если бы той же закономерности подчинялась и Н2О, то соответствующие константы для неё были бы: £пл — 100°С, ^кип— 80°С. В действительности же — это всем известно — для воды tnjIO°C и fKHn 100°С.
О Н2о?
0H2S
H2Se0
“100 -50
С>н2о?
0H2S
т. кип.
Н2О!©
0Н2Те
0 50 100°С
Т. пл.
0Н2О!
4 34Se ©H2Se
52
5 Те 0Н2Те
-100 -50 О
50 100 °C
(вместо +100 °C!) и замерзать при —100 °C (вместо 0°С!). Попробуйте представить себе, что получилось бы, если бы В. вела себя «нормально».
Обратимся к другим аномалиям В. (а их немало).Так, жидкая В. имеет самую высокую теплоёмкость среди всех жидкостей [1 кал/(г-град), или по Международной системе единиц СИ 4,19 кдж/(кг-град)]. Аномально и изменение плотности В. Плотность других жидкостей, как правило, при понижении температуры постепенно возрастает и становится максимальной при замерзании. А плотность В. при охлаждении «нормально» возрастает лишь до +4 °C, достигая 1 г/см3. От +4 °C до 0 °C она немного уменьшается. Плотность же льда резко, скачком уменьшается до 0,91 г/см3. Теплота плавления льда 332,7 кдж/кг (79,4 кал/г) также аномально высока — в 13,5 раза выше, чем, например, у свинца.
Как же объяснить необычные свойства В.? Чтобы ответить на этот вопрос, обратимся к строению В.
Молекула Н2О нелинейна; угол между связями О—Н равен 104,27°. Связи эти ковалентные, но они полярны (атомы Н несут на себе некоторый положительный заряд, атомы О — отрицательный). Поэтому полярна и молекула в целом; она представляет собой диполь. Дипольные молекулы взаимодействуют сильнее, чем молекулы неполярные. Но молекулы В. связаны между собой гораздо прочнее, чем можно было бы ожидать, учитывая лишь физическое взаимодействие диполей. Это объясняется существованием водородных связей. В водородной связи участвуют атом О одной молекулы и атом Н — другой:
Нч /Н
>О. . .Н —Ст Hz
Такие связи гораздо менее прочны, чем ковалентные связи О—Н внутри молекул, и всё же именно благодаря им взаимодействие молекул в В. гораздо сильнее, чем во многих других жидкостях (например, в жидких H2S, H2Se, Н2Те). В процессе теплового движения молекул водородные связи рвутся, но взамен тут же возникают новые. Таким образом, в жидкой В. существует динамическая система межмолекулярных водородных связей. Иначе говоря, молекулы в В. ассоциированы. Именно повышенная прочность связей между молекулами Н2О служит причиной аномально высокой температуры кипения В. Сильное межмолекулярное взаимодействие затрудняет переход молекул из жидкости в пар.
В кристаллах льда тоже существуют водородные связи, но здесь система таких связей статична, а следовательно, ещё более прочна, чем в жидкой В. В этом — причина аномально высокой температуры и теплоты плавления льда.
Рассмотрим теперь структуру льда более внимательно. Вероятно, каждого поражали красота и разнообразие форм снежинок (рис. 2,а). Но при всём разнообразии снежинок их внутреннее строение всегда одинаково. В кристаллах льда каждая молекула Н2О соединена водородными связями с четырьмя соседними. Такая структура ажурна — в ней много «пустот» (рис. 2,6). Вот почему плотность льда сравнительно низка. При плавлении льда часть «пу
ВОДА
100
стот» заполняется «одиночными» и «сдвоенными» молекулами Н2О, уже освободившимися из кристаллической решётки. Поэтому плотность В. выше, чем у льда. Такое увеличение плотности продолжается и при нагревании от 0 до +4 °C; но при более высоких температурах начинает преобладать тепловое движение молекул, расстояния между отдельными молекулами Н2О увеличиваются, и изменение плотности становится «нормальным».
Поскольку тепловая энергия при нагревании В. расходуется не только на ускорение движения молекул Н2О, но и на разрыв водородных связей между ними, то и теплоёмкость В. оказывается столь большой. Кстати скажем, что даже в парах В. при 100 °C эти связи разорваны ещё не полностью, так что из каждых 200 моле-
кул Н2О в среднем 7 попарно связаны между собой. В виде (Н2О)2 В. существует и при растворении её в органических растворителях. Вот как сложна «простая» и обыкновенная В.!
С высокой полярностью молекул Н2О связано большое значение её диэлектрической проницаемости и почти не имеющая себе равных способность растворять другие полярные соединения и вызывать электролитическую диссоциацию кислот, оснований и солей.
И В., и лёд (при их достаточной чистоте) вполне прозрачны и бесцветны. В толстых слоях В. имеет голубоватую окраску, потому что задерживает красную часть спектра световых лучей сильнее, чем синюю (а синюю отражает). Ещё сильнее поглощает В. невидимые инфракрасные (тепловые) лучи — это имеет важнейшее значение для температурного режима нашей планеты.
Так как водород имеет 2, а кислород — 3 стабильных изотопа, существует 9 различных вариантов соединения их в молекулы В. (а с учётом радиоактивных изотопов — 36 вариантов). В природной В. молекулы, состоящие только из «лёгких» изотопов ХН и 16О, составляют 99,73% (см. Водород).
Химические свойства. Молекула Н2О настолько прочна, что разрушить её можно лишь оч^нь энергичным внешним воздействием. Разложение В. по обратимой реакции:
2Н2О^2Н2 + О2
становится заметным лишь при нагревании до 2000 °C (термическая диссоциация). Оно происходит и под действием ультрафиолетовых лучей (фотохимическая диссоциация). Радиоактивное излучение разлагает В. с образованием водорода, кислорода, перекиси водорода и очень активных свободных радикалов.
В.— слабый электролит. При комнатной температуре лишь одна из 10 млрд, молекул диссоциирует по уравнению:
н2о^н++он-
так что при 25 °C в чистой воде концентрации Н+ и ОН- составляют всего 10“7 г-ионов/л. Это соответствует водородному показателю pH=7. При повышении температуры электроли-
Р и с. 2. а — внешний вид снежинки, б — её внутреннее строение.
Многообразие форм снежинок поистине удивительно. Но как бы ни различались снежинки по внешнему виду, их внутреннее атомное строение всегда одинаково. В кристаллической структуре льда каждая молекула Н2О соединена водородными связями с четырьмя соседними. В итоге возникает ажурный пространственный каркас. Обратите внимание на то, что «обычные* ковалентные связи атомов кислорода со «своими» атомами водорода короче, чем водородные связи с Н-атомами соседних молекул Н2О. Это соответствуем большей прочности «обычных» связей по сравнению с водородными.
101
тическая диссоциация В. усиливается; при 700 °C и давлении 130000 атм концентрация Н+ становится такой же, как при обычных условиях у 10%-ной соляной кислоты. Ион водорода Н+ в растворах не существует в свободном состоянии, а присоединяется к молекуле Н2О и образует ион гидроксония Н3О+. Как и все другие ионы, гидроксоний в водной среде, в свою очередь, гидратирован, то есть окружён несколькими молекулами Н2О. Таким образом, в водных растворах наряду с Н3О+ есть более сложные ионы (например, Н3О+-ЗН2О, то есть н,ор.
Сравним, как ведут себя в водных растворах гидраты азота, кислорода и фтора — трёх соседей по II периоду системы Менделеева:
NH3+H20z± NH4+OH-Н2О + Н2О 72: Н3О+ + ОН-HF + H2O 77 H3O+ + F-
Водный раствор аммиака, давая ионы ОН”, имеет основные свойства. HF даёт ионы Н3О+ и потому является кислотой. Ну, а В., образуя и ионы Н3О+ и ионы ОН”, проявляет своеобразную амфотерность.
Все ионы в водных растворах окружены «хороводом» молекул В. Многие вещества при кристаллизации их из водных растворов прочно удерживают одну или несколько молекул Н2О, образуя кристаллогидраты. Связанную таким образом В. называют кристаллизационной. Она сравнительно легко удаляется из кристаллов, особенно при нагревании. Если же В. вступила в химическую реакцию с каким-либо веществом, и при этом образовалось новое соединение, говорят о конституционной В. Так, при добавлении Н2О к СаО образуется Са(ОН)2 и только разложение этой гидроокиси при прокаливании позволяет удалить из неё конституционную В. Адсорбционной называют В., поглощённую твёрдыми веществами с большим числом пор (например, активным углём). Гигроскопической (капиллярной)—В., заполняющую тонкие канальцы в других веществах (например, в почве).
Щелочные и щелочноземельные металлы разлагают В. с выделением водорода уже при обычной температуре, а магний и цинковая пыль — при кипячении. Алюминий реагирует с В. после очистки его от окисной плёнки. Менее активные металлы вступают в реакцию при температуре красного каления:
3Fe + 4Н2О = Fe3O4 + 4Н2
Металлы, стоящие в ряду напряжения после водорода, с В. не реагируют. В. играет большую роль в процессе атмосферной и электрохимической коррозии металлов. Используя В., содержащую «тяжёлый» кислород 18О, удалось показать, что при коррозии железа во влажной атмосфере «ржавчина» получает кислород именно из В., а не из воздуха.
В. взаимодействует со многими неметаллами. Атомарный кислород превращает В. в перекись водорода Н2О2 (см. Водорода перекись). Фтор уже при обычной температуре, а хлор — при
ВОДА
100 °C или на свету разлагает В. с образованием атомарного кислорода:
Н2О + С12 = 2НС1 + О
При комнатной температуре и хлор, и бром, и иод реагируют с В. иначе — по обратимой реакции:
Х2 + Н2О НХ + НХО (здесь X — любой из галогенов). В. окисляет фосфор при нагревании под давлением:
2Р + 6Н2О = 2НРО3 + 5Н2
Известно, что В. используют при гашении пожаров. В то же время горящий уголь часто поливают некоторым количеством В. для усиления горения. Это связано с образованием горючего водяного газа (смеси СО и Н2):
с + н2о-со + н2
Растворимые в В. солеобразующие окислы взаимодействуют с ней, давая кислоты и основания. Многие соли, а также некоторые другие вещества вступают с В. в реакцию .обменного разложения, называемую гидролизом.
Благодаря высокой растворяющей способности и химической активности В. огромное большинство реакций в живой и неживой природе протекает в водных растворах и при участии В. В известном смысле, физика и химия окружающей нас среды — это физика и химия В. Исключительно интересны каталитические свойства В. Не все знают, что даже такие бурно протекающие реакции, как взаимодействие хлора с водородом, горение фосфора и щелочных металлов в кислороде и др., не начинаются, если отсутствуют хотя бы ничтожные количества (следы) влаги.
Можно провести эффектный опыт, показывающий эту способность В. Если смешать порошок алюминия с иодом, то добавление маленькой капли В. вызывает бурную реакцию образования бурого АП3, напоминающую вулканическое извержение (см. рис. к статье Иод).
Вода в природе. В.— самое распространённое вещество на поверхности нашей планеты. Водная оболочка Земли — гидросфера— составляет около 71% земной поверхности. В связанном состоянии В. находится и в земной коре — литосфере, причем подсчитано, что запасы такой В. примерно равны массе свободной В. в гидросфере. Найдено, что 1 км3 гранита при плавлении может выделить 26 млн. т В. Ещё больше «резервы» В., заключённые в более глубоких недрах Земли — в мантии. Считают, что здесь до 13 млрд, км3 В., то есть вдесятеро больше, чем в гидросфере. Но на поверхность вулканы выносят лишь 1 км3 такой В. ежегодно.
В. играла и играет определяющую роль в геологической истории Земли, в формировании её теплового режима, климата и погоды,
ВОДА
102
Рис. 3. Круговорот воды в природе исключительно разнообразен и иа рисунке показаны лишь некоторые его звенья. В левой части рисунка изображён подводный вулкан, который поставляет водяные пары и горячую воду в Мировой океан (1). В правой части наземный вулкан (2) извергает водяные пары в атмосферу, где они включаются в грандиозный круговорот воды земной поверхности. На склоне вулкана фонтанируют горячие источники — гейзеры (3) (у нас они известны на Камчатке).
Огромные массы воды испаряются с поверхности океана (4) и дают начало облакам, которые переносятся ветрами на сотни и тысячи километров. Облака частично снова отдают свою воду океану (5) (обратите внимание на дождь в океане, может быть, это тропический ливень?), другие облака несут влагу на материк (6).
Человек вмешался в круговорот воды, изменил его. Он построил водохранилища на реках (7, 8), оросил сухую степь. На правом берегу реки вы видите зелёйый орошённый оазис, иа левом — жёлтую выгоревшую степь.
Велика роль подземных вод на Земле, все горные породы пронизаны водой (она показана голубыми точками и чёрточками). Поток подземных вод направлен с материков в сторону океана (9). Растворяя известняки, подземные воды образовали пещеры с замысловатыми подземными залами, озёрами и узкими переходами между ними; такая пещера показана в центре рисунка (Ю). Несут свои воды в океан реки (11).
В полярных районах (12), иа вершинах высочайших гор (13) вода находится в твёрдом виде: льды на сотни и тысячи лет изымают Н2О из общего круговорота. Если бы растаяли льды Арктики и Антарктики, уровень Мирового океана поднялся бы на 50 метров, были бы затоплены многие приморские низменности, тот город, который изображён иа берегу океана...
А где же подземное оледенение (вечная мерзлота), где другие стороны великого круговорота воды на Земле, возможно, спросит читатель? Но мы же договорились, что все звенья этого круговорота иа одном рисунке показать невозможно!
круговорота веществ и т. д. И в этом сказали своё слово упоминавшиеся аномалии В. Так, например, сохранение достаточно тёплой В. в пресных водоёмах зимой зависит от того, что при +4 °C В., имеющая при этой температуре максимальную плотность, перемещается в глубину. Образовавшийся лёд из-за меньшей, чем у В., плотности остаётся на поверхности и изолирует эту сравнительно тёплую В. от холодного воздуха (ведь лёд плохо проводит
тепло). Растения, укрытые зимой снеговой шубой, не вымерзают. Благодаря увеличению объёма при замерзании В. участвует в разрушении горных пород (выветривании). В.— решающее условие возникновения и существования жизни на Земле.
Для нормальной жизнедеятельности каждому человеку необходимо в день в среднем 2 л питьевой В. Это значит, что население нашей страны выпивает ежедневно около 0,5 млн. т В. Огромное её количество потребляют и растения. Подсчитано, что для получения 1 т пшеницы
103
нужно около 1500 т В., хлопчатника— 10 000 т. Большая часть этой В., оставив растворённые в ней соли растению, испаряется листьями, давая атмосфере столько В., сколько её несут все реки Земли. Лишь 1,5—2% поступающей в растения В. расходуется в процессе фотосинтеза, но это составляет около 650 млрд, т в год. Земля накопила свои запасы В. в гидросфере за 2,5—3 млрд. лет. Но если бы В., используемая при фотосинтезе, не возвращалась растениями в окружающую среду, то всего через 2 млн. лет они полностью «высушили» бы нашу планету.
Запасы пресной В., содержащей до 0,05% солей, составляют примерно 2% гидросферы. Доступна для использования лишь малая её часть (по некоторым подсчётам, всего около 20000 км3/год). Поэтому особое значение приобретает бережное расходование В. и решение проблемы её очистки.
Получение. Казалось бы, разговор об этом не заслуживает особого внимания: ведь В.— самое распространённое вещество на поверхности Земли. Но всё дело в том, что нас окружает не чистая В., а растворы различных веществ в В. Даже в самой чистой, по житейским понятиям, В. «что-нибудь да растворено...». (См., например, отдельную статью Вода, жёсткость.) Абсолютно же чистую В. получить очень и очень трудно. Дистиллированная вода, полученная конденсацией водяного пара и достаточно чистая для большинства целей, не годится для точных химических исследований. Критерием чистоты В. служит постоянство её свойств, в частности электропроводности. Лишь после 35—40 повторных дистилляций В. в вакууме перестают изменяться её свойства. Посуда для перегонки должна быть сделана из кварца. Самую же чистую В. получают взаимодействием тщательно очищенных водорода и кислорода в присутствии платинового катализатора:
2Н2 + О2 = 2Н2О
Применение. Области применения В.— даже если говорить только о промышленности — настолько обширны, что практически невозможно назвать какой-либо производственный процесс, в котором не используется В. Промышленность нашей страны ежесекундно потребляет столько В., сколько несёт её Волга. А в ближайшие годы В. нужно будет втрое больше. На получение 1 т стали расходуется 150 т В., бумаги — 250 т, синтетических волокон— 4000 т... О применении В. в чисто химических производствах, в качестве непосредственного участника химических реакций, рассказано в соответствующих статьях (Азотная кислота, Серная кислота и др.).
Как ни странно поначалу звучит утверждение, что вода служит архитектуре и искусству, но это действительно так.
Что же ещё может так украсить сад или парк?
ВОДА
Спокойная гладь В. небольшого бассейна или пруда, отражающая деревья и скульптуру, располагает к отдыху и сосредоточенным размышлениям. Летящая, искрящаяся на солнце В. фонтанов и каскадов создаёт приподнятое, праздничное настроение ... Размещая в определённом порядке трубы, из которых бьёт В., и варьируя высоту струи, можно как бы «строить» из В. Иной — фантастический — облик приобретает В. вечером в лучах электрического света. Например, знаменитый главный парк Петродворца под Ленинградом (XVIII — начало XIX вв.) своей славой обязан воде: изящная архитектура и прекрасный, созданный искусными садоводами парк гармонически сочетаются с самой большой в мире системой разнообразных по форме фонтанов и каскадов (около 2000 струй). Весенний день, в который впервые после зимнего периода пускаются фонтаны, отмечается здесь как городской праздник (рис. 4, стр. 104).
В. может быть и материалом скульптуры: во многих странах есть традиция сооружать зимой ледяные изображения героев сказок, легенд. А традиционные жилища эскимосов «иглу» строятся из блоков плотно слежавшегося снега.
Если собрать все научные исследования о В. и процессах, идущих с её участием, получится огромная библиотека. Но и сегодня далеко не всё известно об этом интереснейшем, давно знакомом, но во многом загадочном, таком обильном и таком дефицитном веществе — о простой воде. В. Л. Василевский. ВОДА, жёсткость. Жёсткость воды (Ж. в.) это совокупность свойств, обусловленных содержанием в ней ионов Са2+ и Mg2+. Если концентрация этих ионов велика, то воду называют жёсткой, если мала — мягкой. Вы хотите в жёсткой воде помыть голову с мылом — но пена не образуется. То же и при стирке белья — жёсткость не только ухудшает качество тканей, но и повышает затраты мыла. В обоих случаях мыло (калиевая соль стеариновой кислоты С17НзбСООК) расходуется на связывание ионов Са2+ и Mg2+ и осаждается в виде нерастворимых солей:
2С17Н35СОО- + Са2+ = (С17Н35СОО)2Са
2С17Н35СОО- + Mg2+ = (C17H35COO)2Mg
Пена образуется лишь после полного осаждения этих ионов. (Правда, некоторыми синтетическими моющими средствами можно пользоваться и в жёсткой воде, так как образующиеся при этом кальциевые и магниевые соли растворимы в воде.)
В очень жёсткой воде с трудом развариваются пищевые продукты, а сваренные в ней овощи невкусны; плохо заваривается чай, теряется его аромат. При большом содержании ионов Mg2+ (как в море или океане) вода горьковата на вкус и оказывает послабляющее действие на кишечник. Никто из нас не станет пить такую воду, хотя в санитарно-гигиеническом отношении ионы Са2+ и Mg2+ и не вредны.
Непригодна жёсткая вода для использования в паровых котлах. Растворённые в ней со-
ВОДА
104
Рис. 4. Петродворец. Морской канал.
ли при нагревании и испарении воды образуют на стенках котлов слой накипи, который плохо проводит теплоту. Это ведёт к перерасходу топлива, к преждевременному износу котлов, а иногда из-за перегрева стенок котлов и к аварии. При кипячении жёсткой воды образуется и накипь в чайниках. Не годится жёсткая вода и для обогащения полезных ископаемых методом флотации, где применяются олеиновая кислота и другие флотореагенты, образующие малорастворимые соли с Са и Mg. Наконец, жёсткая вода вредна для металлических конструкций, трубопроводов, кожухов охлаждаемых машин.
Ионы Са2+ обусловливают кальциевую жёсткость, а ионы Mg2+—магниевую жёсткость. Общая Ж. в. складывается из кальциевой и магниевой, то есть из суммарной концентрации ионов Са2+ и Mg2+. По отношению к процессам умягчения воды различают жёсткость карбонатную и некарбонатную. Карбонатная жёсткость вызвана присутствием растворённых гидрокарбонатов кальция Са(НСО3)2 и магния Mg(HCO3)2. При кипячении гидрокарбонаты разрушаются, образующиеся при этом малорастворимые карбонаты выпадают в осадок, и общая Ж. в. уменьшается. Поэтому карбонатную жёсткость называют также временной.
При кипячении ионы Са2+ осаждаются в виде средних карбонатов:
Са2 + + 2НСО? = СаСО3 + Н2О + СО2
а ионы Mg2+— в виде основного карбоната или в виде гидроокиси магния:
2Mg2 + + 2НСО? + 2ОН - = (MgOH)2CO3 + СО2 + Н2О
В свою очередь, ионы ОН” образуются при взаимодействии ионов НСО3 с водой:
НСОГ + Н2О = Н2СО3 + ОН -
Жёсткость, сохраняющаяся после кипячения воды, называется постоянной или не-карбонатной. Она обусловлена растворёнными в воде кальциевыми и магниевыми со-лямй сильных кислот, главным образом сульфатами и хлоридами.
По количественному содержанию ионов Mg2+ и Са2+ природную воду различают как очень мягкую (концентрация этих ионов примерно до 30 мг/л), мягкую (30—80 мг/л), средней жёсткости (80—150 мг/л), жёсткую (150— 250 мг/л) и очень жёсткую (свыше 250 мг/л). Особенно большой жёсткостью отличается вода морей и океанов. (В океанах средняя концентрация Са2+ — 450 мг/л, Mg2+ — 1290 мг/л и общая — 1740 мг/л.) Напротив, многие воды северных рек и рек, имеющих ледниковое питание, очень мягки. Вода для хозяйственных нужд и для питья — водопроводная вода — имеет концентрацию Са2+ и Mg2+ не более 170 мг/л. Для определения жёсткости воды существуют специальные методы.
Очень часто жёсткую воду перед употреблением умягчают. Так, карбонатную жёсткость можно устранить добавлением гашёной извести Са(ОН)2:
Са2 + + 2НСОГ + Са2 + + 2ОН -=2СаСО3 + 2Н 2О Mg2 + + 2НСО- + 2Са2 + + 4ОН - =
= 2CaCO3 + Mg(OH)2 + 2H2O
При одновременном добавлении гашёной извести и соды Na2CO3 можно избавиться от карбонатной и некарбонатной жёсткости (известково-содовый способ). При этом карбонатная жёсткость устраняется известью (см. выше), а некарбонатная — содой:
Са2+ +СО2- = СаСО3 Mg2++CO2- = MgCO3 и далее:
MgCO3 + Са2+ + 2ОН - = Mg(OH)2 + СаСО3 Один из наиболее современных способов ус* транения жёсткости основан на применении катионитов — синтетических ионообменных смол и алюмосиликатов, например Na2[Al2Si2O8-H2O] (см. также Силикаты).
Их состав условно можно выразить формулой Na2R, где Na+— весьма подвижный катион, a R2“— частица катионита, несущая отрицательный заряд, в данном примере [Al2Si2O8*nH2OJ2-. Если пропускать жёсткую воду через слои катионита, то ионы натрия обмениваются на ионы кальция и магния:
Са2 + + Na2R = 2Na + + CaR Mg2+ + Na2R = 2Na+ + MgR
Таким образом, ионы Ca2+ и Mg2+ переходят из раствора в катионит, а ионы Na+— из катионита в раствор,
105
ВОДОРОД
и жёсткость устраняется. После обеднения катионита ионами Na+ катиониты обычно регенерируют. Их выдерживают в растворе NaCl, где происходит обратное замещение — ионы Na+ переходят в катионит, а ионы Са2+ и Mg2+— в раствор:
CaR + 2Na + = Na2R + Са2 +
MgR + 2Na+ = Na2R + Mg2+
После этого регенерированный катионит может быть использован для смягчения новых порций жёсткой воды. Г. П. Хомченко.
ВОДОРОД (лат. Hydrogenium) Н — первый химический элемент периодической системы Менделеева; атомный номер 1, атомная масса 1,0079. Газ без цвета и запаха. В природе встречаются два стабильных изотопа В.—гН (протий) и 2Н, или D (дейтерий), и один радиоактивный — 3Н, или Т (тритий). Искусственно получен также крайне неустойчивый изотоп 4Н.
Основную массу природного В. составляет протий, на 6800 атомов которого приходится лишь 1 атом дейтерия. Однако это соотношение несколько меня-ется в зависимости от конкретных условий. Так, поверхностные воды океана немного обогащены дейтерием, входящим в состав тяжёлой воды D2O, а атмосферные осадки и ледники обеднены им. Это объясняется тем, что Н2О испаряется чуть-чуть легче, чем D2O. Тритий образуется из азота в верхних слоях атмосферы при ядер-ных реакциях под действием нейтронов космических лучей:
VN +
Jn = J2C+?H
Количество трития на Земле ничтожно — во всех водах его содержится около 2—3 кг. При радиоактивном превращении трития (период полураспада Ti/t= 12,262 года) в результате Р~-распада заряд ядра увеличивается на единицу, а масса практически не меняется; так образуется изотоп гелия 3Не.
Водород в природе. В.— самый распространённый элемент космоса. Он составляет около половины массы Солнца и большинства звёзд, основную часть газов межзвёздной среды и туманностей. В недрах звёзд при температурах в сотни миллионов градусов и давлении в миллионы атмосфер В. находится в виде протонов— ядер атомов }Н. Здесь происходит реакция слияния атомных ядер (реакция термоядерного синтеза): из четырёх протонов }Н образуется ядро атома гелия £Не, а также два
позитрона и два нейтрино. Энергия, выделяющаяся при этом, огромна: она позволяет звёздам и Солнцу светить миллиарды лет.
Содержание В. в земной коре 1,5-10-1 % по массе. В. входит в состав самого распространённого на Земле вещества — воды. Общее содержание В. на Земле составляет по массе 1%, а по числу атомов 16%. Содержится он и в соединениях, слагающих угли, нефть, природный газ, глины, а также организмы животных и растений. Однако в свободном состоянии В. встречается редко; небольшие количества его содержатся в вулканических и других природных газах. Ничтожные количества свободного В. (0,0001% по числу атомов) присутствуют в атмосфере. В природных водах ионов Н+ — не более 0,0001 г в 1 л. Но роль их огромна.
Воды с повышенным содержанием ионов В. способны разрушать минералы и даже растворять металлы. Такие воды образуются во многих почвах и болотах, где идёт энергичное разложение органических веществ. Это они делают почвы кислыми, неплодородными. В нашей стране кислые почвы нейтрализуют, добавляя известняк СаСО3 или гашёную известь Са(ОН)2. А в районах действующих вулканов, где с больших глубин поднимаются горячие воды, содержащие минеральные кислоты (соляную, фтористоводородную, борную и другие), иногда текут целые «кислые реки». Так, на Курильских островах есть реки, вода которых — это фактически раствор соляной кислоты с высоким содержанием ионов железа и алюминия.
Историческая справка. В трудах химиков XVI и XVII вв. неоднократно упоминалось о выделении горючего газа при действии кислот на металлы.
В 1766 г. английский учёный Г. Кавендиш собрал и исследовал этот газ и назвал его «горючим воздухом». В 1783 г. знаменитый французский химик А. Лавуазье путём анализа и синтеза доказал сложность состава воды. В 1787 г. Лавуазье определил «горючий воздух» как новый химический элемент и дал ему название hydrogene (по-гречески «хюдор» — вода, «геннао» — рождаю), то есть водород, «рождающий воду». Отсюда и происходит латинское название В.— hydrogenium. В первой таблице атомных весов элементов (1803 г.) вес В., как самого лёгкого из них, был принят за единицу; оказалось, что веса других элементов выражались числами, близкими к целым.
На этом основании английский учёный У. Праут в 1815—16 гг. высказал предположение, что все элементы образованы из В., являющегося первичной материей. Эта гипотеза была оставлена после того, как более точные измерения дали для многих элементов дробные значения атомных весов (шведский химик Я. Берцелиус в 1818 г., швейцарский химик Ж- Стас позднее).
В наши дни, казалось бы, гипотеза Праута возродилась: ядро водорода — протон (по-гречески «прбтос» — первое) действительно входит в состав атомных ядер всех элементов. Однако современное учение о строении атома отнюдь не сводится к догадке Праута, оно неизмеримо шире и глубже (вы убедитесь в этом, прочитав статью Атом).
ВОДОРОД
Атом и молекула водорода. Атом В. состоит из одного ядра (протона) и одного электрона. Это простейший атом, не имеющий аналогов в периодической системе. Теряя электрон, В. образует катион — положительно заряженный ион Н+, то есть, подобно щелочному металлу, проявляет окислительное число 4-1. Однако катион Н+ — «голое» ядро. Эго отличает его от катионов щелочных металлов, вокруг ядер которых расположены электронные оболочки. Иону В., обладающему к тому же очень малым радиусом, ничто не мешает в ходе реакций глубоко проникнуть в электронное облако других атомов, при этом он может соединяться с ними ковалентной связью. Но даже в таком, химически связанном состоянии В. сохраняет способность притягиваться к другим электроотрицательным атомам (например, к кислороду), образуя с ними так называемую водородную связь.
И хотя водородные связи слабее обычных химических связей, они существенно влияют на свойства веществ. Так, благодаря этим связям вода затвердевает при значительно более высокой температуре, чем это предсказывает теория (см. Вода). Водородные связи придают молекулам белков строго определённую пространственную конфигурацию.
Конечно же, никакими такими свойствами щелочные металлы не обладают. Более того, атом В. способен ещё и присоединять электрон, превращаясь в анион, отрицательный ион Н~, и проявляя окислительное число—1. Такой ион имеет электронную оболочку гелия. В этом отношении В. сходен с галогенами, анионы которых также имеют электронные оболочки типа инертных газов. Поэтому в периодической системе элементов В. относят и к I группе, как это сделал Д. И. Менделеев, и к VII группе, где расположены галогены.
Молекула В. состоит из двух атомов, связанных между собой ковалентной связью, которая возникает вследствие обобществления электронов
И- 4-Н. = Н:Н
Атом В., благодаря простоте своего строения, послужил первым объектом, на котором были проверены выводы квантовой механики (см. Атом). А молекула В. послужила первой моделью, на которой проверялись методы квантовой химии, позволяющие вычислять энергии связей между атомами и предсказывать результаты химических превращений.
Физические свойства. В обычных условиях В. имеет плотность 0,0899 г/л; это самый лёгкий газ — он почти в 15 раз легче воздуха! В. сжижается при —252,6 °C и затвердевает при —259,1 °C. Только гелий сжижается при более низкой температуре. Жидкий В.— также самая лёгкая из известных жидкостей: при температуре кипения плотность жидкого В. всего 0,071 г/см3.
106
В. замечателен своей высокой теплопроводностью: нагретое тело, помещённое в атмосферу В., остывает в 6 раз быстрее, чем на воздухе. Это свойство формально роднит В. с металлами. Причина такой высокой теплопроводности В. заключается в том, что его лёгкие молекулы имеют очень большую среднюю скорость теплового движения. И ещё одним удивительным свойством обладает свободный В.: в то время как в воде он растворяется1 очень слабо (около 0,02 объёма на объём Н2О в обычных условиях), его растворимость в некоторых металлах необычайно высока. Например, 1 объём палладия может поглотить до 900 объёмов В.
Растворяют В. и другие металлы, хотя в меньшей мере. Это приходится учитывать в технике. В. заметно (особенно при повышенных температурах) проникает вглубь металла или сплава. Диффузия В. через углеродистый сплав, например через сталь, иногда приводит к разрушению сплава из-за взаимодействия В. с углеродом (в технике этот процесс называют декарбонизацией).
Химические свойства. Химическая активность молекулярного В. в обычных условиях невелика. Но при высоких температурах он распадается (диссоциирует) на атомы, которые чрезвычайно активны. Диссоциация легче происходит в присутствии катализаторов (палладия, платины и др.). Атомарный В. образуется также в различных химических реакциях, например при взаимодействии цинка с соляной кислотой:
Zn 4- 2НС1 = ZnCl2 4- 2Н
Но такой В. существует очень короткое время, а затем соединяется в молекулы Н2. Поэтому в химии часто используют В. именно «в момент выделения» (по-латыни — in statu nascendi), то есть получают его в присутствии того вещества, с которым он должен прореагировать.
В обычных условиях молекулярный В. взаимодействует лишь с наиболее активными элементами: фтором (со взрывом — даже в темноте и при сильном охлаждении), с хлором (только на ярком свету, но лучше при нагревании), с бромом и иодом (неполностью и только при нагревании). Во всех случаях образуются галогеноводороды HX(HF, НС1, НВг, HI), например:
Н2 + С12 = 2НС1
Синтез хлористого водорода из В. и хлора проводится в промышленности.
При нагревании В. взаимодействует со многими элементами. С кислородом образует воду:
2Н2 + О2 = 2Н2О
Горение В. в атмосфере кислорода сопровождается выделением значительного количества теплоты. Смесь В. с кислородом (2 объёма Н2
107
ВОДОРОД
Генри КАВЕНДИШ. Исследовал водород.
Английский физик и химик Генри Кавендиш родился 10 октября 1731 г. в Ницце. По окончании в 1753 г. Кембриджского университета посвятил себя работам в области электричества, теплоты, химии газов. Кавендиш впервые получил в чистом виде «горючий воздух» (водород) и «связываемый воздух» (углекислый газ) и измерил их плотность (1766 г.). Он произвёл очень точные для того времени количественные определения состава атмосферного воздуха (1781 г.) и воды (1784 г.). Пропуская электрические искры через воздух и поглощая продукты реакции (окислы азота) едкой щёлочью, Кавендиш нашёл, что какое-то очень небольшое количество газа не соединяется с кислородом. Только в 1894 г. английский учёный У. Рамзай показал, что этот остаток состоит из аргона (точнее, из его смеси с другими инертными газами).
Кавендиш был членом Лондонского королевского общества, Парижской академии наук и других учёных обществ. По словам одного из его биографов, Кавендиш был «самым богатым среди учёных и самым учёным среди богачей». Но обладая огромным состоянием (около 1 миллиона фунтов стерлингов), учёный жил очень скромно и уединённо. Свои доходы он тратил на научные исследования, которые выполнял в собственной, прекрасно оборудованной лаборатории. Умер Кавендиш 24 февраля 1810 г. в Лондоне. В его память в 1871 г. при Кембриджском университете основана Кавендишская лаборатория. Ею заведовали такие знаменитые физики, как Дж. К. Максвелл, Дж. У. Рэлей, Дж. Дж. Томсон, Э. Резерфорд, У. Брэгг. В ней работали виднейшие физики Англии, США и других стран, в том числе советский физик академик П. Л. Капица.
и 1 объём О2) при поджигании сильно взрывается и поэтому носит название гремучего газа. Взаимодействие В. с кислородом (так же, как и со фтором) относится к классу разветвлённых цепных реакций. Их механизм был изучен советским химиком академиком Н. Н. Семёновым. В ходе таких реакций образуются особые активные частицы — радикалы Н, ОН и О (точка над символом означает неспаренный
электрон, О — бирадикал с двумя неспаренными электронами). Радикалы, реагируя с молекулами исходных веществ, дают воду и ещё некоторое количество радикалов, которых, таким образом, становится всё больше и больше:
н + о2=бн+о
6н + н2=н2о+н б+н2 = 6н+н
и т. д. Скорость такой реакции нарастает, как лавина, и в конце концов может произойти сильный взрыв.
С азотом В. реагирует в присутствии катализаторов по обратимой реакции:
3H2 + N2^2NH3
По этой реакции при нагревании и повышенном давлении в промышленности получают аммиак. При повышенной температуре В. реагирует также с серой, селеном и теллуром.
Взаимодействие В. со щелочными и щелочноземельными металлами приводит к образованию гидридов, например:
о +1-1
2Li + H2 = 2LH
Реакция В. со щелочными металлами служит примером процесса, в котором В. играет роль окислителя. Однако это для него нехарактерно, и в большинстве других случаев Он ведёт себя как типичный восстановитель. Так, со слож
ными веществами В. реагирует только как восстановитель, например:
+ 2 0 0 +1
СиО + Н2 = Си + Н2О
Восстанавливает В. и органические соединения, но только в присутствии катализаторов (Pd, Pt и др.).
Действуя в определённых условиях В. на малоценные твёрдые топлива (бурые угли, горючие сланцы), можно превратить их в жидкую смесь углеводородов, подобную нефти. Жидкое топливо, так называемый с и н-тин, образуется также при нагревании смеси В. и окиси углерода в присутствии катализаторов.
Отметим, наконец, что В. входит в состав кислот. Присутствие ионов Н+ оказывает огромное влияние на ход многих химических и биологических процессов.
Получение. В лабораторных условиях В. получают в аппаратах Киппа по реакции цинка с разбавленной (1:5 по объёму) серной кислотой (рис. 1):
Zn + H2SO4 = ZnSO4 + Н2
Чистый В. можно также получить, пропуская пары воды через раскалённые железные стружки:
4Н2О + 3Fe = Fe3O4 + 4Н2
А в так называемых полевых условиях очень удобно получать В. действием воды на гидрид лития:
LiH + Н2О = LiOH+Н2
Этот способ недёшев, но хорош тем, что из сравнительно малого количества реагента получают большой объём газа. 1 кг LiH даёт около 3 м3 Н2 (воду не следует принимать в расчёт: её практически всегда можно найти на месте).
ВОДОРОД
108
Рис. 1. Получение водорода в аппарате Киппа, а — кран закрыт; б — открыт.
Аппарат Киппа — простой и удобный прибор для получения газов. Он имеется, вероятно, в каждой химической лаборатории. Для получения водорода в аппарат загружают цинк и через верхнее отверстие заливают 20%-ную серную кислоту. Если кран аппарата Киппа открыт, то кислота из верхнего шара по трубе сначала попадает в нижний шар, а затем — в средний, где находится цинк. Начинается выделение водорода:
H2SO4 +Zn = ZnSO4 + Н2
Как только кран закрывают, давление водорода в среднем шаре быстро растёт. Под давлением газа кислота уходит обратно в верхний шар, и выделение водорода прекращается. Снаряжённый реагентами аппарат Киппа всегда готов к употреблению; с его помощью можно несколько часов поддерживать нужный ток газа.
/ Наиболее распространённый способ промышленного получения В.— так называемая конверсия метана (главной составной части природного газа):
СН4 + 2Н2О = 4Н2 + СО2
В. образуется также в результате двух промышленно важных процессов: газификации жидкого и твёрдого топлива. Суть первого процесса — термическое разложение предельных углеводородов нефти (пиролиз) с образованием непредельных углеводородов и В., например:
С5Н12 = С5Н10 + Н2 пентан пентен
Во втором процессе углерод реагирует с водой при нагревании, давая смесь В. и окиси углерода («водяной газ»):
н2о + с = н2 + со
Иногда газификацию ведут в присутствии воздуха или чистого кислорода. В зависимости от
этого состав получаемого газа меняется и его используют либо как топливо, либо как химическое сырьё.
Большие количества свободного В. содержат газы, образующиеся при коксовании каменного угля.
Иногда для получения В. применяют электролиз воды, точнее водных растворов кислот или щелочей (чистая вода проводит электрический ток очень плохо). При электролизе воды наряду с В. получают также кислород.
Газообразный В. хранят под большим давлением в баллонах из стали, окрашенных в зелёный цвет.
Применение. Первоначально В. использовали главным образом для наполнения воздушных шаров и дирижаблей. Однако впоследствии от этого отказались, так как В. чрезвычайно опасен в пожарном отношении. Сейчас водородом наполняют лишь исследовательские шары-зонды, несущие научную аппаратуру. И всё же В. продолжает участвовать в полётах, но не на воздушных шарах, а в ракетах как топливо. Здесь используется способность В. сгорать в атмосфере кислорода с выделением большого количества теплоты. Перспективно «сжигание» В. в так называемых топливных элементах, непосредственно преобразующих химическую энергию топлива в электроэнергию.
Дело в том, что горение — это реакция окисления — восстановления, в ходе которой горючее (у нас В.), окисляясь, отдаёт электроны, а кислород, восстанавливаясь, эти электроны присоединяет:
0 0 +1-2
2Н2 + О2 = 2Н2О
о _ .
4Н —4е —4Н +
о
20+ 4е = 2О2~
Этот процесс в принципе можно осуществить таким образом, что перенос электронов будет происходить не непосредственно от атома к атому, а через проводник электрического тока.
Водород как горючее ... (и сам В., и его смеси). Эта тема обсуждается учёными всего мира как чрезвычайно перспективная. Ведь, помимо того, что у В. большая теплотворная способность, он сгорает «чисто» (превращается в воду) и не загрязняет окружающую среду вредными выхлопными газами. В этом огромное преимущество В. перед бензином, керосином и другим «углеродным топливом».
В недалёком будущем предстоит найти такие водородные смеси, которые можно было бы использовать в уже существующих двигателях внутреннего сгорания, в том числе в автомобильных.
109
ВОДОРОДА ПЕРЕКИСЬ
Взаимодействие В. с кислородом применяется для сварки и резки металлов кислородо-водородным пламенем (температура до 2800 °C). А в так называемых атомарно-водородных горелках, дающих температуру до 4000 °C, используется способность молекулярного В. поглощать большое количество теплоты при диссоциации на атомы, а затем снова её выделять при соединении атомов в молекулы.
Проходя через вольтову дугу, струя В. в таких горелках сильно нагревается причём молекула В. частично диссоциирует на атомы. Столкнувшись с какой-нибудь твёрдой поверхностью, атомы В. снова соединяются в молекулы. Поэтому предмет, внесённый в струю атомарного В., сильно раскаляется: выделяющаяся энергия как бы концентрируется на его поверхности.
Обширны и чисто химические области применения В, (они многообразны, но все связаны с его восстанавливающей способностью). Так, В. используют для синтеза аммиака и хлористого водорода. С помощью В. получают особо чистые металлы, нужные полупроводниковой технике. В. широко применяется в промышленности органического синтеза для восстановления самых разнообразных соединений, для получения метилового спирта, твёрдых пищевых жиров из жидких растительных масел, синтетического жидкого топлива.
Рис. 2. Взрыв дирижабля «Гинденбург». Воспламенившись в закрытом резервуаре, смесь водорода с воздухом может вызвать страшной силы взрыв, подобный тому, который погубил в 1937 г. немецкий дирижабль «Гинденбург».
Большие перспективы открывает применение изотопов В.—дейтерия и трития — в качестве термоядерного горючего. Эти изотопы в будущем обеспечат человечество практически неисчерпаемым источником энергии.
Ядра дейтерия при высокой температуре (сотни миллионов градусов) сливаются в ядра гелия. При этом освобождается огромное количество энергии — около 4,19-1011 кдж (10lf ккал) на каждый килограмм. Содержание дейтерия в природном В. относительно мало (ок. 0,015%). Но из-за очень широкой распространённости В. на Земле запасы дейтерия практически безграничны. К тому же для «выделения дейтерия из обычной воды используют такой сравнительно простой процесс, как электролиз. При электролизе растворов легче разлагается вода, содержащая протий, и остаток обогащается дейтерием. Проводя электролиз много раз, можно получить практически чистый дейтерий.
Следует отметить, что пока термоядерную реакцию удаётся осуществить только при взрывах термоядерных («водородных») бомб; на пути проведения управляемой термоядерной реакции учёным и инженерам предстоит преодолеть ещё много различных трудностей. Ожидают, что осуществить' управляемую термоядерную реакцию удастся в ближайшие 20—30 лет.
Тяжёлая вода уже и сейчас играет важную роль в атомной промышленности: ядра дейтерия служат прекрасным замедлителем нейтронов в атомных реакторах.
Итак, В. в природе выполняет важнейшую роль: он служит «топливом» для звёзд, а его ядра, протоны, являются «кирпичиками», из которых построены ядра других элементов. На Земле В. входит в состав воды, без которой невозможна жизнь. Для современного человечества В.— это сырьё для химической промышленности и вещество, необходимое в технике. А для человечества будущего—практически неиссякаемый источник энергии. В. Е. Жвирблис. ВОДОРОДА ПЕРЕКИСЬ Н 2О2 — соединение водорода с кислородом. Бесцветная вязкая жидкость с неприятным «металлическим» привкусом. Плотность 1,450 г/см*3; /пл—0,43 °C, /кип 150,2 °C (при кипении сильно разлагается). При длительном хранении В. п. медленно выделяет кислород, превращаясь в воду: 2Н2О2 = 2Н2О + О2
Разложение ускоряют следы некоторых металлов (Си, Мп, Fe, Pt и др.) и их катионов, а также облучение, нагревание, электрический разряд. А для того чтобы замедлить разложение В. п., к ней можно добавить немного фосфорной кислоты.
В молекуле В. п. атомы связаны так: Н—О—О—Н; пространственный вид молекулы показан на рисунке.
В.п. обладает слабо выраженной кислотной функцией; при взаимодействии с некоторыми основаниями она образует перекиси:
Н2О2 + Ва(ОН)а = ВаО2 + 2Н2О которые можно рассматривать как соли В.п.
ВОЗДУХ
по
фо
0Н
Строение молекулы перекиси водорода Н2О2. Четыре атома, образующие эту молекулу, не лежат в одной плоскости.
Степень окисления атома кислорода в молекуле Н2О2 равна —1, то есть является промежуточной между наиболее характерными для кислорода значениями, —2 и 0. (Как это рассчитать? Степень окисления водорода в его соединениях с неметаллами всегда равна +1- Исходя из электронейтральности молекулы имеем:
2(+1) + 2х = 0
где х — степень окисления кислорода, отсюда х=—1.) Поэтому В. п. в разных условиях может играть роль как окислителя (что более характерно), так и восстановителя. Уравнения соответствующих реакций окисления-восстановления записывают так:
Н2О2 окислитель
Н2О2 восстановитель
Н2О24-2Н + 4-2е = 2Н2О (в кислой среде)
Н2О24~2е = 2ОН~ (в щелочной и нейтральной среде)
Н2О2 — 2е = 2Н + -[-О2 (в кислой и нейтральной среде)
Н2О2 + 2ОН“—2е = О2 + 2Н2О (в щелочной среде)
Можно заметить, что когда В. п. выступает как восстановитель, продуктом реакции всегда является свободный кислород. Приведём примеры конкретных реакций:
НО -1 -1 о -?
окисли- 2KI+H2O2+H2SO4=I2+K2SO4+2H2O
тель
Н2О2 восстановитель
2КМпО4 + 5Н А + 3H2SO4 = K2SO4 +
+ 2 о
+ 2MnSO4 + 5О2 + 8Н2О
В природе В. п. образуется при окислении многих веществ кислородом воздуха. Следы её всегда содержатся в атмосферных осадках. Образуется В. п. и в организмах животных и растений. Но концентрация её чрезвычайно мала: под действием особых ферментов Н2О2 быстро разлагается и окисляет органические вещества.
Один из видов жуков-бомбардиров использует это как орудие защиты. В одной из его желёз содержится раствор В.п. и органического соединения гидрохинона. В другой — раствор ферментов. При контакте этих растворов В.п. разлагается с образованием О2, а гидрохинон окисляется. При этом выделяется теплота. В минуту
опасности механизм защиты срабатывает, и из желёз жука выбрасывается струя жидкости с температурой около 100 °C.
Водный раствор В. п. получил в 1818 г. французский химик Л. Ж. Тенар, действуя разбавленными и охлаждёнными кислотами на перекись бария:
ВаО2 + H2SO4 = BaSO4 + Н2О2
В промышленности В. п. получают электро; лизом раствора серной кислоты, 1 л которого содержит около 300 г H2SO4. На аноде сначала образуется пероксодисерная (надсерная) кислота H2S2O8, которая затем при действии воды даёт В. п. и серную кислоту:
2H2SO4 H2S2O8 + 2Н ++2ё
H2S2O8 + 2Н2О = 2H2SO4 + Н2О2 Применение В. п. разнообразно. Это мягкий отбеливающий агент для перьев, пуха, волос, тканей. Отбеливая (окисляя краситель), В. п. не портит основного материала. В. п.— и пенообразователь при производстве некоторых пластиков, и искусственный «старитель» вин, и дезинфицирующее средство в медицине.
В искусстве же В. п.— «реставратор» старых картин. Свинцовые белила, взаимодействуя в течение многих лет с сероводородом, содержащимся в воздухе, превращаются в PbS — чёрный сульфид свинца. В. п. переводит чёрный сульфид в белый сульфат, возвращая картине «молодость»:
PbS + 4Н2О2 = PbSO4 + 4Н2О
Продают В. п. в виде 3%- и 30%-ных растворов (последний называется «пергидроль»). В. п. не ядовита, но её концентрированные растворы могут вызвать сильные ожоги. В. К. Бельский. ВОЗДУХ — смесь газов, главным образом азота (78,09% по объёму) и кислорода (20,95%), составляющая атмосферу, В В. постоянно находятся также аргон и другие инертные газы, двуокись углерода, пары воды и др. (подробнее см. Атмосфера}. Средняя молекулярная масса В. 29. Масса 1 л сухого В. при нормальных условиях 1,2928 г; растворимость в воде 29,18 см3/л. При нормальном давлении и температуре —192 °C В. превращается в голубоватую жидкость с плотностью 0,96 г/см3.
С древних времён и до 70-х гг. XVIII в. В. считался одним из первоначал всего существующего («стихией»). Лишь работы К- Шееле (1768—73 гг.), Д. Резерфорда (1772 г.), А. Лавуазье (1772—77 гг.) и Г. Кавендиша (1781 г.) показали сложность состава В. (см. Азот, Кислород). Наличие в В. инертных газов было впервые установлено лишь в 90-х гг. XIX в. Изучение физических свойств В. в XVII — XIX вв. привело к открытию газовых законов, обнаружению атмосферного давления, к широкому применению В. в различных пневматических устройствах. В 1877 г. физикам Л.Кайете (Париж) и А. Пикте (Женева) впервые удалось превра
Ill
тить В. в жидкость. , А в 1895 г. немецким инженером К- Линде была создана первая промышленная установка для сжижения В. Благодаря освоению методов сжижения и разделения В. он стал важнейшим промышленным сырьём.
В., а точнее, содержащийся в нём кислород, необходим для дыхания почти всем животным и растениям (кроме некоторых микробов). Недаром говорят: «Нужен, как воздух!» Человек потребляет ежесуточно как минимум около 1 кг пищи и 2 кг воды. За это же время через лёгкие проходит около 12 кг В., из которого извлекается примерно 700 г кислорода. Это значит, что только население нашей страны потребляет для дыхания 4 млрд, т В. в год.
Воздух в технике. Подсчитано, что 97,6% необходимой человечеству энергии даёт сегодня сжигание различных видов топлива при непременном участии кислорода В. В то же время В.— обязательный технологический компонент многочисленных промышленных производств, требующих участия кислорода (получение серной и азотной кислот и многих органических веществ, выплавка металлов и т. д.). В.— единственный неиссякаемый источник получения связанного азота.
Техника использует и физические свойства В.: его применяют для электро- и теплоизоляции, нагревания или охлаждения различных механизмов и аппаратов. Сжатый В. «работает» в многочисленных пневматических
ВОЗДУХ
устройствах (распылители, воздушные тормоза, отбойные молотки), в автомобильных шинах и мячах, в кораблях и автомобилях «на воздушной подушке»...
Сжижение и разделение воздуха. Ещё французский химик А. Лавуазье разделил В. химическим путём на азот и кислород. Нагревая ртуть в замкнутом сосуде с В., ему удалось связать кислород в окись ртути и получить в остатке «чистый» азот. (Спустя более столетия в этом остатке обнаружили и инертные газы.) Прокаливание окиси ртути позволяло «вернуть» связанный кислород в свободное состояние. Понятно, такой способ разделения В. не годился для промышленного применения — и мал золотник, и дорог! Но уже очень давно был известен метод разделения жидкостей, различающихся температурой кипения. Поэтому успешное разделение В. удалось провести, научившись сначала превращать его в жидкость путём глубокого охлаждения. Как известно, чтобы охладить нагретый предмет, нужно отнять у него тепло путём контакта с другим, более холодным телом. Но ведь в естественных земных условиях даже температура—50 °C это трудно достижимый холод, а В. конденсируется чуть ли не при —200 °C!
Решение нашли на первый взгляд неожиданное: задачу охлаждения В. поручили ... самому В.! Известно, что при сжатии газа затраченная на сжатие работа по
Одна из возможных схем установки для сжижения воздуха. Компрессор (1) сжимает воздух до 40—50 атм. Выделившееся при этом тепло отводится в омываемом водой холодильнике (2). Сжатый воздух дополнительно охлаждается очень холодным воздухом, циркулирующим в холодильниках (3). Около 80% сжатого воздуха поступает в детандер (4). Здесь происходит его. расширение с совершением полезной работы и одновременное охлаждение. Этот охлаждённый воздух и используется в холодильниках (3). Около 20% сжатого воздуха, минуя детандер (4), через узкое отверстие (б) поступает в аппарат (6). За счёт происходящего здесь расширения температура снижается уже до —200°C и происходит сжижение воздуха, который через кран (7) поступает на разделение в ректификационную колонну.
ВОЛЬФРАМ
112
вышает энергию молекул и увеличивает температуру газа. Напротив, быстрое расширение газа (особенно, если газ при этом совершает работу) приводит к потере энергии и охлаждению. Это справедливо для всех газов, составляющих В. (заметим, что водород и гелий следуют этому правилу лишь при достаточно низких температурах). Этот-то принцип «самоохлаждения» расширяющихся газов и лежит в основе сжижения В.
После предварительной очистки от пыли, двуокиси углерода, а также от примесей углеводородов и агрессивных окислов азота и серы В. поступает на сжижение (рис.). Жидкий В. в аппарате 6 состоит из азота (/кип —195,8 °C), аргона (—185,9 °C), кислорода (—182,9 °C). Предоставим жидкий В. самому себе. Слегка нагревшись от окружающей среды, он закипит, причём легче всего будет испаряться азот, так что через некоторое время кипящий жидкий В. будет содержать 50—60% кислорода, а газовая смесь над ним обогатится азотом. Если теперь сконденсировать газовую смесь и повторить испарение, обогащение жидкости кислородом увеличится. В пределе, при достаточном числе таких «повторений», можно добиться полного разделения кислорода и азота, а также выделения инертных газов из смеси. На практике такое многократное «повторение» процесса испарение — конденсация проводят в специальных ректификационных колоннах.
Разделение В.— единственный промышленный источник азота и инертных газов (кроме гелия и радона) и один из важнейших источников чистого кислорода.
В последнее время, в связи с выходом человека в космическое пространство и освоением морских глубин, изучаются и уже используются пригодные для дыхания в этих условиях искусственные газовые смеси (искусственный В.), в которых азот заменён другим газом (подробнее см. Гелий). В. Л. Василевский. ВОЛЬФРАМ (лат. Wolframium) W — химический элемент VI группы периодической системы Менделеева; атомный номер 74, атомная масса 183,85. Тугоплавкий тяжёлый металл светло-серого цвета. В природе встречается в виде смеси пяти стабильных изотопов с массовыми числами 180, 182, 183, 184 и 186; изотопы 182W, 184W и 186W — преобладающие (26,41%; 30,64% и 28,41% соответственно).
Ещё в XIV — XVI вв. мастера, выплавлявшие олово, заметили, что при прокаливании одной из руд с углём много олова теряется, уходя в шлак. Руду назвали Wolf Rahm — «волчья пена». Говорили, что вольфрам «похищает олово и пожирает его, как волк овцу». Теперь мы знаем, что «волк» — это примесь минерала вольфрамита (Fe,Mn)WO4, препятствующая выплавке олова из касситерита SnO2.
Другой минерал В.— «тяжёлый камень» (tungsten) был хорошо известен в Англии и Швеции. В 1781 г. шведский химик К- Шееле выделил из тунгстена • (впоследствии названного шеелитом CaWO4) окисел WO3.
183,85 74
W
5d46s2
ВОЛЬФРАМ
Два года спустя испанские химики братья д’Элуяр выделили WO3 из вольфрамита; они же впервые получили сам металл восстановлением WO3 углём. «Вольфрам» — международное название элемента, однако же в США, Англии, Франции и Италии сохраняется и другое его наименование — «тунгстен».
В.— редкий элемент, по содержанию в земной коре (1,3-10~4% по массе) он стоит лишь на 55-м месте. В свободном состоянии в природе В. не встречается. Вольфрамит и шеелит — наиболее распространённые его минералы. В СССР значительные месторождения вольфрамовых руд обнаружены на Урале, Кавказе, в Киргизии и Забайкалье, а также в Амурской области, на Енисее и в других районах.
В.— самое тугоплавкое (за исключением алмаза) и одно из самых тяжёлых (плотность 19,3 г/см3) простых веществ. Плавится В. при 3410 °C, а кипит при 5900 °C. Чистый металл пластичен, хорошо поддаётся ковке и прокатке. Даже при высоких температурах В. испаряется очень мало (что особенно важно для его применения в электрических лампах накаливания).
В соответствии с положением В. в периодической системе Менделеева его атом имеет шесть валентных электронов (конфигурация 5d46s2, см. А том). В соединениях В. проявляет степени окисления от 4-2 до 4-6 (соответственно валентности от II до VI). Наиболее устойчивы соединения шестивалентного В. По химическим свойствам В. чрезвычайно близок к молибдену. В обычных условиях металл химически устойчив. Но при нагревании выше 400 °C он заметно окисляется на воздухе до WO3. При высоких температурах В. окисляется также в газах — NO2, СО, СО2 и в парах воды, например:
W + 3H2O = WO3 + 3H2
Если бы не газовая коррозия, В. мог бы стать образцовым термостойким конструкционным материалом. Ведь из всех тугоплавких металлов он сохраняет наибольшую механическую прочность при высоких температурах. В. научились защищать и от газовой коррозии — его либо легируют (то есть вводят в него добавки Си, Ni, Nb, Та и др.), либо покрывают защитными тугоплавкими окислами, например А12О3 или ВеО.
Фтор реагирует с В. при комнатной температуре, другие галогены — при высоких. При этом в отсутствии влаги или кислорода обра
113
ВОЛЬФРАМ
зуются галогениды В.: WFe, WC16, WBr5, WI2. А в присутствии следов влаги или О2 — оксигалогениды, например:
W + ЗС12 + Н2О = WOC14 + 2НС1
На холоду В. практически не реагирует ни с одной минеральной кислотой, но легко растворяется в смеси HNO3 и HF. Окись WO3 растворяется в едких щелочах с образованием вольфраматов — солей вольфрамовой кислоты:
WO3 + 2NaOH = Na2WO4 + Н2О
Промышленные руды бедны вольфрамом (всего 2% WO3). Поэтому сначала их обогащают — получают вольфрамитовые и шеелитовые концентраты, содержащие 50—60% WO3. Переработку концентратов проводят в три этапа: получение чистой WO3; восстановление WO3 до металла водородом при 900 °C; превращение порошка В. в компактный металл. Первые два этапа для химика обычны. Но получение компактного В. из его порошка потребовало особой технологии, так как вследствие очень высокой тугоплавкости В. не представляется возможным расплавить и отлить его в форму. На помощь пришла металлокерамика, или порошковая металлургия.
Вообще говоря, методы, близкие к современной порошковой металлургии, то есть минующие процессы литья, применялись ещё с древних времён. И даже в гробницах египетских пирамид найдены драгоценные металлы в виде порошков. А знаменитая железная колонна в Дели (Индия) была изготовлена сваркой в сочетании с ковкой — ведь расплавить железо в то время не могли.
Порошковая металлургия в её современной форме была разработана русским химиком-металлургом П. Г. Соболевским для производства ковкой платины в середине 20-х гг. XIX в. (см. об этом в статье Платиновые металлы). Но особенно актуально встала эта проблема в начале XX в., и именно в связи с необходимостью покорить тугоплавкий В.
По способу, предложенному в 1910 г. американским учёным У. Д. Кулиджем, порошок В. прессуют в шта-бики (брусочки), которые спекают при 1300 °C в атмосфере водорода, а затем пропусканием через штабики электрического тока разогревают их до 3000 °C. Зёрна В. свариваются. Такие заготовки подвергают горячей ковке, после чего их протягивают в проволоку или прокатывают в ленты и листы. Вот сколько операций вместо обычного литья!
И хотя описанный метод получения изделий из В. кажется сухо прозаичным, он тонок до ювелирности, и за ним стоит длинная история научных и технических поисков.
Только после того как была разработана технология получения компактного В., стало возможным промышленное применение чистого металла, главным образом в электротехнике и радиотехнике (нити ламп накаливания, электроды для получения плазмы и атомноводородной сварки, выпрямители высокого напряжения, обмотка высокотемпературных электропечей, катоды и антикатоды в электровакуумных и газоразрядных трубках — этот список может быть продолжен).
Но ещё до разработки метода порошковой металлургии В. этот металл вводили в сталь для повышения её твёрдости и прочности. Было известно, что стали, содержащие В., сохраняют твёрдость при нагревании до 700— 800 °C (вместо 200—250 °C для других; распространённых в то время сталей). Работая резцом из вольфрамовой стали, можно значительно повысить скорость резания: инструмент нагревается до температуры красного каления, но твёрдости не теряет. Сейчас около 85% добываемого В. идёт на производство быстрорежущих инструментальных сталей. В сталь В. вводят в виде сплава с железом — ферровольфрама (25—30% Fe), который получают в электрических печах из вольфрамита (см. Ферросплавы).
Важное значение имеют и сплавы, в которых В. является главным компонентом. Самый известный из сверхтвёрдых сплавов — победит. Он состоит из карбида вольфрама WC (около 90%), зёрна которого сцементированы кобальтом. Победит сохраняет прочность при нагревании до 1000—1100 °C, что позволяет увеличивать скорость резания ещё больше — в десятки раз. Замечательно служит для этой цели и стеллит — сплав трёх металлов (W—Со—Сг). А «тяжёлый сплав» (W—Си—Ni) поглощает радиоактивное излучение в полтора раза лучше свинца и применяется для изготовления защитных устройств.
Находят применение и соединения В. (например, Na2WO4 применяют в производстве утяжелённых огне- и водоустойчивых тканей).
Техническое и хозяйственное значение В. очень велико. Развитие атомной и авиационно-космической техники требует всё возрастающего количества высокотемпературных, прочных, устойчивых материалов. В этом отношении В. весьма перспективен, а потому с уверенностью можно сказать, что В.— один из металлов будущего. 3. А. Старикова.
8 хэш
ГАДОЛЙНИЙ (лат. Gadolinium) Gd — химический элемент III группы периодической системы Менделеева из семейства лантаноидов; атомный номер 64.
ГАЛЛИЙ (лат. Gallium) Ga — химический элемент III группы периодической системы Менделеева; атомный номер 31, атомная масса 69,72. Серебристо-белый металл. В природе элемент встречается в виде смеси двух устойчивых изотопов, 69Ga (60,5%) и 71Ga (39,5%).
Интересна история открытия Г. В конце 1870 г. Д. И. Менделеев на основе периодической системы предсказал существование нескольких элементов и
среди них «экаалюминия».Учёный весьма точно описал,какими свойствами должен обладать этот ещё не открытый тогда элемент, и даже предвидел, что он будет обнаружен спектральным методом. И действительно, в 1875 г. П. Э. Лекок де Буабодран открыл при помощи спектроскопа в пиренейской цинковой обманке новый элемент, который по всем своим свойствам был аналогичен «эка-алюминию» Менделеева. Лекок де Буабодран назвал его галлием в честь своей родины — Франции (лат. Gallia). Примечателен следующий факт. Лекок де Буабодран сначала определил плотность Г. 4,7 г/см3, что не совпало с предположением Менделеева (5,9—6,0 г/см3). Менделеев написал во Францию письмо, отметив это несовпадение. Французский учёный повторил опыты и нашёл свою ошибку. Впоследствии он писал: «Профессор из Санкт-Петербурга, никогда не только не державший галлия в руках, но даже не видавший его спектра, оказался прав. Плотность галлия была действительно 5,9! Я думаю ... нет необходимости настаивать на огромном значении подтверждения теоретических выводов Менделеева». Это был триумф периодического закона!
В земной коре Г. распространён больше, чем хорошо известный свинец. Его содержание составляет 1,9-10”3 % по массе. Однако в природе он очень рассеян, и его единственный минерал галлит CuGaS2 — «минералогическая экзотика». Небольшие запасы этого минерала обнаружены лишь недавно в Республике Заир (Африка). Обычно соединения Г. встречаются в качестве примесей в цинковых и алюминие
вых рудах. Бокситы Греции, например, содержат лишь 300 г Г. на 1 т руды.
Г.— металл, мягкий, как свинец. Среди нескольких десятков металлов лишь три имеют очень низкие температуры плавления. Один из этих металлов — Г. Он превращается в жидкость уже от тепла человеческих рук, так как его /пл всего 29,8 °C [другие два металла, о которых шла речь,— это цезий (28,5 °C) и ртуть (—38,9 °C)]. Однако /кип Г. очень высока, 2230 °C.
У жидкого Г. есть два интересных свойства. Во-первых, он имеет склонность к переохлаждению, то есть расплавленный Г. очень долго не застывает, даже если температура снижается ниже его температуры плавления. Во-вторых, при затвердевании Г. расширяется (из прочих металлов такое свойство имеют ещё висмут и сурьма). Очевидно, все эти аномальные свойства связаны с тем, что кристаллическая решётка Г. сильно отличается от тех, которые обычно имеют металлы.
У атома Г. три внешних (валентных) электрона (конфигурация 4s24p\ см. Атом). В своих соединениях он может проявлять степени окисления + 1, +2 и +3 (первые две, однако, весьма нетипичны); соответственно валентности Г. I, II и III. По химическим свойствам Г. весьма сходен с алюминием. Г. устойчив на воздухе и лишь при нагревании выше 260 °C образует белый порошок окиси:
4Ga + ЗО2 = 2Ga2O3
С галогенами Г. реагирует уже на холоду: 2Ga + 3Cl2 = 2GaCl3
Непосредственно соединяется Г. также с серой и элементами V группы (за исключением азота), образуя соответственно сульфид Ga2S3, фосфид GaP и т. д. Г. устойчив по отношению к воде, но активно реагирует с разбавленными кислотами:
2Ga + 6НС1 = 2GaCl3 + ЗН2
2Ga + 3H2SO4 = Ga2 (SO4)3 + 3H2 Сульфат Г. Ga2 (SO4)3 (как и сульфат алюминия) даёт квасцы, например KGa(SO4)2* 12Н2О. Подобно алюминию, Г. растворяется и в щелочах — с выделением водорода и образованием солей (галлатов).
115
ГАЛОГЕНЫ
Гидроокись Ga(OH)3 устойчива по отношению к воде, но хорошо растворима в кислотах. Как и гидроокись алюминия, она растворяется в щелочах. Таким образом, Ga(OH)3 — типичная амфотерная гидроокись. Она является даже «идеально амфотерной», так как диссоциирует по кислотному и основному типу практически в равной степени.
В периодической системе элементы подгруппы Г. (Г., индий и таллий) следуют непосредственно за переходными металлами, поэтому на свойства этих элементов оказывает влияние заполнение предшествующей d-оболочки (так называемое d-сжатие, аналогичное лантаноидному /-сжатию, см. Лантаноиды). Так, при переходе от А1 к Ga атомный радиус несколько уменьшается, а энергия ионизации несколько возрастает. И поэтому у Ga(OH)3 кислотные свойства выражены более отчётливо, чем у А1(ОН)3. В частности, гидроокись Ga, в отличие от гидроокиси А1, растворяется даже в водном растворе аммиака.
Все соединения Ga(III) в водных растворах подвержены сильному гидролизу; раствор при этом приобретает кислую реакцию.
Окислы Г., соответствующие низшим степеням окисления, можно получить путём восстановления Ga2O3:
Ga2O3 + Н2 = Н2О + 2GaO (серый порошок)
Ga2O3 + 4Ga = 3Ga2O (тёмно-коричневый порошок).
Соединения Ga(I) — сильные восстановители.
Мировое производство Г. не превышает пока нескольких десятков тонн. Его выделяют из
Галлий плавится при температуре чуть ниже 30 °C, поэтому его слиток можно расплавить теплом человеческой руки. Расплавленный металл при спокойном (без перемешивания) охлаждении не застывает немедленно, а может месяцами оставаться при комнатной температуре жидким. Как и ртуть, галлий используют для изготовления термометров. Только таллиевые термометры делают не из стекла, а из кварца, так как предназначаются они для измерения высоких температур (до 1000 °C).
отходов полиметаллических руд (главным образом цинковых и алюминиевых) в виде окиси или хлорида. Подсчитали, например, что при выработке 1 млн. т алюминия из отходов этого производства можно получить 20 т Г. Чистый металл выделяют либо электролизом его солей, либо восстановлением окиси углём или водородом при нагревании:
Ga2O3 + ЗС = 2Ga + ЗСО
Широкого применения Г. пока не нашёл, однако открываются довольно хорошие перспективы его использования в различных областях промышленности и техники. Из всех известных металлов Г. находится в жидком состоянии в самом большом температурном интервале (разность между величинами температур кипения и плавления составляет почти 2200 °C!). Поэтому Г. применяют в качестве термометрической жидкости; им заполняют кварцевые термометры для измерения температуры до 1000 °C. Благодаря этому же свойству Г. используют как теплоноситель в атомных реакторах (см. также Висмут). Г. хорошо смачивает неметаллические поверхности, поэтому его наносят на стекло и получают при этом зеркала, прекрасно отражающие свет и способные выдерживать температуры до 500— 600 °C. Иногда Г. применяют вместо ртути в диффузионных вакуумных насосах.
Со многими металлами Г. легко образует сплавы, как правило, очень легкоплавкие. Например, сплав 82% Ga, 12% Sn и 6% Zn плавится всего лишь при 17 °C. Такие сплавы применяют в сигнальной технике, в ювелирном и зубоврачебном деле.
В последнее время большое внимание привлекают соединения Г. с элементами V группы периодической системы, проявляющие хорошие полупроводниковые свойства. Например, на основе оранжевого фосфида GaP создан полупроводниковый источник красного света. Под действием электрического тока он «загорается» в исключительно короткое время (меньше 10“6 сек), что может оказаться полезным для различных быстродействующих устройств. Арсенид GaAs послужил рабочим веществом первого полупроводникового лазера.
В. К- Бельский. ГАЛОГЕНЫ (устаревшее галоиды) — химические элементы главной подгруппы VII группы периодической системы Менделеева: фтор F, хлор С1, бром Вг, иод I и астат At; неметаллы.
Название «галогены» дано этим элементам за способность образовывать с металлами соли, например NaCl (по-гречески «хальс» — соль, «геннао» — рождаю). Название же «галоиды» (по-гречески «эйдос»— вид, образ, подобие, то есть солеподобные, похожие на соль) —
ГАЛОГЕНЫ
116
неправильно; тем не менее оно прижилось, особенно у химиков-органиков. At — радиоактивный, короткоживущий элемент, его химия изучена недостаточно. По одним свойствам он похож на Г., по другим — на металлы. В этой статье мы его рассматривать не будем.
В свободном состоянии Г. состоят из двухатомных молекул: F2, С12, Вг2 и 12. При нормальных условиях F2 и С12 — газы, Вг2 — жидкость, 12 — кристаллическое вещество. На последней электронной оболочке атомов всех Г. находится по 7 электронов (электронная конфигурация s2p6, см. Атом). Поэтому для достижения устойчивой оболочки ближайшего инертного газа (s2p6) они легко присоединяют один электрон. Следовательно, самая характерная реакция атомов Г.— образование однозарядного аниона X” (так часто обозначают Г. в формулах): о _
Х+1е=Х-например О 0 + -
2Na + Cl2 = 2NaCl
Иными словами, Г. проявляют сильные окислительные свойства. Они соединяются с большинством элементов с образованием галогенидов (см. Фториды. Хлориды. Бромиды и Йодиды). Г. не вступают в непосредственное взаимодействие лишь с кислородом, азотом, углеродом и инертными газами (только фтор реагирует с ксеноном). Связи Г. с активными металлами I и II групп периодической системы носят ионный характер, с прочими элементами — ковалентный. Известны соединения Г. друг с другом. (О таких — межгалогенных соединениях— см., например, в статьях Хлор. Иод.)
Вступая в характерные для всех Г. реакции, отдельные элементы, однако, ведут себя по-разному. При переходе от F к I вследствие нарастания числа электронных оболочек увеличивается размер атома (см. таблицу). Валентные электроны всё дальше удаляются от
ядра, и иод гораздо труднее, чем фтор, присоединяет электрон. Иными словами, при переходе в подгруппе сверху вниз окислительная активность свободного Г. (F2, С12, Вг2, 12) падает (сравните по таблице их электроотрицательности). А поэтому каждый предыдущий Г. (более сильный окислитель) вытесняет последующий из его соединений с металлами и водородом, например:
о -1 о -1 С12 + 2К1 = 12 + 2КС1
В периодической системе Менделеева один лишь фтор при образовании химической связи с другими элементами не позволяет не только отнять, но даже несколько оттянуть от себя электроны (ни одного из семи). Все же остальные Г. предоставляют эту возможность другим, более электроотрицательным элементам (тому же фтору или, например, кислороду). Способность к отдаче электрона при переходе в подгруппе сверху вниз возрастает (сравните энергии ионизации отдельных Г., приведенные в таблице). Поэтому в соединениях с кислородом все Г., кроме фтора, имеют положительные степени окисления, вплоть до максимально возможной для элементов VII группы периодической системы (+7): НОС1, С12О3,СЮ2, КВгО3, + 6 +7
С12О6, К5Юв. В этих соединениях связь X—О ковалентная, электроны атомов Г. лишь оттянуты в сторону кислорода. Но для иода, наименее электроотрицательного Г., известны даже такие соединения, в которых он образует положительно заряженные ионы — катионы. Таковы кристаллы INO3 и IPQ4, в узлах решёток которых расположены ионы 1+ и 13+.
Увеличение размеров атомов Г. при переходе по группе сверху вниз обусловливает закономерное изменение и физических свойств простых веществ. От фтора к иоду растёт плотность, повышаются температуры плавления и
Свойства галогенов
к ф S ф К? Атомный номер Атомная масса Конфигурация внешней электронной обо- »х 5 х * г Си Н — о онный адиус ~ (°А)* Электроотрицательность (условные еди- Энергия иониза-. ции (эв)*** Температура кипения (°C) Температура плавления (°C) Внешний вид
лочки < °— S схХ ницы) ** при нормальных условиях
F 9 18,99840 2$22р*? 0,64 1,33 4,0 15,8 —188,13 —219,61 Бледно-жёлтый газ
С1 17 35,453 3$23р5 0,99 1,81 3,0 13,07 — 33,6 —100,98 Жёлто-зелёный газ
Вг 35 79,904 4s24p® 1,14 1,96 2,8 11,844 58,78 -7,2 Тёмно-бурая жидкость
I 53 126,9045 5s25p® 1,33 2,20 2,5 10,45 184,35 113,5 Чёрно-серые кристаллы
* 1 А=10“8 см. ** Электроотрицательность атома — условная величина, характеризующая относительную способность атома в молекуле приобретать отрицательный заряд. *** Энергия ионизации — энергия, необходимая для отрыва от атома одного электрона (в данном случае первого), выраженная в электронвольтах (эв).
117
ГАФНИИ
кипения, углубляется окраска веществ (это показано в таблице).
Галогеноводороды общей формулы НХ при нормальных условиях — бесцветные газы (кроме HF, который кипит при 19,4 °C). Все они хорошо растворимы в воде, их водные растворы — типичные кислоты. При переходе от HF к HI сила кислот возрастает. Это связано с тем, что с увеличением размера иона Х~ ослабляется связь X—Н, и ион водорода отщепляется легче. По той же причине уменьшается термическая устойчивость галогено-водородов.
Вследствие своей высокой химической активности Г. в природе встречаются только в виде солей, которые и служат сырьём для их промышленного получения. Подробнее о каждом из элементов см. статьи Астат, Бром, Иод, Фтор, Хлор. В. К- Бельский.
ГАЛОИДЫ — устаревшее название галоге-нов.
ГАФНИЙ (лат. Hafnium) Hf — химический элемент IV группы периодической системы элементов Менделеева; атомный номер 72, атомная масса 178,49. Серебристо-белый металл. Природный элемент состоит из шести стабильных изотопов с массовыми числами 174, 176—180. В смеси изотопов преобладает 180Hf (35,22%).
В земной коре Г. распространён примерно так же, как молибден и вольфрам (его содержание Ы0“4% по массе). Он принадлежит к числу рассеянных в природе элементов, собственных минералов не имеет и всегда сопутствует цирконию, гораздо более распространённому. Из-за большого сходства с цирконием Г. был открыт лишь в XX в. Его открытие — одна из блестящих страниц истории периодической системы Менделеева.
Неизвестный элемент с порядковым номером (Z) 72 долго и тщетно искали среди редкоземельных элементов. Однако в 1921 г. знаменитый датский учёный Нильс Бор%, обосновавший связь периодического закона со строением атома, показал, что число редкоземельных элементов, следующих за лантаном, должно быть равно 14 и что, следовательно, элемент с Z=72 находится не в III, а в IV группе системы Менделеева и является аналогом циркония. Следовательно, искать его надлежало в минералах циркония. И уже в 1922 г. в Копенгагене голландец Ц. Костер и венгр Г. Хевеши открыли методом рентгеноспектрального анализа новый элемент в норвежской циркониевой руде, давнему название в честь датской столицы (позднелат. Hafnia).
По внешнему виду Г. похож на сталь. Плотность Г. 13,09 г/см3. Он тугоплавок — /пл 2222 °C; /кип .5400 °C. Металл пластичен, легко поддаётся механической обработке. У атома Г. четыре внешних (валентных) электрона (конфигурация 5d26s2, см. Атом). В соединениях он проявляет обычно степень окисления +4 (ре
же +3 и +2) (соответственно валентности IV» III и II). По химическим свойствам Hf и Zr необычно близки. Это объясняется сходством атомов Zr и Hf и сходством ионов Zr4+ и Hf4+ — у них (соответственно) одинаковое расположение электронов и практически одинаковые радиусы. Последнее обстоятельство связано с так называемым «лантаноидным сжатием» (см. Лантаноиды).
Компактный металлический Г. весьма стоек против внешних воздействий. На воз
духе он покрывается с поверхности тонкой прочной плёнкой окиси НЮ2. Более активен Г. при повышенных температурах: он соединяется с галогенами, азотом, углеродом, бором, кислородом, серой и другими неметаллами. Окись НЮ2, бориды HfB и HfB2, нитрид HfN чрезвычайно тугоплавки (плавятся выше 3000 °C), а карбид HfC имеет температуру плавления 3887 СС. По отношению к воде, щелочам и большинству кислот Г. устойчив.
Он взаимодействует только с кислородными кислотами (окислителями) или вступает в реакции в тех случаях, когда образуются комплексные соединения, например с концентрированной серной кислотой:
Hf + 5H2SO4 = H2[Hf(SO4)3] + 2SO2 + 4H2O
Окись HfO2 — белое тугоплавкое вещество (ZnjI около 3000 °C), нерастворимое в воде и в растворах щелочей. В растворимые соединения её можно перевести сплавлением со щелочами:
HfO2 + 2NaOH = Na2HfO3 + Н2О
При этом получаются соли гафниевой кислоты— гафнаты. Гидроокись Г. (гафниевая кислота) Hf (ОН)4 — амфотерное вещество. (Формулу гидроокиси правильнее писать НЮ2-хН2О, так как соотношение окисла и воды непостоянно.)
Выделяют Г. из отходов производства циркония. За этой фразой скрыт кропотливейший труд, использование наичувствительных методов разделения Zr и Hf, основанных на самых малых различиях свойств этих металлов.
Здесь можно упомянуть, например, фракционную кристаллизацию комплексных соединений K2lZrF6] и K2[HfF6] (гексафторцирконат и гексафторгафнат калия); избирательную экстракцию соединений Zr и Hf органическими растворителями и т. д. Металлический Г. восстанавливают из его соединений чаще всего натрием (реакция идёт при 850 °C):
HfCl4 + 4Na = Hf + 4NaCl
ГЕЛИИ
118
А так как при высоких температурах Г. становится весьма активным, то восстановление проводят в закрытых стальных цилиндрах в атмосфере инертного газа аргона. В последнее время для получения особо чистого металла применяют образование тетраиодида Hfl4 и его термическую диссоциацию (подробнее об этом методе см. в статье Титан).
Производство Г. мало даже в мировом масштабе. Это связано, в первую очередь, с небольшими его запасами и трудностями производства. Однако Г. применяется сейчас во многих областях техники. Он обладает повышенной способностью испускать электроны при нагревании (так называемой эмиссионной способностью). А это в сочетании с высокой тугоплавкостью делает металл пригодным для изготовления катодов электронных ламп и других деталей для рентгене- и радиотехники. Благодаря способности Г. поглощать большие количества газов при высоких температурах, он используется в качестве газопоглотителя (геттера) в вакуумных приборах. Тугоплавкие соединения Г., о которых шла речь выше, входят в состав многих жаростойких материалов. Так, например, сплав 80% TiC и 20% HfC плавится лишь при 4200 °C.
Мы уже рассказали, что Zr и Hf — металлы -близнецы, у них очень много общего. Однако в процессах получения ядерной энергии они ведут себя совершенно противоположно. Цирконий не задерживает «тепловых» нейтронов, а Г., наоборот, сильно поглощает их. А именно тепловые нейтроны и вызывают ядерную реакцию деления урана. На атомных электростанциях применяются оба металла. Цирконий служит конструкционным материалом для самих реакторов. А Г. входит в состав регулирующих стержней, замедляющих или останавливающих реакцию. И поскольку назначения обоих металлов прямо противоположны, задача их разделения приобретает особую остроту. В. К. Бельский.
гашёная Известь — гидроокись кальция Са(ОН)2. См. Известь и Кальций.
ГЕЛИЙ (лат. Helium) Не — химический элемент VIII группы периодической системы Менделеева; атомный номер 2, атомная масса 4,00260; инертный газ.
Элемент состоит из 2 стабильных изотопов: 3Не и 4Не. Содержание 3Не в смеси мало; не превышает 1,3-10~4%. Ядра 2Не, иначе называемые альфа-частицами, или гелионами, чрезвычайно устойчивы. Образование ядер £Не из более простых частиц — ядер водорода (протонов) }Н — сопровождается выделением большого количества энергии. Считают, что эта термоядерная реакция служит одним из главных источников энергии Солнца и других звёзд (см. также Водород). Благодаря этому процес
су на звёздах и в межзвёздном пространстве накапливаются очень большие запасы Г., и на Солнце Г. по распространённости занимает второе место после водорода.
На Земле Г. постоянно образуется при распаде урана, тория и других а-радиоактивных элементов, которые в очень небольших количествах входят в состав различных горных пород, например гранитов. Скорость диффузии Г. через твёрдые тела при обычной температуре крайне низка, и поэтому основная часть образовавшегося при радиоактивном распаде Г. на долгое время оказывается включённой в горные породы. По мере их разрушения Г. постепенно проникает в атмосферу и в подземные газы. Вероятно, в подземные газы, которые служат единственным промышленным источником Г. и где его содержание иногда превышает 10%, попадает и Г., образующийся в расплавленной магме. Несмотря на постоянное поступление новых порций Г. в атмосферу, содержание Г. в ней не увеличивается, так как в верхних слоях атмосферы лёгкие атомы Г. постепенно приобретают вторую космическую скорость и рассеиваются в космическом пространстве.
1 м3 воздуха содержит всего около 5 см3 Г., поэтому неудивительно, что впервые, в 1868 г., француз Ж. Жансен и англичанин Дж. Н. Локьер обнаружили Г. не на Земле, а на Солнце (спектральным анализом). История открытия Г. отражена в его названии (греч. «гёлиос» — Солнце). На Земле Г. был найден английским химиком У. Рамзаем в 1895 г. (см. рис. 2 к статье Атмосфера).
Плотность Г. (0,178 г/л) намного меньше плотности воздуха (1,29 г/л), что позволяет наполнять негорючим Г. воздушные шары, дирижабли и т. д. Подъёмная сила Г. составляет около 92% подъёмной силы водорода (проверьте, пользуясь законом Архимеда, правильно ли это). Однако широкому применению Г. препятствуют его редкость и высокая стоимость. Благодаря хи
мической инертности Г. применяют для создания защитной атмосферы при плавке, резке и сварке активных металлов. Г. менее электропровод ен, чем другой инертный газ — аргон, и поэтому электрическая дуга в атмосфере Г. возникает при более высоких температурах. Г. плохо растворим в воде (при 20 °C в 1 л воды растворяется не более 8,8 мл Г.) и в крови. Благодаря этому свойству Г. помогает водолазам бороться с кессонной болезнью.
119
ГЕРМАНИЙ
Известно, что растворимость газа в жидкости пропорциональна его давлению над жидкостью. При увеличении давления воздуха особенно сильно возрастает растворимость азота в крови. В этом — причина кессонной болезни. При быстром аварийном подъёме водолаза на поверхность давление резко уменьшается, и растворённый в крови азот бурно выделяется, кровь «кипит», пузырится (точно так же, как пузырится только что открытая бутылка с лимонадом). А пузырьки газа вызывают закупорку кровеносных сосудов, иногда приводящую даже к смерти пострадавшего.
Растворимость Г. в крови ничтожна, поэтому водолаза, работающего в атмосфере, где азот заменён Г., можно быстро поднимать на поверхность с глубины 100 м и более. Если этим «гелиевым» воздухом наполнить кабину космического корабля, то полёт в нём станет безопаснее. Непредвиденное нарушение герметичности корабля, сопровождающееся резким падением давления в кабине, не вызовет в этом случае у космонавта кессонной болезни. Кроме того, замена азота гелием создаст дополнительный комфорт для космонавта, так как Г. обладает в 6 раз более высокой теплопроводностью, чем азот, и быстрее отводит избыточное тепло от организма. Гелиево-кислородная атмосфера уже была с успехом испытана в американских космических кораблях.
Нидерландский физик Г. Камерлинг-Оннес в 1911 г. впервые получил жидкий Г. Его точка кипения равна —268,93 °C. Ни одна жидкость, кроме Г., не кипит при столь низкой температуре. Жидкий Г.— предмет огромного числа научных исследований.
Если жидкий Г., состоящий только из 4Не (так называемый Г.-1), охладить ниже —270,97 °C, то его свойства резко изменяются, и полученная жидкость (Г.-II) обладает рядом уникальных физических свойств. Г.-П свободно протекает через мельчайшие отверстия и капилляры, не обнаруживая вязкости. Это явление, Так называемая сверхтекучесть, открыто в 1938 г. советским физиком П. Л. Капицей, а объяснено в 1941—1944 гг. советским физиком Л. Д. Ландау. Плёнки Г.-П быстро перемещаются по поверхности твёрдых тел (сверхползучесть), вследствие чего Г.-П быстро покидает сосуды, в которых он хранился.
Г.— единственный элемент, который в жидком состоянии не отвердевает лри нормальном давлении, как бы глубоко его ни охлаждали. Наименьшее давление перехода жидкого Г. в твёрдый 25 атм, при этом /пл равна —272,ГС.
Жидкий Г. незаменим в технике низких и сверхнизких температур (в так называемой криогенной технике). С помощью жидкого Г. вещество удаётся охладить до температур, отличающихся от абсолютного нуля (—273,16 °C) менее чем на Г. При таких температурах характер движения частиц, составляющих вещество, резко меняется. Тепловое движение частиц при глубоком охлаждении резко ослабевает, почти прекращается. Сильно охлаждённые вещества обладают рядом удивительных особенностей. Так, некоторые металлы и сплавы ниже некоторой температуры (называемой критической) становятся сверхпроводниками. Явление сверхпроводимости открыл в 1911 г. Камерлинг-Оннес. Электрический ток
проходит по сверхпроводникам, не испытывая никакого сопротивления. Учёные провели интересный опыт. В кольце из сверхпроводящего металла (Hg) был возбуждён электрический ток. Два года кольцо охлаждали жидким Г. И всё это время сила тока, протекающего по кольцу, оставалась постоянной.
У явления сверхпроводимости большие прикладные возможности. Уже спроектирована, например, линия передачи электроэнергии на большие расстояния по сверхпроводящему кабелю, охлаждаемому жидким Г. Уже созданы сверхпроводящие магниты (соленоиды из сверхпроводящих обмоток), также охлаждаемые жидким Г. А если бы Г. было много и он был дёшев? Многочисленные линии электропередачи, вся воооще электротехника и радиотехника работали бы без энергетических потерь.
С. С. Бердоносов. ГЕРМАНИЙ (лат. Germanium) Ge — химический элемент IV группы периодической системы Менделеева; атомный номер . 32, атомная масса 72,59. Серовато-белые кристаллы с серебристым блеском. В природе элемент представлен пятью устойчивыми изотопами с массовыми числами 70, 72, 73, 74 и 76, среди которых преобладает 74Ge (36,50%).
В 1870 г. Д. И. Менделеев на основе своего периодического закона предсказал существование и свойства экасилиция — ещё неизвестного аналога кремния. А в 1886 г.
немецкий химик К* Винклер, делая анализ минерала аргиродита, выделил из него металл, который оказался совершенно тождественным экасилицию Менделеева. В честь своей родины Винклер назвал новый элемент германием и впоследствии писал: «Вряд ли может существовать более яркое доказательство справедливости учения о периодичности элементов».
Содержание Г. в земной коре составляет 1,4-10~4% по массе и по распространённости в природе он занимает 53-е место. Г. относится к рассеянным элементам. Собственных минералов у него немного, крупных скоплений они не образуют и промышленного значения не имеют. Почти все они (аргиродит AgGeSe, германит Cu3GeS4) представляют собой смешанные сульфиды Г. и других металлов. Обычно соединения Г. входят в состав других минералов как примесь. Довольно много его (до 1 %) в некоторых месторождениях каменного угля и нефти.
Г. имеет плотность 5,327 г/см3, /пл 937,5 °C, /кип 2700 °C. Он довольно твёрд и очень хрупок. В наше время Г. более всего известен как полупроводник.
ГЕРМАНИЙ
ГЕРМАНИЙ
Кристалл Г. построен так же, как и кристалл алмаза (см. Углерод): каждый атом связан с четырьмя другими атомами ковалентными связями. Но, в отличие от углерода, внешние электроны атомов Г. удерживаются ядрами менее прочно и при нагревании или облучении могут отрываться и переходить в межатомное пространство. Чистый Г. при низких температурах практически не проводит электрического тока и этим отличается от металлов. При нагревании Г. проводит ток тем лучше, чем выше температура. В этом отношении Г. ведёт себя иначе, чем металлы, электропроводность которых с повышением температуры падает. Такое поведение типично для полупроводника.
Каков же механизм прохождения тока в полупроводниках? Он может быть двояким. Если проводимость обусловлена движением в кристалле «полусвободных» электронов, то она называется электронной проводимостью (или n-проводимостью). На том месте, откуда ушёл электрон, остаётся «дырка». В эту дырку может перескочить соседний электрон, тогда на его месте возникнет новая дырка и т. д. Теперь, если наложить электрическое поле, то электроны будут перескакивать не случайно, а преимущественно против поля (к положительному полюсу). А дырки будут перемещаться по направлению поля (к отрицательному полюсу). Таким образом, дырки условно можно считать положительно заряженными носителями тока. Проводимость, обусловленную движением дырок, называют дырочной проводимостью (или р-проводимостью). У совершенно чистого Г. электронная и дырочная проводимости одинаковы, потому что число электронов равно числу дырок.
Однако это соотношение можно изменить искусственным путём. С этой целью введём в Г. примесь элемента V группы, например мышьяка или сурьмы. У них на внешней электронной оболочке находится пять электронов, а у Г. четыре; поэтому общее число электронов в полупроводнике превысит число дырок, и электронная проводимость будет преобладать над дырочной. Наоборот, при введении примеси элемента III группы, например галлия или индия, у которых на внешней оболочке находится три электрона, главную роль будет играть дырочная проводимость. Поэтому полупроводниковые свойства очень сильно зависят от чистоты материала.
В соответствии с положением Г. в периодической системе Менделеева его атом имеет четыре внешних (валентных) электрона (конфигурация 4s24p2, см. А том). В соединениях он проявляет степени окисления +4, +2 и —4 (валентности II и IV). На воздухе он практически не изменяется и лишь при нагревании до 700 °C окисляется до двуокиси GeO2 и окиси GeO. При нагревании Г. довольно легко соединяется с галогенами и серой:
Ge + 2С12 = GeCl4 (бесцветная жидкость)
Ge4-2S = GeS2 (белый порошок)
В ряду напряжений Г. стоит после водорода, поэтому вода и бескислородные кислоты на него не действуют (в отсутствие окислителей). Растворяется Г. в разбавленной азотной кислоте:
3Ge + 4HNO3 + Н2О = 3H2GeO3 + 4NO и особенно хорошо — в царской водке:
3Ge+4HNO3+12HCl=3GeCl4+4NO+8H2O
Со щелочами Г. взаимодействует только в присутствии окислителей (например, перекиси во
120
дорода); при этом образуются соли — гер-манаты:
Ge + 2КОН + 2Н2О = K2GeO3 + ЗН2О
Двуокись GeO2 — вещество белого цвета. Это амфотерный окисел: он растворяется и в кислотах, и в концентрированных растворах щелочей:
GeO 2 + 4НС1 = GeCl4 + 2Н2О
GeO 2 + 2NaOH = Na2GeO3 + H2O
Последняя реакция протекает значительно легче первой, то есть кислотная функция окисла выражена сильнее.
Производные Ge (II) неустойчивы и являются довольно сильными восстановителями. Окись GeO — вещество чёрного цвета, плохо растворимое в воде. Это тоже амфотерный окисел: соответствующая гидроокись растворяется и в кислотах и в щелочах. В последнем случае образуются соли — германиты:
Ge(OH)2 + 2NaOH = Na2GeO2 + 2Н2О
Степень окисления —4 Г. проявляет в соединениях с металлами (германиды), напримерMg2Ge. Как и для углерода и кремния, для Г. характерно образование водородных соединений общей формулы Ge„H2n+2, называемых германами. Например, моногерман можно получить действием соляной кислоты на германид магния:
Mg2Ge + 4НС1 = 2MgCl2 + GeH4
К настоящему времени известны германы вплоть до Ge9H20. Все они представляют собой неустойчивые ядовитые газы.
Несмотря на огромную потребность в Г., мировое производство его пока исчисляется лишь сотнями тонн. Это объясняется прежде всего малым содержанием его в земной коре и чрезвычайной рассеянностью. Добывают Г. из золы каменных углей и отходов производства цветных металлов. Принципиальная схема его получения такова. Обогащённое сырьё обрабатывают соляной кислотой и получают легко летучий GeCl4. Его очищают дистилляцией и подвергают гидролизу:
GeCl4 + 2Н2О = GeO2 + 4НС1
Полученную двуокись восстанавливают водородом при 700—1000 °C
GeO2 4~ 2Н2 — Ge Ч- 2Н2О
Но на этом процесс не заканчивается. Для основного потребителя Г.— полупроводниковой техники — необходим металл очень высокой чистоты: содержание примесей не должно превышать 10“7 % (например, 1 г Ni может «испортить» 10 т Ge). Поэтому металл далее подвергают тщательной очистке специальными методами. О наличии примесей в Г. можно судить по его электропроводности: чем чище Г., тем меньше его электропроводность.
121 ГИДРОСФЕРА
Главным образом благодаря Г. радиотехника и электроника от громоздких ламповых приборов переходят к компактным портативным полупроводниковым, в которых отсутствуют электронные вакуумные лампы с колпаками, анодами, катодами, сетками, занимающими так много места. Недаром говорят, что без Г. не было бы сегодняшней радиолокации. Практически во всех современных электронных приборах от карманного радиоприёмника до больших вычислительных машин вы найдёте крошечные германиевые диоды и триоды (транзисторы).
Важная область применения Г.— инфракрасные детекторы: как наше обычное стеклянное окно прозрачно для световых лучей, так германиевое «окно» прозрачно для лучей инфракрасных. Кристаллы Г. применяют для изготовления термисторов — приборов для очень точного измерения температуры. Их действие основано на том, что высокое электрическое сопротивление Г. при повышении температуры сильно падает (3—6% на 1 °C). Техника не обходит своим вниманием и соединения Г. Двуокись GeO2 — основа для производства оптических стёкол с высокой преломляющей способностью. В. К- Бельский.
ГИГРОСКОПИЧЕСКАЯ ВОДА, см. Вода. ГИДРИДЫ — соединения водорода с другими, более электроположительными элементами (то есть главным образом с металлами). Соединения водорода с теми неметаллами, электроотрицательность которых выше, чем у водорода,— вода Н2О, хлористый водород НС1, сернистый водород H2S, гидридами не называют (таблица электроотрицательностей приведена в статье Металлы).
По типу химической связи и свойствам различают Г. солеобразные, металлические и летучие. К солеобразным относятся Г. щелочных и щелочноземельных металлов (NaH, СаН2). Характер связи в них ионный, водород содержится в форме отрицательного иона Н--. Очень интересно строение металлических Г.— их образуют Sc, Ti, V и многие другие металлы, называемые переходными. Так как атом водорода имеет самые малые размеры среди всех прочих атомов, то он проникает внутрь кристаллической решётки металла и располагается в её пустотах. «Растворяясь» таким образом в металле, как бы внедряясь в него, водород может изменить свойства металла, например повысить его твёрдость и хрупкость. (О переходных металлах см. в статье Закон Менделеева.) Бор, кремний, германий, олово, мышьяк образуют летучие Г.; характер связи в их молекулах ковалентный.
Как способы получения Г., так и области их применения очень разнообразны. Одни Г. получают при нагревании (до 200—600 °C) металлов с водородом (LiH, NaH). Другие — косвенным путём, например действием кислот на некоторые взаимные соединения металлов:
Mg2Sn 4- 4НС1 = 2MgCl2 + SnH4
Иногда Г. образуются как отходы производств. Так, очень ядовитый газ арсин AsH3 выделяется при травлении металлов (Fe, Zn) техническими кислотами (серной, соляной), содержащими примесь As: водород в момент выделения соединяется с мышьяком. На производстве всегда помнят об этом и заботятся о вентиляции и других мерах предосторожности. Применяют Г. и как восстановители в органическом синтезе, и как катализаторы, и для получения особо чистых металлов, и как высококалорийное топливо. Г. многих элементов имеют и собственные названия; об их свойствах и применении см., например, Бороводороды, Кремневодороды. Г. В. Самсонов.
ГИДРОКАРБОНАТЫ — кислые соли угольной кислоты, например NaHCO3. Прежнее название — бикарбонаты. См. также Карбонаты.
ГИДРОСУЛЬФАТЫ — кислые соли серной кислоты, например KHSO4. Прежнее название — бисульфаты. См. также Сульфаты.
ГИДРОСУЛЬФЙДЫ — кислые соли сероводородной кислоты H2S, например Ba(HS)2.
ГИДРОСУЛЬФИТЫ — кислые соли сернистой кислоты, например Ca(HSO3)2. Прежнее название — бисульфиты. См. также Сульфиты. ГИДРОСФЕРА — водная оболочка Земли. Название дано от греч. «хюдор» — вода и «сфайра» — шар.
Если бы приближающиеся к нашей планете космонавты из других миров хотели дать ей имя, они, наверное, назвали бы её не «Земля», а «Вода». Ведь на долю Мирового океана (всех морей и океанов) приходится 70,8% поверхности Земли при средней толщине водного слоя 3,78 км. Все моря и океаны, ледники, реки и озёра, подземные водоёмы и грунтовые воды составляют вместе Г. общей массой 1,5-1018 т и объёмом 1,5 млрд. км3. На долю вод суши приходится около 6% объёма Г., причём подавляющая её часть — подземные и почвенные воды — скрыта от наших глаз. Может показаться неожиданным, что количество воды во всех реках (1200 км3) в 10 раз меньше, чем в атмосфере, и в 5 раз меньше, чем во всех живых обитателях Земли. Около 10% поверхности Земли постоянно занимает лёд (не считая зимнего снежного и ледяного покрова). При равномерном распределении лёд одел бы поверхность планеты панцирем толщиной в 60 м.
Ледяную оболочку Земли часто называют криосферой (греч. «крйос» — холод, лёд). Это — ледники на горных массивах, в Антарктиде и Гренландии, подпочвенный лёд вечной мерзлоты (около 50% всей территории СССР!), а также льды морей и океанов, покрывающие
ГИДРОСФЕРА
122
постоянно около 12% поверхности моря. Огромные запасы пресной воды заключают в себе плавающие ледяные горы — айсберги — целые ледяные острова, достигающие нескольких десятков километров в длину и возвышающиеся над поверхностью океана до 500 м.
Г. имеет огромное значение для формирования кли мата и погоды. Мировой океан — гигантский аккуму лятор тепла. В слое толщиной всего в 1 см поглощаете* 94% солнечной энергии, падающей на поверхность воды. Около 1/ь поглощённого тепла расходуется на испарение воды (450—500 тыс. км3 в год), возвращающейся на поверхность Земли в виде дождя и снега. Если бы годовые осадки были равномерно распределены, то они составили бы слой в 1 м по всей поверхности Земли. Но такой равномерности, конечно, не получается. Средняя температура поверхности океана равна +17,4 °C (от —4 °C в Охотском до+39 °C в Красном море). Это на 3° выше, чем средняя температура у поверхности суши. Такая разница температур вызывает циркуляцию атмосферы, причём суша получает с атмосферными осадками больше воды, чем испаряет. Благодаря этому питаются водой реки и большие количества солей перемещаются с суши в моря и океаны.
Медленно остывая на 1 °C, один объём воды нагревает на 1 °C 3100 объёмов воздуха. Нагреваясь с приходом лета медленнее, чем суша, моря охлаждают атмосферу и делают сезонные изменения погоды менее резкими.
Рис. 1. Вид Земли из Космоса.
«.Если бы приближающиеся к нашей планете космонавты из других миров хотели дать ей имя, они, наверное, назвали бы её не „Земля**, а „Вода**. Ведь на долю Мирового океана приходится 70,8% поверхности Земли.» (стр. 121).
Большое влияние на климат оказывают естественные «холодильники» Земли — полярные льды. «Тепловой экватор» Земли, то есть линия самых высоких температур, проходит много севернее географического экватора (на 10° по долготе) из-за ледяного дыхания Антарктиды.
Г. возникла 2,5—3,5 млрд, лет назад. Предполагают, что при выплавлении мантии — слоя, на котором покоится земная кора,— из глубин земли выходили газы и пары, состоящие из неметаллов и их соединений: Н2, Cl2, N2, СН4, СО2, SO2, инертных газов, NH3, NH4C1, HCl, HF, H2S... Конденсация паров воды и дала начало Мировому океану. Нерастворимые в воде газы — водород, азот, метан, аргон и др.— образовали древнейшую атмосферу Земли, а растворимые газы привели к появлению в океане анионов: SO4“, СО?", Cl", F-... Разрушение горных пород (выветривание) доставляло океану катионы (Na+, Са2+, Mg2+, Li+, К+...).
Выплавление мантии продолжается и в настоящее время. На суше насчитывается до 600 действующих вулканов, а на дне океана — десятки тысяч. Ведь толщина земной коры там всего 5—6 км. Это гораздо меньше, чем на суше (до 40 км). Подсчитано, что вулканы пополняют запасы воды в Г. на 1 км3 в год. Уместно упомянуть, что, кроме воды «земной», поступающей из недр планеты, Земля получает и воду «небесную», в полном смысле слова космическую: поток протонов, идущий от Солнца, в верхних слоях атмосферы захватывает электроны. Образовавшиеся в результате атомы водорода взаимодействуют с атмосферным кислородом, давая воду: 2Н+О=Н2О. Расчёт показывает, что таким путём Земля может получать от 1 до 1,5 км3 воды в год.
Повторяющиеся процессы превращения и перемещения воды на Земле называют круговоротом воды. Вы видите его на рисунке 3 в статье Вода.
В наше время океан содержит около 3,5% растворённых солей: больше всего — галогенидов (88,7% от общего количества), сульфатов (10,8%) и карбонатов (0,3%) натрия, магния, калия, кальция, стронция. Концентрации других солей не превышают 10“6—10“9 % для каждой из них. В воде рек и пресных озёр содержится в среднем около 0,05% солей, причём здесь преобладают карбонаты (до 80% всех солей).
Воды Г. содержат также растворённые газы. Концентрация СО2 в воде в 25 раз выше, чем в воздухе. Известно, что соотношение кислорода и азота в воздухе равно округлённо 21 : 79 (по объёму). В воде (у поверхности) оно меняется (35 : 65), потому что кислород растворяется в ней лучше азота. Это, конечно, не значит, что в воде «легче дышать»: 1 литр воды растворяет лишь 31 мл кислорода, так что
123
ГИДРОСФЕРА
концентрация его в воде в семь раз меньше, чем в воздухе. Но растворённый в воде кислород обеспечивает дыхание многочисленных обитателей Г. Мировой океан, в котором и возникла когда-то жизнь, сейчас является «родным домом» для 10 тыс. видов растений и свыше 150 тыс. видов животных. И именно морские водоросли дают наибольшее количество кислорода, выделяемого в атмосферу растительным миром Земли (см. Биосфера).
Лишь небольшая часть «живого богатства» морей и океанов используется сегодня человечеством. Основную массу живых организмов в Мировом океане составляют не гигантские морские животные — киты. И не 16 тысяч видов морских рыб. 90% биомассы, населяющей моря и океаны, это микроскопические животные и растительные организмы, объединяемые названием планктон. Пока планктон — это богатая белками, углеводами и витаминами пища рыб, моллюсков и даже китов, но не людей или животных суши. Однако вспомните известную одноклеточную водоросль хлореллу. При искусственном выращивании (а такие опыты ведутся во многих странах) хлорелла даёт урожай, измеряемый сотнями центнеров сухой питательной массы с гектара. И эта питательная масса содержит почти впятеро больше белка, чем пшеница.
Недостаточно используются и большие минеральные богатства Г. Общее содержание брома в морской воде в 40 раз выше, чем в земной коре, и составляет 9-Ю13 т, а добыча его— лишь 10б т в год (однако это 70°/о потребностей!). Из 3,8-101в т морской поваренной соли добывают только 8-106 т, что покрывает свыше 25% мировых потребностей. Морская вода уже сегодня даёт сотни тыс. т магния, однако это лишь десятимиллиардная часть его запасов.
Огромные богатства хранятся на дне океана. Большие районы дна покрыты так называемыми железомарганцевыми конкрециями, шариками и лепёшками размером 0,5—20 см. Запасы эти оценивают в сотни и тысячи млрд, т, а ведь конкреции содержат, кроме Fe и Мп, также Си, Ni, Со, редкие элементы. (Кобальта, например, в конкрециях в тысячи раз большеv чем во всех его месторождениях на суше.) Подсчитано, что затраты на организацию добычи этих сокровищ с глубины 4—5 км окупятся в течение нескольких лет. Одна только Австралия добывает со дна океана около 1 млн. т титановых минералов. Подо дном морей скрыты большие нефтяные запасы. Уже в 1970 г. каждая пятая тонна нефти взята со дна морей и океанов. Г. и сейчас — мощный источник энергии, используемый челове-
Рис. 2. Р. Кент. «Ноябрь в Северной Грен? ланди и». Музей изобразительных искусств им. А. С. Пушкина, Москва.
чеством. А после овладения управляемым термоядерным синтезом люди получат до сих пор невиданные количества энергии за счёт использования дейтерия, содержащегося в водах Мирового океана в составе тяжёлой воды.
С развитием промышленности, с ростом использования в сельском хозяйстве ядохимикатов в серьёзную проблему выросло загрязнение гидросферы. Каковы же главные источники её загрязнения? Это — нефтеперерабатывающие и химические, целлюлозно-бумажные и металлургические предприятия. Это — судоходство и сплав леса. Это — и воды, поступающие с сельскохозяйственных угодий и несущие с собой ядохимикаты. Это — и сточные воды городов. В нашей стране принято законодательство и осуществляются эффективные меры по охране природы. К 1980 г. полностью прекратится сброс в гидросферу неочищенных стоков и бытовых сточных вод в бассейне Волги и Урала. Приняты серьёзные меры против загрязнения Каспийского моря и озера Байкал. Новые технологические процессы в промышленности проектируются так, чтобы уменьшить потребление воды и, по возможности, использовать воду в замкнутом цикле, то есть возвращать в производство многократно использованную и затем очищенную воду. Так^ если в 1957 г. свежая вода требовалась нефтеперерабатывающим заводам в количестве 8 м3 на каждую тонну нефти, то в 1960 г. использовалось уже 1,3 м3/т, в 1967 г. — 0,84 м3/т, а на передовых предприятиях — 0,12 м3/т.
ГИПОХЛОРИТЫ
124
Несмотря на обилие воды на Земле, человечество уже сейчас стоит перед трудной проблемой: для самых насущных нужд промышленности и сельского хозяйства нужна пресная вода, а её-то в природе слишком мало. Поэтому особенно большое значение приобретает очистка воды.
Однако, как отмечалось на сессии Верховного Совета СССР в сентябре 1972 г., посвящённой охране природы, какие бы методы получения новых запасов пресной воды ни были бы предложены, рациональное использование воды рек и озёр, вероятно, останется главным направлением решения проблемы.
В. Л. Василевский. ГИПОБРОМЙТЫ — соли бромноватистой кислоты НВгО, например NaBrO. См. Бром. ГИПОСУЛЬФИТ — бытующее название -тиосульфата натрия Na2S2O3. См. Тиосульфаты.
ГИПОХЛОРИТЫ — соли хлорноватистой кислоты НОС1. Известны только Г. щелочных и щелочноземельных металлов — бесцветные вещества, хорошо растворимые в воде, например КОС1, Са(ОС1)2. Как соли слабой кислоты Г. в растворах сильно гидролизованы и имеют щелочную реакцию:
КОС1 + Н2О = НОС1 + к+ + он-
Получить эти соли можно, пропуская хлор в охлаждённый раствор щёлочи:
С12 + 2NaOH = NaCl + NaOCl + Н2О
Выделяющийся газ С12О — причина резкого запаха «хлорки». При действии на неё соляной кислоты выделяется хлор:
Са (ОС1) Cl + 2НС1 = СаС12 + С12 + Н2О
Г., как и хлорноватистая кислота, разлагаются:
2КОС1 = 2КС1 + О2 (на свету)
ЗКОС1 = КС1О3 + 2КС1 (при нагревании)
В. К- Бельский.
ГИПС — минерал состава CaSO4-2H2O (двуводный кристаллогидрат сульфата кальция). В строительстве Г., наряду с другим минералом кальция — ангидритом CaSO4, служит сырьём для получения вяжущих материалов. Такие материалы необходимы при кладке стен из кирпичей или для скрепления отдельных блоков строений. Г. и ангидрит — это так называемые воздушные вяжущие материалы. Они сохраняют прочность на воздухе лишь в отсутствии воды. Поэтому их используют только для внутренней отделки наземных построек. (Есть и другие вяжущие материалы — гидравлические, которые после затвердевания устойчивы в воде, см. Цемент.)
Получение вяжущего строительного материала на основе Г. было известно ещё в глубокой древности. В Древнем Египте за 2,5—3 тыс. лет до н.э. Г. применяли при сооружении пирамид. На Руси его начали использовать для строительства каменных зданий в XII—XIII вв.
Горная порода, состоящая из Г. (гипсового камня), служит сырьём для получения вяжущего материала — алебастра CaSO4-0,5H2O.
Такой раствор, содержащий хлорид и гипохлорит натрия, называется жавелевой водой и служит для отбеливания тканей, выведения пятен и дезинфекции.
При взаимодействии хлора с Са (ОН)2 в растворе получается весьма важный технический продукт хлорная известь (или белильная известь, а в просторечии — «хлорка»):
.О—С1
Са(ОН)2 + С12 = Са< + Н2О ХС1
В промышленности хлорную известь получают пропусканием хлора над сухой гидроокисью кальция. Однако формула СаС1(ОС1) отражает лишь средний состав продукта. На самом деле это смесь СаС12, Са(ОС1)2, Са(ОН)2 и Н2О.
Хлорная известь (белый порошок) обладает сильными окислительными свойствами. Применяется для отбеливания, дегазации, дезинфекции, как средство против отравляющих веществ (ОВ). На воздухе хлорная известь реагирует с углекислым газом:
2СаС1 (ОС1) + СО2 = СаС12 + СаСО3 + С12О
Рис. 1. Музей изобразительных искусств им. А. С. Пушкина в Москве, один из залов. В музее собрана коллекция гипсовых копий (слепков) с выдающихся произведений мирового искусства.
125
ГОРНЫЕ ПОРОДЫ
Рис. 2. Кристалл гипса. В природе минерал гипс часто образует крупные кристаллы-двойники, напоминающие по форме ласточкин хвост.
Обжиг гипсового камня ведут при 140—190 °C:
CaSO4.2H2O^CaSO4.0,5H2O+ 1,5Н2О
Эта реакция обратима. И если алебастр снова смешать с водой, то получится пластичная масса, быстро твердеющая с образованием Г. в виде прочного камня. Такое твердение вяжущих веществ, происходящее в результате присоединения к ним воды, называется гид-ратационным. Оно характерно также для извести и цемента.
При твердении алебастра происходит небольшое увеличение объёма, что очень удобно для получения гипсовых слепков в искусстве, зубопротезировании и др. Слегка расширяясь, Г. точно воспроизводит все мельчайшие детали полой формы. Быстро отвердевающие гипсовые повязки широко применяются в хирургии.
Обжиг Г. при 600—700 °C даёт так называемый ангидритовый цемент, состоящий главным образом из безводного CaSO4. Его иногда называют «мёртвым» Г., так как он проявляет вяжущие свойства лишь в присутствии катализаторов твердения (известь, сульфат и гидросульфат натрия в смеси с железным или медным купоросом). И, наконец, обжигом при 800—1200 °C добиваются «оживления» Г.: при этом CaSO4 частично разлагается и образующаяся известь СаО играет роль катализатора твердения.
При замешивании с водой этот высокообжиговый Г. даёт массу, твердеющую относительно медленно. Поэтому можно до начала твердения («схватывания») успеть утрамбовать получающуюся рыхлую массу, а тем самым уплотнить её и увеличить прочность материала. Изделия, полученные таким образом, чрезвычайно устойчивы к механическим воздействиям (особенно к истиранию), а, кроме того, они тепло- и звуконепроницаемы. Поэтому такой Г. служит, например, материалом для полов.
Полы из гипса, известково-гипсовая штукатурка стен, листы сухой штукатурки, искусственный мрамор, декоративные лепные и скульптурные изделия ... этот перечень можно продолжить. А в смеси с различными заполнителями Г. идёт на изготовление бетонов и цементов. Помимо этого, он используется в производстве бумаги, красок, эмалей, керамических изделий. 3. А. Старикова.
ГЛИНОЗЁМ — то же, что алюминия окись А1аО3. См. также Алюминий.
ГОЛЬМИЙ (лат. Holmium) Но — химический элемент III группы периодической системы Менделеева из семейства лантаноидов; атомный номер 67.
ГОРНЫЕ ПОРОДЫ — плотные или рыхлые геологические тела, слагающие земную кору и состоящие из одного или нескольких минералов. Самая распространённая Г. п.— гранит — содержит в основном зёрна кварца, полевых шпатов и слюд. Эти минералы видны невооружённым глазом. Минеральный состав многих других Г. п. можно установить только под микроскопом. Наука, изучающая Г. п., называется петрографией (от греч. «пётрос» — камень, «графо» — пишу, описываю). Г. п. слагают горы и равнины, дно океанов и морей (рис.). Иными словами, они развиты не только в горах, как можно подумать при буквальном понимании термина «Г. п.»
Большая часть земной коры — это изверженные (магматические) Г. п. Они образовались из огненно-жидкого силикатного расплава, так называемой магмы, при её застывании. Если магма застывала на большой глубине при высоких давлениях, то из неё кристаллизовались Г. п., содержащие крупные минералы, видимые невооружённым глазом. Такие Г. п. называют интрузивными (лат. in-trusio — вторжение). К ним относятся граниты, диориты, габбро и многие другие. Но местами магма изливается по трещинам в земной коре на поверхность земли или на дно морей и океанов. Такая магма называется лавой. На земной поверхности температура и давление низкие, лава застывает быстро с образованием мелкокристаллических пород, называемых излившимися, или эффузивными (лат. effusio — излияние). Среди них наиболее распространены тёмные базальты. Они слагают целые острова в Тихом океане, а в СССР широко распространены в Армении, на Дальнем Востоке и в других районах, где ещё десятки и сотни тысяч лет назад извергались вулканы.
По химическому составу интрузивные и эффузивные Г. п. очень разнообразны — анализ обнаруживает в них 89 химических элементов периодической системы Менделеева. Но всё же в большинстве пород преобладают три элемента — О, Si и А1, составляя вместе более 80% массы Г. п. В базальтах, кроме того, много железа и магния. Большинство металлов находится в изверженных Г. п. в очень малых количествах — тысячных, десятитысячных и ещё меньших долях процента. Например, в базальтах обнаружено около 2-10“3% Си, 4-10“7% Аи, 5-10-б% U.
ГОРНЫЕ ПОРОДЫ
Очень интересны ультраосновные породы— тёмно-зелёные, почти чёрные, изредка встречающиеся на земной поверхности (например, на Урале). Считают, что эти породы застыли из магмы, которая образовалась в слоях, подстилающих земную кору. К этим слоям— таинственной мантии — человеку ещё не удалось проникнуть. Ультраосновные породы сравнительно бедны кремнием (в среднем только 19%, а в гранитах — 32% Si) и очень бедны алюминием (только 0,45% А1), но зато богаты магнием (почти 26% Mg) и часто содержат повышенные количества Ni, Сг, Pt. Недаром Урал
126
славится месторождениями этих ценных металлов! При застывании магмы местами развиваются геохимические процессы, приводящие к концентрации металлов. Часто эти металлы осаждаются из горячих подземных вод, связанных с магмой. Так образуются Г. п., содержащие много минералов Fe, Си, Zn, Со, Mo, Sn. Если из такой Г. п. экономически выгодно добывать металлы, то она называется рудой, а место её скопления — рудным месторождением.
Изверженные Г. п.— прекрасные строительные материалы, особенно они красивы при по-
В сухих степях и пустынях особенно хорошо изучать обнажения горных пород. Ведь там наблюдениям не мешает густая растительность — как у нас в сибирской тайге или в лесных дебрях бассейна Конго и Амазонки. Вы видите на рисунке, что река размыла большую массу горных пород, как бы врезалась в них. Поэтому по обрывам её берега можно судить о геологическом строении массива. В нижней части обрыва залегают светло-серые известняки. Под микроскопом видно (рис. а), что эта порода состоит из остатков скелетов различных мелких животных. Они обитали на дне тёплого моря, бывшего здесь много миллионов лет назад. Но условия изменились, и море ушло. А потом в этом месте протекала большая река и в её долине отлагались красные пески (полуокатанные песчинки при небольшом увеличении показаны на рис. б). Река существовала долго, она успела отложить мощные толщи красных песков и глин. Теперь они обнажаются в обрывах.
Современная небольшая река — возможно, отдалённый потомок той — могучей. И сейчас в русле отлагаются галька и песок — материал будущих осадочных горных пород (конгломератов и песчаников).
Микроскоп — один из самых важных приборов для изучения горных пород, он используется в петрографии уже более 100 лет.
Иногда по внешнему виду трудно определить тип породы, но с помощью микроскопа это сделать легко. На рис. в показан гранит под микроскопом, на рис. г — метаморфическая порода (гнейс).
127
ГРЕМУЧИМ ГАЗ
лировке. Многие замечательные здания и памятники в нашей стране облицованы красным гранитом Карелии и Украины, базальтами Армении.
Г. п. образуются не только на больших глубинах, но и в биосфере — на земной поверхности, на дне морей и океанов. Под влиянием воздуха, воды и живых организмов изверженные Г. п. в биосфере разрушаются, «выветриваются»— они превращаются в песчаные и глинистые частицы.
Химические элементы в биосфере начинают новый сложный цикл миграции, происходит — как говорят геологи — осадочная дифференциация. В реках и морях, там, где течение быстрое, осаждаются пески, часто состоящие из минерала кварца. Они так и называются — «кварцевые пески». А в местах медленного течения или там, где воды застаиваются, осаждаются глинистые частицы, образующие речные, озёрные, морские глины.
И кварцевые пески, и глины — ценные полезные ископаемые. Пески нужны для изготовления стекла и строительных материалов. А обыкновенная глина не менее необходима, чем самые ценные руды металлов: из одних глин изготовляют фарфор, из других кирпич, третьи нужны при бурении нефтяных скважин, в пищевой промышленности и т. д.
Важную роль при образовании Г. п. в биосфере играет разложение остатков растительных и животных организмов. При перегнивании остатков растений образуются угли, а из раковин многих животных — известняки и другие Г. п. (Ещё итальянские учёные эпохи Возрождения знали, что на вершинах Апеннин развиты известняки, состоящие из остатков раковин. Это — свидетельство, что некогда, десятки миллионов лет назад, там простиралось море.) Замечательная особенность биосферы та, что здесь образуются Г. п., состоящие в основном из двух-трёх элементов. Действительно, в углях это в основном С, Н,О, в известняках — С, О, Са (минерал кальцит СаСО3), в скоплениях поваренной соли — Na и С1 (минерал галит NaCl).
Г. п., возникшие в биосфере при осадкообразовании, получили название осадочных. Некоторые из них являются осадочными рудами Fe, Мп, Al, Р и других элементов. Места их скопления называются осадочными месторож
дениями. К ним относятся, например, знаменитые на весь мир месторождения пиролюзита (марганцовых руд) в Грузии (у города Чиату-ра) и на Украине (у города Никополя), месторождения бокситов (алюминиевых руд) в Гвинейской Республике и т. д.
В некоторых участках земной коры происходит опускание осадочных пород на большую глубину, где они подвергаются сильному давлению и нагреванию. Эти процессы называются метаморфизмом (от греч. «метаморфосис» — превращение). Так из песков образуются кварциты, из известняков — мрамор, из глин — глинистые сланцы, и т. д. Изверженные горные породы после своего образования тоже местами метаморфизуются. В результате, например, из гранитов образуются гнейсы. Метаморфические Г. п.— тоже ценные полезные ископаемые. Вспомним, например, о красивых станциях Московского метро — они отделаны мрамором, добытым из различных месторождений СССР.
Мы уже сказали, что металлы добывают из руд. И пока на Земле ещё имеются богатые рудные месторождения, такие Г. п., как граниты, базальты, сланцы, в качестве источника металлов интереса не представляют. Однако учёные предполагают, что месторождения руд многих металлов через десятки (максимум сотни) лет будут исчерпаны. Что ждёт тогда человечество? Оказывается, оснований для опасений нет — источником металлов станут обыкновенные Т. п., количество которых практически неограничено.
Целая армия геологов-петрографов изучает горные породы во всех районах нашей страны.
А. И. Перельман.
ГРАФИТ — одна из аллотропных модификаций углерода.
ГРЕМУЧИЙ ГАЗ — смесь двух объёмов водорода и одного объёма кислорода. При поджигании Г. г. сильно взрывается. В присутствии губчатой платины горит спокойнее. При реакции
2Н2 + О2 = 2Н2О + 572,5 кдж выделяется большое количество теплоты, что позволяет получать высокие температуры (около 2800 °C). Поэтому пламя Г. г. служит для плавки кварца, платины, для газовой сварки и резки металлов и т. д. Для сжигания Г. г. применяют особую горелку, в которой исключается возможность взрыва.
ДИСПРОЗИЙ (лат. Dysprosium) Dy — химический элемент III группы периодической системы Менделеева из семейства лантаноидов; атомный номер 66.
ДИХРОМАТЫ (б и х р о м а т ы) — соли дихромовой кислоты НаСг2О7, например дихромат калия К2Сг2О7.
ЕВРОПИЙ (лат. Europium) Ей — химический элемент III группы периодической системы Менделеева из семейства лантаноидов; атомный номер 63.
ЕДКИЙ НАТР — гидроокись натрия NaOH. См. Натрий.
ЕДКОЕ КАЛИ — гидроокись калия КОН. См. Калий.
ЖЕЛЕЗНЫЕ СПЛАВЫ — сплавы, преобладающим компонентом которых является железо. Наиболее распространённые Ж. с.—железоуглеродистые сплавы: чугун и сталь. Для получения специальных сталей служат ферросплавы. О чугуне, стали и ферросплавах в нашей книге помещены отдельные статьи. Однако есть ещё множество Ж. с. с особыми физическими свойствами; эти сплавы незаменимы во многих областях техники.
Большую группу Ж- с. составляют магнитные сплавы. Известно, например, что в трансформаторах переменного тока перемагничивание сердечника совершается 50 раз в секунду и при этом происходят потери энергии. Чтобы уменьшить их, сердечники трансформаторов изготовляют из так называемых магнитно-мягких материалов, которые легко перемагничиваются; к их числу относятся сплавы Fe с 0,5—4,5% Si и с малым содержанием С. Кроме того, эти Ж. с. обладают высоким электрическим сопротивлением и гасят возникающие в металле сердечника вредные вихревые токи (токи Фуко). Ж. с., содержащие Ni, Со, W, А1, напротив, хорошо сохраняют намагниченность. Их называют магнитно-жёсткими и используют для изготовления постоянных магнитов. В технике используются и магнитострикционные Ж. с., содержащие Ni
и Со. Пластины этих сплавов под действием магнитного поля изменяют свои размеры (удлиняются и укорачиваются). Поэтому, например, сердечник из такого Ж. с. может под действием переменного магнитного поля работать как излучатель ультразвуковых волн.
К Ж. с. с особыми физическими свойствами относят также сплавы с малым коэффициентом термического расширения. Наиболее известны из них инвар (35—37% Ni), платинит (42— 48% Ni) и фернико (32% Ni и 15% Со); остальное — Fe. Эти Ж- с. при нагревании расширяются очень мало (у инвара, например, коэффициент термического расширения почти в 12 раз меньше, чем у стали). Поэтому их применяют в особо точных приборах и для изготовления мер длины.
Мы не имеем возможности рассказать здесь о многих других типах Ж. с. с особыми физическими свойствами (ведь наша Энциклопедия—химическая). Отметим лишь, что на качество этих Ж. с. очень сильно влияют примеси. При получении таких сплавов стараются снизить содержание примесей до минимума, что усложняет технологию их выплавки.
ЖЕЛЕЗО (лат. Ferrum) Fe — химический элемент VIII группы периодической системы Менделеева; атомный номер 26, атомная масса 55,847. Блестящий серебристо-белый металл. В природе элемент состоит из четырёх стабильных изотопов с массовыми числами 54, 56, 57 и 58, среди которых преобладает изотоп 66Fe (91,68%).
Историческая справка.
Первое Ж-, попавшее ещё
в глубокой древности в руки наших предков, было, по-видимому, не земного, а космического происхождения: Ж- входит в состав метеоритов, падающих на нашу планету из космического пространства. Не случайно на «некоторых древних языках Ж. именуется «небесным камнем». Относительно происхождения русского слова
26
55,847
3d64sa
ЖЕЛЕЗО
129
«железо» мнения учёных расходятся и общепринятой точки зрения здесь нет.
Ж. в метеоритах находится в виде металла. Оно сравнительно легко поддавалось обработке, и человек начал изготовлять из него украшения и простейшие орудия. Поскольку нужда в Ж. была постоянной, а метеориты не падали «по заказу», люди стремились научиться извлекать его из земных недр. В земной же коре самородное, металлическое Ж. встречается крайне редко.
Ж- входит в состав многих природных соединений — минералов, из которых слагаются железные руды. Способ получения Ж. из руд был открыт, по-видимому, во 2-м тыс. до н.э. в западной части Азии и вслед за тем распространился в Вавилоне, Египте, Греции. Вначале Ж. получали примитивным, так называемым сыродутным, способом. Для этого куски руды дробили, смешивали с древесным углём и помещали в специальные ямы, вырытые с наветренной стороны на склоне гор или высоких холмов (эти ямы получили название «горнов»). Затем разжигали костёр; при высокой температуре шёл процесс восстановления Ж-— руда превращалась в металл. Температура процесса в этих условиях была ниже температуры плавления Ж-, поэтому получался не жидкий расплавленный металл, а мягкая железная губка; для превращения в монолитный металл и придания нужной формы её подвергали горячей ковке.
По прочности и твёрдости первое рудное Ж. ещё во многом уступало бронзе (см. Медь), а сыродутный процесс был малопроизводительным. Но постепенно технология совершенствовалась, увеличивались размеры горнов, для подачи воздуха начали применяться большие меха, приводимые в движение усилиями сразу нескольких человек. Позднее древние металлурги научились
ЖЕЛЕЗО
путём науглероживания и закалки придавать Ж. нужные свойства. Теперь уже Ж. могло не только успешно конкурировать с бронзой, но и занять главенствующее положение среди материалов, используемых человеком. На смену бронзовому веку пришёл век железный (начало 1-го тысячелетия до н. э.).
Железо в природе. Ж>— один из наиболее распространённых в природе элементов: по содержанию в земной коре (4,65% по массе) оно уступает лишь кислороду, кремнию и алюминию. Учёные подсчитали, что примерно 20 000 000 млрд, т этого элемента сконцентрировано в виде месторождений, пригодных для промышленной разработки.
Важнейшие руды: бурые железняки (основной минерал гидрогетит HFeO2-nH2O, содержащий 63—69% Fe), красные железняки (гематит Fe,Os, 70% Fe), магнитные железняки (магнетит Fe3O4, 72,4% Fe), шпатовые железняки (сидерит FeCOs, 48,2% Fe). Большинство минералов Ж. имеет жёлто-бурый, коричневый, красно-бурый и тёмно-красный цвет.
Не случайно название минерала «гематит» происходит от греческих слов «хайма»(означающего кровь) и «литое»— камень. Старое русское название этого. минерала — «кровавик». Характерно, что и слово «руда», которое когда-то, видимо, относилось только к месторождениям Ж., родственно русскому слову «рдеть» — быть ярко-красным — и украинскому «рудый» — рыжий.
Рис. 1. На Всемирной промышленной выставке в Брюсселе. Атомиум ночью.
«В 1958 г. внимание посетителей Всемирной промышленной выставки в Брюсселе привлекало необычное здание Атомиума. Девять громадных, диаметром 18 м, металлических шаров как бы висели в воздухе: восемь — по вершинам куба, девятый — в центре. Это была модель элементарной ячейки кристаллического а-железа, увеличенная в 165 млрд, раз» (стр. 130).
9 хэш
ЖЕЛЕЗО
130
Наиболее крупные природные запасы руд Ж. находятся в СССР — Курская магнитная аномалия, Криворожское железорудное месторождение, на Урале (горы Магнитная, Высокая, Благодать), в Казахстане (Соколовское и Сар-байское месторождения), на юге Западной Сибири (Горная Шория). Крупными месторождениями Ж. располагают США (район Верхнего озера, Айронспринг, Айрон-Маунтин и др.).
Исследования советских и американских учёных показали, что Ж. в заметных количествах присутствует в лунном грунте. В образцах, доставленных автоматической станцией «Луна-20», Ж- находится в распылённом неокисленном виде. А красноватый оттенок Марса, как полагают учёные, обусловлен тем, что многие горные породы, слагающие эту планету, покрыты окисью Ж.
Физические и химические свойства. Чем объясняется выдающаяся роль Ж. в современной технике? Его широкая распространённость в природе сочетается с очень ценными физическими свойствами. Ж- пластично и легко поддаётся механической обработке (резанию, прокатке, волочению, ковке, штамповке). А путём термической обработки — закалки и отпуска, ему можно придать прочность и твёрдость. Закалке и отпуску, то есть нагреванию и охлаждению при определённом температурном режиме, подвергают в технике не чистое Ж., а сталь — сплав его с углеродом и другими элементами. Основаны же эти процессы на том, что при нагревании и охлаждении Ж. (и сталь) претерпевает полиморфные превращения, на существование которых указал ещё в 1868 г. замечательный русский металлург Д. К. Чернов. В конце XIX — начале XX вв. многими учёными были установлены температурные границы существования, кристаллическая структура и свойства полиморфных модификаций ж.
В 1958 г. внимание посетителей Всемирной промышленнной выставки в Брюсселе привлекало необычное здание Атомиума (рис. 1). Девять громадных, диаметром 18 м, металлических шаров как бы висели в воздухе: восемь — по вершинам куба, девятый — в центре. Это была модель элементарной ячейки кристаллического Ж. (рис. 2,а), увеличенная в 165 млрд, раз. Расположение атомов, соответствующее такой модели, характерно для Ж- в нормальных условиях — эту модификацию Ж. с объёмноцентрированной решёткой называют a-Fe.
Типичное свойство a-Fe — сильный магнетизм. При нагревании до 769 °C Ж. теряет магнитные свойства (эта температура называется точкой Кюри) и остаётся немагнитным при всех
Рис. 2. Кристаллические структуры а- и у-модификаций железа (а и б). Расположение атомов в кристаллах характеризуется трехмерной пространственной периодичностью, иными словами, кристалл имеет решётчатое строение. В кристаллической структуре всегда можно выделить «элементарную ячейку» — параллелепипед, обладающий следующим свойством: весь кристалл можно считать сложенным из таких параллелепипедов, одинаково ориентированных, соприкасающихся целыми гранями и заполняющих всё пространство кристалла. Во всех элементарных ячейках данного кристаллического вещества расположение атомов совершенно идентично.
В простейших случаях элементарная ячейка представляет собой куб. Именно так обстоит дело в кристаллах железа. а — в кубической ячейке а-железа атомы располагаются в вершинах куба и в его центре — такая решётка называется объёмноцентрированной. На рисунке показаны восемь связей, которые образует центральный атом с ближними к нему атомами. Мысленно окружив изображённую ячейку другими такими же ячейками, нетрудно убедиться, что в этой структуре каждый атом имеет 8 ближайших соседей. Такое же строение имеют многие металлы, например ванадий, тантал, a-хром, молибден, барий, щелочные металлы.
б — ячейка ^-железа тоже кубическая, но здесь атомы размещены и в вершинах куба, и в центрах всех его граней — такая решётка называется гранецентрированной. В данном случае каждый атом окружают 12 ближайших соседей (связи на рисунке не показаны во избежание загромождения чертежа). И эта структура весьма типична для металлов; ею обладают медь, серебро, золото, алюминий, платина, палладий и другие металлы.
дальнейших превращениях. Выше769°Си вплоть до 910 °C немагнитное Ж. сохраняет свою кристаллическую структуру. Эту модификацию называют j}-Fe. При 910 °C кристаллическая решётка Ж. перестраивается и становится кубической гранецентрированной: 0-Fe превращается в y-Fe (рис. 2,6). При 1400 °C решётка Ж. вновь становится кубической объёмноцентрированной: y-Fe переходит в б-Fe. Эта мо
131
дификация Ж- сохраняется вплоть до температуры плавления.
Таким образом, у Ж- существуют модификации с широким диапазоном свойств. И, что очень важно, то же относится к сплавам Ж-Меняя режимы термической обработки Ж- и его сплавов, металлу можно придать то или иное строение и изменить тем самым в нужном направлении его твёрдость, пластичность и другие физические характеристики.
Ж. имеет t„, 1539 °C, /кип около 3200 °C, плотность 7,874 г/см3 (при 20 °C).
Конфигурация внешних электронных уровней у атома Ж- 3d’4s2 (см. Атом). В соединениях Ж. проявляет различные степени окисления, из них наиболее устойчивы +2 и +3 (соответственно валентности II и III); максимально возможная степень окисления +6 (валентность VI). Так, с кислородом Ж. образует ряд окислов: FeO, Fe,O3 и Fe3O4 (соединение FeO с Fe3O3). По издавна существующей у нас номенклатуре неорганических соединений их называют соответственно: закись, окись и закись-окись. По новой номенклатуре, более близкой к международной,— оксид железа (II), оксид железа (III), оксид железа (II, III). К соединениям шестивалентного Ж- относятся ферраты — соли не выделенной в свободном состоянии железной кислоты (например, феррат калия K3FeO4).
Ж.— металл средней химической активности. Во влажном воздухе при обычной температуре легко окисляется, покрываясь рыхлой ржавчиной (её состав Fe3O3-nH3O). Вследствие своей пористости ржавчина не препятствует доступу кислорода и влаги к металлу и поэтому не предохраняет его от дальнейшего окисления. По некоторым данным, в прошлом столетии от коррозии гибла чуть ли не половина всего производимого Ж- В наше время успешно разрабатываются методы борьбы с коррозией, и потери от неё удалось снизить до нескольких процентов.
Если Ж- нагреть в сухом воздухе выше 200 °C, оно покроется тончайшей окисной плёнкой, которая защищает металл от дальнейшего окисления при обычных температурах; на этом основан технический метод защиты Ж-— воронение. При нагревании в водяном паре Ж- окисляется с образованием Fe3O4 (или FeO — выше 570 °C) и выделением водорода:
3Fe + 4Н3О = Fe3O4 + 4Н3
Эту реакцию осуществляют, пропуская пары воды через фарфоровую или железную трубку, заполненную стружкой или гвоздями, нагретыми до красного каления. Интересно, что именно эта реакция позволила французскому
ЖЕЛЕЗО
учёному А. Лавуазье установить состав воды. И она же стала основой одного из первых промышленных способов получения водорода (железо-паровой метод).
Гидроокись Fe(OH)2 на воздухе неустойчива; образуется в виде белого осадка при действии едких щелочей или аммиака на водные растворы солей Fe(II) в атмосфере водорода или азота. При соприкосновении с воздухом Fe(OH)a сначала зеленеет, затем чернеет и, наконец, быстро переходит в красно-бурую гидроокись Fe(OH)3. Закись FeO проявляет только основные свойства. Окись FeaO3 амфотерна и потому способна реагировать с основными окислами (например, с MgO). При этом она образует соединения типа FeaO3*MeO — ферриты, широко применяющиеся в радиоэлектронике, автоматике, вычислительной технике (здесь Me — двухвалентный металл, например Ni, Со, Zn, Mg).
Ж. легко вступает в реакцию с галогенами и галогеноводородами, давая соли, например хлориды FeCla и FeCl3. С серой при нагревании Ж. образует сульфиды FeS и FeSa.
Многие соединения Ж. (с О, S и другими элементами) имеют переменный количественный состав (в частности, содержание серы в моносульфиде колеблется от 50 до 53,3 ат.%). Это вызывается дефектами кристаллической структуры вещества. Так, в закиси Ж. часть ионов Fe2+ в узлах решётки замещена ионами Fe3+; чтобы сохранить электронейтральность, отдельные узлы решётки, которые должны принадлежать ионам Fe2+, остаются пустыми, соотношение между числом атомов Fe и О несколько меняется и формула соединения приобретает видРе0,937О. Естественно, для приближённых расчётов это уточнение не принимают во внимание.
В ряду напряжений Ж. расположено до водорода. Оно легко растворяется в разбавленной серной и соляной кислотах с выделением водорода и образованием соответствующих солей:
Fe + 2НС1 = FeCla + Н2
Fe + H2SO4 = FeSO4 + H2
Азотная кислота высокой концентрации пассивирует Ж-, образуя на его поверхности защитную окисную плёнку (поэтому концентрированную HNO3 можно перевозить в железных цистернах). Разбавленная HNO3 растворяет Ж- с образованием ионов Fe2+ и Fe3+ и выделением окиси азота NO и двуокиси азота NO2. В аналитической химии для обнаружения Ж. служит роданид калия KSCN. При его прибавлении к раствору соли Fe(III) жидкость окрашивается в кроваво-красный цвет (рис. 3). Это образуется роданид железа:
FeCl3 + 3KSCN = Fe(SCN)3 + ЗКС1
Реакция настолько чувствительна, что позволяет обнаружить присутствие одной части Fe3+ примерно в миллионе частей воды.
ЖЕЛЕЗО
132
Рис. 3. «Кровь из воды». При сливании разбавленных растворов хлорида железа FeCl3 и роданида калия KSCN образуется кроваво-красный раствор роданида железа Fe(SCN)3. Окраска появляется, даже если в растворе присутствуют ничтожные количества ионов Fe3+, поэтому ее широко используют в аналитической химии для обнаружения железа. Этот опыт любят демонстрировать как пример «чудесных» химических превращений.
Получение и применение. Чистое Ж. получают в сравнительно небольших количествах электролизом водных растворов его солей или восстановлением водородом его окислов. В стадии научной разработки находится способ непосредственного получения Ж. из руд путём электролиза расплавов (подобно алюминию). Получает развитие производство достаточно чистого Ж. прямым восстановлением его из рудных концентратов водородом, природным газом или углём при относительно низких температурах.
Вследствие своей невысокой прочности Ж. используется в технике, как правило, не в чистом виде, а в виде различных по составу и свойствам сплавов. Важнейшие промышленные сплавы Ж.— сталь и чугун (в быту «железными» часто называют стальные или чугунные изделия). Чугун выплавляют в доменных печах, сталь — в кислородных конвертерах, мартеновских и электрических печах. Для производства сталей и сплавов особо ответственного назначения применяются новые процессы — вакуумный и электрошлаковый переплав, плазменная и электроннолучевая плавка и другие. Учёные работают над созданием экономически выгодных способов выплавки стали в непрерывно действующих агрегатах, обеспечивающих высокое качество металла и позволяющих авто
матизировать процесс. Большую группу сплавов Ж. составляют так называемые ферросплавы, которые применяют в металлургии, главным образом при выплавке специальных сталей. Более подробно со сплавами Ж. вы познакомитесь в статьях Чугун, Сталь и Ферросплавы.
Производство Ж. и его сплавов постоянно растёт. В 1974 г. мировая выплавка стали достигла примерно 700 млн. т; из них в СССР выплавлено 136 млн. т.
Применение Ж. насчитывает уже много столетий, но настоящее вторжение Ж. в технику произошло на рубеже XVIII и XIX вв. В 1779 г. через реку Северн (Англия) был построен первый мост из чугуна. В 1818 г. было спущено на воду первое железное судно. Вот что писал в ноябре 1868 г. лондонский «Морской сборник»:
«В Гринкоке ремонтируется сейчас первый в мире железный корабль „Вулкан44. 50 лет тому назад во время пуска его со стапеля народ собрался со всех окрестностей, чтобы посмотреть на чудо — действительно ли корабль, построенный из железа, в состоянии держаться на воде».
Спустя четыре года, в 1822 г., между Лондоном и Парижем начал курсировать созданный в Англии первый железный пароход. Крупным потребителем этого металла стали дороги, названные железными. Первая железная дорога была введена в действие в Англии в 1825 г. Спустя несколько лет железные дороги были сооружены в США, Австрии, Германии, Бельгии, Франции. В 1837 г. начала работать первая в России железная дорога Петербург — Царское Село.
В то же время ещё незадолго до описываемых событий некоторые народы имели о Ж- весьма смутное представление. Знаменитый английский мореплаватель XVIII в. Джеймс Кук рассказывает в своих дневниках, как во время одного из первых путешествий он подарил несколько железных гвоздей жителям тихоокеанских островов. Те долго с недоумением вертели их в руках, а затем, посоветовавшись с верховным жрецом, начали закапывать в землю. Это, в свою очередь, вызвало недоумение у англичан. Тогда туземцы знаками объяснили гостям, что гвозди прорастут и дадут много металлических бананов, с помощью которых их племя победит своих врагов.
Правда, уже вскоре островитяне поняли, что Ж. не следует закапывать в землю, и стали находить ему самое разнообразное применение. Во время последующих путешествий Кук писал об отношении к Ж. жителей Полинезийских островов: «... Ничто так не манило к себе посетителей наших судов, как этот металл; железо всегда было для них самым желанным, самым драгоценным товаром».
Однажды его матросам удалось за ржавый гвоздь получить целую свинью. Из-за этих гвоздей на корабле Кука начали твориться невообразимые вещи: матросы вытаскивали гвозди отовсюду, они даже попытались утащить якорь, чтобы наковать из него новых гвоздей. Кук вынужден был вывесить особый приказ, грозивший самым суровым наказанием каждому, кто решится унести с корабля какой-либо железный предмет.
Всё возраставшая потребность общества в Ж.— строительство железных дорог, корабле
133
ЖЕЛЕЗО
строение, машиностроение, строительное дело, военное дело — обусловила в XIX в. бурное развитие чёрной металлургии (чёрными металлами называют Ж- и его сплавы). Особенно резко увеличилось производство Ж. во второй половине прошлого века, когда были разработаны способы получения литой стали — бессемеровский, томасовский, мартеновский. Характерно, что в наше время на долю Ж- и его сплавов приходится около 95% всех производимых в мире металлов. Машино-
строение и приборостроение, транспорт и энергетика, радиоэлектроника и оборонная тех-
ника — ни одна отрасль современной промышленности немыслима без Ж. Недаром В. И. Ленин назвал этот металл одним из фундаментов цивилизации.
Но не только техника нуждается в Ж. Не будь его на Земле, не смогла бы существовать
жизнь в привычных для нас формах: ведь этот элемент входит в кровь почти всех представителей животного мира нашей планеты. Двухвалентное Ж- содержится в гемоглобине — веществе, обеспечивающем «дыхание тканей».
При недостатке Ж. в организме человек начинает быстро утомляться, возникают головные боли, появляется плохое настроение. Рецепты различных «железных» лекарств были из-
вестны ещё в старину. Многочисленные соединения Ж. широко применяются и в современной медицине.
Ж. необходимо не только животным, но и растениям. Ещё в 1707 г. французский химик Лун Лемери (младший) обнаружил Ж. в золе сожжённых трав. Впоследствии выяснилось, что этот элемент входит в состав всех растений, так как он совершенно необходим для образования хлорофилла. Ж. содержится в дыхательных ферментах и в значительной степени влияет на интенсивность дыхания растений. Любопытно, что только морской планктон потребляет в год полмиллиарда тонн Ж., то есть почти столько, сколько производят его ежегодно все металлургические заводы нашей планеты.
С. И. Венецкий.
О магнетизме железа. Магнитные свойства Ж-были обнаружены ещё в глубокой древности. А его первое «магнитное» применение — в качестве компаса, описано китайским учёным Сума Тзяном более 2 тысяч лет тому назад. Все знают, что магнитный компас служит че
ловечеству до сих пор.
Строго говоря, все вещества и тела природы (от элементарных частиц и атомов до планет, звёзд и галактик) обладают магнитными свойствами, то есть «ощущают» действие магнитного поля или способны сами его создавать. По магнитным свойствам все вещества можно разделить на две большие группы — слабомагнитные и сильномагнитные (или просто магнитные). К слабомагнитным относятся все газы и жидкости, а из простых твёрдых тел, на-.
пример серебро и сера, из сложных — лёд и поваренная соль, кремнезём и резина и т. д. Обнаружить у них проявление магнетизма можно лишь очень тонкими и точными научными методами. Сильномагнитных веществ сравнительно немного: это прежде всего Ж-, никель, кобальт, некоторые лантаноиды', сильномагнитными свойствами обладает ряд сплавов и соединений. Латинское наименование Ж-—ferrum — дало таким веществам общее название— ферромагнетики, а для. комплекса
их свойств — термин ферромагнетизм.
В отличие от всех других веществ, ферромаг-
нетики очень легко и сильно намагничиваются в магнитном поле, сохраняя намагниченное состояние и после выключения поля (одни очень прочно, другие — нет). Те, что долго остаются намагниченными, годятся для изготовления постоянных магнитов. Их примером служат
школьные пособия (U-образные и стержневые постоянные магниты) или магнитная стрелка компаса. Применение ферромагнетиков, разумеется, не ограничивается школьными опытами с железными опилками. Магнитные мате-
риалы постоянно «работают» и в телевизора», и в радиоприёмниках, и в электромоторах, и в трансформаторах, и. вообще в миллионах приборов и устройств радиотехники,
электротехники и т. д.
Причины сильного магнетизма Ж. много столетий казались совершенно загадочными. Они не находили научного объяснения вплоть до 1928 г., когда советский физик Я- И. Френкель и (чуть позднее его) немецкий физик В. Гейзенберг сумели открыть тайну ферромагнетизма. Оказалось, что это явление — квантовое. И, чтобы понять его, необходимы представления квантовой механики, законы которой управляют микромиром. Лишь после «изобретения» и разработки квантовой механики (в 1924—27 гг.) стало возможным объяснить и законы излучения, и магнетизм, и многие другие явления. В потоке открытий тех лет мы выделим открытие спина электрона (сначала в 1925 г. в виде гипотезы для объяснения некоторых особенностей атомных спектров, а затем в 1928 г. строго теоретически). Ока
залось, что электрон, помимо массы и заряда (измеренных ещё в 1911 г.), обладает третьей основной характеристикой — спином. Её можно, весьма и весьма приближённо, трактовать как результат вращения электрона вокруг оси, проходящей через его «центр». Но электрон — это электрический заряд, а его вращение — круговой ток. Поэтому каждый электрон из-за наличия у него спина одновременно является крохотным магнитиком, который весьма условно можно уподобить магнитной стрелке (раз
ЖЕЛЕЗО
134
мером всего лишь 10"13 см!) с полюсами на оси спина.
Ж.— переходный металл, у которого на 3</-уровне есть электроны с неспаренными спинами (см. Атом). А объяснение ферромагнетизма состоит в том, что в Ж. электрическое взаимодействие этих неспаренных &/-электро-нов таково, что их спинам «удобно», точнее энергетически выгодно, располагаться параллельно друг другу. А раз так, то и «магнитные стрелочки» тоже устанавливаются параллельно, например все северные магнитные полюса «вверх», а южные — «вниз». В результате их магнитные силы складываются. У каждого они очень малы, но электронов много: в 1 см3 около 1022 магнитных спинов! Так образуется большой магнит — кусок Ж. Параллельный порядок сохраняется при повышении температуры вплоть до точки Кюри (769 °C). Выше этой температуры ферромагнетизм исчезает, так как тепловое движение (колебания атомов) становится столь интенсивным, что спины уже не могут «держать строй» — ориентация становится хаотической. Тогда говорят, что Ж. размагнитилось. Если затем охладить Ж. ниже точки Кюри, то порядок восстановится.
Надо отметить, что после восстановления параллельного порядка путём охлаждения обычный кусок Ж. (или постоянный магнит) может остаться и размагни
ченным; он намагнитится вновь только после помещения в магнитное поле. Почему? Оказывается, что параллельность спинов восстанавливается не по всему объёму Ж., а по малым объёмам, называемым доменами. В каждом таком домене спины параллельны, но в соседних доменах могут «смотреть» в разные стороны. Для восстановления порядка во всём объёме нужно поместить образец в магнитное поле.
Для полноты картины надо сказать, что к сильно магнитным относят и некоторые вещества, которые «внешне» никакого магнетизма не проявляют. Они называются антиферромагнетиками. В антиферромагнетиках неспаренные спины соседних атомов кристаллической решётки анти параллельны. То есть оси их параллельны, а «смотрят» они в противоположные стороны. В результате компенсации магнитных сил магнетизм во вне не проявляется, хотя внутри кристалла спиновый порядок есть. При повышении температуры выше некоторой критической точки (называемой точкой Нееля) этот порядок разрушается по тем же причинам, что в ферромагнетиках. Примером антиферромагнетиков служат некоторые соединения переходных металлов семейства железа и семейства лантаноидов — таковы окислы (СоО, NiO), галогениды (FeF2, NiCl2), карбонаты (СоСО3).
Наконец, есть и такие вещества, в которых магнитные спины, хотя и смотрят в противоположных направлениях, но не компенсируют друг друга полностью (например, когда «смотрящих вверх» больше, чем «смотрящих вниз»). Тогда опять получается магнит, подобный Ж. пс поведению (но с иным спиновым порядком!). Примерами таких веществ служат фе р р и ты. Из них делают антенны для транзисторов, многие детали в телевизорах и других радиоустройствах, элементы памяти электронно-вычислительных машин и т. п.
А. А. Гусев.
^Железо не только основа всего мира, самый главный металл окружающей нас природы, оно —• основа культуры и промышленности, оно —• орудие войны и мирного труда. И трудно во всей таблице Менделеева найти другой элемент, который был бы так связан с прошлыми, настоящими и будущими судьбами человечества ».
А. Е. Ферсман
(Ферсман А. Е., Занимательная геохимия, 1948, с. 109)
МАНТИЯв
АТМОСФЕРА-
ГИДРОСФЕРА
ЗЕМНАЯ КОРА
2900км
ВНЕШНЕЕ ЯДРО
ВНУТРЕННЕЕ ЯДРО
6371км
5000км
ЗЕМНАЯ КОРА — твёрдая наружная оболочка Земли. Каменный панцирь нашей планеты очень тонок — от 5 км на дне Тихого океана до 70 км под горными массивами на суше (в среднем — всего 0,25% радиуса Земли!). 3. к. находится в постоянном взаимодействии с другими геосферами — атмосферой, гидросферой, биосферой.
Из 105 элементов периодической системы 14 получены искусственно, а количество естественно радиоактивных элементов, астата и франция, исчезающе мало. Таким образом, 3. к. состоит практически из 89 химических элементов. Однако встречаются они не одинаково часто (см. таблицу).
По массе 3. к. более чем на 84% состоит всего из трёх химических элементов — кислорода, кремния, алюминия. А вместе с железом, кальцием, натрием, калием, магнием эти элементы составляют более 99% массы 3. к. Лишь немногие химические элементы встречаются в 3. к. в самородном состоянии (золото, серебро, платина, медь, сера и некоторые другие). Большинство элементов входит в её состав в виде
Рис. 1. Внутреннее строение Земли. Вмасшта-бе всей нашей планеты слой земной коры очень тонок (в среднем всего около 0,25% радиуса Земли, составляющего 6371 км). Соотношение между толщиной земной коры и внутренних слоев Земли на нашем рисунке нарушено — иначе кора выглядела бы тончайшей линией. Под земной корой залегает мантия — до глубины около 2900 км. Почти вся она находится в твердом кристаллическом состоянии и, как полагают, слагается преимущественно из силикатов железа и магния. Ниже мантии лежит ядро. Различают ядро внешнее (до глубины около 5000 км) и внутреннее. По мнению геофизиков, внешнее ядро — жидкое (расплавленное), а внутреннее — твёрдое. Предполагают, что ядро состоит из железа и никеля. Белыми пунктирными линиями на рисунке показаны границы между отдельными слоями. Сами же слои (геосферы) для наглядности условно окрашены в разные цвета. Температура под земной корой предполагается близкой к 600—700 °C, а в ядре — порядка 4000—5000 °C. Плотность земной коры в среднем составляет 2,8 т/м3; мантии 3,3—5,6 т/м3. В ядре плотность резко возрастает, составляя в центре Земли 12,5 т/м3. С глубиной растет и давление — от. 10е н/м2 у верхней границы мантии до 136 109 н/м2 на рубеже между мантией и ядром и до 361 «10е н/м2 в центре Земли.
РАСПРОСТРАНЕННОСТЬ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ В ЗЕМНОЙ КОРЕ
2
3
5
6
И Па ДО ГЛУБИНЫ 16 км (% по массе) Ша IVa Va Via H 1,510'' He
3 Li 3,2’Ю'3 Be 3,8Ю'4 Атомный- номер элемента—" » -74 W 1,3:10'- —Содержание элемента (в% по массе) 5 В l,210'3 6 с 2,ЗЮ'2 ’n l,910'3 8 0 47,0 9 F 6.610'2 10 Ne
Na 2,5 Mg 1,87 П1б IV6 V6 VI6 VII6 । VIII6—1 16 II6 13 Al 8,05 14 Si 29,5 15 p 9,310'2 16 s 4.71O'2 17 Cl l,71O'2 18 Ar
19 К 2,5 20 Са 2,96 21 Sc НО'3 22 Ti 4,510'' 23 V 91O'3 24 Cr 8,310'3 Мп НО'1 26 Fe 4,65 27 Со 1,8-Ю'3 28 Ni 5,8-Ю'3 29 Си 4,710'3 30 Zn 8,ЗЮ'3 31 Ga 1,910 3 32 Ge 1,4-Ю'4 33 As l,71O'4 34 Se 5-Ю'6 35 Br 2,HO'4 36 Kr
37 Rb 1,5Ю'2 38 Sr 3.410'2 39 Y 2,9Ю'3 40 Zr 1,7-Ю'2 41 Nb 210'3 42 Mo 1,MO'4 43 Тс 44 Ru (5Ю'7) 45 Rh (МО'7) 46 Pd 1,ЗЮ'6 47 Ag 7Ю'6 48 Cd 1,ЗЮ'5 49 In 2,5-Ю'5 50 Sn 2,510'4 51 Sb 51O'5 52 Те HO'7 53 1 410'5 54 Xe
55 Cs 3,7Ю'4 56 Ва 6,5Ю'2 57 La* 2,9-Ю’3 72 Hf НО'4 73 Ta 2,51O'4 74 w 75 Re 7-1O'8 76 Os (5-Ю'6) 77 1г (НО'7) 78 Pt (510'7) 79 Аи 4,3-Ю'7 80 Hg 8,ЗЮ'6 81 TI НО'4 82 Pb l,61O'3 83 Bi 91O'7 84 Po (2-1O'14) 85 At 86 Rn (71O''6)
87 Fr 88 Ra (Н0'°) 89 < ** Ас (61О'10) 104 Ku 105 (Ns)
ЛАНТАНОИДЫ
58 Ce 71O'3 59 Pr 91O'4 60 Nd 3,710'3 61 Pm 62 Sm 810'4 63 Eu l,310'4 64 Gd 810-4 65 Tb 4,31O'4 66 Dy 510'4 67 Ho l,710'4 68 Er 3,31O'4 69 Tm 2,710'5 70 Yb 3,310'5 Lu 8-10 5
**АКТИНОИДЫ
90 Th 1,3-1O'3 91 Pa (H0''°) ГО CD s e " о а 93 Np 94 Pu (HO15) 95 Am 96 Cm 97 Bk 98 Cf 99 . Es 100 Fm 101 Md 102 (No) 103
137
ЗОЛОТО
химических соединений, как простых, так и сложных (см. Минералы). Из сложных минеральных агрегатов составлены горные породы.
3. к.— минеральная кладовая человечества, содержащая все необходимые людям в их жизни и производственной деятельности элементы и их соединения.
3. к., как и другие наружные оболочки Земли — атмосфера, гидросфера,—более или менее доступны прямому изучению. Более глубокие недра Земли исследуются лишь косвенными методами. Поэтому обычно, говоря об их строении, составе и свойствах, добавляют слово «предположительно». Об этом надо помнить и рассматривая рисунок 1.
Наиболее достоверные методы исследования глубоких недр Земли — сейсмические. Изучение сейсмических волн (возникающих при землетрясениях) показало, что скорость их распространения в веществе Земли по мере углубления в недра изменяется скачками. Так, удалось установить разделение «твёрдой» Земли на отдельные сферы — 3. к., мантию и ядро — и провести между ними границы. Сделать выводы о химическом составе Земли помогает изучение метеоритов — «небесных камней», которые, как предполагают, подобны веществу глубоких недр Земли. Рассказывает о себе Земля и при вулканических извержениях, когда по каналам и трещинам в 3. к. из недр Земли на её поверхность извергаются лавы, горячие газы и обломки горных пород. В. Л. Василевский.
Распространённость химических элементов в земной коре. Перед вами—несколько необычный вариант системы Менделеева. Здесь наряду с атомным номером элемента приведено число, характеризующее его распространённость в земной коре (в процентах по массе).
Красным цветом отмечены клетки элементов, содержание которых составляет десятки процентов (это — кислород и кремний); оранжевым цветом — элементы, распространённость которых оценивается в целые проценты, зелёным — в десятые, голубым — в сотые доли процента. В синий цвет окрашены клетки тех элементов, содержание которых составляет тысячные доли процента. Клетки остальных элементов оставлены неокрашенными (10~4% и менее). Если распространённость элемента определена недостаточно достоверно; число заключено в скобки. Для инертных же газов (Не, Ne, Аг, Кг н Хе) содержание в земной коре не приводится вообще — настолько неточно оно установлено. Прочерк в клетке сделан у искусственно полученных радиоактивных элементов, которых в естественных условиях в природе вообще нет. Обратите внимание: наиболее распространены химические элементы с небольшими атомными номерами (по 26-й); элементы с чётными атомными номерами, как правило, распространены больше, чем их «нечётные соседи» . Разобраться в причинах этих закономерностей вы сможете лишь после того, как познакомитесь со строением атомного ядра. А пока скажем лишь, что элемент распространён тем больше, чем устойчивее ядро его атома.
Все числа даны по расчётам советского геохимика академика А. П. Виноградова.
НАИБОЛЕЕ РАСПРОСТРАНЕННЫЕ ЭЛЕМЕНТЫ ЗЕМНОЙ КОРЫ
Рис. 2. Земная кора содержит 89 химических элементов. Однако свыше 99?4 её массы приходится на долю восьми элементов (кислород, кремний, алюминий, железо, кальций, натрий, калий, магний).
ЗОЛОТО (лат. Aurum) Au — химический элемент I группы периодической системы Менделеева; атомный номер 79, атомная масса 196,9665. Блестящий жёлтый металл. Элемент представлен одним устойчивым изотопом ™Au.
«Золото было в сущности первым металлом, который открыл человек»,— писал К. Маркс. Раннее знакомство человека с 3. объясняется тем, что оно встречается в природе в свободном виде и прцрлекает глаз необычным красивым жёлтым цветом и блеском. При археологических раскопках часто находят и каменные орудия, и золотые предметы, относящиеся к периоду неолита. В Государственном Эрмитаже (Ленинград) хранятся высокохудожественные золотые изделия скифских ювелиров (VII в. до нашей эры — первые века нашей эры), обнаруженные при раскопках причерноморских курганов. Задолго до нашей эры 3. употреблялось в качестве денег в древнем Китае, Индии, Египте и государствах Месопотамии. За красивый внешний вид и большую химическую стойкость алхимики назвали 3. «царём металлов».
Подчёркивая совершенство 3., алхимики обозначали его знаком Солнца, то есть кругом с точкой в центре Q. Они пытались превратить в 3. «несовершенные» металлы,
золото
138
в частности ртуть. Но все эти попытки оказались бесплодными, так как превратить один элемент в другой химическим путём невозможно. Тайна взаимного превращения элементов разгадана лишь в наше время—и это уже совсем иная,новая область науки — ядерные реакции.В принципе,действительно, можно синтезировать 3., облучая нейтронами изотоп ртути 196Hg. В определённых условиях этим способом можно получить за месяц 1,5 г 3., переработав более 1000 г природной ртути. Но стоимость 3., полученного путём ядерных реак
ций, будет фантастически велика, так что о практическом значении этого способа не может быть и речи.
Золото в природе. Земная кора содержит всего 4,3-10"7% 3. по массе. Это очень мало в относительном исчислении (74-е место среди других элементов) и всё же очень много по абсолютной величине — около 100 млрд. т. В природе 3. чрезвычайно рассеяно. Современная сверхчувствительная аппаратура химиков-аналитиков позволяет обнаружить его всюду — в земле, воде, в организмах растений и животных.
Так, например, 3. нашли в виноградном вине, а 1 т осины содержит ЗмгЗ. В основном 3. находится в природе в свободном виде — это самородное золото, содержащее серебро (от следов до 43%), медь, реже — платиновые металлы и др. Встречается 3. и в виде химических соединений; таков теллурид АиТе2.
Промышленный интерес имеют как коренные месторождения 3., так и россыпи. В коренных месторождениях частицы 3. размером от микрона до миллиметра вкраплены в твёрдые горные породы. При разрушении горных пород 3. вместе с песком и глиной уносится водой в русла рек, где и образуются золотые россыпи. Заметим, что только один Амур выносит в Тихий океан 8,5 т 3. ежегодно. Часто 3. является спутником медных и полиметаллических руд. Промышленными считаются руды, содержащие 2—8 г металлического 3. (и выше) на 1 т руды. В Советском Союзе месторождения золотых руд находятся в Восточной и Западной Сибири, Казахстане, Средней Азии и на Урале. Крупнейшие в мире золотые самородки были найдены в Австралии (от 85,5 до 111,6 кг).
Физические и химические свойства. 3.— очень мягкий (ноготь оставляет на нём след) и тяжёлый (плотность 19,32 г/см3) металл. По ковкости же и тягучести оно превосходит все остальные металлы. Кусочек 3. величиной со спичечную головку можно расплющить в просвечивающий голубовато-зелёным цветом лист
площадью 50 м2. Образец 3. весом 1 г можно протянуть в проволоку длиной 2 км.
Исстари на Руси выделывали «золотую бить» («сусальное золото»). Сначала 3. прокатывали в вальцах до толщины листа писчей бумаги, затем укладывали между тонкими плёнками, снятыми со слепой кишки коровы, и проковывали на наковальне деревянными молоточками. Повторяя эту операцию неоднократно, получали листочки 3. толщиной всего в десятую долю микрона, то есть в 500 атомов (ими покрывали деревянные предметы, кожу и т. д.).
3. плавится при 1064,43 °C, кипит при 2947 °C, образуя жёлто-зелёный тяжёлый пар. Как и другие благородные металлы, 3. довольно летуче и начинает испаряться при температуре ниже точки кипения. Химически 3. весьма инертно, оно замыкает ряд напряжений (ряд активностей) металлов. На воздухе 3. не изменяется даже при нагревании. С кислородом, азотом, водородом и углеродом не реагирует вообще. При комнатной температуре не взаимодействует с галогенами. При нагревании по-
Рис. 1. Золотая маска египетского фараона Тутанхамона (XIV в. до н. э.). Найдена в его гробнице, обнаруженной английским археологом Г. Картером в 1922 г. вблизи древних Фив.
«Лицо было сделано из чистого золота, глаза из арагонита и обсидиана, брови и веки из стекла цвета лазурита. Это лицо напоминало по своей неподвижности маску и в то же время оно былЪ словно живое»
(из книги К. Керама «Боги, гробницы, ученые»).
139
ЗОЛОТО
рошка 3. до 150 °C образуется хлорид АиС13, который при 190 °C выделяет хлор:
2 Au + ЗС12 = 2AuCl3 AuCl3 = AuCl + Cl 2
3. не растворяется в кислотах и щелочах, но поддаётся действию смесей кислот: соляной и азотной (см. Царская водка), серной и азотной, серной и марганцовой:
Au + HNO3 + ЗНС1 = AuCl3 + NO + 2HaO
Из раствора 3. в избытке царской водки при выпаривании выделяются жёлтые кристаллы гидрата золотохлористоводородной кислоты HAuG14-3H2O.
В химических соединениях 3. проявляет степени окисления +1 и +3 (то есть оно одно- и трёхвалентно). Образование высшей валентности объясняется тем, что в реакциях атома 3. участвуют как единственный s-электрон внешней валентной оболочки, так и два d-электрона предыдущей оболочки (конфигурация 5d106s; объяснение дано в статье Атом). Соединения 3. непрочны, легко восстанавливаются до металла:
2AuCl3 + 3SnCl2 = 2Au + 3SnCl4
При этом получается красный или розовый коллоидный раствор 3. («кассиев пурпур»). Так можно обнаружить присутствие соединений 3. в растворе.
3. склонно к комплексообразованию. Водный раствор цианида натрия в присутствии кислорода, реагируя с 3., образует комплексное соединение цианоаурат (I) натрия:
2Au + 4NaCN + =
= 2Na [Au (CN)21 + 2NaOH Продукты реакции растворимы в воде. Эта реакция, позволяющая перевести 3. в виде его соединений в раствор, лежит в основе важнейшего промышленного способа извлечения 37 из руд (цианирование).
Получение и применение. Почти до конца прошлого столетия 3. добывали в основном из россыпей. И в наше время немалую часть 3. извлекают из россыпей с помощью драг. При дражном способе разрыхлённую породу промывают водой на шлюзе (наклонном жёлобе), покрытом перфорированными железными листами, деревянными трафаретами или пеньковыми коврами. Пустая порода уносится сильными струями воды, а тяжёлые крупинки 3. остаются на шлюзе.
Сейчас большая часть 3. добывается не из россыпей, а из коренных месторождений. Предварительно измельчённую твёрдую породу обрабатывают раствором цианида натрия. 3. переходит в раствор в виде комплекса
Рис. 2. Алхимик и его помощник за работой в лаборатории. Человеческие фигуры изображены карикатурно, но всё лабораторное оборудование (тигли и приёмники на полу, мехи для раздувания огня, слесарные инструменты на столах и на задней стене, перегонный куб со шлемом и др.) выписано очень тщательно и реально. Фотография с гравюры на дереве (1520 г.).
Na[Au(CN),l. После фильтрации раствора 3. осаждают цинком:
2Na (Au (CN)21 + Zn = NajZn (CN)J + 2Au Значительную часть 3. добывают в качестве побочного продукта при переработке медных и полиметаллических руд (см. Медь).
Древнейшая область применения 3.— изготовление ювелирных изделий, посуды и утвари, а иногда и скульптурных фигур. В знаменитой гробнице фараона Тутанхамона (рис. 1) найдено много золотых предметов (сокровища из этой гробницы привозили в нашу страну в 1973 г. и, возможно, вам удалось их увидеть). Мягкость, необычайная ковкость и тягучесть дают возможность делать из 3. самые различные вещи — массивные чаши и кубки, украшенные изображением исторических и легендарных персонажей, броши и серьги, свитые из тончайших золотых нитей. Никакой другой металл не может так эффектно сочетаться с драгоценными и поделочными камнями, жемчугом, эмалью. Позолоченные архитектурные детали (лепные или резные из дерева) и позолоченная мебель органически входят в пышное и торжественное убранство прекрасных дворцов XVIII в. Такими дворцами знаменит, например, Ленинград и его окрестности (Зимний дворец в самом Ленинграде, Большой дворец в Петергофе, Большой дворец в Пушкине...).
Для золочения предметов из дерева, кожи и других материалов их поверхность специально подготавливают (грунтуют), затем намазывают клеем и покрывают либо сусальным 3., либо тончайшим золотым порошком. А нанесённый сверху особый лак, образуя прозрачную и
золото
140
Рис. 3. Золочёные купола соборов на территории Московского Кремля.
твёрдую плёнку, защищает позолоту от влаги и механических повреждений.
Изделия из металлов прежде золотили так: на них наносили тестообразную амальгаму 3. (сплав его с ртутью); затем накаливали докрасна; при этом ртуть испарялась (/квп 357,25 °C), а слой 3. оставался.
Такое «золочение через огонь» давало очень прочную позолоту. Однако оно давно оставлено из-за чрезвычайной ядовитости ртутных паров. В 1835—1843 гг. при золочении медных листов для куполов Исаакиевского собора в Петербурге от отравления ртутью погибло 60 рабочих.
Изобретение гальванотехники академиком Б. С. Якоби в 1838 г. позволило со временем отказаться от этого вреднейшего производства. Золочение медных листов для куполов собора Христа Спасителя в Москве (в 1854— 1857 гг.) производилось уже гальваническим способом. Электролитом служил раствор комплексного цианида K[Au(CN)a]. Катодом — листы из меди, анодом — листы из цлатины. Источником тока — батареи из гальванических элементов. При прохождении тока 3. осаждалось на медных листах, платиновые же оставались без изменения. Всего было осаждено 493,14 кг 3. Отличие гальванического золочения в наше время состоит в том, что процесс ведут при повышенной температуре, аноды делают либо растворимые — из тонкого листового 3., либо нерастворимые — из графита, а электроэнергию получают от генераторов постоянного тока.
Гальванотехника позволяет получать золотые покрытия-нужной толщины и оттенка. Присутствие в электролите цианида меди сообщает покрытию красный оттенок, цианида серебра— зеленоватый. Гальваническим золочением нанесены покрытия на каркасы рубиновых звёзд Московского Кремля, изготовленные из нержавеющей стали.
В наши дни золотят ответственные радиодетали, что делает их стойкими против коррозии й устраняет переходные сопротивления в местах контактов. 3. прекрасно отражает свет и тепло, поэтому из него производят тонкие плёнки для линз и отражателей в инфракрасной аппаратуре; позолочены были поверхности нескольких искусственных спутников Земли.
Исключительное значение имеют сплавы 3.
Ведь чистое 3. мягко, прочность его невелика, оно легко истирается; например, монета из чистого 3. при обращении теряет каждое столетие пятую часть своего веса. Добавка меди в 3. делает его более твёрдым и упругим.
Вооружась увеличительным стеклом, легко обнаружить на внутренней поверхности изготовленного в СССР золотого кольца пятико
нечную звезду, на фоне которой изображены серп и молот, а также число, например 583 (могут быть и другие числа). Это оттиск государственного клейма, которое показывает «пробу» изделия. Проба 583 указывает, что сплав содержит 58,3% 3., или 583 г в 1000 г сплава
(остальное медь). Пробой выражают содержание 3. в сплавах, применяемых для изготов-
ления и ювелирных изделий, и медалей, и зубных протезов. В сплаве с платиной и другими металлами 3. идёт на изготовление химически
стойкой аппаратуры.
Среди соединений 3. наиболее важна золотохлористоводородная кислота HAuCI4'3H8O. Она входит в состав электролита ванн для
гальванического золочения, применяется для окраски стекла и фарфора, для установления пробы драгоценного металла. Органические препараты 3. служат для лечения волчанки и ревматического артрита. Таковы разнообразнейшие области применения этого замечательного элемента. Большая же часть добываемого 3. служит одним из средств обеспечения государственной валюты. Ю. И. Романькое.
и
ЙЗВЕСТЬ — продукты обжига (и последующей переработки) известняков, мела и других пород, состоящих главным образом из карбоната кальция СаСО3. Различают И. негашёную и гашёную, И. натровую, а также И. белильную (или хлорную).
Известняки — сырьё для получения И.— широко распространены в природе. Они обычно залегают в виде пластов, составляя приблизительно 20% от общего количества осадочных пород. Кроме главного минерала — кальцита СаСО3, известняки содержат примеси минерала доломита CaCO3’MgCO3, глинистых и песчаных пород и др. Обжигом известняка в специальных печах при температуре около 1000 °C получают окись кальция —негашёную И.:
СаСОз = СаО + СО2 (1)
При её взаимодействии с водой (гидратации) образуется гидроокись кальция —г а ш ё н а я И.:
СаО + Н2О = Са (ОН)2 + 649,5 кдж (2)
При «гашении» выделяется так много теплоты, что вода закипает; отсюда другое, теперь уже мало употребляемое название негашёной И.— «кипелка». Водяные пары разрыхляют И., и она превращается в рыхлый пушистый порошок; поэтому гашёная И. называется ещё и «пушонкой».
И., гашёная и негашёная, с незапамятных времён применяется в строительстве для скрепления камней и кирпичей. Обычно для этой цели приготовляют тестообразную массу из И., песка и воды. Механизм твердения гашёной и негашёной И. несколько различен. Негашёная — СаО затвердевает при взаимодействии с водой (уравнение 2). Это так называемое гид-ратационное твердение (см. также Гипс). Продукт реакции — Са (ОН)2 растворим существенно меньше, чем исходное вещество — СаО. А это — достаточное условие для образования прочного скрепления.
Гашёная И. Са (ОН)2 твердеет под действием СО2 воздуха вследствие образования труднорастворимого кристаллического СаСО3 и высыхания:
Са (ОН) s + СО, = СаСОз + Н ,0 (3)
Из-за постепенного выделения воды в зданиях, где использовали И., довольно долго сохраняется сырость.
В этом отличие И. от цемента. Именно известковой массой пользовались древние китайцы при сооружении башен Великой китайской стены (строилась с IV века до н. э.). На Руси И. впервые применили при постройке Десятинной церкви в Киеве (989 г.). Придавая большое значение строительным работам, Иван Грозный в 1584 г. учредил Каменный приказ, которому предписал заниматься добычей и обработкой строительного камня, обжигом кирпича и изготовлением И.
Под действием большого количества воды и углекислого газа воздуха трудно растворимый карбонат кальция может постепенно превратиться в хорошо растворимый бикарбонат:
СаСОз + Н2О + СО2 = Са (НСО3)2 (4)
Рафаэль. Диспут. Фреска (1509 г.). Фрагмент. Эта фреска украшает один из парадных залов Ватиканского дворца.
«Одно из своих древнейших применений известь нашла во фресковой настенной живописи» (стр. 142).
индий
142
Это приводит к постепенному разрушению сооружений. Поэтому ещё древние мастера старались улучшить водоустойчивость известковых смесей, добавляя к И. цемянку — толчёный кирпич, щебень. В наше время водоустойчивую, так называемую гидравлическую, И. получают из известняков, содержащих много глины (6—20%). В этом случае при обжиге наряду с СаО образуются и более сложные соединения кальция — силикаты (2CaO-SiO2), алюминаты (СаО-А12О3) и ферриты (CaO-Fe2O3). Они-то и придают И. способность сохранять прочность в воде. Поэтому гидравлическую И. используют для строительства сооружений, подвергающихся длительному действию воды.
Одно из своих древнейших применений И. нашла во фресковой настенной живописи.
Фреска — это картина или орнамент, написанные по сырому грунту — известковой штукатурке водяными или приготовленными на известковом растворе красками. На Руси начиная с конца X в. этим способом расписывали церкви (например, в Киеве, Новгороде). Штукатурка для грунта — это особым образом приготовленная гашёная И. Она наносится на стену непосредственно перед росписью. При высыхании фреска покрывается тонкой плёнкой СаСО3 (см. уравнение реакции 3), хорошо предохраняющей изображение от разрушения. Такая живопись требует от художника точного, уверенного письма, ибо грунт быстро высыхает, и тогда уже ничего исправить нельзя. Поэтому художник покрывает грунтом только такой участок стены, какой может расписать за один день. К качеству штукатурки-грунта предъявляли большие требования, готовили его тщательно и долго (в Древней Руси — иногда несколько лет). Но эти усилия с лихвой окупались прочностью фрески, её устойчивостью к изменению атмосферных условий на протяжении многих сотен лет.
Использование гашёной и негашёной И. далеко не исчерпывается строительным делом и искусством: она необходима в металлургии, в химической промышленности, в производстве бумаги, стекла, сахара, кожи, в сельском хозяйстве, для водоочистки и т. д. Мировая выработка И. исчисляется десятками млн. т.
Если при гашении И. заменить воду раствором NaOH, то получится так называемая натровая И.— смесь главным образом Са(ОН)2 (83%) и NaOH (5%). Её приготовляют прибавлением к концентрированному раствору NaOH измельчённой свежепрокалённой СаО и выпариванием полученной массы досуха. Натровую И. широко применяют в лабораториях для поглощения СО2.
При действии газообразного хлора на сухую И.— пушонку, получается хлорная (белильная) И. См. Гипохлориты. 3. А. Старикова. ЙНДИЙ (лат. Indium) In — химический элемент III группы периодической системы Менделеева; атомный номер 49, атомная масса 114,82. Серебристо-белый металл. В природе элемент представлен двумя изотопами: 1131п (4,33%) и 1161п (95,67%). Второй изотоп
очень слабо радиоактивен (период полураспада 6-1014, или 600 тыс. млрд. лет!). Испуская электроны (₽”-частицы), он превращается в один из изотопов олова:
“4n-0-=“6Sn
И. был открыт в 1863 г. немецкими химиками Ф. Райхом и Т. Рихтером.
Исследуя с помощью спектрального анализа цинковые руды из окрестностей Фрейберга, они пытались найти в них незадолго перед этим открытый элемент таллий. В спектре одного из образцов учёные обнаружили две тёмно-синие линии, не принадлежащие ни одному из известных тогда элементов. По цвету линии были похожи на применявшийся ещё в древние времена краситель индиго; отсюда и название нового элемента. Сначала И. считали двухвалентным и его атомный вес принимали равным 75. Только в 1869—70 гг. Д. И. Менделеев и независимо от него немецкие учёные Л. Мейер и Р. Бунзен правильно определили и атомный вес И., и его положение в периодической системе.
Содержание И. в земной коре 2,5-10"6% по массе (63-е место по распространённости). Это элемент крайне рассеянный: его собственные минералы очень редки.
Чистый И.— светлый металл с высокой степенью «серебристости» — его способность отражать световые лучи значительно выше, чем у серебра. И. легкоплавок, его /пл 156,2 °C, /кип2075 °C. Он гораздо мягче свинца — палочка И. оставляет на бумаге ещё более чёткий след, чем свинцовая. Плотность И. 7,362 г/см3. И. и его соединения окрашивают пламя газовой горелки в сине-фиолетовый цвет.
В соответствии с положением И. в периодической системе Менделеева его атом имеет три внешних (валентных) электрона (конфигурация Ss^p1). В соединениях он может иметь степени окисления +1, +2 и +3 (валентности I, II и III); последняя наиболее характерна.
На воздухе И. устойчив, а при нагревании окисляется кислородом до жёлтой окиси 1п2О3; энергично взаимодействует с серой:
2In + 3S = In2S3
При комнатной температуре реагирует с хлором и бромом, образуя 1пС13 и 1пВг3. И. устойчив по отношению к воде, однако в присутствии окислителей медленно корродирует:
41п + ЗО2 + 6Н2О = 41п (ОН)3
И. стоит в ряду напряжений до водорода и легко вытесняет последний из кислот, окисляясь при этом до 1п(+3):
2In + 3H2SO4 = In2(SO4)3 + ЗН2
В щелочах И. практически не растворяется.
143
ИНЕРТНЫЕ ГАЗЫ
Хотя гидроокись 1п(ОН)з и проявляет амфотерные свойства, однако, в отличие от гидроокисей алюминия и галлия, кислотная функция выражена у неё гораздо слабее. Гидроокись 1п(0Н)3 — белое, не растворимое в воде (но хорошо растворимое в кислотах) вещество. Она лишь медленно взаимодействует с концентрированными растворами щелочей, при этом образуются соли — и н даты.
Соединения И. низших степеней окисления довольно неустойчивы. Соли 1п(+2) распадаются с образованием солей 1п(+3) и 1п(+1):
21пС12 = InCl3 + InCi
Такая реакция самоокисления — самовосстановления называется диспропорционированием. Галогениды 1п(+1) и окисел 1п2О — очень сильные восстановители.
Мировое производство И. ничтожно мало — лишь несколько десятков тонн в год. Это связано прежде всего с тем, что соединения И. в природе очень рассеяны, а его запасы незначительны. Получают И. как побочный про^ дукт при переработке сернистых руд цинка, свинца, меди. Соединения И. переводят в раствор и восстанавливают либо цинком:
2In3+ + 3Zn = 2In + 3Zn2+ либо путём электролиза.
Однако скромные масштабы производства И. вовсе не говорят о скромных запросах промышленности. Наоборот, этот металл применяется и будет широко применяться в различных областях техники. Известно, что зеркала из различных материалов неодинаково отражают различные лучи. Вот почему цветные одежды «в зеркале» имеют несколько иную окраску, чем на самом деле. Такой недостаток имеют и серебряные, и ртутные зеркала. И. же наиболее равномерно отражает световые волны всех длин; поэтому и сам металл, и его сплавы с серебром применяют для изготовления различных зеркал в точных приборах и для покрытия рефлекторов. Такое покрытие не тускнеет со временем, и его коэффициент отражения остаётся постоянным. Ввиду стойкости И. по отношению к воздуху и воде он используется и как антикоррозионное покрытие для различных металлов.
Большую роль в технике играют различные сплавы И.
Диапазон их применения — от подшипников до кабинета стоматолога (добавка И. увеличивает прочность сплавов для пломбирования зубов). Введение небольшого количества И. в сплавы меди повышает их стойкость против морской воды. В то же время И.— один из компонентов легкоплавких сплавов, находящих широкое применение и в промышленности, и в быту (см. также Висмут). Так, сплав 24% In и 76% Ga плавится при 16 °C. В медицине вместо гипса иногда пользуются сплавом 18,1% In, 41,0% Bi, 22,1% Pb, 10,6% Sn, 8,2% Cd, имеющим /пл 47 °C.
Большое внимание привлекают соединения И. с элементами V группы периодической системы. Многие из них обладают полупровод
никовыми свойствами: антимонид InSb, арсенид InAs, фосфид InP. Последний применяют в выпрямителях, работающих при повышенных температурах. A InSb и InAs способны изменять своё электросопротивление под действием инфракрасных лучей, что используется в термопеленгации (определение местонахождения предмета по его теплоизлучению). Если же InSb подвергнуть давлению в 30 000 атмосфер, то меняется его кристаллическая структура, и при этом электропроводность возрастает в миллион раз!
В полупроводниковой технике применяются и другие соединения и сплавы И.
В. К. Бельский. ИНЕРТНЫЕ ГАЗЫ (благородные газы) — элементы, образующие главную подгруппу VIII группы периодической системы Менделеева: гелий Не (атомный номер Z=2), неон Ne (Z=10), аргон Аг (Z=18), криптон Кг (Z=36), ксенон Хе (Z=54) и радон Rn (Z=86).
И. г. постоянно присутствуют в воздухе (в 1 м3 воздуха около 9,4 л И. г., главным образом Аг). Состав же воздуха учёные анализировали уже со второй половины XVIII в. Однако обнаружить И. г. долгое время не удавалось. Из-за своей химической пассивности они никак не проявляли себя в обычных реакциях и ускользали из поля зрения исследователей. Только после разработки очень чувствительного метода — спектрального анализа были открыты сначала Не (1868 г.) и Аг (1894 г.), а затем и другие И. г. В начале XX в. человечество с удивлением узнало, что воздух, такой привычный и, казалось, давно изученный, содержит 6 неизвестных ранее элементов.
И. г. находятся в растворённом виде в воде, содержатся в некоторых горных породах. Гелий иногда входит в состав подземных газов. Такие газы и служат его единственным промышленным источником. Ne, Аг, Кг и Хе добывают из воздуха в процессе его разделения на азот и кислород (см. Воздух).
Источником Rn служат препараты урана, радия и других радиоактивных элементов. Хотя все И. г.,кроме Rn, стабильны, их происхождение во многом связано с радиоактивностью. Так, ядра Не, иначе называемые а-частицами, постоянно образуются при радиоактивном распаде урана или тория. 40Аг, преобладающий в природной смеси изотопов аргона, возникает при радиоактивном распаде природного изотопа калия 40К (путём так называемого A-захвата). Наконец, происхождение большей части земных запасов Хе обусловлено, вероятно, самопроизвольным делением ядер урана.
Все И. г. не имеют цвета и запаха. Внешние электронные оболочки атомов И. г. называются завершёнными, так как содержат максимально возможное для соответствующих внеш-
ИНЕРТНЫЕ ГАЗЫ
144
Рис. 1. Постоянный и наведённый диполи. а — молекула НС1 (постоянный диполь), б — недефор-мированные молекулы хлора, в — в электрическом поле молекулы С12 деформировались и стали диполями (поле могут создать соседние молекулы С12).
них оболочек число электронов: 2 у Не и по 8 у остальных И. г. Такие оболочки обладают высокой устойчивостью. С этйм связана, во-первых, химическая пассивность И. г. по отношению к другим элементам. А во-вторых, неспособность их атомов вступать в связь друг с другом, вследствие чего молекулы И. г. одноатомны. И. г., особенно лёгкие, трудно перевести в жидкое или твёрдое состояние. Попробуем разобраться, почему это так.
Молекулы других газов либо представляют собой постоянные диполи (например, молекулы НС1), либо довольно легко становятся диполями (например, молекулы С12). У постоянных диполей (рис. 1,а) «центры тяжести» положительного и отрицательного зарядов постоянно не совпадают между собой. Образование же диполя в молекулах типа С12 связано со смещением в них «центров тяжести» зарядов друг относительно друга под воздействием внешних сил, в частности под действием электрических полей соседних молекул (рис. 1,6 и в). Таким образом, и в молекулах НС1, и в молекулах С12 между разноимёнными полюсами диполей действуют силы электростатического притяжения. При определённых пониженных температурах этих сил оказывается достаточно, чтобы удержать молекулы друг возле друга. Иными словами — чтобы перевести газ в жидкость и далее в твёрдое вещество. Ведь с понижением температуры тепловое движение молекул замедляется, и силы, вызывающие взаимное отталкивание молекул, ослабевают.
У атомов же И. г. расположение электронов вокруг ядер строго сферическое (рис. 2). Поэтому соседние атомы не могут вызвать смещение «центров тяжести» электрических зарядов в атомах И. г. и привести к образованию «наведённого» диполя, как в молекулах хлора. Таким образом, ни постоянных, ни наведённых диполей
в атомах И. г. нет. А раз так, то и силы притяжения между ними при нормальных условиях практически отсутствуют.
Как же тогда объяснить, что И. г. всё-таки удаётся перевести в жидкое состояние (см. таблицу).
Дело в том, что ядра всех атомов постоянно колеблются. Из-за этих колебаний «центры тяжести» положительного и отрицательного зарядов на какие-то мгновения оказываются в разных местах атома И. г. Силы притяжения, возникающие между разноимёнными полюсами таких мгновенных диполей, весьма малы. И только при низких температурах их оказывается достаточно, чтобы вызвать конденсацию И. г.
От гелия к радону температуры плавления и кипения И. г. повышаются. Это связано с тем, что атомы наиболее тяжёлых И. г.— Хе и Rn — содержат много электронов. А большие электронные оболочки легче деформируются, и у атомов скорее возникают мгновенные диполи. Атомы тяжёлых И. г. легче удерживаются друг около друга, и переход газа в жидкость происходит при менее низкой температуре.
Долгое время попытки получить обычные химические соединения И. г., образование которых сопровождалось бы перераспределением электронов между атомами, оканчивались неудачами. И. г. получили даже название «благородных», по аналогии с благородными металлами. Положить конец представлениям об абсолютной химической недеятёльности И. г. удалось канадскому учёному Н. Бартлетту, который в 1962 г. сообщил о синтезе соединения ксенона с гексафторидом платины PtFe. Полученное соединение ксенона имело состав XePtFe. В последующие годы было синтезировано большое число и других соединений Rn, Хе и Кг. Оказалось, что эти И. г. прояв-
оболочки атомов
Рис. 2. Электронные инертных газов.
145
ИНЕРТНЫЕ ГАЗЫ
Уильям РАМЗАЙ — первооткрыватель инертных газов.
Английский химик Уильям Рамзай родился 2 октября 1852 г. в Глазго. Его отец был инженером-железнодорожником. Когда Уильям учился в университете (в Глазго), он, играя в футбол, сломал ногу. Во время лечения юноша увлёкся химией, читая известный в то время учебник Т. Грэма. Высшее образование закончил в Тюбингенском университете (Германия) и в 1872 г. защитил там диссертацию, посвящённую органической химии. Затем преподавал на родине, в университете в Глазго, был профессором в университетах Бристоля и Лондона. Учёным того времени были хорошо известны его исследования по органической и физической химии. В 1894 г. Рамзай совместно с Дж. Рэлеем открыл неизвестную ранее составную часть воздуха — аргон. В 1895 г. он получил гелий. В 1898 г. (вместе со своим сотрудником М. Траверсом) открыл криптон, ксенон и неон. Открытие инертных газов, составивших в то время отдельную (нулевую) группу системы Менделеева, принесло Рамзаю мировую славу. В 1904 г. он удостоен Нобелевской премии. Рамзай был членом Лондонского королевского общества, Академий наук в Париже и в Петербурге и многих других научных обществ.
Не все открытия учёного подтвердились. Оказались ошибочными сообщения о превращении меди в литий под действием радиевого излучения и о существовании ещё одного инертного газа «метаргона» (он оказался смесью аргона с продуктами испарения смазки для стеклянных кранов). По этому поводу Рамзай не без юмора заметил: «Непогрешимым быть невозможно, а в случаях ошибок найдётся всегда очень большое число добрых друзей, которые исправят погрешность».
Рамзай был широко образованным человеком с весьма разносторонним кругом интересов. Он хорошо владел древними и новыми языками Запада, читал по-русски. Большой знаток литературы, живописи и музыки, он и сам неплохо рисовал, играл на фортепиано и на скрипке. Был искусным танцором, увлекался спортом — теннисом, плаванием на яхте, ездой на велосипеде. 23 июля 1916 г. Рамзай умер от рака носовой полости, вызванного радиоактивными излучениями. В те годы от них ещё не умели защищаться.
ляют степени окисления +1, +2, +4, +6 и +8 (валентности I, II, IV, VI и VIII).
Получить соединения более лёгких И. г. пока не удалось. Теоретические расчёты показывают, что соединения Аг, может быть, и будут синтезированы, а вот у Не и Ne их получить нельзя. Относительно высокая химическая активность тяжёлых И. г. по сравнению с лёгкими связана с тем, что притяжение внешних электронов к ядру в атомах тяжёлых И. г. ослаблено. Причиной служит, во-первых, отталкивание внешних электронов со стороны большого числа внутренних. А во-вторых, увеличение расстояния между электронами внешней оболочки и ядром.
Прочность связи электрона в атоме характеризуется энергией ионизации — наименьшей
энергией, которую необходимо затратить для удаления электрона из атома. Как видно из таблицы, в группе И. г. энергия ионизации, соответствующая удалению первого электрона, сильно уменьшается от Не к Rn. Притяжение внешних электронов у атомов тяжёлых И. г. сравнительно невелико, и агенты, обладающие сильной способностью принимать электроны (прежде всего фтор), оказываются в состоянии давать с этими И. г. соединения.
После открытия Бартлетта возник вопрос: а не столкнулись ли химики с какими-то новыми, неизвестными ранее силами? Дальнейшие исследования позволили ответить: нет, ничего необычного здесь нет. Так, многие соединения И. г. построены по ионному типу, давно и хорошо изученному. В частности, в
Свойства инертных газов
Элемент Атомный номер Атомная масса Атомный радиус (А)* Конфигурация внешней электронной оболочки Энергия ионизации (эв) ♦♦ Температура кипения (°C) Температура плавления (°C) Плотность (г/л)
при НС фмальных ус ловиях
Не 2 4,0026 1,40 1s2 24,58 —268,93 —272,6 0,178
Ne 10 20,179 1,54 2s22pe 21,56 —245,9 —248,6 0,900
Аг 18 39,948 1,88 3s23p6 15,76 —185,9 —189,3 1,784
Кг 36 83,80 2,02 4s24p6 14,00 —153,2 —157,1 3,745
Хе 54 131,30 2,16 5s25p« 12,13 —108,1 —111,8 5,851
Rn 86 222 — 6s26p® 10,75 ок. —63 ок. 71 9,73
* 1А = 10“8 см. ** 1 эв =1,6-10-19 дж. ♦** При 25 атм.
10 ХЭШ
иод
146
молекуле XePtFe ксенон играет роль положительного однозарядного иона, а группа PtFe— отрицательного. В большинстве других соединений И. г. (фториды, окислы, соли) связь полярна, причём электроположительным в них всегда является атом И. г. Интересно, что по прочности связи, по расстояниям между центрами атомов в молекулах, по форме молекул фториды ксенона очень похожи на фториды иода.
До сих пор во всех известных реакциях И. г. так или иначе участвует самый активный неметалл — фтор. Одни вещества (фториды И. г.) получают, действуя на И. г. фтором или фторидами металлов (SbF5, PtFe). Другие (например, окислы) образуются при разложении фторидов И. г. водой или растворами щелочей. Теоретические расчёты показывают, что возможно образование соединений с хлором для такого И. г., как Хе. Однако пока попытки получить чистые хлориды Хе оканчивались неудачами. Накопленные сведения по химии И. г. подтверждают, в общем, мнение об их крайне низкой химической активности.
Подробнее о химических соединениях Кг, Хе и Rn рассказано в статьях, посвящённых этим элементам.
Открытие химических соединений И. г. оказалось чрезвычайно важным для решения вопроса об их размещении в таблице Менделеева. Раньше группе И. г. приписывали номер 0. Теперь же в соответствии с максимальной степенью окисления +8, проявляемой Хе (например, известен ХеО4), группу И. г. рассматривают как главную подгруппу VIII группы (см. также Закон Менделеева).
Как и многие другие газы, И. г. способны давать так называемые соединения включения (клатраты). Так, ещё в 1896 г. французский учёный Р. Вийяр получил при замораживании воды в атмосфере аргона вещество состава Аг-6Н2О. Такие соединения возникают за счёт чисто пространственных факторов, без перераспределения электронов. Как же образуются клатраты?
В кристаллических решётках некоторых веществ, например льда, фенола, гидрохинона, имеются полости, по своим размерам примерно равные атомам И. г. Каждая полость образована определённым числом молекул. Атомы И. г. могут внедриться в эти полости, после чего они оказываются как бы запертыми в них (недаром название «клатрат» происходит от лат. clatratus, что означает «закрытый решёткой»). Поскольку каждая полость образована определённым числом молекул, то и соединения включения имеют постоянный состав. Впервые обнаружил существование многих клатратов И. г. советский химик Б. А. Никитин.
Промышленное применение И. г. основано главным образом на их низкой химической активности или на специфических физических свойствах. В последнее время практическое ис-
пользование нашли фториды ксенона. О применении И. г. см. в статьях об отдельных элементах. С. С. Бердоносов.
ИОД (лат. lodum) I — химический элемент VII группы периодической системы Менделеева; атомный номер 53, атомная масса 126,9045; относится к семейству галогенов. Свободный И.— кристаллы чёрно-серого цвета с металлическим блеском. В природе элемент представлен одним устойчивым изотопом 1271.
Когда-то средняя атомная масса природного И. была больше, потому что наряду с 1271 существовал и Р“ - радиоактивный изотоп 12е1 с перио
дом полураспада 1,72-107 лет. Но даже такой огромный промежуток времени очень мал по сравнению с возрастом Земли. И постепенно этот изотоп исчезал, превращаясь в изотоп ксенона 129Хе:
it’I-p-^rXe
Поэтому средняя атомная масса И. уменьшилась, и в периодической системе возникла аномалия: теллур (атомный номер 52) стоит перед И., хотя его атомная масса (127,60) больше.
И. был открыт в 1811 г. французом Б. Куртуа — владельцем небольшой селитроварни. Однажды он заметил, что при добавлении концентрированной серной кислоты к рассолу, оставшемуся после извлечения соды из золы морских водорослей, выделяются фиолетовые пары, по запаху напоминающие хлор. Есть, правда, предание, будто подобный опыт произвёл не сам Куртуа, а ... его кот, случайно опрокинувший склянку с серной кислотой на кучку золы.
В 1813—1814 гг. французский химик Ж--Л. Гей-Люссак и английский химик Г. Дэви установили элементарную природу нового вещества. Оно получило название «иод», от греческого «иоэйдэс» — фиолетовый.
В природе И. из-за большой химической активности в свободном состоянии не встречается. В земной коре его немного — всего 4-10“б% по массе (здесь И. принадлежит 60-е место, тогда как его аналогу—хлору — 17-е). Минералы И.— лаутарит Са(Ю3)2 и дитцеит 7Са(Ю3)2-8СаСгО4 — очень редки. Соли И.— иодиды — содержатся в небольших количествах в гидросфере — морской воде, а также в буровых водах нефтяных скважин. Некоторые морские водоросли и губки обладают удивительной способностью поглощать И. из воды. Особенно активна в этом отношении ламинария — хорошо известная всем морская капуста, содержащая в сухом виде 0,1—0,6% И.; поэтому врачи рекомендуют её как хорошо усвояемую форму И.
147
ИОД
Кристаллы И. имеют плотность 4,94 г/см3; при медленном нагревании и обычном давлении И. из твёрдого состояния сразу переходит в газообразное. Переход вещества из твёрдого состояния в газообразное, минуя жидкое, называется сублимацией, или возгонкой. Этим свойством Й. пользуются для его очистки. Но если нагревать И. быстро, он плавится при 113,5СС и кипит при 184,35°С. И. плохо растворим в воде: при обычных условиях 1 л Н2О растворяет 0,34 г И. (раствор И. в воде называется иодной водой). Значительно больше растворимость И. в сероуглероде и в органических растворителях (бензоле, спирте и др.). Растворы И. в спирте и эфире тёмно-бурого цвета, в бензоле, сероуглероде и хлороформе— фиолетового. Неодинаковой растворимостью Й. в воде и в не смешивающихся с ней жидкостях пользуются для его извлечения (экстракции) из водных растворов.
Проведём такой опыт. Прильём в пробирку с желтоватой иодной водой немного бензола; последний всплывает, образуя на поверхности воды тонкий слой. Взболтаем
Взаимодействие алюминия с иодом. Реакция между этими веществами интересна тем, что её катализатором служит обычная вода. Смесь совершенно сухих мелких порошков алюминия и иода может долго храниться при комнатной температуре безо всяких изменений (на рис. слева). Стоит, однако, прибавить к ней 2—3 капли воды, как через несколько минут начнётся бурная реакция образования иодида алюминия: 2А1+ -г312= 2А113. При этом смесь сильно разогревается и пары иода возгоняются, образуя большое облако (на рис. справа). Поэтому реакцию следует проводить или на открытом воздухе, или в закрытом вытяжном шкафу. Для этого опыта рекомендуется брать алюминий в 3-кратном избытке (по сравнению с требуемым по уравнению реакции). Общая же масса взятых реагентов не должна превышать 10—15 г.
io*
жидкости и, дав им отстояться, увидим, что водный слой стал бесцветным, а бензольный — фиолетовым; почти весь И. из воды перешёл в бензол.
И. даёт ярко-синее соединение с крахмалом (это одна из качественных реакций на присутствие свободного И.).
Так, если капнуть иодной водой на срез картофелины, то на её поверхности появится синее пятно. Образуется новое соединение: молекулы И. проникают между полимерными цепочками молекул крахмала, связывая их между собой. При нагревании окраска исчезает, при охлаждении появляется вновь.
У атома И. семь внешних (валентных) электронов (конфигурация 5s25p6, см. Атом). В своих соединениях он проявляет степени окисления -1 (KI), -1 (НЮ), +3 (1С13),+5 (НЮз) и +7 (К3Ю5) (соответственно валентности I, III, V, VII). Как и для прочих галогенов, для атома И. характерно присоединение одного электрона:
о
I 4- 1е = 1г
Однако, так как И. стоит последним в подгруппе F—С1—Вг—I, он уступает прочим галогенам по химической активности (см. Галогены). И. не реагирует непосредственно с углеродом, азотом, кислородом, серой и селеном; его соединения с этими элементами получены косвенным путём. При нагревании И. взаимодействует с водородом, образуя газообразный иодистый водород. Эта реакция обратима:
Н2+12 ^2Н1
С металлами И. особенно энергично реагирует в присутствии влаги (см. также Вода). Если на смесь порошков алюминия и И. капнуть воды, происходит вспышка:
2А1 + 312 = 2А11з
Этот эффектный опыт изображён на рисунке. И. окисляет и некоторые сложные вещества:
H2S+ I2 = S4-2HI
Во всех этих реакциях получаются соединения 1(—1); это либо HI, либо иодиды.
И. взаимодействует со своими соседями по группе. А так как он из всех галогенов наиболее электроположителен, то в этих реакциях ведёт себя как восстановитель:
0 О +1 - !
I2 + С12 = 2IC1
При растворении И. в воде образуется наряду с иодистоводородной иодноватистая кислота HOI:
124-Н2О = НО1 -г HI
Это амфотерное вещество, способное диссоциировать и как основание и как кислота:
ОН - + I+ HOI Н+ -|- Ol-
иодиды
148
Соединения -I (+1) — иодноватистая кислота HOI и её соли (гипоиодиты) — очень неустойчивы и существуют только в растворе. Из соединений И. со степенью окисления +3 известны, например, фосфат 1РО4, нитрат I(NO3)3, ацетат 1(СН3СОО)3 — твёрдые вещества жёлтого цвета, устойчивые лишь ниже О °C. Все такие соединения очень чувствительны к действию влаги:
51РО4 -г 9Н2О = 12 4- ЗНЮ3 + 5Н3РО4
При взаимодействии И. со щелочами образуются соединения I (4-5), соли йодноватой кислоты НЮ3— йодаты:
312 -4 6NaOH = 5NaI -4 NaIO3 4- ЗН2О
Эти соединения наиболее устойчивы из всех, где И. имеет положительные степени окисления. Кислоту можно получить в свободном состоянии (это бесцветное кристаллическое вещество). А её соли встречаются в природе (например, NaIO3 как примесь к селитре NaNO3). При нагревании йодноватой кислоты выше 240 °C образуется иодноваты-й ангидрид 12О5 (бесцветное кристаллическое вещество). Он применяется для определения угарного газа СО:
5СО -- 12О5 = 12 4 5СО2
По объёму образовавшегося углекислого газа или по количеству выделившегося И. находят содержание СО в газовой смеси (например, в дымовых газах).
Если йодаты окислять хлором в щелочной среде, то образуются перйодаты (соли иодной кислоты) — соединения 1(4-7). Сама же иодная кислота существует в свободном состоянии в виде' кристаллогидрата НЮ4*2Н2О. Соединения 1(4-7) — сильные окислители:
4-7 4-2 +7 -1
5КЮ4 -4- 8MnSO44- 12Н2О = 8НМпО4 - 5KI + 8H2SO4
Добывают И. главным образом из буровых вод нефтяных скважин. Для получения И. из этого сырья у нас в СССР построены два крупных завода — в Сураханах и Нефте-Чале. Способы получения И. основаны на окислении йодидов свободным хлором, нитритом натрия или двуокисью марганца в присутствии кислот:
2KI гС12 = 2КС1 + 12
2KI+MnO24-2H2SO4 =
- K2SO4 4- MnSO4 + 2Н2О + I2
И. и его соединения находят разнообразное применение. Иодид серебра Agl наряду с бромидом серебра AgBr используется в фотографии (см. Серебро). Многие иодсодержащие соединения (особенно иодистоводородная кислота) — реактивы для различных химических синтезов в лабораториях. А в металлургии есть так называемый иодидный способ получения особо чистых металлов (см. об этом в статьях Титан, Цирконий, Гафний).
Самые же большие количества И. потребляет фармацевтическая промышленность: И. содержится во многих лекарственных препаратах — недаром в их названия обычно входит корень «иод» (саиодин, иодол, липииодол...). Всем знаком наш бытовой «иод», которым смазывают ранки для дезинфекции; это 5%-ный раствор И. в спирте. Важное применение находит в медицине и искусственно получаемый
радиоактивный изотоп 1311 (период полураспада 7\2=8 дней). С его помощью изучают и лечат щитовидную железу, проводят диагностику различных заболеваний.
Человеческий организм содержит около 25 мг И. Большая часть его (около 15 мг) сконцентрирована в щитовидной железе, а точнее, в её гормоне — тироксине.
Физиологическая роль И. в организме ещё недостаточно выяснена; но очевидно, что отсутствие И. приводит к тяжёлым заболеваниям, таким, как «зоб» и «кретинизм». Ежедневный приём небольших доз соединений И. помогает избавиться от зоба. В Китае и Японии таких больных издавна лечат золой морских губок. Наиболее богаты И. лук, морская рыба, морская капуста. Пищевая промышленность выпускает иодированную поваренную соль (то есть NaCl с добавкой KI). А в почву тех районов, где И. мало, вносят иодистые удобрения.
В. К- Бельский.
ИОДЙДЫ — соединения иода с другими, более электроположительными элементами.
И. металлов — соли иодистоводородной кислоты — в основном хорошо растворимы в воде (за исключением Agl, Hg2I2, Cu2I2 и Hgl2). Водные растворы И. щелочных и щелочноземельных металлов имеют нейтральную реакцию. У растворов И. прочих металлов реакция кислая, что объясняется гидролизом:
АПз + ЗН2О = А1 (ОН)3 + 3HI
И. неметаллов (за исключением газообразного йодистого водорода HI) — твёрдые вещества. Водой они разлагаются:
Р13-ЗН2О=Н3РОз + ЗН1
Большинство И. хорошо растворяются и в органических жидкостях. И.— сильные восстановители. Например, KI восстанавливает даже такой слабый окислитель, как FeCl3:
4-3 - 1 0 4-2
2FeCl3 + 2КI = I2 + 2КС1 + 2FeCl2
О применении различных И. см. также статьи Иод, Серебро, Калий, Алюминий, Титан.
ИОДИСТОВОДОРОДНАЯ КИСЛОТА — раствор йодистого водорода HI в воде.
ИОДИСТЫЙ ВОДОРОД HI—соединение иода с водородом. Бесцветный газ с резким неприятным запахом; /кип —35,9°C. Реакцию получения И. в. из водорода и иода:
H2 + I2z±2HI проводят над платиновым катализатором при 300—500°C. Реакция обратима: образующийся И. в. термически нестоек и при повышении температуры разлагается на элементы.
И. в. хорошо растворим в воде: при нормальных условиях 1 объём Н2О поглощает 425 объёмов HI. Раствор И. в. в воде называется иодистоводородной кислотой HI. Это .очень сильная кислота (по ряду HF—НО— —НВг—HI сила кислот увеличивается, см.
149
ИТТРИЙ
Галогены). Она активно взаимодействует с металлами, стоящими в ряду напряжений до водорода:
Zn + 2HI = Znl2 + Н2
Интересно, что серебро, стоящее в ряду напряжений после водорода, всё-таки вытесняет его из иодистоводородной кислоты:
2Ag + 2HI = 2AgI + Н2
Эта уникальная реакция возможна вследствие очень малой растворимости Agl в воде и сдвига реакции вправо по мере выпадения Agl в осадок. Соли иодистоводородной кислоты называются иодидами. Сама кислота и её соли — сильные восстановители. Так, при длительном стоянии кислота окисляется даже кислородом воздуха:
4HI + О2 = 212 + 2Н2О
Выделяющийся иод придаёт ранее бесцветному раствору кислоты окраску. При небольших концентрациях 12 раствор желтеет, при значительных — буреет.
Получить И. в. аналогично хлористому водороду по реакции иодида с концентрированной серной кислотой нельзя, потому что выде
ляющийся HI тут же восстанавливает серную кислоту до сероводорода:
H2SO4 + 8HI = H2S + 4I2 + 4Н2О
Удобный лабораторный метод— взаимодействие иодида фосфора с водой:
Р1з+ЗН2О=Н3РОз+ЗН1
А иодистоводородную кислоту получают пропусканием сероводорода в иодную воду: H2S+ I2 = 2HI + S
О применении И. в., кислоты и её солей см. Иод, Йодиды.
ИРИДИЙ (лат. Iridium) Ir — химический элемент VIII группы периодической системы Менделеева; атомный номер 77; относится к семейству платиновых металлов.
ИТТЕРБИЙ (лат. Ytterbium) Yb — химический элемент III группы периодической системы Менделеева; атомный номер 70; относится к семейству лантаноидов.
ЙТТРИЙ (лат? Yttrium) Y — химический элемент III группы периодической системы Менделеева; атомный номер 39. См. Редкоземельные элементы.
„Какой только области механических искусств не нужны знания химии! Могут ли обойтись без нее земледелец, металлургf фармацевт, врач, золотых дел мастер, чеканщик монет и т.п.? Если бы человечеству пришлось избрать из числа всех наук только три и притом применительно к нашим потребностям, следовало бы предпочесть всем другим наукам механику, естественную историю и химию**.
Дени Дидро (1713—1784)
(Собр. соч.» т. 10, М.» 1947, с. 305)
к
112,40 48
Cd
4dl05s2
КАДМИЙ
КАДМИЙ (лат. Cadmium) Cd — химический элемент II группы периодической системы Менделеева; атомный номер 48, атомная масса 112,40. Металл серебристо-белого цвета с синеватым отливом. Природный К. состоит из смеси 8 стабильных изотопов с массовыми числами 106. 108. ПО—114, 116.
Природные соединения цинка всегда содержат К-как примесь. В 1817 г. при ревизии аптек вблизи Магдебурга возникло подозрение, что имевшаяся там окись цинка загрязнена мышьяком. Образцы её, а также карбоната цинка ZnCO3,служившие для получения ZnO по реакции:
ZnCO3 = ZnO + СО2 были посланы на экспертизу в Гёттинген профессору Ф. Штромейеру. Прокалив ZnCO3, он с удивлением заметил, что жёлтый цвет горячей ZnO не исчез при охлаждении (давно было известно, что белая ZnO при накаливании желтеет, а при охлаждении вновь белеет). Штро-мейер, пропустив сероводород в раствор препарата в соляной кислоте, получил оранжево-жёлтый осадок. Это был сульфид неизвестного ранее металла. Штро-мейер назвал его кадмием, от позднелатинского cadmia fossil is («ископаемая кадмия»), как именовался один из цинковых рудных минералов — каламин, или галмей. Коричневая окись CdO, образовавшаяся при прокаливании CdCO3, и была причиной жёлтой окраски холодной окиси цинка.
Земная кора содержит 1,3-10-5% К. по массе (64-е место среди других элементов). В свободном виде К. в природе не встречается. Его минералы — гринокит CdS, монтепонит CdO, отавит CdCO3 — весьма редки. Рудных скоплений, пригодных для промышленной эксплуатации, К. не образует, но он практически всегда содержится вместе с цинком в полиметаллических рудах.
К.— мягкий тяжёлый металл. Он легко куётся и прокатывается. Плотность К. 8,65 г/см3, /пл 320,9 °C, /кип 767 °C. Если палочку чистого К. приложить к уху и изгибать, то слышится треск, вызываемый трением кристаллов металла друг о друга. Это явление свойственно и чистому олову.
Конфигурация последних энергетических уровней атома К. 4d105s2 (см. Атом). Валентными являются два s-электрона последней оболочки. Поэтому в соединениях К. проявляет степень окисления -г2 (валентность II). На воздухе металл тускнеет, покрываясь плёнкой окиси CdO, предохраняющей его от дальнейшего окисления. Благодаря этому К- применяется для нанесения защитных покрытий на другие металлы. При сильном же нагревании К. сгорает тёмно-жёлтым пламенем до CdO — порошка коричневого или бурого цвета. Соединяясь с галогенами, К. даёт бесцветные галогениды — CdCl2 и др. При нагревании порошков К. и серы образуется сульфид CdS жёлтого цвета. В ряду напряжений К. стоит до водорода. Он медленно растворяется в кислотах, например:
Cd-?2HC1-CdCl2T-H2
К действию же щелочей К. (в отличие от своего соседа — цинка) устойчив. Если к раствору соли К. прилить раствор NaOH, то выпадает белый осадок гидроокиси Cd(OH)2, обладающей, в отличие от Zn(OH)2, только основными свойствами.
К. получают как побочный продукт при переработке цинковых, свинцово-цинковых и медно-цинковых руд. Отходы, содержащие 0,2— 7% К-, обрабатывают разбавленной серной кислотой, которая растворяет ZnO и CdO. Из раствора К. осаждают цинковой пылью:
Zn + CdSO4 - Cd + ZnSO4
Образовавшийся губчатый осадок (смесь Cd и Zn) растворяют в разбавленной H2SO4 и из раствора сульфатов выделяют К. электролизом.
С пуском первого уранового реактора К. попал в число стратегически важных материалов. Оказалось, что изотоп 113Cd (а в обычном К. его содержание около 12%) обладает колоссальной способностью поглощать тепловые нейтроны.
В ядерном реакторе стержни, изготовленные из К-, выполняют роль «кочегаров», регулирующих работу «топки». То вводя в реактор стержни, то выводя их, можно уменьшать или увеличивать поток тепловых
151
КАЛИИ
нейтронов, а следовательно, и скорость ядерной реакции. А если реакция развивается слишком бурно, то наготове всегда есть массивный аварийный кадмиевый стоп-стержень, который опускается в реактор под действием собственной тяжести и усмиряет «разбушевавшиеся» нейтроны.
Половина добываемого К. идёт на покрытие деталей (кадмирование) автомобилей, танков, самолётов, морских судов, поверхность которых постоянно подвергается воздействию агрессивной среды. Покрытия из К. и прочнее, и красивее цинковых. К.— необходимый компонент многих легкоплавких сплавов. Например, так называемый сплав Вуда, состоящий из 50% Bi, 25% РЬ, 12,5% Sn и 12,5% Cd, плавится всего лишь при 68 °C. Такие сплавы применяют как легкоплавкие припои. При их использовании отпадает нужда в раскалённом паяльнике и всю работу можно производить при температуре горячей воды.
Упомянутый легкоплавкий сплав был изобретён ещё в 1860 г. мало известным англичанином Б. Вудом; но по недоразумению это изобретение иногда приписывают знаменитому американскому физику Р. Вуду, родившемуся в 1868 г. (мы упомянули о Р. Вуде в статье Литий).
Специалистам-электротехникам хорошо известно, что медные провода постепенно истираются под действием токоснимающих устройств. (Токоснимающие устройства вы видели не раз: это — и трамвайные дуги, и троллейбусные усы. «Снимая ток» с проводов, трамвайная дуга передаёт его мотору.) Чтобы увеличить прочность и стойкость против износа троллейбусных проводов, к меди добавляют около 1 % Cd. При этом электропроводность Си уменьшается всего на 11—12%, зато прочность увеличивается в 1,5 раза, а стойкость против истирания — в 3 раза.
Большое количество К. идёт на изготовление мощных электрических аккумуляторов (наиболее известен из них АКН — аккумулятор кадмиево-никелевый).
Палитра кадмиевых красок.
Сульфид CdS применяется для изготовления красок, долгое время сохраняющих свежесть и яркий блеск (рис.). Сульфид CdS и селенид CdSe обладают полупроводниковыми свойствами. Изготовленные из них фотоэлементы вы можете найти, например, в фотоаппаратах.
Ю. И. Романьков.
КАЛИЙ (лат. Kalium) К — химический элемент I группы периодической системы Менделеева; атомный номер 19, атомная масса 39,098. Серебристобелый металл. Природный К. состоит из трёх изотопов — двух стабильных 39К и 41К (в природной смеси их содержится 93,10 и 6,88% соответственно) и одного радиоактивного 4ОК(0,0119 %).
Изотоп 40К имеет огромный период полураспада,
1 миллиард 320 миллионов лет. Большая часть 40К, испуская Р”-лучи, превращается в изотоп кальция 40Са, а меньшая часть, захватывая электрон, превращается в изотоп аргона ^Аг:
1 ?К гоСа + р
ЯК + е
"Аг + ?
Некоторые соединения К. (например, поташ, добывавшийся из древесной золы) были известны уже в древности; однако их не отличали от соединений натрия. Только в XVIII в. было показано различие между «растительной щёлочью» (поташом К2СО3) и «минеральной щёлочью» (содой Na2CO3). Сам же К. (в свободном состоянии) впервые получил английский учёный Г. Дэви в 1807 г. электролизом влажного едкого кали КОН. Название «калий» происходит от арабского слова «алкали» — что означает «зола растений», а также «щёлочь». Принято и другое название К-, происходящее от поташа: potassium в Англии, Франции, США и potassio в Италии. В начале XIX в. в России К- также называли «по-тассием».
Содержание К. в земной коре составляет 2,5% по массе. По распространённости он занимает 7-е место (после натрия). Вследствие высокой химической активности К. в свободном состоянии в природе не встречается. Его наиболее важные минералы это — сильвин КС1, сильвинит (K,Na)Cl, карналлит KCl-MgCl2-6H2O, каинит КС1 • MgSO4• ЗН2О (см. Минералы).
Богаты К. и многие силикаты (слюды, нефелины, полевые шпаты и др.). Большое количество калийных солей растворено в морской воде и соляных озёрах.
Советский Союз занимает первое место в мире по богатству и качеству залежей калийных солей. Наиболее крупное месторождение находится в районе Соликамска — Березников на Урале. По запасам К. оно далеко превосходит все месторождения других стран, вместе взя
КАЛИИ
152
тые. Впервые в Соликамских залежах поваренной соли (NaCl) К. был обнаружен академиком Н. С. Курнаковым в 1916 г. А в 1927 г. там была начата постройка крупного калийного комбината, который в 1933 г. дал первую продукцию отечественных калийных удобрений. Недавно открыто новое крупное месторождение калийных солей в Белоруссии — на этом месте создаётся прекрасный современный город Солигорск. В Казахстане возле г. Актюбинска находятся огромные запасы полигалитов — смешанных сульфатов калия, кальция и магния.
К. плавится при довольно низкой температуре 63,55 °C, а кипит при 760 °C. Он легче воды (как литий и натрий) — его плотность составляет 0,862 г/см3. К.— хороший проводник тепла и электрического тока. Он очень мягок и легко режется ножом. Пары соединений К. окрашивают пламя горелки в характерный фиолетовый цвет, что позволяет исследователю обнаруживать этот элемент.
Конфигурация внешней электронной оболочки атома К. 4s1. Химически К. более активен, чем его соседи сверху по подгруппе щелочных металлов — лит^ий и натрий. Это объясняется тем, что единственный валентный электрон атома К. удалён от ядра дальше и поэтому энергия, необходимая для его отрыва, значительно меньше. В соединениях К. всегда проявляет степень окисления +1 (валентность 1). Тип связи в соединениях К. с неметаллами — ионный.
К. бурно окисляется на воздухе, образуя с кислородом соединения трёх типов: в небольших количествах окись К2О и перекись К2О2 и в основном надперекись КО2. Поэтому хранить металл надо под слоем керосина или минерального масла, с которыми он не реагирует.
К. соединяется практически со всеми неметаллами. С водородом при 200 °C он образует гидрид:
2К + Н2 = 2КН
Реакции галогенов и К. происходят с воспла-. менением и взрывом, например:
2К + С12 = 2КС1
С серой при слабом нагревании образуется сульфид:
2К + S = K2S
С азотом К. не взаимодействует даже при высоких давлениях и больших температурах, и соединение этих элементов — нитрид К.— можно получить в небольших количествах лишь при электрическом разряде:
6K + N2 = 2K3N
Поэтому азот можно использовать как защитную атмосферу при проведении работ с металлическим К.
Очень энергично реагирует К. с водой: 2К + 2Н2О = 2КОН + Н2
При этом выделяется так много тепла, что образующийся водород воспламеняется. Так как К- легче воды, то он «бегает» по её поверхности, и там, где «пробежал» кусочек металла, вспыхивают искры. Второй продукт этой реакции — одна из самых сильных щелочей — гидроокись калия КОН (едкое кали). При высоких температурах (350—400 °C) К. восстанавливает из SiO2 и силикатов свободный кремний, разрушая стекло. Поэтому в стеклянной посуде проводить реакции с участием металлического К. при такой температуре нельзя.
Очень интересны сплавы К- с натрием, в частности тем, что они в большинстве своём ... жидкие. Например, сплав 77,2% К и 22,8% Na — это серебристо-белая жидкость с температурой плавления —12 °C. Такие сплавы обладают высокой химической активностью и применяются в синтезе органических соединений как восстановители. Впервые система К—Na была исследована русскими учёными Н. С. Кур-паковым и Н. А. Пущиным в 1901 г.
Получить сплав К. и натрия можно уже при простом сдавливании кусочков этих металлов.
Однажды молодой английский химик Т. Э. Торп, по поручению своего учителя Г. Роско, вёз в подарок немецкому учёному Р. Бунзену кристаллы К- и натрия, положив их вместе в банку с керосином. Но при вручении подарка вместо кристаллов в банке оказалась жидкость, похожая на ртуть. (Можно представить себе изумление обоих химиков.)
В промышленности металлический К. получают в небольших количествах, потому что сам металл применяется весьма ограниченно. Его выделяют, действуя на расплавленные КОН или КС1 металлическим натрием:
Na + КОН = К + NaOH Na + КС1 = К + NaCl
Процесс ведут при температуре 450 °C в специальных аппаратах из никеля. При температуре около 1000 °C металлический К- можно получить, вытесняя его кальцием:
Са + 2КС1 = 2К + СаС12
Здесь может возникнуть вопрос — почему Na и Са вытесняют К из его соединений, хотя в ряду напряжений они стоят после него? Но обратитесь к учебнику и вспомните: ряд напряжений применим к реакциям окисления — восстановления, протекающим только в водных растворах.
Чистый К. идёт главным образом на синтез новоксида — надперекиси КО2. Своеобразно применение этого редкого соединения. При
153
КАЛЬЦИЙ
его взаимодействии с углекислым газом выделяется свободный кислород:
4КО2 + 2СО2 = 2К2СО3 + ЗО2
Так происходит «проветривание» подводных лодок и других закрытых помещений особенно ответственного назначения.
Металлический К. намного дороже натрия. Близость же их свойств позволяет во многих случаях заменять К. натрием. Однако есть область, в которой эта замена невозможна.
Подобно азоту и фосфору, К. с полным правом называют «элементом плодородия». И главный его потребитель— сельское хозяйство. Поскольку К. увеличивает способность зелёных растений синтезировать углеводы, потребность в нём повышена у «сахаристых» культур — сахарной свёклы и картофеля. Ежегодно со сбором урожая люди вновь и вновь уносят К. из почвы. Для утоления калийного голода и используют удобрения. Ими служат обычно природные соли К.— сильвин, сильвинит, каинит, а также продукты их переработки — хлорид КО, калийная селитра KNO3, поташ К2СО3, шёнит K2SO4-MgSO4, сульфат K2SO4 и др. Часто К. вносят в почву вместе с другими питательными веществами — в виде комплексных удобрений (см. Минеральные удобрения).
Соединения К- широко применяются и в других областях промышленности и техники. Гидроокись КОН служит электролитом для щелочных аккумуляторов, используется в производстве жидкого мыла. Хлорат КС1О3 (бертоллетова соль) — отличный окислитель, применяется в пиротехнике, а также в лабораториях для получения кислорода по реакции:
2КС1О3 = 2КС1 + 302
Нитрат KNO3 — калийная селитра — важная составная часть чёрного (охотничьего) пороха. О соединениях К-см. также Карбонаты, Нитраты, Силикаты, Сульфаты, Хлориды, Хроматы и т. д.
К.— элемент, обладающий естественной радиоактивностью. Учёные установили, что за счёт радиоактивного распада К. в природе выделяется большое количество тепла: образно говоря, К. не только кормит Землю, но и вносит свой вклад в её обогрев. В. К- Бельский. КАЛИФОРНИЙ (лат. Californium) Cf -радиоактивный химический элемент с атомным номером 98; относится к актиноидам.
КАЛОМЕЛЬ — хлористая ртуть Hg2Cl2. См. Ртуть.
КАЛЬЦИЙ (лат. Calcium) Са — химический элемент II группы периодической системы Менделеева; атомный номер 20, атомная масса 40,08. Серебристо-белый лёгкий металл. Природный К. состоит из смеси 6 стабильных изотопов с массовыми числами 40, 42, 43, 44,46 и 48. Из них наиболее распространён 4(Са (96,97%).
Природные соединения Са — известняк, мрамор, гипс — были известны уже в древности. Издавна в строительном деле применяли и известь (продукт обжига известняка). Долгое время известь считали простым телом — «землёй». Но в 1808 г. английский химик Г. Дэви провёл электролиз слегка увлажнённой гашёной извести и получил на ртутном катоде амальгаму неизвестного металла. Дэви назвал его «кальцием» — от латинского слова calx (родит, падеж calcis) — известь. В России почти до середины XIX в. К. называли «известковием», «известковистостью», «основанием известковой земли».
Содержание К. в земной коре составляет 2,96% по массе. Здесь по распространённости он занимает 5-е место среди элементов и уступает только кислороду, кремнию, алюминию и железу. В океанической воде содержится 0,04% Са. Вследст
вие своей высокой химической активности К. в природе встречается только в виде соединений. Наиболее распространённые минералы К. — кальцит СаСО3 (и его разновидности — исландский шпат и арагонит), ангидрит CaSO4, гипс CaSO4- 2Н2О, флюоритCaF2 (плавиковый шпат). Мел, мрамор, известняк — широко распространённые осадочные горные породы,частично образующие земную кору,—более чем на 90% состоят из СаСО3. Минералы и породы на его основе покрывают около 40 млн. км2 земной поверхности. Главные месторождения известняка в СССР находятся на Украине, в Карелии, в Прибалтике, в Поволжье, на Дону, на западном склоне Урала. Мощные меловые отложения имеются в Поволжье, Белоруссии, Днепровско-Донецкой впадине. Наиболее важные месторождения мрамора красивых расцветок — на Урале, в Карелии, Грузии, Армении, Узбекистане, Забайкалье. Крупнейшие залежи гипса — в Архангельской, Московской, Горьковской областях. Наиболее крупные месторождения флюорита — в Забайкалье.
В природе происходит круговорот К. Известняковые горные массивы, образовавшиеся на протяжении длительных геологических периодов, подвергаются выветриванию — разрушению под действием воды и воздуха. Нерастворимый карбонат СаСО3 постепенно переходит в растворимый гидрокарбонат Са (НСО3)2. Обратимая реакция
СаСО3 + СО2 + Н2О Са(НСО3)2 лежит в основе перераспределения К- по поверхности планеты. Потоки воды приносят К. в моря и океаны. Сюда он приходит в форме растворимого Са(НСО3)2 (сдвиг равновесия
КАЛЬЦИИ
154
Рис. 1. Эта композиция составлена из скелетов коралловых полипов. За свою красивую окраску они издавна ценятся как поделочные камни. Живые же кораллы образуют целые подводные леса, окрашенные в осенние краски — жёлтые, оранжевые, красные, коричневые... Химический состав кораллов сложен и непостоянен. Это в основном карбонат кальция с примесями соединений железа и магния.
вправо). На своём пути природные воды испаряются и теряют углекислый газ и тогда Са(НСО3)2 частично снова переходит в СаСО3 (сдвиг влево).
С этим связано и образование в пещерах удивительно красивых «каменных сосулек» — сталактитов и сталагмитов. При медленном испарении воды в пещерах на их сводах нарастают сталактиты, навстречу им снизу поднимаются сталагмиты. Таким образом, сдвиг реакции вправо обусловливает рассеяние К. в природе, сдвиг влево — его концентрирование.
Но что же происходит с Са(НСО3)2 в море? Мельчайшие живые существа, а также более крупные, вплоть до моллюсков, кораллов и иглокожих, извлекают растворённый карбонат К. из воды для построения своих скелетов и панцирей. Когда животные погибают и опускаются на дно, их известковые панцири и скелеты накапливаются в виде мощных слоёв СаСО3. И если вода отступает, отложения оказываются на поверхности. Так снова концентрируется К... Но воздух и вода опять «работают» против концентрации и «гонят» вечного странника К. по планете.
Для чего же нужен К- живым организмам? Прежде всего, его соли участвуют в построении костного скелета; минеральное вещество костей состоит примерно на 80% из трикальцийфосфата Са3(РО4)2 и на 13% из карбоната СаСО3. Кроме того, К- входит в состав сыворотки крови (в виде солей фосфорной и лимонной кислот), где он играет двоякую роль. Во-первых, ион Саа+ возбуждает сердечную деятельность. А во-вторых, кровь, лишённая
ионов Са2+, не свёртывается на воздухе; при их отсутствии организм погиб бы от потери крови при малейшей царапине.
К.— лёгкий (плотность 1,54 г/см3) и довольно твёрдый металл; его /пл 851 °C, /кип 1482 °C. Летучие соли К. окрашивают пламя газовой горелки в кирпично-красный цвет (по этому признаку элемент может быть обнаружен исследователями).
К. относится к подгруппе щелочноземельных металлов. Конфигурация внешней электронной оболочки его атома 2s2. В подавляющем большинстве своих соединений К. проявляет степень окисления+2 (валентность II). Известны, кроме того, соединения, в которых степень окисления К. равна +1; это так называемые субгало-гениды — CaF, CaCl, СаВг. Они образуются при взаимодействии Са с CaF2, СаС12, СаВг2 в расплавленном состоянии и существуют только при высоких температурах.
Химически К. очень активен. При обычной температуре он легко взаимодействует с кислородом воздуха, а при нагревании даже воспламеняется. Поэтому хранят его в масле или герметически закупоренных сосудах (обычно же куски металла весом до 60 кг помещают в бумажные мешки, вложенные в железные оцинкованные запаянные барабаны).
Рис. 2. «При медленном испарении воды в пещерах на их сводах нарастают сталактиты, навстречу им снизу поднимаются сталагмиты» (стр. 154, слева).
155
КАЛЬЦИИ
К. непосредственно взаимодействует со всеми неметаллами. Из них — с фтором, хлором и бромом уже на холоду:
Са + С1а = СаС12
(с йодом — только в присутствии следов влаги). Расплавленные галогениды образуют с металлическим К. уже упоминавшиеся субга-логениды:
СаС12+Са^ 2СаС1
Реакция обратима, и при медленном остывании субгалогениды распадаются. Выделить их удаётся лишь путём очень быстрого охлаждения (этот процесс химики называют «замораживанием»).
При повышенных температурах К. соединяется с серой (образуется сульфид CaS), фосфором (фосфид Са3Р2), углеродом (карбид СаС2). К. относится к числу тех немногих металлов (см. также Литий), которые способны непосредственно соединяться с азотом (К.— при 500 °C):
ЗСа “I- N 2== Ca3N 2
При взаимодействии с водородом при 300— 400 °C К. образует гидрид СаН2 — соединение, в котором водород выступаете качестве аниона. Гидрид К. реагирует с водой, выделяя такое количество тепла, что происходит самовоспламенение водорода:
СаН2+2Н2О=Са (ОН)а+2На
2Н2 + Оа = 2НаО
К.— сильный восстановитель, он вытесняет многие металлы из их сульфидов, окислов, галогенидов. Так, с его помощью получают чистый уран из тетрафторида урана:
UF4 + 2Са = U + 2CaFa
Однако и сам К. вытесняется из его соединений ещё более активными металлами — восстановителями, например алюминием (см. ниже).
С холодной водой К. взаимодействует сначала быстро, но затем реакция замедляется, так как поверхность металла покрывается плёнкой труднорастворимой гидроокиси Са(ОН)2:
Са + 2НаО = Са (ОН)2 + Н2
С горячей водой К. реагирует очень энергично. Поскольку К. стоит в ряду напряжений до водорода, он б^рно реагирует с разбавленными кислотами:
Са •+ 2НС1 = СаС12 + Н2
В промышленности металлический К. получают либо электролизом (как калий и натрий), либо восстановлением извести алюминием или магнием. Для первого, уже знакомого читателю метода, берут в качестве электролита расплавленный хлорид СаС12. Второй способ со-
Рис. 3. У входа в Одесские катакомбы. В Одесских катакомбах близ Чёрного моря — заброшенных каменоломнях — когда-то добывали известняк-ракушечник, этот созданный природой незаменимый строительный материал. «Республика под землёй», «Подземный фронт» — так называют катакомбы Одессы времён войны с фашистскими захватчиками. Здесь в неимоверно трудных условиях базировались народные мстители —партизаны, отсюда они наносили дерзкие удары врагу. В районе катакомб ныне создан Музей партизанской славы.
стоит в нагревании смеси извести с порошком А1 или Mg в вакууме при 1100—1200 °C:
ЗСаО + 2А1 = А12О3 + ЗСа
СаО + Mg = MgO + Са
Пары К. конденсируются на верхней охлаждаемой части сосуда, в котором ведётся реакция. В последние годы разрабатывается ещё один способ получения К-— термическая диссоциация карбида СаС2 в вакууме при 1750 °C.
Высокая химическая активность не позволяет использовать К. в качестве конструкционного материала. Как активный восстановитель К. служит для получения многих редких и тугоплавких металлов — урана, тория, ванадия, хрома, циркония, бериллия, тантала и др. из их соединений. Благодаря способности сравнительно легко соединяться с кислородом и азотом К. применяют для очистки инертных газов от примесей N2 и О2 и для создания глубокого вакуума в электровакуумных трубках. А способность связывать кислород, серу, фосфор и углерод делает его ценной добавкой к шихте в металлургии при выплавке меди, никеля, бронз, специальных сталей.
Большое применение в технике получили антифрикционные сплавы (от греч. «ацтй» — против й лат. frictio — трение), так называ
КАРБИДЫ
емые кальциевые баббиты — сплавы свинца с0,75—1,1 % Са, 0,65—0,95% Маидо0,05% Li. Они предназначаются для заливки деталей, подвергающихся трению, например подшипников. Магниево-кальциевые сплавы используются в самолёто- и ракетостроении.
Широчайшее применение находят природные соединения К-, среди которых самое распространённое— карбонат СаСО3 (известняк). Значительные количества известняка идут на производство извести, бетона, гипса и цемента.
Упомянем о применении и других соединений К.: флюорит CaF2 — ценный материал для оптической промышленности; карбид СаС2 — идёт на производство ацетилена; гидрид СаН2— сильнейший восстановитель. В сельском хозяйстве применяют нитрат Ca(NO3)2 и ортофосфаты Са3(РО4)2, СаНРО4, Са(Н2РО4)2 как удобрения и подкормку для скота. С хлоридом СаС12 химики постоянно встречаются в лаборатории. Эта соль сильно гигроскопична (поглощает влагу) и её (в виде пористых кусков) помещают в эксикаторы, где держат вещества, «боящиеся» влаги. Список хорошо известных соединений К. можно было бы ещё значительно продолжить. 3. А. Старикова.
КАРБИДЫ — соединения углерода с более электроположительными элементами, главным образом с металлами. Разнообразие строения и свойств и широкий диапазон применения К. привлекают к этому классу соединений всё большее внимание. По типу химической связи между элементами (ионная, ковалентная, металлическая) все К. можно разделить на три основные группы. Характер химической связи обусловливает резкое различие в свойствах К.
1. Ионные (солеобразные) К.— соединения углерода с активными металлами I, II и III групп периодической системы Менделеева (щелочными, щелочноземельными и др.). Эти К. можно рассматривать как продукты замещения водорода металлом в метане СН4 или в ацетилене С2Н2. Такие К. разлагаются водой с выделением соответствующего углеводорода:
А14С3 + 12Н2О = 4А1 (ОН)3 + ЗСН4 СаС2 + 2Н2О = Са (ОН)2 + С2Н2
Из ионных К. наиболее известен карбид кальция СаС2.
2. Ковалентные К.— соединения углерода с кремнием и бором, SiC и В4С. Эти К. устойчивы в различных химически активных средах и при высоких температурах. Они хрупки и очень тверды. Свойства эти объясняются строением К- Так, SiC (карборунд), точнее одна из его многочисленных кристаллических форм, построен как алмаз: в кристаллической решётке алмаза половина атомов С за
156
мещена атомами Si. О карбидах кремния и бора, о структуре алмаза и вообще о сверхтвёрдых материалах рассказано также в статьях Бор, Кремний, Углерод.
3. Мета л л о подобные К-—соединения углерода с переходными металлами (см. Металлы).
Строение таких К- можно представить себе следующим образом: в кристаллическую решётку металла «внедряются» атомы углерода. Химическая связь здесь, как и в самих металлах, осуществляется свободными обобществлёнными электронами (то есть валентными электронами, утратившими связь с отдельными атомами и как бы принадлежащими всей кристаллической решётке). Участие в металлической связи принимают и электроны атомов углерода.
А поскольку К. построены не «обычно», а представляют собой так называемые фазы внедрения, их формулы зачастую не подчиняются привычным значениям валентности (например, VC, Мо2С, Fe3C, Мп3С2 и даже Сг23Св). Такие К- обладают многими свойствами, присущими металлам,— характерным блеском, высокими тепло- и электропроводностью, но отличаются от металлов хрупкостью и твёрдостью.
к. часто встречаются в метеоритах, падающих на Землю из межпланетного пространства. На самой Земле их получают только искусственно. Способы синтеза К. различны: а) взаимодействие элементов при высоких температурах:
4А1 + ЗС = А14С3 б) нагревание окислов с углем:
SiO2 + ЗС = SiC + 2СО
V2O6 + 7С = 2VC + 5СО
в) нагревание металлов в атмосфере углеводородов, например порошка магния в струе ацетилена при 500 °C:
2Mg + С2Н2 = 2MgC2 + Н2
Области применения К. исключительно разнообразны. Назовём лишь некоторые. Так, о том, что ковалентные К.— SiC и В4С — самые известные абразивные материалы, знают многие (в быту и технике они применяются, например, для шлифования). Широко распространены К. в химическом машиностроении. Но вот и менее известные сведения. Из SiC делают нагреватели высокотемпературных электрических печей (силитовые печи) и грозораз-рядники для высоковольтных линий электропередач. А из В4С изготовляют, например, сопла для пескоструйных аппаратов; такие аппараты применяют для очистки литых деталей, для снятия старой краски и грязи с домов и памятников.
Металлоподобные К. наиболее тугоплавки из всех веществ (HfC плавится при 3890 °C, ТаС — при 3887 °C; а у сложного карбида гафния и тантала /пл около 4000 °C, то есть самая высокая из всех известных температур плавления). Металлоподобные К. служат основой особо твёрдых тугоплавких сплавов.
157
КАРБОНАТЫ
Так как сами К- довольно хрупки, то для приготовления таких сплавов к К> часто добавляют порошок вязкого пластичного металла (например, кобальта) и полученную смесь прессуют и спекают. При этом образуются керамико-металлические материалы — керметы. Высокопрочные и тугоплавкие керметы (содержащие К. вольфрама, титана, тантала, ниобия и др.) — лучший материал для резания металлов и бурения горных пород. Благодаря жаропрочности и жаростойкости таких материалов из них изготовляют лопатки газовых турбин и детали реактивных двигателей.
Трудно переоценить роль самого главного К.— Fe3C. Не случайно он назван цементитом. И чугун, и сталь обязаны своей твёрдостью и износостойкостью тому, что в их структуре присутствует Fe3C. Однако углерод, содержащийся в стали, даёт К. не только с железом, но и с легирующими металлами. Одни из таких К. полезны, и их создают специально. Другие— вредны, и с ними борются. Рассказ об этом был бы очень увлекателен, но для малоподготовленного читателя — труден.
Атомная техника, космонавтика, микроэлектроника — таковы самые новые области техники, в которых уже начали использовать ценные свойства К. Г. В. Самсонов.
КАРБОНАТЫ (углекислые соли)— соли угольной кислоты Н2СО3. Поскольку угольная кислота двухосновна, она образует два ряда солей: средние (карбонаты) с анионом СО|“ и кислые (гидрокарбонаты, или бикарбонаты) с анионом НСО^. Без разложения при довольно высоких температурах плавятся только средние К. щелочных металлов. Средние К. остальных металлов при нагревании, не плавясь, разрушаются до соответствующих окислов, например:
MgCO3 = MgO + СО2
Интересно, что средние К. двухвалентной меди й трёхвалентных металлов настолько непрочны, что разлагаются уже при обычных условиях и поэтому вообще не могут быть выделены.
Устойчивость гидрокарбонатов ещё ниже, чем средних К. Уже при небольшом нагревании они разрушаются с образованием средних К. и выделением воды и углекислого газа. Например, гидрокарбонат натрия разлагается несколько выше 100 °C:
2NaHCO3 = NaaCO3 + Н2О + СО2
В воде растворимы только средние К. щелочных металлов и аммония и практически все гидрокарбонаты.
Угольная кислота Н2СО3 — кислота слабая. Поэтому её соли подвержены гидролизу — обратимой реакции с водой:
Na2CO3 + H2O^±NaHCO3 + NaOH
NaHCO3 -HH2O^±H2CO3 + NaOH
или в ионном виде:
СО23"+ Н2О нсо3- + он-
нсо3- 4- Н ,0 Н 2СО3+ОН -
При обычных условиях гидролиз к. протекает главным образом по первой ступени и завершается образованием ионов НСО^. Смоченная такими растворами лакмусовая бумажка синеет, указывая на щелочную реакцию. При действии кислот (даже таких слабых, как уксусная) все К. выделяют двуокись углерода:
СаСО3 4- 2НС1 = СаС12 + Н2О + СО2
В природе К. распространены довольно широко. Именно в их залежах (а не в живом веществе, каменном угле и нефти, атмосфере и гидросфере) сосредоточено основное количество углерода, которым располагает наша планета. Среди природных К- главное место занимает минерал кальцит СаСО3, часто образующий в виде известняка и мела громадные горные хребты. Значительно реже встречается другая форма СаСО3 — белоснежный иди красиво окрашенный примесями мрамор. О пере-
Рис. 1. Фрагменты мраморных плит. Красивым рисунком и окраской мрамор наделён от природы. Особенно же ярко они проявляются от прикосновения человеческих. рук — после полировки.
КАРБОРУНД
158
мещении кальция в виде СаСО3 и Са (НСО3)2 на Земле мы рассказали в статье Кальций. В земной коре находятся и К. многих других металлов: магния, железа, цинка, марганца, свинца, бария. Иногда минералы на основе солей угольной кислоты имеют сложный состав. Таковы, например, доломит MgCO3-CaCO3 и знаменитый малахит СиСО3-Си (ОН)2, зелёный камень, идущий на изготовление украшений.
О том, насколько разнообразны области применения природных К., можно судить хотя бы по СаСО3. Известняк — и хорошо известный строительный камень, и сырьё для производства извести’, мел используют и как полировочный материал, и для получения минеральных красок; а мрамор—для отделочных работ в строительстве и для скульптуры. Доломит MgCO3-CaCO3 и магнезит MgCO3 после обжига дают хорошие огнеупорные материалы. А природные К. свинца, цинка, марганца — ценные руды, из которых выплавляют металлы. Однако многих нужных К. в природе либо нет вообще, либо недостаточно, и человек получает их искусственно. Важнейшие из них —сода (Na2CO3 и NaHCO3) и поташ К2СО3 (см. Калий). Б. А. Поповкин.
КАРБОРУНД — техническое название карбида кремния SiC. См. Карбиды, Кремний. Название «К.» произведено от двух слов: латинского carbo — уголь, и «корунд» — очень твёрдый минерал (состава А12О3).
КАУСТИЧЕСКАЯ СОДА (от лат. caus-ticus — жгучий, едкий) — то же, что едкий натр NaOH. См. Натрий, Сода.
КВАСЦЫ — кристаллогидраты двойных сернокислых ~ солей (сульфатов) типа K2SO4*Al2 (SO4)3-24H2O или, в другой форме записи, К[А1 (SO4)2b 12Н2О.
Чем отличаются друг от друга эти формулы? Только ли тем, что во втором случае все коэффициенты в два раза меньше, чем в первом? И что означают во второй формуле. квадратные скобки?
Первую, наиболее «обычную» формулу надо понимать так: К-—это смешанные, или двойные, соли. И действительно, в водных растворах они диссоциируют так, как будто состоят из отдельных сульфатов:
K2SO4 • A12(SO4)3 • 24Н2О = 2К++2АР+ +4SO£-+24H2O
Формула же с квадратными скобками говорит о том, что К• можно рассматривать как комплексные соединения. Это значит, что у К- в твёрдом состоянии в одних узлах кристаллической решётки находятся катионы калия К+, а в других — сложные анионы [Al (SO4)2] -. И можно даже ожидать, что в специально подобранных растворителях при низких температурах К- будут диссоциировать так, что в раствор перейдут (наряду с катионами К+) именно эти сложные, заключённые в квадратные скобки, анионы. (О комплексных соединениях можно прочитать и в других статьях книги, например Кобальт.)
Кроме алюминиевокалиевых К. (они бесцветны), есть К. и другого состава: калий может быть заменён другим одновалентным катионом (аммонием, цезием, рубидием), а алюминий — трёхвалентными хромом или железом. Таковы, например, светло-фиолетовые железоаммониевые К. (NH4)2SO4-Fe2 (SO4)3-24H2O или тёмно-фиолетовые хромокалиевые К. (их обычно называют просто хромовыми) K2SO4-Cr2 (SO4)3-24H2O.
Все К- в твёрдом состоянии, независимо от того, какие именно элементы входят в их состав, кристаллизуются в формах кубической системы. Структура кристаллов не изменяется, если часть атомов калия заместить, например, аммонием, а хром — железом. Это явление, широко распространённое и среди других классов соединений, называется изоморфизмом (об изоморфизме см. в статье Минералы).
Получить К. нетрудно: они кристаллизуются из растворов, содержащих смесь соответствующих сульфатов. А повторное растворение и кристаллизация позволяют выделить К. в очень чистом виде. На воздухе кристаллы К. выветриваются, то есть постепенно теряют кристаллизационную воду и рассыпаются в порошок. Поэтому сохранить их можно лишь в закрытых сосудах.
Применяют К. издавна:
Так, с алюминиевыми К., как с протравой при крашении тканей, люди познакомились на много веков раньше, чем с самим алюминием. Металл даже назван по латинскому наименованию квасцов — alumen. «Протравливание» ткани, то есть обработка её растворами К-, позволяет красителю прочно удерживаться на ткани. В зависимости от того, какие были выбраны К*, краситель ализарин даёт на ткани различные цвета: с железными — фиолетовый, с хромовыми — коричневый, с алюминиевыми — от жёлтого до красного. А если материал предварительно не обработать К-, ализарин оставит его вообще бесцветным.
159
КИСЛОРОД
Карл Вильгельм ШЕЕЛЕ —один из крупнейших химиков XVIII в. Впервые получил кислород.
Шведский химик Карл Вильгельм Шееле родился 9 декабря 1742 г. в Штральзунде в семье мелкого торговца. После обучения в начальной школе Шееле в возрасте 15 лет покинул родительский дом и отправился в Гётеборг. Там он поступил учеником к аптекарю. В аптеках разных городов Швеции протекала и вся его дальнейшая работа. С 1775 г. и до конца жизни Шееле заведовал аптекой в Чёпинге. Он умер 21 мая 1786 г.
Научные исследования Шееле вёл с самым простым оборудованием (колбы, реторты, пивные стаканы, бычьи пузыри для собирания газов) и в крайне неблагоприятных условиях — то на подоконнике аптечной лаборатории, то в холодном деревянном сарае. Однако благодаря редкой наблюдательности, таланту экспериментатора и настойчивости он обогатил химию множеством важных открытий. Шееле открыл хлор и марганец. Он был непосредственно причастен к открытию бария, молибдена и вольфрама, впервые получив соединения этих неизвестных ещё элементов (окись бария, трёхокиси молибдена и вольфрама). Благодаря его блестящему искусству стали известны мышьяковая, кремнефтористоводородная и синильная кислоты. Он впервые выделил из соков растений органические кислоты (винную, лимонную, яблочную, щавелевую) и глицерин из жрров. Разработал способ получения фосфора из костяной золы. «Он не мог прикоснуться к какому-либо телу без того, чтобы не сделать открытия» — так впоследствии оценили его деятельность потомки.
Крупнейшее достижение Шееле — это открытие «огненного воздуха», то есть кислорода. Шееле получил его в 1768—1773 гг. нагреванием окиси ртути, селитры и других веществ и отметил способность нового газа энергично поддерживать горение. Однако его «Химический трактат о воздухе и огне» с описанием этих опытов вышел в свет только осенью 1777 г., когда уже были опубликованы работы Дж. Пристли и А. Лавуазье. Будучи сторонником гипотезы флогистона, Шееле не смог правильно объяснить роль кислорода в процессах горения и дыхания.
Больше всего Шееле ценил возможность спокойно заниматься химическими экспериментами, которые, по его выражению, «заставляли сердце смеяться». Поэтому он до конца жизни оставался аптекарем, отклонив множество выгодных предложений из различных университетов. Несмотря на своё скромное общественное положение, Шееле был в 1775 г. избран членом Стокгольмской академии наук. Его работы собраны в двух книгах «Полное собрание сочинений по физике и химии», которые вышли уже после смерти учёного. В Стокгольме поставлен памятник Шееле. Он изображён сидящим у химической печи, держа в руке тигельные щипцы.
Хорошо знакома с К- (в частности, с хромовыми) и кожевенная промышленность; здесь К- применяют как дубящие вещества, и недаром «хром» — название одного из лучших сортов кожи. А сам процесс выдерживания кожи в растворе К*, «заквашивание», видимо, и дал К-их русское название. Алюминиевокалиевые К- входят в состав «дубящего фиксажа» — фотографических фиксирующих растворов; К- способствуют уплотнению желатинового слоя, что предохраняет его от царапин и других повреждений. Благодаря вяжущим и антисептическим свойствам алюминиевокалиевые К. применяют в медицине. В. JJ. Василевский
•
КИСЛОРОД (лат. Oxygenium) О — химический элемент VI группы периодической системы Менделеева; атомный номер 8, атомная масса 15,9994. Элемент имеет три устойчивых изотопа: 1в0, 17О, 180, среднее содержание которых
составляет соответственно 99,759%,
0,037%и 0,204%от общего числа атомов К. на Земле. В свободном состоянии К. встреча
ется в виде двух аллотропных модификаций — «обыкновенного» К. О2 и озона О3. При нормальных условиях «обыкновенный» К.— газ без цвета и запаха. Трудно назвать другой элемент, который играл бы на нашей планете такую же исключительно важную роль, как К.
Кислород в природе. К.— всюду. В виде соединений он распространён на земной поверхности так, как никакой другой элемент. Он составляет около % весовых частей воды (88,8% по массе), которая, в свою очередь, занимает почти 3/4 земной поверхности (см. Гидросфера}. В твёрдой наружной оболочке Земли — земной коре на долю связанного К. приходится 47,0% по массе или 58% по числу атомов (это значит, что из каждых 100 атомов земной коры 58 — это атомы К-).
В главных породообразующих минералах, из которых состоят горные породы, слагающие земную кору,— силикатах и алюмосиликатах, большие по размерам ионы О2- расположены так плотно, что касаются или почти касаются друг друга. Остальные наиболее распространённые элементы — кремний и алюминий — образуют небольшие ионы (ион Si4+ — в 3,5, а А13+ — почти в 3 раза меньше размером, чем О2-). Такие ионы
КИСЛОРОД
размещаются в свободном пространстве между ионами О2- так, что, по некоторым подсчётам, на долю К- приходится свыше 3/4 объёма земной коры. Результат почти фантастический!
Вместе с азотом и незначительным количеством других газов свободный К. О2 образует атмосферу, где его содержится 23,15% по массе или 20,95% по объёму — всего же 1,5-1015 т. Несмотря на незначительную растворимость К. в воде, общее количество растворённого в водоёмах свободного К. весьма велико: 1,5-1013 т. Если бы растворённый К. вернулся в атмосферу, то занял бы при нормальных условиях объём свыше 10 млн. км3. Много? Но, как мы видим, это лишь 1% от общего количества свободного К. в атмосфере! К. в связанном состоянии входит в состав таких биологически важных веществ живых организмов, как вода, белки, жиры, углеводы, нуклеиновые кислоты и др. В целом К. составляет 50—85% веса животных и растительных тканей.
О роли К. в природе написаны тысячи книг. Ведь все животные и растения дышат кислородом и вне кислородной атмосферы просто не могут существовать (лишь некоторые микроорганизмы, так называемые анаэробы, живут без К.). Вдыхаемый К. окисляет в организмах углеводы и другие вещества, и в этом процессе биологического окисления как раз и выделяется та энергия, которая расходуется затем на все жизненные проявления. К. выполняет на нашей планете и огромную санитарную роль: на первый взгляд покажется странным, что речь идёт здесь об участии К. в процессах ... гниения. Но ведь именно при гниении К. окисляет остатки погибших животных и растений, и в этом процессе в конечном итоге углерод и водород органических соединений превращаются в двуокись углерода и воду. А процессы горения! Они происходили и происходят на Земле и независимо от человеческой деятельности. Что же касается че
ловека, то для него горение топлива издавна
служит источником и тепла, и энергии, приво-
дящей в движение машины.
Практически весь свободный К. нашей планеты возник и сохраняется благодаря неуто-
мимой работе растений, выделяющих его
процессе фотосинтеза:
6СО2 + 6Н2О + 2,83 кдж
хлорофилл ---------------► (солнечный свет)
— С.Н12О. + 6О2 Q 14 Q I А
Процессы дыхания животных и растений, а
также гниения и горения действуют в направлении, обратном фотосинтезу, и переводят К. атмосферы в связанное состояние:
CeHi2Oe + 6О2 = 6СО2 + 6Н2О + 2,83 кдж
160
Заметим, что общее количество К., которое выделяется растениями нашей планеты при питании — фотосинтезе, несравненно больше того, что потребляет зелёная масса при дыхании. Ежегодно растительный мир Земли возвращает в атмосферу около 400 млрд, т К-, причём, по некоторым подсчётам, «львиную долю» его дают морские водоросли и гораздо меньшую — растения суши.
Подсчитано, что если бы фотосинтез прекратился, то в течение 2000 лет весь К. атмосферы был бы израсходован. Все эти и многие другие окислительно-восстановительные процессы с участием К. в живой и неживой природе и составляют круговорот кислорода в природе
Круговорот кислорода тесно связан с круговоротом углерода — вы убедитесь в этом, изучив рисунок 1 к статье Углерод, Но процессы, идущие в природе с участием кислорода, ещё многообразнее. Вероятно, в круговорот кислорода включены не только внешние сферы Земли — атмосфера, гидросфера, земная кора, но и более глубокая — подстилающая земную кору — мантия.
Сейчас предпринимаются первые попытки «прощупать» вещество мантии — пробурить алмазными бурами скважины в тех местах, где расстояние до мантии минимально. Мощная плавучая буровая установка уже сооружена в Тихом океане. Пройдёт немного времени и в руках исследователей окажется первый столбик (керн) вещества мантии. А тогда представление о круговороте кислорода приобретёт большую законченность и ясность.
Историческая справка. Процессы горения и дыхания издавна привлекали внимание учёных. Первые указания на то, что не весь воздух, а лишь его «активная» часть поддерживает горение, имеются в китайских рукописях VIII в. Много позже итальянский учёный и художник Леонардо да Винчи (1452 г.—1519 г.) рассматривал воздух как смесь двух газов, лишь один из которых расходуется при горении и дыхании. Позднее к разгадке этой тайны близко подходили и другие учёные. Однако окончательное открытие двух главных элементов воздуха— азота и К-, сделавшее эпоху в науке, произошло в конце XVIII в. Элемент К* был почти одновременно получен шведским учёным К- Шееле (1768— 1773 гг.) путём прокаливания селитры (KNO3, NaNO3) и других веществ и английским — Дж. Пристли (1774 г.) при нагревании окиси ртути HgO. В 1772 г. Д. Резерфорд (Англия) открыл азот. А в 1775 г. знаменитый А. Лавуазье (Франция) провёл количественный анализ воздуха и пришёл к выводу, что он «состоит из двух (газов) различного и, так сказать, противоположного характера», то есть из азота и К. На основе широких экспериментальных исследований Лавуазье дал правильное объяснение горения и дыхания как процессов взаимодействия веществ с К- Поскольку К. входил в состав кислот, Лавуазье назвал его oxygenium, то есть «образующий кислоты» (от греч. слов «оксюс» — кислый и «геннбо» — рождаю); отсюда и русское название «кислород».
161
КИСЛОРОД
Джозеф ПРИСТЛИ. Одним из первых получил кислород.
Английский учёный Джозеф Пристли родился 13 марта 1733 г. в Филдхеде, близ Лидса, в семье ткача. По окончании духовной академии в Девентри (1755 г.) жил в разных городах Англии, был священником в общинах диссидентов (лиц, несогласных с учением англиканской церкви), преподавал древние и новые языки, математику, физику. В 1773—1779 гг. был секретарём богатого вельможи лорда У. «Шелборна. Вместе с ним посетил Париж, где познакомился с А. Лавуазье и другими крупнейшими учёными. Позднее обосновался в Бирмингеме, где и вёл исследования в собственной лаборатории.
У властей и духовенства он всегда слыл человеком неблагонадёжным из-за приверженности к идеям философов эпохи Просвещения и постоянной полемики с церковниками. Он восторженно встретил события Великой французской революции, и Национальное (учредительное) собрание присвоило ему звание гражданина Франции. 14 июля 1791 г., во вторую годовщину взятия Бастилии, в Бирмингеме произошли беспорядки, спровоцированные реакционными элементами. Толпа погромщиков ворвалась в жилище Пристли, уничтожила его рукописи, лабораторию, библиотеку и подожгла дом. Это произошло на глазах Пристли, скрывшегося в соседнем здании. Ему пришлось бежать в Лондон, но продолжавшаяся травля вынудила учёного эмигрировать в США. Там он и умер 6 февраля 1804 г. в Нортумберленде.
Своей главной задачей Пристли считал сочинение богословских и философских трактатов. Химией же он, по собственному признанию, занимался как дилетант, не зная и первых её основ. Тем не менее ему принадлежат важные открытия в области пневматической химии (химии газов). В те времена газы считались всего-навсего различными видами воздуха. До работ Пристли были известны только «горючий воздух» (водород, а также смеси горючих газов, выделяющиеся при сухой перегонке дерева, каменного угля и др.) и «связываемый воздух» (двуокись углерода, названная так потому, что её «связывают», то есть поглощают, едкие щёлочи и известь). Пристли, живший одно время в Лидсе, по соседству с пивоваренным заводом, заинтересовался газом, выделяющимся^ при брожении пивного сусла. Он показал тождество этого газа со «связываемым воздухом» и обнаружил (1771 г.), что зелёные растения при действии света делают этот газ пригодным для горения и дыхания. Говоря современным языком, Пристли открыл фотосинтез — усвоение растениями углерода из СО2 и выделение при этом кислорода. В 1774 г. учёный, нагревая в фокусе двояковыпуклой линзы окись ртути, плавающую на поверхности ртути, получил газ, в котором свеча горела необычайно ярким пламенем. Он назвал новый газ «дефлогистированным воздухом». Но ни Пристли, ни К. Шееле, получивший этот газ несколько ранее, не смогли понять значения своего открытия. Величайшее событие тех лет — открытие кислорода — первым оценил А. Лавуазье.
Кроме кислорода, Пристли получил новые газы: окись азота, аммиак, хлористый водород, сернистый. газ и другие. Свои работы по пневматической химии он собрал в трёхтомнике «Опыты и наблюдения над различными видами воздуха». Пристли обладал острой наблюдательностью и был превосходным экспериментатором. Он обогатил химию множеством важных фактов, но не смог ни истолковать, ни обобщить их: до конца жизни он упорно придерживался гипотезы флогистона. Даже в 1800 г., когда она была везде сдана в архив, Пристли опубликовал сочинение «Теория флогистона доказана и сложность воды опровергнута», которое считал вершиной своей научной деятельности. ъ
Пристли был членом Лондонского королевского общества, иностранным членом академий наук в Петербурге и в Париже.
Атом и молекула, кислорода. Мы видели, что из трёх природных изотопов К. содержание лёгкого, 16О, резко преобладает. Это связано с тем, что ядро атома 16О состоит из 8 протонов и 8 нейтронов и его массовое число (16) кратно четырём. Физики установили, что такие ядра обладают особой устойчивостью.
В соответствии с положением К. в периодической системе элементов Менделеева . восемь электронов атома О располагаются на двух оболочках: 2 электрона — на более близкой к ядру и 6 электронов — на внешней (полная электронная конфигурация ls22s22p4; конфи
гурация внешних электронов 2sa2p4, см. Атом). Принимая на внешнюю оболочку недостающие для её завершения 2 электрона, К. проявляет отрицательную степень окисления.
Отрыв электронов с наружной оболочки (что отвечало бы положительной степени окисления) требует такой большой затраты энергии, что при химических реакциях не реализуется. Лишь в исключительных случаях (например, в соединении с фтором OF2) К. имеет степень окисления +2. Валентность К. в соответствии с числом образуемых им химических связей равна II.
1 1 хэш
КИСЛОРОД
162
Молекула «обыкновенного» К- двухатомна (О2).
Ещё не так давно её изображали так О: :О или 0=0 (здесь каждая чёрточка соответствует одной общей паре электронов). Однако было установлено, что кислород парамагнитен, то есть при помещении в поле он намагничивается в направлении поля. Физическая теория говорит, что это возможно лишь в том случае, когда в молекуле имеются непарные электроны : О: О: С такой формулой согласуется и тот хорошо известный химикам факт, что К. во многих случаях активно реагирует целой своей молекулой и образует перекиси (например, водорода перекись Н—О—О—Н). При этом молекула 02 ведёт себя как радикал с непарными электронами.
В приведенной формуле атомы О связаны между собой лишь одной парой электронов. Однако установлено, что связь атомов в молекуле 02 прочнее, чем связь, обусловленная только одной электронной парой. Поэтому и формула : О: О: недостаточно точно выражает строение молекулы 02. Приходится считать, что не только одна общая электронная пара, но и другие электроны каким-то образом помогают связать атомы между собой. Этой сложной теме посвящены многие теоретические и экспериментальные исследования современной неорганической химии.
Физические свойства. При нормальных условиях К. имеет плотность 1,42897 г/л. При —182,9 °C и 760 мм рт. ст. конденсируется в бледно-синюю жидкость (плотность 1,13 г/см3), а при —218,7 °C затвердевает, образуя синие кристаллы.
Долгое время К- относили к «постоянным газам», так как при обычной температуре не могли превратить его в жидкость никаким, даже очень высоким давлением. В дальнейшем выяснилось (Д. И. Менделеев, 1861 г., английский химик Т. Эндрюс, 1869 г.), что для каждого газа существует так называемая критическая температура, выше которой одно только увеличение давления неспособно сблизить отдельные молекулы настолько, чтобы газ сконденсировался. Для К. такая температура лежит около—119 °C, то есть много ниже, чем для Cl2, СО2, SO2 и некоторых других газов, сжижающихся под давлением уже при обычной температуре.
К. сравнительно мало растворим в воде (5 объёмов К. в 100 объёмах воды при О °C). Однако азот, аргон и другие газы, входящие в состав воздуха, за исключением СО2, растворяются в воде ещё хуже К. Поэтому в воздухе, растворённом в воде, относительное содержание К. повышается.
Химические свойства. К.— активный неметалл. Известны его соединения со всеми элементами, кроме лёгких инертных газов— гелия, неона и аргона. С галогенами, криптоном, ксеноном, золотом и платиновыми металлами К. непосредственно не реагирует, так что соединения его с этими элементами получают косвенным путём. Все остальные элементы способны соединяться с К. непосредственно. Почти все эти процессы — реакции окисления, экзотермичны, то есть сопровождаются выделением энергии. Если реакция идёт достаточно быстро, а энергия выделяется в виде тепла и света, процесс называют горением.
Лёгкость взаимодействия металлов с К. различна. Наиболее активны в таких реакциях щелочные металлы, легко окисляются и щелочноземельные металлы. Взаимодействие зависит и от многих других факторов — состояния поверхности металла, степени его измельчения, присутствия примесей. Так, хром или алюминий в обычных условиях окисляются лишь с поверхности, причём образовавшаяся тонкая плёнка окисла (её называют защитной плёнкой) препятствует дальнейшему окислению. Повышение температуры и уменьшение размеров частиц всегда ускоряют реакцию с К- Железо в обычных условиях окисляется медленно. Но раскалённая докрасна железная проволока горит в кислороде:
3Fe + 2О2 = Fe3O4
Приготовленный же специальным образом же
лезный порошок — пирофорное железо — са
мовоспламеняется на воздухе уже при обычной
температуре.
Исключительно важна и роль воды в процессах окисления. Например, даже такой активный металл, как калий, с совершенно лишённым влаги К- не реагирует, но воспламеняется в нём при обычной температуре в присутствии ничтожных количеств паров воды. Вот случай, когда вода не гасит огонь, а вызывает его!
Подсчитано, что в результате коррозии, то есть окисления металлов на воздухе при совместном действии К. и влаги, ежегодно теряется от 1 до 1,5% всего металла, извлечённого людьми из недр Земли и воплощённого в изделиях и конструкциях.
Взаимодействие К. с серой, углеродом, азотом, фосфором происходит при обычных условиях очень медленно (скорость реакции существенно зависит и от того, с каким аллотропным видоизменением имеют дело; так, белый фосфор окисляется значительно легче, чем красный). При повышении температуры скорость реакции возрастает, так что при некоторой, характерной для каждого элемента (и для каждой аллотропной модификации), температуре воспламенения начинается горение. Взаимодействие азота с К.:
N2 + O2 = 2NO
благодаря особой прочности молекулы N2 эн-дотермично (идёт с поглощением энергии) и становится заметным лишь при температуре выше 1200 °C или в электрическом разряде. Большой интерес представляет реакция соединения К. с водородом:
2Н2 + О2 = 2Н2О
(подробнее см. Водород).
К. взаимодействует и со многими сложными веществами. Водородные соединения неметал
163
лов, например сероводород и аммиак, горят в присутствии К.:
2H2S + ЗО2 = 2SO2 + 2Н2О (при избытке К.) 2H2S + О2 = 2S + 2Н2О (при недостатке К.) 4NH3 + ЗО2 = 2N2 + 6Н2О (вотсутствии катализатора).
Окисление аммиака кислородом воздуха в присутствии катализатора даёт окись азота NO. На этом основано производство азотной кислоты. А с реакцией окисления метана мы встречаемся повседневно дома. Вспомните, как вспыхивает голубым пламенем газовая горелка. Это горит смесь метана (главная составная часть природного газа) с воздухом, давая желанное тепло:
СН4 + 20 2 = С02 + 2Н2О
Взаимодействие К. с другими видами топлива (торф, уголь, керосин, бензин) — по сей день важный источник тепла и энергии в промышленности, на транспорте и т. д.
Получение. Источниками К. в промышленности служат вода и воздух. Из воды К. получают электролизом, причём к воде для увеличения её электропроводности добавляют NaOH или H2SO4, которые практически не расходуются. О выделении К- из воздуха см. Воздух. Хранят и транспортируют газообразный К- в стальных баллонах под давлением 150 атм, а в заводских условиях — в специальных резервуарах (газгольдерах). Жидкий К. хранят и перевозят в сосудах с двойными стенками, из пространства между которыми для уменьшения теплопроводности выкачан воздух.
В небольших количествах К. можно получить разложением перманганата калия при нагревании:
2КМпО4 = К2МпО4 + МпО2 + О2
Для этой же цели можно использовать сурик РЬ3О4, окись ртути HgO, бертоллетову соль КС1О3, селитры, например KNO3. К. выделяется при разложении перекиси водорода, особенно бурно в присутствии катализаторов — мелко раздробленной платины, солей железа и др.:
2Н2О2 = 2Н2О + О2
Эта реакция в перспективе может быть использована для снабжения водой и К. экипажа космических кораблей. Подсчитано, что 2 кг 95 %-ной Н2О2 достаточно, чтобы обеспечить минимальную суточную норму воды и К. одному космонавту. А поскольку разложение Н2О2 — процесс экзотермический, выделяющееся тепло (800 ккал/кг) тоже можно использовать.
КИСЛОРОД
Очень удобным источником К. служат перекиси щелочных металлов. Так, на советских кораблях-спутниках «Восток» и «Восход» необходимый для дыхания К. получали по реакции:
4 КО 2 + 2Н2О = 4КОН + ЗО2 Выделяющаяся при дыхании СО2 одновременно поглощалась гидроокисью щелочного металла:
2 КОН + СО2 = К2СО3 + Н2О Суммарное уравнение:
4КО2 + 2СО2 = 2К2СО3 -г ЗО2
Применение. Мировое промышленное потребление К. составляет около 100 тыс. т в день. Известно, что в производстве чугуна и стали в конвертеры, доменные и мартеновские печи вдувается воздух. Однако для металлургических процессов необходим лишь К. Азот воздуха «разбавляет» К., уменьшает скорость реакций, уносит с собой значительное количество тепла. В конвертерной выплавке стали азот попросту вреден, так как поглощается металлом, снижая его качество. Вот почему даже небольшое обогащение воздуха кислородом (до 25—30%) ускоряет металлургические процессы, повышает производительность аппаратуры и качество металла, снижает расход сырья. К. или обогащённый им воздух применяют также при обжиге сульфидных руд в производстве меди, никеля, свинца.
Высокая температура, развивающаяся при горении ацетилена в К. (около 3000 °C), используется для сварки и резания стальных изделий. Если нагреть сталь до достаточно высокой температуры, а затем направить на нагретую поверхность струю чистого К., металл горит. Перемещая струю К-, удаётся разрезать листы из стали и других металлов и сплавов. Жидкий К. используют как окислитель топлива в ракетных двигателях. Пропитанные жидким кислородом угольный порошок, древесные опилки и другие вещества (такие смеси называются оксиликвитами) применяют при взрывных работах.
К. нужен и в медицине: в концентрации 40— 95% им дают дышать при заболеваниях сердца и лёгких. В ряде случаев К. вводят под кожу или дают больным воду, газированную им под давлением. «Тяжёлый» К. 18О применяется в качестве «меченого атома» в научных исследованиях.
К. используют в многочисленных технологических процессах химической промышленности (см. также Воздух}.
Можно ли из 105 известных нам элементов считать какой-либо «главным»? В известном смысле — да. В космосе это — водород, первый по распространённости элемент Вселенной.
11*
КОБАЛЬТ
164
58,9332 27
Со
3d'4s!
КОБАЛЬТ
С него начинаются эволюция звёзд и ядерный синтез более тяжёлых элементов. В условиях нашей планеты главная роль принадлежит К. И не случайно ещё 150 лет назад известный шведский химик Я. Берцелиус сказал: «Кислород является центром, вокруг которого вращается вся химия». В. Л. Василевский. КОБАЛЬТ (лат. Cobaltum) Со — химический элемент VIII группы периодической системы Менделеева; атомный номер 27, атомная масса 58,9332. Серебристо-белый металл со слабым красноватым оттенком. Природный К- представлен одним устойчивым изотопом 59Со.
Ещё в древних Египте и Китае мастера умели готовить красивые смальты — непрозрачное стекло, из которого делали цветную мозаику. Были среди них и смальты удивительно глубокого синего цвета — теперь мы знаем, что их получали сплавлением кобальтовой руды, кварцевого песка и поташа. (Кстати, само слово «смальта», по-немецки Schmalte, происходит от немецкого schmelzen — плавить.) Красивый синий цвет и придавали смальте соли К- По-видимому, синее кобальтовое стекло ценилось в Египте очень высоко. Ваза из него была найдена в гробнице фараона Тутанхамона (XIV век до н. э.). Секрет приготовления таких стёкол был утерян и вновь открыт только в XVI в. знаменитыми мастерами Венеции. Сам же металл впервые выделил шведский химик Г. Брандт в 1735 г. из руд, по виду очень похожих на медные, но не дающих металла при попытках его выплавить. Немецкие рудокопы назвали эти руды «кобольдами» по имени злого и насмешливого горного духа, или гнома, мешающего работать горнякам; отсюда и название элемента.
К. встречается в природе всегда вместе с никелем. Известно свыше 30 минералов, содержащих К.; в основном это мышьяковистые и сернистые соединения. Важнейшие минералы: кобальтовый колчедан (Fe, Co)S2, кобальтовый блеск (кобальтин) CoAsS, шпейсовый кобальт (смальтин) CoAs2. По распространённости в земной коре К. занимает 34-е место, составляя 1,8-10~3% по массе (а на 35-м месте стоит свинец). Обнаружен К- и в железных метеоритах. В Советском Союзе его добывают из полиметаллических руд на Урале и Кольском полуострове. К.— ценный металл; его руды считаются промышленными даже при содержании 0,1% Со.
К.— ковкий, хорошо полирующийся металл с плотностью 8,9 г/см3. Он плавится при 1493 °C и кипит при 3100 °C. По магнитным свойствам К., как и никель, напоминает железо, то есть относится к числу ферромагнетиков (см. Железо). К-— типичный представитель переходных металлов (см. Металлы). Кон
фигурация внешних электронных уровней в его атоме 3d74s2 (см. Атом). По химическим свойствам К- похож на железо, но менее активен. В большинстве своих соединений К. имеет степени окисления 4-2 и +3 (валентности II и 111). При обычной температуре влага и кислород воздуха на компактный металл не действуют, тогда как порошкообразный К. с диаметром частиц менее 5 мк (1 мк=0,001 мм) пирофорен, то есть самовозгорается при соприкосновении с воздухом. При нагревании К. на воздухе до белого каления образуется смешанный окисел чёрного цвета Со3О4 (то есть СоО-Со2О3). Осторожно прокаливая без доступа воздуха карбонат СоСО3, можно получить оливково-зелёный СоО, а разложением нитрата Co(NO3)2 — бурый Со2О3. Окислы К. нерастворимы в воде и в щелочах, а при действии кислот образуют соединения двухвалентного К.:
СоО + 2НС1 = СоС12 + Н2О
Со2О3 + 6НС1 = 2СоС12 + ЗН2О + С12
Таким образом, Со2О3 является сильным окислителем.
Розовую гидроокись Со(ОН)2 получают действием щелочей на соли К.:
CoSO4 + 2NaOH = Со (ОН)2 + Na2SO4
На воздухе она медленно, а под действием сильных окислителей быстро окисляется до бурой гидроокиси Со(ОН)3:
2Со (ОН)2 + NaClO + Н2О = 2Со (ОН)3 + NaCl С галогенами К. соединяется при обычной температуре:
Со + Вг2 = СоВг2
а с другими неметаллами, кроме азота, при нагревании:
Со + S = CoS
Плавиковая кислота на К. не действует, а в разбавленных серной, азотной и соляной кис-
Рис. 1. Палитра кобальтовых красок.
165
КОБАЛЬТ
Рис. 2. Постепенное отщепление воды от СоС12«6НаО при нагревании сопровождается изменением структуры и цвета соединений.
а— Для комплексного соединения [СоС12 (Н2О)4] *2Н2О (розового цвета) показана внутренняя сфера, то есть [СоС12(Н2О)4].
б — Строение розовато-фиолетового СоС12 «2Н2О. в — Строение голубого СоС12.
лотах он растворяется с образованием соединений двухвалентного К., выпадающих из раствора в виде кристаллогидратов красного цвета: CoSO4-7H2O, Co(NO3)i-6H2O и СоС1а-6НаО. Последнее соединение начинает терять кристаллизационную воду даже при слабом нагревании.
При 49 °C от СоС1а-6НаО отщепляются две молекулы воды, при 58 °C — ещё две; при 90 °C — пятая, а при 140 °C— последняя, шестая молекула воды. Цвет соединения при обезвоживании постепенно изменяется, переходя от розово-красного к розовато-фиолетовому и к голубому. Во влажной атмосфере хлорид К. снова присоединяет воду и становится розово-красным.
На этом основано применение разбавленных растворов СоС1а в качестве так называемых симпатических чернил. Текст, написанный бледно-розовым раствором на бумаге жёлтого цвета, не виден, но если бумагу прогреть, то чётко выступят синие буквы.
В чём же тут дело? Почему удаление воды
приводит к таким изменениям цвета соединения? А вот почему. Шестиводный хлорид К. (сульфат и нитрат К., конечно, тоже) представ
ляет собой не «обычное», а комплексное соеди-
ненение, и его формулу правильнее писать так:
[СоС1а(Н2О)41-2НаО
Это означает, что атом Со окружён четырьмя молекулами НаО и двумя атомами С1, то есть как бы «сидит
в домике)», который имеет вид октаэдра (рис. 2, а). Химики говорят: 4 молекулы Н2О входят во внутреннюю сферу комплекса. Две другие молекулы Н2О (в формуле они стоят за квадратными скобками) не принадлежат к числу ближайших соседей Со (внешнесферная вода), и при нагревании они «уходят» первыми; при этом строение комплекса не меняется. Если нагревать дальше, то отщепляются две молекулы воды, уже непосредственно связанные с атомом Со (внутрисферная вода); при этом изменяется строение комплекса — «домик» уже не октаэдр, а тетраэдр (рис. 2,6); меняется и цвет соединения. Теперь у атома Со только четыре ближайших соседа, располагающихся по вершинам тетраэдра. Потом, с отщеплением двух последних молекул Н2О, число соседей атома Со не изменяется, то есть остаётся равным четырём, но каждый атом С! связан с двумя атомами Со и служит между ними «мостиком» (рис. 2, в).
Так, от привычных представлений о простых химических соединениях мы перешли к представлению о более сложных — комплексных соединениях. Число известных сейчас комплексных соединений намного превышает число простых. Что касается К., то у двухвалентного Со (II) ещё можно говорить о простых соединениях, но для трёхвалентного Со (III) простые соединения почти неизвестны, тогда как комплексных получено несколько тысяч. Именно на примере соединений трёхвалентных К. и хрома швейцарский учёный А. Вернер в 1893 г. обосновал, а в последующие годы развил теорию строения этих сложных соединений.
Интересное комплексное соединение — карбонил К. Соа(СО)8 (здесь степень окисления К. равна нулю), можно получить при 120— 200 °C и давлении около 300 атм по реакции:
2 СоСОз + Ю СО = Со2 (СО)8+ 4СО2
Оранжевые кристаллы карбонила К. нерастворимы в воде, но хорошо растворяются в органических жидкостях.
Получение металлического К. из полиметаллических руд, в которых он содержится,— сложный технологический процесс. После обогащения руды и обжига концентрата из него извлекают преобладающий металл данной руды. Остаток растворяют в разбавленной соляной кислоте, из раствора выделяют сопутствующие металлы. В растворе же остаются К. и никель. Для их разделения пользуются тем, что Со2+ легче переходит в Соз+, чем Ni2+ в Ni3+. При добавлении точно рассчитанного количества гипохлорита натрия NaClO в осадок выпадает только Со(ОН)3 (уравнение реакции см. выше), который отфильтровывают. Переосаждение повторяют несколько раз. Вполне чистый К. можно получить либо электролизом его солей (обычно сульфата CoSO4), либо термическим разложением карбонила К.
Долгое время применялись только соединения К.» главным образом для производства кра
КРЕМНЕВОДОРОДЫ
166
сок. Это, прежде всего, знаменитая кобальтовая синяя. И многие другие, различных цветов
и оттенков.
Но только с начала XX в. нашли применение кобальтовые сплавы, и это коренным образом изменило судьбу металла. Первым из таких сплавов был стеллит (сплав К. с хромом и вольфрамом), названный так в 1910 г. его изобретателем американским металлургом Э. Хейнзом по яркому блеску полированных изделий из этого сплава (от латинского stella — звезда). Состав стеллита в зависимости от назначения колеблется в довольно широких пределах (20—35% Сг, 5—20% W, 1—3% Fe, до 2% С, остальное Со). Сплав очень стоек против коррозии и истирания. Вследствие дороговизны стеллита его наваривают на режущие инструменты, на поверхность деталей ма
шин и т. д.
После появления стеллита К. стал одним из
важнейших металлов техники. Детали реактивных двигателей и газовых турбин изготовляют из сплава виталлиума (65% Со, 28% Сг, 3% Ni, 4% Мо), не подвергающегося газовой коррозии даже при 800 °C. К. входит в состав и других интересных и крайне важных для различных областей техники сплавов. Так, в сильно агрессивных средах ещё более стоек, чем платина, сплав, содержащий 13% Si, 7% Сг, 3% Мп? остальное Со.
однажды (в 1957 г.) в нашей печати с
А
Sfifl
щалось о таком интересном событии: при
помощи сильного постоянного магнита, изготовленного из сплава ал ни ко (50% Fe, 24% Со, 14% Ni, 9% Al, 3% Си) в виде прутка, был извлечён гвоздь из бронхов ребёнка и тем спасена его жизнь. Кроме алнико, сейчас получено множество других магнитных материалов с использованием К. Особенно перспективны (хотя ещё и мало изучены) магнитные сплавы и соединения К- с редкоземельными металлами (см. Лантаноиды).
Порошок К. применяется при изготовлении твёрдых Сплавов методом порошковой металлургии. С этой целью порошки карбидов WC или TiC тщательно перемешивают с 3—15% порошка Со, прессуют в стальных формах и затем спекают в защитной газовой среде. Технологию производства сплавов этого типа разработал немецкий учёный К. Шрётер в 1923— 1925 гг. В СССР первый такой сплав — победит (90% WC, 10% Со) был получен в 1922 г. Твёрдые сплавы необходимы для изготовления режущего и бурильного инструмента. Всего же на производство сплавов К. расходуется около 80—90% его добычи. Но, помимо этого, К.— прекрасный катализатор (ускоритель) многих реакций.
К.— очень важный для жизни элемент. При недостатке его в почве, а следовательно, и в растениях, у животных появляется тяжёлое заболевание— анемия. Поэтому К. вводят в состав микроудобрений.
Хорошо известный витамин В12, стимулирующий кроветворные функции костного мозга, содержит 4,5% Со. Благотворно влияет К. и на обмен веществ. С помощью содержащих этот элемент лекарств пытаются победить даже такую серьёзную болезнь, как диабет. Некоторые из очень сложных комплексных соединений К. способны обратимо переносить кислород, подобно гемоглобину (см. Железо). Эти соединения усиленно изучаются.
Для К., как и для других элементов, получены искусственные, не существующие в природе изотопы; все такие изотопы радиоактивны. Искусственный изотоп в0Со (его период полураспада 7\/2=5,27 года) заменил в медицине дорогостоящий радий: «кобальтовая пушка> облучает раковые опухоли, спасая многие и многие жизни.
Добыча К. неуклонно растёт; так, с 1923 г. по 1946 г. она выросла в 30 раз, а за два последние десятилетия ещё в 5 раз. В. И. Белова. КОНСТИТУЦИОННАЯ ВОДА — см. Вода. КРЕМНЕВОДОРОДЫ, силан ы,— соединения кремния с водородом. По строению молекул К. подобны углеводородам. При комнатной температуре моносилан SiH4 и дисилан Si2He —газообразные вещества, остальные К.— до последнего известного Si8H18 — летучие жидкости. К. ядовиты и имеют неприятный запах. Химически они менее устойчивы, чем углеводороды. Так, углеводороды загораются только если их поджечь, а К. самовоспламеняются на воздухе:
2Si2He + 7О2 = 4SiOa + 6Н2О
Вода не реагирует с углеводородами, но разлагает К-, например:
Si2He + 4Н2О = 2SiO2 + 7Н2
Образованием и дальнейшим разложением SiH4 пользуются для получения чистого кремния в промышленности полупроводников.
КРЕМНЕЗЁМ — то же, что кремния двуокись SiO2.
кремниевые кислоты — соединения кремниевого ангидрида 8Ю,сводой, очень слабые кислоты. Соотношение SiO» и Н,0 в К. к. бывает самым различным. Поэтому их состав обычно представляют в виде общей формулы nSiO2-mH2O (например, вместо H4SiO4 пишут SiOf2H,O). Кислоты с разным п легко переходят друг в друга; поэтому выделить их по отдельности не удаётся. Даже угольная кислота вытесняет К- к. из их солей в водных растворах:
K«O-nSiO2 + Н,СО8 = K»CO8 + nSiO2-H2O
Образующаяся К. к. выпадает в виде белого студенистого осадка — геля. При его обез-
167
КРЕМНИИ
Кремнёвый п и столет (Россия XVII в.). Государственная Оружейная палата. Москва.
воживании получается силикагель — бесцветное пористое вещество с очень большой поверхностью, широко применяющееся для адсорбции паров и газов.
Растворимость К. к. в воде очень мала и тем меньше, чем меньше отношение т/п. В растворе присутствует, по-видимому, главным образом SiO2-2H2O — наиболее богатая водой форма. У природных К. к. т/п<^1 и в воде они практически нерастворимы. К их числу относятся минералы опал, трепел, диатомит, кремень. Соли К. к.— силикаты. В воде хорошо растворимы лишь силикаты щелочных металлов с п=1—4.
История кремня — одной из природных К- к.— очень интересна. В эпоху палеолита, то есть 800—1000 тысячелетий тому назад, кремень помог человеку в борьбе за жизнь. Этот твёрдый камень, от которого легко отколоть куски с острыми краями, послужил материалом для первого оружия (наконечников копий и стрел) и первых орудий труда (топоров, ножей и др.). Позднее, когда на смену камню пришли медь, бронза и железо, кремень нашёл использование в виде огнива. При ударе твёрдого кремня о железо от него отрываются мелкие осколки, нагретые до высокой температуры, вследствие быстрого перехода механической энергии удара в тепловую. А раскалённое железо легко загорается на воздухе:
3Fe + 2О2 = Fe3O4
и воспламеняет трут. До изобретения спичек такой способ добывания огня был широко распространён. По этому же принципу устроено и старинное оружие — кремнёвый пистолет, только в нём вместо трута воспламеняется порох.
Твёрдые кремнёвые шары и сейчас используются в мельницах для размола разных материалов. Более важное техническое применение нашли другие природные К. к.— остатки древних мельчайших морских животных и растений — диатомит и трепел. Прокаливанием их получают «белую сажу»—тонкий порошок SiO2, служащий инертным наполнителем при изготовлении резины, смазок, красок и лаков. Трепел, пропитанный нитроглицерином,— это динамит. Вследствие высокой пористости диатомита и трепела их применяют для фильтрования масел.
В крутых обнажениях по берегам рек туристы могут встретить выходы диатомитов: это указание геологам на то, что здесь могут быть ещё не открытые месторождения газа или нефти. Распознать диатомит просто: если
приложить язык к серо-белым хрупким кускам камня, язык «примораживает», как если в зимний день коснуться им железа. Туристам полезно знать ещё об одном соединении К. к.— «растворимом стекле» — бесцветном или чуть желтоватом водном растворе силиката натрия nSiO2’Na2O (где п=2—4). Этот раствор продаётся под названием «конторский клей силикатный». Отправляясь в далёкое путешествие, можно законсервировать свежие яйца, покрыв их тонким слоем такого клея.
Растворимое стекло получают сплавлением SiO2 с содой:
nSiO2 + Na2CO3 = nSiO2-Na2O + СО2
Готовят растворимое стекло, конечно, не для консервации яиц. Им пропитывают бетонные покрытия дорог, увеличивая срок их службы; дерево и текстиль после такой пропитки не возгораются.
Об использовании разнообразных силикатов см. Силикаты, Стекло, Бетон, Цемент.
Некоторые К. к. существуют в газообразной форме. Это их свойство — очень интересное и важное. При 400— 700°C и 5—500 атм SiO2 взаимодействует с водяным паром. Из образующихся К- к. в газообразное состояние переходят SiO2*2H2O, 2SiO2*3H2O и SiO2*H2O. Такой процесс происходит, по-видимому, в глубинах земной коры. Газообразные К- к. поднимаются по трещинам к поверхности земли и попадают в полости горных пород. Оказавшись при более низких температурах и давлениях, они распадаются. При этом образуются кристаллы SiO2, в том числе и кристаллы горного хрусталя. Так в природе происходит своеобразная перегонка вещества.
А. П. Пурмаль.
КРЕМНИЕВЫЙ АНГИДРИД —то же, что кремния двуокись SiO2.
КРЕМНИЙ (лат. Silicium) Si — химический элемент IV группы периодической системы Менделеева; атомный номер 14, атомная масса 28,086. Природный элемент состоит из трёх изотопов: 28Si (92,27%),29Si (4,68%), 30Si (3,05%). Тонкий порошок К. бурого цвета, а кристаллы К.—тёмно-серые с металлическим блеском.
Историческая справка. Двуокись К. SiO2 — самое распространённое химическое соединение на Земле. Обычный песок — кремнезём SiO2, и твёрдая горная порода — кремень nSiO2*/nH2O, знакомы людям уже много тысячелетий. Однако открытие К- как самостоятельного химического элемента произошло лишь в XIX в., позже, чем вольфрама, никеля, урана и других, гораздо более редких элементов.
В 1811 г. во Франции Ж. Л. Гей-Люссак и Л. Ж. Те-нар, нагревая фтористый кремний с калием, получили буро-коричневое вещество — это был К-:
28’086 14
Si
3s’3p=
КРЕМНИЙ
SiF4 + 4К = 4KF + Si
Однако тогда исследователи не поняли, что им удалось открыть новый элемент. Лишь 12 лет спустя знаменитый
КРЕМНИЙ
168
Рис. 1. Природные соединения кремния служили людям уже в каменном веке. Первые ножи и топоры, наконечники стрел и копий делались из острых осколков кремня.
шведский химик Я. Берцелиус при нагревании кремнефтористого калия с калием по реакции:
K2SiF6 + 4К = 6KF + Si получил тот же продукт и показал, что найден новый элемент, назвав его «силиций» (от латинского silex —кремень). В русском языке первоначальное название К. было «кремнезёмий». В 1834 г. академик Г. И. Гесс ввёл более короткое — «кремний» (происходит от греческого слова «кремнос» — утёс, скала).
Применение К- и его сплавов началось сравнительно недавно (см. ниже). Но природные соединения К- служили людям уже в каменном веке. Первые ножи и топоры, наконечники стрел и копий делались из острых осколков кремня (об этом рассказано в статье Кремниевые кислоты). К глубокой древности относят и кустарное . изготовление стекла. Древнеегипетские гончары для придания глиняным горшкам и кувшинам водонепроницаемости глазировали их — перед обжигом покрывали содой и песком. Глазурь по химическому составу очень близка к стеклу. Кто-то из гончаров приготовил, наверное, и самое древнее из дошедших до нас стеклянных изделий — зеленовато-коричневую бусину, найденную при раскопках Фив. Возраст этой бусины 5,5 тысячи лет!
Кремний в природе. В неорганической природе К. играет, пожалуй, столь же важную роль, как углерод в природе живой. По распространённости в земной коре К.— второй (после кислорода) элемент. Если принять всю массу земной коры за 100%, то на долю К. приходится 29,5%; по числу же атомов это составляет около 20,0%. Во всех природных соединениях атом К. связан с атомами кислорода. 12% массы земной коры составляет кремнезём SiO2 (см. Кремния двуокись) и 75% — более сложные кислородные соединения К. — силикаты (глины, полевые шпаты, сланцы, слюды и др.). В 1 л морской и пресной воды содержится около 3 мг К. в форме кремниевых кислот и их солей.
Попадая в ткани растений и отлагаясь в них, соединения К- повышают их жёсткость и способность расти вверх. Не случайно особенно много К. в растениях «высокого роста» — бамбуках, хвощах; зола одного из хвощей содержит до 20% SiO2! Много К. и в стеблях жёсткой осоки (вспомните, как неприятно — точно остриём бритвы — порезаться осокой). А теперь пофантазируем — лишим природные воды К- И тогда травы, кустарники, деревья, бессильные устремиться ввысь, оказались бы стелющимися и ползучими растениями. Общее количество зелёной массы уменьшилось бы из-за этого в несколько раз. Уменьшилось бы и количество вырабатываемого растениями кислорода. А это привело бы к уменьшению числа наземных животных, питающихся растениями и дышащих кислородом. Не исключено, что недостаток растительной и животной пищи на самых ранних этапах истории цивилизации затормозил бы развитие человеческого общества. Три миллиграмма К-в 1 литре воды предотвратили эту печальную картину. Но вернёмся к действительности.
Пониженное содержание К- вызывает задержку роста кукурузы, ячменя, бобовых, табака и других сельскохозяйственных культур. Необходим К- и морским организмам: диатомеи, радиолярии, звёзды, губки содержат довольно много К. Так, скелет некоторых губок на 88% состоит из SiO2. Известно около 2000 видов диатомовых водорослей. Их отложения в местах, бывших некогда морским дном, простираются подчас на сотни тысяч квадратных километров. В организмах высших животных содержание К- намного меньше, чем в растениях, и биохимическая роль его пока неясна.
Физические и химические свойства. Плотность кристаллического К- 2,33 г/см3, /пл 1417°С, а /кип 2600 °C. Расположение атомов Si в кристалле К. такое же, как атомов С в алмазе (см. Углерод),— каждый атом находится в центре тетраэдра и связан ковалентно с четырьмя другими атомами Si. Кристаллическая модификация К., подобная графиту, неизвестна. По твёрдости К. заметно уступает алмазу, а электрическая проводимость К. много больше. Алмаз — диэлектрик, а К.— полупроводник; его удельное сопротивление составляет 2,3-10Б ом-см при комнатной температуре и уменьшается при нагревании.
Проводимость К. увеличивается, если в его кристаллической решётке заместить некоторые атомы Si атомами элементов, принадлежащих соседним группам периодической системы. Замена Si на Р или Bi приводит к возрастанию электронной, а замена Si на А1 или В — дырочной проводимости. (О механизме электронной и дырочной проводимости см. в статье о
Рис. 2. Скелеты морских животных радиолярий. Разнообразные и изящные, они слагаются из кремнезёма SiO2. На рисунке сильно увеличены. В действительности размеры радиолярий бывают в пределах от 40—50 мк до 1 мм.
КРЕМНИИ
170
другом полупроводнике — германии.) Но, конечно, для создания на основе К. полупроводника с определёнными свойствами необходимо предварительно хорошо очистить К. от примесей. Проводимость К. возрастает и при освещении (см. ниже).
В периодической системе элементов К.— ближайший аналог углерода. Конфигурация внешней электронной оболочки атома Si 3s23p2. Иными словами, на внешней оболочке атома находятся два s-электрона и два р-электрона, всего четыре — как и у атома углерода (см. Атом). Поэтому в химии этих элементов есть общие черты.
В большинстве соединений Si (как и С) четырёхвалентен (SiH4, SiF4, SiO2, Si3N4 и др.).
В некоторых соединениях Si (так же, как и С) двухвалентен (например, в SiO).
В кремневодородах атомы Si соединены друг с ApyroM(H3Si—SiH2—SiH3), как и атомы С в углеводородах.
Известны соединения, содержащие и Si, и С. Простейшее из них — карборунд SiC, более сложное имеет формулу H3Si—СН3.
С металлами Si образует тугоплавкие силициды, подобные по составу и свойствам углеродным соединениям — карбидам.
Довольно близки Si и С и по своей способности вступать в химические реакции. При комнатной температуре лишь фтор взаимодействует с тонкоизмельчённым К.: Si4-2F2=SiF4. При 400—600 °C такая реакция идёт и с С12, и с Вг2, а в кислороде К. сгорает:
Si + О2 = SiO2.
С углеродом и азотом К. соединяется лишь при очень высоких температурах. В обычных условиях кислоты не действуют на Si (как и на С).
Есть, однако, в химии этих элементов существенные различия. В отличие от углерода, К. не склонен образовывать соединения, в которых атомы Si соединены в длинную цепь. А чем измеряется эта «склонность»? Той энергией, которая выделяется при возникновении связи между атомами, то есть энергией химической связи. Чем больше выделяющаяся энергия, тем легче образуется химическая связь. И тем устойчивее получающееся соединение. Энергия связи двух атомов углерода в цепи
1111
—С—С—С—С— почти в 2 раза больше, чем для
1111
1111
атомов К. в цепочке —Si—Si—Si—Si—. И если
Illi
у молекул углеводородов цепочка может состоять из практически неограниченного числа атомов С, соединённых друг с другом, то,
в случае кремневодородов, самая длинная цепь включает лишь 8 атомов Si (октасилан Si8H18).
Из курса органической химии можно узнать, что углерод легко образует двойные (—С=С—) и тройные (—С=С—) связи. К- же к образованию химических связей такого типа не склонен.
И ещё одно важное различие углерода и К»: энергия химической связи Si—О намного больше, чем энергия связи С—О. Поэтому, в отличие от углерода, у К- очень распространены и устойчивы соединения, в которых атомы Si
I I
и О связаны в длинные цепи —Si—О—Si—О—.
I I
К ним относятся, например, многочисленные природные и синтетические силикаты.
Сопоставьте сказанное и вам будет понятно, почему соединения углерода и К», аналогичные по составу, могут резко различаться по структуре и свойствам.
Сравним простейшие и наиболее распространённые из них — СО2 и SiO2. В молекуле двуокиси углерода атом С связан с атомами О двумя двойными связями (О=С=О). Каждая молекула СО2 существует самостоятельно и взаимодействует с другими молекулами СО2 очень слабо. Этим и объясняется, что в обычных условиях двуокись углерода — газ.
Атом Si не склонен к образованию двойной связи; энергия же одинарной связи Si—О очень велика. В молекуле двуокиси кремния каждый атом Si связан простыми связями с четырьмя атомами О, а каждый атом О связан с двумя атомами Si, так что образуется полимерное химическое соединение (SiO2)„, структуру которого можно представить как
—Si — О—Si—О —Si—О—
ООО
I
Поэтому при обычных условиях SiO2 — твёрдое и тугоплавкое вещество.
Получение и применение. Выделяют К. из кремнезёма SiO2 — дешёвого и легкодоступного сырья. Прокаливая в электрических печах SiO2 с коксом:
SiO2 + 2С = 2СО + Si
получают технический К- 95—98%-ной чистоты.
Специальными методами выделяют К. высшей степени чистоты, измеряемой «числом девяток». Так, К. марки «6 девяток» содержит один
171
КРЕМНИИ
чужеродный атом на 999 999 атомов К., то есть 99,9999% Si. Для очистки технического продукта К. сначала переводят в легколетучие соединения — SiH4, Sil4, которые очищают многократным сжижением и испарением, а затем разлагают при высокой температуре. Применяют также восстановление SiCl4 или трихлорсилана SiHCl3 водородом или цинком при нагревании. Полученный К. дополнительно очищают методом зонной плавки. Приготовление веществ со степенью чистоты в несколько девяток очень сложно и дорого.
Рассказ о самой современной области применения К. начнём издалека. Вы, наверное, наблюдали за уверенной поступью «Лунохода-2» по скалистым берегам моря Ясности. Когда луноход отправляется в путь, роль его «бензобаков» выполняют аккумуляторные батареи. Но энергии, запасённой на Земле, аппаратам хватает лишь на первое время. Постепенно батареи истощаются и им обязательно нужна подзарядка. На Земле это просто: подключить их к любой электрической цепи. В космосе же электростанции и линии электропередачи пока отсутствуют. И человек обратился... к Солнцу. А на помощь пришли полупроводники, и среди них — высокочистые монокристаллы К.
При поглощении света в кристаллах К. освобождаются электроны. Если соединить освещаемый и неосвещаемый участок К. проводником, то по нему пойдёт ток. На этом эффекте и основано действие полупроводниковых фотоэлементов, превращающих световую энергию в электрическую. Кремниевые фотоэлементы могут превращать в электрическую до 15% падающей на них световой энергии Солнца. А объединённые тысячами на панелях высокочистые кристаллы К. заменяют электростанцию. Такие солнечные батареи и питают аппаратуру искусственных спутников Земли, межпланетных станций, луноходов.
Кремниевые преобразователи солнечной энергии в электрическую начинают применять и на самой нашей планете; особенно перспективна эта новая энергетика в тех районах Земли, где много солнечных дней. Однако такое использование полупроводниковых свойств кристаллов К. не только не единственное, но и не основное. К. (наряду с германием) —главный материал для транзисторов, диодов, термисторов и других полупроводниковых приборов.
Технический К. широко используют в металлургии. Добавки К. в стали и чугуны улучшают их механические свойства. Большие добавки К. придают железу устойчивость к действию кислот. Только НС1 разрушает спла-
Рис. 3. Автоматический самоходный аппарат «Л у и о х о д- 2> . Его доставила на Луну автоматическая станция «Луна-21 > 16 января 1973 г. Солнечные батареи (1), питающие луноход, составлены из кремниевых фотоэлементов.
вы железа с 14—18% К. (ферросилид), а на сплавы с 50% К. и выше (ферросилиций) никакие кислоты не действуют. В производстве стали малые добавки К. (в виде ферросилиция) служат'для раскисления — удаления кислорода из расплавленного металла. Существует специальная отрасль металлургии — силикотермия, подобная алюминотермии (см. Алюминий)} нагревание с К. позволяет получать некоторые металлы из их окислов по реакции:
2МеО + Si = SiO2 + 2Ме
где Me — двухвалентный металл.
Вот уже несколько десятилетий получают и используют карбид кремния SiC — искусственный «полуалмаз» (производство же настоящих искусственных алмазов началось сравнительно недавно, см. Углерод). Распространённое название SiC — карборунд. Карборундовые точильные круги имеются в любой механической мастерской. Самые твёрдые токарные резцы из специальных сплавов легко заточить на карборундовом диске. Тонкий порошок SiC применяют для шлифовки -твёрдых материалов. А среди известных всем «наждачных» шкурок есть и карборундовые — это наклеенные на бумагу или ткань мельчайшие кристаллики SiC. Хотя карборунд уступает по твёрдости алмазу, но он значительно более жаростоек и плавится лишь при 2500 °C. В отличие от алмаза, карборунд, хотя и плохо, но проводит электрический ток; из него делают стержни — нагревательные элементы — электрических печей. Как жаростойкий материал карборунд интересен и для ракетной техники.
КРЕМНИЯ ДВУОКИСЬ
172
Ещё более жаростойки нитрид кремния Si3N4 и силициды тантала, молибдена, рения (см. Нитриды и Силициды).
Год из года ширится применение различных крем-нийорганических соединений. В их числе и полиоргано-силоксаны — вещества, содержащие цепи атомов
R R R
I I I
— Si —О —Si —О—Si — I I I
R R R
(где R — органические радикалы). Эти материалы устойчивы к повышенным температурам, хорошие электроизоляторы, не смачиваются водой. Электрические машины, у которых обмотка и выводы имеют кремний-органическую изоляцию, могут работать, находясь в воде. Кремнийорганические смазочные масла не боятся как повышенных, так и пониженных температур. Даже при охлаждении до —80 °C они почти не меняют своих свойств. Это очень важно на тех высотах, где летают современные реактивные лайнеры и где температура на 50—60° ниже нуля.
Об использовании соединений К. см. также Кремния двуокись, Кремниевые кислоты, Силикаты, Стекло, Бетон, Цемент.
А. П. Пурмаль.
КРЕМНИЯ ДВУОКИСЬ (кремнезём) SiO2 — соединение кремния с кислородом, ангидрид кремниевых кислот. SiO2 — наиболее распространённое в земной коре соединение, составляющее в виде различных минералов 12% её массы. Основной минерал — кварц — встречается повсюду в виде обычного песка (SiO2) с примесями окисей железа и алюминия. А яшма, халцедон, агат, сердолик — так называемые полудрагоценные камни — всё это разновидности кварца. Они отличаются приро
Рис. 1. Горный хрусталь (разновидностькварца) часто встречается в виде друз — группы кристаллов, наросших иа стенки трещин в горных породах (как это изображено на фотографии).
дой и количеством примесей и поэтому имеют различную окраску. Встречаются в природе и гидратные формы К. Д. типа nSiO2-/nH2O (см. Кремниевые кислоты). Наиболее чистая разновидность кварца — это горный хрусталь, прозрачный и бесцветный.
Слово «хрусталь» произошло от греческого «крйсталлос» — лёд. Древние греки, находившие прозрачный хрусталь в горах, считали, что это лёд, замёрзший настолько, что уже не может растаять. Кристаллы горного хрусталя иногда достигают огромных размеров. В 1958 г. в Казахстане был найден кристалл весом 70 т. Малые примеси придают хрусталю различную окраску: фиолетовую (аметист), жёлтую (цитрин), дымчатую (раухтопаз), чёрную (морион). Всего же существует около 400 минералов, состоящих в основном из SiO2 и включающих различные примеси других веществ.
Кристаллическая SiO2 — очень твёрдое, прочное, тугоплавкое вещество (/пл 1610 °C при быстром нагреве, 1723 °C — при медленном). При остывании расплавленной.ЭЮ2 образуется аморфное кварцевое стекло.
Кислоты не действуют на SiO2. Лишь фтористоводородная кислота постепенно растворяет его (см. также Фтористый водород):
SiO2 + 6HF = H2SiF6 + 2Н2О
В растворах щелочей SiO2 медленно переходит в соли кремниевых кислот, например:
nSiO2 + 2КОН = K2O-nSiO2 + Н2О
При сплавлении SiO2 с окислами, гидроокисями, карбонатами металлов образуются силикаты. Так, в процессе стекловарения идут реакции, которые можно выразить суммарным уравнением:
СаСОз + Na2CO3 + 6SiO2 =
= Na2O*CaO-6SiO2 + 2СО2
На варку стекла и производство бетона идут огромные количества кварцевого песка. А чистый кварц незаменим в приборостроении.
Кристаллы кварца обладают замечательным свойством — преобразовывать почти без потерь электрическую энергию в механическую, и наоборот. Такие вещества называют пьезоэлектриками. Под действием электрического поля кварцевая пластинка сжимается, а при снятии напряжения принимает прежние размеры. Таким образом, переменное электрическое поле вызывает колебания кварцевой пластинки, которые передаются окружающей среде — пластинка издаёт звук, служит звуковым генератором. А если частота электрического поля выше20 000 гц, испускается ультразвук. .
173
КРИПТОН
Рис. 2. Ваза из минерала раухтопаза (дымчатого кварца). Эта ваза размером со спичечный коробок, её высота всего лишь 5,5 см.
Кварцевые пластинки работают и в звукоснимателях радиол и проигрывателей. Скользящая по грампластинке игла передаёт механические колебания, связанные с неровностями звуковой дорожки — этого застывшего звука — на кристаллик кварца. Кристаллик трансформирует механические колебания в электрические, они усиливаются специальной электронной схемой и передаются на динамик. Звучит музыка, человеческая речь, пение. Для использования пьезоэлектрических свойств кварца кристаллы должны быть достаточно большими и правильно ориентированными. Потребность техники столь велика, что, кроме природных, в последние годы всё больше используют искусственно выращенные кристаллы кварца.
Из мелких кристаллов кварца и из отходов производства пьезоэлектрических пластин получают кварцевое стекло. Оно прозрачно не только для видимого света, но и для ультрафиолетового, обладающего целебным действием.
При многих заболеваниях врачи назначают больному «кварц», то есть облучение ультрафиолетовым светом ртутно-кварцевых ламп. Электрический разряд, проходя через пары ртути, генерирует ультрафиолетовые лучи, а кварцевое стекло пропускает их почти полностью. Такие лампы можно увидеть в больницах и поликлиниках, профилакториях, детских садах. Это лампы низкого давления. Прочность кварца позволила создать и лампы высокого и сверхвысокого давления, нагревающиеся до нескольких сотен градусов. Заполняются такие лампы парами ртути или инертными газами. Они работают либо в режиме непрерывного электрического разряда, либо в режиме коротких, но мощных разрядов (так называемые импульсные лампы). Яркость излучения этих ламп больше яркости Солнца. Близка к солнечной яркость кварцевых ламп накаливания, заполненных парами иода или брома (так называемые галогенные лампы). Эти новые виды ламп, по-видимому, заменят в скором будущем обычные лампы накаливания. Они просты в эксплуатации, во много раз меньше размером и служат в несколько раз дольше, чем обычные, а свет их по составу близок к солнечному.
Кварцевое стекло устойчиво к действию большинства химических веществ, жаростойко и выдерживает резкие изменения температуры. Поэтому из него делают высококачественную лабораторную посуду. Стакан из кварцевого стекла не треснет, если его раскалить добела и затем бросить в холодную воду. Не пробуйте провести такой опыт с хрустальным бокалом. Хрустальное стекло содержит лишь 55—57% SiO2 и не обладает свойствами кварцевого.
А. П. Пурмаль.
83,80 36
КРИПТОН (лат. Kryptonum) Кг — химический элемент VIII группы периодической системы Менделеева; атомный номер 36, атомная масса 83,80; инертный газ. Атмосферный К. состоит из 6 стабильных изотопов, среди которых преобладает 84Кг (56,9%).
К- открыт в 1898 г. англичанами У. Рамзаем и М. Трэверсом в остатках медленно испаряющегося жидкого воздуха. Учёным пришлось переработать несколько тонн жидкого воздуха, чтобы получить заметные количества К. В память об этих трудностях новый элемент и получил своё название (от греческого слова «кри-птос» — скрытый).
К.— редкий газ. В 1 м3 воздуха содержится всего около 1 см3 К. (весь К., находящийся в воздухе трёхкомнатной квартиры, можно поместить в баночку из-под майонеза); Долгое время для К-, как и для других инертных газов, не могли получить химических соединений с другими элементами. Но в 1962 г. появилось сообщение о синтезе химического соединения ксенона — сенсационное сообщение, заставившее пересмотреть установившиеся взгляды на свойства инертных газов. Это сразу же привлекло внимание химиков всего мира и с тех пор было выполнено множество работ по исследованию взаимодействия К. с фтором, кислородом и другими активными реагентами.
Оказалось, что в очень специфических условиях можно синтезировать фториды К.— KrF2 и KrF4, бесцветные кристаллические вещества. Так, KrF2 образуется при облучении светом ртутной лампы смеси К. и фтора при —250°С. Фториды К. очень непрочны, они разлагаются уже при комнатной температуре. Но вскоре удалось синтезировать и соединение, устойчивое при обычных условиях: им оказался криптонат бария ВаКгО4 — соль, получаемая действием охлаждённого раствора Ва(ОН)2 на KrF4.
КРИПТОН
КСЕНОН
174
Как говорилось выше, концентрация К. в атмосфере мала. Однако масштабы современной промышленности по переработке воздуха столь огромны, что позволяют выделять К- в количествах, пригодных для его промышленного использования. Криптоном часто наполняют электрические лампочки. Тяжёлые атомы К. создают для испарения атомов вольфрама со спиральной нити более сильное препятствие, чем азот или аргон. Поэтому использование К. позволяет, не снижая мощности лампы, значительно уменьшить её объём и увеличить срок службы (о роли инертного газа в электрической лампе см. в статье Аргон}. Разрядные трубки, наполненные К>, испускают чисто белое свечение и применяются для изготовления реклам. Подробнее о химических, а также о физических свойствах К. см. Инертные газы. С. С. Бердоносов.
КРИСТАЛЛИЗАЦИОННАЯ ВОДА, см. Вода. КСЕНОН (лат. Xenonum) Хе — химический элемент VIII группы периодической системы Менделеева; атомный номер 54, атомная масса 131,30; инертный газ. Природный К. состоит из смеси 9 изотопов, среди которых преобладают 129Хе (26,44%), 131Хе (21,18%) и 132Хе (26,89%).
К- открыт в 1898 г. английскими учёными У. Рамзаем и М. Траверсом как небольшая примесь к криптону\ — и
его название (греч. «ксё-нос> — чужой). В воздухе, единственном источнике К., этого элемента меньше, чем золота в морской воде (только 8,7 см3 К. на 100 м3 воздуха).
Среди инертных газов К. оказался пер
вым, поколебавшим представление об абсолютной химической недеятельности элементов, атомы которых имеют завершённую,8-электронную, внешнюю оболочку. Именно для К. в 1961 г. канадский учёный Н. Бартлетт получил первое химическое соединение инертного газа. Сообщение Бартлетта увидело свет в 1962 г. Его блестящий эксперимент состоял в следующем.
В кварцевую ампулу поместили К- (он бесцветен) и гексафторид платины PtFe (тёмно-красный пар), разделённые тонкой перегородкой. Когда перегородку разбили, газы смешались и в ампуле немедленно образовался жёлтый порошок нового вещества. Химический анализ показал, что его формула XePtFe.
Бартлетт не случайно поставил такой опыт. Сначала он изучал взаимодействие PtFe и молекулярного кислорода Оя. Оказалось, что эти вещества дают несколько необычное соединение, содержащее ионизированную молекулу кислорода O2[PtFe]~. Минимальное напряжение, при котором происходит отрыв первого электрона от молекулы Оа, равно 12,2 вольт; это даже несколько
Рис. 1. На фотографии показаны кристаллы XeF4.
больше, чем у К. (12,13 вольт). Поэтому решили проверить, не вступит ли в аналогичную реакцию и К» Предположение оказалось правильным, и открытие XePtFe положило начало новому разделу неорганической хи-" мии — химии инертных газов. Об исключительном внимании к новой теме исследования можно судить хотя бы по такому событию: уже в 1963 г. состоялась конференция по химии инертных газов, на которой исследователи из 30 институтов разных стран сделали около 40 докладов.
В своих соединениях К. проявляет степени окисления +1, +2, +4, + 6 и +8 (кстати, пока не объяснено, почему нет соединений К. со степенями окисления +3, +5 и +7). Партнёрами К. в химических реакциях могут выступать фтор или фториды металлов (RuFe, RhFe, SbF6 и др.), обладающие высоким сродством к электрону — мощной способностью отрывать электроны от других атомов и молекул и перетягивать их на свои электронные орбиты. Со свободным фтором К. даёт XeF2, XeF4, XeFe и (возможно) XeF8.
Кристаллы одного из фторидов К.— XeF4 показаны на рис. 1; строение молекулы XeF4 — на рис. 2.
В сухой атмосфере фториды К- вполне устойчивы и могут сохраняться месяцами. На влажном же воздухе
Рис. 2. Строение молекулы XeF4. Эта молекула представляет собой плоский квадрат, в вершинах которого расположены атомы F, а на пересечении диагоналей — атом Хе.
175
КУРЧАТОВИЙ
они разлагаются: так, действие воды на XeFa приводит к выделению Хе и F2. При взаимодействии XeFe и НаО сначала образуется бесцветная жидкость XeOF4, а затем — XeO2F2, и, наконец, твёрдый прозрачный ХеО3, часто взрывающийся при хранении.
Если на водный раствор ХеО3 воздействовать гидроокисью бария Ва (ОН)2, то образуется ксенат бария Ва3ХеОв. Известны и перксенаты натрия, серебра, а также соответствующая им ксеноновая кислота Н4ХеОв. В состав перксенатов входит обычно несколько молекул кристаллизационной воды: Na4XeOe-2HaO, Na4XeOe«8HaO. Исходя из перксената натрия, можно синтезировать высший окисел ХеО4, устойчивый при О °C только в парах.
В промышленности К. получают из воздуха. Из-за очень низкого содержания газа в атмосфере объём его производства невелик. А между тем К. необходим современной технике: ксеноновая газоразрядная лампа — это один из самых мощных на сегодня источников света, по яркости излучения соизмеримый с Солнцем.
Как же устроена такая лампа? Кварцевая колба с герметически встроенными электродами заполнена К. Между электродами происходит дуговой разряд, электрическая энергия переводит атомы К- в так называемое возбуждённое состояние. Такие атомы и обеспечивают яркое свечение лампы. Ксеноновые газоразрядные лампы ставятся в мощные прожекторы и кинопроекторы. А поскольку спектр их излучения близок солнечному, ксеноновые лампы, имитируя Солнце, помогают выращивать растения в теплицах.
С помощью фторидов К., возможно, удастся решить проблему перевозки агрессивного фтора: XeF2 и XeF4 вполне транспортабельны (например, в обычных ящиках), а выделить из них фтор не представляет большого труда. По той же причине фториды К. перспективны и как фторирующие агенты. ХеО3 может найти применение как взрывчатое вещество, обладающее тем преимуществом, что после его разложения не образуется каких-либо вредных газов. О физических свойствах К. см. Инертные газы. С. С. Бердоносов.
КУРЧАТОВИЙ (лат. Kurtchatovium) Ku — радиоактивный химический элемент IV группы периодической системы Менделеева; атомный номер 104.
Как и другие элементы, расположенные в конце периодической системы, К. не существует в природе и может быть получен только искусственно — по ядерным реакциям.
Задолго до открытия К. среди учёных шла оживлённая дискуссия о свойствах этого «ещё не родившегося» элемента. Большинство исследователей предполагало, что он окажется первым трансурановым элементом, не относящимся к семейству актиноидов. Поэтому ожидалось, что его свойства будут резко отличаться от свойств предшествующих в таблице
(26D Ю4
Ku
6d!7s!
КУРЧАТОВИЙ
Менделеева 14 элементов, составляющих это семейство.
Первые сведения о 104-м элементе получила в 1964 г. группа советских физиков во главе с Г. Н. Флёровым в Объединённом институте ядерных исследований (г. Дубна). При облучении плутония 242Ри сильно разогнанными ядрами неона 22Ne образовались ядра, которые по радиоактивным свойствам отличались от ядер всех известных изотопов. По ряду признаков синтезиро
ванные ядра должны были принадлежать новому элементу с Z=104 (условно обозначим его Э). В этом случае ядерную реакцию его синтеза следовало записать так:
%442Pu + 2oNe = 2™Э + 5Jn (одновременно с ядрами нового элемента в результате ядерной реакции возникало несколько нейтронов). Но, может быть, учёные получили не новый элемент, а всего лишь новый изотоп одного из уже известных актиноидов? Полную ясность могло дать только изучение химических свойств полученных атомов.
Провести такое исследование обычными химическими методами оказалось невозможно. Скорость радиоактивного распада синтезированных ядер очень велика — период полураспада 7\/ всего несколько секунд. Кроме того, в распоряжении исследователей были буквально единичные атомы (по расчётам, 1 атом нового элемента мог образоваться в среднем за 5 часов работы сложнейшей аппаратуры). Поэтому нельзя, например, работать с раствором 104-го элемента — ведь даже простое перемешивание жидкости в пробирке занимает несколько секунд. Никогда раньше подобные трудности не стояли перед химиками. Даже в случае 101-го элемента — менделевия, для которого основные химические свойства были установлены с использованием всего 17 атомов, работать несравненно легче: величина Т»/3 исследуемого изотопа Md превышала 1 час.
Схема прибора для изучения хлорида 104-го элемента — курчатовия. / — поток ядер неона 22 Ne; 2 — плутониевая мишень; 3 — труба; 4 — детектор.
КЮРИИ
176
Задачу решила группа химиков, работавших в Объединённом институте ядерных исследований под руководством чехословацкого учёного И. Звары. Предложенная ими методика была очень остроумной. В её основе лежало предполагаемое различие свойств 104-го элемента и актиноидов, иными словами, различие свойств элементов III и IV групп системы Менделеева. Так, например, хлориды трёхвалентных актиноидов — америция, кюрия и др. (АгпС13, СтОз и т. д.) при обычных условиях крайне труднолетучи (температуры их кипения лежат около 1500 °C), тогда как хлориды четырёхвалентных циркония и гафния (ZrCl4, HfCl4) с заметной скоростью возгоняются уже при 250—300 °C. Эти особенности хлоридов и использовала группа И. Звары для. химической идентификации 104-го элемента. Опыт можно было осуществить за десятую долю секунды. Он состоял в следующем.
Поток ядер иона 22Ne, разогнанных до высоких скоростей на ускорителе, падал на тонкую плутониевую пластиночку — мишень (см. рис.). В исключительно редких случаях столкновение ядер 22Ne с ядрами 242Ри приводило к ядерной реакции (см. выше). За счёт огромной кинетической энергии, которую несло ядро 22Ne, вновь возникшее ядро вылетало из мишени и попадало внутрь прибора. В этот прибор непрерывно подавались пары ZrCl4 и NbCl6 (по своей летучести эти хлориды очень похожи друг на друга).
Избыточная энергия, которой первоначально обладали атомы 104-го элемента, давала им возможность вступать в весьма необычные химические реакции, отбирать атомы хлора у молекул ZrCl4 и NbCl6. В итоге возникал хлорид нового элемента, соответствующий его максимально возможной положительной степени окисления. Полученный хлорид вместе с ZrCl4 и NbCl6 током азота переносился по длинной (4 м) трубе, нагретой до 300 °C. Если хлорид нового элемента похож на хлориды
элементов IV группы и легколетуч, то он не должен оседать на стенках трубы. В таком случае ток газа перенесёт его в специальный детектор, где регистрируются акты распада ядер. Напротив, актиноиды в аналогичных условиях давали труднолетучие хлориды, которые полностью задерживались на стенках трубы и в детектор не попадали.
Этот эксперимент потребовал от исследователей колоссального труда.
Десятки раз меняли они режим нагрева трубы, состав и скорость потока газовой смеси, материал аппаратуры. Опыты продолжались неделями, не прекращаясь ни днём, ни ночью. Наконец весной 1966 г. удалось добиться успеха. В детекторе было зафиксировано за несколько суток 12 актов распада ядер нового элемента. Это дало основание сделать окончательный вывод, что получен не изотоп одного из известных актиноидов, а новый, 104-й элемент. Название ему дали в честь выдающегося советского физика И. В. Курчатова, длительное время возглавлявшего в Советском Союзе работы по ядерной физике.
По новейшим данным, величина 7\/2 изотопа 2б9Ки составляет ок. 4,5 сек. Для другого его изотопа 260Ки (также впервые полученного советскими физиками) величина Ti/2 ок. 0,1 сек. Долгоживущий изотоп 261Ки имеет период полураспада 70 сек.
Изучение К. продолжается, и новые исследования, без сомнения, принесут много интересных сведений об этом элементе.
С. С. Бердоносов. КЮРИЙ (лат. Curium) Cm — радиоактивный химический элемент с атомным номером 96; относится к актиноидам.
«Благодаря открытию явлений радиоактивности мы узнали новый, негаданный источник энергии. Этим источником Явились химические элементы... А теперь перед нами открываются в явлениях радиоактивности источники атомной энергии, в миллионы раз превышающие все те источники сил, какие рисовались человеческому воображению».
В. И. Вернадский (Речь на Общем собрании Академии наук 29 декабря 1910 г., см. Избр. соч., т. 1, 1954, с. 620, 623)
ЛАНТАН (лат. Lanthanum) La — химический элемент III группы периодической системы Менделеева; атомный номер 57. См. Редкоземельные элементы.
ЛАНТАНИДЫ— то же, что лантаноиды. ЛАНТАНОИДЫ (лантаниды) — семейство из 14 химических элементов с атомными номерами Z от 58 до 71. Эти элементы расположены в 6-м периоде системы Менделеева за лантаном и относятся, как и лантан, к III группе. К Л. принадлежат: церий Се (Z=58), празеодим Рг (59), неодим Nd (60), прометий Pm (61), самарий Sm (62), европий Ей (63), гадолиний Gd (64), тербий ТЬ (65), диспрозий Dy (66), гольмий Но (67), эрбий Ег (68), тулий Тш (69), иттербий Yb (70) и лютеций Lu (71). Все Л., кроме радиоактивного прометия, имеют стабильные изотопы и встречаются в природе. Вместе с лантаном, иттрием и скандием Л. образуют группу редкоземельных элементов.
Термин «лантаноиды» в переводе с греческого означает «подобные лантану». Он от
ражает особую близость химических свойств элементов этого семейства и аналогию их с лантаном. В природе Л. обычно встречаются практически в одних и тех же минералах. Поэтому открытие отдельных элементов Л. стало возможным только после того, как химики научились разделять чрезвычайно близкие по свойствам элементы.
История открытия Л. восходит к концу XVIII — началу XIX вёков, когда были обнаружены две «редкие земли» — иттриевая и цериевая (напомним, что «землями» тогда называли порошкообразные, тугоплавкие, нерастворимые в воде окислы). Иттриевая земля была открыта финским химиком Ю. Гадолином в 1794 г. (см. статью Редкоземельные элементы) .Цериевуюзем-л ю открыли в 1803 г. шведские химики Я. Берцелиус и В. Гизингер и немецкий,химик М. Клапрот при анализе «тяжёлого камня из Бастнеса» (минерал бастнезит, по химическому составу — фторкарбонат лантана, церия, празеодима и неодима). Позднее выяснилось, что обе «земли» — смеси окислов.
Подробное исследование цериевой и иттриевой «земель», выполненное шведом К- Мосандером, учеником Берцелиуса, позволило ему в 1838—1843 гг. разложить (как говорят часто, «расщепить») эти земли на 6 составляющих: окислы иттрия, церия, эрбия, тербия, «дидима»
Некоторые свойства лантаноидов
Элемент Атомная масса Число электронов на внешних оболочках Физические свойства Содержание в земной коре, % по массе
4-я 5-я 6-я Плотность, г/см3 Температура, °C
S Р d f s Р d S плавления кипения
Се 140,12 2 6 10 1 2 6 1 2 6,77 795 3470 7-Ю-3
Рг 140,9077 2 6 10 3 2 6 — 2 6,78 935 3130 9-Ю-4
Nd 144,24 2 6 10 4 2 6 —— 2 7,01 1024 3030 3,7.10-3
Pm* — 2 6 10 5 2 6 — 2 7,26 1080 — —
Sm 150,4 2 6 10 6 2 6 — 2 7,54 1072 1900 8-Ю-4
Eu 151,96 2 6 10 7 2 6 — 2 5,26 826 1440 1,3.ю-4
Gd 157,25 2 6 10 7 2 6 1 2 7,89 1312 3000 8-10-4
Tb 158,9254 2 6 10 9 2 6 —— 2 8,27 1356 2800 4,3-10-4
Dy 162,50 2 6 10 10 2 6 2 8,53 1407 2600 5-10-4
Ho 164,9304 2 6 10 11 2 6 — 2 8,80 1461 2600 1,7-10-4
Er 167,26 2 6 J0 12 2 6 2 9,05 1497 2900 3,3-ю-4
Tm 168,9342 2 6 10 13 2 6 — 2 9,33 1545 1730 2,7-10-5
Yb 173,04 2 6 10 14 2 6 —— 2 6,98 824 1430 3,310-5
Lu 174,97 2 6 10 14 2 6 1 2 9,84 1652 3330 8-10-5
* Pm (прометий) радиоактивен; о его свойствах см. статью Прометий.
12 хэш
ЛАНТАНОИДЫ
178
и лантана (в дальнейшем выяснилось, что дидим — это смесь двух элементов — празеодима и неодима). Открытие остальных Л. стало возможным только после того, как в 1860 г. был предложен новый, очень чувствительный метод анализа — спектроскопический.
Каждый шаг вперёд в химии Л. требовал огромного труда исследователей, обширных знаний и высокой техники эксперимента. Единственный метод, который в прошлом веке применяли для разделения Л., состоял в том, что исходный материал сначала растворяли в концентрированной кислоте, а затем многократно осаждали из раствора ту или иную соль. Этот метод называется дробным осаждением или, если образующийся осадок кристал-личен, дробной кристаллизацией. Разделение было возможно благодаря незначительной разнице в растворимости солей разных Л.
Повторяя дробную кристаллизацию сотни, тысячи раз, удавалось отделить Л. друг от друга. Так, для выделения лютеция французу Ж. Урбену пришлось выполнить более 15000 кристаллизаций. 10000 раз повторил дробную кристаллизацию немецкий учёный В. Ноддак, чтобы получить всего 10 мг чистой окиси гольмия. Следует отметить, что эффективность разделения обычно проверяли по результатам спектроскопического изучения, но окончательные выводы делали на основе определения атомной массы выделяемого Л. Атомную массу нужно было определять с такой большой точностью, которую трудно обеспечить даже в настоящее время.
А потому наряду с действительными открытиями Л. в печать попадали и ошибочные сообщения. Ошибались даже такие выдающиеся химики прошлого века, как Мосандер и Лекок де Буабодран. Сообщений о лжеоткры-тиях новых Л. было около сотни. Многие Л. к моменту открытия выделяли с большим количеством примесей, что ещё сильней осложняло дело. Получить чистые Л. оказалось ещё труднее, чем открыть новые. Так, все Л. в виде чистых металлов удалось получить только в 50-е годы нашего века. В настоящее время для разделения и очистки Л. используют такие эффективные методы, как экстракцию и ионообменную хроматографию.
Представление об истории открытия Л. даёт рис. 1.
Распространённость в природе. Из того, что Л-относят к редкоземельным элементам, может сложиться впечатление, будто их запасы ничтожны. Однако в земной коре не так уж мало даже самых малораспространённых Л.— тулия, иттербия, лютеция. По крайней мере, каждого из этих элементов больше, чем такого обычного металла, как ртуть. Наиболее распространены церий и неодим. Доля церия в земной коре выше, чем доля меди, неодима же больше, чем свинца.
Л. образуют около 70 минералов. А как примеси они встречаются почти в 200 других минералах. Тем не менее добыча Л. очень трудна. Объясняется это прежде всего тем, что богатых месторождений Л. практически не существует; находятся Л. обычно в сильно рассеянном состоянии. Одним из главных ис
точников Л. служит минерал монацит — смесь фосфатов Л., иттрия, тория и других элементов. Богатые месторождения монацита имеются в Индии и Бразилии. Большие запасы Л. найдены и в нашей стране.
Физические и химические свойства. В свободном виде Л.— типичные металлы серебристобелого цвета с разными оттенками. Сведения
Рис. 1. К истории
Иттрий
Зрбий
Тербий
К.Мосандер, Швеция, 1843
Иттербий
Зрбий
179
ЛАНТАНОИДЫ
о некоторых физических свойствах Л. приведены в таблице (стр. 177).
Для Л. наиболее обычна степень окисления +3 (валентность III). Только для Се и, в меньшей степени, для Рг, ТЬ и Dy характерна степень окисления +4. Состояние окисления +2 известно для Sm, Ей и Yb. По химическому поведению все Л. очень похожи.
открытия лантаноидов.
Я.Берцелиус,
В.Гизимгер, Швеция ► 1803 М.Клапрот, Германия
К.Мосандер, Швеция, 1839-1843
Дидим
Самарий
Гадолиний
Ж.Мариньяк, Швейцария, 1880
П.З. Лекок де Буабодран, Франция, 1879
Неодим Празеодим
Европий
Самарий
Э.Демарсе, Франция, 1896-1901
^“К.Дуэр фон Вельсбах, Австрия. 1885
Это связано с особенностями строения элект-. ронных оболочек их атомов.
У атомов Л., как и у всех остальных элементов 6-го периода, имеются 6 электронных оболочек. Число электронов на последней (6-й) и предпоследней (5-й) оболочках у всех Л. приблизительно одинаково. А так как именно эти две оболочки определяют химические свойства элементов, то понятно, что все 14 Л. ведут себя в химических реакциях как элементы-близнецы.
Но куда же попадают новые электроны, появляющиеся в атомах с увеличением заряда ядра от +58 (у церия) до +71 (у лютеция)? Ведь при возрастании заряда ядра на единицу в атоме появляется новый электрон, и у самого последнего лантаноида—Lu электронов на 14 больше, чем у Се.
Обычно (у других элементов) новый электрон занимает место на последней или предпоследней электронных оболочках. У лантаноидов же дело обстоит иначе: электроны заполняют сравнительно глубоко расположенную 4-ю оболочку, точнее говоря, её так называемый f-уровень. Как установили физики, /-уровень содержит 14 мест, и эти места остаются незанятыми у предшествующих элементов 4-го, 5-го и 6-го периодов. Заполнение электродами /-уровня 4-й оболочки (то есть 4/-уровня) как раз и происходит у атомов Л. Максимально на этом уровне может разместиться 14 электронов (см. Атом). Поэтому семейство Л. и насчитывает 14 элементов. Аналогичные особенности заполнения электронных оболочек — у актиноидов.
При переходе от La к Lu наблюдается так называемое «лантаноидное сжатие» — закономерное уменьшение радиусов атомов и ионов. Казалось бы, дело должно обстоять иначе: чем больше электронов, тем больше и радиус. Однако вследствие того, что новые электроны Л. попадают на глубоко лежащую электронную оболочку, а число электронов на внешних оболочках остаётся постоянным, сила притяжения внешних электронов возрастает по мере увеличения заряда ядра. В результате радиусы атомов и ионов уменьшаются (рис. 2).
Химические соединения Л. очень сходны между собой. Так, их гидроокиси общей формулы Ме(ОН)3 имеют основной характер, не растворяются в щелочах и легко реагируют с кислотами, например:
Sm (ОН)3 + ЗНС1 = SmCl3 + ЗН2О
При нагревании окислов Л. в смеси окиси углерода СО и хлора С12 можно получить безводные трихлориды, например:
Nd2O3 + ЗСО + ЗС12 = 2NdCl3 + ЗСО2
12*
ЛАНТАНОИДЫ
Если хлор заменить парами брома, образуются трибромиды Л. Тригалогениды Л. имеют высокие температуры плавления (600 °C и выше),
что свидетельствует о ионном характере связи атомов Л. с галогенами. Из хорошо растворимых в воде солей Л. можно отметить хлориды и нитраты. В водных растворах трёхзарядные катионы Се, Gd, Tb, Yb и Lu бесцветны; Рш,
Ей и Ег имеют розовую окраску; Sm, Dy и Но— жёлтую; Рг и Тш — зелёную; Nd — фиолетово-красную. К числу малорастворимых солей Л- принадлежат фториды, карбонрты, фосфаты.
Применение Л. очень разнообразно. Так, сплав различных Л. (так называемый «мишметалл») обладает пирофорными свойствами:
при трении с поверхности сплава летит сноп искр. Поэтому сплав «мишметалла» с железом идёт на изготовление «кремней» для зажигалок. Из этого же сплава можно делать насадки трассирующих пуль. От насадки при трении о воздух летят искры, и весь путь пули (её трасса) становится видимым. Это особенно важно при стрельбе в ночное время.
Многие соли Л. служат катализаторами различных химических реакций. Л. получили широкое применение и в такой новейшей обла
сти техники, как строительство ядерных реакторов.
Использование Л. в реакторостроении связано с тем, что многие их изотопы (в том числе 149Sm, 161Eu, 163Eu, 167Gd) очень сильно поглощают медленные нейтроны, «работающие» в ядерном реакторе. Поэтому Л., у которых имеются такие изотопы, можно применять в защитных материалах, предохраняющих персонал реакторов
180
от вредного нейтронного облучения. Кроме того, эти Л. вводят в состав управляющих стержней ядерных реакторов. Если реакция идёт слишком быстро, стержни опускают в реактор; тогда часть нейтронов поглощается в стержнях и реакция замедляется. При необходимости ускорить ядерную реакцию стержни выдвигают из активной зоны реактора.
Важная область применения многих Л. (Eu, Gd, Tb и других) — получение люминофоров, то есть веществ, способных длительное время светиться после действия на них видимого света или другого электромагнитного излучения. Из люминофоров, содержащих добавки Л., изготовляют экраны цветных телевизоров, светящиеся экраны рентгеновских установок и т. д.
Л. обладают необычными магнитными свойствами. Это связано с тем, что при переходе от элемента к элементу, по мере заполнения /-уровня, не все электроны оказываются спаренными (то есть объединёнными в пары с противоположно направленными спинами). В соответствии с правилом Хунда (см. Атом), число неспаренных электронов у Л. может достигать 7 — именно столько их на /-уровне атома гадолиния. Наличие неспареннрх электронов приводит к тому, что все Л.— сильно магнитные вещества. В частности, Gd по магнитным свойствам напоминает железо — оба эти элемента относятся к числу так называемых ферромагнетиков. У остальных Л. магнитные свойства и атомная магнитная структура сложны и сильно зависят от температуры. Подробнее о магнитных свойствах веществ см. в статье Железо,
Эти особенности Л., свойственные и другим переходным металлам, открыли в последние годы путь к созданию совершенно новых магнитных материалов. Радиотехника, электротехника, приборостроение и создание уникальных электронно-вычислительных машин с огромной ёмкостью «памяти» — эти традиционные и новейшие области техники — уже применяют новые магнитные материалы на основе Л. Среди них особенно интересно соединение са-
ри с. 2. «Лантаноидное сжатие». Так называют уменьшение размера атома элемента — лантаноида с увеличением его атомного номера.
Радиус атома, А °
181
ЛИТИИ
мария с кобальтом SmCo5 — рекордный материал для постоянных магнитов, а также так называемые ферриты-гранаты общей формулы Ln3FeBOi2 (где Ln — лантаноид). Многие учёные и инженеры считают, что Л.— это завтрашний день техники.
О некоторых других применениях Л. говорится ниже, при характеристике отдельных представителей семейства. Сведения о радиоактивном элементе — прометии даны в статье, посвящённой этому элементу.
Церий (лат. Cerium) Се, Z=58.
Название дано в честь астероида Цереры, малой планеты Солнечной системы. Ц.— единственный Л., для которого степень окисления +4 (валентность IV) более характерна, чем +3. Так, при нагревании на воздухе металлический церий сгорает, давая двуокись СеО2; окисел Се2О3 можно получить только в специальных условиях. В щелочной среде ион Се3+ быстро окисляется до иона Се4+; в кислой среде, напротив, Се4+ легко восстановить до Се3+. Церий— главный компонент «мишметалла» (см. выше). Небольшие добавки Се к стали увеличивают её прочность. Соли церия — хорошие катализаторы многих химических реакций.
Празеодим (лат. Praseodymium) Pr, Z=59.
Название элемента составлено из греческих слов «пра-синос»— светло-зелёный и «дйдимос»—близнец. Оно указывает, с одной стороны, на окраску многих соединений Рг, а с другой — на историю его открытия. Рг был получен при разделении «дидима» — смеси Рг и Nd, которую долгое время принимали за индивидуальный элемент.
У Рг необычный для Л. окисел — его состав РгвОп(4РгО2«Рг2О3). В смеси с неодимом Рг используют для получения бесцветных стёкол, хорошо поглощающих ультрафиолетовые лучи.
Неодим (лат. Neodymium) Nd, Z=60.
Название элемента произведено от греческих слов — «нёос» — новый и «дйдимос». Nd используют не только в смеси с Рг. Так, сплавы магния, алюминия или титана с Nd при небольшой плотности обладают повышенной прочностью и могут применяться в самолёто- и ракетостроении.
Самарий (лат. Samarium) Sm, Z—62.
Впервые выделен из минерала самарскита, найденного на Урале и названного в честь начальника штаба корпуса горных инженеров В. Е. Самарского-Быховца. Название дал в 1847 г. немецкий химик Г. Розе, которому Самарский предоставил для исследования значительные количества этого редкого минерала. Как уже говорилось, Sm используют в ядерных реакторах. Об о4ень мощных постоянных магнитах из SmCo6 уже упоминалось.
Европий (лат. Europium) Eu, Z=63.
Происхождение названия этого элемента не нуждается в пояснении. Важная область применения Ей — получение люминофорных материалов (см. выше). Кроме того, Ей часто используют при создании лазеров — мощных источников светового излучения. (О лазерах см., например, в статье Алюминия окись.)
Гадолиний (лат. Gadolinium) Gd, Z=64.
Назван в честь финского учёного, члена-корреспондента Петербургской академии наук Ю. Гадолина. Интересным свойством обладают хлорид и сульфат гадолиния. Если эти соли поместить в магнитное поле (намаг
нитить), а затем поле убрать, то соли, размагничиваясь, охладятся. Повторяя цикл намагничивание—размагничивание много раз, удаётся, например, охладить сульфат гадолиния Gd2(SO4)3«8H2O до сверхнизкой температуры, отличающейся от абсолютного нуля (—273,16 °C) менее чем на 0,001°.
Среди лантаноидов Gd — единственный металл, обладающий при комнатной температуре ферромагнитными свойствами (см. выше).
Тербий (лат. Terbium) Tb, Z=65.
Назван по шведскому городку Иттербю, около которого были найдены сравнительно большие запасы минералов лантаноидов. ТЬ используется для приготовления люминофоров и лазерных материалов.
Диспрозий (лат. Dysprosium) Dy, Z=66.
Название элемента свидетельствует о трудностях, которые пришлось преодолеть при его открытии (по-гречески «диспрбситос» — труднодоступный). По современным данным, Dy не столь уж редкий элемент; его почти в 4 раза больше, чем, например, вольфрама. Dy используется в специальных сплавах, которые обладают магнитными свойствами только при очень низких температурах.
Гольмий (лат. Holmium) Но, Z=67.
Назван в честь столицы Швеции — Стокгольма, который по-латыни называли «Гольмия». Как и другие Л., Но используется в люминофорах.
Эрбий (лат. Erbium) Er, Z=68.
Название произведено от Иттербю (см. выше). Применяется для тех же целей, что и ТЬ, и, кроме того, для приготовления специальных стёкол.
Тулий (лат. Thulium) Tm, Z=69.
Название получил от греческого «Туле» — так античные географы называли крайние северные страны. Широкое применение находит искусственно получаемый радиоактивный изотоп тулия 170Tm. С помощью его у-из-лучения можно осуществлять радиографирование — просвечивание живых материалов, тонких слоёв металла (до 5—6 мм толщиной) (о радиографии см. также в статье Радий). Чувствительность тулиевой радиографии столь высока, что с её помощью удалось, например, прочитать надписи на бронзовом ассирийском шлеме, которые стёрлись и для невооружённого глаза были незаметны.
Иттербий (лат. Ytterbium) Yb, Z=70.
Третий Л., получивший название от города Иттербю. Небольшие количества Yb добавляют к двуокиси циркония при изготовлении специальных жаропрочных материалов.
Лютеций (лат. Lutetium) Lu, Z=71.
Назван в честь Парижа, по-старому «Лютеция». Lu — один из самых дорогих Л., он ценится значительно выше, чем золото и платина. Разумеется, о широком применении столь дорогого вещества пока говорить не приходится.
С. С. Бердоносов. лед — см. Вода.
ЛЙТИЙ (лат. Lithium) Li — химический элемент I группы периодической системы Менделеева; атомный номер 3, атомная масса 6,941. Мягкий серебристо-белый металл. Природный Л. состоит из двух стабильных изотопов: 6Li (7,52%) и 7Li (92,48%).
литии
182
В 1817 г. шведский химик А. Арфедсон, проводя анализ минерала петалита, открыл в нём неизвестную ранее щёлочь. Учитель Арфедсона Я. Берцелиус дал ей название «литион» (от греч. «лйтеос» — каменный), так как, в отличие от едких кали и натра, которые были получены из золы растений, новая щёлочь была обнаружена в минерале. Он же назвал литием металл, являющийся, как тогда говорили, «основой» этой щёлочи. В 1818 г. английский химик Г. Дэви получил Л. электролизом гидроокиси Л. LiOH.
По распространённости в земной коре Л. занимает 28-е место (3,2-10“3% по массе). Для сравнения укажем, что на 26-м месте находится медь. Обнаружен Л. более чем в 150 минералах, из них собственно литиевых — около 30 (сподумен, лепидолит, ам-бигонит, уже упоминавшийся петалит и т. д.). Состав этих минералов сложен, многие из них относятся к очень распростра
нённому в земной коре классу алюмосиликатов. О том, как расшифровать их «непривычные» формулы — сподумен Li[AlSi2Oel, петалит Li[AlSi4Oi0l и другие,— вы узнаете из статей Минералы и Силикаты. Крупнейшие месторождения соединений Л. находятся в Канаде, США, Южной Родезии, Юго-Западной Африке, Бразилии, а также в СССР.
Интересно, что минерал сподумен встречается в природе в виде больших кристаллов массой в несколько тонн. На руднике Этта в США нашли кристалл сподумена в форме иглы длиной 16 м и массой 100 т.
Получение соединений Л. из руд, содержащих обычно от 0,5 до 3,5% Л., технически сложно и связано с кропотливой работой по их обогащению. Свободный металл выделяют электролизом расплавленной смеси хлоридов Л. и калия и другими способами.
Л.— самый лёгкий из всех металлов (его плотность 0,534 г/см3) — в 5 раз легче алюминия и почти вдвое легче воды. Л. плавится при сравнительно низкой температуре 180,5 °C, а кипит при 1317 °C. Металл очень мягок, легко режется ножом, вязок и пластичен. Соединения Л. окрашивают пламя в красивый карминово-красный цвет. Этим весьма чувствительным методом пользуются в качественном анализе для обнаружения Л.
В 1891 г. Р. Вуд (впоследствии известный американский физик, а тогда — молодой выпускник Гарвардского университета) приехал в город Балтимор, чтобы поучиться химии у профессора А. Ремсена. Он поселился в университетском студенческом пансионе. Жильцы пансиона подозревали, что им дают на завтрак блюда из вчерашних объедков. Вуд, оставив на тарелке несколько кусоч
ков мяса, посыпал их хлоридом LiCl — веществом, столь же безвредным, как поваренная соль. На следующее утро Вуд унёс в лабораторию часть поданного на завтрак мясного блюда и в его золе обнаружил Л. по красной линии его спектра. Так была разоблачена не в меру экономная хозяйка пансиона.
Л. принадлежит к подгруппе щелочных металлов — самых активных металлов периодической системы. Конфигурация внешней электронной оболочки атома Л. 2s1. Во всех своих соединениях он проявляет степень окисления + 1 (валентность I).
Л. соединяется с кислородом при комнатной температуре, образуя окись Li2O. Во влажном воздухе Л. медленно покрывается слоем гидроокиси LiOH. При повышенных температурах легко соединяется с галогенами, серой, фосфором, кремнием и другими неметаллами. Л.— один из немногих элементов, непосредственно соединяющихся с азотом. Правда, при комнатной температуре скорость реакции мала, но при нагревании до 250 °C резко возрастает — Л. воспламеняется в азоте:
6Li + N2 = 2Li3N
При нагревании выше 400 °C Л. соединяется с водородом:
2Li + H2 = 2LiH
Гидрид Л. LiH (бесцветные кристаллы) — прекрасный восстановитель, применяющийся в разных областях химической практики. Хорошо зарекомендовали себя как восстановители и смешанные гидриды Л. и других металлов, например литийалюминийгидрид LiAlH4. Гидрид Л. интересен ещё и тем, что 1 кг его, реагируя с водой, даёт около 2800 л водорода:
LiH + Н2О = LiOH + Н2
(В том, что водорода действительно образуется так много, вы убедитесь, выполнив соответствующие расчёты.) Поэтому гидрид Л. можно рассматривать как своеобразный «склад», в котором удобно хранить и транспортировать огнеопасный водород (при этом необходимо предохранять LiH от малейших следов влаги).
Л. стоит первым в ряду напряжений (см. Металлы). Взаимодействуя с водой, Л. вытесняет из неё водород:
2Li + 2Н2О = 2LiOH + Н2
Эта реакция идёт при комнатной температуре, но гораздо менее бурно, чем с прочими щелочными металлами. Минеральные кислоты энергично реагируют с Л. с выделением водорода и образованием соответствующих солей (LiCl, Li2SO4 и т. д.). Хранят этот очень активный металл в герметичной таре под слоем вазелинового масла. А для длительного хранения его
герметично запрессовывают в тонкостенные
лочки из меди или алюминия.
183
Ещё не так давно Л. был сравнительно мало изучен и «мало кому нужен». Интерес же к нему в наши дни огромен. Это объясняется прежде всего тем, что Л. находится вне конкуренции как источник промышленного получения трития (тяжёлого изотопа водорода). А тритий — это главная составляющая водородной бомбы и главное «горючее» для термоядерных реакторов будущего.
При ядерном взаимодействии изотопа eLi с нейтроном *п образуются тритий JH и изотоп гелия <Не. Уравнение этой ядерной реакции записывают так:
|Li + Jn = ?H+jHe
Индексы внизу — это заряды ядер, а индексы вверху — массовые числа изотопов; нейтрон — нейтральная частица с массовым числом 1. В ядерной реакции сохраняются общая сумма зарядов (здесь 3+0= 1+2) и общая сумма атомных масс (6+1=3+4).
В последнее время всё больше и больше Л. требует металлургия. Сплав магния с 10% Л. прочнее, а главное, легче самого магния. Хорошо известны сплавы алюминия и Л.— склерон и аэрон — там Л. всего 0,1%. Сплавы эти, помимо лёгкости, обладают высокой прочностью, пластичностью, повышенной стойкостью к коррозии и очень перспективны для авиастроения. Добавка 0,04% Л. к свинцово-кальциевым подшипниковым сплавам повышает их твёрдость и уменьшает коэффициент трения.
В больших количествах применяются в промышленности и соединения Л. Так, добавка LiOH к электролиту щелочных аккумуляторов (КОН) повышает срок их службы в 2—3 раза и ёмкость на 20%. На основе LiOH и органических кислот (особенно стеариновой и пальми-
ЛЮТЕЦИЙ
типовой) производят морозо- и термостойкие пластичные смазки для защиты металлов от коррозии в интервале температур от —60 до + 150°С.
Галогениды и карбонат Л. применяют в производстве оптических, кислотоупорны^ и других специальных стёкол, а также термостойкого фарфора и керамики, диэлектрических материалов, различных глазурей и эмалей.
Соединения Л. ускоряют (катализируют) некоторые химические реакции. Поднесите к куску сахара зажжённую спичку — он будет плавиться, но не гореть. А если посыпать сахар карбонатом Л., он загорится не хуже бумаги.
Немало и других интересных перспектив открывается перед Л. По значимости в современной технике это один из важнейших редких элементов. В. К. Бельский.
ЛИТОСФЕРА (от греч. «литое» — камень и «сфайра» — шар) — этот термин до последнего времени использовали как синоним термина земная кора. Сейчас в состав Л. обычно включают наряду с земной корой и находящуюся под ней верхнюю часть мантии.
ЛОУРЕНСИЙ (лат. Lawrencium) Lr — название, предложенное для радиоактивного химического элемента с атомным номером 103 из семейства актиноидов. Название и символ элемента № 103 окончательно не утверждены, поэтому в периодической системе Менделеева их помещают в скобках.
ЛЮТЕЦИЙ (лат. Lutetium) Lu — химический элемент III группы периодической системы Менделеева из семейства лантаноидов] атомный номер 71. См. также Редкоземельные элементы.
*Так, новейшая химия занимается главным образом тем, что разъединяет то, что природа объединила. Мы устраняем синтез природы, чтобы изучить её в отдельных элементах».
Иоганн Вольфганг Гёте (1749—1832)
(Избр. философ, произв., М., 1964, с. 306)
м
МАГНЕЗИЯ — обычно то же, что окись магния MgO; см. Магний.
МАГНИЙ (лат. Magnesium) Mg — химический элемент II группы периодической системы Менделеева; атомный номер 12, атомная масса 24,305. Серебристо-белый металл. Природный элемент состоит из смеси трёх стабильных изотопов: 24Mg (78,60%), 28Mg (10,11%) и 24Mg (11,29%).
Минералы, содержащие М. (тальк, магнезит, асбест), были известны и*применялись ещё в глубокой древности. В частности, из волокна асбеста изготовляли фитили для «неугасимых» лампад (древнегреческое слово «асбестос» означает неугасимый, немеркнущий). Однако долгое время магнезит (карбонат М.) ошибочно отождествляли с известняком (карбонатом кальция) и известь СаО с магнезией MgO. Открыть М. помогли исследования эпсомского источника минеральных вод в Англии, обнаруженного в 1618 г. Горькая вода этого источника обладала лечебным (слабительным) действием. Врач Н. Гру в 1695 г. выделил из неё соль, которую назвали английской, или горькой, солью (а минерал того же состава — эпсомитом). Оказалось, что эта соль представляет собой сульфат неизвестного металла. При осаждении её раствора содой Na2CO3 был получей порошок (основной карбонат М.), названный «белой магнезией». Вплоть до конца XVIII в. магнезией именовали самые различные природные и полученные искусственно вещества: магнитный железняк Fe3O4, пирит FeS2, пиролюзит МпО2, некоторые препараты сурьмы, висмута и др. Это название, как считают, произошло от древнего города Магнесия (в Малой Азии), в районе которого имеются залежи магнитного железняка. В 1808 г. английский химик Г. Дэви смешал увлажнённую магнезию — гидроокись М. Mg(OH)2 — с окисью ртути и пропустил через смесь ток от сильной батареи. На катоде выделилась амальгама нового металла, которому Дэви дал название «магнезий» (magnesium). Оно сохранилось в Англии, Германии, Италии, Франции и многих других странах. В России с 1831 г. принято название «магний». В 1829 г. французский химик А. Бюсси получил М., восстанавливая его расплавленный хлорид калием.
В природе М. широко распространён. Его содержание в земной коре несколько меньше, чем калия. и составляет 1,87% по массе (8-е место среди элементов). В свободном состоянии М. в силу своей химической активности не встречается, но зато повсеместно распространены его соединения — ведь почти каждый восьмой минерал содержит М. (!). По своему химическому составу минералы М. весьма разнообразны. Мощные скопления образуют кар-
бонаты М.— магнезит MgCO3 и доломит CaCO3-MgCO3 (из доломита состоят целые горные кряжи, поэтому запасы его практически неисчерпаемы). Широко распространены силикаты М.— оливин, тальк и серпентин (особенно важна для техники одна из разновидностей серпентина — асбест, уже упомянутый выше).
Очень важен для промышленности и один из галогенидов М.— карналлит КСЬ •MgCl2*6H2O, служащий исходным сырьём для получения металла. Огромные запасы этого минерала име
ются у нас на Северном Урале в районе Соликамск — Березники; мощность пластов достигает там 100 м.
Исключительно большое количество солей М. содержится в воде морей и океанов (см. Гидросфера}. Кстати говоря, уже работают заводы, добывающие М. из морской воды.
М. содержится в живых организмах. Одно из самых замечательных веществ в природе — зелёный пигмент растений хлорофилл — содержит около 2,7% М. С помощью хлорофилла совершается один из важнейших природных процессов — фотосинтез (подробнее см. Биосфера. Кислород). Без М. нет хлорофилла, а без хлорофилла была бы невозможна жизнь на нашей планете.
М.— очень лёгкий металл; его плотность 1,739 г/см3 — на г/3 меньше, чем плотность алюминия. По прочности же М. почти в два раза превосходит алюминий. Эти замечательные качества обеспечили сплавам на основе М. одно из первых мест в авиастроении. М.— металл средней твёрдости, он довольно тягуч и его можно прокатать в тонкие листы. Плавится М. при 651 °C, кипит при 1107 °C.
Конфигурация внешней электронной оболочки атома М. 2s2 (см. А том) и в своих соединениях он всегда проявляет степень окисле
185
МАГНИЙ
ния +2 (валентность II). На воздухе поверхность металла приобретает матово-белый оттенок из-за образования окисной плёнки. Эта плёнка защищает металл от дальнейшего окисления даже при нагревании до 350—400 °C.
Тонкую ленту или стружку М. легко поджечь спичкой, но компактный кусок М. от спички не загорается, так как М. обладает большой теплопроводностью, и тепло быстро отводится от места нагрева, распределяясь по всей массе. Горит М. ослепительно ярким белым пламенем, выделяя большое количество тепла (тепла, выделенного при сгорании 20 г М., достаточно, чтобы довести до кипения 1 л ледяной воды). Раньше М. широко применяли в фотографии для яркого освещения объекта («магниевая вспышка»). Интересно, что тушить горящий М. с помощью обычного углекислотного огнетушителя нельзя—М. продолжает гореть и в атмосфере двуокиси углерода:
2Mg + СО2 = 2MgO + С
Окись MgO—белое тугоплавкое вещество, хорошо растворимое в кислотах. Окись М. называют или просто магнезией или жжёной магнезией.
М. весьма бурно реагирует с галогенами, например хлором:
Mg + Cl2 = MgCl2 а при нагревании—с серой, углеродом, азотом и другими неметаллами:
Mg + S = MgS
Mg + 2C = MgC2 3Mg + N2 = Mg3N2 Зеленоватый порошок нитрида Mg3N2 образуется в небольших количествах и при сжигании М. на воздухе. А так как нитрид М. разлагается во влажной атмосфере по реакции:
Mg3N2 + 6Н2О = 3Mg(OH)2 + 2NH3
то окалина М. пахнет аммиаком.
С холодной водой М. реагирует весьма медленно вследствие малой растворимости гидроокиси Mg(OH)2, но из кипящей воды легко вытесняет водород:
Mg + 2Н2О = Mg (ОН)2 + Н2
Гидроокись Mg(OH)2— типичное основание. Легко растворяется М. и в разбавленных кислотах:
Mg + 2НС1 = MgCl2 + Н2
Щёлочи на М. не действуют. Все соли М. бесцветны, неядовиты, в водных растворах горьки на вкус и в большинстве своём хорошо растворимы; труднорастворимы MgCO3, Mg3(PO4)2 и MgF2.
В царской России практически не было магниевой промышленности. Производство М. широко развернулось в нашей стране лишь при Советской власти. Основной способ получения М.— электролиз расплавленного карналлита (см. выше) при температуре, превышающей
точку плавления М. Жидкий М. при электролизе всплывает на поверхность электролита, откуда его периодически удаляют с помощью вакуумных ковшей.
В последнее время М. производят и термическим методом — восстановлением при высокой температуре его окиси:
MgO + C = Mg + CO
Можно восстанавливать смесь СаО и MgO, получаемую обжигом доломита:
CaCO3-MgCO3 = СаО + MgO + 2СО2
А в качестве восстановителя, кроме углерода, применяют и кремний:
2MgO + 2СаО + Si = Са 2SiO4 + 2Mg
М. отгоняется и осаждается в приёмниках, охлаждаемых воздухом. Термическим способом выделяют М. более чистый, чем электролитический. С помощью М. получают трудновосстановимые металлы — титан, цирконий, ванадий:
TiO2 + 2Mg = Ti + 2MgO
Этот метод, называемый магниетермией, аналогичен алюминотермии (см. Алюминий). Порошок М. входит в состав смесей для осветительных ракет и снарядов (вместе с веществами, легко отдающими кислород,— KNO3, КС1О3 и др.).
С активностью М. связана ещё одна важная область его применения. Погрузив лист М. в воду и соединив его проводником с металлической конструкцией, создают по существу огромный гальванический элемент. Магниевый лист (активный электрод) со временем разрушается, а металл самой конструкции сохраняется. Подобную защиту имеют эстакады морских нефтепромыслов из металла и железобетона на Нефтяных Камнях Каспия. См. также Цинк.
В основном же М. идёт на производство магниевых сплавов, обладающих ценнейшими качествами. В состав этих сплавов, кроме М., входят главным образом алюминий, цинк, цирконий. После надлежащей термической обработки эти сплавы приобретают высокую прочность. Они легки (плотность около 1,8 г/см3), не магнитны, не дают искр при ударах и трении, газонепроницаемы, стойки против вибрационных нагрузок. Всё это делает их одним из основных материалов для самолётостроения, транспортного машиностроения, приборостроения и т. п. А так как эти отрасли индустрии широко развиваются, то мировое производство М. из года в год возрастает. Главный недостаток магниевых сплавов — сравнительно низкая коррозионная стойкость. Поэтому детали из сплавов М. защищают от кор
МАНГАНАТЫ
186
розии лакокрасочными покрытиями или искусственным наращиванием защитной окисной плёнки.
Из соединений М. наиболее важна окись MgO. Благодаря своей тугоплавкости (/пл 2800 °C) она идёт на изготовление огнеупорных тиглей, труб, кирпичей. Смесь прокалённой MgO и раствора MgCl2 используют в строительстве под названием «магнезиальный цемент», или «цемент Сореля» (см. Цемент). Из него можно получить лёгкие, огнестойкие и звуконепроницаемые строительные материалы — фибролит (при введении древесных стружек) и ксилолит (с добавкой опилок). Жжёная магнезия применяется в медицине как средство от изжоги и лёгкое слабительное. Перхлорат Mg(С1О4)2 — ангидрон — служит в лабораториях для осушения газов. Он жадно поглощает воду, превращаясь в кристаллогидрат Mg(C104)2-6H20. Это соединение теряет всю воду при нагревании около 240 °C, после чего ангидрон опять пригоден для работы.
Находят применение и природные силикаты М.— тальк и асбест, особенно последний: благодаря огнестойкости, малой теплопроводности и волокнистой структуре асбест служит прекрасным теплоизоляционным материалом.
Ю. И. Романьков. МАНГАНАТЫ — соли марганцовистой кислоты Н2МпО4. Анион МпО|“ придаёт раствору тёмно-зелёную окраску. М. могут проявлять свойства как окислителей, что более характерно (первая реакция), так и восстановителей (вторая):
-1 +6 +4 0
2KI + KfrMnO4 + 2Н2О = MnO2 + I2 + 4КОН + 6 0 +7 -1
2К2МпО4 + С12 = 2КМпО4 + 2КС1
МАРГАНЕЦ (лат. Marganum) Мп — химический элемент VII группы периодической системы Менделеева; атомный номер 25, атомная масса 54,9380. Тяжёлый серебристо-белый металл. В природе элемент представлен одним стабильным изотопом 66Мп.
Минералы М. известны с давних времён. Уже в глубокой древности природное соединение МпО2— пиролюзит (от греч. «пир»— огонь и «люо»— пресекаю) — служил для устранения зелёной окраски стекла, вызываемой примесью закиси железа. Об этом упоминает ещё римский естествоиспытатель Плиний Старший (1 в.н.э.). Этот минерал называли «чёрной магнезией» (см. Магний) из-за внешнего сходства х минералом магнетитом Fe3O4. Но получить из пиролюзита железо не удавалось. Поэтому в XVI или XVII вв.он получил название шап-ganesium (от греческого глагола «манганёйо» — обманываю). Только в 1774 г. шведский химик К- Шееле доказал, что это соединение неизвестного металла. В том же году другой шведский учёный Ш. Ган, накаливая смесь пиролюзита с углём, получил М., загрязнённый углеродом. Первоначальное латинское название manganesium вскоре было сокращено в manganum (по-немецки Mangan,
но по-английски manganese). Русское название «марга нец», возможно, произошло от немецкого Manganerz — пиролюзит (буквально «марганцовая руда»).
М. в природе довольно много. Его содержание в земной коре оценивается в 0,10% по массе, и по распространению здесь он занимает 11-е место среди прочих элементов. В свободном состоянии в природе М. не встречается. Его многочисленные минералы — это главным образом различные окислы. Среди них уже упоминавшийся пиролюзит, псиломелан — МпО2 с различными при
месями, манганит Мп2О3-Н2О, гаусманит Мп3О4, браунит Мп2О3. Совсем недавно учёные обнаружили, что на дне Тихого, Атлантического и Индийского океанов находятся огромные запасы М;— так называемые железо-марганцевые конкреции, занимающие около 10% всей площади океанического ложа. Запасы железо-марганцевых конкреций в океане оцениваются в 1500 млрд, т, в том числе 358 млрд.т Мп, 207 млрд.т Fe, многие млрд.т Al, Mg, Ni, Sn, Си. Возможно, что разработка этих месторождений — дело недалёкого будущего. По разведанным запасам М. и их добыче СССР занимает первое место в мире. Наши главнейшие месторождения М.— Чиатурское на Кавказе и Никопольское на Украине.
Из других минералов М. интересен орлец MnSiO3 (часть Мп2+ может быть замещена Са2+, Fe2+, иногда Mg2+, Zn2+). Цвет его — от вишнёво-красного до малинового и розового; отсюда другое название минерала — родонит (от греч. «родон»—роза и «литое» — камень). Богатое месторождение этого минерала, найденное на Среднем Урале, начали разрабатывать в середине XVIII в. В Петропавловском собо-ре-музее в Ленинграде хранится саркофаг массой 7 т из розового орлеца. Глыба минерала, из которой было изготовлено это произведение искусства, имела массу 47 т! Исключительную красоту камня используют и советские архитекторы и художники. Так, колонны станции метро «Маяковская» в Москве облицованы нержавеющей сталью и малиновым орлецом. Это сочетание даёт удивительный красочный эффект.
По внешнему виду М. похож на железо. Плотность М. 7,4 г/см3, плавится он при 1245 °C, а кипит при 2150 °C. М. имеет 4 аллотропные модификации, обозначаемые греческими буквами а, 0, у, 6. Модификация а
187 МАРГАНЕЦ
устойчива при обычной температуре (вплоть до 705 °C), остальные — при высоких. Модификация а хрупка, у и 6 (отчасти 0) пластичны. М. хорошо поглощает водород (примерно 25 мл Н 2 на 1 г М.), не взаимодействуя с ним химически.
В соответствии с положением М. в периодической системе Менделеева его атом имеет семь внешних (валентных) электронов (конфигурация 3d64s2, см. Атом). В соединениях М. может иметь практически весь «спектр» положительных степеней окисления, от +1 до +7 (соответственно валентности I—VII); наиболее характерны для М. степени окисления +2, +4 и +7. В соединениях с низшей степенью окисления М. напоминает железо, а некоторые его производные с высшей степенью окисления (+7) похожи на соответствующие соединения хлора (так, КМпО4 похож на КС1О4). С увеличением степени окисления возрастают окислительные свойства соединений М. (см. ниже).
М. легко окисляется с поверхности, но образующаяся плёнка окисла предохраняет металл от дальнейшего разрушения. При нагревании М. активно взаимодействует с кислородом (получается смесь окислов разной валентности), с галогенами (образуются производные 2-валентного М., например МпС12), серой (MnS, MnS2), азотом (Mn3N2) и другими неметаллами. В ряду напряжений М. стоит до водорода и вытесняет его даже из воды (если металл брать в виде мелкого порошка). В соляной и разбавленной серной кислотах М. растворяется с выделением водорода и образованием солей Мп(+2).
Окись МпО и соответствующая гидроокись Мп (ОН) 2 обладают основными свойствами. Двуокись МпО2 и соответствующая гидроокись Мп(ОН)4 амфотерны, с преобладанием кислотных свойств. Производные Мп (+4) — соли типа К<МпО4— называются манганитами. Так, закись-окись Мп3О4 (минерал гаусманит) +2 +4 -2
можно рассматривать как соль МПгМпО4, то есть как манганит двухвалентного Мп.
Соединения Мп (+2) — хорошие восстановители:
+ 2 +5
3MnSO4 + 2КСЮ3 + 12КОН = +6 -1
= 3K2MnSO4 + 2КС1 + 3K2SO4 + 6Н2О (эта реакция идёт при сплавлении) + 2 +4
2MnSO4 + 5РЬО2 + 6HNO3 = + 7 + 2 + 2
+ 2НМпО4 4- 3Pb(NO3)2+2PbSO4 + 2Н2О
Соединения Мп (+4) могут выступать и как окислители (например, при взаимодействии с соляной кислотой), и как восстановители
(например, при взаимодействии с бертоллето-вой солью):
+4 -1 +2-1 0
МпО2 + 4НС1 = МпС12 + С12+2Н2О (по этой реакции в лабораториях получают хлор)
ЗМпО2 + КСЮ3 + 6КОН = +6 -1
=ЗК2МпО4 + КС1 + ЗН2О (реакция идёт при сплавлении).
Из соединений Мп (+6) наиболее характерны марганцовистая кислота и её соли манганаты, И кислота, и её соли мало устойчивы. Ндпример, зелёный раствор манганата калия постепенно становится фиолетовым вследствйё превращения его в перманганат; одновременно выпадает гидрат двуокиси М.:
ЗК2МпО4 + ЗН2О = зелёный
= 2КМпО4 + МпО2 • Н2О + 4КОН фиолетовый
Вследствие перемены окраски раствора манганат калия назвали в XVIII в. минеральным хамелеоном. В середине XIX в. хамелеоном начали называть перманганат калия. Весьма важны соединения Мп(+7) — марганцовая кислота, марганцовый ангидрид и перманганаты.
В промышленности М. получают различными способами. Один из них — электролитический—даёт наиболее чистый металл. Этот метод разработан в нашей стране грузинскими учёными во главе с Р. И. Агладзе ещё в 1939 г. Большой интерес, проявляемый к М. химикат ми и металлургами Грузинской ССР, не случаен: ведь Чиатурское месторождение марганцовой руды пользуется общесоюзной и мировой известностью. По методу Агладзе, электролизу подвергают водный раствор MnSO4 с добавкой (NH4)2SO4. Катоды изготовляют из титанового сплава или нержавеющей стали, аноды — из свинца. Чешуйки М. снимают с катодов и переплавляют.
Основной потребитель М.— чёрная металлургия. Почти 95% получаемого М. идёт на раскисление и десульфурацию стали и чугуна и на добавки в специальные стали. М. придаёт им такие ценные качества, как коррозионная стойкость, износостойкость, вязкость и твёрдость. Для введения М. в сталь применяют чаще всего его сплавы с железом — ферромарганец (70—80% Мп, 0,5—7,0% С, остальное — Fe и примеси). Выплавляют ферромарганец в доменных и электрических печах (см. Ферросплавы). Интересно, что из всех химических элементов, которые добавляют в сталь для улучшения её свойств, М. был одним из первых. И именно сталь с повышенным содержанием М. (около 15%) була одной из первых
МАРГАНЦОВАЯ КИСЛОТА
188
легированных сталей. Её изобрёл в 1883 г. английский металлург Р. Гадфилд. Сталь, богатая М., отличается особой износостойкостью. Из неё изготовляют детали, подвергающиеся истиранию при больших нагрузках, Что же это за детали? Один из примеров — особенно ответственные части дробилок, драг, мельниц и других машин, добывающих и перерабатывающих полезные ископаемые. Другой пример — рельсовые крестовины и части стрелочных переводов на железных дорогах и трамвайных путях. Обычная же рельсовая сталь содержит 0,9—1,6% Мп.
До недавнего времени самыми распространёнными нержавеющими сталями были такие, которые содержали 18% Сг и 8% Ni. В СССР разработана и внедрена в промышленность безникелевая нержавеющая сталь, содержащая 14% Сг и 15% Мп.
Очень интересны сплавы М. с медью. Один из них — манганин (ок. 85% Си, 12% Мп, 3% Ni) характеризуется высоким электрическим сопротивлением, почти не зависящим от температуры. Поэтому из манганина изготовляют катушки сопротивления электроизмерительных приборов. Особенно же важно то, что электросопротивление манганина зависит от давления, причём зависимость эта линейная. Поэтому манганин в виде катушек сопротивления применяют в электрических манометрах для измерения очень высоких давлений.
Сплав меди с М. и алюминием (76% Си, 14% Мп и 10% А1) замечателен тем, что он закаляется, как сталь, и ферромагнитен, хотя ни одна из его составных частей заметных ферромагнитных свойств не проявляет.
Интересны соединения М., в которых его степень окисления равна нулю. Это—карбонил Мп2(СО)10 и его производные. Из них особенно важен метилциклопентадиентрикарбонил марганца СН3СбН4Мп (СО)3—жёлтая жидкость, кипящая при 233 °C. Это хороший антидетонатор для моторного топлива, не уступающий широко используемому тетраэтилсвинцу (ТЭС) (см. Свинец). О применении некоторых других соединений М. см. также Перманганаты. Микродозы М. необходимы для жизнедеятельности растений и животных. В. К. Бельский. МАРГАНЦОВАЯ КИСЛОТА НМпО4 — соединение марганца в степени окисления +7, довольно сильная кислота. Существует только в растворе. Анион MnOf придаёт раствору малиново-фиолетовый цвет. Сама кислота и её соли (перманганаты) являются сильными окислителями
МАРГАНЦОВИСТАЯ КИСЛОТА Н2МпО4 — соединение марганца в степени окисления +6, слабая кислота. В свободном виде не сущест
вует, неизвестен и её ангидрид (МпО3). При нагревании М. к. разлагается на марганцовую кислоту и двуокись марганца (реакция самоокисления — самовосстановления):
+6 +7 +4
ЗН2МпО4 = 2НМпО. + МпО. + 2Н,0 Соли М. к. называются манганатами.
МАРГАНЦОВОКИСЛЫЕ СОЛИ — соли марганцовой кислоты; то же, что перманганаты.
МАРГАНЦОВЫЙ АНГИДРИД Мп2О7— ангидрид не существующей в свободном состоянии марганцовой кислоты НМпО4. Зелёно-чёрная маслянистая жидкость. Получается при действии на перманганат калия концентрированной серной кислоты (действующей как водоотнимающее средство) на холоду:
2KMnO4 + H2SO4 = Mn2O7 + K2SO4 + Н2О Выше 55 °C разлагается со взрывом:
2Мп2О7 = 4МпО2 + ЗО2
М. а.— настолько сильный окислитель, что светильный газ, спирт, эфир при соприкосновении с ним сразу воспламеняются.
МЁДНЫЕ СПЛАВЫ — сплавы на основе меди; подразделяются на бронзы, латуни и медно-никелевые сплавы. Все они — твёрдые растворы металлов в меди (о твёрдых растворах см. статью Алюминиевые сплавы).
«Настоящая», известная с древнейших времён бронза — это сплав меди с 8—10% олова.
Видимо, эта бронза была получена случайно в ходе выплавки меди. Большая твёрдость
Рис. 1. Царь-пушка. Отлита в 1586 г. русским мастером А. Чоховым. Царь-пушка предназначалась для обороны Кремля, но из нее никогда не стреляли. Калибр 88,9 см, вес 39,3 т.
189
МЕДЬ
Рис. 2. Царь-колокол. Это гигантский колокол весом почти 202 т отлит в 1733—1735 гг. русскими мастерами отцом и сыном И. Ф. и М. И. Моториными.
и относительная легкоплавкость позволили ей вытеснить медь и положить начало бронзовому веку, который длился свыше двух тысячелетий. До нашего времени дошли прекрасные произведения искусства из бронзы мастеров древнего мира — Египта, Греции, Рима и Китая. В XIX в. оловянистаябронза широко применялась для отливки пушек и колоколов (из нее изготовлены, например, знаменитые Царь-пушка и Царь-колокол в Московском Кремле).
Сейчас оловянистая бронза постепенно вытесняется другими бронзами. Дело не только в том, что их конструкционные свойства лучше, но и в том, что олово дефицитно. Очень прочны алюминиевые бронзы (5—10% А1), идущие на изготовление деталей авиационных двигателей. Из свинцовой бронзы делают подшипники для паровозов, судовых и авиационных двигателей, водяных турбин. Бронзы с небольшим содержанием кремния дёшевы и с успехом заменяют дорогие оловянистые. Исключительно прочны и долговечны бериллиевые бронзы (2% Be). Благодаря своим упругим свойствам они служат материалом для пружин, практически не знающих усталости (выдерживающих до 20 млн. циклов нагрузки). В повседневной жизни мы постоянно встречаемся с монетной бронзой. Это сплав меди с 4,5—5,5% алюминия, из него чеканят разменную монету («медь»).
Латуни — сплавы меди с цинком (до 38% Zn). В специальные латуни, кроме цинка, добавляют алюминий, железо, марганец. Латуни де
шевле бронз, легко обрабатываются, стойки к действию химических реагентов. Они идут на изготовление радиаторных труб, гибких шлангов, патронных гильз. Латуни с добавкой алюминия по внешнему виду похожи на золото, из них изготовляют значки, эмблемы.
В медно-никелевых сплавах основной добавкой является никель. Мельхиор, содержащий 29—33%Ni, устойчив к действию морской воды, перегретого пара и других агрессивных сред. Он применяется в кораблестроении, при изготовлении приборов точной механики, домашней утвари, вилок, ложек, ножей. Мельхиор с содержанием 18—20% Ni идёт на чеканку монет («серебро»). Нейзильберы — сплавы меди, никеля (13,5—16,5%) .и цинка (18—22%) обладают красивым серебристым цветом, стойки в растворах солей и органических кислотах. Они идут на изготовление медицинских инструментов, художественных изделий и бытовой посуды. Константан (сплав Си с 40% Ni) и манганин (сплав Си с 12— 14% Мп и 2—4% Ni) имеют высокое электрическое сопротивление, почти не зависящее от температуры; применяются в электроизмерительных приборах. Ю. И. Романьков.
МЕДЬ (лат. Cuprum) Си — химический элемент I группы периодической системы Менделеева; атомный номер 29, атомная масса 63,546. Металл красного цвета. Элемент представляет собой смесь двух стабильных изотопов с массовыми числами 63 (69,1%) и 65 (30,9%).
Историческая справка. М. известна с древнейших времён. Она встречается в природе в виде самородков, покрытых зелёным или голубым налётом, кра-
сивого красного цвета в изломе, необычайно тяжёлых по сравнению с камнем. Ударами каменного молотка человек придавал кускам М. нужную форму. М. оказалась первым металлом, который заменил камень в первобытных орудиях труда. Это произошло 6000 лет назад (около 4000 лет до н. э.). Так и датируется начало медного века.
В поэме «О природе вещей» знаменитого древнеримского философа и поэта Тита Лукреция Кара говорится:
«Прежде служили оружием руки могучие, когти.
Зубы, каменья, обломки ветвей от деревьев и пламя.
После того была найдена медь...»
В Египте ещё в период Древнего царства из М. выковывали топоры, ножи, булавы и предметы домашней утвари. Камни пирамиды Хеопса (3000 лет до н. э.) обтёсаны медным инструментом. Постепенно египтяне научились не только обрабатывать медные самородки, но и выплавлять М. из руд, добываемых на Синайском полуострове. Иногда попадались руды с примесью олова,
МЕДЬ
190
Рис. 1. Бронзовая статуэтка.
что привело к открытию сплава М. с оловом — бронзы. Бронза оказалась более легкоплавкой и твёрдой, чем сама М. Открытие бронзы положило конец короткому медному веку и начало веку бронзовому, гораздо более длительному (с конца 4-го по 1-е тысячелетие до н. э.).
Древние римляне вывозили медную руду с острова Кипра, отсюда произошло латинское название М. «куп-рум». До нашего времени сохранились прекрасные бронзовые статуи Древнего Египта и Эллады, бронзовые бюсты римских цезарей. Бронза была и остаётся любимым материалом скульпторов всех времён.
Медь в природе. По распространённости в земной коре (4,7-10“3% по массе) М. следует за никелем и занимает всего лишь 26-е место среди других элементов. М. встречается в природе в виде самородков, порой значительных размеров. Так, в 1857 г. в США в районе Великих Озёр был найден самородок массой 420 т. Интересно, что выступающие части его были отбиты ещё каменными топорами. Однако самородная М. в наше время составляет незначительную часть от общего производства металлов. Подавляющая часть М. присутствует в горных породах в виде соединений. Из сотен минералов М. промышленное значение имеют немногие, в частности халькопирит—медный колчедан CuFeS2, халькозин—медный блеск Cu2S, ковеллин CuS, малахит CuCO3«Cu(OH)2 и азурит 2CuCO3-Cu (ОН)2.
Минералы М. ярко и красиво окрашены. Вот как в сказе П. П. Бажова описываются владения Хозяйки Медной горы: «Как комнаты под землёй стали, а стены у них разные. То все зелёные, то жёлтые с золотыми крапинками. На которых опять цветы медные. Синие тоже есть, лазоревые. Одним словом, изукрашено, что и сказать нельзя».
Действительно, кусочки ковеллина (его называют ещё «медный индиго») отливают всеми цветами радуги. Медный блеск имеет синевато-стальной цвет и блестит,
как полированный металл. Халькопирит отливает золотом. Азурит густо-синего цвета со стеклянным блеском, а его разновидность — землистый азурит (или медная синь) ярко-голубого.
Малахит высоко ценили ещё в древности. Цвет малахита меняется от светло-зелёного до атласного тёмнозелёного с разного рода прожилками. Не случайно его назвали по растению «мальва» (по-гречески «малахе») — благодаря сходству минерала с зелёным листом. Малахит и азурит, как правило, встречаются вместе. В СССР их залежи имеются на Урале, Алтае, в Казахстане. В 1835 г. на Урале была найдена глыба малахита весом 250 т.
Русские мастера умели особым способом облицовывать малахитом вазы, столы и колонны зданий («русская мозаика»). Малахитовые вазы высотой в человеческий рост можно увидеть в ленинградском Эрмитаже.
Физические и химические свойства. М.— металл красного, в изломе розоватого цвета, в тонких слоях при просвечивании приобретает зеленовато-голубой оттенок. Плотность М. 8,96 г/см3; /пл 1083 °C, /кип 2600 °C. М.— металл довольно мягкий, весьма ковкий, из него можно прокатать листочки толщиной всего в 2,5 микрона (в 5 раз тоньше папиросной бумаги!). М. хорошо отражает свет, прекрасно проводит электричество и тепло (в этом отношении её превосходит только серебро).
М., серебро и золото составляют побочную подгруппу I группы периодической системы Менделеева*. Со щелочными металлами (основная подгруппа) их сближает лишь способность образовывать одновалентные катионы. Для М. и её аналогов характерно то, что они могут давать соединения с валентностью, превышающей номер своей группы. Кроме того, элементы подгруппы М., в отличие от щелочных металлов, склонны к комплексообразованию, образуют окрашенные соли, то есть проявляют свойства, сближающие их с никелем, палладием и платиной (побочная подгруппа VIII группы).
Такое поведение Си, Ag и Au объясняется конфигурацией внешних электронных оболочек их атомов. У атома М. самая наружная (4-я от ядра) оболочка содержит один s-электрон, ему предшествуют десять d-электронов 3-й от ядра оболочки (конфигурация StPMs1, см. А там).
Атомы М. и её аналогов могут при образовании соединений терять не только самый внешний s-электрон, но один или два d-электрона предшествующей оболочки, проявляя более высокую степень окисления. Для М. окислительное число +2 (валентность II) более характерно, чем +1 (валентность I).
М. химически малоактивна и в чистом, сухом воздухе не изменяется. Однако атмосфера, в которой мы живём, содержит водяные пары и двуокись углерода. Поэтому неудивительно, что, например, произведения скульптуры, изготовленные из М. и бронзы, со временем по
191
МЕДЬ
крываются зеленоватым налётом — «патиной». В обычной атмосфере патина состоит из основного карбоната М. (малахита). В атмосфере, содержащей двуокись серы, медные изделия покрываются основным сульфатом CuSO4-3Cu(OH)2 (по составу это минерал брошантит), а вблизи моря — основным хлоридом CuCl2-3Cu(OH)2 (по составу минерал атакамит). Интересно, что патина образуется только во влажном воздухе (при влажности выше 75%).
Патина придаёт изделиям из М. и бронзы красивый, как говорится, «старинный» вид. А сплошной налёт патины обладает и защитными свойствами, предохраняя металл от дальнейшего разрушения. Но образующаяся плёнка может быть с дефектами и потому недостаточно надёжной. Гораздо прочнее такое же покрытие, нанесённое на металл искусственно.
Один из способов искусственного получения патины таков. Изделие из М. или бронзы обрабатывают серной кислотой и затем выставляют на воздух. Через некоторое время операцию повторяют. Образующийся сульфат М. гидролизуется и постепенно превращается в устойчивую плёнку CuSO4-3Cu(OH)2.
В начале 1974 г. появилось сообщение о том, что распалась дружная квадрига знаменитых венецианских коней, украшающих собор святого Марка. Их возраст исчисляется с IV в. до н. э. И вот первого слева пришлось препроводить в новое «стойло», специально оборудованное в Музее Марчано. Что же произошло? Загрязнения атмосферы постепенно разрушают бронзу скульптуры. В течение веков им не может противостоять естественная патина. И коней приходится отправлять на «клиническое лечение»...
Если быстро погрузить в холодную воду раскалённый докрасна кусок М., то на его поверх
Рис. 2. Малахитовая ваза.
ности образуется ярко-красная плёнка закиси Си2О. При умеренном же нагревании М. на воздухе поверхность её покрывается чёрной окисью СиО. Обычно образцы Си содержат сотые доли Си2О. При нагревании такого металла в атмосфере, содержащей водород и некоторые другие газы (СО, СН4), происходит восстановление Си2О:
Cu2O + Н2 = 2Си + Н2О
Cu2O + СО = 2Си + СО2
Образовавшиеся пары воды и двуокись углерода выделяются из металла, вызывая появление трещин. А это резко ухудшает механические свойства М. («водородная болезнь»).
Гидроокись Си (ОН)^ выпадает в виде объёмистого голубого осадка при действии щелочей на растворы солей двухвалентной М. Это слабое основание, образующее с кислотами соли. Впрочем свежеосаждённая Си(ОН)2 растворяется и в концентрированных растворах щелочей, но её кислотный характер выражен слабо.
Фтор, хлор и бром реагируют с М., образуя соответствующие галогениды двухвалентной М., например:
Си + С12 = СиС12
При взаимодействии иода с нагретым порошком М. получается иодид одновалентной М.:
2Cu + I2 = 2CuI
М. горит в парах серы:
Си + S = CuS
К сере М. проявляет большее сродство, чем к кислороду. На этом свойстве основан пирометаллургический способ получения М.
В ряду напряжений М. стоит после водорода. В бескислородных кислотах она не растворяется. О действии на М. HNO3 и H2SO4 см. в статьях Азотная кислота. Серная кислота. Одну из важнейших солей М.— сульфат получают растворением металла в нагретой разбавленной H2SO4 при продувании воздуха:
2Cu + 2H2SO4 + О2 = 2CuSO4 + 2Н2О
Безводный сульфат М. бесцветен; присоединяя воду, он превращается в медный купорос CuSO4-5H2O, лазурно-синие прозрачные кристаллы. Благодаря свойству сульфата М. из
менять окраску при увлажнении его использу
ют для обнаружения следов воды в спиртах,
эфирах, бензинах и пр.
Получение М.— сложный, многоступенчатый процесс. Ещё в XIX в. М. выплавляли из руд, содержащих 15% и даже больше металла. В на
ше время богатые М. руды практически исчер-
паны. М. получают сейчас из сернистых руд, содержащих всего лишь 1—7% Си. Такую руду
предварительно обогащают, отделяя ценные минералы от основной массы пустой породы.
МЕДЬ
192
Из полученных концентратов М. извлекают главным образом (на 80%) пирометаллургическими методами.
Плавку руд М. вели обычно либо в отражательных, либо в электрических печах. В отражательных — необходимое для плавления тепло получают сжиганием углеродистого топлива (естественный газ, мазут), причём тепло отражается от свода и стен печи, что повышает температуру рабочего пространства. В электрических печах тепло получают при пропускании через расплавленный шлак электрического тока. Но плавка, основанная на внешних источниках теплоты, неэкономична. Поэтому теперь преобладают способы плавки, в которых используется теплота сгорания сульфидов, составляющих основную массу медных концентратов.
При плавке вследствие большего сродства М. к сере, а компонентов пустой породы и железа к кислороду М. концентрируется в сульфидном расплаве (штейне), а окислы образуют шлак. Шлак легче штейна, и потому они легко отделяются друг от друга, как масло и вода. В штейне М. находится в виде сплава сульфидов, главным образом Cu2S и FeS. В специальных конвертерах, наподобие бессемеровских (см. Железо). на поверхность жидкого штейна вдувают воздух. Сначала окисляется сульфид железа, вследствие большего сродства Fe к кислороду; для связывания окислов железа в конвертер добавляют кварц SiO2 — так образуется легко отделяемый конвертерный шлак. Затем окисляется сульфид меди с образованием металлической М. и SO2. Эту черновую М., содержащую 97—99% Си, разливают в формы. При рафинировании, чаще всего электролитическом, из черновой М. извлекают ценные металлы — спутники, в том числе золото, серебро. висмут, никель, кобальт, цинк, а также селен и теллур. Чистота электролитической М.
достигает 99,98%. Чистая М. (не менее 99,9% Си) нужна для электротехники — любые примеси резко снижают электропроводность М.
Для электролитического рафинирования слитки черновой М., имеющие форму толстых досок, подвешивают в ваннах, содержащих раствор медного купороса с добавкой серной кислоты. В тех же ваннах подвешены тонкие листы чистой М. Они служат катодами, а отливки из черновой М.— анодами. При надлежащих условиях (напряжение, плотность тока, состав и температура электролита) во время прохождения тока на аноде про* исходит растворение М.:
Си — 2ё = Си2 +
а на катоде — разряд катионов М.:
Си2+ + 2ё = Си
Примеси, в том числе Ag, Au, Pt, выпадают на дно ванны в виде илообразной массы — анодного шлама (немецкое Schlamm — ил). Его переработка на благородные металлы нередко окупает все расходы по рафинированию черновой М. электролизом.
Производство М.— одно из ведущих в цветной металлургии. Восстановление цветной металлургии в СССР после Гражданской войны ознаменовалось пуском в 1922 г. Кала-тинского (ныне Кировоградского) медеплавильного завода на Урале. Одновременно началась переработка лома и отходов на вторичную М. в Петрограде. В 1925 г. на Урале был пущен крупный Карабашский завод. В годы первой пятилетки вступили в строй медеплавильные заводы Красноуральский на Урале и Карсакпайский в Казахстане. В 1940 г. работали два новых крупных медеплавильных комбината — Балхашский в Казахстане и Меднбгор-ский на Южном Урале. В послевоенные годы пущен новый гигант медной промышленности — Норильский горно-металлургический комбинат. А в 1959 г. на Южном Урале, в связи с осво-
Рис. 3. Город Гай. На Южном Урале освоены месторождения медно-колчеданных руд. Здесь вырос город Гай, построенный молодёжью (1959 г.).
ением месторождения медноколчеданных руд, возник город Гай. И сам город, и его горнообогатительный комбинат построены молодёжью.
Применение. Высокая электро- и теплопроводность М., её пластичность и коррозионная стойкость определили направление использования металла в промышленности. Больше половины всей добываемой М. идёт на производство электрических проводов. Так, чтобы передать трёхфазный переменный ток на расстояние 1 км тремя медными проводами диаметром по 2 мм, необходимо 84 кг М. В электровакуумной технике употребляется так называемая бескислородная (вакуумная) М. высокой чистоты. В технике сильных токов из М. изготов-
ляют проводники с сечениями различной формы.
Высокая теплопроводность и сопротивление коррозии позволяют изготовлять из М. ответ-
ственные детали тепл
менников
холодиль-
ников, вакуумных аппаратов, трубопроводов для перекачки масел и топлив и т. д. А что касается современного автомобиля, то он со-
193
МЕТАЛЛЫ
Александр Александрович БАЙКОВ — один из основателей физико-химической теории металлургических процессов.
Советский металлург и химик Александр Александрович Байков родился 6 августа 1870 г. в городе Фатеже (ныне Курская область) в семье адвоката. Ещё будучи учеником Курской гимназии, он заинтересовался химией и устроил дома более чем скромную лабораторию. В ней он делал простейшие опыты и даже читал Своим товарищам «лекции», сопровождавшиеся демонстрациями. Настольной книгой юноши стали «Основы химии» Д. И. Менделеева. По окончании Петербургского университета Байков в 1895 г. был назначен заведующим химической лабораторией Института инженеров путей сообщения (ныне Ленинградский институт инженеров железнодорожного транспорта). Здесь он близко столкнулся с проблемами производства И испытания рельсов и других металлических изделий, производства цемента. Одновременно он вёл исследования сплавов меди с сурьмой. В 1903 г., после защиты этой работы в качестве диссертации, Байков был избран профессором металлургии и технической химии открытого в 1902 г. Петербургского (ныне Ленинградского) политехнического института.
В этом институте Байков развернул блестящую широкую педагогическую и научную деятельность. Он читал совершенно оригинальные курсы общей металлургии (теория металлургических процессов), металлургии меди и иных, кроме железа, металлов. Он читал также созданный им самим курс металлографии и организовал первую в России учебную металлографическую лабораторию. После Великой Октябрьской революции Байков принял ближайшее участие в восстановлении металлургической промышленности, в создании новых предприятий и производств, в частности высококачественных сталей, новых видов цементов и огнеупоров. Им воспитана большая школа металлургов — исследователей и практиков. Научно-техническая и педагогическая деятельность Байкова получила высокую оценку: он был избран академиком (1932 г.), ему присуждено звание лауреата Государственной премий СССР (1943 г.) и Героя Социалистического Труда (1945 г.). Умер Байков в Москве 6 апреля 1946 г. Институт металлургии Академии наук СССР носит имя А. А. Байкова. Издано 5-томное собрание его трудов.
На фотографии — портрет А. А. Байкова на бронзовой медали.
держит 10—15 кг М.— из М. и её сплавов делают радиаторы, маслоохладители, проводники тока.
Издавна М. применялась в строительстве; древние египтяне строили медные водопроводы. И в наше время свыше 200 т М. пошло на водопроводную систему одного из самых высоких зданий в мире—Эмпайр стейт билдинг (Нью-Йорк). В средние века крыши церквей и замков покрывали листовой М. Знаменитый королевский замок в Эльсиноре (Дания) покрыт кровельной М. В современной архитектуре орнаменты и другие украшения из М., отличающиеся золотистыми или красноватыми тонами, служат прекрасным дополнением к основному материалу.
Около 30—40% М. используют в виде различных сплавов (см. Медные сплавы).
Применение находят и соединения М., главным образом медный купорос и другие соли. Минеральные краски, средства борьбы с вредителями и болезнями растений, катализаторы различных процессов — таков далеко не полный перечень областей их службы.
В растительных и животных организмах М. входит в состав «живых катализаторов» — ферментов и участвует во многих физиологических процессах. М. способствует росту и развитию растений и повышает их устойчивость к холоду. При недостатке М. в почве растения «жухнут», перестают плодоносить. Питаясь такими растениями, домашний скот заболевает анемией и зачастую гибнет. При недостатке М. в организме нарушается обмен веществ и развивается анемия и у человека. Ведь М., точнее, содержащие её ферменты, стимулирует кроветворную деятельность костного мозга и повышает интенсивность окислительных процессов. При
недостатке М. в почве её удобряют сульфатом М. Есть и содержащие М. препараты, которыми лечат человека и животных. Ю. И. Романъков.
МЕНДЕЛЁВИЙ (лат. Mendelevium) Md — радиоактивный химический элемент с атомным номером 101; относится к актиноидам.
МЕТАЛЛЫ — химические элементы, характеризующиеся способностью отдавать внешние (валентные) электроны. В свободном состоянии М. образуют простые тела, для которых характерны следующие свойства: 1) особый, металлический блеск, то есть высокая способность отражать свет; 2) лёгкость пластической деформации, то есть способность под действием внешних сил изменять форму без нарушения сплошности; 3) высокая электропроводность, уменьшающаяся с повышением температуры; 4) хорошая теплопроводность.
Из известных сейчас 105 химических элементов 83 принадлежат к М. и лишь 22 — к неметаллам. Если в длинном варианте периодической системы элементов Д. И. Менделеева провести прямую от бора до астата (см. таблицу на стр. 34—35), то можно считать, что неметаллы расположены на этой прямой и справа от неё, а М.— слева. Впрочем, провести резкую границу между М. и неметаллами вряд ли возможно.
Роль металлов в истории материальной культуры.
Греческое слово «мёталлон» означало первоначально
13 хэш
МЕТАЛЛЫ
194
копи, рудники; отсюда и произошёл термин «металл». В древности и в средние века считалось, что существует только 7 М.: золото, серебро, медь, олово, свинец, железо, ртуть. Это число соотносилось с числом планет, которыми астрономы тех времён признавали Солнце, Луну, Венеру, Юпитер, Сатурн, Марс и Меркурий. По алхимическим представлениям, М. зарождались в земных недрах под влиянием лучей планет и постепенно совершенствовались, превращаясь в конечном итоге в серебро и золото. Наиболее совершенный М.— золото. По верованиям алхимиков, естественный, крайне медленный ход совершенствования неблагородных металлов можно сильно ускорить, прибавляя к ним «философский камень». Это фантастическое вещество было предметом многовековых бесплодных поисков.
В 1789 г. французский химик А. Л. Лавуазье в своём руководстве по химии дал список простых веществ, в который включил все известные тогда 17 М. (Sb, Ag, As, Bi, Co, Cu, Sn, Fe, Mn, Hg, Mo, Ni, Au, Pt, Pb, W, Zn). По мере развития методов химического исследования число известных М. стало быстро возрастать. В первой половине XIX в. были открыты спутники платины (платиновые металлы); получены путём электролиза не
которые щелочные и щелочноземельные М.; положено начало разделению редкоземельных М.; при химическом анализе минералов открыты неизвестные ранее М. В начале 1860-х гг. с помощью спектрального анализа были открыты рубидий, цезий, индий, таллий. Блестяще подтвердилось существование М., предсказанных Д. И. Менделеевым на основе его периодического закона (галлия, скандия и германия). Открытие радиоактивности в конце XIX в. повлекло за собой поиски радиоактивных М., увенчавшиеся полным успехом. Наконец, методом ядер-ных превращений начиная с середины XX в. были получены не существующие в природе радиоактивные М., в том числе и те, что принадлежат к трансурановым элементам.
В истории материальной культуры, древней и новой, М. имеют первостепенное значение. Уже в доисторические времена людям были известны самородные М.— золото, серебро, медь.
В поэме «О природе вещей» древнеримский поэт и философ-матери ал ист Тит Лукреций Кар (1 в. н. э.) пишет:
«Чтобы оружье иметь и орудья для рубки деревьев, Чтобы обтёсывать лес и выстругивать гладкие
брусья, Чтобы буравить, долбить и просверливать в дереве дыры, Это они серебром или золотом делать пытались... Тщетно: слабей была стойкость у этих металлов, и с медью Вровень они не могли выдерживать грубой работы».
Поэт замечает:
«Так обращенье времён изменяет значенье предметов: Что было раньше в цене, то лишается вовсе почёта, Следом другое растёт, выходя из ничтожества к блеску».
На смену меди и бронзе (сплаву меди с оловом; по-латыни и медь, и бронза называются одинаково — aes) пришло железо. В природе в металлическом состоянии железо встречается только в виде железных метеоритов, которые всегда содержат никель (иногда 30%). Возможно, что метеоритное железо, в котором было около 3% никеля, послужило первоначально для изготовления ору-
„Семь металлов создал свет по числу семи планет"
В древности и в средние века были известны только семь металлов.* Это число соотносилось с числом известных тогда планет — Солнцем, Луной, Меркурием, Венерой, Марсом, Юпитером и Сатурном. Под влиянием лучей этих небесных светил, учили алхимики, в недрах Земли зарождаются и совершенствуются металлы. Каждая планета имела свой особый знак, который одновременно служил и символом металла, находящегося под её влиянием. Вот эти знаки:
О—Солнце (золото) С —Луна (серебро)
9 — Меркурий (ртуть)
$ — Венера (медь) 2/.— Юпитер (олово) сГ—Марс (железо) 5—Сатурн (свинец)
Обычно принимают, что приведенные знаки — не что иное, как атрибуты соответствующих богов античной мифологии — жезл Меркурия, зеркало Венеры, трон Юпитера, щит и копьё Марса, коса Сатурна. Впрочем, имеются и другие объяснения.
Миниатюра из алхимической книги изображает семь металлов в недрах Земли в виде семи богов, укрывшихся в пещере. В середине сидит Аполлон, бог Солнца и золота; слева от него — Диана, богиня Луны и серебра; справа — Венера, символизирующая планету Венера и медь. В глубине стоят (слева направо): Юпитер — олово, Марс — железо, Сатурн — свинец и Меркурий — ртуть.
195
МЕТАЛЛЫ
Николай Семёнович КУРНАКОВ. Положил начало новому методу в химии — физико-химическому анализу растворов и сплавов.
Советский физико-химик Николай Семёнович Курнаков родился 6 декабря 1860 г. в Нолинске (ныне Кировская область). Его отец, участник обороны Севастополя (1854—1855 гг.), скончался в 1868 г. от последствий тяжёлой контузии, полученной на войне. Среднее образование будущий учёный получил в Нижегородской военной гимназии, высшее — в Петербургском (ныне Ленинградском) горном институте, который окончил в 1882 г. Химией увлёкся ещё в 12 лет и проделал множество опытов в своей домашней лаборатории. В 1893 г. Курнаков стал профессором химии Горного института. А в 1902 г. Курнаков занял кафедру общей химии в только что основанном Петербургском (ныне Ленинградском) политехническом институте. Здесь он организовал большую, богато оборудованную лабораторию. И здесь же развернулись исследования Курнакова в области металлических сплавов. Для этих исследований он создал новые приборы и новые методы.
Как известно вам из учебника, в начале XIX в. среди учёных утвердилось мнение, будто все химические соединения имеют определённый постоянный состав. Такое утверждение способствовало внедрению в химию атомистических представлений Дж. Дальтона. Впоследствии Курнаков предложил называть эти соединения дальтонидами. На основе богатейшего экспериментального материала Курнаков показал, что существует и другой класс соединений, не подчиняющихся законам постоянства состава и кратных отношений. Он назвал их бертоллидами (в 1912 г.), в память К. Л. Бертолле, который в начале XIX в. доказывал, что состав химических соединений, вообще говоря, должен быть переменным. В начале XX в. Курнаков и его сотрудники открыли лишь несколько бертоллидов среди двойных металлических сплавов. В наше же время известны тысячи неорганических соединений, состав которых не удовлетворяет законам стехиометрии. К таким нестехиометрическим соединениям принадлежат многие гидриды, оксиды, сульфиды, нитриды, карбиды, комплексные соединения, некоторые минералы.
Курнаков положил начало новому методу исследования растворов и сплавов, который в 1913 г. назвал физико-химическим анализом. Этот метод позволяет определять состав веществ, образующихся в растворах и сплавах, не выделяя этих веществ из систем и не делая их анализа. В 1913 г. Курнаков был избран академиком. В 1918 г. по его инициативе был основан при Академии наук Институт физико-химического анализа, который в 1934 г. вошёл в состав Института общей и неорганической химии АН СССР. С 1944 г. этот институт носит имя Н. С. Курнакова — своего основателя и первого директора.
Николай Семёнович всегда говорил, что одна из главных задач науки — её помощь в изучении и освоении природных минеральных богатств страны. Эта сторона его деятельности развернулась только после Великой Октябрьской революции. Сульфат натрия из залива Кара-Богаз-Гол, Соликамские месторождения калиевых солей, тихвинские бокситы, индерские бораты... Для их познания и использования успешно трудились Курнаков и его сотрудники. Они очень много сделали и для организации у нас новых производств — сплавов высокой прочности на основе лёгких металлов (алюминия, магния),. сплавов высокого электрического сопротивления для электроизмерительных и нагревательных приборов, сплавов платиновых металлов. Курнаков воспитал большую научную школу химиков и металлургов. Он награжден премией имени В. И. Ленина (1928 г.) и Государственной премией СССР (1941 г.). Его избранные труды изданы в 3 томах. Н. С. Курнаков умер 19 марта 1941 г. под Москвой.
жия и орудий труда (сплавы железа с содержанием свыше 3% никеля плохо поддаются обработке).
Лукреций пишет:
«Си ль/ железа, потом и меди, были открыты, Но применение меди скорей, чем железа, узнали; Легче её обработка, а также количество больше... Мало-помалу затем одолели мечи из железа, Вид же из меди серпа становился предметом
насмешек»
Уже в ту далёкую эпоху железо (но земного, а не космического происхождения, получаемое восстановлением его руд углём) заняло первое место среди прочих М. по своему значению.
Своё ведущее место железо сохраняет и в наши дни. На его долю приходится около 95% от мировой добычи М. Ну, а другие М.?
13*
В статье Химия служит человеку мы рассказали о том, как стремительно в современной технике возросла роль алюминия и титана. Ядерной энергетике необходимы уран, торий, цирконий, натрий... «Обычной» электротехнике — медь, вольфрам, молибден... А электротехнике будущего, где важная роль отводится сверхпроводникам,— ниобий. Без германия, индия, сурьмы не получили бы сегодняшнего развития полупроводники. Без редкоземельных элементов—электронно-вычислительные устройства, лазеры и другие, самые новые области техники. Вся фото- и кинотехника «работает» на серебре. Это — лишь малый список примеров.
МЕТАЛЛЫ
196
Механическая прочность и химическая стойкость чистых М., их электрические, магнитные и другие свойства (такие, как сопротивление износу, режущая способность) далеко не всегда удовлетворяют запросам техники — гражданской и военной. Но уже очень давно человек узнал на опыте, что металлические сплавы во многих случаях обладают гораздо более ценными свойствами, чем чистые М. Целая эпоха материальной культуры — бронзовый век — характеризуется применением бронзы (сплава меди с оловом) для изготовления оружия и орудий труда. С древнейших времён к золоту и серебру, из которых делали предметы роскоши и религиозных культов и изготовляли монеты, прибавляли медь. Это не только уменьшает расход драгоценного металла, но и продлевает срок службы изделия. Ведь сплавление золота или серебра с медью повышает их прочность, твёрдость и стойкость против истирания.
В конце XV в. для отливки типографских литер стали пользоваться сплавом свинца с сурьмой, гораздо более твёрдым и легкоплавким, чем чистый свинец. А сплав 2 частей свинца с 1 частью олова, плавящийся при невысокой температуре, прочный и хорошо пристающий к поверхности, очень давно служит для пайки металлических изделий. Но и это — лишь малый список примеров. На протяжении более двух тысячелетий путём тяжёлого опыта было изобретено большое число металлических сплавов самого разнообразного назначения. Их состав, способы получения и обработки обычно держались в секрете. Правда, в начале XIX в. благодаря успехам химии уже можно было установить состав любого сплава. Но чтобы получить сплав с желаемыми свойствами, мало знать его состав. Надо изучить и его микроскопическое строение, и условия плавки, отливки, механической и термической обработки... Все эти и многие другие вопросы составляют предмет металловедения—науки, возникшей из потребностей техники в конце XIX в.
Классификация. В соответствии с местом, занимаемым в периодической системе Менделеева, различают М.— элементы главных подгрупп (или непереходные) и М. — элементы побочных подгрупп (или переходные). М. главных подгрупп (подгрупп а) характеризуются тем, что в их атомах происходит последовательное заполнение электронных уровней s и р. В М. побочных подгрупп (подгрупп б) происходит достраивание d- и /-уровней. В соответствии со сказанным, эти металлы делят на d-группу и две /-группы (лантаноиды и актиноиды). (О последовательности заполнения электронных уровней атома рассказано в статьях Атом, Закон Менделеева.)
В подгруппы а входят 22 М.:
Li, Na, К, Rb, Cs, Fr (la) Be, Mg, Ca, Sr, Ba, Ra (Ila) Al, Ga, In, TI (Illa) Ge, Sn, Pb (IVa) Sb, Bi (Va)
Po (Via)
В подгруппы б входят 33 переходных элемента d-группы:
Си, Ag, Аи (16)
Zn, Cd, Hg (116)
Sc, Y, La, Ac (III6) Ti, Zr, Hf, Ku (IV6) V, Nb, Ta, № 105 (V6) Cr, Mo, W (VI6) Mn, Fe, Re (VIM)
Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt (VIII6) и 28 элементов /-группы — 14 лантаноидов и 14 актиноидов.
Такова классификация М.— химических элементов. В технике же издавна сложилась своя классификация. Она далеко не так логична, как химическая,— в основе её лежит то один, то другой практически важный признак. Так, различают М. лёгкие (например, Li, Na, К, Be, Mg, Са, Al, Ti) и тяжёлые (Mn, Fe, Co, Ni, Cu, Zn, Cd, Hg, Sn, Pb и др.). Выделяют группу тугоплавких М. (Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Re) и группу благородных M. (Ag, Au, платиновые металлы). В геологии (да и в технике) принято выделять редкие М. и рассеянные М.
К редким причисляют М., мало распространённые в природе, не образующие значительных рудных скоплений, трудно извлекаемые из природного сырья и мало применяемые в технике. Впрочем, все эти признаки довольно условны. Например, содержание в земной коре титана или циркония, обычно считающихся редкими, значительно выше таких давно известных М., как медь, олово, свинец. Галлия и германия в земной коре во много раз больше, чем сурьмы и висмута, известных уже в средние века. Применение редких М. в технике постоянно возрастает. К редким М. относят лёгкие (Li, Rb, Cs, Be), тугоплавкие (все указанные выше, кроме Сг), рассеянные (Ga, In, TI, Ge, а также Hf и Re), редкоземельные (Sc, Y, La, лантаноиды), радиоактивные (Tc, Pm, Ra, актиноиды).
Железо и сплавы на его основе называют чёрными М.; все прочие М.— цветными. Этими группами классификация М. не исчерпывается. Ведь та или иная область техники всегда может выдвинуть на первый план какие-нибудь особенно важные свойства М.
Физические и химические свойства. Перечисленные в начале статьи характерные физические свойства простых веществ—М. обусловлены их электронным строением. М. можно представить как «жёсткую» кристаллическую решётку из положительных ионов, погружённую в «море» или плотный газ легко подвижных
197
МЕТАЛЛЫ
Значения электроотрицательностей элементов
Н
2 Li Be в C N 0 F
1,0 1,5 2,0 2,5 3,0 3,5 4,0
3 Na Mg Al Si P S Cb.
3 Pt 0,9 1,2 1,5 1,8 2,1 2,5 3,6
о S 4 К Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br
о. О) 0,8 1,0 1,3 1,5 1,6 1,6 1,5 1,8 1,9 1,9 1,9 1,6 1.6 1,8 2,0 2,4 2,8
Е 5 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Те I
0,8 1,0 1,2 1,4 1,6 1,8 1,9 2,2 2,2 2,2 1,9 1.7 1,7 1,8 1,9 2,1 2,5
6 Cs Ba La — Lu Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At
0,7 0,9 1,0-1,2 1,3 1,5 1,7 1,9 2,2 2,2 2,2 2,4 1,9 1,8 1,9 1,9 2,0 2,2
7 Fr Ra Ac Th Pa U Np — No
0,7 0,9 1,1 1,3 1,4 1,4 1,4-1,3
электронов. Эти электроны компенсируют силы электрического отталкивания между положительными ионами и тем самым связывают их в твёрдые тела. Такой тип связи атомов называют мета л л ической связью. Легко подвижные электроны свободно перемещаются в кристаллической решётке М., вследствие чего они хорошо проводят электричество и тепло. Отражательная способность (характерный блеск) также связана с наличием в М. свободных электронов. Структура М. объясняет и их механические свойства, в частности лёгкость пластической деформации. Но вопросы эти — область скорее физики, а не химии, и углубляться в них здесь мы не будем.
М. присущи многие общие химические свойства, обусловленные слабой связью валентных электронов с ядром атома. Так, М. проявляют положительную степень окисления и образуют положительно заряженные ионы (катионы). Окислы и гидроокиси типичных М. имеют основной характер. Взаимодействуя с кислотами, М. дают соли. Эти свойства элементов часто называют металлическими.
Металлические свойства элементов можно сравнивать, сопоставляя их электроотрицательность. Эта величина, выраженная в условных единицах, характеризует способность атома в молекуле (в ковалентной связи) притягивать электроны. Элементу присущи свойства М. в тем большей степени, чем ниже его электроотрицательность. Иными словами, наиболее типичные М.— это наиболее электроположительные элементы. Величины электроотрицательности приведены в таблице (таблица взята из книги Л. Полинга «Общая химия», 3-е издание, Москва, 1974). Она имеет вид длинного варианта периодической системы Менделеева.
Из таблицы 1 видно, что в коротких периодах (втором и третьем) системы Менделеева электроотрицательность возрастает по мере увеличения атомного номера. В длинных перио
дах (с четвёртого по седьмой) наблюдаются отклонения, но общая тенденция повышения электроотрицательности по мере продвижения от щелочного металла к галогену сохраняется. Наименьшую электроотрицательность (0,7) имеют цезий и франций, наибольшую — фтор (4,0). Таблица показывает постепенность перехода от М. к неметаллам.
В пределах подгрупп (а и б) с увеличением атомного номера электроотрицательность в общем уменьшается, хотя и не всегда последовательно. В семействах лантаноидов и актиноидов она сохраняется примерно на одном уровне (1,0 — 1,2 для лантаноидов и 1/3 — 1,4 для актиноидов). Кроме электроотрицательности, для характеристики химических свойств атомов М. служат энергия ионизации и ионный радиус. См. Атом, Закон Менделеева, Щелочные металлы, Щелочноземельные металлы.
Ряд напряжений.
Уже в средние века рудокопы знали, что если железный предмет попадёт в «купоросную воду» медных рудников (раствор медного купороса, образовавшийся при окислении сульфидных медных руд), то его поверхность покрывается красным слоем меди. Алхимики, называр-шие железо Марсом, а медь — Венерой, описывали это явление так: «Вооружённый блистающий Марс бросается в объятия растаявшей в слезах Венеры и при этом краснеет». Были известны и другие подобные факты. Если в разбавленный раствор нитрата серебра влить немного ртути, то через некоторое время можно увидеть нечто поразительное. Как писал немецкий химик второй половины XVIII в. И. X. Эркслебен, на поверхности ртути вырастает «прекрасное серебряного цвета .деревце, которое Дианиным, серебряным, также и философическим древом именуется... Древо сие состоит из хрупкого состава серебра со ртутью и рождается посредством низвержения [осаждения] серебра..., причем . серебро с прочею ртутью в ... амальгаму превращается». Здесь, хотя и упоминается Диана — богиня Луны (серебра), но по существу процесс объяснён, как вытеснение рту-, тью серебра из раствора AgNQ3.
С современной точки зрения в обоих случаях происходит реакция окисления — восстановления. На поверхности железа, погружённого в раствор, содержащий катионы меди Си2+, осаж-
МЕТАЛЛЫ
198
Li К Ва Na Mg Al Mn Zn Cr Fe Cd Co
Ряд напряжений для важнейших ме
дается медь, а железо переходит в раствор в виде катионов Fe2 + :
Cu2+ + Fe = Fe2+4-Cu
Подобный же процесс происходит и в случае «Древа Дианы»
2Ag+ + 2Hg = Hgr+2Ag
В статье Ртуть вы узнаете, почему Hg образует катионы Hg|F, а не Hg+.
Путём опыта установлено, что все М. можно расположить в ряд, каждый член которого отдаёт свои электроны катионам всех последующих его членов. Он называется рядом напряжений или рядом активностей. Приводим его в сокращённом виде и без числовых данных.
Ряд начинается с наиболее активных М. и кончается наименее активными. Все М., стоящие в ряду до водорода, вытесняют его из разбавленных кислот, кроме азотной кислоты; М., расположенные после водорода, не выделяют его из разбавленных кислот.
. М., стоящие слева, вытесняют М., стоящие справа, из растворов их солей (см. выше — ртуть вытесняет серебро из раствора AgNO3). Первые члены ряда выделяют водород из воды (уже при комнатной температуре). А потому их реакции с раствором, содержащим катионы других М., протекают более сложно (читатель, конечно, сообразит, что образуются едкая щёлочь и водород). В сущности говоря, в реальных условиях вытеснение предыдущим М. последующего начинается с магния и алюминия и заканчивается серебром.
Ряд напряжений можно представить и в виде системы химических уравнений:
Ряд напряжений в виде системы химических уравнений
LU ^Li + + e Ni: Ni2+ - F2e
±:K+ + ё Sn ^Sn2+- |-2ё
Ba 4 ± Ba2+ 4-2e Pb Pb2+ +2e
Na 2± Na + Ц- - e H2 ?^2H++2e
Mg? Mg2 + - F2e Sb ^±Sb3+ + 3e
Al < — А13++3ё Bi Bi3+ +3e
Mn ч — Mn2+ +2ё Cu <—- Cu2+ —|- 2e
Zn; — Zn2+ +2e Hg: ^y2Hg?+ + e
Cr * = Cr3++3e Ag; — Ag+ 4- e
Fe + — Fe2+- 4-2ё Pt; pps+ 2]-2ё
Cd + Co4 =tCd2+--— Co2+- H2- Au; Au3 + + Зё
О химических свойствах М. и их зависимости от положения элемента в системе Менделеева подробно рассказано и в статьях об отдельных М. (см., например, Алюминий, Барий,
Бериллий, Ванадий и т. д.), и в более общих (Платиновые металлы, Редкоземельные элементы, Щелочные металлы, Щелочноземельные металлы), и в самой обобщающей — о периодическом законе и системе Менделеева. Коснёмся лишь одного вопроса, мало освещённого в этих статьях.
Металлиды. М. могут вступать в химическое взаимодействие друг с другом. При этом образуются интерметаллические соединения, или металлиды. Некоторые из них можно рассматривать как производные гидридов (водородных соединений) М., в которых водород замещён другим металлом. Примеры: Mg2Sn и Mg2Pb — продукты замещения водорода магнием в гидридах олова H4Sn и свинца Н4РЬ. Но подавляющее большинство металлидов имеет состав, не отвечающий обычным представлениям о валентности, например SnSb, СаРЬ3, Na31Pb8.
Металлиды похожи на чистые М. только по внешним признакам — металлическому блеску и цвету: обычно они серебристо-белые, иногда с сероватым оттенком (лишь очень не? многие имеют золотисто-жёлтый или красноватолиловый цвет). Твёрдость и удельное электрическое сопротивление металлидов, как правило, выше, чем образующих их М., а пластичность — всегда значительно ниже. Например, соединение А12Си, состоящее из очень пластичных металлов — алюминия и меди, настолько хрупко, что его легко растереть в мелкий порошок в стеклянной ступке. Объясняется это тем, что пространственная решётка металлидов построена сложнее, чем у чистых М. При повышении температуры пластичность металлидов обычно повышается.
Всем, кто работает в химической лаборатории, хорошо знакомы обычные методы химического исследования: выделение веществ в чистом виде (возгонкой, перегонкой, перекристаллизацией, фильтрованием) и количественный анализ полученного продукта. Но установить этими методами состав металлидов не удаётся: оказывается, невозможно отделить кристаллы металлида от жидкости, из которой они выделились.. Здесь приходит на помощь физико-химический анализ — метод исследования, основанный и разработанный в России и в СССР Н. С. Курнаковым и его школой. Он позволяет устанавливать состав металлидов, не выделяя
199
МИНЕРАЛЫ
Ni Sn Pb н2 Sb Bi Си Hg Ag Pt Au
таллов в водных растворах при 25 °C.
их в чистом виде и не делая их химического анализа. Но здесь не место рассказывать об этом более подробно.
Несмотря на свои, в общем-то мало удовлетворительные механические свойства, металли-ды находят практическое применение. Например, сплавы на основе олова (изобретённые ещё в 1839 г.) состоят из пластичной мягкой основной массы Sn, в которую вкраплены кристаллики твёрдых металлидов (SnSb, SnCu и др.); их применяют для заливки вкладышей подшипников.. Иногда же именно металлиды (а не сами М.) дают рекордные свойства, необходимые для новой техники. Металл ид SmCo5 оказался наилучшим материалом для сильных постоянных магнитов. А металлид Nb3Ge — рекордным по сверхпроводимости (об этом рассказано в статье Ниобий). Соединения AlSb, InSb и другие широко применяются как полупроводники. Об упрочнении сплавов на основе алюминия при выделении высокодисперсных металлидов рассказано в статье Алюминиевые сплавы.
По мере всестороннего изучения свойств металлидов непрерывно расширяются и области их практических применений. Здесь, как и во многих других случаях, стирается грань между «чистой» и «прикладной» наукой. Академик Н. С. Курнаков всегда говорил, что «чистой» и «прикладной» науки нет, а есть наука и её приложения к практике.
С. А. Погодин. МЕТАФОСФОРНАЯ КИСЛОТА (НРО3)Х — одна из фосфорных кислот.
МИНЕРАЛЕ! — образовавшиеся в природе простые и сложные вещества, находящиеся, как правило, в твёрдом состоянии; составная часть горных пород и руд.
Какие только физические и химические процессы не протекают в окружающей нас природе! Ни один самый сложный химический комбинат (или даже комплекс физических, химических, биологических институтов) не может состязаться по разнообразию и обилию процессов с гигантским «комбинатом» — природой. В одних «цехах» этого комбината идёт испарение воды и осаждение солей (в солёных озёрах), в других — кристаллизация горных пород из силикатных расплавов — магм (в вулканических районах), в третьих — смешение вод разного состава (например, в дель
тах, где пресная речная вода смешивается с солёной морской). Эти и множество других физико-химических процессов приводят к образованию М.
СМ. мы встречаемся постоянно. Всем хорошо известен М. галит — поваренная соль NaCl, целые массивы которой залегают в земных недрах, например у нас в Донбассе. Внимательно приглядевшись к речному песку, мы обнаружим в нём несколько М.— белый твёрдый кварц SiO2, блестящие листочки светлой или тёмной слюды, зёрна полевых шпатов и пр. М. различаются по их физическим свойствам — цвету, блеску, твёрдости, форме кристаллов и другим особенностям.
Изучение М. начинается, как говорят геологи, «в поле», то есть на обнажении горных пород, в карьерах и шахтах рудников, на разведочных скважинах. Один из лучших знатоков М. академик А. Е. Ферсман особое значение при определении М. придавал цвету.
Вот что он писал: «Окраска минералов и пород является одним из замечательнейших и характернейших признаков природных соединений. Её значение, во-первых,— в диагностике, определении минералов, так как всё наше знание и опыт минералога основывается прежде всего на учёте окраски; так, увидев на скале сине-зелёные потёки, минералог правильно делает заключение о рудах меди...». Действительно, по цвету опытный минералог иногда сразу определяет (диагностирует) некоторые М. меди (зелёный малахит, синий азурит), марганца (чёрный пиролюзит), ртути (красная киноварь) и другие.
Но это только первый этап изучения, для полного же определения М. необходимо произвести целый комплекс исследований. Поэтому, аккуратно завернув драгоценные находки, снабдив их этикетками с указанием места нахождения, минералог отправляет образцы в лаборатории на анализ.
Состав, структура и свойства минералов. Обычно с понятием «минерал» связывают представление о твёрдом веществе. Однако В. И. Вернадский, А. Е. Ферсман и другие крупные учёные относят к М. также природные жидкости и газы (природные воды, нефть, воздух, газы земной коры). По химическому составу М. бывают и простыми, состоящими всего из одного элемента, и сложными, содержащими не менее двух химических элементов.
Простых М. сравнительно немного — это, например, алмаз и графит, самородные золото, серебро, медь, сера, платиновые металлы. Не
МИНЕРАЛЫ
200
измеримо больше М.— химических соединений и твёрдых растворов. (Подробно о непривычном для школьника понятии «твёрдые растворы» рассказано в статье Алюминиевые сплавы.) Примером М.— твёрдых растворов служат самородная платина — твёрдый раствор железа и платиновых металлов в платине; железные метеориты — твёрдые растворы Fe — Ni. Изоморфные смеси, о которых рассказано ниже,— тоже твёрдые растворы.
Состав и структуру М. изучают разными методами. Особенно большие успехи были достигнуты в этой области, когда на помощь химическому анализу в начале XX в. пришли рентгеновские лучи. Оказалось, что «кирпичами», из которых построено большинство М., являются положительно и отрицательно заряженные ионы—катионы и анионы, например Na+, Са2+, Fe2+, Fe3+, Al3+, Cl“, NO^, SOJ”. Эти «кирпичи», или структурные единицы, находятся в узлах кристаллической решётки и соединены между собой химическими связями. Для характеристики М. важны не только химические свойства атома или иона, но и его размеры. Известный норвежский геохимик В. М. Гольдшмидт в 1926 г. подсчитал радиусы большинства ионов. С тех пор эти числа неоднократно проверялись и сейчас они известны для большинства ионов (здесь приводятся ионные радиусы по Н. В. Белову и Г. Б. Бокию, принятые у геохимиков). Оказалось, что радиусы ионов составляют около 1-10“8 см (величина 1-10”8всм называется ангстремом и обозначается 1А). Например, ион кислорода О2- имеет сравнительно большие размеры, его радиус равен 1,36 А, у натрия ион Na+ меньше —о 0,98 А, а у бериллия Ве2+ ещё меньше— 0,34 А. Если химический элемент проявляет различную валентность, то ионы его имеют разладе размеры, например у двухвалентного железа Fe2+ радиус иона больше, чем у трёхвалентного Fe3+ (соответственно 0,80 и 0,67 А). Ну а что же даёт минералогу знание размеров ионов?
При образовании М. из растворов или расплавов в них попадают ионы, сходные по размеру; очень условно это можно сравнить с просеиванием песка на ситах. В результате многие М. каких-либо широко распространённых элементов содержат примеси редких элементов, имеющих ионы близкого размера. Такие примеси называются изоморфными. А само явление — замещение ионов или целых молекул в кристаллической решётке другими ионами или молекулами (близкими по размерам) — изоморфизмом. Так, редкий элемент индий содержится в виде изоморфной примеси (сотые доли процента), в минерале цинка сфа
лерите ZnS благодаря близости ионных радиусов Zn2+ и 1п3+ (0,83 и 0,92 А). Всё это очень затрудняет установление формулы М.— ведь в отличие от химика, синтезирующего соединение, минералог в большинстве случаев не может наблюдать образование М., он не знает точно состава исходных расплавов и растворов, их температуры и т. д. Некоторые М. образовались миллионы и даже миллиарды лет назад, на больших глубинах, в условиях,которые мы ещё не всегда себе полностью представляем! И все жё современные физические и химические методы исследования позволяют глубоко заглянуть в М., точно установить его состав и структуру.
Вот как, например, можно записать формулу антофиллита, тонкие волокна которого, напоминающие пучки ниток, часто обнаруживают в изверженных горных породах, испытавших на глубине большое давление и подвергшихся действию горячих вод:
(Mg2 + , Fe2 + )7[Si4O11]e-(OH")2
Минералогам пришлось немало поработать, пока они разобрались в составе этого сложного М. Формула говорит о том, что антофиллит — это силикат магния и железа, каркас которого образует группа [Si4Ou]6- (её поместили в квадратные скобки). В состав М. входят также катионы Mg2 + и Fe2 + , размеры которых близки, в связи с чем они могут замещать друг друга в разных пропорциях. Поэтому в антофиллитах из разных районов содержание Fe2+ и Mg2+ не одинаковое. В М. имеются Н и О, причём они находятся в виде аниона гидроксила ОН~. Таким образом, формулы М. часто бывают гораздо сложнее, чем формулы обычных неорганических химических соединений.
Кстати сказать, в специальной литературе по минералогии формулы М., подобных антофиллиту, упрощают—заряды ионов ставят только для элементов с переменной валентностью:
(Mg, Fe2+)7[Si4On]2(OH)2
(с точки зрения химика такая запись — не вполне строгая).
Итак, ионы близкого радиуса и одинакового знака заряда заменяют друг друга в кристаллической решётке М. Возможно, именно поэтому содержание отдельных элементов в М. сильно колеблется, и они часто не имеют определённого состава. В таких случаях состав М. дают более простой формулой — как, например, для псиломелана — марганцевого М. чёрного цвета:
BaMn2+MnJ+O20-3H2O
В состав псиломелана входят и двухвалентные, и четырёхвалентные катионы марганца. Химический анализ показывает, что в псиломеланах встречаются часто примеси W, Fe, Al и V, которые замещают четырёхвалентный марганец Мп4+, а также Mg, Со, Си, замещающие двухвалентный мар ганец Мп2 + . А вместо Ва в кристаллическую решётку могут входить близкие по размеру катионы Са, Na, Sr и UOf+. Таким образом, зная размеры ионов, а они приводятся во всех учебниках минералогии, можно заранее предсказать элементы-спутники в М.
201
МИНЕРАЛЫ
Александр Евгеньевич ФЕРСМАН — «поэт камня». Один из основателей геохимии.
Александр Евгеньевич Ферсман родился 8 ноября 1883 г. в Петербурге. Отец его, в прошлом архитектор, после русско-турецкой войны 1877—1878 гг. остался военным. Однако в доме поддерживалась необычная для военной среды того времени обстановка, в которой «вольно дышалось и мысли, и искусству». Летом семья переселялась в Крым. Здесь ^^ачалось увлечение мальчика многоцветным и сказочным миром камня. «История камня переплетается с общей историей культуры, науки и искусства,— говорил он впоследствии,— разве не в этом главный ее интерес?»
В 1901—1903 гг. Ферсман учился в Новороссийском университете (в Одессе), в 1903—1907 гг.— в Московском. Здесь его руководителем был В. И. Вернадский, основатель нового направления в минералогии. Вместе с Вернадским Ферсман изучал минералы и их историю, «на атомном уровне», стараясь понять, какие изменения и превращения они претерпели в результате сложных химических процессов, идущих в природе. Вот как вспоминал впоследствии Ферсман свои первые студенческие экскурсии с учителем: «Мы учились понимать историю минерала: его образование..., его гибель в струйках воды, его превращение в новые соединения. Мы учились по-новому смотреть на окружающую нас природу, понимать, что каждый камень связан с природой тысячами нитей, которые тянулись не только к каплям дождя, не только к остаткам древних раковин, но и к современной жизни, к органическим растворам поверхности и к деятельности самого человека». Учение о химическом составе Земли, о законах поведения в ней химических элементов стало содержанием новой науки — геохимии. Ферсману принадлежит первое фундаментальное изложение основ этой науки («Геохимия» в 4 томах, 1933—1939 гг.). Он показал, как периодический закон Менделеева управляет распределением, сочетанием, миграцией и концентрацией химических элементов в природе.
Московский университет Ферсман покинул в 1911 г. вместе с Вернадским и другими передовыми преподавателями, выразив тем самым протест против политики царизма в области просвещения. Дальнейшая научная деятельность Ферсмана была связана преимущественно с Академией наук, действительным членом которой он стал в 1919 г.
Трудно перечислить экспедиции, в которых побывал учёный. Практическое приложение минералогии и геохимии при поисках полезных ископаемых было делом всей его жизни. Он изучал месторождения драгоценных камней и других полезных ископаемых на Урале, в Забайкалье, Средней Азии, Карелии, в Швеции, на острове Эльба и т. д. (1907—1940 гг.). В 1920 г. организовал экспедицию на Кольский полуостров, в Хибинские горы, где в 1926 г. открыл крупнейшее месторождение апатитов. Он был инициатором создания в Хибинах первого в мире заполярного горнопромышленного узла, города, научной базы Академии наук. В реализации этих планов большую помощь оказал ему Сергей Миронович Киров. Промышленное освоение Кольского полуострова было начато в 1929 г. Воздвигнутый здесь город Хибиногорск был в 1934 г. переименован в Кировск.
1925 год. Первая экспедиция в Каракумы. Здесь в самом центре пустыни Ферсман изучает месторождение серы. По его настоянию в пустыне был построен завод, много лет снабжавший нашу промышленность превосходной и дешёвой серой. Ферсман был крупным организатором советской науки, директором многих учреждений Академии наук, в том числе Минералогического и Геохимического институтов. В годы Великой Отечественной войны он организовал и возглавил военно-геологическую комиссию Академии наук. Учёный отличался поразительной разносторонностью и работоспособностью. Он говорил, что рука у него устаёт писать, но голова никогда не устаёт думать.
Он был и известным популяризатором науки, автором увлекательных книг для детей и юношества. Вы легко убедитесь в этом, если раскроете «Занимательную геохимию» или «Рассказы о самоцветах». А «Занимательная минералогия» издавалась у нас и за рубежом более 30 раз. Ферсману принадлежит серия новелл — «Воспоминания о камне», которую его ученик академик Д. И. Щербаков назвал «научной лирикой». Учёный глубоко чувствовал эстетическую сторону своей профессии. «Поэтом камня» назвал его писатель А. Н. Толстой. В молодости он интересовался историей искусства и даже думал выбрать её в качестве своей профессии, чуть не изменив минералогии. В его трудах вы найдёте много интересного о драгоценных камнях, о красках природы, о роли камня в истории культуры.
За научные труды Ферсману были присуждены премия имени В. И. Ленина (1928 г.) и Государственная премия СССР (1942 г.), а также награды за рубежом. А. Е. Ферсман умер 20 мая 1945 г. в Сочи.
МИНЕРАЛЫ
202
Вероятно, всем известен красивый голубовато-зелёный М. бирюза — любимый камень Востока. Бирюзу продавали на всех восточных базарах, её добыча началась ещё за тысячи лет до нашей эры. В странах Средней Азии было множество бирюзовых рудников. В состав бирюзы входят медь, цинк, фосфор, кислород, алюминий, железо, водород. Вот её формула:
(Си, Zn)(Al, Fe3+)6(OH)8[PO4]4«4H2O
Изучение структуры и состава М.— это результат большой совместной работы минералогов, химиков, физиков.
Следующий этап в исследовании — объяснить на основе знаний о составе и структуре свойства М. Благородный голубовато-зелёный цвет бирюзы, вероятно, зависит от меди, так как и многие другие М. этого металла имеют зелёный цвет (вспомним изумительной красоты изделия из зелёного малахита, хранящиеся во дворцах и музеях Ленинграда). Зелёный цвет придаёт М. также хром, и в Минералогическом музее вы можете полюбоваться волконскои-том — редким глинистым М., который так ценится в качестве замечательной зелёной краски. Красивый жёлтый цвет имеют многие М. шестивалентного урана, синий — некоторые М. меди. Иногда цвет М. зависит не от основных ионов, а от примесей. Поэтому минерал одного и того же вида может иметь разный цвет (кварцы — SiO2 бывают белыми, фиолетовыми, розовыми, чёрными).
Но, конечно, не все М. имеют такой сложный состав, как антофиллит, псиломелан или бирюза. Некоторые имеют простую формулу, например мирабилит (глауберова соль) Na2SO4- 10Н2О или кальцитСаСО3, входящий в состав известняков и мраморов.
Распространённость минералов и их классификация. В земной коре известно свыше 2500 М., но исследователи открывают всё новые и новые. И всё же общее число М. вряд ли превысит несколько тысяч. Это намного меньше, чем число неорганических химических соединений, полученных в лабораториях (десятки тысяч!). Одна из причин такого несоответствия — малая распространённость большинства химических элементов в земной коре. Среднее содержание какого-либо элемента в земной коре геохимики назвали кларком — в честь американского химика Ф. Кларка, более 40 лет посвятившего изучению этой проблемы. Одни кларки очень высокие (для кислорода и кремния — десятки процентов, для алюминия, железа и некоторых других элементов— целые проценты). Но большая часть химических элементов в земной коре содержится в очень малых количествах (см. таблицу в статье Земная кора).
Так, среднее содержание в земной коре радия составляет лишь 1-10“10%, очень мало его и
в природных водах. Поэтому на Земле нет радиевых минералов (в лабораториях же соли радия получают). Сера и хлор имеют сравнительно высокие кларки (более 0,01%) и образуют много М., тогда как их химические аналоги теллур, селен, иод относятся к числу редких и имеют мало М. (кларки—менее 0,0001 %).
Основой классификации М. служат химический состав и структура. С этой точки зрения выделяют простые вещества (самородные металлы и неметаллы), сульфиды и близкие к ним М., кислородные соединения (окислы, силикаты, бораты, фосфаты, карбонаты и т. д.), галогениды (хлориды, фториды, бромиды и иодиды), органические соединения. Больше всего М. в группе кислородных соединений. Одних только силикатов минералоги насчитали 375, а ведь к ним надо добавить 260 фосфатов и родственных минералов, 67 карбонатов, 187 окислов и гидроокисей, 125 сульфатов, 42 бората, 14 вольфраматов и молибдатов, 5 хроматов и 8 нитратов. Всего в этой группе около 1000 М.
Различия в кларках определяют и разное число М. у отдельных элементов и их неодинаковое распространение в природе. Самый распространённый элемент в земной коре — кислород (47% по массе), поэтому у него много М.— 1364, и эти М. встречаются повсюду. Золото имеет низкий кларк (4,3-10“7%), у него известно лишь 9 М., встречающихся редко. Класс силикатов составляет 75% земной коры. Особенно распространены кварц SiO2 и полевые шпаты. Доля самородных элементов (0,1%), сульфатов (0,5%), фосфатов (0,7%) в земной коре незначительна (количество М. всюду дано в процентах по массе). Таким образом, большинство М. относится к редким, некоторые же обнаружены лишь в одном-двух местах на Земле. Таков, например, редкий минерал тарапакаит— хромат калия КгСгО4, найденный в пустыне Тарапака в Чили. Самые распространённые на Земле горные породы, такие, как граниты, базальты, глины, известняки, состоят всего лишь из нескольких десятков М.
Происхождение минералов. Поиски полезных М. ведут всюду — ив горах, и в пустынях, и в Арктике, и во влажных тропиках, и на дне морей и океанов. Но для того чтобы найти нужный М., надо знать, каково его происхождение (генезис).
Многие М. образовались при застывании огненно-жидких силикатных расплавов — магм, находящихся обычно на больших глубинах, но изливающихся на поверхность при извержении вулканов. При застывании «кислых магм», содержащих много кислотного окисла SiO2 образуются светлые породы — граниты. Они
203
МИНЕРАЛЫ
состоят из трёх минералов — кварца, полево го шпата и слюды. При застывании «основных магм» (сравнительно бедных SiO2) образуется другая порода — тёмногзелёные базальты, также содержащие полевые шпаты. В них уже нет кварца, но много бутылочно-зелёного М. оливина, богатого магнием и железом (Mg2+, Fe2+)2SiO4. Всего при застывании магмы образуется около 250 М., все они, как правило, имеют сложный состав и содержат, кроме главных элементов, множество элементов-спутников.
Свыше 1000 М. кристаллизуется из природных вод — поверхностных и подземных, холодных и горячих, пресных и солёных. Знаменитый русский геохимик В. И. Вернадский вы
делил более 1500 различных видов вод, и с каждым из них связано образование М.
Если к раствору, содержащему хлорид бария, добавить раствор сульфата натрия, то произойдёт известная реакция:
ВаС12 + Na2SO4 = BaSO4 + 2NaCl
'в результате которой нерастворимый сульфат бария выпадет в осадок. Аналогичная реакция протекает и в природе. Глубинные горячие воды часто содержат хлориды кальция, стронция, бария. Если такие воды по трещинам в земной коре приближаются к поверхности, то уже на небольшой глубине они встречают подземные воды, содержащие сульфат .натрия. При их взаимодействии, как и в лаборатории, осаж-
Минералы из коллекции Минералогического музея имени А. Е. Ферсмана АНСССРв Москве. 1 —кальцит (минерал кальция), найден в пещере в Средней Азии; 2 —целестин (минерал стронция) с серой, за своё сходство с цветком образец был назван Ромашкой, а «сорвана» она была в Италии; 3 — амазонит (полевой шпат, алюмосиликат калия), красивый поделочный камень из Ильменских гор (Урал); 4 — аметист (фиолетовая разновидность кварца), образец с Урала; 5 — отенит (урановый минерал); 6 — малахит (минерал меди), найден в Нижнем Тагиле; 7 — киноварь (минерал, из которого получают ртуть), образец из месторождения в Средней Азии; 8 — сера из Узбекистана.
МИНЕРАЛЬНЫЕ УДОБРЕНИЯ
даются труднорастворимые сульфаты. Так в трещинах появляются жилы голубоватого целестина SrSO4, медово-жёлтого барита BaSO4, белого гипса CaSO4-2H3O. Кальций более распространён в земной коре, чем барий или стронций, и поэтому гипс встречается чаще.
Наконец, надо отметить и важную группу М., образующихся в телах живых организмов (био- ’ литы) — в животных, растениях, микроорганизмах. Из биолитов состоят многие известняки, выходы которых прослеживаются на десятки и сотни километров. Таковы, например, толтры—известняковые гряды на Волыно-По-дольской возвышенности, наследники тёплого миоценового моря, в котором жили моллюски, мшанки, водоросли, кораллы и другие организмы с известковым скелетом. Известняки состоят из кальцита СаСО3.
Минералы в космосе. М. существуют и в космосе. Многие годы единственную информацию о них давали метеориты — обломки небесных тел, которые изредка залетают в земную атмосферу. Здесь они раскаляются и в виде «огненной массы» с огромной скоростью врезаются в земную поверхность, где быстро остывают. По внешнему виду метеориты напоминают обыкновенные камни, и поиски их представляют большие трудности. В нашей стране в таких поисках участвуют наряду с учёными и школьники. Уже обнаружены многие сотни метеоритов, хорошо изучен их состав.
Оказалось, что метеориты содержат М., известные и на Земле,— алмаз, самородные золото и серу, оливин (о котором уже упоминалось), гиперстен, бронзит и другие силикаты. Но в метеоритах были обнаружены и М., чуждые земной коре,— осборнит TiN, шрейбер-зит (Fe, Ni, Со)3Р, ольдгамит CaS, добреелит FeCr2S4 и другие. Всего в метеоритах пока установлено 66 М., то есть в десятки раз меньше, чем на Земле. Особенно своеобразны по составу так называемые железные метеориты, состоящие в основном из железа и никеля [минералы камасит (до 10% Ni), тэнит (34— 36% Ni), октиббегит (60—77% Ni)]. Есть гипотеза, что они образовались при космической катастрофе из ядра какой-то планеты. Предполагают, что и земное ядро, подобно железным метеоритам, состоит из железа и никеля.
Новые сведения о М. космоса поступили от советских и американских учёных, изучивших лунные породы. На поверхности Луны были обнаружены многие М., известные и на Земле,— преимущественно различные силикаты и алюмосиликаты (оливин, пироксены, плагиоклазы и др.). Все знакомые нам лунные М. образовались при высоких температурах в условиях, аналогичных образованию земных базальтов. Изучение лунных М. ещё только начинается. Но уже ясно, что лунные породы и сходны с земными, и отличаются от них. На луне был обнаружен, например, минерал троилит FeS, неизвест
204
ный в земных породах, но установленный в метеоритах. Обнаружено в лунных породах и рассеянное самородное железо, характерное для метеоритов и очень редко встречающееся в земной коре. В ближайшие годы наука раскроет немало тайн минерального состава Луны, Марса и других планет.
Сложен труд минералога! В нашей стране изучают М. особые исследовательские институты, в университетах есть кафедры минералогии, издаются специальные минералогические журналы, есть и Всесоюзное минералогическое общество. Образцы М. нередко путешествуют за тысячи километров, преодолевая путь сначала на оленях или верблюдах, потом на самолётах, поездах — чтобы попасть, например, с Камчатки или из Каракумов в хорошо оснащённые лаборатории научных центров, где их просветят рентгеновскими лучами, положат под электронный микроскоп, «прощупают» микрозондом и т. д. Но часто этот путь бывает ещё сложнее, так как в поле некоторые М. остаются незамеченными из-за очень малого размера кристаллов (например, у минералов глин), из-за отсутствия характерных внешних признаков. Геологи не раз пропускали месторождения из-за того, что они были представлены невыразительными «коварными» М.
Трудно перечислить все производства, где используются минеральное сырьё, минералы. Напомним только, что железо, медь, цинк, уран и другие металлы, уголь, фосфорные и калийные удобрения — всё это представляет собой М. или получено из М. И хотя многие М. синтезируют сейчас в лабораториях и на заводах (например, искусственные алмазы и рубины), основная масса М. добывается пока из природных источников. И тысячи экспедиций ежегодно ищут М. на суше и на море, во всех концах нашей необъятной страны.
А. И. Перельман. МИНЕРАЛЬНЫЕ УДОБРЕНИЯ — природные или синтетические неорганические вещества, содержащие необходимые для питания растений химические элементы.
Питательные вещества поступают в растения через листья и корни. Листья поглощают из воздуха, главным образом, углекислый газ, превращая его с помощью солнечного света в органические вещества (процесс фотосинтеза, см. Биосфера. Кислород. Углерод). А корни берут из почвы воду и растворённые в ней питательные вещества в виде минеральных солей. Можно спросить, зачем же нужны растениям искусственно созданные человеком М. у.? Ведь растения жили, росли и развивались, умирали и рождались вновь задолго до появления человека на земле.
Действительно, в большинстве почв имеются все элементы, необходимые для питания расте
205
ний. Но часто, в так называемых бедных почвах, их содержится недостаточно для того, чтобы получать высокие и устойчивые урожаи. А кроме того, необходимые элементы зачастую находятся в почве в виде таких химических соединений, которые плохо или совсем не усваиваются растениями. Во всех этих случаях в возделываемые почвы надо вносить удобрения. А более общий ответ на вопрос таков: М. у.— это дополнительная подкормка, а чем лучше питаются культурные растения, тем быстрее они растут и лучше развиваются, тем выше урожаи полей, огородов, садов, тем лучше качество зерна, овощей, плодов и других культурных растений.
Какие же именно минеральные соли нужны для их питания? На этот вопрос может ответить химический анализ сухого вещества растений. Оказывается, в состав растений входит приблизительно 45% С, 42% О, 6,5% Н, 1,5% N, а остальные 5% составляют Р, К, Mg, S, Fe, В и другие химические элементы — всего их около 60.
Ещё в 1840 г. немецкий химик Ю. Либих научно доказал потребность растений в минеральном питании и определил необходимые для них питательные элементы. Сейчас наукой точно установлено, что растениям в относительно больших количествах требуются азот, фосфор, калий и в значительно меньших количествах — сера, магний, кальций, бор, марганец, медь, молибден, железо, цинк, кобальт, иод и другие элементы.
Главный источник углерода — углекислый газ атмосферы. Кислород поступает как из атмосферы, так и из почвы через корни (в составе Н2О). Источник водорода — вода. Остальные элементы растения получают через корни в виде минеральных солей. А азот? Ведь его в воздухе свыше 78%. Растения, можно сказать, «купаются» в азоте, но подавляющее большинство их видов, за редчайшим исключением, не умеют усваивать азот воздуха. Они поглощают азот из почвы, а там, где его мало, зачастую испытывают «азотный голод».
В связи с тем, что растениям требуются легко усваиваемые минеральные соли, многие природные удобрения мало пригодны для питания растений и подавляющее большинство их приготавливается искусственным (синтетическим) путём на химических заводах. Вырабатываемые промышленностью М. у. (минеральные туки) делятся на три большие группы: азотные, фосфатные, или фосфорные, и калийные. Вырабатываются также кальциевые, борные, магниевые и другие удобрения. Те из них, что требуются в небольших количествах, называются микроудобрениями.
МИНЕРАЛЬНЫЕ УДОБРЕНИЯ
Различают также простые и комплексные (сложные и смешанные) удобрения. В состав простых входит только один из питательных элементов. В состав комплексных — два, три и более. В сложных удобрениях питательные элементы химически связаны в составе единого вещества. А смешанные удобрения — это механические смеси разных (простых или сложных) удобрений (так называемые тукосмеси).
В зависимости от количественного содержания питательных веществ различают низко-и высококонцентрированные удобрения. Их концентрацию принято выражать в процентах: для азотных — по содержанию азота N, для фосфатных — по содержанию фосфорного ангидрида Р2О5, для калийных — окиси калия К2О. Форма удобрений может быть разной: порошок, гранулы, жидкость. Жидкие М. у. (растворы аммиака, аммонийных солей и т. д.) очень удобны тем, что их можно подавать на поле, как только растение «потребует» подкормки.
Азотные удобрения. Азот входит в состав белка растений и необходим для построения живой клетки. Входит он и в состав хлорофилла — зелёного вещества растений, поглощающего солнечную энергию. Выше было сказано, что, несмотря на изобилие свободного азота в воздухе, растения его не усваивают. В начале XX в. перед человеком встала и вскоре была решена важная научно-техническая проблема — найти способ связывания атмосферного азота в соединения, легко усваиваемые растениями. Наиболее выгодным способом оказался синтез аммиака, перерабатываемого затем в различные растворимые и легко усваиваемые растениями соли.
Промышленность выпускает азотные удобрения в большом ассортименте. Укажем лишь важнейшие.
Аммиачная селитра (нитрат аммония) NH4NO3 характеризуется наиболее высоким содержанием азота — в сумме 35% [в аммонийной (NH^) и нитратной (NO3) формах]. Это наиболее эффективное из простых азотных удобрений. Получают аммиачную селитру взаимодействием аммиака с азотной кислотой:
NH3 + HNO3-NH4NO3
Кальциевая селитра (нитрат кальция) Ca(NO3)2 содержит 13—15% N. Получают её взаимодействием мела или известняка с азотной кислотой:
СаСОз + 2HNO3 - Са (NO3)2 + СО2 + Н2О
Натриевая селитра (нитрат натрия) NaNO3 содержит около 16,5% N, встречается в природе в виде залежей. Крупнейшее её ме-
МИНЕРАЛЬНЫЕ УДОБРЕНИЯ
206
Рис. 3. Ровенский завод азотных удобрений (Украинская ССР). Цех разделения воздуха.
«Огромные количества — больше 50 млн. т азота в год — даёт промышленности разделение воздуха...» (стр. 52).
сторождение находится в Чили (отсюда другое её название — чилийская селитра). NaNO3 извлекают из породы горячим выщелачиванием с последующей кристаллизацией соли. Получают её и синтетически, например нейтрализацией азотной кислоты содой:
Na2CO3 + 2HNO3 - 2NaNO3 СО2 + Н2О
Сульфат аммония (NH4)2SO4 содержит около 21 % N. Его получают поглощением аммиака серной кислотой:
2NH3 + H2SO4 = (NH4)2SO4
Моч евина, или карбамид, (NH2)2 СО содержит около 46% N; это высококонцентрированное простое азотное удобрение синтезируют из аммиака и двуокиси углерода:
2NH3 + СО2 = NH2—СО—ONH4
Полученный в первой стадии карбамат аммония, отщепляя воду, превращается в мочевину:
NH2—СО—ONH4~^NH2—СО—NH2
Фосфорные удобрения. Фосфор входит в состав многих органических соединений в растениях, в том числе и тех, что содержатся в семенах. Фосфорное питание растений регулирует темпы их роста и развития.
Из простых фосфорных удобрений наиболее распространены суперфосфат, преципитат,
и трикальцийфосфатов,
2, апатита, гипса и других
фосфоритная мука. Простой суперфосфат — это смесь монокальцийфосфата Са(Н2РО4)2, ’ ди-СаНРО4 и Са3(РО4)
примесей; содержит примерно 20% Р2О5. Суперфосфат получают обработкой природных фосфоритов и апатитов серной кислотой. При этом фосфор из среднего фосфата (то есть из неусвояемой, нерастворимой формы) переходит в гидрофосфаты (усвояемые, растворимые формы). СССР имеет богатейшие запасы фосфоритов на Кольском полуострове и на юге Казахской ССР. При обработке апатита или фосфорита не серной, а фосфорной кислотой получают концентрированное фосфорное удобрение— двойной суперфосфат, содержащий до 50% Р2О5. При обработке фосфорной кислоты известью получают дикальцийфосфат СаНРО4-2Н2О; такое удобрение содержит 35—40% Р2О5 и называется преципитатом.
В качестве фосфорных удобрений используются также томасшлак (богатый фосфатами
кальция шлак от производства стали по способу английского металлурга С. Дж. Томаса, см. Сталь), фосфоритная мука, костяная мука.
Калийные удобрения. Калий имеет исключительно важное значение для обмена веществ в растительном организме. При недостатке калия растения становятся более восприимчивыми к болезням, уменьшается их способность синтезировать углеводы. В качестве калийных удобрений используются природные (сырые) калийные соли и промышленные концентрированные удобрения. Богатейшее в мире калийное месторождение — Соликамское — находится в СССР. Залежами калийных солей располагают у нас также Белоруссия, Западная Украина. За рубежом крупные месторождения калийных солей расположены в ГДР, Франции, США, Испании.
Из сырых калийных солей наибольшее применение находят каинит KCl-MgSO4-3H2O, карналлит KCl-MgCl2-6H2O, полигалит K2SO4 • MgSO4 • 2CaSO4 • 2Н 2О, лангбейнит
K2SO4-2MgSO4; в них содержится от 10 до 17% К2О.
Концентрированное удобрение — хлористый калий КС1 получают из сильвинита, представляющего собой природную смесь КС1 и NaCl с небольшими примесями солей магния и кальция. Используя различную растворимость содержащихся в сильвините солей, получают продукт, обогащённый КС1 (50—62% из расчёта на К2О). Содержащийся в этом продукте хлор вреден для некоторых растений. В таких случаях используют сульфат калия K2SO4; его получают обработкой хлорида калия или минерала лангбейнита серной кислотой.
207
МИНЕРАЛЬНЫЕ УДОБРЕНИЯ
Из сложных удобрений в СССР наибольшее значение имеют следующие:
Аммофос — смесь моно- и диаммонийфосфата, NH4H2PO4 и (NH4)2HPO4; содержит до 58% Р2О5 и до 15% N; получается при обработке фосфорной кислоты аммиаком.
При обработке фосфатов азотной кислотой получают нитрофосфат — удобрение, содержащее 14—20% N и столько же Р2О5. Если же на образующуюся в начальной стадии смесь подействовать аммиаком, то получится другой тип сложного удобрения — нитроаммофос, где азот содержится в нитратной и аммонийной формах.
Нитрофоска — продукт, содержащий «полный комплект» основных питательных элементов: N, К, Р. В разных сортах нитрофоски может быть различное соотношение элементов, в среднем же — по 11—16% N, К2О, Р2О5. В нитрофоске азот находится и в нитратной форме (KNO3), и в аммонийной (в виде фосфатов аммония), и в аммонийно-нитратной (в виде NH4NO3); фосфор — в виде СаНРО4, NH4H2PO4 и (NH4)2 НРО4; калий в виде КС1 и KNO3. Из всех известных удобрений нитрофоска обладает наибольшей агрономической эффективностью. Смешанные удобрения можно получать в самых различных комбинациях и с добавками всевозможных микроудобрений.
Мировое производство М. у. в 1972 г. составило 79,7 млн. т в пересчёте на питательные вещества (N + P2O5+K2O).
В Советском Союзе промышленность удобрений развивалась ускоренными темпами, особенно за послевоенные годы. Данные об их выработке (в млн. т питательных веществ) таковы:
1940г. 1950г. 1960г. 1970г. 1973г.
0,7 1,2 3,3 13,1 17,4
Постоянное расширение производства и применения М. у. давно стало нашей государственной и партийной политикой.
Дальнейшее увеличение производства М. у. наряду с другими агротехническими мерами обеспечит высокие и устойчивые урожаи сель-скохозяйственных культур.
Для систематического получения высоких урожаев состав необходимых элементов в почве должен постоянно воспроизводиться; ведь с каждым урожаем сельскохозяйственные культуры уносят из почвы миллионы тонн азота, фосфора, калия и значительное количество других элементов. Заменить один элемент другим нельзя. Для каждого растения требуется «свой», нужный ему состав элементов. Сколько и в каком ассортименте они нужны тем или иным
сельскохозяйственным культурам, решает наука, называемая агрохимией. Принцип: «сыпь больше — получишь урожай больше» — совершенно неприемлем и даже вреден. Только научный подход к вопросам питания растений, улучшения структуры и плодородия почв может принести успех.
В разных странах на один гектар почвы вносится различное количество удобрений. Это связано с тем, что почвы отличаются по своему плодородию, структуре, кислотности и т. д.; различна и структура растениеводства (удельный вес отдельных культур в общем объёме посевов). По данным за 1970 г., в СССР, Румынии, Югославии, США, Италии, Канаде в среднем было внесено в почву до 100 кг удобрений на гектар (в пересчёте на 100%-ное содержание питательных веществ). Болгария, Венгрия, Польша, Швеция, Англия, Франция, Чехосло-
Рис. 2. Солигорский калийный комбинат (Минская область). На обогатительной фабрике «Беларуськалий» . Здесь выпускают калийные удобрения.
МОЛИБДЕН
208
вакия, Дания вносят 150—250 кг, а такие страны, как ГДР, ФРГ, Япония, Голландия, Бельгия,— 300—700 кг.
В СССР на основе многолетних полевых опытов и практики колхозов и совхозов определены следующие средние прибавки урожая в центнерах от использования одного центнера минеральных удобрений (в условных туках):
зерно 1,1—1,3
хлопок-сырец 0,7—0,9 сахарная свёкла 6,5—7,0
лён-долгунец 0,2—0,25
картофель 7,1— 7,5 овощи и бахчевые 7,9—12,0 плоды и ягоды 1,7— 2,0 виноград 3,0— 3,2
95,94 42
Мо
4d55s’
МОЛИБДЕН
Участие химии в современном земледелии ис-' ключительно велико и оно неуклонно возрастает с каждым годом (см. также статью Химия служит человеку). В. С. Титов.
МОЛИБДЕН (лат. Molybdaenum) Мо — химический элемент VI группы периодической системы Менделеева; атомный номер 42, атомная масса 95,94. Светло-серый металл. В природе встречается в виде смеси семи стабильных изотопов с массовыми числами 92, 94, 95, 96, 97, 98, 100. Из них наиболее распространён 98Мо (23,75%).
Вплоть до XVIII в. встречавшийся в природе минерал — молибденовый блеск — не отличали от графита и свинцового
блеска, так как по внешнему виду и мягкости они чрезвычайно похожи друг на друга. Эти минералы носили общее название «молибден» (в переводе с греческого «молюбдос» означает свинец). В 1778 г. шведский химик К. Шееле после многократной обработки порошка молибденового блеска азотной кислотой получил молибденовую кислоту (см. ниже). Вскоре восстановлением её ангидрида МоО3 был получен и сам металл#
В природе М. распространён сравнительно мало, несколько меньше, чем его аналог—вольфрам. Содержание М. в земной коре 1,1 • 10“4% по массе (56-е место среди элементов). Из 15 минералов М. молибденовый блеск (современное название молибденит) MoS2 — наиболее важный, имеющий промышленное значение. Крупные месторождения молибденовых руд в Советском Союзе находятся на Кавказе и в Казахстане.
М.— один из самых тугоплавких металлов: по температуре плавления (2620 °C) он уступает только вольфраму, рению и танталу (их /пл составляет 3410 °C, 3180 °C и 2996 °C соответственно); кипит М. около 4800 °C. М.— тяжёлый ме-салл, его плотность 10,2 г/см3. Он более пла-ттичен, чем вольфрам, то есть легче поддаётся ковке и прокатке.
В соответствии с положением М. в периодической системе Менделеева его атом имеет шесть внешних (валентных) электронов (конфигурация 4d55s1, см. Атом). Обычно Мг проявляет степень окисления +6 (валентность VI). Известны и соединения, в которых он имеет степени окисления +2, +3, +4 и +5.
При невысоких температурах М. химически устойчив. Он соединяется только с самым активным галогеном — фтором, образуя летучий фторид MoFe. С хлором и бромом М. взаимодействует при нагревании с образованием МоС1б и МоВг4.
Выше 400 °C М. окисляется кислородом воздуха до трёхокиси М.:
2Мо + ЗО2 = 2МоО3
При 700 °C М. вытесняет Н2 из паров воды, окисляясь до двуокиси:
Мо + 2Н2О = МоО2 + 2Н2
К действию щелочей М. весьма устойчив, а из кислот его растворяют лишь крепкая HNO3 и царская водка. Из двух окислов — МоО2 (тёмно-коричневые кристаллы) и МоО3 (белые с зеленоватым оттенком) наиболее устойчив МоО3— молибденовый ангидрид. Он плохо растворим в воде, но щёлочь и аммиак взаимодействуют с ним с образованием солей молибденовой кислоты Н2МоО4. Состав этих солей — молибдатов—непрост; он выражается общей формулой х-Ме2О-уМоО3-?Н2О, где х<у, Me — металл или группа NHJ\
Наиболее известен парамолибдат аммония 3(NH4)2O-7MoO3-zH2O — одно из самых важных соединений М.
Если ткань, пропитанную бесцветным раствором парамолибдата аммония, поместить в бесцветный раствор хлорида олова SnCl2, подкисленный H2SO4, материал тотчас же окрасится в красивый небесно-голубой или синий цвет. Эта краска называется молибденовой синью и образуется в результате восстановления МоО3 до окисла низшей валентности (восстановителем служит SnCl2). Парамолибдат аммония не только окрашивает ткань, но одновременно и дезинфицирует её (уничтожает микроорганизмы) и тем самым удлиняет срок её службы.
А из растворов, содержащих анион фосфорной кислоты РО|“, парамолибдат аммония осаждает очень малорастворимую соль — фосфомолибдат аммония (NH4)3PO4* 12МоО3*6Н2О ярко-жёлтого цвета. Эта реакция открыта более 100 лет тому назад, и до сих пор её очень любят химики-аналитики: так определяют содержание фосфора в рудах и сплавах, в почвах и удобрениях.
В промышленности М. и его соединения получают из молибденита MoS2. Поскольку содержание М. в рудах очень незначительно (0,1— 1%), руду сначала обогащают. Концентрат, содержащий 47—50% М., подвергают окислительному обжигу:
2MoS2 + 7О2 = 2МоО3 + 4SOa
209
МЫШЬЯК
Полученную трёхокись М. тщательно очищают различными методами от примесей. А затем восстановлением МоО3 в токе сухого водорода при 950—1000 °C получают порошок М.
Для превращения порошка в компактный металл используют способ порошковой металлургии (он описан в статье Вольфрам). Более современные методы — дуговую и электроннолучевую плавку — применяют для получения крупных слитков М. (массой до 2000 кг). При дуговой плавке М. расплавляется в электрической дуге, а при электронно-лучевой — под действием мощного электронного пучка, испускаемого одной или несколькими электронными пушками.
Более 75% М. идёт на производство легированных сталей. Такие стали по сравнению с обычной обладают повышенной твёрдостью, прочностью и упругостью. Они успешно применяются в авиа- и автомобилестроении, для изготовления лопаток турбин и орудийных стволов. Для легирования служит не сам М., а его сплав с железом — ферромолибден (55— 70% Fe), так как получать его значительно дешевле и проще (см. Ферросплавы).
М.— очень важный компонент кислотостойких сплавов. Сплавы Мо—Fe—Ni, содержащие до 28% Мо, обнаруживают необычайную стойкость к действию всех минеральных кислот (кроме плавиковой) при температурах до 100 °C. В аппаратах, изготовленных из таких сплавов,
Электрическая лампа накаливания. 1 —нить «акала из вольфрама; 2 — молибденовые крючки, придающие определённую форму нити накала. Они поддерживают её в течение всего срока службы лампы; -3 — средняя часть электродов, служащих для подачи напряжения. Она сделана из молибдена и вплавлена в стекло.
проводятся многие процессы химической промышленности, протекающие в агрессивных средах. Особенно большое будущее принадлежит жаропрочным молибденовым сплавам. Если алюминиевые сплавы сохраняют прочность вплоть до 350 °G, титановые — до 500 °C, то молибденовые — до температур порядка 1200 °C. Поэтому из них делают, например, наиболее ответственные детали реактивных и ракетных двигателей, работающих при высоких температурах.
Может быть, вы уже слышали о «молибденовом» стекле. А ведь М. в составе такого стекла нет. И получило оно своё название совсем по другой причине. Вольфрам, из которого делают нити накаливания обычных электрических ламп, при нагревании сильно расширяется. И если вольфрамовую проволоку впаять в стеклянный стерженёк в центре лампочки, стекло треснет. М. же имеет коэффициент теплового расширения, равный коэффициенту расширения специального стекла. Поэтому вольфрамовые нити сначала укрепляют на крючках из М., и уже эти крючки впаивают в стеклянный стерженёк. Стекло такого стерженька и называют молибденовым.
В технике используются и некоторые соединения М., прежде всего — чистый дисульфид MoS2. В подшипниках скольжения, в зубчатых передачах и в других трущихся металлических частях различных машин необходимы особые смазки, предупреждающие преждевременный износ металла. MoS2, отличающийся очень низким коэффициентом трения, оказался ценнейшим компонентом таких смазок.
Окислы М. служат катализаторами многих процессов органического и неорганического промышленного синтеза. Особенно ценны такие катализаторы для нефтяной промышленности, так как они не отравляются сероводородом и с их помощью можно перерабатывать сырые нефти любого качества.
А в живых организмах М. катализирует процессы азотного обмена. Поэтому растворимые молибдаты в небольших дозах всегда вводят и в состав микроудобрений.
3. А. Старикова.
МЫШЬЯК (лат. Arsenicum) As — химический элемент V группы периодической системы Менделеева; атомный номер 33, атомная масса. 74,9216. В природе элемент представлен одним стабильным изотопом 76As.
Природные соединения М.— золотисто-жёлтый аурипигмент As2S3 и тёмно-красный реальгар As4S4 — использовались ещё древними народами для приготовления красок; о них упоминается уже в сочинениях Аристотеля (IV в. до н. э.). Соединения М. были известны как очень сильные яды; их называли «арсеникон», что по-гречески значит сильный, мужественный; отсюда и латинское название элемента. В Древней Руси мышьяковыми соединениями травили насекомых и грызунов (русское «мышьяк» идёт от «мышь» и «яд»). Из-за ядовитости М. алхимики изображали его в виде извивающейся змеи с раскрытой пастью.
Прокаливая природные сульфиды М. на воздухе, алхимики получали так называемый белый мышьяк (окисел As2O3). В истории средних веков известны случаи, когда коронованные особы пользовались «белым мышьяком» для тайного уничтожения своих противников. Подбросить отраву в пищу врага считалось простым и безнаказанным способом борьбы, так как устанавливать причину гибели человека тогда ешё не умели.
14 хэш
мышьяк
210
74,9216 33
As
4s24p3 мышьяк
Принято считать, что впервые свободный «металлический» М. получил немецкий алхимик Альберт фон Больд-штедт (около 1250 г.);но, вероятно, М. получали и прежде.
По распространённости в земной коре М. занимает 51-е место среди всех прочих элементов (1,7-10~4% по массе). Самородный (свободный) М. встречается в природе крайне редко. Из 120 минералов, содержащих М., наиболее распространены мышьяковый колчедан (арсенопирит) FeAsS и названные выше красивые минералы аурипигмент и реальгар. Очень важны для промышленного получения М. полиметаллические руды, в которых М. встречается вместе с Au, Ag, Си, Со и другими металлами. Сульфиды М.
часто сопутствуют цинковой обманке и пириту.
Подобно фосфору и многим другим элементам, М. существует в нескольких аллотропных модификациях. При обычных условиях устойчив так называемый серый, или металлический, М.— кристаллы с серебристым металлическим блеском, довольно хорошо проводящие тепло и электричество; они очень хрупки и легко измельчаются в порошок; плотность серого М. 5,72 г/см3. Если серый М. нагреть выше 600°C, то он возгоняется, не плавясь. А если пары его быстро охладить, образуется другая модификация — жёлтый М., прозрачные, мягкие, как воск, кристаллы; по своему виду и свойствам жёлтый М. напоминает белый фосфор, но гораздо менее устойчив. Известны также коричневая, чёрная и другие формы М. Выше 270 °C все эти формы переходят в металлический М. Расплавить М. можно лишь в запаянной трубке под давлением собственных паров, которое при температуре плавления М. (817 °C) равно 36 атм. Ни в воде, ни в органических жидкостях М. не растворяется.
У атома As (аналогично азоту и фосфору) внешняя электронная оболочка содержит пять электронов (конфигурация 4s24p3, см. Атом). В связи с этим в химических соединениях М. проявляет степени окисления +5 (например, в As2O5), +3 (As2O3) и —3 (AsH3); соответственно валентности М. равны V и III.
На воздухе уже при комнатной температуре М. покрывается тонкой окисной плёнкой, отчего его блеск со временем исчезает. При нагревании в кислороде или на воздухе М. горит синим пламенем, окисляясь в мышьяковистый ангидрид:
4As 302= 2As3O3
Порошок М., брошенный в сосуд с хлором, воспламеняется, причём образуется хлорид М.:
2As + ЗС12 = 2AsCl3
Непосредственно соединяется М. с другими галогенами и с серой.
В ряду напряжений М. стоит за водородом и поэтому не вытесняет последнего из кислот. Азотная кислота и царская водка растворяют его, окисляя до мышьяковой кислоты:
3As + 5HNO3 + 2Н2О = 3H3AsO4 + 5NO
Мышьяковистый ангидрид As2O3— твёрдое вещество белого цвета, мало (около 2%) растворимое в воде. Это соединение амфотерно (с небольшим преобладанием кислотных свойств) и взаимодействует как с кислотами, так и со щелочами:
As2O3 + 6NaOH = 2Na3AsO3 + ЗН2О
As2O3 + 6НС1 = 2AsCl3 + 3H2O
Мышьяковистому ангидриду соответствует слабая мышьяковистая кислота, существующая только в растворе,— в ортоформе H3AsO3 и метаформе HAsO2. В растворе эти формы находятся в равновесии:
H3AsO3 HAsO2 + Н2О
которое сильно сдвинуто вправо. Большинство арсенитов — солей мышьяковистой кислоты— является производными метаформы HAsO2.
Высший окисел М. As2O5— мышьяковый ангидрид — может быть получен осторожным нагреванием мышьяковой кислоты H3AsO4, образующейся по приведенной выше реакции (получить As2O5 сжиганием As нельзя). Мышьяковый ангидрид — это белая, стекловидная масса, расплывающаяся на воздухе; соответствующая ему мышьяковая кислота — трёхосновная кислота средней силы, во многом напоминающая фосфорную (см. Фосфорные кислоты).
Соли мышьяковой кислоты — арсенаты и соли фосфорной — фосфаты имеют аналогичные формулы и почти равную растворимость, одно и то же число молекул кристаллизационной воды и одинаковую форму кристаллов. Арсенаты и фосфаты были одним из тех примеров, на которых немецкий минералог Э. Митчерлих открыл в 1819 г. явление изоморфизма. Митчерлих назвал изоморфными такие родственные по химическому составу вещества, которые образуют кристаллы одинаковой формы. Сейчас мы знаем, что в изоморфных кристаллах сходные атомы или ионы могут легко замещать друг друга (см. об этом в статье Минералы).
Однако, в отличие от фосфорной кислоты, мышьяковая проявляет окислительные свойства:
H3AsO4 + 2HI = H3AsO3 + I2 + Н2О
Образование соединений с металлами для М. менее характерно, чем для азота или фосфора, однако подобные соединения известны (на
211
пример, Mg3As2) и называются арсенидами. Из них особенно интересен арсенид галлия GaAs — полупроводник, из которого в 1962 г. был сделан первый полупроводниковый лазер. С водородом М. непосредственно не соединяется, но при действии на арсениды разбавленных кислот выделяется мышьяковистый водород AsH3, иначе называемый арсином.
В промышленности М. получают либо из руд, содержащих его как единственную полезную составную часть, либо попутно — при переработке медных и полиметаллических руд, содержащих М. как примесь. И в том и в другом случае сульфидные руды обжигают в присутствии воздуха (при этом сульфид переходит в окисел):
2AS2S3 Ч- 902 — 2AS2O3 6SO2
А затем окисел восстанавливают до свободного М. нагреванием с древесным углём:
As2O3 + 3C=2As + 3CO
Свободный М. используется главным образом как добавка к некоторым сплавам. Так, меди он придаёт большую стойкость к окислению. М. добавляют и к свинцу, идущему на изготовление дроби: М. не только увеличивает твёрдость свинца, но и повышает поверхностное натяжение его расплава, что способствует получению дробинок сферической формы.
Сам М. не ядовит, но зато чрезвычайно ядовиты все его соединения, растворимые в воде. Соединения М. были одними из самых первых инсектицидов — средств для уничтожения вредных насекомых. А в конце первой мировой войны Германия применила органические соединения М. как химическое оружие — боевые отравляющие вещества (ОВ).
Однако соединения М. не только убивают, но и помогают в борьбе за жизнь (так сложна и многогранна связь биологии и химии). Тысячелетиями страдало человечество от разрушительных инфекционных болезней, одной из которых был сифилис. В 1909 г. крупный не-t мецкий микробиолог П. Эрлих после долгих и кропотливых трудов получил новый лекарственный препарат «606» — органическое соединение М., носившее длинное название «диокси-диаминоарсенобензолдигидрохлорид» (это был 606-й по счёту из испытанных Эрлихом пре
МЫШЬЯКОВИСТЫЙ ВОДОРОД
паратов). Лечебное действие нового вещества оказалось прямо пропорционально длине его названия. Это чудесное лекарство, оказавшееся действенным против сифилиса, возвратного тифа, малярии, сонной болезни, положило начало современной химиотерапии. П. Эрлих назвал его «сальварсан» (по-немецки Sal-varsan, от лат. salvo — спасаю и нем. Arsen — мышьяк). В поэтическом же переводе сальварсан означает: «Да здравствует мышьяк!».
В. К. Бельский.
МЫШЬЯКОВИСТОКИСЛЫЕ СОЛИ—то же, что арсениты. См. также Мышьяк.
МЫШЬЯКОВИСТЫЙ ВОДОРОД (арсин) AsHз— соединение мышьяка с водородом. Бесцветный газ; в чистом состоянии запаха не имеет, но благодаря сопутствующим примесям обычно пахнет чесноком. При 300—400 °C М. в. быстро разлагается:
2AsH3z±2As + 3H2
При этом мышьяк отлагается в виде чёрного блестящего налёта («мышьякового зеркала») на стенках сосуда или трубки, в которой находится газ.
В 1836 г. английский химик Дж. Марш предложил использовать это явление для обнаружения весьма малых количеств As (тысячных долей миллиграмма). А поскольку соединения мышьяка очень ядовиты, обнаружить следы его бывает крайне важно, например при судебной экспертизе. Способ Марша очень прост.
Испытуемый раствор приливают в склянку, в которой идёт выделение водорода, например по реакции Zn с НС1. Если в растворе присутствуют соединения мышьяка, то водород «в момент выделения» восстанавливает их до М. в., например:
As2O3 + 12Н = ЗН2О + 2AsH3
Если подогреть газоотводную трубку, по которой выходит смесь водорода с М. в., последний разлагается, причём на трубке появляется чёрный налёт мышьяка. При более сильном нагревании он испаряется и оседает вновь на более холодных частях стеклянной трубки, в которой ведётся опыт.
М. в. растворим в воде, но химически с ней не реагирует. Он очень токсичен — это один из самых сильных неорганических ядов.
В. К- Бельский.
МЫШЬЯКОВОКИСЛЫЕ СОЛИ—то же, что арсенаты. См. также Мышьяк.
14*
НАТРИЙ (лат. Natrium) Na — химический элемент I группы периодической системы Менделеева; атомный номер 11, атомная масса 22,98977. Серебристо-белый металл. В природе Н. встречается в виде одного стабильного изотопа 23Na.
В 1807 г. в Лондоне на собрании Королевского общества Г. Дэви сообщил о том, что ему удалось разложить действием электрического тока едкое кали КОН и едкий натр NaOH и выделить из них неизвестные металлы, обладающие необычными свойствами. Эти металлы очень быстро окислялись на воздухе, а на поверхности воды плавали, выделяя из неё водород.
Ещё в глубокой древности были известны соединения Н.— поваренная соль (хлорид Н.) NaCl, едкая щёлочь (гидроокись Н.) NaOH и сода (карбонат Н.) Na2CO3. Последнее вещество древние греки называли «нитрон»; отсюда и происходит современное название металла — «натрий». Однако в Великобритании, США, Италии, Франции сохраняется слово sodium — так «окрестил» элемент Г. Дэви (от испанского слова «сода», имеющего то же значение, что и по-русски).
Н.— один из самых распространённых в природе элементов. На его долю в земной коре приходится 2,5% по массе (6-е место среди элементов). Будучи одним из активнейших металлов, Н. в свободном состоянии в природе не встречается. Зато его минералы весьма разнообразны. К их числу относится хорошо знакомая всем поваренная соль NaCl — минерал галит (или каменная соль).
Вот удивительная вещь, столь типичная для химииI Взятые по отдельности, Н. и хлор губительно действуют на организм,- а их соединение — это вещество, без которого невозможна жизнь человека. NaCl обеспечивает постоянство осмотического давления крови и создаёт условия для существования красных кровяных телец — эритроцитов; сохраняет способность к возбудимости мышц, а в желудке образует соляную кислоту, необходимую для переваривания и усвоения пищи. О том, как важна для нас соль, говорят и созданные народом поговорки — «Хлеб да соль», «Съесть пуд соли» и т. д. В год человек потребляет от 3 до 5 кг поваренной соли.
Наша страна исключительно богата залежами каменной соли — таковы районы Березники — Соликамск, Соль-Илецк, Усолье-Сибирское, Сольвычегодск и другие (даже названия городов связаны с солью!). Толщина соляных пластов иногда достигает 1,5 км. Крупные запасы соли есть в ГДР, ФРГ, США, Австрии и
других странах. Но залежами галита не исчерпывается мировой запас NaCl. Огромное количество этого вещества находится в растворённом состоянии в морской воде (общие запасы исчисляются астрономическим числом 3,8-1016 т) и в соляных озёрах. Например, соли озера Баскунчак хватило бы нашей стране для пищевых целей на 400 лет!
Из других важнейших минералов, содержащих Н., назо
вём мирабилит (глауберовасолb)Na2SO4- ЮН2О. Крупнейшие его запасы находятся в заливе Каспийского моря Кара-Богаз-Гол (прочитайте повесть К. Паустовского «Кара-Бугаз»).В Чили добывают нитрат NaNO3—чилийскую селитру (важное минеральное удобрение). У нас в Армении и Сибири есть богатые залежи минерала троны, состава Na2CO3-NaHCO3-2H2O.
Н.—легкоплавкий металл (он плавится при 97,83 °C), кипит при 882,9 °C. Это один из самых лёгких металлов — он легче воды (плотность Н. 0,968 г/см3). Н. и его соединения окрашивают пламя газовой горелки в жёлтый цвет. Эта реакция так чувствительна, что открывает присутствие малейших следов Н. повсюду (например, в комнатной или уличной пыли).
Человек выделяет с потом заметные количества NaCl, в чём легко убедиться, хлопнув ладонями перед газовым пламенем. Как рассказывают, знаменитый немецкий химик Р. Бунзен требовал, чтобы приходящие экзаменоваться студенты предъявляли ему учебник, по которому занимались. К экзамену строгий профессор допускал только тех, чьи учебники при быстром^ раскрывании и закрывании перед газовой горелкой давали достаточно продолжительное жёлтое окрашивание пламени.
Н.— типичный представитель щелочных металлов, один из самых активных элементов периодической системы. Внешняя электронная оболочка атома Na содержит один электрон
213
НАТРИИ
Гемфри ДЭВИ. Путём электролиза впервые получил калий, натрий и другие активные металлы.
Английский химик и физик Гемфри Дэви родился 17 декабря 1778 г. в Пензансе. Сын резчика по дереву. Учился в частной школе, очень похожей на то мрачное заведение, что описано Ч. Диккенсом в романе «Жизнь и приключения Николаса Никльби». Затем работал аптекарским учеником, причём усиленно занимался самообразованием; изучал химию по учебнику А. Л. Лавуазье.
В 1798—1801 гг. Дэви был сотрудником Пневматического института в Клифтоне, где получал различные газы и испытывал на самом себе их действие. Особое его внимание привлекла закись азота, поскольку существовало мнение, будто этот газ — основная причина всех инфекционных заболеваний. Дэви показал ошибочность столь странной гипотезы. Он обнаружил, что вдыхание закиси азота вызывает приятное опьянение и устраняет зубную боль. Опыты Дэви получили широкую известность. Он даже устраивал специальные сеансы для любителей неизведанных ощущений. По свидетельству современника, надышавшись веселящим газом, «одни джентльмены прыгали по столам и стульям, у других развязались языки, третьи обнаружили чрезвычайную склонность к потасовке». Спустя почти полвека, в 1844 г., американский дантист X. Уэллс использовал N2O для обезболивания при удалении зубов. В наши дни смесь закиси азота с кислородом служит для устранения болевых ощущений при хирургических операциях, при инфаркте миокарда.
В 1801 г. Дэви стал ассистентом, а в 1802 г.— профессором Королевского института в Лондоне; здесь его блестящие лекции привлекали большую аудиторию. В прекрасно оборудованной лаборатории института Дэви в 1807— 1808 гг. получил путём электролиза калий, натрий и амальгамы кальция, стронция, бария, магния. В 1808—1809 гг. описал явление дугового электрического разряда, которое назвал вольтовой дугой (впервые дуговой разряд открыл в 1802 г. профессор В. В. Петров в Петербурге). В 1810 г. Дэви доказал элементарную природу хлора, а в 1814 г.— иода.
В 1815 г. он изобрёл лампу с металлической сеткой, позволяющую безопасно вести добычу каменного угля там, где выделяется взрывчатый рудничный газ. Это изобретение сулило учёному большие доходы, но он отказался его патентовать, заявив: «лучшим вознаграждением за мои работы будет сознание того, что я сделал добро мне подобным». Дэви разработал и впервые в истории читал курс сельскохозяйственной химии. Вопреки распространённому тогда мнению, он высказал мысль, что минеральные соли составляют часть пищи для растений. Учёный указывал на необходимость полевых опытов для разрешения вопросов земледелия.
Дэви был президентом Лондонского королевского общества, иностранным членом Петербургской академии наук и Парижской академии наук. Здоровье учёного было сильно подорвано вдыханием ядовитых газов. Он скоропостижно скончался 29 мая 1829 г. в Женеве, во время поездки в Швейцарию.
Для своей будущей эпитафии (надгробной надписи) Дэви оставил такие слова: «Единственной моей целью было служить делу человечества и если это удалось мне, то я чувствую себя с избытком вознаграждённым той приятной мыслью, что такая судьба выпала мне на долю».
(конфигурация 3s1, см. Атом), Свой единственный валентный электрон Н. легко отдаёт и поэтому в своих соединениях всегда проявляет степень окисления +1 (валентность I).
Взаимодействуя с кислородом при разных условиях, Н. образует соединения двух типов — окись Na2O и перекись Na2O2 (Na—О—О—Na).
Как сильный окислитель Na2O2 используют для отбеливания тканей и других материалов. Другая важная область применения перекиси Н. (как и перекиси калия) — регенерация кислорода в закрытых помещениях и приборах (подводных лодках, газоубежищах, изолирующих противогазах и т. д.). Перекись Н. взаимодействует с накапливающейся при дыхании двуокисью углерода, высвобождая кислород:
2Na2O2 + 2СО2 = 2Na2CO3 + О2
Н. непосредственно реагирует со всеми неметаллами. Взаимодействие с водородом, при 400 °C приводит к гидриду:
2Na + H2 = 2NaH
С азотом в тихом электрическом разряде Н. образует нитрид Na3N (с примесью азида NaN3):
6Na + N2 = 2Na3N
При обычной температуре Н. самовоспламеняется в атмосфере фтора и хлора, например: 2Na + Cl2 = 2NaCl
С бромом и иодом реагирует при нагревании. При 800—900 °C соединяется с углеродом, образуя карбид Na2C2. Растирание Н. с серой даёт сульфид Na2S и смесь полисульфидов (Na2S3, Na2S4 и т. д.). Н. энергично реагирует с водой, вытесняя водород:
2Na + 2Н2О = 2NaOH + Н2
Такая реакция происходит даже со льдом при температуре —80 °C, а с тёплой водой идёт со взрывом. Поэтому нельзя тушить водой загоревшийся Н. Недаром в некоторых химических лабораториях над ракови-
нашатырный спирт
214
Схема получения едкого натра и хлора электролизом раствора хлорида натрия. На рисунке показан разрез электролизёра. 1 — слой ртути, служащей катодом; 2 — электролит (раствор NaCl); 3 — бак из шифера; 4 — графитовый анод; 5 — анодная шина; 6 — раствор NaOH, собирающийся в среднем отсеке; 7 — шиферная перегородка; 8 — качающийся механизм.
Раствор NaCl налит в бак 3 из шифера. Он приводится в качательное движение устройством 8. Бак разделён на три отсека двумя шиферными перегородками 7 (диафрагмами), не доходящими до его дна. На дне бака находится слой ртути /, служащей катодом. Графитовые аноды 4 помещают в двух крайних отсеках. При пропускании тока на анодах выделяется хлор, по реакции 2С1 “—2ё=С12- Его отводят по трубам (на рисунке они не показаны). На катоде образуется амальгама натрия, которая разлагается водой с выделением едкого цатра и водорода:
2 (Na, Hg) + 2Н2О = H2+2Hg + 2NaOH
Во время электролиза бак медленно покачивается, благодаря чему амальгама всё время перемещается по дну ванны. Раствор едкого натра, собирающийся в среднем отсеке, содержит неразложившийся хлорид натрия. Для его удаления раствор упаривают, причём почти весь NaCl выпадает в осадок, который отфильтровывают. Раствор едкого натра выпаривают либо до содержания 50% NaOH, либо досуха. Сухой остаток плавят в котлах из щёлочеупорного чугуна (содержащего около 0,5% Ni). Твёрдый технический едкий натр содержит 92—95% NaOH (остальное — примеси Na2CO3 и NaCl).
нами висят шуточные надписи типа: «Не хотите стать уродом — не бросайте натрий в воду!». Хранят Н. под слоем керосина.
Гидроокись NaOH, едкий натр, или, как называют это соединение в технике, каустическая сода (от лат. causticus — едкий, жгучий),— одно из самых сильных оснований. Это важный продукт химической промышленности. Технический продукт, кроме NaOH, содержит примеси (высший сорт до 3% Na2CO3 и до 1,5% NaCl). Большое количество NaOH идёт на приготовление электролитов для щелочных аккумуляторов, производство мыла, красок, целлюлозы. Получают NaOH электролизом водного раствора NaCl (рисунок).
О применении других соединений Н. можно прочитать в статьях Нитраты, Сода, Сульфаты, Фосфаты, Хлориды и т. д.
Использование металлического Н., конечно, далеко не так обширно, как его соединений. Но и здесь есть много интересного. Основной метод получения Н.— электролиз расплавленной поваренной соли. При этом на аноде выделяется хлор, а на катоде — Н. Техническая
трудность, состоящая в близости температуры плавления NaCl (801°С) и температуры кипения Н. (882,9 °C), преодолевается добавлением к NaCl других солей (КС1, NaF, СаС12). Такие смеси плавятся ниже 600 °C, что позволяет вести электролиз при 575—585 °C.
Металлический Н. используется для восстановления чистых металлов из их соединений — калия (из КОН), титана (из TiCl4) и др. В последнее время появился большой интерес к Н. и его сплаву с калием как к теплоносителям для ядерных реакторов — ведь щелочные металлы плохо поглощают нейтроны и поэтому не препятствуют делению ядер урана (об этом см. также в статье Висмут). В мощных экономичных газосветных электролампах, служащих для освещения автострад, пристаней, вокзалов, и т. п., используется ярко-жёлтое свечение паров натрия. В. К- Бельский.
НАШАТЫРНЫЙ СПИРТ — 10-процентный водный раствор аммиака. Прозрачная летучая жидкость с острым запахом и щелочными свойствами. Н. с. дают вдыхать при обмороках.
215
НЕОН
НАШАТЫРЬ — то же, что хлорид аммония NH4C1. См. Аммония ион.
НЕГАШЁНАЯ ИЗВЕСТЬ — окись кальция СаО. См. Известь и Кальций.
НЕМЕТАЛЛЫ — химические элементы, характеризующиеся преимущественной способностью присоединять электроны. В свободном состоянии, в виде простых тел, Н. не имеют физических свойств, присущих металлам. Иногда Н. называют металлоидами (от «металл» и греч. «эйдос» — вид, облик, образ), что неправильно, так как по внешнему виду Н. совсем не похожи на металлы. К Н. относятся водород Н, бор В, углерод С, азот N, кислород О, фтор F, кремний Si, фосфор Р, сера S, хлор С1, мышьяк As, селен Se, бром Вг, теллур Те, иод I, астат At, а также инертные газы — в общей сложности 22 элемента. В химических соединениях атомы Н. воссоздают электронную оболочку инертных газов путём присоединения недостающих электронов.
Наиболее типичные Н.— галогены расположены в периодической системе элементов Менделеева непосредственно перед инертными газами. В отличие от типичных металлов, Н. образуют с кислородом кислотные окислы (ангидриды), гидраты которых—кислоты. С водородом Н. образуют газообразные соединения; водные растворы некоторых из них (галогено-водородов) — сильные кислоты. С металлами типичные Н. дают соединения с ионной связью (например, NaCl, СаО). Соединения Н. между собой характеризуются ковалентной связью (например, C1F, CS2).
В древности были известны только два Н.— углерод и сера. В XIII в. получен мышьяк, в XVII в.— водород и фосфор, в конце XVIII в. открыты кислород, азот, хлор, теллур. В 1789 г. французский химик А. Л. Лавуазье включил их в список неметаллических простых тел. В первой половине XIX в. были получены бром, иод, селен, кремний, бор. Только в конце XIX в. удалось изолировать фтор и открыть инертные газы. Радиоактивный элемент астат в природе не встречается; искусственно (по ядерной реакции) он был получен впервые в 1940 г.
См. также статьи об отдельных элементах. С. А. Погодин.
НЕОДИМ (лат. Neodymium) Nd — химический элемент III группы периодической системы Менделеева из семейства лантаноидов^ атомный номер 60.
НЕОН (лат. Neonum) Ne — химический элемент VIII группы периодической системы Менделеева; атомный номер 10, атомная масса 20,179, инертный газ.
Атмосферный Н. состоит из 3стабильных изотопов с массовыми числами 20, 21 и 22. Наиболее распространён из них 20Ne (90,92% в смеси изотопов).
К открытию Н. привёл спектральный анализ легколетучей фракции воздуха, проведенный в 1898 г. англичанами У. Рамзаем и М. Трэверсом. Двенадцатилет-ний сын Рамзая случайно зашёл в кабинет к отцу в момент проведения опыта и, увидев необычный ярко-красный спектр только что открытого газа, радостно закричал: «Новый! Новый!». Так и был назван элемент (греч. «нёос» — новый).
Н.— редкий газ. В 1 м3 воздуха, который служит его единственным источником, содержится около 16 мл Н. При возбуждении в Н. электрического разряда наблюдается оранжево-красное свечение. Газовую смесь, состоящую из Н., азота и гелия и содержащую до 40 % Н., используют для наполнения рекламных трубок. Газосветные трубки, наполненные Н. и другими инерт
ными газами, украшают в вечернее время улицы современного города. В аэродромных маяках загораются неоновые дуговые лампы. А неоновые люминесцентные лампы светят втрое ярче обычных. На панелях самых разных приборов мерцают оранжево-красные сигнальные лампы. О физических и химических свойствах Н. см. Инертные газы.
Газосветные трубки, наполненные неоном и другими инертными газами, украшают в вечернее время улицы современного города.
НЕПТУНИИ
НЕПТУНИЙ (лат. Neptunium) Np — радиоактивный химический элемент с атомным номером 93; относится к актиноидам.
НЙКЕЛЕВЫЕ СПЛАВЫ —сплавы на основе никеля, а также сплавы, которым никель придаёт особо ценные свойства.
Некоторые Н. с. были известны уже в далёкой древности. Например, найдены монеты Бактрийского царства (II в. н. э.), изготовленные из сплава меди с 20% Ni (интересно, что примерно тот же состав имеет сплав, из которого чеканят современную монету достоинством в 10, 15, 20, 50 коп. и 1 рубль). Сплав пакфонг, состоящий из меди, никеля и цинка, серебряно-белого цвета, выплавлялся в древнем Китае. А природные сплавы железа с никелем — железные метеориты, содержащие обычно от 3% до 30% Ni (иногда и до 77%), несомненно были знакомы уже первобытному человеку.
Промышленное использование Н. с. началось лишь в конце XIX в., когда во Франции возникло производство никеля, основанное на переработке никелевых руд из Новой Каледонии. Исследования шотландского металлурга Дж. Рили (1889 г.) показали, что небольшие добавки никеля (около 3%) повышают механические свойства стали, особенно её вязкость. Броня из никелевой стали при попадании снаряда не растрескивается. Никелевые стали с добавками хрома, молибдена, ванадия сделались одним из важнейших конструкционных материалов, особенно с развитием в начале XX в. авто- и авиастроения.
Подробное изучение физических свойств системы железо — никель, в зависимости от её состава, предпринятое в Международном бюро мер и весов (Севр, близ Парижа) физиком и метрологом Ш. Э. Гильомом и продолженное другими исследователями, дало чрезвычайно ценные результаты. Уже в 1896 г. Гильом нашёл, что коэффициент теплового расширения сплава железа с 36—38% Ni ничтожно мал (1,5-10"6 на 1 °C при температурах от—80 °C до 100 °C). Сплав получил название инвар (от лат. invariabilis — неизменяемый) и стал применяться для изготовления измерительных приборов, размеры которых не должны изменяться при колебаниях температуры. Ещё меньший коэффициент теплового расширения (ниже МО-6 на 1 °C) имеет суперинвар — сплав 64% Fe, 32% Ni, 4% Со.
Много лет тому назад в электрических лампочках проволочки, подводящие ток к угольным или вольфрамовым нитям, делали из платины. Ведь платина и стекло имеют почти одинаковые коэффициенты теплового расширения— около 9,0-10"6 на 1 °C. (О том, почему это так важно, рассказано в статье Молибден.) Но вследствие дороговизны платины она вскоре была заменена платинитом — сплавом железа с 48% Ni (коэффициент линейного расширения тоже около 9,0 • 10“6 на 1 °C). Однако долго ещё находились «предприимчивые» люди, собиравшие перегоревшие лампочки в надежде добыть из них драгоцен
216
ный металл. Легко себе представить их разочарование!
Как неприятно, а порой опасно, когда ваши часы то отстают, то спешат. Происходит это от разных причин. Но одна из главных — колебания температуры, изменяющие модуль ущ ругости часовой пружины. Сплав железа с 34—37% Ni, 7—13% Сг, 0,3% С, называемый элинваром (от лат. elasticitas — упругость, invariabilis—неизменяемый), сохраняет свои упругие свойства при изменениях температуры, а потому является превосходным материалом для часовых пружин.
И никель, и железо — самые типичные ферромагнитные металлы. Их сплавы, в зависимости от состава, имеют магнитные свойства, как говорится, на все вкусы. Здесь и магнитнотвёрдые сплавы, которые намагничиваются (и перемагничиваются) в сравнительно сильных полях и обладают высокой коэрцитивной силой (от лат. coercitio — удерживание; так называется величина, характеризующая способность ферромагнетика сохранять намагниченное состояние). К ним относятся сплавы железа с 14—25% Ni. Введение некоторых добавок улучшает их свойства (магнико — сплав железа с 14% Ni, 8% Al, 24% Со, 3% Си; алии — сплав железа с 25% Ni, 12% Al, 4% Си). Здесь и магнитно-мягкие сплавы, которые намагничиваются и перемагничиваются в относительно слабых магнитных полях. Такие сплавы содержат, кроме железа, от 40 до 80% Ni (и нередко некоторые добавки). Они характеризуются малой коэрцитивной силой и высокой магнитной проницаемостью; отсюда их название пермаллои (от англ, permeability — проницаемость, alloy — сплав). Такие сплавы широко применяются в технике слабых токов — телефонии, телеграфии, радиотехнике, телемеханике. А сплавы железа с 25—27% Ni и 5% Сг или железа с 18% Ni и 8% Сг принадлежат к немагнитным материалам.
Большую группу Н.с. составляют жаропрочные (способные выдерживать механические нагрузки при высоких температурах) и жаростойкие (противостоящие химическому разрушению при одновременном воздействии агрессивных газов и высокой температуры). Жаростойкость этих Н. с. объясняется образованием на их поверхности очень прочной окисной плёнки, защищающей сплав от дальнейшего окисления. Особо ценные свойства имеют нихромы — сплавы никеля с 10—30% хрома. Они жаропрочны, жаростойки, обладают высоким электрическим сопротивлением (то есть примерно в 10 раз большим, чем железо, никель, платина) и малым температурным коэффициентом сопротивления (0,0001—0,0002 на 1 °C; для
217
НИКЕЛЬ
чистых металлов он равен 0,004—0,006 на 1 °C). Рабочая температура этих сплавов в атмосфере воздуха 1000—1110 °C. Они применяются как материал для нагревательных элементов электрических печей. Но прокатка и волочение нихромов затруднительны; поэтому там, где возможно, их заменяют более пластичными ферронихромами — сплавами никеля с 15—30% Сг и 10—20% Fe; их рабочая температура около 900 °C.
Введение некоторых добавок в сплавы типа нихромов улучшает их жаропрочность и жаростойкость. Из таких сплавов на основе никеля назовём инконель (около 15% Сг, до 9% Fe, около 2,5% Ti и 0,9% Nb), н и-моник (20% Сг, 16% Со, 2,3% Ti, 1,9% Al) и хэсталлой (15% Сг, 3,5% Со, 5,0 % Мо, 7,0% Fe, 2,5% Ti, 2,3% Al). Эти (и подобные им Н. с.) служат для изготовления лопаток турбинных и реактивных авиационных двигателей, работающих при 900 °C и выше.
Число Н. с. достигает нескольких тысяч. Мы ограничимся приведенными примерами, но упомянем ещё только о монель-металле. Он назван по имени А. Монеля, президента Канадской международной никелевой компании, в лаборатории которой был разработан в 1905 г. Это «натуральный» сплав, получаемый непосредственно из сульфидных медно-никелевых руд. Кроме никеля, монель-металл содержит 27—29% Си, 2—3% Fe, 1—2% Мп. Он стоек в морской воде, щелочах, органических кислотах, обладает высокой пластичностью и прочностью. Широко применяется для изготовления химической заводской аппаратуры, в кораблестроении, электротехнике, электровакуумной промышленности и других производствах. Если бы никель не был так дефицитен, его сплавы применялись бы ещё шире. О других сплавах никеля и меди см. Медные сплавы. С. А. Погодин. НЙКЕЛЬ (лат. Niccolum) Ni — химический элемент VIII группы периодической системы Менделеева; атомный номер 28, атомная масса 58,70. Серебристо-белый металл с лёгким желтоватым оттенком. Природный Н. состоит из пяти стабильных изотопов — 68Ni, 60Ni, 61Ni, 62Ni и 64Ni, из которых наиболее распространён 68Ni (67,7%).
Название «никель» происходит от известного уже в XVII в. минерала купферникель (арсенид Н., NiAs), часто вводившего в заблуждение горняков внешним сходством с медными рудами. Первая часть слова «купферникель» — название меди, а вторая — имя докучливого «горного духа». Он не прочь позабавиться над горняками, подсунув им вместо меди «пустую породу». Слово «Н.» долго было у горняков бранным. Шведский химик А. Кронштедт, сумевший в 1751 г. впервые выплавить из этой руды небольшой слиток («королёк») металла, назвал его никелем. Открытие нового элемента оспаривалось. Считали, что Кронштедт получил сплав железа и кобальта с мышьяком и серой. Одной из причин сомнений было убеждение в том, что число металлов не может превышать числа планет (см. Металлы). Только в 1804 г., после того как немецкий химик И. Рихтер изучил свойства чистого Н., все признали его новым элементом. (Рихтеру, однако, пришлось 32 раза перекристаллизовать никелевый купорос NiSO4*7H2O, преж-
58,70 28
Ni
3d‘4s!
НИКЕЛЬ
де чем восстановить из него чистый металл.) Кронштедт, умерший в 1765 г., не дождался этого признания.
В земной коре содержится 5,8-10"3% Н. по массе (25-е место среди химических элементов; на26-мместе—медь). Кроме купферникеля NiAs (теперь называемого никелином), наиболее важны: миллерит — сульфид NiS; пентландит (сульфид, в котором Н. частично замещён железом); никелевый блеск, или герсдорфит, NiAsS (смешанный сульфид-арсенид Н.). Встречается в природе и оки
сел NiO (минерал бунзенит). В Советском Союзе месторождения Н. находятся на Урале, в Казахстане, на Сахалине и Кольском полуострове. Среди главнейших металлов современной техники Н. занимает одно из первых мест; его руды считаются промышленными при содержании 0,3% Ni и даже ниже.
Получение Н. из бедных руд — длительный процесс, связанный с их предварительным обогащением. Н. выделяют из руд в виде сульфида, который затем обжигают до окисла; а из окисла NiO восстанавливают металл в дуговых электрических печах. Готовый металл гранулируют выливанием в воду. Но это лишь черновой Н., требующий дальнейшей очистки. Чистый Н. получают электролизом.
Для этого в ванне, наполненной раствором сульфатов Н. и аммония, подвешивают аноды из чернового Н. и катоды из нержавеющей стали или алюминия. При прохождении тока Н. на анодах переходит в катионы Ni2+, которые разряжаются на катодах, где отлагается Н. чистотой до 99,99%. Примеси (As, Pb, Pt, Pd и др.) оседают на дне ванны и перерабатываются отдельно (см. Платиновые металлы).
Очистку Н. производят через очень своеобразное соединение — карбонил никеля Ni (СО)4. Это жидкость, кипящая при 43 °C, очень ядовитая. Её образование позволяет отделять Н. от других металлов (например, меди), которые подобных соединений не дают.
При карбонильном процессе над тонкораздробленным Н. при температуре 80—100 °C пропускают под давлением окись углерода СО; затем образовавшийся Ni(CO)4 разлагают, повышая температуру до 200 °C:
Ni + 4СО 8°~100-^ Ni(CO)4 —22 Ni + 4СО
Окись углерода возвращают в производство. В настоящее время разработаны методы получения Н. карбонильным методом непосредственно из некоторых руд.
Об английском химике и крупном промышленнике Л. Монде, открывшем в 1889 г. карбонил Н., говорили, что он дал крылья тяжёлому металлу. Действительно, Н. тяжелее железа: его плотность 8,9 г/см3. Плавится Н. при
НИЛЬСБОРИИ
218
1453 °C, кипит при ~ 3000 С. Чистый Н.— тягучий, ковкий, хорошо полирующийся металл. По магнитным свойствам Н., как и кобальт, напоминает железо, то есть относится к числу ферромагнетиков (см. Железо).
Конфигурация высших электронных уровней атома Ni 3d84s2 (см. Атом). В соединениях он обычно проявляет степень окисления --2 и только в небольшом числе соединений --3 (соответственно валентности II и III).
В ряду напряжений Н. стоит за железом и кобальтом. При обычных условиях компактный Н. очень стоек против действия воды и воздуха. (Тонкий порошок Н. пирофорен — самовозгорается на воздухе.) В декоративных целях и для защиты от коррозии изделия из менее стойких металлов и сплавов часто покрывают тонким слоем Н.— никелиру-ю т, электролитически осаждая Н. из раствора соли (NH4)2SO4-NiSO4-6Н2О.
Со всеми галогенами и с серой Н. легко соединяется при нагревании. Разбавленные соляная и серная кислоты растворяют Н. очень медленно; разбавленная азотная — легко (в концентрированной HNO3 Н. пассивируется и не растворяется). При взаимодействии с кислотами образуются соли двухвалентного Н.:
3Ni + 8HNO3 = 3Ni (NO3)2 + 2NO + 4Н2О
При упаривании растворов эти соли осаждаются в виде кристаллогидратов зелёного цвета (различных оттенков). Таковы нитрат Ni (КО3)2-6Н2О> хлорид NiCl2-6H2O, сульфат NiSO4-7H2O (никелевый купорос). В растворах и расплавах щелочей Н. устойчив. Зелёная гидроокись Ni(OH)2 осаждается при взаимодействии растворов щёлочи и соли Ni2+:
Ni (NO3)2 4- 2NaOH - Ni (OH)2 + 2NaNO3
Гидроокись Ni(OH)3 в виде чёрного осадка образуется при действии на Ni (ОН)2 сильных окислителей в присутствии щёлочи:
2Ni (ОН)2+ 2NaOH + Вг2 = 2Ni (ОН)3 + 2NaBr
Окись Ni (III) не получена, так как её гидрат отдаёт кислород уже при комнатной температуре, превращаясь в Ni3O4-2H2O. Закись NiO может быть получена в виде серо-зелёного порошка прокаливанием карбоната NiCO3, нитрата Ni (NO3)2-6H2O или гидроокиси Ni(OH)2.
Окисление Ni2+ до Ni3+ электрическим током использовал знаменитый американский изобретатель Т. Эдисон в 1897 г. для железно-никелевого аккумулятора, более лёгкого, чем обычный, свинцовый. В аккумуляторе Эдисона анодом служит гидроокись Н., катодом— железный спрессованный порошок, электролитом — раствор едкого кали. По реакции (стрелка вправо)
Разрядка
2Ni(OH)3 + Fe . * 2Ni(OH).> + Fe(OH)2
Зарядка
получают ток напряжением около 1,8 в. В современных аккумуляторах этого типа железо заменено кадмием.
О роли Н. в современной технике можно сказать очень много. Порошкообразный Н.— весьма распространённый катализатор, то есть ускоритель многих реакций. На изготовление катализаторов расходуется до 10% получаемого металла. Заманчива перспектива применения никелевого катализатора для окисления водорода — топлива энергетики будущего. Значительные количества Н. расходуются для щелочных аккумуляторов и антикоррозионных покрытий. Ковкий Н. в чистом виде идёт на изготовление листов, труб и т. д. Он служит материалом и для специальной химической аппаратуры. Главная же область применения Н., потребляющая 80% всей его добычи, это никелевые сплавы. В. И. Белова.
НИЛЬСБОРИЙ (лат. Nilsbohrium) Ns — название, предложенное советскими учёными для радиоактивного химического элемента V группы периодической системы Менделеева с атомным номером 105. Как и другие элементы, расположенные в конце периодической системы, Н. не существует в природе и может быть получен только искусственно — по ядерным реакциям.
Попытки синтезировать этот элемент были начаты в 1967 г. группой советских физиков в Объединённом институте ядерных исследований (г. Дубна), которую возглавил академик Г. Н. Флёров. Незадолго до этого, в 1964—66 гг., в Дубне был открыт элемент с атомным номером 104 — курчатовий Ku. В периодической системе элементов Ku — первый «трансактиноидный» элемент, расположенный вслед за группой очень похожих друг на друга элементов — семейством актиноидов. Трудности получения 105-го элемента очень напоминали те, что пришлось преодолеть при синтезе его предшественника — курчатовия. Вновь требовалось быстро отделить считанное число атомов нового элемента от милли
105 (Ns)
6d37s2
(НИЛЬСБОРИЙ)
ардов миллиардов атомов, составлявших мишень, которую использовали при проведении ядерной реакции. Затем нужно было за доли секунды изучить основные свойства новых атомов.
Методика, которую применили советские учёные для получения элемента № 105, очень похожа на ту, которая обеспечила им успех при получении курчатовия. Мишень, содержащую несколько миллиграммов изотопа америция 243Ат, облучали ускоренными на циклотроне ионами неона-22. В тех случаях, когда ядра 22Ne «достигали цели» и вступали в ядерную реакцию с ядрами 243Аш (отметим, что таких благоприятных событий было очень мало — всего несколько за многочасовой опыт), образующееся новое ядро вылетало из мишени и попадало на непрерывно движущуюся металлическую ленту (рисунок). Лента доставляла собранные ядра к специальным детекторам-пластинкам из особого фосфатного стекла.
219
НИОБИЙ
Схема установки для получения 105-го элемента. I —поток ядер неона 22Ne; 2 —мишень из америция Ат; 3 — стенка камеры; 4 — непрерывно движущаяся «бесконечная» металлическая лента; 5 — детекторы (пластинки из фосфатного стекла).
По случайному стечению обстоятельств число пластинок (105) соответствовало атомному номеру синтезируемого элемента. Учёные ожидали, что ядра 105-го элемента будут испытывать быстрый радиоактивный распад, и продукты распада оставят на поверхности стёкол следы — маленькие ямочки, которые после специальной обработки пластинок можно увидеть под микроскопом. По этим следам они и надеялись доказать факт образования элемента № 105.
Напряжённая работа, продолжавшаяся в Дубне до февраля 4970 г., принесла нашим физикам успех. Действительно, на стеклянных пластинках удалось обнаружить несколько сотен следов, свидетельствовавших о радиоактивном распаде ядер неизвестного ранее элемента. Было установлено, что ядерная реакция, приводящая к получению нового элемента, следующая:
2^Am + i,Ne = 261105 + 4jn
Период полураспада Ti/2 синтезированных ядер был найден равным 1,8 сек. Название новому элементу советские учёные предложили дать в честь выдающегося датского физика Нильса Бора. Затем в СССР и США были синтезированы и другие изотопы H.: 260Ns (Т*/2=1,6 сек) и 262Ns (Ti/2=40 сек). Отметим, что американские исследователи предложили для элемента № 105 другое название — «ганий» (в честь известного немецкого химика и физика Отто Гана).
Учёные Дубны выполнили и первое исследование химических свойств Н. Для этого была использована та же методика, которая применялась при изучении элемента № 104 (подробно она описана в статье Курчатовий). Оказалось, что высший хлорид Н. NsCl6 довольно легко летуч и по этому своему свойству очень похож на хлорид ниобия NbCl6.
Вероятно, в будущем физики и химики расскажут нам о свойствах Н. ещё много нового и интересного. С. С. Бердоносов.
НИОБИЙ (лат. Niobium) Nb — химический элемент V группы периодической системы Менделеева; атомный номер 41, атомная масса 92,9064. Металл светло-серого цвета, по внешнему виду похожий на платину. Элемент имеет один стабильный изотоп 93Nb.
Н. был открыт в 1801 г. английским химиком Ч. Гетчетом в минерале, найденном в Колумбии, и назван колумбием. Но когда вскоре (1802 г.) был открыт тантал, по свойствам очень близкий к колумбию, эти два элемента стали считать за один.
Индивидуальность колумбия была доказана лишь в 1844 г., когда он был вновь открыт немецким химиком Г. Розе и назван «ниобием» (в честь Ниобы — дочери мифического царя Тантала, чем и подчёркивалось родство свойств Nb и Та). Кстати, до сих пор в химической литературе некоторых стран (в особенности США) Н. часто называют колумбием (символ СЬ).
Н. относится к редким элементам. Среднее содержание его в земной коре 2-10"3% по массе, и по распространённости он занимает 31-е место среди других элементов (Та — 49-е). Вследствие близости физических и химических свойств элементы Nb и Та обычно встречаются вместе, образуя смешанные минералы. Наиболее распространены и важны для промышленности колумбиты и танталиты с общей формулой (Fe, Мп) (Nb, Та)2О6 и лопарит (Са, Се, Na) (Та, Nb)O3 (о том, как понимать такие сложные формулы, см. в статье Минералы).
Чистый Н. обладает высокой прочностью, пластичностью и теплопроводностью. Он легко поддаётся сварке и механической обработке. Плотность Н. 8,57 г/см3. Н.— один из тугоплавких металлов, он плавится только при 2500 °C, а кипит при 4927 °C. Н. служит ценной добавкой к различным сплавам, так как повышает их жаростойкость и устойчивость против коррозии.
В соответствии с положением Н. в периодической системе Менделеева его атом имеет пять внешних (валентных) электрона (конфигурация 4d45s\ см. Атом). В соединениях Н. проявляет чаще всего степень окисления +5, а кроме того, 4-4, 4-3 и 4-2 (валентности Н. соответственно V, IV, III и II). При обычных условиях Н., как и тантал, химически очень стоек. Он начинает окисляться на воздухе только около 200 °C, а с хлором, бромом, иодом, водородом и азотом реагирует при ещё более высоких температурах. И только фтор соединяется с Н. при комнатной температуре. Н. отличается также удивительной стойкостью к действию кислот. Даже царская водка бес
НИТРАТЫ
220
сильна против него. Только смесь фтористоводородной и азотной кислот способна растворить Н.:
3Nb+21HF+5HNO3=3H2NbF7+5NO+10H2O
Однако с расплавленными щелочами Н. реагирует легко и образует ниобаты — соли ниобиевой кислоты. Сама эта кислота — очень слабая, с едва заметными кислотными свойствами, поэтому нередко её называют даже гидроокисью Н. Состав ниобиевой кислоты соответствует формуле Nb2O5-xH2O; выше 400 °C она полностью отдаёт воду, превращаясь в пя-тиокись Nb2O5.
Для получения Н. ниобиево-танталовые концентраты подвергают сложной химической переработке, в результате которой выделяют смесь пятиокисей Н. и тантала. Вы уже знаете, что Nb и Та в природе сопутствуют друг другу и настолько сходны химически, что их долгое время принимали за один металл. Трудности разделения элементов-близнецов особенно велики. Их «обычные» соединения очень похожи друг на друга и надо отыскать соединения необычные (а зачастую и очень сложные), которые обладали бы несхожестью.
Для Nb и Та такие соединения впервые нашёл швейцарский химик Ж- Ш. Мариньяк. Предложенный им (в 1866 г.) способ разделения основывался на различии растворимости (в 10—12 раз!) KaTaF-? и K2NbOF6*H2O в разбавленной плавиковой кислоте. На смену этому способу пришли другие — более эффективные, но и более сложные (мы не будем их описывать).
Для выделения самого металлического Н. его пятиокись восстанавливают сажей в вакууме при высокой температуре:
Nb2O5 + 7С = 2NbC + 5СО
Затем смесь порошков карбида и пятиокиси Н. нагревают в вакууме до 1800—1900 °C:
Nb2O5 + 5NbC = 7Nb + 5СО
Порошок Н. сваривают в компактный металл методом порошковой металлургии (см. Вольфрам) .
Н. применяется в основном в виде сплава с железом — феррониобия (50—60% Nb). Его вводят в специальные стали, устойчивые к действию большинства кислот и других агрессивных сред. Сочетание жаростойкости с химической стойкостью позволяет использовать эти стали для изготовления химической и нефтеперегонной аппаратуры, деталей реактивных двигателей, ракет, газовых турбин.
Совершенно новые области применения Н.— атомная и электрорадиотехническая промышленность. Металлическим Н. и сплавами на его основе иногда покрывают стержни ядерного горючего. Такая оболочка позволяет значительно повысить рабочую температуру вну
три ядерного реактора и благодаря этому увеличить его мощность. А управляет этой сложной техникой электронная аппаратура, в которой не последнюю роль играют микроминиатюрные и ёмкие конденсаторы, изготовленные из Н. и его сплавов.
В статье о гелии мы рассказали об удивительном явлении — сверхпроводимости: ниже определённой температуры некоторые металлы полностью теряют электрическое сопротивление (см. Гелий). Эту температуру называют критической и обозначают Тс. Но у самых чистых сверхпроводящих металлов температура Тс лишь ненамного — на несколько градусов — выше абсолютного нуля. И это чрезвычайно затрудняет их применение. Чем выше Тс, тем лучше — здесь имеет значение каждый градус!
Н. замечателен тем, что некоторые его сплавы и соединения (например, соединение Nb3Sn, сплавы Nb—Zr, Nb—Al—Ge) имеют относительно высокую температуру перехода в сверхпроводящее состояние — Тс в пределах 18—21° К- (Заметим, что у соответствующих чистых металлов эта температура гораздо ниже: у Nb 9,2 °К; у Sn 3,73 °К; у Zr 0,55°К ) Когда же научились делать проволоку из этих сплавов (а это тоже было нелегко — они очень хрупки), стало возможно изготовлять соленоиды со сверхпроводящей обмоткой. Их называют сверхпроводящими магнитами. Магнитное поле внутри такого соленоида очень большое: 50—60 тыс. эрстед! И это далеко не предел — возможно удастся довести его до 300 тыс. эрстед!
Такие магниты портативны, легки и удобны для пользования (вся установка имеет размер школьной парты). Они совсем не похожи на обычные электромагниты, служащие для получения таких же полей, — весьма громоздкие и тяжёлые устройства. Сверхпроводящие магниты уже сейчас широко применяют в научных лабораториях. И не только магниты. Создано множество электронных устройств со сверхпроводящими элементами.
В 1973 г. было найдено рекордное по сверхпроводимости соединение — Nb3Ge. Его 7\.=23,2°К- Это на 3° выше точки кипения жидкого водорода! А ведь водорода на Земле неизмеримо больше, чем гелия. И конечно же, внедрять сверхпроводники, охлаждаемые жидким водородом, много проще и выгоднее, чем те, которые необходимо помещать в жидкий гелий. Правда, пока ещё из Nb3Ge не умеют делать проволоку. Но шаг в будущее сверхпроводимости сделан. И сделан — благодаря ниобию.
НИТРАТЫ (азотнокислые сол и)— соли азотной кислоты HNO3. Н. образуются при взаимодействии металлов, их окисей, гидроокисей и карбонатов с азотной кислотой, например:
3Cu + 8HNO3 = 3Cu(NO3)2 + 2NO + 4Н2О
NaOH + HNO3 = NaNO3 + Н2О
Н.— кристаллические вещества, хорошо растворимые в воде. Н. лития LiNO& и большинство Н. двухвалентных металлов при осаждении из насыщенных водных растворов образуют кристаллогидраты, а на воздухе, поглощая влагу, расплываются. Н. одновалентных металлов (К, Rb, Cs, Ag), а также Т1, Ва и РЬ негигроскопичны и не образуют кристаллогидратов.
221
НИТРИТЫ
При нагревании выше температуры плавления Н. разлагаются. Н. активных металлов (К, Na, Са, Ва) превращаются в соответствующие нитриты. Н. менее активных металлов (Mg, Pb, Fe, Zn, Си) образуют окислы этих металлов, а неактивных (Ag, Au) — свободные металлы. Но все они при разложении выделяют кислород:
2KNO3 = 2KNO2 + O2
2Pb (NO3)2 = 2РЬО + 4NO2 + О2 2AgNO3 = 2Ag + 2NO2 + О2 Поэтому, например, тлеющий кусочек угля вспыхивает ярким пламенем, если его бросить в расплавленный KNO3. Не так давно Н. калия служил для приготовления чёрного пороха (см. Селитры).
Месторождения Н. встречаются редко. Основную же массу весьма необходимых Н. получают на заводах. Наибольшее применение имеют Н. щелочных металлов и аммония, используемые в сельском хозяйстве в качестве азотных удобрений.
НИТРИДЫ — соединения азота с другими, более электроположительными элементами, главным образом с металлами. Получают Н. двумя путями — прямым синтезом (обычно при нагревании):
3Mg+ N2 = Mg3N2
и взаимодействием металла с аммиаком (обычно также при нагревании):
2А1 + 2NH3 = 2A1N + ЗН2
По строению и свойствам Н. можно разделить на следующие группы:
а) Н. щелочных и щелочноземельных металлов. Это типичные соли, в которых между элементами действуют ионные связи. Такие соединения можно рассматривать как производные аммиака, в молекуле которого атомы водорода замещены металлом. Водой эти Н. легко разлагаются с образованием аммиака:
Mg3N2 + 6Н2О = 3Mg(OH)2 + 2NH3
б) Н. металлов III группы периодической системы Менделеева и Н. неметаллов. Здесь связи между атомами элементов ковалентные. Эти Н. (в особенности AIN, BN, Si3N4) исключительно устойчивы к химическим воздействиям, тугоплавки и очень термостойки, то есть способны выдержать частые и резкие смены температур — быстрое нагревание и охлаждение. Поэтому их применяют для приготовления жаропрочных сплавов и для термоизоляции. Особенно интересен Н. бора BN. Он существует в трёх кристаллических модификациях (а, Р и у) и у каждой из них — свои особенности. Так, a-BN напоминает графит: оба вещества
имеют «слоистую» кристаллическую структуру, оба мягки и применяются как «твёрдые смазки» (подробнее см. Бор, Углерод). Благодаря сходству с графитом и белому цвету a-BN называют белым графитом. В ядерной технике a-BN служит надёжной защитой от нейтронного излучения. Другая модификация, P-BN (боразон, или кубонит), имеет алмазоподобную структуру и по твёрдости и другим свойствам подобна алмазу. Боразон производят в больших количествах и наряду с искусственным алмазом используют для шлифования и резания различных материалов — стекла, камня, металла. Модификация y-BN изучена ещё сравнительно мало.
в) К третьей, наиболее многочисленной, группе относятся Н. переходных металлов (см. Металлы). Их строение и свойства определяются тем, что атомы азота «проникают» внутрь кристаллической решётки металла; формулы этих Н. часто не отвечают формальным правилам валентности (например, W2N, Ti3N, Mn4N). Такие Н. во многом подобны металлам — имеют высокие электропроводность и теплопроводность, тугоплавки (например, TiN плавится при 3200 °C, а HfN —при 3380 °C). Поэтому их называют металлоподобными Н: Однако они отличаются от металлов очень высокой твёрдостью и хрупкостью. ‘ Металлоподобные Н. жаропрочны и химически инертны.
Н. образуются на поверхности металлов под действием азота при высоких температурах (500—600°С); этот процесс называется азотированием. Так получают нитридные покрытия, придающие металлическим изделиям твёрдость, износостойкость и коррозионную стойкость. Такие изделия применяются в технике высоких температур, газоту рбиностроении, энергетике. Г. В. Самсонов.
fl НИТРЙТЫ (азотистокислые соли) — соли азотистой кислоты HNO2. Бесцветные кристаллические вещества, хорошо растворимые в воде (за исключением AgNO2). Наибольшее значение имеют Н. щелочных металлов, которые могут быть получены взаимодействием щелочей с азотистым ангидридом N2O3 или с эквимолекулярной смесью окиси и двуокиси азота:
2NaOH + N2O3 = 2NaNO2 4г Н2О
2NaOH 4- NO 4- NO2 = 2NaNO2 + H2O
H. щелочных металлов получаются также при восстановлении или термическом разложении нитратов:
NaNO3 4- Pb = NaNO2 4- PbO 2NaNO3 = 2NaNO2 4- O2
НОБЕЛИЙ
222
При подкислении растворов Н. выделяется слабая азотистая кислота:
NaNO2 + НС1 = NaCl + HNO2
Н. содержат азот в степени окисления +3; поэтому они могут быть как окислителями (восстанавливаясь большей частью до NO), так и восстановителями (окисляясь до нитратов). Главная область применения Н.— промышленность органических красителей. В ней Н. натрия служит для реакции диазотирования ароматических аминов — важной промежуточной ступени в производстве азокрасителей. NaNO2 используется в медицине как сосудорасширяющее средство (в небольших дозах).
НОБЕЛИЙ (лат. Nobelium) No — название, предложенное для радиоактивного химического элемента с атомным номером 102 из семейства актиноидов. Поскольку название и символ элемента № 102 окончательно не утверждены, в периодической системе Менделеева их помещают в скобках.
НОМЕНКЛАТУРА НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ — система рациональных названий сложных неорганических веществ. Лат. слово nomenclatura переводится как перечень имён, названий.
Немного истории. Вплоть до коренного преобразования химии в конце XVIII в. веществам давались произвольные, случайные наименования. Легко летучие жидкости назывались спиртами; к ним относились «соляной спирт» (соляная кислота, то есть водный раствор НС1), «селитряный спирт» (азотная кислота HNO3), «нашатырный спирт» (водный раствор аммиака NH3). Маслообразные жидкие и твёрдые вещества назывались маслами: «купоросное масло» (концентрированная серная кислота H2SO4), «мышьяковое масло» [хлорид мышьяка (III) AsCl3], «сурьмяное масло» [хлорид сурьмы (III) SbCl3]. Нередко вещества получали названия по именам первооткрывателей, как «глауберова соль» (десятиводный сульфат натрия Na2SO4« 10Н2О), открытая немецким химиком XVII в. И. Р. Глаубером. Иногда — по местностям, где вещество было впервые найдено. Примером служит эпсомская соль (семиводный сульфат магния MgSO4«7H2O), обнаруженная в конце XVII в. при выпаривании минеральной воды источника близ местечка Эпсом в Англии.
В старинных названиях порой были указания на вкус веществ, их цвет, запах, внешний вид, целебное действие и др. Ацетат свинца Pb (QHgC^-ЗН2О назывался свинцовым сахаром по своему сладкому вкусу, водные сульфаты железа (II), меди (II) и цинка (FeSO4*7H2O, CuSO4«5H2O, ZnSO4«7H2O) именовались купоросами—зелёным, синим и белым. Под загадочными словами «философская шерсть» скрывалась окись цинка ZnO, получаемая в виде белых хлопьев при сжигании паров цинка. Такие названия, как «Сильвиева противолихорадочная соль» (хлорид калия КС1) или «Гомбергова успокоительная соль» (борная кислота Н3ВО3), указывали на медицинское действие соединений, истинное или мнимое. Наконец, в алхимических трактатах постоянно встречались такие названия, как «серый волк», «красный лев», «зелёный лев», «лилейная невеста», «красный жених», которые с большим трудом поддаются расшифровке.
К концу XVIII в. химикам было известно не более 150—200 соединений, для которых существовали тысячи названий. Дело в том, что одно и то же вещество обычно имело несколько названий (иногда 10 и более). А вместе с тем одним и тем же наименованием обозначались различные вещества. Например, «белый купорос» означал и сульфат цинка (водный и безводный), и бесцветные безводные сульфаты меди и железа.
Попытки положить конец хаотическому состоянию химической номенклатуры делались уже начиная с середины XVIII в. Но они не имели успеха, так как химия того времени была в плену ложных представлений гипотезы флогистона. Только после её ниспровержения трудами знаменитого французского учёного А. Л. Лавуазье и создания им кислородной теории горения (см. Кислород) стала возможной разработка рациональной химической номенклатуры. Это было осуществлено весной 1787 г. комиссией французских химиков под председательством Лавуазье.
Комиссия постановил а, чт о каждое сложное вещество может иметь только одно название. Это название должно отражать состав соединения, быть удобнопроизносимым и не противоречить духу языка. Название соединения составляется из двух слов; одно из них указывает род соединения, другое — его вид.
Например, для соединений металлов с кислородом было принято родовое название оксид (по-русски окись); прибавляя к нему наименования металлов, составляли видовые названия, например оксид цинка, оксид ртути. Соединения неметаллов с кислородом Лавуазье считал кислотами (этот ошибочный взгляд просуществовал до 1860-х гг.), например СО2 — угольная кислота, SO3 — серная кислота, SO2 — сернистая кислота. Родовые названия солей производились от корней латинских названий кислот с добавлением суффиксов -а т (для высшей степени окисления) и -и т (для низшей), например с у л ь ф а ты — соли серной и сульфиты — соли сернистой кислоты. Названия солей составлялись из наименований кислоты (родового) и основания (видового), например карбонат извести СаСО3, сульфат окиси меди CuSO4.
Химическая номенклатура Лавуазье послужила основой для создания национальных химических номенклатур, в частности русской — её основателями были В. М. Севергин (начиная с 1801 г.) и Г. И. Гесс с сотрудниками (1831—1835 гг.). В то время, в соответствии с воззрениями Лавуазье и Берцелиуса, соли считались соединениями оснований (основных окислов) с кислотами (ангидридами кислот). Например, согласно Гессу карбонату кальция СаСО3 придавали формулу СаС, СО2 (между формулами составных частей соли ставили запятую) и название «углекислая известь». В 1861 г. Д. И. Менделеев предложил называть соли прилагательными, составленными из наименований кислоты и металла, например, углекальциевая соль СаСО3, серножелезистая соль FeSO4. (Менделеев и другие учёные того времени рассматривали соли как производные кислот, в которых водород замещён металлом). Наряду с менделеевской номенклатурой для солей применялись и названия, образованные по типу прежних, но в них вместо наименования основания указывался металл (углекислый кальций СаСО3, сернокислый натрий Na2SO4). Номенклатура Д. И. Менделеева была общепринята (с некоторыми поправками) вплоть до начала 1930-х гг., когда большинство отечественных химиков высказалось в пользу так называемых международных названий, заимствованных из западно-европейских языков.
О номенклатуре на современном этапе. Приведём систему названий, принятую в нашей отечественной литературе и, в частности, в
223
НОМЕНКЛАТУРА
Антуан Лоран ЛАВУАЗЬЕ. Положил начало новому периоду развития химии — веку количественных исследований.
Французский химик Антуан Лоран Лавуазье родился 26 августа 1743 г. в Париже. Окончил юридический факультет Парижского университета. В студенческие годы увлечённо изучал естественные науки, особенно физику и химию. В 1766 г. за изыскание наилучшего способа освещения улиц большого города Лавуазье получил от Парижской академии наук золотую медаль. С 1772 г. он — действительный член Парижской академии наук, а в 1785 г.— её директор. По поручению Академии участвовал в разработке многих важных для того времени проблем — воздухоплавания, общественной гигиены и других. В 1775— 1791 гг. был директором Управления порохов и селитр.
В 1768—1791 гг. Лавуазье был членом «Компании откупов» — организации финансистов, которая брала на откуп государственные налоги, то есть вносила ежегодно в казну их сумму, а затем с избытком взимала её с населения; разница же поступала в карман откупщиков. Эта система была предметом справедливой ненависти народа, и во время Великой французской революции откупщики были казнены на гильотине. 8 мая 1794 г. в Париже был казнён и Лавуазье. Знаменитый математик Ж. Лагранж сказал: «Потребовалось лишь одно мгновение, чтобы отрубить эту голову, но может быть и столетия будет мало, чтобы создать подобную ей». В 1796 г. Лавуазье был посмертно реабилитирован.
С начала XVIII в. и вплоть до работ Лавуазье в химии господствовала гипотеза флогистона. По учению немецкого химика Г. Э. Шталя, флогистон — это материальное начало горючести (по-гречески «флогистос» — горючий, воспламеняемый). Он содержится во всех горючих веществах, а также в металлах. При горении дерева, масла и т. п. флогистон выделяется в виде пламени, остаётся же «земля» (зола). Точно так же неблагородные металлы — свинец, олово, медь, железо — при обжигании теряют флогистон и превращаются в порошкообразные вещества — «земли», «окалины», «извести» (по-современному окислы). Итак, учил Шталь, горючие вещества и металлы состоят из флогистона и земли.
Лавуазье первым оценил величайшее событие в химии тех лет — открытие кислорода. Последовательно применяя взвешивание веществ до и после реакции, он показал, что при процессах горения и при обжигании металлов происходит не их разложение на «флогистон» и «землю», а соединение с кислородом. Так Лавуазье, по словам Ф. Энгельса, «...впервые поставил на ноги всю химию, которая в своей флогистонной форме стояла на голове». Посредством точных опытов он доказал сложность состава воздуха и воды, которые до того считались «стихиями», то есть элементами.
В 1786—1787 гг. по инициативе Лавуазье и под его руководством была создана первая рациональная химическая номенклатура, основанная на кислородной теории. Руководствуясь определением простого вещества как такого, которое ещё не разложено на более простые, Лавуазье конкретизировал это понятие. Он составил первый список реальных простых веществ, в который включил все известные тогда металлы и неметаллы. В список вошли и «земли» (окислы кальция, стронция, бария, магния, алюминия и кремния, то есть соответственно известь, стронциан, барит, магнезия, глинозём и кремнезём), но с оговоркой, что они могут оказаться сложными (это подтвердилось в начале XIX в.).
В 1789 г. Лавуазье опубликовал свой классический труд «Начальный учебник химии, изложенной в новом порядке и согласно современным открытиям». Автор определил химию, как науку о составе веществ, об их анализе. В химии началась новая эпоха, названная веком количественных исследований. Направление Лавуазье привело к открытию множества новых веществ, простых и сложных, позволило установить законы постоянных и кратных отношений. А это, в свою очередь, дало экспериментальное обоснование химической атомистике, начатой Дж. Дальтоном и разработанной Я. Берцелиусом.
Воззрения Лавуазье получили всеобщее признание в конце XVIII — начале XIX вв. Лишь очень немногие крупные учёные (среди них Г. Кавендиш и Дж. Пристли) оставались до конца жизни приверженцами гипотезы флогистона. В России распространению новых воззрений активно содействовали академики Я. Д. Захаров, В. В. Петров, В. М. Севергин и А. И. Шерер.
Все свои исследования Лавуазье вёл в собственной лаборатории, которую он, будучи очень богатым человеком, оборудовал точнейшими физическими приборами-весами, термометрами, барометрами, а также аппаратами своей конструкции. Учёный был превосходным экспериментатором и тонким наблюдателем. Его заветы: «стремиться делать хорошо, а не делать много», «упрощать опыты насколько возможно», «никогда не восполнять поспешными заключениями молчания фактов» — могут и в наши дни служить руководящими указаниями для исследователей.
НОМЕНКЛАТУРА
224
этой книге. Основой Н. н. с. служат русские и латинские названия химических элементов. При составлении названий соединений применяются корни латинских названий элементов, отличающиеся от русских (см. таблицу 1).
Таблица 1
Антимон- antimonium — сурьма
Аргент- argentum — серебро
Арсен- arsenicum — мышьяк
Аур- aurum — золото
Гидр- hydrogen i urn — водород
Карб-, карбон- carboneum — углерод
Купр- cuprum — медь
Манган- manganum — марганец
Меркур- mercurius — ртуть
Нитр- nitrogenium — азот
Оке- oxygenium — кислород
Плюмб- plumbum — свинец
Силик- silicium — кремний
Станн- stannum — олово
Стиб- stibium — сурьма
Сульф- sulphur — сера
Тио- греч. тэйон — сера
Ферр- ferrum — железо
А теперь нам снова придётся отвлечься и вернуться к истории — правда уже недавней. К середине XX в. в русской химической литературе— учебной, научной, справочной — одновременно употребляется различная Н. н. с., но ни одна не охватывает всего их многообразия. Кроме того, каждая из применяемых систем имеет свои частные недостатки, греша в основном непоследовательностью. Приблизительно такое же положение сложилось и в иностранной химической литературе.
В целях упорядочения Н. н. с. в международном масштабе Номенклатурная комиссия Международного союза чистой и прикладной химии (ШРАС) в 1959 г. разработала принципы номенклатуры неорганической химии. Ниже мы будем именовать эти правила «МН» (международная номенклатура). В нашей стране наряду с названиями по МН сохраняется и система названий по русской номенклатуре («PH»). В одних случаях — когда побеждают традиции и сам строй русского языка — всё ещё доминирует PH. А благодаря всё более и более расширяющимся международным научным связям, благодаря распространению переводных изданий вступает во всё большие права МН.
А скоро ли закончится этот «переходный» период и мы введём в отечественную литературу единую Н. н. с.? Над этим работают сейчас номенклатурные комиссии и авторы учебников.
Названия неорганических соединений (по международной и русской номенклатурам — МН И PH).
Названия радикалов (атомных групп, переходящих без изменений из одного соединения в другое) составляются из корней латинских названий элементов и суффикса -и л (от греческого слова «хюле»— вещество), например гидроксил ОН, карбонил СО. Термины «аммоний» NH4, «циан» CN, «родан» CNS, «амид» NH2 сохраняются как в русской, так и в международной номенклатурах (PH и МН).
В названиях соединений, состоящих из атомов двух элементов с ионной или полярной ковалентной связью, на первом месте ставится наименование более электроотрицательной части. По МН оно составляется из корня латинского названия элемента и суффикса -и д (от греческого суффикса «-идее», означающего отчество в собственных именах). На втором месте ставится название более электроположительного элемента (в родительном падеже). Его степень окисления указывают двумя способами — либо римской цифрой в скобках (что предпочтительнее), либо приставками, заимствованными из греческих количественных числительных: геми-(полу-), моно- (1), ди- (2), три- (3), тетра- (4), пента- (5), гекса- (6), гепта- (7), окто- (8), эн-неа- (9), дека- (10).
Примеры по МН: NaCl — хлорид натрия; FeCl2 — хлорид железа (II), дихлорид железа; FeCl3— хлорид железа (III), трихлорид железа; Cu2S — сульфид меди (I), гемисульфид меди; CuS — сульфид меди (II), моносульфид меди.
По PH вместо суффикса -и д применяются суффиксы -истый для указания либо единственной, либо низшей степени окисления и -н ы й (иногда -о в ы й, -е в ы й) для высшей степени окисления. Степень окисления обозначают также русскими числительными или же римскими цифрами в скобках. Примеры по PH: NaCl — хлористый натрий; FeCl2— хлористое железо, двухлористое железо, хлористое железо (II); FeCl3— хлорное железо, трёххлористое железо, хлористое железо (III).
Соединения элементов с кислородом, в которых он связан только с более электроположительными атомами, называются по МН о к-сидами, по PH — ок и ела ми, причём в порядке возрастания степени окисления элемента им либо дают названия закись, окись, двуокись, трёхокись, либо указывают римской цифрой степень окисления. Высшие окислы, которые можно получить отнятием воды от кислот, называют ангидридами (МН отменяет этот термин). В таблице 2 сопоставлены названия окислов хрома по МН и PH. Соединения, в которых атомы кислорода связаны и друг с другом, и с атомами электроположительного элемента, называются по МН пероксидами, а по PH —перекисями [пе-
225
НОМЕНКЛАТУРА
Таблица 2
Формула Международная номенклатура (МН) Русская номенклатура (PH)
СгО Оксид хрома (II), моноксид хрома Закись хрома, окись хрома (II)
Сг2О3 Оксид хрома (III), дихром триоксид,гемитри-оксид хрома, сесквиок-сид * хрома Окись хрома, окись хрома (III), полуто-раокись хрома
СгО3 .Оксид хрома (VI), триоксид хрома Хромовый ангидрид, окись хрома (VI), трёхокись хрома
* От лат. sesqui — полтора.
роксид (перекись) водорода Н—О—О—Н, пе-/°
роксид (перекись) бария Ва(^.
Соединения неметаллов с водородом, водные растворы которых имеют характер кислот, называют, сочетая корень русского названия элемента или прилагательного от него со словом «водород» (НО — хлороводород, хлористый водород; H2S — сероводород, сернистый водород). Для прочих простейших соединений неметаллов с водородом сохраняются традиционные (исторические) термины: вода Н2О, аммиак NH3, фосфин РН3, арсин AsH3, метан СН4, силан SiH4, боран ВН3. Соединения водорода с металлами называются гидридами (по МН), например гидрид лития LiH, гидрид (дигидрид) кальция СаН2, иногда — водородистыми металлами (PH).
Основания называются по МН гидроксидами, по PH — гидроокисями. Если металл образует свыше одного основания, указывают степень окисления металла либо римской цифрой в скобках, либо греческой приставкой.
Примеры: Fe(OH)2—гидроксид железа (II), дигидроксид железа; Fe(OH)3— гидроксид железа (III), тригидроксид железа.
Термины «гидрат закиси», «гидрат окиси» выходят из употребления. Для родового обозначения растворимых в воде (сильных) оснований традиционно сохраняется термин «щёлочи».
Для кислот применяется только PH. Бескислородные кислоты называют по соответствующим водородистым соединениям с прибавлением слова «кислота» (бромистоводородная кислота НВг, сернистоводородная кислота H2S). Однако часто пользуются и историческими названиями, такими как плавиковая кислота для HF, соляная — для НС1, синильная — для HCN.
Названия кислородных кислот составляются из корня русского названия кислотообразующе
го элемента и суффиксов -на я, -овая (-евая) (для высшей степени окисления), -истая, -о вата я, -оватистая (для низших степеней окисления в убывающем порядке).
Примеры: НС1О4—хлорная кислота, НС1О3— хлорноватая..., НС1О2— хлористая..., НС1О — хлорноватистая кислота.
Чтобы отличать кислоты, образованные элементом одной и той же степени окисления, но содержащие различное число молекул воды, служат приставки орто- (греч. «ортбс» — ис^ тинный, правильный) для кислоты с наибольшим содержанием воды и мета- (греч. предлог после)—с наименьшим.
Примеры: Н3РО4—ортофосфорная кислота, НРО3— метафосфорная кислота (в последние годы их нередко называют «фосфорная ортокислота» и «фосфорная метакислота»).
Кислоты, содержащие свыше одного атома кислотообразующего элемента в одной и той же степени окисления, называются изополикислотами; число атомов этого элемента указывают русской (иногда греч.) приставкой.
Примеры: H2S2O7 — двусерная кислота, Н2Сг3О10— трихромовая кислота, Н2В4О7— четырёхборная (тетраборная) кислота. Название «пирокислота» выходит из употребления.
В отличие от названий кислот, для наименования их анионов пользуются только* МН (попытки создать PH анионов успеха не имели). Названия анионов кислородных кислот составляют из корня латинского наименования
В школьном химическом кабинете. В школе вы проводите первые химические опыты. И в школе впервые постигаете язык химии — учитесь правильно произносить названия химических соединений, читать и записывать химические формулы и уравнения.
15 хэш
НОМЕНКЛАТУРА
226
кислотообразующего элемента и суффиксов -ат и -и т (соответственно для высшей и низшей степени окисления, например SO*’ — сульфат-ион, SO|” — сульфит-ион).
Если число кислот, образуемых элементом в различных степенях окисления, больше двух, то название аниона (по МН), в котором эта степень наивысшая, имеет суффикс -ат и приставку пер- (лат. приставка per- означает избыток, усиление). Следующие степени окисления в убывающем порядке указываются суффиксами -ат, -ит и, наконец, -ит с приставкой г и п о- (греч. приставка, означающая ослабление качества).
Примеры: С1О4— перхлорат -ион, С1О"^—хло-рат-ион, CIOjt — хлорит-ион, CIO” — гипо-хлорит-ион.
В названия анионов орто- и метакислот включают эти приставки (POJ“ — ортофосфатной, РО7 — метафосфат-ион). Названия анионов кислых солей имеют приставку гидро-(HSOf — гидросульфат-ион, Н2РО4“ — дигид-роортофосфат-ион).
Названия всех солей (по МН) составляют из названий их анионов и катионов (K2SO4—
сульфат калия, K2SO3— сульфит калия,
NaHCO3— гидрокарбонат натрия).
Степень окисления катиона указывают (по МН) римскими цифрами в скобках: FeSO4— сульфат железа (II), Fe2(SO4)3— сульфат железа (III).
Но часто, по сложившейся традиции, названия таких солей образуют от самих кислородных кислот (KNO3— азотнокислый калий, K2SO4— сернокислый калий, K2SO3— сернистокислый калий, К2СО3— углекислый калий). Нередко встречаются нерекомендуемые названия солей, такие как бисульфат (надо гидросульфат) натрия NaHSO4, бихромат (надо дихромат) калия К2Сг2О7, метабисульфит (надо дисульфит) калия K2S2O5, а также старинные наименования: квасцы, купоросы, селитры.
При пользовании МН следует помнить слова А. М. Бутлерова (1859 г.) о русской химической номенклатуре: «Большей частью своей массы она сольётся с общей химической номенклатурой, а русские названия, выработавшиеся в обыденном языке, как были, так и останутся в употреблении у русских химиков».
С. А. Погодин.
«Чтобы приобрести знания..., необходимо соединить между собой точное и определённое понятие с определённым названием. Пренебрежение этим приведёт к тому, что всё множество вещей нас подавит и всякий обмен сведениями прекратится из-за отсутствия общего языка». „ v
Карл Линней (1763 г.)
(Линней К., цитируется по книге «Жизнь науки», 4973, с. 274)
«...Имея ясную теорию, мы всегда можем составить ясную номенклатуру и терминологию: когда этого нет, то наша терминология не пособит учению, а только затруднит его».
Д. И. Менделеев (1857 г.)
(Соч., т. 15, 1949, с. 161)
ОЗОН — аллотропное видоизменение кислорода. В отличие от двухатомной молекулы обычного кислорода (О2), молекула О. трёхатом-на (О3).
Впервые О. был обнаружен в 1785 г. голландским физиком М. ван-Марумом: оказалось, что воздух после пропускания через него электрических искр приобретает характерный запах и способность окислять ртуть при комнатной температуре.
В 1840 г. швейцарский химик X. Ф. Шёнбейн подробно изучил свойства газа, образующегося в этих условиях, и дал ему название «озон» (от греч. глагола «озо» — пахну).
В электрическом разряде О. образуется из обычного кислорода по реакции:
ЗО2 + 285 кдж 2О3
Реакция обратима, образование О. идёт с поглощением теплоты (энергии).
О.— более сильный окислитель, чем «обыкновенный» кислород. О. окисляет почти все металлы кроме Au, Pt, 1г. При действии О. на Ag образуется чёрный оксид AgO:
Ag + О3 = AgO + О,
Многие органические вещества в присутствии О. воспламеняются. Обнаружить присутствие О. в газовой смеси можно (как нашёл Шёнбейн) по характерной реакции — выделению иода из нейтрального раствора иодида калия:
Оз + 2KI + Н2О = I2 + О2 + 2КОН
Свободный поддаёт с крахмальным клейстером синее окрашивание (см. Иод). «Обыкновенный» кислород в реакцию с KI не вступает.
Все замечали, что после сильной грозы воздух особенно свеж и бодрящ. Отчасти это связано с образованием О. при электрических разрядах. Ничтожные его количества всегда содержатся в атмосфере. Но если бы собрать даже весь атмосферный О., он создал бы у поверхности Земли слой толщиной всего в 3—5 мм (а обычный кислород атмосферы дал бы слой в 5,5 км). И всё-таки О. имеет для жизни на Земле немалое значение.
Главную часть энергии солнечного излучения несут световые волны длиной от 1* 10_б до 3-10-4 см. Наиболее короткие из них, от !• 10-6 до 4*10"?, это ультрафиоле
товые лучи. Если бы весь их поток дошёл до Земли, то живые организмы были обречены на смертельные ожоги и гибель. Однако самые энергичные и опасные — так называемые жёсткие — ультрафиолетовые лучи, встречаясь на большой высоте с молекулами О2, вызывают превращение их в О3, расходуя на это свою энергию. Так губительная для жизни часть солнечного излучения задерживается на дальних подступах к Земле. В атмосфере над ночной половиной Земли содержание О. уменьшается, и лишь утреннее Солнце восстанавливает равновесие.
Подражая природе, люди получают О. действием электрического разряда на газообразный кислород (рисунок). При обычной температуре такой способ даёт смесь, содержащую лишь несколько процентов О3.
При охлаждении О3 сжижается при—111,9 °C, а О2 при— 182,9 °C, что позволяет их разделить. Однако получить О.— это только полдела. Не менее трудная задача — сохранить его.
Озонатор. К электродам (а) и (б) подводят высокое напряжение — так, что в пространстве между внутренним и внешним стеклянными сосудами возникает так называемый тихий электрический разряд (то есть разряд без свечения и искр). Проходя через область электрического разряда, кислород О2 частично (на несколько процентов) превращается в озон О3.
15*
ОЛЕУМ
228
Молекула О8 неустойчива и самопроизвольно превращается в О2 с выделением тепла. Скорость этого превращения бывает различной. Контакт О. с ничтожными количествами органических веществ, серы или некоторых металлов настолько ускоряет превращение, что в течение долей секунды происходит взрыв. Наоборот, присутствие небольших количеств азотной кислоты стабилизирует О., а в сосудах из стекла, некоторых пластмасс или чистых металлов газообразный О. при —78 °C практически не разлагается.
Решение проблемы получения и хранения О. высоких концентраций открывает широкие перспективы его применения. Благодаря сильным окислительным свойствам О. можно использовать в производстве многих органических веществ, при получении нитрата аммония из аммиака и т. д. О. убивает микроорганизмы, поэтому его применяют для очистки воды и воздуха. Однако в воздухе допустимы очень малые концентрации О.; при большем содержании он чрезвычайно ядовит. В. Л. Василевский, ОЛЕУМ —100%-ная серная кислота H2SO4, в которой растворён избыток серного ангидрида SO3. Тяжёлая маслянистая жидкость (отсюда название, по-латыни oleum — масло) или (в зависимости от количества растворённого SO3) кристаллическое вещество. Применяется в производстве красителей, взрывчатых веществ и др. (см. также Серная кислота), ОЛОВО (лат. Stannum) Sn — химический элемент IV группы периодической системы Менделеева; атомный номер 50, атомная масса 118,69. Серебристо-белый металл. О.— один из немногих элементов, имеющих большое число стабильных изотопов. Их девять: 112Sn, 114Sn — 120Sn, 122Sn. Наиболее распространён 120Sn, на его долю в природной смеси приходится около 33%. Встре
чается в природе и слаборадиоактивный изотоп 124Sn с периодом полураспада более 1016 лет.
О. принадлежит к металлам, давно известным человечеству. Более чем за 4000 лет до н. э. умели изготовлять сплавы О. с медью — бронзы. Лёгкость выплавки О. из его руд была установлена ещё древними египтянами. Знакомо О. было и финикийцам и карфагенянам, которые изготовляли из него водопроводные трубы. Происхождение названия этого элемента окончательно не установлено. Предполагают, что латинское слово «станнум» происходит от санскритского «ста», что означает прочный, стойкий. А русское слово «олово» связано с материалом сосудов, в которых хранилась «оливина», «ол» (древнеславянские названия пива, браги, мёда).
Рис. 1. Игрушечный оловянный сервиз (2-я половина XIX в.).
Когда-то из олова, послушного легкоплавкого металла, делали игрушки — красивые, изящные, разнообразные. Оловянные солдатики и оловянная посуда были любимыми детскими играми.
По распространённости в земной коре О. стоит всего лишь на 47-м месте (2,5*10“4% по массе). (Для сравнения отметим, что элемент той же группы — свинец занимает 35-е место, а следующее за О. 48-е место занимает уран.) В самородном состоянии металл в заметных количествах не встречается и О. находят главным образом в виде минерала касситерита SnO2 (оловянного камня). Второй по значимости минерал — станнин (оловянный колчедан) Си 2FeSnS4.
Главнейшими поставщиками О. на мировом рынке являются Боливия, Индонезия, Малайзия, Нигерия, Заир, Китай. В нашей стране крупные запасы руд этого металла есть в Восточной Сибири, Казахстане и на Дальнем Востоке.
О.— серебристо-белый блестящий металл, медленно тускнеющий на воздухе. Плавится О. при 231,9 °C, а кипит при 2270 °C. О. мягко и пластично, хорошо куётся и легко прокатывается в тонкие листы.
Если сгибать палочку чистого О., приложив один из её концов к уху, то можно услышать характерный треск, обусловленный трением отдельных кристаллов О. друг о друга. Это явление получило название «крик О.». Интересно, что явление «крика» при сгибании наблюдается ещё у одного металла — кадмия, и тоже только очень чистого. Вот, пожалуй, два известных случая, когда качественный химический анализ можно проводить... на слух.
О. имеет три аллотропные модификации. Наиболее устойчиво так называемое белое О., или P-Sn, с плотностью 7,29 г/см3. При температуре ниже 13,2 °C стабильна другая модификация— a-Sn, или «серое О.» с плотностью 5,85 г/см3. Превращение белого О. в серое (и
229
ОЛОВО
обратный процесс) начинается при 13,2 °C, но протекает с бесконечно малой скоростью. При дальнейшем понижении температуры скорость превращения возрастает и при —33 СС достигает максимума. Поэтому при устойчивых сильных холодах в разных местах оловянных предметов начинается быстрый переход белого О. в серое. Удельный объём при этом переходе резко увеличивается (на 25,6%), и металл превращается в серый порошок. Такой переход считается своеобразной «болезнью» О. и называется оловянной чумой. Интересно, что переход 0->а ускоряется при появлении на белом О. пылинок серого (центров кристаллизации), то есть эта болезнь (как и настоящая чума) «заразна».
Любопытно, что если 0-Sn — типичный металл, то a-Sn — полупроводник. Третья аллотропная модификация — «хрупкое О.» (y-Sn) устойчива только от 161 °C до точки плавления.
Известно много случаев, когда оловянная чума приводила к весьма неприятным последствиям. На морозе металл покрывается серыми пятнами и рассыпается. Так, в 1912 г. из-за разрушения паянных оловом сосудов с жидким топливом погибла экспедиция Р. Скотта к Южному полюсу. Во время отступления наполеоновских солдат из Москвы в 1812 г. оловянные пуговицы на шинелях превратились в серый порошок. В отчёте Русского музея (в Ленинграде) за 1922 год можно прочитать об эпидемии оловянной чумы. «Болезнь» повредила старинные изделия из О., которые хранились в этом музее, не отапливавшемся в годы Гражданской войны. Поэтому чтобы предохранить от разрушения оловянные и паянные оловом предметы, их следует хранить в отапливаемых помещениях. Кроме того, полезно вводить в О. небольшие добавки свинца и висмута, которые замедляют скорость превращения.
У атома О. четыре внешних (валентных) электрона (конфигурация 5s2 5р2, см. Атом). В соединениях О. имеет степени окисления 4-2 и 4 4 (соответственно валентности II и IV), последняя более характерна. При обычных условиях О. устойчиво на воздухе, так как оно покрыто тонкой защитной плёнкой окисла. При продувании струи кислорода над расплавленным металлом О. горит, образуя наиболее устойчивый окисел — двуокись SnO2. Окись SnO можно получить лишь косвенным путём; уже на воздухе она легко окисляется до SnO2. Легко взаимодействует О. с галогенами, образуя галогениды состава SnX4 и SnX2. Примером служит взаимодействие металла с сухим хлором:
Sn 4- 2С12 = SnCl 4
Образующаяся бесцветная жидкость — четырёххлористое О. (тетрахлорид О.) — хороший растворитель для серы, фосфора, иода. Раньше сухим хлором удаляли О. с вышедших из строя лужёных изделий. Сейчас этот способ
мало распространён из-за его вредности и непроизводительности. При нагревании О. с серой и хлоридом аммония получают золотисто-жёлтые кристаллы SnS2 — дисульфида О. Это вещество, известное под названием «с у-сальное золото», входит в состав красок для «золочения» различных изделий — деревянных, гипсовых и др. (Сусальным золотом часто называют и «золотую бить», см. Золото.)
С водородом, азотом, углеродом и кремнием О. непосредственно не реагирует, но соединения этих элементов с О. известны — их получают косвенными путями.
О. устойчиво и по отношению к воде. В ряду напряжений О. располагается до водорода, но почти рядом с ним; поэтому О. лишь медленно растворяется в сильных кислотах:
Sn 4-4НС1 = SnCl4 4-2Н2
Горячую серную кислоту О. восстанавливает до SO2:
Sn 4- 4H2SO4 = Sn (SO4)2 4- 2SO2 4- 4H2O
Разбавленной азотной кислотой О. окисляется до Sn2+:
3Sn 4- 8HNO3 = 3Sn (NO3)2 4- 2NO 4- 4H2O а концентрированной — до оловянной кислоты (её соли называются станнатами):
Sn 4- 4HNO3 = H2SnO3 4- 4NO2 4- Н2О
При реакции О. с царской водкой образуется тетрахлорид О.:
3Sn4-4HNO34' 12НС1 = 3SnCl4 4- 4NO + 8Н2О
Рис. 2. Игрушечный домик с оловянными фигурками. Дамы и рыцари в костюмах XVI в.
осмии
230
Рис. 3. Олово А. Е. Ферсман назвал «металлом консервной банки». Консервную тару изготовляют из белой (лужёной) жести. Это тонкая листовая сталь, покрытая слоем олова.
О. растворяется не только в кислотах, но и в щелочах (при нагревании). В этом случае выделяется водород и образуются соли оло-вянистой кислоты (станниты):
Sn 4- 2КОН - K2SnO2 + Н2
Этим свойством О. пользуются для снятия его с негодных консервных банок.
Двуокись О. проявляет преимущественно кислотные свойства, а у окиси сильно выражены основные. Характерной чертой соединений Sn(-F2) является их сильная восстановительная способность, что часто используется в химическом анализе. Особенно сильны восстановительные свойства О. в щелочной среде. Так, станниты в этих условиях восстанавливают Bi3+ до металла:
3Na2SnO2 + 2Bi (NO3)3 + 6NaOH = 2Bi + + 3Na2SnO3 + 6NaNO3 + 3H2O
Получение О. в принципе довольно просто. Руду (обычно касситерит) сначала обогащают, а затем концентрат нагревают в печи с углём, чтобы восстановить двуокись О. до металла:
SnO2 + 2С - Sn + 2СО
Загрязнённый металл можно очистить либо окислением примесей, либо электролизом. Од
нако половину всей добычи О. составляет так называемый вторичный металл; его получают из отходов белой жести, лома и сплавов, в том числе из негодных консервных банок.
Благодаря устойчивости О. в воздухе, воде, слабых органических кислотах, а также сравнительной дешевизне им покрывают другие металлы для защиты их от коррозии (лужение). О. можно с полным правом назвать «металлом консервной банки». На изготовление белой жести (листовое железо, покрытое О.), из которой делают такие банки, идёт 40% всего добываемого в мире О.
Из остальных областей применения О. назовём лишь самые главные. Чистое О. легко прокатывается в тонкие листы — станиоль, используемые в электрических конденсаторах.
Очень важны многочисленные сплавы О. Это и бронзы (они описаны в статье Медные сплавы), и сплавы для подшипников — баббиты, и типографские сплавы (см. о них также в статье Сурьма). Это и многочисленные припои — мягкие, легкоплавкие сплавы, хорошо смачивающие поверхности большинства металлов (см. Висмут, Кадмий).
Двуокись О. служит составной частью жаростойких эмалей и глазурей; в виде мельчайшего порошка применяется для полировки стекла. Красильщикам хорошо известно так называемое протравное крашение, при котором на ткани предварительно осаждаются вещества, прочно удерживаемые волокнами ткани и хорошо адсорбирующие краски. К таким веществам относится гидрат двуокиси О. х SnO2- у Н2О.
Мы уже упоминали о том, что соединение О. с водородом можно получить косвенным путём. К сожалению, бывают случаи, когда станнометан SnH4 (сильный яд) образуется в старых, залежавшихся консервах. (Оловянное покрытие взаимодействует с органическими кислотами, входящими в состав пищевого продукта.) Образование SnH4 — одна из причин случающегося иногда отравления консервами. В. К. Бельский.
ОРТОФОСФОРНАЯ КИСЛОТА Н3РО4—одна из фосфорных кислот.
ОСМИЙ (лат. Osmium) Os — химический элемент VIII группы периодической системы Менделеева; атомный номер 76; относится к семейству платиновых металлов.
ПАЛЛАДИИ (лат. Palladium) Pd — химический элемент VIII группы периодической системы Менделеева; атомный номер 46; относится к семейству платиновых металлов.
ПЕРМАНГАНАТЫ —соли марганцовой кислоты НМпО4. Хорошо растворимы в воде П. натрия и щелочноземельных металлов, растворимость П. калия, рубидия и цезия значительно меньше. Растворы П. окрашены в фиолетово-малиновый цвет, присущий иону МпО^. П.— одни из самых сильных окислителей. В зависимости от среды раствора (кислая, + 7
нейтральная или щелочная) Мп восстанавливается до разных степеней окисления. В кислой среде продуктом восстановления П.
2 +
являются соли катиона Мп, в нейтральной среде, как правило, получается двуокись марганца МпО2, а в щелочной — соли марганцовистой кислоты Н2МпО4. Приведём примеры взаимодействия П. калия с сульфитом калия в растворах с кислой (а), нейтральной (б) и щелочной (в) средами:
2КМпО4 + 5K+2SO3 + 3H2SO4 = + 2 +6
= 2MnSO4 + 6K2SO4 + 3H2O (a)
+ 7 +4
2KMnO4 + 3K2SO3 + H2O = + 4 +6
= 2МпО2 + 3 K2SO4 + 2 КОН (б)
+ 7 +4
2КМпО4+ K2SO3 + 2КОН = + 6 +6
= 2К2МпО4 + K2SO4 + Н2О (в)
При нагревании П. разлагаются с выделением кислорода и поэтому иногда применяются в лабораториях для его получения:
2КМпО4= К2МпО4+МпО2+О2
П.— важные химические соединения. П. калия, в быту называемый «марганцовкой», используется как дезинфицирующее средство — для полоскания горла при простудах, для приёма внутрь при отравлениях (в очень слабой концентрации). В. К- Бельский.
ПЕРХЛОРАТЫ — соли хлорной кислоты НС1О4. Как правило, П. хорошо растворимы
в воде. При нагревании разлагаются с выделением кислорода, например:
КС1О4 = КС1 + 2О2
В промышленности П. получают электролизом растворов хлоратов. П. калия и аммония применяют в производстве взрывчатых веществ. П. магния Mg(C104)2 (ангидрон) — отличное водоотнимающее средство. По осушающей способности он уступает только фосфорному ангидриду. но выгодно отличается от Р2О5 тем, что может быть использован многократно. Стоит прогреть ангидрон в вакууме, как его осушающая способность восстанавливается.
ПИРОФ0СФОРНАЯ КИСЛОТА Н4Р2О7 — одна из фосфорных кислот.
ПЛАВИКОВАЯ КИСЛОТА (фтористоводородная кислота) — раствор фтористого водорода HF в воде.
ПЛАТИНА (лат. Platinum) Pt — химический элемент VIII группы периодической системы Менделеева; атомный номер 78; относится к семейству платиновых металлов.
ПЛАТИНОВЫЕ МЕТАЛЛЫ — химические элементы: рутений Ru, родий Rh, палладий Pd (лёгкие П. м.), осмий Os, иридий 1г, платина Pt (тяжёлые П. м.), принадлежащие ко второй и третьей триадам VIII группы периодической системы Менделеева. П. м., наряду с серебром Ag и золотом Au, называют благо-
44 Ru 101,07 Ad^s1 РУТЕНИЙ 45 Rh 102,9055 , .8С 1 Аа 5 s РОДИЙ 46 Pd 106,4 4d,05s° ПАЛЛАДИЙ
76 Os 190,2 5d66s2 ОСМИЙ 77 Ir 192,22 5d76s2 ИРИДИЙ 78 Pt 195,09 5d96sl ПЛАТИНА
ПЛАТИНОВЫЕ МЕТАЛЛЫ
232
родными металлами благодаря их высокой химической стойкости, тугоплавкости и красивому внешнему виду.
Распространение в природе и добыча. П. м. относятся к наименее распространённым в природе элементам. Земная кора содержит (в % по массе) 5-Ю"7 Ru, 1 • 10“7 Rh, 1,3- 10“e Pd, 5-10“e Os, 1-Ю"7 1г, 5-Ю"7 Pt. Вследствие своей ничтожной химической активности П. м. чаще всего встречаются в самородном состоянии — обычно в виде сплавов, как между собой, так и с другими металлами. В состав так называемой самородной платины входят, главным образом, минералы ферроплатина (77—81% Pt, 20—14% Fe) и поликсен (80— 92% Pt, 10—6% Fe), кроме того, эти минералы содержат прочие П. м., а также Си и иногда Ni. Все эти металлы образуют с Pt твёрдые растворы. (О том, что такое твёрдые растворы, рассказано . в статье Алюминиевые сплавы.) В них нередко наблюдаются включения осми-стого иридия (твёрдого раствора иридия в осмии); последний встречается и как самостоятельный минерал. Кроме 1г и Os, он содержит Pt, Ru, Rh, Си, Fe. Остальные самородные П. м.— аваит (ок. 77% 1г, 20% Pt), порперцит (8—24% Pd, 75—90% Au), родит (34—43% Rh, 45% Au) и другие — очень редки. Весьма редки и природные химические соединения, образуемые П. м. с серой и мышьяком. В небольших количествах они содержатся в сульфидных полиметаллических рудах (в частности, медно-никелевых). В самородном золоте обычно присутствуют незначительные примеси П. м. (чаще всего Pt и Pd), находящиеся в виде твёрдых растворов.
Существуют первичные — коренные месторождения П. м., и россыпные, образующиеся при разрушении (выветривании) коренных. Некоторые коренные месторождения платины представляют собой громадные массивы дунита, содержащего мелкие зёрна самородной платины. Дунит (назван так по горе Дун в Новой Зеландии) — это горная порода, состоящая из силикатов железа и магния с примеськ! хромистого железняка FeCr2O4 и других тяжёлых минералов. Под действием воздуха, влаги и колебаний температуры дунит превращается в сыпучее тело *— песок. Он переносится водой и постепенно накапливается в оврагах, долинах, на дне рек и ручьёв, образуя россыпные месторождения.
Долгое время россыпные месторождения служили единственным источником добычи Pt и других П. м. Платиноносные пески промывают проточной водой на специальных сооружениях — драгах. Вода уносит лёгкие частицы пустой породы, а самородная платина и со
путствующие ей тяжёлые минералы остаются в виде чёрных шлихов. Их собирают, и на небольших ручных устройствах (вашгердах) отделяют тяжёлые минералы; в результате получают шлиховую платину.
В наши дни главным источником получения П. м. служат сульфидные медно-никеле-вые руды (см. Медь, Никель). Их месторождения находятся в Советском Союзе (Норильский рудный район в Красноярском крае), Канаде (округ Садбери, провинция Онтарио) и других странах.
История открытия. Самородная платина известна издавна. Имеются указания на то, что она служила для изготовления изделий в древних Египте, Эфиопии, Греции и Южной Америке. В XVI в. самородная платина была вторично «открыта» испанскими колонизаторами, грабившими и опустошавшими Южную Америку. Вместе с самородным золотом там встречался очень тяжёлый, белый, тусклый металл, который никак не удавалось расплавить. Испанцы назвали его «платиной» — уменьшительным именем от испанского plata — серебро. В 1744 г. испанский морской офицер Антонио де Ульоа привёз образцы платины в Лондон. Новый металл вызвал живой интерес учёных Европы. Уже в середине XVIII в. платина, которую первоначально считали «белым золотом», была признана самостоятельным металлом.
В 1803 г. английский физик и химик У. X. Волластон обнаружил в самородной платине палладий, получивший это название от малой планеты Паллады (открытой в 1802 г.), и родий, названный так по розово-красному цвету его солей (от греч. «рбдон» — роза). В 1804 г. английский химик Смитсон Теннант, исследуя чёрный порошок, остающийся после растворения самородной платины в царской водке, открыл в нём ещё два металла. Один из них получил название «иридий» вследствие разнообразия окраски его солей (от греч. «ириоэйдёс» — радужный), другой был назван «осмием» по резкому запаху его четырёхокиси (от греч. «осмё» — запах). Наконец, в 1844 г. профессор Казанского университета К- К- Клаус при исследовании остатков от аффинажа (очистки) уральской самородной платины в Петербургском монетном дворе открыл ещё один П. м.— рутений (от позднелатинского Ruthenia — Россия).
По воспоминаниям А. М. Бутлерова, который учился у Клауса, он «имел привычку ... при растворении платиновых руд в царской водке мешать жидкость прямо всеми пятью пальцами и определял крепость непрореагировавших кислот на вкус». Особенно удивляться этому не приходится. Химики старой школы всегда пробовали на вкус все получаемые ими вещества. Вплоть до середины XIX в. указание вкуса было одним из обязательных признаков при описании свойств веществ. По-
Русские платиновые монеты второй четверти XIX в*
233
ПЛАТИНОВЫЕ МЕТАЛЛЫ
Карл Карлович КЛАУС — исследователь платиновых металлов. Открыл рутений.
Русский химик Карл Карлович Клаус родился 22 января 1796 г. в Дерпте (ныне Тарту). Сын художника, он рано потерял родителей и с 14 лет начал трудовую жизнь. Сперва в Дерпте Карл был помощником пекаря, а затем служил в аптеках разных городов, пока не завёл собственную аптеку в Казани (1826 г.). В 1831 г. Клаус поступил на должность «инспектора химического кабинета» Дерптского университета. Одновременно он прошёл университетский курс и вскоре (1837 г.) получил степень магистра философии. Снова переехав в Казань, Клаус с 1839 г. стал профессором Казанского университета по кафедре химии.
Здесь в 1841 г. он занялся исследованием остатков от аффинажа уральской платины. Осенью 1844 г. учёный открыл в них новый металл, который назвал рутением. Сообщив об этом Петербургской академии наук, Клаус послал образцы препаратов рутения в Стокгольм знаменитому шведскому химику Я. Берцелиусу. На первых порах «законодатель химии» заявил, что ему присланы нечистые соединения иридия, но вскоре признал свою ошибку. Поздравляя Клауса с открытием нового элемента, Берцелиус писал: «Ваше имя будет неизгладимо начертано в истории химии». Продолжая изучение платиновых металлов, Клаус сделал множество наблюдений, сохранивших своё значение вплоть до наших дней. В этом можно убедиться, познакомившись с его монографиями — «Химическое исследование остатков уральской платиновой руды и металла рутения» и «Материалы к химии платиновых металлов».
В 1852 г. Клаус занял кафедру фармации в Дерптском университете, где продолжал исследовать платиновые металлы. Клаус впервые обратил внимание на различия реакций платиновых металлов, взятых отдельно, от металлов, находящихся в смеси друг с другом. Он же подметил аналогию химических свойств рутения и осмия, родия и иридия, палладия и платины.
Карл Карлович был крупным специалистом по ботанике. Он неоднократно участвовал в ботанических экскурсиях, описал флору Поволжья. По воспоминаниям современников, учёный то целыми днями безвыходно работал в лаборатории (причём пробовал на вкус все растворы), то неделями просиживал над своими гербариями. А кроме того, Клаус был одарённым художником; ему принадлежат превосходные иллюстрации к описаниям научных экскурсий, в которых он участвовал. В музее при химической лаборатории Казанского университета хранятся приготовленные Клаусом соединения рутения и других платиновых металлов. К. К. Клаус был членом-корреспондентом Петербургской академии наук. Он умер 24 марта 1864 г. в Дерпте.
разительнее всего то, что такие «дегустации» оставались без вредных последствий (исключение составляет шведский химик К. В. Шееле, который, как передают, погиб при попытке определить вкус полученной им безводной синильной кислоты).
Физические и химические свойства. Свойства отдельных П. м. сопоставлены в таблице. П. м. принадлежат к переходным металлам периодической системЬг Менделеева (см. Металлы). Бросается в глаза отсутствие правильности заполнения оболочек в атомах П. м.— и по горизонтали, и по вертикали таблицы (для наиболее сходных между собой пар элементов: Ru — Os, Rh — Ir, Pd — Pt). Это вызвано тем, что с возрастанием заряда ядра и накоплением числа электронов закономерности заполнения внешних электронных оболочек усложняются; сложнее становятся и закономерности в изменений валентности (подробнее см. Закон Менделеева).
Часто недоумевают: почему у палладия — элемента 5-го периода — отсутствуют электроны на 5-й электронной оболочке? Pd представляет собой единственное исключение из правила — общее число электронных оболочек у него равно не номеру периода (как это бывает обычно), а на единицу меньше. Как же это объяснить?
Для всех переходных металлов (и для П. м. в том числе) характерно заполнение электронами d-уровня предпоследней (считая от ядра) оболочки. Максимально
на этом уровне может находиться 10 электронов (см. Атом), причём целиком заполненный d-уровень обладает повышенной устойчивостью. При переходе от Rh к Pd новый электрон должен занять девятое место на 4d-yровне. Для полного же заполнения этого уровня нужен ещё только один электрон. И для атома Pd оказывается энергетически выгодным, чтобы s-электрон 5-й оболочки, имевшийся в атомах Ru и Rh, перескочил на d-уровень 4-й оболочки и занял там последнее свободное место (электронная конфигурация 4d10). В результате общее число оболочек у атома оказывается равным четырём.
Интересно, что такого перескока s-электрона не наблюдается у аналога палладия — платины. Из-за большей удалённости от ядра Sd-уровня (по сравнению с 4d-ypoBHeM), а также по ряду других причин у атома Pt электронная конфигурация Sd^s1 оказывается энергетически более выгодной, чем 5d10.
В компактном виде П. м. (кроме Os) химически неактивны. Однако в мелкодисперсном состоянии, в виде так называемой черни (её получают, восстанавливая П. м. из водных растворов их соединений), П. м. при нагревании легко реагируют с кислородом, серой, галогенами и другими неметаллами, а также с многими металлами. Наиболее легко соединяется с кислородом Os; на воздухе постепенно (а при нагревании очень быстро) образуется высший окисел OsO4, весьма летучий и очень ядовитый (как и его аналог RuO4). При на
ПЛАТИНОВЫЕ МЕТАЛЛЫ
234
гревании на воздухе Ru и Pd до красного каления образуются низшие окислы, соответственно RuO2 и PdO, а в токе О2 или под действием сильных окислителей — высшие окислы RuO4 и PdO2. Высшие окислы — сильные окислители: так, спирт при соприкосновении с RuO4 воспламеняется. При ярко-красном калении Rh на воздухе переходит в Rh2O3, а 1г — в 1гО2; оба окисла при дальнейшем нагревании разлагаются на кислород и металл.
Весьма интересна способность некоторых П. м. (Ru, Pd, Pt) поглощать водород. Особенно это свойственно Pd, 1 объём которого поглощает до 900 объёмов водорода. При этом Pd сохраняет металлический вид, но увеличивается в объёме, растрескивается и становится хрупким.
При нагревании все П. м. реагируют с галогенами, образуя галогениды. Наиболее высокие степени окисления П. м. проявляют во фторидах. Так, для Os получены OsF8, OsFe, OsF5, OsF4, тогда как в соединениях с другими галогенами Os имеет валентности II, III, IV.
Действие кислот на отдельные П. м. различно. Ru и Os не растворимы ни в чистых кислотах, ни в их смесях. Rh и 1г устойчивы к действию чистых кислот, а в царской водке (смесь соляной и азотной кислот), будучи сплавлены с Pt, Zn, Си, они растворяются очень медленно. Pd легко растворим в царской водке, а
также в азотной и горячей серной кислотах: 3Pd + 8HNO3 = 3Pd (NO3)2 + 2NO + 4Н2О
Pd + 2H2SO4 = PdSO4 + SO2 + 2H2O
Ha Pt индивидуальные кислоты не действуют, но при слабом нагревании она растворяется в царской водке с образованием платинохлористоводородной кислоты, в которой Pt четырёхвалентна:
Pt + 6НС1 -г 2HNO3 = Н2 [PtClJ + NO + + NO2+3H2O
Платинохлористоводородная кислота H2[PtCl6] принадлежит к числу координационных, или комплексных, соединений. В формулах таких соединений сложный (комплексный) ион заключают в квадратные скобки. (О комплексных соединениях рассказано, например, в статье Кобальт.)
Платинохлористоводородная кислота выпадает из растворов в виде красно-коричневых кристаллов состава H2[PtCle]-6H2O. Из её солей наибольшее значение для технологии имеет хлороплатинат аммония (NH4)2 [PtCle] — светло-жёлтые кристаллы, мало растворимые в воде. При прокаливании они разлагаются:
2NH4CI
(NH4)2[PtCle] = Pt + 2С12 + 2NH3 + 2HCi
При этом платина остаётся в мелкораздробленном виде («платиновая губка»).
Свойства платиновых металлов
Свойство Рутений (Ruthenium) Ru Родий (Rhodium) Rh Палладий (Palladium) Pd Осмий (Osmium) Os Иридий (Iridium) 1г Платина (Platinum) Pt
Атомный номер . . . 44 45 46 76 77 78
Атомная масса . . . 101,07 102,9055 106,4 190,2 192,22 195,09
Массовые числа при- 96,98,99,100 103 102, 104, 105, 184, 186, 187, 191 (38,5%), 193 194(32,9%), 195
родных изотопов * Конфигурация внеш- 101, 102(31, 61%), 104 106 (27,33%), 108 (26,71%), НО 188, 189, 190, 192 (41,0%) (61,5%) (33,8 0/0), 196, 198; 190 [T1/t= = 9*10хх лет], 192 [7\л=10Ч лет]
них электронных
уровней 4<Г55Х 4d85sx 4d105s° . 5d66s2 5d76s2 5d96sx
Наиболее характерные степени окис-
ления 3, 4, 6, 8 3 2, 3 4, 6, 8 3, 4 2, 4
Цвет Серовато-белый Серебристоголубой Серовато-белый Серовато-белый Серебристо-белый Серовато-белый
Плотность, г/см3 . . 12,2 12,42 11,97 22,5 22,4 21,45
Температура плавле-
ния. °C 2250 1960 1552 3050 2410 1769
Температура кипе-
ния, °C 4900 4500 3980 5500 5300 4530
Пластичность .... Хрупок Ковок при 800—900 °C Ковок и тягуч Хрупок Хрупок Ковка и тягуча
* В круглых скобках указаны процентные содержания наиболее распространённых изотопов; в квадратных — периоды полураспада [7\/ ] слаборадиоактивных природных изотопов платины.
235
ПЛАТИНОВЫЕ МЕТАЛЛЫ
Пётр Григорьевич СОБОЛЕВСКИЙ. Разработал оригинальный способ получения ковкой платины.
Русский металлург и химик Пётр Григорьевич Соболевский родился 15 февраля 1782 г. в Петербурге. Отец его, доктор медицины Г. Ф. Соболевский, был известен как ботаник (в 1799 г. он опубликовал «Петербургскую флору» на латинском языке). Пётр Григорьевич окончил Сухопутный кадетский корпус и недолгое время был офицером (с 1798 по 1804 г.). Молодой военный с большим интересом и рвением изучал естественные и технические науки. В 1811 г. в газетах появилось сообщение об изобретённой им «термолампе» — печи, в которой древесина перерабатывалась на светильный газ, дёготь, «пригорело-уксусную кислоту» (смесь воды с метиловым спиртом, ацетоном и уксусной кислотой) и древесный уголь.
С 1815 г. Соболевский работал инженером на уральских заводах. Здесь он ввёл разные технические усовершенствования (и в частности, передел чугуна в железо пудлингованием). Интересно, что первые русские пароходы, курсировавшие по Каме и Волге с 1817 г., были сооружены по проекту П. Г. Соболевского. А в 1824 г. ему поручили составить проект, а затем и руководить постройкой Соединённой лаборатории Горного кадетского корпуса (ныне Ленинградский горный институт им. Г. В. Плеханова) и Горной аптеки. В 1826 г. «обербергпробирер» Соболевский был назначен управляющим лабораторией. Здесь он разработал оригинальный способ получения ковкой платины, описанный в статье «Платиновые металлы». Спустя 30 с лишним лет — после того, как в 1859 г. стала возможной плавка больших масс платины в кислородноводородном пламени,— изобретение Соболевского применять перестали. Только в 1910 г. оно возродилось в виде порошковой металлургии (о ней рассказано в статье «Вольфрам»), П. Г. Соболевский справедливо считается основоположником этого метода, ныне широко применяемого в новейшей технике. Соболевский был членом-корреспондентом Петербургской академии наук. Он умер 5 ноября 1841 г. в Петербурге. На фото — бюст Соболевского (музей Горного института в Ленинграде).
Получение. Сырьём для получения чистых П. м. служат: 1) самородная и шлиховая платина; 2) осмистый иридий; 3) платиновый лом, то есть непригодные для употребления изделия из Pt; 4) анодный шлам (от немецкого Schlamm — грязь, ил), то есть илообразные осадки, накопляющиеся на дне ванн, в которых производится электролитическая очистка меди и никеля, полученных из сульфидных руд. Разделение П. м. и получение их в чистом виде — аффинаж — очень сложное и трудное дело из-за большого сходства их химических свойств. В течение последних 150 лет было предложено множество способов переработки сырой платины. Все они требуют большой затраты труда, времени и дорогих реактивов, а также высокого мастерства.
Для получения чистой Pt самородную платину, а также платиновые шлихи и лом обрабатывают царской водкой при нагревании. В раствор переходят Pt, Pd, частично Rh и 1г — эти П. м. образуют комплексные соединения H2[PtCl6], H2[PdClJ, Н3|RhCl6] и Н2[1гС16], а также Fe и Си, в виде FeCl3 и СиС12. Нерастворимый в царской водке остаток состоит из осмистого иридия, хромистого железняка, кварца и других минералов.
Из раствора хлористым аммонием осаждают Pt в виде (NH4)2[PtCl6]. Перед этим проводят одну очень важную операцию. Нельзя допустить, чтобы в осадок вместе с Pt выпал 1г, дающий аналогичное нерастворимое соединение (NH4)2[ 1гС16] (остальные П. м. хлористым аммонием не осаждаются). Осаждения 1г избегают прибавлением, например, раствора сахара (способ советского химика академика И. И. Черняева); сахар восстанав-+ 4 +3
ливает 1г до 1г. А соединение (NH4)3[ 1гС1б] растворимо и «не загрязняет» осадка.
Хлороплатинат аммония отфильтровывают, промывают, высушивают и прокаливают. Полученную губчатую платину спрессовывают, а затем расплавляют в кислородно-водородном пламени или в электрической печи, используя ток высокой частоты. Из фильтрата от хлороплатината аммония и из осмистого иридия извлекают остальные пять П. м. путём сложных химических операций.
Как сказано выше, в настоящее время главным источником получения П. м. служат сульфидные медно-никелевые руды. В результате сложной металлургической переработки этих руд П. м. переходят в так называемые черновые металлы — нечистые никель и медь. При последующем электролитическом рафинировании благородные металлы (в том числе П. м.) осаждаются на дне электролитической ванны в виде шлама, который по мере накопления отправляют на аффинаж (аффинаж ведут по приведенной выше схеме).
Применение. Первым практическим применением самородной платины после её открытия оказалась ... подделка золота. Пользуясь тем, что платина довольно легко сплавляется с золотом и почти не влияет на его плотность, некоторые «предприимчивые» люди начали добавлять платину к золоту в количествах, мало изменяющих цвет золота. Чтобы прекратить такое злоупотребление, испанское правительство воспретило добычу и продажу платины и приказало утопить все её запасы. Однако в Англии и Франции велись научные исследования платины и изыскание способов изготовления изделий из неё. Главным препятствием была тугоплавкость платины; удавалось расплавить лишь ничтожные количества её теплом солнечных лучей, сосредоточенных в фокусе огромной двояковыпуклой линзы.
Чтобы расплавить большие массы платины, пришлось -пойти окольным путём. В конце XVIII в. парижский
ПЛАТИНОВЫЕ МЕТАЛЛЫ
236
ювелир Жаннетти пред, ожил сплавлять платину с мышьяком, а затем сильным прокаливанием на воздухе выжигать мышьяк из полученных слитков. После удаления мышьяка слитки подвергались горячей ковке.
Между тем в России произошли события, которые позволили создать первое промышленное изготовление изделий из платины. В 1819—1822 гг. на Урале в золотоносных песках был обнаружен очень тяжёлый серовато-белый металл, который, как показал в 1823 г. В. В. Любарский, оказался самородной платиной. В 1824 г. на Урале было добыто около 33 кг самородной платины (примерно вдвое больше, чем в Южной Америке), а в 1825 г.— уже около 181 кг. К этому времени из-за бездарности царских министров состояние денежных ресурсов. России оставляло желать много лучшего. Очередной министр финансов Е. Ф. Канкрин решил поправить положение выпуском платиновой монеты. Почему так? Потому что Pt считалась гораздо менее блаюродным и более дешёвым металлом, нежели золото (отсюда и названия самородной Pt на Урале — «лягушачье золото», «серебрёц»).
Однако нет худа без добра: при разработке технологии производства этих «дешёвых» денег П. Г. Соболевский и В. В. Любарский создали новый способ получения Pt. Он заключается в том, что губчатую платину, набитую в цилиндрические железные формы, сильно сдавливают на холоду винтовым прессом; полученные цилиндры нагревают до белого каления в течение 36 часов, после чего проковывают в полосы или прутки Уже к концу 1826 г. этим способом было превращено в ковкое состояние около 1590 кг уральской платины. Для сравнения заметим, что в Южной Америке с 1741 по 1825 гг. было добыто всего около 1150 кг платины. Новую методику получения платины использовали без изменений в производстве платиновой монеты, начатом в 1828 г. Выпускались монеты достоинством в 3, 6 и 12 руб. Однако в 1845 г. царское правительство решило прекратить выпуск платиновой монеты, а в 1862 г. продало за бесценок одной английской фирме все накопившиеся на монетном дворе запасы платины и платиновых остатков.
Между тем спрос и цены на платину стали расти, особенно в конце XIX в., когда получил распространение контактный способ производства серной кислоты. Но владельцы уральских платиновых приисков, которые давали до 95% мировой добычи платины, не использовали этой благоприятной конъюнктуры. Вместо того чтобы возобновить аффинаж самородной платины, они стали продавать её за границу. Мало того, сами платиновые прииски постепенно перешли в руки иностранных фирм; разработка месторождений велась хищнически. Только в 1914 г., в начале первой мировой войны, был запрещён вывоз сырой платины, а в 1915—1917 гг. построен аффинажный завод в Екатеринбурге (ныне Свердловск).
Как известно, одним из первых мероприятий Советской власти был Декрет о национализации недр (1917 г.). Вскоре (1918 г.) была введена государственная монополия на добычу, очистку и куплю-продажу платины. Тогда же по инициативе выдающегося химика профессора Л. А. Чугаева был основан при Академии наук Институт по изучению платины и других благородных металлов (в 1934 г. вошёл в состав Института общей и неорганической химии АН СССР). В 1922 г. был организован трест «Уралплатина», в котором сосредоточилась вся деятельность по восстановлению добычи платины, организации её аффинажа и изготовлению платиновых изделий. Работая в тесном
содружестве с Институтом платины, которым руководили Л. А. Чугаев, Н. С. Курнаков и И. И. Черняев, трест успешно справился со всеми стоящими перед ним задачами. Сырьевая база была значительно расширена благодаря открытию П. м. в медно-никелевых рудах Заполярья. В области исследования П. м., их соединений и сплавов советские учёные заняли первое место в мире.
Если в XIX в. практическое применение Pt ограничивалось изготовлением лабораторных принадлежностей (тиглей, чашек, электродов и др.), некоторых заводских аппаратов (например, реторт для концентрации серной кислоты и контактной массы для сернокислотного производства), то в XX в. оно значительно расширилось. В связи с этим платина, которая 100 лет тому назад была в 4 раза дешевле золота, в начале XX в. сравнялась с ним по стоимости. В 1966 г. платина на мировом рынке стоила в 4,3 раза дороже золота. Её мировая добыча составляет несколько десятков тонн в год.
В настоящее время главные области применения П. м. это — промышленность, научные и технические приборы, стоматология и ювелирное дело.
Стойкость к воздействию кислорода воздуха даже при высоких температурах, кислото- и жароупорность делают Pt, Rh и Ir ценными материалами для лабораторной и заводской химической аппаратуры. В химической посуде, изготовленной из Pt, а также из её сплавов с Rh и 1г, работают с агрессивными веществами при довольно высоких температурах. А тигли из Rh и 1г — металлов, ценящихся дороже, чем Pt, применяют для работ с фтором и его агрессивными соединениями, а также для исследований при очень высокой температуре (около 2000 °C). Из сплава 90% Pt+10% 1г изготовлены эталоны метра и килограмма. В частях приборов, где требуется большая твёрдость и стойкость против износа, используют природный осмистый иридий. Очень светлый и нетемнеющий со временем сплав 80% Pd+20% Ag идёт для шкал астрономических и навигационных приборов.
По способности отражать свет Rh лишь немного уступает серебру. Он не тускнеет со временем, поэтому в астрономических приборах предпочитают покрывать зеркальные поверхности родием. Для измерения температур до 1600 °C служат термопары из тонких проволок, изготовленные из Pt и сплава 90%Pt+10% Rh. Более высокие температуры (до 2000 °C) можно измерять термопарой из 1г и сплава 60% Rh + 40% Ir. С помощью платинового термометра сопротивления температуру измеряют с точностью до 0,0001° — значительно точ
237
ПЛУТОНИЙ
нее, чем ртутным термометром. Термометр сопротивления представляет собой длинную (несколько метров) тонкую платиновую проволоку, намотанную на изолятор. Электрическое сопротивление этой проволоки линейно зависит от температуры; поэтому, измеряя величину сопротивления (а сделать это с помощью специальных приборов можно очень точно), определяют и температуру, при которой находится проволока.
П. м., а также и их сплавы катализируют многие химические реакции. Так, в процессах гидрогенизации широко используют Pd, а при получении азотной, серной и уксусной кислот — Pt (или, ещё лучше, её сплав с небольшой добавкой Rh).
Один из сильнейших ядов, не имеющих запаха,— угарный газ СО (см. Углерода окись) легко обнаружить, если внести в газовую смесь полоску фильтровальной бумаги, смоченной раствором PdCl2. Происходит реакция:
PdCl2 + СО + Н2О = СО2 + 2НС1 + Pd
Вследствие выделения мелкораздробленного Pd бумага чернеет.
Сплавы Pt и Pd, которые не темнеют со временем и не имеют привкуса, применяют в зубоврачебной технике. На научные и промышленные цели идёт около 90% всех производимых в мире П. м., остальные 10% поглощает ювелирное производство. Ведь ювелиры давно знают, как Pt и её сплавы заставляют «играть» драгоценные камни, особенно алмазы.
На ордене Ленина—высшей награде СССР — рельефное изображение В. И. Ленина сделано из платины. С. А. Погодин.
ПЛУТОНИЙ (лат. Plutonium) Pu — радиоактивный химический элемент с атомным номером 94; относится к актиноидам. В свободном виде П.— серебристо-белый металл.
Очень интересна история открытия и изучения П. Первый его изотоп, 238Ри, был синтезирован при реакции ядер тяжёлого водорода (дейтерия) с ядрами изотопа урана 238U. Этот синтез провела в 1940 г. группа американских учёных под руководством Г. Сиборга. При радиоактивном распаде 238Ри (период полураспада = 86,4 года) испускаются ядра гелия
(а-частицы). Новый элемент назвали в честь планеты Плутон, так как предшественники П. в таблице Менделеева — уран и нептуний — получили свои названия также в честь планет.
Обычно об открытии нового элемента немедленно сообщается в печати. На этот раз традиция была нарушена. В 1940 г. уже бушевала 2-я мировая война (1939—
1945 гг.). А так как, по расчётам физиков, некоторые изотопы П. могли оказаться пригодными для создания нового оружия невиданной силы, сообщение Сиборга и его коллег об открытии П. было засекречено и увидело свет только после войны, в 1946 г.
В 1941 г. был открыт второй изотоп П., также а-ра-диоактивный — 239Ри; он образуется в результате ряда ядерных превращений при облучении 238U нейтронами. Период полураспада 239Ри достаточно велик (Т^ = =2,4* 104 лет) и его можно накопить в больших количествах. Изучение свойств 239Ри показало, что предсказания физиков верны: П. (наряду с изотопом урана 236U) оказался основным источником ядерной энергии. С помощью только 1 г 239Ри (шарик величиной с горошину) можно получить такое же количество энергии, как при сжигании 4000 кг угля. Самым долгоживущим изотопом П. является а-радиоактивный 244Pu (Ti/2 = =7,5-107 лет). Огромное военное значение 239Ри заставило химиков провести тщательное изучение свойств плутония.
В Советском Союзе первые опыты по его получению были начаты в 1943—1944 гг. Проблемой получения и выделения П. занимались две группы учёных: в Радиевом институте АН СССР в Ленинграде под руководством академика В. Г. Xлопина и в Институте атомной энергии АН СССР в Москве, где работой руководил академик И. В. Курчатов. Впервые в СССР П. был выделен в 1945 г. из облучённого нейтронами урана.
Сначала количества П., имевшиеся в распоряжении учёных, были столь малы, что можно было буквально пересчитать все атомы нового элемента. Однако уже в 1947 г. он был получен в микрограммах. Используя препараты П. массой в миллионные доли грамма, невидимые невооружённым глазом, химики с помощью специальных микрометодов провели изучение П. Изучение столь полное, что через два года о его свойствах стало известно не меньше, чем о свойствах многих обычных металлов. В невероятно короткий срок был разработан технологический процесс извлечения П., и в начале 1949 г. в нашей стране начал выдавать продукцию первый завод по производству П. В том же году в СССР была испытана первая атомная бомба. Монополия США на ядерное оружие была ликвидирована.
Кроме 238Ри и 239Ри, синтезированы и другие изотопы П. Периоды полураспада всех изотопов П. значительно меньше возраста Земли, и поэтому «первичный» П. (то есть тот, который мог существовать в момент формирования нашей планеты) давно распался. Однако тщательные анализы показали, что в природе всегда присутствует небольшое количество П. Это — так называемый вторичный П., постоянно образующийся в урановых рудах при взаимодействии ядер 238U с нейтронами. Нейтроны же частично поступают из космоса, а частично образуются при некоторых ядерных реакциях, самопроизвольно протекающих в земной коре. Содержание П. в урановых рудах ничтожно — его в 400 000 раз меньше, чем такого редкого элемента, как радий. Поэтому, разумеется, весь П., используемый на практике, получают искусственно.
Весьма своеобразны свойства металлического П. Среди других актиноидов он имеет самую низкую температуру плавления, равную 640 °C; /кип 3235 °C. Плотность П. при комнат-' К ИII А
полонии
238
200 400 600’С
Изменение плотности плутония с температурой.
ной температуре 19,8 г/см3. Главная особенность металлического П. состоит в том, что при нагревании от комнатной температуры до плавления он претерпевает необычно много (пять) аллотропных превращений.
Иными словами, у П. существует шесть аллотропных форм; их обозначают буквами греческого алфавита — а, Р, у, д, т) и е. Каждое аллотропное превращение связано с перестройкой атомов в кристаллической решётке П., вследствие чего плотность металла скачкообразно меняется. На рисунке показаны только 4 скачка плотности (4 отрезка, параллельные оси ординат); переход б -> т] не связан с резким изменением плотности.
Совершенно исключительным является тот факт, что при нагревании от 25 до 320 °C плотность П. очень сильно, почти на 20%, уменьшается, а затем, при более высоких температурах, растёт. Металлический П. обладает ещё одной уникальной особенностью. Как известно, металлы при нагревании расширяются. Однако П. при нагревании от 320 до 480 °C не расширяется, а сжимается. Это явление до сих пор не объяснено до конца.
Аномально высокое увеличение плотности П. при охлаждении расплава приводит к тому, что отливки из чистого П., остывшие до комнатной температуры, обязательно растрескиваются. Поэтому изделия, используемые, например, как заряды атомных бомб, изготовляют не из чистого П., а из П., содержащего специальные добавки. Эти добавки при охлаждении ведут себя противоположно самому П. и поэтому препятствуют образованию трещин.
По химическому поведению П. во многом напоминает такие актиноиды, как уран и нептуний. В соединениях он проявляет степени окисления от 4-2 до 4-7 (соответственно валентности от II до VII). Наиболее характерна для П. степень окисления 4-4. Так, при про
каливании на воздухе почти всех его солей образуется окисел РиО2. Получены и два других окисла П.— РиО и Ри2О3. В водных растворах П. проявляет степени окисления от +3 до 4-7. Каждой из них соответствует своя окраска: степени окисления 4-3 — сине-фиолетовая; 4-4 — розовая; 4-5 — бесцветная; +6 — оранжевая и 4-7 — синяя. Интересно, что в одном и том же растворе могут одновременно присутствовать ионы П. в разных степенях окисления. Для выделения П. из облучённого нейтронами урана используют различия в химических свойствах этих элементов. Ри (4-5), в отличие от U и Np, сравнительно легко восстанавливается до Ри (4-4) и трудно окисляется до Ри (4-6).
При выполнении исследований с изотопом 239Ри нужно постоянно контролировать количество взятого П. Если масса П. превысит некоторую предельную величину (так называемую критическую массу), то произойдёт взрыв, аналогичный взрыву атомной бомбы. Величина критической массы 239Ри составляет около 5 кг. Такой кубик П. имеет ребро около 6—7 см.
Кроме военных целей, П. (изотоп 239Ри) применяется как горючее в атомных реакторах, например на атомных электростанциях. Кроме того, на основе изотопа 238Ри изготовляют атомные батарейки (см. также Прометий) со сроком службы 3—5 лет и более. Такие батарейки могут быть использованы как источник энергии (например, в специальных термоэлектрических генераторах, стимулирующих у больных людей работу сердечной мышцы). По масштабам использования П. прочно занимает первое место среди всех искусственно полученных элементов. С. С. Бердоносов.
ПОЛОНИЙ (лат. Polonium) Ро — радиоактивный химический элемент VI группы периодической системы Менделеева; атомный номер 84. Мягкий серебристо-белый металл. Все существующие в природе 7 изотопов П. (их массовые числа 210—212, 214— 216 и 218) радиоактивны, причём распадаются они очень быстро. Наиболее долгоживущий изотоп 209Ро имеет период полурас
пада Ti/2103 года, но он малодоступен. Среди природных изотопов самый долгоживущий — 210Ро (Ti/t = 138,4 суток). Этот изотоп можно выделить из природных минералов в ощутимых (как говорят химики, в «весовых») количествах. С ним и работают обычно исследователи.
(209) 84
Ро
6s26p4
ПОЛОНИЙ
239
ПОЛОНИЙ
Мария СКЛОДОВСКАЯ-КЮРИ. Внесла основополагающий вклад в учение о радиоактивности.
Химик и физик Мария Склодовская-Кюри родилась 7 ноября 1867 г. в Варшаве в семье учителя гимназии. Окончив гимназию в 1883 г. с золотой медалью, она не могла получить высшего образования в царской России (в то время Польша входила в состав Российской империи). В 1891 г. стала студенткой физико-математического факультета Парижского университета. В 1895 г. вступила в брак с видным французским физиком Пьером Кюри. Вместе с ним в 1898 г. она обнаружила излучение тория и открыла два новых излучающих элемента — полоний и радий. Тогда же Мария Кюри назвала «радиоактивностью» способность испускать лучи, создав новый термин, принятый с тех пор в науке. Она же высказала гипотезу о природе радиоактивности, предположив, что это явление присуще атомам элементов. Блестящие открытия явились результатом ни с чем не сравнимого труда. Впоследствии подсчитали, что исследователям пришлось вручную переработать около 11 тонн радиоактивных руд.
В 1903 г. супругам Кюри (вы видите их на фотографии) и А. Беккерелю — пионерам исследования радиоактивности — была присуждена Нобелевская премия по физике. Мария Кюри стала первой женщиной — обладательницей высшей награды за научные достижения.
В 1906 г. от несчастного случая погиб Пьер Кюри. Несмотря на этот страшный удар, Мария выполнила завет своего мужа: «что бы ни случилось, даже если станешь как тело без души, нужно всё так же работать». Только работать она стала ещё больше и самоотверженнее. В 1910 г. вместе с А. Дебьер-ном (он в 1899 г. открыл актиний) Мария Кюри выделила металлический радий и точно определила его атомный вес. За это в 1911 г. ей вторично была присуждена Нобелевская премия, на сей раз по химии. После неё свыше полувека ни один учёный не удостаивался такой чести.
Будучи профессором Сорбонны и директором Института радия в Париже, Склодовская-Кюри исследовала физические и химические свойства радиоактивных веществ и возможности их практического применения. «Она сама отдавалась целиком своему делу и умела добиваться очень многого от других» — так вспоминали о ней современники. Работы Кюри и её школы отличаются глубиной и оригинальностью. Её двухтомный труд «Радиоактивность» переведён на многие языки, в том числе на русский.
Многие академий и учёные общества различных стран мира избирали Марию Склодовскую-Кюри своим почётным членом. Она была с 1907 г. членом-корреспондентом Петербургской академии наук и с 1926 г. иностранным членом Академии наук СССР. Мария Кюри умерла 4 июля 1934 г. в Париже.
Ядра 210Ро, испуская а-частицы (то есть ядра гелия оНе), превращаются в ядра стабильного изотопа свинца:
2182Ро-> 8?6РЬ + *Не
Образующиеся а-частицы обладают большой кинетической энергией, которая при их торможении в веществе переходит в тепло. Длина пути этих частиц составляет всего тысячные доли миллиметра, поэтому почти все они «поглощаются» в самих полониевых препаратах. В результате металлический П. (210Ро) и его соединения разогреваются и имеют температуру выше окружающей.
Первое знакомство с П. совпало с началом новой эпохи в науке — с открытием радиоактивности. В 1896 г. французский физик А. Беккерель обнаружил, что уран постоянно испускает какие-то неизвестные лучи, способные, в частности, вызвать почернение фотоэмульсии. Их и назвали радиоактивными излучениями. Мария Склодовская-Кюри, в то время работавшая у Беккереля, сравнила радиоактивность урановых минералов и чистых солей урана. Оказалось, что радиоактивность минералов значительно выше той, которую можно было ожидать, исходя из известного содержания в них урана. Поэтому исследовательница предположила, что в урановых минералах содержатся элементы, ещё более радиоактивные, чем сам уран.
В деревянном сарае, на скорую руку переделанном в лабораторию, Мария Кюри и её муж Пьер Кюри, пользуясь обычной лабораторной посудой, на простых деревянных столах вручную переработали несколько тонн урановой смоляной руды. Эта титаническая ра-
Рис. 1. Лаборатория Пьера и Марии Кюри. «В деревянном сарае, на скорую руку переделанном в лабораторию, Мария Кюри и её муж Пьер Кюри, пользуясь обычной лабораторной посудой, на простых деревянных столах вручную переработали несколько тонн урановой смоляной руды» (стр. 239).
ПРАЗЕОДИМ
240
бота, вошедшая в историю как образец научного подвига, привела в 1898 г. к открытию двух новых элементов: сначала П., а затем радия. Название П. получил в честь родины М. Склодовской-Кюри — Польши (по-латыни Polonia).
П.— редкий элемент. Его распространённость в земной коре составляет 2-10~14% по массе, а это в 5000 раз меньше, чем радия. Природный П. обычно выделяют из продуктов распада радона—222Rn. Таким путём в 1910 г. был впервые выделен образец, содержавший весовые количества этого редчайшего элемента — около 0,1 мг. В настоящее время миллиграммовые количества П. получают искусственно, в ядерных реакторах, облучая нейтронами висмут.
Плотность металлического П. ок. 9,3 г/см3» /пл 254 °C, а /кип 1162 °C. Конфигурация внешней электронной оболочки атома П. 6s26р4 (см. Атом). По химическим свойствам П. ближе всего к теллуру. Как и теллур, П. проявляет в соединениях степени окисления —2, 4-2, 4-4, 4-6 (соответственно валентности II, IV и VI). Степени окисления —2 соответствует летучий гидрид Н2Ро и соли полониевоводородной кислоты — полониды. Н2Ро образуется при одновременном растворении металлического П. и магния в соляной кислоте:
Ро 4- Mg 4- 2НС1 = MgCl2 4- Н2Ро
Степень окисления 4-2 элемент проявляет в некоторых соединениях с галогенами (РоС12, РоВг2 и др.) и с серой (PoS). Так, РоС12 об-
Рис. 2. Капилляр с фильтром для фильтрования осадков полония. Справа, для. сравнения размеров, иголка с ниткой и спичка.
разуется при растворении П. в соляной кислоте:
Ро 4- 2НС1 = РоС12 4- Н2
Наиболее многочисленны соединения П., отвечающие степени окисления 4-4. Нагревая П. с кислородом при 250 °C, получают РоО2. Известна гидроокись РоО(ОН)2. Соединения П., отвечающие степени окисления4-6, получены то-, лько в индикаторных количествах (то есть недоступных взвешиванию). Это РоО3, соли полониевой кислоты — полонаты (типа К2РоО4) и другие.
Экспериментальная работа с П. сложна и опасна, так как он чрезвычайно токсичен. Многие соединения П. летучи при комнатной температуре и легко загрязняют помещения, где работают с П. Летучесть соединений П. повышается ещё из-за того, что они саморазогреваются (см. выше). Поэтому при работе с П. его помещают в специальные боксы, причём руки защищают толстыми резиновыми перчатками. Исследования свойств П. выполняют на образцах с очень малой массой, используя специальные методы. Так, отфильтровывание осадков, содержащих П., проводят с помощью капилляров диаметром около 0,5 мм, в которые впаян стеклянный пористый фильтр (рисунок). В этих условиях даже простейшая химическая операция — фильтрование — становится чрезвычайно трудоёмкой.
Области применения П. очень интересны. Так, энергию распада 210Ро можно преобразовать в электрический ток. Поэтому 210Ро применяли, например, в электроэнергетических установках спутников «Космос-84» и «Космос-89», где он обеспечивал длительную непрерывную работу различной аппаратуры. В смеси с бериллием 210Ро даёт удобный источник нейтронов, который используют, в частности, для проведения анализа состава различных материалов. Перспективны и другие пути применения полония. С. С. Бердоносов.
ПОТАШ — карбонат калия К2СО3. См. Калий, Карбонаты.
ПРАЗЕОДИМ (лат. Praseodymium) Рг — химический элемент III группы периодической системы Менделеева из семейства лантаноидов; атомный номер 59.
ПРОМЕТИЙ (лат. Promethium) Pm — радиоактивный химический элемент III группы периодической системы Менделеева; атомный номер 61; относится к лантаноидам. Известно около 15 радиоактивных изотопов П., все они получены искусственно.
Попытки обнаружить элемент № 61 в природе предпринимались с 20-х годов XX столетия, но долгое время заканчивались неудачами. Сейчас, когда свойства П. подробно изучены, это не кажется удивительным: периоды полураспада всех изотопов П. по сравнению с возрастом Земли очень малы, и поэтому весь П., даже если он существовал когда-либо на нашей планете, давно распался. Правда, П. постоянно образуется в природе при ядерных реакциях, однако в чрезвычайно малых количествах. Обнаружить его в природе удалось только
241
ПРОТАКТИНИЙ
в 1968 г. с помощью специальных сверхчувствительных методов анализа. В этих опытах учёные имели дело буквально со считанным числом атомов П.
К моменту открытия «земного» П. возраст нового элемента уже составлял 23 года: ещё в 1945 г. американские химики Д. Маринский, Л. Гленденин и Ч. Кориэлл
(145) 61
Pm
4f56s2
ПРОМЕТИЙ
выделили из продуктов деления урана, в отработанных стержнях ядерного реактора, первые пять миллионных долей грамма (5-10“6 г) элемента № 61. Новый элемент назвали в честь легендарного героя древних греков Прометея, который, по преданию, похитил у богов огонь и передал его людям. Название «прометий» служит на-
поминанием о том драматическом пути, который человечеству пришлось пройти, чтобы овладеть энергией атомного ядра. При работе ядерных реакторов образуется довольно много 147Ргп, и его можно выделять в граммовых количествах. Самым устойчивым изотопом является
145Рт (Т»/2 около 18 лет), но он малодоступен.
Металлический П. удалось получить только в 1963 г. Свойства его пока слабо изучены. По оценкам, плотность составляет 7,26 г/см3, температура плавления достигает 1080 °C. В химическом отношении П. ведёт себя как типичный лантаноид, в соединениях постоянно проявляет окислительное число +3 (валентность III). Известны окись Рт2О3, гидроокись Рт(ОН)3,. а также некоторые соли, например хорошо растворимые в воде нитрат Pm(NO3)3 и хлорид РтС13.
Среди искусственно полученных элементов П. по широте применения занимает почётное 4-е место, уступая лишь плутонию, нептунию и технецию. Это связано со свойствами изотопа 147Рт. Во-первых, испускаемые им 0“-частицы (электроны) обладают сравнительно небольшой энергией и поглощаются уже в тонком слое воздуха. А во-вторых, 0“-распад 147Рт, в отличие от большинства других изотопов, испускающих 0“-частицы, не сопровождается губительным у-излучением, которое обладает высокой проникающей способностью. По этим двум причинам для работы с 147Рт не нужны дорогостоящие защитные экраны.
Важно и то, что продолжительность жизни этого изотопа достаточно велика (период полураспада 7\/2=2,7 года).
С помощью 147Рт получают удобные источники постоянного свечения: изотоп добавляют в состав специальных веществ — люминофоров. Испускаемые ядрами 147Ргп 0“-частицы почти полностью поглощаются в люминофоре и вызывают свечение. Светосоставы, содержащие 147Рт, непрерывно светятся в течение нескольких лет; поэтому с их помощью делают ука
затели и надписи в слабоосвещённых местах, например в тёмных участках шахт, и т. д.
А если тоненькую таблетку светящегося состава, содержащего 147Рт, окружить сверху и снизу слоями кремниевого фотоэлемента, способного превращать световую энергию в электрическую (см. Кремний), то получится миниатюрная (размером в канцелярскую кнопку) электрическая батарейка, срок непрерывной службы которой измеряется годами. Такие батарейки получили название атомных. Диапазон использования атомных батареек очень широк: от слуховых аппаратов и до систем энергопитания управляемых снарядов.
С. С. Бердоносов. ПРОТАКТИНИЙ (лат. Protactinium) Ра — радиоактивный химический элемент; атомный номер 91; относится к актиноидам. Блестящий светло-серый металл.
Элемент был открыт в 1913 г., когда К- Фаянс и О. Гёринг в городе Карлсруэ (Германия) обнаружили среди продуктов распада урана изотоп 234Ра, оказавшийся короткоживущим (его период полураспада Т1/2-= 1,18 мин). Тщательное изучение свойств П. стало возможно 5 лет спустя, когда был открыт другой изотоп П.— долгоживущий 231Ра (Л..—32480 лет, масса 231, 0359), постоянно образующийся в природе при распаде урана. Название элемента происходит от двух слов: «прбтос» (по-гречески «первый») и актиний. Оно связано с тем, что П. является родоначальником актиния: 231Ра, испуская а-частицы, превращается в 227Ас.
П. относится к числу наименее распространённых на Земле элементов. На его до
лю приходится менее 1 • 10“10% массы земной коры — примерно столько же, сколько на долю радия. Однако выделение П. в весовых количествах из-за его своеобразных химических свойств связано со значительно большими трудностями, чем выделение радия. Основным природным источником П. служат урановые руды; содержание в них 231Ра достигает 0,34 г на 1 т урана. Первые 2 мг чистого соединения П.— Ра2О5 — удалось получить только в 1927 г. Дальнейшие исследования учёных раскрыли многие свойства П., и в 1960 г. в одной только Англии из отходов переработки урана удалось получить «фантастическое» количество П.— 130 г изотопа 231Ра.
На воздухе поверхность П. быстро тускнеет из-за образования плёнки окисла. Плотность П. 15,4 г/см3, /11Л 1560 °C, /кип 4280 °C. В соединениях П. проявляет степени окисления от + 2 до +5 (соответственно валентности от II
231,0359 91
5f26d'7s2
ПРОТАКТИНИЙ
16 хэш
ПРОТАКТИНИИ
242
до V). В тех случаях, когда валентность П. равна II, III или IV, его соединения сходны с соответствующими соединениями тория и урана\ при этом П. ведёт себя как типичный актиноид. Однако свойства Ра (+5) резко отличаются от свойств 5-валентных актиноидов и больше всего похожи на свойства 5-валентных ниобия и тантала. Так, П. образует высший окисел состава Ра2О5, который при сплавлении со щелочами даёт соли — протактинаты, например NaPaO3, Na3PaO4. Эта особенность химии П. объясняется тем, что у 2-, 3- и 4-валентного П. пятая электронная оболочка атома содержит /-электроны, а 5-валентный П. /-электронов не имеет (см. Атом, Актиноиды).
В водных растворах, содержащих Ра (+5), происходят сложные процессы гидролиза и полимеризации. В результате образуются соединения, которые легко адсорбируются на стенках посуды и на твёрдых частицах примесей, всегда присутствующих в растворе. Поэтому опыты, поставленные с Ра (+5) в растворах, часто оказываются невоспроизводимыми и трудно контролируемыми. П. заслужил славу одного из наиболее коварных и неясных элементов, с чем и связаны особые трудности его выделения. Несмотря на широкий фронт работ по изучению П., в химии этого элемента до сих пор много неясного — такого, что ещё ждёт своих исследователей. С. С. Бердоносов.
«...В преступных руках радий может представить серьёзную опасность, и встаёт вопрос: выиграет ли человечество от познания тайн природы, достаточно ли оно созрело, чтобы ими пользоваться, или это познание обратится ему во вред? Пример открытий Нобеля показателен в этом отношении: мощные взрывчатые вещества позволили человеку выполнять замечательные работы, но они же стали ужасным разрушительным средством в руках великих преступников, толкающих народы к войне. Я отношусь к числу тех, кто думает вместе с Нобелем, что человечество извлечёт больше пользы, чем вреда, из новых открытий».
пьер Кюри
(Кюри Пьер см. в кн.: «Труды Института истории естествознания и техники», т. 19, 1957, с. 166)
РАДИЙ (лат. Radium) Ra — радиоактивный химический элемент II группы периодической системы Менделеева; атомный номер 88. Серебристо-белый блестящий металл. Четыре радиоактивных изотопа Р.— 223Ra, 224Ra, 226Ra и
228Ra — встречаются в природе. Наиболее долгоживущим из них является 226Ra, который можно получить в весовых количествах (его период полураспада Тг/г около 1620 лет); остальные изотопы Р. распадаются значительно быстрее. Атомная масса
226Ra равна 226, 0254. Может показаться странным, что при таких высоких скоростях радиоактивного распада изотопы Р. всё-таки присутствуют в природе — ведь возраст Земли исчисляется миллиардами лет, и, казалось бы, все атомы Р. должны были давно распасться.
Наличие Р. в природе объясняется тем, что его изотопы постоянно образуются при распаде, радиоактивных изотопов урана (235U и 238U) и тория (232Th). Периоды полураспада этих изотопов урана и тория чрезвычайно велики (сотни миллионов лет и более); поэтому можно считать, что количества их на Земле в течение жизни многих человеческих поколений не меняются. Изотопы U и Th дают начало природным радиоактивным рядам, а изотопы Р. являются промежуточными членами этих рядов. В каждом таком ряду устанавливается так называемое радиоактивное равновесие, так что запасы Р. в природе остаются практически неизменными. (Подробнее о радиоактивных рядах см. в статье Уран.)
Открытие Р. вызвало переворот в физике и химии. Хотя Р.— второй (после полония) элемент, обнаруженный благодаря сильной радиоактивности, его значение для науки оказалось значительно большим, чем полония. Это объясняется тем, что соли Р. проще выделить в чистом виде. В 1898 г. пионеры в изучении радиоактивности — Мария Склодовская-Кюри и Пьер Кюри — откры
ли, что при переработке урановой смоляной руды (минерал U3O8) вместе с осадком BaSO4 выпадает новый, сильно радиоактивный элемент. Его назвали «радием» (от лат. radio — испускаю лучи). Уже в 1902 г. супруги Кюри получили препараты, содержащие несколько десятков миллиграммов Р. Для выделения радия они провели тысячи перекристаллизаций водных растворов хлоридов и бромидов Р. и бария. При каждой кристаллизации Р. переходил в осадок чуть-чуть быстрее, чем барий, и это свойство позволило в конце концов получить совершенно чистые препараты радиевых содей. Выделение Р. стало примером упорного, настойчивого труда. Владимир Маяковский с добычей Р. сравнил труд поэта:
«Поэзия —
та же добыча радия.
В грамм добыча, в год труды».
Р.— очень редкий элемент; его содержание в земной коре оценивается в 1-10“10% по массе (80-е место среди прочих элементов). Радиевых минералов не существует. Как продукт радиоактивного распада урана, Р. неизменно присутствует во всех урановых минералах. В среднем они содержат около 3,33-10“7 частей Р. на 1 часть урана, то есть 1 г Р. приходится на 3 т урана в руде. Чтобы выделить
Пробный радиевый завод. Находился на территории химического завода в посёлке Бондюжском (ныне Менделеевск), в трёх километрах от пристани Тихие Горы на Каме. В конце 1921 г. в труднейших условиях голода и хозяйственной разрухи здесь были получены первые в нашей стране препараты радия.
16*
РАДОН
244
1 г Р., надо переработать около 4 т чистейшей урановой смоляной руды и израсходовать около 450—500 т химикалий, 1000 т угля и 10 000 т дистиллированной воды.
Промышленное производство Р. началось в 1902—1903 гг. во Франции, Германии и Англии, а в 1914 г.— в США. В России Р. не добывался, хотя уже в начале XX в. в Ферганской области было открыто месторождение тюямунита — минерала урана. Только при Советской власти, благодаря содействию В. И. .Ленина, был организован пробный радиевый завод, на котором В. Г. Хлопин и И. Я. Башилов разработали технологию извлечения Р. из ферганской руды. По этой технологии с 1922 г. началось производство Р. в СССР.
Благодаря радиоактивному излучению все соединения Р. на воздухе испускают голубое свечение. Температура препаратов, содержащих Р., несколько выше окружающей среды за счёт энергии радиоактивного распада (1 г 226Ra выделяет в 1 час около 582 дж, то есть 139 кал теплоты).
В свободном состоянии Р. впервые получили в 1910 г. М. Кюри и А. Дебьерн, но пока его свойства изучены слабо. По разным источникам, его /пл 700—960 °C, а /кип выше 1140 С.
Р. относится к числу щелочноземельных металлов. Внешняя электронная оболочка его атома содержит два электрона (конфигурация 7s2, см. Атом). Эти электроны Р. легко отдаёт при образовании химических соединений, в которых он всегда имеет степень окисления +2 (валентность II). По химическим свойствам Р. больше всего похож на барий, но ещё более активен химически. На воздухе из-за взаимодействия с азотом он покрывается чёрной плёнкой нитрида Ra3N2. С водой Р. бурно реагирует, выделяя Н2 и давая сильное основание Ra(OH)2. Из солей Р. хорошо растворимы в воде Ra(NO3)2, RaCl2 и RaBr2; наименее растворимы RaSO4 и RaCO3. Из всех соединений Р. путём анализа установлен состав лишь трёх солей: RaCl2, RaBr2 и RaBeF4. Состав остальных соединений пока указывают предположительно по аналогии с солями бария, так что здесь ещё есть над чем поработать!
Изучение Р. сыграло огромную роль в развитии науки о строении и свойствах микромира, положило начало глубокому исследованию радиоактивности. Альфа-частицы, испускаемые 226Ra, имеют высокую энергию. До 1930-х годов они были главными «снарядами» физиков; этими снарядами учёные обстреливали ядра различных элементов периодической системы. Так были сделаны важнейшие открытия «атомного века»: обнаружено существование ядра атома, осуществлено первое искус
ственное ядерное превращение, открыт нейтрон. Долгое время Р. был единственным элементом, радиоактивные свойства которого находили практическое применение. Так, в медицине Р. использовали для лечения рака (радиотерапия).
Под действием радиоактивных излучений ткани, поражённые раковой опухолью, разрушаются значительно быстрее здоровых. Поэтому, действуя радиоактивными лучами на поражённые опухолью участки организма, часто удаётся разрушить злокачественное образование (особенно если лечение начато на ранних стадиях болезни). Шансов на успех тем больше, чем ближе к поверхности организма расположена опухоль, так как иначе излучение может полностью поглотиться, не достигнув цели. Правда, и в этом случае иногда удаётся разрушить опухоль, введя в поражённый ею орган маленькие ампулы с радиоактивным веществом.
В промышленности с помощью Р. можно контролировать качество литых изделий, сварных швов и т. д. (так называемая гамма-дефектоскопия). Если внутри контролируемого изделия имеется пустота или включение шлака (плотность которого ниже, чем металла), то испускаемые радиоактивным изотопом у-лучи, проходя через изделие, поглощаются в дефектных местах слабее, чем в остальных участках. Регистрируя излучение, прошедшее через разные участки изделия, можно обнаружить в нём пустоты и другие дефекты.
В наши дни на смену Р. в подавляющем большинстве случаев пришли искусственно получаемые радиоактивные изотопы *(60Со, 13TCs и другие), которые в тысячи раз дешевле. Поэтому сейчас применение Р. весьма ограничено. В медицине Р. сохраняет некоторое значение как источник радона при лечении радоновыми ваннами: при радиоактивном распаде 22GRa образуется 222Rn:
2|®Ra—> 222Rn + 42He
Радон выделяют, пропуская через растворы солей Р. ток воздуха. В небольших количествах Р. расходуется для приготовления простейших лабораторных источников нейтронов (в смеси с бериллием). С, С. Бердоносов.
РАДОН (лат. Radonum) Rn — радиоактивный химический- элемент VIII группы периодической системы Менделеева; атомный номер 86; инертный газ.Все изотопы Р. радиоактивны, наиболее долгоживущий из них 222Rn имеет период полураспада Ti/2 3,823дня. Несмотря на столь быстрый распад, количество Р. в природе постоянно.
Объясняется это тем, что 3 изотопа Р. образуют-
245
РАДОН
Виталий Григорьевич ХЛОПИН — один из основателей советской радиохимии и радиевой промышленности.
Советский химик Виталий Григорьевич Хлопин родился 26 января 1890 г. в Перми. Сын Г. В. Хлопина, впоследствии известного профессора-гигиениста. В 1908—1912 гг. учился в Петербургском университете. Летние семестры 1910 и 1911 гг. работал в Гёттингенском университете (Германия). Был оставлен при Петербургском университете для подготовки к званию профессора.
Во время войны 1914—1918 гг. при Академии наук В. И. Вернадский и другие виднейшие учёные организовали Комиссию по изучению естественных производительных сил России (КЕПС). Одной из её главных задач была постановка производства радия. Однако осуществить её оказалось возможным только после Великой Октябрьской революции, при личной поддержке В. И. Ленина. Привлечённый В. И. Вернадским к работе в КЕПС Хлопин возглавлял Коллегию по организации и эксплуатации пробного радиевого завода. Заведовать заводом был назначен И. Я. Башилов, который спроектировал всё необходимое оборудование и установил его на территории химического завода в посёлке Бондюжском (ныне город Менделеевск) на реке Каме. 1 июля 1921 г. Хлопин и Башилов запустили опытную установку для извлечения радия, а 1 декабря были получены первые советские препараты радия. Всего (в пересчёте на металл) было получено 12,1 мг радия. Эти препараты Хлопин продемонстрировал на заседании Третьего Менделеевского съезда в мае 1922 г. Хлопин был одним из организаторов основанного в 1922 г. Радиевого института АН СССР и с 1939 г. его директором.
Работа с радиоактивными элементами имеет свои особенности. Во-первых, время существования этих элементов ограничено (они распадаются). А во-вторых, химик обычно работает с весьма малыми их концентрациями. Хлопин изучал очень важный для практики вопрос — как распределяются микроколичества радиоактивных элементов между твёрдой фазой и раствором в процессе осаждения. Он сформулировал правило (носящее теперь его имя), которое легло в основу процессов выделения чрезвычайно малых количеств веществ и сыграло важную роль в развитии радиохимии. В 1939 г. В. Г. Хлопин был избран академиком АН СССР. В 1949 г. ему присвоено звание Героя Социалистического Труда. Трижды он был удостоен Государственных премий СССР. Хлопин умер 10 июля 1950 г. в Ленинграде.
ся при распаде других природных радиоактивных элементов и входят в так называемые радиоактивные ряды. (О них рассказано в статье Уран.) Изотоп 222Rn, возникающий в ряду урана-238 из радия-226, называется собственно «радон»; 219Rn, входящий в ряд актиноурана (изотопа урана-235), называется «актинон»; a 220Rn, принадлежащий к ряду тория-232, называют «торон».
Английский учёный Э. Резерфорд в 1899 г. пришёл к выводу, что препараты тория выделяют неизвестный ранее газ, который он предложил назвать эманацией (от лат. emanat io — истечение). Последующие исследования показали, что эманация — это радиоактивный инертный газ с атомным номером 86. В 1923 г. его решили назвать радоном по имени наиболее долгоживущего изотопа, образующегося из радия.
Распространённость Р. на Земле крайне низка. По содержанию в земной коре он занимает всего лишь 84-е место среди других элементов (7-10"16/6 по массе). Во всей атмосфере содержится только 370 л Р. В атмосферу Р. попадает из горных пород и других объектов, содержащих уран или торий. Йз-за быстрого распада Р. не успевает равномерно перемешаться с другими газами атмосферы, поэтому содержание его над разными участками земной поверхности неодинаково. Больше всего Р. там, где много радиоактивных руд. В 10 км от поверхности Земли Р. примерно в 100 раз меньше, чем у поверхности. Химическая активность Р. весьма низка. При реакции со фтором он даёт фторид, вероятно, состава
RnF2. Изучение химии Р. затруднено из-за его высокой радиоактивности (о физических и химических свойствах Р. см. также Инертные газы). Работа с Р. крайне опасна.
Вызвано это тем, что Р. с воздухом попадает в лёгкие, где многие ядра Р. успевают распасться за время между вдохом и выдохом. К тому же Р. относительно хорошо растворим в воде, а следовательно, и в крови. После распада Р. образуются радиоактивные изотопы полония, висмута, свинца и таллия, которые прочно «прилипают» к внутренним стенкам лёгких и, постепенно накапливаясь, могут вызвать тяжёлые заболевания. Поэтому работа с Р. требует особых мер предосторожности (защита органов дыхания марлевой повязкой-респиратором, мощная вентиляция помещений и т. д.).
В начале XX в., когда проводились первые исследования Р., учёные не подозревали об опасности работы с ним, и за это неведение приходилось подчас расплачиваться жизнью. Так, англичанин У. Рамзай, впервые определивший плотность и атомную массу Р. (для этого он сконструировал сверхчувствительные весы, позволявшие взвешивать весь наличный Р.— около 0,1 мм3, с точностью до десятимиллиардных долей грамма), погиб в 1916 г. от рака лёгких.
Однако, как и озон и многие другие вещества, Р. в небольших концентрациях обладает целебными свойствами. Для лечения обычно используют купание в воде, содержащей Р. (радоновые ванны). Во время приёма такой ванны небольшая часть (1—2%) растворённого в' воде Р. попадает внутрь организма или адсорбируется на коже. Распадаясь, атомы Р.
РЕДКОЗЕМЕЛЬНЫЕ ЭЛЕМЕНТЫ
246
(и продукты его последующего распада) вызывают как бы микроуколы, благотворно влияющие на организм. Раньше для радоновых ванн использовали только природные источники, теперь же такую воду можно получить искусственно в лечебном учреждении.
Многие другие применения Р. основаны на его радиоактивности и химической инертности. Р.— прекрасная газообразная радиоактивная «метка» (меченый атом). Так, с помощью Р. можно проследить за движением газа в домне, обнаружить место утечки из подземного трубопровода и т. д.
Для получения Р. достаточно продуть через водные растворы солей радия какой-либо газ (например, воздух), который захватывает весь образующийся в растворе Р. Отделить Р. от воздуха можно, пропуская газ через ловушки с твёрдой двуокисью углерода — «сухим льдом» (где Р. конденсируется) или через активированный уголь и другие пористые материалы (на которых адсорбируется). С. С. Бердоносов. РЕДКОЗЕМЕЛЬНЫЕ ЭЛЕМЕНТЫ — хи мические элементы: скандий Sc (атомный номер Z—21), иттрий Y (Z=39), лантан La (Z=57) и лантаноиды (Z от 58 до 71). Название «редкоземельные элементы» (в литературе встречаются сокращённые обозначения «РЗЭ», «РЗМ») дано в связи с тем, что эти элементы встречаются в природе сравнительно редко и дают тугоплавкие нерастворимые в воде окислы — по старинной терминологии «земли» (вспомните ещё одну такую «землю» — глинозём А12О3). В периодической системе Менделеева РЗЭ расположены в подгруппе б III группы. Характерная особенность РЗЭ — их совместное присутствие в природе и близость химических свойств.
Первым среди РЗЭ был открыт иттрий; в 1794 г. финский химик Ю. Гадолин выделил из минерала иттер-бита «иттриевую землю» (минерал позднее переименовали в гадолинит). Последующие исследования, тя-
к
Таблица 1. — Сравнение свойств, предсказанных Д. И. Менделеевым для «экабора» и обнаруженных Л. Ф. Нильсоном у скандия
Экабор ЕйВ
Скандий Sc
Атомный вес 44
Состав окиси (ЕкВ)2О3
Плотность окиси 3,5
Окись нерастворима в щелочах
Соли бесцветны
Углекислый и сернокислый экабор нерастворим в воде
Хлористый экабор менее летуч, чем хлористый алюминий
Атомный вес 44,1
Состав окиси Sc2O3
Плотность Sc2O3 3,864
Окись нерастворима в щелочах
Соли бесцветны
Sc2(CO3)3 нерастворим в воде, a Sc2(SO4)3 хорошо растворим в воде
ScCl3 начинает возгоняться при 850 °C, а А1С13 — при 100 °C
нувшиеся почти столетие, показали, что «иттриевая земля» содержит наряду с окисью иттрия примеси других редких земель. Шведский химик Л. Ф. Нильсон в 1879 г. при разделении «иттриевой земли» обнаружил новый элемент — скандий. Это открытие имело особенно важное значение для всеобщего признания периодического закона Д. И. Менделеева. Дело в том, что Менделеев, основываясь на своей периодической системе, в 1870—71 гг. подробно предсказал свойства некоторых тогда ещё не открытых элементов и среди них — «экабора», который должен был занять одну из пустых клеток в III группе системы. Именно этот элемент был открыт Нильсоном и получил название скандия. И почти все его свойства, предсказанные Д. И. Менделеевым, подтвердились (см. таблицу 1). А это во многом способствовало утверждению самого закона.
Значительно раньше, чем скандий, был открыт лантан. Шведский химик К- Мосандер исследовал другую редкую «землю» — «цериевую». В итоге многолетней работы он выделил из неё лантановую «землю» (1839 г.), а затем (1841 г.) разложил последнюю на лантановую и дидимовую (позднее оказалось, что дидим — это смесь празеодима и неодима, см. Лантаноиды).
РЗЭ чрезвычайно рассеяны в земной коре. Но сейчас уже вряд ли следует относить их к мало распространённым. Так, иттрий и лантан занимают по распространённости 28-е и 29-е место. Скандия — меньше (38-е место). Но и его почти в 10 раз больше, чем молибдена, и в 20 раз больше, чем сурьмы.
В природе РЗЭ обычно встречаются вместе, в одних и тех же минералах. Среди них важное значение имеет монацит — смесь фосфатов РЗЭ, тория и других элементов. Sc и Y образуют и самостоятельные минералы, однако они очень редки. Среди иттриевых минералов важно отметить ксенотим YPO4 и обнаруженный советскими геологами в 1961 г. минерал, состоящий из фторидов натрия, кальция и иттрия и названный гагаринитом в честь пионера космоса Ю. А. Гагарина. Из скандиевых минералов наиболее богат Sc тортвейтит Sc2Si2O7, встречающийся в Норвегии и на Мадагаскаре. Этот минерал очень редок; так, в норвежских рудниках с 1950 г. по 1960 г. его добыли всего 50 кг.
В свободном виде Sc, Y и La — металлы серебристо-белого цвета, тускнеющие на воздухе из-за образования плёнки окисла; их физические свойства приведены в таблице 2.
Конфигурации двух внешних электронных оболочек (последней и предпоследней от ядра) у Sc, Y и La аналогичны (см. таблицу 2). В образовании химических соединений принимает «участие d-электрон предпоследней оболочки и два s-электрона последней оболочки. Поэтому в соединениях Sc, Y и La имеют степень окисления 4-3 (валентность III). По реакционной способности и по свойствам соединений РЗЭ ближе к щелочноземельным металлам, чем к алюминию. Например, иттрий (подобно кальцию) в токе хлора возгорается при 200 °C:
2Y 4~ ЗС12 2YC13
РЕНИИ
Таблица 2. — Некоторые свойства Sc, Y и La
Элемент 1 Атомная масса Конфигурация внешних электронных оболочек плотность, г/см3 температура, °C Соде ржание в земной коре, % по массе
плавления кипения
Sc 44,9559 3s2 3pG 3d1 4s2 2,99 1539 2730 1-ю-3
У 88,9059 4s2 4p° 4rf! 5s2 4,48 1509 2930 2,9-10-3
La 138,9055 5s2 5pG 5J1 6s2 6,17 920 3470 2,9 10-3
А лантан (как и Са) при нагревании в азоте даёт нитрид:
2La + N2 = 2LaN
Окислы РЗЭ общей формулы Ме2О3 нерастворимы в щелочах, но легко реагируют с кислотами, например:
Sc2O3 -г 6НС1 = 2ScCl3 + ЗН2О
При взаимодействии с водой окислы дают гидроокиси Ме(ОН)3. Окись лантана реагирует с водой, подобно извести СаО, с выделением большого количества тепла:
La2O3- 3H2O = 2La(OH)3
Растворимость сульфатов Sc, Y и La в воде быстро понижается при переходе от легкорастворимого Sc2(SO4)3 к малорастворимому La2(SO4)3; сходное изменение растворимости наблюдается и у сульфатов щелочноземельных элементов. Из гидроокисей Sc, Y и La наиболее сильными основными свойствами обладает La(OH)3, напоминающая Са(ОН)2.
Рассмотрим некоторые свойства Sc, Y и La (сведения о лантаноидах даны в соответствующей статье).
Скандий (лат. Scandium) Sc, Z-=21. Название получил в честь Скандинавского полуострова — родины первооткрывателя этого элемента Л. Ф. Нильсона. Важная особенность Sc по сравнению с другими РЗЭ — амфотерный характер его гидроокиси Sc(OH)3. В отличие от гидроокисей других РЗЭ, Sc (ОН)3 растворяется не только в кислотах, но и в щелочах; в последнем случае образуются соли — скан-диаты. Sc применяют
в новейшей технике. Он идёт на производство скандиевого феррита, из которого изготовляют элементы памяти электронно-вычислительных машин. Скандиевые ферриты работают лучше, чем ферриты других типов (например, марган-
цевые). Ведутся поиски и других областей применения Sc (например, в металлургии).
Иттрий (лат. Yttrium) Y, Z=39. Назван в честь Иттербю (это — шведский городок, около которого в 1784 г. был впервые найден минерал иттербит). Из иттербита (позднее переименованного в гадолинит) были выделены 4 РЗЭ: иттрий, тербий, эрбий и иттербий (см. Лантаноиды). Очень чистая окись иттрия Y 2О3 используется, как и окись скандия, при изготовлении ферритовых элементов в технике сверхвысоких частот.
А кроме того, на основе Y2O3 готовят вещества, входящие в состав светящихся экранов цветных телевизоров. В последнее время Y стали применять в металлургии при производстве специальных сплавов.
Лантан (лат. Lanthanum) La, Z=57. Название происходит* от греческого слова «ланта-но» — скрываюсь и отражает трудности, которые пришлось преодолеть при его открытии. В современной технике широко используется окись лантана La2O3. Если это вещество добавить к смеси, из которой варят оптическое стекло, то каче
ство фото- и кинообъективов резко улучшится. Из лантанового стекла изготовлен один из лучших советских фотообъективов «Инду-стар-61ЛЗ»; среди кинолюбителей большой популярностью пользуется кинокамера, которая так и называется «Лантан». Есть и другие области применения этого элемента. Так, в металлургии La вместе с церием и другими лантаноидами (их смесь часто называют «мишметаллом») служит присадками к стали, чугуну и сплавам цветных металлов. Мишметалл улучшает механические свойства, коррозионную устойчивость и жаропрочность сплавов.
С. С. Бердоносов.
РЕНИЙ (л ат. Rhenium) Re — химический элемент VII группы периодической системы Менделеева; атомный номер 75, атомная масса 186,207. Светло-серый металл. Природный Р. состоит из двух изотопов: устойчивого 185Re (37,07%) и слаборадиоактивного 187Re (62,93%).
РЕНИЙ
248
Последний с периодом полураспада 1-Ю11 лет испускает 0"-частицы, превращаясь в изотоп осмия:
178'Re = 1?’0s +13-
Существование аналога марганца с атомным весом 190 предсказал в 1871 г. Д. И. Менделеев, который назвал этот элемент «тримарганцем». Но только в 1925 г. немецкие химики Ида Такке и В. Ноддак обнаружили его спектральным методом в минерале колумбите. Назван Р. по имени Рейнской провинции Германии.
75
186,207
5d56s2
РЕНИЙ
Р. принадлежит к редким и рассеянным элементам. Его содержание в земной коре ненамного превышает содержание радия и оценивается 7-10“8% по массе (78-е место по распространённости). Обычно Р. встречается как примесь в минералах других элементов— сравнительно много его в молибдените Мо52(до 1,88%) и колумбите (см. Ниобии). Собственных же минералов Р. пока найдено всего три, и они очень редки. Один из них —джез-казганит CuReS4 впервые найден у нас в городе Джезказган Казахской ССР. Кстати, залежи медной руды на территории Джезказгана были обнаружены ещё в конце XVIII в.
По внешнему виду Р. напоминает платину. При описании его физических свойств можно везде использовать слова «один из самых». И действительно, это один из самых тяжёлых металлов (по плотности 21,03 г/см3 уступает лишь платине, иридию и осмию), один из самых тугоплавких металлов (по температуре плавления 3180 °C уступает лишь вольфраму), один из самых высококипящих металлов (по температуре кипения 5900 °C он уступает только гафнию). Металлический Р. ковок. Он длительно сохраняет высокую прочность даже при таких больших температурах, как 2000СС.
У атома Р. семь внешних (валентных) электронов (конфигурация 5d56s2, см. Атом). Химия Р. довольно сложна и разнообразна, потому что для него известны соединения практически со всеми степенями окисления, от —1 до 4-7, например: Re2O7 (4 7), ReO3 (4-6), ReCl5 (4-5), ReS2 (4-4), ReCl3 (4-3), ReCl2-*4H2O (+2), Re2O (4-1), Re2 (CO)10 (0),
KIRe(CO)5](—1). Наиболее характерны и устойчивы соединения Р. со степенью окисления 4-7 (валентностью VII). Металлический Р. очень стоек по отношению к воздуху и воде. Реакция с кислородом начинается лишь при температурах 300—600 °C, причём образуется смесь окислов Re2O7 и ReO3. При нагревании
Р. взаимодействует с галогенами и серой: 2Re4- 5Cl2 = 2ReCl5
Re 4- 2S - ReS2
В ряду напряжений Р. стоит за водородом и поэтому не вытесняет его из кислот. Он не рас-, творяется даже в кипящих соляной и плавиковой кислотах. Однако азотная кислота легко реагирует с металлом:
3Re 4- 7HNO3 = 3HReO4 + 7ХО 4- 2Н,0
Растворяют Р. также горячая концентрированная серная кислота и перекись водорода: 2Re 4- 7H2SO4 - 2HReO4 - 7SO2 4- 6H2O 2Re 4- 7H2O2 - 2HReO4 4- 6H2O
На P. действуют и расплавленные едкие щёлочи при доступе кислорода:
4Re 4- 4КОН 4- 70 2 - 4KReO4 + 2Н.2О
В результате всех описанных реакций образуется либо рениевая кислота HReO4, либо её соли — перренаты, например KReO4. Рениевая кислота довольно сильная. Она существует только в растворе. В отличие от хорошо известной марганцовой кислоты НМпО4 (см. Марганец), рениевая HReO4 бесцветна и для неё и её солей окислительные свойства не характерны. Ангидрид этой кислоты Re2O7 — светло-жёлтое вещество, хорошо растворимое в воде.
Спрос в промышленности на Р. пока значительно превышает предложение. Связано это прежде всего с огромной трудностью получения Р. Свидетельство этому хотя бы то, что мировое производство Р. пока составляет лишь несколько тонн. А трудности получения, в свою очередь, обусловливаются малыми запасами и распылённостью природных соединений Р. Даже в самых богатых рением молибденовых рудах его содержание не превышает 0,05—0,1%. При окислительном обжиге молибденита MoS2 образуется Re2O7; этот окисел переводят в кислоту HReO4, последнюю превращают в соль — перренат аммония, который восстанавливают до свободного металла водородом при 800 °C: 2NH4ReO4 + 4Н2 - 2Re + Х2 -| 8Н2О
Но это лишь принципиальная схема технологического процесса. А на самом деле путь от руды до металла, осложнённый кропотливой работой по его очистке, значительно длиннее.
От пера авторучки до ракеты — вот современный диапазон применения Р. Он используется везде, где нужны высокие твёрдость и износоустойчивость,— при изготовлении компасов, точнейших весов и других приборов, ответственных деталей сверхзвуковых самолётов и ракет. В обычных осветительных лампах и в фотографических импульсных «лампах-вспыш
249
РТУТЬ
ках» применяются сплавы Р. с вольфрамом — из них нить накаливания более долговечна. Сплав Р. с вольфрамом даёт исключительно ценную термопару, позволяющую точно измерять температуры до 2500 °C. Сплавы Р. с вольфрамом и другими тугоплавкими металлами — молибденом, танталом — отличаются высокой жаропрочностью. Они применяются в авиа-и космической технике. Устойчивость Р. по
отношению к воздуху и воде делает его очень перспективным антикоррозионным покрытием (ренирование). Цистерны и баки, сделанные из ренированных железных листов, применяются для перевозки соляной кислоты. Новая область применения Р.— катализ. Металл оказался очень хорошим катализатором процессов окисления аммиака и метана, гидрирования этилена и спиртов. В. К. Бельский.
РОДИЙ (лат. Rhodium) Rh — химический элемент VIII группы периодической системы Менделеева; атомный номер 45; относится к семейству платиновых металлов.
РТУТЬ (лат. Hydrargyrum) Hg — химический элемент II группы периодической системы Менделеева; атомный номер 80, атомная масса 200,59. Жидкий металл серебристого цвета. Природная Р. состоит из смеси семи изотопов с массовыми числами 196, 198^—202, 204; среди них наиболее распространён изотоп 202Hg (29,8%).
Самородная Р. была известна ещё древним индийцам и китайцам, которые за тысячи лет до н. э. знали её свойство растворять золото и серебро. Древние римляне добывали киноварь (минерал HgS) в горах Испании (Альмаден) из месторождения, не потерявшего промышленного значения и по сей день. Киноварь использовалась как краска, лекарство и космети
ческое средство. Древнегреческий врач и натуралист Диоскорид (I в. н. э.) описывает получение Р. нагреванием киновари на железной сковороде, помещённой в глиняный горшок, покрытый крышкой. По реакции:
HgS + Fe = FeS + Hg
получались пары Р., которые сгущались на холодной крышке в серебристую жидкость. Диоскорид дал ей название «хюдраргирос» (от греч. «хюдор» — вода и «ар-гирос» — серебро), то есть жидкое серебро. Издавна было замечено, что Р. способна растворять многие металлы (этот процесс называют амальгамацией). С помощью амальгамации ещё в Древнем Риме извлекали из руд золото.
Алхимики называли Р. «Меркурием» — по наиболее подвижной планете, носящей имя Меркурия, крылатого вестника богов в античной мифологии. Название «мер-курий» сохранилось для Р. в английском, испанском, французском и итальянском языках. Средневековые алхимики считали Р., серу и соль тремя «первоначалами».
Они верили, что Р. можно превратить в золото. Но, как мы знаем теперь, химическим способом превратить один элемент в другой нельзя. Только развитие ядерной физики позволило учёным получить атомы золота из атомов Р. Однако этот способ чудовищно дорог и, конечно, не применяется для превращения Р. в золото (подробнее о превращениях элементов с помощью ядерных реакций рассказано в статье Элементы химические).
Земная кора содержит всего 8,3-10“6 % Р-(по массе). По распространённости Р. занимает 66-е место среди других элементов. Р.— рассеянный металл. В месторождениях Р. сконцентрировано лишь 0,02°/о от всех известных её запасов в земной коре. Иногда встречается самородная Р., причём чаще её находят в виде сплавов (амальгам) с золотом, серебром и палладием. Основной рудный минерал Р.— киноварь HgS. Наиболее крупные месторождения киновари в СССР находятся в Донбассе, Сибири и Средней Азии.
Р.— единственный металл, жидкий при комнатной температуре (затвердевает при —38,97 °C). Эта блестящая, серебристая жидкость — самая тяжёлая из всех известных жидкостей; её плотность 13,52 г/см3. Р. проводит электрический ток и тепло приблизительно в 50 раз хуже, чем серебро. Сосуд с Р. легко нагреть.— удельная теплоёмкость Р. мала (почти в 30 раз меньше, чем у воды). При нагревании Р. сильно расширяется, что используется для измерения температуры (ртутные термометры). Кипит Р. при 357,25 °C. Пары Р. при высоких температурах излучают голубовато-зелёный свет, богатый ультрафиолетовыми лучами. Такое же излучение происходит и при электрическом разряде в стеклянной трубке, содержащей пары Р. Ртутные кварцевые лампы гораздо экономичнее обычных электрических ламп накаливания. В твёрдом состоянии, при температуре ниже—38,97 °C, Р. легко режется ножом и куётся (это описал ещё в 1759 г. М. В. Ломоносов).
Конфигурация высших энергетических уровней атома Р. 5d106s2. В периодической системе Менделеева Р. завершает побочную подгруппу II группы:'Zn—Cd—Hg. Однако, в отличие от цинка и кадмия, в соединениях Р. бывает не только двухвалентной, но и формально одновалентной.
На самом же деле в последнем случае два атома двухвалентной Р. непосредственно связаны друг с другом: —Hg—Hg—. Поэтому простейшая формула HgX (где X — например, С1) неверна,— следует писать Hg2X2, или X—Hg—Hg—X. При электролитической диссоциации таких солей Р. в раствор переходят двойные двухзарядные катионы Hg2+.
Р.— химически стойкий элемент. В сухом воздухе чистая Р., подобно золоту, платине и другим благородным металлам, не меняет своего внешнего вида. Но если Р. содержит
РТУТЬ
250
Макет химической лаборатории конца XVII— середины XVIII вв. Обратите внимание на приборы для перегонки (перегонные кубы со стеклянными шлемами, к длинному «клюву» которых присоединялись приёмники для собирания жидкостей). Слева на первой полке снизу стоят две колбы с плоским дном и очень узким и длинным горлом. Такие колбы служили для приготовления «самоосаждённой ртути», то есть окиси ртути HgO. С этой целью на дно колбы наливали тонкий слой ртути и нагревали её несколько недель на песчаной бане. При этом пары ртути конденсировались в горле колбы, и жидкая ртуть стекала на её дно. Одновременно ртуть медленно окислялась, превращаясь в красный порошок. По имени английского учёного Роберта Бойля, который в 1673 г. изобрёл эти колбы, они получили название «бойлев ад».
ничтожные количества примесей (например, несколько долей цинка на 100 млн. долей Р.), то её поверхность тускнеет на воздухе, покрываясь плёнкой окислов.
Р. с кислородом образует закись Hg2O, окись HgO и перекись HgO2. Из них наиболее устойчива окись, способная существовать в двух формах — красной и жёлтой.
Красная форма образуется, например, при слабом нагревании нитрата:
2Hg (NO3)2 = 2HgO + 4NO2 + O2
При добавлении щелочей из раствора этой соли выпадает жёлтая окись Р. Обе формы окиси Р. имеют одинаковую кристаллическую структуру. Цвет же их зависит от размеров кристалликов. При поперечнике кристалликов, равном 0,002 мм, цвет HgO жёлтый, при поперечнике 0,01—0,02 мм — красный. Красная окись Р. входит в состав краски для подводных частей морских судов,— она губительно действует на морские организмы. Жёлтая окись Р. применяется в глазных мазях.
Сравнительно легко, гораздо легче своих аналогов — Zn и Cd — Hg. взаимодействует с галогенами. Из её галогенидов наибольший интерес представляют хлористая и хлорная Р. Малорастворимую в воде хлористую Р. Hg2Cl2,
иначе каломель, обычно получают по обменной реакции:
Hg2 (NO3)2 + 2NaCl = Hg2Cl2 + 2NaNO3
Каломель применяют для изготовления каломельных электродов, по отношению к которым измеряют неизвестные потенциалы и других электродов. Хлорная Р. HgCl2, иначе сулема, образуется при нагревании смеси сульфата двухвалентной Р. с поваренной солью:
HgSO4 + 2NaCI = HgCl2 + Na2SO4
В последнем случае пары HgCl2 конденсируются на холодных стенках сосуда, в котором происходит реакция, в виде возгона (по-латыни sublimatum) — отсюда и название «сулема». Это очень сильный яд. Сулему в виде растворов применяют для дезинфекции и для протравливания семян. Интересно, что каломель, в отличие от сулемы, не ядовита.
При растирании порошка серы с Р. образуется чёрный сульфид HgS. Если его нагреть до 350 °C, он переходит в киноварь — красную модификацию. Киноварь применяют в производстве красок.
Р. стоит в ряду напряжений после водорода и поэтому не вытесняет его из кислот. В азотной и концентрированной серной кислотах Р. растворяется. Если взят избыток Р., получается соль Hg|+, а при избытке кислоты образуется соль Hg2+:
Hg + 2H2SO4 = HgSO4 + SO2 + 2H2O
2Hg + 2H2SO4 = Hg2SO4 + SO2 + 2H2O
3Hg + 8HNO3 = 3Hg (NO3)2 + 2NO + 4H2O 6Hg + 8HNO3 - 3Hg2 (NO3)2 + 2NO + 4H2O
Hg2SO4 применяют для изготовления так называемых нормальных элементов, служащих эталоном для точного измерения напряжения. При нагревании смеси Р. со спиртом и крепкой азотной кислотой образуется гремучая Р. Hg(ONC)2. В сухом виде она от удара или трения разлагается со взрывом:
Hg(ONC)2 = Hg + 2CO+N2
Гремучая Р. раньше широко применялась в детонаторах.
Р. и её соединения очень ядовиты. Ещё арабские алхимики и врачи заметили, что змеи и скорпионы покидают жилище, в котором была разлита Р. Её пары даже при комнатной температуре довольно быстро распространяются в окружающем пространстве. Отравление парами Р. сопровождается общей слабостью, острой головной болью, расстройством речи и при длительном воздействии может привести к смерти. Р., вытекшая из разбитого термометра, создаёт большую опасность, так как вследствие своего большого поверхностного натяжения (в шесть с лишним раз выше, чем у воды) металл разделяется на мелкие сферические капельки, которые очень трудно собрать. Но если Р. всё же пролилась, то лучше всего её капельки собрать стеклянной пипеткой с грушей, а затем протереть поверхность, где была Р.,
251
РУБИДИИ
влажным тампоном. В лаборатории пролитую Р. можно собрать медной пластинкой, поверхность которой очищена погружением в азотную кислоту.
В промышленности Р. получают либо нагреванием до 700—800 °C рудного концентрата, состоящего преимущественно из сульфида Р., в токе воздуха, либо нагреванием смеси концентрата с железом или негашёной известью:
HgS + О2 = Hg + SO2 HgS + Fe - Hg + FeS 4HgS + 4CaO = 4Hg + 3CaS + CaSO4 Пары P. отводят в специальные холодильники. Р. очищают от механических примесей фильтрованием через замшу или сукно; она просачивается через натянутый на края приёмного сосуда фильтр под действием силы тяжести.
Хранят и перевозят Р. в стальных баллонах, вмещающих 30—40 кг Р. (сталь ртутью не амальгамируется). В лабораториях Р. очищают перегонкой в вакууме и хранят в запаянных стеклянных ампулах или плотно завинченных стальных баллонах на специальных поддонах.
Высокая плотность, подвижность и химическая стойкость Р. определили её широкое применение в различных областях науки и техники. Так, достаточно столбика Р. высотой всего 760 мм, чтобы уравновесить давление многокилометрового столба атмосферного воздуха. Р. широко используют в барометрах, манометрах, ареометрах, расходомерах, авиационных гироскопах и т. п. Давление меньше 1 атм чаще всего так и измеряют в миллиметрах ртутного столба (мм. рт. ст.). Большой термический коэффициент линейного расширения позволяет применять Р. в термометрах.
Р. нужна в производстве хлора и едкого натра (см. Натрий). При электролизе раствора поваренной соли Р., служащая катодом, циркулирует по дну ванны, что обеспечивает высокую эффективность процесса.
В электротехнике Р. применяется в выключателях, выпрямителях тока, лампах дневного света, кварцевых лампах и др. О соединениях Р. с металлами см. Амальгамы.
Ю. И. Романьков.
РУБЙДИЙ (лат. Rubidium) Rb — химический элемент 1 группы периодической системы Менделеева; атомный номер 37, атомная масса 85,4678. Серебристо-белый металл. Природный Р. состоит из двух изотопов. Из них 85Rb (72,15%) стабилен, a 87Rb (27,85%) радиоактивен (его период полураспада около 50 миллиардов лет). Испуская Р"-частицы, 87Rb превращается в изотоп стронция:
87Rb-+87eSr +13-
Название этого серебристо-белого металла в переводе на русский язык означает «тёмно-красный». Кажется, парадокс?! Но причина такого названия — не цвет ме
талла, а особые линии в красной части спектра, свойственные Р. Он был открыт методом спектрального анализа в минеральных водах Дюркхейма (Германия) немецкими учёными Р. Бунзеном и Г. Кирхгофом в 1861 г. Кстати, Р. Бунзен и получил впервые чистый Р. в 1863 г. восстановлением кислого виннокислого Р. сажей.
Р. достаточно широко распространён в природе. На его долю в земной коре приходится 1,5-10"2% по массе (20-е место среди всех элементов). Но Р. очень рассеян и, в отличие от других щелочных металлов — лития, натрия, калия и даже цезия, не имеет собственных минералов, а всегда, как правило, сопутствует калию или литию. Практическое значение для промышленной добычи Р. имеют такие минералы, как лепидолит (см. Литий) и карналлит (см. Калий). В Соликамске на Урале н аходятся крупнейшие залежи карналлита, и его можно считать перспективным сырьём для выделения Р.
В карналлите Р.— малая примесь (0,04—
0,15%). Но мировые запасы этого минерала столь велики, что общее содержание Р. в подобных месторождениях достигает миллионов тонн. Богаты соединениями Р. воды некоторых минеральных источников.
Р.— вязкий металл, при обычной температуре он выглядит почти как паста. Р. очень легкоплавок (/пл 38,9°С); /кип 703 °C. Металл этот лёгкий, его плотность равна 1,525 г/см3, он очень мягок и легко режется ножом. Пары соединений Р. окрашивают пламя горелки в характерный пурпурово-красный цвет.
Р.— один из самых активных металлов. Он вступает во все реакции, свойственные щелочным металлам, но гораздо более энергично, чем литий, натрий и калий. Конфигурация внешней электронной оболочки атома Р. 5s1 (см. Атом). Его единственный валентный электрон легко отдаётся, и в своих соединениях Р. всегда имеет степень окисления 4-1 (валентность I). Связи атома Р. с другими элементами носят преимущественно ионный характер.
На воздухе Р. мгновенно воспламеняется, его реакция с кислородом идёт даже при очень низких давлениях. При этом образуется, главным образом, надперекись Р.— порошок жёлтого цвета:
Rb 4- О2 — RbO2
Известны также и другие соединения Р. с кислородом: перекиси Rb2O2 и Rb2O3 (Rb2O2-2RbO2) и окись Rb2O, которая может быть получена
РУТЕНИИ
252
лишь в специальных условиях при недостатке кислорода.
Воспламеняется Р. и в атмосфере галогенов: 2Rb + С12 = 2RbCl
При растирании Р. с порошком серы происходит взрыв, и образуется сульфид Rb2S.
С водородом Р. реагирует при повышенных температурах (300—350 °C) в присутствии катализаторов с образованием неустойчивого гидрида RbH. Гидрид Р. энергично взаимодействует с водой, выделяя водород:
RbH + Н2О = RbOH + Н2
С азотом при обычных условиях Р. непосредственно не соединяется, и нитрид Rb3N может быть получен лишь в тихом электрическом разряде. С водой Р. реагирует чрезвычайно бурно, со взрывом:
2Rb + 2Н2О = 2RbOH + Н2
Выделяющийся водород немедленно воспламеняется. Такая реакция проходит даже со льдом при температурах до —108 °C. Образующаяся гидроокись RbOH — очень сильное основание. Её нельзя хранить в стеклянной посуде, так как она разъедает стекло. В стеклянной посуде нельзя работать и с металлическим Р. при повышенных температурах, потому что при 300 °C он разрушает стекло, восстанавливая кремний из SiO2 и силикатов. Р. легко образует сплавы со многими металлами. Одним словом, его нужно очень тщательно ограждать от внешних воздействий. Обычно Р. хранят в запаянных ампулах из особого стекла под слоем тщательно обезвоженного керосина.
Свободный Р. получают восстановлением его хлорида металлическим кальцием в вакууме при 700—800 °C:
2RbCl + Ca = 2Rb + CaCl2 Но этой простой основной стадии предшествует долгая и кропотливая работа по извлечению хлорида Р. из природного минерала карналлита.
До сих пор Р.— редкий элемент не только в природе, но и в промышленности. Круг областей применения этого металла и его соединений пока чрезвычайно узок. Наибольшая часть Р. идёт на изготовление фотоэлементов, работающих в видимой области спектра. Подобно своему аналогу — цезию — Р. используется в качестве газопоглотителя (геттера) при изготовлении вакуумных приборов. Галогениды Р. применяют в медицине как болеутоляющее и успокаивающее средство, а также при лечении некоторых форм эпилепсии.
Однако последние исследования учёных показали, что Р. недолго оставаться «безработным», и его соединения найдут широкое применение в катализе. Добавка соединений Р. в катализаторы намного ускоряет получение синтетической нефти (синтола), метилового спирта, а также стирола и бутадиена — исходных веществ для получения синтетического каучука. Таков далеко не полный перечень возможностей использования Р. и его соединений в будущем. В. К. Бельский.
РУТЕНИЙ (лат. Ruthenium) Ru — химический элемент VIII группы периодической системы Менделеева; порядковый номер 44; относится к семейству платиновых металлов.
«Сложный, длинный путь странствований по расплавам и по растворам прошли химические элементы, прежде чем в горных породах или в минеральных жилах они соединились в твёрдые минералы, наиболее устойчивые химические соединения. На этом длинном пути они испытали много различных превращений. Вместе и неразлучно прошли этот путь только те, которые особенно похожи друг на друга».
Л. Е. Ферсман
(Ферсман А. Е., Занимательная геохимия, 1948, с. 163)
САМАРИЙ (лат. Samarium) Sm — химический элемент III группы периодической системы Менделеева из семейства лантаноидов} атомный номер 62.
СВИНЕЦ (лат. Plumbum) Pb — химический элемент IV группы периодической системы Менделеева; атомный номер 82, атомная масса 207,2. Синевато-серый металл. Природный С. встречается в виде четырёх стабильных изотопов: 204РЬ (1,5%), 206РЬ (23,6%), 207РЬ (22,6%) и 208РЬ (52,3%). Последние три изотопа являются конечными членами радиоактивных рядов распада урана, актиния и тория соответственно (о радиоактивных рядах рассказано в статье Уран).
С.— один из семи металлов (олово, ртуть, зо
лото, серебро, железо и медь), известных человеку с давних времён.Раскопки показали, что ещё за 3—4 тыс.лет до н. э. в Месопотамии и Египте из С. изготовляли монеты, статуэтки, таблички для письма, различные предметы быта. Одно из «семи чудес света» — висячие сады Семирамиды — орошались водой через сложную систему колодцев, трубопроводов и других гидравлических сооружений, сделанных из С.
«Странные капризы» истории — в древности С. был незаменим там, где он сейчас совершенно не употребляется. Ведь вся водопроводная система Древнего Рима тоже была сделана из С. и система эта была весьма разветвлённой. По свидетельству древнеримского инженера и архитектора Витрувия (1 в. до н. э.), точек индивидуального потребления воды в Древнем Риме было более 13,5 тысяч.
До XVII в. С. часто путали с оловом. До сих пор в некоторых славянских языках (польском, чешском) С. называется «олово». О происхождении латинского и других названий С. существуют лишь различные догадки и предположения.
С. по распространённости в земной коре занимает среди других элементов лишь 35-е место (1,6-10“3% по массе). Самородный С. находят крайне редко. Чаще всего элемент встречается в виде сульфида PbS. Этот хрупкий блестящий минерал серого цвета называют галенитом, или свинцовым блеском. Галенит
в месторождениях обычно сопровождается другими минералами С.— они образуются при взаимодействии галенита с кислородом воздуха и двуокисью углерода (см. также Сульфиды). Это — англезит PbSO4 (свинцовый купорос) и церуссит РЬСО3 (белая свинцовая руда). Есть ещё минерал крокоит РЬСгО4 (красная свинцовая руда). Галенит часто встречается совместно со сфалеритом ZnS, халькопиритом CuFeS2, аргентитом Ag2S и другими минералами, образуя полиметаллические руды.
Крупные месторождения С. находятся в Австралии, США, Канаде, Мексике, ГДР, ФРГ и других странах. У нас в СССР залежи руд С. имеются в Казахстане, Восточной Сибири, Северной Осетии и на Алтае. Следы С. как конечного продукта радиоактивного распада всегда содержатся в минералах урана и тория.
С.— тяжёлый металл (плотность 11,34 г/см3). Свежий его срез быстро тускнеет из-за образования на поверхности пдёнки окисла. Плавится С. при 327,4 °C, а кипит при 1725 °C. Это — самый мягкий из обычных металлов: он легко режется ножом и даже царапается ногтем. Его можно без труда прокатать в тонкие листы.
В соответствии с положением С. в периодической системе Менделеева у его атома четыре внешних (валентных) электрона (конфигурация 6s26p2, см. Атом). В соединениях С. проявляет степени окисления +2 и +4 (валентности II и IV). Для С., в отличие от его соседей по IV группе системы — германия и олова, более устойчиво 2-валентное состояние, чем 4-валентное. Поэтому соединения РЬ (+2) не проявляют восстановительных свойств; и, наоборот, соединения РЬ (+4) — сильные окислители.
На воздухе С. покрывается окисной плёнкой, поэтому изделия из него лишены блестящего металлического вида. При нагревании расплавленного С. на воздухе образуется окись РЬО, которая имеет две кристаллические модификации — жёлтую (глёт) и красную (массикот). При нагревании РЬО на воздухе около
СВИНЕЦ
254
500 °C она превращается в РЬ3О4 (сурик) красного цвета. При нагревании с разбавленной азотной кислотой сурик переходит в двуокись С. коричневого цвета:
РЬ3О4 4- 4HNO3 - РЬО2 + 2Pb (NO3)2 - 2Н2О
Можно проделать такой опыт. Асбестовый лист посыпать тонким слоем РЬО2. Под лист поставить горелку так, чтобы остриё пламени касалось центра листа. Через некоторое время станет заметно, что там, где пламя касалось листа, образовался жёлтый кружок РЬО, далее красное кольцо сурика РЬ3О4, и, наконец, по периферии неразложившаяся РЬО2 коричневого цвета:
РЬО2
390-420° 510-550°
--------РЬ3О4---------->
РЬО
При нагревании С. непосредственно соединяется со многими неметаллами, в частности со всеми галогенами, серой, селеном, теллуром. Вода на С. не действует, но в присутствии кислорода медленно разрушает его с поверхности: 2РЬ -4 О2 -4 2Н2О = 2РЬ (ОН)2
В ряду напряжений С. стоит непосредственно перед водородом и практически не вытесняет его из кислот. С холодной соляной и разбавленной серной кислотами С. не реагирует, так как образующиеся РЬС12 и PbSO4 плохо растворимы и покрывают металл защитной плёнкой. В горячей соляной кислоте С. медленно растворяется с образованием комплексного соединения Н[РЬС13]:
РЬ + ЗНС1-Н[РЬС13] + Н2
Горячая концентрированная серная кислота (более 80?^) также растворяет С. При взаимодействии С. с холодной концентрированной азотной кислотой:
РЬ 4 4HNO3 Pb (NO3)2 -г 2NO2 - 2Н2О образуется нитрат С., который не растворим в азотной кислоте; его плёнка предохраняет металл от дальнейшего разрушения. Но в воде Pb(NO3)2 растворим хорошо и поэтому С. легко реагирует с разбавленной азотной кислотой:
ЗРЬ 4- 8HNO3 = ЗРЬ (NO3)2 -!- 2NO 4Н2О
При доступе воздуха С. растворяется в уксусной кислоте с образованием ацетата, дающего тригидрат РЬ (С2Н3О2)2-ЗН,О. Это сладкие на вкус кристаллы, называемые свинцовым сахаром. Но пробовать такой «сахар» ни в коем случае нельзя — он сильно ядовит. Растворяется С. и в щелочах:
РЬ 4- 2КОН = К2РЬО2 -4 Н2
РЬО — амфотерный окисел. При нагревании он взаимодействует со щелочами с образованием солей — пл гамбитов:
РЬО -4 2КОН = КоРЬОо 4- НоО
Однако основные свойства для РЬО более характерны: РЬО -4 2Hi\O3 = Pb(NO3)2 Н.,0
У РЬО2, наоборот, сильнее выражена кислотная функция. Двуокиси С. соответствуют свинцовые кислоты — метасвинцовая Н2РЬО3 и ортосвинцовая Н4РЬО4. Они не выделены в свободном состоянии (известны лишь их соли — плюмбаты). Так, РЬ3О4 можно рассматривать как свинцовую соль ортосвинцовой кислоты (РЬ2+РЬО4“ — ортоплюмбат свинца).
Соединения РЬ(+4) — сильные окислителе. РЬО2 окисляет хлорид-ион до свободного хлора:
+ 4 - 1 +2 0
РЬОо 4- 4НС1 - РЬС12 4- С1, 4- 2Н2О
4-2 4-7
РЬО2 может даже окислить Мп до Мп, то есть является более сильным окислителем, чем перманганат калия: + 4 +2
5РЬО2 4- 2Mn(NO3)2 4- 6HNO3 + 2 +7
= 5Pb(NO3)2 4- 2HMnO4 4- 2Н2О
Полагают, что выплавка С.— один из первых, известных человеку металлургических процессов. Действительно, в принципе, методика получения С. из церуссита РЬСО„ довольно проста. Уже при нагревании около 320°C он разлагается на СО2 и РЬО. Последняя уже при 400—500 °C восстанавливается углём до металла. Более сложна переработка галенитовой руды. Её сначала концентрируют пенной флотацией, а затем нагревают в печи с коксом и известняком. Реагируя с кислородом вдуваемого в печь воздуха, сульфид С. превращается в окись:
2PbS 4- ЗО2 = 2РЬО 4- 2SO2
Одновременно окисляются и примеси. Наконец, раскалённый уголь восстанавливает окисел до металла:
РЬО 4-С = РЬ4-СО
Так получают С., содержащий довольно много примесей. Его очищают электролизом.
Основная масса (25—35%) С. идёт на изготовление пластин для аккумуляторов. Часто С. применяют как материал для реакционных сосудов и камер благодаря коррозионной стойкости металла и лёгкости изготовления из негц, деталей. Так, свинцом покрывают внутренние поверхности химической аппаратуры при производстве серной кислоты. Трубы для перекачки кислот, изоляционный материал для электрических кабелей, свинцовое стекло (хрусталь) — вот далеко не полный перечень областей применения металла.
С. хорошо поглощает радиоактивные излучения и рентгеновские лучи. Поэтому в лабораториях, где работают с радиоактивными веществами, защитные стенки делают из свинцовых кирпичей (каждый такой кирпич весит больше 10 кг).
Многочисленны и очень важны различные сплавы С.— типографские (см. также Сурьма), баббиты и припои (см. также Олово), сплавы
255
СЕЛЕН
для изготовления пуль и дроби. Чистый С. идёт на выработку тетраэтилсвинца (ТЭС) РЬ(С2Н5)4— летучей ядовитой жидкости — отличного антидетонатора, повышающего качество моторного топлива.
Применение соединений С. разнообразно. Это, в первую очередь, различные краски — красный сурик РЬ3О4, жёлтый глет РЬО, свинцовые белила 2РЬСО3* РЬ (ОН)2.
Правда, краски на основе свинцовых белил со временем темнеют — под действием содержащегося в воздухе сероводорода образуется чёрный сульфид PbS. Если же картину осторожно протереть, скажем, перекисью водорода, то чёрный сульфид окислится и перейдёт в белый сульфат PbSO4 и тогда картина снова просветлеет. Таким «чудесным обновлением» икон церковники часто обманывают верующих.
Некоторые растворимые в воде соединения С. используются в медицине как наружные болеутоляющие и противовоспалительные средства. Такова, например, свинцовая примочка — водный раствор основного ацетата С. РЬ (ОН) (С2Н3О2). Однако свинцовая пыль и соединения С. очень ядовиты. Если даже малые дозы С. постоянно попадают в организм, С. накапливается, замещая кальций костного скелета. Максимальное содержание С. в воздухе на промышленных предприятиях не должно превышать 0,00001 мг на 1 л.
В. К. Бельский. СЕЛЁН (лат. Selenium) Se — химический элемент VI группы периодической системы Менделеева; атомный номер 34, атомная масса 78,96. В обычных условиях — серое кристаллическое вещество. Природный С. состоит из шести изотопов: 74Se, 76Se, 77Se, 78Se, 80Se и 82Se. Из них наиболее распространён 80Se (49,8%).
Впервые С. был обнаружен шведским химиком Я. Берцелиусом (1817 г.) в отходах сернокислотного производства. Это было время, когда астрономы открыли планету Урай и несколько так называемых малых планет, в том
числе Цереру и Палладу. Поэтому в названиях открытых в те годы химических элементов — уран, церий, палладий — явно чувствуется «небесное», астрономическое влияние (хотя в основе названий и упомянутых элементов, и небесных тел лежат имена мифологических героев). По аналогии с открытым ранее теллуром, название которого связано с нашей планетой, Берцелиус дал новому элементу имя «селен» (от греч. «селене» — Луна) в честь вечной спутницы Земли.
С. относится к числу рассеянных в природе элементов. Будучи аналогом и постоянным спутником серы, он почти не образует собственных минералов, а входит в небольшом количестве в сульфидные минералы меди, цинка, свинца (см. Сульфиды) или встречается как примесь в самородной сере вулканического происхождения. Содержание С. в земной коре оценивается в 5-10“6 % по массе (68-е место среди прочих элементов).
Подобно сере С. образует несколько аллотропных модификаций. Наиболее устойчивая из них — кристаллический серый С. (плотность 4,807 г/см3; /пл 217°C, /кип 685 °C). Кристаллы его построены из правильных спиралевидных цепей, состоящих из атомов Se (рисунок). В аморфном С. (красного цвета) такие цепи переплетаются хаотически. Элементарный (серый) С.— полупроводник.
Важное и интересное свойство его — увеличение электропроводности при освещении примерно в 1000 раз. Под действием световой энергии часть электронов отрывается с наружной электронной оболочки атомов Se, благодаря чему и возникает возможность прохождения электрического тока. Используя это свойство, создают селеновые полупроводниковые фотоэлементы — приборы, в которых сила тока зависит от освещения. Такой фотоэлемент может, например, включать городское электроосвещение с наступлением сумерек и выключать на рассвете, то есть при определённой естественной освещённости города. (О фотоэлементах рассказано также в статье Кремний.)
По своему химическому поведению С.— неметалл. У его атома, как и у атома серы, шесть внешних (валентных) электронов (конфигурация 4s24p4, см. Атом). В соединениях С. проявляет степени окисления —2, 4-4, 4-6 (валентности II, IV, VI).
При обычной температуре С. реагирует только с фтором, при небольшом нагревании —
Элементарная ячейка кристаллов серого селена имеет форму правильной шестиугольной призмы. (О том, что такое элементарная ячейка, см. подпись к рис. 2 в статье Железо.) Такую элементарную ячейку и кристаллы с такими ячейками называют гексагональными (от греч. «гекса» — шесть, «тонна»—угол). Как видно из рисунка, атомы в сером селене прочными ковалентными связями соединены в спиральные цепи. Между цепями действуют значительно более слабые (остаточные) силы связи.
СЕЛИТРЫ
256
с хлором и бромом. При горении С. на воздухе образуется двуокись SeO2 — белое кристаллическое вещество. Высший окисел — трёхокись SeO3 получается только косвенным путём Прямое взаимодействие С. с металлами приводит к селенидам, например K2Se, Al2Se3. По реакции С. с водородом (300 СС) или при действии разбавленных кислот на селениды образуется газообразный селенистый водород:
K2Se - 2НС1 = H2Se -- 2КС1
H2Se — бесцветный газ с отвратительным запахом, ещё более ядовитый, чем сернистый водород. Растворяясь в воде, он даёт кислоту более сильную, чем сероводородная. H2Se — сильный восстановитель. При окислении H2Se образуется аморфный С.:
H2Se -г S = H2S -г Se
Растворение С. в азотной кислоте даёт селенистую кислоту:
3Se -г 4HNO3 + Н2О = 3H2SeO3 + 4NO
В отличие от сернистой кислоты, H2SeO3 может быть выделена в свободном состоянии. Она проявляет как окислительные, так и восстановительные свойства (последние — в меньшей степени). При действии перекиси водорода превращается в селеновую кислоту:
H2SeO3 + Н2О2 = H2SeO4 + Н2О
Как кислота H2SeO4, так и её соли (селе-наты) очень напоминают по свойствам серную кислоту и сульфаты, а селеновый ангидрид SeO3 подобен серному ангидриду SO3. Однако соединения, содержащие С. в окислительном состоянии +6, более сильные окислители, чем аналогичные соединения серы. Так, селеновая кислота способна окислять хлористый водород:
H2SeO4 + 2НС1 = H2SeO3 + Cl2 + Н2О
Поэтому смесь селеновой и соляной кислот действует подобно царской водке и способна растворять золото и платину. Все соединения С. очень ядовиты.
Ежегодное мировое производство С. составляет несколько сотен тонн. Для получения С. проводят обжиг отходов (шламов), образующихся при очистке меди электролизом. С. превращается в двуокись SeO2, которая легко возгоняется при 315 °C. Восстановление SeO2 сернистым ангидридом даёт аморфный С.:
SeO2 + 2SO2 = Se + 2SO3
Остроумным способом очистки С. является его своеобразная «перекристаллизация». С. растворяют в горячем концентрированном растворе сульфита натрия. Получающаяся соль Na2SSeO3 аналогична тиосульфату натрия
Na2S2O3 (см. Сера), но разлагается при охлаждении, выделяя свободный С.:
нагревание w т _
Na — Ох--------------------->Na—Ох ZO
)S = O + Se
Na—Oz *--------Na—Oz ^Se
охлаждение
Наибольшее применение С. находит в полупроводниковой технике, в частности для изготовления выпрямителей переменного тока. Интересно использование С. в так называемых вентильных фотоэлементах, где он работает не сам по себе, а в виде соединений с каким-либо металлом. Такие фотоэлементы не требуют электроэнергии извне, а сами вырабатывают электрический ток при освещении. Они применяются в фотоэкспонометрах и многих других приборах, регистрирующих освещённость и её изменения. Чувствительность таких приборов очень велика, а сила тока, возникающего при освещении, достаточна, например, для приведения в действие затвора фотоаппарата для автоматической установки в нём диафрагмы.
Использование С. в других областях народного хозяйства невелико. Двуокись SeO2 добавляют в стекло для его окрашивания в красивый рубиновый цвет; её применяют также в органическом синтезе (главным образом как окислитель и катализатор). В. Л. Василевский. СЕЛЙТРЫ, общее название нитратов натрия NaNO3, калия KNO3, аммония NH4NO3, кальция Ca(NO3)2 и бария Ba(NO3)2.
В природе С. (калиевая, натриевая и кальциевая) образуются при разложении различных органических остатков под действием бактерий. Особенно интенсивно идёт этот процесс в сухих, жарких областях Земли, лишённых растительности. Так, при гниении птичьего помёта (гуано) выделяется аммиак, который под влиянием бактерий окисляется кислородом воздуха сначала в азотистую, а затем в азотную кислоту,— из неё и образуются С. Осадки смывают С. в ложбины, и там возникают целые скопления солей. Огромные залежи натриевой С. имеются на высокогорном плато в Чили. Поэтому натриевую С. и называют чилийской. Уже многие миллионы лет этот район представляет собой жаркую, безводную пустыню. Многочисленные скопления С. находятся на узкой полосе вдоль берега, тянущейся с севера на юг на расстояние свыше 700 км между портами Икике и Антофага-ста. Через эти порты идёт вывоз С. в США и другие страны. Чилийская С. содержит примесь (около 0,02%) йодата натрия NaIO3, из которого получают иод.
Калиевая С. уже в VIII в. использовалась в Византии для приготовления зажигательных смесей. С конца
257
СЕРА
XIV в. из неё готовят чёрный порох — смесь тонко измельчённых калиевой С. (75%), древесного угля (15%) и серы (10%). Воспламенение пороха может быть описано такой реакцией:
2KNO3 + S + ЗС = K2S + N2 + ЗСО2
Выделяющиеся газы и выталкивают пулю из ствола ружья. А пороховой дым — это сульфид калия, образующийся в виде мельчайших твёрдых частичек. В наше время чёрный порох используют лишь для снаряжения охотничьих патронов.
Вплоть до 1840-х гг. калиевую С. получали в селитряницах — кучах, сложенных из растительных и животных отбросов, смешанных с известняком, мергелем, старой штукатуркой, соломой и землёй. Кучи время от времени поливали нечистотами. Через 2—3 года на поверхности куч появлялся белый налёт, состоящий главным образом из нитрата кальция. Его выщелачивали водой, полученный раствор обрабатывали поташом К2СО3 (извлечённым из золы). При этом по реакции:
Ca(NO3)2 + К2СО3 = СаСО3 + 2KNO3
из кальциевой С. получали калиевую. Эту «сырую селитру» очищали перекристаллизацией и применяли для изготовления пороха. Кроме того, С. добывали из небольших залежей в Индии и Китае, откуда её ввозили в Европу (поэтому калиевую С. одно время называли индийской).
Первые месторождения натриевой С. на нынешней территории Чили были открыты в начале XIX в. Однако ввоз чилийской С. в Европу ещё в 1830-х гг. был очень невелик. Её применяли главным образом для получения азотной кислоты. (Ведь натриевая С. гигроскопична, и содержащий её порох попросту подмок бы. Для изготовления пороха годится лишь негигроскопичная калиевая С.)
В 1840-х гг. начали вести конверсию (превращение) NaNO3 в KNO3 по реакции:
2NaN О3 + К2СО3 = Na2CO3 + 2KNO3
А с конца 1850-х гг., когда на Стасфуртском месторождении поваренной соли стали добывать дешёвые соли калия— сильвин КС1, карналлит КС1 MgCl2*6H2O и др., Калиевую селитру издали получать по реакции:
NaNO3 + КС1 = KNO3 + NaCl
Растворимость NaCl почти не зависит от температуры, а растворимость KNO3 при охлаждении горячего насыщенного раствора сильно уменьшается, и на дне котла оседают кристаллы KNO3. В начале 1870-х гг. в провинциях Атакама (принадлежавшей Боливии) и Тарапака (принадлежавшей Перу) были открыты новые богатые залежи натриевой С. Чили заявила претензии на обладание этими областями. Вспыхнула Тихоокеанская война 1879—1883 гг., в результате которой Атакама, Тарапака и Антофагаста (принадлежавшая Боливии) перешли к Чили.
А между тем в мире рос спрос на азотные удобрения. Натриевую С. начали применять как удобрение ещё с момента открытия её залежей в Чили. Но скоро и эти мощные её скопления не могли удовлетворить растущего спроса на связанный азот. Надо было найти способы промышленного получения С. из более доступных веществ.
В наше время в промышленности сначала получают натриевую С., действуя окислами азота на раствор едкого натра или соды. При этом образуется смесь нитрата и нитрита натрия; NaNO2 окисляют азотной кислотой в NaNO3. Натриевую С. превращают в калиевую взаимодействием с КО (см. выше).
Аммиачную С. получают нейтрализацией 50—60 %-ной азотной кислоты аммиаком:
nh3 + hno3-nh4no3
Раствор NH4NO3 выпаривают в вакууме, сухой остаток превращают в гранулы (мелкие зёрна).
Аммиачная С.— непременный компонент многих взрывчатых веществ, называемых аммонитами. Однако главная область применения С.— самая мирная: аммиачная, натриевая и калиевая С.— это азотные удобрения, без которых невозможно развитие современного сельского хозяйства (см. Минеральные удобрения).
СЁРА (лат. Sulfur) S — химический элемент VI группы периодической системы Менделеева; атомный номер 16, атомная масса 32,06. Твёрдое хрупкое вещество жёлтого цвета. Природная С. состоит из четырёх стабильных изотопов: 32S (95,02%), 33S (0,75%), 34S (4,21%), 36S (0,02%).
Историческая справка. С. известна человечеству с самых древних времён. Встречаясь в природе в свободном состоянии, она обращала на себя внимание характерной жёлтой окраской, а также тем резким запахом, который сопровождал её горение. Считалось даже, что запах и голубое пламя, распространяющиеся горящей С.,
отгоняют демонов. Сернистый ангидрид SO2—удушливый газ, образующийся при сжигании С., ещё в древности использовался для отбеливания тканей. При раскопках Помпеи нашли картину, на которой изображён противень с С. и приспособление для подвешивания над ним материи. Издавна употреблялась С. и её соединения для приготовления косметических средств и для лечения кожных заболеваний. И очень давно её начали применять для военных целей. Так, в 670 г. защитники Константинополя сожгли арабский флот с помощью «греческого огня»; это была смесь селитры, угля и С. Те же вещества входили в состав чёрного пороха, широко применявшегося в Европе ещё в средние века и до конца XIX в. (см. Селитры).
Серная кислота, одно из самых важных соединений С., была открыта, по-видимому, к X в. Начиная с XVIII в. её производят в промышленных масштабах и вскоре она становится важнейшим химическим продуктом, необходимым и в металлургии, и в текстильной промышленности, и в других, самых различных отраслях хозяйства. В связи с этим начались ещё более интенсивные поиски месторождений С., изучение химии С. и её соединений и совершенствование методов их извлечения из природного сырья.
Русское название элемента происходит от древнеиндийского (санскритского) слова «сира» — светло-жёлтый. Приставка «тио», часто встречающаяся в словах, имеющих отношение к С., идёт от греческого названия С. — «тёйон» (небесный, божественный). Ведь С. издавна была символом горючести; огонь же считался достоянием богов, пока Прометей, как говорит миф, не принёс его людям.
17 хэш
СЕРА
258
Сера в природе. С. относится к весьма распространённым элементам: земная кора содержит 4,7-10“2% С. по массе (15-е место среди других элементов), а Земля в целом — много больше (0,7%). Главная масса С. находится в глубинах Земли, в её мантии — слое, расположенном между земной корой и ядром Земли. Здесь, на глубине примерно 1200— 3000 км, залегает мощный слой сульфидов и окислов металлов. В земной коре С. встречается как в свободном состоянии (самородная сера), так и, главным образом, в виде соединений — сульфидов и сульфатов. Из сульфидов в земной коре наиболее распространены серный колчедан (пирит) FeS2, медный колчедан (халькопирит) FeCuS2, свинцовый блеск (галенит) PbS, цинковая обманка (сфалерит) ZnS. Большие количества С. встречаются в земной коре в виде труднорастворимых сульфатов — гипса CaSO4-2H2O, ангидрита CaSO4 и барита BaSO4. В морской воде растворены сульфаты магния, натрия и калия (см. Гидросфера}.
Интересно, что в древние времена геологической истории Земли (около 800 млн. лет назад) сульфатов в природе не было. Они образовались как продукты окисления сульфидов, когда в результате жизнедеятельности растений возникла кислородная атмосфера Земли (см. также Кислород). В вулканических газах обнаруживают сернистый водород H2S и сернистый ангидрид SO2. Поэтому самородная С., встречающаяся в районах, близких к действующим вулканам (Сицилия, Япония), могла образоваться при взаимодействии этих двух газов:
2H2S + SO2 = 3S + 2H2O
Другие залежи самородной С. связаны с жизнедеятельностью микроорганизмов.
Микроорганизмы участвуют в многообразных химических процессах, которые в целом составляют круговорот С. в природе. При их содействии сульфиды окисляются до сульфатов. Сульфаты поглощаются живыми организмами, где С. восстанавливается и входит в состав белков и других жизненно важных веществ. При гниении отмерших организмов белки разрушаются и образуется сернистый водород, который далее окисляется либо до элементарной С. (так и накапливается самородная С.), либо до сульфатов. Интересно, что бактерии и водоросли, окисляющие сероводород до С., собирают её в своих клетках. Клетки таких микроорганизмов могут на 95% состоять из чистой С.
А можно ли установить, каково происхождение природной самородной С.— вулканическое (по приведенной выше реакции) или биогенное (то есть связанное с жизнедеятельностью организма)? Оказывается можно, и по
могают этому два обстоятельства.w Во-первых, вулканическая С. всегда содержит примесь химического аналога С.— селена; микроорганизмы же избегают включать селен в свой жизненный цикл (из-за его ядовитости) и потому биогенные месторождения С. селена не содержат. Во-вторых, живые существа усваивают «лёгкий» изотоп 32S чуть-чуть предпочтительнее, чем тяжёлый 34S. Поэтому соотношение этих двух изотопов в биогенной С. отличается от среднего природного.
Элементарную С. добывают из самородных руд и другого сырья (см. ниже). Главные месторождения самородной С. расположены в Мексике, США, СССР, Польше, Италии, Японии.
Физические и химические свойства. Молекула С. при обычной температуре и вплоть до 150 °C состоит из восьми атомов, соединённых друг с другом, как показано на рисунке. (В уравнениях химических реакций принято, однако, обозначать молекулу С. просто S, а не S8.) В парах С. при температуре кипения (444,6 °C), кроме молекул S8, существуют также S6, S4 и S2, причём с дальнейшим нагреванием крупные молекулы распадаются, и при 900 °C остаются лишь молекулы S2, которые при ещё более высоких температурах диссоциируют на атомы. При этом изменяется и окраска паров С. (оранжевая — красная — бесцветная).
С. относится к числу элементов, имеющих аллотропные модификации. Молекулы S8 при обычных условиях образуют кристаллы ромбической (октаэдрической) a-S. При быстром нагревании такие кристаллы плавятся при 112,8 °C. Однако если нагревание вести медленно, то начиная с 95,5 °C a-S превращается в другую модификацию, 0-S, называемую также за форму её кристаллов моноклинной (призматической). Такая 0-S плавится при 119,3 °C. Обе модификации имеют и различную плотность (2,07 г/см3 для а- и 1,96 г/см3 для 0-формы). Другие аллотропные видоизменения С. в обычных условиях неустойчивы и легко превращаются в a-S.
Очень интересны свойства расплавленной С. При температуре выше 155—160 °C подвижная светло-жёлтая
Строение молекулы серы S8. Эта молекула представляет собой восьмичленное кольцо, но не плоское, а «гофрированное» (оно напоминает корону).
259
СЕРА
жидкость, состоящая из отдельных молекул S8 (X-S), темнеет, вязкость её вдруг возрастает почти в 20 тыс. раз (при 187 °C), так что расплав теряет подвижность и не выливается из колбы. Считают, что при этом кольца S8 разрываются и происходит полимеризация: nS8-*(S8)„, причём образуются длинные, похожие на спираль молекулы ц-S, содержащие до 10 000 атомов. При дальнейшем нагревании вязкость расплава уменьшается, так как длинные цепи рвутся на более короткие отрезки. Выливая нагретый до 200—300 °C расплав в холодную воду, получают резиноподобную пластическую С., в которой из-за быстрого охлаждения длинные молекулы не успевают «перестроиться» и дать устойчивые кристаллы a-S. Удивительна эластичность пластической С.! Кусок такой С. можно растянуть в 10 раз! Этот «неорганический каучук» был бы замечательным пластическим материалом, но он очень неустойчив: при комнатной температуре уже через несколько дней пластическая С. превращается в хрупкую a-S.
С. нерастворима в воде, но несколько растворима в бензине, спирте и других органических растворителях и хорошо — в сероуглероде CS2. Тепло и электричество С. проводит плохо. Интересно сравнить её с водой, которая, как известно, очень плохой проводник электрического тока. С. проводит ток в 60 млн. раз хуже и является типичным диэлектриком. ‘При трении о кожу палочка С. заряжается отрицательно и может служить источником статического электричества.
В соответствии с положением С. в периодической системе Менделеева её атом имеет шесть внешних (валентных) электронов (конфигурация 3s23p4, см. Атом). При взаимодействии с элементами, сравнительно легко отдающими электроны, С. принимает на внешнюю оболочку недостающие для её завершения 2 электрона и проявляет себя, подобно кислороду, как окислитель; степень окисления С. при этом равна—2. Но благодаря большему радиусу атома С. может и отдавать электроны внешней оболочки, проявляя по отношению к более активным неметаллам свойства восстановителя; максимальная степень окисления С. при этом равна +6. Валентность С. в соединениях меняется от II до VI.
Из всех металлов лишь золото и платина способны противостоять действию С. при всех условиях. Уже при комнатной температуре С. окисляет щелочные металлы, щелочноземельные металлы, медь, серебро, ртуть и образует сульфиды. При нагревании С. реагирует со свинцом, алюминием, железом, цинком. Так, медная спираль, помещённая в пары С., раскаляется и превращается в сульфид меди Cu2S. При поджигании с помощью магниевой ленты смеси порошков С. и алюминия идёт бурная реакция:
2А1 + 3S = A12S3
Интересно, что эту реакцию вызывает и смачивание такой смеси небольшим количеством
тёплой воды, играющей роль катализатора (как и реакция алюминия с иодом).
Из «обычных» неметаллов не реагируют с С. лишь азот и иод. Однако даже с такими активными неметаллами, как кислород, хлор и бром, взаимодействие идёт с достаточной скоростью лишь при нагревании. Сжигание С. на воздухе приводит к получению двуокиси серы, или сернистого ангидрида'.
S + о2 = so2
Высший окисел — серный ангидрид SO3— при этом образуется лишь как небольшая примесь. С водородом С. реагирует при нагревании:
H2 + S^±H2S причём равновесие этой реакции смещено в сторону образования сернистого водорода лишь до 350 °C, а при более высоких температурах H2S разлагается. При нагревании С. реагирует с хлором, давая S2C12 (а в избытке хлора SCI2). По своему строению S2C12 напоминает перекись водорода: С1—S—S—Cl. С бромом образуется только S2Br2. А небольшой по размерам атом фтора даёт с С. даже SF6 — газообразное химически инертное и неядовитое вещество. Все другие галогениды С.— ядовитые жидкости, легко разрушающиеся водой. Фосфор в отсутствие воздуха при нагревании с С. образует сульфиды, например P2S5. Углерод при 800— 900 °C даёт сероуглерод CS2.
Элементарная С. выступает в роли восстановителя по отношению к таким сильным окислителям, как HNO3, КС1О3, КМпО4 и т. д.:
S + 2HNO3 = H2SO4 4- 2NO
В целом же для С. и её соединений характерна цепочка окислительно-восстановительных взаимодействий, в которых степень окисления С. равна —2, 0, +4, —6 (см. схему).
— 2 0 +4 +6
H2S^±S^SO2 so3 (H2SO3) (H2SO4)
и сульфиды и сульфиты и сульфаты
Интересная особенность химии С.— способность атомов S разной степени окисления реагировать между собой. Примером служит взаимодействие С. с концентрированной серной кислотой при нагревании:
+ 6 0 +4
2H2SO4 + S = 3SO2 + 2Н2О
В обратном направлении эта реакция идёт при нагревании SO2 с водой в запаянной трубке. Другой интересный пример — образование тиосульфата натрия при кипячении С. с раствором Na2SO3:
Na2SO3 + S = Na2S2O3
СЕРЕБРО
260
Тиосульфат натрия (старое название — гипосульфит) содержит один атом С. с валентностью VI, другой—с валентностью II.
Na—уЭ Na—o/S^S
Тиосерная кислота, отвечающая этой соли, неустойчива и в водных растворах распадается с выделением сернистого ангидрида и очень мелких частичек элементарной С.
Для химии С. характерна тенденция к образованию связи нескольких атомов S между собой. При нагревании растворов сульфидов с порошком С. образуются полисульфиды:
Na.,S + nS = Na2S„+1
где n=l—8.
Широко известным веществом такого типа ZS является пирит FeS2, или Fe^ | XS
Получение и применение. В промышленности элементарную С. получают и очищают несколькими способами. Самородную С. извлекают с глубины 150—250 м так: в скважину вставляют тройную трубу; через внешнюю полость подают под давлением перегретую до 160—170 °C воду, которая заставляет С. расплавиться. Под давлением сжатого воздуха, поступающего по самой тонкой внутренней трубе, расплавленная С. поднимается на поверхность. Самородную С., значительно загрязнённую пустой породой, выплавляют в специальных чугунных ретортах; пары же С. собирают в больших камерах, где они конденсируются в виде мелкого порошка, называемого ещё со времён алхимиков серным цветом (flores chemici — химические цветы). Если же С. переплавляют и разливают в формы (обычно в виде палочек), её называют чер е н-ковой С.
Элементарную С. можно получить каталитическим окислением сероводорода H2S в присутствии Fe2O3 или А12О3. Источник же сероводорода в промышленности — природные газы и газы, образующиеся при коксовании угля и переработке нефти. Сероводород в промышленных масштабах получают также из гипса CaSO4-2H2O или из барита BaSO4, например так:
BaSO4 + 4С = BaS + 4СО
BaS + СО2 Z Н2О = ВаСО3 + H2S
Ежегодное мировое производство С. исчисляется миллионами тонн. По словам академика А. Е. Ферсмана, «сера—двигатель химической промышленности». И действительно, со времени промышленной революции конца
XVIII — начала XIX вв., вызвавшей строительство крупных химических заводов, серная кислота была основным продуктом химии и применялась в самых разнообразных производствах. По количеству получаемой серной кислоты долгое время судили об уровне химической промышленности страны. И сейчас около половины всей производимой С. расходуется для получения H2SO4. Правда, теперь серную кислоту по большей части получают из другого, более дешёвого сырья (см. Серная кислота).
Кроме того, 25% мирового производства С. идёт на получение гидросульфита кальция Ca(HSO3)2, необходимого в бумажной промышленности. А потребность в бумаге —тоже своего рода двигателе прогресса — неуклонно возрастает. И ещё одно важнейшее применение С. в наши дни делает её крайне необходимой. С.— самый распространённый вулканизующий агент для каучука. Под действием С. каучук приобретает новые, необходимые для его эксплуатации физические свойства, превращаясь в резину.
С. необходима и во многих других отраслях народного хозяйства — в производстве спичек и искусственного шёлка, для борьбы с вредителями сельскохозяйственных культур, для синтеза различных органических веществ и т. д. Очень чистую С. используют в медицине.
В. Л. Василевский.
СЕРЕБРО (лат. Argentum) Ag — химический элемент I группы периодической системы Менделеева; атомный номер 47, атомная масса 107,868. Блестящий белый металл. Природный элемент состоит из смеси двух стабильных изотопов,относительное содержание которых примерно одинаково: 107Ag (51,35%) и 109Ag (48,65%).
С. встречается на Земле в свободном виде, а потому стало известно человеку ещё в доисторические времена, очевидно
вскоре после золота и меди. Уже за 2500 лет до н. э. С., как и золото, использовалось в качестве денег во всех странах, расположенных от Инда до Нила. Вначале С. считалось чрезвычайно редким металлом и ценилось всего лишь в 2,5 раза дешевле золота. К 800 г. до н. э. добыча С. значительно расширилась, особенно в Греции, и оно стало цениться уже в 13, а затем и в 15 раз дешевле золота. Такое соотношение их ценности оставалось и всё последующее время, вплоть до 1874 г., когда вследствие большого притока С. из Америки и Австралии цена на него упала в 3 раза. От красивого белого цвета С. (по-гречески «аргос» — белый, блестящий, сверкающий) произошло его греческое название «аргирос», а от него — латинское «аргентум». Алхимики обозначали С. лунным серпом (см. Металлы).
261
СЕРЕБРО
Серебро в природе. По распространённости в земной коре (7-10“6% по массе) С. занимает всего лишь 67-е место среди других элементов; однако в 20 раз превосходит золото. Встречается С. в виде самородков с примесью золота, ртути и сурьмы; известны серебряные самородки весом до 13,5 тонн. Из минералов С. наиболее важны серебряный блеск Ag2S и роговое С. AgCl. Однако и самородное С., и его собственные минералы редки. Основная масса С. извлекается в качестве побочного продукта при добывании других металлов, главным образом свинца и меди. Крупнейшие месторождения серебряных руд находятся в Мексике и США. В СССР богатые месторождения С. встречаются в Средней Азии, Сибири, на Дальнем Востоке.
Физические и химические свойства. С. сверкает — оно почти полностью отражает падающий на него видимый свет. В этом отношении С. не имеет себе равных среди других металлов. Лучше всех металлов С. проводит тепло и электрический ток. По ковкости же и тягучести оно уступает только золоту. Из С. можно получить лист толщиной всего в четверть микрона. Из 1 г С. можно вытянуть проволоку длиной около 2 км. С. твёрже золота, но мягче меди. Из него легко изготовить изделия любой формы и профиля, правда, они получаются слишком мягкими и быстро истираются при употреблении; чтобы сделать С. более твёрдым, его сплавляют с медью. С.— металл довольно тяжёлый (плотность 10,5 г/см3), плавится оно при 960,8 °C, кипит при 2212 °C.
Как и другие благородные металлы, С. химически малоактивно: в сухом чистом воздухе не изменяется,.не вытесняет водорода из кислот (в ряду напряжений стоит после водорода). Не действуют на С. и расплавленные щёлочи. При плавлении С. растворяет кислород, который выделяется при затвердевании расплава, вследствие чего на поверхности металла возникают как бы маленькие вулканические кратеры— С. разбрызгивается. Поэтому из чистого С. нельзя изготавливать изделия путём отливки (то есть разливая расплавленный металл в формы). Из серебряных же сплавов изделия отливать можно.
Для С. характерно большое сродство к сере. Если потереть металл кусочком серы, то он темнеет из-за образования плёнки сульфида:
2Ag + S = Ag2S
Темнеет С. и на воздухе, содержащем сероводород и другие летучие соединения серы.
Видели ли вы почерневшие «от времени» старинные серебряные подсвечники или давно не чищенные тёмные серебряные ложки? Чтобы удалить чёрный налёт с серебряного предмета, следует протереть его водным раствором тиосульфата натрия Na2S2O3 (1:4), в котором Ag2S растворяется с образованием комплексной соли состава
Русская серебряная чаша XII — XIII вв. Государственный Эрмитаж. Из таких чаш (их называли братинами) на братских дружеских пирушках пили, черпая напиток ковшами.
Na3[Ag(S2O3)2]. Очищенный предмет надо прокипятить в растворе квасцов и тщательно промыть водой — для удаления избытка тиосульфата; без этой операции предмет почернеет ещё больше. Чтобы С. не темнело совсем, его покрывают тончайшими бесцветными плёнками лака, солей или металлов, стойких к действию серы.
Свободные галогены при обычной температуре медленно соединяются с С. Кислоты, содержащие кислород, например кипящая концентрированная серная и холодная разбавленная азотная, растворяют С. (как и медь) с выделением соответственно SO2 и NO:
2Ag + 2H2SO4 = Ag2SO4 + SO2 + 2H2O
3Ag + 4HNO3 = 3AgNO3 + NO + 2H2O
В растворе цианистого натрия в присутствии кислорода воздуха С. (как и золото) растворяется с образованием комплексного соединения:
2Ag + 4NaCN + Ч2О2 + Н2О= = 2Na [Ag (CN)21 + 2NaOH
В царской водке С., в отличие от золота, не растворяется, так как на его поверхности образуется защитная плёнка нерастворимого хлорида AgCl.
В соответствии со своим положением в I группе периодической системы С. в большинстве соединений проявляет степень окисления +1 (одновалентно). Однако есть и производные С. в степенях окисления +2 и +3, например AgO, AgF2, комплексное соединение KlAgFJ. Проявление С. высших степеней окисления объясняется тем, что в реакциях атома С. может участвовать не только единственный валентный s-электрон внешней оболочки, но также один или два d-электрона предыдущей оболочки (конфигурация 4d105s1, объяснение см. в статье Атом). Окислы С. могут быть получены косвенным путём. Закись Ag2O бу
СЕРЕБРО
262
ро-чёрного цвета осаждается при введении ионов ОН" в раствор, содержащий ионы Ag+:
2AgNO3 + 2 КОН = Ag2O + 2KNO3 + Н2О Возможно, что как промежуточный продукт при этом образуется Ag(OH), которую, однако, выделить не удаётся. Если на закись Ag2O подействовать озоном, то образуется окись AgO чёрного цвета, которая разлагается уже при 100 °C, при дальнейшем нагревании взрывается.
Из галогенидов С. в воде растворим фторид AgF. Хлорид, бромид и иодид — AgCl, AgBr и Agl отличаются очень слабой растворимостью в воде и в разбавленных кислотах. Их получают действием соответствующих галогенидов натрия или калия на раствор нитрата С.:
AgNO3 + NaCl = AgCl + NaNO3
Образующиеся галогениды С. выпадают в осадок в виде хлопьев (AgCl — белый, AgBr и Agl — желтоватые). Такие реакции имеют большое значение в аналитической химии — ионы Ag+ служат очень чувствительными реагентами на галогенид-ионы. Наименее растворимая соль С.— сульфид Ag2S. Из растворимых солей С. наиболее распространён нитрат AgNO3; его получают непосредственным растворением С. в азотной кислоте.
Характерная особенность С., как и других элементов побочной подгруппы I группы,— способность образовывать комплексные соединения. Одно из них, Na[Ag(CN)2], мы уже упоминали. Хорошо известно, что AgCl практически нерастворим в воде и кислотах. Однако он растворяется, например, в растворе аммиака, причём образуется комплексное соединение [Ag(NH3)2]CL (О комплексных соединениях можно прочитать в статье Кобальт.)
Получение и применение.
Ещё жители древнего Карфагена умели добывать С. из его руд амальгамацией. Процесс этот основан на способности ртути вытеснять С. из его соединений. Выделенное С. растворяется в избытке ртути с образованием амальгамы. Этот сплав можно легко отделить от пустой породы, а затем удалить ртуть возгонкой. Так получается чистое С. Метод цианирования — извлечение золота и С. из руд с помощью растворов NaCN (или KCN) — был открыт в 1843 г. в России П. Р. Багратионом (племянником прославленного героя Отечественной войны 1812 г.).
Цианирование и сейчас служит основой добычи С. из серебряных руд. Предварительно руду измельчают и обогащают. Затем обогащённый концентрат цианируют — переводят С. в раствор действием NaCN по приведенной выше реакции. Образующийся цианидный комплекс С. разрушают цинковой пылью по обменной реакции:
2Na [Ag (CN)J + Zn = Na2 [Zn (CN)J + 2Ag
Полученный осадок С. отправляют на переплавку, в процессе которой С. очищается от
примесей. Заметим, что С. было первым металлом, полученным в очень чистом состоянии-Это сделал ещё в прошлом веке бельгийский химик Ж- Стас; чистое С. учёный использовал как своеобразный эталон для точного определения атомных весов других элементов.
В наше время основную часть С. добывают в качестве побочного продукта при переработке свинцово-цинковых и медных руд.
Химическая стойкость С. позволяет (точнее, вынуждает — ведь С. дорого и дефицитно) изготовлять из него специальное химическое оборудование и аппаратуру для пищевой промышленности. Значительное количество С. идёт на покрытие радиодеталей ответственного назначения, на серебрение проводов в высокочастотной радиотехнике, стекла и керамики в электронной аппаратуре. Благодаря хорошей способности отражать свет С. издавна применялось для изготовления зеркал, а позже — отражателей прожекторов и телескопов. Однако зеркало из С. легко чернеет от присутствия следов H2S в воздухе; в последнее время для этих целей С. заменяют алюминием.
Первый химический источник электрического тока — знаменитый «вольтов столб» представлял собой чередующиеся диски С. и олова, разделённые прокладкой из сукна, смоченного кислотой. И сейчас немалое количество С. идёт на производство серебряно-цинковых аккумуляторов, мощность которых на единицу массы и объёма является наивысшей среди всех известных аккумуляторов.
С. было первым металлом, пришедшим в медицину. В Древнем Египте лечили язвы и раны, прикладывая к ним пластинки из С. Было замечено, что вода, находясь в сосудах из С., не портится. В XIX в. учёные выяснили, что С. в ничтожнейших концентрациях способно убивать бактерии. Для того чтобы обеззаразить воду ста таких бассейнов, как «Москва», достаточно было бы диспергировать (распылить) в ней при помощи электрического тока 1 г С. (на самом же деле воду в бассейне «Москва» обеззараживают хлором). В медицине издавна использовался и нитрат С. («ляпис»), оказывающий антисептическое действие на ткани.
Нитрат AgNO3 — исходное вещество для получения других соединений С. В лаборатории аналитика AgNO3 служит реактивом на галогенид-ионы, в заводском цехе идёт на приготовление электролита для гальванического серебрения. А без труднорастворимых галогенидов С. невозможно себе представить существование всей современной фото- и кинопромышленности (см. ниже).
Шире, чем само С., применяют его сплавы с другими металлами. Сплавы С. с золотом встречаются в природе в самородном виде и издавна известны под названием «электрум». Из сплавов С. с другими драгоценными металлами и медью изготовляют ювелирные и бытовые из
263
делия, а в некоторых странах чеканят монеты. Эти сплавы, так же как и сплавы золота, стандартизированы ; наиболее распространённая проба С. 875 (см. Золото).
В настоящее время потребность в С. намного превышает его общую мировую добычу.
Ю. И. Романъков.
Серебро и фотография. Фото- и кинопромышленность всего мира потребляют огромные количества С.— тысячи тонн в год. Дело в том, что известны всего три соединения, на которых можно осуществить фотопроцесс: AgBr, AgCl, Agl.
Ещё в XVII в. английский учёный Р. Бойль заметил, что встречающееся в природе «роговое серебро» (минерал кераргирит AgCl) — перламутрово-серая просвечивающая масса, темнеет на свету. Позднее установили причину — под действием света хлорид С. разлагается с выделением металлического С.:
2AgCl - 2Ag Cl, Ещё более высокой чувствительностью к свету обладает бромид AgBr. Это и есть основное и пока незаменимое соединение для производства фото- и киноматериалов (светочувствительность иодида Agl, наоборот, слишком высока, и поэтому с ним трудно работать).
Попробуем внимательно, шаг за шагом, проследить за фотопроцессом. Фотоматериалы готовят так. На подложку (например, на плёнку) наносят слой суспензии мельчайших кристалликов AgBr в желатине и дают массе застыть. Кристаллики AgBr, содержащиеся в фотоматериалах, называют зёрнами; размер зёрен очень мал (как правило, меньше микрона).
Что же происходит с зёрнами AgBr при фотографировании? Очень упрощённо это можно представить себе так. В кристаллической решётке AgBr чередуются ионы Ag+ и Вг-. Свет, попадая на зёрна AgBr, взаимодействует с ионами Вг-, в результате чего возникает свободный электрон и нейтральный атом Вг. В кристаллической решётке всегда есть дефекты — нарушения правильности чередования составляющих её частиц. Роль дефектов могут выполнять атомы других элементов («примесные» атомы), попавшие в решётку при её формировании. Такие дефекты ведут себя как «электронные ловушки» — они захватывают все свободные электроны, оказавшиеся в решётке. В результате ионы Ag+, расположенные около ловушки, получают возможность приобрести недостающие электроны и восстановиться до нейтральных атомов. Так в зерне AgBr возникает «зародыш скрытого изображения», состоящий из 20—30 нейтральных атомов С. Число зародышей тем больше, чем сильнее освещали фотоматериал. Эти зародыши так малы, что увидеть их можно только в самые чувствительные электронные микроскопы. Поэтому сформированное зародышами изображение и называют скрытым изображением.
Теперь задача состоит в том, чтобы скрытое изображение сделать видимым. Для этого облучённый светом фотоматериал проявляют, помещая его в раствор восстановителя. Восстановителями (соединениями, отдающими электроны) служат известные всем фотолюбителям гидрохинон, метол и другие органические соединения. При проявлении переход ионов Ag+ в нейтральные атомы Ag (Ag+~e-:Ag) происходит только в тех зёрнах AgBr, в которых под действием света уже сформировались зародыши скрытого изображения. Важно, что при этом восстанавливаются все ионы Ag+, входящие в состав таких зёрен — нейтральные атомы Ag служат в них как бы катализатором процесса восстановления. Те же зёрна, в которых зародышей нет, в реакцию с проявителем не вступают (разумеется, если проявление идёт не слишком долго, иначе прореагируют все зёрна, и фотоматериал окажется «перепроявленным» — он потемнеет).
СЕРНАЯ КИСЛОТА
После проявления изображение стало видимым, но его необходимо «закрепить» (зафиксировать) — удалить из фотоматерйала непрореагировавшие зёрна AgBr. Для этого фотоматериал помещают в раствор фиксажа — тиосульфата натрия (фотографы часто «по старинке» называют его гипосульфитом). Тиосульфат натрия переводит С. оставшихся зёрен AgBr в комплексное соединение, растворимое в воде:
2\a,S2O3AgBr _ Na3[ Ag(S2O3)2] + NaBr
Оставшиеся на фотоматериале мельчайшие кристаллики металлического С. и создают, видимое изображение.
Полученный так фотоматериал называют негативом, поскольку наиболее тёмные участки в нём находятся в местах, которые подверглись самому сильному действию света и, следовательно, соответствуют самым светлым участкам фотографируемого объекта. Отпечатки с негатива делают, пропуская через него свет. Прошедший свет попадает на фотобумагу. Так как кристаллики С. на негативе не пропускают света, то чёрно-белое изображение на бумаге (позитив) оказывается обратным негативу.
Как уже говорилось, наибольшее значение для производства фотоматериалов имеет AgBr. Для того чтобы изменить светочувствительность материала, к AgBr часто добавляют AgCl или Agl.
Итак, существование современной фото- и кинопромышленности было бы невозможно без С. А между тем это очень редкий металл, запасы его на Земле весьма ограничены. Необходимо всячески беречь С. Эта просьба-предостережение относится и к фотолюбителям. Не выливайте отработанный фиксаж в раковину. Его надо собрать и затем извлечь из него С. Для этого можно, например, подействовать на фиксаж цинковой пылью. Более активный цинк вытесняет С. из комплекса, и оно выпадает в виде чёрного порошка.
Сейчас смывают С. и со старых снимков, и со старых плёнок. И вероятно, фотолюбительство — это единственная область, где потерям С. ещё не поставлены надёжные преграды.
С. С. Бердоносов. СЕРНАЯ КИСЛОТА H2SO4—сильная двухосновная кислота, отвечающая степени окисления серы -;-6. Безводная С. к.— вязкая, похожая на масло бесцветная жидкость. Применение С. к. настолько обширно и разнообразно, что её по праву можно считать главной кислотой промышленности.
Мировое производство С. к. приближается к 110 млн. т в год. Но если бы не сознательная деятельность человека, это вещество встречалось бы на Земле лишь как один из самых редких минералов. Свободная С. к., обычно в небольших количествах, сопровождает залежи пирита FeS2 там, где этот минерал соприкасается с влагой и воздухом (см. Сульфиды). В районе Каракумского месторождения серы С. к. пропитывает окружающие пески. Считают, что она образуется в естественных условиях при медленном окислении серы:
2S н- 30, 2Н2О 2H2SO4
Подобный процесс возможен и в особых микроорганизмах — серобактериях.
Физические и химические свойства. Чистую 100°о-ную С. к. иногда называют моногидратом, так как в ней соблюдается точно эквимолекулярное соотношение серного ангидрида и воды (H2O-SO3). Оговоримся сразу: моногидратом часто называют и высококонцентрированную С. к.— такое употребление этого слова
СЕРНАЯ КИСЛОТА
264
особенно принято у технологов. Называют моногидратом и гидрат С. к. H2SO4-H2O, образующийся при её смешивании с водой. Чистая С. к.— бесцветная маслянистая жидкость с плотностью 1,9203 г/см3; температура кристаллизации +10,45 гС; температура кипения 296,2 С. В парообразном состоянии С. к. диссоциирует:
H2SO4TztH,O + SO3
Благодаря более высокой температуре кипения, сравнительно с другими сильными кислотами, С. к. при нагревании вытесняет эти кислоты из солей:
H2SO4 + 2KNO3 - K2SO4 + 2HNO3
Эта реакция идёт при 100 СС. Газообразные фтористый и хлористый водород С. к. вытесняет уже при обычных условиях:
H2SO4 + NaCl ---- NaHSO4 + НС1
Таким образом, С. к. позволяет получать кислоты — галогеноводородные, борную, фосфорную, азотную — из их солей.
С. к. смешивается с водой во всех отношениях, причём растворение сопровождается значительным выделением тепла. Поэтому если готовить растворы С. к., добавляя воду к концентрированной С. к., первые порции прилитой воды закипают и разбрызгивают С. к. По этой причине всегда добавляют С. к. к воде (но не наоборот), перемешивая жидкость.
Большое сродство к воде позволяет использовать С. к. в качестве высушивающего и водоотнимающего средства. Пропуская через концентрированную С. к. ток влажного воздуха или другого газа, химически не взаимодействующего с ней (СО2, N2, НО), можно практически полностью освободить газ от паров воды. С. к. отнимает воду и от кристаллогидратов: бросьте синий кристалл медного купороса CuSO4-5H2O в С. к.— он быстро обесцветится, теряя воду и превращаясь в бесцветные сульфат CuSO4 или гидросульфат Cu(HSO4)2.
С. к. способна отнимать элементы воды и от органических соединений, содержащих кислород в виде гидроксильных групп —ОН. Так, дегидратация этилового спирта в присутствии концентрированной С. к. приводит к получению этилена или диэтилового эфира:
(H2SO4)
С2Н5ОН----------+ СН2- СН2 4- Н2О
этиловый 4 этилен
спирт
(H2SO4)
2С.»Н5ОН------> СН3СН0—О—СН2СН3 4- Н2О
этиловый 4 диэтиловый эфир
спирт
Сахар, целлюлоза, крахмал и другие углеводы при контакте с С. к. обугливаются — также благодаря обезвоживанию. На коже С. к. оставляет тяжёлые ожоги.
В водных растворах С. к. ступенчато диссоциирует на ионы, причём диссоциация по второй ступени становится заметной лишь при достаточном разбавлении:
H2SO4 zzt Н + + HSO; 2Н + + 5ОГ
Разбавленная С. к. растворяет все металлы, предшествующие водороду в ряду напряжений: о
Zn + H2SO4 = ZnSO4 + Н2 или, в виде ионного уравнения: о о
Zn 4- 2Н+ -Zn2+ + Н2
Таким образом, в разбавленных растворах С. к. окислителем металлов выступают ионы водорода (точнее, гидроксония Н3О+ см. Вода}. В концентрированной С. к. окислителем служит сера, находящаяся в своей высшей степени окисления. Поэтому металлы, кроме Au, Pt, Ru, Rh, Os, Ir, растворяются (окисляются) концентрированной С. к. независимо от их положения в ряду напряжений:
0 +6 42 +4
Си + 2H2SO4 = CuSO4 + SO.» + 2Н2О
Железо представляет лишь кажущееся исключение: при концентрации С. к. более 93% оно не растворяется в кислоте благодаря образованию на его поверхности защитной плёнки окисла Fe2O3. (Разбавленная С. к., напротив, легко растворяет железо — в ряду напряжений оно стоит до водорода.) Свинец, также предшествующий водороду в ряду напряжений, практически не растворяется в разбавленной С. к. из-за защиты его поверхности нерастворимым сульфатом PbSO4. В концентрированной С. к. этот сульфат растворяется *
PbSO4 4- H2so4 - Pb (HSO4)2
Поэтому с концентрированной С. к. свинец реагирует. Устойчив в С. к. чугун, содержащий кремний, благодаря чему из такого чугуна делают аппаратуру на сернокислотных' заводах и ёмкости для хранения и транспортировки С. к.
Окислительные свойства С. к. проявляются не только по отношению к металлам, но и в реакциях с другими восстановителями (S, С, H2S, НВг, HI и т. д.):
2H2SO4 4- С - 2SO2 4- СО2 4- 2Н2О 4-6 Сильные восстановители превращают H2SO4 + 4 о
не только в SO2, но и в элементарную серу (S)
-2 и сернистый водород (H2S).
Будучи двухосновной кислотой, С. к. даёт два ряда солей — средние и кислые (о солях С. к. см. Сульфаты, Квасцы}.
Производство С. к. и её применение. Вот уже около 1000 лет как люди научились получать С. к.
Алхимикам удавалось сделать это ещё в X в. при прокаливании квасцов или купоросов. Отсюда и произошло название купоросное масло, применяемое и сегодня для 92%-ной С. к. Спустя 5 веков алхимики научились
265
СЕРНАЯ КИСЛОТА
готовить С. к. сжиганием серы в смеси с селитрой и растворением полученного серного ангидрида SO3 в воде.
Но понадобилось ещё несколько столетий, чтобы промышленная революция и развитие капиталистического способа производства сделали необходимым промышленное получение С. к. в больших количествах. На рубеже XVIII и XIX веков был создан и усовершенствован нит-розный метод получения С. к. (его камерная, а затем и башенная модификации).
Развитие в XIX в. новых отраслей промышленности — производства синтетических красителей и взрывчатых веществ — требовало С. к. более высоких концентраций, чем давал нитрозный метод (65—78%-ная С. к.). Поэтому в конце XIX в. всё большее распространение получает контактный метод производства С. к., основные принципы которого были установлены ещё в 1831 г. В наше время этот метод стал преобладающим.
Значительные изменения происходили и в сырьевой базе С. к. Уже в 1818 г. для производства С. к. вместо дорогостоящей серы начали использовать пирит. Однако очистка сернистого газа SO2, получаемого обжигом пирита, обходится очень дорого. В наши дни довольно много С. к. производят из отходящих газов цветной металлургии и из сероводорода, одного из отходов нефтехимической и коксохимической промышленности.
В нашей стране впервые производство С. к. началось в 1805 г., однако к началу XX в. царская Россия безнадёжно отставала от развитых капиталистических стран. Если мировое производство С. к. в 1913 г. составляло 6 млн. т, то доля России в нём (0,12 млн. т) была ничтожной. Около 2/3 сырья Россия ввозила из Испании, Португалии, Швеции и Норвегии. За годы Советской власти СССР вышел на второе место в мире после США по производству С. к. В 1974 г. в нашей стране было произведено 16,7 млн. т С. к.
Технологическая схема производства С. к. контактным способом хорошо известна из школьных учебников. В нашей стране используется и другой, так называемый башенный, способ её получения.
На первой стадии, одинаковой для обоих методов, получают сернистый ангидрид SO2. Исходным сырьём может быть, в принципе, любое вещество, содержащее серу: природные сульфиды железа (прежде всего, пирит FeS2), а также сульфиды меди и никеля, сульфидные полиметаллические руды, гипс CaSO4-2H2O и элементарная сера. Всё больше и больше используют газы, которые выделяются при переработке и сжигании горючих ископаемых (угля и нефти), содержащих соединения серы.
Полученный SO2 окисляют до SO3 или до H2SO4, используя для этого в контактном методе кислород воздуха в присутствии твёрдого катализатора (контакта), а в башенном методе — окислы азота. С этой стадии оба метода отличаются друг от друга.
Башенный метод (рис. 1). В специальной окислительной башне 3 смешивают окись азота NO с воздухом в таком соотношении, чтобы половина имеющегося NO превратилась в двуокись азота NO2:
2NO + О2 = 2NO2
Рис. 1. Схема производства серной кислоты башенным способом. В окислительной башне (3) кислород воздуха превращает половину окиси азота NO в двуокись азота NO2, так что газовая смесь содержит равные количества NO и NO2. В башнях (4) и (5) смесь окислов азота поглощается серной кислотой с образованием нитро-зилсерной кислоты. Выходящий из башни (4) раствор, называемый нитрозой, подаётся на орошение башен (/) и (2), куда противотоком поступает сернистый ангидрид SO2 и добавляется вода.
В этих башнях происходит окисление SO2 и образование серной кислоты, имеющей концентрацию около 75%. Выделяющаяся при этом окись азота NO снова поступает в окислительную башню (3), а потери окислов азота с выхлопными газами компенсируются добавлением в башни (/) и (2) азотной кислоты.
СЕРНАЯ КИСЛОТА
266
В результате газовая смесь содержит равные количества NO и NO2. Она подаётся в башни 4 и 5, орошаемые 75%-ной С. к.; здесь смесь окислов азота поглощается с образованием нитрозилсерной кислоты:
NO + NO2 + 2H2SO4 = 2NO (HSO4) + Н2О Раствор нитрозилсерной кислоты в С. к., называемый нитрозой, орошает башни 1 и 2, куда противотоком поступает SO2 и добавляется вода. В результате гидролиза нитрозилсерной кислоты образуется азотистая кислота:
NO (HSO4) + Н2О = H2SO4 + HNO2 Она-то и окисляет SO2 по уравнению:
SO2 + 2HNO2 = H2SO4 + 2NO
В нижней части башен 1 и 2 накапливается 75%-ная С. к., естественно, в большем количестве, чем ее было затрачено на приготовление нитрозы (ведь добавляется «новорожденная» С. к.). Окись азота NO возвращается снова на окисление. Поскольку некоторое количество её теряется с выхлопными газами, приходится добавлять в систему азотную кислоту HNO3, служащую источником окислов азота.
Недостаток башенного метода состоит в том, что полученная С. к. имеет концентрацию лишь 75% (при большей концентрации плохо идёт гидролиз нитрозилсерной кислоты). Концентрирование же С. к. упариванием пред
ставляет дополнительную трудность. Преимущество этого метода в том, что примеси, содержащиеся в SO2, не влияют на ход процесса, так что исходный SO2 достаточно очистить от пыли, то есть от механических загрязнений. Естественно, башенная С. к. бывает недостаточно чистой, что ограничивает её применение.
Контактный метод (рис. 2). Этот метод позволяет получить не только С. к. любой концентрации, но и олеум, то есть раствор серного ангидрида SO3 в С. к., необходимый во многих отраслях промышленности.
Окисление SO2 здесь проводится в контактном аппарате 3 на твёрдом катализаторе — на платине или на сложном катализаторе, содержащем окислы ванадия с добавками SiO2, А12О3, К2О, СаО, ВаО. Наличие в исходном SO2 примесей (As2O3, SeO2 и др.) выводит катализатор из строя («отравляет» его). Поэтому необходима тщательная очистка SO2 — весьма громоздкий процесс (рис. 1 и 2 в статье Сернистый ангидрид). В частности, такая очистка обязательна, если SO2 получен обжигом пирита. Очищенный SO2 подогревается до 440 °C и поступает на окисление (в смеси с воздухом) в первый слой катализатора, где идёт реакция:
SO2 + 1/fi2 SO3 + 96,7 кдж
При прохождении через первый слой около 70% SO2 превращается в SO3 и газовая смесь
Рис. 2. Схема производства серной кислоты контактным способом. Компрессор / подаёт сернистый ангидрид SO2 в смеси с кислородом воздуха в теплообменник 2. Здесь SO2 нагревается до 440 °C, обтекая трубы, по которым проходит горячий серный ангидрид SO3 из контактного аппарата 3. Нагретая газовая смесь (SO2+O2) поступает затем сверху в контактный аппарат 3, где катализатор расположен на решётчатых полках а, между которыми находятся теплообменники (Г. В этих теплообменниках газовая смесь, разогревшаяся до 600 °C в результате реакции в слое катализатора, снова охлаждается до оптимальной температуры и поступает в следующий слой катализатора. В абсорбционных башнях 4 и 5 полученный SO3 поглощается олеумом (раствор 18—19% SO3 в H2SO4) и «моногидратом» (98%-ная H2SO4), так что конечный продукт представляет собой олеум, содержащий 20% SO3.
267
СЕРНИСТЫМ АНГИДРИД
разогревается до 600 °C за счёт теплоты реакции. Поскольку при высокой температуре равновесие реакции сдвигается влево, необходимо охладить смесь, прежде чем подавать её на второй и последующий слои катализатора (обычно их бывает 4—5). Это охлаждение происходит в теплообменниках, которые ставят между слоями. В этих же теплообменниках одновременно идёт нагревание «свежего» SO2 перед его поступлением в первый слой катализатора. Многослойный катализатор позволяет практически нацело превратить SO2 в SO3, который после охлаждения до 80 °C в теплообменнике 2 поступает в абсорбер 4. орошаемый олеумом (раствор 18—19% SO3 в H2SO4), а затем в абсорбер 5, орошаемый 98%-ной кислотой («моногидратом»). С. к. такой концентрации особенно хорошо растворяет SO3. Полученный олеум можно разбавить до любой концентрации С. к.
Мы уже упоминали, что необходимость тщательной очистки SO2 очень усложняет общую схему производства С. к. контактным методом. Можно однако получать SO2 не из пирита, а из такого сырья, которое обеспечивает необходимую чистоту продукта. На коксохимических заводах, например, получается достаточно чистый сероводород H2S. Его смешивают с воздухом и сжигают:
2Н 2S + ЗО2 = 2Н2О + 2SO2
Далее SO2 перерабатывают в С. 1с. по контактному методу без дополнительной очистки. Концентрация С. к. при этом составляет 92—94%.
Часть С. к. остаётся в виде тумана, который приходится улавливать в электрофильтрах или поглощать в башнях, орошаемых моногидратом. Необходимость тщательной очистки SO2 отпадает также при получении его сжиганием природной серы, не содержащей селена.
Несмотря на то, что пирит доступен и дёшев, доля этого минерала как исходного сырья для получения С. к. постепенно снижается.
Около 40% С. к. расходуется для производства удобрений. В химической промышленности С. к. используется для получения других кислот, для производства взрывчатых веществ и моющих средств, лекарственных препаратов и красителей. С. к. участвует в производстве искусственного волокна — вискозного шёлка. Применяется С. к. и в металлургии, и металлообработке. С. к. заполняют свинцовые аккумуляторы (недаром один из её сортов называют «аккумуляторная С. к.»). С. к. расходуется для очистки нефти и нефтепродуктов: здесь С. к. помогает освободиться от вредных примесей — органических соединений серы.
Самая дешёвая, самая доступная и «универсальная» из кислот — С. к., так же необходима технике, как металлы и уголь, нефть и цемент. Вот почему уровень промышленного
развития страны определяется и такими показателями, как производство С. к.и темп его роста. В. Л. Василевский.
СЕРНИСТАЯ КИСЛОТА H2SO3 — слабая двухосновная кислота, отвечающая степени окисления серы +4. Существует только в растворе. Образуется при растворении в воде сернистого ангидрида:
Н2О + SO2 H2SO3
Эта реакция обратима — при кипячении раствора С. к. полностью разлагается. H2SO3 даёт кислые и средние соли (см. Сульфиты). С. к.— сильный восстановитель: при действии окислителей она превращается в серную кислоту. СЕРНИСТОКЙСЛЫЕ СОЛИ — то же, что сульфиты.
СЕРНИСТЫЙ АНГИДРИД [сернистый газ, двуокись серы, оксид серы (IV)], SO2 — ангидрид сернистой кислоты. С. а.— бесцветный газ с резким характерным запахом. Хорошо растворяется в воде (80 объёмов С. а. в одном объёме воды при 0 °C), давая слабую сернистую кислоту:
SO2 + H2O = H2SO3
В присутствии катализаторов при 400— 600 °C реагирует с кислородом, превращаясь в серный ангидрид SO3. Реакция:
2SO2 + О2 = 2SO3
Рис. 1. Схема электрофильтра (одно из звеньев очистки SO2). Источник постоянного тока создаёт между проволочным отрицательным электродом (а) и стенками металлической трубы (б) напряжение до 60 тыс. вольт. Вокруг проволоки (а) возникает «корона» — характерное свечение, связанное с ионизацией газов в электрофильтре. Частицы пыли, сталкивающиеся с ионами, заряжаются отрицательно и притягиваются к стенкам, заряжённым положительно. Пыль периодически стряхивается в бункер (в).
Запыленный газ
в
Очищенный газ
СЕРНИСТЫЙ АНГИДРИД
268
Рис. 2. Схема очистки сернистого ангидрида, полученного обжигом пирита. Газовая смесь, полученная в результате обжига пирита (так называемый обжиговый газ), перед поступлением в контактный аппарат должна быть очень тщательно очищена. В противном случае примеси приведут в негодность катализатор — отравят его. В обжиговом газе почти всегда содержится трёхокись мышьяка А^Оз — сильный каталитический яд. Вредной примесью является и серная кислота, которая образуется при охлаждении из водяного пара Н2О и серного ангидрида SO3, содержащихся в обжиговом газе. А, кроме того, газ должен быть очищен от пыли, которая, оседая на катализаторе, тоже понижает его активность.
На рисунке вы видите систему электрофильтров (1, 4, 6) и башен (2, 3, 5, 7), через которые последовательно проходит обжиговый газ. Электрофильтр даёт наиболее полную очистку газа от твёрдых частиц. Пыль улавливается в электрофильтре /. Как именно он работает, показано на отдельном рисунке (рис. /). Однако, чтобы удалить в электрофильтрах трёхокись мышьяка, необходимо предварительно перевести её из газообразного состояния в твёрдое, а для этого охладить обжиговый газ (его начальная температура около 350 °C). Уже в первой промывной башне 2 газ охлаждается примерно до 75 °C; здесь он соприкасается с 70% -ной серной кислотой, орошающей башню. Во второй промывной башне 3, орошаемой 30% -ной серной кислотой, газ охлаждается примерно до 30 СС.
В промывных башнях происходит образование из SO3 и Н20 серной кислоты в виде тумана — мелких капелек, взвешенных в газовой фазе. Туманообразная серная кислота и трёхокись мышьяка частично улавливаются в промывных башнях, полностью же — в электрофильтрах 4 (А^Оэ) и 6 (H2SO4). Перед поступлением в электрофильтр 6 газ проходит увлажнительную башню <5, орошаемую 5% -ной серной кислотой. Благодаря увлажнению оставшиеся в газе мелкие частицы тумана H2SO4 укрупняются и уже полностью оседают в электрофильтре 6. Наконец, в осушительной башне 7, орошаемой 94—97% -ной H2SO4, обжиговый газ высушивается (ведь концентрированная серная кислота жадно поглощает воду). А затем очищенный газ — сернистый ангидрид — передаётся на окисление в контактный аппарат (он изображён на рис. 2 в статье Серная кислота).
важнейшая в производстве серной кислоты.
С. а.— сильный восстановитель:
SO2 + 2HNO3 = H2SO4 + 2NO2
Напротив, по отношению к сильным восстановителям С. а. выступает в роли окислителя: SO2 + 2H2S = 3S + 2Н2О
С. а. способен реагировать со многими органическими красителями, образуя бесцветные продукты присоединения. Поэтому его часто используют для отбеливания тканей.
В лаборатории С. а. удобно получать нагреванием концентрированной серной кислоты с медными опилками:
Си + 2H2SO4 = CuSO4 + SO2 + 2НаО
или действуя этой кислотой на NaHSO3:
2NaHSO3 + H2SO4 = NaaSO4 + 2H2O + 2SO2
В промышленности большое значение имеет получение С. а. обжигом сульфидов, например: 4FeS2 + 11О2 = 2Fe2O3 + 8SO2
Очистка SO2, полученного обжигом пирита FeS2, очень сложна (рис. 1, 2).
С. а. встречается в природе как один из компонентов вулканических газов (Везувий, Ключевская сопка и др. вулканы). В наше время «соперником» вулканов становится промышленность: уголь и нефть содержат в среднем 1% серы, которая при их сжигании превращается в С. а. Таким образом, с дымовыми газами в атмосферу ежегодно поступает С. а. больше, чем потребляет его промышленность всего мира для производства серной кислоты. Большие количества С. а. выбрасывают в атмосферу и предприятия цветной ме-
269
таллургии. Решение проблемы улавливания и использования С. а., образующегося при обжиге сульфидов меди, цинка, свинца, никеля, имеет огромное народнохозяйственное значение.
С. а. ядовит, поэтому очистка от него промышленных газов важна и с гигиенической точки зрения. С другой стороны, из-за своей ядовитости С. а. с давних пор используется для уничтожения насекомых и микроорганизмов (см. Сера), Даже А. С. Пушкин оставил шутливое упоминание об этом: «...карантинный страж курил жаровней серной».
Сам С. а. и соединения, легко его выделяющие, применяются для предохранения овощей и фруктов от плесени и гнилостных микробов. Как С. а., так и полученный из него гидросульфит кальция Ca(HSO3)2 применяют в производстве целлюлозы из древесины (см. Сульфиты),
С. а. можно использовать и в холодильных установках. Достаточно небольшого давления (3—4 атм), чтобы С. а. превратился в жидкость. При испарении жидкого С. а. расходуется большое количество тепла, и температура понижается до —50 °C.
Несмотря на многообразие путей использования С. а. наибольшие количества его потребляются всё же в производстве серной кислоты. В, Л, Василевский,
СЕРНИСТЫЙ ВОДОРОД (сероводород) H2S — соединение серы с водородом. Бесцветный газ с неприятным запахом тухлых яиц. Растворяется в воде с образованием очень слабой сероводородной кислоты (при обычной температуре в 1 объёме воды растворяются 3 объёма С. в.). Соли сероводородной кислоты называют сульфидами,
В естественных условиях С. в. встречается в вулканических газах, а иногда и в так называемом природном газе, выходящем из недр Земли. Он растворён в водах минеральных источников и в буровых водах нефтяных месторождений. В природе С. в. постоянно образуется при гниении органических остатков животного происхождения. (Поэтому точнее было бы сказать, что тухлые яйца пахнут С. в., а не наоборот.) Много С. в. образуется и в некоторых промышленных химических процессах (при очистке нефтепродуктов, коксовании угля). Всё это приводит к тому, что С. в. постоянно содержится в атмосфере (хотя обычно в небольших количествах).
Известно, что серебряные предметы со временем темнеют из-за медленного взаимодействия с С. в. воздуха (см. Серебро):
4Ag + 2H2S + О2 = 2Ag2S + 2Н2О
СЕРНЫЙ АНГИДРИД
Подобным же образом реагируют в этих условиях с С. в. и другие металлы и их соединения. В частности, применявшаяся в старину белая краска — свинцовые белила 2РЬСО3*РЬ(ОН)2— чернеет из-за превращения основного карбоната свинца в сульфид PbS. Поэтому старые картины и иконы со временем теряют яркость красок. Осторожная обработка поверхности таких картин окислителем (например, перекисью водорода) «обновляет» их — чёрный сульфид PbS окисляется до белого сульфата PbSO4.
С. в.— восстановитель. Не только такие сильные окислители, как азотная кислота или хлор, но и кислород воздуха уже при комнатной температуре легко взаимодействует с ним. Поэтому водные растворы С. в. на воздухе неустойчивы:
2H2S + О2 = 2S + 2Н2О
Окисление С. в. в присутствии катализаторов (гидроокисей железа или алюминия) приводит к получению так называемой газовой серы. Подобный процесс возможен и в естественных условиях. Залежи серы имеются, например, во Франции вблизи от выходов природного газа, содержащего H2S. Сжигание же С. в. в избытке воздуха даёт сернистый ангидрид:
2H2S + ЗО2 = 2Н2О + 2SO2
Эта реакция всё чаще применяется для получения SO2 из H2S (см. Серная кислота),
С. в. можно получить прямым синтезом из элементов:
H2 + S = H2S
Этот способ технически осуществить довольно сложно. Поэтому обычно в лаборатории С. в. получают в аппарате Киппа, разлагая сульфид железа разбавленными кислотами (серной или соляной):
FeS + H2SO4 = FeSO4 + H2S
С. в. крайне ядовит уже в концентрациях 0,1%. Он вреден для организма и в гораздо меньших количествах, если действует длительное время. Однако воды минеральных источников, а также ил лиманов и озёр (лечебные грязи), содержащие небольшое количество С. в., оказывают целительное действие при некоторых болезнях. А кроме того, существуют микроорганизмы, для которых присутствие С. в. жизненно необходимо: они используют реакцию окисления H2S в своём организме как источник энергии^ При этом С. б. окисляется до свободной серы или даже до серной кислоты. В. Л. Василевский, СЕРНИСТЫЙ ГАЗ SO2 — то же, что сернистый ангидрид.
СЕРНОВАТИСТАЯ КИСЛОТА — то же, что тиосерная кислота H2S2O3. См. также Сера.
СЕРНОКИСЛЫЕ СОЛИ — то же, что сульфаты,
СЕРНЫЙ АНГИДРЙД, трёхокись серы, оксид серы (VI), SO3 — ангидрид серной кислоты. При комнатной температуре свежеприготовленный С. а.— бесцветная жидкость, кипящая при 44,7 °C; при 16,8°C она
СЕРОУГЛЕРОД
270
затвердевает в прозрачную кристаллическую массу, которая со временем полимеризуется и становится похожей на волокна асбеста. Твёрдый С. а. существует в нескольких модификациях; та из них, которая плавится при 16,8 °C, имеет формулу (SO3)3; в других модификациях 5О3ещё более полимеризован. Взаимодействуя с водой, С. а. даёт серную кислоту:
SO3 + H2O = H2SO4
Во влажном воздухе С. а. «дымит» — это испаряющийся SO3 образует с влагой капельки тумана серной кислоты. С. а. хорошо растворяется в 100%-ной H2SO4 (см. Олеум). С. а.— сильный окислитель: реагируя с серой, фосфором, углеводородами, восстанавливается до SO2. В лаборатории С. а. можно получить прокаливанием сульфата железа (III):
Fe2 (SO4)3 = Fe2O3 + 3SO3
О получении его в промышленности — см. Серная кислота.
СЕРОВОДОРОД — то же, что сернистый водород.
СЕРОВОДОРОДНАЯ КИСЛОТА — раствор сернистого водорода в воде.
СЕРОУГЛЕРОД CS2 — соединение углерода с серой. Бесцветная легколетучая жидкость. Свежеприготовленный чистый С. обладает приятным эфирным запахом. После длительного стояния на воздухе желтеет и начинает отвратительно пахнуть вследствие частичного разложения под действием света с образованием сероокиси углерода S=C=O. Кипит при температуре 46,26 °C, затвердевает при —112,1 °C. В воде С. практически нерастворим и с ней не взаимодействует. На воздухе чрезвычайно легко воспламеняется и горит светло-синим пламенем:
CS2 + ЗО2 = СО2 + 2SO2
Интересно, что бумага, внесённая в пламя С., даже не обугливается — настолько низка температура, развивающаяся при его горении!
С. прекрасно растворяет смолистые вещества, жиры, масла, каучук, а также серу, фосфор и иод. На этом основано его применение для экстрагирования веществ из природных материалов. Большие количества С. идут на производство вискозы.
Обычно получают С. пропусканием паров серы через слой раскалённого угля:
С + S2 = CS2
При работе с С. нужно помнить, что он очень ядовит и огнеопасен.
СИЛАНЫ — соединения кремния с водородом. См. Кремневодороды.
СИЛИКАТЫ — солеобразные химические соединения, содержащие кремнекислородные кислотные остатки различного состава (SixOv).
С. чрезвычайно широко распространены в природе. Глина, тальк, слюда, сланцы — всё это С. По подсчётам советского геохимика академика А. Е. Ферсмана, на долю С. приходится около трёх четвертей массы земной коры. Приблизительно треть всех известных минералов относится к С. Исключительно велика и роль С. в промышленности. Керамика, кирпич, цемент, бетон, асбест, стекло и многие другие материалы в основном состоят из С.
Поэтому неудивительно, что С. были и остаются предметом пристального внимания учёных-химиков. Вряд ли найдётся другой класс неорганических соединений, представителям которого было бы посвящено такое же количество самых разнообразных исследований. Но есть и ещё одна немаловажная причина того интереса, который вызывают к себе С.,— исключительное своеобразие и разнообразие их строения. Изучение этого вопроса сыграло выдающуюся роль в формировании современных представлений о пространственном расположении атомов и о химической связи в неорганических веществах.
С. нередко обладают весьма сложным составом. В них входят различные металлы (чаще всего Al, Fe, Ti, Zr, Mn, Be, щелочные и щелочноземельные металлы), причём очень часто в веществе содержится 2—3 различных катиона, а иногда и более. Наряду с кремнекислородным остатком, в котором Si нередко бывает частично замещён на А1 или В, в состав С. могут входить и такие анионы как ОН“, F", С1", О2-, СО|", SOJ" и другие. В состав некоторых С. входит кристаллизационная вода.
Свойства С. бывают довольно различны в зависимости от их состава и строения. Часто С. образуют прозрачные и очень красиво окрашенные кристаллы; некоторые из них — драгоценные камни, используемые в ювелирном деле (гранат, топаз, изумруд и др.).Плотность С. обычно колеблется от 2 до 4 г/см3. Твёрдость иногда очень невелика (например, у талька), но бывает и довольно значительной (например, у циркона ZrSiO4). При нагревании С. либо плавятся, либо разлагаются в твёрдом состоянии. Температуры плавления колеблются от 750 до 2000 °C, а иногда бывают и выше.
С. обычно нерастворимы в воде. Исключение составляют С. натрия и калия, получаемые сплавлением SiO2 с соответствующими гидроокисями или карбонатами, например:
SiO2 -f- Na2CO3 — СО2 -Н Na2SiO3
Водные растворы этих солей называют «жид-
271
СИЛИКАТЫ
ким стеклом». В результате гидролиза «жидкое стекло» имеет сильно щелочную реакцию, а С. слабых оснований гидролизуются практически полностью.
Несколько десятков лет назад С. считались солями поликремниевых кислот с общей формулой mH2O*nSiO2 (например, при т=1, п=1 формула имеет вид H2SiO3 — метакремниевая кислота; при /п=2, п=1—ортокремние-вая кислота H4SiO4; при т= 1, п=2 — метадикремниевая кислота H2SiO5 и т. д.). На .этой основе записывались формулы и выводились названия С.
Например, MgCa(SiO3)2 назывался смешанным метасиликатом Mg и Са (минерал диопсид), Be3Al2(SiO3)e — метасиликатом бериллия и алюминия (берилл). Считалось, что в этих С. присутствуют анионы, состав которых определяется указанной формулой, в данном случае — анионы SiO3“. Дальнейшие исследования показали, насколько наивным и неточным был такой подход, который к тому же в более сложных случаях приводил к полной неопределённости. Так, С. состава 3MgO*2SiO2*2H2O была приписана формула Mg3H2(SiO4)2*H2O, в соответствии с которой он представляет собой моногидрат кислой соли ортокремниевой кислоты. Но тому же составу отвечает формула Mg3(Si2O5)(OH)4(n многие другие формулы), согласно которой это — основная соль метади-кремниевой кислоты. Впоследствии оказалось, что именно вторая формула более правильно передаёт строение этого С. (минерал антигорит).
Сложность состава С. и их нерастворимость в воде не позволяли выйти из круга подобных противоречий путём чисто химических исследований. Решение этой проблемы, как и многих других «роковых» проблем химии, было найдено в результате применения физического метода — рентгеноструктурного анализа, позволившего непосредственно «заглянуть» внутрь твёрдых С. По выражению советского учёного академика.Н. В. Белова, «эти рентгеновские анализы в корне изменили здание минералогии, науки, имевшей за своими плечами двухтысячелетнюю историю». Н. В. Белов имеет право на эти слова: именно его работы наряду с предшествующими исследованиями английского учёного У. Л. Брэгга и американского учёного Л. Полинга послужили основой современных представлений о строении С.
Рентгеноструктурный анализ показал, что С., так же как и большинство твёрдых неорганических соединений, никогда не бывают построены из отдельных молекул. Основой их строения является так называемый кремне-кислородный тетраэдр, в центре которого располагается атом Si, а по вершинам — атомы О (рис. 1, а, б). Такие тетраэдры чаще всего имеют общие вершины, то есть общие атомы кислорода. Узор, образуемый соединяющимися тетраэдрами,— его называют кремнекислородным мотивом — может быть очень разнообразным.
Рис. 1, а — Правильный четырёхгранник (тетраэдр) — фигура, которая лежит в основе геометрии силикатов; б" — в силикатах по Вершинам тетраэдров располагаются атомы кислорода (красные шарики), а в центрах тетраэдров — атомы кремния (коричневые шарики). Поэтому тетраэдры в силикатах называют «кремнекислородными» ; в — группировки атомов Si и О, которые встречаются в островных силикатах; г — те же фрагменты изображены в виде тетраэдров с общими вершинами.
СИЛИКАТЫ
272
Первую группу составляют островные С., в которых присутствуют изолированные тетраэдры SiO4 или их сочетания, имеющие конечные размеры (рис. 1, в, г). Как показано на рисунке, тетраэдры могут соединяться по нескольку штук, в частности, могут образовывать замкнутые кольца; при этом возникает анион, в котором все атомы объединены ковалентными связями Si—О. Такие анионы, чередуясь с катионами, образуют кристаллическую структуру. Иногда в её состав входят,
Рис. 2. Цепи и ленты, присутствующие в цепочечных силикатах. Показаны лишь небольшие участки таких цепей и лент в виде тетраэдров, соединённых по вершинам. В действительности же, вытянутые в одном направлении цепи и ленты гораздо длиннее и содержат сотни тысяч кремнекислородных тетраэдров.
кроме того, и другие анионы (например, гидроксильные группы ОН', ионы F“) или нейтральные молекулы воды. Изолированные тетраэдры [SiO4] присутствуют в кристаллических структурах топаза Al2[SiO4](OH)F и гранатов, например А12Са3 (SiO4]3 (состав кремнекислородного мотива принято записывать в квадратных скобках).
Анионы в виде сдвоенных тетраэдров, которым отвечает состав [Si2O7], наблюдаются в кристаллах минерала тортвейтита Sc2[Si2O7]. В структуре бенитоита BaTi [Si3O9] найдены кольца состава [Si3O9], построенные из трёх тетраэдров, а в структуре диоптаза Cue[SieO18]-6H2O— кольца состава [SieOi8], включающие шесть тетраэдров. Такие же кольца присутствуют в кристаллах берилла Be3Al2[SieO18], о котором говорилось выше; нетрудно видеть, что в такой структуре выделение анионов SiOf" лишено физического смысла. Структура миларита КСа2Ве2А1 [Sii2O30] содержит ещё более причудливые фрагменты — сдвоенные шестичленные кольца состава [Si12O30].
Далее следует группа цепочечных С., в структурах которых кремнекислородные тетраэдры образуют бесконечные цепи или ленты (рис. 2). Простейшим примером служит уже упоминавшийся минерал диопсид MgCa[SiO3]2, где тетраэдры объединяются в цепочку из звеньев [SiO3] (отдельные анионы SiOf~ опять-таки отсутствуют).
Более сложное строение имеет минерал тремолит Ca2Mg5[Si4On]2, в котором кремнекислородные тетраэдры складываются в бесконечные ленты, состоящие из шестичленных колец. Такую ленту можно рассматривать и как объединение двух цепей [SiO3] с обобщением части атомов кислорода.
Следующая группа — это слоистые, или листовые, С. Само название подсказывает, что в этих структурах кремнекислородные тетраэдры образуют бесконечные слои, которые, однако, могут иметь весьма различное строение (рис. 3). В структуре каолинита А12 [Si2О5] (ОН)4 — минерала, являющегося
основным компонентом многих глин,— наблюдаются слои, построенные из шестичленных колец и напоминающие в этом отношении слои в графите (см. Углерод).
Наконец, существует группа каркасных С. Чтобы описать их строение, необходимо учесть, что некоторые химические элементы обладают способностью имитировать кремний, частично замещая его в структурах кислородных соединений. Это в первую очередь отно сится к А1 и В. Например, в кристаллах нефелина, в состав которого входят кислород, кремний, алюминий и натрий, атомы А1, так же как и атомы Si, окружены кислородом по тетраэдру. Кремнекислородные тетраэдры [SiO4] сосуществуют с алюмокислородными [А1О4]. Обобщая свои вершины, тетраэдры обоих типов совместно образуют трёхмерный каркас, в пустотах которого находятся атомы Na. В по
273
СИЛИКАТЫ
добных случаях естественно включить алюминий в состав кремнекислородного, а точнее алюмокремнекислородного, мотива. Формула этого каркасного минерала запишется в виде Na [AlSiOJ. Он относится к числу так называемых алюмосиликатов.
От алюмосиликатов принципиально отличаются силикаты алюминия, в которых алюминий обычно окружён атомами О, располагающимися по вершинам октаэдра. Такой алюминий, разумеется, не входит в кремнекислородный мотив, строительными единицами которого служат тетраэдры.
С. алюминия являются гранат Al2Ca3[SiOJ3, топаз Al2[SiO4] (OH)F и другие минералы.
Возвращаясь к алюмосиликатам, отметим, что они совсем не обязательно имеют каркасные структуры. Например, в цепочечной структуре силлиманита присутствуют ленты из четырёхчленных колец (рис. 2) состава [AlSiO5]; в половине тетраэдров атомы Si замещены на атомы А1. Эта структура интересна ещё в одном отношении. В ней наряду с атомами А1, окружёнными кислородом по тетраэдру, присутствуют атомы А1, кислородное окружение которых представляет собой октаэдр и которые, следовательно, не входят в алюмокремнекисло-родный мотив. Из формулы этого минерала Al [AlSiO5], отражающей его строение, видно, что он является алюмосиликатом алюминия.
Каркасный С. данбурит Са [B2Si2О8] служит примером структуры, где в роли имитатора кремния выступает бор.
Даже из этого краткого обзора можно видеть, насколько разнообразным бывает строение С. Сейчас перед учёными стоит задача: найти закономерности, которые обусловливают то или иное расположение атомов в каждом С. На этом пути уже многое сделано.
Так, в работах Н. В. Белова и его учеников было показано, что в структурах С., содержащих крупные катионы, в качестве строительных единиц довольно часто выступают сдвоенные кремнекислородные тетраэдры. Иллюстрацией здесь может служить пара минералов — берилл Be3Al2[SieOi8] и миларит КСа2Ве2А1 |Si12O30]. В первом из этих минералов, где присутствуют лишь небольшие по размерам катионы бериллия и алюминия, кремнекислородный мотив, как уже говорилось, представлен замкнутыми кольцами из шести тетраэдров. Во втором минерале наряду с бериллием и алюминием содержатся значительно более крупные катионы кальция и калия, и кольца строятся из спаренных тетраэдров; в итоге эти кольца оказываются «двухэтажными» (рис. 1, в, г — внизу).
Отыскание подобных закономерностей отнюдь не является самоцелью. Ведь от строения С. (как и других веществ) самым непосредственным образом зависят их свойства, а следовательно, и возможности практического использования. Так, кристаллы слоистых С. легко раскалываются на тонкие пластинки, то есть обладают спайностью. Хороший пример — всем известные слюды. Одна из них, называемая мусковитом, имеет формулу KAl2[AlSi3Oi0] (ОН)2. Со слоистым строением
этого минерала связаны и его высокие электроизоляционные свойства. Напротив, каркасные С. обычно обладают значительной прочностью.
Тесная связь строения и свойств обнаруживается в очень интересной и важной для промышленности группе С., называемых цеолитами. Это — каркасные алюмосиликаты, структуры которых содержат более или менее широкие «каналы». В «каналах» находятся молекулы воды, довольно слабо связанные с каркасом. При осторожном нагревании вода
Рис. 3. Слои кремнекислородных тетраэдров, обнаруженные в кристаллических структурах листовых силикатов. Чтобы составить более полное представление о структуре слоя, следует мысленно продолжить орнамент во все стороны в плоскости чертежа.
18 ХЭШ
СИЛИКАТЫ
274
Рис. 4. В кристаллах гидросодалита и анальцима атомы Si и А1 располагаются по вершинам многогранников, называемых кубооктаэдрами.
а — кубооктаэдр, б — укладка кубооктаэдров в пространстве кристалла. Шестиугольные грани соответствуют кольцам, каждое из которых образовано кремнекислородными тетраэдрами.
постепенно удаляется без разрушения кристаллической структуры. Замечательно, что вслед за тем вода снова может быть поглощена в том же количестве или заменена другими молекулами, например сероводорода или аммиака. Таким образом, кристаллическая структура цеолитов способна играть роль «сита», избирательно пропускающего молекулы не слишком большого размера. В последние годы цеолитные «молекулярные сита» находят широкое практическое применение.
Наиболее обычные природные цеолиты — гидросодалит Na3[AleSieO24] (ОН)2- пН2О, анальцим Na [AlSi2Oe]• Н2О и другие — в сухом, дегидратированном состоянии поглощают молекулы с диаметром до 3—4 А. Их алюмОкремнекислородный каркас схематически изображён на рис. 4. Из рисунка видно, что в каркасе присутствуют полости с «окнами» в виде шестичленных колец из алюмо- и кремнекислородных тетраэдров (присутствие подобных колец, но в качестве изолированных островов, мы отмечали в кристаллах берилла). Размеры этих входных отверстий определяют размеры молекул, способных поглощаться цеолитом.
Другой цеолит — шабазит Na[AlSi2Oe]*3H2O (в этом природном минерале натрий частично замещён кальцием) построен гораздо сложнее (рис. 5). Здесь присутствуют «двухэтажные» шестичленные кольца (подобные кольцам в миларите); сцепляясь между собой, они образуют пустоты, имеющие не только «шестерные», но и «восьмерные» окна. Поэтому шабазит оказывается эффективным ситом и в отношении более крупных молекул; он легко поглощает молекулы метана (диаметром около 4,25 А). Шабазит позволяет, например, выделить из бензина особо высококачественную изооктановую фракцию без примеси
октановой, снижающей качество горючего. Длинные, но «тонкие» молекулы нормального октана поглощаются шабазитом, проходя через восьмерные окна; для более «толстых» молекул изооктана окно слишком мало (рис. 6). (Разделить же изооктан и октан путём обычной перегонки — непростая задача, так как температуры кипения этих веществ очень близки.)
Многократные попытки синтеза шабазита до настоящего времени не привели к успеху. Поэтому исключительную роль сыграло получение синтетических молекулярных сит, по своим качествам не только не уступающих шабазиту, но даже превосходящих его. Это — так называемыё сито А состава Na12[Al12Si12O48b 27Н2О и сито X состава Na3Ca[Al5Si7O24]* 15Н2О. В первом из них, как и в шабазите, содержатся восьмерные окна, но ёмкость полостей больше; поэтому сито А более эффективный поглотитель, чем шабазит. В сите X окна образованы сцеплением двенадцати тетраэдров (диаметр около 13А); в результате это сито способно пропускать весьма крупные молекулы.
Мы рассказали об одном из новейших и далеко не исчерпанных путей практического использования цеолитов. Однако уже давно эти вещества находят другое важное применение. Оно основано на том, что цеолиты легко обменивают содержащийся в них натрий на кальций и магний. Поэтому цеолиты — прекрасное средство для уменьшения жёсткости воды (см. Вода, жёсткость). При фильтровании воды через слой цеолита происходит ионный обмен: кальций и магний поглощаются, а в воду переходит эквивалентное количество натрия. Когда обменная способность цеолита истощается, его регенери-
Рис. 5. Кремнекислородный каркас в шабазите. Показаны только атомы Si и А1, обозначенные чёрными кружками; в кристаллах эти атомы соединяются между собой через атомы кислорода. Закрашенные квадраты — это места примыкания «двухэтажных» шестерных колец к обширной центральной полости. Эта полость имеет большие «окна» в виде восьмерных колец.
275
СОДА
Рис. 6. Разделение компонентов бензина с помощью молекулярных сит. Крупные шары — это атомы кислорода, которые принадлежат тетраэдрам, образующим восьмерное кольцо; атомы Si значительно мельче и не показаны на чертеже. Входящие в состав углеводородных молекул атомы С и Н изображены соответственно чёрным и белым цветом.
а — «неразветвлённая» молекула нормального октана СН3—СН2—СН2—СН2—СН2—СН2—СН2—СН3 проходит через кольцо.
б — для «разветвлённой» молекулы изооктана, например для изомера
СН3
I
СН3 —С —сн2 —сн — сн3 I I сн3 сн3
кольцо слишком узко.
руют, обрабатывая 5—10%-ным раствором поваренной соли. Для смягчения воды обычно используют искусственно получаемые цеолиты (пермутиты), синтез которых осуществляется при добавлении горячего раствора сульфата алюминия A12(SO4)3 к раствору, содержащему жидкое стекло Na2SiO3 и соду Na2CO3.
Чтобы более или менее полно рассказать о разнообразных применениях С., пришлось бы написать целую книгу, и здесь мы вынуждены ограничиться только некоторыми примерами. В заключение отметим, что на силикатных материалах в значительной мере базируется строительная промышленность. Фундамент современного дома образуют огромные бетонные кубы, его стены сложены из кирпича или бетонных панелей, скреплённых цементным раствором, оконные проёмы закрыты стеклом. Бетон, кирпич, цемент, стекло бывают весьма различны по составу, но всегда их основой или важной составной частью служат С. П. М. Зоркий.
силициды — соединения кремния с металлами. В зависимости от структуры и свойств С. можно разбить на две группы. К первой относятся С. щелочных и щелочноземельных металлов (например, Li4Si и Ca2Si). Они разлагаются водой и разбавленными кислотами с выделением силанов:
Ca2Si + 4НС1 = 2СаС12 + SiH4
Наиболее интересна группа металлоподобных С., образованных переходными металлами. В структуре этих С. цепи и слои из атомов кремния размещены в кристаллической решётке металла. Поэтому их формулы часто не соответствуют формальным правилам валентности (например, Mn3Si, ReSi2, Cr5Si3). Это твёрдые вещества с довольно высокими температурами плавления (например, С. тантала TaSi2 плавится при 2200 °C). Очень важно, что С. не окисляются даже при высоких температурах. Так, например, С. молибдена MoSi2 почти не изменяется на воздухе вплоть до 1730 °C, что позволяет использовать его как нагреватель электрических печей.
Интересно объяснение такой устойчивости. При нагревании этого соединения молибден окисляется до МоО3 — летучего окисла, который удаляется с поверхности, а кремний окисляется до SiO2, которая тонким слоем покрывает всю поверхность и защищает её от дальнейшего окисления. Однако при 1730 °C двуокись кремния начинает плавиться; под действием сил поверхностного натяжения расплав собирается в шарик и «обнажает» свежую поверхность MoSi2; после этого силицид катастрофически быстро разрушается.
Образование силицидных слоёв на металлах широко используется для защиты от окисления и коррозии в различных химических реагентах. Многие С. являются полупроводниками.
Г. В. Самсонов.
СИНЙЛЬНАЯ КИСЛОТА (ц и а н и с т о-водородная кислота) — раствор цианистого водорода HCN в воде.
СКАНДИЙ (лат. Scandium) Sc — химический элемент III группы периодической системы Менделеева; атомный номер 21. См. Редкоземельные элементы.
СОДА — общее техническое название различных карбонатов натрия. Различают кальцинированную С. Na2CO3, кристаллическую С. Na2CO3-ЮН2О, пищевую (питьевую или двууглекислую) С. NaHCO3. Несколько особняком стоит каустическая С.— техническое название гидроокиси натрия NaOH.
В природе С. встречается в виде рассолов в некоторых озёрах. Есть у неё и минералы — на-трон Na2CO3- ЮН2О, термонатрит Na2CO3-H2O, трона Na2CO3-NaHCO3-2H2O. Но крупных скоплений эти минералы не образуют. До конца
18*
СОЛЯНАЯ КИСЛОТА
XVIII в. С. получали из золы растений или из содовых озёр. Однако потребность в С. всё росла, и природные источники не могли удовлетворить её.
В связи с этим Парижская академия наук назначила большую премию за разработку промышленного способа получения С. В 1787—89 гг. французский химик Н. Леблан предложил сул ьфатный способ производства С. Первый завод по методу Леблана начал работать во Франции уже в 1791 г. Заключается этот метод в следующем. Поваренную соль нагревают с серной кислотой: 2NaCl + H2SO4 = Na2SO4 + 2НС1 Полученный сульфат натрия прокаливают с углём и известняком; при этом сначала сульфат восстанавливается до сульфида:
Na2SO4 + 4С = Na2S 4- 4СО а затем происходит обменная реакция: Na2S + CaCO3 = Na2CO3 + CaS Плав, содержащий С., сульфид кальция CaS, избыток угля и извести, обрабатывают водой, при этом С. переходит в раствор. Последний затем выпаривают и получают кристаллическую С. Na2CO3* ЮН2О. При этом в качестве побочных продуктов получаются НС1 и CaS, который не имеет никакого промышленного применения. НС1 сперва выпускали через дымовые трубы на воздух, что приводило к гибели растительности вблизи содовых заводов. Затем его начали поглощать водой. CaS образовывал целые горы отвалов, распространявших запах H2S. Способ Леблана теперь — уже далёкое прошлое.
В 60-х гг. XIX в. сульфатный способ получения С. стал уступать место более удобному и экономичному аммиачному способу, предложенному бельгийским инженером Э. Сольвэ. Концентрированный раствор NaCl насыщают аммиаком, а затем углекислым газом. При этом получается гидрокарбонат аммония:
NH3 + Н2О + СО2 = NH4HCO3 Последний, реагируя с NaCl, даёт хорошо растворимый хлорид аммония и малорастворимый на холоду гидрокарбонат натрия:
NH4HCO3 + NaCl = NH4C1 + NaHCO3
При нагревании до 150—200 °C гидрокарбонат натрия разлагается на кальцинированную С., воду и углекислый газ:
2NаНСО3 — Na2CO3 4- Н2О 4~ СО2 который возвращается в производство. Раствор NH4C1 нагревают с известковым молоком и получают аммиак:
2NH4C1 + Са (ОН)2 = СаС12 + 2Н2О + 2NH3 который идёт на насыщение новых порций соляного рассола. В этом процессе аммиак и углекислый газ расходуются только на неизбежные, сравнительно небольшие потери. Конечный продукт по чистоте значительно превосходит С., полученную по способу Леблана. Единственный побочный продукт СаО 2 также находит применение (см. Кальций, Хлориды). Теоретические основы аммиачно-содового процесса, необходимые, чтобы вести его в наивыгоднейших
276
условиях, разработал в 1904 г. русский химик-технолог П. П. Федотьев (о нём рассказано в статье Алюминий). Питьевую С. можно получить пропусканием углекислого газа через холодный раствор Na2CO3:
Na2CO3 4- СО2 + Н2О = 2NaHCO3
Каустическую С. (NaOH) получают обычно электролизом раствора NaCl (см. Натрий) или химическим путём по реакции:
Na2CO3 4- Са (ОН)2 = 2NaOH + СаСО3 Обработанная таким образом кальцинированная С. становится едкой (по-гречески «каусти-кос» — жгучий), отсюда и название этого соединения (в быту часто употребляется сокращённый термин «каустик»).
Трудно найти такую область хозяйства, где бы С. не применялась. Без С. сейчас не могут обойтись стекольная и мыловаренная промышленность, текстильная и целлюлознобумажная; её используют для очистки нефти и стирки белья, для обессеривания чугуна и приготовления шипучих безалкогольных напитков, в производстве огнетушителей и при выпечке пирогов, для получения различных соединений натрия и изготовления красок. У каждой домашней хозяйки всегда есть питьевая С. Врачи прописывают питьевую С. больным с повышенной кислотностью желудочного сока; как смягчающее средство С. входит в состав многих лекарств. В общем С.— один из важнейших продуктов химической промышленности. Её мировое производство оценивается в несколько десятков миллионов тонн. В 1974 г. в СССР произведено 4,5 млн. т кальцинированной и 2,2 млн. т каустической С. В. К- Бельский. СОЛЯНАЯ КИСЛОТА (х л о р истоводородная кислота) НС1 — сильная одноосновная кислота, представляет собой раствор хлористого водорода в воде.
С. к.— бесцветная жидкость с резким характерным запахом хлористого водорода (техническая С. к. имеет желтоватую окраску из-за примесей хлора и солей железа). Максимальная концентрация С. к. около 36%, такой раствор имеет плотность 1,18 г/см3. Концентрированная С. к. «дымит» на влажном воздухе и вот почему. Выделяющийся хлористый водород, попадая в такой воздух, вызывает конденсацию паров воды и образование мельчайших капелек С. к., которые мы и видим как слабый «дым» (точнее этот дым следовало бы назвать туманом).
С. к.— одна из самых сильных кислот; она энергично реагирует с металлами, стоящими в ряду напряжений до водорода, например:
Zn + 2НС1 = ZnCl2 + Н2 Fe + 2HC1 =FeCl2 + H2
277
СТАЛЬ
При этом образуются её соли — хлориды. С сильными окислителями С. к. ведёт себя как восстановитель, например:
МпО2 4- 4НС1 = МпС12 4 С12 — 2Н2О
(эта и подобные ей реакции используются в лабораториях для получения хлора).
Мировое производство С. к. достигает нескольких миллионов тонн в год. Получение С. к. происходит в две стадии. Сначала хлор сжигают в водородном пламени:
Н2-С12 = 2НС1
а затем образовавшийся газ — хлористый водород поглощается водой. С. к.— один из важнейших продуктов химической промышленности. Она идёт на получение хлоридов различных металлов и синтез хлорсодержащих органических продуктов. В металлургии ею обрабатывают руды, а в кожевенной промышленности — кожу перед дублением. С древнейших времён и до наших дней С. к. применяют для травления металлов — для удаления с их поверхности окислов; такая предварительная обработка не-
Дым без огня. «Нет дыма без огня» —утверждает пословица. Но всегда ли это так? Поставим следующий опыт. На дно одного стакана ёмкостью 200—300 мл поместим каплю концентрированного раствора соляной кислоты; на дио другого такого же стакана — каплю концентрированного раствора аммиака. Издали капель не видно, и стаканы выглядят совершенно пустыми. Прижмём теперь стаканы друг к другу, как показано на рисунке. Через несколько минут пространство внутри стаканов заполнится густым белым дымом, состоящим из мельчайших твёрдых частичек хлорида аммония NH4CI. Этот «дым» несколько секунд буквально валит из стаканов, когда их разъединяют. «Дыма» много, а огня-то нет!
обходима при паянии и лужении. Кроме того, С. к.— один из важнейших реактивов в химических лабораториях.
Вследствие высокой химической активности С. к. хранят и перевозят в стеклянных бутылях или в стальных сосудах, покрытых изнутри специальной резиновой плёнкой (гуммированных).
С. к. является составной частью желудочного сока — она участвует в процессе переваривания пищи и убивает различные болезнетворные бактерии (брюшного тифа, холеры и т. п.). При пониженной кислотности желудочного сока врачи прописывают больным приём очень раз-бавленной С. к. внутрь. В. К. Бельский.
СТАЛЬ — сплавы железа с углеродом, обычно содержащие до 1,7% С; такая сталь называется углеродистой. Если сверх того она содержит добавки хрома, марганца, никеля, молибдена, вольфрама и др., её называют специальной, или легированной (от немецкого legieren —сплавлять).
Получение чистого железа очень трудно и дорого; к тому же его механические свойства невысоки. Но уже давно было замечено, что прибавление углерода превращает железо как бы в другие металлы, более пригодные для практических применений. Это С. и чугун.
Итак, углерод — необходимая составная часть С. всех сортов. Углерод обязательно входит и в состав чугуна, однако в больших количествах (обычно около 4%). Главное же качественное отличие С. от чугуна состоит в том, что С., особенно при высоких температурах, способна к пластической деформации. Иными словами, под действием внешних сил С. изменяет свою форму без нарушения сплошности. На этом свойстве основаны ковка и прокатка С. (в горячем состоянии), штамповка изделий и волочение проволоки (в холодном состоянии).
Кроме углерода (и специальных добавок), в С. присутствуют примеси: кремний, марганец, сера и фосфор. Эти примеси попадают в С. в процессе её производства и при современном состоянии металлургии совершенно неизбежны. Примеси кремния и марганца (обычно 0,1 — 0,5% Si и 0,2—0,8% Мп) не оказывают существенного влияния на свойства С. Но даже незначительные, казалось бы, примеси серы и фосфора сильно ухудшают её пластичность. Сера, находящаяся в С. в виде сульфида FeS, вызывает красноломкость, то есть хрупкость в горячем состоянии. Объясняется это явление тем, что FeS образует с железом легкоплавкую смесь (эвтектику, /пл 988 °C), которая при затвердевании С. располагается между её кристаллическими зёрнами. Если такую С. нагреть до температуры, необходимой для начала ковки или прокатки (от 1000 до 1300 °C), то при первом же Обжатии металл даёт трещины. Связь
СТАЛЬ
между его зёрнами становится слабой, подобно связи между песчинками, из которых ребёнок слепил пирожок, замешав песок на воде. В обычных С. допускается до 0,04—0,05% S, а в высокосортных — не более 0,02% S.
Примесь фосфора вызывает хладноломкость, то есть делает С. хрупкой при низких температурах. В начале XX в. на одной из русских железных дорог, в суровую зиму, рельс, содержавший около 0,1% Р, развалился на десятки кусков после прохождения поезда, и только случайно не произошло катастрофы. В углеродистой С. фосфор присутствует в виде хрупкого соединения Fe3P. В обычных сортах С. допускается не более 0,05—0,08% Р, а в высших — до 0,02—0,03% Р.
Свойства С. зависят не только от её состава, но и от тепловых воздействий, которым она подверглась, то есть от термической обработки. Последняя состоит обычно в закалке и отпуске. Закалка — быстрое охлаждение С., нагретой до высокой температуры, повышает прочность и твёрдость С., но делает её хрупкой. Отпуск, то есть последующее нагревание до невысокой температуры, уменьшает хрупкость закалённой С., не понижая существенно её прочности и твёрдости.
Современные способы производства С. описаны в учебнике неорганической химии для 9-го класса. И мы рекомендуем читателю прервать на этом месте чтение статьи и заглянуть в учебник. Вероятно, вам захочется узнать, как же на протяжении многих веков менялось и совершенствовалось производство С. Тогда продолжите чтение этой статьи.
Уже в глубокой древности было известно, что прочность и твёрдость сыродутного железа (см. Железо) сильно повышаются, если его раскалить на древесном угле, а затем быстро охладить. Например, в 9-й песне «Одиссеи» Гомера (VIII—VII вв. до н. э.) читаем:
«Так расторопный ковач, изготовив топор иль секиру, в воду металл (на огне раскаливши его, чтоб двойную крепость имел) погружает, и звонко шипит он в холодной влаге...» (перевод В. А. Жуковского).
Здесь названы три процесса: 1) изготовление железного изделия ковкой; 2) нагревание его на раскалённых углях, в результате чего происходит науглероживание железа (цементация), то есть превращение его поверхностного слоя в С.; 3) закалка стального изделия. (Для ясности заметим, что сыродутное железо углерода почти не содержит и потому закалки не принимает.)
Расплавляя цементованную С. в тиглях, получали литую, так называемую тигельную, сталь. Её производство требовало огромных затрат труда и топлива. Тигельная С., содержащая 1,0—1,5% углерода, служила главным образом для изготовления холодного оружия — мечей, кинжалов, сабель. Её называли булатом (от персидского «пулад» — сталь) или дамасской С. (от города Дамаска, где когда-то процветало производство булатного оружия). Искусством производства такой С. в совершенстве владели мастера древних Индии, Персии, Сирии и некоторых других восточных стран. На поверхности булата красуется узор из чередующихся светлых и тём
278
ных полос. Булатный клинок очень прочный и очень острый, им можно перерубить железный гвоздь и разрезать на лету платок из тончайшей шёлковой материи. Булатную шпагу можно согнуть под углом 90°, и она не ломается. Благодаря столь редкому сочетанию свойств булатное оружие ценилось очень дорого, а способы его изготовления хранились в глубокой тайне.
По-видимому, тайна булата была утрачена в XIII или XIV в. В первой половине XIX в. некоторые учёные, надеясь получить булат, вводили в углеродистую С. небольшие добавки золота, серебра, платины, родия, палладия, хрома... Но булата изготовить так и не удалось, дело было вовсе не в добавках. Вспомните строки М. Ю. Лермонтова (1838 г.):
«Отделкой золотой блистает мой кинжал; Клинок надежный, без порока;
Булат его хранит таинственный закал — Наследье бранного Востока».
Поэт не знал, что в 1837 г. горный инженер П. П. Аносов в результате упорного, почти 10-летнего труда получил на Златоустовском заводе булатную С., из которой были изготовлены две сабли и черкесская шашка. Клинки из аносовского булата обладали всеми свойствами лучших изделий древних оружейников. Многовековая тайна была раскрыта.
В работе «О булатах» (1841 г.) Аносов подробно описал ход своих опытов. Он пришёл к выводу, что булат — это углеродистая С., изготовленная при соблюдении особых условий плавки, ковки и термической обработки («таинственный закал»). Благодаря этим условиям металлу придаются нужные структура и свойства. К тому же заключению пришли и русские металлурги Д. К- Чернов, Н. Т. Беляев и другие, также получившие булат.
В первой половине XIX в. в металлургии С. существенного прогресса не наблюдалось. Для производства С. по-прежнему применялось сварочное железо, которое получали посредством кричного передела чугуна и пудлингованием (см. ниже).
В основе этих способов, так же как и других, разработанных позднее, лежат одни и те же реакции: окисление содержащихся в чугуне примесей и излишнего углерода. В результате окисления образуется сначала СО, а затем СО2 (она улетает в атмосферу) и прочие окислы (они уходят в шлак, который с металлом не смешивается). Окислителями служат здесь и кислород воздуха’, и окислы железа. Закись железа образуется при действии кислорода на расплавленный чугун, другие окислы вводятся в виде железной руды или окалины (Fe2O3, Fe3O4). Окисление закисью железа FeO идёт в такой последовательности:
Si + 2FeO = 2Fe + SiO2
Мп 4- FeO = Fe + MnO C + FeO = Fe + CO
Операция, называемая кричным переделом, состоит в том, что расплавленный чугун под действием струи воздуха окисляется с образованием легкоплавкого шлака, состоящего из силикатов железа Fe2SiO4 и марганца Mn2SiO4 и избытка FeO. Последняя окисляет оставшиеся* примеси и углерод. По мере потери углерода точка плавления металла повышается. Так как температура горна была слишком низкой для расплавления малоуглеродистого железа, оно получалось в виде крицы — губчатой массы, пропитанной жидким шлаком (отсюда пошло и название способа). Раскалённую крицу ковали под молотом, чтобы по возможности удалить шлак.
Кричный передел, известный с XIII или XIV в., был мало производителен и дорог. Более выгодным оказался способ, изобретённый в 1766 г. англичанами братьями Т. и Дж. Кранедж и усовершенствованный в 1784 г. их соотечественником Г. Кортом. Этот способ был назван
279
СТАЛЬ
Павел Петрович АНОСОВ. Открыл секрет получения булатной стали.
Русский металлург Павел Петрович Аносов родился в 1799 г. в Петербурге. Сын секретаря Берг-коллегии (Главного горного управления), он рано потерял родителей и воспитывался у деда, служившего механиком на Камских заводах. В 1810 г. поступил в Горный кадетский корпус (ныне Ленинградский горный институт), где много раз награждался книгами и медалями. По окончании курса Павел Петрович был назначен на Златоустовский завод, основанный ещё при Петре I. Здесь в Златоусте, на Южном Урале, Аносов проработал около 30 лет — от практиканта (1817 г.) до начальника Златоустовских заводов (в 1841 г. произведён в генерал-майоры). А с 1847 г. и до конца жизни он был главным начальником Алтайских горных заводов.
Уже в 1819 г. Аносов составил описание производств Златоустовского завода (опубликовано впервые в 1954 г. по сохранившейся рукописи). Этот труд показал и широкий кругозор молодого инженера, и редкую его способность к обобщению и анализу. И в дальнейшем Аносов всегда совмещал каждодневную работу заводского инженера с большими научными изысканиями. Мировую же известность принесли ему работы, завершившиеся получением булатной стали. Обобщающий труд Аносова «О булатах» (1841 г.) был сразу же переведён на немецкий и французский языки. К тому, что рассказано об этих замечательных работах Аносова в статье «Сталь», добавим лишь слова другого русского учёного и металлурга Н. С. Курнакова — Аносов был «первым исследователем, применившим ещё в 1831 г. микроскоп для изучения структуры полированной и протравленной кислотами стали». Тем самым русский горный инженер положил начало микроскопической металлографии — науке» получившей развитие только в конце XIX — начале XX вв.
Аносов изобрёл способ закалки стальных изделий в струе сжатого воздуха (1827—29 гг.). Он получил литую сталь и отлил из неё артиллерийское орудие весом около 0,6 тонны (1836 г.). Усовершенствовал многие заводские механизмы и печи. Организовал производство огнеупорных тиглей — основного оборудования стале- и золотоплавильного производства того времени. За свои труды инженер Аносов был избран членом-корреспондентом Казанского университета, почётным членом Харьковского университета. Умер Аносов 25 мая 1851 г. в Омске. В нашей стране учреждены премии и стипендии имени Л. П. Аносова, издано собрание его трудов.
• пудлингованием (от английского глагола puddle — перемешивать). Оно велось в пламенных печах, в которых чугун нагревался только пламенем, не соприкасаясь с топливом. Под (дно) такой печи сперва покрывали кварцевым песком, а сверху наваливали чушки чугуна. После расплавления чугуна его вручную, с помощью железных ломов, длиной около 3 м, с загнутым концом, перемешивали с находившимся в печи шлаком, чтобы ускорить процесс окисления. По мере выгорания углерода металл делался тестообразным. И опять-таки получали крицы, которые проковывали для удаления шлака. Пудлингование было сопряжено с немалыми потерями железа в шлаке, большим расходом топлива и огромной затратой тяжёлого ручного труда. К тому же оно не разрешало вопроса о получении С. в количествах, необходимых для быстро возрастающих потребностей железнодорожного транспорта, судостроения, строительной техники, военного дела. Оставляло желать лучшего и качество металла. И сварочное железо, и полученная из него С. были неоднородны по составу и содержали шлаковые включения, ослабляющие прочность изделий (удалить ковкой весь шлак из железной крицы так же невозможно, как выжать всю воду из мокрой губки).
Новые технологические приёмы, позволяющие получать достаточно дешёвую С. хорошего качества и в больших количествах, появились в конце 1850-х — начале 1860-х годов.
Английский изобретатель-самоучка Г. Бессемер не знал ни химии, ни металлургии и наивно полагал, будто вся суть пудлингования заключается в перемешивании чугуна. Возложив эту работу на сжатый воздух, Бессемер поставил в угольный горн тигель с расплавленным чугуном
и через глиняную трубку пустил в металл дутьё. В своей автобиографии Бессемер пишет: «Я ждал, что металл застынет. Каково же было моё удивление, когда появился шум. Пламя вырвалось наружу, появился как бы настоящий вулкан. Крыша моей мастерской загорелась... Когда пламя село, я выпустил металл». Из полученного слитка был прокатан рельс высокого качества.
Однако на первых порах нередко случались неудачи, происходившие из-за незнания химической сущности процесса. Позже было выяснено, что при продувании воздуха через расплавленный чугун сперва образуется закись железа FeO. Она растворяется в жидком металле, окисляя Si, Мп, С по уже известным уравнениям. При этом выделяется столько тепла, что металл нагревается выше 1600 °C и может быть отлит. Однако при бессемеровании вредные примеси — сера и фосфор — полностью остаются в стали. Причина в том, что футеровка (внутренняя огнеупорная обкладка) бессемеровских конвертеров делается из кварцевых огнеупоров, состоящих почти целиком из кремнезёма (такая футеровка называется кислой). А в присутствии SiO2 удаление серы по известной реакции
FeS + СаО = CaS + FeO
СТАЛЬ
280
Рис. 1. Выпуск стали. Здесь этот важнейший металл цивилизации начинает свою трудовую биографию: завтра сталь воплотится в станки и строительные конструкции, рельсы и мосты, автомобили и океанские лайнеры.
невозможно: известь и кварц дадут легкоплавкий силикат CaSiO3, и футеровка будет разъедена.
Но даже при небольшом содержании серы бессемеровская сталь нередко получалась хладноломкой. Чтобы помочь беде, английский металлург Р. Мэшет в 1857 г. предложил прибавлять к С. перед разливкой так называемый зеркальный чугун (содержащий 10—20% Мп и около 4% С). После такой добавки С. прекрасно ковалась и прокатывалась. Выяснилось, что красноломкость бессемеровской С. была вызвана присутствием FeO в металле, продутом до почти полного выгорания углерода (до менее 0,1% С). Прибавление марганца, как говорят, «раскисляет» С., то есть восстанавливает растворённую в ней закись железа:
FeO + Mn = Fe + МпО
МпО и SiO2 образуют соединение MnSiO3, которое уходит в шлак. Прибавление зеркального чугуна понижает содержание серы:
FeS + Mn = Fe + MnS
Одновременно оно позволяет ввести в С. желаемое количество углерода. Раскисление С. перед разливкой — одна из важнейших производственных операций. Для раскисления, кроме зеркального чугуна, применяются ферромарганец, ферросилиций (см. Ферросплавы), алюминий. Высококачественную сталь часто раскисляют силикокальцием (сплав Si с 23—35% Са, 4—8% Fe).
При кислой футеровке невозможно и удаление фосфора. Он присутствует в металле в виде фосфида Fe3P. Последний при продувке сперва превращается в ортофосфат железа (II) Fe3(PO4)2. Но эта соль тотчас же реагирует с кремнезёмом:
2Fe3(PO4)2 + 3SiO2 = 3Fe2SiO4 + 2Р2О5
а Р2О5 восстанавливается железом до фосфида*. Р2О5 + 1 IFe = 5FeO + 2Fe3P
И в результате весь фосфор остаётся в металле.
В 1878 г. стал возможным передел чугунов, богатых фосфором, которые выплавлялись в Англии, Германии и Франции. Англичане С. Дж. Томас и П. Джилкрист предложили вести продувку фосфористых чугунов в конвертерах с футеровкой из основных окислов — магнезии MgO или её смеси с известью (MgO+CaO). Такая «основная» футеровка позволяет удалять из металла почти весь фосфор — Р2О5 образует с известью тетракальциевый фосфат, который
уходит в шлак:
2Fe3P + 5FeO + 4СаО = Са4Р2О9 + 1 IFe
Одновременно по уже известной реакции удаляется и часть серы в виде CaS. При использовании воздуха, обогащённого кислородом до 30— 37%, можно получать С., в которой всего 0,02% Р и 0,02% S. Томасшлак, содержащий 12—20% Р2Об, применяется как 'минеральное удобрение.
Однако бессемеровский и томасовский конвертеры имели существенный недостаток: в них нельзя было использовать с к р а п, то есть стальной металлолом. А поскольку к середине XIX в. в промышленных странах накопилось большое количество скрапа, нужно было искать способы, позволяющие переплавлять его в С. Главная трудность состояла в том, что не было печей, позволяющих выплавлять большие массы С.
В 1864 г. французский металлург П. Мартен предложил получать литую С. в регенеративных пламенных печах, в которых тепло продуктов горения используется для повышения температуры в рабочем пространстве (принцип регенерации тепла был разработан незадолго до этого немецким инженером Ф. Сименсом). Эти печи отапливались газом (позднее нефтью). Газ и необходимый для горения воздух предварительно нагреваются до 1100—1200 °C в регенераторах — камерах с решётчатой насадкой из огнеупорного кирпича. Через них попеременно пропускают то раскалённые отходящие печные газы, то холодные горючий газ и воздух. Это позволяет иметь в рабочем пространстве печи температуру около 1700 °C. По производительности мартеновский процесс уступает бессемеровскому и томасовскому. Но, в отличие от них, он даёт возможность получать С. высокого
281
СТАЛЬ
Дмитрий Константинович ЧЕРНОВ —один из основоположников науки о строении и термической обработке стали.
Русский металлург Дмитрий Константинович Чернов родился 1 ноября 1839 г. в Петербурге в семье мелкого чиновника. По окончании в 1858 г. Петербургского (ныне Ленинградского) технологического института преподавал там математику. С 1866 по 1880 гг. был инженером на Обуховском сталелитейном заводе в Петербурге (ныне завод «Большевик» в Ленинграде). С 1889 г.— профессор металлургии Михайловской артиллерийской академии. Умер 2 января 1921 г. в Ялте.
Завод, на котором работал Чернов, был основан горным инженером П. М. Обуховым в 1863 г. Продолжая деятельность известного металлурга П. П. Аносова, Обухов организовал на Златоустовском заводе производство стальных слитков для изготовления стволов артиллерийских орудий. Это было большим новшеством, так как до того орудийные стволы делались из пушечной бронзы (сплава меди с 9—10% олова). Из-за отдалённости Златоуста и отсутствия железнодорожного сообщения с ним по инициативе Обухова сталелитейный завод был построен в Петербурге. В первые годы работы завода его пушки нередко разрывались при пробной стрельбе. Была даже поставлена под сомнение целесообразность производства стальных орудий. За изучение этого вопроса взялся Чернов. Он установил, что аварийные орудийные стволы имеют в изломе крупнокристаллическую структуру, а исправные — мелкокристаллическую, причём по содержанию углерода и примесей они совершенно одинаковы. В результате длительных наблюдений и опытов Чернов в 1868 г. открыл существование у стали критических температурных точек, которые он обозначил буквами «а» и «Ь». Температуры этих точек зависят от содержания углерода. Сталь принимает закалку только будучи нагрета выше точки «а». В точке «Ь» сталь претерпевает структурное превращение. В то время высокие температуры умели определять лишь по цветам каления, то есть очень неточно. Лишь гораздо позднее было найдено, что точка «а» отвечает температуре 723° С и О,8°/о углерода. Позднее было расшифровано и структурное превращение стали в точке «Ь». Оказалось, что твёрдый раствор углерода в гамма-железе (гранецентрированная кубическая решётка) переходит в твёрдый раствор углерода в альфа-железе (объёмноцентрированная кубическая решётка); температура превращения зависит от содержания углерода. На основе своих наблюдений и выводов Чернов разработал наилучшие условия отливки, ковки и термической обработки стали. С тех пор стальные орудия быстро вытеснили бронзовые и прочно вошли в артиллерию. Труды Чернова положили начало науке о строении стали и о зависимости её строения и свойств от состава, условий литья, механической и термической обработки.
Из других многочисленных работ Чернова упомянем лишь о том, что он показал (в 1876 г.) возможность бессемерования уральских малокремнистых чугунов и предсказал большие преимущества применения кислородного дутья в конвертерном процессе.
В часы отдыха любимым занятием учёного было изготовление смычковых инструментов. В 1911 г. в малом зале Петербургской консерватории состоялся концерт для сравнительного испытания инструментов работы Чернова и старых итальянских мастеров. В газетах того времени писали, что скрипки и альты Чернова по качеству звучания не уступали инструментам Амати, Гуар-нери, Страдивари.
В конце XIX в. работы русского металлурга были лучше известны за рубежом, чем на родине. Так, студент Горного института М. А. Павлов, впоследствии и сам выдающийся металлург, узнал о них из статьи «Железо» во французской «Химической энциклопедии». На родине признание и почести пришли к Чернову только в начале XX в. Он был избран почётным членом Русского технического общества, пожизненным почётным председателем Русского металлургического общества, почётным вице-президентом Английского института железа и стали, почётным членом Американского общества горных инженеров. Американский металлург Г. М. Хау назвал его «отцом металлографии железа» (в 1903 г.). В годы гражданской войны Чернов ответил решительным отказом на приглашение эмигрировать в Англию. Его труды неоднократно переиздавались в СССР и в переводах на иностранные языки.
СТАЛЬ
282
качества не из одних только чугунов определённого состава (содержащих мало серы и фосфора). Годятся разнообразные материалы, в том числе скрап, а также чугун, непригодный для томасирования или для бессемерования.
Мартеновский передел ведётся в печах, под которых выполнен либо из кислых (SiO2), либо из основных (СаО, MgO) огнеупорных материалов, в соответствии с чем процесс называется кислым или основным. Химические реакции, идущие в мартеновских печах, принципиально не отличаются от протекающих в конвертерах Бессемера и Томаса. Всё различие состоит в том, что продолжительность передела в конвертерах измеряется минутами, а плавки в мартеновских печах длятся 6—8 часов. В России первая мартеновская печь начала работать на Сормовском заводе в 1870 г. кислым процессом; первые плавки на основном поду были произведены в России в 1881 г. Ёмкость мартеновских печей составляла тогда всего 1,5—2,5 т. Современные печи имеют ёмкость 300—500 т, а на Магнитогорском комбинате и Ждановском заводе им. Ильича работают мартены-исполины ёмкостью 900 т.
Мартеновский процесс быстро занял главенствующее положение среди способов передела чугуна в С. В конце XIX — начале XX вв. появились электрические печи для выплавки С., которые позволяли получать металл высокого качества. Тем не менее до середины 50-х гг. XX в. в мартеновских печах выплавлялось
Рис. 2. Эти большие шаровые резервуары (газгольдеры) сделаны из стали. В них хранят ценное химическое сырьё, например продукты переработки нефтяного газа: пропан, бутан, изобутан.
примерно 80% мирового производства С. Однако в последующие годы в связи с бурным развитием кислородно-конвертерного процесса доля мартеновского процесса в выплавке С. резко упала (до 40% в 1970 г.).
На целесообразность использования кислорода при выплавке С. указывал ещё в 1876 г. замечательный русский металлург Д. К. Чернов. В 1936 г. советский инженер Н. И. Мозговой впервые применил чистый кислород для продувки жидкого чугуна в конвертере. В промышленном масштабе кислородно-конвертерный процесс был впервые внедрён в Австрии в 1952 г. Замена воздуха кислородом позволила значительно повысить температуру металла при продувке, так как тепло расходуется на нагрев азота, входящего в состав воздуха. Это позволило не только перерабатывать чугуны с низким содержанием кремния и фосфора, но и вводить в конвертер скрап (до 25% от веса чугуна) или руду (до 5%). Кроме того, получаемая при этом С. характеризуется низким содержанием азота (0,002—0,006% N) и по качеству практически не уступает мартеновской. Указанные достоинства кислородноконвертерного процесса в сочетании с меньшими капиталовложениями (на 20—25%), снижением себестоимости С. (на 2—4%), увеличением производительности труда (на 25—30%) и обусловили быстрое внедрение этого способа в сталеплавильную практику многих стран. В СССР на ряде заводов работают мощные кислородные конвертеры ёмкостью 300 т, которые по суточной производительности превосходят 900-тонные мартеновские печи.
Полученную тем или иным способом литую С. выпускают в ковш — огромный стальной резервуар, выложенный внутри огнеупорными материалами. Жидкий металл раскисляют, иногда подвергают вакуумированию (для удаления газов), а затем разливают в чугунные формы — изложницы, в которых металл застывает, образуя слитки различного сечения (квадрат, круг, прямоугольник, многоугольник и т. д.). Затем слитки С. направляют в дальнейший передел.
Около 80% всей выплавляемой С. поступает в прокатные цехи, где стальные слитки превращаются в листы, рельсы, балки, трубы и другие изделия. Остальная С. превращается в изделия посредством ковки, литья, штамповки и обработки резанием. Отливка под давлением (см. Чугун) позволяет получать детали заданных размеров и формы, почти не требующие дальнейшей обработки.
Среди сортов С. очень видное место занимают С., содержащие легирующие добавки, в частности хром, марганец, никель, вольфрам. Положительное влияние хрома
283
СТЕКЛО
на свойства С. подметили ещё в 1820-х гг. в Англии М. Фарадей и Дж. Стодарт, а также П. Бертье во Франции. В 1859—1861 гг. Р. Мэшет в Англии обнаружил, что С. с добавкой вольфрама «красностойка», то есть сохраняет твёрдость и режущую способность при температуре красного каления (около 600 °C).
С 1885 г. во Франции возникло производство никелевой С. В 1889 г., по предложению английского металлурга Р. Гадфилда, из С. с 12—14% марганца и около 1% углерода, оказавшейся весьма стойкой против износа, начали отливать стрелки и крестовины для железнодорожных путей, «щёки» дробилок для руды, камней и др. В конце XIX в. В. Н. Липин организовал в России производство легированной С., в частности вольфрамовой и молибденовой. В 1900 г. на Всемирной выставке в Париже демонстрировалась работа резца из хромовольфрамовой быстрорежущей С., изобретённой в США. Такой резец, раскалённый почти докрасна, снимал стальную стружку в несколько раз быстрее, чем резец из углеродистой инструментальной С. Однако развитие выплавки легированных С. сделалось возможным только после создания промышленности ферросплавов.
В последнее время обнаружено, что небольшие добавки редкоземельных металлов (см. Лантаноиды) к нержавеющим, жаропрочным, быстрорежущим и другим специальным С. улучшают их прочность и стойкость против коррозии, облегчают обрабатываемость давлением и резанием, улучшают поверхность отливок.
Помимо массовых способов производства С., какими являются кислородно-конвертерный, мартеновский и электросталеплавильный, разработан ряд более дорогих и менее производительных способов получения С. (и других сплавов), обеспечивающих, однако, очень высокое качество металла. Это вакуумный дуговой и индукционный переплав, электрошлаковый переплав, плазменно-дуговой переплав и др.
Большие перспективы для производства С. открывает прямое восстановление железа из руд, минуя доменный процесс. Над этой проблемой долго работали учёные и изобретатели, но без особого успеха. Вопрос был решён благодаря применению очищенного природного горючего газа (состоящего из метана СН4 с небольшими примесями других углеводородов и H2S) в качестве восстановителя. Железная руда, обогащённая до 68% Fe (то есть до 97,2% РегОз), в виде небольших шариков («окатышей») поступает в печь, где при температуре ниже точки плавления железа протекает реакция:
4Fe2O3 + ЗСН4 = 8Fe + 6Н2О + ЗСО2
Процесс идёт непрерывно; его продуктом являются губчатые окатыши, содержащие 93% железа. Они служат сырьём для производства стали в мартеновских печах или электропечах. В 1971 г. на окраине Гамбурга был построен завод, ежегодно производящий по описанному новому способу 500 000 т стали и 350 000 т проката. У нас в районе Курска намечается постройка металлургического комбината, также работающего на основе прямого восстановления железа, но во много раз более мощного, чем гамбургский. Следует напомнить, что ещё в 1933 г. советский металлург А. А. Байков закончил свою работу «Физико-химические основы прямого восстановления железа из руд» пророческими словами: «Возможно, что наилучшее решение проблемы прямого получения железа будет достигнуто при помощи природных газов». Осуществление этого прогноза оказалось возможным лишь после создания советской промышленности природного газа, самой мощной в Европе (212,4 млрд, м3 в 1971 г.).
Мировое производство С. достигло около 700 млн. т в год (1974). Выплавка в СССР в 1974 г. составила 136 млн. т.
Русские и советские учёные-металлурги — П. П. Аносов, Д. К. Чернов, В. Е. Грум-Гржимайло, М. А. Павлов, А. А. Байков, Н. Т. Гудцов, И. П. Бардин, М.М. Карнаухов, Н. А. Минкевич, К- П. Григорович, А. М. Самарин и многие другие создали научную основу для могучего роста исследования и производства стали в СССР и воспитали большую школу металлургов, которые продолжают и творчески развивают идеи своих учителей.
С. А. Погодин. СТЕКЛО — аморфный, хрупкий, более или менее прозрачный материал, образующийся при застывании расплавов некоторых веществ или смесей. В «обычном» стекле 68—75% SiO2, 10—17% Na2O, 5—10% СаО, до 4% MgO, до 3% К2О и 1—4% А12О3 и Fe2O3. Такое С. называют силикатным — его основную массу составляет SiO2. Помимо «обычного» — силикатного С., изготовляют и многие другие его сорта. Так, есть С. на основе В2О3, Р2О5, GeO2. Их называют боратными, фосфатными, германатными. В состав таких С. также входят окислы щелочных и щелочноземельных металлов. Назначение же различных С. характеризуют их «потребительские» названия — строительное, оконное, бутылочное, посудное, оптическое, электровакуумное, химико-лабораторное...
Близкий к «обычному» состав имеет С. вулканического происхождения — обсидиан. Это природное С. известно человеку с древнейших времён. Наконечники стрел, ножи и другие колющие и режущие предметы из обсидиана, наряду с орудиями из кремня, были найдены в различных местах земного шара.
Много веков существует и варка С.— стеклоделие. В Древнем Египте оно возникло за 3000 лет до н. э. Цилиндр из светло-голубогоС., найденный в Тель-Асмаре, близ Багдада (современный Ирак), изготовлен в середине 3-го тысячелетия до н. э. Из полупрозрачного зеленоватого С. сделана знаменитая ваза с начертанным на ней именем ассирийского царя Саргона II (VIII в. до н. э.); сейчас она находится в Британском музее, в Лондоне. Однако техника древнего стеклоделия была примитивна. Высоких температур получать не умели, а именно от температуры варки зависит прозрачность С. Плавку вели в небольших глиняных горшках; С. получалось непрозрачным и в очень малых количествах. Чтобы украсить изделия из такого С., к нему добавляли минеральные красители. Это имитировало природные полудрагоценные камни. Из С. в те времена делали вазы, бусы, серьги, браслеты, амулеты...
Непрозрачное С. изготовляют и сегодня. Это и декоративные цветные плитки — смальтовые и марблитовые. Это и молочные, опаловые С. для абажуров, электроламп. При получении такого С. в шихту вводят «глушители» — плавиковый шпат CaF2, криолит Na3[AlF6] и другие. Основная же масса продукции современных стеклозаводов — прозрачное, бесцветное С.
СТЕКЛО
284
Рис. 1. Хрустальная люстра из Останки некого дворца в Москве. Сейчас этот дворец, замечательный памятник русской архитектуры и декоративного искусства, превращён в музей творчества крепостных. Его парадные залы, фойе, галереи отличаются богатым, высокохудожественным убранством. Всё здесь — резьба по дереву, цветной художественный паркет, хрустальные и бронзовые люстры и канделябры — выполнено народными мастерами.
Первые изделия из прозрачного С. были изготовлены мастерами Древнего Рима на рубеже нашей эры. Они овладели методом выдувания полых стеклянных изделий, вызвавшим переворот в технологии стеклоделия.
Каково же строение (структура) С.? Почему одни вещества при застывании их расплава переходят в кристаллическое, а другие — в стеклообразное (аморфное) состояние? Причин много. Одна из них — вязкость расплава. Другая— скорость его охлаждения.
Молекулы в любой жидкости (расплаве) расположены беспорядочно. При затвердевании веществ, расплавы которых имеют малую вязкость, молекулы не только при медленном, но и при быстром охлаждении «успевают» занять свои места в образующейся структуре кристалла. Таких веществ большинство — сахар и соль, железо и медь и многие другие. В расплавах же SiO2, В2О3, Р2ОБ и солей соответствующих кислот находятся большие полимерные ионы и молекулы (полисиликаты, полибораты,
полифосфаты). Вязкость таких расплавов велика и при быстром охлаждении они затвердевают, сохраняя почти ту же неупорядоченную молекулярную структуру, какая была в расплаве.
Если расплав затвердевает, то есть переходит в кристаллическое состояние, то физические свойства (тепло- и электропроводность, вязкость, теплоёмкость и другие) меняются скачком при температуре затвердевания. Переход же расплава в твёрдое стеклообразное состояние происходит постепенно (в интервале температур 1000—1400 °C для обычного С.) и без скачкообразного изменения физических свойств. С точки зрения физика, С.— это «твёрдая жидкость».
Варят С. в ванных печах непрерывного действия. Размер «ванны» таков, что самому крупному киту было бы в ней просторно. В эту печь с одного конца засыпают смесь исходных материалов (шихту). Составляют её из песка SiO2, соды Na2CO3 (или сульфата натрия Na2SO4 и угля), мела (или известняка) СаСО3, магнезита MgCO3, селитры KNO3. Другие материалы (ВаСО3, ZnO, В2О3, А12О3, РЬО) вводят в шихту при варке специальных стёкол. В первой части печи идут реакции разложения и окисления-восстановления:
Na2CO3 = Na2O + СО2 СаСО3 = СаО + СО2 Na2SO4 + С = Na2O + СО + SO2 2KNO3 + ЗС = К2О + 2NO + ЗСО
и другие.
Здесь из SiO2 и окислов металлов образуются силикаты:
mMeO + nSiO2 = /nMeO-nSiO2
Пузырьки газов (СО, СО2, NO, SO2) остаются в вязкой массе С. Их удаление происходит во второй, более горячей части печи (1600°C), когда вязкость С. понижается. В последней же части печи прозрачная масса С. остывает до 1200—1300 °C. При этой температуре вязкость стекломассы такова, что из неё удобно изготовлять изделия. Все процессы управления огромной стекловаренной печью автоматизированы. Лишь специальные С.— оптическое, цветное и некоторые другие — варят в небольших печах.
Из-за того, что расплав С. затвердевает постепенно (иначе говоря — благодаря «послушности» С.), можно вырабатывать изделия из стекломассы самыми различными приёмами, требующими времени,— выдуванием, вытягиванием, прессованием. Производство изделий из С. полностью механизировано. Однако в любом физическом или химическом институте есть стеклодувные мастерские, в которых ручным способом воплощают в стекле самые причудливые замыслы — уникальные научные приборы и установки. Это оказывается возможным благодаря и «послушности» С., и опыту мастеров.
Пластичность раскалённого С.— его ценное технологическое качество. А наиболее важные
285
СТЕКЛО
технические качества С.— прозрачность, прочность и устойчивость к действию химических реагентов. Эти свойства можно менять в широких пределах. Они зависят от состава, способа производства и обработки изделий из С. Так, прозрачность С. определяется температурой варки. Подобно стали, С. можно закалить, нагрев его до 600—650 °C и быстро охладив. Прочность такого С. в 4—5 раз выше, чем у обычного С. Тонкий лист сталинита, положенный на опоры, выдерживает вес человека или даже удар стального килограммового шара, падающего с метровой высоты.
При уменьшении содержания SiO2 до 55— 60% и введении в шихту соединений свинца получают свинцовый хрусталь — С., сильно преломляющее свет и хорошо полирующееся; красота хрустальных изделий, конечно же, зависит от мастерства их отделки. А для повышения химической стойкости С. уменьшают содержание Na2O и добавляют В2О3, А12О3, ZrO2 или увеличивают содержание SiO2. На химических заводах трубы для перекачки кислот, щелочей и других жидкостей практически везде делают из такого С., намного более химически стойкого и долговечного, чем специальные стали и обычные пластмассы.
Можно придать С. любой цвет. Спектр радуги, как известно, составляют семь цветов. Шкала же цветов, предложенная немецким учёным В. Оствальдом, включает 680 всевозможных тонов и оттенков. Для окрашивания С. в стекломассу вводят различные окислы, образующие с SiO2 окрашенные силикаты. FeO придаёт С. зелёный цвет, СоО — синий, МпО2 — фиолетовый. Вводят для окраски и другие соединения — те, что при нагревании разлагаются, выделяя металлы в мелкодисперсном состоянии. Так, для получения красного С. служат соединения золота и меди, которые в процессе варки С. восстанавливаются до металлов. В наши дни цветное С. широко используется в сигнальной технике.
Цветное стекло, по словам М. В. Ломоносова, было главным его увлечением. В первой в России химической лаборатории он сварил более четырёх тысяч опытных стёкол. Эти работы легли в основу заводских методов получения прозрачных и непрозрачных цветных С. Из своего цветного стекла Ломоносов вместе с учениками выложил большую (42 м2) мозаичную картину «Полтавская баталия», украшающую и сейчас здание Академии наук СССР в Ленинграде.
Производятся и другие специальные С. Так, чёрное стекло американского физика Вуда пропускает одни только ультрафиолетовые лучи. Многие минералы и красители, освещённые через такое С., ярко светятся в темноте. Эти красивые эффекты находят применение в таких разных областях, как анализ минералов и
театральная техника. Другие С., напротив, совершенно не пропускают ультрафиолетовых лучей и служат для остекления музеев, библиотек. Третьи пропускают инфракрасные лучи и применяются в оптических устройствах, позволяющих «видеть» в темноте, четвёртые не пропускают их и нужны для операционных светильников и проекционных устройств.
Замечательные свойства придают стеклу добавки окислов редкоземельных элементов. Под действием различных излучений такие С. лю-минесцируют. Вспышками света отмечают они прохождение нейтронов, быстрых электронов, -у-фотонов. Это используется в ядерной физике для регистрации и счёта частиц. Ионы Nd3+, Но3+, Ег3+,Тшз+, Yb3+ иногда называют лазерными. Из С., содержащих эти ионы, изготовляют рабочее вещество лазеров. Перечислить все применения С. невозможно...
Но вернёмся к «массовой» технике, к строительству. Корпусы катеров и лодок, кузовы автомобилей изготовляют из стекловолокна, пропитанного специальными клеями. В наибольшем и всё растущем масштабе используют С. в строительстве. И если раньше его применение ограничивалось лишь оконным С., то сегодня перечень строительных деталей и материалов из стекломассы очень велик: полые кирпичи, оконные пакеты, плитки, сталинитовые двери, звуко- и теплоизолирующая стекловата, стекложелезобетонные блоки, трубы, колонны...
С.— отличный материал для художественного оформления домов, не требующих в буду
Рис. 2. А. В. Стошкус «Земля мать» (1960— 1961 гг.). Галерея витража и скульптуры в Каунасе. Вы, наверное, видели цветные стеклянные витражи, заполняющие оконные проёмы. Проходящий сквозь них свет играет всеми цветами радуги, создавая атмосферу радости. А иногда — сказочности, таинственности...
СТРОНЦИИ
286
щем покраски и ремонтов — ведь материалы из С. не выцветают, не разрушаются микроорганизмами, не боятся влаги или жары.
Из С. сделаны и шпиль Университета на Ленинских горах, и звёзды Кремлёвских башен.
А. П. Пурмалъ. СТРОНЦИЙ (лат. Strontium) Sr — химический элемент II группы периодической системы Менделеева; атомный номер 38, атомная масса 87,62. Серебристо-белый металл, по внешнему виду напоминающий серебро. Природный С. состоит из смеси четырёх стабильных изотопов: 84Sr (0,56%), 86Sr (9,86%), 87Sr (7,02%) и 88Sr (82,56%). Об искусственно полученных радиоактивных изотопах С. см. ниже.
Название С. происходит от деревни Стронциан (Шотландия), вблизи которой был найден минерал, очень похожий на витерит (ВаСО3). Далеко не сразу стало ясно, что в этом минерале — стронцианите (SrCO3) содержится неизвестный прежде
элемент. В виде «стронциановой земли» SrO его открывали несколько раз: в Англии А. Кроуфорд (1790 г.) и Т. Хоп (1792 г.), в Германии М. Клапрот (1793 г.), в России академик Т. Е. Ловиц (1795 г.).
Ловиц много лет коллекционировал и исследовал образцы тяжёлого шпата (BaSO4). В 1795 г. он установил, что тяжёлый шпат содержит в виде примеси «стронциано-вую землю». Ему удалось отделить С. от бария. Для этого их сульфаты были переведены в хлориды, а затем хлориды обработаны абсолютным спиртом: SrCl2*6H2O растворился, а ВаС12«2Н2О остался в виде осадка. Металлический С. (в виде амальгамы) впервые получил в 1808 г. английский учёный Г. Дэви.
Содержание С. в земной коре 3,4-10“2% по массе (по распространённости 16-е место среди элементов). Обычно С. присутствует в различных кальциевых минералах в виде изоморфной примеси (см. Минералы). Всего известно более 20 минералов, содержащих С. Из них только два — целестин SrSO4 и стронцианит SrCO3, имеют промышленное значение для получения С. и его солей. В Советском Союзе месторождения этих минералов обнаружены в Архангельской области, в Верхнем и Среднем Поволжье, в Башкирии, Якутии, Таджикистане, в Крыму. За рубежом основные запасы стронциевых руд находятся в Великобритании, ФРГ, ГДР, США.
Важная особенность С.— его способность накапливаться в живом организме. Будучи по свойствам очень близким к кальцию, С. активно участвует в процессе обмена веществ в организме и откладывается вместе с Са в костной ткани. По данным советского геохимика академика А. П. Виноградова, среднее содержание С. в живом веществе равно 0,002%. Особенно много С. аккумулируют морские организмы — некоторые водоросли, кораллы, иглокожие и др. А простейшие морские жи
вотные — акантарии имеют скелет, состоящий целиком из SrSO4. Минерал целестин, такого же состава, встречается в осадочных породах; он осаждается из вод замкнутых бассейнов. В своей книге «Занимательная геохимия» академик А. Е. Ферсман поэтично написал о том, как за миллионы лет из бесцветных иголочек акантарий выросли сказочно красивые небесно-голубые кристаллы целестина (лат. caelestis — небесный).
С.— лёгкий (плотность 2,63 г/см3), мягкий, ковкий, пластичный металл; он легко режется ножом. Его £пл 770 °C, £кип 1380 °C. Летучие соли С. окрашивают пламя в карминово-красный цвет. Это свойство солей С. сделало их незаменимыми компонентами различных пиротехнических составов’ Красные вспышки праздничных фейерверков; красные огни осветительных и сигнальных ракет; следы, прочерчиваемые в ночном небе трассирующими пулями и снарядами...— всё это дают соединения С.— нитрат Sr(NO3)2, оксалат SrC2O4, карбонат SrCO3. Недаром А. Е. Ферсман назвал С. «металлом красных огней».
С. принадлежит к подгруппе щелочноземельных металлов. Внешняя электронная оболочка его атома содержит два внешних электрона (конфигурация 5s2, см. Атом). Во всех своих соединениях он проявляет степень окисления +2 (валентность II). Занимая промежуточное положение между кальцием и барием, С. чрезвычайно близок к ним по своим химическим свойствам. Одно из немногих различий между элементами состоит в разной растворимости солей: соли С. обычно более растворимы, чем соли бария, но менее — чем соли кальция. Этим и пользуются для разделения этих элементов.
С. химически очень активен. Он быстро окисляется на воздухе при комнатной температуре, покрываясь жёлтой плёнкой окиси SrO и перекиси SrO2, а при нагревании воспламеняется. С. непосредственно взаимодействует со всеми неметаллами. С водородом соединяется при температуре выше 200 °C, образуя гидрид:
Sr + Н2 = SrH2
Нагревание С. в азоте при 400 °C приводит к образованию нитрида:
3Sr + N2 = Sr3N2
При повышенных температурах С. легко соединяется с фосфором, серой, галогенами, углеродом. С водой энергично взаимодействует, вытесняя из неё водород:
Sr + 2Н2О = Sr(OH)2 + Н2
С. образует со многими металлами (Mg, Al, Pb, Sn и др.) сплавы, которые обладают весьма высокой твёрдостью и устойчивы к коррозии, что делает их ценными для электротехники и радиотехники.
287
Промышленная переработка целестина SrSO4 и стронцианита SrCO3 довольно проста. SrSO4 сплавляют с содой. Образующийся карбонат SrCO3 легко растворяется в соляной и азотной кислотах. Металлический С. получают главным образом восстановлением SrO алюминием при нагревании в вакууме до 1100—1150 °C. Пары С. превращаются в кристаллы на холодной поверхности конденсатора, помещённого внутрь стальной реторты, в которой ведётся процесс.
Свободный С. применяется мало. Причина этого — его высокая химическая активность.
Но, с другой стороны, именно высокая активность С. иногда нужна технике. Так, С., подобно кальцию, применяют в металлургии при выплавке меди и бронз. С. связывает серу, фосфор, углерод и повышает текучесть шлака, способствуя тем самым лучшему его отделению от выплавленного металла. Кроме того, С. повышает твёрдость меди без снижения её электропроводности. В электровакуумных трубках и других приборах С. поглощает кислород и азот и создаёт глубокий вакуум.
Значительно шире используют соли С. Из хромата SrCrO4 — лимонно-жёлтого пигмента, готовят краски для живописи. Сульфид SrS обладает способностью фосфоресцировать и потому идёт на приготовление светящихся составов — фосфоров. Такими светящимися красками покрывают ночные знаки на шоссейных и железных дорогах, сигнальные знаки в шахтах. Во многих случаях соли С. оказываются маловыгодными, и они уступают место солям других щелочноземельных металлов (в первую очередь кальция), близким по свойствам, но более дешёвым.
Вместе с тем существует область, в которой С. оказался незаменим и принёс большой экономический эффект. Это — производство глазурей и эмалей для покрытия керамики и металлов. Карбонат SrCO3 и окись SrO заменяют дефицитную, дорогую и к тому же ядовитую окись свинца РЬО, а также и соединения бора. При этом, во-первых, нисколько не теряются ценные качества, характерные для свинцовых и борных эмалей и глазурей (зеркальный блеск, сохранение подглазурных красок, низкая температура отжига). А во-вторых, появляются другие важные свойства: повышенная твёрдость, химическая и термическая стойкость.
Особенно же «прославился» С. благодаря существованию радиоактивного изотопа «строн-ций-90». Изучением свойств этого изотопа занимаются тысячи химиков, биологов, врачей, биохимиков, биофизиков во всём мире. Чем же вызвано столь пристальное внимание к изотопу 90Sr? Свойства, присущие этому изотопу, таковы, что могут быть направлены и на пользу человечеству, и во вред ему. Однако если мирное использование 90Sr более или менее подобно
СТРОНЦИЙ
применению радиоактивных изотопов вообще, то опасность его особая.
Начало повышенного интереса к ^Sr совпало с началом атомной эры — запуском в США первого ядерного реактора (1942 г.) и созданием атомных бомб. После испытания в Аламогордо первой бомбы (16 июля 1945 г.) следующие две бомбы менее чем через месяц былй сброшены и взорваны над японскими городами Хиросимой (6 августа) и Нагасаки (9 августа). Оба города были полностью разрушены, погибли одновременно десятки тысяч людей и все последующие годы росло число человеческих жертв. Большую роль в продолжающейся трагедии играет 90Sr.
Взрыв атомной бомбы — это цепная реакция деления ядер урана или плутония. В результате деления образуется около 200 радиоактивных изотопов. Среди них — изотопы щелочноземельных металлов с такими периодами полураспада: 4бСа — 164 дня, 140Ва — 12,8 дня, 89Sr — 51 день и ^Sr — 27,7 лет.
Напомним, во-первых, что период полураспада — это время, за которое из данного количества радиоактивного изотопа распадается половина, и во-вторых, что С., попавший в организм человека, как уже указывалось выше, не выводится, а остаётся в костях скелета. Нерадиоактивные изотопы С. не очень опасны, но вместе с ними остаётся и накапливается в организме радиоактивный "Sr. Он и ведёт там свою разрушительную работу: излучает бета-частицы (электроны), обладающие заметным ионизирующим действием. 0“-частицы поражают и костную ткань и, что особенно опасно, костный мозг — орган кроветворения.
Атомы "Sr, оказавшиеся в атмосфере, воде и почве после ядерного взрыва, вступают в круговорот веществ на Земле и могут попадать в организм человека при дыхании и приёме пищи и воды.
Изотоп 90Sr долгоживущ — его период полураспада таков, что накопление его в организме может продолжаться десятилетиями. «Стронциевая опасность» — один из важнейших аргументов в борьбе Советского Союза и передовых людей всего мира за запрещение испытаний атомного оружия. Накопление 90Sr в природе наблюдалось в годы интенсивных испытаний ядерного оружия в атмосфере. После заключения в Москве 5 августа 1963 г. Договора о запрещении испытаний ядерного оружия в атмосфере, в космическом пространстве и под водой загрязнённость атмосферы изотопом 90Sr к 1967 г. снизилась в 10 раз по сравнению с 1962 г.
Радиоактивные изотопы С. находят и широкое мирное использование. Та же мощная ионизация, тот же большой период полураспада при соответствующем разумном применении превращают «страшный» 90Sr в верного друга и помощника человека. Это и изотопный индикатор («меченый атом») для исследования7 различных процессов. Это и долгоживущие источники 0"-частиц, с успехом применяемые для лечения некоторых заболеваний.
Есть и ещё одна важная область применения 90Sr. Ионизация воздуха его препаратами приводит к снятию электрических зарядов, возникающих при трении в процессах производства бумаги, тканей, пластмасс и других материалов,
СУЛЬФАТЫ
288
являющихся изоляторами. Образование этих зарядов не только нарушает технологический процесс, но и создаёт столь огромные напряжения — до нескольких тысяч вольт,— что они могут привести к искровому пробою и даже к пожару. Применение пластин, покрытых тонким слоем 90Sr, даёт большой экономический эффект: эти пластины недороги, не требуют громоздкой аппаратуры, просты в эксплуатации, компактны и долговечны.
Вообще же хозяйственная и научная ценность С. и его соединений далеко не исчерпана, и с каждым годом открываются всё новые и новые сферы их применения. 3. А. Старикова. СУЛЕМА — хлорная ртуть, хлорид ртути (II) HgCl2. См. Ртуть.
СУЛЬФАТЫ (с е р н о к и с л ы е сол и)— соли серной кислоты H2SO4. Как сильная двухосновная кислота H2SO4 даёт два ряда солей: средние (нормальные) и кислые (гидросульфаты, прежнее название б и сульфаты). Свойства их во многом различны. Из гидросульфатов в твёрдом состоянии получены немногие (KHSO4, NaHSO4 и др.), а средние С. известны для всех металлов. Они по-разному растворимы, по-разному относятся к нагреванию.
Все гидросульфаты растворимы в воде. Из средних С. малорастворимы CaSO4 и Ag2SO4; практически нерастворимы BaSO4, SrSO4, RaSO4 и PbSO4.
Кислые С. при нагревании, отщепляя воду, переходят сначала в дисульфаты (прежнее название пиросульфаты) Me2S2O7, а затем — в нормальные С. и серный ангидрид:
2NaHSO4 = Na2S2O7 + Н2О Na2S2O7 — Na2SO4 -I- SO3
Из нормальных С. наиболее устойчивы к нагреванию соли щелочных металлов. С. щелочноземельных металлов при температурах выше 1000 °C разлагаются:
CaSO4 = СаО + SO3
С. других металлов разлагаются подобным образом при менее высоких температурах:
Fe2(SO4)3 = Fe2O3 + 3SO3
Серный ангидрид в обоих случаях может частично распадаться:
2SO3 = 2SO2 + О2
Способы получения нормальных С. разнообразны— растворение металлов в разбавленной серной кислоте:
Zn + H2SO4 = ZnSO4 + Н2 разложение солей летучих кислот серной кислотой:
СаСО3 + H2SO4 = CaSO4 + Н2О + СО2 окисление сульфитов или сульфидов и другие.
Кислые С. кристаллизуются из растворов нормальных С. в присутствии избытка H2SO4:
Na2SO4 + H2SO4 = 2NaHSO4
Нормальные С. выпадают из водных растворов обычно с одной или несколькими молекулами кристаллизационной воды. С. двухвалентных Fe, Ni, Со, Мп, Zn кристаллизуются с семью молекулами воды (например, FeSO4-7H2O). Кристаллогидраты такого состава (а также CuSO4«5H2O) называют купоросами. При кристаллизации смесей С. двух металлов из водных растворов нередко образуются комплексные соединения, в частности двойные соли; наиболее интересны из них квасцы.
С. широко распространены в природе и с давних пор служат сырьём для химической и других отраслей промышленности. Наиболее устойчивы в природе труднорастворимые С. двухвалентных металлов — бария, стронция, свинца и кальция: барит BaSO4, целестин SrSO4, англезит PbSO4, гипс CaSO4-2H2O, ангидрит CaSO4, кизерит MgSO4-H2O.
Но и легкорастворимые С. могут выпадать из воды соляных озёр либо при её испарении, либо при понижении температуры. Так, на дно залива Кара-Богаз-Гол (Каспийское море) в зимние месяцы в огромных количествах оседает мирабилит Na2SO4* 10Н2О (глауберова соль). Часть его штормами выбрасывается на берег, где образуются белые валы соли. В летнее время мирабилит в самом заливе вновь переходит в раствор, а выброшенный на берег в верхних слоях обезвоживается, превращаясь в тенардит Na2SO4.
К природным минералам относятся и многие сложные С.: астраханит Na2SO4-MgSO4-4H2O, полигалит K2$O4-MgSO4‘2CaSO4-2H2O и др.
С. различных металлов находят самое разнообразное применение. См. о них в статьях о соответствующих элементах: Алюминий, Железо, Калий,. Кальций, Натрий и т. д.
В. Л. Василевский. СУЛЬФИДЫ — соединения серы с более электроположительными элементами. С. металлов — соли сероводородной кислоты H2S (см. Сернистый водород). КС. неметаллов относится, например, P2S5.
Двухосновной сероводородной кислоте соответствуют два ряда солей: средние С. и кислые С. (гидросульфиды, или, как их называли раньше, бисульфиды). Гидросульфиды известны лишь для щелочных и щелочноземельных металлов (например, NaHS). Как средние, так и кислые С. этих металлов растворимы в воде. С. других металлов в воде нерастворимы — часто они служат примером практически нерастворимых веществ.
Вот сравнение: известно, что хлорид серебра AgCl весьма малорастворим в воде (1 г AgCl в 700 л воды). Однако, чтобы растворить 1 г сульфида серебра Ag2S понадобилось бы воды неизмеримо больше — около 2* 1012 л (то есть 2 км3). Различиями в растворимости С. металлов в воде, разбавленных кислотах или в растворах сульфида аммония пользуются в аналитической химии для качественного определения этих металлов. Анализ удобен и тем, что С. многих металлов имеют характерную окраску.
289
СУЛЬФИТЫ
Поскольку С.— соли очень слабой кислоты, растворимые С. сильно подвержены гидролизу:
Na2S + Н2О = NaOH + NaSH
С. аммония и щелочных металлов реагируют в растворах с серой, образуя-полисульфиды (см. Сера). С.— сильные восстановители. Из окислительно-восстановительных процессов с их участием наиболее важен так называемый обжиг — прокаливание природных С. на воздухе, приводящее к окислам металлов и сернистому ангидриду, например:
2CuS + ЗО2 = 2CuO + 2SO2
Этот процесс в промышленности служит для получения цветных металлов и серной кислоты.
С.— одна из важнейших форм существования серы в природе/ Сульфидные минералы столь многочисленны, что уступают по разнообразию лишь силикатам.
Многие природные С. обладают металлическим блеском. Таковы, например, галенит PbS, антимонит St^Sg, халькозин Cu2S, поэтому их называют также свинцовым, сурьмяным, медным блеском. Сульфидные минералы часто окрашены. Так, издавна известна как красная краска киноварь HgS. А название минерала As2S5 — аурипигмент отражает его красивый золотисто-жёлтый цвет. Разнообразную окраску и металлический блеск имеют также так называемые колчеданы: железный FeS2, магнитный FeS (пирротин), мышьяковый FeAsS (арсенопирит), медный CuFeS2 (халькопирит). Некоторые природные С. называют обманками. Так, цинковая обманка ZnS часто сопутствует галениту PbS, но «обманывает» металлургов — при плавке не даёт свинца. Другое название ZnS — сфалерит происходит от греческого «сфалербс» (обманчивый).
Природные С.— важнейшее сырьё для получения меди, никеля, цинка, свинца, ртути. Обычно они содержат примеси золота, серебра, марганца, а также селена, германия и других рассеянных элементов. По-видимому, химические операции с сульфидными минералами, содержащими примеси благородных металлов, иногда приводили средневековых алхимиков к получению небольших количеств золота и серебра. А это поддерживало упорную веру в возможность «превращения» обычных металлов в благородные. В наше время комплексная переработка сульфидных руд позволяет выделять из них многие металлы, в том числе золото и серебро.
На воздухе природные С. постепенно окисляются и взаимодействуют с двуокисью углерода, превращаясь в другие минералы. Так, пирит FeS2 образует сульфаты и гидроокиси железа. Из медного колчедана на воздухе постепенно образуется сульфат меди, а затем малахит СиСОз-Си (ОН)2, куприт Си2О и самородная медь. Конечно, такие процессы протекают медленно. Но в длительной геологической истории Земли они играли и играют большую роль.
19 хэш
Таким образом, С.— ценнейшее сырьё для металлургии и химии. Есть и другие, менее важные области применения С. Старейшая из них — обработка шкур для удаления волос при выделке кож. Для этой цели, сплавляя поташ или соду с серой, издавна получали смесь сульфида и полисульфидов К или Na — так называемую серную печень.
Приготовленные специальным образом С. щелочноземельных металлов, а также цинка и кадмия светятся под действием облучений различного рода. Поэтому такими веществами покрывают экраны телевизоров, рентгеновских установок, счётчиков радиоактивных излучений и других приборов. В. Л. Василевский. СУЛЬФИТЫ (сернистокислые соли)— соли сернистой кислоты H2SO3. Двухосновной H2SO3 соответствуют два ряда солей: средние С., например Na2SO3, CaSO3, и кислые — гидросульфиты (прежнее название бисульфиты), например NaHSO3, Са (HSO3)2. Гидросульфиты растворимы в воде, а из средних растворимы лишь С. щелочных металлов. Все С. как соли слабой кислоты в растворе сильно гидролизованы:
Na2SO3 + Н2О = NaHSO3 + NaOH
С.— восстановители; при действии окислителей они превращаются в сульфаты. Поэтому водные растворы С. неустойчивы на воздухе:
2Na2SO3 + О2 = 2Na2SO4
(Безводные С. на воздухе не окисляются.) Добавляя Na2SO3 к воде, идущей для питания паровых котлов, удаляют из неё растворённый кислород и таким образом предохраняют котлы от коррозии. При нагревании гидросульфитов щелочных металлов образуются д и с у л fa-фи т ы (прежние названия метабисульфиты, пиросульфиты) — соли не выделенной в свободном состоянии двусернистой кислоты:
2KHSO3 = K2S2O6 + Н2О
При обычной температуре дисульфит калия медленно разлагается:
Кг^гОь = K2SO3 + SO2
С., как и сернистый ангидрид, применяют для отбеливания тканей и для консервирования овощей и фруктов (в их присутствии микроорганизмы гибнут). В растворе, содержащем небольшие количества С., можно хранить, например, очищенный картофель несколько суток. Спасает фрукты от порчи и ди сульфит калия, медленно отщепляющий SO2. Его размещают в виде таблеток на стеллажах, где хранятся виноград, яблоки и т. п. С. натрия входит в состав фотографических проявителей (см. Серебро). Гидросульфит кальция Ca(HSO3)2 в ог-
СУРЬМА
290
121,75 51
Sb
5s25p3
СУРЬМА
ромных количествах применяется. в производстве целлюлозы из древесины (см. Сера).
В. Л. Василевский. СУРЬМА (лат. Stibium) Sb — химический элемент V группы периодической системы Менделеева; атомный номер 51, атомная масса 121,75. Кристаллическая С.— вещество серебристо-белого цвета с синеватым оттенком. Природная С. состоит из двух стабильных изотопов, 121Sb (57,25%) и 123Sb (42,75%).
С. известна с глубокой древности. В странах Востока она употреблялась ещё за 3 тысячи лет до н. э. для изготовления сосудов; её соединения входили в состав красок и косметических средств. Впервые описаны свойства С. и её соединений в книге Василия Валентина «Триумфальная колесница антимония» (1604 г.). Интересно, что этот элемент в настоящее время имеет в разных Языках три различных названия. Русский термин «сурьма» происходит от турецкого слова «siirme», что значит «натирание», «чернение бровей». Теперь, очевидно, станет понятным выражение «насур-мить брови». Для этого служило природное соединение С. Sb2S3 (см. ниже), одна из модификаций которого имеет фиолетово-чёрный цвет. Латинское обозначение «стибиум» происходит либо от греческого слова «стйби» — так называли минерал сурьмяный блеск, либо от слова «стймми» — сурьмяная помада для косметических целей. О происхождении слова «антимоний», принятого в большинстве европейских стран и США, существует много толкований. Об одном из них — «средство против монахов» — очень интересно рассказал Ярослав Гашек в повести «Камень жизни».
С. мало распространена в земной коре — 5- 10“б% по массе (59-е место), хотя известно свыше 90 сурьмяных минералов. Из них важнейшие, имеющие промышленное значение,— сурьмяный блеск (он же антимонит и он же стибнит) Sb2S3, кермезит 2Sb2S3-Sb2O3, сервантит (сурьмяная охра) Sb2O4, валентинит Sb2O3. Крупные месторождения сурьмяных минералов есть в Китае, Чехословакии, Алжире, Боливии, Мексике, Японии и США. В дореволюционной России С. совсем не добывалась, не были известны даже её месторождения. И только при Советской власти поиски этих месторождений и производство С. были развёрнуты в полную силу. Советский Союз располагает несколькими крупными месторождениями С. в частности на Кавказе, в Восточной Сибири и Казахстане.
Кристаллическая модификация С. (серая) образует звездообразные кристаллы; плотность 6,69 г/см3, *пл 630,5 °C, /кип 1635—1645 °C. Она плохо проводит тепло и необыкновенно хрупка — настолько, что её можно легко растереть в порошок в обычной фарфоровой ступке. Из
вестны и аморфные модификации С.— жёлтая и чёрная.
По своему положению в периодической системе С.— аналог азота, фосфора, мышьяка. Но С. отличается от них заметно выраженными металлическими свойствами. У атома С., как и у атомов перечисленных элементов, пять внешних (валентных) электронов (конфигурация 5s25p3, см. Атом). Для С. известны соединения со степенями окисления —3 (SbH3), +3 (Sb2O3) и +5 (Sb2O5). Валентности С. равны соответственно III и V. При обычных условиях на воздухе С. не изменяется и лишь при 800 °C сгорает с образованием Sb2O3. Если порошок С. осторожно всыпать в банку, наполненную хлором, то можно наблюдать яркие вспышки, и банка наполняется дымом (рис. 1).
Легко реагирует С. и с другими галогенами. С водородом С. непосредственно не взаимодействует, и соединение SbH3 (стибин) получается лишь косвенным путём, совершенно так же, как мышьяковистый водород. С кислородом С. образует три окисла: Sb2O3 (ему соответствует сурьмянистая кислота H3SbO3 и соли антимониты), Sb2O4 (окисел смешанных степеней окисления +3 и +5) и Sb2O5 (ему соответствует сурьмяная кислота HSbO3 и соли
Рис. 1. Горение сурьмы в хлоре. Эта реакция — одна из красивейших в неорганической химии. В сосуд с хлором осторожно по трубке небольшими порциями всыпают порошок сурьмы, тщательно растёртой в ступке (если используется сосуд ёмкостью 1 л, то нужно брать 2—2,5 г Sb). Как только крупинки сурьмы попадают в сосуд, они тут же вспыхивают яркими звёздочками, так что зрелище немного напоминает праздничный фейерверк. Сосуд постепенно наполняется белым дымом пентахлорида сурьмы:
2Sb + 5Cl2 = 2SbCl5
291
СУРЬМА
антимонаты). Соли Sb (+3) в растворах подвергаются сильному гидролизу с образованием основных солей:
SbCl3 + 2Н2О = Sb(OH)2Cl + 2НС1
Основные соли, отщепляя воду, переходят в соединения своеобразного одновалентного катиона антимонила, или стибила, SbO+:
Sb(OH)2Cl = SbOCl + Н2О
К солям этого катиона относится и упоминаемый ниже «рвотный камень».
Химики средневековья обнаружили, что в расплавленной С. растворяются почти все металлы (при этом образуются соединения — антимониды, например AuSb2). Металл, «пожирающий» другие металлы,— своеобразный «химический хищник». Очевидно, поэтому сурьмяный блеск изображали в виде волка с разинутой пастью (рис. 2).
В ряду напряжений С. располагается за водородом и поэтому не вытесняет его из кислот. Однако такие кислоты, как азотная, действуют на С. с образованием окисла. При использовании разбавленной HNO3 образуется Sb2O3:
2Sb + 2HNO3 = Sb2O3 + 2NO + H2O
а концентрированной — Sb2O5:
2Sb + 10HNO3 = Sb2O5 + 10NO2 + 5H2O
Оба окисла получаются в виде нерастворимых гидратов с переменным содержанием воды. Со щелочами С. не реагирует.
Металлургия С. довольно проста. Нагревая природную Sb2S3 с железом, получают С. «осадительной плавкой» по реакции:
Sb2S3 + 3Fe=3FeS + 2Sb
Путём окислительного обжига руд С. получают Sb2O3, которую затем восстанавливают углём: Sb2O3 + ЗС = 2Sb + ЗСО
В обоих случаях черновую С. очищают электролизом от примесей Си, Fe, As, S и др.
Область применения С. и её соединений чрезвычайно широка — от театральных занавесов до полупроводников. Главным потребителем чистой С. является типографское производство. В отличие от большинства других металлов, расплавленная С. (как и висмут) при затвердевании расширяется. Благодаря этому свойству она входит важной составной частью в типографские сплавы (гарт — сплав свинца, сурьмы и олова, содержит до 20—23% Sb).
Типографский шрифт отливают так: расплавленный металл (типографский сплав) подают под давлением в литейную форму, на одной из стенок которой выдавлено углублённое изображение буквы или знака (это так называемая матрица). Застывая, сплав, содержащий С., несколько расширяется и поэтому очень точно воспроизво
дит детали матрицы. При печатании краска переносит изображение с металлической буквы (литеры) на бумагу. Понятно, что сплавы, содержащие С., очень удобны и для художественного литья, когда особенно важно сохранить тонкости рисунка.
Широко распространены подшипниковые сплавы (баббиты), состоящие из олова, меди и С. В оловянистых баббитах среди мягкой податливой основной массы твёрдого раствора С. и меди в олове располагаются твёрдые износостойкие кристаллики соединений С. с оловом— SnSb и олова с медью — CuSn. Такое со-
Рис. 2. В средние века золото очищали от примесей (серебра и меди) сплавлением с антимонитом Sb2S3 — природным сульфидом сурьмы. При этом происходило образование сульфидов серебра и меди:
Sb2S3 + 6Ag = 3Ag2S + 2Sb
Sb2S3 + 6Cu = 3Cu2S + 2Sb
Сурьма же давала с золотом соединение AuSb2 — метал-лид (такие соединения описаны в статье Металлы), которое растворялось в избытке золота:
Au Ц- 2Sb = AuSb2
Жидкая масса, вылитая в железную коническую форму, образовывала два несмешивающихся слоя: нижний, состоящий из золота, в котором растворён AuSb2, и верхний, состоящий из смеси Sb2S3, Ag2S и Cu2S. По охлаждении слои разделяли. Затем золото, содержащее AuSb2, расплавляли и на его поверхность направляли струю воздуха из мехов. При этом сурьма превращалась в трёхокись Sb2O3, улетавшую в виде белого дыма. Так получали чистое золото. В XVIII в. этот способ вышел из употребления.
Рисунок из трактата немецкого алхимика Михаила Майера («Бегущая Аталанта», 1618 г.) изображает символически обе ступени процесса. На переднем плане серый волк (антимонит) пожирает царя металлов (золото). На заднем плане волк сгорает в пламени, а царь металлов, невредимый, направляется к лодке, которая должна доставить его во дворец на противоположном берегу.
19*
сухой лед
четание свойств обеспечивает длительную службу и малый коэффициент трения в подшипниках, залитых этим сплавом. Сплавы С. со свинцом идут для изготовления дроби и шрапнели. Из сплавов С. с цезием делают фотоэлементы. Соединения С. с металлами III группы периодической системы (в частности, с галлием и индием) — отличные полупроводники.
Некоторые соединения С. применяют в медицине (например, для лечения сонной болезни, распространяемой мухами це-це). В средние века соединения С. служили отхаркивающим и рвотным средством/При этом давалось вино, выдержанное в сосуде, изготовленном из С. Основным действующим началом здесь служил так называемый «рвотный камень» — смешанная виннокислая соль
292
калия и антимонила K[SbO(C4H2Oe)]. Это средство давно не применяют — оно даёт нежелательные побочные эффекты.
Сернистые соединения С. применяют как огнестойкие добавки к тканям (в театральных занавесях и драпировках, в брезентах) и в спичечном производстве. Та часть спичечной коробки, которую обычно называют «тёркой», содержит (наряду с красным фосфором) Sb2S3; вместе они и сообщают тёрке коричневый цвет. Пиротехника и производство каучука — вот ещё две области применения сурьмы. В. К. Бельский. СУХОЙ ЛЕД — твёрдая двуокись углерода СО2; белая снегообразная масса (см. Углерода двуокись).
«Мы начинаем понемногу понимать весь мир, и нам сейчас кажется, что вся вселенная со всеми своими кометами, звёздами, туманностями и планетами построена довольно однообразно. Одни и те же вещества составляют её основу, двенадцать — пятнадцать химических элементов как бы оказываются главными, а среди них первое место занимают металлы — железо, кремний, магний — и газы — водород, кислород и гелий. Наша Земля лишь кусочек этой вселенной и её законы — законы всего мироздания».
А. Е. Ферсман
(Ферсман А. Е., Занимательная минералогия, Свердловск, 1954, с. 65—66)
ТАЛЛИЙ (лат. Thallium) TI — химический элемент III группы периодической системы Менделеева; атомный номер 81, атомная масса 204,37. Серебристобелый металл. Элемент имеет два устойчивых изотопа, 208Т1 (29,50%) и 206Т1 (70,50%).
Т. был открыт в 1861 г. английским учёным У. Круксом. Целью исследования Крукса был теллур — учёный искал его спектроскопическим методом в отходах серно
кислотных заводов. Обнаруженная в спектре яркая зелёная линия не принадлежала ни одному из известных тогда элементов; так произошло открытие. Название Т. дано от греческого слова «таллос», что означает зелёная ветка. Интересно, что тем же методом и в том же материале почти одновременно с Круксом Т. обнаружил и французский учёный К* О. Лами. Он выделил чистый металл и подробно описал его свойства, но опоздал с сообщением, и приоритет открытия остался за Круксом.
Содержание Т. в земной коре, равное 1 • 10“Л% по массе, больше, чем, например, золота, серебра или сурьмы. Однако собственные минералы Т.— лорандит T1AsS2, круксит (Т1, Си, Ag)2Se, а также совсем недавно (1956 г.) найденный у нас в Узбекистане авиценнит Т12О3 (содержащий до 80% Т.) очень редки. Встречается Т. главным образом как незначительная примесь в различных горных породах. Много Т. (иногда до 0,5—1,0%) концентрируется в серном колчедане (пирите) FeS2, служащем сырьём для производства серной кислоты. Вот почему именно в остатках сернокислотного производства У. Крукс и открыл впервые этот элемент.
Т.— мягкий серебристо-белый металл с сероватым оттенком, по внешнему виду похожий на свинец. Т. легкоплавок, его /пл 303 °C, а /кип 1457 °C; плотность 11,85 г/см3. Т. и его соединения окрашивают пламя газовой горелки в изумрудно-зелёный цвет. Все соединения Т.— сильные яды.
Внешняя электронная оболочка атома Т. содержит три электрона (конфигурация 6s26p\
см. Атом). Т. в соединениях проявляет степени окисления +1 и +3 (валентности I и III).
Какая же из них более устойчива и почему? В периодической системе наблюдается следующая закономерность. В подгруппах p-элементов (то есть в главных подгруппах, где электроны заполняют р-уровни) с увеличением атомного номера доля участия s-электронов в образовании связей уменьшается, и для конечных членов этих подгрупп более устойчивы соединения, образующиеся только за счёт р-электронов, то есть соединения низшей степени окисления. Так, например, в главной подгруппе IV группы для германия и олова более устойчивы соединения Ge(+4) и Sn(+4), а для свинца, стоящего в конце группы,— РЬ (+2). Аналогично и в главной подгруппе III группы. У алюминия, галлия и индия более устойчивы соединения А1 (+3), Ga(+3) и 1п(+3), а для таллия — Т1 (+11-
Химия Т.— это в основном химия однозарядного положительного иона Т1+. Так как TI (+1) более устойчив, чем Т1 (+3), то соединения последнего являются окислителями. Многие соединения Т1 (+1) похожи на соединения щелочных металлов: гидроокись Т1ОН хорошо растворима в воде, а её раствор — сильное основание; хорошо растворимы в воде и многие соли Т. — сульфат, нитрат и карбонат; подобно щелочным металлам Т. образует квасцы, например TlCr(SO4)2- 12Н2О. Однако есть соединения Т1 (+1), похожие на соответствующие соединения элементов побочной подгруппы I группы — меди, серебра и золота: галогениды и сульфиды всех четырёх элементов малорастворимы в воде; галогениды TI (+1) так же, как и галогениды серебра, распадаются под действием света; предполагают, что их можно будет применять в фотографии.
Удивительное сочетание химических свойств является уникальной особенностью Т.
В сухом воздухе Т. покрывается с поверхности тонкой серой плёнкой окисла, а при накаливании сгорает с образованием смеси тёмно-коричневого Т12О и чёрного Т12О3, причём последнего окисла получается больше. Однако перевести целиком Т. в Т12О3 можно лишь действием сильных окислителей, например озона. С водой Т12О образует гидроокись Т1ОН, а Т12О3 в воде не растворяется. Окислы и гидроокиси Т. не проявляют амфотерных свойств и являются основаниями (в отличие от аналогии-
ТАНТАЛ
294
ных соединений В,А1 и других элементов подгруппы).
При нагревании Т. реагирует с серой: 2Т1 + S = T12S
и с галогенами:
2Т1 + С12 = 2Т1С1
В ряду напряжений Т. стоит перед водородом и вытесняет его из кислот с образованием соединений Т1 (+1):
2Т1 + 2НС1 = 2Т1С1 + Н2 2Т1+ H2SO4 = T12SO4 + H2
В щелочах Т. не растворим. В отсутствии окислителей вода не действует на Т.» а в присутствии кислорода окисляет его с поверхности:
4Т1 + О2 + 2Н2О = 4Т1ОН
Соединения Т1 (+3) получают окислением соединений Т1 (+1) сильными окислителями — хлором, царской водкой и другими:
Т1С1 + С12 = T1CL (хлорная вода)
Добыча Т.— сложная и кропотливая работа. Его соединения извлекают из сульфидных руд цветных металлов, где Т. содержится очень мало; затем осаждают галогениды или сульфид Т., из которых металл восстанавливают цинком:
+ 1 0 0- +2
2Т1 + Zn = 2Т1 + Zn
Металлический Т. хранят обычно под слоем парафинового масла для защиты от окисления.
Сам по себе металл в промышленности пока не применяется (хотя есть указания на то, что его можно использовать как катализатор). Однако Т. входит в состав важных сплавов, главным образом с оловом и свинцом. Такие сплавы обладают высокой кислотоупорностью. Так, сплав 70% РЬ, 20% Sn и 10% Т1 достаточно устойчив против такой «кислой» среды, как смесь концентрированных соляной, азотной и серной кислот. Сплав 72% РЬ, 15% Sb, 5% Sn и 8% Т1 по качеству превосходит лучшие подшипниковые сплавы на основе олова. Амальгаму Т. (содержащую 8,5% Т1) используют в термометрах в условиях Крайнего Севера, она затвердевает при температуре —60 °C.
Применяются и соединения Т. Это прежде всего материалы для оптических, люминесцентных й фотоэлектрических приборов. Фотоэлемент с оксисульфидом Т. отличается высокой чувствительностью к невидимым инфракрасным лучам, которые испускает всякий нагретый предмет; это используется в инфракрасной технике. Карбонат Т12СО3 применяют для изготовления сильно преломляющих свет стёкол.
Очень часто разные минералы встречаются в природе вместе. Есть такой способ их разделения: измельчённую смесь минералов помещают в жидкость с большой плотностью. И тогда более тяжёлый минерал тонет, а более лёгкий плавает на поверхности. В качестве таких жидкостей используют растворы солей Т. и органических кислот — муравьиной и малоновой.
Токсические свойства соединений Т. также не остаются без применения: сульфат T12SO4— сильный яд-зооцид, используется в борьбе с грызунами. В. К- Бельский.
ТАНТАЛ (лат. Tantalum) Та — химический элемент V группы периодической системы Менделеева; атомный номер 73, атомная масса 180,9479. Светло-серый металл с синеватым отливом. В природе встречаются два изотопа Т.— стабильный 181Та и слаборадиоактивный 180Та (период полураспада 1012 лет). Содержание стабильного изотопа резко преобладает (99,98% в природной смеси изотопов).
Т. был открыт в 1802 г. шведским химиком А. Г. Экебергом в минералах, найденных в Скандинавии. Твёрдый тугоплавкий окисел нового элемента, полученный Экебергом, ни при каких условиях не реагировал с кислотами. Должно быть, поистине «танталовы муки» испытал учёный в безуспешных попытках растворить окисел, если сам новый элемент он назвал «танталом»! Однако
название было удачным ещё и потому, что отражало неспособность окисла растворяться в кислотах. По преданию, Тантал, мифический фригийский царь, жестоко наказанный за оскорбление, нанесённое богам, не мог утолить жажду, стоя по горло в прозрачной воде. Сам металл (в нечистом виде) впервые был выделен шведским химиком Я. Берцелиусом в 1825 г.
Вскоре после открытия Т. выяснилось, что по своим свойствам он очень похож на «колумбий» (так сначала называли ниобий), открытый незадолго до этого. Стали считать, что Т. и «колумбий» — один и тот же элемент. Только в 1844 г. немецкий химик Генрих Розе показал, что они — два элемента-близнеца с чрезвычайно похожими химическими свойствами. Из-за трудности их разделения окись Т. в чистом виде была выделена лишь в 1866 г., а чистый металл — в 1903 г.
Т.— очень редкий элемент. В земной коре содержится всего 2,5-10“4% Т. по массе, то есть в 8 раз меньше, чем ниобия. О минералах Та и Nb рассказано в статье Ниобий. О главном из них — танталите-колумбите см. также ниже.
Т.— тяжёлый металл, его плотность 16,6 г/см3, то есть почти в полтора раза больше, чем свинца. Плавится Т. при 2996 °C, а кипит при 5300 °C. Чистый металл очень пластичен: его можно раскатать в тончайшие листки. Примеси газов (водорода, кислорода и др.), которые Т. поглощает при нагревании, делают металл хрупким.
295
ТЕЛЛУР
Атом Та имеет пять внешних (валентных) электронов (конфигурация 5d36s2, см. Атом). Максимальная и наиболее распространённая степень окисления Т. в соединениях равна +5 (валентность V). Ещё в середине XIX в. Г. Розе установил, что Т. и ниобий обладают почти одинаковыми химическими свойствами, только Т. ещё пассивнее своего аналога. Поэтому всё то, что было сказано о химии ниобия, относится и к Т. с поправкой на его ещё большую стойкость. При комнатной температуре Т. не реагирует ни с кислородом, ни с галогенами (кроме фтора). Высокая химическая стойкость металла обусловлена образованием на его поверхности тончайшей и очень прочной плёнки пятиокиси Та2О5. Окисление Т. кислородом воздуха становится заметным только при 280 °C (ниобия — около 200 °C). При высоких температурах Т. взаимодействует с хлором, бромом, серой, азотом, углеродом, водяным паром и др. В кислотах и их смесях, за исключением смеси фтористоводородной и азотной кислот, металл не растворяется. Практически не действуют на него и растворы щелочей.
Пятиокись Та2О5 имеет кислотный характер. Ей соответствует слабая танталовая кислота, формулу которой записывают как хТа2О5*г/Н2О. Среди солей танталовой кислоты — танталатов — простейшими являются мета-танталаты щелочных металлов МеТаО3, например КТаО3. В природе встречаются метатанталаты железа и марганца; их смеси с метаниобатами железа и марганца — это и есть минерал танталит-колумбит (Fe, Мп) [(Та, Nb)O3]2. О том, как читать такие формулы, рассказано в статье Минералы.
Основная трудность при получении металлического Т. заключается в очистке его от примеси элемента-спутника (ниобия). Разделение этих элементов подробно описано в статье о ниобии. Чтобы получить металл из промежуточного продукта — Та2О5, пятиокись восстанавливают при высоких температурах углеродом или металлическим натрием.
Одно из важнейших применений Т. основано на его исключительной коррозионной стойкости при умеренных температурах. Химическая аппаратура из Т. (теплообменники, нагреватели, трубопроводы, реакторы и т. п.) на заводах, производящих соляную, серную и другие кислоты, работает десятилетиями без заметного износа, в то время как такая же аппаратура из нержавеющей стали выходит из строя через несколько месяцев.
Сверхпрочная окисная плёнка на поверхности Т. придаёт ему ещё и красивый внешний вид — металл отливает радужными цветами. Поэтому им заменяют платину в корпусах часов, браслетах и других ювелирных изделиях. Очень малые размеры и значительную ёмкость имеют электрические конденсаторы, в которых
Т. служит анодом. По всем характеристикам танталовые конденсаторы превосходят алюминиевые. Т.— важнейший металл для «горячей арматуры» электронных ламп.
Наиболее обширную область применения Т. составляют его сплавы. Сплавы на основе Т. (например, 5—20% Сг, 2—25% W, остальное Та) отличаются большой механической прочностью и стойкостью против газовой коррозии при высоких температурах. Карбид ТаС, очень твёрдый и тугоплавкий (плавится около 3880 °C), входит в состав высокотвёрдых сплавов. Сверхжаропрочные сплавы на основе Т.— перспективные материалы для ракетной и космической техники: так, детали из сплава, содержащего 90% Та и 10% W, сохраняют высокую прочность до температуры 3300 °C.
Есть у Т. ещё одно замечательное свойство. Он «уживается» с живой тканью (не взаимодействует с жидкой средой организма). В восстановительной хирургии листы, фольгу, проволоку из Т. применяют для скрепления костей, нервов, для наложения швов и т. д.
ТЕЛЛУР (лат. Tellurium) Те — химический элемент VI группы периодической системы Менделеева; атомный номер 52, атомная масса 127,60. Серебристо-белое кристаллическое вещество с металлическим блеском. Природный Т. состоит из восьми изотопов с массовыми числами 120, 122, 123, 124, 125, 126, 128 и 130, из которых преобладают последние три —126Те, 128Те и 130Те (соответственно 18,7%, 31,8%, 34,5% в природной смеси изотопов).
В 1782 г. венгерский исследователь Ф. Мюллер (Рейхенштейн) в одной из золотых руд обнаружил примесь, в которой заподозрил присутствие неизвестного ранее элемента. Однако лишь через 16 лет немецкий химик М. Клапрот изучил свойства это
го элемента и назвал его по имени древнеримской богини Tellus, матери Земли. Клапрот дал названия и нескольким другим открытым в то время элементам (урану, титану, цирконию). Сам он писал по этому поводу: «Если для вновь открываемого ископаемого нежелательно подобрать название, указывающее на его своеобразные свойства, я нахожу, что лучше всего подбирать такие названия, которые ничего не говорили бы об этих свойствах и не давали бы поэтому повода для неправильных суждений».
Т. мало распространён в природе (1 • 10“ 7% по массе в земной коре — это всего лишь 75-е место среди других элементов). Однако минералы Т. встречаются чаще, чем минералы его аналога —- селена, хотя содержание последнего в земной коре больше. К числу минералов Т.
ТЕРБИИ
296
относятся, например, многочисленные теллуриды (соединения его с металлами) — калаверит АиТе2, алтаит РЬТе и др. Изредка Т. (вместе с серой и селеном) встречается в самородном состоянии; так, в Японии сера вулканического происхождения содержит 0,17% Т.
По внешнему виду Т. напоминает сурьму. Он имеет плотность 6,25 г/см3; /лл 449,8 °C, /кип 990 °C. Кристаллы его построены из длинных спиралевидных правильно расположенных цепей атомов. Т.— полупроводник.
По химическим свойствам Т.— неметалл. Конфигурация внешней электронной оболочки атома Т. аналогична сере и селену (5$25р4), то есть у Т. тоже шесть внешних (валентных) электронов. В соединениях Т. проявляет те же степени окисления, что и указанные его аналоги: —2, +4, +6 (валентность II, IV, VI). Т. устойчив на воздухе, но при высокой температуре горит с образованием двуокиси ТеО2. С галогенами Т. взаимодействует на холоду. При нагревании реагирует с многими металлами, давая теллуриды, например А12Те3. При действии азотной кислоты Т. превращается в теллуристую, а при действии царской водки или 30%-ной перекиси водорода — в теллуровую кислоту.
Говоря о соединениях Т., нельзя не сравнивать их с аналогичными соединениями серы и селена. Теллуристый ангидрид ТеО2, в отличие от сернистого ангидрида SO2, проявляет некоторую амфотерность (растворяется в сильных кислотах):
ТеО2 + 4НС1 = ТеС14 + 2Н2О
Однако кислотные свойства его более выражены, чем основные. Теллуристая кислота Н2ТеО3 много слабее сернистой кислоты и, подобно H2SO3, неизвестна в свободном состоянии. Соли ее — теллуриты — можно получить по реакции:
ТеО2 + 2NaOH = Na2TeO3 + Н2О
Теллуровая кислота резко отличается от серной и селеновой кислот. Благодаря большому атомному радиусу Т. может координировать вокруг себя не четыре атома кислорода (как сера и селен), а шесть, давая таким образом ортотеллуровую кислоту НвТеОв. Это очень слабая кислота (по силе подобная угольной). Известны средние и кислые ее соли — теллура ты, например AgeTeOe, Na2H4TeOe. Обезвоживанием НвТеОб при 300 °C можно получить теллуровый ангидрид — высший окисел ТеО3.
При разложении теллуридов водой или кислотами образуется теллуристый водород Н2Те — газ ещё более ядовитый чем сернистый водород. Уже при обычной температуре
Н2Те распадается на элементы. Водный раствор его — теллуроводородная кислота имеет более выраженные кислотные свойства по сравнению с сероводородной кислотой; на воздухе в растворе Н2Те очень быстро окисляется и в осадок выпадает свободный Т.
Т., как и селен, добывают главным образом из отходов от очистки меди электролизом. Мировое производство его — десятки тонн в год, то есть много меньше, чем селена. Однако и Т. оказался необходимым и перспективным в новейших областях техники. Его соединения с металлами — теллуриды обладают как полупроводниковыми свойствами, так и высокой чувствительностью к различным излучениям (инфракрасному, рентгеновскому и др.). Благодаря этому теллуриды* можно использовать во многих приборах и конструкциях, например, в телевизионных трубках, дозиметрах и т. д. В металлургии Т. добавляют, главным образом к свинцу, для улучшения его механических свойств и химической стойкости. Используют Т. для придания стеклу коричневой окраски и большего светопреломления.
Интересна и ещё одна область, где проявляются свойства Т.— на этот раз ядерные. При делении урана в атомных реакторах образуется радиоактивный изотоп 13бТе — один из многочисленных «осколков деления». После двух последовательных 0- -распадов 13бТе превращается в изотоп ксенона 13бХе:
135 R“ 135 R- 135
Те —► I Хе.
52 53 54
Изотоп 13бХе жадно поглощает нейтроны и тем ухудшает условия работы реактора: нейтроны попадают не в ту цель, для которой они предназначены,— в ядра урана, а в ядра «захватчика». Естественно, что в конструкциях атомных реакторов эта возможность «отравления реактора» учитывается. в, Л, Василевский,
ТЁРБИЙ (лат. Terbium) Tb — химический элемент III группы периодической системы Менделеева из семейства лантаноидов; атомный номер 65.
ТЕХНЁЦИЙ (лат. Technetium) Тс — радиоактивный химический элемент VII группы периодической системы Менделеева; атомный номер 43. Серебристо-коричневый металл. Т. был первым химическим элементом, созданным руками человека (отсюда и его название: «тех-нетбс», по-гречески означающее искусственный). Известно более 20 радиоактивных изотопов Т., но ни один из них практически не встречается в природе.
Открыть Т. удалось лишь в 1937 г., когда учёные, вооружённые знаниями о строении атомного ядра, начали создавать новые ядра в своих лабораториях. Итальянские физики К- Перрье и Э. Сегре пришли к выводу, что для получения элемента № 43 можно использовать элемент № 42 — молибден. Ядро Мо содержит на 1 протон меньше, чем ядро Тс. Поэтому задача синтеза элемента № 43 может быть сведена к «добавке» протона в ядро Мо.
ТИОСУЛЬФАТЫ
98,9062 43
Тс
4d55s2
ТЕХНЕЦИЙ
С этой целью молибденовые пластинки облучали сильно разогнанными ядрами дейтерия (тяжёлого водорода fH). Ядро дейтерия, называемое дейтроном, состоит из нейтрона и протона. Поэтому после попадания дейтрона в ядро Мо образуется ядро с зарядом большим на единицу, то есть ядро Тс.
В настоящее время Т. в виде изотопа "Тс в сравнительно больших количествах накапливается в отходах (в «шлаке») ядерных реакторов, так что его можно выделять в килограммовых количествах. При радиоактивном распаде ядра "Тс испускают Р“-частицы (период полураспада
=2,12-106 лет) и превращаются в ядра рутения "Ru. Атомная масса изотопа "Тс равна 98,9062.
Температуры плавления и кипения металла очень высоки и составляют соответственно 2140 и 4700 °C. Плотность Т. 11,5 г/см3. При охлаждении ниже—265,5 °C (7,7 °К) Т.переходит в сверхпроводящее состояние, то есть его электрическое сопротивление обращается в нуль (исчезает). (О сверхпрово д и-мости см. в статьях Гелий. Ниобий и др.)
В образовании химических связей Т. могут принимать участие 7 электронов, расположенные на его 4d- и 55-уровнях (конфигурация 4d65s2, см. А том). По химическим свойствам Т. ближе всего к рению, хотя он имеет также много общего и с марганцем. В соединениях Т. проявляет степени окисления от +2 до +7 (валентности от II до VII).
При горении Т. в кислороде образуется светло-жёлтое твёрдое вещество — окисел Тс2О7. Растворение Тс2О7 в воде даёт раствор, выпариванием которого можно получить тёмно-красные кристаллы технециевой кислоты НТсО4. Известно большое число солей этой кислоты — пертехнатов, например КТсО4, CsTcO4, AgTcO4. При нагревании розовых кристаллов пертехната аммония образуется второй окисел Т.— ТсО2, чёрное малолетучее вещество.
Стойкость Т. против коррозии исключительно высока, что позволяет использовать этот металл как конструкционный материал в реак-торостроении. Что же касается пертехнатов, то мы пока что не знаем веществ, которые могли бы конкурировать с ними по антикоррозионным свойствам. Пертехнаты предохраняют стальные изделия от разъедания. Так, если стальной предмет опустить в раствор, 1 л которого содержит всего около 0,01 г КТсО4, то даже через несколько лет на этом предмете не появится и следов окисления. Важно, что антикоррозионные свойства иона ТсО^ проявляются не только при комнатной температуре, но и сохраняются при нагревании до 250 °C. Пока, конечно, за
щищать пертехнатами обычные металлические конструкции слишком дорого. Но ими можно предохранять от коррозии важнейшие узлы ядерных реакторов, точных приборов и т. д. Механизм антикоррозионного действия иона ТсО^ до сих пор окончательно не выяснен и продолжает изучаться. . С. С. Бердоносов. ТИОСЁРНАЯ КИСЛОТА (серноватистая кислота) H2S2O3 — сильная, очень непрочная двухосновная кислота. Она представляет собой маслообразную жидкость, уже при—78 °C начинающую разлагаться на H2S и тритионовую кислоту H2S3Oe. При комнатной температуре Т. к. разлагается так:
H2S2O3 = Н2О 4~ SO2 S
Т. к. можно рассматривать как серную кислоту H2SO4, в молекуле которой один из атомов О замещён атомом S. Отсюда и название кислоты: приставка «тио-» происходит от греч. названия серы «тёйон» (см. Сера). Два атома серы в молекуле имеют различные степени окисления, -» в-2
4-6 и —2 (H2SSO3). Благодаря присутствию —2
S .сама кислота и её соли (тиосульфаты) — сильные восстановители.
ТИОСУЛЬФАТЫ (серноватистокислые соли) — соли тиосерной кислоты H2S2O3. Т. щелочных и щелочноземельных металлов, кроме BaS2O3, хорошо растворимы в воде. Т. других металлов в воде не растворимы (кроме ZnS2O3 и CdS2O3). С тиосульфатом натрия Na2S2O3 они дают растворимые комплексные соли, например Na [Ag(S2O3)].
Известны лишь средние Т., из которых наибольшее значение имеет кристаллогидрат тиосульфата натрия Na2S2O3-5H2O (называемый в обиходе гипосульфитом). Его получают кипячением раствора сульфита натрия с серой: Na2SO3 4- S = Na2S2O3
Как и тиосерная кислота, Т.— сильные восстановители; например, Na2S2O3 легко окисляется хлором до серной кислоты:
Na2S2O34-4Cl24-5H2O=
= 2H2SO44-2NaC14-6HCl
На этой реакции было основано применение тиосульфата натрия для поглощения хлора (в первых противогазах). Несколько иначе происходит окисление Т. слабыми окислителями; при этом образуются соли тетратионовой кислоты, например:
2Na2S2O3 4- I2 = Na2S4Oe 4- 2NaI
Водный раствор тиосульфата натрия применяют в фотографии как закрепитель-фиксаж (см. об этом в статье Серебро).
ТИТАН
298
ТИТАН (лат. Titanium) Ti — химический элемент IV группы периодической системы Менделеева; атомный номер 22, атомная масса 47,90. Серебристо-белый сравнительно лёгкий металл, по внешнему виду похожий на сталь. В природе Т. представлен смесью пяти стабильных изотопов с массовыми числами 46, 47, 48, 49 и 50. Наиболее распространён из них 48Ti (73,99%).
По распространённости в земной коре Т.
занимает среди элементов 9-е место (4,5-10“1% по массе). Когда в одном из минералогических музеев произвели анализ 1000 минералов,то оказалось, что 800 из них содержат Т. Правда, в большинстве минералов Т. находится в очень небольших количествах. Но имеются и весьма богатые им руды и минералы — таковы рутил TiO2, ильменит FeTiO3 и др. По запасам титановых руд Советский Союз стоит на одном из первых мест в мире. Не случайно ильменит получил своё название от Ильменских гор, находящихся на Урале.
Необычно долог путь от открытия?, до его промышленного получения и использования. Почти одновременно и независимо друг от друга английский учёный У. Грегор (1789 г.) и немецкий М. Клапрот (1795—97 гг.) открыли окисел нового металла TiO2; первый — в «чёрном песке» (менаканите, то есть ильмените), второй — в минерале рутиле. Клапрот назвал новый элемент титаном (в греческой мифологии исполинские Титаны — дети богини Земли Геи и бога Неба Урана). В 1825 г. шведский химик Я. Берцелиус впервые выделил сам металл, однако в нечистом виде. В течение последующих 100 лет химики многих стран безуспешно пытались получить Т. в чистом виде, восстанавливая его из соединений при нагревании. Из-за высокой химической активности Т. при повышенных температурах он получался сильно загрязнённым (см. ниже). Небольшое количество Т. высокой чистоты удалось выделить лишь в 1925 г. голландским учёным А. ван Аркелю и И. де Буру методом так называемого иодидного рафинирования (см. ниже).
Лишь после того как удалось получить очень чистый металл, выяснилось, что в нём сочетаются ценнейшие свойства. Он не только лёгок и тугоплавок (плотность 4,5 г/см3, /пл 1665 °C, /кип 3227 °C), но и весьма прочен и пластичен. Будучи всего в 1,5 раза тяжелее одного из легчайших металлов — алюминия, Т. имеет почти в три раза более высокую точку плавления и в 6 раз большую прочность. А кроме того, Т. отличается исключительной химической стойкостью при обычной температуре. Во влажном воздухе, в морской воде, азотной кислоте он противостоит коррозии не хуже нержавеющей стали.
Атом Ti имеет четыре внешних (валентных) электрона (конфигурация 3d24s2, объяснение см. в статье Атом). В большинстве своих соединений Т. проявляет степень окисления +4 (валентность IV). Соединения низших степеней окисления (+3 и особенно +2) редки и значительно менее стабильны. На воздухе Т. покрывается очень прочной плёнкой двуокиси:
Ti + О2 = ТЮ2
Эта плёнка защищает металл от дальнейшего окисления. Поэтому Т. устойчив на воздухе даже при нагревании до 550 °C. При температуре красного каления Т. горит в токе кислорода с образованием TiO2. С галогенами Т. реагирует при повышенных температурах (с фтором при 150 °C, хлором при 300 °C, бромом при 360 °C, иодом при 550 °C), образуя тетрагалогениды:
Ti + 2С12 = TiCl4
Сравнительно легко растворяется Т. в соляной кислоте, особенно при нагревании, с образованием фиолетового трихлорида:
2Ti + 6HCl = 2TiCl3-r3H2
Наилучший растворитель Т. и его труднорастворимых соединений — плавиковая кислота HF.
Двуокись TiO2 амфотерна, причём и основные, и кислотные её свойства выражены очень слабо: она совсем не растворима в воде и разбавленных кислотах и очень незначительно — в растворах щелочей; в раствор может быть переведена продолжительным кипячением с H2SO4. Соли, отвечающие кислотной функции TiO2 — титанаты, получают сплавлением TiO2 с окислами, гидроокисями или карбонатами других металлов:
TiO2 + К2СО3 = K2TiO3 + СО2
Тетрагалогениды Т. легко летучи. Их пары сильно дымят на воздухе из-за взаимодействия с парами воды (гидролиз):
TiCl4 + 2Н2О = TiO2 + 4НС1
При этом образуются очень мелкие частички TiO2, которые взвешены в воздухе в виде тумана. Не только галогениды, но и другие растворимые соединения Т. гидролизуются в растворах. Если гидролиз идёт не до конца, а частично, то образуются соединения, содержащие радикал титанил [TiO]2 + , например оксихлорид титана TiOCl2, титанилсульфат TiOSO4.
Уже в 20-е годы стало ясно, что Т., сочетающий лёгкость, высокие механические и антикоррозионные свойства, может быть полезен в авиации. Но пути его производства ещё небыли разработаны, и Т. оставался дорогим лабораторным металлом. Элементу пророчили печальную судьбу мифологических собратьев (Титаны вели борьбу с Зевсом за обладание небом, но были побеждены и низвергнуты в мрачные подземелья Тартара). Одна из причин такого скептицизма крылась в том, что Т. при высоких тем
299
ТИТАН
пературах жадно поглощает из атмосферы азот и кислород и становится хрупким, малопригодным для практических целей. Поэтому все высокотемпературные процессы в технологии Т. надо вести либо в вакууме, либо в атмосфере инертного газа (аргона), а это требует высокоразвитой техники.
В середине 40-х гг. был разработан химический способ производства Т. Обогащённую титановую руду спекают с коксом при одновременном действии хлора:
ТЮ2 -г 2С + 2С12 = TiCl4 + 2СО
Образующийся газообразный TiCl4 пропускают в реакционные камеры, заполненные расплавленным магнием в защитной атмосфере (аргон). Реакция происходит около 850 °C:
TiCl 4 + 2Mg = Ti + 2MgCl2
Хлорид магния и избыток металлического магния отгоняют нагреванием в вакууме. Т. губчатой массой наполняет реакционные камеры (его так и называют — «губка титана»). Губку переплавляют в атмосфере аргона в компактные блоки или направляют на производство сплавов.
Для приготовления небольших количеств особо чистого Т., идущего в основном для исследовательских целей, применяют иодидное рафинирование. В этом методе используется обратимая реакция:
Ti + 2I2 Til4
Подбирая условия, можно заставить её протекать преимущественно в прямом или обратном направлениях. В реакционную камеру загружают порошок Т. в смеси с иодом, после чего из камеры откачивают воздух и нагревают её до 600 °C. В этих условиях образуется летучий Til4 (прямая реакция). В камере протянута металлическая проволока (чаще всего вольфрамовая), которая нагревается электрическим током до очень высокой температуры, около 1800 °C. Молекулы Til4, попадающие на раскалённую проволоку, распадаются (обратная реакция), и совершенно чистый Т. осаждается на проволоке. Освободившийся иод реагирует с новой порцией неочищенного Т. и т. д. Этот метод применяется также для получения других элементов — Zr, Hf, Th, Si, В.
T.— один из важнейших материалов новой техники, и его значение продолжает расти. В 1947 г. было получено только 2 т технического Т., в 1954 г.— 5000 т, а в 1960 г. мировое производство Т. уже достигло массы порядка 100 000 т. Таких темпов роста не знал даже алюминий.
Наиболее широко сам металл и его сплавы используются в самолёто- и ракетостроении. Сочетание высокой прочности и лёгкости с химической стойкостью делает их незаменимыми для современной лётной техники. Недаром монумент в честь освоения космоса советским народом, установленный на Проспекте Мира в Москве, облицован титановыми листами. Из титановых сплавов изготовляют корпуса ^орских судов, подводных лодок. В химической
Монумент в честь освоения космоса советским народом. Скульптор А. П. Файдыш, архитекторы М. О. Барщ и А. Н. Колчин. Установлен в 1964 г. на Проспекте Мира в Москве. Шлейф и ракета монумента облицованы титаном. Как писала газета «Известия» 18 августа 1964 года, «Звёздный корабль из вечцого металла — титана навсегда поднялся в небо столицы».
Под монументом — в стилобате — Мемориальный музей космонавтики.
промышленности титановые трубопроводы, насосы, реакторы, вентиляторы работают в самых агрессивных средах, значительно превосходя по стойкости другие металлические материалы. Способностью Т. поглощать газы пользуются в вакуумной технике.
Самая лучшая белая краска — титановые белила— производится из TiO2. Хорошо образованные прозрачные кристаллы рутила имеют показатель преломления больше, чем у алмаза, и могут служить ювелирными украшениями. TiCl4 используют как дымообразующее вещество.
Таким образом, история Т.— это яркий пример развития химии и химической промышлен-
ТОРИИ
300
ности; «грязный» и поначалу никому не нужный материал превратился в руках химиков и металлургов в важнейший металл XX века.
Ф. Л4. Спиридонов. ТОРИЙ (лат. Thorium) Th — радиоактивный химический элемент; атомный номер 90, атомная масса 232,0381; относится к актиноидам. Серебристо-белый металл. Природный Т. почти целиком состоит из долгоживущего изотопа 32Th (период полураспада 7\/ж = 1,41 • 1010 лет).
Другие изотопы Т. распадаются значительно быстрее. При радиоактивном распаде 232Th испускает ядра гелия (а-частицы) и даёт начало целой цепочке радиоактивных превращений — так называемому радиоактивному ряду Т. (Подробнее о радио
активных рядах рассказано в статье Уран.) Честь открытия Т. принадлежит шведскому химику Я. Берцелиусу, который в 1828 г. исследовал состав минерала с норвежского острова Лёвен. Из этого минерала учёному удалось выделить новую «землю» — белый порошок окисла неизвестного ранее элемента. «Земля» была названа ториевой по имени древнескандинавского бога грома Тора, а содержащий её минерал получил название торита (он представляет собой силикат состава ThSiO4).
Получение металлического Т. связано с большими трудностями, обусловленными повышенной реакционной способностью нагретого Т. и высокой температурой плавления металла. Первые опыты в этом направлении проделал ещё Берцелиус, но получить чистый металл ему не удалось. Образец чистого металлического Т. был впервые приготовлен только спустя почти столетие, в 1914 г., путём разложения иодида ТЫ4 на раскалённой вольфрамовой проволоке (об этом методе см. в статье Титан). Прошло ещё 25 лет, пока в 1939 г. удалось наладить промышленное производство Т.
Распространённость Т. в природе выше, чем другого радиоактивного элемента — урана. На долю Т. приходится около 1,3-10"3% массы земной коры, то есть почти столько же, сколько такого обычного металла, как свинец. Однако Т. принадлежит к числу рассеянных элементов, и это затрудняет его добычу. Т. обнаружен более чем в 120 минералах, но в подавляющем их большинстве находится как примесь, причём его концентрация составляет не более 0,1%. Только в 6 минералах Т. является основным компонентом. К их числу принадлежит уже упоминавшийся торит, а также торианит — смешанный окисел Т. и урана (Th,U)O2.
Главным Же источником Т. служит монацит— минерал, содержащий фосфаты Т. и редкоземельных элементов, а также окислы железа, кремния и другие примеси. В монаците всегда присутствует гелий (образующийся при
радиоактивном распаде Т.), который выделяется при нагревании минерала. В первую мировую войну немцы, использовавшие гелий для наполнения военных дирижаблей, перевезли из Бразилии в Германию огромное количество монацита; после выделения гелия монацит выбрасывали. Проблема овладения атомной энергией заставила по-новому взглянуть на монацит, и сейчас его ценность определяется содержанием в нём Т. (а также ряда редкоземельных элементов, используемых в промышленности).
В компактном виде Т.— светло-серый металл, тускнеющий на воздухе из-за образования плёнки окисла. Порошкообразный Т. способен самовозгораться на воздухе, поэтому его хранят под керосином. Плотность Т. 11,72 г/см3. Температура плавления чистого металла равна 1750 °C. Температуру кипения до сих пор точно определить не удалось. По разным данным, она составляет 3500—4200 °C.
В настоящее время Т. принято относить к актиноидам. Однако можно сказать, что Т.— «белая ворона» в этом семействе элементов. На 5-й электронной оболочке атома Т., в отличие от атомов других актиноидных элементов, нет так называемого /-электрона (см. статью Атом)\ его внешняя электронная оболочка (6d27s2) похожа на электронные оболочки гафния (5d26s2) и циркония (4d25s2). Поэтому химия Т. имеет много общего с химией элементов IV группы периодической системы — циркония и гафния.
Наиболее типична степень окисления Т. +4 (валентность IV). Соединения четырёхвалентного Т. образуются, в частности, при взаимодействии нагретого металла с кислородом, азотом, хлором, серой и другими элементами. Стружки Т. горят ца воздухе, давая окисел ThO2. Это одно из самых труднолетучих и тугоплавких веществ, поэтому из него изготовляют особо огнеупорные тигли. ThO2, в отличие от ZrO2 и HfO2, обладает сильно основным характером и не реагирует с щёлочами. Пропуская над нагретым до красного каления ThO2 смесь СО и С12, можно получитьТЬС14:
ThO2 + 2СО + 2С12 = ThCl4 + 2СО2
Из водных растворов ThCl4 растворы аммиака или щёлочи осаждают белый желатинообразный осадок гидроокиси Т., состав которой наиболее правильно выражает формула хТЬО2-уН2О. Свежеосаждённая гидроокись Т. легко растворяется в кислотах, давая соответствующие соли.
Особый интерес к Т. связан с перспективами его использования в ядерной энергетике. Тяжёлых ядер, которые способны к цепной реакции деления, приводящей к. выделению огромного
301 ТУЛИИ
количества энергии, не так уж много. Это прежде всего 236U и 239Ри, которые в настоящее время являются основным «горючим» ядерных реакторов и бомб. Однако известно, что цепную реакцию деления можно осуществить и на ядрах 233U, но изотопа 233U в природе нет. Его можно «делать» из Т. Как уже говорилось, природный Т. практически состоит из единственного изотопа 232Th. Если облучать Т. в урановом или плутониевом реакторе нейтронами, то каждое ядро 232Th, поглотившее нейтрон, в конце концов (после цепочки ядерных превращений) даст ядро 233U. Из полученной после облучения смеси можно сравнительно просто отделить 233U. Облучаемый Т. (или его соединения) должен быть очень чистым, так как среди примесей могут оказаться хорошие поглотители нейтронов, что резко снизит выход 233U.
Этот способ получения 233U довольно дорог. Реакторов, использующих 233U, очень мало. По этому пути вынуждены идти страны, желающие развивать ядерную энергетику, но располагающие не ураном, а Т.
Возможно, что Т. как источник ядерного горючего станет очень важным для человечества, когда начнут иссякать запасы урана (как уже говорилось, Т. на Земле в несколько раз больше, чем урана).
Некоторое применение Т. находит в металлургии. Сплавы, содержащие Т., а также цирконий, цинк, марганец и ряд других элементов, характеризуются высокой прочностью и большой химической стойкостью при повышенных температурах. Поэтому эти сплавы применяют для изготовления деталей реактивных двигателей и ракет. Добавки Т. улучшают свойства многих катализаторов, используемых в неорганической и органической химии (например, при окислении SO2 в SO3, при гидрировании углеводородов и т. д.). С. С. Бердоносов. ТРАНСУРАНОВЫЕ элементы — химические элементы, расположенные в периодической системе Менделеева после урана и имеющие атомные номера Z=93 и выше. Изотопы всех известных Т. э. радиоактивны; стабильных
изотопов Т. э. не имеют. В природе найдены ничтожные количества только двух Т. э. — нептуния(2=93) и плутония (2=94). Все Т. э. могут быть получены искусственным путём, с помощью различных ядерных реакций.
Первый Т. э.— нептуний был синтезирован в 1940 г., а к 1975 г. известны Т. э. с Z вплоть до 106. Из них элементы с Z от 93 до 103 относятся к актиноидам; элемент с Z=104—курчатовий является аналогом циркония и гафния, а элемент с Z= 105 (его название «нильсборий» пока не общепринято), вероятно, представляет собой аналог тантала. (О синтезе 106-го элемента сообщили в июле 1974 г. советские физики.) Периоды полураспада Ti/t самых устойчивых изотопов отдельных Т. э. быстро уменьшаются с ростом Z, падая до долей секунды. Поэтому открытие новых Т. э. требует разработки экспрессных методов, позволяющих за доли секунды провести разделение элементов и изучить их основные свойства. Пример такого метода описан в статье Курчатовий.
Физики по ряду причин считают, что в области далёких трансуранов (элементы cZ>108 — 110) возможно появление «островов повышенной устойчивости», то есть групп стабильных или долгоживущих изотопов. По мнению некоторых учёных, величина 7\/г для Т. э. с Z=114 (предположительно — аналога свинца) может оказаться столь большой, что можно ожидать присутствия этого элемента на Земле. Однако пока его поиски успеха не принесли. Если в природе этот Т. э. так и не удастся найти, можно надеяться, что его рано или поздно создадут в лабораториях. В таком случае у химиков будет возможность изучить химические свойства этого таинственного элемента, а у физиков — углубить представления об устойчивости атомных ядер и, следовательно, об их строении.
С. С. Бердоносов.
ТУЛИЙ (лат. Thulium) Tm — химический элемент III группы периодической системы Менделеева из семейства лантаноидов; атомный номер 69.
УГАРНЫЙ ГАЗ — то же, что углерода окись СО.
УГЛЕКЙСЛЫЕ СОЛИ—то же, что карбонаты.
УГЛЕКИСЛЫЙ ГАЗ — то же, что углерода двуокись СО 2.
УГЛЕРОД (лат. Carboneum) С — химический элемент IV группы периодической системы Менделеева; атомный номер 6, атомная масса 12,011. Обычными формами существования элемента У. в свободном состоянии являются два простых тела: алмаз и графит, различающиеся по кристаллическому строению и свойствам (см. ниже). Без преувеличения можно назвать У. одним из самых важных элементов периодической системы:
ведь его органические соединения служат основным строительным материалом для создания всего живого на Земле.
Углерод в природе. На долю У. приходится около 2,3*10-2% от массы земной коры, где по содержанию он занимает лишь 17-е место среди других элементов. Главные углеродсодержащие минералы — природные карбонаты; количество У. в них оценивается в 9,6-1016 т. Много У. (около 1013 т) сосредоточено в горючих ископаемых (каменных и бурых углях, нефти, торфе, горючих сланцах, природных горючих газах), образующих мощные скопления в недрах Земли. Они являются в основном продуктами разложения растений, существовавших на нашей планете в давно минувшие времена.
Свободный У. встречается в природе в виде алмаза и графита. Близки по составу к чистому У. некоторые каменные угли, антрациты, содержащие иногда до 98% С. Алмазы чрезвычайно редки. Даже алмазоносные породы (кимберлиты) содержат не более 0,00009% алмазов. Обычная масса кристаллов алмаза 0,2—
0,4 г. Крупные алмазы очень дороги; и если такой алмаз находят, ему присваивают особое название. Самый большой из всех известных до сих пор — «Куллинан». Его нашли в Южной Африке. Он весил 621,2 г при размерах 10 X X 6,5 х 5 см. В Алмазном фонде СССР хранится один из самых больших и красивых алмазов в * мире — «Орлов» (37,92 г). Наиболее значительные месторождения алмазов находятся в СССР (Якутия, Урал), Африке, Бразилии.
Графит образует скопление плотных чешуйчатых масс, загрязнённых минеральными примесями. В СССР залежи графита находятся на Украине, в Амурской области, Крыму и других местах.
Все живые организмы, составляющие биосферу, построены из соединений У. и в среднем содержат 18% его по массе. Общее же количество У. в них составляет 1012 т. Содержание У. в разных организмах может быть весьма различным.
В виде углекислого газа СО2 (см. Углерода двуокись) У. входит в состав атмосферы Земли. Содержание СО2 в атмосфере равно 0,03% по объёму, что при пересчёте на общую массу У. даёт громадное число — 6-1011 т! В водной оболочке, покрывающей большую часть земной коры,—гидросфере—растворено в 60 раз больше СО2, чем его находится в атмосфере.
Углекислый газ служит основным источником У. для растений. Необходимые органические вещества (углеводы, белки, жиры) растения синтезируют из углекислого газа, воды и минеральных солей с помощью энергии солнечного света. Этот процесс называется фотосинтезом (он описан в статьях Биосфера и Кислород). Животные же получают У., употребляя в пищу растения.
Ежегодно растительный мир суши и океана забирает для своих нужд из воздуха и воды около 1010 т У. Однако атмосфера заметно не обедняется У. благодаря тому, что на Земле происходит беспрерывное выделение углекислого газа при дыхании животных и человека, при гниении органических остатков, брожении, извержении вулканов. Большое количество У.
303
УГЛЕРОД
возвращается в атмосферу вследствие деятельности человека — работы машин, фабрик, заводов. В результате в природе идёт постоянный круговорот углерода (рис. 3).
У. распространён не только на Земле, но и на других космических телах. Простейшие соединения У. (углекислый газ, метан) обнаружены в атмосфере почти всех планет солнечной системы, причём общая масса У. на них больше массы Земли. Так, по данным, полученным с помощью автоматических межпланетных станций, атмосфера Марса состоит в основном из СО2. В атмосфере Солнца У.— 11-й в списке элементов. Спектроскопически присутствие У. и его соединений установлено на далёких звёздах, в кометах и туманностях. Органические вещества, графит и даже алмазы содержатся в некоторых метеоритах.
Одним словом, У. во Вселенной достаточно, чтобы создать «углеродную жизнь» не только в родном нам мире!
Историческая справка. У.— один из немногих элементов, имя первооткрывателя которого неизвестно. Алмаз и графит, не говоря уже об угле, знакомы человечеству с древнейших времён. Замечательные свойства алмаза — его необыкновенно высокая твёрдость и красота — издавна служили богатой пищей для фантастических сказок и суеверных измышлений. Своё название алмаз получил от арабского слова «ал-мас» (твердейший) или от греческого «адамас» (непобедимый, несокрушимый).
Долгое время оставалось непонятным, «из чего же состоит алмаз». Первый шаг к выяснению природы алмаза был сделан в 1694 г., когда флорентийские учёные, пытаясь сплавить несколько мелких алмазов в один крупный, нагрели их солнечными лучами при помощи зажигательного стекла. К своему удивлению, флорентийцы обнаружили, что алмазы исчезли, сгорев на воздухе. Затем знаменитый французский химик А. Лавуазье в 1772 г. показал, что при сгорании алмаза образуется углекислый газ. Окончательно состав алмаза установил в 1797 г. английский химик С. Теннант. Он нашёл, что объёмы
Рис. 1. Геологи отыскали в вечной мерзлоте чудо-камни — алмазы (Якутская АССР, алмазные месторождения открыты здесь в 1954—55 гг.).
Рис. 2. Зёрна алмаза вкраплены в горную породу кимберлит. Такие куски кимберлита найдены на алмазном месторождении в Якутии.
углекислого газа, образующегося при сгорании одинаковых количеств алмаза и угля, равны. Это бесспорно доказало химическое тождество обоих веществ.
Графит был известен в древности, но вплоть до 1780-х гг. его считали разновидностью других, близких по внешнему виду минералов. Во второй половине XVI в. из графита начали делать стержни для карандашей (отсюда происходит и название минерала, от греческого «графо» — пишу).
В 1778 г. шведский химик К- Шееле нашёл, что графит при сильном нагревании с расплавленной селитрой сгорает, образуя углекислый газ. Так было показано тождество химической природы столь не похожих друг на друга веществ — угля, графита и алмаза.
Атом углерода. У. имеет два устойчивых изотопа: 12С и 13С, среднее содержание которых составляет 98,892% и 1,108% от общего числа атомов У. на Земле. Таким образом, в природной смеси изотопов У. резко преобладает более лёгкий 12С, ядро атома которого состоит из 6 протонов и 6 нейтронов. Очень важен радиоактивный изотоп У. 14С, испускающий р~-части-цы и имеющий период полураспада Т>/,= = 5 570 лет. Открытие этого изотопа имело чрезвычайно важные последствия для развития археологии, геологии, палеонтологии и истории! Недаром говорят, что все науки связаны между собой и зависят друг от друга. С помощью 14С учёные смогли довольно точно определять возраст углеродсодержащих пород, стоянок древнего человека, его утвари, остатков животных и растений, давно исчезнувших с лица Земли.
Изотоп 14С постоянно образуется в верхних слоях атмосферы из изотопа азота 14N под действием нейтронов космического излучения. Его очень мало — на каждый миллион молекул атмосферного СО2 приходится лишь один атом 14С. Но так как интенсивность космического излучения остаётся всё время постоянной, то и концентрация 14С в воздухе не меняется (убыль ядер 14С за счёт
УГЛЕРОД
304
Рис. 3. Круговорот углерода в природе. Решающую роль в судьбе углерода на нашей планете играют живые организмы. Они — и могучие двигатели круговорота углерода, и составное звено этого круговорота. Растения моря (1) и суши (2) в результате фотосинтеза связывают двуокись углерода СО2, находящуюся в атмосфере и растворённую в водах океана. В кружке (3) как бы под мощным увеличительным стеклом показаны микроскопические зелёные водоросли — фитопланктон. Они поглощают примерно столько же СО2, сколько вся растительность суши (не правда ли, в это трудно поверить?). Животные, питаясь растениями, тоже включаются в круговорот углерода. В кружке (3) нарисованы мелкие морские животные — зоопланктон, пищей которых служат водоросли. Планктон поддерживает существование более крупных обитателей океана. Некоторые морские животные строят свой скелет из карбоната кальция. После их отмирания образуются целые острова и архипелаги, состоящие из СаСО3. Так живые колонии кораллов постепенно превращаются в коралловые острова (4).
Огромные залежи известняков на суше когда-то, миллионы лет назад, тоже образовались из скелетов морских животных (геологи даже целую эпоху в истории Земли называют меловым периодом). Этот процесс (5) надолго выводит углерод из оборота, и запасы такого «малоподвижного» углерода в миллионы раз превышают количество углерода, находящегося в атмосфере и растворённого в водах Земли (в форме СО2). Отмирая, животные организмы дают начало залежам угля (6), нефти (7), гумуса и торфа. Это тоже малоподвижные формы углерода. В них, как и в залежах известняка, как бы «законсервирована» двуокись углерода.
Но круговорот не был бы круговоротом, если бы такому «консервированию» не противостояли обратные процессы — выделение СО2 в результате тех или иных превращений в живой и неживой природе. При дыхании растения и животные (и люди) выделяют СО2 в атмосферу (8) и воды океана. (Заметим попутно, что одни только люди выделяют в атмосферу 1 млрд, т СО2 в год.) Содержат СО2 и вулканические газы (9), пополняя его запасы в атмосфере. Разложение органических веществ в почве и водах, происходящее с участием микроорганизмов, также возвращает СО2 в оборот (10). Интересно отметить, что содержание СО2 в воздухе, находящемся между частицами почвы, иногда в десятки и сотни раз выше, чем в атмосфере. В кружке (11) показаны микроорганизмы и другие обитатели почвы, участвующие в выделении СО2.
На рисунке вы видите костёр. От примитивного костра до самой современной автомашины или топки ТЭС любой процесс горения — сжигание угля, бензина, древесины — пополняет запасы СО2 в атмосфере. С каждым годом возрастает количество топлива, используемого человеком. Растёт и содержание СО2 в атмосфере (12). Это вызывает определённое беспокойство из-за возможных последствий — нагревания нашей планеты (см. Атмосфера). Однако океан, растворяя «лишнюю» СО2, замедляет её накопление в атмосфере. Заметим, что процесс растворения двуокиси углерода в водах Земли обратим. Стрелкой (13) символически показано, что растворённая в океане СО2 находится в равновесии с атмосферной.
305
УГЛЕРОД
радиоактивного распада компенсируется вновь образующимися ядрами). Из воздуха в результате фотосинтеза 14С постоянно поступает в растения, причём концентрация изотопа 14С среди изотопов углерода во всём живом на планете также постоянна. Как только растение погибает, усвоение У. из воздуха прекращается и количество 14С в остатках начинает уменьшаться в соответствии с законом радиоактивного распада (через каждые 5570 лет число атомов 14С уменьшается вдвое). Таким образом, определив радиоактивность неживого органического вещества и сравнив её с радиоактивностью такого же количества органического вещества живого растения, можно рассчитать время, в течение которого происходил распад 14С. Иными словами — узнать возраст находки.
Атомный номер У. равен 6, то есть его атом имеет шесть электронов, два из которых принадлежат внутренней, а четыре — внешней электронной оболочке (полная электронная конфигурация ls22s22p2, наружной оболочки — 2s22p2, см. Атом). Образование ионных связей для У. нехарактерно. Только в соединениях с наиболее активными металлами, легко отдающими электроны, степень окисления У. может быть равна —4 (например, в карбиде алюминия А14С3). С подавляющим большинством элементов У. даёт прочные ковалентные связи, в которых для образования электронных пар используются все 4 электрона внешней оболочки. Обычно валентность У. равна IV.
Известны также немногочисленные соединения У., где он формально двухвалентен, например окись углерода СО (строение молекулы СО обсуждено в статье Углерода окись). Замечательной особенностью является способность атомов С соединяться между собой с образованием прочных и длинных цепей. Концы цепей могут замыкаться. В таком случае возникают циклы из атомов С. По словам Д. И. Менделее-
Рис. 4. Строение кристаллов графита (а) и алмаза (6). Выберем в структуре алмаза пару атомов, которые находятся на кратчайшем расстоянии друг от друга и связаны прочной ковалентной связью. Мысленно поставим в каждый из этих атомов ножки циркуля, а затем, оставив одну из ножек неподвижной и не меняя раствора циркуля, переместим другую ножку так, чтобы она «воткнулась» в какой-либо другой атом углерода. Действуя таким образом, будем поочерёдно перемещать ножки циркуля и при этом передвигаться по структуре в разных направлениях. В результате мы сможем обойти все атомы в структуре алмаза. Иными словами, какие бы два сколь угодно удалённые друг от друга атома мы ни взяли, всегда можно найти путь, ведущий от одного атома к другому и проходящий только по кратчайшим связям углерод — углерод.
Структура графита даёт пример другого типа. Здесь мы можем легко обойти циркулем все атомы, лежащие в одном слое; переход в другой слой без изменения раствора циркуля окажется невозможным. В отличие от алмаза, графит обладает слоистой структурой.
Пространственный каркас, образуемый прочными связями, обусловливает исключительную твёрдость алмаза. Специфические свойства графита, например его способность легко раскалываться параллельно атомным слоям, также нетрудно связать с его строением.
ва, «ни в одном из элементов такой способности к усложнению не развито в такой мере, как в углероде». Это служит причиной того, что соединения У. разнообразны как ни у одного другого элемента. Их изучение — область органической химии. Сейчас известно около 3 миллионов органических соединений (соединений У.) и всего лишь около 60 тысяч соединений всех остальных элементов.
Физические свойства. Различие аллотропных форм У.— графита и алмаза — является ярчайшим примером влияния кристаллического строения твёрдых тел на их физические свойства. В графите атомы С располагаются параллельными слоями (рис. 4,а). При этом атомы внутри слоя связаны между собой сильнее, чем один слой с другим. Графит можно рассматри-
20 хэш
УГЛЕРОД
306
вать как скопление плоских полимерных молекул, состоящих из атомов С и непрочно наслоенных одна на другую. В кристалле алмаза все атомы С соединены между собой очень прочными связями и образуют в пространстве непрерывный трёхмерный каркас (рис. 4,6). Весь кристалл представляет собой как бы одну гигантскую полимерную молекулу У., не имеющую «слабых мест», так как прочность всех связей одинакова. Особенности структуры определяют основные физические свойства алмаза и графита.
Если алмаз представляет собой бесцветные, прозрачные, сильно преломляющие свет кристаллы и является самым твёрдым из всех найденных в природе веществ, то графит — серо-чёрная, непрозрачная, жирная на ощупь чешуйчатая масса с металлическим блеском, по твёрдости уступающая даже бумаге.
Плотность алмаза при 20 °C равна 3,515 г/см3, графита — 2,265 г/см3. Физические свойства кристалла алмаза (твёрдость, электропроводность, теплопроводность, коэффициент расширения и другие) практически одинаковы по всем направлениям. В графите же эти свойства по разным направлениям — перпендикулярному или параллельному слоям атомов С — сильно различаются. По электрическим свойствам алмаз — изолятор; графит проводит электрический ток, но не так хорошо, как металлы.
Алмаз при нагревании без доступа воздуха выше 1000 °C превращается в графит. Графит при постоянном нагревании в тех же условиях не претерпевает никаких изменений вплоть до 3700 °C, когда он возгоняется, не плавясь. Жидкий У. можно получить только при одновременном действии очень высокой температуры (больше 3700 °C) и давления (больше 105 атм).
У. обладает ещё одной — третьей аллотропной формой, называемой карбином, не встречающейся в природе. Карбин — мелкокристаллический порошок чёрного цвета; в его кристаллической структуре длинные цепочки атомов С (громадные линейные молекулы) уложены параллельно друг другу.
Раньше считали, что древесный уголь, сажа и кокс, близкие по составу чистому У. и отличающиеся по свойствам от алмаза и графита, представляют собой самостоятельную аллотропную форму У., так называемый аморфный углерод. Сейчас с помощью рентгеновских лучей установлено, что эти вещества имеют не аморфную, а кристаллическую природу. Они состоят из очень мелких кристаллических частичек, в которых атомы С расположены так же, как в графите. Однако по традиции до сих пор часто говорят об «аморфном» углероде. Некоторые разновидности «аморфного» У. (например, древесный уголь) проявляют сильные адсорбционные свойства, то есть обладают способностью поглощать своей поверхностью значительные количества различных веществ. Такой уголь называют активным.
Химические свойства. При комнатной температуре У. во всех формах химически инертен, однако при сильном нагревании взаимодействует со многими элементами. Как и следует ожидать, исходя из строения различных форм У., легче всего вступает в реакции «аморфный» У., труднее — графит, наиболее химически устойчив алмаз. При нагревании на воздухе «аморфный» У. загорается при температурах 300—500 °C, графит — при 600—700° С, а алмаз— только при 850—1000° С. При избытке воздуха образуется двуокись У.:
С т О2 - СО2
При недостатке воздуха продуктом реакции является окись У.:
2С + О2 = 2СО
Обе реакции сильно экзотермичны, то есть протекают с освобождением большого количества энергии, выделяющейся в виде тепла и света.
Взаимодействие «аморфного» У. и графита с водородом идёт очень медленно даже при высоких температурах. Ускорить реакцию можно с помощью катализаторов, в частности металлов Ni или Pt. Интересно, что в зависимости от температуры получаются разные продукты. Так, при 600—1000 °C в присутствии катализаторов образуется преимущественно метан:
С4-2Н2 = СН4
а при более высоких температурах (1500— 2000°С) — ацетилен:
2С + Н2 = С2Н2
В продуктах взаимодействия У. с водородом при определённых температурах и давлениях могут присутствовать также и другие углеводороды, например этан С2Нв и бензол С6Н6. Алмаз с водородом не реагирует.
Сера непосредственно соединяется с «аморфным» У. и графитом при 700—800 °C, а с алмазом при 900—1000°С. Продукт реакции — сероуглерод'.
С 4- 2S = CS2
С азотом У. соединяется только при пропускании электрического разряда между электродами, сделанными из угля или графита и находящимися в атмосфере азота. При этом образуется циан\
2C-rN2 = C2N2
Фтор не реагирует с алмазом. Взаимодействие фтора с графитом начинается выше 900 °C, с «аморфным» У. — при комнатной температуре (с образованием CF4 и др). Многие металлы соединяются с У. при очень высоких температурах, давая карбиды.
В многочисленных реакциях с химическими соединениями У. проявляет сильные восстановительные свойства. Большой интерес представ
307
ляет его взаимодействие с водяным паром и углекислым газом:
С + Н2О = СО + Н2 (выше 800 °C)
С + СО2 = 2СО (выше 600 °C)
Образующиеся газообразные продукты хорошо горят, и эти реакции лежат в основе газификации твёрдого топлива — перевода каменного или древесного угля в так называемые водяной и генераторный газы. По реакции У. с водой получают также водород, идущий на синтез аммиака. Столь важными для промышленности оказываются эти — простые с виду — химические реакции.
Особенно ярко восстановительные свойства У. проявляются при взаимодействии с окислами металлов, например:
ZnO + C = Zn + CO (при 1200 °C)
Аналогичным образом У. при высоких температурах восстанавливает до свободных металлов окислы кадмия, меди, свинца, сурьмы. А в иных случаях по реакции У. с окислами металлов (например, кальция, ванадия, тантала) образуются карбиды.
Любые формы У. устойчивы по отношению к щелочам даже при длительном нагревании. Холодные концентрированные кислоты тоже не действуют на У., но при нагревании выше 100°C концентрированные H2SO4 и HNO3 медленно окисляют «аморфный» У. (но не графит или алмаз). Широкоизвестны и такие неорганические соединения У., как карбонаты — соли угольной кислоты Н2СО3 и цианиды — соли цианистоводородной, или синильной, кислоты HCN.
Получение. Большую часть алмазов и графита пока получают переработкой природного сырья. Процесс добывания алмазов требует очень больших усилий — для получения 1 г алмазов нужно переработать в среднем 20 т алмазоносной породы. Извлечённую из недр Земли породу сначала тщательно измельчают и затем промывают. При этом относительно тяжёлые камни, содержащие алмазы, отделяются от лёгких глинистых минералов, составляющих 98% первоначального веса породы. Из тяжёлого остатка алмазы извлекают, например, с помощью специального жирового состава (напоминающего вазелин). Алмазы прилипают к нему, а все остальные камни — нет. Для окончательной очистки алмазы промывают в кислотах и щелочах.
С тех пор как было установлено, что алмаз, графит и уголь имеют один и тот же химический состав, учёные неоднократно пытались превратить невзрачный уголь или графит в блистающие алмазы. Сделать это удалось только в 1955 г. Сейчас производство искусственных алмазов налажено и в нашей стране, и за рубежом.Графит или дру
УГЛЕРОД
гие богатые углеродом материалы нагревают до температур выше 1200 °C под давлением больше 50 тысяч атм в присутствии катализаторов (железо, никель, хром и другие металлы). Вес искусственных алмазов не больше 0,2 г; обычно они сильно окрашены примесями.
Главная проблема при получении чистого графита из природных залежей заключается в отделении графита от других минералов, с которыми он очень тесно срастается. В зависимости от вида руды и от крупности включений графита в породе после измельчения сырья используются разные методы обогащения, например флотация. А затем обогащённый продукт многократно обрабатывают концентрированными кислотами (соляной и фтористоводородной) и нагревают его в электрической печи при температуре 2200 °C. Так и получают чистый графит.
В отличие от алмазов, графит, производящийся сейчас искусственно, часто превосходит естественный по чистоте и однородности. Готовится искусственный графит нагреванием спрессованной смеси кокса или антрацита с каменноугольной смолой в электрических печах при 2200— 3000 °C. Большое применение находит пирографит, отличающийся высокой прочностью. Его получают при высокотемпературном разложении (пиролизе) углеводородов.
Промышленность производит также продукты, близкие по составу к чистому У. и являющиеся «аморфным» У. по строению. Так, кокс получают при нагревании некоторых сортов каменного угля или твёрдых остатков, образующихся при переработке нефти, до высокой температуры без доступа воздуха. Для производства сажи обычно сжигают природный газ при недостатке воздуха. Сажа оседает на металлических листах, с которых её собирают. Нагреванием древесины без доступа воздуха получают высококачественный древесный уголь. Изготовляют и пористый активный уголь, обладающий повышенной способностью к адсорбции. Для этого древесные или ископаемые угли обрабатывают перегретым водяным паром или двуокисью углерода при высоких температурах. При этом из углей удаляются смолистые вещества и увеличивается их пористость.
Применение. Если раньше алмаз ценили прежде всего как драгоценный камень, то сейчас — это важнейший, часто незаменимый, технический материал. Только наиболее крупные и прозрачные алмазы используют для украшений. После огранки и шлифовки они превращаются в бриллианты. Применение алмазов в технике связано прежде всего с их уникальной твёрдостью. С помощью алмазов бурят горные породы, режут, сверлят, шлифуют и гравируют стекло и другие особо твёрдые материалы. Алмазные резцы позволяют обрабатывать детали и инструменты с очень большой точностью.
Область применения графита чрезвычайно широка. Высокая жаропрочность, стойкость к химическим воздействиям, хорошая электропроводность делают графит ценнейшим материалом для современной промышленности. Из него изготовляют литейные формы, плавильные тигли и другие огнеупорные изделия. Искусственным графитом и пирографитом покрывают
20*
УГЛЕРОДА ДВУОКИСЬ
308
камеры сгорания и сопла реактивных двигателей (изнутри), а также рули космических ракет. Многие производства невозможны без химически устойчивых графитовых труб или аппаратов, выложенных изнутри графитовыми плитами. Значительные количества графита потребляет электротехническая промышленность для изготовления электродов, гальванических элементов и аккумуляторов, контактов электрических машин и нагревателей для электропечей. Очень чистый графит применяют в ядерных реакторах для замедления нейтронов. Смешанный с маслом графит служит прекрасным смазочным материалом. Жива и самая древняя «профессия» графита — из него делают стержни карандашей и некоторые краски.
Кокс применяется главным образом при выплавке чугуна из руд в доменных печах, где он одновременно выполняет роль топлива и восстановителя. Около 90% получаемой сажи идёт на нужды резиновой промышленности — добавки сажи повышают прочность и увеличивают срок службы резиновых изделий. Кроме того, из сажи приготовляют некоторые чёрные краски и лаки. Активный уголь нужен для поглощения вредных веществ из газов и жидкостей: им заполняют противогазы, с его помощью очищают жидкости от пахучих и красящих веществ. На адсорбционных свойствах активного угля основано и его применение в медицине.
Когда говорят о химии У. и его соединений, часто имеют в виду только органическую химию. Но не нужно забывать, что и чистый У. и его неорганические соединения имеют первостепенное значение для науки и народного хозяйства. Б. А. Поповкин.
УГЛЕРОДА ДВУОКИСЬ (у гол ь н ы й ангидрид, углекислый газ) СО2 — высший окисел углерода. Бесцветный газ, имеющий слегка кисловатые запах и вкус. У. д. приблизительно в 1,5 раза тяжелее воздуха (плотность СО2 1,98 г/л), и поэтому её можно, как жидкость, осторожно «переливать» из одного сосуда в другой. СО2 неплохо растворима в воде: при 20°C в одном объёме Н2О растворяется 0,88 объёма СО2.
Если У. д. охлаждать при атмосферном давлении, то она, минуя жидкое состояние, затвердевает при температуре —78,515 °C. Образуется белая снегообразная масса, известная под названием «сухой лёд». Жидкую СО2 можно получить только при повышенных давлениях, например сжимая газ при комнатной температуре под давлением 58,46 атм.
У. д.— кислотный окисел, ангидрид угольной кислоты. Растворение СО2 в воде сопровождается обратимой реакцией:
СО2 + Н2О^±Н2СО3
равновесие которой сильно сдвинуто влево: в угольную кислоту при обычной температуре превращается не больше 1 % СО2, растворённой в воде. У. д. энергично взаимодействует с сильными основаниями, давая карбонаты — соли угольной кислоты. На такой реакции основан способ обнаружения У. д. Если газ, содержащий СО2, пропускать в известковую воду, она мутнеет, так как выпадает плохо растворимый карбонат кальция:
Са (ОН)2 + СО2 = СаСОз + Н2О
При дальнейшем пропускании СО2 осадок исчезает вследствие образования растворимого гидрокарбоната кальция:
СаСОз и- Н2О + СО2 - Са (НСО3)2 Химически У. д. довольно инертна. Только наиболее активные металлы при высоких температурах отнимают у неё кислород (полностью или частично). Так, магний сгорает в атмосфере СО2 при 600 °C:
2Mg + СО2 = 2MgO - С
Поэтому горящий магний нельзя гасить углекислотными огнетушителями! Большое значение в металлургии имеет взаимодействие СО2 с углеродом, идущее при пропускании У. д. над раскалённым углём:
СО2 4- С = 2СО
У. д. не поддерживает горения и дыхания. В малых концентрациях она не представляет опасности для человеческого организма. Однако при содержании её в воздухе более 4% по объёму наблюдаются раздражение дыхательных путей, головная боль, шум в ушах, а при концентрации больше 10% могут наступить быстрая потеря сознания и даже смерть.
На Земле У. д. является одной из важных форм существования углерода. Воздух, окутывающий нашу планету, содержит около 2,2-1012 т СО2 (см. Атмосфера). Ещё больше её находится в растворённом виде в водах океанов, морей, озёр и рек (см. Гидросфера).
Поскольку У. д.— конечная форма окисления углерода, она образуется и в больших количествах поступает в атмосферу при сжигании топлива. Содержание СО2 в обычных дымовых газах достигает, например, 15—18% по объёму.
В некоторых вулканических местностях У. д. выделяется из трещин земной коры. В низких местах с плохой вентиляцией СО2 скапливается около поверхности земли в больших концентрациях. Животные, попадая в подобные ловушки, погибают от удушья. Таковы, например, знаменитая «Собачья пещера» около Неаполя в Италии и «Долина смерти» на острове Ява.
Громадное значение имеет У. д. для всего живого на Земле. Она служит главным источником углерода для развития растений, которые поглощают её из атмосферы в процессе
309
фотосинтеза (см. Биосфера, Кислород). Животные, напротив, выделяют при дыхании СО2, представляющую собой продукт «сгорания» пищи в организме. Каждый из нас выдыхает в сутки около 1 кг СО2.
Распространена У. д. и вне Земли. Так, она является основным компонентом атмосфер наших космических соседей — планет Марса и Венеры.
В лаборатории У. д. обычно получают взаимодействием соляной кислоты с мрамором в аппарате Киппа (этот аппарат изображен на рис. 1 к статье Водород)'.
СаСОз + 2НС1 = СаС12 + Н2О + СО2
Основным сырьём для получения СО2 в промышленности служит известняк. Обжиг его при температурах 900—1300 °C даёт два важных продукта — негашёную известь и У. д.:
СаСО3=СаО+СО2
Превращение газообразной У. д. в сухой лёд достигается одновременным охлаждением и сжатием её с помощью мощных компрессоров.
Главный потребитель У. д.— пищевая промышленность. Так, хорошо утоляющая жажду газированная вода — это раствор СО2 в воде, приготовленный под давлением. Как только такую воду наливают из закрытого сосуда в стакан, она пузырится — это выделяется СО2, растворимость которой, как и у всех газов, уменьшается с понижением давления. У. д. газируют также фруктовые и минеральные воды. Большое количество СО2 расходуется при производстве соды и промышленном синтезе мочевины. Жидкой У. д. заряжают огнетушители. Пользоваться ими очень удобно, так как выходящая мощная струя холодного газообразного СО2 эффективно гасит огонь и совершенно не портит окружающие предметы.
Широко применяют сухой лёд. Он используется как замечательный холодильный агент, охлаждающая способность у которого в два раза выше, чем у обычного льда. Особенно ценно для успешного хранения и перевозки скоропортящихся продуктов свойство СО2 задерживать развитие микроорганизмов. Мясо, лежащее на сухом льду, хорошо сохраняется. Применяют сухой лёд и при испытании изделий и приборов, предназначенных для работы «в царствах холода»: Арктике, Антарктиде или стратосфере.
Рассеиванием тонкоизмельчённого сухого льда над облаками можно вызвать «искусственный» дождь. Очень холодные (с температурой —78,5 °C) частички сухого льда способствуют тому, что окружающие их мельчайшие капельки водяного пара облаков кристаллизуются и выделяются в виде снега или дождя. Интересно, что для того, чтобы на землю выпало около 1000 т дождевой воды,нужно затратить всего 200 г сухого льда! Б. А. Поповкин.
УГЛЕРОДА ОКИСЬ
УГЛЕРОДА ОКИСЬ (угарный газ) СО — окисел двухвалентного углерода. Газ без цвета и запаха. Плотность 1,25 г/л. У. о. сжижается при —191,5 °C и затвердевает при —205 °C. В воде плохо растворима (при 20 °C 100 объёмов Н2О растворяют только 2,3 объёма СО).
Формально углерод в СО двухвалентен. Однако элементы в этом соединении связаны прочнее, чем можно было бы ожидать, исходя из наличия двойной связи между ними. Поэтому предполагают, что в молекуле У. о. имеется дополнительная, третья, донорно-акцепторная связь, образованная двумя электронами, принадлежащими атому кислорода — донору (обозначена <-). В этом случае молекулу СО можно изобразить как :С О: (каждая чёрточка символизирует пару электронов).
Являясь несолеобразующим окислом, У. о. не взаимодействует при обычных условиях с водой, кислотами и щелочами. При повышенных температурах СО склонна к реакциям присоединения и к реакциям, в которых она служит восстановителем.
У. о. горит на воздухе синим пламенем, превращаясь в СО2:
2СО + О2 = 2СО2
При этом выделяется большое количество тепла (283 кдж/моль), что делает СО ценным газообразным топливом. С помощью всесильных катализаторов, в данном случае окислов марганца и меди, можно заставить У. о. медленно взаимодействовать с кислородом уже при комнатной температуре. Это свойство используется в противогазах, предназначенных для защиты от СО.
Восстановительные свойства У. о. ярко проявляются в реакциях с окислами металлов, например:
NiO + CO = Ni + CO2 (при 300 °C)
В присутствии катализаторов (платины или активного угля) или под действием прямого солнечного света У. о. соединяется с хлором, образуя чрезвычайно ядовитый газ—фосген, применявшийся в первую мировую войну в качестве боевого отравляющего вещества:
СО + С12 = СОС12
Уникальной является способность У. о. присоединяться при повышенных температурах и давлениях к некоторым металлам с образованием необычных (комплексных) соединений, называемых карбонилами, например:
Ni + 4СО = Ni (СО)4
В природе У. о. практически не встречается. В лаборатории её обычно получают, прибавляя по каплям муравьиную кислоту к концентрированной серной кислоте, нагретой до 100 °C:
НСООН = СО + Н2О
УГОЛЬНАЯ КИСЛОТА
310
Серная кислота отнимает воду от муравьиной кислоты.
У. о. образуется тогда, когда сгорание углерода или его соединений происходит при недостатке воздуха. Поэтому в топочных газах всегда есть примесь СО. Так, в выхлопных газах автомобилей её содержание достигает 2—10% по объёму.
Промышленные способы получения У. о. основаны на взаимодействии раскалённого угля с углекислым газом:
С + СО2 = 2СО (/)
или с водяным паром:
С + Н2О = СО + Н2 (2)
Таким образом производят генераторный (/) и водяной (2) газы, использующиеся как газообразное горючее.
В химической промышленности У. о. служит ценным исходным веществом для синтеза органических продуктов: метилового и других спиртов, различных углеводородов и органических кислот. Путём химических превращений из СО и водорода можно получить искусственный бензин.
Своё второе название «угарный газ» У. о. получила из-за относительно частых случаев отравления ею в быту. Она чрезвычайно ядовита, и тех небольших количеств, которые образуются в печке с преждевременно закрытой заслонкой или при работе плохо отрегулированных газовых горелок, вполне достаточно, чтобы отравиться (как говорят, «угореть»). Первыми признаками отравления У. о., наступающего незаметно, являются головная боль и головокружение (в дальнейшем потеря сознания). Лучшее средство помощи — свежий воздух. Об опасности У. о. для человеческого организма следует знать курильщикам — ведь табачный дым содержит от 0,5 до 1 % СО!
Б. А. Поповкин.
УГОЛЬНАЯ КИСЛОТА Н2СО3 —очень слабая и непрочная кислота. При обычных условиях может существовать только в водном растворе в малых концентрациях. Образуется У. к. при растворении двуокиси углерода в воде по обратимой реакции:
Н2О + СО2 Н2СО3
Её присутствие обнаруживается по слабокислой реакции раствора. При нагревании раствора Н2СО3 полностью разрушается с выделением СО2. У. к. двухосновна и в растворе диссоциирует на ионы:
Н2СО3 Н + + НСО3" 2Н + + СО2" причём диссоциация по второй ступени крайне мала. В соответствии с основностью известны
два ряда углекислых солей: средние — карбонаты и кислые — гидрокарбонаты (или бикарбонаты).
УГОЛЬНЫЙ АНГИДРИД — то же, что углерода двуокись СО2.
УРАН (лат. Uranium) U — радиоактивный химический элемент; атомный номер 92, атомная масса 238,029; относится к актиноидам. Серебристо-белый блестящий металл. Природный У. состоит из смеси трёх изотопов, содержание которых резко различно: на долю самого тяжёлого из них 238U приходится 99,274%, на долю 236U (наиболее важного для атомной энергетики) — в 139 раз меньше, то есть 0,72%; наконец, третий изотоп 234U присутствует в исчезающе малых количествах (около 0,006%). Все природные изото
пы У. при радиоактивном распаде испускают а-частицы (ядра гелия 4Не). Периоды полураспада Ti/2 изотопов 238U и 235U огромны (соответственно 4,51 • 109 и 7,13-108 лет) и сравнимы с возрастом Земли. Это — самые тяжёлые изотопы, сохранившиеся на нашей планете со времён её формирования.
Дочерние ядра, образующиеся в результате распада 238U и 236U, также радиоактивны; распадаясь, они образуют новые радиоактивные дочерние ядра и т. д. Таким образом, 238U и 235U дают начало двум длинным цепочкам радиоактивных превращений — так называемым радиоактивным рядам урана-238 и урана-235. Членами этих рядов являются многие природные радиоактивные изотопы элементов, расположенных в системе Менделеева между свинцом и У. (например, изотопы 226Ra, 222Rn и 210Ро, входящие в ряд У.-238, и изотопы 231Ра, 227Ас и 223Fr, входящие в ряд У.-235). Одним из членов ряда У.-238 является и изотоп 234U (Ti/,=2,48* 105 лет). Скорости образования и распада каждого промежуточного члена ряда равны между собой, поэтому содержание каждого такого изотопа хотя и мало (оно тем ниже, чем меньше 7\2 изотопа), но остаётся постоянным. Оба радиоактивных ряда заканчиваются стабильными изотопами свинца. Таким образом, конечными продуктами превращений ядер 238U и 235U являются РЬ и Не (а-частицы).
Образующийся свинец полностью сохраняется в массе уранового минерала. А так как скорость накопления РЬ известна (её легко рассчитать, зная величины изотопов 238U и 236U), можно, определив содержание РЬ и U в исследуемом минерале, найти возраст минерала. Среди методов, применяемых для определения возраста раз
311
УРАН
личных геологических объектов, этот — один из самых точных и надёжных (о другом методе см. в статье Углерод).
После открытия секретов получения ядерной (атомной) энергии У. по своему значению для человечества занял совершенно исключительное место среди других элементов. Первая электростанция, преобразующая ядерную энергию в электрическую, была создана в СССР (рис. 1). Сейчас слово «уран» знакомо каждому.
А между тем первые страницы биографии У. совершенно обычны. Его природные соединения использовали очень давно — при раскопках Древнего Рима археологи находят стёкла, окрашенные с помощью солей У. Как самостоятельный химический элемент У. был открыт в 1789 г., когда берлинский химик-аналитик М. Клапрот заинтересовался составом смоляной обманки — тяжёлого чёрного минерала, который саксонские горняки считали пустой породой. Клапрот выделил из смоляной обманки чёрное металлоподобное вещество и, приняв его за новый металл, назвал именем планеты Уран, открытой в 1781 г. Однако, как оказалось, Клапрот выделил не металлический U, а его двуокись UO2. Ошибка была обнаружена только в 1841 г., когда французский химик Э. Пел иго получил U в виде металла.
Атомная масса У. долгое время принималась равной 116. Поэтому Д. И. Менделеев, создавая первый вариант периодической системы, поместил U вслед за кадмием, в III группу. Однако позднее Менделеев, анализируя свойства U, пришёл к выводу, что его следует размещать в VI группе, а атомная масса элемента должна быть увеличена в 2 раза. У. занял место в самом конце таблицы, в одной группе с молибденом и вольфрамом. Правильность менделеевского исправления атомной массы U подтвердилась. Подтвердилось и сходство многих химических свойств U и W. Долгое время U оставался в периодической системе на том месте, которое ему отвёл Менделеев. Однако после искусственного синтеза трансурановых элементов выяснилось, что их свойства во многом похожи на свойства У. В настоящее время, в соответствии с актиноидной концепцией американского учёного Г. Сиборга, U чаще всего размещают не в VI группе, а относят к семейству актиноидов.
Впервые повышенный интерес к У. возник в 1896 г., после того как французский физик А. Беккерель открыл его радиоактивные свойства. Однако вскоре оказалось, что радиоактивность радия, всегда содержащегося в урановых минералах, в тысячи раз сильнее, и в течение последующих 40 лет интерес к У. определялся в основном возможностью извлечения из его минералов радия. После переработки минералов большая часть У. попадала в отходы.
Положение коренным образом изменилось в начале 1940-х годов, когда физики открыли, что с помощью изотопа 236U можно осуществить цепную реакцию деления. Реакция деления — это такой ядерный процесс, при котором исходное ядро распадается на 2 осколка примерно одинаковой массы, каждый из которых является ядром какого-то элемента. В каждом акте деления ядер 235U одновременно с образованием двух осколков испускается 2 или 3 нейтрона. Эти нейтроны способны вызвать деление новых ядер 236U. Вновь выделяются нейтроны, число их быстро множится, в ядерную реакцию вовлекаются всё новые и новые ядра — имеет место цепная реакция деления (рис. 2).
Рис. 1. Первая в мире атомная электростанция. Введена в строй 27 июня 1954 г. в городе Обнинске (в сотне километров от Москвы). Даёт электрический ток для промышленности и сельского хозяйства прилегающих районов.
Цепная реакция, начавшись, сама не окончится, пока не будет израсходован весь делящийся изотоп или не будут «убраны» из зоны реакции нейтроны, возникающие при делении ядер 236U. При осуществлении цепной реакции каждый из разлетающихся с большой скоростью осколков обладает огромной кинетической энергией. Обычно именно её и называют атомной, точнее ядерной энергией. Так, если при реакции деления израсходуется 1 г 236U, то количество выделившейся энергии составит 8-Ю17 эрг (а при горении 1 г угля — только 2,9-1011 эрг). Поэтому У.— чрезвычайно перспективный источник энергии.
Следует отметить, что цепная реакция деления может развиваться только в специальных условиях, в частности когда содержание в материале других, неспособных к этой ядерной реакции изотопов, не превышает определённого уровня. В естественной смеси изотопов U содержание 23®U мало. Поэтому необходимо было -научиться разделять изотопы U или хотя бы частично увеличивать содержание 236U в смеси («обогащать» природный У.). Разумеется, провести разделение изотопов химическими методами невозможно, так как химические свойства 236U и 238U совершенно идентичны. Но можно использовать небольшие различия физических свойств, например различие масс изотопов. С этой целью пары гексафторида UFe — легколетучего соединения, пропускают через специальный мелкопористый фильтр. Молекулы UF6, содержащие 236U, проходят через фильтр чуть-чуть быстрее молекул, в состав которых входит238U. Если такое «фильтрование» повторить несколько тысяч раз, то удаётся выделить 236UFe в чистом виде. Легко представить себе, сколь трудоёмким является этот процесс.
Энергию, освобождающуюся при цепной реакции деления, можно выделять либо постепенно — в реакторе, либо мгновенно — в бомбе. Реакция в ядерной (атомной) бомбе нерегулируема, она протекает за ничтожную долю секунды, так что огромное количество тепла выделяется мгновенно и происходит взрыв чудовищной силы. А в ядерных реакторах приме
УРАН
312
няют специальные — обычно кадмиевые — стержни, с помощью которых регулируют скорость деления ядер 236U (кадмий очень сильно поглощает нейтроны). Скорость нейтронов, образующихся при делении ядер U в реакторах, понижают, используя специальные вещества — замедлители (воду, графит, парафин и др.). Сталкиваясь с ядрами атомов водорода и углерода, нейтроны теряют свою энергию, и скорость их резко падает. Замедленные нейтроны взаимодействуют с новыми ядрами 236U значительно чаще, чем быстрые, и поэтому для работы ядерных реакторов можно использовать не чистый 236<U, а естественную смесь изотопов U, обогащённую 236U.
Во многих ядерных реакторах можно получать не только энергию, но и новый делящийся материал — изотоп плутония 239Ри. Образно говоря, такой реактор — это печка, в которую положили берёзовые дрова, а после того, как они сгорели, в топке нашли сосновые дрова, причём их оказалось больше, чем было положено берёзовых. Дело в том, что ядра 238U, содержащиеся в обогащённом U, могут поглощать нейтроны. Последующие ядерные превращения приводят к образованию 239Ри, который, как и 236U, способен к цепной реакции деления (см. Плутоний). Реакторы для производства 239Ри кон
струируют специально; их называют реакторами-размножителями (бридерами). В ближайшие десятилетия бридеры станут основным типом энергетических реакторов. Известно, что кроме 236U и 239Ри, способность к цепному делению обнаруживает также изотоп 233U, не встречающийся в природном У. Его можно получать искусственно, облучая природный торий нейтронами (см. Торий).
Важное значение У. заставило тщательно определить его запасы на Земле. Оказалось, что У.— довольно редкий элемент. На его долю приходится 2,5-10“4% массы земной коры; однако это намного больше, чем содержание таких «обычных» элементов, как ртуть и серебро. Общий запас У. в земной коре около 1,3-1014 т.
Промышленную добычу У. осложняет его сильная рассеянность. Где он только ни присутствует— и в горных породах, и в почвах, и в морской воде, и в человеческом организме. Минералы же, богатые У., встречаются редко. Наиболее велико содержание У. в уже упоминавшейся смоляной обманке (её называют также урановой смолкой) и уранините; эти минералы представляют собой различные окислы У. Важное значение имеют карнотит, эвксенит, фер-гюсонит, самарскит и т. д.— в состав этих
Рис. 2. Начало и развитие цепной реакции деления урана. Большие красные шары — это ядра делящегося изотопа 236U. Осколки красных шаров — так называемые осколочные ядра. Маленькие серые шарики — нейтроны. Первый нейтрон возникает за счёт космического излучения или при спонтанном (самопроизвольном) делении ядер урана.
313
УРАН
минералов, наряду с У., входит большое число других элементов (железо, ванадий, ниобий, титан и т. д.). За рубежом наиболее крупные месторождения У. находятся в Северной Америке (Канада, США), Африке (Заир и ЮАР) и Австралии. Запасы У. в месторождениях капиталистических стран оцениваются около 1 млн. т.
Получение У. из минералов — технологически сложная задача. Её трудно решать ещё и потому, что для атомной энергетики нужен очень чистый У. На последней стадии переработки сырья У. подвергают очистке специальными методами — ионообменными и экстракционными. Весьма сложна и выплавка самого металла. Обычно поступают так. Смесь тетрафторида UF4 и магния загружают в специальные тигли, которые нагревают до 500—700 °C. Смесь в каждом тигле поджигают специальным электрическим запалом, и начинается реакция, сопровождающаяся выделением большого количества тепла:
UF4 + 2Mg = 2MgF2 + U
Расплавленный тяжёлый У. собирается на дне тигля. Из-за высокой реакционной способности нагретого У. процесс ведут в атмосфере инертного газа — аргона.
Свежевыплавленный металл обладает стальным блеском, но постепенно тускнеет на воздухе из-за образования плёнки окисла. Температура плавления У. сравнительно невелика (ИЗО °C), температура кипения около 3800 °C. У.— один из самых тяжёлых металлов (его плотность 19 г/см3).
Химически У. весьма активен. Так, порошок У. загорается при незначительном нагревании на воздухе, давая окисел состава U3O8. Со многими неметаллами У. реагирует при 300— 400 °C. Порошкообразный У. выделяет водород из воды при комнатной температуре, а при нагревании У. реагирует с выделяющимся водородом, давая гидрид UH3.
Химические свойства У. в некоторой степени повторяются у следующих за ним в периодической системе элементов—нептуния, плутония, америция (см. Актиноиды). Такое сход
ство химических свойств объясняется особенностями строения электронных оболочек атомов этих элементов. Есть у У. общие черты и с элементами VI группы — молибденом и особенно вольфрамом. В своих соединениях У. проявляет степени окисления от +2 до +6 (соответственно валентности от II до VI). Наиболее характерны для У. степени окисления -(-б и +4.
Соединения, соответствующие этим степеням окисления, устойчивы как в твёрдом состоянии, так и в водных растворах. Двуокись UO2 имеет основной характер, а трёхокись UO3 — амфотерный. При окислении U (+4) в кислом растворе до U (+6) атом U присоединяет 2 атома кислорода и возникает очень прочный катион UO2 +— так называемый уранил-ион. По своему поведению ура-нил-ион отдалённо напоминает ион аммония*NH4+. Как и ион аммония, уранил-ион с кислотными остатками образует соли, например нитрат уранила UO2(NO3)2, ацетат уранила UO2(CH3COO)2 и т. д. Многие соли уранила хорошо растворимы в воде.
Из соединений шестивалентного У. особенно важен гексафторид UFe — бесцветное твёрдое вещество; способность к сублимации (возгонке) у UFe значительно выше, чем даже у свободного иода, который обычно служит примером легко возгоняющегося вещества. Благодаря высокой летучести UFe его используют при разделении изотопов У. Изучению свойств UFe посвящено большое число научных статей, есть даже толстая книга, которая так и называется: «Гексафторид урана». Вряд ли найдётся много неорганических соединений, которые удостоились такой чести.
Кроме применений, связанных с получением атомной энергии и попутным выделением ряда «осколочных» радиоактивных элементов, например технеция и прометия (эти элементы образуются при делении ядер 236U), существуют и иные области использования У. (металлургия, производство цветного стекла и др.). Однако все они не идут ни в какое сравнение с использованием У. в ядерной технике. Существует предположение, что малые количества У. способствуют усвоению растущим организмом таких элементов, как фосфор, азот и калий, но биологическая роль У. выяснена пока недостаточно. С.С.Бердоносов.
ФЕРМИЙ (лат. Fermium) Fm — радиоактивный химический элемент с атомным номером 100; относится к актиноидам.
ФЕРРОСПЛАВЫ — сплцвы железа с различными химическими элементами, вводимые в жидкую сталь для её раскисления или легирования. Раскисление — это восстановление закиси железа FeO, находящейся в расплавленной стали. Для раскисления применяют Ф., содержащие элементы, которые соединяются с кислородом более активно, чем железо (ферросилиций — сплав Fe и Si, ферромарганец, иногда феррованадий). Эти элементы как бы «отнимают» кислород от закиси железа. Легирование — это придание стали особо ценных свойств путём введения в неё Сг, W, V и т. д. Вводить эти элементы в сталь в чистом виде затруднительно — они медленно растворяются в жидком металле, так как температуры их плавления обычно выше, чем у стали; у Ф. же температура плавления такая же, как у стали, или ниже. Кроме того, чистые легирующие металлы получаются трудно и они дороже, чемФ.
Наиболее часто в металлургии применяются ферросилиций (45 и 75% Si), феррохром (не менее 65% Сг), ферромарганец (75—80% Мп), ферровольфрам (65—80% W), ферромолибден (не менее 55% Мо), феррованадий (35—80% V), ферротитан (40% Ti), феррониобий (30—75% Nb) и др.
Ф. получают из концентратов руд соогветст-вующих металлов и железной стружки. Плавку обычно ведут в дуговых электропечах. Восстановителем окислов металлов служит кокс или древесный уголь. Поэтому полученные в электропечах Ф. всегда содержат С (до 6—8%) и вносимые рудой и . коксом примеси (S, Р, As и т. д.). Некоторые стали, например нержавеющие, должны содержать как можно меньше С. Поэтому многие Ф., полученные в электропечах, подвергают специальной обработке для удаления углерода. А кроме того, есть способы получения безуглеродистых Ф.: восстановителем служит либо Si (силикотермия), либо А1 алюминотермия). Ферромарганец, содержа
щий 70—75% Мп, 6—7% С и 2% Si, иногда выплавляют в доменных печах.
Производство ферромарганца и низкопроцентного ферросилиция (10—15% Si) началось ещё в 1860-х гг. Ф. тогда выплавляли в доменных печах. В начале XX в. появились электропечи для выплавки Ф. В России Ф. (в основном феррохром) впервые начали выплавлять в 1910—1911 гг. на заводе «Пороги» близ Челябинска. Производство Ф. в крупных масштабах началось только в 1930-х гг., когда были построены Запорожский, Зеста-фонский и Челябинский заводы. В последние годы в промышленность Ф. внедрены новые процессы, повышающие качество продукции: вакуумная обработка, продувка кислородом в конвертерах.
Ф. часто называют витаминами стали. Они
придают стали прочность, вязкость, коррозионную стойкость и другие ценнейшие качества.
ФОСФАТЫ — соли фосфорных кислот.
Наибольшее практическое значение имеют ортофосфаты (или просто фосфаты) — соли
ортофосфор ной кислоты Н3РО4.
Кислота эта
трёхосновна и для неё известны три типа солей — однозамещённые (КН2РО4), двухзамещённые (СаНРО4) и трёхзамещённые [Са3(РО4)2]. Однозамещённые Ф. всех металлов
хорошо растворимы в воде, а из двух- и трёхзамещённых растворимы только соли щелочных металлов и аммония. Применение Ф. разнообразно. Так, фосфатами аммония про
питывают ткани, пластики, дерево и другие материалы для придания им огнестойкости. Особенно же велико значение Ф. для сельского хозяйства — ортофосфатами являются важнейшие фосфорные удобрения: простой суперфосфат, двойной суперфосфат, преципитат, аммофос (они подробно описаны в статье Минеральные удобрения).
Метафосфаты — соли метафосфорных кислот, и в особенности гексаметафосфат натрия NaePeOi8 используются для умягчения воды и удаления накипи из паровых котлов. Дело в том, что ионы Са2+ и Mg2+, обусловливающие жёсткость воды, образуют нерастворимые в воде метафосфаты, которые могут быть затем легко
удалены.
Соли полифосфорных кислот — полифосфаты входят в состав синтетических мою
щих средств — также, как умягчители воды.
315
ФОСФОР
ФОСФИДЫ — соединения фосфора с другими, более электроположительными элементами, главным образом с металлами. Важнейшие методы получения Ф.— синтез из компонентов при высокой температуре:
ЗСа + 2Р = Са3Р2
или взаимодействие фосфористого водорода с раскалённым металлом:
2А1 + 2РН3 = 2А1Р + ЗН2
Состав Ф. различен и часто не подчиняется «обычным» правилам валентности (как и состав многих боридов, карбидов и т. д.).
Одни Ф. (Na3P, Са3Р2) легко разлагаются водой с выделением РН3, другие (например, Ф. меди, хрома, железа, марганца) устойчивы даже в концентрированных кислотах. Поскольку Ф. содержат фосфор в степени окисления —3, они являются сильными восстановителями.
В современной технике Ф. интересны как полупроводниковые материалы — таковы Ф. галлия GaP, Ф. индия InP и другие. Есть у них и другие области применения. Так, те Ф., которые под действием воды легко выделяют ядовитый РН3, можно использовать для обеззараживания зернохранилищ, складов и т. д.
ФОСФЙН — то же, что фосфористый водород РН3.
ФОСФИТЫ — соли фосфористых кислот. Существуют однозамещённые и двузамещённые Ф. Многие Ф., например однозамещённый NH4H2PO3 и двузамещённые (NH4)2HPO3 и Na2HPO3, хорошо растворимы в воде. Поскольку Ф. содержат фосфор в степени окисления +3, они легко окисляются до фосфатов’.
КН2РО3 4- С12 + Н2О = КН2РО4 + 2НС1
Во многих химических синтезах Ф. используют как восстановители.
ФОСФОР (лат. Phosphorus) Р — химический элемент V группы периодической системы Менделеева; атомный номер 15, атомная масса 30,97376. Элемент имеет один стабильный изотоп 31Р.
Историческая справка. Открытие Ф. приписывается гамбургскому алхимику X. Бранду. В поисках «философского камня», якобы способного превратить неблагородные металлы в золото, Бранд занимался перегонкой сухого остатка от выпаривания мочи. В приёмнике ока
залось вещество, испускавшее голубоватый свет. Так, совершенно случайно, в 1669 г. был открыт белый Ф.— вещество, вызывавшее свечение. Поначалу у Бранда не было сомнений, что свечение без огня мог дать лишь искомый «философский камень». Однако вскоре он понял,
что никакими другими чудесными свойствами его находка не обладает, и продал секрет другому алхимику. Тот, в свою очередь, перепродал его третьему и т. д. Конец «фосфорному бизнесу» положил английский химик Р. Бойль, который в 1680 г. опубликовал в научном журнале более простую и доступную методику получения Ф. В 1771 г. шведский химик К- Шееле дал способ получения Ф. из костяной золы, применявшийся в промышленности вплоть до начала XX в.
Наименование элемента происходит от греческих слов «фос» — свет и «форбс» — несущий. Название «фосфоры» приписывают всем веществам, светящимся в темноте после предварительного облучения, а само явление холодного свечения называют фосфоресценцией.
Вопрос о распространённости Ф. в природе и в частности о его содержании в живых организмах привлёк внимание учёных ещё в начале XVIII в. А в конце XVIII в. было доказано, что присутствие соединений Ф в почве необходимо для жизни растений. Однако прошло больше 40 лет, прежде чем англичанин Лоуз изготовил суперфосфат (действием серной кислоты на костяную золу) — удобрение, легко усвояемое растениями. В России опыты по использованию фосфорных удобрений были поставлены Д. И. Менделеевым во второй половине XIX в. Однако промышленное производство фосфорных удобрений получило развитие в нашей стране только после Великой Октябрьской революции.
Нахождение в природе. Ф. принадлежит к весьма распространённым в природе элементам. На его долю приходится 9,3-10~2% от массы земной коры (13-е место среди всех элементов). Вследствие высокой химической активности Ф. в свободном состоянии в природе не встречается. Он содержится не менее чем в 190 минералах, из которых главнейшими являются апатиты ЗСа3(РО4)2-СаХ2 (где X — фтор, хлор или ОН-группа) и фосфориты — Са3(РО4)2 с различными примесями. В виде фосфидов железа, кобальта и никеля Ф. встречается в железных метеоритах.
В СССР известны крупные месторождения фосфоритов, а на Кольском полуострове имеются самые большие в мире запасы апатитов. Для переработки этих апатитов по решению Коммунистической партии в 1929—34 гг. построен крупный химический комбинат, пуск которого позволил нашей стране отказаться от ввоза фосфорных удобрений из-за границы.
Тело человека содержит в среднем около 1,5 кг Ф., главным образом в виде фосфатов кальция. Из этого количества 1,4 кг приходится на кости, около 130 г — на мышцы и 12 г — на нервы и мозг. Зубная эмаль по составу и кристаллическому строению соответствует фторапатиту.
А из курсов органической химии и общей биологии вы узнаете следующее.
Нуклеиновые кислоты, которые играют важнейшую роль в передаче наследственной информации, а также другие биологически активные органические вещества содержат Ф. В маленьких внутриклеточных образованиях — митохондриях идут химические реакции, обеспечивающие организм энергией: здесь питательные вещества, поступившие в организм с пищей, окисляются кис
ФОСФОР
316
лородом, пришедшим из лёгких. Большая часть энергии, выделившейся при окислении, запасается молекулами аденозинтрифосфорной кислоты (сокращённо её называют АТФ). Это сложное органическое соединение, содержащее Ф., служит универсальным аккумулятором и распределителем энергии: оно накапливает энергию, заключённую в химических связях питательных веществ, и «распоряжается» ею. Когда организму нужно выполнить синтез белка, мышечную работу, передачу нервных импульсов или любой другой процесс, требующий затраты энергии, молекулы АТФ подвергаются гидролизу. В результате и выделяется необходимая для жизненных процессов энергия. Вообще же в энергетическом обмене организмов принимают участие и другие содержащие Ф. вещества. Для них есть даже особое название: «богатые энергией соединения» (или «макроэргические соединения»).
Подобно азоту, кислороду, углероду, Ф. проходит в природе определённый цикл превращений. Растения извлекают фосфорнокислые соли из почвы, переводят их в сложные фосфорсодержащие органические вещества, которые с растительной пищей попадают в организм животных. После смерти животных и растений их останки попадают обратно в почву, где фосфорсодержащие соединения постепенно распадаются с образованием в конечном счёте солей фосфорной кислоты. Существенную поправку в круговорот фосфора в природе вносит сознательная деятельность человека. Урожаи культурных растений всего мира ежегодно уносят с полей миллионы тонн соединений Ф. А так как природных источников пополнения почвы соединениями Ф. почти не существует, постепенно развивающийся в ней «фосфорный голод» проявляется ещё более остро, чем азотный. Тем важнее встаёт проблема производства и использования фосфорных удобрений.
Физические и химические свойства. Ф.— один из элементов, самых богатых аллотропными модификациями. Белый Ф.— кристаллическое вещество белого цвета (иногда жёлтого, из-за наличия примесей) получается при конденсировании паров Ф. Молекула белого Ф. состоит из четырёх атомов, располагающихся по вершинам правильной пирамиды, все грани которой — равносторонние треугольники; такой многогранник называется тетраэдром (рис.1, а). Плотность белого Ф. 1,83 г/см3, /пл 44,1 °C, /Кип 280,5 °C. Основная характерная черта этой формы — высокая реакционная способность по отношению к другим элементам, особенно к кислороду. Поэтому обычно белый Ф. хранят под водой — он не растворяется в Н2О и не реагирует с ней. На воздухе белый Ф. самопроизвольно воспламеняется при нагревании выше температуры плавления. Медленное окисление его паров сопровождается свечением (вспомним алхимика Бранда). Белый Ф. чрезвычайно ядовит и даёт болезненные, трудно заживающие ожоги. При длительном нагревании
Рис. 1. Строение белого и чёрного фосфора. а — Кристаллы белого фосфора образованы молекулами Р4. Такая молекула имеет вид тетраэдра — правильного многогранника, все грани которого являются равносторонними треугольниками.
б’ — Молочный пакет по форме близок к тетраэдру. Это — своеобразная модель молекулы Р4.
в — В кристаллах чёрного фосфора молекул нет — они состоят из бесконечных слоёв. На рисунке показано строение такого слоя (в несколько идеализированном виде). Как и в молекуле Р4, здесь каждый атом связан с тремя ближайшими «соседями». Но закон соединения атомов таков, что в результате возникает не молекула, а слой. Чтобы разобраться в его строении, полезно обратиться к кристаллической структуре графита (см. Углерод). И в графите, и в чёрном фосфоре слои состоят из шестичленных колец, но в первом случае они плоские, а во втором — особым способом изогнуты, гофрированы.
без доступа воздуха при 280—300 °C белый Ф. переходит в красный Ф.— другую аллотропную модификацию.
Красный Ф. представляет собой аморфный порошок. Его плотность 2,3 г/см3, /пл 590 °C. Этот Ф. менее реакционноспособен, чем белый, так что его удобнее хранить. Абсолютно чистый красный Ф. неядовит, однако в техническом продукте всегда присутствуют ядовитые примеси. При высоком давлении (12 000 атм) и температуре 220 °C из белого Ф. получают чёрный. Эта модификация обладает самой большой плотностью по сравнению с остальными (2,7 г/см3) и наименее реакционноспособна. По внешнему виду и по внутреннему строению чёрный Ф. похож на графит (см. Углерод): кристалл построен из бесконечных слоёв атомов, но только, в отличие от плоских графитовых слоёв, в чёрном Ф. слои «гофрированы» (рис. 1, в). При нормальных условиях чёрный
317
ФОСФОР
Ф. ведёт себя как полупроводник, а при повышенном давлении проводит ток как металл (белый и красный Ф.— изоляторы).
Есть у Ф. и ещё несколько аллотропных модификаций— фиолетовый (металлический), коричневый, алый, стеклообразный чёрный и другие. Все они — аморфные вещества, существующие, как правило, при высоких давлениях.
В соответствии с положением Ф. в V группе периодической системы Менделеева его атом, как и атом азота, содержит на внешней электронной оболочке пять электронов (конфигурация 3s23p3, см.. Атом). Внешние электроны у атома Р связаны с ядром менее прочно, чем у атома N, поэтому Ф. легче отдаёт их в химических реакциях, чем азот. В соединениях Ф. проявляет степени окисления +5, +3 и —3 (соответственно валентности V и III). Химические связи атома Ф. с другими элементами — неметаллами носят ковалентный характер.
Ф. горит на воздухе с образованием фосфорного ангидрида:
4Р + 5О2 = 2Р2Об
а при недостатке воздуха — фосфористого ангидрида Р2О3. Белый Ф. горит в атмосфере галогенов, давая тригалогениды, например:
2Р + ЗС12 = 2РС13
а при избытке галогена — пентагалогениды:
РС13 + С12 = РС1б
Фториды Ф. получают косвенным путём, так как прямое действие фтора на Ф. сопряжено с опасностью взрыва. С серой Ф. взаимодействует при сплавлении с образованием сульфидов различного состава (например, P4S3). Если порошок Ф. нагревать с металлами (обычно нагревание проводят в атмосфере инертного газа, чтобы избежать окисления Ф.), то образуются фосфиды. С водородом Ф. в обычных условиях не реагирует, однако косвенным путём получают фосфористый водород (или фосфин) РН3 — соединение, похожее на аммиак. При нагревании Ф. взаимодействует с крепким раствором щёлочи, при этом выделяется фосфин и образуется двузамещённый фосфит щелочного металла:
2Р + 2NaOH + Н2О = Na2HPO3 + РН3
Ф. растворяется также в окисляющей его разбавленной (около 32%) азотной кислоте:
ЗР + 5HNO3 + 2Н2О = ЗН3РО4 + 5NO
Это удобный лабораторный метод получения фосфорной кислоты.
Необходимо — ради предосторожности — рассказать о реакции Ф. с бертоллетовой солью:
6Р + 5КС1О3 = ЗР2Об + 5КС1
Реакция сопровождается выделением большого количества тепла и света, поэтому её часто проводят юные химики, которые хотят поразить аудиторию сильным хлопком. Следует в самой категорической форме запретить выполнение этой реакции! Взрыв происходит даже при самом слабом трении или нажиме, часто совершенно неожиданно, без видимых причин, в момент приготовления смеси. В результате на кожу, а иногда и в глаза, попадает горящий Ф., который вызывает тяжёлые, трудно заживающие ожоги.
В описанные выше реакции вступают все модификации Ф., однако легче всего они проходят с белым Ф.
Химики давно заворожены удивительным свечением, наблюдающимся при окислении белого Ф. Эта реакция изучалась много и много раз.
В 1926—28 гг. процесс внимательнейшим образом исследовали советские учёные Н. Н. Семёнов и Ю. Б. Харитон (ныне академики, а тогда совсем молодые исследователи-энтузиасты). Были учтены все факторы, влияющие на процесс,— давление, температура, размеры сосуда, примеси... Оказалось, что окисление паров Ф. при низком давлении не подчинялось принятым в то время теоретическим представлениям. Выходили за рамки этих представлений и процессы окисления паров серы, водорода, окиси углерода (тоже при низких давлениях). Как доказал Н. Н. Семёнов, учёные столкнулись с новым типом химических процессов — цепными разветвлёнными реакциями.
О цепных реакциях мы рассказали в статьях Водород, Фтор, Хлористый водород. Для подробного же знакомства с этой областью химии у читателя-школьника ещё нет подготовки. Скажем только, что излучателем (источником фосфоресценции) при окислении паров белого Ф. является, по-видимому, неустойчивая молекула окиси РО. Она не существует при обычных условиях и обнаруживается лишь в парах (с помощью спектрального анализа).
Получение и применение. Основным сырьём для получения Ф. и его соединений служат фосфориты и апатиты. Измельчённая руда смешивается с коксом, который служит восстановителем, и кремнезёмом SiO2, необходимым для связывания в шлак содержащегося в руде кальция. Процесс ведут в электрических печах при температуре 1400—1600 °C. Суммарное уравнение реакции:
Са3(РО4)2 + 3SiO2 + 5С = 2Р + 3CaSiO3 + 5СО Выделяющиеся газы (пары Ф. и СО) отводят в орошаемые водой конденсаторы и затем собирают в приёмнике с водой, под слоем которой расплавленный Ф. и накапливается. Получаемый таким образом белый Ф. затем очищают и перерабатывают — в зависимости от назначения. Для некоторых целей (например, для получения полупроводников) нужен Ф. высокой степени чистоты (не менее 99,999—99,9999%).
Ежегодное производство Ф. велико (по некоторым прогнозам — около 1,3 млн. т в 1975 г.). Большая часть его расходуется на получение так называемой термической фосфорной кислоты, из которой затем производят концентрированные фосфорные удобрения — двойной су-
ФОСФОРИСТЫЕ КИСЛОТЫ
318
Рис. 2. Производство спичек не может обойтись без фосфора.
перфосфат, преципитат, нитрофоску и др. (см. Минеральные удобрения). Довольно много Ф. перерабатывается в полифосфаты, применяемые в синтетических моющих средствах в качестве умягчителей воды. Ещё один важный потребитель Ф.— спичечное производство.
Раньше головки спичек делали из белого Ф., при этом спички зажигались от трения о любую поверхность. Однако вследствие крайней ядовитости белого Ф. от такого способа отказались. Теперь головки спичек делают из смеси окислителя (например, бертоллетовой соли КСЮ3), восстановителя (серы) и различных наполнителей. Такая спичка зажигается лишь при трении о специально подготовленную поверхность — намазку спичечного коробка, в состав которой входит красный Ф. При трении головки о намазку спичечной коробки красный Ф. загорается, окисляясь бертоллетовой солью, и зажигает серу. Первые безопасные спички появились в конце XIX в. в Швеции, поэтому их часто называли шведскими спичками.
Во время первой и второй мировых войн белым Ф. снаряжали зажигательные бомбы и артиллерийские снаряды; кроме того, он использовался в специальных бомбах для образования дымовых завес; при взрывах таких бомб Ф. распыляется и сгорает с образованием фосфорного ангидрида Р2О5, переходящего от влаги воздуха в туман фосфорной кислоты.
В металлургии Ф. применяется как раскислитель (см., например, Железо, Сталь) и как компонент некоторых сплавов. Так, сплавы меди — оловянные бронзы, содержащие в качестве добавки фосфид меди Си3Р, обладают хорошими литейными и механическими свойствами, очень стойки к коррозии и имеют красивый внешний
вид. Фосфиды металлов служат для получения и легирования полупроводниковых материалов.
Особое место среди производных Ф. принадлежит фосфорорганическим соединениям. Большинство из них обладает биологической активностью — широко известны такие инсектициды (средства, применяемые для борьбы с насекомыми), как тиофос, хлорофос, карбофос и другие. Самостоятельный класс веществ составили фосфонитрил хлор иды (PNCl2)n, то есть:
С1 С1
I I
С1 С1
Когда молекулярная масса этого полимера достигает нескольких тысяч, получается каучукоподобное вещество. Такой, единственный в своём роде, «безуглеродный каучук» обладает значительной термостойкостью — начинает разрушаться лишь при температуре около 350 °C; к сожалению, однако, он разлагается под действием воды.
Итак, роль Ф. в природе и народном хозяйстве велика и многогранна. Было бы несправедливо не упомянуть и о той большой роли, которую сыграл Ф. в истории изучения радиоактивности. В 1934 г. известные французские физики Ирэн и Фредерик Жолио-Кюри открыли, что при бомбардировке алюминия а-частицами образуется новый, не существующий в природе радиоактивный изотоп 30Р. Так было открыто новое явление, получившее название искусственной радиоактивности. Получены и другие искусственные радиоактивные изотопы Ф.; важнейший из них 32Р (период полураспада 7^—14 дней} применяется в качестве «меченого атома», например для изучения различных биохимических процессов в организме животных и растений. в. д. Бельский.
ФОСФОРИСТЫЕ КИСЛОТЫ — кислородные кислоты, фосфора, отвечающие его степени окисления 43 и различающиеся между собой соотношением числа молекул фосфористого ангидрида Р2О3 и воды Н2О. При растворении В воде Р2О3 даёт слабую двухосновную орто-фтофосфористую кислоту:
Р2О3 + ЗН2О = 2Н3РО3
Н3РО3 в водном растворе — сильный восстановитель, при' нагревании выделяет водород:
Н3РО3 + Н2О = Н3РО4 + Н2
В свободном состоянии Н3РО3— кристаллическое вещество.При нагревании безводной Н3РО3 происходит так называемая реакция диспропорционирования (самоокисления-самовосстановления):
4Н3РО3 = РН3 + ЗН3РО4
Известны также метафосфориетая НРО2 и пирофосфористая Н4Р2Об кислоты (аналогично фосфорным кислотам). Соли Ф. к.—фосфиты.
319
ФОСФОРНЫЕ КИСЛОТЫ
ФОСФОРИСТЫЙ АНГИДРИД, трёх-окись фосфора, оксид фосфора (III), Р2О3 — ангидрид фосфористых кислот. Исследования показали, что структуре Ф. а. отвечает удвоенная формула Р40б. Ф. а.— белая, похожая на воск, кристаллическая масса, которая плавится при 23,8 °C, превращаясь в бесцветную жидкость. При взаимодействии с холодной водой Р40б даёт Н3РО3 (см. Фосфористые кислоты). Получают Ф. а. окислением белого фосфора при недостатке кислорода.
ФОСФОРИСТЫЙ ВОДОРОД (фосфин) РН3 — соединение фосфора с водородом. Ядовитый бесцветный газ с резким неприятным запахом «гнилой рыбы». На воздухе чистый и сухой Ф. в. загорается при нагревании выше 100—140 °C. Если же (как это часто бывает) Ф. в. содержит примеси дифосфина Р2Н4, он самовоспламеняется на воздухе. Смесь фосфинов иногда выделяется при разложении трупов на кладбищах; вспышки огоньков над могилами, обусловленные самовоспламенением фосфинов на воздухе, раньше очень пугали суеверных людей.
Ф. в. может быть получен действием воды на фосфиды'.
Са3Р2 + 6Н2О = ЗСа (ОН)'2 + 2РН3
Он хорошо растворим в воде, причём химически с ней не реагирует. При взаимодействии Ф. в. с некоторыми сильными кислотами в отсутствии воды образуются соли фосфония (подобные солям аммония), например:
РН3 + Н1 = РН41
Вода разлагает соли фосфония:
РН41 + Н2О = РН3 + [Н3О]+ + I-
ФОСФОРНОКИСЛЫЕ СОЛИ — то же, что фосфаты.
ФОСФОРНЫЕ КИСЛОТЫ — кислородные кислоты фосфора, отвечающие его степени окисления +5 и различающиеся между собой соотношением числа молекул фосфорного ангидрида Р2О5 и воды Н2О. При растворении фосфорного ангидрида в избытке воды образуется ортофосфорная кислота Н3РО4 — часто её называют просто фосфорной. Безводная ортофосфорная кислота — бесцветные кристаллы. Растворение Р2О5 в воде происходит в несколько стадий, см. Фосфорный ангидрид. Из графической формулы Н3РО4
Н —Ох
Н— О-^Р = О н—о/
видно, что она трёхосновна. Её соли — ортофосфаты бывают одно-, двух- и трёхзамещённые (см. Фосфаты).
В лаборатории Н3РО4 получают окислением элементарного фосфора 32%-ной HNO3:
ЗР + 5HNO3 + 2Н2О = ЗН3РО4 + 5NO
В промышленности Н3РО4 получают несколькими методами. Один из них, термический, заключается в восстановлении природных фосфатов до элементарного фосфора с последующим сжиганием его до пятиокиси Р2О5, которая в избытке воды образует фосфорную кислоту. В последнее время внимание химиков-производственников привлёк новый способ синтеза Н3РО4. Окисление фосфора производят водяным паром при температуре 600—900 °C в присутствии катализаторов (ими могут служить платина, цирконий, медь, титан):
2Р + 5Н2О = Р2О5 + 5Н2
В избытке водяных паров фосфорный ангидрид переходит в ортофосфорную кислоту, а выделяющийся при этом водород является ценным побочным продуктом. Ортофосфорная кислота — важнейший полупродукт для производства фосфорных удобрений (см. Минеральные удобрения). Очищенная, или так называемая пищевая, Н3РО4 нужна для приготовления фармацевтических препаратов, кормовых концентратов.
При упаривании ортофосфорной кислоты она, потеряв часть воды, переходит в другую Ф. к.— дифосфорную, или пирофосфорную:
2Н3РО4= Н4Р2О7 + Н2О
Её графическая формула:
О О
н-о II II /О-н
/Р—о—Р С н—ох о—н
Дифосфорная кислота Н4Р2О7
Н4Р2О7 — бесцветные кристаллы. Они хорошо растворяются в воде, давая более сильную кислоту, чем Н3РО4. Дифосфорная кислота—родоначальник целого семейства полифосфорных кислот, для которых характерно наличие нескольких атомов фосфора, связанных в линейную цепочку через кислородные мостики:
н—
н—ох
н—О
Н—О
Трифосфорная кислота Н6Р30ю
Тетрафосфорная кислота НбР4С13
фосфорный ангидрид
320
Для таких полифосфорных кислот наблюдается любопытная закономерность: сила кислоты возрастает с увеличением числа атомов фосфора.
Из школьного курса вам известна мет а фосфорная кислота НРО3. Однако эта привычная формула не отражает действительного строения соединения. На самом деле НРО3 полимеризована и её формулу следует писать (НРО3)„, где и от 3 до 8. Полимерные метафос-форные кислоты представляют собой бесцветную стекловидную массу, хорошо растворимую в воде. Образуются они, например, на первой стадии гидратации фосфорного ангидрида. Выделить же их можно при упаривании водных растворов ортофосфорной кислоты. Метафос-форные кислоты — очень сильные.
Чтобы отличить ортофосфорную кислоту от мета- и пирофосфорной, пользуются реакцией их солей с азотнокислым серебром AgNO3. В присутствии аниона ортофосфорной кислоты образуется жёлтый осадок, в присутствии анионов мета- и пирофосфорной — белый.
ФОСФОРНЫЙ АНГИДРИД, пяти-окись фосфора, оксид фосфора (V), Р2О5 — ангидрид фосфорных кислот. Простейшая формула Р2О5 не отражает истинного строения этого окисла, имеющего несколько модификаций. Так, одна из них — так называемая летучая — имеет состав Р40ю. Схема пространственного строения молекулы Р40ю дана на рисунке. Кристаллическая решётка этой модификации слагается из молекул Р40ю, связанных между собой лишь слабыми межмолекулярными силами, легко разрывающимися при нагревании. Отсюда и летучесть этой разновидности. Другие модификации полимерны. Они образованы бесконечными слоями тетраэдров РО4.
При сгорании фосфора в сухом кислороде или воздухе образуется обильный белый дым Ф. а. Он осаждается на стенках сосуда в виде
Молекулы Р4О10 построены из четырёх тетраэдров РО4, каждый из которых связан с тремя соседними через общие атомы кислорода (на рисунке один из них — верхний — отмечен пунктиром).
белого порошка. При этом получается смесь нескольких разновидностей, легко разделяемых при нагревании. Летучая P4Oi0 осаждается вне зоны нагревания в виде нежной массы мягких кристаллов. В пробирке же остаётся спёкшаяся масса нелетучего Ф. а. Она настолько тверда, что с трудом разбивается молотком. Обе разновидности гидратируются — присоединяют воду. Летучая — мгновенно, с вскипанием, сопровождающимся шипением, а нелетучая — медленно. В конце реакции образуется одно и то же соединение—ортофосфорная кислота Н3РО4, но через неодинаковые промежуточные продукты. Ниже приведена схема гидратации летучей
НО— Р^°^Р — он
+2HgO J \ +НгО
HO-^jP .Р^-ОН
(Z ° ЧО
ОН
I О=Р—ОН
I
о
I О=Р—он
I о
+н2о
Н3РО4 +
ОН I о=р—он о +н>°
I О==Р—он
I он
о—р—он I о
О=Р—он
он I О=Р—он
I о I О=Р—он
I о
О=р—он I он
+нго
4Н3РО4
он
—► 2Н3РО4
321
ФТОР
модификации. Реакция заключается в размыкании связей Р—О—Р. Сначала образуется четырёхосновная тетраметафосфорная кислота (НРО3)4 с кольцеобразным анионом, затем кольцо разрывается, обращаясь в четырёхзвенную цепочку. Далее от неё отщепляется звено за звеном в виде молекул Н3РО4.
Главное применение Ф. а. основано на его способности жадно поглощать воду. По осушающему действию Ф. а. превосходит все остальные вещества. Он отнимает от сложных веществ даже химически связанную воду. Так, например, из безводной хлорной кислоты получают её ангидрид:
4НС1О4 + Р4О10 = (НРО3)4 + 2С12О7
При работе с Ф. а. требуется особая осторожность.
ФРАНЦИЙ (лат. Francium) Fr — радиоактивный химический элемент I группы периодической системы Менделеева; атомный номер 87. Ф.— наименее устойчивый из всех радиоактивных элементов, встречающихся в природе. Единственный его природный изотоп 223Fr имеет период полураспада 22 мин. Другие изотопы Ф., полученные искусственно в различных ядерных реакциях, распадаются ещё быстрее.
Долгое время попытки открытия 87-го элемента оканчивались неудачами. Где только его ни искали: в минералах цезия и редкоземельных элементов (лепидолите и самарските), в солях цезия, в продуктах радиоактивного распада урана и тория! Иногда в печати появлялись сообщения об открытии стабильных изотопов элемента № 87, который одни исследователи называли «виргиний», другие — «Молдавию», третьи — «алкалиний». Однако проверки каждый раз показывали ошибочность этих сообщений. Честь открытия Ф. принадлежит Маргарите Перей (Франция, 1939 г.), которая детально исследовала характер радиоактивного распада ядер изотопа актиния ^Ас. До Перей считалось, что ядра 227Ас способны только к одному типу радиоактивного распада, связанному с испусканием 0“-частиц (при этом возникают ядра изотопа тория 227Th). Перей установила, что ядра 227Ас в 12 случаях из 1000 испускают не 0“-частицы (электроны), как обычно, а а-частицы (ядра гелия) и переходят в ядра элемента № 87
227 Др _. 223РГ | 41-Тр
8 97-1С * 871 1 I 21 1С
Новый элемент Перей назвала в честь своей родины.
Ф.— один из редчайших элементов. В поверхностном слое Земли толщиной 1,6 км его имеется всего лишь 24,5 г. Одно из самых больших количеств Ф., которое когда-либо имели в своём распоряжении исследователи, составляло около
5-10“13 г (изотоп 212Fr) и было получено советскими учёными в Объединённом институте ядерных исслёдований (г. Дубна, СССР) при облучении 1 г урана сильно ускоренными протонами. Из-за отсутствия стабильных и долгоживущих изотопов все свойства Ф. изучены на очень малых,индикаторных, как говорят химики, количествах этого элемента. По оценке, плотность Ф. 2,5 г/см3; температура плавления около 8 °C; температура кипения
около 620 °C. Конфигурация внешней электронной оболочки атома Ф. 7s1 (см. Атом). Исходя из положения Ф. в таблице Менделеева, можно было ожидать, что он представляет собой самый активный щелочной металл. Изучение свойств Ф. подтверждает это предположение. Химическое поведение Ф. больше всего похоже на поведение цезия. Почти все соли Ф. хорошо растворимы в воде, при кристаллизации Ф. хорошо осаждается вместе с солями цезия. Открытие Ф. позволило заполнить одно из последних пустых мест в периодической системе Менделеева между водородом и ураном.
С. С. Бердоносов.
ФТОР (лат. Fluoruni) F — химический элемент VII группы периодической системы Менделеева; атомный номер 9, атомная масса 18,99840; относится к семейству галогенов. Бледно-жёлтый газ с резким запахом. В природе встречается один устойчивый изотоп 19F.
Историческая справка.
Первым известным соединением Ф. был плавиковый шпат, флюорит CaF2. На
звание он получил за способность делать металлургические шлаки легкоплавкими и жидкотекучими (лат. fl ио —теку), что важно для полноты отделения металла от шлака. В 1771 г. перегонкой из смеси CaF2 и H2SO4 шведский химик К- Шееле получил кислоту, которую назвал плавиковой. Французский учёный А. Лавуазье назвал её флуориевой кислотой, полагая, что это соединение неизвестного элемента «флуория» с кислородом. В 1810 г. английский химик Г. Дэви электролизом соляной кислоты получил хлор, доказав тем самым, что эта кислота является соединением хлора с водородом. И тогда Дэви по аналогии высказал предположение, что плавиковая кислота — это соединение неизвестного элемента с водородом. В 1816 г. название «флуор» было заменено на «фтор» (греч. «фторос» — разрушение, гибель).
Получить Ф. в свободном состоянии долго не удавалось из-за его необычайной химической активности.
21 хэш
ФТОР
322
И лишь в 1886 г. французский химик А. Муассан выделил Ф. электролизом безводного фтористого водорода HF, к которому был добавлен бифторид калия KHF2. Ф. очень долго не находил практического применения. Ещё сравнительно недавно в «Технической энциклопедии» (изданной в Москве в 1936 г.) в статье о Ф. говорилось: «Из-за трудности получения и хранения фтор не имеет практического значения для промышленности». Интенсивное развитие химия Ф. получила лишь в годы 2-й мировой войны, когда для создания атомного оружия потребовалось отделение изотопов 236U от значительно более распространённого 238U (см. Уран). Наиболее эффективным оказался метод их разделения, где используется крайне летучий гексафторид урана UFe. А так как по своему разрушительному действию UFe лишь немного уступает Ф., то понадобились новые, сверхстойкие материалы — смазки, затворные и гидравлические жидкости, прокладки, шланги...
Было ясно, что вести поиск надо среди полностью «профторированных» соединений, то есть тех, что уже подверглись действию Ф. и больше ему не поддадутся. У американского химика Д. Саймонса, работавшего с соединениями Ф. ещё до войны, нашлось несколько миллилитров перфторгексана CgF14 (органическое соединение гексан СбН14, в котором все атомы Н замещены F). Как и ожидалось, перфторгексан оказался стабильным к действию UFe. Новые перфторированные вещества в честь Д. Саймонса в американском строжайше засекреченном атомном проекте получили кодовое название «вещества Джо». На основании полифторированных соединений углерода была создана новая область — химия фторорганических соединений. Стойкие и в химическом и в термическом отношении фторированные соединения имели, по выражению Саймонса, «алмазное сердце и шкуру носорога». В создании фторорганической химии в СССР немалая роль принадлежит академику И. Л. Кнунянцу, начавшему свои исследования примерно в то же время, что и американские учёные.
Фтор в природе. Содержание Ф. в земной коре 6,6-10”2% по массе; он — тринадцатый по распространённости элемент (а его аналог хлор— восемнадцатый). В свободном виде Ф. в природе не встречается, тогда как менее активный хлор в виде С12 входит в состав вулканических газов. Основное сырьё для получения Ф. и его соединений — флуорит CaF2, встречающийся в виде кристаллов, окрашенных в фиолетовые, жёлтые, зелёные и другие тона. Иногда, хотя и крайне редко, находят прозрачные бесцветные кристаллы флуорита, представляющие большую ценность для оптической промышленности. Ф. является составной частью природных фосфатов — апатитов и фосфоритов, например 3Ca3(PO4)2-CaF2-CaCl2, а также криолита Na3AlFe.
Небольшое количество Ф. (в виде ионов) содержится в природных водах, причём концентрация его в водах неодинакова в разных районах земного шара. Кроме того, Ф. входит в состав растительных и животных организмов, куда попадает вместе с водой и пищевыми продуктами. А если солей Ф. в питьевой воде слишком мало, то даже развивается заболевание — кариес зубов (его М9жно предотвратить добавкой фтористых солей в питьевую воду). Однако
и высокая концентрация Ф. в воде вредна для здоровья (оптимальная 1—1,5 мг/л). В некоторых растениях, произрастающих в Южной Африке и Австралии, содержится фторацетат калия FCH2COOK — сильный яд. Менее одного грамма свежих листьев таких растений достаточно, чтобы убить, например, барана.
Атом и молекула фтора. В соответствии с положением в системе Менделеева (второй период, седьмая группа) атом F имеет две электронные оболочки, и на второй — наружной — содержится семь электронов. Иными словами, полная электронная конфигурация атома F ls22s22p6, конфигурация внешней электронной оболочки 2s22p6 (см. также Атом). Для воссоздания устойчивой завершённой оболочки ближайшего инертного газа (неон, 2s22p6) атому F недостаёт лишь одного электрона. Поэтому в химических реакциях Ф. активно отнимает электроны у других атомов, молекул, ионов. Ещё большая активность Ф. (по сравнению с хлором и другими галогенами) объясняется тем, что внешняя электронная оболочка его атома находится ближе к ядру и в большей степени испытывает влияние (притяжение) положительного ядерного заряда. Приобретая электрон, атом F превращается в отрицательно заряженный ион F“, который способен соединяться ионной связью с положительно заряженными ионами других элементов. В свободном состоянии Ф. образует двухатомную молекулу F2. Ковалентная связь F—F в этой молекуле очень слаба и легко разрывается в различных реакциях. Ковалентные связи Ф. образует и в соединениях с другими элементами, близкими к Ф. по электроотрицательности, например во фторорганических соединениях (таблица электроотрицательностей приведена в статье Металлы).
Физические и химические свойства. Резкий запах Ф. похож на смесь запахов хлора и озона. При —188,13 °C газ конденсируется в жидкость жёлтого цвета,затвердевающую при —219,61 °C. При обычных условиях плотность газообразного Ф. 1,69 г/л.
В свободном состоянии Ф. обладает исключительно высокой химической активностью; достаточно сказать, что он реагирует почти со всеми химическими элементами. Реакции прямого соединения Ф. сопровождаются выделением большого количества тепла и легко переходят в горение и взрыв. Такова, например, реакция Ф. с водородом:
H2 + F2 = 2HF + 30,91 кдж/моль
Реакция эта, являющаяся примером важного класса разветвлённых цепных реакций, открытых и исследованных советским учёным академиком Н. Н. Се-
323
ФТОР
меновым, протекает по следующему механизму (точка над символами F и Н означает неспаренный электрон):
f2->f+f
f + h2 = hf+ Н
H + F2 = HF+F и т. д.
Реакционную цепь начинают активные частицы — свободные атомы F, в ничтожном количестве имеющиеся в молекулярном F2 или образовавшиеся под действием тепла и света. Число активных частиц, атомов F и Н, остаётся, как мы видим, постоянным и даже может уменьшаться: f+f = f2
н + н = н2 F + Н = HF
Но в результате реакций выделяется тепло, а в реакции Н + F2 = HF + F* его выделяется столь много, что образовавшийся атом F* становится возбуждённым (возбуждённый атом мы обозначили значком *), то есть очень быстрым, очень активным. Энергии возбуждения с избытком хватает, чтобы разбить непрочную молекулу Ф.:
F2+F*= 3F
В этой реакции число активных частиц увеличивается. Частицы эти дают начало новым реакционным цепям (цепи как бы «разветвляются»). Если скорость, с которой растёт число активных частиц, превышает скорость их гибели, то число реакционных цепей и реагирующих молекул возрастает как лавина — происходит взрыв. Если, напротив, скорость гибели активных частиц превышает скорость их роста, реакция затухает. А соотношение скоростей зависит и от давления, и от концентрации реагентов, и от наличия примесей, и даже от размера реакционного сосуда... Вот почему долгое время одни исследователи утверждали, что смесь водорода и фтора взрывается даже при низких температурах, а другим удавалось приготовить смеси, устойчивые при комнатной температуре.
О неразветвлённых цепных реакциях рассказано в статье Хлористый водород.
С щелочными и щелочноземельными металлами Ф. реагирует даже на холоду. Mg, Al, Zn, Fe, Cu, Ni и некоторые другие металлы в этих условиях стойки к действию Ф., так как на их поверхности сразу же образуется слой фторида (например, MgF2, A1F3), служащий защитой от дальнейшего взаимодействия. При нагревании же Ф. реагирует со всеми металлами, в том числе с золотом и платиной. Ф. образует ряд соединений с кислородом: OF2, O2F2, O3F2, O4F2. Это единственные соединения, при образовании которых атомы кислорода отдают электроны; поэтому их правильнее называть не окис-лами фтора, а фторидами кислорода (см. Номенклатура неорганических соединении). Соединения эти крайне неустойчивы и химически активны. O2F2, например, взрывается даже при соприкосновении с твёрдым снегом. При 200— 250 °C Ф. реагирует с С12, образуя C1F и C1F3. Во время 2-й мировой войны трёхфтористый хлор использовался в Германии для снаряжения зажигательных бомб (даже вольфрам и осмий горят в нём). Бром и иод воспламеняются в атмосфере Ф., при этом могут быть получены
BrF, BrF3, BrF5, IF<e; и IF7. С азотом Ф. соединяется лишь в электрическом разряде:
N2 + 3F2 = 2NF3
Образующийся при этом трифторид азота — очень интересное соединение. Раскалённей древесный уголь горит в нём энергичнее, чем в кислороде, а горение водорода в NF3 используют даже для сварки металлов. В отличие от необычайно взрывчатого NC13, трёхфтористый азот стабилен; его можно хранить в стальных баллонах под большим давлением и работать с ним можно при высоких температурах.
Сера реагирует с Ф. на холоду, образуя SF6, SF4 и другие фториды. Ф. как более активный элемент способен вытеснять кислород из окислов и другие галогены из галогенидов. Так, вода, древнейшее средство тушения пожаров, сама горит в струе Ф. бледно-фиолетовым пламенем:
2Н2О + 2F2 = 4HF + О2
Кислород здесь — продукт горения. Ярким пламенем горит в струе газообразного.Ф. «негорючий» асбест. Благодаря своей исключительной активности Ф. разрушил представление о химической пассивности инертных газов (об этом подробно рассказано в статьях Инертные газы и Ксенон).
Получение и применение. Ф. получают электролизом раствора бифторида калия KF-HF в безводном фтористом водороде HF. Вследствие высокой химической активности жидкого и газообразного Ф. аппаратура для работы с ним должна быть изготовлена из никеля или его сплавов; в некоторых случаях пригодны также медь, магний, алюминий, малоуглеродистая сталь. Перед началом работы аппаратура заполняется Ф. под небольшим давлением, поверхность металла фторируется («пассивируемся»), и защитная плёнка фторида предохраняет его от дальнейшей коррозии. Если же целостность покрытия из фторида металла будет по каким-либо причинам нарушена, может произойти самовозгорание. Поэтому ёмкости с Ф. помещают обычно за ограждение, а при работе используют дистанционное управление.
Ф. очень ядовит: вдыхание воздуха, содержащего более 10"4% Ф., смертельно. Строгие меры безопасности следует соблюдать и при работе с фторидами неметаллов — сильными окислителями. Говорят, что «фториды галогенов (C1F3, BrF3, IF5) менее опасны, чем свободный Ф., так же как гремучая змея менее опасна, чем кобра». Исследование биологической активности Ф. и его соединений — одно из важных направлений биологии и химии.
При реакции Ф. с углеводородами, аминами, аммиаком и в особенности с водородом выделяется большое количество тепла, что позволяет применять Ф. в качестве высокоэффективного
21*
ФТОРИДЫ 324
окислителя для ракетных топлив. Газообразный Ф. используют для получения UFe (см. Фториды), для получения SFe (лучшего газообразного диэлектрика) и многих других соединений. В различных областях народного хозяйства применяются также другие соединения Ф., такие как A1F3 и Na3[AlFe] (см. Алюминий), HF (см. Фтористый водород), BF3 (см. Бор).
Особенно же интенсивно развивается в последнее время химия фторорганических соединений. Более крупные по сравнению с водородом атомы F создают вокруг атомов С в органических веществах своеобразный экран, предохраняя углеродную цепь от разрушения при воздействии агрессивных веществ. Кроме того, сама связь С—F более прочна по сравнению со связью С—Н. Всё это делает фторированные углеводороды исключительно химически и термически стойкими. Изделия из политетрафторэтилена (технические названия «тефлон», «фторопласт-4») не разрушаются в кипящей азотной кислоте, серной кислоте, щелочах, хлоре, фтористом водороде. Вот сравнение: электроизоляция из обычных материалов, например из резины или полиэтилена, обеспечивает нормальную работу аппаратуры до 85 °C; обмотка же из фторопласта позволяет увеличить температуру до 200 °C, а кратковременно и до 250 °C.
Тефлон способен резко снижать трение между соприкасающимися поверхностями. Поэтому тефлоновой плёнкой покрывают лыжи самолётов полярной авиации. Да и у горнолыжников на смену тяжёлым деревянным лыжам пришли лёгкие дуралевые (см. Алюминиевые сплавы), покрытые тонким слоем тефлона. Благодаря химической инертности, низким температурам замерзания и абсолютной безвредности для организма фторированные и хлорфторированные метаны и этаны (техническое название «фреоны») служат хладоагентами в холодильниках. Лёгкая сжижаемость газообразных фреонов даёт возможность производить в огромных количествах так называемые аэрозольные упаковки.
Вещество, которое употребляется в распылённом состоянии, например краситель, упаковывается в небольшой баллончик с тонким отверстием, закрываемым клапаном. При нажатии на клапан отверстие открывается, давление в баллончике падает, фреон начинает испаряться, струя его паров с большой скоростью выходит из отверстия, захватывая мелкие капельки распыляемого вещества. Не нужен ни пульверизатор, ни источник сжатого воздуха. В подобных аэрозольных упаковках и у нас и за рубежом продаются красители, инсектициды, репелленты (средства, отпугивающие насекомых, например хорошо известная туристам «дэта»), лаки для женских причёсок и кремы для тортов.
Химия Ф. дала и ряд ценных лекарственных препаратов. Совершенно безвредное обезболивающее (анестезирующее) средство фто-
ротан CF3CHBrCl используется даже в детской хирургии. 5-фторурацил и его производные подавляют рост многих злокачественных опухолей.
Уникальные свойства Ф. и колоссальные успехи в синтезе и изучении его соединений дают нам право утверждать: Ф. окажет человечеству ещё много услуг в самых различных областях практики — от ядерной техники до лекарственной химии. И. И. Кандрор.
ФТОРИДЫ — соединения фтора с другими химическими элементами. Поскольку фтор — самый электроотрицательный элемент, его атом, связанный с любым другим атомом, всегда заряжен отрицательно. Ф. большинства металлов — ионные соединения. Эти Ф. образуют прочные кристаллические, решётки, обладают высокими температурами плавления и являются типичными солями. Одни из них хорошо растворимы в воде (например, фториды Na, К, Ag, Hg), другие — труднорастворимы. Ввиду способности иона фтора F“ к образованию весьма прочной связи с ионом водорода Н4' (об этом рассказано в статье Фтористый водород) Ф. щелочных металлов очень гигроскопичны и легко образуют кислые соли —так называемые бифториды, например KHF2. Очень важным свойством обладают Ф. переходных металлов в их высшем валентном состоянии (о переходных металлах рассказано в статье Металлы). Это — ковалентные соединения. Их особенность — слабое взаимное притяжение молекул и как следствие этого летучесть. На летучести гексафторида урана UFe основано разделение изотопов урана. Ф. неметаллов — типичные ковалентные соединения, в свободном виде это жидкости или газы. Окисляющая способность фтора настолько высока, что он образует устойчивые фториды даже с инертными газами. Ф. неметаллов — сильные окислители. О некоторых из них рассказано в статье Фтор.
И. И. Канд pop.
ФТОРИСТОВОДОРОДНАЯ КИСЛОТА (плавиковая кислота) — раствор фтористого водорода HF в воде.
ФТОРИСТЫЙ ВОДОРОД HF — соединение фтора с водородом. Бесцветная легкоподвижная жидкость с резким запахом. На воздухе Ф. в. сильно дымит, образуя с влагой воздуха мельчайшие капельки раствора. Раствор Ф. в. в воде называется фтористоводородной кислотой (или плавиковой кислотой). Безводный Ф. в. кипит при 19,43 °C, затвердевает при —83 °C; по плотности (0,991 г/см3) и другим физическим свойствам напоминает воду. Подобно воде, Ф. в. полимеризован за счёт водородных связей (в формуле они изображены пунктиром).
325
ФТОРИСТЫЙ ВОДОРОД
F ...F
HZ ХН. н‘‘ н н‘‘
’’F^ ’’F *’F
В статье Вода подробно рассказано о том, что водородные связи служат причиной аномальных свойств Н2О по сравнению со свойствами H2S, H2Se, Н2Те. То же — ив случае Ф. в. По сравнению с другими галогеноводородами у него относительно высокая температура кипения (/КИПНС1 — 85,1 °C, НВг —68,7 °C, HI —35,9 °C), а кислотность и электропроводность ниже.
В отличие от довольно слабой плавиковой кислоты, безводный Ф. в. проявляет сильные кислотные свойства. Он легко реагирует с металлами, стоящими в ряду напряжений до водорода, например:
Zn + 2HF = ZnF2 + Н2
Исключение составляют те металлы, на поверхности которых образуется плёнка нерастворимого фторида, в том числе алюминий, магний, свинец, железо, никель.
Безводный Ф. в. служит хорошим растворителем для многих органических и неорганических соединений. При пропускании электрического тока через растворы органических соединений в HF водород в этих соединениях полностью замещается фтором. Электрохимическое фторирование — один из важнейших методов получения сполна фторированных органических соединений (см. Фтор).
Ф. в. реагирует со многими кислотными окис-лами. Так, с серным ангидридом SO3 он обра
зует фторсульфоновую кислоту HSO3F — одну из самых сильных кислот:
SO3 + HF = HSO3F
При действии Ф. в. на двуокись кремния SiO2 образуется газообразный тетрафторид кремния;
SiO2 + 4HF = SiF4 + 2Н2О
SiF4, взаимодействуя с избытком HF, даёт кремнефтористый водород—непрочное соединение:
SiF4 + 2HF = H2SiFe
Эта реакция используется и для травления стекла, и для удаления песка (SiO2) с металлического литья.
Стеклянные изделия покрывают защитным слоем парафина, на котором процарапывают необходимые рисунки и надписи. А затем их обрабатывают Ф. в. На тех местах, где парафина не было, остаётся матовый рисунок — след стекла, удалённого в виде SiF4 или H2SiF6.
В промышленности Ф. в. получают нагреванием плавикового шпата с концентрированной серной кислотой:
CaF2 + H2SO4 = CaSO4 + 2HF
Важнейшая область применения Ф. в.— производство фтора и фторорганических соединений. С помощью Ф. в. поручают искусственный криолит Na3[AlF6], необходимый для производства алюминия.
Ф. в. и плавиковая кислота очень ядовиты и вызывают болезненные трудно заживающие ожоги, опасные повреждения глаз и верхних дыхательных путей. Работа с ними требует исключительной осторожности (хорошая вентиляция, защитная резиновая одежда, защитные очки). И. И. Кандрор.
«... Хи миЯ часто одаряла меня величайшими наслаждениями познания ещВ неразведанных тайн природы. Она дала мне возможность послужить людям, облегчить их труд, избавить их от некоторых страданий, порой — от гибели. Она помогла мне стать человеком небесполезным для моей Родины.,.'».
Н, Д, Зелинский
(Зелинский Н. Д., Обращение по радио к молодёжи 16 января 1941, см. Собр. трудов, т. 4, I960, с. 578)
ХЛОР (лат. Chlorum) Cl — химический элемент VII группы периодической системы Менделеева; атомный номер 17, атомная масса 35,453; относится к семейству галогенов. Жёлто-зелёный газ с резким запахом. В природе X. представлен двумя устойчивыми изотопами — 35С1 (75,53%) и 37С1 (24,47%).
Историческая справка. Соединения X. известны не одну тысячу лет; трудно установить, когда именно наши предки познакомилась с каменной солью NaCl или нашатырём NH4C1. Но СсМ элемент «молод», ему всего 200 лет. В 1774 г. шведский химик К- Шееле, действуя соляной кислотой на минерал пиролюзит МпО2 (см. Марганец), обнаружил выделение жёлто-зелёного газа с характерным запахом. Далеко не сразу выяснилось, что открыт новый элемент. Так, французский химик А.. Лавуазье считал этот газ окислом некоего неизвестного элемента мурия (от лат. muria — рассол, солёная вода) и называл его окисленной соляной кислотой. И только в 1810 г. английский химик Г. Дэви после многократных тщетных попыток обнаружить присутствие кислорода в исследуемом газе установил, что это вещество — простое. Новому элементу Дэви дал название «chlorine» (от греческого «хлорбс» — жёлто-зелёный). Вскоре французский химик Ж. Л. Гей-Люссак сменил его на «хлор», которое стало принятым всюду, кроме стран английского языка, где сохранилось название, данное Дэви.
Хлор в природе. X. широко распространён в природе; в земной коре его 1,7* 10-2% по массе (18-е место среди всех элементов). Из-за своей высокой химической активности X. встречается в природе не в свободном состоянии, а только в виде соединений с металлами. Наиболее известные его минералы — это галит (каменная, или поваренная, соль) NaCl, сильвин КС1, сильвинит (Na,K)Cl, карналлит КС1 • MgCl 2 • 6Н 2О, каинит КС1 • MgSO4 • ЗН 2О, бишофит MgCl2*6H2O (об этих минералах см. в статьях Натрий, Калий, Магний) и др. Мировые запасы каменной соли в недрах Земли выражаются астрономическим числом 3,5-1015 т. Каждый год добывают десятки млн. т NaCl, из них 5 млн. т идёт в пищу. Об исключительно важном значении этой соли для организма рассказано в статье о натрии. Очень много солей X. растворено в водах океанов, морей, рек и озёр, например в Мёртвом море, озёрах Балхаш, Эльтон, Баскунчак и др. Полный объём солей X. в водах Мирового океана, по оцен
ке некоторых учёных, 22 млн; км3 — это даже трудно себе представить . Испар я ясь, такие .воды оставляют после себя пласты соли и даже целые соляные горы. Например, гора Ходжа-Му-мын на юге Таджикистана, сплошь состоящая из каменной со-
ли, возвышается на 900 м над уровнем моря.
Физические и химические свойства. Молекула X. двухатомна (С12). В обычных условиях газ почти в 2,5 раза тяжелее воздуха (1 л С12 весит 3,214 г). При нормальном давлении X. переходит в жидкое состояние при —33,6 °C, а затвердевает при —100,98 °C; при обычной температуре легко сжижается под давлением. X. растворяется в воде с образованием желтоватой хлорной воды (1 объём Н2О поглощает около 2 объёмов С12 при комнатной температуре). Если хлорную воду сильно охладить, можно увидеть образование жёлтых кристаллов состава С12*7,ЗН2О. Такая на первый взгляд странная формула соответствует строению этого кристаллогидрата: его основу составляет кристаллическая решётка льда, в пустотах которой между молекулами Н2О «внедрились» молекулы С12. Кристаллогидрат X. относится к числу клатратных соединений; см. о них в статье Инертные газы. Хорошо растворим X. в органических растворителях, особенно в гексане CeHi4 и четырёххлористом углероде СС14.
X.— очень ядовитый газ. Он раздражает слизистую оболочку дыхательных путей даже в таких малых концентрациях, как 0,001 мг на 1 л воздуха. Первой помощью при отравлении X. служит вдыхание кислорода, паров спирта с эфиром или аммиака (из его водного раствора— нашатырного спирта).
В соответствии с положением X. в периодической системе его атом имеет 7 внешних (валентных) электронов (конфигурация 3$2ЗрБ, см. Атом). Для достижения устойчивой 8-элек-тронной оболочки ближайшего инертного газа— аргона атому С1 не хватает лишь одного элек-
327
ХЛОР
трона; поэтому X. охотно вступает в реакции, в которых его атом приобретает электрон: о _ - ’
Cl + е = С1
Помимо соединений со степенью окисления атома С1 —1 (NaCl и другие соли — хлориды), известны и такие, где С1 имеет положительные степени окисления: +1, +3, +4, +5, +6 и +7. Естественно, что атом С1 в этих случаях должен быть связан с ещё более электроотрицательными атомами (N, О или F). Таковы, например, + 1 +3 +4 +Ь +6
соединения FC1, NC13, С1О2, КС1О3, С12О6,
NaC104.
X. принадлежит к числу весьма активных окислителей. Он непосредственно реагирует со многими металлами и неметаллами:
2Na+ Cl2 = 2NaCl
2Р + ЗС12 = 2РС13 Н2+ С12 = 2НС1
О получении по этой реакции НО подробно рассказано в статье Хлористый водород. В приведенных примерах X. реагирует с другими элементами как окислитель, отнимающий электрон. И есть только одна единственная реакция свободного X. с другим простым веществом, где X. выступает как восстановитель:
О 0 +3—1
C12 + 3F2 = 2C1F3
(взаимодействие идёт при 280 °C). С кислородом и азотом X. непосредственно не реагирует и его соединения с этими элементами получаются косвенными путями.
Например, СС14 (четырёххлористый углерод) можно синтезировать последовательным замещением атомов Н атомами С1 в метане:
СН4 + С12 = СН3С1 + НС1 (хлористый метил) СН3С1 + С12 = СН2С12 + НС1 (хлористый метилен)
СН2С12 + С12 = СНС13 + HCi (хлороформ) СНС13-|- С12 = СС14 + НС1 (четырёххлористый углерод)
Реакции такого типа называются металептическими, а само последовательное замещение атомов водорода в насыщенном углеводороде (метане, этане и т. д.) атомами галогена получило название металепсии (от греческого «металепсис» — замена).
Хлористый азот NC13 (жёлтая маслянистая жидкость с резким запахом) образуется, в частности, действием X. на насыщенный раствор нашатыря:
NH4C1 + 3Cl2 = NCi8 + 4НС1
Французский химик П. Л. Дюлон, открывший в 1811 г. NC13, не знал о его сильной взрывчатости и жестоко пострадал, лишившись глаза и трёх пальцев.
Особый интерес представляют собой кислородные соединения X. При взаимодействии свободного X. и воды образуется хлористый
водород НС1 и хлорноватистая кислота НОС1: С12 + Н2О Г НС1 + НОС1 х
^2Н+ + С1- + ОС1-
Эта реакция обратима. В состоянии равновесия в растворе присутствуют все четыре вещества. Сдвинуть равновесие вправо можно добавлением щёлочи к раствору. Тогда ионы гидроксила ОН" соединятся с ионами Н+ в молекулы Н2О и концентрация ионов Н+ в правой части уравнения понизится. Чтобы вернуть состояние равновесия, всё больше и больше молекул С12 и Н2О будут вступать во взаимодействие. Если же X. пропускают в раствор щёлочи, то вместо смеси двух кислот образуется смесь их солей — хлорида (NaCl) и гипохлорита (NaOCl):
С12 + 2NaOH = NaCl + NaOCl + Н2О
Далее, от хлорноватистой кислоты (содержащей +1
С1.) можно перейти к кислородсодержащим соединениям, где атом С1 имеет степени окисления +5 и -F7. Нагреванием раствора НОС1 получают хлорноватую кислоту:
+1 +5 -I
ЗНОС1 = НС1О3 + 2НС1
При нагревании солей этой, кислоты — хлора-
Получение хлора в лаборатории. Из капельной воронки небольшими порциями в колбу, где находится двуокись марганца МпО2, добавляют концентрированную соляную кислоту НС1. При осторожном нагревании реакционной смеси выделяется хлор. Его очищают от примеси хлористого водорода, пропуская через промывал-ку с водой (С12 растворяется в воде значительно хуже, чем НС1). Газообразный хлор более чем в 2 раза тяжелее воздуха, поэтому его можно собрать в пробирки так, как это показано на рисунке. Хлор ядовит, поэтому опыт следует проводить в вытяжном шкафу.
ХЛОР
328
тов образуются соли хлорной кислоты — перхлораты:
+ 5-2 +7—2 - 1 О
2КС1О3 = КС1О4 + КС1 + О2 Известна ещё одна кислородсодержащая кис-+ 3
лота — хлористая кислота НСЮ2 и её соли — хлориты. Помимо кислот, известны также и бинарные соединения X. с кислородом.
С12О — окись X., жёлто-коричневый газ с запахом, похожим на запах X., может быть получен взаимодействием С12 и окиси ртути:
С12 + HgO = С12О + Hg
С1О2 — двуокись X., зеленовато-жёлтый газ; получается действием хлора на хлорит натрия:
2NaC102 + Cl 2 = 2NaCl + 2С1О2
С12Об — дымящая жидкость тёмно-красного цвета; образуется при взаимодействии хлоратов с фтором: 2КС1О3 + F2 = 2KF + С12Об
С12О7 — бесцветная жидкость; получается при действии на хлорную кислоту НС1О4 водоотнимающими агентами:
(Р2Об)
2НС1О4—-+С12О7 + Н2О
С12О является ангидридом хлорноватистой кислоты: С12О + Н2О = 2НОС1
С12О7 — ангидрид хлорной кислоты: С12О7 + Н2О = 2НС1О4 А два других окисла ведут себя сложнее, каждый из них— ангидрид одновременно двух кислот:
+ 4 +3 +5
2С1О2 + Н2О = НСЮ? + НС1О3 +6 +5 +7
С12Ов + Н2О = НС1О3 + НС1О4
Последняя реакция становится понятной, если учесть, что С12Ов содержит атомы хлора в разных степенях окисления (+5 и +7): °х+5 +7/^°
2>С1— О—С1=О
По ряду + 1 +3 +5 +7
нею -+ нсю2 нею. нею.
возрастает устойчивость кислот и их солей, усиливаются кислотные свойства, и наоборот, с ростом степени окисления уменьшается окислительная активность.
Получение. Производство хлора — одно из самых крупнотоннажных в химической промышленности. По оценочным данным, в 1975 г. в мире будет добыто около 32 млн. т X. Сущность процессов сводится к следующему. Сильный окислитель, действуя на соединения, содержащие ион С1“, отнимает у него «лишний» электрон; получается свободный хлор:
2NaCl + F2 = 2NaF + Cl2
Самое важное в методике получения — выбор окислителя; выбор же определяется масштабом процесса. В лабораторных условиях, когда нуж
но сравнительно легко и быстро получить небольшие количества X., используются энергичные окислители, например перманганат калия КМпО4, двуокись марганца МпО2 или дихромат калия К2Сг2О7. Реагируя с соляной кислотой, —1
они окисляют С1 до свободного X.:
2КМпО4+16НС1 = 5С12+2КС1+2МпС12+8Н2О
МпО2+4НС1=С12+МпС12+2Н2О
К2Сг2О7+14НС1-ЗС12+2СгС13+2КС1+7Н2О
Естественно, что в промышленных условиях, когда речь идёт о миллионах тонн X., применение больших количеств таких дорогих химических окислителей невыгодно. В этом случае на помощь приходит электрический ток.
В широко распространённом электролитическом методе получения X. исходным сырьём служит природный хлористый натрий, запасы которого практически неисчерпаемы. При электролизе раствора NaCl на аноде идёт окисление иона С1“ до свободного X.:
_ о 2С1- — 2е = С12
а на катоде — восстановление иона Н+ до свободного водорода:
__ о 2Н+ + 2е = Н2
В растворе остаются ионы Na+ и ОН“, то есть едкий натр. Суммарно реакцию электролиза раствора поваренной соли можно записать так:
2NaCl + 2Н.0 — ™ CL + Н2 + 2NaOH
Схему процесса электролиза вы найдёте в статье Натрий. Здесь, образно говоря, «одним ударом убивают сразу трёх зайцев» — по одной реакции одновременно получаются три важнейших продукта химической промышленности — хлор, водород и едкий натр. Процесс этот далеко не прост. Технологам приходится решать множество конструкционных задач, связанных с типом электролизных ванн, видом, формой и расположением электродов, характером подачи тока, методикой отвода продуктов реакции... Полученный X. сжижают и помещают в стальные баллоны под давлением 6 атм.
Применение. Использование X. идёт по двум путям. В первом важны окислительные свойства X., и прежде всего его отбеливающая способность. На отбелку тканей, целлюлозы и бумаги идёт примерно 15—20% получаемого X.
Проделаем такой опыт: в две банки, заполненные сухим X., внесём две тряпочки, испачканные чернилами: одну — сухую, другую — влажную. Через некоторое время оказывается, что на влажной тряпочке следы чернил исчезли, а сухая не изменилась. Влажный X. отбеливает, а сухой нет. Это связано с взаимодействием X. и воды (см. выше); образующийся гипохлорит-ион ОС1~ более сильный окислитель, чем молекулярный С12
329
ХЛОР
Клод Луи БЕРТОЛЛЕ — один из основателей учения о химическом равновесии. Открыл белящее действие хлора.
Французский химик Клод Луи Бертолле родился 9 декабря 1748 г. в селении Таллуар (Савойя). В молодости Бертолле полагал, что его удел — врачевание. В 1770 г. он получил степень доктора медицины Туринского университета. В 1772 г. поселился в Париже, где занял должность врача при дворе герцога Орлеанского. В распоряжении Бертолле оказалась химическая лаборатория, устроенная ещё дедом герцога. В ней он и начал научные работы. Большую роль в становлении Бертолле как химика сыграла его дружба с А. Лавуазье.
В 1785 г., первым из французских химиков, он примкнул к антифлогисти-ческой химии Лавуазье. Вместе с Лавуазье и другими учёными он в 1786— 1787 гг. участвовал в создании новой номенклатуры химических соединений и в 1789 г. в основании существующего и ныне французского химического журнала «Анналы химии». В годы Великой французской революции он много сделал для развития производства селитры, пороха, стали и для организации новых высших школ — Политехнической и Нормальной. Здесь Бертолле читал курсы химии.
В 1798—1799 гг. Бертолле в числе многих учёных был участником Египетской экспедиции Бонапарта и членом организованного в Каире Египетского института. Вместе с французскими солдатами делил тяготы походов и боёв. При виде противника, по команде «Батальонам построиться в каре, учёных и ослов на середину!», войска строились в виде прямоугольников, ограждая тех, кого надо было особенно беречь: учёных и тягловую силу — ослов. По возвращении из Египта Бертолле поселился в парижском предместье Аркёй.
Большую известность доставило Бертолле его предложение (1785 г.) использовать хлор для беления полотна и других тканей. До того сырое полотно, подлежащее белению, расстилали на земле и ждали, пока под действием солнечных лучей и дождей оно не станет чисто белым. Такое беление длилось иногда несколько месяцев и занимало большие пространства, годные для лугов и пашен. Сначала у Бертолле служил для беления газообразный хлор, но в 1787 г. учёный заменил его более удобной белильной жидкостью; она получалась при пропускании хлора в холодный разбавленный раствор едкого кали (или поташа). (При этой реакции образуется гипохлорит калия; он очень легко отдаёт атомарный кислород, который и производит беление.) Этот способ позволил сильно увеличить производительность текстильных фабрик и освободить для сельского хозяйства большие земельные участки.
Пропуская хлор в концентрированный раствор едкого кали, Бертрлле впервые получил хлорат калия — бертоллетову соль. Учёный попробовал заменить ею селитру при производстве пороха, но попытка кончилась взрывом с человеческими жертвами.
Бертолле нашёл, что сероводород состоит из водорода и серы, а синильная кислота — из водорода, углерода и азота. Тем самым он показал существование бескислородных кислот, что получило общее признание лишь около середины XIX в.
Во время пребывания в Египте учёный обратил внимание на образование натрона (природной соды) при действии воды озёр, содержащей хлорид натрия, на известняк. Эти наблюдения заставили его по-новому взглянуть на проблему «химического сродства» (так называли в то время причину, побуждающую вещества к химическому взаимодействию). Химическое сродство считалось избирательным свойством, некоей постоянной величиной, присущей тому или иному веществу. Бертолле показал, что химическое сродство зависит от массы реагирующих веществ и от условий реакции (например, температуры). Об этом он сделал доклад в Египетском институте в июле 1799 г. В работах «Исследования законов сродства» (1801 г.) и «Опыт химической статики» (1803 г.) Бертолле изложил свои взгляды на химические реакции как на равновесный, обратимый процесс, зависящий прежде всего от относительной массы взаимодействующих веществ. Отсюда он сделал вывод, что и состав образующихся соединений должен быть переменным. Бертоллё отстаивал это мнение в длительной полемике с французским химиком Ж. Л. Прустом, утверждавшим, что состав химических соединений постоянен и не зависит от способа их получения. Спор учёных, длившийся с 1801 по 1808 г., закончился установлением закона постоянства состава. Только в начале XX в. Н. С. Курнаков показал, «что обе стороны правы в своих утверждениях, но что точка зрения Бертолле является более общей». Бертолле был членом Парижской академии наук. Он умер 6 ноября 1822 г. в предместье Парижа Аркёй.
ХЛОРАТЫ
330
Известно, что энергичное окисление — это метод уничтожения болезнетворных бактерий, поэтому 4—5% С12 используют для санитарных целей. Примером служит обеззараживание воды (хлорирование), идущей для бытовых нужд (питьевая вода, вода для бассейнов и бань и т. п.).
Другой путь применения X.— синтез раз
личных органических и неорганических соединений. В области неорганической химии —
это синтез хлористого водорода и получение соляной кислоты, синтез хлоридов разных металлов. В области органической химии — полу
чение важнейших растворителей — хлороформа СНС13, дихлорэтана С2Н4С12 и четырёххлористого углерода СС14; синтез действенных инсектицидов и зооцидов (ядов для вредных насекомых и животных) — гексахлорана СвНвС1в, дихлордиметилтрихлорметилметана (а проще, всем известного ДДТ), хлорофоса и др. Примерно 60—70% добываемого X. идёт для получения
таких известных полимеров, как полихлорвинил (винипласт), хлоропреновый каучук, волокно хлорин, искусственной кожи — пави
нола и др.
В этой статье рассказано о самом элементе X. О его не менее интересных соединениях, их свойствах, методах получения и путях использования можно прочесть в статьях: Хлористый водород, Соляная кислота, Царская водка, Хлориды, Хлорноватистая кислота, Гипохлориты, Хлористая кислота, Хлориты, Хлорноватая кислота, Хлораты, Хлорная кислота, Перхлораты. В. К- Бельский.
ХЛОРАТЫ — соли хлорноватой кислоты НС1О3. В воде, как правило, хорошо растворимы. Как и сама кислота, X.— сильные окислители. При нагревании разлагаются:
4КС1О3 = 4КС1 + 6О2
4КС1О3 = КС1 + ЗКС1О4
Первая реакция преимущественно идёт в присутствии катализатора (обычно это двуокись марганца MnO2). X. можно получить, пропуская хлор через горячий раствор щёлочи:
ЗС12 4- 6КОН = 5КС1 4- КСЮз 4- ЗН2О
В промышленности X. получают электролизом горячих растворов хлоридов.
Наибольшее практическое применение имеет хлорат калия КС1О3, обычно называемый бер-толлетовой солью. Большая часть вырабатываемого КСЮз потребляется для производства спичек (см. также Фосфор), пиротехнических составов, взрывчатых веществ, пороха, сигнальных ракет и др. В отличие от КС1О3, хлорат натрия NaC103 имеет гораздо более «мирное» применение. Его растворы в воде —
отличное средство для уничтожения сорняков, им обрабатывают железнодорожное полотно, чтобы избавиться от сорных трав.
Все X. очень ядовиты, они разрушающе действуют на кровь. А если учесть, что сухие смеси X. с легко восстанавливающимися веществами, особенно с органическими, взрываются при ударе, то ясно, что при работе с этими солями надо соблюдать особую осторожность.
ХЛОРЙДЫ — соединения хлора с элементами, имеющими меньшее значение электроотрицательности, то есть со всеми металлами и неметаллами, кроме азота, кислорода и фтора.
- Электроотрицательность хлора равна 2,83, азота 3,07» кислорода 3,50 и фтора 4,10. Поэтому соединения хлора с кислородом и фтором называют не хлоридами этих элементов, а окислами и фторидами хлора. Однако NC13 * принято называть хлоридом азота (III) или хлористым азотом. Если элемент образует с хлором только одно соединение, его называют X. или хлористым, например NaCl — хлорид натрия или хлористый натрий. Если элемент образует с хлором два соединения или больше, то к названию элемента прибавляют в скобках римскими цифрами его валентность, например РС13 — хлорид фосфора (III), РС15 — хлорид фосфора (V).
X. металлов (или соли соляной кислоты) — твёрдые вещества, плавящиеся или возгоняющиеся без разложения. Впрочем некоторые X. при нагревании частично или полностью теряют хлор. Так, PtCl4 превращается в PtCl2 и С12, а АиС13 разлагается на Аи и С12. В воде X. металлов хорошо растворимы, кроме AgCl, CuCl, Hg2Cl2, Т1С1 и РЬС12. Водные растворы X. щелочных и щелочноземельных металлов имеют нейтральную реакцию. Растворы X. прочих металлов имеют кислую реакцию вследствие гидролиза:
А1С13 + ЗН2О = А1(ОН)3 4- ЗНС1
X. неметаллов могут быть газообразными (НС1), жидкими (РС18, SiCl4) или твёрдыми (РС1Б). Все они, кроме НС1, легко гидролизуются:
РС1Б + 4Н2О = Н3РО4 4- 5НС1
X. натрия, калия и некоторые другие встречаются в природе в виде минералов и в растворённом состоянии. Об этом, а также о промышленном применении хлоридов см. в статьях: Натрий, Калий, Магний, Кальций, Барий, Алюминий, Серебро, Цинк, Ртуть.
ХЛОРИСТАЯ КИСЛОТА НС1О2 — одноосновная неустойчивая кислота, в которой хлор имеет степень окисления 4-3. Графическая формула Н—О—С1=О. X. к. существует только в растворе. При хранении разлагается: ЗНС1О2 = НС1 4- 2НС10з
Получается одновременно с хлорноватой кислотой при растворении в воде двуокиси хлора: 2С1О2 4- Н2О = НС1О2 4- НСЮ8
331
ХЛОРИСТЫЙ ВОДОРОД
Сама X. к. практического применения не находит, в промышленности используются её соли — хлориты.
ХЛОРИСТОВОДОРОДНАЯ КИСЛОТА — раствор хлористого водорода НО в воде; то же, что соляная кислота.
ХЛОРИСТЫЙ ВОДОРОД НО — соединение хлора с водородом. Бесцветный газ с резким запахом. При нормальном давлении X. в. переходит в жидкое состояние при температуре —84,8 °C и затвердевает при —114,2 °C. Он несколько тяжелее воздуха— 1 л X. в. весит 1,64 г. Молекула НО очень прочна, лишь при 1500 °C она начинает распадаться на хлор и водород. X. в. хорошо растворяется в воде; при обычных условиях 1 объём воды поглощает около 450 объёмов X. в. Чтобы показать это наглядно, надо собрать прибор, изображённый на рисунке. Раствор НС1 в воде называется соляной кислотой. На влажном воздухе X. в. дымит, образуя с парами воды мелкие капельки соляной кислоты.
В отсутствии влаги X. в. неохотно вступает в химические реакции. При нагревании на воздухе до 400—500 °C в присутствии катализатора CuCl2 X. в. горит:
4НС1 + О2 = 2Н2О + 2С12
X. в. образуется при непосредственном взаимодействии хлора с водородом:
Н2 + С12 = 2НС1
Эта реакция при обычных условиях идёт очень .медленно, но если смесь газов сильно нагреть или осветить прямыми солнечными лучами или вспышкой магния, то происходит взрыв.
Своеобразие процесса заинтересовало учёных. В 1913 г. немецкий химик М. Боденштейн установил, что в пересчёте на один квант световой энергии (его обозначают hv) получается 100 000 молекул НС1. Это положило начало исследованию нового типа процессов, названных цепными реакциями. Вскоре учитель Боденштейна известный учёный В. Нернст объяснил механизм этого процесса. Под действием кванта света (или тепловой энергии) молекула С12 разбивается на два активных атома С1, которые затем реагируют с молекулой водорода с образованием молекулы НС1 и активного атома Н. Этот последний, в свою очередь, взаимодействует с неразло-жившейся молекулой С12, при этом снова получаются молекула НС1 и активный атом С1, который снова реагирует с молекулой водорода, и т. д. (точка над символами С1 и Н означает неспаренный электрон).
СЦ-^С1+С1
С1 + Н2=НС1 + Н
Н + С12 = НС1 + С1
С1+Н2=НС1 + Н и т. д.
Здесь в каждой стадии одна активная частица порождает только одну активную частицу; такие цепные реакции называются неразветвлёнными. О разветвлённых цепных реакциях см. в статьях Фтор и Водород.
Основной промышленный способ получения X. в.— это именно прямой синтез из элементов. Сырьём служат хлор и водород, получаемые одновременно при электролизе раствора поваренной соли (подробнее об этом — в статьях Натрий и Хлор). Синтез проводят в специальных печах, где хлористоводородная смесь после первоначального поджигания спокойно горит; температура пламени порядка 2000 °C. Поскольку начальная стадия реакции идёт очень бурно, печи синтеза служат всего 2—3 года. Большое внимание промышленности привлекает X. в., получаемый как побочный продукт хлорирования органических соединений:
rh + ci2 = rq + hci
(R — органический радикал)
Примером служит хлорирование циклогексана с целью получения гексахлорана:
СбН12 + 6С12 = CeHeCl6 + 6НС1
В связи с тем, что производство органических хлорпроизводных развивается очень быстро, этот способ весьма перспективен.
«Фонтан» на лабораторном столе. Используя очень хорошую растворимость хлористого водорода в воде, можно устроить маленький «химический фонтан» . Для этого в нижнюю склянку А наливают воду, подкрашенную синим лакмусом. Верхнюю Б наполняют сухим хлористым водородом. Затем открывают зажим В и через трубочку Г в склянку А вдувают воздух (например, резиновой грушей). Как только первые капли воды попадут в склянку Б, они мгновенно поглотят хлористый водород и там образуется вакуум. После этого вода устремится в склянку Б и начнёт бить красным фонтаном. Красный цвет фонтанирующей воды объясняется тем, что раствор хлористого водорода в воде является кисло-- той, а лакмус, как известно, в кислой среде становится красным.
ХЛОРИТЫ
332
В лабораторных условиях, когда нужно получить небольшие количества X. в., пользоваться прямым синтезом трудно, невыгодно, да и просто опасно. В этом случае применяют реакцию, известную ещё алхимикам:
NaCl + H2SO4 = НС1 + NaHSO4
Раньше эта реакция лежала в основе промышленного получения X. в. Соль должна быть сухой, а серная кислота концентрированной, иначе образующийся X. в. сразу же растворится в воде.
X. в.— важный продукт химической промышленности. Он идёт главным образом на производство соляной кислоты и синтетических органических хлорсодержащих продуктов и полимеров (например, хлорвинил — мономер для производства полихлорвинила — получают взаимодействием X. в. с ацетиленом).
В. К- Бельский. ХЛОРИТЫ — соли хлористой кислоты НСЮ2. Хорошо растворимы в воде. По окислительной активности занимают промежуточное положение между гипохлоритами и хлоратами (см. Хлор). В кислой среде проявляют сильные окислительные свойства:
4KI + KC1O2 + 2H2SO4 = О - 1
= 2I2 + KC1 + 2K2SO4 + 2H2O
Однако с более сильными окислителями (С12, КМпО4 и др.) могут быть восстановителями:
+ 3 +7
КС1О2 4- 2КОН + 2КМпО4 = + 5 +6
= КС1О3 + 2К2МпО4 + Н2О
Из всех X. практическое использование находит лишь NaC102, применяющийся для дезинфекции, для отбеливания тканей, целлюлозы и бумаги. Его получают по реакции:
2ClO2+2NaOH+H2O2=2NaClO2+2H2O+O2
В твёрдом состоянии все хлориты при нагревании и ударе взрывоопасны:
NaC102 - NaCl + О2
ХЛОРНАЯ ИЗВЕСТЬ (белильная известь)— см. в статьях Хлор, Известь.
ХЛОРНАЯ КИСЛОТА НС1О4 —одноосновная кислота, в которой хлор имеет степень окисления +7. Графическая формула:
Н—О—С1=О
X. к. существует в свободном состоянии — это бесцветная жидкость, дымящая на воздухе. Хорошо растворима в воде. X. к.— самая сильная из всех известных кислот (см. Хлор).
При действии на X. к. * водоотнимающих средств образуется хлорный ангидрид С12О7: 2НС1О4—^С12О, + Н2О
X. к. практически не проявляет окислительных свойств, в отличие от хлорноватистой, хлористой и хлорноватой кислот. Большинство солей X. к.— перхлоратов—хорошо растворимо. Поэтому X. к. используется для растворения металлов, сплавов и минералов.
Получают X. к. действием концентрированной серной кислоты на перхлораты:
КС1О4 + H2SO4 - KHSO4 + НС1О4
С безводной X. к. надо обращаться очень осторожно — она взрывоопасна.
ХЛОРНОВАТАЯ КИСЛОТА НСЮз —сильная одноосновная кислота, в которой хлор имеет степень окисления +5. Графическая формула:
В свободном состоянии X. к. не выделена и существует только в растворе. Максимальная концентрация, которую можно получить выпариванием раствора X. к., равна 40%. Если продолжить выпаривание, X. к. разлагается:
8НС1О3 = 4НС1О4 + 2С12 + ЗО2 + 2Н2О
По многим своим свойствам X. к. напоминает азотную кислоту. В частности, смесь X. к. и соляной кислоты — сильнейший окислитель, по действию очень похожий на царскую водку. При смешении двух кислот происходит реакция:
2НС1О3 + 2НС1 - 2С1О2 + С12 + 2Н2О
Выделяющиеся двуокись хлора и свободный хлор обеспечивают сильное окислительное действие смеси.
Ангидрид X. к.— окисел С12О5 — не выделен. Известны только С1О2— смешанный ангидрид X. к. и хлористой кислоты и С12О6 — смешанный ангидрид X. к. и хлорной кислоты (см. Хлор).
X. к. может быть получена по реакции:
Ва (С1О3)2 + H2SO4 - BaSO4 + 2НС1О3
Её соли называются хлоратами.
ХЛОРНОВАТИСТАЯ КИСЛОТА НОС1 — слабая одноосновная кислота, в которой хлор имеет степень окисления +1. Графическая формула:
Кислота существует только в растворе. Она образуется вместе с соляной кислотой при взаимодействии хлора и воды по обратимой реакции:
НС1 + НОС1
333
ХРОМ
Если в растворе присутствует взвесь окиси серебра, то можно получить раствор чистой X. к.
2С12 + Н2О + Ag2O = 2AgCl + 2НОС1
X. к.— очень неустойчивое соединение и в растворе разлагается по трём различным механизмам:
НОС1 = НС1 + О (1)
2НОС1 = С12О + Н2О (2)
ЗНОС1 = НСЮз + 2НС1 (3) При длительном хранении на свету или в присутствии катализаторов — ионов тяжёлых металлов, происходит реакция 1. Выделяющийся атомарный кислород и обеспечивает сильное окислительное действие X. к. Реакция 2 идёт под действием водоотнимающих средств (например, СаС12); выделяющийся при этом газ С12О является ангидридом X. к. При нагревании раствора протекает реакция 3, при этом наряду с соляной образуется хлорноватая кислота.
X. к.— очень сильный окислитель; например, реагируя с неметаллами, она переводит их в высшие положительные степени окисления:
О +5
5НОС1 + 2Р + ЗН2О = 2Н3РО4 + 5НС1 о +6
ЗНОС1 + S + Н2О = H2SO4 + ЗНС1 О +5
5НОС1 + Вг2 + Н2О = 2НВгО3 + 5НС1
Благодаря сильным окислительным свойствам сама X. к. и её соли — гипохлориты находят применение в качестве отбеливающих и дезинфицирующих средств.
ХРОМ (лат. Chromium) Сг — химический элемент VI группы периодической системы Менделеева; атомный номер 24, атомная масса 51,996. Металл серо-стального цвета. В природе встречается в виде смеси четырёх стабильных изотопов с массовыми числами 50, 52, 53 и 54. Наиболее распространён изотоп 62Сг (83,76%).
X. открыт в 1797 г. французским учёным Л. Вокленом. Этот период в истории химии знамено
вался бурным развитием химико-аналитических исследований минералов. А красная свинцовая руда (минерал крокоит Р,ЬСгО4), найденная в Сибири, привлекала особенное внимание необыкновенной прозрачностью и чудесным красным цветом. Действуя на порошок крокоита поташом, Воклен выделил соль жёлтого цвета (хромат калия):
РЬСгО4 + К2СО3 = К2СгО4 + РЬСО3
При нагревании К2СгО4 с соляной кислотой он получил зелёный раствор СгС13. Дальнейшие опыты привели Вок-лена к окиси Сг2О3, из которой путём сильного нагревания с углём он выделил свободный металл, неизвестный ранее. По предложению французских учёных, новый элемент вследствие разнообразия окраски его соединений был назван хромом (от греч. «хрома» — краска, цвет). Независимо от Воклена в 1798 г. X. был открыт в том же минерале немецким химиком М. Г. Клапротом.
Содержание X. в земной коре 8,3-10“3% по массе, по распространённости среди элементов он занимает 22-е место (на 21-м ванадий, на 23:м цинк). Из минералов X. наибольшее значение имеет буровато-чёрный хромит (хромистый железняк) FeO-Cr2O3. Окраска многих минералов обусловлена присутствием в них X. Так, например, золотисто-зелёный тон драгоценному изумруду (разновидность минерала берилла) или красный — рубину (разновидность минерала корунда А12О3) придаёт примесь окиси Сг2О3. Важнейшие месторождения хромитовых руд в СССР: Урал, Казахстан, Закавказье.
X.— твёрдый, довольно тяжёлый (плотность 7,19 г/см3), пластичный, ковкий металл; плавится при 1903 °C, кипит при 2480 °C. Ничтожные примеси кислорода, азота и углерода резко изменяют физические свойства X., в частности он становится хрупким. Получить X. без этих примесей очень трудно.
В соответствии с положением X. в периодической системе его атом имеет шесть внешних (валентных) электронов (конфигурация 3d64sx, см. Атом). Обычно X. проявляет степени окисления +2, +3, +6 (валентности II, III, VI соответственно).
При невысоких температурах X. химически мало активен (взаимодействует только с фтором). Выше 600 °C реагирует с галогенами, серой, азотом, кремнием, бором, углеродом, кислородом. Взаимодействие с кислородом протекает сначала довольно активно, затем, однако, резко замедляется, так как поверхность металла покрывается тонкой, чрезвычайно устойчивой окисной плёнкой, препятствующей дальнейшему окислению. При 1200 °C плёнка начинает разрушаться, и окисление снова идёт быстро. При 2000 °C X. воспламеняется в кислороде с образованием тёмно-зелёной окиси Сг2О3.
Металл растворяется в НС1 и H2SO4 с выделением водорода. При взаимодействии с соляной кислотой без доступа воздуха образуется дихлорид:
Сг + 2НС1 = СгС12 + Н2
В присутствии кислорода взаимодействие приводит к трихлориду:
2Сг + 6НС1 + О2 = 2СгС13 + Н2 4- 2Н2О Если же металл погрузить на некоторое время в HNO3 (концентрированную или разбавленную),
ХРОМ
334
то он перестаёт растворяться в НС1 и H2SO4, не изменяется при нагревании с галогенами и т. д. Это явление—пассивирование, объясняется образованием на поверхности металла защитного слоя, состоящего главным образом из окиси X.
Соединения двухвалентного X.— сильные восстановители, чрезвычайно неустойчивые в присутствии влаги и кислорода. Практического значения они пока не имеют. Напротив, соединения трёхвалентного X. устойчивы на воздухе и в растворах. Окись Сг2О3 образуется при нагревании некоторых соединений шестивалентного X., например:
4СгО3 = 2Сг2О3 + 30 2
(NH4)2Cr2O7 = Сг2О3 + Na + 4Н2О
Последняя реакция и есть знаменитый «вулкан», который часто показывают на уроках химии. Мелкий порошок дихромата аммония всыпают в фарфоровый тигель, сверху добавляют несколько кусочков магниевой стружки и поджигают их. При сгорании Mg развивается высокая температура. Начинается разложение дихромата, которое протекает с выделением тепла и постепенно захватывает всё большие и большие количества соли. В конце концов реакция идёт всё более и более бурно — появляются искры, пламя, летит рыхлый и лёгкий «пепел» — порошок Сг2О3. Он образует целую «гору», постепенно скрывая под собой тигель,— типичное извержение вулкана в миниатюре.
Формулу гидроокиси Сг(ОН)3 правильнее писать Сг2О3-пН2О, так как это — соединение переменного состава (выделены гидроокиси с п от 1 до 9). Это амфотерное соединение, легко растворяющееся в кислотах и щелочах:
Сг(ОН)3 + ЗНС1 = СгС13 + ЗН2О
Сг(ОН)3 + NaOH = NaCrO2 + 2Н2О
NaCrO2 и другие соли хромистой кислоты НСгО2 называются хромитами. Хромовые квасцы — двойной сульфат KCr(SO4)2- 12Н2О — по составу и кристаллическому строению вполне подобны, алюминиевым квасцам, но отличаются от них тёмно
фиолетовым цветом (см. Квасцы). Прочие соли
трёхвалентного X. окрашены в самые различ
ные оттенки всех цветов радуги (кроме синего
и голубого). В качестве хромофоров («носите-
лей цвета») они входят в состав керамических,
живописных и печатных красок. Для X.,
особенно трёхвалентного, известно большое число комплексных соединений (об этом классе соединений рассказано в статье Кобальт). Соединения шестивалентного X.— трёхокись СгО3 (хромовый ангидрид), соли хромовых кислот (хроматы) — сильные окислители.
Соединения X., особенно шестивалентного,
ядовиты. Поражают они главным образом верхние дыхательные пути, лёгкие и глаза. Поэтому работа с ними требует осторожности.
Сырьём для промышленного получения X. служит хромистый железняк. Его химическая переработка приводит к окиси Сг2О3. Восстановление Сг2О3 с помощью алюминия (алюминотермия) или кремния (силикотермия) даёт металлический X. невысокой степени чистоты:
Сг2О3 + 2А1 = А12О3 4- 2Сг 2Сг2О3 + 3Si = 3SiO2 + 4Сг
Более чистый металл получают электролизом концентрированных растворов хромового ангидрида. В металлургии, где расход X. для легирования сталей очень велик, используют не сам X., а его сплав с железом — феррохром (не менее 65% Сг). Феррохром выплавляют из смеси хромита с коксом в электрических дуговых печах (см. Ферросплавы).
Основной потребитель X.— металлургия. Сталь при добавлении X. становится гораздо более стойкой к действию химических реагентов (коррозионно-стойкой); повышаются и такие важные свойства стали, как прочность, твёрдость и износостойкость. X.— обязательная составляющая нержавеющих, кислотоупорных, жаропрочных сталей. Электролитическое покрытие- хромом железных изделий (хромирование) также сообщает им устойчивость к коррозии. При этом преследуются ещё и чисто декоративные цели — тонкий слой X., нанесённый на подслой меди и никеля, придаёт изделиям красивый голубоватый долго сохраняющийся блеск.
Хромированные поверхности современного автомобиля совсем не подвержены коррозии и долго сохраняют красивый голубоватый блеск.
335
ХРОМПИК
Семейство хромовых сплавов весьма многочисленно. Нихромы (сплавы с никелем) и хромали (с алюминием и железом) устойчивы в интервале 1000—1300 °C, обладают высоким электросопротивлением и используются для изготовления нагревателей в электрических печах сопротивления. Стеллит — сплав X. (20—25%), кобальта (45—60%), вольфрама (5—20%), железа (1—3%) — очень твёрд, стоек против износа и коррозии; применяется в металлообрабатывающей промышленности для изготовления режущих инструментов. Хромомолибденовые стали используются для создания фюзеляжей самолётов. X. входит и в состав очень многих марок специальных сталей.
Старейшие потребители соединений X.— текстильная и кожевенная промышленность. Здесь соли X. используются и как красители, и как составная часть дубильных растворов (отсюда название «хромовые сапоги»). В России московский аптекарь Гельм впервые стал готовить Сг2О3 для раскраски фарфоровой посуды (1811 г.), а позже К2СгО4 и К2Сг2О7 для ситцепечатания.
Хромит служит для изготовления огнеупорного хромитового и хромомагнезитового кирпича. Такой кирпич химически пассивен, устойчив выше 2200 °C, хорошо выдерживает резкие колебания температур; он используется для кладки сталеплавильных печей. Своды из хромомагнезитового кирпича выдерживают вдвое больше плавок, чем своды из кварцевого («динасового») кирпича — классического огнеупорного материала. Окись Сг2О3 — полировальный материал, прекрасный катализатор для многих органических реакций. Сг2О3 является основной примесью к А12О3 при выращивании кристаллов рубина, используемого в лазерных устройствах, в часовой и ювелирной промышленности (о лазерах рассказано в статье Алюминия окись). 3. А. Старикова.
ХРОМАТЫ — соли хромовых кислот. Соли монохромовой кислоты Н2СгО4 называют монохроматами (или просто хроматами), соли дихромовой — дихроматами (бихроматами). Монохроматы обычно окрашены в жёлтый цвет. Они устойчивы только в щелочной среде, а при подкислении превращаются в оранжевокрасные дихроматы:
2Na2CrO4 + H2SO4 = Na2Cr2O7 + 4~ Na2SO4 4~ H2O
В лабораториях для мытья химической посуды часто применяется хромовая смесь.
Растворимые в воде X. натрия и калия применяют в текстильном и кожевенном произ
водстве (см. Хром), как консерванты древесины (они уничтожают древесные грибки). Нерастворимые X. некоторых металлов — прекрасные художественные краски. Это и жёлтые кроны (PbCrO4v ZnCrO4, SrCrO4), и красный свинцово-молибденовый крон (содержит РЬСгО4 и МоСгО4) и многие другие. Богатством оттенков — от розово-красного до фиолетового славится SnCrO4, используемая в живописи по фарфору.
ХРОМОВАЯ СМЕСЬ — сернокислотный раствор дихромата калия или натрия, используемый для мытья стеклянной химической посуды в лаборатории. Наиболее часто применяется раствор, содержащий по массе приблизительно 12 частей К2Сг2О7, 70 частей воды и 22 части концентрированной H2SO4.
ХРОМОВОКИСЛЫЕ СОЛИ—то же, что хроматы.
ХРОМОВЫЕ КИСЛОТЫ — кислоты хрома, отвечающие его степени окисления 4-6 и различающиеся соотношением числа молекул СгО3 и Н2О. Существуют только в виде растворов. При растворении хромового ангидрида СгО3 образуется монохромовая кислота Н2СгО4 (обычно её называют просто хромовой). Подкисление раствора или увеличение содержания в нём СгО3 приводит к кислотам общей формулы пСгО3-Н2О. При п=2, 3, 4 это, соответственно, ди-, три- и тетрахромовые кислоты. Самая сильная из них — дихромовая 2СгО3-Н2О, то есть Н2Сг2О7. X. к. и их соли — сильные окислители, ядовиты.
X. к. входят в состав электролита, который используют для получения хрома или для хромирования различных металлических изделий. ХРОМОВЫЙ АНГИДРИД, трёхокись хрома, оксид хрома (VI), СгО3 — окисел шестивалентного хрома. Тёмно-красные игольчатые кристаллы. При нагревании около 200 °C разлагаются с выделением кислорода и образованием Сг2О3. X. а.— сильный окислитель; если на кирпич насыпать немного X. а. и покапать на него спиртом, вспыхивает яркое пламя (опыт «низведения огня с неба»). При растворении X. а. в воде образуется хромовая кислота Н2СгО4. Получают СгО3 действием избытка концентрированной H2SO4 на насыщенный водный раствор дихромата натрия:
Na2Cr2O7 4- 2H2SO4 = 2СгО3 4- 2NaHSO4 4- Н2О
Больше всего X. а. расходуется для электролитического хромирования (см. Хром). X. а.— одно из самых токсичных соединений хрома. ХРОМПИК — техническое название дихроматов калия и натрия, К2Сг2О7 и Na2Cr2O7>
ЦАРСКАЯ ВОДКА — смесь одного объёма азотной кислоты HNO3 и трёх объёмов соляной кислоты НС1; обе кислоты концентрированные. Ц. в.— один из сильнейших окислителей, она действует на металлы значительно агрессивнее, чем каждая из составляющих её кислот. Так, в Ц. в. растворяются даже золото й платина:
Au + HNO3 + ЗНС1 = AuCl3 + NO + 2Н2О 3Pt + 4HNO3 + 12НС1 = 3PtCl4 + 4NO + 8Н2О Кстати, именно потому, что в ней растворяется «царь металлов» — золото, эта смесь и получила у алхимиков название «Ц. в.».
В чём причина такой активности смеси азотной и соляной кислот? Дело в том, что азотная кислота, сама будучи очень сильным окислителем, окисляет соляную кислоту до свободного хлора. В момент выделения хлор представляет собой не молекулы С12, а атомы С1. Этот атомарный хлор и является активным действующим началом Ц. в.:
HNO3 + ЗНС1 = 2С1 + NOC1 + 2Н2О
ЦЕЗИЙ (лат. Caesium) Cs — химический элемент I группы периодической системы Менделеева; атомный номер 55, атомная масса 132,9054. Светлый металл с золотисто-жёлтым оттенком. Имеет один стабильный изотоп 133Cs.
В 1846 г. немецкий химик К- Платтнер исследовал состав минерала поллуцита с острова Эльбы, считая, что в нём присутствует калий. Учёный был весьма озадачен, обнаружив, что окончательный подсчёт результатов по
казал нехватку калия в 7,25%. В 1860 г. немецкие учёные Р. Бунзен и Г. Кирхгоф, исследуя с помощью спектроскопа минеральные воды из Дюркгейма в Баварии, обнаружили две яркие линии в синей части спектра, принадлежащие новому элементу. Поэтому он и был назван «цезий», от латинского прилагательного caesius — «небесно-голубой». (Ц. был первым элементом, открытым с по-
мощью спектрального анализа.) А спустя несколько лет было доказано, что большая часть «недовеса» в анализе Платтнера — это Ц.
Ц.— редкий элемент. На его долю в земной коре приходится всего 3,7-10“4% по массе — это 45-е место среди всех элементов. Однако, в отличие от своего соседа по группе щелочных металлов — рубидия, Ц. имеет собственные минералы: очень редкий авогадрит (K,Cs)[BF J и имеющий промышленное значение поллуцит (поллукс) (Cs, Na) [ Al2Si2O6] • nH 2О. (Понять эти формулы вам поможет статья Минералы.) Месторождения поллуцита есть в США, Швеции, Юго-Западной Африке, Канаде и у нас в Казахстане. В небольших количествах Ц. содержится как примесь в минералах лития и калия, в морской воде и минеральных водах.
Ц. плавится при 28,5 °C, он мог бы превратиться в жидкость от тепла человеческих ладоней (однако такого опыта лучше не делать — Ц. химически очень активен). Кипит Ц. при 705 °C. Он представляет собой вязкий, мягкий, как воск, и довольно лёгкий металл (плотность Ц. 1,90 г/см3). Соединения Ц. окрашивают пламя горелки в фиолетовый цвет.
По своему положению в периодической системе Ц.— самый активный из всех имеющихся в природе металлов. Атом Cs теряет свой единственный валентный электрон (конфигурация внешней оболочки 6s1) легче, чем атомы всех других щелочных металлов, проявляя в соединениях всегда степень окисления +1 (валейт-ность I). На воздухе Ц. немедленно воспламеняется и сгорает с образованием надперекиси:
Cs + О2 = CsO2
Нормальная окись Ц. Cs2O .может быть получена лишь окислением в особых условиях при недостаточном количестве кислорода. Так же бурно, со взрывом Ц. реагирует с галогенами, серой, фосфором:
2Cs + С12 = 2CsCl
2Cs + S = Cs2S 5Cs + 2P = Cs5P2
337
При повышенных температурах Ц. взаимодействует с водородом, давая гидрид:
2Cs + Н2 = 2CsH
С азотом Ц. реагирует в поле электрического разряда с образованием нитрида Cs3N. При температурах выше 600 °C Ц. разрушает стекло, восстанавливая кремний из SiO2 и силикатов. Если маленький кусочек Ц. бросить в воду, то немедленно произойдёт взрыв (воспламенение выделяющегося водорода); Ц. реагирует со взрывом даже со льдом при низкой температуре, —116 °C:
2Cs + 2Н2О = 2CsOH + Н2
По этой реакции образуется гидроокись CsOH— самое сильное основание. Оно настолько активно взаимодействует с двуокисью углерода и стеклом, что его нужно защищать от них серебром или платиной.
После знакомства со столь бурными реакциями Ц. с окружающей средой естественно возникает вопрос — как же его хранить? Ц. для этого . . . загрязняют, то есть сплавляют с другими металлами. Сплавы Ц. не так реакционноспособны и хранить их удобнее. А при необходимости из этих легкоплавких сплавов Ц. всегда можно отделить отгонкой в вакууме.
Получить свободный Ц. довольно трудно. Впервые его выделил в 1882 г. шведский химик К. Сеттерберг электролизом смеси цианидов CsCN и Ba(CN)2. В 1890 г. русский химик Н. Н. Бекетов предложил восстанавливать Ц. из его гидроокиси магнием при высоких температурах. Однако наиболее распространён сейчас метод, предложенный в 1911 г. французским химиком А. Акспилем и заключающийся в вытеснении Ц. из его хлорида металлическим кальцием в вакууме при 700 °C:
2CsCl + Са = 2Cs + СаС12
Ц. применяется в различных областях современной науки и техники. По чувствительности к свету он превосходит все другие металлы: электрон из него выбивается даже инфракрасными лучами. Поэтому Ц.— ценнейший материал для изготовления фотоэлементов. Восприимчивость Ц. к инфракрасным лучам используется при изготовлении специальных оптических прицелов к снайперским винтовкам для ведения огня в ночных условиях.
Подобно рубидию, Ц. применяется при изготовлении радиоламп как внутренний газопоглотитель (геттер). Ц. и ряд его соединений служат катализаторами. Ц. значительно усиливает каталитическое действие железа при синтезе аммиака, a CsOH — превосходный катализатор в процессе получения муравьиной кислоты.
Большое внимание уделяется в наши дни исследованию Ц., находящегося в состоянии
ЦЕМЕНТ
ионизированного газа — «цезиевой плазмы». Ионные ракетные двигатели основаны на ускорении ионов и выбросе их в пространство для создания реактивной тяги. Так как Ц. (и рубидий) очень легко ионизируется при довольно низких температурах и обладает достаточной массой, он перспективен в этой новейшей области. Но путь от расчётов и лабораторных опытов до работающих устройств, по-видимому, окажется длинным. В. Д'. Бельский.
ЦЕМЕНТ (от лат. caementum — щебень, битый камень) — порошкообразный вяжущий материал, образующий при смешении с водой тестообразную массу, самопроизвольно затвердевающую в прочное камневидное тело. Ц., затвердевающие только на воздухе, называются воздушными. Ц. же, способные к затвердеванию и на воздухе и в воде, называются гидравлическими. В последнее время термин «цемент» обычно относят только к этому классу Ц. Важнейшие из них — портландцемент, пуццолановые, шлаковые, глинозёмистые Ц.
Пуццолановый Ц. назван так по местности Поц-цуоли близ Неаполя, где около 1000 лет до н. э. древние римляне добывали изверженные породы, служившие добавкой к известковым строительным «растворам».
Портландцемент был впервые приготовлен в начале XIX в. После нашествия Наполеона и пожара 1812 года в Москве развернулось широкое строительство. Одним из ведущих специалистов Военно-рабочей бригады, участвовавшей в 1817—21 гг. в восстановлении Москвы, был инженер Е. Г. Челиев (Челидзе). Он открыл и внедрил в практику новое вяжущее вещество — искусственный гидравлический Ц., разработал также способ получения Ц. из глины и известняка, сконструировал печь для обжига, предложил составы смесей. Результаты своих работ Челиев опубликовал в книге «Полное наставление как производить дешёвый и лучший мертель или цемент...», которая вышла в свет в Москве в марте 1825 г.
Несколько ранее, в ноябре 1824 г., местная газета города Лидса в Англии сообщила, что «каменщик города Д. Аспдин получил патент на цемент, похожий на порт-ландский камень» (самый распространённый в то время строительный камень, который добывали около г. Порт-ланда). Аспдин в своём патенте ограничился рекомендациями — «беру определённые количества», «выжигаю до возможно более полного удаления углекислоты»... В 1826 г. в Англии началось производство нового Ц., быстро получившее дальнейшее развитие. На родине Асп-дина была установлена мемориальная доска в честь «изобретателя, сделавшего весь мир своим должником». А в феодально-крепостнической России изобретение Челиева было забыто.
В 1867 г. М. Сорель во Франции получил магнезиальный Ц. взаимодействием окиси магния MgO с концентрированным раствором хлорида магния MgCl2.
С тех пор технология получения Ц. значительно усовершенствовалась. В наше время производится несколько десятков видов одного только портландцемента: белый, термостойкий, волокнистый, декоративный и т. д. Производство Ц.— сложнейший физико-хими
22 хэш
ЦЕРИИ
ческий процесс, требующий знания коллоидной химии, минералогии, кристаллохимии и др. Существенный вклад в науку о Ц. внесли А. А. Байков, П. А. Ребиндер и другие советские учёные, развившие теорию твердения минеральных вяжущих веществ.
Сырьём для получения Ц. служат известняковые породы, глины, богатые SiO2, промышленные отходы (шлак, зола). Их смесь подвергают обжигу при температуре около 1450 °C в цилиндрических стальных вращающихся печах до 230 м длиной и до 7 м диаметром. Печи выложены внутри огнеупорным кирпичом. Из печи выходит так называемый клинкер — тёмно-серые, внешне однородные шарики. После тщательного измельчения клинкера в шаровых мельницах получается тончайший порошок Ц., готовый к употреблению.
Выбор температурного режима и соотношения компонентов в исходной смеси, введение специальных добавок позволили получать Ц. с заданными свойствами.
Каков же механизм твердения Ц.? Этот процесс состоит из нескольких стадий. Сначала цементный порошок замешивают с небольшим количеством воды и получают густую пасту. Крупинки Ц. частично растворяются и, взаимодействуя с водой, дают новые химические соединения — гидраты. Гидраты менее растворимы в воде, чем исходные вещества, поэтому они выпадают из пересыщенного по отношению к ним раствора. Множество мельчайших кристалликов постепенно разрастается и соединяется между собой. Так образуется пространственная сетка-каркас, которая пронизывает весь объём цементного теста. Первоначально сетка чрезвычайно тонка и хрупка, но постепенно она становится более плотной и прочной из-за роста уже возникших и выделения новых кристаллов гидрата. Таким образом «цементное тесто» превращается в цементный камень.
Ц.— основной строительный материал, служащий для возведения наземных, подземных и гидротехнических сооружений. Теперь уже невозможно представить себе сколько-нибудь значительную стройку без Ц., бетона и железобетона (в эти два материала Ц. входит как основной компонент, см. Бетон). По выпуску Ц. Советский Союз занимает первое место в мире. 3. А. Старикова.
ЦЁРИЙ (лат. Cerium) Се — химический элемент III группы периодической системы Менделеева из семейства лантаноидов], атомный номер 58.
ЦИАН (д и ц и а н) C2N2 или (CN)2— соединение углерода и водорода. Графическая формула N=C—C=N. Бесцветный горючий
338
очень ядовитый газ с резким запахом. Название «циан» происходит от греческого «кюа-неос» — тёмно-синий; соединения Ц. впервые были получены из некоторых синих красок (берлинской лазури и др.). Отсюда и старое русское название Ц.— «синерод». По некоторым химическим свойствам Ц. напоминает галогены (роль атома галогена играет одновалентный радикал CN). Ц. используется в химической промышленности и в лабораторной практике для различных синтезов.
ЦИАНИДЫ — соли цианистоводородной кислоты HCN. Ц. щелочных и щелочноземельных металлов хорошо растворимы в воде, а Ц. тяжёлых металлов, как правило,— плохо. Будучи солями очень слабой кислоты, Ц. в растворе сильно гидролизованы:
NaCN + Н2О HCN + NaOH
(равновесие обратимой реакции сдвинуто вправо). В ионной форме уравнение записывают так:
CN- + H2Oz± HCN + ОН-
Растворы Ц. пахнут цианистым водородом и имеют щелочные свойства.
В практическом отношении наиболее важны Ц. натрия и калия. Их получают взаимодействием щелочей с цианистым водородом или другими, более сложными способами. Обе соли, особенно NaCN, применяются при добыче золота, которое они растворяют в присутствии кислорода воздуха по реакции:
4Au + 8NaCN + 2Н2О Н- О2 -= 4Na[Au(CN)2] + 4NaOH Na[Au(CN)2] — комплексное соединение, анионы CN- входят во внутреннюю сферу комплекса. Образование таких соединений для Ц. очень характерно. (Прочитать о строении комплексных соединений можно в статье До-бальт.) Как и цианистый водород, Ц. чрезвычайно ядовиты.
ЦИАНИСТОВОДОРОДНАЯ КИСЛОТА (с и-нильная кислота) — раствор цианистого водорода HCN в воде.
ЦИАНИСТЫЕ СОЛИ — то же, что цианиды. ;
ЦИАНИСТЫЙ ВОДОРОД HCN — соединение водорода, углерода, азота. Графическая формула Н—C=N. Бесцветная легколетучая жидкость с характерным запахом, напоминающим запах горького миндаля. Кипит при 25,7 °C, затвердевает при —14 °C. С водой Ц. в. смешивается во всех отношениях, образуя цианистоводородную кислоту (синильную кислоту), очень слабо диссоциирующую:
HCN^=±H+ + CN-
339
ЦИНК
Цианистоводородная кислота настолько слаба, что вытесняется из своих солей — цианидов даже угольной кислотой:
2KCN + Н2СО3 = К2СО3 + 2HCN
Получить HCN можно действием кислот на цианиды. Ц. в. служит сырьём для производства ценных полимерных материалов.
Ц. в. необычайно ядовит даже в очень малых дозах.
ЦИНК (лат. Zincum) Zn — химический элемент II группы периодической системы Менделеева; атомный номер 30, атомная масса 65,38. Металл сине-вато-белого цвета. Природный элемент— это смесь 5 стабильных изотопов с массовыми числами 64, 66, 67, 68 и 70; наиболее распространён 64Zn (48,89%).
Хотя цинковые руды были известны людям ещё с давних времён и древние греки, египтяне, римляне
умели изготовлять сплав Ц.с медью—латунь,однако чистый металл выделить долгое время не удавалось. Дело в том, что восстановление окиси Ц. (ZnO) углём до металла происходит при 1000—1100 °C, а кипит Ц. при 906°C. Образующийся пар Ц. при соприкосновении с воздухом воспламеняется и опять превращается в ZnO. Мастера древних Индии и Китая научились конденсировать пары Ц. без доступа воздуха в глиняных сосудах и таким образом получать его в слитках. В Европе металлический Ц. стал известен лишь в средние века. Но вплоть до второй половины XVII в. его привозили из Индии. А в 1746 г. немецкий химик А. С. Маргграф разработал способ получения Ц. прокаливанием смеси его окиси с углём без доступа воздуха в глиняных огнеупорных ретортах с последующей конденсацией паров Ц. в холодильниках.
Происхождение названия «цинк» неясно. Этот термин встречается в трудах швейцарского врача и химика Т. Парацельса (1530 г.) и других учёных XVI— XVII ₽в.
Земная кора содержит 8,3-10"3% Ц. по массе. Это 23-е место среди других элементов (на 22-м находится хром). Важнейший минерал Ц.— сфалерит (или цинковая обманка) ZnS — входит в состав многих сульфидных руд. При постепенном окислении сульфида в природе образовались цинкит ZnO, смитсонит ZnCO3 и другие минералы Ц. Обычно цинковые руды— полиметаллические; они содержат также минералы меди, свинца и других металлов. И — что очень важно — цинку в минералах сопутствуют кадмий (0,1—0,3% в сфалерите), германий, индий, галлий... Эти ценнейшие редкие элементы извлекают попутно при добыче Ц. В нашей стране крупные месторождения цинковых руд находятся на Алтае, Северном Кавказе и в других районах.
Ц.— металл средней твёрдости. Плотность его 7,13 г/см3. Ц. легко плавится (/пл 419,5 °C) и принадлежит к числу наиболее летучих металлов (/кип 906 °C). Ц. высокой чистоты пластичен, то есть его можно ковать и прокатывать в тонкую фольгу. Технический же металл становится пластичным лишь при нагреве до 100—150 °C.
Конфигурация высших энергетических уровней атома Zn 3d104s2. Степень окисления Ц. в соединениях равна +2 (валентность II). При хранении на воздухе Ц. тускнеет благодаря образованию тонкого, но плотного слоя окисла, который предохраняет металл от дальнейшего окисления. В атмосфере же влажного воздуха, особенно в присутствии СО2 или SO2, Ц. разрушается уже при комнатной температуре. При достаточном нагревании на воздухе Ц. сгорает ярким голубовато-зелёным пламенем, образуя окись ZnO белого цвета. Сухие фтор, хлор и бром не взаимодействуют с Ц. на холоду. Но в присутствии паров воды Ц. может воспламениться в галогене, образуя галогенид (например, хлорид ZnCl2). Смесь цинкового порошка с серой при нагревании реагирует со взрывом. Сульфид ZnS — очень интересное соединение. Если к нему добавить ничтожные примеси меди или некоторых других металлов, то после предварительного освещения оно некоторое время светится в темноте (проявляет люминесцентные свойства). Этим пользуются для приготовления светящихся составов.
В ряду напряжений Ц. стоит значительно раньше водорода и вытесняет его из таких кислот, как соляная, разбавленная серная и т. д. По реакции:
Zn 4-2НС1 = ZnCl2 + Н2 в лабораториях получают водород (см. рис. 1 к статье Водород). При растворении Ц. в концентрированной серной кислоте обычно выделяется двуокись серы:
Zn + 2H2SO4 = ZnSO4 + SO2 + 2H2O
Концентрированная азотная кислота взаимодействует с Ц. с выделением окислов азота (обычно NO2), а разбавленная — с образованием либо окислов азота (NO, N2O), либо аммиака NH3, который тотчас же превращается в нитрат аммония:
4Zn + 10HNO3 = 4Zn(NO3)2 + NH4NO3 + 3H2O
Ц., в отличие от своих аналогов — кадмия и ртути, взаимодействует с крепкими растворами щелочей:
Zn + 2NaOH = Na2ZnO2 + Н2 При этом образуются цинкаты — соли, в которых Ц. входит в состав аниона. Гидро-
22*
цинк
340
Рис. 1. Гальванический элемент. На рисунке показан гальванический элемент с цинковым и медным электродами, опущенными соответственно в растворы сульфата цинка ZnSO4 и сульфата меди CuSO4. Название «гальванический» дано в честь итальянского профессора Л. Гальвани (1737—1798 гг.), который обнаружил, что мышцы лацки лягушки, подвешенные за нерв к железной решётке на медном крючке, сокращаются при соприкосновении с железной решёткой. Это открытие физиолога первым правильно объяснил другой итальянский учёный, физик А. Вольта (1745—1827 гг.). Вольта пришёл к выводу, что сокращение мышц вызывает электрический ток, образующийся в цепи, которая содержит электроды из двух разных по своей активности металлов — железа и меди. Исследования Вольты показали, что более эффективен как источник тока элемент с цинковым и медным электродами (типа показанного на рисунке). До изобретения динамомашины гальванические элементы были единственными источниками электрического тока, использовавшимися в лабораториях. С их помощью, например, англичанин Г. Дэви впервые получил в свободном состоянии натрий, калий и другие активные металлы.
окись Zn(OH)2 амфотерна. В избытке щёлочи она растворяется с образованием цинката, например:
Zn(OH)2 + 2NaOH = Na2ZnO2 + 2Н2О
Соли Zn (+2) бесцветны и хорошо растворимы в воде, за исключением ZnF2, ZnS, ZnCO3.
Интересно, что весьма чистый Ц. растворяется в кислотах очень медленно. Если же в Ц. имеется примесь менее активного металла, например меди, то скорость растворения резко возрастает. Чтобы было понятно, почему так происходит, расскажем сначала о том, как используют Ц. в гальванических элементах.
Представим себе, что в левом колене Н-образного сосуда (см. рис. 1) находится цинковый стерженёк, погружённый в раствор ZnSO4, а в правом колене — медный стерженёк, погружённый в раствор CuSO4. В средней
части сосуда имеется пористая перегородка, препятствующая перемешиванию растворов CuSO4 и ZnSO4. Снаряжённый так сосуд представляет собой гальванический элемент — прибор, с помощью которого химическую энергию можно превратить в электрическую. Каким образом? Примем во внимание, что в атоме Zn 2 внешних электрона связаны с ядром сравнительно слабо, а в атоме Си внешние электроны связаны значительно прочнее. Поэтому атомы Zn могут довольно легко терять 2 электрона и переходить со стерженька в раствор в виде ионов Zn2+:
Zn = Zn2+ +2ё
а ионы Си2+, находящиеся в растворе, стремятся приобрести 2 электрона, после чего нейтральные атомы меди отлагаются на медном стерженьке:
Си2+ +2ё = Си
Однако если стерженьки в приборе электрически не соединены между собой, то ни растворения Zn, ни осаждения Си практически не происходит. Ионы Zn2+ не смогут далеко уйти от «своего» стерженька (их будут удерживать отрицательно заряженные электроны), а ионам Си2+ неоткуда взять недостающие электроны. Иное дело, если стерженьки соединить проводником. Тогда электроны с цинкового стерженька будут непрерывно переходить на медный, в левом колене прибора начнёт растворяться Ц., а в правом — отлагаться медь. И так до тех пор, пока весь Ц. не растворится или не израсходуется весь сульфат меди в растворе. И всё это время между стерженьками гальванического элемента (их называют электродами) будет течь электрический ток.
Если на электроды рассматриваемого гальванического элемента взглянуть с точки зрения связи между атомами в металлах, то оказывается, что в металлическом Ц. концентрация свободных электронов выше, чем в металлической меди. Возникающий электрический ток стремится выровнять концентрацию свободных электронов в электродах.
Вернёмся теперь к вопросу: почему добавки меди к Ц. ускоряют реакцию Ц. с кислотой (например, соляной)? В том случае, если используется очень чистый Ц., водород выделяется прямо на его поверхности. Слой газообразного водорода быстро покрывает поверхность Ц. тонкой плёнкой, и доступ к нему кислоты оказывается затруднённым. Реакция протекает очень медленно. Иное дело— Ц., содержащий примесь менее активного металла (такого, как медь). Некоторые мельчайшие участки металла, состоящие из атомов меди, всегда окажутся у поверхности загрязнённого Ц. В этом случае Zn и Си образуют так называемую гальваническую пару, которая «работает» по тому же принципу, что и гальванический элемент. Можно сказать, что небольшие участки Zn и Си образуют своего рода гальванические микроэлементы (рис. 2). Атомы Zn, находящиеся у поверхности, будут терять электроны и переходить в раствор. Освобождающиеся электроны попадут на медные участки. Ионы водорода Н + будут подходить к этим участкам (их притягивают отрицательно заряженные электроны) и разряжаться около их поверхности. В результате выделение водорода происходит не на поверхности Ц., а на поверхности меди. Переходу ионов Zn2+ в раствор ничто не мешает, и реакция протекает с высокой скоростью.
Полиметаллические руды, из которых добывают Ц., содержат лишь 1—3% Zn. После их обогащения получают цинковые концентраты (48—58% Zn) и одновременно концентраты пиритные, медные и свинцовые. Переработка цинкового концентрата начинается с обжига:
2ZnS + ЗО2 = 2ZnO 4- 2SO2
341
ЦИРКОНИЙ
Полученные при обжиге газы, содержащие 4— 6% SO2, направляют в производство серной кислоты. От ZnO к Zn идут двумя путями. Первый путь—пирометаллургический—существует издавна. ZnO нагревают с коксом при 1250— 1350 °C в специальных огнеупорных ретортах:
ZnO + С = Zn + СО
Пары Ц. конденсируются в приёмниках. Жидкий металл отливают в чушки или плитки.
Второй путь—гидрометаллургический. Окись ZnO, полученную обжигом концентрата, растворяют в серной кислоте. А затем очищенный раствор ZnSO4 подвергают электролизу; Zn осаждается на катоде.
Рис. 2. Гальванический микроэлемент. Такой элемент «работает» на поверхности цинка, опущенного в раствор серной кислоты, если цинк содержит небольшую примесь меди. Чистый цинк реагирует с серной кислотой очень медленно. (Поэтому в снаряжённом им аппарате Киппа водород почти не выделяется.) Для ускорения реакции достаточно добавить в кислоту несколько капель раствора сульфата меди CuSO4. Сначала ионы меди Си2+ получают от нейтральных атомов цинка электроны:
о
Zn — 2ё = Zn2+
о
Си2+ + 2ё = Си
Нейтральные атомы меди переходят на поверхность цинка и образуют на ней мельчайшие участки металлической меди.
На рисунке и показан одни из таких участков (сильно увеличенный). Электроны, освобождающиеся при переходе иоиов Zn2+ с поверхности металлического цинка, поступают на медные участки. А иоиы Н+ подходят к этим участкам и разряжаются около поверхности меди. Цинковая поверхность остаётся всё время свободной, и ионы цинка легко переходят в раствор. Наблюдается бурное выделение водорода:
Zn + H2SO4 = ZnSO4 + н2
Почти половина производимого Ц. идёт на защиту стали от коррозии. Для покрытия слоем Ц.— цинкования — изделия из стали очищают от ржавчины и погружают в ванну с расплавленным Ц. Этот процесс — горячее цинкование — основан на том, что жидкий Ц. легко образует сплавы с железом. Применяют и «холодное», то есть электролитическое, цинкование (так защищают, например, проволоку).
Поскольку в ряду напряжений Ц. стоит до железа, при контакте обоих металлов в коррозионной среде разрушается Ц. Поэтому Ц. для защиты сталей предпочтительнее, чем, например, олово. Ведь если в защитном оловянном покрытии появится отверстие, то в коррозионной среде начнёт разъедаться сталь. А цинковое покрытие продолжает защищать сталь даже после того, как оно уже сильно повреждено.
Важное практическое применение имеют сплавы Ц. с другими металлами, прежде всего с медью (это латуни, описанные в статье Медь). А в полиграфии зарекомендовали себя сплавы Zn с А1, Си и Mg, из них отливают всевозможные шрифты. В последние годы цинком защищают стартовые конструкции при запуске ракет. Тепло реактивной струи ракеты частично поглощается при испарении цинкового покрытия, и стартовая конструкция не разрушается. Разнообразно применение соединений Ц. Сульфид Ц. входит в состав литопона — белой краски, получаемой по реакции:
ZnSO4 + BaS = BaSO4 + ZnS
Она дешевле свинцовых белил и не темнеет в присутствии сероводорода (см. Свинец), к тому же Ц. неядовит. Окись Ц. применяют как белый пигмент и как наполнитель в резиновой промышленности. Необходима она и в косметике, и в медицине (мази, пасты, присыпки при кожных заболеваниях). На люминесцентных свойствах ZnS основано одно из самых современных применений элемента: ZnS в смеси с CdS входит в состав покрытий для экранов телевизоров.
ЦИРКОНИЙ (лат. Zirconium) Zr — химический элемент IV группы периодической системы Менделеева; атомный номер 40, атомная масса 91,22. Блестящий твёрдый металл, похожий на сталь. Элемент состоит из смеси 5 стабильных изотопов с массовыми числами 90, 91, 92, 94 и 96. Из них наиболее распространён 90Zn (51,46%).
По содержанию в земной коре (1,7-10“ 2% по массе) Ц. занимает 19-е место и превосходит такие обычные металлы, как медь, цинк, олово, никель и свинец, но в 25 раз уступает титану. Породы, содержащие Ц., встречаются в Индии, Австралии, США и других странах, а у нас —
ЦИРКОНИЙ
342
на Украине, в Казахстане, Сибири. Народную легенду о кроваво-красном эвдиалите, одном из красивейших минералов Ц., записал выдающийся советский учёный А. Е. Ферсман в своей книге «Воспоминания о камне». Самые богатые минералы Ц.— бадделеит ZrO2 и циркон ZrSiO4. Желтовато-красные прозрачные кристаллы циркона называются гиацинтами, это очень дорогие и редкие драгоценные камни. Во всех природных соединениях Ц. всегда содержится примесь гафния (до 4—6%).
Ц. был открыт в 1789 г. немецким химиком М. Клапротом в драгоценном камне с острова Цейлон — цирконе, откуда и получил своё название (в пе
реводе с персидского — «золотистый»). Клапрот выделил лишь окисел нового элемента ZrO2, а в 1829 г. шведский химик Я. Берцелиус впервые получил сам металл в виде порошка, сильно загрязнённого примесями. Попытки выделить чистый металлический Ц. предпринимались неоднократно, но долгое время были безуспешными. В этом отношении истории титана и Ц. очень похожи — чистый Ц. также был получен только в 1925 г. методом иодидного рафинирования (см. Титан). Получавшийся до тех пор загрязнённый примесями Ц. был хрупок и не проявлял каких-либо важных для техники свойств.Тем удивительнее свойства, обнаруженные у чистого Ц.
Плотность Ц. 6,45 г/см3, /пл 1852 °C, /кип 3580—3700 °C. Сочетание сравнительно малой плотности с очень высокой тугоплавкостью и хорошей электропроводностью сделало Ц. ценным конструкционным материалом. Одно из самых важных его свойств — большая химическая стойкость.
В периодической системе элементов Ц. и следующий за ним гафний принадлежат к подгруппе титана (Ti—Zr—Hf). Атомы всех трёх элементов имеют четыре внешних (валентных) электрона (у Zr электронная конфигурация высших энергетических уровней 4d25s2, см. Атом). Но Zr и Hf имеют, кроме того, почти равные радиусы ионов, что объясняется так называемым «лантаноидным сжатием» (см. Лантаноиды). Это приводит к необычайному их химическому сходству, большему, чем у любой другой пары элементов-аналогов. В соединениях Ц. обычно проявляет степень окисления 4-4 (валентность IV). Природные соединения Ц. низших валентностей неизвестны, их получают в лабораториях довольно сложными методами. На Ц. не действуют вода, разбавленные кислоты и щёлочи. Он растворяется лишь в расплавах щелочей, во фтористоводородной кислоте, в царской водке, например: 3Zr 4- 4HNO8 4- 12НС1 = 3ZrCl4 4- 4NO + 8Н2О
На воздухе Ц. окисляется только с поверхности:
Zr 4- О2 = ZrO2
Образующаяся прочная поверхностная плёнка ZrO2 хорошо предохраняет его от дальнейшего окисления. Напротив, порошок Ц. в этих условиях горит, выделяя огромное количество тепла и расплавляя всё, с чем соприкасается. При повышенных температурах Ц. сильно поглощает газы (О2, N2, Н2, СО2) и становится хрупким. В таких же условиях Ц. соединяется с галогенами (хлором, бромом, иодом), например:
Zr 4- 2С12 = ZrCl4
При соприкосновении с парами воды образующиеся тетрагалогениды сразу гидролизуются по обратимой реакции:
ZrCl4 4- Н2О ZrOCl2 4- 2НС1
Т ехнологи я промышленного производства Ц. очень сложна. После обогащения руды и очистки полученных концентратов выделяют тетрахлорид ZrCl4, который нагревают в вакууме при 500 °C с металлическим натрием:
ZrCl4 4- 4Na = Zr 4- 4NaCl
Наиболее чистый Ц. получают (как и титан) иодидным рафинированием.
Ц. и его сплавы применяются в различных областях народного хозяйства. Из них изготовляют камеры сгорания и сопла реактивных двигателей, аппараты и трубопроводы для агрессивных жидкостей в химических производствах. В виде легирующих добавок Ц. значительно улучшает свойства многих металлов и сплавов. Так, легированная им сталь становится материалом для броневых плит, а кислотоупорность титана в присутствии Ц. увеличивается в 70 раз. Из Ц. делают хирургические инструменты и даже, пользуясь тем, что он не ржавеет, протезы некоторых частей человеческого организма, например участки кровеносных сосудов.
Многие соединения Ц. имеют сверхвысокие температуры плавления — таковы карбид ZrC (/пл 3530 °C), борид ZrB2 (3000 °C), нитрид ZrN (2980 °C). Огнеупорная керамика из этих материалов выдерживает огромные тепловые нагрузки. Но, пожалуй, самая важная, уникальная способность Ц. состоит в том, что он очень мало поглощает нейтроны (см. Уран). Благодаря этому Ц. служит материалом для всевозможных конструкций и деталей ядерных реакторов. Но, как нарочно, его химический близнец, гафний, наоборот, поглощает нейтроны настолько сильно, что Ц. даже с небольшой примесью гафния тормозит ядерные реакции и его нельзя употреблять в атомной энергетике. Поэтому так важна задача разделения Zr и Hf.
343
ЧУГУН
Михаил Александрович ПАВЛОВ —один из основоположников современной теории доменного процесса.
Советский металлург Михаил Александрович Павлов родился 21 января 1863 г. в посёлке Божий Промысел (ныне влился в Ленкорань Азербайджанской ССР) в семье кубанских казаков. Потеряв родителей в раннем детстве, остался на попечении деда. Окончил реальное училище в Баку в 1880 г. и поступил в Горный институт в Петербурге. Проходя студенческую практику на Юзовском заводе (ныне Донецкий металлургический завод имени В. И. Ленина), решил посвятить себя доменному производству, чтобы понять, что именно происходит внутри доменной печи (тогда этого никто толком не знал). По окончании курса, в 1885 г., Павлов работал инженером на заводах Вятского горного округа, а с 1896 г.— на Сулинском заводе (ныне Ростовская область), где поставил, впервые в России, доменную плавку на антраците. Для изучения доменного дела он побывал в Швеции и США. Павлов преподавал во многих высших учебных заведениях, особенно долго (в 1904—1941 гг.) он был профессором Петербургского (Ленинградского) политехнического института.
Первой крупной работой Павлова было «Исследование плавильного процесса доменных печей Климковского завода» (1902 г.), в котором он подробно изучил влияние различных факторов на ход доменного процесса. Павлов составил и издал ценные пособия по металлургии — атласы конструкций доменных и мартеновских печей, руководства для определения их рациональных размеров, руководства для расчёта доменных шихт. На его совершенно оригинальном курсе «Металлургия чугуна» (3 тома) воспитывались многие поколения советских доменщиков. В годы пятилеток Павлов активно участвовал в создании новых типов мощных доменных печей. В 1932 г. избран академиком. Он удостоен Государственных премий СССР (1943 и 1947 гг.) и звания Героя Социалистического Труда (1945 г.). М. А. Павлов умер 10 января 1958 г. в Москве. На фотографии — портрет М. А. Павлова на медали, отлитой в 1925 г. из чугуна (даты на медали: с 1885 г, Павлов — инженер, с 1900 г.— профессор).
Для разделения используют, например, различную растворимость комплексных солей этих металлов или различную летучесть их галогенидов. Однако до сих пор процессы разделения очень трудоёмки и дороги. Дальнейшее расширение использования Ц. сдерживается пока его высокой стоимостью. Химики должны разработать лучшую и более дешевую технологию производства и очистки Ц.
Ф. М. Спиридонов. ЧУГУН — сплав железа с углеродом (обычно около 4% С), содержащий примеси кремния, марганца, фосфора, серы, а иногда и других элементов (хрома, никеля, титана, ванадия); в отличие от большинства сортов стали, Ч. не выдерживает ковки. Состав Ч. зависит от состава шихты, то есть сырых материалов, загружаемых в доменную печь. (О выплавке Ч. рассказано в учебнике неорганической химии для*9-го класса средней школы; перед чтением настоящей статьи рекомендуем возобновить в памяти этот раздел.)
В истории материальной культуры выплавка Ч., развитие доменного производства — одна из самых интересных, хотя и трудных страниц. Шаг за шагом, сначала методом случайных наблюдений, проб и ошибок, а затем— научных исследований, накапливались здесь знания и технические усовершенствования.
Прочитав в этой книге статью о железе, вы узнаете, что в давние времена металлурги не умели получать его в жидком состоянии. В пе
чах, что сооружали в древности, температуру, близкую к точке плавления железа (1539 °C), невозможно было достигнуть. Удавалось получить лишь так называемое мягкое сварочное железо, легко поддающееся деформации ковкой. Но в странах Средиземноморья, по-ви-димому в IV—VI вв. до н. э., было сделано такое наблюдение. Если увеличивать высоту сыродутного горна и в смесь железной руды и раскалённого древесного угля подавать больше воздуха (то есть повышать температуру в печи), то образуется жидкий металл, который собирается внизу горна. Сейчас мы знаем, что при высокой температуре углерод древесного угля соединяется с железом, образуя цементит Fe3C (карбид железа), который растворяется в железе и снижает температуру его плавления с 1539 °C до 1145 °C (при 4,3% Спо массе). В застывшем виде этот металл оказался твёрдым и хрупким, ковать его было нельзя; поэтому вначале его считали отходом производства [по-английски и сейчас Ч. называется «пиг айрон» (pig iron) — «свинское железо»]. Но позднее из Ч. стали отливать сковороды, котлы для варки пищи и даже артиллерийские орудия.
Начало более широкого применения Ч. относится к середине XIV в., когда появился кричный способ передела Ч. (см. Сталь). Тогда же в Западной Европе возникла выплавка Ч. в «домницах» — доменных печах небольшой высоты (до 5 м) и малой производитель
ЧУГУН
344
ности (до 1,6 т Ч. в сутки). В России первые доменные печи появились в XVII в. (близ Тулы и в Олонецком крае). На Урале выплавка Ч. началась в 1701 г. и к середине XVIII в. Россия по производству Ч. обогнала Англию и Швецию.
Термины «домница» и «доменная печь» происходят от славянского «дмати» — дуть. В домницы сверху загружали железную руду и древесный уголь. Снизу мехами подавали дутьё (воздух) по трубе, вставленной в отверстие («фурму»). Окислы железа, содержащиеся в руде, восстанавливались древесным углём до железа. Эти реакции известны из школьного учебника, и мы лишь напомним их. Углерод (в то время из древесного угля) окисляется кислородом воздуха:
С + О2 = СО2; С + СО2 = 2СО Образующаяся при этом окись углерода, а также сам углерод восстанавливают окислы железа:
3Fe2O3 -J- СО = 2Fe3O4 СО2
Fe3O4 + СО = 3FeO + СОа
FeO + СО = Fe + СО2 FeO + C = Fe + CO Как уже было сказано, раскалённое железо,
соприкасаясь с углём, превращалось в жидкий Ч., стекавший в нижнюю часть печи — горн. Однако в руде имеются не только окислы железа, но и пустая порода, содержащая в основном тугоплавкие вещества: кремнезём SiO2 и глинозём А12О3. В сыродутных и кричных горнах SiO2 соединялся с закисью железа FeO по реакции:
2FeO + SiO2 = Fe2SiO4
Полученное соединение образовывало с избытком кремнезёма SiO2 легкоплавкий жидкотекучий шлак.
Для перевода в шлак SiO2 и А12О3 в доменную шихту стали добавлять известняк. Образующаяся при его разложении известь СаО даёт с SiO2 и А12О3 соединения, смесь которых плавится около 1250—1300 °C.
Уже в первой половине XVIII в. в Англии вследствие быстрого истребления лесов на смену древесному углю пришло новое топливо — кокс, получаемый спеканием каменного угля. Если бы кокс не появился, то, вероятно, всех лесов Земли не хватило бы для снабжения доменных печей топливом. Но новое топливо принесло и новые трудности. Прежде всего, ведение доменного процесса на коксе потребовало более мощных воздуходувных машин. Только в 1782 г. в доменных цехах появились воздуходувки, приводимые в действие паровыми машинами, что позволило значительно увеличить производительность доменных печей.
Рис. 1. Доменный цех Донецкого металлургического завода им. В. И. Ленина. Все знают, что чугун и сталь — это основа машиностроения и строительства, и что чёрная металлургия — это база для технического оснащения всех отраслей народного хозяйства.
345
ЧУГУН
Иван Павлович БАРДИН — один из создателей современной чёрной металлургии.
Советский металлург Иван Павлович Бардин родился 13 ноября 1883 г. в селе Широкий Уступ (ныне Саратовской области) в семье бедного деревенского портного. В 1903 г. он поступил в Институт сельского хозяйства в Ново-Александрии (ныне Пулавы, Польша), откуда был исключён1 за участие в студенческих волнениях. В 1910 г. окончил Киевский политехнический институт. Будучи студентом, под влиянием профессора В. П. Ижевского увлёкся металлургией. Но получить работу на отечественных заводах Бардину не удалось — мешала «политическая неблагонадёжность». В поисках заработка он отправился в США. Однако там, несмотря на диплом инженера, ему пришлось зарабатывать на жизнь тяжёлым физическим трудом — сборщиком сельскохозяйственных машин, подручным кузнеца, рабочим прокатного цеха. Вернувшись на родину, он в 1912 г. устроился чертёжником на Юзовском (ныне Донецком металлургическом) заводе. Там он встретился с замечательным металлургом М. К. Курако, который, не имея специального образования, был начальником доменного цеха. Бардин стал его помощником, а после ухода Курако — начальником цеха. В 1923 г. Бардин был командирован в Германию, Бельгию и Англию. По возвращении работал на важных металлургических предприятиях страны.
В 1929—1936 гг. Бардин был одним из руководителей строительства Кузнецкого металлургического комбината. Здесь в труднейших условиях, при морозах ниже —30 ° С, на пустом месте было создано одно из крупнейших советских предприятий чёрной металлургии. В 1937 г. Бардин был переведён в Москву, где занимал руководящие должности по чёрной металлургии. В 1932 г. он избран академиком, в 1939 г. стал директором Института металлургии АН СССР. Во время Великой Отечественной войны он руководил работами Академии наук на нужды обороны. Бардин получил Государственные премии СССР за работы по мобилизации ресурсов Урала (1942 г.) и по применению кислорода для интенсификации мартеновского процесса (1949 г.); Ленинскую премию (1958 г.) — за создание первых установок для непрерывной разливки стали. В 1945 г. удостоен звания Героя Социалистического Труда. Умер в Москве 7 января 1960 г. Центральный научно-исследовательский институт чёрной металлургии в Москве носит имя И. П. Бардина. Изданы 2 тома его трудов.
Другую трудность вызвало содержание серы в коксе (в древесном угле серы нет). Присутствие серы в Ч. вредно, поскольку высокое её содержание ухудшает жидкотекучесть и механические свойства Ч., делает отливки из него пузыристыми. В передельном же Ч., идущем на производство стали, должно быть не выше 0,05% S. Для уменьшения содержания серы в Ч. его выплавку ведут на сильнооснбвном шлаке, богатом известью. При этом происходит реакция:
FeS + СаО + С = Fe + CaS + СО
Сульфид кальция уходит в шлак.
Кокс — самая дорогая составная часть доменной шихты. Для его производства необходимы особые сорта каменных углей, хорошо спекающиеся, бедные золой и серой. Ещё более дорогим был древесный уголь. Поэтому вполне понятно стремление всячески уменьшить расход топлива в доменных печах. В 1829 г. начали подавать в доменные печи воздух, предварительно пропущенный через чугунные трубы, нагреваемые каменноугольными топками. Уже сравнительно невысокий (150—300°C) подогрев дутья существенно сократил расход горючего. И ещё он уменьшился после того, как (с 1832 г.) воздухонагреватели начали отапливать колошниковым газом, то есть отходящим газом доменных печей. В 1857 г. английский инженер Э. А. Каупер сконструировал воздухонагреватель, в котором насадка из огнеупорного кирпича попеременно то нагревается пламенем колошникового газа, то отдаёт накопленное ею тепло дутью (воздуху), затем поступающему в доменную печь. Применение горячего дутья позволило значительно сократить расход кокса. В наше время, с 1950-х гг., с целью экономии кокса в доменные печи стали вдувать природный газ, состоящий преимущественно из метана СН4, а позднее — мазут, угольную пыль. Это одновременно улучшило ход восста
новления руды и повысило производительность печи. Температура дутья обычно достигает 1000—1200 °C. Обогащение дутья кислородом повышает температуру и значительно ускоряет работу печи.
Постепенное истощение залежей богатых железных руд привело к применению руд, содержащих меньше железа и больше пустой породы, что увеличивало массу шлака и расход топлива. В XIX в. были построены первые рудообогатительные фабрики, где из руды водой отмывали пустую породу. А в XX в. на обогатительных фабриках, в специальных агрегатах — сепараторах железная руда отделялась от пустой породы с помощью электромагнитов. Однако для лучшего их разделения руду приходилось дробить. При этом получались мелкие, даже пылеватые рудные концентраты. Для загрузки в печь они непригодны, так как идущий навстречу шихте поток газов переносит пыль в газопроводы и газоочистители. Поэтому были изобретены способы окускования железных руд: брикетирование, агломерация и окомкование (производство окатышей). Рудные материалы поступают теперь в печь в виде кусков, и вынос пыли незначителен. «.
Основную.массу Ч., выплавляемого в доменных печах, составляет белый, или передельный, Ч., служащий сырьём для производства стали. На долю литейного Ч. приходится примерно 10% металла, получаемого в доменных печах; он идёт на отливку изделий.
В литейном, или сером, Ч. углерод присутствует преимущественно в виде графита, чем объясняется тёмносерый, почти чёрный, цвет излома этого Ч. Для него также типично высокое содержание кремния (2—3%), который сообщает Ч. жидкотекучесть, то есть способность
ЧУГУН
346
хорошо заполнять литейные формы. Однако восстановление Si из SiO2 требует высокой температуры. Поэтому при производстве литейного Ч. увеличивается расход кокса. В настоящее время, чтобы получить литейный Ч., в передельном Ч. растворяют ферросилиций — сплав Fe и Si (см. Ферросплавы).
Литейный Ч. обычно содержит 0,5—1,2% Мп, а также 0,03—0,08% Р- Более высокое содержание фосфора делает Ч. хрупким, однако в Ч., предназначенном для художественного литья, бывает и больше фосфора, так как он увеличивает жидкотекучесть сплава. Серы допускается до 0,03%, об её вредном действии сказано выше.
Серый Ч. применяется в машиностроении для производства фасонных изделий посредством литья в песчаные или металлические формы (кокили). В последние десятилетия получил распространение способ литья под давлением: жидкий Ч. (или другой металл, сплав) поступает под давлением (от 20 до 1500 атм) в стальную пресс-форму. После затвердевания и охлаждения получается плотная отливка с гладкой поверхностью, обычно требующая лишь минимальной отделки.
Механическая прочность и химическая стойкость серого Ч. невысоки. Они могут быть значительно улучшены введением некоторых добавок. Такой легированный Ч. служит для изготовления изделий, подвергающихся сильным и продолжительным механическим, тепловым и химическим воздействиям. Например, хромистый Ч. (26—36% Сг) очень прочен при температурах до 500 °C, стоек против окисления на воздухе до 1100°С, а также в кислотах (из него изготовляют химическую аппаратуру, печную арматуру, трубопроводы). Алюминиевый Ч., или чу га ль (20—24% А1), стоек против окисления до 1000 °C (он идёт на детали печей к топок). Сил ал (5—
Рис. 2. Решётка Летнего сада в Ленинграде работы архитекторов Ю. М. Фель те на и П. Е. Егорова (1784 г.). Эта ажурная чугунная ограда, отделяющая сад от набережной Невы, принадлежит к шедеврам не только русского, но и мирового искусства. Чёткий узор решётки оживлён изящными бронзовыми позолоченными вставками.
Рис. 3. П. К. Клодт. Лошади. Отливка 1915 г. Каслинский завод. Из собрания Свердловской картинной галереи.
Город Касли (Челябинская область, РСФСР) издавна славится художественным чугунным литьём (чугунолитейный завод был основан здесь ещё в 1747 г.). «Каслинское литьё» — архитектурные украшения и настольная скульптура — не раз получали высокую оценку на международных и отечественных выставках.
6% Si) обладает жароупорностью до 800 °C; ферро си л ид, или термисилид (14—18% Si), жароупорен до 900°C и устойчив против всех минеральных кислот, кроме HF и НС1; добавка 3,5—4,0% Мс значительно повышает его стойкость против НС1. Марганцовый Ч. (7— 12% Мп) практически немагнитен и имеет высокое электросопротивление (140—160 мком*см), он применяется в электротехнической аппаратуре.
Низкая прочность и хрупкость серого Ч. в значительной степени связаны с тем, что в нём графит присутствует в виде сравнительно крупных, пластинчатых выделений. Превращение их в мелкие образования приближённо сферической формы («сфероидизация» графита) сопровождается значительным повышением прочности Ч. и улучшением его пластичности. Такая сфероидизация графита может быть достигнута введением в жидкий Ч. 0,15% мишметалла (технический сплав 52% Се, 18% Nd, 5% Рг, 24% La и примесей). Эта добавка также улучшает прочность и пластичность Ч.; дальнейшие добавки мишметалла (до 0,75%) повышают жидкотекучесть, горячую ковкость и стойкость против окисления.
Доменный шлак состоит в основном из SiO2, А12О3, СаО и MgO. Шлак либо увозят в отвал, либо гранулируют, то есть выливают в воду, где он застывает в виде небольших кусочков. Гранулированный шлак применяют в строительстве для сооружения дорог, для производства теплоизолирующих материалов — шлаковой ваты и шлаковой пемзы. При плавке высокоглинозёмистых руд получаются шлаки с повышенным содержанием А12О3; из них делают шлаковый цемент.
Современный доменный цех — совокупность различных сооружений и установок. Запасы сырых материалов — руды, кокса и флюса, хранятся в бункерных эстакадах высотой 10 м и более. Из них шихтовые материалы поступают на автоматические весы, а затем в специальные вагонетки (скипы). Они по наклонному подъёмнику доставляют шихту в засыпной аппарат, равномерно распределяющий руду, кокс и флюс по сечению печи. Все
347
ЧУГУН
эти работы полностью автоматизированы. Доменная печь снабжена множеством приборов, показывающих уровень шихты, температуру и давление дутья, температуру и состав колошникового газа. Показания приборов позволяют следить за работой печи и устранять все отклонения от нормы. Из горна периодически выпускают Ч. и шлак. Ч. поступает по желобам в ковши ёмкостью 100 т и выше, которые отвозят по рельсовым путям в передельный цех. Литейный Ч. отливают в «чушки» на разливочных машинах.
Доменная печь работает непрерывно несколько лет (в среднем около 5, иногда 8— 10), после чего её ставят на капитальный ремонт.
Доменный процесс — один из сложнейших и самых «капризных» в металлургии. Изучение теории процесса началось ещё в середине XIX в. Большой вклад внесли в неё русские и советские учёные А. А. Байков, М. А. Павлов, И. П. Бардин и многие другие.
М. А. Павлов так вспоминает о своей студенческой практике в доменном цехе Юзовского (ныне Донецкого) металлургического завода в 1884 г.
«Около домен „колдовал44 старый мастер англичанин, который, словно по наитию, добавлял разные материалы в шихту или снимал их. Он ничего не объяснял и, может быть, не смог бы объяснить, если бы захотел. В доменную печь нельзя заглянуть; никто не видит, что в ней делается, но инженер должен это знать, чтобы управлять процессом. Как же это сделать? Я ходил и ходил вокруг доменной печи, делая записи веса материалов, загруженных в печь, описывая внешний вид шлака и чугуна, просил у И. Юза анализы руд, но постоянно наталкивался на множество непонятных, необъяснимых для меня явлений в ходе печи.
— Ну, нет, этого я так нс оставлю. Я тебя буду понимать, — мысленно говорил я, обращаясь к домне, как к живому существу.
И вот, кончается моя долгая жизнь, а я всё ещё работаю над решением этой задачи».
На основе широких научных исследований удалось создать высокомеханизированные и автоматизированные доменные печи, дающие необходимый народному хозяйству чугун. Если в 1913 г. Россия занимала 5-е место в мире погодовой выплавке Ч. (4,2 млн. т), то уже в 1940 г. СССР вышел на 3-е место (15 млн. т), в 1947 г.— на 2-е место (после США), а в 1970 г. обогнал США. В 1974 г. выплавка Ч. в нашей стране составила 99,9 млн. т, то есть увеличилась почти в 7 раз по сравнению с той, которая была до Великой Отечественной войны.
В феврале 1973 г. на Новолипецком металлургическом заводе задута доменная печь с годовой производительностью 2,2 млн. т чугуна (или около 6 тыс. т в сутки). Её полезный объём 3200 м3. А доменная печь № 9 Криворожского металлургического завода имени В. И. Ленина, задутая в конце 1974 г., имеет полезный объём 5000 м3 и даёт в сутки 11 тыс. т чугуна. Это не только самая большая в мире печь, но и самая совершенная. Управление всеми механизмами — дистанционное, плавильный процесс регулируется с помощью электронно-вычислительных машин, дутьё нагревается до 1450 °C. Ежегодно она будет производить 4 млн. т чугуна, то есть примерно столько, сколько все доменные печи России в 1913 г. С. А. Погодин.
«Научные открытия и успехи не происходят вдруг; они накопляются мало-помалу; нужно много работы для того, чтобы могло появиться лицо, охватывающее запас накопившихся знаний, умеющее добыть из этого запаса новую крупную идею, закон, управляющий совокупностью явлений».
Д. И. Менделеев
(Соч., т. 24, 1954, с. 235)
9
ЩЕЛОЧНОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ — химические элементы, входящие в главную подгруппу II группы периодической системы Менделеева: кальций Са, стронций Sr, барий Ва и радий Ra. Гидроокиси этих элементов — сильные основания — похожи по свойствам на гидроокиси щелочных металлов', окислы же их тугоплавки и напоминают окислы алюминия и некоторых тяжёлых металлов (такие окислы прежде называли «землями»). Отсюда и название «Щ. м.».
Важной особенностью Ra является его радиоактивность; о радиоактивных свойствах Ra подробно рассказано в статье, посвящённой этому элементу.
Щ. м.— серебристо-белые, блестящие, мягкие металлы, быстро тускнеющие на воздухе. Соли щ. м. окрашивают бесцветное пламя горелки в характерные цвета: Са — кирпичнокрасный, Sr — карминово-красный, Ва — желтовато-зелёный. Это используют для качественного обнаружения элементов. Атомные радиусы элементов главной подгруппы II группы (см. таблицу) значительно меньше радиусов соседних щелочных металлов. Это связано с возрастанием зарядов атомных ядер и полным заполнением электронами s-уровней внешних оболочек. Поэтому металлические связи вЩ. м. более прочны, чем в щелочных металлах. А это проявляется в более высоких точках плавления и кипения и больших значениях плотностей у щ. м.
Щ. м. близки друг другу по химическим свойствам, что объясняется сходством строения их атомов. Внешняя электронная оболочка атома содержит два электрона (конфигурация s2); ей предшествует устойчивая оболочка инертного газа (s2p6)- Поэтому атомы Щ. м. легко отдают два своих валентных электрона, переходя в ионы Ме2+:
Me — 2е — Ме2+
В соединениях они проявляют степень окисления -г2 (валентность II). Однако вследствие уменьшения атомного радиуса по сравнению со щелочными металлами отрыв электронов происходит уже с большим трудом, о чём свидетельствуют большие величины энергии ионизации (см. таблицу). Химические связи Щ. м. с элементами-неметаллами носят преимущественно ионный характер.
Щ. м.— сильные восстановители. Они принадлежат к числу наиболее активных металлов и располагаются в начале ряда напряжений (см. Металлы). Щ. м. разлагают воду с выделением вод о р ода:
Me - 2Н2О - Me (ОН)2 -г Н2
Они непосредственно взаимодействуют почти со всеми неметаллами. При сжигании Щ. м. на воздухе образуются их окислы МеО. Соответствующие гидроокиси Me (ОН)2 — сильные основания, растворимые в воде (сила основания и растворимость увеличиваются при переходе от Са к Ва). В отличие от щелочных
Основные константы элементов главной подгруппы II группы периодической системы (щелочноземельных металлов, бериллия и магния)
Элемент Атомный номер Атомная масса Конфигурация внешней электронной оболочки Атомный радиус (A)* Ионный радиус (A)* Энергия ионизации I (эв) ** Энергия ионизации II (эв)** Температура плавления (°C) Температура 1 кипения (°C) Плотность (г/см3)
Be 4 9,01218 2s2 1,13 0,31 9,32 18,21 1284 2450 1,848
Mg 12 24,305 3s2 1,60 0,65 7,64 15,03 651 1107 1,739
Са 20 40,08 4s2 1,97 0,99 6,11 11,87 851 1482 1,54
Sr 38 87,62 5s2 2,15 1,13 5,69 11,03 770 1380 2,63
Ва 56 137,34 6s2 2,21 1,35 5,21 10,00 710 1640 3,76
Ra 88 226,0254 7s2 2,35 1,44 5,277 10,144 960 1140 5,5
* А — ангстрем;- 1 А=10~8 см. ** эв — электронвольт; I—для 1-го электрона, II—для 2-го электрона.
349
металлов, у Щ. м. есть довольно много малорастворимых солей: сульфаты, фосфаты, карбонаты и др. В водных растворах соли, содержащие ионы Ме2+, гидролизу не подвержены.
Кроме упомянутых четырёх элементов, в главную подгруппу II группы входят бериллий Be и магний Mg; однако их, как правило, не относят к Щ. м.
Бериллий по химическим свойствам больше напоминает алюминий, чем своих соседей по подгруппе. Дело в том, что ион Ве2+ очень малых размеров (сравните в таблице ионные радиусы). Поэтому Ве2-к взаимодействует с анионами гораздо сильнее, чем ионы Щ. м., и для него характерно образование ковалентных связей. Гидроокись Ве(ОН)2, как и гидроокись А1 (ОН)3, проявляет заметные амфотерные свойства. Второй элемент подгруппы — магний, тоже отличается от Щ. м. Так, его гидроокись Mg(OH)2 слаборастворима в воде (как у Be), а сульфат MgSO4, наоборот, хорошо растворим (в этом отношении Mg близок к цинку, стоящему в побочной подгруппе). Однако, чтобы показать ход изменения свойств по II группе системы, мы включили в таблицу и эти элементы.
Все Щ. м. довольно широко распространены в природе. Вследствие высокой химической активности они никогда не встречаются в свободном состоянии, а только в виде соединений. Как правило, все Щ. м. получают электролизом их расплавленных солей (обычно хлоридов) или гидроокисей.
Подробно об отдельных элементах см. Кальций, Стронций, Барий, Радий. См. также статью Закон Менделеева. В. К- Бельский. ЩЕЛОЧНЫЕ МЕТАЛЛЫ — химические элементы, составляющие главную подгруппу I группы периодической системы Менделеева: литий Li, натрий Na, калий К, рубидий Rb, цезий Cs и франций Fr. Франций радиоактивен, причём все его изотопы очень быстро распадаются. Поэтому Fr в весомых количествах не выделен, и его свойства изучены мало.
Гидроокиси Щ. м.— самые сильные осно-вайия — называются едкими щелочами; отсюда и название этой подгруппы элементов. Щ. м.— серебристо-белые блестящие металлы, быстро тускнеющие на воздухе. Они очень
ЩЕЛОЧНЫЕ МЕТАЛЛЫ
мягки (легко режутся ножом). Соли Щ. м* окрашивают бесцветное пламя горелки в ха" рактерные цвета: Li — красный, Na — жёлтый» К, Cs — фиолетовый, Rb — пурпурово-красный. Это используют для качественного обнаружения элементов.
Физические свойства Щ. м. закономерно изменяются от Li к Cs (см. таблицу). Температуры плавления металлов (кстати, весьма низкие у всех Щ. м.) и кипения убывают, а плотности возрастают при переходе от Li (он почти в два раза легче воды) к Cs (почти в два раза тяжелее её). Такая закономерность связана с кристаллическим строением металлов. Кристаллические решётки всех Щ. м. одинаковы по своему типу, но расстояния между центрами атомов в решётках изменяются. Это объясняется тем, что атомные радиусы при переходе от Li к Cs увеличиваются (см. таблицу): у каждого последующего элемента на одну электронную оболочку больше, чем у предыдущего, и атомы становятся больше. Связи между атомами ослабляются, и решётку становится легче разрушить. Поэтому и понижаются температуры плавления. Плотности же металлов от Li к Cs увеличиваются оттого, что масса атомов возрастает значительнее, чем объём.
По химическим свойствам все Щ. м. очень близки. Это объясняется сходством строения их атомов. Внешняя электронная оболочка содержит один электрон (конфигурация s1); ей предшествует устойчивая оболочка инертного газа (s2p6)- Поэтому атомы Щ. м. легко отдают свой единственный валентный электрон, переходя в ионы Ме+:
Me—1ё = Ме+
В соединениях они проявляют степень окисления + 1 (валентность I).
Вследствие увеличения атомного радиуса и увеличения числа электронных оболочек при переходе от Li к Cs уменьшаются силы электростатического взаимодействия между ядром и валентным электроном, а следовательно, умень
Основные константы щелочных металлов
Элемент Атомный номер Атомная масса Конфигурация внешней электронной оболочки Атомный радиус (A)* Ионный радиус (А)* Энергия ионизации (эв) ** Температура плавления (°C) Температура кипения (°C) Плотность (г/см3)
Li 3 6,941 2s1 1,55 0,60 5,390 180,5 1317 0,534
Na 11 22,98977 3s1 1,89 0,95 5,138 97,83 882,9 0,968
К 19 39,098 4s1 2,36 1,33 4,339 63,55 760 0,862
Rb 37 85,4678 5sl 2,48 1,48 4,176 39 696 1,532
Cs 55 132,9054 6s1 2,68 1,69 3,893 28,5 705 1,90
• о
* А — ангстрем; 1 А=10-8 см. ** эв — электронвольт.
ЭЙНШТЕЙНИЙ
350
шается энергия, необходимая для его отрыва (обратите внимание на уменьшение приведённых в таблице энергий ионизации). Иными словами, отрыв электрона легче проходит для каждого последующего Щ. м., чем для предыдущего, то есть активность вступления Щ. м. в химические реакции возрастает в последовательности Li, Na, К, Rb, Cs. Связи Щ. м. с элементами-неметаллами носят преимущественно ионный характер. Все Щ. м.— сильные восстановители. Они располагаются в начале ряда напряжений (см. Металлы) и разлагают воду с выделением водорода:
2Ме + 2Н2О = 2МеОН + Н2
Они быстро окисляются на воздухе, взаимодействуют непосредственно почти со всеми неметаллами и многими металлами. Поэтому Щ. м. хранят под слоем органических жидкостей. Соли Щ. м. хорошо растворимы в воде; исключение составляют LiF, Li2CO3 и Li3PO4, а также соли хлорной кислоты МеС1О4 (где Me—К, Rb, Cs, Fr).
Вследствие высокой химической активности Щ. м. встречаются в природе не в свободном виде, а только в соединениях. Получают Щ. м. электролизом их расплавленных хлоридов (Li, Na, К), нагреванием хлоридов с кальцием в вакууме (Rb, Cs) и другими способами. Подробно об отдельных элементах см. Литий, Натрий, Калий, Рубидий, Цезий и Франций. См. также статью Закон Менделеева. В. К, Бельский.
ЭЙНШТЕЙНИЙ (лат. Einsteinium) Es— радиоактивный химический элемент с атомным номером 99; относится к актиноидам.
ЭЛЕМЕНТЫ ХИМИЧЕСКИЕ — составные части простых и сложных веществ; каждый Э. х. представляет собой совокупность атомов определённого вида, обладающих одним и тем же зарядом ядра. В химии Э. х. принято называть просто элементами. В 1975 г. известно 106 Э. х.; из них 89 обнаружены в природе, а остальные получены искусственно. (Элемент № 106 синтезирован советскими физиками в 1974 г.) Э. х.— это как бы кирпичики разных сортов, из которых построено всё многообразие окружающего мира. Э. х. иногда сравнивают с буквами алфавита, из ограниченного числа которых можно составить сотни тысяч различных слов.
Каждый Э. х. имеет особое название. На разных языках название одного и того же элемента может и записываться, и звучать совершенно по-разному. Так, элемент, известный нам как олово, англичане называют tin (произносится «тин»), а немцы — Zinn (произносится «цинн»). Чтобы облегчить общение хими
ков разных стран, с давних времён принято давать Э. х. латинские названия, например олово по-латыни называется Stannum (произносится «станнум»). Когда-то латынь была международным языком науки, и такие названия были понятны учёным всех наций. В наши дни латынь знают немногие, но традиция сохранилась: каждому новооткрытому элементу обязательно дают латинское название. Например, название элемента менделевия (данное в честь великого русского учёного) по-латыни пишется Mendelevium, звучит «менделевиум». От латинских названий элементов происходят и сокращённые обозначения Э. х.— их символы. По предложению шведского химика Я. Берцелиуса— законодателя химии первой половины XIX в., символы состоят из первой буквы латинского названия элемента, за которой часто пишут ещё одну из следующих букв. Первая буква всегда прописная, вторая — строчная. Так, символ углерода С (лат. СагЬо-neum), кальция Са (лат. Calcium), кобальта Со (лат. Cobaltum) и т. п. Но запись СО, состоящая из двух прописных букв, относится не к элементу, а к химическому соединению (окиси углерода), в состав которого входит 1 атом углерода (символ С) и 1 атом кислорода (символ О). Такую запись называют ф о р м у-л о й химического соединения. Символы большинства элементов произносят так же, как названия этих элементов звучат по-русски, например Be — бериллий, F—фтор, Ва — барий, W — вольфрам. Исключения (случаи, когда произношение символа не совпадает с русским названием элемента) приведены в таблице.
Произношение символов некоторых элементов
Элемент Символ Произношение символа Элемент Символ Произношение символа
Водород Н аш Медь Си купрум
Углерод (2 це Мышьяк As арсеникум
Азот N эн Серебро Ag аргентум
Кислород О о Олово Sn станнум
Кремний Si силициум Сурьма Sb стибиум
Фосфор Р ПЭ Золото Au аурум
Сера S эс Ртуть Hg гидраргирум
Железо Fe феррум Свинец Pb плюмбум.
Каждый Э. х. занимает определённое место в периодической системе элементов Д. И. Менделеева (см. Закон Менделеева). Атомный номер Э. х. в периодической системе равен электрическому заряду ядра его атома, выраженному в единицах заряда протона. Поэтому атомный номер однозначно характеризует элемент. В свою очередь, заряд ядра равен числу содержащихся в нём протонов; иными
351
ЭЛЕМЕНТЫ ХИМИЧЕСКИЕ
Йенс Якоб БЕРЦЕЛИУС — законодатель химии первой половины XIX в.
Шведский химик и минералог Йенс Якоб Берцелиус родился 20 августа 1779 г. в селе Веферсунда в крестьянской семье. Во время обучения в Упсальском университете юноша живо интересовался химией, однако окончил университет (1802 г.) со степенью доктора медицины. И всё же былое увлечение не прошло даром. Редкий талант и завидное трудолюбие вскоре выдвинули Берцелиуса в число крупнейших исследователей-химиков. С 1807 г. он профессор университета в Стокгольме, с 1810 по 1832 г.— Медико-хирургического института там же.
Основная историческая заслуга Берцелиуса — экспериментальное обоснование и развитие химической атомистики. Он очень точно для своего времени определил атомные веса почти всех известных тогда элементов, установил состав и формулы около 2000 соединений. Берцелиус создал химическую символику (1814 г.) и номенклатуру, принципами которых мы продолжаем пользоваться. Он открыл химические элементы церий, селен и торий; впервые получил в свободном состоянии кремний, цирконий, титан и тантал. Берцелиус положил начало методике количественного анализа органических соединений.
Шведский учёный развивал прогрессивные для своего времени идеи об электрической природе химического сродства. По мнению Берцелиуса, в молекуле химического соединения различные части связаны между собой силами электрического притяжения. Электрохимическая теория Берцелиуса сильно повлияла на развитие всей химии XIX в.
Авторитет Берцелиуса в научном мире был необычайно высок, недаром современники величали его патриархом химии. В возрасте чуть более тридцати лет он стал президентом Стокгольмской академии наук, а с 1818 г. долгие годы оставался её непременным секретарём. Был иностранным членом многих академий наук, в том числе Петербургской, Парижской и Лондонского Королевского общества.
Из года в год, на протяжении 27 лет (начиная с 1821 г.), Берцелиус публиковал фундаментальные «Обзоры успехов физики и химии» — своеобразную критическую энциклопедию физических и химических исследований. Сейчас кажется удивительным, что это было под силу одному человеку, даже такому гению как Берцелиус. Но «Обзоры» появлялись регулярно, и, если можно так выразиться, дирижировали развитием химии. Многотомный «Учебник химии» Берцелиуса был непревзойдённым в первой половине XIX в. и переводился почти на все европейские языки.
Работал Берцелиус в своей домашней лаборатории, занимавшей две небольшие комнаты, одна из которых служила также кухней. Жил очень скромно, был прост в обращении, пользовался любовью и уважением своих учеников. Он умер 7 августа 1848 г. в Стокгольме. Его научная школа вышла далеко за границы Швеции.
словами, ядра всех атомов данного Э. х. содержат одно и то же число протонов (масса протона приблизительно равна 1 углеродной единице, его заряд равен +1, см. Атом). Например, в ядре каждого атома углерода С (атомный номер 6) 6 протонов. Кроме протонов, в состав атомных ядер входят и нейтроны, причём число нейтронов в атомных ядрах одного и того же Э. х. может быть различным (масса нейтрона также приблизительно равна 1, но он не имеет заряда). Атомы, ядра которых содержат одинаковое число протонов и разное — нейтронов, называют изотопами; каждый Э. х. имеет по несколько изотопов (природных или полученных искусственно). У каждого элемента массы атомов его изотопов несколько различаются между собой. Встречающиеся в природе Э. х. имеют определённый изотопный состав, а процентное содержание отдельных его изотопов называется относительной распространённостью изотопов данного Э. х.
Нужно иметь в виду, что символом изотопа служит символ соответствующего Э. х., но при обозначении изотопа вверху слева указывают его массовое число, то есть общее число протонов и нейтронов в ядре, а внизу слева — атомный номер, то есть число протонов в ядре. Так, углерод имеет два природных изотопа с массовыми числами 12 и 13, что записывают соответственно и 13С. Изотопам же водорода }Н, |Н, ?Н, свойства которых из-за значительных различий масс различаются между собой довольно сильно, даны индивидуальные названия и символы (соответственно, протий Н, дейтерий D и тритий Т). Часто при обозначении изотопов атомный номер не указывают, например пишут просто 12С и 13С. Иногда массовое число и атомный номер пишут справа, например Q2.
Важная характеристика Э. х., встречающихся в природе,— атомная масса элемента (средняя масса атома элемента). Напомним, кстати, что атомную массу принято выра
ЭЛЕМЕНТЫ ХИМИЧЕСКИЕ
352
жать в углеродных единицах массы; одна углеродная единица массы составляет х/12 часть массы атома изотопа углерода 12С. Если изотопный состав элемента известен, то его атомную массу можно найти как среднюю величину масс атомов всех его изотопов с учётом распространённости изотопов. Так, природный хлор состоит из двух изотопов — 35С1 и 37С1, массы которых равны соответственно 34,964 и 36,961, а относительные распространённости составляют 75,53 и 24,47%. Атомная масса элемента хлора равна:
34,964x75,53 + 36,961 x24,47 о г-
-----------ioo-----------= 35>453
Все вещества, образованные Э. х., можно подразделить на две большие группы: на вещества простые и сложные. Простые образуются путём сочетания атомов одного и того же Э. х., сложные состоят из атомов нескольких Э. х. В простом веществе атомы Э. х. могут быть либо химически связаны между собой (примерами служат алмаз, графит, кислород, озон и т. д.), либо находиться в свободном, химически не связанном состоянии (пары некоторых металлов и инертные газы). Сложными называют только такие вещества, в молекулах которых атомы различных Э. х. связаны между собой химической связью. Так, сульфид железа FeS, где между атомами серы и железа в кристаллах существует химическая связь,— сложное вещество, а порошок, состоящий из железных опилок и элементарной серы,— смесь двух простых веществ.
Простое вещество — это форма существования Э. х. в свободном состоянии. Одному элементу часто соответствует несколько простых веществ (то есть несколько аллотропных форм, или аллотропных модификаций). Так, элементу углероду соответствует два природных простых вещества — алмаз и графит, элементу кислороду — также два — кислород и озон. В первом случае аллотропия обусловлена раз
ным характером химической связи атомов С в алмазе и в графите, во втором — неодинаковым числом атомов О в молекулах О2 и О3.
Названия Э. х., за исключением углерода, совпадают с названиями соответствующих им простых веществ (по крайней мере, с названием одной из аллотропных форм). Поэтому понятия «элемент химический» и «простое вещество» часто путают. Отметим основные различия между ними. Каждому Э. х. присущи: строго определённый заряд ядра его атомов (атомный номер), атомная масса, природный изотопный состав, степени окисления, в которых элемент даёт химические соединения, и некоторые другие характеристики. Любое простое вещество обладает определённой плотностью, температурами плавления и кипения, растворимостью и многими другими свойствами, которые различны для разных простых веществ. Характеристики элемента относятся к его отдельным атомам, а свойства простого вещества присущи не отдельным атомам, а их совокупности. При указании элемента остаётся непонятным, о каком же именно простом веществе идёт речь, а при указании простого вещества всегда совершенно ясно, из атомов какого элемента оно состоит. Э. х. известно 106, а число простых веществ превышает 500. И Э. х. и простые вещества принято подразделять на металлы и неметаллы.
Важнейшая особенность Э. х.— их сохранение при химических реакциях. В ходе реакций разрываются связи между атомами в молекулах исходных веществ и образуются новые связи между атомами, в результате чего возникают молекулы новых веществ. При этом происходит лишь перераспределение электронов внешних электронных оболочек; заряды атомных ядер не изменяются, остаются практически постоянными и массы атомов. Таким образом, химические реакции сопровождаются не превращениями Э. х., а перегруппировкой атомов в молекулах. На рисунке показан
Одно из важнейших свойств элементов — их сохранение при химических превращениях. Примером служат реакции с участием меди. Красные медные опилки (1) при нагревании на воздухе превращаются в чёрный порошок — окись меди СиО (2). Реакция окиси меди с разбавленной серной кислотой даёт голубой раствор сульфата меди CuSO4 (3). Электролизом раствора сульфата меди (4) можно получить металлическую медь, отлагающуюся на графитовом электроде в виде тонкой красной плёнки (5). Медь реагирует с влажным хлором при комнатной температуре, давая коричнево-бурый хлорид меди СиС12 (6). При растворении хлорида меди в большом количестве воды получают небесно-голубой раствор (7). Если в этот раствор добавить порошок железа (8), то более активное железо вытеснит медь из хлорида и можно вновь получить порошок меди (9).
В центре рисунка изображена медная медаль с барельефом дракона, кусающего свой хвост. У алхимиков этот дракон служил символом химических превращений. По краю медали по-латыни написано «De nihilo nihil fit» — «Из ничего не создаётся ничто». Этой фразой древний философ Тит Лукреций Кар в I веке до н. э. кратко выразил мысль о сохранении материи.
В наши дни физики-ядерщики сумели осуществить превращения одних химических элементов в другие. Так, облучая изотоп меди 65Си ядрами тяжёлого водорода — дейтерия, можно из меди получить цинк:
29С11 + 1Н = JjjZn + 2дП
353
ЭЛЕМЕНТЫ ХИМИЧЕСКИЕ
23 ХЭШ
ЭЛЕМЕНТЫ ХИМИЧЕСКИЕ
354
пример: сохранение Э. х. меди в химических процессах (исходное вещество — красная медь; после осуществления всех процессов она получена вновь).
Представление о сохранении Э. х. в химических реакциях возникло в результате длительного развития знаний человека о природе. Сильным толчком к формированию этих представлений был, вероятно, крах многочисленных попыток алхимиков средневековья разработать способ превращения в золото свинца или какого-либо другого относительно дешёвого «сырья». Собственно, во времена алхимиков не существовало ещё ясного понимания того, что же следует называть Э. х. Обычной была точка зрения, согласно которой четырьмя элементами Вселенной считались огонь, воздух, вода и земля, а всё в окружающем мире представляло собой сочетания этих «элементов». Горький опыт алхимиков, убедившихся в том, что попытки получить золото из не содержащих его источников — пустая затея, оказался чрезвычайно важным для формирования и развития действительно научных представлений об Э. х.
В 1661 г. английский химик Р. Бойль сформулировал понятие о химическом элементе, как о веществе, не разложимом на химически более простые части. Конечно, такая формулировка была ещё далека от совершенства (в ней не разграничивались, например, понятия простого вещества и элемента). Но учёный верно определил важнейшую особенность элементов: их химическую неразложимость, иными словами, сохранение их при химических реакциях.
Большой вклад в развитие учения об Э. х. внёс М. В. Ломоносов, который первым в середине XVIII в. стал считать элементами атомы, из которых состоят вещества. Так, в 1748 г. он писал, что молекулы воды состоят из совокупности её атомов (тогда воду считали простым веществом).
В 1789 г. А. Л. Лавуазье впервые дал список Э. х. (он называл их «простыми телами»), то есть веществ, которые к тому времени не были разложены на более простые. Вывод о том, что каждому Э. х. соответствует определённый вид атомов, был окончательно утверждён в науке благодаря трудам английского физика и химика Дж. Дальтона. В начале XIX в. Дальтон разработал первую таблицу атомных весов (масс) нескольких элементов (Н, N, С, О, Р, S), причём за единицу был принят атомный вес водорода.
Успехи в изучении строения атома позволили в начале XX в. прийти к выводу, что старые представления об абсолютной неизменяемости,
неуничтожимое™ Э. х. нуждаются в поправках. Действительно, существуют такие процессы, при которых перестройку претерпевают и атомные ядра. Превращения, в которых ядро изменяет свой заряд (или заряд и массу) называются ядерными реакциями.
В природе ядерные реакции происходят при распаде радиоактивных ядер таких Э. х., как радий, торий, уран и др., при бомбардировке атмосферы и поверхности земли космическими лучами; сложные ядерные реакции идут в недрах Солнца и других звёзд. Физики научились искусственно воспроизводить многие ядерные реакции—не только природные, но и такие, о наличии которых в природе нам ничего не известно (например, цепные ядерные реакции с участием ядер изотопов 235U и 239Ри в ядерных реакторах). Для осуществления и исследования ядерных реакций широко применяются ускорители заряженных частиц — мощные установки, в которых, например, ядра водорода (протоны или дейтроны) разгоняют до очень высоких скоростей и бомбардируют ими «мишень» — вещество, в котором хотят вызвать ядерную реакцию. Именно этим путём был впервые получен технеций — неизвестный ранее Э. х. с атомным номером 43. При бомбардировке молибденовой мишени дейтронами происходит ядерная реакция:
УМо + *Н = 9|Тс + 2Jn
Как видно из уравнения, кроме ядер нового элемента JgTc, освобождаются нейтроны Jn (по два на каждое образующееся ядро технеция). С помощью искусственно проведенных ядерных реакций были впервые получены такие элементы, как прометий, астат, нептуний, и все остальные Э. х. с более высоким атомным номером, чем у урана. Все эти элементы относятся к числу радиоактивных (радиоактивными называют Э. х., у которых все изотопы радиоактивны).
Широкое практическое использование различных Э. х. в виде простых веществ и соединений требует чётких знаний о том, как и где Э. х. располагаются в земной коре, каковы их общие запасы. Изучением этих вопросов занимается, в частности, геохимия (подробно они освещены в статьях Земная кора, Атмосфера, Гидросфера).
Большое научное значение имеет и проблема изучения распространённости Э. х. в космосе, во всей Вселенной. Как установили учёные-астрохимики и астрофизики, распространённость элементов в космосе существенно отличается от их распространённости в земной коре. Если в земной- коре преобладают кислород, кремний и алюминий, то в космосе наиболее
355
ЭРБИИ
распространены самые лёгкие Э. х.— водород и гелий. С повышением атомного номера Э. х. их космическая распространённость, в общем, резко снижается. Однако кривая зависимости космической распространённости Э. х. от их атомного номера носит не плавный характер, а имеет несколько максимумов. Максимумы приходятся на элементы (точнее говоря, на определённые изотопы этих элементов) с высокой устойчивостью ядер. Детально причины
повышенной устойчивости (и, следовательно, распространённости в космосе) тех или иных ядер выяснены ещё не до конца; много остаётся неясного и в представлениях о том, как Э. х. возникали и возникают во Вселенной.
г С. С. Бердоносов.
ЭРБИЙ (лат. Erbium) Er — химический элемент III группы периодической системы Менделеева из семейства лантаноидов; атомный номер 68.
^Пойдём ныне по своему отечеству... Станем искать металлов, золота, серебра и прочих; станем добираться отменных камней, мраморов, аспидов и даже изумрудов, яхонтов и алмазов. Дорога будет не скучна... А металлы и минералы сами на двор не придут,— требуют глаз и рук к своему прииску... Итак, не должно сомневаться о довольстве всяких минералов в Российских областях, но только употреблять доброе прилежание с требуемым знанием*.
М. В. Ломоносов
(Поли. собр. соч., т. 5, 1954, с. 620,621)
23*
ХИМИЯ СЛУЖИТ ЧЕЛОВЕКУ
Наш земной шар — это гигантский универсальный химический комбинат, действующий вот уже миллиарды лет*. В результате сложных реакций огненная масса шара превращалась в твердь — земную кору. Разнообразные химические взаимодействия, и прежде всего четырёх элементов — углерода, водорода, азота и кислорода, привели к зарождению простейших органических соединений. Постепенно усложняясь, такие соединения дали начало жизни на Земле. Растительный мир нашей планеты — исполинская фабрика фотосинтеза, снабжающая атмосферу свободным кислородом О2. Этой фабрике обязан своим существованием животный мир.
Многие века человек использовал для целей своих всё то, что могла дать природа: строил жилища из дерева, делал орудия труда из камня и посуду из глины, одевался в шкуры животных, силой ветра приводил в движение мельницы и корабли. Начиная с глубокой древности люди наблюдали взаимодействия самых разнообразных веществ и сил природы. И уже тогда стремились разгадать её тайны. Обугливая дерево, обжигая глину, выплавляя металлы из руд, выделяя лекарственные вещества из растений, наши давние предки, сами того не подозревая, знакомились с химическими процессами. Можно считать поэтому, что химия — одна из древнейших областей познавательной деятельности человека. Первые практические сведения по химии появились, как полагают, в Древнем Египте. Эти знания держались в строжайшем секрете и считались «священным искусством». Становление же химии как науки о превращении веществ началось во второй половине XVII века.
А уже в середине XVIII века великий Ломоносов писал: «Широко распростирает химия руки свои в дела человеческие... Куда ни по
* Это положение — одно из самых общих — иллюстрируют многие и многие страницы нашей Энциклопедии. Поэтому-то оно совсем не случайно открывает Введение к нашей книге и повторено здесь, в заключительной статье.
смотрим, куда ни оглянемся, везде обращаются пред очами нашими успехи ее прилежания». Могущество химии в наши дни неизмеримо возросло.
Мы носим одежду и обувь, сшитые из синтетической ткани и искусственной кожи и окрашенные химическими красителями. Мебель и посуда, бумага и лекарства, словом, неперечислимое множество обычных вещей, делаются с помощью химии. Без неё не было бы ни фотографии, ни кино, ни телевидения. Но химия не только изменила и украсила наш быт. Она создала материалы, из которых конструируют самолёты и автомобили, электронно-вычислительные машины и синхрофазотроны, протезы суставов и кровеносных сосудов. Химии принадлежит почётное место и среди наук, подготовивших возможность межпланетных путешествий; она дала ракетам и космическим кораблям топливо и конструкционные материалы.
Если бы всего 100 лет назад кто-нибудь сказал, что из бревна можно сделать шёлковую ткань, а из газа — ботинки или небьющуюся стеклянную посуду, такого человека сочли бы праздным фантазёром. Сейчас всё это — реальность, не вызывающая удивления. Вещества й материалы, которые рождаются в результате чудесных химических превращений, можно назвать «второй природой». Но далеко не всем известно, как происходят эти чудесные превращения.
На вопрос, как заставить вещества взаимодействовать между собой, отвечает наука химия. Вопрос же, где и каким образом такие взаимодействия осуществляются в больших масштабах, следует адресовать химической промышленности.
Химическая промышленность
В литературе, особенно популярной, понятия «химическая наука», «химическая промышленность», «химическая технология» часто объединяются одним термином «химия». Это удобно, но, конечно, не совсем точно. А что же означает каждое из этих понятий?
357
Михаил Васильевич ЛОМОНОСОВ — учёный-энциклопедист.
Михаил Васильевич Ломоносов родился 19 ноября 1711 г. в деревне Дени-совке бывшей Архангельской губернии (ныне село Ломоносове Архангельской области). Умер 15 апреля 1765 г. в Петербурге. С его биографией вы познакомились, когда изучали русскую литературу XVIII в. Физик и химик, геолог и металлург, астроном и географ, историк и экономист, филолог, оратор, художник и поэт, он, по словам А. С. Пушкина, «...был первым нашим университетом». В. Г. Белинский назвал Ломоносова отцом русской литературы. С неменьшим правом можно сказать, что он был и отцом русской науки, в частности отечественной химии. Ломоносов был избран членом академий наук в Петербурге (1745 г.), Стокгольме (1760 г.) и Болонье (1764 г.).
Химия того времени, по выражению Ф. Энгельса, была ещё «в пелёнках». Она считалась искусством разлагать «смешанные тела» на их «начала», получаемые при действии постепенно усиливающегося нагревания. Главными целями химии были приготовление лекарств и выплавка металлов из руд. Поэтому она находилась в руках врачей, аптекарей и металлургов.
Как химик Ломоносов значительно опередил свой век. Получив хорошую подготовку по математике и физике в Марбурге (Германия), он уже в своих первых работах (1739—1741 гг.) исходил из гипотезы корпускулярного строения материи и последовательно прилагал эту гипотезу к объяснению химических явлений. Представления Ломоносова об «элементах» (атомах) и «корпускулах» (молекулах) очень близки к определениям этих понятий, принятым много десятилетий спустя, в 1860 г., на международном съезде химиков в Карлсруэ. Весьма примечательно, что в работе 1741 г. (опубликована лишь в 1904 г.) Ломоносов предвосхитил законы постоянных и кратных отношений, открытые значительно позже — в начале XIX в.
В 1748 г. в письме к знаменитому математику, члену Петербургской академии наук Л. Эйлеру Ломоносов сформулировал свой закон сохранения материи и движения (опубликован в 1760 г. на латинском и русском языках). Закон Ломоносова гласит: «...Все перемены, в натуре случающиеся, такого суть состояния, что сколько чего из одного тела отнимется, столько присовокупится к другому; так, ежели где убудет несколько материи, то умножится в другом месте... Сей всеобщий естественный закон простирается и в самые правила движения...».
В 1748 г. Ломоносов, преодолев большие трудности, добился постройки химической лаборатории при Академии наук. Здесь он вёл теоретические и прикладные исследования и положил начало новой области химических знаний — физической химии. Этот термин существовал и ранее. Например, по определению И. Ф. Генкеля, у которого учился Ломоносов, он означал «искусство основательно исследовать тела и разумно разлагать их» (1741 г.). Ломоносов же полагал, что «физическая химия есть наука, объясняющая на основании положений и опытов физики то, что происходит в смешанных телах при химических операциях» (1752 г.). Это определение кажется написанным не в середине XVIII в., а на полтора столетия позже.
В 1752—1753 гг. Ломоносов прочитал студентам курс физической химии с демонстрацией опытов и практическими занятиями. Он составил обширные программы измерений свойств растворов, но успел выполнить лишь часть намеченных исследований. Не успел он и закончить труд «Введение в истинную физическую химию», о чём писал с большой горечью. Слишком уж был он загружен работами по истории и географии России, писанием должностных од и многими другими делами и обязанностями.
Из работ прикладного характера особенно важны изыскания Ломоносова, касающиеся составов для цветных стёкол (свыше 4000 опытов). По рецептам учёного были изготовлены стёкла для его высокохудожественных мозаичных картин, в частности для «Полтавской баталии» (ныне находится в здании Академии наук в Ленинграде). В 1753—1755 гг. Ломоносов устроил в Усть-Рудицах стекольную фабрику, которая вырабатывала изделия из цветных стёкол, а также стёкла самых разнообразных оттенков для мозаики.
В 1764 г. Ломоносов в проекте устава Академии наук писал, «химик... имеет в своем ведомстве и расположении химическую лабораторию..., и в оной старается во-первых доискиваться новых приращений сея науки, особливо в показании причин в химических переменах, дабы сию науку вяще и вяще приближать к физике и, наконец, поставить оную с нею в равенстве, притом не оставлять и других трудов химических, кои простираются до дел практических, в обществе полезных, чего от химии ожидают краски, литейное дело, медицина, экономия и прочее».
Эти мысли — своего рода научное завещание Ломоносова. Отыскание причин химических превращений и объяснение их с точки зрения законов физики составляет и в наши дни главную задачу химико-теоретических исследований. А приложение их к потребностям практики — другая, не менее важная сторона деятельности химика. В своём служении народу советские химики следуют примеру и заветам великого учёного Михаила Васильевича Ломоносова.
358
Рис. 1. Московский Государственный университет имени М. В. Ломоносова, химический факультет, одна из учебных лабораторий. Здесь воспитываются будущие учёные-химики. «Создание любого вещества или материала начинается с науки» (стр. 3$8).
Создание любого вещества или материала начинается с науки (рис. 1). Промышленность реализует научные открытия и помогает оценить их пользу; она ставит перед наукой всё новые и новые задачи. Наука же неустанно совершенствует промышленность, направляет её по пути прогресса. Хорошо сказал об этом Менделеев, прославившийся не только как учёный, но и как борец за развитие отечественной экономики: «Промышленность и истинная наука друг без друга не живут, друг от друга получают силу, и этот союз родит блага, без него не веданные».
•Но научные открытия не могут быть сразу же использованы промышленностью. Необходимо создать промышленный способ реализации того или иного открытия. Иными словами, предложить технологию процесса. Наука, разрабатывающая промышленные методы превращения исходных веществ (сырья) в вещества с новым составом, строением и свойствами (продукты), называется химической технологией. Основная задача химической технологии — создание таких процессов, которые позволяли бы получать высококачественную продукцию с наименьшими затратами человеческого труда, сырья, энергии и времени.
Дать точное определение понятию «химическая промышленность» практически невозможно. В широком смысле слова — это совокупность производств, использующих химическую технологию.
Но химические процессы господствуют в самых разнообразных сферах созидания «второй природы»: в металлургии и нефтепереработке, в производстве строительных материалов и бумаги, в текстильной и пищевой индустрии... Действительно, что такое выплавка чугуна в доменных печах? Конечно же, химический процесс: железная руда, кокс, флюс и воздух превращаются в вещества нового состава и качества — чугун, шлак, колошниковый газ. В основе этого процесса лежат химические реакции — восстановление окислов железа до металла, шлакообразование и получение смеси газов (СО, СО2, N2). На химических процессах основано и получение цветных металлов. Тем не менее производство любых металлов относят не к химической, а к металлургической отрасли индустрии. Заводы же, вырабатывающие целлюлозу и бумагу, принадлежат промышленности целлюлозно-бумажной, а стекло и цемент — промышленности строительных материалов.
Таким образом, термин «химическая промышленность» носит условный характер. Он значительно уже, чем понятие «химическое производство», и его границы очерчены под влиянием многих факторов — технологических, исторических и даже юридических.
В этой связи понятие «химическая продукция» (означающее, в широком смысле слова, продукты химического превращения) тоже приобретает условный характер. Это надо иметь в виду, чтобы вернее и глубже разбираться во взаимоотношениях химии со всеми остальными отраслями народного хозяйства. Кстати, часто один и тот же химический продукт производят заводы разных отраслей промышленности: серную кислоту, минеральные удобрения, бензол, аммиак вырабатывают и химические, и металлургические предприятия.
Народное хозяйство нашей страны — цепь взаимосвязанных звеньев. Промышленность — важнейшее и ведущее звено народного хозяйства. Её продукция составляет около двух третей так называемого общественного продукта и приносит более половины национального дохода. Одна из важнейших её отраслей — химическая промышленность. В наши дни она выпускает множество различных продуктов, которые классифицируют по свойствам, методам получения, назначению и объединяют в несколько больших групп. В соответствии с этими группами формируются и отдельные отрасли химической индустрии.
Горно-химическая промышленность, как показывает само название, занимается добычей я переработкой горно-химического сырья: фосфоритов, апатитов, калийных солей... Здесь освобождают руды и минералы от пустой породы и получают концентраты —продукты, обогащённые полезным веществом. Они же, в свою очередь, служат сырьём для других химических предприятий.
Основная химическая промышленность, используя главным образом природное минеральное
359
сырьё и различные отходы других производств, даёт народному хозяйству самые необходимые неорганические вещества. Она вырабатывает минеральные удобрения (туковая промышленность), серную кислоту (сернокислотная), кальцинированную соду (содовая), хлор, соляную кислоту и едкий натр (хлорная промышленность). Даже из такого перечня ясно, что эта отрасль является фундаментом, то есть основой (отсюда и название), всей химической промышленности. Множество химических продуктов на той или иной стадии их получения обязательно соприкасалось с кислотой или щёлочью. Надевая платье из вискозного шёлка или из найлона, любуясь цветными фотографиями, вдыхая аромат духов, мы, конечно, забываем о «прозаических» H2SO4, HQ, NaOH, Na2CO3... А ведь создать без них эти красивые вещи было бы невозможно. Недаром кислоты, щёлочи, сода производятся миллионами тонн.
Промышленность тяжёлого, (или основного) органического синтеза вырабатывает важнейшие соединения: углеводороды, спирты, альдегиды, кетоны, кислоты, эфиры, амины; органические продукты, содержащие хлор, фтор, фосфор, кремний и прочие элементы. Все эти и многие другие вещества служат исходными продуктами для производства пластмасс, синтетических каучуков, лаков и красок, химических волокон, моющих средств, ядохимикатов. «Синтез» — слово греческое; оно означает соединение, сочетание, созидание. Органический синтез — величайшее завоевание химии. Его роль в созидании «второй природы» огромна.
Производство полимерных материалов—• сравнительно новая, бурно развивающаяся отрасль химической промышленности. С её продуктами, самыми яркими образцами «второй природы» — пластмассами, синтетическими каучуками, химическими волокнами, сегодня знакомы все.
Промышленность пластических масс и синтетических смол выпускает сотни тысяч материалов и изделий, без которых трудно сейчас представить себе жилищное строительство, авиа-, судо-, автомобиле-и машиностроение, электротехнику, быт... Производство пластмасс, как, впрочем, и всех вообще полимерных материалов, включает две стадии. Одна из них — «чисто химическая», где путём органического синтеза получают высокомолекулярные соединения — полимеры. Другая — переработка полимеров в изделия — напоминает машиностроение. Здесь работают мощные гидравлические прессы и машины, которые формуют пластмассы (так называемые литьевые и экструзионные машины).
Заводы этой отрасли вырабатывают также синтетические смолы, необходимые для производства клеёв, химических волокон, искусственной кожи, строительных и электротехнических материалов...
Промышленность синтетического каучука даёт жизнь чуть ли не 50 тысячам различных изделий. Это автомобильные, авиационные, мотоциклетные и велосипедные шины (шинная промышленность), эластичные конвейерные ленты, шланги, прокладки, обувь, игрушки (резиновая промышленность). Не будет преувеличением сказать, что без продукции резиновой и шинной промышленности остановились бы автомобили, перестали бы работать электродвигатели, компрессоры, насосы и, конечно, не летали бы самолёты (в самолёте ТУ-104 — около 9 тыс. резиновых деталей общей массой 2,5 т). Короче, прекратилась бы жизнь огромного машинного парка на суше, на воде и в воздухе. Нетрудно вообразить себе последствия такой катастрофы.
Промышленность химических волокон выпускает продукцию двух видов. В одном случае химической переработке подвергают древесину, хлопок или другие дары «первой природы», уже содержащие готовый полимер — целлюлозу. Её нужно прежде всего извлечь, а затем и «облагородить», то есть придать ей, тоже путём
химических превращений, ценные качества. Производство волокон на основе целлюлозы освоено у нас в годы первых пятилеток. Всем хорошо известен вискозный и ацетатный шёлк (вспомните — «рубашка из бревна»...). В другом же случае сначала синтезируют сами полимеры и на их основе получают волокна, которые так и называют «синтетические». Это и капрон, и лавсан, и нитрон, и их зарубежные аналоги, носящие различные фирменные названия. Сырьём же для синтеза полимеров служат продукты переработки нефти и нефтяных газов («ботинки из газа»...). Химические волокна идут не только на одежду. Из них изготовляют и прочный авто- и авиакорд, и рыболовные сети, и всякие другие ткани технического назначения.
Волокна можно делать и из стекла. Как это ни странно, хрупкое стекло становится гибким и прочным, когда его превращают в тонкую нить. Стеклянные волокна вырабатываются на заводах промышленности стекловолокна. Так появляются прочные материалы — стеклопластики.
Промышленность органических красителей (иначе анилино-красочная промышленность) создаёт всё разнообразие красителей для тканей, кожи, Меха, синтетических материалов. Из всех отраслей химической индустрии эта, пожалуй, наиболее традиционная для химии. Ведь над проблемой получения синтетических красителей, более дешёвых, чем природные, успешно работали ещё в XIX в. крупнейшие химики-органики (вспомните знаменитого Н. Н. Зинина). Вокруг этой проблемы складывались целые научные школы и у нас, и в других странах.
Современные анилино-красочные заводы вырабатывают, помимо красителей, и вещества, придающие тканям специальные свойства: несмачиваемость, несминаемость.
Лакокрасочная промышленность выпускает свыше двух с половиной тысяч разновидностей лаков, красок, грунтовок, шпаклёвок. Они служат и для защиты от атмосферного воздействия, и для декоративной отделки всевозможных машин, приборов, предметов быта, изготовленных из металла, железобетона, дерева, пластмасс.
Хорошее лакокрасочное покрытие — это прежде всего прочная плёнка. До 30-х годов XX в. плёнкообразующими веществами служили растительные масла (льняное, конопляное) или природные смолы (канифоль, янтарь). Сейчас их сменили синтетические материалы, которые-не только дешевле, но и совершеннее природных по своим качествам.
Химико-фотографическая промышленность вырабатывает кинофотоплёнку и магнитные ленты, фотобумагу и фотопластинки, химикаты для фото-и кинодела. Надо ли рассказывать о её роли в жизни современного общества?
В последние годы бурно развивается самая молодая отрасль химической промышленности — нефтехими я. Раньше из нефти получали, главным образом, топливо (бензин, керосин, мазут). В послевоенные годы положение резко изменилось. В нашей стране освоение крупных нефтяных месторождений Башкирии, Татарии, Куйбышевской области, Сибири дало возможность направить значительную часть нефти на химическую переработку для получения таких ценных продуктов, как бензол, этилен, пропилен, этиловый спирт, фенол, ацетон, нефтяные парафины, жирные кислоты и высшие жирные спирты. Сбывается мечта Д. И. Менделееву о тонкой химической переработке нефти (ему принадлежат мудрые и саркастические слова: «нефть — не топливо, топить можно и ассигнациями...»). Помимо самой нефти, в последние годы для получения органических продуктов всё шире используются её спутники — газы нефтедобычи и нефтепереработки.
К химической индустрии относится и асботехниче-ская промышленность, выпускающая асбестовые ткани, шнур, волокна, набивки, тормозные ленты. Есть ещё и сажевая промышленность; она вырабатывает сажу, без которой не может обойтись производство шин и прочих резиновых изделий.
Такова современная классификация нашей химической промышленности. В некоторых странах к отраслям химической промышленности относят, сверх того, и получение мыла, парфюмерии, лекарственных препаратов, целлюлозы, бумаги... А в других странах, напротив, производство шин и резино-технических изделий, пластмассовых изделий и химических волокон в число отраслей химической промышленности не включают.
Химическое производство, в принципе, отлично от механической обработки материалов. В самом деле: вот цех, где из стального слитка изготовляют рельсы или трубы. Конечно, они не похожи на первоначальный слиток, но и в своей новой форме металл остаётся тем же металлом. Химия превращает ядовитый газ фосген в безвредный поролон — мягкий и лёгкий материал; взрывоопасный газ ацетилен — в негорючее прочное вещество, способное поглощать из морской воды золото; известный всем порошок борной кислоты — в боразон, соревнующийся по твёрдости с алмазом.
В химической индустрии большие количества сырья, полупродуктов и готовых продуктов приводятся в движение сравнительно малыми затратами человеческого труда. Тут нет ни тележек (конечно!), ни даже электрокар и мостовых кранов. Мощные компрессоры сжимают до высокого давления огромные объёмы газов; жидкости перетекают по разветвлённым трубопроводам; сыпучие вещества перемещает пневмотранспорт. Оттого и внешне химический завод резко отличается от любого другого. Когда вы подъезжаете к промышленному району и видите огромные башни, вертикальные колонны с множеством перекрещивающихся трубопроводов, громадные металлические этажерки, а на них — реакторы, аппараты, ёмкости,— значит, перед вами открывается панорама химического завода (рис. 2). Но рядом с цехами, где установлена аппаратура для проведения главного химического процесса, много цехов вспомогательных; они обеспечивают основные цеха водой, паром, электроэнергией, тарой... А кроме того, они очищают вредные стоки и газообразные выбросы главного производства.
Последнее чрезвычайно важно. Нельзя допускать, чтобы реки, водоёмы и атмосфера загрязнялись опасными химическими веществами. Сейчас ни один новый завод не строится без очистных сооружений. Ими оснащаются и старые заводы. Столь же важно, чтобы ничто не пропадало зря. Учёные и инженеры непрерывно совершенствуют химическую техноло
360
гию, добиваясь полной утилизации побочных продуктов основного производства.
Одного взгляда на современный химический завод достаточно, чтобы понять: химическая промышленность в наши дни может успешно развиваться только на базе передовой машинной техники. Необходима сложная аппаратура для химических реакций: могучие компрессоры и насосы, ректификационные колонны, смесители для твёрдых, жидких и газообразных веществ, фильтрующие устройства, газоразделительные и выпарные аппараты, кристаллизаторы, электролизёры, дробилки, литьевые машины и многое другое. Поэтому развитие химической индустрии во многом зависит от успехов химического машиностроения. Открытия учёных, сделанные ими в лабораториях, могут оказаться бесплодными, если инженеры-машиностроители не создадут необходимой заводской аппаратуры.
Химические процессы сложны. Они требуют большой точности в соблюдении рабочего режима — так называемых технологических параметров (температуры, давления, скорости реакции). Поэтому химия нуждается в разнообразных, подчас весьма тонких приборах. Современные химические процессы поддаются почти полной автоматизации. У аппаратов и машин обычно не видно людей: управление ведётся с центрального пульта (рис. 3). В жизнь предприятия прочно входит и электронно-вычислительная техника. Инженер-химик нашего времени должен знать не только свою науку, но хорошо владеть основами конструирования машин, разбираться в теплотехнике, электротехнике, радиоэлектронике.
Основные этапы развития химической промышленности в нашей стране
Химическая промышленность дореволюционной России. Химические промыслы возникли в России давно, в XII—XIII вв. А во времена Ивана Грозного Россия уже производила поташ, селитру и серу, порох, дёготь и смолу, минеральные краски. Неверно, что химические промыслы были занесены в Россию из Западной Европы: так, русские научились получать серу из колчеданов ещё в середине XVII в.— раньше, чем шведы, которых в Европе считают пионерами в этой области. Россия в тот период даже экспортировала в страны Западной Европы поташ, смолу, дёготь, рыбий клей, а позднее и хромовые соли, железный купорос, скипидар, канифоль, фосфор. Эти и другие продукты высоко ценились на европейском рынке. Благую роль в развитии химических производств сыграли «аптечное дело» и «пробирное искусство», совершенствованию которых Пётр I уделял большое внимание. Русские аптеки того времени явились зародышем будущих исследовательских лабораторий. Однако перерастание примитивных химических промыслов в настоящую промышленность как в Западной Европе, так и в России зависело прежде всего от развития главных потребителей химии—тяжёлой и лёгкой индустрии.
Сооружение первых русских химических предприятий заводского типа относится к началу XVIII в. В 1720 г.
361
Рис. 2. Химический комбинат в Сумгаите (Азербайджанская ССР).
«Когда вы подъезжаете к промышленному району и видите огромные башни, вертикальные колонны с множеством перекрещивающихся трубопроводов, громадные металлические эта-жерки, а на них— реакторы, аппараты, ёмкости, — значит, перед вами открывается панорама химического завода» (стр. 360).
близ Москвы был построен сравнительно крупный химический завод, где производились железный купорос, азотная кислота, краски, скипидар и канифоль. В 1805 г. возникло первое производство серной кислоты. В 1823 г. в районе г. Грозного было налажено небольшое нефтеперегонное производство, а в 1859 г. в окрестностях Баку начал перерабатывать нефть уже достаточно крупный по тому времени завод. Стали строиться небольшие предприятия по выпуску минеральных удобрений. Первый суперфосфатный завод был введён в действие в 1868 г. в г. Ковно (ныне Каунас).
В 70-х годах XIX в. в России появляется целый ряд химических заводов, иные из которых действуют и сейчас. Среди них Кусковский на окраине Москвы, Бондюжский на Каме, Березниковский на Урале, Тентелевский в Петербурге. К началу первой мировой войны в стране насчитывалось 46 сернокислотных заводов, но все они вместе давали лишь 120—150 тыс. т H2SO4 в год. (В 1974 г. в СССР её было выработано около 16,7 млн. т.) Крупного производства минеральных удобрений царская Россия не знала: полукустарные предприятия в 1913 г. дали в общей сложности около 90 тыс. т удобрений, в основном суперфосфата. (В 1974 г. в нашей стране произведено около 80,3 млн. т минеральных удобрений.) Три содовых завода — Березниковский, Донецкий и Славянский — давали примерно 160 тыс. т кальцинированной соды. (В 1974 г. её годовая выработка составила около 4,5 млн. т.) Промыш
ленное производство органических продуктов находилось совсем в зачаточном состоянии: два завода красителей, несколько мелких фабрик фармацевтических препаратов, кустарные лесохимические промыслы. Пластмассы вырабатывались на одном заводе в г. Орехово-Зуево, а вискозное волокно выпускала только небольшая фабрика в Мытищах.
Помещичье-буржуазный строй не способен был преодолеть общую экономическую отсталость России. В ключевых отраслях русской экономики — в металлургии, машиностроении господствовал иностранный капитал. Не избежала этой участи и химическая промышленность; более 50% акций принадлежало в ней французским, бельгийским, английским фирмам. Эти фирмы всячески препятствовали развитию самостоятельной русской химической промышленности. Между тем в России были исключительно благоприятные возможности для создания своей химии.
Во-первых, в недрах страны таились богатейшие залежи всего необходимого сырья. Во-вторых, русская химическая наука начиная со времён М. В. Ломоносова занимала передовые рубежи в этой области знания. В конце XIX и начале XX вв. в России работали Д. И. Менделеев, А. М. Бутлеров, Н. Н. Бекетов, Н. С. Курнаков, Н. А. Меншуткин, С. В. Лебедев, Н. Д. Зелинский, В. И. Вернадский, А. Е. Фаворский, Д. Н. Прянишников и другие выдающиеся учёные. Однако, как это хорошо
362
Рис. 3. Щёкинский химический комбинат (Тульская область, РСФСР). Центральный пульт управления цеха синтеза метанола. На современных химических предприятиях работа разнообразных аппаратов (дозаторов, фильтров, отстойников, смесителей, реакторов и т. д.) автоматизирована.
«У аппаратов и машин обычно не видно людей: управление ведётся с центрального пульта» (стр. 360).
известно, многие крупные изобретения русских химиков из-за отсталости царской России воплощались в жизнь прежде всего за рубежом.
Приведём лишь два примера.
В 1837 г. академик Петербургской академии наук Б. С. Якоби открыл способ осаждения металлов из растворов их солей при помощи электрического тока. Он назвал свой метод гальванопластикой. Это открытие сразу же получило промышленную реализацию за рубежом, разумеется, без ссылок на приоритет Якоби. Только во второй половине XIX в. по его способу начали в России золотить купола церквей.
Открытый Н. Н. Зининым в 1842 г. метод получения анилина из нитробензола, положивший начало анилинокрасочной промышленности, был впервые реализован в Германии. Правда, в этом случае немецкий химик А. В. Гофман признал, что «если бы Зинин не сделал ничего более, ... то и тогда его имя осталось бы записанным золотыми буквами в истории химии».
Эти примеры не единичны.
Царское правительство не заботилось об освоении сырьевых ресурсов страны. Огромные природные богатства лежали в недрах земли даже неразведанными. Достаточно сказать, что потребность в таких элементах, как бор, алюминий, висмут, сурьма, молибден, вольфрам, бром, иод, кобальт, никель, удовлетворялась до первой мировой войны только путём импорта. Но уже в начале войны, в январе 1915 г., группа выдающихся учёных, сознавая, что военная блокада обречёт страну на экономическую изоляцию, внесла в Академию наук предложение об организации постоянной Комиссии по изучению естественных производительных сил России (сокращённо — КЕПС). Вскоре такая комиссия была создана, на
меченные ею задачи были обширны и неотложны. КЕПС потребовала, в частности, организации научно-исследовательских институтов в России. В то время они насчитывались единицами, а существовавшие при Академии наук лаборатории были очень и очень бедны. Так, химическая лаборатория Академии наук имела всего двух штатных и двух нештатных сотрудников. Работа КЕПС не встретила ни понимания, ни поддержки со стороны правительства. Царские чиновники боялись любых проявлений общественной инициативы. К тому же во главе КЕПС стоял опальный учёный, бросивший вызов самодержавному строю, бывший профессор Московского университета В. И. Вернадский.
Деятельность КЕПС широко развернулась лишь после Великой Октябрьской социалистической революции. В. И. Ленин поставил перед комиссией ответственную задачу: как можно скорее составить план экономического подъёма страны.
Химическая промышленность после Великой Октябрьской революции и в период первых пятилеток. Молодой Советской республике пришлось создавать химическую промышленность практически заново; скудное наследие царской России было почти полностью уничтожено за годы первой мировой и гражданской войн. Ещё в 1918 г., определяя пути строительства социализма в нашей стране, Ленин указывал, что необходимо прежде всего взяться за создание мощной тяжёлой промышленности: металлургии, машиностроения, химической индустрии. Детище Ленина — план ГОЭЛРО, принятый в 1920 г. VIII Всероссийским съездом Советов, предусматривал восстановление всего народного хозяйства; а в области химической промышленности намечалось в течение примерно 10 лет превзойти довоенный уровень в 2,5 раза.
В 1927—29 гг. выпуск химической продукции уже превзошёл довоенный уровень. Развернулись плодотворные поиски источников сырья. Именно в те годы были открыты знаменитые залежи калийных солей в районе Соликамска и месторождения апатитов и фосфоритов на Кольском полуострове. Было организовано производство синтетического аммиака на Чернореченском химическом заводе (г. Дзержинск), пластмасс на Охтинском (г. Ленинград) и Орехово-Зуевском заводах, лаков и красок в Ростове-на-Дону, Ленинграде и Ярославле. Тогда же подверглись реконструкции и многие старые химические предприятия.
Стране нужны были квалифицированные кадры инженеров и техников. Молодёжь тянулась к знаниям. Но специализированных вузов и даже техникумов не хватало. В 1920 г. на базе среднего Промышленного училища было создано первое отраслевое химическое высшее учебное заведение — Московский химико-технологический институт им. Д. И. Менделеева. Самостоятельные химические факуль
363
теты возникли в семи университетах и политехнических институтах. За это же время были организованы научно-исследовательские учреждения, такие как Институт чистых химических реактивов в Москве (1918 г.), Российский институт прикладной химии (Петроград, 1919 г.), Научный институт по удобрениям (Москва, 1919 г.), Государственный радиевый институт (Петроград, 1922 г.). В 1918 г. в Москве появилась Центральная химическая лаборатория ВСНХ, преобразованная через тринадцать лет в Физико-химический институт имени Л. Я. Карпова, видного деятеля революционного движения, одного из организаторов советской химической промышленности. Созданные в те годы институты стали впоследствии крупными центрами научной жизни страны.
За годы первых пятилеток построено около 70 новых химических заводов. Среди них — комбинат «Апатит» в Хибинских горах, Соликамский калийный комбинат, азотно-туковые заводы в Бобриках (ныне Новомосковский комбинат), Березниках и Горловке. Триумфом молодой советской химии был пуск в 1932 г. первого в мире завода синтетического каучука (СК) по методу академика С. В. Лебедева (наша каучуковая индустрия опередила тогда Германию на пять лет, а Канаду и США — на десять). В том же 1932 г. вошёл в строй второй завод СК — в Воронеже, а через год ещё один — в Ефремове. В течение 1929—37 гг. выпуск химической продукции увеличился в 10 раз. Наша промышленность начала вырабатывать многие продукты, которые раньше ввозились из-за границы.
Но индустриализация страны и подъём колхозного сельского хозяйства требовали всё больше и больше химических продуктов. И XVIII съезд партии объявил третью пятилетку (1938—42 гг.) пятилеткой химии. Осуществление намеченных задач было прервано вторжением в нашу страну фашистских орд. Многие промышленные центры были разрушены. Значительную часть заводов удалось эвакуировать в восточные районы страны, и там они продолжали выпуск продукции для нужд фронта. Тем не менее причинённый войной ущерб оказался велик. В 1945 г. страна вырабатывала значительно меньше химических продуктов, чем до войны.
Химическая промышленность в послевоенный период. С окончанием Великой Отечественной войны химическую индустрию надо было быстро перестроить на удовлетворение потребностей мирного времени. В результате четвёртой пятилетки (1946—50 гг.) производство химической продукции превысило уровень 1940 г. почти в два раза. В это вре
мя стали вырабатываться новые виды пластмасс и химических волокон, важные ядохимикаты и много других, прежде неведомых синтетических продуктов. В течение 1951— 1955 гг. выпуск химической продукции увеличился ещё в два с лишним раза. Было построено большое количество крупных предприятий с усовершенствованной технологией и новым, более мощным оборудованием.
В 1957 г. валовая продукция химической промышленности увеличилась по сравнению с довоенным 1940 годом более чем в 5 раз. По объёму выпуска химической продукции Советский Союз занял второе место в мире после Соединённых Штатов Америки, опередив Англию, Францию, ФРГ, Японию. Но, как и в годы первых пятилеток, химия всё-таки оставалась отстающей отраслью нашего народного хозяйства. А селу требовались во всё возрастающих количествах удобрения и ядохимикаты, строительству и машиностроению — резиновые изделия и пластмассы, лаки и краски и другие новейшие материалы; людям в их каждодневной жизни—красивая, дешёвая одежда, меха, обувь, предметы домашнего обихода из синтетических веществ. Развитие же некоторых отраслей современной техники стало просто немыслимым без разнообразных продуктов химического синтеза.
Поэтому-то руководство Коммунистической партии Советского Союза приняло в мае 1958 г. специальное постановление «Об ускорении развития химической промышленности и особенно производства синтетических материалов и изделий из них для удовлетворения потребностей населения и нужд народного хозяйства». Чрезвычайный XXI съезд партии воплотил это постановление в цифры семилетнего плана развития народного хозяйства на 1959—65 гг.
Химическая промышленность в 1 9 5 9—6 5 гг. Основные идеи нового подхода к развитию химии заключались в следующем. Прежде всего признано было необходимым расширить выпуск предметов народного потребления — одежды, обуви, изделий хозяйственного и культурно-бытового назначения, а для этого значительно увеличить выработку синтетических смол, пластических масс, химических волокон. А чтобы дать населению достаточное количество продуктов сельского хозяйства, намечено было повысить производство минеральных удобрений. Вторая важная идея предусматривала решительный перевод химической промышленности на новую сырьевую базу — нефть, нефтяные газы и природный газ вместо каменного угля, пищевого и растительного сырья. Без новых источников сырья химия, особенно органическая, не могла бы
364
успешно развиваться. Третьей идеей было создание крупных производственных комбинатов с полной (комплексной) переработкой сырья. И наконец, общей характерной чертой нового подхода явился курс на широкую химизацию все) о народного хозяйства (см. раздел Химизация народного хоз я й ст в а). Выполнение такой громадной программы потребовало расширения исследований в области теоретической и прикладной химии. И, одновременно,— расширения конструкторских работ в сфере создания новых машин, аппаратов, приборов и средств автоматизации для химической промышленности.
В течение короткого времени были организованы новые научно-исследовательские и про
ектные институты. За годы семилетки свыше 500 заводов и крупных химических производств построено заново, а большинство действующих предприятий подверглись реконструкции на новой технической основе. В Башкирии, Татарии, в Поволжье, Южном Казахстане, Узбекистане, Закавказье и Сибири появились новые центры химической индустрии. Практически заново создана была химическая промышленность в Белоруссии и Прибалтике. На всё это потребовались огромные средства — за 7 лет около 9 млрд, рублей вложено государством в развитие химии. Это в 2,3 раза больше, чем за все предыдущие годы Советской власти.
В итоге за 7 лет производство минеральных удобрений выросло в 2,5 раза. Сельское хо-
Рост производства основных неорганических продуктов.
0.28 16650
4483
Млн.т Минеральные удобрения 55,4
ШО' 1950 I960 1970 1974
365
зяйство стало получать около 30 млн. т удобрений в год. Много ли это? Судите сами: дореволюционная Россия потребляла их ежегодно примерно 250 тыс. т.
Выработка пластмасс и синтетических смол возросла более чем в 3 раза, а синтетических волокон — в 6 раз.
Химическая промышленность в восьмой (1966—70 гг.) и девятой (1971— 1975гг.)пятилетках и дальнейшие перспективы её развития. 500 заводов и крупных производств, построенных за 1959— 1965 гг.,— это было много. Но опять-таки недостаточно. И снова строительство, снова реконструкция... В восьмой и девятой пятилетках химическая индустрия продолжала раз
виваться более быстрыми темпами, чем вся наша промышленность в целом.
До сих пор мы говорили об относительном увеличении продукции, не указывая абсолютных размеров производства. А они интересны. Ведь можно вместо всего только 1 тонны выработать 5 тонн и по праву отметить увеличение производства в 5 раз (хотя разница будет не так уж велика). Совсем другое дело, когда прирост даже на 1% составляет тысячи или миллионы тонн. Это характеризует огромную работу больших коллективов людей, а за многолетний промежуток времени — нашу историю, историю трудовых подвигов народа.
Об этом колоссальном ррсте советской химической индустрии расскажут диаграммы.
Рост - производства важнейших синтетических продуктов и изделий.
2487
471
1974
366
В обозримом будущем нашей химической промышленности предстоит новый качественный скачок. Что же будет отличать её от недавнего прошлого? Во-первых, высокопроизводительные и высокоэкономичные процессы, осуществляемые в агрегатах большой мощности. А во-вторых, полностью автоматизированные предприятия и производства с замкнутым технологическим циклом, исключающим отходы. А ведь исключить отходы — это значит: и сберечь ценное сырьё (отходы одного производства могут служить сырьём для другого), и не допустить загрязнения среды, в которой мы живём.
И, конечно же, мы ждём от химической промышленности дальнейшего резкого улучшения качества продукции и расширения её ассортимента — для всемерного удовлетворения нужд народного хозяйства.
О том, какую роль играет химия в народном хозяйстве, рассказано в следующем разделе статьи.
Химизация народного хозяйства
Химизация народного хозяйства — это широкое и эффективное использование химической технологии и химических продуктов во всех областях материального производства, это мощный рычаг технического прогресса во всех отраслях народного хозяйства и роста народного благосостояния.
Слово «химизация» впервые ввёл в 1924 г. академик Д. Н. Прянишников применительно к сельскому хозяйству, по сходству с термином «механизация».
Рассказ о химизации народного хозяйства мы тоже, вслед за академиком Прянишниковым, начнём с производства пищевых продуктов.
Химизация сельского х оз я й ств а. Наши далёкие предки не пахали землю, не сеяли хлеба, не выращивали овощей; они не знали того, что мы сейчас называем сельским хозяйством. Земля дарила людям плоды дикорастущих растений, охотой и рыболовством они добывали пищу. Прошли сотни, тысячи лет, пока человек сделал величайшее открытие: из попавшего в почву зерна вырастает растение, приносящее много зёрен. Примерно 6 тыс. лет тому назад человек стал переходить от кочевого к осёдлому образу жизни: появилось земледелие. И хотя с тех пор постоянно увеличивались посевные площади и развивалось животноводство, человечество на протяжении всей своей истории находилось в постоянном страхе перед неурожаем и голодом.
Население земного шара быстро растёт. 9—10 тыс. лет назад на Земле жило, по-видимому, не более одного миллиона человек, к началу нашей эры — около 200 млн., сейчас — 3 миллиарда с лишним, а к 2000 году, по прогнозам учёных, на Земле будут жить 6—6,5 млрд, человек. Вместе с тем научные расчёты показывают, что нет оснований думать, будто Земля не сможет обеспечить такое количество людей продовольствием. Если использовать все современные возможности науки и техники, то наша планета способна прокормить десятки миллиардов человек.
Как же можно увеличить производство пищевых продуктов? Есть несколько путей.
Один из них — расширение посевных площадей. Сейчас из 13,5 млрд, гектаров земной поверхности только 10% используется под пашни, сады, плантации. Другой путь — использование огромных продовольственных ресурсов Мирового океана. Сейчас эта огромная кладовая практически ещё не затронута. Так, океаны и моря Южного полушария дают всего 2% мирового улова рыбы, хотя водная поверхность в этом полушарии вдвое больше, чем в Северном.
Есть и пути, связанные с химией. И может быть, самые перспективные. Это, в первую очередь, повышение плодородия земли. Возможности здесь поистине огромны. Если бы урожайность продовольственных культур удалось повсеместно поднять до уже достигнутого сегодня передового уровня, то пищевые ресурсы увеличились бы в 4—5 раз. Ведь, по оценке учёных, примерно 70% земли во всём мире обрабатывается до сих пор примитивными орудиями, без использования новейших научных достижений агротехники. Далее — химический синтез пищевых продуктов уже сегодня перестал быть фантастикой. А в будущем этот путь откроет почти неограниченный источник продовольствия. И кто знает, может быть уже скоро начнут работать заводы, дающие «зелёное золото» — зелёную массу, содержащую сахар, белки, крахмал, масла...
Интенсификация сельского хозяйства во многом связана с успехами химии. Немного истории. Хотя человек занимался земледелием несколько тысяч лет, тайна плодородия земли была разгадана сравнительно недавно. В 1840 г. немецкий учёный Ю. Либих в своём труде «Химия в приложении к земледелию и физиологии» выдвинул идею, что важным элементом питания всех зелёных растений являются неорганические — минеральные соли. Чтобы сохранить плодородие почвы, надо внести в неё такое количество минеральных солей, которое взяли растения. Основы теории Либиха оказались правильными. Появилась новая наука — агрохимия, развитая в России трудами А. Н. Энгельгардта, Д. И. Менделеева, А. Е. Зайкевича, П. А. Костычева, К. А. Тимирязева, Д. Н. Прянишникова.Сейчас установлено, что для питания растений нужны прежде всего фосфор, калий, азот, а в очень небольших количествах ещё и марганец, бор, магний, железо, иод...
Даже не зная ничего достоверного о механизме питания растений, люди издавна удобряли землю навозом, золой, торфом. Но только химия научилась производить вещества, со
367
держащие все элементы, необходимые для питания растений,— минеральные удобрения.
Удобрения восстанавливают плодородные почвы и повышают урожайность полей. Многочисленные опыты и расчёты показывают, что каждая тонна минеральных удобрений позволяет получить с 1 гектара дополнительный урожай в 6—8 т зерна, или 6—10 т хлопка-сырца, или 6—7 т сахарной свёклы, или 60— 100 т картофеля. Каждый час труда, затраченный одним рабочим на заводе минеральных удобрений, экономит около 25—30 часов труда человека, занятого в сельском хозяйстве. В 1975 г. у нас будет выработано 90 млн. т удобрений (в условных единицах).
Но получить высокий урожай — это ещё не всё. Необходимо его сохранить. По данным Организации Объединённых Наций, вредные насекомые, сорные травы и болезни растений ежегодно уничтожают почти третью часть мирового урожая. На нашей планете насчитывается полтора миллиона видов насекомых и микроорганизмов. Около 70 тыс. из них — активные вредители сельского хозяйства. Они необычайно плодовиты. Так, одна пара бабочек озимой совки даёт за лето три поколения и может наплодить миллионы прожорливых гусениц. Это уже астрономические числа.
Современная химия даёт эффективное оружие в борьбе с вредителями сельского хозяйства — ядохимикаты, иначе называемые пестицидами (название происходит от двух латинских слов: pestis — зараза и caedo — убиваю). Вот их неполный перечень: гербициды (herba — трава, сорняк) — применяются для борьбы с сорняками; инсектициды (insectum — насекомое) — для борьбы с вредными насекомыми; фунгициды (fungus — гриб) — против грибковых заболеваний растений; зооциды (греч. «збон» — живое существо, животное) — для уничтожения вредных грызунов. Химический состав таких веществ весьма различен. Это и относительно простые неорганические соединения — медный купорос CuSO4-5H2O, арсенаты кальция и натрия Ca3(AsO4)2 и Na3AsO4, цианамид кальция CaCN2, фосфид цинка Zn3P2, это и очень сложные органические препараты, содержащие хлор, фосфор, серу.
Всем нам нравится полевой василёк. А знаете ли вы, что этот невинный голубой цветок может унести с гектара за сезон 60—70 кг азота, 20—30 кг Р2О5, около 100 кг К3О и от 250 до 1000 л воды (на 1 кг сухого цветка). Это наносит прямой урон пшенице, которая, не в пример васильку, кормит человека. На помощь приходят гербициды. Они в 20 с лишним раз сокращают затраты человеческого труда на прополку.
В истории было немало случаев, когда саранча или колорадский жук начисто уничтожали урожаи. Сейчас
Рис. 4. Весеннее опрыскивание садов.
на страже урожая стоят инсектициды: порошки для опыления (дусты), растворы и эмульсии для опрыскивания. Опыление и опрыскивание производятся не только наземным способом, но и с самолётов (рис. 4).
Растения, как и животные, при плохом уходе болеют. Возбудители их болезней — грибки, вирусы, микробы. Сегодня массовое распространение получило химическое «протравление» семян зерновых культур перед севом. Фунгицидами у нас повсеместно обрабатываются плодовые деревья, особенно виноградники и цитрусовые, чтобы обезопасить их от грибковых заболеваний.
Среди пестицидов есть и химические препараты, позволяющие удалить листья с растений,— дефолианты (по-латыни — folium — лист, de — приставка, означающая удаление). Оказывается, что если перед созреванием хлопчатника удалить его листья, быстрее созревают и раскрываются коробочки, а главное — при этом можно ввести машинную уборку.
В среднем каждый рубль, затрачиваемый на производство ядохимикатов, сохраняет урожай на 16—20 рублей. А для цитрусовых — даже на 40—70 рублей.
Пестициды приносят человеку очевидную пользу. Но не оказывают ли они, кроме того, вредного воздействия на природу? Этот естественный вопрос возник уже тогда, когда ядохимикаты стали применять впервые. В большинстве своём пестициды при разумном использовании безвредны для теплокровных животных. Но тщательные наблюдения показали, что они бывают страшны для полезных насекомых, которые под их воздействием погибают вместе с вредными. При этом нарушаются веками установившиеся законы равновесия в живой природе. Так, улетают в другие места насекомоядные птицы, а это, в свой черёд, ведёт к усиленному размножению уцелевших насекомых, что вскоре вызывает необходимость более энергичного применения ядохимикатов. Многие учёные полагают, что химические яды вредно действуют на процессы почвообразования и опасно загрязняют водоёмы.
Не следует ли прекратить применение химических средств защиты растений? Нет, это привело бы к резкому снижению урожайности. Но надеяться только на химию, по-видимому, действительно нельзя. Наиболее перспективна идея комплексных систем борьбы с вредителями. В таких системах агротехнические и химические методы сочетаются с биологическими. Ведь у вредителей есть и естественные враги. И можно создавать такие условия (искусственные ландшафты), которые менее благоприятны для размножения вредителей и более благоприятны для роста численности их врагов.
Значительно повышается роль химии в животноводстве. С древних времён голод был страшным бичом не только человека, но и домашних животных. Химия помогает повысить питательность естественных кормов. К ним добавляются искусственные белки, синтетические аминокислоты, витамины, а в последнее время — сверх того антибиотики и биостимуляторы роста. Если к одной тонне обычного корма для свиней добавить 0,8 кг синтетических аминокислот, то привес животных увеличится на 20—25%, и доход от дополнительно полученного мяса в 8—10 раз превысит стоимость химических добавок.
В любом живом организме непрерывно протекают многообразные превращения различных химических соединений. Белки, жиры и углеводы расщепляются на свои составные части; некоторые соединения окисляются, другие восстанавливаются; происходит не только распад, но и синтез веществ. Все эти явления изучает бурно развивающаяся в наши дни биохимия. Многие химические процессы, происходящие в живых организмах, можно было бы осуществить в лабораториях, но для этого потребовалась бы в одном случае высокая температура, в другом — высокое давление, не говоря уже об иных специальных условиях. В организмах же эти реакции идут легко и быстро, как бы сами по себе. Весь секрет в том, что там действуют особые катализаторы — ферменты (или энзимы). Они обладают способностью не только ускорять реакции, но и придавать им нужное направление.
Ферменты — это вещества белковой природы. Важные энзимы содержатся в быстро размножающихся микроорганизмах. Их строение и биохимию, жизнедеятельность и наследственность изучает микробиология. Накопленные ею знания уже сегодня приносят человеку неоценимую пользу. Знаете ли вы, что сыр, кефир, кумыс и многие другие пищевые продукты получают в результате микробиологических процессов?
Существует разветвлённая микробиологическая промышленность. Из растительного сырья она вырабатывает спирты, антибиотики, витамины, ферменты и — что особенно важно для повышения продуктивности животноводства — кормовые дрожжи для скота. Совсем недавно была открыта возможность получать их из углеводородов нефти. Эти дрожжи называют БВК — белково-витаминные концентраты. В СССР первое опытное производство
368
БВК возникло ещё в 1963 г., а сейчас для этой цели строятся крупные заводы.
Климатические и почвенные условия в разных уголках нашей страны неодинаковы. Особенно неблагоприятны северные районы. Но и тут помогает химия. Грунт защищают от морозов синтетической светопрозрачной плёнкой, и в таких своеобразных теплицах круглый год выращивают огурцы, капусту, морковь, помидоры. А как удобны для орошения полей пластмассовые трубы... Они легки и прочны. Передовые хозяйства нашей страны применяют их уже довольно широко.
Химия решает и ещё одну важную народнохозяйственную задачу. Какой смысл тратить зерно, картофель* свёклу для производства технического этилового спирта, если его можно получать синтетическим путём из нефти? Зачем расходовать растительные масла для приготовления лаков и олиф, когда вместо них можно успешно применять синтетические смолы? Незачем тратить животные жиры для выработки мыла, а казеин из молока — для приготовления клея, когда и для того, и для другого существуют прекрасные синтетические заменители. Использование пищевого сырья на технические нужды уже сейчас резко сокращается, а скоро будет полностью прекращено.
Приведенных примеров достаточно, чтобы убедиться, какие неисчерпаемые возможности открывает химия для решения проблемы питания на нашей планете.
Химия в промышленности и строительстве. Пожалуй, одна из древнейших ветвей химии — металлургия. Начало бронзового века историки относят к 4-му тысячелетию до нашей эры. Тогда был открыт химический процесс выплавки меди посредством нагревания смеси её руды с древесным углём; одновременно стали выплавлять серебро, свинец, олово. В начале 1-го тысячелетия до н. э. начался железный век. С тех пор железо стало главным металлом — главным материалом, без которого просто невозможно представить себе человеческую цивилизацию. Железо в рудах находится в соединении с кислородом. И потому металл выделяют из руд восстановлением. Роль восстановителя выполняет углерод — кокс. В этом химическая суть сложного доменного процесса. Мы напоминаем об этом потому, что и другие металлы, как правило, встречаются на Земле в виде окислов или солей, и получение металлов из руд почти всегда основано на ряде последовательных химических превращений.
Вот относительно молодой для техники, но стремительно завоевавший широчайшее при
369
знание лёгкий металл — алюминий, ставший сейчас основным материалом в авиации и электропромышленности. Алюминия на Земле много, но встречается он лишь в связанном состоянии. Конечный процесс выделения металла из природного сырья — электролиз А12О3. Чтобы получить пригодную для этого процесса окись алюминия (глинозём), приходится подвергать руду очень сложной химической переработке.
Металл новейшей техники — титан — вполне оправдывает своё громкое имя. Реактивной авиации требуется лёгкий металл типа алюминия, однако гораздо более прочный, да притом и жаростойкий. Титан удовлетворяет этим требованиям. Он всего в полтора раза тяжелее, но зато в шесть раз прочнее алюминия. И, в отличие от стали, не поддаётся коррозии. Титан также встречается на Земле в соединениях с кислородом. Из руд вначале получают двуокись титана TiO2, затем тетрахлорид TiCl4 и, наконец, сам металл. Впрочем, не «наконец»: чтобы выделить чистый титан, используют ещё один, очень тонкий химический процесс, называемый иодидным рафинированием.
Химия создала и целую подотрасль металлургической промышленности — гидрометаллургию. Как извлечь из руд нерастворимые в воде золото, соединения серебра, меди, цинка, молибдена, вольфрама, урана и многих других металлов? Руду обрабатывают кислотами и щелочами, цианидами и другими химическими реагентами. Металлы надо сначала перевести в растворимые соединения, чтобы затем осадить их из раствора либо электролизом, либо иными методами.
Огромное будущее принадлежит химии полупроводников. Полупроводники — это материалы-революционеры: они коренным образом меняют традиционную технику. (Достаточно сравнить ламповый и транзисторный приёмники.) Для полупроводников, да и для многих других отраслей новой техники, необходимы вещества высокой степени чистоты. Так, материалы для радиоэлектроники (кремний, германий, арсенид галлия...) или для атомной энергетики (уран, цирконий, бериллий, графит...) не должны содержать примесей посторонних элементов в сумме более 10" 4— 10" 6% (а по отдельным — особенно мешающим примесям—даже 10~9%). Такие сверхчистые вещества получают разнообразными, часто очень сложными химическими методами. Работу ведут в особых помещениях с «элементостерильным» климатом: воздух в лабораториях специально очищен, а мебель и оборудование изготовлены из стекла, фарфора, пластмассы.
Прочитайте в нашей книге статьи о «старых» и «новых» металлах, статьи о полупроводникахч И вы убедитесь, что в технологии их получения, разделения и очистки царит химия.
Металлы — основа нашей промышленности, особенно машиностроения. И железный век всё ещё продолжается.
Академик А. Е. Ферсман как-то сказал: «Будущее за другими металлами, а железу будет отведено почётное место старого, заслуженного, но отслужившего своё время материала. Но до этого будущего ещё далеко... Железо — пока основа металлургии, машиностроения, путей сообщения, судостроения, мостов, транспорта ...».
Однако благодаря успехам химии в XX в. созданы и совершенно новые материалы, которые часто успешно конкурируют с металлами: это — полимеры. И по словам академика Н. Н. Семёнова, «наиболее характерными материалами будущего будут... полимеры, то есть пластмассы, синтетическое волокно, синтетический каучук, кожа, мехит. п...». Несомненно, что ещё в этом веке перечисленные материалы станут доминирующими и в промышленности, и в строительстве, и в бь1ту.
Новые неорганические и органические полимерные материалы обладают сочетанием удивительных свойств, отличающих их от классически традиционных материалов. Они легки и достаточно прочны; способны выдерживать весьма высокие и очень низкие температуры^ не поддаются сильному электрическому воздействию и защищают от вредных радиоактивных излучений; отражают или пропускают электромагнитные и звуковые волны; не подвергаются заметным изменениям в агрессивных средах. И при всём этом из них сравнительно легко изготовлять самые различные изделия. Такие материалы позволяют развить совершенно новую технику. Они незаменимы в ракето-, самолёте-, судо- и приборостроении, в электротехнике и атомной энергетике.
Мы действительно живём в начале эры полимеров. Будущее их огромно. Представление о них дают хотя бы такие примеры.
Ещё на заре полимерной химии было изобретено небьющееся органическое стекло. А в последнее время получен совершенно новый армированный пластмассовый материал — стеклопластик. Хрупкое стекло, вытянутое в нити и спрессованное вместе со смолами в монолит, оказалось в 3—4 раза прочнее обычной стали и не менее прочным, чем её лучшие легированные сорта. К тому же стеклопластик в 4 раза легче стали и не подвергается коррозии. Из него можно делать и железнодорожные вагоны, и корпуса кораблей, и даже ракеты. Пластмассовый корпус значительно уменьшает вес судна и позволяет увеличить скорость корабля. Он не требует окраски, не ржавеет, не обрастает морскими организмами.
Широко известно, что царская водка — смесь крепкой азотной и соляной кислот— растворяет почти все металлы,
24 хэш
370
включая золото и платину. А химики-полимерщики создали материал, устойчивый к действию даже этой чрезвычайно агрессивной смеси — фторорганическое соединение тефлон. Эта пластмасса служит первоклассным материалом для изготовления самой химической аппаратуры.
Решить задачу получения пресной воды из морской мечтал ещё древнегреческий философ Аристотель. История мореплавания знает тысячи случаев, когда люди в море погибали от жажды! Новые полимеры, ионообменные смолы (иониты), способны опреснять морскую воду. Более того, тот или иной ионит может избирательно поглощать лишь отдельные соли. Купаясь в море, мы и не подозреваем, что погружаемся в очень разбавленный раствор золота — принимаем «золотую ванну». Есть такие ионообменные смолы, которые улавливают морское золото. Правда, этот способ добычи драгоценного металла пока экономически не выгоден. А вот для очистки воды от многих солей иониты уже нашли широкое применение: опресняя воду, они избавляют котлы от накипи и тем самым позволяют экономить тысячи тонн топлива.
В век электричества огромное значение имеют электроизоляционные материалы. Есть пластмассы, значи; тельно превосходящие природные материалы по своим электроизолирующим свойствам. Их применение более чем в 5 раз повышает срок службы электрических машин, значительно увеличивает их мощность и снижает вес. Современная кабельная промышленность почти целиком базируется на изоляции из полимеров — полиэтилена и поливинилхлорида.
Всё шире использует пластмассы строительная индустрия. Это и линолеум для полов, и плёночные материалы для обоев. Из пластмасс делают трубы, санитарнотехнические изделия, тепло- и звукоизоляционные материалы. Древесностружечные и древесноволокнистые плиты, изготавливаемые из отходов деревообработки и синтетических смол,—замечательный материал для внутренней отделки зданий.
Но вот над чем стоит задуматься. Бытовавшее ещё недавно мнение, что пластмассы — лишь заменители металла, дерева, стекла и других традиционных материалов, теперь безнадёжно устарело. Да, действительно, вкладыш подшипника можно сделать или из латуни или из текстолита, шестерню — или из стали или из полиформальдегида. Такая замена технически и экономически выгодна, однако всё-таки это лишь замена. А вот когда дело доходит до небьющегося стекла для самолётов, до тефлона, до ионитов, до кремнийорганическбй изоляции..., тогда оказывается, что в природе вообще нет материалов с подобными свойствами. Здесь уж пластмассы выступают не как заменители, а как неповторимые материалы.
Производство пластмасс — самая динамичная отрасль химической индустрии. Мировая выработка этих материалов с 1913 г. по 1970 г. увеличилась с 50 тысяч до 25 млн. т — в 500 раз! По некоторым прогнозам, она увеличится к 1976 г. ещё почти в два раза, а к 1981 г.— в 3,5—4 раза. Уже в 1974 г. производство пластмасс и синтетических смол в Советском Союзе составило около 2,5 млн. т.
Оглядитесь вокруг повнимательнее. И вы Сами найдёте множество доказательств того, что современная химия проникла во все отрасли
народного хозяйства. Идёт повсеместная химизация во всех сферах созидания. Мы ощущаем сегодня этот процесс даже в быту...
Химия — источник увеличения товаров народного потребления. Было время, когда человек непосредственно почти «не держал в руках» плоды химического производства. Подчёркивая это, Д. И. Менделеев в своё время писал: «Отличительную черту огромного большинства химических заводов составляет отсутствие их применения в обыкновенном, домашнем обиходе».
Да, так было 75—100 лет назад. Но теперь положен ие измен и л ось.
Побывайте с экскурсией на заводах химических волокон или пластмасс. Вас прежде всего пригласят «на выставку» — в зал, где экспонируется продукция завода. Вы увидите бобины различных волокон, а рядом изделия из них — элегантные шубки, искусственный каракуль, ковры, кофточки, носки, ботинки, плащи, тарелки, скатерти, умывальники, подносы, рыболовные лески и множество других красивых и нужных вещей. Всё это — предметы домашнего обихода или товары народного потребления. Теперь мы ощущаем успехи химии непосредственно на себе — в своей каждодневной жизни.
Получение химических волокон — яркий пример того, как в новейшую эпоху материалы создаются новым способом, принципиально отличным от способов, существовавших ранее. Столетиями человек, чтобы сшить одежду, сначала тяжёлым трудом выращивал на полях лён, коноплю, хлопок, постоянно завися от капризов природы. Химия освобождает человека и от этого тяжёлого физического труда, и от этой зависимости. Химические волокна вырабатываются на заводах во всё возрастающих количествах. В 1970 г. их мировое производство превысило 6 млн. т.
Всего производится около 25 видов различных волокон: шерстеподобные, несминаемые, эластичные, сверхпрочные, негорючие... Интересно такое сравнение: чтобы выработать тонну хлопкового или льняного волокна, надо затратить 1400—1700 человеко-часов, а на тонну вискозного штапельного волокна — всего только 150 человекочасов. При выработке натуральной шерсти затрачивается в 20—30 раз больше труда, чем в производстве шерстеподобных волокон — нитрона и лавсана. А для естественного и искусственного шёлка это различие ещё больше.
Однако, создавая для тканей сначала отдельные волокна, человек продолжает копировать природу. Но вполне резонно спросить: зачем продавливать через тончайшие отверстия жидкую полимерную массу и получать нити, которые затем нужно ещё сплетать и ткать по принципам дедовской технологии? Не разумней ли сразу получить «нетканую ткань»—пористо-волокнистую плёнку со всеми достоинствами нынешних тканей, но без их недостатков? Это была бы настоящая техническая революция. Над этой проблемой работают ныне учёные и решение её наверняка не за горами.
371
Полимеры дали нам и искусственную кожу. Сейчас это уже не кирза, из которой в недавнем прошлом шили солдатские сапоги. Это кожа, которую не отличишь от натуральной ни внешне, ни по свойствам. Точнее, по свойствам она часто лучше натуральной. Сейчас в нашей стране ежегодно изготовляют столько синтетической кожи, что это эквивалентно забою 50 млн. голов рогатого скота! Затраты труда на выработку этой кожи в 10 раз меньше по сравнению с натуральной.
Плащи, скатерти, гардины, абажуры, детские игрушки, стиральные порошки, духи, тысячи мелочей галантереи и множество других веществ, облегчающих домашний труд и украшающих быт,— всё это нам даёт химия. Это — химизация повседневной жизни во плоти.
Химия на службе здоровья человека. Средняя продолжительность жизни человека за последние 100 лет значительно выросла, прежде всего — в экономически развитых странах. Как показывает статистика, резко сократилась детская смертность. И взрослых, но ещё сравнительно молодых людей перестали уносить страшные эпидемические болезни — тиф, холера, оспа, чума. Медицина не могла бы достичь таких успехов без прогресса химии. В чём же заслуга химии? Прежде всего в том, что она создала обилие синтетических лекарственных препаратов.
...Человек заболел, поднялась температура. Врач прописал аспирин. Всем известно, что аспирин — жаропонижающее и болеутоляющее средство. Но только немногие знают, что аспирин, или ацетилсалициловую кислоту, получают путём последовательных химических превращений, в начале которых стоит, уголь или нефть.
...Человек стал быстро утомляться, жизненный тонус понижен. Врач рекомендует витамины (недаром латинское слово vita означает «жизнь»). Хорошо известно, что витамин С, или аскорбиновая кислота, содержится в плодах шиповника. Но химия делает медицину независимой от лекарственных растений. Ведь из картофельного или кукурузного крахмала можно получить глюкозу, а из глюкозы — путём тонких химических превращений — аскорбиновую кислоту.
Сложным органическим синтезом производят сейчас подавляющее большинство лекарств, витаминов, гормонов...
Но сколько ещё работы предстоит учёным! Мы много говорили о полимерах. Белки живых организмов — это тоже полимеры, только очень сложные. В природе их синтезом управ
ляют нуклеиновые кислоты (сокращённо их называют ДНК и РНК), тоже сложные макромолекулы — носители наследственности. Поиски путей искусственного синтеза белков, создание технологии, подражающей природе (или даже более совершенной!), это — грандиозная проблема. С её разрешением появятся и новые мощные источники пищи и чудодейственные медицинские препараты. Будут побеждены многие страшные болезни, увеличится продолжительность жизни человека.
Совместные работы химиков и медиков привели к созданию синтетических кровезаменителей, к применению пластмасс в хирургии и протезировании. Сейчас уже не редкость — пластмассовые протезы костей, суставов, пищевода, мочеточников и жёлчных протоков. А из тефлона и найлона изготовляют протезы кровеносных сосудов. Вместо хирургического шёлка служат нити из химических волокон, которые не надо удалять после операции — они сами рассасываются. А в лёгочной хирургии применяют пористые полимерные материалы.
Словом, и здесь идёт благой процесс химизации, облегчающий борьбу за жизнь человека и за его долголетие.
* * *
Итак, химия — мощный рычаг технического прогресса, источник народного благосостояния, один из фундаментов, на котором зиждется человеческая цивилизация. И недаром развитие химической науки и промышленности, всемерная химизация народного хозяйства — это, как указано в Программе КПСС, одно из необходимых условий построения материально-технической базы коммунизма.
Пройдут многие годы, а влияние химии — древней, но совсем не стареющей науки,— на самые различные сферы жизни будет возрастать всё больше и больше.
Вдохновенно и образно сказал о химии Максим Горький устами одного из героев пьесы «Дети солнца»: «...Это изумительная наука, знаете! ...Её зоркий, смелый взгляд проникает и в огненную массу солнца, и во тьму земной коры, в невидимые частицы вашего сердца..., в тайны строения камня и в безмолвную жизнь дерева. Она смотрит всюду и, везде открывая гармонию, упорно ищет начало жизни... И она найдёт его, она найдёт!»... В. С. Титов.
24*
АЛФАВИТНО-ПРЕДМЕТНЫМ УКАЗАТЕЛЬ
А
Абразивные материалы 156
Аваит 232
Авиали 61, 62, 63
Авиценнит 293
Авогадрит 336
Автотермичные процессы 55
Агат 172
Агрохимия 7, 366
Аденозинтрифосфорная кислота
(АТФ) 316
Адсорбционная вода 101
Азиды 51
Азот 47, 15, 73, 111, 112, 119, 160,
163, 205, 221, 350
— атом 50
— в природе 47
— водородные соединения 50
— двуокись 49, 51, 53
— жидкий 52
— закись 51, 97, 213
— изотопы 47, 52
— историческая справка 47
— кислородные соединения 51, 52, 55
— молекула 49
— окисление 162, 163
— окислы 48, 51
— окись 49, 51, 53
— получение 52
— применение 52
— пятиокись 55
— свободный 47, 48, 49
— связанный 47, 48, 49, 69, 205
— трёхокись 51, 52
— трифторид 323
— трихлорид 323
— физические свойства 49
— химические свойства 49
Азотирование 221 f
Азотистая кислота 52, 51, 221, 222
Азотистоводородная кислота 51
Азотистокислые соли 52, 221
Азотистый ангидрид 52, 51, 221
Азотная кислота 52, 49, 71, 72, 163, 220, 222, 336
— получение и применение 54
— физические и химические свойства 52
Азотнокислые соли 55, 220
Азотные удобрения 47, 55, 71, 72, 205, 221, 257
Азотный ангидрид 55, 51
Азотфиксирующие бактерии 48, 49
Азурит 190
Айсберги 122
Аквамарин 80
Аккумулятор 262, 267
— Эдисона 218
Аккумуляторная серная кислота 267
Активные атомы 323, 331
Активные частицы 107, 323, 331
Актиниды 56
Актиний 56, 196, 197, 241, 321
Актиноиды 56, 30, 37, 42, 176, 179, 196, 197, 311
Актинон 245
Акцептор 70
Алебастр 124, 125
Александрит 80
Ализарин 158
Алкалиген 47
Алкалиний 321
Аллотропия 162, 258, 306, 316, 317, 352
Алмаз 60, 90, 156, 168, 171, 204, 221, 302, 303, 305, 306, 307
Алии 216
Ал нико 166
Алтаит 296
Алунит 64
Алхимия 5, 6, 137, 139, 194, 197, 209, 222, 249, 250, 260, 264, 289, 291, 315, 332, 336, 339, 354
Альбит 64
Альфа-железо 129, 130, 281
Альфа-частица 11, 12, 56, 60, 75, 81,
91, 118, 143, 239, 244, 300, 310, 321
Алюминаты 65, 142
Алюминиевая промышленность 67
Алюминиевая фольга 67
Алюминиевое зеркало 67
Алюминиевые бронзы 189
Алюминиевые квасцы 60, 63, 158, 159
Алюминиевые сплавы 60, 63, 209
Алюминиевый провод 67
Алюминиевый чугун 346
Алюминий 63, 32, 114, 159, 196, 197,
293, 369
-г- взаимодействие с иодом 146
— галогениды 64, 65
— гидроокись 65, 67, 115
— историческая справка 63
— карбид 65
— нитрат 65
— нитрид 65
— окисление 162
— окись 67, 65, 66, 125
— получение 66
— применение 66
— распространённость в природе 64
— силикаты 273
— субгалогениды 66
— субфторид 66
— субхлорид 66
— сульфат 65, 67
— сульфид 65
— физические свойства 64
— фторид 65
— химические свойства 64
— хлорид 65, 67
Алюминотермия 67, 314, 334
Алюмокислородный тетраэдр 272, 274
Алюмокремнекислородный каркас 274
Алюмокремнекислородный мотив 273
Алюмосиликаты 64, 159, 182, 273
Амазонит 203
Амальгамация 68, 249, 262
Амальгамы 68, 262, 286, 294
Америций 69, 56, 57, 58, 59
Аметист 68, 172, 203
Амиды 71
Аминокислоты 47
Аммиак 69, 47, 48, 50, 54, J07, 214, 221
— молекула 69
— получение 71
— применение 71
— физические свойства 69
— химические свойства 69
Аммиакаты 70
Аммиачная вода 72
Аммиачная селитра 205, 257
Аммоний 224
— динептунат 59
— ион 72, 70, 313
— карбонат 72
— нитрат 72, 205
— нитрит 72
— парамолибдат 208
— соли 70
— фосфаты 314
— фосфомолибдат 208
— хлорид 47, 69, 72
— хлороплатинат 234, 235
Аммониты 257
Аммофос 207, 314
Анальцим 274
Анаэробы 160
Ангидриды 224
Ангидрит 124, 153, 258, 288
Ангидритовый цемент 125
Ангидрон 186, 231
373
Англезит 253, 288
Ангстрем 13
Анилино-красочная промышленность 359, 362
Анионы 13, 66, 106, 122, 157, 158, 200, 270, 272
— названия 225, 226
Анод 140
Анодирование 64
Анортит 64
Антигорит 271
Антимон- 224
Антимонаты 291
Антимониды 143, 291
Антимоний 290
Антимонил 291
Антимонит 97, 289, 290, 291
Антимониты 290
Антиферромагнетики 134
Антифрикционные сплавы 155
Антофиллит 200
Апатиты 315, 322
Арагонит 153
Аргент- 224
Аргентит 253
Аргиродит 119
Аргон 72, 73,-77, 107, 118, 143, 145
Аргоновые трубки 72
Арматура 82
Арсен- 224
Арсенаты 73, 210, 211
Арсениды 73, 115, 211
Арсеникон 209
Арсениты 73, 210, 211
Арсенопирит 210, 289
Арсин 73, 121, 211
Асбест 184, 186
Асботехническая промышленность 360
Ассоциация молекул 99
Астат 73, 115, 116, 136, 215, ,354
Астраханит 288
Атакамит 191
Атмосфера 73, 47, 83, 84, 118, 121, 135, 137, 160, 227, 302, 308
Атмосфера (единица давления) 74
Атмосферное давление 74
Атом 11—28, 106, 116, 350, 351, 352
— вероятность нахождения электрона^ 23, 24, 28
— водорода 15, 16, 17, 24, 25, 26, 105, 122
— возбуждённые состояния 16
— история проблемы И
— кислорода 161
— масса 13, 352 {
— многоэлектронный 17
— модель И, 13
— основы современной теории строения 20
— периодическая система и строение атома 37
— планетарная модель 11, 13, 20, 21, 24
— размеры 13
— «траектория электрона в атоме» 24, 25, 28
— углерода 303, 305
— фтора 322
— электронная конфигурация 19, 42, 43, 45, 116, 145, 234
— электроотрицательность 50, 116, 121, 197
— энергетические уровни, распределение электронов 16, 18, 19, 20, 22, 26, 37
— энергия ионизации 13, 32, 116, 145, 348, 349
— ядро 12, 13, 23, 26, 44, 45, 57,
137, 144, 244, 296, 297, 311, 350, 351
Атомарно-водородная горелка 109
Атомная масса 13, 44, 116, 145, 177, 234, 348, 349, 351, 352, 354
Атомная электростанция 311
Атомная энергия 311
Атомное ядро 12, 13, 23, 26, 44, 45, 57, 137, 144, 244, 296, 297, 311, 350, 351
— заряд 13, 29, 44, 45, 350, 351
— радиус 13
— реакция деления 311
Атомный вес 6, 15, 43, 44, 262, 354
Атомный номер 13, 26, 29, 36, 42, 44, 116, 137, 145, 180, 197, 234, 348, 349, 350
Атомный радиус 12, 13, 116, 145, 348, 349
Атомный спектр 13, 33
Аур- 224
Аурипигмент 209, 210, 289
Аффинаж 235, 236
Ацетилен 163, 306
Ашарит 89
Аэрозольные упаковки 324
Аэрон 183
Б
Баббиты 156, 230, 291
Бадделеит 342
Базальт 125, 202, 203
Бактерии 48, 49, 86, 88, 258, 263, 330
«Бариевая каша» 80
Барий 79, 13, 159, 196, 197, 244, 2§6, 348
— амальгама 69
— борид 91
— гидроокись 79
— карбонат 79, 80
— криптонат 173
— манганат 80
— нитрат 79, 80
— окись 79
— перекись 79
— платиноцианид 80
— сплавы 80
— сульфат 79, 80
— сульфид 79
— хлорат 79
— хлорид 79
— хромат 79, 80
Барит 79, 204, 258, 260, 288
Баритовая вода 79
Бастнезит 177
«Белая сажа» 167
Белильная известь 124, 141, 142, 329
Белково-витаминные концентраты 368
Белый графит 221
Бензин
— разделение компонентов 275
Бензол 306
— экстракция брома бензолом 93 Бенитоит 272
Берилл 80, 271, 272, 273, 333
Бериллат-ион 81
Бериллаты 30, 81
Бериллиевые бронзы 189
Бериллий 80, 30, 38, 39, 196, 197, 240, 244, 348, 349
— атом 19
— атомный радиус 13
— в смеси с актинием 56
— гидроокись 81
— окись 81
Берклий 81, 56, 58, 59
Бертоллетова соль 153, 317, 329, 330
Бертоллиды 195
Бессемеровский процесс 279
Бета-частицы 56, 57, 241, 287, 297
Бетон 81, 125, 275
Бикарбонаты 83, 121, 157, 310
Биолиты 204
Биомасса 123
Биосфера 83, 77, 126, 136, 302
Биохимия 7
Бирюза 202
Бисмит 97
Бисульфаты 89, 121, 288
Бисульфиды 288
Бисульфиты 89, 121, 289
Битумы 82
Бифториды 324
Бихроматы 89, 128, 335
Бишофит 326
Благородные газы 143
Благородные металлы 89, 196(, 232, 261
«Бойлев ад» 250
Бокситы 63, 64, 127
Болонские светящиеся камни 79
Бор 89, 30, 95, 121, 299
— атом 19
— галогениды 89, 90
— гидриды 92
— карбид 91
— нитрид 90, 91, 221
— фторид 89
Бора теория строения атома 11, 14, 17, 20, 21, 24
Боразон 90, 221
Бораны 91, 92
Бораты 91, 89, 202
Бориды 91, 89
Борин 92
Борирование 91
Борная кислота 91, 89
Борный ангидрид 89, 91
Бороводороды 92, 91
Браунит 186
Бридер 312
Бриллианты 307
де-Бройля соотношение 22
Бром 93, 13, 115, 116, 123
Броматы 95, 94
Бромид-ион 94
Бромиды 95
Бромистоводородная кислота 95, 94
Бромистый водород 95
Бромистый этил 94
Бромная вода 95, 93
Бромноватая кислота 95, 94
Бромноватистая кислота 95, 94, 124
Бромсодержащие органические соединения 94
Бронза 129, 188, 190, 194, 196, 228, 281
Брошантит 191
374
Булат 278
Булатная сталь 278, 279
Бунзенит 217
Бура 95, 89 :
Бурые железняки 129
Бурый газ 51, 53
Буферные растворы 92
Быстрорежущая сталь 283
В
Валентинит 290 Валентность 30 Валентные оболочки 19, 37 Валентные электроны 29, 43, 116, 156 Ванадаты 96
Ванадий 96, 185, 196, 197
— окислы 96
— пятиокись 96, 97
— сплавы 97
Венера, планета 194
— атмосфера 78
Вернадского закон 88
Вероятность нахождения электрона
23, 24, 28
Веселящий газ 97, 212
Ветер 76
Взрывчатые вещества 47, 257
Виргиний 321
Висмут 97, 114, 196, 197, 291
— гидроокись 98
— окисел 98
— сплавы 98
Висмутаты 98
Висмутин 97
Висмутовая кислота 98
Висмутовая охра 97
Висмутовая спираль 98
Висмутовый блеск 97
Висмутовый теплоноситель 98
Виталлиум 166
Витерит 79
Вода 98, 105, 106, 121, 163, 203, 223
— в природе 101
— жёсткость 103, 274
— обеззараживание 262, 330
— опреснение 370
— осветление 67
— получение 103
— пресная 102, 103, 122, 124
— применение 103
— строение 99
— физические свойства 99
— химические свойства 100
«Вода» (элемент) 6, 354
Водоотнимающие средства 231, 264 Водород 105, 15, 77, 100, 163, 350 — атом 15, 16, 17, 24, 25, 26, 105, 122 — атомный радиус 13 — атомный спектр 16 — в природе 105, 122 — жидкий 106 — изотопы 13, 105, 109 — иодистый 148, 147 — историческая справка 105 — молекула 105 — перекись 109, 163 — получение 107 — применение 108 — физические свойства 106 — химические свойства 106 — хлористый 106
Водородистые металлы 225
«Водородная болезнь» 191
Водородная связь 99, 100, 106
Водяной газ 101, 310
Водяной пар 74
Возбуждённые состояния 16, 19
Воздух НО, 74, 75, 107, 143, 160, 223, 304, 308
— в технике 111
— жидкий 173
— загрязнение 77, 89
— искусственный 112, 119
— разделение 111
— сжижение 111, 112
«Воздух» (элемент) 6, 354
Воздушные вяжущие материалы 124
Возникновение жизни 77, 356
Возраст геологических объектов — определение 305, 310
Волна — частица 14, 21, 22, 24, 28
Волновая механика 21, 22—см. также
Квантовая механика
Волновая функция 23, 25
Волновое уравнение 23, 24, 25, 26
Волокна химические 359, 365, 370
«Волчья пена» 112
Вольтов столб 262
Вольфрам 112, 113, 159, 196, 197, 209
— карбид 113, 157
— окись 112
— сплавы 113, 249
Вольфраматы 113, 202
Вольфрамит 112
Вольфрамовая кислота 113
Вороненйе 131
Временная жёсткость воды 104
«Вторая природа» 7, 358
Вуда сплав 98, 151
«Вулкан» (опыт) 101, 334
Вулканические газы 268, 269, 304,
308, 322
Вулканы 122
Выветривание 102, 122, 127, 153, 232
Вяжущие строительные материалы 124, 125
Г
Гагаринит 246
Габбро 125
Гадолиний 114, 177, 181
— сульфат 181
Гадолинит 246, 247
Газгольдер 282
Газированная вода 309
Газификация твёрдого топлива 108, 307
Г азобетон 82
Газопоглдтители 252, 337
Газоразрядные лампы 173, 175
Газосветные лампы 214, 215
Газы 112, 161
Галенит 253, 254 , 258, 289
Галит 114, 127, 199, 212, 326
Галлий 114, 39, 44, 194, 196, 197, 293
— арсенид 115, 211
— гидроокись 115
— жидкий 114
— сплавы 115
— фосфид 115, 315
Галмей 150
Галогенид-ионы 262
I
Галогениды 50, 64, 65, 116, 122
Галогенные лампы 173
Галогеноводороды 106, 117, 215
Галогены 115, 26, 30, 94, 215
Галоиды 117, 115
Гальваническая пара 340
Гальванический микроэлемент 66, 340, 341
Гальванический элемент 340
Гальванопластика 362
Гальванотехника 140
Гамма-дефектоскопия 244
Гамма-железо 130, 281
Гамма-лучи 244
Ганий 219
Гарт 291
Гаусманит 186, 187
Гафнаты 117
Гафниевая кислота 117
Гафний 117, 45, 196, 197, 299, 342
— борид 91
— гидроокись 117
— карбид 117, 156
— окись 117
Гашёная известь 118, 141
Гекса- 244
Гексагональные кристаллы 255
Гексахлоран 330, 331
Гелиево-кислородная атмосфера 119
«Гелиевый» воздух 119
Гелий 118, 11, 14, 42, 76, 77, 143, 144,
145, 300
— атом 18, 25
— жидкий 119
Гелионы 118
Гель 166
Гематит 129
Геми- 224
Генераторный газ 310
Геологическая история 77, 101
Геосферы 135, 137
Геохимическая деятельность организмов 88
— человека 88
Геохимия 7, 126, 201, 354
Гепта- 224
Гербициды 367
Германаты 120
Германиды 120
Германий 119, 39, 44, 121, 194, 196
197, 293
— двуокись 120, 121
— окись 120
Германит 119, 120
Германы 120
Герсдорфит 217
Геттеры 252, 337
Гиацинт 342
Гигроскопическая вода 101
Гидр- 224
Гидравлическая известь 142
Гидравлические вяжущие материалы 124
Гидразин 50
Гидразоний 50
Гидратационное твердение 125, 141
Гидриды 121, 198, 225
Гидро- 226
Гидрогетит 129
Гидрокарбонаты 121, 83, 157, 310
Гидроксиды 225
Гидроксиламин 50
375
Гидроксоний 101
Гидролиз 101
Гидрометаллургия 369
Гидроокись 202, 225, 348, 349
Гидросодалит 274
Гидросульфаты 121, 89, 288
Гидросульфиды 121, 288
Гидросульфиты 121, 289
Гидросфера 121, 76, 77, 83, 101, 136,
137, 159, 302, 308
Гипо- 226
Гипобромиты 124, 94, 95
Гипоиодиты 148
Гипосульфит 124, 260, 263, 297
Гипохлорит-ион 328
Гипохлориты 124, 333
Гипс 124, 153, 204, 258, 260, 288
Главное квантовое число 16, 18, 19
Глазурь 168
Глауберова соль 202, 212, 222, 288
Глёт 255
Глина 127, 270, 272
Глинозём 125, 63, 67
Глиций 80
Глубокое охлаждение 119
Глюциний 80
Гнейс 126, 127
Гниение 160
Гольмий 125, 177, 181
Горение 108, 160, 162, 223
— сурьмы в хлоре (опыт) 290
Горно-химическая промышленность 358
Горные породы 125, 137, 199, 202
Горный хрусталь 172
Горючий воздух 105, 107, 161
Гранат 270, 272, 273
Гранит 125, 126, 127, 202
Графит 127, 90, 272, 302, 303, 305,
306, 307, 308, 316, 345, 346
— искусственный 307
Гремучий газ 127, 106
«Греческий огонь» 257
Гринокит 150
Грязи целебные 86
д
Дальтониды 195
Дальтонизм 15
Данбурит 273
Двуокись 224
Двухромовая кислота 89
Двухромовокислые соли 89
ДДТ 330
Дейтерий 13, 105, 109, 123, 297
Дейтрон 297, 354
Дека* 224
Декарбонизация 106
Денитрификаторы 48
Денитрификация 49
Дефлогистированный воздух 161
Дефолианты 367
Джезказганит 248
Ди- 224
Диатомит 167
Диборан 92
Дибромэтан 94
Дидим 39, 177, 178, 181, 246
Димеризация 51
Динамит 167
Диопсид 271, 272
Диоптаз 272
Диорит 125
Диполь 99, 144
Диспрозий 128, 39,' 177, 181
Диспропорционирование 143, 318
Диссоциация 100, 101
Дистиллированная вода 103
Дисульфаты 288
Дисульфиты 289
Дитцеит 146
Дифосфорная кислота 319
Дифракция 22
Дихлорэтан 330
Дихроматы 128, 89, 335
Дихромовая кислота 89, 128, 335
Дициан 49, 338
Длина волны 22
Добреелит 204
Дождь 122
— искусственный 309
Доломит 141, 158, 184
Доменный процесс 343, 344, 347, 358
Донорно-акцепторная связь 70, 309
Древесноволокнистые плиты 370
Древесностружечные плиты 370
Дробная кристаллизация 178
Дробное осаждение 178
Дунит 232
Дуралюмин 61
«Дырки» 120
Дырочная проводимость 120
Дыхание 86, 123,133, 160
Е
Европий 128, 177, 181
Едкие щёлочи 212, 349
Едкий натр 128, 214
Едкое кали 128, 152, 212
Ж
Жаропрочные сплавы 62, 216, 249
Жаростойкие сплавы 157, 171, 216
Железные метеориты 128, 204, 216, 315
Железные сплавы 128, 132
Железный колчедан 289
Железо 128, 84, 194, 195, 196,-197,
204, 264, 277, 283, 314, 343, 350, 368
— в природе 129
— гидроокись 131
— закись 131
— историческая справка 128
— карбид 157, 343
— магнетизм 133
— окисление 162
— окислы 131
— окись 131
— пирофорное 162
— получение 132
— применение 132
— роданид 131
— сварочное 278, 279
— сульфиды 131, 132
— сыродутное 278
— физические свойства 130
— химические свойства 130
Железоаммониевые квасцы 158
Железобетон 82
Железомарганцевые конкреции 123, 186
Железо — никель система 216
Жёсткая вода 103
Жёсткость воды 103
Жесть белая 230
Живое вещество 84, 86, 88
Жидкая углерода двуокись 309
Жидкие удобрения 205
Жидкий азот 52
Жидкий водород 106
Жидкий воздух 111, 112
Жидкий галлий 114
Жидкий гелий 119
Жидкий кислород 162, 163
Жидкое стекло 270, 271
Жидкое топливо 107
Жолиотий 60
3
Завершённые электронные оболочки 143, 144, 145
Закалка 130, 278, 279, 281
Закись 224
Заряд ядра 13, 29, 44, 45, 350, 352, 354
Защитная окисная плёнка 64, 81, 131, 162, 186, 210, 216, 253, 295, 298, 342
Зелёные растения 84, 86, 102
«Земли» 63, 153, 177, 223, 246, 348
Земля, планета
— 1 атмосфера 73
— вид из Космоса 122
— строение 136
«Земля» (элемент) 6, 354
Земная кора 136, 76, 83, 101, 122, 125, 126, 127, 159, 183, 202, 270
Зеркало металлическое 67, 143, 236, 262
Золото 137, 68, 89, 190, 194, 196, 197, 232, 235, 249, 261, 289, 291, 350, 354
— в природе 138
— историческая справка 137
— получение 1&9
— применение 130
— физические свойства 138
— химические свойства 138
Золотохлористоводородная кислота 140
Золочение 139, 140
Зоомасса 84
Зоопланктон 304
Зооциды 330, 367
И
Изверженные горные породы 125, 126, 127
Известняк 126, 127, 141, 153, 156, 157, 158, 204, 304, 309
Известь 141, 125, 153, 309
Изоморфизм 158, 200, 210
Изополикислоты 225
Изотопный состав элементов 351
Изотопы 13, 57, 59, 234, 351
— распространённость 351
Изумруд 80, 270, 333
Ильменит 298
Импульсные лампы 173
Инвар 128, 216
376
Инверсия структурная 69
Ин даты 143
Индиевые зеркала 143
Индий 142, 194, 196, 197, 293
— гидроокись 143
— сплавы 143
— фосфид 315
Индийская селитра 47, 257
Инертные газы 143, 43, 44, 74, 77, 107, 110, 112, 173, 174, 215
Инконель 217
Инсектициды 73, 330, 367
Интерметаллические соединения 62, 198
Интрузивные горные, породы 125 Инфракрасное излучение 74, 75, 76, 100, 337
Инфракрасные детекторы 121 Иод 146, 13, 44, 45, 73, 115, 116, 202 Иодаты 148
Иодидное рафинирование 298, 299, 342
Йодиды 148, 147, 149
Иодированная поваренная соль 148 Иодистоводородная кислота 148, 147, 149
Йодистый водород 148, 147
Иодная вода 146
Иодная кислота 148
Йодноватая кислота 148
ИодноватисТая кислота 147, 148
Йодноватый ангидрид 148
Ион аммония 72, 70
— гидроксония 101
Ионизация 13, 75, 116, 145
— воздуха 287
Ионная связь 156, 215, 221, 322, 348, 350
Ионные карбиды 156
Ионные ракетные двигатели 337 Ионный радиус 116, 200, 348, 349 Ионообменные смолы 370
Ионосфера 74
Ионы 101, 103, .104, 105, 197, 200, 340, 341
— многозарядные 57
Иридий 149, 89, 196, 197, 231, 232, 234 — осмистый 232, 236
Искусственный дождь 309
Исландский шпат 153
Испанские белила 98
Итмид 97
Иттербий 149, 177, 181, 247 «Иттриевая земля» 177, 246 Иттрий 149, 196, 197, 246, 247
К
Кадмиевые краски 151
Кадмий 150, 196, 197
— «крик» 228
— селенид 151, 256
— сульфид 150, 151
Кадмирование 151
Каинит 151, 153, 206, 326
Калаверит 296
Каламин 150
Калиевая селитра 153, 256, 257
Калий 151, 13, 19, 72, 196, 197, 213, 251, 349
— атом 42
— гидроокись 128, 152, 153
— дисульфит 289
— дихромат 335
— манганат 187
— надперекись 152
— нитрат 153
— окисление 162
— перманганат 98, 231
— сплавы с натрием 152
— сульфат 153, 206
— фтор ацетат 322
— хлорат 153, 330
— хлорид 153, 206
— хромат 202
— цианид 338
Калий — натрий система 152
Калийные соли 151
Калийные удобрения 153, 206
Калифорний 153, .56, 58, 59
Каломель 153, 250
Кальциевая жёсткость воды 104
Кальциевая селитра 205, 256, 257
Кальций 153, 13, 19, 196, 197, 204, 286, 348
— амальгама 69
— гидрид 156
— гидроокись 141
— гидросульфит 260, 289
— карбид 155, 156
— карбонат 141, 153, 154, 156, 304, 308
— нитрат 156
— ортофосфаты 156
— сульфит 269
— хлорид 156
— цианамид 49
Кальцинированная сода 275, 276, 364
Кальцит 127, 141, 153, 157, 202, 203, 204
Камасит 204
Каменная соль 212, 326
Каолин 64
Каолинит 272
Капиллярная вода 101
Карб- 224
Карбамид 72, 206'
Карбиды 156, 91, 97, 113, 170, 295, 306
Карбин 306
Карбон- 224
Карбонатная жёсткость воды 104
Карбонаты 157, 121, 122, 202, 287, 302, 307, 308, 310
Карбонилы металлов 165, 188, 217, 309
Карборунд 158, 156, 170, 171
Каркасные структуры 272, 273
Карналлит 151, 184, 206, 251, 252, 257, 326
Карнотит 312
Каслинское литьё 346
Кассиев пурпур 139
Касситерит 112, 228, 230
Катакомбы 155
Катиониты 1(Й
Катионы 13, 66, 106, 122, 200, 226, 270, 272
Катод 140
Каустик 276
Каустическая сода 158, 214, 275, 276
Квантовая механика 21, 22, 24, 25, 26, 28, 33, 45
Квантовые числа 17, 19, 25
Кванты 14, 16
Кварц 172, 199, 202, 203
Кварцевые пески 127
Квасцы 158, 63, 114, 226, 288, 293
Кераргирит 263
Кермезит 290
Керметы 157
Кернит 89
Кессонная болезнь 118, 119
Кимберлит 302, 303
Киноварь 203, 249, 250, 289
Киппа аппарат 108
Кислород 159, 15, 73, 75, 77, 84, 86, 111, 112, 122, 202, 213, 227, 350
— атом и молекула 161
— в природе 159
— жидкий 162, 163
— изотопы 159, 163
— историческая справка 160
— получение и применение 163
— физические свойства 162
— химические свойства 162
Кислородное дутьё 345
Кислородно-конвертерный процесс 282
Кислородные воды 86
Кислоты 222
— названия 225
Кислые почвы 105
«Кислые» реки 105
Кларки 202
Клатраты 145
Клей силикатный конторский 167
Климат 122
Клинкер 338
Клубеньковые бактерии 49
Кобальт 164, 123, 133, 196, 197
— гидроокись 164
— изотопы 166
— карбонил 165
— окислы 164
— хлорид 165
Кобальтин 164
Кобальтовая пушка 166
Кобальтовое стекло 164
Кобальтовые краски 164, 166
Кобальтовые сплавы 166
Кобальтовый блеск 164
Кобальтовый колчедан 164
Ковалентная связь 70, 92, 100, 106, 116, 156, 215, 221, 255, 322
Ковалентные карбиды 156
Ковеллин 190
Кокс 307, 308, 344, 345, 368
Колумбит 219, 248, 294
Колчеданы 289
Комплексные соединения 158, 165,
234, 254, 261, 262, 288, 309, 338
Конвертер 280, 282, 314
Конденсатор 295
Константан 189
Конституционная вода 101
Координационная связь 70
Кораллы 154, 304
Коренные месторождения 138, 232
Корпускулы 354
Коррозия металлов 101, 131, 162, 297, 298
Корунд 68, 333
Космическая пыль 74, 75
Космические лучи 74
377
Космос
— вид Земли из Космоса 122
— минералы в Космосе 204
— элементы химические, распространённость 354, 355
Космохимия 7
Коэрцитивная сила 216
Красители органические 359
Красноломкость стали 277, 280
Красностойкость стали 283
Красные железняки 129
Кремень 167, 168
Кремневодороды 166, 93, 170
Кремнёвый пистолет 167
Кремнекислородные кислотные остатки 270
Кремнекислородный каркас 274
Кремнекислородный мотив 271, 272, 273
Кремнекислородный тетраэдр 271, 272, 274
Кремнезём 166, 167, 170, 172
Кремниевые кислоты 166, 168
Кремниевые фотоэлементы 171
Кремниевый ангидрид 167, 166
Кремний 167, 121, 159, 166, 299, 350
— в природе 168
— двуокись 172, 166, 167, 170
— историческая справка 167
— карбид 156, 158, 171
— получение 170
— применение 170
— физические свойства 168
— химические свойства 168
Кремнийорганические соединения 172
«Крик олова» 228
Криогенная техника 119
Криолит 65, 66, 322, 325
Криосфера 121
Криптон 173, 73, 143, 145
— фториды 173
Криптоновые лампы 174
Кристаллизационная вода 101, 288
Кристаллическая сода 275
Кристаллическая структура 90, 130, 131, 158, 255, 272, 273, 274, 305, 306, 316
Кристаллогидраты 101, 158, 264, 288, 326
Критическая масса 238
Критическая температура 162, 281
Крица 278, 279
Кричный передел 278
Кровавик 129
«Кровь из воды» 132
Крокоит 253, 333
Кроны 335
Круговорот азота в природе 47, 48
— воды в природе 102, 122
— кальция в природе 153
— кислорода в природе 160
— серы в природе 258
— углерода в природе 303, 304
— фосфора в природе 316
Круксит 293
Ксенон 174 , 73, 143, 144, 145, 173
— изотопы 296
— фториды 145, 146, 174
Ксеноновые лампы 175
Ксенотим 246
Ксилолит 186
Кубонит 221
Кубооктаэдр 274
«Купоросное масло» 222, 264
Купоросы 222, 226, 264, 288
Купр- 224
Куприт 289
Купферникель 217
Курчатовий 175, 30, 60, 218, 301
— изотопы 176
Кюри точка 130, 134
Кюрий 176, 56, 57, 58, 59
Л
Лава 125
Лавсан 359, 370
Лазер 68, 181, 211, 285
Лакокрасочная промышленность 359
Лакриматоры 94
Ламинария 146
Лампа газоразрядная 173, 175
— газосветная 214, 215
— кварцевая ртутная 249
— криптоновая 174
— ксеноновая 73, 175
— люминесцентная 215
— накаливания электрическая 72, 113, 173, 174, 209, 216
— неоновая 215
Лангбейнит 206
Лантан 177, 56, 196, 197, 246, 247
— борид 91
Лантаниды 177
Лантаноидное сжатие 115, 117, 179, 180, 342
Лантаноиды 177, 30, 37, 39, 42, 57,
58, 196, 197, 246
— гидроокиси 179
— история открытия 177
— окислы 177, 178, 180, 181
— применение 180
— распространённость в природе 178
— физические свойства 178
— химические свойства 178
Латунь 188, 189, 339
Лаутарит 146
Легирование стали 314
Легированная сталь 188, 209, 277, 283, 342
Легированный чугун 346
Лёгкие металлы 196
Легкоплавкие сплавы 151
Лёд 99, 100, 102, 121
Лепидолит 251
Лестничная таблица периодической системы элементов 36, 37
Летучая щёлочь 47
Лечебные грязи 269
Листовые структуры 272, 273
«Лисьи хвосты» 55
Литий 181, 13, 30, 196, 197, 251, 349
— атом 19
— взаимодействие с азотом 182
— гидрид 182
— нитрид 49
Литийалюминийгидрид 182
Литопон 341
Литосфера 183, 83, 101
Литьё 346
Лопарит 219
Лорандит 293
Лоуренсий 183, 56, 58, 60
Лужение 230
Луна 194
Лунные минералы 204
Люминесцентные лампы 215
Люминесценция 75, 339, 341
Люминофоры 180, 241
Лютеций 183, 177, 178, 181
«Лягушачье золото» 236
Ляпис 262
м
Магма 125, 126, 199, 202, 203
Магматические горные породы 125, 126, 127
Магналий 61
Магнезиальный цемент 186, 337
Магнезит 158, 184
Магнезия 184, 185, 186
Магнетизм железа 133
Магнетит 129, 186
Магниевая вспышка 185
Магниевая жёсткость воды 104
Магниево-кальциевые сплавы 156
Магниевые сплавы 185
Магниетермия 185
Магний 184, 13, 32, 196, 197, 308, 348, 349
— амальгама 69
— гидроокись 185
— нитрид 185
— окись 184, 185, 186
— перхлорат 186, 231
Магнико 216
Магнитное квантовое число 17, 19
Магнитно-жёсткие сплавы 128
Магнитно-мягкие сплавы 128
Магнитные вещества 180
Магнитные материалы 133, 134, 166, 180, 181
Магнитные свойства 133, 134, 216
Магнитные сплавы 128, 216
Магнитный железняк 129, 184
Магнитный колчедан 289
Магнитострикционные сплавы 128
Мазер 69
Макроэргические соединения 316
Малахит 158, 190, 191, 203, 289
Манган 224
Манганаты 186, 187, 188
Манганин 188, 189
Манганит 186
Манганиты 187
Мантия 76, 122, 126, 136, 160, 258
Марганец 186, 67, 84, 158, 159, 196, 197
— закись-окись 187
— карбонил 188
— окислы 186, 187
Марганцовая кислота 188, 187, 231
Марганцовистая кислота 188, 186, 187, 231
«Марганцовка» 98, 231
Марганцовокислые соли 188
Марганцовый ангидрид 188, 187
Марганцовый чугун 346
Марс, планета 194
— атмосфера 78
Мартеновский процесс 280, 282, 345
«Масла» 222
Массовое число 351
Медная синь 190
Медно-никелевые руды 232, 236
378
Медно-никелевые сплавы 188, 189
Медные сплавы 188
Медный блеск 190, 289
Медный индиго 190
Медный колчедан 190, 258, 289
Медный купорос 191, 193, 264
Медь 189, 194, 196, 197, 289, 350
— в природе 190
— гидроокись 191
— историческая справка 189
— окислы 191
— получение 191
— применение 192
— сульфат 191, 193, 340
— физические свойства 190
— химические свойства 190
Международная номенклатура неорганических соединений 224
Межгалогенные соединения 94, 116
Мезосфера 74
Мейера таблица элементов 44
Мел 157, 158
Мельхиор 189
Менделевий 193, 31, 44, 56, 57, 58, 60
Менделеева закон 29—45, 6, 114, 119, 194, 201, 246
Мер кур- 224
Меркурий 194, 249
Мета- 225
Метаалюминиевая кислота 65
Метабисульфиты 289
Метабораты 95
Метаборная кислота 91
Метакремниевая кислота 271
Металепсия 327
Металептические реакции 327
Металлиды 62, 198
Металлическая связь 156, 197
Металлические свойства 30, 36,43, 197
Металлический блеск 193, 198, 289
Металловедение 196,
Металлография 279, 281
Металлоиды 215
Металлокерамика 113
Металлоподобные карбиды 156
Металлоподобные нитриды 221
Металлоподобные силициды 275
Металлургия 163, 187, 192, 193, 279, 280, 281, 282, 283, 343, 344, 369
Металлы 193, 5, 29, 30, 36, 42, 44, 126, 127, 171, 215, 217, 264, 288, 340, 348, 349, 352, 369
— классификация 196
— коррозия 101, 131, 162, 297, 298
— окисление 162
— пассивирование 53
— переходные 30, 42, 121, 134, 180
— платиновые 231
— соединения с водородом' 121
— физические свойства 196
— химические свойства 196
— щелочноземельные 348
— щелочные 349
Метаморфизм горных пород 127
Метан 303, 306
— конверсия 108
— окисление 163
Метаниобаты 295
Метаргон 147
Метасвинцовая кислота 254
Метатанталаты 295
Метафосфаты 314
Метафосфор истая кислота 318 Метафосфорная кислота 199, 314, 320 Метеориты 75, 128, 129, 137, 156, 164, 194, 200, 204, 216, 303, 315
«Мефитический воздух» 47 Меченый атом 52, 246, 287, 318 Миграция химических элементов 84, 127
Микробиологическая промышленность 368
Микроорганизмы 86, 88, 258 , 263, 269, 289, 304, 309
Микроудобрения 91, 205, 209
Микроэлементы 91 Миларит 272, 273 Миллерит 217 Минерализация 86 Минералогия 87, 201, 204, 270 Минералы 199, 124, 125, 270 — в Космосе 204 — возраст, определение 310 — классификация 202 — происхождение 202 — распространённость 202 — состав, структура и свойства 199 Минеральная щёлочь 151 Минеральные удобрения 204, 361, 364, 367
Мирабилит 84, 202, 212, 288 Мировой океан 121, 122, 123 Митохондрии 315 Мишметалл 180, 247, 346 Молдавий 321 Молекула — азота 50 — аммиака 69 — ассоциация 99 — водорода 105 — воды 99, 100 — димеризация 51 — кислорода 162 — серы 258 — фтора 322 Молекулярная биология 28 Молекулярные сита 274 Молекулярные часы 69 Молибдаты 202, 208 Молибден 208, 159, 196, 197, 296 — дисульфид 209 — окислы 208, 209 — силицид 275 Молибденит 208, 248 Молибденовая кислота 208 Молибденовая синь 208 «Молибдёновое» стекло 209 Молибденовые сплавы 209 Молибденовый блеск 208 Монацит 178, 246, 300 Монель-металл 217 Монетная бронза 189 Моно- 224 Моногидрат 263, 264, 267 Монохроматы 335 Монохромовая кислота 335 Монтепонит 150 Морион 172 Мочевина 71, 206 Мрамор 127, 153, 157, 158 Мур ий 326 Мусковит 273 Мышьяк 209, 121, 350 — трёхокись 268
Мышьяковая кислота 210
— соли 73
Мышьяковистая кислота 210
Мышьяковистокислые соли 211, 73,
210
Мышьяковистый ангидрид 210
Мышьяковистый водород 211, 73
«Мышьяковое зеркало» 211
«Мышьяковое масло» 222
Мышьяковокислые соли 211, 210
Мышьяковый ангидрид 210
Мышьяковый колчедан 210, 289
Мягкая вода 103
н
Наждак 68
Натриевая селитра 205, 256, 257
Натрий 212, 13, 153, 196, 197, 349
— амальгама 68
— гексаметафосфат 314
— гидрокарбонат 157
— гидроокись 128, 214
— дихромат 335
— карбонаты 275
— метаборат 89
— перекись 213
— сплавы с калием 152
— тиосульфат 124, 259, 260, 261, 263, 297
— хлорат 330
— хлорид 212, 214, 326, 328, 330
— цианид 338
Натровая известь 142
Натрон 275, 329
Наука и промышленность 358
Нашатырный спирт 214, 69, 70, 222, 326
Нашатырь 215, 69, 72, 326
Негашёная известь 215, 141
Нейзильбер 189
Нейтрон 351, 12, 13, 57, 75, 81, 91,
98, 240, 244, 301, 311, 312, 342
Неметаллы 215, 5, 29, 30, 44, 193, 352
Неодим 215, 39, 177, 178, 181
Неон 215, 19, 143, 145
Неоновые лампы 215
Неоновые трубки 215
Неопределённости принцип 24, 25
Неорганическая химия 9
Неорганические продукты 364
Неорганические соединения
— номенклатура 222
Нептуний 216, 56, 58, 301, 354
Нептуноил-ион 59
Нержавеющая сталь 188, 314
Нефелин 64, 272
Нефтехимия 359
Нефть 123
«Низведение огня с неба» 335
Никелевые сплавы 216
Никелевые стали 216
Никелевый блеск 217
Никелевый купорос 218
Никелин 217
Никелирование 218
Никель 217, 133, 196, 197
— арсенид 217
— атом 19, 20
— карбонил/217
Нильсборий 218, 30, 301
Нимоник 217
379
Ниобаты 220
Ниобиевая кислота 220
Ниобиево-танталовые концентраты 220
Ниобий 219, 196, 197, 294, 295
Нитр- 224
Нитратные удобрения 205
Нитраты 220, 52, 202, 221, 256
Нитриды 221, 71
Нитриты 221
Нитрификация 49
Нитроаммофос 207
Нитроген 47
Нитроза 265, 266
Нитрозилсерная кислота 265, 266
Нитрозоний 51
Нитрон 359, 370
Нитроний 51
Нитрофоска 207
Нитрофосфат 207
Нихромы 216, 335
Нобелий 222, 56, 58, 60
Номенклатура неорганических соеди-
нений 222, 351
Нуклеиновые кислоты 315, 371
Ньюлендса таблица элементов 44
О
Обеззараживание воды 262, 330
Обжиговый газ 268
Облака 76
Обманки 289
Обсидиан 283
«Огонь» (элемент) 6, 354
Одлинга таблица элементов 44
Озон 227, 75, 159, 245
Озонатор 227
Окисление 108, 162
— биологическое 160
— степень 36, 37, 42, 43, 57, 58, 60,
116, 224, 226, 234, 352
Окисление-восстановление 197
Окислительно-восстановительная
зональность 88
Окислы 36, 202, 224
Окисная защитная плёнка 64, 81, 131,
162, 186, 210, 216, 253, 295, 298, 342
Окись 222, 223, 224
Оке- 224
Оксиды 222, 224
«Октавы» 44
Октан 274, 275
Октиббегит 204
Окто- 224
Олеум 228, 267, 270
Олово 228, 121, 194, 196, 197, 253,
293, 350
— двуокись 230
— галогениды 229
— «крик» 228
— сплавы 199
Оловянистая бронза 189, 318
Оловянистая кислота 230
Оловянистые баббиты 291
Оловянная кислота 229
«Оловянная чума» 229
Оловянный камень 228
Оловянный колчедан 228
Ольдгамит 204
Опал 167
Опреснение воды 370
Опрыскивание растений 367
Оптический квантовый генератор 68
Опыление растений 367
Орбитальное квантовое число 17, 19
Органическая химия 6, 19, 305, 363
Органические вещества
— геологическая роль на Земле 86
Органический синтез 359
Орлец 186
Орто- 225
Ортоалюминиевая кислота 65
Ортоборная кислота 91
Ортоклаз 64
Ортокремниевая кислота 271
Ортосвинцовая кислота 254
Ортотеллуровая кислота 296
Ортофосфаты 314, 319
Ортофосфористая кислота 318
Ортофосфорная кислота 230, 314, 319, 320
Осадочные горные породы 126, 127
Осадочные месторождения 127
Осадочные руды 127
Осборнит 204
Осветление воды 67
Осколочные ядра 311, 312, 313
Осмий 230, 89, 196, 197, 231, 232, 233, 234, 248
Осмистый иридий 232, 236
Основания 29, 225
Отавит 150
Отенит 203
Отпуск 130, 278
Отравляющие вещества 211, 309
Охрана природы 78, 89, 103, 123, 124, 360
Очистка воды 370
п
Пакфонг 216
Палладий 231, 89, 106, 196, 197, 231, 232, 233, 234, 255
Парамагнетизм 162
Парижская зелень 73
Пассивирование металлов 53
Пассивность 334
Патина 191
Патронит 96
Паули принцип 17, 18, 26
Пенобетон 82
Пента- 224
Пентландит 217
Пер- 226
Пергидроль 110
Перекиси 162, 163, 224
Переходные металлы 30, 42, 121, 134, 180
Период полураспада 56, 57, 234, 287
Перйодаты 148
Периодическая система элементов Д. И. Менделеева 18, 26, 28, 72, 116, 117, 137, 145, 197, 350
— значение и история 43
— и строение атома 37
— лестничная таблица 36, 37
Периодический закон Д. И. Менделеева 29—45, 6, 114, 119, 194, 201, 246
Периодичность свойств элементов 19, 29—37, 44, 45, 119
Периодичность энергии ионизации атомов 32
Периоды в ряду элементов 19, 29, 30, 33, 42
Перксенаты 175
Пермаллой 216
Перманганаты 231, 187, 188
Пермутиты 275
Пероксиды 224
Перренаты 248
Пертехнаты 297
Перфтор гексан 322
Перхлораты 231, 328, 332
Пески 126, 127
Пестициды 367
Песчаник 126
Петалит 182
Петрография 125
Пирит 84, 86, 184, 258, 260, 263, 265,
267, 268, 289, 293
Пирографит 307
Пирокислота 225
Пироксилин 55
Пиролиз 108
Пиролюзит 127, 184, 186, 326
Пирометаллургия 192
Пиросульфаты 288
Пиросульфиты 289
Пирофюрное железо 162
Пирофосфористая кислота 318
Пирофосфорная кислота 231, 319, 320
Пирротин 289
Плавиковая кислота 231, 321, 324
Плавиковый шпат 153, 321, 325
Плакировка 66
Планетарная модель атома 11, 13,
20, 21, 24
Планка постоянная 14
Планктон 123, 133, 304
Пластическая деформация 277
Пластические массы 359, 370
Пластичность 193, 198, 277
Платина 231, 196, 197, 200, 216, 231,
232, 234, 236
Платинит 128, 216
Платиновая губка 234
Платиновые металлы 231, 89, 194, 196
— применение 235
— распространение в природе и добыча 232
— физические и химические свойства 233
Платинохлористоводородная кислота 234
Плодородие почвы 47, 48
Плутоний 237, 56, 57, 58, 301
— изотопы 237, 312
— использование в ядерной энергетике 238, 301
Плюмб- 224
Плюмбаты 254
Плюмбиты 254
Пневматическая химия 47, 161
Победит 113, 166
Поваренная соль 199, 212, 214, 326, 328
— иодированная 148 1
Подземные воды 101, 121
Подземные газы 118
Полевые шпаты 64, 203
Полезные ископаемые 127
Полиборные кислоты 91
380
Полигалит 206, 288
Поликремниевые кислоты 271
Поликсен 232
Полимербетон 82
Полимерные материалы 359
Полимеры 330, 359, 369
Полиметаллические руды 253
По^иорганосилоксаны 172
Полисульфиды 260
Политетрафторэтилен 324
Полифосфаты 314
Полифосфор ные кислоты 314, 319
Полихлорвинил 332
Поллукс 336
Поллуцит 336
Полонаты 240
Полониды 240
Полоний 238, 12, 56, 73, 196, 197, 243
Полупроводники 73, 89, 115, 120, 143, 170, 171, 195, 211, 255, 292, 296, 315, 369
Полярное сияние 75
Порох 257
Порошковая металлургия 113, 166
Порперцит 232
Портландцемент 337
Порядковый номер элемента 13
«Постоянные» газы 162
Потассий 151
Поташ 240, 151, 153, 158
Празеодим 240, 39, 177, 181
Праута гипотеза 105
Преципитат 206, 314
Приближённые методы 26
«Примесные» атомы 263
Припои 230
Проба благородных металлов 140, 263
п-Проводимость 120
р-Проводимость 120
Прометий 240, 177, 354
Промышленность и наука 358
Простое вещество 49, 352
Протактинаты 242
Протактиний 241, 42, 56, 58
Протий 13, 105
Протон 12, 13, 29, 75, 105, 118, 351, 354
Протравливание семян 367
Протравливание ткани 158
Протуберанцы 76
Псиломелан 186, 200
Пудлингование 279
Пуццолановый цемент 337
Пушонка 141
Пьезоэлектрики 173
р
Радий 243, 12, 56, 80, 196, 197, 202, 239, 240, 241, 245, 311, 348
— изотопы 243
Радикалы химические 107, 224
Радиоактивное равновесие 243
Радиоактивность 11, 12, 239, 243, 311, 318
Радиоактивные ряды 243, 245, 253, 310
Радиоактивные элементы 56, 196, 354
Радиография 181
Радиоизлучение 75, 76
Радиолярии 168
Радиотерапия 244
Радиохимия 245
Радон 244, И, 143, 144, 145 — изотопы 244
Радоновые ванны 244, 245, 246 «Разрешённые» орбиты 15, 16, 20, 21 Раскисление стали 314
Распространённость изотопов 351, 352 — минералов 202
— элементов химических 137,351,354
Рассеянные металлы 196
Растворимое стекло 167
Растворы твёрдые 200
Растения — химический состав 205 Растительная щёлочь 151 Раухтопаз 172
Рафинирование металлов 192, 299, 342
Рвотный камень 291, 292
Реакции химические 329, 352, 354
Реальгар 209, 210
Редкоземельные элементы 246, 117, 177, 195, 196
Резерфорда модель атома 11, 15, 20, 21
Резиновая промышленность 359
Рениевая кислота 248
Рений 247, 196, 197
— сплав с вольфрамом 249
Ренирование 249
Рентгеновское излучение 74
РЗМ 246
РЗЭ 246
Роговое серебро 261, 263
Родий 249, 89, 196, 197, 231, 232, 234, 236
Родит 232
Родонит 186
Россыпные месторождения 138, 232
Ртутное зеркало 143
Ртутно-кварцевые лампы 173
Ртутный термометр 249, 251
Ртуть 249, 93, 194, 196, 197, 262, 350
— гремучая 250
— окись 250
— сплавы 68
— хлористая 153, 250
— хлорная 250
Рубидий 251, 13, 194, 196, 197,337,349
— гидроокись 252
— надперекись 251
Рубин 68, 204, 333, 335
Руда 126, 127, 129, 138
Рудные месторождения 126
Русская номенклатура неорганических соединений 224
Рутений 252, 89, 196, 197, 231, 232, 233, 234, 297
Рутил 298, 299
Ряд активностей 198
Ряд напряжений 197, 198
Ряды радиоактивные 243, 245, 253, 310
С
Сажа 307, 308
Сажевая промышленность 360
Сальварсан 211
Самарий 253, 39, 177, 181
— соединение с кобальтом 181
Самарскит 181, 312
Самоокисление-самовосстановление 318
САП 62
Сапфир 68
Сассолин 89, 91
Сатурн, планета 194
— атмосфера 78
Сверхползучесть 119
Сверхпроводимость 119, 220, 297
Сверхпроводники 119, 195
Сверхпроводящие магниты 220
Сверхтекучесть 119
Сверхчистые вещества 369
Свет 13, 14, 76, 160
Свинец 253, 158, 194, 196, 197, 264, 293, 296, 350
— азид 51
— изотопы 253
— нитрат 254
— окись 253, 254
— сплавы 254
— сульфат 255
— сульфид 253
Свинцовая бронза 189
Свинцовая примочка 255
Свинцовая пыль 255
Свинцовое стекло 254
Свинцовые белила ПО, 255, 269
Свинцовые кислоты 254
Свинцовые руды 253
Свинцовый блеск 253, 258, 289
Свинцовый кирпич 254
Свинцовый купорос 253 «Свинцовый сахар» 222, 254 Свинцовый хрусталь 285
Свойства веществ
— и «дегустация» 232, 233
Связь химическая 19,20,21, 116, 156, 170, 197, 200, 215, 221, 270, 348, 350 352
Селен 255, 90, 202, 258, 295, 296
— двуокись 256
Селенаты 256
Селениды 151, 256
Селенистая кислота 256
Селенистый водород 99, 256
Селеновая кислота 256
Селеновый ангидрид 256
Селитротвор 47
Селитры 256, 47, 226
Селитряницы 257
«Селитряный спирт» 222
Сера 257, 84, 202, 203, 261, 296, 350
— в природе 258
— водородные соединения 99
— газовая 269
— галогениды 259
— двуокись 74, 267
— историческая справка 257
— окисление 162
— получение 260
— применение 260
— самородная 258, 260
— трёхокись 269
— физические свойства 258
— химические свойства 258
— черенковая 260
Сервантит 290
Сердолик 172
Серебро 260, 89, 190, 194, 195, 196, 197, 269, 289, 350
— бромид 95, 263
— в природе 261
— галогениды 262
— и фотография 263
— иодид 148
381
— нитрат 262
— окислы 261
— получение 262
— применение 262
— сплавы 262
— сульфид 261, 262, 288
— физические свойства 261
— химические свойства 261
— хлорид 288
Серебряное зеркало 143
Серебряно-цинковый аккумулятор 262
Серебряный блеск 261
Серная кислота 263, 89, 228, 257, 260, 268, 270, 288, 289, 364
— производство и применение 264
— физические и химические свойства 263
«Серная печень» 289
Сернистая кислота 267, 89, 121, 289
Сернистокислые соли 267, 289
Сернистый ангидрид 267, 257, 258, 259, 265, 269, 289
Сернистый водород 269, 270, 84, 86,
258, 259, 260, 264, 270
Сернистый газ 269, 78, 265, 267 — очистка 267, 268
Серноватистая кислота 269, 297
Серноватистокислые соли 297
Сернокислые соли 269, 288
Серный ангидрид 269, 263, 265
Серный колчедан 258, 293
Серный цвет 260
Серобактерии 263
Сероводород 270, 269, 84, 86, 258,
259, 260, 264, 267, 269, 270
Сероводородная кислота 270, 121, 269, 288
Сероводородного заражения зона 86
Сероводородные воды 86
Сероуглерод 270, 259, 306
Серпентин 184 d-Сжатие 115 /-Сжатие 115, 117
Сидерит 129
Силал 346
Силаны 270, 166
Силик- 224
Силикагель 167
Силикатное стекло 283
Силикаты 270, 64, 136, 142, 151, 159,
168, 170, 173, 184, 202, 204
— алюминия 273
Силикотермия 171, 314, 334
Силициды 275, 170
Силумин 61
Сильвин 151, 153, 257, 326
Сильвинит 151, 153, 206, 326
Символы изотопов 351
— элементов 350
Симпатические чернила 165
Синерод 338
Синильная кислота 275, 307, 338
Синтетическая кожа 371
Синтетические волокна 359, 370
Синтетические лекарственные препараты 371
Синтетические продукты и изделия 365
Синтетические смолы 359, 365
Синтетический каучук 359
Синтин 107
Сита молекулярные 274
Скандиаты 247
Скандиевые ферриты 247
Скандий 275, 19, 38, 44, 194, 196, 197, 246, 247
Склерон 183
Скрап 280
Сланцы 270
Сложные вещества 352
Сложные удобрения 205, 207
Слоистая структура 272, 273, 305, 316
Слюда 270, 273
Смальта 164
Смальтин 164
Смарагд 68
Смешанные удобрения 205, 207
Смитсонит 339
Смоляная обманка 312
Снежинки 99, 100
Сода 275, 151, 158 , 212
Соединения включения 145
Солеобразные карбиды 156
Соли 222
— названия 226
Солнечная батарея 171
Солнечная корона 76
Солнечное излучение 75
Солнце 74, 75, 76, 78, 105, 118, 194
— атмосфера 78
Соляная кислота 276, 222, 330, 331, 336
«Соляной спирт» 222
Сохранение элементов в химических реакциях 352, 354
Спектр атомный 13, 33
Спин 18, 133, 134
Спиновое квантовое число 17, 19
«Спирты» 222
Спичечное производство 318
Сплав Вуда 98, 151
Сплавы 195, 196, 277
Сподумен 182
Среда окружающая
— борьба с загрязнением 77, 78, 89, 108, 123, 124, 360
Сродство к электрону 13
Сталагмиты 154
Сталактиты 154
Сталеалюминиевый кабель 67
Сталь 277, 60, 61, 97, 113, 128, 130, 132, 157, 163, 187, 188, 209, 216, 335, 342, 344
— легирование 314
— раскисление 314
Станиоль 230
Станн- 224
Станнаты 229
Станнин 228
Станниты 230
Станнометан 230
Старение металлов 61, 62
Стекло 283, 168, 173, 275
— Вуда 285
— травление 325
Стекловолокно 359
Стеклоделие 283
Стеклопластики 359
Стеллит 113, 466, 335
Стиб- 224
Стибил 291
Стибин 290
Стибнит 290
Стратосфера 74, 75
Стронцианит 286, 287 «Стронциановая земля» 286
Стронций 286, 13, 196, 197, 203, 251, 348
— изотопы 286, 287
Стронций-90 287
Субгалогениды 66, 154, 155
Сулема 288, 250
Сульф- 224
Сульфат аммония 206
— калия 206
Сульфаты 288, 84, 86, 89, 121, 122, 158, 202, 222, 258, 264, 269, 289
Сульфиды 288, 30, 84, 86, 258, 260, 265, 269
Сульфиты 289, 89, 121, 222, 267
Суперинвар 216
Суперфосфат 206, 314, 315, 361
Сурик 254
Сурьма 290, 114, 196, 197, 350
— горение в хлоре 290
Сурьмяная охра 290
Сурьмяная кислота 291
Сурьмянистая кислота 290
«Сурьмяное масло» 222
Сурьмяный блеск 97, 289, 290, 291
Сусальное золото 229
Сухой лёд 292, 308, 309
Сфалерит 253, 258, 289, 339
т
Таллий 293, 194, 196, 197
— галогениды 293
— гидроокись 293
— окислы 293
— сплавы 294
— сульфат 294
Тальк 184, 186, 270
Тантал 294, 196, 197, 219, 220
— карбид 156, 295
— пятиокись 295
— сплавы 295
Танталаты 295
Танталит-колумбит 295
Танталиты 219
Танталовая кислота 295
Танталовые конденсаторы 295
Тарапакаит 202
Твёрдые растворы 61, 200
Твёрдые смазки 221
Твёрдые сплавы 166
Теллур 295, 44, 45, 202, 240, 255, 293
Теллураты 296
Теллуриды 296
Теллуристая кислота 296
Теллуристый ангидрид 296
Теллуристый водород 99, 296
Теллуриты 296
Тёллуровая кислота 296
Теллуроводородная кислота 296
Теллуровый ангидрид 296
Температура
— атмосферы 75, 76, 77, 78
— измерение 121
— низкие и сверхнизкие 119, 220
Тенардит 288
Теплоизоляционные материалы 370
Тербий 296, 177, 181, 247
Термисилид 346
Термистор 121
Термическая диссоциация 100
382
Термическая обработка металлов 62, 278, 281
Термонатрит 275
Термопара 249
Термосфера 74, 75
Термоэлектронная эмиссия 91
Термоядерная реакция 109
Термоядерное горючее 109
Термоядерный реактор 183
Термоядерный синтез 105, 123
Тетра- 224
Тетраборан 92
Тетраборная кислота 91, 95
Тетраметафосфорная кислота 321
Тетратионовая кислота 297
Тетрафосфорная кислота 319
Тетраэдр 72, 316, 320
— алюмокислородный 272, 274
— кремнекислородный 271, 272, 274
Тетраэтилсвинец 94, 188, 255
Тефлон 324, 370
Технециевая кислота 297
Технеций 296, 196, 197, 354
— окислы 297
Тинкал 89, 95
Тио- 224
Тиосерная кислота 297, 260, 269
Тиосульфаты 297, 124
Типографские сплавы 291
Титан 298, 123, 185, 196, 197, 369
— двуокись 298
— сплавы 97
— тетрагалогениды 298
Титанаты 298
Титанил (радикал) 298
Титановая губка 299
Титановые белила 299
Титановые сплавы 209, 299
Толтры 204
Томасовский конвертер 280
Томасшлак 206, 280
Томсена — Бора таблица элементов 36, 37
Топаз 270, 272, 273
Топливные элементы 108
Топливо 107, 108, 163
Торианит 300
«Ториевая земля» 300
Торий 300, 56, 58, 245, 299
— гидроокись 300
— изотопы 243
— использование в ядерной энергетике 300, 301
— окисел 300
— сплавы 30'1
Торит 300
Торон 245
Тортвейтит 246, 272
Травление металлов 277
— стекла 325
«Траектория электрона в атоме» 24, 25, 28
Трансактиноидные элементы 218
Трансурановые элементы 301, 57, 311
Тремолит 272
Трепел 167
Трёхокись 224
Трёхцентровая связь 92
Три- 224
«Тримарганец» 248
Тритий 13, 75, 105, 109, 183
Трифосфорная кислота 319
Троилит 204
Трона 212, 275
Тропопауза 74
Тропосфера 74, 75, 83
Тугоплавкие металлы 67, 196
Тугоплавкие сплавы 156
Туки 205
Тукосмеси 205
Тулий 301, 177, 181
— изотоп 181
Тунгстен 112
Тэнит 204
Тюямунит 244
Тяжёлая вода 109
Тяжёлый камень 112
«Тяжёлый сплав» 113
Тяжёлый шпат 79, 286
У
Угарный газ 302 , 309
Углеводороды 166
Углекислые соли 302, 157
Углекислый газ 302, 308, 77, 78, 84,
107, 170, 204, 205,303, 304, 306, 310
Углерод 302, 6, 15, 170, 277, 350
— аморфный 306, 307
— атом 19, 303
— в природе 302
— двуокись 308, 77, 78, 84, 107, 170, 204, 205, 302, 303, 304, 306, 310
— изотопы 303, 305
— историческая справка 303
— окисление 162
— окись 309, 237, 302, 306
— получение 307
— применение 307
— сероокись 270
— физические свойства 305
— химические свойства 306
— четырёххлористый 327, 330
Углеродная единица 13, 352
Углеродное топливо 108
Уголь 127, 302, 307
— активный 306, 307, 308
— древесный 306, 307, 343, 344
— каменный 302, 307, 344, 345
Угольная кислота 310, 121, 157, 307, 308
Угольный ангидрид 310, 308
Удобрения 52, 71, 72, 91, 204, 205, 209, 257, 317
«Удушливый воздух» 47
Ультраосновные породы 126
Ультрафиолетовое излучение 74, 75, 76, 173, 227, 249, 285
Уран 310, 12, 56, 57, 58, 255, 301
— гексафторид 313, 322, 324
— изотопы 243, 310, 311, 312, 313
— использование в ядерной энергетике 301, 311
— окислы 313
Уранил-ион 313
Уранинит 312
Урановая смолка 312
Ускорители заряженных частиц 354
ф
Фазы внедрения 156
Фергюсонит 312
Ферменты 368
Фермий 314 , 56, 57, 58, 59
Фернико 128
Ферр- 224
Ферраты 131
Ферриты 131, 134, 142
Ферриты-гранаты 181
Ферриты скандиевые 247
Феррованадий 96, 97, 314
Ферровольфрам 113, 314
Ферромагнетизм 133, 134
Ферромагнетики 133, 180, 216, 218
Ферромарганец 187, 314
Ферромолибден 209, 314
Феррониобий 314
Ферронихромы 217
Ферроплатина 232
Ферросилид 171, 346
Ферросилиций 171, 314, 346
Ферросплавы 314, 67, 128
Ферротитан 314
Феррохром 314, 334
Фибролит 186
Физико-химический анализ 195, 198
Физическая химия 6
Физические приближения 26
«Философская шерсть» 222
«Философский камень» 5, 194, 315
Фитопланктон 304
Флогистон 161, 222, 223
Флуор 321
Флуоресценция 80
Флюорит 153, 156, 321, 322
Фольга алюминиевая 67
Формула химического соединения 350
Фосген 309 _
Фосфаты 314, 202, 210, 315, 319, 322
Фосфиды 315, 115, 318, 319
Фосоин 315, 317, 319
Фосфиты 315
Фосфонитрилхлориды 318
Фосфония соли 319
Фосфор 315, 350
— изотопы 318
— историческая справка 315
— нахождение в природе 315
— окисление 162
— получение 317
— применение 317
— пятиокись 320
— ' сульфид 259
— трёхокись 319
— физические свойства 316
— химические свойства 316
Фосфоресценция 315, 317
Фосфористые кислоты 318, 315
Фосфористый ангидрид 319
Фосфористый водород 319, 315, 317
Фосфоритная мука 206
Фосфориты 315, 322
Фосфорная кислота 210, 317, 319
Фосфорнокислые соли 319
Фосфорные кислоты 319, 314
Фосфорные удобрения 206, 314, 315, 316, 317
Фосфорный ангидрид 320, 231
Фосфорорганические соединения 318
Фосфоры 287, 315
Фотоматериалы 263
Фотоны 14, 16
Фотосинтез 77, 78, 84, 88, 102, 103, 160, 161, 204, 304, 309, 356
Фотохимическая диссоциация 100
383
Фотоэлементы 171, 255, 256, 292, 294, 337
Франций 321, 56, 73, 136, 196, 197, 349
Фреоны 324
Фреска 141, 142
Фтор 321, 13, 115, 116, 175
— атом и молекула 322
— в природе 322
— историческая справка 321
— получение 323
— применение 323
— физические свойства 322
— химические свойства 322
Фторапатит 315
Фториды 324, 174, 175, 323
Фторирование 325
Фтористоводородная кислота 324, 231
Фтористый водород 324
Фтороборная кислота 90
Фторопласт-4 324
Фторорганические соединения 322, 324
Фторсульфоновая кислота 325 Фунгициды 367
X
Халцедон 172
Халькозин 190, 289
Халькопирит 190 253, 258, 289
Химизация народного хозяйства 366
Химизация сельского хозяйства 366
Химико-фотографическая промышленность 359
Химическая продукция 358, 362, 363, 364, 365
Химическая промышленность 356
Химическая технология 358 , 366
Химические процессы 358 , 360
Химические свойства элементов 19,29, 30, 32, 33, 37, 42, 44, 45, 49
Химические средства защиты растений 367, 368
Химический завод 360, 361, 363
х<Химический фонтан» 331
Химическое машиностроение 360
Химическое производство 358 , 360
Химическое сродство 329, 351
Химия 5, 356
Химия в промышленности и строительстве 368
Химия инертных газов 143, 174
Химия — источник увеличения товаров народного потребления 370
Химия на службе здоровья человека 371
Химия служит человеку 356—371, 7, 208
Хладноломкость 278
Хлор 326, 13, 115, 116, 159, 202
— атомарный 336
— в природе 326
— историческая справку 326
— окислы 328
— получение 328, 187, 277
— применение 328
— физические свойства 326
— химические свойства 326
Хлораты 330, 327, 328, 332
Хлорвинил 332
Хлорелла 123
Хлориды 330, 277, 349, 350
Хлорирование воды 330
Хлористая кислота 330, 328, 332
Хлористоводородная кислота 331, 276
Хлористый азот 327
Хлористый водород 331, 106, 276, 327
Хлориты 332, 331
Хлорная вода 326
Хлорная известь 124, 141, 142, 329
Хлорная кислота 332, 231, 328, 350
Хлорноватая кислота 332, 327, 330, 333
Хлорноватистая кислота 332, 124, 327, 328
Хлорофилл 160, 184, 205
Хлороформ 327, 330
Хром 333, 67, 196, 197
— окисление 162
— окислы 333, 334, 335
Хромали 335
Хроматы 335, 202, 334
Хромирование 334, 335
Хромистый железняк 333, 334
Хромистый чугун 346
Хромит 333, 335
Хромитовый кирпич 335
Хромиты 334
Хромовая кислота 335
Хромовая смесь 335
Хромовокислые соли 335
Хромовые квасцы 158, 159, 334
Хромовые кислоты 335
«Хромовые сапоги» 335
Хромовые сплавы 335
Хромовый ангидрид 335, 334
Хромокалиевые квасцы 158
Хромомагнезитовый кирпич 335
Хромомолибденовые стали 335
Хромофоры 334
Хромпик 335
Хрупкость стали 277, 278
Хрусталь 172, 173, 284, 285
Хэсталлой 217
ц
Царская водка 336, 54, 139, 256, 369
Цвет минералов 199, 202
Цветное стекло 285, 357
Цветные металлы 196
Цезиевая плазма 337
Цезий 336, 13, 194, 196, 197, 252, 321, 349
Целестин 203, 204, 286, 287, 288
Целлюлоза 359
Цемент 337, 125, 275, 346
Цемент Сореля 186
Цементация 278
Цементит 157, 343
Цементный бетон 82
Цеолиты 273, 274
Цепные неразветвлённые реакции 331
Цепные разветвлённые реакции 107, 317, 322, 323
Цепные реакции 107
Цепные ядерные реакции 311,312, 354
«Цериевая земля» 177, 246
Церий 338, 39, 177, 178, 181, 255
— борид 91
Церуссит 253, 254
Циан 338, 49, 306
Цианиды 338 , 49, 307, 339
Цианирование 139, 262
Цианистоводородная кислота 338, 275, 307
Цианистые соли 338
Цианистый водород 338, 275
Циклотрон 60
Цинк 339, 158, 196, 197
— в гальванических элементах 340
— окись 222, 339, 341
— сульфат 340
— сульфид 339, 341
Цинкаты 339
Цинкит 339
Цинкование 341
Цинковая обманка 258, 289, 339
Циркон 270, 342
Цирконий 341, 117, 118, 185, 196, 197, 299
— борид 342
— карбид 342
— нитрид 342
Цитрин 172
ч
Частицы элементарные 11, 12, 21, 22,24,28,56,57,60, 75, 81,91,118, 143, 239, 241, 244, 285, 287, 297, 300, 310, 321, 354
Чёрная магнезия 185, 186
Чёрная металлургия 187, 344, 345
Чёрные металлы 133, 196
Чернь платиновых металлов 233
«Четыре элемента» 6, 354
Чилийская селитра 47, 206, 212, 256, 257
Чугаль 346
Чугун 343, 128, 132, 157, 163, 264, 277, 278, 279, 282, 308
— зеркальный 280
ш
Шабазит 274
Шеелит 112
Шёнит 153
Шинная промышленность 359, 365
Шлак 279, 280, 321, 346
Шлам 235
Шпатовый железняк 129
Шпейсовый кобальт 164
Шредингера уравнение 23, 24, 25, 26
Шрейберзит 204
щ
Щелочноземельные металлы 348, 42, 101, 121
Щелочные металлы 349, 26, 30, 42, 101, 121, 190
э
Эвдиалит 342
Эвксенит 312
Эйнштейний 350, 56, 57, 58, 59
Экаалюминий 39, 44, 114
Экабор 38, 44, 246
Экаиод 73
Экакадмий 39
Экасилиций 39, 44, 119
Экзосфера 74, 75
Экзотермические реакции 162
384
Экстракция 93
Электроды 340
Электроизоляционные материалы 370
Электролиз 63, 64, 65, 68, 185, 187, 213, 214, 217, 341
— воды 108
Электролизёр 66
Электролит 66, 140, 214
Электролитическая диссоциация 100, 101
Электролитическое рафинирование 192
Электромагнитное излучение 74, 75
Электронная единица 13
Электронная конфигурация 19, 42, 43, 45, 116, 145, 234, 348, 349
Электронная пара 162
Электронная плотность 23, 28
Электронная подоболочка 18
Электронная проводимость 120
«Электронная этажерка» 19, 20
Электронное облако 23, 24, 25, 28, 43
Электронное состояние 17, 18, 19, 23, 25, 26
Электронные ловушки 263
Электронные оболочки 12, 13, 17, 18, 37, 42, 45, 50, 75, 116, 179, 190, 215, 233, 246, 247, 348, 349
— завершённые 143, 144, 145
Электронный октет 42, 50
Электронный слой 18
Электроны 11—28, 37, 42, 43, 57, 58, 91, 105, 108, 116, 120, 133, 179, 197, 233, 321, 340
— вероятность нахождения 23, 24, 28
— волна — частица 14, 24, 28
— «траектория в атоме» 24, 25, 28 s-Электроны 17, 19, 23, 25, 42, 58, 177, 190, 196, 233, 293
р-Электроны 17, 19, 25, 42, 58, 177, 196 293
d-Электроны 17, 42, 58, 177, 190, 196 233
/-Электроны 58, 177, 179, 180, 196
Электроотрицательность 50 116,121, 197
Электропроводность металлов 64
Электрофильтр 267
Электрум 262
Элемент №102 56, 58, 60, 222
Элемент №103 56, 57, 58, 60, 183
Элемент №104 175, 176, 218, 301
Элемент №105 196, 218, 301
Элемент №106 29, 30, 301, 350
«Элементы» Вселенной 6, 354
Элементы химические 350, 5, 11, 13, 15, 18, 19, 26, 49, 84, 127, 197
— периодический закон Д. И. Менделеева 29—45
— распространённость 137, 351, 354
Элинвар 216
Эманация 245
Эмиссия электронная 118
Эндотермические реакции 162
Энергетические подуровни 18, 20
Энергетические уровни 16, 18, 19, 20, 22, 26, 37
Энергетический обмен организмов 316
Энергия ионизации 13,32, 116, 145, 348, 349
Энергия электрона 23, 24
Энзимы 368
Эннеа- 224
Эрбий 355, 177, 181, 247
Эритроний 96
Этан 306
Эффузивные горные породы 125
ю
Юпитер, планета 194
— атмосфера 78
Я
Ядерная энергия 311
Ядерные реакции 354, 12, 57, 59, 73, 75, 81, 91, 98, 118, 138, 151, 175, 180, 183,218, 219,311,312, 342
Ядерный реактор 81, 91, 98, 109, 115, 118, 150, 180, 214, 238, 241, 287, 296, 300, 311, 312, 342, 354
Ядерный теплоноситель 98 •
Ядохимикаты 367
Ядро атомное 12, 23, 26, 57, 144, 137, 244, 296, 297
— заряд 13, 29, 44, 45, 350, 351, 352, 354
— радиус 13
— реакция деления 311
Ядро Земли 136, 137
Яшма 172
Неорганическая химия. Энциклопедия школьника. Гл. ред. И. П. Алимарин. М., «Советская Энциклопедия», 1975.
384 с. с ил.
Сдано в набор 25 сентября 1974 г. Подп. к печ. 25 апреля 1975 г. Т-00130.
Тираж 101 тыс. экз. Формат 84х1081/1в. Бумага офсетная № 1. Объём 24 физич. печ. л., 40,32 усл. п. л., 46,89 уч.-издат. л. Цена 2 р. 69 к.
Издательство «Советская Энциклопедия», Москва, 109817, Ж-28, Покровский бульвар, 8
Набрано в Ордена Трудового Красного Знамени Первой Образцовой типографии им. А. А. Жданова «Союз-полиграфпрома» при Государственном комитете Совета Министров СССР по делам издательств, полиграфии и книжной торговли. Москва, Ж-54, Валовая, 28. Заказ № 1762.
Отпечатано на Ордена Трудового Красного Знамени Калининском полиграфкомбинате «Союзполиграф-прома» при Государственном комитете Совета Министров СССР по делам издательств, полиграфии и книжной торговли, г. Калинин-24, пр. Ленина, 5. Зак. 2012
Школьные учебники (((Р
SHEBA.SPB.&U/SHKOLA