/











































































































































































































































































































































































































































































































































Автор: Ахметов Т.Г. Порфирьева Р.Т.
Теги: неорганическая химия общая и неорганическая химия неорганические вещества химическая технология
ISBN: 5-06-004333-9
Год: 2002




Текст
химическая
технология неорганических веществ
ВНИМАНИЕ!
много опечаток
УДК 546 ББК 24.1
X 46
Авторы:
Т. Г. Ахметов, Р. Т. Порфирьева, Л. Г. Гайсин, Л. Т. Ахметова, А. И. Хацрииов
Рецензенты:
проф К Ф Ткачев, Ю С Плышевский (гл специалисты УНИХИМ г Екатеринбург), кафедра «Технология неорганических веществ» РХТУ им Д И Менделеева (зав каФ проф А И Михайличенко)
Химическая технология неорганических веществ: В 2 кн. X 46 Кн. 2. Учеб, пособие / Т. Г. Ахметов, Р. Т. Порфирьева, Л. Г. Гайсин и др.; Под ред. Т. Г. Ахметова.— М.: Высш, шк., 2002.— 533 с.: ил.
ISBN 5-06-004333-9 (кн. 2)
В книге даны сведения по технологии соединений мышьяка, серы, железа, хрома, кобальта, хлора, фтора, брома, иода, марганца и никеля. Рассмотрены вопросы промышленной безопасности и санитарно-технических норм описанных производств Приводится описание физико-химических основ и конкретных способов их получения
Книга может быть полезной преподавателям и студентам кафедр химической технологии неорганических веществ, электрохимии, охраны труда и безопасности жизнедеятельности
Для студентов высших учебных заведений, обучающихся по направлению «Химическая технология неорганических веществ»
ISBN 5-06-004333-9 (кн. 2)
ISBN 5-06-004147-6 © ФГУП «Издательство «Высшая школа», 2002
Оригинал-макет данного издания является собственностью издательства «Высшая школа», и его репродуцирование (производство) любым способом без согласия издательства запрещается
Предисловие
Настоящая книга предназначена в качестве учебного пособия для студентов высших учебных заведений специальности 250200 — химическая технология неорганических веществ
В процессе подготовки учебного пособия авторы исходили из требований государственного образовательного стандарта (СП 01, раздел СД 02) к данной специальности и использовали опыт преподавания этой дисциплины в высших учебных заведениях России.
В настоящем учебном пособии (вторая книга) дается теоретическое обоснование и практическое построение производственных процессов по технологии соединений мышьяка, серы, железа, хрома, кобальта, хлора, фтора, брома, иода, марганца и никеля. При этом подробно рассматриваются различные пути по интенсификации производственных процессов и улучшению качества производимой продукции. В этих же разделах описаны особенности структуры и физико-химические свойства химических веществ и способы получения целевых продуктов поскольку без определенных знаний основных особенностей структуры и свойств трудно, а иногда и невозможно, организовать производство.
Параллельно с этим курсом студенты изучают ряд других дисциплин по технологии и оборудованию заводов, поэтому в данном учебном пособии подробно не рассматривается оборудование заводов неорганических веществ, а также не описываются конструктивные детали и механические расчеты специального оборудования. В учебном пособии особое внимание уделено химическим и физико-химическим процессам, протекающим в процессе производства неорганических веществ, рассматриваются пути интенсификации производственных процессов и улучшения качества производимой продукции.
Предлагаемая вниманию читателей вторая книга является продолжением учебного пособия «Химическая технология неорганических веществ», изданного издательством «Химия» (г. Москва) в 1998 г. для студентов высших учебных заведений обучающихся по специальности химическая технология неорганических веществ.
В предлагаемой книге мы сохранили преемственность с ранее изданными книгами (учебниками и учебными пособиями) по данной специальности. В данное пособие, кроме технологии целевых продуктов, включены и способы получения базисных элементов, оксидов, гидроксидов, нитридов, силицидов, боридов, карбидов и т.п. Это диктовалось необходимостью более глубокого понимания происходящих физико-химических процессов.
В книге представлены результаты исследований в области технологии неорганических веществ последних лет в России и за рубежом, включая результаты исследования самих авторов.
Гл. 1 пособия написана Л.Г. Гайсиным и Р.Т. Порфирьевой, гл. 2 — Р.Т. Порфирьевой, гл. 3—Л.Т. Ахметовой, гл. 4 — А.И. Хацрино -вым и Р.Т. Порфирьевой, гл. 5—7 — Т.П Ахметовым и Л.Т. Ахметовой, гл. 8 — А.И. Хацриновым, гл. 9 — Л.Т. Ахметовой, гл. 10 — Л.Г. Гайсиным, гл. 11—А И. Хацриновым и гл. 12 — Л.Г. Гайсиным. Содержание и текст всех разделов пособия в целом авторы обсуждали вместе.
Авторы выражают благодарность Л.П. Мухамадеевой, Я.М. Каримову, М М. Зариповой п <Д.М. Бажановой за огромную оформительскую работу, проведенную при подготовке данного пособия к печати.
Считаем также приятным долгом выразить глубокую благодарность профессорам А.И. Михайличенко, Ю.И. Шумяцкому и И. А Петропавловскому (РХТУ им Д.И. Менделеева), К.В. Ткачеву и Ю.С. Плышевскому (УНИХИМ, г.Екатеринбург) за большой труд по рецензированию и ценные замечания, способствовавшие улучшению качества рукописи. Авторы благодарны также профессору И.Н. Андрееву за оказанную помощь.
Авторы
ГЛАВА 1
МЫШЬЯК И ЕГО СОЕДИНЕНИЯ
Содержание мышьяка в земной коре составляет 1,710'4% (масс.). Он относится к рассеянным элементам, однако образует свыше 160 собственных минералов. Практически не встречается в природе в самородном виде. Наиболее распространенными минералами, имеющими промышленное значение, являются арсенопирит FeAsS, реальгар As4S4 и аурипигмент AS2S3. Промышленное значение имеют мышьяковые руды, содержащие не менее 2—5% мышьяка, богатые мышьяком считаются руды с содержанием мышьяка 25—35%. Значительные запасы мышьяка концентрируются также в рудах цветных металлов.. Он генетически ассоциируется с рудами W, Sn, Pb, Sb, Zn, Си, Ni и Со. Почти со всеми этими металлами мышьяк образует минералы — простые и сложные арсениды (сперрилит РЬАзг, шмальтин Со-As3, теннатит 3Cu2S-As2S3 и др.). Мышьяк встречается также в месторождениях благородных металлов — золота и серебра.
Свойства. Атом мышьяка имеет конфигурацию внешней электронной оболочки- 4s24/23; степени окисления -3, +3 и +5; энергии ионизации при последовательном переходе от As0 к As5+ соответственно равны 9,815, 18,62, 28,34, 50,1 и 62,6 эВ; электроотрицательность по Полингу — 2,1; атомный радиус 0,148 нм, ковалентный радиус 0,122 нм, ионные радиусы (в скобках указаны координационные числа) As3+ 0.072 нм (6), As5+ 0,047 нм (4), 0,060 нм (6), As3’ 0,191 нм.
Мышьяк существует в нескольких аллотропических формах, из которых наиболее устойчив серый, так называемый металлический. Мышьяк (a-As) с ромбоэдрической кристаллической решеткой, а = = 4135 нм, a = 54,13°, z = 2, пространственная группа ТЗти (в гексагональной установке а = 0,376 нм, с = 1,0548 нм), плотность 5,74 г/см3. В процессе быстрой конденсации паров мышьяка на поверхности, охлаждаемой жидким газом, получают прозрачные, мягкие как воск кристаллы желтого мышьяка с кубической решеткой, с плотностью около 2,0 г/см3. При нагревании и на свету желтый мышьяк быстро переходит в серый. Известны также нестабильные аморфные формы мышьяка, например черный мышьяк с плотностью около 4,7 г/см3, который образуется в токе водорода. При нагревании выше 270° С он переходит в серый мышьяк. Серый мышьяк имеет вид се-
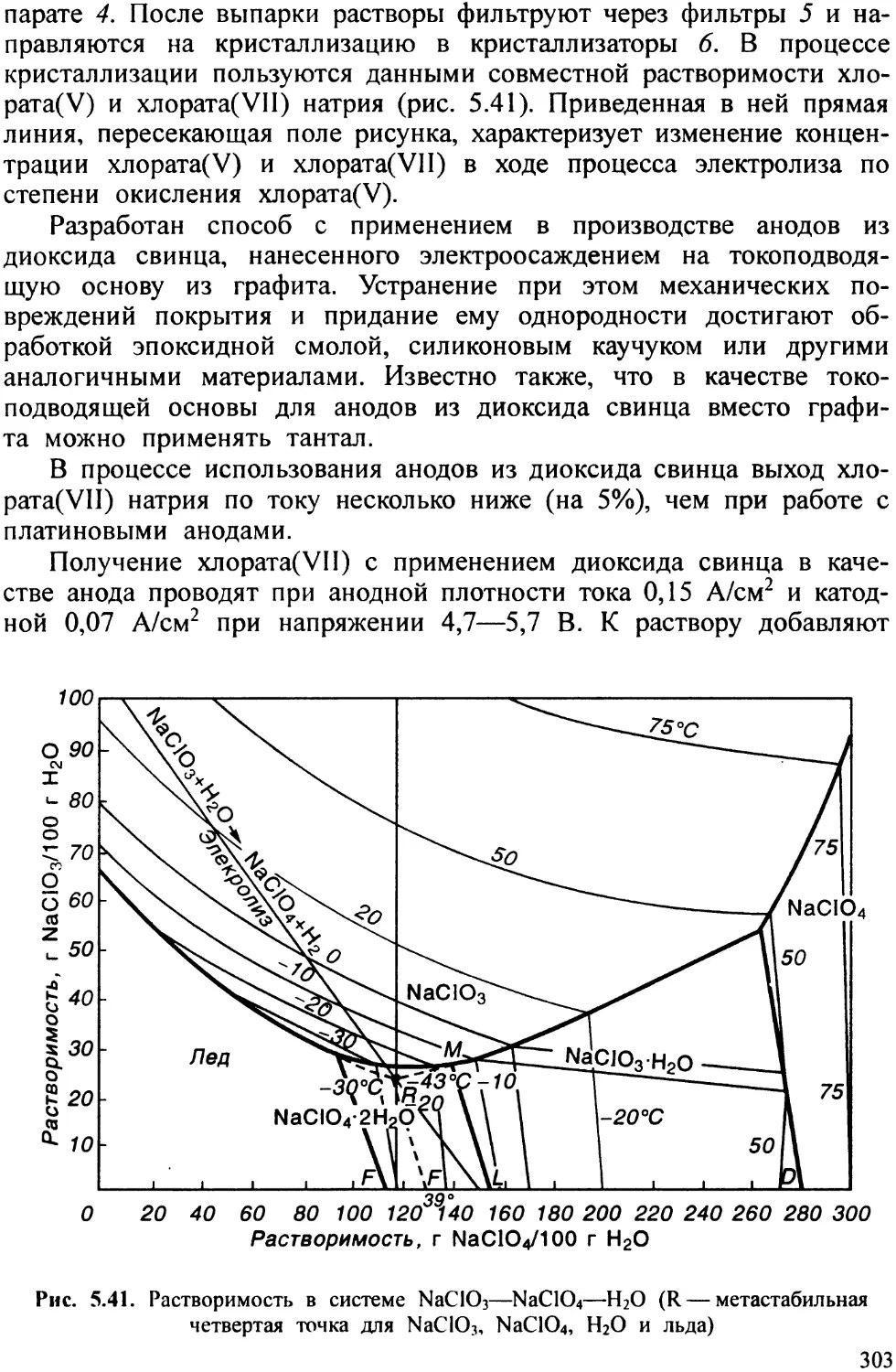
Рис. 1.1. Двойной слои а юмов в решетке металлического мышьяка
ческого металла. Кристаллизуется он в слоистой решетке (рис. 1.1). Каждый слой состоит из атомов, расположенных в двух параллельных плоскостях. Таким образом, каждый атом образует три пирамидальные (ковалентные) связи с тремя наиболее близкими атомами (2,51 А) из параллельной плоскости этого же слоя. У каждого атома имеется и три более отдален
ных атома (3,15 А) в плоскости среднего слоя. Электрическая проводимость мышьяка меньше, чем у истинных металлов (4% электрической проводимости серебра).
Температура возгонки кристаллического мышьяка 615° С; плотность жидкого 5,24 г/см3 (817° С); Ср = 25,05 Дж/(моль-К); ДЯп°л = = 28 кДж/моль, ДЯ",1Г = 150 кДж/моль (для As4); 5298 = 35,6 Дж/(моль-К); уравнение температурной зависимости давления пара: Igp (мм рт. ст) = 11,160-7357/Г (623—1090К); температурный коэффициент линейного расширения 4 10’6 К'1 (293 —573К); /крит = = 1400° С, /?крит = 22,0 МПа, <7крит = 2,65 см3. Пары мышьяка бесцветны до 800° С, состоят из молекул As4, выше 1700° С —из As2, а в интервале 800—1700° С —из смеси As2 и As4. Серый мышьяк очень хрупок, разрушается по спайностям; твердость по Бринеллю около 1500 МПа, твердость по Моосу 3,5. Мышьяк диамагнитен, обладает металлической проводимостью 3,3-10’5 Ом-см, температурный коэффициент расширения 3,9-10'3 К’1 (273—373 К).
Мышьяк химически активный металл. На воздухе он легко окисляется. При нагревании порошкообразный мышьяк воспламеняется и горит голубым пламенем, в среде фтора и хлора самовоспламеняется.
Получение. Металлический мышьяк получают из его сесквиокси-да восстановлением последнего углеродсодержащими восстановителями (чаще всего древесным углем) по схеме
As2Oi + ЗС -> 2As + ЗСО
Полученный мышьяк очищают сублимацией.
Для синтеза полупроводниковых соединений мышьяк получают из предварительно очищенных гидрида или хлорида мышьяка хими
ческим осаждением из газовой фазы. Для этого арсин разлагают при 300—400° С в токе водорода или аргона по схеме
AsH3 н- > As + 1,5Н2
2AsH3 —2As + ЗН2
Образующийся мышьяк отлагается в виде черного зеркала на стенках сосуда или трубки, в которой находится газ. Хлорид мышьяка восстанавливают водородом высокой чистоты по схеме
2AsCh + ЗН2----> 2As + 6НС1
Наиболее чистые сорта мышьяка получают, сочетая дистилляцию и кристаллизацию. Рассмотренные процессы проводят при 815—850° С и давлении 4—6 МПа. Температура превращения кубической и монокристаллической модификации (арсеномит <-> клаудетит) при давлении 1 атм равна 240±30° С.
Применение. Элементный мышьяк применяется в виде добавок к сплавам (на основе меди, палладия и олова) и полупроводниковым материалам. Мышьяк особой чистоты применяют для синтеза важнейших полупроводниковых соединений.
1.1. ОКСИДЫ МЫШЬЯКА
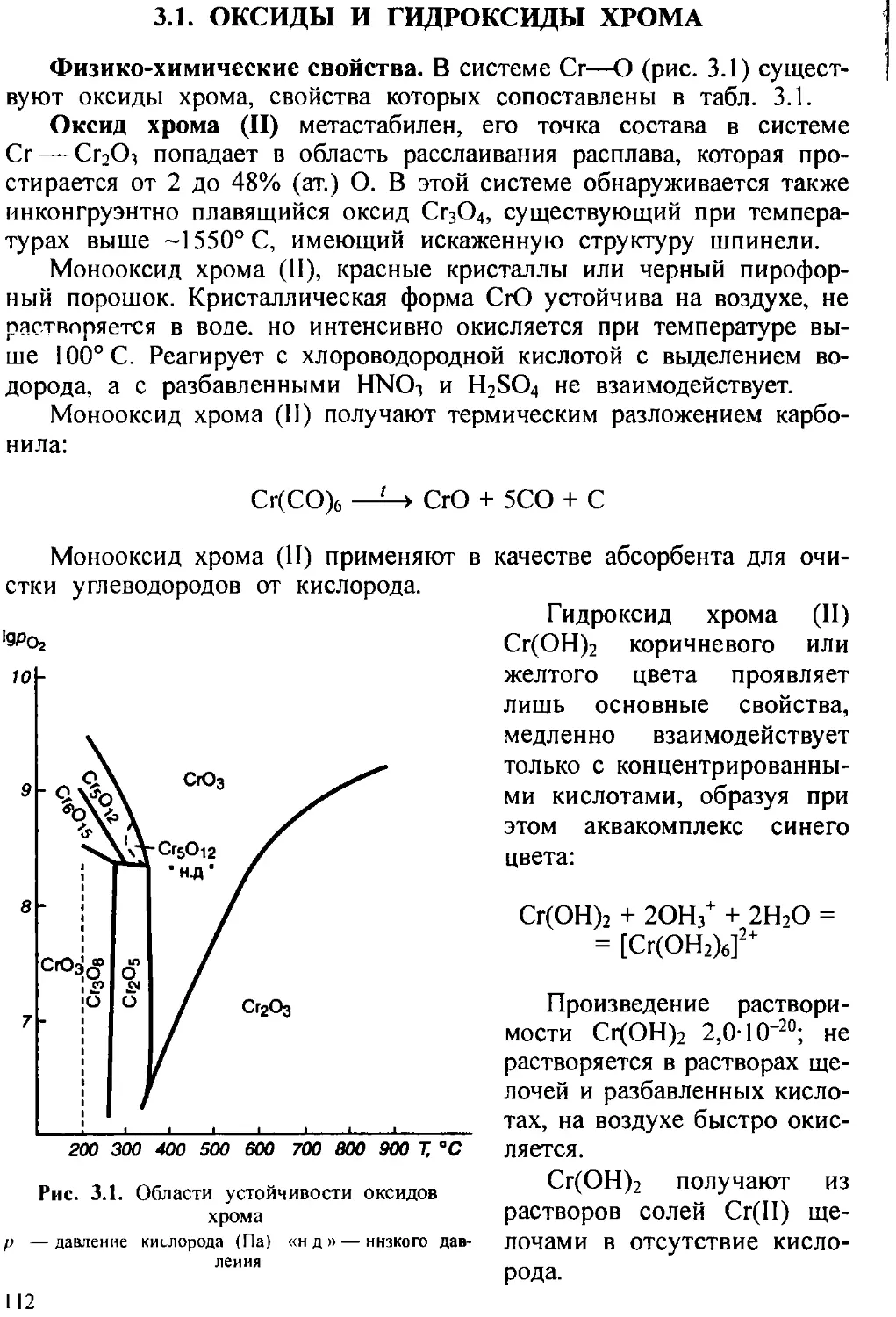
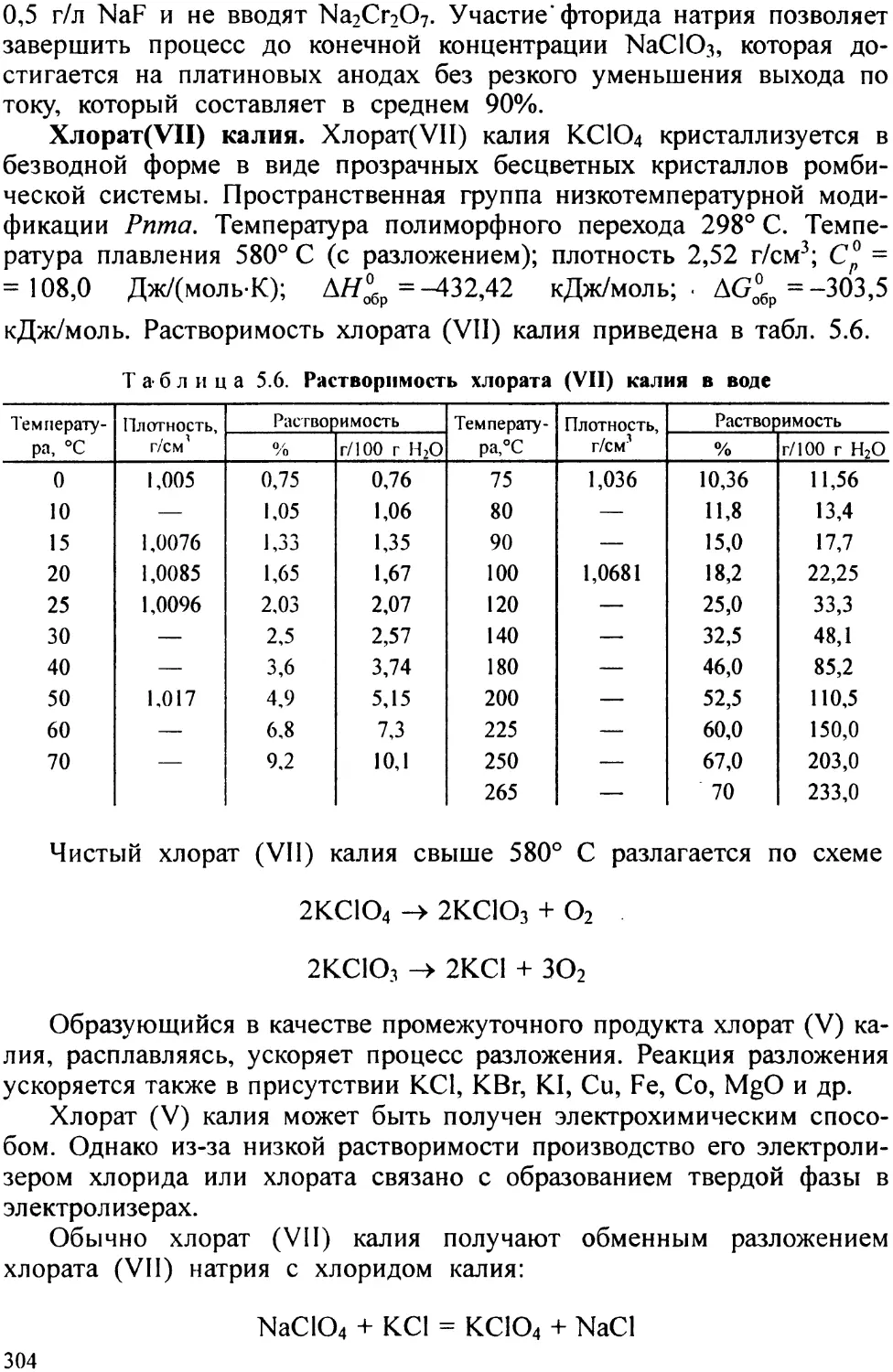
В настоящее время известны следующие оксиды мышьяка: 1) монооксид AsO — газообразное вещество, образующееся при электрическом разряде в парах триоксида мышьяка при пониженном давлении; 2) диоксид мышьяка As2O4 — кристаллическое вещество, образующееся при спекании мышьяковистого (As2O3) и мышьякового ангидридов (As2O3) при 280° С в присутствии паров воды. Представляет из себя бесцветные кристаллы ромбической сингонии (а = 0,8566 нм, b = 0,7271 нм, с = 0,5236 нм, z = 4, пространственная группа Рпат). Выше 500° С он диспропорционирует на As2O3 и As2O5; 3) устойчивые формы оксида мышьяка в газовой фазе — сесквиоксид As2O3. До 300° С основная форма в газовой фазе — димер, выше этой температуры он заметно диссоциирован, а при температурах выше 1800° С газообразный оксид практически состоит из мономерных молекул As2O3 с температурой кипения 461°С.
Термодинамические свойства оксидов мышьяка приведены в табл. 1.1.
Газообразная смесь As2O3 и As4O6 образуется при окислении мышьяка кислородом, при окислительном обжиге сульфидных минера-
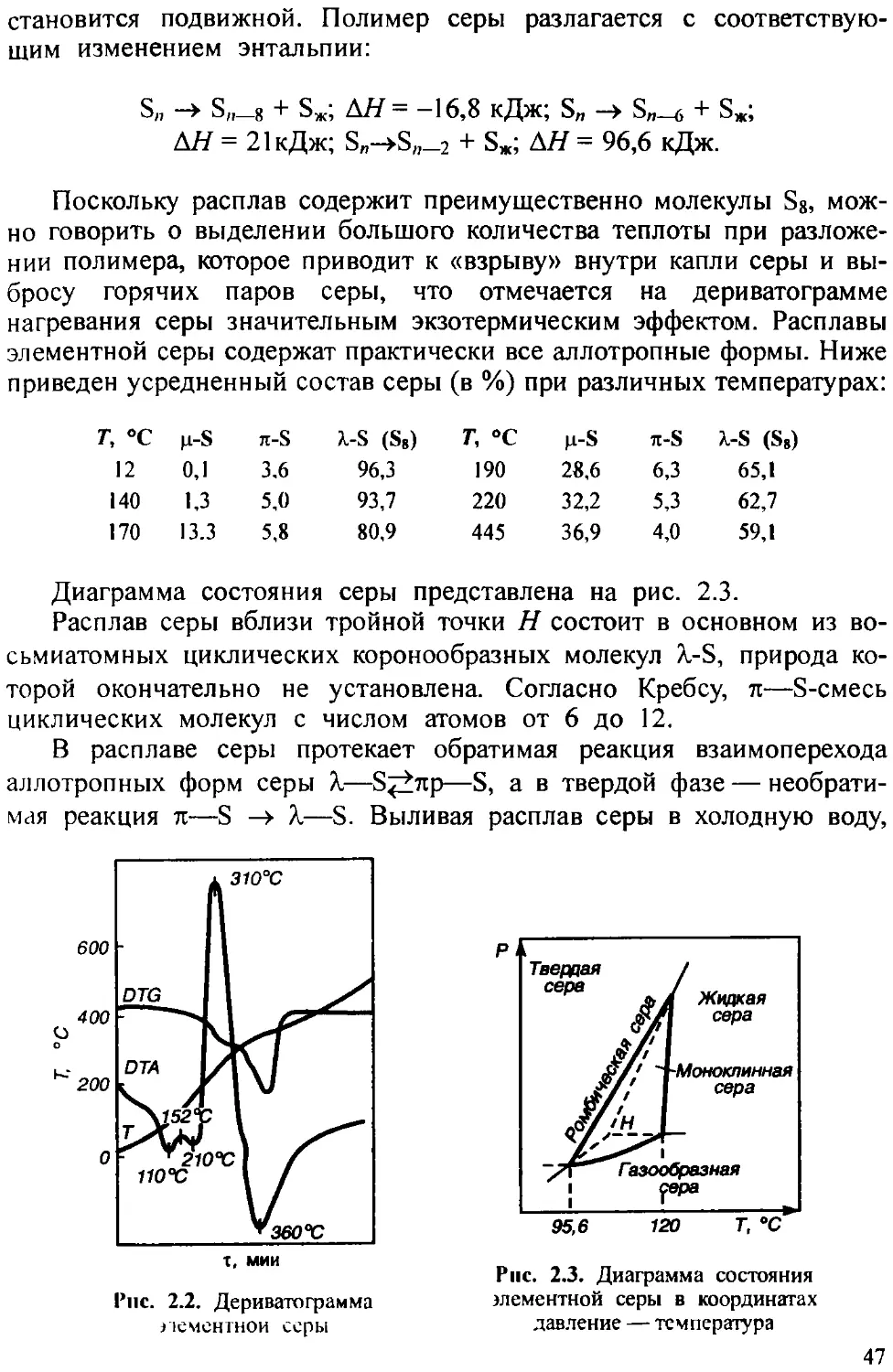
лов мышьяка (арсинопирита), руд цветных металлов и полиметаллических руд.
В процессе конденсации пара AS2O3 (АздОб) выше 310° С (рис. 1.2) образуется стекловидная форма A.S2O3; плотность 3,74 г/см1; растворимость 1,82 г в 100 г воды при 25° С. При конденсации пара ниже 310° С образуется бесцветная поликристаллическая кубическая модификация (а = 1,1075 нм, z = 8, пространственная группа FcRniy, температура плавления 278° С; плотность 3,87 г/см1; растворимость 2,05 г в 100 г воды при 25° С; в природе арсенолит (табл. 1.2).
Таблица II Термодинамические свойства оксида мышьяка
Соединение VкДж/моль S22h, Дж/(моль К) С„ Дж/(мольК)
AsO (г) -57,45 230,17 32,34
As4O6 (г) -1230,13 — —
Арсенолит (к) -1334,73 233,47 203,77
Клаудетит (к) -1331,59 245,19 221,76
As2O4 (к) -799,58 — —
As2O, (к) -926,44 105,44 116,53
I а б л и ц а 12 Фазовые превращения As2O3 (As4O6)
Тип фазового перехода МПа г, °C А//оо,„ кДж/моль AS, Дж/(моль К)
Арсенолит
к->ж 48,54 88,2
к->г 4,75 10 1 278 104,60 189,96
ж->г 56,10 101,67
Клаудетит
к->ж 45,19 76,99
К ✓ Г 9,9 10 ’ 314 101,26 172,38
Ж“^Г 56,06 95,40
Ж->Г 0,1 461 56,06 76,57
Арсенолит устойчив ниже 13° С, около 221—223° С медленно начинает превращаться в моноклинную модификацию (а = 0,5339 нм, b = = 1,2984 нм, с = 0,45405 нм, b = 94,27°, z = 4, пространственная группа Р2/п\, температура плавления 314° С; плотность 4,15 г/см1; растворимость 2,1 г в 100 г воды при 25° С; в природе — минерал клаудетит.
Все формы сесквиоксида мышьяка хорошо растворимы в кислотах и щелочах.
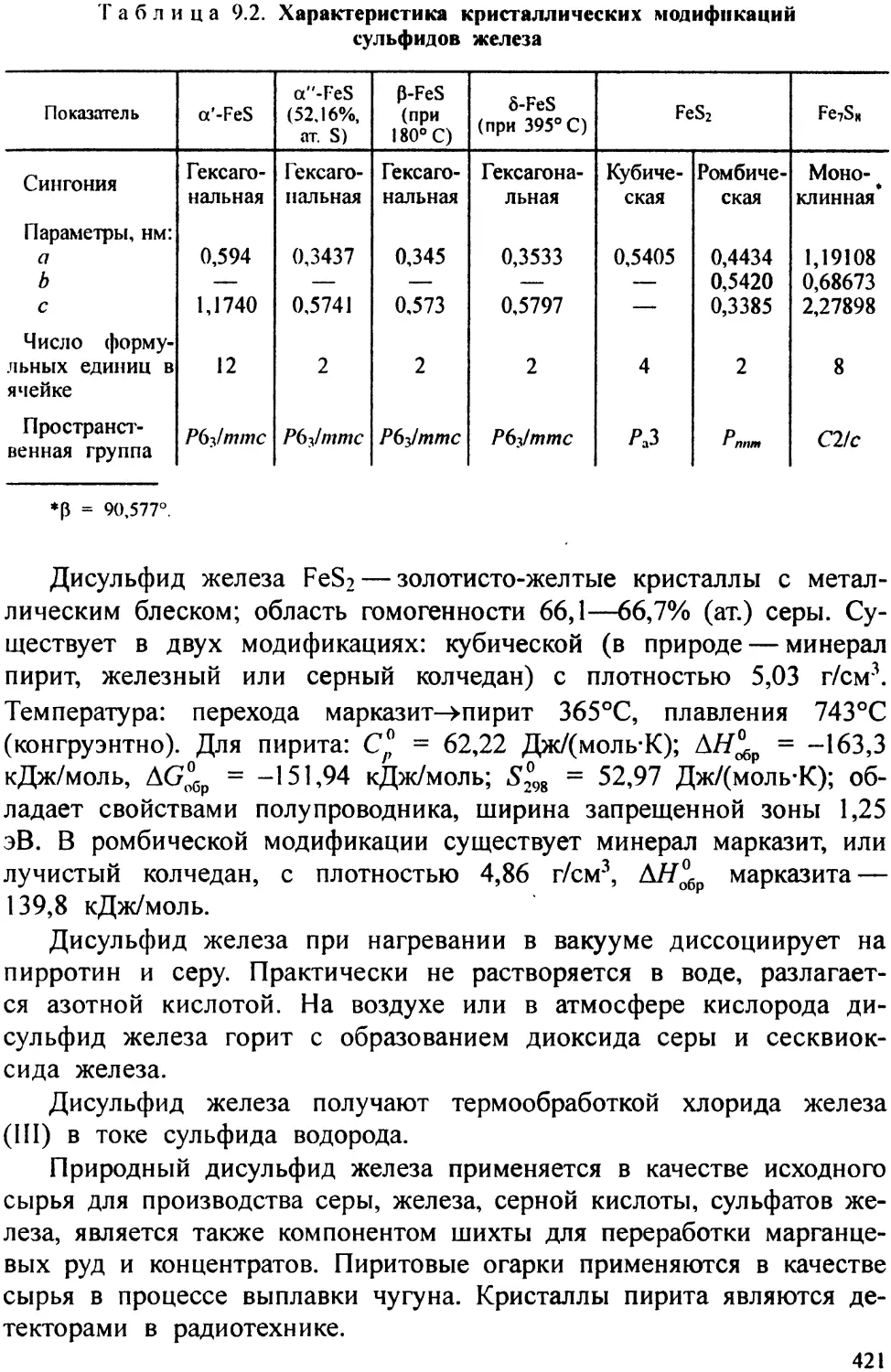
Оксид As (V) (мышьяковый ангидрит) As2O5 — бесцветные кристаллы ромбической сингонии (а = 0,8454 нм, b = 0,8645 нм, с =
= 0,4629 нм, z = 4, пространственная группа 7>2l2i2i); плотность 4,1 г/см\ растворимость 65,8 г в 100 г воды при 25°С. При нагревании As3O5 диссоциирует на As4O6 (газ) и О3; при 730° С суммарное давление диссоциации 0.1 МПа.
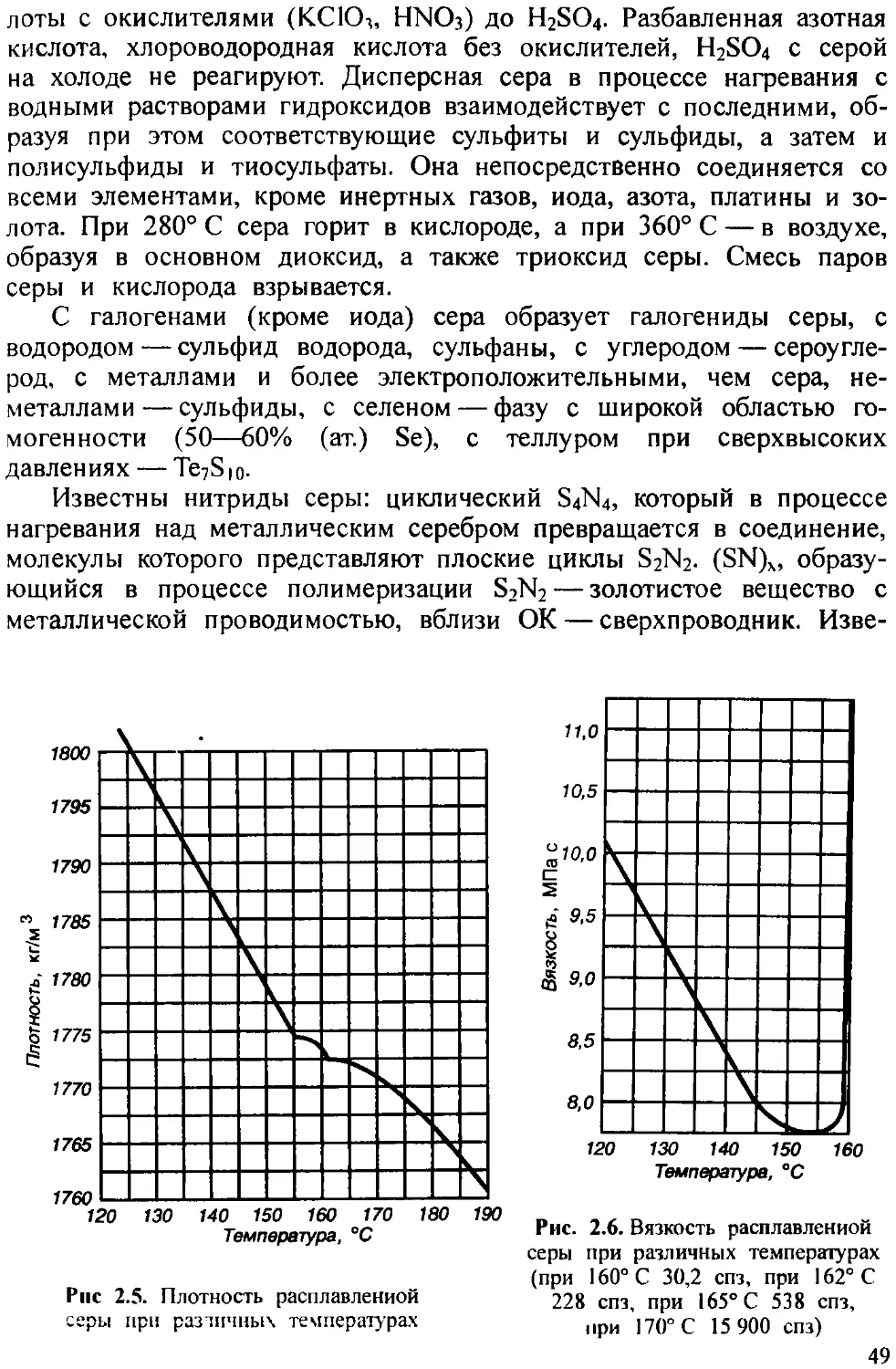
Пентаоксид мышьяка (мышьяковый ангидрид) AS2O5 можно получить нагреванием на воздухе сесквиоксида мышьяка до 600—700° С (рис. 1.3).
Технология сесквиоксида мышьяка. Сесквиоксид мышьяка (мышьяковистый ангидрид) плохо растворим в воде. Растворимость при 0° С равна 1,2 г, а при 100° С — 6 г в 100 г воды. Водные растворы его имеют кислую реакцию. Будучи амфотерным оксидом, As2Oi реагирует и с щелочами, и с кислотами. Однако его кислотные свойства превалируют над щелочными.
Образующаяся при растворении AS2O3 в воде и выделяемая из соединений мышьяковистая кислота H3ASO3 стабильна лишь в водных растворах, в которых она диссоциирует по схеме
As’"1 + ЗОН # As(OH), = HiAsO, Н+ + AsO“ + Н2О
Константы диссоциации H3ASO3 при 20° С: К\ = 4-10’10; Ку = = 7-10'и; Ку = 4-10'14. Первая константа диссоциации Аз(ОН)з = 5-10'16.
Рис. 1.3. Зависимость степени окисления АвгО, в As2O, от температуры
Растворы мышьяковистого ангидрида легко восстанавливаются до элементного мышьяка или до арсина AsH3 (7КИП = -62,5 °C). Давление пара AsH3 (в мм рт. ст.) может быть определено по уравнению Igp = -1403,32 Г1 - 9,43935 Ig7 + 0,0080377 + 28,82835.
Мышьяковистая кислота легко окисляется в мышьяковую азотной кислотой, галогенами и другими окислителями, однако устойчива по отношению к кислороду. Ее производные значительно легче окисляются свободным кислородом особенно в щелочной среде и в присутствии катализаторов — солей меди, марганца, иода и др. Раствор арсенита натрия с молярным соотношением As2O3:NaOH, равным 1:6, с концентрацией As2O3 около 14—17 г/л, содержащий 5 г/л солей меди, при барботаже через него воздуха (5—10 л/г через 100 мл раствора), при 60° С окисляется кислородом воздуха в течение одного часа на 40—45%. Степень окисления возрастает с повышением температуры до 70—75° С, с увеличением молярного соотношения As2O3:NaOH и уменьшается с понижением концентрации As2O3 Реакция окисления As2O3 хроматом калия при 30° С, pH 8,3—10,6 и ионной силе раствора 1,75 имеет первый порядок относительно концентраций каждого из обоих реагентов. При pH > 9 скорость реакции не зависит от концентрации Н+, а при pH < 9 — увеличивается с ростом концентрации Н+ При pH 9,1 константа скорости реакции К = 1,61-10'1 л/(г-моль-с).
С некоторыми галогенидами мышьяковистая кислота образует в водных растворах комплексные соли переменного состава (MX«As2O3, где X — галоген), разлагающиеся при кипячении. С хлоридами натрия или аммония подобные комплексы не образуются.
При окислительном обжиге мышьяксодержащих руд протекают следующие реакции с образованием сесквиоксида мышьяка:
2As2S3 + 9О2 = 2As2O3 + 6SO2
2As2S2 + 7О2 = 2As2O3 + 4SO2
2FeAsS + 5Н2О = As2O3 + Fe2O3 + 2SO2
Образующийся сесквиоксид мышьяка отходит из печей в виде паров (рис. 1.4) вместе с другими газообразными продуктами обжига, при охлаждении которых превращается в тонкодисперсную пыль, выделяемую из газов в пылеуловителях (циклонах). Побочной реакцией обжига является окисление сесквиоксида мышьяка до его пентаоксида
As2O3 + О2 = As2O5
давление пара которого очень мало, вследствие чего он переходит в огарок
Сесквиоксид мышьяка, выгруженный из пылеуловителей, содержит различное количество примесей, что зависит в основном от состава исходного сырья. При чистоте, соответствующей требованиям потребителей, его выпускают в виде товарного продукта — белого мышьяка. При наличии загрязнений исходное сырье (серый или черный мышьяк) подлежит рафинированию путем повторного обжига при температурах более низких, чем при обжиге руды.
Для термообработки мышьяксодержащих руд и концентратов в производстве используются механические печи различных конструкций — одноподовые с вращающимся подом или гребками и многоподовые. Применяются также отражательные и муфельные печи, которые обеспечивают получение более чистого продукта. Преимуществом муфельных печей является то, что в них получаются более чистые и концентрированные газы сесквиоксида мышьяка.
Степень использования исходного сырья зависит от размеров его частиц, состава, температуры процесса и продолжительности нахождения в реакционной зоне. Размер частиц (зерен) исходного сырья поддерживают в пределах 3—5 мм. Обжиг концентрата или руды в более измельченном виде приводит к спеканию массы исходного сырья. Для избежания этого в руду добавляют карбонат кальция (известняк). Однако это снижает процент использования исходного сырья, так как при этом образуется арсенат кальция и равновесие реакции сдвигается в сторону окисления сесквиоксида мышьяка в пентаоксид.
Аналогичный процесс происходит и при обжиге концентратов, содержащих значительное количество оксидов металлов, таких, как свинец, магний, железо, кальций и др. Для исключения образования пентаоксида мышьяка и арсенатов указанных металлов в исходную
шихту добавляют уголь для восстановления арсенатов и проведения процесса при более высокой температуре. В этом случае обжигаются и скородитовые руды, имеющие общую формулу FeAsO4 • 2Н2О, в которой мышьяк находится в виде пентаоксида.
В процессе обжига смешанных руд, содержащих скородит и арсенопирит, параллельно с основными реакциями обжига протекает реакция восстановления пентаоксида мышьяка из скородита арсенопиритом по уравнению
5As2O5 + 2FeAsS = 6As2O3 + Fe2O3 + 2SO2
Этот процесс дает возможность уменьшить количество угля в исходной шихте.
Серосодержащие руды подвергаются окислительному обжигу при 500—550° С, арсенопиритовые руды и концентраты обжигают при 600—700° С, восстановительный обжиг (в присутствии углерода) скородитовых и смешанных руд проводят при 800—900° С. Отходы производства в виде огарка, содержащего ценные металлы и до 0,5—1,5% сесквиоксида мышьяка, подлежат дальнейшей переработке для их извлечения.
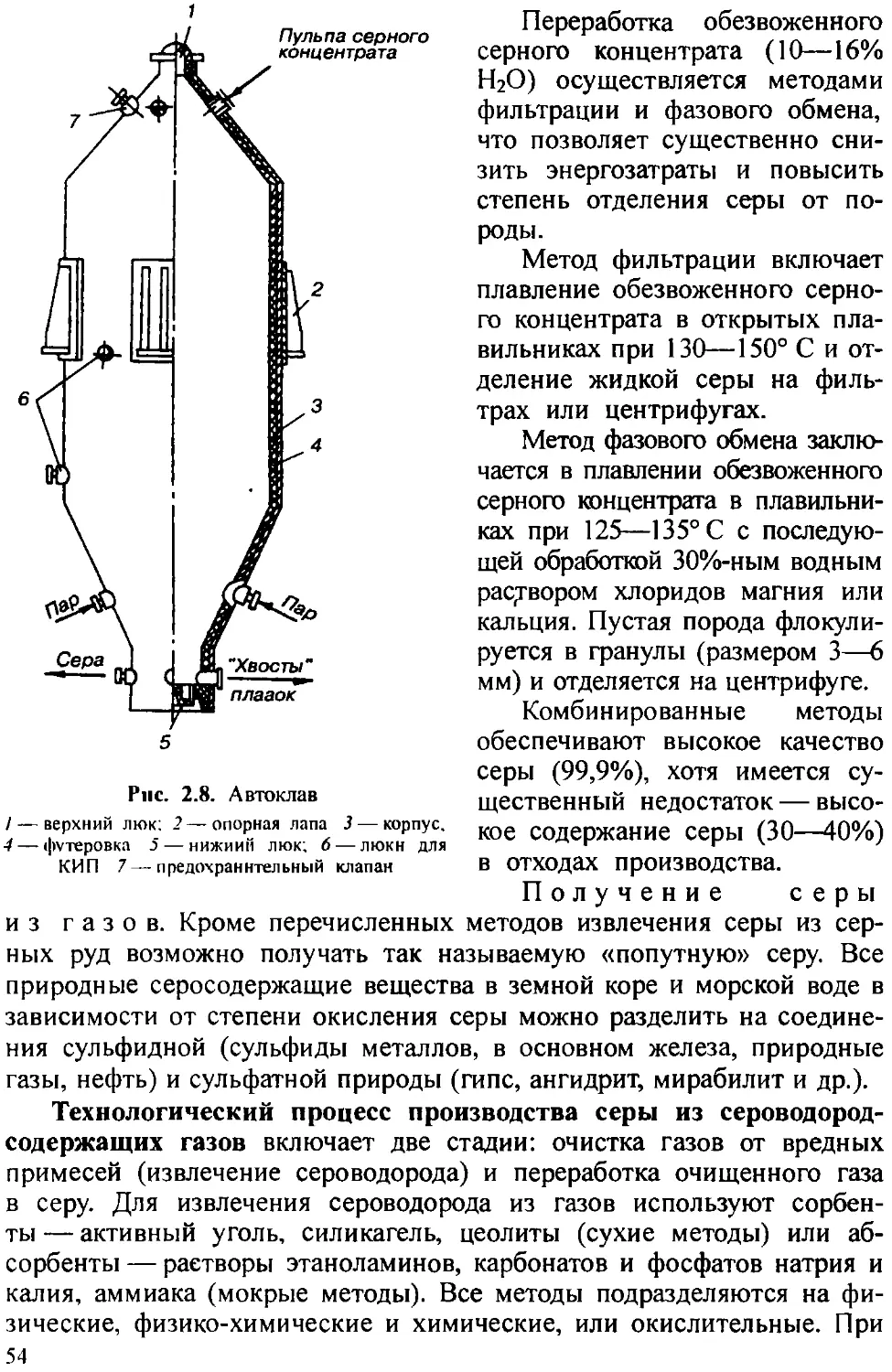
Обжиговые газы из печи, содержащие 50—85 г/м3 сесквиоксида мышьяка, очищаются от пыли в сухих электрофильтрах. Температура газов при входе в электрофильтры 450—500° С, а при выходе — около 35О°С. Дальнейшее охлаждение газов до 80—110° С с одновременной кристаллизацией твердого сесквиоксида мышьяка производят в холодильниках разных конструкций (осадительных камерах, полых башнях, рукавных фильтрах или электрофильтрах, а иногда совмещая несколько этих аппаратов), в которых осаждается основная масса целевого продукта— сесквиоксида мышьяка. Остатки целевого продукта улавливают в оросительных башнях или в мокрых электрофильтрах, орошаемых водой. Образующуюся суспензию из электрофильтров отжимают на фильтрах ФПАКМ, а получающийся при этом осадок сушат в сушилках с кипящим слоем.
По рассмотренной технологии получают технический белый мышьяк, содержащий 90—95% As2O3. Для получения более чистых сортов продукта, содержащего более 97% основного вещества, его подвергают возгонке при 500—600°С и вторично улавливают. Степень извлечения сесквиоксида мышьяка при рафинировании составляет около 85% от исходного продукта. Остатки после рафинирования, содержащие до 50% As2O3, передают для смешения с исходным сырьем, идущим на обжиг для получения технического продукта.
Стекловидный или аморфный сесквиоксид мышьяка получают путем постепенной возгонки белого мышьяка при 500—600° С и по-12
следующей его конденсации. Стекловидный белый мышьяк получают также прессованием при 150° С рафинированного белого мышьяка под давлением до 2500 атм.
1.2. МЫШЬЯКОВАЯ КИСЛОТА
Мышьяковая (ортомышьяковая) кислота H3AsO4-0,5H2O— бесцветные кристаллы с температурой плавления 36°С (процесс плавления происходит с разложением), растворимая в воде (88% масс., при 20°С), гигроскопична. В водных растворах — трехосновная кислота:
= 5,6-10'\ = 1,7-10'7 и К1Ц = 3,0-10'12. При нагревании около
100°С теряет воду, превращаясь в пиромышьяковую кислоту H4AS2O7, а при более высоких температурах переходит в метамышьяковистую кислоту HAsO3.
Ортомышьяковую кислоту применяют для получения пентаоксида мышьяка, арсенатов (V), мышьякорганических соединений и в качестве антисептика для древесины. Она является промежуточным продуктом при получении арсенатов (III) и других соединений.
Получение ортомышьяковой кислоты азотио-кислотиым способом производится по следующему уравнению:
3As2O3 + 7Н2О + 4HNO, = 6H3AsO4 + 4NO
В настоящее время это наиболее распространенный метод получения ортомышьяковой кислоты в химической промышленности независимо от того, что технология связана с выделением оксида азота. Это в свою очередь требует регенерации азотной кислоты из оксидов азота, образующихся при ее восстановлении. Процесс регенерации осложняется сильным пенообразованием, вызванным участием содержащихся в исходном сесквиоксиде мышьяка примесей и вследствие обильного выделения газов в результате реакции. Процесс осложняется и тем, что образующийся оксид азота восстанавливает часть исходной азотной кислоты до диоксида азота 2HNO3 + NO = 3NO2 + Н2О. В результате процесс в реакторе идет по уравнению
As2O3 + 2Н2О + 2HNO, = 2H,AsO4 + NO + NO2
Поэтому расчеты по оптимальному соотношению между исходными сесквиоксидом мышьяка и азотной кислотой ведут по последнему уравнению. Научно и экспериментально установленной оптимальной концентрацией исходной азотной кислоты является 30—32% HNOi. При этой концентрации исходной кислоты получается 50—58%-ная ортомышьяковая кислота. В связи с тем, что 32%-ная
13
азотная кислота не обеспечивает необходимой скорости процесса, применяют катализаторы, в качестве которых служат иодо- и хлороводородная кислоты (Н1 и НС1). Реакцию между азотной кислотой и сесквиоксидом мышьяка ведут с 2—3%-ным избытком последнего. По окончании процесса окисления оставшийся осадок Аз20з возвращают в начало процесса.
Выделяющиеся оксиды азота (nNO + 7nNO2) абсорбируют водой, а образующуюся 28—32%-ную азотную кислоту возвращают на смешение с реакционной кислотой и применяют на стадии окисления.
Процесс в производстве ведут в следующем порядке. В реактор, изготовленный из нержавеющей стали и снабженный паровой рубашкой, а также мешальным устройством, заливают 30—32%-ную азотную кислоту в количестве, равном '/, от необходимого для одной операции. Кислоту нагревают до 75—78°С и постепенно загружают сесквиоксид мышьяка В качестве катализатора в реактор вводят незначительные количества иода (0,5 г на 1 т As2Oj). Остальную часть расчетного количества азотной кислоты вносят постепенно в течение 3—4 ч. К концу процесса реакционная масса занимает около половины объема реактора. В процессе реакции происходит сильное вспенивание массы, для уменьшения которого рекомендуется вводить в аппарат сульфонафтеновые кислоты (контакт Петрова) из расчета 30—40 г на 1 т. После 5—6 ч, когда заканчивается подача расчетного количества сесквиоксида мышьяка, температуру в реакторе повышают до 90—99° С, а перемешивание продолжают в течение 5—6 ч для полного восстановления азотной кислоты.
Полученная в реакторе ортомышьяковая кислота с концентрацией 700 г/л As2Oi отстаивается для отделения твердых примесей, в том числе от исходного избыточного сесквиоксида мышьяка, после чего раствор фильтруется через фильтры ФПАКМ.
Существует и непрерывный способ окисления исходного сесквиоксида мышьяка азотной кислотой в двух последовательно и каскад-но расположенных реакторах с метальными устройствами. Способ позволяет достичь более высокого выхода ортомышьяковой кислоты, чем периодический. При этом способе обеспечивается спокойное ведение процесса, он исключает резкие изменения скорости реакции и переброс реакционной массы, а также облегчает регенерацию азотной кислоты из оксидов азота ввиду постоянства их концентрации.
Представляется перспективным проведение процесса окисления сесквиоксида азотной кислотой и кислородом под давлением. Скорость процесса окисления сесквиоксида мышьяка и степень регенерации азотной кислоты прямо пропорциональны давлению кислорода. Установлено, что процесс идет с наибольшей скоростью при 80°С и сильном перемешивании реакционной массы.
14
1.3. АРСЕНАТЫ
Арсенаты — соли кислородсодержащих кислот мышьяка. Различают арсенаты — (V) и арсенаты (III). Известны арсенаты, в которых, одновременно присутствуют As (III) и As (V), например Cr2As'" As/O|2.
Арсенаты (V) и арсенаты (III) — производные соответствующих ортомышьяковых (H3AsO4 и H3AsO3), метамышьяковых (HAsO3 и HAsO2) И ПОЛИМЫШЬЯКОВЫХ (H4AS2O7 И H4AS2O5, H5AS3O10, H2As4O7) кислот. В свободном виде из приведенных кислот получена лишь H3AsO4 Наиболее широко применяются ортоарсенаты (V) и метаарсенаты (III), часто называемые просто арсенатами. В основе структуры арсенатов (V) лежат тетраэдры AsO4, причем ортоарсенаты обычно построены из изолированных тетраэдров, а метаарсенаты имеют цепочечную или циклическую структуру (кольца из трех или четырех тетраэдров, соединенных вершинами). В основе структуры арсенатов (III) лежат сплюснутые пирамиды AsO3, которые могут соединяться в цепи через атомы кислорода.
Существуют также гидро- и дигидроарсенаты (V), которые при нагревании отщепляют воду и превращаются соответственно в пиро-и метаарсенаты (V). В качестве промежуточных продуктов могут быть получены и другие соединения, например:
NaH2AsO4 —^^->Na2H2As2O7 Ч35°С >Na3H2As3Oi0 —230°С >NaAsO3
При гидролизе мета- и пироарсенатов (V) образуются гидроарсенаты (V). При высоких температурах (около или выше температуры плавления) метаарсенаты (V) разлагаются до пироарсенатов (V), которые далее могут переходить в ортоарсенаты (V), обладающие наибольшей термической устойчивостью. При термическом разложении последних могут образовываться оксоарсенаты (V). Безводные арсенаты (III) при 300—500° С диспропорционируют с образованием арсенатов (V) и As. Метаарсенаты (III) могут предварительно разлагаться до пиро- или ортоарсенатов (III) с отщеплением As2O3.
Арсенаты щелочных металлов, а также гидроарсенаты (V) щелочноземельных и некоторых тяжелых металлов растворяются в воде, арсенаты (III) щелочноземельных металлов малорастворимы в воде, а все остальные арсенаты практически нерастворимы в воде. Арсенаты разлагаются минеральными кислотами и растворами щелочей. Арсенаты (111) в щелочных растворах — сильные восстановители.
Арсенаты известны для всех металлов, кроме золота и металлов платиновой группы. Щелочные металлы образуют безводные, мета-, пиро- и ортоарсенаты (V), плавящиеся конгруэнтно, и тиоарсенаты — инкою руэнтно. Щелочноземельные и другие металлы в 15
степени окисления +2 в отсутствие воды образуют мета-, пиро- и ортоарсенаты (V), а также оксоарсенаты (V), например состава бМеО • AsiO5. Из арсенатов (III) чаще других встречаются Mn(AsO2)2 и M^CAsCh),.
Для элементов в степени окисления +3 характерно образование ортоарсенатов (V). Арсенаты (III), выделенные из водных растворов, обычно имеют состав М(АзО2)зиН2О, полученные в отсутствие воды — MAs(X Для элементов степени окисления +4 характерны гидроарсенаты (V) и пироарсенаты (V). Примеры арсенатов (V) металлов в степени окисления +5: NbOAsO4 • 4Н2О и NbO(HAsO4)3 • 5Н2О.
Существуют двойные арсенаты, например MIMnAsO4, MiM1i1(AsO4)2; MiMi,i(AsO4)2. Из арсенатов, содержащих другие анионы, наиболее изучены М5"(А5О4)зХ, где X—ОН', F', СГ и др.
Известно около 85 природных минералов, относящихся к арсенатам (V): скородит FeAsO4 -2И2О, эритрин Co3(AsO4)2 -8Н2О, оливенит Cu2AsO4(OH), миметизит Pb5(AsO4)Cl и др. Природные арсенаты (III) — очень редкие минералы, например армангит Мп3(АзОз)2.
Арсенаты получают в основном взаимодействием оксидов мышьяка или растворов мышьяковых кислот с оксидами, гидроксидами или карбонатами металлов, арсенаты тяжелых металлов — реакцией их солей с МазАзОд или NaAsO2 (иногда ЫзАзО4 или LiAsO2) в растворе. Арсенаты (V) получают также окислением арсенатов (III).
Арсенаты — диэлектрики или полупроводники с большой шириной запрещенной зоны. Многие, например KH2AsO4 или арсенаты редкоземельных элементов,—сегнето- и пьезоэлектрики. Некоторые арсенаты — антиферромагнетики. Среди арсенатов есть и твердые электролиты, например Ag7I4AsO4, NaZr2(AsO4)3.
Гидроарсенат (V) натрия Na2HAsO4 • 7Н2О — бесцветные кристаллы; температура плавления 57° С; плотность 1,874 г/см1; растворимость в воде при 25° С 28,3% (масс.); температура обезвоживания гидрата 56—100° С; температура разложения 220° С; растворим в спирте и глицерине, нерастворим в эфире.
Получают термообработкой смеси As2O3 + МаМОз с последующим гидролизом и перекристаллизацией из водного раствора или окислением раствора NaAsO2 кислородом воздуха в присутствии сульфата меди либо анодным окислением.
Применяется в качестве инсектицида и антисептического средства.
Дигидроарсенат (V) калия KH2AsO4 — бесцветные кристаллы, решетка тетрагональная {а = 0,7629 нм, с = 0,71612 нм, z = 4, пространственные группы ./42с/); плотность 2,88 г/см3; температура разложения 211° С. Растворим в воде 25° С 28,3% (масс.); а также в спирте, глицерине, не растворяется в эфире Арсенаты аммония, рубидия и цезия применяются в качестве материала нелинейной оптики. 16
Гидроарсеиат (V) свинца PbHAsO4— белые кристаллы; плотность 6,05 г/см\ температура разложения 400° С; растворимость в воде (25° С) 2-10‘4% (масс.).
Арсеиат (V) кобальта Co3(As04)2 • 8Н2О— розовые кристаллы; плотность 3,178 г/см’; температура обезвоживания гидрата 120—400° С; температура разложения -1000° С. Является пигментом для росписи на стекле и фарфоре, компонентом шихты в производстве цветного стекла.
Технология арсената (III) натрия. Арсенат (III) натрия — NaAsO2, твердое вещество с плотностью 3,49 г/см5, с температурой разложения 550° С. Растворимость в воде при 25° С составляет 61% (масс.). Арсенат (III) натрия выпускают в виде водной пасты, содержащей 18% влаги и не менее 52% основного вещества.
Арсенат (III) натрия в промышленности получают действием водных растворов карбоната натрия на сесквиоксид мышьяка по схеме
As2O3 + 2Na2CO, + Н2О = 2Na2HAsO, + 2СО2
Фактически технический продукт состоит из смеси солей натрия мета- и ортомышьяковой кислот, процесс при этом проходит по схеме
As2O3 + 3Na2CO, = 2Na3AsO3 + ЗСО2
As2O3 + Na2CO3 + 2H2O = 2NaH2AsO3 + CO2
As2O, + Na2CO3 - 2NaAsO2 + CO2
Процесс ведут в реакторах, снабженных паровой рубашкой и метальным устройством. Исходные водные растворы, состоящие из карбоната натрия с концентрацией 30—35% Na2CO3, содержащие в своем составе гидроксид натрия до 20—25% от массы исходного карбоната, в течение 45—60 мин постепенно дозируют расчетное количество сухого сесквиоксида мышьяка. Процесс ведут при 90—95° С. После внесения исходных видов сырья перемешивание массы продолжают в течение 3—4 ч. При этом особо контролируют температуру процесса, так как при более низких температурах (ниже 80° С) прекращается растворение сесквиоксида мышьяка. Более высокие температуры (выше 95° С) приводят к выбросам реакционной массы из-за обильного выделения диоксида углерода. Конец реакции характеризуется исчезновением пены и началом спокойного кипения реакционной смеси.
Упаривают растворы в тех же реакторах в течение 18—20 ч. Конец процесса определяется по содержанию в суспензии воды, которой должно быть не более 18%. Образующаяся суспензия приобретает консистенцию сиропа большой вязкости. Поскольку арсенат (III) натрия обычно применяют в виде суспензий, его выпускают в виде 17
товарного продукта с влажностью 18% Н2О. После затаривания продукта в стальные барабаны он застывает в виде пасты.
На производство одной тонны арсената (III) натрия технической квалификации расходуется 528 кг сесквиоксида мышьяка (в пересчете на 100% As2O3), 237 кг карбоната натрия (в пересчете на 95% Na2CO3), 50 кг гидроксида натрия (в пересчете на 92% NaOH), 12 мГкал пара, 32 кВт-ч электроэнергии и 3,2 м3 воды.
Полученный таким способом пастообразный продукт имеет низкое качество по неоднородности своего состава, что затрудняет его дозировку у потребителей. Кроме этого, затвердевший в стальных барабанах продукт трудно выгружать. Поэтому более рациональна организация производства порошкообразного или гранулированного продукта. Для этих целей раствор арсената (III) натрия после упарки до содержания 20—25% воды направляют на сушилку в кипящем слое для получения порошкообразного продукта или в вертикальные распылительные грануляторы для получения гранул.
Сухой кристаллический арсенат (III) натрия получают взаимодействием сесквиоксида мышьяка со смесью NaOH и Na2CO3 по схеме
2As2O3 + 2NaOH + Na2CO3 = 4NaAsO2 + H2O + CO2
В процессе смешения исходных реактивов при общем содержании воды 30—35% в температурном интервале 60—70° С образуется суспензия, нагревая которую до 85° С получают студенистую массу черного цвета. Полученная масса направляется на сушку (для получения порошка) или на грануляцию (для получения гранул заданных размеров). Оба процесса проводят при 160—200° С. Проведение процесса грануляции или сушки в аппаратах с кипящим слоем заметно снижает температуру. Можно получить и чешуйчатый продукт, содержащий до 3% влаги, проводя процесс в вакуум-вальцовой сушилке.
Технология арсената (III) кальция. Технический продукт— смесь метаарсената (III) Ca(AsO2)2 и ортоарсената (III) Ca3(AsO3)2. Чистый арсенат (III) кальция разлагается при 500°С, растворимость в воде (25°С) 0,09% (масс.). Применяется в качестве инсектицида, является бактерицидным средством, а также компонентом смеси для протравливания семян и необрастающих красок для судов.
Арсенат (III) кальция получают, растворением сесквиоксида мышьяка в известковом молоке по реакциям
As2O, + Са(ОН)2 = Ca(AsO2)2 + Н2О
As2O + ЗСа(ОН)2 = Ca3(AsO3)2 + ЗН2О
При этом арсенат (III) кальция выделяется в осадок. Гидроксид кальция (известь), применяемый для получения арсената (III) кальция 18
(и других мышьяксодержащих препаратов), должен легко реагировать (гаситься) водой и содержать не менее 92% активного оксида кальция (или 70% активного оксида кальция в исходной пушонке). В настоящее время в химической промышленности применяются два способа производства: мокрый и полусухой.
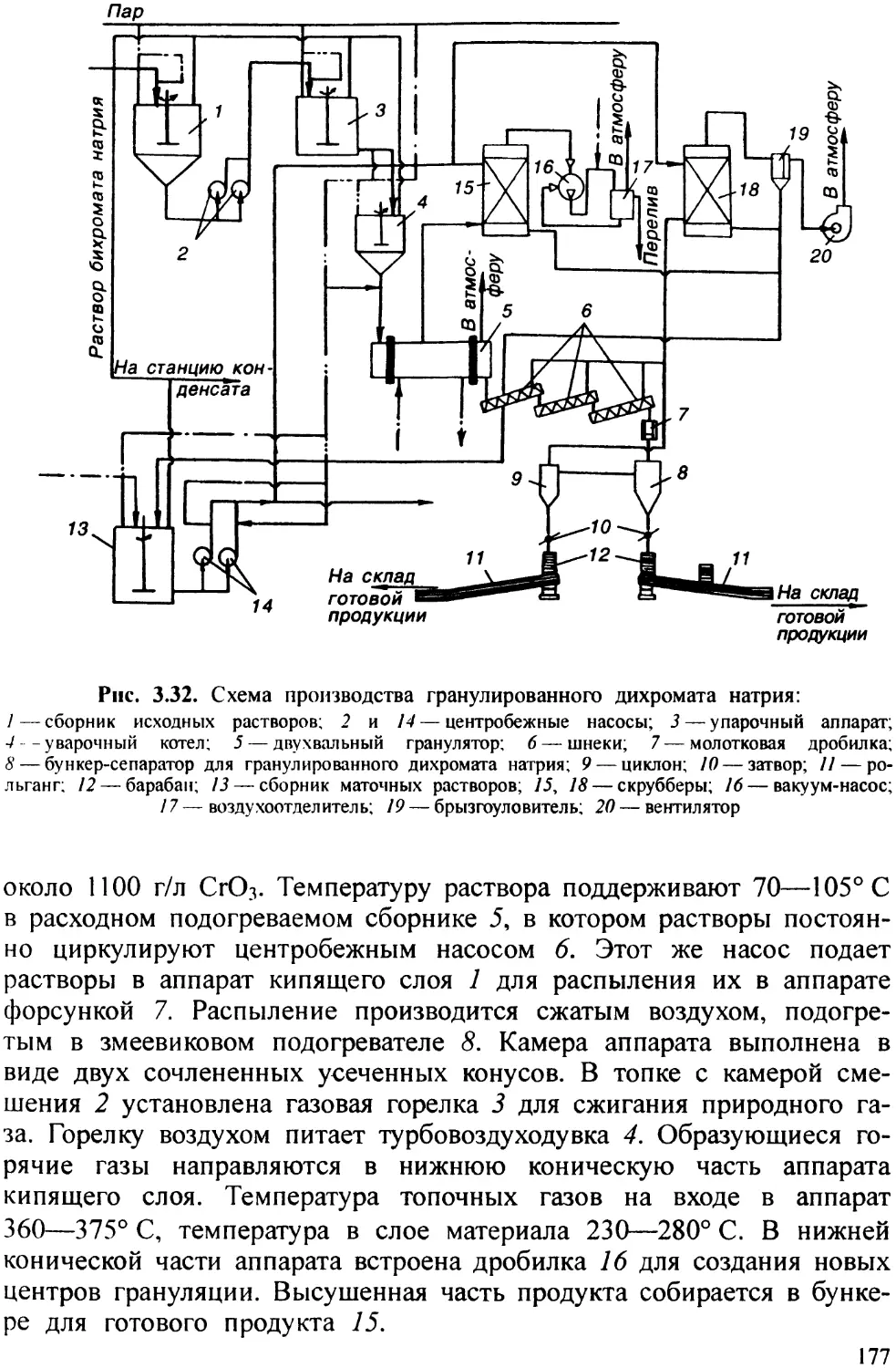
Мокрый способ. Согласно существующей схеме получения (рис. 1.5), в реактор 1 с мешалкой и паровым барботером загружают известковое молоко, нагревают острым паром до 85—90° С и при работающей мешалке вводят сесквиоксид мышьяка. Исходное количество известкового молока загружают с 10—15%-ным избытком против стехиометрических расчетов.
Полученная суспензия направляется на барабанный вакуум-фильтр 8, откуда маточные растворы (фильтрат) поступают в вакуум-сборник 9 и в дальнейшем используются для приготовления известкового молока. Отфильтрованная от маточных растворов паста арсената (III) кальция с влажностью не более 30% направляется на
Рис. 1.5. Схема получения арсенита кальция мокрым способом
/ — реактор. 2 — сборник для суспензии оксида и гидроксида кальция, 3 — мерник для суспензии оксида и гидроксида кальция -/ — бункер для белого мышьяка, 5 — бункер-дозатор 6—рукавный фильтр для улавливания пыли 7 — вакуум-насос, 8 — вакуум-фильтр 9— вакуум-сборник для фильтрата; /0 — барометрический конденсатор; // — шнековый питатель, /2— сушилка; 13— шнек-холодильник /4 — бункер для арсенита /5 — мокрый пылеуловитель, 16— скруббер /’ — вентилятор IS — мезьничный бункер 19 — центробежная вакуум-мельница 20—расфасовочный бункер 21 — шнек 22—расфасовочная машина 23 — бункер целевого продукта
19
сушку в барабанную сушилку 12 с наружным обогревом топочными газами. Водяные пары из барабанной сушилки 12 отсасываются вентилятором 17 и подаются в пылеуловитель 15, орошаемый водой, и в скруббер 16. Разрежение на выходе паров из сушилки 12 поддерживают не ниже 5 мм водяного столба. Высушенный продукт с влажностью не более 1% проходит через шнек 21, корпус которого охлаждают снаружи водой, поступает в мельничный бункер 18, откуда пневмотранспортером подается в центробежную вакуум-мельницу 19. Размолотый продукт направляют на расфасовку 22. Расфасованный в полиэтиленовую тару продукт — арсенит (III) кальция — направляют на склад готовой продукции.
На производство 1 т технического арсенита (III) кальция мокрым методом расходуют: 0,678 т сесквиоксида мышьяка (100% AS2O3), 0,335 т СаО (100%) в виде известкового молока, 0,3 т условного топлива, 350 кВт-ч электроэнергии и 25 м3 воды.
Полусухой способ (рис. 1.6) отличается от предыдущего тем, что смешение исходной извести-пушонки с сесквиоксидом мышьяка проводят с ограниченным количеством воды. Исходные виды сы-
Рнс. 1.6. Схема получения арсената (111) кальция полусухим способом
20
рья в сухом виде загружают в вакуум-сушилку 6, представляющую из себя неподвижный горизонтальный цилиндрический аппарат, вдоль оси которого вмонтирован вал мешалки с гребками. Снаружи корпус сушилки имеет паровую рубашку. Затем в сушилку заливают воду в количестве, необходимом для разбавления извести-пушонки в отношении 2'1. Массу перемешивают в течение часа без подачи пара и при отключенном вакууме. Конец реакции контролируют по анализу пастообразной массы, которая не должна содержать более 0,5% свободного As2Oi, после чего для высушивания образующейся пасты включают паровой обогрев и пускают вакуум-насос. Через каждые 30 мин направление вращения вала мешалки автоматически изменяется, причем изменяется и перемешивание полупродукта в сушилке — от середины к краям или от краев к середине. Постепенно теряя влагу, паста через 3—4 ч загустевает и комкуется. Крупные комья при дальнейшей сушке раскалываются на более мелкие, чему способствует раздавливание их свободно лежащими в сушилке трубами, передвигаемыми гребками. Сушка продолжается до содержания в продукте 1—1,2% влаги. Выгружаемый из сушилки продукт поступает на размол и расфасовку.
Существует и другой вариант полусухого способа получения арсенита (III) кальция, согласно которому смешение исходных видов сырья-сесквиоксида мышьяка и извести-пушонки производят в реакторе в течение 10—15 мин, после чего в реактор заливают воду (60—70° С) и перемешивают массу около 4 ч. Полученную пасту высушивают в барабанной сушилке, обогреваемой топочными парами, имеющими на входе 700—800° С, на выходе 130—160 °C.
Полусухой способ экономичнее мокрого вследствие большей простоты технологии, отсутствия операции фильтрации, меньших потерь и меньшего количества вредных выбросов. На производство 1 т технического арсенита (III) кальция полусухим методом расходуют: 0,64—0,66 т сесквиоксида мышьяка (100% As2O0, 0,35—0,365 т СаО (100%) в виде извести-пушонки, 0,35 т условного топлива или 6 т пара, 260 кВт ч электроэнергии и 25 м3 воды.
Технология арсенита (III) — ацетата меди. Арсенит (III) — ацетат меди (парижская или швейнфуртская зелень) Cu4(AsO2)6 (СНзСОО)2 — изумрудно-зеленое твердое вещество; плотность 3,2 г/см3; нерастворимо в воде и спирте. Водой постепенно гидролизуется с отщеплением As2O4, из 0,25 г парижской зелени и 100 мл воды в суточном стоянии суспензии в раствор переходит —0,005 г Аз20з, те около 3—4% общего количества мышьяка. Плотность технической парижской зелени колеблется от 2,8 до 1,3 г/см3, а насыщенная масса от ~0,3 г/см3 у очень дисперсных образцов до 1 г/см3 у образцов грубого помола. Обладает очень высокой атмосферостой костью (за исключением сульфида водорода), легко растворяется в кислотах, аммиаке, очень ядовита.
21
Медный
Рис. 1.7. Схема производства парижской зелени.
Арсенит (III) — ацетат меди — применяется в качестве инсектицида и фунгицида, является компонентом необрастающих красок для судов, пигментом для художественных красок.
Сырьем для получения парижской зелени являются: сесквиоксид мышьяка, карбонат натрия, гидрат сульфата меди и уксусная кислота. Схема производства приводится на рис. 1.7.
В первой операции в водной среде получают арсенит (III) меди:
As2Ch + На2СОз = 2NaAsO2 + СО2
К полученному раствору добавляют уксусную кислоту, которая, нейтрализуя избыток карбоната натрия, образует ацетат натрия:
Ма2СОз + 2СЖСООН = 2CH3COONa + СО2 + Н2О
Затем в реакторе 8 осаждают парижскую зелень при смешении полученного раствора, содержащего арсенит (III) меди и ацетат натрия, с горячим (90—95° С) раствором сульфата меди (400 г/л):
6NaAsO, + 2CH3COONa + 4CuSO4 = = 3Cii(AsO2)2 • Си(СНзСОО)2 + 4Na2SO4 22
Для полноты реакции и полного удаления из раствора ионов меди избыток уксусной кислоты нейтрализуют карбонатом натрия, осадок отфильтровывают на фильтрах 10, тщательно промывают на нутч-фи-льтре 12. Полученную пасту, содержащую —35% влаги, сушат в вакуум-сушилках 12 в течение 10—12 ч до влажности 1% и измельчают в мельнице 21. На производство 1 т парижской зелени расходуют: 0,575 т сесквиоксида мышьяка (100% As2O2), I т медного купороса (100% C11SO4 • 5Н2О), 0,415 т карбоната натрия и 0, 127 т уксусной кислоты (100%), 420 кВт-ч электроэнергии, 8 т пара и 55 м1 воды.
Более экономичный способ получения парижской зелени, в котором промежуточным продуктом является карбонат меди, называется полусухим. По этому способу взаимодействием раствора сульфата меди (390—410 г/л C11SO4) с раствором карбоната натрия (260 г/л Na2CO2) при 70° С получают осадок дигидроксокарбоната меди:
2CuSO4 + 2Ма2ССЬ + Н2О = СиСО, • Си(ОН)2 + 2Na2SO4 + СО2
После отстоя растворов и их фильтрации полученный осадок дигидроксокарбоната меди промывают водой репульпацией, после чего суспензию, содержащую не более 0,2% сульфата натрия, загружают в реактор, куда предварительно заливают 3/4 расчетного количества уксусной кислоты и загружают всю требуемую навеску сесквиоксида мышьяка с добавкой молотого каолина (50—100 кг на 800 кг As2Oj). После смешения реагентов в реактор постепенно вносят суспензию гидрата-карбоната меди. После окончания процесса выделения диоксида углерода и вспенивания реакционной массы к ней добавляют остаток уксусной кислоты и подогревают массу до 70° С. Подачей острого пара повышают температуру до 90° С и массу перемешивают около 18 ч с постепенной передачей ее на сушку после завершения процесса образования парижской зелени (в течение 2,5—3 ч) по схеме
2СиСО, • Си(ОН)? + 3As2O2 + СН3СООН = = Си(СЖСОО)2 • 3Cu(AsO2)2 + СО2 + Н2О
Сушка продукта производится в две стадии: 1) до влажности 35—40% в гребковой двухвальной сушилке воздухом с температурой 70—80° С, 2) до влажности не более 1,2% в вакуум-сушилке. -При этом исходный пастообразный продукт становится сыпучим. Высушенный продукт размалывают и расфасовывают.
На производство I т целевого продукта рассмотренным способом расходуют. 0,56 т сесквиоксида мышьяка (100% As2O2), 0,982 т медного купороса (100% CuSO4 • 5Н2О), 0,447 т карбоната натрия (95% Na2CCh), 0,116 т уксусной кислоты (100% СН2СООН), 5,6 мГкал пара, 282 кВт-ч электроэнергии и 50 м’’ воды. Этот способ обеспечива-23
ет получение целевого продукта 1-го сорта из сесквиоксида мышьяка, содержащего всего 80—85% As2O4.
Существует технология щелковской зелени, отличающейся от парижской зелени тем, что понижает содержание As2O4 (до 34—35%) и СиО (до 19—20%) и соответственно увеличивает расход продукта при применении. Получение щелковской зелени аналогично получению парижской лишь с той разницей, что взамен карбоната натрия используют гашеную известь. Процесс образования щелковской зелени состоит из следующих реакций:
As2O4 + Са(ОН)2 = Ca(AsO2)2 + Н2О
2СЖСООН + Са(ОН), = Са(СН3СОО)2 + 2Н2О
3Ca(AsOi)? + Са(СН4СОО)2 + 4CuSO4 = = 3Cu(As62)2 • Cu(CH3COO)2 + 4CaSO4
Поскольку в результате последней реакции в растворе не остается примесей, то из технологического цикла исключается промывка пастообразного осадка, что сокращает общую продолжительность производственного процесса.
На получение 1 т целевого продукта расходуют: 0,35 т сесквиоксида мышьяка (100%), 0,148 т оксида кальция в виде пушонки, 0,6 т медного купороса (100% CuSO4-5H2O) и 0,075 т уксусной кислоты (100%).
1.4. АРСЕНАТ (V) КАЛЬЦИЯ
Арсенат (V) кальция Ca4(AsO4)2-5H2O имеет плотность 2,70 г/см\ растворимость его в воде составляет 0,013% (масс.) (25° С), температура обезвоживания 135—310° С.
В системе CaO—As2C>5—Н2О при 17° С устойчивым являются следующие соединения-
при pH'
от 2 до 6 .... от 4,6 до 6.8 от 6,8 до 7,8 от 7,8 до 9 6 более 9.6
CaH4(AsO4)2
Ca2H2(AsO4)2 2Н2О
Ca2H2(AsO4)2 4Н2О 5СаО 2As,O, ЮН2О
Ca3(AsO4)2 _тН2О 4СаО As2O, 5Н2О
4CaO-As2O5-5H2O обезвоживается при нагревании до 200—215° С, а при 350° С превращается в твердый раствор Са(ОН)2 в дигидрате трикальцийарсената 3CaO-As2O5-2H2O, стойкий вплоть до 500° С. 24
Промышленные образцы арсената (V) кальция, которым обычно приписывают формулу пентагидрата тетракальцийарсената 4CaOAs2Os‘5H2O или Ca3(AsO4)2Ca(OH)2-4H2O, меняют состав в зависимости от технологического регламента получения: молярное отношение CaO.As2O3 колеблется обычно в пределах 3,3—3,9.
При взаимодействии раствора гидроксида кальция (не суспензии) с ортомышьяковой кислотой выделяются прямоугольные призмы 4CaO-As2O5’5H2O и ромбические пластинки 3CaOAs2O510H2O. Последний метастабилен при избытке Са(ОН)2 в системе. Устойчивой фазой при 40—60° С являются твердые растворы Са(ОН)2 в дигидрате трикальцийарсената 3CaO As2Os-2H2O.
Водная суспензия 4CaOAs2O5-5H2O имеет значение pH 9,6, а суспензия 3CaO As2O5-10H2O — 9,1. Эти соединения хорошо растворяются в воде при pH 7,5—10. Водные же суспензии 3CaOAs2O5-2H2O и твердых растворов гидроксида кальция в этой соли имеют pH в пределах 8,3—8,6 и относительно мало растворимы.
По другим данным, при 35° С в системе СаО—As2O5—Н2О стабилен Ca3(AsO4)2; при CaO:As2O5>3 осадок состоит из Ca3(AsO4)2 и Са(ОН)2. При 62° С установлено существование Ca3(AsO4)2 2Н2О и 4CaO As2O5 хН2О, а в области 4>CaO:As2O5>3—твердых растворов Са(ОН)2 в дигидрате трикальцийарсената. При 90° С стабильны Ca3(AsO4)2 и 3Ca3(AsO4), Ca(OH)2
Гидроарсенат (V) кальция в водной суспензии гидролизуется:
CaHAsO4 + 2Н2О -> Са(ОН)2 + H3AsO4
Избыток Са(ОН)2 предотвращает переход мышьяка в раствор. Практически при длительном хранении или нахождения препарата на воздухе содержащийся в нем диоксид углерода карбонизирует известь, в результате чего гидролиз интенсифицируется и появляется свободная мышьяковая кислота. В отличие от долго хранившегося продукта свежеполученный арсенат (V) кальция, состоящий в основном из основной соли, выделяет водорастворимый мышьяк медленно и в небольших количествах, чем объясняется его безопасность для растений. Склонность препарата к гидролизу, а также к слеживанию в основном обусловливается гигроскопичностью арсената кальция. На рис. 1.8 приведены данные о степени содержания влаги в препаратах арсената (V) кальция в зависимости от относительной влажности воздуха. Гигроскопичность арсената (V) кальция увеличивается при содержании в нем примесей NaOH или NaCI (даже в количествах всего 0,5%). Однако распыливаемость препарата незначительно зависит от его влажности, если она не превышает 5%.
Арсенат (V) кальция, согласно требованиям действующего стандарта (ГОСТ), должен содержать 38—42% As2O5, не более 1% 25
201--1---1---1--1---1---1--1----
0 2 4 6 a 10 12 14
Содержание влаги в пределах, %
Рис. 1.8. Зависимость влажности арсената кальция от относительной влажности воздуха
AsjOi, не более 0,6% суммы водорастворимых AS2O5 и As2O3, не менее 2% щелочи (в пересчете на СаО) и не более 1% влаги. Требования к дисперсности продукта такие же, как и для других мышьяковых препаратов: 96% продукта должно проходить через сито 6400 отв/см2, при этом максимальный размер частиц не должен быть больше 85 мкм, что обусловлено склонностью продукта к флокуляции. Практически средний диаметр частиц выпускаемого химической промышленностью арсената (V) кальция 8—17 мкм, а максимальный около 40 мкм.
На основе арсената (V) кальция в смеси с наполнителем — глиной или каолином (в количестве 45%), с добавкой 3% асидола называют меритолом.
Препарат купфермеритоль — комплексная соль арсенатов (V), (III)
кальция и меди, разбавленная образующимся в процессе производства гипсом. Купфермеритоль получают, добавляя раствор медного купороса в смесь водных суспензий арсената (V) кальция и извести-пушонки. Образующуюся суспензию превращают на фильтре в пасту, которую затем сушат, размалывают и гидрофобизируют асидолом. Препарат содержит по 18—20% AS2O5 и СиО, не менее одного процента свободного гидроксида кальция, не более 0,5% As2O3 и 0,6% водорастворимых As2O5 + As2O3, не более 1% влаги и 2—3% асидола.
Применяется препарат, называемый лондонским пурпуром, представляющий собой смесь арсената (III) и арсената (V) кальция. Получают его в виде отхода от производства фуксина и некоторых других красителей и кроме других примесей содержит краситель, придающий ему пурпурный цвет. Состав некоторых образцов лондонского пурпура: 26,8—28% общего мышьяка (As), 4,5—5,8% As2O3, 35,6—37,4% AS2O5, 0,12—0,4% водорастворимого мышьяка.
Способы получения арсената (V) кальция. Разработаны несколько способов получения арсената (V) кальция, имеющих промышленное значение. Все они основаны на окислении соединений мышьяка (III): 1) каталитическое окисление растворов арсената (III) кислородом воздуха в щелочной среде; 2) осаждение арсената (V) из мышьяковой кислоты, полученной окислением сесквиоксида мышьяка; 3) электрохими-26
ческое окисление арсената (ill); 4) окисление арсената (III) кальция воздухом при высоких температурах (термический способ).
Кроме перечисленных способов существует способ получения арсената (V) кальция из окисленных мышьяковых руд, заключающийся в выщелачивании As2Os раствором гидроксида натрия.
Технология арсената (V) кальция каталитическим окислением арсената (III) воздухом. Каталитический способ производства арсената (V) кальция основывается на окислении арсената (III) натрия в арсенат (V) натрия кислородом воздуха в присутствии катализатора—медного купороса—с последующим осаждением арсената (V) кальция обработкой раствором известкового молока (рис. 1.9).
Взаимодействие арсената (V) натрия с водной суспензией гидроксида кальция протекают в несколько стадий:
As2O, + 6NaOH = 2Na3AsO3 + ЗН2О (1)
2Na3AsO3 + О2 = 2Na3AsO4 (2)
Na3AsO4 + Ca(OH)2 + 4H2O = CaNaAsO4-4H2O + 2NaOH (3)
2CaNaAsO4-4H2O + Ca(OH)2 + 2H2S = Ca3(AsO4)210H2O + 2NaOH (4)
Ca3(AsO4)2-10H2O + Ca(OH)2 = Ca3(AsO4)rCa(OH)2-4H2O + 5H2O (5)
Согласно технологической схеме, в стальной реактор 1 с мешалкой и барботером для подачи острого пара из сборника 3 заливают расчетное количество маточных растворов и из сборника 2 растворы гидрооксида натрия. Указанные растворы вносят в реактор в таком соотношении, чтобы концентрация гидрооксида натрия была в пределах 75—85 г/л NaOH. В подогретую в реакторе 1 острым паром до 80—90° С жидкость при работающей мешалке вносят из бункера 4 через автодозаторы 5 сесквиоксид мышьяка в количестве, отвечающем молярному отношению As2O3:NaOH от 1:6 до 1:6,5. После перемешивания в течение 30 мин при 85—95° С по реакции (1) образуется раствор арсената (III) натрия с концентрацией не менее 50 г/л As2O3. В полученном растворе отношение NaOH:As2O3 должно находиться в пределах 1.21—1,31. Общая продолжительность процесса образования арсената (III) натрия составляет 1,5—2 ч. В полученный раствор вводят твердый или в виде водного раствора медный купорос (в количестве 2—3,5% от участвующего в реакции 100%-ного сесквиоксида мышьяка), который является катализатором при окислении арсената (III) в арсенат (V). После растворения медного купороса в растворе реакционную массу из реактора 1 перекачивают в
Рис. 1.9. Технологическая схема производства арсената кальция каталитическим способом
1 — реактор, 2 — мерник раствора NaOH. 3— резервуар-отстойник для маточных растворов, 4—разгрузочный бункер для белого мышьяка 5— ве-
совой бункер. 6, 51— рукавные фильтры; 7, 39, 41 — вакуум-насосы, 8, 11, 19, 20, 31, 33—центробежные насосы 9 — отстойник для арсенита на-
ные вакуум-фильтры, 30, 36—вакуум-сборники 32, 34—смесители 37, 38—барометрические конденсаторы 40—приямок 42 — шнек-питатель 43 — сушилка; 44, 49—вентиляторы, 45— пылеуловитель 47—шнековый холодильник 48 — подпечный бункер 50 — мельничный бункер 52—центробежная вакуум-мельница 53 — расфасовочный бункер, 54 — шнек 55 — аппарат расфасовки 56— барабан для целевого продукта
окислитель 12. При переработке сесквиоксида мышьяка, содержащего значительное количество примесей в виде сульфидов мышьяка и других, замедляющих процесс окисления, раствор арсената (III) натрия направляют на предварительный отстой для отделения шлама, а затем в окислитель. В этом случае раствор сульфата меди добавляют к арсенату (III) натрия в окислителе.
Окислитель представляет собой стальную цилиндрическую емкость с коническим дном, снабженную барботерами для подачи воздуха и пара. В раствор с температурой 65—85° С, через барботер пропускают воздух в течение 1,5—4 ч. Отходящий из окислителя воздух проходит ловушку 13 и промывной скруббер 14, затем выбрасывается в атмосферу. Окисление арсената (III) в арсенат (V) идет по реакции (2).
Окончание реакции определяют по содержанию в реакционной массе сесквиоксида мышьяка. Оно должно быть меньше 1 г/л.
Полученный раствор арсената (III) натрия передают в осадитель 17, представляющий собой стальной цилиндрический резервуар с метальным устройством и паровой рубашкой. В подогретый до 85° С раствор при перемешивании вводят из мерника известковое молоко, содержащее 160 г/л СаО в рассчитанном по содержанию As2O3 в растворе. Подогревают массу в реакторе до 95—98° С. Расчеты по осаждению арсената (V) кальция ведут по уравнению
2Na3AsO4 + 4Са(ОН)2 = Ca,(AsO4)2 • Са(ОН)2 + 6NaOH
Осаждение считается законченным, когда содержание As2O3 в жидкой фазе (в растворе) становится меньше 1 г/л, а в твердой фазе суспензии содержится 38—42% As2O5 (в пересчете на сухое вещество). В случае необходимости состав реакционной массы корректируют, вводя недостающий реагент. Описанный режим не является оптимальным. Экспериментально установлено, что для получения арсената (V) кальция высокого качества и с максимальным выходом процесс осаждения следует вести при энергичном перемешивании при температуре не ниже 90° С, причем мышьяксодержащий раствор необходимо прибавлять к известковому молоку медленно. Молярное соотношение СаО : As2O5 в исходных растворах должно быть равным 4, или по массе — 0,97. Практически берут 0,93—0,97 масс.ч СаО на I масс.ч. As2Os. Применяемая известь должна быть высокого качества и свежегашеная.
По окончании процесса осаждения прекращают перемешивание и подогрев. Раствору, содержащему регенерированный гидроксид натрия, дают отстояться. Осветленный раствор, содержащий 65—75 г/л гидроксида натрия, направляют на приготовление арсената (III) натрия. Оставшуюся в осадителе суспензию арсената (V) кальция для отмывки от щелочи перемешивают с водой и подвергают двукратной
фильтрации на барабанных или ленточных вакуум-фильтрах 29 непрерывного действия На первом фильтре осадок промывают водой. Полученный фильтрат, разбавленный промывной водой, содержащий около 40 г/л гидроксида натрия, используют для гашения извести. Осадок с первого фильтра репульпируется с водой в реп^льпаторе 32 с мешалкой и поступает на второй барабанный вакуум-фильтр 35, после которого паста арсената (V) кальция, содержащая до 30% влаги, направляется на сушку. Сушка производится в барабанных сушилках 43 до содержания влаги не более 1% в целевом продукте.
Следует отметить, что сушка пасты арсената (V) кальция более рациональна не непосредственно дымовыми газами, а с помощью наружного обогрева. Это позволило бы избегать перегрева продукта и снижения его токсичности. На некоторых заводах после предварительного высушивания пасты до содержания 10—14% влаги окончательную сушку производят в вакуум-сушилке с паровым обогревом.
Выходящий из сушилки арсенат (V) кальция охлаждают в шнековом холодильнике 47, а затем пневмотранспортом передают в бункер готовой продукции 53, из которого передается на расфасовку.
На производство I т арсената (V) кальция описанным способом расходуют: 0,35 т сесквиоксида мышьяка (100% Аз20з), 0,48 т извести (100% СаО), 0,077 т гидроксида натрия (92% NaOH), 0014 т медного купороса (100% CuSO4-5H2O), а также 2,5 т пара и 22,6 м1 воды.
Получение арсената (V) кальция нейтрализацией мышьяковой кислоты известью (рис. 1.10) имеет преимущества перед другими способами. Полученный этим способом арсенат (V) кальция имеет повышенную дисперсность и пригоден для авиационного опрыскивания концентрированными суспензиями. Способ основан на нейтрализации мышьяковой кислоты известковым молоком:
2H3ASO4 + 4Са(ОН)2 = Ca<AsO4)2 • Са(ОН)2 + 6Н2О
Реакцию проводят в цилиндрическом вертикальном реакторе 2, снабженным паровой рубашкой и мешалкой. В него заливают известковое молоко с концентрацией 200 г/л, подогревают до 80—90° С и подают при постоянном перемешивании 10—12%-ный раствор мышьяковой кислоты. Процесс осаждения арсената (V) кальция продолжается 40—60 мин после внесения мышьяковой кислоты. Полученную суспензию арсената (V) кальция направляют на барабанный вакуум-фильтр Фильтрат и промывную воду используют для разбавления исходной концентрированной кислоты и для приготовления известкового молока. Для получения легко фильтруемого осадка процесс осаждения проводят при 50—60° С. При этом вместо 30
Рис. 1.10. Технологическая схема мышьяковой кислоты и арсената (V) кальция азотно-кислотным способом-
1— бункер, 2 —реактор для получения мышьяковой кислоты, 3 — холодильник, 4, 5 —отстойники 6 — нутч-фильтр, 7 — сборник; 8 — мерник, 9 — окислительная башня, 10, 11 — абсорберы, /2 13 — сборник азотной кислоты; 14— приемник азотной кислоты; 15, 16 — мерник, 17—реактор-осадитель; 18 — вакуум-фильтр 19— сборник фильтрата, 20 — сушилка, 21, 23 — бункеры
22 — мельница, 24 — камера расфасовки, 25 — фильтр
тетракальцийарсената образуются кристаллы твердого раствора гидроксида кальция в дигидрате трикальцийарсената.
Паста арсената кальция, снимаемая с поверхности барабана фильтра, имеет влажность 30% и поступает в вакуум-сушилку с паровым обогревом или в барабанную сушилку, обогреваемую снаружи топочными газами. Выходящие из сушилки пары проходят циклон, где уносимая пыль улавливается при барботаже через воду. Выгружаемый из сушилки продукт пневмотранспортом передается в бункер, из которого поступает на размол 22, а затем на расфасовку. Пыль арсената (V) кальция улавливается в фильтрах 25. Воздух, отсасываемый из этих фильтров ротационными вакуум-насосами, промывается водой, которую периодически возвращают в цикл на разбавление исходной мышьяковой кислоты и приготовление известкового молока.
На 1 т целевого продукта расходуют: 0,37 т сесквиоксида мышьяка, 0,07 т азотной кислоты, 0,5 т извести, 0,02 кг иода, 70 м1 воды, 3 т пара, 540 кВт-ч электроэнергии.
Хлорный способ получения арсената (V) кальция. Применяя в качестве окислителя элементный хлор, хлорируют суспензию сесквиоксида мышьяка в воде, но смесь Н3РО4 и HCI не разделяют.
Хлорируют суспензию As2O3 в воде (185 кг As2Oi на 1 м1 воды) Процесс проводится в футерованных кислотоупорными плит-31
ками хлораторах с гуммированными мешалками. Процесс протекает 1 по реакции 1
As2Ch + 2СЬ + 5Н2О = 2H,AsO4 + 4HCI + 66,4 ккал. ]
В образующуюся смесь разбавленных кислот в процессе хлори- 1 рования (3—4 ч) непрерывно вводят известковое молоко. В хлорато- * рах поддерживают температуру в пределах 80—90° С, а дозировку j известкового молока производят по величине pH реакционной массы । с таким расчетом, чтобы нейтрализовать лишь хлороводородную кислоту. По окончании реакции раствор содержит около 200 г/л мышьяковой кислоты и хлорид кальция, образовавшийся при нейтрализации хлороводородной кислоты. Раствор отстаивается от нерастворимых примесей, введенных с известковым молоком. После подогрева до 80—90° С раствор сливают тонкой струей в известковое молоко для осаждения арсената (V) кальция. Процесс осаждения заканчивается при pH 10. Такая последовательность осаждения обеспечивает получение осадка необходимой структуры.
Процесс осаждения арсената (V) кальция возможен и карбонатом кальция, но реакция идет медленнее, чем с известью.
Осадок арсената (V) кальция отделяют от маточного раствора на барабанных вакуум-фильтрах. Отфильтрованная паста содержит около 30% влаги и высушивается на вальцовых, барабанных, вакуумных сушилках или в печах с кипящим слоем.
Существуют и другие способы получения арсената (V) кальция.
При термическом способе получения арсената (V) кальция путем смешения сесквиоксида мышьяка, извести и воды полученная паста содержит до 40% влаги, которая после высушивания и термообработки при 500—600° С содержит смесь арсената (III) кальция с избытком извести При дальнейшей термообработке в окислительной атмосфере (в струе воздуха или топочных газов, не содержащих восстановителей) образуется арсенат (V) кальция.
Способ получения арсената (V) кальция непосредственно из окисленных скородитовых мышьяковых руд, содержащих 18—27% мышьяка, а также из бедных руд с содержанием 7—15% As предусматривает обработку измельченной руды раствором гидроксида натрия при 90—92° С. При этом пентаоксид мышьяка переходит в раствор в виде арсената (V) натрия. Например, если мышьяк содержится в руде в виде минерала скородита (Fe2OyAs2O5-4H2O или FeAsO4-2H2O), то протекает следующая реакция:
FeAsO4 • 2Н,0 + 3NaOH = Na4AsO4 + Fe(OH)3 + 2Н2О
Концентрация применяемого для выщелачивания руды раствора гидроксида натрия может колебаться от 5 до 40% в зависимости от
химического и минералогического состава руды. Извлечение пентаоксида мышьяка в раствор составляет 95—100%. После выщелачивания и отстаивания суспензии осветленный раствор арсената (V) натрия перерабатывают на арсенат (V) кальция по известному способу — осаждением его известковым молоком. Известковое молоко с концентрацией 20% СаО готовят из свежегашеной извести и осаждение ведут при 90—95° С, приливая горячий раствор арсената (V) натрия к горячему раствору известкового молока:
2NaiAsO4 + 4Са(ОН)2 = Ca,(AsO4)i + Са(ОН)2 + 6NaOH
Осадок арсената (V) кальция после отделения от маточного раствора промывают водой до полного удаления образующегося гидроксида натрия, сушат при 120° С и измельчают. При значительном содержании водорастворимого пентаоксида мышьяка в препарате к нему прибавляют гидроксид кальция.
Полученный продукт содержит 39—40% общего As2O5, 0,15% водорастворимого As2C>5, 0,15—0,2% свободного СаО и имеет объемную массу 0,45 т/м1.
1.5. АРСЕНИДЫ
Арсениды — соединения мышьяка с более электроположительными химическими элементами. Они известны для всех металлов и полуметаллов, кроме сурьмы, висмута, свинца и таллия.
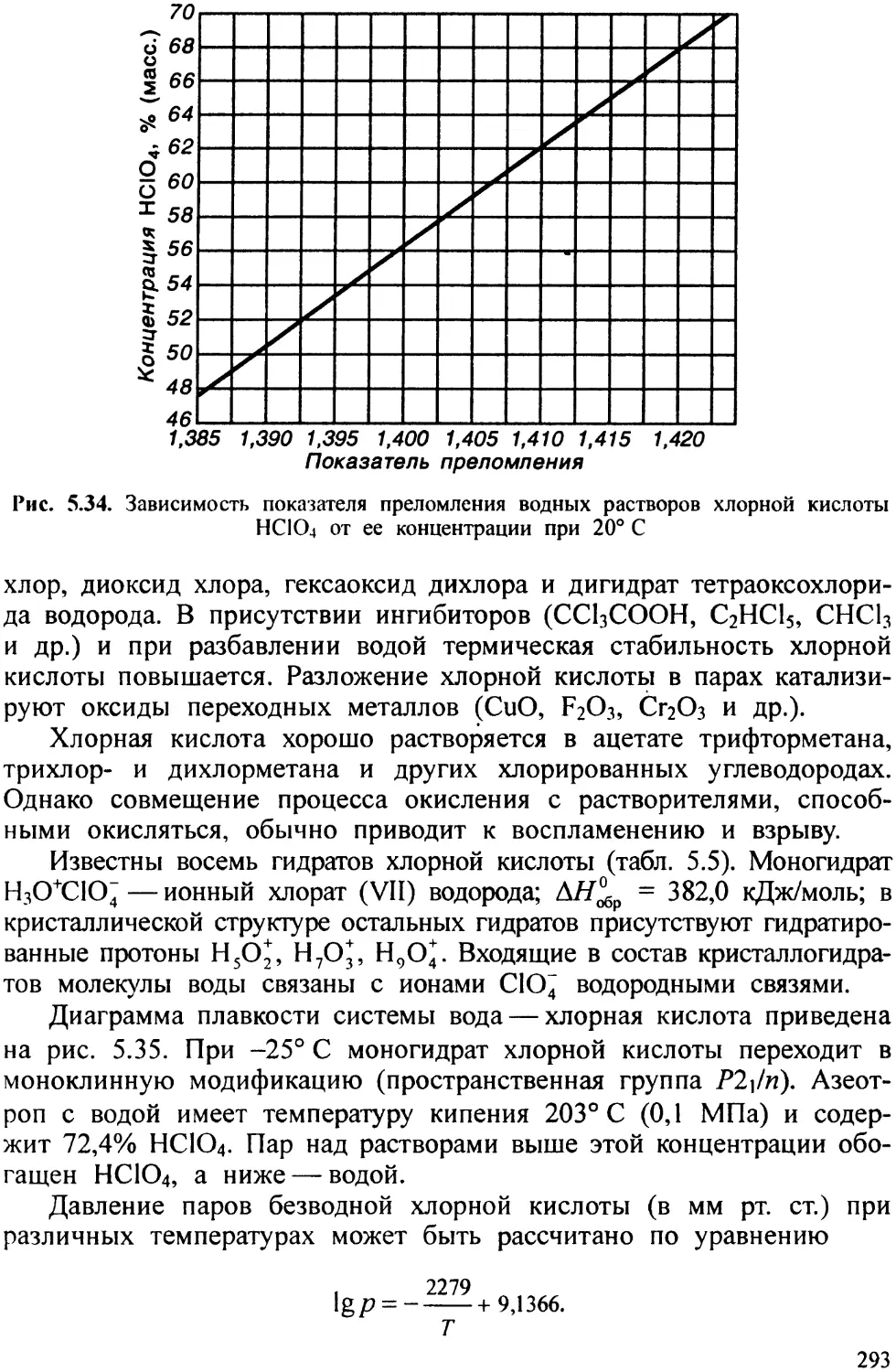
Арсениды —кристаллические высокоплавкие соединения с металлическим блеском, обычно серебристо-белого или светло-серого цвета (редко желтого или красного). Они обладают полупроводниковой, полуметаллической, а низшие арсениды — металлической проводимостью. Некоторые арсениды переходных металлов, например Cr2As и Fe2As, являются антиферромагнетиками. Высокими точками Нееля отличаются CrAs(823 К), Mn2As(580 К). Арсенид марганца — ферромагнетик. Некоторые арсениды (MoAs2, Pd2As) при Г<1 К становятся сверхпроводниками.
Щелочные металлы образуют арсениды типа MAs и MgAs, но для натрия известен пентаарсенид NaAsj, а для кальция — диарсенид CaAs2. Элементы подгруппы 16 медь образуют плавящиеся конгруэнтно арсенид СщАз наряду с низшими (CugAs, CueAs) и высшими (CuAs). Элементы II группы обычно образуют плавящиеся конгруэнтно арсениды типа MtAs2 и высшие MAs2(Be, Cd, Zn), MAs, где M — щелочноземельный металл.
Элементы подгруппы II 1а, кроме таллия, образуют плавящиеся конгруэнтно моноарсениды MAs, кристаллизующиеся в структуре
сфалерита, являющиеся полупроводниками. Для бора известен низший арсенид B6As.
Элементы подгруппы IVa, кроме углерода и свинца, образуют плавящиеся конгруэнтно MAs. Для кремния и германия известны соединения MeAs2, для олова — Sn3As4. Для элементов подгруппы титана характерны соединения M4As, MAs, MAs2, а переходные металлы V—VII групп образуют арсениды состава M3As, M2As, Me5As2, MAs и MAs2. Наибольшее число арсенидов известно для ванадия (7) и для никеля (8), а для рения и осьмия — лишь по одному (Re3As7 и OsAs2).
Существуют также двойные арсениды: MM’As (NaCdAs и FeMnAs), MM'2As2 (CaNi2As2 и др.), M"M,vAs2 (CdGeAs2). Известны и тройные интерметаллические соединения и соли со сложными анионами, например XAs4 (X = Ge, Si, Zn, Co и др.), способными образовывать цепочечные, слоистые и каркасные структуры. Большинство этих соединений применяется в качестве полупроводников.
Арсениды щелочных металлов гидролизуются водой с выделением AsH3, арсениды щелочноземельных металлов с водой реагируют слабо, а с разбавленными кислотами легко. Арсениды же тяжелых металлов с водой практически не взаимодействуют, реагируют с кислотами, а при сплавлении — с щелочами. С увеличением содержания мышьяка‘в молекуле химическая стойкость арсенидов повышается. При действии окислителей или нагревании в воздухе арсениды окисляются до арсенатов (III) или сесквиоксида мышьяка. Высшие арсениды при нагревании отщепляют часть As.
Известно около 25 природных минералов, относящихся к арсенидам, важнейшими из которых являются: смальтин CoAs3.x, кобальтин CoAsS, никелин NiAs, лелингит FeAs2, арсенопирит FeAsS, сперрилит PtAs2.
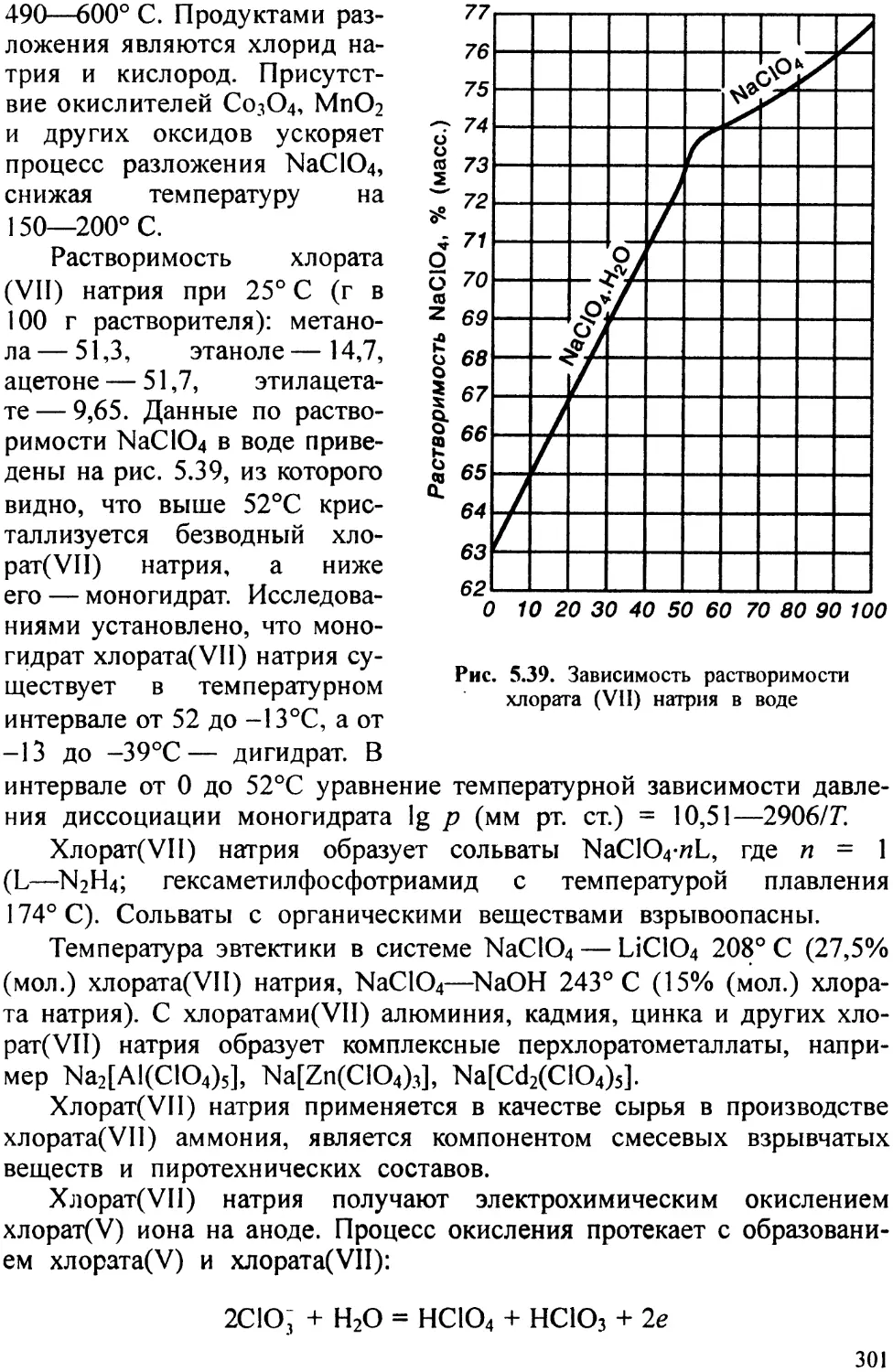
Арсенид галлия GaAs — темно-серые с фиолетовым оттенком и металлическим блеском кристаллы, решетка кубическая типа сфалерита (а = 0,565321 нм), температура плавления 1238° С; плотность 5,317 г/см1 (20° С), жидкого 6,00 г/см3 (1238° С); С° = 46,9 Дж/(моль'К); &Н°Л = 105,6 кДж/моль, ДЯ0°6р = -74,1 кДж/моль; 8°^ = 64,27 Дж/(мольК); температурный коэффициент объемного расширения 5,93-Ю^К'1 (0—252° С); теплопроводность 150 Вт/(м-К) при 27° С; е = 13,2. Полупроводник; при 27° С ширина запрещенной зоны 1,428 эВ, подвижность электронов 8500 см2/(В-с), дырок 400 см2/(В-с); эффективная масса электронов проводимости те = О,О65тло, дырок тр = = 0,5 тлп, где тио — масса свободного электрона.
Арсенид галлия устойчив к кислороду воздуха и парам до 600° С, слабо реагирует с серной и хлороводородной кислотами с выделением AsH3, под действием азотной кислоты пассивируется, разлагается в растворах щелочей.
34
Арсенид галлия является одним из полупроводниковых материалов для интегральных микросхем, фотоприемников, СВЧ- и фотодиодов, транзисторов, инжекционных лазеров, оптических фильтров и модуляторов лазерного излучения, солнечных батарей и др.
Арсенид галлия получают сплавлением галлия с мышьяком под давлением паров мышьяка (около 100 кПа). Микрокристаллы выращивают методами зонной плавки, направленной кристаллизации под давлением паров мышьяка или вытягиванием из-под слоя флюса сесквиоксида бора под давлением аргона (150 кПа). Эпитаксиальные пленки, а также мелкокристаллический арсенид галлия получают путем химических транспортных реакций с водородом в качестве газа-носителя:
2GaAs + 2HCI # 2GaCl + 2As + Н2 (900->750° С)
при этом GaAs получают в горячей зоне (900° С) по реакции
2Ga + 2AsCh + ЗН2 -> 2GaAs + 6HCI
Процесс ведут также при 700° С:
Са(С2Н5)з + AsH3 = GaAs + ЗС2Н6
Арсенид галлия в виде пленок получают также из его растворов в расплавленном галлии. Для легирования монокристаллов и пленок используют добавки Те, Se, S, Sn, Zn, Cd и Се. Монокристаллы с высоким р (I08 — 109 Ом см) получают с использованием добавок железа или хрома.
Арсенид алюминия AlAs — серые кристаллы с металлическим блеском, решетка кубическая типа сфалерита (а = 0,5662 нм); неустойчив во влажном воздухе, водой и разбавленными кислотами разлагается с выделением AsH2. Полупроводниковый материал для солнечных батарей, компонент твердых растворов с GaAs и другими соединениями типа a"'Bv, используемых в лазерах, фотодиодах и др.
Арсенид никеля NiAs — бронзово-желтые или светло-красные кристаллы с металлическим блеском, решетка гексагональная (а = = 0,3963 нм, с = 0,5049 нм, z = 2, пространственная группа Рб^/ттсу, обладает металлической проводимостью. Перспективен в качестве компонента эвтектических композиций с GaAs и InAs для приборов, действие которых основано на магниторезистивном эффекте, для детекторов инфракрасного излучения и др. Перспективные полупроводниковые материалы—Zn2As2, Cd2As2, CaAs2, а также арсе-ипды типа CdSiAs2
35
Арсениды получают: сплавлением мышьяка с соответствующим металлом в вакууме, инертной атмосфере, под давлением пара мышьяка или под слоем флюса, например В2О3, а также действием пара мышьяка на металлы. Для получения пленок или мелких кристаллов используют химические транспортные реакции. Арсениды могут быть получены: I) взаимодействием AsCl3 с металлами; 2) взаимодействием AsHi с оксидами, растворами солей соответствующих металлов; 3) сплавлением мышьяка с галогенидами металлов; 4) восстановлением арсенатов (V) и арсенатов (III) металлов водородом; 5) взаимодействием мышьяка с растворами соответствующих металлов в жидком аммиаке.
1.6. ХАЛЬКОГЕНИДЫ МЫШЬЯКА
В системе As—S известны сульфиды AS2S5, As2S3 (минерал аурипигмент); As4S4 (минерал реальгар) и As4S3 (минерал диморфит); в системе As—Se — селениды As2Se3 и As4Se4; в системе As—Те известен один теллурид — As2Te3. Структура и свойства халькогенидов мышьяка приведены в табл. 1.3 и 1.4, а уравнения температурной зависимости— давления пара над жидкими и твердыми халькогенидами мышьяка—приведены в табл. 1.5.
Халькогениды мышьяка устойчивы на воздухе, не растворяются в воде, хорошо растворяются в щелочах и при нагревании в азотной кислоте. Все обладают полупроводниковыми свойствами, прозрачны в ИК-области спектра.
Сесквисульфид мышьяка AS2S3 растворяется в жидком аммиаке, а также в водных растворах сульфидов и полисульфидов щелочных металлов с образованием соответствующих дитиоарсенатов (III) MAsS2 и тиоарсенатов (V) M3ASS4. Сгорает на воздухе, давая As2O3 + SO2; окисляется HNO3. Моносульфид AsS существует в виде тетрамера AS4O4, в парах начинает диссоциировать около 800° С, а при 1000° С димерен (As2S2); растворим в водных растворах сульфидов щелочных металлов.
Пентасульфид мышьяка AS2S5 кристаллизуется при 300—600° С и 4—5 ГПа; температура плавления 190° С (инконгруэнтно). Кристаллический OC-AS4S4 получают синтезом из элементов. При вакуумной перегонке он переходит в P-As4S4, а при 400—700° С и 4—7 ГПа образуется еще одна модификация As4S4 (температура перехода р—>а 250° С).
Стеклообразные сульфиды мышьяка устойчивы к кристаллизации. Например, кристаллический a-As2S3 получают термообработкой стекла при 280° С (в течение 10 сут), p-As2S3—при 400—700° С и 4—5 ГПа (температура перехода Р~>а 210° С), y-As2S-, — при 700—1000° С и 4—5 ГПа. 36
I абл и ца 13 Характеристика кристаллических модификаций халькогенидов мышьика
(-оедине- Сингония Параметры элементной ячейки Про-странст-венная группа формульных еди- Цвет
а, нм b нм с. нм град
Л—AsiSi Моноклинная 1,147 0,957 0,424 90,45 Р2, /п 4 Желтый
As2S, Ромбиче- 1,037 0,990 0,866 - - — »
A—As4S-, » 0,912 0,799 1,010 — Р„та 4 Ораиже-во-желтый
А—As4S4 Моноклинная 0,927 1,350 0,656 106,61 /*2, /п 4 Красно-коричневый
P-As4S4 » 0,9957 0,9935 0,8889 102,48 С2!с — »
a-AsiSej » 1,2053 0,9890 0,4277 90,47 П, !п Коричневый
a-As4Se4 » 1,0000 1,386 0,669 113,2 Р2> In 4 Темно-серый
a-AsiTe! » 1,4440 0 405 0,992 97 С2/т 4 »
Измельченный стеклообразный As2Se3 кристаллизуется интенсивно при 330° С, превращаясь в a-форму, а p-форма образуется при давлении 3—7 ГПа. Аналогично получают и кристаллический а- и P-As4Se4.
Таблица 14 Свойства халькогенидов мышьяка
Показатель As2S, As4S4 As2Se2 AS4SC4 As2Te2
7П1 °C 310 321 370 295 385
7 °C 708 534 861 572 1027
7CTtK1 °C 180 — 180 170 110—115
Плотность, г/см’ 3.48* при 19°С 3,506 (а) 3.254 (р) 4,60* - 6,23 5,53*
С", Дж/(моль К) 115,6 188,4 121,50 — 127,57
ДН".р, кДж/моль -159,1 -269,6 -102 -154 -37,6
ЛИ",, кДж/моль 28,7 — 40,8 — 46,8
Д/7"с„, кДж/моль 86.1 70,3 108,1 — 116,0
S’",w, Дж/(моль К) 163,7 254,1 194,5 253,7 226,5
Ширина запрещенной юны (300 К), эВ 2,56* — 1,7* — 0,48
р Ом см 101’* - I012 10”’ - 4 2 104*
* Для стекла. Для стеклообразного As2S2 температурный коэффициент линейного расширения а = 23,9 Ю'К1, г = 7 9 />,, " = 2,58 Для стеклообразного As2Sei а = 21,6 НГ'ТС1, е = 10,2, " = 2 78, подвижность дырок 2 10'см2/(В с)
** Для кристаллическою As.Tc подвижность электронов 170 см2^(В с), дырок—80 см2/(Вс)
37
Теллурид в кристаллической a-форме образуется при синтезе из элементов, P-As2Fei — при 450° С и 8—9 ГПа (температура перехода а—>р 200—290° С)
Стекла из халькогенидов мышьяка применяют в качестве полупроводниковых материалов в электронике, оптике, бессеребряной фотографии, электрофотографии, запоминающих устройствах. Их используют для изготовления волоконных световодов для ИК-области спектра. Реальгар является компонентом пиротехнических смесей, добавкой при литье дроби, его применяют также для удаления волос с кожи. Аурипигмент и реальгар — пигменты, применяющиеся для приготовления красок для живописи.
Таблица 15 {качения коэффициентов в уравнении 1g р (мм рт. ст.) = -А/Т + В
Соединение A в Интервал температур, К Основные компоненты насыщенного пара
As4S4 (ж) 3670 7,13 573—773 As4S4
As4S4 (тв) 6238 11,337 432—548 As4S4
As4Se4 (ж) 4022 6,875 758—999 —
As4Se4 (тв) 7842 14,170 469—543 As4Se4
As2S3 (ж) 4307 7.253 729—966 —
As2S3 (тв) 8794 13,987 497—569 —
As2Sc, (ж) 5683 7.89 926—1132 —
As2Se, (ib) 9092 13,417 541—626 As2, As2Se3, AsSe, As2Se2, Se<„ As4
As2Tc, (ж) 6050,4 7.5488 1052—1200 As2, As4, Te2
As2Te3 (тв) 7590 1051 493—713 As2, As4, Te2
/К — жидкость, тв — твердое
1.7. ГАЛОГЕНИДЫ МЫШЬЯКА
Рассмотрим наиболее изученные галогениды мышьяка (табл. 1.6).
I аблица 16 Свойства галогенидов мышьяка
Показатель AsF, AsF, AsCh AsBr, As.1,
Д,„ °C -78,9 -5.94 -16 31 141
rk„„. °C -52.8 57,8 1,30 221 371
Плотность, г/см’ 7,71 10 ’ 3,01 2,17 3,54 4,39
ДА/,",, кДж/моль II 5 10,4 10,1 11,8 21,8
Д/7,”,,, кДж/моль — 33,5 38,1 41,8 56,5
Д/7'^,. кДж/моль -1236,7 -957 -315,5 -199 -64,8
Д"м, Дж/(моль К) 309 181,2 212,5 161 213
38
Пеитафторид мышьяка AsF5 — бесцветный газ (см. табл. 1.6); уравнение температурной зависимости давления пара над жидким AsF5: Igp (мм рт. ст.) = 7,845—1093,7/(193—220 К); растворяется в воде и растворах щелочей (со значительным выделением теплоты), в диэтиловом эфире, этаноле и бензоле. С сухим стеклом на холоде не реагирует. в присутствии влаги легко его разъедает. Обугливает бумагу и сахар. При нагревании реагирует с медью, цинком и железом, особенно легко — со свинцом и ртутью. При нагревании реагирует с фторидами щелочных металлов, образуя при этом растворимые в воде гексафторарсенаты (V) M[AsF6]; из водных растворов выделен также K2[AsF7]H2O. С фторидами Си, Zn, Fe образует соединение M[AsF7].
Пентафторид мышьяка получают фторированием элементного мышьяка и применяют в качестве катализатора процессов полимеризации, а также для получения компонента электролитов источников тока Li[AsF6],
Пеитахлорид мышьяка AsCls; температура плавления — 40° С, нестоек, разлагается при 50° С за несколько минут. Существуют хлоропроизводные As(V). Оксихлорид AsOCl3, образующийся при действии О3 на AsCI3, при низких температурах малоустойчив, но продукт его разложения As2O3CI4 устойчив до 150° С.
Пентахлорид мышьяка получают при УФ-облучении раствора трихлорида мышьяка в жидком хлоре.
Существуют двойные хлориды As(V), например SbAsCIjo и GaAsCl8, содержащие катион [AsCl4]+ тетраэдрического строения.
Трифторид мышьяка AsF3— бесцветная, подвижная, дымящая на воздухе жидкость; уравнение температурной зависимости давления пара над жидким AsF3: 1g р (мм рт. ст.) = 61,38—4150/7’—18,26 lg Т-, растворяется в воде с разложением, этаноле, диэтиловом эфире, бензоле. При нагревании реагирует с обычным и кварцевым стеклами. Присоединяет бром, иод и аммиак. Энергично реагирует на холоде с хлоридами кремния, фосфора и серы. С фторидами щелочных металлов образует фторарсенаты (III) M[AsF4], с SbF3 — соединение [AsF2]+[SbF4]'. При действии элементного хлора на трифторид мышьяка образуется димерный дихлоротрифторид мышьяка, имеющий строение [AsC14]+[AsF6]‘; температура плавления 160° С (с разложением) При его нагревании в вакууме получают мономерный AsC12F3 с температурой плавления 75° С, а также другие малоустойчивые хлор-фториды As(V).
В производстве трифторид мышьяка получают действием серной кислоты на смесь сесквиоксида мышьяка с дифторидом кальция, а также термообработкой As2O3 с KHF2
Трихлорид мышьяка AsCl3 — бесцветная маслянистая жидкость, дымящая на воздухе, образует кристаллы с перламутровым блеском; ^равнение температурной зависимости давления пара: 1g р (мм рт. 39
ст.) = 7,953—2043/7(323—373 К); растворим в воде (53%, масс.), в разбавленных растворах гидролизуется с образованием мышьяковистой кислоты; растворим в хлороводородной кислоте, с повышением концентрации НС1 растворимость AsCI3 снижается (растворимость в 11 М HCI 200 г/л), снижается также и степень гидролиза. Трихлорид мышьяка отгоняется из соляно-кислого раствора. При пропускании окислителей (С12, О2 и др.) в раствор трихлорида мышьяка в хлороводородной кислоте As(III) переходит в As(V).
С большинством органических растворителей трихлорид мышьяка смешивается во всех отношениях с образованием продуктов присоединения. Хорошо растворяет серу, фосфор и хлориды многих металлов. С' хлоридами одновалентных металлов образует хлорарсенаты (III) M[AsC14], с хлоридами рубидия и цезия — M3[As2C19], а аммиаком— AsCI3-4NH3. Пары трихлорида мышьяка восстанавливаются водородом до металлического мышьяка. 2AsCl3 + 3H2>2As + 6НС1. Трихлорид мышьяка восстанавливается также щелочными металлами, Mg, Zn, Al при нагревании, реагирует с медью, никелем, оловом, свинцом и другими металлами:
AsCh + 3Na = As + 3NaCl
2AsCl3 + 3Mg = 2As + 3MgCl2
2AsCl3 + 3Zn = 2As + 3ZnCI2
Трихлорид мышьяка получают действием хлора на элементный мышьяк или газообразного НО на As2O3 при 180—200° С:
2As + 3CI2 = 2AsCl3
As2O3 + 6HCI = 2AsCI, + ЗН2О
а в промышленности — хлорированием сесквиоксида мышьяка в расплаве серы. Очищают ректификацией, адсорбцией, экстракцией, термообработкой и т.д.
Трихлорид мышьяка применяют для получения мышьяка высокой чистоты, полупроводниковых соединений мышьяка, в производстве медицинских препаратов и для получения других его соединений.
Бромид мышьяка AsBr3— бесцветные кристаллы ромбической сингонии (<я = 1,051 нм, Ъ = 1,207 нм, с = 0,431 нм, z = 4, пространственная группа Гигроскопичен, расплывается на воз-
духе. Растворим в воде (с разложением), диэтиловом эфире, бензоле и сероуглероде.
40
Получают бромид мышьяка взаимодействием брома с мышьяком, а также брома со смесью сесквиоксида мышьяка и серы по уравнениям:
2As + 3Br2 = 2AsBn
AS2O3 + 3Br2 + S = 2AsBr3 + SO3
Трииодид мышьяка Asl3 — оранжево-красные или рубиново-красные кристаллы гексагональной сингонии (а = 0,721 нм, с = 0,2145 нм, z = 6, пространственная группа R3). В расплавленном состоянии коричнево-красный; уравнение температурной зависимости давления пара: 1g р (мм рт. ст.) = 30,148—4897/Т + 7,0 1g Г; при хранении на воздухе постепенно окисляется; растворим в воде с разложением, этаноле, диэтиловом эфире, бензоле, ксилоле, сероуглероде. С иодидами калия, рубидия и цезия образует иодарсенаты (III) М-фАзД?].
Трииодид мышьяка получают действием раствора иода в диэтиловом эфире и сероуглероде на мышьяк, а также осаждением его иодидом калия из соляно-кислого раствора трихлорида мышьяка.
Трииодид мышьяка применяют в процессе синтеза полупроводниковых соединений мышьяка.
Все тригалогениды мышьяка в парообразном состоянии мономер-ны. Их молекулы имеют строение тригональной пирамиды с атомом мышьяка в вершине. При их окислении или взаимодействии с As2O3 образуются малоустойчивые оксигалогениды AsOX, а гидратированные окси галоген иды получают при неполном гидролизе тригалогенидов мышьяка.
В процессе сплавления тригалогенидов с халькогенидами мышьяка получают халькогенгалогениды типа АзЭХ. Это вещества— красно-коричневого или черного цвета, в большинстве легко образующие стекла.
При переходе от хлора к иоду температура плавления As3X повышается. Они устойчивы на воздухе и применяются в качестве компонентов халькогенидных стекол и стеклообразных полупроводников.
1.8. ГИДРИД МЫШЬЯКА
Гидрид мышьяка (гидрид, мышьяковистый водород) AsH3, газ без цвета и запаха (имеет чесночный запах, обусловленный продуктами его частичного окисления). Молекула гидрида мышьяка пирамидальная с атомом мышьяка в вершине; углы HAsH 92,08°, длина связи AsH 0,1511 нм. Температура плавления гидрида -117° С, кипения -62,5° С; плотность газа 3,5 г/л; жидкого 1,766 г/см3 при -И 1,8° С и 1,64 г/см3 при -64,3° С; С° = 38,7 Дж/(молыК);
41
АН°п = 1,2 кДж/моль; AH,“n = 16,7 кДж/моль; АЯ^р = -66,4 кДж/моль;
= 223,0 Дж/(моль-К); уравнения температурной зависимости: давления пара (мм рт. ст.) над твердым веществом 1g р = = 6265,9г1—260,99 + 98,51 1g Г + 39,6 • 1О'3Д141,6—159,8 К), над жидкостью 1g р = 6640,56г1—388,57 + 171,53 1g Г—183,39-10’3Д 166,5—210,7 К), вязкости т| = 0,4984-10'7Г,’°15 Па-с (213—273 К). Растворимость при 25° С (см3/см3): в воде — 0,28, ацетоне — 9,3, хлороформе—10,9, нитробензоле — 11,72, вазелиновом масле — 22,7.
Гидрид мышьяка, будучи неустойчивым соединением, медленно разлагается на элементы при комнатной температуре, быстро и нацело при 500° С:
2AsH3 —2As + ЗН2
Термическое разложение гидрата мышьяка с осаждением мышьяка на горячей поверхности в виде зеркала используют для обнаружения мышьяка даже в незначительных количествах (проба Марша). При обычной температуре гидрид мышьяка не окисляется кислородом. С элементным хлором взаимодействует с воспламенением уже при 196° С, с бромом и иодом — при комнатной температуре с образованием соответствующих галогенидов:
2AsH3 + ЗС12 = 2AsCl3 + ЗН2
2AsH3 + 3Br2 = 2AsBr3 + ЗН2
2AsH3 + 3I2 = 2AsI3 + ЗН2
Реагируя с серой, образует сульфид водорода и мышьяка по схеме
2AsH3 + 3S = 2As + 3H2S
С сухим хлоридом водорода и разбавленной хлороводородной кислотой не реагирует, с концентрированной хлороводородной кислотой образует трихлорид мышьяка с выделением водорода:
AsH3 + ЗНС1 = AsCl3 + ЗН2
При действии концентрированной серной кислоты гидриды мышьяка переходят в металлический мышьяк и высшие арсины. Окислители в водных растворах окисляют гидриды мышьяка до сесквиоксида, а в растворах солей никеля и кобальта образуются арсениды этих металлов. Щелочные металлы в аммиачном растворе образуют с гидридами мышьяка металларсины, например NaAsH2 и Na2AsH. 42
Выделены также аммиакаты, например NaAsH2-2NI-h и LiAsH2-4NH3. Металларсины с метилхлоридом дают моно- и диметиларсины.
Гидрид мышьяка получают кислым гидролизом арсенидов магния, цинка и других, восстановлением соединений мышьяка водородом, взаимодействием галогенидов или других соединений мышьяка с Li[A1H4], Na[BH4] или другими гидридами, например:
AsCh + Li[AlH4] + HCI AsH-, + LiCl + A1C1,
NaAsO2 + Ma[BH4] + HCI ?№-» AsH, + NaBO2 + NaCI + H2
Гидрид мышьяка применяют для легирования полупроводниковых материалов, а также для получения мышьяка высокой чистоты.
ГЛАВА 2
СЕРА И ЕЕ СОЕДИНЕНИЯ
2.1. ЭЛЕМЕНТНАЯ СЕРА
Свойства и применение серы. Сера —элемент VI группы Периодической системы, атомный номер 16, атомная масса 32,066. Конфигурация внешней электронной оболочки атома 3s23p\ Наиболее характерные степени окисления -2, +4, +6. Энергии ионизации при последовательном переходе от S0 к S6+равны соответственно 10,3601; 23,35; 34,8, 47,29 и 88,0 эВ. Атомный радиус 0,104 нм.
Основной характеристикой атома серы, существенно определяющей особенности процессов образования, типы химической связи и физико-химические свойства сульфидных фаз, является его акцепторная способность, вызванная стремлением к достройке оболочки до конфигурации s2pb, присущей инертным газам и отвечающей минимальной энергии. Эта особенность атома серы обусловливает значительную долю ионной связи Me—S во многих сульфидах, а также образование атомами серы ковалентно связанных групп S,„ в частности определяет склонность к образованию полисульфидных фаз.
Сера — довольно распространенный элемент, среднее содержание ее в земной коре 0,05% (масс.), в воде морей и океанов — 0,09%. Она встречается в виде самородной серы, пирита FeS2, антимонита SbiS^, галенита PbS, киновари HgS, сфалерита и вюрцита ZnS, кове-лина CuS, аринтита Ag2S, барита BaSO4, ангидрита CaSO4, гипса CaSO4-2H2O. Кроме того, сера присутствует в виде соединений в углях, сланцах, нефти, природных газах. В природе сера встречается в виде четырех изотопов: 32S, 33S, 34S, 36S.
Серу добывают более чем в 50 странах мира. Чаще ее производят в элементном виде. Около половины элементной серы производят из самородных руд, а остальную часть — из газов переработки нефти, сульфитных руд и природных серосодержащих газов. Основными производителями серы являются США, Канада, Польша, Мексика, Ирак и Франция. Серу вырабатывают на Ченур-Кояшском серном руднике в Крыму, на Каракумских серных заводах, на руднике Шор-Су, на Куйбышевском и Гаурдакском серных предприятиях. Огромные залежи серы открыты в Предкарпатском сероносном 44
Рис. 2.1. Различные структурные модификации
а — S7 б — ромбическая S«. в — S|(l, г— S|2
бассейне. Намечается развитие сероносного района Курильских островов и Камчатки.
Элементная сера обладает способностью образовывать большое число различных аллотропных модификаций. Это объясняется высокой способностью атомов серы соединяться друг с другом с образованием кольцевых или цепных молекул. Наиболее стабильной температуры 95,39° С является ромбическая a-сера. Элементарная ячейка ее кристаллов состоит из 16 мо
лекул циклооктасеры Sg. Природная сера практически полностью состоит из a-серы. Выше 95,39° С ромбическая a-сера переходит в моноклинную P-модификацию. Элементарная ячейка кристаллов P-серы состоит из шести молекул Sg. Как и все кристаллы моноклинной сингонии, кристаллы p-серы растут преимущественно в одном направлении и имеют игольчатую форму. Третьей кристаллической модификацией циклооктасеры является моноклинная у-се-ра Ее элементарная ячейка состоит из четырех молекул Sg.
На рис. 2.1 изображены различные структурные модификации серы S7, Sg, Sio и S|2 Известно, что кристаллическая структура а- и p-серы построена из неплоских восьмичисленных циклических молекул в виде короны и отличается взаимной ориентацией этих молекул в кристаллической решетке. Длина связи в молекуле серы составляет 0,25 нм, валентный угол —108°.
Сера легко образует циклические молекулы с разным числом атомов п Для циклических молекул найдены следующие значения энергии связи S—S:
п 456789 10 11
£, кДж/моль 207.9 238,2 257,3 255,7 262,0 295,5 256,7 259
В настоящее время получены метастабильные модификации от $6 до Siq. Свойства различных аллотропных форм серы приведены в табл. 2.1.
45
При быстром охлаждении бензольного раствора серы получена так называемая «перламутровая сера», содержащая в структуре цикла S8, отличающаяся по конфигурации от молекул а- и Р-серы S8. Известна ромбоэдрическая сера, образованная молекулами S6 (конфигурация в виде кресла) с расстоянием S—S, равным 2,06А, и валентным углом 102°.
В высокосимметричной молекуле S|2 (длина связи S—S 2,05А, валентный угол 106,5°) шесть атомов кольца (а) компланарны, три атома (Ь) лежат выше, а три (с) — ниже плоскости шестиугольника.
Все промежуточные формы S„ (и = = 3—20) переходят в ромбическую модификацию циклооктасеры через стадию полимерной серы. Образование полимерных групп S„ возможно в результате приобретения одним атомом серы конфигурации s2p6, а другим — sp\
s2pA + s2pA> sp' + s2p6
с непрерывным обменом конфигурациями между атомами и осуществлением связи между ними парой переходящих от атома к атому электронов.
При нагревании выше 120° С (рис. 2.2) сера переходит в жидкое состояние. При 159,4° С она имеет наименьшую вязкость. При 159,4° С скачкообразно происходит полимеризация и циклические молекулы превращаются в полимерные цепи. Вязкость резко увеличивается. Повышение вязкости происходит из-за возрастания цепи полимера. При 187° С расплав темно-коричневого цвета практически не текуч. При нагревании выше 187° С цепи разрываются, укорачиваются и жидкость вновь
46
становится подвижной. Полимер серы разлагается с соответствующим изменением энтальпии:
S„ —> S„_8 + 8Ж; Д//= -16,8 кДж; S„ —> S„_6 + 8Ж; ДЯ = 21 кДж; S„->S„_2 + 8Ж; ЛЯ = 96,6 кДж.
Поскольку расплав содержит преимущественно молекулы Sg, можно говорить о выделении большого количества теплоты при разложении полимера, которое приводит к «взрыву» внутри капли серы и выбросу горячих паров серы, что отмечается на дериватограмме нагревания серы значительным экзотермическим эффектом. Расплавы элементной серы содержат практически все аллотропные формы. Ниже приведен усредненный состав серы (в %) при различных температурах:
У, °C ц-S п-S X-S (Sg) Тч °C ц-S л-S X-S (Sg)
12 0,1 3.6 96,3 190 28,6 6,3 65,1
140 1,3 5,0 93,7 220 32,2 5,3 62,7
170 13.3 5,8 80,9 445 36,9 4,0 59,1
Диаграмма состояния серы представлена на рис. 2.3.
Расплав серы вблизи тройной точки Н состоит в основном из восьмиатомных циклических коронообразных молекул Х-S, природа которой окончательно не установлена. Согласно Кребсу, л—S-смесь циклических молекул с числом атомов от 6 до 12.
В расплаве серы протекает обратимая реакция взаимоперехода аллотропных форм серы X—8^±лр—S, а в твердой фазе — необратимая реакция л—S —> X—S. Выливая расплав серы в холодную воду,
1’пс. 2.2. Дериватограмма цементной серы
Рис. 2.3. Диаграмма состояния элементной серы в координатах давление — температура
47
получают аморфную пластическую серу. Эти формы состоят из длинных, нерегулярно расположенных зигзагообразных цепей. При 20—95° С аморфная сера превращается в a-серу, при 96—100° С — в P-серу. Кроме описанных модификаций серы известны также co-S — гексагональная, p-S — ромбоэдрическая, 5-S — моноклинная и др. Однако все они термодинамически неустойчивы.
Сера способна возгоняться. Уже при температуре 7° С создается заметное давление ее паров над твердой серой. При 350° С наблюдается резкое увеличение давления паров серы, а при 444,6° С сера закипает; АНИСП = 9,2 кДж/моль.
Пар содержит молекулы S„, где п = 2—12. При 150° С в парах наблюдают молекулы S8, 8б и S7; при 444,6° С — S8, S7, S2; при 700° С — S8, S6, S7, S7, S7; выше 730° С преобладают молекулы S2, выше 1500° С — одноатомная сера. Масс-спектрометрическое исследование показывает, что сера в ромбоэдрической модификации испаряется в виде Se, а при температуре выше точки кипения — в виде S8.
Сера не растворяется в воде, плохо растворима в этаноле, гептане, гексане, лучше — в толуоле, бензоле (рис. 2.4, а) и сероуглероде (рис. 2.4, б). Плотность серы при различных температурах приведена на рис. 2.5, а изменение вязкости при различных температурах — на рис 2.6. Лучшими растворителями серы являются жидкий аммиак (под давлением), хлорид серы (S2C12), которые широко применяются в промышленности в качестве растворителей серы.
Расплавленная сера при 120° С окисляется концентрированной серной кислотой до диоксида серы, а смесью хлороводородной кис-
48
лоты с окислителями (КСЮз, HNO3) до H2SO4. Разбавленная азотная кислота, хлороводородная кислота без окислителей, H2SO4 с серой на холоде не реагируют. Дисперсная сера в процессе нагревания с водными растворами гидроксидов взаимодействует с последними, образуя при этом соответствующие сульфиты и сульфиды, а затем и полисульфиды и тиосульфаты. Она непосредственно соединяется со всеми элементами, кроме инертных газов, иода, азота, платины и золота. При 280° С сера горит в кислороде, а при 360° С — в воздухе, образуя в основном диоксид, а также триоксид серы. Смесь паров серы и кислорода взрывается.
С галогенами (кроме иода) сера образует галогениды серы, с водородом — сульфид водорода, сульфаны, с углеродом — сероуглерод, с металлами и более электроположительными, чем сера, неметаллами— сульфиды, с селеном — фазу с широкой областью гомогенности (50—60% (ат.) Se), с теллуром при сверхвысоких давлен иях — Te7S 10.
Известны нитриды серы: циклический S4N4, который в процессе нагревания над металлическим серебром превращается в соединение, молекулы которого представляют плоские циклы S2N2. (SN)X, образующийся в процессе полимеризации S2N2 — золотистое вещество с металлической проводимостью, вблизи ОК — сверхпроводник. Изве-
Рнс 2.5. Плотность расплавленной серы при различных температурах
Рис. 2.6. Вязкость расплавленной серы при различных температурах (при 160° С 30,2 спз, при 162° С 228 спз, при 165° С 538 спз, при 170° С 15 900 спз)
49
стны также соединения, содержащие серу в катионной форме: Sjh, S 2qT и др. Циклические молекулы S,, могут быть лигандами, как, например, в соединении K2[Pt(S5)3]-
Основным потребителем серы является сернокислотная промышленность. Элементная сера практически вытеснила пирит и другие виды сырья. Широко используется сера в целлюлозно-бумажной промышленности, для производства искусственных волокон, сероуглерода, красителей. В сельском хозяйстве сера применяется в качестве фунгицида для борьбы с вредителями хлопчатника, картофеля, винограда. Сера является сырьем для производства полисульфидов. В последнее время она широко применяется для получения композиционных материалов, придавая им износоустойчивость, коррозионностойкость, механическую прочность и термостойкость.
Способы получения элементной серы. Методы извлечения серы из руд делятся на две группы: геотехнологические и наземные.
Выбор метода получения природной серы зависит от химического состава сырья, условий его залегания в земной коре и экономичности метода.
Сущность геотехнологических методов заключается в переводе серы в расплав (или раствор) с последующим подъемом его на поверхность сжатым воздухом. Впервые метод подземной выплавки серы был предложен в 1891 г. Г. Фрашем. Серу плавят при 113—И 9° С нагнетанием в серный пласт перегретой воды и с помощью сжатого воздуха поднимают наверх, где она собирается в емкости и подается на фильтр для очистки от минеральных примесей. В настоящее время разработаны новые технологии выплавки серы: высокочастотная (сера плавится от теплоты, выделяемой переменным электрическим полем высокой частоты в пласте), подземная газификация серы (при использовании теплоты, выделяющейся от сжигания серы непосредственно в пласте). Геотехнологические методы достаточно экономичны при определенных условиях залегания серы в земной толще (водонепроницаемость пород кровли и подошвы, достаточная мощность рудного пласта, определенные структурные особенности серы и др.).
Наземные методы. Предполагают карьерную добычу серной руды с последующей ее переработкой. Наземные методы делятся на прямые (непосредственно из руды) и комбинированные (с предварительным обогащением исходного сырья). Из всех промышленных методов переработки серных руд комбинированные методы наиболее эффективны.
Термический метод, заключающийся в выплавке серной руды и возгонке серы в печах или автоклавах, ввиду значительной коррозии и аппаратурных трудное!ей в настоящее время не применяется. 50
Методы извлечения серы из руд
Производство природной серы
Геотехнологические. Непосредственно на месте залегания руды Наземные. Из предварительно добытой руды
Подземная выплавка серы перегретой водой (метод ПВС) Прямые методы
—| Термический [
Подземная выплавка серы токами высокой частоты —| Экстракционный |
Z
Подземная газификация серы Комбинированные методы
Из обезвоженных концентратов (10-16% Н2О)
Подземная возгонка, экстракция серы
1
Из сгущенных концентратов (50-60% Н2О) Фильтрационный
|
Флотационноавтоклавный Метод фазового обмена
Экстракционный способ. Экстракционные способы основаны на высокой растворимости серы в различных растворителях. Измельченную руду обрабатывают растворителем и затем из раствора выделяют серу различными способами. В качестве растворителей используют сероуглерод, нефтепродукты, ароматические углеводороды, хлорпроизводные алифатических углеводородов и др.
Высокая растворимость серы в сероуглероде предполагает высокое качество извлекаемой серы, однако ввиду взрыво-, пожароопасности и 51
токсичности метод не находит широкого применения. Использование нефтепродуктов (например, керосина) в качестве растворителя определяется высоким извлечением серы из руды и дешевизной растворителя, однако огне- и взрывоопасность производства также ограничивают использование таких растворителей.
Из ароматических органических соединений для экстракции серы чаще всего используют бензол и его хлорпроизводные: толуол, ксилол, нафталин, но и они не нашли широкого применения по причине пожароопасности и дороговизны. Экстракция серы хлорпроизводными алифатических углеводородов в последнее время привлекает внимание как отечественных, так и зарубежных специалистов ввиду пожаро- и взрывобезопасности процесса. Известны и широко применяются методы извлечения серы из руды экстракцией ее тетрахлорэтиленом, тетрахлорметаном, трихлорметаном и др. Указанные растворители позволяют извлекать серу высокой чистоты из серных руд с низким ее содержанием (8—15,6%).
Известен способ получения серы экстракцией водными растворами сульфидов. Переход серы в раствор при этом происходит не за счет физического растворения, а в результате образования полисульфидов, которые при нагревании разлагаются, выделяя серу:
Na2S + S„ ^±Na2S„+I
Na2S„ > । + 2HC1 = (и + 1)S + 2NaCI
В основе процесса экстракции серы раствором сульфида аммония лежат реакции:
(NH()2S + (х- 1)S = (NH4)2Sx
(NH4)2S —<-> (х - 1 )S + 2NH3 + H2S
2NH-) + H2S -> (NH4)2S
Несмотря на высокую степень извлечения серы, доступность и дешевизну экстрагента, метод не нашел повсеместного применения ввиду токсичности сульфида водорода и аммиака, а также низкой скорости и периодичности процесса.
Комбинированные методы. Широкое применение нашли комбинированные методы, включающие флотационное обогащение серной руды и последующее извлечение серы из полученного кон центрата.
При флотационно-автоклавном методе (рис. 2.7) нагревание сгущенного концентрата острым паром производится в автоклавах (рис. 2.8). Серный концентрат, содержащий 40% серы, насосами подается в сгуститель 2, где происходит сгущение суспензии, и далее в виде суспензии (43—45% твердого) поступает в автоклав 7. Туда же че-52
Рнс. 2.7. Схема переработки флотационного концентрата автоклавным способом
реагентов
рез дозировочные бачки 19, 20 и емкость 21 поступают реагенты: растворы полифосфата натрия и кальцинированной соды и керосин. Нагревание острым паром под давлением 0,5—0,66 МПа (температура 210—220° С) производится в течение 1—1,5 ч до избыточного давления в автоклаве 0,28—0,32 МПа и температуры 140° С. При этом происходит нагрев массы, расплавление серы, ее коалесценция, отделение серы от пустой породы, которая всплывает в верхнюю часть автоклава, а сера плотностью 1,78 г/см3 оседает в нижней части. Процесс разделения серы и пустой породы осуществляется введением реагентов (Na5P3Oio + ИагССЬ, керосин), назначение которых сводится к изменению избирательной смачиваемости минералов пустой породы и повышению критического влагосодержания. Серу отстаивают 2—5 мин и выпускают через серные иглы в отстойник. В автоклав вновь подается пар для перемешивания «хвостов», которые поступают в сгуститель 11 и после сгущения отправляются на перефлотацию. Продолжительность процесса 2,7 ч. Степень извлечения серы составляет 70—80%. Несмотря на недостатки (периодичность процесса, большой расход дорогостоящих реагентов и др.), флотационно-автоклавный метод в настоящее время является основным в производстве серы.
53
Пульпа серного концентрата
плааок
Рис. 2.8. Автоклав
верхний люк; J — опорная лапа 3 — корпус.
•футеровка 5 — нижний люк; 6 — люкн для КИП 7 — предохранительный клапан
из газов. Кроме перечисленных
Переработка обезвоженного серного концентрата (10—16% Н2О) осуществляется методами фильтрации и фазового обмена, что позволяет существенно снизить энергозатраты и повысить степень отделения серы от породы.
Метод фильтрации включает плавление обезвоженного серного концентрата в открытых плавильниках при 130—150° С и отделение жидкой серы на фильтрах или центрифугах.
Метод фазового обмена заключается в плавлении обезвоженного серного концентрата в плавильниках при 125—135° С с последующей обработкой 30%-ным водным раствором хлоридов магния или кальция. Пустая порода флокулируется в гранулы (размером 3—6 мм) и отделяется на центрифуге.
Комбинированные методы обеспечивают высокое качество серы (99,9%), хотя имеется существенный недостаток — высокое содержание серы (30—40%) в отходах производства.
Получение серы методов извлечения серы из сер
ных руд возможно получать так называемую «попутную» серу. Все природные серосодержащие вещества в земной коре и морской воде в зависимости от степени окисления серы можно разделить на соединения сульфидной (сульфиды металлов, в основном железа, природные газы, нефть) и сульфатной природы (гипс, ангидрит, мирабилит и др.).
Технологический процесс производства серы из сероводородсодержащих газов включает две стадии: очистка газов от вредных примесей (извлечение сероводорода) и переработка очищенного газа в серу. Для извлечения сероводорода из газов используют сорбенты — активный уголь, силикагель, цеолиты (сухие методы) или абсорбенты — растворы этаноламинов, карбонатов и фосфатов натрия и калия, аммиака (мокрые методы). Все методы подразделяются на физические, физико-химические и химические, или окислительные. При 54
сухих физических методах достигается высокая степень очистки, однако они громоздки, требуют большого количества сорбента и применяются для очистки газов с низким содержанием сульфида водорода (до 0,5%). В промышленности применяют в основном физико-химические и окислительные методы.
Физико-химические методы основаны на обратимости процессов хемосорбции сероводорода из газов различными химическими веществами (или их растворами) и включают физическое поглощение H2S из газа.
Промышленные схемы очистки газов от сульфида водорода водными растворами этаноламинов могут быть одно- и двухступенчатыми.
По одноступенчатой схеме газ подается в абсорбер, орошаемый 15%-ным водным раствором моноэтаноламина. Взаимодействие абсорбентов с сероводородсодержащими газами описывается следующими уравнениями:
2RNH2 + H2S (RNH3)2S
2R2NH + H2S (R2NH2)2S
(RNH,)2S + H2S <2 2(RNH3)HS
(R2NH2)2S + H2S # 2(R2NH2)HS
где R = —CH2—CH2—OH.
Отработанный раствор, содержащий сульфид водорода, направляется в теплообменник, где подогревается до 100° С, и далее в регенератор. Регенерированный раствор охлаждается до 25—40° С и вновь направляется на поглощение сульфида водорода, а выделившийся сульфид водорода — на получение серы.
На рис. 2.9 приведена схема двухступенчатой очистки природных газов от сероводорода моноэтаноламиновым методом. Природный газ, очищаемый от сульфида водорода и СО2, поступает в абсорбер 1 первой ступени, где промывается 18—25%-ным раствором моноэтаноламина. Насыщенный сульфидом водорода раствор направляется в экспандер 2, где при снижении давления из раствора выделяются углеводородные газы. После теплообменника 5 нагретый до 100—105° С раствор подается в отгонную колонну 4 первой ступени, где его регенерируют глухим паром. Регенерированный и охлажденный до 20—25° С раствор возвращается в абсорбер 1.
Газ из абсорбера 1 поступает в абсорбер 5, в котором осуществляется вторая ступень очистки 7—10%-ным раствором моноэтаноламина. Регенерация использованного раствора происходит аналогично 55
Рис. 2.9. Схема двухступенчатой газовой очистки природных газов от сульфида водорода моноэтаноламииовым методом
регенерации в первой ступени при 120—125° С, что позволяет провести более глубокую регенерацию.
К достоинствам этого метода можно отнести доступность, высокую реакционную способность, стабильность и дешевизну этаноламинов; к недостаткам — энергоемкость процесса, необходимость дальнейшей переработки сероводорода в серу и коррозию оборудования.
Для получения серы из газов используется также вакуумкарбо-натный метод, когда в качестве абсорбента применяется 12—15%-ный раствор карбонатов калия или натрия:
К2СО, + H2S # КНСО3 + KHS
Но ввиду невысокой степени очистки этот метод применяется лишь для грубой очистки небольших объемов газа.
Для дальнейшей переработки полученного сульфида водорода в элементную серу применяется процесс Клауса (рис. 2.10).
Сульфид водорода из установки сероочистки разделяется на два потока. Большая его часть (82%) поступает в топку реактора-генератора 1, где окисляется при 900—1300° С кислородом воздуха:
2H2S + О2 = 2S + 2Н2О
2H2S + ЗО2 = 2SO2 + 2Н2О
В котловой части реактора 1 продукты горения охлаждаются до 300° С, при этом образуется водяной пар с р = 1,3 МПа. Далее в конденсаторе-генераторе 2 первой ступени газы охлаждаются до 155—160° С, и пары серы полностью конденсируются. Образующаяся сера стекает в хранилище, а газовая фаза направляется в камеру смешения подогревателя 3 первой ступени. В топку подогревателя 3 56
Рис. 2.10. Схема получения элементной серы методом Клауса / — реактор-генератор. 2, 5 — конденсаторы-генераторы, 3, 6 — подогреватели 4, 7 — конверторы 8 — конденсатор-экономайзер 9 — сероуловитель, 10 — печь дожита. //—дымовая труба
подается вторая часть газа (10%) и воздух. Газовая смесь нагревается до 280° С и поступает в конвертор 4 первой ступени, заполненный бокситовым катализатором, где протекает следующая реакция:
О2 + 2H2S = 2S + 2Н2О
Образующиеся пары серы в виде молекул Sg и S6 конденсируются в конденсаторе-генераторе 5 второй ступени при 155—165° С, и жидкая сера стекает в хранилище, а газовая фаза подается в конвертор 7 второй ступени.
В топку подогревателя 6 подают оставшееся количество газа (8%). В конденсаторе-экономайзере 8 газы из конвертора 7 охлаждаются до 140° С, сера конденсируется и стекает в хранилище, а газовая смесь поступает в печь дожита 10, где при 600° С полностью окисляется до диоксида серы. Из печи дожита 10 через трубу 11 газы выбрасывают в атмосферу.
Получение серы из газов методом Клауса применяется лишь при высоком содержании H2S в газе (более 30%). При низком содержании H2S количества выделяемой теплоты недостаточно для ведения автотермичного сжигания сероводорода.
Недостатками метода являются многостадийность процесса, энергоемкость и наличие выхлопных газов, содержащих до 1% сернистых соединений.
Исследован процесс прямого неполного каталитического окисления сульфида водорода кислородом с использованием в качестве катализаторов синтетических цеолитов и боксита. Установлено, что для газов, содержащих менее 5% H2S, необходимо одноступенчатое окисление. Газы, содержащие более 5% H2S, целесообразно подвергать двухступенчатому окислению. Газы смешиваются со стехиометрическим количеством воздуха Xi и подаются в реактор с катализатором. При этом образуется сера, диоксид серы и вода. На выходе из реактора газ разделяется на два потока. Первый поток поступает во вто-
57
рой контактный аппарат, а второй поток — на конденсацию серы в холодильник-конденсатор, после чего также поступает в реактор второй ступени, где оставшийся сульфид водорода доокисляется при 350° С. В реактор дополнительно подается воздух в количестве Х2. Степень превращения сульфида водорода в серу достигает 99%, если соотношение Х2/Х, = 0,3-ь0,35.
При мышьяково-содовом способе получения серы из газов в качестве абсорбента применяют раствор оксидно-мышьяково-натриевой соли Na4As2S5O2. Свежий поглотительный раствор получают при взаимодействии оксида мышьяка с раствором карбоната натрия:
2Na2CO3 + As2O3 + Н2О 2Na2HAsO3 + 2СО2
Затем раствор обрабатывают сульфидом водорода и кислородом:
2Na2HAsO3 + 5H2S Na4As2S5 + 6Н2О
Na4As2S5 + О2 Na4As2S5O2
Очищаемый от сульфида водорода газ промывают в скрубберах поглотительным раствором:
Na4As2S5O2 + H2S Na4As2S6O + H2S
Na4As2S6O + H2S ^±Na4As2S7 + H2O
В регенераторах раствор продувается сжатым воздухом, в результате чего тиоарсенат натрия окисляется с выделением серы:
2Na4As2S7 + О2 2Na4As2S6O + 2S
2Na4As2S6O + О2 2Na4As2S5O2 + 2S
Степень извлечения серы этим способом составляет 90—98%. Полученная сера содержит в качестве примеси мышьяк и ее можно применять как фунгицид.
Предварительной обработкой растворов диоксидом серы при мышьяково-содовой очистке до содержания свободного диоксида серы в нем 1—2 г/л можно получить чистую серу без примесей мышьяка. На процесс поглощения H2S и регенерацию раствора сильно влияет величина pH, которую поддерживают в пределах 7,8—7,9.
К недостаткам метода относят высокий расход карбоната натрия (400—500 кг на 1 т серы), наличие токсичных веществ в растворе, чувствительность процесса к ряду примесей.
58
Указанные недостатки отсутствуют в щелочно-гидрохиноновом методе очистки газов от сульфида водорода, окислителями которого являются хинон и его производные. Метод применяется при очистке газов с большим содержанием кислорода (до 21%).
Метод включает три основные стадии. На первой стадии происходит хемосорбция H2S из газа:
Ма2СО3 + H2S # NaHS + NaHCO3
На второй стадии гидросульфид натрия вступает в реакцию с хиноном:
NaHS+NaHCO3 + O
О ->Na2CO3 + HO
>—OH+S
При этом регенерируется карбонат натрия, восстанавливается катализатор и выделяется элементная сера.
Третья стадия — регенерация поглотительного раствора
НО
ОН+Ог -► О
О+Н2О
и отделение полученной серы. Происходит флотация серы, которая в виде пены собирается в верхнем слое раствора и после фильтрации направляется на дальнейшую переработку. Часть полученного на первой стадии гидросульфида натрия окисляется до тиосульфата натрия:
2NaHS + 2О2 = Na2S2O3 + Н2О
Часть поглотительного раствора ввиду загрязнения его тиосульфатом натрия сбрасывается в стоки и заменяется новым.
Одним из первых процессов, в котором был использован гидрохинон, является метод «Перокс», где в качестве абсорбента применяют водный раствор аммиака с 0,3% гидрохинона.
Дальнейшим развитием метода «Перокс» явился метод «Стред-форд», где промышленные газы промываются водным щелочным раствором натриевой соли антрахинон 2,6-дисульфокислоты (АДК) с добавлением соединений металлов переменной валентности, например ванадата натрия:
2NaVO3 + 2NaHS + Н2О = Na2V4O9 + 2S + 4NaOH
Совместное применение АДК с ванадатом натрия позволяет в 40—50 раз ускорить реакцию окисления адсорбированного H2S, снижает скорость образования тиосульфата натрия.
59
В Японии разработан метод «Такахакс», заключающийся в использовании в качестве поглотительного раствора натриевой соли 1,4-нафтахинон-2-сульфокислоты. При этом происходят следующие реакции:
H2S + NH4OH = NH4HS + Н2О
H2S + Na2CO3 = NaHS + NaHCO,
Хинон регенерируют продувкой кислородом:
ОН О
Известен так называемый «горячий поташный метод», основанный на поглощении сульфида водорода 25—35%-ным раствором карбоната калия при 107—116° С.
Методы извлечения серы из серосодержащих газов. Технология извлечения серы из концентрированных серосодержащих газов [>7% (об.) SO2] основана на восстановлении серы различными восстановителями (С, СО, СН4, Н2, H2S и др.). Процесс проводят либо при высокой температуре (1100—1300° С), либо в присутствии катализаторов (сульфиды или оксиды железа, активированный глинозем, силикагель и др.). 60
12
Рис. 2.11. Схема получения элементной серы из концентрированных сернистых газов
1—дозатор. 2— печной агрегат, 3 — котел-утилизатор, 4— электрофильтр 5 // — теплообменники 6 8—контактные аппараты 7 — котел низкого давления, 9 — скруббер, 10 — сборник
12 — центробежный насос
На рис. 2.11 приведена схема процесса получения элементной серы из концентрированных серосодержащих газов, где в качестве восстановителя используется бензин (фирма «Оутокумпу», Финляндия). Печной агрегат для взвешенной плавки 2 имеет три отделения: плавильную шахту, отстойник и выходную шахту — аптейк. Сухой пиритный концентрат плавится при температуре 1250° С при сжигании мазута. Воздух для сжигания мазута подается в строго дозированных количествах. Твердый остаток мазута после горения (штейн) накапливается в отстойнике печи. Газы проходят над штейном и содержат 1,4—12,8% (об.) SO2 перед входом в аптейк, в котором поддерживается температура 1350° С и за счет впрыскивания бензина восстановительная атмосфера. Происходит восстановление гидроксида серы до серы. Газовая фаза охлаждается в утилизаторе 3 и электрофильтре 4. Далее в контактном аппарате 6 (узел Клауса), заполненном катализатором на основе глинозема, при 450° С протекают следующие реакции:
COS + Н2О (пар) = H2S + СО2
2COS + SO2 = 3S + 2СО2
После охлаждения в котле 7 газ направляют во второй контактный аппарат 8, где при 200—250° С диоксид серы взаимодействует с сульфидом водорода с образованием серы и воды. Выхлопные газы после обезвреживания выпускают в атмосферу. В качестве восстановителя в последнее время широко применяется природный газ. Восстановление диоксида серы идет по уравнению
3SO? + 2СН4 = 2СО2 + 2Н2О + 2H2S + S
61
Рис. 2.12. Окислительная печь Клауса для регенерации элементной серы-
/ — горелка: 2— скоба 3 — люк. 4—крышка, 5 — футеровка 6 — кожух. 7—газоход. 8— сифон для слива элементной серы 9 колосниковая решетка; 10—катализатор
После конденсации образующихся паров серы сернистые соединения газовой фазы перерабатываются в серу на установке Клауса (рис. 2.12). Использование в качестве катализаторов дунита, мучайско-го боксита, гипса, активного глинозема позволяет снизить температуру восстановления SO2 метаном с 1250—1300° С
до 900° С (низкотемпературный каталитический процесс). Степень превращения SO2 достигает 96—98%.
В двухстадийном комбини-
рованном процессе получения серы из кислородсодержащего (до 10% О2) газа металлургического производства частично восстановленный при 1250° С диоксид серы
подвергают каталитическому восстановлению конвертированным газом на алюмокобальтовом концентрате при 250—500° С, что позволяет значительно сократить расход восстановителей и повысить степень извлечения серы до 94—95%.
Для улавливания диоксида серы из отходящих газов их пропускают через скруббер, орошаемый плавом состава: ЫСОз—32%, Na2CO—33%, К2СОз—35% при температуре 427° С. Уловленный в виде сульфитов и сульфатов газ подается на восстановление СО и Н2 при 600° С. Полученный H2S перерабатывается до серы.
Производство серы из сернистых газов технически затруднено, поэтому вначале очищают газы от SO2 его поглощением различными растворами с последующей переработкой до SO2, серы и серной кислоты. Существует множество методов очистки газов от SO2: известняковые, водные, аммиачные, сульфит-гидросульфитные, цинковые, фосфатные, каталитические и др. Водный метод применяется в Японии и Скандинавии (поглотитель — морская вода). В США разработан комбинированный содово-электролитический способ поглощения SO2, заключающийся в поглощении диоксида серы содовым раствором, осаждении образующегося сульфата натрия и восстановлении его до серы. Этот метод отличается сложностью и дороговизной аппаратурного оформления, что ограничивает его использование. Другой фирмой США, а также НИОГАЗ предложено в качестве поглотительного раствора использовать растворы сульфит-бисульфатов щелочных и щелочноземельных мс!аллов, а в качестве восстановителей — водород, оксид углерода, сульфид водорода и др. Известен так называемый «жидкофазный про-62
цесс Клауса», когда средой для взаимодействия SO2 и H2S служат растворы кислот — серной, фосфорной, борной, органических кислот и др. При избытке H2S образуется сера:
2H2S + SO2 # 3S + 2Н2О
при избытке SO2 — тиосульфат и политионат:
4HSO; + 2H2S 3S2O3- + 2Н+ + ЗН2О
3Hso; + h2s s4o^ + зн2о
При абсорбции SO2 возможно частичное окисление сульфитов до сульфатов кислородом обжиговых газов.
На рис. 2.13 приведена технологическая схема получения элементной серы из бедных SO2 газов жидкофазным сернокислотным способом. Газ с температурой 170° С и содержанием 0,1—0,5% SO2 смешивается с сульфидом водорода в отношении H2S:SO2 = 2:1 в смесителе 1 и поступает в абсорбционную колонну 2, орошаемую 80%-ным раствором серной кислоты при температуре 135—140° С. Концентрация SO2 в газе после абсорбции составляет 0,01—0,02% и газ выбрасывают в атмосферу. Образующаяся при взаимодействии SO2 и H2S сера (процесс Клауса) поступает в декантатор 3, где собирается в нижнем слое и периодически перемешивается. Серную кислоту и золу направляют в отстойник 5, а затем — в барабанный вакуум-фильтр 6. Серную кислоту вновь возвращают на орошение абсорбированной колонны. Серу направляют потребителям в качестве товарного продукта.
В качестве поглотительных растворов возможно использование нефтяных сульфоксидов, водных растворов солей К2НРО4 и К4Р2О7 и др.
Другие методы получения серы. Получение серы из пирротиновых концентратов. Пирротиновые концентраты содержат соединение железа с серой Fe„S(rn — пирротин (магнитный колчедан). Процесс извлечения серы из пирротинового концентрата весьма сложен и состоит из
80%-ная H2SO4
Сера
Рис. 2.13. Схема получения элементной серы из газов с низким содержанием диоксида серы жидкофазным серно-кислотным способом / — смеситель, 2 — абсорбционная колонна 3 — декантатор. 4 — центробежные насосы 5 — отстойник 6—вакуум-фильтр 7, 8 — сборники этементной серы
63
нескольких стадий: выщелачивания руды, осаждения сульфидов, ав- 1 токлавной дезинтеграции, флотационного разделения и, наконец, ав- | токлавной выплавки серы. Несмотря на технологическую сложность | переработки пирротинового концентрата, метод широко применяется в промышленности, и количество получаемой серы исчисляется сотнями тысяч тонн в год. Выщелачивание руды можно производить различными методами. Известен способ сернокислотного выщелачивания:
MS + H2SO4 = MSO4 + H2S, где М = Си, Со, Ni.
Осуществляют также окислительное автоклавное выщелачивание по уравнению
MS + n(NH3) + 2О2 = M(NH3)„SO4.
Предложено получать серу выщелачиванием сульфидных руд FeCl3:
CuFeS, + FeCI3 = CuCl + 4FeCI2 + 2S
FeS + 2FeCI3 = 3FeCI2 + S
В отечественной промышленности используют метод автоклавного окислительного выщелачивания:
4Fe„S„+ I + ЗиО2 = 2«Fe2O3 + 4(и + 1 )S; MS + 2О2 = MSO4
Процесс проводят при ПО—112° С в автоклавах, снабженных мешалками. Окислителем служит кислородно-воздушная смесь (55—65% ! О2) с парциальным давлением 0,9—1,0 МПа. В автоклав вводятся по- J верхностно-активные вещества — ЦИАТИМ-209 и сульфитно-дрожже- : вая бражка. Продуктами выщелачивания являются парогазовая смесь, ! которую выбрасывают в атмосферу, и окисленная суспензия концентра- ' та, которая направляется на осаждение сульфидов. Осаждение сульфи- 1 дов металлов происходит при 90° С в результате обработки растворов металлическим железом и серой:
MSO4 + Fe + S = FeSO4 + MS
На следующей стадии — сульфидной флотации — происходит отделение основной части железа и серосодержащих соединений. Температура процесса 40° С, pH 3—4. Твердая фаза суспензии «хвостов» содержит гидратированные оксиды железа, жидкая — кислый раствор сульфата железа (II). Концентрат содержит элементную серу с сульфидами цветных металлов. Пульпа серо-сульфидного концентрата на-64
правляется на автоклавную дезинтеграцию. Процесс протекает в автоклавах при 20—30° С в присутствии гидрофилизатора — сульфида натрия, после чего серо-сульфидный концентрат подвергается серной флотации с применением флотоагентов (керосина, нефти, спиритов Си—Ci6 и др.). «Хвосты» флотации — медно-никелевый концентрат—являются конечным продуктом химического обогащения поли-минеральных руд Серный концентрат направляют на производство серы автоклавным методом. В процессе автоклавной дезинтеграции суммарные потери серы составляют 7—10% за счет растворения некоторого количества серы в щелочном растворе. Если в качестве гидрофилизатора использовать сульфид кальция или известково-серный отвар, потери серы снижаются до 1%.
Производство серы из сульфатов металлов приобретает все большую актуальность. Природные сульфаты характеризуются достаточно высоким содержанием серы (18—26%). Получение ее из сульфатов возможно путем восстановительного процесса:
1) термическое разложение сульфатов без добавок при 1500° С
2CaSO4 = 2СаО + 2SO2 + О2
2) термическое разложение в присутствии веществ (кремнезема, глинозема, оксида железа и др), понижающих температуру восстановления;
3) восстановление сульфатов различными восстановителями (оксидом углерода, сульфидом водорода, водородом, метаном и др.).
Разработана технология восстановления сульфата кальция углеродом:
CaSO4 + 2С = CaS + 2СО2
CaS + 3CaSO4 = 4СаО + 4SO2
CaS + 2SO2 = CaSO4 + 2S
За рубежом получают диоксид серы, восстанавливая CaSO4 смесью СО и Н2 при 1320° С. Предложено получать сульфид водорода двухстадийным способом: на первой стадии сульфат кальция восстанавливается до сульфида продуктами конверсии метана при 600—1000° С, на второй — образовавшийся сульфид кальция разлагается Н2О и СО2 по уравнению
CaS + Н2О + СО2 = СаСОз + H2S
Химическая технология неорганических веществ, кн 2
Повышение давления до 0,3—1,0 МПа позволяет совместить стадии и получать сульфид водорода при 400—450° С; его направляют на производство серы.
Предложен метод получения серы из сульфата кальция восстановлением его природным газом при 1000—1200° С и обработкой образующегося сульфида кальция хлоридом водорода:
CaSO4 + СН4 = CaS + СО2 + 2Н2О; CaS + 2НС1 = СаС12 + H2S
Образовавшийся сульфид водорода обрабатывают гидрохиноном до получения серы.
Известен способ производства серы восстановлением смеси природных сульфатов натрия и кальция. Восстановление природным газом проводится в шахтной печи при температуре 950° С с добавлением в шихту SiO2:
Na2SO4 + CaSO4 + 6SiO2 + 3/2CH4 = = 2S + Na2OCaO-6SiO2 + 3/2CO2 + 3H2O
При этом достигается 98,8—98,9%-ное восстановление сульфатов.
Получение серы из углей. Сера является нежелательной примесью углей, используемых в коксохимической промышленности и для газификации. В настоящее время серу получают из коксового газа, содержащего до 5—20 г/м3 H2S, либо из дымовых газов ТЭЦ, содержащих SO2. Улавливание серы из коксовых газов осуществляют вакуум-карбонатным или мышьяково-содовым способами. Улавливание SO2 из дымовых газов ТЭЦ производится путем поглощения растворами или твердыми адсорбентами (оксидами железа, аммония, марганца, карбонатами кальция, магния, алюминатом натрия и др.). В настоящее время широко используется в США метод поглощения известняком:
СаСО3 + 2SO2 + Н2О = Ca(HSO3)2 + СО2
Ca(HSO3)2 + СаСО3 = 2 CaSO3 + СО2 + Н2О
2 CaSO3 + О2 = 2 CaSO4
Известковым способом улавливают 90% SO2.
Известен способ улавливания SO2 аммиаком с получением сульфата аммония:
SO2 + 2NH, Н2О = (NH4)2SO3; 2(NH4)2SO3 + О, - 2(NH4)2SO4 66
Наиболее перспективен магнезитовый способ, при котором протекают следующие реакции:
MgO + SO2 + 6Н2О = MgSOr6H2O
2MgO + 2SO2 + O2 + 14H2O = 2(MgSO4-7H2O) MgSCh = MgO + SO2; 2MgSO4 + C = 2MgO + 2SO2 + CO2
Для обжига смеси сульфита и сульфата магния применяют вращающиеся печи с кипящим слоем. Степень очистки дымовых газов составляет 90%.
Получение серы из нефтей. Серу из нефтей получают методом гидроочистки, заключающимся в гидрировании сероорганических соединений с образованием углеводорода и сульфида водорода по уравнениям:
RSH + Н2 = RH + H2S
или
RSR + Н2 = RSH + RH; RSH + Н2 - RH + H2S
или
+ Н2 = СН3(СН2)2СН3 + H2S
Образующийся сульфид водорода удаляют из газов нефтепереработки этаноламином и направляют на получение серы по методу Клауса.
Широко используется метод газификации высокосернистого нефтяного топлива с одновременной сероочисткой горючего газа. Поглощение сульфида водорода ведут в присутствии щелочноземельных металлов, марганца или железа при 800—1000° С. При этом образуются сульфиды соответствующих металлов. Затем осуществляется регенерация отработанного реагента при 950—1000° С. Обжиговый газ, содержащий 10—13% SO2, идет на производство серной кислоты или серы, а реагент возвращается в производство. Степень улавливания H2S составляет 90—95%.
Выпускается комовая, жидкая и гранулированная сера. Комовая сера получается путем розлива жидкой серы тонким слоем на открытых площадках и дальнейшего разбивания застывшего пласта. Жидкую серу хранят в специальных резервуарах с электрическим подогревом. Она удобна для транспортировки и специальных хранилищ. Гранулированная сера (размер гранул 0,5—6 мм) обладает преиму-
ществами перед другими видами товарной серы, так как удобна и безопасна при хранении и транспортировке. Кроме перечисленных видов серы выпускаются также молотая, полимерная, чешуйчатая, пластинчатая, медицинская, особо чистая сера и сера в отливках. Молотая сера является продуктом размола комовой и используется в шинной, резинотехнической промышленности, в сельском хозяйстве. Среди препаратов серы для сельского хозяйства наибольшее применение получил 80%-ный смачивающийся порошок серы, получаемый доизмельчением молотой серы до размера частиц не более 20 мкм с последующей обработкой поверхности частиц гидрофилизаторами. Иногда используется сера (3—5 мкм), образующаяся при очистке газов от сероводорода жидкостно-окислительными методами.
Производство полимерной серы обусловлено потребностями резиновой промышленности. Существует несколько способов получения полимерной серы.
Получение серы из расплавов. Расплав серы при 160—365° С, содержащий стабилизаторы (галогены, хлориды алюминия, гексахлорпараксилол), регулирующие процесс полимеризации, быстро охлаждают до полного отверждения. Отвержденные расплавы размалывают и удаляют аллотропные формы путем экстракции растворителями (сероуглеродом, толуолом, перхлорэтиленом), после чего полимерную серу высушивают и затаривают. Изменяя температуру расплава и количество вводимого стабилизатора, можно изменять молекулярную массу целевого продукта.
Получение полимерной серы при взаимодействии SO2 с H2S сводится к тому, что газ, содержащий H2S и SO2, пропускают через реактор барботажного типа. Средой для проведения реакции служат вода или предельные спирты с добавлением минеральных кислот. Продукт отделяют от жидкой среды и выдерживают 3—14 сут до полной полимеризации. Затем размалывают и экстракцией удаляют растворимые аллотропные формы.
Получение серы охлаждением паров серы в жидких средах— тетрахлорметане, хлороформе. Образующуюся серу сушат, размалывают и очищают от растворимых аллотропных форм так же, как и в первых двух методах.
Получение чешуйчатой серы. Расплавленную серу при 135° С подают в ванну, в которой горизонтально установлен барабанный кристаллизатор, охлаждаемый изнутри водой. Кристаллизатор погружен на 5 мм в жидкую серу и вращается с частотой 8—10 об/мин. Сера, застывшая на его поверхности, срезается ножом и в виде чешуек подается на склад.
Пластинчатая сера выпускается в виде плиток толщиной до 5 мм и характеризуется высоким сопротивлением измельчению. Производство плиточной серы осуществляют путем охлаждения расплавлен-68
ной серы на специальной транспортной ленте, которая движется над открытыми баками с охлаждающей водой. При этом сера на транспортере застывает монолитным слоем и под действием собственной тяжести разламывается на пластины.
Медицинская сера используется в производстве фармацевтических и косметических препаратов. Изготовляют ее путем тонкого помола серы высокой чистоты в токе инертного газа. Сера в отливках получается заливкой расплавленной серы в специальные формы.
Сера особой чистоты содержит примесей не более 1-Ю’4—1-10'5% (масс.). Она применяется в электронной и оптической технике, для синтеза кристаллических сульфидов кадмия, галлия и др. Для получения серы особой чистоты применяются различные методы. Химический метод заключается в обработке жидкой серы смесью концентрированных азотной и серной кислот и многократной промывке серы перегретой водой, что позволяет получить серу, содержащую не более 2-10’4% (масс.) примесей. Разработаны способы очистки серы от примесей с применением хлороводородной кислоты и поверхностно-активных веществ. Такая же степень очистки достигается при использовании дистилляционных методов. Изучены теоретические основы применения метода противоточной кристаллизации для получения серы особой чистоты Исследована кинетика взаимного перехода X-S^m-S в твердой и жидкой фазах. Примеси кремния, железа, никеля, марганца, алюминия, свинца и некоторых других элементов присутствуют в виде взвешенных частиц. По методу противоточной кристаллизации возможна глубокая очистка серы от примесей до содержания металлов 2 10'4%, мышьяка <1 10'5%, органических примесей 210'6%.
Широко используются комбинированные методы, включающие противоточную кристаллизацию из расплава и дистилляцию с малой скоростью испарения. При применении этих методов содержание примесей в конечном продукте составляет 2,5-10‘4% (масс.).
Предложен метод получения кристаллической серы охлаждением расплава серы в инертной жидкости (перегретая вода) при интенсивном перемешивании. Этот способ позволяет получать чистую серу, используемую резиновой промышленностью и для производства серной кислоты.
Серосодержащие композиционные материалы. В настоящее время производство природной и вторичной серы непрерывно возрастает. Увеличивается доля попутной серы, полученной при очистке нефти, природного газа, топочных газов и других промышленных выбросов. В ряде стран (Канада, Россия, Польша) производство серы постепенно превышает ее потребление, что приводит к значительному снижению ее стоимости. Поэтому разработка технологий серосодержащих материалов и композиций является актуальной, а их производство становится экономически обоснованным.
69
Сера — активный реакционноспособный и структурообразующий элемент. Она вступает в химическое и физико-химическое взаимодействие со многими неорганическими и органическими соединениями, образуя химические соединения и композиции с новыми свойствами.
Серосодержащие композиционные материалы имеют ряд ценных механических свойств — высокую прочность, стойкость к истиранию, долговечность и т. д. Сера нерастворима в воде и неорганических кислотах (кроме HF и Н2СгО4), поэтому материалы из нее обладают водонепроницаемостью и кислотостойкостью. Перечисленные свойства серы определяют использование ее для производства коррозионно-и водостойких материалов и композиций широкого назначения.
В настоящее время определились два основных направления применения серы—для получения серных вяжущих и для производства гидроизоляционных композиций.
Серное вяжущее можно использовать вместо цемента для получения бесцементных композиций. Технология производства серного вяжущего значительно проще цементной технологии. Одним из явных преимуществ серного бетона по сравнению с цементным является быстрый набор прочности. Серный бетон достигает проектной прочности (30—70 МПа) за несколько часов при любой влажности и температуре окружающей среды. По сравнению с цементом аналогичных марок при изготовлении серного вяжущего снижаются энергозатраты в 1,5—2,5 раза, удельные капитальные затраты на организацию производства до 50%, себестоимость в 2,5—3 раза. Композиции представляют собой искусственный каменный материал, образовавшийся в результате твердения отформованной смеси. В качестве наполнителей используют андезитовую муку, кварцевый песок, отходы производства. При добавлении цветных минеральных красителей можно получать материалы различной цветовой гаммы.
Природная сера — кристаллическое вещество. При охлаждении расплава серы происходят процессы кристаллизации и перекристаллизации. Изменение плотности в процессах кристаллизации обусловливает усадочные деформации композиции за счет возникновения растягивающих напряжений. Деформация вызывает микро- и макротрещины. Для уменьшения хрупкости и устранения перечисленных недостатков в композиции вводят различные пластификаторы — тиокол-резенит, термопрен, резиновую крошку, атактический полипропилен, нафталин, дициклопентадиен и т.п.
Интересными особенностями обладают материалы, полученные при использовании полимерной серы. Полимерная сера является аморфно-кристаллическим полимером и подобно каучуку ее можно вулканизировать мышьяком, фосфором и т.д. В полимерной сере имеются спиралеобразные цепи, состоящие до 104 —106 атомов серы Такая сера не растворима в органических растворителях и обла
дает по сравнению с кристаллической серой более высокой адгезией к минеральным наполнителям и пластичностью. При твердении композиций, содержащих полимерную серу, в материале практически не возникает внутренних напряжений. Для стабилизации полимерной серы в твердом состоянии используют различные стабилизаторы структуры — галогены, красный фосфор, сосновое масло и др.
Перспективным для использования серы является получение серных композиций, содержащих высокомолекулярные модификаторы и наполнители. В качестве таковых можно использовать битум или битуминозные породы. При добавлении серы в битуминозную породу при температурах выше 200° С возможно химическое взаимодействие, образование конденсированных соединений типа асфальтенов и соответственно обогащение битума необходимыми конденсированными структурами. Происходит «сшивание» углеродных скелетов и- образование трехмерных структур. Весьма эффективно для получения серосодержащих композиционных материалов также использование отходов низкомолекулярного полиэтилена с молекулярной массой 2000—8000. Как известно, низкомолекулярный полиэтилен, полученный при повышенных температурах, имеет разветвленную структуру и значительное количество двойных связей С=С, что определяет его высокую реакционную способность. Высокие прочностные свойства и водостойкость таких композиций объясняются получением однородной структуры с более плотной упаковкой в результате химического взаимодействия серы с битумной составляющей и низкомолекулярным полиэтиленом, увеличением адгезии между молекулами вяжущего и минерального наполнителя, а также силами взаимодействия между атомами самой серы, когда увеличивается участие прочных ковалентных связей S—S.
Такие композиционные материалы могут успешно применяться в качестве облицовочных плит и гидроизоляционных материалов в химических производствах, для устройства автодорог даже в суровых климатических условиях.
2.2. ОКСИДЫ СЕРЫ
Сера образует следующие оксиды: S2O, SgO, S2O7, 8бО7, S6O, S7O2, SO2 и SO3. Кроме SO2 и SO7 остальные оксиды неустойчивы.
S2O — газ; С® = 44,3 Дж/(моль-К); АЯ°6р = -108,9 кДж/моль;
= 267,3 Дж/(моль-К). Монооксид дисеры образуется в процессе пропускания диоксида серы в тлеющем разряде.
Кристаллический SsO образуется в процессе взаимодействия полисульфида водорода (FhSu) с дихлоридом оксосеры SOCI2.
Жидкий тетраоксид серы (SO4) с температурой плавления 3° С (с разложением) получают из диоксида серы и кислорода в тлеющем разряде при -190°С.
Жидкий S2O2 с температурой плавления 0° С, ДЯ°6р = -811,7 кДж/моль получают в процессе окисления диоксида серы озоном или разложения SO4. Соединения SO4 и S2O7 содержат пероксидную группировку—О—О— Формально им соответствует пероксомоно-серная кислота (кислота Каро) H2SC>5 (температура плавления 47° С, с разложением) и пероксосерная кислота H2S20s (температура плавления 65° С, с разложением), которые образуются в процессе электролиза серной кислоты или гидросульфатов при 'Производстве пероксида водорода.
Диоксид серы — бесцветный газ с удушливым запахом, с температурой плавления -73° С, кипения -10° С. Плотность жидкого SO2 при -8° С равна 1,46 г/см\ а при 0° С и 760 мм рт. ст. 2,93 г/л . Теплота испарения (Дж/кг) составляет: при -10° С 389,65; при 0° С 380,08; при 20° С 362,54; при 30° С 353,08. Давление паров над жидким SO2 при 20° С 330,26, а при 50° С 841,13 кПа. Средняя теплоемкость газообразного SO2 в интервале 0 — 100° С равна 0,6615 Дж/кг. Молекула диоксида серы имеет форму треугольника, в котором расстояние S—О равно 1,43 нм, а угол О—S—О составляет 119°.
В молекуле SO2 между атомом серы и каждым атомом кислорода имеется одна ст-связь и делокализованная л-связь. Молекула диоксида серы имеет форму
° °
119,5°
Термодинамические характеристики SO2: ДЯ298 = -297 кДж/моль; А5298 = 248,2 Дж/(моль-К); AG298 = -300 кДж/моль.
При обычных условиях SO2 легко растворяется в воде: в 1 объеме воды растворяется 40 объемов диоксида серы с выделением теплоты в количестве 34,4 кДж/моль. При 10° С и атмосферном давлении насыщенный раствор содержит примерно 15%, а при 20° С—10% SO2. В таких растворах находится гидрат SO2-7H2O. При нагревании водных растворов выделяется весь растворенный диоксид серы. Спектральные исследования растворов показали, что значительная часть SO2 растворена в воде и лишь небольшое количест
во его содержится в виде сернистой кислоты H2SO2, которая образуется при взаимодействии SO2 с водой по схеме
SO2 + Н2О # H2SO3 <2 н+ + HSO; 2Н+ + SO,'
Молекула SO2 имеет форму пирамиды с треугольным основанием и атомом серы в вершине.
Диоксид серы может проявлять как окислительные, так и восстановительные свойства. В присутствии катализатора он окисляется до триоксида кислородом воздуха:
2SO2 + О2 = 2 SO,
В присутствии же сильных восстановителей, например сульфида водорода, восстанавливается до элементной серы:
SO2 + 2H2S = 3S + 2Н2О
Потенциал ионизации диоксида серы составляет 12,34 эВ.
Частоты колебаний диоксида серы в различных его состояниях приведены в табл. 2.2.
Жидкий диоксид серы является сильным ионизирующим растворителем. Соединения ковалентной природы растворяются в жидком SO2 лучше, чем соединения ионного типа.
Таблица 22 Частоты колебаний диоксида серы в различных состояниях
Изотопный состав Состояние SO2 Частота колебаний, см 1
V[ v2 v.
SI6O2 Газообразное 1151 518 1362
Твердое (-180° С) 1147 521 1330
Водный раствор 1157 — 1332
SI6OIXO Газообразное 1122 507 1341
S'*02 » — — 1316
Давление паров SO2, рассчитанное по уравнению
Igp = -1867,52/7-0,0158657 + 0,000015574 Г1 + 13,07540,
при различных температурах составляет:
Г, К 243 253 273 283 293
р, мм рт ст 284,8 530,6 1159,6 1714 2456
Жидкий S02 широко применяется в качестве растворителя, обладающего значительным температурным интервалом. Он является хорошим растворителем органических веществ, особенно ароматических углеводородов и олефинов. Жидкий SO2 с водой смешивается не полностью. По данным К. Виккерта, существует соединение SO2H2O, а растворимость воды в жидком диоксиде серы при 22° С составляет 2,3 г/100 г SO2.
Диоксид серы образует устойчивые сольваты со многими галогенидами щелочных металлов. В литературе известно существование таких соединений, как SbCl2-SO2, SrBr4-SO2, (TiCl4)2-SO2, A1C13 SO2. Спектроскопическими исследованиями установлено, что аммиак в жидком диоксиде серы образует соединение (NH3)2SO2+SO2“. Однако авторы допускают возможность образования и таких соединений, как NH+4HSO~ (в присутствии влаги в SO2), и образование кислоты HSO2NH2.
Жидкий диоксид серы — растворитель с низкой диэлектрической проницаемостью.
Способы получения диоксида серы. Сырьем для получения диоксида серы являются: сера, колчеданный концентрат, сероводород, сульфаты железа, гипс, ангидрит, фосфогипс, алунит и др. (см.: Васильев В.Т., Отвагина М.И. Технология серной кислоты. М.: Химия, 1985. САЗ).
Технологические схемы получения диоксида серы основываются на следующих реакциях:
4FeS2 + 11О2 = 2Fe2O3 + 8SO2 (ДН = 3415,7 кДж/моль);
3FeS, + 8О2 = Fe3O4 + 6SO2 (ДЯ = 2438,2 кДж/моль);
2H2S + ЗО2 = 2SO2 + 2Н2О (ДЯ= 1030 кДж/моль);
S + О2 = SO2 (ДЯ = 293 кДж/моль);
CaSO4 + 2С = CaS + 2СО2 (ДЯ = 377,1 кДж/моль);
2CaSO4 + С = 2СаО + 2SO2 + СО2 (ДЯ = 578 кДж/моль);
2CaSO4 = 2СаО + 2SO2 + О2 (ДЯ = -491 кДж/моль).
Значения парциального давления газов, образующихся при разложении CaSO4 и CaS + 3CaSO4, приводятся в табл. 2.3. 74
1 аблица 23 Парциальное давление газов при разложении CaSO4 и CaS + 3CaSO4
CaSO4
CaS + 3CaSO4
1180
1240
1280
/’so.-.s..,. ГПа
3,2
9,6
17,9
Г, °C
1000
1060
1100
ГПа 61,3 184,0 310,6
CaSO4
rr or*
' ГПа
1340 41,6
1370 63,3
Получение из колчеданов. В качестве сырья для получения диоксида серы используются серный колчедан порошковый, серный колчедан флотационный (КСФ-1, КСФ-2 и КСФ-3) и пиритный концентрат. Содержание серы в них колеблется в пределах 35—50,5%.
Для обжига колчеданов применяются следующие виды печей: I) механические; 2) аэрофонтанные; 3) с кипящим слоем; 4) комбинированные.
Обжиг колчеданов —довольно сложный процесс. Сложность обусловлена содержанием в исходном сырье большого количества примесей. К ним относятся: оксиды кремния и алюминия, сульфиды мышьяка, селена, теллура и других рассеянных элементов (индия, рения, таллия, сурьмы, висмута и др.), флюорит, карбонаты и сульфиты магния, кальция, сульфиды цветных металлов (меди, цинка и свинца). Количество примесей в колчедане зависит от его месторождения и технологии предварительной обработки на предприятиях цветной металлургии.
Из приведенной на рис. 2.14 дериватограммы флотационного колчедана видно, что при 340° С начинается выделение элементной серы из дисульфида железа по схеме
FeS2 = FeS + S-103,9 кДж.
Появление после указанной температуры экзотермического пика показывает, что выделяющаяся в виде паров сера начинает гореть (окисляться):
S + О2 = SO2 + 362,4 кДж.
Выделение серы в виде паров и нарастание интенсивности горения продолжается до 420° С, после чего интенсивность реакции уменьшается до 502° С.
Не слишком быстрое замедление экзотермического процесса при 502—530° С, по-видимому, объясняется выделением серы из содержащихся в исходном сырье арсенопирита и антимонита:
2FeAsS + 5О2 = Fe2O2 + As2O2 + 2SO2
2Sb2St + 9O2 = 2Sb2O, + 6SO2
75
Рис. 2.14. Дериватограмма флотационного колчедана
Рис. 2.15. Зависимость относительной константы скорости диссоциации дисульфида железа от температуры при различной степени выделения серы (числа у кривых)
Небольшой экзотермический пик при 590° С, очевидно, соответствует началу процесса разложения полупродукта сульфида железа:
2FeS + О, = Fe2Oi + 2SO2; 4FeS + 7О2 = 2Fe2O3 + 4SO2
Разложение сульфида железа сопровождается плавлением его при 675° С. При этом происходит перестройка кристаллической решетки сульфида железа. Процесс продолжается до 750° С с меньшей убылью массы сульфида железа в единицу времени. Имеющийся небольшой пик при 730° С, по-видимому, объясняется выделением элементной серы из образующихся в ходе основного процесса сульфидов, а возможно, и дисульфидов. После 750° С процесс продолжается с уменьшением массы образца в более замедленном режиме, что объясняется реакциями примесей исходного сырья, протекающими по следующим уравнениям:
СаСОз + SiO2 = CaOSiO2 + СО2
2FeS2 + СаСО, + SiO2 = CaOFe2O3-SiO2 + 4SO2 + CO2
2FeS2 + SiO2 + 4O2 = 2FeOSiO2 + 4SO2 76
Ряд подобных реакций с примесями сырья можно продолжить.
Из дериватограммы (см. рис. 2.14) видно, что образование FeSC>4 в процессе обжига колчедана по реакции FeS2 + ЗО2 = FeSO4 + SO2 при 250—300° С исключается. Приведенная на рис. 2.15 зависимость подтверждает данные дериватограммы.
В получаемом из дисульфида железа газе содержится и некоторое количество триоксида серы, образующегося при 600—800° С в условиях каталитического воздействия образующегося оксида железа по схеме
Fe2O3
2SO2 + О2 2SO3
Содержание триоксида в условиях обжига исходного сырья находится в пределах 0,3—0,5% (рис. 2.16). С целью снижения содержания триоксида серы в обжиговом газе рекомендуют проведение процесса при 850—900° С с дальнейшим быстрым снижением температуры до 400—425° С.
На рис. 2.17 приведена технологическая схема обжига колчедана в механических печах с получением диоксида серы. Согласно схеме, флотационный колчедан из бункера 1 поступает в шнековую дробилку 2, откуда ковшовым элеватором подается в сито 3. Мелкие частицы колчедана через автоматические весы 6 ковшовыми элеваторами 7 передаются в механическую печь 8. Крупные частицы колчедана возвращаются на дополнительное измельчение в валковую дробилку 5 и направляются в начало процесса. Образующийся в печах огарок охлаждается в шнековом транспорте 10 и транспортером 11 загружается в тележку для вывозки в специальное хранилище. Обжиговый газ после печи проходит через батарею циклонов 72, где подвергается грубой очистке от пыли, и направляется ' ”
еле электроочистки газ очищается от селена, мышьяка и других примесей и после осушки серной кислотой направляется на производство сульфитных солей.
В настоящее время для обжига колчедана в основном применяются печи с кипящим слоем (КС), интенсивность работы которых в 8—10 раз выше, чем у механических печей. При обжиге в печах КС на 1 т обжигаемого концентрата получается 0,8 т пара, а концентрация обжигового газа составляет 8—15% SO2 вместо 4,0—4,5% в механических печах.
Согласно приведенной на рис. 2.18 технологической схеме исходный флотацион-
Рис. 2.16. Зависимость между содержанием диоксида и триоксида серы в обжиговом газе из печи КС
Рис. 2.17. Схема процесса обжига флотационного колчедана в механических печах
Рис. 2.18. Схема процесса обжига флотационного колчедана в печах КС
' — воздуходувка 2 — горелка J — вывод топочных газов, 4 — печь КС, 5 — котел-утилнзатор, 6—циклоны 7 — барабанный холодильник, 8 — бункеры для огарка
ный концентрат поступает в печь КС 4. Необходимое количество воздуха для создания кипящего слоя и горения колчедана подается воздуходувкой 1 через дутьевую решетку. Образующийся в печи обжиговый газ проходит через котел-утилизатор 5, в котором не только охлаждается, но и очищается от крупных частиц выносимого из печи огарка. После котла-утилизатора через систему циклонов 6 обжиговые газы (10—14% диоксида серы) направляются на тонкую очистку в электрофильтрах. Огарок, образующийся в печи КС, постоянно выгру-78
Рис. 2.19. Печь КС
/ — корпус, 2 — шамотная футеровка, 3 — диатомовая футеровка, 4 — загрузочное устройство' 5 — форкамера, 6—колосниковая решетка, 7—камера, 8—люк для выгрузки огарка, 9— смотровое окно Ю — отверстие для горелки // — газоход
жается в бункер 8. Оседающая в котле-утилизаторе и в циклонах пыль огарка также собирается в бункерах, откуда направляется во вращающийся барабанный холодильник 7. Охлажденный огарок вывозится в огаркохранилище.
На рис. 2.19 приводится устройство печи КС конструкции ГИПРОХИМа. Печь КС для обжига флотационного колчедана состоит из металлического корпуса 1, футерованного изнутри шамотным 2 и диатомовым 5 кирпичом. Печь имеет в нижней части загрузочное устройство 4 для непрерывной загрузки колчедана. В нижней части имеется люк 8 для выгрузки огарка. В верхней части печи имеется газоход /7 для выхода сернистых газов. Печь имеет форкамеру 5, решетку провальной форкамеры для колосников б, смотровое окно 9, окно для горелки или форсунки 10. Печь снабжена также камерой для подачи воздуха к решетке форкамеры 5.
Для обжига флотационного колчедана применяются также печи других конструкций К ним относятся: печь КСЦВ (с кипящим слоем и с циклоном возврата), разработанная в НИУИФе и ГИПРОХИМе; печь ДКСМ (с двумя кипящими слоями Малеца), которая совмещает процесс обжига с охлаждением образующегося диоксида серы; трубчатые печи и т.д.
Получение из элементной серы. Простой на первый взгляд процесс получения диоксида из элементной серы по реакции
S + о2 -> so2
имеет свои тонкости, связанные с физико-химическими свойствами исходной элементной серы.
79
Процесс горения серы может служить примером цепной разветвленной реакции:
Sg—>S? + S; S + О2—>SO + О; О + Sg—>S + SO + Se
SO + O2^SO2 + О; О + SO2->SO3 и т.д.
Элементную серу как в отечественной, так и в зарубежной промышленности сжигают в жидком состоянии. Наиболее распространенными и совершенными являются циклонные и камерные печи.
Как показано на рис. 2.20, сера поступает в плавильник 1, где она расплавляется. Из плавильника сера погружным насосом 2 направляется в фильтр 5, после которого отфильтрованная сера поступает в промежуточную емкость 3. Из емкости 3 другим погружным насосом 2 сера перекачивается в форсунку 6. Погружные насосы снабжены электродвигателями 4. Форсунка 6 смонтирована в переднем конце циклонной печи 7. Процесс распыления осуществляется подачей в серную линию сухого воздуха. Образующийся в печи газ SO2 с другого конца печи по газоходу передается в котел-утилизатор 8, после чего направляется на производство сульфитных солей.
На рис. 2.21 приводится конструкция двухкамерной печи ЦКТИ-НХЗ для сжигания серы. Циклонная печь для сжигания серы имеет горизонтальный цилиндрический корпус 7, изготовленный из стали и футерованный огнеупорным кирпичом 2. Печь снабжена форсункой жидкого топлива 5 для пуска печи, передним смотровым окном 4, форсункой для распыления серы 6, боковым смотровым окном 3, заслонкой для регулирования подачи воздуха 8 и каналом для подачи воздуха 7. Печь установлена на опорах 9.
Получение из сульфида водорода. Сульфид водорода— бесцветный газ с характерным запахом. Интересно, что с повы-
Рис. 2.20. Схема сжигания технической серы в расплавленном состоянии.
/ — плавильник серы 2 — погружные насосы. 3— промежуточная емкость, 4 — электродвигатель 5 — фильтр 6 — форсунка; 7 — печь, 8 — котел-утилизатор
80
Рис. 2.21. Циклонная печь для сжигания серы
шением концентрации H2S в атмосфере запах его ослабевает, и наоборот. Способность образовывать водородные связи у сероводорода выражена слабее, чем у воды, в результате чего сероводород в обычных условиях существует в виде газа. Температура плавления равна 83° С, а температура кипения составляет 60,2° С. Плотность жидкого H2S при 60° С равна 0,96 г/см\ а газа H2S при 0° С и 760 мм рт. ст. — 1,5392 г/л. В 1 л воды при 20° С и 760 мм рт. ст. растворяется 2,67 л H2S (3,85 г). С повышением температуры растворимость сульфида водорода падает и при 100° С составляет лишь 0,81 л.
В жидком состоянии H2S ионизируется:
H2S ...H2S SH\ + SH'
В воде H2S ионизируется значительно лучше:
H2S ...Н2О ОН+з + SH
81
Частоты колебаний (см-1) H2S имеют следующие значения:
V, v2 v2
Гаюобразный H2S 2615 1183 2627
Твердый H,S 2532 1186 2544
Самым перспективным источником диоксида серы является сероводород. Это связано не только с увеличением добычи нефти и газа, но и с более сильной загрязненностью их сульфидом водорода в более глубоких слоях земли. Как известно, в земле глубже 15 км под природным газом и нефтью находится 100%-ный сульфид водорода. В настоящее время природные газы некоторых месторождений России (например, в Оренбурге) содержат значительное количество сульфида водорода. В некоторых газовых месторождениях за рубежом содержание сульфида водорода достигает 40%.
Для получения диоксида серы сульфид водорода сжигают в печах. Процесс сжигания протекает по реакции
2H2S + ЗО2 = 2SO2 + 2Н2О + 1038 кДж/моль.
Печь представляет собой стальной цилиндрический котел, футерованный огнеупорным кирпичом. Сульфид водорода поступает через горелку в верхнюю часть печи, где смешивается с воздухом; горит факелом. Ввиду взрывоопасности и токсичности исходных и образующихся газов печь оборудуется мембранным клапаном, который при нарушениях поступления исходного сульфида водорода, воздуха, при изменениях их соотношения, а также при угасании пламени автоматически останавливает процесс горения. Температуру в печи поддерживают в пределах 900—1000°С. Для более полного горения сульфида водорода в печи поддерживают избыток воздуха. Обжиговые газы охлаждаются в котлах-утилизаторах до 450° С, после чего направляются на дальнейшую переработку.
В последние годы для сжигания сульфида водорода пользуются комбинированным агрегатом типа ПКС-10/40 (печь-котел для сжигания сульфида водорода). ПКС представляет собой вертикальный водотрубный котел, оборудованный двумя форсунками для подачи исходного сульфида водорода. Котел имеет форсунку для пуска агрегата, двойной кожух, внутри которого проходит поступающий в печь воздух. Обжиговый газ в зависимости от концентрации исходного сульфида водорода содержит от 4 до 12,5% SO2.
Интенсивность горения сульфида водорода определяют по уравнению
Z = Q/V,
82
где 1—напряжение топочного пространства, кДж/(моль-ч); Q — количество выделяющейся в топке теплоты, кДж/ч; V—объем топки, м3.
При расчетах печей для сжигания сероводорода значение 1 принимается равным 600—800 кДж/(м3-ч).
Другие способы получения. Значительный интерес как с экономической, так и с экологической точки зрения представляет применение выхлопных газов, содержащих 1—1,5% диоксида серы. К ним относятся отходящие газы теплоэлектроцентралей, химических и металлургических производств, котельных установок и агломерационных цехов. В настоящее время ежегодный выброс диоксида серы в мире в пересчете на 100%-ный SO2 составляет около 100 млн.т/год, в том числе лишь в США в 1990 г. выбросы составили 55 млн.т.
Разработано большое число способов улавливания диоксида серы из выхлопных газов. К ним относятся: 1) контактное окисление SO2 на катализаторах до SO, с дальнейшим использованием последнего в производстве серной кислоты; 2) методы с применением различных катализаторов и сорбентов; 3) методы очистки выхлопных газов оксидом алюминия, гидроксидов и карбонатов металлов; 5) озонно-каталитический метод и др.
К первой группе методов очистки отходящих газов относится метод Панелека (США), согласно которому дымовые газы очищаются от пыли в электрофильтрах при температуре газа 485° С с коэффициентом улавливания 99,9% SO2. Очищенные газы, проходя контактный аппарат (ванадиевая контактная масса), окисляются до триоксида. Степень окисления диоксида составляет 90%.
Подобный же способ разработан в Токийском институте технологии; согласно этому способу, газы до контактных аппаратов подвергаются тонкой очистке от пыли в пылеуловителях.
Разработана схема очистки выхлопных газов ТЭЦ, предусматривающая дополнительный подогрев газов, поступающих в контактный аппарат.
Предложен метод, в основе которого лежит каталитическое окисление диоксида серы до триоксида серы на оксид или триоксид железа при температуре дымовых газов 500—600° С. При этом оксид железа играет не только роль катализатора, но и вступает в реакцию с образующимся триоксидом серы по схеме
Fe2O, + SO, -> FeSO4
Образующийся сульфат железа при температуре 700° С разлагается по реакции
FeSO4 -> SO, + FeO
83
При этом триоксид серы направляется на производство серной кислоты, а оксид железа—в начало процесса для повторного использования в качестве катализатора.
Разработан способ, по которому очищенные от пыли отходящие газы при повышенной температуре пропускаются через слой активного угля, орошаемого водой. При правильно выбранных значениях времени контакта между газом и углем, а также температуры реакции значительная часть (до 90%) диоксида серы переходит в триоксид.
В Германии и Голландии применяется способ «сульфицид» (Лурги), основанный на улавливании SO2 из отходящих газов сернокислотных цехов, производства диоксида титана и дымовых газов ТЭС. Согласно этому способу, запыленный выхлопной газ поступает в скруббер Вентури, орошаемый 20%-ной серной кислотой. Газы с температурой 60—70° С поступают в реактор, в котором в насыщенной водяным паром атмосфере на углеродсодержащих катализаторах протекает процесс взаимодействия диоксида серы с кислородом и водой:
2SO, + 2Н2О + О2 = 2H2SO4
Адсорбированную серную кислоту вымывают водой до 10—20%-ной концентрации и передают в сборник циркуляционной кислоты скруббера Вентури. Способ обеспечивает улавливание 96% диоксида серы, содержащегося в исходных дымовых газах.
По способу фирмы «Мицубиси» (Япония) в выхлопные газы, содержащие 0,13% диоксида серы, 0,001% SO-) и 150 мг/м1 пыли, в адсорберах распыляется порошкообразный оксид марганца. Диоксид и триоксид серы реагирует с оксидом марганца по схеме
SO2 + MnO = MnSCh; SO, + MnO = MnSO4
2MnSO, + O2 = 2MnSO4
Затем газ, пройдя через циклонные электрофильтры, выбрасывается в атмосферу. Выбрасываемый газ содержит не более 0,013% SO2 и менее 60 мг/м1 пыли. Осаждаемая в циклонах и электрофильтрах пыль состоит из непрореагировавшего оксида марганца и образующегося сульфата марганца. Из этой смеси при перемешивании ее водой готовят суспензию. При этом сульфат марганца образует водные растворы, а оксид марганца в воде не растворяется. Образующуюся суспензию обрабатывают аммиаком по схеме
MnO + MnSO4 + Н2О + NH, -> (NH4)2SO4 + MnO + Н2О 84
Оксид марганца на фильтрах отделяют от водных растворов сульфата аммония и после сушки направляют на повторное использование в адсорберах. Из растворов кристаллизуют сульфат аммония, который после сушки выпускается в виде товарного продукта. Маточные растворы сульфата марганца используют повторно для растворения образующегося в адсорберах сульфата марганца.
Предложен ряд способов сухой очистки газов от диоксида серы непосредственно в адсорберах с применением для этой цели сухого тонкоизмельченного хлорида натрия, карбоната лития, доломитовой пыли, силикагеля, аммиака, адсорбентов, насыщенных иодом и содержащих углерод.
Методы очистки отходящих газов от диоксида серы с применением суспензий различных веществ предусматривают использование в качестве последних аммиака, оксидов магния, кальция, цинка и ароматических аминов (ксилидина, диметиланилина и др.).
При магнезитовом способе улавливания диоксида серы из выхлопных газов протекают следующие реакции:
MgO + Н2О = Mg(OH)2; Mg(OH)2 + SO2 = MgSO3 + H2O
MgSO3 + H2O + SO2 t Mg(HSO3)2
Mg(HSO3)2 + Mg(OH)2 = MgSO3 + H2O
2MgSO3 + O2 = 2MgSO4
Образующиеся кристаллы MgSO3-6H2O путем фильтрации выделяют из суспензии, сушат и подвергают термообработке при 850—900° С по схеме
MgSO3-6H2O -> MgSO3 + 6Н2О; MgSO3 -> MgO + SO2
2MgSO4 -> 2MgO + 2SO2 + O2
При этом целевой продукт — диоксид серы — направляется на использование по назначению, а оксид магния возвращается в виде Mg(OH)2 на орошение абсорбера. Степень очистки выхлопных газов от диоксида серы составляет 90—95%.
Технологический процесс очистки газов от диоксида серы с применением оксида цинка протекает в две стадии. На первой стадии диоксид серы улавливается водными растворами сульфита натрия по схеме
Na2SO3 + Н2О + SO2 - 2NaHSO3
85
На второй стадии образующийся гидросульфит натрия реагирует с суспензией оксида цинка:
2NaHSO3 + ZnO + 1,5Н2О = ZnSO3-2,5H2O + Ma2SO3
Образующиеся водные растворы сульфита натрия направляются в начало процесса — на орошение в абсорбер. Осадок подвергается сушке и термообработке при 300—500° С по схеме
ZnSOr2,5H2O —ZnO + SO2 + 2,5H2O
Образующийся оксид цинка возвращается в абсорбер.
В процессе абсорбции диоксида серы образуются сульфат-ионы в системе. Их периодически удаляют из системы в виде солей кальция (CaSO4, CaSO40,5H2O и CaSO4-2H2O) путем обработки раствора оксидом или гидроксидом кальция.
Известковый метод очистки выхлопных газов от диоксида серы предусматривает проведение следующих химических реакций:
СаО + Н2О = Са(ОН)2 (1)
Са(ОН)2 + SO2 = CaSO3 + Н2О (2)
2CaSO3 + О2 = 2CaSO4 (3)
для этих целей возможно и применение известняка по схеме
СаСО3 + SO2 = CaSO3 + СО2
Из уравнений (1)—(3) видно, что лишь уравнение (2) описывает процесс поглощения диоксида серы. Оксид и гидроксид принимают лишь одну молекулу SO2. Однако в присутствии воды протекают следующие реакции:
СаО + Н2О + 2SO2 = Ca(HSO3)2
СаСО3 + 2SO2 + Н2О = Ca(HSO3)2 + СО2
CaSO3 + SO2 + Н2О = Ca(HSO3)2
При этом образуется водорастворимая соль Ca(HSO3)2, которая в жидком виде выводится из системы абсорбции и разлагается соляной кислотой:
Ca(HSO3)2 + 2HCI = НС1 = SO2 + Н2О + СаС12
86
Приведенный метод позволяет очистить дымовые газы от диоксида серы на 95% и от пыли на 99%.
Аммиачно-циклический способ основан на поглощении диоксида серы из отходящих газов путем введения в систему аммиака. Процесс ведется в водной среде по схеме
2NH-) + Н2О + SO2 = (NH4)2SO3
(NH4)2SO3 + Н2О + SO2 = 2NH4HSO3
Образующиеся в абсорберах водные растворы сульфита и гидросульфита аммония разлагаются кислотами по схеме
2NH4HSO3 + (NH4)2SO3 + H2SO4 = 2(NH4)2SO4 + 2Н2О + 2SO2
2NH4HSO3 + (NH4)2SO3 + 4HN0, = 4NH4NO3 + 3H2O + 3SO2
NH4HSO3 + (NH4)2SO3 + H3PO4 = (NH4)3PO4 + 2H2O + 2SO2
При этом в зависимости от температуры процесса возможно образование NH4H2PO4 и (NH4)2HPO4.
Образующиеся по приведенным уравнениям реакций сульфат, нитрат и фосфаты аммония являются хорошими компонентами жидких комплексных удобрений. Из этих растворов путем их фильтрации и кристаллизации можно получить соответствующие чистые соли.
Анализ приведенных способов получения диоксида серы показывает, насколько большими потенциальными возможностями они обладают. Способы позволяют на их основе организовать производство всевозможных сульфитных солей, сжиженного диоксида серы и всевозможных серосодержащих соединений.
Большое применение в производстве диоксида серы имеют сульфаты щелочноземельных металлов, таких, как сульфаты кальция, магния и бария, запасы которых значительны в природе. К ним же можно отнести и фосфогипс, образующийся в производстве фосфорной кислоты экстракционным методом.
Разложение сульфатов указанных металлов обычно проводят в барабанных печах непрерывного действия или в печах с кипящим слоем. Если эти соли находятся в виде мелкого порошка, то используют и печи с фонтанирующим слоем
При этом протекают следующие реакции их разложения:
2BaSO4—>2ВаО + 2SO2 + О2
2MgSO4—>2MgO + 2SO2 + О2
2CaSO4—>2СаО + 2SO2 + O2
87
С целью снижения температуры процесса и его ускорения в исходную шихту вводят кокс:
2MgSO4 + С = 2MgO + 2SO2 + СО2
2CaSO4 + С = 2СаО + 2SO2 + СО2
Такую же цель преследует введение в состав шихты оксидов железа, кремния, а также оксидов и сульфидов щелочноземельных металлов:
3BaSO4 + BaS = 4ВаО + 4SO2
6MgSO4 + 2MgO = 8MgO + 6SO2 + 3O2
2CaSO4 + 2SiO2 = 2CaOSiO2 + 2SO2 + O2
2CaSO4 + Fe2O3 = 2CaOFe2O3 + 2SO2O2
3CaSO4 + CaS = 4CaO + 2SO2
Приведенные схемы в технологическом аспекте вполне реальны. Имеются теоретические сведения о них в литературе; некоторые из них уже применяются в производственных условиях как у нас в стране, так и за рубежом. Экономичность их использования в производстве предопределяется следующими факторами: 1) наличие местного сырья; 2) местное потребление образующихся целевых продуктов и полуфабрикатов; 3) комплексное использование сырья и образующихся материалов.
Триоксид серы. Триоксид серы SO3 (оксид серы (VI)) — бесцветная жидкость, молекула имеет плоскотреугольное строение:
j!V О о
Расстояние О—О 0,248 нм, ц = 0. Температура кипения 44,7° С; ?крит = 218,3° С. Значения плотности триоксида серы при различных температурах приведены на рис. 2.22, а свойства различных форм приведены в табл. 2.4.
88
Таблица 24 Свойства триоксида серы
Показатель Жидкость Пар a-SO, 0-SO, Y-SO,
°C — — 16,79 32,5 62,2(0,24 МПа)
Плотность, г/см1 1,97 — 1,995 — —
С' Дж/(моль К) 180 50,21 — — —
А//".,, кДж/моль -438,5 -395,8 -450,6* -451,7 -455
Дж/(моль К) 122,6 256,7 88,3 79,5 52
* При 289 85K
Диэлектрическая проницаемость 3,11 (18° С). В процессе охлаждения ниже i6,79° С— прозрачные кристаллы, структура которых построена из кольцевых или открытых зигзагообразных молекул:
°^6и« О .0
О 14 нм^ S S<5
0^1 1^0
^=0
(SO3)3 (тример)
Кристаллическая решетка ромбическая (oc-SOt, а = 1,07 нм, b = 1,93 нм, с = 5,30 нм, z = 12, пространственная группа Рпа2). Полимеризуется триоксид серы в присутствии следов влаги. При этом образуется вначале P-SO-» — бесцветные шелковистые асбестоподобные кристаллы моноклинной сингонии, структура которых состоит из длинных зигзагообразных цепей —S02—О—SO2—, на концах которых имеются группы ОН. В процессе дальнейшей полимеризации P-форма переходит в моноклинную у-форму. Синтезирована также 5-форма S02. Данные о фазовых переходах приведены в табл. 2.5.
(SO3)<x (полимер)
жидкого триоксида серы в температурном интервале от 0 до 45° С
89
Уравнение температурной зависимости давления пара lgp(nG/) = = -А/Т + В. Значения коэффициентов А и В приведены в табл. 2.6.
Переходы а->Р, р—>у, у->8 протекают медленно, P-форма может быть превращена в а лишь через паровую фазу (в отсутствие влаги) в процессе конденсации паров ниже 16,8°С.
Термическая диссоциация триоксида по схеме 2SCh -> 2SO2 + О2 начинается при температуре 450° С, процесс заканчивается полностью лишь при 1200° С.
Таблица 25 Характеристика фазовых переходов триоксида серы
Фазовый переход Температура °C Давление гПя Энтальпия, кДж/моль
а жидкость 16,79 204,6 5,61
Жидкость пар 16,79 204,6 44,22
а nap 16,79 204,6 49,82
Р жидкость 31.5 501,6 12,1
Жидкость пар 31,5 501,6 42,64
Р 7? пар 31,5 501,6 54,4
у жидкость 62,2 2395 30,1
у пар 62,2 2395 68,3
Жидкость пар 62,2 2395 37,2
Таблица 26 Значения коэффициентов А и В
Форма SO, В Температура, К
Д 2680 11,56 215—290
В 2680 12,08 220—287
Г 3610 14,13 241—324
Жидкость 2230 10,01 335—492
Гидроксид серы дымит на воздухе, бурно реагирует с водой с образованием серной кислоты и выделением большого количества тепла. Давление паров жидкого триоксида серы растет с повышением температуры (см. рис. 2.22). Триоксид серы энергично реагирует с оксидами, гидроксидами, солями, образуя сульфаты соответствующих металлов. С хлороводородной кислотой образует хлорсульфоновую кислоту, а со фтороводородной кислотой — фторсульфоновую HSO3F. Является сильным окислителем. Окисляет серу, фосфор, углеводороды и др., восстанавливаясь при этом до диоксида серы. Реакционная способность триоксида серы повышается в ряду 3<у<Р<а. 90
Триоксид серы является основным полуфабрикатом в производстве серной кислоты и олеума. Его применяют для получения HCO3CI, SeO3, SO2CI2 и других в качестве сульфирующего агента.
Сульфид водорода. Сульфид водорода (сероводород) H2S — бесцветный газ с резким запахом тухлых яиц.
Молекула H2S имеет угловую форму, длина связи S—Н 0,1336 нм, угол HSH 92,06°. Плотность 1,538 г/л (25° С); С°р = 34,23 Дж/(моль-К); ДН^р = -20,5 кДж/моль; 52°98 = 205,68 Дж/(моль-К); энергия диссоциации 724,0 кДж/моль; /крИт = 100,4° С, ^крит = 9,01 МПа, <7крит = 0,349 г/см1. Легко сжижается в бесцветную жидкость (при 0° С и давлении 1,02 МПа). Для жидкого сульфида водорода: температура кипения 60,35° С, плотность 0,938 г/см’ (-81° С), 0,964 г/см' (-60° С), Ср =68,0 Дж/(моль-К) (200 К). На рис. 2.23 приведено изменение давления паров жидкого сульфида водорода в зависимости от температуры. Уравнения температурной зависимости давления пара: lgp(MM рт. ст.) = -1538,5/7+ 26,826 + 9,081Ig7—6,4487 (195<7<530К), ДЯ°п = 18,7 кДж/моль (-60,35° С).
Рис. 2.23. Итменение давления паров жидкого сульфида водорода от температуры
Сульфид водорода при -146,95° С образует бесцветные кристаллы кубической модификации, а ниже 169,65° С переходит в тетрагональную форму (ДЯперехода = 1,53 кДж/мОЛЬ).
При обычной температуре сульфид водорода устойчив, в вакууме начинает диссоциировать выше 500° С, а при -1690° С разлагается полностью.
Растворимость газообразного сульфида водорода в воде (% масс., 0,1 МПа): 0,694 (0° С), 0,378 (20° С), 0,232 (40° С), 0,076 (80° С). Образующийся водный раствор — сероводородная кислота — слабая кислота (К] = 9,5-10'8, К2 = 110'14), образующая соли (сульфиды и гидросульфиды). Существует клатрат H2S6H2O. Сульфид водорода растворяется в органических растворителях лучше, чем в воде.
Собственная ионизация сульфида водорода H2S в жидком состоянии (2H2S^>H3Sk + SH’) очень низка (К = 3-10'33).
Сульфид водорода — сильный восстановитель. В процессе нагревания на воздухе постепенно окисляется, а при 250° С воспламеняется и горит с образованием диоксида серы (при избытке кислорода) или серы (при недостатке кислорода). Сульфид водорода легко окисляется в водном растворе кислородом и галогенами. Наиболее сильные окислители, например азотная кислота, хлор, окисляют сульфид водорода до серной кислоты.
Сульфид водорода реагирует со значительным количеством металлов и их оксидов. В процессе нагревания в присутствии влаги и воздуха образуются сульфиды металлов. Сульфид водорода с серой образует сульфоны (полисульфиды):
Na2S + (п- 1)S = Na2S„
Персульфид-ионы имеют цепочечное строение:
Персульфиды (полисульфиды) проявляют как окислительные, так и восстановительные свойства; а также диспропорционируют:
I 2-2 -2
Na2S2 + SnS = SnS2 + №28 (S2 — окислитель);
4FeS2 + 11О2 = 2Fe20i + 8SO2 (S2 — восстановитель);
I -20-2
Na2S2 = Na3S +8(82 — окислитель, восстановитель).
92
Сульфид водорода встречается в природе в основном в месторождениях нефти и газа, а также в вулканических газах и водах минеральных источников. Он постоянно образуется в процессе разложения белковых веществ.
Сульфид водорода получают в качестве побочного продукта в процессе очистки нефти, природного и промышленного газов. Сульфид водорода применяют в качестве сырья в производстве элементной серы и серной кислоты.
Серная кислота. Физико-химические свойства и применение. Серная кислота H2SO4 (тетраоксосульфат водорода), бесцветная маслянистая жидкость без запаха, замерзающая при 10,4° С. В твердом и жидком состоянии молекулы H2SO4 связаны водородными связями. Жидкий H2SO4 — ионизирующий пястворитепь его собственная ионизация выражается уравнением
H2SO4....H2SO4 SO4H; + HSO4, К = 2,7-10’4.
В водных растворах тетраоксосульфат водорода — сильная двухосновная кислота (К\я= 1-1 ПО3, К2л = 1,2-10’2). Длины связей в молекуле S=O 0,143 нм, S—ОН 0,154 нм, угол HOSOH 104°, OSO 119°. Кипит с разложением, образуя азеотропную смесь (98,3% H2SO4 и 1,7% Н2О с температурой кипения 338,8%, табл. 2.7). Серная кислота, отвечающая 100%-ному содержанию H2SO4, имеет состав (%): H2SO4 99,5, HSO4 0,18, H3SO4 0,14, 0,09, H2S2O7 0,04, HS2O7 0,05. Серная кислота
смешивается с водой и SO3 во всех соотношениях. В водных растворах серная кислота практически полностью диссоциирует на Н+, HSO4 и SO4’. Образует гидраты H2SO4 «H2O, где п = 1, 2, 3, 4 и 6, 5.
Растворы триоксида серы с серной кислотой называются олеумом. Они образуют два соединения: H2SO4SO3 и H2SO4-2SO7. Олеум содержит также пиросерную кислоту, образующуюся по реакции H2SO4 + SO,->H2S2O7.
Температура кипения водных растворов серной кислоты повышается с ростом ее концентрации и достигает максимума при содержании 98,3% H2SO4 (рис. 2.24, табл. 2.7). Температура кипения олеума с увеличением содержания SO3 понижается (рис. 2.25). При повышении концентрации водных растворов серной кислоты общее давление пара над растворами понижается и при содержании 98,3% H2SO4 достигает минимума. С увеличением концентрации триоксида серы в олеуме общее давление пара над ним повышается. Давление пара над водными растворами серной кислоты и олеума вычисляется по уравнению 1g /ХЛа) - А-В/Т + 2,126. При этом величины коэффициентов А и В зависят от концентрации серной кислоты. Пар над водными растворами серной кислоты состоит из смеси паров воды, серной кислоты и триоксида серы. При этом состав пара отличается от состава жидкости при всех концентрациях серной кислоты, кроме соответствующей азеотропной смеси.
93
98,3%
Рис. 2.24. Температура кипения водных растворов H2SO4 при I атм (состав жидкости и пара)
1325(36)
Рис. 2.25. Температура кипения олеума при 1 атм (состав жидкости и пара)
Таблица 2 7
Свойства H2SO4 и ее сосдииеиий
Показатель H2SO4x хб5Н2О H2SO4x х4Н2О H,SO4x хЗН2О H2SO4x х2Н2О H2SO4x хН2О H2SO4 H2SO4x xSO, H2SO4x x2SO,
Содержание H2SO4 при температуре плавления,% (масс ) 45,58 57,64 65,79 73,13 84,48 100 110,1 113,9
Содержание свободного SO,,% - - - - - - 44,95 62,0
Cm, °C -52,88 -28,36 -36,39 -39,47 8,48 10,31 35,85 1,2
Скип, °C 117,9 134,0 152,9 174,8 223,0 279,6 89,8 61,0
1.3623 1,4726 1,5622 1,6471 1,7738 1,8305 1,9890 2,0010
С", Дж/(моль К) 570,7 382,5 319,2 261,0 240,1 138,9 274,6 421,6
кДж/моль 34,29 30,66 24,01 18,25 19,45 10,73 — —
52,ж, Дж/(моль К) 588.29 414,87 345,62 276,58 211,64 156,9’ — —
11, 10 3 Пас 3 022 4,752 7,199 11,073 19,724 24,740 53,6 45,8
р, Ом см 1,645 2,417 3,588 5,682 10,214 74,074 555,500 5666,670
*Для газа S'(.„ -299,17 ДжДмоль К) АН'^ =-732,74 кДж/моль Для жидкости АН^р =-813,99 кДж/моль, AG"р = - 690 14 кДж/моль, А//’ бесконечного разбавления серной кислотой 92 1096 кДж/моль
94
С повышением температуры усиливается диссоциация серной кислоты. Уравнение зависимости константы равновесия 1пХ'р = = 14,74965- 6,714641п(298/Г - 8,10161 • 1О4/2—-9643,04/7-9,4577- 1O‘Y + + 2,19062-10’6Т2. При нормальном давлении степень диссоциации: Ю’\373 К), 2,5(473 К), 27,1(573 К), 69,1(673 К). Плотность 100%-ной серной кислоты определяют по уравнению d= 1,8517—1,1-10'3/ + + 2-10’6/2г/см\ Для определения плотности и концентрации серной кислоты пользуются номограммой, приведенной на рис. 2.26. С повышением концентрации растворов серной кислоты их теплоемкость уменьшается и достигает минимума для 100%-ной серной кислоты (рис. 2.27, а), а теплоемкость олеума с повышением содержания SO^ увеличивается (рис. 2.27, б).
В процессе повышения концентрации и понижения температуры теплопроводность X уменьшается: X = 0,518 + 0,0016/—(0,25 + + Г/1293)с/100, где с—концентрация серной кислоты, %.
На рис. 2.28 приведены показатели изменения вязкости с повышением концентрации соответственно серной кислоты и олеума при 20° С.
Серная кислота является сильным окислителем. Особенно при повышении температуры. Она окисляет иодид водорода и частично бромид водорода до свободных галогенов, углерод — до его диоксида, окисляет также многие металлы (Си, Hg и др.). При этом серная кислота восстанавливается до диоксида серы, а наиболее сильными восстановителями — до элементной серы и сульфида водорода.
Разбавленная серная кислота взаимодействует со всеми металлами, находящимися в электрохимическом ряду напряжений левее водорода, с выделением Н2. Окислительные свойства для разбавленной кислоты не характерны
Способы получения серной кислоты. Наибольшее применение имеют системы получения серной кислоты из газов от сжигания элементной серы или обжига колчедана. В производстве применяется в основном контактный метод производства серной кислоты. Схема получения серной кислоты контактным способом из колчедана приведена на рис. 2.29, согласно которой исходный колчедан поступает в обжиговую печь 2, в нижнюю часть которой вентилятором 7 подают воздух.
Горение колчедана является сложным физико-химическим процессом. Первой фазой процесса является его термическая диссоциация по схеме:
2FeS2->2FeS + S2 (4)
Исследованиями установлено, что при 680° С происходит окончательное разрушение кристаллической решетки исходного пирита, а при 800° С он имеет формулу FeSioe-
95
5
Р/5 so3, %
Рис. 2.27. Теплоемкость серной кислоты (а) и олеума (б) при 20° С
?ч Si сх| с?
18
16
14
12
10
8
О 1020 30 40 50 60 70 60 90100
Концентрация H2SO4, %
Рис. 2.28. Зависимость вязкости водных растворов серной кислоты (а) и олеума (б) от концентрации при 20° С
Кислота Олеум
на склад на склад
Рис. 2.29. Схема производства серной кислоты из колчедана контактным способом
7, 12 — вентиляторы 2 — печь КС 3— котел-утилизатор 7 —циклон 5— сухие электрофильтры, б — первая промывная башия 7—вторая промывная башня. 8— мокрые электрофильтры 9— увлажнительная башня 10— продукционная башня 11—холодильник; 13— теплообменник /•/ — контактный аппарат; 15— ангидридный холодильник (экономайзер) 16 — олеумный абсорбер; 17—моногидрагный абсорбер 18 — оросительные холодильники. 19— трубчатый холодильник 20— сборники кислоты
В процессе при низких температурах (250—300° С) допускают возможность окисления исходного пирита с образованием сульфата железа:
FeS2 + ЗО2 = FeSO4 + SO2 (5)
а выше 300° С происходит разложение FeSO4:
FeSO4 -> FeO + SO2 + l/2O2 (6)
При температуре выше 300—400° С и достаточном количестве кислорода реакция горения колчедана выражается суммарным уравнением
4FeS2 + 1Ю2 = 2Fe2O3 + 8SO2 (7)
а при недостатке кислорода (в слабоокислительной среде) уравнением
3FeS2 + 8О2 = Fe2O4 + 6SO2 (8)
FeS2 разлагается при 647° С и, не плавясь, переходит в FeS, который разлагается при 800° С, значительно низкой, чем температура плавления (1195° С).
Медленное окисление колчедана начинается при 170—260° С. Низший предел температуры воспламенения определяется началом отщепления первого атома серы по уравнению (4) и воспламенением серы:
S + О2 = SO2 (9)
Температура воспламенения зависит также от минералогического состава исходного сырья, примесей, степени измельчения колчедана и концентрации кислорода в газовой фазе, влажности исходного воздуха и образующегося газа.
В процессе обжига пирита стадия отгонки первого атома серы и окисление образующегося FeS„ практически проходят одновременно. Это объясняется тем, что в результате отделения серы FeS„ остается пористым с высокоразвитой поверхностью контакта.
На ход процесса обжига оказывают влияние и примеси исходного пирита. К ним относятся: породообразующие примеси; сернистые соединения элементов подгрупп мышьяка; сульфиды цветных металлов; селениды и теллуриды и соединения других рассеянных элементов. Примеси в процессе обжига вступают в соответствующие реакции:
МеСО3 + MeSO4 = MeSO4 + МеО + СО2
CaCOi + SiO2 = CaSiO2 = CaSiO3 + CO2
99
CaF2 + H20 = СаО + 2HF
СаО + SiO2 = CaSiO,
SiO2 + 4HF = SiF4 + 2H2O
2FeAsS + 5O2 = Fe2O3 + As2O3 + 2SO2
Sb2S3 + 4,5O2 = Sb2O3 + 3SO2
В реакции входят также сульфиды висмута, меди, цинка, свинца и др.
При низких температурах сульфиды в начале процесса окисляются до сульфатов, которые при повышенной температуре разлагаются с выделением оксидов и диоксида серы. В результате эти металлы выходят из печи в виде сульфатов или соответствующих оксидов.
Рис. 2.30. Схема основных систем сухих электрофильтров
100
Из рассеянных элементов, содержащихся в колчеданах, главными являются селен и теллур. Они из зоны обжига уносятся как в виде элементов, так и в виде их оксидов.
Образующийся в печи КС 2 огарок направляется в огаркохранилище, а газы — в котел-утилизатор 3, в котором за счет теплоты газов вырабатывается пар. Далее газ, проходя циклон 4, очищается от пыли, направляется в сухие электрофильтры 5 (рис. 2.30), после которых поступает в промывное отделение. Мокрую очистку газа от остатков
Рис. 2.31. Первая промывная башня
пыли и вредных для ванадиевых катализаторов примесей проводят в промывных башнях первой ступени 6 мокрых
электрофильтров 8.
Промывная башня первой ступени (рис. 2.31) представляет собой
полую стальную колонну, внутри выложенную рольным свинцом, кислотостойкими материалами — полиизобутиленом, фаолином и футерованную кислотоупорным кирпичом. При повышенном содержании фтора ее футеруют углеграфитовыми блоками.
Для повышения равномерного распределения газа внутри башни из кислотоупорного кирпича выкладывают сужающееся устройст-
во — пережим.
Кислоту для орошения подают центробежным насосом из сборника в коллекторы 2, расположенные на крышке башни, откуда она поступает в 10—20 распылителей. В некоторых системах при необходимости дополнительно устанавливают распылители на боковой поверхности башни или располагают их в 2—3 яруса по высоте. Обычно применяют центробежные, ударные, щелевые и другие типы распылителей.
Вторая промывная башня 7 (рис. 2.29) заключена в стальной корпус, выложенный кислотостойкими или фарфоровыми кольцами Раши-га для увеличения поверхности контакта газа и жидкости. При этом снизу укладывают несколько рядов крупных колец 80—120 мм, а затем на весь объем загружают более мелкие кольца размером 50 мм. Незаполненное пространство над насадкой имеет высоту около 1 м, в котором размещены распылители кислоты. Насадка располагается на ко-
101
/ — корпус
Газ Кислота
Рис. 2.32. Вторая промывная башня
5 — решетка.
лосниковой решетке, опирающейся на столбики из кирпича. На входе имеется газовая коробка, а на входе кислоты, для ее равномерного поступления и распределения, установлен напорный сборник. Кислота вытекает из башни через нижний штуцер в сборник, центробежным насосом через холодильники подается на орошение. Обычно устанавливают два насоса с самостоятельными кислотопроводами.
Увлажнительная башня (см. рис. 2.29) имеется на ряде действующих заводов, а для вновь проектируемых установок ее не предусматривают, поскольку работу промывных отделений перевели на испарительный режим.
Затем газ освобождают от влаги в сушильной башне 10, а от брызг и тумана серной кислоты — в брызгоуловителе 11. Сушильные башни орошают серной кислотой по циклу: сборник — насос — холодильник — башня — сборник.
Очищенный и осушенный сернистый газ вентилятором 12 подают в контактное отделение. При этом все оборудование до нагнетателя работает под разрежением, а после нагнетателя — под давлением.
102
В контактном отделении диоксид серы нагревается в теплообменнике 13 до температуры зажигания катализатора и поступает на окисление в контактный аппарат 14. Процесс окисления диоксида серы в триоксид
SO2 + 0,5О2 # SO,
обратимый и степень окисления газа любого состава определяются температурой и парциальными давлениями компонентов реакции.
Окисление диоксида серы проводят гетерогенно на ванадиевых катализаторах, которые вытеснили ранее применяемые платиновые.
Чистый пятиоксид ванадия обладает слабой каталитической активностью. Активность его резко возрастает в присутствии солей щелочных металлов. Их прометирующая роль обусловлена образованием низкоплавких пиросульфованадатов. Активный компонент в условиях катализа находится в расплавленном состоянии. Исследования V2O5—K2SO4 и V2O5—K2S2O7 показали, что в катализаторах близок состав 1,2 V2O5K2OSO3 или K2O-2SO3 и наличие эвтектики с температурой плавления ниже 430° С в первой системе.
Схема процесса может быть сформулирована следующим образом:
I )2 V5+ + О2’ + SO2 2V4+ + SO,
2)2 V4+ + 1/2O2->2V5+ + О2’
В первой стадии достигается равновесие, а вторая стадия является медленной и определяет скорость процесса.
В процессе контактирования, т.е. окисления на слоях катализатора, температура его повышается, поэтому газ охлаждают между слоями в промежуточных теплообменниках, а из последнего слоя образующийся триоксид серы направляют во внешний теплообменник и ангидридный холодильник 15 и далее на абсорбцию SO3 в олеумный 16 и моногидратный 17 абсорберы. Далее, после очистки от брызг и тумана серной кислоты в брызгоуловителе 11, выхлопные газы выбрасывают в атмосферу с содержанием 0,15—0,3% SO2.
По аналогичной технологии получают серную кислоту из серы в гех случаях, когда сера содержит вредные для катализатора примеси, например мышьяк. При использовании свободной от подобных вредных примесей серы применяют короткую схему получения серной кислоты. Согласно схеме, расплавленную отстоянную и отфильтрованную серу подают на сжигание в печь, куда поступает воздух, осушенный в сушильной башне. Образующийся при этом диоксид серы после печи охлаждают в котле-утилизаторе и пароперегревателе от I 000—1200 до 400—450° С и направляют в контактное отделение.
103
Окисление диоксида серы производят в пятислойном контактном аппарате. Реакционный газ охлаждают после I слоя в пароперегревателе, после II слоя — в теплообменнике, после III и IV слоев — поддувом воздуха. После V слоя газ охлаждают во внешнем теплообменнике и ангидридном холодильнике и направляют в абсорбционное отделение, аналогичное приведенной выше схеме.
С целью снижения выбросов в атмосферу оксидов серы, повышения степени извлечения целевого продукта и снижения расходов исходного сырья и себестоимости серной кислоты разработана и применяется технологическая «схема с двойным контактированием (ДК). Сущность метода заключается в проведении процесса окисления диоксида с.епы с выявлением образовавшегося триоксила серы в пополнительном абсорбере. На первой стадии абсорбирует SO2, а выходящую из промежуточного абсорбера газовую смесь нагревают в теплообменниках и направляют во вторую стадию контактирования. Затем газ вновь охлаждают и абсорбируют триоксид серы.
Метод ДК одновременно со снижением содержания диоксида серы в выхлопных газах позволяет перерабатывать более концентрированные газы. Остаточное содержание диоксида серы в выхлопных газах составляет 0,02—0,05%.
Перспективным способ производства серной кислоты является нитрозный метод ее получения.
В нитрозном процессе окисление диоксида серы осуществляется оксидами азота, которые являются переносчиками кислорода. При этом высшие оксиды азота —NO2 и N2O2— восстанавливаются до оксида азота NO, который вновь окисляется до высших оксидов. Существенную роль при этом играет нитроза, представляющая собой раствор оксидов азота в водных растворах серной кислоты. В процессе растворения в водных растворах в зависимости от степени их окисления образуют нитрозилсерную кислоту
N2O, + 2H2SO4 = 2HNSO5 + Н2О
или нитрозилсерную и азотную кислоты
2NO2 + H2SO4 = HNSO5 + HNO3
Схему кислотообразования в нитрозном процессе объясняют следующим образом:
SO2(r) SO2(p-p)
HNSO5 + Н2О H2SO4 + HNO2
104
HNO2 + SO2(p-p) -> H2SO4 + 2NO(p-p)
NO(p-p) ЫО(газ)
2NO(r) + O2(r) -> NO2 (r)
(NO + NO2) (r) # (NO + NO2) (p-p)
(NO + NO2) (p-p) + H2SO4 # HNSO5 + HNO2
Принципиальная схема производства серной кислоты нитрозным методом приведена на рис. 2.33, согласно которой исходный горячий диоксид серы поступает в первую башню, предназначенную для выделения оксидов азота из азотной кислоты, орошающей башню. Процесс называется денитрацией серной кислоты, а башня 1 называется денитрационной. Около 35% денитрированной кислоты, вытекающей из этой башни, передается на склад в качестве целевого продукта. Оставшаяся часть (2/3) кислоты направляется на орошение последней башни 4.
Денитрационная башня орошается небольшим количеством серной кислоты. Последняя сильно нагревается, что ускоряет процесс выделения оксидов азота. Одновременно с денитрацией кислоты в башне 1 диоксид серы частично абсорбируется серной кислотой и окисляется оксидами азота. По характеру протекающих процессов первую башню схематически делят на три зоны. В нижней зоне башни происходит упаривание серной кислоты с выделением водяных паров в газовую фазу, в средней зоне в результате наибольшего ее разбавления из нитрозы выделяются оксиды азота, в верхней зоне
конденсируются поступающие снизу пары воды. Таким образом, происходит разбавление нитрозы и частичное окисление растворяющегося в ней диоксида серы. Перечисленные процессы трудно строго разделить по зонам, поскольку они частично совмещаются друг с другом Кроме того, в первой башне из газа улавливаются остатки пыли, поглощаются диоксид селена и сесквиоксид мышьяка, конденсируются пары серной кислоты, образующиеся из триоксида серы,
на склад
Рис. 2.33. Принципиальная схема производства серной кислоты нитрозным способом
105
присутствующего в обжиговом газе. При этом образуется серно-кислотный туман, который частично поглощается в первой башне, а большая часть его поступает в последующие башни, в которых вследствие значительной суммарной поверхности частиц тумана существенно влияет на протекающие в башнях процессы.
Целевой продукт в башенных системах отводят лишь из денитра-ционной башни, в которой улавливаются все примеси обжигового газа. Поэтому башенная кислота обычно загрязнена мышьяком, селеном, огарковой пылью и другими примесями.
Процесс абсорбции диоксида серы из обжигового газа серной кислотой и окисление диоксида серы нитрозой происходит в башне 2. В этой башне образуется около 70—80% серной кислоты. Поэтому ее называют продукционной башней. Процесс кислотообразования происходит по всей высоте башни 2. Основное количество диоксида серы окисляется в ее нижней части. Оксиды азота, выделяющиеся из нитрозы в процессе окисления диоксида серы, частично поглощаются в верхней части башни, орошающей ее нитрозой. Однако значительная часть оксидов поступает в смеси с газовым потоком в окислительную башню 3. Здесь орошается оксида азота столько, сколько требует соотношение между оксидом и диоксидом азота, благоприятное для поглощения их в абсорбционной башне.
В окислительной башне 3 оксид азота (II) окисляется кислородом, содержащимся в газе. Степень окисления оксидов азота в этой башне регулируют, пропуская часть газа по байпасу. Из окислительной башни газ поступает в абсорбционную башню 4, в которой оксиды азота поглощаются орошающей ее серной кислотой.
В процессе охлаждения обжигового газа и образования серной кислоты выделяется значительное количество теплоты. Поэтому в де-нитрационной и продукционных башнях орошающая кислота нагревается и до возврата на орошение ее охлаждают в холодильниках 5.
В процессе производства кислоты имеют место потери оксидов азота с отходящими газами, с продукционной кислотой и др. Для восполнения этих потерь в продукционную башню 2 вводят азотную кислоту. Вода в систему вводится через денитрационную 1 и продукционную 2 башни.
В современных башенных системах достигнута высокая интенсивность, равная 220—250 кг/(м3 сут) 100%-ной серной кислоты. Расход азотной кислоты составляет 15—18 кг/т. Целевой продукт выпускается с минимальным содержанием оксидов азота (0,03%). Плотность орошения башен составляет 10—15м3/(м2ч).
В НИУИФе разработан безнитрозный цикл орошения, обеспечивающий практически полное окисление диоксида серы. Метод основан на контакте безнитрозных 12—50%-ных растворов серной кислоты с нитрозными газами, выходящими из продукционной зоны. 106
Скорость процесса окисления диоксида серы при использовании без-нитрозного цикла в 2—3 раза выше, чем скорость процесса окисления нитрозами. Окислителями SO2 являются оксиды азота нитрозных газов, вступающие в контакт с водными растворами серной кислоты, или азотная кислота, вводимая в эти растворы.
Разработан способ улавливания оксидов азота выхлопных газов башенного процесса, основанный на поглощении N2O? серной кислотой, a NO2 водой. Разработана также технология башенной серной кислоты с концентрацией 90,5—92,5%-ной H2SO4 путем использования теплоты обжигового газа для упаривания 75%-ной башенной кислоты.
Разработанный в НИУИФе способ применения кислорода вместо воздуха резко снизит удельные капиталовложения, снижая при этом себестоимость продукции.
ГЛАВА 3
ХРОМ И ЕГО СОЕДИНЕНИЯ
Содержание хрома в земной коре 0,035% (масс.), а в воде океанов и морей — 2-10'5 мг/л. Известно более 40 хромсодержащих минералов, из которых для извлечения хрома и его соединений применяют хромшпинелиды (Mg, Fe) (Cr, Al, Fe)2O4, основой которых является хромат (HI) FeCr2O4.
Хромат (III) FeCr2O4 обычно состоит из 35—50% Сг2О3, 15—25% FeO + Fe2O3, 15—20% А12О3, 5—15% MgO, 3—8% SiO2, 1—3% СаО. Бедные руды, содержащие менее 35% Сг2О3, называются железохромовыми. Потребителям хром содержащее сырье поступает в виде руды или концентрата, получаемого обогащением бедных руд. К хром содержащим минералам относятся: крокоит РЬСгО4, вол-конскоит 'Cr2Si4Oio (ОН)2а?Н2О, уваровит Ca3Cr2(SiO4)3, вокеленит Pb2Cu[CrO4]PO4, феникохроит Pb3O(CrO4)2. В метеоритах обнаружены сульфидные минералы хрома.
Природный хром состоит из смеси четырех изотопов: 50Сг (4,35%), 52Сг (83,79%), 5,Сг (9,50%) и 54Сг (2,36%). Конфигурация внешних электронных оболочек атома 3(/5451; степени окисления +2, +3, +6, реже +4, +5 и +1; энергия ионизации при переходе от Сг° к Сг+6 6,766, 16,49, 30,96, 49,1, 69,3 и 90,6 эВ; атомный радиус 0,127 нм, ионные радиусы, в нм (в скобках указаны координационные числа): для Сг2" 0,073 (6), Сг3+ 0,0615 (6), Сг4+ 0,041 (4), 0,055 (6), Сг5+ 0,0345 (4), 0,049 (6) и 0,057 (8), для Сг6+ 0,026 (4) и 0,044 (6).
Хром — голубовато-белый металл. Кристаллическая решетка объемно-центрированная кубическая (а = 0,28845, z = 2, пространственная группа При 312 К (точка Нееля) переходит из парамаг-
нитного в антиферромагнитное состояние. Еще один переход (без изменения структуры) фиксируется при 170—220 К. Температура плавления 1890° С, температура кипения 2680° С, плотность 7,19 г/см1; С° = 23,3 Дж/(моль-К); АЯПЛ = 21 кДж/моль, АЯИСП - 338 кДж/моль; 52°98 = 23,6 Дж/(моль-К).
Ю8
Уравнения температурной зависимости давления пара для твердого хрома 1g/? (мм рт. ст.) = 11,454 — 22 598/7"—0,406 IgT + 0,781 Т (298—2163 К), для жидкого хрома 1g/? (мм рт. ст.) = 9,446—18204/Т+ + 0,114 IgT (2163—2950 К).
Хром парамагнитен, его магнитная восприимчивость +3,49'109. Твердость хрома по Бринеллю 1060 МПа. Хром технической чистоты хрупок, выше 200—250° С приобретает пластичность.
Хром устойчив на воздухе (но в тонкоизмельченном виде пиро-форен) и к действию воды. В процессе нагревания в кислороде до 900° С сгорает с образованием сесквиоксида. Растворяется в хлорированной и разбавленной серной кислотах. В концентрированных !1NO$, НСЮ4, Н,РО4 и под действием окислителей хром пассивируется. Пассивный хром очень устойчив. Водные растворы щелочей на хром не действуют, расплавы щелочей в отсутствие воздуха медленно реагируют с выделением водорода.
Фтор действует на хром при температуре выше 350° С. Сухой хлор начинает реагировать с хромом выше 300° С, а влажный хлор — с 80° С. При температуре красного каления хром реагирует с бромом, иодом, фторидом и хлоридом водорода.
С водородом хром непосредственно не реагирует. Растворимость водорода в хроме 0,44% (ат.) при 800° С, но электролитический хром может содержать значительно большие количества — до 300 объемов на объем металла.
Поглощением азота тонкоизмельченным порошком хрома при 800—1000° С получают нитрид хрома CrN, а при 1200—1300° С — Cr2N. Эти нитриды получают также действием аммиака на хром при 850° С. Образующиеся нитриды, особенно CrN, обладают высокой химической стойкостью. Они применяются в качестве компонентов твердых сплавов, являются и катализаторами. Мононитрид хрома применяется в качестве полупроводникового материала для термоэлектрических генераторов.
Хром сплавляется с бором, углеродом и кремнием с образованием соответствующих боридов, карбидов и силицидов. Они применяются в качестве компонентов твердых, жаростойких сплавов, износоустойчивых и химически стойких покрытий.
Хром с оксидами углерода не взаимодействует.
Сера в виде паров при температурах выше 400° С с хромом образует целый ряд сульфидов от CrS до Cr5Sg. Сульфиды образуются также под действием сульфида водорода (-1200° С) и паров сероуглерода на хром. В процессе сплавления с Se хром образует селениды, аналогичные по составу сульфидам. Теллуриды хрома имеют состав от СгТе до СгТе-,. Металлы, взаимодействуя с сеск-вихалькогенидами Сг2Хз, образуют халькогенохроматы (III). Боль-
109
шинство из халькогенохроматов (III) обладают полупроводниковыми свойствами и являются либо ферромагнетиками, либо антиферромагнетиками. Соединения с однозамещенными металлами состава MCrFe2 большей частью имеют ромбоэдрическую решетку типа NaHF2. Для калия, рубидия и цезия известны также соединения состава МСг2Х4, которые имеют структуру шпинели. Некоторые из них при высоких температурах и давлениях переходят в структуру типа NiAs. Для редкоземельных элементов известны также соединения типа МСгХ3
Для хрома характерна способность к образованию комплексных соединений в разных степенях окисления. Образование комплексов стабилизирует низкие степени окисления хрома. Сг(1) известен всегда в виде комплексов, например КДСДСЫ^ЫО]. Соединения Сг(П) неустойчивы, поскольку они сильные восстановители, поэтому легко окисляются на воздухе. Их водные растворы сохраняются лишь в инертной атмосфере. Из растворов кристаллизуются гидраты, например С^СЮДгбНгО. Из комплексов Сг(П) самый распространенный K4[Cr(CN)6],
Наиболее устойчивы соединения Сг(Ш). В водных растворах катион Сг(П1) существует в виде инертного аквакомплекса [Сг(Н2О)6]3+ с незначительной скоростью обмена молекул воды на другие лиганды. В результате этого соли в растворах и в кристаллическом состоянии существуют в виде изомеров, содержащих приведенный гексааквакатион, входящий во внутреннюю сферу комплекса. Известно значительное число комплексов Сг(Ш) с координационным числом 6 и октаэдрической конфигурацией, в большинстве химически инертных. К их числу относятся комплексы с нейтральными лигандами и с разнообразными анионами (галогенидные, цианидные, роданидные, сульфатные, оксалатные и др.). Характерны поли-ядерные формы комплексов с гидроксидными, кислородными, ам-минными, роданидными мостиками.
Соединения Cr(IV) немногочисленны — это простые и комплексные галогениды, а также хроматы (IV).
Cr(V) участвует в оксогалогенидах (CrOF3), в хроматах (V) и в галогенохроматах типа (М2'[СгОС15] или M[CrOF4],
Cr(VI) образует многочисленные хроматы. В растворах он может присутствовать в виде ионов СгО3Х , где X — галоген, СгОз(О8О,)з и т.д. Соединения Cr(Vl) — сильные окислители.
Получение хрома. Непосредственной металлургической переработкой необогащенных хром содержащих руд (хромита) способом восстановительной плавки в электропечах получают феррохром с содержанием 60—70% хрома, применяемый в черной металлургии. ПО
Существующие способы получения соединений хрома из феррохрома основаны на окислительном обжиге исходного хромата в смеси с карбонатом натрия и доломитом при 1100—1200° С. Образующийся при этом Ма2СгО4 выщелачивают водой и после очистки раствора от алюминия обработкой H2SO4, СО2 или Na2Cr2O7 из раствора кристаллизуют либо Na2CrO4, либо Na2Cr2O7, либо содержимое раствора перерабатывают на другие соединения хрома Необходимый для получения металлического хрома его сеск-виоксид получают восстановлением щелочного раствора Na2CrO4 элементной серой в процессе кипячения или в автоклавах при 140—160° С по реакции
4Na2CrO4 + 6S + (2и + 1)Н2О = 2(Сг2Оу«Н2О) + 3Na2S2O3 + 2NaOH
Далее раствор подкисляют кислотой и проводят вторую стадию восстановления с получением хромихромата или гидроксида Сг(П):
10 Na2CrO4 + 3Na2S2O3 + 7H2SO4 + (2и + 7)Н2О 2(2Сг2О3СгО3-иН2О) + 13Na2SO4
Путем сушки и термообработки гидроксидных осадков получают сесквиоксид хрома.
' Металлический хром обычно производят восстановлением Сг2О3 алюминием или элементным кремнием. Реже применяется процесс восстановления углеродом, в процессе которого получается хром с большим содержанием углерода. Разработаны также процессы восстановления трихлорида хрома магнием. Более чистый хром получают электролизом либо сернокислых растворов СгО3, либо хромоаммониевых квасцов.
Металлический хром рафинируют обработкой его водородом при высоких температурах, вакуумной дистилляцией, зонной плавкой, иодидным рафинированием. Особо чистый хром получают термическим разложением органических хромсодержащих комплексов, например бис-(этилбензол)хрома с последующей водородной очисткой металла.
Хром применяют в металлургии в качестве компонента сталей различного назначения. Различные соединения хрома применяют в качестве огнеупорных материалов, пигментов, дубителей кожи, протрав при крашении, реактивов, магнитных материалов и др. Соотношение областей применения: металлургия — 75%, огнеупоры — 10%, прочие — 15%.
ill
3.1. ОКСИДЫ И ГИДРОКСИДЫ ХРОМА
Физико-химические свойства. В системе Сг—О (рис. 3.1) существуют оксиды хрома, свойства которых сопоставлены в табл. 3.1.
Оксид хрома (II) метастабилен, его точка состава в системе Сг — Сг2О3 попадает в область расслаивания расплава, которая простирается от 2 до 48% (ат.) О. В этой системе обнаруживается также инконгруэнтно плавящийся оксид Сг3О4, существующий при температурах выше -1550° С, имеющий искаженную структуру шпинели.
Монооксид хрома (II), красные кристаллы или черный пирофорный порошок. Кристаллическая форма СгО устойчива на воздухе, не растворяется в воде, но интенсивно окисляется при температуре выше 100° С. Реагирует с хлороводородной кислотой с выделением водорода, а с разбавленными HNO-, и H2SO4 не взаимодействует.
Монооксид хрома (II) получают термическим разложением карбонила:
Сг(СО)6 —СгО + 5СО + С
Монооксид хрома (И) применяют в качестве абсорбента для очи-
стки углеводородов от кислорода.
Рис. 3.1. Области устойчивости оксидов хрома
р —давление кислорода (Па) «н д » — низкого давления
И2
Гидроксид хрома (II) Сг(ОН)2 коричневого или желтого цвета проявляет лишь основные свойства, медленно взаимодействует только с концентрированными кислотами, образуя при этом аквакомплекс синего цвета:
Сг(ОН)2 + 2ОН3+ + 2Н2О = = [Сг(ОН2)6]2+
Произведение растворимости Сг(ОН)2 2,0-1 О*20; не растворяется в растворах щелочей и разбавленных кислотах, на воздухе быстро окисляется.
Сг(ОН)2 получают из растворов солей Сг(П) щелочами в отсутствие кислорода.
Таблица 3.1. Свойства оксидов хрома
Показатель СгО Сг3О4 Сг2О3 СгО2 СГ5О12 Сг2О5
Цвет Красный Оранжевый Зеленый Черный Черный Черный
Сингония Параметры ячейки, нм Кубическая Тетрагональная нальная Тетрагональная Ромбическая Моноклин-
а 0,412 0,62026 0,49576 0,4422 1,2044 1,201
ь — — — — 0,8212 0,852
с — 0,85386 1,35874 0,2918 0,8177 0,929
Р, град — — — —I — 92,0
Z 4 8 6 2 4 —
Пространственная группа Fm3m — ЯЗс Р^г/тпсс РЬсп —
°C 1550 1705* 2334** — — —
Плотность, г/см3 — — 5,21 4,95 3,68 —
С", Дж/(моль-К) — — 119 — — —
кДж/моль -335 -1447 -1141 -588,3 -2890 -1200
S'2.„, Дж/(моль-К) 61 150,5 81 48,1 281 116
•Инконгруэнтно ♦* Температура кипения 3000° С
Сг.О15 Ст3Оц СгОз
Черный Коричневый Красный
Ромби- Ромби- Ромби-
ческая ческая ческая
0,847 1,201 0,8525
1,290 2,660 0,4755
1,008 0,782 0,4743
— — —
4 — —
Стоп - С2ст
3,34 2,82
— — 58
— -1791 -590
— 183 73,2
Рис. 3.2. Зависимость степени окисления сесквиоксида хрома до его триоксида от продолжительности термообработки шихты
К); парамагнетик, при 32 К (точка
Оксид хрома (III) (сеск-виоксид) Сг20з (минерал эс-колаит) имеет структуру типа корунда (a-форма). Его цвет меняется от светло-зеленого у тонкодисперсного материала до почти черного у крупных кристаллов. В процессе нагревания зеленый цвет обратимо переходит в коричневый. Существует аморфный Сг20з, а также метастабильная кубическая у-форма со структурой типа шпинели (а = = 0,836 нм) коричневого цвета. Описана также тетрагональная модификация (а = = 0,9480 нм, с = 0,5160 нм), устойчивая выше 1000° С. Сг2Оз заметно летуч выше 1200°С; испаряется конгруэнтно, с диссоциацией в па-
рах; уравнение температурной зависимости давления пара 1g р (мм рт. ст.) = = 10,62—25 300/Т (1504—1821 Нееля) переходит антиферромаг-
нитное состояние, АЯ перехода 0,80 кДж/моль. Полупроводник, ширина запрещенной зоны ~3,4 эВ; твердость по шкале Мооса 9. На рис. 3.2 приведена зависимость степени окисления Сг2Оз до СгО3 от продолжительности термообработки.
Сесквиоксид существует в виде гидратов с содержанием 5,3 и 1 молекулы воды на одну молекулу Сг2О3 (рис. 3.3).
Старение сесквиоксида связано с увеличением количества гидратной воды и размера частиц, изменением цвета и снижением растворимости в кислотах и особенно в щелочах.
Наиболее сильное влияние на старение гидрата оказывает нагрев, особенно под давлением. При этом уменьшается содержание гидратной воды вплоть до образования моногидрата сесквиоксида хрома Сг2Оз-Н2О, являющегося пределом, к которому стремится гидрогель в
процессе старения.
Сг20з химически малоактивен. Не растворяется в воде и органических растворителях, не взаимодействует с водными растворами 114
щелочей. Растворяется лишь в сильных кислотах при продолжительном нагревании, окисляется и переходит в раствор под действием горячих растворов персульфатов и хлоратов, а также 70%-ной HCIO4. В процессе спекания с оксидами или карбонатами металлов образует хромиты:
2КОН + Сг2О3 = 2КСгО2 + Н2О
Na2CO3 + Cr2O3 = 2NaCrO2 + СО2
Образующиеся оксохроматы ти-
+2 Рис. 3.3. Диаграмма
па М (СгО2)2 ЯВЛЯЮТСЯ координаци- состав — температура для системы онными полимерами, т.е. смешан- сесквиоксид — вода
ными оксидами (типа шпинели
MgAl2O4). В их кристаллах атомы М(П) находятся в тетраэдрическом, а атомы хрома (III) в октаэдрическом окружении атомов кислорода. Сесквиоксид хрома взаимодействует с хлором в присутствии углерода при 650—850° С, а выше 1500° С восстанавливается до металла действием Н2, С, СО, Si, Al, Са, Mg и т.п.
Известен ряд гидроксидов Сг(1П). В процессе реакции водных растворов солей Сг(1П) с щелочью или аммиаком образуется гелеобразный осадок Сг(ОН)3пН2О:
Сг3+ + ЗОН’ = Сг(ОН)3
Образующийся осадок имеет переменный состав Сг2О3иН2О. Это многоядерный слоистый полимер, в котором роль лигандов играют ОН и ОН2, а роль мостиков — ОН-группы.
Его состав и структура зависят от условий получения. При хранении и в процессе нагревания Сг2О3иН2О вследствие замены связей Сг—ОН—<3г на связи Сг—О—Сг теряет активность. Свеже-осажденный Сг(ОН)3 (Сг2О3иН2О) хорошо растворяется в присутствии кислот и щелочей, которые вызывают разрыв в связи в слоистом полимере:
сг(он)3 + зон; = [Сг(он2)6]3+
Сг(ОН)3 + ЗОН’ = [Сг(ОН)6]3’
115
OH2 H OH2 H OH2
I L
H2o—Cr Cr Cr,
/\^O^ I
/ I
HO OHH H OH2
Получение гидроксида хрома (III) и его переход в катионные и анионные комплексы выражают следующим суммарным уравнением:
[Сг(ОН2)6]’‘ Сг(ОН), 7^^- [Сг(ОН6)]3
Растворимость гидроксида хрома в воде 1-10'7% (масс.) при 25° С. Окраска (голубая, зеленая, зеленовато-черная или фиолетовая), а также его химическая активность зависят от условий осаждения. Он легко растворяется в минеральных кислотах и растворах щелочей, образуя соответствующие соли. По мере старения осадков их реакционная способность снижается. В процессе высушивания осадков образуется гидрат Сг(ОН)зЗН2О, который может быть получен в кристаллическом виде; структура гексагональная (а = 1,230 нм, с = 0,487 нм); плотность 1,64 г/см1. Малоустойчив, при хранении, а также при нагревании до 70° С переходит в аморфный гидрат Сг2От5Н2О, устойчивый до 85° С. При дальнейшем нагревании получают аморфный гидроксид Сг(ОН)з. Кристаллизуется он в структуре типа байерита А1(ОН)з, решетка гексагональная (а = 0,5288 нм, с = 0,4871 нм, г = 2); плотность 2,9 г/см1; обезвоживается при 150° С.
Известен также гидрат — изумрудная зелень или зелень Гийе — состава Сг2ОунН2О, где п = 1,5—2; не растворяется в минеральных кислотах, растворах щелочей, легко растворяется в растворах СгОз. Устойчив на воздухе. В процессе нагревания до -200° С переходит в Сг2ОзО,5Н20. Однако в процессе хранения на воздухе снова ее поглощает. Плотность обезвоживается при температуре около 600° С.
Оксогидроксид хрома СгООН (или Сг2Оз Н2О) известен в виде нескольких модификаций. а-Модификация (минерал гримальдит) образует кристаллы голубовато-серого, голубовато-зеленого или коричнево-красного цвета. Имеет ромбоэдрическую структуру (а = 0,2960 нм, с = 1,329 нм, z = 3, пространственная группа /?3ш); плотность 116
4,12 г/см1. P-Модификация зеленого цвета (минерал гвианаит) имеет ромбическую решетку (<я = 0,4861 нм, Ъ = 0,4292 нм, с = 0,2960 нм, г = 2, пространственная группа Рптп)\ плотность 4,57 /см1. Третья модификация (минерал бресуэллит) красно-коричневого цвета имеет ромбическую структуру типа диаспора (а = 0,449 нм, b = 0,986 нм, с = 0,297 нм, z = 4, пространственная группа РЪпт). Описана также изумрудно-зеленая P-модификация, тоже ромбическая, со структурой типа бемита (пространственная группа Стет). Есть сведения в литературе о существовании еще одной, кубической, модификации серо-фиолетового цвета.
Оксогидроксид хрома устойчив на воздухе, практически не реагирует с минеральными кислотами и растворами шелочей Обезвоживание a-формы происходит при 370—440° С, Р-модификации — при 480—550° С, у-модификации — при 520—570° С. СгООН образуется в процессе неполного обезвоживания гидроксида хрома (III), является полуфабрикатом в производстве сесквиоксида хрома.
Диоксид хрома СгО2 кристаллизуется в структуре типа рутила. По некоторым данным, имеет область однородности (отношение О.Сг от 1,901 до 2,013), ферромагнетик, обладает металлической проводимостью; при 110° С переходит в парамагнитное состояние (без изменения структуры). При 510° С разлагается до Сг2О3. На воздухе устойчив, в процессе длительного кипячения в воде диспропор-ционирует по уравнению
ЗСгО, + 2Н2О = 2СЮОН + Н2СгО4
Хлороводородная кислота окисляет ее с выделением С12.
Известна также аморфная парамагнитная форма СгО2 коричневого или черного цвета, содержит следы воды. В процессе обезвоживания разлагается. В качестве гидрата диоксида можно рассматривать и гидроксохромат [Сг(ОН)2]2СгО4, который осаждается из растворов, содержащих Сг(1П) и Cr(VI) при pH 3—4; коричневое рентгеноаморфное вещество, содержащее дополнительно 5—5,5 молекул воды.
Существуют гидроксохроматы Cr(III) — это Сг(ОН)2(НСгО4)-4Н2О, превращающийся при хранении с маточным раствором в СгО2-2Н2О, а также Сг5(ОН)з(СгО4)3 пН2О. Вещества эти аморфные, легко растворяются в разбавленных минеральных кислотах и растворах щелочей. В процессе термообработки они обезвоживаются и при 250°С разлагаются. Гидроксохроматы хрома (III) общей формулы хСг2О3>СгОзпН2О называют хромихроматами.
Оксид хрома Сг5О12 — хромат (VI) хрома (III). Полагают, что это фаза переменного состава, чья область однородности лежит в интервале составов СгО24 СгО2 48 (по другим данным от СгО2,з85 до 117
Сг243о). He растворяется в воде, растворяется в разбавленных минеральных кислотах.
Оксид хрома Сг2О5 содержит хром в двух степенях окисления, его структурная формула Сг2(СгО4)2(Сг2О7). Описаны две модификации— моноклинная антиферромагнитная с температурой Нееля ниже 80 К и существующая при высоких давлениях ромбическая, для которой предложена формула Сг6О15. Черные пластинчатые кристаллы. Не реагирует с водой и разбавленными кислотами, растворяется в концентрированной серной кислоте при комнатной температуре. Получают термическим разложением СгО3 или Cr3Og в атмосфере кислорода при 270—300° С.
В качестве гидрата Сг2О7 принято рассматривать гидроксодихромат Сг(ОИ)Сг2О7-2Н2О, осаждающийся из водных растворов. Коричневое аморфное вещество, растворимое в разбавленных кислотах и щелочах; при 100—160° С он обезволивается и при 250—350° С разлагается.
Оксид Cr3Og [Сг2(Сг2О7)2(Сг3Ою)]— мелкие темно-коричневые кристаллы; антиферромагнетик, температура Нееля ~80 К; легко разлагается водой; получают разложением СгО3 на воздухе (270—300° С) в атмосфере кислорода (240° С) или гидротермальным его разложением при 270° С.
Декахромат хрома Сг2(СгюО31)3, или CrO2 906, является первичным продуктом термического разложения СгО3. Черный аморфный порошок с плотностью 2,88 г/см\ при 240° С начинает разлагаться с образованием СгО3 и Cr3Og.
Триоксид хрома СгО3_красные или фиолетово-красные кристаллы. Под давлением 11,5 ГПа и температуре 23° С переходит в другую модификацию. Температура плавления 180—202° С; заметно испаряется с диссоциацией в парах, уравнение температурной зависимости давления пара 1g р (мм рт. ст.) = 20,14—10 300/Т (448—468 К); полупроводник. Малоустойчив, начинает разлагаться при комнатной температуре. В процессе медленного нагревания триоксид хрома выделяет кислород с образованием промежуточных продуктов (см. рис. 3.3). Триоксид хрома очень сильный окислитель. Гигроскопичен, расплывается на воздухе. Хорошо растворяется в воде. Растворимость в воде растет с повышением температуры (рис. 3.4). Растворяясь в воде, образует хромовые кислоты, существующие лишь в водных растворах. Водные растворы триоксида хрома начинают кипеть лишь выше 102° С (рис. 3.5, а), замерзать ниже -7° С (рис. 3.5, б). Триоксид хрома хорошо растворяется в азотной кислоте (рис. 3.6).
Между хромовой Н2СгО4 и дихромовой Н2Сг2О7 кислотами в растворе сосуществует равновесие, сдвигающееся в процессе разбавления раствора в сторону образования хромовой кислоты. Обе эти кислоты сильные, они полностью диссоциированы по первой ступени. В Н8
концентрированных сильнокислых растворах СгОз образуются также ионы трихромовой СгзОю и тетрахромовой Сг4О^ кислот.
При действии на растворы триоксида хрома серы, бора, углерода и некоторых металлов (Mg, Zn, Си, Мо и др.) в процессе нагревания происходит полимеризация с получением хромовых полимеров — от вязкотекучих до смолоподобных и стекловидных. Образующиеся полимеры рентгеноаморфны, молекулярная масса 103—104. В этих процессах ионы металлов входят в состав полимеров, а неметаллы большей частью оказывают лишь каталитическое действие. Смолы обратимо растворяются в воде. Полимеры, кроме Cr(VI), содержат также Cr(III), Cr(IV), Cr(V).
Известны также соединения с содержанием кислорода больше, чем в СгО3: пероксиды, например синие перхромат Cr2(CF2Oio)3 и гидропероксид СгО(О2)2Н2О.
Способы получения триоксида хрома. Физико-химические основы процесса. Триоксид хрома получают разложением хромата (VI) натрия серной кислотой при температуре около 200° С:
Na2Cr2O7 + 2H2SO4 = 2CrO3 + 2NaHSO4 + H2O
Образующиеся при этом в жидком состоянии триоксид хрома и гидросульфат натрия практически не растворимы друг в друге. Однако легко отделяются друг от друга механически из-за разницы их плотностей. Получаемый гидросульфат натрия используют для перевода хромата (III) натрия в хромат (VI) натрия:
2Na2CrO4 + 2NaHSO4 = Na2Cr2O7 + 2Na2SO4 + H2O
119
Таким образом, основными видами сырья являются: концентрированный раствор хромата (III) натрия (не менее 925 г/л) и техническая серная кислота, содержащая не менее 92, 5% H2SO4.
Кинетика разложения СгО3 в двойной системе H2SO4—Na2SO4 приведена на рис. 3.7 Составы гидросульфатного плава в процессе выплавки СгСЬ лежат на диаграмме близко от ординаты, соответствующей составу Na2SO4H2SO4.
Рис. 3.5. Температура кипения (а) и замерзания (б) водных растворов триоксида хрома
120
Концентрация HNO3, % (масс.)
Рнс. 3.6. Растворимость триоксида хрома в водных растворах азотной кислоты при 25° С
Время, мин
Рнс. 3.7. Кинетика процесса разложения СгО3 в присутствии гидросульфата натрия и серной кислоты при 215° С:
/—100% H2SO4 2 —NaHSO4 + 35% H2SO4, 3 — NaHSO4 + 20% H2SO4, 4 —NaHSO,,
5 — NaHSO4 + 5,7% Na2SO4, 6 — NaHSO,+ 18% Na2SO4
Жидкий гидросульфат натрия водяного пара'
Температура, °C 188
Давление пара, мм рт ст 18
обладает значительным давлением
193 203 220 230
20 27 49 58
Выделяя пары воды, он медленно переходит в дисульфат натрия:
2NaHSO4 Na2S2O7 + Н2О
121
Плотность жидкого гидросульфата натрия при 200° С равна 1,99, а у СгОз — 2,25—2,30 г/см\ Такая разница в значениях плотности обусловливает интенсивное расплавление СгОз и гидросульфата натрия в процессе отстаивания. На скорость этого процесса влияют также вязкость и поверхностное натяжение расплавов. Низкая растворимость NaHSC>4 в расплаве СгОз способствует получению продукта высокой чистоты (99,4—99,8 СгОз).
В процессе выплавки СгОз протекают побочные реакции, снижающие степень использования исходного сырья и качество целевого п родукта:
1. Термическая диссоциация СгО3 с образованием фазы переменного состава в пределах от СгОз до СгО29б, а затем декахромат хрома СгОгЗО СгО3:
32 СгО3 = Сг2(Сг10Оз1)з + 1,5 О2
или в общем виде
(т + 2)СгОз = Сг2О3тиСгОз + 1,5О2
Экспериментально установлены следующие значения степени раз-
ложения в зависимости от температуры:
Температура, °C ........
Степень р<и;южения за 30 мин, %
Степень разложения за 60 мин, %
200 210 230 250
0,06 0,28 1,02
0,07 0,17 0,55 2,00
Изучен процесс разложения СгОз действием H2SC>4, NaHSO4 и их смесей. При этом показано, что заметная реакция СгО3 с концентрированной серной кислотой начинается при 135° С и при 190° С протекает энергично со значительным разогревом смеси. Скорость разложения СгОз заметно быстро растет с увеличением содержания свободной H2SO4. Одновременно в продуктах реакции повышается значение отношения нерастворимой хромисульфатсерной кислоты к хромисульфату (см. рис. 14.7). В смеси СгОз и NaHSO4 + 20% H2SO4 за 20 мин разлагается при 215° С 10% СгОз. Повышение температуры до 240—260° С увеличивает скорость разложения СгОз примерно в 2,5 раза.
2. Разложение СгОз серной кислотой или гидросульфатом натрия с образованием сульфата хрома и нерастворимой в воде натриевой соли хромисульфатсерной кислоты, накапливающихся в гидросуль-фатном плаве:
4СгОз + 6H2SO4 = 2Cr2(SO4)3 + ЗО2 + 6Н2О
Cr2(SO4)3 + xNaHSO4 = 0,5xNa2SO4-Cr2(SO4)3’yH2SO4 + (0,5х-y)H2SO4 122
3. Образование хлороводородной кислоты, хлорида хромила хрома из NaCI, содержащегося в хроматных (VI) растворах:
NaCl + H2SO4 = NaHSO4 + НС1
2HC1 + CrO, = CrO2Cl + H2O
6HC1 + 3H2SO4 + 2CrO, = 3C12 + Cr2(SO4), + 6H2O
Хлорид хрома, на выходе из реактора, гидролизуется конденсирующимися в газоходах парами воды:
СгО2С12 + 2Н2О = 2HCI + Н2СгО4
4. Образование гидросульфата натрия из Na2SO4, содержащегося в хроматных (VI) растворах:
Na2SO4 + H2SO4 = 2NaHSO4
Получение триоксида хрома. Согласно существующей технологии, исходные растворы хромата (VI) натрия упаривают в противоточных выпарных установках (рис. 3.8) с принудительной циркуляцией. Упаренные растворы содержат СгО3 не менее 925 г/л. Они должны быть нейтральными и прозрачными с содержанием нерастворимого остатка не более 0,3 г/л.
Упаренные растворы поступают в реактор для выплавки триоксида хрома (рис. 3.9), представляющий собой стальной цилиндрический аппарат 1 с вогнутым дном. Верхняя часть реактора расширена с целью создания резервной емкости 2 на случай сильного вспенивания реакционной массы. Раствор рабочей частью установлен внутрь электропечи 4 и зафутерован легковесным шамотным кирпичом 5. На футеровке по изоляторам закреплены нагревательные элементы 6. С целью постоянного перемешивания реакционной массы реактор снабжен рамной мешалкой 3, вращающейся электродвигателем 7 через редуктор 8.
Параллельно с растворами хромата (VI) натрия в реактор дозируется серная кислота. В первом периоде процесса, занимающем до 60% общего времени операции, происходит выпаривание воды, поступающей с сырьем и образующейся при реакции взаимодействия исходных Ма2Сг2С>7 с H2SO4. Реакционная масса вспенивается вследствие плохой очистки исходных хроматных растворов от нерастворимых примесей. Бурное смешивание массы уменьшают путем введения добавок хроматного плава или кислоты. В конце периода интенсивное кипение массы прекращается, поэтому объем массы уменьшается. Во втором 123
Рис. 3.8. Схема трехкорпуспои вакуум-выпарной сиоемы.
периоде процесса, занимающем примерно 20% всей продолжительности операции, происходит плавление гидросульфата натрия (при 186° С) и триоксида хрома (при 196—197° С). Температуру реакционной смеси доводят до 200—205° С. Небольшой перегрев выше температуры плавления триоксида хрома необходим для полноты разделения NaHSO4 и СгО-,. Недопустимо поднимать температуру выше 205° С, поскольку при этом ускоряется процесс разложения СгО-, и ухудшается отстаивание из-за интенсивного выделения кислорода. Серную кислоту вводят в систему на 5—10% меньше против стехиометрического количества. Поэтому в конце процесса, когда начинает плавиться СгОч, осторожно добавляют остальное количество серной кислоты с таким расчетом, чтобы общий расход ее составлял -105% от стехиометриче-ски необходимого количества. Добавление кислоты заканчивают при изменении цвета застывшей пробы гидросульфата натрия. Он должен 124
быть слабооранжевого цвета. Зеленый оттенок показывает на избыток, а красный — на недостаток кислоты. Наличие небольшого количества свободной кислоты в гидросульфате натрия способствует полноте отделения гидросульфата натрия от триоксида хрома благодаря уменьшению вязкости и плотности NaHSO4.
При достижении 200—205° С интенсивный нагрев прекращают, расплав спускают в отстойник, в котором автоматически поддерживается температура 198—200° С. С такой температурой масса выгружается в гранулятор, состоящий из двух плотно соприкасающихся между собой полых стальных барабанов, охлаждаемых изнутри водой и заключенных в стальной кожух. Масса из отстойника поступает в гранулятор по трубе. Барабаны приводят во вращение от электродвигателя через редуктор и вращаются в противоположном направлении. Каждый барабан оборудован ножом для снятия
в
Рис. 3.9. Реактор для получения триоксида хрома и гидросульфата натрия' / — реакционная часть, 2 — резервная часть, 3 — рамная мешалка, 4 — электропечь к реактору, 5 — огнеупорная футеровка, 6 — нагревательные элементы, 7 — электродвигатель, 8— редуктор мешалки
триоксида хрома и для снятия прилипше-
го материала из их поверхности. В процессе вращения барабана на их поверхности прилипает целевой продукт, который режется ножом,
переходит в шнеки, которые передают материал на транспортер с одновременным измельчением крупных чешуек. Гранулятор имеет поддон, который заполняется водой или раствором Na2Cr2O4 для смачивания наружной поверхности барабанов. Отработанный раствор из поддона периодически направляют в травочник.
Образующиеся на стадии выплавки газы и пары СгО2С12, С12, HCI, СгОд из реакторов направляют на установку по их обезвреживанию. Установка состоит из двух последовательно соединенных скру-берров с керамическими кольцами Рашига и орошаемых в противотоке распылителями циркулирующим раствором карбоната натрия или маточными растворами тиосульфата натрия — отходом производства сесквиоксида хрома.
В скруберрах кислые газы и пары взаимодействуют с карбонатом натрия с образованием соответствующих солей:
CrO,CI, + 2Na2COi = Na2CrO4 + 2NaCI + 2СО2
Cl, + Na,CO, = NaOCl + NaCl + CO,
125
Полученный целевой продукт содержит 99,2—99,3% СгО3, 0,16—0,20 SO*" и 0,17%
нерастворимого остатка.
Существует также схема получения триоксида хрома (рис. 3.10), согласно которой исходную серную кислоту из сборника 1 дозируют в реактор 4 через дозаторы 2. Параллельно в реактор дозируется хромат (VI) натрия дозатором 3. Реактор снабжен метальным устройством и имеет коническую нижнюю часть,
Рис. 3.10. Схема получения триоксида хрома кУДа ПОДЭЮТ исходные ВИ-/ — сборник серной кислоты; 2 3, 7 — дозаторы, ДЫ СЫрЬЯ. Коническая 4 - реактор 5 • противень для СгО, 6 — роликовый ЧЭСТЬ реактора обогревается транспортер 8—злеватор 9 —бункер Ю — сборник
гидросульфата натрия // — охлаждающие вальцы РЯДОМ НебоЛЫНИХ гаЗОВЫХ горелок, что обеспечивает равномерное нагревание до 200° С. Реактор одновременно является и отстойником.
Серную кислоту расходуют в количестве 106—110% от стехиометрического.
Образующиеся растворы гидросульфата натрия переливаются в сборник 10, затем на охлаждающие вальцы 77, в которых они охлаждаются и в виде чешуек тарируются. Триоксид хрома из реактора передается в противень 5 для СгОз и роликовым транспортером загружается в бункер 9 целевого продукта. Согласно технологии, продолжительность одной операции равна 2 ч.
Разработан метод ускорения процесса выплавки СгОз заменой серной кислоты олеумом и использованием высококонцентрированного плава хромата (VI). При удельном расходе хромата (VI) натрия (67,1% СгОз) 1,53 т, избытке кислоты 5%, концентрации плава хромата (VI) 62% СгОз и кислоты 92,5% H2SO4 приходится выпарить в реакторе на 1 т целевого продукта (СгОз) -480 кг воды [в том числе -300 кг из плава исходного хромата (VI)]. С применением безводного хромата (VI) и 20%-ного олеума, подогретого до 150° С, процесс выпарки воды практически не требуется и реакция становится автотермической.
Разработан способ производства СгОз разложением хроматов (VI) натрия и калия серной кислотой с кристаллизацией СгОз из раствора:
Na2Cr2O7(K2Cr2O7) + 2H2SO4 + Н2О = 2H2CrO4 + 2NaHSO4(KHSO4)
126
Разработан также способ разложения серной кислотой хроматов металлов (Са, Sr, Ва, РЬ), образующих нерастворимые сульфаты:
МеСгОд + H2SO4 = MeSO4 + H2CrO4
Недостатком этих способов является образование отходов нерастворимого сульфата, загрязненного хроматом.
Предложен способ разложения хромата (III) и хромата (VI) калия, натрия и кальция азотной кислотой:
Na2CrO4 + 2HNO3 2NaNO3 + H2CrO4
Na2Cr2O7 + 2HNO, + H2O 2NaNO3 + 2H2CrO4
В отличие от способов разложения серной кислотой азотокислотное разложение обеспечивает получение ценного отхода — нитрата натрия. Однако основной трудностью этого способа является разделение СгО3 и NaNO3. Предложено хромат (VI) натрия разлагать 62—67%-ной азотной кислотой, а выпавший нитрат натрия отделить от маточного раствора, содержащего 50% СгО3 и 6—10% NaNO3. Нитрат натрия, промытый двукратно водой, содержит всего 0,3% СгО3. Маточный раствор упаривают в вакуум-выпарном аппарате (при температуре ниже 70—80° С) до кристаллизации ~70% СгО3, который затем отделяют от растворов в центрифуге, промывают и сушат. Полученный целевой продукт содержит более 99,5% СгО3 и следы NaNO3. Выход СгО3 и NaNO3 составляет -95%. Нитрат натрия можно удалить из маточного раствора обработкой растворами гексафторосиликата водорода:
2NaNO3 + H2SiF6 = 2HNO3 + Na2SiF6
CrO3 получают разложением хромата (VI) натрия гексафторосиликатом водорода:
Na2Cr2O7 + H2SiF6 + Н2О = Na2SiF6 + 2H2CrO4
Целевой продукт СгО3 получают концентрированием фильтрата после отделения Na2SiFf,.
Известен способ гидролитического расщепления тригидрата хромата (VI) кальция СаСг2О7-ЗН2О при 105—125° С в кристаллизационной воде:
СаСг2О7-ЗН2О СаСгО4 + СгО3 + ЗН2О
127
Предложен способ перевода хромата (III) кальция в хромат (VI) диоксидом углерода или серной кислотой, получая при этом триоксид хрома из хромата (VI) кальция, полученного из хроматного плава (спека) известково-хроматовой шихты.
Разработан способ получения триоксида хрома окислением его гидроксида кислородом Для этого из раствора сульфата хрома водным раствором NaOH осаждают гидроксид хрома и добавляют 0,5% антрахинон а-сульфоновой кислоты и 10% активированного угля в качестве катализатора. При этом в атмосфере кислорода при достаточно длительном освещении дневным светом происходит количественное окисление до Н2СгО4.
Триоксид хрома получают окислением хромсульфатных растворов диоксидом свинца. При этом кислые растворы сульфата хрома подвергают кипячению с избытком активированного диоксида свинца до перехода хрома (III) в хром (VI) по уравнению
Cr2(SO4)3 + ЗРЬО2 + 5Н2О = 2СгО3 + ЗРЬО + 3H2SO4 + 2Н2О
Отфильтрованный оксид свинца просушивают и подвергают термообработке в смеси с карбонатом натрия в сильном токе воздуха. Образующийся плюмбат натрия гидролизуют и РЬО2 возвращают на начало процесса:
2РЬ0 + 2Na2CO3 + О2 = 2Na2PbO3 + СО2
Na2PbO3 + И2О = РЬО2 + 2NaOH
Окисление Cr2(SO4)3 возможно и диоксидом марганца:
Cr2(SO4)3 + ЗМпО2 + 2И2О = 2Н2СгО4 + 3MnSO4
Триоксид хрома получают анодным окислением соединений трехзамещенного хрома. Отработанный раствор, содержащий —100 г/л Сг2О3 и,350 г/л H2SO4, подвергают при 30—50° С электролизу в ванне с диафрагмой и свинцовым анодом.
Процесс у анода:
2Сг3' + 7Н2О = СгэО^ + 14Н+ + бе
Процесс у катода:
6Н+ + бе = ЗН2
128
Свинец в качестве анода имеет большое значение в процессе: образующийся на его поверхности РЬО2 каталитически разлагает получающийся сначала на аноде Н2О2, который в кислой среде восстанавливает хроматы. Процесс ведут при напряжении на ванне около 3,5 В, плотности тока на аноде 120—300 а/м2 и более высокой плотности на катоде. Это снижает степень восстановления Выход по току достигает 85—90%. Расход электроэнергии составляет -2600 кВт ч на 1 т СгО2.
Предложен также способ получения СгОч электролизом раствора хромата (VI) аммония с применением анода из феррохрома. Разработан также способ получения триоксида хрома электролизом раствора Na2CrO4. Метод основан на переносе хромат-иона к аноду и иона натрия к катоду. При этом параллельно получают гидроксид натрия и водород у катода и кислород у анода.
Способы получения сесквиоксида хрома. Сесквиоксид хрома, согласно требованиям существующего ГОСТ 2912—83, производится в трех видах: «металлургический», «пигментный», «часовой». Согласно требованиям ГОСТа, содержание основного вещества находится в пределах 98—99% Сг20ч, влаги — 0,15%, нерастворимых в воде веществ — 0,3—0,5%. Остальные требования отличаются в зависимости от вида продукта. Так, для «металлургического» сесквиоксида хрома требуется отсутствие мышьяка, цинка, олова, свинца, сурьмы, кадмия и висмута, в то время как для других видов продукта указанные примеси допустимы. Для «пигментного» продукта регламентируются pH водной вытяжки, красящая способность от эталона, цвет и оттенок, а также укрывистость и т.д. Для «часового» и «металлургического» видов не нормируется остаток на ситах и т.д.
Широко применяемыми в промышленности способами получения сесквиоксида хрома являются: 1) восстановление хромата (VI) натрия в растворе элементной серой (хроматно-серный способ); 2) термическое разложение триоксида хрома до сесквиоксида.
Разработаны также и другие способы, некоторые из них применяются в производстве.
Хроматно-серный способ. Хроматно-серный способ имеет два варианта с одностадийным восстановлением и упрощенный — с двухстадийным восстановлением.
По первому варианту сначала восстанавливают щелочной раствор Na2CrO4 элементной серой при кипячении:
4Na?CrO4 + 6S + (2и + 1 )Н2О = (1)
= 2(Сг2ОчпН2О) + 3Na2S2O4 + 2NaOH
129
Образующийся после отделения гидроксида хрома щелочной раствор тиосульфата натрия разлагают серной кислотой:
3Ma2S2O4 + H2SO4 = 3Na2SO4 + 4S + H2O (2)
Серу отделяют и возвращают на восстановление исходного хромата (VI) натрия, а раствор сульфата натрия перерабатывают на товарный продукт.
По упрощенной схеме щелочной раствор тиосульфата натрия не отделяют, а используют для восстановления хромата (VI) натрия по суммарной реакции
10Na2CrO4 + 3Na2S2O, + 7H2SO4 + 2иН2О = (3)
= 2(2Сг2ОзСгО,иН2О) + 13Na2SO4 + 7Н2О
При этом схема упрощается за счет исключения передела регенерации элементной серы и ряда операций по переработке гидроксида хрома.
Преимущество хроматно-серного способа заключается в использовании недорогостоящего исходного сырья — раствора хромата (VI) натрия.
Физико-химические основы производства. Необходимость некоторого количества гидроксида натрия в начале процесса диктуется тем, что процесс восстановления, приведенный в уравнении (1), фактически проходит в несколько стадий и является автокаталитическим. Исходя из химизма образования тиосульфата натрия при взаимодействии серы с гидроксидом натрия предложена следующая схема реакций:
18ОН + 9S = 3SOj“ + 6S2’ + 9Н2О (4)
3SO*’ + 3S = 3S£>t (5)
8СЮ^ + 6S2“ + (11 + 4н)Н2О = 4(Сг2Ог«Н2О) + ЗБ/)," + 22ОН~ (6)
Суммарная реакция
8СЮд + 12S2 + (2 + 4и)Н2О = 4(Сг2ОуиН2О) + бЗзО; + 40 Н (7)
Как видно, уравнение (7) аналогично уравнению (1), но выражено в ионной форме.
По другим данным процесс гидролиза серы при кипячении с водой идет с образованием сульфоксиловой кислоты H2SO2, разлагающийся в щелочном растворе с образованием тиосульфат-иона. Поэто-130
му считается вероятной следующая схема процесса восстановления хромата серой:
I2OH + 12S = 6S2’ + 6H2SO2 (8)
6H2SO, + 6ОН = ЗЗгО*" + 9Н2О (9)
При этом сульфит-ион может образоваться в результате побочной реакции:
2CrO; + 2ОН + 3H2SO2 + иН2О = Сг2О3иН2О + 3SO* + 4Н2О (10)
Кроме указанных реакций, в некоторой степени протекает реакция
8Na2CrO4 + 3Na2S + (8 + 4и)Н2О = (11)
= 4(Сг2О3Н2О) + 3Na2SO4 + 16NaOH
При избытке серы маточные растворы имеют желто-бурый цвет полисульфидов натрия, образующихся по реакциям:
6NaOH + 2(п + 1)S = 2Na2S„ + Na2S2O3 + ЗН2О (12)
Na2S + (и—1)S = Na2S„ (13)
Далее, в процессе смешения суспензии после первой стадии восстановления с раствором Ма2СгО4 происходит окисление полисульфидов натрия до тиосульфата натрия:
8Na2CrO4 + 3Na2S2 + (8 + 4и)Н2О = (14)
= 4(Сг2О3«Н2О) + 3Na2S2O3 + 16NaOH
Осаждающийся из щелочного раствора гидроксид хрома содержит значительное количество неотмываемой водой щелочи (7—8% NaOH в пересчете на сухое вещество), возможно, адсорбированной. В процессе окислительной термообработки, в процессе работы по варианту одностадийного восстановления, гидроксид хрома переходит в сесквиоксид хрома, а примеси (адсорбированная щелочь и неотмы-тые соли натрия) переходят в окислительной среде в хромат и соответственно сульфат натрия (часть сульфата натрия также дает хромат), которые затем выщелачивают из плава:
Сг2О3иН2О = Сг2О3 + иН2О (15)
8NaOH + 2Сг2О3 + ЗО2 = 4Na2CrO4 + 4Н2О (16)
4Na2SO4 + 2Сг2О3 + О2 = 2Na2CrO4 + 4SO2 (17)
131
В результате реакций (16) и (17) в хромат переходит 10—13% оксида хрома.
В связи с поглощением диоксида серы из дымовых газов, в процессе очистки их от пыли, скрубберная суспензия может стать кислой согласно реакции
7Na2CrO4 + 6SO2 + nH2O = (18)
= Na2Cr2O7 + 2Сг2ОуСгОуиН2О + 6Na2SO4
Образующаяся суспензия, снижая щелочность серной суспензии, может привести к торможению процесса восстановления Na2CrO4 в реакционном каскаде С целью предотвращения этого поддерживают небольшую щелочность скрубберной суспензии путем добавления к ней щелочного тиосульфатного раствора.
В процессе восстановления хромата тиосульфатом в зависимости от условий (pH, температура, массовое отношение Na2CrO4:Na2S2C>3, равное т в исходном растворе) могут протекать реакции неполного восстановления хромата с образованием гидратированных хроми-хро-матов или полного восстановления с образованием гидроксида хрома, а также реакции неполного окисления тиосульфата с образованием политионатов.
При т = 1,67-ь2,73, pH 5—6 и 150° С протекает реакция полного восстановления:
8Na2CrO4 + 3Na2S2O3 + 5H2SO4 + (4и—5)Н2О = (19)
= 4(Сг2От«Н2О) + HNa2SO4
, 162 8 „
(реакции отвечает т =-----= 2,73).
158 3
В действительности реакция идет между ионами СгтО,', образующимися в процессе взаимодействия хромата и кислоты, и ионами S2O, и Н+:
2СГ2О^ + 3S2O,’ + 2Н+ + (4и-1 )Н2О = 4(Сг2ОгиН2О) + 6SO‘~ (20)
При ти>3,35, те значительном избытке Сг6+, протекает реакция неполного восстановления:
при 100° С
12 Na2CrO4 + 3Na2S2O4 + 9H2SO4 + (4и-9)Н2О = (21)
= 4(Сг2О3СгОзиН2О) + 15Na2SO4
а при 150° С — реакция (3).
132
В качестве примера реакции неполного окисления тиосульфата натрия приведем:
2Na2CrO4 + 6Na2S2O3 + 5H2SO4 = (22)
= Cr2O3«H2O + 3Na2S4O6 + 5Na2SO4 + (5-и)Н2О
В процессе окисления автоклавной обработки суспензии после второго восстановления достигается практически полное окисление тиосульфата и политионатов до сульфат-ионов.
Политионаты окисляются по уравнению
7Сг2О,” + 3S4O*“ + (7л + 2)Н2О = (23)
= 7(Сг2О3-иН2О) + 12SOf + 4Н+
Необходимость полного окисления восстановителей до выпарки раствора сульфата натрия трактуется исключением процесса восстановления хромата в процессе выпаривания с образованием хро-ми-хроматов, приводящих к повышенному расходу гипохлорита кальция и загрязнению товарного сульфата натрия.
Осадок со второй стадии восстановления кроме гидратированных хроми-хроматов содержит небольшое количество основных сульфатов хрома, образующихся в процессе взаимодействия хроми-хроматов и гидроксида хрома с серной кислотой.
Процессы, происходящие при термообработке по упрощенной схеме, выражаются уравнениями:
4(2Сг2О3-СгО3иН2О) = 10Сг2О3 + ЗО2 + 4иН,О (24)
2Cr2(SO4)3 = 2Сг2О3 + 6SO2 + ЗО2 (25)
Кроме основных реакций, связанных с непосредственным получением сесквиоксида хрома, по уравнению (2) при разложении серной кислотой тиосульфатного раствора протекают побочные реакции:
2NaOH + H2SO4 = Na2SO4 + 2Н2О (26)
Na2SO3 + H2SO4 = Na2SO4 + SO2 + H2O (27)
Na2S + H2SO4 = Na2SO4 + H2S (28)
В условиях местного перекисления протекают реакции:
S2O; + 2Н+ = Н2О + S + SO2 (29)
2S20j + 2Н+ = S3O^ + H2S (30)
133
Образующийся диоксид серы взаимодействует с H2S и Na2S2O3 с выделением элементной серы:
2H2S + SO2 = 3S + 2Н2О (31)
гМа^О, + SO2 = 2Na2SO4 + 3S (32)
Помимо реакции (30) могут протекать реакции образования политионатов, например:
2S2O“” + 3SO2 = S,O*” + S40jr” (33)
5S2O;” + 6H = 2S£>7 + 2(5-x)S + 3H2O (где x = 3-=-6) (34)
+S = Sx+IOr (35)
При изменении pH среды политионаты могут разлагаться:
SVO^ = SO,” + SO2 + (x-2)S (36)
Протекание реакций определяется относительным количеством и концентрацией кислоты. Разрушение политионатов щелочью исходного тиосульфатного раствора или карбонатом натрия идет по следующим реакциям, где х = 3-ьб:
2Na2SvO6 + 6NaOH = (10—2x)Na2SO, + (2х—5)Na2S2O, + ЗН2О (37)
2Na2SvO6 + ЗМа2СО3 = (10—2x)Na2SO, + (2х—5)Na2S2O3 + ЗСО2 (38)
Получение сесквиоксида хрома одностадийным восстановлением (рис. 3.11). Согласно схеме получения с термообработкой отхода — тиосульфатного раствора — исходную серную суспензию готовят путем измельчения исходной элементной серы в шаровой мельнице в растворе хромата (VI) натрия. Полученную серную суспензию классифицируют в гидроциклоне.
Серную суспензию смешивают с суспензией регенерированной серы (из тиосульфатного раствора) и оборотной пульпой из скрубберов печей (термообработка гидроксида хрома) и образующуюся смесь направляют в каскад реакторов, где при температуре кипения раствора хромата (VI) натрия (102—103° С) происходит процесс восстановления серой по реакции (1)
Образующийся гидроксид хрома отделяют от тиосульфатного раствора на барабанных вакуум-фильтрах с промывкой пасты горячим конденсатом.
134
Раствор Na2CrO«
Сера
Упаковка сесквиоксида
Рис. 3.11. Принципиальная схема получения сесквиоксида хрома хроматно-сернь способом (вариант одностадийного восстановления)
Промытая паста гидроксида хрома подвергается окислительной термообработке. Процесс проводится во вращающихся барабанных печах с доведением температуры плава примерно до 1150° С и выше. При этом гидроксид хрома переходит в сесквиоксид хрома, а адсорбированная щелочь и содержащие серу соединения — в Na2CrO4 и Na2SO4.
Плав сесквиоксида хрома выщелачивают и полученный слабый раствор Na2CrO4 отделяют фильтрацией в две стадии на барабанных вакуум-фильтрах.
Промытую пасту сесквиоксида хрома высушивают в барабанных сушилках с наружным обогревом. Сухой сесквиоксид упаковывают в стальные барабаны
Образующиеся тиосульфатные растворы делят на две части. Часть растворов смешивают с маточным раствором, полученным после отделения кристаллов сульфата натрия на последней стадии переработки. Образующуюся смесь подогревают до кипения (-100° С) и направляют в каскад реакторов в смеси с дозированным количеством серной кислоты. В реакторах происходит разложение тиосульфата натрия [см. реакцию (2)].
Полученную в каскаде реакторов суспензию в сульфатном растворе сгущают в отстойнике Дорра, а сгущенную суспензию смешивают с дозированным количеством раствора хромата (VI) натрия и направляют на восстановление хромата.
Нейтрализованный раствор отделяют на фильтре ФПАКМ от серы и гидроксидов, выпавших в процессе нейтрализации. Осветленный раствор концентрируют в вакуум-выпарной установке. Выпавшие кристаллы сульфата натрия отделяют от маточного раствора на центрифуге, а при необходимости сушат в барабанной сушилке. Сульфат натрия является товарным продуктом, а маточный раствор направляют на процесс разложения.
Получение сесквиоксида хрома двухстадийным восстановлением представлено на рис. 3.12. Схема упрощена за счет исключения по-слепрокалочных стадий: выщелачивания, фильтрации и сушки сесквиоксида хрома. Это возможно, так как вторая стадия восстановления проводится в слабокислой среде и поэтому выпадающий в осадок гидратированный хроми-хромат в своем составе не содержит адсорбированной щелочи и после тщательной промывки и термообработки получают кондиционный металлургический сесквиоксид хрома.
Согласно схеме, часть исходного хромата (VI) натрия восстанавливают в автоклаве жидкой серой [см. реакцию (1)].
Во второй стадии восстановления хромата натрия суспензию гидроксида хрома в щелочном тиосульфатном растворе от первой стадии восстановления смешивают с оставшейся частью раствора Ма2СгО4 и последний восстанавливают при температуре кипения путем добавле-136
ния серной кислоты. Отфильтровывают выпавший хроми-хромат и процесс восстановления хромата (VI) натрия в фильтрате [см. реакцию (3)] завершают примерно при 150° С в каскаде автоклавов.
Гидратированные хроматы, полученные во второй стадии восстановления, отделяют двухстадийной фильтрацией от раствора сульфата натрия, который используют в производстве Ма2Сг2О7 для промывания сульфата натрия.
Раствор Na2CrO4
-.кРу^.Рная.> Г Второе восстановление суспензия L
Первая фильтрация
Репульпация
Фильтрат II
фильтрация
-J
Прокаливание
улавливание
-j Смешение
J Серная
Первое восстановление
| Сесквиоксид хрома ~
Фильтрат I __________________ клавирование
Фильтрация на фильтрпрессе
Рис. 3.12. Принципиальная схема получения сесквиоксида хрома хроматно-серным способом (вариант двухстадийного восстановления)
137
Полученные гидратированные хроми-хроматы подвергают термообработке с получением товарного продукта [см. реакции (24) и (25)]. Перед затариванием продукт охлаждают в аппаратах кипящего слоя.
Для создания оптимальных условий восстановления УНИХИМом рекомендованы следующие параметры:
Число реактивов в каскаде pH реакционной среды
в I реакторе во II реакторе . ..........
в 111 реакторе в IV реакторе
Продолжительность пребывания в каждом реакторе, мин
Температура реакциоппон массы. °C
Температура исходного раствора, °C Концентрация исходного раствора, г/л Na2CrO4 Массовое отношение Na1CrO4‘Na2S2O4 в исходном растворе
7,0±0,1
6,0±0,1
5,5±0,1
5.5+0,1
30—60
100—105
50—60
130—160
Получение сесквиоксида хрома термическим разложением его триоксида. Из приведенной принципиальной схемы получения сесквиоксида хрома термическим разложением его триоксида (рис. 3.13) видно, что она состоит из следующих основных стадий: 1) термообработка триоксида хрома в барабанной вращающейся печи; 2) выщелачивание плава и отделение водорастворимых солей от сесквиоксида хрома; 3) сушка сесквиоксида хрома.
Стадии 2 и 3 проводят в основном так же, как и в процессе получения сесквиоксида хрома хроматно-серным способом.
Изучение системы СгО-,—Сг2О4 показывает, что термическое разложение триоксида хрома, выражаемое суммарным уравнением:
4СгО, = 2Сг20з + ЗО2
в действительности протекает через ряд промежуточных стадий.
По литературным данным, в процессе разложения триоксида хрома образуются следующие химические соединения с типичными термо- и рентгенограммами:
1) декахромат хрома Сг2(СгюО-!1)з = СгО2 90б (серого цвета, плотность 2,88 г/см3);
2) дихромат хрома Сг2(Сг2О7)з = Сг8О21 = СгО2 625 (черного цвета, плотность 3,07);
3) монохромат хрома Сг2(СгО4)4 = Сг5О,2 = СгО240 (черного цвета, плотность 3,29).
В системе имеются также две узкие гомогенные области в пределах составов: первая — от СгО2 до СгО2% и вторая — от СгО, 56 до СгО, 50. 138
Рис. 3.13. Принципиальная схема получения сесквиоксида хрома термическим разложением его триоксида
Время, мин
Рис. 3.14. Термограмма и политерма
Имеющиеся результаты хорошо иллюстрируют приведенные термограмма и политерма разложения (рис. 3.14) и диаграмма состав — температура при различной продолжительности обработки (рис. 3.15).
Большой эндотермический эффект (рис. 3.15, кривая 7), начинающийся при 202° С, соответствует процессу плавления СгОз. Кривая выделения кислорода (рис. 3.15, кривая 2) показывает, что разложение СгОз начинается еще незадолго до его плавле
ния (участок ав кривой). В процессе дальнейшего повышения температуры до 280° С равномерно
увеличивается вязкость расплава, показывающая полимеризацию жидкой фазы переменного состава в пределах от СгОз до СгО2 96, которая одновременно разлагается с образованием твердого серого продукта состава Сг02,доб-
Большой экзотермический эффект от 280 до 365° С, сопровождаемый интенсивным процессом выделения кислорода (рис. 3.15, участок вг кривой 2), соответствует разложению декахромата хрома с образованием черного крупнопористого продукта состава СгО2 625. Более детальное рассмотрение показывает, что процесс на этом участке протекает в две стадии, с промежуточным образованием газообразного СгОз (выделяется красный дым) по схеме
СгО2,90б(тв) -> CrO2 62s(tb) + СгОз(г) + О2; СгОз(г) = СгО2>625(тв) + О2
Небольшой экзотермический эффект в температурных пределах 365—410° С, сопровождаемый выделением О2 (рис. 3.15, участок гд кривой 2), соответствует разложению бихромата хрома с образованием черного монохромата Сг024о (сопровождается незначительным выделением газообразного СгОз, который разлагается на СгО2л0 и О2). Последний эндотермический эффект, начинающийся при 465° С, отвечает разложению СгО2,40 Д° СгО, 56, т.е. до сесквиоксида хрома, содержащего небольшой избыток кислорода. На участке де кривой 2 (рис. 3.15), отвечающем данному эффекту, наблюдается очень сильное выделение кислорода. Полное удаление активного кислорода наступает лишь при температуре выше 800° С.
140
600
Cr5Oi2
150 —i----1-----u------1-----1------1-----1----1---
3,0 2,8 2,6 2,4 2,2 2,0 1,8 1,6 1,5
СгОз Число атомов О на 1 атом Сг Сг2Оз
Рис. 3.16. Диаграммы состав — температура для системы триоксид хрома — сесквиоксид хрома в неравновесном состоянии, полученные при различной продолжительности обработки (препарат выдерживался при каждой температуре) / — 30 мин 2 — 4 ч 3 — 8 ч -I—15 ч 5 — при непрерывном нагревании со скоростью
6 град/мин
Термографические данные согласуются с диаграммами (см. рис. 3.15), из которых видно значительное влияние фактора времени на температурные границы существования разных фаз в системе СгОз—Сг20з. чем продолжительнее время обработки, тем ниже температурная область существования отдельных фаз.
Подводя итоги, можно сказать, что термическое разложение СгОз протекает примерно по схеме
СЮ, СгО296—СгО2,7 285-300'с > СгО2625 34°-370'с->
34°-37°.Т....» СгО2 40 45o-49o;-q-» СгО.,56 СгО, 50
141
Сесквиоксид хрома несколько летуч при 1000°С в окислительных условиях, по всей вероятности, из-за образования СгО3.
В исходном жидком триоксиде содержится некоторое количество NaHSO4 и нерастворимого кислого сульфата хрома. Последний может образоваться в процессе термообработки NaHSO4 и оксидов хрома. Во избежание загрязнения целевого продукта серой к триоксиду хрома рекомендуется добавлять перед вводом его в печь карбонат натрия, который переводит гидросульфат в сульфат натрия, а нерастворимый сульфат хрома (III) в его оксид и сульфат натрия, выщелачиваемый из плава водой:
Cr2(SO4)3 + 3Na2CO3 = Cr2O3 + 3Na2SO4 + 3CO2
Вместо карбоната натрия можно применить и хромат (VI) калия:
Cr2(SO4)3 + 7К2Сг2О7 = 4К2СгО4 + 3K2SO4 + 6Сг2О3 + 7,5О2
Другие способы получения сесквиоксида хрома. Сесквиоксид хрома для полировальных паст ГОИ и часовой промышленности состоит из порошкообразного сесквиоксида хрома (72—86%), связанного смесью парафина, стеарина, олеиновой кислоты и керосина. Размер частиц полировального сесквиоксида хрома составляет 0,4—1,2 мкм.
Сесквиоксид хрома для полировальных паст получают восстановлением хромата (VI) натрия или калия элементной серой:
Na2Cr2O7 + S = Cr2O3 + Na2SO4
Реакция протекает автотермически. Размолотую в шаровой мельнице смесь Na2Cr2O7 и серы поджигают в футерованной кирпичом восстановительной камере, под которой выложен чугунными плитами. Температура поддерживается выше 100° С. Полученный плав измельчают и выщелачивают в мельнице мокрого помола, и оксид хрома тщательно отмывают горячей водой от водорастворимых солей. Отмытый сесквиоксид хрома классифицируют отмучиванием в воде. Различные по крупности фракции дают и различные марки полировального сесквиоксида хрома.
Сесквиоксид получают восстановлением хромита (VI) калия древесным углем:
K2Cr2O7 + 2С = Сг,О3 + К2СО3 + СО
При этом исходный мелкопористый хромат (VI) калия и подсушенный уголь (в соотношении 2:1) размалывают в шаровой мельни-142
це. К полученной смеси добавляют хромат (VI) калия из расчета содержания в шихте 9—11% угля и направляют шнековым смесителем на восстановление в цепную печь непрерывного действия типа аглоленты. При 300—400° С смесь в печи загорается. Продолжительность процесса ~5 мин. Образующийся плав выщелачивают. Пасту Сг20з, полученную после выщелачивания и двухстадийной фильтрации, высушивают в барабанной сушилке с наружным обогревом. Сухой продукт обезуглероживают окислительной термообработкой в электропечи при 650—700° С, а затем репульпируют, вновь отфильтровывают, промывают от Na2CrO4 и высушивают. Растворы К2СОз (первый фильтрат с 30—50% К2СО3) перерабатывают на поташ.
Сесквиоксид хрома получают восстановлением хромата (VI) натрия хлоридом аммония:
Na2Cr2O7 + 2NH4CI = Сг2О3 + 2NaCl + N2 + 4Н2О
Исходную смесь раствора Na2Cr2O7 (850—1080 г/л СгОз) с измельченным хлоридом аммония восстанавливают в барабанной вращающейся печи при 900—1000° С. Полученный плав выщелачивают. Сесквиоксид хрома промывают водой и высушивают.
Сесквиоксид получают восстановлением кристаллического хромата (VI) натрия аммиаком:
2Na2CrO4 + 2МН3 = 2Ма2ОСг2О3 + N2 + ЗН2О
Образующийся хромит (III) натрия разлагают водой с участием СО2:
2Ма2О Сг2Оз + 3 И2О + 2СО2 = 2Сг(ОН)3 + 2Иа2СОз
Рекомендуется восстановление хромата (VI) натрия углем при 700—800° С по схеме
4Ма2СгО4 + ЗС = 2 Ма2Сг2О4 + 2Па2СОз + СО2
Термодинамически более вероятна реакция восстановления Na2CrO4 оксидом углерода:
2Na2CrO4 + ЗСО = Na2Cr2O4 + Па2СОз + 2 СО2
Для разрушения хромата (III) натрия и получения в процессе восстановления углеродистыми и другими восстановителями относительно чистого оксида хрома рекомендуется добавление в шихту соединения алюминия, образующего алюминат натрия:
2Ма2СгО4 + 4А1(ОН)з + ЗСО = Сг2Оз + 4МаА1О2 + 6Н2О + ЗСО2
143
Соединение алюминия регенерируют из раствора взаимодействием с гидрокарбонатом натрия:
NaAIO2 + Н2О + NaHCO3 = Na2CO3 + А1(ОН)3
Рекомендован способ восстановления хромата (VI) натрия при 650—700° С смесью серы и угля с одновременным получением сульфида натрия:
2Na2CrO4 + 2S + 5С = Cr2O3 + 2Na2S + 5СО
Восстановление хромата (VI) калия красным фосфором дает почти теоретический выход Сг2О3 по реакции
3K2Cr2O7 + 2Р = 2К3РО4 + ЗСг2О3 + 2О2
Сесквиоксид получают также восстановлением твердых хроматов (VI) калия водородом:
K2Cr2O7 + ЗН2 = Cr2O3 + 2КОН + 2Н2О
2К2СгО4 + ЗН2 = Cr2O3 + 4КОН + Н2О
Разработан способ получения сесквиоксида восстановлением раствора Na2CrO4 сульфидом натрия:
8СЮ; + 6S2" + (I I +4и)Н2О = 4(Сг2О3пН2О) + ЗЗэО^ + 22ОН"
В нагретый до 100° С раствор Na2CrO4 (не более 100—200 г/л) вводят раствор Na2S в молекулярном отношении Na2CrO4 : Na2S = 8 : 7 и полученную суспензию выдерживают при 100—106° С в течение 2 ч. В литературе имеются сведения о возможности получения сесквиоксида хрома по следующим реакциям:
8Na2CrO4 + 3Na2S + 20Н2О = 8Cr(OH)3 + 3Na2SO4 + 16NaOH
2Na2CrO4 + 3Na2S + 8H2O = 2Cr(OH)3 + lONaOH + 3S
В УНИХИМе (Л.Г.Булатова и Я.Е.Вильнянский) изучили процесс взаимодействия хлоридов хрома с кислородом и водяным паром. По полученным ими данным трихлорид хрома переходит при 500° С в токе воздуха в сесквиоксид хрома в течение 1ч по реакциям:
4СгС13 + ЗО2 = 2Сг2О3 + 6С12
4СгС12 + ЗО2 = 2Сг2О3 + 4С12 144
Кинетические исследования показали, что реакция трихлорида хрома с кислородом имеет автокаталитический характер и скорость ее удовлетворительно выражается топокинетическим уравнением Колмогорова— Ерофеева. При 350—400° С процесс протекает в кинетической области, при 500—700° С — в диффузионной. При 350—450° С реакция идет с выделением наряду с хлором значительного количества хлорида хромила; в интервале 500—700° С количество последнего незначительно, а при 800° С он снова появляется в результате реакции кислорода в холодной зоне с возгоном СгС13. Оптимальная температура процесса 560—600° С (в атмосфере кислорода).
3.2. ХРОМАТЫ
Хроматами называют соли хромовых кислот. Наиболее широко встречаются хроматы (VI) — производные триоксида хрома (СгОз) желтого или красного цвета (табл. 3.2). Получают их взаимодействием растворов Н2СгО4 или СгО3 с оксидами, гидроксидами, карбонатами металлов, осаждением из растворов солей при действии хроматов натрия или калия, а также окислением соединений, содержащих хром в низших степенях окисления. Существует несколько относящихся к хроматам минералов, например крокоит РЬСгО4. Хроматы (VI) обычно изоморфны с соответствующими сульфатами. Анион СгО^ имеет тетраэдрическое строение, энергия образования Гиббса AG° = -730 кДж/моль.
Хроматы щелочных металлов и магния хорошо растворимы в воде, у хроматов щелочно-земельных металлов растворимость сильно уменьшается от кальция к барию. Растворы хроматов имеют щелочную реакцию. Хроматы тяжелых металлов в воде практически не растворяются, в особенности соли Pb, Bi, Ag, Hg. Хроматы редкоземельных элементов плохо растворяются, причем соединения элементов иттриевой группы водой разлагаются с образованием основных солей. Хроматы щелочных и щелочно-земельных металлов плавятся без разложения, а хроматы остальных металлов разлагаются до плавления.
В процессе подкисления растворов хроматов образуются дихроматы, что сопровождается переходом их окраски в оранжевую. Дихро-мат-анион Сг^О, построен из двух хромкислородных тетраэдров с общей вершиной по атому кислорода, как показано ниже:
О
О
145
Соединение Сингония
КСгО4 (NH^CrO, (NH4)2Cr2O7 CuCrO4 Ag2CrO4 СаСгОч Ромбическая Моноклинная Ромбическая Тетрагональна
SrCrO4 Моноклинная
BaCrO4 ZnCrO4 Ромбическая
a-PbCrO4 Моноклинная
P-PbCrO4 PbjCrOj CoCrO4 Ромбическая Моноклинная Ромбическая
Таблица 3.2. Свойства некоторых хроматов (VI)
Параметры кристаллической решетки Пространственная группа Плотность, Дж/(мольх хК) дя:,.. кДж/моль Дж/(мольх хК)
£7, НМ 6, нм с, нм Р, град
0,593 1,042 0,763 — 4 Ртсп 2,73 146 -1408 200
0,615 0,630 0,776 115,22 — — 1,89 — -1163 657
0,774 0,754 1,376 93,70 2 C2Jc 2,15 293 -1810 —
0,5433 0,8968 0,5990 — 4 Стет 4,04 — — —
1,0063 0,7029 0.5540 — 4 Рпта 5,62 142 -732 218
0,725 — 0,634 — 4 J4lamd 3,12 113 — 131
0,6794 0,7131 0,6489 102,90 — — 3,895 — — —
0,9105 0,5541 0,7343 — 4 Рпта 4,50 — -1429 172
0,5505 0,8383 0,6219 — 4 Стет 3,40 — — —
0,710 0,740 0,680 77,50 4 РЪ/с* 6,12 — -925 167
0,867 0,559 0,719 — 4 Рита — — — —
1,4018 0,5683 0,7143 115,23 4 СИт 6,27 — — —
0,5516 0,8336 0,6262 — 4 Стет 4,08 — — —
Дихроматы щелочных, щелочно-земельных и большинства тяжелых металлов легко растворимы в воде. Трудно растворимы дихроматы серебра, таллия. Растворы дихроматов имеют кислую реакцию. Дихроматы менее термически устойчивы, чем хроматы (VI).
Для щелочных (кроме лития) и некоторых других металлов известны темно-красные трихроматы М2Сг30ю и коричнево-красные тетрахроматы М2Сг20н. Для многих металлов получены оксохроматы и гидрооксохроматы (основные оксохроматы). Оксо- и гидрооксохроматы тяжелых металлов в воде не растворяются. В литературе приводится значительное количество двойных хроматов, например KZn2(CrO4)2OHH2O, хромато-сульфатов и т.д. Для некоторых металлов известны хромато-дихроматы, например Bi2(CrO4)2Cr2O7.
Действием пергидроля на водные растворы хроматов в зависимости от условий получают пероксохроматы: синие — состава М2Сг2О12 или красные — состава M^CrOg. Синие пероксохроматы при ударе, нагревании или соприкосновении с концентрированной серной кислотой взрываются. Красные пероксохроматы при обычной температуре довольно устойчивы, а при нагревании разлагаются.
Хромат натрия Na2CrO4 желтые кристаллы; до 423° С устойчива орторомбическая модификация (а = 0,717 нм, Ъ = 0,924 нм, с = 0,588 нм, z = 4, пространственная группа РптЪ), выше 423° С — гексагональная; ДЯперехода = 9,62 кДж/моль; температура плавления 794° С; плотность 2,72 г/см1; С° = 142,1 Дж/(молыК); ДЯ^р =-1343 кДж/моль, ДЯ° =24,69 кДж/моль; 52°98 = 176,6 Дж/(моль-К).
Растворимость хромата натрия в воде 45,8% (масс.) (25° С); ДЯ растворения для бесконечно разбавленного раствора -1362,4 кДж/моль (рис. 3.16). Из водных растворов ниже 19,52° С кристаллизуется декагидрат (ДЯ0°бр = -4276,9 кДж/моль), в интервале 19,52—26,6° С — гексагидрат, в интервале 26,6—62,8° С — тетрагидрат (ДЯо6р = -2526,5 кДж/моль), выше 62,8° С обезвоживается. В водном растворе хромата (VI) натрия существует равновесие
Сг2О, + Н2О 2НСгО; 2СЮ^ + 2Н+
При действии кислот хромат натрия переходит в дихромат Na2Cr2O7. При медленном добавлении концентрированной серной кислоты к почти насыщенному водному раствору хромата натрия образуются многоядерные анионы изополикислот: СгзО^ и Сг4О^3.
Хромат натрия — сильный окислитель. Он восстанавливается сульфидом водорода в щелочной среде до Наз[Сг(ОН)б], серой—до Сг20з. С кремнием при нагревании образует CrSi2, при сплавлении с РЬО—РЬСгО4.
147
Хромат натрия — полуфабрикат в процессе получения СггОч, служит окислителем в органическом синтезе и производстве красителей, протравой в процессе крашения тканей.
Хромат (VI) калия К2СгО4— кристаллы лимонно-желтого цве-। а, при 668° С ромбическая форма переходит в гексагональную красную, Д//перечода = 10,0 кДж/моль; температура плавления 973° С, Д//П1 = 33,0 кДж/моль; растворимость в воде 38,96% (масс.) (20° С); не растворяется в этаноле, эфире. Данные о растворимости при различных температурах приведены на рис. 3.17.
Хромат (VI) калия — в природе минерал тарапакоит — применяется в качестве протравы при крашении, дубителя в кожевенной промышленности. Является отбеливателем для масел, воска, протравы для семян, окислителем в органическом синтезе, реагентом в хроматографии.
Хромат (VI) калия получают взаимодействием К2Сг2О7 и К.2СОз в водном растворе. Образующиеся растворы упаривают и кристаллизуют К2СгО4 Кристаллы отделяют от маточных растворов и сушат.
Хромат (VI) аммония (NH4)2CrO4 — золотисто-желтые кристаллы. Растворимость в воде 27,2% (масс.) при 25° С, малорастворим в этаноле и ацетоне. Температура разложения хромата (VI) аммония 180° С. В процессе хранения на воздухе постепенно выделяет аммиак и превращается в дихромат.
Рис. 3.16. Растворимость хромата (VI) натрия Na2CrO4 в воде при различных температурах
Рис. 3.17. Растворимость хромата (VI) калия в воде при различных температурах
148
Хромат (VI) аммония получают взаимодействием водных растворов аммиака с СгО7 или (NH4)2Cr2O7.
Хромат (VI) аммония применяется в качестве протравы при крашении тканей, дубящего вещества и компонента светочувствительного слоя фотоматериалов.
Дихромат аммония (NH^CrjO?— оранжево-красные кристаллы, разлагающиеся выше 170° С, а при 240° С —со вспышкой.
Дихромат аммония хорошо растворяется в воде, особенно с повышением температуры (рис. 3.18)
Дихромат аммония получают взаимодействием водных растворов NH4C1 с Na2Cr2O7
Рис. 3.18. Растворимость дихромата аммония (NH4)2Cr2O7 в воде
Ха2Сг2О7 + 2МН4С1 = (NH4)2Cr2O7 + 2NaCI
Образующийся дихромат аммония отделяют от хлорида натрия, кристаллизуют, отделяют от маточных растворов, кристаллы промывают и сушат.
Дихромат аммония является окислителем в органическом синтезе, отбеливателем для жиров, воска, парафина. Применяется также в качестве компонента светочувствительного слоя фотоматериалов, исходного вещества для синтеза оксидов хрома, катализаторов, для получения фоторезисторов, фотосенсибилизаторов Он является компонентом пиротехнических составов, консервантом для древесины.
Хромат (VI) кальция СаСгО4 — желтые кристаллы. При 50° С становится красно-коричневым (при охлаждении окраска восстанавливается). Температура плавления около 1150° С (под давлением О2), а при атмосферном давлении температура плавления 1022° С (с разложением). Растворимость в воде 2,27% при 19° С, 0,79% при 70° С. Из растворов кристаллизуются СаСгО4-2Н2О, СаСгО4Н2О и СаСгО40,5Н2О, обладающие существенно большей растворимостью и полностью обезвоживающиеся при -400° С. Растворяется в этаноле. СаСгО4 получают взаимодействием растворов Ма2Сг2О7 с Са(ОИ)2:
Ма2Сг2О7 + 2 Са(ОИ)2 = 2 CaCrO4 + 2NaOH + Н2О
а также взаимодействием Na2CrO4 с СаС12:
Na2Cr2O7 + СаС12 = CaCrO4 + 2NaCI
149
Образующийся осадок хромата (VI) кальция отделяют от растворов хлорида натрия и подвергают термообработке.
Хромат (VI) кальция — минерал хроматит — применяется в качестве компонента шихты в процессе алюмотермического получения хрома, пигмента, ингибитора коррозии, компонента покрытий для легких сплавов. Он является деполяризатором для батарей, окислителем в процессе получения хрома.
Хромат (VI) бария ВаСгО4— желтые кристаллы с температурой плавления 1380° С, практически не растворяется в воде и органических растворителях, разлагается минеральными кислотами.
ВаСгО4 получают взаимодействием хлорида бария с дихроматом калия. Хромат (VI) бария применяют в качестве окислителя в процессах синтеза красителей, компонента взрывчатых веществ, пиротехнических составов, антикоррозионных покрытий на стали, составов головок спичек, пигмента (баритового желтого) для керамики.
Хромат (VI) цинка — лимонно-желтые кристаллы; выше 440° С разлагается; практически не растворяется в воде; образует кристаллогидраты с 7, 2 и 1 молекулами воды; в процессе кипячения с большим количеством воды гидролизуется с образованием гидроксохро-матов.
Хромат (VI) цинка получают взаимодействием Zn(OH)2 с К2Сг2О7. Применяется в качестве пигмента, компонента антикоррозионных покрытий.
Хромат (VI) свинца РЬСгО4 — желтые или оранжево-красные кристаллы. При нагревании при 707° С хромат (VI) свинца переходит в желтую ромбическую, а при 783° С — в красную тетрагональную модификации. Температура плавления 844° С (с разложением). В воде и органических растворителях практически не растворяется. Растворяется в азотной кислоте, концентрированных растворах щелочей.
Хромат (VI) свинца получают взаимодействием водных растворов нитрата или ацетата свинца с хроматами натрия, калия или NH*.
Хромат (VI) свинца применяется в качестве пигмента (желтый крон), окислителя в органическом синтезе, компонентом материалов термохромных датчиков
Оксохромат свинца РЬ2СгО4 — оранжевые или красные кристаллы, применяется в качестве пигмента (оранжевый крон), является материалом для термисторов.
Хроматы (V) значительной частью черного, голубовато-черного или темно-зеленого цвета, содержат тетраэдрический анион СЮ’ Для щелочных металлов получены соединения типа М3СгО4 и МСг3О8, для щелочноземельных металлов типа М3(СгО4)2, М2СГ2О7 и М5(СгО4)3ОН, а для редкоземельных элементов— типа МСгО4. 150
Хроматы (V) малорастворимы в воде, а в процессе кипячения с водой гидролизуются с диспропорционированием на Сг(1П) и Cr(VI).
Хроматы (V) получают термическим разложением хроматов (VI) в смеси с оксидом или нитратом соответствующего металла, гидротермальным путем, взаимодействием оксидов при высоких давлениях.
Хроматы (IV) представляют собой сложные оксиды. В структуре некоторых из них имеется тетраэдрический анион СЮ*\ Хроматы (IV) преимущественно зеленого цвета. Для щелочных металлов известны хроматы типа М?СгОз и М4СгО4, для щелочноземельных — типа МСгОз, М2СгО4, М3СгО5 и М4СгО6.
Хроматы (III) представляют собой сложные оксиды (табл. 3.3). Хроматы (III) металлов в степени окисления +1 имеют состав МСгО2 (метахроматы), получены также М3СЮ3 (ортохромиты). Для металлов в степени окисления +2 характерны соединения МСг2О4, многие из которых имеют структуру типа шпинели. Редкоземельные элементы, кроме скандия, дают соединения МСгО4 со структурами типа перовскита. Хроматы (III) получают термообработкой сесквиоксида с оксидами, карбонатами и другими соединениями металлов, а также восстановлением или термическим разложением хроматов (VI) и дихроматов (VI).
Из растворов, получающихся в результате растворения гидроксида хрома (III) в щелочи, кристаллизуются гидроксохроматы (III) состава МСг(ОН)4, МзСг(ОН)6 и М4Сг(ОН)7. Для щелочноземельных металлов получены гидроксохроматы типа М3Сг2(ОН)2 со структурой типа граната. Хроматы (III) в основном тугоплавки, химически стойки.
Хромат (III) магния MgCr2O4 — темно-зеленые, серо-зеленые или красные кристаллы. В природе минерал магнезиохромит.
MgCr2O4 получают в процессе нагревания MgCr2O7 или термообработкой оксида магния с сесквиоксидом хрома с добавкой сесквиоксида бора
MgCr2O4 применяют в качестве компонента огнеупоров и кристаллизатора в органическом синтезе.
Хромат цинка (III) ZnCr2O4 — светло-зеленые кристаллы, получают термообработкой смеси нитратов цинка и хрома или термическим разложением дихромата цинка.
Применяют хромат (III) цинка в качестве катализатора в органическом синтезе и компонента керамических датчиков влажности.
Хромат (III) железа (II) FeCr2O4 — черные или темно-коричневые кристаллы (минерал хромит).
Получают хромат (111) железа (II) в процессе нагревания смеси FeCI3 + К2Сг2О7 или термообработкой смеси сесквиоксидов железа и хрома с добавкой борной кислоты.
Природный хромат (III) железа (II) является основным видом сырья для получения хрома и его соединений.
151
Таблица 3 3.
Соединение Сингония Параметры кристаллической решетки, нм
67, НМ Ь нм с, нм
NaCrOj Тригональная 0,297 — 1,594
CuCrO? » 0,2975 — 1,7096
CuCr2O4 Тетрагональная 0,8532 — 0,7788
MgCr2O4 Ромбическая 0,8329 — —
CaCrjOj » 0,903 1,058 0,296
ZnCr2O4 Кубическая 0,8337 — —
CdCr2O4 » 0,8584 — —
MnCr2O4 » 0,8437 — —
FeCr2O4 » 0,8420 — —
CoCr2O4 » 0,8342 — —
NiCr2O4 » 0,8316 — —
YCrO3 Ромбическая 0,5242 0,5520 0,7534
LaCrO3 » 0,5514 0,5478 0,7752
CeCrOj » 0,5475 0,5475 0,7740
SmCrOj » 0,5367 0,5508 0,7643
Свойства некоторых хроматов (1П)
г Пространственная группа Температура плавления, °C Плотность, Дж/(моль К) кДж/моль Дж/(моль К)
3 Rim — 4,39 — — —
3 Rim — 5,49 — -674 —
— JA}lamd — — — -1288 —
8 Fd3m 2350 4,43 127 -1787 106
— — 2170 4,98 — -1841 —
8 Fd3m — 5,29 — -1550 —
8 » — 5,79 — -1438 —
8 » — 4,97 — — —
8 » 2153 5,1 — -1452 146
8 » — 5,14 — -1430 138
8 » — 5,24 — -1382 119
6 Pbnm 2430 5,76 97 -1555 97
6 » 2510 6,75 109 — 112
6 » 2300 6,84 118 -1352 119
6 » 2400 7,28 106 — —
Известны также хроматы, содержащие хром в двух степенях окисления, в частности +3 и +4 (например, Sr4Cr2O9), +4 и +6 [Са5(СгО4)ч] или +5 и +6 [Са10(СгО4)7]. Их иногда называют хроми-то-хроматами, а состав выражают формулой тиМОиСгОзрСг2Оз.
Способ получения хромата натрия. Основным способом получения хромата натрия (Na2CrO4) является метод, основанный на переработке хромитовых руд, содержащих (в %):
Cr2Oi SiO2 Рвобщ А12О3 MgO
45—63,5 0,5—12 10—22 8,5—15 11—18
Обычно в рудах содержится менее 1,3% СаО, до 0,04% фосфора и серы, в незначительных количествах Ni, Со, Мп, Ni, V и другие элементы.
Хромат натрия получают методом высокотемпературной окислительной термообработки шихты, состоящей из хромита, карбоната натрия, доломита и иногда небольшого количества выщелоченной массы. Термообработанную массу выщелачивают, а полученный раствор хромата натрия отделяют от нерастворимого остатка.
На рис. 3.19 приведена принципиальная схема получения хромата натрия, согласно которой она состоит из следующих стадий производства.
Подготовка сырьевых материалов. Комовую хромовую руду и доломит подвергают измельчению в щековых дробилках, сушат во вращающихся сушильных барабанах, а затем размалывают в шаровых мельницах, работающих в замкнутом цикле с сепаратором. Карбонат натрия, имеющий комки, измельчают в молотковой дробилке.
Приготовление шихты. Измельченные хромит и доломит, карбонат натрия и пыль отходящих газов печей дозируют синхронно работающими автоматами-дозаторами и смешивают в двухвальном лопастном смесителе.
Окислительный обжиг шихты. Шихту дозируют автоматами-дозаторами во вращающиеся барабанные печи, в которой, перемещаясь навстречу топочным газам, она нагревается до 1150—1200° С. Химизм процесса окислительного обжига хромита невозможно описать одной или несколькими простыми реакциями. Процесс является очень сложным, многокомпонентным. В нем при указанных температурных интервалах наравне с газовой и жидкой фазами имеется значительное число твердых фаз, состав которых зависит от режима термообработки. Условно окислительную термообработку хромита железа в присутствии карбоната натрия можно представить уравнениями реакций:
4(FeOCr2O1) + 8Na2CO, + 7О2 = 8Na2CrO4 + 2Fe2O3 + 8СО2
4(MgOCr2Oi) + 8Na2CO, + 6O2 = 8Na2CrO4 + 4MgO + 8CO2
153
Рис. 3.19. Принципиальная схема получения хромата натрия
Параллельно идут реакции, в результате которых появляются многие другие соединения, в первую очередь ферриты, алюминаты, силикаты натрия и кальция и хроматы кальция.
Оксид железа, находящийся в шихте в свободном состоянии и в составе хромшпинелида, взаимодействует с оксидами натрия, кальция и магния с образованием ферритов и алюмоферритов. При этом образуется также феррат (IV) натрия:
2Ма2ОРе2Оз + бШСОз + О2 = 4Na4FeO4 + 6СО2
который растворяется в жидкой фазе прокаливаемой массы и окисляет хром (III). Это интенсифицирует окислительный обжиг, поскольку, расходуясь на окисление хромита, многократно регенерируясь, фер-155
рат служит переносчиком кислорода из газовой фазы к поверхности зерен исходного хромита.
Процесс окисления Сг(1П) в Cr(IV) идет последовательно через Cr(IV) и Cr(V). Наличие в плаве хроматов (V) натрия и кальция установлено; они растворены в жидкой фазе шихты и участвуют в передаче кислорода, образуясь в результате окисления хромата (III) хроматом (VI), они затем окисляются газообразным кислородом до хромата (VI).
Процесс образования хромата (VI) из хромата (III) является эндотермическим, требующим затраты значительных количеств теплоты. Чем выше температурный уровень термообработки и чем больше избытка кислорода в печи, тем быстрее идет процесс окисления.
Хромат натрия в процессе термообработки образуется при сравнительно низких температурах и с карбонатом натрия образует эвтектический расплав, содержащий 6£,5% Na2CrO4. Появление жидкой фазы в системе Na2CrO4—Ма2СО4 возможно уже при 655° С. В процессе дальнейшего нагревания реакционной массы жидкая фаза обогащается карбонатом натрия, причем состав расплава в начале изменяется от эвтектической точки вдоль линии ликвидуса по направлению к точке плавления карбоната натрия (рис. 3.20). Однако одновременно с возрастающей скоростью процесса происходит образование хромата натрия, замещающего карбонат натрия в жидкой фазе. Карбонат натрия расходуется не только на основную реакцию, но и на реакции с другими компонентами шихты на образование феррита, алюмината и силиката натрия. Последние взаимодействуют в окислительной атмосфере с хромитами железа и магния с образованием хромата натрия:
2MgCr2O4 + 4Na2AI2O4 + О2 = 4Na2CrO4 + 2MgO + 4AI2O,
FeCr2O4 + 2Na2AI2O4 + l,75O2 = 2Na2CrO4 + 0,5Fe2O, + 2A12O,
2MgCr2O4 + 4Na2Fe2O4 + 3O2 = 4Na2CrO4 + 2MgO + 4Fe2O3
FeCr2O4 + 2Na2Fe2O4 + l,75O2 = 2Na2CrO4 + 2,5Fe2O3
FeCr2O4 + 2Na2SiO4 + I,5O2 = 2Na2CrO4 + 0,5Fe2SiO4 + 1,5SiO2
Хромат натрия не реагирует с оксидами магния, алюминия и железа, с алюминатом и ферритом натрия вплоть до 1100—1200° С. Активно ведут себя по отношению к хромату натрия кремнезем и силикат натрия. В процессе нагревания до 1100° С смеси Ма2СгО4 и SiO2 образуется хромосиликат натрия зеленого цвета.
Диаграмма плавкости системы Na2CrO4—NaSiO, приведена на рис 3.21. В системе, содержащей менее 40% метасиликата, образуется лишь эвтектика (температура плавления 770° С) с содержанием 10% Na-jSiCh 156
Рнс. 3.20. Изменение состава жидкой фазы в системе Na2CrO4—Na2CO4 с повышением температуры
1100
Рис. 3.21. Диаграмма плавкости системы Na2CrO4—Na2SiO3 при разных температурах
По мере реакции карбоната натрия происходит растворение новых его количеств В результате этих реакций твердый карбонат натрия исчезает задолго до достижения шихтой температуры плавления карбоната натрия. Полный переход твердого карбоната натрия в раствор выражен изломом кривой (см. рис. 3.20, точка А).
Диаграмма (рис. 3.22) условно учитывает возможность растворения в жидкой фазе других ингредиентов термообрабатываемой шихты, поэтому на ней жидкая фаза в конце процесса термообработки состоит из чистого хромата натрия. Однако в жидкой фазе содержится некоторое количество хорошо растворимого в хромате натрия хромата кальция. При 1000° С растворимость хромата кальция в хромате натрия достигает 80%.
В процессе термообработки доломита происходит его двухступенчатое разложение. Первая ступень отвечает частичному разложению доломита по реакции
CaMg(CO,)2 = СаСОз + MgO + СО2
вторая—диссоциации образовавшегося СаСО3:
СаСОз = СаО + СО2
Температура, при которой рсо = 1атм, составляет для CaMg(CO3)2 — 730° С, МёСОз — 640° С и СаСОз — 910° С.
157
Оксид и карбонат кальция взаимодействуют с кислородом и сеск-виоксидом хрома или хромитом по следующим уравнениям:
4СаСО3 + 2Сг2О3 + ЗО2 = 4СаСгО4 + 4СО2
4СаСОз + 2MgCr2O4 + ЗО2 = 4CaCrO4 + 2MgO + 4СО2
2СаСО3 + FeCr2O4 + 1,75О2 = 2CaCrO4 + 0,5Fe2O3 + 2СО2
4СаО + 2Сг2О3 + ЗО2 = 4СаСгО4
4СаО + 2MgCr2O4 + ЗО2 = 4CaCrO4 + 2MgO
2СаО + FeCr2O4 + 1,75О2 = 2CaCrO4 + 0,5Fe2O3
Однако в плаве растворено незначительное количество хромата кальция, поскольку растворенные ионы кальция вступают в реакции, приводящие к образованию нерастворимых соединений кальция — силикатов, ферритов, алюминатов и др. В шихте появляется значительное количество хромата кальция лишь при большом недостатке карбоната натрия в шихте.
На рис. 3.22 приведена диаграмма плавкости системы СаСгО4 + + Na2CO3 = Na2CrO4 + СаСО3 при невысоких температурах и в атмосфере диоксида углерода до начала процесса разложения карбоната ка-
Рмс. 3.22. Диаграмма плавкости системы CaCrO4 + Na2CO3 = Na2CrO4 + СгСО3 при невысоких температурах в атмосфере диоксида углерода
158
льция. В этих условиях система является трехкомпонентной (составы даны в молярных процентах). В равновесии с плавом находятся следующие твердые фазы: СаСгОд, СаСО3, Na2Ca(CO3)2 и твердые mNa2CO3rcCaCO3 и /7Na2CrO4 ^СаСгОд. На диаграмму нанесены изотермы и обозначены температурные минимумы (точки плавления двойных и тройных эвтектик) и максимумы. Как видно из приведенных данных, появление жидкой фазы в печи термообработки возможно уже при 645°С. С повышением температуры состав жидкой фазы меняется. Параллельно меняется и состав реагирующих твердых фаз, диссоциирует карбонат кальция. В системе Ь^СгОд— СаСгОд имеется максимум при 812°С, соответствующий образованию твердых растворов. Двойная эвтектика содержит 48,4 мол.% СаСгОд, плавится при 740°С и состоит из твердого раствора (Na2, Са)СгОд и чистого хромата кальция. Наиболее легкоплавкие смеси расположены вдоль пограничных линий, соединяющих точки тройных эвтектик (668, 645 и 648°С) и точку двойной эвтектики карбоната натрия и хромата натрия (655°С). Из этих смесей образуется жидкая фаза в шихте, содержащей карбонат натрия, ниже 700°С. В процессе термообработки известковых шихт (без карбоната натрия) образование хромата кальция ниже 800°С протекает медленно, а при более высоких температурах образуются хромито-хроматы, снижающие выход хромата.
Содержащиеся в исходной хромитовой руде примеси существенно не влияют на предельную степень окисления сесквиоксида хрома. От присутствия оксидов кремния, алюминия и железа степень окисления Сг2О3 не уменьшается, если термообрабатываемая шихта содержит оксид кальция в количестве, достаточном для связывания кислотных оксидов. Однако примеси влияют на скорость процесса окисления хрома — снижение содержания сесквиоксида хрома, т. е. повышение содержания примесей (SiO2, Fe2O3 и AI2O3) приводит к увеличению продолжительности термообработки исходных хромитовых шихт в производственных условиях.
Выщелачивание плава. Выходящий из печи плав пропускают через валковую дробилку, а затем выщелачивают неконцентрированными растворами хромата (со второй стадии фильтрации) в шаровой мельнице. В мельнице получают шламовую суспензию с Т:Ж~1:3. При этом часть суспензии должна давать остаток на сите с отверстиями 0,075 мм не более 25—30% (на сите с отверстиями 0,06 мм не более 50%), а жидкая часть суспензии содержит 150—200 г/л СгО3. Температура суспензии поддерживается в пределах 70—90° С. Плотность шламовой суспензии поддерживают постоянной автоматическим регулированием подачи неконцентрированных растворов в мельницу.
Процессы выщелачивания и сатурации являются ответственными, поскольку на этой стадии производства имеют место высокие потери хрома.
159
В процессе выщелачивания плава твердый раствор хроматов, карбонат и алюминат натрия переходят в раствор, а остальные минеральные фазы остаются в твердой части шламовой суспензии и подвергаются гидратации.
При воздействии воды на трикальцийсиликат он расщепляется по уравнению
3CaOSiO2 + лН2О = ЗСа(ОН)2 + SiO2(n-3)H2O
В насыщенных растворах Са(ОН)2 процесс идет по уравнению
3CaOSiO2 + лН2О = Са(ОН)2 + 2CaO-2SiO2(n-l)H2O
В процессе гидратации выделяется свободный гидроксид алюминия:
3(5СаО-ЗА12О3) + 42Н2О = 5(ЗСаОА12О3-6Н2О) + 4(А12О3-ЗН2О)
В твердом растворе вся или часть воды может заменяться на SiO2 с образованием гидроградиентов по реакции
ЗСаОА12О3-6Н2О + 2CaOSiO2H2O = - 3CaO Al2O3 SiO2-4H2O + 2Са(ОН)2 + Н2О
Характерной реакцией гидроалюминатов и гидроферритов кальция является образование комплексных солей двух типов (X = SO^", СЮ’-, СО’-):
3CaO AI2Oy3CaX-304-32H2O и ЗСаО А12ОгСаХ Ю4-12Н2О
Образование сульфоалюминатов кальция происходит при действии на гидроалюминаты растворов CaSO4 и Na2SO4. Низкосульфатная форма 3CaO Al2O3 CaSO412H2O метастабильна и с течением времени переходит в высокосульфатную форму 3CaOAl2O3-3CaSO4*31H2O. Высо-косульфатная форма полностью гидролизуется в воде по уравнению
3CaOAl2O3-3CaSO4-31H2O =
= ЗСа(ОН)2 + 3(CaSO4-2H2O) + 2А1(ОН)3 + 19Н2О
В присутствии растворимых карбонатов или солей магния Са(ОН)2 переходит в менее растворимый СаСО3 или соответственно Mg(OH)2, что способствует гидролизу.
160
Из изложенного видно, что в процессе гидратации хроматного плава возможно образование Са(ОН)2, коллоидных гидросиликатов кальция переменного состава, коллоидных гидроксидов алюминия и железа, кристаллических гидроалюминатов и гидроферритов кальция или их твердых растворов, гидрогранатов Mg(OH)2 и хромато-алюминатов кальция. Вследствие малой скорости гидратации и образования защитных пленок коллоидных продуктов гидратации в шламе должны оставаться значительные количества исходных минеральных фаз плава. В раствор переходят хроматы натрия и кальция, карбонат и алюминат натрия, в незначительном количестве силикат натрия. Ионы кальция, перешедшие в раствор, реагируют с ионами ОН"1 и СО^- с образованием труднорастворимых Са(ОН)2 и СаСОз:
СаСгО4 + 2NaOH Са(ОН)2 + Na2CrO4
CaCrO4 + Na2CO3 СаСОз + Na2CrO4
В процессе сатурации, очевидно, протекают реакции:
Na2O Al2O3 + СО2 + ЗН2О <^Na2CO3 + 2А1(ОН)3
Са(ОН)2 + СО2 = СаСОз + Н2О
Са(ОН)2 + Na2CO3 СаСОз + 2NaOH
а также, в некоторой степени, реакции карбонизации Mg(OH)2, гидросиликатов, гидроалюминатов, гидроферритов, хромато-алюминатов кальция.
Фильтрация шламовой суспензии. Суспензию после сатурации подвергают двухстадийной фильтрации на барабанных вакуум-фильтрах. Фильтрат первой стадии фильтрации с содержанием 130 г/л СгОз обычно является целевым продуктом производства. Отмытый горячей водой шлам второй стадии фильтрации репульпируют оборотной водой и направляют в шламонакопитель.
Получение кристаллического хромата натрия. Хроматные растворы, полученные описанным способом, очищают от алюминия, фильтруют и упаривают в многокорпусной ваку-ум-выпарной установке. Из концентрированного раствора кристаллизацией при охлаждении или уваркой содержания воды порядка 30% получают соответственно кристаллический или плавленый четырехводный кристаллогидрат.
Дихромат натрия. Физико-химические свойства. Дихромат натрия Ыа2Сг2О7 — оранжевые ромбические кристаллы, расплывающиеся на воздухе. Плотность 2,5 г/см3; температура А 161
о Химическая технология неорганических веществ, кн. 2
плавления 356,7°С; в процессе нагревания при 400°С разлагается с образованием Na2CrO4 и Сг2Оз, энтальпия разложения равна 9,6 ккал/г-моль; слабо парамагнитен (2,1-10‘7); в процессе нагревания при 84,6°С двуводный продукт переходит в безводное состояние. Хорошо растворяется в воде (рис. 3.23). Из водных растворов образуется кристаллогидрат Na2Cr2O7-2H2O, представляющий ярко-оранжевые продолговатые призмы, расплывающиеся на воздухе. Сингония V(a:b:c = = 0,5912:1,0000:0,5698, Р = 94°55’). Плотность 2,348.
Температура замерзания:
Na2Cr2O7-2H2O, % (масс.)... 10 20 30 40 50 60 69
Температура, °C........... -1,7 -3,6 -6,0 -9,6 -15,1 -26,6 -48,2
0 насыщенных растворов
Температура, °C 30 40 50 60 80
/?н {), мм рт. ст. 17,26 29,61 50,30 82,49 199,0
Величина pH:
Na2Cr2O7-2H2O, г/л ... 20 10 5 2 1 0,5 0,25 0,125
pH ... 3,87 4,01 4,13 4,33 4,48 4,63 4,78 4,90
Плотность при 25—30° С:
Na2Cr2O7-2H2O, % (масс.) .. 10,06 20,47 30,71 40,03 50,51 70,08
Теплоемкость, кал/(гград) .. 0,912 0,832 0,771 0,711 0,647 0,544
Вязкость Na2Cr2O7-2Н2О% (масс.): 10 при 25° С — 0,96 сп; 50 при 25° С —2,41 сп; 60 при 25° С —3,94 сп; 70 при 75° С —2,44 сп.
Технический дихромат натрия производят согласно действующему ГОСТ 2651—84 (табл. 3.4).
Таблица 3.4. Состав технического дихромата натрия (по ГОСТ 2651—84)
Показатель Кристаллический безводный Гранулиро-ванный безводный Гранулированный водный Плавле-ный
Высший сорт Первый сорт I сорт II сорт
Триоксид хрома, не менее 75,5 75,0 74,0 72,5 71 70
или Na2Cr2O7, не менее 98,9 98,25 — — — —
Сульфаты (SO4), не более 0,1 0,2 0,5 0,5 0,7 0,7
Хлориды (С1), не более 0,002 0,003 0,005 0,005 0,005 0,005
Нерастворимый в воде остаток, не более 0,1 0,02 0,05 0,03 0,05 0,05
pH водного раствора Не ниже 3,5
162
Рис. 3.23. Диаграмма растворимости дихромата натрия Na2Cr2O7 в воде при различных температурах
Физико-химические основы получения дихромата натрия. Наиболее широко применяемым способом получения дихромата натрия является переработка растворов (желтых щелоков) в Na2CrO4 путем их очистки, упарки, травки серной кислотой, концентрирование образующихся растворов (красных щелоков) Na2Cr2O7, очистка от сульфата натрия и выделение кристаллов или гранул дихромата натрия.
Исходные желтые щелоки Na2CrOv, загрязненные алюминатом натрия (до 5 г/л А12О3) и нерастворимым остатком (до 1 г/л), обрабатывают серной кислотой или раствором дихромата натрия. При этом кислота (или дихромат) вступает в реакцию с имеющимися в исходном растворе ионами ОН-, СО^’, АЮ2, СгО^" в определенной последовательности. В начале очистки ионами Н+ кислоты (или ионами НСгО;) нейтрализуются ионы ОН- и ионы СО3"
ОН’ + Н+ = Н2О
или
он’ +нсго; -Сго^ +н2о
СО;’ + н+ = нсо;
163 ь*
или
со2’ + нею; = нсо; + ею*’
Затем нейтрализуются ионы НСО; и АЮ;:
нсо; + н+ = н2со3 = н2о + со2 или
нсо; + нею; = ею*- + н2о + со2
АЮ; + Н+ + Н2О = А1(ОН)3
ИЛИ
аю; + нею; + н2о = А1(ОН)3 + сю;2
После протекания приведенных выше реакций при дальнейшем добавлении серной кислоты происходит переход иона Cr^^" в Сг2О2. Суммарные уравнения реакций в процессе подтравки серной кислотой:
2NaOH + H2SO4 = Ma2SO4 + 2Н2О
Na2CO3 + H2SO4 — Na2SO4 + H2O + CO2
2NaAlO2 + H2SO4 + 2H2O = Na2SO4 + 2A1(OH)2
Рис. 3.24. Изменение pH раствора Na2Cr2O4 в зависимости от количества серной кислоты
Протекание приведенных реакций связано с изменением pH раствора. На рис. 3.24 приведена кривая изменения величины pH раствора хромата калия в зависимости от количества введенной серной кислоты. Резкие изменения pH соответствуют: первый (сверху) — нейтрализация свободной щелочи (NaOH и Na2CO3), второй — нейтрализации гидрокарбоната и алюмината, третий — переводу Сг2О;;~ в Сг2О2". Переход алюмината в его гидроксид заканчивается при pH 8,2. Эту величину в производстве выдерживают довольно точно, поскольку при недо-траве в растворе остается алюминат, а при повышенном перетраве разрушается филь-
164
троткань на фильтрах. При перекислении растворов до pH 5 гидроксид алюминия переходит в раствор в повышенных количествах.
Процесс подтравки проводят при 80° С в реакторах при непрерывном перемешивании с медленным введением в систему серной кислоты (-75% H2SO4) или раствор дихромата натрия до содержания в растворе 3—5 г/л Na2Cr2O7. Для получения крупнозернистого осадка подачу серной кислоты или раствора Na2Cr2O7 производят медленно. Заданное значение pH поддерживается с точностью ±0,2 pH.
Полученную суспензию гидроксида алюминия в растворе Na2CrO4 фильтруют. Осадок гидроксида алюминия промывают и фильтр разгружают. Промытый осадок содержит около 15% А1(ОН)3 и 4—5% Na2CrO4.
Очищенный от алюминия раствор Na2CrO4 концентрируют на ва-куум-выпарной установке до концентрации 350 г/л СгО3. На некоторых заводах эта стадия отсутствует. Достоинством схемы с упаркой желтых растворов является возможность использования для этой операции выпарных аппаратов без принудительной циркуляции.
Далее желтые щелока обрабатывают в реакторе серной кислотой для перевода хромата натрия в его дихромат:
2 Na2CrO4 + H2SO4 = Na2Cr2O7 + Na2SO4 + H2O
На некоторых заводах вместо серной кислоты применяют гидросульфат натрия:
2 Ма2СгО4 + 2NaHSO4 = Na2Cr2O7 + 2Na2SO4 + Н2О
Существует также способ перевода хромата натрия в его дихромат углекислотным методом:
2Na2CrO4 + 2СО2 + Н2О Na2Cr2O7 + 2NaHCO3
При этом исходный раствор Na2CrO4 обрабатывают диоксидом углерода под давлением, а выпадающий в осадок гидрокарбонат натрия превращают в «желтый» карбонат натрия, используемого в процессе окислительной термообработки хромита.
В аммиачно-углекислотном методе применяется двухстадийный процесс:
Ма2СгО4 + 2NH3 + 2СО2 + 2Н2О 2NaHCO3 + 2(NH4)2CrO4
(NH4)2CrO4 + Na2CrO4 ±^Na2Cr2O7 + 2NH3 + H2O
165
Фтороводородный способ:
2Na2CrO4 + 2HF = Na2Cr2O7 + 2NaF + H2O или
2Na2CrO4 + NaAlO2 + 6HF = Na2Cr2O7 + Na3AlF6 + 3H2O
Электролитический метод. При электролизе раствора Na2CrO4 переводится в дихромат по схеме
2Н2О 2Н+ + 2OFT
2СЮ4" + 2Н+ СГ2О2’ + Н2О
Согласно способу, выход по току для насыщенных растворов Na2CrO4 достигает 92—95% при 15—25° С и 75% при 50—60° С.
Раствор Na2CrO4 обрабатывают при 70—80° С газообразным хлором. При этом наряду с дихроматом натрия образуется ценный хлорат натрия:
6Na2CrO4 + ЗС12 = 3Na2Cr2O7 + 5NaCl + NaClO3
Проведены также работы по изучению процессов травки хлороводородной кислотой и гексафторосиликатом водорода.
Анализ приведенных способов перевода монохроматов в дихроматы показывает, что все они основаны на существующем в растворах равновесии между ионами СЮ4~, HCiO4 и Сг2О? :
2СгО4“ + 2Н+ 2НСгО" СГ2О2’ + Н2О
Таким образом, в результате гидролиза в растворе К2СгО4 содержатся ионы СЮ4“, НСгО4 и ОН-. В процессе добавления в раствор кислоты НА вследствие диссоциации последней в нем появляются также ионы Н+ и А'. Очевидно, что система будет стремиться к состоянию равновесия, определяемому константами равновесия протекающих между ионами обратимых реакций, определяющих растворимость компонентов. При расчете равновесий между хроматами и кислотами можно допустить, что все соли как сильные электролиты полностью диссоциированы и в растворе отсутствуют гидродихро-матные ионы НСг2О7 •
Предполагая, что все компоненты системы хорошо растворимы, т.е. не выпадают в осадок, ионный состав и степень перехода определяются следующими уравнениями. 166
Константа диссоциации травящей кислоты:
к _ [tf][A ] (39)
1 [НА] •
Константа диссоциации гидрохроматного иона: [ЬГЦСгОГ] (40)
2 [HCrOj ] ’
Константа гидролиза дихроматного иона:
[НСгО;]2 (41)
3 [Сг2О2-]
при этом К2 = 0,9-10'7—1,26-1 О'6, а К3 = 0,91-lQ-2—3,03-Ю'2 (при 25° С). Если обозначить начальную молярную концентрацию кислоты а
и хромата с, то
а = [НА] + [А’], (42)
с = [СЮ42-] + [НСЮ;] + 2[Сг2О2-]. (43)
Условие электронейтральности раствора (концентрацией Н+ и ОН" можно пренебречь) отвечает уравнению
[Ме+] = 2[СЮ2 ] + 2[Сг2О2 ] + [ НСЮ;] + [А’] = (44)
= [СЮ>] + с + [А-],
откуда, учитывая, что [Ме+] = 2с, получаем:
с = [ СгО4~] + [А~]. (45)
Степень превращения:
x^^-[CrOj-] } [СгОд ] (46)
с с
Совместное решение уравнений приводит к выражению
2К,(\-хУ{а-сх)2 +КхК2К3х(\-х\а-сх) = К22К3сх3, (47)
из которого х можно определять методом постепенного приближения или графически. На рис. 3.25 приведены кривые, показывающие зависимость степени перехода х Ь^СгОд в Na2Cr2O7 от IgATi, т.е. от 167
константы диссоциации травящей кислоты, для а = с = 1 (кривая 7) и а = с = 3 (кривая 2) при К2 = 10'7 и К3 = 0,02 и для а = с = 3 при К2 = IO’7 и = 1 (кривая 3).
Допущение отсутствия ионов дихромата в растворе (пренебречь ими можно при величине А*з>1) приводит к упрощенному уравнению для расчета степени перехода:
_ А,(а + с)- у1к^(с- а)2 + 4КхК2ас (48)
Х~ 2(К-Кг)с ’
из которого видно, что степень перехода практически не зависит от концентраций, а лишь от молярного отношения кислоты и хромата. В случае, когда а = с, уравнение (48) после преобразований дает:
(49)
На рис. 3.25 приведена рассчитанная по этому уравнению зависимость степени перехода от -Ig^ при К2 - 10'7 (кривая 7) и 8-10'7
(кривая 2).
В случае производства травки хорошо растворимой кислотой, но с образованием при этом малорастворимой соли, выпадающей в твердую фазу (например, при травке фтороводородной кислотой), то
ионный состав и степень перехода определяются уравнениями (39) и (40) (наличием можно пренебречь) и уравнением, характери-
Рис. 3.25. Зависимость степени
перехода хромата в дихромат от константы диссоциации травящей кислоты
зующим растворимость МеА. По закону действующих масс при наличии:
МеА Ме+ + А“ (твердая фаза) (раствор)
твердой фазы произведение растворимости является константой: ПР =
= [Ме+][А-].
В первом приближении можно принять линейную зависимость растворимости МА в хроматно-дихроматных растворах от степени перехода. По значениям для крайних точек можно найти:
[А-] =
ПР ПР f, ПР>
= —+10-— х
[КГ] 2с \ 2с J
168
или
[А ] = b\-bix, (50)
, ПР х. . ,
где о, =-----растворимость МеА в хроматном; b — в дихроматном
2с
растворе, a - b—by.
Уравнение (43) в данном случае будет иметь вид
[НА] + [А] = (51)
где у—степень перехода кислоты в виде соли МА в твердую фазу.
Поскольку [М+] = 2с—уа, то из условия электронейтральности раствора следует:
с+[НА] = а + [СЮ*“]. (52)
Совместное решение уравнений приводит к квадратному уравнению:
*,(1-л)(а-сх) (53)
2 b-b2x ’
если Z?2 = 0 (растворимость МеА постоянна) и а = с, то
_ 2/С,с+ К2ЬХ - + (54)
Х~ 2К^с '
На рис. 3.25 нанесена рассчитанная по уравнению (54) кривая 3 степени перехода для а = с, by - 0,05с и Кг = 10‘7. В процессе травления монохроматного раствора фтороводородной кислотой можно принять Ку = 10‘3 и by = 0,05с, при этом уравнение (54) даст х = 0,998.
Если травление производят малорастворимой кислотой (например, угольной), то можно приближенно принять [НА] = const и [А“] = = const = b. Тогда
[Ft ][СгО^ ] ^[НА][СгО^-] _ £,[НА](1-л) 2 [НСгО“] [A ][HCrO;] (b{ + b2x)x ’
откуда
_ЩАХ_ (55)
a\[ha]+/c2V
169
Пока насыщение раствора солью МеА еще не достигнуто, можно принять: [А“] + [НСгО4] = сх. Тогда
^/С,[НА](1-х)
и
-К,[НА]+7^,2[НА]2 + 4£,КДНА]
(56)
Если применяемая для травки кислота (или ангидрид) газообразна, то при равновесии [НА]газ [НА]раствор на степень перехода должно влиять давление. Если для первого приближения допустить, что система подчиняется закону Генри, то [НА] = Кр, где р — парциальное давление кислоты или ее ангидрида в газовой фазе.
Уравнение (55) идентично выражению, подтвержденному Я.Р. Гольдштейном экспериментально в процессе травления диоксидом углерода:
Y _ [Н^]
На процесс травления газообразной кислотой (или ангидридом) при наличии равновесия [НА]2 [НА]р_р.
Допуская в первом приближении, что система подчиняется закону Генри, то [НА] = кр, где р — парциальное давление кислоты или ее ангидрида в газовой фазе. Подставив это значение [НА] уравнения (55) и (56), находим зависимость х от давления; при наличии МеА в твердой фазе
(57)
*Лр + М’
а при отсутствии МеА в твердой фазе
_ -к,кг + ^к2к2Р2 + 4к,к2кср (58)
Х ~ 2Кгс
Из уравнения (57) следует
Х = 1+АА (59)
К.Л’
следовательно, с повышением давления степень перехода Na2CrO4 в Ма2Сг2О? растет. 170
Влияние температуры сказывается, главным образом, на изменении К
растворимости НА и МА, поскольку отношение — можно принять не
К,
зависящим от температуры. Вследствие того, что растворимость газов сильно уменьшается с повышением температуры, а растворимость солей обычно увеличивается, то, как видно из уравнения (59), степень перехода с повышением температуры должна уменьшаться.
Когда все компоненты хорошо растворимы, степень перехода х не зависит от концентрации. По-другому обстоит дело при малорастворимой кислоте. Поскольку Ь\ сильно уменьшается с ростом концент-ПР
рации (й, = —), то, как видно из уравнения (57), значение х должно 2с
существенно повышаться с ростом концентрации при наличии МеА в твердой фазе (изменением [НА] с концентрацией можно пренебречь). Это подтверждено экспериментально при травке хромата диоксидом углерода (рис. 3.26) (точка пересечения кривых соответствует выпадению NaHCO3).
Что касается влияния «силы» травящей кислоты, то с уменьшением константы диссоциации кислоты до К\ = 10‘4 степень перехода снижается с 1 до 0,97—0,99%; при К] = 10’5 х = 0,91—0,97%; с дальнейшим снижением К\ до 10‘9 значение х падает до 0,09—0,2 и асимптотически приближается к нулю.
Полученные дихроматные растворы после травления обрабатывают хлорной известью или гипохлоритом кальция для окисления содержащихся в растворе хроми-хроматов:
2(Сг2О3тиСгО3) + ЗСа(ОС1)2 = ЗСаС12 + (2т + 4)СгО3
Выпаривание растворов дихромата натрия проводят в многокорпусных (обычно число корпусов не превышает пяти) вакуум-выпар-ных аппаратах.
Исходный хроматный раствор (прозрачный и свободный от алюминия) концентрируют со 100—НО до 300—350 г/л СгО3.
Данные по совместной растворимости хромата и сульфата натрия в воде (рис. 3.27) позволяют найти предельную концентрацию хромата натрия, при которой начинает выпадать в осадок сульфат натрия. На рис. 3.28 растворимость представлена как функция массового отношения Na2SO4/Na2CrO4 в исходном растворе. После подтравки указанное отношение составляет ®0,1. Этому отвечает предельная концентрация Na2CrO4 около 39%. Обычно выше этой концентрации хроматные растворы не упаривают.
Слабый дихроматный раствор, полученный в процессе травления, содержит 250—300 г/л СгО3, значительное количество сульфата на-
171
Концентрация хрома, моль
Рис. 3.26. Изотермическая кривая степеней перехода для разных концентраций хроматов
Рис. 3.27. Совместная растворимость сульфата и хромата натрия в воде: / — 33° С; 2 — 55,07° С
трия и некоторое количество хлорида натрия. С целью выделения из системы основной части сульфата натрия раствор концентрируют до содержания 560—600 г/л СгО3. В дальнейшем его упаривают после отделения сульфата натрия. При значительных концентрациях раствора дихромата натрия выпадают мелкие кристаллы сульфата натрия, которые трудно отделить и отмыть от раствора.
Политермическая диаграмма растворимости в системе Ыа2Сг2О7—Na2SO4—Н2О приведена на рис. 3.29. Поверхность ACEHF соответствует растворам, насыщенным Na2Cr2O7*2H2O; поверхность FHC'A', CBDE и ED'B'CH—растворам, насыщенным соответственно Na2Cr2O7, Na2SO410H2O и Na2SO4. Дихроматами раствор после травления насыщен сульфатом натрия (состав его соответствует, например, при 98° С точке А); в процессе концентрирования раствора Na2SO4 выделяется в осадок и состав его перемещается по линии КС в направлении к эвтонической точке С', в которой раствор насыщен дихроматом и сульфатом натрия. В высококонцентрированных растворах содержание сульфата натрия весьма мало: например, точка С' отвечает содержанию 79,76% Na2Cr2O7 и 0,98% Na2SO4. Следовательно, в процессе выпаривания в осадок выделяется почти весь сульфат натрия. Это подтверждается также следующими данными о растворимости Na2SO4 в дихроматных растворах при 98° С:
Содержание дихромата натрия
(в пересчете на СгО3), г/л......... 300 370 550 680 980 1100
Содержание Na2SO4. г/л.............. 143 86 34 23 12 8
Растворы упаривают в противоточных двух- или трехкорпусных вакуум-выпарных установках до концентрации 730—760 г/л
Na2Cr2O7, 130—340 г/л Ma2SO4 и -10 г/л NaCl. 172
Осадок Na2SO4 отделяют от растворов дихромата натрия в фильтрующих центрифугах. Кристаллы сульфата натрия сушат в противоточных вращающихся барабанных сушилках при 350—500° С.
Дихроматные растворы, отфильтрованные от сульфата натрия, концентрируют с -39% СгО3 (51% Na2Cr2O7) до 57% СгО3 в таких же вакуум-выпарных аппаратах.
Упаренный раствор поступает в отстойники, в которых он отстаивается от кристаллов Na2SO4 и NaCl. После отстоя 1,5 ч в растворах с содержанием 1000 г/л СгО3 остается взвешенного сульфата натрия -10 г/л и более. С целью достижения лучшей очистки от Na2SO4 и NaCl раствор, предназначенный на выработку товарного дихромата натрия, охлаждают перед отстаиванием до 80—85° С при работающей мешалке. Из рис. 3.30 видно, что при указанной температуре растворимости Na2SO4 и NaCl в насыщенном растворе дихромата натрия близки к минимуму. Охлаждением раствора перед отстаиванием получают стандартный продукт. Процесс отстоя продолжается в течение 2—3 ч, после чего раствор охлаждают до 80° С и отстаивают еще более 6 ч.
Осветленный раствор декантируют, а отстой разбавляют водой (-1:1), перемешивают и передают на первую стадию дихроматных растворов.
Рис. 3.28. Предельное содержание хромата натрия в хроматно-сульфатных растворах
Рис. 3.29. Равновесие в системе
Na2Cr2O7—Na2SO4—Н2О
173
140
60
0
100
80
8*120
1 » I--1—I--L—X
40 80 120 Температура, °C
Температура, °C
6
a
Рис. 3.30. Растворимость в насыщенном растворе Na2Cr2O7: a — Na2SO4; 6 — NaCl
Отстоявшийся раствор с концентрацией -75% уварива-
ют до товарного продукта в плавильных (уварочных) котлах или в выпарных аппаратах с выносной греющей камерой.
Производство дихромата натрия. Дихромат натрия в производстве получают по схеме, приведенной на рис. 3.31. Исходный раствор хромата натрия центробежным насосом 2 из сборника 1 перекачивают через подогреватели 3 в подтравочник 7, в котором раствор обрабатывают серной кислотой из напорного сборника 5. Из подтравочников раствор с осадком выпавшего гидроксида алюминия перекачивают в рамный фильтр-пресс 6. Образующийся в фильтре шлам репульпируется в репульпаторе 7 и центробежным насосом 2 перекачивается на шламовые пруды. Часть отфильтрованного раствора самотеком поступает в отстойник 9, а часть направляется в сборники 8. Промытый горячей водой гидроксид алюминия — «хромаль» — репульпируют оборотной водой и откачивают на шламовые пруды; концентрированные промывные воды направляют в отстойники 9, а слабые воды перекачивают на погаску плава.
Из отстойников 9 осветленный раствор передают через спиральные подогреватели 10 на вакуум-выпарную установку 11. Упаренный раствор из последнего корпуса перекачивают через промежуточные сборники 8 в травочники 14, в которых хромат переводят в дихромат обработкой серной кислотой. После травки раствор обрабатывают в травочнике суспензией гипохлорита кальция.
Стравленный раствор поступает через промежуточные смесители в последний (по ходу сокового пара) корпус четырехкорпусной выпарной установки 16. Упаренный раствор с кристаллами Na2SO4 направляют из первого корпуса выпарной установки через самоиспари-тель 18 в баки-питатели центрифуги 79. 174
Рис. 3.31. Схема получения дихромата натрия:
1 — сборник; 2 — центробежный насос; 3— трубчатый подогреватель; 4—подтравочник; 5—напорная емкость; 6—фильтр-пресс, 7 — сборник-ре-пульпатор; 8— сборник; 9 — отстойник; 10 — спиральный подогреватель; 11 — вакуум-выпарная установка желтых щелоков (П и IV корпуса не показаны); 12 — барометрический конденсатор; 13— вакуум-насос; 14 — травочник; 15— смеситель; 16, 17—вакуум-выпарная установка красных щелоков (Ц корпус не показан); 18— с амоис паритель; 19—центрифуга; 20 — тарельчатый питатель; 21 — барабанная сушилка; 22 — шнек; 23 — бункер;
24 — пневмонасос; 25—промывная колонна; 26 — дымосос; 27 — отстойник; 28 — монтежю; 29—выпарная установка (продукционная)
Осадок сульфата натрия, отделенный на центрифуге 79, промывают горячей водой и передают ленточным транспортером в тарельчатый питатель 20 сушильного барабана 21. Высушенный сульфат натрия подают шнеком 22 и элеватором в бункер 23, а из него двухкамерным пневматическим насосом 24 — на склад готовой продукции. Сушат сульфат топочными газами от сжигания мазута или природного газа. Отходящие из сушилки газы очищают от пыли в насадочной промывной колонне 25, орошаемой в цикле водой до насыщения ее сульфатом.
Маточные растворы с центрифуг 19 передают через промежуточные смесители 15 в трехкорпусную вакуум-выпарную установку 77. Упаренный раствор из первого корпуса установки поступает через самоиспаритель 18 в отстойник 27, в котором отстаивается от кристаллов сульфата натрия и хлорида натрия (последний выпадает при охлаждении раствора до ~80° С). Осветленный раствор спускают через гребенку в монтежю 28, откуда сжатым воздухом его передают в уварочные котлы (на схеме не показаны) либо в однокорпусную выпарную установку 29. В уварочных котлах и на установке 29 раствор уваривают до готового продукта — плавленого дихромата натрия, который сливают в стальные барабаны, передаваемые по рольгангам на упаковку и на склад готовой продукции.
На рис. 3.32 приведена схема производства гранулированного дихромата натрия. Отстоявшийся в сборнике 7 раствор дихромата натрия с концентрацией 1100—1150 г/л СгОз центробежным насосом 2 перекачивается в упарочный аппарат 3, в котором раствор упаривается до концентрации 67—68% СгО3. Далее раствор упаривается до концентрации 72% СгО3 в уварочном котле 4. Плав гранулируют под вакуумом в охлаждаемом водой двухвальном грануляторе 5. Гранулированный продукт системой шнеков 6 передают в молотковую дробилку 7, в которой продукт измельчают до величин частиц не более 15 мм. В бункере-сепараторе 8 крупная фракция отделяется от мелкой, которая улавливается в циклоне 9. Через затворы 10 указанные фракции гранулированного дихромата натрия поступают на упаковку в стальные барабаны 12, поступающие постоянно по рольгангу 77. Упакованная продукция передается на склад готовой продукции.
Промывкой в скрубберах 15 и 18 маточным раствором в закрытом цикле улавливают пыль дихромата натрия с последующим использованием закрепленных промывных растворов. Отходящая из системы улавливания пыль, проходя через брызгоуловитель 19, вентилятором 20 выбрасывается в атмосферу.
На некоторых заводах успешно работает схема гранулирования дихромата натрия из раствора в псевдоожиженном слое (рис. 3.33). На гранулирование поступает дихроматный раствор с концентрацией 176
Пар
Рис. 3.32. Схема производства гранулированного дихромата натрия:
/—сборник исходных растворов; 2 и 14 — центробежные насосы; 3— упарочный аппарат; 4- -уварочный котел; 5 — двухвальный гранулятор; 6—шнеки; 7—молотковая дробилка; 8 — бункер-сепаратор для гранулированного дихромата натрия; 9 — циклон; 10 — затвор; // — рольганг; 12 — барабан; 13 — сборник маточных растворов; 15, 18 — скрубберы; 16 — вакуум-насос;
17 — воздухоотделитель; 19 — брызгоуловитель; 20 — вентилятор
около 1100 г/л СгОз. Температуру раствора поддерживают 70—105° С в расходном подогреваемом сборнике 5, в котором растворы постоянно циркулируют центробежным насосом 6. Этот же насос подает растворы в аппарат кипящего слоя 7 для распыления их в аппарате форсункой 7. Распыление производится сжатым воздухом, подогретым в змеевиковом подогревателе 8. Камера аппарата выполнена в виде двух сочлененных усеченных конусов. В топке с камерой смешения 2 установлена газовая горелка 3 для сжигания природного газа. Горелку воздухом питает турбовоздуходувка 4. Образующиеся горячие газы направляются в нижнюю коническую часть аппарата кипящего слоя. Температура топочных газов на входе в аппарат 360—375° С, температура в слое материала 230—280° С. В нижней конической части аппарата встроена дробилка 16 для создания новых центров грануляции. Высушенная часть продукта собирается в бункере для готового продукта 75.
177
Рис. 3.33. Схема грануляции дихромата натрия в псевдоожиженном слое:
/ — аппарат кипящего слоя; 2 — топка с камерой смешения; 3— газовая горелка; 4 — турбовоздуходувка; 5—расходный обогреваемый сборник раствора дихромата натрия; 6 — центробежный насос для циркуляции и подачи раствора; 7 — форсунка для распыления раствора; 8—змеевиковый воздухоподогреватель; 9—циклон для очистки газов от пыли; 10—форсунки, распиливающие слабый раствор для улавливания мелкой пыли; // — каплеуловитель; 12— дымосос; /3 — сборник для слабого раствора дихромата натрия; 14 — центробежный насос; 15 — бункер для готового продукта; 16 — встроенная дробилка для создания центров грануляции
Отходящие из аппарата газы проходят через циклон 9 для очистки газов от пыли, далее в газоходе из форсунок 10 орошаются слабым раствором, подаваемым центробежным насосом 14. проходят через каплеуловители 11. в которых улавливается мелкая пыль, и вентилятором 12 выбрасываются в атмосферу. Из каплеуловителя 11 слабые растворы дихромата натрия собираются в сборнике 13.
На получение 1 т дихромата натрия расходуют 0,393 т серной кислоты (в пересчете на моногидрат), 1,029 т хромата натрия, 0,005 т гипохлорита кальция, 140 кВт ч электроэнергии, 7,7 т пара. При этом на 1 т целевого продукта параллельно получают 0,515 т сульфата натрия. На гранулированный продукт дополнительно расходуется около 90 кВт ч электроэнергии и 0,3 т пара.
178
Дихромат калия. Физико-химические свойства. Дихромат калия К2СГ2О7 существует в трех модификациях.
0С-К2СГ2О7 — оранжево-красные, устойчивые на воздухе, пластинчатые или призматические кристаллы. Сингония VI, а = 7,50, b = 7,38, с = 13,40; а = 82°, р = 96,13°, у = 90,15°. Элементарная ячейка содержит четыре молекулы; плотность 2,676 г/см3.
Выше 241,6° устойчив Р-К2СГ2О7; сингония V, а = 7,47, b = 7,35, с = 12,97, а = 91,55°. Плотность 2,10 г/см3. Температура плавления 398° С. Начинает разлагаться при -500° С. В процессе нагревания начинает разлагаться выше 610° С. Ср = 48,53 + 67,110‘V (от 111 до 241° С) и Ср = 55,01 + 43,7-10'3 Т (от 290 до 385° С); ДЯ0°бр =-486,4 кДж/моль, ДЯП°Л = 8,770 кДж/моль, Д//ПеР.(а->р)= 0,366 кДж/моль. Слабо парамагнитен. На рис. 3.34 приведены данные о растворимости К2Сг2О7 в воде.
Дихромат калия растворим в жидком SO2 и хлориде фосфора, нерастворим в жидком аммиаке и этиловом спирте.
Технический дихромат калия должен удовлетворять требованиям существующего ГОСТ 2652—84:
Показатель Высший сорт I сорт II сорт
К2Сг2О7, % не менее 99,6 99,3 99,0
Хлориды (С1), % не более 0,07 0,2 0,4
Сульфаты (SO4), % не более 0,03 0,08 Не нормируется
Влага (Н2О), % не более 0,05 0,1 0,2
Нерастворимый в воде остаток, 0,08 0,15 0,2
% не более
Рис. 3.34. Растворимость дихромата калия К2СГ2О7 в воде
179
Физико-химические основы процесса получения дихромата калия. Процесс основан на реакции дихромата натрия с хлоридом калия или сульфатом калия:
Na2Cr2O7 + 2КС1 = K2Cr2O7 + 2NaCl (60)
Na2Cr2O7 + K2SO4 = K2Cr2O7 + Na2SO4 (61)
Фактически в обеих способах процесс протекает не в четырехкомпонентной системе, выражаемой уравнением (60) или (61), а в пятикомпонентной системе
К', Nal I СггО,”, SO*’, СГ—Н2О (62)
Это вызвано наличием в исходном растворе дихромата натрия Na2SO4 и NaCl, которые накапливаются в маточном растворе в результате цикличности производства. Таким образом, теоретической основой процесса обменного разложения являются равновесия, существующие в пятикомпонентной системе [см. уравнение (62)]. Эта система, а также четырехкомпонентная система К+, Na^lCr^?", SO4—Н2О изучены при 25—98° С в УНИХИМе Л.А. Боровских и Я.Е. Вильнянским.
Здесь приводятся квадратные диаграммы растворимости четырехкомпонентной хлоридной систему (рис. 3.35) и сульфатной системы (рис. 3.36). Состав солевой части жидкой фазы на диаграммах выражен в молекулярных (ионных) процентах; содержание воды в моль Н2О на 100 моль солей видно по изогидратам.
Рис. 3.35. Изотермы системы:
а — Na2Cr2O7 + 2КС1 # К2Сг2О7 + 2NaCl при 25° С; б— Na2Cr2O7+2KCl £ K2Cr2O7+2NaCl при 100°С
180
и 10 20 30 40 50 60 70 80 90 100
Рис. 3.36. Диаграмма растворимости четырехкомпойентной взаимной системы КагСггСЬ+КгЗОд КгСггС^+МагЗОд при 25° С
Из диаграммы хлоридной системы видно, что стабильной парой солей в системе являются К2Сг2О7 и NaCl. Наибольшим полем кристаллизации и, следовательно, наименьшей растворимостью как при 100° С, так и особенно при 25° С обладает К2Сг2О7. Сульфатная система осложнена образованием твердых растворов гла-зерита 3K2SO4 Na2SO4 и Na2SO4. Поле этих твердых растворов занимает, особенно при 98° С, большую часть диаграммы.
Циклический процесс производства К2Сг2О7 обменным разложением (циклические процессы непригодны из-за потерь хрома с маточными растворами) должен удовлетворять следующим требованиям: 1) отсутствие совместного осаждения К2Сг2О7 и натриевой соли; 2) получение достаточно крупных, хорошо фильтруемых и отмываемых кристаллов; 3) высокая производительность цикла, т. е. высокий съем кристаллов целевого продукта с единицы массы или объема маточных растворов в цикле; 4) минимальное удельное (на 1 т К2Сг2О7) количество выпариваемой воды.
Сравнение графоаналитических расчетов ряда циклов хлоридной и сульфатной систем на основе диаграмм растворимости показывает, что в циклах с большим выходом К2Сг2О7 приходится нередко выпаривать больше воды. Поэтому выбор оптимального цикла производят на основании технико-экономического расчета с учетом конкретных условий.
На рис. 3.37 показано построение некоторых циклов в хлоридной системе. В качестве примера рассмотрим построение цикла EGHE.
Насыщенный дихроматом калия и хлоридом натрия раствор Е охлаждают от 100 до 25° С. Кристаллизацию К2Сг2О7 (луч кристаллиза-
ции BEG) доводят до точки G, немного не доходящей до изотермы 25° С, во избежание выпадения NaCl и загрязнения им дихромата калия. Кристаллы дихромата отделяют от маточного раствора, последний нагревают и выпаривают при 100° С до достижения точки Н. При этом выпадает в осадок NaCl. Точка Н находится как пересе-
Рис. 3.37. Изображение циклических методов получения дихромата калия на диаграмме системы Na2Cr2O7+2KCl K2Cr2O7+2NaCl
чение луча кристаллизации ML с прямой АЕ. Цикл замыкается про-
182
цессом разбавления раствора Н смесью КС1 и Na2Cr2O7, взятых в эквивалентных количествах (точка Л), с получением раствора Е. Осадок отделяют от раствора Е.
Выход К2СГ2О7 ( в г-экв) за цикл легко определяется графически: если провести прямую ЕЕ\9 параллельную прямой GN, то отрезок NE дает выход Na2Cr2O7 — 0,242 г-экв или относительный выход 0 242
——100 = 46,1 %. Количество выпариваемой воды на 1 кг К2СГ2О7 со-0,525
ставляет, по данным графоаналитического расчета, 1,80 т.
Аналогично рассчитывают цикл KGLK, отличающийся от предыдущего тем, что из маточного раствора отделяют выпавший NaCl перед смешением с исходными реагентами, дает более высокий выход КэСг2О7 за цикл, а именно 0,346 г-экв или —•—100 = 58,6% и несколько 0,590
большее количество выпариваемой воды (0,87 т на 1 т К2Сг2О7).
Из диаграммы (рис. 3.37) видно, что оптимальным по выходу кристаллов за один цикл является проведение процесса на стабильной диагонали: отношение расстояния между изотермами 100 и 25° С к длине луча кристаллизации, соответствующее выходу кристаллического продукта, является максимальным. Исходя из этого обстоятельства, в УНИХИМе (Я. Е. Вильнянский и 3. С. Банных) разработан относительно простой метод графического расчета процесса обменного разложения (рис. 3.38).
Стабильной паре солей соответствует трехкомпонентная система К2Сг2О7—NaCl—Н2О. Для ее изображения использован прямоугольный треугольник, в котором составы приведены в массовых процентах и расчеты изменения фазового состава производятся по правилу рычага. На диаграмме нанесены изотермы растворимости при 25 и 100° С по данным Робертсона. Максимальный съем К2Сг2О7 с единицы массы раствора обеспечивает эвтонический раствор (точка Е\) изотермы 100° С. Луч кристаллизации К2Сг2О7 при охлаждении раствора от 100 до 25° С KEjP пересекает изотерму (25° С) в точке Р, дающей состав маточного раствора. Количество горячего раствора, необходимое для получения 1000 кг К2Сг2О7, равно 1000-^- = 4227 кг. Получится маточного раствора 4277—1000 =
РЕ,
= 3277 кг. К маточному раствору добавляют 100022L21 - 890,5 кг 294,21
Na2Cr?O7 (в виде раствора) и 1000 2 74,55 -506,8 кг КС1. В результате 294,21
обменного разложения получают 1000 кг К2Сг2О7 и 890,5 + + 506,8—1000 = 397,3 кг NaCl. Сумме продуктов обменного разло-183
N WO к (NaCl)
Рис. 3.38. Циклический процесс получения дихромата калия
жения соответствует на диаграмме точка О ( 1000 100 = 71,54% 1397,3
К2СГ2О7). Найденное количество NaCl осаждают из раствора выпариванием смеси раствора Р и свежей загрузки. Составу выпаренной смеси соответствует точка U9 лежащая на пересечении прямой РО и луча кристаллизации хлорида натрия NEj. Принимая, что с раствором Ка2Сг2О7 вводится минимальное количество воды, достаточное лишь для получения при обменном разложении раствора, насыщенного К2СГ2О7 при 100° С, свежая загрузка отобразится точкой R, лежащей на пересечении прямой ОН с изотермой 100° С, а смесь маточного раствора со свежей загрузкой (точка S) на пересечении прямой RP и HU.
PS
Количество раствора R равно 3277-----=2994 кг, а масса всех ма-
RS
териалов, поступающих на обменное разложение (без промывных вод): 3277 + 2994 = 6271 кг. Хлорид натрия выделяют изотермическим выпариванием раствора (точка S). Количество выпариваемой во-SU
ды: 6271— = 1603 кг. Получатся суспензии NaCl (точка U) ни
6271—1603 = 4668 кг и горячего раствора (точка Еу) 4668—397 = = 4271 кг (расхождение с ранее найденной величиной 0,15%).
Далее цикл, изображаемый многоугольником EjPSUEi, возобновляется.
184
В растворе (точка Е\) содержится 29,50% К2СГ2О7. Следовательно, выход ЮСГ2О7 составляет —5^— = 79,4%. Он лучше, чем в приве-4271 0,295
денных выше циклах. Однако с таким выходом, по-видимому, работать затруднительно из-за образования слишком мелких кристаллов К2СГ2О7.
Для пятикомпонентной системы К+, Na^CnC?-, SO4 , Cl—Н2О диаграммы растворимости сложны и затруднительны для графоаналитических расчетов. По диаграммам растворимости пятикомпонентной системы определено влияние Na2SO4 на обменное разложение с КС1. Найдено, что при содержании SO^" в системе более 14% от суммы анионов SO4" + 2С1~ условия процесса значительно ухудшаются. При большом содержании SO^- снижается выход К2СГ2О7 за цикл, возможно загрязнение продукта глазеритом и возможны потери калия в виде гла-зерита с хлоридом и сульфатом натрия. По заводским данным, моляр-
SCT'
ное отношение ----------- равно 0,10—0,14. Поэтому производствен-
SO^’ + 2С1 ’
ные условия отражают удовлетворительно диаграмма (рис. 3.39), полученная сечением изотермы пятикомпонентной системы плоско-
SO42'
стью, соответствующей отношению --------------, равному 0,10.
SO2' + 2C1“
Получение дихромата калия. Согласно схеме производства дихромата калия (рис. 3.40), в аппарат 4 вводят маточный раствор из сборника 33. Одновременно в аппарат 4 из сборника 1 через приемный сборник 2 перекачивают центробежным насосом 9 отмеренное количество и подогретый в емкости 3 раствор дихромата натрия. В аппарат 4 из
Рис. 3.39. Диаграмма пятикомпонентной системы К+, Na+||Cr2O5’, SOj , Cl — — Н2О (при 10% SOj ) при 25 и 98° С
185
Рис. 3.40. Схема производства дихромата калия:
7 — сборник растворов дихромата натрия; 2, 8 — сборники; 3 — расходная емкость; 4—реактор обменного разложения; 5— тельфер; 6, 15, 23 — сборники; 7 — кристаллизатор; 9 — центробежный насос; 10, 12, 26 — питательные сборники; 77, 27 — центрифуги, 13 — элеватор; 14— бункер; 76 — тарельчатый питатель; 77—шнек; 18— сушилка; 19 — барабаны с дихроматом калия; 20—калорифер; 21, 24—вентиляторы; 22 — скруббер; 25— емкость для оборотной воды; 28 — репульпатор; 29 — фильтр; 30—емкость, 31 — емкость для промывной воды; 32—сборник серной кислоты; 33 — сборник для маточных и промывных вод
сборника 32 поступает расчетное количество серной кислоты. Исходные растворы вводят в систему в таком количестве, чтобы концентрация СгОз в смешанном растворе достигла 260—280 г/л. Раствор нагревают до 105° С и вводят отмеренное количество (тельфером 5) хлорида калия в количестве, необходимом для перевода дихромата натрия в дихромат калия. Затем раствор обрабатывают хлорной известью или гипохлоритом кальция с целью осветления и дают раствору отстояться от кристаллов NaCl.
Отстоявшийся раствор спускают из аппарата 4 в сборник 6, куда предварительно залито небольшое количество «скрубберной воды» из сборника 23 для снижения концентрации NaCl и предотвращения его выпадения при кристаллизации дихромата калия. Из сборника 4 раствор центробежным насосом 9 перекачивают через расходный сборник 12 в охлаждаемый водой барабанный, вращающийся кристаллизатор 7. Образующаяся в кристаллизаторе суспензия кристаллов К2СГ2О7 в маточном растворе, проходя приемную емкость 8 и питательный сборник 10, направляется на центрифугу 11. Отжатый и промытый в центрифуге холодной водой дихромат калия передают элеватором 13 в бункер 14, а маточный раствор и промывную воду направляют через сборник 15 и 33 на обменное разложение. Маточный раствор возможно очистить от нерастворимых примесей, пропуская их через фильтр 29.
Из бункера 14 кристаллы сырого дихромата калия через тарельчатый питатель 16 и шнек 17 подают в барабанную сушилку 18, обогреваемую воздухом, подогретым в паровом калорифере 20. Сухой дихромат калия упаковывают в стальные барабаны 19. Отсасываемый из сушильного барабана воздух очищают от дихроматной пыли в системе, состоящей из скруббера 22, сборника 23 для растворов, вентилятора 24 и центробежных насосов 9. Воздух после системы очистки направляется на дополнительную газоочистку.
Осадок, остающийся в аппарате 4 после спуска осветленного раствора и состоящий в основном из хлорида натрия, репульпируют маточным раствором и откачивают через питательную емкость 26 на центрифугу 27. Отжатый осадок промывают оборотной водой из емкости 25, содержащей 3—5 г/л СгОз. Промытый осадок репульпируют оборотной водой в репульпаторе 28 и откачивают в отстойный пруд, а маточный раствор и промывную воду возвращают через сборники 31 и 33 на обменное разложение. При необходимости оборотный раствор дотравливают серной кислотой из емкости 32.
Рассмотренный способ обеспечивает использование хрома -99,0%. Потери с хлоридом натрия достигают 0,5—0,75%, а с хлоридом хромила (при дотравлении) 0,05%.
На получение 1 т целевого продукта расходуют: 1,004 т дихромата натрия (67,1% СгОз), 0,542 т хлорида калия (95% KCI), 0,025 т серной кислоты (100% H7SO4), 0,002 т хлорной извести, 300 кВт-ч 187
электроэнергии, 3,4 т пара, 8,2 м3 свежей воды, 70 м3 оборотной воды и 0,4 пог. м бельтинга.
Отходы, содержащие 1—2% К2Сг2О7, 33—49% Na2SO4 и 50—65% NaCl, могут быт применены в керамической и стекольной промышленности.
3.3. ГАЛОГЕНИДЫ ХРОМА
Физико-химические свойства галогенидов хрома. Дифторид хрома CrF2 при давлении 2,7 ГПа превращается в ромбическую модификацию, а при 27—29 ГПа — в тетрагональную (табл. 3.5); плохо растворим в воде, не растворяется в этаноле. Окисляется на воздухе. Медленно реагирует с минеральными кислотами. С фторидами щелочных металлов образует комплексы МСгРз и M2CrF4.
Трифторид хрома СгРз не растворяется в воде, этаноле, растворах аммиака, плохо растворяется в кислотах. В процессе нагревания на воздухе переходит в Сг2О2; гидролизуется водяным паром выше -500° С. С фторидами щелочных металлов при сплавлении образует МзСгРб и М5СгзР]4 зеленого цвета; ферромагнетик. Ниже 15° С устойчив нонагидрат СгРз-9Н2О (фиолетовый), растворимость в воде 0,9% при 0° С, легко обезвоживается до гексагидрата (фиолетовый, трудно растворяется в воде). Выше 20° С из водных растворов кристаллизуется зеленый тригидрат СгРз-ЗН2О, его растворимость в воде 3,46% (масс.) при 20° С, растворы склонны к пересыщению. Тригидрат, не растворимый в воде, для которого предложена формула [Сг(Н2О)б]СгР6, образуется в процессе нагревания гексагидрата при 60—70° С.
Трифторид хрома получают взаимодействием гидроксида или солей хрома (III) с фтороводородной кислотой:
Сг(ОН)3 + 3HF = CrF3 + ЗН2О
Cr2(SO4)3 + 6HF = 2CrF3 + 3H2SO4
Трифторид хрома применяется в качестве протравы при крашении тканей, является пигментом.
Тетрафторид хрома CrF4 — коричневое аморфное вещество с температурой плавления 277° С, температурой возгонки 295° С; плотность 2,90 г/см3; = -1200 кДж/моль. В процессе действия воды диссоциирует на соединения хрома (III) и хрома (IV), разъедает стекло, с щелочными металлами образует соединения типа М2СгРб.
Тетрафторид хрома получают в процессе фторирования порошка хрома:
Cr + 2F2 = CrF4
188
Таблица 3.5. Свойства галогенидов и оксогалогенидов хрома
Показатель CrF2 CrF3 CrF6 CrO2F2 СгС12 СгС13 СгВг2 СгВг3 CrJ2 CrJ3
Цвет Синевато-зеленоватый Зеленоватый Огненно-красный Фиолетовокрасный Бесцветный Красно-фиолетовый Бесцветный Темно-зеленый, черный Коричнево-красный, бледно-серый Черный
Сингония Моноклинная Тригональная Ромбическая Моноклинная Ромбическая Моноклинная Моноклинная Тригональная Ромбическая Тригональная
Параметры ячейки, нм: а 0,4732 0,602 0,55 0,568 0,665 0,5959 0,711 0,6308 0,3915 0,6859
b 0,4718 — 0,74 0,492 0,599 1,0321 0,3649 — 0,7560 —
с 0,3505 1,73 1,63 0,904 0,348 0,6114 0,6217 1,835 1,3553 1,988
Р, град 96,52 — — 93 — 108,48 93,88 — — —
Z 2 6 8 4 2 — 2 6 4 6
Пространственная группа P2Jc R3c Ртсп P2i/c Рппт С2!т — R3 Стс2\ Р3212
Температура плавления, °C 894 1100 102 31,6 824 1152 842 — 856 857
Температура кипения, °C 1820 1400 117 29,6* 1330 947* — 800* 1248 —
Плотность, г/см3 4,11 3,78 — 3,21 2,88 3,03 4,36 4,25 5,20 4,92
с;:, Дж/(моль«к) 59 79 — — 71 92 — 96 — —
ДЯП. , Оор’ кДж/моль -775 -1159 -1466 -880 -396 -570 -298 -400 -157 -205
АН™, кДж/моль 19 42 19,3 25,9 37 — — — — —
ДЯисп, кДж/моль 251 201 35,6 60,5** 198 288** — — — —
кДж/(моль-К) 86 94 209 — 116 125 134 150 154 —
♦Температура возгонки: **ДЯ|ЮЗГ
При фторировании под давлением получают пентафторид CrF5 и лимонно-желтый гексафторид CrF6, который легко диспропорциони-рует в вакууме при 100° С.
Диоксидифторид хрома (хромилфторид) СгС>2р2 на свету медленно полимеризуется, превращаясь в серое вещество с температурой плавления -200° С; водой легко гидролизуется; разрушает стекло, кварц, бурно окисляет органические вещества; с хроматами образует фторохроматы, например КСгОзЕ
Диоксифторид хрома получают в процессе нагревания дихромата калия с дымящей серной кислотой и дифторидом кальция.
Диоксифторид хрома применяют в качестве реагента в органическом и неорганическом синтезе.
Дихлорид хрома СгСЬ относительно устойчив в сухом воздухе, с кислородом начинает реагировать при -900° С, быстро увлажняется и окисляется в присутствии паров воды. Из водных растворов (голубых) кристаллизуются кристаллогидраты: до 38° С — темно-синий тетрагидрат, в интервале 38—51° С — его темно-зеленая модификация, при 51 —83° С — бледно-голубой тригидрат, выше 83° С — бледно-зеленый дигидрат, который обезвоживается при 113°С. Дихлорид хрома трудно растворяется в этаноле, не растворяется в эфире. С хлоридами щелочных металлов образует соединения типа МзСгСЬ, МСгС13, М2СгС14.
Дихлорид хрома применяют для получения хрома высокой чистоты и в качестве реагента в аналитической химии.
Трихлорид хрома СгС13 известен в тригональной модификации; антиферромагнетик. С кислородом начинает реагировать при 350° С, с водородом — при 515° С, восстанавливаясь до СгС^, а выше 700° С — до металлического хрома. Трихлорид хрома не растворяется в воде, а в присутствии восстановителей (СгСЬ) хорошо растворяется, давая темно-зеленые растворы. Из водных растворов ниже 30° С кристаллизуется темно-зеленый СгС13-1ОН2О, выше 30° С — гексагидрат, для которого известны четыре изомерные формы. В процессе обезвоживания гексагидрата получают коричневый тригидрат, фиолетовый дигидрат и красные кристаллы СгС13-1,5^0 и СгСЬДбНгО. Процесс обезвоживания сопровождается гидролизом. При сплавлении с хлоридами щелочных металлов СгС1з образует хлорохроматы преимущественно типов М3СгС1б и М3СГ2С19.
Трихлорид хрома применяют в процессе хромирования стальных изделий, для получения металлического хрома. Кристаллогидрат хлорида хрома применяют в качестве протравы в процессе крашения. Он является компонентом электролитов.
Тетрахлорид хрома СгС14 устойчив лишь в парах, мо-ЖС1 быть сконденсирован при резком охлаждении, но разлагается уже при -80° С.
190
Из оксохлоридов хрома наиболее важен хромилхлорид СЬСгОг — вишнево-красная жидкость с температурой плавления -96° С. Температура кипения СгО2С12 116° С; = -570,3
кДж/моль; дымит на воздухе, под действием света разлагается с выделением хлора; сильный окислитель и хлорирующий агент. Он со многими органическими веществами реагирует со взрывом; бурно разлагается водой с образованием Н2СгО4 и НС1. Растворяется в тетра- и трихлориде метана, сероуглероде, бензоле. Хорошо растворяет хлор; горит в сухом аммиаке.
Дибромид хрома СгВг2 энергично окисляется на воздухе. В отсутствии кислорода дает голубые водные растворы, образует гексагидрат. Дибромид хрома получают восстановлением СгВгз водородом и др.
Трибромид хрома СгВгз в процессе нагревания окисляется, медленно растворяется в горячей воде, быстро — в присутствии восстановителей. Известно несколько форм гексагидрата трибромида хрома. Получают взаимодействием паров Вг2 с порошком хрома.
Дииодид хрома Сг12 хорошо растворяется в воде и в отсутствие кислорода (голубые растворы) образует гексагидрат. Дииодид хрома получают взаимодействием металлического хрома с йодом. Применяется в качестве пигмента для стекла.
Трииодид хрома Сг!з в процессе нагревания отщепляет иод, с трудом растворяется в воде, легко — в присутствии Сг12. Из водных растворов кристаллизуется темно-фиолетовый гексагидрат.
Трииодид хрома получают из простых веществ.
Йодиды хрома являются полуфабрикатом в процессе иодидного рафинирования хрома.
Способы получения хлоридов хрома. Хлорид хрома (II) получают воздействием хлорида водорода на металлический (в виде порошка) хром:
Cr + НС1 = СгС12 + Н2
или же восстановлением при 600—700° С трихлорида хрома водородом:
2СгС13 + Н2 = 2СгС12 + 2НС1
Хлорид хрома (III) получают селективным хлорированием хромитов и феррохрома в присутствии восстановителей при 800—1000° С (рис. 3.41). Процесс хлорирования брикетов руды, полученных путем размола исходных руды и углеродистого восстановителя со смешением их и формированием, ведут в шахтных печах. Разработан также процесс гранулирования смеси сульфит-целлюлозными растворами. Образующиеся гранулы сушат и подвергают хло-191
Хромовая руда Углеродистый
или концентрат восстановитель
Продукт - безжелезистый Хлорный Хлорное Промывание
хромовый концентрат хром железо воды
Рис. 3.41. Принципиальная схема получения трихлорида хрома из хромовой руды
рированию. В процессе хлорирования брикетов или гранул образуется хлорид хрома (III), наряду с которым и другие летучие и нелетучие хлориды: AICI3, SiCl4, MgCl2 и др. При небольшом избытке хлора низшие хлориды хрома и железа (СгС12 и FeCl2) не образуются. В отсутствие восстановителя хлорируется лишь оксид железа (II). Образующиеся летучие хлориды могут быть разделены дробной конденсацией вследствие различного давления их пара.
Хлорид хрома (III.) отделяют от других хлоридов, используя различную их летучесть. В процессе получают водонерастворимый безводный СгС1з. Добавкой реагентов (Al, Zn, ZnSO4 и особенно СгС12) его переводят в раствор, а из раствора получают растворимый в воде гексагидрат хлорида хрома (III). Растворение СгС1з можно производить и без катализаторов — приведением водной суспензии в соприкосновение с катодом или нагреванием под давлением при 150—250° С.
Описано получение безводного СгС1з из сесквиоксида хрома в присутствии углеродистого материала. Реакция протекает около 1020° С по уравнению
Сг2О3 + 2,4С + ЗС12 = 2СгС1з + 1,8СО + 0,6СО2
Разработана технология трихлорида хрома путем обработки водных растворов хромонатриевых квасцов хлоридом бария:
Na2Cr2(SO4)4 + 4ВаС12 = 4BaSO4 + 2NaCl + 2CrO3 192
Образующиеся при этом растворы путем фильтрации освобождают от нерастворимого сульфата бария. Далее растворы фильтруют, упаривают и выделяют из них кристаллы трихлорида хрома.
Хлорид хрома СгС1у6Н2О (ГОСТ 4473—84 на продукт реактивной квалификации) получают восстановлением раствора триоксида хрома в 35%-ной хлороводородной кислоте этиловым спиртом (в конце процесса пироксидом водорода), упаркой растворов до плотности 1,54 г/см3 и кристаллизацией. В УНИХИМе разработан вариант применения древесных опилок в качестве восстановителя.
Трихлорид хрома получают хлорированием феррохрома (углеродистого) при 950—1000° С на 100% и с большей скоростью.
В вертикальном реакторе, в процессе подачи исходного хлора в среднюю зону, процесс можно вести с получением хлорида хрома (II) в расплаве (в приемнике из нижней зоны) по уравнению
-Cr7C3 +С1, =CrCl, +-С 7 7
и хлорид хрома (III) в возгоне:
-Сг7С3 +1,5С12 =СгС12 +-С 7 7
В процессе хлорирования с избытком хлора получаются газообразные хлориды хрома (III) и железа (III), а без избытка хлора получается расплав хлорида хрома (II). При температуре приемника не ниже 815° С образуется также хлорид железа (II). Отношение СгС12:СгС1з в расплаве растет с повышением температуры.
Из-за значительной разницы в летучести хлориды хрома могут быть практически полностью отделены от хлоридов железа. При хлорировании феррохрома с получением дихлоридов проще схема производства, меньше расход хлора и расход электроэнергии на последующий электролиз при производстве металлического хрома.
В качестве хлорирующего агента можно использовать и хлороводород. Равновесная степень превращения НС1 в процессе взаимодействия с Сг2Оз в условиях атмосферного давления невелика. При осуществлении процесса в шахтной печи при 950—1000° С и притоке газа и феррохрома получают расплав СгС12 с содержанием FeCl2 не менее 1%. Рекомендовано двухстадийное хлорирование хромита хлоридом водорода, а затем хлором. Термообработкой брикетов из хромита, углерода и хлорида натрия при 800° С и пропусканием хлора, с последующим выщелачиванием плава неконцентрированной хлороводородной кислотой получают в шламе после выщелачивания фио-
193
7 Химическая технология
неорганических веществ, кн. 2
летового цвета хлорид хрома (III). В процессе термообработки хлориды железа, алюминия и кремния улетучиваются, а хлориды магния и кальция выщелачиваются.
Разработан способ получения хлорида хрома (III) путем хлорирования феррохрома в расплаве FeCl2—СгС12 при 800° С и ВаС12—СаС12 при 900° С.
Хлорид хрома (III) может быть получен растворением гидроксида хрома в хлороводородной кислоте, например из гидроксида, полученного растворением феррохрома в хлороводородной (или серной) кислоте с последующим осаждением карбонатом или гидроксидом железа (II), и из гидроксида хрома (III), полученного восстановлением хроматных растворов тиосульфатом натрия.
3.4. СУЛЬФАТЫ ХРОМА
Физико-химические свойства сульфатов хрома. Сульфат хрома (III) Cr2(SO4)3— красновато-коричневые кристаллы тригональной сингонии (<7 = 0,812 нм, с = 2,186 нм), плотность 3,012 г/см3; С ° =281 Дж/(моль К); ДН^р = -3308 кДж/моль; 52°98 = 288 Дж/(моль-К); парамагнетик.
Сульфат хрома (III) в процессе нагревания выше 350° С разлагается:
3Cr2(SO4)3 - •—-> 2CrO(SO3)2 + 2Cr2O3 + 5SO2 + ЗО2
а выше 640° С до Сг2О3:
4CrO(SO4)2 640°c-> 2Сг2О3 + 8SO2 + 7О2
Сульфат хрома (III) не растворяется в холодной воде и разбавленных кислотах, плохо растворяется в этаноле.
Известен кристаллогидрат Cr2(SO4)3-18Н2О — сине-фиолетовые кристаллы с температурой плавления -80° С; плотность 1,70 г/см3; Ср° = 934 Дж/(моль-К); АЯ^р=-8317 кДж/моль; 52°98 = 1035 Дж/(моль-К); растворимость в воде (в пересчете на безводную соль) 45,4% (масс.) при 20° С, растворяется в этаноле. В процессе плавления превращается в зеленый декагидрат, при 100° С переходит в пентагидрат, а при 440° С — обезвоживается полностью (процесс не сопровождается гидролизом). При 700—735° С сульфат хрома (III) разлагается с образованием сесквиоксида:
2Cr2(SO4)3 -> 2Сг2О3 + 6SO2 + ЗО2 194
Известны также фиолетовые гидраты с 17, 16, 15 и 14 молекулами воды. В процессе нагревания фиолетовых водных растворов до 70—80° С они становятся зелеными, после чего из них могут быть выделены зеленые гидраты, в которых часть анионов SO^- находится во внутренней сфере комплекса СгОз(ОЗОз)2".
Известны зеленые кристаллы с 15 молекулами воды (плотность 1,867 г/см3), с 8, 6, 5 и 3 (плотность 2,429 г/см3) молекулами воды и др. Стеклообразные или аморфные гидраты хорошо растворяются в воде, не растворяются в этаноле.
Сульфат хрома (III) с сульфатами щелочных металлов и NH^ образует квасцы. Из концентрированных сульфатных растворов выделены кислые сульфаты Сг2(8О4)зН28О4-16Н2О (фиолетовый и зеленый) и др., которые иногда рассматривают как хромсерные кислоты. Они хорошо растворимы в воде.
КСг(8О4)2- 12Н2О — темно-фиолетовые кристаллы (в проходящем свете — рубиново-красные). Показатель преломления света 1,4814; плотность 1,826 г/см3; температура плавления 89° С; С" = 161,8 Дж/(моль-К). В процессе нагревания ниже 100° С отщепляет шесть молекул воды, в интервале 120—200° С — 4Н2О, а остальные 2Н2О — 200—450° С.
Растворимость в воде при 25° С — 243,9 г/л; при 18° С — 282,0 г/л. Не растворяется в этаноле. Фиолетовый раствор становится зеленым при 50—70° С; теплота растворения равна (при 20° С) 9,50 ккал/г-моль, pH раствора фиолетовых квасцов 2,8.
NaCr(SO4)2-12H2O не кристаллизуется. При нагревании до 100° С теряет 8Н2О. Упругость диссоциации (12Н2О—>6Н2О): при 50° С 40, при 80° С -241 мм рт. ст. Растворимость в воде при 18° С -948 г/л (при содержании фиолетовой модификации 51,7%).
Сульфат хрома (И) Сг8О4-5Н2О— синие кристаллы триклинной сингонии (а = 0,724 нм, b = 1,094 нм, с = 601 нм, а = 125,32°, р = = 97,63°, у = 94,32°); в сухом состоянии устойчив на воздухе; растворяется в воде (17,3% (масс.) при 0° С), растворы легко окисляются кислородом воздуха. В процессе нагревания обезвоживается при 30—265° С с последовательным образованием тетра-, ди- и моногидрата, а безводный CrSO4 разлагается при ~500°С:
2Сг8О4—>Сг2О3 + SO2 + SO3
С сульфатами щелочных металлов образует двойные соли типа шенита М2Сг(8О4)2-6Н2О.
195
Сульфат хрома (II) получают растворением хрома в серной кислоте:
Cr + H2SO4 = CrSO4 + Н2
Получают также восстановлением растворов Cr2(SO4)3 электролитически или цинком:
Cr2(SO4)3 + Zn = 2CrSO4 + ZnSO4
Применяют в качестве восстановителя в органическом синтезе и поглотителя кислорода из газов.
Способы получения сульфатов хрома. Наравне с сульфатом хрома (III) широко применяется и гидроксосульфат хрома.
Гидроксосульфат хрома (III) получают так же, как и хромовые квасцы, восстановлением дихромата натрия диоксидом серы и солями сульфитного ряда (сульфит, гидросульфит и тиосульфат натрия) или органическими веществами (сахар, глюкоза, крахмал, мука, глицерин, формалин и др.).
В качестве восстановителей применяли патоку и глюкозу:
8Na2CnO7 + 24H?SO4 + С]?Н22Оц(2СбН12Об) = = 16Cr(OH)SO4 + 8Na2SO4 + 12СО2 + 27Н2О(28Н2О)
Реакция идет с частичным образованием промежуточных продуктов окисления углеводов, в основном органических кислот (щавелевой и др.) и альдегидов. С повышением температуры ускоряется реакция и меньше образуется промежуточных продуктов.
В процессе восстановления диоксидом серы реакция идет по уравнению
Na2Cr2O7 + 3SO2 + Н2О = 2Cr(OH)SO4 + Na2SO4
Гидроксосульфат хрома (III), полученный по приведенным' реакциям, имеет основность 1/3 или 33,3%. Действительный состав продукта значительно сложнее, чем показывает формула Cr(OH)SO4, вследствие имеющих место процессов гидролиза при кислотности растворов pH ~3,5. Состав и структура комплексных ионов хрома зависят от характера восстановителя. Так, при восстановлении глюкозой получаются преимущественно катионные комплексы хрома, а при восстановлении опилками — ионные. В процессе взаимодействия Na2Cr2O7 с SO2 при повышенной температуре образуется комплексный сульфат хрома [Cr2(OH)2SO4(OH2)6]SO4; при комнатной температуре наряду с сульфатокомплексами образуются дитионатохромкомп-196
лексы, поскольку SO4 окисляется частично до дитионовой кислоты H2S2O6, не стойкой при нагревании. Na2SO4 повышает стабильность растворов гидроксосульфатов хрома.
Процесс восстановления органическими веществами проводят в освинцованном реакторе, снабженном метальным устройством. Обычно к раствору дихромата натрия добавляют серную кислоту в количестве, соответствующем необходимой основности продукта, а затем медленно, во избежание кипения и вспенивания, добавляют восстановитель для полного восстановления.
Сухой хромовый экстракт получают также восстановлением раствора дихромата натрия (~35О г/л) в две стадии: сначала — гидролизом опилок в 60—65%-ной серной кислоте и затем — глюкозой или патокой. Образующийся раствор упаривают до концентрации -300 г/л Сг2Оз и сушат при 95—110° С до порошкообразного состояния (<30% Сг2О3).
В процессе восстановления диоксидом серы газ из серной горелки пропускают через башню с керамической насадкой противотоком к раствору дихромата натрия. После окончания восстановления через раствор пропускают острый пар в течение примерно 1,5 ч до полного удаления избытка диоксида серы и требуемого «старения раствора».
Получаемый сиропообразный раствор плотностью 1,38—1,50 г/см3 высушивают в распылительной сушилке, обеспечивающей образование хорошо растворимого аморфного продукта зеленого цвета. Продукт упаковывают во влагонепроницаемые бумажные мешки. Производят основной сульфат хрома, содержащего 24—25% Сг2Оз и имеющего основность от 33 до 52%. Примерный состав продукта с основностью 33,3% при восстановлении органическими соединениями (в %):
Сг20з SO3 Na2SO4 Н2О Прочие
24 25 23 24 4
Продукт с основностью -33%, содержащий 21,5—22,0% Сг2Оз, не растворимых в воде (при 60° С) не более 0,2% и Ре20з не более 0,1%, производят восстановлением дихромата калия формалином:
2К?СгэО7 + 6H2SO4 + ЗНСНО = = 4Cr(OH)SO4 + 2K2SO4 + ЗСО2 + 7Н2О
Процесс восстановления ведут в реакторе, футерованном кислотоупорными плитками, при 60—70° С (не выше 90° С) в течение —10 ч. Полученный раствор (плотность 1,6 г/см3) высушивают в распылительной сушилке из нержавеющей стали дымовыми газами, разбавленными воздухом, с температурой не выше 200°С. При этом 197
способе на получение 1 т продукта расходуют 454 кг К2Сг2О7, 81 кг KHSO4, 352 кг H2SO4 и 46 кг НСНО.
Рекомендовано комбинирование основных солей хрома (III) с растворимыми солями щелочно-земельных элементов, цинка, алюминия или магния, дающие возможность получения растворимых препаратов высокой основности (66,7%) приготовлением двойной магний-хромовой соли по уравнению
MgCr2O7 + 3SO2 + Н2О = MgSO4Cr2(OH)2(SO4)2
В УНИХИМе (М.Т.Серебренникова с сотр.) разработан непрерывный способ производства сухого хромового дубителя с основностью 40—42% восстановлением диоксидом серы растворов монодихромата натрия с концентрацией СгОз около 300 г/л при 60—70° С в противоточном аппарате, состоящем из трех последовательно соединенных горизонтальных реакторов с метальным устройством. Процесс протекает автотермично по реакции (п = 0,15-ь0,3)
2Na2Cr?O7 + ШСгО4 + (7,5 + 2,5h)SO2 + (3—2,5и)Н2О = = 2,5Cr2(SO4)i,8(OH)2,4.2/?(SO3> + 3Na2SO4
Восстановленный раствор с концентрацией 15—17% Сг2Оз нагревают до -100° С для освобождения его от диоксида серы, а затем чешуируют или сушат при распылении. В процессе чешуирования получают товарный продукт, содержащий ~23% Сг2О3, а при сушке—27,5% Сг2О3.
Сульфат хрома (III) получают также восстановлением раствора Na2CrO4 диоксидом серы с последующей упаркой растворов, отделением выпавших кристаллов Na2SO4 и окислением сульфита в сульфат.
Восстановление протекает по уравнению
6Na2CrO4 + 15SO2 = 2Сг2(8Оз)з 4" Сг2(8О4)з 4" 6Na2SO4
Затем сульфат хрома (III) разлагают серной кислотой:
Сг2(8Оз)з + 3H2SO4 = Сг2(8О4)з + 3SO2 + ЗН2О
Способы получения хромовых квасцов. Хромовые квасцы MeCr(SO4)212H2O, где Me — Na, К, NH4, получают восстановлением соответствующих дихроматов в водном растворе в присутствии серной кислоты или взаимодействием раствора сульфата хрома с соответствующим сульфатом. 198
В процессе получения квасцов из дихроматов применяют те же восстановители, что и при получении сульфата хрома (III). Предложен также метод восстановления оксидами азота или нитрозилсерной кислотой с получением азотной кислоты:
K2Cr2O7 + 4H2SO4 + 6МО2 + 23Н2О = 2[KCr(SO4)2-12H2O] + 6HNO3
Ma2Cr2O7 + H2SO4 + 3HSO4NO + 23Н2О = = 2[NaCr(SO4)2-12H2O] + 3HNO3
Хромонатриевые квасцы. Хромонатриевые квасцы получают восстановлением подкисленного серной кислотой раствора Na2Cr2O7 (1100—1200 г/л) при температуре не выше 60—65° С древесными опилками и в конце реакции мукой:
4Na2Cr2O7 + 16H2SO4 + С6Н10О5 + 75Н2О = = 8[NaCr(SO4)2-12H2O] + 6СО2
В порядке усовершенствования способа в качестве восстановителя применяли торфяную газогенераторную смолу, что ускоряет процесс восстановления и улучшает качество продукта. Операция длится до 5—6 сут, после чего жидкий продукт сливают в деревянную тару, в которой он превращается в пастообразную массу. Целевой продукт содержит 16—20% Сг2О3 и не более 1,5% свободной серной кислоты.
Хромокалиевые квасцы. Хромокалиевые квасцы получают восстановлением раствора К2Сг2О7 в серной кислоте диоксидом серы:
K2Cr2O7 + H2SO4 + 3SO2 + 23Н2О = 2[KCr(SO4)2-12H2O]
Для восстановления возможно использовать отходящие газы контактного серно-кислотного производства, а также производства сульфитных солей.
Хромокалиевые квасцы получают из сточных вод после окисления органических веществ (дихроматом калия), а также восстановлением дихромата формальдегидом:
2К2Сг2О7 + 8H2SO4 + ЗНСОН + 37Н2О = 4[KCr(SO4)212H2O] + ЗСО2
В футерованном реакторе растворяют 5 т К2Сг2О7 в 7 м3 воды, добавляют 7 т серной кислоты (плотность 1,84 г/см3), после чего постепенно вводят 2550 кг 30%-ного формалина. Температуру в реакторе поддерживают в пределах 35—40° С. По окончании процесса ре-199
акционную массу охлаждают до 20—25° С. Для повышения выхода продукта поддерживают 5%-ный избыток серной кислоты. Твердую часть реакционной массы отжимают и промывают в центрифуге. Полученный целевой продукт практически не содержит железа и содержит -15% Сг2Оз.
Хромокалиевые квасцы технической квалификации производят восстановлением древесными опилками. На 1 т К2Сг2О7 расходуется - 1,1 т H7SO4 и -0,13 т опилок и получают -3 т квасцов с содержанием 98—98,5% KCr(SO4)2-12H2O.
Продолжительность реакции восстановления при 50—55° С составляет 8 ч. Восстановленный раствор (плотность 1,33—1,38 г/см3) после отстоя передается в кристаллизатор. Продолжительность процесса кристаллизации -20 сут. Кристаллы промывают холодной водой и высушивают при температуре не выше 35° С.
Процесс кристаллизации хромокалиевых квасцов проводят и во вращающихся барабанных кристаллизаторах. Возможно обезвоживание растворов хромокалиевых квасцов в распылительной сушилке.
Хромоаммонийные квасцы. Существует в основном два способа, разработанных для электролитического получения металлического хрома. По первому способу восстанавливают хромат натрия диоксидом серы, и электролит готовят добавлением серной кислоты и сульфата аммония к полученному раствору сульфата хрома (после кристаллизации и отделения сульфата натрия). По второму способу перерабатывают хромит или высокоуглеродистый феррохром непосредственно в хромоаммонийные квасцы.
Более просто получение хромоаммонийных квасцов из феррохрома. Процесс получения электролита состоит из следующих стадий.
1. Растворение феррохрома в серной кислоте, к которой добавлен маточный раствор хромоаммонийных квасцов и отработанный анолит, содержащий сульфат аммония, серную и хромовую кислоты. Хром (VI) анолита предварительно восстанавливают диоксидом серы в насадочной башне. Продолжительность процесса выщелачивания феррохрома составляет 48 ч, а процесс проводится в футерованном кислотоупорным кирпичом стальном реакторе. Образующийся после выщелачивания раствор разбавляют в охладительном сборнике маточным раствором соли Мора, охлаждают примерно до 80° С и отделяют от кремнийсодержащего твердого остатка на гуммированных фильтрах. Остаток промывают водой и отводят в шламонакопитель. Фильтрат поступает в футерованный сборник для кондиционирования, снабженный паровой рубашкой, где при повышенной температуре в течение 2 ч происходит стабилизация зеленой модификации соли NH4Cr(SO4)212H2O (конденсирование раствора).
200
2. Кондиционированный раствор охлаждают в гуммированном ва-куум-кристаллизаторе до 5° С, и выпавшие кристаллы сульфата железа (III) отделяют на барабанном вакуум-фильтре. Сырой сульфат железа (II) растворяют в промывной воде (от промывки кремнийсодержащего остатка) и добавляют к раствору сульфат аммония в количестве, необходимом для получения соли Мора (NH4)2Fe(SO4)2-6H2O. Полученный раствор охлаждают в вакуум-крис-таллизаторе, и выпавшую соль Мора отфильтровывают на вакуум-фильтре и высушивают в барабанной сушилке. Продукт применяют в качестве удобрения.
3. Фильтрат после отделения сырого сульфата железа (II) направляют через контрольный фильтр в стальные с коррозионно-устойчивым покрытием внутри сгустители-кристаллизаторы, рассчитанные на 10-дневный запас. Параллельно в сгустители-кристаллизаторы вводят католит, содержащий сульфат аммония и хром (II). В сгустителях-кристаллизаторах при 30° С зеленая модификация квасцов переходит в фиолетовую и выпадают в осадок кристаллы хромоаммониевых квасцов. Выход кристаллов составляет около 80%. Кристаллы отфильтровывают и промывают на горизонтальном вакуум-фильтре. Промытые кристаллы растворяют в горячей воде, и полученный электролит направляют через контрольный фильтр в электролитические ванны.
В УНИХИМе разработан способ получения хромоаммонийных квасцов и хромоаммиачного электролита восстановлением растворов хромата натрия диоксидом серы и древесным углем с выводом сульфата натрия в виде мирабилита и циркуляцией сульфата аммония в технологическом цикле. Процесс (в цикле с электролитическим получением металлического хрома) состоит из следующих стадий.
1. Восстановление части исходного раствора Na2CrO4 (210—240 г/л) диоксидом серы:
2Na2CrO4 + (3 + h)SO2 + (2-и)Н2О = 2Na2SO4 + Cr2SO4(SO3)w(OH)4_2/?, где п - 14-1,25.
2. Смешение раствора гидросульфат-сульфита хрома с маточным раствором хромоаммонийных квасцов, кристаллизация мирабилита и отделение его от раствора.
3. Восстановление второй части раствора Na2CrO4 и шестизамещенного хрома анолита (со стадии электролита) размолотым древесным углем в присутствии серной кислоты, поступающей с анолитом:
Na2Cr2O7 + (NH4)2Cr2O7 + 8H2SO4 + ЗС =
= 2Cr2(SO4)3 + Na2SO4 + (NH4)2SO4 + 3CO2 + 8H2O
201
Восстановление древесным углем предотвращает накопление в системе H2SO4. Оптимальные условия восстановления: значительный избыток свободной серной кислоты; нагревание раствора до 102—106° С и затем до температуры кипения 115—120° С; стехиометрическое количество угля. Возможно восстановление и древесными опилками.
4. Смешение восстановленного раствора (см. стадию 3) с маточным раствором после отделения мирабилита. При этом протекает реакция
Cr2SO4(SO3)„(OH)4_2,7 + 2H2SO4 = Cr2(SO4)3 + hSO2 + (4-и)Н2О
Образующийся диоксид серы отдувают из раствора доведением его до кипения.
5. Кристаллизация хромоаммонийных квасцов из смеси раствора после стадии 4 (охлажденного до 50—60° С) и части католита, содержащего (NH4)2SO4 и CrSO4, и отделение выделившихся кристаллов. Двузамещенный хром, вводимый с католитом, ускоряет переход зеленой модификации квасцов в кристаллизующуюся фиолетовую (кристаллизация продолжается в течение 1—2 сут).
6. Приготовление электролита из раствора хромоаммонийных квасцов и другой части католита.
ГЛАВА 4
ФТОР И ЕГО СОЕДИНЕНИЯ
Физико-химические свойства фтора. Содержание фтора в земной коре 0,065% (масс.). В природе встречается лишь в виде соединений. Основным фторсодержащим минералом является флюорит (плавиковый шпат) CaF2. Общее содержание фтора в промышленных рудах флюорита 122 и в перспективных — 236 млн. т. К фторсодержащим минералам относятся также криолит Na3AlF6, хиолит 5NaF-3AlF3, селлаит MgF2, иттрфлюорит Са]_.vYxF2_х. Изоморфно замещая ионы ОН-, О2- и другие, фтор входит также в состав многих минералов, например топаза Al2SiO4(OH,F)2, амблигонита LiAlPO4(OH,F), бастнезита (Ce,La)CO3F, апатита и фосфоритов Ca5(PO4)3(OH,F), слюд и гидросиликатов. Кроме флюорита промышленное значение имеют апатит и фосфориты, из которых фтор получают в виде химических соединений в больших количествах. Соединения фтора содержатся во многих подземных водах.
В небольших количествах фтор входит в состав живых организмов, так в организме человека 2,6 г фтора, из которых 2,5 г в костях. Он участвует в процессах образования зубов и костей, в обмене веществ и в активации некоторых ферментов. Нормальной дозой суточного поступления в организм человека считается 2,5—3,5 мг. Повышенные или пониженные количества фтора вызывают различные заболевания.
Фтор состоит из одного стабильного нуклида 19F. Конфигурация внешней электронной оболочки атома 2s22p5, степень окисления -1, а энергия ионизации при последовательном переходе от F0 к F7+ соответственно равна 1681, 3375, 6046, 8409, 11 024, 15 164 и 17 868 кДж/моль. Фтор — самый электроотрицательный элемент, его электроотрицательность по Полингу 3,98.
Молекула свободного фтора двухатомна, межатомное расстояние 0,14165 нм.
Фтор — бесцветный газ с сильным запахом, напоминающим запах хлора. Температура кипения —188,20° С, а плавления — 219,70° С. Плотность газа при 0° С и 0,1 МПа 1,693 кг/м3, жидкости—1516 кг/м3 (-188° С). АНПЛ = 0,5104 кДж/моль, АЯИСП = 6,544 кДж/моль; С ° = 31,34 Дж/(моль К); 52°98 = 202,68 Дж/(моль-К).
203
В твердом состоянии при обычном давлении фтор образует две кристаллические модификации: ниже -227,60° С a-форму с моноклинной решеткой (плотность 1,97 г/см3 при -250° С), выше — P-форму с кубической решеткой (а = 0,667 нм, плотность 1,70 г/см3).
Плотность жидкого фтора описывается уравнением d = = 1,907—2,20-10-3Г—2,948-10-5Г2 г/см3 (67<Г<103 К); вязкость — уравнением г| = 2,43-10'7ехр( 196/7) Па-с; уравнение температурной зависимости давления пара над жидким фтором
1п(р/ртрПа) = 7,89592346Х + 3,38765063Jf2—1,34590196^ + + 2,73138936Х(1—JQ1’4327,
где X = (1—7"тр/7)/(1—7"тр/Гкрит); Гтр и ртр — соответственно температура и давление в тройной точке; ГКрИТ — критическая температура.
Фтор растворяется в некоторых фторидах, хлор- и фторуглеродах. Жидкий фтор неограниченно смешивается с жидким кислородом и озоном. Энергично реагирует с водой:
2F2 + 2Н2О = 4HF + О2
Он не образует кислородных кислот. Фтор бурно реагирует с водородом, образуя фторид водорода:
F2 + Н2 = 2HF
Фтор является сильнейшим окислителем и фторирующим реагентом. Благодаря высокой энергии связи элемент — фтор во фторидах и низкой энергии диссоциации F2 многие реакции фторирования простых веществ, оксидов, галогенидов и других соединений необратимы, сопровождаются выделением значительного количества теплоты и образованием фторидов элементов в высших степенях окисления. В атмосфере фтора «горят» такие стойкие вещества, как стекло (в виде ваты):
SiO2 + 2F2 = SiF4 + О2
Исключительно активно проходит взаимодействие фтора с большинством простых веществ. С серой и фосфором фтор реагирует даже при температуре жидкого воздуха (-190° С):
S + 3F2 = SF6, ДЯ2°98 = -1207 кДж;
2Р + 5F2 = 2PF5, ДЯ°8 = -3186 кДж.
204
Фтор окисляет даже некоторые так называемые инертные газы:
Хе + 2F2 = XeF4, ДЯ2°98 = -252 кДж.
Непосредственно фтор не реагирует лишь с Не, Ne и Аг.
Однако по кинетике взаимодействия со фтором поведение веществ может резко отличаться. Большинство реакций имеют цепной характер. Они самопроизвольно инициируются даже при комнатной или более низкой температуре и протекают со вспышкой или со взрывом, а в потоке— с возникновением пламени. К таким реакциям относятся фторирование водорода и водородсодержащих веществ (углеводородов, Н2О, NH3, Ы2Н4, HCI, HBr, HCN и др.), серы и ее производных (SO2, SOCI2, S4N4, SBr2 и др.), Si, Р и др. При комнатной температуре со фтором способны реагировать щелочные металлы, некоторые галогениды, гидраты солей. Однако реакции фтора со значительной частью простых веществ и неорганических соединений протекают лишь при их активации, достигаемой повышением температуры или давления.
Оксиды металлов и многие соли более устойчивы к действию фтора, чем сами металлы. Взаимодействие оксидов может сопровождаться образованием на промежуточных стадиях оксифторидов.
Наиболее устойчивы к действию фтора благородные газы, азот, кислород, алмаз, СО, СО2, сапфир, алунд и некоторые виды стеклоуглерода.
Получение и применение фтора. Получение свободного фтора включает добычу и обогащение флюоритовых руд, серно-кислотного их разложения с выделением и очисткой безводного фторида водорода, его электрохимическое разложение.
Электрохимическое разложение фторида водорода осуществляют тремя способами: низкотемпературным (15—50° С в смеси HF с KF), среднетемпературным (70—120° С, расплав КН2Рз) и высокотемпературным (245—310° С, расплав KHF2). В настоящее время в промышленности применяют среднетемпературный способ.
Стандартный потенциал разложения фторида водорода в расплаве КН2Рз равен 2,9 В. Промышленные электролизеры работают обычно при 80—105° С, напряжении 8,5—12,0 В и анодной плотности тока 70—180 мА/см2. Их мощность по току достигает 11 кА, опытных образцов— 15 кА, что соответствует производительности 7—10 кг Р2/ч.
Электролизеры представляют собой стальные или монелевые ванны с размещенными на крышке угольными анодами и расположенными между анодами стальными катодами. В некоторых конструкциях между катодами и анодами имеются перфорированные диафрагмы, предотвращающие смешивание и взаимодействие выделяющихся фтора и водорода. Современные электролизеры снабжены системами непрерывной подачи в них фторида водорода, поддержа-205
ния постоянной температуры, отвода выделяющихся фтора и водорода. Отбор фтора проводят с помощью специальных коробчатых сборников, «колоколов», расположенных на крышке и погруженных в расплав таким образом, что они окружают верхнюю часть анодов. В процессе электролиза на поверхности угольных анодов пассивируют слой фторидов графита CFY, что вызывает «анодный эффект» — резкое повышение напряжения и его скачки. Этот эффект подавляют введением в электролит добавок, использованием анодов особой конструкции и пульсирующего тока. Свежезагружаемый электролит тщательно обезвоживают, проводя электролиз примеси влаги при низком напряжении. Для снижения температуры процесса разрабатывают электролиз смеси КЬЬРз—NH4HF2—НЕ
Фтор рафинируют методами селективной сорбции примесей (HF на гранулированном пористом NaF), вымораживания примесей, сжижения фтора, химического и физического связывания примесей. Глубокую очистку фтора проводят низкотемпературной ректификацией или его хемосорбцией, например путем образования и термического разложения K2NiF4. В лабораторных условиях пользуются твердыми источниками, например MnF4 в процессе нагревания до 200° С выделяет около 15% фтора от своей начальной массы. Получают также реакцией
2K2MnF6 + 4SbF5 = 4KSbF6 + 2MnF3 + F2
Применяют также пиротехнические источники фтора, содержащие соли тетрафтораммония.
Свободный фтор применяют в качестве фторирующего реагента в производстве гексафторида урана, галогенфторидов, тетра- и гексафторидов серы, трифторида бора, фторидов азота, фторидов графита, высших фторидов металлов (WF6, МоРб, ReF6), фторидов благородных газов, фторорганических производных и др. Атомный фтор используют в химических лазерах на HF и DF, для синтеза KrF2, фторидов кислорода и др.
4.1. ФТОРИД ВОДОРОДА И ФТОРОВОДОРОДНАЯ КИСЛОТА
Фторид водорода HF — бесцветный газ или подвижная жидкость, дымящая на воздухе. Температура плавления -83,36° С; температура кипения 19,52° С; температура критическая 230° С; плотность критическая 0,29 г/см3, критическое давление 6,49 МПа; С° = 29,14 Дж/(моль-К); Д/7°бр = -273,3 кДж/моль; 52°98 = 173,675 Дж/(моль-К); АЯ'сп = 7,489—ЛЗ,9(Т—292,69) кДж/моль (250<Т<390 К); уравнение температурной зависимости давления пара над фторидом водорода 206
Igp(na) = 0,21317—918,24/7" + 3,215421g/1, где 193<7"<378 К. Плотность жидкого HF близка к плотности воды: d = 1,0020—2,2625-10‘3Z + + 3,125-10'6/ г/см3, где -74</<42° С, d*2 = 0,98. Чистый жидкий HF
практически не ионизирован, р = 10'60м-см(0° С).
Характерное свойство HF — склонность к ассоциации. Средняя степень ассоциации в жидкости п = 6. В газовой фазе могут присутствовать линейные и циклические олигомеры с п = 4, 6—12. Наиболее устойчив циклический (HF)6. В газообразном, жидком состоянии, также в водных растворах молекулы фторида водорода ассоциированы (рис. 4.1).
Фторид водорода хорошо растворим в воде. Водные растворы его называют фтороводородной кислотой. Равновесные концентрации фторида водорода в газовой фазе над фтороводородной кислотой приведены на рис. 4.2 и 4.3.
Температуры кипения в системе HF—НзО и равновесные составы пара при атмосферном давлении приведены на рис. 4.4. Как видно из диаграммы, в системе HF—Н2О имеется азеотропная смесь, содер-
жащая -38% HF и кипящая при 109° С (0,101 МПа).
Фторид водорода является хорошим растворителем для многих веществ. Наибольшая растворимость характерна для пентафторида сурьмы (смешивается неограниченно), фторида таллия (580 г в 100 г
HF при 12° С) и фторид цезия (199 г в 100 г HF при 10° С). Растворимость фторидов щелочных металлов (кроме LiF) в HF в 2—4 раза ниже, чем в воде, BaF2, PbF2 и CoF2 — в сотни, BeF2 — в тысячи, а AgF — в сотни тысяч раз больше, чем в воде. Это связано с сильной сольватацией фторид-иона в HF.
Фторид водорода
реагирует со многими простыми веществами с образованием фторидов, с оксидами — оксифторидов и фторидов, замещает галоген в галогенидах металлов, с фторидами щелочных и других одновалентных металлов образует гидрофториды металлов, с фторидами и оксидоф-торидами многих элементов в присутствии воды — фторсодержащие кислоты, например H2SiF6, HBF4 и др.
Рнс. 4.1. Зависимость средней молекулярной массы и давления насыщенного пара фторида водорода от температуры
207
Фторид водорода получают реакцией флюорита с серной кислотой:
CaF2 + H2SO4 = 2HF + CaSO4
Реакцию проводят при 120—180° С. Образующиеся реакционные газы очищают от пыли, конденсируют из них HF и подвергают двухступенчатой ректификации. Применяется также способ получения HF серно-кислотным или термическим разложением NH4HF2:
2NH4HF2 + H2SO4 = 4HF+ (NH4)2SO4
NH4HF2 —2HF + NH3
Фторид водорода применяют в качестве катализатора гидрирования, дегидрирования, алкилирования в органической химической технологии, реагента в производстве хладонов и фторопластов, тетрафтора урана, трифторида алюминия и др. Фторид водорода является исходным сырьем для получения фтора и фторсульфоновой кислоты.
HF в кислоте, %
Рис. 4.2 Концентрация HF в газовой фазе над фтороводородной кислотой
Рис. 4.3. Зависимость давления пара HF (Р, кПа) над фтороводородной кислотой от температуры. Содержание НЕ %: /—41,2; 2 — 50,9; 3 — 55,1;
4 — 60,52; 5 — 65,45; 6 — 70,23
208
Фтороводородная кислота — водный раствор фторида водорода. В системе HF—Н2О (рис. 4.5) могут существовать три-соединения: H2O’HF(H3O+F'), Н2О-2НР(НзО+НР“) и H2O4HF, температура плавления которых соответственно 36, 78 (с разложением) и -100° С. Температура плавления эвтектической смеси Н2О—H2OHF -72° С; для азеотропной смеси (38,2% (масс.) HF) температура кипения 114,5° С. При всех составах, отличных от азеотропного, фтороводородная кислота испаряется инконгруэнтно. Равновесное давление паров HF и Н2О над фтороводородной кислотой описывается уравнением 1g р(Па) = = A-BIT. Значения коэффициентов приведены в табл. 4.1.
Таблица 4.1. Значения коэффициентов А и В при различных концентрациях фтороводородной кислоты
Концентрация HF,% (масс.) Pin Р\\го
А В А В
10 10,168 2610 10,919 2225
20 10,621 2605 10,867 2235
30 10,759 2500 10,768 2247
50 10,449 2120 10,738 2386
70 10.437 1830 — —
Для растворов HF + яН2О ДЯ^р = -317,74 кДж/моль (п = 1), -322,03 кДж/моль (п = 10), -322,36 кДж/моль (п = 100) и -335,65
Рис. 4.4 Температура кипения и состав пара в системе HF—Н?О при 0,101 МПа:
Рис. 4.5. Диаграмма плавкости системы HF— Н2О
/— состав жидкости; 2 — состав пара
209
Рис. 4.6. Зависимость плотности фтороводородной кислоты от концентрации HF (%)
кДж/моль (п = оо). Фтороводородная кислота содержит ионы Н+, F-, HF", Н3О+, (HF)„F" и молекулы H3O(HF)„F" с п = 1—4. Плотность фтороводородной кислоты при 25° С меняется от 1,00132 г/см3 (20 моль/дм3) до 1,07099 г/см3 (11,40 моль/дм3).
Фтороводородная кислота— сильная кислота, р/Са 3,14; образует соли— фториды, реагирует со многими веществами, в том числе с такими оксидами, как SiO2, В2О3, Nb2O5, Те2О5, МоО3, WoO3 и силикатами.
На рис. 4.6 приведены данные по изменению плотности фтороводородной кислоты в зависимости от ее концентрации, а на рис. 4.7 — номограмма для определения парциального давления HF над фтороводородной кислотой.
Фтороводородную кислоту получают растворением HF в воде, водной абсорбцией газообразных продуктов серно-кислотного разложения CaF2, пирогидролизом CaF2, UF3 и фторидов других металлов, флюоритовых руд и фторсодержащих отходов.
Фтороводородная кислота является сырьем в производстве A1F3, UF3 и других неорганических фторидов, компонентом растворов трав
ления стекла, кварца, материалов микроэлектроники, металлов и неко
торых сплавов.
Фтороводородную кислоту (техническую), согласно действующему ГОСТ 2567—73, выпускают марок А и Б с содержанием не менее 70 и 40% HF и не более 0.5 и 0,02% H2SiF6, 0,1 и 0,02% H2SO4. Кислоту перевозят в гуммированных железнодорожных цистернах и в полиэтиленовых сосудах. Тарой для жидкого фторида водорода, содержащего HF не менее 99,9% (высший сорт) или 99,87% (1-й сорт), являются стальные баллоны, контейнеры и специальные железнодорожные цистерны, рассчитанные на эксплуатацию в условиях повышенного давления.
Технология фторида водорода и фтороводородной кислоты. В молекуле F2 связь слабая (160 кДж/моль), а связь Н—F — прочная. Это может служить энергетической основой протекания реакций фторирования в виде цепной раз-210
ветвленной реакции фтора из элементов
Н2 + F2. Это дает возможность получать хлорид по следующей схеме, состоящей из стадий:
F2 + стенка -> 2F#
F2 + Н2 -> Н + HF + F
F* + Н2 -> HF* + Н-
Н- + F2 -> HF* + F-
HF* 4- H2 -> HF + H2*
H2* + F2 -> Н- + HF + F.
H2* + M -> H2 + M
Н' + О2 + М —> но2’ + м
Н*(или F«) 4- стенка -> обрыв
Зарождение цепи
Продолжение цепи Передача возбуждения Разветвление
Дезактивация
Обрыв цепей
t, °C
25 -
30 -
HF, % (масс.) 30 -п
2—*
60-
р, мм рт. ст.
Р 30
- 20
1.
Г 6
- 4
~ 2
г- 1 h
Г-0.4
Г 0-2
70 -
J
Рис. 4.7. Номограмма для определения парциального давления HF над фтороводородной кислотой
211
Наиболее распространенным в промышленности способом получения фторида водорода является процесс разложения природного флюорита с 90—92%-ной серной кислотой:
CaF2 + H2SO4 = 2HF + CaSO4
Процесс проводят при 120—180° С в барабанных вращающихся печах непрерывного действия.
Параллельно с основным процессом протекают и побочные реакции, вызванные примесями исходного сырья:
СаСОз + H2SO4 = CaSO4 + СО2 + Н2О
М2О3 + 3H2SO4 = M2(SO4)3 + ЗН2О
SiO2 + 4HF = SiF4 + 2Н2О и др.
В промышленности существуют три способа получения фтороводородной кислоты:
1) водные растворы HF;
2) 100%-ный жидкий HF;
3) слабые водные растворы из отходов (в основном) переработки выхлопных газов HF в процессах переработки флюорита.
Получение жидкого (100%-ного) фторида водорода и фтороводородной кислоты. Для получения жидкого фторида водорода необходимо иметь высококонцентрированный и не содержащий значительных количеств тетрафторида кремния газ. Поэтому в производстве в качестве исходного сырья применяют плавиковый шпат (флюорит) или его концентрат с содержанием 96—97% CaF2 и минимальное количество диоксида кремния.
На рис. 4.8 приведены показатели выхода жидкого фторида водорода в зависимости от концентрации газа в процессе охлаждения его от 80 до -15° С. Как видно из рисунка, выход жидкого фторида водорода резко снижается с понижением концентрации газа HF и при 54% достигает нуля. Из газа, содержащего менее 54% HF, можно конденсировать жидкий фторид водорода лишь при температурах ниже -15° С, что в производстве нерационально.
Наличие в исходном газе незначительных количеств воды резко снижает степень выхода жидкого фторида водорода. Содержание воды в газе зависит от наличия примесей в исходном сырье, разлагающихся с выделением воды, уравнения реакций, приведенных выше. Вследствие этого при получении жидкого фторида водорода качество исходного сырья имеет особое значение. Поэтому в производстве наилучшим способом отделения воды является дробная конденсация фторида водорода из газа. 212
Поскольку фторид водорода смешивается с водой в любых отношениях, образуя так называемые высококонцентрированные фтороводородные кислоты, содержащие 80% HF и более, то одним из способов получения жидкого фторида водорода может являться метод ректификации таких кислот. В этом случае в кубе ректификационной колонны остается постоянно кипящая смесь, а концентрированный фторид водорода, отходящий из колонны, может быть сконденсирован в жидкий продукт.
Согласно приведенной на рис. 4.9 схеме получения безводного фторида водорода, исходный
Рис. 4.8. Степень выхода жидкого фторида в зависимости от концентрации исходного газа
плавиковый шпат, содержащий не
менее 97% CaF2 и не более 1% SiO2 и 1% СаСОз, при необходимости сушат в барабанных сушилках. Сухой продукт дробят на щековых, конических и валковых дробилках и измельчают на шаровых мельницах до тонкости, при которой на сите с 3000 отв/см2 остается не более 10% материала.
Исходный порошкообразный флюорит из приемного бункера элеватором 5 поднимается в верхний бункер 7, откуда шнековым транспортером 8 загружается в бункер-питатель 9. Исходная серная кислота из оборудованного воздушным клапаном 2 сборника 1 центробежным насосом перекачивается в напорную емкость 10, откуда через ротаметр 11 подается в смеситель-питатель. Одновременно в питатель непрерывно дозируется исходный флюорит. Из смесителя-питателя шнекового типа, в котором продолжительность пребывания массы составляет 5—6 мин, шихта (смесь флюорита с серной кислотой) поступает в печь 6.
Для получения HF применяют барабанные вращающиеся печи непрерывного действия. Барабан из СтЗ толщиной 12—16 мм заключен в кирпичную кладку и обогревается природным газом. Шихта из смесителя поступает в печь через отверстие в неподвижной загрузочной головке (снабженной также патрубком для отвода газа) и по мере протекания процесса превращается в рассыпчатый сухой материал, который удаляют через наклонный желоб в задней крышке печи. Реакция протекает наиболее интенсивно в передней части печи. Эта часть барабана печи защищена вставленной внутрь антикоррозийной броней из малоуглеродистой стали толщиной 14 мм. Материал брони — сталь ЭИ-533(Х23Н23МЗДЗ) или ЭИ-643(Х23Н28МЗДЗТ).
213
кислота
Рис. 4.9. Схема получения безводного жидкого фторида водорода:
7 — сборник кислоты. 2 — воздушный клапан; 3— центробежный насос; 4— бункеры; 5 — элеватор. 6— печь разложения плавикового шпата; 7 — дымовая труба. 8 — шнековый транспортер; 9— бункер питателя; 10— напорный сборник; 11 — ротаметр; 12 — циклонный пылеуловитель;
13 — скруббер; 14, 16, 18 — абсорбер; 75 — холодильник; 77 — холодильник; 19—сборник; 20—вентилятор; 27 — ректификационная колонна;
22 — дефлегматор
Продолжительность пребывания реакционной массы в печи составляет 55—60 мин при температуре в передней части печи 160—180° С, а в задней — 220—280° С. Отходящий из печи газ имеет температуру 120—140° С и содержит до 80% НЕ Выгружаемые из печей отходы производства состоят из -80% СаБОд, 2—6% CaF2 и 10—12% серной кислоты. Отходы нейтрализуют карбонатом кальция.
Газообразный HF, выходящий из печи, по газоходу проходит через пылеуловитель 12, промывается серной кислотой в скруббере 73, в котором при помощи центробежного насоса 3 через холодильник 75 циркулирует серная кислота и направляется в абсорбционную колонну 14, выпускающую 80%-ный НЕ Остаток газа направляется в абсорбционную колонну 16, выпускающую 60%-ную фтороводородную кислоту. После циркуляции насосом 3 в системе холодильник 77 — абсорбер 16 выпускается 60%-ная фтороводородная кислота. Далее газ из абсорбера 16 направляется в абсорбционную установку 18, выпускающую 30%-ную фтороводородную кислоту по той же технологии, как и в абсорбционных установках 14 и 76.
Выхлопные газы перед выпуском в атмосферу промывают водными растворами гидроксида натрия в колонне с коксовой насадкой. 80%-ная фтороводородная кислота подвергается ректификации в медной колпачковой колонне 27 и через дефлегматор 22 выходит в виде товарного продукта. 60%-ная фтороводородная кислота возвращается на питание абсорбционных колонн, где концентрируется до 80% НЕ
Для очистки жидкого фторида водорода от незначительных количеств воды его обрабатывают 90—100%-ной серной кислотой в экстрактивно-дистилляционной колонне. В верхнюю часть колонны подают серную кислоту, а в среднюю часть колонны — жидкий фторид водорода. Давление в колонне поддерживают в пределах 0,35—0,7 атм, а температуру в верхней части — 27—50° С, в нижней части -150—180° С. С верхней части колонны отводится безводный HF, а с нижней — 80%-ная серная кислота.
Повышенная температура в нижней части колонны и разбавление серной кислоты обусловливают гидролиз образующейся в небольшом количестве фторсульфоновой кислоты.
Безводный фторид водорода можно получить ступенчатым гидролизом жидкой фторсульфоновой кислоты:
HSO3F + Н2О = HF + H2SO4
При этом количество воды, добавляемой на каждой ступени, должно быть меньшим, чем необходимо для полного разложения HSO3F.
Получение фтороводородной кислоты отличается от способа получения жидкого фторида водорода в основном системой абсорбции фторида водорода. В этом случае процесс абсорбции фторида водо-215
рода проводят в системе, состоящей из нескольких абсорберов (7—8 абсорберов), заполненных коксом, угольными кольцами или деревянными рейками. Корпуса абсорберов гуммируют и футеруют в два слоя угольными блоками на бакелитовой замазке. В настоящее время абсорберы изготовляют из фторопласта. Абсорберы, работающие при температуре ниже 80° С, изготовляют из органического стекла или полиэтилена. Их можно изготовлять также из графита.
Процесс абсорбции проводят следующим образом. Последний абсорбер орошается водой, и получаемая неконцентрированная кислота передается на орошение предыдущих абсорберов противотоком газу. Из первой по ходу газа абсорбера вытекает товарная кислота. Для просасывания газов через систему за последним абсорбером устанавливают химзащищенный вентилятор.
Кислота циркулирует с помощью химзащищенных центробежных насосов. Как видно из рис. 4.10, для получения стандартной фтороводородной кислоты, содержащей 40% HF, при 25° С требуется, чтобы концентрация HF в поступающем на абсорбцию газе была выше 7 мг/л, а при 50° С — выше 30 мг/л. Орошение абсорберов водой регулируют таким образом, чтобы в товарной кислоте содержалось 12—18% НЕ
В абсорберах параллельно с процессом поглощения HF происходит конденсация SiF4. Концентрация SiF4 в технической фтороводородной кислоте из абсорберов зависит от содержания диоксида кремния в исходном плавиковом шпате и колеблется от 2 до 20%. Очистку фтороводородной кислоты от SiF4, образующую гексафторосиликат натрия, производят обработкой ее карбонатом натрия в химзащищенных реакторах. Образующийся по реакции
2HF + Si F4 + Ма2СО3 = Na2SiF6 + Н2О + СО2
Рис. 4.10. Растворимость гексафторосиликата натрия в фтороводородной кислоте при различных температурах
труднорастворимый гексафторосиликат натрия выделяется в осадок, который отделяют отстаиванием или отфильтровывают на вакуум-фильтре и промывают водой. Промывные воды смешивают с фтороводородной кислотой, направляют на получение фторидов.
Применение избытка карбоната натрия для осаждения гексафторосиликата натрия не допускается, поскольку при этом нейтрализуется часть HF:
216
H2SO4 + Na2CO3 — Na2SO4 + H2O + CO2
2HF + Na2CO3 = 2NaF + H2O + CO2
При расходе стехиометрического количества карбоната натрия, необходимого для очистки растворов от гексафторосиликата водорода, приведенные реакции не имеют решающего значения, поскольку растворимость гексафторосиликата натрия меньше, чем растворимость фторида и сульфата натрия. Поэтому содержащийся в растворе гексафторосиликат водорода осаждает Na2SiF6 из этих солей по схеме
2NaF + H2SiF6 = Na2SiF6 + 2HF
Na2SO4 + H2SiF6 =: Na2SiF6 + H2SO4
В очищенной кислоте содержится небольшое количество H2SC>4, NaF и незначительное количество Na2SiF6, зависящее от концентрации HF и температуры (см. рис. 15.10). Содержание Na2SiF6 несколько больше соответствующего его растворимости.
С целью получения фтороводородной кислоты (не содержащей H2SiF6 и Na2SiF6) печной газ очищают от тетрафторида кремния до процесса абсорбции HF путем поглощения SiF4 раствором K2SO4.
На получение 1 т 100%-ного HF расходуют: 2,55 т флюорита (100% CaF2), 3,75 т серной кислоты (100% H2SO4).
Жидкий фторид водорода применяется в качестве растворителя спиртов, альдегидов и эфиров и катализатора для процессов полимеризации, изомеризации и алкилирования, в частности в процессе получения высокоактивных моторных топлив. Значительные количества безводного газообразного и жидкого фторида водорода применяют в процессах получения фторзамещенных органических соединений— фторуглеродов, используемых в качестве теплоносителей, диэлектриков, средств огнетушения, смазочных веществ, а также для изготовления термо- и химически стойких пластических масс (фторопласты), хлорсодержащие фторуглероды (фреоны). Кислоту применяют в производстве фторидов титана, циркония, ниобия, тантала, ванадия и др. Высшие фториды кобальта CoF3, марганца MnF3, церия CeF4, свинца PbF4 и серебра AgF2 являются катализаторами в процессах фторирования углеводородов. В процессах получения каучуков, фармацевтических препаратов и других в качестве катализатора используют фторид бора. Катализаторами служат также фториды сурьмы, ртути и цинка. Фторид водорода применяют для извлечения урана из фосфатов и для получения фторидов урана.
Значительное количество солей фтора используется в металлургии. Около 70% добываемого дифторида кальция (флюорита) применяют в качестве флюса в мартеновских и электрических печах. Чистые сорта фторида кальция применяются в оптических приборах и т. д.
217
4.2. ТЕХНОЛОГИЯ ФТОРИДОВ
Наиболее широко применяемыми в народном хозяйстве являются фториды натрия, калия, аммония, кальция, алюминия, кремния и др.
Фторид натрия. Фторид натрия NaF — бесцветные кристаллы с кубической решеткой (а = 0,46344 нм, z = 4, пространственная группа Fm3m) с температурой плавления 996° С, кипения 1770° С, имеет плотность 2,766 г/см3. С" = 46,82 Дж/(моль-К); А//о°бр = -576,6 кДж/моль, ДС7°бр = -542,6 кДж/моль, ДЯП°Л = 34,25 кДж/моль, ДЯ^ЗГ = 280,7 кДж/моль (ОК), ДЯВ°ОЗГ = 176 кДж/моль; 52°98 =51,6 Дж/(моль-К). Растворимость фторида натрия в воде (41,5 г/л при 20° С), безводном HF (30,1 г в 100 г при 11° С); ДЯ° растворения для бесконечно разбавленного водного раствора 0,943 кДж/моль.
Фторид натрия реагирует с фторидами и оксифторидами металлов III—VIII групп, бериллием и магнием, образуя фторометаллаты. С фторидом водорода и его водными растворами образует NaHF2.
Встречается в природе в виде минерала виллиомита.
Фторид натрия применяют в производстве алюминия и фторида водорода (для получения 1Чаз[А1Р6] и NaHF2), в качестве компонента составов для очистки и алитирования металлов, флюсов для сварки, пайки и переплавки металлов, стекол, эмали, керамики, огнеупоров, кислотоупорного цемента, термостойких смазок, составов для травления стекол, зубной пасты, твердых электролитов, консерванта древесины, инсектицида, реагента для фторирования воды, сорбента для поглощения гексафторида урана из газовых потоков и в процессе очистки UF6 или WF5. Применяется также в качестве реагента в процессе получения фтороуглеводородов, таких, как компонент специальных сортов бумаги, ингибитор брожения, огнезащитных составов и средств пожаротушения.
Способы получения фторида натрия. Фторид натрия получают нейтрализацией фтороводородной кислоты техническим карбонатом натрия. При этом нейтрализуются и примеси других кислот:
2HF + Ма2СО3 = 2NaF + Н2О + СО2
H2SiF6 + Na2CO3 = Na2SiF6 + Н2О + СО2
H2SO4 + Na2CO3 = Na2SC>4 + Н2О + СО2
После нейтрализации кислот образовавшийся гексафторосиликат натрия при повышенной температуре разлагается карбонатом натрия:
Na2SiF6 + 4Na2CO3 + 2Н2О = 6NaF + SiO2 + 4NaHCO3 218
При повышенной температуре образующийся гидрокарбонат натрия разлагается по схеме
2NaHCO3 = Na2CO3 + H2O + CO2
Зависимость скорости процесса разложения Na2SiF6 карбонатом натрия при различных температурах приведена на рис. 4.11, из которой видно, что при содержании в растворе 150,7 г/л Na2CO3 гексафторосиликат натрия при 90—95° С разлагается практически
полностью.
При введении карбоната натрия в раствор технической фтороводородной кислоты вначале нейтрализуется гексафторосиликат водорода, а образующийся гексафторосиликат натрия выделяется в осадок. В процессе разложения кристаллов Na2SiF6 после нейтрализации раствора на их поверхности образуется тонкая пленка гидрогеля диоксида кремния, затрудняющая доступ карбоната натрия. В результате процесс разложения резко замедляется даже при интенсивном перемешивании реакционной массы. Для исклю
чения этого явления процесс ведут при постоянной щелочности
реакционной массы. При этих условиях Na2SiF6 не образуется. В
реактор вводят лишь меньшую часть фтороводородной кислоты и расчетное количество карбоната натрия. Количество вводимой кислоты должно быть достаточным для того, чтобы реакционная мас-
са была подвижной. Через определенный период вводят всю расчетную массу кислоты. При этих условиях загрузки исходных реагентов гексафторосиликат натрия не выделяется в осадок, а полностью разлагается карбонатом натрия.
Образующи йся труднора-створимый фторид натрия выделяется в осадок в виде кристаллов. Осадок фторида натрия отфильтровывают, промывают водой от маточного раствора и сушат.
На получение 1 т фторида натрия расходуют: 0,45—0,46 т HF в виде фтороводородной кислоты и 1,08—1,10 т карбоната натрия.
Рис. 4.11. Кинетика процесса разложения гексафторосиликата натрия в растворе карбоната натрия (150,7 г/л Na2CO3) при различных температурах
219
Фторид натрия получают термическим разложением гексафторосиликата натрия во вращающейся барабанной печи при 600—620° С по реакции
Na2SiF6 = 2NaF + SiF4
Процесс продолжается 20 мин, а степень разложения исходного сырья составляет 98—100%. Выделяющийся SiF4 абсорбируется водой и перерабатывается на Na2SiF6. На этом принципе основана циклическая схема производства фторида натрия термическим методом. Получаемый целевой продукт содержит 85—88% NaF, до 1,5% Na2SiF6 и 8,5—10% нерастворимого в воде остатка, состоящего в основном из SiO2 Расход исходного гексафторосиликата натрия при однократном использовании его составляет 2,34 т на 1 т фторида натрия, а при замкнутом процессе с использованием выделяющихся газов — 0,83 т с учетом 10% потерь от массы исходного сырья. В процессе термообработки предварительно гранулированного сырья потери от уноса пыли значительно снижаются. Концентрация образующегося в процессе термообработки тетрафторида кремния в газе составляет 2,5—8,6%. При дальнейшем увеличении концентрации SiF4 процесс разложения исходного сырья замедляется и качество целевого продукта ухудшается.
При абсорбции выделяющегося SiF4 водой получают чистый гексафторосиликат водорода с концентрацией 35—39% H2SiF6.
Гексафторосиликат натрия получают карбонатно-термическим способом по схеме
Na2SiF6 = 2NaF + SiF4
SiF4 + 2Na2CO3 = 4NaF + SiO2 + 2CO2
Na2SiF6 + 2Na2CO3 = 6NaF + SiO2 + 2CO2
При этом шихта, состоящая из 47% гексафторосиликата натрия и 53% карбоната натрия, увлажняется до содержания 25% воды. Увлажнение производится для уменьшения уноса материалов с выхлопными газами из печи и снижения температуры процесса. Шихта подвергается в барабанной вращающейся печи термообработке при температуре 580—600° С.
Образующийся продукт охлаждают в шнековом водяном холодильнике, размалывают и упаковывают в фанерные барабаны. Целевой продукт содержат 70—74% NaF.
На 1 т 70%-ного фторида натрия расходуют 0,675 т карбоната натрия (100% Na2CO3), 0,61 т гексакремнесиликата натрия, 0,16 т условного топлива, 180 кВт-ч электроэнергии и 40 м3 воды. 220
Видоизмененный вариант способа получения фторида натрия из отходящих газов основывается на реакции
SiF4 + 2Na2CO3 + 2Н2О = 4NaF + SiOr2H2O + 2СО2
Как видно из уравнения, процесс осаждения фторида натрия осуществляется в одну стадию. Однако при этом получается смесь NaF с SiO2-2H2O, полное разделение которой в производственных условиях затруднено.
Поташный метод производства фторида натрия заключается в конверсии гексафторосиликата калия маточным раствором карбоната калия, разделении образующегося раствора фторида калия и выделившейся кремниевой кислоты, конверсии фторида калия карбонатом натрия с осаждением NaF.
Согласно схеме, гексафторосиликат калия получают осаждением его хлоридом калия из раствора гексафторосиликата водорода:
H2SiF6 + 2КС1 = K2SiF6 + 2НС1 (1)
или абсорбцией газообразного тетрафторида кремния раствором карбоната калия:
3SiF4 + 2К2СО3 = 2K2SiF6 + 2СО2 + SiO2 (2)
Дальнейшие стадии процесса проходят по уравнениям:
K2SiF6 + 2К2СО3 = 6KF + SiO2 + 2СО2 (3)
6KF + 3Na2CO3 ЗК2СО3 + 6NaF (4)
Образующийся раствор карбоната калия полностью возвращается на стадии (2) и (3). В варианте применения хлорида калия для получения фторида кремния ~30—35% этого раствора необходимо перерабатывать в товарный карбонат калия.
По обоим вариантам технологии образуется осадок аморфной кремниевой кислоты (гидрат диоксида кремния), который после промывки и сушки является побочным продуктом, называемым белой сажей.
Процесс разложения гексафторосиликата калия раствором карбоната калия протекает с высокой скоростью при температуре кипения смеси 103—113° С. Исходный гексафторосиликат при этом разлагается практически полностью при небольшом (5%) избытке карбоната калия.
221
На рис. 4.12 приведена часть диаграммы водной системы 2KF + Na2CCh 2NaF + К2СОз при 75° С. Для процесса осаждения фторида натрия имеют значение разрезы диаграммы данной системы через поверхность насыщения NaF при различном соотношении в растворе К2СОз и KF. Как видно из приведенной диаграммы, экспериментально определены два таких разреза для температуры 75° С при отношениях К2СОз.КР, равных 2,2 и 3,4 (кривые I и II). Они показывают, что по мере роста концентрации К2СОз + KF происходит полное высаливание NaF до начала кристаллизации К2СОз-1,5Н2О. Эти данные позволяют организовать технологию с полным выделением фторида натрия путем выпарки раствора карбоната калия в присутствии фторида калия.
Рис. 4.12. Диаграмма взаимной водной системы 2KF + Na2CO3 2NaF + Na2CO3 при 75° С в прямоугольных координатах (разрез по оси NaF — К2СО3 и области в координатах 2NaF—К2СО3—2KF):
/ — разрез через поверхность насыщения NaF при отношении K2COvKF=2,2; II— то же, при K2COvKF = 3,4
222
Фторид калия. Фторид калия KF — бесцветные кристаллы с кубической решеткой (а = 0,5347 нм, z = 4, пространственная группа Fm3m). Температура плавления 858° С, кипения 1510° С. Плотность 2,505 г/см3. С° = 49,04 Дж/(моль-К); ДЯ„°Л = 29,4 кДж/моль (1131 К), АН,"П = 171 кДж/моль (1780 К), ДЯ°р =-565,1 кДж/моль, AG^p =-536,4 кДж/моль; = 66,52 Дж/(моль-К).
Уравнения температурной зависимости давления пара (в Па): Igp = 23,123—11 990/7—3,5251gT(1262—1447 К), Igp = = 10,143—9160/7(1624—1776 К).
Растворимость в воде: 45% (масс.) при 18,6° С, в жидком HF—36,5 г в 100 г при 8° С. Гигроскопичен, образует дигидрат с температурой плавления 42° С и тетрагидрат с температурой плавления 18,7° С. Образует также соединения KFH2O2 и KF-2H2O2 (температура плавления 55° С) и гидрофториды. С фторидами металлов образует комплексные соединения (фторометаллаты), например K2(ZrF6).
Фторид калия встречается в природе в виде редкого минерала ка-роббита.
Фторид калия применяют для синтеза гидрофторидов калия, гексафторофосфата калия [K(PF6)] и фторометаллатов калия. Фторид калия является фторирующим реагентом в органическом синтезе, компонентом кислотоупорных замазок, специальных видов стекла, флинака (эвтектическая смесь KF, NaF и LF).
Фторид калия получают по реакциям:
HF + КОН = KF + Н2О
NH4F + КОН = KF + NH4OH
2HF + К2СО3 = 2KF + Н2О + СО2
Образующиеся растворы фильтруют, упаривают. Из упаренных растворов выделяют кристаллы, которые отжимают на центрифуге, промывают и сушат.
Фторид аммония. Фторид аммония NH4F — бесцветные кристаллы с гексагональной решеткой типа вюрцита (а = 0,4439 нм, с = 0,7165 нм, z = 2, пространственная группа Рбтс). Температура разложения фторида аммония с выделением аммиака и образованием гидрофторида аммония равна 167° С:
2NH4F = NH3 + NH4HF2
Плотность фторида аммония 1,002 г/см3; Ср°= 65,27 Дж/(моль-К), = -465,9 кДж/моль; 52°98 = 71,96 Дж/(моль-К). Растворимость в
223
воде 45,38% при 25° С. При температуре 27,4° С образует ЫНдР-ЬЬО, а ниже 0° С — твердые растворы со льдом; плохо растворяется в органических растворителях, очень хорошо — в жидком фториде водорода. С NH4HF2 образует эвтектику (температура плавления 109,3°С), с многими элементами, оксидами и солями —фторо- или оксофторометаллаты аммония.
Гидродифторид аммония NH4HF2 — бесцветные слабо гигроскопические кристаллы с ромбической решеткой (а = 0,840 нм, b = 0,816 нм, с - 0,367 нм, z = 4, пространственная группа Ртап)\ температура плавления 126,45° С, температура кипения -238° С (с разложением); плотность 1,505 г/моль (20° С); С° = 106,6 Дж/(моль-К); АЯП°Л = 19,1 кДж/моль; Д//°сп = —188 кДж/моль; АЯ0°бр = -804 кДж/моль; = = 115,51 Дж/(моль-К).
Гидрофторид аммония растворяется в воде (41,5% при 25° С) и жидком фториде водорода, а в этаноле и ацетоне не растворяется.
Фторид и гидрофторид аммония являются фторирующими агентами в производстве LiF, BeF2, A1F3 и других фторидов, компонентами растворов для очистки котлов и труб от накипи, флюоритовых концентратов— от кварца и кальцита, составов для травления полупроводников, стекла и некоторых металлов, в процессе получения HF через NaHF2, для гидротермального выращивания монокристаллов кварца и аметиста.
Известны также три гидротетрафторид аммония NH4H3F4 (температура плавления 23,4° С) и пентагидрогексофторид NH4H5F6 (температура плавления 7,8° С), разлагающиеся выше 20° С до NH4HF2 и НЕ
Способы получения. Фториды аммония получают из выхлопных газов или из продуктов улавливания гексафторосиликата или фторида аммония, легко перерабатываемых в другие соединения фтора. Растворы фторида аммония могут быть использованы в процессах получения практически всех солей фтора.
При нейтрализации аммиаком или его водными растворами отходящих фторидных газов протекают следующие реакции:
HF + NH3 = NH4F
(NH4)2SiF6 + 4NH3 + (n + 2)H2O = 6NH4F + SiO2HH2O
2NH4F = NH4FHF + NH3
4NH4FHF + SiO2 = 4NH4F + SiF4 + 2H2O
2NH4F + SiF4 = (NH4)2SiF6
224
Для получения фторосиликата аммония растворы нейтрализуют аммиаком до pH 2^-3, упаривают до 28—30%, кристаллизуют. Кристаллы отделяют на центрифугах и сушат.
Кристаллический фторид аммония получают из его водных растворов путем их упарки до насыщенного состояния и кристаллизации. Кристаллы отделяют от растворов и сушат при 35—40° С. Маточный раствор возвращают на процесс упарки или на начало процесса.
В качестве технического продукта производят не фторид аммония, а его смеси с гидрофторидом аммония NH4HF2. В процессе упарки раствора NH4F без возврата аммиака получается плав, состоящий из смеси NH4HF2 (70—75%) и NH4F (16—20%), который в процессе охлаждения на холодильных вальцах превращается в твердый чешуйчатый продукт — гидрофторид аммония.
Фторид кальция. Фторид кальция CaF2 — бесцветные кристаллы. До 1151° С существует a-CaF2 с кубической решеткой (а - 0,54626 нм, z = 4, пространственная группа Fm3m\ а выше 1151° С — разупорядо-ченная модификация тетрагональной сингонии; температура плавления 1318° С, температура кипения около 2530° С, плотность 3,181 г/см3; С ° = 67,03 Дж/(моль-К); ДЯП°Л = 30 кДж/моль (1691 К), ДЯи°сп = 305 кДж/моль (2800 К), ДЯв°озг = 436 кДж/моль (ОК), ДЯо°бр = -1221 кДж/моль, ДС0°бр =-1168 кДж/моль; S2°98 = 68,45 Дж/(моль-К). Растворимость в воде 16 мг/л при 18° С. Выше 800° С взаимодействует с парами воды с образованием фторида водорода; пирогидролиз наиболее эффективен в расплаве фторида кальция в смеси с диоксидом кремния. В процессе нагревания взаимодействует с неорганическими кислотами. Реакцию с концентрированной серной кислотой при 130—200° С используют для промышленного получения фторида водорода.
Фторид кальция встречается в природе в виде минерала флюорита (плавиковый шпат). Фторид кальция является компонентом металлургических флюсов, специальных стекол, эмалей, керамики, оптический и лазерный материал. Флюоритовые концентраты являются основным сырьем для получения фторида водорода.
Способы получения. Синтетический фторид кальция отличается от природного необогащенного плавикового шпата тонкодисперсной структурой. В процессе взаимодействия HF, SiF4 с СаСОз или СаО в первый период реакции образуется гексафторосиликат кальция, а далее — фторид кальция по схеме
2HF + SiF4 + СаСОз — CaSiF6 + Н2О + СО2
CaSiF6 + 2СаСО3 = 3CaF2 + SiO2 + 2СО2
2HF + SiF4 + ЗСаСОз = 3CaF2 + SiO2 + ЗСО2 + Н2О
8 Химическая технология неорганических веществ, кн. 2
225
Процесс проводят при температуре 70—80° С в течение 2—3 ч с постоянным перемешиванием реакционной массы. В результате реакции образуется суспензия с массовым отношением Т:Ж, равным 1:4. Осадок фторида кальция получается тонкодисперсным и трудно отстаивающимся. Фильтруемость осадка зависит от соотношения HF и SiF4 и качества используемого карбоната кальция, который применяют тонко измельченным. Чем больше HF в растворе, в процессе нейтрализации следует использовать молотый известняк, поскольку применение мела в этих условиях приводит к образованию трудно фильтрующегося осадка. Чем больше в смеси SiF4, тем легче фильтруется полученный осадок. Лишь при отношении HF:SiF4, меньшем 1:12, применяют мел, позволяющий достичь полной нейтрализации кислоты.
При непосредственном поглощении выбросов, содержащих фторидные газы, водной суспензией мела возможно нейтрализовать практически весь карбонат, поддерживая в конце процесса в суспензии концентрацию СаСО3 -0,5 г/л для предотвращения проскока фторсодержащих газов.
Осажденный фторид кальция отжимают на центрифуге отстойного типа. В отфугованной массе остается 60—65% влаги. Суспензию фторида кальция сушат с получением тонкодисперсного продукта с величиной частиц -5 микрон. Сухой продукт содержит -63% CaF2, 24—30% SiO2, 10% СаСО3 и 3% прочих примесей. При использовании в качестве сырья свежеосажденного оксида кальция (извести) выход CaF2 составляет по фтору 85—92%, по оксиду кальция 74—87%.
Фторид кальция с содержанием основного вещества 94% получают путем термической диссоциации CaSiF6 при 380—400° С:
CaSiF6—>CaF2 + SiF4
Разработан способ получения фторида кальция поглощением фторидсодержащих газов растворами фторидов щелочных металлов с последующей конверсией образовавшихся кремнефторидов гидроксидом натрия или калия и каустификацией раствора фторида гидроксидом кальция:
SiF4 + 2MF = M2SiF6
M2SiF6 + 4МОН = 6MF + Si(OH)4
4MF + 2Ca(OH)2 = 2CaF2 + 4MOH
В этом процессе гидроксид не расходуется, кроме технологических потерь, так как часть получаемого во второй стадии раствора фторида возвращают на абсорбцию, а часть направляют на каустификацию. Процесс каустификации проходит с большой скоростью и 226
практически заканчивается в течение одного часа. Конверсия фторида кремния также проходит с достаточной скоростью и полнотой. Получаемые шламы хорошо отстаиваются.
Разработан также способ улавливания HF известняком из газов, выделяющихся в производстве обесфторенного плавленого фосфата. При этом выходящие из печи газы очищают от пыли. Газы с температурой 100—450° С пропускают через башню со слоем известняка 2,5—3,0 м (размер кусков 6—40 мм). При концентрации HF, соответствующей 0,15—6 г/м3 фтора, степень поглощения составляет 95—96%. Периодически часть известняка (~40%) заменяют свежим. Фторид кальция накапливается в мелочи, содержащей 80—95% CaF2.
Фторид алюмшния и криолит. Фторид алюминия AIF3 — бесцветные кристаллы. В обычных условиях устойчива a-модификация с тригональной решеткой (а = 0,503 нм, а = 58,50° , z = 2, пространственная группа 702); плотность 1,882 г/см3. Известна также устойчивая до 710—720° С у-модификация с тетрагональной решеткой (а = 0,354 нм, с = 0,600 нм). Температура возгонки 1270° С; Ср° = 75,10 Дж/(моль-К); АЯв°озг = 272 кДж/моль, А770°бр = -1510 кДж/моль; 5298 = 66,48 Дж/(моль-К); уравнения температурной зависимости давления пара:
Igp (Па) = 16,565—16 967/Г (980<Г<1123 К),
Igp (Па) = 14,719—14 974/Г (1027<Г<1184 К).
Фторид алюминия плохо растворяется в воде (0,41% (масс.) при 25° С), лучше растворяется в водных растворах фторида водорода, не растворяется в органических растворителях. Образует кристаллогидраты с 1, 3 и 9 молекулами воды. Известны также кристаллогидраты: AlFr3,5H2O и AIF30,5H2O.
В процессе нагревания фторид алюминия гидролизуется парами воды, медленно взаимодействует с концентрированной серной кислотой и расплавами щелочей.
Фторид алюминия применяется в качестве электролита (5—7%) в производстве алюминия. Он входит в состав флюсов, эмалей, стекол, глазурей, керамики, покрытий сварочных электродов. Является катализатором в органическом синтезе. Обратимую реакцию AIF3 с AI используют для очистки металла.
Гексафтороалюминат натрия Na3AlF6 — бесцветные кристаллы; температура плавления 1011° С (с разложением); плотность низкотемпературной a-модификации (криолита) 2,97 г/см3; С°=216 Дж/(моль-К); АНо°бр =-3312 кДж/моль, AG0°6p = -3148 кДж/моль; 5298 = 238 Дж/(моль-К).
227
8*
Гексафтороалюминат натрия плохо растворяется в воде (0,42% (масс.) при 25° С) и органических растворителях. Взаимодействует с концентрированной серной кислотой, при нагревании гидролизуется парами воды. Расплав Na3AlF6 электропроводен и является хорошим растворителем для сесквиоксида алюминия и других оксидов.
В системе NaF—AIF3—Н2О при 25° С могут существовать две твердые фазы — при концентрации NaF меньше 1,4% конгруэнтно растворяющаяся NaMAI4F23 или 3Na3AlF6-2NaF-AlF3, а при концентрации NaF больше 1,4% инконгруэнтно растворяющийся криолит Na3AlF6. Кроме криолита, фторида натрия и алюминия образует хио-лит 5NaF-3AlF3.
Криолит — редкий минерал в природе. Он применяется в качестве основного компонента (80—85%) электролита в производстве алюминия. Он является металлургическим флюсом, компонентом стекол и эмалей, наполнителем резин.
Промежуточным продуктом (полуфабрикатом) в производстве AIF3 является гексафтороалюминат аммония (NH4)3A1F6— бесцветные кристаллы; плотность 1,837 г/см3 (20° С); А/70°бр =-3060 кДж/моль. При нагревании разлагается последовательно до NH4AIF4 и AIF3. Растворяется в воде (7,66% (масс.) при 25° С), не растворяется в органических растворителях. Получают взаимодействием А1(ОН)3 и А12Оз с NH4F или NH4HF2.
Согласно существующему ГОСТ 19181—78, фторид алюминия производят высшего, первого и второго сортов. ГОСТ регламентирует содержание в целевом продукте соответственно по сортам: основного вещества не менее 93,88—98%; не более 4,7 и 7% AI2O3; 0,3, 0,4 и 0,5% суммы оксидов (SiO? и РегОз); 0,05, 0,1 и 1% Р2О5; 0,5, 0,7 и 1% SO;“.
Продукт высшего сорта получают взаимодействием AI2O3 или А1(ОН)з с раствором HF:
А120з + 6HF + mH20 = 2AlF37nH2O + ЗН2О
А1(ОН)з + 3HF + mH20 = A1F3wH2O + ЗН2О
Образующуюся суспензию отделяют от воды, сушат и продукт подвергают термообработке при 500—600° С до AIF3.
Фторид алюминия получают также действием А12Оз газообразного фторида водорода при 450—600° С по схеме
А12Оз + 6HF = 2AIF3 + ЗН2О
228
По разработанному в УНИХИМе способу фторид алюминия получают путем перемешивания при 85° С смеси гидроксида алюминия с HF и SiF4 по реакции
2А1(ОН)3 + 2HF + SiF4 = 2A1F3 + SiO2-2H2O + 2Н2О
Раствор отделяют от кремнегеля фильтрованием. Отфильтрованный раствор содержит 8—14% A1F3, который охлаждают с целью кристаллизации A1F3-3H2O. Осадок тригидрата отделяют и, нагревая его до 500—700° С, получают безводный фторид алюминия.
Фторид алюминия получают также по следующим уравнениям:
А1(ОН)3 + 6NH4F = (NH4)3A1F6 + 3NH3 + Н2О
А1(ОН)3 + 3NH4HF2 = (NH4)3A1F6 + ЗН2О
Образующийся гексафтороалюминат аммония разлагают путем термообработки в температурном интервале 450—550° С по уравнению
(NH4)3AIF6 -> A1F3 + 3HF + 3NH3
Выделившиеся газы возвращаются на начало процесса для получения NH4F по схеме
NH3 + HF = NH4F
Гексафторосиликат натрия получают осаждением по реакции
2HF + SiF4 + 2NaCl = Na2SiF6 + 2HCI
Как видно из приведенных на рис. 4.13 данных, присутствие в растворе избытка хлорида натрия значительно снижает растворимость Na2SiF6. Так, при 25° С она равна 0,78%. Введение в раствор 2% NaCl снижает содержание Na2SiF6 до 0,1%, а при 10% NaCl и больше в растворе остаются лишь сотые доли процента Na2SiF6. Поэтому хлорид натрия вводят с расчетом, чтобы в образующемся маточном растворе после осаждения целевого продукта было около 2% NaCL
Образующиеся кристаллы целевого продукта отделяют от маточного раствора и геля диоксида кремния в гидроциклонах или отстаиванием.
Сгущенную суспензию после декантации раствора и кремнегеля обрабатывают раствором карбоната натрия для нейтрализации хлороводородной кислоты. Избыток карбоната натрия не допускают, поско-229
Рис. 4.13. Изотермы растворимости в системе Na2SiF6—NaCl—Н2О при 25 (/) и 50° С (2)
льку это ведет к потере целевого продукта вследствие перехода его в более растворимый фторид натрия.
Кристаллы целевого продукта отжимают и промывают в центрифуге до остаточного содержания NaCl и НС1 в продукте не более соответственно 0,2 и 0,02%. Кристаллы после центрифуги содержат 10—13% Н2О, их сушат при 400° С во вращающихся барабанных сушилках до влажности менее 1%. Образующиеся в процессе сушки комки продукта разламывают в вакуумных мельницах ударно-центробежного типа. Измельченный продукт отсасывается из кожухов мельниц, осаждается и собирается в циклонах.
Отходом производства гексафторосиликата натрия является слабый раствор хлороводородной кислоты, содержащий 3—4% HCI, 2% NaCl и 0,15% Na2SiF6.
Синтетический гексафтороалюминат натрия (криолит) получают действием карбоната натрия или фторида натрия на раствор А1(ОН)з или А120з в HF:
2А1(ОН)з + 3Na2CO3 + 12HF = 2Na3AlF6 + 6Н2О + ЗСО2
А12О3 + 3Na2CO3 + I2HF = 2Na3AlF6 + 6Н2О + ЗСО2
а также взаимодействием водных растворов AIF3 и NaF по схеме
AIF3 + 3NaF = Na3AlF6
Во всех способах образующуюся суспензию отделяют от воды, полученный пастообразный продукт промывают, сушат и при необходимости просеивают.
ГЛАВА 5
ХЛОР И ЕГО СОЕДИНЕНИЯ
5.1. ХЛОР
Среднее содержание хлора в земной коре составляет 1,7-10‘2% (масс.). Большие запасы хлора в мировом океане (среднее содержание 18,83 г/л), а в подземных рассолах он находится в виде хлорида натрия (50—240 г/л). В земной коре хлор встречается в виде галита NaCl, карналлита KClMgCl2-6H2O, сильвина КС1, сильвинита NaCI KCl, каинита KCl-MgSO4-3H2O, бишофита MgCl2-6H2O, тахгид-рида 2MgCl2 CaCl212H2O. Менее, распространенными минералами являются: кераргирит AgCl, бисмоцелит BiOCi, псевдокотуннит К2РЬС14, баумлерит 2КС1СаС12. Хлор содержится также в силикатных метеоритах (0,09%), в железных (0,36% в виде трихлорида железа), в вулканических газах (1,3% в виде элементного хлора, хлоридов водорода и натрия и др.). Содержание хлора в человеческом теле составляет 0,25% (0,45% от сухой массы); в плазме крови (0,32—0,37%), а в растениях (в зависимости от состава почвы): в табаке — 2,3%, в моркови -1,5%, в зерновых — 0,05%, в картофеле — 0,03%.
Природный хлор состоит из смеси двух изотопов 35С1 (75,77%) и 37С1 (24,23%); ядра обоих изотопов имеют электрический квадрупо-льный момент.
Конфигурация внешней электронной оболочки атома хлора 3s23p5; степени окисления -1 (хлориды), +1 (гипохлориты), +3 (хлориты), +5 (хлораты) и +7 (перхлораты); потенциал ионизации при последовательном переходе от С1° к С1+7 12,96776, 23,805, 39,90, 53,50, 67,80, 96,7 и 114,27 эВ; электроотрицательность по Полингу 3,16; ковалентный радиус С1° 0,099 нм; ионные радиусы (в скобках указаны координационные числа) СГ 0,167 нм (6), С15+ 0,026 нм (3), С17+ 0,022 нм (4), 0,041 нм (6).
Молекула хлора двухатомна, длина связи (в газе) т = 0,1987 нм. Сродство молекулы хлора к электрону 2,45 эВ, потенциал ионизации 11,48 эВ. Энергия диссоциации С12 239,240 кДж/моль, равновесная термическая диссоциация на атомы определяются константой А°(Г) = = р2(С1)//?(С1)?), где р — давление; IgA0 = -6,8257(1000 К), 0,2660 (2000 К), 1,9617 (3000 К).
231
Физико-химические свойства хлора. Хлор — желто-зеленый газ с резким удушающим запахом, с температурой плавления 100,98° С. Хлор кипит при температуре -33,97° С. Критические константы: температура 143,75° С, давление 7977,3 кПа, плотность 0,573 г/см3. Уравнение температурной зависимости плотности в интервале от -90 до 80° С d = 1,6583346—0,002003753/(/ + 80) + 0,0545596743(/ + 80)2 г/см3, при 25° С плотность 3,214 г/см3, плотность твердого при -195° С 2,13 г/см3. На рис. 5.1 приведены данные по изменению плотности в зависимости от температуры. Для молекулы Ch’ С* = 33,949 Дж/(моль-К), ДЯПЛ = 6,757 кДж/моль, АЯИСП = 22,43 кДж/моль, 52°98 = 222,965 Дж/(моль-К); для атома С1 (газ) С ° =21,838 Дж/(моль-К), ДЯ0°бр = 121,302 кДж/моль, S298 = 165,076 Дж/(моль-К); для иона СГ (газ): С ° = 20,786 Дж/(моль-К), 5298 = 153,346 Дж/(моль-К), ДЯ0°бр = -233,670 кДж/моль, ДЯ0°бр СГ (в воде) -167,080 кДж/моль. Зависимость давления паров жидкого хлора от температуры приведена на рис. 5.2, уравнение температурной зависимости давления пара In р = А + BIT + С1пГ + DT + + E(F— T)/FT(205—417 К), где А = 62,402508, В = -4343,5240, С = = -7,8661534, D = 1,0666308-10"2, Е = 95,248723, F = 424,90; при 20° С давление пара Ch 0,669 МПа; т| = 4,88-1 О'4 Па-с, уравнение температурной зависимости т| = [0,00585(1 + 0,05878/—0,05392/2)]-105 Па-с (от -34 до -77° С); у-10’5Н/см 31,61 (-61,3° С), 28,38 (-44,5° С), 25,23 (-28,7° С), уравнение температурной зависимости у = [21,70(1—0,007742/)]-105
-50-40-30-20-10 О 10 20 30 40 50 60 70 80 90 100 Температура, °C
Рис. 5.1. Плотность жидкого хлора при различных температурах
232
Н/см; 8 = 1,00152 (25° С); 2,147 (-65,15° С), 2,088 (-45,25° С), 2,051 (-22,0° С), 1,968 (0°С), 1,54 (142° С). Электродный потенциал С12 (газ 0,1 МПа); теплопроводность 0,079 Вт/(м-К) при 273 К. Хлор кристаллизуется при -160° С в ромбической решетке, а = 0,624 нм, b = 0,448 нм, с = 0,826 нм, z = 4, пространственная группа Стеа, т = 0,1980 нм.
Хлор хорошо растворяется в неполярных жидкостях, а в воде— слабее. Растворимость,% (масс.): в тетрахлориде углерода— 16,4 (0°С), 8,46 (25° С), бензоле —24,7 (10° С), 18,5 (20° С), 14,7 (30° С), воде—1,44 (0°С), 1,07 (6° С), 0,828 (15° С), 0,711 (20° С), 0,449 (40° С), 0,329 (60° С). Как видно из данных рис. 5.3, растворимость хлора в концентрированных растворах хлорида натрия в несколько раз ниже, чем в воде. В водном растворе хлора устанавливается равновесие:
С12 + Н2О т» НС1О + СГ + Н+
Из водного раствора кристаллизуется клатрат С12-6Н2О, давление диссоциации которого равно 0,1 МПа при 9,6° С. С ионом С1“ молекулы С12 образуют в водном растворе ионы CI3 по следующей реакции:
С12 + СГ<±С1" (/<=0,19).
Жидкий хлор является хорошим растворителем хлоридов, например: ВС13—65,5%(-136,4° С), SiCl4 28,8% (0° С), TiCl4 74,9% (20° С) % (масс.).
Хлор является одним из наиболее химически активных элементов таблицы Менделеева. Он непосредственно взаимодействует со всеми
Рис. 5.2. Зависимость давления паров жидкого хлора от температуры
Рис. 5.3. Растворимость хлора в воде и в растворах хлорида натрия при различных температурах
233
металлами и большинством неметаллов, образуя при этом соответствующие хлориды. Лишь в реакциях хлора с кислородом, азотом и ксеноном требуются индивидуальные методы их активации путем ультрафиолетового облучения или электроразрядом. Процесс хлорирования серы и фтора проводят при комнатной температуре, кремния при 200° С, железа 215° С, меди и алюминия при 240° С, титана -250° С, серебра 260° С, тантала -380° С, молибдена -420° С, а остальные металлы (Ni, Mg, Pt, W и Cr) в пределах 520—680° С. Реакционная способность оксидов металлов по отношению к хлору значительно ниже, чем у соответствующих металлов, и убывает в ряду: Na2O, Ag2O, СаО, PbO, CdO, МпО, NiO, ZnO, FeO, MgO, Ре2Оз, ZnO2, TiO2, А120з и SiO2. В присутствии углерода температура процесса хлорирования оксидов снижается.
Высокая химическая активность объясняется легкостью образования атомов хлора из его молекул, а также высоким сродством атома хлора к электрону и высокой энергией связи хлора с большинством элементов. Стабильные соединения хлора — хлориды, гипохлориты, хлориты, хлораты, перхлораты. Установлено, что действительный заряд на атоме хлора во всех соединениях этих классов по абсолютной величине значительно ниже формального. Вследствие высокого сродства атома хлора и хлоркислородных радикалов к электрону хлор бывает анионом, входит в состав аниона (СЮ', СЮ;, СЮ^ и СЮ4) или является лигандом в комплексных анионах ВС1;, TiCl^- и т.п.
Применение хлора. Мировое производство хлора в настоящее время составляет около 30 млн. т в год. Применяется он почти во всех отраслях народного хозяйства.
Потребителями хлора в химической промышленности являются производство окислительно-отбеливающих веществ (гипохлориты натрия, лития, кальция, хлорная известь, жидкий хлор), синтетической хлороводородной кислоты, хлоридов многих элементов (Al, Fe, Zn, Ti, Si, AS, P и др.). Большие количества хлора расходуются на производство хлорорганических продуктов: поливинилхлорида, хлорного жаучука, растворителей, продуктов хлорирования метана, углеводородов парафинового и ароматического рядов, хлорсодержащих средств защиты растений от вредителей сельского хозяйства и болезней. Значительное количество хлора расходуется на производство этиленгликоля, глицерина, сульфанола и других продуктов.
Доля хлора, расходуемого на различные нужды, в большинстве стран колеблется в пределах (%):
Производство химических продуктов: органических............................... 50—85
неорганических........................... 10—15
Целлюлозно-бумажная промышленность........ 2—15
Очистка воды и санитарные нужды......... 2—10
Прочие.................................... 5—15
234
Кроме приведенных данных, хлор применяется в производстве искусственных волокон, глинозема, мыла и других областях народного хозяйства.
СПОСОБЫ ПОЛУЧЕНИЯ ХЛОРА И ГИДРОКСИДА НАТРИЯ
В мировой практике производства хлора и гидроксида натрия существуют три способа их получения: 1) способ с твердым катодом и фильтрующей диафрагмой; 2) метод с ионообменной мембраной; 3) способ с ртутным катодом.
Физико-химические свойства гидроксида натрия. Гидроксид натрия NaOH — бесцветные кристаллы. До 299° С устойчива ромбическая модификация (а = 0,33994 нм, с = 1,1377 нм), а выше 299° С — моноклинная; ДЯ полимерного перехода 5,85 кДж/моль. Температура плавления 323° С, а кипения 1403° С. Плотность 2,02 г/см3; С° = 59,54 Дж/(мольК); АЯ^р = -425,88 кДж/моль, АЯ°ЗГ = = 239,335 кДж/моль; Д//”л = 7,8 кДж/моль; S^g = 64,43 Дж/(мольК). Показатели преломления: меньший Np = 1,457, средний Nm = 1,470, больший Nf, = 1,472.
Гидроксид натрия растворяется в метаноле -23,6%, масс. (28° С), этаноле -14,7%, масс. (28° С). Данные по растворимости NaOH в воде -52,2% масс. (20° С). ДЯ растворения для бесконечно разбавленного водного раствора -44,45 кДж/моль. В жидком аммиаке практически не растворяется. На рис. 5.4 приводится номограмма для определения плотности и концентрации водных растворов гидроксида натрия. Из водных растворов (рис. 5.5) при 12,3—61,8° С кристаллизуется моногидрат (сингония ромбическая; температура плавления 65,1° С; плотность 1,829 г/см3; ДЯ0°бр = 734,96 кДж/моль), а в интервале от -28 до -24° С — гептагидрат, от -24 до -17,7° С — пентагидрат, от -17,7 до -5,4° С — тетрагидрат (a-модификация), а при -5,4—12,3° С—NaOH-3,5H2O, температура плавления 15,5° С). Известен также метастабильный P-NaOH-4H2O. При дальнейшем нагревании выше 61,8° С кристаллизуется безводный гидроксид натрия. На рис. 5.6 приведены температуры кипения водных растворов гидроксида натрия и чистой воды при одинаковом давлении их насыщенных паров (на кривых показано количество NaOH, % (масс.) в растворах, приходящееся на 1% (масс.) воды).
Гидроксид натрия — сильное основание. Со спиртами образует алкоголяты. Расплавленный гидроксид натрия растворяет металличе-235
t, °C
70
60
50
40
30
20
10
0 -J
Рис. 5.4. Номограмма для определения давления водяных паров над водными растворами гидроксида натрия
Рис. 5.5. Температура кристаллизации водных растворов гидроксида натрия
ский натрий и гидроксид натрия. Разрушает материалы органического происхождения, например бумагу, кожу и др.
Гидроксид натрия выпускается промышленностью в виде водных растворов марки I (45% NaOH) и марки II (42% NaOH). Производится также в твердом виде с 96%-ным содержанием NaOH.
Гидроксид натрия применяют для очистки нефти и масел; в производстве бумаги, мыла, искусственных волокон; в качестве осушающего агента газов и органических жидкостей. Водные растворы являются электролитами в воздушно-цинковых элементах.
Способ получения хлора и гидроксида натрия с твердым катодом и фильтрующей диафрагмой (рис. 5.7). Согласно схеме, в электролизер, в анодную его часть, вводят с расчетной скоростью раствор хлорида натрия. Раствор фильтруется через диафрагму 3 в катодное пространство, откуда и выводится. При этом на аноде 2 протекает основная реакция окисления ионов хлора:
СГ-е -> 0,5С12 (1)
а также побочные реакции, главная из которых разряд молекул воды:
Н2О-2е -> 0,5О2 + 2Н+ (2)
На твердом электроде образуется молекулярный кислород, а на графитовом за счет окисления углерода получается диоксид углерода:
2Н2О + С-4е -» СО2 + 4Н+ (3)
237
Рис. 5.6. Температура кипения водных растворов гидроксида натрия и чистой воды при одинаковом давлении их насыщенных паров
Рис 5.7. Схема диафрагменного электролизера для получения хлора и гидроксида натрия:
./ — катод, 2 — анод, 3 — диафрагма
Образующиеся на аноде газы выводятся из анодного пространства, регенерируются гидроксид-ионы, которые с ионами натрия образуют гидроксид натрия:
Н2О-е 0,5Н2 + ОН’ (4)
Na+ + ОН" NaOH (5)
Гидроксид-ионы, несущие отрицательные заряды, под действием градиента электрического потенциала стремятся к аноду и, проникая в анодную зону, повышают pH анолита, способствуя ускорению побочных реакций и реакций хлора с раствором анолита.
Для уменьшения проникновения гидроксид-ионов в анодное пространство и подавления побочных реакций на аноде и в анолите применяют фильтрацию раствора через диафрагму от анолита к катоду.
Плотность тока гидроксид-ионов в диафрагме зависит от градиента электрического потенциала и концентрации гидроксид-ионов в ней и от скорости противотока анолита, т.е. от толщины диафрагмы и концентрации гидроксида натрия в католите. 238
В анолите электролизера растворяется хлор, и при этом может протекать ряд реакций в объеме:
С12 + 2ОН" СЮ" + СГ + Н2О (6)
НСЮ + ОН СЮ" + Н2О (7)
СЮ + 2НСЮ #СЮ; + 2Н+ + 20" (8)
Реакции (6) — (8) протекают с заметной скоростью в условиях диафрагменного электролиза, т.е. при температуре анолита 90—100° С, концентрации хлорида натрия выше 4-103моль/м3 при pH анолита более 4.
При нейтрализации гидроксид-ионов в диафрагме вблизи анодной стороны при минимальной толщине диафрагмы ее диффузионное сопротивление будет полностью использовано для уменьшения потока гидроксид-ионов. Если толщина реальной диафрагмы меньше, чем для условий электролиза, то будет снижаться выход по току хлора и гидроксида натрия, снижение концентрации хлора в анодном газе и загрязнение раствора католита хлоридными соединениями. При достаточной толщине диафрагмы повышение температуры должно приводить к повышению выхода по току хлора и гидроксида натрия, а снижение концентрации питающего раствора хлора, к уменьшению выхода по току за счет повышения растворимости хлора в анолите.
Из схемы, поясняющей степень влияния толщины диафрагмы на показатели процесса электролиза, видно, что до достижения предельной концентрации гидроксида натрия и толщины диафрагмы гидроксид-ионы нейтрализуются внутри диафрагмы на некоторой небольшой толщине — в зоне нейтрализации с образованием воды и хлороксидных продуктов, уносимых потоком раствора в католит (рис. 5.8). Основная же часть ионов гидроксида натрия нейтрализуется ионами водорода, образовавшимися в результате разряда молекул воды на аноде.
В процессе уменьшения притока раствора через диафрагму возрастает концентрация гидроксида в католите, например от скат до с'кат-При этом зона нейтрализации смещается в сторону анолита на величину Дх (рис. 5.8, а), возрастает толщина диффузионной щелочной зоны и величина потока гидроксид-ионов сравняется с потоком нейтрализующих компонентов в анолите, который фильтруется навстречу гидроксид-ионам. Выход по току гидроксида натрия незначительно возрастает за счет уменьшения скорости противотока.
Если толщина диафрагмы для данных условий электролиза отвечает ее минимальному значению, то в процессе увеличения концент-239
б
Рис. 5.8. Схема, поясняющая влияние толщины диафрагмы на показатели электролиза:
а — зона нейтрализации внутри диафрагмы, б—нейтрализация гидроксида натрия происходит в аноде
рации гидроксида натрия от скет до с'кат возрастает градиент концентрации и перенос гидроксид-ионов в анодное пространство, в котором пройдут реакции образования хлороксидных продуктов и соответствующее снижение анодного выхода по току.
Известно, что образование хлорноватистой кислоты и гипохло-рит-ионов в диафрагменных процессах может происходить в пространственно разделенных зонах. Так, при рН<4 гипохлорит-ионы практически не образуются и между хлором и гидроксид-ионом протекает реакция
С12 + ОН’ НС1О + СГ (9)
При более высоких значениях pH протекает реакция по уравнению (6) и образуются гипохлорит-ионы, а также реакция (8), приводящая к образованию хлорат-ионов. Поэтому считается целесообразным снижать значение pH питающего рассола введением в него хлорида водорода, заменяя реакции (6), (7) и (9) реакцией ионов водорода и гидроксид-ионов с образованием воды. Это способствует повышению качества целевых продуктов. Нейтрализация в анолите гидроксид-ионов хлоридов водорода исключает образование хлорноватистой кислоты и нейтрализацию вновь образующихся гидроксид-ионов в диафрагме или в католите, повышая тем самым катодный выход по току гидроксида и анодный выход по току хлора.
Значительное влияние на выход по току в анодном процессе может оказать материал электрода и условия электролиза — плотность 240
тока, концентрация хлорид-иона в анолите и pH, а также состав анодного газа и ток, расходуемый на выделение кислорода. В электролизерах с фильтрующей диафрагмой применяют графитовые или титановые с электрокаталитическим покрытием аноды. Их готовят из искусственного графита. Для этого из смеси нефтяного кокса, антрацита и каменноугольной смолы сначала спрессовывают аноды нужной формы (обычно в виде прямоугольных плит), обжигают при 1000—1200° С, а затем после пропитки маслопеском проводят графи-тацию при температурном интервале 2500—2700° С, переводя уголь в графит.
Титановые аноды готовят следующим образом: после предварительной подготовки титана наносят на его поверхность растворы, содержащие соединения рутения и титана, например Ru(OH)CI3 или TiCl4. После высушивания раствора на поверхности титана аноды нагревают при температуре 400—470° С. При этом поверхность покрывается твердым раствором оксидов титана и рутения по следующей суммарной реакции:
Ru(OH)Cl3 + TiCl4 + ЗН2О -> RuO2TiO2 + 7НС1
На рис. 5.9 и 5.10 представлены поляризационные кривые и выход по току кислорода на анодах из разных материалов, характеризующих зависимость поляризации и состав газа от материала электрода и состава раствора. Из рис. 5.10 видно, что на ход процесса особенно большое влияние имеет pH раствора. Так, при pH раствора хлорида натрия >3 увеличивается потенциал ПТА (платиновое гальваническое покрытие) из-за изменения состава оксидов на поверхно-
Рис. 5.9. Зависимость потенциала анодов в растворе хлорида натрия с концентрацией 300 г/л. а при 90° С от плотности тока:
/ ОРТА: 2 - платинированный титан при pH 3. 3—графит и платинированный титан при рН>3
Рис. 5.10. Выход по току кислорода в зависимости от pH раствора NaCl при плотности тока 2 кЛ/м2 и температуре 80° С:
/ — ОРТА (оксиды рутения и титана), 2 — платинированный титан
241
Рис. 5.11. Влияние кислотности раствора хлорида натрия на скорость разрушения графитового анода:
/ — новый графитовый анод; 2 — бывший в работе графитовый анод
сти анода. С повышением pH раствора возрастает скорость побочных процессов на катоде. Существенно зависит от pH раствора скорость разряда воды на анодах из графита и соответственно разрушение анодов [см. реакцию (3)], приводящее также к механическому выкрашиванию частиц графита (рис. 5.11).
Технологическая схема получения хлора, гидроксида натрия и водорода диафрагменным способом (рис. 5.12) предусматривает стадии растворения исходного хлорида натрия 7, содово-каустическую очистку полученных растворов 2, нейтрализацию и фильтрацию полученного рассола 3 и электролиз растворов 4. Полученные растворы гидроксида натрия упаривают
5, а хлор охлаждают 8, сушат 9 и сжижают 70. Водород охлаждают 72 и направляют потребителям.
Исходный раствор хлорида натрия должен содержать > 310 г/л NaCl, <51О"3 г/л ионов кальция, <1-10‘ 3 г/л ионов магния, <5 г/л ионов сульфата, 0,05—0,1 г/л NaOH и 0,3—0,4 г/л Na2CO3. Кроме того, рассол для диафрагменного электролиза должен иметь прозрачность 98% и содержание иойов аммония не более 10 мг/л.
С целью получения растворов хлорида натрия, отвечающих необходимым требованиям, исходные рассолы подвергают очистке по определенной схеме (рис. 5.13).
Согласно схеме очистки, исходный сырой рассол с концентрацией 310—315 г/л хлорида натрия поступает в сборник 7, затем подогревается в теплообменнике 2 и центробежным насосом 3 через воздухоотделитель в осветлитель 10. Оборотный рассол с концентрацией 305—310 г/л NaCl подают двумя потоками, один из которых проходит через карбонизатор 4 в сборник карбонизированного рассола 5 и далее в воздухоотделитель осветлителя, а второй — непосредственно в осветлитель.
Для очистки исходного рассола от примесей кальция и магния применяют содово-каустический метод, основанный на образовании малорастворимых соединений кальция и магния. В процессе очистки протекают следующие реакции:
СаС12 + Na2CO3 = СаСО3 + 2NaCl
СаЗОд + Na?CO3 = СаСОз + Na2SO4
242
Са(НСО3)2 + 2NaOH = СаСО3 + Na2CO3 + 2Н2О
MgCI2 + 2NaOH = Mg(OH)2 + 2NaCl
MgSO4 + 2NaOH = Mg(OH)2 + Na2SO4
Одновременно осаждается гидроксид железа.
Процесс очистки проводят обычно в осветлителе 10. Возможно провести его и в карбонизаторе оборотного рассола 4.
В осветлитель 10 подают также гидролизованный полиакриламид (ПАА) и флокулянт, улучшающий и ускоряющий процесс осветления очищаемого рассола. Растворы ПАА готовят в аппара-те-гидролизере б, куда вводят товарный продукт (8% гель) для обработки при повышенной температуре электролитическими растворами. В процессе обработки 20—40% амидных групп ПАА замещается на гидроксильные и возрастает при этом коагулирующая способность ПАА. Гидролизованный ПАА разбавляют очищенным рассолом до концентрации 0,2—0,3%.
Обратный рассол
Рис. 5.12. Принципиальная схема производства гидроксида натрия, хлора и водорода:
/ — растворение хлорида натрия; 2 — содово-каустическая очистка рассола; 3— нейтрализация рассола; 4— электролиз; 5 — упарка растворов; 6 — сборники; 7—приготовление обратного рассола; 8 — охлаждение хлора; 9 — осушка хлора; 10 — сжижение хлора; 11— сборники жидкого хлора; 12 — холодильники водорода
243
Щелока Пар
Рис. 5.13. Схема очистки рассола для диафрагменного электролиза:
/ — сборник сырого рассола; 2— подогреватель (теплообменник); 3 — центробежные насосы; 4, 5 — сборник оборотного карбонизованного рассола; 6 — автоклав для гидролиза полиакриламида (ПАА); 7—растворитель гидролнзованного полиакриламида; 8— емкость для раствора полиакриламида; 9—дозатор; /0 — осветлитель с шламовым фильтром; // — сборник осветленного рассола; 12 — насос: 13 — фильтр; 14 — смеситель; 15 — напорная емкость хлороводородной кислоты; 16—нейтрализатор рассола; 17 — напорная емкость; 18— подогреватель (теплообменник) рассола;
19— сборник шлама; 20 — фильтры; 21 — сборник для очистки очищенного рассола
Процесс разбавления ПАА проводят в сборнике 7, из которого раствор передают в напорную емкость S, а оттуда дозатором 9 в осветлитель 10. Осветленный рассол поступает в сборник осветленного рассола 77, откуда центробежным насосом 12 перекачивается на фильтр 73, в смеситель 14 на нейтрализацию избыточной щелочи хлороводородной кислотой, подаваемой из сборника 75, и далее через сборник 16 и емкость 17 и подогреватель 18 — на электролиз.
Отделяемый в осветлителе 10 шлам выводят в сборник 79, откуда подают на фильтры 20. Осадок с фильтров направляют в шламонакопи-тель, а отфильтрованный раствор возвращают на начало процесса.
Процесс электролиза в производстве проводится в диафрагменном электролизере, приведенном на рис. 5.14. Электролизер состоит из корпуса 7, крышки 2, катодного элемента 3, анода 4, коллектора электрощелоков 5 и сливного устройства 6 (рис. 5.15).
Электролизер обеспечивает получение электрощелочи, содержащей 120—140 г/л гидроксида натрия, 170—210 г/л хлорида натрия и до 7 г/л сульфата натрия. При этом содержание в электрощелочи хлората (V) натрия не превышает 0,3 г/л, а гипохлорит и карбонат натрия отсутствуют. 244
Состав анодного газа — хлоргаза, выходящего из электролизера, должен находиться в пределах, % (об.): С12 — 96,5—98,0;
О?—1,0—2,5; Н2— 0,1—0,5; СО2 — 0,1—0,3. Водород, выводимый из электролизера, должен составлять не менее 99,9% (об.), хлор в нем должен отсутствовать.
Рассол подают в электролизер из расчета 1,1-10’3—1,3-10’3 м3/ч на каждые кА нагрузки, что обеспечивает при оптимальной температуре электролита 90—95° С получение электрощелочи с расчетной концентрацией.
Важно поддерживать высокий уровень анолита в электролизере. Уровень анолита должен быть выше верхней части катода с нанесенной диафрагмой не менее чем на 0,3 м (см. рис. 5.14), поскольку при этом обеспечивается равномерная фильтрация через диафрагму. Для изменения протекаемости диафрагмы можно уменьшать уровень анолита, наклоняя вертикальную трубу сливного устройства, которая шарнирно соединена с выводным патрубком электрощелочи (см. рис. 5.15). Уровень анолита контролируют поплавковым уровнемером в крышке электролизера, температуру — по термометру, скорость подачи рассола — ротаметром.
Технология хлора и гидроксида натрия с ионообменной мембраной состоит из стадий подготовки и очистки исходного рассола, электролиза и упарки раствора гидроксида натрия. Этот способ отличается от диафрагменного и ртутного глубокой очисткой рассола от
Рис. 5.14. Диафрагменный электролизер: / — корпус катода; 2 — крышка; 3 — катодный элемент; 4 — анод; 5 — коллектор электроще-локов; б — сливное устройство
Рис. 5.15. Сливное устройство:
/ — стенка катода; 2 — внутренняя труба; 3 — наружная труба; 4 — шарнир; 5 — прерыватель струи (тока)
245
солей тяжелых и щелочноземельных металлов, повышенным подкислением анолита и наличием стадии доупарки гидроксида натрия до отсутствия в нем хлоридов.
На рис. 5.16 приведена примерная схема промышленной установки (фирма Асахи Кемикл), которая состоит из трех отделений: I—рассольного, II—электролиза и III—выпарки.
Исходные растворы хлорида натрия (рассол) поступают в реактор 7, в котором производят глубокую очистку их от.солей жесткости до содержания 0,2 мг/л с помощью фосфат- и фтор-ионов при рН>10. При этом концентрация фосфат-ионов поддерживается на уровне 0,1—1,0% (масс.), фтор-ионов — 0,01—1,00% (масс.). Для улучшения процесса осаждения в качестве «затравки» в растворы вводят фосфат калия, фторапатит или оксид кальция в количестве 1—100 мг/л (в пересчете на гидроксид), после чего растворы фильтруют через фильтр 2 с намывным слоем целлюлозы.
Растворы после фильтрации подвергаются более глубокой очистке от ионов кальция и тяжелых металлов на ионообменных смолах в колонне 3. В качестве ионообменников применяют слабокислый ионит из сополимера на основе акрилата или метакрилата. Применяют также неорганические сорбенты, на поверхность которых осажде-
Рис. 5.16. Схема мембранной установки процесса получения хлора и гидроксида натрия:
/ — реактор для химической очистки рассола; 2 — фильтр; 3— колонна ионообменной очистки; 4— башня обесхлоривания; 5—электролизер; 6 — сборник анолита; 7— сборник католита; 8 — установка синтеза хлорида водорода; 9 — стадия сжижения хлора; 10 — система очистки;
// — барометрический конденсатор; 12, 13, 14—выпарные аппараты
246
ны ионогенные группы, являющиеся сильными комплексообразовате-лями, хелатные смолы и др.
Обычно такую очистку проводят на установке, срстоящей из нескольких колонн, в которых попеременно протекают три процесса: собственно очистка рассола, регенерация и подготовка колонны к работе. При этом в процессе поддерживают оптимальные значения pH, температуру среды; скорость процесса очистки рассола на ионообменных смолах достигает нескольких десятков м3/(ч-м3 смолы). Качество получаемого рассола — менее 0,01 мг/л солей жесткости.
Разработан также метод тонкой очистки рассола экстракцией в жидкой фазе (Италия, фирма Де Нора) органическими хелатными соединениями, растворенными в керосине или газолине с добавкой, увеличивающей полярность растворителя. Процесс проводят на установке, представляющей собой каскад из четырех смесителей-декантаторов, в которых извлекают примеси, промывают рассол, регенерируют и промывают адсорбент.
Извлечение примесей из рассола осуществляют в первом смесителе при pH 10—11 и объемном отношении рассола и органической фазы 1:1. Для удаления хелатного агента, увлеченного рассолом, последний промывают растворителем при pH 4—5 и равном объемном отношении органической и водной фаз. Очищенный и промытый рассол направляют в отпарную колонну для более полного удаления растворителя. Рассол перед подачей в электролизер 5 пропускают через колонну с активированным углем (на рисунке не показано) для удаления остатков хелатного агента и растворителя, после чего его подкисляют.
Для регенерации хелатного агента и вывода кальция и магния из системы в смеситель вводят 18%-ную хлороводородную кислоту при объемном соотношении водной и органической фаз 1:1. В жидкофазном методе достигается более глубокая очистка исходного рассола с одновременным уменьшением расхода химических реагентов. Однако этот способ требует применения более сложного оборудования и расхода пара и при этом в рассольную систему вводятся органические примеси, загрязняющие систему.
Очищенные рассолы направляются в сборник анолита б, откуда рассол (анолит) циркулирует через электролизер 5. Часть анолита далее направляют в виде обедненного рассола в отделение рассола. Полученный в электролизере газообразный хлор направляется в скруббер (на рисунке не показан), в котором он осаждается очищенным рассолом из отделения очистки рассола. Часть охлажденного газообразного хлора направляется на установку синтеза хлороводородной кислоты 5, а оставшуюся часть направляют на станцию осушки и сжижения 9. Давление газообразного хлора контролируют регулятором давления, установленным на трубопроводе подачи хлоргаза.
247
Католит из электролизера направляют в сборник католита 7, откуда часть его поступает в выпарные аппараты 12—14 для рекуперации теплоты в качестве теплоносителя и возвращаются в сборник 7.
Особенностью технологии мембранного электролиза является работа по замкнутому рассольно-анолитному циклу. Это означает, что попадающие с рассолом примеси и образующиеся в нем в результате электролиза побочные продукты будут постепенно накапливаться, если их не выводить из системы или не разрушать. Поэтому для исключения накопления нежелательных анионов после обесхлоривания отработанного анолита в вакууме химически разрушают хлораты и выводят из системы сульфаты.
Образующийся в электролизере водород направляется вместе с католитом в сборник, в котором отделяется от раствора и подается в скруббер. В скруббере водород охлаждается водой, после чего часть его направляется на установку синтеза хлороводородной кислоты S, а часть — в систему очистки 10. Давление водорода контролируют регулятором давления на трубопроводе подачи водорода.
Часть католита (раствора гидроксида натрия) подают в отделение выпарки, где его упаривают до концентрации 50%.
Температура в электролизере регулируется изменением тепловой нагрузки выпарных аппаратов, а также охлаждением католита.
Газообразный хлор из сборника анолита 6 и водород из сборника католита 7 подают на установку синтеза хлороводородной кислоты, в которой водород сжигают в хлоре с образованием хлорида водорода. Абсорбируя хлорид водорода водой, получают хлороводородную кислоту, которую частично расходуют для нейтрализации ионов ОН-, мигрирующих из католита в анолит.
Полученные в мембранных электролизерах растворы гидроксида натрия содержат незначительное количество хлорида натрия (0,03% масс.). Для концентрирования растворов гидроксида натрия применяют систему выпарки с рекуперацией тепла, выделяющегося при электролизе. В такой выпарной системе католит можно концентрировать до 35% (масс.), а при давлении небольшого количества пара в последний корпус выпарной системы получают 50%-ный гидроксид натрия. Применение выпарки с рекуперацией теплоты позволяет в два раза сократить расход пара по сравнению с обычной выпаркой.
Согласно схеме (рис. 5.17), растворы гидроксида натрия из сборника католита поступают последовательно в выпарные аппараты 1 — 3. Большая часть католита циркулирует в качестве теплоносителя в теплообменниках, нагревая растворы гидроксида натрия, проходящие через выпарные аппараты. Испаряемая влага в виде пара поступает в теплообменники, в которых нагревает растворы гидроксида натрия. Пар, образовавшийся в выпарных аппаратах 4 и 5, направляют в барометрический конденсатор 7, где он конденсируется. Таким образом, теплота, 248
образующаяся в процессе электролиза, переносится католитом и используется дважды для концентрирования растворов гидроксида натрия.
Образующийся в последнем выпарном аппарате пар передают на начало схемы выпарки для концентрирования растворов гидроксида натрия из сборника. Пар из испарителя нагревает растворы гидроксида натрия в теплообменнике выпарного аппарата 6, а образующиеся при этом пары воды из выпарного аппарата поступают в барометрический конденсатор и конденсируются. Таким образом, тепло пара, направляемого в теплообменник конечного выпарного аппарата 7, используют трижды для концентрирования растворов гидроксида натрия. Сконденсировавшийся в теплообменниках пар используют в качестве технической воды для подачи в сборник католита.
Растворы гидроксида натрия после аппаратов первой и второй ступени передают в конечный выпарной аппарат, где их концентрация повышается до 50% (масс.). Пар из конечного выпарного аппарата используют в выпарных аппаратах 1 и 6 трехкратно.
Разработана также схема производства хлора и чистого гидроксида натрия способом электролиза с катионообменной мембраной. Схема включает стадии предварительной очистки рассола карбонатно-гидро-ксидным (содово-каустическим) методом, глубокой очистки исходного рассола фосфатным методом и на ионообменных смолах, электролиза с биполярными и монополярными электролизерами, обесхлорирования анолита, удаление хлоратов из анолита, вывода сульфатов из цикла, до-насыщения анолита твердым хлоридом натрия или концентрирования анолита выпариванием.
Создана стойкая в условиях электролиза катионообменная мембрана нужной сегментивности и способы ее модификации, освоены не разрушаемые аноды для мембранного процесса электролиза, отработаны биметаллические композиции сталь — титан, созданы промышленные конструкции электролизеров на общую нагрузку 400—700 кА.
Электролизеры фильтр-прессного типа выполняют как с биполярными, так и с монополярными ячейками. Эти электролизеры имеют следующие технико-экономические показатели:
Нагрузка на одну ячейку, кА......... 6—10
Напряжение, В....................... 3,7—4,0
Выход по току, %.................... 85—90
Концентрация, %: растворов NaOH..................... 20
хлора.............................. 98
Мембранный электролизер при повышенном давлении имеет ряд преимуществ по сравнению с проведением процесса при нормальном давлении.
249
Основным же преимуществом процесса под повышенным давлением является снижение напряжения на электролизере. Зависимость напряжения на электролизере с мембраной Нафион-315 от температуры приведена на рис. 5.18. Процесс ведут при плотности тока 4 кА/м2; концентрации NaCl 2,5 н.; содержании кислоты в анолите 0,05 н. НС1; давлении в катодной камере выше, чем в анодной, на 10 кПа (~0,1 кгс/см2).
Из приведенных данных следует, что в процессе увеличения давления в электролизере существует критическое значение температуры, выше которой напряжение на электролизере начинает возрастать, что может нарушить целостность мембран. Повышение давления позволяет сдвинуть это значение в область более высоких температур. Так, при нормальном давлении (кривая 7) повышение температуры более 85° С вызывает заметный рост напряжения, а при давлении ~0,3 МПа температура возрастает до 120° С без увеличения напряжения на электролизере (кривая 6).
Ранее отмечалось, что при повышенных плотностях тока в двухслойных мембранах возможно вскипание раствора и разрушение мембран. Повышение давления позволяет увеличить плотность тока на мембране, а следовательно, и продолжительность электролизера без этих нежелательных последствий.
Рис. 5.17. Схема процесса выпарки растворов гидроксидов натрия:
1, // — соответственно первый и второй корпус выпарных аппаратов; Ill—конечный корпус аппарата; 1-6 — выпарные аппараты;
7 — конденсатор
Рис. 5.18. Зависимость падения напряжения в электролизере от температуры при различных давлениях (в МПа) в катодной камере: /—0,10; 2 — 0,13; 3 — 0,15; 4 — 0,20; 5 — 0,25; 6 — 0,30
250
Получение продуктов электролиза под давлением позволяет сократить или исключить затраты на транспортирование хлора и водорода. Можно также существенно уменьшить затраты на сжижение хлора.
С повышением температуры процесс электролиза можно эффективно использовать джоулево тепло, выделяющееся в электролизере, например, для выпарки растворов гидроксида натрия. Может быть использована также теплота хлора и водорода.
На рис. 5.19 приведена схема производства хлора и гидроксида натрия мембранным способом при повышенном давлении. Согласно схеме, в электролизере 1 анодное пространство отделено от катодно-
Рпс. 5.19. Схема процесса получения хлора и гидроксида натрия мембранным способом под давлением:
/ — электролизер; 2— сборник; 3, -/—колонны; 5—аппарат для обесхлоривания; 6 — аппарат для донасышения; 7 — аппарат для очистки рассола; 8 — сборник католита; 9, 10—колонны
251
го катионообменной мембраной. Сборник анолита 2 имеет перегородку, отделяющую концентрированный рассол с низкой температурой от разбавленного анолита с высокой температурой. В колонне 3 хлоргаз охлаждают холодным насыщенным раствором.
Расход воды в рассольно-анолитном цикле за счет переноса воды вместе с ионами натрия через мембрану восполняется подачей холодной воды. Вода охлаждает хлоргаз в колонне 7, в процессе которого совершается регенерация теплоты. Подогретая же вода далее направляется в колонну обесхлоривания 5.
Для разрушения накапливающегося в системе при электролизе гипохлорита в линию горячего анолита подают хлороводородную кислоту; для поддержания pH анолита на низком уровне хлороводородную кислоту можно подавать в линию циркуляции анолита или в сборник 2. Растворенный хлор выделяется в колонне обесхлоривания при пониженном давлении. Рассол после обесхлоривания и охлаждения донасыщают хлоридом натрия в аппарате 6. Рассол очищают в отделении очистки рассола 7 от примесей кальция, магния, сульфата и др., хлор возвращается с помощью газодувки в колонну 3.
В сборнике католита 8 перегородка разделяет разбавленный раствор NaOH с низкой температурой и концентрированный раствор NaOH с высокой температурой. В колонну 9 подают водород, где он охлаждается водным раствором гидроксида натрия, поступающим с трехстадийной выпарки. Для поддержания необходимой концентрации водных растворов гидроксида натрия в катодный цикл подают
холодную чистую воду.
Способ получения хлора и гидроксида рах с ртутным катодом. Способ получения
Рис. 5.20. Схема электролитической ванны с ртутным катодом:
/ - электролизер; 2 — разлагатель амальгамы; 3 — ртутный насос; 4 — графитовый катод; 5 — амальгамный анод; 6 — ртутный катод; 7 — анод
натрия в электролизе-хлора и гидроксида натрия в электролитических ваннах с ртутным катодом в отличие от других способов обеспечивает получение непосредственно из ванны чистого раствора гидроксида натрия.
На рис. 5.20 приведена схема электролитической ванны с ртутным катодом, согласно которой ванна состоит из электролизера 7, разлагателя амальгамы 2 и ртутного насоса 3. В электроли-
252
зер подают хлорид натрия и ртуть. В процессе электролизера на аноде 7 выделяется хлор и образуются сопутствующие примеси, а на ртутном катоде 6—натрий, образующий с ним сплав — амальгаму натрия. Обедненный хлоридом натрия раствор, хлоргаз и амальгаму выводят из электролизера. Амальгама натрия попадает в разлагатель 2, в который одновременно подают воду. В разлагателе имеется графитовый катод 7, который электрически накоротко замкнут с амальгамой, являющейся в разлагателе анодом 5. В результате электрохимического процесса в разлагателе образуется концентрированный раствор гидроксида натрия. Ртуть из разлагателя ртутным насосом перекачивается в электролизер.
В электролизере протекают следующие процессы. Суммарная химическая реакция в электролизере:
NaCl + nHg -> NaH&, + 0,5С12
где NaHg„ — амальгама натрия.
На аноде, как и в диафрагменном электролизере, происходит процесс окисления ионов хлора:
СГ - е -> 0,5С12
На катоде восстанавливаются ионы натрия с образованием сплава с ртутью:
Na++ е + nHg -> NaHg„
Благодаря образованию сплава электродный потенциал смещается на величину AGaM/F и становится в случае электролиза раствора хлорида натрия почти на 1 В положительнее потенциала выделения металлического натрия, здесь AGaM — изобарно-изотермический потенциал образования амальгамы, Дж/моль Na+; F—постоянная Фарадея, Клмоль"1.
Наряду с выделением металлического натрия, образующего амальгаму на ртутнрм катоде, возможно выделение водорода, равновесный потенциал которого положительнее стационарного потенциала амальгамного электрода. Однако заметному выделению водорода на ртутном катоде препятствует высокое перенапряжение этой реакции на ртути.
Приведенные на рис. 5.21 поляризационные кривые процессов выделения натрия и водорода на ртути из раствора хлорида натрия показывают, что в результате высокого перенапряжения процесса выделения водорода на ртутном катоде плотность тока выделения водорода невысока и составляет незначительную долю от плотности тока 253
Рис. 5.21. Поляризационные кривые зависимости выделения натрия и водорода на ртутном катоде в водном растворе хлорида натрия. Поляризационные кривые при выделении:
/ — натрия; 2 — водорода; 3 — суммарная поляризационная кривая
разряда ионов натрия. Это обеспечивает возможность выделения натрия на ртутном катоде с высоким выходом по току при потенциалах отрицательнее -1,8 В.
Хлор, выделяющийся на аноде, растворяется и реагирует с раствором хлорида натрия:
С12,р + Н2О = Н+ + СГ + нею
нею + ОН’ = СЮ’ + Н2О
СЮ’ + 2НСЮ = сю; + 2Н+ + 2СГ
На катоде по реакции Н2О + е —> 0,5Н2 + ОН" образуются гидроксид-ионы, вступающие в реакцию с хлорноватистой кислотой и нейтрализующие ионы водорода: ОН" + Н+ —> Н2О.
Наряду с выделением водорода на ртутном катоде протекают реак
ции восстановления хлора и хлороксидных соединений, приводящие
к снижению выхода по току натрия:
С12,р + 2е —> 2СГ (Ю)
НСЮ + 2е-> СГ + ОН’ (Н)
СЮ; + ЗН2О + бе -> СГ + 6 ОН’ (12)
Реакции (11) и (12) приводят к подщелачиванию прикатодного слоя электролиза. При этом реакции (10) и (12) снижают выход по току щелочного металла. Кроме перечисленных реакций в процессе может иметь место химическое взаимодействие амальгамы натрия с кислым раствором в электролизере, приводящее к образованию водорода. Однако обычно скорость реакции меньше скорости других побочных реакций.
Таким образом, на ртутном катоде в процессе электролиза протекает реакция выделения натрия и побочные реакции выделения водорода и восстановления растворенного хлора и хлороксидных соединений.
При электролизе с ртутным катодом в электролизере отсутствует диафрагма и водород, образующийся на катоде, попадает полностью 254
в хлоргаз. Поэтому по содержанию водорода в хлоргазе рассчитывают плотность тока выделения водорода.
Амальгама натрия, поступившая в разлагатель из электролизера, разлагается в результате реакции с водой:
NaHg„ + Н2О -> NaOH + 0,5Н2 + nHg (13)
Поскольку гетерогенная химическая реакция амальгамы натрия с водой протекает сравнительно медленно, ее катализируют, помещая в разлагатель графитовую насадку, имеющую электрический контакт с амальгамой. При этом образуется короткозамкнутый гальванический элемент, в котором на графитовом катоде идет процесс выделения водорода:
Н2О + ё-> 0,5Н2 + ОН" (14)
а на амальгамном аноде — растворение натрия:
NaHg„ - е -> Na+ + nHg (15)
В объеме раствора протекает химическая реакция образования гидроксида натрия:
Na++ ОН"-> NaOH (16)
Электроны, освободившиеся в результате реакции (15), через контакт ртути с графитом перетекают на графит, где участвуют в процессе выделения водорода.
В соответствии с реакцией (13) э.д.с. амальгамного элемента будет равна
£=£,дл^,
Г ^Н,()
где Eq — стандартная э.д.с., В; о—активность молекул воды в
концентрированном растворе гидроксида натрия в разлагателе, кмоль/м3.
При разложении амальгамы в короткозамкнутом элементе э.д.с. расходуется на создание определенной плотности тока разложения. Установлено, что интенсифицировать процесс разложения амальгамы можно путем подбора материала насадки и снижения величины перенапряжения водорода.
255
Рис. 5.22. Принципиальная схема производства хлора и гидроксида натрия в электролизерах с ртутным катодом:
/ — ванна с ртутным катодом; / — электролизер; 2 — разлагатель амальгамы натрия; 3 — насос; 4 — узел дехлорирования анолита; 5 — сатуратор; 6 — узел очистки анолита; 7 — узел очистки рассола; 8—выпарка очищенного рассола; 9 — узел подготовки воды; 10— теплообменник;
11— фильтр; 12 — охлаждение водорода; 13— отмывка водорода; 14—абсорбер
На рис. 5.22 приведена схема производства хлора и гидроксида натрия в электролизерах с ртутным катодом, в которой основным аппаратом является электролитическая ванна с ртутным катодом.
Ртутный катод включает электролизер 7, разлагатель амальгамы натрия 2 и ртутный насос 3. Исходный раствор хлорида натрия с концентрацией 305—310 г/л вводят в электролизер. В процессе электролиза происходит снижение концентрации исходных растворов хлорида натрия, и из электролизера вытекает раствор анолита, содержащий 260—270 г/л хлорида натрия. Раствор донасыщают чистым выпарным хлоридом натрия в сатураторах 5 (аппараты, в которых через слой выпарного чистого хлорида натрия проходит обедненный электролит). Раствор после донасыщения снова поступает в электролизер. Перед процессом донасыщения анолит частично освобождают от растворенного хлора — дехлорируют путем введения хлороводородной кислоты до pH 2—3. При этом реакция смещается влево
С12 + Н2О НС1О + Н+ + СГ
256
Так удаляют значительную часть растворенного хлора в колоннах вакуумирования узла дехлоратора 4 или отдувают хлор из раствора воздухом.
Чистую выпаренную соль получают на выпарных установках 8 из очищенного от кальция, магния и других примесей раствора хлорида натрия. Для этого используют содово-каустический способ очистки 7, аналогичный очистке исходного рассола для диафрагменного электролиза.
После донасыщения выпаренной солью раствор поступает в электролизеры с ртутным катодом. Для исключения накопления в рассольном цикле вредных для электролиза примесей часть раствора (-20—30%) выводят на очистку на установке б, в которой раствор подщелачивают. В необходимых случаях добавляют карбонат натрия до концентрации 50—150 г/л и фильтруют. В процессе из раствора осаждается железо в виде гидроксида железа (III), магний — в виде гидроксида и кальций — в виде карбоната. Очищенный и отфильтрованный раствор возвращают в рассольный цикл электролиза.
Таким образом, стадия очистки и донасыщения раствора хлорида натрия в рассольно-анолитном цикле электролиза, при которой растворенный хлор удаляется не полностью, позволяет исключить заметное осаждение ртути из раствора хлорида натрия. Это обеспечивает минимальные технологические потери ртути в рассольно-анолитном цикле, поскольку вся ртуть, растворенная в анолите, вытекающем из электролизера, в виде HgCh снова возвращается в электролизер.
При работе электролизера на анодах выделяются хлор и кислород или диоксид углерода в зависимости от вида используемых анодов. Параллельно с анодным газом смешивается водород, образующийся на ртутном катоде. При заданных условиях электролиза хлоргаз содержит 0,5% водорода, а при нарушениях циркуляции ртути либо попадания в раствор или ртутный катод железа и примесей (так называемых амальгамных ядов — хрома, ванадия и некоторых других) возможно усиленное выделение водорода. Это вызывает снижение выхода по току натрия на катоде, качества хлоргаза и за счет подщелачивания раствора резко повышает содержание растворенного хлора в анолите, что может в дальнейшем нарушить стадию очистки раствора.
Выводимый из электролизера хлоргаз охлаждают, осушают и сжижают.
В разлагатели амальгамы подают очищенную воду, а из разлага-телей выводят два продукта: концентрированный раствор гидроксида натрия и газообразный водород, насыщенный парами воды и ртути.
Воду перед подачей в разлагатель фильтруют от взвесей и отделяют побочные катионы и анионы на ионообменных смолах в отделениях водоподготовки 9.
9 Y 257
у Химическая технология
неорганических веществ, кн. 2
Выводимый из разлагателя амальгамы раствор гидроксида натрия имеет температуру 100—110° С, содержит растворенное и диспергированное железо в количестве 5-10'4—5-10'3% и ртуть. Для снижения содержания примесей раствор гидроксида охлаждают водой в поверхностных теплообменниках 10 до 35—40° С, после чего фильтруют на ФПАКМ 11 и направляют в сборники-хранилища.
Водород, выводимый из разлагателей амальгамы, охлаждают в холодильниках, устанавливаемых обычно на разлагателях амальгамы, а затем в холодильниках водорода, устанавливаемых на рядовых коллекторах водорода 12 rq 20° С. При этом из него конденсируется вода и ртуть.
Содержание ртути в водороде после охлаждения не превышает 14 мг/м3. Водород дополнительно промывают в отмывочных колоннах 13 анолитом из ванн с ртутным катодом либо хлорной водой, полученной при охлаждении хлоргаза, а затем раствором гидроксида натрия и водой для удаления следов активного хлора. Водород одновременно отмывается от ртути, которая растворяется в хлорной воде или анолите с образованием дихлорида ртути. Остаточное содержание ртути в водороде после отмывки —0,1 мг/м3.
При необходимости глубокой очистки водорода от паров ртути до значения ПДК 0,01 мг/м3 водород пропускают через абсорбер 14 с твердым поглотителем ртути — активированным углем.
Важным вопросом в производстве является очистка сточных вод от ртути. Сточные воды, содержащие ртуть, образуются в процессах промывки оборудования, в результате проливов технологических растворов, в процессе промывки насадки разлагателей амальгамы, в виде конденсата при охлаждении водорода и регенерации ртути из шламов промывной водой.
ПДК ртути в сточных водах установлена 5-10‘6г/м3. Поэтому очистка сточных вод ртути является неотъемлемой частью производства.
На рис. 5.23 приведена схема очистки сточных вод от ртути. Согласно схеме, ртутьсодержащие сточные воды поступают в сборник-усреднитель 7, после чего их фильтруют на песчаном или другом фильтре 2 от взвешенных частиц. После фильтров вода, содержащая ртуть, поступает в сборник 5, в котором подщелачивается до pH 10—11, а затем в хлоратор 4. В хлоратор вводят газообразный хлор, который с гидроксидом натрия образует гипохлорит натрия, окисляющий содержащуюся в воде ртуть до его дихлорида HgCh. После полного перевода всей ртути в дихлорид растворы подкисляют в реакторе 5 до pH 3—4 с таким расчетом, чтобы обеспечить оптимальные условия дехлорирования в колонне 6 с активированным углем. После дехлорирования ртуть сорбируют из воды на ионообменной смоле в колонне 7. Очищенную воду направляют в сборник S, откуда по мере накопления откачивают центробежным насосом 9. 258
Рис. 5.23. Схема очистки сточных вод от ртути;
/ — сборник-усреднитель; 2— фильтр; 3 — сборник; 4 — хлоратор; 5 — реактор; б—колонка-а сорбер; 7—колонна с ионообменной смолой; 8 — сборник очищенной воды; 9—насос
Для очистки от ртути применяют анионообменную смолу на основе винилпиридина ВП-1-АП, которую после насыщения ртутью выгружают из колонн и направляют на переработку. Регенерацию ее от ртути производят, промывая хлороводородной кислотой, а полученный элюат возвращают в цикл электролиза.
В производстве применяют ванны с ртутным катодом со скрубберными и горизонтальными скрубберами.
Хлоргаз, получаемый в электролизерах с графитовыми анодами, имеет состав: 99,0% С12; 0,6% СО2; 0,2% Н2, остальное — воздух. При использовании металлооксидных анодов получают продукт, содержащий 98,5% С12; 1,0% О2; 0,2% СО2; 0,1% Н2, ос зльное — воздух.
Из разлагателя амальгамы выводят раствор гидроксида натрия, содержащий 42—50% NaOH, 0,01—0,05% NaCl, 0,2% Иа2СОз и 3 мг/м3 ртути.
5.2. ОКСИДЫ ХЛОРА
Хлор образует следующие оксиды: монооксид (ди лороксид, ге-миоксид) С12О, диоксид С1О2, перхлорат хлора (дих..сротетраоксид) С12Од, триоксид (дихлорогексаоксид) С12Об и оксид хлора (VII) (дихлорогептаоксид) С12Оу.
Все оксиды хлора имеют резкий запах. Они термически и фотохимически нестабильны и склонны к взрывному распаду, все имеют положительное значение АНОбр-
Монооксид хлора С12О — газ желто-оранжевого цвета со слабым зеленоватым оттенком, а в жидком состоянии — красно-коричневый. Длина связи С1 — О 0,1700 нм, угол ОСЮ 111°, ц = 2,6О-1О‘зоКл-м (табл. 5.1); уравнение температурной зависимости давления пара Igp (мм рт. ст.) = 7,87—1373/5 (173—288 К).
259
9*
Таблица 5.1. Свойства оксидов хлора
Показатель С12О С1О2 С12О4 С12О6 С12о7
Тт, °C -120,6 -59 -117 3 -93,4
Ткип* °C 2,0 9,7 44,5 203* 87
Плотность, г/см3 — 1,653 (5° С) 1,82 (0° С) 2,023 (3,5° С) 1,805** (25° С)
С”, Дж/(мольК) 47,81 42,00 86,03 — —
Д/Лр, кДж/моль 81 93 180 310 240
Д//исп , кДж/моль 26 26,3 30 40 34
5^, Дж/(мольК) 267,84 256,77 327,23 — —
♦Расчетная. ** 2,83 г/см^ при -160° С.
Гемиоксид хлора растворяется в воде с образованием оксохлората водорода НС1О (хлорноватистая кислота) по схеме
Ci2O + Н2О = 2HCIO
Растворимость гемиоксида хлора (г в 100 г воды при 0° С): 33,6 (2,66 кПа), 52,4 (6,65 кПа). В процессе хранения при 60—100° С гемиоксид хлора распадается (термический распад) в течение 12—24 ч, а выше 110° С — через несколько минут взрывом. При этом освещение ускоряет процесс распада и повышает вероятность взрыва.
Гемиоксид хлора с хлоридами образует оксихлориды:
TiCl4 + С12О = ТЮС12 + 2С12
ТаС15 + С12О = ТаОС13 + 2С12
AsCI3 + 2С12О = AsO2Cl + ЗС12
а с диоксидом азота образует смесь NO2C1 и NO3CI с N2O5 — чистый NO3C1:
2NO, + С12О = NO2CI + NO3CI
N2O5 + С12О = 2NO3CI
При фторировании гемиоксида хлора дифторидом серебра получают трифтороксид хлора, а реакцией с пентафторидом мышьяка или пентафторидом сурьмы — соли хлорила CIO^MF^.
Гемиоксид хлора получают пропусканием хлора, разбавленного азотом, над HgO или реакцией хлора с влажным карбонатом натрия. 260
Гемиоксид применяют в качестве хлорирующего агента при хлорировании насыщенных органических соединений.
Диоксид хлора СЮ2 — газ желтого цвета, в жидком состоянии ярко-красного цвета, в твердом — красновато-желтый. Длина связи CI—О 0,1475 нм, угол ОСЮ 117° ; уравнение температурной зависимости давления пара 1g р (мм рт. ст.) = 7,7427—1275,1/Г(226—312 К) (рис. 5.24), растворимость в воде 26,1 г/л (25° С, 20,68 кПа), растворяется в тетрахлориде углерода, тетраоксохлорате (VII) водорода (хлорной кислоте) и уксусной кислоте. В индивидуальном состоянии взрывоопасен, при 30—50° С процесс распада идет с измеримой скоростью, а выше температуры 50° С после периода индукции взрывается.
Диоксид хлора в щелочной среде диспропорционирует на СЮ2 и ClOj, а в присутствии пероксида водорода образуется СЮ2 и выделяется кислород.
Рис. 5.24. Давление паров диоксида хлора CIO2 в зависимости от температуры
261
Диоксид хлора восстанавливается иодидами, арсенидами, оксидом свинца, сернистой кислотой, аминами до хлорит-иона. С диоксидом азота и пентаоксидом диазота образует NO3CI, а с NOC1—NO2CI. Он фторируется фторидами серебра и брома или разбавленным фтором до CIO2F.
Диоксид хлора применяют вместо хлора как экологически более безопасный продукт для отбеливания древесной суспензии, целлюлозы, синтетических волокон, а также для очистки питьевой и технологической воды, обеззараживания сточных вод.
Перхлорат хлора (дихлортетраоксид) С12О4 (СЮСЮз) — светло-желтая жидкость, а в кристаллическом состоянии почти бесцветен.
Триоксид (дихлоргексаоксид) С^Об — жидкость ярко-красного цвета, в твердом состоянии — оранжевый, в процессе охлаждения окраска ослабевает. В газообразном состоянии или в виде жидкости молекулы имеют строение O3CI—О—СЮз, а в кристаллическом состоянии СЮ2СЮ4. Кристаллы триоксида хлора моноклинной сингонии с пространственной группой С*, z = 4. Давление пара 39,9 Па (0°С), 133 Па (19° С).
В температурном интервале 0—10° С триоксид медленно разлагается по схеме СЬОв -> 2002 + О?, а выше 20° С в продуктах распада появляются Ck С водой реагирует со вспышкой, образуя НСЮз и НС1О4. С хлоридами, бромидами, нитридами образует перхлораты, например с NOC1 дает NOCIO4, с N2O5 — NO2C1O4, с AICI3 — C1O2[A1(C1O4)4], а с FeCh — ClO2[Fe2(ClO4)7]. При нагревании в вакууме приведенные комплексы отщепляют СЬОб и переходят в несольватированные перхлораты А1(С1О4)з, Ре(С1О4)з.
Триоксид хлора получают реакцией озона с диоксидом хлора или действием фтора на хлораты металлов по схемам:
6С1О2 + 2Оз = ЗС12О6
Mg(ClO3)2 + F2 = СЬОб + Mgp2
Триоксид хлора применяют в качестве исходного сырья в процессе получения безводных перхлоратов в лабораторных условиях.
Оксид хлора (VII) (хлорный ангидрид, дихлорогептаоксид) CI2O7 — бесцветная жидкость, чувствителен к удару и трению. Молекула имеет строение О3С1—О—СЮз с длиной связи С1—О 0,1709 нм, в группах СЮз — 0,1405 нм, угол C1OCI 118,6°, ОСЮ 115,2° ц = = 2,40-10"30 Кл-м.
Кристаллы оксида хлора (VII) моноклинные (пространственная группа С2/с). Уравнение температурной зависимости давления пара lgp(MM рт. ст.) = -7J96—\T1QIT.
262
Неограниченно растворим в тетрахлориде углерода, хорошо растворим в хлорной кислоте. С водой оксид хлора (VII) не смешивается, на границе раздела фаз реагирует с образованием НСЮ4. Реакция сильно экзотермична (Д//2°98 реакции -211 кДж/моль), поэтому разогревание слоя CI2O7 может привести к взрыву. Процесс распада оксида хлора (VII) в газе на хлор и кислород протекает с измеримой скоростью в температурном интервале 100—120° С, однако с повышением давления CI2O7 выше 13,3 кПа приобретает взрывной характер. Жидкий оксид хлора (VII) устойчив до 60—70° С. Присутствие низших оксидов хлора ускоряет процесс его распада.
Для жидкого оксида хлора (VII) характерны реакции с образованием ковалентных соединений с группой СЮ3. С аммиаком в тетрахлориде углерода он образует NH4HNCIO3 и NH4CIO4, с алкилами— соответственно RHNCIO3 и R2NCIO3, с SbF5—SbOF3 и FC1O3; а с пентаоксидом диазота в тетрахлориде углерода — NO2CIO4.
Оксид хлора (VII) получают действием пентаоксида ди фосфора или олеума на хлорную кислоту или электролизом раствора хлорной кислоты на платиновых электродах ниже 0° С. При этом оксид хлора (VII) накапливается в анодном пространстве. Чистый CI2O7 получают в процессе нагревания в вакууме некоторых перхлоратов, например Nb(ClO4)5, MoO2(ClO4)2.
Способы получения диоксида хлора. Диоксид хлора получают в производстве действием восстановителей (SO2, NO2, метанола и др.) на подкисленный раствор хлората щелочного металла, в процессе нагревания смеси хлората с влажной щавелевой кислотой, а также действием хлора на хлориты (диоксохлораты (III)).
Особое внимание при разработке способов получения диоксида хлора обращают на обеспечение безопасности процессов и подбор стойких материалов для аппаратуры, которую изготовляют из кислотоупорной керамики, стекла, кварца, фосфора и пластмасс (органическое стекло, винипласт, фторопласт и др.), а в необходимых случаях и свинец.
Все существующие способы получения диоксида хлора основываются на процессе восстановления хлората по схеме
2НС10з + 2НС1 = 2СЮ2 + С12 + 2Н2О
или
сю; + 2Н+ + е = С1О2 + Н2О
Для разложения исходного хлората с образованием хлорноватой кислоты в производстве применяют концентрированный раствор серной кислоты (8—10 н. раствор).
263
Необходимая для протекания реакции хлороводородная кислота образуется в результате взаимодействия хлорноватой кислоты с восстановителем. В качестве восстановителей применяют диоксид азота, оксид углерода, сульфат марганца и др. В промышленных условиях освоено применение диоксида серы метанола, а в случае применения хлороводородной кислоты восстановители не требуются.
Процесс получения диоксида хлора восстановлением хлоратов хлоридами в присутствии серной кислоты состоит из трех одновременно протекающих реакций:
2С1О“ + 2СГ + 4Н+ = 2С1О2 + С12 + Н2О (17)
5СЮ; + СГ + 6Н+ = 6С1О2 + ЗН2О (18)
СЮ; + 5СГ + 6Н+ = ЗС12 + ЗН2О (19)
Реакция (1) является превалирующей, поскольку протекает с большей скоростью (в 100—1000 раз), чем другие. В процессе восстановления хлоратов хлоридами теоретически активный хлор не расходуется, поскольку параллельно с диоксидом хлора образуется молекулярный хлор, обладающий также активными окислительными свойствами. Целевой продукт — диоксид хлора — получается в виде разбавленного газа (до 10% СЮ2) или водного раствора.
Выход диоксида хлора зависит от степени протекания побочной реакции:
С1О2 + 4Н+ + 5е = СГ + 2Н2О
Для предотвращения побочной реакции отвод продуктов реакции из реактора производят струей инертного газа или воздуха.
Сернисто-кислотный способ получения диоксида хлора основан на реакции хлората натрия с серной кислотой с образованием хлорноватой кислоты
2NaC103 + H2SO4 = 2НСЮ3 + Na2SO4
и хлороводородной кислоты при взаимодействии хлорноватой кислоты с диоксидом серы с дальнейшим восстановлением НСЮ3 хлороводородной кислотой:
НС1О3 + 2Н2О + 2SO2 = НС1О + 2H2SO4
НСЮ + H2SO3 = НС1 + H2SO4 264
При температуре до 20° С и pH в пределах 1,0—3,5 лимитирующей скорость процесса стадией является реакция НС Юз с H2SO3. Избыток последней по сравнению с необходимым количеством для образования хлороводородной кислоты реагирует с диоксидом хлора с выделением хлороводородной и серной кислот.
Согласно схеме получения диоксида хлора по сернисто-кислотному способу (рис. 5.25), применяют раствор хлората натрия с концентрацией 45—47% NaClO3 и 75%-ную серную кислоту. Исходные реагенты— NaCIO3 и H2SO4 — подаются в емкости-дозаторы 7 и 2. В нижней части (на дне) реакторов 3 и 4 установлены стальные круги 5 с отверстиями для подачи смеси воздуха с диоксидом серы. Образующийся при этом диоксид хлора направляется в промывную колонну 6, в которой он путем промывки водой очищается от хлора, поскольку растворимость диоксида хлора в воде превышает растворимость хлора в десять раз и практически не зависит от концентрации хлора. Состав азеотропной смеси в системе диоксид хлора— хлор в водной среде при 10°С отвечает примерно 90% (мол.) CIO2 и 10% (мол.) С12. При температурах 25 и 35° С содержание хлора в азеотропе соответственно растет до 20 и 30% (мол.). Соотношение диоксида хлора и хлора в газе тем выше, чем ниже концентрация применяемого раствора хлората натрия. На этом основаны способы получения диоксида хлора с применением разбавленных растворов, содержащих 50—60 г/л NaClO3. Температуру реакции поддерживают в пределах 75—90° С. Отвод теплоты реакции сопровождается испарением воды, уносимой инертным газом.
Получают диоксид хлора в аппарате, приведенном на рис. 5.26. Согласно технологии, исходный раствор хлората натрия поступает в основной реактор 3, проходя скруббер 2, в котором взаимодействует с хлором. Это повышает эффективность процесса на 1—2%. С целью увеличения поверхности контакта фаз в качестве пенообразователя в реакционную смесь вводят водные растворы сульфата натрия. Процесс останавливают при достижении остаточной концентрации хлората натрия 10—20 г/л. Переработку хлората натрия, содержащегося в отходящей жидкости, производят во вспомогательном реакторе 6. При смешении продукта из обоих реакторов получают газ с содержанием 10% диоксида хлора. При введении в реакционную массу хлорида натрия в количестве 5—10% от массы хлората натрия выход диоксида хлора составляет 95—97%. Добавление исходного хлората к циркулирующей жидкости после вывода сульфата натрия из системы 5 позволяет поддерживать постоянную концентрацию хлората в рабочем растворе. Операции вывода и ввода производятся непрерывно.
265
Рис. 5.25. Схема получения диоксида хлора по сернисто-кислотному способу: /, 2—емкости-дозаторы; 3, 4—реакторы; 5 — сборник диоксида хлора: 6 — промывная колонна; 7 — поглотитель хлора, 8 — вентилятор; 9 — насос
NaCIO3
Рис. 5.26. Реактор непрерывного действия для получения диоксида хлора по серно-кислотному способу: / — переход газа на абсорбцию; 2 — скруббер; 3 — основной реактор; 4 — подача охлаждающей воды; 5 — вывод отработанного раствора;
6 — дополнительный реактор
Рассмотренный способ получения диоксида хлора является сравнительно экономичным по капитальным затратам, он прост и удобен в эксплуатации.
Существует также видоизмененный вариант этого способа, по которому процесс осуществляют без серной кислоты в присутствии 5—6% (от хлората натрия) дихромата натрия в качестве катализатора с использованием непрореагировавшего хлората после вывода из системы сульфата натрия.
Хлороводородный способ получения диоксида хлора основан на непосредственном взаимодействии основных реагентов — хлората натрия и хлороводородной кислоты:
2NaClO3 + 4НС1 = 2С1О2 + С12 + 2NaCl + 2Н2О
Механизм процесса представляется следующими реакциями:
NaClO3 + НС1 = НС1О3 + NaCl
НСЮз + НС1 = нсю2 + нею
НСЮ2 + НСЮз = 2СЮ2 + Н2О
нею + НС1 = С12 + Н2О
266
Незначительные количества ионов марганца и серебра катализируют реакцию.
Образующийся раствор хлорида натрия используют для электролитического получения хлората натрия. Это позволяет исключить побочные солевые продукты и ускорить реакцию образования диоксида хлора, не доводя ее до до конца. Отработанный раствор, содержащий непрореагировавший хлорат натрия, циркулирует через электролитические ванны, в которых хлорид натрия окисляется в его хлорат.
Представляет большой промышленный интерес получение диоксида хлора по рассмотренному способу с применением хлората натрия и газообразного хлорида водорода. Процесс с лучшим выходом целевого продукта проходит в температурном интервале от -10 до 0° С. Процесс можно осуществить в трубах, загруженных хлоратом натрия и погруженных в охлаждающую жидкость. Предварительная добавка к исходному хлорату пористого материала (пемзы, обожженной глины или силикагеля) препятствует спеканию массы и обеспечивает удаление NaCl после реакции.
В связи с более полным использованием исходного сырья целевой продукт имеет низкую себестоимость. Однако он содержит до 50% (масс.) хлора. Последний отделяют от диоксида хлора известными способами.
Хлоридный способ. Диоксид хлора получают взаимодействием хлората и хлорида в серно-кислотной среде по следующим уравнениям:
Са(СЮ3)2 + СаС12 + 2H2SO4 = 2С1О2 + С12 + 2CaSO4 + 2Н2О
2NaClO2 + 2NaCl + 2H2SO4 = 2С1О2 + Cl2 + 2Na2SO4 + 2Н2О
Эффективность процесса в значительной мере определяется молярным отношением хлората и хлорида. В процессе хлорирования известкового молока (водной суспензии оксида и гидроксида кальция) получается раствор, содержащий 150—200 г/л хлората кальция и 5 моль хлорида на 1 моль хлората. Выход диоксида хлора в процессе восстановления хлората кальция в присутствии 93%-ной серной кислоты при 35—40° С в полузаводских условиях составил в среднем 70%, а степень использования хлората 98,5%. С учетом утилизации хлора в виде перхлората процесс получения диоксида хлора в этих условиях протекает практически без потерь.
Для производства 1 т диоксида хлора по рассмотренному способу расходуется: 4,8 т оксида кальция (85% СаО), 3,6 т хлора (100%) и 4,4 т серной кислоты (93% H2SO4).
Растворы, полученные хлорированием смесей карбонатов натрия и калия, содержат наряду с хлоратом натрия некоторое количество 267
хлората калия. Концентрация хлората натрия в образующихся растворах не превышает 140 г/л при соотношении хлората натрия к хлориду, равном 1:3. Восстановление хлората в этих растворах при оптимальном соотношении серной кислоты к хлорату, равном 3,26:1, позволяет использовать хлорат на 98,3% и достичь 80%-ного выхода диоксида хлора.
Оптимальными условиями получения диоксида хлора из раствора хлората натрия и хлорида натрия являются: концентрация хлората в растворе — 350 г/л, концентрация серной кислоты -93% H2SO4, температура 35° С, соотношение хлората к хлориду—1:1,05 и соотношение серной кислоты к хлорату — 2,7. При этих условиях технологического процесса выход диоксида хлора составляет 96%.
На 1 т диоксида хлора расходуется: 1,75 т хлората натрия, 6,0 т хлорида натрия и 4,35 т серной кислоты (93% H2SO4).
Способ получения диоксида хлора из хлорита натрия базируется на процессе хлорирования хлорита по схеме
2NaClO2 + С12 = 2С1О2 + 2NaCl
Способ применяют в ограниченных масштабах, поскольку в связи с использованием сравнительно недорогостоящих методов получения диоксид хлора является полуфабрикатом в производстве хлорита натрия. Однако при выработке небольших количеств диоксида хлора способ является простым и удобным. Способ может быть осуществлен как сухим, так и мокрым путем. По сухому способу в вертикальную башню загружают сухой технический хлорит натрия. Снизу в башню подводят воздух, содержащий около 2% хлора, а сверху отводят смесь воздуха с диоксидом хлора (до 4% С1О2). Сухой способ позволяет получить С1О2 в «твердом» виде на носителе и избежать коррозии. Однако способ представляет собой значительную опасность вследствие возможности неуправляемого процесса образования взрывчатой смеси при быстром протекании процесса. Для предотвращения перегрева в ходе реакции к исходному хлориту добавляют соли, содержащие значительное количество кристаллизационной воды, например Na2SO410H2O. По мокрому же способу хлор взаимодействует в башне с раствором хлорита натрия. Через раствор (~ 0,3 кг NaClO? в 1 л воды) пропускают воздух, содержащий ~3% С12. В выходящем газе содержится около 4% С1О2 и 1% С12.
Диоксид хлора может быть получен из хлоритов в процессе взаимодействия хлорита с сильными кислотами по схеме
5NaClO2 + 4Н+ = 4С1О2 + 2Н2О + NaCl + 4Na+ 268
Поскольку наравне с этой реакцией протекает ряд побочных, во избежание взрыва хлорит вводят в реакционную среду в виде его водного раствора. Выход диоксида хлора составляет 60—80%. Действием персульфатов на хлориты получают диоксид хлора в количестве 96% от теоретического. В процессе электролиза хлоритов получают диоксид хлора с выходом 97%.
Предложен также способ получения диоксида хлора действием трихлорида азота в смеси с разбавителем на сухой твердый хлорит натрия.
5.3. КИСЛОРОДНЫЕ КИСЛОТЫ ХЛОРА
Хлор образует ряд кислородных кислот: хлорноватистую НС1О, хлористую HCIO2, хлорноватую НСЮз и хлорную HCIO4. Все они являются сильными окислителями. Активность их возрастает от хлорноватистой к хлорной кислоте. Окислительная же их активность увеличивается в обратном порядке — от хлорной к хлорноватистой, которая является наименее устойчивой. В ряду СЮ“ — СЮ;—СЮ;—СЮ" по мере увеличения степени окисления хлора возрастает устойчивость анионов. Это объясняется тем, что в процессе перехода от СЮ" к СЮ; увеличивается число электронов, участвующих в образовании связей. Более устойчив ион СЮ;, в котором все валентные электроны атома хлора принимают участие в образовании связей:
О
ЧС1
0 ч. / 0
Полагают, что в ряду СЮ“— СЮ;—СЮ;—СЮ; возрастает роль тс-связывания. Поэтому, если в СЮ" порядок связи равен 1, то в ионе СЮ4 он составляет 1,5. Повышение порядка связи СЮ" соответствует увеличению средней энергии связи и уменьшает межъядерное расстояние. Вследствие повышения устойчивости в ряду С1О“ — сю;—СЮ;—сю; уменьшается окислительная активность. Поэтому гипохлориты вступают в окислительно-восстановительное взаимодействие в основной среде, а хлораты в растворах окисляют лишь в сильнокислой среде.
Хлорноватистая кислота и ее соли. Хлорноватистая кислота НСЮ — слабая одноосновная кислота (р/Са 7,537). Существует в водных растворах с максимальной концентрацией -30% и в газообразном состоянии. Длина связи С1 — О в молекуле Н — О — С1 0,1689 нм, связи Н—О 0,0974 нм, угол НОС1 104°. Для газа С ° = 37,284
269
Дж/(моль-К); Д//О°бр = -90 кДж/моль, AG^p = -71 кДж/моль; 52°98 -= 236,474 Дж/(моль-К); для бесконечно разбавленного водного раствора Д//°бр = -124,7 кДж/моль. Для водного раствора НС1О характерны равновесия: НС1О«^Н+ + ОСГ и 2HClO<t CI2O + Н2О. В растворе с расчетной концентрацией НСЮ 4 моль/л фактическая концентрация НСЮ 3,606, СЮ" 3,7-104 С12О 1,93-10"1 моль/л; pH 3,43. По мере разбавления раствора содержание СЮ" и С^О убывает, pH возрастает, а фактическая концентрация НСЮ приближается к расчетной. Парциальное давление НСЮ над 0,1 М водным раствором 230 Па.
Скорость и направление распада НСЮ в водных растворах зависят от pH, температуры, концентрации, чистоты и освещения. В сильнокислой среде (рН<3) при комнатной температуре происходит медленный распад по схеме
4НС1О -> 2СЬ + О2 + 2Н2О (20)
В процессе подкисления хлороводородной кислотой наблюдается быстрая реакция, равновесие которой сдвинуто вправо:
НСЮ + НС1 -> С12 + Н2О (21)
а в интервале pH 3,0—7,5 идет автокаталитический процесс:
2НСЮ -> О2 + 2НС1 (22)
Свет ускоряет реакцию (22), причем видимый свет оказывает более сильное влияние, чем ультрафиолетовый.
При pH 6—8 в присутствии в растворе НСЮ и СЮ“ в соизмеримых количествах происходит процесс диспропорционирования:
2НСЮ + СЮ’ -> СЮ’ + 2Н+ + 2СГ (23)
До 70° С вклад этой реакции невелик, но при более высоких температурах она становится преобладающей. Введение в раствор ионов переходных металлов, например Со2+, Ni2+, Cu2+, существенно ускоряет реакцию (22), причем повышение pH ослабляет каталитическое действие. Из соединений платиновых металлов каталитическое действие оказывают лишь соединения иридия, направляющие распад по уравнению (23).
Гипохлориты — соли хлорноватистой кислоты. К ним относят также неорганические ковалентные соединения, содержащие группу ОС1, связанную с электроотрицательным радикалом, например SO2FOC1, NO2OCI, гемиоксид СЮС1; эфиры, например (СН3)3СОС1. 270
Гипохлориты металлов, за исключением гипохлорита кальция,— неустойчивые соединения. В свободном виде они почти не Изучены. Большинство из них выделено лишь в виде кристаллогидратов, а многие из них существуют только в водных растворах.
Гипохлориты щелочных и щелочноземельных металлов хорошо растворимы в воде (г в 100 г): LiClO 72 (20° С), NaClO 53,4 (20° С), Са(С1О)2 33,3 (25° С). LiClO кристаллизуется в виде моногидрата, NaClO образует гидраты с 5 (температура плавления 24,5° С) и 2,5 молекулами воды (температура плавления 57,5° С). Для Са(С1О)2 известны три-, ди- и моногидраты.
В безводном состоянии гипохлориты нестабильны и взрывоопасны. Гидраты гипохлоритов более устойчивы, однако в обычных условиях самопроизвольно разлагаются, например LiC10H20 теряет 2% активного хлора за 53 сут, a NaC105H20 — 30% за 40 сут.
В табл. 5.2 приведены термодинамические свойства гипохлоритов.
Таблица 5.2. Термодинамические свойства гипохлоритов
Соединение кДж/моль ДС^р, кДж/моль
LiClO -388,5 -328,9
NaClO -350,4 -298,7
КСЮ -362,3 -319,1
CsClO -368,1 -328,2
Mg(ClO)2 -688,1 -530,5
Са(С1О)2 -763,2 -626,0
Гипохлориты в водном растворе — сильные окислители. При значении рН<4 они окисляют Г до 12, при pH 5—7 — до Ю^, а при рН>7 — до ю;.
Нитриты в водных растворах окисляются до нитратов:
2NaNO2 + Mg(ClO)2 = 2NaNO3 + MgCl2
Диоксид хлора в нейтральной среде окисляется до CIO3:
2С1О2 + 2NaClO = 2NaClO3 + Cl2
Сульфиты окисляются до сульфатов:
Na2SO3 + КСЮ = Na2SO4 + КС1
Цианиды окисляются до оксоцианидов:
Ca(CN)2 + 2КС1О = Ca(OCN)2 + 2KCI
271
Формиаты и оксалаты окисляются гипохлоритами до карбонатов и r.ri.
Наиболее изученными соединениями являются ковалентные гипохлориты — трифторметилгипохлорит CF3OC1 (температура плавления -142° С, температура кипения 46° С); пентафторгипохлорит серы SF5OCI (температура кипения 9° С); фторосульфурилгипохлорит SO2FOC1 (температура кипения 43,4° С); нитрилгипохлорит NO2OCI (температура плавления 107° С, температура кипения 22° С); перхло-рилгипохлорит CIO3OCI (температура плавления 117° С, температура кипения 44,5° С). Все приведенные соединения очень гигроскопичные летучие вещества, разлагающиеся при 40—100° С. Под действием озона CIO3OCI окисляется до (С1О2)+ (С1О4)‘, NO2OCI — до (NO2)+ (С1О4)‘, SO2FOCI—до (С1О2)+ (SO3F)-.
В процессе взаимодействия ионных и ковалентных гипохлоритов с пероксидом водорода в щелочном растворе, например
NaOCl + Н2О -> Н2О + NaCl + О2
кислород выделяется не в обычном триплетном состоянии, а в возбужденном синглетном. Энергия возбуждения составляет 0,98 эВ. Генерируемый приведенным способом синглетный Д'-кислород применяют в моющих химических лазерах непрерывного действия для передачи энергии возбуждения атомам иода.
Гипохлориты натрия, лития и кальция производят в промышленном масштабе взаимодействием газообразного хлора с водным раствором или суспензией соответствующего гидроксида с последующим выделением кристаллизацией. Производят гипохлориты в виде кристаллогидратов, основных солей и водных растворов. В лабораторных условиях получают: НСЮ — пропусканием хлора через суспензию HgO или В12Оз в воде, а также гидролизом С12О и нейтрализацией раствора НСЮ. Большинство ковалентных гипохлоритов получают действием C1F на соответствующие безводные кислоты или оксиды.
Растворы ионных гипохлоритов применяют в процессах отбеливания тканей, бумаги и целлюлозы, дегазации взрывчатых веществ и в качестве дезинфицирующего средства, в том числе для обеззараживания сточных вод, а также в органическом синтезе при получении трихлорметана, хлорпикрина и др.
Хлористая кислота и ее соли. Хлористая кислота НС1О2 — слабая неустойчивая кислота, при 25° С р7<а = 1,94; АЯо°бр в бесконечно разбавленном водном растворе -53,6 кДж/моль. В течение 9 ч при 40° С и pH 4 распадается на 7%, а при pH 2 — на 80%:
4НСЮ2 -> НСЮз + 2С1О2 + НС1 + Н2О
272
Поэтому хлористая кислота хотя и получена в свободном виде, цо обычно получается в водных растворах. Константа диссоциации ее равна 1,0710'2 при температуре 18° С. Образование хлористой кислоты происходит в значительных количествах лишь в оильнокис-лой среде (рН<3). Однако и при этом в растворе наряду с хлористой кислотой находится и диоксид хлора.
Щелочные растворы хлоритов в темноте вполне устойчивы даже при кипячении. Однако под действием света идет следующая реакция:
600’ -> 2СЮ; + 4СГ + ЗО2
В кислой среде происходит фотохимический распад по схеме
1 осю’ -> 2сю; + бег + зо2 + 200; + зо2
За 30 мин облучения при pH 8,94 степень распада составляет 50%, а при pH 4,76 — 80%.
Хлориты — окислители. Окислительно-восстановительный потенциал растет с уменьшением pH раствора. В кислой среде ClOj окисляет Вг" до Вг2 по схеме
2NaBr + Н2О + КСЮ = Br2 + 2NaCl + 2КОН
NO’ до NO;’:
KNO2 + NaCIO = KNO3 + NaCl
NO до NO2 (но не реагирует c N2O), a H2O2 до O2. Взаимодействие с иодом протекает по схеме
I2 + 10СЮ’ + 4H+ -> 2Ю; + 6СЮ2 + 4СГ + 2Н2О
По реакции СЮ2 с С12 также образуется СЮ2 и СГ. При рН>11 гипохлорит-ион быстро и количественно окисляет СЮ2 до СЮу. Поэтому хлориты отсутствуют в продуктах реакции С12 с водными растворами щелочей. Однако уже при рН<9 окисление гипохлоритом идет лишь до СЮ2:
2NaClO2 + NaCIO + Н2О -> 2СЮ2 + NaCl + 2NaOH
Этой реакцией пользуются в некоторых методах хлоритного беления.
273
Термическая стабильность хлоритов щелочных металлов растет не от лития к цезию, а наоборот, что отличает хлориты от большинства солей кислородных кислот хлора. В процессе медленного нагревания/и в изотермических условиях происходит сильно экзотермическое диспропорционирование без выделения газа (ЗМС1О2 2МС1Оз + MCI); LiClO2 разлагается при 150—180° С, КС1О2— при 135—160° С, RbCIO2 — при 100—140° С, CsClO2 — при 50—100° С. В процессе быстрого нагревания образуются MCI и О2, а в присутствии оксидов переходных металлов или примеси органических веществ реакция может принять взрывной характер.
Хлорит натрия NaClO? — кристаллы моноклинной сингонии (пространственная группа 12/а); плотность 2,468 г/см3; ЛН^р = -311,3 кДж/моль; растворимость в воде при 25° С 75,8 г в 100 г, в этаноле — 6 г/л. Кислые водные растворы разлагаются с повышением температуры и уменьшением pH среды. Достаточно устойчивы щелочные растворы.
Хлорит натрия кристаллизуется из щелочного раствора в виде безводной соли и тригидрата NaClO2-3H2O, переходящего в безводную соль при 37,4° С. При нагревании до 175° С хлорит натрия разлагается с выделением кислорода по схеме
NaClO2 -> NaCl + О2
Реакция разложения идет с большой скоростью вплоть до взрыва.
В слабощелочных растворах, содержащих не более 1 г-моль/л NaClO?, хлорит натрия в процессе кипячения не разлагается. В более концентрированных растворах он разлагается по реакциям:
2NaClO? = 2 NaClO3 + NaCl
NaClO? = NaCl + O2
Константы скорости этих реакций равны соответственно при 103° С — 0,65-10’6 и 1,210’7; при 83° С — 1,6-ИУ7 и 0,2-10 8.
Хлорит натрия получают обработкой диоксида хлора восстановителями в щелочной среде:
2CIO? + 2ОН" = С1О2 + сю; + Н2О
или взаимодействием ее с пероксидами:
2С1О2 + Н?О? + 2ОН~ = 2СЮ; + 2Н?О + О2
274
Полученный раствор хлорита после фильтрации выпаривают и выделяют из них кристаллы. Благодаря низкой растворимости NaClO? в присутствии NaClO3 в твердую фазу выкристаллизовывается хлорит натрия.
В процессе выпаривания раствора происходит частичное разложение хлорита с образованием хлората и хлорида. Для уменьшения потерь хлорита натрия растворы упаривают под давлением около 100 мм рт. ст. при температуре кипения 50—55° С. При этих условиях степень разложения хлорита натрия в течение 24-часового выпаривания не превышает 0,5% при содержании 1,5 — 2 г-моль/л NaC102 в растворе. Процесс разложения растворов достигают добавлением к ним пиро- или полифосфата, а также пергидроля.
Очистку хлорита натрия от хлорида и хлората натрия при необходимости осуществляют обычной перекристаллизацией или высаливанием целевого продукта из раствора бензолом с дальнейшей сушкой. Очищают раствор от NaClO3 также добавлением в реакционную массу водорастворимых соединений щелочно-земельных металлов или гидроксида калия для осаждения менее растворимого хлората калия.
Получаемый хлорит натрия содержит 70—75% NaClO2, 10—11% NaCl и 10—12% Na2CO3.
Разработан способ получения хлорита натрия с высоким выходом (94—98%), в котором в качестве восстановителей используют химические вещества — иодид натрия, гидроксид хрома (III), гемицеллюлозу, а также взаимодействием диоксида хлора с водным раствором тетрагидробората натрия.
Хлорноватая кислота и ее соли. Триоксохлорат водорода НС1О3 выделен в свободном состоянии. Хлорноватая кислота НС1О3 известна лишь в водном растворе с максимальной концентрацией до 40%. Попытка получения более концентрированной кислоты вакуумной отгонкой приводит к ее разложению, а иногда к взрыву. р7<а = 2,7; бесконечно разбавленного водного раствора -95,23 кДж/моль; при 18° С плотность 7,23%-ного раствора 1,0421, 13,57%-ного— 1,0829, а 25,87%-ного— 1,1713 г/см3. Хлорноватая кислота по своим свойствам напоминает азотную кислоту, в частности ее смесь с хлороводородной кислотой — сильный, типа «царской водки», окислитель.
Поскольку триоксохлорат водорода — сильный окислитель, он легко восстанавливается до хлорида водорода:
нсю3 + 3no2 на + 3no;
НС1О3 + 6НВг -> НС1 + ЗВг2 + ЗН2О
275
В слабокислой среде хлорноватая кислота восстанавливается по схеме
НСЮз + H2SO3 -> HCI + H2SO4 + Н2О
а при пропускании диоксида серы, разбавленного воздухом, в сильнокислый раствор идет реакция:
2НСЮз + H2SO3 -> 2СЮ2 + H2SO4 + Н2О
Хлораты (V) — солц хлорноватой кислоты. Анион хлоратов ClOj имеет структуру тригональной пирамиды, длина связей CI—О 0,1507 нм, угол ОСЮ 106°. Анион СЮ3 не образует ковалентных связей через атом О и не склонен образовывать координационные связи. Существует несколько соединений, относящихся к хлоратам (V), в которых группа СЮз связана ковалентно через атом хлора, например FCIO3, NC1O;’, C6H5C1O3 и др.
Хлораты (V) — сильные окислители как в растворе, так и в твердом состоянии. Смеси безводных хлоратов (V) с серой, углем и другими веществами при быстром нагревании и ударе детонируют.
Хлорат-ион (V) окисляется до хлората (VII) лишь электрохимическим путем или под действием XeF2.
Большинство хлоратов металлов находятся в виде гидратов. В безводном состоянии выделены хлораты (V) щелочных и щелочноземельных металлов, серебра, таллия (II), свинца (II), а также NH4CI3, М(СНз)4СЮз и т.п. Хроматы (V) металлов переменной валентности обычно неустойчивы и склонны к взрывному разложению.
Все хлораты щелочных металлов (табл. 5.3) разлагаются экзотермически на МО и О2 с промежуточным образованием перхлоратов (перхлоратов (VII)). Оксиды металлов переменной валентности — МпО2, Ре2Оз, СоО, CuO, NiO и др., а также Na2O2 катализируют распад хроматов (V), снижая в процессе температуру разложения на 100—200° С. Выше 300° С хроматы (V) щелочных металлов имеют небольшое собственное давление пара и могут быть возогнаны.
Хлорат (V) натрия — бесцветные кристаллы кубической сингонии (а = 0,6575 нм, z = 4). Имеет пирамидальное строение, длина связи С1—О 0,1502 нм, угол ОСЮ 106,8°.
При быстром охлаждении расплава хлората (V) натрия кристаллизуются две метастабильные модификации: фаза II — при 230—255° С и моноклинная фаза III — при 255—260° С.
При непрерывном нагревании хлорат (V) натрия разлагается в интервале 390—520° С по приближенному уравнению:
10NaCIO3 -> 4NaCI + 6ЫаСЮ4 + ЗО2 + 146 кДж.
276
Таблица 5.3. Свойства хлоратов (V) щелочных металлов
Показатель LiClOi NaCICh КСЮз ИЬСЮз CsClO,
Сингония — Кубическая Моноклинная Гексагональная Гексагональная
Пространственная группа — Р2.3 Р2\!т R3m R3m
Гпл. °C 129 263 357 342 388
Температура начала 367 465 472 480 483
разложения, °C Плотность, г/см3 2,631 2,493 2,338 3,203 —
с;:, Дж/(моль-к) — 100,1 100,3 103,2 104,6
ДА/‘’,р, кДж/моль -368,3 -357,7 -389,1 -394,3 -406,8
Растворимость в воде, г в 100 г 406,3 95,89 8,58 6,61 7,60
В присутствии катализаторов (Мп, СО3О4 и др.) температура распада снижается до 200—400° С, при этом в продуктах реакции уменьшается доля NaClOzi.
Растворимость хлората (V) натрия при 25° С (г в 100 г растворителя) (рис. 5.27): в воде— 100,1, этилендиамине — 52,8, ДМФА — 23,4, моноэтаноламине -19,7, ацетоне — 0,094. Температура эвтектики с водой [39,7% хлората (V) натрия] — 17,8° С, с LiClO3 [31,0% хлората (V) натрия] — 107,1° С.
Хлорат (V) натрия в кислом растворе окисляет NO2 до NOj, Fe2+до Fe3+, CF до CIO2 и С12. В водном растворе хлорат (V) натрия окисляется до хлората (VII) натрия (электролитически или при действии XeF2).
Хлорат (V) натрия применяют в качестве окислителя в различных пиротехнических составах. Он является кислородоносителем в пиротехнических источниках газов («кислородные свечи»), дефолиантом. Применяется также в качестве сырья в производстве хлоратов (VII) и диоксида хлора.
Хлорат (V) калия КСЮз (бертолетова соль) образует бесцветные моноклинные пластинчатые кристаллы плотностью 2,344 г/см3; негигроскопична, гидратов не образует. При нагревании до 250° С переходит из моноклинной в ромбическую модификацию. Температура плавления 370° С, а при 400° С разлагается с выделением кислорода. В присутствии некоторых оксидов (MnO2, Fe2O3 и др.) хлорат (V) калия разлагается при 150—200° С. С органическими и легко окисляющимися веществами образует чувствительные к механическим воздействиям (трению, удару) и нагреванию взрывчатые смеси. Чувствительность к взрыву возрастает в присутствии бромата калия, а смеси хлората (V) калия с аммонийными солями, аминами, гидразинами могут самовозгораться в процессе хранения. Растворимость КС1О3 в воде приведена в табл. 5.4 и на рис. 5.28.
277
Рис. 5.27. Растворимость хлората (V) натрия в воде при различных температурах
Таблица 5.4. Растворимость хлорида калия в воде
Температура, °C Растворимость Температу-ра, °C Растворимость
г/100 г Н2О % г/100 г Н2О %
0 1,021 3,3 3,21 60 1,115 23,8 19,4
10 1,033 5,2 5,01 70 1,139 30,2 23,4
20 • 1,045 7,3 6,96 80 1,169 37,6 27,4
25 1,051 8,6 8,00 90 1,192 46,0 31,6
30 1,058 10,1 9,30 100 1,219 56,75 35,9
40 1,073 13,9 12,1 104 1,230 60,0 37,6
50 1,092 18,5 15,6
Хлорат (V) калия выше 220° С реагирует с газообразным аммиаком по следующей схеме:
4КСЮ3 + 2NH3 = 2KNO3 + 2КС1 + С12 + ЗН2О + 1,5О2
Хлорат (V) калия применяется в производстве спичек и в пиротехнике. В производстве спичек применяется так называемая пудри-рованная соль, получаемая размолом кристаллов бертолетовой соли. 278
Рис. 5.28. Зависимость растворимости хлората (V) калия в воде от температуры
Способы получения хлората (V) натрия. Хлорат (V) натрия получают электрохимическим методом. При электролизе концентрированного раствора хлорида натрия без разделения электродных продуктов хлорат натрия образуется как в результате химического взаимодействия в растворе первичных продуктов электролиза, так и их электрохимического окисления.
Первичные процессы на электролизах приводят к образованию на аноде хлора и на катоде гидроксида с выделением водорода:
2СГ-2е = С12 (24)
2Н2О + 2е = 2ОН" + Н2 (25)
В результате смешения анодных и катодных продуктов на первой стадии в зависимости от pH образуются гипохлориты или хлорноватистая кислота:
С12 + 2ОН" = СЮ" + СГ + Н2О (26)
С12 + ОН" = НСЮ + СГ (27)
В процессе получения хлората (V) натрия происходит разрядка ионов СЮ на аноде:
СЮ -е СЮ (28)
279
Превращение СЮ в ионы хлората изображается суммарным уравнением:
6СЮ’+ ЗН2О 2CIO’ + 6Н+ + 4СГ + 1,5О2 (29)
Процесс электрохимического окисления СЮ" в СЮ; представляется в виде
6СЮ’ + 6ОН + бе = 2СЮ; + 4СГ + ЗН2О + 1,5О2 (30)
Доля, приходящаяся на электрохимический процесс, в общем процессе образования хлората зависит от концентрации ионов гипохлорита вблизи анода. Ее можно уменьшить, изменяя pH электролита, его температуру, удельный объем электролита на единицу нагрузки, а также ограничивая или затрудняя диффузию ионов гипохлорита к аноду.
Образовавшиеся в растворе гипохлорит и хлорноватистая кислота взаимодействуют между собой с получением хлората по схеме
2HCIO + СЮ’ = СЮ; + 2СГ + 2Н+ (31)
или
нею + 2СЮ’ = СЮ; + 2СГ + Н+ (32)
Протеканию реакций (31) и (32) благоприятствует слабокислая реакция электролита и повышение температуры.
Максимальная скорость реакции (31) имеет место при pH 7, а реакция (9) — при pH 7,6. Константы скорости реакции (31) и (32) близки между собой.
В процессе получения хлората, согласно его химическому механизму, отсутствуют потери выхода по току на выделение кислорода и достигается высокий выход хлората по току.
Протеканию реакции по химическому механизму способствуют повышение концентрации NaCl в прианодном пространстве, поддержание pH на оптимальном уровне, повышение температуры, затруднение процессов диффузии ионов гипохлорита к аноду и, возможно, быстрый вывоз образовавшегося гипохлорита и хлорноватистой кислоты из межэлектродного пространства электролитической ячейки.
Одновременно с основными процессами получения хлората (V) на катоде, аноде и в объеме электролита протекают побочные процессы.
На катоде может происходить восстановление гипохлорита и хлората (V) до хлорида:
СЮ + Н2О + 2е -> СГ + 2ОН’ (33)
СЮ; + ЗН2О + бе -> СГ + 6ОН’ (34)
280
На аноде возможен разряд ионов ОН или молекул воды с выделением кислорода:
2Н2О - 4е -> О2 + 4Н+ (35)
На платиновых, магнетитовых и пероксидно-свинцовых анодах возможно дальнейшее окисление полученного хлората (V) до хлората (VII):
СЮ. + Н2О - 2е—> СЮ; + 2Н+ (36)
На графитовых анодах не достигается потенциал, необходимый для протекания реакции (13), поэтому окисления хлората (V) до хлората (VII) практически не наблюдается.
На ход процесса и выход хлората (VII) влияет также pH электролита, его температура, наличие в нем тяжелых металлов, оптимальная концентрация хлорида натрия в электролите.
Для снижения потерь тока на восстановление к электролиту добавляют соли хрома. При восстановлении хромовой соли на катоде образуется слой из смеси ионов гипохлорита и хлората (V).
В производстве хлората (V) натрия существуют различные варианты непрерывной схемы электролитического окисления NaCl в NaClO3. Поскольку с ростом концентрации хлората (V) и соответственно при снижении концентрации хлорида ухудшается выход по току, работу электролизеров организуют по каскадной схеме. Обычно применяют каскад из 4—6 электролизеров, поскольку при дальнейшем увеличении их числа в каскаде повышение суммарного выхода по току невелико.
В зависимости от условий получения кристаллов хлората (V) натрия различают две технологические схемы производства: 1) с выпаркой растворов хлората (V) натрия; 2) с высаливанием хлората (V) натрия его хлоридом.
Согласно первому способу (рис. 5.29), исходные растворы хлорида натрия готовятся и очищаются в сборнике 7 с мешалкой, куда подаются исходный хлорид натрия, вода, пар, маточные растворы, карбонат натрия и при необходимости гидроксид натрия. Полученный при этом раствор очищается от ионов кальция карбонатом натрия, а от ионов магния — гидроксидом натрия. При необходимости электролит очищают от сульфатов путем осаждения их хлоридом бария, после чего фильтруют через фильтр 2. Отфильтрованные растворы центробежным насосом 3 перекачивают в напорную емкость 7, в которой в растворы вводят необходимое количество хлороводородной кислоты и хроматов.
281
Пар
Рис. 5.29. Схема производства хлората (V) натрия с применением выпарки:
/ — сборник; 2 — фильтр; 3—центробежный насос; 4—напорная емкость; 5 — электролизеры; 6 — сборник электролита; 7 — реактор; 8 — второй корпус выпарной установки; 9 — первый корпус выпарной установки; 10, 11 — фильтры; 12 — сборник упаренных растворов; 13 — вакуум-кристаллизатор. 14 — пароэжекторная установка; 15— фильтр; 16 — сборник маточных растворов; 17 — бункер NaCl; 18 — сушилка. 19, 20 — колонны; 21 — вентилятор; 22 — контактный аппарат;
23 — холодильник; 24 — компрессор
Готовый электролит, содержащий примерно 280 г/л NaCl, 40—80 г/л NaClO3, 3—6 г/л Na2Cr2O7, при pH 6—7 пропускают через каскад из 4—6 электролизеров 5, включенных последовательно по электрическому току и по току жидкости.
Состав электролита, вытекающего из последнего электролизера каскада, зависит от степени превращения хлорида в хлорат (V). При применении графитовых анодов остаточное содержание хлорида натрия обычно составляет не менее 100—120 г/л. В этих условиях содержание хлората (V) натрия составляет 350—375 г/л. Обычно для снижения расхода графитовых анодов и повышения выхода по току остаточное содержание NaCl поддерживают до 130—150 г/л. В результате частичного выделения хлора в процессе электролиза повышается pH электролита. С целью поддержания оптимальных условий процесса в электролизеры добавляют 10%-ный раствор хлороводородной кислоты до содержания активного хлора в электролизере 3—4 г/л.
Основное количество гипохлоритов превращается в хлораты (V) за счет самоокисления хлората (V) водорода в сборнике электролита б. Для ускорения процесса по реакциям (8) и (9) электролит подогревают, после чего подщелачивают в аппарате 7 и проводят химическое обесхлоривание добавлением восстановителей (формиата натрия карбамида или других реагентов).
Для выделения хлората (V) в твердом виде растворы упаривают в двухкорпусном вакуум-выпарном аппарате до содержания в них хлората (V) натрия 850—950 г/л. При этом основная часть хлорида натрия выпадает в виде кристаллов и после фильтрации через фильтры 10 остаются в фильтре, а далее возвращаются вновь в производство для приготовления исходного раствора, поступающего на электролиз. Концентрированные растворы хлората (V) натрия фильтруются через фильтры 77 и направляются в сборник 12 упаренных растворов, после которого — в вакуум-кристаллизаторы 73, оснащенные пароэжекторной установкой 14. Суспензия из кристаллизатора центробежным насосом 3 передается в фильтр 15 хлората (V) натрия. Образующиеся кристаллы хлората (V) натрия собираются в бункер 17 и передаются на сушку в вакуум-сушилку 18. Образующиеся маточные растворы в кристаллизаторе 73 и фильтре 75 направляются на смешение в сборник 7.
Выделяющийся в электролизерах 5 водород поступает в колонну 19 для отмывки его гидроксидом натрия и тиосульфата натрия от хлора, после чего он во второй колонне 20 отмывается водой. Чистый водород вентилятором 21 передается в контактный аппарат 22 для каталитической очистки от брызг щелочи, после чего охлаждается в холодильнике 23 и направляется в компрессор 24.
Согласно второму способу (рис. 5.30), исходные растворы, поступающие из сборника 7 в каскад электролизеров 2, получают донасы-283
Рис. 5.30. Схема производства хлората (V) натрия без выпарки:
1 — сборник исходного раствора; 2 — каскад электролизеров; 3 — напорная емкость хлороводородной кислоты; 4 — сборник электролитических растворов, 5—центробежный насос; 6 — теплообменник электролитических растворов; 7—колонны очистки; 8—аппарат дообезвреживания и нейтрализации; 9,10— дозаторы; 77 —сборник фильтрации: 12, 15 — фильтры; 73 — сборник промывной воды; 14 — сборник дофильтрации; 16 — сборник очищенных растворов; 17 — теплообменник; 18— смеситель, 19—отстойник; 20 — промежуточный сборник; 21 — кристаллизатор; 22 — центрифуга; 23— холодильник: 24 — сборник маточных растворов; 25—сборник промывных вод; 26 — смеситель; 27 — сборник-смеситель исходного раствора
щением чистым хлоридом натрия маточных растворов в сборнике-смесителе 27. Образующийся электролит содержит -200 г/л NaCl, -340 г/л NaClOs и 3—8 г/л КагСгОд. pH, равный 5,6—5,8, в электролизерах каскада поддерживают добавлением 10%-ной хлороводородной кислоты из напорной емкости 3.
Из каскада электролизеров при pH 5,8—6,2 получают растворы, содержащие 520—540 г/л NaClOs, 100—110 г/л NaCl, 3—8 г/л Na2Cr2O4 и ~3 г/л NaClO.
Ниже приведен примерный состав электролита по электролизерам в четырех- и шестиступенчатом каскаде (в г/л):
Ступени каскада При четырехступенчатом каскаде: I II III IV V VI
NaCl 172 149 126 105 — —
NaClO3 При шестиступенчатом каскаде: 380 430 480 530 —
NaCl 180 165 150 135 120 105
NaClO? 362 395 429 462 499 530
Образующиеся в каскаде электролизеров растворы с содержанием хлора 3—4 г/л поступают в сборник 7, в котором за счет реакции (8) содержание хлора снижается до 1,5—2,0 г/л. Для дальнейшего разрушения активного хлора растворы подогревают в теплообменнике 6 до 65—75° С и пропускают через две или три колонны 7, продуваемые для перемешивания раствора сжатым воздухом. В конце процесса (за 4—6 ч) содержание активного хлора снижается примерно до 100 мг/л. Окончательное обесхлорирование проводят химическим способом в аппарате 8, вводя в растворы из дозатора 9 формиат натрия или другие восстановители. После полного обесхлоривания растворы подщелачивают введением гидроксида натрия из дозатора 10 до содержания последнего в растворах 0,5—1,5 г/л и центробежным насосом 5 перекачивают в сборник 77, откуда они через фильтр 12 направляются в сборник промывной воды 13 и сборник дофильтрации 14. Далее растворы фильтруют вторично на фильтре 15 и собирают в сборник очищенных растворов 76. Вторичная фильтрация проводится для окончательной очистки раствора от графитового шлама. Она обеспечивает снижение содержания шлама в растворах до 9—12 г/л.
Отфильтрованные растворы подогревают в теплообменнике 77, в смесителе 18 донасыщают хлоридом натрия, отстаивают в отстойнике 19 и через промежуточный сборник 20 перекачивают центробежным насосом 5 в кристаллизатор 27. Кристаллизатор 27 охлаждается холодильной установкой 23. Образующуюся суспензию кристаллов разделяют, а образующиеся кристаллы хлората (V) натрия промыва-285
ют водой в центрифуге 22. Целевой продукт направляют на упаковку, а маточные растворы и промывные воды собирают соответственно в сборниках 25 и 24. Смесь промывных вод и маточные растворы подогревают в теплообменниках, смешивают в смесителе 26. донасы-щают хлоридом натрия и передают в сборник-смеситель 27 исходного раствора для производства.
В производстве хлората (V) натрия для ведения технологического процесса пользуются изотермой растворимости в системе NaClO3—NaCl—Н2О в присутствии 10 г/л Na2CrO4 и 1 г/л NaOH (рис. 5.31).
Способы получения хлората (V) калия. Хлорат (V) калия получают обменным разложением хлората (V) натрия и хлорида калия по уравнению
NaC103 + КС1 = КС1О3 + NaCl
При этом хлорат натрия получают по одной из описанных ранее схем, а электролитические растворы после очистки от активного хлора и удаления взвеси продуктов разрушения анодов поступают на обменное разложение с хлоридом калия.
Согласно схеме производства хлората калия (рис. 5.32), маточные растворы после отделения хлората (V) калия донасыщают в аппарате 1 хлоридом натрия для восполнения его механических потерь в производстве. Теоретически хлорид натрия не должен расходоваться, поскольку его количество, необходимое в процессе получения хлората (V) натрия в электролизерах, компенсируется количеством NaCl, образую-
Рис. 5.31. Изотермы растворимости в системе NaCIO3—NaCl—Н2О в присутствии 10 г/л Na2CrO4 и 1 г/л NaOH:
/—область ненасыщенных растворов; //—область растворов, насыщенных по NaCl; /// — область растворов, насыщенных по NaCi^b
ЭОЛ
Рис. 5.32. Принципиальная схема производства хлората калия обменным разложением NaC103 с КС1:
/ — аппарат для донасыщения рассола хлоридом натрия; 2 — центробежный насос; 3 — реактор; 4—фильтр; 5 — напорная емкость; 6— сборник хлороводородной кислоты; 7— каскад электролизеров; 8—реактор; 9— фильтр; 10—реактор обменного разложения; // — кристаллизатор;
12— сгуститель; 13— центрифуга; 14 — сборник маточных растворов
щимся по реакции обменного разложения. Далее растворы в реакторе 3 очищаются от солей кальция и магния внесением карбоната и гидроксида натрия, а при необходимости и хлорида бария. Очищенные растворы фильтруются в фильтре 4, в напорной емкости 5 корректируют pH растворов и содержание в них хроматов, после чего растворы поступают в каскад электролизеров 7. При необходимости растворы подкисляют хлороводородной кислотой из сборника 6.
Электролитические растворы после электролизеров обесхлорива-ются в подогреваемом реакторе S, при необходимости нейтрализиру-ются гидроксидом натрия, отфильтровываются через фильтр 9 и поступают на обменное разложение с хлоридом калия в реакторе 10. Реакционная масса направляется в кристаллизатор 77, после которого в сгуститель 12. Сгущенные кристаллы направляются в центрифугу 73, а растворы из сгустителя и центрифуги собираются в сборнике маточных растворов 77, откуда последние центробежным насосом 2 перекачиваются в аппарат для донасыщения исходного рассола 7.
287
Кристаллы хлората (V) калия после центрифуги при необходимости сушатся в вакуум-сушилках или направляются на упаковку.
Хлорат (V) калия получают также известковым методом. При этом хлорированию подвергают известковое молоко (смесь оксида-гидро-ксида кальция в воде) с концентрацией 130—140 г/л СаО. Реакция протекает до полного исчезновения свободной извести по схеме
2Са(ОН)2 + 2С12 = Са(С1О)2 + СаС12 + 2Н2О
В процессе дальнейшего хлорирования образующаяся при гидролизе хлора хлорноватистая кислота окисляет гипохлорит кальция в хлорат (V):
Са(С1О)2 + 4НС1О = Са(С1О3)2 + 4НС1
Образующаяся хлороводородная кислота также реагирует с гипохлоритом, переводя его в хлорид кальция и выделяя при этом ма-лодиссоциированные молекулы НСЮ:
2Са(С1О)2 + 4НС1 = 2СаС12 + 4НС1О
Хлорноватистая кислота окисляет Са(С1О)2. Процесс продолжается до полного перехода гипохлорита в хлорат (V). Следовательно, хлорноватистая кислота является лишь катализатором — переносчиком кислорода.
Суммарная реакция второй стадии процесса выражается уравнением
ЗСа(С1О)2 = Са(С1О3)2 + 2СаС12
Общим уравнением процесса образования хлората (V) в процессе хлорирования известкового молока является
6Са(ОН)2 + 6С12 = Са(С1О3)2 + 5СаС12 + 6Н2О
Уравнение показывает, что в процессе хлорирования лишь 1/6 часть гидроксида превращается в хлорат (V), а 5/6 переходит в хлорид. Этим объясняют невыгодность прямого получения хлората (V) калия хлорированием гидроксида калия по реакции
6КОН + ЗС12 = КСЮз + 5КС1 + ЗН2О
поскольку при этом -83% ценного гидроксида калия, получаемого гидролизом КС! с такой большой затратой энергии, вновь превраща-288
ется в хлорид калия. При хлорировании известкового молока с последующим обменным разложением Са(С1Оз)2 и КС1 расходуется значительно менее ценный и более доступный гидроксид кальция. Важным фактом является и то, что исследованиями установлена максимальная скорость процесса при pH раствора 7—7,4. Окислителями же в этом процессе могут быть как НСЮ, так и СЮ". Процесс образования хлората (V) можно выразить следующими реакциями:
2НСЮ + СЮ" = СЮ; + 2Н+ + 2С1"
2СЮ" + НСЮ = СЮ; +Н+ + 2СГ
С повышением температуры ускоряется процесс окисления гипохлорита хлорноватистой кислотой. Однако повышение температуры вызывает и частичное разложение гипохлорита с потерей кислорода. Оптимальной температурой процесса хлорирования принято считать 75—80° С, при которой потеря кислорода невелика. Для хлорирования можно применять хлор любой концентрации.
На рис. 5.33 приведена клинографическая проекция на горизонтальную плоскость диаграммы растворимости в системе Са(С10з)г + + 2КС1 = 2КС1О3 + СаС12 при 0° С. Поле кристаллизации хлората (V) калия занимает почти всю диаграмму и его окаймляют узкие поля остальных солей. Стабильной является пара КСЮз + СаСЬ, поскольку линия соприкосновения их полей пересекает среднюю линию АОС. Поля Са(СЮ3)2-2Н2О и КС1 не соприкасаются, и эти соли не могут находиться совместно в твердой фазе.
Вертикальная плоскость АОС делит диаграмму на две части. В левой растворы состоят из КСЮз, СаСЬ и Са(СЮз)2, а в правой — из
Рис. 5.33. Диаграмма состояния системы Са(СЮ3)2 + 2КС1 = 2КС1О3 + СаС12 при 0° С (концентрации солей выражены в молях на 100 моль воды)
10 Химическая технология неорганических веществ, кн. 2
289
КСЮз, СаС12 и КС1. По линии АОС растворы состоят лишь из СаС12 и КСЮз. Получаемые в процессе хлорирования известкового молока растворы содержат на 1 моль Са(СЮз)2 5 моль СаС12, и их фигуративные точки располагаются на прямой OR в системе СаС12—Са(С1Оз)2—Н2О. Для любой точки этой прямой, например а, соотношение
Са(С1О3)2 : СаС12 = - = т 5
В процессе растворения хлорида калия состав раствора перемещается параллельно оси ОБ и достигает линии АОС в точке С, когда количество КО эквивалентно хлорату (V) кальция. В производстве обычно КС1 и состав раствора перемещаются в правую часть диаграммы, например в точку р. После выделения КСЮз фигуративная точка маточного раствора несколько поднимается по оси СаС12 вследствие добавочно образующегося по реакции обменного разложения хлорида кальция. Таким образом, состав конечного раствора изображается точкой Ь\ находящейся в правой части диаграммы.
Согласно схеме получения хлората (V) калия, исходное известковое молоко загружается в вертикальный абсорбер 7, залитый известковым молоком. Хлор барботируется в известковое молоко через барботажную трубу. Внутри абсорбера имеется стальной змеевик, по которому циркулирует холодная вода для отвода тепла реакции. Сухой хлор подают в абсорбер по стальной химзащищенной трубе.
Процесс хлорирования в абсорбере проводят до получения в растворах 76—80 г/л Са(С1Оз)2, 230—240 г/л СаС12 и не более 3—4 г/л Са(С1О)2. Выход хлората (V) по поглощенному хлору составляет -95%. Для удаления растворенного хлора прохлорированный раствор продувают воздухом, после чего с целью очистки от гипохлорита к горячему раствору вводят водный раствор аммиака, тиосульфата натрия или органические вещества (муку, древесные опилки и т.п.). Процесс очистки производят в больших реакторах с метальным устройством и паровыми змеевиками для поддержания температуры раствора 60—70° С.
После процесса очистки растворы фильтруют через фильтр. Образующийся при этом шлам промывают для уменьшения потерь хло-рата(У). Промывные воды направляют на процесс приготовления известкового молока.
Очищенные от шлама растворы упаривают в вакуум-выпарном аппарате и направляют в реактор 6 для обменной реакции с хлоридом калия. Обменную реакцию ведут при повышенной температуре. Полученные растворы выпаривают в вакуум-выпарном аппарате. Выпаренные растворы, содержащие 160—170 г/л КСЮз, 450—460 г/л СаС12, направляют на кристаллизацию. В производстве применяются 290
барабанные кристаллизаторы с водным или воздушным охлаждением до 15—20° С. После отжима основной массы кристаллов хлората (V) калия в центрифугах образующиеся при этом маточные растворы подвергаются вымораживанию при -10, -20° С. Кристаллы сушат в вакуум-сушилках. Для получения более чистых сортов хлорат (V) калия подвергают перекристаллизации.
Существуют также и видоизмененные варианты описанной технологии. Так, из прохлорированного раствора до реакции обменного разложения можно выделить значительную часть содержащего в растворах хлорида кальция. Для этого растворы охлаждают до 10° С. Поскольку растворимость Са(С1Оз)2 больше, чем у хлорида кальция, последний выделяется из растворов в виде гексагидрата хлорида кальция (СаС12-6Н2О). Имеется и другой вариант осаждения хлорида кальция. При этом в растворы вводят известковое молоко. В процессе охлаждения в осадок выделяется оксихлорид примерного состава ЗСаОСаС1215Н2О.
Существует вариант производства КСЮз, при котором разложение между КО и Са(СЮз)2 производят до операции выпарки и из выпаренного раствора охлаждением до 30—35° С выделяют до 80% имеющегося в системе КСЮз. Образующуюся суспензию разделяют на центрифуге. Хлорат (V) калия, содержащий 75—80% КСЮз и 5—6% СаС12, направляют на перекристаллизацию, а маточный раствор (~40 г/л КСЮз и 460—470 г/л СаС12) — на дополнительное охлаждение. В процессе охлаждения маточного раствора от 30 до -2, -3° С в нем остается ~5% КСЮз (от первоначального его количества). Остающийся после вымораживания раствор, содержащий 470—489 г/л СаС12 и не более 14 г/л КСЮз, является отходом производства, используемым для получения хлорида бария методом Дюфло, или перерабатывается в товарный хлорид кальция.
По более совершенному способу хлорат (V) калия получают путем проведения процессов хлорирования и обменной реакции в одном аппарате. При этом исходный хлорид калия подают в реактор с известковым молоком с 15—20%-ным избытком по отношению к СаО для обеспечения более полного выделения хлората (V) калия. Первая стадия процесса — образование гипохлорита — не изменяется от присутствия хлорида калия. Вторая стадия — переход гипохлорита в хлорат (V) и хлорид в присутствии хлорида калия — характеризуется устойчивым пенообразованием. Разрушение пены производят введением на ее поверхность избыточного маточного раствора второй кристаллизации. Образующийся хлорированный раствор направляют на выпарку и ее переработку по описанной выше технологии.
На производство 1т хлората (V) калия по известковому методу расходуют: 2,15—2,17 т извести (100% СаО), 2,2—2,3 т хлора 291
ю*
(100%), 0,9—0,98 т хлорида калия (100% КС1), 32—34 кг мелассы (концентрация 4,6%), 400 кВт-ч электроэнергии, 600—610 м3 воды и 7,0—7,1 мгкал пара. В качестве отхода получается около 3 т СаС12 в виде растворов.
Хлорат (V) калия может быть получен и другими способами, в основе которых лежат хлорирование суспензии Mg(OH)2, СаСОз, ZnO и других оксидов, гидроксидов и карбонатов с дальнейшим обменным разложением полученных хлоратов с КС1 или другими солями калия, например K7SO4, К2СОз.
Хлорная кислота и ее соли. Хлорная кислота НСЮ4— сильно дымящая на воздухе бесцветная летучая жидкость. В жидком HCIO4 молекулы димеризованы за счет водородной связи. В парах мономер-на. Длины связей С1—ОН 0,1635 нм, С1=О 0,1408 нм, О—Н 0,098 нм, углы ОСЮ 112°, НОСЮ 106°. Температура плавления -101° С, температура кипения 106° С (с разложением); плотность 1,7608 г/см3; уравнение температурной зависимости давления пара 1g р (мм рт. ст.) = 8,175—2007/Т; зависимость показателя преломления водных растворов хлорной кислоты НС1О4 от ее концентрации при 20° С приводится на рис. 5.34; С? = 120,5 Дж/(моль-К); ДН0°бр = -40,4 кДж/моль, Дб0°бр = -78,5 кДж/моль; S2°98 = 188,4 Дж/(моль-К); р = = 1,351-Ю2 Ом-см; 8 = 118 (298 К); т] = 0,795-Ю'3 Па-с.
Тетраоксохлорат (VH) водорода хорошо растворим в воде. Водный раствор называется хлорной кислотой:
н2о...нсю4 #он; + сю;
Хлорная кислота является одной из наиболее сильных кислот. При 25° С константа равновесия Аа0,7-106. Известны многочисленные оксохлораты (VII). Кристаллогидрат НСЮд-НгО является перхлоратом оксония [ОНз]СЮ4.
Жидкая HCIO4 частично димеризована. Для нее характерно равновесие автодедигратации:
ЗНСЮ4 Н3О+ + сю; + С12О7
Если пары НСЮ4 сконденсировать ниже 0° С, равновесие устанавливается в течение нескольких часов. Присутствие небольшой равновесной концентрации CI7O7 (-0,16 М) определяет низкую термическую стабильность жидкого тетраоксохлората водорода. В парах, в которых равновесие полностью сдвинуто влево, разложение идет при 200—350° С, а в жидкой фазе — при 57—77° С. Пар над 100%-ной НСЮ4 содержит 11% (мол.) С12О7 и 89% НСЮ4. Продуктами термического разложения хлорной кислоты являются: кислород, 292
Рис. 5.34. Зависимость показателя преломления водных растворов хлорной кислоты НСЮ4 от ее концентрации при 20° С
хлор, диоксид хлора, гексаоксид дихлора и дигидрат тетраоксохлорида водорода. В присутствии ингибиторов (CCI3COOH, C2HCI5, СНС13 и др.) и при разбавлении водой термическая стабильность хлорной кислоты повышается. Разложение хлорной кислоты в парах катализируют оксиды переходных металлов (CuO, F2O3, СГ2О3 и др.).
Хлорная кислота хорошо растворяется в ацетате трифторметана, трихлор- и дихлорметана и других хлорированных углеводородах. Однако совмещение процесса окисления с растворителями, способными окисляться, обычно приводит к воспламенению и взрыву.
Известны восемь гидратов хлорной кислоты (табл. 5.5). Моногидрат H3OTIO4—ионный хлорат (VII) водорода; ДЯ^р = 382,0 кДж/моль; в кристаллической структуре остальных гидратов присутствуют гидратированные протоны ИД, Н7Оз, НдО^. Входящие в состав кристаллогидратов молекулы воды связаны с ионами СЮ^ водородными связями.
Диаграмма плавкости системы вода — хлорная кислота приведена на рис. 5.35. При -25° С моногидрат хлорной кислоты переходит в моноклинную модификацию (пространственная группа Р2]/и). Азеотроп с водой имеет температуру кипения 203° С (0,1 МПа) и содержит 72,4% НСЮ4. Пар над растворами выше этой концентрации обогащен НС1О4, а ниже — водой.
Давление паров безводной хлорной кислоты (в мм рт. ст.) при различных температурах может быть рассчитано по уравнению
. 2279
lg/7 = ----+ 9,1366.
293
Таблица 5.5. Свойства гидратов хлората (VII) водорода
Число молекул воды Сингония Пространственная группа Температура плавления, °C
0,25 — — 73,0*
1,0 Ромбическая Рпта 49,90
2,0 » Рпта -20,6
2,5 Моноклинная Р2}!с -32,1
3,0 Ромбическая РЬса -40,20
3,5 » РЬса -45,60
4,0 — — -57,7*
5,5 Кубическая — -50,4
’Инконгруэнтпо.
Состав пара над растворами хлорной кислоты низких концентраций (ниже 72%) обогащен парами воды, а при высоких концентрациях— оксидом хлора (VII). При концентрации 72,4% хлорная кислота образует с водой азеотропную смесь. На рис. 5.36 приведена зависимость состава пара над водными растворами хлорной кислоты различной концентрации. Отклонение состава пара от состава жидкости в области концентраций, близких к 100%, объясняется наличием равновесия между хлорной кислотой, оксидом хлора (VIJ) и моногидратом хлорной кислоты:
ЗНСЮ4 # С12О7 + НСЮ4 Н2О
Рис. 5.35. Диаграмма плавкости системы вода—хлорная кислота
294
Хлорная кислота является одной из сильнейших неорганических кислот. В ее среде соединения кислотного характера ведут себя как основания, присоединяя протон и образуя катионы ацилперхлоратов, например Р(ОН)4СЮ4, 8е(ОН)зС1О", NO^CIO’. В безводной НСЮд, а также в растворах щелочных хлоратов (VII) и СЬО? возможен синтез хлоратов (VII) большинства металлов в не-сольватированном состоянии.
Концентрированная хлорная кислота — сильнейший окислитель, контакт ее со значительным количеством органических материалов приводит к воспламенению и взрыву. Окислительная активность хлорной кислоты с концентрацией менее 72% значительно ниже, а термическая устойчивость выше, чем у 95 — 100%-ной HCIO4.
Водные растворы тетраоксохлората водорода применяют в аналитической химии для растворения металлов, «влажного сожжения» органических веществ и в качестве стандарта в ацидиметрии, а также в качестве компонента полировальных ванн для металлов.
Способы получения хлорной кислоты. Хлорную кислоту получают хлороводородным способом, заключающимся в вытеснении хлорной кислоты из хлоратов (VII) натрия по реакции
NaCIO4 + НС1 = НС1О4 + NaCl
Образующийся при этом хлорид натрия осаждается. Применяемый в качестве сырья xnopaT(VII) натрия содержит не более 0,5% хлорита (NaClO2).
В процессе взаимодействия хлората (VII) натрия с хлороводородной кислотой с концентрацией 35% НС1 или газообразным хлоридом водорода получают раствор, содержащий ~32% HCIO4, 8% НС1, 6% NaCl и непрореагировавшего хлората (VII) натрия и 54% Н2О. Выделяющийся в осадок хлорид натрия отфильтровывают и возвращают
на процесс электролиза для получения хлората (V) и хлората (VII).
Отфильтрованный раствор подвергают трехстадийной выпарке. В стадии выпарки происходит дораз-ложение исходного хлората (VII) натрия и отгоняется газообразный хлорид водорода, который подвергается абсорбции с получением хлороводородной кислоты. Оставшийся раствор содер
Рис. 5.36. Зависимость состава пара над водными растворами хлорной кислоты от состава жидкой фазы
295
жит около 40% хлората (VII) водорода, 0,8—0,9% хлороводорода, 7,0—7,5% хлорида натрия и 52,5% воды. Плотность раствора 1,395 г/см3. Во второй стадии выпарки отгоняется в основном вода, а оставшийся хлорид натрия реагирует с хлоратом (VII) водорода. Раствор с плотностью 1,58 г/см3 состоит из 57% НС1О4 -0,05% НС1, 10,5% ЫаС1О4 и 32,5% Н2О, который подвергают третьей выпарке при 200—270° С. При этом из раствора выкристаллизовывается NaClO4, возвращаемый на начало процесса, и в системе образуется азеотропная смесь, содержащая до 72,4% НСЮ4 с температурой кипения 203° С. После соответствующей очистки от примесей получают продукт, содержащий около 70—72% НС1О4. Безводный НС1О4 получают перегонкой 72%-ной хлорной кислоты в присутствии 15—20%-ного олеума при 50° С под давлением 1 мм рт. ст.
Существует также способ получения хлорной кислоты путем окисления водных растворов хлоратов (VII) щелочных и щелочноземельных металлов серной кислотой:
КС1О4 + H2SO4 = НС1О4 + KHSO4
Образующуюся при этом хлорную кислоту отгоняют под вакуумом. Способ целесообразен лишь в процессе получения безводного хлората (VII) водорода.
Водные растворы хлорной кислоты получают при реакции хлората (VII) калия с гексафторкремнием водорода по схеме
КС1О4 + HSiF6 = НС1О4 + KSiF6
После отделения образующегося осадка труднорастворимого гекса-фторокремнида калия фильтрованием хлорная кислота может быть концентрирована дальнейшей перегонкой азеотропной 72%-ной кислоты.
Для получения хлорной кислоты может быть использована и реакция хлората (VII) бария с серной кислотой:
Ва(С1О4)2 + H2SO4 = 2НС1О4 + BaSO4
Образующуюся кислоту отделяют от сульфата бария фильтрацией.
Разработан способ получения хлорной кислоты электролизом хлороводородной кислоты. Процесс осуществляется электролизом водного раствора хлора в присутствии хлорной кислоты. При этом на аноде протекают единовременно процессы окисления растворенного в воде хлора, образование хлората (V) водорода с дальнейшим окислением его в хлорат^! I), а на катоде происходит выделение водорода. Суммарный процесс представляется уравнением
С12 + 8Н2О = 2НС1О4 + 7Н2
296
Способ обеспечивает уменьшение расходов исходных материалов и числа стадий процесса, необходимых для образования целевого продукта. Поэтому считается более предпочтительным окислять хлор в растворе хлорной кислоты, чем окислять на аноде хлороводородную кислоту. В последнем варианте имеется дополнительная электрохимическая стадия окисления иона хлора. Кроме того, раствор больше загрязняется хлорноватой кислотой. Образование хлорной кислоты при электролизе хлороводородной кислоты происходит при применении растворов с концентрацией 0,1 н. НС1 при плотности тока на гладком платиновом аноде 4000 А/м2. В этих условиях выход по току в расчете на хлорную кислоту составляет 40 — 45%.
В производстве хлорной кислоты электролитическим окислением хлора применяют 40%-ные растворы НСЮд, которые насыщают при -5° С хлором, концентрация которого составляет 3 г/л. Процесс проводят в электролизере фильтр-прессного типа с диафрагмой из пластмассовой сетки. Рамы электролизера выполнены из пластмассовой сетки (из поливинилхлорида), аноды — из платиновой фольги, а катоды — из серебра. Электролизеры рассчитаны на нагрузку 4,4 В и выход по току 60%. Отбор кислоты производится из катодного пространства. Этим приемом удается электрохимически осадить на катоде некоторое количество платины, переходящее в раствор вследствие коррозии.
На катоде выделяется водород, содержащий до 5—7% хлора. Его отводят из катодного пространства, а из анодного пространства — газообразные продукты окисления, в которых содержится 25—27% хлора.
Кислоту из электролизера отгоняют в обогреваемой паром колонне, в которой она очищается от воды, хлорида водорода, образовавшегося при гидролизе хлора и непрореагировавшего водорода. Целевой продукт содержит 68 — 72% НСЮд.
Хлорат (VII) аммония. Хлорат (VII) аммония NH4CIO4 — бесцветное кристаллическое вещество. Кристаллизуется из водных растворов в виде бесцветных кристаллов орто- и ромбической структуры. Ниже 238° С устойчива ромбическая модификация (а = 0,9225 нм, b = 0,5815 нм, с = 0,7456 нм, z = 4, пространственная группа РптаУ, плотность 1,952 г/см3; С ° = 128,1 Дж/(моль-К), ДЯ^р = -295,2 кДж/моль. Выше 238° С устойчива кубическая форма {а = 0,763 нм, пространственная группа FniSmy плотность 1,732 г/см3 (при 270° С); ДЯ полиморфного перехода 9 кДж/моль. С повышением давления температура перехода растет, достигая 300° С при 0,4 ГПа.
Данные по растворимости хлората(УП) аммония приведены на рис. 5.37. Растворимость хлората (VII) аммония при 25° С (г в 100 г растворителя): в метаноле — 6,85; этаноле — 1,90; ацетоне — 2,26.
297
Растворяется также в расплаве хлората(УП) лития. Образующаяся при этом эвтектика NH4CIO4—LiClO4 имеет температуру плавления 182° С и содержит 32,6% хлората(УП) аммония.
В процессе нагревания выше 150° С хлорат(УП) аммония заметно разлагается, выше 250° С он возгоняется с диссоциацией на аммиак и хлорную кислоту (при 270° С давление диссоциации ~13 Па). В интервале 150—300° С изотермическое разложение хлората (VII) аммония идет автокаталитически и практически прекращается после распада около 20% исходного продукта по схеме
4NH4CIO4 —> 2С12 + 2N2O + ЗО2 + 8Н2О
Хлорат(УП) аммония выше 300° С разлагается до конца. Процесс разложения идет до конца и не автокаталитически:
2NH4CIO4 —РИ?че300°С...> С|2 + 2N0 + 02 + 4f42O
Кубическая модификация немного стабильнее ромбической, вследствие чего в области температур фазового перехода (235—250° С) скорость разложения снижается с повышением температуры. Присутствие паров хлората (УП) водорода ускоряет процесс разложения, а паров
аммиака — замедляет.
Рис. 5.37. Зависимость растворимости хлората (VII) аммония в воде от температуры
Хлорат (VII) аммония применяют в качестве окислителя твердых ракетных топлив и смесевых взрывчатых веществ.
Разработано несколько способов получения хлората (VII) аммония. Наиболее распространенный метод получения хлората (VII) аммония заключается в обменном разложении водных растворов хлората (VII) натрия аммонийными солями различных кислот. Один из вариантов этого способа сводится к обменному разложе-
298
нию водных растворов хлората (VII) натрия хлоридом аммония по реакции
NaClO4 + NH4CI = NH4CIO4 + NaCl
После охлаждения образовавшегося раствора выделяются кристаллы хлората (VII) аммония, а хлорид натрия остается в растворе. При упарке маточных растворов хлорид натрия отделяется в виде кристаллов, которые после промывки от хлората (VII) аммония применяют в производстве хлоратов (VII).
Метод видоизменен путем замены хлорида аммония хлороводородной кислотой и аммиаком. Согласно способу, растворы хлората (VII) обрабатывают хлороводородной кислотой или хлоридом водорода и аммиаком по схеме
NaCIO4 + HCI + NH3 = NH4CIO4 + NaCl
Согласно приведенной на рис. 5.38 схеме производства, из сборника 1 растворы хлората^!!) натрия поступают в реактор 2, в котором очищаются от хроматов и тяжелых металлов обработкой их хлоридом бария и карбоната натрия. После чего в реакторе 3 они очищаются от хлоратов и поступают в реактор 7, в котором происходит основной процесс, а именно реакция образования хро-MaTa(VII) аммония. После охлаждения смеси в кристаллизаторе 5 образующиеся кристаллы xpoMaTa(VII) аммония отжимаются в первой центрифуге 6. Кристаллы направляются в сборник 9 с мешалкой на растворение маточными растворами из второй центрифуги 6. Образующаяся смесь маточных растворов с кристаллами направляется на вторичную кристаллизацию в кристаллизатор 10, после охлаждения в котором кристаллы отжимают во второй центрифуге 6 и направляют на сушку.
Маточные растворы из первой центрифуги 6 упариваются в ваку-ум-выпарном аппарате 7. Образующиеся при этом кристаллы хлорида натрия отжимаются в центрифуге 8.
Разработан способ обменного разложения хлората^!!) натрия с гидрокарбонатом аммония по схеме
NaClO4 + NH4HCO3 = NH4CIO4 + NaHCO3
Выделяемый при этом гидрокарбонат натрия выпускают в виде товарного продукта или перерабатывают в карбонат натрия:
2NaHCO, —1-> Na2CO3 + Н2О + СО2
299
Рис. 5.38. Схема производства хлората (VII) аммония по обменной реакции между хлоратом (VII) натрия, аммиаком и хлороводородной кислотой:
/ — сборник МаСЮд из цеха электролиза; 2, J, 4 — реакторы; 5, 10 — кристаллизаторы; б, 8 — центрифуги; 7—вакуум-выпарной аппарат; 9 — сборник с мешалкой
Вместо гидрокарбоната аммония двойное разложение растворов NaClC>4 можно производить газообразным диоксидом углерода и аммиаком
2МаСЮ4 + 2 NH3 + СО2 + 2Н2О = 2 NH4CIO4 + Na2CO3
После отделения кристаллов хлората(УП) аммония маточные растворы обрабатывают диоксидом углерода. При этом образующийся гидрокарбонат выпадает в осадок.
Предложен способ получения хлората(УП) аммония путем обработки исходных растворов хлората(УИ) натрия фосфорной кислотой и аммиаком:
МаСЮ4 + Н3РО4 + 2NH3 = NH4CIO4 + 2NaNH4HPO4
Хлорат(УП) натрия. Хлорат(У) натрия NaClO4 — бесцветные кристаллы. До 306° С устойчива ромбическая модификация — фаза I (а = 0,7085 нм, D = 0,6526, с = 0,7048, пространственная группа плотность 2,495 г/см3), выше ЗО|5° С — кубическая фаза IV (при 315° С а = 0,708 нм; пространственная группа ОА5); АЯ перехода I<±JV 1,30 кДж/моль. Существуют две фазы хлората (VII) натрия высокого давления — фаза II со структурой типа AgMnO4, устойчивая при давлении 2,3—3,0 ГПа, и фаза III со структурой барита, устойчивая при давлении выше 3,0 ГПа. Температура плавления хлората (VII) натрия 469° С (с разложением); С ° = 110,3 Дж/(моль-К), ДЯ^р = “383,5 кДж/моль, S2°98 = 143,9 Дж/(моль-К).
Вблизи температуры плавления начинается медленный процесс разложения. Процесс полного разложения происходит в интервале 300
490—600° С. Продуктами разложения являются хлорид натрия и кислород. Присутствие окислителей С03О4, МпО2 и других оксидов ускоряет процесс разложения ЫаСЮд, снижая температуру на 150—200° С.
Растворимость хлората (VII) натрия при 25° С (г в 100 г растворителя): метанола — 51,3, этаноле — 14,7, ацетоне — 51,7, этилацетате— 9,65. Данные по растворимости ЫаСЮд в воде приведены на рис. 5.39, из которого видно, что выше 52°С кристаллизуется безводный хло-рат(УП) натрия, а ниже его — моногидрат. Исследованиями установлено, что моногидрат хлората(УП) натрия существует в температурном интервале от 52 до -13°С, а от -13 до -39°С— дигидрат. В
О 10 20 30 40 50 60 70 80 90 100
Рис. 5.39. Зависимость растворимости хлората (VII) натрия в воде
интервале от 0 до 52°С уравнение температурной зависимости давления диссоциации моногидрата Ig р (мм рт. ст.) = 10,51—2906/Г.
Хлорат(УП) натрия образует сольваты КаСЮд-иЬ, где п = 1 (L—N2H4; гексаметилфосфотриамид с температурой плавления 174° С). Сольваты с органическими веществами взрывоопасны.
Температура эвтектики в системе ИаСЮд — ЫСЮд 208° С (27,5% (мол.) хлората(УП) натрия, NaClO4—NaOH 243° С (15% (мол.) хлората натрия). С хлоратами(УП) алюминия, кадмия, цинка и других хло-рат(УП) натрия образует комплексные перхлоратометаллаты, например Na2[Al(C104)5], Na[Zn(CIO4)3], Na[Cd2(CIO4)5].
Хлорат(УП) натрия применяется в качестве сырья в производстве хлората(УП) аммония, является компонентом смесевых взрывчатых
веществ и пиротехнических составов.
Хлорат(УП) натрия получают электрохимическим окислением хлорат(У) иона на аноде. Процесс окисления протекает с образованием хлората(У) и хлората(УП):
2С1О3 + Н2О = НСЮ4 + НСЮз + 2е
301
Одновременно на катоде происходит разряд ионов водорода и образование щелочи, реагирующей с НСЮз и НСЮ4:
2Н2О + 2е = Р2 + 2ОН"
нсю4 + НСЮз + 2ОН" = сю; + сю; + 2Н2о
Суммарный процесс представляется следующей схемой:
сю; + н2о = сю; + н2
Электролиз проводят с применением в качестве анодов платиновых сеток, а в качестве катодов — перфорированных никелевых пластинок.
В качестве катодных материалов могут быть использованы нержавеющая сталь и графит. С целью избежания потерь тока в результате частичного разряда ионов ОН- поддерживают небольшую кислотность электролита в пределах 0,1—0,15 г/л НС1. При избытке же кислоты может произойти разложение хлората (V) СЮ;, а также разряд ионов хлора на аноде.
При концентрации электролита 500—700 г/л ИаСЮз, температуре 40—60° С, анодной плотности тока 3000—7000 А/м2 и катодной 100—2000 А/м2, напряжении 6—10 В, начальный выход по току составляет 95—98%. К концу процесса при концентрации NaClO4, равной 900—1000 г/л, и ЫаСЮз ниже 50 г/л выход по току снижается до 40—50%. В среднем выход по току составляет около 85%.
Установлено, что оптимальной плотностью тока на аноде должны быть 0,4—0,5 А/см2. В процессе проведения электролиза с применением платиновых анодов и стальных катодов для исключения восстановления катодов в электролит добавляют 3—4 г/л Ка2Сг2О2. В этом варианте максимальный выход по току достигается при 30—40° С.
Согласно технологической схеме, приведенной на рис. 5.40, исходные растворы после перемешивания с маточными растворами в сборнике 1 подвергаются электролизу в каскаде электролизеров 2, после чего собираются в сборнике 3 и упариваются в выпарном ап-
Маточник
Рис. 5.40. Принципиальная схема производства хлората (VII) натрия:
/ — смеситель растворов; 2 — каскад перхлоратных электролизеров; 3 — сборник растворов; 4 — выпарной аппарат; 5 — фильтры; 6 — кристаллизатор; 7—центрифуга; 8 — сушилка
302
парате 4. После выпарки растворы фильтруют через фильтры 5 и направляются на кристаллизацию в кристаллизаторы 6. В процессе кристаллизации пользуются данными совместной растворимости хло-para(V) и хлората(УП) натрия (рис. 5.41). Приведенная в ней прямая линия, пересекающая поле рисунка, характеризует изменение концентрации хлората(У) и хлората(УП) в ходе процесса электролиза по степени окисления хлората(У).
Разработан способ с применением в производстве анодов из диоксида свинца, нанесенного электроосаждением на токоподводящую основу из графита. Устранение при этом механических повреждений покрытия и придание ему однородности достигают обработкой эпоксидной смолой, силиконовым каучуком или другими аналогичными материалами. Известно также, что в качестве токоподводящей основы для анодов из диоксида свинца вместо графита можно применять тантал.
В процессе использования анодов из диоксида свинца выход хло-рата(УП) натрия по току несколько ниже (на 5%), чем при работе с платиновыми анодами.
Получение хлората(УП) с применением диоксида свинца в качестве анода проводят при анодной плотности тока 0,15 А/см2 и катодной 0,07 А/см2 при напряжении 4,7—5,7 В. К раствору добавляют
Рис. 5.41. Растворимость в системе NaC103—NaC104—Н2О (R — метастабильная четвертая точка для NaCIO3, NaC104, Н2О и льда)
303
0,5 г/л NaF и не вводят Na2Cr2O7. Участие’фторида натрия позволяет завершить процесс до конечной концентрации NaClCh, которая достигается на платиновых анодах без резкого уменьшения выхода по току, который составляет в среднем 90%.
Хлорат(УП) калия. Хлорат(УП) калия КСЮд кристаллизуется в безводной форме в виде прозрачных бесцветных кристаллов ромбической системы. Пространственная группа низкотемпературной модификации Рпта. Температура полиморфного перехода 298° С. Температура плавления 580° С (с разложением); плотность 2,52 г/см3; С° = = 108,0 Дж/(мольК); Д7/0°бр =-432,42 кДж/моль;. AG0°6p =-303,5 кДж/моль. Растворимость хлората (VII) калия приведена в табл. 5.6.
Таблица 5.6. Растворимость хлората (VII) калия в воде
Температура, °C Плотность, г/см3 Растворимость Температу-ра,°С Плотность, г/см3 Растворимость
% г/100 г Н2О % г/100 г Н2О
0 1,005 0,75 0,76 75 1,036 10,36 11,56
10 — 1,05 1,06 80 — 11,8 13,4
15 1,0076 1,33 1,35 90 — 15,0 17,7
20 1,0085 1,65 1,67 100 1,0681 18,2 22,25
25 1,0096 2,03 2,07 120 — 25,0 33,3
30 — 2,5 2,57 140 — 32,5 48,1
40 — 3,6 3,74 180 — 46,0 85,2
50 1,017 4,9 5,15 200 — 52,5 110,5
60 — 6,8 7,3 225 — 60,0 150,0
70 — 9,2 10,1 250 — 67,0 203,0
265 — 70 233,0
Чистый хлорат (VII) калия свыше 580° С разлагается по схеме
2КСЮ4 -> 2КС1О3 + О2
2КСЮ3 -> 2КС1 + ЗО2
Образующийся в качестве промежуточного продукта хлорат (V) калия, расплавляясь, ускоряет процесс разложения. Реакция разложения ускоряется также в присутствии КС1, KBr, KI, Си, Fe, Со, MgO и др.
Хлорат (V) калия может быть получен электрохимическим способом. Однако из-за низкой растворимости производство его электролизером хлорида или хлората связано с образованием твердой фазы в электролизерах.
Обычно хлорат (VII) калия получают обменным разложением хлората (VII) натрия с хлоридом калия:
NaClO4 + КО = КСЮ4 + NaCl
304
Исходные растворы готовят с содержанием 600 г/л NaC104, фильтруют и направляют в кристаллизатор с параллельной подачей растворов хлорида калия. Температуру процесса поддерживают в зависимости от концентраций исходных реагентов, чтобы хлорат (VII) калия оставался в растворе, а значительная часть образовавшегося хлорида выпадала в осадок. Процесс ведут, пользуясь данными табл. 5.7.
Таблица 5.7. Совместная растворимость КСЮ4 и NaCl
Плотность, г/см Концентрация, г/100 г Н2О Твердая фаза Плотность, г/см Концентрация, г/100 г Н2О Твердая фаза
КСЮ4 NaCl КСЮ4 | | NaCl
При о°с При 75° С
1,005 0,76 0 КС1О4 1,036 11,52 0 КС1О4
1,214 1,01 35,86 КС1О4 + + NaCl 1,207 9,32 41,47 КС1О4 + + NaCl
1,209 0 35,9 NaCl 1,176 0 37,9 NaCl
При 25° С При 100° С
1,013 2,07 0 КС1О4 1,068 22,2 0
1,207 2,22 35.73 КС1О4 + + NaCl 1,116 19,75 10,44 КС1О4
1,198 0 36,3 NaCl 1,153 17,6 20,75 ►
При 50° С 1,19 15,92 30,81
1,017 4,95 0 КС1О4 1,216 14,51 38,37 КС1О4 + + NaCl
1,205 4,5 36,52 КС1О4 + + NaCl 1,201 11,99 62,99' NaCl
1,185 0 36,8 NaCl 1,183 5,1 38,87
1,164 0 40,00 NaCl
После отделения NaCl растворы охлаждают и отфильтровывают кристаллы хлората (VII) калия. Далее кристаллы сушат и выпускают в виде товарного продукта. Выход целевого продукта составляет 95%. Товарный продукт содержит до 99,5% КС1О4 и не более 0,01—0,04% влаги.
ГЛАВА 6
БРОМ И ЕГО СОЕДИНЕНИЯ
6.1. БРОМ
Содержание брома в земной коре составляет 1,6-10"4% (масс.) (1015—1016т). Бромсодержащими минералами являются: бромаргирит AgBr и эмболит Ag(Cl,Br). Однако и они в настоящее время практически исчерпаны. Бром в природе находится в рассеянном состоянии. Соединения брома содержатся в морской воде (0,065%, масс.), рассолах соляных озер (до 0,2%) и в подземных рассолах нефтеносных районов (до 0,1%). Бор содержится также в минерале галите NaCl (0,005—0,03%, масс.), сильвине KCI (0,02—0,1%), карналлите КС1 • MgCl2 • 6Н2О (0,1 —0,39%), бишофите MgCI2 • 6Н2О (до 0,6%).
Свойства. Бром имеет атомную массу 79,904. Состоит из двух стабильных изотопов: 79Вг (50,56%) и 81 Вг (49,44%). Конфигурация внешней электронной оболочки 4s24/?5; степени окисления -1, +1, +3, +5 и +7; энергия ионизации при последовательном переходе от Вг° до Вг7+ соответственно 11,88, 21,80, 35,90, 47,3, 59,7, 88,6, 109,0, 192,8 эВ; атомный радиус 0,119 нм, ионные радиусы Вг(6), Вг3+(4), Вг5+(3), Вг7+(6), Вг7+(4) соответственно 0,182, 0,073, 0,045, 0,053, 0,039 нм.
Молекула брома двухатомна, длина связи в молекуле 0,228 нм; энергия диссоциации 190,0 кДж/моль, степень диссоциации 0,16% при 800° С и 18,3% при 1284° С. В парах брома обнаружено существование молекулы Вг4.
Бром Вг2 — тяжелая красновато-бурая жидкость с резким запахом, образующая желто-бурые пары. Температура пларления -7,25° С, а кипения—58,78° С. Плотность жидкого брома 3,187(1-0,001086 t) г/см3.
Зависимость плотности и вязкости жидкого брома от температуры характеризуется следующими данными:
/, °C.................. О 10 20 25 30 40 50 58,78
Плотность, г/см3.... 3,187 — 3,1226 3,1055 3,0879 — — 3,009
Вязкость! О'3, Па с. 1,253 1,107 0,991 0,942 0,897 0,817 0,746 0,693
306
Зависимость вязкости (в Па-с) от температуры (Г, ° С) описывается следующим уравнением:
1,241 10"3
~ 1 + 0,01225/+2,721-КГ6/2’
Уравнение температурной зависимости давления пара над жидким бромом:
1g/?(Па) = -2047,75/7-0,0061007+ 0,9589 IgT + 10,76568;
С;? = 75,69 Дж/(моль-К); АЯП°Л = 10,58 кДж/моль, ДЯи°сп = 30,86 кДж/моль; 52°98 = 152,0. Плотность жидкого брома 3,1055 г/см3.
При температуре -7,2° С бром образует красно-коричневые кристаллы со слабым металлическим блеском, которые при температуре -252° С становятся бесцветными. Кристаллизуется в ромбической системе (а = 0,448 нм, b = 0,667 нм, с = 0,872 нм, z - 4, пространственная группа Сета). Плотность твердого брома 4,073 г/см3 (-7,3° С).
В табл. 6.1 приведены значения равновесного давления паров брома при различных температурах.
Таблица 6.1. Равновесное давление паров брома
/, °с | р, кПа /, °C | р. кПа | 1 °C | А кПа 1 °с | р, МПа
Твердое состояние Жидкое состояние
-80 0,0034 0 8,76 40 28,8 110,3 0,981
-60 0,0475 И 10,48 50 32,4 139,8 1,962
-40 0,396 20 14,7 58,78 98,1 174,0 3,925
-20 2,280 25 18,6 216,0 0,196
-7,3 5,900 30 23,2 243,5 0,490
Во всех фазах — газовой, жидкой и твердой — молекулы брома двухатомны. Диссоциация молекул брома происходит лишь при высоких температурах. Так, при 727° С бром диссоциирует на 0,57%, а диссоциирует полностью лишь при температуре выше 2000° С. Процесс диссоциации брома на атомы ускоряется под действием света и электрического тока.
Бром растворяется в воде в пределах 3,58 г в 100 г при 20° С (рис. 6.1). В присутствии же хлоридов, особенно бромидов, растворимость брома повышается, а в присутствии сульфатов — понижается. При температуре ниже 5,84° С из водных растворов осаждаются гранатово-красные кристаллы октагидрата Вг2 • 8Н2О. Растворимость 307
же воды в жидком броме составляет 0,05% (масс.). В водных растворах бром частично гидролизуется:
Вг2 + Н2О НВгО + Н+ + ВГ
константа гидролиза брома:
[НВгО][ГГ][Вг~] [Вг2]
при 0° С равна 0,7 • 10‘9 , при 20° С —5,2 • 10‘9 и при 35° С— 11,3 • 10‘9 Насыщенный раствор брома в воде называется бромной водой. По своей реакционной способности бром занимает промежуточное положение между хлором и иодом. С другими галогенами образует неустойчивые, но отличающиеся высокой химической активностью BrF3 (температура плавления 8,8° С, кипения 127° С), BrF5 (температура плавления -61,3° С, кипения -40,5° С), ВгО (температура плавления — около -66° С, кипения — около -5° С с разложением) и IBr (температура плавления 41° С, а кипения 116° С). С кислородом и азотом бром непосредственно не реагирует даже при повышенных температурах, а нестойкие кислородные и азотсодержащие Вг2О, ВгО2> Br3C\ NBr • 6NH3 получают косвенными методами. Бром не реагирует также с углеродом.
Бром — сильный окислитель. Однако при действии более сильных окислителей на бромид выделяется элементный бром:
Вг + С12 1т Br2 + 2С1
Рис. 6.1. Растворимость брома в воде
308
В избытке же галогена реакция идет дальше:
Вг2 + С12 2ВгС1
С солями кислородных кислот галогенов направление реакций галогенов и галогенидов зависит от pH среды. В кислой среде реакция с галогенидами протекает с образованием элементных галогенов:
Вг" + ВгО" + 2Н* = Вг2 + Н2О
5Вг” + ВЮ3’ + 6Н+ = ЗВг2 + ЗН2О
В нейтральной и щелочной среде образуются кислородные соединения брома:
Br + СЮ" BrO“ + С1"
Br + ЗСЮ- ВгОз“ + ЗС1-
В водной суспензии элементная сера окисляется бромом:
S + 4Н2О + 3Br2 = H2SO4 + 6НВг
Бром в водных растворах окисляет сульфиты и тиосульфаты до сульфатов:
SO*" + Вг2 + Н2О = SO*’ + 2Н+ + 2Вг
S2O*’ + 4Br + 5Н2О = 2HSO; + 8Н+ + 8Вг
В нейтральной среде нитриты окисляются бромом до нитратов:
NO" + Br2 + Н2О = NO’ + 2ВГ+ 2Н+
Концентрированная азотная кислота окисляет бромиды до нитро-зилбромида:
2HNO3 + 6Вг + 6Н+ = 2NOBr + 2Вг2 + 4Н2О
Аммиак при взаимодействии с бромом образует азот:
2NH3 + 3Br2 = N2 + 6Н+ + 6ВГ
Гидразин окисляется бромом до азота:
N2H4 + 2Вг2 = N2 + 4Вг + 4Н+
Фосфиты и арсенаты (III) окисляются бромом до фосфатов и арсенатов (V). Соли муравьиной и щавелевой кислот окисляются бромом до диоксида углерода:
НСОО- + Вг2 = СО2 + Н+ + 2Вг
С,©*’ + Вг2 = СО2 + 2Вг~
Оксид углерода взаимодействует с бромом:
СО + Вг2 = СОВг2
309
При действии оксида марганца на растворы бромидов выделяются свободные галогены:
МпО" + 2Вг + 4Н+ = Вг2 + Мп2+ + 2Н2О
а при избытке окислителя и в присутствии концентрированной хлороводородной кислоты образуется ВгС1:
2MnO; + 5Br + 5С1- + 16Н+ = 5ВгС1 + 2Мп2+ + 8Н2О
Бром окисляет также и соли двузамещенного железа:
2Fe2+ + Br2 = 2Fe3+ + 2Вг
Гексаферроцианиды (II) окисляются бромом до гексаферроцианидов (III):
2[Fe(CN)6]4- + Br2 = 2[Fe(CN)6]3‘ + 2Вг
Бром образует ряд кислородсодержащих кислот.
В процессе растворения брома в воде происходит реакция
Вг2 + Н2О НВгО + НВг
Образующаяся бромноватистая кислота НВгО обладает слабыми кислотными свойствами, но является сильным окислителем. Она существует лишь в водных растворах, под действием света разлагается на бромид водорода и кислород, является слабой кислотой (p/Ca = 8,69); при стоянии диспропорционирует.
Соли бромистой кислоты — гипобромиты — устойчивы в щелочных растворах; при рН< 9 и в процессе нагревания переходят в броматы; в щелочной среде являются сильными окислителями. Получают гипобромиты взаимодействием элементного брома со щелочами на холоду:
Br2 + 2NaOH = 2NaBrO + Н2О
Гипобромит натрия получают в виде пента- и октагидратов, а калия — в виде тригидрата (КВгО • ЗН2О); желтые кристаллы; разлагаются около 0° С; применяются в аналитической химии в качестве реагента для определения аммиака и аминов.
Соли НВгО2 (бромиты) — кристаллы; устойчивы при обычных условиях; при нагревании разлагаются; в нейтральной среде — слабые окислители. Получают взаимодействием брома со щелочами при строго контролируемых значениях pH, температуре и концентрации; ЫаВгО2 • ЗН2О — желтые кристаллы; обезвоживаются в вакууме при 310
200° С; выше 200° С разлагаются на NaBr и О2; применяются для удаления крахмала из тканей.
Соли бромноватой кислоты (р/Са = 0,7) — (броматы) триоксобро-маты(У) — бесцветные кристаллы; при нагревании разлагаются по схеме М(ВгОз)2 МО + Вг2 + 2,502. Применяются в броматометрии. Ва(ВгОз)2 кристаллизуется из растворов в виде дигидрата; растворяется в воде (0,657 г безводной соли в 100 г при 20° С); получают обменной реакцией броматов щелочных металлов с хлоридом бария:
2КВгОз + ВаС12 = Ва(ВгО3)2 + 2КС1
Применяется для получения бромноватой кислоты.
Бромная кислота НВгОд, тетраоксобромат (VII) в свободном состоянии не выделена. Получены его водные растворы; сильный окислитель. Соли (перброматы) — кристаллы, стабильные в обычных условиях. Выше 250° С разлагаются по схеме
2КВгО4 -> 2КВгО3 4- О2
хорошо растворяются в воде; средние окислители; перброматы получают окислением броматов натрия и калия, окислением броматфто-ром в щелочной среде.
Оксиды брома получают косвенными методами, так как они при обычных условиях не реагируют с кислородом.
Оксид брома Вг2О8, образующийся при действии озона на бром, представляет собой бесцветные кристаллы, устойчивые лишь при низких температурах или в избытке озона. Твердый коричневый оксид брома ВЮ2, устойчив лишь ниже -40° С, получают действием тихого электрического разряда на охлажденную смесь брома и кислорода. Медленное нагревание В1О2 в вакууме выше -40° С приводит к образованию оксида Вг2О бурого цвета.
Известны и неустойчивые соединения брома с галогенами, получающиеся при непосредственном взаимодействии элементов. Наиболее важным из них является трифторид брома ВгРз (светло-желтая жидкость с температурой плавления 8,8° С, кипения 127° С) и пятифторид брома BrF5 (бесцветная жидкость с температурой плавления -61,3° С, кипения 40,5° С), отличающиеся весьма высокой химической активностью. Нагревая в запаянной трубке смеси серы с бромом, получают бромид серы S2Br2 — гранатово-красная, маслянистая жидкость с температурой плавления -40° С, кипения 54° С. Бромид серы растворим в тетрахлориде углерода, дисульфида углерода, бензоле; разлагается водой:
2S2Br2 + 2Н2О = SO2 + 3S + 4НВг
311
Известен также бромид тионила SOBr2, получаемый действием диоксида серы на бром в присутствии РС1з или при взаимодействии SOCI2 и НВг:
SOC12 + 2HBr = SOBr2 + 2НС1
Применение. Элементный бром применяется в качестве исходного сырья для получения бромсодержащих неорганических и органических соединений. Технический бром производят трех сортов, содержащих не менее 99,2—99,9% брома. Существующий ГОСТ 454—70 регламентирует содержание в нем примесей хлора, сульфатов, влаги, органических и нелетучих веществ. Жидкий бром транспортируют в стальных баллонах емкостью 40—320 л, футерованных свинцом. Тарой для малых количеств брома служат толстостенные стеклянные бутыли с притертыми пробками, защищенные от света.
Способы получения брома. Основными источниками для получения брома являются рассолы соляных озер и буровые воды нефтеносных районов, содержащие 0,2—3,5 кг брома в 1 м3, а также маточные растворы, образующиеся в процессе переработки карналлита и сильвинита, содержащие 0,6—45 кг брома в 1 м3. Растворы кроме бромидов содержат: СГ, SO4", НСО3, COj-, Na+ , КГ, Са2+, Mg2+, Sr2+, а также Г, В4О?~, HS", S2", NH4+, Fe2+, Мп2+ и органические вещества. Основные затруднения в производстве создают присутствующие в исходных рассолах HS“, S2-, NH4+, Fe2+, Мп2+ и органические вещества. Они взаимодействуют с хлором, вводимым для окисления бромидов. Органические вещества затрудняют извлечение брома, снижают выход целевого продукта и загрязняют его образующимися хлор- и броморганическими веществами.
Применяемые в химической промышленности способы получения брома из природных вод основаны на окислении бромидов до элементного брома с дальнейшим выделением его из системы. Окисление бромидов в основном производят хлором. Извлечение же элементного брома из системы осуществляют отгонкой паром; десорбцией воздухом или инертным газом. Разработаны способы экстракции несмешиваю-щимися водой растворителями, а также осаждения нерастворимых соединений и адсорбции.
Отгонка элементного брома с водяным паром непосредственно из рассолов рентабельна лишь при более высоких концентрациях целевого продукта в рассоле (выше 10 г/см3). При меньших концентрациях брома в рассолах повышается себестоимость продукта из-за резкого повышения расхода пара.
Способ воздушной десорбции обеспечивает получение брома из рассолов с невысоким его содержанием. При этом необходимы дополнительные расходы реагентов на улавливание брома из бромовоздушной смеси, 312
а также дополнительная производственная 1ерация — переработка полученного полуфабриката на элементный бром.
Получение брома из рассолов отгонкой паром. Схема получения брома из рассолов паровым способом приведена на рис. 6.2. Исходный бромсодержащий рассол в теплообменнике 4 нагревается теплом маточных растворов рассола до 80—90° С, поступает в верхнюю часть колонны 7. Колонная система Кубиерского представляет собой цилиндр высотой 6 м, сложенный из толстых (18—20 см) гранитных, андезитовых или бештаунитовых плит с сечением 1,5x0,8 м. Внутри она разделена горизонтальными перегородками на 7—8 камер. Для сообщения камер между собой в перегород
Рис. 6.2. Схема получения брома паровым способом:
/ — колонна паровой отгонки; 2 — конденсатор; 3 — холодильник; 4 — теплообменник; 5 — рафинер; 6 — испаритель; 7 — холодильник
ках имеются отверстия, перекрытые керамическими колпачками, образующими гидрозатворы. Для прохода газа в двух отверстиях каждой перегородки
установлены широкие керамические трубы. В камерах горизонтально
уложены керамические плиты с отверстиями, служащими насадкой.
Газообразный хлор подают в третью камеру снизу, а в нижнюю— водяной пар давлением 1,5—2 атм для нагрева рассола до
106—120° С. Пар поступает по тому же трубопроводу, что и хлор. В
результате смешения хлор растворяется в рассоле, вытесняет из его солей бром (рис. 6.3), переходящий в газовую фазу.
В колонне хлорирования происходят сложные химические процессы. Суммарная реакция окисления бромида хлоридом описывается уравнением
2Вг + С12 Вг2 + 2СГ
(1)
Образующийся элементный бром реагирует с хлором:
Вг2 + Cl2 2ВгС1
(2)
313
Реакция (1) протекает в две стадии:
Вг + С12 *5 BrCl + СГ (3)
Br+ BrCl Br2 + CP (4)
Бром, хлорид брома и хлор реагируют с ионами СГ:
Вг2 + СГ Вг2СГ (5)
BrCl + СГ <5: BrCl; (6)
С12 + СГ СЦ (7)
Бром реагирует также с ионами Вг:
Вг2 + Вг Вг3~ (8)
В процессе взаимодействия свободных галогенов с водой возможны также реакции гидролиза:
Вг2 + Н2О «7 Н+ + Вг + НВгО
BrCl + Н2О Н+ + СГ + НВгО С12 + Н2О Н+ + СГ + НСЮ
В результате процесса взаимодействия хлорида с бромидом, со
держащимся в исходном рассоле, получается система, содержащая Br2, BrCl, С12, Вг2СГ, BrCl’, Вг;, С1“, НВгО и НСЮ.
Кроме того, содержащие-
Экв. хлора/ экв. брома
Рис. 6.3. Зависимость степени окисления бром-иона от дозировки хлора (эквиваленты) при рН<4
ся в исходном рассоле ионы НСО3 и СО,- приводят в процессе хлорирования (около 100° С) к образованию хлоратов:
ЗСО3’ + ЗС12 =
= СЮ; + 5СГ + ЗСО2
бнсо; + зс12 =
= сю; + 5сг + бсо2 + зн2о
Образующиеся хлораты при высоких значениях pH
314
не являются окислителями, и израсходованный на их образование хлор теряется. Для снижения расхода хлора исходные рассолы предварительно подкисляют до pH 2,5—6,0 серной (возможно, и хлороводородной) кислотой, вводя ее в рассолопровод после подогревателей или в поглотительную колонну (рис. 6.4).
При подкислении рассолов содержащийся в них сульфид водорода окисляется хлором:
H2S + 4СЬ + 4Н2О = H2SO4 + 8НС1
Окисляются также карбонаты и гидрокарбонаты по схеме
2МН4НСОз + ЗС12 = 6НС1 + N2 + 2СО2 + 2Н2О
Кроме того, хлороводородная кислота образуется в результате разложения хлорноватистой кислоты каталитическим действием присутствующего в рассоле железа:
2НС1О 2НС1 + О2
Выходящую из верхней части колонны смесь паров брома, водяного пара и избыточного хлора направляют в конденсатор 2 (см. рис. 17.2). Сконденсировавшийся бром и бромная вода самотеком поступают в водяной холодильник 3, где бром, имеющий большую плотность, собирается на дне и непрерывно отводится из нижней части холодильника 3 в рафинер 5 для очистки от хлора.
Бромная вода из верхней части колонну паровой отгонки 7.
В рафинере 5 производится ректификация брома-сырца от избыточного хлора. Отогнанный хлор, содержащий некоторое количество паров брома, направляют в головную камеру, отделенную от колонны гидравлическим затвором, или же в специальную колонну, где они смешиваются поступающим рассолом. Туда же направляют и газы, не сконденсировавшиеся в холодильнике.
Отработанный рассол, содержащий свободные галогены, обезвреживают, пропуская че-
Рис. 6.4. Зависимость содержания хлора в бромовоздушной смеси (в % к сумме Вг2+С12) от степени окисления бром-иона и pH рассола:
/ — pH 6; 2 —pH 5; 3 — рН<4
315
рез слой железных стружек или же добавляя растворы диоксида серы или тиосульфата натрия для связывания свободных галогенов, рассол нейтрализуют карбонатом натрия или оксидом кальция. Нейтрализованный рассол (маточные растворы иодо-бромного производства) проходит через теплообменники, где его теплота используется для подогрева свежего раствора.
Получение брома из рассолов воздушно-десорбционным способом. Воздушно-десорбционный способ применяют в основном для извлечения брома из рассолов (рапа соленого озера, буровая вода или маточные растворы производства калийных солей), содержащих от 0,065 до 1 кг/м3 брома, и более концентрированных рассолов (рис. 6.5).
Исходный рассол из сборника 1 центробежным насосом 2 по гуммированному трубопроводу подается на верх десорбера б. Одновременно в этот же трубопровод подается из фильтра 3 хлор и из напорной емкости 4 дозатором 5 серная кислота. Рассол непрерывно стекает по насадке десорбера 6 навстречу потоку воздуха, засасываемого из атмосферы вентилятором 75, установленным в конце системы. Освобожденный от брома рассол из нижней части десорбера поступает в нейтрализатор 7, в котором обрабатывается тиосульфатом натрия и известковым молоком и перекачивается в хранилище.
Бромовоздушную смесь, образующуюся в десорбере б, направляют в хлороочистительную колонну S, орошаемую сверху неконцент-
Рис. 6.5. Схема получения брома воздушно-десорбционным способом:
/ — сборник; 2, 10, 12 — центробежный насос; 3 — фильтр; 4—напорная емкость; 5 — дозатор; б — десорбер; 7 — нейтрализатор; 8—хлороочистительная колонна; 9 — емкость для хлороочистительного раствора; 11 — абсорбер; 13 — отстойник; 14 — восстановитель; 15 — вентилятор
316
рированным раствором бромида железа (III). Содержащийся в бромовоздушной смеси хлор взаимодействует с бромидом орошаемого раствора по реакции
2FeBr3 + ЗС12 = 2FeCl3 + ЗВг2
Очищенная от хлора бромовоздушная смесь направляется в абсорбер 11, в котором бром поглощается раствором бромида железа (II):
2FeBr2 + Br2 = 2FeBr3
Образующийся раствор бромида железа (III) направляют на восстановление железными стружками по схеме
2FeBr3 + Fe = 3FeBr2
Часть восстановленного раствора направляют на поглощение брома и на хлороочистку в колонну 8, а оставшуюся часть используют на переработку и получение элементного брома и бромидов. При необходимости раствор бромида железа упаривают до состояния его плава.
Отработанный воздух после абсорбера 11 при необходимости возвращают в десорбционную башню 6. Это диктуется необходимостью охлаждения рассола в зимний период работы, а также исключения выброса в атмосферу абгазов, содержащих свободные галогены.
Отработанный рассол обрабатывают раствором тиосульфата натрия для связывания свободных галогенов, а также известковым молоком для нейтрализации остаточной кислоты.
Получение элементного брома из концентрированных рассолов. Согласно существующей технологии, бромсодержащим сырьем является бромид железа (II), представляющий собой кристаллы зеленого цвета, образующий кристаллогидраты в гексагональной системе (рис. 6.6). Кристаллы имеют плотность 4,64 г/см3. Безводный продукт плавится при 684° С. Насыщенный раствор бромида железа (II) кипит при 132° С.
Для выделения элементного брома в качестве окислителя применяют хлор.
При непрерывном способе получения брома исходные водные растворы бромида железа (II) из сборника поступают в подогреватель, обогреваемый глухим паром, откуда передаются сверху реакционной колонны в среднюю часть, в которую подается хлор, а снизу колонны — пар. Образующиеся по реакции 2FeBr2 + ЗС12 = 2FeCl3 + 2Вг2 пары брома выходят из верхней части колонны, охлаждаются в конденсаторе-холодильнике и после охлаждения поступают в бромоотделитель, после которого жидкий бром направляется в рафинер, где обрабатывается 317
FeBr2, % (масс.)
Рис. 6.6. Диаграмма растворимости бромида железа (II) в воде
при 60° С раствором бромида железа (III). В процессе обработки брома в рафинере бромидом железа (III) содержащийся в броме остаток хлора реагирует с последним по схеме
ЗС12 + 2FeBr3 = 2FeCl3 + 3Br2
Отработанный раствор из рафинера смешивается с раствором бромида железа (II), поступающим в колонну. Раствор бромида железа (II) для предотвращения выпадения гидроксида железа подкисляют хлороводородной кислотой.
Бром из рафинера поступает в холодильник, оттуда — в сборник жидкого брома. Из сборника бром передается в осушитель, в котором осушивается серной кислотой, поступающей из сборника-мерника. После осушки серной кислотой бром направляется в сборник осушенного брома, а далее в контейнер. Отработанная серная кислота собирается в сборник отработанной кислоты, а далее отправляется потребителям.
Способы поглощения брома из бромовоздушной смеси. Для улавливания брома из бромовоздушной смеси применяют химические поглотители. Полученные в результате полупродукты перерабатывают в жидкий бром или его соединения.
Поглотители брома делятся на две группы: щелочные и восстанавливающие.
К щелочным восстановителям относятся растворы гидроксидов и карбонатов натрия, калия и гидроксид кальция. Согласно реакции брома, с ними образуются смеси бромидов и кислородных соединений брома.
К восстанавливающим поглотителям относятся элементное железо, растворы бромида железа, диоксид серы и его растворы, растворы аммиака, карбамида, уротропина- в растворах гидроксидов, карбонатов и известковом молоке и др. Продуктами взаимодействия брома с восстанавливающими поглотителями являются водные растворы бромидов или бромоводородной кислоты.
Разработан также процесс поглощения брома концентрированными растворами бромидов при 15—20° С. Такие растворы хорошо поглощают элементный бром с образованием ионов Вг“. В процессе 318
нагревания раствора до 110° С бром отгоняется и конденсируется. Способ осуществлен в процессе получения брома из маточника (рапа Мертвого моря), содержащего 1—1,2% MgBr2.
Поглощение брома растворами бромида железа. Процесс поглощения элементного брома из бромовоздушной смеси растворами бромида железа (II) основан на реакции
2FeBr2 + Вг2 = 2FeBr3
Далее образующийся бромид железа (III) восстанавливают порошком элементного железа:
2FeBr3 + Fe = 3FeBr2
Процесс поглощения брома проводят в аппаратах, насаженных кольцами Рашига 50x50 мм высотой слоя 3—4 м. Исходные растворы бромида железа (II) имеют концентрацию брома 200—400 кг/м3. Раствор циркулируют многократно с плотностью орошения 1,5 — 3 м/ч до 80—95%-ного перехода исходного бромида железа (II) в бромид железа (III). С достижением концентрации брома 400—550 кг/м3 отработанный раствор выводят из системы.
Исследования кинетики процесса абсорбции брома в газовой фазе показали, что скорость процесса поглощения зависит от концентрации брома и поглотителя и описывается уравнением хемосорбции:
G = KF т(Рг — гс\ где G — масса поглощенного вещества, кг; F—поглощающая поверхность, м2; т — время, ч; Рг — концентрация (парциональное давление) поглощаемого вещества в газовой фазе, Па; К и г — константы; с — концентрация поглотителя.
В процессе поглощения брома растворами бромида железа (II) в результате теплоты реакции (84,4 кДж/моль) несколько повышается температура раствора, в результате чего затрудняется процесс поглощения влаги из воздуха и образуются высококонцентрированные растворы бромида железа (III).
В образующихся растворах Fe3+ восстанавливают до Fe2+ порошком элементного железа, для чего растворы подогревают до 30—35° С, затем за счет теплоты реакции (63,6 кДж/моль FeBr2) температура их повышается до 50—60° С. Для проведения реакции берут 5—7-кратный избыток порошка. Процесс продолжается 3—5 ч, в течение которых 97—99% Fe3+ переходит в Fe2+.
Поглощение брома проводят растворами, содержащими 700—750 кг/м3 FeBr2. Образующиеся растворы подвергают кристаллизации FeBr2-6H2O. Полученные кристаллы отделяют от маточных растворов 319
путем центрифугирования. Образующиеся маточные растворы возвращают на поглощение брома.
Поглощение брома растворами гидроксидов и карбонатов щелочных металлов. Процесс поглощения брома проводят растворами гидроксида натрия по реакциям:
2NaOH + Вг2 = NaBrO + NaBr + Н2О
6NaOH + 3Br2 = NaBrO3 + 5NaBr + ЗН2О
Последняя реакция протекает при более высоких температурах.
При поглощении брома растворами карбоната натрия при комнатной температуре протекают следующие реакции:
2Na2CO3 + Br2 + Н2О = NaBr + NaBrO + 2NaHCO3
2NaHCO3 + Br2 = NaBr + NaBrO3 + H2O + CO2
а при повышенных температурах—
6Na2CO3 + 3Br2 + 3H2O = 5NaBr + NaBrO3 + 6NaHCO3
6NaHCO3 + 3Br2 = 5NaBr + NaBrO3 + 3H2O + 6CO2
Приведенные уравнения дают представление только о конечных продуктах процесса. Образующийся диоксид углерода повышает концентрацию водородных ионов в растворе. В этих условиях бром реагирует не полностью, так как устанавливается равновесие:
ВгО" + Н+ НВгО
НВгО" + Вг + Н+ Вг2 + Н2О
Смещению равновесия в системе способствует также образование трибромид-иона Вг3". Чтобы продолжить процесс поглощения брома, необходимо исключить JiBrO из системы, превращая гипобромит в бромат. Оптимальные уЯовия для этого превращения: pH 9—11 и повышенная температура? .
В процессе поглощения брома концентрированными растворами гидроксидов щелочных металлов возможно выпадение броматов .в твердую фазу. Пользуясь диаграммами растворимости в системах NaBr—NaBrO3—Н2О (рис. 6.7) и КВг—КВгО3—Н2О (рис. 6.8), мож-320
но определить предельно допустимые концентрации исходного гидроксида, при которых броматы в твердую фазу не выпадают.
В процессе поглощения брома концентрированными растворами карбоната натрия при 60—70° С возможно выпадение гидрокарбоната в твердую фазу.
Предельную концентрацию раствора карбоната натрия, из которого в процессе поглощения брома не будет выкристаллизовываться гидрокарбонат, можно рассчитать, пользуясь диаграммой растворимости в системе NaBr— —NaBrCh—NaHCO3—Н2О при 25 и 35° С (рис. 6.9). Как видно из диаграммы, при 25° С эта концентрация равна 8,3%. В действительности же эта концентрация раствора карбоната натрия будет еще ниже, поскольку гидрокарбонат натрия может образовываться также при поглощении диоксида углерода из воздуха. При 70° С гидрокарбонат не выпадает в осадок, поскольку образующийся диоксид углерода моментально выделяется в газовую фазу.
Исследованиями установлено, что скорость процесса абсорбции брома растворами гидроксида и карбоната натрия определяется сопротивлением газового пограничного слоя и пропорциональна концентрации брома в газо-
11 Химическая технология неорганических веществ, кн. 2
Рис. 6.7. Диаграмма растворимости в системе NaBr—NaВгО,—П2О при температуре:
/ -10; 2 — 25; 3 — 45. -I - 80° С
Рис. 6.8. Диаграмма растворимости в системе КВг—КВгО?—1ЬО при 25° С
Рис. 6.9. Диаграмма растворимости в системе NaBr—NaBrCb—NaHCO?—1ЬО при 25 и 35° С
321
вой фазе. Она не зависит также от концентрации поглотителя. Скорость процесса абсорбции брома растворами карбоната натрия имеет максимальное значение при 50° С и не зависит от концентрации.
Поглощение брома известковым молоком. Поглощение брома известковым молоком отличается от поглощения растворами гидроксида и карбоната щелочных металлов тем, что поглотителем является суспензия. Растворимость гидроксида кальция составляет 0,165 г на 100 г воды. В процессе поглощения брома известковым молоком количество необходимого в растворе гидроксида кальция по мере его расхода постоянно пополняется в результате растворения твердого гидроксида. Процесс растворения проходит с большей скоростью, поскольку поверхность растворяющихся частиц очень велика (~103 м2 на 1 м3 суспензии) и во много раз больше поверхности раздела фаз, на которой происходит процесс поглощения. Вследствие этого известковое молоко имеет свойства гомогенного поглотителя с постоянной концентрацией ионов ОН" в жидкой фазе.
В случае накопления бромида кальция в получаемых растворах возможно выпадение в твердую фазу оксибромидов кальция состава СаВг2-ЗСаО16Н2О и др. Для избежания этого процесса пользуются разбавленными растворами (около 5% известкового молока). Поглощение проводят в колоннах с барботажными полками.
Поглощение бройа щелочными растворами восстановителей. Поглощение брома из бромовоздушной смеси щелочными растворами восстановителей обеспечивает получение бромидов, минуя стадию вторичного получения элементного брома из полуфабрикатов. Поскольку существующие технические требования к бромидам предусматривают минимальное содержание в них хлора, необходима эффективная очистка бромовоздушной смеси от хлора.
Согласно существующей технологии, для улавливания брома применяют щелочные растворы аммиака. Концентрацию аммиака поддерживают минимальной (-0,05 кг/м3). Концентрация бромида в поглощающем растворе составляет 500—520 кг/м3. Многократную циркуляцию бромсодержащих растворов производят в абсорбере. В циркулирующий раствор бромида непрерывно подают расчетное количество растворов щелочи и аммиака на соответствующее количество поступающего брома. Образующийся бромид по мере накопления выводится из системы, а для устранения этого недостатка поглощение брома ведут щелочным раствором, к которому добавляют аммиак для разложения гипобромита.
При высокой щелочности растворы поглощают из воздуха значительное количество диоксида углерода, что приводит к накоплению карбоната и гидрокарбоната в растворе. Обычно растворы содержат до 16 322
кг/м3 карбоната натрия и 3—4 кг/м3 гидрокарбоната. Растворы очищают от них, обрабатывая перед процессом упарки жидким бромом.
При поглощении брома смесью растворов карбоната натрия с аммиаком ступенчато происходят следующие реакции:
6Na2CO3 + 2NH3 + 3Br2 = 6NaBr + 6NaHCO3 + ЗН2О + N2
6NaHCO3 + 2NH3 + 3Br2 = 6NaBr + 3CO2 + 6H2O + N2
Возможность процесса поглощения брома растворами гидрокарбоната, содержащими аммиак, объясняется присутствием в растворе ионов ОН". В этих условиях коэффициент хемосорбции возрастает с увеличением концентрации карбоната натрия. При высокой концентрации карбоната натрия в поглощающем растворе возможно осаждение гидрокарбоната в твердую фазу. Максимальная концентрация карбоната натрия в растворе может составлять 100—110 кг/м3. В этих условиях получаются растворы, содержащие около 100 кг/м3 брома. Более концентрированные растворы (до 200 кг/м3 брома) образуются в процессе поглощения брома циркулирующим раствором бромида натрия, в который непрерывно вводят карбонат натрия и аммиак с одновременным выводом из системы определенного количества раствора бромида и натрия. При установившемся режиме раствор имеет следующий состав: NaBr — 200, Na2CO3 — 8—10, NH3—1,5—2 кг/м3. Содержание же при этом BrOj не превышает 0,05 кг/м3.
При поглощении брома известковым молоком в присутствии аммиака процесс проводят в барботажных колоннах, имеющих четыре полки. Раствор перетекает с полки на полку по переливным трубкам. Процесс орошения проводят раствором, содержащим 5—5,5% активного оксида кальция, к которому непрерывно добавляют 10—25%-ный раствор аммиака. Полученные растворы бромида кальция содержат 140-—170 кг/м3 иона брома. Недостатком способа является потеря брома в виде аэрозоля бромида аммония, а также образование осадка в результате взаимодействия гидроксида кальция с диоксидом углерода.
Поглощение брома аммиаком происходит как в жидкой, так и в газовой фазе. Общая эффективность процесса зависит также от скорости улавливания аэрозоля бромида аммония поглотительным раствором. Процесс изучен в лабораторных условиях при использовании насадки из колец Рашига слоем 3 м. Абсорбер орошался раствором бромида аммония концентрацией 120—380 кг/м3, к которому вводили аммиак в количестве, соответствующем количеству поступающего брома. При этом установлено, что скорость абсорбции мало зависит не только от скорости газа (сог), но и от плотности орошения (сож) и концентрации бромида аммония в растворе. Суммарная скорость процесса описывается уравнением
и*
323
___ ii л л 0,96 0,296 / 1 \О,55 v 11,44 о)г о)ж ( 1 ’
где cNH4Br в моль/л.
Степень поглощения составляла 98—99%, а коэффициент абсорбции — 49,3 — 57,3 м/ч.
Разработан процесс поглощения брома раствором аммиака в растворе бромида кальция. Образующийся при этом бромид аммония превращается в бромид кальция при взаимодействии поглотительного раствора с карбонатом кальция.
Аэрозоль не образуется в случае применения восстановителей, не содержащих летучих продуктов, например уротропин и карбамид.
С уротропином происходит следующая реакция:
1 8Вг2 + 36КОН + М4(СН2)б = ЗбКВг + 2N2 + 6СО2 + 24Н2О
Образующийся раствор, содержащий 500—520 кг/м3 бромида калия, имеет желтую окраску, поскольку присутствие уротропина препятствует полному осаждению железа и других металлов в щелочной среде.
Поглощение брома раствором карбоната натрия в присутствии карбамида происходит по следующему уравнению:
3Br2 + (NH2)2CO + 6Na2CO3 = 6NaBr + 6NaHCO3 + CO2 + N2
Способ не применяется в производстве из-за выпадения гидрокарбоната в твердую фазу, низкого использования карбоната натрия и низкой концентрации брома в получаемых растворах (не выше 80—85 кг/м3).
Поглощение брома диоксидом серы. Процесс поглощения брома диоксидом серы и его водными растворами (сернистой кислотой) основан на реакции
Br2 + SO2 + 2Н2О = H2SO4 + 2НВг
Процесс поглощения можно проводить как в газовой, так и в жидкой фазе. В процессе взаимодействия в газовой фазе образуются мельчайшие частицы жидкости (капли), состоящие из H2SO4 и НВг, которые улавливаются в брызгоуловителях. В жидкой фазе бром улавливают циркулирующим раствором НВг и H2SO4 с последующим взаимодействием растворенного брома (Вг2, Вг3, Вг5“) с сернистой кислотой в растворе. Абсорбция брома водными растворами бромидов связана с резким понижением давления его пара вследствие образования комплексных соединений (рис. 6.10).
Проведение процесса взаимодействия брома с водными растворами бромидов является более целесообразным в газовой фазе, поскольку при этом требуется меньший объем аппаратуры, упрощается регулиро-324
вание процесса и снижается расход диоксида серы, который составляет 0,5—0,6 т на 1 т элементного брома.
Взаимодействие между диоксидом серы и элементным бромом происходит с большой скоростью. Процесс осуществляется в газоходе между десорбером и абсорбционной колонной. Диоксид серы дозируют в избытке (5—10%) в автоматическом режиме. Процесс регулируют потенциометрическим газоанализатором.
Улавливание брызг смеси кислот проводится в насадочной колонне, орошаемой циркулирующей смесью кислот. Колонна имеет одинаковый с десорбером
Рис. 6.10. Зависимость коэффициента распределения брома от содержания Вг" в растворе при 0, 25 и 50° С
диаметр и высоту слоя насадки
4—6 м. Для полного улавливания остатков смеси кислот и избыточного диоксида серы устанавливают второй скруббер, орошаемый морской водой (до 10% воды, поступающей на переработку), которую затем возвращают в процесс. Исходящая из брызгоуловителя жидкость поступает в десорбционную башню. Степень абсорбции брома составляет 98,5—99%. Раствор содержит 10—20% НВг, 8—18% H2SO4 и до 1% НС1. Концентрация бромида водорода в растворе зависит от концентрации брома в бромовоздушной смеси и
давления паров воды над рассолом.
В условиях работы в пленочном режиме на лабораторной установке коэффициент абсорбции брома (Р) может быть рассчитан из уравнений
Nu = —; Nua = 0,149 Rer0’54 ReM0’62. pD
где 1 — характеристическая длина, D — коэффициент диффузии.
Коэффициент абсорбции бромида водорода в условиях полузавод-ской установки составляет от 31,6 до 41,0 м/ч.
Растворы смеси кислот направляют на производство из них элементного брома хлорированием или бромоводородной кислоты перегонкой. Обезбромленный раствор, содержащий серную и хлороводородную кислоты, направляют на подкисление поступающей морской воды. Таким образом, удвоенный расход хлора для получения элементного брома данным способом компенсируется уменьшением или исключением расхода кислоты по сравнению с щелочным поглощением.
325
Получение элементного брома ионообменным способом. Процесс получения элементного брома с помощью ионообменных смол основан на их способности селективно сорбировать элементный бром из растворов. Окисленные растворы (рассолы), содержащие бром, контактируют с анионитом, который после насыщения бромом обрабатывают диоксидом серы или сульфитом натрия для восстановления в бромид. Образующийся бромид десорбируют концентрированным водным раствором хлорида натрия. Раствор, отделенный от анионита, хлорируется, после чего образующийся элементный бром отгоняется водяным паром при 80—90° С. Пары брома конденсируются в жидкий бром. Степень сорбции брома анионитами составляет 95—97% и не зависит от температуры ( в пределах 2—50° С) и его концентрации в растворе. В этом преимущество ионообменного способа перед воздушно-десорбционным. Этот метод более перспективен и был шире исследован для сорбции иода, поэтому свойства ионитов и теория метода подробно описаны в гл. 18.
Наиболее сильной поглотительной способностью брома из водных растворов обладают аниониты в СГ-ионной и Вг“-ионной формах. Сорбция связана с образованием в фазе смолы комплексов ионов С1Вг2", Вг3", Вг5 и т. д. Образование ионов такого типа в смоле доказано ИК-спектроскопическими исследованиями анионитов, насыщенных бромом по характерной полосе Вг3". Для сорбции брома применяют сильноосновные аниониты АВ-17 (Россия) и близкие по свойствам к ним JRA-400 (Германия), DOWEX 1x8 ( США), JMAC-54, Kastel А300 ( Италия).
Предельное насыщение анионита в Br-ионной форме отвечает формуле Вг7", для анионита в Cl-ионной форме оно ниже. Равновесие первой стадии между анионитом и раствором брома в хлорид-ном растворе описывается уравнением
[Вг2СГ]цм[СГ]р [Вг2СГ]р[СГ]см ’
где индексы см и р относятся к смоле и раствору.
Ниже приводятся значения константы равновесия в зависимости от концентрации NaCl в растворе:
Концентрация NaCl, г-экв/л. 0,5 1,0 2,0 4,0
/СЮ*3...................... 4 6,25 6,9 19,2
Для анионита АВ-17 в Br-ионной форме константы ионообменного равновесия равны:
^5{.Ы?Г.к=211О;
[Вг;ивг-]„ [BrJJBr-L
1,059 106.
326
Процесс сорбции брома из растворов хлорида натрия может быть также описан уравнением
г = г
Х\+Кс'
где Г — насыщение ионита, г/г; Г*, — предельное насыщение ионита, г/г; К—константа равновесия, мг-экв'1 • л; с — концентрация брома в жидкой фазе, г/л.
Ниже приведены значения константы равновесия (К) и насыщения ионита (Г) в зависимости от концентрации NaCl:
Концентрация NaCl, г-экв/л........
С , г/г...........................
К, мг-экв’1 - л...................
0,5 1,0 2,0 4,0
0,430 0,514 0,521 0,865
21,0 13,0 10,4 5,1
Количество сорбированного брома при одинаковой равновесной
концентрации его в растворе снижается при увеличении концентрации хлорида. Изотермы сорбции брома анионитом АВ-17 из растворов хлорида натрия различной концентрации показаны на рис. 6.11. Количество сорбированного брома не зависит от температуры в пределах 0—50°С.
Извлечение брома из рассолов
с помощью анионитов состоит из следующих стадий: подкисление рассола; выделение элементного брома действием хлора; адсорбция брома ионитом; десорбция брома из ионита; получение элементного брома из концентрата.
В процессе применения для окисления бромида хлора в количестве, близком к эквивалентному, получается равновесная смесь, содержащая Br2, BrCl и Вг. В процессе ионообменного извлечения брома из хлоридных рассолов, когда такой раствор взаимодействует с анионитом, сорбция Вг2 и BrCl идет с одинаковой скоростью, вследствие чего смещение установившегося равновесия и продолжение реакции окисления не происходит, как это
Рис. 6.11. Изотермы адсорбции брома анионитом АВ-17 в СГ-форме при 20° С из растворов хлорида натрия различной концентрации (в г-экв/л):
/ — 0,01; 2 — 0,1; 3 — 0,5; 4—1,0; 5 — 2,0; 6 — 4,0
327
бывает в процессе воздушной десорбции. В связи с этим предложено принять для окисления бромида избыток хлора и сорбировать его в виде ВгС1, что позволяет повысить степень извлечения брома.
Предложено также предварительно насыщать анионит хлоридом брома и затем пропускать через него рассол, содержащий Вг", с тем, чтобы в фазе смолы происходила реакция: Вг + BrCl = Вг2 + С1“.
Процесс сорбции брома на ионообменной смоле может быть проведен со стационарным или взвешенным слоем смолы.
Процесс поглощения брома в аппарате со стационарным слоем проводят в закрытом аппарате с избыточным давлением до 0,6 МПа. Такое давление необходимо для прохождения рассола с высокой скоростью через фильтры для отделения от него взвешенных частиц и через слой зернистого ионита.
Рапа из сборника фильтруется через фильтр из кварцевого песка и поступает в хлоратор, куда вводят серную кислоту и хлор. Выделяющийся элементный бром адсорбируют в адсорбере, представляющем собой цилиндрический аппарат диаметром 3,4 м и высотой 5,2 м, заполненный анионитом, высота слоя которого составляет 2,5 м. Рассол через слой анионита пропускают со скоростью около 33 м/ч. Отработанную рапу из адсорбера через смолоуловитель направляют на нейтрализацию, а затем на сброс. Содержание свободного брома в отработанной рапе в начале цикла равно нулю, а в конце — 0,02 кг/м3 (при начальном содержании его в поступающей рапе 0,27—0,28 кг/м3).
Контролируют процесс по потенциалу платинового электрода, опущенного в отработанную рапу. В этих условиях насыщение ионита бромом достигает примерно 0,25 кг/кг. В процессе окисления бромида на 87—90% степень поглощения ионитом равна 95—97%, а общий выход — степень — извлечения 80—82%.
Процесс десорбции брома из анионита проводят в том же аппарате, что и адсорбцию. Чтобы не нарушить непрерывность процесса, устанавливают два параллельно работающих адсорбера. Для десорбции брома используют растворы сульфита и хлорида натрия. Сульфит является восстановителем, переводящим Вг2 в Вг, а хлорид вводится для вытеснения образовавшегося Вг2 в жидкую фазу. Концентрация сульфита натрия в растворе составляет 50 кг/м3, NaCl — 100 кг/м3.
Применяют многократную циркуляцию промывных растворов с использованием разбавленных растворов для приготовления десорбирующего раствора и для подкйсления исходной рапы. Полученный раствор с концентрацией Вг 60—65 кг/м3 направляют на выделение элементного брома. После проведения 150 циклов работы сорбционные свойства анионита существенно не меняются. Расход анионита в процессе многократного использования определяется его потерями в результате измельчения и уноса с отработанной рапой, что составляет 15—30 кг на 1 т брома.
328
При конструировании аппаратов для сорбции брома во взвешенном слое необходимо учитывать, что плотность набухшего анионита АВ-17 в Cl-ионной форме (1,13 г/см3) ниже плотности промышленных рассолов. Однако по мере насыщения бромом она быстро увеличивается, достигая значений 1,258—1,267 г/см3 при содержании брома 0,23 кг/кг смолы. Поэтому аппарат изготовляют трехсекционным, причем диаметры конусов увеличиваются снизу вверх. Скорость процесса сорбции и динамическая обменная емкость возрастают по мере увеличения исходной концентрации брома и определяются, главным образом, скоростью подвода брома к поверхности раздела фаз.
Разработан способ сорбции брома в открытом канале, в котором турбулентным потоком движется хлорированная морская вода. В поток непрерывно вводят смолу. Канал имеет длину 1,35 км; продолжительность пребывания воды в канале составляет 15—30 мин при использовании смолы с размером частиц 0,3—0,8 мм и 5—15 мин для смолы с размером частиц 0,07—0,15 мм. В другом конце канала имеется отстойная зона, откуда суспензию насыщенного ионита подают на вакуум-фильтры.
Процесс десорбции брома из анионита может быть осуществлен и окислением бромида хлором до BrOj при 100° С.
Способы очистки брома. В броме-сырце после холодильников бромной колонны содержатся 2—5% хлора, а иногда и больше, органические примеси и вода.
Бром очищают от хлора двумя способами: ректификацией и обработкой растворами бромидов. Значительную часть хлора очищают ректификацией, которую проводят в установке, соединенной непосредственно с окислительной колонной. Поэтому хлор, поступающий в систему в составе брома-сырца, а также некоторое количество его, уносимое отгоняемым хлором, возвращают в систему. Пользуясь диаграммой кипения системы бром—хлор (рис. 6.12), можно рассчитать ректификационную колонну для отделения хлора от брома.
Для ректификации брома используют колонны (рафинеры), изго-
Рис. 6.12. Диаграмма кипения в системе бром—хлор при различном давлении (в кПа):
/ — 98,1; 2 — 66,6; 3 — 40,0; 4—13,3
329
товленные из керамики, стекла или титана, заполненные кольцами Рашита на перфорированных решетках. Рафинер соединяется с обратным холодильником и испарителем. Простейший испаритель представляет собой змеевик из керамики или стекла, погруженный в ванну с горячей водой.
Бром-сырец поступает в верхнюю часть рафинера, стекает по насадке, смешивается с парами, поднимающимися с нижней части испарителя, очищаясь при этом от содержащегося в нем хлора. Хлорид брома в жидкой и газовой фазах в значительной степени диссоциирован, поэтому отгонка хлора из брома-сырца не представляет особых затруднений.
Очищенный от хлора бром поступает в испаритель, где он частично испаряется, а оставшееся количество через холодильник стекает в сборник целевого продукта Изменяя температуру в ванне испарителя, регулируют интенсивность процесса испарения брома, а также эффективность процесса ректификации. Дополнительное фракционирование хлор-бромной смеси проводят в обратном холодильнике. Газовая смесь возвращается из обратного холодильника в колонну с отношением Вг2/С12 = 0,7—0,9. Схема обеспечивает снижение содержания хлора в целевом продукте до 0,2—0,3%. Более полная очистка достигается в ректификационных колоннах, работающих с подачей сырца в среднюю часть колонны. При необходимости бром направляют на повторную ректификацию во вторую колонну. Освобождение брома от хлора достигается и в процессе выдерживания его при температуре, близкой к температуре кипения. Особо чистый бром получают сублимацией его ниже температуры замерзания и фракционированной конденсации.
Бром освобождается полностью от хлора в процессе промывания его раствором одного из бромидов, например бромида калия или бромида железа (III), а также перегонкой брома с растворами бромидов или в процессе фильтрации его через слой сухого бромида. Эффективным способом очистки является пропускание паров брома при температуре выше температуры его кипения через раствор бромида.
В броме-сырце содержится растворенная и эмульгированная вода. Эмульгированная вода после отстоя находится сверху и может быть отделена без особых трудностей. Растворимость воды в жидком броме равна 0,046%, поэтому частичное ее удаление происходит в процессе отстоя жидкого брома, если температуру раствора поддерживать несколько ниже температуры кипения брома (около 50° С). В течение 16 ч содержание воды в броме снижается до 0,003%, а после двукратной дистилляции оно составляет 0,0025—0,003%. Для полного обезвоживания бром промывают 70—80%-ной серной кислотой. Высушивают бром, пропуская его через насадочную колонну, заполненную кислотой. Эффективная очистка достигается и в процессе пропускания паров брома через слой серной кислоты.
330
Применяется способ обезвоживания брома пропусканием его через слой тщательно очищенных железных стружек. Железо реагирует с бромом лишь в присутствии воды с образованием гидроксида железа (II). Недостатком способа является возможность загрязнения брома бромидом железа (И).
Обезвоживают бром стружками металлического магния. Образующийся бромид магния в броме не растворяется. Из химических осушителей можно применять плав бромида кальция и перхлорат магния (ангидрон). Осушители регенерируют термообработкой в вакууме до 170—240° С.
Предложено также для осушки брома применять естественные (морденит, клиноптилит) и искусственные цеолиты (молекулярные сита) с диаметром пор 0,3—0,5 нм. Регенерацию сорбента проводят при нагревании его до 200—350° С в потоке сухого воздуха. Осушка производится путем фильтрации брома через слой сорбента. Продолжительность защитного действия зависит от скорости фильтрации. Остаточное содержание влаги составляет 0,002—0,006%.
В броме, получаемом из рассолов, всегда содержится примесь органических веществ, образующихся в результате их бромирования. Температура кипения органических примесей выше температуры кипения брома и в процессе перегонки они накапливаются в хвостовых фракциях. Основной примесью брома является бромоформ. Обнаружены также тетрабромид углерода, бромдихлорметан, дибромхлорметан и др.
Бром от органических веществ очищают путем его ректификации, промывая серной кислотой, и термическим их разложением в присутствии катализаторов (активированный уголь, содержащий до 10% оксида железа) и без них, а также в присутствии водяного пара. Процесс термообработки с разложением примесей проводят при 425—625° С. Присутствующий хлор реагирует с органическими веществами с образованием хлорида водорода, который отмывают водой.
6.2. БРОМИД ВОДОРОДА И БРОМОВОДОРОДНАЯ КИСЛОТА
Физико-химические свойства. Бромид водорода НВг — бесцветный газ с резким запахом, сильно дымящий на воздухе, плавится при температуре -86,9° С, кипит при 66,7° С. Плотность жидкого бромида водорода 2,17 г/см3 (-68° С); = 2,3 кДж/моль; Д/7°сп = 17,61
кДж/моль; ДЯ^р=-34,1 кДж/моль;. 52°98 = 198,6 Дж/(моль-К); Гкрит = = 90,0° С; ркрит = 8,54 МПа; давление пара (в МПа): 1,73 (12° С), 2,41 (26° С), 4,07 (50° С). Термически устойчив: степень диссоциации до радикалов при 1000° С 0,5%, а при 1220° С— 1,1%. Связь Н—Вг поляр-331
ная, дипольный момент 0,79-10‘30 Кл-м. Хорошо растворяется в воде (в г 100 г воды): 221 (0° С), 193 (25° С), 130 (100° С). Температура кипения азеотропной смеси (47,63% НВг) 124,3° С.
Водный раствор бромида водорода — одна из самых сильных кислот, в растворе бромид водорода полностью диссоциирован; d^20 для растворов различной концентрации: 1,0723 (10%), 1,2580 (30%), 1,5173 (50%), 1,7675 (65%). Сильный восстановитель. Окисляется медленно, выделяя элементный бром при хранении на воздухе.
Бромид водорода и бромоводородная кислота применяются в процессе получения органических и неорганических соединений брома. Бромид водорода применяют также в качестве катализатора органических реакций. Разбавленные водные растворы бромида водорода используются в медицине.
Способы получения бромида водорода и бромоводородной кислоты. Бромид водорода и его водный раствор — бромоводородную кислоту — получают в основном двумя способами: 1) из элементов; 2) кислотным разложением бромидов.
Получение бромида водорода из элементов. В обычных условиях бром с водородом не реагируют. Реакция между ними начинается лишь при температуре около 200° С. Однако и при 300° С реакция идет с небольшой скоростью, а при температуре красного каления реакция проходит в сопровождении желто-зеленого пламени и может протекать со взрывом.
Реакция брома с водородом имеет цепной характер и может состоять из следующих стадий:
1. Инициирование (термическое):
Br2 + М К» > 2Br- + М
2. Продолжение цепи
Вг- + Н2 —НВг + Н-
Н • + Вг2 > НВг + Вг-
Н- + НВг —£-> Н2 + Вг-
3. Обрыв цепи
Вг- + Вг- + М —> Br2 + М
lg Ка = 12,8 - 190/0, Ко = 6 -10'9 (200° С);
1g К, = 11,25-80,6/0, К = 2-102 (200° С);
332
Igtf2 = 11,33-5,4/0, K2 = 5 • IO10 (200° С);
lg К3 = 10,39-6,5/0, К2 = 5 • 109 (200° С);
lgtf4 = 9,12-8,9/0, K< = 1,3-1010 (200° C);
Условие длинных цепей: [H2][Br • ] = (A?2[Br2][H] + АГз[НВг])[Н • ].
В начальный момент, когда [НВг] = О, [Н-] / [Вг] = / ^[Вгз]
= К\ / Ki =10'8 при [Н2] = [ Вг2], и, следовательно, обрывом цепей по реакциям: Н-+Н-+МиН*+Вг-+ М можно пренебречь.
Условие стационарности: Ко [Br2][ М] = 2/<4[Вг-]2[ М], откуда [Вг • ] = (/<о[Вг2]),/2(27<4)-,/2, [Н • ] = ^[Н2][Вг- ](/С2[Вг2] + А^НВг])"1.
Скорость реакции:
W = Aj[H2][Br* ] + 7<2[Вг2][Н • ] - tf3[HBr][H • ]
В производственных условиях бромид водорода из элементов получают сжиганием водорода в парах брома в реакционной колонне, снабженной специальной горелкой. Смесь исходных газов пропускают через катализатор. В качестве катализатора применяют платиновую сетку (при 700—800° С), платинированный асбест или силикагель (при 200—300° С) или древесный уголь (при 600° С и выше). Разработаны способы применения в качестве катализаторов сесквиоксида хрома, оксида магния и дибромида кальция.
Исходную бромоводородную смесь готовят, пропуская через жидкий бром тщательно очищенный от кислорода водород. Состав газа регулируют изменением температуры брома и скорости движения потока водорода. С целью полного превращения брома в бромоводород избыток водорода в исходной смеси должен быть не более 15%.
В процессе получения решающее значение имеет отвод образовавшегося бромида водорода из реакционной зоны, так как катализаторы, в частности платина, могут провоцировать обратную реакцию.
Исходный водород тщательно высушивают и освобождают от примеси кислорода. Стадию катализа проводят со стационарным или взвешенным слоем катализатора. Реактор изготовляют из кварца или никеля. Образующийся бромид водорода после реактора охлаждают до 50—55° С до конденсации паров брома, после чего он высушивается и поступает на конденсацию. Полученный жидкий бромид водорода затаривается в стальные баллоны.
Пары брома улавливают медными стружками, нафталином, влажным красным фосфором и др. В качестве высушивающих агентов применяют ангидрон (перхлорат магния), безводный бромид кальция, пентаоксид фосфора, сесквиоксид алюминия и силикагель.
333
Поглощая газообразный бромид водорода водой, получают 48%-ную бромоводородную кислоту, а очистку от брома производят поглощением анионообменными смолами.
Синтез из элементов применяют и при получении бромида дейтерия путем проведения реакции без катализатора при 750—800° С с избытком исходного элементного брома.
Получение бромоводородной кислоты обработкой бромидов кислотами. Бромоводородную кислоту получают в процессе обработки бромидов 30—40%-ной серной кислотой по схеме
2NaBr + H2SO4 = 2НВг + Na2SO4
Применение более концентрированной серной кислоты вызывает окисление образующегося бромида водорода. Процесс окисления можно предотвратить путем введения хлорида олова (II) или подобного восстановителя. В производстве процесс окисления бромида водорода исключают путем строгого соблюдения соотношения между бромидом водорода и водой, а также точным дозированием исходной серной кислоты. В этом случае удобнее пользоваться бромидами металлов, образующими йерастворимые сульфаты, например
CaBr2 + H2SO4 == 2HBr + CaSO4
Если вместо серной кислоты для разложения бромида применять сиропообразную фосфорную кислоту, то можно получать бромоводород с хорошим выходом и без побочных продуктов:
ЗКВг + Н3РО3 = ЗНВг + К3РОз .
Другие способы получения бромоводорода и его водных растворов. Газообразный бромид водорода получают из бромоводородной кислоты, применяя в качестве обезвоживающего агента бромид кальция (СаВг2) или бромид лития (LiBr), а также концентрированную серную кислоту.
Существует способ получения бромоводородной кислоты из водных растворов бромидов (KBr, NaBr), основанный на пропускании их через катионит в Н^-форме. Известны способы получения бромоводородной кислоты взаимодействием элементного брома с элементной серой и ее соединениями.
При действии элементного брома на водную суспензию элементной серы при 70° С энергично идет реакция:
3Br2 + S + 4Н2О = 6НВг + H2SO4
334
При этом вначале образуется S2Br2, который разлагается водой в процессе дальнейшего бромирования:
S2Br2 + 8Н2О + 5Вг2 = 12НВг + 2H2SO4
Диоксид серы, реагируя с бромом в водной среде, также образует бромоводород:
SO2 + Вг2 + 2Н2О = 2HBr + H2SO4
Данная реакция используется в промышленности в процессе поглощения брома из бромовоздушной смеси диоксидом серы. Из полученной смеси бромид водорода выделяют перегонкой.
Бромид водорода получают реакцией элементного брома с сульфидом водорода:
3Br2 + 2H2S = 4HBr + S2Br2
Образующийся дибромид серы в дальнейшем гидролизуется.
Бромид водорода получают в процессе гидролиза бромида алюминия при 200—300° С:
2А1Вг3 + ЗН2О = 6НВг + А12О3
Эту реакцию применяют также для получения бромида дейтерия.
В лабораторных условиях бромид водорода получают бромированием органических веществ, например тетрагидронафталина:
СюН |2 + 4Br2 = CioHgBr4 + 4 НВг
Для лабораторного применения бромид водорода получают реакцией элементных фосфора и брома по реакциям:
3Br2 + 2Р = 2РВг3
РВгз + ЗН2О = ЗНВг + Н3РО3
6.3. БРОМИДЫ
Бромиды — соединения брома с другими элементами. Бромиды металлов, такие, как NaBr, КВг, в большинстве случаев типичные соли. Бромиды неметаллов (CBr4, SiBr4) — легкоплавкие, легколетучие вещества с молекулярной решеткой, в воде они либо труднорастворимы, либо моментально разлагаются. Некоторые из бромидов неметаллов являются бромангидридами (РВг3, ВВг3). К ним относятся также бромиды высоковалентных металлов (TiBr4j SnBr4, АбВгз).
335
Промежуточное положение между типичными солями и бромангид-ридами занимают бромиды железа, циркония, гафния и др.
Наиболее широко применяемыми в народном хозяйстве бромидами являются бромиды натрия, калия, аммония, кальция, железа.
Бромшд натрия. Бромид натрия NaBr — бесцветные кристаллы кубической сингонии (а = 0,59772 нм, z = 4, пространственная группа ЕтЗт); плотность 3,21 г/см3; температура плавления 747° С; С° = 51,38 Дж/(моль-К), ДЯо°бр =-361,16 кДж/моль, АЯВ°ОЗГ = 217 кДж/моль (ОК), ДЯП°Л = 26,23 кДж/моль; 5298 = 86,93 Дж/(моль-К); уравнение температурной зависимости давления пара:
Igp (мм рт. ст.) = -12 100/Г + 20,691 + 3,529 IgFnpn (1270-1443 К);
в насыщенном паре бромида натрия при 747° С содержится около 28 молярных процента димера. Гигроскопичен.
Растворимость (в 100 г): в воде — 80,1 (0° С), 2,32 (20° С); метаноле— 17,3 (0°С), 16,8 (20° С); глицерине — 38,7(20° С). Растворяется также в пиридине, жидком аммиаке, плохо — в ацетоне.
Бромид натрия ниже 50° С из водного раствора образует дигидрат— бесцветные кристаллы моноклинной сингонии (а = 0,6757 нм, b = 1,0431 нм, с = 0,6573 нм, Р = 113,49°, z = 4, пространственная группа P2i/a); = 36,34 Дж/(моль-К); АЯо°бр = -951
кДж/моль; 5298 = 175 Дж/(моль*К); выше 50° С обезвоживается, а ниже 40° С переходит в пентагидрат (рис. 6.13).
Бромид натрия с фтором и хлором образует соответственно NaF и NaCl, а с сесквиоксидом бора — ВВг3. У бромида натрия выделены аддукты — NaBr-AlBr3 и NaBr-ScBr3.
Бромид натрия применяется в качестве оптического материала, седативного вещества в медицине, противовалирующей добавки к проявителю и для изготовления фотоэмульсий.
Получение бромшда натрия. Бромид натрия получают действием брома на растворы гидроксида натрия в присутствии аммиака, а также обменной реакцией между бромидом железа и сульфата натрия.
Широко применяемым способом получения бромида натрия является взаимодействие брома с раствором гидроксида натрия (или карбоната натрия) в присутствии восстановителей, например аммиака, карбоната аммония, формальдегида, муравьиной кислоты, карбамида или уротропина:
3Na2CO3 + 2NH3 + 3Br2 = 6NaBr + 3CO2 + 3H2O + N2
Na2CO3 + 2HCOONa + 2Br2 = 4NaBr + 3CO2 + H2O
336
Процесс ведут в закрытом эмалированном аппарате, в который заливают расчетное количество раствора карбоната или гидроксида натрия. При постоянной работе метального устройства в растворы вводят восстановитель, а затем под слой смеси приливают бром. Ввиду экзотермич-ности процесса на крышке реактора, во избежание потери брома, устанавливают ловушки, заполненные железными стружками или орошаемые растворами карбоната натрия.
Образующиеся растворы фильтруют от нерастворимого остатка и взвешенных частиц, упаривают до содержания в них не более 15—20% маточных растворов. Суспензию подают в центрифугу, где производят отделение кристаллов при температу-
Рис. 6.13. Зависимость растворимости бромидов натрия, калия и аммония в воде от температуры
ре выше 70° С.
Для получения 1 т продукта рассмотренным методом расходуют: 0,8—0,82 т брома, 0,45—Ч),48 т гидроксида натрия, 0,07—Ю,08 т аммиака или 0,23—0,25 т 100%-ной муравьиной кислоты.
Бромид натрия получают из растворов бромида кальция, образующегося в процессе абсорбции брома известковым молоком в присутствии аммиака. К раствору бромида кальция (140—170 г/л Вг) добавляют сульфат натрия (или его карбонат):
СаВг2 + Na2SO4 = 2NaBr + CaSO4
Образующуюся суспензию перемешивают в реакторе в течение 1—2 ч, отфильтровывают дигидрат сульфата кальция. Растворы бромида натрия упаривают до плотности 1,40—1,42 г/см3 и вторично фильтруют от осадка, выделяющегося в процессе упарки (CaSO4-2H2O, СаСОз). Затем раствор упаривают до образования густой массы, отделяют кристаллы на центрифугах. Образующиеся маточные растворы возвращают в начало процесса.
Разработаны способы получения бромидов, основанные на реакции обменного разложения легко доступных бромидов с оксидами, гидроксидами и солями других металлов. К ним относится получение бромидов из бромида железа, бромида аммония, бромида алюминия, а также из смеси бромоводородной и серной кислот, получаемых в процессе сорбции брома из бромовоздушной смеси.
337
Получение бромида натрия из его хлорида и бромоводородной/ кислоты |
NaCl + НВг = NaBr + НС1 I
основано на более низкой температуре кипения азеотропной смеси НС1—Н2О (110° С), чем смеси НВг—Н2О (126° С). Процесс ведут при 110° С, добавляя к твердому хлориду натрия бромоводородную кислоту в избытке (20% от стехиометрического). В конце процесса в реакторе остается твердый бромид натрия, а хлороводородная кислота испаряется в виде газа и после конденсации может быть использована для подкисления исходных бромсодержащих рассолов. Целевой продукт после промывки водой содержит 0,5—0,8% NaCl. Выход по НВг составляет 96—98%.
Описанные способы получения бромида натрия аналогичны для получения бромида калия.
Бромид калия. Калия бромид КВг — бесцветные кристаллы с кубической гранецентрированной решеткой типа NaCl (а = 0,6596 нм, z = 4, пространственная группа Fm3rn). При 298° С и 1,7 ГПа образуется кубическая модификация типа CsCl; плотность 2,75 г/см3; температура плавления 734° С, кипения 1455° С; Cj = 52,47 Дж/(моль-К); Д/7П°Л = 25,52 кДж/моль, АЯ ° =-393,45 кДж/моль; S2°98 = 95,92 Дж/(моль-К).
Растворимость бромида калия в воде ( г в 100 г ): 54,0 (0° С), 65,6 (20° С), 105,0 (100° С). Температура плавления эвтектики (45,7 г КВг в 100 г воды) 12,6° С. Температура кипения насыщенного раствора (-110 кг КВг в 100 г воды) 112° С. Плотность 10%, 20%, 30% и 40%-ных во- г дных растворов соответственно 1,0740, 1,1601, 1,2593 и 1,3746. Растворяется в жидком аммиаке, метаноле, этаноле, глицерине и эфире. Обра- * зует непрерывный ряд твердых растворов с КС1.
Бромид калия применяют в.качестве седативного средства в медицине, является компонентом травителя в процессе гравирования, изготовления фотоэмульсий и в качестве противовуалирующего вещества в фотографии. Монокристаллы бромида калия применяют в процессе изготовления призм для ИК-спектроскопии. Порошкообразный бромид калия является матрицей при снятии ИК-спектров твердых веществ.
Получение бромида калия. Существует значительное число способов получения бромида калия. Все они идентичны (лишь с незначительными отклонениями) способам получения бромида натрия.
Получают бромид калия формиатным способом. Он основан на реакции
2КОН + НСООН + Вг2 = 2КВг + Н2О + СО2
338
К исходному раствору гидроксида калия с содержанием 290—310 кг/м3 КОН приливают расчетное количество муравьиной кислоты, после чего медленно, при интенсивном перемешивании, приливают бром. Вследствие теплоты реакции температура процесса повышается до 75—85° С. По этой причине в процессе применения более концентрированных растворов гидроксида калия возможна кристаллизация бромида калия в реакторе. Выделяющийся диоксид углерода в начальной стадии реакции связывается щелочью с образованием карбоната, а затем и гидрокарбоната. В конце реакции гидрокарбонат вступает в реакцию, которая сопровождается бурным выделением диоксида углерода. Конец реакции определяют тем, что раствор становится нейтральным
Бромид натрия этим способом не получают, поскольку гидрокарбонат натрия, образующийся в процессе реакции, выпадает в осадок и затем медленно реагирует с бромом.
В процессе получения бромидов из бромида железа(П) к концентрированному раствору последнего (около 40%) в процессе нагревания и перемешивания добавляют карбонат калия или натрия до щелочной реакции раствора. Реакция идет по уравнениям:
FeBr2 + К2СО3 + Н2О = 2KBr + Fe(OH)2 + СО2
FeBr2 + Na2CO3 + Н2О = 2NaBr + Fe(OH)2 + СО2
В присутствии воздуха гидроксид железа(П) легко окисляется до гидроксида железа(Ш). Раствор после отстоя отделяют от осадка. Осадок промывают водой (при необходимости дистиллированной). Промывные воды (концентрированные) добавляют к полученному раствору, а слабые растворы используют для промывки следующих партий гидроксида железа.
Применяется также способ получения бромида калия взаимодействием раствора гидроксида калия с бромом:
6КОН + ЗВг2 = КВгОз + 5КВг + ЗН2О
с последующим восстановлением образующегося бромата калия углем при высокой температуре. Получение бромида калия по этому способу проще, чем бромида натрия, поскольку бромид калия во всех температурных интервалах выделяется из растворов в безводной форме. Для получения 1 т бромида калия расходуют: 0,70—0,72 т брома, 0,54—0,58 т гидроксида калия, 0,06—0,065 т аммиака.
Бромид аммония. Бромид аммония NH4Br кристаллизуется в безводной форме в кубической системе. Плотность 2,40 г/см3; тепло-339
та образования ДЯ^ =-64,61 ккал/моль; при 394,6° С возгоняется, а при 542° С разлагается; не гигроскопичен, устойчив на воздухе и свету лишь в чистом состоянии.
Бромид аммония получают взаимодействием водного раствора аммиака и брома. Согласно технологии, жидкий бром при интенсивном перемешивании и сильном охлаждении постепенно прибавляют к водным растворам аммиака. При этом происходит реакция
3Br2 + 8NH4OH = 6NH4Br + N2 + 8Н2О
Реакцию проводят с избытком аммиака во избежание образования устойчивого ЫНдВгз. К концу реакции в растворе должно содержаться 2—3 кг/м3 свободного аммиака. В случае появления в растворах кристаллов их растворяют, дополнительно вводя в систему воду.
Бромид аммония получают также в процессе улавливания брома из бромовоздушной смеси аммиаком.
Получают бромид аммония обменной реакцией между бромидом железа и аммиаком, а также бромидом кальция и карбонатом аммония.
Отфильтрованные растворы бромидов упаривают для получения кристаллов. Процесс обычно ведут в эмалированных реакторах до получения суспензии (Т:Ж = 8:1). Суспензию бромида калия и бромида аммония загружают в кристаллизатор и охлаждают до 20° С. После отделения кристаллов на центрифуге их сушат при температуре около 100° С.
6.4. БРОМАТЫ НАТРИЯ И КАЛИЯ
Броматы М, соли НВгО3— бесцветные кристаллы; устойчивы при обычных условиях. Броматы щелочных металлов в процессе нагревания разлагаются по схеме
2МВгО3 -> 2МВг + ЗО2
Для NaBrO3 температура плавления 380° С; плотность 3,34 г/см3; растворимость в воде (г в 100 г) — 36,4 (20° С), 90,8 (100° С). Для КВгОз плотность 3,25 г/см3; разлагается при 400° С с выделением кислорода; растворимость в воде (г в 100 г) -6,9 (20° С), 49,7 (100° С).
Броматы натрия и калия получают путем введения брома в концентрированные растворы гидроксида калия или натрия, например КОН (-300 г/л). В начальный период процесс ведут при 45—50° С, а в конце процесса температуру повышают до 95° С. Реакции идут по уравнениям:
6КОН + ЗВг2 = 5КВг + КВгОз + ЗН2О
340
6NaOH + 3Br2 = 5NaBr + NaBrO3 + 3H2O
Промежуточными продуктами этих реакций являются гипобромит и бромит, которые затем диспропорционируют на бромат и бромид. С повышением температуры в конце реакций скорость процессов возрастает. Однако процесс идет не до конца. Для разложения остатков гипобромита и бромита в конце процесса бромирования к реакционной массе добавляют водные растворы аммиака. Полученный раствор, содержащий 520 кг/м3 бромида и ПО кг/м3 бромата, охлаждают до 20° С. Образующиеся кристаллы отделяют от маточного раствора на центрифуге. Практически в осадок выпадает до 90% содержащегося в растворе бромата калия. Далее бромат калия, содержащий примесь бромида, очищают путем перекристаллизации. Бромат натрия получают таким же способом.
Маточный раствор после кристаллизации бромата используют для получения бромида. Для этого содержащийся в нем остаток бромата восстанавливают. Процесс бромирования гидроксида ведут при сильном охлаждении, при определенной концентрации (40% NaOH или 53% КОН) и температуре — 5° С, в твердую фазу выпадает NaBr*2H2O и соответственно КВг. Броматы калия и натрия получают также из растворов, содержащих бромид и бромат кальция, образующиеся в процессе абсорбции брома известковым молоком. Бромат калия из этой смеси осаждают хлоридом калия:
Ca(BrO3)2 + 2КС1 = 2КВгО3 + СаС12
Для получения бромата натрия раствор обрабатывают карбонатом или сульфатом натрия:
Ca(BrO3)2 + Na2CO3 = 2NaBrO3 + СаСО3
Ca(BrO3)2 + Na2SO4 = 2NaBrO3 + CaSO4
Броматы получают химическим или электрохимическим окислением бромидов. Согласно химическому способу, бромиды или элементный бром окисляют хлорной известью или хлором по уравнениям:
2КВг + ЗСа(С1О)2= 2КВгО3 + ЗСаСЬ
Вг2 + 5С12 + 12КОН = 2KBrO3 + 10КС1 + 6Н2О
Электрохимический способ получения бромата сводится к анодному окислению бромида. В процессе можно применять не только 341
растворы бромида, но и растворы, образующиеся при взаимодействии щелочи с бромом.
Процесс электролиза ведут без диафрагмы при 60—70° С, высокой плотности тока (1500 А/м2) и напряжении 4,5—5 В. Аноды изготовляют из платины, графита, диоксида свинца, оксида родия, а катоды — из нержавеющей стали. Для предотвращения катодного восстановления к электролиту добавляют 0,2% (масс.) бихромата калия. При охлаждении электролита из него выкристаллизовывается бромат. Для снижения растворимости бромата в раствор вводят бромид или гидроксид. Маточный раствор после отделения кристаллов целевого продукта возвращают на электролиз, добавив к нему бром. Выход по току составляет 80—96%.
ГЛАВА 7
ИОД И ЕГО СОЕДИНЕНИЯ
7.1. ИОД
Содержание иода в земной коре 4-10‘5% (масс.) (1014—1015т). Иодсодержащие минералы — иодаргирит Agl, лаутарит Са(Юз)2 крайне редки. Иод в природе находится в рассеянном состоянии в магнетических и осадочных горных породах (10‘4—10’5%, масс.), так как он легко выщелачивается из них водами. Основные запасы иода находятся в подземных водах (0,01—0,1 и выше) нефтяных и газовых месторождений, в селитренных отложениях (до 1%). В природе встречается лишь один стабильный изотоп |271.
Мировые запасы иода в промышленных месторождениях составляют 2,6 млн.т. Мировое производство иода составляет около 15 тыс. т/год (1981).
Свойства. Иод имеет атомную массу 126,9045. Конфигурация высшей электронной массы атома 5s25p5. Степени окисления: -1, +1, +3, +5 и +7. Энергия ионизации 1°—>1+—>12+ соответствует 10,45136 и 19,100 эВ. Атомный радиус 0,136 нм, ионный радиус (в скобках указано координационное число) Г 0,206 нм (6), 15+ 0,058 нм (3), 0,109 нм (6), 17+ 0,056 нм (4), 0,067 нм (6). Молекула двухатомна; длина связи 0,266 нм; энергия диссоциации 148,826 кДж/моль; степень диссоциации 2,8% при 727° С и 89,5% при 1727° С.
Иод — черно-серые кристаллы с фиолетовым металлическим блеском, имеющие форму пластин или листочков. Легко возгоняется до начала плавления, пары окрашены в фиолетовый цвет, имеют резкий запах. Кристаллическая решетка ромбическая {а = 0,4792 нм, b = 0,7271 нм, с = 0,9803 нм, пространственная группа Стеа, z = 4 молекулы 12). Температура кипения 185,24° С, плавления — 113,60° С.
Плотность твердого иода 4,940 г/см3 (25° С), жидкого — 3,960 г/см3 (при 120° С). Критические константы: температура 546° С, давление 11,4 МПа, плотность 1,64 г/см3.
Для твердого 12: С® = 54,44 Дж/(моль-К), АЯП°Л = 15,67 кДж/моль (386,75 К), ДЯ°П =41,96 кДж/моль (475,5 К), 52°98 = 116,13
Дж/(моль-К). Для газообразного иода: Ср° = 20,79 Дж/(моль-К), ДЯ^. = 106,762 кДж/моль, = 180,67 Дж/(моль-К); уравнение тем
343
пературной зависимости давления пара над твердым иодом: 1g р (в мм рт. ст.) = 3594,03/Г + 0,00044347^2,9759 lg Т + 21,91008; теплопроводность твердого иода 0,449 Вт/(м2-К) при 300 К, жидкого = = 0,116 Вт/(см2-К) при 386,8 К. Вязкость жидкого иода при 126° С 2,286-Ю'3 Па-с, а газообразного при 126° С 17,85-10'6 и 36,0-10'6 при 523° С; показатель преломления 1,98 (114° С). Нормальный электродный потенциал в водном растворе Eq (h/Г) составляет + 0,535 В.
В табл. 7.1 приведены изменения равновесного давления паров иода с повышением температуры.
Таблица 7.1. Равновесное давление паров иода при различной температуре
Л °C Л Па II '.“С 1 Р. Па II Л °C 1 1 р, кПа /,°С 1 р-кпа
Твердое состояние Жидкое состояние
-50 0,0049 20 25,9 60 0,574 120 14,6
-40 0,025 30 67,6 80 2,01 140 28,9
-20 0,40 40 137 100 6,05 160 52,4
0 2,97 50 279 114,5 12,0 180 90,2
10 10,75 — — — — 184,9 98,1
Иод является неполярным гидрофобным элементом. Диаграмма растворимости иода в воде приведена на рис. 7.1. Термодинамические свойства водных растворов иода:
до;; ь (тв) 16,4, ь (Г) 3,0 кДж/моль
д//: .х ь (тв) 0 «2 (г) 106,8 кДж/моль
ь (тв) 137 12 (Г) 123 кДж/моль
Поскольку иод является неполярным веществом, поэтому он растворим в неполярных органических растворителях. Растворимость иода в предельных углеводородах относительно невелика. Однако она возрастает при увеличении числа атомов углерода. Более высокая растворимость иода в нафтеновых и особенно ароматических углеводородах. Хорошо растворяется иод и в галогенопроизводных углеводородах [в % (масс.) при 25° С]:
2,2-Диметилбутан . 1,369 Хлороформ ... 3,10
н-Гептан 1,702 1,2-Дихлорэтан .. 5,43
Циклогексан.- .. 2,719 Сероуглерод .. 16,92
Бензол .. 14,09 * Этанол ... 21,33
н-Ксилол .. 16,55 Диэтиловый эфир ... 25,20
Тетрахлорид углерода . 1,90 Нитробензол ... 5,06
Растворимость иода в полярных органических соединениях, содержащих кислород, азот и серу, выше его растворимости в неполярных растворителях.
344
Из неорганических растворителей практическое значение для растворения иода имеют концентрированные минеральные кислоты, например в 100 г 83%-ной серной кислоты растворяется около 0,36 иода.
По своей реакционной способности иод уступает фтору, хлору и брому. Со многими веществами — углеродом, азотом, кислородом, серой и селеном — непосредственно не взаимодействует. С водородом, кремнием, бором и многими металлами реагирует лишь при повышенных температурах.
Одним из характерных химических свойств иода является то, что он сильный окислитель.
Рис. 7.1. Растворимость иода в воде
Поэтому окислительно-восстановительные реакции иода, протекающие в
водных растворах, играют значительную роль в его технологии. Реакции взаимного вытеснения галогенов
2Г + С12 12 + 20"
2Г + Вг2 12 + 2Вг“
используются в промышленности для перевода иода из ионного состояния в молекулярное.
В избытке галогена реакция идет по схеме
12 + 5С12 + 6Н2О = 210; + 10СГ + 12Н+
В нейтральной и щелочной средах образуются кислородные соединения иода:
12 + 500" 210" + 5СГ + 2Н+
12 + 7С12 + 16ОН" 2Ю4 + 14СГ + 8Н2О
С озоном в водных растворах в кислой среде иодиды реагируют, выделяя иод:
2KI + О3 + Н2О = I2 + 2КОН + О2
а при действии пероксида водорода на нейтральные растворы йодидов галоген не выделяется:
Н2О2 + Ю" = Н2О + Ю"
345
Сульфид водорода и сульфиды окисляются иодом до элементной серы
H2S + Ь = 2HI + S
Реагируя с тиосульфатом, иод образует иодид-ион
2820з“ + I2 = S4O*~ + 2Г
Йодиды окисляются концентрированной серной кислотой:
2Г + 2H2SO4 I2 + 2HSO; + H2O
В нейтральной среде нитриты металлов окисляются до их нитратов:
no; +12 + н2о = no; + 2Г + 2Н+ а азотистая кислота окисляет иодиды до элементного иода 2HNO2 + 2Г + 2Н+ = 12 + 2Н2О + 2NO
Концентрированная азотная кислота окисляет иод до йодноватой кислоты
I2 + 10HNO3 = 2НЮ3 + 10NO2 + 4Н2О
Гидразин окисляется иодом до азота
N2H4 + 2I2 = N2 + 4Г + 4Н+
Фосфиты и арсенаты окисляются иодом до фосфатов и арсенатов.
Соли муравьиной и щавелевой кислот окисляются до диоксида углерода
нсо; + 12 = СО2 + Н+ + 2Г
С2О; + 12 = 2СО2 + 2Г
Дихроматы окисляют иодиды до элементного иода
2Сг2О,' + 2Г = ЗСЮ^ + СгО2 + 12 346
В кислой среде с иодидами реагируют и хроматы и дихроматы С12О;’ + 6Г + 14Н+ = 2Сг3+ + 312 + 7Н2О 2СЮ;“ + 6Г + 16Н+ = 2Сг3+ + 312 + 8Н2О
В процессе реакции оксида марганца и перманганата с водными растворами иода в кислой среде выделяется свободный иод:
МпО2 + 2Г + 4Н+ = 12 + Мп2+ + 2Н2О 2МпО; + ЮГ + 16Н+ = 512 + 2Мп2+ + 8Н2О в избытке перманганата иодид окисляется до йодата:
2МпО4 + Г 4- 2Н+ = ГО; + МпО2 4- Н2О
В процессе реакции солей железа (III) с иодидами выделяется элементный иод
2Fe3+ + 2Г = 2Fe2+ + 12
Элементный иод выделяется также в процессе реакции солей ме-ди(П) на иодиды
2Си2+ + 2Г= I2 + 2Си+
Водные растворы иода проводят электрический ток, что связано с их частичным гидролизом:
12 4- Н2О <5 Г 4- н+ 4- HJO
Параллельно с этим образуются ионы трииодида:
12 + Г^П
В процессе гидролиза межгалогенных соединений более легкий атом галогена превращается в галогенид-ион, а более тяжелый — в галогеноватистую кислоту, например
IC1 4- Н2О СГ 4- Н+ 4- НЮ
Образующаяся иодноватистая кислота является нестойким соединением и постепенно подвергается диспропорционированию:
ЗНЮ -> ГО; + 21" 4- ЗН+
347
Таким образом, гидролиз иода протекает по уравнению
ЗЬ + ЗН2О±^Ю; + 5Г + 6Н+
Степень гидролиза иода в насыщенном растворе составляет около 0,7%. Образующаяся в процессе гидролиза иода иодноватистая кислота является слабой кислотой. Значение рЛ*а для НЮ равно «4-Ю"13. В присутствии ионов ОН“ она переходит в Ю“. В щелочной среде ион Ю“ достаточно стоек, однако в область промежуточных значений pH он легко превращается в IOj. Установлено, что этот переход идет в две стадии с участием на первой стадии молекул НЮ
НЮ + Ю" 10' + Н+ + Г
КХ + Ю“ ю; + Г
Вторая стадия процесса гидролиза галогенов протекает со сравнительно небольшой скоростью, которая может быть измерена аналитическими методами. Установлено, что в буферных растворах при концентрации 12, равной 1,15- 10 4М (30 г/м3), pH 6—8 и ионной силе ц = 0,05 Н, скорость гидролиза иода [в л/(моль-мин)] описывается уравнением
-^[12]_к[М[0Н“]
где К—функция pH и температуры.
Применение. Иод является сырьем в производстве неорганических и органических иодсодержащих соединений. Применяется в качестве акцептора водорода в процессе дегидрирования предельных углеводородов и катализатора в органическом синтезе. Широко применяется в медицине в качестве антисептика и антитириоидного средства в виде иодной настойки (5- и 10%-ный раствор в спирте) или раствора Лиголя (водный раствор смеси иода и иодида калия) и органических соединений (йодоформ, иодгност, саиодин, иодол, сер-гозин и др.). В промышленности иод применяют в процессе иодид-ного рафинирования металлов, например титана, циркония, гафния. Около 70% иода перерабатывают в иодиды, применяющиеся кроме медицины в фотографии, лабораторной практике (метод иодометрии), в сельском хозяйстве и для иодирования пищевой соли. Искусственные радиоактивные изотопы I25I {V11 = 60,2 сут), 1311 (Г172 = 8,05 сут), |321 (Г172 = 2,26 г) применяют в медицине для диагностики заболеваний и лечения щитовидной железы. 348
Согласно существующему ГОСТ 545—71, иод технической квалификации первого и второго сортов должен содержать соответственно не менее 99 и 97,5% основного вещества и не более 0,010 и 0,015% хлора и брома, 0,1 и 0,2% органических веществ и 0,05 и 0,15% золы.
7.2. ТЕХНОЛОГИЯ ИОДА
В природе существует непрерывный круговорот иода. Он совершает круговорот в основном в виде элементного иода, что объясняется способностью иона иодида окисляться кислородом воздуха до элементного иода. Это показывает и тот факт, что иод встречается в морской воде в нескольких видах, например 1°, Г, Г и 15+.
Содержание иода в морской воде составляет 0,01—0,07 мг/л, а в морских илах — 0,002—0,01%. В водах, сопутствующих нефти, содержится 0,01%, а в чилийских отложениях селитры — около 1% иода. Концентрация иода в фосфоритах достигает 280 мг/кг, а в бурых углях — до 6 мг/кг.
В хлориде натрия, полученном из морской воды, содержится 0,1 мг/кг иода, в природном хлориде натрия — 0,25 мг/кг, а в калийных солях — до 0,06 мг/кг. В доменной пыли содержится до 0,014% иода, а в воде, сконденсированной в процессе охлаждения коксового газа, — до 0,2 г/л.
Анализ показывает, что в настоящее время более пригодным видом сырья остаются буровые воды нефтяных месторождений, а также твердые отходы калийной промышленности.
Физико-химические основы получения иода. Важной стадией технологии иода является процесс окисления исходных растворов (сырья). В качестве окислителя обычно применяют хлор.
Обычно получение иода из исходных минерализованных вод осуществляют после извлечения из них брома. В процессе получения брома содержащийся в растворе иодид окисляется до йодата. Для получения элементного иода растворы после извлечения брома обрабатывают соответствующим восстановителем.
Окисление иодида хлором. Иодиды легко окисляются до элементного иода хлором. В процессе достаточного подкисления, исключающего гидролиз иода, реакция протекает в две стадии:
Г + Cl22С1 + СГ
Г + IC1 12 + СГ
2Г + С12 12 + 2СГ
349
Зависимость термодинамической константы равновесия суммарной реакции определяют по табличным значениям термодинамических функций:
1g К = 9183,2/(7—4,897).
Константа равновесия при 298 К составляет 8,3-1025 и при 313 К — 2,75-1024. Константы равновесия первой и второй стадий оценивают, воспользовавшись имеющимися данными о константе диссоциации 1С1 в хлороводородной кислоте:
[12 ч-12СГ][С12ч-С1-] ’Cl [IC1+IC1']2
Исследованиями процесса окисления иодида хлором установлено, что при концентрации 10—100 г/м3 показано, что протекание реакции в присутствии большого количества хлорида существенно отличается от протекания ее в бессолевых процессах. В отсутствии солей окисление иодида протекает нацело при pH 1—8 и расходе хлора около 103% от стехиометрии. Концентрация выделенного иода при pH ниже 5 остается постоянной, а при более высоких значениях pH постепенно снижается в результате гидролиза. При высоких концентрациях хлорида полное окисление иодида возможно лишь в сильнокислой среде. При рН<3 и стехиометрическом количестве хлора окисление Практически полное и для повышения его необходим избыток хлора. Установлено, что максимальная степень окисления достигается при pH 5. Снижение степени окисления и повышенный расход окислителя обусловлены частичным переокислением иодида до йодата.
Экспериментально установлено, что реакция протекает меньше, чем за 0,07 с. Установлено также, что продолжительность реакции соизмерима с продолжительностью смешения реагентов в скоростных смесителях. При этом показано, что быстрая галогенизация (—0,01 с) позволяет достичь более высокой степени окисления и меньшего расхода окислителя, чем при обычном перемешивании. Показано, что в 2 н. NaCl при pH 7,3 в процессе окисления хлорной водой достигается 98%-ный выход иода при расходе хлора около 1,1 г-экв, что на 9% выше, чем при простом перемешивании. Показано, что при быстрой гомогенизации переокисленные формы иода (1О“ и Ю^) образуются лишь после окисления всего иодида, тогда как в процессе медленного перемешивания IO3 появляется до образования расчетного количества иодида. Это объясняется переокислением иодида в результате местных очагов повышенной концентрации окислителя. При pH 4,9 скорость образования переокисленных форм иода возрастает 350
и независимо от уменьшения периода гомогенизации избежать переокисления с хлорной водой не удается (рис. 7.2).
Используемые для получения иода буровые воды содержат значительное количество различных органических и неорганических веществ, окисляющихся хлором. Окисление некоторых из них предшествует окислению иода, а другие, например нафтеновые кислоты, окисляются медленно, а выделенный иод постепенно взаимодействует с ними. Поэтому для исключения потерь иода в результате его восстановления в производстве процесс ведут с переокислением иодида так, чтобы по мере восстановления иода образующийся иодид снова окислялся.
Окисление иодида гипохлоритом. Реакция окисления иодида гипохлоритом протекает медленнее, чем окисление хлором. Реакция значительно ускоряется в условиях быстрого перемешивания реагентов.
В буферных растворах, содержащих значительные количества хлоридов, гипохлорит окисляет иодид до элементного иода при рН<4, а при pH 5—7 — до Ю^, при рН>7 — до IO4 и температуре 20—50° С:
ПТППт° тс Ё=3г
Рис. 7.2. Характер окисления иода хлорной водой (pH 7,3) в процессе механического перемешивания (а) в тангенциальном смесителе при различной продолжительности смешения (б — 0,1 с;
в — 0,05 с; г — 0,01 с)
осг + г -> ог + сг
ОГ + Г + 2Н+ -> 12 + Н2О
При концентрации иод-иона 2-10'4 г-экв/л водородных ионов, присутствующих в буферном растворе, достаточно для поддержания этой реакции. С увеличением pH максимальная степень окисления иодида до элементного иода падает, но не в такой степени, как при окислении хлорной водой. Расход же окислителя при максимальной степени окисления меньше, чем при окислении хлором. Снижение продолжительности гомогенизации также улучшает выход элементного иода (с 94 до 97%) и снижает расход окислителя, но ниже, чем при окислении хлорной водой. Явление переокисления иода наблюдается лишь после израсходования всего иодида. Различие в активности хлорной извести и гипохлорита объясняется тем, что образова-351
ние ионов йодата требует участия ионов Ю“ и молекул НЮ. Последние образуются лишь в процессе окисления хлорной водой, в то время как окисление гипохлоритом — ионы Ю“.
Окисление иодида нитритом. Процесс окисления иодида до йодата возможен лишь в сильнокислой среде. В нейтральной или щелочной среде реакция не идет, следовательно, окислителем является не ион нитрита, а свободная серная кислота. Реакция идет по уравнению
2HNO2 + 2Г + 2Н+ = I2 + 2Н2О + 2NO
Как известно, азотистая кислота является слабой кислотой (Ка = = 4,0-10'4). В кислой среде при рН<2 в растворах присутствует свободная азотистая кислота, а в нейтральной и щелочной средах (рН>5) — ион нитрита. В 'промежуточной области одновременно существуют HNO2 и NO~. Азотистая кислота в растворах нестабильна и медленно разлагается с выделением NO2 и NO.
В присутствии сильных кислот реакция между нитритом и иодидом протекает мгновенно, а в буферных растворах при pH 4,5 скорость реакции пропорциональна концентрации нитрита и иодида. При увеличении их концентрации реакция каталитически ускоряется. При избытке нитрита степень окисления не увеличивается, а с повышением температуры скорость окисления иодида нитритом снижается.
Растворимость кислорода в исходных рассолах достаточна для окисления содержащегося в них иодида до элементного иода (2,5-1О'3 г-экв/л). Растворимость оксида азота в рассолах несколько ниже (0,6-10'4 г-моль/л). Кинетические исследования показывают, что скорость окисления I" в присутствии оксида азота не зависит от концентрации растворенного кислорода и зависит от концентрации NO, Г и pH раствора:
2
= 1,24 • 109 [Г ]2 [NO]16 [Н+ ]'’3. dt
Процесс протекает й две стадии. В первой стадии оксид азота окисляется кислородом до промежуточного соединения (вероятно, HNO2), а во второй стадии происходит окисление иодида соединением HNO2. Вторая стадия является лимитирующей, определяющей суммарную скорость всего процесса по схеме
NO + NO -> N2O2 (быстро протекающая реакция)
N2O2 + О2 -> N2O4 (медленно протекающая реакция)
352
N2O4 + 2NO + 2H2O —> 4HNO2 (быстро протекающая реакция)
2HNO2 + 2Г + 2Н* -> Ь + 2NO + Н2О (медленно протекающая реакция)
При сопоставимых концентрациях NaNC>2 и NO скорости реакции окисления иодида примерно равны. При pH 1,5 в 2,5 н. NaCl и концентрации иодида 50 г/м3 в течение 15 мин реакция протекает на 95,5 — 96%.
Окисление иодида озоном. Использование озонированного воздуха в качестве окислителя исключает применение химических реагентов в процессе выделения иода из буровых вод. В слабокислой среде (кислотность 1—3 мг-экв/л) достигнуто окисление 79 — 84% содержащегося в буровых водах иодида. В лабораторных условиях из буровых вод получен иод путем окисления исходного иодида озоном до йодата с последующей реакцией последнего с иодидом в кислой среде.
Воздушно-десорбционный способ получения иода. Воздушно-десорбционный способ получения иода из буровых вод идентичен и основан на том же принципе, что и способ извлечения брома, и отличается лишь отсутствием стадии очистки от хлора. Это дает возможность предприятиям использовать оборудование как для получения иода, так и брома.
Схема процесса состоит из стадий: очистка буровой воды от примесей; подкисление буровой воды; выделение иода; десорбция иода воздухом; абсорбция иода из иодовоздушной смеси; выделение элементного иода из концентратов, полученных в процессе поглощения иода.
Воздушно-десорбционный способ может быть использован и для извлечения иода из щелочных вод. Система отличается лишь тем, что из нее исключается стадия подкисления.
Воздушно-десорбционный способ получения иода прост и менее трудоемок, аппаратура компактна, что позволяет легко автоматизировать технологический процесс. Способ обеспечивает получение качественного целевого продукта. Эффективность производства повышается особенно при использовании исходных растворов с повышенным содержанием в них иода и проведение процесса при повышенных температурах.
Согласно схеме получения иода (рис. 7.3), исходные отфильтрованные иодсодержащие рассолы (буровые воды) из приемной емкости 1 перекачиваются насосом 2 в нижнюю часть хлоратора 3. Туда же из напорной емкости 4 поступает серная кислота и хлорная вода, образующаяся в смесителе 6 путем смешения воды из приемной емкости 7, и отфильтрованный в фильтре 5 хлор. В нижнюю часть хлоратора поступает также серная кислота из напорной емкости 4. Подкисление буровой воды ведут серной или хлороводородной кислотой. Независимо от 353 12 Химическая технология неорганических веществ, кн. 2
вида кислоты подкисление исходной буровой воды проводят до pH 2—3,5. Затем вводят окислитель — хлорную воду.
Хлоратор 3 представляет собой полый цилиндрический аппарат, в котором для перемешивания установлены диафрагмы. В процессе хлорирования соблюдается хорошее перемешивание реагентов и небольшая концентрация окислителя — не более 2 г/л активного хлора. Превышение указанного предела приводит к локальному переокисле-нию Г до IO3.
Окисленные в хлораторе 3 буровые воды поступают в десорбционную башню S, где растворенный иод извлекается встречным потоком воздуха. Процесс десорбции осуществляют в башнях с насадкой при скорости воздуха 0,5—1 м/с (считая на полное сечение башни). Количество затрачиваемого воздуха зависит от давления пара иода над рассолом и всегда меньше, чем больше содержание иода в буровой воде, выше температура и ниже ее соленость (рис. 7.4).
Содержание иода в отходящей из десорбера 8 паровоздушной смеси колеблется в пределах от 0,05 до 0,25 мг/л, при этом расход воздуха в 1,1—1,8 раза больше теоретического. Степень десорбции обычно составляет 92 — 97%.
В составе образующегося в десорбере иодовоздушной смеси кроме иода содержатся и другие вещества, обладающие заметным давлением пара над рассолом. В процессе извлечения иода из кислой буровой воды в иодовоздушной смеси содержатся диоксид углерода (в
Рис. 7.3. Схема получения иода воздушно-десорбционным способом:
/ — приемная емкость; 2, 13— насос; 3— хлоратор; 4, 7 — емкость; 5 — фильтр; 6 — смеситель;
8 — десорбер; 9 — сборник; 10— абсорбер; 11 — сборник для смеси кислот; 12 — вентилятор;
13 — центробежный насос
354
результате разрушения карбонат- и гидрокарбонат-ионов) и нафтеновые кислоты. Содержание диоксида углерода зависит от количества карбонатов в исходной буровой воде, а содержание нафтеновых кислот достигает 0,5 кг на 1 кг иода. В случае извлечения иода из щелочной буровой воды количество диоксида углерода и нафтеновых кислот в иодовоздушной смеси относительно невелико.
Процесс извлечения иода из иодовоздушной смеси производят введением в смесь диоксида серы в присутствии паров воды:
SO2 + I2 + 2Н2О = 2HI + H2SO4
Рис. 7.4. Изменение коэффициента распределения иода между газовой фазой и водными растворами солей и водой в зависимости от температуры
Процесс практически осуществляют передачей иодовоздушной смеси в абсорбер 10. При этом введение диоксида серы в газоход производят перед абсорбером 10, который изнутри заполнен керамическими кольцами Рашига и частично стеклянной ватой. С целью уменьшения потерь иода насадка должна быть влажной и потому башня периодически, а при необходимости и непрерывно орошается циркулирующим раствором смеси кислот по системе абсорбер 10 — сборник для смеси кислот 11 — центробежный насос 13 — абсорбер 10. Плотность орошения не превышает 1 м3/(м2-ч).
Образующаяся смесь кислот HI и H2SO4 окисляется кислородом воздуха с выделением свободного иода, а с целью исключения его потерь расход диоксида серы повышают до 170—250% от стехиометрических расчетов. Содержание свободного иода в циркулирующем растворе не должно превышать 0,1 г/л. Концентрация образующейся смеси кислот (80—120 г/л HI, 55—90 г/л H2SO4) зависит от давления водяного пара в иодовоздушной смеси. Коэффициент абсорбции иодида водорода в зависимости от плотности орошения (м/ч) и скорости воздуха Wr (м/с) равен
Ка =27,1^жХл>
где т = 0,05—0,06 и п = 1,1—1,2.
Повышение температуры и концентрации смеси HI и H2SO4 в абсорбционной башне выше указанного предела увеличивает потери 355
12*
иода по причине роста давления пара иодоводорода над раствором. Содержащиеся в иодовоздушной смеси нафтеновые кислоты практически не улавливаются сорбентом и по известным данным содержание их в растворе по отношению к иоду равно 2—4:1000, в то время как в газовой фазе это соотношение равно 15—45:100.
В дальнейшем иод выделяют из раствора путем введения окислителей (хлора, йодата, хлората). Выход иода составляет 95—98%.
Получающиеся после выделения иода маточные растворы с содержанием 25—35 г/л НС1 и 80—120 г/л H2SO4 используют для подкисления исходной буровой воды.
Сорбент также может быть использован для получения иодоводо-родной кислоты или ее солей. Прямое разделение HI и H2SO4 дистилляцией невозможно, так как в процессе нагревания смеси до 30—60° С протекают следующие реакции:
2Н1 + H2SO4 = I2 + SO2 + 2Н3О
6HI + H2SO4 = 3I2 + S + 4Н2О
8HI + H2SO4 = 4Ь + H2S + 4Н2О
Для получения чистой иодоводородной кислоты смесь обрабатывают хлоридом бария:
HI + H2SO4 + ВаС12 = HI + BaSO4 + 2НС1
Образующийся сульфат бария отфильтровывают и промывают. Водный раствор HI и НС1 подвергают фракционной дистилляции. В первой фракции, кипящей ниже 125—126° С, содержится практически весь HCI, 0,002—0,04% 12 и 0,3—0,9% HI. Кубовый остаток имеет состав 55—56% HI, 0,1—Ю,2% НС1. Выход иодида водорода достигает 96—98%.
Степень извлечения иода из подкисленных буровых вод составляет 77—92%.
Разработан процесс, в котором восстановителем является сульфид водорода. Процесс поглощения иода ведут 20%-ным раствором иодоводородной кислоты (рис. 7.5), которая поглощает иод с образованием полииодидов I’, затем их восстанавливают сульфидом водорода:
i; + H2S = 3r + S + 2H4
Побочным продуктом в этом случае является элементная сера, которую отделяют от раствора путем фильтрации. Расход сульфида водорода в 1,9—2,5 раза больше теоретического. Это объясняется 356
получать иодид калия непосредствен-
но из иодовоздушной смеси. Образующийся по реакции гидрокарбонат калия хорошо растворим в воде и не выпадает в осадок. Поскольку раствор иодида калия является хорошим поглотителем иода, то поглощение можно вести непосредственно этим раствором, добавляя к раствору щелочь и пероксид водорода для восстановления поглощенного иода и превращения его в иодид калия.
Угольно-адсорбционный способ извлечения иода из рассолов основывается на безнапорной фильтрации рассола через слой зернистого угля. Вместо активированного угля можно использовать иониты, крахмал и некоторые высокомолекулярные соединения.
Угольно-адсорбционный способ извлечения иода состоит из следующих стадий: подкисление буровой воды для нейтрализации ее естественной щелочности и создания необходимой кислотности; выделение элементного иода с помощью окислителей; адсорбция иода из раствора активированным углем; десорбция иода с угля раствором щелочи; выделение иода-сырца из концентратов под действием кислоты и окислителя (рис. 7.6).
Для адсорбции иода применяют активированный уголь КАД, получаемый специальной обработкой каменноугольного обогащенного полукокса • перегретым паром при температуре 800—1000° С. Применяемый в технологии уголь КАД имеет насыпную плотность 0,350—0,400 г/см3 с размерами частиц 1,0—3,5 мм. Влажность угля 10%, зольность 9—14%. Уголь имеет несколько видов пор: микропоры (10‘3—10‘4 см), переходные поры (10‘5—10"6 см) и микропоры. Число микропор в угле не влияет на величину абсорбции, но определяет ее скорость. Радиус микропор имеет тот же порядок, что и молекула. Микропоры представляют из себя не случайные промежутки между кристаллитами углерода, а свободные объ-357
H2SO4
Уголь на реактивацию
Рнс. 7.6. Схема получения иода угольно-адсорбционным методом:
/ — приемный сборник; 2, //, /2, /4, /9 — центробежный насос; 3, 5 — напорная емкость;
4 — хлоратор; б — смеситель; 7 — адсорбер; 8 — приемник; 9 — отмывочный котел; 10 — растворитель; 13 — отстойник; 15 — очистительный узел; /б, 17 — емкость; 18 — угольный фильтр
емы в атомной структуре. В процессе адсорбции они заполняются поглощенным веществом. Эффективная удельная поверхность микропор равна 350—850 м2/ч.
Буровые воды (рассолы) из сборника 1 центробежным насосом 2 перекачиваются в напорную емкость 3, откуда направляются в хлоратор 4 на хлорирование. После хлорирования рассолы в смесителе 6 смешиваются с серной кислотой, подаваемой из напорной емкости 5.
В процессе хлорирования подкисленных (серной кислотой) рассолов образуется свободный иод по схеме
2Г + С12 = 12 + 2СГ
5Г + Юз + 6Н+ = 312 + ЗН2О 2Г 4- СЮ’ 4- 2Н+ = 12 + СГ + Н2О
358
После смесителя 6 рассолы направляются в каскадный адсорбер 7, в котором адсорбцию иода осуществляют активированным углем. Адсорбер 7 представляет собой вертикальный цилиндрический аппарат, изготовленный из железобетона с кислотоупорным покрытием, имеющий ложное дно, на котором помещается слой угля.
Диаметр адсорберов 1,9—3,5 м, высота 2,8—4 м. В адсорбер загружают от 4 до 20 м3 угля, степень заполнения 60—80%. Адсорберы устанавливают группами (по три-четыре аппарата). Процесс осуществляется противотоком: через слой свежего угля протекает исходная вода, прошедшая другие адсорберы, а через насыщенный уголь — поступающая буровая вода. С целью обеспечения самотека адсорберы размещают уступами или создают разность уровней. В процессе насыщения угля в первом по ходу буровой воды адсорбере уголь выгружают, а на его место загружают уголь из следующего адсорбера и т.д.
Для интенсификации процесса уголь в адсорбере перемешивают сжатым воздухом. Вода проходит через адсорберы снизу вверх, в результате чего она более равномерно распределяется по слою угля, предотвращается его слеживание и загрязнение. Скорость пропускания воды при работе с чистыми растворами 4—5 м/ч, с загрязненными— до 3 м/ч. Продолжительность пребывания воды в группе адсорберов для чистой воды составляет около 1 ч, а для загрязненных—1,5 ч. *
Для правильного ведения процесса адсорбции необходимо своевременно выгружать и перегружать уголь, не допуская потерь иода с отработанной водой и обеспечивая равномерный выпуск готового продукта. Уголь выгружают для отмывки после того, как степень поглощения иода в первом адсорбере уменьшится до 20—25%. При работе с чистой водой уголь может регенерировать 4—6 раз, а при работе с загрязненными водами — 2—3 раза. После такого количества регенераций активность угля значительно снижается и его направляют на реактивацию.
Загрузку и выгрузку угля проводят гидротранспортом по гидропроводам в виде пульпы (Т:Ж = 1:4—6).
Существенным недостатком данного способа является способность активированного угля поглощать нафтеновые кислоты. Количество их, адсорбируемое углем, может достигать 10—15% от массы угля, т.е. почти столько же, сколько сорбируется иода из загрязненных вод. В процессе отмывки иода с угля гидроксидами щелочных металлов нафтеновые кислоты растворяются в них, поэтому требуется специальная очистка растворов гидроксидов для получения чистого иода.
Отработанные растворы после адсорберов 7 направляются на производство брома, а иодсодержащий уголь передается в сборник-приемник S, откуда в отмывочный котел 9, в который из растворителя 10 на-359
сосами 11 перекачивают растворы гидроксида натрия. В отмывочный котел передают также уголь из угольных фильтров 18 и приемника 8.
С целью десорбции иода уголь в котле 9 промывают горячей водой для удаления солей и кислоты, а затем нагревают с раствором щелочи при 90—99° С в течение 2—6 ч. Раствор сливают, а уголь промывают водой до полного удаления раствора. Промывные воды в зависимости от концентрации иода в них либо присоединяют к первому раствору, либо используют для промывки следующих порций угля и приготовления раствора щелочи. Промывка длится 36—48 ч.
Остаточное содержание иода в отмытом угле до 0,1%, а щелочи до 0,4%. Отмытый уголь после обработки кислотой и нитритом в очистительном аппарате 15 возвращают на адсорбцию.
В процессе отмывки иода гидроксидом натрия наряду с реакцией
312 + 6МаОН = 5NaI + NaIO3 + ЗН2О
идут восстановительные процессы, в результате чего в щелочах содержится меньше йодата натрия, чем должно быть по уравнению реакции. Обычно в производстве в иодат превращается 2—4% иода вместо расчетных 16,7%. Растворы содержат также нафтеновые кислоты и остатки гидроксида натрия (0,4—2,4 кг/м3). Содержание иода в получаемых щелоках в пределах 20—35 кг/м3.
Теоретический расход гидроксида натрия на обмывку 1 т иода с угля составляет 0,315 т. Фактический же расход значительно больше, поскольку часть гидроксида расходуется на побочные процессы: нейтрализацию кислоты, растворение нафтеновых кислот, содержащихся в угле, и т.п. Практический расход гидроксида натрия в процессе работы на чистых водах—1,0—1,2 т, а на загрязненных — до 2 т на 1 т целевого продукта.
Разработан способ отмывки иода с угля растворами карбоната и сульфита натрия. Карбонат натрия в качестве реагента для отмывки иода имеет то преимущество, что он отмывает уголь не только от нафтеновых кислот, но и от гипса:
CaSO4 + Na2CO3 + СО2 + Н2О = Са(НСО3)2 + Na2SO4
При отмывке сульфитом натрия идет реакция
12 + Na2SO3 + Н2О = 2NaI + H2SO4
Значительный интерес представляет способ получения кристаллического иода без участия реагентов на отмывку его с угля. В процессе нагревания угля, насыщенного иодом, без доступа воздуха при атмосферном давлении или в вакууме до 650—700° С иод в значительной 360
степени возгоняется и может быть собран в виде кристаллов в охлаждаемом холодильнике. Часть иода при этом восстанавливается до иодида и может быть отмыта водой. В процессе же перегонки с перегретым паром достигается более полная отгонка иода при 325—420° С. Уголь после термической десорбции обладает высокой активностью.
В процессе электрохимической десорбции иода насыщенный уголь подают в катодное пространство электролизера и заливают раствором электролита, например сульфата натрия. Электролиз ведут при повышенной температуре, так что иод, переходящий в раствор на катоде и затем выделяющийся на аноде, возгоняется.
Ионитный способ получения иода в отличие от угольно-адсорбционного способа имеет следующие преимущества:
1) высокая сорбционная емкость ионита по иоду (до 350—400 кг/м3 по сравнению с 50 кг/м3 для угля КАД); 2) многократное использование анионита в процессе сорбции иода; 3) селективность способа сорбции иода; 4) возможность проведения процесса сорбции при пониженном расходе окислителя и меньшей кислотности; 5) возможность полной механизации и автоматизации производства; 6) способ может быть применен и для буровой воды с низкой температурой и высокой концентрацией иода.
Согласно существующей технологии, исходная иодсодержащая вода предварительно очищается от соединений железа, отстаивается или фильтруется, затем хлорируется для перевода Г в 12. В качестве анионита применяют смолу АВ-17 в СГ-форме. Хлорированная и подкисленная вода поступает в один из двух попеременно действующих адсорберов. В адсорбере происходит сорбция элементного иода на ионообменной смоле. Отработанную воду через смолоуловитель направляют на сброс. После насыщения иодом смолу, не выгружая из адсорбера, обрабатывают раствором сульфита натрия для восстановления элементного иода до иодида, который десорбируют раствором хлорида натрия, доокисляют оставшийся на смоле иодид подкисленным раствором нитрита натрия, а затем промывают водой. Концентрированные и слабые растворы, получаемые в процессе десорбции, собирают в сборниках. Неконцентрированные растворы из сборника передают на извлечение иода.
Получение элементного иода из концентратов. При получении элементного иода из концентратов в качестве окислителей применяются хлор и хлорат калия.
При использовании чистых растворов пользуются хлором (рис. 7.7). Процесс окисления проводят в слабокислой (до 0,5 г-экв/л) или нейтральной среде. Реакция протекает со значительной скоростью и количественно. Однако при быстром окислении иод выделяется в виде мелких кристаллов, сильно адсорбирующих нафтеновые кислоты. Поэтому, применяя загрязненные растворы, пользуются 361
медленно действующими окислителями — хлоратом калия или йодатом натрия. Последний получают путем частичного окисления хлором иодида натрия, содержащегося в концентрате:
Nal + ЗС12 + ЗН2О = NaIO3 + 6НС1
Окисленную часть концентрата смешивают с неокисленной:
NalO3 + 5NaI + 6НС1 = 3I2 + 6NaCl + 3H2O
Процесс окисления иодида хлоратом калия идет по реакции
КС1О3 + 6NaI + 3H2SO4 = 3I2 + КС1 + 3Na2SO4 + 3H2O
Процесс окисления йодатом натрия проводят при кислотности растворов 0,6—0,75 г-экв/л, а хлоратом калия— 1,0—1,5 г-экв/л и температуре 40—50° С. Для ускорения реакции окисления иодида хлоратом калия в процесс вводят катализаторы — хлорид железа (III), соли ванадата аммония и др. Катализаторы ускоряют процесс в 4—6 раз.
При окислении хлоратом калия и йодатом натрия получают крупные кристаллы иода, которые быстро оседают, легко отделяются от
Рис. 7.7. Область расположения изотермы адсорбции иода углем КАД из промышленных рассолов:
А — изотерма адсорбции из его водных растворов; Б—область расположения адсорбции иода из промышленных рассолов; В—пределы измерения концентрации иода в промышленных рассолах
маточных растворов и не захватывают органические примеси.
Конец процесса выделения иода определяют анализом маточных растворов, в которых должно оставаться не более 0,6 кг/м3 иод-иона. Конец процесса определяют по значению окислительно-восстановительного потенциала, который в момент окончания окисления должен равняться 485 мВ.
На рис. 7.8 приводится схема выделения и очистки иода, согласно которой исходные иодидные концентраты подогревают в подогревателе 7, после чего в смесителе 2 смешивают с отработанным поглотительным раствором. Смесь растворов фильтруется в фильтре 5, хлорируется в реакторе 4, после чего отстаивается в отстойнике 5, откуда суспензия кристаллов направляется в напорную емкость S, а маточные растворы собираются в емкости 6, из которой перекачиваются на адсорбцию. Сус-
362
NaOH
Рис. 7.8. Схема выделения и очистки иода:
1— подогреватель; 2 — смеситель; 3— фильтр; 4 — реактор; 5 — отстойник; 6 — емкость для маточного раствора; 7, 77, 16—насосы; 8, 15 — напорные емкости; 9 — центрифуга; 10 — сборник; 12, 20, 23 — бункера; 13 — электротельфер; 14 — переносной бункер; 77 — скруббер (абсорбер);
18, 25 — вентилятор; 19—циклон; 21 — питатель; 22 — сушилка; 24 — калорифер
пензия кристаллов направляется в центрифугу 9. Отжатые кристаллы выгружаются в бункер 12, а оттуда в передвижной бункер 14. Промывные воды после центрифуги 9 собираются в сборник 10, откуда насосом 11 перекачиваются в адсорбер, где они смешиваются с маточными растворами из емкости 6. Иод из бункера 14 подается с помощью тельфера 13 в бункер 20 для загрузки через питатель 21 в барабанную вращающуюся сушилку 22, в которой сушится воздухом при 50° С, подаваемым вентилятором 25 через теплообменник 24. Сухой продукт из сушилки 22 собирается в бункере 23, откуда направляется на упаковку.
Горячая газовоздушная смесь из сушилки 22, проходя циклон 19, направляется в скруббер 17, где очищается от твердых частиц орошением раствором гидроксида натрия из напорной емкости 75. Растворы циркулируют с помощью центробежного насоса 16. Часть отработанного поглотительного раствора по мере необходимости перекачивается насосом 16 в смеситель 2.
Для выделения иода из концентратов используют также электрохимическое окисление.
363
Технический иод, согласно действующему ГОСТ 545—76, производят двух сортов (А и Б) с содержанием основного вещества 99 и 97%. Согласно требованиям ГОСТа, лимитируется содержание органических веществ не более 0,08 и 0,15%, хлора — 0,01 и 0,015%, а также для сорта А влаги не более 0,8%.
В процессе регенерации иода из иодида водорода, образующегося в качестве побочного продукта, в некоторых процессах органического синтеза для окисления применяют кислород. 10—30%-ный водный раствор иодоводородной кислоты окисляют при ПО—130° С в присутствии небольшого количества азотной кислоты или поверхностно-активных веществ типа алкилсульфокислот. Газообразный иодид водорода окисляют под давлением при 50° С или при 200—400° С, пропуская через слой веществ с развитой поверхностью.
Способы очистки иода. Получаемый из производственных концентратов иод по содержанию примесей не отвечает требованиям, предъявляемым к продукции реактивной квалификации. Разработано значительное число способов очистки иода: сублимация, плавление и фильтрация, направленная кристаллизация, промывка кристаллов с последующей сушкой, ректификация, плавление под слоем серной кислоты, экстракция из растворов органическими растворителями, перекристаллизация из органических растворителей с последующей сушкой.
Наиболее эффективным способом очистки иода от примесей является сублимация. Процесс проводят при атмосферном давлении или в вакууме (50—100 Па) в стальных, чугунных или керамических ретортах. В испарителе поддерживают температуру плавления 114—180° С, а иногда ниже (80—90° С). Образующиеся пары иода конденсируют при 40—60° С. Установлено, что интенсивность испарения иода пропорциональна разности концентрации паров непосредственно над зеркалом испарения и выше него и зависит от интенсивности конвекционных токов. Разность концентраций растет с повышением температуры.
Процесс проводят, поддерживая температуру паров иода ниже, чем температуру его плавления. В этих условиях иод осаждается в виде крупных кристаллов, легко отделяющихся от стенок аппарата.
Процесс сублимации иода проводят в печах, загруженных стальными кюветами или керамическими тиглями с иодом. Печи обогреваются газами, образующимися при горении газа. Пары иода, выходящие из печи, конденсируются в керамических или асбоцементных холодильниках, охлаждаемых воздухом. Испарители иода изготовляют из сплава ниобия с танталом, а конденсатор — из титана.
Для получения более чистого иода его многократно перегоняют. Загрузку и перегрузку иода из одного испарителя в другой и выгрузку на 364
кристаллизацию ведут в расплавленном состоянии. В некоторых производствах иод-сырец смешивают с реагентами, связывающими влагу и органические примеси (до 8—12% оксида кальция), пары иода пропускают через фильтры, задерживающие механические примеси.
Плавление исходного иода-сырца осуществляют в токе перегретого водяного пара под давлением 0,15—0,25 МПа. При этом образуются два слоя — жидкий иод и водный раствор. Воду сливают, а плав охлаждают на вращающихся ность валка иод срезают ножом.
Концентрация H2SO4, % (масс.)
Рис. 7.9. Растворимость иода в серной кислоте
валках, а налипший на поверх-
Плавление иода под слоем 90—93%-ной серной кислоты при 120—150° С дает возможность извлечь из иода воду и разрушить органические вещества (рис. 7.9). Расход серной кислоты 2—3 т на 1 т иода. Остывший плав дробят, промывают водой для удаления остатков серной кислоты и сушат.
Для получения более чистых сортов иода исходное сырье возго
няют в вакууме или перекристаллизовывают из его раствора тетрахлоридом углерода. Предложено также высаливание иода из раствора в этаноле, экстракция иода из растворов иодида калия с помощью органических растворителей.
7.3. ИОДИД ВОДОРОДА И ИОДОВОДОРОДНАЯ КИСЛОТА
Иодид водорода HI — бесцветный удушливый газ, сильно дымящий на воздухе. Длина связи 0,1609 нм, энергия диссоциации 294,66 кДж/моль; ц = 0,448-10‘30 Кл-м. Температура плавления — 50,8° С, температура кипения — 35,36° С. Плотность жидкого иодида 2,86 г/см3 (-50,8° С); Гкрит = 150,7° С, ркрит = 8,31 МПа; С° =29,16 Дж/(моль-К); ДНлл = 2,86 кДж/моль, ДНи°сп = 19,77 кДж/моль, ДН°- = 26,36 кДж/моль; 52°98 = 206,48 Дж/(моль-К); давление пара (в МПа): 0,402 (0° С), 3,817 (100° С). Растворимость в воде (в 100 г при 1013 гПа): 234 (10° С), 132,5 (127° С); водных растворов HI: 1,1649 (20%, масс. HI), 1,4029 (40%), 1,5600 (50%), 1,7700 (60%); температура кипения азеотропной смеси с водой (56%, масс. HI) 127° С; сильный окислитель.
Иодид водорода термически не стабилен, при повышенных температурах диссоциирует по обратимой реакции на элементы.
365
Степень диссоциации 22,9% при 227° С и 30,1% при 727° С. Под действием света и длиной волны менее 400 нм разлагается. Сильный восстановитель.
Водный раствор иодида водорода (иодоводородная кислота)— бесцветная жидкость с резким запахом. Сильная кислота, p7<a~ll. На воздухе, и особенно при действии света, медленно окисляется с выделением элементного иода. Производится промышленностью кислота, содержащая 47% иодида водорода.
Иодид водорода может быть получен прямым синтезом из элементов. Проведенные исследования показали, что реакция между иодом и водородом идет с участием свободных атомов иода и имеет цепной характер.
При обычных условиях иод не реагирует с водородом. Однако при 100° С начинается медленная реакция между ними, что связано с более легкой диссоциацией молекулы иода на атомы по схеме Ь>21.
Далее цепь продолжается по схеме
Н2 +1- >Н1 + Н-
Н+12—^->Н1+1-
При стехиометрическом соотношении реагентов реакция образования иодида водорода протекает при 175° С на 90%, при 700° С — 78%. Следовательно, для проведения реакции с достаточно высоким выходом необходимо вести процесс при не очень высоких температурах, применяя для ускорения реакции катализаторы. В настоящее время в производстве иодоводорода в качестве катализатора применяют платинированный асбест.
Водород очищают от кислорода и сушат. Он проходит через испаритель с иодом. Температуру в испарителе иода поддерживают 100—160° С для перевода иода в газовую фазу. Далее смесь проходит через слой катализатора, нагреваемого до 100—400° С. Реакция образования иодида водорода эндотермическая, поэтому процесс подогрева постоянен.
Газы, выходящие из реакционной зоны, охлаждают до -20° С, при которой иодоводород освобождается от непрореагировавшего иода, так как он конденсируется. Окончательная очистка смеси от иода достигается в процессе взаимодействия его со ртутью. Для отделения избыточного водорода иодид водорода конденсируют с помощью жидкого воздуха или азота.
Для получения иодоводородной кислоты иодид водорода абсорбируют водой. 366
Иодид водорода получают путем гидролиза иодида фосфора (Р13), который в свою очередь получают при взаимодействии иода с красным фосфором непосредственно в реакционном сосуде.
Другой способ получения иодида водорода основан на взаимодействии иода с сульфидом водорода. Реакцию проводят в водной среде:
H2S + I2 = 2HI + S
Образующуюся кислоту перегоняют, отбирая фракцию, кипящую при 125—127° С (56% HI).
Иодоводородная кислота образуется также при использовании сульфида водорода в процессе поглощения иода из иодовоздушной смеси и в смеси с серной кислотой при использовании для той же цели диоксида серы. Для получения из этой смеси чистую иодоводо-родную кислоту сульфат-ион осаждают в виде BaSO4. Смесь хлороводородной и иодоводородной кислот разделяют ректификацией, поскольку азеотропная смесь НС1—Н2О кипит при 108,7° С, а смесь HI—Н2О —при 126,5° С.
Иодид водорода получают также по схеме
3FeI2 + 4Н2О = Fe3O4 + 6HI + Н2
В литературе описаны процессы, в которых иодид водорода получают фотохимическими реакциями с нитритом лития и сульфатом железа (II):
LiNO2 + Н2О + I2 = LiNO3 + 2HI
2FeSO4 + 2Н2О + I2 = 2FeSO4(OH) + 2HI
7.4. ПОЛУЧЕНИЕ ИОДИДОВ
Иодиды одно- и двухзамещенных металлов — соли. Все они, кроме Agl, Cu2I2 и Hg2I2, хорошо растворимы в воде.
Из неорганических соединений иода наибольшее значение имеют иодиды натрия, калия и аммония. Иодиды образуют бесцветные кристаллы. Иодид натрия кристаллизуется в кубической системе с плотностью 3,67 г/см3; плавится при 651° С. Растворимость его в воде показана на рис. 7.10. Образует кристаллогидраты NaF2H2O и NaI-5H2O. Сильно гигроскопичен. При длительном хранении кристаллы его окрашиваются в желтый цвет из-за выделения свободного иода.
367
Йодиды калия и аммония кристаллизуются лишь в безводной форме в кубической системе. Плотность иодида калия 3,13 г/см3, плавится при 723° С. Плотность иодида калия 3,13 г/см3.
Получение иодидов щелочных металлов и аммония. Получение иодидов щелочных металлов непосредственным взаимодействием иода с растворами соответствующих гидроксидов в химической промышленности не применяется из-за трудности отделения их от йодатов. Из диаграмм растворимости в системе Nal—NalOa—Н2О (рис. 7.10, а) видно, что в чистом виде можно выделить лишь иодат калия и натрия. Йодиды же всегда будут загрязнены иодатами, для удаления последних необходимо применять многократную перекристаллизацию.
Исходя из изложенного, иодиды получают такими способами, в которых иод взаимодействует с гидроксидами или карбонатами в присутствии восстановителей, обеспечивающих превращение йодата в иодид. Предпочтение отдают таким восстановителям, при взаимодействии которых с иодом и щелочью не образуется твердых побочных продуктов, например пероксида водорода, муравьиной кислоты, формальдегида и гидразина по реакциям:
2МОН + 12 + Н2О2 = 2MI + 2Н2О + О2
М2СО3 + 12 + НСООН = 2MI + 2СО2 + Н2О
10 20 30 40 50 60 70
Nal, % б
Рис. 7.10. Изотермы растворимости в системе: а — KI—КЮ3—Н2О;
б — Nal—NaIO3—Н2О
368
2М2СО3 + 2I2 + НСОН = 4MI + ЗСО2 + Н2О
4МОН + 2I2 + N2H4 = 4М1 + 4Н2О + N2
При использовании в качестве восстановителя пероксида водорода готовят раствор иода в растворе иодида металла и к нему прибавляют щелочной раствор пероксида водорода с температурой 8—10° С, охлаждая реакционную массу так, чтобы температура ее не превышала 20° С. В полученном растворе снова растворяют иод и добавляют раствор щелочи, повторяя эту операцию до получения раствора, содержащего 550 кг/м3 MI, 3 кг/м3 МОН и не содержащий МЮз.
В процессе восстановления муравьиной кислотой последнюю прибавляют к исходному раствору иода в растворе гидроксида металла при температуре -70° С.
В процессе получения иодидов кроме приведенных восстановителей можно применять сульфид водорода, металлическое железо и т.п.:
I2 + 2МОН + H2S = 2MI + 2Н2О + S
3FeI2 + 8МОН + I2 = Fe3O4 + 8MI + 4Н2О
На некоторых заводах вместо Fel2 используют Fe3I8 и реакцию ведут по схеме
Fe3I8 + 8МОН = Fe3O4 + 8MI + 4Н2О
Fe3I8 + 4М2СО3 = Fe3O4 + 8MI + 4СО2
Процесс получения иодидов ведут в реакторах из нержавеющей стали с мешалкой и паровой рубашкой. Исходный раствор карбоната или гидроксида нагревают до кипения, а затем медленно вводят раствор в течение 3—6 ч (с целью получения легко фильтруемого осадка Fe3O4, последний отделяют фильтрацией растворов). Осадок промывают до полного извлечения иодидов.
Концентрации исходных растворов МОН и Ме2СОз, а также оптимальные условия кристаллизации иодидов из растворов могут быть определены с помощью диаграмм растворимости в системах Nal—Ка?СОз—МаНСОз—Н2О (рис. 7.11), KI—К2СОз—КНСО3—Н2О (рис. 7.12), KI—КОН—К2СОз—Н2О (рис. 7.13) и KI—Nal—Н2О (рис. 7.14). Полученные рассмотренными способами растворы иодидов освобождают от избытка щелочи, добавляя Рез18 или HI, очищают от SO42' с помощью гидроксида бария и упаривают. Дополнительную очистку растворов от других примесей проводят обработкой их активированным углем, ионообменными смолами и др.
369
Na2CO3
Рис. 7.11. Растворимость (мол.%) в системе Nal—NaHCO3—Na2CO3—Н2О при 25° С. Поля кристаллизации: / —Nal-2H2O; 2 —Na2COvH2O;
3 — Na2COr7H2O; 4 — Na2COv 10H2O;
5 — NaHCOrNa2COvH2O (трона); 6 — NaHCCh
Рис. 7.12. Растворимость (мол.%) в системе К!—КНСО3—К2СО3—Н2О при 0—50° С
Рис. 7.13. Растворимость (мол.%) в системе KI—КОН—К2СО3—Н2О при 0 и 50 °C. В поле кристаллизации гидроксида калия твердая фаза при 0°С КОН-2Н2О, при 50° С КОН Н2О; точка превращения около 32 — 33° С
Рис. 7.14. Растворимость в системе KI—Nal—Н2О при 0, 25 и 50° С
Йодиды получают из смеси иодоводородной и серной кислот, образующихся в процессе поглощения иода из иодовоздушной смеси диоксидом серы. С этой целью путем обработки смеси хлоридом бария серную кислоту переводят в хлороводородную:
H2SO4 + ВаС12 = 2НС1 + BaSO4
370
а образующейся смесью (HCI + HI) обрабатывают хлорид калия или хлорид натрия:
КС1 + HCI + HI = KI + 2НС1
Хлороводородную кислоту при температуре выше 110° С отгоняют, и в остатке получается чистый иодид калия или натрия. Этот способ может быть использован для очистки иодидов от хлоридных и бромидных примесей.
Существует способ получения иодидов из рассмотренной же смеси кислот, основанный на осаждении из нее иодида меди и переработки ее в целевые иодиды действием гидроксидов.
Изучены несколько способов получения иодида аммония, легко разлагающегося при высоких температурах с образованием водорода:
2NH4I —2NH3 + 12 + Н2
Иодид аммония получают при действии аммиака и диоксида углерода на раствор иодида калия в 87,5%-ном изопропиловом спирте:
2NH3 + СО2 + Н2О + 2KI = 2NH4I 4- К2СО3
Получают иодид аммония действием иодида калия на нитрат аммония:
KI + NH4NO3 = NH4I + KNO3
7.5. ПОЛУЧЕНИЕ ЙОДАТОВ И ПЕРЙОДАТОВ щелочных металлов
Иодаты — соли йодноватой кислоты НЮ3, представляющей бесцветные стекловидные кристаллы с температурой плавления выше 110° С. В процессе нагревания триоксоиодат(У) водорода разлагается:
2НЮз —1-+ 12О5 + Н2О
Триоксоиодат(У) водорода относится к типу кислоты ЭО2(ОН)„ с К. = 1,7-10~! и рЯа = 0,77.
Водные растворы йодноватой кислоты получают окислением иода хлором, концентрированной азотной кислотой или другими сильными окислителями с последующей отгонкой воды в вакууме.
371
При действии концентрированной (98—99%-ной) азотной кислоты на иод получается йодноватая кислота в виде белого осадка:
3I2 + 10HNO3 = 6НЮ3 + 10NO + 2Н2О
Йодноватая кислота используется для звукопроводов акустоопти-ческих устройств.
Соли йодноватой кислоты — йодаты — разлагаются при температуре лишь выше 400° С. Они обладают сильными окислительными свойствами. При взаимодействии с иодидами в присутствии кислот йодаты выделяют элементный иод.
Иодат калия КЮз— бесцветные кристаллы; растворим в воде (8,13 г в 100 г при 20° С). Встречается в природе как примесь к чилийской селитре, а №Юз — в виде самостоятельного минерала лаутарита. При сильном нагревании йодаты разлагаются, выделяя кислород. Иодат калия применяется в качестве реактива в иодатометрии.
Для получения йодатов применяют способы химического или электрохимического окисления иодидов или элементного иода. В качестве же окислителей применяют хлор, триоксохлорат(У) калия (бертолетову соль), манганат(УП) калия (перманганат, марганцово-кислый), концентрированную азотную кислоту и т.д.
Окисление иода триоксохлоратом(У) калия проводят концентрированной хлороводородной кислотой при нагревании. При этом происходит реакция
l2 + HCI + 2КС1Оз = KIO3HIO3 + С12 + КС1
В процессе охлаждения полученного раствора дииодат калия КЮз-НЮз выпадает в осадок, который после перекристаллизации из воды переводят в иодат действием гидроксида калия:
КЮз-НЮз + КОН = 2КЮз 4- Н2О
Из водных растворов получают кристаллы.
Окисление манганатом (VII) калия идет по реакции
312 4- 10КМпО4 4- 2Н2О = 6KIO3 4- 10MnO2 + 4КОН
Перйодаты — соли иодной кислоты HIO4. Она известна в виде дигидрата бесцветных кристаллов с температурой плавления 122° С, выше которой разлагается с выделением кислорода; относится к кислотам типа ЭО (ОН)„; слабая кислота. Образует соли: метапериодаты MIO4, ортопериодаты M3IO5, парапериодаты МзН2Ю6. 372
Перйодаты получают окислением иодидов или же йодатов при температуре кипения по реакциям:
NaIO3 + ЗМаОН + С12 = Na2H3IO6 + 2NaCl
NaIO3 + 4NaOH + Cl2 = Na3H2IO6 + 2NaCI + H2O
KIO3 + 2KOH + Cl2 = KIO4 + 2KC1 + H2O
Образующиеся перйодаты относительно трудно растворимы в растворах хлоридов и при охлаждении переходят в кристаллическое состояние. В качестве окислителей используют также триоксохло-par(V) калия и натрия, персульфаты калия и натрия. Перйодаты получают также электролитическим окислением йодатов.
ГЛАВА 8
МАРГАНЕЦ И ЕГО СОЕДИНЕНИЯ
8.1. МАРГАНЕЦ
Содержание марганца в земной коре составляет 0,1% (масс.), в океанической воде — 2 • 10‘7%. Он входит в состав значительного количества минералов, преимущественно в виде оксидов. В табл. 8.1 приводятся наиболее распространенные природные соединения марганца.
Таблица 8.1. Минералы, содержащие марганец
Минерал Химическая формула Плотность
Пиролюзит р-МпО2 4,75- -5,00
Вернадит МпО2/?Н2О 3,0—3,2
Курнакит (браунит) ЗМп20зМп810з 4,7- -4,9
Манганит Р-МпООН(Мп2Оз-Н2О) 4,2- -4,4
Манганозит МпО
Пирохроит и бескремит МпОН2О (а и р)
Родохрозит МпСОз 3,4- —3,6
Родонит MnSiO3 3,5- -3,7
Гаусманит Мп3О4 (а и р) 4,7- -4,9
Гидрогаусманит Мп3О4/?Н2О (а и р)
Биксбиит (Мп,Ге)20з
Псиломелан /иМО /?МпО2хН2О, где [М = Ba,Ca,K,Mn(II)] 2,3- -4,7
Гроутит /-МпООН 4,15—4,35
Марганец в природе имеет один устойчивый изотоп 55Мп. Конфигурация внешней электронной оболочки атома степень окисления от +2 до +7, наиболее устойчивы и распространены соединения Мп(П) и Mn(VII); энергии ионизации при последовательном переходе от Мп° к Мп2+ соответственно равны 7,435 и 15,6401 эВ; электроотрицательность по Полингу 2,5; ионные радиусы Мп2+ 0,080 нм (4), 0,089 нм (5), 0,104 (7), Мп7+ 0,039 (4), 0,060 нм (6).
Марганец — серебристо-белый металл. Известно четыре кубические кристаллические модификации: ниже 710° С устойчива а-форма, 374
при 710-1090° С — P-форма, при 1090—1137° С — у-форма, а при охлаждении до комнатной температуры у-форма переходит в тетрагональную кристаллическую модификацию. Ниже -173° С марганец анти-ферромагнитен, при более высоких температурах парамагнитен; магнитная восприимчивость +9,6-10"6. Модификации а, Р и 5 хрупкие, а у-Mn пластичен, однако после деформации медленно разрушается; твердость по шкале Роквелла а-Мп 70, у-Мп 20.
Марганец легко окисляется на воздухе, образуя выше 800° С окалину, состоящую из внутреннего слоя МпО и внешнего слоя Мп3О4. Ниже 800° С образуется Мп2Оз, а при температурах ниже 450° С — МпО2. Поглощает водород (до 60 см3 на 100 г Мп) с образованием твердых растворов.
Марганец взаимодействует с галогенами, давая дигалогениды МпХ2 — кристаллы, хорошо растворимые в воде (кроме MnF2). Взаимодействует при нагревании с серой, азотом, фосфором, углеродом, кремнием и другими неметаллами. Известны нитриды марганца: MnN6, Mn5N2, Mn4N, MnN, Mn6N5, Mn3N2 и MnJSI, где x = 9,2—25,2. Сульфид марганца MnS существует в трех кристаллических модификациях (Р-форма — минерал алабандин); плохо растворимы в воде и может быть осажден из растворов солей двузамещенного марганца сульфидами щелочных металлов.
Марганец с фосфором образует фосфиды: МпР, МпРз, Мп2Р, МпзР, МпзР2 и Мп4Р. С углеродом образует карбиды: МпзС, Мп5С2, Мп15С4, Мп23Сб и Мп2С7.
С кремнием марганец образует силициды: MnSi, MnSiij, Mr^Si и Mn5Si3.
Марганец взаимодействует с минеральными кислотами, образуя соответствующие соли, а из растворов солей двухвалентного марганца при pH 8,7 осаждается слабое основание Мп(ОН)2. Еще более слабые основные свойства проявляет нерастворимый в воде Мп(ОН)з. Известны неустойчивые соли ортомарганцоватистой Н4МпО4 и марганцоватистой Н3МпО4 кислот.
Марганец получают методами металлотермии (карбо-, силико-или алюмотермия) рудных концентратов, а также химическим способом — выщелачиванием марганца серной кислотой с последующим электролитическим восстановлением сульфата марганца.
В производстве исходные руды предварительно обогащают, а при необходимости подвергают их термообработке (обжигу) для разложения карбонатов, восстанавливают (для перевода в соединения Мп2+) или сплавляют с кварцитом.
Карботермическим восстановлением обычно получают высокоуглеродистый ферромарганец (содержание углерода до 6—8%), алюмо-термическим — чистый марганец.
375
Наиболее чистый марганец, содержащий всего до 0,1% примесей, получают электрохимическим способом электролизом растворов сульфата марганца, содержащих сульфат аммония при pH 8,0—8,5.
Марганец применяется для раскисления, десульфурации и легирования сталей t (более 90% производимого марганца). Для удаления из стали серы и кислорода из металла марганец вводят в виде высокоуглеродистого ферромарганца (8—9 кг на 1 т стали) для легирования стали в виде средне- и малоуглеродистого ферромарганца. Марганец является компонентом сплавов цветных металлов, сплавов с алюминием и магнием. Он придает стали, алюминию и магнию твердость, прочность и устойчивость к коррозии. Из марганца производят его сплавы для создания защитных антикоррозионных покрытий на металлах.
8.2. ХИМИЧЕСКАЯ ПЕРЕРАБОТКА МАРГАНЦЕВЫХ РУД
Марганец в природе встречается лишь в виде его соединений (см. табл. 8.1) и в основном в марганцевых и железных рудах осадочного происхождения. Руды подразделяют на три класса: марганцевые— при содержании марганца более 40% и железа менее 10%; железомарганцевые — 5—40% марганца и 10—35% железа; марганцовистые железные руды при содержании марганца менее 5%.
Важнейшей является пероксидная руда, представленная пиролюзитом, псиломеланом, манганитом и браунитом и содержащая 60—70% диоксида марганца. Основной примесью, как обычно, является диоксид кремния. Получаемый после мокрого обогащения руды концентрат содержит до 96% диоксида марганца.
Карбонатные марганцевые руды представляют собой манганокаль-циты и кальциевые родохрозиты. При термообработке руды содержащиеся в ней карбонаты марганца и кальция диссоциируют с образованием соответствующих оксидов.
Для переработки бедных марганцевых руд, содержащих 26—28% марганца, 2—6% и более железа, 30—40% диоксида кремния и 0,05—0,1% серы, разработаны химические способы.
Одним из широко применяемых методов переработки бедных марганцевых руд является сернисто-кислотный способ. Этот способ, применяемый во многих странах, основан на разложении бедных марганцевых руд диоксидом серы с последующим пиролизом образующегося сульфата марганца.
В процессе насыщения диоксидом серы водной суспензии пиро-люзитовой руды или шлама образуется дитионат марганца:
MnO2 + 2SO2 = MnS2O6
376
который с течением времени, и особенно при повышении температуры, разлагается с образованием сульфата марганца и выделением диоксида серы:
MnS2O6 = MnSO4 + SO2
Протеканию процесса с ростом количества сульфата марганца способствуют побочные реакции, к которым относятся: окисление диоксида серы до триоксида
2SO2 + МпО2 —> МпЗгОб
а также взаимодействие диоксида марганца с его дитионатом в присутствии SO2:
MnO2 + MnS2O6 = 2MnSO4
При соблюдении постоянной продолжительности процесса изменение отношения Т:Ж в пределах от 1:2 до 1:10 не влияет на количество переходимого в раствор марганца. Однако при концентрированных суспензиях уменьшается степень перехода в раствор железа и других примесей, особенно фосфора, которые снижают степень фильтруемости образующихся растворов. С повышением концентрации исходного диоксида серы степень перехода марганца в раствор возрастает. При соотношении Т:Ж = 1:10, отношении Mn:SO2, равном 1:3, концентрации диоксида серы -14% и температуре процесса 80° С в течение 90 мин достигается 94—95%-ный перевод марганца в раствор.
Переработку манганитовых руд, содержащих сесквиоксид марганца, ведут комбинированным методом. В первой стадии с целью перевода сесквиоксида марганца в его диоксид руду обрабатывают серной кислотой:
МП2О3 + H?SO4 — MnSO4 + MnO2 + Н2О
Выделившийся при этом диоксид марганца перерабатывается в сульфат марганца обработкой диоксидом серы.
В процессе насыщения диоксидом серы водной суспензии манганитовой руды (без предварительной серно-кислотной обработки) образуются сульфит и сульфат или дитионат марганца по следующей схеме:
Мп?Оз + 2SO2 = MnSCh + MnSO4
Мп2Оз + 3SO2 = MnSO3 + MnS2O6
377
Поскольку образующийся при этом дитионат марганца разлагается до сульфата, а сульфит марганца окисляется содержащимся в исходном газе кислородом или кислородом воздуха, то в результате процесса получается раствор сульфата марганца. Манганитовые руды реагируют с диоксидом серы со значительно меньшей скоростью, чем пиролюзит-ные. Дополнительное измельчение исходных манганитовых руд ускоряет процесс их растворения, причем содержание в растворе дитионата увеличивается с одновременным уменьшением концентрации сульфата марганца. Такой процесс объясняется разностью скоростей процессов растворения руды и разложения образующегося дитионата марганца. В отличие от пиролюзитной руды при разбавлении суспензии из манганитовой руды (в пределах Т:Ж = 1:2 до 1:10) степень извлечения марганца увеличивается за счет роста количества растворенного диоксида серы в газе и соответственно в растворе ускоряется процесс разложения дитионата. В процессе обработки исходной водной суспензии манганитовой руды (Т:Ж = 1:10) диоксидом серы (16,2—17,6% SO2) при отношении Mn:SC>2, равном 1:1,9, и температуре 80° С в течение 3 ч достигается 90%-ное извлечение марганца в раствор.
В процессе обработки диоксидом серы водной суспензии карбонатной марганцевой руды образуется сульфит, который, окисляясь содержащимся в исходном газе кислородом или воздухом, переходит в сульфат по схеме
МпСОз + SO2 = MnSO3 + СО2
2MnSO3 + О2 = 2MnSO4
В данном процессе дитионат практически не образуется. Однако при 18—20° С реакция исходного диоксида серы с суспензией карбонатной руды идет медленно. Для ускорения этого процесса сырье предварительно обжигают. Для некоторых видов карбонатных руд характерно интенсивное разложение и при 20° С, а при 80° С достигается высокая степень извлечения марганца. Наилучшие же результаты получаются при Т:Ж = 1:10. Степень разложения исходного сырья зависит от структуры его строения. Поэтому схема получения и условия переработки карбонатных руд могут быть разными, если даже исходное сырье имеет одинаковый химический состав.
Процесс сульфатизации исходного диоксида марганца может быть произведен сульфатом железа (II) или пирита по схеме
4МпО2 + 4FeSO4 = 4MnSO4 + 2Fe2O3 + О2
8MnO2 + 4FeS2 + 11О2 = 8MnSO4 + 2Fe2O3
378
Процессы проводятся термообработкой исходных смесей при 550—600° С во вращающихся печах в течение 2—3 ч. В этих условиях степень извлечения марганца из исходного сырья для первой реакции составляет 98—99%, а для второй реакции — 89—90%.
При сульфатизации соединений марганца диоксидом серы при повышенных температурах оказалось, что процесс сульфатизации сесквиоксида марганца протекает с заметной скоростью и при 450° С. Скорость процесса резко повышается в интервале 450—600° С, а затем до 750° С практически не изменяется. Увеличение концентрации диоксида серы в исходном газе ускоряет процесс сульфатизации исходного сесквиоксида марганца (рис. 8.1). Процесс сульфатизации природной карбонатной марганцевой руды диоксидом серы протекает аналогично, однако скорость процесса сульфатизации растет во всем интервале температур особенно заметно выше 600° С (рис. 8.2).
Бедные руды оксидов марганца обогащают сульфидным способом, используя для этого газы промышленных предприятий, содержащие сульфид водорода.
Образующийся сульфид марганца обычно переводят в сульфат су-льфатизирующим обжигом или обработкой серной кислотой.
Алюмосиликатные марганцевые руды при высоких температурах восстанавливают в присутствии сульфата или карбоната кальция.
Рис. 8.1. Степень сульфатизации сесквиоксида за 30 мин при различных температурах и концентрациях диоксида серы в газе (цифры на кривых — концентрация диоксида серы в %)
Рис. 8.2. Изменение степени сульфатизации природной карбонатной марганцевой руды за 30 мин при различных температурах и концентрациях диоксида в газе (цифры на кривых — концентрация диоксида серы в газе в %)
379
Присутствие карбоната кальция предотвращает выделение сульфида водорода в процессе последующего выщелачивания. Марганец из бедных руд извлекают также термообработкой исходного сырья с добавкой сульфата или хлорида аммония. Существует также способ извлечения марганца термообработкой исходного сырья в псевдоожиженном слое при 750° С.
Из растворов, образующихся в процессе извлечения марганца из его руд, получают концентрат высокого качества осаждением его в виде карбоната. Осаждение проводят раствором карбоната натрия. Процесс осуществляют непрерывным способом путем подачи исходных растворов в нижнюю часть реактора, а образующийся карбонат марганца отбирают из верхней части. Суспензия перемешивается при 55° С и pH 5,7—7,2 в течение 3—5 ч, чем достигается значение плотности кристаллов образующегося карбоната марганца 1,0—1,3 г/см3. Суспензия из реактора передается в сгуститель Дорра, фильтруется и промывается на ФПАКМ, после чего направляется на сушку.
Существует способ извлечения марганца из бедных карбонатных руд путем обработки их водной суспензии диоксидом углерода под давлением. При этом давление диоксида углерода регулируют так, чтобы карбонат марганца полностью перешел в раствор в виде гидрокарбоната, а количество воды в системе должно быть достаточным, чтобы после снижения давления до одной атмосферы в процессе обратного осаждения карбоната марганца перешедшие в раствор гидрокарбонаты железа, кальция и другие остались в растворе.
Давление диоксида углерода и минимальное количество воды могут быть вычислены для данной руды с помощью следующих констант равновесии при 25° С:
МпСОз = Мп2+ + СО2’ FeCOj = Fe2+ + СО2’ К\ = [Мп2+][СО2’] = 5,05-10'10; К2 = [Fe2+][CO2’] = 2,1110'11;
СаСОз = Са2+ + СО2’ К2 = [Са2+][СО2] = 4,82-10’9;
СО2’ + Н2СО3 = 2НСО; [НСО;]2 4 [Нгсо3нсогг
СО2 + Н2О = Н2СО3 [Н2СО3] ^5 / J со,
Вследствие незначительной растворимости компонентов, по утверждению авторов, можно вместо активностей воспользоваться молярными концентрациями. 380
Константы диссоциации Н2СОз:
[FT][HCO;]
1 [Н2СО3]
= 4,52-10-7,
JH*][CO;] " [нсо;]
= 5,59-Ю"11.
Вследствие незначительной диссоциации иона гидрокарбоната допускают, что все ионы карбоната превращаются в ионы гидрокарбоната.
Отсюда
^ЛЛ=[Мп?НСО;1г=М8.10-7, (1)
К2К^К5 = [ре^Цн9°П2 = 5,76 • 10-9, /со. (2)
= [Са'ИНС0^ = 1,32 • 10-6. /со. (3)
Например, если руда содержит 32,9% МпСОз, 23,0% РеСОз и 28,6% СаСОз, то в 1 м3 руды находится 2,86 моль МпСОз, 1,98 моль РеСОз и 2,86 моль СаСОз. Обозначим через х — кг воды на 1 м3 руды, необходимое для растворения всего СаСОз, т.е. 2,86 моль Са2+, ПРИ Рсо = 'атм- Тогда [Са2+] = и [НСОз‘] = где — чис-х х
ло молей Mn2+ + Fe2h + Са2+, остающихся в растворе, из которых число молей Мп2+ обозначим у, а число молей Fe2+ — z. Подставив эти значения в уравнения (1)—(3) и решив их, находим у = 0,299, z = =0,0125 и х = 478 кг. Давление СО2, необходимое для полного растворения 2,86 моль.
Мп2+ в 478 кг воды можно рассчитать из системы уравнений (1) и (2). Обозначим количество растворяющихся молей Fe2+ в 1 кг воды через г. Тогда
2,86 2,86 2,86 ,
478 2 478 + 478 +Г)1 = j 3 g. j Q"7
381
„ 2,86 2,86 .
rf2(47R+ 47«+Г)} о
-----478 478-------- 5,76-Ю-9.
/с<>2
Решив эту систему уравнений, находим г - 2,5-10’4, т.е. вместе с МпСОз растворится 2,5-10"4-478 = 0,119 моль РеСОз, и /СОо = 29,7 атм. Поскольку степень превращения СО2 при 25° 082%, то необходимое для процесса давление диоксида углерода равно 36 атм. Полученный из одного кг руды раствор, отфильтрованный от нерастворимых веществ, будет содержать 2,86 моль МпСОз, 2,86 моль СаСОз и 0,119 моль РеСОз в виде гидрокарбонатов. В результате понижения давления до 1 атм из раствора выделится в осадок 2,86-0,299 - 2,56 моль МпСОз и 0,119—0,0125 = 0,107 моль РеСОз. Кальций полностью остается в растворе. Степень извлечения из руды марганца составит ттуЮО = 89,5%, а железа ^^100 = 5,4%.
2,86 1,98
8.3. ОКСИДЫ И ГИДРОКСИДЫ МАРГАНЦА
Марганец образует следующие оксиды: МпО, Мп2О3, МпО2, МП3О4, Мп2О2 и Mn5Og. Все оксиды, кроме MfyOj, — нерастворимые в воде кристаллы.
В процессе термообработки высшие оксиды выделяют кислород с образованием низших оксидов:
P-MnO2 5b0°c > P-Mn2O3 —Р-МП3О4 -1|80'с > у-Мп3О4
МпО-ОН 260°с > а-Мп2О3 600*с > Р-Мп2О3 970°с > Р-Мп3О4
На рис. 8.3 приведены значения равновесных давлений (lgpOi) при диссоциации отдельных фаз элементного марганца. Линии 7, 2, 3 и 4 делят диаграмму на поля устойчивости отдельных фаз — элементного марганца, оксида марганца, оксида марганца (II, III), сесквиоксида и диоксида марганца.
При термообработке на воздухе или в атмосфере кислорода выше 300° С оксид и сесквиоксид окисляются до диоксида марганца.
Монооксид марганца МпО (минерал манганозит) до -155,3° С — устойчивая гексагональная модификация, а выше — кубическая (табл. 8.2). Он является полупроводником, антиферромагнетиком с точкой Нееля 122 К. Магнитная восприимчивость монооксида +4,85-10‘3(293 К). 382
1-103
Рис. 8.3. Зависимость lgpo> от у- при диссоциации оксидов марганца по реакциям:
/ — 4МпО2 = 2Мп2О3 + О2;
2 — 6Мп20з = 4Мп3О4 + О2;
3 — 2Мп364 = 6МпО + О2;
4 — 2Мп + О2 = 2Мп + О2
Монооксид марганца обладает слабоосновными свойствами, при нагревании восстанавливается водородом и активными металлами до элементного марганца:
МпО + Н2 -> Мп + Н2О
при взаимодействии оксида магния с минеральными кислотами образуются соли двухвалентного марганца слабо-розовой окраски. Розовая окраска кристаллогидратов и растворов солей Мп2+ обусловлена ионом [Мп(Н2О)4]2+:
MnO + H2SO4 = MnSO4 + Н2О
MnO + 2НС1 = МпС12 + Н2О
MnO + 2HNO3 = Mn(NO3)2 + Н2О
Таблица 8.2. Свойства оксидов марганца
Показатель МпО Р-МпО2 Мп2О.з а-МпзО4 Мп(ОН)2 у-MnO (ОН)
Цвет Серо- Коричне- Коричне- Коричне- Бледно- Черный
зеленый во-черный во-черный во-черный розовый
Сингония Кубиче- Тетрагона- Кубиче- Тетрагона- Гексагональ- Ромбиче-
ская льная ская льная ная ская
Параметры 0,453 0,927(b) 0,287
решетки, нм: а с 0,4448 0,438 0,286 0,941 0,575 0,942 0,334 0,468
Число фор-
мульных единиц в ячейке 4 2 16 4 — 4
Пространственная группа Fm3m PJmnm 1аЗ 14\lamd Сзт Р hnm
Г ° С ' пл, 1569* 585** 100** 1564** — —
Плотность, г/см3 5,18 5,026 4,50 4,856 3,258 4,2
с;:, Дж/(мольК) 44,1 54,0 107,5 139,3 — 62,8
кДж/ моль -385,1 -521,5 -957,7 -1387,6 -700**** —
С ° Дж/ (моль-К) 61,5 53,1 110,4 154,8 — —
*Д//'’П 43,9 кДж/моль, “Грнп (с выделением О2), "*Д//® = 127,6 кДж/моль, ““Дб^, =
= -618,7 кДж/мол ь.
383
С гидроксидом натрия при 700—800° С и избытке кислорода образуется Na3MnO4 по схеме МпО + 3NaOH + 0,7502 = Na3MnO4 + 1,5 Н2О, а при действии на оксид марганца (NH^S образуется сульфид марганца.
Оксид марганца МпО получают в производстве терморазложением при 300° С в инертной атмосфере по следующим схемам:
Мп(ОН)2 МпО + Н2О
Мп(С2О4) -> МпО + СО2 + СО
Мп(МО3)2 -> МпО + 2NO2 + 0,5О2
МпСОз -> МпО + СО2
Оксид марганца получают также контролируемым восстановлением диоксида и сесквиоксида марганца водородом или оксидом углерода при 700—900° С.
Оксид марганца применяется в качестве компонента ферритов и других керамических материалов, шлака для десульфуризации металлов, микроудобрений, является катализатором процесса дегидрогенизации пиперидина, а также антиферромагнитным материалом.
Сесквиоксид марганца М112О3 существует в а- и Р-модификаци-ях с температурой перехода ос —> Р 600° С; парамагнетик, парамагнитная восприимчивость 4-1,41 10‘5 (293 К).
При 300° С восстанавливается до монооксида по схеме
МП2О3 + Н2 = 2МпО 4- Н2О
При нагревании с алюминием восстанавливается до элемента:
Мп2О3 4- 2А1 = 2Мп 4- А12Оз
Сесквиоксид марганца реагирует с разбавленными азотной и серной кислотами с образованием диоксида марганца и соответствующих солей по схеме
Мп2Оз 4- H2SO4 = MnSO4 4- МпО2 4- Н2О
МП2О3 4" 2HNO3 = Мп(ЫОз)2 4- МпО2 4- Н2О
Сесквиоксид марганца получают терморазложением манганита:
2МпООН —Мп2О3 + Н2О 384
Оксид марганца (II,III) МпзО4 (минерал гаусманит); а-МпзО4 при 1160° С переходит в |3-МпзО4 с кубической кристаллической решеткой; А/7° перехода ос —> (3 20,9 кДж/моль; парамагнетик, магнитная восприимчивость +1,24-10’5 (298 К). Проявляет химические свойства, подобные оксиду и сесквиоксиду марганца.
Оксид марганца (VII) МП2О7 (гептаоксид димарганца, марганцевый ангидрид), высший оксид марганца — маслянистая жидкость, зеленая в отраженном и темно-синяя в проходящем свете с температурой плавления 5,9° С и плотностью при 20° С 2,40 г/см3. При повышении температуры до 55° С начинает переходить на a-Mn2O3 с выделением кислорода, после чего, быстро разогреваясь с достижением температуры около 90° С, взрывается. При этом скорость детонации около 400 м/с, а чувствительность к удару такая же, как у гремучей ртути. При взаимодействии с влагой воздуха разлагается до диоксида марганца с выделением кислорода и озона. Однако при отсутствии влаги может сохраняться в закрытом сосуде при — 10° С длительное время. Мп2О7 — сильный окислитель, он окисляет даже NaCl и Nal до NaClO3 и ИаЮз. При контакте с оксидом марганца (VII) горючие вещества воспламеняются.
Оксид марганца (VII) получают взаимодействием КМпО4 с серной кислотой на холоду по схеме
2КМпО4 + H2SO4 = Мп2О7 + K2SO4 + Н2О
Оксид марганца Мп2п (Мп1уО4)з — твердое вещество, не растворимое в воде. Может быть получен окислением МпО или МпзО4; легко разлагается на диоксид марганца и кислород.
Из гидроксидов марганца стехиометрическими соединениями являются лишь Мп(ОН)2 и МпО(ОН), а другие представляют собой гидратированные оксиды переменного состава, близкие по своим химическим свойствам соответствующим оксидам. Кислотные свойства гидроксидов увеличиваются с возрастанием степени окисления марганца: Мп(ОН)2<МпО(ОН)<МпО2-хН2О<МпзО4-хН2О.
Гидроксид марганца Мп(ОН)2 практически нерастворим в воде; основание средней силы; растворим в растворимых солях аммония; на воздухе окисляется до МпО2хН2О.
Гидроксид МпО(ОН) известен в двух модификациях; при 250° С в вакууме обезвоживается до у-Мп20з; нерастворим в воде. Природный манганит не входит в реакцию с азотной и слабой серной кислотами, медленно реагирует с SO2 H2O, а искусственно полученный легко реагирует с минеральными кислотами. МпО(ОН) легко окисляется кислородом до Р-МпО2.
385 13 Химическая технология неорганических веществ, кн. 2
8.4. ДИОКСИД МАРГАНЦА
Физико-химические свойства. Диоксид марганца МпО2— самое распространенное соединение марганца в природе. Наиболее устойчива P-модификация (минерал пиролюзит). Известны также ромбическая у-МпО2 (минерал рамсделит), а также а, 5 и 8, рассматриваемые как твердые растворы различных форм диоксида марганца. Парамагнетик, магнитная восприимчивость +2,28-10"3 (293 К). Диоксид марганца — нестехиометрическое соединение, в его решетке всегда наблюдается недостаток кислорода, амфотерный оксид. Он образует с минеральными кислотами соли четырехзамещенного марганца, легко разлагающиеся водой с выделением гидратированного диоксида марганца, а в качестве кислотного оксида — соли марганцовистой кислоты, манганиты. В зависимости от способа образования диоксид марганца существует в различных модификациях, отличающихся физическими свойствами. В процессе нагревания диоксид марганца отщепляет кислород и переходит последовательно в сесквиоксид, оксид марганца (II,Ill) и в оксид марганца МпО. При этом температуры перехода диоксида в сесквиоксид несколько различны в зависимости от модификаций исходного МпО2. Однако общий вид изобары диссоциации меняется незначительно и остается подобным изображенным на рис. 8.4. Переход МпО2 в Мп2Оз и другие оксиды происходит в состоянии твердых растворов оксидов.
Диоксид марганца при 170° С восстанавливается водородом:
МпО2 + Н2 = МпО + Н2О
При сплавлении со щелочами или основными оксидами диоксид марганца играет роль кислотного оксида и образует манганиты:
МпО2 + СаО = СаМпОз
При нагревании с кислотами диоксид марганца проявляет окислительные свойства:
MnO2 + 4НС1 = МпС12 + С12 + 2Н2О
При взаимодействии МпО2 с наиболее сильными окислителями образуются производные Mn(VI) и Mn(VII):
ЗМпО2 + КСЮз + 6КОН = ЗК2МпО4 + КС1 + ЗН2О
2МпО2 + ЗРЬО2 + 6HNO3 = 2НМпО7 + 3Pb(NO3)2 + 2Н2О 386
Применение диоксида марганца. Широкое использование диоксида марганца в различных отраслях народного хозяйства базируется на его хороших абсорбционных свойствах. Как природный пиролюзит, так и полученный в промышленности искусственный диоксид марганца применяются в качестве деполяризаторов в гальванических элементах, в процессе изготовления промышленных противогазов — в качестве поглотителя оксида угле
Рис. 8.4. Изобара диссоциации диоксида марганца
рода, в качестве низкотемпературных катализаторов для некоторых химических процессов.
Диоксид марганца хорошо поглощает пары ртути. В процессе промывки водной суспензии диоксида марганца при pH 2—5 в двухполочном пенном аппарате из газа улавливается до 99% содержащихся в нем ртутных паров. Количество поглощенной ртути достигает 20% от массы сухого диоксида марганца в суспензии. Поглощенная ртуть извлекается, а диоксид марганца регенерируется термообработкой его при 500° С в течение двух часов. Восстановленная пероксидная марганцевая руда используется в качестве катализатора в процессе селективного окисления примеси оксида углерода в азотоводородной смеси, идущей на синтез аммиака.
Из оксидов марганца с добавлением оксидов редкоземельных элементов получают монокристаллы, обладающие пьезоэлектрическими и ферроэлектрическими свойствами.
Диоксид марганца применяется в производстве стекла, в керамической промышленности для изготовления глазури для придания изделиям пурпурного или коричневого оттенка, а также для приготовления фиолетово-черной эмалевой краски и эмалирования изделий из железа. Диоксид марганца применяют также для получения его соединений, сиккативов, как деполяризатор в сухих элементах, в качестве реагента для обнаружения хлор-иона, окислителя в гидрометаллургии цинка, меди, урана. Он является компонентом катализатора в гапколитовых патронах и др.
Искусственно получаемый диоксид марганца электрохимическим путем и специальной обработкой пиролюзита — ГАП (гипховский активированный пиролюзит) используют в основном в химических источниках тока как обладающую хорошими деполяризующими свойствами. Емкость гальванических элементов, изготовленных на ГАПе, на 15—20% выше, чем элементов, изготовленных на электролитическом 387
13*
диоксиде марганца. Особенно ценным свойством элементов, изготовленных на ГАПе, является их срок службы в течение длительного времени — до двух лет.
ГАП для элементной промышленности, согласно МРТУ 6-09 № 2939—66, должен содержать (в %): диоксида марганца — не менее 70, сесквиоксида марганца — не более 15, сесквиоксида железа— не менее 1,5, никеля — 0,03, кобальта — 0,004, меди — 0,03, сульфатов— 0,7, влаги — 4,0 и нерастворимого остатка в хлороводородной кислоте остатка — не более 12,0%.
Электролитический диоксид марганца должен содержать не менее 90% диоксида марганца и не более: 8% Мп20з, 4% гигроскопической влаги, 0,6% сульфатов, 1,5% Ре2Оз; величина pH — не ниже 3,6. Через сито №021 должно проходить 95% материала, через сито № 007 — не менее 65%.
Способы получения диоксида марганца. Существует много способов получения диоксида марганца. В производстве диоксид марганца получают разложением нитрата и гидроксида марганца при 200° С на воздухе, восстановлением перманганата марганца в нейтральной среде, а также электролизом солей двузамещенного марганца и др.
Получение диоксида марганца электрохимическим способом. Электролитом в процессе получения диоксида марганца электрохимическим способом является раствор, содержащий 300—350 г/л сульфата марганца и 180—200 г/л серной кислоты.
Процесс электролиза проводится при 20—25° С в химзащищен-ных ваннах, со свинцовыми электродами, при напряжении в ванне 3—3,5 В и при плотности тока на аноде 500 А/м2 и на катоде 1000—1200 А/м2. В ваннах большой мощности требуется установка охлаждаемых анодов. В результате окисления на аноде двузамещенного марганца образуется диоксид марганца, а на катоде выделяется водород. Общий процесс протекает по уравнению
MnSO4 + 2Н2О = МпО2 + H2SO4 + Н2
При 18° С нормальный потенциал окисления Мп2+—>Мп3+ равен + 1,511 В, а для Mnu—>Мп4+—1,642 В, т.е. на аноде наиболее вероятно образование Мп3+. В сильнокислом растворе образование Мп4+, требующее более высокого потенциала, происходит после завершения превращения Мп2+ в Мп3+. В слабокислом же растворе имеет место равновесие:
2Мп3+±; Мп2+ + Мп4+
которое сдвигается вправо вследствие гидролиза
Mn(SO4)2 + 2Н2О = MnO2 + 2H2SO4
388
Поэтому образование MnO2 происходит без окисления Мп3+ на аноде, где окисляется лишь Мп2+. В результате выход диоксида марганца по току зависит от кислотности раствора, которая повышается в процессе электролиза по мере уменьшения концентрации Мп2+. В сильнокислом же растворе, в котором концентрация Мп3+ и Мп4+ велика, происходит частичное обратное восстановление на катоде, доходящее до 25%. В слабокислом растворе анод покрывается рыхлой коркой, образующейся в результате гидролиза диоксида марганца, затрудняющего доступ Мп2^ к аноду и понижающего перенапряжение выделения кислорода, в результате чего усиливается его образование. Уменьшение концентрации Мп2+ также снижает выход по току. Поэтому отбираемый из ванн и направляемый на выщелачивание оксида марганца электролит содержит 50—60 г/л MnSO4 и до 450 г/л H2SO4. Сопоставление с начальным составом электролита показывает, что окисление на аноде достигает 80—85%.
Потери от катодного восстановления можно уменьшить применением графитовых катодов, а также полностью устранить, закрывая катод диафрагмой из ткани, стойкой в кислом электролите. Однако диафрагма загрязняется диоксидом марганца, что усложняет ее использование. Электролит необходимо очищать от примесей железа, меди, кобальта и других металлов, понижающих выход по току. Известно, что в присутствии в электролите 0,05 г/л железа или меди выход по току падает приблизительно на 30%. Рекомендуется применение порошковых (угольных) катодов.
Исследованиями процесса электролиза проточного раствора химически чистых MnSO4 и H2SO4 в ванне со стальными катодами, анодами из сплава свинца с 4% сурьмы и с хлопчатобумажной диафрагмой установлено, что выход по току снижается от 95,5 до 39,4% при повышении анодной плотности тока от 418 до 2650 А/м2 и возрастает от 39,4 до 79,1% с увеличением температуры анолита от 26,5 до 73° С. Максимальный выход по току был достигнут при содержании в электролите 207 г/л MnSO4.
Раствор электролита выпускают из ванны вместе с осадком диоксида марганца и передают в гуммированную центрифугу, где осадок отделяют от электролита и промывают водой. В связи с сильной адсорбционной способностью диоксида марганца его отмывка требует значительного количества воды. После центрифуги диоксид марганца высушивают от остаточной влаги и адсорбционной воды в сушилках при температуре не выше 100° С. Температура сушки регламентируется во избежание ухудшения адсорбционных свойств целевого продукта.
Известен и другой вариант получения диоксида марганца электрохимическим способом, согласно которому получение электролитического диоксида марганца ведут в ваннах с графитированными угольными электродами. Исходным сырьем является суспензия гид-389
роксида марганца (И), которую растворяют в серной кислоте (в кислом отработанном электролите) с добавкой пероксида водорода для восстановления диоксида марганца по схеме
MnO2 + H2SO4 + Н2О2 = MnSO4 + 2Н2О + О2
После фильтрации растворов от нерастворимого остатка (состоящего в основном из сульфата кальция) получают электролит, содержащий 100 г/л сульфата марганца, 20 г/л серной кислоты и менее 0,03 г/л железа и меди. Электролиз проводят в температурном интервале 85—95° С, при анодной плотности тока 100 А/м2 и катодной 100—140 А/м2 и напряжении 2,5—3 В. Выход по току 90—93%, а расход электроэнергии 2600 кВт-ч на 1 т продукта. Использование анодов одноразовое (продолжительность процесса электролиза 800—1000 ч), а катодов — 5000 ч. Образующийся в процессе электролиза диоксид марганца осаждается плотным слоем на анодах — графитированных угольных стержнях, укомплектованных в анодных рамах. По окончании процесса электролиза рамы с осадком промывают и демонтируют, после чего их комплектуют новыми электродами и загружают в ванны. Осадок целевого продукта, снятый с анодов, дробят, размалывают, промывают водой, высушивают и упаковывают.
Получение активированного пиролюзита. Исходным сырьем для получения ГАП (гипховский активированный пиролюзит, а ГИПХ — государственный институт прикладной химии) является наиболее высококачественный пиролюзит, содержащий 80—90% диоксида марганца. Исходный пиролюзит подвергают термообработке во вращающихся печах при 800—950°С. При этом диоксид марганца переходит в низшие оксиды, преимущественно в сесквиоксид марганца. О качестве материала судят по молярному отношению МпО:МпО2. Наилучшим считается соотношение 1:1, при котором МпО2 восстанавливается до Мп2Оз. На практике это соотношение меняется от 0,96 до 1,1.
Исходную руду после термообработки выщелачивают 17—18%-ной серной кислотой при 80° С. При этом серная кислота реагирует с сеск-виоксидом марганца с образованием сульфата и диоксида марганца:
Мп2Оз + H2SO4 = MnO2 + MnSO4 + Н2О
Образующийся сульфат марганца переходит в раствор, а диоксид марганца остается в осадке.
Выщелачивание плава проводят в растворителях типа Чанкса, состоящих из двенадцати ячеек с методическим выщелачиванием исходного плава и ростом концентрации растворов сульфата марганца из ячейки в ячейку. Чем полнее извлекается из плава диоксид мар-390
ганца, тем более высокого качества получается ГАП. Образующийся осадок ГАП промывают водой, нейтрализуют и высушивают при температуре не выше 100° С.
Отходом производства ГАПа является раствор сульфата марганца, который используют в качестве электролита в электролитическом производстве диоксида марганца, а также в производстве металлического марганца и для получения кристаллического сульфата или карбоната магния.
Получение активного диоксида марганца из бедных руд. Некоторые бедные марганцевые руды обладают высшей дисперсностью и при их переработке получают активные марганцевые материалы. При обработке этих руд минеральными кислотами получают соответствующие соли, из которых получают целевые продукты.
Азотно-кислотная переработка бедных марганцевых руд состоит в следующем. Исходную руду измельчают в шаровых мельницах до частиц размером 0,25 мм и подвергают термообработке во вращающихся печах. После охлаждения их в реакторах обрабатывают азотной кислотой. При этом образуется нитрат марганца по схеме
MnO + 2HNO3 = Mn(NO3)2 + Н2О
Полученные растворы фильтруют от нерастворимого остатка. Фильтрат упаривают при 116° С до 65%-ного твердого вещества. Затем в обогреваемой паром двухвальцовой сушилке нитрат марганца разлагают, причем для получения у-МпО2, используемого для изготовления сухих элементов, температура разложения не превышает 350°С. Процесс разложения идет по схеме
Mn(NO3)2 35?*с > MnO2 + 2NO2
Выделяющиеся оксиды азота, азотной кислоты и пары воды возвращаются на начало процесса для нитрации исходной руды, а диоксид марганца размалывают, промывают и сушат.
При проведении процесса разложения растворов нитрата марганца во вращающихся пламенных печах обеспечивается регенерация оксидов азота в азотную кислоту в пределах 95%.
Предложен также способ регенерации диоксида марганца из растворов, содержащих сульфаты аммония и марганца, получающихся в процессе окисления диоксидом марганца органических соединений. При этом исходные растворы обрабатывают известковым молоком:
MnSO4 + (NH4)2SO4 + Са(ОН)2 = Mn(OH)2 + NH3 + Н2О + CaSO4
391
Выделяющийся аммиак улавливают, а осадок гидроксида марганца растворяют в азотной кислоте.
После отделения гипса путем фильтрации растворы выпаривают до получения сухого нитрата марганца, из которого термообработкой при 200—300° С получают диоксид марганца.
Диоксид марганца получают из низкосортных марганцевых руд обработкой их концентрированной хлороводородной кислотой. Максимальное извлечение марганца из предварительно обожженной при 800° С руды достигается в течение 20 мин. При содержании в исходной руде 36,6% марганца и 30,4% железа в раствор переходит около 70% марганца, а остаток содержит 55,4% железа и 9,25% марганца. Гидролиз полученного раствора хлорида марганца при 10-кратном разбавлении водой в присутствии воздуха обеспечивает выпадение осадка, содержащего 85% МпО2 и практически не содержащего железа.
Диоксид марганца получают при окислении хлором в щелочной среде осадка гидроксида марганца.
Диоксид марганца, пригодный для деполяризаторов в сухих батареях, получают обработкой соляно-кислой вытяжки из восстановленного природного пиролюзита, содержащего 5—25% марганца в виде его хлорида и 0,5—5% свободной хлороводородной кислоты, щелочным раствором гипохлорита в процессе охлаждения. В конце процесса выделения осадка диоксида марганца pH смеси должен быть в пределах 2—5. Образующийся осадок диоксида марганца промывают водой и выдерживают длительное время в воде.
Разработан способ переработки сульфата марганца в его хлорид эквимолярным количеством хлорида кальция:
MnSO4 + СаС12 = МпС12 + CaSO4
После фильтрации осадка сульфата кальция обрабатывают раствор хлорида марганца избыточным количеством гидроксида кальция:
МпС12 + 2Са(ОН)2 -> Мп(ОН)2 + СаС12 + Са(ОН)2
В полученную суспензию Мп(ОН)2 и избытка Са(ОН)2 в растворе СаС12 пропускают воздух (кислород) для перевода Мп2+ в Мп4+. Для завершения процесса окисления суспензию хлорируют, и образующийся диоксид марганца фильтрацией отделяют от раствора хлорида кальция.
В присутствии водяного пара при -600° С твердый хлорид марганца гидролизуется с образованием оксидов по схеме
2МпС12 + ЗН2О Mn2O3 + 4НС1 + Н2
ЗМпС12 + 4Н2О Mn3O4 + 6НС1 + Н2
392
или с большей скоростью в присутствии паровоздушной смеси:
4МпС12 + 4Н2О + О2 2Мп2О3 + 8НС1
6МпС12 + 6Н2О + О2 2Мп3О4 + 12НС1
При 600° С степень превращения в паровой среде в течение 2,5 ч достигает 73%, а в паровоздушной среде за 2 ч — 99%. С повышением температуры скорость гидролиза увеличивается. Получаемые продукты являются смесью Р»Мп2Оз и Р-МпзОд.
При переработке бедных марганцевых руд с применением водных растворов аммиака исходные руды, содержащие МпО, обрабатываются ~ 18%-ным водным раствором аммиака, содержащим ~2% сульфата, хлорида, нитрата или карбоната аммония. При этом образуется раствор комплексной марганцево-аммонийной соли с концентрацией в пересчете на Мп(ОН)2 ~10 г/л. Процесс выщелачивания ускоряется в присутствии восстановителей, например сульфида аммония (в количестве 3—15 кг на 1 т руды). Из отфильтрованного раствора диоксидом углерода осаждают карбонат марганца. Температура процесса выше 60° С, а молярное отношение СО2:Мп в растворе больше 1,75. Полученный карбонат марганца переводят в его диоксид термообработкой суспензии при 150—315° С в течение нескольких часов.
8.5. СУЛЬФАТ МАРГАНЦА
Сульфат марганца существует в виде безводной соли и одно-, ди-, тетра-, пента- и гептагидратов сульфата марганца.
Физико-химические свойства сульфата марганца и его гидратов. Свойства сульфата марганца MnSO4 и его гидратов приведены в табл. 8.3.
Сульфат магния безводный плавится при 700° С; Ср - 100,24 Дж/(моль-К); AG^p = -958,11 кДж/моль; ниже 11 К (точка Нееля) антиферромагнетик, а выше — парамагнетик, магнитная восприимчивость + 1,366-1 О’6 (293 К). Около 850° С разлагается на МпзО4, SO3 и SO2. Растворимость в воде (г в 100 г): 52,9 (0° С), 64,8 (25° С), 35,5 (100° С), 20,5 (120° С), 3,9 (160° С).
Моногидрат MnSO4H2O образуется в процессе нагревания высших гидратов при -200° С, а из водных растворов осаждается в виде кристаллов в температурном интервале 150—160° С.
393
Таблица 8.3. Свойства сульфата марганца и его гидратов
Показатель MnSO4 MnSO4H2O MgSO4-4H2O MgSO4-5H2O
Цвет Бесцветный Светло-розовый Светло-розовый Розовый
Сингония Ромбическая Моноклинная Моноклинная Триклинная
Параметры элементарной ячейки, нм:
а 0,5267 0,7758 0,594 0,637
b 0,8046 0,7612 1,376 1,077
с 0,6848 0,7126 0,801 0,613
угол, град — 115,70 (р) 90,80(р) 98,8(a) 109,9(р) 77,8(у)
Пространственная группа Стет АНа Р2х!п Р\
Число формульных единиц в ячейке 4 4 4 2
Показатели преломления:
№ 1,632 1,521 1,517
№Р 1,595 1,519 1,510
М, 1,562 1,511 1,498
Плотность, г/см2 3,25 2,95 2,107 2,103
обр, кДж/моль -1066,7 -1387,4 — -2555,5
5° 2‘)Х, Дж/(моль-К) 112,6 155,65 — 370,6
Дигидрат MnSO4-2H2O кристаллизуется из водных растворов при 25—40° С, а тетрагидрат MnSO4-4H2O — при 26—27° С. В кристаллогидрате MnSO4-4H2O группа SO42' играет роль связующего мостика между атомами Мп(И):
394
Пентагидрат MnSO4- 5Н2О имеет температуру плавления 55° С (инконгруэнтно).
Гептагидрат MnSO4-7H2O— розовые кристаллы; температура плавления 8,6° С (инконгруэнтно); плотность 2,09 г/см3; ЛН0°бр = -3136,3 кДж/моль; устойчив ниже 8,6° С.
Сульфат марганца образует аммиакат с шестью, а моногидрат с пятью молекулами аммиака.
Известны двойные сульфаты М^Од-МпЗОд, а также их ди-, тетра- и гексагидраты.
Сульфат марганца встречается в природе в виде минералов сми-кита MnSO4H2O, маллардита MnSO4-7H2O, джококуита MnSO4-5H2O и илезита MnSO4-4H2O.
Известен сульфат марганца (III) Mn2(SO4)3 — зеленое аморфное или кристаллическое вещество; малоустойчив, разлагается при 160° С. Гидролизуется водой, растворяется в серной кислоте; сильный окислитель.
Известны также Mn2(SO4)-H2SO4- 4Н2О, двойные соли M'Mn(SO4)2, M'Mn(SO4)212H2O, где М' — Rb, Cs и некоторые другие металлы.
Получение сульфата марганца. Сульфат марганца получают из его растворов, образующихся при растворении в серной кислоте природного диоксида марганца и бедных марганцевых руд. Разработаны и другие способы, базирующиеся на предварительном восстановительном обжиге марганецсодержащей руды. К ним относится аммиачный способ, в котором выщелачивание восстановленной руды производится сульфатом аммония. Другие способы основаны на обработке серной кислотой марганцевой карбонатной руды.
Получение сульфата марганца из карбонатной руды обработкой ее разбавленной серной кислотой (отношение Mn:H2SO4 = 1:2,1, Ж:Т~3) в присутствии катализатора KI проводят при pH 2—3, температуре 80—90° С и при перемешивании смеси воздухом до полного выщелачивания марганца. Образующуюся горячую суспензию фильтруют. Растворы сульфата марганца направляют на получение кристаллического продукта, а осадок промывают холодной водой и промывную воду возвращают на начало процесса.
Выделение кристаллического сульфата марганца из его водных растворов осуществляется упаркой и кристаллизацией или высаливанием концентрированной серной кислотой.
Ввиду того, что растворимость сульфата марганца с повышением температуры от 27 до 100° С понижается, кристаллизация охлаждением концентрированного раствора оказывается невозможной. Изотермическая кристаллизация в процессе упарки из раствора воды затруднена из-за нарастания на греющих поверхностях выпарного 395
аппарата толстого слоя («шуба») соли. Процесс резко снижает коэффициент теплопередачи. Кроме того, в процессе выпарки образуются мелкие кристаллы MnSO4-H2O, затрудняющие в дальнейшем процесс их фильтрации и отделения от маточных растворов.
Для исключения этих трудностей процесс кристаллизации сульфата марганца осуществляют путем нагревания растворов. Растворимость сульфата марганца выше 100° С резко снижается (рис. 8.5). При этом в твердой фазе находится одноводная соль МпБОд-НзО. Если раствор содержит 20% MnSO4, то при нагревании его до 160° С в твердую фазу выделится 86%, а при 180° С — 94% от общего количества MnSO4.
Выделение сульфата марганца может быть осуществлено нагреванием раствора до 160° С в автоклаве острым паром, имеющим давление 7—8 атм. После 15-минутной выдержки при этой температуре в маточном растворе остается 38% сульфата марганца. Затем маточный раствор сливают через фильтр, находящийся внутри автоклава, а в автоклав загружают новую партию свежих растворов. После двух-или трехкратной кристаллизации соль выгружают.
Образующиеся в производстве маточные растворы применяют для разбавления серной кислоты, идущей в производство ГАПа. Поскольку в исходном растворе сульфата марганца содержится около 1% MgSO4, то в маточном растворе будет накапливаться сульфат магния. Присутствие сульфата магния в солевой системе благоприятствует процессу кристаллизации сульфата марганца. Одна
Рис. 8,5. Растворимость сульфата марганца в воде при 120—200° С
ко часть маточного раствора по мере накопления сульфата магния должна выводиться из технологического цикла.
Растворимость сульфата марганца сильно падает в присутствии в системе серной кислоты. Установлено, что при 25° С при содержании в системе 40,8% H2SO4 растворимость MnSO4 составляет 3,8%, а при 62% H2SO4 — 0,5%. При концентрации H2SO4 ниже 62% твердой фазой является MnSO4H2O, а при более высокой ее температуре в твердую фазу осаждается комплексное соединение MnSO4-H2SO4 H2O.
На основании приведенных данных предложен способ осаждения сульфата марганца из раствора. К раствору вводят серную кислоту до ее концентрации 40% H2SO4. Этим технологическим приемом достигают выпадения в осадок около 80%
396
сульфата марганца. Отделенные от маточных растворов в центрифуге кристаллы сульфата марганца направляются на сушку, а растворы — на повторное выделение из них кристаллов путем высаливания.
8.6. КАРБОНИЛ МАРГАНЦА
Карбонил марганца — кристаллы золотисто-желтого цвета с температурой плавления 154—155° С и плотностью 1,75 г/см3. Медленно разлагается на свету, на воздухе — при 110° С, при 50° С сублимируется в вакууме. Растворяется в органических растворителях, умеренно — в углеводородах, хорошо — в тетрагидрофуране, не растворяется в воде. ИК-спектр имеет характеристические частоты СО 2045,8; 2014,7; 1983,8 см'1. Структурная формула карбонила марганца Mn2(CO)i0:
СО СО со со
СО----Мп---Мп-----СО
/\ /\ со со со со
По химическим свойствам Мп2(СО)ю— типичный представитель карбонилов металлов. Связь Мп—Мп легко разрывается под действием галогенов с образованием Mn(CO)5-Hal (Hal — хлор, иод, бром) и амальгамы натрия с образованием NaMn(CO)s, который при действии кислот переходит в НМп(СО)5.
Карбонил марганца получают взаимодействием безводного хлорида марганца с оксидом углерода под давлением более 10 МПа в присутствии избытка металлоорганических соединений — А^СдН^з и др.
Карбонил марганца применяется в процессе получения порошка марганца и нанесения марганцевых покрытий.
8.7. КАРБОНАТ МАРГАНЦА
Карбонат марганца МпСО3— бледно-розовые кристаллы, приобретающие коричневый оттенок в результате окисления в присутствии кислорода и воды; кристаллическая решетка гексагональная (а _= = 0,4771 нм, с = 1,5664 нм, z = 6, пространственная группа ЯЗс; плотность 3,62 г/см3; С? = 94,8 Дж/(моль-К); АЯ0°бр = -881,7 кДж/моль; 53°98 = 109,5 Дж/(моль-К); ниже 32,14 К (точка Нееля) антиферромагнетик, а в интервале 32,14 — 64,5 К (точка Кюри) ферромагнетик. Растворимость осажденного карбоната марганца 0,0065 г в 100 г воды, при 20°С образует геми- , моно- и дигидраты.
397
Карбонат марганца при нагревании выше 250° С разлагается до СО2 и МпО и высших оксидов марганца.
При нагревании в присутствии кислорода образуется Мп3О4 или МпО2.
Карбонат марганца является полуфабрикатом в процессе получения его оксида, а также солей Мп(П) из марганецсодержащих руд. Получают карбонат марганца также действием гидрокарбоната натрия на водные растворы солей Мп(П).
Карбонат марганца применяется в качестве пигмента (марганцевый белый) и компонента термоиндикаторных покрытий, а также в процессе получения сиккативов, ферромарганца и зеркального чугуна.
8.8. ХЛОРИД МАРГАНЦА
Марганец образует дихлорид и трихлорид. Наиболее изученным из них и широко применяемым в народном хозяйстве является дихлорид.
Дихлорид марганца существует как в безводном состоянии, так и в воде двух кристаллогидратов: МпС12-2Н2О и МпС12-4Н2О. В кристаллогидратах с меньшим числом молекул воды роль лигандов помимо ОН2-групп играют их анионные составляющие. В обоих приведенных кристаллах октаэдрическое окружение Мп(П) возникает за счет молекул воды и ионов хлора:
МпС12-2Н2О
С1
С1
ОН2
МпС12-4Н2О
Дихлорид марганца — амфотерное соединение, розовые кристаллы тригональной сингонии с параметрами решетки: а = 0,620; угол 34,53° (а)._ Число формульных единиц в ячейке 1; пространственная группа R3m\ температура плавления 650° С; температура кипения 1240° С; плотность 2,977 г/см3; С°р = 72,93 Дж/(моль-К); ДЯ^р = = —481,2 кДж/моль; &Н''„ - 37,6 кДж/моль; ДЯ°СП = 148 кДж/моль; 52% = 118,2 Дж/(моль К).
Дихлорид марганца применяют для получения пигментов, в качестве катализатора в органическом синтезе, реагента для обнаружения 398
S2Og_, IO4, для обработки семян с целью ускорения роста растений и в качестве репера в физико-химическом анализе.
Способы получения хлорида марганца (II). Основными методами получения хлорида марганца (II), широко применяемыми в нашей и зарубежной практике, являются растворение в хлороводородной кислоте марганцевой руды, пиролюзита, карбоната, оксида и гидроксида марганца с получением его хлорида по схеме
МпО2 + 4НС1 = МпС12 + С12 + 2Н2О
МпСОз + 2НС1 = МпС12 + Н2О + СО2
МпО + 2НС1 = МпС12 + Н2О
Мп(ОН)2 + 2НС1 = МпС12 + 2Н2О
В результате приведенных реакций наиболее чистые растворы хлорида марганца образуются из гидроксида и карбоната марганца.
В процессе обработки сырой или восстановленной руды хлороводородной кислотой в раствор переходят соли тяжелых и других металлов. Очищают растворы путем обработки их избытком исходной руды. Находящиеся в начале процесса хлориды железа, алюминия и других металлов осаждаются в виде их гидроксидов в образующейся щелочной среде:
2А1С13 + ЗМпО + ЗН2О = 2А1(ОН)3 + ЗМпС12
2FeCl3 + ЗМпО + ЗН2О = 2Fe(OH)3 + ЗМпС12
Дополнительно очищают растворы от меди, никеля, кобальта и других примесей, нейтрализуя их аммиаком. При этом образуются легко фильтруемые сложные аммиакаты, от которых освобождаются фильтрацией. Растворы при необходимости очищают от тяжелых металлов путем обработки их сульфидом натрия.
Отфильтрованные от всех примесей растворы хлорида марганца выпаривают до насыщения, после чего охлаждают до 20° С. Образующиеся кристаллы МпС12-4Н2О отделяют в центрифугах от маточных растворов и промывают холодной водой. Разработан также способ получения тонкодисперсного целевого продукта непосредственно из растворов в одну стадию — сушкой их в сушилках с фонтанирующим слоем. Для получения максимального количества кристаллов МпС12-4Н2О растворы подкисляют хлороводородной кислотой, т.е. высаливают кристаллы с возвратом образующихся слабых кислых растворов на начало процесса.
399
Получают хлорид марганца (II) путем промывания 10%-ной хлороводородной кислотой шлама производства перманганата калия. Промытый шлам обрабатывают 30%-ной хлороводородной кислотой. Bz результате экзотермической реакции и нагревания реакционной массы до 90° С 95% содержащегося МпСЬ превращается в его хлорид.
Процесс сопровождается выделением хлора. Образующийся раствор хлорида марганца нейтрализуют карбонатом или гидроксидом марганца, фильтруют и высушивают в распылительной сушилке воздухом при 150° С. Целевой продукт содержит 82% МпСЬ-
Раствор хлорида марганца (II) может быть получен взаимодействием сульфата марганца (II) с СаС12 или обработкой раствором хлорида кальция твердого карбоната марганца по реакции
МпСОз (кр) + СаСЬ (раств) МпСЬ (раств) + СаСО3 (кр)
Исследования проводились, используя синтетический карбонат марганца. Оказалось, что с повышением концентрации исходного хлорида кальция равновесная концентрация хлорида марганца резко возрастает. Так, для 100° С при концентрации хлорида кальция 5,5 и 7,5 г-мол/л концентрация хлорида марганца (II) равна соответственно 79,2 и 146,5 г/л; для 200° С при концентрации хлорида кальция 4 и 5 г-мол/л концентрация хлорида марганца (II) соответственно 98,8 и 162 г/л. Смещение равновесия реакции в концентрированных растворах хлоридах кальция в сторону образования МпСЬ объясняется образованием прочных гидратов кальция. При концентрации СаСЬ 4—5 г-мол/л при 100° С за 8 мин 65% МпСОз превращается в его хлорид, за 15 мин — 85%, за 60 мин — 90%. Лимитирующей стадией процесса является образование промежуточного соединения — гидроксикарбоната марганца Мп(ОН) (НСО3) за счет присоединения молекул воды к карбонату марганца.
Получают хлорид марганца (II) хлорированием элементным хлором пористых руд, кристаллического пиролюзита и^и оксидной марганцевой руды при 550—600° С в присутствии восстановителей. После охлаждения плава хлорид марганца (II) выщелачивают водой и выделяют из них кристаллы целевого продукта. Растворы очищают от примесей железа путем фильтрации их через толстый слой активированного пиролюзита с диаметром частиц 1—1,5 мм или добавлением к раствору сырой карбонатной марганцевой руды с последующим отфильтровыванием осадка.
Получают хлорид марганца (II) из его оксида обжигом пирофорной смеси марганцевой руды (содержащей МпСЬ) с ферромарганцем и древесными опилками. Если ферромарганец выполнял в процессе роль восстановителя, то опилки обеспечивали необходимую порис-400
тостб. Спекшийся расплав измельчали и растворяли в хлороводородной Цислоте. Растворы хлорида марганца очищали от железа и других примесей и получали из них кристаллы тетрагидрата хлорида марганца (II).
8.9. НИТРАТ МАРГАНЦА (II)
Нитрат марганца (II) Mn(NO3)2— бледно-розовые гигроскопичные кристаллы с кубической решеткой; ДН^р = -574,6 кДж/моль. Растворимость в воде (г в 100 г): 102,0 (0° С), 157,1 (25°^ 428,0 (50° С) и 498,8 (75° С). Из водных растворов в зависимости от концентрации кристаллизуются гекса-, тетра-, ди- или моногидраты. При нагревании выше 180° С разлагается до оксидов марганца.
Гексагидрат Мп(ЫО3)2-6Н2О — розовые гигроскопичные кристаллы. Существет в трех модификациях с температурами перехода -37 и -5° С. Температура плавления 25,3° С; Д/7п°л = 40,2 кДж/моль; для аморфного гексагидрата в АЯо°бр = -2331 кДж/моль. При нагревании до 110° С гексагидрат переходит в дигидрат, который выше 170° С разлагается до диоксида марганца.
Безводный нитрат марганца получают: 1) реакцией марганца или его хлорида с М2О4 или смесью N2O4 с эти л ацетатом; 2) реакцией диоксида марганца с расплавом нитрата аммония; 3) дегидратацией кристаллогидратов нитрата марганца в вакууме (~10 Па) ниже 170° С.
Гидраты нитрата марганца получают взаимодействием марганца, его диоксида или карбоната с разбавленной азотной кислотой. Технологические схемы идентичны с технологией сульфата марганца (II).
8.10. МАЖЕФ
В процессе фосфатирования стальных изделий применяются препараты: порошок Паркера, дигофат, мажеф и др. Все они в основном состоят из фосфатов марганца и железа. Они дают более прочные покрытия, чем марганцевые, при минимальной -длительности процесса фосфатирования.
Наибольшее применение в промышленности получили препараты, состоящие из монофосфатов марганца и железа (мажеф) с соотношением между ними 10:14-15:1.
Исходным сырьем для получения необходимых фосфатов может быть фосфорная кислота или суперфосфат. Дигидроортофосфат марганца Мп(Н2РО4)2 назван дигофатом, который можно получить из фосфорной кислоты и сульфата марганца или восстановленного пи-401
ролюзита. Разработан способ получения дигофата из фосфорной кислоты и ферромарганца.
В процессе фосфатирования металлов происходит разложение хорошо растворимых фосфатов марганца на более основные форфаты:
Мп(Н2РО4)2 МпНРО4 + Н3РО4
ЗМпНРО4 ^Мп3(РО4)2 + Н3РО4
Образующиеся фосфаты кристаллизуются как на поверхности металла в виде пленок, так и в растворе (шлам).
Появление фосфатной пленки на металле является вторичным процессом, которому предшествует растворение металла и связан-
ные с этим изменения состава слоя раствора, прилегающего к анодным и катодным участкам поверхности металла. Фосфатная пленка стойка не только к влиянию атмосферных осадков, она
служит хорошим грунтом для последующего нанесения органических защитных покрытий.
Для выяснения состава осадков, выделяющихся из водных растворов фосфатов марганца, пользуются диаграммой % растворимости
системы МпО—Р2О5—Н2О (рис. 8.6), согласно которой при 100° С в твердую фазу сначала выделяется метастабильный тригидрат моногидроортофосфата, который переходит полностью в стабильную при этой температуре безводную соль лишь в течение нескольких суток. Поскольку процесс фосфатирования длится всего 40—60 мин, то ме-
тастабильный МпНРО4-ЗН2О не успевает разложиться и таким образом входит в состав фосфатной пленки. Вторым ее компонентом является безводный гидрофосфат марганца МпНРО4.
601- Мп3(РО4)2°
Р2О5, %
Рис. 8.6. Стабильная изотерма 100° С системы МпО—Р2О5—Н2О
Марганцевый препарат для фосфатирования сталей (мажеф), содержащий монофосфаты марганца и железа, может быть получен следующими способами.
1. Растворением в избытке горячей (90° С) фосфатной кислоты оксида марганца, восстановленной углеродом из пиролюзита, в присутствии железа. Полученный раствор отфильтровывают от нерастворимого остатка, упаривают до плотности 1,67—1,69 г/см3 и охлаждают для кристаллизации монофосфата мар
ганца с содержанием незначительного количества монофосфата железа.
402
2, Растворением ферромарганца в фосфорной кислоте с последующей упаркой полученных растворов и кристаллизацией соли.
3. Получением кислых фосфатов марганца и железа из сульфатов марганца и железа и тринатрийфосфата.
Первый способ позволяет получить продукт, загрязненный растворимыми в фосфорной кислоте примесями, содержащимися в исходном пиролюзите. Второй способ связан с необходимостью упарки фосфорно-кислых растворов. Третий способ лишен этих недостатков. Согласно способу, исходные растворы сульфата марганца, сульфата железа (II) и тринатрийфосфата смешивают при 60—70° С. В результате протекают реакции:
3MnSO4 + 2Na3PO4 = Mn3(PO4)2 + 3Na2SO4
3FeSO4 + 2Na3PO4 = Fe3(PO4)2 + 3Na2SO4
Образующиеся фосфаты марганца (III) и железа (III) выделяются в объемистый осадок, который отфильтровывают в барабанных вакуум-фильтрах и промывают водой до полного исчезновения сульфатов, а затем отжимают под гидравлическим прессом от избыточной влаги и высушивают при температуре до 300° С.
Полученный порошок, содержащий 30—35% Мп и 2—3% Fe, смешивают с фосфорной кислотой, количество которой должно быть достаточным для перевода тризамещенных фосфатов в однозамещенные:
Мп3(РО4)2 + 4Н3РО4 = ЗМп(Н2РО4)2
Fe3(PO4)2 + 4Н3РО4 = 3Fe(H2PO4)2
При этом происходит частичное образование МпНРО4:
Мп3(РО4)2 + Мп(Н2РО4)2 = 4МпНРО4
Ввиду экзотермичности этих реакций образующаяся смесь быстро разогревается, а по охлаждении застывает в однородную мелкокристаллическую массу, которую высушивают при температуре не выше 80—100° С и размельчают. Полученный препарат содержит 17—19% Мп, 1,5—2,5% Fe и 49—52% Р2О5. По минеральному составу мажеф — порошок, состоящий в основном из монофосфата марганца, небольших количеств фосфатов железа и дифосфата марганца.
403
8.11. МАНГАНАТЫ
Манганаты — соли несуществующих в свободном состоянии кислот марганца. Известны неустойчивые соли ортомарганцовистой (НдМпОд) и марганцовистой (Н3МпО4) кислот. Наиболее важны в народном хозяйстве соли марганцоватой кислоты НзМпОд — манганаты и марганцевой кислоты НМпО4 — перманганаты. Манганаты содержат тетраэдрические анионы МпОд, MnOj" или МпОд“.
Перманганаты. Перманганаты [манганаты (VII)] — соли марганцевой кислоты НМпО4; известны для щелочных и щелочноземельных металлов, аммония, серебра и алюминия. Фиолетово-черные кристаллы (табл. 8.4) растворимы в воде. Для перманганатов лития, натрия и кальция известны кристаллогидраты. Твердые манганаты (VII) термически неустойчивы, медленно разлагаются с выделением кислорода уже при комнатной температуре, скорость разложения возрастает в присутствии воды и при нагревании. В присутствии восстановителей могут разлагаться со взрывом. Перманганаты — очень сильные окислители. В щелочной среде МпО< восстанавливается до МпО*", в нейтральной — до МпС>2, в кислой — до Мп(П). А
Таблица 8.4. Свойства некоторых манганатов
Соединение Сингония Температура разложения, °C Плотность, г/см3 Растворимость в воде, г в 100 г
AgMnO4 Моноклинная 160 4,27 0,92
NH4MnO4 Ромбическая 70 2,22 8,00
CsMnO4 » 250 3,60 0,23
Ca(MnO4)2-4H2O Моноклинная 130—140 2,50 3,88
Ba(MnO4)2 Ромбическая 100 3,77 72,4
K2MnO4 » 600 2,80 —
Na2MnO4 » 300 2,68 —
BaMnO4 » — 5,20 —
Na3MnO4 Кубическая 1250 2,61 Разлагается
Перманганаты применяют в аналитической химии для определения металлов, оксалатов, иодидов, бромидов и других соединений.
Марганцевую кислоту получают действием на перманганаты концентрированной серной кислотой. Кислота существует лишь в подкисленных водных растворах. Сильный окислитель. Разлагается до МП2О7 и Н2О.
Манганаты (VI) — соли марганцевой кислоты Н2МпО4; известны лишь для щелочных металлов и бария. Зеленые кристаллы. При нагревании разлагаются до марганца (V), диоксида марганца и кислорода. Окислители в нейтральной среде восстанавливаются до Mn(IV) 404
и до Мп(П) — в кислой. Сильные окислители, окисляют Mn(Vl) до Mn(VlI). В нейтральном и кислом растворах возможно диспропорционирование Mn(VI) до Mn(VII) и Mn(IV). При действии кислот на Mn(VI) образуется марганцевая кислота Н2МпО4, которая быстро разлагается на МпО2 и НМпОд. Mn(Vl) легко образует двойные соли с сульфатами и хроматами металлов. Mn(VI) получают в промышленности анодным окислением марганца в расплаве щелочи или диоксида марганца.
Манганаты (V) (гипоманганаты) — соли марганцовистой кислоты Н3МпО4; известны лишь для лития, натрия, калия и бария. Зеленые кристаллы. В водном растворе быстро диспропорционируют Mn(V) —> Mn(VI) + МпО2 и идет реакция с перманганатами МПО4" + + МпО" -> 2МпО4-.
Манганаты (V) получают пропусканием кислорода через расплав смеси диоксида марганца с гидроксидом марганца (атомное отношение Na:Mn = 3:1) при 800° С либо восстановлением растворов манганатов (VII) сульфитом натрия.
Соединения типа CaMnO3, Mg6MnO8 и другие представляют собой не соли, а двойные оксиды (CaO-MnO2, 6MgO-MnO2); нерастворимы в воде.
Из манганатов наибольшее практическое значение имеет перманганат калия; К2МпО4 — полуфабрикат в процессе получения КМпО4 из МпО2; ВаМпО4 (касселева или марганцевая зелень) — зеленый пигмент, применяемый для фресковой живописи; Ва3(МпО4)2— голубой пигмент для пластмасс, художественных красок и эмалей; Са3(МпО4)2-5Н2О применяют для стерилизации питьевой воды. Манганаты (V) входят в состав малярной краски — марганцевой синей.
Получение перманганата калия. Перманганат калия получают в промышленности реакцией диоксида марганца с гидроксидом калия и электрохимическим способом.
Перманганат калия в процессе щелочного разложения получают в две стадии. В первой стадии получается манганатный плав, содержащий К2МпО4. Во второй стадии манганат окисляется в перманганат.
Манганатный плав получают сплавлением пиролюзита с гидроксидом калия в присутствии воздуха по схеме
2MnO2 + 4КОН + О2 = 2К2МпО4 + 2Н2О
Тонко измельченный в шаровой мельнице пиролюзит высокого качества смешивается с 50%-ным раствором гидроксида калия. Далее смесь сплавляется при 200—270° С, так как более высокие температуры приводят к разложению образовавшегося манганата с выделе-405
нием кислорода. Разложение К2МпО4 при 475—960° С в атмосфере кислорода или азота протекает в основном по схеме
ЗК2МпО4 = 2К3МпО4 + МпО2 + О2
при этом ~8—10% манганата разлагается по схеме
2К2МпО4 = 2К2МпО3 + О2
Образующийся по первой реакции диоксид марганца теряет часть кислорода и находится в плаве в виде оксидов с суммарным составом МпО]^ 75.
Получение манганатного плава проводят во вращающихся барабанных печах непрерывного действия. В печь непрерывно вводят смесь молотого пиролюзита и 85%-ного гидроксида калия с температурой 250° С. Суспензия поступает в печь с температурой 350° С. Поскольку применяют печи с внутренним обогревом, имеющие, например, кольцевую горелку для сжигания газообразного топлива, а в центре пламени — сопло для подачи суспензии, исходная смесь спекается, не контактируя со стенками печи. После первой печи плав — гранулят для завершения реакции — направляют во вторую печь — «печь дожигания», по которой масса перемещается при температурном интервале 140—250° С в течение четырех часов. Вторую часть печи обогревают газами, отходящими с первой стадии процесса термообработки. Газы содержат 8—30% (об.) О2 и 10—35% (об.) Н2О. Полученный манганат-ный плав направляется на получение перманганата калия.
При получении плава содержащиеся в исходном пиролюзите примеси влияют на физические свойства плава. Так, сесквиоксид железа особых осложнений при проведении процесса не вызывает, а оксиды алюминия и кремния образуют с исходным гидроксидом калия растворимые (низкоплавкие) соединения, что приводит к увеличению объема массы плава.
Разработан способ получения манганитного плава повышенного качества. По этому способу исходный молотый пиролюзит смешивают с расплавленным 75—85%-ным гидроксидом калия, а полученную смесь гранулируют на валках. Гранулированный манганитный плав сушат при 160—180° С (при температуре ниже температуры его размягчения), что обеспечивает однородность плава. Плав окисляют воздухом, при этом манганит практически полностью переходит в манганат. Полученный плав содержит 60—65% К2МпО4, 12—13% МпО2 и 8—9% КОН + К2СО3.
При подаче суспензии пиролюзита в 80%-ном гидроксиде калия на наружную поверхность вращающихся в разные стороны вальцов, обогреваемых изнутри топочным газом, продолжительность пребыва-406
ния суспензии на вальцах при 350—400° С составляет одну минуту. Плав непрерывно соскабливается с поверхности вальцов стационарно прикрепленными ножами. Производительность вальцов ~50 кг/(м2-ч), промышленные агрегаты с поверхностью 5 м2 производят до 1000 т целевого продукта в год.
Возможно получение манганата из пиролюзита электрохимическим способом с применением в качестве электролита расплавленного гидроксида калия, в котором пиролюзит находится во взвешенном состоянии. Процесс электролиза проводят при 195—200° С. Выход не превышает 60% от теоретического. Повышение избытка гидроксида калия в получаемом полуфабрикате затрудняет дальнейшее электрохимическое окисление К2МпО4 в КМпО4.
Переход манганата в перманганат происходит даже при простом кипячении водного раствора по реакции
ЗК2МпО4 + 2Н2О = 2КМпО4 + MnO2 + 4КОН
Процесс заметно ускоряется при обработке раствора диоксидом углерода:
ЗК2МпО4 + 2СО2 = 2КМпО4 + МпО2 + 2К2СО3
Образующийся карбонат калия обрабатывают известью для регенерации гидроксида калия:
К2СО3 + СаО + Н2О = 2КОН + СаСО3
Получение перманганата этим способом оказывается невыгодным, так как значительная доля исходного манганата превращается в диоксид марганца.
Окисление манганата хлором по реакции
2К2МпО4 + С12 = 2КМпО4 + 2КС1
также экономически невыгодно, поскольку регенерация гидроксида калия из его хлорида, например электролизом, является дорогим процессом.
В настоящее время перевод манганата в перманганат осуществляется электрохимическим окислением. При этом на аноде образуется перманганат калия:
МпОд~— е = МпО"
а на катоде — гидроксид калия и водород
407
2Н2О + 2е = Н2 + 2ОН"
Процессы, происходящие в электролизере, могут быть схематически выражены суммарным уравнением:
2К2МпО4 + 2Н2О = 2КМпО4 + 2КОН + Н2
Манганатный плав выщелачивается в резервуарах с мешалкой маточным раствором, полученным после электролиза. Растворение манганата при 70° С продолжается около 1,0—1,5 ч. Отстоявшийся раствор направляется на электролиз, а шлам поступает в барабанные вакуум-фильтры, где отделяется от раствора и возвращается на процесс получения манганатного плава. Шлам содержит 35—50% МпО2 (не прореагировавшего при получении манганата) и другие примеси, перешедшие из исходного пиролюзита. По мере накопления перешедших из пиролюзита примесей шлам вывозится в шламонакопитель.
Процесс электролиза проводится в ваннах, представляющих собой стальной цилиндрический резервуар с коническим днищем, по которому уложен змеевик для регулирования температуры. Ванна снабжена мешалкой и спускным краном. Железные аноды расположены внутри ванны в виде нескольких концентрических цилиндров на расстоянии 100 мм друг от друга. Применяют также никелевые аноды. Между анодами находятся катоды — железные стержни диаметром 20—25 мм. Общая поверхность катодов приблизительно в 10 раз меньше поверхности анодов, что уменьшает потери от катодного восстановления. Плотность тока на аноде 60—70 А/м2, на катоде -700 А/м2. Анодные и катодные пластины опираются на стеклянные или фарфоровые изоляторы. Диаметр ванны 1,3—1,4 м, высота цилиндрической части 0,7—0,8 м, конической части 0,5 м. В ванну вмещается 900—1000 л раствора электролита. Электролиз проводится при 60° С. Напряжение на ванну в начале электролиза составляет -2,7 В, нагрузка—1400—1600 А. В конце процесса электролиза напряжение возрастает до 3 В, а сила тока несколько падает. Ванны работают сериями, по нескольку штук. Число ванн в серии определяется характеристикой генератора постоянного тока. Расход энергии на 1т КМпО4 составляет 700 кВт-ч.
Процесс электролиза ведут без диафрагмы, поскольку она засоряется диоксидом марганца, незначительное количество которого образуется в процессе. Поэтому выход по току зависит, главным образом, от степени обратного восстановления перманганата на катоде. Большая щелочность электролита препятствует использованию добавок для образования на катоде защитной пленки. Уменьшению выхода по току способствует также выделение на аноде кислорода 408
и обратный переход КМпО4 в К2МпО4 по причине высокой концентрации гидроксида калия:
4КМпО4 + 4КОН = 4К2МпО4 + 2Н2О + О2
Реакция каталитически ускоряется присутствующим в электролите диоксидом марганца. Увеличению выхода по току способствует низкая анодная плотность тока и искусственное перемешивание электролита, уменьшающее концентрационную поляризацию на аноде; при перемешивании в прианодном слое создается более высокая концентрация К2МпО4, понижается анодный потенциал, в результате чего уменьшается выделение кислорода.
Выход по току и степень окисления увеличиваются при электролизе насыщенного раствора К2МпО4 в присутствии кристаллов. Такой раствор содержит примерно 180 г/л К2МпО4, 30—40 г/л КМпО4,150 г/л КОН и 50 г/л К2СО3. Процесс электролиза продолжается до снижения концентрации К2МпО4 до 15—30 г/л. Образующийся КМпО4 плохо растворим, поэтому частично выделяется в осадок в виде кристаллов. По окончании процесса раствор электролита в смеси с кристаллами перманганата калия поступает в стальные охлаждаемые с помощью водяных рубашек холодильники с метальными устройствами. После полной кристаллизации выпавшие кристаллы отжимают на центрифуге и промывают водой. Маточные растворы и промывные воды после центрифуги направляются на выщелачивание манганатного плава. Маточные растворы содержат 23 г/л КМпО4, 16 г/л К2МпО4, 210 г/л КОН и 60 г/л К2СО3.
После промывки на центрифуге и высушивания получается загрязненный технический перманганат калия, содержащий 80—95% КМпО4, примеси диоксида марганца, манганата калия, сульфатов, карбоната и гидроксида калия. Для получения качественного целевого продукта образующиеся кристаллы растворяют в чистой воде при 85° С, фильтруют и из этих растворов путем охлаждения получают чистые кристаллы. Кристаллы отфильтровывают и сушат.
Часть маточных растворов после кристаллизации перманганата калия, содержащих кроме перманганата и щелочей алюминаты, ванадаты и другие примеси каустифицируют известью [СаО или Са(ОН)2] и отфильтрованные растворы направляют на выщелачивание манганата.
Разработан также способ утилизации маточного раствора восстановлением КМпО4 и К2МпО4 раствором формалина до диоксида марганца. Оставшийся после отделения диоксида марганца раствор гидроксида и карбоната калия нейтрализуют азотной кислотой и получают нитрат калия.
Разработан способ непосредственного получения перманганата калия анодным растворением марганца в щелочном электролите, содержащем гидроксид или карбонат калия, в процессе электролиза с ано-409
дами из ферромарганца, с ~70% Мп и 1—6% углерода. Процесс электролиза идет по общему уравнению:
2Mn + 2ОН" + 6Н2О = 2МпО; + 7Н2
При содержании в аноде меньше 44% Мп перманганат не образуется. Катод изготовляют из материала, который устойчив в щелочном растворе перманганата меди. Процесс электролиза можно осуществлять без диафрагмы или с диафрагмой из асбестового полотна. В последнем случае катодное восстановление уменьшается и выход по току значительно больше. Оптимальная температура электролита 16—18° С. Повышение температуры приводит к увеличению степени превращения перманганата в манганат. Электролит содержит 20—30% КОН или К2СОз. Электролизу препятствует образующаяся на ферромарганцевом аноде оксидная пленка, повышающая потенциал, особенно при малой концентрации щелочи в электролите. В процессе применения анодов из силикомарганца пассивирующая пленка образуется только при малых концентрациях электролита и высоких плотностях тока. Высокие концентрации электролита приводят к появлению растворимых соединений железа, образующихся при повышенном потенциале.
Оптимальная анодная плотность тока при использовании в качестве электролита раствора, содержащего 300 г/л К2СОз, 16—18 А/дм2, а при 200—250 г/л КОН — 30—40 А/дм2. Выход по току не превышает 50%, а выход продукта (степень перехода растворенного марганца в перманганат) 80—85%; расход электроэнергии 12 кВт-ч на 1 кг КМпО4. Продукт электролиза — КМпО4— получается в виде мелких кристаллов, смешанных с большим количеством электролитического шлама. Электролит охлаждают, отделяют от осадка на барабанном вакуум-фильтре в центрифуге и возвращают в процесс. Осадок обрабатывают горячей водой для извлечения КМпО4, который затем кристаллизуют. Фильтрация горячей (70—90° С) электролизной суспензии для отделения шлама перед кристаллизацией перманганата позволяет получить чистый продукт (до 90,7% КМпО4).
8.12. МАРГАНЦЕВЫЕ ПИГМЕНТЫ
Из марганцевых пигментов наибольшее значение имеют марганцевая голубая и марганцевая фиолетовая. Кроме того, представляет интерес диоксид марганца в качестве составной части термостойкого черного пигмента и марганцевая зеленая.
Марганцевая голубая. Марганцевая голубая по химическому составу представляет собой изоморфную смесь бария манганата (V) (гипоманганата) Ва3(МпО4)2 и сульфата бария BaSO4. Цвет — небес-410
но-голубой; плотность 4,4 г/см3; насыпная плотность 2 г/см3; pH водной вытяжки 8,5. Обладает хорошей светостойкостью (8 баллов), высокой атмосферо- и термостойкостью (до 400° С), стойкостью к действию воды, органических растворителей и щелочей. Она неустойчива к действию кислот, а также водных растворов сульфатов. Не разлагается концентрированной хлороводородной кислотой.
Марганцевая голубая применяется в качестве голубого пигмента для пластмасс, художественных красок и эмалей, является щелочестойким пигментом для окраски портландцемента. Она входит в состав малярной краски — марганцевой синей.
Физико-химические основы технологии. В промышленности марганцевую голубую получают термообработкой смеси нитрата и сульфата бария с соединениями марганца — диоксидом, карбонатом, манганатом бария с последующей отмывкой полученной массы от не прореагировавших соединений.
Цвет и свойства марганцевой голубой зависят от качества исходных реагентов, их массовых соотношений и технологических условий получения пигмента. Наиболее определяющими для качества целевого продукта является соотношение в исходной шихте основных видов сырья — соединений марганца и нитрата бария, а также температурный режим термообработки. Так, ярким цветом и высокой стойкостью обладают пигменты, полученные из шихты с низким содержанием марганца и высоким содержанием нитрата барйя. Оптимальная температура термообработки шихты 700—750° С с определенной выдержкой. При обработке полученной массы соляной кислотой из нее удаляются оксиды бария и марганца.
При получении марганцевой голубой в процессе взаимодействия с нитратом бария оксиды марганца низшей валентности окисляются в высшие оксиды по уравнению
МпО + иВа(ЫОз)2 -> МпОх + иВаО + 2иЫО
Образующиеся оксиды МпОх обладают кислотными свойствами, поэтому реагируют со свободным оксидом бария с образованием манганата бария, который с сульфатом бария образует изоморфную смесь, являющуюся искомым пигментом.
В водных растворах соли — манганаты (V) — устойчивы лишь в присутствии сильных щелочей. В нейтральных и слабощелочных растворах соединения манганатов (VI) легко разлагаются с образованием соединений марганца (VI) и марганца (VI), а в кислых растворах— на соединения марганца (III) и марганца (VII). В сухом виде манганаты (гипоманганаты) устойчивы на воздухе, свободном от влаги и диоксида углерода, или при высоких температурах. Экспериментально установлено, что стойкость гипоманганата может быть повы-
411
шена совместным осаждением его с фосфатом, арсенатом и вольфраматом натрия, с которыми он образует изоморфные смеси, устойчивые в водных растворах, даже в отсутствие щелочей. Такие соединения получают термообработкой при 750—800° С смеси, состоящей из 1 моль Na3PO410H2O с 0,01 моль Мп(СН3СОО)2- Изоморфные смеси получают также с фосфатом кальция. Образующиеся соединения не растворяются в воде.
Таким образом, синий и сине-зеленый цвет марганцевой голубой можно объяснить присутствием в составе пигмента гипоманганата бария Ва3(МпОд)2. Манганат (V) бария устойчив лишь в виде изоморфной смеси с сульфатом бария при избытке последнего и при соблюдении условий технологии. Так, при повышенном содержании марганца в исходной шихте, малой реакционной способности компонентов шихты и недостаточно высокой температуре термообработки образуются неустойчивые пигменты.
Роль исходного нитрата бария в процессе синтеза пигмента заключается не только в окислении соединений марганца низшей валентности, но и в создании сильнощелочной среды:
2Ba(NO3)2 -> 2ВаО + 4NO + ЗО2
Для создания щелочной среды непосредственно в самом начале процесса заменяют часть нитрата бария на его гидроксид.
Состав пигмента представляют общей формулой Ва3(МпО4)2 • • nBaSO4 • /лВаО, где п = 10 4-16, а тп = 1 4-3. Предполагают, что в процессе синтеза пигмента образуется не нейтральный гипоманганат бария Ва3(МпО4)2, а основная соль Ba3(MnO4)2-mBaO.
Схема получения марганцевой голубой. Процесс получения пигмента состоит из следующих технологических операций: 1) приготовление шихты; 2) термообработка шихты и ее размол; 3) обработка полученной массы хлороводородной кислотой; 4) промывка и фильтрование пигмента; 5) сушка пигмента и его размол.
Согласно технологической схеме (рис. 8.7), исходные компоненты сырья — нитрат и сульфат бария, соответствующее соединение марганца— смешивают в смесителе 7. При необходимости в систему добавляют воду. Образующаяся смесь направляется для термообработки в печь 2, где в течение 2—3 ч подвергается термообработке при 700—720° С. Конец реакции, т.е. конец процесса термообработки, определяют по синему цвету реакционной массы. В процессе термообработки исходной шихты протекает реакция образования пигмента:
MnO2 + 6BaSO4 + 6,5Ba(NO3)2 —>
-> 4ВаО + 13NO + 9,5О2 + Ba1,5MnO4-BaO-6BaSO4
412
Рис. 8.7. Схема получения марганцевой голубой:
/ — смеситель; 2 — печь термической обработки; 3— шаровая мельница; 4 — реактор для обработки шихты; 5 — барабанный вакуум-фильтр; 6 — вакуум-сушилка; 7 — узел размола и упаковки
пигмента
После выгрузки шихты из печи ее охлаждают, измельчают и направляют в реактор 4 для обработки хлороводородной кислотой. В этой операции удаляется избыток оксида бария в виде его хлорида, а также другие возможные примеси: не прореагировавшего диоксида марганца, свободные манганат и гипоманганат марганца. После обработки кислотой в реакторе 4 осветлившуюся часть дополнительно промывают водой, добавляя восстановители, например два раза водой с 3% (масс.) нитрата натрия, а затем два раза с 0,5—0,1% (масс.) щавелевой кислоты. Промывку суспензии продолжают до отсутствия иона хлора в промывной воде. Иногда суспензию пигмента после первых промывок стабилизируют не только введением восстановителей, но и нейтрализацией щелочами, например гидроксидом бария. Промытый пигмент фильтруют на барабанных вакуум-фильтрах 5, сушат в вакуум-сушилке 6, после чего сухой продукт размалывают в дезинтеграторе 7 и упаковывают.
8.13. МАРГАНЦЕВАЯ ФИОЛЕТОВАЯ
Свойства и применение. Марганцевая фиолетовая является двойной солью пирофосфата марганца (III) и аммония MnNH4P2O70,5H2O. Промышленностью выпускаются также виды и сорта продукта, в которых часть марганца (~0,3 моль) замещена ионами трехзамещенных металлов Fe3+ и А13+. Цвет пирофосфата — фиолетовый с красноватым оттенком, а цвет замещенных пирофосфатов — фиолетовый с синеватым оттенком. Плотность 2,6—2,7 г/см3, дисперсный состав: 50—60% частиц размером до 2 мкм, 70—80% — до 1 мкм. Светостойкость марганцевой фиолетовой — 7 баллов в полном тоне и 6 — при разбеле с диоксидом титана.
413
При обычной температуре марганцевая фиолетовая устойчива к действию воды и незначительно разлагается разбавленными минеральными кислотами, а при нагревании до 90—100° С частично разлагается водой и полностью — кислотами. В присутствии окислителей (особенно нитрата аммония) марганцевая фиолетовая устойчива к действию воды и уксусной кислоты, незначительно разлагается на холоду концентрированной азотной кислотой. Легко разлагается щелочами, даже разбавленными. При нагревании до 300° С разлагается.
Марганцевая фиолетовая применяется для производства масляных, эмульсионных красок и нитрокрасок.
Получение. Основными видами сырья в производстве марганцевой фиолетовой являются: нитрат или диоксид марганца, фосфорная кислота и фосфат аммония. Процесс ведут при 280—300° С. В некоторых производствах целевой продукт получают и без применения фосфорной кислоты. Взамен нитрата и диоксида марганца применяют его карбонат или сульфат. В этом случае в состав исходной шихты вводят окислитель. Лучшим окислителем считается нитрат аммония. В настоящее время широко применяется вариант получения марганцевой фиолетовой термообработкой диоксида или нитрата со смесью фосфорной кислоты и фосфата аммония (одно- или двузамещенного) в следующем мольном соотношении: МшНзРОд^Нд^НРОд = 1:1,5:1,5.
Полученные из нитрата марганца пигменты отличаются чистым и насыщенным сине-фиолетовым цветом. В состав исходной шихты вводят нитрат или сульфат железа из расчета 0,3 моль на 1 моль МпС>2.
Получение марганцевой фиолетовой состоит из следующих операций: 1) приготовление и термообработка шихты; 2) выщелачивание плава; 3) промывка пигмента; 4) сушка пигмента; 5) измельчение целевого продукта и упаковка.
Смесь исходных видов сырья размельчают в шаровой мельнице и смешивают с фосфорной кислотой. Термообработка шихты связана с определенными трудностями, так как при 180—220° С она расплавляется и превращается в вязкую массу, трудно поддающуюся перемешиванию. Поэтому процесс проводится в реакторе из нержавеющей стали с ленточным мешальным устройством для перемешивания вязкой массы. Продолжительность подъема температуры до 280—300° С составляет около 3 ч. После достижения необходимой температуры массу выдерживают полтора часа.
После процесса термообработки плав охлаждают до 120° С и заливают водой для выщелачивания избыточного количества фосфорной кислоты и фосфата аммония. Полученную суспензию отжимают и промывают на барабанном вакуум-фильтре. Образующуюся пасту пигмента сушат в вакуум-сушилках при 100—120° С и измельчают.
414
ГЛАВА 9
ЖЕЛЕЗО И ЕГО СОЕДИНЕНИЯ
9.1. ЖЕЛЕЗО
Железо — очень распространенный элемент в природе. Содержание железа в земной коре составляет 4,65% (масс.). Известно свыше трехсот минералов, которые составляют месторождения железных руд. Руды, содержащие железа не менее 16%, имеют промышленное значение. Важнейшие рудные минералы железа приведены в табл. 9.1.
Таблица 9.1. Минералы, содержащие железо
Минерал Химическая формула Содержание железа, %
Магнетит (магнитный железняк) Fe3O4 72,4
Гематит (железный блеск, крас- Fe?Oi 70
ный железняк)
Гетит a-FeO(OH) или Fe2O3H2O
Лепидокрокит y-FeO(OH) 62,0
Гидрогетит (лимонит) Fe2O3*H2O 62,0
Сидерит FeCO3 48,2
Ильменит FeTiO3 36,8
Различают следующие основные типы железных руд.
Бурые железняки-руды гидроксидов железа (III) (главный минерал — гетит) содержат до 66,1% Fe; имеют осадочное происхождение.
Гематитовые руды или красные железняки (главный минерал— гематит), содержат до 50—65% Fe. Для них характерно залегание богатых руд поверх мощных толщ бедных (30—40% Fe) магнетитовых кварцитов.
Магнетитовые руды, или магнитные железняки (главный минерал — магнетит), содержат 45—60% Fe. Верхние горизонты магнетитовых рудных тел обычно частично окислены до гематита (полумартиты и мартиты).
Силикатные руды, содержащие до 25—40% Fe осадочного происхождения, используемые для выплавки чугуна, относятся к группе зеленых слюд-хлоридов. Главные минералы — шамозит Fe4(Fe,Al)2[/U2Si20io](OH)8 и тюрингит (Mg,Fe\5 АЬ^^^АЬ^Ою] (ОН^-иГБО — содержат до 42% Fe.
415
Считается перспективным использование и бедных железных горных пород, а также железомарганцевых конкреций.
Железо состоит из четырёх стабильных изотопов: 54Fe (5,84%), 56Fe (91,68%), 57Fe (2,17%), 58Fe (0,31%). Конфигурация внешних электронных оболочек 3rf’4s2, степени окисления +2 и +3 (наиболее характерны), +1, +4, +6, +8; энергия ионизации при последовательном переходе от Fe° к Fe5+ 7,893, 16,183, 30,65, 57,79 эВ; электроотрицательность по Полингу 1,8; атомный радиус 0,126 нм, ионные радиусы (в нм, в скобках указаны координационные числа) для Fe2+ 0,077 (4), 0,092 (6), 0,106 (8), для Fe3+ 0,063 (4), 0,079 (6), 0,092 (8).
Железо — серебристо-белый пластичный металл. В обычном давлении существует в четырех кристаллических модификациях (рис. 9.1). До 917° С существует a-Fe с объемно-центрированной кубической решеткой (а = 0,286645 нм, z = 2, пространственная группа 1тиЗти); a-Fe ферромагнитно, но при 769° С (точка Кюри) переходит в парамагнитное состояние без изменения сингонии и других свойств, кроме магнитных; А/7°перехода = 1,72 кДж/моль. Парамагнитное железо (£-Fe) устойчиво в температурном интервале 769—917° С. В интервале 917—1394° С существует y-Fe с гранецентрированной кубической решеткой (при 950° С а = 0,3656 нм, z = 4, пространственная группа Fm3m)\ Д/Тперехода Р~>У 0,91 кДж/моль. Выше 1344° С существует 8-Fe.
Температура плавления железа 1535° С (ДЯП°Л = 16,6 кДж/моль), температура кипения 2750° С. Плотность (в г/см3): a-Fe 7,87 (20° С), 7,67 (600° С); y-Fe 7,59 (1000° С); 5-Fe 7,409; жидкого металла 7,024 (1538° С), 6,962 (1600° С), 6,76 (1800° С).
Железо — металл средней химической активности. Железо в жидком состоянии неограниченно растворяет Al, Си, Мп, Ni, Со, Si, Ti, хорошо растворяет V, Cr, Pt, ограниченно — Mo, Sn, С, S, Р, As, Н2, N2, О2, не растворяет Pb, Ag, Bi. С углеродом образует твердые растворы внедрения — феррит и мартенсит с a-Fe, аустенит с y-Fe. В
Рис. 9.1. Полиморфные превращения железа
416
зависимости от содержания углерода в железе различают: мягкое железо (< 0,2% С), сталь (0,2—1,7% С) и чугун (1,7—5% С).
В сухом воздухе около 200° С на поверхности железа образуется тонкая оксидная пленка, защищающая металл от дальнейшего окисления. Выше 200° С скорость процесса коррозии железа увеличивается, образуется слой ржавчины; внутренняя зона состоит из вюстита FevO (х = 0,89—0,95), а поверх него слой РезОд, а затем Ре20з.
Железо не растворяется в воде и холодных растворах щелочей, реагируя же с разбавленными кислотами и горячими концентрированными растворами щелочей, образует соли железа (И). Концентрированные HNO3 и H2SO4 пассивируют железо, образуя на поверхности оксидную пленку, не растворимую в кислотах.
Металлическое железо получают металлургическим способом, который включает операции дробления, измельчения, обогащение магнитной сепарацией до содержания Fe 64—68%, получения концентрата (74—83% Fe) и плавкой. Основную массу железа выплавляют в виде чугуна и стали. Технически чистое железо, или армко-железо (0,02% С, 0,035% Мп, 0,14% Сг, 0,02% S, 0,015% Р), выплавляют из чугуна в сталеплавильных печах или кислородных конвертерах.
Чистое железо получают: 1) восстановлением оксидов железа твердым (коксик, каменноугольная пыль), газообразным (Н2, СО, их смесь, природный конвертированный газ) или комбинированным восстановителем; 2) электролизом водных растворов или расплавов солей железа; 3) разложением пентакарбонила железа (карбонильное железо).
Сварочное, или кричное, железо получают окислением примесей малоуглеродистой стали железистым шлаком при 1350° С или восстановлением из руд твердым углеродом.
Губчатое железо (97—99% Fe) получают восстановлением оксидов железа при 750—1200° С. Полученный пористый агломерат частиц железа, пирофорен, в горячем состоянии легко поддается обработке давлением.
Карбонильное железо (до 0,00016% С) получают терморазложением Fe(CO)5 при 300° С в среде аммиака с последующим отжигом в среде водорода при 500—600° С. Образующийся порошок с размером частиц 1—15 мкм перерабатывается методом порошковой металлургии.
Особо чистое железо получают зонной плавкой.
Технически чистое железо является материалом для сердечников электромагнитов и якорей электромашин, а также пластин аккумуляторов.
Карбонильное железо применяют в процессе нанесения тончайших пленок и слоев на магнитофонные ленты, в качестве катализатора, антианемического средства и др.
Из губчатого железа выплавляют высококачественные стали. Железный порошок применяют в процессе сварки и для цементации меди. Искусственные радиоактивные изотопы 55Fe (Г1/2 = 2,6 ч) и 59Fe (Г1/2 = 45,6 сут) являются изотопными индикаторами.
417
14 Химическая технология
неорганических веществ, кн. 2
9.2. КАРБОНИЛЫ ЖЕЛЕЗА
Известны три карбонила железа: одноядерный — пентакарбонил железа, двухъядерный — нонакарбонилдижелезо, трехъядерный — додекакарбонилтрижелезо.
Пентакарбонил железа Fe(CO)5—желтая летучая жидкость. Молекула имеет конфигурацию тригональной бипирамиды (рис. 9.2). В молекуле карбонила железа группы СО связаны с атомом железа через атомы углерода: электронная пара от атома углерода передается атому железа, образуя a-связь, а d-электроны металла переходят на вакантные разрыхляющие л-орбитали СО (л-связь). При этом принимают, что СО является донором двух электронов, а металл находится в нулевой степени окисления.
Температура плавления пентакарбонил железа — 20° С, температура кипения 102,5° С (с разложением); d420 = 1,46; давление пара 27,93 ГПа (20° С). В процессе нагревания при 60° С начинает разлагаться с выделением СО и железа. Растворяется в органических растворителях; диамагнетик; ядовит.
В ИК-спектрах имеются характеристические полосы 1800—2200 см’1 (валентные колебания группы СО).
Пентакарбонил железа окисляется кислородом воздуха, кислотами, галогенами, а также хлораминами и др.
При взаимодействии пентакарбонила железа с кислотами образуются соответствующие соли [FeSO4, FeCl2, Ре(ЫОз)2].
При взаимодействии с бромом происходит следующая реакция:
Fe(CO)s + Br2 —> [i/wc-Fe(CO)4Br2] + СО
Щелочными металлами пентакарбонил железа восстанавливается до карбонилферратов:
Рис. 9.2. Строение молекулы пентакарбонила железа Fe(CO)5
Fe(CO)5 + 2Na -> Na2[Fe(CO)4] + СО
Реагирует с гидроксид-ионом:
Fe(CO)5 + 2ОН" -> [FeH(CO)4] ’ + НСО’
Группы СО в пентакарбон и л железе могут замещаться на лиганды — фосфины, фосфиты, амины, изонитрилы, непредельные органические соединения и др., например
Fe(CO)5 + СН2 = СНСН =СН2 4
-> [Ре(т]-С4Н6)(СО)з] + 2СО
418
Пентакарбонилжелезо Fe(CO)s получают реакцией СО с железом при 180—200° С и давлении 16—20 МПа. Обычно применяют губчатое железо или сплав железа с серой (5—7%). Продукт огнеопасен и токсичен.
Нонакарбонилдижелезо Fe2(CO)9 (формула 1)
ОС СО СО
\| / 3FeC ос\ / с(А ^ос ОС —~Fe - СО
\Х/ ос
СО СО
II
образуется при облучении пентакарбонилжелеза ультрафиолетовым светом, это двухъядерное соединение со связью Fe—Fe, в котором атомы железа связаны как непосредственно, так и через СО-мостики. Продукт представляет золотисто-желтые кристаллы, температура разложения 100° С. Плохо растворим в органических растворителях. Медленно окисляется на воздухе. Легко диссоциирует по схеме
Fe2(CO)9 -> Fe(CO)5 + Fe(CO)4
Выделяющийся при этом тетракарбонил железа энергично присоединяет различные лиганды, образуя комплексы [Fe(CO)4L], где L — фосфины, ненасыщенные органические соединения и др. Реагирует также с анионом ОН, образуя [Fe(CO)8]2‘. Нонакарбонилдижелезо получают фотолизом Fe(CO)5.
Додекакарбонилтрижелезо Рез(СО)]2 {формула II) — черно-зеленые кристаллы с температурой разложения 140—150° С, температурой возгонки 70° С (с разложением). Незначительно растворяется в органических растворителях. При стоянии в комнатных условиях медленно разлагается с образованием пирофорного Fe.
Додекакарбонилтрижелезо получают действием диоксида марганца на [FeH(CO)4p, соляной или муравьиной кислоты на [РезН(СО)п], пероксида водорода и перманганата калия на [Fe(CO)4]2‘ , а также в процессе нагревания Fe2(CO)9.
Карбонилы железа применяют в производстве порошка карбонильного железа, применяющегося для изготовления магнитов, магнитофонных лент, фасонных деталей; в производстве железооксидных пигментов; для получения сесквиоксида железа для ферритов, в качестве катализаторов в органическом синтезе. Карбонилы железа являются основным исходным сырьем в синтезе железоорганических соединений.
419
14*
9.3. СУЛЬФИДЫ ЖЕЛЕЗА
Железо образуют следующие сульфиды: FeS, Fe7Sg, РезБд, Fe2Ss и FeS2. Fe7Sg существует в моноклинной и гексагональной модификациях, устойчив до 220° С. РезЗд — минерал смитит, кристаллы с ромбоэдрической решеткой. Известен малоустойчивый оксид Ре23з с кубической решеткой типа шпинели. Получают сухим способом из тонкоизмельченного железа и сульфида водорода при 100° С или сплавлением сульфида железа (II) с элементной серой. При хранении во влажном воздухе он гидролизуется и окисляется по следующей схеме:
Fe2S3 + 1,50? + ЗН2О = 2Fe(OH)3 + 3S
Моносульфид железа FeS — коричневые или черные кристаллы; нестехиометрическое соединение, при 743° С область гомогенности 50—55,2% (ат.) серы. Существует в нескольких кристаллических модификациях— а1, а п, р и 8 (табл. 9.2). В процессе нагревания до 138° С а'-форма переходит в P-форму (ДЯ°перехода 2,39 кДж/моль, а при 325° С P-форма переходит в 8-форму (АН перехода 0,50 кДж/моль). Температура плавления моносульфида железа 1193° С (FeS с содержанием S 51,9%, ат.), ДНП°Л = 32,37 кДж/моль; плотность 4,79 г/см3; для a-FeS (50%, ат. S): Ср° = 50,58 Дж/(моль-К); ДН^р =-100,5 кДж/моль, ДС^р = -100,9 кДж/моль; 52°98 = 60,33 Дж/(моль К). При нагревании в вакууме выше 700° С отщепляет серу, давление диссоциации 1g р (мм рт. ст.) = —15 695/7" + 8,37. Модификация 8 парамагнитна, a', Р и а" антиферромагнитны, твердые растворы или упорядоченные структуры с содержанием серы 51,3—53,4% (ат.)—ферро- или ферримагнитны.
Моносульфид железа практически не растворяется в воде (6,210‘4%, масс.), разлагается в разбавленных минеральных кислотах с выделением сульфида водорода. На воздухе влажный монооксид железа легко окисляется до сульфата железа (II). Встречается в природе в виде минералов пирротина (магнитный колчедан FeSj.i^) и троплита (в метеоритах).
Моносульфид железа получают путем термообработки молярной смеси порошкообразного металлического железа с серой при -600° С или взаимодействием сульфида водорода или серы с сесквиоксидом железа при 750—1050° С. Получают также реакцией сульфидов щелочных металлов или аммония с солями железа (II) в водном растворе.
Сульфид железа применяют для получения сульфида водорода, может быть использован в процессе концентрирования цветных металлов.
420
Таблица 9.2. Характеристика кристаллических модификаций сульфидов железа
Показатель a’-FeS a"-FeS (52,16%, ат. S) p-FeS (при 180° С) 6-FeS (при 395° С) FeS2 FciSk
Сингония Гексаго- Гексаго- Гексаго- Гексагона- Кубиче- Ромбиче- Моно-
Параметры, нм: нальная нальная нальная льная ская ская клинная
а 0,594 0,3437 0,345 0,3533 0,5405 0,4434 1,19108
b — — — — — 0,5420 0,68673
с Число форму- 1,1740 0,5741 0,573 0,5797 — 0,3385 2,27898
льных единиц в ячейке 12 2 2 2 4 2 8
Пространственная группа ♦р = 90,577° Рв^ттс P&Jmmc Рб^/ттс Pfhjmmc ЛЗ Р 1 ппт С2!с
Дисульфид железа FeS2 — золотисто-желтые кристаллы с металлическим блеском; область гомогенности 66,1—66,7% (ат.) серы. Существует в двух модификациях: кубической (в природе — минерал пирит, железный или серный колчедан) с плотностью 5,03 г/см3. Температура: перехода марказит-спирит 365°С, плавления 743°С (конгруэнтно). Для пирита: С ° = 62,22 Дж/(моль-К); ДН^р = -163,3 кДж/моль, ДСо°бр = -151,94 кДж/моль; 52°98 = 52,97 Дж/(моль-К); обладает свойствами полупроводника, ширина запрещенной зоны 1,25 эВ. В ромбической модификации существует минерал марказит, или лучистый колчедан, с плотностью 4,86 г/см3, Д//°бр марказита — 139,8 кДж/моль.
Дисульфид железа при нагревании в вакууме диссоциирует на пирротин и серу. Практически не растворяется в воде, разлагается азотной кислотой. На воздухе или в атмосфере кислорода дисульфид железа горит с образованием диоксида серы и сесквиоксида железа.
Дисульфид железа получают термообработкой хлорида железа (III) в токе сульфида водорода.
Природный дисульфид железа применяется в качестве исходного сырья для производства серы, железа, серной кислоты, сульфатов железа, является также компонентом шихты для переработки марганцевых руд и концентратов. Пиритовые огарки применяются в качестве сырья в процессе выплавки чугуна. Кристаллы пирита являются детекторами в радиотехнике.
421
9.4. НИТРАТЫ ЖЕЛЕЗА
Нитраты железа (II) и (III) существуют в основном в виде кристаллогидратов. К ним относятся гексагидрат нитрата железа (II), нонагидрат нитрата железа (III) и гексагидрат нитрата железа (III).
Гексагидрат нитрата железа (II) Fe(NO3)2-6H2O — светло-зеленые кристаллы с ромбической решеткой. Температура плавления 60,5°С. Хорошо растворяется в воде, причем растворимость его растет с повышением температуры. Растворимость в воде (г в 100 г, в расчете на безводную соль): 71,03 (0°С), 86,95 (24°С) и 166,67 (60,5°С). В равновесии с насыщенным водным раствором существует в интервале от -12 до 60,5°С и концентрации раствора в пределах 39—69,5%. При более низких температурах и концентрациях растворов устойчив нонагидрат нитрата железа (II). Безводный продукт, а также его гидраты неустойчивы, поэтому легко окисляются на воздухе.
Нонагидрат нитрата железа (III) — гигроскопические светло-фиолетовые кристаллы с моноклинной решеткой (а = 0,140 нм, b =0,970 нм, с = 1,103 нм, Р = 95,52°, пространственная группа Р2|/с); температура плавления 50,1 ° С; плотность 1,81 г/см3; АЯ^р =-3338,8 кДж/моль. При нагревании плавится. Процесс плавления сопровождается частичным разложением исходного продукта с выделением азотной кислоты и последующим кипением раствора при 125° С. Растворимость в воде (г в 100 г, в расчете на безводную соль): 67,08 (0° С), 82,48 (20° С) и 104,83 (40° С). В водном растворе гидролизуется. Растворяется также в ацетоне, эфире, этаноле.
Гексагидрат нитрата железа (III) — бесцветные гигроскопичные кристаллы с кубической решеткой; кристаллизуется из водных растворов, содержащих не менее 57% (масс.) HNO3 и 28% Fe(NO3)3; плотность 1,68 г/см3; растворяется в воде, ацетоне и этаноле.
Гидраты нитрата железа (III) получают в результате реакции железных порошков или стружек с 20—30%-ным раствором азотной кислоты в токе воздуха по схеме
2Fe + 6HNO3 + 12Н2О + 1,5О2 -> 2Fe(NO3}r6H2O + ЗН2О
Получают гидраты нитрата железа (III) также обменной реакцией между сульфатом железа (III) и нитратом бария.
Гидраты нитратов железа (II) и (III) применяются в качестве коагулянта в процессе очистки сточных вод, протравы при крашении шерсти, а также для синтеза оксидных катализаторов.
422
9.5. ФОСФАТЫ ЖЕЛЕЗА
Существуют ортофосфаты железа (II) и (III). Ортофосфаты железа (II) образуют кристаллогидраты с четырьмя и восемью молекулами воды, существуют также в безводном состоянии. Ортофосфат железа (III) существует в основном в виде его дигидрата. Известны конденсированные фосфаты железа (III).
Ортофосфат Fe(II) (в природе минерал графтонит) Fe.^PO^ — кристаллы (табл. 9.3); получают осаждением из водных растворов сульфата железа (II) фосфатом натрия при 60—80° С с последующим обезвоживанием образующихся кристаллогидратов. Кристаллизуется из водных растворов в форме октагидрата Рез(РО4)2*8Н2О (в природе — минерал вивианит), который при нагревании теряет четыре молекулы кристаллизационной воды и переходит в тетрагидрат Рез(РО4)2*4Н2О, который полностью обезвоживается при 180° С. Известны и кислые фосфаты железа (II), например РеНРС^иНгО, где п = 1, 2, а также фосфаты РепзРе1П(РО4)з и конденсированные состава Fe(PO4)2. Все фосфаты железа (И) при нагревании частично окисляются кислородом воздуха до фосфатов железа (III); в воде нерастворимы, но растворимы в минеральных кислотах.
Ортофосфат железа (III) FePO4 — желтые кристаллы; низкотемпературная модификация при 600° С переходит в гексагональную (а = = 0,504 нм, с = 0,562 нм), а выше 950° С — в ромбическую типа штренгита; = -1287,8 кДж/моль; не растворяется в воде; растворяется в хлороводородной и серной кислотах. Из водных растворов кристаллизуется в виде дигидрата FePO4-2H2O, который при температуре выше 250° С полностью обезвоживается; в природе — минералы штренгит (ромбический) и меташтренгит (моноклинный).
Ортофосфат железа (III) получают взаимодействием дигидрофосфата натрия (ЫаН2РО4) с водным раствором соли железа (III). Он образуется также как защитное покрытие в процессе фосфатирования стали и чугуна. Кислый фосфат железа (III) имеет состав Ре(Н2РО4)з-2Н2О.
Известны конденсированные фосфаты железа (III) РеНгРзОнгиНгО, где п = 0, 1, 2, а также линейные и циклические состава Ре(РОз)з, в которых анион представляет собой циклы Р4О*2" или Р6О*8 либо имеет линейное цепочечное строение (РО3)х. Циклические полифосфаты около 1000° С превращаются в линейные, которые плавятся при 1250° С с частичной потерей Р2О5, а при дальнейшем нагревании при 1300—1400° С дают смесь Ре4(Р2О7)з и линейного Ре(РОз)з
Известны двойные, ди- и тетрациклофосфаты, соответствующие МТеРгО? и МТе(РОз)4, где М'—щелочной металл.
423
Таблица 9.3. Характеристика фосфатов железа
Показатель Fei(PO4)2-8H2O Fe3(PO4)2-4H2O Fe3(PO4)2 FePO4-2H2O
Цвет Бледно-голубой Розовый
Сингония Моноклинная Моноклинная Моноклинная Ромбическая Моноклинная
Параметры элементарной ячейки, нм: 1,0039 1,045 0,881 1,005 0,532
а 1,3388 0,465 1,1169 0,992 0,987
b 0,4657 0,925 0,6145 0,874 0,869
с 104,3 100,55 99,6 — ~90
Р, град Число формульных 2 2 4 4
единиц в ячейке Пространственная группа 1 Нт Р2\1а Р2х1с — Р2\1т
Плотность, г/см2 2,58 — — 2,58 2,74
9.6. ХЛОРИДЫ ЖЕЛЕЗА
Железо с хлором образуют хлориды железа (II) и (III).
Дихлорид железа — бесцветные кристаллы, желтеющие на воздухе вследствие окисления; кристаллическая решетка гексагональная (а = 0,357 нм, с = 1,751 нм, z = 3); температура плавления 677° С, температура кипения 1026° С; плотность 3,162 г/см3; уравнение температурной зависимости давления пара Igp (в мм рт. ст.) = 26,53—9475 Г1—5,231g7(950—1299 К); С ° = 76,29 Дж/(моль-К), ДЯ°р = 342,9 кДж/моль, = 43,04 кДж/моль, ДЯ°СП = 126,48 кДж/моль; S2°98 = = 117,9 Дж/(моль-К).
Дихлорид железа растворяется в воде (35,3% (масс.) при 7° С, 38,4% при 20° С, 48,3% при 96° С), этаноле, ацетоне, нерастворим в диэтиловом эфире. В водных растворах слабо гидролизован. Из водных растворов при 12,3° С кристаллизуется FeCl2-6H2O, в интервале 12,3—76,5° С—FeCI2-4H2O, выше 76,5° С — FeCl2-2H2O, который при 120°С переходит в FeCl2 H2O.
Тетрагидрат дихлорида железа FeCl2-4H2O — прозрачные голубовато-зеленые кристаллы с моноклинной решеткой (а = 0,591 нм, b = 0,717 нм, с = 0,844 нм, £ = 112,17°, пространственная группа Р2|/с); плотность 1,937 г/см3; при 220° С и в инертной атмосфере обезвоживается.
Безводный хлорид железа FeCI2 в процессе нагревания в воздухе окисляется с образованием смеси FeCl3 и FeOCl, а выше 300° С осушенным водородом восстанавливается до металла. Кристаллы FeCl2 имеют слоистую структуру (рис. 9.3).
424
Дихлорид железа с аммиаком образует аммиакаты типа FeCl272NH3 (п = 1, 2, 6, 10), а с хлоридами щелочных металлов и аммония — комплексные хлориды, например NH4[FeCl3].
Безводный дихлорид железа FeCl2 получают действием газообразно
OFe Qci
Рис. 9.3. Структура слоя FeCl2
го хлорида водорода на
стружку или порошок металлического железа при 500° С по схеме
Fe + 2НС1 = FeCl2 + Н2
или восстановлением хлорида железа (III) водородом при 500° С:
2FeCl3 + Н2 = 2FeCl2 + НС1
а растворы хлорида железа (II) растворением металлического железа в хлороводородной кислоте:
Fe + 2НС1 = FeCl2 + Н2
Дихлорид железа является основным исходным сырьем в производстве трихлорида железа, применяется в качестве катализатора в органическом синтезе, является компонентом антианемических препаратов в медицине.
Трихлорид FeCh — темно-красные кристаллы с зеленоватым оттенком, решетка гексагональная (а = 0,606 нм, с = 1,74 нм, z = 6); температура плавления 309° С, температура кипения 320° С; плотность 2,898 г/м3; 4ТИТ = 650—700° С, ^крит = 4,5—5,0 МПа; уравнение температурной зависимости давления пара Igp (в мм рт. ст.) = = 55,898—10 7547х1—12,641g7(298—580 К); С° = 94,84 Дж/(моль-К); ДЯ° = 502,1 кДж/моль, = 42,91 кДж/моль, АЯИ°СП = 25,20 кДж/моль; = 142,3 Дж/(моль К). В парах до 440° С или в растворенном состоянии в сероуглероде трихлорид железа существует в виде димера:
425
а при 750° С в избытке хлора — в виде мономера:
Растворяется в воде (47,9% (масс.) при 20° С, 84,6% при 100%) и при этом в воде образуются октаэдрические комплексы [Fe(OH2)6]3+, т.е. происходит следующее:
Растворяется в метиловом и этиловом спиртах, ацетоне, диэтиловом и диизопропиловом эфирах, слабее — в сероуглероде и бензоле.
Из насыщенных водных растворов кристаллизуются кристаллогидраты FeCI27?H2O, где п = 2,0;. 2,5; 3,5; 6,0. Образующийся кристаллогидрат РеС13-6Н2О— желто-коричневые кристаллы с моноклинной решеткой (а = 1,189 нм, b = 0,705 нм, с = 0,599 нм, Р= 100,5°, z = 2, пространственная группа С2/т).
Безводный хлорид железа (III) при нагревании до высоких температур в инертной атмосфере разлагается по схеме
2FeCI3
2FeCl2 + С12
426
в токе сухого водорода выше 100° С восстанавливается до FeCI2
2FeCI3 + Н2 -> 2FeCl2 + 2НС1
а при более высокой температуре восстанавливается до металла
2FeCl3 + ЗН2 -> 2Fe + 6НС1
Безводный хлорид железа (III) окисляет KI, SnCI2, H2S, Mg, Са, Al и др. По этим реакциям практически во всех случаях окисления происходят окислительно-восстановительные процессы, например
2FeCl3 + 6KI = 2FeI2 + I2 + 6КС1
С сесквиоксидом железа он образует оксохлорид FeOCl:
Fe2O3 + FeCl3 = 3FeOCl
Трихлорид железа — жесткая кислота Льюиса. С аммиаком образует [Fe(NH3)6]Cl3:
FeCl3 + 6NH3 = [Fe(NH3)6]Cl3
с пиридином FeCl3-4C5H5N, с этанолом FeCl3-2C2H5OH, с хлоридами металлов первой и второй группы, а также аммония образует комплексные хлориды, например NH4[FeCl4], K[FeCl4], Ca[FeCl4]2 и т.д.
Трихлорид железа встречается в природе в виде минерала мо-лезита.
Безводный хлорид железа (Ill) в производстве получают взаимодействием сухого хлора с железом при 400—500° С
2Fe + ЗС12 = 2FeCl3
обработкой сесквиоксида железа при 500° С
Fe2O3 + 6НС1 = 2FeCl3 +• ЗН2О
а также хлорированием водных растворов дихлорида железа:
2FeCl2 + С12 = 2FeCl3
По последнему способу трихлорид железа получают в виде его водных растворов.
Трихлорид железа является сырьем для получения других соединений, хлорирующим агентом как в технологических процессах, так 427
и в жилищно-комунальном хозяйстве, протравой при крашении тканей, катализатором и реагентом в органическом синтезе, процесса травления печатных плат, реагентом для обнаружения SCN“, Sn2+, Н2О7, для определения фенолов и енолов.
9.7. СУЛЬФАТЫ ЖЕЛЕЗА
Изучены физико-химические свойства и способы получения сульфатов железа (II) и (III), существующих в виде безводных солей и их кристаллогидратов, широко применяемых в народном хозяйстве (табл. 9.4).
Таблица 9.4. Свойства сульфатов железа
Показатель FeSOi FeSO4-7H2O FeSO4-4H2O FeSO4H2O Fe2(SO4)_, Fe2(SO4)v9H2O
Сингония Ромбиче- Моноклин- Моноклин- Моноклин- Моно- Гексагональ-
ская ная ная ная клинная ная
Параметры элементарной ячейки. 0.525 1,402 0,5979 0,7624 0,8296 1,085
нм; 0,798 0,650 1,364 0,7468 0.8515 —
и h 0,659 1,101 0,798 0,7123 1,160 1,703
о с Р, град —' 105,57 90,43 115,86 90,5 —
Число формульных единиц в ячей- 4 4 4 4 4 —
ке
Пространственная группа Ст /ст Р2,/с Р2\/т А2/а Р2\/т —
Т °C 1 ПЛ, V -700°С 64 — — Выше 600° С* —
Плотность, г/см2 3,14 1,898 — — 3,097 2,1
С”, Дж/(мольК) 100,7 394,9 — — — —
кДж/моль -923,2 -3014 — — -2605 —
5^, Дж/(моль К) 107.6 409,5 — — 259,2 —
‘Температура разложения. “Известна также гексагональная модификация.
Сульфат железа (II). Физико-химические свойства. Сульфат железа (II) FeSO4 — бесцветные гигроскопичные кристаллы, растворимые в воде. Из водного раствора при температурах от —1,82 до 56,8° С кристаллизуется голубовато-зеленого цвета гептагидрат, в интервале 56,8—64° С — тетрагидрат, а выше 64° С — моногидрат белого цвета (рис. 9.4).
Гептагидрат сульфата железа (II) FeSO4*7H2O (техническое название — железный купорос, в природе — минерал мелантерит) — голу-428
бовато-зеленые кристаллы; растворяется в воде (14,91% (масс.) FeSC>4 при -1,8° С, 21,01% при 20° С, 35,06% при 56,7° С), метаноле, этаноле, глицерине; на воздухе выветривается, вследствие образования на поверхности Fe(OH)SO4 окрашивается в желтый цвет. При нагревании на воздухе в результате дегидратации и окисления гептагидрата образуются FeSO4 H2O, FeSO4, Fe(OH)SO4,
Рис. 9.4. Политерма растворимости в системе FeSO4—Н2О
Процесс
Fe2(SO4)3, Fe2O3 и SO3.
Тетрагидрат FeSO4*4H2O — зе-
леные кристаллы, а моногидрат FeSO4‘H2O — бесцветные.
обезвоживания моногидрата FeSO4 H2O сопровождается разложением
с выделением диоксида серы и с одновременным образованием основного сульфата железа (II), а дальнейшее повышение температуры сопровождается окислением Fe(II) в Fe(III) с образованием Fe2O(SO4)2- Давление диссоциации моногидрата сульфата железа (II)
FeSO4*H2O FeSO4 + Н2О составляет 0,4 мм рт. ст. при 50° С, 0,8 мм рт. ст. при 70° С и 3,8 мм рт. ст. при 90° С. В процессе термообработки в обычных условиях полностью обезвоживается при 300° С (с частичным разложением).
Растворимость сульфата железа (II) в воде снижается в присутствии серной кислоты, которая высаливает его кристаллогидраты из растворов. При этом уменьшается и предельная концентрация раствора, соответствующая точке перехода гептагидрата сульфата железа (П) в его моногидрат. В системе FeSO4—H2SC>4—Н2О (рис. 9.5) при 50° С существуют области кристаллизации стабильных FeSO4*7H2O, FeSO4-4H2O, FeSO4 H2O и FeSO4 и метастабильных FeSO4-7H2O и FeSO4-4H2O. При значениях концентрации серной кислоты, находящихся за стабильной областью существования гепта- и тетрагидрата, из пересыщенных растворов сначала выделяется метастабиль-ный гептагидрат, который переходит в метастабильный тетрагидрат с более повышенной скоростью, чем последний переходит в стабильный моногидрат. При 60° С гептагидрат сульфата железа (II) не образуется, а в температурном интервале 70-—90° С не выделяется также и тетрагидрат. В интервале 50—90° С при концентрации серной кислоты меньше 27,2% температурный коэффициент растворимости FeSO4H2O отрицателен, а при более высокой концентрации серной кислоты — положителен (в пределах 50—70° С температур-429
Рис, 9.5. Зависимость растворимости FeSO4-7H2O от содержания в растворе свободной H2SO4 при различных температурах
ный коэффициент снова становится отрицательным при концентрациях выше 76% H2SO4). При высоких температурах растворимость сульфата железа (II) в серной кислоте резко снижается. Так, при 200—240° С в растворе, содержащем -91% H2SO4, находится всего 0,2—0,3% FeSOi. При этом сульфат железа (II) частично переходит в сульфат железа (III) по реакции
2FeSO4 + 2H2SO4 =
= Fe2(SO4)s + SO2 + Н2О
Растворимость же сульфата железа (III) в концентрированной серной кислоте до температуры 200° С составляет всего около 0,1%.
Давление диссоциации в процессе термообработки сульфата железа (II) по уравнению
2FeSO4 Ре20з + SO2 + SO3
при 614° С равно 254 мм рт. ст., а при 550° С — 600 мм рт. ст.
При отсутствии кислорода сульфат железа (И) разлагается с достаточно высокой скоростью и полнотой при 700° С. В токе же воздуха процесс его разложения значительно ускоряется в присутствии восстановителей — угля и пирита. При взаимодействии сульфата железа (II) с FeS или FeS2 (700—800° С) образуются Ре3С>4 и SO2 по схеме
2FeSO4 + FeS = РезО4 + 3SC>2
Смесь из 82,3% моногидрата сульфата железа (II), 15,2% дисульфида железа и 2,5% угля разлагается в токе воздуха при 600° С за 40—60 мин на 98%. С увеличением содержания кислорода в газовой фазе скорость разложения сульфата железа (II) уменьшается в отсутствие восстановителей (колчедана, угля), но возрастает в их присутствии. Это объясняется тем, что восстановители, сдвигая реакцию SO3 SO2 + 1/2Ог вправо, ускоряют процесс диссоциации сульфата железа (II). 430
Сульфат железа (II) при не очень высоких температурах лишь частично разлагается до оксида железа (II):
2FeSO4 + С = 2FeO + СО2 + 2SO2
В присутствии воздуха или водяного пара сульфат железа (II) конвертирует хлориды щелочных металлов в сульфаты по схеме
FeSO4 + 2NaCl = FeCl2 + Na2SO4
В присутствии кислорода в водном растворе происходит постепенное окисление FeSO4. В присутствии же серной кислоты и с повышением ее концентрации скорость процесса окисления ферросуль-фата в феррисульфат значительно уменьшается. Процесс окисления сульфата железа (II) ускоряется при пропускании через раствор газа, содержащего диоксид серы и кислород. При этом происходит образование сульфата железа (III):
2FeSO4 + О2 + SO2 = Fe2(SO4)3
В процессах гидрометаллургии цинка окисление Fe2+ производят при его концентрации в растворе около 2 г/л Fe2+ (в виде FeSO4) и рН~5—5,2 при наличии твердой фазы из продуктов гидролиза, окисленных соединений железа. В этом случае значительную роль в процессе окисления Fe2+ играет адсорбция кислорода осадками оксидных соединений железа.
Существуют также способы бактериального окисления солей двухвалентного железа в кислых растворах.
Применение сульфата железа (II). Сульфат железа (II) применяется в качестве компонента электролита в гальванотехнике, в сельском хозяйстве для борьбы с вредителями садов и слизнями. Его применяют также для уничтожения мхов, лишайников и грибных спор.
Гептагидрат сульфата железа (II) используют и для питания растений. Железо необходимо растениям в качестве катализатора в процессе образования хлорофилла. Как известно, при недостатке железа растения заболевают хлорозом и листья теряют зеленую окраску. Кроме этого, железо входит в состав многих окислительных ферментов и играет решающую роль в процессе дыхания растений, применяется в качестве консерванта древесины, является хорошим фунгицидом.
Сульфат железа (II) — сильный восстановитель, является антиане-мическим средством и сырьем в производстве пигментов и минеральных красок. Сульфат железа (II) применяют в текстильной промышленности, в фотографии и для приготовления чернил. Отходы производства сульфата железа (II) перерабатывают в изоляционный 431
материал — феррон или ферригипс, представляющие собой смесь гидроксидов железа (II) и гипса с наполнителем.
Сульфат железа (II) применяется в аналитической химии в качестве реактива для определения NO3, МпО“ и Се4+.
Способы получения сульфата железа (II). В настоящее время сульфат железа в промышленности получают из колчеданных огарков и травильных растворов.
Получение гептагидрата сульфата железа (II) из колчеданных огарков. Сырьем для получения гептагидрата сульфата железа (II) является колчеданный огарок, охлажденный без доступа воздуха, обладающий магнитными свойствами и содержащий в своем составе некоторое количество сульфида железа (II). Огарок состоит из 45% сесквиоксида железа, 25% оксида железа (II) и 10—15% сульфида железа (II).
В процессе обработки исходного сырья серной кислотой образуются сульфаты железа (II) и (III) по схеме
Fe2O3 + 3H2SO4 = Fe2(SO4)3 + ЗН2О
FeO + H2SO4 = FeSO4 + Н2О
FeS + H2SO4 = FeSO4 + H2S
Выделяющийся сульфид водорода реагирует с сульфатом железа (III), восстанавливая последний до сульфата железа (II) по реакции
Fe2(SO4)3 + H2S = 2FeSO4 + H2SO4 + S
При недостатке сульфида железа (II) в исходном сырье-колчедане восстановительный процесс проводят элементным железом:
Fe2(SO4)2 + Fe = 3FeSO4
Процесс разложения огарка серной кислотой в производственных условиях проводят в химзащищенных реакторах, оснащенных метальным устройством. Реакция продолжается в течение 5—10 мин, после чего процесс перемешивания останавливают, а реакционную массу выдерживают в реакторе около 30 мин. За этот период реакционная масса превращается в твердый пористый продукт, представляющий обводненный сульфат железа (II). Далее продукт растворяют в воде. Полученные водные растворы сульфата железа (II) отстаивают, и чистые растворы направляют на кристаллизацию. Процесс кристаллизации проводят в вакуум-кристаллизаторах. Образующаяся водная суспензия сульфата железа (II) в центрифугах типа ФГП разделяется на кристал-432
лы целевого продукта и маточные растворы. Кристаллы гептагидрата сульфата железа (II) при необходимости сушат, а маточные растворы возвращают на начало процесса для смешения со свежеприготовленными растворами сульфата железа (II).
Исследован процесс взаимодействия сульфидов железа серной кислотой и установлено, что реакция исходного сульфида железа (И) в его сульфат протекает очень энергично даже при невысоких температурах (50—60° С), а реакция дисульфида железа с серной кислотой начинается лишь с достижением 200° С, идет медленно с выделением серы и ее диоксида. Для ускорения процесса и более полного протекания реакции температуру повышают до 300° С и поддерживают значительный избыток серной кислоты.
Гептагидрат оксида железа (II) может быть получен из колчеданного огарка сульфатизацией его диоксидом серы с одновременным окислением в водной среде:
2Fe2O3 + 6H2SO4 + ЗО2 = 2Fe2(SO4)3 + 6Н2О
Fe2(SO4)3 + SO2 + 2Н2О = 2FeSO4 + 2H2SO4
Получение гептагидрата сульфата железа (II) из травильных растворов. Травильные растворы образуются в процессе обработки (травления) серной кислотой стальных изделий для удаления с них окалины (оксидов железа) перед дальнейшей обработкой их поверхности. Для травления применяют 20—25%-ную серную кислоту, а отходы (травильные растворы) содержат 15—20% сульфата железа (II) и 2—10% свободной серной кислоты.
Разработана технология сульфата железа (II) из этих растворов путем их охлаждения до -5, -10° С или выпаркой их с последующей кристаллизацией при охлаждении до 20—25° С. Возможно также высаливание кристаллов серной кислотой (см. рис. 9.5), и после отделения кристаллического осадка маточный раствор возвращают на процесс травления железа.
Выделение гептагидрата сульфата железа (II) из травильных растворов производят охлаждением их воздухом или водой, или в ваку-ум-кристаллизационных установках. Предназначенную для травления металла серную кислоту обычно используют в качестве высаливающего агента.
Процесс регенерации травильных растворов путем их нагревания более экологичен. В этом варианте сульфат железа (II), растворимость которого с повышением температуры выше 64° С уменьшается, выделяется в виде моногидрата сульфата железа, а маточный раствор возвращается на процесс травления. Из раствора, насыщенного 433
FeSO4-H2O, можно выделять данное соединение в твердую фазу высаливанием, добавляя при этом 65—85% FeSO4-7H2O.
Если выделение гептагидрата сульфата железа (II) производится охлаждением отработанного травильного раствора, содержащего а% FeSC>4 , а в остающемся маточном растворе b% FeSC>4, то из 1000 кг травильного раствора будет получено 1,83 х кг FeSO4-7H2O или х кг FeSO4, причем
(а-6)1000
Х~ 100-1,836’
10х а степень извлечения FeSO4 из травильного раствора будет ----------%.
а
Если производить охлаждение отработанного травильного раствора с одновременным введением в кристаллизатор т кг серной кислоты на 1000 кг травильного раствора, то
_ (а- 6)1000- Ьт
Х~ 100-1,83* '
Если отработанный травильный раствор плотностью 1,275 г/см3, содержащий 300 г/л или 23,5% FeSO4 и 1—2% H2SO4, охладить до -30° С, он станет насыщенным (см. рис. 9.5, табл. 9.5). При дальнейшем охлаждении до 3—4° С концентрация маточного раствора станет равной 14% сульфата железа (II). При этом из 1 т травильного раствора будет получено 234 кг FeSC>4-7H2O, а степень извлечения FeSO4 составит 54%.
Таблица 9.5. Растворимость FeSO4 в водных растворах серной кислоты
Температура, °C Растворимость FeSO4 (%) при содержании в растворе H2SO4 (%)
0 2 4 7 10 15 20 35
0 13,6 12,9 12,0 10,8 9,5 7,2 5,5 3,4
5 15,2 14,4 13,5 12,0 10,7 8,0 6,5 4,7
10 17,2 16,1 15,2 13,7 12,3 10,1 8,2 6,6
20 20,9 19.7 18,4 16,8 15,7 13,3 11,2 9,5
25 23,0 21,4 20,4 18,6 17,3 15,0 12,8 11,4
30 24,8 23.1 22,0 20,5 19,2 16,9 14,8 13,2
Если же охладить раствор лишь до 30° С, но с одновременной добавкой в кристаллизатор 0,25 т 75%-ной серной кислоты на 1 т отработанного травильного раствора для повышения кислотности маточного раствора до -16% (200 г/л H2SO4), то из 1 т травильного раствора будет получено 75 кг FeSO4-7H2O, а степень извлечения FeSO4 составит в 434
одном цикле 17,5%. Однако при высаливании серной кислотой с одновременным охлаждением до 3—4° С выход гептагидрата сульфата железа (II) из 1 т травильного раствора составит около 290 кг, а степень извлечения сульфата железа в одном цикле будет около 67%. В процессе работы на замкнутом цикле, т.е. с возвратом всего маточного раствора на травление, весь образующийся сульфат железа (II) может быть выделен в виде гептагидрата сульфата железа (II).
Наиболее эффективным методом выделения сульфата железа (II) из травильных растворов является вакуум-кристаллизация, которая применяется в настоящее время во всех производствах. При остаточном давлении в кристаллизаторе 25—10 мм рт. ст. раствор кипит при 5—10° С. Охлаждение раствора достигается за счет его самоис-парения. Вакуум создается в аппаратах с помощью пароструйных эжекторов, в которых смешивается пар высокого и низкого давлений. Применение многоступенчатых эжекторов повышает их коэффициент полезного действия и значительно сокращается расход пара. Сократить расход пара можно ступенчатым испарением раствора при разных температурах.
В растворе, поступающем на кристаллизацию из травильных ванн с температурой 63—65° С, содержится -10% H2SO4 и 18% FeSC>4. В кристаллизатор добавляют серную кислоту в количестве, необходимом для получения маточного раствора с концентрацией -23% H2SO4. Получающийся маточный раствор содержит 6,9—7% FeSO4. При этом на 1 т расходуемой на травление 76%-ной серной кислоты получается 8,7—8,8 т травильного раствора, из которого испаряется в процессе кристаллизации 0,73 т воды. Теоретический выход гептагидрата сульфата железа (II) на 1 т перерабатываемой серной кислоты равен 2,16 т. С учетом потерь раствора с металлом, с парами воды и других практический выход составляет 1,8—2 т.
При непрерывном процессе вакуум-кристаллизации расход пара значительно сокращается. Он зависит от числа корпусов установки. В четырехкорпусной установке он составляет -0,5 т на 1 т гептагидрата сульфата железа (II). На рис. 9.6 приведена схема четырехкорпусной непрерывной кристаллизационной установки. Охлаждение раствора происходит в четырех последовательно соединенных кристаллизаторах. Кристаллизаторы снабжены якорными или пропеллерными мешалками. Аппараты изготовлены из нержавеющей стали, внутренняя поверхность которых гуммируется для защиты от коррозии.
Смесь отработанного травильного раствора и свежей серной кислоты охлаждается в первом корпусе от 60 до 40° С, во втором — до 30° С, в третьем —до 22° Сив четвертом — до 10° С. Пары из первого и второго корпусов поступают в главный конденсатор, разделенный по высоте на две части, работающие под разным давлением (45 и 34 435
Рис. 9.6. Вакуум-кристаллизационная установка непрерывного действия:
/ — мерник отработанного травильного раствора; 2 — мерник серной кислоты; 3 — вакуум-кристаллизатор; 4 — главный эжектор первой ступени сжатия; 5 — главный конденсатор; 6, 7, 8 — эжекторы второй, третьей и четвертой ступеней сжатия; 9 — промежуточный сдвоенный конденсатор; 10— барометрический сборник; 11— водоотделитель; 12 — конденсационный горшок;
13— насос; 14 — сборник суспензии кристаллов; 15 — центрифуга
мм рт. ст.), что позволяет эффективнее использовать охлаждающую воду. В нижнюю часть конденсатора входит пар из первого корпуса, в верхнюю — из второго. Пары из третьего и четвертого корпусов вначале сжимаются до 34 мм рт. ст. в первой ступени сжатия в двух параллельно работающих эжекторах, а затем идут в верхнюю часть главного конденсатора. Несконденсировавшаяся часть пара, выходящая из основного конденсатора, после сжатия эжекторами второй ступени направляется последовательно на промежуточную конденсацию, а затем на сжатие в третью ступень, на дополнительную конденсацию и, наконец, после сжатия в четвертой ступени выбрасывается в атмосферу. Выходящая из четвертого корпуса кристаллизационной установки суспензия поступает на центрифугирование для отделения кристаллов гептагидрата сульфата железа (И) от маточного раствора.
Сульфат железа (III) Fe2(SO4)3— светло-желтые гигроскопичные кристаллы (см. табл. 9.4). Растворяется в воде, из водного раствора кристаллизуется в виде кристаллогидратов Ре2(8О4)зиН2О, где п = 12, 10, 9, 7, 6 и 3. Растворяется также в ацетоне, нерастворим в этаноле.
Нонагидрат Рег^ОДз ^НэО — желтые кристаллы; в природе — минерал кокимбит; растворяется в воде (440 г в 100 г воды при 20° С), этаноле, гидразине. Не растворяется в ацетоне. В водных растворах си-436
льно гидрализован. При нагревании до 98° С нонагидрат превращается в тетрагидрат, при 125° С — в моногидрат, а при 175° С — в безводный Fe2(SO4)3, который выше 600° С разлагается по схеме
Fe2(SO4)3 ^‘с > Fe2O3 + 3SO3
Fe2(SO4)3 образует с аммиаком аммиакат:
Fe2(SO4)3 + 12NH3 = Fe2(SO4)3-12NH3
с сульфатом щелочных металлов и аммония образует квасцы M2ISO4Fe2(SO4)312H2O, а также комплексные сульфаты состава M'3[Fe(SO4)3]3H2O.
Сульфат железа (III) восстанавливается углеродом до сульфата железа (II) по схеме
Fe2(SO4)3 + С = 2 FeSO4 + СО2 + SO2
Степень превращения Fe2(SO4)3 в FeSO4 возрастает с повышением температуры, продолжительности взаимодействия и увеличением площади поверхности частиц исходного угля. Так, при 300° С и диаметре частиц угля меньше 3 мм степень превращения за 60 мин составляет 53%, за 90 мин — 76%, а за 120 мин — 90%.
Сульфат железа (III) восстанавливается также диоксидом серы до сульфата железа (II) с одновременным образованием свободной серной кислоты:
Fe2(SO4)3 + SO2 + 2Н2О = 2 FeSO4 + 2H2SO4
Образующийся по этой реакции Fe2+ вновь окисляется в присутствии диоксида серы и кислорода до Fe3+. При необходимости взаимодействие FeSO4 с SO2 может быть направлено в сторону образования Fe2(SO4)3 или H2SO4. На этом основан каталитический метод окисления диоксида серы в водном растворе в присутствии ионов железа. При 80° С, концентрации диоксида серы в газе 3% (а также и при меньших концентрациях), отношении SO2 : О2, равном 1:4, и содержании Fe2+ в растворе 30 г/л можно получать растворы, содержащие 20—25% H2SO4.
Сульфат железа (III) применяют для декапирования нержавеющих аустенитных сталей, сплавов меди с алюминием, в качестве компонента электролита в гальванотехнике, раствора в гидрометаллургии меди, для получения квасцов, в производстве пигментов Fe2O3, в ка-437
честве коагулянта при очистке воды, а также в качестве протравы при крашении.
Сульфат железа (III) в виде его водных растворов получают взаимодействием сесквиоксида железа с 75—80%-ной серной кислотой.
Растворы сульфата железа (III) получают также окислением серного колчедана азотной кислотой по схеме
2FeS2 + IOHNO3 = Fe2(SO4)3 + H2SO4 + 4Н2О + 10NO
Получают сульфат железа (III) и обработкой колчеданного огарка серной кислотой при повышенной температуре. При этом исходный просеянный огарок предварительно обрабатывают концентрированной азотной кислотой (8—10*кг на одну тонну исходного огарка). Окисленный огарок перемешивают и выдерживают перед реакцией в течение 5—6 ч.
9.8. ОКСИДЫ И ГИДРОКСИДЫ ЖЕЛЕЗА
Оксид железа (II). Железо образует следующие оксиды: FeO — оксид железа (II), Ре2Оз — сесквиоксид, РезО4 — оксид железа (II, III). Существуют следующие гидроксиды: Fe(OH)2; Fe(OH)3 и FeO(OH) (табл. 9.6).
Связь между оксидами и гидроксидами железа показана в приведенной схеме:
Fe(OH)3 -н2о a-FeO(OH) y-FeO(OH) .о/ 400 °C ’ a-Fe2O3 y-Fe2O3 Fe(OH)2 J/ О2+Н2О Fe Вакуум 250“С >1400 “С ' Fe3O4 ' FeO Окисление Окисление
438
Таблица 9.6. Свойства оксидов и гидроксидов железа
Показатель FeO Ре2Оз Fe3O4 FeCOHh Fe(OH)3 a-FeO(OH)
а Y
От тем-
Цвет Черный но-красного до черно-фи- Коричневый Черный Бледно-зеленый Красно-коричневый Коричневый
олетового
Сингония Кубическая Тригональная Кубическая Кубическая Гексагональная — Ромбическая
Параметры решетки, нм: а b с 0,4357 0,542 (а = 55,28°) 0,8339 0,839 0,324 0,447 0,46 0,994 о,з
Число формульных единиц в ячейке 4 2 — 8 1 — 4
Пространственная группа Fm3m R3c Fd3m Fd3m P3ml — Pbnm
Т °C 1369* 1565 (с разл.) — 1594** 150—200*** 350—400*** 136***
Плотность, г/см3 5,7 5,24 4,4-4,88 6,18 3,4 2,44—360**** 4,28
С", Дж/(мольК) — 113,91 — 144,63 97 — —
ДЯ"р, кДж/моль -272,4 -825,4 — -1120 -570 -824,5 -559,8
кДж/моль — -743,3 — -1016,9 -487,1 -697,6 —
Дж/(моль-К) 53,9 87,53 — 146,7 88 106,8 —
*Д#^=32,2 кДж/моль (1380° С, фаза Fe(vw7O). = 138,3 кДж/моль (1596С).***Темперазура разложения. ♦♦♦♦Для Fe2O3-xH2O.
Оксид железа (II) FeO (в технике — вюстит). Имеет структуру хлорида натрия или оксида магния. Ионы О2" имеют плотнокубическую упаковку с ионами Fe2+ в октаэдрических пустотах между ними. В кристаллической решетке вюстита имеются вакантные узлы, и его состав отвечает формуле FexO, где х = 0,89—0,95; уравнение температурной зависимости давления разложения: lgp(O2 в мм рт. ст.) = -26 730/Г + 6,43(7>1813 К). В воде практически не растворяется, хорошо растворяется в кислотах, а также в растворах щелочей. Легко окисляется, пирофорен. После термообработки при высоких температурах снижаются его химическая активность и пирофорность.
Оксид железа с точной формулой FeO фактически не существует, а всегда имеет промежуточный состав от FeOios до FeO]j9 (при 1400° С). Отсюда в решетке не хватает 5—15% катионов Fe2+, эквивалентное количество которых превращено в катионы Fe3+.
Три оксида железа—у-РезОз, РезОд и FeO — при различных температурах находятся в равновесии с кислородом, причем возможен переход одного оксида в другой, например РеО-»РезОд—>Ре2Оз (что можно написать и так: FeO—>FeOi,33—>FeOi 5). Эти превращения происходят следующим образом: решетки FeO или РезОд, которые являются плотными решетками ионов О2’, содержащих в октаэдрических пустотах ионы железа, растягиваются благодаря присоединению снаружи ионов О2’, образующихся из молекул О2. Электроны, необходимые для ионизации кислорода, дают ионы Fe2+, которые при этом переходят в ионы Fe3+. Одновременно ионы Fe3+ мигрируют в решетке, распределяясь равномерно по вновь образовавшимся октаэдрическим пустотам. Обратный процесс перехода оксида Ре2Оз при нагревании в РезОд и FeO состоит в отрыве с поверхности кристалла некоторых атомов кислорода после того, как они отдали один электрон ионам Fe3+, которые переходят в ионы Fe2+ и распределяются в решетке.
Твердые ионные соединения, если их решетки не имеют дефектов, не проводят электрического тока. При высоких температурах такие соединения становятся проводниками, так как электроны способны перемещаться в решетке. Кристаллы с дефектами структур в решетках являются полупроводниками. Кристаллы, содержащие катионы одного и того же элемента в различных степенях окисления, например Fe2+ и Fe3+, проводят электрический ток лишь в результате перехода электронов от одного иона к другому. Для того чтобы соединение, не проводящее электрического тока, стало электропроводным, достаточно незначительного отклонения состава от стехиометрического. Даже самые устойчивые оксиды, такие, как оксид алюминия, при высоких температурах становятся полупроводниками. Электропроводность таких полупроводников меняется в зависимости от парциального давления кислорода, с которым они находятся в равновесии. Особый случай представляет магнетит РезОд, в котором ионы Fe2+ и Fe3+ в соотноше-440
нии 2:1 распределены в пустотах плотной решетки ионов О2" (шпинельная решетка). Поэтому магнетит является почти таким же хорошим проводником электрического тока, как металл.
В природе иоцит — чрезвычайно редкий минерал.
Оксид железа (II) при нагревании реагирует с водой с выделением водорода. В водных растворах оксид железа при взаимодействии с разбавленными кислотами образует катионный аквакомплекс по схеме
FeO + 2OHJ + ЗН2О = [Fe(OH2)6]2+
Оксид железа (11) получают в виде черного порошка в процессе термического разложения оксалата железа в атмосфере азота или без доступа воздуха по схеме
FeC2O4-3H2O —FeO + ЗН2О + СО2 + СО
Получают также в процессе восстановления сесквиоксида железа водородом или оксидом углерода по уравнениям:
Fe2O3 + Н2 = 2FeO + Н2О
Fe2O3 + СО = 2FeO + СО2
Сесквиоксид железа Ре2Оз существует в трех полиморфных модификациях: а-форма — минерал гематит, у-форма— маггемит (оксимагнетит) и 8-форма с тригональной кристаллической решеткой; температуры перехода а —> у 677° С, у -> 8 777° С; АН перехода а —> у 0,67 кДж/моль. Для модификации a-Fe2O3 уравнение температурной зависимости давления разложения: Igp (О2, в мм рт. ст.) = -10 291/Г + + 5,75lg7—1,0910'3Г—0,75-1057^—12,33; растворяется в хлороводородной и серной кислотах, слабо — в азотной кислоте; парамагнетик, точка Нееля 953 К. Модификации у- и 8-форм ферромагнитны.
Оксиды железа Fe2O3 применяются в качестве сырья для производства металлического железа, являются минеральными пигментами. a-Fe2O3 является компонентом футеровочной керамики, цемента, термита, поглотительной массы для очистки газов, полирующего материала, применяется для получения ферритов, в качестве пигментов. y-Fe2O3 применяется в качестве рабочего слоя магнитных лент.
Физико-химические основы получения сесквиоксида железа — красного железооксидного пигмента. Красный железооксидный пигмент — сесквиоксид железа — получают в производстве терморазложением сульфата железа (II) при 700—800° С, или гидроксида железа (III), или оксида железа (II, III) при 600—700° С.
441
Гептагидрат сульфата железа (II) в процессе нагревания (рис. 9.7) обезвоживается и разлагается с выделением сесквиоксида железа и оксидов серы. Процесс отщепления молекул воды происходит в температурном интервале 68—73° С, а с достижением 120—125° С в исходном продукте остается лишь одна молекула воды. Исходный гептагидрат полностью теряет седьмую молекулу кристаллизационной воды в температурном интервале 288—309° С.
В процессе термообработки происходит следующее:
FeSO4-7H2O -> FeSO4H2O + 6Н2О
4FeSO4 H2O + О2 -> 2Fe2O3 + 4Н2О + 4SO3
Процесс разложения происходит также в неокислительной среде. Суммарное уравнение реакции в этих условиях будет протекать по схеме
6FeSO4 —> 2Fe2O3 + Fe2(SO4)3 + 3SO2
а затем
Fe2(SO4)3 —> Fe2O3 + 3SO3
Результаты исследования процессов, происходящих при термообработке, показали, что они более сложны. В окислительной среде моногидрат сульфата железа (II) при повышении температуры окисляется до основного сульфата железа (III):
4FeSO4-H2O + О2 -> 4Fe(OH)SO4 или Fe2O3-2SO4-2H2O
Рис. 9.7. Термограммы гептагидрата сульфата железа (II): а—в воздушной среде; 6 — в среде аргона
442
Соединение Ре2Оз-28О4иН2О является наиболее устойчивой и распространенной солью железа. В процессе получения оно обычно кристаллизуется с пятью молекулами воды. Процесс его окисления начинается при 90° С.
При 420—450° С процесс окисления моногидрата сульфата заканчивается и окисляется уже обезвоженная соль:
12Ре8Од + ЗО2 —> 4Fe2(SO4)3 + 2Ре2<Эз
При температуре около 500° С происходит параллельный процесс обезвоживания основной соли:
6Fe(OH)SO4 -> 2Fe2(SO4)3 + Fe2O3 + ЗН2О
При 650—700° С сульфат железа (III) разлагается до его оксида с выделением триоксида серы, разлагающегося в этих условиях до диоксида серы и кислорода.
Параллельный процесс разложения сульфата железа (II) происходит также в неокислительной среде и связан с окисляющим действием триоксида серы, который переходит при этом в диоксид:
4FeSO4 4FeO + 4SO3
4FeSO4 + 2SO3 -> Fe2(SO4)3 + SO2
4FeO + 2SO3 —> 2Рв2Оз + 2SO2
6FeSO4 —> 2Ре2Оз + Fe2(SO4)3 + 3SO2
Затем протекает реакция разложения сульфата железа (III):
Fe2(SO4)3 —> РегОз + 380з
Обобщенные результаты реакций разложения и окисления могут быть представлены уравнениями следующих реакций:
FeSO4-7H2O —6^7-3--^-> FeSO4 H2O + 6Н2О (1)
4FeSO4 H2O + О2 |20'125-^ 4Fe(OH)SO4 или 2Ре2Оз48Оз-иН2О (2)
Ре8О4-Н2О —2-8^’с- > FeSO4 + Н2О (3)
12FeSO4 + ЗО2 > 4Fe2(SO4)3 + 2Ре2Оз (4)
6Fe(OH)SO4 ^~5^> 2Fe2(SO4)3 + Fe2O3 + 3H2O (5)
443
6FeSO4
650-750* С
> Fe2(SO4)3 + 2Ре20з + 3SO2
(6)
Fe2(SO4)3.....^-7А9:с„> Fe2O3 + 3SO3
(7)
Как видно из приведенных уравнений, при термообработке гептагидрата сульфата железа (II) в неокислительной среде процессы обезвоживания и разложения протекают по реакциям (1), (3), (6) и (7).
На рис. 9.8 приведены зависимости констант скорости процесса разложения сульфатов железа в начальной и конечной фазах при 650—800° С.
Степень разложения во времени и состав выделяющихся газов для FeSO4*H2O, Fe(OH)SO4 и Fe2(SO4)3 при 650—700° С в окислительной и неокислительной среде показана на рис. 9.9. В производственных условиях степень окисления сульфата железа (II) в процессе обезвоживания зависит от температурного режима и продолжительности нагрева, а также количества участвующего в реакции воздуха. Установлено, что при 300° С в течение одного часа степень окисления в неподвижном слое массы реагентов составляет 1,6%, а в кипящем слое, при слабом кипении, — 4,5, умеренном — 30, а в сильном -37%. В процессе же энергичного кипения массы в течение одного часа степень окисления составляет при 150° С — 3, при 200° С — 9, при 250° С — 35, а при 300° С — 37%. Полное же окисление массы С.
Поскольку окисление реакционной массы не доходит до конца, процесс разложения в производственных условиях протекает по всем приведенным выше реакциям. В результате частичного спекания сульфата железа в процессе термообработки диффузионные явления играют значительную роль в течение всего процесса, включая и область разложения его до 90%. Поэтому процесс разложения исходного сырья в малоподвижном слое, например во вращающихся печах, протекает весьма медленно при 700° С и лишь в интервале температур 700—900° С сильно вырастает (при 900° С — в три раза по сравнению со скоростью процесса разложения при 700° С). Однако при высоких температу-
происходит лишь при 500
^103
Рис. 9.8. Зависимость константы скорости разложения сульфатов железа от температуры: / —FeSO4; 2 — Fe>(SO4)<;
3 — Fe(OH)SO4
444
Рис. 9.9. Процесс разложения сульфатов железа:
а—при 650° С в воздухе; б — при 650° С в токе азота; в — при 700° С в воздухе. / — FeSO4 Н2О;
2 — Fe(OH)SO4; 3 — Fe2(SO4),
pax заметно ухудшается цвет образующегося сесквиоксида железа, что связано с резким увеличением размера частиц (от 0,2 до 2—3 мкм). Из приведенных на рис. 20.10 данных видно резкое снижение выше 800° С удельной поверхности образующегося а-Ре20з.
а-Ре20з получают в производстве и термообработкой гидроксида железа(Ш) или оксида железа (II, III). В процессе термообработки они переходят в а-Ре20з, который в этих процессах образуется при сравнительно низкой температуре (около 300° С). Однако а-Ре2О3 свои пигментные свойства приобретает лишь при 600—700° С, что связано с
его кристаллизацией.
В качестве исходного сырья обычно применяют сульфат или хлорид железа (II). Их осаждают карбонатами натрия или кальция в виде карбонатов железа (II) или известковым молоком до гидроксида железа (II). Путем окисления последних получают а-Ре2Оз по уравнениям:
2FeSO4 + Na2CO3 -> 2FeCO3 + + 2Na2SO4
2FeCi2 + 2СаСОз -* 2РеСОз + + 2CaCI2
Рис. 9.10. Зависимость удельной поверхности а-РезО4 от температуры термообработки
445
2FeCl2 + 2Ca(OH)2 -> 2Fe(OH)2 4- 2СаС12
4FeCO3 + 2H2O + O2 -> 2Fe2O3H2O + 4CO2
4Fe(OH)2 + O2 -> 2Fe2O3 H2O + 2H2O
Процессы, происходящие в процессе термообработки:
Fe2O3 xH2O -> Fe2O3 + хН2О или 4(FeO-FeiO3) + О2 -> 2Fe2O3
В процессе термообработки сохраняется форма и в значительной степени размеры кристаллов исходных веществ.
На рис. 9.11 приведены термограммы гидроксида железа (III) и оксида железа (II, III), из которых видно, что процесс дегидратации гидроксида железа (III) начинается при 320° С и заканчивается при 400° С. Процесс окисления Ре3Од начинается при 200° С. При этом первый, более широкий экзотермический эффект связан с топохимическим однофазным окислением Ре3Од в y-Fe2O3, а второй — с переходом y-Fe2O3 в a-Fe2O3.
В процессе термообработки гидроксид железа (III) изменяет свою удельную поверхность в зависимости от температуры термообработки (рис. 9.12). Величина удельной поверхности проходит через резко выраженный максимум как в случае термообработки образцов в условиях вакуума, так и при атмосферном давлении. Появление максимума объясняется отторжением молекул воды из объема гидрокси-
Рис. 9.11. Термограммы гидроксида железа (III) (/) и Fe?O4 (2)
Температура, °C
Рис. 9.12. Удельная поверхность a- Fe(OH)3 после термической обработки в течение одного часа: 1 — в вакууме, 2 — при атмосферном давлении (цифры на кривых — остаточное содержание воды в образцах, %)
446
да, что способствует разрушению и разрыхлению структуры, а также кристалломорфным переходам гетита в гематит. В процессе нагревания при более высоких температурах (>240° С) удельная поверхность уменьшается, что связано с образованием новой кристаллической решетки a-Fe2O3. Такой процесс наблюдается и при нагревании Fe3O4.
Разработан способ получения а-Ре2Оз минуя термообработку путем окисления металлического железа кислородом воздуха в растворах сульфата железа (II) с применением специальных зародышей.
Разработан также способ получения а-Ре2Оз из водных суспензий гидроксида железа (III) в автоклаве под давлением и при температуре 170—190° С.
Получение сесквиоксида железа. Процесс получения a-Fe2O3 в производственных условиях состоит из следующих стадий: обезвоживание гептагидрата сульфата железа (II); термообработка обезвоженного продукта; промывка; мокрый помол с классификацией частиц; сушка оксида железа.
Процесс обезвоживания необходим потому, что в процессе обезвоживания гептагидрата сульфата железа (II) происходит плавление его в собственной кристаллизационной воде, в результате чего сульфат железа (II) образует твердую спекшуюся массу. Чтобы этого избежать, исходный гептагидрат смешивают с моногидратом сульфата железа (И) с таким расчетом, чтобы общее количество выделяемой кристаллизационной воды не превышало 4 моль на один моль FeSO4. Подготовленная смесь плавится в печи при 350—400° С, образующийся продукт, моногидрат сульфата железа (III), измельчают на валковой дробилке.
Термообработку моногидрата сульфата железа (II) проводят при 700—725° С для получения пигмента с желтоватым оттенком и при 775—825° С для получения пигмента с синеватым оттенком. Процесс термообработки ведут до разложения сульфата железа (II) на 90—93% практически до содержания водорастворимых солей в пересчете на Fe2(SO4)3 7% в условиях, близких к кинетической области, так как в этих условиях процесс термообработки протекает значительно интенсивнее и снижается возможность ухудшения цвета в результате перекала. При термообработке к исходному моногидрату сульфата железа (II) обычно добавляют 4—5% восстановителя, в частности мазута.
Аппаратурное оформление процессов обезвоживания и термообработки примерно одинаковое: вращающиеся барабанные печи, футерованные шамотным или хромомагнезитовым кирпичом. На некоторых заводах вместо вращающихся печей применяют конвейерные печи или печи с кипящим слоем. Печи обогреваются газообразными продуктами сжигания мазута или природного газа в топке печи.
447
Внутренний диаметр вращающихся печей для обезвоживания и термообработки 1,5—2 м, а длина печи 14 м. В процессе обезвоживания температура газов у входа в печь составляет 550—600° С, а у выхода — до 220° С. В процессе термообработки температура поддерживается в пределах: на входе в печь — 900—1100° С, а на выходе—400—450° С.
Недостатком барабанных вращающихся печей является то, что в них происходит процесс частичной грануляции материала, это приводит к замедлению процесса и затрудняет диффузию газов от поверхности частиц через слой материала.
Термообработка в кипящем слое является более прогрессивным методом, позволяет интенсифицировать операции обезвоживания и разложения сульфата железа (II), повысить содержание диоксида серы в отходящих газах и получить продукт в виде однородной массы. Во избежание загрязнения целевого продукта в производстве пользуются топливом, не содержащим серы, а процесс сгорания проводят в высокоэффективных топках беспламенного горения, в которых обеспечивается полнота сгорания. Печи оборудуются также улавливающими системами, в которых оседают мелкие частицы, уносимые выхлопными газами (-15—20%).
Для обезвоживания используется гептагидрат сульфата железа (II), который с помощью шнека, охлаждаемого водой, подается в реактор с кипящем слоем моногидрата сульфата железа (II). Размер частиц моногидрата 0,2—0,6 мм, скорость газов в печи поддерживается равной 0,25—0,35 м/с, температура в слое 200—300° С, температура под подиной должна быть не выше 500° С во избежание образования значительного количества сульфата железа (III). Высота слоя составляет 1,8—2,0 м, перепад давления в кипящем слое -850—950 мм рт. ст.
Безводный сульфат железа с частью газов через переливную трубу подают в циклон, а оттуда в загрузочный бункер печи термообработки.
Безводный сульфат железа (II) помимо моногидрата содержит значительное количество окисленных соединений железа.
Процесс разложения обезвоженного сульфата железа (II) в кипящем слое происходит с большей скоростью уже при температуре в слое 650—700° С и ускоряется с повышением температуры, скорости давления газа в печи. Увеличение интенсивности процесса происходит лишь до некоторого предела, выше которого изменения уже незначительны.
Значительный технико-экономический интерес представляют также работы по получению а-Ре2Оз разложением растворов гептагидрата сульфата железа (II) в распыленном состоянии.
а-Ре2Оз получают также в прокалочной печи конвейерного типа. Конвейер состоит из ряда тележек, расположенных на колосниковой 448
решетке непрерывной ленты, движущейся по замкнутому рельсовому пути. Под верхней рабочей ветвью конвейера расположены вакуум-камеры, над которыми находится прокалочная печь. Гептагидрат сульфата железа (II) из бункера поступает в тележки, которые заполняются слоем толщиной в 150—250 мм. Смесь продуктов сгорания с воздухом из рабочей камеры печи просасывается вентилятором через слой гептагидрата, находящегося в тележках, и поступает сначала в вакуум-камеры, а затем — в сборник на газоочистку. В начальной стадии нагрева гептагидрат плавится, кипит, обезвоживается и спекается, образуя пористую массу. Дальнейшие процессы при нагреве до температуры разложения и разложение сульфата железа (II) происходят на сильно развитой поверхности и весьма интенсивно. При выходе из печи основная масса прокаленного продукта (около 75%) снимается ножом, а остальное количество ссыпается с тележек в бункер. Оптимальная температура обработки 700—750° С достигается при температуре рабочих газов под слоем сульфата железа (II) 800° С. Продолжительность термообработки 100—200 мин. При длине рабочей части колосникового полотна 12 м, ширине 1,4 м, площади -17 м2 производительность печи составляет -1700 кг/ч, площади -17 м2 производительность печи составляет -1700 кг/ч по гептагидрату сульфата железа (II) или 400 кг/ч по прокаленному продукту.
Термообработанный материал состоит преимущественно из крупных агрегатов (1—3 мм), он хорошо отстаивается и его промывают декантацией с применением сгустителей непрерывного действия. Обычно выполняют четырехкратную промывку с репульпацией сгущенного осадка в емкостях с метальным устройством, соблюдая Т:Ж = 1:10. Первые промывки производят при температуре не выше 60° С во избежание образования в результате гидролиза основных сульфатов железа, которые удаляются с большим трудом. При последней промывке добавляют незначительное количество карбоната натрия (15 кг на 1 т целевого продукта) для осаждения остаточного сульфата железа (III). Для получения тонкодисперсного продукта, свободного от крупных спекшихся частиц, промытый осадок подвергают мокрому помолу в шаровых мельницах. Суспензию после размола разбавляют водой до соотношения Т:Ж = 1:10 и подвергают гидросепарации. Верхний слой с гидроциклона сгущают в отстойниках, фильтруют на барабанных вакуум-фильтрах, сушат в сушилках барабанного типа.
а-БегОз получают также термообработкой гидроксида железа (III) и РезО4. Осажденный гидроксид железа (III) после промывки отжимают на барабанных вакуум-фильтрах. Образующаяся паста с содержанием влаги 30—45% загружается в барабанные вращающиеся печи. Температура входящих в печь газов 750—800° С, выходящих — 250—300° С. Период прохождения массы через печь составляет около трех часов.
449
15 Химическая технология
Производительность печи -18—20 т в сутки. Прокаленная масса после выгрузки из печи охлаждается в холодильном барабане, размалывается в роликокольцевой мельнице.
a-Fe2O3 получают сжиганием пентакарбонила железа по схеме
4Fe(CO)5 + 13О2 = 2Fe2O3 + 20СО2
Процесс получения a-Fe2O3 состоит в распылении пентакарбонила воздухом и сжигании его во взрывной камере с температурой 700° С. Полученный сесквиоксид отличается очень высокой степенью дисперсности.
Разработан способ получения a-Fe2O3 минуя термообработку путем окисления металлического железа кислородом воздуха в растворах сульфата железа (II) с применением специальных зародышей.
Разработан также способ получения a-Fe2O3 из водных суспензий гидроксида железа (III) в автоклаве под давлением и при температуре 170—190° С.
Оксид железа (II, III). Оксид железа (II, III) — соединение формулы Fe3O4, или FeOFe2O3, Рен(РеП1О2)2 (минерал — магнетит) — представляет собой смешанный оксид FeO и Fe2O3, в кристаллической решетке которого содержатся оба типа ионов железа Fe2+ + Fe3+ с таким же кубическим расположением, как в шпинелях, с которыми он изоморфен. В процессе нагревания разлагается; при 627° С ос-форма переходит в P-форму; уравнение температурной зависимости давления разложения: Igp (О2 в мм рт. ст.) = -33 265/7" + + 13,37 (7>843 К); ферромагнетик, точка Кюри 900 К; отличается высокой электрической проводимостью.
Fe3O4 растворяется в кислотах с образованием солей Fe(II) и Fe(III). Прокаленный при 1200—1300° С природный магнетит практически не растворяется в кислотах и их смесях. Не растворяется также в азотной кислоте и аммиаке. При нагревании на воздухе окисляется до сесквиоксида железа.
Fe3O4 является исходным материалом для изготовления электродов, используемых при электролизе хлоридов щелочных металлов, компонентом активной массы щелочных аккумуляторов, цветного цемента, футеровочной керамики и термита, широко применяется в качестве пигмента.
Получение оксида железа (II, III) основано на способности гидроксидов железа (II) и (III) вступать между собой в реакцию с образованием Fe3O4 по следующей схеме:
2Fe(OH)3 + Fe(OH)2 = FeOFe2O3 + 4Н2О 450
Существование этой реакции обусловлено кислотным характером гидроксида железа (III) и основным характером гидроксида железа (II). Предполагают, что вначале образуются продукты присоединения гидроксидов, которые затем дегидратируются по схеме
2Fe(OH)3 + Fe(OH)2 = Fe(OH)2-2Fe(OH)3 = = FeOFe2O3-HH2O = FeOFe2O3 + иН2О
Значительное влияние на состав и свойства смешанного гидроксида оказывают условия его образования. В зависимости от условий смешанный гидроксид получается в виде непрочного, но активного соединения, адсорбирующего значительное количество воды и легко окисляющегося кислородом воздуха в оксид или гидроксид железа (III). Такой оксид железа образуется в процессе взаимодействия гидроксида железа (II) со свежеосажденным аморфным гидроксидом железа (III).
Известны два мокрых способа получения Fe3O4: 1) взаимодействие соли железа (II) с гидроксидами или карбонатами и окисление выпадающего в осадок Fe3O4; 2) окисление металлического железа ароматическими нитросоединениями в водном растворе солей железа.
Первый способ заключается в окислении гидроксида железа (II) кислородом воздуха в гидроксид железа (III), который вступает в реакцию с еще не окислившимся гидроксидом железа (II) с образованием смешанного оксида железа:
4Fe(OH)2 + 2Н2О + О2 = 4Fe(OH)3
2Fe(OH)3 + Fe(OH)2 = FeOFe2O3 + 4H2O
Гидроксид железа (II) можно заменить карбонатом железа, который при нагревании гидролизуется, образуя гидроксид железа (II), окисляющийся затем в Fe3O4 по схеме
FeCO3 + Н2О = Fe(OH)2 + СО2
6Fe(OH)2 + О2 = 2Fe3O4 + 6Н2О
С целью ускорения процесс окисления проводят при нагревании. По данным некоторых исследователей, процесс окисления может быть ускорен добавлением к гидроксиду железа (II) небольших количеств нитрита натрия или хлорида цинка.
Получаемый по этому способу Fe3O4 обладает высокой стойкостью, не окисляется кислородом воздуха и отличается высокими пигментными свойствами.
451
15*
Образование Fe3O4 при окислении металлического железа ароматическими нитросоединениями в растворе электролита начинается с процесса получения гидроксида железа (II), который вступает во взаимодействие с вновь образующимся в результате коррозии гидроксидом железа (III):
2Fe + 2Н2О + О2 = 2Fe(OH)2
4Fe(OH)2 + 2Н2О + О2 = 2Fe(OH)3
2Fe(OH)3 + Fe(OH)2 = Fe3O4 + 4H2O
Образование Fe3O4 в процессе окисления металлического железа происходит лишь в среде электролитов, не гидролизующихся в присутствии железа.
На состав и свойства Fe3O4 влияют условия проведения реакции. Поэтому для получения высококачественного пигмента процесс окисления следует вести при строго установленных условиях.
Существует также способ получения Fe3O4 в восстановительном процессе из гидроксида и оксида железа (III). Процесс восстановления ведется газообразными восстановителями.
Моногидрат оксида железа (1П). FeO(OH) — гетит, или Fe2O3 H2O, липидокрокит y-FeO(OH).
Существует в трех полиморфных модификациях: наибольшее значение имеет a-форма (гетит). Свето-, атмосфере- и щелочестойкость очень высока. Растворяется в минеральных кислотах. Нерастворим в уксусной кислоте. При нагревании выше 180—200° С теряет гидратную воду и переходит в сесквиоксид железа, при 275—300° С этот переход происходит очень быстро. у-Форма моногидрата оксида железа FeO(OH) — соединение, устойчивое при кипячении в воде при 100° С; в автоклаве при 105—115° С полностью переходит в a-FeO(OH). В процессе термообработки при 250° С переходит в y-Fe2O3. В последнее время установлено существование и 8-формы.
Способы получения моногидрата оксида железа (III). Моногидрат оксида железа (III) получают: 1) окислением воздухом раствора соли железа (II) в присутствии металлического железа при постепенном добавлении гидроксидов щелочных металлов; 2) окислением металлического железа ароматическими нитросоединениями; 3) окислением водной взвеси свежеосажденного гидроксида или карбоната железа (II).
Окисление раствора соли железа (II) воздухом. Получение моногидрата оксида железа (III) окислением воздухом водного раствора соли железа (II) в присутствии металлического железа состоит из 452
следующих операций: приготовление раствора гептагидрата сульфата железа (II); приготовление раствора карбоната натрия или гидроксида (аммония, натрия или кальция); приготовление зародыша; синтез моногидрата оксида железа (III), промывка осадка целевого продукта; отжим и сушка.
Для получения моногидрата оксида железа (III) применяют реакторы с разной емкостью от 25—30 до 150 м3. Реакторы футеруют кислотоупорным кирпичом или плитками, изготовленными из кислотоупорных материалов. Реактор разделен решеткой на две зоны; над решеткой в цилиндрической части помещается металлическое железо, в нижней части (конической), под решеткой, находится устройство для глухого и острого пара, а также воздушный барботер.
В качестве сырья обычно применяют отходы жести тарных цехов, состоящие из стали марки 3. Они имеют сильно развитую поверхность, из-за чего ускоряется процесс окисления.
Зародыши готовят окислением взвеси свежеосажденного гидроксида (иногда карбоната) железа (II) в растворе сульфата железа (II) кислородом воздуха или хлоратом калия при комнатной температуре или при слабом нагреве. Для осаждения гидроксида железа (II) применяют гидроксид кальция и натрия или аммиак. Процесс окисления считают завершенным при pH около 4. При 20° С получается зародыш светлого, при 30° С — среднего, а при 40—50° С — темного оттенка пигмент. Зародыш синтезируют или в отдельной емкости, после чего направляют в реактор, или в самом реакторе, куда перекачивают растворы сульфата железа (II) и аммиака, а выделившийся осадок окисляют воздухом. Зародыш обычно берут в таком количестве, чтобы его содержание в целевом продукте было около 10%.
В реакторе происходят следующие химические реакции:
4FeSO4 + 2Н2О + О2 = 4Fe(OH)SO4
Fe(OH)SO4 + 2NH3 + 2Н2О = FeO(OH) + (NH4)2SO4 + H2O
Кислотность раствора является следствием частичного гидролиза соли железа (III):
Fe(OH)SO4 + 2Н2О = Fe(OH)3 + H2SO4
При данном способе применяются исходные растворы с начальной концентрацией сульфата железа (II) 100—120 г/л. По принятому технологическому режиму допустимая концентрация сульфата аммония считается -200 г/л.
453
16 Химическая технология
Процесс осаждения проводят до содержания сульфата железа (П) в растворе 20—30 г/л; конечная концентрация осадка в суспензии — 50—60, а сульфата аммония — 70—80 г/л.
Скорость образования целевого продукта определяется в значительной степени скоростью барботирования воздуха, которая составляет 100—200 м3/ч.
Процесс получения целевого продукта в присутствии металлического железа состоит в окислении кислородом воздуха сульфата железа (II). Роль металлического железа заключается в нейтрализации кислоты и сохранении постоянной концентрации в растворе сульфата железа (II):
Fe + H2SO4 = FeSC>4 + Н2
Необходимая кислотность среды достигается за счет того, что гидролиз превалирует над реакцией нейтрализации. Сульфат железа (II) при этом играет роль переносчика кислорода по схеме
2Fe + 2Н2О + [2FeSO4 + О2] = 2Fe(OH)2 + 2FeSO4
4Fe(OH)2 + О2 = 4FeO(OH) + 2H2O
Синтез целевого продукта проводится в каскадной батарее из трех реакторов. В первый реактор непрерывно подают раствор сульфата железа (II) (110—120 г/л) и зародыша (15—18 г/л). Из третьего реактора непрерывно сливают готовую суспензию. Температура растворов во всех реакторах 75—80° С. Окисление ведут воздушно-ам-миачной смесью при pH 3,5—4,0.
Фильтрование, промывку и сушку целевого продукта проводят по общеизвестным приемам.
Маточный раствор, содержащий 200 г/л сульфата аммония и 10—15 г/л сульфата железа (II), упаривают и применяют в виде удобрения. Первые промывные воды, содержащие 10—15 г/л сульфата аммония, используют для приготовления раствора сульфата железа (II). Вторые и третьи промывные воды направляются на первую и вторую промывки.
Окисление металлического железа ароматическими нитросоединениями. По этому способу металлическое железо окисляют нитробензолом в водных растворах в присутствии хлороводородной кислоты или хлорида железа (II), образующегося в результате растворения исходного железа в хлороводородной кислоте. Изменив условия реакции, по этой схеме можно получить оксиды железа в виде тонкодисперсных образований; при этом помимо РезО4 можно получить и гидроксид железа (III). Как известно, нитробензол не реагирует с со-454
лями оксида железа (II). Образование оксидов железа можно представить следующей схемой, в которой указаны лишь основные направления реакции:
^FeO(OH) РеОРе2Оз = РезОд Ре2Оз
Fe -> Fe(OH)2 -> [Fe(OH)2 Fe(OH)3] -+ Fe(OH)3
Fe(OH)3 Fe2O3-HH2O
Согласно этой схеме, в начале процесса металлическое железо переходит в гидроксид железа (II), который затем окисляется в соответствующий оксид в зависимости от условий. Образование РезОд, Ре2Оз и FeO(OH) происходит в результате окисления гидроксида железа (II) сначала в активный гидроксид железа (III), который, вступая во взаимодействие с не окислившимся гидроксидом железа (II), образует непрочные продукты присоединения типа Fe(OH)2-Fe(OH)3 или, что то же, РеОРе2Оз*иН2О. Эти продукты затем либо дегидратируются и переходят в прочное соединение РезОд, либо окисляются в неустойчивый гидроксид железа (III) или Ре2Оз*Н2О.
Для получения светло-желтого пигмента — Ре(ОН)з — в качестве электролитов применяют соли алюминия, хрома, олова, церия и некоторых других металлов. Причина специфического влияния этих электролитов обусловлена способностью их растворов гидролизоваться почти полностью в присутствии металлического железа:
2А1С1з + 3Fe + 6Н2О = 2А1(ОН)3 + 3FeCl3 + ЗН2
Следовательно, в процессе окисления металлического железа нитробензолом в присутствии этих электролитов в реакции участвует не сам электролит, а продукт его гидролиза, т.е. гидроксид железа (III) или основная соль металла и раствор соли железа (II).
Кристаллический гидроксид железа (III) образуется лишь в присутствии гидроксида трехзамещенного металла, например А1(ОН)3 или Сг(ОН)з, и соли железа (II), например FeCl2. Такое действие гидроксидов трехзамещенных металлов объясняется их амфотерностью и свойствами зародышей. Реагируя как вещества, обладающие кислотным характером, с гидроксидом железа, они тем самым предупреждают взаимодействие гидроксидов железа (II) и (III) и исключают образование РезО4, FeO и Fe(OH)2. В результате этих реакций гидроксид железа (II) окисляется в кристаллический гидроксид железа (III).
Наибольшее значение в качестве электролита имеет хлорид алюминия, его гидролиз протекает примерно на 90%. Гидроксид алюминия стабилизируется остатком хлорида алюминия и переходит в ко-455
16*
лоидный раствор. Примерный состав соединения, образующегося при стабилизации, А1С1з 5А1(ОН)з.
Окисление карбоната или гидроксида железа (II). Карбонат и гидроксид железа (II) получаются в процессе взаимодействия соли железа (II) с карбонатом натрия или гидроксидами щелочных металлов в виде осадка белого или серо-зеленого цвета. Окисление производится барботированием воздуха при комнатной температуре или хлоратом калия при 50—60° С:
РеЗОд + Na2CO3 = РеСОз + Na2SO4
4FeCO3 + иН2О + О2 = 2Fe2O3nH2O + 4СО2
Количество гидратной воды в осадке может колебаться в пределах 12—15%, что соответствует 1,0—1,5 моль Н2О на один моль Fe2O3. Кристаллическая структура этих гидратов — гётит.
9.9. ЖЕЛЕЗНАЯ ЛАЗУРЬ
Физико-химические свойства, применение. Железная лазурь (ми-лори, берлинская лазурь, парижская синяя) — неорганический синий пигмент состава Fe4+[Fe2+(CN)6]3 • К4[Ре(СЫ)б] • иН2О, где х = 0,3—0,8, х = 12—24. Железная лазурь кристаллизуется в кубической системе (а = 0,510 нм). В железной лазури может содержаться 4—10% К; 56—68% Fe(CN)6; 19—22% железа; 3—17% воды.
Цвет железной лазури может изменяться от темно-синего до черного, от синего до голубого. Оттенок — красноватый с медным брон-зящим блеском; красноватый без медного блеска и зеленоватый.
Состав различных видов железной лазури:
Темная Светлая Гэлубая
К 4,04 8,95 10,0
Fe(CN)6 56,8 60,3 67,3
Н2О:
удаляемая при 60° С 4,87 1,95 1,35
удаляемая при 120° С 11,87 4,34 2,52
Плотность, г/см' 1,92 — 1,80
Удельная поверхность, м2/г.... 100 200 — 2550
Маслоемкость, г/100г. 60 — 30
456
Железная лазурь не растворяется в воде, слабых кислотах, органических растворителях, легко разлагается щелочами до гексацианоферрата калия и Fe2O3H2O. Устойчива при нагревании до 180° С, а при 280° С разлагается с выделением цианида водорода, оксида железами) и аммиака.
Концентрированная серная кислота разлагает лазурь, особенно при температуре кипения по схеме
Fe4[Fe(CN)6]3 + 18H2SO4 + 18Н2О = = 2Fe2(SO4)3 + 9(NH4)2SO4 + 18СО + 3FeSO4
Железная лазурь образует коллоидные растворы при обработке ее водными растворами щавелевой и винной кислот, тартрата аммония или гексацианоферратов щелочных металлов. Под действием хлороводородной кислоты коллоидные растворы разрушаются.
Железная лазурь применяется для производства масляных, эмалевых, нитролаковых и других красок, в том числе полиграфических. Непригодна для водоэмульсионных и других красок, имеющих щелочную среду, и для окраски по известковой штукатурке.
Физико-химические основы получения железной лазури. В процессе взаимодействия гексацианоферрата (III) с солями железа (III) и гексацианоферрата (II) с солями железа (II) образуются нерастворимые соединения интенсивно-синего цвета:
3K4[Fe(CN6)] + 4FeCl3 = Fe4[Fe(CN)6]3 + 12КС1 берлинская лазурь
3K3[Fe(CN6)] + 3FeCl2 = Fe3[Fe(CN)6]2 + 6KC1 турнбулева синь
При избытке гексацианоферрата (II) или гексацианоферрата (III) берлинская лазурь и турнбулева синь образуются в растворимой форме, причем состав обоих соединений одинаковый:
K4[Fe(CN6)] + FeCI3 = KFe[Fe(CN)6] + ЗКС1 (растворимая турнбулева синь)
K3[Fe(CN6)] + FeCl2 = KFe[Fe(CN)6] + 2КС1 (растворимая берлинская лазурь)
Результаты исследований показали, что турнбулева синь и берлинская лазурь полностью идентичны KFe[Fe(CN)6]. Их кристаллы образованы полимерными ионами [Ре2(СЫ)б]“ и ионами К+.
Согласно данным ренгеноструктурных исследований, турнбулева синь и берлинская лазурь имеют одинаковую кубическую решетку с атомами железа в эквивалентных положениях. Установлено, что их 457
у-резонансные спектры одинаковы (рис. 9.13). В их кристаллах атомы Fe(II) окружены атомами углерода, а атомы Fe(III) — атомами азота цианидных групп и образуют полимерный координационный ион [Fe2(CN)6]“ со структурой типа ЯеОз (рис. 9.14), в междоузлиях которой расположены ионы К+.
Железную лазурь получают взаимодействием соли железа (И) с гексацианоферратом (III) калия с последующим окислением образующегося белого осадка (так называемого белого теста) в кислой среде. Соли железа (II) способны образовывать смешанные гексацианоферраты с однозамещенными катионами, при этом содержание последних может быть весьма значительным. По активности образовывать смешанные гексацианоферраты однозамещенные катионы располагаются в следующий ряд:
Cs>Rb>NH4>K>Na>Li
Тяжелые металлы редко образуют «чистые» гексацианоферраты состава Men2[Fe(CN)6]. Такие соли возможно получить при взаимодействии солей тяжелых металлов с K4[Fe(CN)6], Li4[Fe(CN)6] и иногда с Na4[Fe(CN)6]-K4[Fe(CN)6] образует смешанные гексацианоферраты с солью железа (II) по следующей схеме:
2FeSO4 + xK4[Fe(CN)6] = Fe2[Fe(CN)6]-(x— l)-K4[Fe(CN)6] + 2K2SO4
Состав белого теста, т.е. соотношение между Fe2[Fe(CN)6] и K4[Fe(CN)6], зависит от соотношения между исходными реагентами Fe2SO4 и K4[Fe(CN)6] и от температуры процесса осаждения: чем мень
Рис. 9.13. У-СнСКТрЫ бср.!! П1СКОИ лазури (а). турнбулевоп сини (о) и co.icii К Ее | п
Рис. 9.14. Координационная решсчка кристаллов берлинской лазчрн
45Х
ше избыток исходного сульфата железа и выше температура процесса осаждения, тем больше гексацианоферрата калия в белом тесте.
Установлено, что в практике производства значение х находится в пределах от 0,3 до 0,8. Процесс образования белого теста может быть представлен следующими уравнениями:
2FeSO4 + 2K4[Fe(CN)6] = Fe2[Fe(CN)6]-K4[Fe(CN)6] + 2K2SO4
2FeSO4 + l,5K4[Fe(CN)6] = Fe2[Fe(CN)6]-0,5K4[Fe(CN)6] + 2K2SO4
Белое тесто легко получается на воздухе:
2Fe[Fe(CN)6]*K4[Fe(CN)6] + О2 + 2Н2О = = Fe4[Fe(CN)6]3-K4[Fe(CN)6] + 4КОН
Как видно из уравнения, железо (II) переходит в железо (III) и связывает большее число групп Fe(CN)6, а часть калия переходит в раствор в виде гидроксида. В связи с этим, для исключения присутствия щелочи, процесс окисления проводят в кислой среде, а иногда с добавкой солей аммония, которые также способны нейтрализовать щелочь по схеме
NaOH + NH4C1 = NaCl + NH3 + H2O
Процесс окисления белого теста позволяет получать железную лазурь с повышенным (до 20%) содержанием калия, что невозможно в процессе осаждения гексацианоферрата калия непосредственно солью железа (III). Так, при стехиометрическом соотношении реагентов 3K4[Fe(CN)6] + 4FeCl3 образуется чистый гексацианоферрат с незначительным содержанием калия. Чистые гексацианоферраты в отличие от смешанных содержат значительное количество прочно удерживаемой воды, менее стойки к действие разбавленных кислот и к нагреву, менее интенсивны и укрывисты. "Это в значительной мере обусловливает низкие пигментные свойства' и малую устойчивость железной лазури, получаемой прямым методом.
Процессы, происходящие при окислении белого теста, в действительности более сложны, так как при этом протекают и ионообменные реакции.
Зависимость состава железной лазури от условий получения белого теста показана ниже:
Температура осаждения, °C....... 20 60 60 60 60
Продолжительность кипячения, ч. — — — 5 5
Количество добавляемой H2SO4,%
(масс.) от FeSO4................. — — — 18 200
459
Состав железной лазури, %:
К 4,04 5,97 8,95 10,1 10,0
Fe(CN)6 56,8 57,8 60,3 61,8 67,3
Fe экстрарадикальное 19,8 19,7 19,7 19,5 19,53
Удаляемая Н2О:
при 60° С 4,87 3,28 1,95 1,97 1,35
» 120° С 11,37 6,38 4,34 4,59 2,53
С повышением температуры процесса осаждения, продолжительности кипячения и кислотности среды содержание калия в лазури увеличивается, а воды уменьшается. Кроме всего увеличивается и содержание Fe(CN)6 в лазури. В процессе нагревания вода постепенно испаряется, а полностью удаляется лишь при разрушении лазури.
Количество калия, групп Fe(CN)6 и воды, входящих в состав лазури, находится в определенной зависимости: чем выше содержание групп Fe(CN)6, тем больше в лазури калия и меньше воды. Однако количество калия ограничено и не превышает 10—11% даже в том случае, когда число групп Fe(CN)6 продолжает увеличиваться.
Ниже приведен состав натриевой железной лазури, полученной в различных технологических условиях:
Температура осаждения, °C ... 20 60 60
Продолжительность кипячения, ч — — 5
Состав железной лазури, %:
Na .... 2,26 3,82' 3,26
Fe(CN)6 ... 54,8 59,3 ‘ 58,6
Fe экстрарадикальное .... 20,5 19,8 20,4
Удаляемая Н2О:
при 60° С .... 4,04 3,33 2,97
» 120°С 12,76 11,12 8,18
Состав белого теста: Fe2[Fe(CN)6]-0,5Na4[Fe(CN)6] при температуре осаждения 25° С, а при температуре осаждения 75° С Fe2[Fe(CN)6] 0,66Na4[Fe(CN)6]. При избытке соли железа (II) содержание Na4[Fe(CN)6] снижается и при большем избытке составляет 0,36 и 0,43 моль на 1 моль Fe2[Fe(CN)6] при температурах процессов осаждения 25 и 75° С соответственно. Таким образом, в процессе окисления гексацианоферрат (III) способен образовывать смешанные соединения даже с гексацианоферратом натрия, в то время как в процессе осаждения гексацианоферрата натрия непосредственно солью железа (III) при стехиометрическом соотношении реагентов получается лишь соль Ред[Ре(СК)б]з без примеси соли натрия. Это подтверждает большую способность иона Fe2+ к образованию смешанных гексацианоферратов по сравнению с ионом Fe3+. 460
Аммониевую лазурь получают взаимодействием соли железа (II) с гексацианоферратом калия или натрия в присутствии солей аммония [NH4CI или (NH4)2SO4]. Способ основан на способности аммоний-иона вытеснить калий- или натрий-ионы из железной лазури. Обычно в производстве пользуются недорогостоящим гексацианоферратом натрия. Количество исходных солей аммония берут из расчета 15—20% (масс.) от исходного гексацианоферрата натрия. В процессе получения аммониевой лазури в кислой среде или при избытке в растворе солей аммония образуется лазурь, содержащая, помимо групп NH4, всего 0,2—0,3% натрия; при осаждении в нейтральной среде и без избытка солей аммония в лазури остается до 1,0—1,2% Na. Содержание в лазури NH4-rpynn обычно эквивалентно содержанию калия. Исключение составляет лишь аммониевая лазурь, полученная в процессе кипячения белого теста в сильнокислой среде. В этом варианте содержание аммониевых групп оказывается больше.
Ниже приведен состав нескольких видов аммониевой лазури:
Темная Средняя Светлая Гэлубая
Na 0,2 0,6 0,55 Следы
Fe(CN)6 58,6 62,2 67,9 67,7
Fe экстрарадикальное 20,6 21,2 19,9 19,7
NH4 3,0 4,7 4,8 7,4
Удаляемая Н2О:
при 60° С 4,2 3,67 1,7 1,7
» 120° С 10,0 7,3 3,5 3,7
Железную лазурь применяют для производства масляных, эмалевых, нитролаковых и других красок, в том числе полиграфических. Непригодна она для водоэмульсионных и других красок, имеющих щелочную среду, и для окраски по известковой штукатурке.
Получение железной лазури. Сырьем в производстве железной лазури являются гексацианоферрат калия или натрия, сульфат железа (II), окислители (КСЮз или К2СГ2О7), кислоты (серная или хлороводородная) и соли аммония [NH4C1, (№{4)2804]. Количество кислоты и окислителя берут обычно несколько меньше к требуемому по расчету для окисления железа (II) в железо (III) по уравнению
6FeSO4 + КСЮз + 3H2SO4 = 3Fe2(SO4)3 + КО + ЗН2О
В процессе окисления хромпиком необходимо учитывать расход кислоты для перевода калия и хрома в растворимое состояние:
2К2Сг2О7 + 8H2SO4 = 2K2SO4 + 2Сг2(8О4)з + 8Н2О + ЗО2
461
Обычно применяют меньшее количество кислоты с таким расчетом, чтобы хром частично или полностью оставался в составе лазури в виде гидроксида (1—9%):
6Ре8Од + К2СГ2О7 + 4H7SO4 + 72Н2О — = 3Fe2(SO4)3 + K2SO4 + 4Н2О + Сг2О3иН2О
Процесс получения железной лазури состоит из следующих стадий: приготовление растворов гептагидрата сульфата железа (II) и синькали (или синьнатра); осаждение белого теста, его промывка, нагревание и кипячение; окисление белого теста; промывка пигмента и его фильтрование; сушка и размол пигмента.
Гептагидрат сульфата железа (II) растворяют при 50—60° С в небольшом количестве воды, после чего добавляют воду из расчета получения раствора, содержащего 150—200 г/л сульфата железа (II). Полученные растворы фильтруют.
Синькали (или синьнатр) растворяют также в небольшом количестве воды и разбавляют до получения раствора, содержащего 200 г/л. Хлорат калия ввиду его пожароопасности хранят в емкости, залитой холодной водой.
Белое тесто осаждают в реакторе, куда вначале заливают раствор сульфата железа (II), который разбавляют водой и при энергичном перемешивании с раствором синькали. Выпавший осадок при перемешивании выдерживают в течение одного часа.
Избыток синькали после осаждения белого теста недопустим, так как [Ре(СЫ)б]4‘, обладающий высоким зарядом, стабилизирует железную лазурь, образуя прочный неоседающий золь.
По окончании процесса осаждения белого теста его окисляют или подвергают промывке, нагреву и кипячению. Для промывки белое тесто разбавляют водой, дают отстояться в течение 20—24 ч, после чего осветлившийся слой сливают с осадка, а вместо него приливают воду. Обычно белое тесто промывают один раз, а при необходимости до 4—5 раз.
Нагрев и кипячение проводят острым или глухим паром, при этом продолжительность операции зависит от марки железной лазури. Для получения особо светлых видов лазури к белому тесту перед кипячением добавляют соответствующее количество кислоты.
Готовое белое тесто охлаждают до 60—70° С добавлением воды, после чего при перемешивании добавляют серную или хлороводородную кислоту и раствор перхлората калия или хромпика.
Процесс окисления продолжается около часа. Конец реакции определяют по отсутствию в маточном растворе солей железа (II). 462
Железную лазурь в реакторе отмывают от водорастворимых солей. Для полного растворения водорастворимых солей осадок промывают 10—12 раз. Для ускорения промывки применяют высокомолекулярные водорастворимые флокулянты.
Лазурь фильтруют на фильтр-прессах типа ФПАКМ и листовых вакуум-фильтрах. После промывки суспензию лазури фильтруют и полученную пасту, содержащую около 55% воды, сушат при 60—70° С, после чего размалывают.
Для сушки пасты лазури применяются гребковые вакуум-сушилки и сушилки с формующим устройством. Размол лазури производится в ударно-центробежных мельницах.
Технологическая схема производства железной лазури приведена на рис. 9.15.
Синькали, гептагидрат сульфата железа (И) и перхлорат калия растворяются в аппаратах 7, 2 и 10. Растворы сульфата железа (II) в синькали для очистки фильтруют на рамных фильтрах 7. Из сборников 5 и 6 чистые растворы сульфата железа (И) и синькали поступают в реактор 14, где синтезируется белое тесто. После разбавления, перемешивания, отстоя, декантации, нагрева и кипячения белое тесто окисляют раствором перхлората калия или хромпика в присутствии хлороводородной или серной кислоты при повышенной температуре.
По окончании синтеза лазурь промывают репульпацией с применением листовых вакуум-фильтров 77. Промытая лазурь из последнего фильтра переносится с помощью тельфера 16 к загрузочному бункеру 18 и сбрасывается в него после отключения вакуума. Отфильтрованную пасту загружают в гребковую вакуум-сушилку 24 и высушивают под вакуумом. Высушенная лазурь транспортируется из сушилки шнеком 25 и элеватором 27 на размольную установку, состоящую из бункера 28 с питателем 29 и мельницы 30. Размолотую лазурь упаковывают в тару и вывозят на склад готовой продукции.
Лазурь, увлекаемая в процессе сушки парами воды, улавливается в сухом уловителе 23, а затем в мокрой ловушке 41, из которой суспензия возвращается в репульпатор 19. Вакуум для фильтрования и сушки лазури создается вакуум-насосами 40 и 43. Пары воды, удаляемые в процессе сушки лазури под вакуумом, поступают в барометрический конденсатор 38, орошаемый водой, и через гидравлический затвор 39 выводятся в канализацию. Сушилку в первой стадии сушки обогревают паром, а в последней стадии — горячей водой из бойлера 22 во избежание перегрева.
463
Рис. 9.15. Технологическая схема производства железной лазури
Вода
9.10. ГЕКСАЦИАНОФЕРРАТЫ (II, III)
Известны гексацианоферраты (II, III) аммония, калия, натрия, кальция, железа и других элементов.
Гексацианоферрат (II) (желтая кровяная соль) K4[Fe(CN)6]-3H2O— светло-желтые кристаллы с тетрагональной решеткой (а = 0,939 нм, с = 3,376 нм, z = 8, пространственная группа 14j/a); плотность 1,853 г/см3 при 17° С. Ниже -26° С (точка Кюри) переходит в моноклинную модификацию (а = 0,934 нм, b = 1,687 нм, с = 0,934 нм, Р = 90° , z = 4, пространственная группа С2/с); сегнетоэлектрик. Для тетрагональной модификации С? = 482,4 Дж/(моль-К); ДЯо°бр = 1471,5 кДж/моль; 52°98 = 592,9 Дж/(моль К). Выше 87,3° С превращается в безводную соль (плотность 1,935 г/см3). Растворимость в воде (г в 100 г): 31,5 (25° С), 48,3 (50° С), Д/^раств 55,0 кДж/моль. Практически не растворим в эфире, этаноле, пиридине, анилине и этилацетате. В вакууме выше 600° С разлагается на KCN, Fe и (CN)2, а в инертной атмосфере — на KCN, Fe3C, К и N2.
Разлагается концентрированной хлороводородной кислотой с выделением белого осадка H4[Fe(CN)6] по схеме
K4[Fe(CN)6] + 4НС1 = H4[Fe(CN)6] + 4КС1
Реагирует с разбавленной серной кислотой по схеме
2K4[Fe(CN)6] + 6H2SO4 = K2Fe[Fe(CN)6] + 6KHSO4 + 6HCN
С солями М(П) и М(1П) образует малорастворимые соединения, в частности с солями железа (III) дает синий осадок приблизительного состава KFin[Fe1I(CN)6] (берлинская лазурь или турнбулева синь). В водных растворах окисляется хлором по схеме
2K4[Fe(CN)6] + С12 = K3[Fe(CN)6] + 2НС1
Гексацианоферрат (II) калия получают из содержащих цианидные соединения отходов после очистки газов на газовых заводах. Отходы обрабатывают водной суспензией гидроксида кальция, а полученный фильтрат, содержащий Ca2[Fe(CN)6], перерабатывают:
Ca2[Fe(CN)6] + 2КС1 = K2Ca[Fe(CN)6] + СаС12
K2Ca[Fe(CN)6] + К2СО3 = K4[Fe(CN)6] + СаСО3
Гексацианоферрат (II) калия получают также взаимодействием суспензии FeS водным раствором KCN.
465
Гексацианоферрат (II) калия применяют в производстве пигментов, цианистых соединений, ферритов, цветной бумаги, крашении шелка, в качестве компонента ингибирующих покрытий и при цианировании сталей, для выделения и утилизации радиоактивного цезия, в качестве реагента для обнаружения ионов железа (III), цинка и меди (II). Является электролитом в хемотронных приборах.
Гексацианоферрат (III) калия (красная кровяная соль) Кз[Ге(СЫ)б]—темно-красные кристаллы с моноклинной решеткой (а = 1,342 нм, b = 1,040 нм, с = 0,838 нм, Р = 90° , z = 4, пространственная группа Р2|/с); плотность 1,845 г/см3; С° = 316,5 Дж/(моль-К); АЯ0°бр= 253,5 кДж/моль; £2°98 = 420,9 Дж/(моль-К). Растворимость в воде (в г 100 г): 40,8 (15,6° С) и 58,7 (37,8° С); в водном растворе на свету постепенно переходит в K4[Fe(CN)6]. В растворах щелочей окисляет сульфид водорода до элементарной серы, иодид водорода до иода, оксид свинца до его диоксида, аммиак до азота и солей аммония, вольфрама до WoO2'4. С солями железа (II) образует темно-синий осадок турнбулевой сини. С концентрированной серной кислотой реагирует, образуя при этом FeH(SO4)2, KHSO4, NH4HSO4 и СО.
Гексацианоферрат (III) калия получают окислением K4[Fe(CN)6] хлором:
2K4[Fe(CN)6] + С12 = 2K3[Fe(CN)6] + 2КС1
Процесс протекает как в растворе, так и при действии газообразного хлора на твердую соль. В случае растворов соли разделяют кристаллизацией. Вместо хлора можно использовать и бром.
Гексацианоферрат (III) калия применяют в качестве компонента тонирующих, отбеливающих, усиливающих, ослабляющих растворов в фотографии, компонентом электролита в гальванопластике, является электролитом в хемотронных приборах, реагентом для обнаружения Fe2+, Lir и Sn2+.
ГЛАВА 10
КОБАЛЬТ И ЕГО СОЕДИНЕНИЯ
10.1. КОБАЛЬТ
Содержание кобальта в земной коре 4-10‘3% (масс.), а в воде океанов— 0,005 мг/л. Известно более 30 минералов кобальта. Важнейшим из них является: каролит C11C02S4, линнеит C03S4, кобальтин CoAsS, скуттерудит C0AS3, шмальтин C0AS2, асболан СозО47иМпО2иН2О, эритрин Co3(AsO4)2-8H2O, сферокобальтит С0СО3. Спутниками кобальта в природе являются: никель, железо, медь и марганец. Кобальт добывают из медно-никелевых, пиритных, мышьяковисто-кобальтовых руд и др. Перспективным источником кобальта являются марганцевые конкреции на дне океанов.
Природный кобальт состоит из двух изотопов — 59Со (99,83%) и 57Со (0,17%). Конфигурация внешних электронных оболочек 3s23p63(/4.s2; степень окисления -1, 0, +2, +3, редко +1, +4 и +5; энергии ионизации при последовательном переходе от Со° к Со3+ соответственно 7,866, 17, 057 и 33,50 эВ; сродство к электрону 0,94 эВ; электроотрицательность по Полингу 1,9; атомный радиус 0,125 нм, ионный радиус (координационное число 6) 0,079 нм для Со2+, 0,069 нм для Со3+ и 0,067 нм для Со4+.
Кобальт — серебристо-белый, слегка желтоватый металл с розоватым или синеватым отливом. При комнатной температуре устойчива a-модификация с гексагональной решеткой, а = 0,2505 нм, с = 0,4089 нм, z = 2, пространственная группа Р631ттс\ при 427° С. а-Со переходит в 0-Со с кубической гранецентрированной решеткой, а = 0,3544 нм, z = 4, пространственная группа Fniim', ДЯ^реХ0Да = 0*45 кДж/моль. Температура плавления 1494° С, температура кипения 2960° С; плотность 8,90 г/см3; С" = 24,8 кДж/(моль-К); Д/7П°Л = 16,3 кДж/моль, Д//°сп = 375 кДж/моль; 52°Q8 =30,1 Дж/(моль-К); уравнения температурной зависимости давления пара: для твердого кобальта Igp (гПа) = = 12,68—24 126/7-0,114 lg7 + 0,874-1037 (298—1767 К), для жидкого кобальта Igp (гПа) = 11,547—20 695/7—0,608 lg7(1767—3230 К); температурный коэффициент линейного расширения 13,6-10^(303—673 К); теплопроводность 69,5 Вт/(м-К); р = 6,5-Ю’9Ом-м, температурный коэффициент р = 6,510'3К‘1(273—473 К); ферромагнетик, точка Кюри 467
1121° С. Модуль нормальной упругости 270 ГПа, праст = 130 МПа; относительное удлинение 2,5%; твердость по Бринеллю для отожженного образца 470—1230 МПа.
Стандартный электрический потенциал кобальта — 0,29 В. Компактный кобальт устойчив на воздухе, выше 300° С покрывается оксидной пленкой, тонкодисперсный — пирофорен.
Кобальт не взаимодействует с водой, водными растворами щелочей, карбоновых кислот, слабо реагирует с разбавленными минеральными кислотами. В концентрированной азотной кислоте он пассивируется. Металлический кобальт заметно поглощает водород, не образуя соединений. Гидриды кобальта СоН2 (устойчив до 45° С) и СоН (устойчив до 150° С) получают косвенным путем. Кобальт медленно растворяет азот, образуя твердый раствор (до 0,6% по массе азота при 600° С). С аммиаком образует нитриды — C03N при 250—300° С и Co2N при 380—500° С. Расплав кобальта реагирует с углеродом, образуя карбид Со3С, который разлагается в процессе кристаллизации. Карбид Со2С получают действием на металл оксида углерода или метана при 225° С, который разлагается в вакууме при 300° С. В процессе сплавления кобальта с кремнием образуются силициды: Co2Si, CoSi и CoSi2 с температурой плавления соответственно 1332, 1465 и 1327° С, с бором — бориды Со3В, Со2В и СоВ с температурой плавления соответственно 1180, 1280 и 1460° С. Взаимодействует с элементным фосфором или фосфином, образуя фосфид Со2Р с температурой плавления 1386° С. Получены также фосфиды кобальта СоР и СоР3.
В процессе сплавления кобальта с серой получают моносульфид P-CoS — серебристо-белые кристаллы с металлическим блеском, решетка гексагональная (а = 0,3385 нм, с = 0,5213 нм, z = 2, пространственная группа P6jJmmc)\ плотность 5,45 г/см3; температура плавления 1180° С; температура воспламенения около 680° С (при величине зерна 0,1—0,2 мм); растворяется в азотной кислоте, а в присутствии пероксида водорода — также в хлороводородной кислоте. В природе P-CoS — минерал джепурит. Известна также неустойчивая ромбоэдрическая у-модификация CoS. Под действием сульфида водорода или сульфида аммония на нейтральные водные растворы кобальта выпадает черный осадок аморфного а-CoS, хорошо растворимый в разбавленных минеральных кислотах, кристаллизующийся при непродолжительном хранении в P-CoS. В процессе диссоциации CoS в атмосфере сульфида водорода образуется CoqOs с кубической кристаллической решеткой; температура плавления 835° С (инконгруэнтно); плотность 5,24 г/см3. Известны также высшие сульфиды: Со35д — серебристо-белые с розоватым оттенком кристаллы со структурой типа шпинели (а - 0,941 нм), плотность 4,86 г/см3 (минерал 468
линнеит); C0S2 (а = 0,564 нм), плотность 4,73 г/см3(минерал каттие-рит); C02S3 — кристалл с кубической решеткой.
В процессе сплавления кобальта с селеном образуется селенид CoSe с температурой плавления 1055° С (минерал фребольдит), а также диселенид кобальта CoSe2, плавящийся инконгруэнтно при 960° С (минерал трогталит). При сплавлении кобальта с теллуром получают теллурид кобальта Со3Те4 с температурой плавления 1010° С и СоТе2, плавящийся инконгруэнтно при 749° С. Кобальт сплавляется также с мышьяком и сурьмой. К настоящему времени известны следующие арсениды кобальта: CoAs с температурой плавления 1180° С; C05AS2, C02AS, плавящиеся инконгруэнтно соответственно при 923, 958 и 1014° С; C02AS3, C0AS2 и CoAs3 (в процессе нагревания отщепляют As). Арсениды кобальта хорошо растворяются лишь в азотной кислоте и царской водке.
Антимониды кобальта: CoSb (температура плавления 1202° С, плотность 8,24 г/см3), CoSb2 (плотность 8,24 г/см3) и CoSb3 (плотность 7,62 г/см3), плавящиеся инконгруэнтно соответственно при 919 и 859° С. CoSb обладает металлической проводимостью, CoSb2 и CoSb3 являются полупроводниками с шириной запрещенной зоны соответственно 0,2 и 0,5 эВ.
Силикат кобальта Co2SiO4 — фиолетовые кристаллы с ромбической решеткой (а = 0,599 нм, b = 0,477 нм, с = 1,6027 нм, z = 4, пространственная группа Р,нспУ, температура плавления 1415° С; плотность 4,63 г/см3. Известен также метасиликат CoSiO3, устойчивый до 1000° С. Силикат кобальта и калия K2CoSi3O8 — основа синего стекла (смальты).
Получение кобальта. Кобальтсодержащие руды или концентраты подвергают гидрометаллургической переработке с применением серной кислоты или водных растворов аммиака либо вначале перерабатывают пирометаллургическим способом. Пирометаллургическим способом в качестве полуфабрикатов получают сульфидный или металлический сплав, обогащенный кобальтом, который переводят в раствор гидрометаллургическими способами, например выщелачиванием серной кислотой, анодным разложением. От примесей кобальт очищают фракционным окислением и гидролитическим осаждением (удалением железа, марганца, мышьяка и частично меди), цементацией (удаление меди и золота), а также экстракцией. Для разделения кобальта и никеля первый обычно осаждают действием хлората натрия либо хлора или других окислителей в щелочной среде.
Металлический кобальт получают: восстановительной плавкой Со3О4, образующегося при переработке гидроксида кобальта (III); электролизом водных растворов сульфата или хлорида кобальта; восстановлением водородом под давлением из аммиачных растворов. Рафинирование кобальта проводят электролизом водного раствора сульфата кобальта в присутствии ортоборной кислоты. Кобальт высокой 469
чистоты получают экстракционной очисткой с использованием кобальтовых солей жирных кислот (кобальтового мыла), с помощью которых извлекают из растворов сульфата кобальта примеси железа, меди, цинка, никеля и др.
Кобальт в основном (около 65%) применяется в качестве компонента сплавов. Около 10% производимого кобальта расходуется для производства катализаторов, около 10% — для изготовления пигментов. Кобальт является также компонентом стекол и керамики, а его соединения применяются для подкормки животных и в качестве микроудобрений. Он входит в состав витамина В^. Радиоактивный изотоп 60Со (Г1/2 = 5,27 года, источник у-излучения) получил широкое применение в медицине в борьбе с раком («кобальтовая пушка»).
10.2. ОКСИДЫ И ГИДРОКСИДЫ КОБАЛЬТА
Кобальт образует оксиды и гидроксиды: монооксид СоО, оксид кобальта (II, III) СозОд, сесквиоксид СО2О3, гидроксид кобальта (II) Со(ОН)2, гидроксид кобальта (III) Со(ОН)3.
Монооксид кобальта. Монооксид кобальта (II) СоО — серые, коричневые или оливково-зеленые кристаллы с кубической решеткой (a-форма а = 0,4258 нм, z = 4, пространственная группа Fm3w); плотность 6,47 г/см3. При температуре 985° С переходит в Р-модификацию, аналогичную по структуре a-форме, но меньшей плотности. Температура Нееля 290 К; при этой температуре переходит в тетрагональную модификацию (при — 180° С а = 0,42552 нм, с = 0,42058 нм); Д/7° перехода 0,30 кДж/моль; температура плавления 1810° С; С° = 55,2 Дж/(моль-К); Д/7^р = -239 кДж/моль; 52°98 = 52,7 Дж/(моль-К); уравнение температурной зависимости давления диссоциации Igp (гПа) = = 11,15—25 200/7" (100—1500 К). Полупроводник p-типа; ширина запрещенной зоны 0,65 эВ.
Монооксид кобальта практически не растворяется в воде (3,1-10‘4%, масс.) и органических растворителях. Растворяется в расплавах щелочей, а при доступе воздуха — в водном растворе аммиака. В процессе нагревания на воздухе выше 390° С переходит в оксид кобальта (11, III). Реагирует с минеральными кислотами, образуя при этом соответствующие соли. В процессе термообработки не пассивируется. Легко восстанавливается до металла действием водорода выше 120° С и оксидом углерода выше 200° С по схеме
СоО + Н2 —Со + Н2О
СоО + СО --^’с > Со + СО2
470
С кислородом образует нестехиометрические фазы.
Монооксид кобальта получают термическим разложением гидроксида кобальта (II) или гидроксокарбоната в инертной атмосфере, а также восстановлением или термическим разложением оксида кобальта (И, III) по схеме
Со(ОН)2 —СоО + Н2О
Со2СО3(ОН)2-хН2О —2СоО + СО2 + (х + 1)Н2О
СО3О4 + Н2 —> ЗСоО + Н2О
2СО3О4 —-—> бСоО + О2
Монооксид кобальта является сырьем в производстве катализаторов и ферритов, применяется в качестве компонента твердых электролитов, пигментов для керамики, стекла и фарфора.
Оксид кобальта (II, III). СО3О4 — серые или черные кристаллы с кубической решеткой типа шпинели Со+2Со+3О4 (а = 0,8086 нм, z = 8, пространственная группа 7\/Зш); плотность 6,073 г/см3; С° = 122,8 Дж/(моль-К); АЯо°бр = 887 кДж/моль; £2°98 = 102,9 Дж/(моль-К). Температура Нееля 40 К. В процессе нагревания выше 900° С разлагается до монооксида, уравнение температурной зависимости давления разложения Igp (гПа) = 17,37—17 700/Г (1033— 1143 К).
Не растворяется в воде и органических растворителях. Практически не реагирует с минеральными кислотами. Разлагается в расплавленных щелочах. С кислородом образует нестехиометрические фазы. Легко восстанавливается водородом и оксидом углерода. Сильный окислитель. При взаимодействии СО3О4 с кислотами выделяется кислород:
2СоСо2О4 + 12Н+ + 30Н2О = 6[Со(ОН2)6]2+ + О2
а при взаимодействии концентрированной НС1—СЬ.
Оксид кобальта (II, III) получают термообработкой гидроксидов, гидрооксокарбоната или нитрата кобальта на воздухе около 700° С.
Оксид кобальта (II, III) является исходным сырьем для производства металлического кобальта, монооксида кобальта, катализаторов, компонентов шихты для изготовления специальной керамики, кобальтсодержащих пигментов, реактивом для анализа органических веществ.
Сесквиоксид кобальта. Сесквиоксид кобальта СО2О3 — серое, темно-коричневое или черное скрытокристаллическое вещество; содержание кислорода в нем обычно немного меньше стехиометрического. Структура гексагональная (а - 0,464 нм, с = 0,567 нм, z = 2);
471
ДЯ^ = -577 кДж/моль. В воде и органических растворителях практически не растворяется. С кислотами реагирует слабо. Выше 300° С разлагается до Со3О4.
Сесквиоксид кобальта в производстве получают термообработкой гидроксидов или нитрата кобальта до 300° С.
Сесквиоксид кобальта применяют в качестве катализатора в органическом синтезе, адсорбента для газовой хроматографии, для получения ферритов, является пигментом для эмалей и глазурей, а также материалом катодов для гальванических элементов с неводными растворами электролитов.
Гидроксид кобальта (II) Со(ОН)2 — синее (a-форма) или розовое скрытокристаллическое либо фиолетовое кристаллическое (P-форма) вещество. а-Форма [иногда для нее приводят формулу ЗСо(ОН)2-2Н2О] метастабильна и при «старении» переходит в Р-фор-му. Структура обеих модификаций гексагональная; для а: а = 0,309 нм, с = 0,8 нм; для Р: а = 0,3173 нм, с = 0,4640 нм, z = 1, пространственная группа Р3т\9 плотность 3,60 г/см3; ДЯ^Р = -540 кДж/моль; £°98 = 83 Дж/(моль-К). Оксид кобальта (II) практически не растворим в воде (210‘4%, масс.) и органических растворителях. Амфотерное соединение, проявляющее преимущественно основные свойства. Растворяется в водных растворах аммиака. Реагирует с минеральными кислотами. С концентрированными водными растворами щелочей образует гидроксокобальтаты, например Ыа2[Со(ОН)6]. Све-жеосажденный гидроксид кобальта (II) легко окисляется на воздухе. В процессе нагревания при 110° С переходит в СоО(ОН). При нагревании без доступа воздуха a-форма выше 150° С дегидратируется, а P-форма не теряет воду полностью даже при 300° С.
Гидроксид кобальта (II) получают реакцией гидроксида натрия или аммиака в водной среде с солью кобальта по схеме
СоС12 + 2NaOH = Со(ОН)2 + 2NaCl
CoSO4 + 2NH3 + 2Н2О = Со(ОН)2 + (NH4)2SO4
Гидроксид кобальта (II) применяют для получения солей кобальта и катализаторов.
Гидроксид кобальта (III) Со(ОН)3 или СоО(ОН)Н2О— темно-коричневое скрытокристаллическое вещество; по структуре аналогичен сесквиоксиду кобальта; плотность 4,29 г/см3. Содержание воды обычно немного меньше стехиометрического; может содержать избыточное количество активного кислорода благодаря примеси гидроксида СоО2иН2О. Д//0°б = -725,5 кДж/моль; 52°98 = 100 Дж/(моль-К). При нагревании до 100° С частично дегидратируется с образованием 472
СоО(ОН). Разлагается на свету. Гигроскопичен. Не растворяется в органических растворителях, практически не растворим в воде (3-1 О’40/©, масс.). Медленно реагирует с азотной и серной кислотами (с выделением кислорода), легче — с водородной кислотой с выделением элементного хлора и с сульфитом водорода. Реагирует также с фтороводородной кислотой и с сульфидом водорода.
Гидроксид кобальта (III) получают окислением хлором, хлоратом, пероксидом водных растворов солей кобальта в присутствии щелочи.
Гидроксид кобальта (III) является полуфабрикатом в производстве оксида кобальта (II, III) и солей кобальта (III).
Гидроксооксид кобальта (III) СоО(ОН) — черные кристаллы; существует в двух модификациях — тригональной (ромбоэдрической) с плотностью 4,72 г/см3_(а = 0,2851 нм, с = 1,3150 нм, z = 3, пространственная группа RSm) и ромбической типа диаспора, образующейся при высоких температурах и давлениях (а = 0,4353 нм, b = = 0,9402 нм, с = 0,2840 нм, z = 4, пространственная группа РЬПП1)\ Л//0°бр = -435 кДж/моль. Не растворяется в воде. В процессе нагревания при температуре 300° С обезвоживается с потерей части кислорода. Реагирует с хлороводородной кислотой с выделением хлора. С азотной и серной кислотами реагирует с большим трудом. Не реагирует также с водными растворами щелочей и аммиака.
Гидроксооксид кобальта (III) получают нагреванием гидроксидов кобальта до 100° С с доступом воздуха, а также действием пероксида водорода на водную суспензию гидроксида кобальта (II) при нагревании в присутствии щелочи. Является компонентом катализаторов, пигментом, а также полуфабрикатом в процессе получения оксида кобальта (II, III).
10.3. КАРБОНИЛЫ КОБАЛЬТА
Известны три карбонила: октакарбонилдикобальт, додекакарбонилтетракобальт и гексадекакарбонилгексакобальт.
Октакарбонилдикобальт Co2(CO)s — оранжевые кристаллы; температура плавления 51 ° С (с разложением). В кристаллах его молекула состоит из двух фрагментов: Со(СО)з, соединенных связью Со—Со и двумя мостиковыми группами СО:
17 Химическая технология неорганических веществ, кн. 2
473
В молекуле карбонила кобальта группы СО связаны с атомом кобальта через атомы С: электронная пара от атома С передается атому кобальта, образуя сг-связь, а d-электроны металла переходят на вакантные разрыхляющие л-орбитали СО (л-связь). Кобальт координирует такое число групп СО, чтобы его электронная оболочка достраивалась до оболочки стоящего за ним благородного газа. При этом принимают, что СО является донором двух электронов, а кобальт находится в нулевой степени окисления.
В растворах эта форма находится в равновесии с другой формой — (СО)4Со—Со(СО)4, не содержащей мостиковых групп СО.
Растворяется в органических растворителях, но не растворяется в воде. Окисляется кислородом воздуха, а также галогенами, серной и азотной кислотами до соединений Со (II) и СО. Восстанавливается щелочными металлами до [Со(СО)4]“ и [Соз(СО)ю]‘, водородом — до [СоН(СО4)]. Известны также соли K[Co(CO4),NH4[Co(CO) 4].
Октакарбонилдикобальт получают взаимодействием смеси водорода и оксида углерода с гидроксокарбонатом кобальта или оксида углерода с Со(ОСОСНз)2 в гексане в температурном интервале 150—180° С и при повышенном давлении, а также реакции металлического кобальта с оксидом углерода.
Октакарбонилдикобальт применяют для получения компактного и порошкообразного кобальта высокой чистоты, для нанесения покрытий кобальта на металлы, пластмассы, керамику, в качестве катализатора в органическом синтезе.
Додекакарбонилтетракобальт Со4(СО)]2 — черные кристаллы с температурой плавления 60° С (с разложением) и температурой возгонки 90° С/1 мм рт. ст. (с разложением). Растворяется в органических растворителях незначительно. Медленно окисляется кислородом воздуха. Получают путем нагревания октакарбонилдикобальта при 50° С в инертной атмосфере.
Гексадекакарбонилгексакобальт Co6(CO)i6 — черные кристаллы. Растворяется в углеводородах. Получают окислением [СОб(СО)]5]2- и [Со6(СО)|4]4“, образующихся в процессе восстановления Со4(СО)]2 щелочными металлами в тетрагидрофуране.
10.4. ГАЛОГЕНИДЫ КОБАЛЬТА
Кобальт образует ди- и трифториды, дихлорид, дибромид и диио-дид, свойства которых приведены в табл. 10.1.
Дифторид кобальта CoF2— розовые кристаллы с тетрагональной решеткой (а = 0,46951 нм, с = 0,3179 нм, z = 2, пространственная группа РА/тпт); = 44,8 кДж/моль, АНи°сп = 201 кДж/моль (2012 К); AGo°6p = -620,7 кДж/моль; 52°98 = 81,96 Дж/(моль К). Раство-474
римость в воде -14,5 г/л при 25° С. Образует ди- и тетрагидраты, из растворов фтороводорода осаждается в виде CoF2*5HF*6H2O.
Дифторид кобальта получают взаимодействием оксида или карбоната кобальта (II) с газообразным фторидом водорода при нагревании
СоО + 2HF = CoF2 + Н2О
СоСОз + 2HF = CoF2 + Н2О + СО2
термическим разложением NH4C0F3
NH4C0F3 —CoF2 + HF + NH3
осаждением из водных растворов солей кобальта (II) при действии фтороводородной кислоты
СоС12 4- 2HF = C0F2 + 2НС1
Дифторид кобальта является полуфабрикатом при синтезе трифторида кобальта, комплексных фторидов кобальта (III) и кобальта (IV). Он применяется в производстве катодов в химических источниках тока, в качестве компонента лазерных материалов и катализатора в производстве фторуглеводородов.
Таблица 10.1. Свойства галогенидов кобальта
Соединение Температура плавления, °C Температура кипения, °C Плотность, г/см3 с;. Дж/(моль-К) °’,Р кДж/моль
CoF2 ИЗО 1740 4,46 68,78 -665,7
C0F3 — — 3,89 — -783,2
СоС12 740 -1050 3,356 78,49 -310
CoBr2 678 — 4,909 79,66 -216
Со12 516 760 5,68 — -85,8
Трифторид кобальта C0F3 — коричневые, зеленовато-коричневые или светло-коричневые кристаллы с ромбоэдрической решеткой (а = = 0,5279 нм, а = 56,97°, z = 2, пространственная группа Юс). Возгоняется без разложения. Водой и ее парами гидролизуется. В органических растворителях не растворяется. Электролитическим окислением суспензии дифтора кобальта в 40%-ном водном растворе фторида водорода получен CoF2-3,5H2O. В процессе нагревания при 300—400° С при взаимодействии трифторида кобальта с органическими веществами атомы водорода и хлора замещаются на атом фтора. Трифторид кобальта фторирует ненасыщенные соединения.
17*
475
Трифторид кобальта получают взаимодействием металлического кобальта или дифторида кобальта со фтором или трифторхлоридом при 200—300° С по схеме
2Со + 3F2 = 2C0F3
2C0F2 + F2 = 2C0F3
2Со + 2C1F3 = 2CoF3 + С12
6CoF2 + 2C1F3 = 6C0F3 + С12
Трифторид кобальта применяется в качестве реагента и катализатора в производстве фтороуглеводородов.
Дихлорид кобальта СоС12 — голубые гигроскопичные кристаллы. Существует в двух полиморфных модификациях. Температура перехода 680° С; ДЯ;Л = 38 кДж/моль; ДЯ°СП = 14,5 кДж/моль; AGL = -267 кДж/моль; 52°98 = 109,6 Дж/(моль-К). Растворяется в воде (51,3 гв 100 г при 20° С). В процессе нагревания или под действием водоотнимающих средств (СаС12, спирт и др.) окраска кристаллогидратов дихлорида кобальта меняется, что связано с изменением характера координации лигандов и появлением многоядерных комплексов. Процесс нагревания кристаллогидратов дихлорида кобальта сопровождается следующим изменением их состава и окраски:
СоС12-6Н2О -5-^-> СоС12-4Н2О СоС12-2Н2О ?°-с->
красно-коричневый темно-красный фиолетовый
—^->СоС12-Н2О • СоС12 сине-фиолетовый розовый
Растворяется в метаноле и этаноле с образованием синих растворов.
Дихлорид кобальта получают взаимодействием кобальта с элементным хлором, взаимодействием кобальта, оксида кобальта, гидроксида кобальта или солей кобальта (II) с хлороводородной кислотой с последующей дегидратацией в вакууме при температуре 150° С или обработкой SOC12 по схеме
Со + С12 = СоС12
Со + 2НС1 = СоС12 + Н2
СоО + 2НС1 = СоС12 + Н2О
476
Со(ОН)2 + 2НС1 = СоС12 + 2Н2О
СоСОз + 2НС1 = СоСЬ + Н2О + СО2
Дихлорид кобальта и его гидраты — протравы в процессе крашения тканей, микродобавки в корм скоту, компоненты растворов для нанесения покрытий кобальта на металлы. Дихлорид кобальта применяется также в процессе получения катализаторов и в индикаторах влажности.
Дибромид кобальта СоВг2 — зеленые гигроскопичные кристаллы. Существуют в двух полиморфных модификациях, температура перехода 375° С, Д/70 перехода 1,67 кДж/моль; Д//°сп = 205 кДж/моль (951 К). Растворяется в воде (199,1 г в 100 г при 60° С), этаноле, эфире и других растворителях. Образует моногидрат СоВг2-Н2О (на воздухе устойчив до 130° С, температура плавления 238° С, с разложением), дигидрат СоВг2-2Н2О (устойчив до 100° С, температура плавления 160° С, с разложением), тетрагидрат (температура плавления 62° С, с разложением) и гексагидрат с температурой плавления 43,6° С. Получают взаимодействием кобальта с бромом при нагревании. Дибромид кобальта применяют для получения катализаторов, а также в индикаторах влажности.
Дииодид кобальта Со12 — коричнево-красные гигроскопичные кристаллы с гексагональной решеткой. Известна также желтая модификация, получаемая в процессе перегонки в вакууме. Растворяется в воде (237,7 г в 100 г при 0° С). Образует гидраты с 9, 6, 4 и 2 молекулами воды.
Дииодид кобальта получают взаимодействием кобальта с иодидом водорода при 400—500° С. Применяют в основном в индикаторах влажности.
В водном растворе получены также хлориды, бромиды и иодиды кобальта (III), а в виде комплексных соединений — фториды и хлориды Со(Ш) и Co(IV).
10.5. КАРБОНАТЫ КОБАЛЬТА
Существуют два карбоната кобальта СоСОз и Со2(СОз)з, а также гидроксокарбонаты.
Карбонат кобальта (II) СоСОз — красные или розовые кристаллы с ромбоэдрической решеткой (для гексагональной установки: а_ = = 0,4675 нм, с = 1,5135 нм, z = 6; пространственная группа Юс); плотность 4,13 г/см3; С ° = 79,9 Дж/(моль-К); ДЯ^р = 730,5 кДж/моль; 5^8 = 87,9 Дж/(моль-К). При нагревании до температуры выше 350° С разлагается на оксид кобальта и диоксид углерода.
477
Практически не растворяется в воде (2,7-10“3%, масс.) и органических растворителях. Энергично реагирует с минеральными кислотами, образуя при этом соответствующие соли кобальта (II). В природе СоСОз — минерал сферокобальтит.
Карбонат кобальта (II) получают реакцией между гидрокарбонатом натрия на водный раствор сульфата кобальта (II) под давлением диоксида углерода
CoSO4 + 2NaHCO3 = СоСОз + Na2SO4 + Н2О + СО2 анодным растворением металлического кобальта в растворе гидрокарбоната натрия, разложением аммиачно-карбонатных комплексов кобальта при пропускании диоксида углерода.
Карбонат кобальта (II) является сырьем для получения других соединений кобальта, кобальтовых ферритов и катализаторов.
Известны также гекса- и тригидрат карбоната кобальта (II).
Карбонат кобальта (III) Со2(СО3)з — твердое оливково-зеленое нестойкое вещество. Получают окислением СоСОз или взаимодействием водного раствора карбоната аммония с трифторидом кобальта.
Существуют гидроксокарбонаты кобальта различного строения: Со2СО3(ОН)2 хН2О, Соз(СОз)2(ОН)2’5Н2О или Со5(СО3)2 (ОН)6 Н2О, образующиеся при действии карбоната натрия на водные растворы солей кобальта (II). Они представляют собой скрытокристаллические красно-фиолетовые вещества. Не растворяются в воде и органических растворителях. Разлагаются в кипящей воде, энергично реагируют с кислотами. В процессе нагревания обезвоживаются и выше 250° С разлагаются на карбонат и оксид кобальта (II).
Карбонаты и оксокарбонаты кобальта являются сырьем при получении оксида и солей кобальта, пигментов и катализаторов. Они применяются также в качестве компонентов шихты для цветной керамики, термочувствительных красок и микродобавок к кормам в животноводстве.
10.6. НИТРАТЫ КОБАЛЬТА
Нитрат кобальта (II) Со(1ЧОз)2 — розовые кристаллы с кубической решеткой (а = 0,741 нм); Д//Л = 421,5 кДж/моль. Растворяется в воде (г в 100 г): 83,94 (0° С), 100, 43 (25° С) и 184,82 (70° С). Из водных растворов в зависимости от концентрации и температуры образуются кристаллогидраты ди-, тетра- и гексагидраты. Растворяется также в ацетонитриле, ди мети л сульфокси де, этилацетате. В процессе нагревания водных растворов нитрата кобальта (II) при дегидратации образуются соответствующие гидраты с 1, 3 и 5 молекулами воды. При дальнейшем нагревании последних безводный нитрат кобальта 478
(II) не образуется, а происходит разложение с выделением азотной кислоты и образованием вначале гидроксосолей, а затем оксида кобальта (II). Нитрат кобальта (II) образует комплексы типа R2[Co(NO3)4], где R— органический радикал, щелочной металл.
Гексагидрат нитрата кобальта (II) — розовые кристаллы; выше 20° С устойчива a-форма с моноклинной решеткой (а = 0,142 нм, b = 0,614 нм, с = 1,266 нм, Р = 112,79° С; z = 4, пространственная группа Л/by температура плавления 55° С; плотность 1,89 г/см3; С° = 412,1 Дж/(молыК); АЯП°Л = 41,0 кДж/моль; ДЯ^р = 2208,6 кДж/моль; 52°98 = 473,9 Дж/(мольК). Хорошо растворяется в воде, ацетоне, спирте и других растворителях. Для тетрагидрата температура плавления 83,5° С, С? = 326 Дж/(моль-К), ДЯ0°бр = 1629,2 кДж/моль; для дигидрата АН^р = 1020,8 кДж/моль.
Нитрат кобальта (II) получают реакцией металлического кобальта или хлорида кобальта (II) со смесью N2O4 с этил ацетатом, а его гидраты — взаимодействием гидроксида кобальта (II) или оксидов кобальта с разбавленной азотной кислотой.
Нитрат кобальта (И) и его гидраты применяют для получения оксидных катализаторов, синтеза других солей кобальта, в том числе №4[Со(1ЧОз)б] — реактива на калий-ион.
Нитрат кобальта (III) Со(МОз)з—кристаллы с триклинной решеткой (а = 0,567 нм, b = 0,744 нм, с = 0,833 нм, а = 103,7°, 0 = = 102,1°, у = 106,7°, z = 2, пространственная группа Р1); при 40° С сублимируется в вакууме (-1,3 Па). Реагирует с водой с выделением кислорода. Растворяется в тетрахлоруглероде и гексане. Сильный окислитель. Бурно реагирует с эфирами, кетонами, нитрилами.
Образует комплексы. Аквакомплексы кобальта (III) не стабильны, так как являются сильными окислителями:
[Со(ОН2)6]3+ + 1ё = [Со(ОН2)6]2+, Е°298 = 1,81 В.
Ионы [Со(ОН2)б]3+ окисляют даже воду.
Из катионных комплексов для кобальта (III) очень устойчивы многие аминокомплексы. Для гексаамин-иона [Со(МНз)6]3+ не наблюдается диссоциация В растворах. Он не реагирует с хлороводородной кислотой и медленно реагирует с сульфидом водорода и гидроксидом натрия. Среди кобальтатов (III) многочисленны устойчивые соединения типа М^[Со(СЫ)6] и Мз‘[Со(К02)б].
Взаимодействие производных кобальта (II) с KCN в присутствии окислителя приводит к образованию гексацианокобальтата (III) калия Кз[Со(С1Ч)б]. Выделена также сильная трехосновная кислота H3[Co(CN)6].
479
Кроме катионных и анионных комплексов для кобальта (III) известны многие нейтральные комплексы, которые можно рассматривать в качестве полуфабрикатов в процессе перехода от катионных к анионным комплексам, например при переходе от [Co(NH3)6]3+ к [Co(NO2)6]3-: [Co(NH3)6]CI3, [Co(NH3)5(NO2)]C12, [Co(NH3)4(NO2)2]CI, [Co(NH3)3(NO2)3], K[Co(NH3)2(NO2)4], K2[Co(NH3)(NO2)5], K3[Co(NO2)6].
Нитрат кобальта (III) получают взаимодействием трифторида кобальта с N2O5.
10.7. СУЛЬФАТЫ КОБАЛЬТА
Сульфат кобальта (II) CoSO4 существует в трех кристаллических модификациях: a-форма сиренево-розового цвета, решетка ромбическая (а = 0,671 нм, b = 0,845 нм, с = 0,465 нм, z = 4, пространственная группа Рпта), плотность 3,71 г/см3; Р-форма— сиреневого цвета, решетка ромбическая, температура перехода а—>Р 440° С, Д/7° перехода 6,7 кДж/моль; моноклинная модификация устойчива при высоких давлениях. Для a-CoSO4: С° = 103,2 Дж/(моль-К);
= -889 кДж/моль; 5298 = 117 Дж/(моль-К). В процессе нагревания выше 700° С разлагается до оксида кобальта (II) с выделением кислорода и диоксида серы. Очень гигроскопичен.
Растворяется в воде (27,2% (масс.) при 25° С, 38,2% при 70° С), метаноле (0,418%), этаноле (0,017%), хорошо растворяется также в глицерине. Из водных растворов кристаллизуются гидраты с 7, 6, 4, 2 и 1 молекулами воды.
Гептагидрат (кобальтовый купорос, минерал биберит) CoSO4-7H2O — красные кристаллы с моноклинной решеткой (а = 1,545 нм, b = 1,308 нм, с = 2,004 нм, Р = 104,67°, z = 16, пространственная группа Р2|/с); плотность 1,95 г/см3; С° = 390,5 Дж/(моль-К); ДН0°бр = = 2982 кДж/моль; 5298 = 406,1 Дж/(моль-К). Известны также две мета-стабильные модификации гептагидрата — ромбическая и моноклинная. В процессе нагревания до 43° С гептагидрат переходит в гексагидрат (минерал мурхаузит) CoSO4-6H2O— оранжево-красные кристаллы с моноклинной решеткой; плотность 2,029 г/см3; Ср° = 353,6 Дж/(моль-К); ДНо°бр = -2677 кДж/моль; 5298 = 367,8 Дж/(моль К). В процессе термообработки при 71° С гексагидрат переходит в моногидрат (решетка моноклинная, плотность 3,075 г/см3), который при 250° С обезвоживается полностью. Тетра- и дигидраты метастабильны
Сульфат кобальта (И) в присутствии серной кислоты образует комплексные анионы и, в частности, [Co(SO4)2]2‘. Известны также двойные соли с сульфатами щелочных металлов у аммония M2Co(SO4)2-6H2O 480
типа шенитов и др. В процессе взаимодействия водных растворов сульфата кобальта (II) с основаниями образуются различные гидроксосу-льфаты, например Со5(8О4)2(ОН)б-5Н2О или Co4SO4(OH)6-4H2O.
Сульфат кобальта (II) получают взаимодействием оксида или гидроксида кобальта (II) с серной кислотой по схеме
Со + H2SO4 = COSO4 + Н2
СоО + H2SO4 = СоЗОд + Н2О
Водные растворы сульфата кобальта (II) применяют в процессе гидрометаллургической переработки кобальтсодержащего сырья с целью извлечения кобальта. Он является сырьем для получения других соединений кобальта, компонентом электролитов в процессе кобаль-тирования пигментов для стекла и керамики, осушителем.
Сульфат кобальта (III) 002(804)3-18Н2О — голубовато-зеленые или синие кристаллы, разлагающиеся выше 30° С, а в водном растворе — уже при 0° С. Более устойчив в водных растворах серной кислоты.
Сульфат кобальта (III) — сильный окислитель: вытесняет хлор из хлорида водорода, иод из иодида калия, окисляет этиловый спирт и муравьиную кислоту. С сульфатами калия, рубидия, цезия и аммония получены кобальтовые квасцы МеСофОдЬ’ПНгО:—не устойчивые в воде темно-синие кристаллы.
Сульфат кобальта (III) получают анодным окислением или озонированием водного раствора сульфата кобальта (II) в серной кислоте.
Сульфат кобальта (III) применяют в качестве титриметрического реагента при определении нитритов и др.
10.8. КОБАЛЬТОВЫЕ ПИГМЕНТЫ
Все соединения кобальта (II) окрашены. Ионы кобальта относятся к числу ионов, меняющих свое координационное число в зависимости от природы химического соединения. Установлено, что ионы кобальта (И) координируются с 4 или 6 ионами кислорода и в соответствии с этим окрашивают кристаллы и растворы в синий, сине-зеленый, зеленый или фиолетовый цвет.
Соединения кобальта (II), применяемые в качестве пигментов, относятся к соединениям следующих типов: 1) шпинели строения АХ2О4 и AZO4, где А—Со (часто с примесью цинка и магния), X—А1 и Cr, Z—Sn и Ti; 2) смешанные оксиды СоОхМеО; 3) смешанные силикаты кобальт-цинка; 4) соли кобальта (фосфаты и арсенаты кобальта).
481
10.9. АЛЮМИНАТ КОБАЛЬТА
Физико-химические свойства алюмината кобальта. Алюминат кобальта СоАЬОд — синие кристаллы со структурой типа шпинели (а = 0,809 нм, z = 8, пространственная группа Fd3m); плотность 4,44 г/см3; температура плавления I9600 С. Не растворяется в воде и водных растворах щелочей, разлагается в горячей хлороводородной и концентрированной серной кислотах.
Алюминат кобальта применяется в качестве пигмента (синь Тена-ра) для художественных красок, пластмасс, стекла и керамики.
Синие и сине-зеленые кобальтовые пигменты образуются в температурном интервале 1100—1300° С, что соответствует правилу Таммана, согласно которому обмен катионами в твердом теле происходит с измеряемой скоростью лишь после достижения температуры, составляющей 2/3 температуры плавления. Процесс образования шпинелей происходит на поверхности оксидов путем диффузии участвующих в нем ионов в противоположных направлениях. Скорость и полнота процесса зависят от температуры. Однако очень высокая температура приводит к спеканию частиц и ухудшению пигментных свойств, в связи с чем используются другие пути ускорения процесса. Значительную роль играет активное состояние исходных оксидов. Так, процесс образования алюмината кобальта — СоАЬОд — из оксидов ускоряется при температуре перехода у = А12Оз -> а-А12Оз (-1000° С), что может быть объяснено значительно большей подвижностью и реакционноспособностью А12Оз в момент переориентировки составляющих его частиц. Такая же закономерность наблюдается при замене СоО на Со3О4. Это объясняется образованием в процессе реакции СоО в более активном состоянии. Предварительный нагрев исходных оксидов при повышенной температуре резко замедляет реакцию. На рис. 10.1 приведены данные по степени влияния различных факторов на скорость процесса образования алюмината кобальта.
Хорошие результаты получают из-за высокой дисперсности исходных видов сырья и их тщательного перемешивания. Такие же результаты получают и в процессе термообработки осажденного карбоната кобальта (П) и гидроксида алюминия, а также в процессе термообработки смеси сульфата кобальта с алюмоаммониевыми квасцами по схеме
СоСОз + 2А1(ОН)з = СоА12О4 + СО2 + ЗН2О
COSO4 + 2NH4A1(SO4)212H2O = COAI2O4 + 2NH3 + 5SO2 + 13Н2О
В процессе термообработки исходные виды сырья плавятся в своей кристаллизационной воде, а при термообработке обезвоженных 482
солей в реакцию вступают оксиды в момент диссоциации и их образования, при котором их активность наибольшая.
На полноту и скорость реакции положительное влияние оказывает участие в реакционной смеси избытка оксида алюминия, что связано с замещением в шпинели трех ионов кобальта (II) на два иона алюминия (III) и образованием в решетке дополнительных вакансий; катионы при этом частично расположены беспорядочно и способны диффундировать с большей скоростью.
Технология алюмшната кобальта. Сырьем для получения алюмината кобальта являются сесквиоксид, гидроксид, сульфат, алюмокалиевые и алюмоаммо-нийные квасцы и сесквиоксид, сульфат, гидроксокарбонат и нитрат кобальта. Основные стадии получения алюмината кобаль-
Рис. 10.1. Степень влияния различных факторов на скорость процесса образования шпинели из оксидов кобальта и алюминия:
1 — СоО+А12Од (предварительная термообработка исходных оксидов при 800° С в течение 4 ч) в атмосфере азота в течение 15 мин; 2 — СоЮд+АЬОз (800° С, 4 ч) в атмосфере азота 15 мин; 3 — CO3O4+AI2O3 (800° С, 4 ч и в атмосфере кислорода—15 мин); 4 — СоО+А12Оз (800° Сив атмосфере азота — 5 мин); 5 — СоО+А12Оз (1100° С, 4 ч) в атмосфере азота в течение 15 мин)
та — приготовление шихты и ее термообработка.
Исходную шихту получают: 1) механическим смешением исход-
ных реагентов с последующим тонким размолом смеси реагентов, обычно мокрым, с последующей сушкой; 2) расплавлением реагентов в кристаллизационной воде с последующим обезвоживанием; 3) совместным осаждением карбонатов и гидроксидов из растворов смеси реагентов в воде с последующими промывкой и сушкой.
При механическом смешении используют оксиды и гидроксиды технической квалификации, обладающие низкой реакционной способностью и дисперсностью. Поэтому все исходные компоненты подвергают длительному (10—20 ч) мокрому размолу в шаровой мельнице (соблюдая соотношение материал : шары : вода « 1:1:0,8). Далее смесь высушивают до состояния порошка или пасты и подвергают термообработке при 1300—1350° С в течение 3—5 ч. Термообработанная масса обычно получается спекшейся и ее подвергают длительному (20—24 ч) мокрому размолу в шаровых мельницах, после чего сушат, измельчают на дезинтеграторах или шаровых мельницах и фасуют.
Для получения шихты в виде расплава применяют соли алюминия, плавящиеся в кристаллизационной воде: и
483
NH4A1(SO4)212H2O. В расплаве растворяются все составные части шихты и образуется гомогенная смесь. При работе на этих видах сырья все его компоненты загружают в алюминиевый реактор с электрообогревом, нагревают до 300° С, перемешивают до образования гомогенной расплавленной массы и ее обезвоживания, затем шихту охлаждают, выгружают и измельчают на дробилке или рифленых вальцах и подвергают термообработке при 1200—1300° С в течение 2,5—3 ч. Далее массу также подвергают мокрому размолу в шаровых мельницах, промывают, сушат и дезинтегрируют.
Цинкат кобальта. Физико-химические свойства цинката кобальта. Цинкат кобальта (зеленый кобальт) по своему химическому составу представляет собой твердый раствор оксида кобальта (II) в оксиде цинка CoOxZnO.
Цинкат кобальта — зеленые кристаллы с гексагональной решеткой. Плотность 5,460 г/см3, обладает значительной свето-, атмосферо-и термостойкостью. Легко разлагается в серной и хлороводородной кислотах. Цвет зеленого кобальта колеблется от светло- до темно-зеленого и зависит от соотношения оксида кобальта (II) и оксида цинка. Чем больше в пигменте содержится оксида кобальта (II), тем цвет его темнее. Промышленность производит лишь несколько видов зеленого кобальта, например темный, приблизительного состава CoO!5ZnO и светлый — состава CoO*50ZnO.
Зеленый кобальт применяется для ответственных и термостойких покрытий, окрашивания пластмасс и изготовления художественных красок.
Технология цинката кобальта. Сырьем для производства цинката кобальта (зеленого кобальта) служат цинковые белила (оксид цинка с примесями), гептагидрат сульфата цинка и оксиды, сульфат, нитрат, карбонат кобальта и др.
Оксид кобальта (II) и оксид цинка кристаллизуются в различных кристаллических системах: СоО — в кубической, a Zn — в гексагональной. Поэтому они могут образовывать твердые растворы лишь в особых условиях.
Оксид кобальта (II) в процессе нагревания претерпевает ряд сложных превращений. В процессе разложения нитрат кобальта (II) переходит в сесквиоксид, который при 705° С переходит в смешанный оксид СоОСо2Оз (СО3О4). В температурном интервале 869—1008° С СО3О4 полностью диссоциирует с образованием оксида кобальта (II).
При совместной термообработке смеси оксида кобальта (II) и оксида цинка с достижением температуры 800° С исходный оксид кобальта (II) переходит в сесквиоксид, после чего реагирует с оксидом цинка, образуя кобальтат цинка ZnOCo2O3 (ZnCo2O4) — продукт черного цвета с кубической решеткой типа шпинели. Образующаяся смесь из 60% СоО и 40% ZnO после термообработки полностью со-484
стоит из соединения ZnCo2O4. При меньшем содержании оксида кобальта образуются продукты темно-зеленого цвета — смесь ZnCo2O4 с оксидом цинка.
В процессе термообработки до 900—1000° С соединение ZnCo2O4 диссоциирует на СоО и ZnO. При этом в зависимости от соотношения количеств оксидов кобальта и цинка получаются соединения различного типа. При содержании оксида кобальта (II) до 30% образуется твердый раствор оксида кобальта (11) в оксиде цинка малахитово-зеленого цвета, кристаллизующийся в гексагональной системе, характерной для оксида цинка. При содержании оксида кобальта (II) более 70% получается розовый твердый раствор оксида цинка в оксиде кобальта (II), кристаллизующийся в кубической системе, характерной для оксида кобальта (II). При содержании оксида кобальта (II) между 30 и 70% получается смесь соединений, кристаллизующихся в различных системах.
В качестве зеленого пигмента применяют твердые растворы оксида кобальта (II) в оксиде цинка, кристаллизующиеся в гексагональной системе, поскольку лишь эти продукты обладают насыщенным зеленым цветом. Содержание оксида кобальта (II) в соединениях, применяемых в качестве пигментов, значительно ниже 30% и обычно не превышают 12—15%.
Зеленый кобальт получают в производстве термообработкой при 1000—1100° С смеси диссоциирующих солей кобальта-сульфата, карбоната и других солей с различными количествами цинковых белил.
Шихту приготавливают смешением цинковых белил с сульфатом кобальта (II). Наилучшие результаты получаются при мокром смешении, при этом образуются основные соли типа Zn(OH)2CoSO4. Для этого к цинковым белилам, размешанным в небольшом количестве воды, добавляют раствор сульфата кобальта (II), содержащий 250 г/л CoSO4’7H2O, и перемешивают в двухлопастном смесителе в течение 3—4 ч. Раствор сульфата кобальта приливают к суспензии цинковых белил медленно, поскольку в противном случае из-за слишком быстрого образования основных солей получаются сцементированные кусочки и крупинки.
После перемешивания до отсутствия комков образовавшуюся густую суспензию выгружают и сушат при любой температуре, но не выше 800° С. После сушки твердые куски шихты измельчают на дробилке или вальцах и направляют на термообработку.
Термообработку полуфабрикатов проводят в печах при 1000—1100° С. При термообработке сульфат кобальта (II) разлагается и в результате взаимодействия оксида кобальта (II) с оксидом цинка образуется зеленый пигмент.
После термообработки полученную массу размельчают в шаровых мельницах и подвергают тонкому размолу с сепарацией.
485
Фосфаты кобальта. Фосфаты кобальта известны в качестве двух типов пигментов темно-фиолетового кобальта (фосфата кобальта) и светло-фиолетового кобальта (фосфат кобальт-аммония).
Фосфат кобальта Со3(РО4)2 — фиолетовые кристаллы с моноклинной решеткой; плотность 2,584 г/см3; температура плавления 1160° С, не растворяется в воде, растворяется в минеральных кислотах, разлагается в водных растворах щелочей; образует октагидрат красного цвета. В фосфатах координационное число кобальта соответствует 6.
Светло-фиолетовый кобальт представляет собой моногидрат фосфата кобальт-аммония состава CoNH4PO4-H2O. Пигмент отличается лессирующей способностью, ограниченно стоек к действию света и весьма чувствителен к нагреванию (заметно изменяет свой цвет уже при 100° С), растворяется в кислотах, разлагается щелочами.
Фосфат кобальта и моногидрат фосфата кобальта-аммония являются пигментами для масляных художественных красок (кобальт темно-фиолетовый и кобальт светло-фиолетовый) и пластмасс.
Получение фосфата кобальта. Темно-фиолетовый кобальт получают термообработкой при 900—1100° С октагидрата фосфата кобальта, образующегося в процессе взаимодействия сульфата кобальта (II) с фосфатом натрия по схеме
3CoSO4 + 2Na3PO4 + 8Н2О = Со3(РО4)2-8Н2О + 3Na2SO4
Со3(РО4)2-8Н2О = Со3(РО4)2 + 8Н2О
При термообработке октагидрата фосфата кобальта (II) при температуре 130—225° С удаляется кристаллизационная вода (рис. 10.2). Содержание Р2О5 в продукте увеличивается с 27,38 до 38,6%, а масса образца уменьшается на 28,3%. При этом безводный ортофосфат кобальта переходит в аморфное состояние (его коэффициент преломления 1,535). В интервале температур 495—555° С аморфный фосфат переходит в кристаллическое состояние. Этот переход фиксируется на термограмме в виде экзотермического эффекта, а на ИК-спект-ре — расщеплением основной полосы, характерной для фосфатов (900—1200 см"1), на ряд менее интенсивных полос (рис. 10.3). При 1145—1155° С наблюдается экзотермический эффект, показывающий процесс плавления Со3(РО4)2.
В некоторых производствах темно-фиолетового кобальта в качестве сырья применяют гидрофосфат натрия, так как в этом случае после термообработки образуется пигмент более чистого фиолетового цвета:
3CoSO4 + 4Na2HPO4 + 8Н2О = Со3(РО4)2-8Н2О + 2NaH2PO4 + 3Na2SO4
Осаждение октагидрата фосфата кобальта в производстве проводят при 50—60° С, смешивая растворы сульфата кобальта и гидро-486
Рис. 10.2. Термограмма октагидрата Рис. 10.3. ИК-спектр фосфата кобальта:
фосфата кобальта / — аморфный продукт; 2 — кристаллический
продукт
фосфата натрия (200—250 г/л). На одну масс, часть гептагидрата сульфата кобальта (II) расходуется 1,7 масс, часть Na2HPO4*12H2O. Лучшие результаты достигаются в процессе добавления раствора сульфата кобальта к растворам фосфата натрия. При этом вначале образуется осадок сиреневого цвета, который при перемешивании переходит в легко осаждающийся кристаллический розовый осадок, который промывают декантацией или на фильтре, сушат, размалывают и подвергают термообработке при 800—900° С.
После термообработки целевой продукт размалывают, промывают горячей водой на фильтрах или центрифугах, сушат и подвергают тонкому размолу с сепарацией.
Получение мюногидрата фосфата кобальт-аммюния. Моногидрат фосфата кобальт-аммония получают взаимодействием сульфата кобальта (II) с фосфатом аммония или смесью двузамещенного фосфата аммония и аммиака:
CoSO4 + (NH4)2HPO4 + NH3 + Н2О = CoNH4PO4H2O + (NH4)2SO4
Технологический процесс получения светло-фиолетового кобальта сводится к приливанию концентрированного раствора сульфата кобальта, содержащего 250 г/л CoSO4-7H2O, к концентрированному раствору фосфата аммония при 40—45° С.
Реакция среды в процессе осаждения — слабощелочная. Оптимальная температура 40—50° С. При 20° С осадок имеет бледно-розовый цвет, а при 60° С принимает интенсивный фиолетовый оттенок.
Полученный осадок промывают водой при температуре 50° С декантацией или на фильтре, а затем пигмент высушивают при 40—50° С.
487
ГЛАВА 11
НИКЕЛЬ И ЕГО СОЕДИНЕНИЯ
11.1. НИКЕЛЬ
Содержание никеля в земной коре составляет 8-10’3% (масс.), а в воде океанов — 0,002 мг/л. В свободном состоянии никель в природе не встречается, а содержится в различных минералах.
Известно более 50 минералов никеля. Важнейшими из них являются: миллерит NiS, никелин NiS, хлоантит NiAs2, герсдорфит NiAsS, ульманит NiAsSb, пентландит (Fe,Ni)9S8, гарниерит (Ni,Mg)3Si4Oio(OH)io,4H20, ревдинскит (непуит) (Ni,Mg)3Si2O5(OH)4, аннабергит ^(AsO^ SbhO. В основном никель добывают из сульфидных медно-никелевых и из силикатно-окисленных руд. Мировые запасы никеля на суше исчисляются в 70 млн.т.
Никель может существовать в виде стабильных изотопов. Так, природный никель состоит из пяти изотопов: 58Ni (67,88%), 60Ni (26,23%), 61 Ni (1,19%), 62Ni (3,66%) и 64Ni (1,04%).
Свойства. Атом никеля имеет электронную конфигурацию 3s2 Зр6 3d* 4s2; степени окисления +2, редко +1, +3 и +4; энергия ионизации № —> Ni+ -> Ni2+ -> Ni3+ 7,634, 18,153 и 35,17 эВ; электроотрицательность по Полингу 1,80; атомный радиус 0,124 нм, ионный радиус (в скобках указаны координационные числа) Ni2+ 0,069 нм (4), 0,077 нм (5), 0,083 нм (6).
Никель — серебристо-белый металл. Кристаллическая решетка гранецентрированная кубическая (а = 0,35238 нм, z = 4, пространственная группа Fm3rn). Температура плавления 1455° С, а кипения 2900° С; плотность 8,90 г/см3; Ср° = 26,1 Дж/(моль К); АЯП°Л = 17,5 кДж/моль; ДЯ°СП = 370 кДж/моль; 52°98 = 29,9 Дж/(моль-К); уравнение температурной зависимости давления пара для твердого никеля Igp (гПа) = 13,369—23 013/Т + 0,5201gT + 0,395Т (298—1728 К), для жидкого Igp (гПа) = 11,742—20 830/Т + 0,6181gT (1728—3170 К); ферромагнетик, точка Кюри 631 К, твердость по Бринеллю (отожженного) 700—1000 МПа. Чистый никель — пластичный материал, легко обрабатывается как в холодном, так и горячем состоянии, поддается прокатке, волочению и ковке. 488
Никель, химически малоактивный элемент. Однако тонкодисперсный порошок, полученный восстановлением соединений никеля водородом при низких температурах, пирофорен.
При хранении на воздухе никель покрывается тонкой защитной пленкой оксида. Не взаимодействует ни с водой, ни с влагой воздуха. При нагревании до 800° С начинается процесс его оксидирования. С серной, фосфорной, хлороводородной и фтороводородной кислотами никель реагирует очень медленно. На него не действуют органические кислоты, особенно в отсутствие воздуха. Никель хорошо реагирует с разбавленной азотной кислотой, а с концентрированной пассивируется. Никель не реагирует с растворами и расплавами щелочей и карбонатов щелочных металлов, не реагирует также с жидким аммиаком. Водные же растворы аммиака корродируют никель.
Никель в высокодисперсном состоянии является активным катализатором в процессах синтеза в органической химии. В этих целях применяют скелетный никель (никель Ренея), получаемый сплавлением с алюминием или кремнием с последующим выщелачиванием щелочью, или никель на носителе.
Никель поглощает водород и образует с ним твердые растворы, а с азотом не реагирует вплоть до 1400° С, не реагирует также с аммиаком, а высокодисперсный никель при 300—450° С образует с ним нитрид Ni3N.
Никель в расплаве растворяет углерод, образуя карбид Ni3C, который при кристаллизации расплава разлагается с выделением графита. При сплавлении с кремнием образует силициды Ni5Si2, Ni2Si, NiSi, Ni5Si2, Ni3Si, Ni2Si, NiSi и др., с бором образует бориды с различными соотношениями составляющих, с селеном — селениды NijSey, с теллуром — теллуриды (Ni/ГеД а с парами фосфора — фосфиды NijrPy. Синтезированы также арсениды никеля.
Никель в процессе реакции с фосфорной кислотой образует фосфат Ni3(PO4)2, а также пирофосфат Ni2P2O7. Получены также арсенаты (V) [Ni3(AsO4)2-8H2O], силикаты (Ni2SiO4) и алюминаты — NiAl2O4 (никелевая шпинель).
Важнейшими комплексными соединениями никеля являются аммины. Наиболее характерны гексааммины и акватетраммины с катионами соответственно [Ni(NH3)6]2+ и [Ni(H2O)6]2+. Кроме них возможны смешанные акваамминокомплексы [Ni(H2O)6-n(NH3)/7]2+, где и=1—6 (рис. 11.1). Они образуются в растворах при аммиачной переработке никелевых руд.
При растворении Ni(CN)2 в растворах цианидов щелочных металлов образуются цианоникелаты K2[Ni(CN)4]n K4[Ni(CN)6], а в процессе восстановления K2[Ni(CH)4] в аммиачных растворах могут быть получены комплексы Ni(O) и Ni(l), например K4[Ni(CN)4], K3[Ni(CN)4], K4[Ni2(CN)6], легко окисляющиеся на воздухе. Дицианиды никеля 489
х 14
<ь
ti О Ф
8-
6-
4 -2-
200 400 600 800 1000 1200
к, нм
11.1. Электронные спектры поглощения ионов vnu \ 12+ (гх!;/к1и_\.12+ /о\ гы;/лм\_12+
Рис. 1
[Ni(OH,)6]2+ (/), [Ni(NH,)6l2+ (2), [Ni(en)3]2+ (3)
з Е
1
Ni(CN)2 являются полимерами сетчатого строения с квадратной структурной единицей.
В комплексах Ni(III) и Ni(IV) координационное число никеля равно 6, например Кз[ЬйР6], K2[NiF6], которые получаются при действии фтора на смеси хлоридов никеля и калия; сильные окислители. Из других типов известны
соли гетерополикислот, например (NH4)6H7[Ni(MoO4)6]-5H2O.
Получение. Никельсодержащие руды перерабатывают пиро- и гидрометаллургическим способами. При переработке силикатно-окисленных руд (не поддаются обогащению) применяют восстановительную плавку с получением ферроникеля, который далее подвергают продувке в конвертере с целью рафинирования и обогащения или плавку на штейн с серосодержащими добавками (FeS2 или CaSO4). С целью удаления железа штейн продувают в конвертере, а затем подвергают дроблению и термообработке. Из образовавшегося оксида никеля восстановительной плавкой получают металлический никель.
Никелевые концентраты, получаемые при обогащении сульфидных руд, плавят на штейн с последующей продувкой в конвертере. Из медно-никелевого штейна после его медленного охлаждения флотацией выделяют концентрат NiaS2, который аналогично штейнам из окисленных руд обжигают и восстанавливают.
Процесс гидропереработки сульфидных никелевых материалов (концентратов, штейнов) заключается в автоклавном окислительном выщелачивании либо водными растворами аммиака (при низком содержании кобальта), либо серной кислотой. Из аммиачных растворов после осаждения и разделения сульфида меди никель осаждают водородом под давлением. Для разделения никеля, кобальта и меди из аммиачных растворов применяют также экстракционные способы с использованием хелатообразующих экстрагентов.
Автоклавный процесс окислительного выщелачивания с получением сульфатных растворов применяют как к обогащенным материалам (штейнам) с переводом никеля и других металлов в раствор, так и к бедным пирротиновым Fe7Sg концентратам. В последнем случае окисляется преимущественно пирротин, что позволяет выделить элементную серу и сульфидный концентрат, переплавляемый далее на никелевый штейн. 490
Процесс рафинирования обычно проводят электролизом в сульфатных и сульфатно-хлоридных растворах с очисткой электролита от меди (цементацией на активном никелевом порошке), железа и кобальта (окислением и осаждением гидроксокарбонатом никеля), цинка (ионным обменом) и т.п.
Чистый никель получают также (в виде порошка или дроби) термическим разложением тетракарбонила никеля по схеме
Ni(CO)4 —*-> Ni + 4СО
Применение. Основная масса производимого никеля является компонентом легированных сталей, жаростойких, сверхтвердых, магнитных, коррозионно-стойких сплавов. Металлический никель — конструкционный материал для химической аппаратуры и ядерных реакторов, для аккумуляторных электродов, материал покрытий на стали, чугуне, алюминии и других металлах.
Никель является микроэлементом для млекопитающих и растений. В организме взрослого человека содержится 5—13,5 мг никеля.
11.2. ОКСИДЫ И ГИДРОКСИДЫ НИКЕЛЯ
Из оксидов и гидроксидов известны монооксид (а- и Р-формы), гидроксид Ni(OH)2, гидроксооксид P-NiO(OH) и смешанный гидро-ксидооксид состава ЬНзО2(ОН)4.
Монооксид (минерал бунзенит) — нестехиометрическое кристаллическое соединение NiOr, где х~1. В зависимости от способа получения и состояния (условий) оксида цвет может меняться от светло- до темно-зеленого и далее черного; до 252°С устойчив a-NiO, кристаллы гексагональной сингонии (а = 0,295 нм, с = 0,724 нм); С° = 44,3 Дж/(моль-К); АЯ^р = 239,7 кДж/моль; 52°98 = 38,0 Дж/(моль*К). Выше 252° С устойчив P-NiO, кристаллы кубической сингонии; параметры элементарной ячейки (а = 0,417 нм); число формульных единиц в ячейке — 2; пространственная группа Fm3m\ плотность 7,45 г/см3. Температура плавления 1682° С, однако уже выше 1230° С начинается диссоциация NiO на элементный никель и кислород. Обладает слабоосновными свойствами; в воде не растворяется. Водородом, углеродом, металлическим магнием и алюминием восстанавливается до элементного никеля. С оксидами других металлов образует оксиды, например типа шпинели BaNiOj.
Монооксид никеля получают термообработкой соли двухвалентного никеля или его гидроксида при 1000—1100° С или оксидированием металлического никеля при нагревании на воздухе или в среде кислорода.
491
Монооксид никеля применяют для получения солей двухвалентного никеля и никельсодержащих катализаторов и ферритовых порошков, в качестве пигмента для стекла, глазурей и керамики.
Из водных растворов солей Ni(II) при действии растворимых в воде металлов осаждают гидроксид никеля:
Ni(NO3)2 + 2NaOH = Ni(OH)2 + 2NaNO3
Гидроксид никеля Ni(OH)2 осаждается в виде объемного зеленого геля, образующегося при стоянии кристаллической гексагональной сингонии; параметры элементарной ячейки (а = 0,312 нм, с = 0,450 нм); число формульных единиц в ячейке — 4, пространственная группа C3w; плотность 3,65 г/см3; = 80,0 Дж/(моль-К); ДЯ^р = = 543,85 кДж/моль (для свежеосажденного ДЯ^р = 531,7 кДж/моль, ДС?0°5р = 477,32 кДж/моль). Произведение растворимости в водном растворе 2-Ю’16; легко растворим в аммиаке с образованием амминокомплексов; неамфотерен; при 230—250° С разлагается до оксида никеля. Применение гидроксида никеля в щелочных железо-никелевых или кадмий-никелевых аккумуляторах основано на электрохимическом окислении Ni(OH)2 в щелочном растворе.
Существование высших оксидов никеля нельзя считать твердо установленным, хотя и имеются сведения о получении оксида никеля состава NiO| 33.2,0 окислением NiO, NiCl2, Ni(OH)2 кислородом при 200—400° С или солей Ni(II) бромом в щелочном растворе. Однако индивидуальность выделенных фаз как химических соединений определенного состава не доказана. Предполагают, что механизм образования подобных фаз сводится к диффузии атомов никеля из объема к поверхности и связыванию их кислородом в неупорядоченные фазы Nix (х>1,0). Имеются сведения о выделении гидроксида Ni(OH)3 при окислении Ni(OH)2 хлором или бромом в щелочном растворе. Однако индивидуальность Ni(OH)3 нельзя считать доказанной, поскольку выделенное соединение Ni(III) из щелочного раствора идентифицировались либо как двойной оксид MMiO2, либо как гидроксидооксид NiO(OH).
Гидроксид никеля (HI) NiO(OH) — черные кристаллы гексагональной сингонии; параметры элементарной ячейки: а - 0,304 нм, с = = 0,482 нм; пространственная группа СЗти; плотность 4,35 г/см3; ДЯ^р = 385,2 кДж/моль; существует в двух модификациях у и р. y-NiO(OH) получают окислением металлического никеля смесью пероксида с гидроксидом натрия, a P-NiO(OH)— обработкой щелочного раствора нитрата никеля бромом или электролизом этого раствора. 492
11.3. СУЛЬФАТ НИКЕЛЯ
Существует безводный сульфат никеля NiSO4, а также в виде кристаллогидратов — NiSO4-7H2O, a-(NiSC>4-6H2O), P-fNiSCVefyO) и одноводного NiSCVbhO (табл. 11.1). При температуре выше 700° С NiSC>4 разлагается по схеме
NiSO4 700^ -> NiO + SO3
давление диссоциации 7,5 кПа (760° С); гигроскопичен; растворимость (г в 100 г): в воде —21,4 (°C), 29,94 (30° С), 33,39 (50° С), 43,42 (99° С); в метаноле — 0,001 (15° С); в этаноле — 0,017 (15° С).
Таблица 11.1. Свойства сульфата никеля и его гидратов
Показатель NiSO4 NiSO4-7H2O a-(NiSO4-6H2O) 0-(NiSO4-6H2O) NiSO4H2O
Цвет Лимон- Изумруд- Голубова- Зеленый Светло-зе-
но-желтый но-зеленый то-зеленый леный
Сингония Ромбическая Ромбическая Тетрагональная Моноклинная Ромбическая
Параметры решет-
ки, нм: 0,51596 1,18 0,6779 0,984* 0,7398
а 0,78362 1,20 — 0,717 0,7591
b 0,63378 0,680 1,824 2,40 0,6831
с
Число формульных единиц в ячей- 4 4 4 8 4
ке
Пространственная группа Стет Р2(2 j2 />4,2,2 С2/с АНа
Плотность, г/см3 3,68 1,949 2,037 2,0 3,357**
С;;, Дж/(мольК) 97,8 364,8 328,7 343,4 171,60
кДж/моль -874,1 -2979,4 -2685,70 -2690,10 —
S"W4 Дж/(моль-К) 103,9 379,2 334,7 306,1 —
’Значения параметров даны для минерала; 0 = 97,5°.
’’Рентгеновская плотность.
Сульфат никеля образует гидраты, важнейшим из них является сульфат никеля NiSO4*7H2O, кристаллизующийся из водных растворов в интервале от -3,4 до 31,5°; температура плавления 31,5° (инконгруэнтно); показатели преломления: больший = 1,4925, средний Nm = 1,4893, меньший Np = 1,4693; растворимость в воде 101 г в 100 г (20° С); растворим в этаноле.
Из водных растворов при 31,5—53,6° С кристаллизуется а-форма гексагидрата NiSO4-6H2O, а при 53,6—84,8° С — его P-форма. Выше 84,8° С устойчив моногидрат NiSO4’H2O, который обезвоживается при 493
нагревании до 280° С. Гидраты с 2, 3, 4, 5 молекулами воды метастаби-льны, кристаллизуются из водных растворов при 90—98° С.
Сульфат никеля образует двойные соли где
М — К, Rb, Cs, NH4+, аммиакаты NiSO4*6NH3, NiSO4-4NH3-2H2O и др.
В системе NiSO4—NaOH—Н2О выделяются осадки основных сульфатов никеля, состав которых определяется концентрацией растворов. При добавлении 0,1н. раствора гидроксида натрия к раствору сульфата никеля с концентрацией, меньшей 0,1М, образуется осадок гидроксида никеля. В интервале концентраций 1 М сульфата никеля осаждается 3NiSO4-4Ni(OH)2, а при дальнейшем увеличении концентрации NiSO4 вначале выделяется 3NiSO4*2Ni(OH)2, постепенно переходящий в 3NiSO4-4Ni(OH)2. По другим данным, при приливании раствора NaOH к раствору сульфата никеля образуется осадок NiSO4*4Ni(OH)2, а при обратном порядке сливания растворов — NiSO4*5Ni(OH)2. Четырехосновный сульфат никеля неустойчив в воде и растворах гидроксида натрия, поэтому переходит в гидроксид никеля (II).
В системе NiSO4—H2SO4—Н2О образуются комплексные ионы [Ni(SO4)2]2-.
При подкислении аммиачного раствора сульфата никеля серной кислотой от pH 7,1—8 до pH 4 при 55—60° С образуется никельам-монийсульфат Ni(NH4)2(SO4)2. В присутствии же в этой системе кобальта в процессе снижения значения pH до 6,2 выделяется в осадок почти весь никель (практически свободный от кобальта) в виде Ni(NH4)2(SO4)2. При дальнейшем снижении pH оставшегося раствора до 4 в осадок выделяется оставшийся никель и практически весь кобальт в виде Ni(NH4)2(SO4)2 и Co(NH4)2(SO4)2.
В системе NiSO4—Co(SO4)2—Н2О образуются смешанные кристаллы.
В системе NiSO4—Na2SO4 образуются твердые растворы в широкой области со стороны сульфата натрия и три двойные соли: 3Na2SO4-NiSO4, Na2SO4 NiSO4 и Na2SO4-3NiSO4. Имеются две эвтектики: при 41% NiSO4 — 671° С и при 55% NiSO4 — 700° С.
Гидраты сульфата никеля встречаются в природе в виде минералов: моренозит NiSO4-7H2O, ретгерсит a-(NiSO4-6H2O) и никельгекса-гидрит P-(NiSO4’6H2O).
Никелевый купорос, а также смесь гепта- и гексагидратов применяют для получения чистого электролитического никеля, других соединений никеля и никельсодержащих катализаторов. Никелевый купорос, а также M'Ni(SO4)2 (где М — К, NH4) являются компонентами электролитов в гальванотехнике для нанесения покрытий на металлы. В смеси с некоторыми препаратами сульфат никеля применяют в качестве фунгицида.
Способы получения сульфата никеля. В производстве сульфат никеля выпускают двух марок: НС-0 — для получения никельсодер-494
жащих продуктов реактивной квалификации и НС-1—для процессов никелирования, аккумуляторной, жировой и парфюмерной промышленности. Согласно существующему ГОСТ 2665—73, сульфат никеля должен содержать не менее 20,6% никеля вместе с кобальтом, а при отсутствии последнего — не менее 20,3% никеля. Допустимые количества примесей указаны в табл. 11.2.
Таблица 11.2. Максимально допустимые количества примесей в техническом сульфате никеля (%)
Марка Со РЬ Zn Fe Си Са Mg Cl Нерастворимый остаток
НС-0 0,2 0,001 0,004 0,001 0,001 0,08 0,03 0,01 0,03
НС-1 — 0,001 — 0,002 0,002 0,2 0,03 0,1 0,05
Сульфат никеля получают взаимодействием элементного никеля, его оксида, гидроксида и гидроксокарбоната с серной кислотой
Ni + H2SO4 = NiSO4 + Н2
NiO + H2SO4 = NiSO4 + Н2О
Mi(OH)2 + H2SO4 = NiSO4 + 2H2O
Ni2CO3(OH)2 + 2H2SO4 = 2NiSO4 + 3H2O + CO2
Купорос получают растворением никеля в разбавленной серной кислоте, содержащей некоторое количество азотной кислоты, с последующей нейтрализацией гидроксокарбонатом никеля и упариванием образующихся растворов.
Гептагидрат NiSO4-7H2O получают также из маточных сульфатных растворов кобальтового производства и из растворов при рафинировании меди.
Получение сульфата никеля из маточных растворов кобальтового производства. Маточные растворы кобальтового производства содержат около 30 г/л никеля. Они образуются при растворении в серной кислоте зеленых гидратов, получаемых при электролитическом растворении никель-кобальтовых анодов или при растворении кобальтового шпур-штейна в атмосфере кислорода под давлением (в автоклаве).
Согласно технологической схеме (рис. 11.2), сульфатные растворы из сборника 1 дозируются дозатором 3 в реактор 4. При непрерывном перемешивании в реактор 4 дозируются водные растворы карбоната натрия из сборника 2:
NiSO4 + Na2CO3 = NiCO3 + Na2SO4
495
Рис. 11.2. Технологическая схема получения никелевого купороса из маточных растворов кобальтового производства:
/ — сборник маточного раствора кобальтового производства; 2 — сборник раствора карбоната натрия; 3 — дозатор; 4—реактор; 5—центробежный насос; 6 — отстойник Дорра; 7 — промежуточная емкость; 8 — репульпатор; 9, 12 — фильтр; 10—реактор; 11 — реактор-нейтрализатор; 13 — ва-куум-выпарной аппарат; 14 — вакуум-кристаллизатор; 15 — центрифуга
Реакционную смесь в реакторе 4 нагревают до. 80° С и перемешивают воздухом. Полученная суспензия перекачивается центробежным насосом 5 в отстойник 6, из которого после отстоя растворы через промежуточную емкость 7 перекачиваются в бассейн сточных вод, а суспензия карбоната никеля отжимается на первом барабанном вакуумном фильтре 9, откуда через репульпатор 8, где его смешивают с чистой водой, передается во второй барабанный вакуум-фильтр 9. Промытый осадок со второго вакуум-фильтра поступает в реактор 10, а промывные воды перекачиваются в бассейн сточных вод.
В реакторе 10 суспензия карбоната никеля при 80—85° С обрабатывается крепкой серной кислотой по схеме
NiCO3 + H2SO4 = NiSO4 + Н2О + СО2
Полученные растворы, содержащие 160 г/л никеля, очищают в реакторе 11 от примесей железа, меди и кобальта путем подачи влажного карбоната никеля. При этом образуются нерастворимые в воде карбонаты по схеме
FeSO4 + NiCO3 = FeCO3 + NiSO4
CuSO4 + NiCO3 = CuCO3 + NiSO4
CoSO4 + NiCO3 = CoCO3 + NiSO4
496
Растворы фильтруют от примесей через фильтр ФПАКМ 12 и направляют на вакуум-выпарной аппарат 13. Упаренные растворы перекачивают в вакуум-кристаллизаторы 14, откуда их направляют в непрерывно действующую центрифугу типа НГП 75. Отжатые и промытые кристаллы никелевого купороса ЬПЗОд-ТЬЬО с влажностью 3—5% укупоривают или предварительно брикетируют на гидравлических прессах в блоки. Часть маточных растворов, содержащих 120—140 г/л никеля, добавляют к раствору, получаемому после растворения карбоната никеля в серной кислоте в реактор 10, другую часть присоединяют к исходному раствору (в реактор 7), поступающему из кобальтового производства.
Получение сульфата никеля из растворов медно-электролитных производств. В процессе электролитического растворения медных анодов и осаждения меди на катоде электролитом служит раствор медного купороса, содержащий 30—45 г/л меди. В электролите кроме меди содержатся примеси — сульфаты никеля, железа, цинка, кальция и сесквиоксид мышьяка. При рафинировании меди некоторые из этих примесей практически полностью переходят в раствор, другие — в шлам, а остальные — частично в шлам, частично в раствор. Полностью в раствор переходят металлы более электроположительные, чем медь. К ним относятся никель, цинк и железо. Эти металлы не осаждаются на катоде и постепенно накапливаются в растворах, что приводит к снижению растворимости сульфата меди и к снижению эффективности процесса электролиза. Для уменьшения в электролите указанных примесей растворы периодически выводят из системы электролиза, а взамен добавляют к электролиту серную кислоту. Выведенные из системы растворы, содержащие никель, цинк и железо, регенерируют.
В процессе растворения 250 т анодов в сутки, содержащих 0,1% никеля, в раствор переходит 250 кг Ni, а суточный вывод из системы— 10 м3 раствора с содержанием в нем 25 г/л никеля или 25 м3 раствора с содержанием 10 г/л Ni. Согласно технологическому регламенту, раствор, подлежащий регенерации, содержит меди — 40, серной кислоты— 180, никеля — 15, железа — 3 и мышьяка -7 г/л. Растворы нейтрализуют медным скрапом в затравочной башне при продувке воздухом и после упарки кристаллизуют из них медный купорос. Полученный при этом маточный раствор упаривают с последующей кристаллизацией загрязненного медного купороса, который в дальнейшем очищают перекристаллизацией. Вторичный маточный раствор, содержащий около 50 г/л Си, 40 г/л H2SO4 и 50 г/л Ni, подвергают упарке и охлаждают для кристаллизации никелевого купороса. Полученный продукт отвечает требованиям действующего ГОСТ. Оставшийся маточный раствор подвергают вторичной упарке и кристаллизации. Полученный при этом загрязненный сульфат нике-
497
ля направляют на вторичную кристаллизацию, а конечный маточный раствор после упарки до содержания в нем 1200 г/л H2SO4 и очистки от примесей возвращают на процесс электролиза меди.
Получение сульфата никеля растворением никеля в серной кислоте основывается на реакции:
Ni + H2SO4 = NiSO4 + Н2
Реакцию ведут в присутствии азотной кислоты. В производстве на 1 кг исходного элементного никеля берут 5 кг 33—35%-ной серной кислоты и 120—150 г азотной кислоты (в пересчете на 100%). Процесс ведут в реакторах из нержавеющей стали при 60° С и постоянном перемешивании реакционной массы. Образующийся раствор сульфата никеля путем фильтрации очищают от нерастворивше-гося остатка и при необходимости очищают от примесей железа и меди, сопровождающих исходный никель. С этой целью в растворы вводят металлический никель в виде порошка, который вытесняет медь из раствора, а железо осаждают действием черного гидроксида никеля (основной карбонат никеля) в присутствии кислорода воздуха.
Растворы сульфата никеля могут быть очищены от примесей железа и меди ступенчатым осаждением их гидроксидов аммиаком с образованием осадка гидроксида железа при pH 3 и гидроксида меди при pH 4,5. Процесс осаждения рассматриваемых гидроксидов облегчается при введении перед каждой ступенью нейтрализации некоторого количества затравки соответствующего гидроксида (железа или меди).
Разработан также метод очистки сульфата никеля от примесей рассматриваемых металлов противоточной экстракцией, экстрагентом при этом служит Ni-мыло.
Очистка раствора сульфата никеля от ионов цинка может быть осуществлена анионообменным методом с применением смолы — во-фатита MD. Смолу предварительно обрабатывают сульфидом водорода для насыщения ионами S2-. При обработке сульфидом водорода под давлением 19 ат концентрация содержания серы достигает 0,58 г на 100 мл смолы. В результате ионного обмена при pH 3,2 протекает следующая реакция:
82-вофатит + Zn2+ + SO^" = ZnS + SO^" + вофатит
В этом процессе 80—90% выделяющегося сульфида цинка задерживается в свободном объеме ионообменной колонны, равном 60—70% от объема смолы. Остальное количество ZnS улавливается неполярным фильтром — вофатитом F.
Ионообменные смолы могут быть использованы и для очистки раствора сульфата никеля, полученного из отработанных катализа-498
торов. Для этого смолу предварительно обрабатывают щелочью. При пропускании через такую смолу кислого водного раствора, содержащего ионы железа и никеля, в результате ионного обмена pH раствор повышается до 3—5 и из раствора выделяется только железо. Наиболее эффективно Fe3+ отделяется при pH 4,5. Такое значение pH достигается при обработке щелочных смол раствором хлорида аммония.
11.4. ГАЛОГЕНИДЫ НИКЕЛЯ
К галогенидам никеля относятся его хлориды, фториды, бромиды и иодиды, а также двойные галогениды и их кристаллогидраты. В табл. 11.3 приведены свойства безводных галогенидов никеля.
Таблица 11.3. Свойства безводных галогенидов никеля
Показатель NiF2 NiCl2 NiBr2 Nil2
Сингония Тетрагональная Тригональная Тригональная Тригональная
Параметры решетки, нм: а с 0,46505 0,30836 0,354 1,732 0,3714 1,830 0,3895 1,963
Пространственная группа Pbmnm R3m R3m R3m
Гпл, °C 1452 1009 963 797
Гкип или Тюзп °C 1474 970 919 —
Плотность, г/см3 4,72 3,54 5,45 5,83
С", Дж/(моль К) 64,1 71,7 — —
Д7/(^, кДж/моль -658 -304 -214 -82,4
Дж/(моль-К) 73,6 98,1 129 158
ДН“п, кДж/моль — 77,3 — —
Д//,"Г1Г, кДж/моль 284 225 224,6 —
Растворимость в воде при 25° С,% (масс.) 3,86 39,7 56,7 59,7
Дифторид никеля NiF2— зеленовато-желтые кристаллы типа рутила (Т1О2); уравнение температурной зависимости давления пара: 1g р (мм рт. ст.) = 20,28—14 650/7" + 3,02 IgT (298—1220 К); при комнатной температуре устойчив на воздухе, при термообработке окисляется до оксида никеля:
NiF2 + О2 — NiO + F2
не гигроскопичен; растворимость в воде не зависит от температуры.
499
Дифторид никеля получают фторированием оксида, хлорида никеля или других соединений никеля по схеме
2NiO + 2F2 -> 2NiF2 + О2
NiCl2 + F2 -> 2NiF2 + Cl2
Образующиеся растворы очищают отстоем и фильтрацией от нерастворимых веществ и из водных растворов получают кристаллы NiF2-4H2O.
Тетрагидрат дифторида никеля NiF2-4H2O — светло-зеленые кристаллы плотностью 2,219 г/см3. В процессе нагревания теряет одну молекулу воды, полностью обезвоживается лишь в токе фторида водорода при 350—400° С.
Безводный дифторид никеля применяют в качестве катализатора фторирования, для изготовления катодов в химических источниках тока, в качестве компонента лазерных материалов.
Трифторид никеля NiFa образуется в виде розового осадка при электролизе расплава KHF2; сильный окислитель, разлагается водой, выделяя из нее кислород.
Известны производные тетрафторида никеля NiF4, в частности K2[NiF6]: розовые или темно-красные кристаллы кубической сингонии; разлагается во влажном воздухе; водой гидролизуется; получают фторированием смеси хлоридов калия и никеля; применяют в качестве фторирующего реагента.
Дихлорид NiCl2, дибромид NiBr2 и дииодид Nil2 кристаллизуются в ромбоэдрической решетке типа CdCl2: для NiBr2 известна также другая модификация со структурой типа Cdl2; при термообработке на воздухе окисляются до NiO; безводные соли могут быть получены действием соответствующих галогенов на элементный никель, а также обезвоживанием кристаллогидратов.
Дихлорид никеля — золотисто-желтые или желто-коричневые кристаллы; уравнение температурной зависимости давления пара: 1g р (мм рт. ст.) = 21,88—13 300/7" + 2,68 IgT" (298—1220 К); гигроскопичен; растворяется в воде, этаноле и гликоле. Из водных растворов в интервале от -53,4 до 28,8° С кристаллизуется гексагидрат, при 28,8—64,3° С — тетрагидрат, при 64,3—117,9° С — дигидрат.
Гексагидрат NiCI2-6H2O — кристаллы от светло- до темно-зеленого цвета моноклинной сингонии {а = 1,023 нм, b = 0,705 нм, с = = 0,657 нм, Р = 122,16°, пространственная группа С2т\9 плотность 1,91 г/см3; в сухом воздухе выветривается, а во влажном расплывается. Согласно результатам структурных исследований, в ряде гексагидратов NiX2-6H2O октаэдрическая координация атома никеля (II) со-500
здается за счет двух атомов X (С1,Вг) и четырех молекул воды [Ni(OH2)4X2]-2H2O. Оставшиеся две молекулы воды связывают между собой молекулы [Ni(OH2)4X2].
Тетрагидрат NiCI2-4H2O— кристаллы от желтовато-зеленого до темно-зеленого цвета тетрагональной сингонии {а = 0,662 нм, с = 1,323 нм, пространственная группа Р4тт)\ плотность 1,72 г/см3; в сухом воздухе выветривается.
Интересно строение NiCl2. Этот кристаллогидрат представляет собой гексахлороникелат (II) гексаакваникеля (II) [Ni(OH2)6]2 [NiCU].
Дигидрат NiCl2’2H2O — бледно-желтые или коричневато-желтые кристаллы моноклинной сингонии (а = 0,697 нм, b = 0,690 нм, с -= 0,881 нм, Р = 91,5°, пространственная группа 12/лтг): плотность 2,56 г/см3; при 140° полностью обезвоживается.
Кристаллы дигидратов хлоридов никеля и бромидов никеля имеют цепную структуру:
он2 он2 он2
I I ^С1 I ^С1
I СГ | xcr | xcr
OH2 OH2 OH2
В отличие от [Ni(OH2)6]2+ аминокомплекс [Ni(NH3)6]2+ легко образуется, например, при взаимодействии твердого хлорида никеля с аммиаком:
MiCl2 + 6NH3 = [Ni(NH3)6]Cl2
Кроме [Ni(OH2)6]2+ и [№(МНз)6]2+ возможны аквааминокомплексы [Ni(OH2)6.„(NH3)„]+2, где п = 1—6. Замена лигандов Н2О на лиганды NH3 приводит к изменению окраски комплексов от ярко-зеленого до синего цвета. Это объясняется увеличением параметра расщепления А (изменением энергии d—d-переходов), что приводит к сдвигу полос поглощения в сторону меньших длин волн. Еще больший сдвиг полос поглощения наблюдается у этилендиаминовых комплексов [Ы!(еп)з]2+.
Хорошая растворимость гидроксида никеля (II) в присутствии аммиака и солей аммония объясняется образованием аммиакатов:
Ni(OH)2 + 6 NH3 = [Ni(NH3)6](OH)2
501
Существует значительное число двойных хлоридов никеля (NiSnC16*6H2O, K2NiCl4-6H2O и др.). Важнейшим из них является NH4NiC13*6H2O, получаемый кристаллизацией из хлороводного раствора смеси NiCl2 с NH4C1 при ~20° С; голубовато-зеленые кристаллы; растворяется в воде; при 70° С обезвоживается, а выше 170° С разлагается.
Безводный NiBr2— желто-коричневого цвета, безводный Nil2 — стально-серый или черный; гигроскопичны, растворяются в воде, метаноле, этаноле; из водных растворов кристаллизуются в виде гексагидратов. NiBr2-6H2O— зеленые кристаллы; при 28,5° С теряет три молекулы воды, при -200° С обезвоживается. NiI2-6H2O — голубовато-зеленые кристаллы; при 49° С теряет две молекулы воды, а при 140° С обезвоживается.
NiCl2 и его гидраты применяют в качестве компонентов электролитов для рафинирования никеля и для никелирования, для получения никелевого порошка, в качестве катализатора хлорирования органических соединений, а безводный хлорид никеля — в качестве компонента термочувствительных красок.
Дибромид никеля применяют для получения катализаторов и термочувствительных красок.
11.5. ТЕТРАКАРБОНИЛ НИКЕЛЯ
Тетракарбонил никеля Ni(CO)4 (рис. 11.3) — бесцветная жидкость с температурой кипения 42,2° С, плавления—17,2° С. Растворяется в органических растворителях, но нерастворим в воде. При нагревании выше 36° С начинает разлагаться с образованием никеля:
Ni(CO)4 Ni + 4СО
Смеси тетракарбонила никеля с воздухом самовоспламеняются и самопроизвольно взрываются, если концентрация тетракарбонила выше 4% по объему. Медленно окисляется в воздухе. Энергично окисляется галогенами, азотной кислотой и царской водкой до соединений Ni(II). Восстанавливается, образуя карбонилникелат-анионы. Например, при взаимодействии тетракарбонила никеля со щелочными металлами или с их амальгамами в ТГФ или с метанольными растворами щелочей образуются смеси анионов [Ni5(CO)i2]2- и [Ni6(CO)i2]2-
В промышленности тетракарбонил никеля получают из никельсодержащего сырья и оксида углерода под давлением 20 МПа при 180—250° С. Получают также из никельсодержащего сырья, предварительно обработанного водяным газом, и СО без давления при 502
50—60° С. Сырьем служат губчатый и компактный никель, природные руды, сплавы Ni—Fe, никелевый штейн, анодный скрап. В лабораторных условиях тетракарбонил никеля синтезируют взаимодействием свежеприготовленного высокодисперсного никеля с оксидом углерода при 100° С или солей никеля (II) с
оксидом углерода в присутствии восстановителей или без них.
Очищают технический продукт дистилляцией или ректификацией.
Путем газофазной конденсации никеля с оксидом углерода при низких температурах в
лом никеля получен гептакарбонилдиникель — Ni2(CO)7.
Рис. 11.3. Строение молекулы тетракарбонила никеля Ni(CO)4
смеси с тетракарбони-
Тетракарбонил никеля применяют для получения никеля высокой
чистоты, для нанесения пленок никеля на металлы, керамику, пластмассы и др., в качестве катализатора в органическом синтезе и при получении полиформальдегида.
11.6. СУЛЬФИДЫ НИКЕЛЯ
Сульфиды никеля встречаются в природе в виде минералов: миллерита (желтый никелевый колчедан) у-NiS, хизлевудита Ni3S2, год-левскита Ni7S6, полидимита Ni3S4, ваэсита NiS2, цетландита Ni5-xFe4+xS8, где 0<х<0,5.
Наиболее изученными сульфидами никеля являются приведенные в табл. 11.4 сульфиды.
Моносульфид NiS — нестехиометрическое соединение, для него, как и для других сульфидов никеля, характерна определенная область гомогенности; существует в двух кристаллических модификациях у- и P-NiS; температура перехода у—>Р снижается от 389° С (при содержании 50% (ат.) серы) до 282° С (-50,2% (ат.) серы).
Сульфид Ni3S7 при 555° С переходит в высокотемпературную фазу Ni3±rS2 с широкой областью гомогенности. Сульфид Ni3O4 при 365° С, не плавясь, образует NiS2 и P-NiS. Синтезированы нестехиометрические соединения ЬПб85, которые существуют при 397—573° С, после чего образуют Ni3+YS2 и р-NiS. Сульфид Ni7O6 при 399° С также разлагается на Ni6Ss и P-NiS.
Все сульфиды никеля практически не растворимы в воде и органических растворителях. Разлагаются лишь азотной кислотой и царской водкой. При нагревании в вакууме выше 500° С диссоциируют с образованием паров серы и фаз, обогащенных никелем. При нагревании на воздухе окисляются.
503
Таблица 11.4. Свойства сульфидов никеля
Показатель y-NiS P-NiS NiS2 Ni3S2 Ni3S4 Ni6S5 Ni7S6
Цвет Латунножелтый Черный . Фиолетовосерый Латунножелтый Серебристо-серый Бронзовый Темно-желтый
Сингония Ромбоэдрическая Гексагональная Кубическая Ромбоэдрическая Кубическая Ромбическая Ромбическая
Параметры ячейки,
нм:
а 0,563 0,343 0,518 0,4080 0,946 1,122 0,918
b — — — — — 1,656 1,126
с — 0,534 — — — 0,327 0,946
угол, град 116,60 — — 89,41 — — —
Число формульных единиц в ячейке 3 2 4 1 8 4 —
Пространственная группа R3m РЬз!ттс РаЗ R32 Fd3m — —
Гпл. °C — 996 1007 808 356 573 399
Плотность, г/см3 5,4 5,6 4,45 5,82 4,78 — —
с;;, Дж/(мольК) 47,11 — — 117,73 — 258,6 —
ДЯ^, кДж/моль -94,1 — -134,0 -192,6 -326,4 -494,4 -502,4
Дж/(моль-К) 52,99 — 67,8 134,0 171,3 294 —
Область гомогенности, ат. %S 50,0—50,1 50,1—52 — 36,5—44 — 45,3—46 —
(282° С) (800° С) — (635° С) — (524° С) —
Сульфид никеля получают взаимодействием никеля с серой при нагревании в вакууме; NiS и NiS2— термообработкой смеси оксида никеля (или карбоната никеля) с серой и карбонатом калия. NiS получают также осаждением сульфидом водорода из водных растворов солей никеля в слабокислой среде. При этом вначале образуется черный осадок аморфного сульфида никеля (а-NiS), растворимого в разбавленной хлороводородной кислоте, который затем переходит в P-NiS, не растворимый в разбавленной хлороводородной кислоте.
Ni3S2 и Ni6S5 применяются в качестве компонента никелевого файнштейна — промежуточного продукта переработки на Ni силикат-но-никелевых и других руд.
Ni, NiS и Ni3S2 применяют в качестве катализаторов в процессах гидрогенизации и дегидрогенизации, a NiS2 является катализатором в процессах органического синтеза.
11.7. КАРБОНАТ НИКЕЛЯ
Карбонат никеля NiCO3— зеленые (плотность 4,39 г/см3) или желтые (плотность 4,36 г/см3) кристаллы с ромбоэдрической решеткой (а = 0,4597 нм, с = 1,4725 нм, z = 6, пространственная группа 7?3с), обе формы по структуре считаются идентичными, с; = 86,2 Дж/(моль-К); ДН0°бр = 694 кДж/моль; 52°98 = 86,2 Дж/(моль-К); ниже 22,2 К (точка Нееля) антиферромагнетик. При термообработке выше 300° С разлагается с выделением оксида никеля и диоксида углерода. Практически не растворяется в воде (9,3*10‘3% масс.), в водных растворах карбонатов и гидрокарбонатов щелочных металлов и в органических растворителях. Не разлагается в кипящей воде, устойчив к действию нагретых концентрированных азотной и хлороводородной кислот, царской водки. Очень медленно растворяется в концентрированном водном растворе аммиака.
Карбонат никеля получают взаимодействием водных растворов гидрокарбоната натрия (NaHCO3) с хлоридом никеля с выдержкой в автоклаве под давлением (12 МПа) диоксида углерода при 250° С (зеленая форма) или при -180° С (желтая форма), термообработкой суспензии гидрооксокарбоната никеля под давлением (от 5 до 30 МПа) СО2 при 100—450° С, с дальнейшим обезвоживанием гидратов карбоната никеля.
При этом процессы идут по реакциям:
2NaHCO3 + NiCl2 = NiCO3 + 2NaCl + H2O + CO2
Ni(CO3)^Ni(OH)2 + CO2 = 2NiCO3 + H2O
18 Химическая технология
505
Гексагидрат карбоната никеля NiCO3-6H2O (минерал хеллиэ-рит) — голубые или светло-зеленые кристаллы с триклинной решеткой; плотность 1,97 г/см3; неустойчив при хранении на воздухе.
Легко реагирует с разбавленными минеральными кислотами:
NiCO3-6H2O + 2НС1 = NiCl2 + СО2 + 7Н2О
NiCO3-6H2O + H2SO4 = MiSO4 + 7Н2О
При нагревании легко теряет воду и диоксид углерода:
NiCO3-6H2O —NiO + СО2 + 6Н2О
При -140° С превращается в гидроксокарбонат:
2NiCO3-6H2O —> NiCO3Ni(OH)2 + СО2 + 11Н2О
Гексагидрат карбоната никеля получают электролизом воды, насыщенной диоксидом углерода, с никелевыми электродами.
Существуют также NiCO3 H2O, NiCO3-2H2O и NiCO3-3H2O.
Гидрокарбонат Ni(HCO3)2 — светло-зеленые кристаллы с кубической решеткой (а = 0,8383 нм); плотность 2,9 г/см3; получают кратковременной обработкой гидроксокарбоната никеля водой и диоксидом углерода в автоклаве при 220—250° С по схеме
NiCO3-Ni(OH)2 + Н2О + ЗСО2 = 2Ni(HCO3)2 + Н2О
При действии карбонатов или гидроксокарбонатов щелочных металлов на растворы солей Ni(ll) образуются водные гидроксокарбона-ты никеля переменного состава с отношением Ni(CO3): Ni(OH)2 в зависимости от условий от 5:1 до 1:4 и даже 1:21. Гидроксокарбонаты образуются также при разложении никелевых аммиачно-карбонатных растворов:
2[Ni(NH3)6]CO3 + 2Н2О NiCO3Ni(OH)2 + СО2 + Н2О + 12NH3
При действии кислорода и влаги на тетракарбонил никеля также образуется гидроксокарбонат по схеме
2Ni(CO)4 + 5О2 + Н2О = NiCO3Ni(OH)2 + 7СО2
При обработке элементного никеля на воздухе аммиачными растворами карбоната аммония получается также гидроксокарбонат никеля:
2Ni + (NH4)CO3 + 2Н2О = NiCO3Ni(OH)2 + 2NH4OH 506
Гидроксокарбонаты никеля — аморфные желатинообразные вещества. Могут быть получены и в виде относительно хорошо фильтрующихся осадков. В зависимости от условий процесса сушки могут быть получены различные виды гидроксокарбонатов — от тяжелой плотной стеклоподобной массы до легкого тонкого землистого порошка; цвет — от бесцветного до темно-зеленого.
Структура гидроксокарбонатов построена из чередующихся слоев Ni(OH)2 и NiCOj. Практически не растворим в воде, легко растворяется в разбавленных минеральных кислотах, плохо — в водных растворах гидрокарбоната натрия, аммиачных растворах (МНд^СОз. При нагревании частично обезвоживается при температурах от 100 до 210° С, остаточная вода и диоксид углерода отщепляются при 250—420° С.
В литературе описаны условия получения гидроксокарбонатов определенного состава, например с отношением NiCO3:Ni(OH)2:H2O, равным 1:4:1, 1:2:4, 3:4:12, 1:1:1, 5:2:7 и т.д. К их числу относится и М!зСОз(ОН)4-4Н2О (минерал заратит) — травянисто-зеленые кристаллы с кубической решеткой (а = 0,615 нм, z = 4); плотность 2,649 г/см3.
В технической литературе под карбонатом никеля обычно понимают гидроксокарбонаты. Соединение, близкое по составу к Т^СОзСОН^-НгО, промежуточный продукт при извлечении никеля из окисленных руд аммиачно-карбонатным методом. Гидроксокарбонаты применяют для очистки электролита в процессе рафинирования никеля, для получения сульфата и других солей никеля (II), никельсодержащих катализаторов и в качестве пигмента для керамики и стекла.
11.8. НИТРАТ НИКЕЛЯ
Нитрат никеля Ni(NCh)2— зеленые кристаллы кубической сингонии (а = 0,73 нм); С ° = 86 Дж/(моль-К); АЯ^р = 401,5 кДж/моль. При нагревании разлагается:
Ni(NO3)2 —Ni(NO2)2 зоо-азо- с. > Ni0* (х = 1—1 5)
Растворимость в воде (г в 100 г Н2О): 79,2 (0° С), 100 (25° С), 139,2 (50° С), 180,1 (75° С). Растворяется в ацетонитриле, ДМФА, ДМСО и других полярных растворителях. Из водного раствора кристаллизуются гидраты нитраты никеля, содержащие 9, 6, 4 и 2 молекулы воды при концентрациях исходного раствора соответственно 36—39, 40—58, 60—66 и 68—70% (масс.).
Гексагидрат Ni(NO3)2-6H2O — зеленые кристаллы триклинной сингонии {а = 0,7694 нм, b = 1,1916 нм, с = 0,5817 нм, а=102,3°, р=102,5°, у = 105,9°, пространственная группа Р1). Температура 507
18*
плавления 54° С (инконгруэнтно); плотность 2,04 г/см3; = 462,3 Дж/(моль-К); ДЯП°Л =41,4 кДж/моль, АЯ^р = 2215,0 кДж/моль; 52°98 = = 511,3 Дж/(моль-К).
При нагревании гидраты нитрата никеля вначале частично дегидратируются, свыше 150° С разлагаются с образованием гидроксосо-лей переменного состава. Конечный продукт разложения—NiOx
Ni(NO3)2-6H2O NiNO3 + 6Н2О 15О‘С > xNi(OH)2NiNO3 -+ NiOx
Безводный нитрат никеля получают растворением хлорида никеля в смеси N2O4 или Ы2О5 с органическим растворителем с последующей отгонкой из реакционной зоны смеси оксидов азота и растворителя. Кристаллогидраты нитрата никеля синтезируют обработкой элементного никеля, его оксида, гидроксида или карбоната разбавленной азотной кислотой по схеме
Ni + 2HNO3 = Ni(NO3)2 + Н2
NiO + 2HNO3 = Ni(NO3)2 + H2O
Ni(OH)2 + 2HNO3 = Ni(NO3)2 + 2H2O
NiCO3 + 2HNO3 = Ni(NO3)2 + H2O + CO2
Из полученных растворов путем их фильтрации, упарки и кристаллизации получают целевой продукт.
Нитрат никеля применяют для получения других соединений никеля, а также никельсодержащих катализаторов в качестве компонента электролитов для нанесения никелевых покрытий на металлы, является шихтой в производстве цветной керамики.
ГЛАВА 12
САНИТАРНО-ТЕХНИЧЕСКИЕ ХАРАКТЕРИСТИКИ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ И ПРОМЫШЛЕННАЯ БЕЗОПАСНОСТЬ
12.1. СОЕДИНЕНИЯ МЫШЬЯКА
Основными соединениями мышьяка, широко применяемыми в народном хозяйстве, являются: оксиды (III) и (V), гидроарсенит натрия, метаарсенит кальция, метаарсенат — ацетат меди, гидроарсенат натрия, ортоарсенат кальция, сульфиды мышьяка (I) и (III).
Соединения мышьяка действуют на нервную систему, стенки сосудов, вызывают увеличение проницаемости и паралич капилляров. Вследствие нарушения механизмов регуляции процессов кровообращения и нервно-трофических процессов развиваются некро-биотические поражения в печени, сердце, кишечнике, почках, а также трофические кожные заболевания и поражения ногтей. Первичный механизм токсического действия As(III) связывают с блокированием SH-групп тиолсодержащих ферментов. Нарушается жировой и углеводный обмен, понижаются окислительные процессы в тканях. Воздействие мышьяка на дигидролипоатдегидрогеназы, липоамидоксиредуктазы и ряд коферментов нарушает окислительное декарбоксилирование пировиноградной и а-кетоглутаровой кислот. Мышьяк рассматривают и как физиологический антагонист иода, селена и, возможно, фосфора. Он вызывает злокачественные новообразования, оказывает в эксперименте тератогенное действие. Соединения мышьяка(Ш) значительно токсичнее, чем мышьяка(У). Токсичность зависит также от растворимости. Плохорастворимые соединения, например сульфиды и сам мышьяк, малоядовиты.
Различают три формы острого отравления: 1) при поступлении яда в желудок (например, при отравлении пестицидами) наиболее обычна желудочно-кишечная форма; 2) при больших дозах развивается паралитическая форма. Токсическая доза 0,01—0,05 г (при повышенной чувствительности 0,001 г), смертельная доза 0,06—0,20 г 509
(иногда переносятся и большие дозы); 3) в процессе вдыхания пыли мышьяковистых соединений (например, при работах по сухому протравливанию семян).
Хронические отравления человека сопровождаются желудочно-кишечными расстройствами, поражением слизистых оболочек и кожи: слезотечением, светобоязнью, отеком век, конъюнктивитом, помутнением стекловидного тела и роговицы, сухостью в носу и зеве, насморком, иногда изъязвлением и даже прободением носовой перегородки, стоматитом, ларингитом, трахеитом, бронхитом.
Поражение нервной системы: снижение работоспособности, нарушение мышления, запоминания и речи, головные боли. При этом возможны депрессия, галлюцинации, раздражительность. Снижение половой активности. Полиневрит.
Нередки дегенеративные изменения во внутренних органах, в особенности в печени, почках и сердце.
Соединения мышьяка поступают в организм при вдыхании, заглатывании пыли и через кожу. В крови мышьяк избирательно накапливается в эритроцитах. Мышьяк(У) в тканях медленно восстанавливается до более токсичного мышьяка(Ш). Мышьяк обнаруживается во всех органах, но количественные взаимоотношения зависят и от пути поступления яда в организм, и от времени его определения после отравления. Концентрация мышьяка в паренхиматозных органах со временем уменьшается, а в эктодермальных тканях, напротив, увеличивается. Выделение происходит весьма интенсивно, в основном с мочой. В норме мышьяк обнаруживается во всех тканях, содержание в моче колеблется от 0,003 до 0,15 мг/л.
При остром отравлении через рот применяют рвотные, промывание желудка теплой водой, взвесью жженой магнезии (20 г на 1 л воды). Затем больному дают по чайной ложке микстуры (к 100 ч. раствора сульфата железа плотностью 1,43 прибавляют 300 г холодной воды) через каждые 10—12 мин до прекращения рвоты. После этого промывают желудок. Внутривенно 20 мл 25—40%-ного раствора глюкозы с аскорбиновой кислотой (500 мг) и витамином В] (50 мг). Капельные клизмы из 5%-ной глюкозы. Физический раствор под кожу. Камфара, кофеин. Кислородная терапия. Подвергшиеся острому отравлению подлежат госпитализации.
Предельно допустимая концентрация для оксидов мышьяка (III) и (V) 0,3 мг/м3, для арсената кальция 1 мг/м3, а для арсената свинца 0,15 мг/м3. При работе с пылевидными соединениями мышьяка применяют респираторы «Лепесток» и др., а также защитные очки.
510
12.2. СОЕДИНЕНИЯ ХРОМА
Биологическая роль хрома в организме точно не установлена. Присутствие хрома в растениях, в тканях животных и человека позволяет считать его микроэлементом. Металлический хром и соединения хрома (II) менее токсичны, токсичны соединения хрома (III), наиболее ядовиты соединения хрома (VI).
Соединения хрома вызывают местное раздражение кожи и слизистых оболочек, приводящее к их изъязвлению, а в процессе вдыхания аэрозолей — к прободению хрящевой части носовой перегородки, поражению органов дыхания, вплоть до развития пневмосклероза. Общетоксическое действие сказывается в поражении печени, почек, желудочно-кишечного тракта, сердечно-сосудистой системы. Независимо от пути введения в первую очередь поражаются почки — сначала канальцевый аппарат, затем сосудистая сеть с преимущественным поражением клубочков. И хром(Ш), и хром(У1) изменяют активность ферментов и угнетают тканевое дыхание. Аллергическое действие проявляется приступами, сходными с бронхиальной астмой и развитием кожной сенсибилизации, являющейся причиной «хромовых экзем». Имеются работы, свидетельствующие о более высокой заболеваемости и смертности от рака органов дыхания и пищеварения среди работающих на хромовых производствах.
В процессе вдыхания тумана Н2СГО4 в концентрации 20—30 мг/м3 появляются острые отравления с преимущественным поражением глубоких дыхательных путей. Известны острые, в том числе и смертельные отравления К2СЮ4 и К2СГ2О7. У пострадавших острая недостаточность почек с анурией, гиперкалиемией, ацидозом и азотемией. При концентрации СгОз 0,2 мг/м3 те или иные поражения носовой полости встречаются у всех занятых в производстве, в 2/3 случаев процесс доходит до прободения носовой перегородки. Порог раздражающего действия аэрозоля хрома (VI) для наиболее чувствительных лиц 0,0025 мг/м3.
Среди рабочих хромовых производств отмечена повышенная заболеваемость раком легких. В США смертность от рака легких среди этого контингента в 25—29 раз выше, чем в других слоях населения.
В процессе нанесения на кожу и слизистых оболочек растворимые соединения хрома (СГ2О3, хроматы, дихроматы и др.) оказывают раздражающее и прижигающее действие. Соединения хрома (VI) вызывали изъязвления, соединения хрома (III) язв не вызывали. Нанесение на кожу 50- и 25%-ных растворов К2СГ2О7 приводило к гибели 91,6% животных. Скорость проникания через кожу соединений хрома (III) и хрома (VI) при низких концентрациях одного порядка. При более высоких концентрациях соединения хрома (VI) всасывались вдвое быстрее.
511
Установлено, что хром (VI) в организме частично восстанавливается в хром (III). В плазме крови хром (III) связывается с белками. Хром обладает сродством к легочной ткани, но параллельно накапливается также в ретикулоэндотелиальной системе печени, поджелудочной железе и костном мозге. В легких у лиц, работающих с хромсодержащими соединениями, хром обнаружен через много лет после прекращения работы.
При поражении слизистой носа — после тщательной механической очистки и промывания водой с мылом — после работы и перед сном смазывать носовые ходы топленым салом, смесью ланолина с вазелином, рыбьим жиром, 0,25—0,5%-ной хлораминовой мазью, приготовленной на свином сале, или желтой ртутной мазью. При кашле — кодеин с карбонатом натрия, дионин и др.
Предельно допустимая концентрация для сесквиоксида хрома, хроматов, дихроматов (в пересчете на СгОз) 0, 01 мг/м3, для хромоаммониевых квасцов (в пересчете на СгОз) 0,02мг/м3, для гексагидрата хлорида хрома (в пересчете на СгОз) 0,01 мг/м3, для оксида хрома 1мг/м3.
В качестве индивидуальной защиты в производстве соединений хрома применяют респираторы типа «Лепесток», «Астра-2», РУ-60М или изолирующие шланговые противогазы с механической подачей воздуха. Перчатки резиновые, латексные. Спецодежда — комбинезон с капюшоном из плотной ткани.
12.3. СОЕДИНЕНИЯ ФТОРА
Фтор. Раздражающие свойства фтора в несколько раз выше, чем фторида водорода. Резорбтивное действие объясняют возможностью фтора вступать в свободнорадикальные реакции с тканями организма. Длительное воздействие опаснее, чем повышение концентрации.
При 0,015 мг/л ощущается лишь запах; 0,023—0,038 мг/л вызывают раздражение глаз и носа. Отмечено слабое привыкание к раздражающему эффекту. В США принято считать переносимыми следующие концентрации: 0,039 мг/л — 5 мин, 0,023 — 15 мин, 0,016 — 30 мин, 0,0078 — 60 мин. Концентрация 0,077 мг/л требует защиты органов дыхания и глаз.
Соприкосновение кожи с газом на 2 с вызывает термический ожог II степени; воздействие 0,15—0,30 мг/л приводит к раздражению открытых участков кожи.
При внутривенном введении ,8F в организм его не находили в почках, печени и щитовидной железе. В хронических опытах отложение в зубах и костях увеличивается по мере увеличения длительности отравления. Метаболизм F2 отличается от метаболизма НЕ 512
Предельно допустимая концентрация при 8-часовом воздействии 0,15—0,20 мг/м3. При воздействии в течение 60 мин допускается 1,55 мг/м3.
В производстве фтора применяют изолирующие шланговые противогазы с подачей чистого воздуха. Спецодежда из стекловолокна или неопреновой резины; защитные очки из оргстекла.
Фторид водорода и фтороводородная кислота сильно раздражают верхние дыхательные пути. При высоких концентрациях — раздражение глаз и слизистой оболочки носа; слезотечение, блефароспазм, слюнотечение; могут развиться медленно заживающие изъязвления, конъюнктивы глаз, слизистых оболочек носа, полости рта, гортани и бронхов, гнойный бронхит, носовые кровотечения. Иногда рвота, колики, симптомы действия на центральную нервную систему, ощущение удушья, приступы тетании. Сердечно-сосудистые повреждения: изменение проводимости, острая дилатация сердца, нарушение коронарного кровообращения, падение кровяного давления, выраженная недостаточность кровообращения. Функциональные заболевания печени; возможно развитие токсического гепатита. Нефропатия. Увеличение содержания гемоглобина и эритроцитов в крови, замедленная РОЭ, лейкопения, нейтропения, относительный лимфоцитоз. Исходом отравлений могут быть бронхиты, пневмосклероз, бронхоэктазы, дистрофические изменения миокарда, поражение печени. При высоких концентрациях— спазм гортани и бронхов. Хроническое отравление может вызываться даже небольшими концентрациями за счет фтор-иона, обладающего высокой токсичностью.
В производстве имели место отравления при 6—10-минутном пребывании в атмосфере, содержащей 0,40—0,43 мг/л. Максимальная концентрация, выносимая долее Imhh, — 0,1 мг/л; при 0,05 мг/л — значительное раздражение слизистых оболочек; раздражение заметно и при 0,025 мг/л, хотя вдыхание может продолжаться без особого труда несколько минут; порог раздражающего действия 0,008 мг/л. Порог рефлекторной реакции, оцениваемый по изменению световой чувствительности глаза, составляет 0,00003 мг/л.
В производственных условиях при 0,002—0,003 мг/л болезненных явлений не наблюдалось. При концентрациях, близких к 0,005 мг/л, у рабочих обнаружены функциональные изменения центральной нервной системы и поражения зубов.
Фторид водорода на кожу действует сильно прижигакЯце, вызывая пузырьковые дерматиты, трудно заживающие язвы. Ощущение боли наступает лишь при контакте с очень крепкими растворами.
Предельно допустимая концентрация фторида водорода 0,5 мг/м3.
К индивидуальной защите относятся: фильтрующий противогаз марки В, при наличии аэрозоля кислоты в воздухе — с фильтром. Для поглощения фторида водорода применяют ионитовое волокно. В 513
производстве фторида водорода носят резиновые перчатки, фартуки, сапоги, защитные очки из оргстекла.
Фториды металлов являются протоплазматическими ядами, действующими главным образом на ферменты. Они блокируют также SH-группы. Полагают, что в присутствии фосфора фтор вступает в комплексные соединения с магнием, марганцем, железом и другими биоэлементами. В результате нарушается обмен, особенно углеводный. К тому же фтор осаждает кальций, что приводит к нарушениям кальциевого и фосфорного обмена. Конкурируя с иодом, может вытеснить его из иодорганических соединений. Процесс острого отравления фтором сопровождается действием на центральную нервную систему и мускулатуру, а также желудочно-кишечный тракт. При хроническом отравлении основные изменения происходят в костях и зубах. Наряду с этим наступают сосудистые нарушения, поражения верхних дыхательных путей, пищеварительного тракта, нервной системы и кожи. Изменения в костях у человека, обычно изменения типа остеосклероза. Тяжесть токсического действия фторидов зависит от их растворимости в биологических средах. Оба эти показателя уменьшаются в ряду HF>NaF>AlF3. При их совместном действии имеет место суммарный токсический эффект. Токсичность фторидов обусловлена не только действием фтор-иона, но и токсичностью катиона, например BaF2, CuF2, PbF2, CrF2.
Предельно допустимая концентрация для фторидов 1 мг/м3 (в пересчете на HF), а для BaF2 — 0,2 мг/м3.
В производстве фторидов применяют респиратор «Лепесток», резиновые перчатки, шлем для защиты от пыли, брезентовые пылезащитные костюмы.
Гексафтороалюминат натрия (Na3AlF6, криолит) обладает токсическим действием подобно фторидам. Природный криолит несколько токсичнее искусственного.
Из желудочно-кишечного тракта всасывается 77% от принятой дозы. В процессе работы с криолитовой пылью в организм рабочего поступает ежедневно 0,2—0,35 мг фтора на 1 кг массы тела. На рентгенограмме изменения в костях были обнаружены в среднем через 24 года, а усиленное обезызвествление костей наступало через 11 лет. У рабочего персонала, вдыхавшего пыль криолита, отмечались пневмокониотические изменения.
Предельно допустимая концентрация при изолированном присутствии в воздухе рекомендуется 0,5 мг/м3.
Кремнефтороводородная кислота. Прием внутрь 1 г кремнефтороводородной кислоты (H2SiF6-2H2O) вызывает смерть. Местное действие подобно действию фтороводной кислоты, но слабее. После применения состава для чистки стекол, содержащего 5—6% H2SiF6-2H2O, в кистях рук чувствуется жжение, ощущение «ползания мурашек», затем 514
появляются признаки воспаления кожи, синие пятна на ногтях. Через 4 дня сходит кожа, через 10 дней — ногти.
Гексафторосиликат натрия. Токсическое действие гексафторосиликата натрия (Na2SiF6) сходно с действием фторидов. Раздражающее действие (слезотечение, конъюнктивит, насморк, носовые кровотечения, явления трахеобронхита, тошнота, рвота, понос, колики, острые и хронические дерматиты). При случайном отравлении через рот — резко выраженные признаки поражения желудочно-кишечного тракта. Возможно наступление паралича сосудодвигательного центра, коллапс и смерть от остановки дыхания. Смертельная доза 0,4—4,0 г. Хроническое отравление проявляется симптомокомплек-сом, характерным для действия фторидов.
12.4. СОЕДИНЕНИЯ ХЛОРА
Соединения хлора. У работающих в атмосфере с небольшим содержанием хлора повышается сопротивляемость к гриппозной инфекции. Однако при ощутимой концентрации хлора и большой длительности контакта могут поражаться глаза и органы дыхания в виде гипертрофических воспалений слизистой оболочки носа и хронических бронхитов. У работающих с хлором наблюдаются также кариес зубов, воспаление десен, желудочно-кишечные расстройства, анемия, головные боли, бессонница.
Иногда хлор может вызвать острый дерматит и даже экзему.
Вдыхание высоких концентраций хлора может привести к моментальной смерти рефлекторного происхождения (рефлекторное торможение дыхательного центра). Концентрация хлора в помещении 0,001—0,006 мг/л оказывает на человека заметное раздражающее действие; 0,1—0,2 мг/л опасны для жизни уже при получасовом воздействии.
При отравлении хлором человека необходимо вынести на свежий воздух. Обеспечить покой и согревание тела.
ПДК хлора в воздухе 1 мг/м3.
К индивидуальным средствам защиты относятся промышленные фильтрующие противогазы марки В, резиновые перчатки, обувь и передники.
Оксид хлора(1) — гемиоксид С12О — сильно раздражает глаза.
Диоксид хлора (С1О2) — раздражающий газ, поражает верхние и глубокие дыхательные пути. При остром отравлении — раздражение слизистой глаз, верхних дыхательных путей, кашель, затруднения дыхания, слабость, головокружение, рвота. Симптомы отравления продолжаются более недели. Последствием могут быть конъюнктивит, бронхит астмоидного характера и эмфизема легких. Возможно нару-515
шение психики у рабочих, подвергавшихся воздействию небольших концентраций Ch и СЮ2.
Предельно допустимая концентрация 0,1 мг/м3. Индивидуальная защита — промышленные противогазы марки В, резиновые перчатки, обувь и передники.
Гипохлорит, хлораты (V) и (VII) натрия и калия — см. Хлор.
12.5. СОЕДИНЕНИЯ БРОМА
Бром. Бром в парообразном состоянии действует на живой организм сходно с хлором, но слабее. Это объясняется его резорбтивным действием.
В процессе вдыхания небольших концентраций бром вызывает кашель, увеличенную секрецию слизистых оболочек, носовые кровотечения, чувство стеснения в груди, головокружение, головную боль, иногда рвоту и понос, боли в теле, сыпь на туловище и конечностях, исчезающую на следующий день. При более высоких концентрациях— коричневую окраску слизистой оболочки рта, конъюнктивиты, характерный запас вдыхаемого воздуха, насморк, слюнотечение, кашель, удушье, спазмы голосовой щели, охриплость, бронхит; позже иногда более тяжелые симптомы. Раздражающее действие на слизистые оболочки носа и зева отчетливо проявляются при концентрации 0,013—0,032 мг/л. Считают работу невозможной даже при 0,004 мг/л. При 0,011—0,023 мг/л отмечено сильное удушье даже при кратковременном действии, а при 0,005—0,036 мг/л (в присутствии НВг) без противогаза нельзя выдержать даже 2—3 мин.
Жидкий бром действует на кожу прижигающе и окрашивает ее в желтый цвет. Язвы, вызванные бромом, плохо заживают. При попадании брома на кожу быстро обмыть ее водой и смазать мазью с гидрокарбонатом натрия. Для замещения и выведения брома из организма принимать хлорид натрия по 10—30 г 4 раза в день.
При введении кроликам меченого брома больше всего его обнаруживается в гипофизе, а затем в щитовидной железе, семенниках, печени, крови, мозге и мозжечке. Выделяется 82Вг в основном почками.
Предельно допустимая концентрация 0,5 мг/м3.
В производстве брома обслуживающий персонал пользуется противогазом марки В или шланговыми противогазами с принудительной подачей чистого воздуха. Применяют герметичные защитные очки.
Бромид водорода и бромоводородная кислота. Порог раздражающего действия бромида водорода при однократном вдыхании животными 0,026 мг/л. При вдыхании 0,066 мг/л бромида водорода нарушаются функции центральной нервной системы и терморегуляция, однако бромид водорода действует в 2—3 раза слабее, чем элементный бром. 516
Бромиды, вводимые крысам в желудок в высоких дозах, вызывают обильное мочеиспускание, частый и жидкий стул, адинамию, клинические судороги. Для мышей ЛД50 = 3,2 г/кг, для крыс—-3,1, для кроликов — 4,0, для морских свинок — 5,5 г/кг. Общее действие слабое и в производственных условиях острого отравления бромид вызывать не может. Известна способность брома замещать иод в щитовидной железе, что отражается на состоянии основного обмена. Возможность подобного действия исключать нельзя при длительном поступлении бромидов в организм.
12.6. СОЕДИНЕНИЯ ИОДА
Иод. Токсическое действие паров иода заключается в раздражении слизистых оболочек (см. Хлор). В процессе поступления внутрь больших количеств иода обнаруживается и общее действие на кровь, железы, значительно сильнее, чем действие иона иода.
В процессе действия паров иода наступает кашель, насморк, слезотечение, конъюнктивит, опухание околоушных желез, головные боли, шум в ушах, головокружение, двойное видение. В тяжелых случаях— также рвота, понос, белок и гемоглобин в моче, задержка мочи, одышка. Имеют место спазм голосовой щели, смерть. После отравления долго сохраняются слабость, пониженная сопротивляемость организма. Есть случаи образования метгемоглобина и выделение его в моче. При хроническом отравлении — катар слизистых оболочек носа, зева; исхудание; желтоватая окраска зубов. Порог раздражающего действия 0,0015—0,002 мг/л. При 0,003 мг/л работа невозможна. У некоторых людей, особенно страдающих базедовой болезнью, чувствительность к иоду резко повышена. В процессе получения иода из буровых вод нефтяных месторождений в воздухе содержится 0,1—0,002 мг/л. При 0,05 мг/л операторы пользуются противогазами. Более 80% рабочих жалуются на охриплость голоса, кашель с мокротой, болезненность носа и выделение слизи. Диагностированы риниты, назофарингиты, ларингиты, бронхиты. При большом стаже — эрозии и атрофические явления в слизистой оболочке носа. Максимальная вентиляция легких и минутный объем легких снижены. При обследовании 85 человек почти у всех обнаружено функциональное расстройство центральной нервной системы по типу астеновегетативного синдрома.
Иод вызывает дерматит. У животных, находящихся на территории иодного завода, изменения со стороны век, конъюнктивы, хрусталика, сосудов сетчатки. У рабочих при действии паров иода — катаральный конъюнктивит.
Иод — биоэлемент. Ежедневная потребность организма в нем 60—100 мкг. В крови находили 15 мкг. %. Наибольшее количество — в 517
щитовидной железе (1—10 мг). Выделяется из организма главным образом через почки (15—16 мкг/сут), в меньшей степени — с испражнениями и потом. Фтор, поступая в организм в больших количествах, способствует энергичному выделению из него иода.
Противоядием иода является тиосульфат натрия, применяемый в виде ингаляций 5%-ного раствора и внутривенно (30—50 мл 10—20%-ного раствора).
Предельно допустимая концентрация 1 мг/м3. При повышенной температуре окружающего воздуха рекомендовано 0,3—0,5 мг/м3.
В производстве иода пользуются фильтрующим противогазом марки В, герметичными очками ПО-3, перчатками и спецодеждой.
Иодид водорода и иодоводородная кислота. Токсическое действие иодида водорода и иодоводородной кислоты подобно действию хлорида водорода и хлороводородной кислоты, но слабее.
Йодиды и йодаты. Йодиды и йодаты обладают раздражающими свойствами. При смертельном отравлении через рот белых мышей описаны поражения пищеварительного тракта, выраженная форма гемолиза эритроцитов, гемоглобинурия, жировая дегенерация печени и миокарда, распространенный некроз извитых канальцев почек с отложением гемоглобиновых цилиндров. Йодиды калия и натрия менее токсичны, чем йодаты калия и натрия. В производственных условиях действие йодатов обычно сочетается с влиянием элементного иода.
В производственных условиях обслуживающий персонал носит противогаз марки В от иодида водорода и респираторы от аэрозолей иодидов, герметичные очки.
12.7. СОЕДИНЕНИЯ МАРГАНЦА
Широко применяемыми в народном хозяйстве соединениями марганца являются его оксиды (II), (II, III), (III) и (IV), хлорид, сульфат и карбонат.
Соединения марганца — сильные яды, действующие на центральную нервную систему, вызывая в ней тяжелые органические изменения, главным образом экстрапирамидный симптомокомплекс. В тяжелых случаях — картина паркинсонизма. В патогенезе отравления играет роль влияние марганца на синтез и обмен катехоламинов. Большие дозы угнетают рефлекторную возбудимость спинного мозга, а также ацетилхолинэстеразу.
Марганец, будучи биоэлементом, принимает участие в окислительно-восстановительных процессах, в фосфорилировании, входит в состав аргиназы, кокарбоксилазы, оксидаз, щелочных фосфотаз и других, участвует в синтезе витаминов С и Вь возможно, Е. Усиливает обмен белков, обладает симпатотропным и липотропным дейст-518
вием. Образует комплекс с у-глобулином крови человека, названный транс-манганином. Введенный извне марганец, накапливаясь в митохондриях, изменяет каталитические, энергетические, обменные процессы в клетках; нарушает лизосомальные ферменты; угнетает аденозинфосфатазы, глюкозо-6-фосфатазу, сукцинатдегидрогеназу и др. Повышает уровень сахара и молочной кислоты в крови. В эксперименте сдвиги в этих системах проявляются раньше, чем клинические и морфологические признаки отравления. Известно стимулирующее действие марганца на гемо- и эритропоэз, сменяющееся угнетением их. Есть также указания на мутагенный эффект марганца, а также на гонадотоксическое действие. Вдыхание пыли оксидов марганца в эксперименте, а также у человека вызывает особую форму пневмокониоза— манганокониоз. Известна также высокая заболеваемость воспалением легких рабочих, вдыхавших пыль марганцевой руды. Наибольшее число профессиональных отравлений возникало при контакте с высшими оксидами. При любых вариантах поступления соединений марганца особо резкие нарушения обнаруживаются в головном мозге, особенно в области стриопаллидарной системы. Они сходны с наблюдаемыми при гипоксии. Вторичные же повреждения развиваются в результате локальных расстройств мозгового кровообращения. Дегенеративные изменения обнаруживаются также в печени, почках, реже в сердечной мышце.
Как правило, отравление соединениями марганца развивается в результате хронического воздействия. Картина отравления зависит от поражения нервной системы. По течению и тяжести хронические отравления разделяют на три стадии. Первая, начальная, обычно характеризуется функциональными поражениями центральной нервной системы; иногда изменения со стороны желудка, симптомы полиневрита. Жалобы на головную боль, головокружение, утомляемость, сонливость, отсутствие аппетита, изжогу, боли в конечностях, парестезии и судороги в них, иногда на боли в области сердца, половую слабость. Объективно-повышенная возбудимость мышц; часто расширение глазных щелей и резкое мигание, учащение пульса; увеличение щитовидной железы; явления гипо- или анацидного гастрита; легкие полиневритические расстройства типа вегетативного неврита конечностей; некоторая неловкость в движениях.
Во второй стадии — при дальнейшем прогрессировании заболевания, а иногда без предварительных симптомов — выявляются признаки начальной токсической энцефалопатии. Могут сохраняться все указанные выше явления, но усиливается гипомимия и мышечный тонус (или он ослабляется); изменения в технической сфере (необоснованная веселость и т.д.). Полиневритические явления усиливаются с поражением как чувствительной, так и двигательной сфер. Повышается уровень гемоглобина и эритроцитов в крови. В крови также снижение содержа-519
ния белка, изменение соотношений белковых фракций в сыворотке крови; повышен уровень холестерина и Р-липопротеинов.
Третья стадия наиболее тяжелая — «марганцевый паркинсонизм» — может являться вслед за более ранними признаками или без них. Клиническая картина напоминает паркинсонизм после эпидемического энцефалита. Характерны расстройства эмоциональной сферы, интеллекта. Иногда афазия из-за ограничения движения языка и мышц челюсти.
Человек получает с пищей -4 мг марганца в сутки. Выделение с мочой составляет 0,01 мг/л. В производственных условиях происходит как вдыхание, так отчасти и заглатывание пыли. Всасывание из легких протекает очень активно. Из крови, где марганец находится в виде белкового комплекса с у-глобулинами (трансманганина), он быстро выделяется и накапливается в печени, почках, а также в железах внутренней секреции. Накопление в разных отделах головного мозга неодинаково и находится на более низком уровне, чем в прочих органах, но фиксация в мозгу, по-видимому, значительно более прочная. Из тканей слабо связанный марганец быстро выделяется, а прочно связанный с внутриклеточными микромолекулами более медленно.
Предельно допустимая концентрация для марганца как аэрозоля конденсации (в пересчете на Мп) 0,03 мг/м3, для марганца — аэрозоля дезинтеграции 0,2 мг/м3, при одновременном воздействии марганца и фтора 0,15 мг/м3.
В производстве применяют противопылевые респираторы «Лепесток», «Астра-2». При большой концентрации аэрозоля — промышленный противогаз с коробкой БКФ и фильтром. Спецодежда из бумажной ткани.
12.8. СОЕДИНЕНИЯ ЖЕЛЕЗА
Основными соединениями железа, широко применяемыми в народном хозяйстве, являются оксиды железа (II), (III), сульфат и хлорид железа (II), а также его карбонил.
Токсическое действие железа и его неорганических соединений. Соединения Fe(II) обладают некоторым общим токсическим действием: у белых крыс, кроликов при поступлении через рот — параличи, смерть в судорогах. Хлориды железа токсичнее сульфатов. Соединения Fe(III) менее ядовиты, но действуют прижигающе на пищеварительный канал и вызывают рвоту. Аэрозоли железа и его оксидов, руд и других соединений Fe в процессе длительного воздействия откладываются в легких и вызывают сидероз — разновидность пневмокониоза с относительно доброкачественным течением. Различают так называемый «красный сидероз», вызываемый сесквиоксидом 520
железа, и «черный сидероз», возникающий от вдыхания пыли Fe, его карбонатов и фосфатов. Сидероз характеризуется малым количеством жалоб, удовлетворительным общим состоянием, длительным сохранением трудоспособности, редко сочетается с туберкулезом. Возможны также бронхиты, начальная эмфизема; сухой плеврит.
Описан случай «железной лихорадки» у электросварщиков после работы в плохо вентилируемом помещении, в атмосфере пыли и паров, содержащих Fe. Симптомы — усталость, потливость, повышение температуры до 38,3—39,3° С, лейкоцитоз.
Утверждают, что пыль чистых оксидов железа или железа не фиб-рогенна. Однако у рабочих, имеющих контакт только с чистой пылью Fe (без примеси S1O2), обнаружены изменения, сходные с картиной силикоза I—II степени. У рабочих, занятых измельчением красковых руд в производстве сурика, пневмокониоз в 2,5 раза чаще, чем в железоруд-пых шахтах. Пыль конвертерного производства (85—93% оксидов железа, 3,4 — 5,2% общего SiC>2, 1—2,1% свободного SiCh, не более 0,5% Сг, Мп, V и S) вызывает пневмокониоз, напоминающий пневмокониоз сварщиков. Пневмокониоз был выявлен при добыче железной руды, где рабочие подвергаются воздействию пыли РегОз и SiC>2.
Считается, что железо всасывается в желудке лишь после окисления в Fe2+ и образования белкового комплекса — ферритина. При введении крысам в желудок 59Ре20з через 4 ч в желудочно-кишечном тракте еще находилось 99,3% 59Fe. При вдыхании 59Ре2Оз в легких крыс через 4 ч обнаружено 52—62%, в желудочно-кишечном тракте— 33%, в крови, печени, почках—1% от введенной дозы.
ПДК оксида железа с примесью оксидов марганца до 3%, чугун, чугун в смеси с электрокорундом до 20%, легированные стали и их смеси с алмазом до 5% — 6 мг/м3; оксид железа с примесью фторидных или 3—6% марганцевых соединений, железный агломерат — 4 мг/м3. В США для растворимых солей железа принята ПДК 1 мг/м3 (по Fe), для аэрозоля оксида железа — 5 мг/м3. Для феррита бария рекомендуется 2 мг/м3, а содержащих до 19% оксидов марганца ферритов—1,5 мг/м3.
В качестве индивидуальной защиты от железосодержащих пылей в производстве применяют респираторы «Лепесток» и др.; защитные герметические очки и спецодежду из пыленепроницаемой ткани.
Пентакарбонил железа Fe(CO)5 сильно ядовит при вдыхании, введении внутрь или всасывании через кожу. Вызывает острый отек легких независимо от пути введения. Предполагают, что выделение происходит через легкие. Смертельная доза для кроликов при введении через рот или внутривенно 1,75 мг/кг. При вдыхании паров в течение 30 мин для белых мышей ЛК50 = 2,19, а для белых крыс 0,91 мг/л. В производственных условиях отравления протекали сходно с картиной интоксикации оксидом углерода.
521
Предельно допустимая концентрация в США установлена 0,8 мг/м3.
В производстве применяют противогаз марки П-2, который защищает одновременно от СО и Ре2Оз. При привесе коробки противогаза 25 г не защищает от СО; при привесе 45 г противогазная коробка негодна и должна немедленно ^сменяться. Предусматривают меры, предотвращающие выделение в воздух Fe(CO)5, а также СО. Дегазацию производят окислителями (КМпОд, хлорамином). Рекомендуется проведение периодических медицинских осмотров работников производства не реже одного раза в 12 месяцев.
12.9. СОЕДИНЕНИЯ КОБАЛЬТА
В народном хозяйстве широко применяются металлический кобальт, оксиды кобальта (II), (II, III) и (III), сульфат, хлорид, ацетат и тетракарбонил кобальта.
Токсическое действие кобальта и его неорганических соединений. Кобальт — важный биологический элемент. В малых дозах в организме он активизирует ряд ферментов, регулирующих тканевое дыхание, кроветворение и другие процессы, а в больших дозах угнетает. Угнетающее действие связано с образованием комплексов кобальта с SH-группами энзимов, способностью тормозить процесс переноса электронов по дыхательной цепи и окислительное фосфорилирование. В результате влияния на тканевое дыхание развивается гистотоксическая гипоксия. Токсические дозы кобальта угнетают гемопоэз. Поли-глобулия сходна с развивающейся на высоте. Считают, что для ее развития необходимо присутствие в организме достаточного количества меди. Влияние кобальта на кроветворение объясняют возникающей тканевой гипоксией, угнетением дыхательной функции форменных элементов крови, мобилизацией железа для улучшения синтеза гемоглобина, стимуляцией костного мозга или эритропоэтического фактора. Под влиянием кобальта изменяется строение и функция щитовидной железы вследствие общего нарушения окислительных процессов, а также нарушаются каталитические реакции в самой железе, блокируется тирозиниодиназа, поглощение и окисление неорганического иода; кобальт связывает SH-группы эпителия и коллоида.
Избыток кобальта в организме влияет на сердечно-сосудистую систему, расширяет сосуды, снижает кровяное давление; избирательно поражает сердечную мышцу. Дефицит белка усиливает токсическое действие кобальта. При длительном вдыхании кобальта или его оксидов возникают воспалительные и склеротические изменения в легких. Комплексные соединения кобальта действуют сходно с его солями (хлорид, сульфат и т.д.). Так, при введении под кожу 15—25 мг хлорида кобальта на 9—13-й день у кроликов — одышка, цианоз, снижение окислительных процессов в миокарде. У собак однократное введение хлорида 522
кобальта вызывало синусовую тахикардию. При однократном введении в трахею крыс 25—50 мг оксида кобальта(П) быстро развиваются массовые кровоизлияния и отек легких, смерть на 1—2 сутки. При таком же введении сесквиоксид кобальта вызывает воспалительно-пролиферативную инфильтрацию вокруг скопления пыли и инфильтрацию межальвеолярных перегородок. Вдыхание аэрозоля 1 %-ного раствора СоСЬ, Со[Со — ЭДТА] и Na2[Co — ЭДТА] в течение трех часов вызвало у крыс и морских свинок отек легких. Ежедневное введение кобальта в виде его нитрата по 3 мг/кг шесть раз в неделю в течение 50—60 дней у кроликов повышало число эритроцитов на 89%, а содержание гемоглобина — на 73%.
Мелкие частицы кобальта вызывают острый дерматит в виде многочисленных несливающихся красных капсул, узелков и отека; иногда поверхностные изъязвления. Сам металлический кобальт — слабый аллерген и редко бывает причиной контактного дерматоза.
Предельно допустимая концентрация для кобальта и его оксида 0,5 мг/м3. Защитными средствами органов дыхания в производстве соединений кобальта являются респираторы «Астра», «Лепесток» и др.
Тетракарбонил кобальта Со(СО)4 подобно другим карбонилам металлов вызывает раздражение глубоких дыхательных путей. Менее токсичен, чем карбонил никеля. Разлагаясь с большой скоростью на воздухе, по-видимому, в какой-то период действует в виде паров, а затем в виде продуктов разложения — мелкодисперсного аэрозоля неорганических соединений кобальта и сорбированных на нем паров тетракарбонила кобальта, оксида углерода и карбонил-гидрида кобальта [Со(СО)4Н].
Процесс вдыхания паров у человека вызывает слабость, тошноту и затруднение дыхания. При более высоких концентрациях — расширенные и суженные, неравномерные зрачки, двойное видение, снижение корнеальных и сухожильных рефлексов, отклонение языка, а иногда судорожные подергивания. Описаны также случаи отека легких. Симптомы поражения центральной и вегетативной нервной системы исчезали в зависимости от тяжести через 3—4 или 10—15 дней.
При хронических отравлениях среди работающих много ринитов, ринофарингитов, понижение обоняния. Бывают также признаки поражения миокарда, тенденция к анемизации.
Концентрация 0,0008 мг/л (по кобальту) вызвала прижигание конъюнктивы и роговицы крыс. Однократное и особенно повторное нанесение тетракарбонила кобальта на кожу крыс в количестве 1,0—1,5 мг/см2 вызывало изъязвление.
После однократного вдыхания в легких задерживается ~5%, которые удаляются в течение 2 недель.
Предельно допустимая концентрация 0,01 мг/м3 (по кобальту).
523
В производстве тетракарбонила кобальта применяют специальный фильтрующий противогаз марки П-2. При более высоких концентрациях — шланговые противогазы с подачей свежего воздуха или изолирующие кислородные приборы.
12.10. СОЕДИНЕНИЯ НИКЕЛЯ
Наиболее широко применяемыми в народном хозяйстве соединениями никеля являются металлический никель, оксиды никеля (II), (III), гидроксиды никеля (II) и (III), сульфат, хлорид, нитрат и сульфиды— NiS, Ni2S, NisS2, Ni3S4, Ni6S5, а также его тетракарбонил.
Токсическое действие никеля и его неорганических соединений. Никель активирует или угнетает ряд ферментов: фаргиназу, карбоксилазу, 5-нуклеозидфосфатазы и др. Он влияет на дефосфорилирование аминотрифосфата. В крови человека никель связывается преимущественно с у-глобулином сыворотки. После введения хлорида никеля кроликам в сыворотке крови обнаружен белок — никелоп-лазмин, идентифицированный как агмикроглобулин. Однако, по другим данным, 90% никеля в крови кроликов через 24 ч связывается с альбуминами, лишь незначительная часть поступившего NiCl2 выявлена во фракциях а2-глобулина. В организме никель образует комплексы с биокомплексонами. Никель имеет особое сродство к легочной ткани, в эксперименте при любом пути введения поражает ее. Оказывает влияние на кроветворение и углеводный обмен. Металлический никель и его соединения вызывают образование опухолей у животных, а также профессиональный рак. Канцерогенное действие никеля связывают с нарушением метаболизма клеток. Соли никеля вызывают поражение кожи человека с развитием повышенной чувствительности к металлу.
Смертность от рака легких, полости носа и его пазух составляет 35,5% всех смертей рабочих, занятых электролизом и рафинированием никеля. На первом месте был рак легких, на втором — желудка. Наиболее часто страдали работавшие при пирометаллургических процессах в обжиговосстановительных цехах (стаж 12—23 года, концентрации пыли колебались в пределах порядка 10—102мг/м3; в ней содержалось 70% Ni в виде сульфидов, NiO или металлического никеля). Высока смертность от рака в цехах электролиза при наличии В воздухе аэрозолей NiCb и NiSOzi. Средний стаж работы у умерших от рака легких 7—13 лет. от рака желудка—10—14 лет.
Считают, что никель не обладает прямым раздражающим действием на кожу. Однако у никелировщиков, работающих на производстве Ni электролизом и имеющих контакт с его солями, наблюдается 524
«никелевая экзема», «никелевая чесотка»: фолликулярно расположенные папулы, отек, эритема, пузырьки.
Никель и его соединения — сильные сенсибилизаторы.
Никель поступает в организм человека через желудочно-кишечный тракт. При этом всасываются не только соли, но и высокодисперсный металл и оксиды. В крови никель образует комплексные с белками плазмы — никелоплазмин. Никель, поступивший в результате вдыхания или через рот, распределяется в тканях более или менее равномерно. Однако в дальнейшем проявляется тропность никеля к легочной ткани.
Предельно допустимая концентрация оксида никеля (II), оксида никеля (III), сульфида никеля (в пересчете на никель) 0,5 мг/м3. Соли никеля в виде гидроаэрозоля (в пересчете на никель) 0,0005 мг/м3. Аэрозоль медно-никелевой руды — 4 мг/м3. Для аэрозолей файнштейна, никелевого концентрата, пыли электрофильтров никелевого производства 0,1 мг/м3.
В качестве защитных средств в производстве применяются респираторы, изолирующие, шланговые противогазы или респираторы. Пользуются защитной пастой ИЭР-2, ланолино-касторовой мазью и др.
Тетракарбонил никеля Ni(CO)4 раздражает глубокие дыхательные пути, вызывая пневмонию и отек легких независимо от пути поступления в организм. Значительное общетоксическое действие направлено на нервную систему. Ингибирует аминопиридиндеметилазу, цито-хром-450, триптофанпирролазу, бензпиренгидролазу. Угнетает синтез РНК, белков, что, возможно, связано с подавлением активности РНК-полимеразы в ядрах клеток. Полагают, что влияние на индукцию ферментов в какой-то степени связано с канцерогенным действием тетракарбонила никеля. Пока не установлено, действует ли тетракарбонил никеля целой молекулой или продуктами разложения. Однако после вдыхания или введения в вену тетракарбонил никеля выделяется с выдыхаемым воздухом, а также циркулирует в крови какое-то время как таковой. Тетракарбонил никеля подвергается внутриклеточному метаболизму с образованием Ni и СО. Клеточными окислительными системами Ni окисляется в Ni2+ и частично связывается с нуклеиновыми кислотами, он имеет особое сродство к РНК; часть его транспортируется в плазму. СО образует карбоксигемоглобин и в конечном итоге выдыхается. Очень незначительная часть тетракарбонила никеля окисляется до СО2. Даже при смертельных концентрациях или дозах и полном и быстром разложении тетракарбонила никеля образовавшийся СО не может дать выраженной картины отравления.
Предельно допустимая концентрация 0,0005 мг/м3.
В производстве при повышенных концентрациях тетракарбонила никеля пользуются фильтрующим промышленным противогазом 525
марки П-2. Его защитное действие рассчитано на 2,5 ч при концентрации тетракарбонила никеля 0,005 мг/л и СО 0,1 мг/л. В присутствии СО допускается привес коробки противогаза на 45 г, после чего она уже непригодна. В случае резко повышенных концентраций применяют шланговые противогазы. Применяют также специальную одежду и перчатки.
12.11. СЕРА И ЕЕ СОЕДИНЕНИЯ
Элементная сера. В обычных условиях токсические свойства элементной серы выражены очень слабо. Поэтому острые отравления в производстве не наблюдались. Однако при концентрации серы в рабочей зоне 30—40 мг/м3 у обслуживающего персонала кроме жалоб на головную боль и утомляемость наблюдалось раздражение верхних дыхательных путей, гастриты, снижение кровяного давления и моноцитов в крови. При длительной работе зарегистрированы также случаи костных изменений в черепе, воспалительные процессы в лобных пазухах и костях верхней челюсти. Отмечены также повышенная заболеваемость рабочих хроническими бронхитами, эмфиземой легких, а также изменения в сердечной деятельности. При многолетней работе в производстве серы возможен тиопневмокониоз.
Пыль серы вызывает конъюнктивит, а у особо чувствительных людей — экзему.
Механизм отравления серой зависит от среды. Так, в щелочной среде: S + OH_->S2O4’ + Н^О; в присутствии кислорода: 82Оз"+О2-> -> SO^" + SO2; в присутствии различных микробов: S + HR ->H2S + R.
Таким образом, сера в организме человека может превращаться в диоксид серы или сероводород и далее в результате окислительно-восстановительных процессов переходит в серную кислоту.
Диоксид серы SO2 действует раздражающе на верхние (а при более высоких концентрациях и длительном контакте) и на более глубокие дыхательные пути, что объясняется поглощением диоксида серы влажной поверхностью слизистых оболочек и последующим образованием сернистой и серной кислот:
SO2 + Н2О = H2SO3, 2H2SO3 + О2 = 2H2SO4
Указанные кислоты помимо местного поражения (кислотные ожоги) слизистых оболочек приводят к нарушению обменных и ферментативных процессов в организме. Так, экспериментально установлены изменения в углеводном обмене, угнетение окислительных процессов в головном мозгу, печени, селезенке, мышцах, нарушения белкового обмена, торможение окислительного дезаминирования ами-526
нокислот и окисления пировиноградной кислоты, а также снижение содержания в организме витаминов В и С. Обнаружено вредное воздействие диоксида серы на кроветворные органы и кости.
Экспериментально установлен нижний порог чувствительности запаха диоксида серы, равный 0,006—0,003 мг/л, концентрация 0,05 мг/л диоксида серы вызывает раздражение глаз, 0,02—0,03 мг/л — раздрае жение в горле, а 0,05 мг/л — кашель. При концентрации 0,06 мг/л у человека начинается сильное чихание и кашель. Концентрацию 0,12 мг/л диоксида серы в рабочей зоне человек может выдержать 3 мин, а 0,3 мг/л — лишь 1 мин.
В процессе хронического отравления диоксидом серы наблюдаются ухудшения обоняния и понижение вкусового восприятия, хронические, чаще атрофические, риниты, катары верхних дыхательных путей, фарингиты, ларингиты, бронхиты, часто обостряющиеся до состояния астмы, отмечаются желудочно-кишечные расстройства, конъюнктивит, разрушение зубов. Для начального периода хронического отравления характерна склонность к заболеваемости гриппом, гастритом, язвенным болезням, а также одышка, головные боли, слабость и утомляемость.
При остром отравлении необходимо вынести пострадавшего на свежий воздух и промыть глаза и нос 2%-ным раствором карбоната натрия. При кашле дать кодеин или дионин и провести тепловлажные ингаляции 2—3%-ным раствором гидрокарбоната натрия. Для частичного выведения из организма диоксида серы необходимо дать отхаркивающие препараты и теплое молоко с боржоми, можно с медом и маслом, поставить горчичники на область трахеи.
ПДК диоксида серы в производственных помещениях составляет 10 мг/м3, для триоксида серы—1 мг/м3.
К индивидуальным средствам защиты в производстве относятся фильтрующий противогаз марки В и специальный противогаз марки С. При одновременном присутствии наряду с диоксидом серы монооксида углерода рекомендуют применять противогазы марки МПС и МПС-Ф.
Серная кислота. Концентрированная серная кислота вызывает сильный химический ожог из-за денатурации белка. Она быстро проникает в глубь ткани с образованием белого струпа, переходящего в темно-коричневый. При отпадании струпа обнажается глубокая язва с резко очерченными краями и светло-красным дном. При больших поверхностных поражениях бывают смертельные исходы. При соприкосновении с 3%-ным раствором серной кислоты наблюдаются разрыхление кожи на руках, изъязвления, хронические гнойнички около ногтей и т.п.
У обслуживающего серно-кислотное производство персонала наблюдаются заболевания слизистой полости рта, разрушение зубов
527
(главным образом резцов), атрофические изменения слизистой верхних дыхательных путей, бронхиты, пневмосклерозы, гастриты, язвенная болезнь, кожные заболевания. Результаты некоторых исследований свидетельствуют о функциональных изменениях нервной и сердечно-сосудистой систем, а также о нарушении печени.
При остром отравлении парами серной кислоты у работников наблюдаются раздражения верхних дыхательных путей, насморк, чихание, кашель, затруднение дыхания, спазм голосовой щели, жжение в глазах, покраснение конъюнктивы глаз. При более высокой концентрации паров серной кислоты, а также диоксида серы в рабочей зоне могут появиться кровавая мокрота, рвота, а впоследствии тяжелые болезни бронхов и легких.
Раздражающее действие паров и тумана серной кислоты проявляется уже при концентрации 0,001 мг/л. У здоровых людей в закрытом помещении рефлекторные изменения дыхания отмечались при концентрации тумана или паров 0,00035—0,005 мг/л.
Рекомендуемая литература
Авербух Т. Д., Павлов П. Г. Технология соединений хрома.— Л.: Химия, 1967. 376 с.
Амелин А.Г. Производство серной кислоты.— М.: Химия, 1967^*472 с.
Ахметов Т.Г., Бусыгин В.М., Гайсин Л.Г., Порфирьева Р.Т. Химическая технология неорганических веществ.— М.: Химия, 1998^488 с.
Ахметов Н. С. Общая и неорганическая химия.— М.: Высшая школа, 2001. 743 с.
Беленький Е. Ф., Рискни И. В. Химия и технология пигментов.— Л.: Химия, 1974. 672 с.
Борисов В. М., Ажикнна Ю. В., Гальцев А. В. Физико-химические основы получения сложных фосфорсодержащих удобрений.— М.: Химия, 1983. 378 с.
Васильев Б.Т., Отвагина М.И. Технология серной кислоты.— М.: Химия, 1985> 384 с.
Вредные вещества в промышленности. Т. 3. Справочник для химиков, инженеров и врачей / Под ред. Н. В. Лазарева и И. Д. Гадаскиной.— Л.: Химия, 1977. 607 с.
Габриелова М. Г., Морозова М. А. Производство неорганических ядохимикатов.— М.: Химия, 1964. 327 с.
Gmelins handbook of inorganic chemistry, system. № 5. F. Fluorine. Suppl. V. 2. The element, B., 1980.
Гольдинов A. JI., Копылев Б. А., Абрамов О. Б., Дмитриевский Б. А. Комплексная азотно-кислотная переработка фосфатного сырья.— Л.: Химия, 1982. 207 с.
Денисов Е. Т. Кинетика галогенных химических реакций.— М.: Высшая школа, 1978. 367 с.
Downs A. J., Adams С. J. The chemistry of chlorine, bromine, iodine and astatine. Oxford, 1975.
Егоров Л. Ф., Осыка В. Г., Пивоваров М. Д. Технология получения бромжелеза из природных рассолов.— М.: Химия, 1979. 197 с.
Ж и гач А. Ф., Стасиневич Д. С. Химия гидридов.— Л.: Химия, 1969. 676 с.
Запольская М. А., Зенкевич Н. Г., Комарова Е. Г. Физико-химические свойства фтористого водорода.— М.: Химия, 1977. 119 с.
Зимин В.М., Камарьяи Г.М., Мазанко А.Ф. Хлорные электролизеры.— М.: Химия, 1984^304 с.
Кононов А. В., Стсрлнн В. Н., Евдокимова Л. И. Основы технологии комплексных удобрений.— М.: Химия, 1988. 319 с.
Ксензенко В. И., Стасиневич Д. С. Химия и технология брома, иода и их соединений.— М.: Химия, 1995. 432 с.
Кубасов В.Л., Банников В.В. Электрохимическая технология неорганических веществ.— М.: Химия, 1989, 288 с.
Мельник Б. Д., Мельников Е. Б. Краткий инженерный справочник по технологии неорганических веществ.— М.: Химия, 1968. 432 с.
529
Набиев М. Н. Азотнокислотная переработка фосфатов.— Ташкент.: ФАН, 1976, В 2-х т. Т. I, II. 820 с.
Некрич М. И., Ковалев М. П., Черняева Ю. И. Общая химическая технология.— Изд-во Харьковского университета, 1969. 336 с.
Познн М. Е. Технология минеральных солей.— Л.: Химия, 1974. В 2-х ч. Часть I и II. 1558 с.
Позин М. Е. Физико-химические основы неорганической технологии.— Л.: Химия, 1985. 384 с.
Поз и и М. Е. Технология минеральных удобрений.— Л.: Химия, 1989. 352 с.
Раков Э. Г. Химия и технология неорганических фторидов.— М.: Химия, 1990. 197 с.
Рысс И. Г. Химия фтора и его неорганических соединений.— М.: ГХИ, 1956. 113 с.
Роде Т. В. Кислородные соединения хрома и хромовые катализаторы.— Изд-во АН СССР, 1962. 198 с.
Ролстен Р. Ф. Иодидные металлы и иодиды металлов / Пер. с англ. М., 1968.
Ротинян А. Л., Филатов В. П., Цибитов Г. В. Оптимизация производства хлора.— М.: Химия, 1980/272 с.
Соколовский А. А., Уняианц Т. П. Краткий справочник по минеральным удобрениям.— М.: Химия, 1977. 376 с.
Сухотин А. М. Справочник по электрохимии.— Л.: Химия, 1981 488с.
Фримантл М. Химия в действии. В 2-х т. Т. I, И.— М.: Мир 1991.
Харлампович Г. Д., Залевский А. А., Новиков А. А., Поляков Ю. Д. Развитие промышленности минеральных удобрений.— М.: Химия, 1984. 352 с.
Химическая энциклопедия. Изд-во Большая Российская энциклопедия.— М., 1988—1999.
Якименко Л. М. Производство хлора, каустической соды и неорганических хлор-продуктов.— М.: Химия, 1974 600 с.
Якименко Л. М. Получение водорода, кислорода, хлора и щелочей.— М.: Химия, 1981 280 с.
ОГЛАВЛЕНИЕ
Предисловие..........................................................3
Глава 1. Мышьяк и его соединения.....................................5
1.1. Оксиды мышьяка..................................................7
1.2. Мышьяковая кислота.............................................13
1.3. Арсенаты.......................................................15
1.4. Арсенат (V) кальция............................................24
1.5. Арсениды..................................................... 33
1.6. Халькогениды мышьяка...........................................36
1.7. Галогениды мышьяка............................................ 38
1.8. Гидрид мышьяка.................................................41
Глава 2. Сера и ее соединения.......................................44
2.1. Элементная сера................................................44
2.2. Оксиды серы....................................................71
Глава 3. Хром и его соединения.....................................108
3.1. Оксиды и гидроксиды хрома.....................................112
3.2. Хроматы.......................................................145
3.3. Галогениды хрома..............................................188
3.4. Сульфаты хрома................................................194
Глава 4. Фтор и его соединения.....................................203
4.1. Фторид водорода и фтороводородная кислота.....................206
4.2. Технология фторидов...........................................218
Глава 5. Хлор и его соединения.....................................231
5.1. Хлор..........................................................231
5.2. Оксиды хлора..................................................259
5.3. Кислородные кислоты хлора.....................................269
Глава 6. Бром и его соединения.....................................306
6.1. Бром..........................................................306
6.2. Бромид водорода и бромоводородная кислота.....................331
6.3. Бромиды.......................................................335
6.4. Броматы натрия и калия........................................340
Глава 7. Иод и его соединения......................................343
7.1. Иод...........................................................343
7.2. Технология иода...............................................349
531
7.3. Иодид водорода и иодоводородная кислота.......................
7.4. Получение иодидов.............................................
7.5. Получение йодатов и перйодатов щелочных металлов..............
Глава 8. Марганец и его соединения.................................
8.1. Марганец......................................................
8.2. Химическая переработка марганцевых руд........................
8.3. Оксиды и гидроксиды марганца..................................
8.4. Диоксид марганца..............................................
8.5. Сульфат марганца..............................................
8.6. Карбонил марганца.............................................
8.7. Карбонат марганца.............................................
8.8. Хлорид марганца...............................................
8.9. Нитрат марганца (II)..........................................
8.10. Мажеф........................................................
8.11. Манганаты....................................................
8.12. Марганцевые пигменты.........................................
8.13. Марганцевая фиолетовая.......................................
Глава 9. Железо и его соединения...................................
9.1. Железо........................................................
9.2. Карбонилы железа..............................................
9.3. Сульфиды железа...............................................
9.4. Нитраты железа................................................
9.5. Фосфаты железа................................................
9.6. Хлориды железа................................................
9.7. Сульфаты железа...............................................
9.8. Оксиды и гидроксиды железа....................................
9.9. Железная лазурь...............................................
9.10. Гексацианоферраты (II. 111)..................................
Глава 10. Кобальт и его соединения.................................
10.1. Кобальт......................................................
10.2. Оксиды и гидроксиды кобальта.................................
10.3. Карбонилы кобальта...........................................
10.4. Галогениды кобальта..........................................
10.5. Карбонаты кобальта...........................................
10.6. Нитраты кобальта.............................................
10.7. Сульфаты кобальта............................................
10.8. Кобальтовые пигменты.........................................
10.9. Алюминат кобальта............................................
Глава 11. Никель и его соединения..................................
11.1. Никель.......................................................
11.2. Оксиды и гидроксиды никеля...................................
11.3. Сульфат никеля...............................................
11.4. Галогениды никеля............................................
11.5. Тетракарбонил никеля.........................................
11.6. Сульфиды никеля..............................................
11.7. Карбонат никеля..............................................
11.8. Нитрат никеля................................................
532
Глава 12. Санитарно-технические характеристики неорганических веществ и промышленная безопасность..................509
12.1. Соединения мышьяка ...........................................509
12.2. Соединения хрома..............................................511
12.3. Соединения фтора..............................................512
12.4. Соединения хлора..............................................515
12.5. Соединения брома..............................................516
12.6. Соединения иода..............................................517
12.7. Соединения марганца..........................................518
12.8. Соединения железа............................................520
12.9. Соединения кобальта..........................................522
12.10. Соединения никеля............................................524
12.11. Сера и ее соединения.........................................526
Рекомендуемая литература............................................529
Учебное издание
Ахметов Тимерхан Габдуллович Порфирьева Резида Тимерхановна Гайсин Ленар Гайнуллович Ахметова Лилия Тимерхановна Хацринов Алексей Ильич
ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ Кинга 2
Редактор Т. С. Костян Художник К Э. Семенков Художественный редактор Ю. Э. Иванова Технический редактор Л. А. Овчинникова Корректор Г. И Буханова Компьютерная верстка Н. И. Журавлевой
Лицензия ИД № 06236 от 09.11.2001
Изд. № Х/Е-247’. Сдано в набор 25.06.2001. Подл, в печать 15.05.2002
Формат 60x88’/16. Бум. офсетная. Гарнитура «Таймс». Печать офсетная Объем: 32,83 усл. печ. л., 33,33 усл. кр.-отт., 32,86 уч.-изд. л.
Тираж 5000 экз. Заказ № 1257
ФГУП «Издательство «Высшая школа»
127994, Москва, ГСП-4, Неглинная ул., д. 29/14 Тел. (095) 200-04-56
E-mail: info@v-shkola.ru http://www.v-shkola.ru
Отдел реализации: тел.: (095) 200-07-69, 200-59-39, факс: (095) 200-03-01 E-mail: sales@v-shkola.ru
Отдел «Книга-почтой»: тел. (095) 200-33-36. E-mail: bookpost@v-shkola.ru
Отпечатано во ФГУП ИПК «Ульяновский Дом печати» 432980, г. Ульяновск, ул. Гончарова, 14