/




































































































































































































































































































































































































































































































































































































































































































Автор: Ахметов Т.Г. Порфирьева Р.Т.
Теги: неорганическая химия общая и неорганическая химия неорганические вещества химическая технология
ISBN: 5-06-004244-8
Год: 2002




Текст
технология
неорганических
х .жЖимическая
технология неорганических веществ
Под редакцией академика АНТ РТ, профессора Т.Г. Ахметова
В двух книгах книга 1
Допущено
Министерством образования Российской Федерации в качестве учебного пособия для студентов высших учебных заведений, обучающихся по специальности "Химическая технология неорганических веществ" направления подготовки дипломированных специалистов "Химическая технология неорганических веществ и материалов ”
льду
Москва "Высшая школа" 2002
УДК 546 ББК 24.1
X 46
Авторы:
Т. Г. Ахметов, Р. Т. Порфирьева, Л. Г. Гайсин, Л. Т. Ахметова, Я. М. Каримов, А.И. Хацринов
Рецензенты:
проф. К. Ф. Ткачев, Ю. С. Плышевский (гп. специалисты УНИХИМ, г. Екатеринбург), кафедра «Технология неорганических веществ» РХТУ им. Д. И. Менделеева (зав. каф. проф. А. И. Михайличенко)
Химическая технология неорганических веществ: В 2 кн. Кн. 1.
X 46 Учебное пособие / Т. Г. Ахметов, Р. Т. Порфирьева, Л. Г. Гайсин и др.; Под ред. Т. Г. Ахметова.— М.: Высш, шк., 2002.— 688 с.: ил.
ISBN 5-06-004244-8 (кн. 1)
В книге даны сведения по технологии соединений натрия, калия, меди, стронция, цинка, бо-ра, алюминия, свинца, титана, азота, фосфора. Рассмотрены вопросы промышленной безопасности и санитарно-технических норм описанных производств. Приводится описание физико-химических основ и конкретных способов их получения.
Книга может быть полезной преподавателям и студентам кафедр химической технологии неорганических веществ, электрохимии, охраны труда и безопасности жизнедеятельности.
Для студентов высших учебных заведений, обучающихся по направлению «Химическая технология неорганических веществ».
УДК 546
ББК 24.1
Учебное издание
Ахметов Тимерхан Габдуллович, Порфирьева Резида Тимерхановна, Гайсин Ленар Гайнулович Ахметова Лилия Тимерхановна, Каримов Ягафар Мухтарович, Хацринов Алексей Ильич
ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
Книга 1
Редактор Т. С. Костян. Художник И.Д. Драгунская.
Художественный редактор Ю. Э. Иванова. Технический редактор Л. А. Овчинникова.
Корректор Г. Н. Буханова. Компьютерная верстка Н. И. Журавлевой
Лицензия ИД № 06236 от 09.11.2001. Изд. № Х/Е-247. Сдано в набор 25.06.2001
Подп. в печать 22.03.2002. Формат 60x88 716. Бум. офсетная. Гарнитура «Таймс» Печать офсетная. Объем: 42,14 усл. печ. л., 42,64 усл. кр.-отт., 43,30 уч.-изд. л.
Тираж 5000 экз. Заказ № 1258
ФГУП «Издательство «Высшая школа». 127994, Москва, ГСП-4, Неглинная ул., д. 29/14
Тел. (095) 200-04-56
E-mail: info@v-shkola.ru http://www.v-shkola.ru
Отдел реализации: тел.: (095) 200-07-69, 200-59-39, факс: (095) 200-03-01 E-mail: sales@v-shkola.ru
Отдел «Книга-почтой»: тел. (095) 200-33-36. E-mail: bookpost@v-shkola.ru
Отпечатано во ФГУП ИПК «Ульяновский Дом печати». 432980, г. Ульяновск, ул. Гончарова, 14
ISBN 5-06-004244-8 (кн.1)
ISBN 5-06-004147-6
Оригинал-макет данного издания является собственностью издательства «Высшая школа», и его репродуцирование (производство) любым способом без согласия издательства запрещается
© ФГУП «Издательство «Высшая школа», 2002
Предисловие
Отсутствие современных пособий по технологии неорганических веществ для подготовки специалистов химической промышленности и необходимость систематизации результатов научно-технических исследований и достижений в этой области за последние 20 лет обусловили издание учебного пособия «Химическая технология неорганических веществ», выпущенного издательством «Химия» в 1998 году. Предлагаемая вниманию читателей книга представляет продолжение предыдущего учебного пособия, охватывает технологии неорганических соединений, не вошедших в него, и предназначена в качестве учебного пособия для студентов высших учебных заведений по специальности 250200 «Технология неорганических веществ». При подготовке книги авторы исходили из требований государственного образовательного стандарта к данной специальности и использовали опыт преподавания этой дисциплины в высших учебных заведениях России.
Настоящее пособие не имеет аналогов в отечественной и зарубежной химической литературе. Оно охватывает все разделы неорганической химической технологии. В нем собраны и систематизированы разбросанные в литературе данные по физико-химическим свойствам как самого химического элемента, так и его оксидов, гидроксидов, карбонатов, сульфидов, хлоридов, сульфатов и т.д. с дальнейшим изложением физико-химических основ технологий. Представлены результаты исследований в области технологии неорганических соединений последних лет в России и за рубежом, в том числе результаты исследований самих авторов.
В первой книге приведены данные по технологии соединений натрия, калия, меди, стронция, цинка, бора, алюминия, свинца, титана, азота и фосфора. В последнем разделе приводятся сведения по санитарно-техническим требованиям, предъявляемым к современному химическому производству.
Гл. 1 пособия написана Л.Г.Гайсиным и Я.М.Каримовым, гл. 2 — Т.Г.Ахметовым и Л.Т.Ахметовой, гл. 3 — Я.М.Каримовым и Л.Т.Ахметовой, гл. 4 — Л.Г.Гайсиным и Я.М.Каримовым, гл. 5 и
3
6 — Л.Т.Ахметовой и А.И. Хацриновым, гл. 7 — Т.Г.Ахметовым и Р.Т.Порфирьевой, гл. 8 — Л.Г.Гайсиным и Л.Т.Ахметовой, гл.
9 — Р.Т.Порфирьевой, гл. 10 — Л.Т.Ахметовой и А.И. Хацриновым, гл. 11—Р.Т.Порфирьевой и А.И. Хацриновым, гл. 12 — Л.Г.Гайсиным и Я.М.Каримовым. Содержание и текст всех разделов пособия в целом авторы обсуждали вместе.
Авторы выражают благодарность Минзиле Минзагитовне Зариповой, Асии Мансуровне Бажановой, Наталье Николаевне Бирюлиной за огромную работу, проведенную при подготовке данного пособия к печати.
Считаем также приятным долгом выразить глубокую благодарность профессорам А.И.Михайличенко (зав. кафедрой ТНВ М), Ю.И.Шумяцкому, И.А.Петропавловскому, С.А.Анурову, которые выполнили большой труд по рецензированию и сделали несомненно ценные замечания, способствующие улучшению качества рукописи.
Авторы будут благодарны всем читателям за конкретные замечания и пожелания, которые будут учтены в дальнейшем.
Авторы
ГЛАВА 1
НАТРИЙ И ЕГО СОЕДИНЕНИЯ
1.1. НАТРИЙ
Содержание натрия в земной коре составляет 2,64% (масс.). В водах океанов, морей и рек натрий содержится в виде растворимых солей в количестве около 2,9% при общей концентрации солей в морской воде 3,5—3,7%. Наличие натрия установлено также в атмосфере Солнца и межзвездном пространстве. В природе натрий встречается лишь в виде его солей. Важнейшими минералами являются: галит (каменная соль) NaCl, мирабилит (глауберова соль) НагЗОгЮНгО, тенардит Na2SC>4, чилийская селитра NaNO3, криолит Na3[AlF6], трона NaHCO3-Na2CO3-2H2O, бура (тинкал) ИагВ^-ЮНгО, альбит Na[AlSi3Og], нефелин Na[AlSiO4]. Соединения натрия входят в состав живых организмов, главным образом в виде его хлорида.
Свойства. Натрий имеет атомную массу 22,98977. В природе один стабильный изотоп 23Na. Конфигурация внешней электронной оболочки 3s1; степень окисления +1; энергия ионизации Na°->Na+->Na2+ 5,13915 и 47,304 эВ; атомный радиус 0,192 нм, ионный радиус 0,116 нм (координационное число 6), 0,153 нм (12).
Натрий — серебристо-белый металл, в тонких слоях с фиолетовым оттенком. Выше -222° С устойчива модификация с кубической решеткой (а - 0,4291 нм, z = 2, пространственная группа 1тЗт); при -222° С переходит в гексагональную форму (а = 0,3767 нм, с = 0,6154 нм, z = 2, пространственная группа бР^ттс). Плавится при 97,86° С, а кипит при 883,15° С; плотность 0,96842 г/см3(19,7° С), уравнения температурной зависимости плотности: (7=0,9725-20,11 -MF4#-!,5-Ю'7? г/см3 (98—1370° С); С ° = 28,23 Дж/(моль К), для газа 20,79 Дж/(моль-К); = 2,6 кДж/моль, АЯ°ип = 88,99 кДж/моль, ДН^. = 107,5 кДж/моль (298,15 К); = 51,30
Дж/(моль-К), для газа 52°98 = 153,61 Дж/(моль-К).
В газообразном состоянии натрий состоит из частиц Na и Na2. Содержание Na2 (Л//°Обр для газа 142,3 кДж/моль) увеличивается с ростом температуры: 0,8% (600 К), 1,3% (650 К), 2,5% (750 К).
Натрий очень активный элемент. На воздухе он окисляется до оксида Na2O или пероксида Na2O2. Энергично реагирует с водой с выделе-5
нием водорода и образованием гидроксида натрия. При большой поверхности контакта реакция идет со взрывом. Натрий воспламеняется и горит в атмосфере фтора и хлора при обычной температуре. С бромом реагирует лишь при нагревании, а с иодом в обычных условиях не реагирует. Энергично реагирует с серой, селеном и теллуром, образуя халькогениды составов Na2X, NaX, NaX2 и др. Благородные газы незначительно растворяются в твердом и жидком натрии, при 200° С натрий поглощает водород, образуя гидрид. Натрий с азотом в электрическом разряде дает нитрид Na3N или азид NaN3. Натрий реагирует с разбавленными кислотами, образуя соли. Легко растворяется в жидком аммиаке, образуя раствор синего цвета с металлической проводимостью, содержащий катионы натрия и сольватированные электроны; взаимодействуя с аммиаком при 300—400° С или в присутствии катализатора в процессе охлаждения до -30° С, натрий образует амид NaNH2; в интервале 800—900° С натрий с углеродом образует карбид натрия (Na2C2), а реакцией с ацетиленом при 98° С получают NaCsCH и этилен. С графитом натрий образует соединения с большим избытком углерода типа Ci20Na, C64Na, C36Na. Натрий образует также интер-металлиды с Ag, Au, Cd, Ga, Tl, Sn, Pb, Sb, Bi, К и Cs, a co ртутью— амальгамы — интерметаллиды состава NaHg2, NaHg4 и др. Процесс проводят при постепенном введении натрия в ртуть, находящуюся под слоем керосина или минерального масла.
Получение. Металлический натрий получают: 1) электролизом расплава хлорида натрия. С целью снижения температуры плавления электролита в исходное сырье вводят хлориды калия или кальция, фторид кальция; 2) электролизом расплава гидроксида натрия и др. Процесс электролиза хлорида натрия проводят в электролизерах с диафрагмой. Аноды изготовляют из графита, а катоды — из меди или железа. Образующийся металлический натрий очищают от примесей хлоридов, оксидов, кальция и углерода введением в расплав натрия смеси NaOH—Na2CO3—NaCl или пероксида натрия с дальнейшей обработкой расплава металлическими литием, титаном или сплавом Ti—Zr, низшими хлоридами титана и вакуумной дистилляцией.
Применение. Натрий и его сплавы с калием применяются в качестве жидких теплоносителей в ядерных реакторах. Натрий в виде его паров применяют для наполнения газоразрядных ламп, а сплавы со свинцом — в производстве тетраэтилсвинца и подшипников. Натрий применяют в качестве модификатора алюминиевых и других сплавов, восстановителя в металлургическом производстве титана, циркония и тантала. Он является катализатором в процессе получения бутадиенового каучука. Амальгама натрия является исходным сырьем для получения гидроксида натрия высокой чистоты; изотоп 24Na применяется в процессе радиологического лечения некоторых 6
форм лейкемии и в диагностических целях. Изотоп 22Na является позитронным источником.
Натрий металлический хранят под слоем инертной обезвоженной жидкости (керосин, очищенное минеральное масло и т.п.), перевозят в запаянных металлических емкостях.
1.2. ОКСИДЫ НАТРИЯ
Для натрия образование пероксидных соединений более характерно, чем для других щелочных металлов. Поэтому, реагируя с кислородом, натрий образует не оксид, а пероксид:
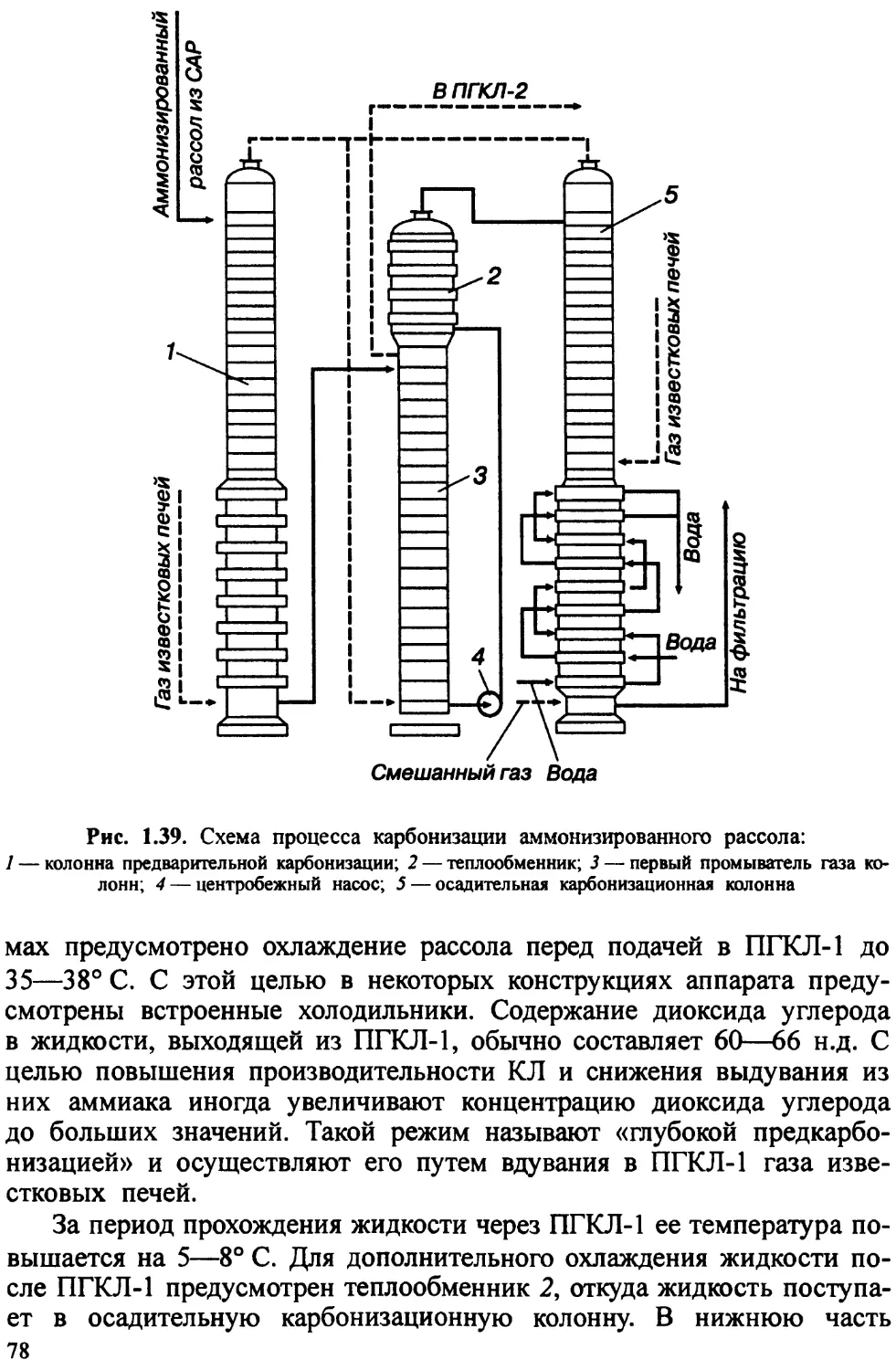
2Na + O2=Na2O2
а оксид получают восстанавливая пероксид металлическим натрием:
Na2O2 + Na = NaO
Из оксидов натрия известен также малостойкий озонид NaO3.
Оксид натрия Na2O существует в а-, Р- и у-модификациях. а-Форма, бесцветные кристаллы кубической сингонии (а = 0,556 нм, z-4, пространственная группа Fm3m) при 750° С переходит в кубическую p-модификацию, которая в свою очередь при 970° С переходит в кубическую у-модификацию.
Оксид натрия плавится в инертной атмосфере при 1132° С. Процесс плавления оксида натрия сопровождается сублимацией. В газообразном состоянии частично диссоциирует на натрий и кислород.
а-Модификация оксида натрия: плотность 2,37 г/см3, С° = 69,0 Дж/(моль-К), АЯХ =-414,8кДж/моль, AG^p =-376,1 кДж^оль, АЯ^, =36,0 кДж/моль, - 75,3 Дж/(моль-К).
Оксид натрия реагирует:
с водой Na2O + Н2О = 2NaOH
с серой 4Na2O + 4S = 3Na2S + Na2SO4
с диоксидом углерода Na2O + СО2 = Na2CO3
с аммиаком Na2O + NH3 = NaNH2 + NaOH
с водородом Na2O + Н2 = NaH + NaOH
с хлором 2Na2O + Cl2 = 2NaCl + Na2O2
Оксид натрия в процессе нагревания на воздухе до 400° С образует пероксид 2Na2O + О2 = 2Na2O2.
Оксид натрия получают по следующим реакциям, протекающим при нагревании:
5NaN3 + NaNO3 = 3Na2O + 8N2
7
2NaOH + 2Na = 2Na2O + H2 3NaN3 + NaNO2 = 2Na2O + 5N2 2NaNO2 + 6Na = 4Na2O + N2
Оксид натрия хранят в безводном бензоле и применяют в качестве исходного сырья для различных синтезов.
Пероксид натрия Na2O2, бесцветные кристаллы. Технический пероксид натрия слабо-желтого цвета, что показывает присутствие в составе пероксида натрия примеси NaO2. Существует в а- и 0-моди-фикациях. До 512° С существует p-Na^ с гексагональной кристаллической решеткой (а = 0,6208 нм, с = 0,4469 нм, z = 3, пространственная группа Р62т). Выше 512° С устойчив a-Na2O2, ДЯ перехода Р 5,36 кДж/моль; плотность 2,61 г/см3; С® = 89 Дж/(моль-К); ДЯ£,= 17 кДж/моль, =-512 кДж/моль; 5^8=94 Дж/(моль-К).
Пероксид натрия — сильный окислитель, при 311 — 400°С начинает выделять кислород, а при 540° С бурно разлагается. Растворяется в воде с образованием NaOH, Н2О и О2. Образует кристаллогидраты Na2O2-2H2O, Na2O2-4H2O, Na2O2-8H2O и Na2O2-2H2O2-4H2O. Реагирует с влагой и диоксидом углерода воздуха:
2Na2O2 + Н2О + СО2 = 2NaOH + Na2CO3 + О2
cN2O4 Na2O2 + N2O4 = 2NaNO3 cNO Na2O2 + 2NO = 2NaNO2 c SO2 Na2O2 + SO2 = Na2SO4
Пероксид натрия получают в производстве окислением расплавленного натрия кислородом воздуха, после чего очищают его от диоксида углерода и сушат.
Пероксид натрия применяют в подводных лодках и дыхательных приборах изолирующего типа для регенерации воздуха от диоксида углерода по реакции
2Na2O2 + 2СО2 = 2Na2CO3 + О2
Водные растворы пероксида натрия применяют для отбеливания бумаги, тканей и целлюлозы.
Надпероксид натрия (супероксид натрия, гипероксид натрия) NaO2, желтые кристаллы; выше -50° С устойчива фаза I кубической сингонии (а = 0,549 нм, z = 4, пространственная группа Fmlm), в интервале от -50 до -77° С — кубическая фаза II (а = 0,546 нм, простран-8
ственная группа РаЗ), в интервале от -77 до -230°С — ромбическая фаза III (при -100°С а = 0,426 нм, b = 0,554 нм, с - 0,334 нм, пространственная группа Рппт), ниже -230°С — фаза IV; плотность 2,21 г/см3; С° = 71,88 Дж/(моль-К), =-261 кДяЛюль, =-219 кДж/Ьюль.
В процессе непрерывного нагревания со скоростью 5 град/мин над-пероксид натрия около 100° С начинает разлагаться с выделением кислорода, основная часть которого выделяется с достижением 240—300° С. Твердым продуктом распада является пероксид водорода. На процесс распада влияет дефектность кристаллов исходного продукта.
Надпероксид натрия растворяется в жидком аммиаке (~0,5 г в 100 мл). При -32,5° С из раствора выпадает осадок — аммиакат:
NaO2 + 2NH3 = NaOr2NH3
Растворение надпероксида натрия в воде сопровождается выделением кислорода и образованием гидроксида натрия и пероксида водорода:
2NaO2 + 2Н2О = 2NaOH + Н2О2 + О2
В присутствии катализатора (МпО2 и др.) или в процессе нагревания реакция с водой протекает по схеме
4NaO2 + 2Н2О = 4NaOH + ЗО2
Надпероксид натрия — сильный окислитель. В обычных условиях в присутствии паров воды он реагирует с диоксидом серы:
Na2O2 + SO2 = Na2SO4
с диоксидом азота:
Na2O2 + 2NO2 = 2NaNO3
В процессе нагревания при 100—180°С надпероксид натрия поглощает оксид углерода, образуя карбонат:
Na2O2 + СО = Na2CO3
В присутствии паров воды при комнатной температуре диоксид углерода полностью вытесняет активный кислород из надпероксида натрия:
2Na2O2 + 2СО2 — 2Na2CO3 + О2
9
В отсутствие влаги эта реакция идет лишь при температуре выше 100° С.
Надпероксид натрия в промышленности получают медленным нагреванием пероксида натрия от 200 до 450° С при давлении кислорода 10—15 МПа. Разработан также способ получения надпероксида натрия окислением кислородом металлического натрия в среде 1,2-диметокси-этана в присутствии флуорена или бензофенона при обычных условиях.
Надпероксид натрия применяется в качестве компонента систем для регенерации кислорода в замкнутых помещениях.
1.3. ХЛОРИД НАТРИЯ
Хлорид натрия (поваренная соль, каменная соль) NaCl, бесцветные, мало гигроскопичные кристаллы с кубической гранецентрированной решеткой (а = 0,56402 нм, z = 4, пространственная группа Fm3m)\ плотность 2,161 г/см3 (20° С); температура плавления 801° С; температура кипения 1413° С; С° = 50,50 Дж/(моль-К); АЯП°Л= 28,20 кДж/моль, ДН^р=-411,26 кДж/моль; 5^ =72,15 Дж/(моль-К). В равновесном па
ре содержится 83 моль % NaCl и 17% Na2C12. Растворимость в воде (г в 100 г): 35,68 (10° С), 35,87 (20° С), 36,80 (50° С), 38,12 (80° С). При температуре выше +0,15°С хлорид натрия существует в виде безводной соли. Как видно из рис. 1.1, дигидрат хлорида натрия кристаллизует-
Рис. 1.1. Растворимость в системе NaCl — Н2О
ся в интервале от -21,2 до 0,15°С. Плотность дигидрата НаС1-2НгО 1,6 г/см3; давление водяного пара над ним изменяется от 91,77 (-21,2° С) до 462,84 Па (0,15° С). Насыщенный водный раствор (28,41% (масс.) NaCl) кипит при 108,7° С.
Хлорид натрия применяют в качестве исходного сырья более чем 1500 производств различных веществ и материалов. Хлорид натрия — пищевой продукт, консервирующее средство, применяется в качестве сырья в производстве карбоната натрия, хлора, гидроксида натрия, хлорной извести и др. Мировое производство хлорида натрия в 1980 г. составляло около 175 млн. т/год, а средняя годовая норма пищевого потребления хлорида натрия на одного человека составляет 8—10 кг, а общее потребление, включая производственное, в отдельных странах достигает 25—75 кг в год.
ю
В качестве корма в животноводстве расходуется 5—10% произведенного хлорида натрия.
По способу добычи и обработки пищевая поваренная соль делится на: 1) мелкокристаллическую — выварочную; 2) молотую разных видов (каменная, самосадочная) и различной крупности помола; 3) немолотую — комовую (глыба), дробленку и зерновую (ядро); 4) иодированную. Требования к качеству пищевой соли регламентируются ГОСТ 13830—68 (табл. 1.1).
Таблица 1.1. Состав пищевой соли (в %) по ГОСТ 13830—68
Сорт Содержание NaCl в сухом веществе, не менее Содержание не растворимых в воде веществ в сухом веществе, не более Максимальное содержание влаги Предельное содержание в сухом веществе
Са2+ Mg2+ so? Fe2O3
Экстра 99,7 0,03 0,1 0,02 0,01 0,16 0,005
Высший 98,4 0,16 Каменная соль 0,25 садочная и само- д^ садочная ’ выварочная 5,0 0,35 0,05 0,8 0,005
Первый 97,7 0,45 Каменная 0,25 садочная и само- ^д садочная ’ выварочная 5,0 0,5 0,1 1,2 0,01
Второй 97,0 0,85 Каменная 0,25 садочная и самоса- , g дочная 9 выварочная 6,0 0,65 0,25 1,5 0,01
Способы получения хлорида натрия. Основными сырьевыми источниками хлорида натрия являются: 1) пласты и штоки «каменной» соли; 2) океанская и морская вода, озерные рассолы; 3) растворы хлорида натрия в соляных источниках и грунтовых водах; 4) солончаки; 5) возгоны хлорида натрия на кратерах вулканов и в трещинах лавовых потоков.
Крупные месторождения хлорида натрия: Илецкое — в Оренбургской области, его пласт разведан на глубину 1,5 км; Усольское — в Иркутской области. В Прикаспийской низменности насчитывается около 2000 озер, в том числе Баскунчак и Эльтон. Баскунчак имеет овальную форму площадью около 115 км2. При этом запасы соли в озере за счет вод впадающих в него источников возрастают ежегодно более чем на 800 тыс.т или на ~100 т в час. Наибольшее количество хлорида натрия находится в морской воде.
Методы добычи хлорида натрия-сырца разделяются на следующие основные группы: 1) добыча хлорида натрия (каменной соли);
п
2) добыча самосадочной соли из соленых озер; 3) добыча садочного хлорида натрия бассейновым способом из морской и озерной вод; 4) получение выварочного хлорида натрия путем выварки ее из естественных и искусственных рассолов.
Для технических целей применяют каменную и самосадочную соль, а для пищевого хлорида натрия — выварочную, самосадочную и садочную соль. Кроме этого производят специальные сорта соли— иодированную, брикетированную и исслеживающуюся, а также хлорид натрия высокой чистоты (99,9% и выше NaCl).
Каменная саль встречается в природе в виде пластов, штоков и линз, достигающих толщину сотен и тысяч метров. Обычно она имеет плотное компактное строение и сопровождается залежами и мощными толщами ангидритовых, гипсовых и других пород. Поэтому каменная соль содержит такие примеси, как глину, ангидрит, битумы и др.
Подземную добычу каменной соли ведут обычно на глубине до 500 м. Разработку осуществляют в камерах до 30 м высотой без креплений.
Самосадочная соль, добыча которой составляет половину добываемой соли, имеет различный минеральный состав. Обычно она кроме хлорида натрия содержит хлориды калия, магния, а также сульфаты магния и кальция.
В процессе испарения рапы из хлорид-сульфатных озер вначале выделяются карбонаты кальция и магния, затем гипс, а далее хлорид натрия. Состав некоторых образцов самосадочной и бассейной поваренной соли приведен в табл. 1.2.
Таблица 1.2. Состав образцов самосадочной и бассейной поваренной соли (в %)
Соль NaCl СаС12 MgCl2
Крымская бассейная 98,16—99,17 —— 0,05—0,4
Туркменская самосадочная (оз.Куули) 96,7—98,3 — 0—1,14
Баскунчакская самосадочная 97,22—99,32 0—0,24 0,48
Павлодарская самосадочная 97,91—99,22 — 0,01—0,44
Соль CaSO4 MgSO4 Нерастворимый осадок
Крымская бассейная 0,3—0,95 0—0,3 0,05—0,14
Туркменская самосадочная (оз.Куули) 0,24—1,27 0—0,59 0,19—0,5
Баскунчакская самосадочная 0—0,85 0-0,25 0,07—0,6
Павлодарская самосадочная 0,01—0,43 0,03—0,24 0,6—0,7
12
Получение выварочной соли. Сырьем для получения выварочной соли являются естественные рассолы, добываемые из недр земли. Они отличаются сравнительно высокой концентрацией хлорида натрия и небольшим содержанием примесей. Обычно исходный рассол содержит (в г/л): NaCl — 280—310; MgCl2 и MgSO4 — 0,2—4; CaSO4 — 5—6; СаС12— 0,2—0,8, плотность рассола равна 1,19—1,20 г/см3 (при 15° С).
Для получения более чистого хлорида натрия, а также предотвращения процесса инкрустации аппаратов от накипи рассолы перед выпаркой очищают от ионов Са2+, Mg2* и SC>42‘.
Существует несколько способов очистки рассолов: 1) очистка карбонатом натрия, осаждением из рассолов ионов кальция и магния; 2) известково-содовый, введением в рассол извести и карбоната натрия, очищая одновременно от солей кальция и магния; 3) извест-ково-сульфатно-содовый, осуществляемый в две стадии. В первой стадии в исходный рассол вводят сульфат натрия и известь, освобождая рассол тем самым от растворимых солей магния и кальция. Во второй стадии рассол освобождается от гипса путем карбонизации его диоксидом углерода или введением карбоната натрия.
В процессе очистки рассола этим способом идут следующие реакции:
Na2SO4 + Са(ОН)2 # CaSO4 + 2NaOH
MgCl2 + 2NaOH Mg(OH>2 + 2NaCl
Первая стадия
MgSO4 + 2NaOH -> Mg(OH)2 + Na2SO4
CaCl2 + NaOH -> Са(ОН)г + 2NaCl
Вторая стадия
2NaOH + CO2 = Na2CO3 + H2O
CaSO4 + Na2CO3 = CaCO3 + Na2SO4
Очистка исходных рассолов карбонатом натрия наиболее проста. Однако она пригодна лишь для рассолов, содержащих незначительные количества магний-иона, так как Mg2+ трудно осаждается карбонатом натрия. Очистка этим способом протекает наиболее удовлетворительно лишь при температуре около 100° С. Преимущество данного способа— в больших скоростях процесса отстоя рассола от осадка.
При замене части карбоната натрия его гидроксидом очистка дает хорошие результаты и без нагревания рассола — в процессе перемешивания в течение 30—60 мин. Концентрация катионов Са2+ и Mg2* в растворе NaCl может быть снижена до 30—40 ммоль/л.
13
Наилучшим способом, обеспечивающим интенсивную очистку рассолов при минимальном расходе карбоната натрия (~2—2,5 раза меньше, чем по известково-содовому способу), является известково-сульфатно-содовый способ, широко применяемый на современных предприятиях. В процессе выпарки рассолов морского типа при температуре кипения под атмосферным давлением с достижением насыщения кристаллизуется хлорид натрия. На рис. 1.2 приведена равновесная диаграмма растворимости при 100° С в водной системе 2NaCl + MgSO4 = Na2SO4 + MgCl2, состоящей из основных компонентов морской воды. Фигуративная точка солевой массы жидкой фазы по мере кристаллизации хлорида натрия движется из начального положения 1. В стабильной области кристаллизации осаждается около 70% NaCl и левеита — Na2SO4-MgSO4-2,5H2O (рис. 1.2, точка 2). В процессе дальнейшего выпаривания вместо смеси галита и левеита продолжает кристаллизоваться лишь галит в метастабильной области. Поле кристаллизации находится выше пунктирной линии. Задержка выделения сульфатов вследствие достаточно большой стойкости ме-тастабильного состояния повышает общую степень извлечения хлорида натрия при кипении раствора до 91—96%.
Выпаривание исходных рассолов осуществляют в чренах или в ва-куум-выпарных аппаратах. На чренных установках очистку рассола от примесей производят в процессе упаривания. Хлорид натрия получается в виде более крупных кристаллов, чем при вакуумной выпарке. Для выпарки хлорида натрия в вакуум-выпарных установках необходима предварительная очистка рассола от соединений кальция и магния.
Выпарной чрен представляет собой открытый прямоугольный резервуар с размерами: длина 15—20 м, ширина 8—10 м, высота 0,4—0,5 м. В процессе нагревания рассола в чрене до 80° С из него выделяются
MgSO4
Рис. 1.2. Изотерма растворимости системы NaCl—MgSO4—Н2О при 100° С
Рис. 1.3. Растворимость CaSO4 в насыщенном растворе NaCl
14
сульфид водорода и другие растворенные газы, а в осадок выпадает сульфат кальция (рис. 1.3). При температуре кипения (108° С) гидрокарбонат кальция разлагается до карбоната, который выделяется в осадок. Осадившиеся твердые примеси удаляются специальными гребками. С достижением насыщения начинает кристаллизоваться хлорид натрия. Соли магния остаются в растворе. Для получения мелких кристаллов целевого продукта температуру поддерживают в пределах 90—100° С, а крупнокристаллическую соль получают при 50—-60° С.
Кристаллы соли выгребают механизированными гребками и отжимают в центрифугах до влажности 3—5% или высушивают в сушилках.
На рис. 1.4 представлена схема механизированной чренной выпарки хлорида натрия. Насосом 2 нагнетают воду из реки или озера через наружные трубы скважины 1. За счет разности давления из скважины по ее внутренней трубе рассол поступает в наземный резервуар 3. Из резервуара 3 рассол насосом 4 перекачивают в бак 5, откуда регулируемым самотеком рассол поступает в круглый чрен с механизированной выгрузкой хлорида натрия. Параллельно с исходным рассолом в чрен поступают маточные растворы, образующиеся в процессе отжима кристаллов в центрифуге 14. Маточные растворы собираются в сборнике 16, откуда часть их направляется в бак 5 для смешения со свежими рассолами, после чего смесь поступает в круглый чрен. Часть маточных растворов из сборников 15 и 16 направля-
Рис. 1.4. Схема механизированной чренной выварки хлорида натрия:
1 — скважина; 2, 4 — насосы; 3 — резервуар; 5 — бак; б, 13 — сигнальные лампы; 7 — вентиль; 8— скребок; 9 — топки; 10 — мешалка; 11 — солесборник; 72, 18— вакуум-монтежю; 14—центрифуга; 75, 16— сборники; 77 — транспортер; 19—весы; 20 — ловушка; 27 — барометрический конденсатор; 22 — ресивер; 23 — вакуум-насос
15
ются также в вакуум-монтежю 18. Пары из вакуум-монтежю, пройдя через барометрический конденсатор 21 и ловушку 20, охлаждаются и через рессивер 22 под действием вакуума, создаваемого вакуум-насосом 23, удаляются в атмосферу. Образующиеся кристаллы в вакуум-монтежю загружаются в центрифугу 14, откуда отжатые кристаллы хлорида натрия подаются на сушку. Режим работы, особенно температура процесса в вакуум-монтежю, регулируется автоматически, что показывается сигнальными лампами 6 и 13.
Получение хлорида натрия вымораживанием. Хлорид натрия получают из концентрированных рассолов путем кристаллизации. При низких температурах из насыщенных рассолов кристаллизуется дигидрат хлорида натрия NaCl-2H2O. Для получения хлорида натрия его дигидрат извлекают из рапы. При повышении температуры воздуха выше +0,15° С он разлагается с переходом в чистый NaCl (см. рис. 1.1).
Дигидрат хлорида натрия выделяется в зимний период в соляных источниках, а также во многих озерах. Он практически не содержит примесей, и его вымораживание из рассола является одним из методов получения чистого хлорида натрия. В процессе плавления 1 т дигидрата при 25° С получают 481,5 безводного хлорида натрия с выходом 77,8%.
Получение хлорида натрия из галитовых отходов. В процессе переработки сильвинита с получением хлорида натрия в качестве отхода получается до 80% от массы исходного сырья, загрязненного примесями хлорида натрия. Отходы содержат до 92—96% NaCl, 1,2—2,5% КС1, 0,6—2% CaSO4, 0,05—0,2 MgCh и 0,3—3% не растворимых в воде веществ. Разработан наиболее простой и экономичный способ получения хлорида натрия из рассматриваемых отходов путем их промывки насыщенным раствором NaCl на реечном классификаторе. Полученный продукт содержит до 98% NaCl с содержанием до 0,3—0,32% КС1.
Разработан способ получения более чистой продукции растворением отходов, химической очисткой полученного рассола и его вакуум-выпар-кой. Разработан также способ получения хлорида натрия флотацией отходов, который имеет значительные преимущества перед вакуум-выпар-кой, так как не требует расхода дорогостоящего пара. Согласно способу, из отходов флотируют примеси. Одновременно флотация обеспечивает получение продукта с высоким содержанием основного вещества (99,7%) NaCl, но образующийся продукт загрязнен флотореагентами.
Разработан способ получения хлорида натрия хорошего качества путем промывки галитовых отходов от переработки верхнекамских сильвинитов. Согласно этой схеме (рис. 1.5), отходы смешиваются в смесителе 1, перекачиваются насосом 2 на дуговые сита 3, 5, а также на планфильтр 4 для отделения средней фракции размером 3±0,5 мм, содержащей наименьшее количество примесей. Эта фрак-16
ция при Ж:Т, равном 0,7, дробится на дробилке 7, смешиваясь с рассолами в смесителе 8, насосом 9 перекачивается на дуговые сита 10, после которых крупные частицы направляются в дробилку 7, а соответствующие помолу 0 с дуговых сит 10 поступают в трехступенчатые каскадные контактные чаны 12, в которых перемешиваются 30—40 мин. При этом растворимые составляющие переходят в раствор, а затем суспензия обесшламливается в гидросепараторах 13 и 75, просеиваясь на расположенном между ними дуговом сите 14. Загрязненный рассол направляют в отстойник 19, а сгущенная пульпа из горизонтальной мешалки 16 поступает в центрифугу 17. Отжатые кристаллы хлорида натрия транспортером 18 передают на сушку, а маточные растворы направляют в отстойник 19, откуда в сборник 20, из которого насосом 21 перекачиваются в напорный бак 11 на смешение с растворами, идущими на вакуум-выпарку.
Получаемый хлорид натрия по качеству соответствует первому сорту пищевой соли. Он содержит (в %): NaCl — 98,5—99,0; CaSO4—0,5—0,6; MgCl2 — 0,02—0,07; нерастворимый остаток -0,3—04; КС1 — отсутствует.
Техническая поваренная соль может быть получена и из отходов флотации хлорида кальция из сильвинита.
Рис. 1.5. Получение хлорида натрия первого сорта промывкой галитовых отходов: 1, 6 и 8 — смеситель; 2, 9 и 21 — насос; 3, 5, 10 и 14 — дуговые сита, 4 — планфильтр; 7 — дробилка; 11 — напорный бак; 12 — контактные чаны; 13 и 15 — гидросепараторы; 16 — горизонтальная мешалка; 17 — центрифуга; 18— транспортер; 19 — отстойник; 20 — сборный бак
17
Получение хлорида натрия высаливанием из раствора и кристаллизацией. Технологический процесс заключается в смешении рассолов с реактивами (MgCl2, СаС12), их фильтрации, охлаждении и кристаллизации, отделении выпавших кристаллов хлорида натрия и их сушке. В этом способе отсутствует потребность в топливе или паре на упарку растворов, а также предварительная очистка исходного рассола.
В процессе смешения 1 м3 рассола, содержащего 20,7% NaCl, 1,3% MgSO4, 4,3% MgCl2, 73,7% Н2О (пл. 1,23 г/см3), с одного кубометра хлоридомагниевого рассола, содержащего 27,6% MgCl2, 4,4% MgSO4, 1% NaCl, 67,0% Н2О (пл. 1,25 г/см3), выход кристаллизующегося хлорида натрия составляет около 78 кг.
Разработан способ перекристаллизации каменной соли, позволяющий производить чистый хлорид натрия смешением каменной соли с маточным раствором, остающимся после второй кристаллизации. Солевую суспензию обрабатывают острым паром, конденсация которого приводит к растворению кристаллов соли при 100—105° С. Нерас-творившаяся часть, содержащая примеси, отделяется в отстойнике, а горячий раствор направляют на кристаллизацию в две стадии — при охлаждении его до 80° С, а затем до 50° С. Соль из кристаллизаторов отжимают на центрифугах и высушивают.
Другой вариант заключается в обработке каменной соли циркулирующим насыщенным расвором хлорида натрия при температуре от 0 до -20° С. При этом соль переходит в дигидрат NaCl-2H2O, который отфильтровывают и нагревают. Дигидрат плавится в своей кристаллизационной воде и выделяется чистая соль. Маточные растворы возвращают на начало процесса.
Хлорид натрия сорта «Экстра» получают из сильно загрязненной ангидритом каменной соли перекристаллизацией из растворов хлорида кальция. Каменную соль перемешивают в течение 30 мин с исходным раствором, содержащим 24% СаС12. В кристаллизаторе раствор охлаждается от 112 до 20° С. Выход хлорида натрия из 1 м3 раствора составляет около 50 кг. Получаемая после промывки соль содержит 99,2% NaCl. Расход СаС12 (100%) при 50-кратном использовании составляет 11 кг на 1 т получаемой соли.
1.4. СУЛЬФАТ НАТРИЯ
Сульфат натрия Na2SO4, бесцветные кристаллы, известен в четырех полиморфных модификациях (табл. 1.3); показатели преломления: меньший Np= 1,469, средний Nm= 1,476, больший Ng =1,481.
Сульфат натрия встречается в природе в виде минералов: тенардита Na2SO4, мирабилита Na2SO4-10H2O, глауберита Na2Ca(SO4)2, вантгоффита NaeMg(SO4)4, глазерита Na2K«(SO4)4, астраханита Na2Mg(SO4)2-4H2O. 18
Тенардит при нагревании претерпевает ряд полиморфных превращений, а при 890° С плавится. Природный тенардит, полученный из горячего насыщенного раствора и высушенный при 110°С, имеет структуру, отличающуюся от переплавленного тенардита. Для последнего характерны следующие фазовые переходы:
хт ОГЛ 180-200-С . о 240-С . Х1 с _ 570-600-С . „
a-Na2SO4 < p-Na2SO4 < > y-Na2SO4 < 8-Na2SO4
В процессе нагревания тенардита, выделившегося из водных растворов, происходят следующие превращения:
Т-^о^ y-Na2SO4 < 57°-600'с > 3-Na2SO4
а в процессе охлаждения:
8-Na2SO4 < 57°~60°,С >Y-Na,SO4
p-Na2SO4 > a-Na2SO4 ->Т
Переход a-Na2SO4 в тенардит при обычных условиях происходит медленно в присутствии влаги. Полное обезвоживание сопровождается переходом в y-Na2SO4. С K2SC>4, Li2SC>3, Na2COa и рядом других соединений Na2SC>4 образует непрерывный ряд изоморфных твердых растворов, а с сульфатами скандия и иттрия — двойные соли, с сульфатом стронция — эвтектику.
Сульфат натрия кристаллизуется из водных растворов при температуре от 32,384° С до 233° С в ромбической системе, а выше 233° С — в моноклинной. Ниже 32,384° С выделяются прозрачные моноклинные кристаллы Na2SO4-10H2O (мирабилита), от — 3,5 до 24,25° С — метастабильный гептагидрат Na2SO4-7H2O (рис. 1.6). При 32,384° С мирабилит инконгруэнтно плавится — разлагается на безводный сульфат натрия и его насыщенный раствор. Растворимость сульфата натрия в воде (табл. 1.4) с повышением температуры от 32,384 до ~120°С уменьшается, затем возрастает, а выше 233° С резко снижается, приближаясь к нулю при критической температуре воды 365° С. Процесс растворения безводного сульфата натрия в воде сопровождается выделением теплоты вследствие гидратации, а растворение мирабилита протекает с поглощением теплоты, затрачиваемой на разрушение гидратных связей.
19
Таблица 1.3. Свойства сульфата и гидросульфата натрия
Показатели Na2SO4 Na2SO410H2O NaHSO4 NaHSOrHjO
Сингония Ромбическая Ромбическая Гексагональная Ромбическая Моноклинная Триклинная Моноклинная
Параметры элементарной ячейки, нм:
а 0,5863 0,933 0,5405 0,69666 1,1512 0,7005 0,8213
b 1,2304 0,548 — 0,89511 1,037 0,7125 0,7812
с 0,9821 0,702 0,724 0,56109 1,2847 0,6712 0,7805
угол, град — — — — 107,789 95,93(a) 92,31(р) 75,52(j) 120,04
Число формульных единиц в ячейке 8 4 2 4 4 4 4
Пространственная группа Fddd — Рбзтс — Р2,с РТ Да
Температура фазового превращения, °C 185* 241* >597* 884*** 1429**** (с разл.) — 186*** 58,5***
Плотность, г/см3 2,663 — — 2,696 1,465 2,476 2,103 (13,5°С)
С°, Дж/(моль-К) 128,04 — — — 549,4 — —
кДж/моль -1387,9 0,3** 10,9** — -4329,6 -1133 -1421,5
5^, Дж/(моль-К) 149,58 — — — 592,0 126 167
* Температура полиморфного перехода. "ДЯ полиморфного перехода. «"Температура плавления. ""Температура кипения.
Таблица 1.4. Растворимость сульфата натрия в воде
Температура, °C Концентрация насыщенного раствора, % (масс.) Твердая фаза Темперазу-ра, °C Концентрация насыщенного раствора, % (масс.) Твердая фаза
-0,6 1,96 Лед 40 32,5 Na2SO4 (ромб.)
-1,2 3,85 Лед + Na2SO410H2O 50 31,9 —
0 4,5 Na2SO410H2O 70 30,5 —
10 8,2 — 100 29,9 —
15 11,7 — 120 29,5 —
20 16,1 — 140 29,6 —
25 21,9 — 233 32,0 Na2SO4 (poM6.) + Na2SO4 (моноклинная)
30 28,8 — Na2SO4 (моноклинная)
32,4 33,2 Na2SO410H2O + + Na2SO4 (ромб.) 280 25,3
Кривая давлений водяного пара над мирабилитом (рис. 1.7), которая показывает, что при уменьшении влажности окружающего воздуха меньше давления диссоциации кристаллы мирабилита теряют кристаллизационную воду и покрываются слоем безводного сульфата натрия.
Рис. 1.6. Диаграмма растворимости в системе Na2SO4— Н2О
Рис. 1.7. Давление диссоциации Na2SO410H2O
Давление пара насыщенных растворов сульфата натрия при высоких температурах:
t°C........... 200 250 300 350 362
р, кгс/см2.... 14,3 38 85 167 195
Растворимость сульфата натрия в насыщенных водных растворах хлорида натрия с повышением температуры непрерывно возрастает вплоть до температуры плавления безводной эвтектики NaCl — Na2SO4 (рис. 1.8). При этом максимум давления пара эвтонических растворов тройной системы Na2SO4 — NaCl — Н2О (228—230 кгс/см2) намного (на 170—175 кгс/см2) ниже максимума давления пара насыщенных растворов системы NaCl — Н2О (рис. 1.9). На рис. 1.10 приведены изотермы растворимости в системе Na2SO4—NaCl — Н2О. При 25° С эвто-нический раствор, равновесный с Na2SO4 и NaCl, содержит 6,85% Na2SO4 и 23,14% NaCl, а в равновесии с Na2SO4 и Na2SO4-10H2O находится раствор, содержащий 15,61% Na2SC>4 и 14,4% NaCl.
Растворимость мирабилита в системе Na2SO4—NaOH — Н2О при 25° С имеет минимум (рис. 1.11) и затем плавный подъем к точке перехода Na2SO4-10H2O в Na2SO4, соответствующей содержанию в растворе 7,73% NaOH. Растворимость Na2SO4 при 25° С при увеличении концентрации NaOH от 0 до 48% уменьшается от 21,8 до 0,45%.
Гидросульфат натрия NaHSO4, бесцветные кристаллы, претерпевает. два полиморфных превращения при 140 и 170°С. Показатели преломления: Np= 1,43, № = 1,46, №=1,47. Растворимость в воде 22,2% (масс.) (25° С), 33,3 (100° С), в этаноле—1,4% (25° С).
Гидросульфат натрия образует моногидрат NaHSO4-H2O — бесцветные гигроскопические кристаллы. В процессе нагревания моногидрат переходит в Na2S2O7 по схеме:
NaHSO4 H2O NaHSO4 + Н2О 2NaHSO4 —Na2S2O7 + Н2О
Моногидрат получают растворением мирабилита в 20%-ной серной кислоте с последующим упариванием на водяной бане и охлаждением
Na2SO410H2O + H2SO4 = 2NaHSO4H2O + 9Н2О
а безводную соль получают кристаллизацией из расплава смеси сульфата натрия с концентрированной серной кислотой.
Сульфат натрия применяется в целлюлозно-бумажной и стекольной промышленности, в производстве моющих средств, в цветной металлур-22
NaCl
40
30
20
S ©
Na2SO4
Рис. 1.8. Политерма растворимости в системе Na2SO4_NaCl— Н2О при температурах от 100 до 700° С
NaCl
Критическая точка воды
NaCl
NaoSO
00
H20
Эвтоники NaCI+Na2SO4
400 600
Температура,
О 200
350
300
NagSO, 10 -
Рис. 1.10. Растворимость
в системе Na2SO4— NaCl — Н2О при температурах от 0 до 100° С: Г — галит; М — мирабилит; Т — тенардит
Рис. 1.9. Кривые зависимости давления водяного пара от температуры растворов, насыщенных Na2SO4, NaCl и смесью солей (эвтоника)
Рис. 1.11. Растворимость в системе Na2SO4 — NaOH — Н2О при 25° С
гии, текстильной и кожевенной промышленности. Сульфат натрия является сырьем в производстве силикатов натрия, серной кислоты, сульфата аммония, сульфида, карбоната и гидроксида натрия. Глауберова соль является слабительным средством в медицине. Гидросульфат натрия — флюс в цветной металлургии, а также приме
няется в качестве реагента для перевода труднорастворимых оксидов в растворимые сульфаты.
Сульфат натрия выпускается согласно требований существующего ГОСТ 6318—88, приведенных в табл. 1.5.
Таблица 1.5. Требования к химическому составу сульфата натрия
Высший сорт I сорт II сорт
Сульфат натрия, не менее 99,3 97,5 94,0
Нерастворимый остаток в воде, не более 0,5 1,5 4,5
Хлориды в пересчете на NaCl, не более 0,2 1,0 2,0
Сульфат кальция, не более 0,05 0,5 1,0
Железо (Ре20з), не более 0,01 0,01 0,03
Влага, не более 0,5 3 7
[Приведенные нормы (в %), за исключением влаги, относятся к сухому веществу.]
Способы получения мирабилита. Мирабилит в мировой практике получают главным образом бассейным методом. В зависимости от масштабов производства бассейная система может быть организована одной котловинбй или системой бассейнов. Например, для выделения мирабилита из рапы оз. Кучук в период зимней кристаллизации пользуются единственным бассейном, соединенным с озером каналом, который применяется как для подачи рассолов из озера, так и для обратного слива маточных растворов с производства.
Бассейный метод основан на использовании солнечной энергии для испарения воды в летний период и природного холода для кристаллизации солей в интервале от -20 до 20° С.
Из диаграммы растворимости в системе NaCl—MgSO4—Н2О (рис. 1.12) следует, что точки составов воды океана (7), Средиземного (2) и Черного морей (3), залива Сиваш (4) и Сакского озера (5) не соответствуют условиям получения значительных количеств мирабилита бассейным способом, так как образуются растворы, насыщенные хлоридом натрия, которые в процессе дальнейшего испарения и будут 24
МдС12- 6Н2О МдС12
Рис. 1.12. Диаграмма растворимости в системе NaCl — MgSO4 — Н2О:
при 25° С (сплошные линии) и 0° С (пунктирные линии)
бассейновым способом путем комплекс-
кристаллизоваться в каче- Na2SO4__________________ MgSO4
стве первой твердой фа- 100| °
зы. В процессе дальнейшего концентрирования воды Аральского моря (7) получается раствор, насыщенный астраханитом, а воды оз. Балхаш (S) — тенардитом. Следовательно, из всех перечисленных вод в летний период можно получить сульфат натрия лишь из воды оз. Балхаш. Практический выход тенардита из 1м3 балхашского рассола составляет 110—120 кг, а степень использования сульфат-иона всего 50—55%; при зимней садке мирабилита степень использования SO2' достигает 90—95%.
Мирабилит получаку
ной переработки межкристальных рассолов. Наравне с мирабилитом получают хлорид натрия, эпсомит и бишофит. Согласно технологической схеме, пять последовательно соединенных основных бассейнов действуют: для обогащения рассолов сульфат-ионом; зимней кристаллизации продукционного пласта мирабилита; летней кристаллизации галита; зимней кристаллизации эпсомита; концентрирования рассолов с удалением значительной части оставшихся примесей солей калия, натрия и магния. Конечный хлормагниевый рассол хранят в бассейне.
Один цикл технологии переработки длится два года. Стадия обогащения рассолов вследствие сравнительно высоких зимних температур (0° С) и для получения пласта мирабилита толщиной не менее 40 см при относительно небольшом выходе мирабилита (менее 100 кг/м3) из первичных рассолов.
Процесс обогащения обычно ведут кристаллизацией мирабилита из первичных рассолов и растворением этого пласта в летнее время в следующем объеме тех же рассолов.
На рис. 1.13 показано направление лучей кристаллизации на солевой проекции диаграммы системы Mg2*, Na+, СГ, SO^’ и НгО.
25
Рис. 1.13. Направление лучей кристаллизации в процессе бассейной переработки межкристальных рассолов залива Кара-Богаз-Гол
В диаграмме средний солевой состав межкристальных рассолов отображен точкой 1. В процессе охлаждения первичных рассолов в бассейне-обогатителе получают кристаллы мирабилита и маточный раствор; состав его характеризует точка 2. В летний период образующийся мирабилит растворяется в первичных рассолах, которые не насыщены при
температуре выше 15° С. В результате смешения образуется рассол (его солевой состав характеризует точка 3), который передают в последующий бассейн, где в зимний период получают продукционный пласт мира
билита и маточные растворы, отвечающие точке 2. Эти рассолы направляют в галитовый бассейн, в котором за летний период испарения кристаллизуется галит.
Параллельно жидкая фаза постепенно меняет солевой состав (луч 2-+4). Из оставшейся жидкой фазы (обедненной галитом) после разбавления водой в течение зимы кристаллизуется эпсомит. В процессе его кристаллизации солевой состав жидкой фазы изменяется (луч 4->5). При этом основная часть солей представлена хлоридом магния, но положение точки 5 указывает на заметное содержание ионов
натрия и хлора.
За период дальнейшего летнего испарения воды из этого рассола (луч 5->6) выделяется хлорид натрия и незначительное количество кизерита. Конечный продукт бассейной переработки рассолов в предельном случае может иметь состав эвтоники (MgCh—NaCl—MgSO4-H2O). В табл. 1.6 приведены примерные составы жидких фаз, соответствующие процессу последовательной кристаллизации.
На некоторых заводах мирабилит получают из межкристальных рассолов (рис. 1.14), для чего рассолы в скруббере 2 очищают от сульфида водорода сжатым воздухом, подаваемым вентилятором 1. В верхнюю часть скруббера 2 подаются рассолы. Выдуваемый воздухом сульфид водорода направляют на переработку. 26
Рис. 1.14. Схема получения искусственного мирабилита из межкристальных рассолов: 1— вентилятор; 2— скруббер с насадкой; 3— теплообменник; 4 — кристаллизатор; 5 — дисковый вакуум-сгуститель; 6—делитель суспензии; 7—смеситель; 8— ресивер; 9 — вакуум-насос;
10— центрифуга; 11 — транспортер; 12 — барометрическая емкость; 13 — насос
Таблица 1.6. Средние составы рассолов иа различных стадиях их бассейновой переработки
Стадии бассейновой перера-ботки Состав, % Состав, индексы Иене-ке Плотность, кг/м3
NaCl Na2SO4 MgCb Н2О so? Mg2* н2о
Исходные межкристальные рассолы 14,1 5,5 7,8 72,6 16,0 34,0 16,7 1232 (15°С)
Обогащенные межкристальные рассолы 13,0 8,7 7,2 71,1 24,8 30,4 16,0 1251 (15°С)
Маточный мирабилитовый раствор 15,1 3,0 8,3 73,7 8,9 36,9 17,3 1226 (0°С)
Галитовый рассол 5,6 7,3 16,4 70,7 21,5 82,9 14,0 1273 (25°С)
Эпсомитовый рассол 6,0 3,6 17,6 72,8 П,2 80,7 15,2 1251 (2,5°С)
Кондиционный маг- ний-хлоридный рассол 2,0 4,5 27,0 66,5 11,1 95,5 10,9 1316 (25°С)
Очищенные от сульфида водорода рассолы после теплообменника 3, обогреваемого маточными растворами, из барометрической емкости 12 направляются в кристаллизатор 4. В кристаллизаторе процесс охлаждения рассолов и компенсация теплоты (процесс экзотермический) происходят за счет испарения жидкого аммиака в аппаратах с теплопередающими стенками и отчасти рекуперации холода маточных растворов, остающихся после отделения мирабилита. Межкристальные растворы охлаждаются до 10° С в результате рекуперации холода сбросного раствора и дальнейшего охлаждения жидким аммиаком до 0
27
(-1° С) в первых кристаллизаторах и образованием кристаллической массы в следующих. Жидкий аммиак поступает с аммиачно-холодильной станции обычного типа.
Суспензия из кристаллизаторов 4 поступает в дисковый вакуум-сгуститель 5 на сгущение в смесителе 7, после чего твердую фазу отжимают в центрифуге 10. Образующиеся в смесителе 7 растворы и маточные растворы из центрифуги подают в барометрическую емкость 12, откуда, проходя теплообменник 3, направляются на начало процесса — в бассейны. Отжатый в центрифуге 10 мирабилит транспортером 11 передается на производство безводного сульфата натрия.
В процессе переработки сырого мирабилита в безводный сульфат натрия значительную роль играют характер и количество в нем примесей. С целью стабилизации качественного состава мирабилита его промывают на стадии фильтрации. Из данных по промывке мирабилита охлажденной водой видно, что применение промывной жидкости в количестве 10—15% от массы мирабилита обеспечивает получение продукта, содержащего менее 0,1% хло-рид-иона (рис. 1.15). Фильтрат же промывных вод возвращают на стадию кристаллизации мирабилита.
Способы получения сульфата натрия. Значительное количество производимого сульфата натрия получают обезвоживанием мирабилита. Качество целевого продукта зависит от методов его получения. Например, в процессе получения продукта во вращающихся барабанных сушилках получается комкующийся и слеживающийся сульфат натрия, а в процессе обезвоживания в распылительных сушилках— пушистый, легкий сульфат с насыпной массой 450—500 кг/м3.
Различие методов обезвоживания исходного сырья приводит также к неодинаковому химическому составу целевого продукта.
Разработано множество способов обезвоживания мирабилита в заводских условиях. В основу их положены следующие стадии и процессы: 1) плавление исходного мирабилита; 2) упарка растворов; 3) автоклавирование; 4) сушка воздухом и дымовыми газами; 5) высаливание; 6) обезвоживание с применением легколетучих растворителей и др.
Количество промывной жидкости, ма/1000кг
Рис. 1.15. Зависимость остаточного содержания хлорид-иона в мирабилите от объема промывной воды
28
Получение сульфата натрия методом плавления. В основе этого способа лежит плавление мирабилита при 32,4° С с дальнейшим разложением по схеме
Na2SO410H2O = Na2SO4 + ЮН2О
Освобождающаяся кристаллизационная вода растворяет часть сульфата натрия с образованием насыщенного раствора, а 35,8% сульфата натрия остается в твердой фазе и может быть выделен известными технологическими приемами из системы. На этих принципах основана технология сульфата натрия из мирабилита. Согласно технологической схеме (рис. 1.16), исходный мирабилит транспортером 11 подается на плавитель 7, соединенный циркуляционным насосом 12 с выносной греющей камерой 2. На столик плавителя синхронно поступает циркулирующая суспензия с температурой 75—85° С. В процессе плавления исходного мирабилита в твердую фазу выделяется до 40% сульфата натрия (при теоретических расчетах 45,9% Na2SO4 при 90° С). Необходимая для проведения процесса плавления теплота поступает от вторичного пара последнего корпуса выпарных аппаратов 6.
11
Рис. 1.16. Схема получения сульфата натрия плавлением мирабилита:
1— плавитель; 2 — выносная греющая камера; 3 — сгуститель; 4— смеситель; 5 — центрифуга;
6— выпарные аппараты; 7— вращающаяся барабанная сушилка; 8 — циклон; 9 — мокрый скруббер; 10 — дымосос; 11— транспортер; 12 — центробежный насос
29
Образующаяся суспензия сульфата натрия, содержащая 15% твердой фазы, поступает в сгуститель 3, откуда сгущенная часть, проходя через смеситель 4, загружается в центрифугу 5. Жидкая фаза, образующаяся в процессе плавления, направляется в выпарные аппараты 6. На выпарку направляют и маточные растворы после центрифуг 5.
Сырой сульфат натрия, полученный в процессе фильтрации суспензий всех стадий переработки, поступает во вращающуюся барабанную сушилку 7, обогреваемую топочными газами. От пылевидного сульфата натрия воздух из сушилки 7 очищается в циклонах 8, мокром скруббере 9 и через дымосос 10 выбрасывается в атмосферу. Целевой продукт транспортером 11 передается на склад готовой продукции.
Рассмотренный способ получения сульфата натрия обеспечивает низкий коэффициент использования исходного сырья — мирабилита. Согласно этой технологии, на 1 т целевого продукта расходуется около 4,5 т мирабилита, что почти в два раза превышает теоретические расчеты.
Получение сульфата натрия методом плавления — выпаривания. Способ выделения безводного сульфата натрия из растворов, получаемых при плавлении мирабилита, путем выпаривания этих растворов в многокорпусных выпарных установках экономически предпочтительнее способа повторной кристаллизации мирабилита и его плавления.
Аналогичная установка описана на рис. 1.17. Корпуса (2 и 3) работают под разрежением. Температура кипения раствора в корпусе 2 — 80, в корпусе 3 — 65° С. Греющая камера корпуса 2 обогревается свежим паром, вторичный пар из этого корпуса подают в греющую камеру корпуса 3. Растворы, содержащие некоторое количество твердой фазы (в виде слива из сгустителя) и предназначенные для дальнейшей переработки, подают параллельно в оба корпуса. Исходный мирабилит плавится в плавителе 1 за счет теплоты суспензии, вытекающей из корпусов 2 и 3 выпарной установки и пара корпуса 3, который подается в вынесенную греющую камеру 6 плавителя. При недостатке теплоты на этой стадии в установке предусмотрена подача свежего пара в греющую камеру плавителя. В процессе работы температура в плавителе 1 поддерживается 50° С.
Образующийся в плавителе 1 раствор сульфата натрия направляется в сгуститель 4. Кристаллы безводного сульфата натрия отжимаются и промываются в центрифуге 5. Сульфат натрия после центрифуги направляют на сушку, а маточные растворы — в плавитель 1.
В зависимости от качества примесей маточные растворы передают на стадию кристаллизации мирабилита и далее удаляют призе
Рис. 1.17. Схема конверсии мирабилита
меси в виде фугата после фильтрации мирабилита. Способ обеспечивает получение 1 т сульфата натрия при расходе 2,3 т исходного сырья — мирабилита. Целевой продукт содержит 99,9% основного вещества.
Для получения сульфата натрия из мирабилита путем плавления— выпаривания для выпарки используют высокоэффективные аппараты погружного горения (АПГ). В этом процессе испарение воды происходит при контакте с горячими газами, образующимися за счет сжигания газообразного или жидкого топлива в специальных горелках. Отсутствие теплопередающих стенок обеспечивает постоянство коэффициентов теплопередачи, а большая площадь соприкосновения теплоносителя и выпариваемого раствора — высокие скорости испарения жидкости.
АПГ поддерживают температуру отходящих паров и газов в пределах 75—90° С и обеспечивают возможность применения низкосортного топлива.
Согласно технологии (рис. 1.18), горячие растворы мирабилита из плавителя 1 подают в аппараты погружного горения 2, в которые вместо горючего материала направляют газы, отходящие из сушильного барабана. При этом одновременно обеспечивается: утилизация теплоты отходящих газов, улавливание уносимого сульфата натрия и очистка выбросов, получение более концентрированной суспензии для дальнейшего упаривания. Температура греющих газов на входе 260—280° С, а на выходе — 65—70° С. Из аппаратов ПГ 2 суспензия 31
Рис. 1.18. Схема конверсии мирабилита в сульфат натрия с использованием аппаратов погружного горения:
1 — плавитель; 2 — аппарат погружного горения; 3 — отстойник ; 4 — центрифуга; 5 — барабанная сушилка
перекачивается в отстойник 3, откуда твердая фаза направляется в центрифугу 4, а жидкая фаза — в аппарат погружного горения на упарку. Отжатый в центрифуге сульфат натрия поступает во вращающуюся барабанную сушилку 5, откуда сухой сульфат натрия направляется на упаковку. Целевой продукт — сульфат натрия — содержит 99,3—99,9% основного вещества.
Получение сульфата натрия методом высаливания. Из расплавленного мирабилита или сульфатосодержащих растворов сульфат натрия выделяют высаливанием. Процесс высаливания основан на понижении растворимости сульфата натрия со смещением переходной точки другими веществами. Наиболее экономичным высаливающим реагентом является хлорид натрия, хотя и изучены технологические условия применения для этих целей аммиака, гидроксида натрия, смешанных солей (MgSO4-7H2O + NaCl), смеси NaCl и КС1, сульфида натрия и др.
Способ высаливания отличается от метода выпаривания простотой оборудования, низкой удельной металлоемкостью и энергоемкостью производства.
Наиболее широко применяемым в производстве высаливающим реагентом является хлорид натрия. На рис. 1.19 приведена политер-мическая диаграмма равновесия для системы Na2SO4—NaCl—Н2О. Из диаграммы видно, что в отсутствие хлорида натрия точка перехода мирабилита в сульфат натрия находится при 32,38° С. При добавке хлорида натрия температура перехода снижается и становится равной 17,9° С, при которой содержание хлорида натрия в растворе 32
достигает 22,3%. В тройной точке А в солевой массе эвтони-ческого раствора 25,4% Na2SO4 и 74,6% NaCl, а раствор содержит 7,57% Na2SO4, в то время как в точке В в насыщенном растворе содержится 33,2% Na2SC>4. Ниже 17,9° С выделение в осадок безводного сульфата натрия не происходит, а с повышением температуры поле кристаллизации Na2SC>4 несколько расширяется, в результате че
Рис. 1.19. Проекционная политермическая диаграмма равновесия в системе Na2SO4 —NaCl —Н2О
го степень его высаливания увеличивается. Так, при 50° С в отсутствие хлорида натрия растворимость сульфата натрия составляет 31,9%, а эвтонический раствор содержит 23,89% хлорида натрия и всего 5,17% сульфата натрия.
Таким образом, добавление к растворам сульфата натрия NaCl приводит к кристаллизации сульфата натрия, что позволяет получить в безводном виде значительную долю сульфата. После отделения сульфата натрия отстаиванием и фильтрованием остающийся маточный раствор может быть подвергнут выпариванию для кристаллизации из него NaCl, возвращаемого на высаливание. Отфильтрованный сульфат натрия для удаления из него примеси хлорида натрия промывают насыщенным раствором сульфата натрия (в 1,5-кратном количестве к твердой фазе).
На рис. 1.20 приведена схема получения сульфата натрия высаливанием его из мирабилита хлоридом натрия. Исходный мирабилит подогревается в шнековом плавителе 2, в котором процесс
плавления происходит за счет теплоты циркулирующих растворов через кожухотрубный теплообменник 3, питаемый паром низкого давления. Расплавленная суспензия (насыщенный рассол) поступает в сгуститель 2, из которого сгущенную часть направляют на разделение в центрифугу 4. Маточные растворы (жидкую фазу) частично используют в качестве теплоносителя в процессе плавления, а часть направляют на вторую стадию процесса высаливания. На этой стадии происходит растворение оставшегося твердого хлорида натрия, разбавление и вытеснение высокохлоридного маточного раствора, поступающего в смеси с твердой фазой. Для проведения первой стадии применяют шнековый смеситель, для второй — вертикальный аппарат, работающий в режиме противотока.
2 Химическая технология неорганических веществ, кн. 1
33
Раствор
Рис. 1.20. Схема получения сульфата натрия из мирабилита путем высаливания хлоридом натрия:
/ — плавитель; 2, 7—сгустители; 3— теплообменник; 4— центрифуга; 5, 6 — реакторы высаливания; 3 — барабанная вращающаяся сушилка
Исходный хлорид натрия поступает в реактор основной стадии высаливания 6, в котором растворяется маточными растворами, поступающими из реактора 5, и в виде растворов направляется в сгуститель 7. После отстоя растворы хлорида натрия передаются в реактор 5. Образующаяся в реакторе 5 суспензия сульфата натрия направляется в центрифугу 4, а жидкая часть — в реактор 6.
Отжатые в центрифуге 4 кристаллы сушатся в барабанной вращающейся сушилке 8.
Полученный продукт содержит, %:
Na2SO4 NaCl MgSO4 CaSO4 K2SO4 H.O. H2O
Без промывки .. 98,23 0,85 0,45 0,23 0,12 0,08 0,04
Промытый водой.... 99,4 0,10 0,28 0,13 — 0,05 0,04
В НИОХИМ разработан способ высаливания сульфата натрия гидроксидом натрия, основанный на значительно большей энергии гидратации гидроксид-иона по сравнению с сульфат-ионом. В табл. 1.7 приведены данные по растворимости сульфата натрия в водных растворах гидроксида натрия при температурах в интервале 0—80° С.
34
Таблица 1.7. Растворимость сульфата натрия в водных растворах гидроксида натрия
Концентрация NaOH, % Температура, °C
50 70 90 100 120 140
0 46,8 43,9 42,7 42,4 41,8 42,0
5 28,4 26,5 24,9 24,5 23,5 22,2
15 11,7 11,0 10,7 10,0 9,6 10,1
25 3,7 3,5 3,3 2,9 2,6 2,6
35 1,9 1,6 1,5 1,3 1,3 1,3
45 0,6 0,6 0,6 0,6 0,6 0,6
55 0,3 0,3 0,3 0,2 0,2 0,3
Номограмма, связывающая степень высаливания безводного сульфата натрия при 60° С из его водного насыщенного раствора растворами гидроксида натрия различной концентрации, представлена на рис. 1.21. При смешении 0,4—1 объема 40—50%-ного раствора гидроксида натрия с 1 объемом насыщенного раствора сульфата натрия степень высаливания безводного сульфата составляет 85—90%, а раствор после отделения осадка сульфата содержит 20—25% NaOH и 2,5—5% Na2SO4. Конечную жидкую фазу для ее возврата в процесс упаривают.
В процессе непосредственного смешения исходного мирабилита с раствором гидроксида натрия образуется тонкодисперсный (а<100 мкм) труднофильтруемый продукт. Это связано со значительным пересыщением раствора. С целью получения более крупных кристаллов процессы плавления мирабилита и высаливания сульфата натрия из раствора исходный гидроксид натрия вводят медленно и при ин-
Рис. 1.21. Зависимость степени высаливания сульфата натрия от концентрации гидроксида натрия
2*
35
тенсивном перемешивании. Более крупные кристаллы получают при 70—80° С. Для этого в растворы вводят затравочные кристаллы. Получаемые в центрифугах кристаллы промывают плавильным раствором.
Содержащиеся в исходном мирабилите примеси соединений магния, сесквиоксидов, кремневой кислоты и другие в растворе гидроксида натрия образуют гелеобразные осадки, которые затрудняют процессы отстоя и фильтрации. С целью исключения этого явления примеси должны быть отделены до процесса высаливания.
Разработан также способ высаливания сульфата натрия из его растворов применением аммиака (тригидрида азота). При этом расход теплоты в два раза меньше, чем в процессе плавления мирабилита и выпаривании воды из образующегося раствора.
Растворимость сульфата натрия в водных растворах аммиака сильно уменьшается. В результате в процессе взаимодействия регенерированной аммиачно-паровой смеси температурой 60° С и мирабилитом температурой 10° С выход сульфата натрия достигает 90—95% без дополнительного нагревания или охлаждения реакционной массы.
Разработаны способы вытеснения сульфата натрия из его насыщенных растворов с применением органических растворителей, например этанола, метанола, пропилового спирта или ацетона. Способ состоит в пропускании паров спирта через слой мирабилита, при котором последний плавится и из расплава выделяется кристаллический продукт.
В процессе переработки 1 т мирабилита теоретического состава необходимо выпарить 0,559 т воды, на что обычными методами затрачивается 0,3 Гкал. В процессе смешения жидкой фазы, остающейся после плавления мирабилита с 0,3 т этилового спирта, и последующем отделении твердой фазы и дистилляции водно-спиртового раствора для возврата осадителя в процесс требуется затратить всего 0,064 Гкал. Кроме того, при этом улучшается качество целевого продукта, так как спирт не вносит примесей, а также резко уменьшается инкрустация поверхности выпарных аппаратов.
Сульфат натрия получают в качестве побочного продукта в производстве хлороводородной кислоты. Наиболее качественный сульфат натрия получают из отходов осадительных ванн производства вискозы и целлофана. В производстве синтетических жирных кислот получают сульфат натрия с высоким содержанием основного вещества.
1.5. ФОСФАТЫ НАТРИЯ
К фосфатам натрия относят ортофосфаты, пирофосфаты (дифосфаты) и конденсированные фосфаты натрия. Они представляют кристаллические или стеклообразные вещества, хорошо растворимые в воде, образуют кристаллогидраты (табл. 1.8, 1.9 и рис. 1.22)
Пирофосфат натрия ИадРгО? полиморфен, тригидрофосфат натрия NaHjPiO?, дигидрофосфат натрия ИагНгРгО? и гидропирофосфаты 36
натрия Na^HPjO? разлагаются выше 250° С с образованием конденсированных фосфатов натрия.
Конденсированные фосфаты натрия содержат группировку РО"з. Полифосфаты (КаРОз)оо имеют линейное цепочечное строение, циклофосфаты (NaPO3)„, где п-3, 4, 12, — циклическое.
Тетрациклофосфат Na4₽40i2 может иметь конформацию кресла или ванны. Безводная соль существует лишь в конформации ванны.
Олигомерный трифосфат Na5P3Oio, имеющий линейное цепочечное строение, существует в двух модификациях, температура перехода 1417° С.
Ортофосфаты натрия применяются в производстве фармацевтических препаратов, в медицине, фотографии и гальванопластике.
Фосфаты натрия широко применяют в качестве компонентов моющих средств, в качестве умягчителя воды, детергенты для очистки металлов, поверхностно-активных веществ в производстве цементов и при бурении нефтяных скважин. Они являются компонентом синтетических моющих средств (с добавкой трифосфата натрия). В процессе устранения жесткости воды применяют дегидратированные фосфаты натрия, образующие комплексы с магнием, барием, кальцием и другими металлами. Фосфаты натрия применяют также в процессе обогащения руд, в производстве фосфатных стекол, красок, в пищевой промышленности в качестве разрыхлителей теста, для гомогенизации и улучшения качества сыров, колбас, сгущенного молока.
37
Таблица 1.8. Свойства орто- и пирофосфатов натрия
Показатель Na3PO4 Na3PO412H2O NaH2PO4-2H2O NaH2PO4H2O Na2HPO412H2O
Сингония Параметры ячейки, нм: — Тригональная Ромбическая Ромбическая Ромбическая
а — 1,202 — — —
b — — — —
с — 1,266 — — —
Угол, град — — — — —
Число формульных единиц в ячейке — — — — —
Пространственная группа — Р3с\ — — —
Тод,°C 1340 73,3—76,7 60 100 35,1
Плотность, г/см3 2,536 1,62 1,91 2,04 1,52
ДЯобр, кДж/моль -1922,8 -4471,6 — — -5293,5
Растворимость в воде, г в 100 г 12,1(20°С) 94,6(100°С) 1,5(0°С) 157(70°С) Хорошо растворяется 59,9(0°С) 427(100°С) 87,4(34°С)
Показатель Na2HPO4-7H2O Na2HPO4-2H2O Na4P2Ch Na^OrlOHiO Na3HP2Or9H2O МагНЛОтбНгО
Сингония Моноклинная Ромбическая — Моноклинная Моноклинная Моноклинная
Параметры ячейки, нм:
а — 1,034 — 1,696 0,859 1,411
b — 1,364 — 0,696 3,165 0,703
с — 1,698 — 1,485 0,613 1,350
Угол, град — — — 111,68 113,7 117,6
Число формульных единиц в ячейке — — — 4 4 4
Пространственная группа — Р222 — 2!с Р2уа СИс
Гпл/С 48,1 95 895 93,8 — 220
Плотность, г/см3 1,679 2,066 2,534 1,815—1,836 1,836 —
ДЯобр, кДж/моль -3817,6 -2341,6 -3180,1 -6137,1 -4747,2 —
Растворимость в воде, г в 100 г 104(40°С) 100(50°С) 3,16(0°С) 5,41(0°С) Хорошо рас- 6,9(0°С)
117(80°С) 40,26 (100°С) 93,11 (100°С) творяется 35(40°С)
Таблица 1.9. Характеристика конденсированных фосфатов натрия
Показатель (NaPO3) оо I (NaPO3)xII (Na2PO3) » Ш Na2P3O<>-6H2O Na3P3CM,5H2O Na3P3O9
Сингония Моноклинная Моноклинная Моноклинная Моноклинная Ромбическая Ромбическая
Параметры решетки, нм:
а 1,530 1,212 1,137 0,950 1,105 0,793
b 0,696 0,620 0,601 1,103 2,070 1,314
с 0,705 0,699 0,763 0,601 0,801 0,775
Угол, град 93,30 92 85,7 91 — —
Число формульных единиц в ячейке 12 8 8 2 8 4
Пространственная группа P2i/n Р2г/п P2i/n Р11 Л2122 Ртсп
Показатель Na2HP3O9 Na4P4Oi24H2O-M Na4P4Oi2-4H2O-T Na2H2P4O12 Na5P3Oio I NajP3 Ок, П
Сингония Ромбическая Моноклинная Моноклинная Моноклинная Моноклинная Моноклинная
Параметры решетки, нм:
а 0,772 0,967 0,6652 1,974 0,961 1,600
Ь 0,676 1,236 0,9579 1,479 0,534 0,524
с 0,711 0,617 0,6320 0,703 1,973 1,125
Угол, град — 92,3 107,0 90,0 112 93,0
Число формульных единиц в ячейке 2 2 1 8 4 4
Пространственная группа Р1 Р2\!а РТ Р1 С2/с С2/с
Примечание. I — соль Маддрела; П — соль Курроля А; Ш — соль Курроля В.
Моно- и динатрийфосфаты применяют для пропитки тканей и дерева для придания им огнестойкости. При повышенных температурах они разлагаются:
2NaH2PO4 = Na2H2P2O7 + Н2О
2Na2HPO4- 12Н2О = Na4P2O7 + 25Н2О
Образующиеся при этом пиро- и конденсированные фосфаты являются легкоплавкими веществами, покрывающими нагреваемую поверхность тонкой пленкой, защищающей ее от воспламенения.
Согласно ГОСТ 451—41, качеству динатрийфосфата предъявляются требования, представленные в табл. 1.10.
Таблица 1.10. Требования к качеству гидрофосфата натрия (содержание компонентов, %)
Наименования Сорт
I II III
Na2HPO412H2O, не менее 96 92 88
Сульфат (в пересчете на SO3), не более 0,1 1,0 2,0
Железо, не более 0,02 — —
Нитраты (в пересчете на NO3), не более 0,003 — —
Тяжелые металлы группы сероводорода, не более 0,002 — —
Мышьяк, не более 0,001 — —
Хлориды (в пересчете на С1), не более 0,07 — —
Не растворимый в воде остаток, не более 0,02 — —
Технология фосфатов натрия. В производстве фосфатов натрия для получения более чистого продукта применяют термическую фосфорную кислоту. На некоторых заводах разработаны и применяются способы получения чистых солей из фосфорной кислоты при разложении фосфатов серной кислотой, а также из суперфосфата.
Дигидроортофосфат получают нейтрализацией 25%-ной фосфорной кислоты раствором карбоната натрия:
2Н3РО4 + Na2CO3 = 2NaH2PO4 + Н2О + СО2
Образующиеся растворы фильтруют и выпаривают до плотности 1,5 г/см3. Из более концентрированных растворов может быть выкристаллизован моногидрат дигидроортофосфата натрия (NaH2PO4 H2O). Поэтому при использовании кислоты, содержащей выше 40% Н3РО4, исходные виды сырья вводят в реактор-нейтрализатор попеременно. В начале процесса нейтрализации кислоты карбонатом натрия получают 40
раствор Na2HPO4, в который вводят избыток Иа2СОз, после чего смесь нейтрализуют введением необходимого количества фосфорной кислоты. Процесс нейтрализации проводят при 85—95° С. С целью удаления ионов SO42’ из экстракционной кислоты к ней добавляют кальцийсодержащие вещества (СаО, СаСОз, двойной суперфосфат) и производят последующее катионирование кислоты. Нейтрализацией карбонатом натрия кислоты, очищенной от сульфат-ионов, получают концентрированные растворы фосфата натрия. Из образующихся растворов кристаллизуют NaH2PO4-2H2O. Для получения безводной соли раствор нагревают до 100° С и путем охлаждения осаждают NaH2PO4. Разработан также способ гранулирования мононатрийфосфата из раствора в распылительной сушилке.
Процесс получения фосфатов натрия из суперфосфата основан на обработке его водным раствором сульфата натрия. При этом в жидкой фазе образуется дигидроортофосфат:
Са(Н2РО4)2 + Н3РО4 + Na2SO4 + 2Н2О = 2NaH2PO4 + Н3РО4 + CaSO4-2H2O
Образующийся кислый раствор отфильтровывают от нерастворимого остатка суперфосфата и вновь выделившегося гипса. После упарки отфильтрованных растворов из них кристаллизуют дигидроортофосфат.
Гидроортофосфат и ортофосфат натрия получают нейтрализацией ортофосфорной кислоты в две стадии: в первой стадии карбонатом натрия до гидроортофосфата, а во второй — гидроксидом натрия до ортофосфата натрия:
Н3РО4 + Na2CO3 = Na2HPO4 + СО2 + Н2О
Na2HPO4 + NaOH = Na3PO4 + Н2О
В процессе получения гидроортофосфата натрия для более полного разложения карбоната натрия поддерживают некоторый избыток фосфорной кислоты, который нейтрализуется затем маточным раствором. Температуру в реакторе поддерживают в пределах 98—100° С. Раствор дигидроортофосфата после фильтрации перерабатывают на кристаллический дигидроортофосфат.
Более чистые сорта гидроортофосфата (безводный, двух- или семиводный в зависимости от температуры) выделяются в процессе взаимодействия технически чистого дигидроортофосфата аммония с карбонатом натрия:
NH4H2PO4 + Na2CO3 = Na2HPO4 + СО2 + NH3 + Н2О
Карбонат натрия дигидроортофосфат аммония вводят в раствор Na2HPO4, насыщенный при температуре реакции. При этом выделя-41
ются газообразный аммиак и диоксид углерода, а в твердую фазу кристаллический Na2HPO4.
Для получения ортофосфата натрия раствор гидроортофосфата нейтрализуют гидроксидом натрия
Na2HPO4 + NaOH = Na3PO4 + Н2О
За счет теплоты реакции нейтрализации температура раствора поднимается до 112° С. При концентрации исходной ортофосфорной кислоты 25—29% Р2О5 (экстракционная) растворы дигидроортофосфата или ортофосфата натрия до кристаллизации из них соли охлаждением предварительно упаривают. В процессе с применением концентрированной (~45% Р2О3) термической ортофосфорной кислоты растворы не упаривают, а направляют непосредственно на кристаллизацию. После охлаждения нейтрализованных растворов до 30° С ортофосфат кристаллизуется в виде двенадцативодных кристаллогидратов, после чего их отделяют от маточных растворов в центрифуге и высушивают. Двенадцативодный кристаллогидрат гидроортофосфа-та плавится в своей кристаллизационной воде при 60°С, а ортофосфат — при 70° С. Это осложняет процесс высушивания продукта без выделения кристаллизационной воды. В производстве процесс осуществляется в стадии получения растворов гидроортофосфата концентрации 19,8% и ортофосфата натрия 18,7% Р2О3, которые в процессе охлаждения до 60° С полностью затвердевают в распылительной башне в гранулированный или на охлаждаемых вальцах в чешуйчатый продукт. С целью уменьшения слеживания ортофосфат натрия дополнительно охлаждают воздухом в шнеках или вращающихся барабанах.
Разработан способ получения ортофосфата натрия введением к исходным дигидроортофосфату и гидроортофосфату гидроксида или карбоната натрия или основных солей с последующей термической обработкой смеси. В процессе смешения порошкообразного гидроортофосфата и жидкого технического гидроксида натрия образуется паста, которую перерабатывают в обогреваемом паром аппарате типа вальцовой сушилки. Прилипающий к горячей поверхности вальцев ортофосфат натрия срезают с поверхности барабана ножом и охлаждают.
В процессе получения ортофосфата натрия применяется экстракционная кислота, предварительно очищенная от кремнефторида карбонатом натрия:
H2SiF6 + Na2CO3 = Na2Sip6 + СО2 + Н2О
После фильтрации растворов от кремнефторида натрия первый ион водорода ортофосфорной кислоты нейтрализуют раствором кар-42
боната натрия до pH 4,2—4,4. При этом из раствора осаждаются фосфаты железа и аммония в форме легко фильтруемых осадков.
Используя экстракционную фосфорную кислоту, получают ортофосфат натрия с содержанием 19—19,2% Р2О5, 0,6—0,8% SO3, 0,1—0,4% NaOH и ~0,2% фтора.
Разработан способ получения ортофосфата натрия реакцией фосфата алюминия с гидроксидом натрия или спеканием с содой:
Д1РО4 + 3NaOH = Na3PO4 + А1(ОН)3
2AIPO4 + 3Na2CO3 = 2Na3PO4 + А12О3 + ЗСО2
Ортофосфат натрия выделяют из щелочного раствора в твердую фазу.
Разработан также способ получения ортофосфата натрия и серной кислоты электролизом раствора сульфата натрия и экстракционной ортофосфорной кислоты.
1.6. КОНДЕНСИРОВАННЫЕ ФОСФАТЫ НАТРИЯ
В процессе дегидратации ортофосфатов натрия образуются различные конденсированные фосфаты натрия со структурой, аналогичной силикатам, поскольку координационное число как кремния, так и фосфора по отношению к кислороду всегда равно четырем. При этом один из четырех атомов кислорода связан с пятизамещенным атомом фосфора двойной связью и поэтому не может быть связующим звеном между двумя атомами фосфора.
На рис. 1.23 схематически показаны условия образования различных фосфатов и их превращений в процессе нагревания на воздухе дигидрата дигидрофосфата натрия (основные продукты помещены в прямоугольниках).
Широко известный стекловидный фосфат, неправильно называемый гексаметафосфатом (соль Грэма), вследствие цепного строения относится к полифосфатам (а не метафосфатам, как считали ранее).
Конденсированные фосфаты, имеющие разветвленную цепь, содержат один или несколько атомов фосфора, которые связаны через атомы кислорода с тремя другими атомами фосфора, например:
ООО
II II II
МО-Р-О-Р-О-Р-ОМ
I I I
ООО
I I I
МО-Р-О-Р-О-Р-ОМ
II II II
ООО
43
Рис. 1.23. Образование различных соединений фосфора и их превращения в процессе нагревания на воздухе NaH2PO4-2H2O
Среди конденсированных фосфатов наиболее широко применяется безводный триполифосфат натрия. Полифосфаты являются солями очень сильных кислот, но их водные растворы имеют нейтральную реакцию. В воде устойчивы растворы лишь солей щелочных металлов. В нейтральном водном растворе конденсированные фосфаты устойчивы при комнатной температуре, а в кислой среде при температуре выше 60° С они легко гидролизуются, образуя промежуточные соединения до ортофосфата. В щелочных растворах они разлагаются до смеси полифосфатов (три- и тетрафосфатов).
Полифосфаты, а также относящаяся к этой группе не растворимая в воде соль Маддреля (NaPOj) являются солями кислот, приведенных в табл. 1.11. Цепи полифосфорных кислот имеют у каждого атома фосфора сильнокислую группу ОН и на концах цепи по две слабокислых ОН-группы. В соответствии с этим растворы полифосфатов, полученных из первичных ортофосфатов, имеют слабокислую реакцию, а растворы их «нейтральных солей» — слабощелочную реакцию. Слабокислые, нейтральные и щелочные растворы полифосфатов при обычной температуре являются стойкими, но выше 60° С, особенно в кислой среде, они гидролизуются.
Широкое практическое применение полифосфатов натрия основано на их способности связывать кальций и магний, умягчая тем самым воду. Это связано с тем, что они обладают свойствами ионообменников. 44
Например, триполифосфат натрия (Na5P3Oio) образует с солями жесткости соль СагИаРзОю, осаждающуюся при достаточной концентрации ионов кальция в растворе. Он способен связать до 10—11% кальция и 6,4% магния от своей массы. Конденсированные фосфаты натрия способны связывать до 12—18% кальция или 2,9—3,8% магния.
Другим, не менее важным свойством конденсированных фосфатов натрия, особенно триполифосфатов, является способность пептизировать суспензию и снижать вязкость, это свойство широко применяется в процессе флотации руд. С участием полифосфатов натрия значительно повышаются моющие свойства органических поверхностно-активных веществ. Конденсированные фосфаты используют непосредственно при стирке белья и поверхностной чистке одежды. Триполифосфат применяется в качестве составной части (до 85%) большинства моющих средств, а тетранатрийфосфат добавляют к обычному мылу.
Таблица 1.11. Полифосфорные кислоты Н„+2Р»Озл+1
Кислота Число групп ОН
сильнокислых слабокислых
Моноортофосфорная Н3РО4 1 2
Дипирофосфорная Н4Р2О7 2 2
Трифосфорная Н5Р3О10 3 2
Тетрафосфорная ЩР4О13 4 2
Полифосфорная кислота, соответствующая соли Маддреля H„ + 2P„O3«+i п 2
Триполифосфату натрия, выпускаемому промышленностью, согласно ГОСТ 13493—68, предъявляются определенные требования (табл. 1.12).
Таблица 1.12. Требования к качеству триполифосфата натрия
Компоненты Из термической кислоты Из экстракционной кислоты
I сорт II сорт I сорт II сорт
Внешний вид Белый порошок Порошок белого цвета с желтоватым или сероватым оттенком Белый порошок Белый порошок с желтоватым или сероватым оттенком
Содержание Р20з в %, не менее 56,5 54,0 53,5 51,5
Содержание ИазРзОю в %, не более 92,0 90,0 75,0 60,0
Содержание нерастворимого МазРзОю в %, не менее 10,0 12,0 12,0 20,0
pH 1%-ного раствора 9,3—9,8 9,3—9,8 Не нормируется Не нормируется
45
Технология конденсированных фосфатов натрия. Широко применяемым соединением конденсированных фосфатов натрия является триполифосфат натрия. Сырьем для его производства являются как термическая, так и экстракционная фосфорная кислота, которую предварительно очищают от примесей.
Согласно существующей технологии получения триполифосфата, исходную фосфорную кислоту нейтрализуют карбонатом натрия до образования раствора с отношением 5Na2O-3P2Os:
6Н3РО4 + 5Na2CO3 + иН2О = 4Na2HPO4 + 2NaH2PO4 + (и + 5)Н2О + 5СО2
Образующийся раствор выпаривают, а получающуюся смесь сухих солей подвергают дегидратации путем термообработки в температурном интервале 340—400° С. Способ обеспечивает получение низкотемпературной модификации Na5P3Oio, устойчивой ниже 450° С. Продукт отличается от высокотемпературной модификации, устойчивой выше 450°С. Он не слеживается во влажной среде и не комкует-ся при растворении в воде. Для стабилизации низкотемпературной формы триполифосфата применяют добавки небольших количеств (до 1%) карбамида, азотной кислоты, нитрата аммония и др. Они способствуют также более полному переходу гидро- и дигидрофосфатов в триполифосфат и придают белизну целевому продукту.
Установлено, что добавка к исходным реактивам 1—2% нитрата аммония действует каталитически на процесс образования целевого продукта, протекающий при более низкой температуре. При этом получается низкотемпературная модификация Na5P3Oio, отличающаяся хорошей растворимостью.
Ортофосфаты NaH2PO4 и Na2HPO4 в процессе термообработки реагируют по схеме
2Na2HPO4 + NaH2PO4 = ^Р2О7 + NaPO3 + 2Н2О
Процесс протекает с наибольшей скоростью при 185—220° С. Триполифосфат натрия частично образуется при быстрой первичной дегидратации ортофосфатов до 230°С. Основная же масса олигомерного трифосфата натрия получается по реакции полуфабрикатов:
Na4P2O7 + NaPO3 = NasP3Oio
Оптимальной температурой процесса является 290—310° С.
Процесс дегидратации исходных ортофосфатов натрия с образованием триполифосфата продолжается 15—30 мин.
Процесс выпарки растворов ортофосфатов натрия с получением сухих солей, а также их дегидратацию осуществляют в одном совме-46
щенном аппарате. Процесс проводят во вращающихся барабанных печах, которые могут быть использованы для совмещенной сушки исходного раствора и кальцинации исходных ортофосфатов. В этом варианте производства для исключения налипания материала на стенки барабану упаренный раствор, содержащий около 32% Р2О5, смешивают с ретуром целевого продукта (в соотношении по массе 3:3,5:1 при влажности смеси 10—12%) или распыляют раствор на слой сухого (материала.
Разработан способ получения триполифосфата во вращающемся барабане, разделенном кольцевой перегородкой на две зоны. В первой зоне печи исходный раствор, вводимый в барабан посредством распыляющих форсунок, высушивается, а во второй протекают дальнейшие процессы. Барабан обогревается изнутри газовыми горелками.
Разработан также способ получения триполифосфата натрия в распылительных сушилках, представляющих собой вертикальную цилиндрическую башню. Исходный раствор ортофосфатов подается под давлением и распыляется в потоке горячих газов, поступающих в верхнюю часть башни температурой 400—700° С и отходящих температурой 100—200° С. Из сушилки целевой продукт с влажностью менее 5% поступает для термообработки во вращающуюся или неподвижную печь, снабженную горизонтальными лопастными мешалками, служащими и для передвижения материала. Термообработка сухих ортофосфатов натрия может быть осуществлена также в распылительной сушилке совместно с процессом сушки целевого продукта.
В производстве триполифосфата натрия применяют два способа: 1) в совмещенном процессе с применением сушильно-прокалочной распылительной печи и 2) с раздельной сушкой исходного раствора в распылительной сушилке и термообработкой сухих ортофосфатов в кальцинаторе специальной конструкции — турбокальцинаторе.
При совмещенном процессе стадии сушки раствора, термообработки сухих ортофосфатов натрия, а также охлаждения продукта происходят в одном аппарате — сушильно-прокалочной печи.
Сушильно-прокалочная печь (рис. 1.24) представляет собой цилиндрическую башню 1, футерованную изнутри огнеупорным кирпичом. В нижней части печь имеет две прокалочные 5, 6 и две холодильные 10, 11 полки. Верхние прокалочные полки 5, 6 изготовлены из чугуна или жаропрочной стали, а нижние 10, 11 — из стали 3. Исходный раствор распыляют пневматическими форсунками 4 из желоба 2 и коллектора 3 под давлением 5—7 ат. Горячие газы с температурой 800—900° С, образующиеся при сжигании газа, поступают в нижнюю часть печи из кольцевого дымохода 14 через шлицы 8. Основная масса газа выходит в полость между прокалочными полками 5 и 6, а часть газа поступает в зону над верхней полкой 5.
47
2
3
2
Рис. 1.24. Сушильно-прокалочная печь:
1 — башня; 2 — желоб; 3 — коллектор для пара; 4 — форсунка; 5 и 6 — прокалочные полки; 7 — корпус; 8 — шлицы; 9 — скребки; 10, 11 — полки охлаждения; 12 — приводной механизм:
13 — вал; 14 — дымоход
Сухой и частично прокаленный продукт перемещается скребками 9 к центру и ссыпается на полки 5 и 6. Скребки 9 и вал 13 изготовляются полыми. Для их охлаждения через них продувают воздух. С прокалочцых полок 5, 6 материал при температуре около 400° С поступает на холодильные полки 10, 11, по которым пропускают охлаждающую воду. На выходе из башни продукт имеет температуру 30—40°С, а топочные газы выводятся с температурой 140—200° С.
На рис. il .25 приведена схема получения триполифосфата натрия в совмещенной процессе с применением распылительной сушильно-про-калочной пени. Согласно схеме, исходная фосфорная кислота из сборника 1 и карбонат натрия из бункера 2 поступают в реактор 3. Образующиеся растворы из реактора 3 центробежным насосом 19 фильтруют через фильтр 4 и собирают в сборнике 5. Отфильтрованные растворы центробежными насосами 19 непрерывно питают выпарной аппарат 6. Упаренные до 60%-ной концентрации раствор из сборника 7 центробежным насосом 8 передают в напорный сборник 18, где они подогреваются и поступают в сушильно-прокалочную печь 17, где происходит выпаривание раствора, сушка, дегидратация исходных ортофосфатов и охлаждение триполифосфата. Температура в сушиль-но-прокалочной печи поддерживается теплом из газовой топки 16. Отработанные газы вместе с пылью проходят батарейный циклон 13 для очистки от пыли. Уловленная в бункере 14 пыль через мигалки 15 возвращается на первую прокалочную полку печи 17. Проходящие из циклона 13 газы дымососом 12 дополнительно очищаются от пыли в скруббере 9, орошаемом разбавленным исходным раствором, и, пройдя циклонный брызгоуловитель 10, выбрасываются в атмосферу.
Полученный в печи целевой продукт после грубого дробления на дробилке 20 пневмотранспортом подается в классификатор 21, откуда крупные частицы поступают на дополнительное измельчение в мельницы тонкого помола. Мелкий порошковидный материал улавливается в циклонах 22 и поступает в бункер готового продукта 24.
Производимый из экстракционной фосфорной кислоты триполифосфат натрия содержит примеси исходного сырья. К тому же эти примеси в процессе получения целевого продукта действуют как каталитические пассиваторы на процесс дегидратации. Поэтому исходную кислоту, содержащую ~20% Р2О5, 0,4—-0,6% SO3 и 1,5—1,8% фтора, нейтрализуют карбонатом натрия до pH 7. Образующийся осадок, состоящий в основном из фосфатов, сульфатов и кремнефторида натрия, отделяют на фильтре, а очищенный раствор упаривают в распылительной сушилке. При этом содержащиеся в растворе моно- и дигидрофосфат натрия (в молярном соотношении 1:2) дегидратируются при 400° С за счет тепла отходящих газов. Полученный продукт содержит 54,3—55% Р2О5 (в том числе 53,2—54,3% в полиформе и 0,4—-0,9% в ортоформе), 2,8—3,5% Na2SC>4 и 1—1,5% NaF.
49
Рис. 1.25. Схема получения триполифосфата натрия в сушильно-прокалочной печи:
1— сборник фосфорной кислоты; 2 — бункер; 3 — реактор; 4— фильтр; 5— сборник раствора; 6 — выпарной аппарат; 7—сборник; 8— центробежный насос; 9—скруббер. 10—брызгоуловитель; 11— сборник; 12—домосос;73 —циклон; 14— бункер; 15— мигалки; 16 — газовая топка; 17—сушильно- прокалочная печь; 18— напорный сборник; 19—центробежный насос; 20 —^шек ^-^хцассификатор; 22 — циклон; 23—вентилятор; 24 — бункер " —_____.__
Разработан способ использования экстракционной фосфорной кислоты, содержащей ~30% Р2О5 и 1,8—2,0% SO3, 0,2% СаО, 0,7% фтора, 0,4% хлора, 0,3% AI2O3, 0,3% MgO и 0,3% РегОз, путем обработки известняком, а затем концентрированным раствором карбоната натрия. После отделения гипса и кремнефторида натрия к раствору добавляют карбонат бария и нейтрализуют его карбонатом натрия с последующей переработкой в целевой продукт.
Образующийся в процессе очистки экстракционной фосфорной кислоты шлам, состоящий в основном из фосфатов железа и алюминия, используют в производстве гранулированного суперфосфата. Иногда получают комплексное удобрение смешением шлама с нитратом аммония и калийной солью.
Пищевой гексагидрат триполифосфата натрия получают из технического продукта, растворяя его в воде при 60—70° С. Получаемый при этом 17—20%-ный раствор Na5P3Oio отделяют от нерастворимых примесей. Вакуум-кристаллизацией при 60° С (с целью предотвращения процесса гидролиза) из раствора получают кристаллы чистого триполифосфата натрия, которые отделяют в центрифугах от маточных растворов, промывают и высушивают. Во избежание перехода триполифосфата в пиро- или ортоформы гексагидрат сушат горячими газами, содержащими водяной пар при парциальном давлении последнего около 200 мм рт. ст. и относительной влажности менее 48%.
Стекловидный фосфат натрия (соль Грэма) получают нагреванием и расплавлением при 620—700° С дигидрофосфата натрия с последующим быстрым охлаждением образующегося плава. Для получения продукта в виде тонких чешуек расплав выливают между вращающимися охлаждаемыми вальцами. Целевой продукт содержит около 69,6% Р2О5. Его раствор имеет pH около 5,2. Свойства продукта — содержание основного вещества, устойчивость и степень полимеризации зависят от температуры плавления и скорости кристаллизации плава.
Разработан способ непосредственного получения конденсированного фосфата из элементного фосфора и карбоната натрия в камерах для сжигания фосфора, минуя операции приготовления исходных растворов и их упаривание. Согласно этому способу, при 600—700° С образуются твердые продукты с соотношением Na2O : Р2О5 от 1:1 до 1:2, в том числе и 5:3:
Р4 + 5О2 + 0,5Na2CO3 = (0,5Na2O)-2P2O5 + 0,5СО2
Пирофосфат натрия получают дегидратацией моногидрофосфата натрия при 350—400° С:
2Na2HPO4 = Na4P2O7 + Н2О
51
В присутствии незначительных количеств дигидрофосфата натрия снижается температура и увеличивается скорость конденсации моногидрофосфата. Скорость процесса образования пирофосфата натрия зависит также от молярного отношения Na2O: Р2О5. Она имеет минимальное значение при 170° С при отношении Na2O : PjOs, равном 1,5, по причине образования соли ИаНгРО^НагНРОд , устойчивой до температуры превращения ее в пирофосфат. Добавка же ~1% нитрата аммония к массе безводного NaHPO4 ускоряет в два раза процесс образования пирофосфата при 300° С.
Пирофосфат натрия можно получать в таком же аппаратурном оформлении, как и триполифосфат с соответствующим изменением режима по процессу нейтрализации кислоты, упарке раствора и температуре термообработки. Термическую фосфорную кислоту (70—80%) нейтрализуют карбонатом натрия до pH 8,8—9,0. Раствор кипятят для удаления диоксида углерода, выпаривают до концентрации моногидрофосфата натрия 48—50 % и смешивают с 3—4 масс, частями оборотного продукта (до содержания влаги в смеси 10—12%). Смесь нагревается топочными газами до 360—400°С во вращающемся барабане. Спекшиеся частицы размером до 10 мм после охлаждения измельчают. Безводный пирофосфат натрия содержит 52—53,5% общего Р2О5, в том числе 50,5—52% в пироформе и 0,9% в ортоформе.
Аналогичным методом получают пирофосфат и из экстракционной фосфорной кислоты.
Пищевой пирофосфат натрия ИагНгРгО? получают медленной дегидратацией дигидрофосфата натрия при 225—250° С или обработкой при более высокой температуре, но в течение короткого времени:
2NaH2PO4 = Na2H2P2O7 + Н2О
Выше 250° С образуется не растворимая в воде соль Маддреля, ниже 220° С сильно снижается степень конверсии ортофосфата в пирофосфат. Целевой продукт содержит до 63,9% Р2О5, а его водный раствор имеет pH 4,2.
Тринатрийфосфат получают взаимодействием тетранатрийфосфата с хлороводородной кислотой:
Na4P2O7 + НС1 = Na3HP2O7 + NaCl
Исходный технический пирофосфат натрия растворяют при 20—25° С в воде и фильтруют. Из осветленного раствора, содержащего 20—22% Na4P2O7, охлаждением до 10° С выделяют кристаллы Na^OrlOfyO. После отделения кристаллов на центрифуге их обрабатывают 35%-ной НС1 при 35° С в реакторе, снабженным мешалкой и паровой рубашкой. В полученном растворе концентрация солей должна быть не выше 35%. 52
В процессе охлаждения рассолом до 0—1° С из растворов выделяются кристаллы Na3HP2O7 • 9Н2О, которые отделяют от маточного раствора (хлорида натрия) центрифугированием. Целевой продукт содержит: 31—33% Р2О5, 0,005—0,01% не растворимых в воде веществ, 0,5—0,6% хлоридов (в расчете на хлор), менее 0,15% SO3, менее 0,01% железа.
1.7. ГИДРИДЫ И БОРГИДРИДЫ НАТРИЯ
Натрий образует гидрид и боргидрид. Гидрид натрия является типичным ионным (сомлеобразным) соединением. Боргидриды металлов— двойные комплексные гидриды бора и металлов с общей формулой Ме(ВН4)„.
Гидрид натрия — гигроскопичный белый или серовато-белый порошок. При атмосферном давлении и температуре 420° С он разлагается без плавления. Ниже приведены некоторые основные свойства гидрида натрия.
Температура плавления............
Плотность.......................
Изменение плотности при изменении температуры от 20 до 400° С......
Теплота образования (298,1 К)....
Энтропия (298,1 К)..............
Коэффициент линейного расширения (20—400°С).......................
-860°С
1,396 г/см3
1,370—1,275 г/см3
56,5 кДж/моль
48,6 кДж/(моль-К) (823 К), 11,4 э.е.
64,0-1 О'6 град "*
Гидрид натрия нерастворим в обычных инертных органических растворителях. Он растворяется лишь в расплавленном гидроксиде натрия, сплаве калий-натрий, а также в эвтектической смеси LiCl-KCl-NaCl (t ПЛ = 352°С). Растворимость гидрида натрия в металлическом натрии при температуре выше 500° С составляет 5% (масс.).
Гидрид натрия получают гидрированием металлического натрия при 350°С и отложении образующегося гидрида в виде тонких белых иголок на холодных частях прибора.
Пленки чистого гидрида натрия получают в процессе действия водорода на натриевое зеркало.
Взаимодействие натрия с водородом идет с заметной скоростью при температуре около 200° С. Поэтому реакцию проводят с расплавленным натрием. При этом давление водорода должно быть выше давления диссоциации гидрида натрия при данной температуре.
Гидрид натрия производят в виде порошка или суспензий в минеральном масле. Для получения продукта в виде порошка процесс гидрирования расплавленного натрия ведут при повышенных давлениях, применяя интенсивное перемешивание с целью разрушения образую-53
щейся пленки гидрида на поверхности натрия. Процесс проводят во вращающихся автоклавах, заполненных шарами или стержнями, а также специальных мешалок, обеспечивающих сдирание образующейся корки со стенок аппарата.
На некоторых предприятиях исходный расплавленный натрий для увеличения поверхности контакта с водородом диспергируют на поверхности инертного твердого материала, в качестве которого удобно пользоваться гидридом натрия.
Реакция металлического натрия с водородом в присутствии инертного носителя может быть проведена и во взвешенном слое.
В качестве диспергирующих материалов, добавляемых к исходному металлическому натрию, применяются минеральные масла или некоторые углеводороды (антрацен, фенантрен), высшие жирные кислоты и их соли (стеариновая или олеиновая кислота, стеараты натрия и магния). Обычно количество диспергирующей добавки составляет 0,1—1% от массы загрузки. При работе с диспергирующими добавками удобна добавка целевого продукта. Реакцию проводят при 300—380° С и давлении до 10 атм.
Суспензии гидрида натрия отстаиваются медленно, легко перекачиваются насосами и дозируются. Они не воспламеняются на воздухе и безопасны в обращении. Работа с ними не требует специальных приспособлений.
Непрерывный способ получения масляной суспензии гидрида натрия состоит из двух однотипных вертикальных цилиндрических реакторов, соединенных последовательно. В первый реактор непрерывно загружают расплавленный натрий, вводят водород под давлением 10,5 ат и нагретое масло. Температура в реакторе (313° С) автоматически поддерживается на заданном уровне. Суспензия гидрида натрия, содержащая необходимое количество металлического натрия, самотеком перетекает из верхней части первого реактора в нижнюю часть второго реактора. Вместе с суспензией передается и непрореагировавший водород. Из второго реактора суспензия целевого продукта попадает в хранилище. Технология обеспечивает 98,7%-ное использование исходного сырья и получение суспензии, содержащей 23,5% NaH.
Гидрид натрия является сильно реакционноспособным соединением. Он разлагается водой, выделяя при этом водород:
NaH 4-Н2О = NaOH 4-Н2
Реагирует со спиртами:
NaH + С2Н5ОН = NaOC2H5 + Н2
Гидрид натрия взаимодействует с карбоновыми кислотами, например с муравьиной:
NaH + НСООН = NaCOOH 4- Н2
54
В процессе реакции с гидразином образует гидразид:
NaH + N2H4 = NaN2H3 + Н2
При действии ацетилена на аммиачную суспензию гидрида натрия образуется ацетиленид натрия:
NaH + C2H2 = C2HNa + H2
С циклопентадиеном гидрид натрия реагирует по схеме
NaH + C5H6 = C5H5Na + H2
При действии хлорида водорода на гидрид натрия реакция протекает по уравнению
NaH + HCl->NaCl + Н2
а процесс сопровождается воспламенением и взрывом.
Реакция, приводящая к выделению водорода, идет между гидридом натрия и сульфидом водорода:
2NaH + H2S = Na2S + Н2
С расплавленной серой гидрид натрия реагирует очень энергично:
16NaH + Sg = 8Na2S + 8Н2
При нагревании гидрида натрия в токе сухого диоксида углерода образуется оксалат натрия:
2NaH + 2СО2 = Na2C2O4 + Н2
При нагревании гидрида натрия с оксидом углерода происходит следующая реакция:
NaH + 2СО = HCOONa + С
Диоксид серы реагирует с гидридом водорода по схеме
2NaH + 2SO2 = Na2S2O4 + Н2
При взаимодействии с хлоридом цинка гидрид натрия образует следующее соединение:
NaH + 2ZnCl2 = NaH-2ZnCl2
55
Гидрид натрия, подобно щелочным металлам и алкаголятам, является эффективным агентом реакций конденсации карбонильных соединений. Он является катализатором процесса восстановления ароматических углеводородов, процесса полимеризации бутадиена, полимеризации олефинов при низком давлении.
Гидрид натрия может служить для получения водорода, а также может быть применен для обессеривания нефтепродуктов.
Борогидрид натрия. Физико-химические свойства борогидрида натрия. Борогидрид натрия (тетрагидри-доборат натрия) NaBH4, бесцветные кристаллы с кубической гранецентрированной решеткой (а = 0,66164 нм, z = 4, пространственная группа Fm3m). Ниже -80°С переходит в тетрагональную модификацию (а = 0,4354 нм, с = 0,5907 нм). Плотность 1,074 г/см3; насыпная масса 0,3—0,4 г/см3; температура плавления 5О5°С (с разложением); Ср°=2,3 кДж/(кг-К); ДН^р=-190 кДж/моль, ДС2°98 =-119,5 кДж/моль; S°9g = 101,4Дж/(моль-К). Показатель преломления 1,574.
Борогидрид натрия в сухом воздухе стоек до температуры 300° С, выше которой начинается его окисление с выделением метабората натрия. Быстрый окислительный процесс начинается при 420—450° С. В вакууме борогидрид стоек до 400° С, при нагревании выше которой начинается медленное выделение водорода. При 500° С борогидрид натрия превращается в темно-коричневую жидкость, выделяющую водород. Быстрое разложение с образованием металлического натрия, бора, водорода и следов диборана начинается выше 550° С.
Борогидрид натрия гигроскопичен, во влажном воздухе превращается в гидрат. При повышенных температурах (выше 50° С) в результате взаимодействия с парами воды он начинает разлагаться с выделением водорода и следов диборана. Процесс разложения ускоряется присутствующим в воздухе диоксидом углерода.
Борогидрид натрия хорошо растворяется в полярных растворителях, не растворяется в диэтиловом эфире, диоксане, углеводородах. Растворимость (г в 100 г): в воде — 40,6 (0° С), 56 (25° С). Из воды ниже 36,4° С борогидрид натрия кристаллизуется в виде NaBH^HzO, а выше 36,4° С — безводный NaBH4 (рис. 1.26).
Жидкий аммиак является лучшим растворителем борогидрида натрия, поэтому он применяется в производстве для извлечения борогидрида из реакционных смесей, а также для очистки готового продукта перекристаллизацией.
В системе NaBH4—NH3 образуются аммиакаты NaBH4-3NH3 и NaBH4-4,5NH3, конгруэнтно плавящиеся при -16 и -20,5° С (рис. 1.27). 56
Содержание NaBH4, % (масс.)
Рис. 1.26. Диаграмма системы
NaBH, —Н20
NaBH4-3NH3
Содержание NaBH4, % (масс.)
Рис. 1.27. Диаграмма системы
NaBH4 — NH3
NaBH4-6N2H4 | NaBH4N2H4 |
NaBH4-2N2H4 NaBH4 0,5N2H,
Содержание NaBH4, %(масс.)
Рис. 1.28. Диаграмма системы
NaBH4 —N2H4
В системе NaBH4—N2H4 (рис. 1.28) обнаружены четыре соединения: NaBH4-2N2H4, конгруэнтно плавящиеся при 53° С; NaBH40,5N2H4, разлагающиеся по перитектической реакции при 56° С; остальные два соединения образуются в твердой фазе.
Борогидриды применяют в качестве селективного восстановителя групп С = О, С = N, NO2 в производствах антибиотиков, витаминов и стероидных препаратов. Применяют также в процессах получения бороводородов, борогидридов и гидридов других металлов, катализаторов гидрирования, для нанесения металлических покрытий на поверхность различных материалов. Они служат также в качестве порооб-разовагеля для пластиков.
Способы получения борогидридов натрия можно разделить на две группы.
К первой группе относятся способы, основанные на взаимодействии гидрида натрия (или натрия) и водорода с различными соединениями бора:
4NaH + BX^NaBFL, + 3NaX
4Na + 2H2 + BX3->NaBH4 + 3NaX
где X — О, F, Cl, алкоксил (OCH3), PO4~ и т. п.
Вторая группа способов основана на использовании диборана:
3NaX + 2B2H6->3NaBH4 + ВХ3
3NaOCH3 + 2B2H6->3NaBH4 + В(ОСН3)3 4NaH + B2H2->2NaBH4
58
Однако из многочисленных способов получения борогидрида натрия в промышленности применяются два способа: 1) взаимодействие гидрида натрия с метилборатом; 2) действием водорода и металлического натрия на боросиликат, полученный сплавлением декагидрата тетрабората натрия (буры) с оксидом кремния.
Взаимодействие гидрида натрия с метилборатом зависит от температуры и соотношения исходных реагентов. При низкой температуре протекает реакция
NaH + В(ОСН3)з = №ВН(ОСН3)з
При высокой температуре процесс идет по реакции
4NaH + В(ОСН3)з = NaBH4 + 3NaOCH3
При 200° С образующийся триметоксиборгидрид натрия реагирует с гидридом натрия:
NaBH(OCH3)3 + 3NaH = NaBH4 + 3NaOCH3
При 230° С имеет место процесс плавления триметоксиборгидри-да натрия, сопровождающийся разложением:
4NaBH(OCH3)3->NaBH4 + 3NaB(OCH3)4
№В(ОСНз)4->№ОСНз + В(ОСН3)з
Образующийся тетраметоксиборат натрия реагирует с гидридом натрия при 270—290° С:
NaB(OCH3)4 + 4NaH = NaBH4 + 4NaOCH3
Наиболее удобным является проведение реакции с суспензией гидрида в минеральном масле. Суспензия гидрида в отличие от порошкообразного гидрида безопасна в обращении. Она легко дозируется и перекачивается, что делает возможным создание непрерывного процесса. Масло поглощает теплоту реакции. Гидрид натрия, суспензированный в минеральном масле, реагирует с метилборатом в 10 раз быстрее, чем в сухом виде. Высокая скорость реакции снимает необходимость избытка (10—50%) исходного гидрида натрия.
Взаимодействие суспензии гидрида натрия с метилборатом проводят при атмосферном давлении, а реакцию с сухим гидридом — под давлением 10 ат. Выход при работе с масляной суспензией составляет 95%, в то время как с сухим гидридом натрия — 76—87%. При-59
менение масляной суспензии позволяет провести процессы в том же аппарате, в котором ведут реакцию с метилборатом.
Из полученной реакционной массы борогидрид натрия экстрагируют жидким аммиаком. Продукт аммиачной экстракции составляет 88—92%. Для экстракции применяют также изопропиламин и диглим. Процесс экстракции ведут в вертикальных аппаратах с мешалками. Полученную суспензию метилата натрия в аммиачном растворе борогидрида натрия фильтруют. Прозрачный раствор концентриуют в испарителе, а затем упаривают досуха.
Для извлечения же борогидрида натрия из масляной суспензии пользуются растворителями, не смешивающимися с маслом. Суспензию смешивают с аммиаком. После отстоя образуются два слоя: верхний— аммиачный раствор борогидрида натрия и нижний — масляная суспензия метилата натрия.
Борогидрид натрия получают также взаимодействием гидрида натрия с боратами в присутствии диоксида кремния и других связующих добавок. Установлено, что в присутствии связующих соединений образуется борогидрид натрия по реакции
4NaH + 2В20з = NaBH4 + 3NaBO2
Реакция гидрида натрия с метаборатом протекает с хорошим выходом.
Взаимодействие безводного тетрабората натрия, металлического натрия и диоксида кремния с водородом при 450—500° С и давлении 3 ат по уравнению
Na2B4O7 + 7SiO2 + 16Na + 8Н2 = 4NaBH4 + 7Na2SiO3
позволяет получить борогидрид натрия с выходом 90%.
Скорость реакции значительно повышается, если исходный тетраборат натрия предварительно сплавить с диоксидом кремния (кварцевый песок) в молярном отношении Na2B4O7: SiO2 = 1:7 при 1000—1100° С. При этом получается хрупкое, легко размалываемое боросиликатное стекло. Его измельчают и проводят реакцию.
Из полученной реакционной массы, содержащей около 13% NaBH4 целевой продукт извлекают жидким аммиаком. Поскольку процесс проходит без образования побочных продуктов, растворимых в аммиаке, целевой продукт содержит 96—98% основного вещества.
Процесс в производственном масштабе состоит из нескольких стадий. На первой стадии шихту подогревают до 350° С. На второй стадии ее обрабатывают натрием и водородом при давлении 2—6 ат. Температуру при этом поддерживают, применяя охлаждение, в пределах 300—400° С. На этой стадии реакция проходит - на 50%, оста-60
льное количество натрия превращается в гидрид. На третьей стадии реакционную массу выдерживают в атмосфере водорода при 420—450° С для завершения реакции. Процесс проводят в реакторах шнекового типа, причем для второй стадии применяют двухзаходные шнеки, которые обеспечивают хорошее перемешивание массы и исключают ее комкование и налипание на стенки. Шнек устанавливается с некоторым уклоном. Натрий вводят в точке поступления шихты в реактор второй стадии или в нескольких точках по длине этого реактора. Реакционная масса должна быть измельчена. Размеры частиц исходного тетрабората натрия должны быть не более 300 мкм, а частиц диоксида кремния 200—300 мкм. Использование диоксида кремния с размерами частиц менее 60 мкм приводит к восстановлению его до элементного кремния.
Полученная реакционная масса по описанному способу может быть использована также и для получения водных растворов борогидрида натрия. С этой целью ее обрабатывают водной суспензией оксида кальция при 60° С. При этом получается силикат кальция, выпадающий в осадок. После его фильтрации получают раствор, содержащий: NaBH4 — 38 г/л, NaOH—117 г/л и Na2SiO3—12,3 г/л. Раствор упаривают в вакууме при давлении не выше 100 мм рт. ст. Полученные растворы содержат: NaBH4—105,8 г/л, NaOH — 330,5 г/л и Na2SiO3—12,2 г/л. Раствор содержит 92% от загруженного количества NaBH». Осаждение борогидрида натрия ведут также и смесью гидроксида кальция с его хлоридом.
Борогидрид натрия получают также способами, основанными на следующих реакциях:
NaBO2 + 2СаН2 = NaBH4 + 2СаО
NaAlH4 + B(OR)3 = NaBH4 + A1(OR)3 NaAlH4 + BF3 = NaBH4 + A1F3 NaAlH4 + NaBF4 = NaBH4 + NaAlF4 NaAlFLi + BH3-N(C2H5)3 = NaBH4 + A1H3N(C2H5)3
3NaBO2 + 4A1 + 6H2 = 3NaBH4 + 2A12O3
NaBO2 + 2Mg + 2H2 = NaBH4 + 2MgO 3NaB(OCH3)4 + 4A1 + 6H2 = 3NaBH4 + 4A1(OCH3)3
Na3BO3 + Si + 2H2 = NaBH4 + Na2SiO3 3NaBO2 + 4A1 + 2Na2O + 6H2 = 3NaBH4 + 4NaAlO2 NaBO2 + Si + Na2O + 2H2 = NaBH4 + Na2SiO3
61
NaBO2 + FeSi + Na2O + 2H2 = NaBH4 + Na2SiO3 + Fe
2NaH + B2H6 = 2NaBH4
3NaOCH3 + 2B2O6 = 3NaBH4 + B(OCH3)3
Получаемый приведенными способами борогидрид натрия обычно содержит значительное количество примесей. Поэтому они практически являются полуфабрикатами. Разработаны способы очистки целевого продукта методами перекристаллизации, обработкой дибораном, промывкой эфиром и др.
1.8. КАРБОНАТ НАТРИЯ
Физико-химические свойства и применение. Карбонат натрия (кальцинированная сода) Na2CO3 — бесцветные кристаллы с температурой плавления 858° С (Д/Лш 28 кДж/моль). Гигроскопичен.
До 350° С существует a-модификация (табл. 1.13), в интервале 350—479° С — Р с моноклинной кристаллической решеткой, а выше 479° С — гексагональная у-модификация (а = 0,5215 нм, с = 0,6584 нм, z = 2, пространственная группа Рбзтс). а Р и р ^2 у
соответственно 0,80 и 2,1 кДж/моль.
Растворимость карбоната натрия в воде представлена на диаграмме (рис. 1.29). Как видно из диаграммы, ниже 32° С из водных рас
Рис. 1.29. Диаграмма системы
Na2CO3 —Н2О
творов кристаллизуется декагидрат, в интервале 32—35° С — гептагидрат, а выше 35° С — моногидрат карбоната натрия. Установлено также, что выше 112,5° С карбонат натрия существует в виде бесцветной соли. Водные растворы карбоната натрия имеют сильнощелочную реакцию.
Гидрокарбонат (питьевая или пищевая сода) NaHCO3 — бесцветные кристаллы, хорошо растворимые в воде. Растворимость гидрокарбоната натрия (%, масс.): 8,8 при 20° С, 14,1 при 60° С.
Трона (сесквикарбонат, полуторная сода) Na2CO3-NaHCO3-2H2O встречается в природе. Получают путем кристаллизации из смеси горячих растворов карбоната и
62
гидрокарбоната или пропусканием диоксида углерода в раствор карбоната натрия. В процессе охлаждения раствора трона кристаллизуется в форме тонких блестящих игл. Трона очень устойчива, не гигроскопична и не слеживается. Хорошо растворяется в воде без выделения и без поглощения теплоты.
Таблица 1.13. Свойства карбоната и гндрокарбоиата натрия
Показатель a-Na2CO3 Na2CO310H2O Na2COr7H2O На2СОзН2О NaHCOj
Сингония Моноклинная Моноклинная Ромбическая Ромбическая Моноклинная
Параметры решетки, нм:
а 0,8907 1,2754 1,448 1,072 0,751
b 0,5239 0,9009 1,950 0,5249 0,970
с 0,6043 1,260 0,7016 0,6469 0,353
угол, град 101,35 115,85 — — 93,31
Число формульных единиц в ячейке 4 4 8 4 4
Пространственная группа С2 или Ст Сс РЬса РЬса2} P2i/c
Плотность, г/см3 2,509 (25° С) 1,44 (16° С) 1,51 (20° С) — 2,159 (20° С)
С°р, Дж/(мольК) 112,30 550,32 418,4 145,6 88,2
5°298, Дж/(МОЛЬ-К) 135,00 564,00 426,8 168,15 101,3
А//°обР, кДж/моль -1129,19 -4070,03 -3200,3 -1430,01* -947,68
Карбонат натрия применяется в процессе получения мыла и других моющих средств, гидроксида натрия и других соединений натрия, в процессе варки целлюлозы, для обработки бокситов в производстве алюминия, для нейтрализации кислых компонентов при очистке нефтепродуктов, для получения пигментов. Он является компонентом шихты в производстве стекла.
Гидрокарбонат натрия применяют в качестве источника СО2 в процессе выпечки хлеба и изготовления кондитерских изделий, безалкогольных напитков, искусственных минеральных вод, в качестве лекарственных средств. Является компонентом огнетушащих составов.
Декагидрат карбоната натрия входит в состав стиральных порошков, применяемых в прачечных и в шерстомойках.
Существует также пероксокарбонат (гидропероксосольват карбоната натрия, перкарбонат натрия, коммерческое название — «персоль») Na2COj-l,5H2O2— бесцветные малогигроскопичные кристаллы ромбической сингонии (а = 0,91824 нм, Ь= 1,57513 нм, с = 0,67272 нм, 63
z = 4, пространственная группа Aba2). В кристаллической решетке ионы СОз2" связаны с молекулами Н2О2 посредством водородных связей в слои, между которыми находятся ионы Na+; Д770обр = -1474 кДж/моль, энергия отрыва 1,5 молекул Н2Ог (газ) от молекулы Na2CO3 140,7 кДж/моль.
Пероксокарбонат натрия конгруэнтно растворяется в воде; растворимость (г в 100 г): 11,8 (0°С) и 14,7 (20° С). Насыщенный водный раствор (pH 10,8) неустойчив при хранении (в течение суток практически полностью теряет активный кислород).
При постоянном нагревании распад пероксокарбоната натрия с сильным тепловыделением происходит при 140° С. В сухом хранилище при комнатной температуре пероксокарбонат натрия может храниться в течение нескольких месяцев без потери активного кислорода. Влага же заметно ускоряет процесс распада. Небольшие примеси соединений железа, марганца, меди и других переходящих металлов катализируют процесс распада пероксокарбоната натрия, а добавка силикатов, трилона Б и других комплексонов, связывающих ионы тяжелых металлов в прочные комплексы, ингибируют процесс разложения.
Пероксокарбонат натрия получают путем кристаллизации его из водного раствора карбоната натрия и пероксида водорода. Содержание пероксида водорода в растворе должно быть не менее 2%. Получают также орошением сухого карбоната натрия концентрированным раствором пероксида водорода с последующей сушкой при 40—60° С.
Пероксокарбонат натрия применяют в качестве отбеливателя в составе синтетических моющих средств, в текстильной и химической промышленности для окисления красителей и расшлихтовки тканей, а также в качестве дезинфицирующего, бактерицидного и деконтаминирующего средства.
Для стабилизации промышленного пероксокарбоната натрия и увеличения сроков его хранения в состав синтетических моющих средств вводят глицин, полиэтиленгликоль, гексациклофосфат натрия, бораты, которые также придают продукту форму гранул и покрывают их тонким слоем нерастворимой соли — карбоната, сульфата или силиката бария, кальция или магния.
Получение карбоната натрия аммиачно-хлоридным способом. В мире производство карбоната натрия базируется на четырех способах: аммиачный (из хлорида натрия методом Сольвэ); из природных залежей троны; из нефелинов и карбонизацией гидроксида натрия.
Производство карбоната натрия аммиачно-хлоридным способом является сложным замкнутым циклом химического комплекса. Основные реакции аммиачно-хлоридного производства имеют вид: 64
СаСОз СаО + СОг---------
I 1 **
СаО + Н2О = Са(ОН)2
-------------f-------
NaCl + NH3 + СО2 + Н2О NH4CI + NaHCO3 —|
।
Са(ОН)2 + 2NH4CI = 2NH3 + СаС12 + Н2О
| I ...---------------
2NaHCO3 = Na2CO3 + Н2О + СО2 ---
(1)
(2)
(3)
(4)
(5)
Для осуществления основной реакции (3) раствор хлорида натрия насыщают аммиаком и диоксидом углерода. При этом гидрокарбонат выпадает в осадок, который после отделения от жидкой фазы подвергают термообработке по реакции (5) и получают карбонат натрия. Регенерацию аммиака из маточного раствора, содержащего хлорид аммония, осуществляют по реакции (4) при взаимодействии последнего с гидроксидом кальция. Источником диоксида углерода и оксида кальция являются продукты термообработки карбоната кальция по реакции (1) и последующего гашения оксида кальция до его гидроксида по реакции (2).
Основными видами сырья в производстве карбоната натрия являются раствор хлорида натрия (рассол), аммиак и карбонат кальция, а технология состоит из следующих стадий: 1) очистка растворов хлорида натрия; 2) обжиг известняка; 3) приготовление известкового молока; 4) аммонизация рассола; 5) карбонизация аммонизированного рассола; 6) фильтрация гидрокарбоната натрия; 7) регенерация аммиака; 8) кальцинация гидрокарбоната с получением карбоната натрия.
Очистка раствора хлорида н а т р и я. Раствор хлорида натрия добывают выщелачиванием каменной соли в подземных скважинах методом гидровруба. Раствор должен быть близким к насыщению NaCl. В производстве обычно применяют растворы с концентрацией 305—310 г/л,- которые содержат соединения кальция, магния и калия.
Содержание иона кальция в неочищенном растворе обычно колеблется в пределах 0,5—2,3 г/л, а иона магния — 0,02—2,1 г/л. Небольшое содержание иона калия (до 6—8 г/л) считается безвредным для производства.
Исходный сырой рассол очищают от кальций- и магний-ионов обработкой его карбонатом натрия и гидроксидом кальция (известковым молоком) по реакциям:
СаС12 + Na2CO3 = СаСОз + 2NaCI
MgCl2 + Са(ОН)2 = Mg(OH)2 + СаС12
3 Химическая технология 65
неорганических веществ, кн. I
Рис. 1.30. Очистка раствора хлорида натрия:
1, 2, 8, 9 — сборники; 3 — смеситель; 4 — расширительный бачок; 5 — реактор; 6 — отстойник; 7 — резервуар; 10—центробежный насос
Согласно технологической схеме (рис. 1.30), исходные рассолы из сборника 1 дозируются в смеситель 3, в который из сборника 2 вводят водную суспензию гидроксида кальция (известковое молоко) из гасительного отделения известковых печей. Исходные реагенты — раствор карбоната и суспензия гидроксида кальция — в сборниках 1 и 2 соответственно смешиваются с очищенным рассолом. Исходный рассол по рас-солопроводам из сборника 9 центробежным насосом 10 перекачива
ется, проходя расширительный бачок 4, поступает в реактор 5, в котором очищается от кальций- и магний-ионов поступающей смесью, образующейся в смесителе 3. Обрабатывают рассолы в реакторе 5 при оптимальной температуре 17±3°С. Далее рассолы с осадком поступают в отстойник 6, откуда рассол непрерывно удаляется через желоб в резервуар очищенного рассола 7, расположенный по периферии. Шлам из нижней части отстойника 6 периодически спускается в сборник 8, в который поступает шлам из реактора 5.
Осветленный рассол из отстойника 7 центробежным насосом 10 перекачивают на процесс абсорбции, а шлам из сборника 8, где он разбавляется водой, центробежным насосом 10 перекачивается в шламопроводы дистиллерной жидкости.
Очищенные растворы содержат не более 0,02 г/л кальций-иона, 0,007 г/л магний-иона, 0,2—0,3 г/л На2СОз, 0,05—0,09 г/л NaOH.
Обжиг известняка. Диоксид углерода и оксид кальция получают термообработкой (обжигом) карбонатного сырья (известняка или мела) в известково-обжигательных печах шахтного типа. Необходимая для обжига теплота выделяется в процессе сгорания топлива, добавляемого в определенной пропорции к загружаемому сырью.
Разложение исходного карбоната кальция (известняка) идет по реакции
СаСОз СаО + СО2 - 178 кДж
66
На рис. 1.31 приведена зависимость равновесного давления диоксида углерода (рсо,) над поверхностью СаСОз от температуры, из которой видно, что до 600—700° С давление увеличивается незначительно, а затем резко возрастает. При работе известковых печей максимальное парциальное давление диоксида углерода в печном газе составляет 0,04 МПа. Равновесное давление СО2 достигает этой величины при температуре около 800° С. При этой температуре практически начинается процесс разложения исходного карбоната кальция. Однако с целью повышения интенсивности
Рис. 1.31. Зависимость равновесного давления диоксида углерода от температуры
процесса и степени диссоциации температуру в печах поддерживают практически в пределах 1100—1200° С.
Давление диоксида углерода может меняться в зависимости от степени дисперсности СаО и СаСОз. Тонкодисперсные образцы разлагаются при 882—895° С, крупнокристаллические образцы — при 911—921 ° С, а известняки разлагаются при 890—916° С.
В процессе обжига карбонатного сырья разлагается также карбонат магния, обычно содержащийся в известняке в количестве 0,5—3%:
MgCO3 MgO + СО2 —121 кДж
Процесс протекает при 402—480° С, а усиливается примерно при 700° С.
Лимитирующей стадией процесса является процесс образования кристаллических зародышей оксида кальция. Скорость разложения зависит также от процесса теплопередачи. Эта зависимость описывается уравнением (в интервале 1025>12>1214° С при СО2<100%):
бшс = 740 + 0,1481г + 0,13 СО2
В процессе обжига наряду с основным процессом протекает ряд побочных реакций. Образующийся оксид кальция реагирует с содержащимися в исходном сырье примесями:
СаО + SiO2 = CaSiO3
ЗСаО + 2SiO2 = 3CaO-2SiO2
3»
67
СаО + Fe2O3 = CaFe2O4
ЗСаО + А12О3 = ЗСаОД12Оз
Соединения оксида кальция с оксидами алюминия и железа наиболее легкоплавки, чем оксид кремния, и приводят к оплавлению извести.
Водяные пары оказывают каталитическое действие на процесс диссоциации СаСОз в области низких температур (650—750° С), водяной пар адсорбируется на поверхности кристаллов, облегчая выход активированных ионов за пределы кристаллической решетки.
Общепринято, шахтную известковую печь по характеру протекающих в ней термических процессов делить на три последовательные теплообменные зоны (рис. 1.32, 1.33). В верхней части печи (зоне подогрева) происходит подсушка сырья и топлива, нагревание исходной шихты (смеси известняка с коксом) до температуры начала разложения карбоната кальция
(-850° С) за счет теплоты горячих газов, движущихся противотоком из зоны обжига. На границе этих зон температура газов достигает 900—1000° С. При дальнейшем перемещении шихты вниз протекает горение топлива и разложение СаСОз. В конце зоны обжига температура кускового материала и газов достигает максимума (1100—1200° С), а процесс разложения исходного сырья останавливается. Поэтому зону, в которой происходит разложение СаСОз и переход в газовую фазу СО2, называют зоной обжига.
В нижней части зоны обжига процесс горения топлива про-
Концентрация СО^ОъСО, %
б
Рис. 1.32. Изменение температуры и состава газа и материала по высоте печи: a — схема расположения теплообменных зон в шахтной печи; б — температура и состав по высоте печи;
1 — СО2 от горения топлива; 2 — СО2 суммарная; 3 — температура газа; 4 — температура материала
бе
должается, а выделяющаяся теплота расходуется на нагревание воздуха, поступающего из зоны охлаждения. В соответствии с этим зоной горения называют зону, в которой идет расход кислорода на процесс сжигания топлива. Высота зоны горения зависит в основном от диаметра среднего куска топлива (см. рис. 1.33), равномерности распределения топлива в слое, концентрации кислорода и от вида твердого топлива. Наилучшее использование тепла в печи достигается при соотношении средних размеров кусков топлива и известняка от 1:1 до 1:2. Поэтому исходный кокс имеет размер 30—80 мм, а антрацит марки АК «крупный» в виде кусков размером 50—100 мм в поперечнике. Размер же кусков карбонатного сырья составляет в на некоторых заводах 30—150 мм.
Рис. 1.33. Продолжительность процесса обжига СаСО3 в зависимости от размеров кусков
основном 40—120 мм, а
Согласно приведенной на рис. 1.34 схеме получения оксида каль-
ция и диоксида углерода, исходный известняк в смеси с твердым топливом — шихта при помощи загрузочного устройства 1 поступает в печь 2. Образующийся в печи 2 оксид кальция (известь) непрерывно выгружается механизмом 3 в ковшовый транспортер 4, который
Рис. 1.34. Схема получения извести:
1 — загрузочный механизм; 2 — печь; 3 — выгружной механизм; 4 — ковшовый транспортер; 5 — бункеры; 6 — общий коллектор; 7—абсорбер; 8 — электрофильтр
69
загружает оксид кальция в бункеры 5, откуда оксид кальция направляется в гасители извести. Диоксид углерода, образующийся в печи 2, через общий коллектор 6 подается в абсорбер 7 для промывки. Промытые газы проходят через электрофильтр 8 и направляются в компрессорное отделение.
Получение суспензии оксида-гидроксида кальция (известкового молока). Суспензию оксида-гидроксида кальция (известковое молоко) получают смешением оксида кальция (извести) избытком воды:
СаО + иН2О->СаО + Са(ОН)з + (п -1) Н2О
Оксид кальция растворяется в воде. При температуре 0° С растворяется 0,185 г на 100 г воды, а при 100° С — всего 0,077 г на 100 г Н2О. Кинетика процесса гидратации, физико-химические свойства получаемой суспензии, выход готового продукта зависят от следующих факторов: вид, структура и химический состав исходного карбоната кальция; температура и продолжительность термообработки исходного сырья; наличие примесей в исходном сырье и топливе; гранулометрический состав применяемого оксида кальция, продолжительность и условия его хранения; температура воды; водо-известковое отношение (Н2О:СаО); способ и интенсивность перемешивания в процессе гидратации; применение добавок и т.д.
На рис. 1.35 приведены кинетические кривые процесса растворения в воде оксида кальция, полученного при различных температурных условиях обжига известняка. Данные показывают, что гашение обожженной извести с частицами размером 10—20 мм протекает в 1,5 раза быстрее, чем гашение оксида кальция, частицы которого имеют размер 60—80 мм.
Рис. 1.35. Кинетические кривые процесса растворения оксида кальция:
1 — мягкообожженного; 2 — среднеобожженного; 3 — жесткообожженного 70
На кинетику процесса растворения оксида кальция в воде влияют также примеси исходного сырья. Так, сесквиоксид железа, диоксид титана и сесквиоксид хрома замедляют процесс гидратации, а добавка 5% оксида магния снижает скорость реакции на 33%. Существенно замедляет процесс и сульфат кальция. Ускоряет процесс присутствие в системе электролитов (рис. 1.36).
Согласно технологической схеме (рис. 1.37), процесс растворения оксида кальция с получением водной суспензии оксида и гидроксида кальция осуществляют следующим образом.
Исходный оксид кальция (известь) из бункера 1 дозируется барабанным питателем 2 во вращающийся барабанный гаситель 3, в который непрерывно поступает подогретая в эжекционном водоподог-ревателе-конденсаторе 4 вода (обычно для этого используют очищенную хозяйственно-фекальную воду, осветленную часть дис-тиллерной жидкости, маточные растворы дистилляции слабой жидкости и слабую известковую суспензию, полученную в процессе помола и промывки мелких отходов гашения). Подогретая в конденсаторе 4 вода после смешения в коллекторе подается в гаситель 3 и на промывку мелких отходов гашения в шаровую мельницу 6. В конце гасителя 3 смонтирован барабан 7, который имеет вытяжную трубу 8 для выхода паров из системы.
Выходящая из гасителя 3 суспензия распределяется в сортировочном барабане 7 на три фракции: крепкую известковую суспензию, содержащую инертные примеси и недиспергировавшиеся зерна оксида кальция, мелкие и крупные отходы гашения. Крепкая суспензия оксида кальция проходит через сито первой по ходу материала секции сортировочного барабана 7 и после тонкой очистки на вибраци-
т, мин
Рис. 1.36. Кинетика процесса седиментации гидроксида кальция с применением электролитов:
1— 1%-ный раствор NaCl; 2 — Н2О; 3— дистиллерная жидкость, очищенная от SQJ"; 4— 1%-ный раствор Na2SO4; 5 — дистиллерная жидкость
71
Рис. 1.37. Схема процесса приготовления суспензии оксида и гидроксида кальция: 1 — бункер; 2 — барабанный питатель; 3— гаситель оксида кальция; 4 — эжекционный водоподог-реватель-конденсатор; 5 — коллектор горячей воды; 6 — шаровая мельница; 7 — сортировочный барабан; 8 — вытяжная труба; 9 — вибрационные сита; 10 — сборник; 11 — насос; 12 — ленточный конвейер; 13 — скребковый транспортер; 14 — односпиральный классификатор; 15 — сборник слабой известковой суспензии; 16 — сборник шлама
онном сите 9 поступает в сборник 10, оборудованный мешалкой, откуда насосом 11 перекачивается в отделение дистилляции. Твердый остаток поступает во вторую секцию вибрационного сита 9, где его промывают подаваемой в сортировочный барабан 7 горячей водой. Частицы оксида кальция менее 15 мм (мелкие отходы гашения) проходят сквозь сито 9, а крупные куски недокала и топлива, отмытые от известковой суспензии, направляются ленточным конвейером 12 на склад сырья и топлива.
Мелкие отходы гашения отделяют на вибросите 9 от слабой известковой суспензии и по распределительному желобу подают в сборник, снабженный перемешивающим устройством, затем скребковым транспортером 13 направляют в шаровую мельницу 6 мокрого помола. Для разбавления шлама используют горячую воду или дис-тиллерную жидкость. Слабую известковую суспензию, получаемую в процессе размола, гашения и диспергирования оксида кальция, очищают на классификаторах 14 и подают через распределительный желоб в сборник 15. Шлам из классификатора 14 в сборнике шлама 16 72
репульпируют дистиллерной жидкостью и откачивают на стадию перекачки промстоков или на фильтрацию, где смешивают со шламом после дистилляции.
В табл. 1.14 приведен аналитический состав водной суспензии смеси оксида и гидроксида кальция.
Таблица 1.14. Аналитический состав водной суспензии смеси оксида и гидроксида кальция
Суспензия СаО и Са(ОН)2 Шлам из классифи-кагора, % (масс.) Крупные отходы, % (масс.)
% (масс.) н.д.
СаО (акт) 23,5 215,0 25,0 2,0
СаО (неакт) 1,5 13,7 14,0 7,0
MgO 0,3 3,8 0,8 0,3
СаСОз 2,0 11,8 23,2 83,0
R2O3 0,2 — 3,0 1,0
SiO2 + Н.О.* 1,7 — 4,0 4,5
СаС12 2,2 10,1 — —
NaCl 1,1 4,8 — —
Н2О 67,5 952,1 30,0 1,0
*Н.О.— нерастворимый остаток.
Аммонизация рассола. Исходное сырье — рассол до насыщения диоксидом углерода в карбонизационных колоннах в отделении адсорбции насыщают аммиаком и частично диоксидом углерода из парогазовой смеси отделений дистилляции, карбонизации и газовоздушной смеси вакуум-фильтров.
Основной задачей отделения абсорбции является получение аммонизированного рассола с передачей его в отделение карбонизации рассолов следующего состава:
Содержание, н.д.: аммиака......... 100—106
хлорид-ионов... не менее 89
Температура, °C. 28—32
Кроме того, отделение адсорбции должно обеспечивать поглощение аммиака из выхлопных газов производства карбоната натрия.
Для насыщения рассола применяют парогазовую смесь, поступающую из отделения дистилляции (свыше 80% от всей массы) и содержащую аммиак и пары воды. Количество последних зависит от температуры смеси. Обычно в производстве температуру смеси поддерживают не ниже 58° С. При более низких температурах происходит процесс
73
кристаллизации карбонатов аммония NH4HCO3 и (NH4)2CO3, образующихся по схеме:
NH3 + Н2О + CO2->NH4HCO3
2NH3 + Н2О + CO2->(NH4)2CO3
Однако в опубликованных в последние годы работах утверждают, что основной реакцией, протекающей в процессе аммонизации, считается реакция аммиака с диоксидом углерода по уравнению
2NH3 + СО2 NH2COONH4
Образование карбамата аммония создает более благоприятные условия в процессе абсорбции аммиака и диоксида углерода в результате снижения равновесного давления NH3 и СО2 над раствором. Растворы охлаждают, так как процесс поглощения аммиака рассолом сопровождается выделением теплоты.
Процесс абсорбции диоксида идет значительно медленнее, чем аммиака. В связи с этим аппаратуру рассчитывают так, чтобы обеспечивать поглощение заданного количества аммиака и максимально возможного количества диоксида углерода, которое кроме температурного режима определяется присутствием аммиака в рассоле по всей поверхности аппаратов.
Согласно схеме процесса абсорбции, приведенной на рис. 1.38, очищенный рассол из отделения рассолоочистки поступает в напорную емкость 3, откуда самотеком поступает в аппараты абсорбции. Около 75—80% рассола из емкости 3 поступает в промыватель воздуха фильтров (ПВФЛ) 4, где поглощается аммиак из воздуха, проходящего через фильтрующую ткань вакуум-фильтров и содержащего около 0,5—1,0% (масс.) аммиака. Из ПВФЛ рассол поступает во второй промыватель газа колонн (ПГКЛ-2) 7, где абсорбируется аммиак из отходящих газов отделения карбонизации, содержащих до 10% NH3. Остальные 20—25% очищенного рассола из напорной емкости 3 поступают в промыватель газа абсорбции (ПГАБ) 1, в котором абсорбируется аммиак из отходящих газов отделения абсорбции, содержащих до 5% NH3. Одновременно поглощается некоторое количество диоксида углерода, содержащегося в выхлопных газах. Газ из ПВФЛ поступает на вакуум-насос 5, создающий разрежение на вакуум-фильтрах, и выбрасывается в атмосферу. Газ из ПГАБ, содержащий не менее 75% СО2, поступает в вакуум-насос 2, откуда он передается на смешение с диоксидом углерода из содовых печей (в ПГСП), а затем — на карбонизацию. Выхлопные же газы из ПГКЛ-2 выбрасываются в атмосферу. Газ из ПГАБ, содержащий не менее 75% СО2, вакуум-насосом 74
Рис. 138. Схема процесса абсорбции:
1 — промыватель газа абсорбции; 2, 5 — вакуум-насосы; 3 — напорная емкость; 4 — промыватель воздуха фильтров; 6 — холодильник газа дистилляции; 7 — второй промыватель газа колонн;
8 — сепаратор-брызгоуловитель; 9 — сборник аммонизированного рассола; 10, 12 — оросительные холодильники; 11 — постамент-резервуар; 13 — второй абсорбер; 14 — первый абсорбер
передается на смешение с диоксидом углерода содовых печей (в ПГСП), а затем на процесс карбонизации. Выхлопные газы из ПГКЛ-2 выбрасываются в атмосферу.
Рассол после промывки выхлопных газов из ПГКЛ-2 и ПГАБ поступает на основную операцию — абсорбцию аммиака из газа дистилляции, осуществляемую в две ступени — в первом абсорбере (АБ-1) 14 и во втором абсорбере (АБ-2) 13. Две ступени абсорбции диктуются необходимостью в промежуточном охлаждении рассола, нагревающегося в процессах растворения и взаимодействия аммиака с диоксидом углерода и конденсацией водяного пара, поступающего с газом дистилляции. После первого абсорбера нагретый до 60—65° С рассол охлаждают до 28—32° С в оросительном холодильнике 12, после чего он поступает во второй абсорбер, где также нагревается до 65° С, и поэтому, до поступления в сборник аммонизированного рассола (САР), вторично охлаждается в оросительном холодильнике 10. С целью обеспечения самотека рассола на оросительный холодильник второй абсорбер и стоящие на нем аппараты расположены на постаменте-резервуаре 11.
Газ из отделения дистилляции охлаждается и осушается в холодильнике газа дистилляции (ХГДС) 6 охлаждающей водой, а затем, пройдя сепаратор-брызгоуловитель 8, противотоком к рассолу проходит последовательно второй и первый абсорберы, где аммиак поглощается практически полностью. Вместе с аммиаком из газа поглощается значительная часть диоксида углерода. Неабсорбированная часть газов идет в ПГАБ 1. В процессе охлаждения газа дистилляции в ХГДС водяные газы конденсируются, образуя конденсат, содержащий до 150—200 н.д. аммиака и диоксида углерода. Аммиак из таких жидкостей, называемых «слабыми», регенерирует на отдельной установке. Получаемый же при этом газ, содержащий аммиак, диоксид углерода и водяные пары, после охлаждения до 58—60° С поступает в первый абсорбер.
Во всех аппаратах отделения абсорбции жидкости и газы движутся по принципу противотока, что обеспечивает наиболее полное извлечение абсорбируемых компонентов из газа.
Карбонизация аммонизированного рассо-л а. Процесс насыщения аммонизированного рассола диоксидом углерода, в результате которого образуется гидрокарбонат натрия, является основным процессом в производстве карбоната натрия. Образование гидрокарбоната натрия идет по реакции
NaCl + NH3 + СО2 + Н2О NaHCO3 + NH4C1
Процесс ведут ступенчато. В начале процесса аммонизированный рассол, частично обработанный в отделении абсорбции диоксидом углерода, обрабатывают отходящим из известковых печей газом в ко-76
донне предварительной карбонизации (КЛПК), затем в первом про-мывателе газа колонн (ПГКЛ-1) — газами, отходящими из осадительных колонн, и, наконец, в осадительных колоннах (КП), в нижнюю часть которых вводят смешанный газ (65—75% СОг), а в среднюю часть — газ известково-обжигательных печей.
На первой стадии карбонизации большая часть диоксида углерода реагирует с аммиаком:
2NH3 + СО2 -> NH2COONH4
Образующийся карбамат аммония гидролизуется NH2COONH4 + Н2О NH4HCO3 + NH3 с образованием перенасыщенного по HCOj раствора. Из пересыщенного раствора кристаллизуется гидрокарбонат натрия:
NH4HCO3 + NaCl NaHCO3 + NH4C1
Отделение карбонизации (рис. 1.39) комплектуется сериями колонн. В состав серии входят осадительные карбонизационные колонны 5, первый промыватель газа колонн 3 и теплообменник 2. Периодически каждую из осадительных карбонизационных колонн (КД) ставят на промывку, тогда она выполняет функции колонны предварительной карбонизации 1.
Аммонизированный рассол из сборника аммонизированного рассола (САР) поступает в колонну предварительной карбонизации (КЛПК). Одновременно в колонну подается газ известковых печей, содержащий 38—40% (масс.) СО2. В КЛПК параллельно осуществляется промывка колонны от гидрокарбоната натрия, осаждающегося на внутренних поверхностях аппарата, и предварительная карбонизация аммонизированного рассола. Из КЛПК раствор, содержащий 55—60 н.д. диоксида углерода с температурой 42—46° С, подается в первый промыватель газа колонн (ПГКЛ-1). Одновременно в промыватель поступает газ из КЛПК и осадительных колонн. В ПГКЛ-1 улавливается аммиак, отдутый карбонизующим газом из КЛПК. Основное назначение аппарата — более глубокое, чем в КЛ, извлечение диоксида углерода из карбонизующего газа. Это наравне с повышением общей степени использования диоксида углерода в процессе способствует снижению выдувания из КЛ аммиака. Более полному выполнению ПГКЛ-1 своих технологических функций способствует снижение температуры жидкости. При этом снижается равновесное давление NH3 и СО3 над жидкостью, что повышает эффективность процесса их абсорбции. Поэтому в некоторых технологических схе-77
Рис. 1.39. Схема процесса карбонизации аммонизированного рассола:
1 — колонна предварительной карбонизации; 2 — теплообменник; 3 — первый промыватель газа колонн; 4 — центробежный насос; 5 — осадительная карбонизационная колонна
мах предусмотрено охлаждение рассола перед подачей в ПГКЛ-1 до 35—38° С. С этой целью в некоторых конструкциях аппарата предусмотрены встроенные холодильники. Содержание диоксида углерода в жидкости, выходящей из ПГКЛ-1, обычно составляет 60—66 н.д. С целью повышения производительности КЛ и снижения выдувания из них аммиака иногда увеличивают концентрацию диоксида углерода до больших значений. Такой режим называют «глубокой предкарбо-низацией» и осуществляют его путем вдувания в ПГКЛ-1 газа известковых печей.
За период прохождения жидкости через ПГКЛ-1 ее температура повышается на 5—8° С. Для дополнительного охлаждения жидкости после ПГКЛ-1 предусмотрен теплообменник 2, откуда жидкость поступает в осадительную карбонизационную колонну. В нижнюю часть 78
колонны подается смешанный газ (первый ввод), содержащий диоксид углерода 70—80% (масс.), а в среднюю часть — газ известковых печей (второй ввод). Газ из осадительной карбонизационной колонны 5 направляется в ПГКЛ-1, а суспензия — в отделение фильтрации.
Карбонизационные колонны работают сериями, чтобы обеспечить непрерывность потока суспензии, направляемого в отделение фильтрации. В промышленности наибольшее распространение получили серии, состоящие из четырех карбонизационных колонн, из которых три работают в качестве осадительных, а одна — колонна предварительной карбонизации.
Обычно в производстве существуют следующие нормы технологического режима процесса карбонизации:
Содержание СО2 в жидкости, н.д.: выходящий из КЛПК.................... не выше 60
выходящий из ПГКЛ-1................. 60—76
Температура жидкости, поступающей в осадительную карбонизационную колонну, °C 42-—46
Содержание общего аммиака в суспензии, выходящей из КЛ, н.д................... 92—97
Прямой титр суспензии, н.д............ 23—27
Содержание хлорид-иона в суспензии, вы-
ходящей из К Л, н.д.................... не менее 95
Температура суспензии, °C............. не выше 32
Содержание остаточной влаги в гидрокар-
бонате натрия, %....................... не выше 18
Фильтрация суспензии гидрокарбоната натрия. В производстве карбоната натрия процесс фильтрации используется для отделения кристаллов гидрокарбоната натрия от маточных растворов. Получаемые при этом кристаллы направляют в отделение кальцинации, а маточный раствор с промывной водой — в отделение дистилляции для регенерации аммиака.
На рис. 1.40 представлена типовая схема отделения фильтрации, согласно которой суспензия из карбонизационных колонн по трубопроводам поступает в емкость 1, которая представляет собой открытый прямоугольной формы сосуд. Из емкости 1 суспензия по общему трубопроводу поступает в корыто вакуум-фильтров 2. При этом количество суспензии регулируется дроссельными заслонками 5. На барабанных вакуум-фильтрах происходит разделение суспензии. Осадок снимается с барабана фильтра и по желобу поступает на транспортер, направляющий гидрокарбонат в отделение кальцинации.
Смесь воздуха и фильтровой жидкости, прошедшая через фильтровальную перегородку, поступает в сепаратор 4, где разделяется.
79
Рис. 1.40. Схема отделения фильтрации гидрокарбонатной суспензии:
1 — емкость; 2 — вакуум-фильтры; 3 — дроссельные заслонки; 4 — сепараторы; 5, 8, 10 — насосы;
6 — сборник фильтрованной жидкости; 7 — буферный сборник; 9 — сборник жидкости; 11 — промыватель воздуха фильтров
Отфильтрованная жидкость из сепаратора 4 поступает в сборный коллектор, а по нему — в сборник фильтрованной жидкости 6, из которого центробежным насосом 5 подается в отделение дистилляции. Из сепараторов 4 воздух отсасывается через общий вакуумный коллектор вакуум-насосом 10 и выбрасывается в атмосферу.
Между сепараторами вакуум-фильтров и вакуум-насосов устанавливают промыватель воздуха фильтров 77, предназначенный для улавливания содержащихся в воздухе аммиака и диоксида углерода.
Раствор из сборника 9 используется для промывки осадка гидрокарбоната натрия на фильтрах. Для регенерации фильтровальной ткани на фильтры с помощью воздуходувки подается сжатый воздух. С целью поддержания постоянного уровня в корыто фильтра подается несколько большее количество суспензии, чем может переработать фильтр. Избыток ее из емкости 7 перетекает через перелив по трубопроводу в вакуумный сборник 7, снабженный мешалкой. Из сборника 7 насосом 8 суспензию перекачивают в емкость 7. Процесс ведут так, чтобы в сборник 7 поступало минимальное количество суспензии.
В процессе фильтрования имеют место потери гидрокарбоната натрия в результате его частичного растворения в промывной воде и 80
прохода в виде мелких кристаллов через фильтрующую перегородку, а также в результате частичного протекания обратной реакции:
NaHCO3 + NH4CI NaCl + NH4HCO3
Потери в процессе фильтрации увеличиваются при повышении температуры промывной воды выше 45° С и ее количества, а также при нарушении целостности фильтрующей перегородки. В табл. 1.15 приведены данные по зависимости выхода карбоната натрия, количества испаряемой влаги и расхода теплоты на стадии кальцинации от содержания влаги в гидрокарбонате натрия.
Таблица 1.15. Зависимость показателей работы отделения кальцинации от
содержания влаги в гидрокарбонате натрия после < шльтрации
Содержание влаги, % Количество удаляемой влаги, кг на 100 кг готовой продукции Относительный расход тепла для испарения воды на стадии кальцинации, % Выход карбоната натрия* из 100 кг влажного гидрокарбоната натрия
22,23 45,0 139,7 49
20,75 41,3 126,1 50
19,16 37,5 114,7 51
17,58 33,8 100,0 52
15,99 30,2 91,7 53
14,31 26,5 80,3 54
*Из 100 кг влажного гидрокарбоната натрия теоретически максимальный выход карбоната натрия— 63 кг.
Дистилляция аммиака и диоксида углерода. В отделении дистилляции карбоната натрия производится полное отделение аммиака и диоксида углерода из маточного раствора и возвращение их в отделение абсорбции. При этом содержание аммиака должно быть 51—53% (масс.), диоксида углерода — 26—28% (масс.). Температура парогазовой смеси поддерживается в пределах 58—60° С.
В процессе дистилляции протекают реакции разложения карбонатных солей аммония за счет нагревания растворов и реакции разложения связанного аммония при взаимодействии его с суспензией гидроксида кальция. Образующиеся аммиак и диоксид углерода выделяют отгонкой из раствора при повышении его температуры.
Процесс десорбции из фильтровой жидкости осуществляют в дистилляционной колонне, состоящей из дистиллера (ДС), теплообменника дистилляции (ТДС) и конденсатора-холодильника газа дистилляции (КХДС). Обычно нагрев ведется паром. При 35—40° С начинается разложение гидрокарбоната аммония и выделение диоксида углерода:
NH4HCO3->NH3 + СО2 + Н2О
81
При повышении температуры фильтрового раствора до 65—76° С начинает диссоциировать карбонат аммония:
(NH4)2CO3 -> 2NH3 + СО2 + Н2О
Образующийся аммиак в растворе отрицательно влияет на скорость и полноту выделения диоксида углерода, поскольку растворимость последнего растет в присутствии аммиака. С повышением температуры до 80—90° С в теплообменнике дистилляции (ТДС) резко возрастает скорость разложения карбоната. С приближением к точке кипения содержащийся в системе диоксид углерода десорбируется практически полностью.
В КХДС и ТДС кроме приведенных уравнений протекает также следующая реакция:
NaHCOj + NH4CI -> NaCl + NH4HCO3
Фильтровый раствор из ТДС направляется в реактор с мешалкой (смеситель), куда одновременно вводят также водную суспензию гидроксида кальция, между которыми происходит реакция регенерации аммиака из хлорида аммония:
2NH4C1 + Са(ОН)2 = СаС12 + 2NH3 + 2Н2О
Кроме того, в смесителе происходит реакция между карбонатом аммония и гидроксидом кальция:
(NH4)2CO3 + Са(ОН)2 = СаСО3 + 2NH3 + 2Н2О
Помимо фильтровой жидкости в отделение дистилляции поступает незначительное количество слабой жидкости, содержащей в растворенном виде гидрокарбонат и карбонат натрия. В процессе повышения температуры гидрокарбонат натрия диссоциирует с выделением диоксида углерода:
2NaHCO3 -> Na2CO3 + СО2 + Н2О
Последний реагирует с хлоридом аммония, образуя при этом аммиак:
Na2CO3 + 2NH4CI = 2NaCl + 2NH3 + СО2 + Н2О
Согласно приведенной на рис. 1.41 схеме отделения дистилляции, маточный раствор из вакуум-фильтров поступает в конденсаторную 82
На абсорбцию
Рис. 1.41. Схема отделения дистилляции фильтровой жидкости:
7— дистиллер; 2 — теплообменник дистилляции; 3 — конденсатор-холодильник газа дистилляции; 4— смеситель; 5 — первый испаритель; 6 — второй испаритель; 7—парозжектор
На абсорбцию
часть конденсатора-холодильника газа дистилляции (КХДС), где нагревается до 70—78° С и поступает в верхнюю царгу теплообменника дистилляции 2, откуда он движется сверху вниз, соприкасаясь с поднимающейся в противоположном направлении парогазовой смесью.
В ТДС происходит разложение карбонатов аммония, содержащихся в жидкости. Образующийся при этом диоксид углерода практически полностью отгоняется из раствора. Из ТДС жидкость, нагретая до 95—98° С, направляется в смеситель 4, в который при 90—96° С поступает суспензия гидроксида кальция, содержащая 200—240 н.д. активного СаО.
Из нижней части смесителя 4 суспензия поступает в верхнюю часть дистиллера 1 и движется в нем сверху вниз, постепенно теряя аммиак в результате контактирования с поднимающейся вверх парогазовой смесью. Дистиллер 1, как и ТДС, состоит из отдельных царг, между которыми установлены контактные элементы. Суспензия из дистиллера при 108—115° С поступает в первый испаритель 5, а затем во второй испаритель 6. После испарителей суспензия проходит пескоотделитель (на схеме не показан), где отделяются крупные взвешенные частицы. Затем ее разбавляют холодной водой для лучшей работы насосов и перекачивают в обширный земляной отстойник-накопитель («белое море»).
Ниже приведен технологический режим работы отделения дистилляции:
Температура газа, °C: после КХДС.............................. 58—60
после КХДСЖ............................ 58—60
Содержание СО2 в жидкости после ТДС. н.д. не более 1 Давленйе в нижней царге, МПа:
ДС..................................... не более 0,168
ДСЖ.................................... не более 0,115
Содержание, н.д.: хлорид-ионов в суспензии после ДС....... не менее 61
аммиака в суспензии после ДС............ не более 0,1
Прямой титр жидкости, н.д.:
после ДСЖ-2............................ не более 0,1
после ДСЖ-1............................ 1—3
Содержание, н.д.: активного СаО в суспензии после ДС 1,5—2,5 хлора в флегме конденсаторной части
КХДС при подаче в колонну ДСЖ.......... не более 1
Отделение кальцинации г идро к арб онат а натрия. Процесс кальцинации гидрокарбоната натрия является за-84
ключительной стадией в производстве технического карбоната натрия. Он сводится к термическому разложению гидрокарбоната натрия до карбоната:
2NaHCO3 -> Na2CO3 + СО2 + Н2О
Влажный технический гидрокарбонат, поступающий на процесс кальцинации, имеет следующий состав (%, масс.): NaHCO3 — 76—84; Na2CO3 — 2—3; NaCl — 0,2—0,4; NH4HCO3 —1—2; (NH4hCO3—1; H2O — 14—20. Наличие влаги в гидрокарбонате натрия снижает сыпучесть. Влага, представляющая собой насыщенный раствор гидрокарбоната натрия, в процессе кальцинации, а именно при контакте с горячей поверхностью, интенсивно испаряется. В результате выделяющаяся твердая фаза, кристаллизуясь, образует плотно прилипающую к поверхности кальцинатора корку, называемую в производстве «козел». С целью исключения этого явления в производстве исходный влажный гидрокарбонат натрия перед подачей в кальцинатор смешивают с сухим карбонатом натрия (содой). При этом образуется новая твердая фаза— трона—NaHCO3 Na2CO3-2H2O. В процессе смешения свободная влага переходит в кристаллизационную, и продукт становится сыпучим:
NaHCO3 + Na2CO3 + 2H2O->NaHCO3Na2CO3-2H2O
Термографическими исследованиями установлено, что трона в процессе нагревания при 111 ° С теряет кристаллизационную воду:
3(Na2CO3 NaHCO3-2H2O) -► Na2CO3-3NaHCO3 + 2Na2CO3 + 6Н2О
а с достижением температуры 127° С происходит разложение двойной соли по реакции
2(Na2CO3-3NaHCO3) = 5Na2CO3 + ЗСО2 + ЗН2О
являющейся наиболее медленной, лимитирующей процесс кальцинации.
Содержащийся в исходной смеси карбамат натрия-аммония легко разлагается в присутствии водяного пара:
2NH2COONa + Н2О = Na2CO3 + 2NH3 + СО2
В данном процессе выделяется около половины общего количества аммиака, а остальное количество аммиака выделяется в процессе разложения гидрокарбоната натрия, содержащегося в троне.
Карбонат аммония разлагается и в процессе нагревания:
(NH4)2CO3 ->2NH3 + СО2 + Н2О
85
Хлорид аммония реагирует с гидрокарбонатом натрия:
NH4C1 + NaHCO3 = NaCl + NH3 + Н2О + СО2
Следовательно, в процессе кальцинации карбонат натрия образуется из троны и гидрокарбоната натрия. Кинетика процесса определяется соотношением количеств троны и гидрокарбоната натрия в исходной смеси, влажностью гидрокарбоната натрия, а также качеством их смешения. Практически показано, что печь карбоната натрия, или паровой кальцинатор, работают нормально при влажности исходной шихты 6—8%. Исходя из этого количество ретурного карбоната натрия рассчитывают по формуле
/?=^ВЛ.ЖМ—1,
где R — расход ретурного карбоната натрия, кг/кг влажного гидрокарбоната натрия; —влажность сырого гидрокарбоната натрия, %; ^см— приведенная влажность смеси, поступающей на кальцинацию, на практике обычно принимают FFCM = 7%.
Процесс кальцинации осуществляется в печах с ретурным или безретурным питанием и паровых кальцинаторах.
Согласно схеме получения карбоната натрия (рис. 1.42), отмытый на фильтрах влажный гидрокарбонат натрия с общего ленточного транспортера 10 плужковым сбрасывателем 7 подается в бункер 6 вибропитателя 5, откуда вибропитателем и ленточным транспортером 4 через ячейковый питатель 3 подается в смеситель 2. В смеситель одновременно поступает ретурный карбонат натрия и карбонат натрия, отделяемый от газов процесса кальцинации в циклоне 11.
Подготовленную в смесителе 2 карбонатно-гидрокарбонатную смесь (трону) направляют в межтрубное пространство барабана кальцинатора 1.
Кальцинируемая масса из-за вращения и наклона кальцинатора перемещается вдоль оребренных труб в направлении выгрузки, контактируя с теплопередающей поверхностью. В результате тепловой обработки троны образуется карбонат кальция и газы кальцинации (СО2, Н2 и NH3). Карбонат натрия ячейковым питателем 15 выводится из кальцинатора и поступает на систему транспортеров 8, 9 и 16. С наклонного транспортера 8 питателем производится отбор карбоната натрия в смеситель. Основная масса карбоната натрия транспортерами 9 и 14 передается на склад.
Газы, образующиеся в процессе кальцинации, удаляются из кальцинатора через смеситель 2, в котором компрессором создается вакуум. До компрессора газы проходят сухую очистку в циклонах 11 и мокрую в цеховом коллекторе газа кальцинации 12 и промы-86
Рис. 1.42. Схема отделения кальцинации:
1 — паровой кальцинатор; 2 — питающий смеситель; 3, 75 — ячейковые питатели; 4, 10 — ленточные транспортеры; 5 — вибропитатель; 6 — печка-бункер; 7 — плужковый сбрасыватель; 8, 9, 14, 16 — транспортеры; 11 — циклон; 72 — коллектор газа кальцинации; 73 — сепаратор; 77—сборник конденсата; 18— центробежные насосы; 19 — сборник слабой жидкости; 20 — холодильник газа кальцинации; 27 — редукционная охладительная установка (РОУ); 22 — промыватель газа кальцинации; 23 — сборник промывной жидкости
вателе 22. Перед промывателем газы кальцинации охлаждаются в холодильнике 20.
На процесс орошения в коллектор газа кальцинации подают так называемую слабую жидкость, образующуюся в процессе конденсации водяных паров в холодильнике газов кальцинации 20. Жидкость, соприкасаясь с газом, поглощает частично аммиак и пыль карбоната натрия, стекая после этого в сборник 19.
В холодильнике 20 газ проходит сверху вниз по межтрубному пространству, а в трубах противотоком движется охлаждающая вода. С целью исключения закристаллизовывания трубок холодильника и лучшей промывки газа от пыли карбоната натрия межтрубное пространство орошается неконцентрированной жидкостью. В промывате-ле 22 с целью дополнительного охлаждения и полной отмывки от карбоната натрия и аммиака газ орошается водой.
Кальцинатор обогревается водяным паром высокого давления. Перед подачей в кальцинатор он проходит редукционную охладительную установку (РОУ), в которой температура его снижения до 270° С, а давление — до 3 МПа. В трубках кальцинатора пар конденсируется, отдавая теплоту кальцинируемому материалу. Конденсат из кальцинатора выводится в сборник конденсата 17 и далее в расширители, где преобразуется в пар низкого давления.
87
Технологическая схема отделения кальцинации карбоната натрия при использовании печей карбоната натрия с ретурным питанием аналогична рассмотренной. При использовании печей с безретурным питанием исходный влажный гидрокарбонат натрия подается в барабан печи специальным забрасывателем. Смешение его с карбонатом натрия производится внутри печи, поэтому из технологической схемы исключается смеситель и упрощается транспортирование карбоната натрия.
Одним из основных аппаратов в производстве карбоната натрия является печь карбоната натрия. На рис. 1.43 показана огневая печь карбоната натрия с безретурным питанием. Топочные газы, получаемые в топке 1 путем сжигания твердого, жидкого или газообразного топлива, обогревают снаружи вращающийся барабан 2 печи карбоната натрия, изготовленный из стали 3. С обоих концов барабан имеет цилиндрические горловины 13, присоединяемые к барабану через конические царги 10, которые снабжены ободами 8 с опорными бандажами 9, на которых барабан опирается с обеих сторон на стальные ролики 4 с подшипниками 5. Задние ролики снабжены ребордами, фиксирующими положение конца барабана. Привод барабана осуществляется через венцовую шестерню 12 от электродвигателя через редуктор. Торцевая сторона горловины задней загрузочной камеры закрыта крышкой с центральным отверстием для ввода в печь выгрузочного шнека 14. Горловина передней части печи входит в неподвижную загрузочную камеру. Уплотнение горловины сальниковое.
Загрузочная камера имеет штуцер для отвода газов, образующихся в процессе кальцинации. К фронтальной стенке загрузочной каме-
Рис. 1.43. Безретурная печь для обжига карбоната натрия:
/ — топка; 2 — барабан печи; 3 — горелка; 4 — ролики; 5 — подшипники; 6 — загрузочная камера;
7 — забрасыватель гидрокарбоната; 8 — ободы; 9 — опорные бандажи; 10 — коническая царга;
11 — цепь; 12 — венцовая шестерня; 13 — цилиндрическая горловина; 14 — выгрузной шнек
88
ры снаружи крепится забрасыватель гидрокарбоната натрия, а с внутренней стороны — цепь 11 для очистки внутренней поверхности барабана от возможных отложений карбоната натрия, а также для перемешивания и дробления комков карбоната натрия. Загрузка гидрокарбоната натрия производится забрасывателем 7.
В последние годы в процессе кальцинации начали применять и паровые кальцинаторы. В процессе кальцинации в качестве теплоносителя используют пар высокого давления, что дает возможность создать аппараты с развитой поверхностью теплообмена, которая достигает 2500—3000 м2, в то время как в печах карбоната натрия — лишь 150—200 м2. Однако высокая стоимость пара по сравнению с жидким и газообразным топливом экономически не позволяет применять паровые кальцинаторы.
В качестве обобщения изложенного материала по отдельным разделам призводства на рис. 1.44 приведена принципиальная схема получения карбоната натрия.
Согласно схеме, очищенный рассол из сборника 1 подается центробежным насосом 2 в напорную емкость 3, постоянный уровень рассола в которой поддерживается поплавковым регулятором 4. Из напорной емкости рассол распределяется на три параллельных потока.
Первый (основной) поток, составляющий примерно 75% всего расходуемого рассола, поступает во второй промыватель газа колонн 5. Далее поток проходит последовательно через все шесть царг про-мывателя сверху вниз навстречу хвостовым газам, поступающим под давлением 140—150 мм рт. ст. в нижнюю часть аппарата из первого промывателя газа карбонизационных колонн 25. Во втором промыва-теле газа колонн 5 рассол абсорбирует аммиак и частично диоксид углерода из газов, после чего газ, пройдя ловушку б, орошаемую водой (для улавливания брызг рассола), выбрасывается в атмосферу.
Второй поток, составляющий около 15% от всего потребляемого рассола, поступает во вторую барботажную секцию (считая сверху) промывателя воздуха фильтров 7, в котором улавливается аммиак из воздуха вакуум-фильтров 34. Промыватель воздуха фильтров имеет четыре барботажные тарелки и работает под разрежением 350—380 мм рт. ст. Рассол проходит через аппарат сверху вниз, а воздух — снизу вверх, после чего отсасывается вакуум-насосом 12 и выпускается в атмосферу.
Третий поток в количестве около 10% от общего расхода рассола поступает во вторую сверху барботажную секцию промывателя газа абсорции 9, имеющую также четыре барботажные тарелки. Аппарат работает при разрежении 250—280 мм рт. ст. В нем улавливаются остатки аммиака, содержащиеся в хвостовых газах станции абсорции. Газ поступает в нижнюю часть промывателя 9 из первого абсорбера 75 и движется вверх навстречу потоку рассола.
89
На выходе из промывателя газ содержит до 70% диоксида углерода. После брызгоотделителя 10 он подается вакуум-насосом 11 в холодильник газа содовых печей 41, в котором объединяется с основным потоком концентрированного диоксида углерода, используемого для карбонизации аммонизированного рассола. В верхние барботажные секции этих промывателей для улавливания брызг рассола подают воду, которую затем направляют в отстойники. Операция проводится с целью предупреждения отложения солей (инкрустации) в трубопроводах, по которым воздух и газ из этих двух промывателей поступают в вакуум-насосы.
Второй поток рассола, выходящий из промывателя воздуха фильтров 7, смешивается с третьим потоком рассола, выходящим из промывателя газа абсорбции 9, и по U-образной трубе стекает в третью сверху барботажную бочку второго промывателя газа колонны 5, где он соединяется с основным потоком рассола.
Выходящий из второго промывателя газа колонн 5 рассол поступает самотеком по U-образной трубе в верхнюю барботажную секцию первого абсорбера 15. Абсорбер имеет шесть барботажных тарелок и работает при разрежении 200—250 мм рт. ст.
В первом абсорбере происходит абсорбция аммиака и частично диоксида углерода из газа, поступающего из второго абсорбера 16. В первый абсорбер поступает также газ со станции переработки так называемых «слабых жидкостей». Первый абсорбер, как и все аппараты станции абсорбции, работает по принципу противотока. После первого абсорбера рассол содержит 70 н.д NH3 и 24 н.д. СО*'.
Вследствие экзотермичности реакции поглощения аммиака температура рассола в первом абсорбере поднимается до 70° С. Поэтому выходящий из первого абсорбера аммонизированный рассол поступа-
Рис. 1.44. Схема производства карбоната натрия по аммиачному способу:
1, 20, 38, 42 и 68— сборники; 2, 21, 23, 27, 37, 43, 44, 58, 67 и 80—центробежные насосы; 3, 13, 18, 35, 40, 46, 51, 60, 69 — напорные емкости; 4 — поплавковый регулятор; 11, 50, 70, 71 — поплавковые регуляторы; 5 — второй промыватель газа карбонизационных колонн; 6 — ловушка-уловитель; 7 — промыватель воздуха фильтров; 8 — холодильник газа дистилляции; 9 — промыватель газа абсорбции; 10 — брызгоуловители; И и 12 — вакуум-насосы; 14, 17, 78 — регуляторы подачи; 75 — первый абсорбер; 16 — второй абсорбер; 19 — оросительные холодильники; 22 — емкость; 24 — колонна предварительной карбонизации; 25 — первый промыватель газа карбонизационных колонн; 26 — сборник пред карбон изованной жидкости; 28 — осадительная карбонизационная колонна; 29, 30 и 31 — компрессоры; 32 — мерные колонны; 33 — корыто; 34 — вакуум-фильтр; 36, 62 и 64 — транспортеры; 39 — промыватель газа содовых печей; 41 — холодильник газа содовых печей;
45 — холодильник газа малой дистилляции; 47 — конденсатор малой дистилляции; 48 — малый дистиллер; 49 — холодильник; 52 — конденсатор дистилляции; 53 — газоотделитель; 54 — теплообменник дистилляции; 55 — дистиллер; 56 — испаритель; 57 — пескоуловитель; 59 — смеситель; 61 — элеватор; 63 — циклон; 65 — вертикальный смеситель; 66 — содовая печь; 72 — приемный бункер; 73 — вращающееся сито; 74 — вибрационное сито; 75 — подогреватель; 76 — гаситель извести; 77—шаровая мельница мелкого помола; 79 — сборник шлама; 81 — бункер; 82 — ковшовый конвейер; 83 — электрофильтр; 84 — загрузочное устройство; 85 — ячейковый выгружатель;
86 — кольцевой транспортер; 87 — «улита»; 88 — вентилятор; 89 — известково-обжигательная печь
91
ет самотеком в оросительные холодильники (устанавливаемые обычно вне здания), в которых он охлаждается водой до 30° С.
Охлажденный аммонизированный рассол поступает самотеком в верхнюю барботажную секцию второго абсорбера 16. Число барботажных тарелок во втором абсорбере — три. Рассол проходит последовательно все три барботажные бочки сверху вниз. При этом происходит абсорбция рассолом аммиака и частично диоксида углерода из газа, поступающего со станции дистилляции в нижнюю секцию второго абсорбера и проходящего через аппарат снизу вверх.
Потери аммиака в производстве компенсируются добавкой водных растворов аммиака. При этом 20%-ные водные растворы из заводских хранилищ подаются центробежным насосом в напорную емкость 13, откуда через регулятор подачи 14 поступают в верхнюю барботажную секцию второго абсорбера 16.
Вследствие выделения значительного количества теплоты в процессе поглощения аммиака рассолом температура последнего во втором абсорбере также повышается до 70° С. Поэтому выходящий из второго абсорбера аммонизированный рассол, содержащий 100—102 н.д. NH3 и 35—37 н.д. СО,', проходит самотеком через охлаждаемые водой оросительные холодильники 19, в которых температура рассола снижается до 30—35°С. Охлажденный аммонизированный рассол далее поступает в запасные сборники 20, откуда он направляется на карбонизацию.
С целью предохранения аппаратуры станции абсорбции от коррозии в первый абсорбер подается в небольших количествах раствор сульфида натрия (из расчета 0,75 кг 62%-ного сульфида натрия на 1 т карбоната натрия). Раствор сульфида натрия готовится в емкости 22 на очищенном рассоле с добавкой конденсата, после чего полученный раствор перекачивают центробежным насосом 23 в напорную емкость 18, из которой с помощью регулятора подачи 17 он дозируется в трубопровод, подающий рассол из второго промывателя газа колонн 5 в первый абсорбер 15.
Процесс карбонизации аммонизированного рассола производят в три стадии: в колонне предварительной карбонизации 24, в первом промывателе газа колонн 25 и осадительных карбонизационных колоннах 28, в которых в основном происходит осаждение гидрокарбоната натрия. После трех-четырех дней работы в осадительных колоннах накапливается гидрокарбонат и их поочередно переключают на промывку. Промывка колонн производится пропусканием через них аммонизированного рассола с одновременной подачей в колонну диоксида углерода известково-обжигательных печей. Таким образом, промывка колонн осуществляется в процессе предварительной карбонизации. В качестве колонны предварительной карбонизации поочередно используется переключаемая на промывку осадительная колонна. 92
На станции карбонизации аммонизированный рассол из сборников 20 центробежным насосом 21 подается в колонну предварительной карбонизации 24. В нижнюю часть этой колонны компрессором 31 подается охлажденный до 30° С и очищенный в электрофильтре 83 диоксид углерода известково-обжигательных печей 89, содержащий 37—39% СО2. Процесс карбонизации проводят до содержания в растворе 51—53 н.д. COj'. Температура жидкости при этом достигает 39—40° С.
Выходящий из колонны предварительной карбонизации 24 раствор самотеком поступает в верхнюю часть первого промывателя газа колонн 25. Аппарат представляет собой заполненный коксом скруббер. В нижнюю часть аппарата поступают хвостовые газы из осадительных карбонизационных колонн 28 с содержанием 8—10% СО2. Газ в промывателе 25 движется вверх навстречу раствору, отдавая ему часть содержащегося диоксида углерода. После первого промывателя газа колонн 25 в газе остается 3—4% СО2 и вместе с газом из колонн предварительной карбонизации 24 он поступает на окончательную промывку свежим очищенным рассолом во второй промыватель газа колонн 5.
После этого газ с содержанием 2—3% СО2 выбрасывается в атмосферу.
Раствор из первого промывателя газа колонн 25 с содержанием 58 — 60 н.д. СО}’ собирается в сборнике предкарбонизационного раствора 26, откуда перекачивается центробежным насосом 27 в осадительные карбонизационные колонны 28. Карбонизационные колонны группируются обычно сериями. В каждую серию входят по четыре осадительных колонны и одна колонна предварительной карбонизации.
В нижнюю часть осадительных карбонизационных колонн подается диоксид углерода. При этом газ подается на два входа: в нижнюю секцию осадительной колонны компрессором 30 подается концентрированный газ с содержанием 65% СО2, на второй вход (на высоте 7—9м от низа колонны) компрессором 29 подается газ известково-обжигательных печей с содержанием 37—39% СО2. Концентрированный газ получается смешением 90%-ного газа содовых печей и 38%-ного газа известково-обжигательных печей.
В нижней части карбонизационных колонн имеется по десять холодильных секций, в трубное пространство которых подается охлаждающая вода, благодаря чему температура выходящего из осадительных колонн раствора снижается до 26—27° С.
Воду на охлаждение подают двумя параллельными потоками в первую и вторую (считая снизу) холодильные секции карбонизационной колонны. Один поток охлаждающей воды проходит через все нечетные холодильные секции, а второй поток — через все четные холодильные секции колонны.
93
Охлажденный раствор осадительных колонн с температурой 26—27° С, содержащий 70—72 н.д. связанного в NH4C1 аммиака, вместе с выпавшим в осадок гидрокарбонатом натрия непрерывно выдавливается из осадительных колонн 28 в мерники колонн 32 и из расположенного под ними корыта 33 самотеком направляется на вращающиеся барабанные вакуум-фильтры 34.
На вакуум-фильтрах осадок гидрокарбоната натрия отделяется от маточного раствора и отмывается от хлоридов натрия и аммония. Процесс промывки производится слабым раствором, поступающим из промывателя газа содовых печей 39 в напорную емкость 55, а оттуда в промывные устройства вакуум-фильтров. Вакуум на фильтре поддерживается вакуум-насосом.
Фильтровый раствор отделяется от воздуха в сепараторе фильтров и поступает по U-образной трубе в сборник 38, откуда он подается на стадию регенерации аммиака. Воздух, отделенный в сепараторе фильтра от маточного раствора, проходит через промыватель воздуха фильтров 7, в которых свежий очищенный рассол улавливает из него аммиак. Из промывателя 7 воздух отсасывается вакуум-насосом 12 и выбрасывается в атмосферу.
Сырой гидрокарбонат натрия с вакуум-фильтров подается скребковыми транспортерами 36 и 64 в вертикальный смеситель 65, расположенный непосредственно над загрузочными устройствами вращающихся содовых печей 66. Из вертикального смесителя 65 сырой гидрокарбонат натрия поступает в барабанный ячейковый питатель содовых печей, снабженный метательной лопаткой, вращающейся со скоростью 200 об/мин. Этой лопаткой гидрокарбонат натрия забрасывается внутрь барабана содовой печи на глубину 6—9 м и смешивается с находящимся в барабане карбонатом натрия. Барабанная печь вращается со скоростью 4—5 об/мин. Под передней частью содовой печи (со стороны загрузки гидрокарбоната) расположена топка, в которой сжигается топливо. Топочные газы омывают снаружи барабан содовой печи, имеющей обмуровку.
Образующийся в процессе разложения гидрокарбоната натрия в печи карбонат натрия специальными разгрузочными ковшами и выгрузочным шнеком выводится из содовой печи с температурой 140—170° С, поступает в собирающий шнековый транспортер 62 и подается в элеватор 61, а затем в последующие механизмы, транспортирующие его на склад готовой продукции.
Выделяющиеся в процессе разложения гидрокарбоната в содовой печи газы, содержащие диоксид углерода, водяные пары, а также незначительное количество аммиака, удаляются из передней части барабанной печи и поступают в циклон 63, в котором улавливается увлеченная газами пыль карбоната натрия. Осевшая в циклоне пыль карбоната натрия расположенными внутри циклона рамной мешалкой 94
и гребками передвигается к выгрузочному отверстию на дне циклона, закрытому сигнальным клапаном. С приближением гребков к отверстию клапан автоматически открывается, и пыль карбоната натрия по течке возвращается в содовую печь.
Выходящий из циклонов газ поступает в коллектор газа содовых печей, а оттуда в холодильник газа содовых печей 41. С целью ликвидации процесса забивания коллектора частично уносимой из циклона пылью карбоната натрия его орошают слабым раствором с помощью специальных брызгальных устройств. Раствор подается центробежным насосом 43 из сборника 42, расположенного под холодильником газа содовых печей 41.
В холодильнике газа содовых печей 41 газ движется в межтрубном пространстве сверху вниз. Охлаждающая вода поступает в виде двух параллельных потоков и движется по трубам снизу вверх. С целью очистки труб холодильника от оседающей на них пыли карбоната натрия и повышения коэффициента теплопередачи межтрубное пространство холодильника орошается слабым раствором карбоната натрия, подаваемым центробежным насосом 43 из сборника 42. Орошающая жидкость в смеси с конденсатом, образующимся в холодильнике в результате конденсации водяных паров, возвращается в сборник 42. Собирающийся в сборнике избыток слабых растворов центробежным насосом 43 перекачивается на станцию переработки слабых растворов. В холодильник газа содовых печей 41 подается также газ с концентрацией до 70% СОг, отсасываемый вакуум-насосом 11 из промывателя газа абсорбции 9.
Завершающая очистка газа содовых печей от пыли карбоната натрия и аммиака производится в промывателе газа содовых печей 39, в нижнюю часть которого поступает газ из холодильника 41. Промыватель газа содовых печей 39 представляет собой скруббер с насадкой, орошаемой водой или слабыми растворами, содержащими карбонат натрия, из которых аммиак отогнан на станции переработки слабых растворов.
Слабые растворы, содержащие карбонат натрия, направляются из промывателя газа содовых печей 39 в напорную емкость 35 для промывки гидрокарбоната натрия на вакуум-фильтрах.
После промывателя газа содовых печей 39 газ смешивается с расчетным количеством газа известково-обжигательных печей и поступает на компрессор 30, которым он нагнетается в нижнюю часть осадительных карбонизационных колонн 28.
Фильтровая жидкость из сборника 38 центробежным насосом 37 перекачивается в напорную емкость фильтровой жидкости 51. Для поддержания постоянного уровня в напорном сборнике последний снабжен поплавковым регулятором 50. Из напорного сборника фильтровая 95
жидкость поступает в конденсатор дистилляции 52. Поток жидкости в конденсаторе регулируется с помощью ограничительной шайбы.
Далее фильтровая жидкость поступает в трубное пространство верхней теплообменной секции конденсатора 52 и последовательно проходит через все теплообменные секции. Поступающий в конденсатор из теплообменника дистилляции 54 газ движется в межтрубном пространстве конденсатора дистилляции снизу вверх противотоком к фильтровой жидкости и подогревает ее от 26—27° С до 72—73° С. При этом содержащиеся в фильтровой жидкости карбонатные соли аммония частично разлагаются. Для отделения жидкости от газа, выделившегося в процессе разложения этих солей, жидкость после каждой теплообменной секции конденсатора дистилляции поступает в специальный газоотделитель 53, разделенный перегородками соответственно числу теплообменных секций конденсатора дистилляции.
Газ из газоотд ел ителя 53 присоединяется к основному потоку газа, выходящему из верхней секции конденсатора дистилляции. В конденсаторе дистилляции температура газа снижается с 83—85°С до 70—71° С.
Дальнейшее охлаждение газа производят в холодильнике газа дистилляции 8, через который газ проходит по межтрубному пространству. При этом в трубное пространство подается охлаждающая вода, которая проходит через все секции холодильника. Газ охлаждается до 56—58° С и поступает в нижнюю секцию второго абсорбера 16.
Конденсат, образующийся в межтрубном пространстве конденсатора дистилляции 52 и холодильника газа дистилляции 8 с растворившимися в нем карбонатными солями аммония, выводят из этих аппаратов и направляют на станцию переработки слабых растворов.
Подогретая в конденсаторе дистилляции 52 фильтровая жидкость поступает самотеком в теплообменник дистилляции 54, расположенный непосредственно под конденсатором дистилляции. Теплообменник дистилляции представляет собой аппарат барботажного типа с двенадцатью барботажными тарелками. Газ поступает в нижнюю секцию теплообменника дистилляции из ее смесителя и движется навстречу потоку жидкости.
В этом аппарате происходит дальнейшее разложение карбонатных солей аммония, содержащихся в фильтровой жидкости. При этом температура раствора повышается до 90—92° С.
Из теплообменника дистилляции 54 фильтровая жидкость, освобожденная от карбонатных солей аммония и содержащая аммиак в основном в виде хлорида аммония, поступает самотеком в смеситель дистилляции 59, снабженный ментальным устройством. Насосом 67 через напорную емкость 60 в смесь вводится водная суспензия оксида— гидроксида кальция. Хлорид аммония разлагается, а выделив-96
шийся при этом аммиак в большей своей части остается растворенным в жидкости. Часть аммиака переходит в газообразное состояние и присоединяется к потоку газа и пара, проходящему через газовую (верхнюю) часть смесителя дистилляции 59 при движении из дистиллера 55 в теплообменник дистилляции 54.
Выходящий из нижней части смесителя дистилляции 59 раствор самотеком поступает в верхнюю барботажную секцию дистиллера 55 и проходит сверху вниз через все двенадцать барботажных секций дистиллера. В нижнюю секцию дистиллера 55 пар подается навстречу жидкости, из-за чего происходит полная отгонка аммиака из раствора. Пользуются обычно отработанным паром турбин и компрессоров.
Дистиллерная жидкость, имеющая после отгонки аммиака температуру 107—110° С, поступает в испаритель дистилляции 56, в котором за счет снижения давления происходит выделение из раствора значительного количества пара, используемого на станции переработки разбавленных растворов.
После испарителя 56 дистиллерную жидкость разбавляют водой, подаваемой для снижения температуры растворов непосредственно в трубопровод. После пескоуловителя 57 жидкость насосом 58 удаляют в отброс.
Содержащийся в слабых растворах аммиак также регенерируют и используют в процессе аммонизации рассола. Поскольку в растворах аммиак находится исключительно в виде карбонатных солей, процесс проводят лишь подогревом паром.
Основными источниками слабых растворов являются:
1. Холодильник 41, в котором происходит конденсация водяных паров, содержащихся в газах содовых печей. Образующийся конденсат содержит растворенные карбонаты аммония и натрия.
2. Конденсат дистилляции 52 и холодильник газа дистилляции 8, в котором конденсируются водяные пары, содержащиеся в газе, выходящем из теплообменника дистилляции 54. Образующийся в этих аппаратах конденсат содержит растворенные карбонат аммония и аммиак.
Переработка неконцентрированных растворов осуществляется следующим образом. Раствор из напорной емкости 46 поступает в трубное пространство верхней теплообменной секции конденсатора малой дистилляции 47. Подогрев раствора происходит за счет теплоты газа и пара, поступающих из малого дистиллера 48 и движущихся в межтрубном пространстве снизу вверх. Газ из конденсатора малой дистилляции 47 поступает в расположенный непосредственно над ним холодильник газа малой дистилляции 45. Газ проходит через межтрубное пространство, а в трубное пространство подается охлаждающая вода. Охлажденный до 56—58°С газ направляется в третью (сверху) барботажную секцию первого абсорбера 75.
4 Химическая технология 97
неорганических веществ, кн. I
Подогретый в конденсаторе малой дистилляции 47 неконцентрированный раствор поступает самотеком в малый дистиллер 48, представляющий собой скруббер с деревянной хордовой насадкой. В нижнюю часть аппарата навстречу потоку неконцентрированного раствора поступает пар из испарителя дистилляции 56, к которому добавляется отработанный пар от турбин. Под действием этого происходит полное разложение карбонатно-аммонийных солей, содержащихся в неконцентрированных растворах. Освобожденный от аммиака и диоксида углерода раствор поступает в холодильник жидкости 49, в котором он охлаждается водой, а затем центробежным насосом 44 направляется в напорную емкость 40, а далее он поступает в промыватель газа содовых печей 39.
Водный раствор оксида — гидроксида кальция, необходимый для регенерации аммиака из фильтровой жидкости, готовят следующим образом. Добытый в карьере известняк или мел подается по подвесной канатной дороге в бункер известково-обжигательной печи. Сюда же доставляется кокс или антрацит, который добавляется в каждую вагонетку известняка (мела) в количестве 7,5—10% от массы загруженного в нее сырья. Количество загруженного топлива зависит от влажности исходного карбонатного сырья и от высоты печи. Шихта загружается в известково-обжигательную печь 89 через центральное загрузочное устройство 84, снабженное двойным затвором. Расчетное количество воздуха для горения топлива подается в печь вентилятором 88. Образующийся в процессе обжига известняка (мела) диоксид углерода (37—39% СО2) поступает с температурой 80—100° С в скрубберную часть электрофильтра 83, заполненную хордовой насадкой и орошаемую водой. При этом газ охлаждается до 30° С и поступает в электрофильтр для удаления пыли и брызг воды, увлеченных из скрубберной части фильтра. Выходящий из электрофильтра газ подводится по газовому коллектору к компрессорам, подающим его в карбонизационные колонны.
Выгрузку образующегося оксида кальция (извести) из печи проводят непрерывно с помощью расположенного в нижней части печи вращающегося пода (улиты) 87, кольцевого транспортера 86 и барабанного ячейкового выгружателя 85.
Оксид кальция поступает непосредственно в приемную воронку пластинчатого или ковшового конвейера 82, которым он доставляется в запасные и расходные бункера 81 гасителей извести. Из расходного бункера 81 оксид кальция с помощью лоткового питателя подается в горизонтальный вращающийся гаситель оксида кальция 76. Процесс гидратации (частичной) оксида кальция производится теплой водой, для чего используется отработанная вода, поступающая в напорную емкость 69, снабженную поплавковым регулятором 70. Перед поступлением в гаситель извести 76 оксида кальция теплая вода из напор-98
ного сборника дополнительно подогревается в подогревателе 75 за счет теплоты отходящих из гасителя водяных паров. Уровень жидкости в подогревателе 75 поддерживается поплавковым регулятором 71. Дозировка воды в гаситель оксида кальция производится с помощью регулятора подачи 78.
Образовавшаяся в гасителе водная суспензия оксида — гидроксида кальция отделяется от крупных кусков недопала во вращающемся сите 73, вмонтированном в торце гасителя извести 76. Отделившиеся в сите крупные куски недопала промываются водой и собираются в приемном бункере 72, откуда они выгружаются в вагонетки, доставляющие недо-пал к известково-обжигательным печам для повторного обжига.
Окончательная очистка водной суспензии оксида — гидроксида кальция производится на вибрационном сите 74, в котором суспензия очищается от песка и мелких непогасившихся кусков оксида кальция, представляющих собой в основном перепал. Очищенная на вибрационном сите 74 суспензия оксида — гидроксида кальция с концентрацией СаО 200 н.д. поступает в сборник 68, снабженный вращающейся мешалкой. Из сборника суспензия насосом 67 передается в напорную емкость 60, а оттуда в смеситель дистилляции 59.
Отделившиеся на вибрационном сите 74 мелкие отбросы промывают водой и подают в шаровую мельницу 77 мокрого помола. Полученный в результате помола осадок поступает в сборник 79, снабженный вращающейся мешалкой, куда параллельно поступает для разбавления осадка отработанная вода из скрубберной части электрофильтра 83. Разбавленный шлам откачивается из сборника 79 насосом 80 в шламоотстойник.
4*
ГЛАВА 2
КАЛИЙ И ЕГО СОЕДИНЕНИЯ
2.1. КАЛИЙ
Содержание калия в земной коре составляет 2,41% (масс.). Основные калийсодержащие минералы и их составы приведены в табл. 2.1.
Таблица 2.1. Состав и свойства наиболее распространенных калийных минералов
Минерал Состав Содержание K2O, % Плотность, г/см3 Твердость
Сильвин КС! 63 1,97—1,99 2
Сильвинит KCINaCI 22—25 2,10—2,13 2
Карналлит KClMgCl2-6H2O 17 1,57 2
Каинит KClMgSO4-3H2O 19 2,07—2,10 2—3
Шенит K2SO4MgSO4-6H2O 23 2,8 2,5
Лангбейнит K2SO4-2MgSO4 23 2,8 3—4
Полигалит K2SO4MgSO4-2CaSO4-2H2O 16 2,7 2,5—3,0
Алунит (K,Na)2SO4Al2(SO4)3-4Al(OH)3 23 2,6—2,8 3,5—4,0
Калушит K2SO4CaSO4H2O 2,58—2,60 2,5
Глазерит 3K2SO4Na2SO4 2,70 3
Нефелин (K,Na)2OAl2O3-2SiO2 6—7 2,60 5—6
Свойства. Калий имеет атомную массу 39,0983. Состоит из двух стабильных изотопов: 39К (93,259%) и 41К (6,729%), а также радиоактивного изотопа 40К (Tie = 1,32-109 лет). Конфигурация внешней электронной оболочки 4s1; степень окисления +1; энергия ионизации К°->К+->К2+ соответственно 4,34070 эВ и 31,8196 эВ; атомный радиус 0,2313 нм, ионный радиус (в скобках указано координационное число) К+ 0,151 нм (4), 0,152 нм (6), 0,160 нм (7), 0,165 нм (8), 0,178 нм (12).
Калий — мягкий серебристо-белый металл с кубической решеткой (а = 0,5344 нм, z = 2, пространственная группа Jm3m). Температура плавления 63,51 °C, а кипения — 761 °C; плотность 0,8629 г/см3; С° = 29,60 Дж/(моль-К); ЬН°т = 2,33 кДж/моль, ДЯ^Г = 89,0 кДж/моль; 5°98 = 64,68 Дж/(моль-К); уравнение температурной зависимости давления пара (в мм рт. ст.): Igp = 7,34-4507/Т(373 -474 К). 100
Калий — химически очень активный элемент. Легко взаимодействует с кислородом воздуха, при котором образуются следующие соединения:
4К + О2 = 2К2О
2К + О2 = К2О2
К + О2 = КО2
При нагревании калия на воздухе он загорается. С водой и разбавленными кислотами взаимодействует со взрывом и воспламенением. В процессе взаимодействия с серной кислотой в зависимости от соотношения исходных реагентов и условий проведения последняя восстанавливается до сероводорода, элементной серы и диоксида серы, а калий образует различные кислородсодержащие соединения. Азотную кислоту калий разлагает до NO, N2O и N2. При 200—350°С калий реагирует с водородом по следующей схеме:
2К + Н2 = 2КН
Элементный калий в атмосфере фтора воспламеняется, взрывается при соприкосновении с бромом и растирании с иодом. Слабо взаимодействует с хлором. С серой, селеном и теллуром калий реагирует при слабом нагревании:
16К + S8 = 8K2S
2К + Se = K2Se
2К + Те = К2Те
В процессе нагревания с элементным фосфором в атмосфере азота калий образует соответствующие фосфиды:
12К + Р4 = 4К3Р
8К + 5Р4 = 4К2Р5
С графитом при 250—500° С калий образует слоистые соединения состава С8К—С6оК, а с диоксидом углерода реагирует при 10—30° С. Калий при температуре выше 350—400° С разрушает стекло и платину.
Калий растворяется в жидком аммиаке (35,9 г в 100 мл при -70° С), анилине, этилендиамине, тетрагидрофуране и диглиме с образованием растворов с металлической проводимостью. Раствор калия в аммиаке имеет темно-синий цвет, а в присутствии платины и
101
следов воды разлагается, образуя KNH2 и Н2. Калий с азотом не реагирует. В процессе взаимодействия калия с NH4N3 в жидком аммиаке образуется азид калия.
Калий не растворяется в жидких литии, магнии, кадмии, цинке, алюминии и галлии и не реагирует с ними. С натрием калий образует интерметаллид KNa2, плавящийся инконгруэнтно при 7°С. С рубидием и цезием образует твердые растворы, для которых минимальные температуры плавления составляют соответственно 32,8° С (81,4%, масс. Rb) и -37,5°С (77,3% Cs). С ртутью калий образует амальгаму, содержащую два меркурида — KHg2 и KHg с температурой соответственно 270 и 180° С. С таллием калий образует КТ1 (7™, = 335° С), с оловом— K2Sn, KSn, KSn2 и KSn4, со свинцом — КРЬ и фазы состава К2РЬз, КРЬг и КРЬ4, с сурьмой — КзБЬ и KSb (температура плавления соответственно 812 и 605° С), с висмутом — K3Bi, K3Bi2 и KBi2 (температура плавления соответственно 671, 420 и 553° С).
Калий энергично взаимодействует с оксидами азота, а при высоких температурах — с оксидом и диоксидом углерода. Восстанавливает В20з и SiO2 соответственно до В и Si, оксиды Al, Hg, Ag, Ni и другие — до свободных металлов, сульфаты, сульфиты, нитраты, нитриты, карбонаты и фосфаты металлов — до оксидов или свободных металлов. Со спиртами калий образует алкоголяты, с галогеналкилами и га-логенарилами — соответствующие калийалкилы и калийарилы.
В процессе взаимодействия калия с избытком водорода при 300—400° С получают гидрид калия КН, являющийся сильным восстановителем в неорганических и органических синтезах. КН — бесцветные кристаллы с кубической решеткой (а = 0,570 нм, z = 4, пространственная группа Fm3m); плотность 1,52 г/см3; температура плавления 619° С.
Гидрид калия разлагается при нагревании на элементы, сильный восстановитель. Воспламеняется во влажном воздухе в среде фтора и хлора. Энергично реагирует с водой:
КН + Н2О = КОН + Н2
При нагревании с азотом и аммиаком образует KNH2, с H2S — K2S и Н2, с расплавленной серой — K2S и H2S, с влажным диоксидом углерода — НСООК, а с оксидом углерода — НСООК и С.
Металлический калий получают взаимодействием металлического натрия с КОН при 380—450°С или КС1 при 760—890° С. Процесс проводят в атмосфере азота. Взаимодействие жидкого натрия с гидроксидом калия производят противотоком в тарельчатой колонке из никеля. В процессе пары натрия пропускают через расплав КС1. Образующийся сплав K-Na ректифицируют и получают калий с содержанием примесей (%, масс.): 1-10’3 Na и 1-10"* С1.
102
Калий получают также вакуум-термическим восстановлением К.С1 карбидом кальция, сплавами Si-Fe или Si-Al при 850—950° С и остаточном давлении 1,33—13,3 Па; термообработкой К2СГ2О7 с цирконием при 400° С; электролизом гидроксида калия с железным катодом; электролизом хлорида калия или его смеси с карбонатом калия с жидким свинцовым катодом при 680—720° С с последующим разделением калия и свинца вакуумной дистилляцией; электролизом 50%-ного раствора KNH2 в жидком аммиаке при 25° С и давлении 0,75 МПа с амальгамой калия в качестве анода и катодом из нержавеющей стали. При этом образуется 30%-ный раствор калия в жидком аммиаке, который выводится из аппарата для отделения калия.
Металлический калий применяют в качестве материала электродов в химических источниках тока; компонента катодов — эмиттеров фотоэлементов и термоэмиссионных преобразователей, а также фотоэлектронных умножителей. Калий является геттером в вакуумных радиолампах, активатором катодов газоразрядных устройств. Сплав калия с натрием является теплоносителем в ядерных реакторах. Радиоактивный изотоп калия 40К применяется для определения возраста горных пород (калий-аргоновый метод). Искусственно полученный изотоп 42К(Т]/2 12,52 года) является индикатором в медицине и биологии. Калий является важнейшим элементом для питания растений.
2.2. ОКСИДЫ И ГИДРОКСИД КАЛИЯ
Калий образует оксид КгО, пероксид К2О2, надпероксид КОг и озонид КО3.
Оксид калия К2О—бесцветные кристаллы. До 372°С устойчива модификация с кубической решеткой (а = 0,644 нм, 2 = 4, пространственная группа Fm3m), а выше 372°С—с гексагональной. ДЯ®ерехода = 4,1 кДж/моль. Кристаллическая решетка приведена на рис. 2.1. Температура плавления 740° С; плотность 2,32 г/см3; С® = 72 Дж/(моль-К); ДЯ° = 27 кДж/моль, ДЯ^р = -361,7 кДж/моль; S°gg = 96 Дж/(моль-К).
Оксид калия на воздухе расплывается, поглощает диоксид углерода, образуя карбонат калия:
К2О + СО2 = К2СОз
Бурно взаимодействует с водой с образованием гидроксида калия:
К2О + Н2О = 2КОН
«К* ф0-
Рис. 2.1. Кристаллическая решетка К2О
103
Реагирует с водородом при 250° С:
К2О + Н2 = 2КОН
С аммиаком образует гидроксид и KNH2:
К2О + NH3 = КОН + KNH2
Энергично реагирует с галогенами, расплавленной серой и разбавленными кислотами. Оксид калия при нагревании с В2О3, А12О3 и SiO2 образует соответствующие бораты, алюминаты и полисиликаты, с NO2 — смесь KNO3 и KNO2.
Оксид калия получают взаимодействием металлического калия с гидроксидом калия, пероксидом калия, нитритом или нитратом калия при нагревании:
2К + 2КОН = 2К2О + Н2
2К + К2О2 = 2К2О
2KNO2 + 6К = 4К2О + N2
Оксид калия получают также окислением металлического калия, растворенного в жидком аммиаке, кислородом. Оксид калия применяется в качестве активатора катализатора — губчатого железа в процессе получения аммиака.
Пероксид калия К2О2— бесцветные кристаллы с ромбической решеткой (а - 0,6736 нм, Ъ - 0,7001 нм, с = 0,6479 нм, z = 4, пространственная группа Риш); Тт = 545° С; 5 = 2,40 г/см3; С ° = 90,8 Дж/(мольК); ДЯ^ =20,5 кДж/моль, ДЯо°6р = -443,0 кДж/моль; = 117 Дж/(моль-К).
Пероксид калия на воздухе мгновенно окисляется до КО2; энергично взаимодействует с водой с образованием гидроксида калия и кислорода:
4КО2 + 2Н2О = 4КОН + ЗО2
С диоксидом углерода образует К2СО3 и О2:
4КО2 + 2СО2 = 2К2СО3 + ЗО2
Пероксид калия получают пропусканием дозированного количества кислорода через раствор калия в жидком аммиаке, а также разложением КО2 в вакууме при 340—350° С. Пероксид калия применяют для освежения воздуха в подводных лодках и в дыхательных прибоин
pax, используемых пожарниками и водолазами. За основу очистки положены следующие реакции:
К2О2 + СО -> К2СО3
К2О2 + СО2 -> К2СО3 + 1/2О2
Надпероксид калия КО2 — желтые кристаллы с тетрагональной решеткой (а = 0,5704 нм, с - 0,6699 нм, z = 4, пространственная группа 14/wwn; плотность 2,158 г/см3); выше 149°С переходит в кубическую модификацию (а ~ 0,609 нм); температура плавления 535°С; С® = 77,5 Дж/(моль-К); ДЯ^р = -283,2 кДж/моль, ДЯ^, = 20,6 кДж/моль; = 125,4 Дж/(моль-К).
Согласно рентгеноструктурных исследований кристаллов, надпероксид калия содержит однозамещенный ион Oj:
О “О
Это подтверждено тем, что кристаллический надпероксид КО2 парамагнитен и обладает магнитной восприимчивостью, равной 2 магнетонам Бора, что соответствует одному неспаренному электрону. На рис. 2.2 приведена кристаллическая решетка надпероксида калия.
Надпероксид калия — сильный окислитель. Взаимодействуя с водой, образует КОН:
4КО2 + 2Н2О = 4КОН + ЗО2
С диоксидом серы он образует сульфат калия: Рис- 2.2.
Кристаллическая
2КО2 + SO2 = K2SO< + О2 решетка КО2
105
С концентрированной серной кислотой надпероксид калия образует озон:
2КО2 + H2SO4 = K2SO4 + Н2О + Оз
Надпероксид калия с аммиаком образует КОН, N2 и Н2О:
2 КО2 + 2NH3 = 2КОН + N2 + 2Н2О
Смесь надпероксида калия с графитом взрывается. Надпероксид калия получают сжиганием металлического калия в воздухе, обогащенном влажным кислородом, при нагревании до 75—80° С:
К + О2 = КО2
Озонид калия КОз — красные кристаллы с тетрагональной решеткой (а = 0,897 нм, с = 0,7080 нм, пространственная группа J4/wcm); плотность 1,99 г/см3. Озонид устойчив лишь при хранении в герметически закрытых сосудах ниже 0° С. При более высокой температуре разлагается до КО2 и О2; С ° = 75 Дж/(моль-К); = 105 Дж/(моль-К); парамагнетик.
Растворимость озонида калия в жидком аммиаке (г в 100 г): 14,82(-35° С), 12,00(-63,5° С), эвтектика NH3—KO3 (5 г в 100 г NH3) имеет температуру плавления -80° С; при длительном хранении аммиачные растворы разлагаются. Растворяется в фреонах. КОз — сильный окислитель. Мгновенно реагирует с водой уже при 0° С, образуя КОН и О2; с влажным диоксидом углерода образует смесь К2СОз с КНСОз.
Получение озонида калия. 1. Взаимодействием смеси Оз и О2 с КОН или КО2:
2КОН + 2,5О2 = 2КО3 + Н2О
2КО2 + О2 = 2КО3
Процесс ведут при температуре ниже 0° С с последующей экстракцией жидким аммиаком.
2. Озонированием суспензии надпероксида калия или гидроксида калия во фреоне 12.
Озонид калия применяют в качестве компонента составов для регенерации воздуха в замкнутых системах (шахты, подводные лодки, космические корабли).
Гидроксид калия КОН — бесцветные кристаллы; до 247° С устойчива моноклинная модификация, а выше 247° С — кубическая типа NaCl (а = 0,533 нм, z = 4, пространственная группа Fm3m); 106
ДЯ° од» ~ 5,6 кДж/моль; плотность 2,044 г/см3; температура кипения 1325° С (табл. 2.2).
Гидроксид калия растворяется в воде (49,4% по массе при 0° С), этаноле (27,9% при 28° С), метаноле (35,5% по массе при 28° С). Сильно гигроскопичен.
Гидроксид калия — сильное основание, с диоксидами углерода, серы и азота, а также с сульфидом водорода реагирует во влажной среде:
КОН + СО2 = КНСОз
КОН + SO2 = KHSO3
КОН + H2S = KHS + Н2О
2КОН + 2NO2 = KNO3 + KNO2 + Н2О
Таблица 2.2. Свойства гидроксида калия и его кристаллогидратов
Показатель КОН КОН-4Н2О КОН-2Н2О КОНН2О
Температура плавления,0 С 405 -33,5 33,0 150
С", Дж/(моль К) 64,9* — 125,5 96,2
АЯ^, Дж/моль 9,4 26,3 — 9,6
АЯ^>р, кДж/моль -424,7 — -1052,0 -753,5
Дж/(моль-К) 78,9 — 159 117
♦Для кубической модификации: Ср = 49,4 Дж/(моль-К); А/7^. = 197,1 кДж/моль.
Реагирует также с фтороводородной кислотой и оксидом углерода. С бромом и хлором гидроксид калия реагирует лишь при нагревании выше 600° С. КОН в расплавленном состоянии реагирует с бериллием, алюминием, галлием, цинком, оловом и их оксидами и гидроксидами с образованием оксометаллатов, например КАЮ2, K2ZnO2, выделяя при этом соответственно водород или воду. Водные же растворы гидроксида калия с приведенными выше металлами образуют гидроксокомплексы, например Кз[А1(ОН)б], К2[Ве(ОН)4], выделяя при этом водород. Водные растворы гидроксида калия и его расплав взаимодействуют с бором, кремнием, германием и их оксидами и кислотами, образуя соответственно КВО2, К[В(ОН)4], К2В4О?, полисиликатов и полигерманатов. С гидроксидом лития дает KOH-2LiOH с температурой плавления 313° С (с разложением), с гидроксидом натрия — твердые растворы и эвтектику с температурой плавления 170° С; 50 мол. % КОН с гидроксидом рубидия — непрерывный ряд твердых растворов, с гидроксидом бария — твердые растворы и фазу состава КОН-4Ва(ОН)2 с температурой плавления 107
390° С, с карбонатом калия — эвтектику с температурой плавления 360° С; 22% по массе К2СОз.
Гидроксид калия получают: 1) электролизом водных растворов хлорида или карбоната калия с железным (диафрагменный метод) или ртутным катодом, с использованием ионообменных мембран; 2) взаимодействием водного раствора карбоната кальция и гидроксида кальция или реакцией концентрированного раствора сульфата калия с гидроксидом бария по схеме
К2СО3 + Са(ОН)2 = 2КОН + СаСОз
K2SO4 + Ва(ОН)2 = 2КОН + BaSO4
Образующийся фильтрат (раствор гидроксида калия) после отделения соответственно СаСОз или BaSO4 упаривают в никелевых реакторах под вакуумом, для удаления следов влаги гидроксид калия выдерживают при 360—400° С в вакууме; 3) фильтрованием водного раствора сульфата калия через анионит в ОРТ-форме.
Гидроксид калия применяют в производстве жидкого мыла, мерсеризованного хлопка, соединений калия, в щелочных аккумуляторах. Он является абсорбентом сульфида водорода, диоксидов серы и углерода, осушающим агентом для аммиака, гемиоксида азота, фосфина и других не реагирующих с ним газов.
2.3. ХЛОРИД КАЛИЯ
Физико-химические свойства, применение. Хлорид калия КС1, бесцветные кристаллы с кубической решеткой (а = 0,629 нм, z = 4, пространственная группа Fm3m); при 298° С и 1,95 МПа образуется кубическая модификация типа CsCl. Температура кипения 1500° С; плотность 1,989 г/см3; С° = 51,30 Дж/(моль-К); ДЯ^ = 26,32 кДж/моль, ДЯ^р = -436,49 кДж/моль; S°9g = 82,57 Дж/(моль-К). Температура плавления 776° С.
Растворимость КС1 в воде (г в 100 г): 28,1(0° С), 34,3(20° С), 40,3(40° С), 56,2(100° С). Политермическая диаграмма растворимости в системе КС1—NaCl—Н2О приведена на рис. 2.3. Температура плавления эвтектики (24,6 г КС1 в 100 г воды) -10,7° С. Температура кипения насыщенного водного раствора (58,4 г КС1 в 100 г Н2О) 108,59° С. Плохо растворим в аммиаке и метаноле.
Хлорид калия является основным продуктом калийной промышленности. Около 95% хлорида калия применяется в качестве удобрения, а остальное количество перерабатывают в соединения калия, 108
Рис. 2.3. Политермическая диаграмма растворимости в системе КС!—NaCl—Н2О
применяемые в различных отраслях народного хозяйства (КОН, К2СО3, KNO3, K2SO4, KCN, КСЮз и др.).
Согласно требований действующего ГОСТ 4568—83 (табл. 2.3), предусматривается производство хлорида калия марок К (кристаллизацией из растворов), Ф (флотационным обогащением калийных руд) для технических целей и гранулированного или крупнокристаллического продукта для сельского хозяйства. Хлорид калия, выпускаемый для сельского хозяйства с целью исключения слеживаемости, обрабатывается алифатическими аминами или другими кондиционирующими добавками.
Таблица 2.3. Требования к качеству хлорида калия
Показатель Технический Для сельского хозяйства
К Ф
1-й сорт 12-й сорт 1-й сорт | 2-й сорт 1-й сорт | 2-й сорт
Содержание в пересчете на сухое вещество, %-.
КС1, не менее 98 95±1 95±1 91±1 95±1 1 91±1
NaCl, не более 1,4 4,5±1 4,5±1 7±1 Не нормируется
Влаги, не более 1,0 1,0 1,0 1,0 1,0 1 1,0
не растворимого в воде остатка, не более 0,2 Не нормируется Не нормируется
Гранулометрический состав, %:
Гранулы размером 4 мм, не более Не нормируется 5 5
от 1 до 4 мм, не менее » 90 90
менее 1 мм, не более » 5 5
109
Рис. 2.4. Растворимость в системе КС1—NaCl—Н2О при 25 и 100° С
Способы получения хло- ; рида калия. Хлорид калия j производят в основном двумя способами: 1) галургическим и 2) флотационным.
Галургический способ (растворением и раздельной кристаллизацией) основан на свойствах системы КС1—NaCl—Н2О (см. рис. 2.3), а именно на различной растворимости в воде хлоридов калия и натрия. Из данных рис. 2.4 видно, что в эвтоническом растворе при понижении температуры содержание хлорида натрия увеличивается. Фигуративная точка системы, соответствующей составу эвтоническо-
го раствора Еюо при 100° С, в процессе охлаждения раствора, насыщенного К.С1 и NaCl, в осадок выпадает в основном хлорид калия, в растворе остается хлорид натрия.
В процессе охлаждения эвтонического раствора Еюо от 100 до 25° С в результате осаждения хлорида калия состав раствора будет меняться — его фигуративная точка переместится вдоль луча кристаллизации Сп от Еще до п. Если после отделения КС1 в виде осадка раствор снова нагреть до 100° С, то он будет сильно ненасыщенным КС1 и лишь немного недонасыщенным NaCl. Если же этим раствором обработать исходный сильвинит, то в нем будет растворяться преимущественно КС1. После отделения твердого остатка NaCl вновь будет образовываться горячий эвтонический раствор Еюо, из которого в процессе охлаждения выделяется КС1. Применяя такой циклический процесс, осуществляют в производстве разделение сильвинита на КС1 и NaCl.
Полное выделение хлорида калия из сильвинита обеспечивается при условии соответствия количества вводимого в систему хлорида калия количеству циркулирующего раствора. Если принять округленно состав исходного сильвинита 25% КС1 и 75% NaCl (точка S), то точка смесей его с маточным раствором п расположатся на линии nS. Необходима такая смесь, которая полностью разделялась бы на раствор Eioo и твердый NaCl. Этому условию соответствует смесь состава К, лежащего на пересечении линии nS и ЕюоЛ. Следовательно, для полного растворения хлорида калия и получения при этом
ПО
эвтонического раствора Еюо необходимо обрабатывать сильвинит таким количеством маточного раствора состава п, чтобы отношение количеств n:S равнялось отношению отрезков SK.Kn.
При работе с сильвинитом, загрязненным карналлитом, в процессе рециркуляции раствора в нем постоянно накапливается хлорид магния. В присутствии в системе хлорида магния растворимость хлорида натрия снижается, в результате чего в процессе загрязнения раствора хлоридом магния сверх определенного предела в целевой продукт начинает переходить значительное количество хлорида натрия. Это приводит к снижению содержания хлорида калия в целевом продукте. Поэтому по мере накопления в системе хлорида магния раствор выводят из процесса. С целью обновления раствора проводят двухступенчатую упарку. При этом после первой стадии упарки отделяют хлорид натрия пищевой кондиции, а после второй стадии — сильвинитовую суспензию, которую после сгущения возвращают в технологический цикл, а хлорид магния выводят из системы.
Процесс получения хлорида калия рассматриваемым способом включает следующие основные стадии:
1) измельчение сильвинитовой руды;
2) водное выщелачивание (растворение) хлорида калия из сильвинита при температуре 90—98° С маточным раствором. При этом соотношение между количествами исходного сильвинита и маточного раствора поддерживают таким, чтобы растворялся лишь КС1, a NaCl оставался в осадке;
3) отделение раствора от шлама, состоящего из NaCl и глинистых веществ; промывка шлама и глинистых веществ;
4) кристаллизация хлорида калия;
5) отделение кристаллов от маточного раствора и их сушка;
6) нагревание маточного раствора до 99° С и возвращение его в процесс растворения исходного сырья-сильвинита.
Согласно схеме получения хлорида калия (рис. 2.5), измельченная сильвинитовая руда из бункера 1 через дозаторы 3 ленточным транспортером загружается в шнековый растворитель 5. В этом растворителе руду выщелачивают маточным раствором, поступающим из шнекового смесителя 6. Растворы после смесителя, проходя через трубчатый подогреватель 7, нагреваются до 105—115° С. Растворители 4 и 5 не оснащены нагревательными устройствами, поэтому потери теплоты в растворителях компенсируются подачей в аппараты острого пара, В шнековом растворителе 4 раствор и руда перемещаются по принципу прямотока, а в шнековом растворителе 5 — противотоком. Принцип противотока не применяется во избежание образования мелких кристаллов NaCl, высаливаемых из раствора после растворения в нем К.С1. Шлам, выходящий из шнекового растворителя 5, с целью повышения степени извлечения хлорида калия и для 111
Рис 2.5. Схема производства хлорида калия галургическим методом:
1 — бункер с питателем; 2 — ленточный транспортер; 3 — дозатор; 4,5 — шнековые растворители; 6 — шнековый смеситель; 7 — трубчатый подогреватель; 8— отстойник-сгуститель; 9 — смеситель; 10 — центробежные насосы; 11 — сборник шлама; 12 — вакуум-фильтр; 13— вакуум-котел; 14, 16 — сборники для промывной воды; 15 — барометрический сборник; 17—барометрический конденсатор смешения; 18 — брызгоуловитель; 19, 21 — вакуум-насосы; 20 — поверхностные конденсаторы; 22 — дополнительные конденсаторы; 23 — пароструйные эжекторы; 24 — конденсаторы смешения; 25 — барометрический сборник; 26 — горизонтальные вакуум-кристаллизаторы; 27, 30 — смесители; 28 — центрифуга; 29 — сгуститель; 31, 32, 35 — сборники; 33 — барабанная сушилка; 34 — циклон; 36—вертикальный вакуум-кристаллизатор (I ступень); 37 — вентилятор
рекуперации теплоты дополнительно обрабатывают раствором при 70° С в шнековом смесителе 6. Образующийся при этом шлам промывают горячей водой на непрерывно действующем горизонтальном вакуум-фильтре 12. Промытый шлам содержит 5—6% воды и до 2,5% хлорида калия.
Насыщенный горячий (97—107° С) раствор, полученный в шнековых растворителях 4, 5 и смесителе 6, направляют на осветление в шестиконусный отстойник-сгуститель 8. В первых двух конусах происходит выделение солевого шлама, который центробежным насосом 10 возвращают в шнековый растворитель 5, а в остальных конусах осаждаются глинистые частицы. Глинистый шлам периодически выводят в емкость с мешалкой 9, где его обрабатывают горячей водой (Т:Ж = 4:1) и передают центробежным насосом 10 на противоточную промывку в шнековый растворитель 5. Промытый глинистый шлам центробежным насосом 10 выводят в шламокопитель, а промывные воды центробежным насосом 10 возвращают на растворитель в шнековый смеситель 6. С целью ускорения процесса осаждения глинистого шлама применяют коагулянты — щелочной раствор крахмала, водные растворы тилозы или полиакриламида. Эффективность полиакриламида применительно к глинистому шламу в 20 раз выше по сравнению с крахмалом. Разработан новый высокоэффективный коагулянт — сополимер акриламида с акриловой кислотой.
Процесс кристаллизации КС1 из осветленного насыщенного раствора производят в многокорпусных вакуум-кристаллизационных установках 36, 26. Применяются 14-ступенчатые (корпусные) ВКУ В процессе 14-ступенчатого охлаждения скорость охлаждения растворов составляет около 2° С в 1 мин, перепад температур в каждой ступени не превышает 4—5° С. Это обеспечивает получение кристаллов КС1 размерами не менее 0,15 мм. С целью укрупнения кристаллов КС1 в качестве регуляторов в растворы вводят соли свинца, пектин, первичные алифатические амины и др.
В первом кристаллизаторе 36 разрежение составляет 59—64 кПа, а в последнем 26 — 98—100 кПа. Вакуум в системе создается пароструйными эжекторами 23, отсасывающими из кристаллизаторов паровоздушную смесь. Эжекторы установлены на поверхностных конденсаторах 20, в которые подают пар давлением 600—700 ГПа. Соковый пар на первых девяти ступенях (корпусах) ВКУ конденсируются в поверхностных конденсаторах с помощью маточных растворов. При этом раствор нагревается за счет теплообмена с соковым паром до 65—75° С. Раствор перед подачей на растворение сильвинита подогревают дополнительно до 113—115° С в трубчатом подогревателе 7, обогреваемом паром под давлением 200—300 кПа.
Соковый пар из последующих пяти ступеней ВКУ конденсируется в конденсаторах смешения 24, орошаемых водой и снабженных эжек-113
торами 23. Обе линии конденсаторов объединены дополнительными конденсаторами смешения 22, оборудованными пароструйными эжекторами 23 и соединенными с вакуум-насосом 21. Конденсат из конденсаторов смешения, а также дополнительных конденсаторов используют для промывки отвала из растворителей и глинистого шлама.
Суспензия из последнего корпуса вакуум-кристаллизатора 26 по барометрической трубе непрерывно вводится в вакуум-сборник 31, откуда перекачивается в шестиконусный сгуститель 29. Осветленный маточный раствор возвращают в систему растворения. Сгущенную суспензию хлорида калия (Ж:Т = 1:2) из конусов сгустителя 29 перекачивают в емкость с мешалкой 27, откуда она самотеком поступает на центрифугу 28.
В производстве хлорида калия применяют центрифуги полунепрерывного действия типа ФГ-1800 или более производительные центрифуги непрерывного действия 2ФГП-2К-1200Н.
Для снижения слеживаемости КО при хранении и перевозках в суспензию перед центрифугированием добавляют кондиционирующую добавку — 1 %-ный водный раствор гидрохлоридов первичных жирных аминов С]б — С20 из расчета 180—200 г/т целевого продукта.
Кристаллы целевого продукта сушат в прямоточных барабанных сушилках 33 до остаточной влаги 0,5—1,0%. Температура топочных газов на входе в сушилку — 800—900° С, а на выходе—140—160° С. Влагосъем в таких сушилках составляет 35—45 кг/(м3-ч). Применение в производстве взамен барабанных сушилок аппаратов кипящего слоя (рис. 2.6) позволяет повысить удельный влагосъем до 160—260 кг/(м3-ч).
Согласно практических данных, в настоящее время по рассмотренному способу на получение 1 т целевого продукта — хлорида калия (95% КС1) — расходуют около 5 т сильвинита. Количество галитовых отходов на 1 т продукта составляет 2,5—3,5 т. Отходы содержат 91—95% NaCl, до 0,2% MgCl2, 1,2—3,5% КС1, 0,6—2% CaSO4, до 4% нерастворимого остатка и ~6% Н2О.
Флотационный способ получения хлорида калия. Способ пенной флотации, основанный на разделении хлоридов калия и натрия, содержащихся в исходной руде с предварительным выделением глинистого шлама, широко применяется в калийной промышленности. Варианты существующих технологических схем отличаются друг от друга в зависимости от содержания примесей в исходных рудах и от степени их предварительного измельчения, а также температуры процесса.
Флотационное разделение минералов основано на способности смачиваемости его составляющих, в данном процессе хлоридов калия и натрия. В процессе флотации плохо смачиваемые частицы выносятся на поверхность жидкости вместе с пузырьками воздуха.
114
Рис. 2.6. Сушилка кипящего слоя для сушки хлорида калия:
I — форсунка; 2 — топка; 3 — растопочный клапан; 4—разбрасыватель; 5— смотровое стекло; 6 — осветительный фонарь; 7—камера сушилки; 8 — газораспределительная решетка; 9 — разгрузочное устройство; 10— жаропрочный стакан
Прилипание твердой частицы к пузырьку воздуха — процесс самопроизвольный, протекающий с уменьшением энергии Гиббса поверхности. Для условий пенной флотации процесс подчиняется приближенному уравнению:
AG = аж.г(1-со80),
где аж.г— энергия Гиббса 1 см2 поверхности раздела жидкость— газ; 0 — краевой угол (угол смачивания).
Работа, которую может совершить система в процессе вытеснения воздухом жидкости с 1 см2 твердого тела на стадии прилипания частиц к пузырькам, равна AG. При условии полного смачивания твердых частиц хлорида калия водой 0 = 0 и, следовательно, AG = 0. Процесс прилипания возможен лишь при 0>О. Чем гидрофобнее частицы, тем больше значение 0 и тем быстрее и прочнее они прилипают к пузырькам. Краевой угол 0 зависит от структуры твердого вещества, которая определяет характер связей, обнажающихся на твердой поверхности, образующейся в процессе дробления. Если на поверхности частицы имеются некомпенсированные сильные (ионные, ковалентные, металлические) связи, то она хорошо смачивается водой. Если же связи слабые (молекулярные) или компенсированные сильные — поверхность гидрофобна.
В процессе слипания твердой частицы с пузырьком воздуха проис
ходит разрушение гидратных слоев на их поверхностях в точке касания. На это затрачивается кинетическая энергия движущихся частицы и пузырька. Изменение значения энергии Гиббса при уменьшении толщины 8 жидкой прослойки между частицей и пузырьком при их сближении показано на рис. 2.7. По мере уменьшения значения 8, вода из жидкой прослойки удаляется легко (участок кривой 1—2). После процесса смешения гидратных оболочек части-
Рис. 2.7. Изменение значения энергии Гиббса в процессе сближения частицы с пузырьком воздуха
цы с пузырьками расход энергии на удаление воды резко возрастает и значение энергии Гиббса G прослойки увеличивается (участок 2—З). В процессе дальнейшего сближения частицы с пузырьком (участок 3—4) G резко уменьшается. Это показывает, что гидратная прослойка становится неустойчивой, разрывается и в результате частица прилипает к пузырьку. Поскольку на поверхности частицы остается прочно связанный с ней очень тонкий слой воды— молекулярных размеров, удаление которого потребовало бы значительно боль
116
шой затраты внешней энергии, сопровождалось бы значительным ростом G (участок 4—5).
С целью интенсификации процесса флотации применяют флотационные реагенты. В соответствии с их назначением реагенты делят на собиратели, обеспечивающие прилипание флотируемых частиц к пузырькам, на пенообразователи и на модификаторы (регуляторы), создающие оптимальные условия ведения процесса. Применяют также реагенты, усиливающие действие собирателей (активаторы) и ухудшающие или полностью исключающие флотацию (депрессоры или подавители).
В процессе флотации калийных солей наиболее эффективны катионные собиратели — первичные алифатические амины, получаемые из аммиака и нефтепродуктов (парафинов и др.). Обычно применяют их соли, более растворимые в воде (ацетаты и хлориды), например ацетат октадециламин, гидрохлорид амина и др.
В процессе флотации реагенты адсорбируются на поверхности сильвина, делая ее несмачиваемой. На кристаллах галита амины адсорбируются менее прочно, чем в сильвине, поэтому поверхность остается гидрофильной. Вследствие этого такие кристаллы не прилипают к пузырькам воздуха и выпадают в осадок, образуя галитовые хвосты.
Эффективность процесса флотации зависит от температуры, определяемой составом коллектора (собирателя). Сверхоптимальная температура увеличивает адсорбцию собирателя на галите, что нарушает селективность процесса флотации. Практически установлено, что максимальная скорость процесса флотации сильвина при использовании октадециламина достигается при 30—35° С. Эффективность же разделения минералов обеспечивается при pH среды 5—6.
Процесс флотации сильвина осуществляют без введения пенообразователей, поскольку растворы, насыщенные KCI и NaCl, способны легко вспениваться при барботаже воздуха, особенно при участии в нем собирателей. Однако дополнительное введение в систему собирателей (соснового масла или спиртов диоксанового, пиранового ряда и др.) способствует повышению дисперсности пузырьков воздуха и устойчивости пены, гидрофобизации поверхности флотируемых частиц минерала.
Глинистые шламы флотируют анионным собирателем ФР-2 (продукт окисления уайт-спирита) с добавлением керосина для улучшения технологических свойств пены и раствора полиакриламида, в присутствии которого происходит коагуляция шлама, что снижает расход реагентов.
Схемы флотации сильвинитовых руд отличаются методом обработки глинистых шламов: обогащение с предварительной флотацией глинистых шламов и обогащение с депрессией глинистых шламов, а также степенью их измельчения.
117
Флотационное обогащение сильвинитовых руд состоит из следующих операций: 1) дробление и измельчение исходной руды до размеров частиц, обеспечивающих образование механической смеси входящих в состав руды минералов; 2) предварительное удаление глинистого шлама из руды и его подавление (депрессия) в процессе основной флотации; 3) основная флотация с выделением целевого продукта (КС1) в пенный продукт и последующей перечисткой полученного концентрата; 4) перечистка глинистого шлама с целью снижения потерь целевого продукта; 5) обезвоживание хвостов, шлама и концентрата методом сгущения и фильтрования, переработка влажного концентрата в целевой продукт (сушка и для мелких фракций — гранулирование). Возврат в систему маточных растворов.
Мелкозернистая флотация — обогащение мелкозернистой фракции (0,8 мм) — включает трехстадийное обесшламливание в гидроциклонах, смешение с депрессором — карбоксиметилцеллюлозой и флотацию с собирателем — гидрохлоридом амина и вспенивателем— сосновым маслом. После основной флотации и перечисток концентрат обезвоживают на центрифуге и сушат в аппарате кипящего слоя.
В процессе флотации крупнозернистых калийных руд получают концентрат с содержанием 93% основного вещества при степени извлечения 90—92% КС1. На получение 1 т 95%-ного КС1 расходуют: 5,0—5,2 т (в пересчете на 22%) КС1; 0,520—0,780 кг гидрохлорида амина; 1,040 кг смеси мазута и солярового масла (2:1); 2,6—2,8 кг топочного мазута; 60—70 кВт-ч электроэнергии и 4—5 м3 свежей воды.
Основными аппаратами в процессе получения хлорида калия являются флотационные машины и дуговое сито.
Флотационная машина механического типа «Механобр-7ВМ» для мелкозернистого типа флотации (рис. 2.8) состоит из нескольких (до 20) металлических камер, соединенных друг с другом. Суспензия в аппарат поступает в камеры по трубе 7, через которую под воздействием вращающегося импеллера 6 засасывается воздух благодаря интенсивному механическому перемешиванию. Мелкие пузырьки воздуха вместе с суспензией выбрасываются в камеру, где к ним прилипают гидрофобные частицы сильвина, и всплывают на поверхность, образуя слой минерализованной пены. Образующаяся пена лопастями пекогонов 4 сгребается в общий желоб 5. Оставшиеся в суспензии гидрофильные частицы руды (галитовые хвосты) последовательно проходят все камеры и выводятся через сливное отверстие на дальнейшую переработку.
Флотационная машина с кипящим слоем (рис. 2.9) состоит из 6—12 камер. Число камер зависит от их объема. Суспензия подается в наклонную решетку 7, имеющую живое сечение 25—30%, равномерно распределяется по всей ширине камеры 3 и поступает в зону повышение
1 2
3
Рис. 2.8. Флотационный аппарат Рис. 2.9. Пневмомеханическая
«Механобр-7ВМ»: флотационная машина с кипящим слоем:
1 — всасывающая труба; 2 — шибер; 3 — при- 1 — наклонная решетка; 2 — полый вал; вод импеллера; 4 — пекогоны; 5 — желоб; 3 — камера; 4, 5 — штуцеры; 6 — горизонталь-6 — импеллер; 7 — статор ная решетка; 7—патрубок; 8— лопасти стато-
ра; 9 — диспергирующее устройство; 10 — ста-тор; 11— импеллер; 12 — аэролифт
ной аэрации, создаваемую аэролифтом 12. Часть сильвина флотируется в пенный продукт. Несфлотированные частицы передвигаются по наклонной беспровальной решетке 2, периодически перемешиваются восходящими аэрированными потоками суспензии, обеспечивающими дополнительную флотацию сильвина. Часть несфлотированного материала поступает на горизонтальную решетку 6 (живое сечение 15—20%), на которой под действием аэрированных потоков, подаваемых импеллером 11, создается кипящий слой, из которого дофлотиру-ются частицы полезного минерала.
Хвосты флотации выводятся через патрубок 7. В флотационной машине кипящего слоя имеются два контура циркуляции и аэрирования суспензии. Один создается работой аэролифта под воздействием сжатого воздуха, другой — импеллером 11 и диспергирующим устройством 9 в виде полого усеченного конуса с рифленой гофрированной или ребристой поверхностью.
В процессах (основном и контрольном) флотации крупнозернистого сильвина применяют также и машины пенной сепарации ФПС-16. Пенной флотацией называют процесс подачи обработанной реагентами суспензии сверху на пенный слой. При этом гидрофобные частицы минерала задерживаются пеной, а гидрофильные увлекаются жидкостью, поступающей с суспензией сверху и с воздушными пузырьками снизу.
119
Машина ФПС-16 (рис. 2.10) представляет собой пирамидальную камеру 1 с вершиной в нижней части. Суспензия поступает на пенный слой, образуемый в результате подачи сжатого воздуха через резиновые трубчатые аэраторы, которые расположены в два ряда на расстоянии 18—20 мм друг от друга. Число перфораций в трубах — до 60 отверстий на 1 см, избыточное давление воздуха 1,18-105 Па, расход воздуха — до 2 см3/мин. При этом гидрофобные частицы остаются в пенном слое и удаляются через сливной порог 6, а нефлотированные частицы опускаются вниз и выгружаются через разгрузочное устройство 8, оборудованное шланговым затвором в виде черновых хвостов, которые подвергаются дальнейшей обработке. Производительность двухсекционной машины ФПС-16 при крупности кусков до 3 мм составляет 100 т/ч калийной руды.
Дуговое (вогнутое) сито (рис. 2.11) применяют в процессах обезвоживания и классификации мелких частиц. Вогнутая поверхность сита с радиусом кривизны 1,50 и 0,55 м (соответственно площадь
Рис. 2.10. Пневматическая флотационная машина для пенной сепарации:
/—камера; 2—трубчатые аэраторы; 3, 5 — брызгала; 4 — загрузочное устройство; 6 — сливной порог; 7 — желоб; 8 — разгрузочное устройство
120
Рис. 2.11. Дуговое (вогнутое) сито:
7, 2 — крышки; 3 — корпус; 4 — колосниковая решетка сита; 5 — прижимной сегмент; 6 — шибер; Л-радиус кривизны
просеивания 2,0 и 0,95 м2) образуется металлическими колосниками 4, поставленными на ребро. Размер щели регулируется расстоянием между колосниками. Просеиваемый материал движется поперек установленных колосников со скоростью до 3 м/с. При большой скорости движения в щель проходят лишь частицы размером в 1,5—2 раза меньше ширины щели. Удельная производительность сит на 1 м2 рабочей поверхности составляет 200—250 м3/ч суспензии.
2.4. СУЛЬФАТ КАЛИЯ
Физико-химические свойства, применение. Сульфат калия K2SO4, бесцветные кристаллы ромбической сингонии (а = 0,742 нм, b = 1,001 нм, с = 0,573 нм, z = 4, пространственная группа Риат; плотность 2,66 г/см3). Выше 584° С устойчива гексагональная модификация (а = 0,5947 нм, с = 0,8375 нм, z = 2, пространственная группа Ptymmc, плотность 2,26 г/см3), ДЯ° перехода 8,0 кДж/моль; температура плавления 1074° С, температура кипения ~2000°С; С ° = 131,4 Дж/(моль-К); А#™ = =36,3 кДж/моль; ДЯ^р =-1437,7 кДж/моль; 5298 = 175,56 Дж/(моль-К).
Растворимость сульфата калия в воде (г в 100 г): 7,35(0° С), 11,1(20° С), 14,8(40° С), 24,2(100° С).
Температура плавления эвтектики (7,1 г K2SO4 в 100 г Н2О)—1,51° С. Плотность 5%- и 10%-ных водных растворов соответственно 1,0393 и 1,0817.
Сульфат калия образует двойные соли, в том числе квасцы. Соли 2K2SO4 • Na2SO4 (глазерит), K2SO4 • MgSO4 6Н2О (шенит), K2SO4 • MgSO4 • 4Н2О (леонит), K2SO4 • 2MgSO4 (лангбейнит), K2SO4 • MgSO4 • 2CaSO4 • 2H2O (полигалит), K2SO4-CaSO4-H2O (сингенит), a также K2SO4 (арканит) встречаются в природе в месторождениях калийных солей. Сульфат калия содержится также в водах соленых озер.
Гидросульфат калия KHSO4 — бесцветные кристаллы ромбической сингонии (а = 0,840 нм, b = 0,979 нм, с = 1,893 нм, z = 16, пространственная группа РЬса). Температура плавления — 222°С; плотность 2,4 г/см3; ДЯ^р =-1163,3 кДж/моль; S2°98 = 142,2 Дж/(моль-К). Выше температуры плавления образует смесь K2SO4 и K2S2O7. Гигроскопичен, растворяется в воде.
Сульфат калия применяют в качестве удобрения, в процессах получения квасцов и других соединений калия. Является компонентом шихты в производстве стекла.
Гидросульфат калия применяют в качестве компонента флюса в металлургии и сульфирующего агента в производстве красителей, в процессах получения сложных удобрений, а также в аналитической химии для перевода труднорастворимых соединений в легкорастворимые.
121
Способы получения сульфата калия. Существуют две группы способов получения сульфата калия: 1) основанные на переработке полиминеральных руд, содержащих сульфат калия галургическими, флотационными или комбинированными методами; 2) основанные на конверсии хлорида калия серной кислотой и сульфатами натрия, магния, аммония, кальция и др.
Известны и другие способы получения сульфата калия, например в процессе комплексной переработки нефелинов и алунитов и др. Сульфат калия получают также в процессе флотационного обогащения лангбейнитовых и каинито-лангбейнитовых руд.
Способ переработки полиминеральных руд. Физико-химические основы. Полиминеральные каинито-лангбейнитовые руды содержат минералы каинит KCl-MgSO4-3H2O и лангбейнит K2SO4-2MgSO4. Производство на их основе заключается в разделении компонентов растворов выщелачивания исходной руды с получением сульфата калия или калимагнезии, основной частью которой является шенит K2SO4-MgSO4-6H2O. Соотношение между выходом сульфата калия и калимагнезии зависит от химического состава исходной руды и заданного объема производства сульфата натрия и бишофита MgCl3-6H2O. Доля калимагнезии в производстве целевой продукции растет при уменьшении выхода сульфата натрия и бишофита.
В процессе переработки руды происходит конверсия лангбейнита по схеме
2(K2SO4 • 2MgSO4) + 2KCI + 18Н2О = 3(K2SO4 • MgSO4 • 6Н2О) + MgCI2
В процессе выщелачивания каинита образуются шенит и хлорид магния:
2(КС1 • MgSO4 ЗН2О) = K2SO4 • MgSO4 • 6Н2О + MgCl2
Если в растворимой части руды кроме каинита содержится и сильвин, находящийся в эквимолекулярном соотношении с каинитом (KCl:MgSO4 = 1,24), то сульфат калия можно получить и в результате реакции:
КС1 + КС1 • MgSO4 • ЗН2О = K2SO4 + MgCl2 + ЗН2О
При содержании сильвина в исходной руде больше эквимолекулярного по отношению к каиниту, сульфат магния недостаточен для конверсии всего хлорида калия, содержащегося в руде, в его сульфат. Избыток хлорида калия при этом выделяется отдельно:
2КС1 + КС1 • MgSO4 ЗН2О = КС1 + K2SO4 + MgCl2 + ЗН2О
122
Образующийся и накапливающийся в системе хлорид магния выводят из системы с маточными растворами и используют в производстве металлического магния или других продуктов.
Калимагнезию, полученную конверсией лангбейнита, после обезвоживания и сушки выпускают в качестве удобрения, содержащего «31% К2О. Она может быть переработана в сульфат калия по схеме
K2SO4 • MgSO4 • 6Н2О + 2КС1 = 2K2SO4 + MgCl2 + 6Н2О
Переработка лангбейнитовых руд. Приведенная на рис. 2.12 схема переработки каинито-лангбейнитовых руд состоит из следующих основных стадий: 1) получение основных продуктов — сульфата калия или калимагнезии; 2) флотационное разделение труднорастворимых калийных минералов и галита; 3) регенерация калийных солей
Полиминеральная руда
Рис. 2.12. Принципиальная схема комплексной переработки полиминеральных руд
123
из маточных растворов; 4) получение в качестве полуфабриката искусственного карналлита и переработка его в бишофит.
I стадия. Исходная руда измельчается до частиц размером 5 мм и поступает на растворение в три последовательно расположенных горизонтальных растворителя с рамными мешалками по противоточно-прямоточной схеме: руда поступает в первый горячий (105—110° С) раствор — в третий растворитель, в каждом растворителе руда и раствор передвигаются прямоточно. Твердая фаза из одного растворителя в другой транспортируется наклонными ковшовыми элеваторами. Растворитель представляет собой смесь маточного раствора с промывными водами. Перед поступлением в реактор ее подогревают в трубчатых теплообменниках.
В процессе растворения в жидкую фазу переходят легко растворяющиеся минералы: сильвин, шенит, каинит и карналлит. Не растворимые в воде полигалит, лангбейнит, галит из последнего растворителя выводятся на планфильтр. Глинистые шламы в виде тонкодисперсной взвеси выносятся из растворителей с раствором.
Насыщенный раствор первого растворителя отстаивают в отстой-нике-солеотделителе. Образующийся сгущенный солевой шлам направляют в третий растворитель для повторного выщелачивания. Слив отстойника-солеотделителя отстаивают от глинистого шлама в отстойнике, используя в качестве коагулянта полиакриламид. Сгущенный глинистый шлам отмывают от солей калия по противоточной схеме промывки и направляют в шламонакопитель.
Отстоенный насыщенный раствор поступает в девятиступенчатую вакуум-кристаллизационную установку, в которой охлаждается от 65 до 32° С. Далее растворы охлаждают до 20° С в шести поверхностных кристаллизаторах, охлаждаемых водой и аммиаком (последний корпус). В процессе охлаждения раствора происходит кристаллизация ше-нита. Из последнего корпуса кристаллизатора суспензию кристаллов шенита направляют в сгуститель-солеотделитель, где и осаждается основное количество кристаллов шенита. Слив из солеотделителя через дополнительный отстойник выводят на ВКУ, где он подогревается до 45—50° С в поверхностных конденсаторах и разделяется на два потока. Первый поток, проходя трубчатые теплообменники, возвращается на растворение руды, а второй направляют на регенерацию калийных солей (III стадия производства).
Сгущенная шенитовая суспензия из солеотделителя и отстойника фильтруется на барабанных вакуум-фильтрах. Полученный продукт сушат и выпускают в качестве калимагнезия или двухстайным разложением шенита получают сульфат калия.
В последнем случае на первой стадии шенит разлагают в реакторе водой разбавленным сульфатным раствором, поступающим со второй стадии в течение 30—60 мин. При этом получают сульфат калия 124
и концентрированный сульфатный раствор. Его отделяют от шени-то-сульфатной суспензии в сгустителе-солеотделителе, отстойнике и передают на кристаллизацию в ВКУ в смеси с насыщенным раствором из отделения растворения руды. Сгущенная шенито-сульфатная суспензия в течение 10—15 мин доразлагается водой на второй стадии процесса. При этом образуются кристаллы сульфата калия. Кристаллы освобождают от растворов на барабанных вакуум-фильтрах и направляют на сушку.
II стадия. Нерастворенная часть исходной руды из растворителей содержит 3,5—4,0% К2О. Кроме того, остаток содержит: 17% лангбейнита, 9,2% полигалита, 53,3% галита, 7,4% кизерита, 3,9% каинита, 1,8% ангидрита и 7,3% глины.
С целью дополнительного извлечения калийно-магниевых солей осадок (Ж:Т = 3,5:1) измельчают и подвергают флотации. Во флотационной машине происходит разделение минеральных составляющих твердой фазы: калийные минералы переходят в пенный продукт, а галит и основная масса глинистого шлама передаются из флотома-шины в сгуститель-солеотделитель. Продолжительность процесса флотации составляет 25 мин. После сгущения хвосты обезвоживают на барабанных вакуум-фильтрах и направляют в шламонакопитель.
Пенный продукт после двух перечисток сгущают до соотношения Ж:Т = 1,44-1,6, фильтруют и направляют в растворитель, в котором флотационный концентрат растворяется водным раствором сульфата магния. Нерастворившийся полигалитовый остаток после сгущения и фильтрации высушивают в сушилках кипящего слоя и выпускают в виде товарного продукта — калийно-магниевого удобрения, содержащего до 10—13% К2О.
Насыщенный лангбейнитовый раствор из первого растворителя флотационного концентрата после отделения в отстойнике-осветлителе глинистых шламов поступает на первую стадию производства кристаллизации шенита.
III стадия. С целью регенерации калийных солей и извлечения хлорида магния, накапливающихся в маточном растворе, все избыточные маточные растворы подвергают четырехстадийной вакуум-выпарке с последовательным выделением хлорида натрия и калийно-магниевой соли. Затем растворы нагревают до 95—100° С и обрабатывают 25%-ным раствором хлорида кальция. При этом ионы SO«' переходят в гипс, осадок которого направляют в шламонакопитель.
IV стадия. Растворы из гипсового сгустителя упаривают в аппаратах погружного горения (АПГ) при 120—125° С до их концентрации 31—32% MgCI2. Упаренные растворы охлаждают в поверхностных кристаллизаторах до 35° С, в которых происходит кристаллизация смеси галита и карналлита. Суспензию сгущают, отделяют
125
смесь на центрифуге, маточные растворы передают в кристаллизаторы, в которых происходит конверсия карналлита в каинит:
KClMgCl2 • 6Н2О + MgSO4 = КС1 • MgSO4 • ЗН2О + MgCl2 + ЗН2О
Слив сгустителя — осветленный раствор карналлита — проходит четвертую стадию выпарки при 140—145° С до образования плава бишофита MgCl2 • 6Н2О. Плав охлаждают на вальцах и в виде че-шуированного продукта направляют на склад готовой продукции.
Существует способ переработки лангбейнитовых руд, богатых хлоридами калия и натрия. Для переработки лангбейнита на сульфат калия исходное сырье измельчается на молотковых дробилках, работающих в замкнутом цикле с вибрационным грохотом, где классифицируется по классу 1,6 мм. Крупный класс репульпируется с холодной водой во вращающемся барабане с целью отмывки галита и обезвоживается в гидроциклоне. Пески гидроциклона смешиваются с классом 1,6 мм и подаются в два последовательно работающих классификатора. Вода для растворения галита подается во второй классификатор, куда поступает лангбейнит из первого классификатора. Слив первого классификатора после отделения в гидроциклоне и отстойнике мелких кристаллов сбрасывают. Отмытая руда, выходящая из второго классификатора и содержащая 96—98% лангбейнита, обезвоживается в центрифуге и сушится в барабанных сушилках. Высушенный лангбейнит содержит 22% К2О, 18% MgO и не более 2,5% хлора.
Получение сульфата калия из отмытой от хлорида натрия лангбейнитовой руды проводят по схеме
K2SO4 • 2MgSO4 + 4КС1 3K2SO4 + 2MgCl2
Образующийся хлорид магния выводится из системы в виде концентрированного раствора в смеси с небольшими количествами хлоридов натрия и калия.
Схема получения сульфата калия из лангбейнита приведена на рис. 2.13, согласно которой измельченная до крупности 0,1 мм лангбейнитовая руда, возвратные смешанные соли и раствор хлорида калия в расчетных количествах вступают в реакцию в кас-кадно расположенных из пяти взаимосвязанных реакторов 2, оснащенных мешалками. Для поддержания необходимой температуры процесса все реакторы оснащены внешними (водяными) и внутренними (паровыми) змеевиками. Образующийся в реакторах 2 сульфат калия обезвоживается в центрифугах непрерывного действия 3 и сушится в барабанных вращающихся сушилках 5. Маточные растворы, образующиеся в центрифуге, выпариваются в аппаратах погружного горения 7 до концентрации 67% его первоначальной 126
Рис. 2.13. Схема переработки лангбейнита на сульфат калия:
1 — бункер для лангбейнита; 2 — реактор; 3 — центрифуга; 4 — бункер для сульфата калия; 5 — вращающаяся барабанная сушилка; 6 — емкость для сульфатного раствора; 7 — аппарат погружного горения; 8 — кристаллизатор; 9 — отстойник; 10 — фильтр; 11 — растворитель хлорида калия
массы. Количество испаряемой воды должно быть таким, чтобы в процессе последующего охлаждения раствора до 30° С не наступила кристаллизация галита.
В процессе упарки растворов сульфата калия при 70° С и выше происходит кристаллизация лангбейнита и хлорида калия. Горячая суспензия этих солей охлаждается до 30° С в двухкорпусном ваку-ум-кристаллизаторе 8. При этом лангбейнит перекристаллизовывается в леонит, а при достаточно высокой концентрации раствора возможно образование каинита. Выделившиеся в процессе упарки и охлаждения соли сгущаются в отстойнике Дорра 9, фильтруются на барабанном вакуум-фильтре 10 и направляются на стадию конверсии лангбейнита. Маточные растворы после барабанного вакуум-фильтра направляются в реакторы. По мере накопления хлорида магния в маточных растворах последние выводятся из системы.
Существует также способ получения сульфата калия восстановлением лангбейнита. При этом предварительно отмытая от хлорида натрия сырая лангбейнитовая соль смешивается с углем или коксом в барабанном смесителе (92% лангбейнита и 8% угля) и смесь подвергается термообработке в шахтной печи при 800—900° С:
K2SO4 • 2MgSO4 + 2С = K2SO4 + 2MgO + 2СО + 2SO2
Из полученного плава сульфат калия выщелачивают при 95—98° С водой. Оксид магния отделяется на фильтре, а раствор сульфата калия упаривается. Из упаренных растворов выделяют кристаллы.
Сульфат калия можно получить из алунита K2SO4 • А12(8О4)з • •4А1(ОН)з, в котором теоретически содержится 23% K.2SO4. В про-127
изводстве исходную руду дробят, измельчают в шаровых мельницах, сушат и восстанавливают элементной серой при температуре 600—700° С:
K2SO4 • А12(8О4)з-4А1(ОН)з + 3S + 1,5Q> = K2SO4 + ЗА12О3 + 9SO2 + 6Н2О
Образующийся плав растворяют в воде. Суспензию фильтруют, фильтрат упаривают и выделяют из него кристаллы сульфата калия, которые сушат. Образующийся водонерастворимый сесквиоксид алюминия используют в производстве металлического алюминия.
Маточные растворы от выщелачивания сульфата калия после фильтрации возвращают в технологический цикл.
Конверсионные способы получения сульфата калия. Конверсионные методы получения сульфата калия основаны на реакции его хлорида с соединениями, содержащими су-льфат-ион, в основе которых могут быть практически использованы сульфат натрия (мирабилит), сульфат магния (эпсомит), сульфат железа, серная кислота и т.д.
Теоретические основы процесса получения сульфата калия методом конверсии сульфата натрия (мирабилита) хлоридом калия разработаны во ВНИИГе. Из взаимной солевой системы (рис. 2.14)
2КС1 + Na2SO4 1; K2SO4 + 2NaCl
видно, что образование глазерита происходит при взаимодействии мирабилита, хлорида калия и раствора А, получаемого при последующей переработке глазерита на сульфат калия. Точка О, характеризующая состав смеси мирабилита, хлорида калия и раствора А, находится в поле глазерита на луче, соединяющем полюс глазерита G1 и точку т, в котором раствор насыщен тремя твердыми фазами— тенардитом, галитом и глазеритом. При таком положении точки О обеспечивается максимальный выход глазерита. Образующийся глазерит обрабатывается хлоридом калия с получением сульфата калия и раствора А, возвращаемого на первую стадию процесса. Установлено, что в процессе конверсии мирабилита конечным продуктом может быть смесь сульфата калия и глазерита. С целью исключения этого явления и получения лишь сульфата калия предложено упарить избыточное количество воды на первой стадии конверсии. Оптимальной температурой, обеспечивающей максимальный выход сульфата калия, является 25° С. Продолжительность процесса на первой стадии составляет 1 ч, во второй стадии — 30 мин. Глазерит, образующийся на первой стадии конверсии, представляет собой твердый раствор, обогащенный сульфатом натрия. Индекс его по калию составляет 64—66 против теоретического, равного 75. С целью уве-128
личения индекса по калию разработан способ, сущность которого сводится к следующему: 1) глазерит с пониженным содержанием калия обрабатывают раствором А, получаемым при последующем разложении его на сульфат калия; 2) получаемый на стадии обогащения глазерита маточный раствор направляется на получение дополнительных количеств глазерита.
Глазеритовый раствор (рис. 2.14, точка т) выводится из процесса конверсии. При этом извлечение калия в продукт не превышает 75%. Поэтому с целью повышения выхода калия разработаны варианты утилизации глазеритового раствора: 1) упарка с выделением хлорида натрия; 2) упарка с выделением смеси хлорида натрия и сульфата натрия; 3) упарка глазеритового раствора в смеси с раствором А с выделением из системы смеси солей калия.
На основе изучения диаграммы растворимости взаимной системы 2КС1 + NazSCh K2SO4 + 2NaCl при различных температурах (рис. 2.15) предложен новый метод утилизации глазеритового раствора. Фигуративная точка т состава глазеритового раствора при 0° С и ниже находится в поле кристаллизации мирабилита. При этом состав раствора будет изменяться по лучу кристаллизации мирабилита т—1. Мирабилит отделяется от раствора и направляется на первую стадию процесса конверсии. Фигуративная точка J, показывающая состав охлажденного раствора, находится на изотерме 100° С в поле кристаллизации галита, в результате в процессе упарки раствора в твердую фазу будет переходить хлорид натрия. Состав жидкой фазы при этом будет изменяться по лучу кристаллизации хлорида натрия до точки 2, где раствор насыщается хлоридом натрия и глазеритом.
Рис. 2.14. Диаграмма растворимости солей в системе КС1—Na2SO4—Н20 при 25° С
5 Химическая технология неорганических веществ, кн. 1
Рис. 2.15. Диаграмма процесса переработки глазеритового раствора при охлаждении его до 0° С
129
Процесс упарки прекращают, раствор отделяют от кристаллов хлорида натрия, а затем раствор охлаждают до 0° С. Поскольку точка 2, изображающая состав упаренного раствора, находится в поле глазерита, то в процессе охлаждения раствора в твердую фазу будет переходить глазерит. При этом состав жидкой фазы изменяется по лучу кристаллизации глазерита 2—3. Далее происходит совместная кристаллизация глазерита и хлорида калия, причем состав жидкой фазы изменяется по линии 3—1. После отделения калийных солей, направляемых на конверсию, оставшийся раствор вновь может быть
направлен на выпаривание и получение хлорида натрия.
Разработан также вариант схемы переработки глазеритового раствора, по которому упаренный раствор 2 после отделения хлорида натрия направляется на первую стадию конверсии вместе с мираби
литом, полученным в процессе охлаждения глазеритового раствора. Метод позволяет существенно сократить объемы растворов, циркули
рующих в системе «охлаждение — выпаривание» в процессе переработки глазеритового раствора. На рис. 2.16 изображен ход получения сульфата калия из мирабилита и
Рис. 2.16. Схема процесса конверсии мирабилита хлоридом калия с утилизацией глазеритового раствора охлаждением до 0°С:---------------
изотермы 0°С;----------изотерма 100°С:
1 — раствор, образующийся после охлаждения глазеритовых растворов и отделения мирабилита; 2 — упаренный раствор после отделения хлорида натрия; 3 — смесь упаренного раствора 2 с мирабилитом, поступающая на первую стадию конверсии; 4 — смесь хлорида калия с раствором после обогащения гпа-зерита; А — маточный раствор сульфата калия; G1 — глазерит
хлорида калия с утилизацией глазеритового раствора.
Схема получения сульфата калия конверсией сульфата натрия хлоридом калия (рис. 2.17). Хлорид калия и сульфат натрия из бункеров 1 подаются через тарельчатые питатели в реактор конверсии 3 с рамной мешалкой. Одновременно в реактор в расчетном соотношении поступают сульфат натрия (мирабилит) и упаренный раствор, полученный на стадии утилизации маточных растворов, а также серная кислота для нейтрализации карбоната натрия, содержащегося в исходном сульфате натрия. Процесс получения глазерита проводится при 20—25° С.
Повышение температуры несколько повышает скорость процесса, однако при этом существенно увеличивается содержание в растворе глазерита ионов калия и сульфата, что приводит к увеличе-
но
Вода
Na2SO41QH2O
Фильтрат
Фильтрат
Мирабилит
K2SO4 на сушку
со
Е
Н го
5
8
Глазеритовый раствоЬ I
Рис. 2.17. Схема получения сульфата калия конверсией сульфата натрия хлоридом калия:
7 — бункеры для исходного сырья; 2 — сборник серной кислоты; 3, 6, 9 — реакторы конверсии; 4, 7, 10, 15, 19 — отстойники; 5, 8, 11, 20, 21 — центрифуги; 12, 17 — теплообменники; 13 — поверхностный кристаллизатор; 14 — холодильная аммиачная установка; 16—вакуум-фильтр; 18— ВКУ
Обогащенный глазерит
Арканитовый
Фильтрат
J 17
NaCl на сушку
нию нагрузки на отделение утилизации маточных растворов. Процесс производится непрерывно и продолжается 40—60 мин. Образующуюся в реакторе глазеритовую суспензию направляют в сгуститель Дорра 4, в котором при скорости восходящего потока 1 м/ч суспензия осветляется. Суспензия, сгущенная до соотношения Ж:Т = 0,7:1,2, через промежуточный горизонтальный аппарат с метальным устройством поступает на центрифугу 5. Отфильтрованный на центрифуге глазерит направляется на стадию обогащения, где он обрабатывается раствором, полученным на второй стадии конверсии. Процесс обога-131
щения глазерита проводится при 20—25° С в течение 60 мин в реакторе 6, аналогичном реактору первой стадии.
Суспензия обогащенного глазерита направляется в отстойник Дорра 7, где она осветляется при скорости восходящего потока 2,5 м/ч. Сгущенная суспензия с соотношением Ж:Т = 0,7:1,0 подается на центрифугу 8. Фильтрат из центрифуги возвращается в отстойник, а осветленный раствор собирается в сборник и затем подается на первую стадию конверсии. Отфильтрованный на центрифуге 8 обогащенный глазерит направляется в реактор 9, куда одновременно со второй стадии конверсии поступает 28%-ный раствор хлорида калия. Температура суспензии (смеси) в реакторе поддерживается автоматически на заданном уровне подачей пара в рубашку аппарата. Процесс конверсии продолжается 50—60 мин при 20—25° С. Повышение температуры процесса от 25 до 50° С существенно не сказывается на скорости реакции, но при понижении температуры возможна кристаллизация хлорида калия из раствора, что приводит к загрязнению целевого продукта хлоридом калия и глазеритом.
Полученная в реакторе суспензия сульфата калия осветляется в отстойнике Дорра 10. Осветленный раствор направляется на стадию обогащения глазерита, а сгущенная суспензия сульфата калия отжимается в центрифуге. Кристаллы сульфата калия промываются на центрифуге водой и направляются на сушку.
Для повышения степени извлечения калия и сульфат-иона глазе-ритовый раствор обессульфачивают и выпаривают. Процесс обессуль-фачивания раствора проводят охлаждением его до 0—5° С. С этой целью глазеритовый раствор, полученный на первой стадии конверсии, направляется в теплообменник 12 для предварительного охлаждения холодным мирабилитовым маточным раствором и далее в каскад поверхностных кристаллизаторов 73 с аммиачным охлаждением, в которых происходит кристаллизация мирабилита. Образующаяся суспензия сгущается в отстойнике Дорра до соотношения Ж:Т = 3:4 и обезвоживается на дисковом вакуум-фильтре 16. Кек фильтра— мирабилит с незначительными примесями хлоридов калия и натрия — подается на первую стадию конверсии. Осветленный мирабилитовый маточник отдает свой холод исходному глазеритовому раствору в теплообменнике 12, нагревается далее в теплообменнике 17 за счет горячего упаренного раствора и поступает на двухкорпусную выпарную установку 18.
Суспензия хлорида натрия, получаемая на выпарной установке, сгущается в отстойнике Дорра 19 и центрифугируется. Хлорид натрия поступает на сушку. Осветленный раствор из отстойника Дорра охлаждается в теплообменнике 17 и направляется на первую стадию конверсии.
132
Способ получения сульфата калия из его хлорида основан на реакции хлорида калия с сульфатом магния (эпсомитом):
2КС1 + MgSO4 MgCl2 + K2SO4
На рис. 2.18 представлена диаграмма растворимости солей в этой системе с учетом свойств системы К+, Mg7+ ||СГ, SO,', Н2О. Согласно данных системы, процесс проходит в две стадии. В первой стадии процесса кристаллизуется шенит. Из диаграммы следует, что для достижения максимального выхода шенита точка С состава исходной смеси должна лежать на луче кристаллизации ШР, идущем из полюса шенита Ш в точку Р, положение которой соответствует составу маточного раствора, насыщенного шенитом, сильвином и каинитом. Раствор Р — раствор шенита — выводят из цикла, а шенит обрабатывают хлоридом калия в водной среде с образованием сульфата калия и маточного раствора А, насыщенного хлоридом калия, сульфатом калия и шенитом. Этот раствор полностью используют в первой стадии конверсии, и, следовательно, процесс замыкается. Однако для обеспечения полного замкнутого цикла необходимо введение части эпсомита на вторую стадию или части хлорида калия (--1/3 расчетного количества) на первую стадию. Для получения высококачественного сульфата калия (~52% К2О) необходимо применять хлорид калия с высоким содержа
нием основного вещества.
Способ получения сульфата калия взаимодействием сульфата магния с хлоридом калия был разработан ВНИИГ с проведением основ-
ной реакции при 25° С в продукта (полуфабриката) — шенита. Процесс образования шенита длится 30 мин, а процесс разложения его водным раствором хлорида калия с образованием сульфата калия — 90 мин. Было установлено также, что получение целевого продукта может быть эффективным лишь при взаимодействии 95%-ного шенита с техническим хлоридом калия, содержащим 95% KCI.
две стадии с получением промежуточного
Рис. 2.18. Растворимость в водной системе 2КС1 + MgSO4 # K2SO4 + MgCI2 при 25° С
Согласно схеме получения сульфата калия
133
(рис. 2.19), исходное сырье со складов подается в цех ленточными транспортерами. В процессе транспортировки сырье очищают от металлических предметов в магнитном сепараторе. Исходное — эпсомит и хлорид калия — подвергают грохочению с целью отделения крупных фракций (+2 мм), которые измельчают в дезинтеграторах. Мелкие фракции сырья подаются в соответствующие бункеры: хлорид калия в 1 и эпсомит во 2, откуда через дозаторы 3 и 4 соответственно исходные компоненты передаются в смесители 5. В смесители же поступает раствор сульфата со второй стадии конверсии. Они снабжены метальным устройством со скоростью вращения не менее 200 об/мин. Продолжительность процесса смешения компонентов составляет 15—20 мин. Согласно технологии, предусмотрены два смесителя, первый из которых загружается исходными компонентами, а второй выгружает их в реактор 6. Образующаяся в реакторе суспензия непрерывно откачивается в отстойник 7, работающий в режиме гидросепаратора. Скорость восходящего потока в гидросепараторе за счет возврата фильтрата и части маточного раствора составляет 3 м/ч. При этом в слив отстойника выводится мелкодисперсный шлам, содержащийся в эпсомите, который затем удаляется из раствора в процессе контрольной фильтрации. Отфильтрованный раствор шенита поступает в буферную емкость 10, а далее на выпарную станцию 11. Кек с фильтров ФПАКМ направляется на промывку в аппарат 8 для уменьшения потерь калия с пропитывающим раствором.
Сгущенная суспензия шенита из отстойника 7 направляется на центрифугирование в фильтрующие центрифуги 9 с ножевым съемом осадка. Влажный шенит транспортером подается в бункер шенита 24 и далее на вторую стадию конверсии, аппаратурно-технологическая схема которой выполнена аналогично первой стадии технологии. Далее суспензия сульфата калия сгущается в отстойнике 19, отжимается на центрифуге 9, после чего сульфат калия сушат во вращающейся барабанной сушилке 17. Слив отстойника — сульфатный раствор— фильтруется на фильтрах ФПАК, освобождаясь от мелких глинисто-солевых шламов, направляется на первую стадию конверсии. Глинисто-солевые шламы подаются в аппараты 8 на противоточную промывку.
Для повышения коэффициента извлечения калия из исходного хлорида калия соли калия регенерируются из шенитового маточного раствора, выводимого из схемы после первой стадии конверсии. Для этого раствор из промежуточной емкости 10 подается на упарку в аппарат погружного горения 12, в котором выпаривается 15—20% воды. Выделяющиеся при этом в твердую фазу соли, состоящие в основном из галита, сгущаются в отстойнике 13, фильтруются в центрифуге 9 и направляются на склад. Упаренный раствор охлаждается в теплообменнике 14 морской водой от 105 до 30° С и поступает 134
Ряс. 2.19. Схема получения сульфата калия из эпсомита и хлорида калия:
7, 2, 24— бункеры для хлорида калия, эпсомита и шенига соответственно; 3, 23— дозаторы хлорида калия; < 22 — дозаторы эпсомита и шенита соответственно; 5, 21 — смесители; 6, 75, 20—реакторы; 7, 73, 19—отстойники; 8— аппарат с перемешивающим устройством; 9 — центрифуга; 10— буферная емкость; 77, 18— выпарные станции; 72 — аппарат погружного горения; 14 — теплообменник; 76 — аппарат кипящего слоя; 17—барабанная сушилка
в реактор 15, где он перемешивается с эпсомитом. В процессе перемешивания с раствором эпсомит конвертируется содержащимся в нем избыточным хлоридом калия в шенит, который сгущается в отстойнике 13, фильтруется на центрифуге 9 и направляется на сушку в аппарат кипящего слоя 16. Осветленный раствор шенита направляется в эпсомитовый бассейн.
2.5. КАРБОНАТ КАЛИЯ
Карбонат калия (поташ) К2СО3, бесцветные кристаллы моноклинной сингонии (а = 0,564 нм, b = 0,980 нм, с = 0,688 нм, В = 98,8°, z - 4, пространственная группа P2i/c); плотность 2,44 г/см3. При нагревании до 420°С переходит в гексагональную модификацию (при 425°С а = 0,571 нм, с = 0,717 нм, плотность 2,27 г/см3); температура плавления 891 °C; С ° = 114,43 Дж/(моль-К); = -1151,5 кДж/моль, АЯ^,= 30,5 кДж/моль; S°98 = 155,50 Дж/(моль-К). Растворимость в воде (г в 100 г): 105,5 (0°С), 110,5 (20° С), 155,7 (100° С), t/f 10, 20, 30, 40 и 50%-ных водных растворов соответственно 1,0904, 1,1898, 1,2756, 1,4141 и 1,5404. Образует гидраты с 5, 1,5 и 0,5 молекулами воды, полностью обезвоживающиеся при 150—160° С (рис. 2.20).
Карбонат калия сильно гигроскопичен. Равновесная относительная влажность над К2СО3-1,5Н2О при 25°С составляет 43%, на воздухе карбонат калия расплывается. В водных растворах реагирует с диоксидом углерода, образуя КНСОз, а с диоксидом серы — KHSO3 и СО2. Поглощает из воздуха диоксид углерода и превращается в гидрокарбонат.
Гидрокарбонат калия — бесцветные кристаллы моноклинной сингонии плотностью 2,17 г/см3. Растворимость в воде 33,3 г в 100 г при 20° С. При 100—120° С или кипячении водных растворов разлагается на карбонат натрия и диоксид углерода. Гидрокарбонат получают взаимодей-
Рис. 2.20. Политерма системы
К2СО5 — Н2О
ствием диоксида углерода с водными растворами карбоната калия.
Карбонат калия применяют в качестве удобрения в производстве медицинского, оптического, электровакуумного и художественного стекла, а также хрусталя. Применяется для производства некоторых солей, фармацевтических препаратов, жидкого калийного мыла, в процессе крашения и отбелке тканей, для промывания шерсти, для изготовления печатных красок
136
из индатреновых красителей. Его применяют для очистки промышленных газов от сульфида водорода, особенно в коксохимической промышленности в процессе очистки коксового газа.
К качеству выпускаемого промышленностью карбонату калия, согласно ГОСТ 10690—63, предъявляются требования, приведенные в табл. 2.4.
Таблица 2.4. Требования к качеству карбоната калия (содержание в %)
Поташ кальцинированный Поташ полутораводный
сорт 1 сорт 2 сорт 1 сорт 2
Карбонат калия, не менее 98 93 97,5* 92,5*
Натрия в пересчете на Na2CO3, не более 0,9 4,0 0,9 6,0
Хлоридов в пересчете на хлор, не более 0,07 1,5 0,07 2,0
Сульфатов в пересчете на SOj", не более 0,4 1,0 0,5 1,5
Железа в пересчете на Fe2O3, не более 0,005 Не определяется 0,005 Не определяется
Не растворимого в воде остатка, не более 0,1 » 0,1 »
*В пересчете на прокаленное вещество.
Способы получения карбоната калия. Основным и перспективным способом получения карбоната калия является способ производства его в процессе комплексной переработки нефелинов. Нефелин является минералом из группы каркасных силикатов. Химическая формула его KNa3(AlSiO4)4, он имеет кристаллическую структуру типа тридимита (модификации ЗЮг). Искаженные шестичленные кольца из связанных вершинами тетраэдров SiO4 и АЮ4 представляют собой каркас, в пустотах которого располагаются ионы щелочных металлов. Плотность нефелина 2,550-2,665 г/см3.
В чистом виде нефелин содержит до 33,2% AI2O3 и поэтому является ценным сырьем в производстве глинозема. В процессе получения глинозема (AI2O3) из нефелина образуются растворы следующего состава (в г/л): ИагСОз—157; К2СО3 — 62; K2SO4 (включая неокислен-ные соединения серы) — 16; собственно K2SO4—15; R2O3 — 0,1; СГ — 0,07. Эти растворы перерабатывают на содово-поташную смесь и поташ. Способ основан на фазовых равновесиях во взаимной системе
К2СО3 + Na2SO4 1Ча2СОз + K2SO4
изучен в широком диапазоне температур и концентраций (рис. 2. 21).
Исходные растворы обычно содержат некоторое количество гидрокарбонатов калия и натрия (свыше 7 г/л в пересчете на NaHCO3), присутствие которых в системе приводит к значительной коррозии аппаратуры и загрязнениям карбоната калия оксидами железа. Предварительная нейтрализация исходных растворов гидроксидом натрия замедляет про-137
KfiO,.96
Рис. 2.21. Диаграмма водной системы KjCOj+NaiSOi NaiCCh+KjSC^ при 75° С (способ вторичных проекций)
цесс коррозии оборудования и содержание железа в карбонате калия резко снижается (с 0,06 — 0,14% до 0,001—0,002%).
Карбонат калия получают из содово-поташных растворов по следующей схеме (рис. 2.22). Предварительно концентрируют исходные растворы в системе вакуум-выпарных аппаратов с предварительным подогревом раствора в теплообменниках. Концентрация раствора на первой стадии повышается с 200—240 до 400—440 г/л. На второй стадии при 130—132° С выпаривают в однокорпусных вакуум-выпарных аппаратах с выносной греющей камерой до точки насыщения раствора солями Na2COj, K2SO4 и КгСОз-ИагСОз. При этом концентрация солей в растворе достигает 750 г/л. В процессе второй выпарки выделяется смесь из карбоната натрия и сульфата калия, которая отделяется на центрифугах и высушивается. Примерный состав смеси (%): Na2CO3 — 79,06; К2СО3 —11,27; K2SO4 —9,28, 0,01% Fe2O3 и 0,31% потери при прокаливании.
Маточный раствор после второй стадии выпарки смешивают с маточным раствором следующей стадии и упаривается при 105—112° С до насыщения солями K2SO4, КгСОз-ЫагСОз и К2СОз-1,5НгО. Такой раствор содержит 1060 г/л К2СОз, 28,8 г/л Иа2СОз и 45,2 г/л K2SO4.
На третьей стадии выпарки в осадок выпадает смесь солей K2SO4 и КгСОз'ИагСОз, которая отделяется на центрифуге, образующиеся кристаллы растворяются в конденсате, а образующиеся при этом растворы смешивают с исходным содово-поташным раствором.
Раствор карбоната калия после третьей выпарки охлаждают до 45—50°С для выделения К2СОз-1,5Н2О, который отжимают на центрифуге и направляют на упаковку, а маточный раствор возвращают на третью выпарку.
138
Исходный щелок
Рис. 2.22. Принципиальная схема получения чистого карбоната калия из содово-поташных растворов глиноземного производства с окислительной термообработкой карбоната калия
Продукт имеет средний состав (в % на сухое вещество)
К2СО3...............97,63 СГ...................0,07
Na2CO3...............0,82 А12О3...............0,043
K2SO4 „би............0,68 Fe2O3...............0,002
K2SO4................0,46 Hz°.................17’22
Разработан способ гранулирования карбоната калия, содержащего 5—-6% гигроскопической влаги в тарельчатом грануляторе, на который подают смесь К2СОз-1,5Н2О, мелкую и крупную фракцию карбоната калия после измельчения гранул и воду в соотношении 1:0,45—0,8:0,12—0,2. При этих данных гранулы высушиваются в барабанной сушилке до полного обезвоживания без уменьшения прочностных характеристик гранул.
В циркулирующих в технологической системе растворах постепенно накапливаются ионы хлора и неокисленные соединения серы. Для выделения соединений хлора раствор карбоната калия периодически охлаждают до температуры ниже 45° С, выделяя при этом загрязненный хлором (до 1%) карбонат калия, а очищенный фильтрат вновь направляют на третью выпарку. С целью очистки карбоната калия от неокисленных соединений серы карбонат калия промывают конденсатом.
Согласно данным диаграммы (см. рис. 2.21), точка состава нейтрализованного гидроксидом натрия начального раствора Н (If) лежит в поле кристаллизации карбоната натрия. Следовательно, карбонат натрия будет первой твердой фазой, выпадающей в процессе изотермической выпарки. На этом этапе карбонат натрия может быть выделен наиболее полно (свыше 80%) при 150° С. При дальнейшей выпарке солевой состав жидкой фазы изменяется вдоль изотермы от точки М (Af) до точки Е (Е) и затем до точки F (F). На этом пути в твердую фазу выделяется смесь карбоната натрия и глазерита. В точке Е (£') глазерит взаимодействует с карбонатом калия, переходя в карбонат натрия и сульфат калия. В конце процесса в твердую фазу выделяется карбонат натрия и сульфат калия. В точке F (F) раствор насыщен карбонатом натрия, сульфатом калия и двойной солью (табл. 2.5).
Из приведенных в табл. 2.5 данных следует, что K2SO4 выделяется наиболее полно в процессе проведения второй выпарки при температуре не выше 75° С, поскольку уже при 100° С содержание сульфата калия в жидкой фазе начинает увеличиваться. После отделения карбоната натрия, загрязненного сульфатом калия, маточный раствор F (Е), имеющий плотность 1,52 г/см3, подвергается дальнейшей выпарке. На третьей стадии выпарки состав жидкой фазы изменяется вдоль изотермы от точки F (F) до точки G (G') (табл. 2.6). 140
Таблица 2.5. Состав растворов в точке F (F) насыщения солями Na2CO3 • хН2О + KNaCO3 • уН2О + K2SO4 при различных температурах
г, °C Состав раствора, % (масс.) Состав раствора в пересчете на сухое вещество, % (масс.) Твердые фазы
К2СО3 Na2CO3 K2SO4 К2СО3 Na2CO3 K2SO4
25 17,02 21,00 0,75 54,16 43,90 1,94 Na2CO3-7H2O + Na2CO3K2CO3-6H2O + K2SO4
35 45,60 6,70 Следы 87,20 12,80 Следы Na2CO3H2O + Na2CO3K2CO3 + K2SO4
50 45,10 7,22 » 86,30 13,70 » Na2CO3H2O + Na2CO3K2CO3 + K2SO4
75 44,20 8,14 » 84,50 15,50 » Na2CO3H2O + Na2CO3K2CO3 + K2SO4
100 43,59 9,18 0,31 82,12 17,30 0,58 Na2CO3H2O + Na2CO3K2CO3 + K2SO4
150 45,82 8,95 0,74 82,50 16,10 1,40 Na2CO3 + Na2CO3K2CO3 + K2SO4
Таблица 2.6. Состав растворов в точке G (G') насыщения солями К2СО3 • Na2CO3 • уН2О + K2SO4 + К2СО3 • 1,5Н2О при различных температурах
/, °C Состав раствора, % (масс.) Состав раствора в пересчете на сухое вещество, % (масс.) Твердые фазы
К2СОэ Na2CO3 K2SO4 К2СО3 Na2CO3 K2SO4
25 48,10 5,02 0,63 89,45 9,35 1,17 K2CO3Na2CO3- 6Н2О + K2SO4 + К2СО31,5Н2О
35 49,95 4,17 Следы 92,28 7,72 Следы K2CO3-Na2CO3 + K2SO4 + К2СО31,5Н2О
50 52,10 3,12 » 94,20 5,80 » K2CO3*Na2CO3 + K2SO4 + К2СО31,5Н2О
75 56,00 2,01 » 96,55 3,45 » K2CO3Na2CO3 + K2SO4 + K2CO31,5H2O
100 59,38 1,03 0,07 98,18 1,70 0,12 K2CO3Na2CO3 + K2SO4 + K2CO31,5H2O
150 66,81 1,15 0,25 97,94 1,70 0,36 K2CO3Na2CO3 + K2SO4 + K2CO31,5H2O
Из табл. 2.6 видно, что наиболее целесообразна выпарка на третьей стадии при 100° С. При этом жидкая фаза содержит 98,18% карбоната калия в пересчете на сухое вещество.
Выделившуюся в осадок в процессе третьей выпарки загрязненную сульфатом калия двойную соль отделяют и возвращают на смешение с первоначальным раствором. Дальнейший процесс переработки маточного раствора зависит от способов освобождения раствора от накапливающихся примесей — в основном иона хлора и неокис-ленных соединений серы.
Получение карбоната калия высокого качества достигается путем систематического вывода маточных растворов после кристаллизации целевого продукта. Образующийся маточный раствор высушивают для получения карбоната калия, загрязненного неокисленными соединениями серы. Маточный раствор после третьей выпарки и отделения двойной соли дополнительно выпаривают при 75—100° С, в результате из системы частично выделяется К2СОз- 1,5Н2О. Жидкую фазу охлаждают до 25° С, в процессе чего кристаллизуется дополнительное количество К2СО3- 1,5Н2О.
Смесь обоих осадков кристаллического карбоната калия промывают насыщенным раствором карбоната калия, полученным в предыдущей операции. Установлено, что промывка идет полнее при предварительном перемешивании осадка с частью промывного раствора. Вторичную же промывку кристаллов производят непосредственно на фильтре или центрифуге. По этой схеме получается кристаллический карбонат калия, содержащий (в % на сухое вещество): К2СОз — 99,0, Na2CO3—0,3, K2SC>4—0,2, окисляемых соединений (в пересчете на K2SO3) —0,05, Fe2O3 —0,001.
Существует процесс получения высококачественного карбоната калия окислением соединений пероксидом водорода, персульфатами, воздухом в присутствии катализаторов и путем окислительной термообработки (прокалки) карбоната калия.
Схема получения карбоната калия с окислительной термообработкой (см. рис. 2.22) идентична с другими вариантами вплоть до третьей выпарки и отделения двойной соли с дальнейшим высушиванием и прокалкой раствора в течение трех часов при 600° С с доступом воздуха. В этих условиях степень окисления соединений серы до сульфата калия достигает 96—98%. Термообработанный карбонат калия растворяют в воде при 25° С с таким расчетом, чтобы образовавшийся сульфат калия остался нерастворенным. Раствор отфильтровывают от сульфата калия и упаривают при 75° С и охлаждают до 25° С. После промывки кристаллический карбонат калия направляют на упаковку, а маточный раствор возвращают в процесс.
Полученный по описанной схеме карбонат калия имел следующий аналитический состав (в % на сухое вещество): К2СОз — 99,4, 142
Na2CO3— 0,2, K2SO4 — 0,05, окисляемых соединений (в пересчете на K2SO3) —до 0,01, Fe2O3 — до 0,001, Сг2О3 — 0,00004, V2O5 — 0,0001.
Существует несколько способов получения карбоната калия.
Магнезиальный способ получения карбоната калия основывается на процессе карбонизации под давлением 0,5—1,8 МПа суспензии'активного карбоната магния в водном растворе хлорида калия: \
3(MgCO3-3H2O) + 2КС1 + СО2 2(KHCO3 MgCO3-4H2O) + MgCl2
Осаждаемую двойную соль Энгеля отделяют от раствора хлорида магния и разлагают водой или суспензией гидроксида магния при нагревании:
2(KHCO3MgCO3-4H2O) + Mg(OH)2 К2СО3 + 3(MgCO3-3H2O) + Н2О
Полученная суспензия фильтруется. Активный карбонат магния возвращается в процесс, а раствор карбоната калия упаривается, и из концентрированных растворов выделяют его кристаллы, которые после сушки упаковывают в виде товарного продукта.
Карбонат калия получают также по способу, основанному на следующих реакциях:
H2SiF6 + 2КС1 K2SiF6 + 2НС1
K2SiF6 + 2К2СО3 6KF + SiO2 + 2СО2
6KF + 3Na2CO3 ЗК2СО3 + 6NaF
Образующийся раствор карбоната калия отделяется от кристаллов фторида натрия, после чего часть его возвращается в систему, а часть перерабатывается на товарный продукт.
Карбонат калия получают также путем термообработки смеси сульфата калия с гидроксидом кальция в присутствии угля:
К280д + 2С + СаСО3 = К2СО3 + 2СО2 + CaS
Из образующегося плава карбонат калия выщелачивают маточными растворами.
Известен также триметиламиновый метод переработки хлорида калия на его карбонат, заключающийся в обработке смеси растворов хлорида калия и триметиламина диоксидом углерода в процессе перемешивания под давлением:
КС1 + N(CH3)3 + СО2 + Н2О КНСО3 + N(CH3)3-HC1
143
В процессе охлаждения полученного раствора выделяются кристаллы гидрокарбоната калия. Триметиламин снова переводят в свободный триметиламин: /
2[N(CH3)3 • НС1] + Са(ОН)2 = 2N(CH3)3 + СаС12 + 2Й2О
Формиатный способ получения карбоната калия/сводится к обработке сульфата калия суспензией гидроксида кальций в присутствии оксида углерода: ’
K2SO4 + Са(ОН)г + 2СО # CaSO4 + 2НСООК
Отфильтрованный от сульфата кальция и от других примесей раствор упаривают до получения плава, который окисляется воздухом при 800° С во вращающейся барабанной печи:
2НСООК + О2 = К2СО3 + Н2О + СО2
Разработаны способы получения карбоната калия, которые практически потеряли промышленное значение. К ним относятся: получение карбоната калия из растительной зоны, бардяного угля, золы от сжигания паточно-спиртовой барды, золы подсолнечника и т.д.
2.6. НИТРАТ КАЛИЯ
Нитрат калия (калийная селитра) KNO3 — бесцветные кристаллы. Ниже температуры 128,8° С устойчива a-модификация с ромбической решеткой (а = 0,6431 нм, Ь = 0,9164 нм, с = 0,541 нм, z = 4, пространственная группа РтпЬ, плотность 2,11 г/см3), которая выше 128,8° С переходит в гексагональную модификацию Р; АН перехода а—>р 5,04 кДж/моль. В процессе охлаждения a-фазы может образовываться тригональная у-фаза (а = 0,4365 нм, а = 76,56° С, z = 1, пространственная группа R3m) с температурой плавления 334,5° С; С° = 95,06 Дж/(молыК); АН^ = 9,8 кДж/моль, АН^ = 181 кДж/моль, АН^ = = -494,0 кДж/моль; = 132,9 Дж/(моль-К).
В процессе нагревания нитрат калия плавится при 334° С, а выше 338° С разлагается двухступенчато:
2KNO3 -> 2KNO2 + О2
2KNO2 -> К2О + NO2 + NO
144
Растворимость нитрата калия в воде (г в 100 г): при 20° С — 31,6; 30°С — 45,6; 40°С — 63,9; 60°С—109,9; 100°С —243,6; 114°С — 312. Плотность (с/425) 10- и 20%-ных водных растворов нитрата калия соответственно 1,0627 и 1,1326. Температура кипения насыщенного водного раствора 118° С, температура плавления эвтектики (12,2 г KNO3 в 100 г вод!?!) — 2,85° С.
Смеси нитрата калия с органическими веществами легко воспламеняются и горят.
Нитрат калия в природе образуется в процессе разложения органических веществ в результате жизнедеятельности нитрифицирующих бактерий.
Нитрат калия применяется в качестве удобрения, в производстве дымного пороха и пиротехнических составов, спичек, стекла, а также в пищевой промышленности для консервирования мясных продуктов.
Согласно ГОСТ 19790—74, к качеству нитрата калия предъявляются следующие требования (в %):
Сорт 1 Сорт 2 Сорт 3
KNO3 (в пересчете на сухой продукт), не менее 99,9 99,85 99,7
Влага, не более 0,08 0,1 0,2
Хлориды (в пересчете на NaCl), не более 0,03 0,1 Не нормируется
Карбонаты (в пересчете на К2СО3), не более 0,02 0,04 »
Не растворимый в воде остаток, не более 0,03 0,03 »
Не растворимый в хлороводородной кислоте про- 0 005 0,02
каленный остаток, не более //
Окисляемые вещества, не более 0,01 Не нормируется »
Соли кальция и магния (в пересчете на кальций), не более 0,02 0,025 »
Нитрат калия, применяемый в производстве порохов, должен содержать соли кальция и магния не более 0,002%.
Конверсионный способ получения нитрата калия основан на реакции:
КС1 + NaNO3 = KNO3 + NaCl
растворимости КС1—NaNO3—K.NO3—NaCl при 5,25, 50 и 100° С. Из диаграммы (рис. 2.23) видно, что при 5—25° С растворимость солей калия значительно меньше, чем солей натрия, в результате чего в конце процесса кристаллизации нитрат калия занимает значительную часть площади квадрата. С достижением температуры 100° С резко растет поле кристаллизации хлорида натрия. Если приготовить 145
раствор эквимолекулярной смеси К.С1 и NaNOj при 100° С, то фигуративная точка а солевой массы такого раствора, лежащая на пересечении диагоналей квадрата, окажется в поле кристаллизации NaCl. При выпаривании из этого раствора воды при 100° С с достижением насыщения начнется кристаллизация NaCl, а состав солевой массы раствора будет изменяться по линии ab /
В точке b раствор станет насыщенным также и КС1. Поэтому если выделившиеся к этому моменту кристаллы NaCl отделить, а затем охладить раствор, то точка Ь окажется в поле криЬталлизации KNO3 — эта соль и в процессе охлаждения будет осаждаться, причем состав раствора будет изменяться по линии Ьс. [
Поскольку расстояние между точками а и b невелико, то в процессе выпаривания воды из раствора, содержащего эквимолекулярные количества NaNOj и К.С1, осадится лишь незначительное количество NaCl, и раствор станет насыщенным также и КС1. Это приводит к снижению выхода кристаллического K.NO3 в процессе охлаждения раствора. Из диаграммы видно, что для увеличения количества отделяемого NaCl и повышения степени выхода KNO3 необходимо вводить в исходный раствор NaNOs в избытке. Максимальный выход получается, если к концу выделения NaCl раствор насыщен тремя солями — NaCl, К.С1 и KNO3, т.е. солевая масса его изображается точкой Ег- В таком режиме технологии после отделения выделившегося NaCl кристаллизация KNO3 в процессе охлаждения раствора идет по наиболее длинному пути E2d, который обеспечивает максимальный выход продукта.
Наиболее рациональным и экономичным является проведение обменного разложения NaNOj и КС1 в циклическом процессе, проводя процесс кристаллизации NaCl при выпаривании из системы воды при постоянном давлении и переменной температуре. Описание и расчет такого цикла могут быть произведены с помощью комбинации изотермического и изобарического сечений диаграммы. На рис. 2.24 приводится пример оптимального цикла для случая, когда процесс кристаллизации нитрата завершается при 50° С. Точка а на изотермическом сечении характеризует состав маточного раствора после кристаллизации KNO3 на отрезке Ъа в процессе охлаждения. В начальный период кристаллизации солевому составу раствора соответствует точка а. Перед кристаллизацией к раствору добавляют соответствующее количество воды до насыщения его хлоридом натрия только при заданной температуре конца кристаллизации (50° С). Раствор в точке Ь образуется в результате упарки воды и кристаллизации NaCl из кипящего раствора (точка с), которая принадлежит к изобарическому сечению диаграммы и находится на луче выпаривания ВсЬ. Исходный же раствор для выпаривания (точка с) образуется 146
Рис. 2.23. Изотермы растворимости в водной системе КС1 + NaNO3 = KNO3 + NaCl при 5, 25, 50 и 100° С
Рис. 2.24. Оптимальный цикл конверсии КС1 + NaNO3 = KNO3 + NaCl в диаграмме с изотермическим (50°С) и изобарическим (0,1 МПа) сечениями
в процессе смешения маточного раствора (точка а) с эквимолекулярной смесью КС1 и NaNO3.
Следовательно, цикл осуществляется по треугольнику cba. Чем выше солевой коэффициент цикла, т.е. отношение массы полученного KNO3 к массе выпаренной воды, тем меньше расход энергии на выпаривание. Чем выше конечная температура кристаллизации KNO3, тем меньше затраты на охлаждение раствора. Наиболее экономичными являются циклы с температурными интервалами от точки кипения до конечной температуры кристаллизации, находящейся в пределах 50—25° С. При этом для луча упаривания ВЬ оптимальные соотношения К+ : NO, находятся в пределах 0,69—0,96.
В производственных условиях используют 40—50%-ные растворы нитрата натрия, а хлорид калия содержит 95—98% КС1.
Процесс получения нитрата калия состоит из следующих стадий: 1) растворение хлорида калия в растворе нитрата натрия; 2) фильтрация раствора смеси; 3) обменная реакция КС1 и NaNO3 и кристаллизация NaCl; 4) фильтрация раствора нитрата калия (для отделения выпавшего в осадок NaCl); 5) кристаллизация нитрата калия; 6) сушка и упаковка целевого продукта.
Согласно схеме, приведенной на рис. 2.25, получение смеси раствора К.С1 и NaNOs производят в растворителях 1, снабженных паровым змеевиком и метальным устройством, при 60—70° С. Раствор фильтруют на фильтре 2 и подают в реактор 3, снабженный паровым обогревом.
С целью ускорения процесса конверсии раствор перемешивают механической мешалкой или сжатым воздухом, подводимым в ниж-147
Рис. 2.25. Схема получения нитрата калия конверсионным способом:
1 — растворитель; 2, 11 — фильтры; 3 — реактор; 4 — нутч-фильтр; 5 — сборники; 6—напорная емкость; 7, 10 — кристаллизаторы; 8, 12 — центрифуги; 9 — аппарат «распарки»; 13 — вращающаяся барабанная сушилка; 14 — сборники маточных растворов
нюю часть аппарата. Процесс конверсии проводят при 125—130° С до достижения плотности растворов 1,64—1,68 г/см3. Для полного использования исходного калийсодержащего сырья и увеличения выхода целевого продукта процесс ведут с избытком нитрата натрия (сверх стехиометрического количества).
Свыше 70% хлорида натрия, образующегося в процессе обменного разложения, выделяется из раствора в виде шлама, который отделяют на центрифуге или фильтре 4. Промывкой шлама в центрифуге холодной водой содержание KNO3 в нем снижают с 15—20 до 1—3%. Промытый осадок — хлорид натрия — является отходом производства.
Фильтрат и промывные воды собирают в сборнике 5, в котором во избежание процесса кристаллизации нитрата калия поддерживают температуру в пределах 90—105° С. 148
Из приемных сборников 5 горячий раствор нитрата калия поступает в кристаллизатор 7. Температура процесса кристаллизации 25—30° С. Выделяющиеся кристаллы нитрата калия отжимают на центрифуге 8, кристаллы промывают маточными растворами после второй кристаллизации до содержания в кристаллах не более 1,5% NaCl. После растворения кристаллов, полученных в аппарате 9 («распарка»), в раствор добавляют вторичный маточный раствор до плотности растворов 1,54—1,56 г/см3 при 90—100° С. Образующийся раствор фильтруют на фильтре 11 и направляют на повторную кристаллизацию в кристаллизатор 10. Суспензию охлаждают до 50—60° С, центрифугируют, полученные кристаллы промывают чистой холодной водой и сушат во вращающейся барабанной сушилке 13 горячим воздухом (100—110° С).
Сухой нитрат калия, содержащий не более 0,1% влаги, просеивают на односитовом грохоте и упаковывают в пятислойные влагонепроницаемые крафтцеллюлозные мешки, пропитанные битум-автоловой смесью.
На производство 1 т нитрата калия конверсионным методом расходуют: 0,93 т NaNOj, 0,92 т КС1, 0,05 т NH4NO3, 130 кВт-ч электроэнергии, 11 т пара и 103 м3 воды.
Получение нитрата калия из хлорида калия и азотной кислоты основывается на реакции хлорида калия с азотной кислотой:
КС1 + HNO3 = KNO3 + НС1 (1)
2КС1 + 3NO2 + Н2О = 2KNO3 + 2НС1 + NO (2)
Кроме основных реакций при повышенных температурах протекают побочные реакции:
ЗНС1 + HNO3 # NOC1 + С12 + 2Н2О (3)
НС1 + 2NO2 HNO3 + NOC1 (4)
Реакция 1 при низких температурах (25—60° С) протекает с образованием нитрата калия, а реакция 2 также начинается при низких температурах, но она легко обратима. Однако при 100° С и выше равновесие ее почти полностью сдвинуто в сторону образования нитрозилхлорида и хлора. Этому способствует также повышение концентрации кислот в растворе. С повышением концентрации кислот и температуры давление паров НС1 и HNO3 над раствором увеличивается, что и является причиной образования значительных количеств нитрозилхлорида и хлора. В процессе применения 30—40%-ной исходной азотной кислоты и температуре ниже 60°С потери азота в виде нитрозилхлорида незначительны и хлор накапливается в растворе в виде хлороводорода. Путем охлаждения раствора можно выделить значительное количество KNO3, а маточный раствор при этом может быть 149
возвращен в систему. В процессе накопления значительных количеств хлороводородной кислоты в системе перед возвратом раствора в процесс необходимо отгонять из него часть хлорида водорода. Отгоняемые пары конденсируют в виде хлороводородной кислоты.
Нитрат калия получают из хлорида калия и азотной кислоты при повышенной температуре (рис. 2.26). В футерованный фторопластом реактор 1 вносят расчетное количество азотной кислоты, маточный раствор от предыдущей операции и загружают кристаллический хлорид калия. Реакционную массу перемешивают сжатым воздухом при постоянном нагревании острым паром до 75—85° С. Выделяющиеся в ходе реакции газы и водяные пары отсасывают из реактора воздушным эжектором в абсорбер (абсорбционное отделение не показано).
В конце реакции раствор, содержащий в среднем 520 г/л KNOj, 35—65 г/л HNO3 и 120—140 г/л НС1, поступает в кристаллизатор 4. Образующиеся в процессе охлаждения до 25—30° С кристаллы нитрата калия отделяют на центрифуге 5, промывают, сушат в барабанной сушилке 6 и направляют на упаковку.
Маточный раствор с промывными водами содержит 90—110 г/л KNO3, 20—40 г/л HNO3 и 70—80 г/л НС1. Часть растворов возвращают в реактор, оставшаяся часть может быть нейтрализована гидроксидом калия, и после упарки, кристаллизации и центрифугирования из нее выделяют нитрат калия.
Образующийся в реакторе 1 нитрозилхлорид может быть использован в процессе синтеза капролактама.
Рис. 2.26. Схема получения нитрата калия из хлорида калия и азотной кислоты: 1 — реактор; 2 — центробежный насос; 3 — сборник маточных растворов; 4 — кристаллизатор; 5 — центрифуга; б — вращающаяся барабанная сушилка; 7 — холодильник для воздуха; 8 — компрессор
150
2.7. ФОСФАТЫ КАЛИЯ
К фосфатам калия относятся соли ортофосфорной кислоты — дигидрофосфат К.Н2РО4 и дидейтерофосфат K.D2PO4, гидрофосфат К2НРО4 и фосфат К3РО4. Существуют также конденсированные фосфаты — циклофосфаты с циклическим анионом (КРОз)х, где х = 3, 4, 6 и линейные полифосфаты Кп+гРиОзп+ь
Физико-химические свойства. Дигидрофосфат и дидейтерофосфат — бесцветные кристаллы. Выше точки Кюри (для К.Н2РО4 — 151,18° С, для KD2PO4 — 53,19°С) устойчива тетрагональная модификация, ниже точки Кюри — ромбическая. Сегнетоэлектрики.
Для дигидрофосфата калия максимальная спонтанная поляризация 5,1 мкКл/см2. Выше 250° С дигидрофосфат плавится с разложением, образуя (КРОз)/
хКН2РО4 25О'С > (КРОз> + хН2О
Растворяется в воде (25,1 г в 100 г при 25° С), практически не растворяется в этаноле. Температура плавления эвтектики с водой (13,2% по массе КН2РО4) — 2,62° С.
Монокристаллы КН2РО4 и KD2PO4 являются модуляторами и преобразователями частоты в лазерной технике. КН2РО4 применяется в производстве пекарских порошков, пищевых дрожжей, жидких моющих средств, является компонентом минеральных удобрений, эмульгатором пищевых продуктов и составов для очистки нефтепродуктов, умягчителем воды.
Гидрофосфат К2НРО4, бесцветные гигроскопические кристаллы. Выше 250° С превращается в полифосфаты. Хорошо растворяется в воде и этаноле. Образует кристаллогидраты с 6 и 3 молекулами воды. Гексагидрат гидрофосфата при 14,3° С переходит в тригидрат, который при 48,3° С обезвоживается. Температура плавления эвтектики гексагидрата (36,8% по массе К2НРО4) с водой —13,5° С.
Дигидрофосфат калия применяют для приготовления буферных растворов, в качестве питательной среды в процессе выращивания плесневых грибов, продуцирующих пенициллины. Он является компонентом пищевых дрожжей, антифризов и лекарственных средств.
Фосфат калия, бесцветные гигроскопические кристаллы. Растворяется в воде (79,2 г в 100 г при 7,5 °C, 148,1 г при 45,1° С). Из водных растворов фосфата калия кристаллизуются нона-, гепта-, три- и сесквигидраты, плавящиеся инконгруэнтно соответственно при — 6,6, 47,5, 156,5 и 203° С.
В процессе сплавления с Ы3РО4 образует ЫзРО4-2КзРО4 (температура плавления 833° С, с разложением), а с Na3PO4 — непрерыв-151
ный ряд твердых растворов с минимальной температурой плавления 1240° С (65 мол. % К3РО4).
Фосфат калия применяют в качестве компонента электролитов в процессе получения каучуков методом эмульсионной полимеризации.
Трициклофосфат (КРОз)з, диполифосфат К4Р2О7 и триполифосфат К5Р3О10 — бесцветные гигроскопичные кристаллы, хорошо растворимые в воде, образуют кристаллогидраты.
Конденсированные фосфаты калия являются компонентами жидких моющих средств, антиоксидантами и бактерицидами в процессе консервирования мясных продуктов, добавками к жидкостям для бурения, краскам, ингибиторам коррозии.
Способы получения фосфатов калия. Дигидро-, гидрофосфат и фосфат калия могут быть получены в процессе нейтрализации фосфорной кислоты гидроксидом или карбонатом калия по схеме
Н3РО4 + КОН = КН2РО4 + Н2О
Н3РО4 + 2КОН = К2НРО4 + 2Н2О
Н3РО4 + ЗКОН = К3РО4 + ЗН2О
2Н3РО4 + К2СО3 = 2КН3РО4 + Н2О + СО2
Н3РО4 + К2СО3 = К2НРО4 + Н2О + СО2
2НзРО4 + ЗК2СОз = 2КзРО4 + ЗН2О + ЗСО2
Однако в промышленности минеральных удобрений этот способ не применяют из-за высокой стоимости исходных реагентов.
Разработан двухстадийный процесс получения дигидрофосфата калия. В качестве исходных видов сырья применяют хлорид калия, серную кислоту и фосфорит. В первой стадии процесса хлорид калия смешивают с избытком серной кислоты, а полученную смесь подвергают термообработке при 80—100° С. При этом происходит реакция с выделением хлорида водорода:
KCI + H2SO4 = KHSO4 + НС1
Образующийся хлорид водорода направляется на производство хлороводородной кислоты, а гидросульфат калия нейтрализуется фосфатом:
2KHSO4 + Саз(РО4)2 + H2SO4 —*-> 2КН2РО4 + 3CaSO4 • 2Н2О
Образующийся гипс отфильтровывают и отмывают водой, а раствор упаривают до концентрации 50—60% КН2РО4. Последний высаливают из системы метанолом. Маточный раствор подвергают дис-152
тилляции для отчистки метанола, возвращаемого на начало процесса, и получения фосфорной кислоты, содержащей -54% Р2О5. Степени использования фосфора и калия в процессе переработки марокканских фосфоритов составляют соответственно 92 и 90—95%; целевой продукт содержит (в %): К2О 30; Р2О5 46; А1 0,6; Fe 0,6; F 1,5; SO*' 3.
Получаемый в процессе дегидратации дигидрофосфата калия при 320° С конденсированный фосфат калия (КРОз)х содержит 57—58% Р2О5 и около 38% К2О [теоретически в (КРОз)х должно содержаться 50,13% Р2О5 и 39,87% К2О]. В процессе медленного охлаждения получается не растворимый в воде стекловидный плав; при быстром — образуется продукт, в котором часть P20s находится в водорастворимой, другая часть — в цитратно-растворимой форме. Растворимость конденсированного фосфата калия в воде определяется его физическим состоянием и составом (кристаллический продукт — соль Курроля менее растворим, чем стекловидный). Для получения продукта, содержащего весь фосфор в водорастворимой форме, производят неполную дегидратацию или дегидратацию в присутствии незначительного количества различных добавок, например хлоридов или сульфатов щелочноземельных металлов, Ре20з.
Конденсированный фосфат калия является высокоэффективным, негигроскопичным и исслеживающимся удобрением. Он не токсичен для семян, его водные растворы можно аммонизировать, получая смесь фосфатов калия и аммония.
Конденсированный фосфат калия получают разложением хлорида калия фосфорной кислотой около 900° С или оксидом фосфора (V) при 1000— 1050°С.
Существует также способ получения конденсированного фосфата калия разложением хлорида калия термической и экстракционной фосфорной кислотой, содержащей 23% P20s. Процесс осуществляют при 60—70° С. Образующуюся суспензию (-56% Н2О) сушат, а затем массу подвергают термообработке при 350—370°С. При этом получается продукт, содержащий 54% Р2О5 (в цитратно-растворимой форме), 35—39% К2О и около 0,3% хлора. Выделяющийся в газовую фазу хлорид водорода абсорбируют водой с получением 16—18%-ной хлороводородной кислоты.
Разработан также способ с применением упаренной (50% Р2О5) экстракционной фосфорной кислоты. При этом проводят двухступенчатое разложение исходного хлорида калия. При температуре в первой ступени около 300° С и во второй 700° С получают плав, содержащий 57% Р2О5 и 35% К2О.
ГЛАВА 3
МЕДЬ И ЕЕ СОЕДИНЕНИЯ
Содержание меди в земной коре составляет (4,7—5,5)-1О‘3% по массе. Для нее характерны и месторождения гидротермального происхождения. В морской воде содержание меди составляет 3-10'7% по массе, а в речной—Т10"7%.
В земной коре медь содержится в основном в виде сульфидов (свыше 90% мировых запасов и добываемой меди) и в виде кислородных соединений. Среди более 250 минералов наиболее важными являются: халькопирит CuFeS2, ковеллин CuS, халькозин Cii2S, борнит Cu5FeS4, куприт, малахит СиСОз • Си(ОН)2, хризоколл CuSiO3 • 2Н2О и др. Медные руды кроме меди содержат в своем составе золото, серебро, цинк, свинец, селен, железо, никель, молибден, рений и в зависимости от месторождений платиновые металлы.
Ионы меди участвуют во многих физиологических процессах живого организма, вследствие чего содержание меди в живых организмах составляет 210"4% по массе, а в крови человека — около 0,001 мг/л.
Медь в природе существует в виде двух стабильных изотопов 63Си (69,09%) и 65Си (30,91%). Конфигурация внешней электронной оболочки атома 3cZ104s’; степени окисления +1, +2, редко +3, +4; энергия ионизации Си0 -> Си+ -> Си2+ -> Си3+ соответственно равны 7,7264, 20,2921, 36,83 эВ; электроотрицательность по Полингу 1,9; атомный радиус 0,128 нм, ионные радиусы (в скобках указаны координационные числа) Си+ 0,060 нм (2), 0,074 нм (4), 0,091 нм (6), Си2+ 0,071 нм (2), 0,079 нм (5), 0,087 нм (6).
Медь — пластичный, розовато-красный металл с металлическим блеском, тонкие пленки меди при просвечивании имеют зеленовато-голубой цвет. Кристаллическая решетка гранецентрированная кубическая, а = 0,36150 нм, z - 4, пространственная группа Fm3m. Температура плавления 1083,4° С, температура кипения 2567°С; плотность 8,92 г/см3, жидкий при 1100° С — 8,36 г/см3, при 200°С — 8,32 г/см3, рентгеновская плотность 8,9331 г/см3; С° = 24,44 Дж/(моль-К), уравнение температурной зависимости в интервале 154
248—1356,9 К: С° = 4,187(5,41 + 1,4-7-10’3Т) Дж/(моль-К); АН™ =
= 13,02 кДж/моль, скрытая АЯ„л = 205 кДж/моль, АЯИсп = 304,8 кДж/моль; 5298 = 33,15 Дж/(моль-К). Медь диамагнитна.
Медь растворяет водород, который ухудшает ее механические свойства («водородная болезнь»). В сухом воздухе медь практически не окисляется. В процессе нагревания тускнеет в результате образования оксидной пленки. Взаимодействие меди с кислородом воздуха
начинается при 200° С по схеме: Си—>Си2О—>СиО, где fi—тем-t2
пература до 377° С, а /2—температура выше 377° С. Во влажном воздухе в присутствии диоксида углерода на поверхности меди образуется зеленоватая пленка Си(ОН)2 • СиСОз, в присутствии диоксида серы — пленка СиБОд • ЗСи(ОН)2, а в среде сульфида водорода— черная пленка CuS.
Медь не реагирует с водородом, азотом, углеродом и кремнием. В процессе пропускания аммиака над раскаленным металлом образует CU3N, а в аналогичных условиях при контакте с парами серы, селена и теллура на поверхности образуются соответствующие сульфиды, селениды и теллуриды.
Соли меди (1) и (2) с рядом молекул и ионов (NH3, CN-, СГ и др.) образуют устойчивые комплексные соединения типа (МНд^СиВгз), Кз[Си(СМ)д], К2(СиС1д) и аммиакаты.
Основными видами сырья для получения меди являются сульфидные, а реже смешанные руды. В связи с невысокой концентрацией меди в рудах (0,5—1,2%) и полиметалличностью их подвергают флотационному обогащению, параллельно с медным получают и другие концентраты, например цинковый, никелевый, молибденовый, пиритный и свинцовый. В этом процессе содержание меди в медных концентратах доходит до 18—45%. Основное же количество меди получают пирометаллургическим способом, включающим следующие технологические процессы: обжиг концентрата, плавку, конвертирование и рафинирование.
Широкое применение меди в народном хозяйстве обусловлено рядом ее ценных свойств: высокая электрическая проводимость, пластичность и теплопроводность. Более 50% металлической меди применяется в процессе изготовления проводов, кабелей, шин, токопроводящих частей электроустановок. Более 30% меди применяют в виде сплавов (бронза, латунь, мельхиор и др.), широко применяемых при изготовлении художественных изделий, а также в виде фольги. Около 10—12% меди применяют в медицине, сельском хозяйстве, в качестве пигментов, катализаторов, в гальванотехнике и др.
155
3.1. СУЛЬФИДЫ МЕДИ
Медь образует несколько сульфидов, в том числе нестехиометрических (табл. 3.1).
Таблица 3.1. Характеристика кристаллических модификаций сульфидов меди
Показатель CuS a-Cu2S p-Cu2S y-Cu2S Си 1,978 Cul>96S Cui,8S CU7S4
Гексаго- Ромби- Гексаго- Куби- Ромби- Тетраго- Куби- Ромби-
Параметры элементарной ячейки, нм: нальная ческая нальная ческая ческая нальная ческая ческая
а 0,3796 1,1190 0,389 0,5735 2,692 0,39962 0,5564 0,789
b — 2,728 — — 1,571 — — 0,784
с 1,636 1,341 0,668 — 1,356 1,1287 — 1,101
Число формульных единиц в ячейке 6 96 2 4 — 4 4 4
Пространственная группа Р(у$1ттс А Ь2т Ptymmc Fm3m Ртпт P432j2 Fm3m Рпта
Плотность, г/см3 4,68 5,81 5,78 5,60 5,60 5,770 5,60 5,70
ДЯ^5р, кДж/моль -53,2 -79,6 5,61 1,21 — -74,5 -72,18 —
Моносульфид меди CuS — сине-черные кристаллы; температура плавления 502° С (инконгруэнтно); С® = 47,86 Дж/(моль-К); 52°98 = 66,6 Дж/(моль К); в процессе нагревания в вакууме около 300° С разлагается до гемисульфида и элементной серы по схеме
2CuS —Cu2S + S
а на воздухе окисляется до оксида меди (II):
2CuS + ЗО2 —> 2CuO + 2SO2
а в присутствии влаги в воздухе до сульфата меди (II):
CuS + 2О2 н-° > CuSO4
Сульфид меди (I) (гемисульфид) Cu2S — черновато-серые кристаллы; существует в трех модификациях — а, 0, у; температуры перехода а->0 103° С, 0-> у 437° С (при содержании 33,33 атомных процента меди); для a-Cu2S: С ° = 76,37 Дж/(моль-К); = 121,0 Дж/(моль К); для y-Cu2S: С® = 85,08 Дж/(моль-К); температура плавления 1129° С; АЯп°л = 11,3 кДж/моль. y-Cu2S в процессе нагревания 156
в вакууме выше 700° С диссоциирует с образованием металлической меди и паров элементной серы, уравнение температурной зависимости давления пара серы: 1g р(мм рт. ст.) =-15 845/Т + 9,11; произведение растворимости 2,5-1 О'48. В процессе нагревания все модификации сульфида меди (I) окисляются до C11SO4, СиО и SO2.
Для гемисульфида характерна нестехиометрия. Область гомогенности P-C112S 33,33—33,44 атомных процента меди (93—103°С), у-СигЗ 33,33—36,4 атомных процента меди (507° С). Известны также нестехиометрические фазы: Cu2-xSc СКх<0,02, 0,03^х^0,04, 0,04<х<0,1; Cui 8S.
Сульфид Cuj gS существует в нескольких кристаллических модификациях, некоторые из них метастабильны; ниже 33° С разлагается на C117S4 и Cui;96S. Фазы в области составов C112S — Cu] 8S — полупроводники p-типа. Ионную проводимость имеет также P-C112S. C112S и CuS диамагнитны. Ширина запрещенной зоны для a-Cu2S 1,26 эВ (80° С), P-Cu2S 1,7—2 эВ, y-Cu2S 1,3 эВ.
Известен также малоустойчивый дисульфид меди с кубической кристаллической решеткой (а = 0,580 нм, z = 4, плотность 4,24 г/см3).
Все известные сульфиды меди практически не растворяются в воде, разбавленных серной и хлороводородной кислотах. Растворяется в царской водке и азотной кислоте.
Сульфиды меди встречаются в природе в виде минералов ковеллина CuS, халькозина (медного блеска) a-Cu2S, 0C-CU2S + P-CU2S, джар-леита CU197S, дигенита Cui.gS, анилита CU7S4, халькопирита CuFeS2, борнита CusFeS4.
Сульфиды меди получают из металлической меди и элементной серы в процессе нагревания в вакуумированных кварцевых ампулах, а сульфид меди (II) также осаждением сульфидом водорода из водных растворов солей меди (II) в слабокислой среде.
Монокристаллы сульфида меди (I) выращивают зонной плавкой.
Ковеллин, халькозин, борнит и халькопирит являются сырьем в производстве металлической меди и ее сульфата. Сульфид меди (II) применяется в качестве пигмента в красках, а сульфид меди (I) является компонентом медного штейна в процессе получения меди пирометаллургическим способом, а также компонентом полупроводниковых сплавов.
3.2. ОКСИДЫ И ГИДРОКСИДЫ МЕДИ
Медь легко реагирует с кислородом, образуя два оксида: гемиок-сид Си2О, оксид СиО и соответствующие гидроксиды.
Оксид меди (I). Оксид меди (I) (гемиоксид) Си2О — красновато-коричневые кристаллы с кубической решеткой (а = 0,4270 нм, z = 2, пространственная группа РпЗт); в кристаллах CU2O имеет место линейно-тетраэдрическая координация атомов (рис. 3.1);
157
плотность 6,1 г/см3; температура плавления 1242° С; С° = 63,7 Дж/(моль-К); ДЯ^, = 64,3 кДж/моль; ДЯ^р =-173,3 кДж/моль; 5298 = 93,0 Дж/(моль-К). В воде не растворяется и не реагирует. Заметно взаимодействует с гидроксидом калия и натрия по схеме
©Си @0 Си2О + 2NaOH + Н2О = 2Na[Cu(OH)2]
Рис 3.1. Структура Си2О (куприт)
Для гемиоксида характерно диспропорционирование:
2Cu+ = Cu2+ + Си0
Оксид меди (I) восстанавливается водородом, оксидом углерода, активными металлами до металла:
Cu2O + Н2 = 2Си + Н2О
Cu2O + СО = 2Си + СО2
Cu2O + Mg = 2Cu + MgO
Cu2O + Zn - 2Cu + ZnO 3Cu2O + 2A1 = 6Cu + A12O3
В процессе нагревания оксид меди (I) окисляется кислородом:
2Си2О + О2 —4СиО
С иодидом и бромидом водорода гемиоксид образует соответствующие соли:
Cu2O + 2HJ = 2CuJ + Н2О
Cu2O + 2HBr = 2CuBr + Н2О
В разбавленной серной кислоте гемиоксид образует сульфат меди (II) и металлическую медь:
Си2О + Н280д = Си + СиЗОд + Н2О *
В водных растворах аммиака образует аммиакат:
Cu2O + Н2О + 4NH3 = 2[Cu(NH3)2]OH
158
В водных растворах взаимодействует с гидроксидами натрия и калия:
Cu2O + 2NaOH + Н2О = 2Na[Cu(OH)2]
Cu2O + 2КОН + Н2О = 2K[Cu(OH)2]
Гемиоксид меди встречается в природе в виде минерала куприт.
Гемиоксид меди получают электролизом раствора хлорида натрия с использованием медных электродов. В технологическом процессе на катоде выделяется водород, образуются ионы ОН”, а на аноде медь растворяется с образованием ионов Си+. В результате вторичного взаимодействия образуется гемиоксид:
2Cu+ + 2ОН~ = Си2О + Н2О
Гемиоксид осаждается в виде порошкообразной безводной массы. Процесс ведут при температуре не выше 70° С во избежание образования гидроксида меди (I). Напряжение на ванне ~46В, катодная и анодная плотности тока 200 А/м2. Выход по току составляет 99%, расход электроэнергии 1600 кВт ч на одну тонну получаемого геми-оксида меди.
Полученный целевой продукт промывают водой, затем этиловым спиртом и высушивают в вакуум-сушилке.
Гемиоксид меди получают путем нагревания оксида меди (II) при 1100° С, а также восстановлением сульфата меди (II) глюкозой или гидразином в щелочной среде.
Гемиоксид меди применяют в качестве пигмента в производстве стекла, керамики и глазурей. Он является компонентом специальных красок, защищающих подводную часть судна от обрастания, а также в качестве фунгицида.
Оксид меди (II). Оксид меди (И) СиО — черные кристаллы с моноклинной решеткой (а = 0,46837 нм, b = 0,34226 нм, с = 0,51288 нм, Р = = 99,54°, z = 4, пространственная группа С2/с); плотность 6,51 г/см3; температура плавления 1447°С (под давлением кислорода); Ср° = 42,3 Дж/(мольК); A/7n°n = 55,7 кДж/моль; ДЯ^р = -162,1 кДж/моль; S°9t = = 42,7 Дж/(моль-К).
Оксид меди (II) не растворяется в воде. В процессе нагревания до 1100° С оксид меди (II) разлагается по схеме
4СиО —•—> 2Си2О + О2
1100°С
159
Активные металлы, водород, углерод, диоксид углерода, аммиак и алканы восстанавливают оксид меди (II) до металлической меди:
CuO + Mg = Си + MgO СиО + Zn = Си + ZnO СиО + Нг = Си + НгО СиО + С = Си + СО СиО + СО = Си + СОг
С кислотами оксид меди (II) образует соответствующие соли меди (II):
CuO + H2SO4 = CuSO4 + НгО
В водных растворах аммиака образует аммиакат:
CuO + Н2О + 4NH3 = [Cu(NH3)4](OH)2
В процессе сплавления со щелочами оксид меди (И) образует купраты:
2CuO + 2КОН = 2КСиО2 + Н2
В природе оксид меди (II) — минерал тенорит.
Оксид меди (II) в производстве получают взаимодействием сульфата меди (II) с гидроксидом натрия или калия при 80° С:
CuSO4 + 2NaOH = Cu(OH)2 + Na2SO4 + H2O
или реакцией сульфата меди (II) с водным раствором аммиака:
CuSO4 + 2NH3 + 2Н2О = Cu(OH)2 + (NH4)2SO4
Образующийся по обоим уравнениям гидроксид меди (II) после промывки разлагают термообработкой при 200° С по схеме
Cu(OH)2^>CuO + H2°
В лабораторных условиях оксид меди (II) получают путем окисления металлической меди на воздухе при 400—500° С:
2Cu + О2 = СиО
160
Оксид меди применяют в качестве пигмента для стекла, керамики, эмалей, для приготовления электролитов в гальванотехнике, а также для получения оксидных катализаторов.
Гидроксид меди (II) Си(ОН)2 — голубое кристаллическое и аморфное вещество; кристаллическая решетка ромбическая (а -= 0,2949 нм, b = 1,059 нм, с = 0,5256 нм, z = 4); плотность 3,368 г/см3; С° = 96,2 Дж/(моль-К); ДЯ^р = -444,4 кДж/моль; AG^p =-359,4 кДж/моль; 52°98 = 83,7 Дж/(моль-К). В воде практически не растворяется (5-10’4%, масс, при 20° С).
В процессе нагревания выше 70—90° С гидроксида меди (II) или его водных растворов разлагается до СиО и Н2О. Произведение гидратированного оксида меди (II), соответствующее равновесию
СиО • иН2О + Н2О 1; Си2+ + 2ОН"+ иН2О
при 25 28 С, равно 1,5’10 , а ПРси<он)2 ~ 4,8’10 , ПРСи^Ощ2.Н2О — = 2,010’19.
Гидроксид меди (II) с кислотами образует соответствующие соли меди (II):
Cu(OH)2 + 2НС1 = СиС12 + 2Н2О
Cu(OH)2 + H2SO4 = CuSO4 + 2Н2О
Из катионных комплексов меди (II) характерны комплексы с азотсодержащими лигандами, например [Cu(NH3)4(OH2)2]2+ и хелатный [Cu(en)2(OH2)2]2+. Амминокомплексы образуются реакцией аммиака с водными растворами солей меди. На рис. 3.2 показано содержание амминокомплексов меди (II) в водном растворе Cu(OH2)g+ + H3N в зависимости от концентрации последнего. Образованием аммиакатов объясняется
растворение гидроксида меди (II) в водных растворах аммиака:
Cu(OH), + 4H,N + 2Н2О = = [Cu(H3N)4(OH2)2](OH)2
Для меди (II) характерны и анионные комплексы — купраты (II) вида M2[Cu(OH)4], которые легко разлагаются в водных растворах, что свидетельствует о слабых кислотных свойствах гидроксида меди (11).
ЫНзМ
Рис 3.2. Содержание акваамминокомплексов в растворе Cu(OH2)*++ H3N в зависимости от концентрации аммиака
6 Химическая технология неорганических веществ, кн. I
161
Гидроксид меди (II), взаимодействуя с сульфидом водорода, образует сульфид меди:
Cu(OH)2 + H2S = CuS + 2Н2О
В процессе пропускания диоксида углерода через раствор гидроксида меди (II) образуются моноклинные кристаллы синего цвета, основные карбонаты меди (II) (азурит, медная лазурь):
ЗСи(ОН)2 + 2СО2 = 2СиСО3 • Си(ОН)2 + 2Н2О
При изменении условий проведения реакции образуются моноклинные темно-зеленого цвета кристаллы основного карбоната меди (малахит):
2Си(ОН)2 + СО2 = СиСОз • Си(ОН)2 + Н2О
Гидроксид меди (II) с серной кислотой образует основные соли типа CuSO4 • 3Cu(OH)2 или CuSO4 • 2Си(ОН)г.
Кристаллический гидроксид меди (II) получают введением эквивалентного количества гидроксида натрия или калия в аммиачный раствор сульфата меди (II):
[Cu(H3N)4]SO4 + 2NaOH = Cu(OH)2 + 4H3N + Na2SO4
3.3 ХЛОРИДЫ МЕДИ
Медь образует хлориды меди (I) и (II), а также основной хлорид меди (II).
Монохлорид меди (I) CuCl — бесцветные кристаллы; до 408° С устойчива кубическая модификация (минерал нантокит; а = 0,5416 нм, z = 4, пространственная группа F43zn; плотность 4,14 г/см3); выше 408° С переходит в гексагональную модификацию (а = 0,391 нм, с = 0,642 нм, z - 4, пространственная группа Рбз/пс); АЯ° перехода 4,0 кДж/моль; температура плавления 430° С, температура кипения 1490° С; С° = 48,5 Дж/(моль-К); АЯ^ = 10,2 кДж/моль; АЯИ°СП = = 35,4 кДж/моль, АЯ^р =-137,26 кДж/моль; = 87,0 Дж/(моль-К). В газовой фазе существует в виде циклических тримеров. Растворимость в воде 0,0062 г в 100 г. Растворяется в концентрированной хлороводородной кислоте:
CuCl + НС1 = H[CuCl2]
162
H[CuCl2] + HC1 = H2[CuCl3]
H2[CuCl3] + HC1 = H3[CuCl4]
Растворяется в водных растворах аммиака с образованием [Cu(NH3)2]C1.
Растворяется также в диэтиловом эфире. Устойчив в сухом воздухе, но окисляется и гидролизуется во влажном воздухе с образованием основных хлоридов меди:
4CuCl + О2 + 2Н2О = 2СиС12 • Cu(OH)2
Легко окисляется кислородом воздуха, переходя в более устойчивый хлорид:
4CuCl + О2 + 4НС1 = 4СиС12 + 2Н2О
Для хлорида меди (I) характерно диспропорционирование:
2CuCl = СиС12 + Си
Хлорид меди (I) получают восстановлением кислого раствора хлорида меди (И) при избытке металлической меди:
Си + СиС12 = 2СиС1
в качестве восстановителей могут быть применены также гидразин, глицерин, диоксид серы, металлический цинк и алюминий.
Монохлорид меди является полуфабрикатом в производстве металлической меди. Он является поглотителем газов в процессе очистки ацетилена, а также оксида углерода в газовом анализе, катализатором в органической технологии, антиоксидантом для растворов целлюлозы.
Дихлорид меди СиС12 — темно-коричневые кристаллы с моноклинной решеткой (а = 0,685 нм, b = 0,330 нм, с = 0,670 нм, Р = 121° С, z = 2, пространственная группа С2/т~у, температура плавления 596° С; в процессе термообработки при 993° С разлагается по схеме
2CuC12 —2СиС1 + С12
плотность 3,386 г/см3(25° С); С° = 71,9 Дж/(моль-К); ДЯ^р =-215 кДж/моль; S°9S = 108,1 Дж/(моль-К); растворяется в воде, этаноле, ацетоне. Растворимость в воде (г в 100 г): 69,0(0° С), 74,5(20° С) и 98,0(80° С). При температуре ниже 15° С из водных растворов осаждается тетрагидрат — СиС12 • 4Н2О, в интервале 15—26° С — тригид
рат, при 26—42° С — дигидрат СиС12 • 2Н2О (минерал эрнохаль-цит) — синие кристаллы с ромбической решеткой (пространственная группа Ратп)\ плотность 2,54 г/см3; = -818,6 кДж/моль; ДСг^р =-660,1 кДж/моль; при 100° С в токе сухого хлорида водорода обезвоживается. Легко восстанавливается до Cu(I) и Си. Образует комплексные ионы типа [Си(ЫНз)4]2+, [CuBr4]2’ и М[СиС1з].
Хлорид меди (И) получают растворением оксида или карбоната меди (II) в хлороводородной кислоте:
CuO + 2НС1 = СиС12 + Н2О
СиСОз + 2НС1 = СиС12 + Н2О + СО2
а также обменной реакцией сульфата меди (II) с хлоридом бария:
ВаС12 + CuSO4 = CuCl2 + BaSO4
Хлорид меди (II) применяется в процессах омеднения металлов, в качестве катализатора крекинг-процесса, декарбоксилирования, окислительно-восстановительных реакций органических веществ, в качестве протравы в процессе крашения тканей.
Основной хлорид меди (II) CuCl2’Cu(OH)2—желто-зеленые кристаллы; при 250° С разлагается с выделением воды. Основной хлорид СиС12 • ЗСи(ОН)2 • хН2О, где х = 0-3, сине-зеленые кристаллы; не растворяется в органических растворителях, плохо — в воде; применяется в качестве пигмента и фунгицида.
Гидроксид меди (II) получают также взаимодействием кипящего раствора нитрата меди (II) с твердым гидроксидом натрия:
Cu(NO3)2 + 2NaOH = Cu(OH)2 + 2NaNO3
при этом образуется некристаллизующийся гель гидроксида меди (II).
Гидроксид меди (II) применяют в качестве пигмента для стекла, эмалей и глазурей, протравы в процессе крашения, стабилизатора нейлона, фунгицида, для приготовления реактива Швейцера (используемого в качестве сырья в производстве медно-аммиачных волокон), получения солей меди и в качестве препарата для предотвращения обрастания судов.
Гидроксид меди (I) СнОН не получен в виде индивидуального соединения. В процессе взаимодействия солей меди (I) с гидроксидами щелочных металлов в водном растворе образуется гидратированный оксид Си2О • хН2О, а из раствора выделяется лишь оксид меди (I). Однако в процессе растворения оксида меди (I) в растворах гид-164
роксидов щелочных металлов образуются комплексные гидроксиды меди (I) состава М[Си(ОН)2], где М — щелочной металл, например
Cu2O + 2NaOH + Н2О = 2Na[Cu(OH)2]
3.4. СУЛЬФАТЫ МЕДИ
Медь образует сульфаты, гидраты сульфатов и основной сульфат.
Физико-химические свойства. Сульфат меди (II) C11SO4, кристаллы; парамагнетик, магнитная восприимчивость + 1,33-10’3, ниже 54,ЗК (точка Нееля) — антиферромагнетик; растворимость (г в 100 г): в воде—14,3(0°С), 23,05(25°С), 39,5(60°С), 75,4(100°С), в метаноле 1,04(18° С); плотность водных растворов (г/см3): 1,040(4% по массе C11SO4), 1,131(12%), 1,206(18%). Гигроскопичен. Образует ряд гидратов (табл. 3.2). Важнейшим из них является пентагидрат сульфата меди (II) (медный купорос, хальконтит) C11SO4 • 5Н2О.
В пентагидрате сульфата меди (II) вокруг меди координированы четыре молекулы воды в плоскости и две SO ^-группы по оси. Пятая молекула воды играет роль мостика, объединяющего водородными связями молекулы Н2О в плоскости и SO^-rpynny:
Пентагидрат сульфата меди (II) образует асимметричные ярко-синие кристаллы триклинной конфигурации (рис. 3.3). Как видно из 165
Рис. 3.3. Структура кристалла пентагидрата сульфата меди CuSO4 • 5Н2О
рис. 3.3, атом меди октаэдрически окружен шестью атомами кислорода, четыре из которых принадлежат молекулам Н2О, а два (заштрихованные)— ионам SO4". Пятая молекула Н2О (в центре рисунка) соединена четырьмя водородными связями с атомами кислорода молекул воды и сульфат-ионов (атомы водорода на рисунке не видны).
Пентагидрат сульфата меди (II) при 53,7° С претерпевает полиморфный переход; при 105° С отщепляет
две молекулы кристаллизационной воды, при 150° С обезвоживается до моногидрата, а при 250° С переходит в безводную соль, которая
при дальнейшем повышении температуры разлагается по реакции
2CuSO4 = SO3 + CuO • CuSO4
а при дальнейшем нагревании до 900° С полностью разлагается по схеме
2CuSO4 —2CuO + 2SO2 + О2
Давление водяного пара над кристаллогидратами сульфата меди (II) показано на рис. 3.4.
Сульфат меди (II) образует основные сульфаты CuSO4 • 3Cu(OH)2 • • иН2О, где п = 0, 2, 5, двойные соли M2Cu(SO4)2 -6Н2О и аммиакаты [Cu(NH3)4]SO4 • хН2О.
Сульфат меди (II) встречается в природе в виде минералов халь-кокианита CuSO4, халькантита Си-SO4 • 5Н2О, бонаттита CuSO4 • ЗН2О, бутита CuSO4 • 7Н2О, брошантита CuSO4 • 3Cu(OH)2 и др. (табл. 3.2).
Пентагидрат сульфата меди (II) применяют в качестве протравы в процессе крашения текстильных материалов, компонента электролита при рафинировании металлической меди, пестицида, антисепти-166
Рис. 3.4. Давление водяного пара над кристаллогидратами сульфата меди:
1 — CuSO4 • 5Н2О # CuSO4 • ЗН2О + 2Н2О
2 — CuSO4 -3H2O # CuSO4 H2O + 2Н2О
3 — CuSO4 • 5Н2О # CuSO4 • Н2О + 4Н2О
ческого и вяжущего лекарственного средства, пигмента в красках, депрессора в процессе флотации, для получения других соединений меди, усиления и тонирования отпечатков в фотографии.
Сульфат меди (I) CU2SO4—светло-серые кристаллы; плотность 3,605 г/см3; ДЯ^р = -749,7 кДж/моль; устойчив в сухом воздухе около 20° С, в процессе нагревания до 200°С окисляется до оксида и сульфата меди (II); в воде разлагается до металлической меди и сульфата меди (II); сильный восстановитель; получают взаимодействием оксида меди (I) с (СНз^ЗС^.
Таблица 3.2. Свойства сульфата меди (II), его гидратов и основного сульфата
Показатель C11SO4 CuSO4-5H2O CuSO4-3H2O CuSO4H2O CuSCHCiXOHh
Цвет Бесцветный Лазурносиний Голубой Бесцветный Зеленый
Сингония Ромбическая Триклинная Моноклинная — Моноклинная
Параметры элементарной ячейки, нм:
а 0,839 0,614 0,559 — 1,304
b 0,669 1,074 1,303 — 0,983
с 0,483 0,599 0,734 — 0,601
угол, град — 82,3(a) 107,4(р) 102,7(у) 97,3(р) — 103,27(р)
Число формульных единиц в ячейке 4 2 4 — 4
Пространственная группа Рпта Р1 Р2,/а — P2j/c
Т °C 1 ПЛ» 200 95,88* 116,6* 313* 300*
Плотность, г/см3 3,603 2,284 2,7 3,15 3,78
С“, Дж/(моль К) 98,94 281,4 205,2 130,34 —
кДж/моль -771,45 -2280,99 -1686,69 -1085,90 -2181,1
5^, Дж/(моль-К) 109,3 300,6 217,64 149,89 370,49
*Инконгруэнтно.
Способы получения сульфата меди. В настоящее время в промышленности применяются следующие способы получения сульфата меди (II): 1) из медного лома и медных отходов; 2) из оксида меди (II); 3) сульфатизирующим обжигом сульфидов меди; 4) электролитическим рафинированием меди.
Обычно промышленность производит пентагидрат сульфата меди (II), при необходимости из которого получают другие сульфаты меди (II).
Получение сульфата меди (II) из медного лома. Исходную металлическую медь применяют в виде пористых 167
гранул, предварительно очищенных от сопутствующих металлов (Fe, Zn, Al, Pb и др.).
Гранулированную медь растворяют в разбавленной серной кислоте в реакционных башнях (рис. 3.5). Исходная кислота смешивается с маточными растворами сульфата меди (II). Процесс растворения протекает в присутствии воздуха. В процессе смешивания кислород воздуха растворяется в кислоте, диффундирует к поверхности меди и окисляет ее до гемиоксида меди:
4Cu + О2 — 2СизО
Гемиоксид растворяется в серной кислоте:
CibO + H2SO4 = CU2SO4 + Н2О
Образующийся сульфат меди (I) легко окисляется в сульфат меди (II):
2CU2SO4 + 2H2SO4 + О2 = 4C11SO4 + 2Н2О
Рис. 3.5. Изотермы растворимости медного купороса в присутствии свободной серной кислоты
Скорость реакции лимитируется скоростью первой стадии процесса — окислением металлической меди до ее гемиоксида, что объясняется низкой скоростью процесса растворения кислорода в растворе и его медленной диффузией к поверхности гранул меди. Процесс значительно ускоряется после появления в растворе сульфата меди. В результате диспропорционирования
Си + Си2+ = 2Си+
сульфат меди (II) восстанавливается металлической медью до сульфата меди (I), который затем окисляется растворенным в растворах кислородом до сульфата меди (II). Следовательно, сульфат меди (II) является переносчиком кислорода.
168
Температуру в реакционной башне поддерживают в пределах 80—85°С. В процессе окисления меди расходуется около 25% кислорода, поступающего в башню с воздухом, расход которого составляет около 1000 м3 на тонну пентагидрата сульфата меди (II).
Растворимость кислорода снижается с повышением концентрации сульфата меди (II) в растворе. В результате этого при повышении концентрации сульфата меди (II) скорость процесса растворения меди сначала растет за счет каталитического действия сульфата меди (II), а затем снижается вследствие недостатка кислорода. Установлено, что наибольшая скорость процесса растворения при условии поддержания концентрации 120 г/л CuSO4 (для раствора, содержащего ПО г/л H2SO4). С увеличением концентрации серной кислоты растворимость кислорода в ней уменьшается, но повышаются ее окислительные свойства. Процесс растворения меди значительно ускоряется также в присутствии в растворе ионов железа в результате диспропорционирования
4Fe2+ + О2 + 4Н+ = 4Fe3+ + 2Н2О
2Cu + 4Fe3+ = 2Cu2+ + 4Fe2+
Образующиеся ионы Fe2+ вновь окисляются в Fe3+ и являются ка-тализатором процесса. Установлено, что доля растворяющейся меди под действием ионов Fe3+ в растворе, содержащем около 110 г/л H2SO4, 60 г/л CuSO4 и 20—22 г/л FeSO4, составляет около 60% от всего количества меди, перешедшей в раствор.
Существенным в данной стадии технологии является и обеспечение равномерного орошения (смачивания) гранул исходной меди раствором, так как при неполном орошении образующаяся оксидная пленка растворяется не полностью, в результате образуется основной сульфат меди CuSO4 • 2Cu(OH)2, который вследствие своей низкой растворимости кристаллизуется из раствора.
Согласно технологической схеме, приведенной на рис. 3.6, гранулы меди ковшовым элеватором 1 загружают в реакционную башню 2. Одновременно башню орошают маточными растворами из сборника 4. При этом растворы проходят через бак с постоянным уровнем 3. Смешение маточных растворов с исходной серной кислотой из расходной емкости 5 производится в сборнике 4. Образующиеся в башне 2 растворы сульфата меди (II) насосом 7 постоянно перекачиваются во вращающийся барабанный кристаллизатор S, охлаждаемый вентилятором 9. Суспензия кристаллов сульфата меди (II) из кристаллизатора 8 передается в сборник 10, откуда суспензия загружается в центрифугу 11. Отжатые в центрифуге 11 кристаллы сульфата меди (II) элеватором 12 загружаются в сушил-
169
Рис 3.6. Схема производства сульфата меди (II) из гранулированной меди:
1,12— ковшовые элеваторы; 2— башня; 3— напорный бак; 4 — сборник; 5 — емкость для серной кислоты; 6, 7 — центробежные насосы; 8 — вращающийся кристаллизатор; 9, 14 — вентиляторы;
10 — сборник; 11 — центрифуга; 13 — сушилка; 15 — калорифер; 16 — бункер
ку 13 для сушки, а маточные растворы самотеком поступают в сборник 4 для смешения со свежеприготовленными растворами сульфата меди (II). Высушенный целевой продукт из сушилки 13 поступает в бункер готового продукта 16.
В маточном растворе постепенно накапливаются примеси железа. Содержание железа в кристаллах пентагидрата сульфата железа (II) обычно уменьшают предварительным окислением Fe2+ в Fe3+ в растворах. Окислителями являются воздух, азотная кислота, пероксид водорода. Степень очистки повышается в 2—4 раза в процессе добавки в раствор незначительного количества фторида водорода, приводящего к образованию фторидных комплексов Fe3+
На производство одной тонны пентагидрата сульфата меди (II) расходуют 0,27—0,29 т металлической меди и 0,39—Ь,40 т серной кислоты (100%).
Получение сульфата меди (II) из ее оксида базируется на оксиде меди. Оксид меди в свою очередь получают из отходов медеплавильных заводов. Отходы содержат сульфид меди (I), до 10% металлической меди и 0,5—3% железа. Общее содержание меди в отходах составляет 75—78%.
Для окисления сульфида меди (I) в оксид меди (II) отходы измельчают и подвергают термообработке в печах. Разрез одной из многих применяемых в промышленности печей приведен на рис. 3.7. Из рисунка видно, что печь имеет два неподвижных 1 и два подвижных пода 2. Подвижный под оборудован гребками 3. Верхняя часть печи завершается сводом 4. Подвижные поды 2 вращаются вокруг вертикальной оси, совпадающей с осью печи. Они опираются на ролики и опоясаны зубчатыми кольцами, с помощью которых приво-170
Рис 3.7. Печь для обжига отходов производства меди:
1 — неподвижный под; 2 — подвижный под; 3 — гребки; 4 — верхний свод
дятся во вращение. В процессе вращения подвижных подов материал перемещается по сводам от периферии к центру или в обратном направлении и пересыпается со свода на свод. Печь разогревается газом, поступающим под нижний свод печи. Обжиг массы проводят с добавкой 1,75—2,0% угля или кокса, обеспечивающего снижение температуры воспламенения сульфида меди и ускорение процесса его окисления. Температурный режим в печи поддерживается за счет теплоты, выделяющейся в процессе горения исходного сырья и угля. Температуру поддерживают в следующих пределах: 670—700° С на первом поде (сверху), 740—760° С на втором, 650—675° С на третьем и 450—475° С на четвертом.
171
В процессе термообработки содержащийся в исходном сырье су- ] льфид меди (I) окисляется до оксида меди (II) по реакции
Cu2S + 2О2 = 2CuO + SO2 ]
Некоторая часть диоксида серы в связи с присутствием в исходном сырье железа каталитически окисляется до триоксида, который сульфатизирует оксид меди. В результате в продукте обжига исходного сырья, кроме основного продукта — оксида меди, содержится и некоторое количество сульфата меди. Поэтому общая схема процесса может быть описана уравнением
4Cu2S + 9О2 = 4CuO + 2СиО • CuSO4 + 2SO2
С меньшим количеством исходного кислорода и при плохом перемешивании реакционной массы может образоваться и некоторое : количество оксида меди (I):
2Cu2S + ЗО2 = 2Cu2O + 2SO2
Полученный плав имеет состав: 87—90% СиО, 8—10% Cu2S и 2,0—2,5% S. Плав просеивают для отделения от спекшихся комочков и направляют в реактор для растворения в серной кислоте, а крупные частицы после размола в дезинтеграторе возвращают в начало процесса для добавки к исходному сырью.
Реакторы изготовляют из нержавеющей или черной стали (Ст.З), покрывая их с обеих сторон химзащитными материалами, например свинцом, кислотоупорными плитками, поланом или фторопластом.
В начале процесса в реактор при постоянно работающей мешалке заливают маточный раствор от предыдущей операции, содержащий около 28% сульфата меди (II), затем серную кислоту до получе- ] ния раствора с концентрацией 15—20% H2SO4. Массу подогревают до кипения острым или глухим паром. В кипящий раствор вводят небольшими порциями расчетное количество порошкообразного плава оксида меди. Процесс растворения ведут до образования раствора, содержащего 43% сульфата меди и 3—4% серной кислоты.
Оксид меди (II) легко растворяется в серной кислоте, а содержащиеся в исходном сырье металлическая медь и сульфид меди (I) практически не растворяются в серной кислоте, вследствие чего образуется нерастворимый осадок. В осадке содержится и не растворимый в серной кислоте оксид меди (I). Таким образом, в сухом веществе шлама содержится около 50% меди, а также сопровождающие ее золото и серебро. Шлам возвращают в медеплавильные заводы для переработки, а растворы сульфата меди после отстоя направляют на кристаллизацию. 172
Отжатые и промытые в центрифугах кристаллы пентагидрата сульфата меди (II) сушат, упаковывают и выпускают в виде товарного продукта.
Разработан более экономичный способ получения сульфата меди из медьсодержащих отходов медеплавильных заводов, в котором взамен серной кислоты применяют выделяющийся из медеплавильных печей диоксид серы.
Технология сульфата меди (II) сводится к реакции оксида меди в водном растворе сульфата меди с газом, содержащим небольшие количества диоксида серы и кислорода при 85—95° С.
Процесс образования сульфата меди (II) протекает в результате двух независимо идущих реакций. Первая заключается в окислении исходного диоксида серы кислородом в присутствии каталитически действующих ионов меди до серной кислоты:
2SO2 + О2 + 2Н2О = 2H2SO4
которая растворяет оксид меди (II) до сульфата меди (II)
H2SO4 + CuO = CuSO4 + Н2О
Параллельно протекающая вторая реакция заключается в частичном восстановлении диоксидом серы двухзамещенной меди в однозамещенную с образованием плохо растворимой в воде соли Шевреля — комплексной соли сернистой кислоты меди (I) и (II) Cu(CuSO3)2 • 2Н2О или CuSO3 • Cu2SO3 • 2Н2О по схеме
3CuSO4 + 3H2SO3 + ЗН2О = CuSO3 Cu2SO3 • 2Н2О + 4H2SO4
Образовавшаяся соль в отсутствие кислорода в процессе кипячения суспензии разлагается с выделением оксида меди (I):
3(CuSO3 • Cu2SO3 • 2Н2О) = CuSO4 + 2Cu2O + 5SO2
Далее под действием диоксида серы и кислорода в результате образования серной кислоты оксид меди (I) снова переходит в раствор, а осадок соли Шевреля постепенно превращается в сульфат меди (II):
CuSO3 • Cu2SO3 • 2Н2О + SO2 + 2О2 = 3CuSO4 + 2Н2О
Процесс перехода соли Шевреля в сульфат меди происходит в две стадии:
2(CuSO3 • Cu2SO3 • 2Н2О) + ЗО2 = 2Си(ОН)2 • CuSO4 + 3CuSO4 + 2Н2О
2Cu(OH)2 • CuSO4 + 2H2SO4 = 3CuSO4 + 4H2O
173
Таким образом, в результате приведенных процессов из суспензии исчезают все твердые фазы — CuO, CuSO3 • Cu2SO3 • 2Н2О и Си(ОН)2 • СиЗОд и она превращается в водный раствор CuSO4. Из полученного раствора при охлаждении до 20° С выпадают кристаллы пентагидрата сульфата меди (II), которые отжимают, промывают в центрифугах и сушат.
Разработан также способ получения сульфата меди (II) сульфати-зирующим обжигом отходов медеплавильных заводов. Способ более прогрессивен по сравнению с предшествующими, так как в процессе участвует содержащаяся в отходах сера, что позволяет снизить расход серной кислоты почти в два раза. Для достижения этой цели исходное сырье подвергают сульфатизирующему обжигу, т.е. длительной термообработке при 400—500° С при достаточном избытке кислорода. В этих условиях реакции
2SO2 + О2 2SO3 CuO + SO3 CuSO4
смещены вправо и в результате 60—70% сульфидной серы переходит в сульфатную, что соответствуют превращению 30—35% меди в ее сульфат. Для обработки исходного сырья расходуется в полтора раза меньше серной кислоты, чем при окислительном обжиге. Процент извлечения повышается до 90%.
Механизм процессов, происходящих при сульфатизирующем обжиге, можно характеризовать следующими стадиями.
Имеющийся в отходах сульфид меди (I) окисляется частично до оксида и сульфата меди (II):
2Cu2S + 5О2 = 2CuSO4 + 2CuO
Параллельно с этим процессом происходит окисление исходного сульфида меди (I) до диоксида серы и оксида меди (И):
2Cu2S + ЗО2 = 2Cu2O + SO2
2Cu2O + О2 = 4CuO
Оксид меди (II) реагирует с триоксидом серы, образующимся при каталитическом окислении диоксида серы в присутствии содержащегося в исходном сырье оксида железа (III), частично сульфатизирует-ся по реакциям:
CuO + SO3 = CuSO4
4CuSO3 = 3CuSO4 + CuS(II) 174
с последующим окислением образующегося сульфида меди:
CuS + 2Ог — C11SO4
Из приведенных уравнений реакций видно, что для полной суль-фатизации исходного сырья необходимо обеспечить достаточно высокую концентрацию кислорода в газовой фазе. Это достигается путем использования обогащенного кислородом воздуха или применением добавок, обладающих высоким равновесным давлением кислорода в температурных условиях обжига.
Проведенными исследованиями установлено, что степень перехода серы в ее диоксид значительно возрастает при участии оксида меди (II). Из данных рис. 3.8 видно, что с увеличением внесенного в сульфид меди (I) ее оксида заметно повышается степень перехода серы в диоксид. Зависимость степени перехода сульфидной серы в сульфатную от температуры выражается кривой с максимумом (рис. 3.9), что объясняется разницей в скоростях образования и разложения сульфата меди и промежуточных продуктов при различных температурах. Степень окисления сульфида меди при 450° С в течение одного часа без добавки оксида меди составляет 29,5%. С повышением температуры выше 450° С без изменения условий степень перехода сульфида серы в сульфат резко падает и при 750—800° С практически равна нулю. В присутствии же оксида меди (II) увеличивается степень перехода сульфидной серы в сульфатную, а температура, соответствующая максимуму процесса сульфатообразования, сдви-
Рис. 3.8. Степень перехода серы в ее диоксид в зависимости от температуры:
1 — без добавки СиО; 2 — при добавке 10% СиО; 3 — при добавке 25% СиО
Температура,
Рис 3.9. Влияние температуры на переход Cu2S в CuSO4 при различном содержании оксида меди (II) в смеси. Содержание СиО в смеси (в %): /—0; 2—10; 3 — 25; 4 — 50
175
гается в сторону более высоких температур. Так, при добавке 25% оксида меди (II) в температурном интервале 500—550° С в течение одного часа 35—40% сульфидной серы переходит в сульфатную. В этом случае общее количество окислившейся серы достигает 90—95%.
Сульфатизация сульфида меди (I) концентрированной серной кислотой до температуры 300° С протекает по уравнениям реакций:
C112S + 2H2SO4 = CuS + CUSO4 + SO2 + 2НгО
CuS + 2H2SO4 = CuSO4 + SO2 + S + 2Н2О
S + 2H2SO4 = 3SO2 + 2H2O
Наибольший выход сульфата меди (II) достигается при 200° С. При более высоких температурах серная кислота улетучивается с частичным разложением.
В процессе перехода сульфида меди (I) в сульфат меди (II) в процессе автоклавного выщелачивания исходного сырья слабой серной кислотой в присутствии кислорода параллельно протекают следующие реакции:
2Cu2S + 5О2 + 2H2SO4 = 4CuSO4 + 2Н2О (1)
CU2S + О2 + 2H2SO4 = 2CUSO4 + S + 2Н2О (2)
Скорость процесса растворения сульфида меди (I) пропорциональна давлению кислорода в степени 0,5. Оптимальными условиями для осуществления процесса по реакции (1) являются: давление кислорода 4 атм, концентрация серной кислоты 0,01 моль/л, температура 140° С.
Получение сульфата меди (II) из колчеданных огарков. В колчеданных огарках медь содержится в виде сульфидов, оксидов, сульфита, сульфата и халькопирита, что затрудняет полное ее извлечение.
Сульфат и сульфит меди выщелачивается водой, а оксид меди — серной кислотой. Сульфиды меди, которые составляют около 20% меди, не растворяются ни в воде, ни в серной кислоте. Однако они переходят в раствор и в процессе нагревания реагируют с сульфатом или хлоридом железа (III) по схеме
CuS + Fe2(SO4)3 = CUSO4 + 2FeSO4 + S
Cu2S + 2Fe2(SO4)3 = 2CuSO4 + 4FeSO4 + S 176
Образующийся раствор сульфата меди (II) очищают от железа (II) путем окисления его пиролюзитом или кислородом воздуха и обработкой сульфата железа (III) известняком:
6FeSO4 + О2 = 2Ре2(80д)з + 2FeO
Fe2(SO4)3 + ЗСаСОз + ЗН2О = 2Fe(OH)2 + 3CaSO4 + ЗСО2
Халькопирит переводят в водорастворимый хлорид железа (III) путем хлорирования:
2CuFeS2 + 7С12 = 2CuC12 + 2FeCl3 + 2S2C12
Наиболее разработанными способами получения сульфата меди (II) из огарков являются водно-кислотное выщелачивание и хлорирующий обжиг с участием хлорида натрия.
В процессе выщелачивания огарка 1%-ным раствором серной кислоты в течение одного часа при соблюдении отношения Т:Ж= 1:3 в раствор переходит от 60 до 80% меди. В результате шестикратного выщелачивания получают растворы, содержащие 15—18 г/л меди. Растворы очищают осаждением соединений железа (5—6 г/л Fe) карбонатом кальция и последующим окислением хлором. Сульфат меди получают из этих растворов упаркой их до концентрации 80—85 г/л меди с последующей кристаллизацией целевого продукта.
Для перевода не растворимых в воде соединений меди применяют хлорирующий обжиг огарка с хлоридом натрия при высоких температурах:
CuS + 2NaCl + 2О2 = CuCl2 + Na2SO4
К исходному огарку в зависимости от содержания в нем меди добавляют 4—5% хлорида натрия. Полученную смесь измельчают на вальцовой мельнице до размера частиц не более 2 мм и обжигают в механической колчеданной печи (например, печь ВХЗ) при 550—600° С. Топливом для создания указанной температуры в печи является газ. На некоторых заводах в качестве топлива применяют серу или серный колчедан. По этой технологии исходную шихту готовят с соблюдением молярного отношения ее составляющих Си : 3S : 6NaCl. Исходное медьсодержащее сырье состоит из оксидов железа, хлоридов меди (I) и (II), оксида и сульфата меди, а также Na2SO4. В процессе обжига шихты часть хлорида натрия реагирует с серой и водяными парами, образуя хлорид водорода. В выхлопных газах кроме хлорида водорода содержится некоторое количество хло-177
ра, ди- и триоксида серы, водяных паров и соединения мышьяка. Газы после печи поступают в полую башню, орошаемую водой, в результате чего получают 10%-ную хлороводородную кислоту, содержащую хлор и серную кислоту, используемую в дальнейшем в процессе выщелачивания обожженного огарка.
Колчеданные огарки, содержащие 0,5—1,0% меди, хлорируют элементным хлором, а измельчение водорастворимых солей меди из обожженного огарка производят методическим выщелачиванием при 20—25° С, получаются неконцентрированные растворы сульфата меди (II) или хлорида меди (II). Поскольку концентрирование этих растворов путем упарки экономически нецелесообразно, медь выделяют из них методом цементации. Процесс заключается в вытеснении меди из растворов железными стружками или железным ломом по схеме
Cu2+ + Fe = Fe2+ + Си
Электродный потенциал меди значительно выше, чем железа. В растворах, содержащих ионы Си2+ или Fe2+, при обычной температуре и давлении водорода 1 атм он равен для меди + 0,348, а для железа 0,448. В связи с этим железо вытесняет медь из раствора в виде тонкого металлического шлама, называемого цементной медью.
Процесс цементации меди проводят в химзащищенном реакторе, куда вносят очищенный от грязи и ржавчины железный лом, после чего в реактор заливают исходный разбавленный раствор сульфата меди. Для полноты осаждения меди раствор не должен содержать значительных количеств серной кислоты, оптимальная концентрация ее должна быть около 0,05% или около 5-10’5 г-моль/л. При этих условиях железо практически не растворяется в серной кислоте и обеспечиваются оптимальные условия для полного осаждения меди из раствора до содержания ионов меди примерно 5-10’5 г-ион/л.
Образующийся в процессе цементации раствор сульфата железа сливают, а в реактор заливают свежие растворы сульфата меди (II). Описанную операцию повторяют 10—12 раз, после чего выгружают осевшую на дно реактора цементную медь, которую промывают от частиц железа 10—15%-ной серной кислотой. После полной отмывки железа цементную медь промывают водой от кислоты.
Промытая цементная медь получается в виде пасты красновато-бурого цвета. Она содержит 65—70% меди, до 35% влаги и около 1% примесей и перерабатывается в пентагидрат сульфата меди (II) по известным способам. 178
Разработан также способ получения меди из разбавленных растворов сульфата меди путем обработки их слабой аммиачной водой. При этом образуется осадок основного сульфата меди, который после фильтрации обрабатывают серной кислотой для получения сульфата меди (II). При этом процесс протекает по схеме
2CuSO4 + Н2О + NH3 = Cu(OH)2 • CuSO4 + NH4SO4
Cu(OH)2 • CuSO4 + H2SO4 = 2CuSO4 + 2H2O
В случае присутствия в исходных растворах, кроме меди, ионов железа и никеля разработан способ ступенчатого осаждения их аммиаком в процессе нейтрализации раствора последовательно до pH 3; 4,5 и 6.
Получение сульфата меди из отходов медеэлектролитных производств. В процессе электрохимического рафинирования меди применяется электролит, содержащий 30—45 г/л меди в виде сульфата меди (II) и -200 г/л свободной серной кислоты. Кроме этого в электролите присутствуют примеси сульфатов никеля, цинка, кальция и железа, а также сесквиоксид мышьяка и др. Согласно существующей технологии, по мере накопления указанных примесей и переходом в раствор меди часть электролита должна выводиться из системы. Наиболее экономичным способом рекуперации выводимого из цикла электролитного раствора является переработка его на сульфат меди.
Для осуществления процесса рекуперации растворов серную кислоту, содержащуюся в этих растворах, нейтрализуют медьсодержащими материалами, например катодным скрапом, гранулированной катодной и анодной медью, шлаком анодных печей или стружкой. Процесс нейтрализации кислоты производят циркуляцией исходного раствора через слой одного из перечисленных медьсодержащих материалов. Одновременно в растворы барботируют воздух, необходимый для окисления меди в процессе ее растворения в серной кислоте. Процесс окисления проводят при 70—80° С. При достижении содержания свободной серной кислоты в растворе до 0,5% его направляют на упарку. В процессе упарки осаждаются соли железа и кальция. После упарки раствор охлаждают в кристаллизаторах, где выпадает сульфат меди в виде пентагидрата. Из маточного раствора извлекают сульфат никеля.
Содержащиеся в меди примеси в процессе рафинирования переходят в шлам. Остающиеся в шламе значительные количества меди извлекают путем выщелачивания серной кислотой при 85° С с одновременным барботированием воздуха.
179
3.5. КАРБОНАТЫ МЕДИ
Известны карбонаты для Cu(I) и Cu(II).
Карбонаты меди (II) получают в процессе обменных реакций в водных растворах между солями меди (II) и карбонатами других металлов. Вследствие сильного гидролиза осаждаются гидрооксокарбонаты:
2CuSO4 + 2Na2CO3 + Н2О = CuCO3 • Cu(OH)2 + 2Na2SO4 + CO2
3CuSO4 + 3Na2CO3 + H2O = 2CuCO3 • Си(ОН)г + 3Na2SO4 + CO2
Состав осаждающихся карбонатов меди (II) зависит от температуры процесса и концентрации исходных видов сырья.
Карбонат меди (II) получают обработкой основных карбонатов диоксидом углерода под давлением 4,6 МПа при 180° С:
СиСО3 • Си(ОН)2 + СО2 = 2СиСО3 + Н2О
2СиСО3 • Си(ОН)2 + СО2 = ЗСиСО3 + Н2О
Гидрооксокарбонаты легко реагируют с минеральными кислотами с образованием соответствующих солей меди (II) и выделением диоксида углерода:
СиСО3 • Си(ОН)2 + 2H2SO4 = 2CuSO4 + СО2 + Н2О 2СиСО3 • Си(ОН)2 + 6HNO3 = 3Cu(NO3)2 + 2СО2 + 4Н2О
СиСО3 • Си(ОН)2 + 4НС1 = 2СиС12 + СО2 + ЗН2О
В процессе обработки гидроксокарбонатов избытком диоксида углерода в растворе гидроксида щелочного металла образуются комплексные карбонаты типа К2[Си(СО3)г] • ЗН2О.
Под действием водного раствора аммиака гидрооксокарбонаты образуют аммиакаты, а с цианидами металлов дают цианокомплексы меди (II).
Дигидрокарбонат меди СиСО3 • Си(ОН)2 — темно-зеленые кристаллы с моноклинной решеткой (а = 0,9502 нм, b = 1,1974 нм, с = = 0,3240 нм, Р = 98,75°, пространственная группа Р2]/а); плотность 4,03 г/см3; АЯ^Р =-1051,0 кДж/моль, AG^, =-900,9 кДж/моль. В воде практически не растворяется. В процессе нагревания до 200°С разлагается по схеме
СиСОз • Си(ОН)2 —2ОО'С > 2СиО + Н2О + СО2
В природе встречается в виде минерала малахита.
180
Дигидрокарбонат меди (II) является сырьем в производстве других соединений меди. Плотный малахит является ценным поделочным камнем, а землистый малахит и мелкие скопления чистого минерала применяют для изготовления краски «малахитовая зелень».
Дигидроксодикарбонат 2СиСО3 • Си(ОН)2 — синие кристаллы с моноклинной решеткой (а = 0,5008 нм, b = 0,5844 нм, с = 1,0336 нм, 0 = 92,45°, пространственная группа Р2\!а)\ плотность 3,78 г/см3; ДЯ°5Р = -1631,3 кДж/моль, Дб^р = -1431,1 кДж/моль. В воде практически не растворяется. В процессе нагревания выше 220° С разлагается до СиО, СО2 и Н2О. В природе — минерал азурит.
Дигидроксодикарбонат меди (II) применяется в качестве компонента пиротехнических составов. Минерал азурит применяют для получения меди, а также в производстве синей краски.
Имеются сведения о существовании кристаллического карбоната меди (I) желтого цвета с плотностью 4,4 г/см3.
3.6. НИТРАТ МЕДИ
а-Форма нитрата меди — синие гигроскопичные кристаллы с ромбической решеткой (а = 1,12 нм, 0 = 0,505 нм, с = 0,828 нм, z = 4, пространственная группа Pmn2i); при ПО—120° С переходит в 0-форму, выше 170° С разлагается до оксида меди (II); сублимируется при 190° С и давлении около 16 кПа; при быстром нагревании плавится при 255° С (с разложением); ДЯ^ЗГ = 65,3 кДж/моль; ДЯЛ ~ -303,1 кДж/моль. Растворимость в воде (г в 100 г): 84(0° С), 150(25° С) и 182(60° С). Из водного раствора в зависимости от его концентрации кристаллизуется нона-, гекса- и тригидраты нитрата меди. Известны также кристаллогидраты с 2,5 и 1,5 молекулами воды.
Гидраты нитрата меди (II) — гигроскопичные вещества синего цвета.
Гексагидрат Cu(NO3)2 • 6Н2О выше -5° С существует в виде устойчивой модификации с триклинной решеткой (а = 0,591 нм, b = 0,777 нм, с = 0,543 нм, а = 97,65°, 0 = 93,88°, у = 72,53°, z = 1, пространственная группа Р1); температура плавления 24,4° С; ДЯ° = -2108,7 кДж/моль, Дб° =-1564,8 кДж/моль, ДЯ^, = -36,36 кДж/моль.
Тригидрат Cu(NO3)2 • ЗН2О при 114° С плавится, выше этой температуры переходит в гидроксосоли, а выше 20° С разлагается до оксида меди (II).
Гидраты Cu(NO3)2 • 2,5Н2О и Cu(NO3)2 • 1,5Н2О — кристаллы моноклинной сингонии, z = 8, пространственная группа соответственно СИ с и 12/а; для Cu/NO^ • 1,5Н2О а = 2,22 нм, b = 0,490 нм, с = 1,54 нм, 181
р = 48°; для Cu(NO3)2 • 2,5Н2О а = 1,64539 нм, b = 0,49384 нм, с = = 1,59632 нм, р = 93,764°.
Безводный нитрат меди (II) получают при растворении металлической меди в смеси N2O4 с этилацетатом. Процесс идет в две стадии. В первой стадии образуется комплекс Cu(NO3)2 • N2O4, который в процессе нагревания до 80° С образует безводную соль Cu(NO3)2. Гидраты нитрата меди получают по схеме
Си + 2HNO3 + Н2О = Cu(NO3)2 + Н2О + Н2
СиО + 2HNO3 + Н2О = Cu(NO3)2 + 2Н2О
Нитрат меди (II) применяют для получения чистого оксида меди (II), медьсодержащих катализаторов и в качестве фунгицида, а также протравы в процессе крашения тканей.
ГЛАВА 4
СТРОНЦИЙ И ЕГО СОЕДИНЕНИЯ
4.1. СТРОНЦИЙ
Содержание стронция в земной коре составляет 3,410’2% (масс.), в океанических водах— 11,097 млн. тонн (8,1 мг/л). В свободном состоянии стронций в природе не встречается, а содержится в различных минералах. Стронций образует около 40 минералов, из которых промышленное значение имеют целестин SrSQ, и стронцианит SrCOj. Присутствует в качестве изоморфной примеси в различных бариевых, кальциевых и магниевых минералах, а около 24% общих запасов стронция содержится в природных минерализованных водах. Среднее содержание стронция в почвах 0,035% (масс.), в речной воде 0,08 мг/л. Часть содержащегося в океанических водах стронция (около 24%) концентрируется в виде железомарганцевых конкреций (4900 т в год).
Природный стронций состоит из четырех стабильных изотопов: 88Sr (82,56%), 86Sr (9,86%), 87Sr (7,02%), 84Sr (0,56%). Поперечное сечение захвата тепловых нейтронов для природной смеси изотопов 1,21-10'28 м2. Конфигурация внешней электронной оболочки атома 5.?2; степень окисления +2, очень редко +1; энергии ионизации Sr° -> Sr+ -> Sr2+ соответственно равны 5,69410 и 11,0302 эВ; электроотрицательность по Полингу 1,0; сродство к электрону— 1,51 эВ; атомный радиус 0,215 нм, ионный радиус Sr2* 0,132 нм (6), 0,140 нм (8), 0,150 нм (10), 0,158 нм (12).
Свойства. Стронций — мягкий серебристо-белый металл, ковкий и пластичный. Существует в трех полиморфных модификациях: до 2310 С устойчив a-Sr с кубической гранецентрированной решеткой типа Си, а = 0,6085 нм, z = 4, пространственная группа Fm3m, плотность 2,63 г/см3; в температурном интервале 231—623° С — (J-Sr с гексагональной решеткой типа Mg, а = 0,431 нм, с - 0,705 нм, z = 2, пространственная группа 1тЗт. Температура плавления 768° С, температура кипения 1390° С; С ° = 26,79 Дж/(моль К); = 8,2 кДж/моль,
АЯ„°СП = 133,8 кДж/моль, АЯ^Г = 160,5 кДж/моль; 5°98 = 55,70 Дж/(моль-К); парамагнетик, магнитная восприимчивость +1,05-10’9; пластичен.
183
Стронций — химически активный металл, по химическим свойствам сходен с кальцием и барием. Стандартный электродный потенциал Sr2+/Sr° — 2,89 В. При обычных условиях металлический стронций окисляется кислородом воздуха и покрывается пленкой из оксидов SrO ’ и SrO2, при нагревании воспламеняется. Энергично реагирует с водой | и образует гидроксид с выделением водорода. Элементный стронций | взаимодействует с галогенами. При повышенных температурах строк- | ций реагирует с диоксидом углерода: 5Sr + 2СО2 = SrC2 + 4SrO, а при ? 300—400° С с водородом образует гидрид. При нагревании с элемент- j ной серой стронций образует сульфид и полисульфиды SrS„ (п = 2—5), ) с селеном и теллуром — селенид (SrSe) и теллурид (SrTe), с азо- ; том — нитрид Sr3N2, с углеродом — карбид (SrC) и перкарбид (SrC2), с газообразным NH3 — амид (Sr(NH2)2).
Стронций в расплавленном состоянии образует однородные расплавы (растворы) со многими металлами. С кальцием и барием дает j непрерывный ряд твердых растворов, образует интерметаллиды типа < SrMg2, SrAl, SrAU и др. '
Стронций растворяется в разбавленных минеральных кислотах с образованием соответствующих солей и выделением водорода. Концентрированная серная кислота со стронцием образует сульфат стронция, диоксид серы, сульфид водорода и серы, а концентрированная азотная кислота — нитрат стронция и NO. При растворении стронция в жидком аммиаке может быть получен аддукт Sr(NH3)6. Хорошо растворяются в воде галогениды, ацетат и некоторые другие соли стронция, плохо растворяются сульфат, фторид, карбонат, оксалат, арсенат (III), хромат, иодат, фосфат и молибдат стронция.
Сульфат стронция SrSC>4 — бесцветные кристаллы ромбической сингонии (табл. 4.1); выше 1157° С переходит в гексагональную P-SrSC>4, АН перехода а->0 10 кДж/моль; выше 1580° С разлагается с образованием оксидов стронция, диоксида серы и кислорода; растворимость в воде 0,0113 г в 100 мл при 0°С; получают осаждением из водных растворов солей и щелочей стронция сульфатом натрия; наполнитель при изготовлении красок и резины, утяжелитель в буровых растворах.
Гексаферрит SrO • 6Fe2O3 получают спеканием смеси Fe2O3 и SrO или SrCO3 при 1000° С ; магнитный материал.
Гидрид стронция SrH2 — бесцветные кристаллы ромбической сингонии, выше 355° С существует P-SrH2, АН полиморфного перехода 7,2 кДж/моль; получают восстановлением оксида стронция водородом при 700—800° С или из элементного стронция и водорода.
Сульфид стронция SrS — бесцветные кристаллы кубической сингонии; разлагается водой; получают при нагревании эквимолярной смеси стронция и элементной серы, восстановлением сульфата 184
стронция углеродом или водородом; компонент люминофоров, фосфоресцирующих составов. Применяется в качестве средства для удаления волос в кожевенной промышленности.
Получение. Металлический стронций получают электролизом расплава хлорида стронция (85% SrCl2 + КС1 или NH4C1). Процесс ведут на никелевом или железном катоде при 800° С. Исходный хлорид стронция получают обработкой сульфида стронция хлороводородной кислотой. Сульфид стронция в свою очередь получают из сульфата его восстановлением углеродом или водородом при 1100—1300° С. Полученный этим способом стронций содержит 0,3—0,4% калия.
Таблица 4.1. Характеристика соединений стронция
Соединение Температура плавления, °C Плотность, г/см1 Ср”- Дж/(моль • К) ДЯ" , кДж/моль 9° °298> Дж/(моль • К)
a-SrH2 1050 — 44 -180 52
SrS -2227 3,64—3,79 48,70 -480 68,2
a-SrSO4 1607 3,973 102 -1458,5 121
SrO 6Ре20з 1390 — — -5802 581
SrO2 >900 4,59 — -636,6 —
SrCrO4 1283 3,84 — -1434 111,6
Для металлотермического восстановления SrO2 до Sr используют алюминий, кремний или ферросилиций. Процесс ведут при 1000° С в вакууме в стальной трубке. Элементный стронций получают также восстановлением хлорида стронция металлическим магнием в атмосфере водорода.
Стронцианитовый концентрат разлагают обжигом при 1200° С с последующим выщелачиванием оксида стронция водой или минеральными кислотами либо непосредственным растворением стронцианита в азотной или соляной кислоте.
Стронций может быть получен при нагревании в вакууме гидрида стронция при 1000° С , Sr3N2 (140—150° С) или 8г(ННз)6. Образующийся стронций очищают перегонкой в вакууме.
Радиоактивные изотопы стронция образуются при делении 235U. Изотоп 89Sr (Г]/2 50,5 сут) получают также в реакторах из стабильного стронция по реакции 88Sr (п, у) 89Sr или в циклотроне: 88Sr (d, р) 89Sr.
Применение. Металлический стронций применяют в технологии раскисления меди и бронзы, в качестве легирующих добавок к сплавам Mg, Al, Ni, Си, в качестве геттера в электровакуумной технике. Более широко применяются в народном хозяйстве его соли и, в частности, в производстве оптических стекол, стекол для кинескопов, электронных трубок, в пиротехнических составах (дают пламя карминно-красного цвета), фосфоресцирующих составах, в производстве 185
ферромагнитных люминесцентных материалов, эмиссионных покры- | тий радиоламп и т.д. |
Соли стронция, в том числе радиоактивного, применяют в тера- | пии кожных болезней, соли жирных кислот — при изготовлении кон- I систентных смазок. 90Sr — источник у-излучения. а
Хранят стронций в закрытой стеклянной посуде под слоем керосина. '
4.2. ГАЛОГЕНИДЫ СТРОНЦИЯ ‘
Галогениды стронция SrX2, где X = F, Cl, Br, I, бесцветные кристаллы (табл. 4.2). Фторид стронция SrF2 плохо растворим в воде (0,1173 г при 17,4° С и 0,1193 г в 1 л раствора при 27,4° С) и разбавленных минеральных кислотах, растворим в горячей хлороводо- , родной кислоте. В криолитовых копях Гренландии найден минерал : ярмит NaF • 3S1F2 • 3A1Fj.
Хлорид стронция SrCh растворим в воде (34,6% масс, при 20° С, * 50,2% при 100° С); температура кипения насыщенного раствора ) 117° С, из водных растворов ниже 61,34° С кристаллизуется гекса- j гидрат SrCl2 • 6Н2О, расплывающийся во влажном воздухе; при более высоких температурах гексагидрат превращается в SrCh • 2Н2О и далее в SrCl2 • Н2О; при нагревании до 250° С полностью обезвоживается; в отличие от СаС1г • 6Н2О гексагидрат SrCl2 плохо растворим в этаноле (3,64% масс, при 6° С), что используется для их разделения.
Бромид стронция SrBr2 гигроскопичен; растворимость, % масс.: в воде —50,6 (20° С), 69,0 (100° С), этаноле —39,3 (30° С). Ниже 88,62° С из водных растворов кристаллизуется SrBr2 • 6Н2О, а выше этой температуры — SrBr2 • Н2О; гидраты полностью обезвоживаются при 345° С.
Иодид стронция Srl2 имеет растворимость, % (масс.): в воде — 64,0 (20° С), 79,3 (100° С), этаноле —4,3 (39° С), 4,7 (82° С). Ниже 83,9° С из водных растворов кристаллизуется Srl2 • 6Н2О, а выше этой температуры — Srl2 • 2Н2О.
Галогениды стронция образуют двойные соли, аммиакаты и другие аддукты. К ним относятся SrCh • SrF2 (температура плавления 962° С), 2КС1 • SrCh (температура плавления 597° С), SrCl2 • 8NH3, 2КВг • SrBr2 (температура плавления 574° С), Srl2 • 4SO2, Srl2 • 8NH3. Из водных растворов кристаллизуются SrX2 • SrO • 9Н2О, где X = С1, Вг, а также Srl2 • 2SrO • 9Н2О. Эвтектика LiCl — SrCh (48 мол.% SrCh) образуется при 492° С.
Галогениды стронция получают реакцией стронция с галогенами или оксида стронция (либо SrCO3) с галогеноводородными кислотами. В газовой фазе существуют моногалогениды стронция SrX. 186
Таблица 4.2. Свойства галогенидов стронция
Показатель SrF2 SiCl2 SrCl2 • HzO SrCl2 • 6Н2О SrBr2 SrBrj-IbO SrBr2 • 6Н2О Srl2 Srl2 • 61^0
Сингония Кубическая Ромбическая Кубическая Моноклинная Тригональная Тетрагональная Ромбическая Тригональная Ромбическая Тригональная
Параметры элементарной ячейки, нм:
а 0,5796 0,3789 0,69767 1,171 0,7956 1,1633 1,149 0,8221 1,522 0,8604
b — 0,6306 — 0,639 — — 0,4291 — 0,822 —
с — 0,7428 — 0,667 0,4116 0,7155 0,9257 0,4154 0,790 0,4268
Число формульных единиц в ячейке 4 4 4 4 1 10 4 1 8 1
Пространственная группа Fm3m Pmnb Fm3m СИ с Р321 Р4/л Рпта Р321 РЬса Р321
°C 1477 — 874 — — 657 —— 88,62 538 —
Ткжь °C 2460 — 1300 — — — — — — —
Плотность, г/см3 4,18 — 3,085 (25° С) 2,6715 (25° С) 1,954 (20° С) 4,216 — 2,358 4,549 4,415
Показатель преломления 1,435 1,482 1,64986 1,5942 1,5948 1,6172 1,3560 1,4856 1,58 — 1,540 1,576 — 1,632 1,649
С", Дж/(моль-К) 70,0 — 75,61 — — 76,53 — — 77,40 —
кДж/моль —1228,0 — -833,20 -1184 — -717,00 — — -562,00 —
Дж/(моль • К) 82,13 — 114,85 — — 143,14 — — 159,00 —
Бромид и фторид стронция являются оптическими материалами. Фторид стронция применяется в качестве компонента специальных стекол, люминофоров, лазерного материала, SrJ2— люминесцентный материал в сцинтилляционных счетчиках; SrCl2 — компонент пиротехнических составов (придает пламени карминово-красный цвет), его применяют в холодильной промышленности, медицине, косметике.
Получение хлорида стронция. Сырьем для получения хлорида стронция является целестин (SrSO4) и кокс или уголь. Основой технологии являются следующие химические реакции:
SrSO4 + 4С « SrS + 4СО (1)
SrS + 2НС1 = SrCl2 + H2S (2)
Согласно принципиальной схеме производства (рис. 4.1), исходный целестин, содержащий до 95% сульфата стронция и другие примеси (CaSO4, BaSO4, Fe2O2 и др.), измельченный в шаровых мельницах и высушенный, смешивается с коксом (углем) и в смеси с ним гранулируется. Гранулы смеси поступают в печь с кипящим слоем на восстановительный процесс. Полученный в печи плав (в виде гранул) сульфида стронция, проходя через циклон 2 и фильтр 3 (частично в виде пыли),
Рис 4.1. Принципиальная схема получения хлорида стронция:
/ — печь КС; 2— циклон; 3 — фильтр; 4 — шаровая мельница мокрого помола; 5 — реактор; 6 — промыватель шлама; 7 — сборник декантата; 8— фильтр-пресс (ФПАКМ); 9 — резервуар для нейтрализации растворов; 10—вакуум-выпарной аппарат; 11 — барабанный кристаллизатор;
12 — приемник кристаллического хлорида стронция; 13 — центрифуга
188
поступает в шаровую мельницу 4 на выщелачивание. Растворителем плава служат промывные воды из следующей операции. Образующаяся в шаровых мельницах водная суспензия сульфида стронция поступает в реактор 5, в котором происходит основная реакция образования хлорида стронция [см. уравнение (2)]. Образующиеся при этом растворы хлорида стронция направляются в сборник декантата 7, далее на фильтрацию на фильтры 8, а шлам для дополнительной промывки в промыватель шлама 6, откуда в шламонакопитель завода.
Сульфид водорода, выделяющийся в процессе реакции в реакторе 5, откачивается вентилятором в отделение, где из него получают сульфогидрат натрия:
NaOH + H2S -► NaHS + Н2О
Отфильтрованные растворы хлорида стронция упаривают в трехкорпусном вакуум-выпарном аппарате 10 и направляют на кристаллизацию в барабанный вращающийся кристаллизатор 11. Образующаяся суспензия кристаллов, проходя через приемник (сборник) кристаллического хлорида стронция, поступает в непрерывно действующую автоматическую центрифугу типа ФГП-800 и ФГ-1200. Полученные кристаллы хлорида стронция при необходимости сушат в барабанных сушилках или в сушилках в кипящем слое. Поскольку обычно хлорид стронция выполняет роль сырья (в данном случае полуфабриката) для получения других соединений стронция, его не сушат. Обычно в виде товарного продукта выпускают хлорид стронция двуводный (дигидрат) или одноводный (моногидрат), а по описанной технологии получается гексагидрат хлорида стронция. При необходимости по требованиям потребителей выпускают гексагидрат хлорида стронция, так как он по описанной технологии получается шестиводный.
4.3. КАРБОНАТ СТРОНЦИЯ
Физико-химические свойства карбоната стронция. Карбонат стронция SrCOj, бесцветные кристаллы; до 925° С существует а-моди-фикация с ромбической кристаллической решеткой типа арагонита (а = = 0,5107 нм, b = 0,8414 нм, с = 0,6029 нм, пространственная группа Ртсп, плотность 3,785 г/см3); выше 925° С стабильна гексагональная P-модификация структурного типа кальцита (пространственная группа F3c); при 1416° С и давлении СО2 2 МПа образуется у-модификация предположительно кубической сингонии; температура плавления 1494° С; С ° = 82,42 Дж/(моль-К), А/Л, = 1227,0 кДж/моль. АЯ перехода а-»р 16,7 кДж/моль, АЯПЛ = 40 кДж/моль; S®98 = 97,2 Дж/(моль К).
189
Карбонат стронция при нагревании разлагается на оксид стронция и диоксид углерода, уравнение температурной зависимости давления диссоциации: lgPC0 (Па) = 12,767-11549/Т (1093-1193К), lgPC02 (Па) = 11,942- 10579ЛГ (1203—1323 К). Мало растворим в воде (2-10‘3г в 100 г при 25° С). При избытке диоксида углерода в растворе превращается в гидрокарбонат Sr(HCO3)2.
Карбонат стронция применяется в качестве одного из исходных компонентов при изготовлении оксидных катодов электровакуумных приборов, высокотемпературных сверхпроводников, в производстве некоторых сортов стекол (в том числе для кинескопов цветных телевизоров), эмалей, глазурей.
Способы получения карбоната стронция. Основные способы получения карбоната стронция основаны на реакции обмена водорастворимых солей стронция карбонатом натрия или аммония. К водорастворимым солям относятся его сульфид, хлорид и нитрат. Одним из возможных видов сырья может быть оксид и гидроксид стронция.
При этом процессы проходят по следующим реакциям:
SrS + Na2CO3 = SrCO3 + Na2S
SrCl2 + Na2CO3 = SrCO3 + 2NaCl
Sr(NO3)2 + (NH4)2CO3 = SrCO3 + 2NH4NO3
Sr(OH)2 + Na2CO3 = SrCO3 + 2NaOH
Sr(OH)2 + CO2 = SrCO3 + H2O
SrO + H2O + CO2 = SrCO3 + H2O
Получение карбоната стронция из его хлорида. Процесс основан на обменной реакции между хлоридом стронция и карбонатом натрия:
SrCl2 + Na2CO3 = SrCO3 + 2NaCl
Процесс состоит из следующих стадий: 1) приготовление и очистка растворов хлорида стронция; 2) подготовка и очистка растворов карбоната натрия; 3) проведение обменной реакции и сгущение суспензии; 4) разделение и отмывка пасты карбоната стронция; 5) сушка пасты карбоната стронция; 6) затаривание сухого продукта.
Рассмотрим схему производства карбоната стронция из его хлорида (рис. 4.2). Карбонат натрия доставляют пневмотранспортом из силосных башен в бункер 2, откуда он через дозатор 2 поступает в растворитель 3, снабженный метальным устройством. Одновременно в растворитель 3 заливают отмеренное количество раствора карбона-190
Отфильтрованные растворы SrCh
На станцию нейтрализации
Рис 4.2. Схема получения карбоната стронция из его хлорида:
1 — бункер, 2 — дозатор; 3 — растворитель карбоната натрия; 4 — реактор для обработки растворов карбоната натрия оксохлоридом кальция; 5 — ФПАКМ; 6 — напорная емкость растворов карбоната натрия; 7 — напорная емкость растворов хлорида стронция; 8 — реактор; 9,7/— репульпа-торы; 10— барабанные вакуум-фильтры (БОК или БШР); 12 — сушилка с кипящим слоем (КС);
13— центробежный насос
та натрия. Температуру в растворителе поддерживают 60—65° С. Растворы в реакторе 4 обрабатывают хлорной известью, фильтруют на фильтре 5 в напорную емкость 6. Одновременно в напорную емкость 7 поступают отфильтрованные растворы хлорида стронция.
Обменную реакцию проводят в реакторе 8 при температурном интервале 85—95° С. Образующуюся водную суспензию карбоната стронция из репульпатора 9 насосом перекачивают на первый барабанный вакуум-фильтр 10, откуда отжатая паста поступает в репуль-патор 11, где она перемешивается очищенной водой (конденсат), и смесь направляется во второй барабанный вакуум-фильтр 10. Промытая и отжатая на фильтре паста с влажностью 30% направляется в сушилку с кипящим слоем 12.
Образующиеся в процессе промывки карбоната стронция водные растворы хлорида натрия направляются на станцию нейтрализации. Полученный карбонат стронция упаковывается в бумажную тару с полиэтиленовым вкладышем внутри.
Получение карбоната стронция из его гидроксида (оксида). Технология основана на единовременной реакции перехода оксида стронция в его гидроксид, который с диоксидом углерода образует карбонат стронция по схеме (рис. 4.3):
SrO + Н2О = Sr(OH)2 Sr(OH)2 + СО2 = SrCO3 + Н2О
Исходный оксид стронция растворяется в емкости 1 водой. Полученные растворы гидроксида стронция фильтруются на фильтре 2 и направляются в сборник фильтрованных растворов 3, откуда поступа-191
2
Выхлопные пары
Рис. 4.3. Схема получения карбоната стронция из его оксида:
1 — емкость для растворения оксида стронция; 2 — фильтр; 3 — сборник растворов гидроксида стронция; 4 — абсорбер; 5,7— репульпаторы; 6 — барабанный вакуум-фильтр; 8—сушилка кипящего слоя
ют в абсорбер 4 на орошение диоксидом углерода. В абсорбере 4 происходит процесс образования суспензии карбоната стронция. Образующаяся в абсорбере 4 суспензия карбоната стронция поступает в репульпатор 5, откуда — на барабанный вакуум-фильтр 6. Отжатая на фильтре 6 паста карбоната стронция влажностью 30% поступает в репульпатор 7, где она перемешивается с очищенной водой, а образующаяся суспензия поступает в сушилку КС. После сушки сухой продукт идет на упаковку.
4.4. НИТРАТ СТРОНЦИЯ
Нитрат стронция 8г(МОз)2, бесцветные кристаллы кубической сингонии (а = 0,781 нм, z = 4, пространственная группа Тл6); температура плавления 645° С; плотность 3,0 г/см3; С° = 149,91 Дж/(моль-К); ДЯ^р =-984,08 кДж/моль; S2°98 = 194,6 Дж/(моль-К); растворимость в воде (в 100 г): 70,5 (20° С ), 90,0 (40° С) и 98,0 (80° С). Из водных растворов до 29° С при концентрации 47,0% (масс.) кристаллизуется тетрагидрат 5г(ЬЮз)2-4Н2О, а выше — безводный нитрат стронция; температура плавления криогидрата—5,4° С (24,7% по массе нитрата стронция). Тетрагидрат — бесцветные кристаллы моноклинной сингонии, плотность 2,2 г/см3; ДЯ^р =-2161,0 кДж/моль; 5298 = 364 Дж/(моль К); выше 100° С отщепляет воду с образованием безводного нитрата стронция. В водных растворах нитрат стронция диссоциирует, слабо гидролизуется и образует нитратные комплексы [8г(Н2О)з (^1Оз)з]. Получают нитрат свинца растворением Sr, SrO, Sr(OH)2 или SrCCh в растворах HNO3. 192
Нитрат стронция — компонент пиротехнических составов для сигнальных, осветительных и зажигательных ракет, окрашивающий пламя в коричнево-красный цвет, компонент составов для обезвреживания выхлопных газов автомобилей.
Способы получения нитрата стронция. Нитрат стронция получают растворением элементного стронция, его оксида, гидроксида, сульфида, карбоната в азотной кислоте. Очевидна возможность получения нитрата стронция путем растворения природного стронцийсодержащего сырья — стронцианита (8гСОз) в азотной кислоте с дальнейшей очисткой получаемых растворов от примесей известными технологическими приемами. Перечисленные способы основываются на следующих реакциях:
Sr + 2HNO3 = Sr(NO3)2 + Н2
SrO + 2HNO3 = Sr(NO3)2 + Н2О
Sr(OH)2 + 2HNO3 = Sr(NO3)2 + 2H2O
SrS + 2HNO3 = Sr(NO3)2 + H2S
SrCO3 + 2HNO3 = Sr(NO3)2 + H2O + CO2
Нитрат стронция можно получить из его водорастворимых солей, например
SrCl2 + 2NaNO3 = Sr(NO3)2 + 2NaCl
Sr(OH)2 + 2KNO3 = Sr(NO3)2 + 2KOH
SrS + Са(ЫОз)2 = Sr(NC>3)2 + CaS и т.д.
Получение нитрата стронция из стронцианита (рис. 4.4) основано на реакции
SrCO3 + 2HNO3 = Sr(NO3)2 + Н2О + СО2
Измельченный до состояния порошка природный стронцианит из сушилки 1 поступает в промежуточный бункер 2, откуда через дозатор 3 подается в реактор 4. Одновременно в реактор 4 поступают маточные растворы и азотная кислота. Образующийся в реакторе водный раствор нитрата стронция фильтруется в фильтре 6. Образующийся шлам направляется в шламонакопитель, а растворы поступают в реактор 7 для подщелачивания гидроксидом натрия с целью осаждения гидроксидов кальция и железа. Обработанный гидроксидом натрия 7 Химическая технология 193
неорганических веществ, кн. 1
Рис 4.4. Схема получения нитрата стронция из строцианита:
7 — сушилка; 2 — промежуточный бункер; 3— дозатор; 4, 7, 9 — реактор; 5 — насос; б, 8 — фильтр; 10 — вакуум-кристаллизационная установка; 77 — сборник; 12—концентратор;
13 — центрифуга
осадок после перемешивания при 85—95° С передается в фильтр 8 и дозируется в реактор 9, куда добавляется необходимое количество азотной кислоты. Полученные в реакторе 9 растворы нитрата стронция передаются в трехкорпусную вакуум-кристаллизационную установку 10. Образующиеся в кристаллизационной установке смесь кристаллов с растворами, проходя сборник 11, поступают в концентратор 12, откуда суспензия кристаллов направляется в центрифугу 13, а растворы поступают в реактор 4. Целевой продукт — нитрат стронция — направляется на упаковочный конвейер, а при необходимости на сушку.
В процессе получения нитрата стронция, а именно на стадии кристаллизации нитрата стронция, в системе накапливается нитрат бария. Это связано с большей растворимостью гидроксида бария (101,4 г/л), чем гидроксида стронция (47,7 г/л). Поэтому с ростом содержания гидроксида бария маточные растворы периодически выводят из системы с дальнейшим использованием их для получения кристаллического гидроксида или карбоната бария с обратным возвратом в содержащие нитрат стронция растворы в основную систему.
Другие приведенные способы получения нитрата стронция идентичны с рассмотренной схемой.
194
4.5. ОКСИД И ГИДРОКСИД СТРОНЦИЯ
Оксид стронция SrO, бесцветные кристаллы с кубической кристаллической решеткой типа NaCl (а = 0,5083 нм, пространственная группа Fm3m\, плотность 4,7 г/см3; температура плавления 2600 °C; С° = 44,98 Дж/(моль-К); = 590,5 кДж/моль, А/7ПЛ = 70 кДж/моль; S °98 = 55,44 Дж/(моль-К). Соотношение атомов кислорода и стронция в оксиде стронция незначительно отклоняется от стехиометрического в сторону избытка стронция или кислорода при термообработке кристаллов соответственно в парах стронция или кислороде. Избыток стронция достигает 1-Ю"4 моль (в моле оксида стронция) при 1140—1320° С, а избыток кислорода — от 5-Ю’3 до 1-Ю’3 моль кислорода в интервале 700—1150° С при давлении кислорода 105 Па.
При термообработке оксида стронция в среде кислорода при высоком давлении образуется гидроксид стронция; давление диссоциации пероксида стронция: 1,01-105 Па (365° С), 5,07-Ю6 Па (500° С), 1.01-107 Па (600° С).
При взаимодействии с водой оксид стронция образует гидроксид Sr(OH)2 — бесцветные гигроскопичные кристаллы тетрагональной сингонии; температура плавления 535° С; плотность 3,63 г/см3; С° = = 92,06 Дж/(моль-К); ДЯ^р = 965 кДж/моль; 52°98 = 94 Дж/(моль-К). В воде образует кристаллогидраты Sr(OH)2-nH2O, где п = 1,8. Октагидрат гидроксида стронция — бесцветные кристаллы тетрагональной сингонии (а = 0,897 нм, с = 0,1115 нм); плотность 1,90 г/см3; может находиться в равновесии с насыщенным водным раствором гидроксида стронция, при нагревании в сухом воздухе переходит в моногидрат, при 100° С в безводный гидроксид, а при 400° С переходит в оксид стронция; растворимость октагидрата стронция в воде (в 100 г в расчете на оксид стронция): 0,35 (0° С); 0,69 (20° С); 2,18 (50° С); 24,2 (100° С). В процессе обработки кристаллического гидроксида стронция пероксидом водорода образуется октагидрат пероксида стронция.
Оксид стронция применяется в качестве компонента оксидных катодов (эмиттеров электронов в электровакуумных приборах), стекла кинескопов цветных телевизоров (поглощает рентгеновское излучение), эмалей и глазурей, высокотемпературных сверхпроводников, пиротехнических составов; его применяют для выделения сахара из патоки (гидроксид), в качестве исходного сырья для получения элементного стронция.
7*
195
4.6. ТИТАНАТ СТРОНЦИЯ
Титанат стронция SrTiOj, бесцветные кристаллы с кубической решеткой типа перовскита (а - 0,3904 нм, пространственная группа РтЗт); при температуре Кюри (10 К) переходит в тетрагональную модификацию (пространственная группа Р4/ттт); ниже 100 К происходит двойникование (образование областей с различной ориентацией кристаллической структуры), двойники, параллельные в направлении [011], с шириной 10—50 мкм являются сегнетоэлектрическими доменами (области однородной спонтанной поляризации); температура плавления 2040° С; плотность 5,11 г/см3; С° = 98,27 Дж/(моль-К); ДН^р= -1668,5 кДж/моль; 5°98 = 108,7 Дж/(моль-К); показатель преломления п™ = 2,37 (X = 700,0 нм) уменьшается при нагревании, температурный коэффициент = 6,2-10’5 К’1 (1 = 546,0 нм); температурный коэффициент линейного расширения 9,2 10'6 К'1 (298 К), g при 20° С для поликристаллических образцов 270, для монокристаллов 300—400; tgS = 2,5-1 О’4 (20° С); спонтанная поляризация Ps 1,5-10'6 Кл/см3 (1,4 К) при напряженности электрического поля Ес - 670 В/см; при 4,2 К остаточная поляризация 1-10'6 Кл/см2 при Ес = 300—500 В/см; в ИК-спектре максимум поглощения при X = 5000 см'1. Титанат стронция не растворяется в воде, растворяется в горячей концентрированной серной кислоте.
Титанат стронция получают спеканием индивидуальных оксидов стронция и титана при 1200—1300° С или соосажденных труднорастворимых соединений стронция и титана (например, карбонаты, сульфаты и др.) выше 1000° С. Монокристаллы титаната свинца выращивают из растворов в расплаве, методом Вернейля, пленки — катодным распылением, вакуумным напылением.
Титанат стронция применяют в технике СВЧ в качестве диэлектрических антенн, фазовращателей, параметрических усилителей, генераторов гармоник [керамика (Ва^з 8г0>27)Т1Оз]. Пленки используют в не- ! линейных конденсаторах, для изготовления миниатюрных емкостных термодатчиков, датчиков ИК-излучения [пленка (ВаобЗгозэтЬаооозУГЮз], слоистых структур металл — диэлектрик — полупроводник — диэлектрик — металл (линии задержки, фотоприемники, запоминающие устройства и др.).
ГЛАВА 5
ЦИНК И ЕГО СОЕДИНЕНИЯ
5.1. ЦИНК
Содержание цинка в земной коре 7-10’3% (масс.), в океанической воде 0,01 мг/л. Известно более 70 цинксодержащих минералов, важнейшими из которых являются: сфалерит (цинковая обманка) — кубическая модификация ZnS, его светлая разновидность — клейофан, черная— марматит; вюрцит — гексагональная модификация ZnS; смитсонит ZnCCh; каламин Zn^OH^SiiCh-HjO; цинкит ZnO; виллемит Zn2SiO4; франклинит ZnFe2O4. Минералы цинка обычно ассоциированы с минералами свинца и меди в полиметаллических рудах. Постоянными примесями цинка в рудах являются кадмий, индий, германий, галлий и таллий.
Природный цинк состоит из пяти стабильных нуклидов: 64Zn (48,6%), 66Zn (27,9%), 67Zn (4,1%), 68Zn (18,8%) и 70Zn (0,6%). Известен ряд радиоактивных нуклидов, важнейшим из которых является 65Zn с Т\/г 244 сут.
Конфигурация внешних электронных оболочек атома цинка Зс/10 4s2; степень окисления +2; энергия ионизации при последовательном переходе от Zn° к Zn3+ 9,39, 17,96 и 39,70 эВ; сродство к электрону 0,09 эВ; электроотрицательность по Полингу 1,66; атомный радиус 0,139 нм, ионный радиус Zn2+ 0,060 нм (4), 0,068 нм (5), 0,0740 нм (6), 0,090 нм (8) (в скобках указано координационное число).
Цинк — голубовато-белый металл. Кристаллическая решетка гексагональная полуупакованная, а - 0,26649 нм, с = 0,49468 нм, г = 2, пространственная группа C&mmnr, температура плавления 419,58° С; температура кипения 906,2° С; плотность 7,133 г/см3; С? = 25,4 Дж/(моль-К); А//9, = 7,2 кДж/моль; ДЯ°СП = 115,3 кДж/моль, =41,6 Дж/(моль К); уравнения температурной зависимости давления пара: 1g р (мм рт. ст.) = 10,084—6910/Г+0,192 1g Г+0,524-10'2Т (298—692,7 К), 1g р (мм рг. ст.) = 8,242—6294/7’—0,015 IgT7 (692,7—1164 К), температурный коэффициент линейного расширения гб.О Ю^К-1 (273—373 К); теплопроводность 116,0 Вт/(м-К); р = 5,92 мкОм см, температурный коэффициент р = 3,7- 10'3К'’ (298—398 К). Ниже температуры 0,825 К цинк — сверхпроводник. Диамагнитен, магнитная восприимчивость 0,17510’9.
197
Цинк хрупок при комнатной температуре, а при нагревании до 100—150° С становится пластичным и прокатывается в тонкие листы и проволоку. При 200—250° С снова становится хрупким и может быть истолчен в порошок. Компактный цинк тускнеет при хранении на воздухе, покрываясь тонким слоем оксида цинка. Влажный воздух, особенно в присутствии диоксида углерода, постепенно разрушает цинк при комнатной температуре. При сильном нагревании на воздухе цинк сгорает с образованием оксида. В процессе красного каления цинк реагирует с парами воды с образованием оксида цинка с выделением водорода.
Цинк технической чистоты взаимодействует с кислотами с образованием соответствующих солей и с растворами щелочей, образуя гидроцинкаты, например Na2[Zn(OH)4], растворимые в водных растворах аммиака и солей аммония, хлорида железа (III), вытесняет медь, кадмий и другие более электроположительные металлы из водных растворов их солей. Цинк высокой чистоты практически не реагирует ни с кислотами, ни с растворами щелочей. Он не взаимодействует с водородом, однако водород незначительно растворяется в цинке при повышенных температурах. Электролитический цинк может содержать до 1 см3 Н2 на 1 г металла. Цинк с азотом не реагирует, с аммиаком при 550—600° С образует устойчивый на воздухе, но разлагающийся водой нитрид цинка. Кипящий цинк растворяет незначительное количество углерода. Получены тройные карбиды, например Ni3ZnC. В расплавленном цинке растворяется до 15% элементного фосфора. При действии паров фосфора на цинк в процессе нагревания образуются фосфиды Zn3P2 и ZnP2. Получены также арсениды аналогичного состава, а при сплавлении с сурьмой — антимониды Zn3Sb2, Zn4Sb3 и ZnSb. Все соединения цинка с фосфором, мышьяком и сурьмой являются полупроводниками.
Соли цинка в водных растворах сильно диссоциированы и вследствие гидролиза имеют кислую реакцию. В процессе действия растворов щелочей и аммиака осаждаются, начиная с рН~5, гидроксосо-ли, например Zn2(OH)2SO4, переходящие в гидроксид Zn(OH)2, который растворяется в избытке осадителя.
Для цинка характерны комплексы с аммиаком. Сухие соли цинка поглощают до шести молекул NH3. Гидроксид и соли цинка растворяются в водных растворах аммиака с образованием комплексных катионов, содержащих от 1 до 6 молекул аммиака. Из растворов в кристаллическом виде выделены [Zn(NH3)2]X2 и [Zn(NH3)4]X2. Труднорастворимый цианид цинка Zn(CN)2 с избытком щелочных металлов образует легкорастворимые комплексы M2[Zn(CN)4] и M[Zn(CN)3], сульфит-комплексы M2[Zn(SO3)4] и M[Zn(SO3)4], Хорошо растворимые в воде тиосульфат и тиоцианат цинка также образуют соответствующие комплексы [Zn(S2O3)2]2' и [Zn(SCN)4]2'.
198
Фосфид цинка (табл. 5.1) разлагается горячей водой и кислотами с выделением РН3. Применяется в качестве зооцида.
Антимонид цинка не растворим в воде и органических растворителях, медленно реагирует с минеральными кислотами. Монокристаллы ZnSb выращивают направленной кристаллизацией, зонной плавкой или вытягиванием по Чохральскому. Он является полупроводником с шириной запрещенной зоны &Е 0,61 эВ, применяется в .качестве материала для термоэлектрических генераторов.
Кобальтат цинка (зелень Ринмана) состава от ZnCo2O4 до ZnCoO2 представляют собой зеленые кристаллы со структурой типа шпинели. Получают их спеканием оксидов цинка и кобальта или термообработкой совместно осажденных гидроксокарбонатов. Применяется в качестве пигмента для керамики.
Борат Zn[B2O3(OH)5]-H2O в процессе термообработки обезвоживается до Zn2B60n, который при 610° С разлагается. Получают взаимодействием ортоборной кислоты с гидроксидом или гидроксокарбона-том цинка. Борат и его гидраты применяются в качестве антипиренов для тканей и бумаги, пигментов в лакокрасочных покрытиях, являются компонентами люминофоров, а также флюсов в процессах пайки и сварки металлов.
Карбонат цинка ZnCCh при нагревании выше 150° С разлагается. Не растворим в воде (5,7Ю‘5%, масс.) и в органических растворителях, а при кипячении в воде превращается в гидроксокарбонат. Хорошо растворяется в кислотах, растворах щелочей и солей аммония.
Карбонат цинка получают по реакции водных растворов сульфата цинка с гидрокарбонатом калия, насыщенным диоксидом углерода с выдержкой на холоде. Карбонат цинка в природе — минерал смитсонит.
Гидрокарбонат цинка имеет переменный состав. Выделены Zn2(OH)2CO3-2H2O, Zn4(OH)6CO3-nH2O, гп5(ОН)б(СОз)2 (минерал гидроцинкит) и др. Растворимость в воде ~1-10’3% масс. При нагревании до 140° С разлагается. Получают кипячением водных растворов карбоната натрия и сульфата цинка.
Карбонаты цинка применяются в процессах получения других соединений цинка.
Ортосиликат цинка Zn2SiO4 не растворяется в воде и органических растворителях, растворяется в 20%-ной фтороводородной кислоте, разлагается хлороводородной кислотой.
Ортосиликат цинка получают взаимодействием: 1) тетрахлорида кремния с оксидом цинка; 2) обжигом смеси диоксида кремния, хлорида цинка и хлорида натрия в присутствии паров воды; 3) гидротермальным синтезом из оксида цинка и диоксида кремния в водных растворах карбоната натрия. Ортосиликат цинка в природе — минерал виллемит. Применяется в качестве люминофора.
199
Таблица 5.1. Свойства некоторых соединений цинка
Показатель ZnjPa ZnSb Z11AI2O3 Zn[B2O3(OH)5]-•Н2О ZnCO3 Zn2SiO4 Zn3(PO4)2 Zn3(AsO4)2-4H2O ZnWO4
Цвет Серо-стальной Серый Зеленый Бесцветный Бесцветный Бесцветный Бесцветный Бесцветный Бесцветный
Сингония Тетрагональная* Ромбическая Кубическая Ромбическая Тригональная Тригональная Моноклинная*** Триклинная**** Моноклинная*****
Параметры решет-
ки, нм:
а 0,8097 0,6212 0,80883 0,755 0,465 1,394 0,814 0,599 0,469
b — 0,7741 — 0,895 — — 0,563 0,763 0,574
с 1,145 0,8115 — 1,010 1,503 0,9309 1,504 0,543 0,496
Число формульных единиц в ячейке — 8 8 4 6 18 4 1 2
Пространственная группа Ptynmc РЬса Fd3m Рпта R3c R3 С2/с — РИс
Т °C 1193 546 1950** — — 1512 1060 — 1200
Плотность, г/см3 4,54 6,36 4,58 2,44 4,40 4,103 4,0 3,79 7,79
С", Дж/(моль-К) — — — — 80,1 123,3 — — 126
кДж/моль 190,6 -16,8 -2067 — -818 -1641 -2900 — -1230,6
S"w, Дж/(моль-К) — — — — 82,4 131,4 — — 129,9
♦Температура перехода 880° С. **Температура разложения. ***р = 105,13°.
При 940° С переходит в другую моноклинную форму. ****а = 94,18°, р = 91,12°, I = 92,6°. ♦♦♦♦♦р = 89,5°.
Ортофосфат цинка Zn3(PO4)2 не растворяется в воде и органических растворителях. Растворяется в разбавленных кислотах, водных растворах щелочей и аммиака. Тетрагидрат при 110° С теряет две молекулы воды, а при 210° С обезвоживается полностью.
Ортофосфат цинка получают взаимодействием водных растворов сульфата цинка и гидрофосфата натрия или растворением оксида цинка в ортофосфатной кислоте. Безводный продукт получают взаимодействием оксида цинка с гидрофосфатом аммония. Тетрагидрат ортофосфата цинка — в природе минерал гопеит. Является компонентом композиционных и смазочных материалов, катализаторов органического синтеза, антикоррозионных пигментов, люминофоров и цинковых удобрений.
Ортоарсенат цинка Zn3(AsO4)2-4H2O при нагревании до 150° С переходит в моногидрат, а при 290° С обезвоживается полностью. Известен также октагидрат — в природе минерал кёттигит. Не растворяется в воде и органических растворителях, растворяется в кислотах, водных растворах щелочей и аммиака.
Ортоарсенат цинка получают взаимодействием растворов сульфата цинка и Na2HAsO4. Применяется в качестве антисептика для древесины, инсектицида и компонента необрастающих красок.
Получение цинка. Исходным сырьем в производстве металлического цинка являются сульфидные цинковые и полиметаллические руды. В производстве применяют как гидрометаллургические, так и пирометаллургические методы получения цинка. Около 85% производимого цинка получают гидрометаллургическим способом, по которому исходные цинковые концентраты после флотационного обогащения для удаления серы обжигают в печах кипящего слоя или во взвешенном состоянии. Образующийся огарок обрабатывают отработанным электролитом, содержащим серную кислоту. Полученный водный раствор сульфата цинка очищают от железа обработкой оксидом цинка или избытком исходного огарка (стадия «нейтрального выщелачивания»). Совместно с железом осаждаются As, Sb, Al, In, Ga и др. Примеси меди и кадмия, а также никеля отделяют действием цинковой пыли с получением так называемого медно-кадмиевого кека. Примесь кобальта удаляют осаждением а-нитрозо-Р-нафтолом либо этил-ксантогенатом натрия или калия. От хлора очищают либо сульфатом серебра, либо сульфатом меди и цинковой пылью.
Из очищенного раствора цинк осаждают электролитическим путем на алюминиевых катодах. Отработанный электролит возвращают на выщелачивание, остатки от выщелачивания («цинковые кеки») обычно содержат значительные количества цинка в виде малорастворимых сое-201
динений (феррит, свинец и другие металлы). Эти кеки либо дополнительно выщелачивают более крепкой серной кислотой, либо подвергают вельцеванию — обжигу в смеси с коксом в барабанных вращающихся печах при 1200° С, в результате чего образуются возгоны оксидов цинка и свинца — «вельц-оксиды». Эти возгоны перерабатывают гидрометаллургически, подобно вышеописанному, в отдельной технологической схеме с попутным выделением концентрата индия и других редких элементов.
Пирометаллургическое производство цинка также начинают окислительным обжигом, но с получением кускового материала — либо обжигом на ленточной агломерационной машине, либо спеканием порошкообразного огарка. Агломерат в смеси с углем или коксом восстанавливают при температуре выше температуры кипения цинка. Для этих целей используют либо ретортные печи (при этом цинк отгоняется, но шихта полностью не расплавляется), либо шахтные или электрические рудно-термические печи, в которых шихта полностью расплавляется. Во всех случаях пары металлического цинка конденсируются. Наиболее летучая фракция, обогащенная кадмием — так называемая пуссьера,— собирается отдельно и перерабатывается для извлечения кадмия. Твердые остатки в ретортных печах — «раймов-ки» — перерабатываются, например вальцеванием.
Для очистки цинка применяют ликвацию (отделение свинца и железа), двухступенчатую ректификацию (отделение свинца и кадмия, а также меди, железа и др.) и химические методы, в частности удаление свинца действием металлического натрия и удаление железа действием алюминия под слоем флюса. Цинк, полученный гидро-металлургичеким путем, переплавляют с добавлением флюса NH4CI, что способствует удалению Т1 и др.
Цинк высокой чистоты получают дистилляцией в инертной атмосфере или в вакууме, ректификацией и зонной перекристаллизацией в атмосфере аргона. Разработан также метод электролитического рафинирования, в частности с амальгамными электродами.
Применение. Цинк используют в качестве антикоррозионного покрытия железа и стали. Листовой цинк применяют в аккумуляторных и сухих элементах, в типографском деле. В металлургии цинк применяют для отделения свинца от серебра и золота. Цинковая пыль используется для выделения кадмия, индия, золота и т.п. из растворов цементацией. Цинк применяется также в качестве восстановителя в органическом синтезе, в производстве пигментов (цинковые белила). Примерное распределение цинка по областям применения (в %): цинкование и покрытие сплавами цинка — 45; химические источники тока — 20; латуни и бронзы—15; сплавы на основе цинка — 12; пигменты и пр.— 8.
202
5.2. ОКСИД И ГИДРОКСИД ЦИНКА
Оксид цинка ZnO, бесцветные кристаллы гексагональной сингонии (а = 0,32495 нм, с = 0,52069 нм, z = 2, пространственная группа P&Jmc) с плотностью 5,7 г/см3; при нагревании принимает желтую окраску. Плавится под давлением 52 ат при 2000° С. Температура возгонки 1800° С; С° = 40,28 Дж/(моль-К); ДЯ^р=-350,80 кДж/моль; 52°98 = 43,67 Дж/(моль-К); показатели преломления 2,015 и 2,068. Восстанавливается до металлического цинка углеродом и его оксидом, водородом, метаном, карбидом кальция и ферросилицием при нагревании до 1000° С. Оксид цинка амфотерен. С щелочами образует гидроксо-цинкаты, например Na[Zn(OH)3], Na2[Zn(OH)4], Ba2[Zn(OH)6], с растворами солей — простые или двойные гидроксосоли Zn(OH)NO3, Zn5(OH)8C12-2H2O, Zn2Cr(OH)6NO3-xH2O. В процессе термообработки с диоксидом кремния и сесквиоксидом бора — стекловидные Zn2SiO4 и Zn(BO2)2, с оксидами металлов — цинкаты, например CoZnO2-
Оксид цинка встречается в природе в виде минерала цинкита. Применяется в качестве белого пигмента для красок (цинковые белила), является активатором вулканизации и наполнителем в резиновой промышленности, компонентом косметических препаратов — кремов, пудры, лекарственных средств (мазей, паст, присыпок при кожных заболеваниях), зубных цементов. Применяется также в качестве катализатора синтеза метанола, полупроводникового материала и компонента люминофоров.
Гидроксид цинка Zn(OH)2 — бесцветные кристаллы или аморфное вещество, существующее в виде пяти полиморфных модификаций, из которых устойчивой является лишь ромбическая сингония. Малорастворим в воде, амфотерен.
Оксид и гидроксид цинка легко растворяются в кислотах с образованием соответствующих солей. Гидроксид цинка растворяется в водных растворах аммиака, причем его растворимость возрастает с увеличением концентрации аммиака вследствие образования комплексных гидроксидов [Zn(NH3)2](OH)2. В процессе растворения гидроксида цинка в разбавленных растворах гидроксида натрия образуются ионы HZnO'2 и ZnO2’2. При 25° С первая константа кислотной диссоциации Zn(OH)2[Zn(OH)2(ra) = Н+ + ZnO‘2] равна 1,2-10'17. Вторая константа диссоциации [Zn(OH)2(™) = 2и + ZnO2’2] равна 2,210’30. Соответственно константа диссоциации HZnO’2[HZnO‘2 = = Н+ + ZnO2‘2] равна 1,83-10‘13.
В процессе нагревания гидроксид цинка при 35° С начинает разлагаться на оксид цинка с выделением воды. Процесс протекает интенсивно при 100—160° С, а заканчивается лишь при 200° С.
203
Гидроксид цинка применяют для получения различных соединений цинка.
Способы получения оксида цинка. Существует несколько способов получения оксида цинка. Они отличаются друг от друга в зависимости от вида и качества исходного сырья, аппаратурного оформления, а также назначения целевого продукта. Обычно оксид цинка получают из металлического цинка, из его карбоната, нитрата, оксалата и оксидов.
Получение оксида цинка из металлического цинка. Оксид цинка, получаемый из металлического цинка, выпускается различных марок с содержанием основного вещества от 98 до 99,7%. Основными примесями являются оксиды кадмия и свинца (0,015—0,3%), водорастворимые соли (0,1—0,5%) и металлический цинк (0,04—-0,06%). Оксид цинка, используемый в качестве цинковых белил для масляных красок, содержит 91—95% основного вещества и значительное количество оксидов свинца и водораствори-1 мых солей, хотя и цинковые белила выпускаются нескольких сортов: с малым (5—6%) и большим содержанием свинца (15—20%), а в США выпускаются белила с содержанием до 35% свинца.
Практически установлено, что оптимальный размер частиц оксида цинка, применяемых в качестве белил для лакокрасочной промышленности 0,4—0,6 мкм и игольчатая форма частиц, повышает ат-мосферостойкость пленок.
Оксид цинка, полученный из цинксодержащего сырья, имеет частицы игольчатой формы (рис. 5.1).
В промышленности выпускаются специальные марки оксида цинка, различающиеся по среднему размеру частиц и соответственно физико-химическим свойствам, а также по составу.
Процесс получения оксида цинка из металлического цинка состоит из следующих основных стадий: испарение цинка; окисление паров цинка; охлаждение взвеси оксида цинка в газах; осаждение оксидов цинка из газов.
б) в)
Рис. 5.1. Микрофотография оксида цинка (хЮООО), полученного:
а — из металлического цинка; б — из цинкосодержащего сырья; в — с частицами игольчатой формы 204
В стадии испарения цинка применяются муфельные или вращающиеся барабанные печи.
Сырьем для получения оксида цинка является металлический цинк. Обычно применяют электролитический цинк марки ЦО с содержанием основного вещества 99,975%, марки Ц1—99,95% и в отдельных случаях марки Ц2—98,7% цинка.
Рис. 5.2. Муфель с постоянным уровнем цинка:
1 — цилиндрическая часть муфеля; 2 — передняя торцевая часть муфеля; 3 — перегородка; 4 — отверстие в перегородке; 5 — задняя глухая торцевая стенка муфеля; 6 — отверстие для загрузки чукового цинка
Плавку металлического цинка и его испарение проводят в муфелях (рис. 5.2), представляющих собой цилиндр 1 из огнеупорного материала. С одного конца муфель закрыт стенкой 5, а другой конец имеет полукруглое отверстие 2, через которое загружают металлический цинк и из которого выходят пары цинка. Постоянство величины зеркала испарения обеспечивается в круглом муфеле. В тыльной части муфеля устанавливают перегородку 3 с небольшим отверстием снизу 4. Отверстие 6 служит для загрузки цинка. Такой способ загрузки исходного цинка улучшает технологию оксида цинка в муфельных печах. Обычно используют печи, в которых муфели установлены в один или два ряда. На рис. 5.3 приведены план и разрезы печи с 18 муфелями, расположенными в один ряд. Существуют также печи с большим числом муфелей (до 28). Газы от сгорания
топлива поступают в печное пространство с температурой
1300—1350° С и отходят от печей с температурой 1000—1100° С. С повышением температуры в печи естественно повышается и ее производительность. Для равномерности нагревания муфелей продуктами сгорания газа последний сжигается в горелках, расположенных с обоих торцов печи.
Продукты сгорания газа уходят в дымоход через колодец, находящийся в центре пода. Цинковые пары, попадая в окислительный колодец, загораются. Для образования цинковых белил с частицами игольчатой формы в муфель и в окислительный колодец подводят газ.
Положительным качеством этих печей является то, что в них па
ры цинка и оксида цинка не соприкасаются с продуктами сгорания топлива, вследствие чего целевой продукт получается высокой чистоты. Образовавшаяся взвесь оксида цинка попадает в общую для всех колодцев уравнительную камеру 5 (рис. 5.4), расположенную рядом с печью. Каждый окислительный колодец 3 соединяется с уравнительной камерой отдельным коротким соединительным рукавом 4.
205
Рис. 5.3. Муфельная печь для производства оксида цинка
Рис 5.4. Схема получения оксида цинка муфельным способом:
1 — муфель; 2 — муфельное пространство; 3 — окислительный колодец; 4 — соединительный рукав; 5 — уравнительная камера; 6— продуктопровод; 7 — эксгаустер; 8 — рукавный фильтр;
9— шнек; 10 — элеватор; 11—упаковочная машина; 12 — вентилятор вытяжной
В уравнительной камере взвесь оксида цинка в газах, поступающая из всех окислительных колодцев, перемешивается и ее средняя температура обычно поддерживается ~800° С. Уравнительная камера, расположенная рядом с печью, выложена из огнеупорного кирпича. Ее длина равна длине печи. Дно камеры состоит из ряда бункеров.
Поскольку уравнительная камера является более расширенным участком пути, по которому движется взвесь оксида цинка в воздухе, скорость последней в уравнительной камере снижается, в результате чего более грубодисперсные частицы выпадают из воздушного потока и осаждаются в количестве до 1,5% в бункерах, камеры которых могут быть соединены общим шнеком 9, позволяющим периодически разгружать их.
Из уравнительной камеры взвесь оксида цинка в воздухе после охлаждения направляется для улавливания его в систему рукавных фильтров 8 с автоматическим встряхиванием. Оксидопровод 6, соединяющий уравнительную камеру с системой фильтров, служит для охлаждения взвеси оксида цинка в воздухе, поскольку при температурах выше 100° С может воспламеняться ткань фильтров.
Для снижения температуры оксидопровод делают длиной 250—350 м и прокладывают его снаружи здания. Эксгаустер 7 транспортирует взвесь оксида цинка в газах по оксидопроводу. При необходимости для снижения температуры взвеси в оксидопровод до эксгаустера через патрубок, снабженный шибером, засасывается необходимое количество холодного воздуха. При этом увеличивается фильтрующая поверхность. Охлажденная примерно до 100° С взвесь оксида цинка в газах поступает в рукавные фильтры 8 с автоматическим встряхиванием.
207
В процессе прохождения воздуха через ткань фильтра содержащие- I ся в воздухе частицы оксида цинка задерживаются, а воздух проходит I в металлический кожух и вентилятором 12 выбрасывается в атмосферу. 1
Оксид цинка, осевший на фильтрующей перегородке, встряхива- I нием последней периодически выгружается в днище фильтра, откуда системой шнеков 9 и элеватора 10 их подают в бункер упаковочной Я машины И. II
В некоторых производствах, применяющих в качестве исходного I сырья металлический цинк с большим содержанием свинца, приме- 1 няется схема, позволяющая разделять улавливаемые частицы оксида 1 цинка по содержанию в них оксида свинца. |
Разработан способ коагуляции взвеси оксида цинка в низкочас- I тотном акустическом поле. Согласно схеме, от основного оксидопро- I вода взвесь белил в воздухе с концентрацией 20—40 г/м3 подается в I камеру озвучивания. Электросиреной создаются мощные акустиче- I ские колебания. Камера озвучивания сконструирована по типу цикло- I на, из которого взвесь выводится через центральную трубу к цикло- j I нам и далее к контрольным фильтрам. В качестве источника звука I частоты 400 Гц/с используется модификационная электросирена 1 С-43, установленная над камерой и отделенная от ее рабочего обье- j ма звукопроводящей пленкой. Для звукоизоляции электросирены и 1 камеры озвучивания применяют асбест. |
Производимый оксид цинка, согласно требованиям существующе- 1 го ГОСТ 3640—85, должен содержать не менее 99—99,7% целевого ] продукта, оксида свинца от 0,015 до 0,3, потери при прокаливании 1 (П.П.П.) не более 0,3. 1
В производстве оксида цинка применяются также и барабанные '•< вращающиеся печи, идентичные широко применяемым в химической 1 промышленности. <
Получение оксида цинка из цинксодержащего сырья. В процессе получения оксида цинка по способу Витериля в качестве исходного сырья применяют окисленные цинковые руды (смитсонит ZnCO3) и различные цинксодержащие отходы, содержащие 30—50% оксида цинка. Исходное сырье в смеси с углем в процессе термообработки при 1300° С восстанавливает оксид цинка до металлического цинка, образующегося в парообразном состоянии. Далее пары цинка окисляются кислородом воздуха с получением оксида цинка.
Карбонат цинка (смитсонит), применяемый в качестве исходного сырья, при 300° С разлагается с образованием оксида цинка:
ZnCO3 зоо'с > ZnO + СО2 208
Выход суспензии
Рис. 5.5. Печь непрерывного действия с цепной решеткой:
1 — цепная колосниковая решетка; 2 — бункер для угля; 3— камера розжига; 4 — бункер для шихты; 5— резервуар для воды; 6 — камера печи; 7—окислительная камера; 8—шламонакопитель; 9 — дутьевые коробки; 10—бункеры для продуктов, проходящих через решетку; 11 — патрубок
для вывода шлаков
Принципиальная схема печи непрерывного действия приведена на рис. 5.5, согласно которой на колосниковую решетку 1 из бункера 2 загружаются угольные (или антрацитовые) брикеты или гранулы с толщиной слоя 10 см. В процессе движения решетки уголь поступает в камеру розжига 3, в которую постоянно поступает воздух. В камере температура при сгорании брикетов достигает 1250—1300° С. Газы, образующиеся в камере 3 при частичном сгорании угля, выбрасываются в атмосферу. При дальнейшем движении колосниковой решетки 1 в следующей камере на подушку из раскаленного угля из бункера 4 поступают брикеты из шихты или гранул, образующие слой толщиной 12—15 см. Брикеты состоят приблизительно из 20% угля и 80% цинксодержащего сырья. С целью получения прочных, не пылящих брикетов в процессе брикетирования в шихту добавляют раствор сульфита натрия.
Рабочая зона колосниковой решетки заключена в камере печи 6, в которой происходит процесс восстановления оксида цинка в пары цинка при 1100° С. Образующийся на цепной решетке шлак сбрасывается специальным шлакоснимателем 8 через патрубок 11.
Во избежание зашлаковыва-
Рис. 5.6. Схема получения оксида цинка из агломерированного цинкосодержащего сырья на установке с шахтной электропечью непрерывного действия:
1 — бункер для кокса; 2—бункер для агломерата; 3 — печь термообработки; 4 — форсунка; 5 — питатель; 6 — электроды; 7 — водоохлаждаемый обод; 8 — шлаки; 9 — вращающийся разгрузочный стол; 10 — отверстия для подачи воздуха (воздуховоды); 11 — транспортер для удаления шлаков; 12 — камера окисления; 13 — вентилятор; 14 — циклон; /5 — рукавные фильтры
ния боковых стенок камеры в них размещены трубы, в которых циркулирует химически очищенная вода, поступающая из резервуара 5. Необходимый для горения угля и окисления паров цинка воздух подается через дутьевые коробки 9. Значительная часть паров цинка окисляется в камере 6, а образующаяся в ней взвесь оксида цинка в газах поступает в окислительную камеру 7, из которой затем с воздухом направляется в улавливающую систему.
Оксид цинка из обожженных цинковых концентратов получают в шахтных электропечах. При этом обожженный сульфид цинка подвергают двукратной термообработке с целью максимального освобождения исходного сырья от свинца, кадмия и серы. Полученный агломерат содержит 56% цинка, 0,004% свинца, 0,003% кадмия и 0,1% серы.
210
В шахтную электропечь непрерывного действия (рис. 5.6) загружают шихту, состоящую из 70% агломерата и 30% кокса. Кокс из бункера 1 и агломерат из бункера 2 поступают в печь термообработки 3, из которой шихта, нагретая до 800° С, питателем 5 подается в шахтную дуговую электропечь. Из печи пары цинка и оксид углерода с температурой около 900° С поступают в камеру окисления 12. Взвесь полученного оксида цинка в газообразном состоянии вентилятором 13 подается в циклон 14, а затем в рукавные фильтры 15, а оттуда на упаковку. Шлаки, скапливающиеся на вращающемся разгрузочном столе 9, сбрасываются на транспортер 11. Производительность описанной печи по оксиду цинка составляет 30 т/сут. Общая потребляемая мощность печи 3000 кВт. Химический состав (в %) оксида цинка, полученного на описанной установке:
Оксид цинка 98—99,5 Водорастворимые соединения 0,7
Свинец» Кадмий <о,з 0,01 Остаток, не растворимый в НС1 0,15
Хлор Триоксид 0,02 0,35 Остаток при прокаливании 0,3
Регулируя температуру процесса окисления от 200 до 1000° С, при необходимости меняют размер частиц оксида цинка от 0,01 до 1,0 мкм.
5.3. ХАЛЬКОГЕНИДЫ ЦИНКА
Халькогениды цинка (табл. 5.2) при атмосферном давлении существуют в виде двух модификаций — стабильной кубической со структурой типа сфалерита (рис. 5.7, а) (пространственная группа F43m, z = 4) и метастабильной гексагональной типа вюрцита (рис. 5.7, б) (пространственная группа РбЗтс, z = 2).
Возможно получение кристаллов со структурами, включающими кубические (трехслойные) и гексагональные (двухслойные) упаковки. При высоких давлениях халькогениды цинка переходят в другие кубические модификации со структурой типа CsCl. Халькогениды цинка могут иметь отклонения от стехиометрии. Они испаряются конгруэнтно с диссоциацией в парах на компоненты.
Модификации халькогенидов цинка, существующие при атмосферном давлении,— широкозонные полупроводники. У них наблюдается пьезоэлектрический эффект. Модификации высокого давления обладают металлической проводимостью. Халькогениды цинка обладают высокой чувствительностью к электромагнитным волнам, вплоть до самих коротких, поэтому они применяются в качестве люминофоров, сцинтилляторов, материалов ЙК-оптики и т.п.
211
a)
Рис. 5.7. Кристаллическая структура с тетраэдро-тетраэдрической координацией атомов:
а — сфалерит; б — вюрцит
Сульфид цинка ZnS не плавится, однако с сульфидами других металлов (PbS, Cu2S, FeS и др.) образует легкоплавкие штейны. Температура воспламенения на воздухе 755° С. В процессе нагревания ZnS сублимируется, например при 1000° С за 30 мин потеря в массе составляет 0,4%. Под давлением 10’4 мм рт. ст. и 900° С в течение одного часа испаряется -23% сульфида цинка, а при 975° С — 78%. В присутствии восстановителей наряду с сублимацией имеет место также и восстановление сульфида цинка. Скорость процесса сублимации снижается, а скорость процесса восстановления повышается с ростом давления оксида углерода. В среде же водорода, с повышением давления водорода, возрастают и скорость сублимации, и скорость восстановления. Это объясняется степенью скорости диффузии молекул сульфида цинка от твердой поверхности в газовый объем. Для сульфида цинка уравнения температурной зависимости давления пара: для сфалерита 1g р (мм рт. ст.) = 10,571—13846/Г (1095—1435К); для вюрци-та 1g р (мм рт. ст.) = 9,842—13026/Г (1482—1733 К). Переход в кубическую фазу III (а = 0,499 нм при -18 ГПа) наблюдается при давлении 16,4 ГПа, а обратный переход — при 10—11 ГПа.
Сульфид цинка в виде белого аморфного осадка, легко дающего коллоидные растворы, образуется в процессе действия сульфида водорода или сульфида аммония на нейтральные растворы солей цинка. Свежеосажденный сульфид цинка растворяется в сильных минеральных кислотах, но не растворяется в растворах щелочей, аммиака, сульфидов щелочных металлов. После отстаивания свежеосажденный сульфид цинка постепенно кристаллизуется, что ведет к уменьшению растворимости в кислотах. Растворимость сульфида цинка (в форме сфалерита) в воде -6-10"6% по массе. При хранении во влажном воздухе и в виде водной суспензии сульфид цинка окисляется до сульфата. В органических растворителях не растворяется.
212
Таблица 5.2. Свойства халькогенидов цинка
Показатель ZnS ZnSe ZnTe
Цвет Бесцветный Желтый Красный
Сингония Кубическая* Гексагональная Кубическая* ♦ Гексагональная Кубическая Гексагональная
Параметры решетки, нм:
а 0,54109 0,38225 0,5656 0,3996 0,6085 0,4310
с — 0,62613 — 0,6626 — 0,7090
1820 (0,37 1575 (0,053 1305***(0,064
Tw °C МПа) МПа) МПа)
Т °C 1178 1185 — — — —
Плотность, г/см3 4,09 4,08 5,42 — 5,72 —
С", Дж/(мольК) 45,5 — 50,0 — 49,7 —
АЯждг, кДж/моль 149,8 265,3 — — — —
кДж/моль -205 -192 -164 — -119,2 —
Дж/(мольК) 57,7 — 84 — 92 —
Теплопроводность, Вт/(см-К) 0,026 — 0,19 — 0,18 —
Коэффициент преломления 2,37 — 2,66 — 2,99 —
Ширина запрещенной зоны, эВ 3,7 3,8 2,7 — 2,24 —
Эффективная масса:
электронов 0,27 — 0,17 — — —
дырок 0,58 — 0,6 — 0,6 —
Подвижность, CM^fB-c):
электронов 200 140 530 — 340 —
дырок 5 — 28 — ПО —
♦Температура полиморфного перехода 1175° С, ЛН перехода 1,35 кДж/моль. ♦♦Температура полиморфного перехода 1145° С, ДН перехода 0,96 кДж/моль. ♦♦♦ДЯ1И = 65 кДж/моль.
Сульфид цинка получают из элементов в режиме самораспростра-няющегося высокотемпературного синтеза:
Zn + S — ZnS
а также гидротермальным методом:
3ZnO + 4S + 2NH3 + Н2О = 3ZnS + (NH4)2SO4
Образующийся при этом сульфид цинка из водных растворов осаждают действием тиомочевины, а из слабокислых растворов (pH 2—3) — действием сульфида водорода.
Монокристаллы сульфида цинка выращивают: 1) из расплава методом направленной кристаллизации под действием аргона, из раствора в расплаве, например хлорида свинца, осаждением из газовой фазы — в результате возгонки, взаимодействующих паров компонентов или транспортными реакциями с 12 или NF^Cl в качестве носителя; 2) гидротермальным методом — из раствора Н3РО4 или КОН. Пленки сульфида цинка выращивают обычно напылением.
Сульфид цинка применяют в качестве компонента белого пигмента, он является люминофором для экранов электронно-лучевых и рентгеновских трубок, сцинтилляторов, полупроводниковым материалом и т.п. Природные минералы сфалерит и вюрцит являются основ
ным сырьем для извлечения металлического цинка.
Дисульфид ZnS2 — кристаллы с кубической структурой типа пирита (рис. 5.8) (а = 0,5942 нм, z = 4, пространственная группа РаЗ) с плотностью 5,56 г/см3.
Дисульфид цинка получают взаимодействием сульфида цинка с се-
рой под давлением 6,5 ГПа и 400—600°С.
<9zn Qs
Рис. 5.8. Структура ZnS2
Селенид цинка ZnSe — кристаллы кубической модификации. Уравнение температурной зависимости давления пара: 1g р (мм рт. ст.) = 9,436—12140/Г (952—1209 К). Под давлением 13,5 ГПа переходит в кубическую металлическую модификацию (а = 0,511 нм). Влажный ZnSe очень чувствителен к действию воздуха. Высушенный или полученный сухим способом устойчив
на воздухе, окисление его с улетучиванием SeO2 начинается при 300—500° С. Разлагается разбавленными кислотами с выделением H2Se.
214
Селенид цинка получают:
1. Взаимодействием металлического цинка с селеном или сульфида цинка с селенистой кислотой:
Zn + Se = ZnSe
ZnS + HiSeCh = ZnSe + H2O + SO2
а образующуюся смесь подвергают термообработке при 600—800° С.
2. В процессе нагревания ZnS с SeO2 или смеси ZnO, ZnS с селеном:
ZnS + SeO2 = ZnSe + SO2
2ZnO + ZnS + 3Se = 3ZnSe + SO2
3. Термообработкой смеси ZnO с селеном и щавелевой кислотой:
ZnO + Se + НООС—COOH = ZnSe + Н2О + 2СО2
Монокристаллы селенида цинка выращивают направленной кристаллизацией расплава под давлением, осаждением из газовой фазы— возгонкой, взаимодействием паров компонентов или транспортными реакциями. Пленки получают из газовой фазы.
Селенид цинка применяется в качестве лазерного материала и компонента люминофоров (в природе — минерал штиллеит).
Диселенид цинка ZnSe2 со структурой типа пирита (см. рис. 5.8) (а = 0,62930 нм) получают из элементов под давлением 6,5 ГПа при 1000—1300° С.
Теллурид цинка ZnTe в зависимости от способов получения — серый порошок, краснеющий в процессе растирания, или красные кристаллы. При всех изменениях температуры образуется метастабильная гексагональная модификация кристаллов. Получают лишь в газовой фазе. Уравнение температурной зависимости давления пара для кубической модификации: 1g р (мм рт. ст.) = 9,718—11513/7’ (918—1095 К). Под давлением 8,5—9 ГПа превращается в кубическую фазу III, которая при 12—13,5 ГПа переходит в гексагональную металлическую модификацию IV со структурой типа 0-Sn (металл). Устойчив на воздухе. Порошкообразный теллурид цинка разрушается водой. В органических растворителях не растворяется. Разлагается минеральными кислотами с выделением НгТе.
Теллурид цинка получают сплавлением компонентов в атмосфере инертных газов, а также нагреванием смеси оксида цинка с теллуром и щавелевой кислотой:
ZnO + Те + НООС—COOH = ZnTe + Н2О + 2СО2
215
Монокристаллы теллурида цинка выращивают направленной кристаллизацией расплава или вытягиванием по Чохральскому. Применяют также осаждение из газовой фазы — путем возгонки, взаимодействием паров компонентов или транспортными реакциями. Пленки получают из газовой фазы.
Теллурид цинка является материалом для фоторезисторов, приемников ИК-излучения, дозиметров и счетчиков радиоизлучения, люминофором и полупроводниковым материалом, в том числе в лазерах.
5.4. СУЛЬФАТ ЦИНКА
Физико-химические свойства. Сульфат цинка ZnSO4, бесцветные кристаллы ромбической сингонии (а - 0,6731 нм, b = 0,8581 нм, с = 0,4760 нм, z = 4, пространственная группа Рпта); плотность 3,54 г/см3; С ° = 99,14 Дж/(моль-К); АЯ^р =-982,01 кДж/моль; 5298 = 110,62 Дж/(моль-К); АЯ^р =-982,01 кДж/моль; 52°98 = 110,62 Дж/(моль-К).
В процессе нагревания до 600—800° С разлагается до SO3 и оксосуль-фагов (2ZnSO4ZnO и ZnSO4 ZnO), а выше 930° С образуется ZnO. Процесс разложения сульфата цинка ускоряется при добавке 1—2% Ре20з.
Сульфат цинка растворяется в воде и глицерине. Растворимость в воде растет почти линейно с температурой от 27,6% масс, при -7° С до 41,4% при 39° С. В этом интервале температур кристаллизуется гептагидрат ZnSO4-7H2O (цинковый купорос) с ромбической решеткой. При 39—70° С кристаллизуется гексагидрат, растворимость сульфата цинка при этом
47,7% (70° С). Выше 70° С обра-зуется моногидрат ZnSG^-HiO, растворимость сульфата цинка падает до 44% (105° С). Моногидрат обезвоживается при 238° С. Известны также нестабильные тетра-, ди- и гептагидраты (моноклинной сингонии). Насыщенный водный раствор содержит при 0°С—29,4%, при 25° С—36,7%, при 75° С—40,9, при 99° С — 37,7% ZnSO4. Водные растворы сульфата цинка, не содержащие свободной кислоты, при хранении мутнеют вследствие выделения осадка основного сульфата цинка 3Zn(OH)s-ZnSO4-4H2O.
Рис. 5.9. Растворимость в системе
ZnSO4—H2SO4—Н2О. Твердые фазы: acd — ZnSO4-6H2O; выше bed—ZnSO4—Н2О
216
В системе Z11SO4—H2SO4—Н2О при температуре 20° С и общей концентрации сульфата и кислоты 0,25—0,5 г-мол/л образуются комплексные ионы [Zn(SC>4)2]2'. На рис. 5.9 приведена диаграмма растворимости в этой системе, которая позволяет рассчитать выход кристаллов ZnSO4'7H2O.
ЩЛ (Л-л)-100
Из 100 кг исходного раствора кристаллизуется х=----------кг
100- 1,78л
ZnSO4 или 1,78 xZnSO4-7H2O, где А и а — содержание сульфата цинка в растворе до и после кристаллизации, % (масс.).
Изучена система ZnSO4—(NH4)2SO4—Н2О и показано, что при этом образуется двойная соль ZnSO4-(NH4)2SO4-H2O. При 50° С поле ее кристаллизации больше, чем при 35° С. В процессе нагревания до 360° С соль полностью обезвоживается, а выше 370° С разлагается.
В системе ZnSO4—Na2SO4—Н2О образуются двусторонние ограниченные твердые растворы ZnSO4-Na2SO4-4H2O с сульфатами натрия и цинка.
В системе ZnSC>4—(ЫН^ЗОд—Na2SC>4—Н2О при 35° С могут существовать твердые фазы: ZnSO4-(NH4)2SO4-6H2O (наименее растворимое), ZnSO4-Na2SO4-4H2O, Na2SO4-(NH4)2SO4-4H2O, ZnSO^THjO, Na2SO4 и (NH4)2SO4 и др.
Сульфат цинка применяют в производстве вискозы, в качестве микроудобрения и добавки к кормам в сельском хозяйстве, как флотореагент для пропитки древесины, отбеливания бумаги. Он является компонентом глазных капель и электролита в процессе получения металлического цинка электролизом.
Согласно ГОСТ 8723—83, цинковый купорос должен содержать цинка не менее 21,8—22,5% и не более следующих примесей: железа (в пересчете на FeO) — 0,02—0,1; серной кислоты свободной— 0,05—0,1; не растворимых в воде веществ — 0,04—0,3; хлоридов— 0,2—0,3; марганца (в пересчете на МпО) — 0,04—0,2, меди, свинца, кадмия и никеля (в пересчете на РЬ) — 0,01—0,03%.
Способы получения сульфата цинка. Сульфат цинка получают из вторичного сырья и из цинксодержащих руд в процессе обработки их серной кислотой.
В отходах от переработки лома цветных металлов содержится 45—62% цинка. Хорошим сырьем для производства сульфата цинка являются поддувальные отходы в процессе получения оксида цинка (цинковых белил).
Потенциальным источником цинкового сырья являются колчеданные огарки. Целесообразность извлечения цинка из огарков диктуется не только для производства соединений цинка, но и 217
необходимостью очистки огарков от цинка перед переработкой их в металлургии.
Наиболее распространенным способом получения цинкового купороса является растворение в серной кислоте материалов, содержащих цинк и его оксид. Сопутствующие примеси меди, железа, свинца, никеля, олова и другие обусловливают необходимость специальной очистки растворов.
По лучение сульфата цинка из цинксодержащих материалов. Исходное сырье, содержащее цинк и его оксид, размалывают в шаровой мельнице, а затем растворяют в 18—25%-ной серной кислоте. Процесс проводят в реакторах, футерованных кислотоупорным материалом и снабженных метальными устройствами.
Количество теплоты, выделяющееся в процессе предварительного разбавления исходной серной кислоты до необходимой концентрации, а также теплота экзотермических реакций
ZnO + H2SO4 = ZnSO4 + Н2О + 105 кДж
Zn + H2SO4 = ZnSC>4 + Н2 + 167,6 кДж обеспечивают самопроизвольное повышение температуры реакционной массы до 80—100° С без дополнительного подогрева.
С целью ускорения процесса нейтрализации серной кислоты, который замедляется к концу реакции, в реактор загружают избыток цинксодержащего сырья. Образующиеся основные соли цинка разлагают добавкой серной кислоты в конце процесса. Процесс останавливают и фильтруют через барабанный вакуум-фильтр, а в реактор с остаточным сырьем вводят новую порцию серной кислоты и вносят цинксодержащее сырье.
При содержании в сырье значительных количеств свинца и олова, не переходящих в раствор в процессе серно-кислотной переработки, твердый остаток после промывки направляют на переработку для извлечения этих металлов. При содержании в исходном сырье олова образуется плохо фильтрующийся осадок. Поэтому в процессе переработки отходов, содержащих олово, отделяют нерастворившийся остаток не фильтрацией, а отстаиванием. При разбавлении образующейся суспензии до плотности растворов 1,20—1,21 г/см3, что соответствует содержанию в растворе 200—250 г/л сульфата цинка, процесс отстаивания идет с достаточной скоростью. Полное отстаивание достигается лишь в нейтральной среде. В процессе отстаивания кислых растворов в сливе содержатся коллоидные частицы. При продолжительности отстаива-218
ния 10—15 ч получается суспензия с отношением Т:Ж в пределах от 1:1,2 до 1:1,5 (рис. 5.10).
Отфильтрованный раствор содержит 400—420 г/л сульфата цинка и примеси сульфатов железа (II), меди (II), кадмия и никеля, концентрация которых зависит от содержания их в исходном сырье. Очистку раствора сульфата цинка от этих примесей производят в несколько стадий. В первой стадии очищают раствор от железа, вводя в систему окислитель для перевода Fe2+ в Fe3+. В качестве окислителя применяют гипохлорит натрия. После окисления железо (III) осаждают гидроксидом кальция в виде основных солей оксида железа (III). Избыток гидро-
Рис. 5.10. Зависимость скорости осаждения от плотности суспензии
ксида кальция не допускают, поскольку
он осаждает и основные соли цинка. Произведения растворимости: Zn(OH)2— 1,80-10’14 и Ре(ОН)г— 1,64-10‘14 почти равны, а произведение растворимости Ре(ОН)з значительно меньше и равняется 1,1-10’36.
После осаждения соединений железа раствор нагревают до кипения для разрушения небольшого избытка гипохлорита натрия и фильтруют. Осадок от фильтров, как и от предыдущей фильтрации, промывают на фильтре водой для извлечения из него сульфата цинка, а полученные слабые растворы направляют на разбавление исходной серной кислоты в реакторе. После очистки от железа раствор очищают от меди (II), никеля и кадмия. Очистку производят в течение 4—-6 ч путем интенсивного перемешивания раствора с добавлением к нему цинковой пыли. Потенциал у цинка более электроотрицательный, чем у меди, никеля и кадмия. Поэтому металлический цинк вытесняет перечисленные металлы из растворов их солей по реакциям:
Zn + C11SO4 = Си + ZnSO4 + 209,*08 кДж
Zn + NiSO4 = Ni + ZnSO4 + 87,6 кДж
Zn + CdSO4 = Cd + ZnSO4 + 52,4 кДж
В полученном солевом составе наиболее полно с достаточной скоростью протекает цементация меди. Выделение из раствора никеля и кадмия проходит со значительными трудностями, связанными с 219
тем, что они близко стоят в ряду напряжений. При повышенной температуре и интенсивном перемешивании протекает обратный процесс— окисление и растворение кадмия. Одновременно для достижения полноты цементации никеля требуются значительный избыток цинковой пыли и высокая температура.
Используемую для цементации цинковую пыль предварительно обрабатывают небольшим количеством слабой серной кислоты для снятия с поверхности металла тонкого слоя оксида. Выпавшие в виде тонкого шлама металлы отделяют от раствора на фильтре, после чего раствор подвергают вторичной очистке от железа, которое в небольших количествах содержится в цинковой пыли. Отфильтрованный после вторичной очистки раствор содержит 300—350 г/л сульфата цинка и до 10 г/л СГ.
Полученный раствор сульфата цинка содержит небольшие количества основных солей сульфата цинка. Для их разрушения в очищенный раствор сульфата цинка вводят необходимое количество серной кислоты, после перемешивания его направляют на кристаллизацию цинкового купороса или используют в других производствах.
Процесс сушки обычно проводят в сушилках в кипящем слое.
На производство 1 т цинкового купороса расходуют: 0,47 т цинка (в пересчете на металл) и 0,72 т серной кислоты (100%). Из твердого остатка после фильтров извлекают свинец и олово.
Разработан также способ получения сульфата цинка из отходов (пыли), получаемых в процессе вторичной переработки цветных металлов, содержащих 65—70% цинка, 1—6% олова, 7—10% свинца и около 0,6% меди. При этом суспензию пыли в маточном растворе подвергают «нейтральному» выщелачиванию — обработке серной кислотой при 90° С и при продувке барботирующим воздухом. Количество добавляемой серной кислоты недостаточно для извлечения из исходного сырья всего оксида цинка и в раствор переходит лишь около 90% ZnO. Избыточное присутствие оксида цинка и окисление Fe2+ в Fe3+ кислородом воздуха обеспечивают хорошую очистку раствора от железа. Величину pH раствора регулируют в пределах значений, позволяющих исключение процесса осаждения меди, перешедшей в раствор из исходного сырья.
После «нейтрального» выщелачивания суспензия сгущается в отстойниках Дорра, и сгущенная суспензия поступает на «кислое» выщелачивание, а слив из аппарата Дорра проходит контрольную фильтрацию и очищается от меди цементацией цинковой пылью при 60—70° С в аппарате с мешалкой. После отделения цементной меди на фильтре полученный раствор содержит 130—140 г/л цинка в виде его сульфата и направляется на выпарку в двухкорпусный выпарной аппарат, работающий под давлением. Давление в первом и втором 220
корпусах составляет соответственно 3 и 1,5 ат, а температура кипения раствора в них 140 и 120° С. Концентрация выходящих из второго корпуса выпарного аппарата растворов соответствует отношению ZnSO4:H2O, равному 1:5—6. Суспензию направляют на поверхность вращающегося барабана, охлаждаемого изнутри циркулируемой водой. Образующийся на поверхности барабана чешуйчатый продукт состоит в основном из ZnSO4-7H2O и соответствует требованиям существующего стандарта.
«Кислое» выщелачивание суспензии, сгущенной после «нейтрального» выщелачивания, также проводят серной кислотой при 90° С и с продувкой — перемешиванием воздухом. При этом оксид цинка переходит в раствор с образованием водных растворов, содержащих сульфат цинка (~100 г/л в пересчете на цинк) и 1 г/л серной кислоты. Образующиеся растворы после фильтрации применяют для приготовления суспензии исходной пыли, а отделенные на фильтре свинцово-оловянные кеки направляют на дальнейшую переработку.
Получение сульфата цинка из отходов производства вторичной меди. Неиспользуемые отходы в производстве вторичной меди содержат 35—45% оксида цинка, до 10% оксидов меди (I) и меди (II), а также до 20% диоксида кремния. Отходы не применяют для выплавки меди из-за образования вязких шлаков и настылей, а для производства цинкового купороса— по причине высокого содержания меди.
Производство цинкового купороса из приведенных выше отходов базируется на переводе большей части оксида цинка в раствор, а оксида меди в осадок по реакции
C11SO4 + ZnO = ZnSO4 + CuO
Процесс осуществляют в две стадии, проводимые в двух самостоятельных реакторах. В первом реакторе происходит образование конечного раствора, содержащего 45—47% сульфата цинка, направляемого на кристаллизацию. В этот реактор поступает содержащий медь нейтральный 37%-ный раствор из второго реактора, цинковое сырье и 40—80% от общего количества потребляемой серной кислоты, необходимого для сульфатизации оксидов цинка, свинца и кальция (без учета расхода серной кислоты на взаимодействие с оксидами меди, железа (II) и железа (III), которые остаются в кеке в виде оксидов). Твердый остаток (промежуточный кек) из первого реактора обрабатывают серной кислотой во втором реакторе, в котором из него извлекается остаток оксида цинка и растворяется некоторое количество оксида меди. Оптимальная температура во втором реакторе 70° С. Основная масса меди, поступающей в со-221
6,6<
6
£
Рис. 5.11. Зависимость степени очистки растворов ZnSC>4 от меди от продолжительности процесса (при 50 и 70° С)
Рис. 5.12. Зависимость содержания меди в кристаллах сульфата цинка от продолжительности первого выщелачивания при 50, 60 и 70° С
Содержание F© в кристаллах, %
Рис. 5.13. Зависимость содержания железа в кристаллах сульфата цинка от концентрации его в исходных растворах
ставе исходного сырья, выводится из вторичного реактора с конечным кеком (осадком), который с целью уменьшения потерь растворимого сульфата цинка промывают водой. Полученную промывную воду возвращают во второй реактор. Содержание цинка в остаточном кеке колеблется в пределах 6—8%, а выход кека -55% от массы исходной сухой пыли. Степень извлечения цинка составляет 90—92%.
Интенсивность реакции оксида цинка с сульфатом меди снижается с увеличением концентрации сульфата цинка в исходном растворе. Так, при повышении концентрации с 31 до 36% ZnSC>4 продолжительность процесса увеличивается в два раза. Таким способом можно получить рас-
222
твор сульфата цинка, содержащий всего 0,05 г/л меди, без затраты металлического цинка на очистку раствора (см. рис. 5.11 и 5.12).
Очистка растворов от железа основана на предварительном окислении Fe2+ в Fe3+ и взаимодействии сульфата железа (III) с оксидом цинка:
Fe(SO4)3 + 3ZnO + ЗН2О = 2Fe(OH)3 + 3ZnSO4
Процесс окисления проводят, барботируя воздух через раствор. При этом кислород воздуха в нейтральной среде энергично окисляет Fe2+. На рис. 5.13 представлена зависимость содержания железа в кристаллах цинкового купороса от концентрации его в исходном растворе. Кристаллы, полученные из растворов с одинаковым содержанием железа и меди, загрязнены железом приблизительно в 10 раз больше, чем медью.
5.5. ГАЛОГЕНИДЫ ЦИНКА
Галогениды цинка — бесцветные кристаллы тетрагональной сингонии (табл. 5.3).
Фторид цинка ZnF2 кристаллизуется в структурном типе рутила. При высоких давлениях получены также моноклинная модификация кристаллов со структурой типа ZnO2, кубическая сингония типа флюорита и ромбическая модификация со структурой типа а-РЬО2. Уравнение температурной зависимости давления пара:
1g р(мм рт. ст.) = 26,90—13650/7 + 5,031g71(l 148—1778 К).
Фторид цинка гигроскопичен, слабо растворим в воде (1,62% при 20° С), не растворяется в этаноле, разлагается горячими минеральными кислотами. Существует один кристаллогидрат с четырьмя молекулами воды плотностью 2,567 г/см3, обезвоживающийся при температуре выше 120° С. При гидролизе растворов фторида цинка образуются гидро-ксофториды, например Zn(OH)F. В процессе термообработки на воздухе и в токе водяного пара ZnF2 превращается в ZnO.
Фторид цинка получают действием фтороводородной кислоты на карбонат цинка:
ZnCO3 + 2HF = ZnF2 + СО2 + Н2О
Получают также взаимодействием водных растворов фторида натрия и сульфата цинка:
ZnSO4 + 2NaF + 4Н2О = ZnF2-4H2O + Na2SO4
223
Образующийся тетрагидрат фторида цинка обезвоживают.
Фторид цинка является материалом для лазеров, компонентом люминофоров, глазурей, эмалей, специальных стекол, консервантом для древесины, протравой при крашении, раствором для оцинкования.
Бромид цинка ZnBr2 кроме тетрагональной модификации (см. табл. 5.3) существует в ромбоэдрической со структурой (а = 0,392 , нм, с ~ 1,873 нм, z = 3, пространственная группа ЛЗот). При высо- ' ких давлениях найдены еще две модификации. Образовывает стекло с плотностью 3,77 г/см3.
Бромид цинка летуч и возгоняется. Хорошо растворяется в воде (82,5% по массе при 25° С, 86,6% при 80° С). Из водных растворов при температурах от - 8 до 35° С кристаллизуется дигидрат ZnBr2-2H2O (температура плавления 37° С), ниже - 8° С тригидрат ZnBr2-3H2O, а выше 35° С существует безводный ZnBr2. Кристаллогидраты, как и безводный бромид, расплываются на воздухе. В процессе гидролиза растворов бромида цинка образуются гидроксобро-миды различного состава, например Zn5(OH)gBr2. Бромид цинка хорошо растворим в этаноле, хуже в эфире, пиридине и хинолине. Не растворим в сероуглероде.
Таблица 5.3. Свойства галогенидов цинка
Показатель ZnF2 ZnBr2 Znl2 ZnCl2
Параметры* решетки, нм:
а 0,47034 1,140 1,227
с 0,31335 2,180 2,354 Описано в
Число формульных единиц в ячейке 2 32 32 тексте
Пространственная группа PMmnm \4i/acd \4\/acd
Т °C 1 пл» 890 402 446 283
Т °C 1505 670 727 732
Плотность, г/см3 4,94 4,20 4,736 2,91
С°, Дж/(моль К) 65,6 66 — 71,38
кДж/моль -764 -329,7 -208,2 -415,33
5^, Дж/(моль К) 73,7 136 161 111,54
кДж/моль 41,8 15,7 17 23,03
АЯИСП, кДж/моль 185 109,6 96,2 —
♦ Тетрагональная сингония.
Бромид цинка получают взаимодействием цинка с бромом в водном растворе.
Бромид цинка применяют в качестве реагента в органическом синтезе. Он является компонентом стекол для ИК-оптики. Концентриро-224
ванные растворы бромида цинка применяют для наполнения радиационных экранов.
Иодид цинка Znl2— весьма гигроскопичен, на свету желтый. Кроме тетрагональной известны ромбоэдрическая модификация со структурой CdCl2 (а = 0,425 нм, с = 2,15 нм), хорошо растворим в воде (81,2% при 18° С и 83,1% при 80° С), в этаноле. Растворяется также в эфире, ацетоне, глицерине и пиридине. В сероуглероде не растворяется. В процессе термообработки на воздухе превращается в оксид цинка:
2ZnI2 + О2 = 2ZnO + 2I2
Устойчивая фаза в водных растворах ниже 0°С — дигидрат иоди-да цинка с температурой плавления 27° С. Выше 0° С существует безводный иодид цинка. Существует также тетрагидрат ZnI2-4H2O, превращающийся при -7° С в дигидрат.
Иодид цинка получают из элементов в воде с последующим выпариванием и высушиванием. Получающийся продукт очищают сублимацией.
Иодид цинка применяют в качестве катализатора в органическом синтезе. Он является реагентом в аналитической химии и антисептиком в медицине.
Хлорид цинка ZnCl2, бесцветные, очень гигроскопические кристаллы. Известен в трех модификациях: a-ZnCh — тетрагональной сингонии (а = 0,540 нм, с = 1,035 нм, z = 4, пространственная группа /42J), Р-моноклинной сингонии (а = 0,654 нм, b = 1,131 нм, с = 1,233 нм, р = 90°, z = 12, пространственная группа Р2\!п) и у-тетрагональной сингонии (а = 0,370 нм, с = 1,067 нм, z = 2, пространственная группа Ptynmc).
Хлорид цинка хорошо растворяется в воде (432 г в 100 г воды при 25° С и 614 г при 100° С). Растворяется в эфире, этаноле, глицерине, ацетоне.
Вследствие гидролиза водный раствор хлорида цинка имеет кислую реакцию. При частичной нейтрализации образуются основные хлориды: Zn(OH)Cl, Zn5(OH)8C12-2H2O и др. Известны пять кристаллогидратов ZnC12 «H2O, где и = 4, 3, 2,5, 1,5 и 1. Существуют комплексные хлориды MfZnCh], M2[ZnC14], M4[ZnCU], где М = NH4, К, Na, Cs. Существуют также аммиакаты [Zn(NH3)4]C12-H2O, [Zn(NH3)6]C12 и др.
В промышленности хлорид цинка получают действием хлороводородной кислоты на вторичное сырье, обожженную руду или в процессе хлорирования колчеданных огарков серно-кислотных производств. Получают хлорид цинка также в процессе термообработки при 420° С гранулированного металлического цинка в токе хлора, 8 Химическая технология 225
неорганических веществ, кн. 1
действием хлора на нагретые до 700° С оксид или сульфид цинка, реакцией цинка с хлороводородной кислотой. Хлорид цинка очищают сублимацией при 600—700° С в токе хлора.
Хлорид цинка применяют в качестве антисептика для древесины, в процессе изготовления пергамента, для очистки поверхности металлов перед пайкой, в качестве компонента электролита для гальванических покрытий в сухих элементах и как протраву в процессе крашения.
Получение хлорида цинка из цинксодержащих отходов основывается на обработке цинксодержащих отходов хлороводородной кислотой. Кислоту применяют технической квалификации с концентрацией 27—28% НС1. Обработку отходов кислотой проводят в химзащищенных резервуарах, в которые загружают исходное сырье и заливают кислоту. Перемешивание массы производят сжатым воздухом или с помощью мешалок. На некоторых заводах несколько каскадно расположенных резервуаров, в которых растворы хлорида цинка перетекают из одного в другой резервуар, в процессе которого концентрация ZnCl2 повышается до 45%.
Содержащиеся в исходном сырье примеси соединений свинца, диоксида кремния в процессе растворения в хлороводородной кислоте образуют осадки, откладывающиеся на поверхности частиц растворяющегося материала, содержащего цинк, в виде непроникаемой корки. Для исключения этого процесса исходное сырье в измельченном виде подвергают предварительному оксилительному обжигу при 300—500° С.
Реакции растворения цинка и оксида цинка в хлороводородной кислоте экзотермичны и протекают весьма энергично. Однако общая продолжительность процесса достигает 10 ч и более, поскольку к концу он замедляется.
В процессе выделяются значительные количества водорода:
Zn + 2НС1 = ZnCl2 + Н2
Образующиеся растворы хлорида цинка очищают от железа путем окисления с помощью введения в систему окислителей, не загрязняющих растворы продуктами окисления. В качестве окислителей применяют газообразный хлор, хлорную известь или бертолетову соль. Железо (III) осаждают из раствора в процессе нейтрализации кислотности добавкой водной суспензии оксида и гидроксида кальция или ее смеси с хлорной известью. Раствору дают отстояться от осадка и нерастворимой смеси — песка и других примесей, после чего фильтруют или декантируют. Отфильтрованные растворы хлорида цинка выпаривают в чугунных котлах, обогреваемых топочными газами. Процесс выпарки производят при 220—250° С. Полученный плав, содержащий 9—12% воды, заливают в стальные барабаны, в которых целевой продукт затвердевает.
226
На 1 т 45%-ного раствора хлорида цинка расходуют 0,23—0,25 т металлического цинка (96%-ного) или соответствующее количество отходов, содержащих цинк, и 0,9—1 т 27,5%-ной хлороводородной кислоты. На 1 т плавленого хлорида цинка расходуют 2,4 т 45%-ного раствора хлорида цинка и 0,5 т угля.
5.6. ЦИНКОВЫЕ ПИГМЕНТЫ
В качестве пигментов применяются два типа хроматов цинка: грунтовый цинковый и малярный цинковый кроны.
Грунтовочный цинковый крон представляет собой хромат цинка состава «ZnO CrO3 wH2O, в котором среднее значение п = 4, а т = 3, т.е. соединение состава 4ZnOCrO3-3H2O.
Малярный цинковый крон — соединение основных хроматов цинка и хромата калия общего состава 4ZnO-xCrO3-x/4K2O-3H2O, где х = 4—2,5. Наибольшее значение имеет соединение состава 4ZnO-4CrO3K2O-3H2O.
Двойные хроматы цинка и калия частично растворяются в воде, причем растворимость их растет с повышением температуры. В кислотах растворяются легко и полностью.
Плотность цинкового крона 0,341—0,359 г/см3, насыпная плотность 0,09 г/см3, удельная поверхность 5 м2/г.
В процессе нагревания до 150° С (рис. 5.14) цинковый крон не претерпевает изменений, а при 280—300° С он разлагается, превращаясь в массу темно-синего или черного цвета. При этом находящийся в составе трехосновного хромата ZnCrC>4-3Zn(OH)2 гидроксид цинка переходит в оксид, хромат цинка ZnCrO4 распадается на ZnO и Сг2О3, часть оксида цинка остается в неизменном виде, а часть соединяется с сесквиоксидом хрома, образуя хромат цинка ZnCr2O4.
Двойные хроматы цинка и калия 3ZnCrO4-K2CrO4-nZn(OH)2‘xH2O разлагаются таким же образом, за исключением хромата К2С1О4, который остается без изменения.
В процессе обработки термообработанного двойного хромата цинка и калия водой весь монохромат калия легко переходит в раствор.
Рентгенографические исследования цинковых кронов показали, что крона предельного состава:
1
Время
Рис. 5.14. Термограммы цинкового крона состава 4ZnO-4CrOrK2O-3H2O (а) и 4ZnO CrO3-3H2O (б)
8*
227
д
9
7
5
3
1
1,0 1,2 1,4 1,6 1,8 2,0 2,2 2,4 2,6 2,8 3,0 3,2 3,4 3,63,8 4,0 4,2 4,4
ьщ|пг| и и || ill г и I iiiriiiiiiin
7------zZZZ.ZZZZZ_’ ZZZZZ---------Z_ZZZ—Z—zzz
5 zzzzzezzzzzzzzz: zzzzzzzzzzzzzzzzzzzzz з ZZIZZ IZzz zzzzzzzzzzzzzzzzzzzzzzzzzzzz ' lllllllllllllllllllllllllllllllllllllllll 1,0 1,2 1,41,6 1,82,02,22,42,62,83,03,23,43,63,84,04,24,4
в
б
а
Рис. 5.15. Расположение и интенсивность интерференционных линий на рентгенограммах хроматов цинка:
a — состав 4ZnO-4CrO3K2O-3H2O, полученный из ZnCI2 обработкой К2СгО4; б — тот же состав, но полученный из ZnO обработкой K2Cr2Ch и HCI; в — состав 4ZnO4Cr2OrNa2O-3H2O, полученный из ZnO обработкой Na2Cr2O? и HCI; г — состав 4ZnO CrOy3H2O, полученный из ZnO обработкой CrO% д — тот же состав, но полученный обработкой сложного хромата цинка и калия горячей водой
4ZnO-4CrO3'K2O-3H2O и 4ZnO-CrO3-3H2O следует рассматривать в качестве самостоятельных химических соединений, поскольку из рентгенограммы (рис. 5.15) сильно различаются между собой. Крон же, содержащий калий хотя бы в незначительном количестве, не отличается по строению от крона предельного состава 42пО-4СгОз К2О ЗН2О. Замена же в цинковом кроне калия натрием резко изменяет характер кристаллической решетки.
Процесс получения цинкового крона состоит из следующих стадий: диспергирование оксида цинка (цинковых белил) в воде; приготовление водного раствора хромпика или триоксида хрома; обработка водной суспензии оксида цинка кислотой и раствором хромпика или триоксидом хрома; фильтрование и промывка осадка; сушка, размол и упаковка пигмента.
В качестве исходного сырья для получения цинкового крона применяют оксид цинка (цинковые белила), хромпик калиевый, триоксид хрома, серную и хлороводородную кислоты. Рецептуру для производства кронов рассчитывают по соответствующим уравнениям реакции. Например, рецептуру для малярного цинкового крона 4ZnO-4CrO3-K2O-3H2O составляют по уравнению:
4ZnO + 2К2Сг2О7 + 2НС1 + Н2О = 4ZnO-4CrO3K2O-3H2O + 2КС1
Грунтовочный цинковый крои получают также обработкой оксида цинка триоксидом хрома. Цинковый крои получают при следующем соотношении между реагентами:
4ZnO + СгОз + ЗН2О = 4ZnOCrO3-3H2O
5ZnO + СгОз + 4Н2О = 4ZnOCrO3-3H2O + Zn(OH)2
ГЛАВА 6
БОР И ЕГО СОЕДИНЕНИЯ
6.1. БОР
Содержание бора в земной коре составляет 5-10’3% (масс.), а в воде океанов -4,6 мг/л. В свободном состоянии бор в природе не встречается, а содержится лишь в виде кислородных соединений примерно в 80 минералах.
Кроме сведений, приведенных в табл. 6.1, бор в виде буры и борной кислоты содержится в рассолах из нефтяных скважин, а также других буровых вод. Кроме того, бор концентрируется в рассолах озер и морей. Бор в виде борной кислоты содержится в некоторых горячих источниках, откуда он выделяется в виде паров, нагретых до 180—190° С (табл. 6.1). Борная кислота встречается также в извержениях вулканов в виде минерала сассолин. Потенциальными видами боратового сырья являются и сопочные грунты, содержащие до 0,5% триоксида бора.
Свойства. Бор имеет атомную массу—10,81. Природный бор состоит из двух стабильных изотопов — В * 10В (19,57%) и ПВ (80,43%). Поперечное сечение захвата тепловых нейтронов |0В 310'25 м2, nB 4-Ю’32 м2. Степень окисления +3, редко +2; атомный радиус (нм): металлического — 0,091; ковалентного — 0,088, условный радиус иона Э3+ 0,025 нм; энергия ионизации при последовательном переходе от В0 к В5+соответственно 8,29811; 25,156; 37,920; 259,300 и 340,130 эВ.
Свойства соединений бора обусловлены электронной конфигурацией атома бора в возбужденном состоянии характером химической связи, возникающей на основе зр2— ^-гибридизации
2Р
т
В трехзамещенном состоянии бор образует координационно-не-
насыщенные соединения, свободная 2р-орбиталь имеет возможность
230
принять неподеленную электронную пару и образовать четвертую химическую связь, выступая в качестве акцептора электронов. Таким образом, в зависимости от условий бор может образовывать как три равноценные ковалентные связи, расположенные в одной плоскости под углом 120°, 5р2-гибридизация, или четыре ковалентные связи, направленные по вершинам тетраэдра, $р3-гибридизация. Обладая малыми размерами и высоким потенциалом ионизации, атом бора заметно отличает
Рис. 6.1.
Икосаэдрическая группировка атомов Bn
ся по прочности связи валентных электронов от остальных членов третьей группы элементов таблицы Менделеева, что и обусловливает его особые химические свойства и способность формировать сложные по строению полиборатные ионы.
Бор — бесцветное, серое или красное кристаллическое или черное аморфное вещество, тугоплавок (температура плавления — 2300° С), диамагнитен, обладает полупроводниковыми свойствами (ДЕ = 1,55 эВ), известно более 10 аллотропных модификаций (табл. 6.2). Образование той или иной модификации и их взаимные переходы определяются температурой, при которой получают бор: при 600—800° С образуется аморфный продукт (плотность 2,35 г/см3; Д/7° перехода аморфной модификации в Р-ромбоэдриче-скую составляет 5,02 кДж/моль), до 1000° С — а-ромбоэдрическая модификация (красные кристаллы), до 1200° С — наиболее устойчивая Р-ромбоэдрическая форма, до 1500° С — тетрагональные модификации. Расплав обычно кристаллизуется в р-ромбоэдрическую модификацию, в которую переходят и другие формы выше 1500° С. В интервале 1000—1500° С можно одновременно получить смесь всех модификаций, кристаллические решетки которых построены, и икосаэдров Bn — полиэдрических электроно-дефицитных структур (рис. 6.1), содержащих наряду с двухэлектронными двухцентровыми химическими связями В—В многоцентровые двухэлектронные связи, как показано ниже:
231
Таблица 6.1. Борсодержащие
Название Химическая формула Состав, %
B2O, R2O RO H2O 5102
Ашарит (ссайбелеит) Бораты 2MgOB2O3(MgHBO3) 41,4 — 47,9 10,7 —
Борацит MgCl2-5MgO-7B2O3 (MgsBiOuCl) 62,2 — 25,7 — —
Боронатрокальцит (улексит) Na2O 2CaO-5B2O316H2O (NaCaB5O9-8H2O) 43,0 7,6 13,8 35,6 —
Гидроборацит CaOMgO-3B2O3-6H2O (CaMgB6On-6H2O) 50,5 — 23,3 26,2 —
Иньонит 2CaO-3B2O313H2O (Ca2B6On13H2O) 37,6 — 20,2 42,2 —
Колиборит K2O-4MgOllB2O318H2O (KMg2BnO9-9H2O) 60,6 7,4 6,4 25,6 —
Кернит (разорит) Na2O-2B2O3-4H2O (Na2B4O7-4H2O) 50,9 22,7 — 26,4 —
Колеманит 2CaO-3B2O3-5H2O (Ca2B6OH-5H2O) 50,8 — 27,3 21,9 —
Людвигит (MgFe2+ )2Fe3+ BO5 п,з — 13,1 — —
Пандермит (прицеит) 2CaO-3B2O3-3H2O 49,8 — 32,1 18,1 —
Тинкал (бура) Na20-2B203 10H20 (Na2B4O710H2O) 36,5 16,3 — 47,2 —
Тинкалкернит Na2O-2B2O3-5H2O (Na2B4O7-5H2O) 47,8 21,3 — 30,9 —
Данбурит Боросиликаты CaO’B2O3*2SiO2 [CaB2(SiO4)2] 28,3 — 22,8 — 48,9
Датолит 2CaOB2O3-2SiO2H2O [CaB(OH)SiO4] 21,8 — 35,1 5,6 37,5
Свойства Р-ромбоэдрического бора: температура плавления 2074° С; температура кипения 3658° С; Cj* = 11,09 Дж/(моль-К); Д#™ = 50,2 кДж/моль; ДЯВоЗГ = 560 кДж/моль (ОК); ДЯИСП = 512 кДж/моль; 5298 = 5 ,9 Дж/(моль-К) [для газа 153,2 Дж/(моль-К)]; уравнение температурной зависимости давления пара 1g р (атм) = = 7,239-28,840/7’ (1781-2152 К); температурный коэффициент линейного расширения (4,8—7,0)-10’6К‘1(293—1300 К); теплопроводность при 300 К 2,6-10'3 Вт/(м К); дебаевская температура 1220 К; р(0,7-^,0)-109 МОмм (88 К), 105 МОм-м (200 К), 0,05 МОм-м (500 К). Бор — полупроводник p-типа; ширина запрещенной зоны по данным электрических и оптических измерений соответствует 1,42 и 1,53 эВ; дырочная проводимость 55-104, электронная 104м2(В с); постоянная Холла 7-10'3м3/Кл (298 К); концентрация 232
минералы
Кристаллоформа, внешний вид Цвет Твердость по шкале Мооса Плотность, кг/м3 Место нахождения
Спутано-волокни-стая структура Белый, желтый 3,0—3,5 2630—3300 Индер, Калифорния, Норвегия
Вкрапления кристаллов в массе солей Бесцветный, белый 7,0 2910—2970 Индер, Страссфурт
Конкреции волокнистых кристаллов Бесцветный, белый 2,5 1955 Калифорния, Индер, Аргентина
Тонкозернистый, игольчатые кристаллы Бесцветный, белый, розоватый 3,0 2167 Индер, Калифорния, Страссфурт
Таблитчатые кристаллы, сферолиты Бесцветный, белый 2,0 1875 Калифорния, Индер
Мелкокристаллический, зернистый Бесцветный, красноватый 4,0—4,5 2128 Индер, Страссфурт
Волокнистый Бесцветный, белый 2,5 1908 Калифорния, Аргентина
Зернистый, сферолиты Бесцветный, серый 4,2 2423 Калифорния, Турция, Индер
Зернистый, сферолиты Темно-зеленый 5,0 4400 Румыния, Якутия, Калифорния
Мелоподобный Белый 3,0—3,5 2420 Калифорния, Индер
Призмы или таблитчатые кристаллы Бесцветный, белый, сероватый 2,0—2,5 1715 Турция, Калифорния, Южная Америка, Тибет
Тонкозернистый порошок Бесцветный — 1880 Калифорния
Призматические кристаллы Бесцветный, желтоватый 7,0—7,5 2970—3020 Памир, Дальний Восток
Призматические кристаллы Бесцветный, зеленоватый 5,0—5,5 2900—3000 Дальний Восток, Кавказ
собственных носителей 5-1014(433 К) и 9-1019 м"3 (1073 К). Бор диамагнитен, магнитная восприимчивость 0,78-10’9 (298 К). Для монокристаллов показатель преломления 3,44 (при 1 = 0,45 мкм), коэффициент поглощения 10’2m'’ (при X = 1,3—3,8 мкм).
Бор по своей твердости занимает второе место после алмаза. Он очень хрупок, в пластическое состояние переходит лишь при 2000° С.
Получение. Впервые бор был получен в 1808 г. Гей-Люссаком и Л.Тенаром, независимо от них — Г. Дэви. В настоящее время применяются следующие методы получения бора.
233
Таблица 6.2. Характеристика кристаллических модификаций бора
Показатель Ромбоэдрические Тетрагональные
в ромбоэдрической упаковке в гексагональной упаковке
а ₽ а 3 а ₽
Параметры решетки, нм: а 0,506 1,014 0,491 1,094 0,875 1,012
с — — 1,257 2,381 0,506 1,414
а 58,06° 65,28° — — — —
Число формульных единиц в ячейке 12 105 36 324 50 192
Пространственная группа R3m R3m — — Р^г/ппт Р4,22
Межатомное расстояние В—В, нм 0,177 0,165—0,183 0,177 0,165—0,183 0,175—0,185 —
Плотность, г/см3 2,45—2,46 2,31—2,35 — — 2,36—2,37 2,36—2,37
Разложение боратов концентрированной серной кислотой при температуре около 100° С с последующей фильтрацией полученной суспензии от нерастворимого остатка. Полученный фильтрат охлаждают. Образующаяся ортоборная кислота (Н3ВО3) выпадает в осадок, который подвергают термообработке до 235° С с получением В2О3. Аморфный бор получают методом металлотермии — восстановлением В2О3 магнием, калием, натрием, кальцием, цинком или железом по схеме
В2О3 + ЗиМе ->2В + ЗМе„О
Кристаллический бор получают восстановлением галогенидов бора (в основном ВС1з или BF3) водородом
2ВС1з + ЗН2 = 2В + 6НС1
2BF3 + ЗН2 = 2В + 6HF
Разложение галогенидов или гидридов бора при 1000—1500° С по схеме
2ВС1з —<-> 2В + ЗС12
В2Н6 2В + ЗН2
Электролиз расплавленных фтороборатов — Na[BF4] или K[BF4].
Разложение бромида бора на танталовой или вольфрамовой нити при 1300° С в присутствии водорода. Способ обеспечивает по-234
лучение чистого кристаллического продукта (содержание примесей менее 0,05%).
Разлагаются В2Н6 и В1з при 700—1000° С. Получают бор также методом зонной плавки или вытягиванием монокристаллов.
Применение. Бор применяется в качестве добавки в его сплавы с железом (ферробор). Присутствие даже незначительных количеств бора в составе сплавов повышает их механические и антикоррозионные свойства и обуславливает их мелкозернистость. Кроме того, бором насыщают поверхность стальных изделий, что позволяет улучшить их коррозионные свойства и механическую прочность. Аморфный бор применяют в качестве упрочнителя композиционных материалов (в виде волокон), полупроводника для изготовления терморезисторов, счетчиков тепловых нейтронов, преобразователей тепловой энергии в электрическую. Бор и его сплавы применяются в качестве нейтронопоглощающих материалов в процессе изготовления регулирующих стержней ядерных реакторов.
6.2. БОРНЫЕ КИСЛОТЫ
Борные кислоты в свободном состоянии существуют в виде орто-борной Н3РО3 и метаборной НВО2 кислот.
Ортоборная кислота (оксоборат водорода) Н3ВО3 — в безводном виде бесцветные кристаллы, решетка слоистая, расстояние между слоями 0,318 нм, триклинная (а = b = 0,704 нм, с = 0,656 нм, а = 101,10°, Р = 92,30°, у = 120,00°, z = 4, плотность 1,46 г/см3). При давлении выше 4200 МПа образуется модификация с ромбической решеткой, выше 5700 МПа — с тетрагональной. При медленном нагревании (в режиме работы дериватографа) ортоборная кислота при 84—96° С переходит в метаборную кислоту по схеме
Н3ВО3 |0?££-> НВО2 + Н2О
а НВО2 — в борный ангидрид
2НВО2 щ-1Я>,<? > В2О3 + Н2О
Процесс плавления ортоборной кислоты при 170,9° С сопровождается его разложением; С° =81,3 Дж/(моль-К); ДЯ^ =22,34 кДж/моль; ДЯИСП = 98,1 кДж/моль; АЯ^Г = 63,97 кДж/моль; ДЯ^, = 1094,9 кДж/моль; 5°98 = 88,74 Дж/(моль-К). Растворимость в воде 2% при 0° С; 4% при 20° С и 28,7% при 100° С, в глицерине (19,48% при 235
25° С), метаноле (21,0%), этаноле (11,2%), ацетоне (0,65%). Практически не растворим в эфире, в жидком аммиаке, диоксане, пиридине.
Ортоборная кислота — весьма слабая кислота, слабее угольной кислоты и сульфида водорода. Первая константа диссоциации (Н3ВО3 Н2ВО3 + Н+) имеет примерно такое же значение, как и соответствующая константа цианида водорода. В отличие от обычных кислот ее кислотные свойства обусловлены не отщеплением протона, а присоединением гидроксил-ионов:
В(ОН)3 + НОН # [В(ОН)4]‘ + Н+, рК = 9,15.
Однако в присутствии органического вещества, содержащего гид-роксил-ион в составе борной кислоты, раствор приобретает свойство . сравнительно сильной одноосновной кислоты, титруемой водным раствором щелочи с фенолфталеином в качестве индикатора. Подоб- i ное повышение кислотности можно видеть на примере комплекса ? борной кислоты с глицерином: |
НОН2ССН НО\ /ОН НО—СН2
I + /В^ + I
сн2—о но ' он—СНСН2ОН-----►
НОН2ССН —О\ /О—сн2
1 /вС 1
сн2—о о — СНСН2ОН
+ ЗН2О
1. [В5Ов(ОН)4] 2. [ВзО3(ОН)4] 3. [В5О6(ОН)5]2’ 4. [B4O5(OH)J2’
5. [В^вСОН)/ 6. [^О3(ОН^]2’ 7. (В^ОН^Г 8. [B(OH)4]-
Рис. 6.2. Структуры полиборэт-ионов
236
С увеличением концентрации ортоборной кислоты в растворе образуются полиборатные анионы. Наиболее вероятными структурами являются полиионы (рис. 6.2).
Метаборная кислота НВОг — бесцветные кристаллы. Существует в трех модификациях: наиболее устойчивая у-форма с кубической решеткой (а = 0,887 нм, z = 24), плотность 2,486 г/см3, температура плавления с разложением 236° С, ДН^, = - 804,6 кДж/моль; [3-форма с моноклинной решеткой, плотность 2,045 г/см3, температура плавления с разложением 200,9° С, ДЯ^р = -794 кДж/моль; a-форма с кубической решеткой, плотность 1,784 г/см3, температура плавления с разложением 176° С АЯ^р=-789,3 кДж/моль. Температура перехода его в Р-форму 160° С. В процессе смешивания с водой метаборная кислота переходит в ортоборную.
В водных растворах предполагается существование полиборных кислот общей формулы H3m.2n Вж О3ш.„.
Способы получения борной кислоты. Борную кислоту получают из датолитового сырья, из боратных руд, а также путем переработки других видов сырья.
Для разложения борсодержащего сырья могут быть применены следующие виды реагентов: все минеральные кислоты, включая угольную, а также диоксид углерода. В процессе щелочной обработки все щелочи, щелочные соли.
Получение из датолитового сырья. Датолитовая руда содержит 45—50% основного минерала датолита. Сопутствуют ему кальцит (СаСО3), волластонит (CaO-SiO2), гранат (3CaO-Fe2O3-3SiO2), геденбергит (CaO-FeO-2SiO2), гизенгерит (Fe2O3-xSiO2yH2O), кварц (SiO2). С целью уменьшения расхода серной кислоты на побочные процессы исходную руду обогащают до 72—76%-ного содержания основного вещества.
Датолит легко разлагается в серной кислоте (96—100% за один час) при 95° С. На разложение одной тонны сырья расходуется 550 кг серной кислоты с концентрацией 46%. Содержащиеся в исходном сырье примеси — сопутствующие минералы в указанных условиях разлагаются менее активно, так кальцит на 47—50%, гранат на 12—20%, геденбергит на 2—4%. Волластонит разлагается так же, как и датолит. Установлено, что при более низких концентрациях исходной серной кислоты датолит в пределах 25—95° С имеет сравнительно невысокие скорости разложения [0,03-10'5—2,69-10"5 г/(см2-с)] по сравнению с кальцитом, у которого при тех же условиях скорость разложения в 4—8 раз выше, чем у датолита (рис. 6.3). Поэтому процесс разложения проводят при более высоких концентрациях, при 237
(1)
(2)
(3)
(4)
которых скорость разложения кальцита резко снижается, а датоли- j та — уменьшается незначительно.
При обработке датолитового концентрата серной кислотой протекают следующие реакции:
2СаОВ2Оз-28Ю2-Н2О + 2H2SO4 + aq = — 2Н3ВО3 + 2CaSO4*«H2O + 2SiO2-mH2O
CaO-SiO2 + H2SO4 + aq = СаЗС^иНгО + ЗЮг’тиНгО
СаСОз + H2SO4 + aq = СаЗС^иНгО + CO2 + «НгО
ЗСаОРе2Оз-ЗЗЮ2 + 6H2SO4 + aq = = 3CaSO47?H2O + Fe2(SO4)3 + ЗЗЮг-тиНгО
CaO-FeO-2SiO2 + 2H2SO4 + aq = CaSO4nH2O + FeSO4 + 2SiO2-mH2O (5) ;
Образующийся в процессе разложения геденбергита сульфат же- < леза (II) окисляется кислородом воздуха: '
4FeSO4 + 2H2SO4 + О2 = 2Fe2(SO4)3 + 2Н2О
(6)
Рис. 6.3. Скорость процесса разложения минералов в растворах серной кислоты при 98° С: 1 — гидроборацит; 2 — улексит; 5 — колеманит; 4 — гипс; 5 — кальцит; 6 — ангидрит
Приведенные реакции показывают, что в результате серно-кислотного разложения датолитового концентрата образуются борная кислота, сульфат кальция, гидратированный диоксид кремния и сульфат железа (III).
Процесс получения борной кислоты включает следующие стадии: приготовление водной суспензии датолитового концентрата; дозировка серной кислоты и суспензии датолитового концентрата; разложение датолитового концентрата; вызревание реакционной массы в камере; репульпация и охлаждение камерного продукта; выщелачивание; нейтрализация; противоточное ступенчатое фильтрование с промежуточными промывками шлама; подкисление раствора и осаждение из него дигидрата сульфата каль-
238
ция; контрольное фильтрование; вакуум-кристаллизация; сгущение суспензии борной кислоты; фильтрование бората кальция.
Схема получения борной кислоты приведена на рис. 6.4, согласно которой осадок водной суспензии датолитового концентрата поступает в сборник 1, где смешивается с маточным раствором борной кислоты. Обращающаяся суспензия и исходная серная кислота (~93%-ная) дозируются в вертикальный смеситель 2, аналогичный применяемым в суперфосфатном производстве. Образующаяся смесь поступает в суперфосфатную камеру непрерывного действия 3, в которой она загустевает. Вырезанная ножом из камеры масса репульпи-руется оборотным раствором в аппарате с мешалкой 5 и охлаждается во флотационной камере 6 до 35—55° С аэрацией воздухом, после чего полученная суспензия нагревается до 80—90° С.
Для нейтрализации свободной серной кислоты, ионной коагуляции кремниевой кислоты, осаждения оксидов железа и алюминия в суспензию вводят борат кальция до pH 5—5,2. Нейтрализованная суспензия фильтруется на барабанных или патронных вакуум-фильтрах 7, 8. Твердые отходы промываются и удаляются гидротранспортом в шла-мохранилище или передаются в цех по производству борогипса. Промывную воду направляют на репульпацию камерного продукта.
Концентрированный фильтрат, содержащий 10—11% борной кислоты, фильтруют на фильтре 12, подкисляют серной кислотой в сборнике 13, где их выдерживают при 90—95° С, после чего фильтруют на фильтр-прессах 14. Освобожденный от дигидрата сульфата кальция раствор передают в вакуум-кристаллизаторы 16. При охлаждении в вакуум-кристаллизаторах до 15—20° С выпадают кристаллы борной кислоты. Образующуюся суспензию сгущают в гидроциклонах, а кристаллы борной кислоты отделяют от маточных растворов на горизонтальных автоматических центрифугах 18. Кристаллы после промывки в центрифугах конденсатом или обессоленной водой сушат в горизонтальных вращающихся сушилках 21 горячим воздухом или в трубе-сушилке при режимах, предотвращающих переход ортобор-ной кислоты в метаборную.
Маточный раствор борной кислоты после центрифуг частично возвращают на приготовление суспензии в реактор с метальным устройством 23, куда подают известковое молоко. При pH 11,2—11,4 бор из раствора осаждают в виде бората кальция, который отделяют от раствора на патронном 7 и дисковых 8 вакуум-фильтрах и используют для нейтрализации суспензии камерного продукта. Маточный раствор бората кальция направляют на промывку шлама на стадии фильтрования взамен свежей воды, а также на выщелачивание камерного продукта.
Описанный способ обеспечивает получение борной кислоты марок Б и В (табл. 6.3) по существующему ГОСТ 18704—78.
239
Рис. 6.4. Схема получения борной кислоты из датолитового концентрата:
1 — сборник; 2 — смеситель; 3— суперфосфатная камера; 4, 22 — транспортер; 5 — аппарат с мешалкой; 6—флотационная камера; 7, 8—вакуум-фильтры; 9—рессивер; 10— репульпатор; 11, 73, 75, 19 — сборники фильтрата; 72, 14 — фильтр-прессы; 16 — вакуум-кристаллизаторы; 77—сборник суспензии; 18 — центрифуга; 20— бункер; 27 — сушилка; 23— реактор
Таблица 6.3. Качество отечественной борной кислоты
Показатель В пересчете на Марки, ГОСТ 18704—78
А Б В
сорт 1 сорт 2
Содержание борной Н3ВО3 99,9 99,9 99,6 98,6
кислоты, % Содержание, %, не более: хлоридов сульфатов сг SO2\ 0,0001 0,0005 0,001 0,008 Не норк 0,2 !ируется 0,5
железа Fe 0,0003 0,001 0,002 0,003
тяжелых металлов Pb 0,0005 0,001 0,001 Не нормируется
не растворимого в н.о. 0,005 0,01 0,04
воде остатка кальция Са 0,002 0,005 Не норми-
мышьяка As 0,0001 0,0002 руется »
фосфатов роЛ 0,001 0,001 »
Получение из боратовых руд. Основными борными минералами являются ашарит и гидроборацит, улексит и иньоит. Примесями являются дигидрат сульфата кальция, карбонаты кальция и магния, глинистые минералы (гидрослюда, монтмориллонит) и кварц.
В процессе серно-кислотной обработки борсодержащего сырья происходят следующие реакции:
2MgOB2O3H2O + 2H2SO4 = 2Н3ВО3 + 2MgSO4
CaOMgO-3B2O3-6H2O + 2H2SO4 + H2O = 6H3BO3 + CaSO4 + MgSO4
CaCO3 + H2SO4 = CaSO4 + CO2 + H2O
MgCO3 + H2SO4 = MgSO4 + CO2 + H2O (FeAl^Oj + 3H2SO4 = (Fe,Al)2(SO4)3 + 3H2O
В раствор переходят борная кислота, сульфат магния и частично сульфаты железа и алюминия, а дигидрат сульфата кальция полностью выпадает в осадок.
Исследованиями кинетики и механизма процесса серно-кислотного разложения ашарита и гидроборацита определены следующие оптимальные условия: температура не ниже 80° С; продолжительность перемешивания не менее 30 мин; размеры частиц исходного сырья 1 мм.
Гидроборацит разлагается серной кислотой с более высоким эффектом, чем ашарит. Экспериментально установлено, что кривые ско-241
Рис 6.5. Влияние температуры на степень разложения гидроборацита
Время, мин Рис. 6.6. Влияние продолжительности перемешивания на степень разложения ашарита серной кислотой
рости растворения кальциевых минералов (гидроборацита, иньонита, улексита, колеманита и др.) в серной кислоте имеют максимумы, а вид кинетических кривых аналогичен изотермам растворимости сульфата кальция в водных растворах серной кислоты. Данные позволили предположить о наличии на поверхности растворяющихся частиц боратов пленок из дигидрата сульфата кальция.
Изучено также влияние температуры на степень разложения гидроборацита (рис. 6.5) и влияние продолжительности перемешивания на степень разложения ашарита серной кислотой (рис. 6.6).
Основным вопросом в процессе серно-кислотной переработки боратового сырья является выделение с высоким выходом образующейся борной кислоты из раствора, содержащего сульфат магния.
Определение выхода В20з в процессе кристаллизации борной кислоты из растворов, содержащих сульфат магния, можно производить графически с помощью диаграммы растворимости (рис. 6.7) либо рассчитывать исходя из уравнения материального баланса процесса кристаллизации. В первом варианте количество борной кислоты (в кг на 100 кг исходного раствора) определяют как отношение длины отрезка луча кристаллизации борной кислоты от точки исходного раствора до соответствующей изотермы (а) к общей длине луча кристаллизации, т.е. от 100%-ного содержания Н3ВО3 до пересечения с изотермой (с). Выход В20з (%) в твердую борную кислоту вычисляют по формуле
° 56,4 1ЛА
т|=----------100,
с [В2О3]Н
где [В2Оз]н — начальная концентрация В20з в растворе. 242
В точку 100% Н3ВО3 (56,4%В2Оз)
Рис. 6.7. Диаграмма для расчета политермической кристаллизации борной кислоты. Точки составов кристаллогидратов: А — MgSO4-7H2O; Б—MgSO4-6H2O;
В — MgSO4H2O
Количество выкристаллизовавшейся борной кислоты во втором случае определяют по формуле
56,4—[В2О3]М
где [В20з]м — концентрация В20з в маточном растворе, %.
Борную кислоту из боратовых руд получают следующим образом. Исходную руду дробят на щековой, а затем на молотковой дробилке и передают в шаровую мельницу на мокрый помол. В шаровой мельнице руду смешивают с маточными растворами в отношении Т:Ж = 1:1—1,5. Руду измельчают до величины частиц, не превышающей 1 мм. Полученную пульпу разбавляют маточными растворами и направляют в первый из четырех реакторов. В первый реактор по ходу поступает непрерывно расчетное количество серной кислоты. Образующаяся реакционная масса из четвертого реактора поступает 243
на ленточный вакуум-фильтр. Осадок-шлам на фильтре промывается горячей водой и удаляется гидротранспортом в шламонакопитель. Фильтрат подвергается контрольному фильтрованию на автоматическом фильтр-прессе (ФПАКМ) и поступает в трехкорпусные вакуум-кристаллизаторы, где охлаждается до 15—20° С. Для удержания в растворе оксидов железа и алюминия раствор, поступающий в кристаллизатор, должен быть кислым (0,2—0,6% свободной серной кислоты). Кристаллы борной кислоты отделяют от растворов на центрифуге ФГЛ-1800 и промывают холодной водой или конденсатом. Кристаллы сушат на барабанной вращающейся сушилке горячим воздухом при 90—100° С.
Маточный раствор и промывные воды возвращают в процесс на стадию мокрого помола исходной руды и разбавление пульпы перед разложением. Значительную часть маточного раствора, содержащего 1,3—2,5% В2О3, 20—22% MgSC>4 и 0,2—0,5% H2SO4 выводят из технологической системы производства борной кислоты, нейтрализуют оксидом магния и подвергают сушке в распылительных сушилках с получением боромагниевого удобрения.
С учетом переработки в перспективе бедных боратовых руд в УНИХИМе под руководством К.В.Ткачева разработан флотационный метод получения борной кислоты.
В отличие от существующих традиционных методов во флотационном (рис. 6.8) весь маточный раствор, образующийся на стадии кристаллизации борной кислоты, возвращают на стадию разложения исходной руды. В результате этого раствор, поступающий на вакуум-кристаллизацию, концентрируется сульфатом магния и в процессе его охлаждения происходит совместная кристаллизация борной кислоты и гептагидрата сульфата магния. Суспензию из вакуум-кристал-лизаторов подвергают флотации, в процессе чего борная кислота поступает в пенный продукт, а сульфат магния — в хвосты. Пенный продукт основной флотации проходит три перечистки. В третью пе-речистную флотацию поступает маточный раствор с центрифуги, на которой отделяется борная кислота. Конечный концентрат репульпи-руется водой от промывки борной кислоты и увлеченные в пенный продукт кристаллы сульфата магния растворяются. Борную кислоту отделяют на центрифуге, сушат и упаковывают. Хвосты флотации сгущают, отделяют гептагидрат сульфата магния на центрифуге и подсушивают его для удаления поверхностной влаги.
Флотационный метод позволяет реализовать в производстве замкнутый по растворам технологический процесс, повысить степень извлечения борного ангидрида в целевую борную кислоту до 85% и получить параллельно с основным продуктом и побочный — гептагидрат сульфата магния.
244
Боратовая руда
Серная кислота
Маточный раствор и промывная вода
Маточный раствори промывная вода
Н3ВО3 MgSOr7H?O
Рис 6.8. Схема получения борной кислоты флотационным способом
Отходом в процессе переработки боратовых руд серно-кислотным способом является шлам, который может быть легко переработан на борогипс. В УНИХИМе проведены исследования, показавшие возможность использования его в качестве добавки в процессе помола цементного клинкера.
Получение из других видов сырья. Разработан способ получения борной кислоты из синтетического бората кальция СаО-ВгОз-иНгО из необогащенной датолитовой руды углекислотным способом. Для этого разлагают технический борат кальция, содержащий 42—44% сесквиоксида бора. Суспензию бората кальция готовят перемешиванием его с маточными растворами, образующимися в процессе кристаллизации борной кислоты, и содержащими около 2,7% сесквиоксида бора. При этом соблюдается массовое отношение маточного раствора к борату кальция, которое составляет 6:1. Процесс обработки исходной смеси серной кислотой проводят при 90—95° С. Продолжительность процесса разложения — 30—40 мин. Дозировку серной кислоты регулируют по значению pH реакционной среды, которое поддерживают в пределах 2—4.
Перспективным видом сырья для производства борной кислоты являются данбуритовые руды и концентраты, практически не растворимые в серной кислоте и других кислотах. В УНИХИМе разрабо-245
тан способ получения борной кислоты путем предварительной переработки данбурита при 980—1000° С в течение 15—20 мин.
В процессе термообработки данбурит разлагается по схеме
СаО В2О3 • 2SiO2 -> СаО • В2О3 + 2SiO2
Полученная масса обрабатывается серной кислотой. Степень разложения при этом составляет не менее 90%, а дальнейшая переработка сырья идентична производству борной кислоты из датолитового сырья.
В УНИХИМе разработан непрерывный процесс серно-кислотной переработки колеманита по схеме
2СаО • ЗВ2О3 + 2H2SO4 + 7Н2О = 6Н3ВО3 + 2CaSO4
В процессе стабилизированной подачи колеманита тарельчатым питателем серную кислоту дозируют в первый реактор трехкаскадных реакторов. Значение pH в реакционной массе поддерживают в интервале 1,0—2,0. В третьем реакторе процесс проводят при pH 4,9—5,2, подавая колеманитовую руду для осаждения примесей железа в виде его гидроксида. Затем суспензию фильтруют, подкисляют серной кислотой до pH 2,5—3,0 и выдерживают при 90° С в течение 30—40 мин для кристаллизации дигидрата сульфата кальция. После фильтрации растворы охлаждают до 20° С. Образующиеся кристаллы отфильтровывают, промывают холодной водой и сушат.
Шлам репульпируют горячей водой, фильтруют, промывают и выводят из системы. Полученные промывные воды смешивают с маточными растворами и направляют в первый реактор разложения.
Борную кислоту можно также получить из колеманита обработкой его водной суспензии диоксидом углерода по схеме
2СаО • ЗВ2О3 + 2СО2 + 9Н2О = 6Н3ВО3 + 2СаСО3
При этом исходную руду подвергают термообработке при 550—600° С, просеивают через сито 1,25 мм. Полученный полупродукт разлагают в автоклаве при парциальном давлении угольной кислоты 0,2 МПа. Образующийся раствор после разложения содержит 5,5—6,0% сесквиоксида бора и минимальное количество примесей вследствие избирательного действия угольной кислоты. Способ обеспечивает степень извлечения бора ~93%.
Разработан и применяется в промышленности способ получения борной кислоты из колеманита разложением его карбонатом аммония. 246
6.3. ОКСИДЫ БОРА
Известны следующие оксиды бора: сесквиоксид (В20з), монооксид (ВО), диоксид дибора (В2О2), полимерный оксид (В2О2)Х, гемиок-сид (В2О). Известны также ВО2 (ДЯ^р для газа 271,8 кДж/моль), В6О и другие оксиды.
Физико-химические свойства оксидов бора. Сесквиоксид бора В20з — бесцветное стеклообразное или кристаллическое вещество.
Стеклообразный оксид (плотность 1,812 г/см3; ДЯ^, =-1254,9 кДж/моль) имеет слоистую структуру с расстоянием между слоями 0,185 нм; атомы бора расположены внутри равносторонних треугольников ВОз; длина связи В—О 0,145 нм; плавится при 325—450° С; обладает высокой твердостью.
Кристаллический сесквиоксид в обычных условиях существует в виде модификации гексагональной решетки (а = 0,433 нм, с = 0,832 нм, z = 3, пространственная группа Ccm2i; плотность 2,46 г/см3), которая при 400° С и 2200 МПа переходит в моноклинную. Температура плавления 450° С, температура кипения 2250° С; С® = 62,76 Дж/(моль-К); ДЯ^ = 24,56 кДж/моль, ДЯ®ет=433 кДж/моль (ОК), АЯ^р = -1273,5 кДж/моль, ДС^ = -1178 кДж/моль, ДЯ„ерех(ща кристал-лический->стеклообразный 18,6 кДж/моль; S®98 = 53,97 Дж/(моль-К).
Сесквиоксид В20з термически стоек, не восстанавливается углеродом вплоть до 1000° С. В присутствии углерода в процессе нагревания с галогенами образует галогениды, с азотом — нитрид, энергично взаимодействует с водой, образуя борную кислоту (Н3ВО3). В расплавленном состоянии растворяет оксиды многих металлов. Применяется в качестве компонента в производстве специальных стекол, керамики и эмали.
Монооксид ВО существует при высоких температурах в газообразном виде; ДЯ®6р = 9,9 кДж/моль, энергия диссоциации 879 кДж/моль.
В процессе нагревания сесквиоксида с элементным бором при 1000° С в парах образуются линейные молекулы диоксида дибора О = В—В = О. Установлено, что прочность связи В—В превышает 418 кДж/моль; ДЯ^р =-461,7 кДж/моль. В процессе охлаждения паров диоксид дибора диссоциирует с образованием В20з и В, а при быстром охлаждении до 300° С может быть получен аморфный и очень реакционноспособный полимерный оксид (В2О2)Х.
В процессе взаимодействия бора с сесквиоксидом под давлением около 6000 МПа при 1500° С образуется кристаллический ге-миоксид бора В2О со слоистой структурой типа графита; ДЯ^р для газа 271,8 кДж/моль.
Сесквиоксид В20з гигроскопичен. При соприкосновении с влагой воздуха на его поверхности образуется борная кислота.
247
Рис 6.9. Система В2О_?— Н2О
Получение сесквиоксида бора. Сесквиоксид бора получают в процессе обезвоживания борной кислоты. Из диаграммы системы В2О3—Н2О (рис. 6.9) видно, что борная кислота Н3ВО3 плавится конгруэнтно при 170,9° С, в которой Н3ВО3 теряет воду и переходит в метастабильную ромбическую модификацию у-НВО2. В процессе дальнейшего повышения температуры в интервале 158,5—176° С у-НВО2 переходит в моноклинную модификацию Р-НВО2, а в интервале 169,6—200,9° С — в стабильную кубическую модификацию метаборной кислоты а-НВО2.
Безводный ромбоэдрический сесквиоксид бора кристаллизуется и пла-
вится при 450° С. Установлено также, что сесквиоксид бора кристаллизуется лишь из расплавов, контактирующих с а-НВО2. При атмосферном давлении кристаллическая форма В2О3 получается лишь в процессе дегидратации Н3ВО3 в присутствии затравки а-НВО2. Изученные системы показывают, что для получения стеклообразного и кристаллического сесквиоксида бора требуются различные условия и соответственно способы.
Кристаллический сесквиоксид получают дегидратацией борной кислоты при 200° С в вакууме над оксидом фосфора (V).
Кристаллический продукт получают также медленным нагреванием борной кислоты при 225—230° С в открытом сосуде при атмосферном давлении и перемешивании. В процессе таких технологических условий происходит постепенная дегидратация и при содержании в расплаве 8—15% воды (соответствует 40—73% остаточной НВО2) — кристаллизация, начинающаяся через несколько суток.
В УНИХИМе разработан способ получения пористого сесквиоксида бора обезвоживанием борной кислоты при абсолютном давлении около 6,6 кПа и температуре 250° С. Образующиеся куски пористого продукта разламывали и использовали для получения аморфного продукта, который содержит (%): В2О3 — 96; Fe — 0,003—0,004; нелетучие вещества — 0,02; Н2О — по разности.
Процесс дегидратации интенсифицировали путем повышения температуры до 280—300° С и снижения остаточного давления до 1,3—2,0 кПа. В этих условиях получен хорошо сыпучий продукт с насыпной массой 600—700 кг/м3. Процесс дегидратации борной кислоты прово-248
дят в вакуум-сушильном шкафу. Использованние механической сушилки типа Венулет обеспечило достижение степени использования исходного сырья 98—99%. В температурном интервале 186—225° С, остаточном давлении 0,9—1,3 кПа и продолжительности процесса дегидратации 4 ч получен порошкообразный продукт, содержащий 98,67% основного вещества и с насыпной массой 800 кг/м3. Разработан непрерывный процесс под вакуумом с удельной производительностью аппарата 192 кг/(м3 сут). Выход сесквиоксида бора составляет 98%. Расход электроэнергии 5 кВт-ч на 1 кг целевого продукта.
Приведенные результаты показывают, что система В2О3—Н2О может находиться в метастабильном состоянии. При давлении водяного пара и борной кислоты в газовой фазе 1,3—2,0 кПа чистая борная кислота переходит в сесквиоксид, минуя стадию образования НВО2 по схеме: 2Н3ВО3 = В2О3 + ЗН2О. При общем же давлении паров от 1,7 до 6,7 кПа дегидратация Н3ВО3 идет с образованием промежуточных соединений НВО2 и гидрата неопределенного состава:
84-96* С 118-143* С 145-150* С
Н3ВО3 = НВО2 + Н2О; НВО2 = хВ2Оз*уН2О = хВ20з + уН2О
При общем давлении паров в пределах 8,5—12,6 кПа процесс дегидратации борной кислоты происходит с образованием метаборной кислоты и воды, после удаления которой получаются вязкие, вспучивающиеся жидкости.
В УНИХИМе изучен процесс получения сесквиоксида бора под атмосферным давлением на модельной вращающейся печи. При постепенном повышении температуры со скоростью нагревания не более 7° С в мин, продолжительности процесса 40—60 мин и температуре на выходе из печи 260—300° С получен порошкообразный продукт с содержанием 98—99% В20з. При этом установлена возможность интенсификации процесса путем внесения в исходное сырье (Н3ВО3) 30—50% сесквиоксида бора. Процесс апробирован в печах кипящего слоя.
В связи со значительной вязкостью расплава плавленый сесквиоксид бора получают при температуре выше 800° С. Во избежание загрязнения металлами (из материалов аппаратов), особенно железом и кобальтом, процесс плавки проводят в специальных печах, подобных тем, в которых получают безводную плавленую буру. Основными принципами таких печей являются: осуществление непосредственного контакта между теплоносителем и слоем загружаемой борной кислоты, минуя теплопередачу через стенку и соприкосновение с охлаждаемой рубашкой. Это достигается сжиганием топлива в форсунке, расположенной над исходным сырьем в перевернутой топке, окруженной цилиндрической охлаждающей воздушной камерой.
249
Под плавильной печи, куда непрерывно подается исходная борная i кислота, имеет форму чаши и снабжен водяной рубашкой. В центре ! пода расположено кольцевое отверстие для непрерывного стока рас- I плавленного сесквиоксида бора, который стекает по слою загрузки в 1 выпускное отверстие, попадает на охлаждаемые валки и далее посту-| пает на измельчение и рассев. Полученный продукт представляет со-1 бой белый порошок, гранулометрически характеризующийся остатком! на сите 0,1 мм не более 2%.
Качество плавленого сесквиоксида бора реактивной квалификации ’ в соответствии с существующим ГОСТ 10068—78 должен отвечать •! определенным требованиям (табл. 6.4).
Таблица 6.4. Состав сесквиоксида бора (реактивного) по ГОСТ 10068—78
Показатель Чистый для анализа Чистый
Содержание оксида бора, %, не менее 98,5 98,0
Содержание, %, не более Веществ, не летучих при обработке 0,2 0,3
этиловым спиртом
Сульфатов 0,1 0,02
Хлоридов 0,2 0,005
Железа 0,002 0,005
Магния 0,0005 0,0001
Свинца 0,002 0,005
Кальция 0,002 0,010
6.4. ТРИГАЛОГЕНИДЫ БОРА
Галогениды бора все бесцветны. Молекулы ВНа1з имеют форму плоского треугольника с атомом бора в центре (.$р2-гибридное состояние бора). В обычных условиях BF3— газ, BCI3 и ВВгз — жидкости, a BI3 — твердое вещество.
В твердом состоянии галогениды имеют молекулярные решетки. В соответствии с увеличением длины связи и уменьшением ее энергии в ряду BF3—ВС1з—ВВгз—В13 устойчивость уменьшается.
Тригалогениды бора являются сильными акцепторами электронной пары, например BF3 присоединяет молекулы воды, аммиака, эфира, спирта и др.
В процессе гидролиза проявляется кислотная природа галогенидов бора, который протекает необратимо с образованием кислот:
ВС1з + ЗН2О = Н3ВО3 + ЗНС1
250
Наиболее изученными и широко применяемыми галогенидами бора являются трифторид и трихлорид бора.
Трифторид бора. Трифторид бора BF3 — бесцветный дымящий во
влажном воздухе и удушливым запахом газ. Молекула плоская, длина связи В—F 0,1295 нм, угол FBF 120°. Энергия диссоциации 1920 кДж/моль. Температура плавления —128,36° С, а кипения—100,3° С. Плотность газообразного 0,00307 г/см3 (при 20° С), а твердого—1,87 г/см3 (-130° С). С°=50,5 Дж/(моль-К); =4,62 кДж/моль, ДЯ“СП = 17,1 кДж/моль, ДЯ^р=-1119 кДж/моль, =254,3 Дж/(моль-К).
Трифторид бора растворяется в воде с частичным гидролизом (332,1 гв 100 г Н2О при 0° С, 772 МПа), а также во многих органических растворителях. При растворении в воде образуется ВЁз-Н2О (температура плавления 10,2° С, температура разложения 20° С):
F
1 /н F— В — Of
I
F
С аммиаком трифторид бора образует твердое вещество BF3NH3 (температура плавления 163° С)
F н I I F—В—N—Н I I F Н
хорошо растворимое в воде без заметного гидролиза.
Трифторид бора растворяется в водных растворах фтороводородной кислоты:
BF3 + HF = H[BF4]
Трифторид бора образует неустойчивые выше 20° С моногидрат ВРз-Н2О (температура плавления 10,18° С) и дигидрат BF3-2H2O (температура плавления 6,36° С). Трифторид бора — сильная кислота Льюиса. Образует аддукты со спиртами, эфирами, альдегидами, кетонами, органическими соединениями, минеральными и органическими кислотами, например эфират (C2H5)2-O-BF3. Бесцветная жидкость с температурой плавления — 60,4° С, кипения 125° С (с разложением), гидролизующаяся водой. Анизолят С6Н5ОСН3 • BF3 — бесцветная жидкость с температурой плавления 2° С и плотностью 1,207 г/см3. Разлагается при 25° С, гидролизуется водой, растворяется в 251
анизоле. Применяется в качестве рабочего тела в процессе разделения изотопов бора. Трифторид бора получают: 1) взаимодействием элементов 2В + 3F2 = 2BF3; 2) обработкой концентрированной серной кислотой смеси дифторида кальция (флюорита) с сесквиок-сидом бора по реакции
3CaF2 + В2О3 + 3H2SO4 = 2BF3 + 3CaSO4 + ЗН2О
Способ не обеспечивает чистоту целевого продукта по содержанию в нем тетрафторида кремния (до 2—3 мол. % SiF4).
Более чистый по содержанию соединений кремния продукт получают заменой плавикового шпата (флюорита) фторборатом натрия, аммония или калия:
6NaBF4 + В20з + 6H2SO4 = 8BF3 + 6NaHSO4 + ЗН2О
Получают также по реакциям:
Na2B4O7 • ЮН2О + 6CaF2 + 8H2SO4 = =4BF3 + 2NaHSO4 + 6CaSO4 + 17H2O
Na2B4O710H2O + 12HF = Na2O(BF3)4 + 16H2O
Na2O(BF3)4 + 2H2SO4 = 4BF3 + 2NaHSO4 + H2O
Фторид бора высокой чистоты получают по реакции
C6H5N2BF4 —*-> C6H5F + N2 + BF3
В процессе приведенных реакций вместо Na2B4(>7 может быть использован СаВО4, а вместо натриевых солей можно использовать аммониевые соли, например:
6NH4HF2 + 4НзВО3 = 4NH3 + ПН2О + (NH4)2O(BF3)4
(Ш4)2О(ВРз)4 + 3H2SO4 = 4BF3 + 2(NH4)HSO4 + H2SO4 • H2O
Трифторид бора можно также получить взаимодействием борной и фторсульфоновой кислот:
Н3ВО3 + 3SO3HF = BF3 + 3H2SO4
Трифторид бора применяют как катализатор многих органических реакций, в качестве наполнителя в счетчиках нейтронов.
Трихлорид бора BCI3 — бесцветный, дымящийся во влажном воздухе газ. Температура плавления—107,3° С, а кипения—12,6° С. 252
Плотность 1,343 г/см3 (11° С); уравнение температурной зависимости давления пара: 1g р (мм рт. ст.) = 2115 Г1 — 7,04 1g Г + 27,56 (186—285,5К); Ср°= 62,65 Дж/(моль-К); АЯ^П = 23,86 кДж/моль, АЯ^р ~ -404,5 кДж/моль; 52°98 = 289,35 Дж/(моль-К), молекула плоская, длина связи В—С1 1,73 нм.
Трихлорид бора растворяется в тетрахлориде углерода, тетрахлориде титана и тетрахлориде кремния. Гидролизуется в воде, образуя борную кислоту. Восстанавливается воздухом около 880° С по схеме
2ВС13 + ЗН2 = 2В + 6НС1
а при 450° С в присутствии катализатора (Си—А1) восстанавливается до гексагидрида бора:
2ВС13 + 6Н2 = В2Н6 + 6НС1
Фтор полностью вытесняет хлор из молекулы ВС1з:
2ВС1з + 3F2 = 2BF3 + ЗС12
а в процессе взаимодействия BCI3 с бромидами и иодидами бора при 20° С интенсивно образуются смешанные галогениды.
ВС1з — кислота Льюиса, образует аддукты с эфирами, аминами, хлоридами других элементов и некоторыми гидридами. Кислотный характер трихлорид бора проявляет и в процессе взаимодействия с жидким аммиаком:
BCI3 + 6NH3 = B(NH2)3 + 3NH4C1
Трихлорид бора реагирует со спиртами с образованием эфи-ров-боратов B(OR)3.
Трихлорид бора получают хлорированием бора, его сплавов при 1000° С или раскаленной смеси сесквиоксида бора (или Na2B4O7) с углем по следующей схеме:
2В + ЗС12 = 2ВС13
AICI3 + BF3 = ВС13 + A1F3
Трихлорид бора применяют для получения элементного бора высокой чистоты, борогидридов (В2Нб), борорганических и других соединений бора, а также в качестве катализатора процесса катионной полимеризации, реагента для удаления нитридов, карбидов и оксидов из расплавов алюминия, магния, цинка и меди.
253
6.5. НИТРИД БОРА
Нитрид бора существует в аллотропических модификациях. В процессе взаимодействия простых веществ образуется a-модификация с гексагональной атомно-слоистой структурой типа графита (рис. 6.10). Температура плавления 3000° С; С® = 19,71 Дж/(моль-К); ДЯ^=81 кДж/моль, ДЯ^р = -250,5 кДж/моль; = 14,81 Дж/(моль-К); давление пара над твердым нитридом бора при 2000 К 5 Па; полупроводник (табл. 6.5).
При высоких давлениях a-BN (рис. 6.11) переходит в кубическую (типа сфалерита, рис. 6.12) форму 0-BN и метастабильную, серого цвета, гексагональную (типа вюрцита) форму y-BN (рис. 6.13), которые по твердости близки к алмазу.
Нитрид бора устойчив в атмосфере кислорода до 700° С, разлагается горячими растворами щелочей с выделением аммиака. Наиболее реакционноспособным является a-BN.
При комнатной температуре со фторидом водорода образует NH4BF4] по схеме
BN + 4HF = NH4[BF4]
со фтором реагирует по схеме 2BN + 3F2 = 2BF3 + N2.
a-BN получают взаимодействием В2О2 с NH3 при 2000° С в присутствии восстановителя (обычно углерода), а также плазмохимическим методом путем подачи бора в струю азотной плазмы при 5000—6000 К или пиролизом смеси летучих соединений бора и азота при 1300—2300 К. (3-Модификация нитрида бора образуется из a-модификации выше 1350° С и давлениях выше 5 ГПа в присутствии щелочных или щелочно-земельных металлов или их нитридов, а также без них при более высоких давлениях (6—13 ГПа). Модификацию y-BN получают также из a-BN в основном с помощью ударного сжатия при давлениях выше 13 ГПа.
Рис 6.10. Структура a-нитрида бора
Рис 6.11. Диаграмма состояния нитрида бора
254
Рис 6.12. Кристаллическая структура атомов сфалерита
Рис 6.13. Кристаллическая структура атомов вюрцита
Таблица 6.5. Свойства нитрида бора
Показатель Кристаллические модификации
a ₽ Y
Цвет Кристаллическая решетка Параметры решетки, нм: а с Число формульных единиц в ячейке Пространственная группа Плотность, г/см3 Микротвердость, ГПа Модуль Юнга, ГПа Температура Дебая, К: по оси а по оси с Белый Гексагональная 0,2504 0,6661 2 Ptymmc 2,29 0,1—0,7 34—87 2000 410 Черный Кубическая (типа сфалерита) 0,3615 4 F43m 3,45 60—98 840 1720 Серый Гексагональная (типа вюрцита) 0,2550 0,4230 2 Pfymc 3,40 49—57 790 1460 1460
Нитрид бора a-BN применяется в производстве высокоогнеупорных материалов, термостойкого волокна, в качестве сухой смазки в подшипниках, является полупроводником или диэлектриком. Нитрид бора, обогащенный изотопом f0B, является поглотителем нейтронов в ядерных реакторах. Р- и y-BN применяются в качестве сверхтвердых абразивных материалов.
6.6. БОРАТЫ
Бораты (оксобораты) — соли борных кислот: метаборной НВОг, ортоборной Н3ВО3, а также не существующих в свободном состоянии полиборных Нзт.2ПВтОзт.п. По числу содержащихся в молекуле бората атомов бора делятся на моно-, ди-, тетра-, гексабораты и т. д.
255
В —О О-В' О7 'в о 'в-о'о-в о о о о 1111
Рис 6.14. Структура боратов
Структуры боратов включают борокислородные группировки, содержащие от одного до шести, а иногда и девять атомов бора
(рис. 6.14).
Массдоли В2О3> %
Рис 6.15. Диаграмма плавкости системы Na2O—В20з
Координационное число атомов бора три (борокислородные треугольные группировки) или четыре (тетраэдрические группировки). Борокислородные комплексы являются основой не только островных, но и более сложных структур — цепочечных, слоистых и каркасных полимеризованных.
Способы сочетания треугольных ВОз, тетраэдрических ВО4, а также треугольных и тетраэдрических структурных единиц между собой очень разнообразны. О разнообразии составов безводных оксоборатов можно судить по диаграмме плавкости системы Na2O — В2О3 (рис. 6.15).
Бораты — бесцветные аморфные вещества или кристаллы. Сведения об их свойствах приведены в табл. 6.6.
256
Таблица 6.6. Свойства неорганических боратов
Соединение Плотность, г/см’ Растворимость в воде (25° С), % (масс.) Температура обезвоживания,0 С Температура плавления безводной соли, °C Температура боратовой перегруппировки, °C
При родные бораты
Бура Na2B4O710H2O 1,71 2,5 40-400 742 —
Ашарит Mg2B2O5*H2O 2,72 0,14 615—660 1340 —
Гидроборацит CaMgB6Oir6H2O 2,17 0,22 170—440 — 780
Калиборит KMg2B, 1О9-9Н2О 2,13 1,6 100—600 815 650
Колеманит Са2В6О1г5Н2О 2,42 0,26 150—600 950 710
Пандермит Са4ВюО19‘7Н2О 2,42 0,18 120—600 — -800
Синтетические бораты
Натрия метаборат NaBO2-4H2O 1,91 20,2 58—306 968 —
Калия метаборат КВО2-4/ЗН2О 2,23 42,7 100—250 950 —
Калия пентаборат КВ5О8-4Н2О 1,74 3,4 50—350 780 457
Аммония тетраборат (NH4)2B4O7-4H2O — 8,76 87 — 192 (с разл.)
Магния метаборат Mg(BO2)2-8H2O 2,29 0,1 80—350 988 760
Кальция метаборат Са(ВО2)2-2Н2О 2,60 0,25 350—500 1154 700
Бария гексаборат ВаВбОю’4Н2О 2,3 0,52 180—450 — 670
Свинца метаборат РЬ(ВО2)2Н2О 4,9 — 160 600 540
6.7. ТЕТРАБОРАТ НАТРИЯ
Наиболее широко применяемым боратом в народном хозяйстве является тетраборат натрия КагВдО?. Существует в виде дека-, пента-, тетра-, ди-, моногидрата и безводной соли (табл. 6.7).
Декагидрат тетрабората натрия NazB^-lOHjO (минерал бура или тинкал) или Na2[B4O5(OH)4]-8H2O состоит из анионов объединенных 9 Химическая технология 257
неорганических веществ, кн. I
он
он
в цепи посредством водородных связей. Между цепями находятся ионы натрия и молекулы воды, которые, в свою очередь, связывают их между собой как за счет притяжения ионов друг к другу, так и через водородные ионы.
Декагидрат тетрабората натрия в сухом воздухе постепенно выветривается; температура плавления 60,8° С (инконгруэнтно, с образованием тетрагидрата). В процессе нагревания на воздухе декагидрат переходит в метастабильный пентагидрат НагВ^-бНгО (минерал тинкалконит), имеющий строение Na2[B4O5(OH)4]-3H2O; гигроскопичен; в процессе поглощения влаги из воздуха переходит в декагидрат, а в процессе нагревания до 136° С — в тетрагидрат Na2B4O7-4H2O (минерал кернит). Тетрагидрат имеет строение Na2[B4O6(OH)2]-3H2O. Из водных растворов он кристаллизуется выше 60° С. В процессе нагревания до 161° С тетрагидрат переходит в дигидрат (минерал метакернит), имеющий строение НагПМЖОН)*]. При 260° С образуется аморфный моногидрат, который при 380° С переходит в безводный стеклообразный Na2B4O7
Безводный тетраборат натрия Na2B4O7 имеет метастабильные Р (ромбическая) и у (моноклинная) модификации с температурой плавления соответственно 664 и 710° С; температура кипения 1575° С; в процессе охлаждения расплав легко образует стекла с плотностью 2,36 г/см3; в присутствии паров воды испаряется в виде смеси НВОг с NaBO2; расплав буры легко растворяет оксиды металлов; гигроскопичен. Растворимость в воде 3,1% (25° С), 9,5% (50° С). Водные растворы тетрабората натрия обладают буферными свойствами. Растворяется при 25° С в этаноле — 0,05%, ацетоне — 0,006%, не растворяется в диэтиловом эфире, глицерине, а с метанолом образует борорганические соединения.
Декагидрат тетрабората натрия (бура) является сырьем в производстве борной кислоты и других соединений бора, компонентом флюсов для сварки и пайки металлов, шихты для глазурей, эмалей, стекла и керамики, моющих средств, электролитов для осаждения никеля и других металлов, протравой в процессе крашения, антисептиком, консервирующим средством для обработки кож и пропитки древесины. Он применяется также в качестве микрокомпонента хи-258
мических удобрений; исходным сырьем в процессе получения гербицидов, антифризов, изоляционных материалов, крахмала и клеев.
Таблица 6.7. Свойства тетрабората натрия и его кристаллогидратов
Показатель a-Na2B4O7 Na2B4OrlOH20 Na2B4Or5H2O Na2B4Or4H2O
Сингония Параметры решетки, нм: Триклинная Моноклинная Тригональная Моноклинная
а 0,65445 1,18790 1,109 0,70172
b 1,058 1,06440 — 0,91582
с 1,04855 1,22012 2,107 1,56774
а, град 93,279 — — —
Р, град 94,870 108,617 — 108,861
у, град 90,843 — — —
Число формульных единиц в ячейке 4 4 9 4
Пространственная группа Р\ С2/с Я32 Р2\!с
Плотность, г/см3 2,279 1,705 1,88 1,903
1,471 1,446 1,401 1,445
Nm 1,493 1,468 — 1,473
1,528 1,472 1,474 1,488
, N,„ и Ng— соответственно меньший, средний и больший показатели преломления.
Способы получения тетрабората натрия. Тетраборат натрия в производственных условиях получают перекристаллизацией природной буры или кернита, взаимодействием природных боратов и, в частности, боронатрокальцита, ашарита и других боратов со смесью карбоната и гидрокарбоната натрия, действием карбоната натрия на метаборат натрия.
Требования к качеству производимой в нашей стране буры приведены в табл. 6.8.
Таблица 6.8. Качество технической и пищевой буры (в %) по ГОСТ 8429—69
Показатель Техническая Пищевая
Na2B4O7>He менее 49,5 51,5
Не растворимый в воде остаток, не более 0,2 0,02
Na2SO4, не более 0,4 0,1
Na2CO3, не более 0,4 Отсутствует
Хлориды в пересчете на хлор, не более Не нормируется 0,005
Тяжелые металлы группы дисульфида » 0,01 0,004 0,001
водорода в пересчете на свинец, не более \\
Железо, не более
Мышьяк, не более »
Остаток после просева, не более 10
на сите 0,15 мм на сите 0,6 мм » Отсутствует
9*
259
Получение декагидрата тетрабората натрия из борной кислоты. Буру производят из борной кислоты взаимодействием ее с карбонатом натрия по реакции
4Н3ВО3 + Na2CO3 + 4Н2О = Na2B4O7 • ЮН2О + СО2
Исходная борная кислота содержит примеси сульфатов магния, железа и алюминия, которые параллельно с борной кислотой реагируют с карбонатом натрия по следующим уравнениям:
4MgSO4 + 4Na2CO3 + aq = 3MgCO3 • Mg(OH)2-nH2O + 4Na2SO4 + CO2
Рег(8О4)3 + 3Na2CO3 + 3H2O = 2Fe(OH)3 + 3Na2SO4 + 3CO2
A12(SO4)3 + 3Na2CO3 + 3H2O = 2A1(OH)3 + 3Na2SO4 + 3CO2
Образующийся при этом сульфат натрия переходит в раствор, а гидрат гидроксокарбоната магния и гидроксиды алюминия и железа (III) выпадают в осадок.
В процессе реакции исходный карбонат натрия дозируют до 2% избытка по стехиометрическим расчетам. Избыток карбоната натрия обеспечивает более полное использование исходной борной кислоты. При избытке карбоната натрия протекает нормальный процесс кристаллизации целевого продукта — декагидрата тетрабората натрия.
В УНИХИМе изучена система, отвечающая полю кристаллизации декагидрата тетрабората натрия (рис. 6.16). Согласно диаграмме системы, эта область находится между полями кристаллизации пентабората и метабората натрия. Кривые соответствуют составам растворов, насыщенных тетраборатом натрия при разных температурах. Выше кривых находится область ненасыщенных растворов, а ниже — суспензии кристаллов тетрабората натрия в насыщенном растворе. Прямая ОА (рис. 6.16) соответствует стехиометрическому отношению се-сквиоксида бора и оксида натрия. Как видно из диаграммы, в растворах с таким отношением растворимость декагидрата тетрабората натрия при 20—60° С наименьшая. Чем больше отклоняется отношение B2O3:Na2O в системе от стехиометрического для тетрабората натрия, тем выше его растворимость. С повышением температуры растет и растворимость тетрабората натрия.
Поскольку исходный карбонат натрия взят с избытком сверх расчетов 2%, а также возвратом маточных растворов на начало процесса, в производственных растворах постоянно присутствует карбонат натрия.
С целью уточнения влияния соды на ход технологического процесса, а также качества целевого продукта в УНИХИМе изучена растворимость в системе Na2SO4—Na2B4C>7—Н2О в присутствии 4% 260
N^O, %
Рис. 6.16. Область кристаллизации декагидрата тетрабората натрия в системе Na2O—В2О3—Н2О
Рис 6.17. Растворимость в системе Na2SO4—Na2B4O7—Н2О в присутствии Na2CO3 (4%)
Na2CC>3 (рис. 6.17). Изотермы растворимости показывают, что значительная часть ветви кривой, отвечающей состоянию насыщения сульфатом натрия при 90° С, при 35° С находится в поле кристаллизации декагидрата тетрабората натрия, а при 15° С — в поле совместной кристаллизации целевого продукта и сульфата натрия. Отсюда следует, что в процессе охлаждения растворов до 35° С, насыщенных сульфатом натрия, из них будет кристаллизоваться ИагВ^ЧОНгО без примеси сульфата натрия. Следовательно, для получения качественного ИагВдОтЧОНгО из растворов, насыщенных сульфатом натрия, не следует их охлаждать ниже 35° С.
Схема получения декагидрата тетрабората натрия из борной кислоты состоит из следующих основных стадий: растворение борной кислоты; растворение карбоната натрия; получение раствора тетрабората натрия; контрольное фильтрование; кристаллизация; отделение кристаллов тетрабората натрия от маточного раствора; сушка кристаллов. Согласно технологии (рис. 6.18), исходные борную кислоту и карбонат натрия растворяют в растворителях 1 и 2 с метальными устройствами. Растворителем является смесь маточных растворов с промывными водами, температуру которой в процессе растворения поддерживают в пределах 60—70° С. Концентрацию растворов карбоната натрия поддерживают 18—20%, а борной кислоты -10—12% (масс.). В процессе подачи растворов борной кислоты в реактор регулируют ее дозировку 261
Рис. 6.18. Схема получения декагидрата тетрабората натрия:
1 — растворитель борной кислоты; 2 — растворитель соды; 3— реактор; 4, 7—сборники;
5 — фильтр-пресс; 6 — репульпатор; 8— вакуум-кристаллизатор; 9 — сборник суспензий;
10— сборник маточного раствора; 11— центрифуга; 12 — транспортер; 13 — элеватор; 14 — сушильный барабан; 15 — бункер; 16 — узел упаковки
во избежание сильного вспенивания смеси в результате бурного выделения диоксида углерода из реакционной массы.
Реакционная масса при постоянном перемешивании подогревается глухим паром до 90—97° С в течение 30—60 мин. Образующийся раствор тетрабората натрия направляется в сборник 4 и фильтруется на автоматическом фильтр-прессе 5 (ФПАКМ). Образующийся в процессе фильтрации шлам содержит в основном карбонат кальция или магния (в зависимости от исходной борной кислоты) и механические примеси. Шлам в репульпаторе 6 разбавляют водой или маточным раствором и направляют в шламонакопитель или на стадию мокрого помола исходного сырья производства борной кислоты.
Фильтрат с содержанием тетрабората натрия 13±1% и карбоната натрия 1,5 ± 0,5% с температурой 90—95° С направляют в промежуточный сборник 7, откуда — в кристаллизатор 8. В кристаллизаторе растворы при интенсивном перемешивании охлаждают до 25—40° С. Продолжительность охлаждения в зависимости от температуры охлаждающей воды составляет 2—5 ч.
После кристаллизаторов суспензию направляют на центрифугу 11, где после отделения маточного раствора от кристаллов последние промывают холодной водой. Маточный раствор и промывную воду направляют на начало процесса для растворения исходной борной кислоты, а кристаллы тетрабората натрия — в сушильный барабан 14. Сушильным агентом служит горячий воздух с температурой 170—240° С.
262
Высушенный декагидрат тетрабората натрия элеватором 13 подают в бункер 75 готового продукта, а оттуда — в узел упаковки 16. Продукция упаковывается в четырехслойные бумажные мешки с полиэтиленовым вкладышем.
Отходами производства декагидрата тетрабората натрия является шлам и часть маточного раствора. Шлам в количестве 25 кг на одну тонну вырабатываемого целевого продукта содержит механические примеси и некоторое количество карбонатов кальция или магния, образовавшихся в результате реакции борной кислоты с карбонатом натрия. Кроме этих примесей шлам содержит Na2B4O7-10H2O и Na2CO2. Шлам разбавляют водой, перемешивают, а полученные растворы направляют на стадию мокрого помола боратовой руды в производстве борной кислоты.
Получение декагидрата тетрабората натрия из боратов. Разработан способ получения декагидрата тетрабората натрия из боросодержащего минерала улексита, содержащего 40—42% В2О3 реакцией его со смесью карбоната и гидрокарбоната натрия по схеме
2(Na2O • 2СаО • 5В2О3 • 16Н2О) + Na2CO3 + 4Na2HCO3 = = 5Na2B4O7 + 4СаСО3 + СОг + 34Н2О
Процесс проводят в две стадии. В первой стадии при 90—95е С с применением маточных растворов, содержащих 130—185 г/л тетрабората натрия и 20—35 г/л карбоната натрия.
Образующийся раствор отстаивается и отфильтровывается от не-растворившегося остатка породы и образующихся в ходе реакции карбоната кальция и гидроксида железа (III). Фильтрат содержит 290—325 г/л тетрабората натрия и 4—10 г/л карбоната натрия. Осадок в реакторе размешивают с водой, а образующуюся суспензию перекачивают в автоклав для вторичной реакции со смесью карбоната и гидрокарбоната натрия.
Процесс в автоклаве проводят при 120—130° С. Образующуюся суспензию фильтруют. Отмытый в фильтре шлам, содержащий до 1—3% сесквиоксида бора, транспортируют в шламонакопитель. Фильтрат направляют на первый реактор, а промывную воду — в автоклав.
При охлаждении до 45—48° С фильтрата, полученного в первой стадии процесса, кристаллизуют стандартный декагидрат тетрабората натрия.
Разработан также способ получения декагидрата тетрабората натрия из кернита. Согласно технологии, кернитовую руду (~25% В2О3) выщелачивают в реакторах при 90—95° С маточным раствором после кристаллизации. Образующийся при этом раствор направляют в вакуум-кристаллизаторы. Полученная суспензия в центрифуге разделяется 263
на кристаллы и маточные растворы. Кристаллы сушат в турбосушилках, состоящих из расположенных одна над другой вращающихся тарелок. Высушиваемые кристаллы декагидрата тетрабората натрия пересыпаются с тарелки на тарелку.
Если в исходном керните содержатся бораты кальция, то процесс в реакторе-автоклаве ведут в присутствии карбоната натрия, переводящего борат кальция в борат натрия.
Декагидрат тетрабората натрия получают также из гидроборацита. Согласно технологии, исходный концентрат гидроборацита обрабатывают в реакторе смесью маточного раствора с растворами от промывки кристаллов декагидрата тетрабората натрия в центрифуге и от промывки шлама. Затем в реактор вводят расчетное количество смеси карбоната и гидрокарбоната натрия. Суспензию в реакторе перемешивают в течение 2—3 ч при 95—99° С, после чего массу пропускают через ленточный фильтр. Образующийся шлам промывают холодной водой и направляют в шламонакопитель, а фильтрат после контрольной фильтрации передают в кристаллизаторы, а затем в центрифугу и на сушку.
Декагидрат тетрабората натрия из гидроборацитового концентрата можно производить и по кислотно-щелочному способу — с предварительным получением из него борной кислоты и последующей обработкой ее карбонатом натрия.
Для переработки ашаритовых боратов, плохо реагирующих с водным раствором карбоната натрия, предложен известково-содовый способ, при котором в реакционной массе образуется гидроксид натрия. Для осуществления этого способа исходный боратовый концентрат, частицы которого проходят через сито 0,15 мм, подвергают в течение двух часов термообработке при 600—650° С, а затем обрабатывают в течение 3,5 ч в реакторе смесью оксида кальция и раствора карбоната натрия при температуре кипения. Суспензию фильтруют, образующийся шлам подвергают вторичному разложению во втором реакторе, а затем и в третьем, после чего шлам, отфильтрованный и промытый горячей водой, направляют в шламонакопитель. В процессе выщелачивания в третьем реакторе шлам разлагают промывными водами, во втором — фильтратом от суспензии из третьего, а в первом — свежим раствором карбоната натрия в фильтрате от суспензии из второго реактора.
Фильтрат от суспензии из первого реактора перерабатывают в декагидрат тетрабората натрия, для чего содержащийся в нем метаборат переводят в тетраборат путем обработки раствора диоксидом углерода и кипячением, а затем направляют на кристаллизацию.
Другой способ сводится к термообработке измельченной ашарито-вой руды с оксидом кальция при 600—700° С в течение 1 ч. Охлажденный плав разлагают в реакторах раствором карбоната натрия с 264
добавлением маточных растворов и промывных вод. После фильтрации и промывки шлам может быть подсушен и использован в качестве борного удобрения или добавки к удобрениям. Фильтрат обрабатывают диоксидом углерода, после чего кристаллизуют декагидрат тетрабората натрия.
Разработан также способ переработки боратов, содержащих кальцит, путем термообработки их в смеси с сульфатом натрия. В процессе термообработки колеманита происходит следующая реакция:
2(2СаО • ЗВ2О3 • 5Н2О) + 3Na2SO4 = 3Na2B4O7 + 3CaSO4 + СаО + ЮН2О
Декагидрат тетрабората натрия выщелачивается водой из полученного плава, а из растворов после фильтрации кристаллизуют целевой продукт.
6.8. БОРАТЫ КАЛЬЦИЯ
Бораты кальция — бесцветные кристаллы, плохо растворимые в воде. В настоящее время известно около тридцати боратов кальция. Свойства основных соединений, образующихся в системе СаО—В20з—Н2О, приведены в табл. 6.9.
Гексаоксоднборат трикальция Саз(ВОз)2 получают при температуре выше 165° С гидротермальным методом, взаимодействием оксида кальция и сесквиоксида бора, а пентаоксодиборат дикальция Са2В2О5 гидротермальным способом при 285° С.
Дигидроксотетраоксодиборат дикальция Са2[В2О4-(ОН)]ОН выделяют из растворов в щелочной среде; выше 270° С — разлагается; из боратов кальция наименее растворим в воде (0,02% по массе в пересчете на В20з); в природе — минерал сибирскит.
Тетраоксодиборат (метаборат) кальция СаВ2О4 получают реакцией гидроксида кальция с борной кислотой в воде в виде ди-, тетра- и гексагидратов; в природе — минералы кальциборит (безводный), урал-борит (дигидрат), фроловит (тетрагидрат), пентагидроборацит (пентагидрат) и др.
Дигидрат тетраоксодибората кальция СаВ2О4-2Н2О получают в промышленности осаждением гидроксидом кальция при pH 11,8 и 20° С из растворов, образующихся после разложения диоксидом углерода боросиликатного сырья.
Гидроксопентаоксотриборат кальция Са[ВзО;(ОН)] — аналог природного минерала фабианита; пьезоэлектрик. Колеманит обезвоживается при 150—600° С.
265
Таблица 6.9. Свойства боратов кальция
Показатель о CQ б О б Са[В2О4 (ОН)]ОН <5 о О X О CQ «? О Са[В,О5 (ОН)] Колеманит Са[В3О4(ОНЬ]-ЗН2О Иньонит Са[ВА (ОН)5]-4Н2О Пандермит Са2[В50я (ОНЫ-2Н2О
Сингония Гексагональная Моноклинная Моноклинная Ромбическая Моноклинная Ромбическая Моноклинная Моноклинная Моноклинная
Параметры элементарной ячейки, нм: а 1,497 0,672 0,622 0,799 0,697 0,874 1,053
b — 0,5157 0,543 1,162 0,673 1,347 1,126 1,207 —
с — 0,7200 0,356 0,429 0,797 0,439 0,610 0,841 —
угол, град — 92,91 92,9 — 103 — 110,12 112,87 —
Число формульных единиц в ячейке 6 — 2 4 — 4 4 4 —
Пространственная группа Т^С R3c 1479 Р2х/а 1298 Р2\!а 420 Риса 1154 Р2\!с 350—400 Pbnh P2Ja 950 Р2Х1Ь 80—320 800
Плотность, г/см3 3,46 2,58 — 2,696 1, 870 2,72 2,42 1,875 2,415
кДж/моль -3429 -2650 — -2031 — — -3469 -4670 —
Бораты кальция (природные и синтетические) являются компонентом в производстве стекла, эмалей, керамики, микроудобрений. Природные бораты кальция применяются в качестве основного вида сырья в производстве борной кислоты.
Технология боратов кальция. Одним из самых широко применяемых соединений бора является борат кальция. К его качеству, согласно существующего ОСТ 6-08-9—79, предъявляют следующие требования, приведенные в табл. 6.10.
В УНИХИМе разработан углекислотный метод переработки минерала датолита в процессе термообработки с образованием новых фаз, содержащих бор, способных разлагаться угольной кислотой. В процессе разложения датолита угольной кислотой в водный раствор переходит борная кислота, а примеси исходного сырья остаются в твердой фазе. Однако при этом получают растворы борной кислоты невысокой концентрации. Поэтому малорастворимый борат кальция СаОВ2Оз-6Н2О осаждают из растворов при обработке известковым молоком.
Технология состоит из следующих стадий: термообработки датолитового сырья, углекислотного разложения продуктов термообработки и осаждения бората кальция.
Таблица 6.10. Состав бората кальция по ОСТ 6-08-9—79
Показатель Марка А Марка Б
Внешний вид Массовая доля В2О3, %, не менее Массовое отношение В2О3:СаО, не менее Массовая доля А12О3, %, не более Массовая доля Fe2O3, %, не более Массовая доля SiO2, %, не более Насыпная плотность, кг/м3, не менее Остаток на сите №1,25К по ГОСТ 3584—73, % Порошок белого цвета 44,25 1,22 0,08 0,05 0,4 500 Отсутствует Порошок белого цвета с сероватым оттенком 43,0 1,18 Не нормируется 0,1 0,6 400 Не нормируется
Термическая обработка датолитового сырья. Основными рудообразующими минералами датолитовых руд являются: датолит 2CaO B2O3-2SiO2 H2O, кальцит СаСОз, волластонит CaO SiO2, гранат 3CaO-Fe2O3-2SiO2, пироксены (ряд диоксид — геден бергит) CaOFeO-2SiO2, гизенгерит.
В процессе термообработки руд входящие в их состав минералы претерпевают следующие изменения.
Датолит. Боросиликаты, к которым относится и датолит, практически не взаимодействуют с угольной кислотой. Приведенная на
267
Рис 6.19. Тройная система плавкости SiO2—B2Oj—СаО. Обозначения: С—СаО;
В—В2О3
рис. 6.19 диаграмма плавкости системы СаО — В20з— SiO2 показывает, что в процессе нагревания обезвоженного датолита (точка D) в твердую фазу выделяется диоксид кремния (линия DE), затем силикат кальция CaO-SiO2 (линия Е13) и, наконец, устанавливается рав
новесие (точка 72), где присутствует твердая фаза, включающая SiO2,
CaOSiO2, СаО-ВгОз и расплав. Вновь образующийся диборат каль-
Рис. 6.20. Термограмма минерала датолита
ция (СаО В2Оз) может взаимодействовать со слабыми кислотами, в том числе и угольной, в результате чего появляется возможность избирательного извлечения бора в водный раствор.
Согласно приведенной на рис. 6.20 термограмме, датолит до 700° С не претерпевает изменений, а выше этой температуры (первый эндотермический эффект) происходят изменения, связанные с удалением конституционной во
268
ды. Процесс заканчивается при 800° С. В процессе термообработки до 800° С минерал не теряет кристаллической структуры. Несмотря на это, содержание углекислоторазложимого (у.р.) бора не превышает 17%. Это, по-видимому, объясняется тем, что в процессе обезвоживания параллельно образуется соединение с большей реакционноспо-собностью, чем у необезвоженного минерала (табл. 6.11).
Процесс обезвоживания датолита идет по реакции
2СаО • В2О3 -2SiO2 • Н2О ,0°-800'с > 2СаО • В2О3 • 2SiO2 + Н2О
тепловой эффект которой равен 50,699 кДж/моль. При 920—930° С (второй эндотермический эффект) датолит разлагается с образованием дибората кальция СаО-В2О3, силиката кальция p-CaO-SiQj с Ng = 1,632, Np = 1,615, кварца a-SiO2 с Nm = 1,543, кристобалита SiCh по реакции
2СаО • В2О3 -2SiO2 —92°-98О'с > СаО • В2О3 + СаО • SiO2 + SiO2
с тепловым эффектом 26,816 кДж/моль.
С достижением температуры 950° С содержание углекислоторазложимого (у.р.) бора достигает максимального значения. При 1000° С в кристаллической фазе присутствует лишь моносиликат кальция в двух модификациях: высокотемпературной (псевдоваллостонит) и низкотемпературной (волластонит). Диборат кальция и кварц образуют стекловидную фазу, которая не кристаллизуется в процессе охлаждения, в результате чего ее реакционная способность по отношению к угольной кислоте несколько снижается. При 1150° С все минеральные фазы плавятся, образуя боросиликатное стекло.
Высокая степень перехода a-бора в углекислоторазложимую форму достигается при продолжительности термообработки в течение 3—10 мин (рис. 6.21).
40 - мин
900 940 ЭВО 1020
900 940 980 1020 1060 t,°C
Рнс 6.21. Зависимость степени перехода a-бора от температуры и продолжительности термообработки
269
Таблица 6.11. Результаты термообработки датолита при различных температурах
Температура термообработки,0 С Потери при тер-мообработке, % Содержание в термообработанном датолите, % Степень разложения датолита угольной кислотой, %
в2о, (к.р.) В2О, (ур.) SiO2 (своб.)
20 — 21,35 1,13 0,91 —
500 0,18 21,45 1,22 1,18 0,42
600 0,33 21,32 1,44 1,20 1,46
700 2,40 21,90 4,10 1,30 13,70
820 5,90 22,22 5,00 1,30 17,40
900 6,40 22,52 4,70 1,27 16,00
920 6,50 22,52 17,61 10,57 73,10
950 6,50 22,67 21,70 16,04 90,60
1000 6,50 23,13 21,16 7,45 86,70
1150 6,50 22,70 16,90 Не опр. 74,50
Кальцит. Карбонат кальция, входящий в состав датолитового сырья, в процессе термообработки диссоциирует с образованием оксида кальция, который реагирует с продуктами терморазложения датолита— диборатом кальция СаО-В2О3 и оксидом кремния с образованием дикальциевого бората и силиката кальция по схеме
СаО + СаО • В2О3 = 2СаО • В2О3
СаО + SiO2 = CaSiO3
Рис. 6.22. Термограммы датолитовых руд: а — датолит-геденбергитовая; б—датолит-гранатовая; в—да-толит-геденбергит-гранаговая; г — дагол ит-кварц-карбонагная
Гранат. Гранат в процессе термообработки (до 1020° С) не претерпевает никаких изменений. С оксидом кальция гранат не взаимодействует.
Геденбергит. Геденбергит в процессе повышения температуры от 980 до 1020° С окисляется кислородом воздуха по реакции
4(СаО • FeS • 2SiO2) + О2 = = ЗСаО • Fe2O3 • 3SiO2 +
+ CaO-Fe2O3 + 5SiO2
В процессе термообработки датолитовых руд основными процессами являются обезвоживание и разложение датолита. На термограммах четырех разновидностей исходных руд (рис. 6.22) имеют место эндотермические эффекты при 730—740° С и 920—945° С, за исключением кварц-карбонатной руды, в которой эндотермический эффект появляется при 860° С, что связано с разложением кальцита. Высокая степень разложения продуктов термообработки руд достигается при температуре второго эндотермического эффекта (табл. 6.12).
Таблица 6.12. Степень перехода сесквиоксида бора в углекнслоторазложимую форму в продуктах после термической обработки руд
Руда Содержание в продуктах после термообработки, % Степень перехода в у.р.-форму, %
В2О3 (общ) BA (у.р)
Датолит-кварц-карбонатная 15,07 14,82 98,34
Датолит-гран атовая 10,26 8,49 82,74
Датолит-геденбергитовая 11,64 9,40 80,75
Датолит-геденбергит-гранатовая 13,20 11,93 90,96
Датолит 22,30 22,21 99,59
Углекислотное разложение продуктов термообработки. Суспензия, образующаяся в процессе мокрого помола термообработанного датолитового сырья, направляется на карбонизацию дымовыми газами известковых печей. Основная реакция процесса описывается уравнением
СаО • В20з(расплав) + ЗН2О + СО2 = СаСОз + 2Н3ВО3
В процессе одновременно с борной кислотой в раствор переходит и оксид кальция, концентрация которого повышается с возрастанием содержания борной кислоты. При этом pH раствора снижается с 10—11 до 6—7.
Равновесные состояния сесквиоксида бора и оксида кальция в жидкой фазе и состав твердых фаз в системе СаО—В20з—СОг—Н2О при разных парциальных давлениях диоксида углерода приведены на рис. 6.23. Из рис. 6.23 видно, что в равновесных условиях в процессе разложения боратов кальция угольной кислотой максимально возможная концентрация сесквиоксида в растворе при использовании диоксида углерода с парциальным давлением 0,01 Па (10% СО2) составляет 2,5%; при 0,03 МПа (30%) — 2,86%; при 0,1 МПа (100%) — 3,55% В20з. При избыточном парциальном давлении СО2 0,3 МПа концентрация В20з равна 4,06%. В равновесии с раствором в твердой фазе наряду с карбонатом кальция содержится неразложи-271
мый угольной кислотой гексаборат кальция. Образование последнего может происходить двумя путями: 1) взаимодействием образующейся в растворе борной кислоты с непрореагировавшим расплавом бората кальция по реакции
СаО • В20з(расплав) + 4Н3ВО3 = СаО • ЗВ20з • 5Н2О + Н2О
2) в условиях, близких к равновесным, после израсходования дибората кальция возможна следующая реакция:
СаСОз(тв) + 6Н3ВО3 # СаО-ЗВ2О3 • 5Н2О(тв) + СО2 + 4Н2О
Состав образующихся боратов можно рассмотреть из диаграммы растворимости системы СаО—В20з—Н2О при 80 и 25° С (рис. 6.24). При отсутствии угольной кислоты наряду с борной кислотой и гидроксидом кальция в твердой фазе могут существовать гексабораты кальция 1:3:5, 2:3:9 и диборат кальция 1:1:4. Поскольку поля существования гексаборатов кальция граничат, при растворении бората 1:3:5 в растворе может образоваться лишь борная кислота в равновесии с боратом 2:3:9.
Исходя из вышеизложенного, процесс разложения датолитового расплава угольной кислотой можно разделить на два периода. В первом периоде при недостатке угольной кислоты реакция протекает на поверхности частичек расплава. Борат кальция реагирует с водным раствором диоксида углерода с одновременным образованием борной кислоты в растворе и малорастворимого карбоната кальция. Этот кинетический период реакции характерен невысокой концентрацией се-сквиоксида бора в растворе, представленной в основном борной кислотой и равновесным с ней ионом B(OH)V
После повышения концентрации сесквиоксида бора в растворе и полимеризации ионов начинается реакция между СаО-В2Оз (расплава) и Н3ВО3. Наступает диффузионный период реакции. Таким образом, скорость процесса лимитируется процессами диффузии.
В производственных условиях процесс осуществляют во флотационной машине. На рис. 6.25 показана схема работы машины, используемой в виде блока аппаратов. Диоксид углерода засасывается импеллерами флотационной машины из коллектора газа и распределяется в виде мелких пузырьков по всему объему каждой камеры. Суспензия поступает на карбонизацию непрерывно. Продолжительность стадии карбонизации определяется суммарным временем пребывания суспензии в аппаратах всех стадий. Обычно она колеблется от 2 до 4 ч. 272
Рис 6.23. Изотерма растворимости системы СаО—В2О3—СО2—Н2О при 80° С:
АВС и D — эвтонические точки при pCOj 0,01; 0,03; 0,1 и 0,3 МПа соответственно
Рис. 6.24. Растворимость в системе СаО—В2О3—Н2О при 25° С (-----------)
и 80° С(-------)
Рис. 6.25. Флотационная машина
Осаждение дибората кальция. Борат кальция осаждается по реакции
2Н3ВОз + Са(ОН)2 + aq = СаО • В20з • «Н2О + aq
В производственных условиях процесс осуществляют в реакторе (рис. 6.26), в котором непрерывно отделяется кристаллический борат кальция от аморфной фазы, состоящий из двух зон — гравитационного
273
Раствор Са(ОН)2
отстаивания (разделения) и зоны кри- > сталлизации. Боросодержащие растворы взаимодействуют с известно- | вым молоком в зоне кристаллизации с образованием кристаллической и ' аморфной фаз. Образующаяся суспен- ? зия в восходящем потоке поступает в разделительную зону 4, в которой кристаллический продукт, обладающий более высокими по сравнению с аморфной фазой скоростями отстаивания, отделяется и возвращается в зону кристаллизации 5, а мелкодисперсные примеси выводятся из процесса с маточным раствором. Необходимое соотношение дозируемых реагентов, которое регулируется по значению pH 3 в реакционной зоне, а также фиксиро
Рис. 6.26. Реактор осаждения бората кальция:
1 — центробежный насос; 2 — успокоители; 3 — датчик рН-метра; 4 — разделительная зона; 5 — зона кристаллизации
ванные линейные скорости потоков в реакторе обеспечивают полное отделение кристаллического бората кальция от аморфного, а также однородность химического и гранулометрического составов продукта.
На рис. 6.27 приводится схема получения технического бората кальция. Дробленая руда поступает в расходный бункер исходного сырья 1, откуда вибрационным питателем 2 через ленточный автодозатор 3 подается во вращающуюся печь 4, где руда подвергается термообработке топочными газами от сжигания газа (мазута). Отходящие газы из печи проходят систему пылеочистки 5 и дымососом 6 выбрасываются в атмосферу.
Термообработанная руда поступает в мельницу мокрого помола 7, куда подают одновременно, растворы после второй стадии углекислотного разложения расплава. Мельница работает синхронно со спиральным классификатором 8, слив суспензии из которого направляют на первую стадию разложения во флотационную машину 9. Суспензия после разложения поступает самотеком в сгуститель 10, откуда осветленная часть, содержащая растворенную борную кислоту, сливается в сборник 14, а сгущенную суспензию через промежуточный сборник 11 перекачивают в корыто дискового вакуум-фильтра 12. Фильтрат направляется в сборник 14, а шлам после репульпации раствором третьей стадии разложения — во флотационную машину 15. Суспензия на выходе из флотационной машины направляется в сгуститель 16. Осветленный слив собирается в сборнике 19, а сгущенную часть транспортируют на дисковый вакуум-фильтр 18, откуда фильтрат поступает в сборник 19 и затем перекачивается на помол термообработанной руды в мельницу 7 и спиральный классификатор 8.
274
В атмосферу
Газ известковых печей
Рис 6.27. Схема получения технического бората кальция
Шлам после фильтра 8 репульпируют маточным раствором бората кальция из сборника 33 и подают на третью стадию разложения в флотационную машину 20. Суспензия после разложения поступает в сгуститель 21. Слив со сгустителя собирается в сборнике 24, а сгущенная часть через промежуточный сборник 22 и оттуда на дисковой фильтр 23. Фильтрат направляют в сборник 24 и оттуда на репу-льпацию шлама после первой стадии фильтрования, а шлам репульпируют водой и через хвостовой сборник 36 пульпы откачивают на шламохранилище.
Растворы после первой стадии разложения из сборника 11 подвергают контрольному фильтрованию на фильтр-прессе 25 и после охлаждения в теплообменнике 27 направляют на процесс осаждения бората кальция в реактор 28. В реактор же по заданному значению pH, измеряемому рН-метром 31, поступает через дозирующий клапан 30 известковое молоко. В реакторе 28 происходит осаждение продукта и разделение кристаллической и аморфной фаз. Последняя со сливом маточного раствора поступает в сборник 33, а сгущенную суспензию кристаллов отфильтровывают на ленточном фильтре 32. Фильтрат смешивают со сливом в сборнике 33 и направляют на ре-пульпацию шлама после второй стадии разложения, а влажный борат кальция — в сушильный барабан 34, откуда целевой продукт поступает в бункер 35 и на расфасовку.
6.9. ПЕРБОРАТ НАТРИЯ
Перборат натрия — комплексное соединение с формулой Naj[B2(O2)2(OH)4] • 6Н2О, что соответствует наименованию дипероксо-дибортетрагидроксогидрат натрия. Для технического продукта название «перборат натрия» и упрощенные формулы, встречающиеся в литературе (НаВОз-4НгО и ИаВОг-НгОг-ЗНгО), остаются наиболее применительными, поэтому используются и в этом учебном пособии.
Перборат натрия представляет собой кристаллический порошок белого цвета. Кристаллизуется в триклинной сингонии. Температура плавления 65±О,5°С, плотность 1731 кг/м3. Теоретическое содержание активного кислорода 10,38%. Теплота растворения в воде при 16,1° С — 48,416 кДж/моль. В процессе хранения в герметичной упаковке и комнатной температуре в течение года теряет всего 0,1% активного кислорода. В процессе сушки пербората натрия над фосфорным ангидридом или концентрированной серной кислотой при обычной температуре теряет три молекулы воды с образованием соединения ИаВОг-НгОг, содержащего 16% активного кислорода и способного разлагаться со взрывом. Дальнейшее обезвоживание приво-276
дит к образованию смеси NaBO2H2O2, NaBO2-xH2O и комплекса [(NaBO2)2O2], который бурно реагирует с водой с выделением кислорода и образованием метабората натрия.
В процессе упарки при низких температурах смеси пербората натрия с 30%-ным пероксидом водорода получено соединение с четырьмя молекулами пероксида водорода.
Растворимость пербората натрия в воде незначительна. В 100 г воды при 20° С растворяется около 2 г, а при 38° С около 3 г NaBO2-4H2O. В процессе растворения пербората натрия в воде гидролизуется с выделением пероксида водорода:
NaBO2 • Н2О2 ЗН2О -> NaBO2 + Н2О2 + ЗН2О
В процессе нагревания водного раствора пербората натрия до 50—60° С кислород выделяется с заметной скоростью, а при 100° С — бурно.
В кристаллическом безводном метаборате обнаружены циклические триборатные группы:
Перборат натрия широко применяется в качестве отбеливателя, окислителя и составной части синтетических моющих средств.
Способы получения пербората натрия. В химической промышленности применяют два способа получения пербората натрия: химический и электрохимический. Химический метод основывается на реакциях боросодержащего. Перборат натрия можно получить, используя в качестве исходного боросодержащего сырья борную кислоту:
Н3ВО3 + NaOH = NaBO2 + 2Н2О (6.1)
2Н3ВОз + Na2CO3 + Са(ОН)2 = 2NaBO2 + СаСОз + 2Н2О (6.2)
4Н3ВОз + Na2CO3 + 2NaOH = 4NaBO2 + СО2 + 7Н2О (6.3)
буру (тетрабората натрия):
Na2B4O7 + 2NaOH = 4NaBO2 + Н2О (6.4)
Na2B4O7 + Na2CO3 + Ca(OH)2 = 4NaBO2 + CaCO3 + H2O (6.5) природный колеманит:
2CaO • 3B2O3 + 3Na2CO3 + Ca(OH)2 = 6 NaBO2 + 3CaCO3 + H2O (6.6)
2CaO • 3B2O3 + 3Na2CO3 + 2NaOH = 6NaBO2 + 3CaCO3 + H2O (6.7) синтетический борат кальция:
СаО • В2О3 + Na2CO3 = 2NaBO2 + СаСО3 (6.8)
В качестве окислителя могут быть использованы пероксиды металлов, персоли, пероксид водорода, а также кислород воздуха.
В производстве обычно применяют 27—30%-ный раствор пероксида водорода, а в качестве боросодержащего сырья — концентрированный раствор метабората натрия. Процесс осуществляется по реакции
NaBO2 + Н2О2 + ЗН2О = NaBO2 • Н2О2 • ЗН2О (6.9)
Водный раствор метабората натрия, полученный по одной из реакций (6.1) — (6.8), очищают от примесей, вызывающих разложение пероксида водорода и целевого продукта. Для этого обычно применяют соединения магния, алюминия, цинка в виде их гидроксидов, силикатов, хлоридов или сульфатов. Их вводят в систему в процессе нагревания с последующим отстаиванием, а при необходимости и фильтрованием выпавшего осадка.
Химический метод получения пербората натрия сводится к получению раствора метабората натрия путем обработки исходного боросодержащего сырья карбонатом или гидроксидом натрия с последующим введением пероксида водорода (обычно в виде пергидроля). В качестве исходного боросодержащего сырья применяются синтетический борат кальция и борная кислота.
На рис. 6.28 приведена принципиальная схема получения пербората химическим способом. Растворы метабората натрия готовят путем разложения бората кальция гидроксидом или карбонатом натрия в реакторах при 70—95° С. Образующиеся при этом карбонаты сорбируют примеси тяжелых металлов. Процесс проводится при pH 10,4 и продолжается 30—60 мин. С применением в качестве исходного сырья бората кальция процесс проводят в каскаде реакторов, а отде-278
Рис. 6.28. Схема получения пербората натрия химическим методом
ляют и отмывают образующийся карбонат кальция на дисковых фильтрах в несколько стадий с промежуточной репульпацией.
В процессе с использованием борной кислоты и гидроксида натрия примеси, образующиеся в виде гидроксидов (железа, алюминия и др.), осаждают отстаиванием или фильтрованием. Роль сорбента выполняют соли магния, введенные в систему в качестве стабилизаторов в процессе получения метабората натрия. Концентрацию метабората натрия поддерживают на уровне 16%. Перед обменной реакцией растворы должны иметь температуру 15—20° С. Это диктуется избежанием потерь активного кислорода. В систему вносят стабилизатор — силикат магния или натрия, сульфат или хлорид магния, сульфаты алюминия, кадмия и др. Их количество колеблется в зависимости от качества исходного пероксида водорода. Для пероксида водорода, полученного электрохимическим методом, требуется в 1,5—3 раза меньше стабилизатора. Обычно количество стабилизатора составляет в пределах 5—15 кг на одну тонну целевого продукта.
Перборат натрия получают смешением растворов пероксида водорода и метабората натрия с одновременным охлаждением исходных реагентов до 10—30° С. Параллельно с основной реакцией протекает
279
Рис. 6.29. Растворимость в системе NaBO2—NaBO3 • 4Н2О—Н2О
процесс кристаллизации, теплота которой равняется 40,2 кДж/моль. Чем ниже температура в реакторе, тем ниже содержание целевого продукта в маточном растворе, растворимость которого в системе NaBC>2—NaBC>2 -НгСЪ—НгО минимальна в интервале концентраций метабората натрия в растворе от 3 до 7% (рис. 6.29).
Суспензия пербората натрия с достижением заданной температуры имеет отношение Т:Ж около 1:3. В случае применения в качестве исходного сырья бо-
рата кальция, маточный раствор после отделения кристаллов направляют на растворение карбоната натрия и далее на разложение бората. В случае же применения
борной кислоты и раствора щелочи часть маточного раствора выводят из системы на специальную переработку, а другую часть — на стадию получения раствора метабората натрия.
Осадок пербората натрия сушат воздухом, нагретым до 120—130° С; температура продукта на выходе из сушилки 40—45° С. Процесс сушки проводят в аппаратах кипящего слоя, трубе-сушилке или вращающейся барабанной сушилке.
Карбонат кальция (мел) сушат обычно в распылительной сушилке.
Электрохимический метод получения пербората натрия. Перборат получают этим методом, используя как анодный, так и катодный процессы. Катодный метод основан на
реакции катодного восстановления растворенного в электролите кислорода до пероксида водорода. В процессе катодного восстановления достигается высокий выход по току — 80% при равновесном содержании пероксида водорода в электролите 0,5% и плотности тока на катоде 0,002 А/см2.
Перборат натрия получают анодным окислением при электролизе раствора смеси тетрабората натрия и карбоната или гидроксида натрия на платиновом аноде.
Технологическая схема электрохимической стадии получения пербората натрия, усовершенствованная в УНИХИМе, приведена на рис. 6.30. Систему аппаратов (электролизеры, газоотделитель, теплообменник, кристаллизатор) единовременно при пуске заполняют электролитом— раствором, содержащим метаборат, карбонат, гидрокарбонат натрия и добавки. При включении тока в электролизерах 1 280
Электролизные газы
Рис 6.30. Принципиальная схема электрохимической стадии получения пербората натрия:
7 — электролизер; 2 — эрлифт; 3 — газоотделитель; 4 — теплообменник; 5 — кристаллизатор; 6 — смеситель; 7 — центрифуга
образуются перборат натрия (в растворе) и электролизные газы (водород и кислород):
rcNa+ + иВО7 + (4п + 1) Н2О -> «(NaBO2 • Н2О2 • ЗН2О) + 1/2Н2О2 + Н2
Уровни установки аппаратов выбраны с учетом обеспечения непрерывной циркуляции электролита в системе аппаратов электролиза и кристаллизации продукта.
В газоотделителе 3 электролизные газы отделяются от электролита, разбавляются воздухом до взрывобезопасной концентрации (менее 2% Н2) и системой вентиляции выбрасываются в атмосферу.
Отделенный от газов электролит с содержанием до 1—1,2% растворенного пербората натрия охлаждают в теплообменнике < доводя его до перенасыщенного состояния по перборату, и направляют в кристаллизатор 5. Кристаллизатор состоит из двух зон: кристаллизации и отстаивания. В зоне кристаллизации во взвешенном слое находятся кристаллы целевого продукта. Отношение Т:Ж в этой зоне составляет 1:3 — 5. В зоне отстаивания происходит разделение твердой и жидкой фаз — кристаллы пербората натрия поступают в зону кристаллизации, а электролит — в смеситель 6. Выходящий из кристаллизатора электролит в результате выпадения в твердую фазу пербората снижает концентрацию. Поэтому для корректировки его состава в смеситель 6 дозируется необходимое количество концентрированного метаборат-карбонатного раствора. Скорректированный электролит из смесительной воронки поступает в электролизер 2, и цикл замыкается.
Накапливающийся в зоне кристаллизации перборат натрия выводят в виде суспензии на центрифугу. Маточные растворы (электролит) направляют на приготовление метаборат-карбонатного раствора, а влажный продукт — на сушку и упаковку.
281
6.10. ТЕХНОЛОГИЯ БОРАТОВ МЕТАЛЛОВ
В УНИХИМе разработан метод получения боратов металлов в «сгущенном слое», по которому процесс проводят при комнатной температуре путем внесения соответствующего оксида или карбоната металла в суспензию борной кислоты. Борная кислота по мере расходования на реакцию растворяется, сохраняя при этом заданную концентрацию насыщенного раствора и постоянство значения pH. Способ позволяет получать бораты как щелочных и щелочноземельных, так и тяжелых металлов.
Технологическая схема получения боратов металлов в «сгущенном слое» представлена на рис. 6.31. В реактор 5 подают твердую борную кислоту из бункера 3 дозатором 4 и маточный раствор или воду (при получении боратов щелочноземельных металлов). В напорной емкости 9 готовят суспензию борной кислоты, которую также вводят в реактор 5. Из бункера 1 вводят дозатором 2 оксид или карбонат (а в некоторых случаях борат) соответствующего металла.
282
После перемешивания, продолжительность которого устанавливают опытным путем, суспензию бората щелочного металла передают на центрифугу 6, а образующиеся влажные кристаллы в бункер 10 и питатель 11, откуда — в барабанную сушилку 13. Маточный раствор собирают в сборник 7 и далее насосом 8 перекачивают в смеситель 9. В варианте получения боратов щелочноземельных металлов суспензию боратов с влажностью 25—30% питателем 12 направляют в барабанную сушилку 13. Целевой продукт сушат горячим воздухом, нагреваемым в калорифере 14. Отходящий из сушилки воздух очищают от пыли в циклоне 77, а орошаемым маточным раствором — в скруббере 18. Целевой продукт собирается в бункере 22, откуда направляется на упаковку.
Пентаборат калия КВ5О8 • 4Н2О (К2О • 5В20з • 8Н2О или 1:5:8) — белый кристаллический продукт плотностью 1,74 г/см3; растворимость в воде 3,4% (масс.) при 25° С; температура обезвоживания 50—350° С; температура плавления безводной соли 780° С; температура боратовой перегруппировки 457° С.
Из изотермы растворимость системы К2О—В2О3—Н2О при 30° С (рис. 6.32) следует, что во избежание загрязнения целевого продукта борной кислотой маточный раствор после кристаллизации должен содержать не менее 3% В2О3 и 1% К2О. Концентрация сесквиоксида бора в исходном растворе поддерживается на уровне 10—12%.
Пентаборат калия получают реакцией борной кислоты с карбонатом калия при 90—95° С по реакции
IOH3BO3 + К2СО3 = 2КВ5О8 + СО2 + 15Н2О
Образующиеся растворы фильтруют, охлаждают в вакуум-кристаллизаторе, а суспензию разделяют на центрифуге. Кристаллический борат калия КВ5О8 сушат воздухом с температурой 100—130° С. Маточный раствор направляют на растворение исходной борной кислоты.
В УНИХИМе разработан способ получения пентабората калия разложением бората кальция карбонатом калия и диоксидом углерода двухстадийным процессом. На первой стадии водную суспензию бората кальция разлагают карбонатом калия и содержащими диоксид углерода дымовыми газами. При этом образуется хорошо растворимый мета-стабильный борат калия по реакции
5(СаВ2О4 • иН2О) + 2К2СОз + ЗСО2 + aq=
= 5СаСОз + 2КВ5О8 + aq
Рис 632. Изотерма системы К2О—В20з—Н2О при 30° С
283
После фильтрации от карбоната кальция раствор обрабатывают диоксидом углерода:
4КВ5О8 + 2СО2 + aq = 2КВ5О8 • 8Н2О + 2КНСО3 + aq
Кристаллы целевого продукта после отжатия на центрифуге сушат. Раствор гидрокарбоната калия используют для приготовления суспензии исходного бората кальция.
На рис. 6.33 приведена схема получения пентабората калия, согласно которой исходные борат кальция и карбонат калия из бункеров 1 и 3 дозаторами 2 и 4 передают в смеситель 5, куда направляют также маточный раствор из сборников 18, 32 и 37 для репульпации. Подогретую до 90—95° С суспензию передают во флотационную машину 7 для разложения диоксидом углерода, который вводят в виде дымовых газов (8—10% СО2) печей термообработки исходного сырья или известковых печей (30%СО2). Далее суспензия самотеком поступает в сборник 8, в котором подогревается глухим паром до 80—90° С, а затем фильтруется на барабанном вакуум-фильтре. Осадок карбоната кальция с фильтра репульпируют горячей водой в репульпаторе 11, отделяют на вакуум-фильтре 16 и направляют на сушку. Фильтрат с фильтра 16 направляют в сборник 18 и подают в смеситель 5.
Продукционный фильтрат с фильтра 10 собирают с сборнике 14 и подают на контрольное фильтрование в фильтр-пресс 22 (ФПАКМ). Фильтрат из сборника 23 направляют в теплообменник 25 для охлаждения до температуры не выше 30° С. Охлажденный раствор из сборника 26 поступает в карбонизационную колонну 28. Процесс карбонизации осуществляют очищенными от механических примесей дымовыми газами или газами известковых печей, содержащими диоксид углерода. В процессе карбонизации раствор кристаллизуется; суспензию пентабората калия собирают в сборник 29, откуда насосом 30 подают в сгуститель 31. Сгущенную суспензию разделяют на центрифуге 36. Маточные растворы собирают в сборнике 37 и далее используют для репульпации, а влажные кристаллы пентабората калия — в бункере 40 и затем сушат в барабанной вращающейся сушилке 45. Процесс сушки осуществляют горячим воздухом при 100—130° С. Высушенный продукт рассеивают на грохоте 46. Мелкая и крупная фракции продукции направляются в сборник 23 на растворение, а товарная продукция — на упаковку.
Метаборат натрия. Тетрагидрат метабората натрия — NaBO2 • 4Н2О — представляет собой бесцветные кристаллы; плотность 1,91 г/см3; температура обезвоживания 58—306° С; растворимость в воде 20,2% (масс., при 25° С); температура плавления безводной соли 968° С; в процессе полного обезвоживания образуется аморфный продукт; легко разлагается кислотами; взаимодействует с растворами щелочей, карбонатов и гидрокарбонатов щелочных металлов.
284
Рис. 633. Схема получения пентабората калия
В основе технологии метабората натрия лежит растворимость солей в системе Na2O—В2О3—Н2О при разных температурах. Изотермы системы метабората натрия при 20 и 60°С приведены на j рис. 6.34. Прямая линия соответствует составам со стехиометриче- * ским для метабората натрия отношением Na2O : В2О3. В качестве исходного борсодержащего сырья применяют борную кислоту, синтетические бораты кальция и натрия. В процессе обработки боратов кальция определенную часть гидроксида натрия замещают карбонатом, добавляемым в достаточном количестве для перевода всего исходного кальция в виде карбоната в осадок.
УНИХИМом разработана технология кристаллического метабората с применением в качестве исходного сырья технического бората кальция. При этом установлены оптимальные условия процесса разложения бората кальция карбонатом натрия по реакции
СаО • В2О3 • 2Н2О + Na2CO3 + 6Н2О = NaBO2-4H2O + СаСО3 + 4Н2О
Температура разложения 90°С, отношение Т:Ж = 1:4, начальная концентрация растворов карбоната натрия 30%.
Исследованиями механизма разложения исходного бората кальция в растворах карбоната натрия установлено, что процесс протекает по кинетической области и подчиняется уравнению
т = рЯ(Ькс^гС0)), где т — продолжительность процесса полного растворения частиц исходного бората кальция радиусом R, с; р — плотность, кг/м3; к — константа скорости реакции, м/с; b — стехиометрический коэффициент бората кальция (в уравнении реакции равен 1); cn.,co3 — концентрация раствора карбоната натрия, кг/м3.
’ Основой технологии тетрагидрата метабората натрия является система взаимной растворимости NaBO2—Na2CO3—Н2О (рис. 6.35). Содержание компонентов в маточном растворе определяется составом эвтонического раствора при 25°С (точка Е): NaBO2 —12,57%; Na2CO3 — 21,25%.
С учетом внесения стехиометрических количеств исходных бората кальция и карбоната натрия на образование целевого продукта на диаграмме смесь исходных веществ условно обозначена точкой 100% NaBO2. На растворение подают смесь маточного раствора и воды, соответствующую на диаграмме точке П. Образующийся раствор (точка Р: NaBO2—30% и Na2CO3 —11%) охлаждается до 25°С, в результате чего происходит процесс кристаллизации твердого NaBO2 • 4Н2О, и образуется маточный раствор эвтонического состава (точка £). Существующими методами можно 286
Na2O, %
Рис 6.34. Изотерма системы Na2O—В2О3—Н2О при 20 и 60° С
Рис 6.35. Изотермы системы NaBOj—Na2COr~Н2О при 25 и 90° С
определить количество маточного раствора, образующегося в результате кристаллизации 1 т NaBO: • 4НгО; (РК1ЕР) у = 1,044 т и тем самым найти количество начального раствора 1,044 + 1,000 = 2,044 т и воды, необходимой для компенсации потерь в результате образования кристаллогидрата: (ПЕЮП)' 1,044 = 0,521 т.
В реальном технологическом цикле приведенные цифры корректируются с учетом стадии разложения исходного бората кальция и отделения шлама, а также потерь воды в результате испарения в процессах вакуум-кристаллизации, фильтрования и сушки целевого продукта.
6.11. БОРОВОДОРОДЫ
Бороводороды (бораны, гидриды бора), соединения бора общей формулы В„Нт (где п = 2—20), а т равно и + 4 или п + 6. Свойства некоторых бороводородов приведены в табл. 6.13.
Известны также октабораны B8Hi2, В8Н|4, В8Н]6 и В8Н18, нанобо-ран В9Н5, гексадекаборан В^Нго, октадекаборан В18Н22, эйкозобораны B2oHi6 и B2oH26. Из выделенных в свободном состоянии боранов В2Н6, В4Н10 — газы, В5Н9, ВбНю — жидкости, а ВюНм и выше — кристаллы.
287
Таблица 6.13. Свойства бороводородов
Соединение Температура плавления, °C Температура кипения, °C Плотность, г/см3 кДж/моль ДЯ° , crop» кДж/г
жидкого твердого
Диборан 0,447 0,557
В2н6 -165,5 -92,5 (-112°С) (-183° С) 36,60 -72,4
Тетраборан 0,56 0,59
В<Н10 -120,8 16,1 (-35° С) (-70°С) 57,68 -70,4
Пентабораны 0,65 0,63
в5н9 46,7 60 (0°С) (-46,7°С) 59,92 -69,2
ВзНц -122 63 — — 66,88 -69,2
Гексабораны ВбН1() ВбН12 -65 -82 108 80—90 0,69 (0°С) — 71,06 110,77 -66,9
Декабораны 99,7 213 0,78 0,94 -66,88 -66,3
В1()Н14 (100°С) (20° С) 108,68 (для газа)
0,87 145,5
B10H16 81 — — (20° С) (для газа) —
Для молекул бороводородов характерен дефицит электронов, высокие координационные числа атомов бора (до 7), наличие мостиковых связей В—Н—В, существование кластерных группировок из соединенных друг с другом атомов бора, которые в молекулах объединены в полиэдрические группировки — фрагменты икосаэдра (В ]2) или октаэдра (Be). Длины связей В—В 0,169—0,187 нм, В—Н в концевых группах 0,119—0,120 нм, во фрагментах В—Н—В 0,133—0,143 нм.
Бороводороды бесцветны, обладают резким неприятным запахом. Они легко смешиваются друг с другом, растворяются в большинстве органических растворителей.
Химически очень активны, особенно члены ряда В„НЛ + б- На воздухе окисляются до сесквиоксида бора, а при п = 2—6 самовоспламеняются. С водой образуют борную кислоту и выделяют водород. Легче всего разлагается водой диборан, который очень быстро гидролизуется при комнатной температуре. Спирты реагируют с бороводородами аналогично воде, с образованием водорода и алкилборатов. Бороводороды вступают в электрофильное и нуклеофильное замещение:
В.оН.4 + 2(CH3hS -» B10H12[S(CH3)2]2 + Н2
Бороводороды склонны к образованию соединений, содержащих комплексные анионы [ВН4]\ [В3Н8]~, [В9Н14], [ВюНвГ и др. Пи-288
ролиз бороводородов сопровождается полимеризацией и конденсацией образующихся низших гидридов, в процессе которого связи В—В не разрываются.
Получение бороводородов. Основным способом получения бороводородов является конверсия диборана по следующей схеме:
100°С,Н2
В соединении В2Н6 концевые группы ВН2 лежат в одной плоскости, атомы же бора и водорода в них образуют двухэлекгронные и двухцентровые связи. Два центральных атома водорода объединены с атомами бора трехцентровыми двухэлектронными связями В—Н—В, образующимися в результате перекрывания лр3-гибрид-ных орбиталей двух атомов бора и 5-орбитали атома водорода. Эффективный заряд на мостиковых атомах водорода — 0,22, на атомах бора +0,22. В молекуле В4Ню шесть двухцентровых связей В—Н, четыре трехцентровые В—Н—В и одна двухцентровая В—В. Молекулы В5Н9 и ВюНм, кроме связей описанных типов, содержат и трехцентровые связи В—В—В.
Согласно литературным данным, тетраборан образуется в процессе разложения пентабората В5Н11 в вакууме при -20° С в среде диглима, BgH]2 — в процессе взаимодействия диборана с пентабораном в вакууме, а В9Н15 — в процессе каталитической конверсии В5Нц при 0°С в присутствии гексаметилентетраамина. В2оН1б и В20Н26 получают в процессе пиролиза декаборанов соответственно при 350 и 100° С, а В1бН2о и В)8Н22 — при разложении (100° С) соответственно (СНз)28В9Н1з и (С2Н5)2ОВ9Н1з.
Способы получения диборана. Существует значительное число способов получения диборана. Они могут быть разделены на три группы: восстановление соединений бора водородом; восстановление галогенидов и других соединений бора простыми и комплексными гидридами; разложение боргидридов металлов и других производных бороводородов.
Ю Химическая технология 289
неорганических веществ, кн. I
Диборан получают по следующим реакциям:
2ВС13 + 6Н2 45°-с > В2Н6 + НС1
Си—AI
2BR3 + 6Н2 |40~160‘с > В2Н6 + 6RH 20—30 МПа
2BF3 + 6LiH згс > В2Н6 + 6LiF эфир
4BF3 + 3Na[BH4] 2О'С > 2В2Н6 + 3Na[BF4]
диглим
4BF3 + 3Li[AlH4] —2В2Н6 + 3LiF + A1F3
эфир
2Na[BH4] + H2SO4 ~l(>'c > B2H6 + Na2SO4 + 2H2
СбН5С1
2Na[BH4] + I2 20-6 > B2H6 + 2NaI + H2
ДИГЛИМ
ГЛАВА 7
АЛЮМИНИЙ И ЕГО СОЕДИНЕНИЯ
7.1. АЛЮМИНИЙ
Содержание алюминия в земной коре составляет 8,8% (масс.). По распространенности в природе занимает четвертое место среди всех элементов (после О, Н и Si) и первое место среди металлов. В свободном виде алюминий в природе не встречается, а содержится в различных минералах.
Важнейшими минералами, содержащими алюминий, являются: боксит, алунит (квасцовый камень) и нефелин.
Боксит представляет собой смесь гидроксидов алюминия — диаспора и белита А1ООН и гиббсита (гидраргиллита) А1(ОН)з. Алунит (Na, K)2SO4-A12(SO4)3-4A1(OH)3. Нефелин (Na, K)2O-Al2O3-2SiO2. В природе существует один стабильный изотоп 27А1.
Атом алюминия имеет электронную конфигурацию 3s23p; степень окисления +3, менее характерны +1 и +2 (лишь в газовой фазе выше 800° С); энергия ионизации А1°->А1+->А12+->А13+ соответственно 5,984, 18,828, 28,44 эВ; сродство к электрону 0,5 эВ; атомный радиус 0,143 нм. Ионный радиус А13+ (в скобках указаны координационные числа) 0,053 нм (4), 0,062 нм (5), 0,067 нм (6).
Свойства. Алюминий — серебристо-белый легкий металл; кристаллическая решетка кубическая гранецентрированная (а = 0,40403 нм, z = 4, пространственная группа Fm3m). Температура плавления 660° С, температура кипения около 2452° С; плотность алюминия 99,996%-ной чистоты 2,6989 (20° С) и 2,289 (1000° С) г/см3; С°= 24,35 Дж/(моль-К); ДЯ® = 10,9 кДж/моль; ДЯ®П = 302,13 кДж/моль; S®8 = 28 Дж/(моль-К); давление пара (Па) 0,266 (660° С), 13,3 (1123° С), 133 (1279° С); температурный коэффициент линейного расширения 24,58-10V (20—200° С; теплопроводность 1,24-10‘3 Вт/(м-К); R = 0,0265 мкОм-м; для алюминия 99,85%-ной чистоты т| (Н-с/м2) 2-10'3(800° С), 1,5 10’3(900° С), 1,З Ю’3 (1000° С); у (на границе с Аг) 0,86 Н/м (700—750° С). Слабо парамагнитен. Стандартный электродный потенциал А13+/А1°-Ц663 В в кислой среде и -2,35 В в щелочной. Модуль упругости 7-103 МПа; для отожженного металла твердость ю* 291
по Бринеллю 170 МПа, для холоднокатаного 270 МПа, (Траст соответственно 50 и 115 МПа, относительное удлинение 49 и 5,5%. При охлаждении ниже 120 К прочностные свойства алюминия в отличие от большинства металлов возрастают, а пластические не изменяются.
На воздухе алюминий покрывается тонкой прочной беспори-стой пленкой оксида, защищающего металл от дальнейшего окисления и обусловливающий его высокую коррозионную стойкость. По этой же причине алюминий не реагирует с концентрированной азотной кислотой. Алюминий легко взаимодействует с разбавленными хлороводородной, серной и азотной кислотами, образуя соответствующие соли — хлорид, сульфат и нитрат алюминия. Алюминий легко реагирует со щелочами, образуя соответствующие алюминаты.
Алюминий выше 200° С реагирует с элементной серой, давая сульфид AI2S3 — бесцветные кристаллы; температура плавления 1120° С, температура возгонки 1550° С (в токе азота); ДЯ^р—723 кДж/моль; разлагается водой и кислотами.
Алюминий при 500° С с элементным фосфором образует фосфид А1Р — желтовато-серые кристаллы, устойчивые до 1000° С; ДЯ^р —121 кДж/моль; разлагается минеральными кислотами и щелочами.
При взаимодействии расплавленного алюминия с бором образуются бориды А1Вг и AIB12 — желто-серые или коричневые кристаллы; температура плавления 2200° С; не разлагаются водой и кислотами.
С рядом металлов и неметаллов алюминий образует сплавы, в которых содержатся интерметаллические соединения — алюминиды, весьма тугоплавкие и обладающие высокой твердостью и жаропрочностью, например CuAh, CrAl7, TiAh, FeAh и др. Алюминиды выполняют роль модификаторов сплавов и придают изделиям высокие механические свойства.
Получение. Металлический алюминий получают электролизом раствора глинозема (технический А120з) в расплавленном криолите Na3[AlFe] при 960—970° С. Состав электролита: 75—90% масс. Na3[AlF6], 5—12% AIF3, 2—10% CaF2, 1—10% А12О3, с молярным отношением NaF:AlF3 = 2,20—2,85. Промышленный комплекс по производству алюминия включает и получение глинозема из алюминиевых руд, криолита и других фторидов, углеродистых анодных и футеровочных материалов и собственное электролитическое получение алюминия.
Процесс электролиза проводят в аппаратах, катодом в которых является подина ванны, а анодом — предварительно обожженные уголь-292
ные блоки или самообжигающиеся электроды, погруженные в расплавленный электролит. В расплаве протекают следующие реакции:
Na3[AlF6] 3Na++ 2F’ + A1F’4
A1F’4 *5 F + A1F3
AIF3 *5 F + AlF+2 aif+2 F + A1F2+ A1F2+ F + Al3+ AI2O3 A1O+ + A10’2; AIO 2 Al3 + + 2O2‘
AIO+ Al3+ + O2'; Al3+ + 3e -> Al 2O2' - 4e -► O2
Жидкий алюминий накапливается на подине ванны, на аноде выделяется кислород, образующий с его материалом оксид и диоксид углерода. На получение 1 т чернового алюминия расходуется 1,92—1,95 т AI2O3, 0,5—0,6 т анодного материала. Алюминий отбирают из электролизера один раз в 1—2 сут.
Алюминий высокой чистоты (не более 0,05% примесей) получают электролитическим рафинированием чернового алюминия, содержащего до 1% примесей. В качестве электролита чаще всего применяют расплав NaifAlFg], BaCh (до 60%) и NaCl (до 4%). Для получения алюминия особой чистоты (не более 0,001% примесей) применяют зонную плавку или химическую транспортную реакцию:
2А1(ж) + AlF3(r) 3AlF(r)
Применение. Алюминий используют в основном для получения алюминиевых сплавов. Чистый алюминий — конструкционный материал в строительстве. Его применяют также для изготовления токопроводящих и других изделий в электротехнике, для нанесения покрытий на стальные изделия и повышения их коррозионной стойкости. Из алюминиевого сплава был изготовлен корпус первого искусственного спутника Земли.
Алюминий в виде порошка и гранул является раскислителем чугуна и стали, восстановителем оксидов в производстве металлов, например хрома, марганца и кальция методом алюмотермии. Он применяется в качестве компонента твердых ракетных топлив, пиротехнических 293
составов и взрывчатых веществ. Алюминий в виде пудры и пасты применяется в качестве лакокрасочных материалов, а также как газообра-зователь в производстве ячеистых бетонов.
7.2. ОКСИД АЛЮМИНИЯ
Оксид алюминия А120з, бесцветные кристаллы; температура плавления 2044° С; температура кипения 3530° С. Стабильная до 2044° С кристаллическая модификация оксида алюминия — а-А12Оз (корунд), (рис. 7.1); решетка ромбоэдрическая, а = 0,512 нм, а = 55,25° (для гексагональной упаковки а = 0,475, с = 1,299 нм, пространственная группа z = 2); плотность 3,99 г/см3; ДЯ^ = 111,4 кДж/моль; уравнения температурной зависимости: теплоемкости С ° = 114,4 + + 12,9-10-3Т-34,3-105Г2 Дж/(моль-К) (298<Г<1800 К), давления пара 1g р = -54 800ДТ + 1,68) (до ~35ОО К); температурный коэффициент линейного расширения (7,2—8,6)-10’6К‘1 (300<Г<1200 К); теплопроводность спеченного при 730° С образца 0,35 Вт/(моль-К); твердость по Моосу 9; показатель преломления для обыкновенного луча «о 1,765, для необыкновенного пе 1,759 (табл. 7.1).
Таблица 7.1. Термодинамические свойства оксидов алюминия
Оксид CJ, Дж/(моль К) S^, Дж/(моль К) кДж/моль
а-А12О3 79,033 50,92 -1675,6
у-А120з 82,72 53,3 -1657
А1О (г) 30,883 218,275 67,011
А1О2 (г) 44,479 257,658 -81,259
А12О (г) 51,267 256,946 -144,143
А12О2 (г) 60,960 280,209 -405,697
• д/ 0>о
Рис 7.1. Структура а-А120з 294
Модификация а-А12Оз встречается в природе в виде минерала корунда, который часто содержит в растворенном виде оксиды других металлов, придающих ему различную окраску. Корунд может быть получен искусственно в результате термического разложения ромбической модификации А1ООН—диаспора или полиморфных переходов метастабильных форм А120з (у, г) и т.д.), которые образуются при термическом разложении кристаллических модификаций гидроксида алюминия А1(ОН)з — гиббсита и байерита и А1ООН — белита. Эти процессы могут быть представлены следующей схемой:
Гиббсит 250-300°^ Х.Д12О3 ж-жЧ ае-А12О3 1200°с
ОКОЛО 180°
_ 300-500°С А1 п 880°С 5 А1 ~ Ю50 С 12Оо°С Ai л
Бемит ► y-A12O3 "1 8-А120з——►0-А12О3—ot“Al203
180°
Байерит —£ г|(или т]+у)- А12О3 Диаспор
390-400°С
инертен и негигроско-
Модификация у-А120з имеет тетрагональную кристаллическую решетку типа шпинели (а = 0,562 нм, с = 0,780 нм); плотность 3,3—3,4 г/см3; содержит структурно связанную воду в количестве 1—2%. Существует также аморфный оксид алюминия — алюмогель, образующийся при обезвоживании гелеобразного гидроксида алюминия и представляющий собой пористое, а иногда прозрачное вещество.
Оксид алюминия не растворяется в воде, хорошо растворим в расплавленном криолите. Содержание в растворе криолита (92—94%) и оксида алюминия отвечает эвтектическому составу (рис. 7.2). Это дает возможность проводить процесс электролиза при более низкой температуре (800—1000° С, хотя температура плавления криолита 1100° С).
Оксид алюминия амфотерен. С аммиаком и аммиачной водой не реагирует. Химическая активность синтетического оксида алюминия сильно уменьшается с повышением температуры его получения. Природный и искусственный (образовавшийся выше 1200° С) корунд на воздухе при обычных условиях химически пичен. Около 1000° С интенсивно взаимодействует со щелочами и карбонатами щелочных металлов, образуя алюминаты:
2КОН + А12О3 = К2А12О4 + Н2О
Na2CO3 + А120з = Na2Al2O4 + СО2
Медленно реагирует с диоксидом кремния и кислыми шлаками с образованием алюмосиликатов. При сплавлении взаимодействует с KHSO4. Корунд, образовавшийся из диаспора при 500—600° С, взаимодействует также с растворами кислот и щелочей. Алюмогель и у-А120з, полученные при обжиге
295
о 10 юо
NagAIFe AI2O3
Масс, доли А12О3, %
Рис 7.2. Фрагмент диаграммы плавкости системы NasAlFe—AI2O3
гидроксидов алюминия при ~550° С, весьма гигроскопичны и химически активны, реагируют с растворами кислот и щелочей.
Сырьем для получения оксида алюминия являются бокситы, нефелины, алуниты и др. При соотношении в рудах AhOjiSiC^ > 6—7 их перерабатывают по способу Байера (основной метод), при Al2Oj:SiO2 < 6 (высококремнистое сырье) — спеканием с известью и карбонатом натрия.
По способу Байера измельченный в шаровых мельницах боксит выщелачивают в автоклавах оборотным маточным раствором алюмината натрия (после выделения из него части оксида алюминия) при 225—250° С. При этом алюминий переходит в раствор в виде алюмината натрия. В случае применения бокситов, содержащих гиббсит, выщелачивание можно производить при 105° С и обычном давлении в аппаратах с мешалкой. Растворы алюмината натрия разбавляют водой, отделяют шлам и разлагают в аппаратах с мешалкой или эрлифтом. При этом выделяется около половины образовавшегося гидроксида алюминия. Его отфильтровывают и подвергают термообработке во вращающихся печах или в кипящем слое при 1200° С. При этом образуется глинозем с содержанием 15—60% а-А12О3. Маточный раствор упаривается и направляется на выщелачивание новых партий боксита.
По другому способу получения оксида кремния высококремнистую руду (нефелин и др.) измельчают в шаровых мельницах, смешивают с карбонатом натрия и известняком в смесителе и подвергают термообработке во вращающихся печах. Полученную массу выщелачивают водным щелочным раствором в выщелачивателях. Образующийся при этом раствор алюмината натрия в вакуум-фильтре отделяют от шлама, затем освобождают от диоксида кремния, осаждая его в автоклаве при давлении около 0,6 МПа, а затем известью в реакторе при атмосферном давлении, и разлагают алюминат газообразным диоксидом углерода в другом реакторе. Образующийся гидроксид алюминия отделяют от раствора на фильтре и подвергают термообработке при 1200° С. В процессе переработки нефелина кроме глинозема получают карбонат натрия и цемент.
В производстве глинозема из алунитов параллельно получают серную кислоту и сульфат калия. При этом алунитовую руду подвергают термообработке при 500—580°С в восстановительной атмосфере, а полученный расплав обрабатывают раствором гидроксида натрия по способу Байера.
Монокристаллы выращивают зонной плавкой по методу Вернейля.
Синтетический а-АЬОз является сырьем в производстве металлического алюминия. Высокая прочность связи А1—О—А1 и плотная кристаллическая структура предопределяют большую энтальпию об-296
разования, высокую температуру плавления, его твердость и огнеупорность. Поэтому он широко применяется в качестве огнеупорного и абразивного материала в виде корундовых кругов и наждака. Его применяют в производстве керамических резцов, электротехнической керамики. Монокристаллы применяются в качестве лазерного материала, опорных камней часовых механизмов, ювелирных камней. Алюмогель, у-А120з и его смесь с т|-А120з являются адсорбентом для осушки газов (Н2, Аг, С2Н2) и жидкостей (ароматических углеводородов, керосина и др.), в хроматографии, носителями катализаторов (Со-МоОз, Pd, Pt).
7.3. ГИДРОКСИД АЛЮМИНИЯ
Гидроксид алюминия А1(ОН)з — бесцветное твердое вещество.
При действии щелочей на гидроксид алюминия ионы ОН' постепенно замещаются в аквокомплексах [А1(ОН2)б]3+ молекулами воды:
[А1(ОН2)6]3+ + ОН' = [А1(ОН)(ОН2)5]2++ Н2О
[А1(ОН)(ОН2)5]2++ ОН' = [А1(ОН)2(ОН2)4]++ Н2О
[А1(ОН)2(ОН2)4]++ ОН' = [А1(ОН)з(ОН2)з]0 + н2о
Одновременное протекание процесса полимеризации с образованием многоядерных комплексов приводит к выпадению осадка переменного состава А^Оз-иНгО
он, н |/°\ Н2О А1^ /\V но он \/л м ч у /\ «/ но^ ^онно^ н . /°\|> ,А1Ч УАЕ 1 \о/| \ он2 н он2 н о ОН^ \ н он2 ^рн
297
9 ai Qoh
Рис 7.3. Структура гиббсита А1(0Н)з
При стоянии осадок, постепенно выделяя воду, переходит в кристаллический гидроксид алюминия и теряет свою химическую активность — стареет.
Известны четыре кристаллические модификации (табл. 7.2) — моноклинный (у) и триклинный (у) гиббсит, или гидраргиллит, байерит (а) и нордстран-дит (Р), а также аморфный (гелеобразный) гидроксид алюминия переменного состава AI2O371H2O. Существуют также две кристаллические модификации оксигидроксида А1ООН — белит и диаспор. Все модификации гидроксида и оксигидроксида встречаются в природе в виде минералов, являющихся составной частью бокситов.
Гидроксид и оксигидроксид не растворяются в воде, типичные амфотерные соединения, с кислотами образуют
соли трехзамещенного алюминия, а со щелочами — алюминаты. Химическая активность убывает в следую
щем ряду: амфотерный гидроксид алюминия—>байерит->гиббсит-»бе
мит-> диаспор.
у-Гиббсит при 180° С разлагается до бемита, а выше 250° С до А120з. Структура бемита приводится на рис. 7.3. С°= 91,97 Дж/(моль-К); ДЯ^р = 1295,3 кДж/моль; 52°98 = 68,44 Дж/(моль-К). Он является промежуточным продуктом в производстве AI2O3 и алюминия по способу Байера. Может быть получен также при медленном пропускании диоксида углерода через растворы алюмината натрия по схеме
2NaA102 + ЗН2О + СО2 = 2А1(ОН)3 + Na2CO3
у-Гиббсит является полуфабрикатом в производстве других соединений алюминия, антипиреном лакокрасочных материалов и пластмасс, компонентом зубных паст, применяется в медицине в качестве обволакивающего и адсорбирующего средства.
Байерит при 180° С переходит в бемит, а выше 230° С в AI2O3. Образуется в процессе старения гелей гидроксида алюминия. Получают также в процессе пропускания диоксида углерода через растворы алюмината натрия. Применяют в качестве сырья в процессе получения п-АкОз. 298
Таблица 7.2. Свойства кристаллических модификаций гидроксида и оксигидроксида алюминия
Показатель Гиббсит Байерит Нордстрандит Бемит Диаспор
Кристаллическая решетка Параметры решетки, нм: Моноклинная Триклинная Моноклинная Триклинная Орторомбическая Орторомбическая
а 0,864 1,733 0,501 0,889 0,297 0,440
b 0,507 1,008 0,868 0,500 1,223 0,943
с 0,972 0,973 0,476 1,020 0,370 0,284
а — 94,17° — 92,56° — —
Р 94,34° 92,13° 90,27° 110,23° — —
У — 90,00° — 90,32° — —
Пространственная группа С5 2И — С52Л л77 2h г\16 о 2h
Число формульных единиц в ячейке 4 16 2 4 2 2
Плотность, г/см3 2,35—2,42 — 2,53 2,44 3,01—3,06 3,44
Твердость по Моосу 2,5—3,5 — — 3 3,5-40 6,5—7,0
Нордстрандит образуется в процессе старения гелей гидроксида алюминия.
Аморфный гидроксид алюминия получают осаждением его в виде гелей из растворов солей алюминия при пропускании аммиака на холоду или добавлением водных растворов аммиака или щелочей.
Бемит получают из гиббсита термообработкой его на воздухе при 180° С Или нагреванием водных суспензий в автоклаве при 200—250° С. Применяют бемит для получения у-А^Оз.
Диаспор при термообработке выше 450° С переходит в а-А^Оз.
7.4. ГАЛОГЕНИДЫ АЛЮМИНИЯ
К наиболее изученным и применяемым в народном хозяйстве галогенидам алюминия относятся его хлорид и фторид.
Хлорид алюминия А1С1з — белые дымящие на воздухе кристаллы с моноклинной решеткой (а = 0,591 нм, b = 1,024 нм, с = 0,616 нм, 0 = = 108,65°, z = 4, пространственная группа С2/ти); температура возгонки 180° С; тройная точка: температура 192,5° С, давление 0,228 МПа, плотность 2,44 г/см3; A77M = 35,3 кДж/моль, ДЯ^МГ = 115,7 кДж/моль (табл. 7.3).
299
Для А1(Ш) характерны координационные числа 6 и 4, что соответствует окта- и тетраэдрическому строению структурных единиц. В кристаллическом трихлориде алюминия его координационное число 6. При растворении его в воде образуются акваионы [А1(ОН2)б]3+, а при растворении в неполярных растворителях или в процессе возгонки образуются молекулы А12С16:
По причине бинарности и типичности вышеуказанных координационных чисел соединения трехвалентного алюминия в обычных условиях полимерны.
Таблица 7.3. Свойства хлорида алюминия
Показатель А1С1з А12СЦ
кристаллы газ газ
С°, Дж/(моль К) 91,1 71,9 157,2
кДж/моль -705,1 -584,1 -1293
5^, Дж/(моль-К). 109,3 314 472,7
300
На рис. 7.4 приводится строение молекул галогенида алюминия.
Хлорид алюминия в жидком состоянии и в парах до 440° С существует в виде димера, который при 440—800° С сосуществует с мономером. При 800—1000° С стабилен лишь мономер.
Хлорид алюминия растворяется в воде (44 г
QA1 QHal
Рис. 7.4. Строение молекулы Д12На1б
в 100 г при 25° С), спирте (100 г в 100 г при 12,5° С), ацетоне, три- и дихлорэтане, диэтиленгликоле, фосгене, нитробензоле, эфире, СС1д, сероуглероде и др.; не растворим в бензоле и толуоле. Из водных растворов кристаллизуется в виде желтовато-белого, расплывающегося на воздухе гексагидрата. В воде постепенно гидролизуется. Хлорид алюминия — кислота Льюиса. Образует комплексные соединения с аммиаком, хлоридами и оксихлоридами (PCI5, POCI3, NaCl и др.), а также со многими органическими веществами. Взаимодействует с расплавом алюминия, образуя монохлорид А1С1, существующий лишь в газовой фазе.
В литературе описаны следующие основные хлориды алюминия: AbOvHCl (А1ООН А1ОС1); А12(ОН)5С1-2Н2О [А1(ОН)г'А1(ОН)2С1-2Н2О]; А1С13-8А1(ОН)3-37,5Н2О; 2А1С13-7А1(ОН)3-18Н2О; А1С13-4А1(ОН)3-4Н2О; А1С13-4А1(ОН)3-2Н2О и А1С13-7А1(ОН)314Н2О.
При изучении процесса гидролиза основного хлорида алюминия А1С13-4А1(ОН)3-7,5Н2О, заканчивающегося образованием гидраргиллита или смеси его с байеритом (в зависимости от степени разбавления раствора), установлены промежуточные составы 2А1С1313А1(ОН)3-19Н2О и А1С13-9А1(ОН)3-(12,5±3)Н2О, в которых по мере гидролиза увеличивается число молекул А1(ОН)3, приходящихся на одну молекулу А1С13.
Отмечается различная степень полимеризации основного хлорида алюминия. Более подробно этот вопрос изучен в зависимости от соотношения (w)NaOH:AlCl3. Оказалось, что при и до 2,1 состав осадка остается постоянным и соответствует формуле А15(ОН)цС14 [иначе А1С13-А1(ОН)2С1-А1(ОН)2], а при увеличении п до 2,75 он изменяется от А15(ОН)цС14 до АЦ(ОН)11С1, т.е. осадок становится менее загрязненным анионами исходной соли алюминия. Наблюдаемое изменение состава основных хлоридов при повышении pH объясняется различной степенью полимеризации иона А1(ОН)2, который переходит в устойчивую форму А1(ОН)*2. Повышение значения pH от 4 до 7 способствует дальнейшей полимеризации по схеме
А110(ОН)22 -> А124(ОН)£ -» А154(ОНС
причем отношение величины заряда полимеризованного иона к числу атомов алюминия в нем уменьшается от 1 до 0,33. В этом интер-301
вале значения pH возникает значительное число различных по составу рентгеноаморфных гидроксидов алюминия, и лишь при pH 7—11 появляется устойчивая кристаллическая модификация — гидраргиллит.
Хлорид алюминия получают в производстве действием элементного хлора в присутствии восстановителя (углерод, оксид углерода) на обезвоженный каолин или боксит, а в лабораторных условиях взаимодействием хлора или хлороводорода с металлическим алюминием. Гексагидрат хлорида алюминия получают реакцией гидроксида алюминия с концентрированной хлороводородной кислотой с последующим высаливанием его из раствора хлороводородной же кислотой.
Хлорид алюминия применяется в качестве катализатора в органическом синтезе (в реакциях Фриделя — Крафтса), является полуфабрикатом в производстве металлического алюминия методом электролиза, реагентом для извлечения алюминия из сплавов и получения алюминия высокой чистоты. Гексагидрат хлорида алюминия применяют для очистки сточных вод, обработки дерева и т.д.
Фторид алюминия. AIF3, бесцветные кристаллы, существующие в нескольких модификациях. При обычных условиях устойчива а-модифи-кация с тригональной решеткой {а = 0,5039 нм, а 58,50°, z = 2, пространственная группа /?32); плотность 1,882 г/см3. Известна устойчивая до 710—720° С у-модификация с тетрагональной решеткой (а = 0,354 нм, с= 0,600 нм). Температура возгонки 1270° С; С° = 75,10 Дж/(моль-К); ДЯ^ЗГ=272 кДж/моль, ДЯ^р =-1510 кДж/моль; 5°^= 66,48 Дж/(моль-К); уравнения температурной зависимости давления пара:
1g р (Па) = 16,565—16 967/7’(980<7’<1123К);
1g р (Па) = 14,719—14 974/Т(1027<Г<1184К).
Фторид алюминия имеет координационную решетку (рис. 7.5). Он резко отличается по своим свойствам от остальных галогенидов, и прежде всего сравнительно слабой реакционной способностью.
Фторид алюминия плохо растворим в воде (0,41% (масс.) при 25° С), лучше растворяется в растворах фторида водорода, не растворяется в органических растворителях. Образует кристаллогидраты с 1, 3 и 9 молекулами воды. В процессе нагревания гидролизуется парами воды, слабо реагирует с концентрированной серной кислотой, разлагается растворами и расплавами щелочей.
Фторид алюминия AIF3 получают в процессе пропускания сухого газообразного фторида водорода над алюминием или оксидом алюминия при 450-—600° С по схеме
2А1 + 6HF = 2A1F3 + ЗН2
А120з + 6HF = 2A1F3 + ЗН2О 302
Рис 7.5. Координационная решетка A1F3
О Na 9 Al Qf
Рис 7.6. Структура гексафторалюмината натрия
AIF3 получают также взаимодействием водных растворов фторида водорода с А12Оз или А1(ОН)з по схеме
А120з + 6HF = 2A1Fj • ЗН2О
А1(ОН)з + 3HF = AIF3 • ЗН2О
Образующийся кристаллогидрат выделяют из растворов, сушат и обезвоживают термообработкой при 500—600° С.
В промышленности широко применяется способ получения фторида алюминия взаимодействием гидроксида алюминия с водным раствором фторида аммония:
А1(ОН)з + 3NH4F = AIF3 + 3NH4OH
или реакцией гидроксида алюминия с безводным NH4HF2
А1(ОН)з + 3NH4HF2 = (NH4)3[A1F6] + ЗН2О
Образующийся гексафторалюминат аммония подвергается термообработке при 450—550° С с получением высушенного трифторида алюминия.
Трифторид алюминия применяется в качестве компонента электролита (5—7%) в производстве металлического алюминия. Он входит в состав флюсов, эмалей, стекол, глазурей, керамики и покрытий сварочных электродов, является катализатором в органической химической технологии.
303
Трифторид алюминия, как и все галогениды алюминия, легко образуют галогеноалюминаты типа М3 AlFs, М2 AIF5, М A1F4. Наиболее устойчивым из них является гексафторалюминат натрия Na3AlF6. Его бесцветные кристаллы островного типа образованы ионами Na+ и A1F63’ (рис. 7.6).
Гексафторалюминат натрия плавится при 1011° С с разложением NaaAlFe —> 3NaF + AIF3, плотность его 2,97 г/см3; С® =216 Дж/(молыК); ДЯ0°6р =-3312 кДж/моль; AG^p=-3148 кДж/моль; 52°98 = 238 Дж/(моль-К); плохо растворим в воде (0,42% (масс.) при 25° С) и органических растворителях; взаимодействует с концентрированной серной кислотой; при нагревании гидролизуется парами воды. Расплав криолита обладает электрической проводимостью и является хорошим растворителем для AI2O3 и других оксидов.
Криолит в промышленности получают из фторида натрия и сульфата алюминия:
12NaF + A12(SO4)3 = 2Na3AlF6 + 3Na2SO4
Применяются и другие способы: действием карбоната натрия или фторида натрия на водный раствор гидроксида алюминия или AI2O3 в HF; взаимодействием водных растворов фторидов алюминия и натрия; взаимодействием гидроксида алюминия и карбоната натрия или алюмината натрия с водным раствором фторида аммония.
Криолит — редкий в природе минерал. Он применяется в качестве компонента (80—85%) электролита в производстве алюминия, металлургического флюса, компонента стекол и эмалей, наполнителя резины.
Гексафторалюминат аммония (ЫН4)з[А1Р6] — бесцветные кристаллы; плотность 1,837 г/см3 (20° С); ДЯ^р=-3060 кДж/моль; при нагревании разлагается постадийно до NH4[A1F4] и AIF3; растворяется в воде (7,66% (масс.) при 25° С), не растворяется в органических растворителях. Получают взаимодействием А1(ОН)з и AI2O3 с NH4F или NH4HF2- Является полуфабрикатом в производстве фторида алюминия.
7.5. АЛЮМОСИЛИКАТЫ
Алюмосиликаты — природные и синтетические силикаты, содержащие в составе комплексные анионы алюминия и кремния. Типичные примеры комплексных анионов — [AlSiO4]', [AlSi206]’,[AlSi4Oio]', [AhSijOio] • В алюмосиликатах алюминий играет такую же роль, как и кремний. Эти два элемента образуют смешанное соединение— алюмосиликат. Их можно рассматривать в качестве чистых си-304
ликатов, в которых часть кремнекислородных тетраэдров SiO*' заменена алюмокислородными тетраэдрами АЮ*’. Это частичное замещение в силикатах атомов кремния на атомы алюминия создает по причине их различных валентностей избыточный отрицательный заряд, компенсируемый внедрением в кристаллическую решетку алюмосиликатов катионов Na+, К+, Mg2* или Са2+, а иногда Ва2+ и Li+ . Таким образом, частичное замещение атомов кремния на атомы алюминия в SiO2 (Si4Og) дает алюмосиликатные ионы типа [AlSi3Og]’, [AHSijOg]2'. При замене же атомов кремния в ионах силикатов слоистой структуры SijO2- (или Si4O^’) атомами алюминия образуются слоистые алюмосиликатные ионы, например, типа [AlSi3Oio]5'.
Алюмосиликаты распространены в природе и составляют до 50% массы земной коры. Наиболее распространенными в природе являются полевые шпаты. Основные из них — минералы ортоклаз K[AlSi3Og], альбит Na[AlSi3Og] и анортит CaJAhSiiOg]; глинистые минералы — каолинит A12(Si2Oj)(OH)4, галлуазит (H2O)4[A12(Si2OsXOH)4], гидромусковит (иллит) Kx(H20)x[A12(AlSi3Oio)(OH)2.x-(H20)J и др.; минералы группы слюд, например мусковит KAl2[(AlSi3Oio)’(OH)2]. В настоящее время практическое значение имеет нефелин Na[AlSiO4], который перерабатывается с получением алюминатов калия и натрия, оксидов и гидроксидов алюминия, а также карбонатов и гидрокарбонатов калия и натрия.
Алюмосиликаты природного происхождения, не содержащие кристаллизационной воды и ОН-групп,— тугоплавкие, термостойкие и не растворимые в воде вещества. Они разлагаются лишь фтороводородной кислотой и расплавами гидроксидов и карбонатов щелочных металлов. Некоторые алюмосиликаты взаимодействуют с диоксидом углерода и водой, в результате чего выветриваются с образованием кварца и других минералов.
Синтетические алюмосиликаты получают гидротермальным синтезом, термообработкой А12О3 и SiO2 с оксидами или карбонатами металлов.
Природные алюмосиликаты применяют в качестве компонента шихты в производстве керамики, стекла и цементов. Слюды являются электро- и теплоизоляционными материалами.
Синтетические алюмосиликаты применяются в производстве керамических материалов. Особый практический интерес представляют цеолиты Мг/л АкОухЗЮг'уНгО, где п — степень окисления щелочного или щелочноземельного металла М. Они применяются в качестве адсорбентов в хромотографии, а также при очистке, осушке и разделении газов. Они являются реагентами в процессе умягчения воды, катализаторами, носителями катализаторов и др.
Алюмосиликат натрия (ультрамарин). Физико-химические свойства. Ультрамарин представляет собой алюмосиликат натрия, содержащий сульфид и полисульфид натрия. В зависимо-305
сти от способов его получения состав его меняется, поэтому в общем виде может быть представлен следующей формулой: (Na2O-Al2O3-wSiO2)xNa2S„.
С изменением значений т, х и п ультрамарин меняет свой цвет. Такое свойство позволило использовать его в качестве пигмента разных цветов (синий, фиолетовый и красный). Установлено, что значения т = 2,5—3,0 и х = 2,5—3,0 соответствуют ультрамарину синего цвета. Состав же пигмента насыщенного цвета, освобожденного отмучиванием от примесей, представляется формулой (Na2O,A12O3'2,8SiO2)2j5’Na2S5.
Для объяснения переменного состава ультрамаринов их представляли в виде смеси следующих индивидуальных соединений:
(ТЧагО • А12О3 • 2SiO2)3Na2S2 или NagA16Si6S2O24
зеленый малосернистый и малокремнистый;
(Na2O • А120з • 2SiO2)3Na2S или Na7A16Si6S2O24
синий малосернистый или малокремнистый;
(Na2O AI2O3 • 2SiO2)3Na2S4 или NagAlgSigS4O24
синий многосернистый и малокремнистый;
(Na2O-A12O3-3SiO2)2Na2S4 или Na6A14Si6S4C>2o
синий многосернистый и многокремнистый.
Однако выделить индивидуальные соединения не удалось и их существование остается гипотетическим.
Предложена следующая формула синего ультрамарина:
2(Na2O-Al2O3-3SiO2)3Na2S4
Принято считать алюмосиликат натрия скелетом, каркасом ультрамариновой структуры, а полисульфидную часть — ядром.
Рентгенографическими исследованиями различного состава и цвета установлено, что все они имеют одинаковую кристаллическую структуру, такую же, как натуральный ультрамарин из ляпис-лазури (синий драгоценный камень Lapis Lasuli). Основой их является простая анионная решетка [AUSisC^] , но в различных образцах степень изоморфного замещения Al Si неодинакова. Поэтому в более общем виде формулу решетки можно представить как [Al6+lSi6.IO24](6+x>_, где х может иметь положительное или отрицательное значение. Заряд анионов ней-306
трализуется ионами натрия. Было также установлено, что на местах дефектов решетки находятся полисульфиды натрия. Таким образом, решетка ультрамарина может быть изображена в виде сетки из тетраэдров SiO4 и АЮд, образующих замкнутые внутренние полости, в которые помещаются ионы натрия, серы (S2‘, S2‘ и 82или SO2 ). При этом часть ионов натрия закреплена, а другая часть ионов натрия, а также ионы серы находятся в блуждающем состоянии. Ультрамарины всех марок в отличие от цеолитов не содержат воды. Их цвет обусловлен серой. В центре элементарной ячейки содержится 2—4 атома серы. Часть серы в ультрамарине содержится в виде ионов серы (при действии кислот выделяется сульфид водорода), а другая часть является нейтральной серой (может быть удалена при нагревании в вакууме).
Содержание серы в ультрамарине составляет 11—12%, а содержание оксида кремния должно быть высоким, но не более 2,7—2,8 моль на 1 моль AI2O3, что соответствует так называемому силикатному модулю (массовому отношению 8Ю2/А120з) 1,60—1,65. Промышленность выпускает высококремнистые и высокосернистые ультрамарины. Плотность ультрамарина 2,2—2,7 г/см3, его насыпная плотность — 300—450 г/см3 в свободном состоянии и 550—800 г/см3 в утрамбованном виде.
Ультрамарин стоек к нагреванию до 300° С, при более высоких температурах, а именно при 350° С, отщепляет свободную серу, а при 690° С — связанную в виде полисульфидов серу.
Предположительно установлены причины изменения цвета ультрамарина. Цвет ультрамарина обусловлен двумя факторами: строением кристаллической решетки и характером связи в ней между серой и натрием. Варьирование цвета ультрамарина при сохранении его кристаллической решетки объясняют изменением характера связи между натрием и серой, что замечено в процессе обработки ультрамарина кислотой. При этом установлено, что белый ультрамарин всю содержащуюся в нем серу выделяет в виде сульфида водорода, зеленый ультрамарин половину серы выделяет в виде сульфида водорода, а половину — в виде сульфидной серы. Из синего ультрамарина выделяется лишь 25% серы в виде сульфида водорода, а 75% — в виде элементной серы. Отсюда следует, что в белом ультрамарине сера с натрием образует моносульфид (Na2S), в зеленом — дисульфид (Na2S2), в синем — тетрасульфид (№28д), а в фиолетовом и красном связь между натрием и серой отсутствует.
Физико-химические основы технологии. Исходным сырьем для получения ультрамарина являются: силикат алюминия, элементная сера, карбонат натрия, кремнезем и восстановитель. В качестве силиката алюминия применяют каолин: А12Оз*28Ю2-2Н2О. Имеющиеся две молекулы воды в исходном каолине удаляются при температуре распада кристаллической решетки 500—600° С в виде па-307
ров, необходимых для протекания промежуточных реакций в технологическом процессе.
Состав исходной шихты для производства ультрамарина следующий (%, масс.): каолина — 30, карбоната натрия — 30, элементной серы — 30, кремнезема—5 и восстановителя — 5, что соответствует отношению Al2C>3:SiO2:Na2CO3:S = 1:2,8:2,4:8. "
При термообработке исходной шихты при более низких температурах (200—400° С) образуется полисульфид натрия по схеме
3№2СОз + 10S — 2Na2S4 + Na2S2O2 + ЗСО2
Тиосульфат натрия при нагревании неустойчив и при 302—343° С разлагается:
Na2S2O3 —'—> Na2SO3 + S
При дальнейшем повышении температуры происходят следующие реакции:
Na2SO3 + 2С = Na2S + СО + СО2
Na2S + nS - Na2S„
Во всех стадиях процесса с повышенной температурой возможны реакции:
2Na2S2O3 + SO2 = 2Na2SO4 + 3S Na2S + Na2S2O3 Na2S2 + Na2SO2 Na2S2O3 + O2 —> Na2SO4 + SO2 so2 + c = s + co2 Na2SO4 + 2C = Na2S + 2CO2
2 Na2S2O3 + 3S = 2Na2S2 + 3SO2
Na2S2 + S = Na2S2 и т.д.
Образующиеся в рассмотренных химических реакциях полисульфиды натрия играют решающую роль во всех стадиях процесса: создают восстановительную среду, участвуют (хотя и косвенно) в процессе образования алюмосиликата натрия и выполняют свою прямую функцию продукта присоединения. Возможно также предположить, что из-за низкой температуры своего плавления (200—250° С) он 308
способствует некоторому уплотнению реакционной массы, что ускоряет протекание рассмотренных выше реакций.
Процесс образования алюмосиликата натрия начинается при температуре его обезвоживания и разрушения кристаллической решетки исходного каолина, проходит интенсивно и полностью заканчивается при -500° С. Реакция, возможно, протекает при участии гидроксида натрия, образующегося в результате разложения полисульфида парами воды, выделяющимися в процессе обезвоживания каолина:
Al2O3-2SiO2-2H2O —!-> Al2O3-2SiO2 + 2Н2О
Na2S„ + 2Н2О -> 2NaOH + H2S + (п— 1)S
С гидроксидом натрия реагирует продукт разложения каолина:
Al2O3-2SiO2 + 2NaOH = Na2OAl2O3-2SiO2 + Н2О
а выделяющийся диоксид серы в восстановительной среде образует элементную серу:
2H2S + SO2 = 3S + 2Н2О
Возможно также, что и исходный неразложившийся каолин реагирует непосредственно с образующимся гидроксидом:
2NaOH + Na2OAl2O3-2SiO2 = 2Na2OAl2O3-2SiO2H2O
В каолине, так же как и в алюмосиликате натрия, образующееся в ходе реакции на его основе мольное соотношение SiO2:Al2O3 составляет 2:1, а силикатный модуль— 1,18. Повышенное содержание диоксида кремния в исходной шихте способствует ее слабому спеканию, что благоприятствует протеканию процесса в твердой фазе. С целью повышения кремнистости ультрамарина в состав исходной шихты в производстве вводят наиболее активные формы кремнезема (диатомит или инфузорную землю). Процесс присоединения полисульфида натрия к алюмосиликату натрия происходит в интервале -670—730° С с возрастающей скоростью: -30% при 670° С, 70% при 700° С, а полностью — при 730° С. При этом возрастает содержание натрия и серы в целевом продукте. Завершающая стадия процесса — образование ультрамарина— происходит в интервале 730—-750° С и требует выдержки реакционной массы в этом температурном интервале -3—4 ч. Это связано с необходимостью повышения кристалличности и получения целевого продукта с необходимым дисперсным составом. Рентгеноструктурными исследованиями показано, что в продуктах термообработки
до температуры -500° С наблюдаются интерференционные линии исходного каолина, при 500—600° С — лишь кварца, при 670° С — смесь кристобалита, нефелина и ультрамарина (-30% каждого компонента), при 700° С — смеси ультамарина (-75%) и нефелина (-25%), при 730° С — ультрамарина с незначительной примесью кварца, а после выдержки при 730° С — ультрамарина.
Основные процессы, происходящие в процессе термообработки, можно представить в следующем виде: образование полисульфида натрия (200—400° С), обезвоживание и разложение каолина (450—500° С), образование алюмосиликата натрия (500° С), взаимодействие его с полисульфидом натрия и повышение кремнистости алюмосиликата (650—700° С), присоединение полисульфида натрия и постепенное образование ультрамарина (670—730° С) и резкая кристаллизация целевого продукта (730—750° С).
Чистый алюмосиликат натрия, полученный из водных растворов алюмината и силиката натрия состава Na2OA12O3-2,9SiO2, т.е. примерно такого же, как и при синтезе ультрамарина, аморфен и переходит в кристаллическое состояние лишь при высоких температурах (-1000° С) и более длительном нагреве. Процесс кристаллизации ускоряется значительным избытком полисульфида натрия, сильнощелочной средой и определенным температурным режимом.
Производство ультрамарина состоит из следующих стадий: 1) приготовление шихты; 2) термообработка шихты; 3) оксидирование зеленого полуфабриката; 4) промывка синего полуфабриката и его переработка в целевой продукт.
В качестве сырья в производстве применяют каолин, серу, карбонат натрия, инфузорную землю и пек. Состав шихты из перечисленных видов сырья, их исходное соотношение меняются в зависимости от их качества, метода производства и тех требований, которые предъявляются потребителями к целевому продукту. Пределы изменения шихты приведены в табл. 7.4.
Таблица 7.4. Состав применяемой для производства ультрамарина шихты
Наименование сырья Содержание в шихте,% (в расчете на 100%-ное вещество) Содержание основного вещества в сырье,%
I II
Каолин 30,3 25,6 96,9
Сера 29,4 34,6 99,5
Карбонат натрия 28,9 28,8 97,7
Инфузорная земля 5,5 7,5 86,9
Пек 3,6 3,5 97,8
В производстве применяют каолин, не содержащий заметных количеств примесей, особенно железа, присутствие которого контролирует-310
ся и в составе инфузорной земли. В качестве восстановителей применяют каменноугольный пек, древесный уголь и канифоль. Карбонат натрия не должен содержать его гидрокарбоната, выделяющего при нагревании пары воды, которые могут разрушать полисульфиды натрия. С целью лучшего измельчения и смешивания исходных компонентов их применяют лишь сухими, а в процессе сушки каолина при необходимости удаляют не менее половины его гидратной воды. Инфузорную землю после сушки измельчают в шаровых мельницах или дезинтеграторах. Смешение и размол составных частей шихты проводят в шаровых мельницах.
Полученную шихту подвергают термообработке в печах с обращенным пламенем, т.е. с отводом дымовых газов в нижней части печи. Процесс термообработки делится на три периода.
Первый период характеризуется образованием сульфидов натрия и повышением температуры в печи до 450° С. Он характеризуется равномерным нагреванием всех участков печи и отсутствием значительных количеств свободного кислорода. Режим регулируется подачей воздуха в печь.
Второй период характеризуется реакцией между полисульфидами с алюмосиликатом. В результате этой реакции образуется алюмосиликат натрия, содержащий полисульфиды. В зависимости от качества шихты, если она имеет высокую реакционную способность, в течение 2—3 ч при 725—750° С завершается процесс образования ультрамарина по всей реакционной массе. При термообработке же шихты с низкой реакционной способностью температуру реакционной массы поддерживают в пределах 770—780° С. При этом процесс образования ультрамарина продолжается в течение 6—9 ч.
Третий период термообработки является завершающим и состоит в оксидировании зелено-синего ультрамарина в синий, а избыточных полисульфидов и тиосульфата до сульфата натрия. Этот процесс, называемый томлением, протекает в период охлаждения печи.
Образующийся в процессе термообработки полуфабрикат представляет собой смесь ультрамарина (70—80%), сульфата натрия и тиосульфата, а в недоокисленном продукте содержится некоторое количество полисульфидов натрия.
С целью повышения качества целевого продукта освоен способ двухстадийного (раздельного) проведения технологических операций обжига (термообработки) и оксидирования. Приемущество этого способа проявляется в том, что при этом процесс оксидирования происходит при постоянном перемешивании массы зеленого полуфабриката.
По рассматриваемой технологии процесс обжига исходной шихты проводится в шахтной печи (рис. 7.7), состоящей из блока вертикальных камер 1, обогреваемых через боковые стенки дымовыми газами. Газы поступают из выносной топки, в которой сжигается природный
311
газ, движутся по вертикальным дымоходам, соединенным снизу и сверху в коллекторы. Движение газов по системе топка — печь — дымоходы осуществляется с помощью дымососа. Газы из реакционного пространства по мере их накопления и повышения давления удаляются через трубу, соединяющую камеру с атмосферой.
А-А Б-Б
Рис 7.7. Камерная (шахтная) печь для обжига ультрамарина:
1 — реакционная камера; 2 — греющий канал; 3 — загрузочный шлюзовой затвор; 4 — скользящий затвор для выгрузки; 5 — сборник-коллектор для отходящих газов; б — механизм затворов; 7 - кожух механизма; 8 — муровочное отделение; 9 — отверстие для взятия пробы; 10 — отверстие для термопары; И — керамические шиберы; 12 — бункер для выгрузки готовой продукции; 13 — отверстие для вывода реакционных газов
312
Шихту в камеры загружают сверху с помощью питателя, а зеленый полуфабрикат выгружают снизу через подвижной под. Длительность цикла составляет 45 ч.
Выгружаемый из печи зеленый полуфабрикат через специальный герметический затвор направляется в печь для окисления, которая представляет собой цилиндрический аппарат с метальным устройством. Процесс окисления происходит при постоянном перемешивании и непрерывной подаче воздуха в реакционную зону печи. Продолжительность процесса 4—6 ч. По окончании процесса окисления продукт с помощью метального устройства подается к разгрузочному люку, расположенному в нижней части печи.
Разработан более эффективный способ получения ультрамарина, при котором обжиг проходит во вращающейся муфельной печи, футерованной фасонными огнеупорными блоками. Печь снабжена герметическими устройствами непрерывной загрузки шихты и выгрузки зеленого ультрамарина (полуфабриката), а также для отвода реакционных газов, состоящих из сульфида водорода и диоксида углерода. Отдельные звенья печи хорошо уплотнены. Уплотнения оснащены отсасывающими устройствами, исключающими попадание сульфида водорода в рабочее помещение.
Муфельная печь обогревается продуктами сжигания природного газа. Скорость нагрева шихты составляет 100—150° С, что обеспечивает достаточно полное протекание всех рассмотренных раньше реакций при соответствующих температурах. Продолжительность термообработки при 750° С составляет 3—4 ч. Зеленый полуфабрикат, получающийся в печи обжига, направляется в окислительную печь. Полуфабрикат при этом охлаждается до 400° С.
Окислительная печь представляет собой металлический вращающийся барабан, разделенный на несколько секций. Барабан находится в огнеупорной камере, обогреваемой продуктами горения природного газа, сжигаемого в выносной топке. Зеленый полуфабрикат поступает в первую секцию печи, в которую по полой рубашке питателя подается и воздух для оксидирования, затем в следующую секцию, в которую также подается воздух с продуктом. Путем независимой регулировки поступления воздуха и теплового режима в секциях печи создаются условия для управления как первой активной, так и второй завершающей стадией процесса окисления.
Процессы обжига шихты были изучены в лабораторных условиях на установке периодического действия. Процесс обжига шихты проводили в закрытом тигле и в трубчатой печи, герметически закрытой с одного конца и присоединенной к системе улавливания отходящих газов с другого. При этом определяли влияние на качество целевого продукта состава шихты, степени измельчения и смешения исходных 313
компонентов, температурного режима процесса обжига, состава образующейся газовой смеси и условий окисления полуфабриката.
Было установлено, что наибольшее количество исходной серы внедряется в ядро при силикатном модуле 1,6 (рис. 7.8). Качество ультрамарина повышалось при увеличении степени измельчения исходной шихты и тщательности ее перемешивания. Зависимость содержания серы в ядре от степени измельчения составляющих шихты приведена на рис. 7.9. При изучении температурного режима процесса обжига установлено, что от продолжительности выдержки при температуре образования ультрамарина зависит содержание серы в ядре ультрамарина (рис. 7.10).
Схема получения ультрамарина приведена на рис. 7.11, согласно которой инфузорная земля из бункера 1 поступает в барабанную сушилку 2. После сушки она размалывается в шаровой мельнице 3 и поступает в шаровую мельницу 4. Туда же добавляется расчетное количество сухого каолина, серы, карбоната натрия и каменноугольного пека. Сухая и измельченная шихта поступает в бункер 5, откуда смесь транспортируется в печь для обжига 7. После обжига полученная масса оксидируется в печи 8. Ультрамариновый полуфабрикат выщелачивается водой, промывается репульпацией на вакуум-фильтрах 72, 14 и направляется для получения ультрамарина для малярных работ или продукта высшего сорта.
Для получения ультрамарина для малярных работ полуфабрикат сушат в барабанной сушилке 75, размалывают в трубчатой мельнице 77, упаковывают в мешки и направляют на склад. Для получения ультрамарина высших сортов промытый продукт подвергают мокрому помолу на шаровой мельнице 19, классифицируют в гидроциклоне 27, после коагуляции фильтруют на листовом вакуум-фильтре 23, затем сушат в гребковой сушилке 25, упаковывают в мешки и направляют на склад.
Рис 7.8. Зависимость содержания серы в ультрамариновом ядре 8Я от значения силикатного модуля исходной шихты а
314
Рис 7.9. Зависимость содержания серы в ультрамариновом ядре 8Я от степени измельчения исходной шихты
Продолжительность, с
Рис 7.10. Зависимость содержания серы в ультрамариновом ядре S, от продолжительности выдержки при температуре образования ультрамарина
Сера Каолин
Рис 7.11. Схема получения ультрамарина:
1, 5, 9, 16, 24, 26, 28 — бункер; 2, 15 — барабанная сушилка; 3, 4 — шаровая мельница; 6 — транспортное средство с полуфабрикатом; 7—печь для обжига; 8— печь для окисления; 10, 11, 20 — смеситель; 12, 14—вакуум-фильтр; 13, 18—репульпатор; 17 — трубчатая мельница; 19 — шаровая мельница мокрого помола; 21 — гидроциклон; 22 — реактор для кипячения и коагуляции; 23 — листовой вакуум-фильтр; 25— гребковая вакуум-сушилка; 27—смесительный барабан; 29 — автомат для мелкой фасовки; 30—весы; 31 — мешкозашивочная машина; 32 — погрузчик
Для получения ультрамарина высших сортов промытый продукт подвергают мокрому помолу на шаровой мельнице 19, классифицируют в гидроциклоне 21, после коагуляции фильтруют на листовом вакуум-фильтре 23, затем сушат в гребковой сушилке 25, упаковывают в мешки и направляют на склад.
7.6. СУЛЬФАТ АЛЮМИНИЯ
Физико-химические свойства. Сульфат алюминия А12(8О4)з, белые кристаллы с орторомбической решеткой; плотность 2,71 г/см3; С® = 259,6 Дж/(моль-К); АЯ^р = -3444,1 кДж/моль; AG^ = -3102,9 кДж/моль; 5^, = 239,4 Дж/(моль-К). Выше 580° С разлагается до у-А12Оз и SO3. Растворимость в воде при 0° С равна 23,2, 26,6 (20° С), 34,2 (50° С), а при 100° С — 47,1% (масс.). Плохо растворим в спиртах. Гигроскопичен.
Из водных растворов кристаллизуются бесцветные кристаллы А12(8О4)з-18Н2О моноклинной решеткой; температура плавления 86,5° С (с разложением); плотность 1,690 г/см3; А/7^=78,8 кДж/моль, АЯ^р = -8905,7 кДж/моль. При 150° С теряет 4 молекулы кристаллизационной воды, при 160° С — 8, при 250° С — 15, а при 420° С образуется безводный сульфат алюминия.
С сульфатами щелочных металлов и аммония сульфат алюминия образует алюминиевые квасцы, имеющие общую формулу M2SO4A12(SO4)3-24H2O или MA1(SO4)212H2O.
Алюмоаммониевые квасцы NH4A1(SO4)2-12H2O — бесцветные кристаллы с плотностью 1,64 г/см3, плавящиеся при 93,5° С без разложения. В процессе нагревания до 120° С теряет 10 молекул воды, а при 200° С образуется безводная соль. Растворимость при 0° С — 2,6, при 80° С —35, 2 г NH4A1(SO4)-12H2O в 100 г воды.
Алюмокалиевые квасцы КА1(8О4)г-12Н2О — бесцветные кристаллы с плотностью 1,76 г/см3, плавящиеся при 92° С. Растворимость при 0° — 2,96, при 20° С — 5,75, при 70° С — 35,9, при 90° С — 109, при 100° С —154 г KA1(SO4)2 в 100 г воды. Безводная соль гигроскопична, имеет плотность 2,75 г/м3, на воздухе расплывается.
Алюмонатриевые квасцы NaAl(SO4)212H2O — бесцветные кристаллы с плотностью 1,675 г/см3, плавящиеся при 61° С. Растворимость при 0° С —37,4, при 20° С —40,8. при 40° С —44,3 г NaAl(SO4)2 в 100 г воды. В системе Na2SO4 — А12(8О4)з — Н2О при 0° С эвтониче-ский раствор состава (в %, масс.): 3,74 — Na2SO4 и 21,58 — A12(SO4)3 находится в равновесии с твердыми фазами: Na2SO4 и инконгруэнтно растворимых квасцов Na2SO4 Al2(SO4)3-24H2O. Точке перехода соответствует состав раствора 1% Na2SO4 и 27,12% А12(8О4)з, равновесный с твердыми фазами Na2SO4 Al2(SO4)3-24H2O и А12(8О4)з-18Н2О. 316
Рис. 7.12. Двухъядерные комплексы: октаэдры объединены посредством двух мостиковых атомов алюминия
Гидролиз солей алюминия и, в частности, сульфата алюминия сопровождается сложными процессами образования двухъядерных гидроксоак-вокомплексов А12(ОН)г4+, связующими мостиками которых являются ОН-группы, а центральным атомом октаэдрических комплексов алюминий (рис. 7.12). Стадии гидролиза А12(8О4)з можно описать уравнениями:
А12(вО4)з + 2Н2О = A12(SO4)2(OH)2 + H2SO4
A12(SO4)2(OH)2 + 2Н2О = A12(SO4XOH)2 + H2SO4
A12(SO4)(OH)4 + 2H2O = 2A1(OH)3 + H2SO4
A12(SO4)3 + 6H2O = 2А1(ОН)з + 3H2SO4
Образующиеся основные соли алюминия образуют очень нестойкие в воде истинные или коллоидные растворы. При хранении или при повышении температуры они разлагаются с образованием осадка переменного состава, переходящего в устойчивый комплексный ион [A1x(OH)/H2O)J(^. В системе А1(ОН)3—A12(SO4)3—Н2О (рис. 7.13) существуют твердые фазы: 4А12О3-38Оз-24Н2О, ЗА12О3-48Оз-31Н2О (выделяющийся ниже 56° С), А12Оз*8Оз*3,5Н2О — выделяющийся при кипячении раствора нерастворимый осадок и ЗА12Оз-58О3К2О10Н2О
(где R—Na, К, NH4) — образующийся в виде накипи при кипячении в присутствии солей щелочных металлов. Под щелочностью понимается отношение количества гидроксида алюминия к общему его количеству в системе.
В процессе термообработки безводный сульфат алюминия при 610° С начинает разлагаться с выделением триоксида серы по схеме
A12(SO4)3 -» A12O(SO4)2 + SO3
Рис. 7.13. Растворимость в системе
А1(ОН)3 —A12(SO4)3 —Н2О
317
При 810° С происходит дальнейшее разложение
AI2O(SO4)2 -> A12O(SO3)2 + О2
а при 910° С образуется А120з по схеме
A12O(SO3)2 -> А12О3 + 2SO2
Образующийся при этом сесквиоксид алюминия из-за своей значительной дисперсности и дефектности кристаллов обладает повышенной химической активностью и способностью к спеканию.
В процессе разложения основных алюмоаммониевых квасцов в кипящем слое процесс обезвоживания происходит при 460—520° С, а процесс их разложения с выделением диоксида серы — при -1100° С. Практически установлено, что в процессе термообработки при 1000° С в течение 5,5 мин образуется продукт, содержащий 90%, а в течение 12 мин — 95% сесквиоксида.
Применение сульфата алюминия. Сульфат алюминия широко применяется в качестве коагулянта для обработки питьевых и промышленных вод. Коагулирующие свойства обусловлены образованием коллоидного гидроксида и основных сульфатов алюминия в результате его гидролиза. В процессе очистки воды гидроксид и основные сульфаты захватывают находящиеся в воде коллоидные частицы примесей в воде и осаждают их в виде хлопьев. Кроме того, значительные количества сульфата алюминия применяются в целлюлозно-бумажной промышленности для проклеивания бумаги, в текстильной промышленности в качестве протравы в процессе крашения хлопчатобумажных, шерстяных и шелковых тканей, для консервирования деревянных изделий, в производстве древесно-волокнистых плит, в промышленности искусственных волокон.
Значительное количество сульфата алюминия применяется в производстве квасцов. Алюмоаммониевые квасцы являются сырьем для получения синтетического корунда, а алюмокалиевые квасцы применяют в мясоперерабатывающей, кожевенной и текстильной промышленности. Разработаны также способы применения сульфата алюминия и квасцов в качестве фунгицидов.
К сульфату алюминия и квасцам, выпускаемым промышленностью согласно существующих стандартов, предъявляются требования, приведенные в табл. 7.5.
Разработан способ получения оксихлоридов алюминия, в частности пентаоксихлорида А12(ОН)5С1, диссоциирующий также в воде на А12(ОН)5 + и СГ. Получают его действием NaOH на А1С13 или обработкой гидроксида алюминия неконцентрированной хлороводородной кислотой. Полученный продукт содержит 80% А12(ОН)5С1 и соответственно более 40% А12О3, что значительно больше, чем в сульфате алюминия. 318
Таблица 7.5. Требования к химическому составу сульфата алюминия и квасцов технической квалификации (в %)
Технический сульфат алюминия Квасцы алюмокалиевые технические (ГОСТ 15028—69)
Очищен-ный (ГОСТ 5740—71) Очищенный (ГОСТ 12966—67) Неочищен-ный (ГОСТ 5055—49)
I сорт II сорт III сорт высший сорт I сорт II сорт
Д120з, не менее 16,3 15,0 14,5 13,5 9 10,6 10,5 10,3
H2SO4 (свод.), не более Отсутс. 0,05 0,10 0,10 2
Железо (Fe2O3), не более 0,02 0,04 0,10 1,5 0,8 0,0015 0,002 0,0035
Аз20з, не более 0,001 0,003 0,003 0,003 0,003
Нерастворимый остаток, не более 0,3 0,5 0,7 1,0 2,3 0,03 0,035 0,1
Способы получения сульфата алюминия. Существует значительное число способов получения сульфата алюминия. Это связано с различием применяющихся в производстве видов исходного алюминийсодержащего сырья и различными требованиями к качеству продукта.
Получение коагулянта (неочищенного сульфата алюминия). Сырьем для получения коагулянта является каолин — тонкодисперсная глинистая порода, состоящая в основном из каолинита AJ2Si2O5(OH)4 (А120з— 39,5; SiO2 — 46,54; Н2О—13,96% масс.).
Исходное сырье — каолин — подвергается термообработке в барабанных вращающихся печах при 700—800° С. В процессе термообработки исходное сырье освобождается полностью от исходной воды и после охлаждения до 50—80° С поступает в реактор, где обрабатывается 65—67%-ной серной кислотой при 105—110° С в течение 6—8 ч. В процессе реакции поддерживают 6—8%-ный избыток серной кислоты.
Образующуюся в реакторах массу сливают в кристаллизационный лоток — плоскую прямоугольную плиту с бортами 0,25—0,5 м, футерованную кислотоупорными плитками. Для облегчения удаления затвердевшей массы коагулянта из кристаллизаторов процесс разложения каолина проводят избытком серной кислоты, нейтрализуемой в конце процесса каолином. Автоматически действующая машина для снятия сульфата алюминия с плоских кристаллизационных столов представляет собой сдвоенную пластинчатую цепь с ножами и гребками. Перемещаясь непрерывно поперек кристаллизатора, машина срезает целевой продукт слой за слоем и передает его на ленточный транспортер, расположенный вдоль кристаллизатора.
Разработан более совершенный способ получения неочищенного сульфата алюминия практически полным разложением исходного сы-319
рья, поддерживая значительный избыток исходной серной кислоты, заметно сокращая тем самым продолжительность реакции с последующей нейтрализацией избытка кислоты нефелиновой мукой по схеме
Al2O3-2SiO22H2O + 3H2SO4 = A12(SO4)3 + 2SiO2 + 5H2O <
(Na, K)2OAl2O3-2SiO2 + 4H2SO4 = (Na, K)2SO4-A12(SO4)3 + 2SiO2 + 4H2O *
Увеличение избытка исходной серной кислоты и повышение температуры процесса за счет взаимодействия кислоты с водой ускоряют протекание реакции разложения каолина. Образующиеся согласно реакции по второму уравнению квасцы благоприятствуют продукту схва-тываться в плотную прочную массу. Процесс схватывания и прочность массы прямо пропорциональны величине избытка исходной кислоты и соответствующему расходу нефелиновой муки на ее нейтрализацию.
Степень извлечения сесквиоксида алюминия из исходного каолина зависит от его качества, концентрации и количества серной кис* лоты. Отмученный каолин, содержащий 35% сесквиоксида алюминия и 47,5% диоксида кремния, высушенный и измельченный до размера частиц 0,5—1,0 мм, реагирует с максимальной скоростью в избытке 54—55%-ной серной кислоты. Обработка же каолина двойной по отношению к стехиометрической нормой серной кислоты в течение двух часов при 105—120° С позволяет перевести в раствор 85% исходного сесквиоксида алюминия. С применением более концентрированной серной кислоты реакция замедляется в результате образования корок на частицах исходного сырья труднорастворимого безводного сульфата алюминия или же кислых сульфатов и увеличения сопротивления диффузии.
В процессе получения неочищенного сульфата алюминия из каолина и нефелиновой муки в автоклав закачивают воду и серную кислоту с расчетом получения 75—76%-ной серной кислоты. После загрузки расчетного количества каолина в течение 15—35 мин при непрерывном перемешивании температуру 100—110° С в реакторе поддерживают подачей пара в автоклав. По окончании реакции реакционную массу, содержащую 4,5—7,5% сесквиоксида алюминия и 20—25% свободной кислоты, разбавляют водой и вводят в котел (отдельными порциями по 5—10 кг) нефелин в течение 15 мин. Массу перемешивают в течение 2—3 мин, после чего ее сливают на кристаллизационный стол.
По этому способу степень перехода сесквиоксида алюминия из каолина в его сульфат составляет 65—75%, а из нефелина около 90%. Расходные коэффициенты составляют: 0,17 т каолина (с 15%-ной влагой), 0,18 т сухой нефелиновой муки, 0,31 т серной кислоты, 0,21 т пара, 2,5 кВт-ч электроэнергии и 0,12 м3 воды на 1 т получаемого целевого продукта.
320
Разработан и применяется в промышленности способ получения неочищенного сульфата алюминия (коагулянта) из нефелина.
Согласно действующей в производстве технологии, смешение крепкой серной кислоты и нефелинового концентрата производят в двух соединенных последовательно каскадно-расположенных реакторах, снабженных трехлопастными мешалками. В первый реактор непрерывно подают серную кислоту и нефелиновый концентрат. Образующаяся в первом реакторе густая реакционная масса непрерывно перетекает во второй реактор, откуда выходит из нижней части реактора через гидравлический затвор в ковшовый дозатор. В пульпе после второго реактора должно содержаться от 1,5 до 4% избыточной серной кислоты. Экспериментально установлено, что замедление взаимодействия нефелина с концентрированной серной кислотой (более 63% H2SO4) объясняется недостатком воды в жидкой фазе. С наибольшей скоростью нефелин реагирует с 47—73%-ной серной кислотой (рис. 7.14). Поэтому из ковшового дозатора реакционная масса поступает в шнек-реактор, куда подают воду из расчета разбавления серной кислоты до 70—73% основного вещества. Продолжительность нахождения реакционной массы в шнеке-реакторе составляет 28—30 с, а степень разложения исходного нефелина повышается до 85—88%. Из реактора сухая рассыпная масса с температурой 80—100° С поступает на склад, где целевой продукт охлаждается и дозревает в течение 2—4 сут. На производство одной тонны целевого продукта рассмотренным способом расходуется: 0,32 т нефелинового порошка с влажностью до 1% и 0,378 т серной кислоты (100%).
Получение очищенного сульфата алюминия. Очищенный сульфат алюминия получают растворением в серной кислоте гидроксида алюминия по схеме
2А1(ОН)3 + 3H2SO4 = A12(SO4)3 + 6Н2О
Процесс ведут в реакторе, футерованном кислотоупорным кирпичом по слою диабазовой плитки загружая в него одновременно исходные гидроксид алюминия, серную кислоту и воду. Расчеты
количества исходных реактивов производят в стехиометрическом соотношении, соответствующем содержанию в целевом продукте -90% A12(SO4)3-18H2O и
-10% свободной воды. Перемешивание реакционной массы проводят острым паром, поддерживая посто-
11 Химическая технология неорганических веществ, кн. 1
20 30 40 50 60 70 80
Концентрация серной кислоты, %
Рис. 7.14. Влияние концентрации серной кислоты на скорость процесса разложения нефелина
321
янную температуру в реакторе ПО—120° С. Процесс заканчивается в течение 20—30 мин. При этом количество свободной серной кислоты в реакционной массе станет меньше 0,1%. Образующуюся реакционную массу, содержащую 13,5—15,0% сесквиоксида алюминия (в виде сульфата алюминия), для ускорения процесса последующей кристаллизации охлаждают в реакторе до 95° С, продувая через массу в течение 10 мин сжатый воздух. После охлаждения массу сливают на кристаллизационный стол, оборудованный автоматической машиной для срезки застывшего продукта. Процесс кристаллизации плава на столе-кристаллизаторе продолжается ~50 мин и примерно столько же времени требуется для извлечения продукта из кристаллизатора площадью 32—34 м2 и емкостью 6 т.
По описанной технологии на одну тонну целевого продукта расходуется 0,142 т гидроксида алюминия (в пересчете на сесквиоксид алюминия) и 0,40 т серной кислоты (100%).
Разработан и внедрен в производство способ кристаллизации, согласно которого процесс кристаллизации проводят на охлаждаемой изнутри наружной поверхности горизонтального вращающегося барабана. Барабан частично погружен в находящийся в поддоне плав сульфата алюминия, имеющий температуру 90—100° С. Кристаллизаторы облегчают условия труда, обеспечивают непрерывность режима производства и улучшают физические свойства целевого продукта. Срезаемый с поверхности барабана ножом чешуйчатый продукт, содержащий 13,5—14,0% сесквиоксида алюминия, при хранении слеживается. Исслеживающийся продукт получают путем повышения содержания А120з до 15,3—15,8%. При длине барабана вальцев 2,2 м и диаметре 1,8 м (поверхность теплообмена 12,4 м3), при производстве продукта с содержанием 13,5—14,0% А120з число оборотов барабана составляет 4,3 в минуту и средняя рабочая производительность вальцев равна 2,4 т/ч.
Для получения исслеживающегося продукта смешивают суспензию гидроксида алюминия с 60%-ной серной кислотой, взятой в количестве 95—97% от стехиометрического и образующуюся смесь с температурой 100° С для кристаллизации направляют на холодильные вальцы.
Разработан способ непрерывного получения сульфата алюминия, по которому водная суспензия гидроксида алюминия и серная кислота в стехиометрическом отношении подаются с большой скоростью дозирующими насосами в смесительные форсунки реактора, в котором масса находится не менее 30 с, после чего она охлаждается до температуры ниже 100° С в проточном холодильнике и продавливается через сопла и прорези для образования мелкогра-нулированного продукта.
322
Разработан способ получения кристаллического сульфата алюминия высокой чистоты выдерживанием в отдельном сборнике суспензии, полученной в процессе упарки раствора, до ее охлаждения, с целью образования более крупных частиц примесей. После отделения примесей массу охлаждают
Рис. 7.15. Растворимость сульфата алюминия в серной кислоте при 25° С
для кристаллизации.
Растворимость сульфата алюминия в присутствии серной кислоты резко понижается (рис. 7.15), а это явление открывает возможность высаливания кристаллического сульфата алюминия из растворов концентрированной серной кислотой.
Разработано значительное число способов очистки растворов сульфата алюминия путем внесения в них окислителей (МпО2, Н2О2, С12, Вг2 и др.) для окисления соединений железа Fe2+ в Fe3+ с последующим выделением Fe3+ в виде не растворимых в воде соединений, например Fe(OH)3, Fe4[Fe(CN)6]3.
Производство очищенного сульфата алюминия может быть орга
низовано с использованием алюминиевого концентрата, получаемого при предварительном обогащении каолина или глин. За основу технологии принято разложение кремнийсодержащих соединений алюминия диоксидом серы по следующей схеме:
А12О3 + ЗН2О + 6SO2 = 2A1(HSO3)3
А12О3 + 3SO2 = A12(SO3)3
Процесс проводится в присутствии воды. Поэтому первой стадией процесса является образование промежуточного соединения H2SO3, которое во второй стадии реагирует с исходным сесквиоксидом алюминия.
Разложение каолина сернистой кислотой при 25—65° С и атмосферном давлении протекает медленно, а степень перехода сесквиоксида алюминия в раствор в течение 5—7 ч не превышает 8,5%. В этих условиях степень извлечения оксида железа составляет около 22,5%.
Увеличения скорости процесса разложения каолина и степени перехода А120з в водорастворимое состояние достигают повышением давления и температуры в аппарате. При давлении 20 атм и температуре 170° С степень перехода сесквиоксида алюминия в раствор из водной суспензии каолина с отношением по массе Т:Ж, равным 1:10, в течение 8—10 ч достигается 95—96%.
н*
323
Полученную суспензию фильтруют, освобождают от нерастворимого осадка, представляющего собой в основном кремнезем, а полученные чистые растворы гидролизуют:
A12(SO3)3 + ЗН2О = 2А1(ОН)3 + 3SO2
A1(HSO3)3 + ЗН2О = А1(ОН)3 + 3SO2 + ЗН2О
Экспериментально установлено, что в процессе гидролиза образуются основные сернисто-кислые соли алюминия различного состава: A1(OH) (HSO3)2, A1(OH)2 (HSO3), A1(OH)SO3, A12(OH)4SO3 и др. Некоторые из образующихся основных солей трудно растворимы в сернистой кислоте и не подвергаются дальнейшему гидролизу, в результате этого продукт гидролиза состоит из гидроксида и основных солей алюминия и может служить для получения сульфата алюминия с одновременным повышением концентрации диоксида серы.
Процесс упрощается обработкой отфильтрованных растворов су-льфит-гидросульфита алюминия серной кислотой по схеме
A12(SO3)3 + 3H2SO4 = A12(SO4)3 + ЗН2О + 3SO2
Образующийся высококонцентрированный диоксид серы возвращается в начало процесса.
Сульфат алюминия получают также из растворов, образованных растворением боксита в серной кислоте. Известен также способ получения сульфата алюминия с утилизацией травильных растворов.
Коагулянт может быть получен из золы некоторых углей, содержащей А12О3, например в золе подмосковных углей содержится 25—35% А12О3, 8—15% Fe2O3 и 43—60% SiO2. Из золы углей получают коагулянт, содержащий до 8—10% R2O3. Однако извлечение А12О3 из золы составляет всего в пределах 60—80%. Это объясняется реакцией содержащего сесквиоксида алюминия с диоксидом кремния с образованием силикатов алюминия в топках.
Получение квасцов. Сырьем для получения алюмокалиевых квасцов являются: сульфат алюминия и сульфат калия. Для получения алюмокалиевых квасцов KA1(SO4)2-24H2O в концентрированные водные растворы сульфата алюминия, содержащие 8% А12О3, при постоянном перемешивании вносят расчетное количество сульфата калия. После перемешивания в течение одного часа при 80—90° С гомогенные растворы направляют на вакуум-кристаллизацию. Образующиеся кристаллы целевого продукта отделяют от маточных растворов и промывают водой в непрерывно действующих центрифугах типа ФГП. Маточные же растворы и промывные воды из центрифуг возвращают на начало процесса для разбавления растворов сульфата алюминия 324
перед внесением в них сульфата калия. Растворы возвращают на начало процесса до накопления в них содержания сульфата железа до концентрации 1,1% в пересчете на сесквиоксид железа. После этого их применяют для разбавления исходной серной кислоты.
Разработан способ получения алюмокалиевых квасцов путем взаимодействия каолина с серной кислотой, нейтрализации образующейся массы с последующим внесением в реактор сульфата натрия из расчета получения растворов алюмонатриевых квасцов. Образующиеся растворы алюмонатриевых квасцов разбавляют до плотности 1,33 г/см3, фильтруют от осадка диоксида кремния, охлаждают, после чего вносят в них насыщенные растворы хлорида калия. В процессе реакции образуется осадок менее растворимых при низких температурах кристаллов алюмокалиевых квасцов.
В промышленности широко применяется видоизмененный вариант описанной технологии. Полученный из суспензии гидроксида алюминия и серной кислоты раствор смешивают в реакторах со стехиометрическим количеством 15%-ного раствора сульфата натрия и 25%-ным раствором хлорида калия. При этом протекает следующая реакция:
A12(SO4)3 + 2КС1 + Na2SO4 = 2KA1(SO4)2 + 2NaCl
Реакционная смесь нагревается до 70° С, после чего образующийся раствор фильтруют через автоматический фильтр-пресс ФПАКМ от нерастворимого остатка. Отфильтрованные растворы направляются в четырехкорпусную вакуум-кристаллизационную установку, в последнем корпусе которой поддерживается температура около 10° С, что обеспечивает кристаллизацию около 86% содержащихся в растворах квасцов. Образующаяся суспензия кристаллов, проходя сгустители и дозаторы, поступает в центрифуги типа ФГП. Далее кристаллы квасцов подвергаются сушке во вращающихся барабанных сушилках воздухом с температурой 120—130° С, а маточный раствор возвращается на начало процесса для приготовления растворов сульфата алюминия.
По описанной технологии на производство одной тонны целевого продукта расходуется: 0,14 т гидроксида алюминия (100%), 0,23 т хлорида калия (100%), 0,192 т сульфата натрия (100%), 0,40 т серной кислоты (100%), 0,015 т карбоната натрия для нейтрализации остаточной кислотности исходного сульфата натрия, 168 кВт-ч электроэнергии, 60 м3 воды и 1,27 мГк пара.
Широко применяется в промышленности способ получения квасцов из нефелина. При взаимодействии нефелина с серной кислотой образуется смесь калиевых и натриевых квасцов по реакции
(К, Na)2O-Al2O3-2SiO2 + 4H2SO4 + 20Н2О = = (К, Na)2SO4-Al2(SO4)3-24H2O + 2SiO2
325
Согласно технологии, в кипящую серную кислоту с концентрацией 40—50% сульфата водорода в реактор постепенно дозируют исходный i нефелин. По мере загустевания образующейся массы сульфатов алюминия, натрия и калия раствор разбавляют водой до плотности раство- 1 ра 1,16—1,23 г/см3, после чего направляют на фильтрацию. Отфиль- | трованные от нерастворимого осадка растворы подвергают выпарке, i Упаренный раствор охлаждают в лотках, в которых происходит про- ' цесс полной кристаллизации квасцов. Недостатком технологии является неудовлетворительная фильтруемость суспензии квасцов, что объясняется присутствием геля кремниевой кислоты, проходящей раствором через фильтр. С целью получения легкофильтруемого от геля раствора процесс разложения исходного нефелина проводят 74—76%-ной серной кислотой при повышенной температуре. Это позволяет ликвидировать процесс образования геля. В результате дегидратации кремниевой кислоты она переходит в коллоидную форму и не затрудняет процесс фильтрации. Помимо этого, при более низкой концентрации исходной кислоты процесс разложения протекает со значительной скоростью (см. рис. 7.14), что затрудняет осуществление процесса фильтрации. В процессе с применением серной кислоты с концентрацией выше 76% реакция резко замедляется. Исходная кислота берется в количестве 83—88% от стехиометрических расчетов. Температура реакционной массы к концу процесса повышается до 140° С. В результате реакции образуется неплотная пористая масса, из которой квасцы легко выщелачиваются водой. Оптимальное соотношение воды и нефелина в процессе выщелачивания квасцов составляет 2:1.
Получаемые выщелачиванием плава растворы квасцов упаривают до концентрации сесквиоксида алюминия 10,5—12,5%, а затем, охлаждая растворы, выделяют кристаллы. В процессе упарки растворов имеет место сильный гидролиз квасцов, в результате чего образуется осадок основных солей и кислотность раствора повышается. Для исключения процесса образования целевого продукта с повышенной кислотностью (0,1%) в конце процесса упарки производят нейтрализацию раствора гидроксида алюминия нефелином или карбонатом натрия. Средний состав получаемого описанным способом целевого продукта следующий: 40,2% А^БО^з (12% AI2O3), 1,7% Ре2(80д)з (0,7% Ре20з), 11,3% Na2SO4, 4,6% К2§Од, 40,4% кристаллизационной воды и 1,8% примесей.
Калиевые квасцы считаются значительно ценным продуктом, чем натриевые, так как они в отличие от последних могут быть получены более чистыми, что важно для многих потребителей. Поэтому раздельное получение их признано более рациональным. Разная степень растворимости натриевых и калиевых квасцов (рис. 7.16) при различных температурных режимах позволяет выделить до 90% калиевых квасцов в твердую фазу. Оставшийся после выделения в оса-326
док калиевых квасцов раствор, содержащий в основном натриевые квасцы, может быть переработан на твердый продукт выпаркой и охлаждением. Из этих же растворов можно выделить до 90% алюминия в виде калиевых квасцов путем обработки их хлоридом калия по следующей схеме:
NaAl(SO4)2 + КС1 KA1(SO4)2 + NaCl
Разработан также процесс получения квасцов из нефелина путем обработки водной суспензии исходного сырья диоксидом серы:
(K,Na)2O A12O3-2SiO2 + 8SO2 + 4Н2О NaHSO3 + KHSO3 + 2A1(HSO3)3 + 2SiO2
KAI(SO4)2, г на 100 г Н2О
Рис. 7.16. Изотермы растворимости КА1(8О4)2 в растворах NaAl(SO4)2
(20, 30, 40 и 50° С)
Процесс проводится при соотношении Т:Ж в суспензии, равном 1:5. При 20° С в течение 3 ч степень извлечения сесквиоксида алюминия из исходного сырья составляет около 90%. Для получения хорошо фильтрующегося осадка диоксида кремния с исключением процесса гелеобразования реакцию диоксида серы с водной суспензией нефелина проводят ступенчато, с постепенным увеличением объема жидкой фазы разбавлением водой или маточными растворами. В начале процесса величина Т:Ж должна быть равной 1:1, а в конце— 1:5. Полученный раствор фильтруют и перерабатывают в квасцы.
Образующийся раствор упаривают и при охлаждении выделяют из них кристаллический продукт, а газообразный диоксид серы направляется на производство серной кислоты или приготовление из него жидкого SO2.
Алюмоаммониевые квасцы (NH4)2SO4-A12(SO4)3-24H2O получают в производстве путем смешения охлажденных растворов сульфата алюминия, содержащих около 24—25% основного вещества с растворами сульфата аммония и концентрацией 35% (NH4)2SO4. С целью более полного осаждения алюмоаммониевых квасцов берут 10—12%-ный избыток сульфата аммония по отношению к стехиометрическому количеству согласно уравнению:
A12(SO4)3 + (NH4)2SO4 + 24Н2О = (NH4)2SO4-A12(SO4)3-24H2O
В результате экзотермичности реакции растворы разогреваются до 50—60° С. С целью увеличения выхода кристаллов целевого продукта их охлаждают до 15—20° С. При необходимости получения более качественного продукта проводят его перекристаллизацию.
327
7.7. ГИДРИДЫ АЛЮМИНИЯ
Физико-химические свойства. Гидрид алюминия А1Нз — кристаллическое вещество в виде бесцветных полимеров; плотность 1,45 г/см3; температура плавления 100—150° С; температура образования (298 К)—11,4 кДж/моль; энтропия (298 К) 7,18 э.е.; теплоемкость 40,2 кДж/(моль-К); энергия связи А1—Н 331 кДж/моль; кристаллизуется в гексагональной системе, но в зависимости от условий синтеза тип решетки может быть различным.
Для полимерного гидрида алюминия (рис. 7.17) предложена циклическая формула, в которой атомы алюминия соединены между собой водородными мостиками:
Твердый гидрид алюминия начинает разлагаться на элементы в зависимости от степени полимеризации при 100—160° С.
Гидрид алюминия А1Нз является амфотерным соединением, которое может выступать как в качестве донора электронных пар (основное соединение), так и в качестве акцептора (кислотное соединение):
А1Н3 + ЗВНз = А1(ВНд)з основной тетрагидроборат
КН + А1Н3 = К[А1Н4] кислотный тетрагидроалюминат
Гидрид алюминия растворяется с образованием в простых эфирах (метиловый, этиловый), в диметиловом эфире диэтиленгликоля, тетрагидрофуране, диоксане, а также в аминах (триметиламин, триэти-ламин). Не растворим в углеводородах. Растворы гидрида алюминия в 328
этиловом эфире при стоянии посте- ।
пенно выделяют полимер (А1Нз)„, ко- РгЛГ
торый больше уже не может быть пе-
реведен в раствор. у Qh
Наилучшим растворителем гидрида qIjQ
алюминия является тетрагидрофуран: в /~\А1
100 г его растворяется при 20° С 5 г QT|Q
А1Нз. Образующиеся при этом раство- ~
ры гидрида алюминия более стойки и выделяют полимер лишь при нагрева- /->ХТлл нии, а полученный полимер может GT I'* быть растворен В тетрагидрофуране Рис. 7.17. Фрагмент структуры или диоксане. Процесс растворения со- гидрида алюминия (А1Н3)„ провождается деполимеризацией.
В растворах гидрид алюминия обычно малостоек и при нагревании до 50° С начинает разлагаться с выделением водорода и образованием элементного алюминия. Добавка тетрагидрофурана, диоксана или триметиламина стабилизирует эфирные растворы гидрида алюминия как против разложения, так и против полимеризации. Процесс разложения гидрида алюминия в растворах ускоряется примесями металлов.
Гидрид алюминия является сильно реакционноспособным соединением. Он энергично реагирует с кислородом, бурно разлагается водой и взаимодействует со многими органическими и неорганическими веществами.
Диоксид серы восстанавливается гидридом алюминия уже при—100° С; в зависимости от соотношения исходных реактивов и условий проведения реакции могут быть получены элементная сера, сульфид водорода и сульфид алюминия.
Из реакций обмена водорода на галоген можно отметить образование диборана при реакции гидрида алюминия с хлоридом бора:
2А1Н3 + 2ВС13 = В2Н6 + 2 А1С13
получение силана из тетрахлорида кремния:
4А1Н3 + 3SiCl4 = 3SiH4 + 4А1С13
получение фосфина из трибромида фосфора:
А1Н3 + РВг3 = РН3 + А1Вг3
Трихлорид титана реагирует с гидридом алюминия в эфирном растворе с образованием хлоралюмогидрида титана TiCl(AlH4)2.
Разложение гидрида алюминия с выделением водорода происходит при действии воды, спиртов, галогенводородов и азотистоводо-
329
родной кислоты, аммиака, гидразина и т. д. Эти реакции идут уже при низких температурах.
Наличие вакантных орбит в электронной оболочке приводит гидрид алюминия к комплексообразованию, что затрудняет получение гидрида алюминия, свободного от исходного растворителя. По этой причине некоторые из его комплексов могут быть получены в кристаллическом состоянии при охлаждении растворов. Например, с этиловым эфиром образуется соединение (СгНз^СМАШз.
С аммиаком гидрид алюминия образует два типа компонентов: А1Н3 • NH3 и А1Н3 • 2NH3. Образующиеся комплексы при нагревании разлагаются с выделением водорода:
А1Н3 • NH3 -> Н2 + ^/(AIH2NH2)„ (при -40° С)
(A1H2NH2)„ -» (А1Н = NH)„ + иН2 (при -40-20° С)
(А1Н = NH)„ -» (A1N)„ + пН2 (при 150° С)
По химическим свойствам аминные комплексы сходны с самим гидридом алюминия: они легко окисляются и гидролизуются (дымят на воздухе).
Получение гидрида алюминия. Гидрид алюминия получают из алюмогидридов лития или натрия. Взаимодействие алюмогидрида лития с хлоридом алюминия в эфирном растворе при комнатной температуре протекает по уравнению
3LiAlH4 + А1С13 = 4А1Н3 + 3LiCl
Образующийся в растворе полуфабрикат — эфират гидрида алюминия А1Н3 • 2(С2Н5)2О — в зависимости от условий выпадает в осадок в виде твердого бесцветного полимера. Эфир может быть отогнан в вакууме, но не полностью. После отгонки эфира остается твердый продукт, которому приписывают строение алюмогидрида алюминия А1(А1Н4)з.
Процесс упарки растворов гидрида алюминия проводят с большой осторожностью, так как может произойти его разложение. Присутствие диоксида углерода также не допускается, так как он в эфире в процессе упаривания растворов гидрида алюминия может привести к взрыву.
Для многих целей нет необходимости выделять гидрид алюминия из раствора. В этом случае получают растворы с повышенной стабильностью путем добавки комплекообразователей более сильных, чем эфир, например тетрагидрофурана или триметиламина либо хлорида 330
алюминия. Если вместо хлорида алюминия применять его бромид, образуются очень стабильные растворы.
Гидрид алюминия получается в эфирном растворе при действии металлического алюминия на алюмогидрид лития в присутствии хлорида ртути.
Гидрид алюминия получают также из алюмогидридов других металлов (натрия, кальция, магния), но из-за нерастворимости их гидридов в этиловом эфире процесс ведут в тетрагидрофуране.
Гидрид алюминия может быть получен при многократной обработке галогенида или алкилгалогенида алюминия диалкилалюминий-гидридом:
А1С1з + 3A1R2H -> А1Н3 + 3A1R2C1;
2AIRCI2 + 3A1R2H -> А1Н3 + 4A1R2C1
Реакцию проводят в среде какого-либо углеводорода, например гексана, при 50—60° С. Полимерный гидрид алюминия, не растворимый в гексане, выпадает в осадок. Полученный продукт, однако, содержит примесь галогена и алкильных групп. С целью получения более чистого продукта диалкилалюминийхлорид, остающийся в осадке после декантации жидкости, отгоняют в пленочном испарителе при 50° С и разрежении 10‘2—10’3 мм рт. ст.
Гидрид алюминия образуется также при взаимодействии тримети-лалюминия с водородом в тлеющем разряде.
Алюмогидриды (тетрагидридоалюминаты, аланаты), комплексные гидриды, содержащие [А1Н4]‘, легко окисляются кислородом, энергично взаимодействуют с водой, а также растворами кислот и щелочей, выделяя водород. Алюмогидриды с аммиаком, галогенидами металлов и неметаллов образуют сложные комплексы. Восстанавливают органические соединения по кратным связям углерода с другими элементами, не затрагивая связи С=С. Применяют алюмогидриды в качестве селективных восстановителей и источника водорода для получения гидридов элементов, а также при нанесении металлических покрытий.
Алюмогидриды щелочных и щелочноземельных металлов (табл. 7.6) — бесцветные кристаллы, разлагающиеся при нагревании (алюмогидрид бериллия разлагается в эфире уже при 10° С). Они растворимы в диглиме и моноглиме, а алюмогидриды лития и магния также в эфире.
С органическими растворителями образуют сольваты. Особенно прочны сольваты алюмогидридов магния и бериллия, полное разложение которых происходит лишь при распаде самих соединений. Свободные алюмогидриды выделяют из растворов в эфире и тетрагидрофуране отгонкой растворителя, а из растворов в тетрагидрофу-331
ране и диглиме высаливанием эфиром или толуолом. Алюмогидриды получают взаимодействием гидрида металла с хлоридом алюминия в полярном органическом растворителе, а также прямым синтезом элементов при 100—200° С и повышенном давлении водорода в сольватирующих растворителях или в среде углеводородов в присутствии катализатора (алюминийорганические соединения). Существует также способ получения обменной реакцией галогенидов или борогидридов металлов с алюмогидридом лития или натрия по реакции
Li[AlH4] + М -> М[А1Н4] + Li, где М = Na, К, Rb и Cs
Таблица 7.6. Свойства алюмогидридов
Соединение Плотность, г/см3 кДж/моль Температура разложения, °C S^, Дж/(моль К)
Li[AlH4] 0,917 -107,1 150—170 78,7
Na[AlH4] 1,28 -113,1 220—230* 124,0
К[А1Н4] 1,33 -166,0 276—296 129,0
Cs[A1H4] 2,84 -165,0 294—325 150,8
MgtAlHJ 1,05 -234 118 —
CafAirU] — -184,3 230 —
♦Температура плавления 185±5°С.
В качестве смешанного гидрида бора-алюминия можно рассматривать гидридоборат алюминия АЦВН^з. Связь атомов алюминия и бора осуществляется через водород двумя трехцентровыми двухэлектронными связями.
К алюмогидридам относятся также гидриды, содержащие ион [А1Н6]3’, например Na3[AlH6] с плотностью 1,48 г/см3 и температурой разложения 260° С, сложные комплексы, образуемые гидридоалюми-натами щелочных и щелочноземельных металлов, например растворимые в эфире Li2[Mg(AlH4)4], Li[Mg(AIELLO].
7.8. НИТРАТ АЛЮМИНИЯ
Нитрат алюминия A1(NO3)3, бесцветные, дымящиеся на воздухе кристаллы, с температурой плавления 66° С (с разложением), плотностью 1,89 г/см3 и ДЯ^р =— 927 кДж/моль. Возгоняется в вакууме при 50° С, при более высокой температуре разлагается до оксинитрата, а затем и до сесквиоксида алюминия:
4A1(NO3)3 —<-> A14O(NO3)6 + 3NO2 + 5,5O2
A14O(NO3)6---> 2А12О3 + 6NO2 + 0,5О2
332
Нитрат алюминия взаимодействует с эфиром и бензолом со взрывом. Гигроскопичен. Из водных растворов при 25° С кристаллизуется нонагидрат А1(ЫОз)з-9Н2О, представляющий из себя бесцветные, расплывающиеся на воздухе кристаллы с моноклинной решеткой (а = 1,086 нм, b - 0,959 нм, с = 1,383 нм, 0 = 95,15°, z = 4, пространственная группа Plyla), с температурой плавления 73,6° С и ДЯ^р = -3757 кДж/моль.
При нагревании выше 73,6° С нонагидрат переходит в окта-, а затем гексагидрат. При дальнейшем повышении температуры выделяется азотная кислота и оксиды азота с образованием сначала гидроксо-нитрата алюминия, а при достижении 200° С — сесквиоксида алюминия. Нонагидрат хорошо растворим в воде (73,9 г в 100 г при 20° С в расчете на безводный нитрат алюминия), спирте.
Безводный нитрат алюминия получают реакцией бромида алюминия с избытком пентаоксида азота (N2O5) с последующим взаимодействием образовавшегося полуфабриката NChfAlfNCh)^ с хлоридом алюминия. Нонагидрат нитрата алюминия получают действием разбавленной азотной кислоты на гидроксид или оксид алюминия по схеме
А1(ОН)3 + 3HNO3 + 6Н2О = А1(МО3)з • 9Н2О
А120з + 6HNO3 + 6Н2О = 2A1(NO3)3 • 9Н2О
Нитрат алюминия применяют в виде нонагидрата или водных растворов (протрава при крашении тканей), для получения катализаторов и в качестве высаливателя в процессе экстракции соединений актиноидов.
7.9. НИТРИД АЛЮМИНИЯ
Нитрид алюминия A1N, бесцветные кристаллы с гексагональной решеткой типа вюрцита (а = 0,3111 нм, с = 0,4975 нм, z = 2, пространственная группа Рб^тс); температура плавления 2430±200° С; плотность 3,12 г/см3; С ° =30,1 Дж/(моль-К); ДЯ^ около 68 кДж/моль ДЯ^ =-319 кДж/моль; />298 = 20,2 Дж/(моль-К); теплопроводность 30,14 Вт/(м К) при 473 К и 5,86 Вт/(м-К) при 1000 К; температурный коэффициент линейного расширения 4,03-Ю^К’^ЗОО—500 К), 5,64-10‘бК‘1(500—1300 К); р = 2 109(298 К) и 7-Ю5 Омм (870 К); 8 = 8,5. Полупроводник, ширина запрещенной зоны выше 5 эВ. Твердость по Моосу 9, микротвердость 12 ГПа, модуль упругости 350 ГПа (300 К), 2S0 ГПа (1673 К).
Нитрид алюминия окисляется на воздухе выше 900° С. С минеральными кислотами на холоде практически не взаимодействует, горя-333
ними кислотами разлагается медленно. При нагревании реагирует с растворами щелочей с выделением аммиака. С водяным паром образует гидроксид алюминия, с хлором — хлорид алюминия. Устойчив к действию расплавов алюминия, меди, олова и кальция.
Способы получения нитрида алюминия. 1. Взаимодействием элементного азота с алюминиевой пудрой около 1200° С по схеме
2А1 + N2 = 2A1N
2. Реакцией сесквиоксида алюминия с аммиаком около 1000° С:
А12О3 + 2NH3 = 2A1N + ЗН2О
3. Восстановлением сесквиоксида алюминия углеродом в атмосфере азота при 1600—1850° С:
А12О3 + ЗС + N2 = 2A1N + ЗСО
4. Разложением в газовой фазе A1C13-NH3 выше 1100° С с последующим осаждением на подложку из кварца, корунда, графита, молибдена или вольфрама:
А1С1з • NH3 '|^'с-> AIN + ЗНС1
Изделия из нитрида алюминия получают в основном горячим прессованием его порошка при 2000—2100° С и 30 МПа.
Нитрид алюминия применяют (иногда в смеси с BN или SiC) в качестве огнеупорного материала для футеровки ванн, электролизеров, печей, тиглей и др., для изготовления чехлов для термопар, для нанесения коррозионно-стойких и износостойких покрытий на сталь, графит и др.
7.10. ФОСФАТЫ АЛЮМИНИЯ
Ортофосфат алюминия А1РОд — бесцветные кристаллы, имеющие четыре устойчивые кристаллические модификации: до 580° С устойчив а-берлинит с гексагональной решеткой (а = 0,49429 нм, с = = 1,09476 нм, z = 3, пространственная группа Р3]21); плотность 2,64 г/см3; С ° =93,2 Дж/(моль-К); ДЯ^р =- 1733 кДж/моль; AG^ =-1617 кДж/моль; 5®98 = 90,8 Дж/(моль-К). При 580—705° С — 0-берлинит с гексагональной решеткой (а = 0,5038 нм, с = 1,1063 нм, z = 6; АЯ° перехода а->Р 1,30 кДж/моль); при 705—1047° С — гексагональная модификация (А/7° перехода 1,09 кДж/моль); выше 1047° С кубическая (а = = 0,71956 нм, z = 4). Известны также две метастабильные модификации. 334
Ортофосфат алюминия устойчив до 1650° С. Не растворим в воде, реагирует с минеральными кислотами и щелочами.
Монокристаллы берлинита, являющиеся пьезоэлектриками, выращивают гидротермальными методами при 130—180° С из растворов сесквиоксида алюминия в концентрированной фосфорной кислоте (6 моль/л) при атомном отношении А1:Р = 2,3—4,5.
Из водных растворов солей алюминия при действии эквивалентного количества ортофосфата щелочного металла в водном растворе при pH 4,0—4,5 осаждается аморфный фосфат А1РО4*хН2О— катализатор дегидратации спиртов, гидратации непредельных углеводородов, изомеризации эпоксисоединений в альдегиды и др. В процессе нагревания аморфного фосфата вместе с маточником в запаянной ампуле при 100—110° С кристаллизуется дигидрат с ромбической кристаллической решеткой (в природе минерал варисцит) либо с моноклинной решеткой (метаварисцит).
Кислые ортофосфаты А1Нз(РО4)2-ЗН2О, А1Н3(РО4)2-2Н2О и А1(Н2РО4)з Н2О выделены из систем А1РО4—Н3РО4—Н2О, образующих вязкие пересыщенные растворы, в которых атомное отношение Р:А1 может достигать ~2,3. Такие растворы используют в качестве алюмофосфатной связки в производстве безобжиговых керамических материалов, а также для получения покрытий на металлах. В смесях с корундом, кварцем и другими наполнителями они образуют на холоде прочную монолитную массу, которая при нагревании подвергается поликонденсации с образованием водостойких стеклообразных полифосфатов [А1(РОз)з]х, обладающих хорошей адгезией к металлам и керамическим материалам.
ГЛАВА 8
СВИНЕЦ И ЕГО СОЕДИНЕНИЯ
8.1. СВИНЕЦ
Содержание свинца в земной коре 1,6-10‘3% (масс.), в водах Мирового океана 0,03 мкг/л (41,1 млн. т), в речных водах 0,2—8,7 мкг/л. Известно около 80 минералов, содержащих свинец. Он в основном сопутствует силикатам и сульфидам, а также содержится в некоторых углях. Основным минералом является галенит (свинцовый блеск) PbS. Некоторое промышленное значение имеют англезит (PbSO4) и церуссит (РЬСОз). Свинец является конечным продуктом радиоактивного распада урана и тория. Поэтому он содержится в урановых и ториевых минералах. Известны также минералы, содержащие свинец. К ним относятся: пироморфит РЬ5[(РО4)зС1], миметезит Pbs[(AsO4)3Cl], крокоит РЬСгО4, вульфенит РЬМоО4 и штольцит PbWO4 и др.
Руды свинца содержат медь, цинк, кадмий, благородные металлы, висмут, теллур и другие ценные элементы. Природный фон в атмосфере 210'9—5 ГО'4 мкг/м3. В теле человека содержится 7—15 мг свинца.
Свойства. Свинец — металл синевато-серого цвета, кристаллизуется в гранецентрированной кубической решетке, а = 0, 49389 нм, z - 4, пространственная группа Fm3m. Он — один из легкоплавких металлов; температура плавления 327,50° С, кипения —1751° С; плотность, г/см3:11,3415 (20° С), 10,686 (327,6° С), 10,078 (850° С); С° = 26,65 Дж/(моль-К); ДЯ°Л = 4,81 кДж/моль, ДЯ°СП = 177,7 кДж/моль; S2°9g = = 64,80 Дж/(моль-К). Свинец — плохой проводник тепла и электричества; теплопроводность 33,5 Вт/(м К); температурный коэффициент линейного расширения свинца чистотой 99, 997% в интервале температур 0—320° С описывается уравнением: а = 28,15-10Л + 23,6- IO’9?0 С'1; при 7,2 К он переходит в сверхпроводящее состояние. Он диамагнитен.
Свинец мягок, пластичен, легко прокатывается в тончайшие листы. Твердость по Бринеллю 25—40 МПа. Значительно повышают его твердость и прочность такие металлы, как натрий, кальций и магний, но они уменьшают его химическую стойкость. Медь повышает антикоррозионную стойкость свинца к действию серной кислоты. При добавке сурьмы возрастает твердость и его кислотоупорность к H2SO4. Понижают кислотоупорность свинца висмут и цинк, а кад-336
мий, теллур и олово повышают его твердость и сопротивление усталости. Со свинцом практически не реагируют азот, оксид и диоксид углерода, кислород, диоксид серы и водород.
Свинец химически довольно инертен. Стандартный электродный потенциал свинца — 0,1265В для РЬ°/РЬ2+. В сухом воздухе не окисляется, во влажном — тускнеет, покрываясь пленкой оксидов, переходящей в присутствии диоксида углерода в основной карбонат 2РЬСОз РЬ(ОН)2. При комнатной температуре свинец не реагирует с разбавленной серной и хлороводородной кислотами, так как образующиеся на его поверхности труднорастворимые пленки сульфата и хлорида свинца препятствуют дальнейшему его растворению. Концентрированная серная кислота (>80%) и хлороводородная кислота при нагревании взаимодействуют со свинцом с образованием водорастворимых Pb(HSO4)2 и Н4[РЬСЦ]. Свинец устойчив по отношению к фтороводородной кислоте, водным растворам аммиака и щелочей и к многим органическим кислотам. Свинец растворяется в азотной и уксусной кислотах с образованием нитрата РЬ(140з)2 и ацетата свинца РЬ(СизСОО)2. Свинец заметно растворяется также в лимонной, муравьиной и винной кислотах. РЬ4+ — сильный окислитель, поэтому не существуют иодид и бромид свинца. При разряде свинцового аккумулятора РЬ4+ также служит окислителем:
Pb + РЬО2 + 2H2SO4 = 2PbSO4 + 2Н2О
При взаимодействии оксидов Pb(IV) и РЬ(П) с расплавами оксидов, гидроксидов и карбонатов образуются соли — соответствующие плюмбаты (IV) и плюмбаты (II), например Na2PbO3 и Na2PbO2. Свинец медленно растворяется в концентрированных растворах щелочей с выделением водорода и образованием М4[РЬ(ОН)6].
При нагревании свинец реагирует с галогенами, фтороводородной кислотой и с серой.
Получение. Основным сырьем для получения свинца являются сульфидные полиметаллические руды ( содержащие 1—5% свинца), из которых селективной флотацией получают концентраты. Концентрат обычно содержит 40—75% свинца, 5—10% цинка, до 5% меди, а также благородные металлы и висмут. Обычно около 90% свинца получают по технологии, включающей следующие стадии: алгомери-рующий обжиг сульфидсодержащих концентратов, шахтная восстановительная плавка агломерата и рафинирование черного свинца. В настоящее время разрабатывается технология автогенных процессов плавки, позволяющих использовать теплоту сгорания сульфидов.
Агломерирующий обжиг при традиционном производстве свинца проводят на прямолинейных машинах с дутьем воздуха или путем просасывания его. При этом PbS окисляется преимущественно в 337
жидком состоянии: 2PbS + ЗО2 = 2РЬО + 2SO1. В шихту добавляют флюсы (SiOi, СаСОз, РегОз), которые реагируют между собой и с РЬО, образуют жидкую фазу, цементирующую шихту. В готовом агломерате свинец в основном концентрируется в свинцово-силикатном стекле, занимающем до 60% объема агломерата. При этом оксиды цинка, железа, кремния и кальция кристаллизуются в форме слож-i ных соединений, образуя жаропрочный каркас. Эффективная рабочая площадь агломерационных машин 6—95 м2.
В готовом алгомерате содержится 35—45% свинца и 1—2% серы, часть которой находится в виде сульфатов. Далее свинцовый агломерат направляют на восстановительную плавку в шахтных печах. При этом роль восстановителя исполняет кокс, расход которого к массе агломерата составляет 8—14%.
В основе автогенных процессов выплавки свинца лежит экзотермическая реакция
PbS + О2 -> Pb + SO2 состоящая из двух стадий:
2PbS + ЗО2 -> 2РЬ0 + 2SO2
PbS + 2РЬ0 -> ЗРЬ + SO2
Преимущества автогенных способов перед традиционной технологией: исключается агломерационный обжиг, устраняется необходимость разложения исходного концентрата флюсами, что снижает выход шлака, используется теплота от горения сульфидов и частично исключается расход кокса, повышается извлечение диоксида серы с газами.
Полученный тем или другим способом черновой свинец содержит 93—98% основного вещества и следующие примеси: Си (1—5%), Sb, As, Sn (0,5—3%), Al (1—5 кг/т), Au (1—30%), Bi (0,05—0,4%).
Очистку чернового свинца производят пирометаллургически или электролитически, после чего рафинированный свинец разливают в чушки (около 30 кг) либо блоки (~1 т).
Применение. До 45% свинца от общего количества расходуется на производство свинцовых аккумуляторов; до 20% — на изготовление проводов и кабелей и покрытий к ним; 5—20% — на производство тетраэтилсвинца. Свинец применяют для изготовления футеровки, труб и аппаратуры в химической промышленности. В строительстве свинец применяют в качестве изоляции, уплотнителя швов, стыков, в том числе при создании сейсмостойких фундаментов. В военной технике свинец применяют для изготовления шрапнели и сердечников пуль. Свинцовые экраны служат для защиты от радиоактивного и рентгеновского излучений.
338
8.2. ОКСИДЫ СВИНЦА
Существуют следующие оксиды свинца: оксид РЬО, диоксид РЬО2, ортоплюмбат (IV) свинца (II) РЬзО4, сесквиоксид свинца РЬ20з, а также РЬ^Оп и РЬ|2О|9. Все оксиды свинца, кроме РЬО, при термообработке переходят в РЬО с выделением О2. Области их существования приведены на рис. 8.1, а свойства—в табл. 8.1.
Оксид свинца РЬО (рис. 8.2) существует в двух модификациях: низкотемпературной — а-РЬО (минерал глет) и высокотемпературной— Р-РЬО (минерал массикот). Их составы могут отклоняться от стехиометрических, границы областей гомогенности показаны на рис. 8.3. Полиморфный переход 0->а протекает медленнее, чем а->р, поэтому Р-РЬО может существовать при комнатной температуре в ме-тастабильном состоянии, однако при растирании переходит в а-РЬО. Испаряется РЬО конгруэнтно, в основном в виде РЬгО2 и РЬ4О4. Уравнение температурной зависимости давления пара над твердым Р-РЬО: 1g /?(Па) = 13, 71—13, 86-103/7(887—1151 К). Обладает полупроводниковыми свойствами в зависимости от состава (в пределах области гомогенности имеет электронный и дырочный тип проводимости). В процессе термообработки на воздухе РЬО окисляется по схеме
РЬО -3-3££ -> РЬ,2О9.-> Pbi20i7.......> РЬ3О4
В среде восстановителей (Н2 или СО) РЬО восстанавливается до элементного свинца:
РЬО + Н2 = РЬ + Н2О
РЬО + СО = РЬ + СО2
Рис 8.1. Диаграмма давления
О2— температура системы РЮ2
Рис. 8.2. Структура оксида свинца
339
Таблица 8.1. Свойства оксидов свинца
Показатель а-РЬО Р-РЬО а-РЬ1Оз р-РЬгОз а-РЬОз Р-РЬОг РЬ12О19 РЬ2О3 РЬиО17
Цвет Красный Желтый — Красный Коричневый Черный Черный Черный Черный
Сингония Тетрагона- Ромбиче- Ромбиче- Тетрагона- Тетрагона- Ромбиче- Моноклин- Моноклин- Моноклин-
льная ская ская льная льная ская ная ная ная
Параметры элементарной ячейки, нм:
а 0,396 0,5482 0,91152 0,88129 0,4959 0,4986 0,7735 0,7006 0,778
b — 0,4497 0,84696 — — 0,5965 1,0836 0,5632 1,098
с 0,500 0,5891 0,65646 0,65661 0,3384 0,5486 1,1505 0,3909 1,148
Число формульных единиц в ячейке 2 4 4 2 2 — — — —
Пространственная группа PJnmm РЬст РЬат Ptymbe Р42/7ЛЛ7Л — — — —
Температура фазового 550—590 887 -90
превращения, °C
Плотность, г/см3 9,40 9,63 — 8,92 9,35 9,55 9,58 10,05 —
С°, Дж/(мольК) 46,41 — — 146,7 61,0 — — 107,7 —
ДЯ^,, кДж/моль -218,6 0,2 — -720,0 276,0 — — -491,7 —
S'w Дж/(мольК) 67,84 — — 211,3 71, 92 — — 151,9 —
Ширина запрещенной зоны, эВ 2,0 2,7 — 2,1 — — — — ——
Подвижность электронов, см^Вс) 120 5—50 — 0,6—3,0 — — — —
Растворимость (%, масс.) в воде при 20° С: а-РЬО—0,6-10’3, 0-РЬО—1,2-10“3. Хорошо растворим в азотной кислоте и в водном растворе НСЮд, хуже — в хлороводородной и серной кислотах из-за образования труднорастворимых хлорида (РЬС12) и сульфата (РЬЗОД Растворяется в растворах щелочей, образуя при этом гидроксоплюмбаты (II) типа Na2[PbCOH)4].
Способы получения. Оксид свинца может быть получен несколькими способами.
1. Оксид свинца получают кипячением гидроксида свинца РЬ(ОН)2 в водном растворе гидроксида натрия, причем при большом избытке NaOH полу-
чают а-РЬО, а при меньшем — Р-РЬО. При длительном же кипячении смеси происходит переход Р~>а. *
2. Осаждением оксида свинца путем реакции его ацетата в гид-
т,к
1100
1000
900
800
700 600
110-4 о l.fO-з 3 10*
* хрь хо ---►
Рис 8.3. Диаграмма температура—концентрация оксида РЬО; хрь и —концентрация избытка РЬ и О2, в мол. долях; L\ и L2 — расплав
роксиде натрия:
Pb(CH3COO)2 + 2NaOH = РЬО + 2CH3COONa + Н2О
3. Термическим разложением карбоната, нитрата и основного карбоната свинца:
РЬСОз —РЬО + СО2
Pb(NO3)2 —РЬО + 2NO2
2РЬСОз-РЬ(ОН)2 —РЬО + 2СО2 + Н2О
4. Оксидированием расплава 650—700° С элементного свинца, а также оксидированием свинецорганических соединений в потоке кислорода или воздуха по схеме
2РЬ + О2 = 2РЬО
4РЬ(СН3СОО)2 + ЗО2 —4РЬ0 + 6СО2 + 6Н2О
По последнему способу получают наиболее высокие сорта оксида свинца.
341
Технология глета-оксида свинца оксидированием свинца. Процесс оксидирования чистого свинца по схеме 2РЬ + О2 = 2РЬО подчиняется параболической закономерности g-k^Tt при температурах
до 550° С и определяется скоростью диффузии ионов кислорода. При более высоких температурах облегчается доступ кислорода к металлу и процесс подчиняется линейной закономерности g = kt.
Значительное влияние на кинетику процесса оксидирования свинца оказывают примеси. Существенным является не только вид примесей, но и их количество, а также температура процесса. Установ-
лено, что тормозящее действие на скорость процесса оказывают олово, цинк и особенно алюминий. Участие в процессе всего лишь 0,02% алюминия полностью прекращает окисление исходного свинца.
Практически не влияют на ход оксидирования свинца такие примеси, как висмут, серебро и медь. Участие в процессе оксидирования свинца щелочных и щелочноземельных металлов, наоборот, ускоряет процесс, особенно магний (рис. 8.4)
Неодинаковое влияние примесей на скорость оксидирования свинца обусловлено различием степени их сродства к кислороду и структурой образуемой оксидной пленки, т.е. отношением мольных объемов оксидов и соответствующих металлов.
Схема производства оксидирования элементного свинца с получением оксида свинца (глета) состоит из следующих операций (рис. 8.5):
плавление исходного свинца в плавильной печи; оксидирование свинца
Содержание элементов, %(масс.)
Рис 8.4. Зависимость скорости процесса окисления свинца при 500° С от содержания в нем различных примесей
в окислительной печи, пневмотранспортировка глета-полуфабриката; отделение глета-полуфабриката от воздуха; сепарация и размол; упаковка.
Оксидирующая (плавильная) печь (рис. 8.6) состоит из чаши 1, которая зафутерована огнеупорным кирпичом, крышки 3, вала 4 с лопастной мешалкой 2 и приводной части. Вал 4 приводится во вращение (290—310 об/мин) от электродвигателя.
При нормальном ходе процесса подача в печь теплоты извне не производится, так как количество выделяемой теплоты реакции достаточно для поддержания температуры процесса в пределах 450—480° С. Воздух, необходимый для процесса окисления исходного свинца, подается в печь непрерывно вентилятором со скоростью 800—1100 м3/ч.
342
Рис. 8.5. Схема получения глета и сурика:
1 — плавильная печь; 2 — топка; 3 — окислительная печь; 4 — вентилятор; 5 — наклонная шахта; 6 — сухая уловительная камера; 7 — шнек; 8— мокрая уловительная камера; 9— центробежные насосы; 10— сгуститель; 11 — диафрагменный насос; 12 — печь второго обжига; 13, 19, 21, 23, 28, 29 — бункер; 14— питатель; 15, 24 — сепаратор; 16, 25 — дезинтегратор; 17, 26—вентилятор; 18, 27 — циклон; 20, 30 — упаковочная машина; 22 — печь для окисления глета в сурик
Для регулирования температуры в реакционном объеме в печь подают воду со скоростью до 80 л/ч. Соотношение воды и воздуха, подаваемых в реактор, может меняться в зависимости от назначения целевого продукта.
Расплавленный свинец непрерывно поступает в печь через воронку б, лопастью приводится во вращательное движение, дробится и растекается жидким слоем по внутренней поверхности печи. При этом создается значительная поверхность оксидирования свинца. Образующийся глет-полуфабрикат, а также мелкие частицы недоокис-ленного свинца в виде взвеси с воздухом выносятся из печи через отверстие 5 в вытяжную шахту.
На скорость процесса окисления свинца и на некоторые свойства получаемого глета-полуфабриката влияет изменение в составе газовой среды и наличие примесей в исходном сырье-свинце.
Рис 8.6. Оксидирующая печь
344
Свойства получаемого глета-полуфабриката значительно изменяются при незначительных отклонениях в режиме процесса оксидирования. К ним относятся: постоянство температуры, а необходимым его условием является равновесие теплоты, выделяемой при образовании оксида свинца и расходуемой на данной операции (с отходящими газами, на испарение подаваемой воды и через стенки печи). С увеличением подачи воздуха или воды снижается температура в оксидирующей печи, т.е. нарушается тепловой баланс, повышается унос крупных частиц глета-полуфабриката, содержащего значительную долю исходного сырья — элементного свинца.
Недостатком работы оксидирующей печи является налипание образующегося глета и исходного свинца на внутренние поверхности печи, что приводит к уменьшению реакционного объема и изменению теплопередачи печи, нарушению теплового баланса. Кроме того, в процессе работы в печи накапливаются крупные частицы глета, которые не уносятся воздушным потоком.
Согласно технологической схеме (см. рис. 8.5), исходное сырье— элементный свинец — в виде металлической чушки загружается в плавильную печь 7, вмурованную в топку 2, плавится при температуре 450° С и самотеком поступает в окислительную печь 3. Воздух в печь подается вентилятором 4. Образующийся глет-полу-фабрикат, содержащий до 15% неоксидированного свинца, уносится через имеющую наклон шахту 5 в сухую уловительную камеру 6. В шахте происходит частичное дооксидирование металлического свинца. Она выполняет роль и сепаратора. Крупные частицы глета и свинца теряют скорость и возвращаются в плавилку. Осаждающийся в сухой уловительной камере глет-полуфабрикат (около 90%) выводится из камеры шнеком 7. Воздух очищается от пыли в мокрой камере 8. Распыливаемая в верхнюю часть камеры оборотная вода смешивается с частицами глета-полуфабриката и образовавшаяся суспензия непрерывно перекачивается насосом 9 в сгуститель 10. а паровоздушная смесь выбрасывается в атмосферу. Суспензию глета-полуфабриката, выгружаемую из сгустителя, либо перерабатывают в цехе и получают глет или сурик, либо выпускают в виде товарного продукта, используемого в качестве сырья в других производствах свинецсодержащих соединений.
Для получения стандартного оксида свинца РЬО (глета) глет-полу-фабрикат подвергается дополнительному оксидированию в печах вторичной термообработки 72. Муфельная печь 72 снабжена тихоходным метальным устройством. Обогрев печи осуществляется продуктами сгорания жидкого или газового топлива до 600—700° С. Печь работает в двух режимах: периодически и непрерывно. Производительность печи зависит от количества содержащегося в глете-полуфабрикате металлического свинца.
345
Рис. 8.7. Оксидирующая печь для получения глета:
7 — корпус печи; 2 — горизонтальная мешалка; 3 — устройство для загрузки свинца;
4— устройство для выгрузки глета
После печи глет поступает на размольно-сепарационную установку, состоящую из сепаратора 15, дезинтегратора 16, вентилятора 17 и циклона 18. Кондиционный целевой продукт — глет — из бункера 19 поступает на упаковочную машину 20.
Вместо оксидирующих печей для получения глета предложено использовать оксидирующий аппарат (рис. 8.7), снабженный двумя горизонтальными мешалками 2. Применяются в производстве глета и вращающиеся барабанные печи с наружным обогревом, оборудованные внутренними ребрами и частично заполненные шарами из жаропрочной стали для измельчения свинца. Общим для всех используемых реакторов является оксидирование исходного металлического свинца кислородом воздуха и пневмовыгрузка целевого продукта.
Предложены также изменения в аппаратурном оформлении и других стадий производства. Вместо сухой уловительной камеры предложено использование циклонов или системы охладительных бункеров, которые позволили бы классифицировать целевой продукт по дисперсному составу и содержанию металлического свинца. Вместо мокрой уловительной системы используют скрубберы или другие аппараты тонкой очистки воздуха.
Разработан принципиально новый способ получения глета окислением свинца в парообразном состоянии, согласно которому расплавленный свинец поступает на разогретую до 1500° С решетку печи, мгновенно испаряется и оксидируется. Полученный оксид свинца отводится через холодильную башню в осадительную камеру. Предложено также использовать в качестве источника теплоты вольтовую дугу, а в целях сокращения энергозатрат — более полное использование теплоты реакции процесса оксидирования свинца. Отличительной особенностью данного метода является получение сверхтонкодисперсного продукта с низким содержанием металлического свинца в целевом продукте.
Разработан и получил развитие процесс холодного оксидирования свинца на стадии его измельчения. С этой целью используют ситовые мельницы с периферической разгрузкой, циклонные, вихревые или ко-346
нические мельницы с пневмовыгрузкой. Исходный свинец загружают в виде шариков диаметром 20 мм. В некоторых производствах работают мельницы, подобные мельницам вихревого типа с цилиндрическим барабаном и прямоточной подачей воздуха через барабан.
Подаваемый в мельницы воздух одновременно является оксидато-ром свинца, транспортирующим средством, хладогентом, т.е. отводит избыточную теплоту, выделяемую в процессе оксидирования свинца.
Температура в мельницах поддерживается в диапазоне 85—180° С и зависит от типа мельницы и требований, предъявляемых к целевому продукту. Получаемый рассмотренным способом свинцовый порошок оценивается содержанием в нем оксида свинца, фракционным составом и формой (строением) зерен. Оксид свинца, образующийся в процессе получения свинцового порошка, представляет собой только а-РЬО. Концентрация оксида свинца в порошке составляет 65—70% и зависит от режима работы мельницы. В процессе хранения полученного продукта при доступе воздуха содержание в нем оксида свинца растет. За сутки содержание оксида свинца повышается в пределах 0,2—0,5%.
Форма зерен свинцового порошка зависит от способа удаления порошка из мельницы и режима ее работы. При соударении шариков и ударах их о стенку мельницы пластический свинец подвергается деформации и на поверхности шарика появляются тончайшие лепестки металлического свинца, покрытые с поверхности оксидом свинца. Повышение температуры уменьшает деформацию свинца и ускоряет его оксидирование и разрушение, поэтому лепестки постепенно отделяются от шарика, и если порошок быстрее удаляется от зоны измельчения, то его зерна сохраняют форму лепестков. В случае задержки лепестков в барабане мельницы зерна измельчаются, оксидируются и уплотняются. Порошок в таких условиях становится более оксидированным, с большей насыпной плотностью.
Дисперсность (фракционный состав) колеблется в широких пределах. Отдельные частицы достигают 200—300 мкм, основная же часть (до 90%) не превышает 40 мкм.
Свинцовый порошок является полуфабрикатом для производства глета. С целью уменьшения содержания металлического свинца свинцовый порошок подвергают дополнительному оксидированию. Получаемый глет подвергают сепарации и после соответствующего измельчения грубой фракции выпускают целевой продукт.
Оксид свинца (глет) применяют в производстве сурика, других соединений свинца, свинцовых стекол (хрусталь, флантглас) и глазурей, при росписи стекла и фарфора, при изготовлении олиф, в качестве фоточувствительного материала в видиконах.
Ортоплюмбат (IV) свинца (II). Ортоплюмбат (IV) свинца (II) (свинцовый сурик) РЬзОд можно рассматривать в качестве смешанно-347
го оксида. В разном валентном состоянии атомов свинца в РЬ3О4 можно убедиться проведением его реакции с азотной кислотой:
Pb3O4 + 4HNO3 = 2Pb(NO3)2 + PbO2 + 2Н2О
Как видно, при этом происходит обменная реакция и образуются производные РЬ(П) и Pb(IV). Свинцовый сурик (РЬ2РЬО4) (рис. 8.8) существует в двух модификациях: низкотемпературной (a-форма) и высокотемпературной (P-форма). При нагревании выше 570° С он окисляется до оксида свинца.
Уравнение температурной зависимости давления разложения 1g р О2(Па) = 15,0—8,70 103/Т. В воде Р-РЬ3О4 плохо растворим, хорошо растворяется в расплавленном нитрате натрия, водном растворе НС1О4. Получают Р-РЬО при 500° С в потоке воздуха.
Применяют сурик в производстве антикоррозионных грунтовок, аккумуляторов, оптического стекла и хрусталя, а также керамических красок.
В производстве оптического стекла и хрусталя, т. е. стекол с высоким показателем преломления, а также электровакуумного и рентгеновского стекла, сурик применяется в качестве основного вида сырья. Некоторые марки стекол содержат до 60—70% свинца. Требования к качеству свинцового сурика особенно высоки в связи с тем, что присутствие в его составе даже тысячных долей процента таких элементов, как хром, медь, кобальт, придают стеклу нежелательную окраску.
Применение свинцового сурика в качестве пигмента основано на его высоких антикоррозионных свойствах, атмосферостойкости и высокой адгезии лакокрасочных систем, пигментированных суриком. В верхних слоях наружных покрытий цвет сурика под действием сульфида водорода и солнечного света очень быстро меняется, поэтому его применяют для грунтовок.
Высокие антикоррозионные свойства сурика объясняются следующими факторами:
1) сурик в качестве основного оксида свинца создает щелочную среду и ингибирует электрохимическую коррозию стали;
2) являясь окислителем, в момент образования оксида железа
348
©Pbf/iO Q Pb(ii) Q)o
Рис. 8.8. Структура сурика РЬ3О4
(II) переводит ее в оксид железа (III), который при этом создает плотное покрытие, препятствующее развитию коррозии;
3) оксид свинца, присутствующий в составе сурика, реагирует со свободными кислотами связующего, образуя мыла, обладающие водоотталкивающими свойствами, и придает пленке водостойкость.
Технология получения свинцового сурика основана на процессе оксидирования оксида свинца кислородом, независимо от способа его получения, по реакции
6РЬО + О2 = 2РЬ3О4
Процесс оксидирования начинается не в любой точке кристалла оксида свинца, а в наиболее активных центрах (ребра, трещины, поврежденные места кристаллической поверхности) и развивается по межфазной поверхности раздела сурик — оксид свинца. Процесс оксидирования свинца ускоряется при добавлении к нему сурика. Значительная инертность некоторых образцов оксида свинца по отношению к процессу оксидирования объясняется отсутствием активных центров (совершенством поверхности частиц). Термообработка оксида свинца без доступа воздуха резко снижает его активность, видимо, вследствие энергетического выравнивания поверхности частиц РЬО. Измельчение же труднооксидируемых образцов заметно увеличивает скорость процесса оксидирования.
С повышением температуры процесса скорость оксидирования возрастает лишь до известного предела. С максимальной скоростью процесс оксидирования оксида свинца протекает при 500° С, т.е. около температуры структурного превращения (а-РЬО Р-РЬО), при дальнейшем повышении температуры скорость процесса оксидирования падает (рис. 8.9, а).
b 800 cL 2 600 2
g 400
о
40 ‘ do ' l£o ' 1бо ' 200 Время, мин
Рис 8.9. Зависимость скорости оксидирования р-РЬО (фракция > 10 мкм) от температуры (а) и от парциального давления при 480° С (фракция 3 мкм) (б)
349
Увеличение парциального давления кислорода ускоряет процесс оксидирования оксида свинца. Эта зависимость в пределах 150—760 мм рт. ст. незначительно отклоняется от линейной (рис. 8.9, б). С повышением температуры давление диссоциации сурика увеличивает- ? ся в соответствии с уравнением J
1g р = -7089, 5/Г + 1,791 lgF+ 2,16-lQ-4 Т+ 2,8.
Следовательно, повысив давление, можно расширить температурный диапазон процесса получения сурика, что, в свою очередь, ускоряет реакцию процесса за счет увеличения процесса диффузии. На- ; пример, при повышении температуры от 480 до 560° С и давления ’ кислорода до 1 кгс/см2 (или воздуха до 5 кгс/см2) скорость реакции > возрастает в 3,5 раза в результате увеличения концентрации кислорода J и в 2,5 раза вследствие повышения коэффициента диффузии, в резуль- i тате чего в целом скорость реакции возрастает примерно в 9 раз.
По способности к оксидированию 0-РЬО более инертен, чем а-РЬО.
В литературе описывалась невозможность процесса прямого оксидиро- 1 вания Р-РЬО на воздухе и неизбежность промежуточной стадии перехода Р-РЬО -> а-РЬО, а последний принадлежит к одной и той же крис- 1 таллической сингонии с РЬ3О4 и, следовательно, при оксидировании а-РЬО не требуется необходимой перестройки кристаллической решетки, что и служит одной из причин большей активности а-РЬО. Помимо этого, следует учитывать, что только псевдотетрагональная а-РЬО спо- ) собна растворять кислород в процессе оксидирования, что, в свою оче- 5 редь, может отразиться на ее последующем оксидировании. j
Исследованиями электрической проводимости оксидов свинца при 350—450° С установлено, что у а-РЬО она на два порядка выше, чем у Р-РЬО. На этом основании высказано предположение, что различная активность двух структур оксида свинца обусловлена разной степенью упорядоченности их кристаллических решеток, т. е. концентрацией и подвижностью дефектов.
Технология сурика состоит из нескольких основных операций: оксидирование глета-полуфабриката; сепарация и размол; упаковка.
Печь периодического действия для оксидирования глета-полуфабриката в сурик (рис. 8.10) состоит из чугунного корпуса 9 с плоским точеным подом 5. По поверхности пода движутся гребки 4, укрепленные на водилах 3. Нижняя поверхность гребков пришлифована к поду. Форма гребков такова, что они лишь перемешивают сурик. Воздух, необходимый для оксидирования глета-полуфабриката в сурик, поступает в печь через канал 2 и отверстие 1. Для разгрузки печи открывают пробку, и гребки постепенно смещают весь сурик в выгрузочное отверстие 10.
350
В печь с диаметром 3,5 м загружают в один прием 4 т глета-полуфабриката. Глет-полуфабрикат загружают в печь при температуре не выше 300° С. В противном случае вследствие интенсивного процесса оксидирования свинца резко повышается температура и образуются комки глета, которые внутри не оксидируются. Температуру в печи поднимают постепенно в течение восьми часов до оптимальной, после чего следует выдержка (оксидирование). Весь цикл в печи продолжает-
Рис. 8.10. Печь для оксидирования глета-полуфабриката в сурик
351
ся обычно 15—20 ч (в зависимости от требуемого содержания РЬзОДЯ Отходящий воздух из печи направляется в пылеуловительную систему..»
Согласно схеме, глет-полуфабрикат из сухой пылеуловительноц! камеры системой внутрицехового транспорта подается через загрузи зочный бункер в печь. По окончании процесса, который определяют! анализом сурика на содержание РЬОз, его направляют в размольЛ но-сепарационную установку. Целевой продукт передается в бункер цЛ далее на упаковочную машину. Я
Недостатком данной технологии является ее периодичность. Раз-! работай способ получения сурика из глета в печи, работающей под 1 давлением, что позволяет сократить продолжительность процесса ок- я сидирования глета. 1
Схема производства приведена на рис. 8.11. Согласно схеме, глет | подается в бункер 2 из литой стали. Его емкость обеспечивает су- | точную работу установки, а толщина стенок — работу под давлением -до 25 кгс/см . Из бункера глет поступает в печь оксидирования 7, j представляющую собой два последовательно работающих обогревав-мых шнека, также рассчитанных на работу под давлением. В печи
Рис. 8.11. Схема производства сурика под давлением
352
поддерживают температуру 450—550° С. Сурик из печи выгружают в бункер 6. После заполнения глетом-полуфабрикатом бункера 2 систему герметизируют заглушками на загрузочном патрубке бункера 6. Воздух для оксидирования глета-полуфабриката подают компрессором 4 через подогреватель 5. Обработанный воздух из печи поступает в верхнюю часть бункера 2 и далее направляется на упаковочную машину 8. Требуемое содержание основного вещества в сурике регулируется продолжительностью пребывания (60 мин) материала в печи, что достигается изменением числа оборотов шнеков.
Недостатком способа является периодичность процесса и наличие в схеме аппаратов, работающих под давлением. Этих недостатков лишена технология сурика в горизонтальной вращающейся печи, снабженной перегородками в виде широких продольных лопаток. Способ получения сурика из металлического свинца заключается в первоначальном испарении его с использованием вольтовой дуги или в трубчатой печи, и дальнейшим окислением полученных паров воздухом или чистым кислородом. Отличительной особенностью сурика, полученного оксидированием парообразного свинца, является повышенная дисперсность (размер частиц менее 1 мкм), высокое содержание основного вещества в целевом продукте, т.е. РЬзО4, и низкая насыпная плотность (—1000 кг/см3).
Диоксид свинца РЬОг существует в двух модификациях — а-РЬО2 и Р-РЬО2. а-РЬОг при термообработке разлагается с потерей кислорода с полным переходом в оксид свинца по схеме
РЬО2 ..> Pbi20i9 35°’с > РЬ12О17 38О‘С > РЬ3О4 57О'С > РЬО
Диоксид свинца практически не растворим в воде. В растворах щелочей растворяется с образованием гидроксоплюмбатов (IV):
РЬО2 + 2NaOH + 2Н2О -> Na2[Pb(OH)6]
При термообработке с основными оксидами образуют плюмбаты (IV):
РЬО2 + 2Na2O —Na4PbO4
РЬОг—сильный окислитель. При нагревании с серной кислотой реагирует с выделением кислорода:
2 РЬО2 + 2H2SO4 = 2PbSO4 + О2 + 2Н2О
а с хлороводородной кислотой выделяет хлор:
РЬО2 + 4НС1 = РЬС12 + 2Н2О + С12
12 Химическая технология неорганических веществ, кн. 1
353
При растирании с элементной серой или красным фосфором вое- 1 пламеняется. |
Диоксид свинца получают оксидированием ацетата свинца хлор- | ной известью по схеме $
С1
РЬ(СН3СОО)2 + Са(ОС1)2 -> РЬО2 + Са(СН3СОО)2 + С12
Получают диоксид свинца также электрохимическим окислением солей двухвалентного свинца, разложением РЬ3О4 в азотной кислоте.
Диоксид свинца применяют в свинцовых аккумуляторах и в качестве окислителя-компонента головок спичек.
Другие оксиды свинца — РЬгОз, РЬ^Ор, Pbj20i9 — по своим свойствам близки к РЬ3О4. Оксиды Pb220i7 и Pbi20i9 получают термическим разложением диоксида свинца по схеме
РЬО2...Pb120i9 35О'С > Pbi2O17
Получают их также окислением свежеприготовленного оксида свинца по следующей схеме
РЬО ззо’с > РЬ12О,9 37О-с-> Pbi20i7
Сесквиоксид свинца получают гидротермальным разложением его диоксида в водном растворе гидроксида натрия при 250° С или оксидированием оксида свинца при 600° С и давлении кислорода 0,4 ГПа.
8.3. ТИТАНАТ СВИНЦА
Титанат свинца РЬТЮз—кристаллы темно- и светло-желтого цвета, с тетрагональной искаженной решеткой (а = 0, 3904 нм, с - 0, 4150 нм, z = 1, пространственная группа PMmmrri), при повышении температуры параметр а увеличивается, а с уменьшается. При температуре Кюри (766 К) титанат свинца переходит в кубическую модификацию типа перовскита (пространственная группа РтЗт). При нагревании выше 400° С имеет место полисинтетическое двойникование (образование областей с различной ориентацией кристалла) с образованием сегнетоэлектрических доменов по направлениям [101], [011], у которых ширина 0,1—10 мкм. Выше 500° С домены исчезают. Температура плавления 818° С, плотность 7,3 г/см3; сегнетоэлектрик; показатель преломления и20 2,65—2,75; двулучепреломление при 400° С и А 589,6 нм; в ИК-спектре полосы поглощения 590 и 420 см'*(по другим данным 575 и 405 см'1); е монокристаллов 30—170 (20° С), 10 000 (495° С), порис-354
той керамики 50, при добавлении 1—2 мол.% диоксида марганца е 210—280; tgS (1—7)10‘2 (20° С для монокристалла): коэффициент электромеханической связи К = 4—5%; р=1012—5-1013 Ом см.
Титанат свинца не растворим в воде, растворяется в горячей концентрированной серной кислоте.
В производстве титанат свинца получают:
1) термообработкой (спеканием) оксида свинца и диоксида титана (в виде керамики)
хРЬО + уГЮг —хРЬО • yTiO2 (хРЬ • уТЮ3)
2) совместным соосаждением из водных растворов гидроксидов, оксалатов или карбонатов с последующей термообработкой их выше 1000°С
Pb(NO3)2 + 2NaOH = Pb(OH)2 + 2NaNO3
PbO2 + 2NaOH + H2O = Pb(OH)4 + 2NaOH
далее
Pb(OH)4 + TiCO3 1000‘c > PbTiO3 + 2H2O + CO2
Монокристаллы титаната свинца выращивают из раствора в расплаве состава РЬО—0,15 ТЮ2—0,20 В2О3 при 1000° С, а пленки толщиной 0,1—0,3 мкм — катодным распылением.
Титанат свинца широко применяется для получения керамики на основе твердых растворов Pb(Ti, Zr)O3 и Pb(Ti, La)O3. Применяют титанат свинца также для исправления акустоэлектронных (в качестве преобразователя электрической энергии в акустическую, и наоборот), оптоэлектронных (элементы оптической связи) и запоминающих устройств, а также в СВЧ, дефектоскопии.
8.4. ХРОМАТ СВИНЦА
Хромат свинца РЬСгО4 имеет плотность 5,9—6,1 г/см3; малорастворим в воде (0,17—0,20 мг/л), частично растворим в разбавленных кислотах. В концентрированных кислотах (HCI, HNO3) он растворяется полностью:
PbCrO4 + 2НС1 = РЬС12 + СгО3 + Н2О
РЬСгО4 + 2HNO3 = Pb(NO3)2 + CrO3 + Н2О
12*
355
а в уксусной кислоте не растворяется. При действии разбавленных гидроксидов и карбонатов щелочных металлов хромат свинца превращается в одноосновный хромат:
2 РЬСЮ4 + 2NaOH = PbCrO4PbO + Na2CrO4 + Н2О
2 PbCrO4 + 2Na2CO3 + H2O PbCiO4 • PbO + 2NaHCO3 + Na2CrO4
Под действием концентрированных растворов щелочей хромат свинца переходит в оксид свинца, а под действием карбонатов — в карбонат свинца:
PbCrO4 + 2NaOH = PbO + Na2CrO4 + Н2О
PbCrO4 + Na2CO3 = PbCO3 + Na2CrO4
Технология хромата свинца. Хромат свинца получают взаимодействием водного раствора любой водорастворимой соли свинца с хроматом (II) и хроматом (IV) щелочного металла:
Pb(NO3)2 + K2CrO4 = PbCrO4 + 2KNO3
2РЬ(СН3СОО)2 + К2Сг2О7 + Н2О = 2 РЬСгО4 + 2СН3СООК + 2СН3СООН
Образующийся по этим реакциям хромат свинца выпадает в виде светло-желтого осадка, который в маточном растворе при промывке и в процессе сушки темнеет и становится темно-желтым с красноватым оттенком, не изменяя при этом свой химический состав.
Потемнение хромата свинца после осаждения связано с неустойчивостью ромбической структуры и ее перекристаллизацией под действием воды, света и температуры.
Хромат свинца встречается в природе в виде минерала крокоита моноклинной системы. При осаждении из растворов хромат свинца получается также в виде кристаллов моноклинной системы. Однако установлено, что он может существовать в виде кристаллов ромбической и тетрагональной систем.
Позже в литературе было сообщение о диморфизме хромата свинца и его способности кристаллизоваться в моноклинной и ромбической системах.
При изучении превращений, происходящих при нагревании и охлаждении хромата, сульфата и молибдата свинца и их смесей, было установлено, что хромат свинца триморфен, причем каждая форма— ромбическая, моноклинная и тетрагональная — устойчива в определенном температурном интервале.
356
Экспериментально установлено, что светло-желтый хромат свинца, образующийся при осаждении из растворов, является ромбической структурой и его потемнение связано с неустойчивостью этой структуры и перекристаллизацией ее в моноклинную.
Исследованы также условия получения сухого хромата свинца ромбической структуры. Результаты их полностью подтвердили полиморфизм хромата свинца, условия получения отдельных структур, а также их свойства.
Ромбическая структура окрашена в лимонно-желтый цвет, моноклинная— в темно-желтый, а тетрагональная — в красный.
На рис. 8.12 приведены рентгенограммы хромата свинца двух структур. Константы кристаллической решетки для моноклинной структуры: а = 6,82А, b - 7,48А, с = 7,16А, 0 = 102,33°.
Установлена склонность хромата свинца к образованию изоморфных смесей, т. е. он может образовывать смешанные кристаллы с рядом однотипно построенных соединений: BaSO4, CaSO4, РЬМоО4, PbSO4 и др.
Существует также оксихромат свинца РЬСгО4-РЬО с плотностью 0,675—0,698 г/см3. Термостоек, при нагревании до 600° С не разлагается. Растворимость оксихромата свинца примерно в 5 раз меньше, чем у хромата, а именно ~0,05 мг/л. В неорганических кислотах и концентрированных растворах щелочей растворяется полностью. К действию разбавленных растворов щелочей стоек. Уксусная кислота выщелачивает из оксихромата оксид свинца, оставляя при этом без изменения хромат.
I I
Рис 8.12. Рентгенограмма системы РЬСгО4:
1 — моноклинная; 2 — ромбическая
357
Оксихромат свинца получают следующими способами: 1) осаждением основной соли свинца хроматом щелочного металла или нейтральной соли свинца смесью хромата щелочного металла и гидроксидом натрия; 2) обработкой хромата или сульфата-хромата свинца гидроксидом натрия; 3) термообработкой смеси хромата свинца с его оксидом. При этом реакции происходят по следующим схемам:
РЬ(СН3СОО)2 • PtKOHh + Na2CrO4 = РЬСЮ4 • РЬО + 2CH3COONa + Н2О
2Pb(NO3)2 + Na2CrO4 + 2NaOH = PbCrO4 • РЬО + 4NaNO3 + H2O
2PbCrO4 + 2NaOH = PbCrO4 • PbO + Na2CrO4 + H2O
PbCrO4 • PbSO4 + 2NaOH = PbCrO4 • PbO + Na2SO4 + H2O
PbCrO4 + PbO —!-> PbCrO4 • PbO
Образование оксихромата свинца в водной среде происходит при pH раствора не ниже 9; при pH 8 осаждается нейтральный хромат свинца. Повышение pH не препятствует протеканию реакции, так как оксихромат стоек к действию щелочей.
В производстве оксихромат получают из основных солей свинца осаждением их смесью дихромата и щелочи:
2[РЬ(СН3СОО)г • РЬО] + Na2Cr2O7 + 2NaOH = = 2(PbCrO4 • PbO) + 4CH3COONa + H2O
или обработкой сульфохромата свинца гидроксидом натрия по схеме
РЬСгО4 • PbSO4 + 2NaOH = PbCiO4 • PbO + Na2SO4 + H2O
Оксихромат свинца можно получить из тонкодисперсного глета (РЬО) без применения кислот и щелочей. При медленном приливании водного раствора дихромата натрия к суспензии РЬО при температуре выше 40° С происходит гетерогенная реакция с образованием многоосновного хромата:
2(х + 1)РЬО + Na2Cr2O7 + Н2О = 2(xPbOPbCrO4) + 2NaOH
Если исходить из ромбической структуры оксида свинца (глета), то при такой топохимической реакции разрушаются кристаллы РЬО (поскольку основной хромат, как известно, имеет тетрагональную структуру) и реакция протекает достаточно глубоко. В случае РЬО 358
тетрагональной структуры образуются значительно более основные хроматы (х > 50). При реакции РЮ (ромбической) с СгОз в тех же условиях образуется одноосновный хромат РЬО-РЬСгО4.
Средний хромат образуется при обработке многоосновного хромата смесью дихромата с хлороводородной кислотой по реакции
2(хРЬО • РЬСгО4) + xNa2Cr2O7 + 2хНС1 = 2(х + l)PbCiO4 + 2NaCl + хН2О
Этот способ экономически более выгодный, однако его практическое применение ограничено из-за отсутствия надежных вариантов (путей) получения чисто ромбического глета.
8.5. ГАЛОГЕНИДЫ СВИНЦА
Дигалогениды свинца РЬХ2 (табл. 8.2) — кристаллы, при обычной температуре устойчивы в сухом воздухе, а во влажном воздухе с повышением температуры гидролизуются. При нагревании на воздухе окисляются до оксигалогенидов типа РЬгОХ2 и РЬ5О4Х2, при этом способность к окислению повышается в ряду F<Cl<Br<I. Растворимость в воде растет в присутствии одноименных ионов X" в связи с образованием комплексных анионов [РЬХз]~ и [РХ4]~. Легко образуют смешанные галогениды, например PbXF.
Галогениды свинца получают осаждением их из водных растворов солей двухвалентного свинца растворами, содержащими Х“ по схеме
РЬ(СН3СОО)2 + 2НВг = ЗРЬВг2 + 2СН3СООН
РЬСОз + 2HF = PbF2 + СО2 + Н2О
РЬО + 2НС1 = РЬС12 + Н2О
Полученные растворы галогенидов свинца очищают перекристаллизацией из подкисленных водных растворов.
В природе встречаются редкие минералы котунит РЬС12 и матлокит PbCIF.
Тетрафторид PbF4 — бесцветные кристаллы тетрагональной сингонии (а = 0,424 нм, с = 0,8030 нм, пространственная группа 14/тти); температура плавления около 600° С; плотность 6,7 г/см3; =-942,1 кДж/моль; при нагревании разлагается с выделением фтора и фазы приблизительного состава PbF3.
Тетрафторид свинца получают взаимодействием дифторида свинца с элементным фтором.
359
Таблица 8.2. Свойства дигалогенидов свинца
Показатель a-PbF2 P-PbF2 РЬС12 РЬВг2 РЬ12 PbCIF
Цвет Бесцветный Бесцветный Бесцветный с шелковистым Бесцветный с шелковистым Золотисто-желтый Бесцветный
блеском блеском
Сингония Ромбическая Кубическая Ромбическая Ромбическая Гексагональная Тетрагональная
Параметры элементарной ячейки, нм:
а 0,6454 0,5942 0,7623 0,8054 0,4557 0,4106
b 0,7863 — 0,9048 0,9537 — —
с 0,3899 — 0,4535 0,4726 0,6979* 0,7230
Число формульных единиц в ячейке 4 4 4 4 1 2
Пространственная группа Рпаш Fm3m Рпаш Рпаш P3ml PMnmm
Гпл, °C 447** 822 501 373 412 603
Тт °C — 1292 953 893 872 —
Плотность, г/см3 8,37 7,68 5,85 6,67 6,16 —
С“, Дж/(моль-К) 74,1 — 77,0 80,6 — —
ДНпл, кДж/моль 2,1 11,9 23,9 20,8 21,1 36,8
ДНкнп, кДж/моль — 160,3 129,0 118,0 100,5 —
ДНобр, кДж/моль -677,0 — -360,0 -281,9 -175,4 -534,2
5^, Дж/(мольК) 113,1 — 134,4 161,8 175,5 —
Растворимость в воде (г в 100 г):
20° С 0,064 — 0,98 0,87 0,069 0,07
100° С — — 3,25 4,75 0,436 —
'Для основной формы 2Я-РЫ2 в небольших количествах получены политипные модификации 4Я-РЫ2, 6Я-РЫ2 и др. "Температура превращения а->0.
Тетрахлорид свинца РЬС14— желтая жидкость с температурой плавления — 7° С, плотностью 3,18 г/см3 (0°С); ДЯ^р=-328,9 кДж/моль. При 100° С взрывается. С хлороводородной кислотой образует H2PbCU:
РЬС14 + 2НС1 = Н2РЬС16
Тетрахлорид свинца получают взаимодействием [(C2H5)3NH]2PbC16 с концентрированной серной кислотой. В лабораторных условиях получены комплексные соединения M'JPbXe], где X = Cl, F. Тетрабромид и тетраиодид свинца пока не получены.
Дифторид свинца PbF2 применяют в качестве твердого электролита (P-PbF2; вместо чистого дифторида применяют также PbSnF4) для изготовления катодов в химических источниках тока, в качестве компонента керамики, эмалей, лазерных материалов, а также расплавов для выращивания монокристаллов оксидов металлов, в качестве материала оптических покрытий.
Дихлорид свинца РЬС12 является полуфабрикатом в производстве металлического свинца и активатором люминофоров на основе сульфида цинка.
Фторохлорид свинца PbCIF и дииодид свинца РЫ2 применяются в аналитической химии при гравиметрическом определении свинца или фтора.
8.6. СУЛЬФАТЫ СВИНЦА
Сульфат свинца (И) PbSO4 — бесцветные кристаллы. До 878°С сульфат свинца (И) существует в a-форме с ромбической решеткой (а = 0,8480 нм, b = 0,5938 нм, с = 0,6958 нм, z = 4, пространственная группа Рпта), а выше указанной температуры существует в P-форме с моноклинной решеткой. АН полиморфного перехода 17,0 кДж/моль; температура плавления 1170° С. Процесс плавления сопровождается его разложением с выделением диоксида серы, кислорода, оксисульфатов свинца, а затем и его оксида. Плотность P-PbSO4 6,35 г/см3; С ° = 103,29 Дж/(моль-К); ДЯ^ = 40,2 кДж/моль; ДЯ^> = = -921,20 кДж/моль; = 148,71 Дж/(моль-К); показатели преломления: больший Ng 1,8937, средний Nm 1,8826, меньший Np 1,8771. Растворимость (г в 100 г): в воде -0,0045 (25° С); 0,0057(50°С); в растворе (5% воды — 95% H2SO4) -0,080 (25° С). Растворим в водных растворах ацетата и тартрата аммония, щелочей, горячей серной кислоте. Не растворяется в этиловом спирте.
361
Сульфат свинца (II) восстанавливается углеродом и водородом до его сульфида. Реагирует с сульфидом свинца с образованием элементного свинца и диоксида серы. Эта реакция является основной в процессе выплавки металлического свинца из сульфидных руд.
Сульфат свинца (II) образует основные сульфаты — 3PbO-PbSO4-H2O; PbOPbSO4 и др., а также двойные сульфаты [КгРЬ^О^] и др. В природе встречается минерал англезит a-PbSO4.
Сульфат свинца (II) получают:
1) осаждением из растворов солей РЬ(П) ионами SOj":
Pb(CH3COO)2 + H2SO4 = PbSO4 + 2СН3СООН
2) оксидированием сульфида свинца пероксидом водорода:
PbS + 4Н2О2 = PbSO4 + 4Н2О
Сульфат свинца (II) применяют для заполнения ячеек пластин в свинцовых аккумуляторах, является компонентом лаков и пигментов. Такие соединения, как PbOPbS04 и 3PbO-PbSO4-H2O, применяют в качестве добавок к цинковым и свинцовым белилам.
Сульфат свинца (IV) Pb(SO4)2 — кристаллы желтоватого цвета. При нагревании разлагается. Гидролизуется водой с образованием РЬ(ОН)г8О4, а затем РЬО2-хН2О, плохо растворим в горячей концентрированной серной кислоте, является сильным окислителем, образует двойные сульфаты М’2 РЬ(8О4)з.
Сульфат свинца (IV) получают в виде осадка при электролизе 80%-ной серной кислотой со свинцовыми электродами.
8.7. ХАЛЬКОГЕНИДЫ СВИНЦА
К халькогенидам свинца относятся его сульфиды, селениды и теллуриды.
Сульфид PbS, селенид PbSe и теллурид РЬТе — серебристо-серые кристаллы кубической сингонии (типа NaCl, z = 4, пространственная группа Fm3m), а при давлении выше 2,4—4,2 МПа устойчива ромбическая модификация (типа SnS, пространственная группа Рстп). Основные свойства халькогенидов свинца приведены в табл. 8.3.
В парах халькогенидов свинца обнаружены PbX (X = S, Se, Те), РЬ2Х2, РЬ, Х2, РЬХ2. Уравнения температурной зависимости давления пара в условиях конгруэнтного испарения: 1g ррьз(атм) = -11 597/Т + + 6,61; 1g ppbse(aTM) = -11 032/Т + 7,204; 1g />рьте(атм) = -11180/Т + 7,45. Халькогениды свинца не растворяются в воде, разлагаются азотной и серной кислотами. 362
Таблица 8.3. Свойства халькогенидов свинца
Показатель PbS PbSe РЬТе
Параметр кристаллической решетки, нм 0,593 0,6126 0,6460
Плотность, г/см3 7,61 8,26 8,24
Гпл, °C 1113 1080 924
С", Дж/(моль-К) 49,50 50,21 50,54
ДЯПл, кДж/моль 36,5 35,6 41,1
ДЯобр, кДж/моль —99,6 -99,2 -68,6
^298» Дж/(мольК) 91,2 102,5 110,0
Температурный коэффициент линейного расширения, Ю’ЧС1 20,3 19,4 19,8
Теплопроводность, Вт/(м-К) 2,5 1,6 2,0
Ширина запрещенной зоны £ пр 300 К, эВ 0,41 0,29 0,32
ДЕ/ДТ, КГ" эВ/К 4,0 4,0 4,5
Эффективная масса: 0,08 0,04 0,02
электронов 0,08 0,03 0,02
дырок
Подвижность носителей тока, см2/(В с) при 300 К:
электронов 6102 1103 2103
дырок 6102 1103 8103
8 172 206 400
Халькогениды свинца встречаются в природе в виде минералов галенита (свинцовый блеск) PbS, клаусталита PbSe, алтаита РЬТе.
Халькогениды свинца получают взаимодействием расплава или пара свинца с халькогеном Pb + X —РЬХ, осаждением из водных растворов РЬ(П) серо- или селеноводородном: РЬС12 + H2S = = PbS + 2НС1 или Pb(CH3COO)2 + H2Se = PbSe + 2CH3COOH, а также реакцией свинецорганических соединений с халькогенорганиче-скими соединениями. Монокристаллы выращивают осаждением из газовой фазы, из расплава, методом Бриджмена — Стокбаргера или зонной плавкой под давлением пара халькогена.
Состав халькогенидов свинца в зависимости от условий получения отклоняется от стехиометрического в сторону избытка халькогена (от 0,995 до 0,5005 молярных долей свинца или до -1-10’3 молярных долей избыточного компонента, рис. 8.13). С изменением состава халькогенидов свинца наблюдается изменение типа основных носителей заряда, поэтому халькогениды свинца могут быть полупроводниками п- или р-типа.
Халькогениды свинца и их твердые растворы являются хорошими полупроводниковыми материалами в электронике и радиотехнике, например: PbSe — материалы для фоторезисторов, фотоприемников и излучателей в ИК-диапазоне, активная среда в инжекционных лазерах; РЬТе — фотопроводник при низких температурах, материал для оптики.
363
8.8. НИТРАТ СВИНЦА
Нитрат свинца Pb(NO3)2, бесцветные кристаллы кубической сингонии (а = 0,784 нм, z = 4, пространственная группа РаЗУ, плотность 4,538 г/см3; ДЯобр = -451,75 кДж/моль; 5^, = 217,9 Дж/(моль-К). При нагревании выше 200°С начинает разлагаться без плавления с выделением диоксида азота и кислорода и последовательным образованием оксонитратов РЬ(КОз)2,2РЬО, РЬ(НОз)г-5РЬО и оксида РЬО при 500—550°С. Растворимость в воде (г в 100 г): 45,5 (10°С), 58,5 (25° С), 91,6 (60° С) и 116,4 (80° С). Хорошо растворим в гидразине, аммиаке, пиридине и формамиде, не растворяется в этаноле, ацетоне и неполярных растворителях. В водных растворах диссоциирован, при избытке нитрат-ионов образует нитратокомплексы [РЬ(ЬЮз)з]’, [РЬ(НОз)4]2' и [РЬ(ИОз)б]4'. Степень гидролиза нитрата свинца в растворе составляет 0,05—0,15. При повышении pH раствора образуются гидроксонитраты переменного состава РЬ(ОН)л(КОз)>,, некоторые из них выделены и в твердом состоянии. Получен также ряд безводных и гидратированных оксонитратов хРЬ(ЫОз)2;уРЬО-2Н2О.
В лаборатории нитрат свинца получают растворением свинца, оксида и карбоната свинца в азотной кислоте:
Pb + 2HNO3 = РЬ(ЬЮз)2 + Н2
РЬО + 2HNO3 = PbCNOjh + Н2О
РЬСОз + 2HNO3 = Pb(NO3)2 + Н2О + СО2
а также электролизом свинцовых анодов в растворе нитрата натрия.
В промышленности нитрат свинца получают растворением гранул элементного свинца в слабой азотной кислоте.
Нитрат свинца ограниченно применяют для получения других соединений свинца. Он является компонентом пиротехнических составов. 364
ГЛАВА 9
ТИТАН И ЕГО СОЕДИНЕНИЯ
9.1. ТИТАН
Содержание титана в земной коре составляет 0,57% по массе. По распространенности в природе занимает десятое место среди других элементов таблицы Менделеева, но в свободном виде практически не встречается. Хотя и известно более ста минералов, содержащих титан, но не все они в настоящее время имеют промышленное значение.
Разнообразие титансодержащих минералов обусловлено близостью величин ионных радиусов и энергетических потенциалов титана и ряда других распространенных в природе химических элементов, что приводит к их взаимному замещению. Важнейшими минералами являются.
1. Рутил TiO2, некоторые кристаллические модификации которого называются анатазом и брукитом. Рутил — наиболее богатый титаном вид сырья. В чистом виде встречается в природе редко. Обычно содержит до 92—99% диоксида титана, оксидов железа (II и III), а иногда олово, хром, ниобий и тантал. Рутил в смеси с лейкоксеном является основой титансодержащих песчаников, содержащих в своем составе кроме диоксидов титана и кремния лейкоксен, кварц, сидерит и хлорит.
2. Ильменит FeTiOj (47,3% FeO, 52,7% TiO2). Обычно часть железа изоморфно замещается магнием или марганцем с образованием гейкелита MgO • TiO2 и пирофанита МпО • ПОг.
3. Титаномагнетит FeTiOj + FeO • РегОз.
4. Перовскит СаТЮз.
5. Титанит (сфен) CaTiOSiCU. Аналитическая характеристика титансодержащих видов сырья приводится в табл. 9.1.
В титансодержащем сырье в зависимости от месторождений в небольших количествах содержатся: ванадий, сера, фосфор, оксиды хрома, калия, натрия, циркония, а также золото (в титансодержащих песках), марганец, ниобий и др.
Надо отметить, что такие титанаты, как перовскит и ильменит, не являются, как считали ранее, солями гипотетической титановой кислоты Н2ТЮз. Рентгенографические исследования кристаллов этих солей показали, что ильменит состоит из ионов Ti4+, Fe2+, О2‘, правильно располо-365
женных в кубической решетке. Эта решетка изоморфна с решеткой гематита РегОз, в которой половина атомов железа замещена на атомы титана или точнее два иона Fe3+ замещены ионами Ti4+ и Fe2+. Это объясняет, почему ильменит часто содержит больше железа, чем соответствует формуле FeTiOj, а гематит может содержать до 7% титана.
Таблица 9.1. Химический состав титаисодержящего сырья (%)
Составляющие Ильменит Титаномагнетит Перовскит Сфен
TiO2 38,5—61 8—11 44—47 30—33
FeO 9—31 30—34 — 2—4
Fe2O3 18—26 46—50 4,5—6 —
MgO 0,5—4,5 1,5—3 2,5 1
СаО 0,1—1,0 0,6—1,5 34,6—36,8 27—29
SiO2 0,5—9,0 1,5—4,5 5,7—7,8 27—29
A12O3 1—4 3—6 0,9 1,5—2,5
Свойства. Атом титана имеет электронную конфигурацию Зс? 4s2; степени окисления +4, +3, +2: энергия ионизации при последовательном переходе от Ti° к Ti4 равна соответственно 6,82; 13,58; 27,47; 43,2 эВ; сродство к электрону 0,39 эВ; электроотрицательность по Полингу 1,5; радиус атома 0,149 нм, ковалентный радиус 0,132 нм.
Титан — серебристо-белый легкий металл, существует в двух кристаллических модификациях: a-Ti с гексагональной плотноупако-ванной решеткой и (J-Ti с кубической обьемно-центрированной решеткой. Температура перехода из одной модификации в другую 883° С, АН перехода 3,8 кДж/моль. Температура плавления титана 1671° С, а кипения — 3260° С. Плотность a-Ti и p-Ti равна соответственно 4,505 (20° С) и 4,32 (900° С). Титан обладает высокими механическими характеристиками, слабо зависящими от температуры и сильно — от чистоты и способов предварительной обработки. На воздухе титан покрывается защитной оксидонитридной пленкой, обеспечивающей его высокой коррозионной стойкостью к воздуху (до 500° С), морской воде, разбавленным растворам серной и хлороводородной кислотам, а также щелочей, хлоридов и влажному хлору. Азотная кислота пассивирует титан. Титан при 25° С реагирует с концентрированными серной и хлороводородной кислотами, а также с горячими трихлор- и трифторуксусными, концентрированными фосфорной, щавелевой и муравьиной кислотами.
Фтороводородная кислота реагирует при 25° С с титаном независимо от концентрации, а присутствие фторид-ионов способствует растворению титана и в других кислотах из-за образования комплексного аниона [TiF6]2'. Порошок титана растворяется в концентрированных растворах щелочей с выделением водорода и образованием 366
ортотитановой кислоты TiO2 2Н2О. При 1200° С компактный титан загорается на воздухе и в атмосфере азота. Стружка и порошок титана пирофорны.
Получение. Чистый металлический титан получить относительно трудно по причине его активной реакции как с обычными восстановителями (С, А1), так и с малоактивными газами как азот и водород, с которыми он образует соединения внедрения — TiN и TiH. Получают металлический титан путем восстановления тетрахлорида титана магниетермическим способом: TiCU + 2Mg = 2MgCl2 + Ti. Полученную в ретортах титановую губку очищают в вакууме при 960° С.
Получают титан натрийтермическим восстановлением тетрахлорида титана с последующей отмывкой губки от хлорида натрия слабым раствором хлороводородной кислоты. Полученный при этом порошкообразный титан переплавляют.
Получают титан электрохимическим путем из тетрахлорида титана в расплаве хлоридов, а также плазмохимическим восстановлением тетрахлорида титана. Очень чистые сорта титана получают из тет-раиодида титана при высокой температуре в вакууме: T1I4 -> Ti + 212.
Применение. Металлический титан применяют для получения легких прочных сплавов с алюминием, ванадием, молибденом, хромом и др. Титан широко применяется в ракето- и самолетостроении, в химическом машиностроении, энергетике, кораблестроении и для опреснителей воды.
9.2. ОКСИДЫ ТИТАНА
Известно до пятнадцати оксидов титана. Последовательные стадии окисления титана кислородом выражают следующей схемой:
Ti + О2 —> Ti—О —> TifiO —> Ti2O —> Ti3O —>
—> Ti3O2 —> TiO —> Tig O5 —> Ti2O3 —> TiO2
Установлено, что по мере увеличения содержания кислорода доля металлической связи падает, а ковалентной — увеличивается. Если Ti6O и Ti3O— типичные металлические соединения, то ТЮ2 — преимущественно ковалентное соединение. На рис. 9.1 приведена диаграмма состояния системы титан — кислород.
Согласно литературным данным, низшие оксиды титана являются продуктами упорядочения твердого раствора О2 в a-Ti, максимальная концентрация которого 31,9% (ат.) кислорода (О). При длительном обжиге образуются фазы Ti3O (20—30% ат. О) с ромбоэдрической кристаллической решеткой (при 25% ат. О) и TiO ( ~25—33,4% ат. О). Промежуточный оксид Ti3O2 — фаза с гексагональной решеткой.
367
Рис. 9.1. Система титан — кислород
Монооксид TiO — моноклинные кристаллы в зависимости от незначительных примесей от золотисто-желтого до коричнево-фиолетового цвета со структурой типа NaCl со статистическим распределением вакансий Ti и О. TiO испаряется конгруэнтно, в воде не растворяется. Растворяется в разбавленных хлороводородной и серной кислотах. При нагревании на воздухе окисляется. Монооксид титана получают взаимодействием металлического титана с его диоксидом. Применяется в качестве компонента катализаторов как в органической, так и в неорганической химической технологии.
Гидроксид титана (II) Ti(OH)2 получают действием водного раствора аммиака на растворы солей Ti(II). Полученный при этом синий, коричневый или черный осадок, постепенно светлеет из-за разложения по схеме: Ti(OH)2 -> TiO2 + Н2.
Гидроксид титана (II) является сильным восстановителем, легко растворяется в кислотах, при нагревании на воздухе окисляется.
Сесквиоксид Ti2O3. Кристаллы темно-фиолетового или черного цвета со структурой типа корунда. При нагревании до 200° С переходит в другую гексагональную модификацию. При испарении диссоциирует по схеме
Ti2O3 —> TiO + TiO2
368
Окисляется на воздухе лишь при нагревании до очень высоких температур по схеме
Ti2O3 —•—> 2TiO2 О2
С водой и минеральными кислотами сесквиоксид титана не реагирует, а при нагревании растворяется в концентрированной серной кислоте с образованием раствора фиолетового цвета Ti2(SO4)3, а при сплавлении с гидросульфатом калия образуется титанилсульфат калия:
Ti2O3 + 4KHSO4 = 2K2TiO(SO4)2 + 2Н2О
При термообработке с оксидами или карбонатами щелочных, щелочноземельных и других металлов сесквиоксид титана образует двойные оксиды:
К2СО3 + Ti2O3 —> K2OTi2O3 + СО2
СаО + Ti2O3 CaO Ti2O3
Сесквиоксид титана получают по схеме
2ТЮ2 + Н2 —1220^ Ti2o3 + Н2О
Гидроксид титана (III) Ti(OH)3 — осадок вишнево-красного, коричневого, синего или черного цвета, который постепенно белеет в результате окисления водой, легко окисляется на воздухе, растворяется в минеральных кислотах.
Оксид Ti30s или Ti111 TiIV О5 — моноклинные кристаллы голубого или голубовато-черного цвета плотностью 4,29 г/см3.
Согласно литературным данным, в области от Tii/75 до Tii^ существует гомологическая группа фаз TinO2w_i, где п = 4—9 (фазы Маг-нелли), кристаллизующиеся в триклинной решетке.
9.3. ДИОКСИД ТИТАНА
Широко применяемым в народном хозяйстве оксидом титана является его диоксид. Ему принадлежит основная технологическая линия переработки сложного состава сырья (см. табл. 9.1), охватывающая почти все' приемы и операции неорганической химической технологии.
Физико-химические свойства. Чистый диоксид титана — бесцветные кристаллы, которые желтеют при нагревании, но обесцвечиваются после охлаждения. Известен в виде нескольких модификаций. Кроме 369
Рис. 9.2. Структура октаэдра TiOs
рутила (кубическая сингония), анатаза (тетрагональная сингония) и брукита (ромбическая сингония), известных в виде минералов, получены две модификации высокого давления: ромбическая IV и гексагональная V. Брукит при всех условиях метастабилен. При нагревании анатаза и брукит необратимо пре-
вращаются в рутил соответственно при 400—1000° С и ~750° С. Основой структур этих модификаций являются октаэдры TiOe, т.е. каждый ион Ti4+ окружен ионами О2', а каждый ион О2- окружен тремя ионами Ti4+ (рис. 9.2).
Диоксид титана не растворяется в воде и разбавленных минеральных кислотах (кроме фтороводородной) и разбавленных растворах щелочей. С пероксидом водорода образует ортотитановую кислоту Н4ТЮ4:
TiO2 + 2Н2О2 = Н4ТЮ4
Медленно растворяется в концентрированной серной кислоте
TiO2 + 2H2SO4 = Ti(SO4)2 + 2Н2О
в концентрированных растворах щелочей
ТЮ2 + 2NaOH = Na2TiO3 + Н2О
в насыщенном растворе гидрокарбоната калия
ТЮ2 + 2КНСО3 = K2TiO3 + Н2О + 2СО2
При нагревании диоксид титана с аммиаком образует нитрид титана 2TiO2 + 2NH3 —<-> 2TiN + ЗН2О + '/2О2
При сплавлении с оксидами, гидроксидами и карбонатами образуются титанаты и двойные оксиды:
TiO2 + ВаО = ВаО • TiO2 (BaTiO3)
TiO2 + ВаСО3 = BaOTiO2 + СО2 (BaTiO3)
TiO2 + Ba(OH)2 = BaOTiO2 + H2O (BaTiO3)
370
Водородом, углеродом и активными металлами (Mg, Са, Na) диоксид титана при нагревании восстанавливается до низших оксидов, а с хлором при нагревании в присутствии восстановителей (углерода) образует тетрахлорид титана.
Гидроксид TiO2-«H2O в зависимости от условий его осаждения может содержать переменное число связанных с титаном ОН-групп. Полученный при невысоких температурах ТЮ2-лН2О (a-форма) хорошо растворяется в разбавленных минеральных и сильных органических кислотах, но практически не растворяется в растворах щелочей, легко пептизируется с образованием устойчивых коллоидных растворов. После сушки на воздухе образует белый порошок плотностью 2,6 г/см3, приближающийся по составу к формуле ТЮ2 • 2НгО (ортотитановая кислота). При длительной сушке в вакууме она постепенно обезвоживается, приближаясь по составу к формуле TiO2-H2O (метатитановая кислота).
Диоксид титана широко применяется в качестве белого пигмента в лакокрасочной промышленности, в целлюлозно-бумажной промышленности, в производстве синтетических волокон, пластмасс, резиновых изделий, в производстве керамических диэлектриков, термостойкого и оптического стекла, белой эмали, в качестве компонента обмазки электродов для электросварки и покрытий литейных форм.
Технология диоксида титана из ильменитового концентрата. Технология диоксида титана из ильменитового концентрата состоит из трех этапов: 1) получение растворов сульфатов титана; 2) гидролиз раствора сульфатных солей титана; 3) термообработка образующегося диоксида титана.
Растворы сульфата титана получают разложением ильменитовых концентратов, содержащих в своем составе ильменит (РеТЮз) и продукты его лейкоксенизации (выветривания) — FeO, Ре20з и ТЮг, а также титанидные шлаки в серной кислоте. Скорость процесса зависит от минералогического состава сырья, степени измельчения, температуры, концентрации и количества серной кислоты.
Научными исследованиями и практическим путем установлены оптимальные условия проведения процесса разложения ильменита серной кислотой. К ним относятся: жидкофазный процесс под давлением около 40 атм при 200—220° С. При твердофазном способе разложения концентрированной серной кислотой под атмосферным давлением температура процесса достигает 200—220° С за счет экзотермической реакции, что позволяет в течение небольшой продолжительности достичь оптимальной степени разложения исходного сырья.
371
Рис. 9.3. Технологическая схема подготовки растворов TiSCU
от поз. 24
В процессе разложения ильменитового концентрата применяется 85—89%-ная серная кислота. Количество исходной кислоты должно быть достаточным для образования сульфатов титана, железа и других металлов, содержащихся в исходном сырье. Каждый вид сырья требует подбора определенного режима разложения. Обычно применяют тонкомолотый концентрат с остатком 2—5% на сите № 0045 для легкорастворимых и до 1—2% для труднорастворимых концентратов или шлаков.
Согласно технологической схеме подготовки растворов сульфатов титана (рис. 9.3), подсушенный в барабанных сушилках 3 или в сушилках кипящего слоя ильменитовый концентрат размельчают в шаровых мельницах непрерывного действия 8, работающих в замкнутом цикле с сепаратором 9. Разложение концентрата серной кислотой проводят в реакторе 16, представляющем собой цилиндрический аппарат с коническим дном, футерованный двумя слоями кислотоупорных плиток. К конусу реактора подведены две трубы, одна их которых присоединена к небольшому коллектору и предназначена для подачи в реактор сжатого воздуха, пара и воды, а другая—для вывода из реактора рабочего раствора. К крышке реактора присоединена вентиляционная труба большого диаметра и высоты для удаления газов и паров в атмосферу.
Для получения растворов сульфата титана в реактор дозируют 92—94%-ную серную кислоту, включают сжатый воздух и 373
при постоянном перемешивании загружают ильменит. При этом смесь должна поместиться лишь в корпусной части аппарата. После загрузки исходных видов сырья массу подогревают в течение 2—3 мин до 80—90° С путем подачи небольшого количества острого пара из расчета снижения концентрации серной кислоты до 85—88%. При этой температуре начинается экзотермическая реакция, в результате чего в течение нескольких минут температура повышается до 180—220° С, происходит интенсивное разложение исходного ильменита и протекает основная реакция с выделением большого количества газов и пара. Наблюдается сильное вспенивание с увеличением объема реакционной массы.
Основными процессами, протекающими в аппарате, являются следующие реакции:
TiO2 + H2SO4 -> TiOSO4 + Н2О
FeO + H2SO4 FeSO4 + Н2О
Fe2O3 + 3H2SO4 -► Fe2(SO4)3 + 3H2O
A12O3 + 3H2SO4 -> A12(SO4)3 + 3H2O
MgO + H2SO4 -» MgSO4 + H2O
MnO + H2SO4 -> MnSO4 + H2O
CaO + H2SO4 —> CaSO4 + H2O
V2O5 + 5H2SO4 -> V2(SO4)5 + 5H2O
Надо отметить, что для характеристики количества кислоты, приходящейся на долю титана, введен термин «активная кислота», который понимают как разницу между общим количеством расходуемой кислоты и количеством кислоты, необходимой для образования средних солей примесных металлов. Это соотношение называют кислотным фактором F:
(Нг5О4).„_ 270г/л_ ТЮ2 150 г/л А
где 270 г/л и 150 г/л — значения концентраций серной кислоты и диоксида титана в растворе.
Иногда применяют и условную характеристику — кислотность раствора — отношение разности между кислотным фактором и мас-374
совым отношением HzSO^.TiOz в титанилсульфате к этому же массовому отношению. Величину К выражают в процентах:
Х^00,
где 1,225 — массовое отношение H2SO4:TiO2 в титанилсульфате.
Исследование системы TiO2—H2SO4 (рис. 9.4) показало, что она состоит из следующих соединений: TiOSO4-2H2O; TiOSO4 • Н2О; TiOSO4; TiOSO4 • Нг8О4-2Н2О; TiOSO4-H2SO4-H2O и Ti(SO4)2. Моногидрат и безводный титанилсульфат следует рассматривать в качестве продуктов дегидратации дигидрата, а остальные соли — в качестве присоединения к ним серной кислоты.
По эффективности растворения в воде и в разбавленной серной кислоте сульфаты располагаются в следующий ряд: Ti(SO4)2; TiOSO4 H2SO4H2O; TiOSO4H2SO4-2H2O; TiOSO4-2H2O; TiOSO4H2O; TiOSO4.
Данные о концентрациях серной кислоты и температурных интервалов, при которых гидраты диоксида титана ТЮ2 Н2О и некоторые сульфаты титана существуют в виде равновесной твердой фазы, приведены в табл. 9.2.
Таблица 9.2. Интервалы температур и концентрации серной кислоты, при которых существуют твердые фазы
Твердая фаза Z,°C H2SO4,%
TiO2H2O 100 До 39,0
TiOSO4H2O 100 42,5—76,0
TiOSO4H2O 125 49,0—84,0
TiOSO4H2O 150 63—76
TiOSO4H2O 175 71,5—75,0
TiOSO4H2SO4-2H2O 100 78—82
TiOSO4H2SO4 H2O 100—150 83—95
TiOSO4 150—225 76,5—100
Ti(SO4)2 100—250 95—100
Из данных табл. 9.2 видно, что титанилсульфаты образуются из нагретых выше 100° С растворов сульфата титана при значительном избытке в них серной кислоты.
После окончания интенсивной реакции через несколько минут масса в реакторе затвердевает в виде плава. Для получения пористого плава во время затвердевания через массу пропускают сжатый воздух, после чего оставляют массу на 2—3 ч для охлаждения и «вызревания». Установлено, что в процессе кислотной обработки 95—97% содержа-375
Рис 9.4. Система H2SO4—TiO2
щегося в исходном ильмените титана входит в реакцию, в том числе 85—87% во время основной реакции и 8—10% — в период вызревания.
После охлаждения полученного плава до 80—90° С проводят вы- ‘ щелачивание сульфатных солей водой при энергичном перемешивании сжатым воздухом. Процесс выщелачивания проводят при интервале 65—70° С, так как снижение температуры приводит к замедлению процесса, а повышение уменьшает стабильность раствора. Получаемые растворы в основном содержат сульфаты титана и железа (II и III), поэтому процесс восстановления осуществляют до полного перехода Fe3+ в Fe2+ и частично Ti4+ в Ti3+, (в пределах 3—5 г/л). Наличие в растворе указанного количества Тг предотвращает образование Fe3+ при дальнейших операциях. Восстановление производят путем погружения в растворы корзины из нержавеющей стали, заполненной железными стружками. Процесс проводят в интервале 70—75° С при слабом перемешивании раствора воздухом.
Состав полученных растворов после выщелачивания и восстановления колеблется в зависимости от качества исходного сырья. При использовании концентратов коренных месторождений растворы обычно содержат 120—130 г/л сульфата титана (в пересчете на ТЮ2), 90—100 г/л сульфата железа (в пересчете на Fe), 200—250 г/л активной кислоты, 20—25 г/л нерастворимого остатка (шлама). Отношение Fe:TiO2 = 0,90—0,95, a H2SO4’.TiO2 = 1,9—2,0. Повышение концентрации сульфата титана затруднено из-за сильной вязкости раствора и предельного содержания в нем сульфата железа (II). При работе со шлаками или с богатыми концентратами содержание диоксида титана может достигать 220—240 г/л, а активной кислоты— 450—470 г/л.
Растворы сульфата титана после выщелачивания и восстановления освобождают от шлама, представляющего из себя смесь кремниевой кислоты с непрореагировавшей частью исходного сырья. Около 50% шлама представляет из себя тонкодисперсную взвесь и осаждается лишь при коагуляции. В качестве коагулянтов применяют поливиниловый спирт, сульфанол, альбумин, некаль и др. Отстаивание можно заменить фильтрацией. Процесс фильтрации про-376
водят на барабанных вакуум-фильтрах с намывным слоем из древесной муки. Отфильтрованный раствор направляют на кристаллизацию. Шлам после промывки направляют в шламонакопитель, а промывные воды (разбавленный раствор) применяют для выщелачивания плава в реакторе. Во избежание кристаллизации купороса температуру раствора в течение всего процесса отделения шлама поддерживают в пределах 60—65° С.
Выделение из раствора гептагидрата сульфата железа (II) FeSO4-7H2O проводят в непрерывно действующих вакуум-кристалли-заторах. Процесс основан на уменьшении растворимости гептагидрата сульфата железа при охлаждении. Растворимость FeSO4 зависит от концентрации в растворе сульфата титана и температуры. Для растворов с концентрацией 120 г/л диоксида титана и 240 г/л серной кислоты растворимость сульфата железа (II) в зависимости от температуры характеризуется следующими показателями:
Температура, °C.............................. 30 20 14 10 5 0 -6
Растворимость FeSO4 (в пересчете на Fe), г/л. 88 70 48 43 35 25 14
В производстве процесс кристаллизации FeSO4-7H2O достигается охлаждением растворов от 50—60 до 10—15° С испарением воды под вакуумом. После кристаллизации железного купороса получаются растворы с повышенной концентрацией диоксида титана вследствие связывания части воды гептагидрата сульфата железа (II) FeSO4 • 7Н2О. Примерный состав такого раствора (в г/л):
TiO2................................................... 140—150
FeSO4 (в пересчете на Fe)............................. 30—35
Кислотный фактор F..................................... 1,9—2,1
Суспензия из вакуум-кристаллизатора направляется на непрерывно действующую центрифугу с пульсирующей выгрузкой осадка типа АГ или ФГП, где железный купорос отделяется от раствора и промывается.
Растворы сульфата титана после центрифуг направляются на контрольную фильтрацию, которую обычно проводят на непрерывно действующих автоматических фильтрах типа ФПАКМ. При контрольной фильтрации в растворы вносят незначительные количества древесной муки, а процесс фильтрации проводят при 30—40° С. Растворы сульфата титана после контрольной фильтрации должны содержать не более 0,04—0,06 г/л взвешенных частиц. В случае отделения шлама фильтрацией на барабанных вакуум-фильтрах с намывным слоем контрольная фильтрация обычно не требуется.
377
Вакуум
Рис. 9.3. Технологическая схема гидролиза раствора сульфатных солей титана:
56— бункер древесной муки; 57— реактор-выщелачиваггель; 58— реактор; 59, 60— барабанные вакуум-фильтры; 61 — емкости; 62 — сборник пром-вод; 63— смеситель; 64— сборник; 65 — смеситель; 66 — сборник растворов от фильтров; 67—реактор; 68— сборник гидролизной кислоты; 69 — отстойник; 70 — смеситель суспензии; 71— вентилятор; 72 — пылеуловитель; 73— пылеуловитель печи; 74—вращающаяся печь; 75 — топка; 76 — шнек; 77—бункер; 78 — емкость-ковшик; 79—весы; 80 — бункер целевого продукта; 81 — циклон; 82—вентилятор; 83 — сборник целевого продукта; 84 — пневмотранспорт целевого продукта; 86 — бункер; 87—емкость-ковшик; 88 — вентилятор; 89 — бункер; 90 — пылеуловитель; 91 — вентилятор; 92 — бункер; 93 — реактор; 94 — растворители; 95 — дозатор; 96 — промежуточная емкость; 97—фильтр; 98 — емкость; 99 — дозаторы; 100 — реактора; 101 — отстойник; 102 — смеситель; 103 — барабанный вакуум-фильтр; 104 — фильтры; 105 — сборник; 106 — смеситель; 107—барабанный вакуум-фильтр; 108 — сушилка; 109 — бункер; НО — сушилка; 111 — циклон; 112 — бункер; 113 — узел упаковки; 114 — пылеуловитель; 115 — смеситель
Отфильтрованные растворы упаривают до содержания в них солей титана в пересчете на TiO2 220—240 г/л в вакуум-выпарных аппаратах непрерывного действия. Во избежание преждевременного гидролиза растворов процесс упарки ведут при температуре не выше 60° С. В процессе упарки плотность раствора повышается от 1400—1510 до 1550—1630 кг/м3.
Технология гидролиза раствора сульфатных солей титана. Процесс гидролиза методом введения зародышей состоит из нескольких операций: приготовление зародышей; гидролиз раствора сульфата титана; отделение гидролизной кислоты; промывка продукта гидролиза.
Согласно технологической схеме (рис. 9.5), зародыши готовят в реакторе, снабженном пропеллерной мешалкой и кожухом для охлаждающей жидкости (воды), из концентрированных растворов сульфата титана. Исходные растворы разбавляют водой до содержания солей титана в пересчете на ТЮг 40—50 г/л (плотность 1100 кг/м3) и нейтрализуют, медленно добавляя при постоянном перемешивании раствор гидроксида натрия (~50 г/л NaOH) до pH 3 (кислотный фактор 0,2—0,3). Суспензию подогревают примерно до 60° С, прекращают перемешивание и массу оставляют для вызревания при указанной температуре на несколько часов, после чего опять включают мешалку и подачей холодной воды в рубашку аппарата суспензию зародышей охлаждают до 20—25° С.
Гидролиз проводят в стальных, футерованных диабазовыми плитками 379
реакторах, снабженных освинцованными мешалками и змеевиками для нагрева и охлаждения реакционной смеси.
Гидролизу подвергают полученные после упарки растворы сульфата титана следующего состава (в г/л):
ТЮ2................................................. 220—240
Fe.................................................. 40—50
Ti3+................................................ 2—3
Кислотный фактор F.................................. 1,9—2,1
Процесс гидролиза проводят в реакторе, подогреваемом глухим паром. После доведения температуры растворов до 60° С, вводят суспензию зародышей в количестве 0,2—0,5% (в пересчете на ТЮг) от объема исходных растворов. Раствор нагревают до кипения (107—110° С в зависимости от концентрации раствора), выдерживают несколько часов, после чего разбавляют их горячей водой и вновь кипятят. Разбавляют раствор при степени гидролиза 70—75%. При этом количество добавляемой воды ~ 35—40% по отношению к начальному объему раствора. Продолжительность каждой стадии технологии соблюдается строго по регламенту. Она колеблется в некоторых пределах и зависит от сорта и марки получаемого диоксида титана и концентрации исходных растворов. Степень гидролиза составляет 96—97%.
Растворы сульфатов титана обладают рядом специфических особенностей, которые отличают их от обычных растворов — истинных и коллоидных. Их свойства зависят не только от химического состава, но также от условий получения. В процессе глубокой упарки образуются не кристаллические соли, а аморфная стеклообразная масса. Они не коагулируют в процессе высаливания кислотой, способны к обменным реакциям, образованию двойных солей (титанатов), восстановлению титана до низшей валентности и к образованию пероксидных соединений. Аномальное состояние титана в растворе проявляется также в возможности получения концентрированных растворов титанилсульфата, несмотря на его слабую растворимость в серной кислоте средней и высокой концентрации.
Таким образом, растворы титана обладают комплексом свойств, характерных для истинных и коллоидных растворов. Считается, что соли титана в растворе могут находиться в ионном и молекулярном состоянии, в связи с сильной склонностью к гидролизу — в виде гидроксокомплексов общей формулы ТЮ(ОН)<2~Х> (х— определяется химическим составом и состоянием раствора), а также в виде сульфатных комплексов, образующих коллоидные частицы. 380
*вог
§
§ °,
40
20
Ti(SO4)’’
TiO(OH)2
TiOSO4 TiO(SO4)2 TiO2*
0,01 0,1 1 2 3 5 710
Содержание H2SO4, мольАз
Рис. 9.6. Диаграмма состояния титана в серной кислоте
серной кислоты в
Состав ионов и их соотношение существенно зависят от pH среды, который в технологии определяется кислотным фактором и концентрацией растворов. Из приведенной на рис. 9.6 диаграммы состояния неконцентрированных растворов сульфатов титана видно, что в широком интервале концентраций
комплексы титанила: TiO(OH)+, TiO2+, TiOSCU, TiO(SO4 )*. Комплексные соединения титана проявляют склонность к полимеризации. В растворах с низкой концентрацией титана присутствуют в основном моноионы, с повышением концентрации титана (>0,4 моль/л) наступает полимеризация с образованием ди-, три- и полиионов. В растворах титановых солей с концентрацией 0,4—1,4 моль/л димеры находятся в равновесии с мономерами, а в растворах с концентрацией 2,5 моль/л число мономеров в комплексе увеличивается до 8—9.
Полиионы могут достигать размеров коллоидных частиц, являющихся зародышами и ускоряющих процесс гидролиза. Процесс гидролиза растворов сульфатов титана, естественно, сопровождается снижением содержащихся в растворе солей титана, находящихся в молекулярно-дисперсном состоянии, повышением концентрации коллоидных частиц и образованием нерастворимого осадка (TiO2).
При взаимодействии титанилсульфата и воды образуется многоядерный гидрокомплекс
TiOSO4 +2Н2О
Этот комплекс диссоциирует на поликатион титана — Ti — О — Ti — О — и анионы SO4‘. Однако диссоциация практически не доходит до конца и в результате гидролиза получается многоосновная соль, которую называют метатитановой кислотой, что не соответствует составу продукта процесса гидролиза. Поэтому так называемую метатитановую кислоту следует рассматривать как полимер, 381
в котором атомы титана через кислородные мостики связаны с конечными гидроксо- и сульфогруппами:
SO—Ti—б—Ti—О
Процесс гидролиза сульфатов титана с образованием его полиионов объясняет поведение титана в растворе, а именно высокое содержание свободной кислоты, замедленность ионных реакций для значительной доли титана в растворе (~50—60%). Процесс зависит также от температуры. В растворе сульфата титана высокой концентрации (~200 г/л ТЮг) при повышении температуры до 90° С степень полимеризации снижается с 8—9 до 5—6. Понижение степени гидролиза в начальный период нагревания, т.е. до начала процесса выделения осадка, связано со значительным повышением концентрации свободной кислоты при постоянной концентрации титана в растворе (рис. 9.7).
Вследствие уменьшения агрегативной устойчивости гидратирован
ного поликатиона при нагревании выделяется осадок в виде основной гидроксосоли с конечными гидроксо- и сульфогруппами.
Состав и свойства гидратов диоксида титана также находятся в прямой зависимости от условий их получения. При осаждении их из водных растворов солей титана щелочами без подогрева при различных значениях pH (начало осаждения при pH 1,0—1,5 и конец при pH ~3) выпадает гелеобразный осадок гидроксида примерного состава Ti(OH)4. Он хорошо растворяется в органических и разбавленных
неорганических кислотах, в щелочах, а также в растворах солей титана. Образующиеся при этом растворы имеют явно выраженный коллоидный характер. Такие же гидроксиды получаются при разбав-
лении растворов водой на холоду.
Рис 9.7. Зависимость концентрации серной кислоты в процессе гидролиза от концентрации Ti(IV)
При «старении» в процессе кипячения водной взвеси, нагревания сухого остатка, длительном хранении гидроксид теряет гелеобразный характер, а также способность легко растворяться в слабых кислотах и растворах титана. Это объясняется тем, что свежеосажденный гидратированный ион титана (a-форма) имеет относительно большое число ОН-групп. Поэтому они более реакционноспособны, чем «состарившиеся» осадки (0-форма), в которых ОН-группы замещены на кислородные мо-
382
стики. Поэтому «состарившийся» осадок растворяется лишь в 60—70%-ной серной кислоте при 130—140° С. Считается, что примерный состав этой формы гидроксида TiO(OH)2. Подобные соединения получаются при гидролизе путем кипячения.
Гелеобразный продукт называют обычно ортотитановой кислотой Н4ТЮ4 (ТЮ2-2НгО), а «состарившийся» — метатитановой кислотой Н2ТЮ3 (ТЮг-НгО). Рентгенографические исследования показывают, что ортотитановая кислота аморфна при получении ее из ТЮЦ и очень слабо кристаллична при получении из ТЮБОд.
Исследования состарившегося продукта ПМР и ИК-спектроскопия показали наличие в его составе группы ОН-. Поэтому ему приписывают следующее полимерное строение:
он он он
I I I
-Ti— о-Ti— о-Ti— О—
ООО
-Ti—О--Ti—О--Ti—О-
I I I
он он он
где Ti4+ : ОН" =1:1.
Исследования методом ЯМР свежеосажденного продукта показали, что в нем отношение Ti4+ : ОН" = 2:1, т.е. его состав Ti4O7(OH)2. Избыточная вода в гидратах имеет адсорбционный характер.
Продукт, получающийся в процессе гидролиза кипячением, в начальный период содержит две структурно-связанные гидроксильные группы с одним атомом титана. Однако при дальнейшем кипячении гидроксильные группы легко отщепляются и метатитановая кислота переходит в гидратированный диоксид титана ТЮг • «Н2О.
Продукты гидролиза обладают высокой адсорбционной способностью, адсорбируют газы — аммиак, сульфид водорода, ионы РО43’, соли железа (III). Особенно энергично адсорбируют продукты термического гидролиза.
Гидраты диоксида титана легко пептизируются, образуя прозрачные, слегка опалесцирующие золи. Процесс происходит под действием хлороводородной, азотной, муравьиной, уксусной кислот (положительные золи), щавелевой, лимонной, яблочной кислот (отрицательные золи). Действие пептизирующих агентов избирательно. Так, хлорид железа (III) действует, наиболее сильно на продукт термического гид-383
ролиза, хлорид аммония — на продукт щелочного гидролиза, ацетат хрома — на продукт гидролиза на холоду при разбавлении водой. Гидрозоли диоксида титана обычно очень устойчивы. Например, золи ТЮг при их пептизации хлоридом железа (III) в присутствии лимонной кислоты выдерживают разбавление в 3000—4000 раз, в то время как обычные золи мутнеют и теряют устойчивость при 15—20-кратном разбавлении. Золи отличаются своеобразным отношением к электролитам. Так, сульфаты натрия, аммония и железа (II) не вызывают коагуляции даже насыщенного раствора. Порог коагуляции для других электролитов весьма значителен: — 762,
NaNO3 —635, KNO3 —367 мг-экв/л
При добавлении к некоторым золям концентрированной хлороводородной кислоты часть диоксида титана выпадает в осадок, который рассматривается как коллоидная форма ТЮг- При нагревании золи превращаются в гель, который при охлаждении до комнатной температуры самопроизвольно разжижается.
Из диаграммы состояния ТЮг — H2SO4 (см. рис. 9.4) видно, что при 100° С растворы сульфата титана в значительной степени гидролизуются при малой концентрации титана (5 —10 г/л) и высокой концентрации серной кислоты (до 40%-ной H2SO4). При гидролизе имеет место длительный индукционный период, во время которого образуются центры кристаллизации, после чего начинается процесс гидролиза с выделением гидроксида титана в осадок. Общая продолжительность процесса, особенно при работе с концентрированными растворами, достигает около 8 ч.
Процесс гидролиза может быть интенсифицирован и проведен в строго регулируемых условиях добавлением в растворы зародышей, представляющих собой золь гидроксида титана. Зародыши (мицеллы) сравнительно однородны по размерам (0,03—0,08 мкм) и содержат микрокристаллы анатаза (их размер ~ 2 нм). Микрокристаллы обладают сетчатой структурой. Зародышам приписывают цепочечное строение с кислородными связями —О—Ti—О—Ti—О—. Механизм действия зародышей объясняют переносом структурных информаций на молекулярном уровне. Зародышевая цепь соединяется с гидроксильными группами растворенных полимеров титана с образованием определенных структур. Цепь полимера объединяется с цепью зародыша при конденсации ОН" с образованием кислородных мостиков.
Вероятно, в этом процессе имеет место и молекулярный, и кристаллохимический механизм. Действие зародышей можно представить как взаимодействие поверхности частиц гидрозоля диоксида титана с частицами раствора (многоядерными гидроксокомплексами титана) по координационному механизму. При этом частицы раствора уменьшают свой удельный заряд за счет оксоляции, приближаясь по строению к частицам гидрозоля (молекулярный механизм), после чего 384
происходит кристаллический рост самой зародышевой частицы, уплотнение и структурное упорядочение (кристаллохимический механизм). Чтобы гидролиз был воспроизводим по дисперсности и составу продуктов, в растворе должно присутствовать определенное количество (на массовую единицу диоксида титана) зародышей, причем до начала процесса гидролиза растворы должны быть свободны от коллоидных примесей. Поэтому на практике каждый новый процесс гидролиза протекает по-разному.
В процессе гидролиза происходит следующее. До начала кипения в период нагревания раствора значительная часть серной кислоты, связанной с титаном, освобождается. Затем довольно быстро возрастает содержание коллоидной части в растворе и через некоторое время начинается выделение гидроксида титана в осадок. Выделение осадка протекает без отщепления кислоты. В табл. 9.3 приведены данные о состоянии раствора в начальный период процесса гидролиза. Предполагается, что в процессе гидролиза не образуются новые частицы, а растут частицы, введенные в виде зародышей, за, счет присоединения молекул гидроксида титана из раствора. При размере частиц ~0,01 мкм содержание коллоидной части в растворе максимально. В этот период на частицах начинает осаждаться аморфный гидроксид титана, который агрегирует и выпадает в осадок, начинает происходить формирование и рост частиц гидролиза.
Чем меньше количество зародышей, тем меньше образуется коллоидного гидроксида титана и тем крупнее частицы гидролизата:
Количество зародышей, %.................... 0,1 0,2 0,3 0,5
Максимальное количество коллоидной части, % 19 21 25 34
Таблица 9.3. Изменение состояния раствора сульфата титана в процессе гидролиза
Время от начала гидролиза, мин Характеристика раствора Концентрация TiO2, г/л
в растворенном состоянии в коллоидной форме в осадке
4 Прозрачный 104 21,1 0
5 » 97 28,2 0
6 » 92 33,0 0
7 Слегка мутный 84 40,8 0
8 » 78 46,8 0
9 Заметно мутный 70 55,0 0
10 Мутный 71 53,6 0
11 Сильно мутный 88 41,9 0
12 Белый 45 31,6 48,2
13 » 43 26,6 55,2
13 Химическая технология неорганических веществ, кн. 1
385
0 2 4 6 8
Время, ч Рис 9.8. Влияние кислотности растворов сульфата титана на скорость гидролиза
ров. Зависимость скорости
Рис. 9.9. Влияние температуры и кислотного фактора F раствора сульфата титана на скорость
Исходя из сказанного, в производст- 1 ве количество зародышей подбирают таким образом, чтобы получить про- i дукт с заданными физико-химическими характеристиками. Процесс гидролиза является важной операцией, определяю- , щей качество целевого продукта — ди- оксида титана. Состав и свойства про- I дуктов, которые могут образоваться в процессе гидролиза, разнообразны. Поэтому при проведении процесса необходимо поддерживать в установленных пределах концентрацию солей титана и кислоты, температуру, продолжительность, стабильность качества растворов, их плотность и вязкость, содержание в них примесей и ряд других парамет-процесса гидролиза от температуры и
кислотного фактора показана на рис. 9.8 и 9.9. Из данных видно, что при 70° С гидролиз не происходит, при 90° С протекает весьма слабо и лишь при 100° С и выше — со значительной скоростью. Это относится к стабильным растворам, а коллоидные (нестабильные) растворы гидролизуются очень быстро, несмотря на низкие температуры и высокий кислотный фактор.
Процесс гидролиза состоит из трех периодов: подогрев раствора до кипения, кипение и разбавление суспензии. Подогрев не должен быть длительным во избежание потери раствором стабильности и частичного растворения зародышей. В процессе кипячения растворов происходит формирование частиц гидролизата и выделение его в I осадок. Разбавление имеет целью снижение концентрации кислоты в конце процесса гидролиза (~до 300 г/л H2SO4) и повышение таким образом степени гидролиза. Разбавление не должно начинаться раньше, чем исчезнет из раствора коллоидный гидроксид титана.
В результате гидролиза 95—96% титана оседает в виде многоос-
386
новной соли. При этом серная кислота и примеси — соли железа (И), алюминия (III), магния (II), марганца (II) и другие — остаются в растворе. Следует также учитывать значительную адсорбционную способность продукта гидролиза, особенно в стадии формирования, которая проявляется даже в сильнокислой среде. Недопустимо присутствие в гидролизуемых растворах солей железа (III), которые адсорбируются продуктами гидролиза почти необратимо. Лимитируется также содержание фосфорной кислоты (в производстве рутила), так как она замедляет процесс перекристаллизации анатаза в рутил. В качестве примера приводится состав (%) промытых и высушенных продуктов гидролиза:
тю2........................................
so3........................................
Н2О(гидратная).............................
Н2О(несвязанная)...........................
Итого:
81,5
4,4
7,1
6,7
99,7
76,7
7,6
7,6
7,9
99,8
72,8
7,3
13,0
6,7
99,8
74,8
7,0
9,6
8,5
99,8
Продукт гидролиза состоит из агрегатов размером -0,6—0,7 мкм. Каждый агрегат образуется примерно из 100 мицелл размером 0,060 — 0,075 мкм, а каждая мицелла — из 20 микрокристаллов размером -2 нм, аналогичным зародышевым. Агрегаты сохраняют свою структуру в течение дальнейшей обработки. Монокристаллы связываются в мицеллы ионами SO^ и ОН". Пигментные свойства определяются размерами мицелл. Агрегаты весьма сильно коагулированы, из-за чего возможна технологическая обработка гидролизата — промывка и фильтрование. В табл. 9.4 приведены данные по изменению размеров частиц в зависимости от состава гидролизуемого раствора.
Для получения пигментного диоксида титана применяют растворы с концентрацией солей титана (в пересчете на TiC^) 200 — 220 г/л и кислотным фактором F = 1,9-—2,1. Растворы с меньшей концентрацией, например 120—130 г/л TiC>2, образуют гидролизаты, которые при термообработке переходят в крупнодисперсный диоксид титана с низкими пигментными свойствами. Это объясняется значительной склонностью к агрегации мелких кристаллитов и рано наступающей коагуляцией, препятствующей росту частиц.
Исходные зародыши в производстве получают путем нейтрализации растворов сульфата титана гидроксидом натрия.
Суспензия, полученная в процессе гидролиза, охлаждается в реакторе до 70—75° С и фильтруется на барабанном вакуум-фильтре при необходимости с намывным слоем из древесной муки или на фильтрах типа ФПАКМ. Гидролизная кислота, отделенная при фильтрации с содержанием 20—25% H2SO4, направляется на переработку, а осадок промывается методом многоступенчатой репульпации очищенной водой
или конденсатом на барабанных вакуум-фильтрах или ФПАКМ. После промывки суспензия диоксида титана подвергается отбелке-очистке от примесей соединений железа и других металлов. Для этого суспензию, содержащую около 300 г/л диоксида титана, обрабатывают в реакторе 5—10%-ной чистой (аккумуляторной или реактивной квалификации) серной кислотой с добавлением металлического цинка в виде пыли или гранул в количестве около 0,5% (в расчете на ТЮ2), нагревают до 90—95° С и выдерживают при этой температуре до частичного перехода титана (0,5 г/л) в раствор в виде Ti2(SO4)j. При этом примеси железа, хрома, ванадия восстанавливаются и переходят в раствор.
Таблица 9.4. Изменение размеров частиц диоксида титана в зависимости от состава гидролизуемого раствора
Концентрация TiO2 в исходном растворе, г/л Концентрация свобод-ной H2SO4, г/л Степень полимеризации Радиус частиц гидролизата Размер частиц TiO2
при 20° С при 90° С кристаллитов, нм агрегатов, мкм кристаллитов, нм агрегатов, мкм
50 50-100 1 1 5—6 0,8—1,0 50 0,8—1,0
100 100-120 2 1—2 5—6 0,8—1,0 50 0,8—1,0
150 150 3—4 2—3 5—7 0,6—0,7 50 0,6—0,7
200 200-250 8—9 5—6 8—12 0,2—0,3 50 0,2—0,3
После отбелки осадок промывают до содержания в промывных водах менее 0,005% солей железа в пересчете на Fe, разбавляют до концентрации TiO2 300 г/л и направляют на солеобработку.
Солеобработка заключается во введении различных добавок (минерализаторы) с целью ускорения процессов кристаллизации и перекристаллизации анатаза в рутил (рутилизирующие добавки), а также улучшения и стабилизации цвета и придания конечному продукту тех или иных свойств. Основными добавками при получении анатаза являются сульфат и карбонат калия, а также фосфорная кислота, а при получении рутила — метатитановая кислота, не содержащая сульфогрупп, сульфат или оксид цинка, оксиды или соли некоторых других металлов, способствующих рутилизации. Водорастворимые добавки (K2SO4, ZnSO4) вводят в избыточном количестве с учетом потерь в процессе фильтрации. Отфильтрованный на вакуум-фильтре осадок диоксида титана после введения добавок насосом перекачивается в отделение термообработки.
Применяется в производстве и другой метод гидролиза сульфата титана, при котором зародыши образуются в процессе разбавления исходных концентрированных растворов солей титана. Согласно методу, раствор упаривают до содержания солей титана 240—260 г/л (в пересчете на TiO2) и частично разбавляют в определенных условиях для образования зародышей, которые в дальнейшем участвуют в процессе гидролиза.
388
Термообработка продукта гидролиза, т.е. гидратов диоксида титана, по данным дериватографических исследований происходит в следующем порядке: в вакууме они полностью теряют воду при 77° С; на воздухе основная часть воды удаляется при 150—200° С. Эндотермический эффект для ТЮ2-Н2О при 145° С соответствует переходу TiO2-2H2O -у TiO2-H2O + Н2О, а моногидрат обезвоживается при 310° С с образованием кристаллического TiO2.
Лабораторные исследования механизма образования диоксида титана показали, что вода удаляется в интервале температур 200—300° С, а разложение основной массы солей — при 500—700° С. Остающиеся группы SO*' придают продуктам термообработки кислый характер. После термообработки при 800—1000° С в кристаллической решетке продукта остается еще значительное количество ионов сульфата, составляющих 0,1—0,3% (в пересчете на SO3) (табл. 9.5).
При термообработке происходит и рост кристаллов внутри мицеллы с образованием одного кристалла, размеры которого определяются размерами первичной мицеллы. Рост кристаллов заканчивается около 750° С. В это время, видимо, изменяется механизм роста кристаллов, они объединяются с образованием частиц.
Таблица 9.5. Изменение свойств продукта гидролиза при его термообработке
Температура, °C Потери при термообработке, % Содержание SOi, % Плотность, г/см3 Температура, °C Потери при термообработке, % Содержание so, % Плотность, г/см3
300 9,4 6,4 3,66 850 0,3 0,3 3,87
400 7,6 6,3 3,69 900 0,1 0,1 3,85
500 5,4 4,3 3,76 950 0,1 0,1 3,97
550 3,2 2,7 3,82 1000 0,1 0,1 4,19
600 2,2 1,6 3,83 1050 0,1 0,07 4,19
650 1,6 1,1 3,84 1100 — — 4,19
700 0,9 0,6 3,85 1150 — — 4,19
750 0,6 0,3 3,87 1200 — — 4,23
800 0,5 0,3 3,87 — — — —
В результате увеличения размеров кристаллов сильно снижается химическая и адсорбционная способность продуктов термообработки и при этом увеличивается также их кристалличность и изменяются технические характеристики. В табл. 9.6 приводятся экспериментальные данные для продуктов, полученных из растворов с концентрацией диоксида титана 120 г/л.
389
Диоксид титана с низким содержанием триоксида серы и слабой химической активностью получается лишь в результате длительной । термообработки. При этом возможно его спекание, в результате чего могут образоваться крупные частицы, пожелтение продукта, ухудшение пигментных свойств и дисперсного состава. Для исключения этих процессов предложено в качестве стабилизирующих добавок 1 внести в смесь двух- или трехвалентные анионы SO^’ и РО’\ Они одновременно стабилизируют структуру анатаза, т.е. затрудняют ее переход в рутильную. Положительный эффект сохранения анатаза оказывает добавка до 0,5 — 0,6% фосфорной кислоты. Она способствует лучшему формированию частиц, препятствует их росту и ускоряет процесс удаления триоксида серы. Ряд соединений, выполняя роль минерализаторов, способствуют росту частиц TiO2 и удалению SO3. Наиболее эффективен сульфат калия. Введение 0,5 —1,0% K.2SO4 (по отношению к ТЮ2) снижает температуру образования анатазного диоксида титана до 800 — 850° С. Образующийся при этом титанат калия гидролизуется и повышает pH диоксида титана до 8,5.
Таблица 9.6. Изменение размеров кристаллов и дисперсности частиц в зависимости от температуры их термообработки
Температура, °C Размер кристаллов, нм Удельная поверхность, м2/г Размер частиц, мкм Температура,0 С Размер кристаллов, нм Удельная поверхность, м2/г Размер частиц, мкм
200 11 25 0,45 600 22 21 0,44
300 11 23 0,45 700 35 13 0,47
400 15 22 0,43 800 60 8 0,50
500 17 22 0,47 900 100 5 0,50
Наличие в продукте гидролиза связанной серной кислоты предотвращает переход структуры анатаза в структуру рутила, вызывает необходимость применения высоких температур и значительной продолжительности термообработки, что приводит к потере пигментных свойств. В последние годы путем применения МТК, свободной от ионов серы (получают окислением металлического титана пероксидом водорода в аммиачном растворе), при термообработке образует ТЮ2 системы анатаз, которая быстро переходит в рутил при 730° С (рис. 9.10). Установлено также, что введение в состав МТК незначительного количества (до 1%) некоторых соединений (рутилизирующих добавок) MgO, AI2O3, ЗЬгОз, В120з, ванадатов, боратов и особенно ZnO — значительно снижает температуру перехода анатаза в рутил. Большое влияние на процесс оказывает не содержащий SO*’ тонкодисперсный гидроксид титана. Он при сравнительно низких температурах образует кристаллическую структуру рутила и является как бы матрицей, облегчающей перестройку структуры анатаза, получаемой в процессе тер-390
мообработки продуктов гидролиза, в структуру рутила. Вводится в количестве 1—4% (в пересчете на TiO2). Путем подбора количества МТК, не содержащий ионов SO4, и других рутилизирующих добавок (в основном оксид цинка) температуру перекристаллизации анатаза в рутил можно снизить до 820—830° С и получать рутильный диоксид титана с высокими пигментными характеристиками.
Рутилизирующие добавки получают гидролизом разбав-
Рис 9.10. Влияние температуры термообработки на скорость перекристаллизации чистого анатаза в рутил
ленных растворов хлорида титана (10—30 г/л) при 80—85° С в течение 10—15 мин. Иногда перед гидролизом нейтрализуют часть тетрахлорида титана (25—50%) гидроксидом или карбонатом натрия. В качестве МТК, не содержащей сульфат-ионы, может быть применена многоосновная соль, полученная гидролизом сульфатных солей титана; ее обрабатывают гидроксидом натрия, после чего отмывают от сульфатов и пептизируют хлороводородной кислотой. Зародыши
можно получить также из высокощелочных титанатов.
Поливалентные анионы, например РОд3“, в заметных количествах
задерживают переход анатаза в рутил, поэтому в производстве рутила применяются концентраты с небольшим содержанием фосфатов.
Процесс термообработки продукта (гидроксида титана) непрерывно подается во вращающуюся барабанную печь длиной 40—60 м, внутренним диаметром 2—3 м, со скоростью вращения 0,2—0,4 об/мин и с углом наклона 1,5—2,0° С. Коэффициент заполнения печи -10%, производительность 1,5—3,5 т/ч. Продолжительность пребывания продукта гидролиза в печи -8—15 ч. Продукт гидролиза в виде пасты при непосредственном соприкосновении с продуктами горения газа обезвоживается по схеме:
Ti(OH)2 -> TiO2 H2O -> ТЮ2
Температура обработки при получении анатаза -900° С, рутила— 800—850° С. Температура газов на входе в печь 900—1000° С, на выходе 350—400° С; при более низкой температуре на выходе может происходить конденсация паров серной кислоты в загрузочной камере. Удаление воды из пасты заканчивается примерно на первой четверти длины печи от места загрузки, а удаление основной части 391
диоксида серы происходит до начала образования частиц диоксида I титана, т.е. до 800° С.
Отходящие из печи газы, содержащие диоксид серы, пары воды и незначительное количество диоксида титана, поступают в мокрый уловитель, а далее в электрофильтры.
На качество диоксида титана в значительной мере влияет температура термообработки. Для регулирования температуры в 5—6 точках, расположенных по длине печи, устанавливаются термопары, соединенные с терморегулятором. Первая термопара расположена близко от выхода продукта (~1,5 м) для контроля температуры в интервале последних 50° С, когда происходит окончательное образование частиц диоксида титана, а последняя — на расстоянии 6—8 м от места загрузки пасты (гидроксида титана). Процесс термообработки непрерывно контролируется по интенсивности цвета диоксида титана, а в производстве рутила — по степени рутилизации, которая для конечного продукта должна составлять 95—98%, поскольку дальнейшее повышение степени рутилизации требует длительной выдержки, приводящей к ухудшению пигментных свойств.
Диксид титана после термообработки в печи охлаждают сначала в холодильном барабане до 250—300° С, а затем в скребковом транспортере с водяным охлаждением до 50—70° С и направляют на дальнейшие операции: дезагрегацию спекшихся частиц, а при необходимости его применения в лакокрасочной промышленности, поверхностную обработку для улучшения его пигментных свойств. Продукт, состоящий из прочных агрегатов, дезагрегируется на роликокольцевой мельнице.
В производстве других марок диоксида титана рутильной и анатазной структуры проводится дополнительная обработка, включающая мокрый размол в замкнутом цикле с классификатором в присутствии пептизаторов (жидкое стекло, трифосфат натрия, гидроксид натрия), осаждение на частицах диоксида титана гидроксидов алюминия и цинка, силиката алюминия и других соединений, а также промывку, фильтрацию, сушку и микропомол на струйной мельнице. В процессе микропомола при необходимости вводят поверхностно-активные вещества (нафтенаты, фталаты, олеаты). Осажденные вещества и ПАВ модифицируют поверхность пигмента, улучшают его интенсивность, укрывистость, стойкость к атмосферным воздействиям пигментированных им пленок, что связано с резким понижением его фотохимической активности, устраняют абразивность рутильной структуры. Определенным подбором добавок перед термообработкой, а также условий дополнительной обработки удается в значительной степени варьировать свойства диоксида титана. Содержание основного вещества (ТЮг) в необработанном продукте 98 — 98,5%, в обра-392
ботанном — 93—95%. Содержание неорганических веществ (в %) составляет примерно:
Рутил Анатаз
ZnO....................................... 0,0—1,0 —
А120з..................................... 1,0—3,5 1,0—2,5
SiO2...................................... 0,5—2,0 0,0—0,5
Технология переработки отходов производства диоксида титана. При гидролизе растворов сульфата титана в производстве на одну тонну производимого диоксида титана образуется около двух тонн гидролизной серной кислоты (в пересчете на 100%-ную). Она сильно загрязнена примесями. Ниже приводится состав гидролизной кислоты (в пересчете на металлы, в г/л):
Н28О4общ............... 200—500
Fe..................... 20—50
Ti.................... 0,5—10
Al.................... 0,5—10
Mg.................. 0,5—10
Mn.................. 0,5—10
V................... 0,1—2
Cr................... 0,1—2
Существует технология переработки гидролизной кислоты двухступенчатой упаркой растворов в аппаратах погружного горения с фильтрацией их после каждого аппарата АПГ.
Получение концентрированной кислоты упаркой затруднено, так как при концентрации серной кислоты выше 60% имеющиеся в исходной кислоте соли выпадают в виде объемного осадка, заполняющего почти весь объем аппарата упарки. Поэтому проводят двухступенчатую упарку: сначала упаривают растворы до концентрации 45 — 50% H2SO4, охлаждают в холодильнике и фильтруют с отделением основной массы солей. Отфильтрованные растворы направляются на второй аппарат погружного горения, где они упариваются до концентрации 75% H2SO4, затем повторно фильтруются от солей. Полученную кислоту применяют в производстве химических удобрений, а шлам используют для извлечения из него ванадия, хрома и титана или в производстве строительных материалов.
В производстве диоксида титана серно-кислотным способом на одну тонну продукции образуется около 3,5—4,5 т гептагидрата сульфата железа (II), примерный состав которого (масс. %) приведен ниже:
H2SO4 2,0
FeSO4...................... 47,2
Fe2(SO4)3............... 4,1
MgSO4................... 1,0
TiOSO4.............. 3
MnSO4................. 0,3
VOSO4............... 0,1
CaSO4............... 0,3
Железный купорос используется в производстве красных и желтых железооксидных пигментов и частично при осадочном способе синтеза черных железооксидных пигментов.
393
Технология диоксида титана из тетрахлорида титана. Разработаны три способа получения диоксида титана из его тетрахлорида: 1) гидролиз водного раствора его тетрахлорида; 2) парофазный гидролиз тетрахлорида титана; 3) термообработка тетрахлорида в токе кислорода.
Гидролиз водных растворов тетрахлорида. Технология основана на реакции:
TiCl4 + 2Н2О = TiO2 + 4НС1
Однако кажущийся простым на первый взгляд процесс очень сложный и ступенчатый. При внесении тетрахлорида в воду происходит бурная, сильно экзотермическая реакция, иногда со взрывом, с выделением объемистого осадка. Исследованиями установлено, что вначале образуются гидраты TiC14-5H2O и TiC14-2H2O, а затем начинается процесс гидролиза с постепенной заменой атомов хлора ОН-группами по следующей схеме:
TiCl4-5H2O -> TiCl3(OH)-4H2O -> TiCl2(OH)2-3H2O -> TiCl(OH)3-2H2O -► Ti(OH)4 H2O
Гидратированный ион Ti4+ вследствие большого заряда в растворах существовать не может. Поэтому его растворимые производные сильно гидролизуются. Гидролиз протекает с образованием разнообразных многоядерных комплексов. В качестве первой стадии гидролиза TiCh можно рассматривать его координационное насыщение до TiCU-2H2O с последующим отщеплением молекул хлороводорода:
Н2О он +4НС1
,он
но 1 он2
он
На последующих стадиях образуются полимерные гидроксо- и оксопроизводные:
Z|Y|X
—О—Ti —
394
При нагревании процесс гидролиза происходит быстро, на холоду могут быть выделены промежуточные продукты.
При гидролизе раствора тетрахлорида титана в хлороформе с добавлением воды получены соединения состава Т1зО2С12-4Н2О; Т1бО5С114-13Н2О; Т1зО2(ОН)2С1б-8Н2О. Дробное отношение Cl:Ti, по мнению исследователей, позволяет считать, что выявленные продукты имеют полимерный характер.
Для приготовления водного раствора тетрахлорида титана рекомендуется применять разбавленную хлороводородную кислоту. При этом уменьшается разогревание раствора и устраняется преждевременный гидролиз. Это позволяет получить прозрачные растворы тетрахлорида титана высокой концентрации — до 300—350 г/л (в пересчете на ТЮ2).
Для получения диоксида титана гидролизом при кипячении рекомендуется использовать растворы ИСЦ с концентрацией 100—150 г/л (в пересчете на TiO2) с внесением до 5—8% (в пересчете на TiO2) зародышей. При этом может быть применен технический тетрахлорид титана.
Продукт гидролиза представляет собой гидроксид титана полимерного строения с незначительным содержанием остаточного хлора. При термообработке продукта гидролиза с добавкой минерализаторов (1—2% К280д) получается TiO2 в рутильной форме с высокой дисперсностью (около 90% частиц размером менее 1 мкм) и хорошими пигментными свойствами — высокой интенсивностью и укрывисто-стью (30—31 г/м2). Однако продукт обладает низкой атмосферостой-костью и значительно уступает рутилу, получаемому из сульфатных растворов титана.
Жидкофазный гидролиз основан на гидролизе тетрахлорида титана путем взаимодействия его паров с парами воды при нагревании. Рекомендуются как сравнительно низкие температуры — 300—400° С, так и высокие — 900—1100° С.
Исследования парофазного гидролиза в интервале температур 25—700° С показали, что при температуре около 100° С образуются оксо- и гидроксохлориды состава TiOCl2 и Ti(OH)2Cl2. При более высоких температурах содержание этих соединений снижается, однако они остаются в продуктах гидролиза в больших количествах даже при 500° С. Наибольшее значение имеет метод гидролиза при высокой температуре (900—1000° С). При этом получается диоксид титана с незначительным содержанием хлора и с лучшими пигментными свойствами. При парофазном гидролизе вначале образуются мельчайшие кристаллы диоксида титана, которые при дальнейшем нагревании приобретают округлую форму.
395
Суммарная реакция парофазного гидролиза:
TiCl4 + 2Н2О = TiO2 + 4НС1
Процесс, пО'Видимому, протекает ступенчато с образованием в качестве промежуточного продукта оксохлорида титана по схеме
Tid4 + Н2О -> TiOCl2 + 2НС1; TiOCl2 + Н2О -> TiO2 + 2НС1
Для полноты реакции необходим избыток паров воды в пределах 10—15%. Для образования тонкодисперсного продукта рекомендуется разбавление паров исходного тетрахлорида титана инертным газом, обычно азотом в отношении 6—12 объемов азота на один объем тетрахлорида титана. Температура реакции и продолжительность пребывания частиц в зоне высокой температуры должны строго регулироваться, так как при низкой температуре в продукте остается значительное количество хлора, а при длительном нагревании частиц происходит их укрупнение и ухудшение пигментных свойств. Для осуществления оптимальных условий технологии требуется не только подвод теплоты в зону реакции, но и предварительный нагрев исходных реагентов до высокой температуры.
Для получения диоксида титана применяется предварительно очищенный тетрахлорид титана и дистиллированная вода. При 700° С образуется анатазный диоксид титана, а при 800° С — смесь с преобладанием анатаза. Испарение исходного тетрахлорида титана и воды производится в титановых испарителях.
Полученный в реакторе диоксид титана выносится с парогазовой смесью в пылевую камеру, а затем в электрофильтр, где улавливается 99% целевого продукта. Температура на входе в электрофильтр 400° С, а на выходе она должна быть не менее 150° С.
Очищенная парогазовая смесь, содержащая хлорид водорода, не прореагировавшие пары воды и азот, поступает в холодильник для конденсации концентрированной хлороводородной кислоты.
По некоторым данным, диоксид титана хорошего качества может быть получен без перегрева исходных компонентов.
Одним из вариантов парофазового гидролиза является обжиг тетрахлорида титана в факеле водорода. В начале процесса происходит горение водорода, а затем взаимодействие образующихся паров воды с тетрахлоридом титана по схеме
2Н2 + О2 = 2Н2О; TiCl4 + 2Н2О = ТЮ2 + 4НС1
При работе по этой технологии значительно облегчается подвод в реактор теплоты, необходимой для высокотемпературного гидролиза. 396
Способ парофазного гидролиза тетрахлорида титана прост и обеспечивает получение его диоксида с высокими пигментными свойствами.
Сжигание тетрахлорида приобрело большое значение в связи с высоким качеством получаемого пигмента, резким сокращением количества отходов производства и сбрасываемых кислых стоков по сравнению с серно-кислотным способом. Основное его преимущество перед парофазным способом — возможность использования оборотного хлора непосредственно или после регенерации из разбавленной газовой смеси для хлорирования титансодержащего сырья. При этом создается замкнутый технологический процесс по хлору.
Взаимодействие тетрахлорида титана с кислородом протекает по уравнению
TiCl4 + О2 = ТЮ2 + 2С12
Реакция необратима, потому что диоксид титана не реагирует с хлором в отсутствие восстановителя. Процесс окисления проводится с некоторым избытком кислорода (10—15%). Для сжигания тетрахлорида титана можно было бы применять кислород воздуха, однако при этом происходило бы сильное разбавление оборотного хлора и вследствие наличия в воздухе паров воды образование хлорида водорода. В связи с этим сжигание проводится в токе осушенного кислорода. Термодинамически наиболее вероятно образование рутила при окислении тетрахлорида титана. Однако на практике при недостаточно высоких температурах (1000—1100° С) образуется анатаз и лишь при 1300° С — рутил. Для получения TiO2 рутильной структуры рекомендуется введение в состав исходного тетрахлорида титана некоторых добавок (А1С1з, низших оксидов титана и др.) в количестве 1—3%. Действие добавок объясняется тем, что они образуют мельчайшие твердые частицы А120з, которые служат центрами кристаллизации для диоксида титана, облегчая процесс образования новой фазы. Рутилизации способствует также присутствие в реакционной смеси паров воды, однако при этом происходит образование хлорида водорода. По некоторым литературным данным, тетрахлорид кремния стабилизирует анатазную структуру и улучшает дисперсность.
Взаимодействие газообразного тетрахлорида титана с кислородом начинается при 600—700° С и ускоряется при повышении температуры. В общем виде процесс можно рассматривать как образование конденсационных аэрозолей из находящихся в метастабильном состоянии пересыщенных паров. Ввиду малого давления пара диоксида титана и высокой степени пересыщения возникшие молекулярные комплексы интенсивно растут в результате соударения между собой и с маточными молекулами.
397
На рис. 9.11, 9.12 показана зависимость скорости реакции от температуры. Из рис. 9.11 видно, что в интервале температур 600—1100° С реакция удовлетворительно подчиняется линейному закону, т. е. она лежит в кинетической области, в которой скорость процесса в основном определяется скоростью химического взаимодействия. Оптимальной температурой реакции является 1000—1200° С; при более низкой температуре скорость реакции недостаточна, а более высокая температура вызывает дополнительные трудности в аппаратурно-технологическом оформлении процесса. Кроме этого изменение температуры в ту или другую сторону связано с ухудшением качества получаемого продукта.
Механизм процесса кристаллизации частиц диоксида титана объясняется образованием из газовой фазы тонкодисперсных первичных кристаллов, являющихся центрами, на которых в дальнейшем происходит конденсация TiO2 из газовой же фазы. Установлено, что как первичные частицы, так и продукты конденсации имеют правильную кристаллическую форму. Некоторые исследователи определяют состояние диоксида титана в момент образования в газовой фазе как «парообразное».
Частицы диоксида титана после образования во избежание дальнейшего роста и агрегации подвергаются резкому охлаждению («закалке»). Процесс агрегации частиц диоксида титана связан с наличием на их поверхности электростатических зарядов, естественных для любых аэрозолей
Процесс получения диоксида титана вследствие высокой скорости реакции связан со значительными трудностями. К ним относятся: 1)
Рис. 9.11. Зависимость скорости реакции TiCU+Oz TiO2+2C12 от температуры
Рис 9.12. Зависимость логарифма скорости реакции TiCU+Oz -> ТЮ2+2С1г от обратной абсолютной температуры
398
возможность образования целевого продукта с дефектной кристаллической решеткой; 2) под действием высоких температур возможен чрезмерный рост частиц, их агрегация и спекание.
Для исключения этих явлений необходимо строго регулировать параметры синтеза и продолжительность пребывания продукта реакции в зоне высоких температур. По результатам некоторых исследований регулирование процесса может быть достигнуто разбавлением исходного сырья (TiCU) инертными газами и, в частности, азотом. Однако при этом образующийся газ растворяется азотом и требуется регенерация хлора.
Значительные трудности создаются при оформлении технологического процесса, что связано: 1) с коррозирующим свойством сильного окислителя — хлора при высокой температуре; 2) с недостаточной механической прочностью материалов (графита, полиграфита, алунда, керамики, кварца); 3) с высокой температурой и резкими ее колебаниями; 4) с отложениями на стенках реакционной камеры образующегося диоксида титана; 5) с зарастанием коммуникаций образующимися продуктами.
Реакция окисления исходного тетрахлорида титана экзотермическая. Однако количество выделяющейся в результате реакции теплоты недостаточно для самопроизвольного протекания реакции. Поэтому для поддержания нормального хода реакции применяют следующие меры: 1) исходный кислород и пары тетрахлорида титана подогреваются до высоких температур, с тем чтобы их реакция протекала при необходимой температуре (1200° С); 2) реакцию между исходными видами сырья проводят в присутствии горючих газов; 3) осуществляют внешний нагрев реактора.
Аппарат для производства состоит из: 1) испарителя исходного сырья (тетрахлорид титана); 2) перегревателей паров тетрахлорида титана и кислорода; 3) горелок реакционных аппаратов; 4) улавливающих устройств. Для перегрева исходного сырья применяют аппараты различного типа с графитовыми нагревателями, а для перегрева кислорода — трубы из жаропрочной стали, обогреваемые силитовыми стержнями. Пары тетрахлорида нагревают до 500—1000° С, а кислород — до 700—1000° С.
Смешение газов проводят в горелках (форсунках), а сжигание — в реакционной камере. Конструкция форсунок имеет решающую роль в производстве, так как для быстрого протекания реакции и синтеза тонкодисперсного диоксида титана необходимо интенсивное смешение исходных реагентов при входе в реакционную камеру. Значение имеет также направленность паров кислорода с тетрахлоридом титана и соотношение кинетической энергии их истечения из сопла форсунки.
В качестве горючих газов в производстве применяют природный газ, пары жидкого топлива и оксид углерода. При использовании 399
природных газов или паров жидкого топлива целевой продукт получается с хорошими пигментными свойствами, что, по-видимому, связано с присутствием в реакционной зоне паров воды, способствующих рутилизации. Недостатком способа является образование в продуктах сгорания хлорида водорода, загрязняющего хлор. Указанных недостатков лишен способ, при котором в качестве горючего применяется оксид углерода, который образуется в результате восстановления диоксида углерода коксом в газогенераторе. Оксид углерода предварительно очищается от механических примесей и обезвоживается. Для процесса его берут в количестве 0,25—1,00 моль на 1 моль исходного тетрахлорида титана. В процессе подачи в реактор паров тетрахлорида титана, кислорода и оксида углерода температура в зоне горения составляет 1200—1400° С. Газовая фаза после реакции содержит 60—70% хлора, 5—6% кислорода и 35—40% диоксида углерода. Хлор после реактора поступает на регенерацию (абсорбция жидким монохлоридом серы). В результате образуется жидкий раствор, состоящий из моно- и дихлорида серы по схеме
С12 + 3SC1 = SCI + 2SC12
Десорбция хлора происходит при нагревании:
2SC1 + SC12 = 2С12 + 3S
В процессе регенерации отходящий хлор освобождается концентрированной серной кислотой, обезвоживается от капелек и фильтруется. Процесс абсорбции проводится в ряду последовательно соединенных колонн. В связи с экзотермичностью процесса смесь постоянно охлаждается. Хлор улавливается полностью, а отходящие пары и газы выбрасываются через трубу. Обогащенная хлором смесь моно- и дихлорида серы направляется в десорбционные колонны, в которых в процессе кипячения происходит выделение хлора. Образующийся монохлорид серы охлаждается водой и хладагентом, а пары чистого хлора проходят через теплообменник и конденсируются.
Наибольшее значение в качестве источника нагревания в последние годы приобретают плазмотроны, с помощью которых кислород может быть нагрет до 3000—5000° С. На практике они работают при 1500—2000° С. Для получения плазмы может быть использована вольтова дуга или индукционный (высокочастотный) способ нагрева. На рис. 9.13 и 9.14 приведены схемы индукционного и дугового плазмотронов.
Индукционные плазмотроны (рис. 9.13) обеспечивают высокую чистоту ионизированного кислорода. Мощность дуговых плазмотронов (рис. 9.14) достигает нескольких тысяч киловатт, и в результате эрозии 400
Рис 9.13. Дуговая горелка высокого давления (водяное охлаждение не показано):
1 — дуговая камера; 2 — изолятор; 3 — катод; 4 — кварцевое окно; 5 — реактор; 6 — медная труба; 7—закалочное сопло с внутренним диаметром 1,5 мм; 8 — сопло-анод; 9—изолятор
электродов (вольфрам, медь, углерод), особенно в среде кислорода, происходит загрязнение диоксида титана оксидами металла. С целью уменьшения окисления электродов применяются завесы из инертного газа, электроды из титана, алюминия и циркония.
При работе с плазмотронами диоксид титана получается сжиганием в струе ионизированного кислорода перегретых паров, холодных паров или даже жидкого тетрахлорида титана.
Применение плазмотронов в качестве мощных источников энергии для осуществления процесса разложения тетрахлорида титана значительно упрощает аппаратурное оформление процесса, позволяя исключить из схемы перегреватели исходных тетрахлорида титана и кислорода. При этом становится возможным в более широких пределах варьировать температурные условия получения диоксида титана и его физико-химические свойства.
Диоксид титана, синтезируемый хлорным методом, содержит в своем составе некоторое количество адсорбированного хлора (0,1—0,3%), придающего целевому продукту кислый характер. Освобождение продукта от хлора проводится термообработкой его при температуре 400° С или за счет пульсации осушенного воздуха высокого давления.
Рис 9.14. Индукционная плазменная горелка: 1 — пусковой угольный электрод; 2 — вход газа; 3 — латунный держатель; 4 — кварцевая труба; 5 — обмотка; 6 — ярко светящаяся плазма; 7 — хвост факела
401
Диоксид титана, синтезируемый по хлорному способу, выпускает- ! ся рутильной структуры, отличающейся высокими пигментными , свойствами — его разбеливающая способность доходит до 2000 ! условных единиц. |
Основными технологическими операциями получения диоксида I титана сжиганием перегретых паров тетрахлорида титана в среде ' диссоциированного кислорода являются: испарение исходного тетрахлорида титана и его перегрев; нагревание (диссоциация) кислорода; сжигание перегретых паров исходного сырья; «закалка» пылегазовой смеси; выделение диоксида титана из пылегазовой смеси; удаление из целевого продукта адсорбированного хлора, его компрессирование и очистка.
Тетрахлорид титана испаряется при 136° С. Незначительные количества хлоридов других металлов, применяемых для модифицирования, добавляются к исходному тетрахлориду титана или в жидком виде до его испарения или в парообразном виде. Пары TiCU направляют в перегреватель, где они нагреваются до 600—1200° С. Кислород нагревается в плазмотроне. Перегретые пары тетрахлорида титана поступают в форсунку плазмотрона водоохлаждаемого реактора, а в центральный канал форсунки поступает кислород в 10—15%-ном избытке против стехиометрии. Процесс разложения тетрахлорида титана происходит при 1100—1300° С. Образующаяся при этом пылегазовая смесь в камере закалки охлаждается оборотным хлором до 500—600° С. Уловительная система после плазмотрона состоит из пылевой камеры, циклона и рукавных фильтров. В пылевой камере осаждается 30—50% образовавшегося диоксида титана. Смесь при этом охлаждается до 200—250° С. Дополнительное осаждение целевого продукта происходит в циклоне, а полное — в рукавных фильтрах.
Для удаления адсорбированного хлора целевой продукт подвергается термообработке при 300—400° С. Некоторые сорта диоксида титана, получаемого по хлорному способу, выпускаются без дополнительной обработки. При необходимости целевой продукт подвергается поверхностной обработке таким же образом, как и по серно-кислотному методу.
Хлор охлаждается в скруббере до 30—40° С с помощью серной кислоты, а затем поступает в компрессор. Отвод теплоты от серной кислоты проводится с помощью воды и рассола. Охлажденный хлор, содержащий 60—70% С12, подается на хлорирование титансодержащего сырья для получения тетрахлорида титана.
В производстве диоксида титана из его тетрахлорида образуются отходы: 0,35 т 10%-ной хлороводородной кислоты и 0,31 т раствора гипохлорита кальция на одну тонну производимого целевого продукта. 402
9.4. ХЛОРИДЫ ТИТАНА
Титан образует три хлорида: ди-, три- и тетрахлорид титана. Наибольшее значение в народном хозяйстве имеет тетрахлорид титана.
Дихлорид титана — коричневые или черные кристаллы. Сингония тригональная. Параметры элементарной ячейки: а = 0,3568 нм; с = = 0,5887 нм. Уравнение температурной зависимости давления пара: 1g р (мм рт. ст.) = 9,593—10 230/Д753—883К). Тт = 1035° С, Ттп = 1515° С; плотность = 3,13 г/см3; С ° =69,8 Дж/(моль-К); ДЯпл = 37,7 кДж/моль, АЯИСТ ~ 147 кДж/моль; АЯобр = -516 кДж/моль; = 87 Дж/(моль-К). При 800—850° С в вакууме диспропорционирует по схеме
2TiCl2 = Ti + TiCl4
Сильный восстановитель; на воздухе окисляется; реагирует с водой, выделяя водород, метанолом и этанолом; плохо растворим в сероуглероде, СНС1з, диэтиловом эфире.
Получают восстановлением тетрахлорида титана металлическим титаном (TiCl + Ti = 2Т1С12), алюминием (TiCl4 + Al = TiCl2 + AICI3), водородом (TiCl4 + Н2 = TiCl2 + 2HC1) или диспропорционированием TiCl3 (2TiCl3 = TiCl2 + TiCl).
Дихлорид титана применяется в аналитической химии органических веществ (нитро-, нитрозо- и других органических соединений).
Трихлорид титана — темно-фиолетовые или черные кристаллы. Известно несколько политипов, а также коричневая ^-модификация (образуется при восстановлении тетрахлорида титана алкилалюмини-ем), которая после термообработки при 250—400° С превращается в фиолетовую модификацию. Сингония — тригональная. Параметры элементарной ячейки: а = 0,6133 нм; с = 1,753 нм. Уравнение температурной зависимости от давления пара: 1g р(мм рт.ст.) = 21,47—9620/Т + + 3,271g71(298—800 К). Температура плавления 920° С; плотность— 2,656 г/см3; С® =97,1 Дж/(моль-К); АНщ, = 37,7 кДж/моль; ДЯИСП = 205 кДж/моль; АНобр ~ -720 кДж/моль; S°9g = 140 Дж/(моль-К). В парах присутствует в основном Ti2Cl6; при 440° С начинает диспро-порционировать по схеме: 2TiCh = TiCl2 + TiCl. Во влажном воздухе расплывается, быстро окисляется и гидролизуется: 2TiCh + 2О2 = = 2ТЮ2 + ЗС12. Сильный восстановитель. Легко растворяется в воде и этаноле с образованием фиолетовых растворов; из водных растворов могут быть выделены TiCly6H2O, а также тетрагидрат (зеленого цвета). Водные растворы на свету постепенно окисляются и обесцвечиваются.
403
Трихлорид титана получают восстановлением его тетрахлорида водородом (при 800° С), элементным титаном (около 600° С), алюминием или кремнием.
Трихлорид титана применяется в качестве компонента катализаторов процесса полимеризации олефинов. Водные растворы трихлорида титана применяют в аналитической химии.
Тетрахлорид титана — бесцветная прозрачная жидкость, дымящая на воздухе. Сингония — моноклинная. Параметры элементарной ячейки, нм: а = 0,970; Ъ = 0,648; с = 0,975. Уравнение температурной зависимости: плотности d = 1,7609—0,00166f—8,1-IO"7/2 + 1,58-10’9/3 г/см3 (от -22 до 135° С), давления пара 1g р(мм рт.ст.) = 7,682—1964/7(249-409 К) (рис. 9.15). Температура плавления — 24,1° С; температура кипения— 136,3° С; плотность—1,73 г/см3; Cj = 145,3 Дж/(моль-К); Д/Тпл = 10,0 кДж/моль; А77^р = -804 кДж/моль; S®98 = 252,5 Дж/(моль-К);
в парах не диссоциирует и не разлагается вплоть до высоких температур; /крит 365° С, Ркрит 5,01 МПа. Выше 500—600° С окисляется кислородом воздуха до TiO2, в присутствии паров воды образуются также оксихлориды ТЮС12, Ti2O-Cl2. Бурно реагирует с водой с образованием TiO2«H2O, в качестве промежуточных продуктов образуются TiC14-5H2O и гидрооксохлориды Ti(OH)„C14.„-xH2O. При очень медленном добавлении воды с тщательным перемешиванием и охлаждением можно получить устойчивые концентрированные растворы тетрахлорида титана. Растворяется в этаноле и диэтиловом эфире. Восстанав
ливается водородом и активными металлами до дихлорида и трихлорида титана, а затем до элементного титана. Связь титана с атомами
хлора в молекуле тетрахлорида титана имеет ковалентную природу.
Рис. 9.15. Зависимость давления пара над жидким TiCl4
Он химически активен и реагирует со многими неорганическими и органическими веществами при комнатной температуре, при нагревании с тщательно осушенным воздухом не реагирует, а с влажным воздухом образует белый дым вследствие образования твердых продуктов гидролиза.
Тетрахлорид титана растворяет С12 (7,6% масс, при 20° С) или незначительно— НС1. Смешивается во всех соотношениях с жидким НС1, а также с хлоридами Sn, С, Si. Растворяется в хлороводородной кислоте при пропускании газообразного НС1 с получением
от температуры ярко-желтого раствора гексахлортитано-
404
вой кислоты Н2Т1С1б. С разбавленной серной кислотой образует TiOSO4, а с концентрированной — H2SO4.
Технология тетрахлорида титана. Основным способом получения тетрахлорида титана является метод хлорирования титаносодержащего сырья по схеме
TiO2 + 2С12 = TiCU + О2
Рутил, освобожденный от пустой кремнистой породы, является наиболее концентрированным титансодержащим сырьем. Он содержит от 91 до 99% ТЮ2 с небольшими примесями циркония, ниобия, ванадия, хрома, железа, кремния и алюминия. Поэтому получение тетрахлорида титана из этого вида сырья проще, чем при использовании ильменита. Однако рутил более дорогостоящий и менее распространенный вид сырья.
Прямое хлорирование ильменита затрудняется образованием дополнительного количества хлорного железа. По этой причине ильменит до процесса хлорирования подвергают обезвоживанию.
Обезвоживание ильменита проводят прямым восстановлением ильменита восстановительной плавкой или его карбонизацией. Прямое восстановление ильменита осуществляют углеродом, водородом или природными газами в циклонных печах или в печах с кипящим слоем при 900—1200° С:
FeTiO3 + С = Fe + TiO2 + СО
FeTiO3 + Н2 = Fe + TiO2 + Н2О
3FeTiO3 + СН4 = 3Fe + 3TiO2 + CO + 2H2O
Получающееся по этим реакциям металлическое железо отделяется от диоксида титана электромагнитной сепарацией. Остатки железа удаляются при обработке раствором хлорного железа. Очищенный диоксид титана после сушки направляется на производство тетрахлорида титана.
Разработан способ восстановления ильменита в присутствии небольших количеств шлакообразующих веществ. Восстановлением железотитанового сырья коксом в присутствии сульфата кальция при 1100—1200° С получают концентрат с содержанием 90—95%-ного основного вещества.
В процессе восстановительной плавки происходит избирательное восстановление ильменита углеродом. Процесс проводят в электроду-говой печи при 1400—1500° С. При этом получаются богатые титаном шлаки и чугун с содержанием около 1% углерода и 0,5% серы.
405
Образующиеся титанистые шлаки содержат 65—90% ПО2, 0,5—3% FeO, 1—7% SiO2, 0,5—6% А12О3, 0,1—3,5% СаО, 5,5—20% MgO, 0,15—0,4% V2O5, 0,02—0,2% влаги.
Процесс плавки можно вести и в присутствии флюсов (оксида кальция, оксида магния и др.) при более низких температурах. Однако в этом случае затрудняется процесс последующего хлорирования титанистых шлаков по причине образования пленки СаС12, MgCl2 и др.
Представляет интерес восстановление титанового сырья при 1200—1400° С коксом в отсутствие флюсов с получением смеси низших оксидов титана с незначительным содержанием карбида кальция. Полученная смесь хлорируется при более низких температурах (200—500° С), чем шлаки, содержащие диоксид титана.
Разработан способ переработки ильменитового концентрата и других титаножелезистых материалов в сульфиды титана, хлорирующиеся при 175° С. Перспективным способом получения сульфидов титана является плавка титанового сырья на штейн, который получается при термообработке титанового сырья с коксом и пиритом при 1500° С. Полученный при этом целевой продукт содержит 37—45% Ti (в виде TiS, Ti2S3, TiS2, TiO и др.), 10—20% Fe (в элементном виде и в виде TiFeS2), 20—30% S и 1,5—3,5 С.
Одним из способов переработки ильменита является его карбонизация, которая происходит в процессе восстановительной плавки при 1800—2200° С и увеличения содержания углерода в исходной шихте. При этом выплавляется чугун и образуется карбид титана по схеме
FeTiO3 + 4С = ПС + Fe + ЗСО
Образующаяся при плавке в присутствии воздуха пористая масса представляет собой твердый раствор карбида и нитрид титана Ti (С, N) или оксикарбонитрид Ti (С, N, О). После измельчения и отделения основной массы железа (чугуна) электромагнитной сепарацией и обработкой хлороводородной кислотой, в которой карбид и нитрид титана не растворяются, полученный полупродукт направляют на хлорирование.
Технология тетрахлорида титана из рутилового концентрата и титанистых шлаков. При использовании в качестве титансодержащего сырья рутилового концентрата или продуктов обезжелезивания ильменитового концентрата, хлорированию подвергают содержащийся в них диоксид титана по схеме:
ПО2 + 2С12 = ПС14 + О2
406
Реакция протекает с небольшой скоростью даже при высоких температурах (800—1000° С).
С целью ускорения процесса, снижения его температуры в шихту вносят уголь. В присутствии углерода процесс проводят при 600—800° С. Скорость реакции при этом считается оптимальной. При этом в зависимости от температуры процесса получения тетрахлорида титана в газовой фазе образуется оксид углерода (выше 600° С), диоксид углерода (ниже 600° С). При более высоких температурах (900—1000° С) помимо оксида углерода образуется некоторое количество фосгена. Процесс в целом проходит по схеме
TiO2 + 2С12 + 2С = TiCl4 + 2СО
TiO2 + 2С12 + С = TiCl4 + СО2
TiO2 + 4С12 + 2С = TiCl4 + 2СОС12
Установлено, что в диапазоне температур 600—800° С скорость образования фосгена незначительна, а в процессе хлорирования выше 1300° С газообразные продукты состоят в основном из оксида углерода (2/3 объема) и тетрахлорида титана (1/3 объема).
Скорость процесса хлорирования зависит в основном от размеров частиц исходного сырья, скорости и плотности потока хлора и температуры. Процесс хлорирования исходного диоксида титана
ускоряется в присутствии катализатора— диоксида марганца в количестве 0,1% от массы шихты. Каталитический эффект выше при более низких температурах. Степень перехода диоксида титана в его тетрахлорид в лабора
торных условиях при 400° С достигает 70% (в течение 4 ч) вместо 15% без катализатора.
Изучен процесс проведения хлорирования диоксида титана в состоянии исходной шихты в виде брикетов. Установлено, что скорость процесса хлорирования брикетов из диоксида титана и 26% нефтяного кокса зависит от массы брикетов (рис. 9.16). Для брикетов цилиндрической формы глубина
Рис 9.16. Зависимость степени хлорирования диоксида титана от массы брикетов при 700° С
407
хлорирования х, выражающая толщину слоя брикета в см, вступившего в реакцию за период т, может быть вычислена по уравнению
х=(А)х(1_з/]-^)
где Ро—начальная масса брикета, г; р — плотность брикета, г/см3; т| — степень хлорирования.
Величина х вычисляется в виде полуразности определяющих диаметров брикетов 1/2(До — Д) до реакции (До) и оставшегося (Д) к моменту т. Принято, что скорость процесса хлорирования пропорциональ-на внешней поверхности брикета, т.е. — = kS, где S—переменная dx
величина внешней поверхности без учета пористости. Поскольку dP = dx 1
р&аЬс, то — = — к (где к—удельная скорость процесса хлорирования) и, dx р
следовательно, глубина хлорирования прямо пропорциональна времени. С уменьшением размера исходных брикетов увеличивается общая реакционная поверхность, в результате чего ускоряется процесс хлорирования. Однако с уменьшением размеров брикетов по ходу реакции в аппарате возрастает гидравлическое сопротивление (рис. 9.17). При 550° С процесс при малой линейной скорости хлора из кинетической области переходит в диффузионную, для которой эффективная энергия активации реакции равна 157 кДж/моль. Скорость хлорирования пропорциональна линейной скорости хлора в степени 0,43 и возрастает пропорционально содержанию хлора в газовой фазе при концентрациях выше 30% (рис. 9.18).
Рис 9.17. Влияние температуры на скорость хлорирования
Рис 9.18. Зависимость скорости хлорирования от концентрации хлора в газовой фазе
408
Рис 9.19. Схема хлоратора типа шахтной электропечи
Процесс хлорирования титансодержащего сырья проводят в шахтных электрических печах периодического действия (рис. 9.19).
Исходные брикеты шихты загружаются в печь предварительно высушенные, обожженные и от-грохоченные. Они состоят из смеси титансодержащего сырья и кокса с соотношением от 4:1 до 1:1, а также связующего (пека, смолы и сульфитных растворов). Предварительный обжиг брикетов производится для удаления летучих веществ и производится при 500—800° С в зависимости от вида связующей добавки. Шихта содержит 20% углерода.
Для хлорирования применяется хлор из анодных отсеков магниевых электролитов или оборотный хлор — газ производства диоксида титана с содержанием 60—70% (об. % основного вещества). Подают хлор в печь по фурмам, расположенным под углом выше нижнего ряда электродов. Реакция хлорирования экзотермическая. Газообразные продукты хлорирования в смеси с непрореагировавшим (избыточным) хлором выходят из верхней части печи и направляются в систему пылеочистки, поступают в конденсационную систему, состоящую из скруббера и трубчатых холодильников, в которых происходит ожижение и отделение тетрахлорида титана.
В процессе хлорирования образуется не только тетрахлорид титана, но и хлориды сопутствующих примесей. По склонности к хлорированию компоненты исходного титансодержащего сырья располагают в следующий ряд:
CaO>MnO>FeO>MgO>TiO2>Al2O3>SiO2
В процессе хлорирования оксиды, стоящие в ряду перед диоксидом титана, хлорируются нацело, оксиды же алюминия и кремния— примерно на 40%. Температура плавления и кипения некоторых примесных хлоридов и их растворимость в тетрахлориде титана приведены в табл. 9.7.
409
Таблица 9.7. Температура плавления, кипения примесных хлоридов и их растворимость в TiCU
Показатель VOCb SiCU А1С13 FeCl3 МпС12 MgCl2
Т °C 1 пл» -77 -69 193 303 660 704
Т °C 1 кип» 127 57 180 319 1190 1413
Растворимость в TiCl4 Неограниченная (сублимируется) 0,15 0,005 1,7-10’5 <0,01
указанных примесей
Процесс очистки тетрахлорида титана от проводят по следующей схеме:
Технический TiCl 4
NaCl
Си
Н2О
Очистка от Al и V
Cu-V-кек
Первая ректификация
На извлечение ванадия ▼
Вторая ректификация
Суспензия оксохлоридов Очищенный T1CI4 Дистиллят технического SiCI4
В сгуститель
На термообработку
На очистку
От высококипящих хлоридов тетрахлорид титана очищается конденсацией. При этом хлориды магния, кальция, марганца и железа (II) улавливаются в одном конденсаторе, а хлориды железа (III) и алюминия в другом. Тетрахлориды титана и кремния, образующие непрерывный ряд жидких смесей (рис. 9.20) с растворимыми примесями, улавливаются в конденсаторе, орошаемом техническим тетрахлоридом титана. Полученный технический тетрахлорид титана, содержащий ~1% твердых веществ и растворенные примеси, очищается от алюминия и ванадия путем смешения их соединений 410
незначительным количеством воды и восстановителей, в качестве которых применяются металлические натрий и медь. При этом происходит избирательный гидролиз алюминия и восстановление ванадия. В результате образуется твердое вещество, состоящее из оксохлорида алюминия и меднованадиевого пека, от которых жидкий тетрахлорид титана освобождается путем отстоя и фильтрации. В конце процесса тетрахлорид титана подвергается ректификации для освобождения от тетрахлорида кремния. Примерное содержание примесей в очищенном тетрахлориде титана приводится ниже:
Si V Fe Al ТЮС12
До очистки 0,3 0,1 0,02 0,1 0,5
После очистки 0,006 0,004 0,004 0,004 0,001
За последние годы в России разработаны и предложены другие способы получения тетрахлорида титана, направленные на повышение его качества, снижение себестоимости, упрощение технологических приемов, улучшение условий труда и др.
Разработан способ получения тетрахлорида титана хлорированием титансодержащего сырья в потоке хлора и оксида углерода.
Испытаны и описаны способы проведения хлорирования в печах
с кипящим слоем.
Заслуживает внимания постадийное проведение процесса хлорирования в двух или нескольких хлораторах. При этом в первом аппарате осуществляют хлорирование при пониженной температуре с образованием легколетучих хлоридов и образующихся газов. После отделения хлоридов газы направляют во второй аппарат.
Разработан процесс получения тетрахлорида титана хлорированием измельченных титановых шлаков и кокса, взвешенных в расплаве хлоридов и, в частности, карналлита, а также смесь хлорида калия и натрия или чистые хлориды, например расплав хлорида натрия для хлорирования смеси диоксида титана и древесного угля при 900° С. Внесение в расплав около двух процентов хлорного железа интенсифицирует массопе-ренос хлора к поверхности частиц диоксида титана. Экспериментально установлено, что
Рис 9.20. Диаграмма температур кипения в системе TiCl4—SiCl4
411
количество хлора, транспортируемого растворенным хлорным желе- я зом от поверхности пузырька к твердой хлорируемой поверхности, Я примерно в сто раз больше количества растворенного хлора, транс- 1 портируемого через расплав. Аналогично действует и добавка в рас-Я плав хлорида алюминия. I
Тетрахлорид титана высокой чистоты (99,999% основного веще- Я ства) можно получить путем перегонки очищенного продукта под а разряжением, а также путем очистки его дистилляцией в присутст- 1 вии незначительного количества минеральных масел (0,1—0,3% 1 масс.) с последующей отдувкой инертным газом. 1
Перспективными технологиями тетрахлорида титана являются хлорирование карбида титана при 300—400° С; низших оксидов титана при 200—500° С; сульфидов титана при 200—250° С.
S
9.5. ТИТАНАТЫ 1
Титанаты являются солями несуществующих титановых кис- 1 лот — метатитановой Н2ТЮ3, ортотитановой Н4ТЮ4 и полититановых I H2„TimO2m+n Обычно они состоят из высших оксидов титана с основными оксидами. Поэтому особенно разнообразны титанаты щелочных и щелочноземельных металлов. Из других металлов большое число титанатов известно у висмута (от Bi^TiOzo до Bi2Ti40n).
Для металлов в степенях окисления +1 и +2 характерно образование мета-, орто- и полититанатов. Титанаты МпТЮз имеют главным образом структуры ильменита РеТЮз или перовскита СаТЮз. М"ТЮ4 имеют кубическую структуру типа обратной шпинели. Металлы в степени окисления +3, в частности РЗЭ, образуют большей частью оксотитанаты МегТЮз и дититанаты МгЛгО?, в степени окисления +4 — ортотитанаты МТЮ4. Известны многочисленные двойные титанаты, например NaBiTi2O6. Значительное число титанатов являются фазами переменного состава.
Титанаты щелочных металлов растворяются в разбавленных кислотах, а водой гидролизуются. Остальные титанаты не растворимы в воде и разбавленных кислотах, а при нагревании разлагаются в крепкой серной кислоте. Титанаты имеют довольно высокие температуры плавления, являются диэлектриками, большинство из них отличаются высокой диэлектрической проницаемостью. Титанаты металлов в степенях окисления +2 и +3, а также многие двойные титанаты — сегнетоэлектрики. Важнейшими из них являются титанаты бария, свинца и стронция (рис. 9.21).
Титанаты встречаются в природе в виде минералов, например ильменит (РеТЮз), перовскит (СаТЮз), гейкилит (MgTiCh), лопарит (ТЧаСеЪгОб) и др. Они являются компонентами титанистых шла-412
Рис 9.21. Диаграмма плавкости системы ВаО—ТЮ2
широко применяются следующие
ков — полупродуктов при переработке некоторых типов титановых руд.
Получают титанаты путем термообработки диоксида титана с оксидами, карбонатами или гидроксидами соответствующих металлов, термообработкой совместно осажденных гидроксидов, карбонатов, оксалатов.
Титанаты (III) изучены мало. Для щелочных металлов известны МТЮг, для щелочноземельных — МИгОд, для РЗЭ — MTiO3. Они не растворяются в воде, разлагаются минеральными кислотами. При нагревании на воздухе окисляются до титанатов (IV)-
В народном хозяйстве доволы титанаты.
Титанат никеля NiTiO3 (NiO-TiO2), твердый раствор оксида никеля с титанатом цинка — Zn2TiO4-2NiO (2ZnO-TiO2-2NiO), твердый раствор оксидов никеля, сурьмы и титана состава: ТЮг — 85%; БЬгОз — 15%; NiO — 3%, что соответствует формуле Ti020,07Sb2030,05NiO.
Титанат кобальта состава TiO2 0,5CoO (Г1О2 — 68,1%, СоО—31,9%), титанат кобальта-магния — TiO2-0,lCoO-0,05MgO; TiO2-0,3CoO-0,4MgO. Ти-танаты-алюминагы кобальта—ИОг-ОДСоО-ОДМгОз; Г1С>2-0,5СоО-0,5А12Оз-
Титанаты никеля не взаимодействуют при кипячении в 5—10%-ных растворах хлороводородной, серной и азотной кислот, а также в 5%-ном гидроксиде натрия, стойки к высоким температурам (до 800° С).
Экспериментально установлено, что в бинарных системах NiO — ТЮ2, SIJ2O3 — TiO2, NiO — SIJ2O3 при термообработке происходит ряд процессов. Исходный оксид никеля при всех соотношениях с диоксидом титана образует лишь одно соединение NiTiO3. Триоксид сурьмы при концентрации до 6,5 мол.% с диоксидом титана образует твердый раствор, кристаллизующийся в рутильной системе, а при концентрации более 6,5 мол.% образуется новая фаза следующего состава: 4ТЮг-38Ь20з. При термообработке же оксида никеля с триоксидом сурьмы она окисляется до пентаоксида и образуется сурьманат никеля NiSb2O6, кристаллизующийся в трирутильной системе, близкой по структуре к рутилу (постоянные решетки: а = 4,63А, с = 3,06А).
413
142 140 138 136 134 132 130
2Q град
Рис 9.22. Дифрактограмма рутильного диоксида титана (7) и титаната никеля (2)
Изучена тройная система ТЮг—xNiO—.ySbjOj, где х =
= 0,02—0,30 и у = 0,05—0,3. Установлено, что в ней при значении х<0,18 наблюдаются следующие закономерности по фазовому и структурному составу образующихся при термообработке соединений: 1) при соотношении NiO: SbjOs^l образуется рутил и фаза 4Т1Ог-38ЬгОз; 2) при соотношении NiO:Sb2O3>l образуется рутил и титанат никеля (рис. 9.22). Считается, что в рутиле растворяются не исходные оксиды — NiO и 8Ь20з, а продукт их взаимодействия NiSb2O6, структура которого сходна с рутилом, причем при этом имеет место образование твердого раствора доменного типа, в котором целые области (домены) ТЮг заменены NiSb20e.
Высокими техническими свойствами обладают только твердые растворы NiSbzOe в ТЮг, свободные от таких примесей, как NiTiCh, соединения 4ТЮ2-38Ь2Оз, твердого раствора ЗЬгОз в рутиле, оксида никеля, анатаза, а также свободного NiSb2O6. Продукт, свободный от примесей, получается лишь при высокой реакционной способности исходных составных частей (в основном диоксида титана) и тщательном перемешивании исходных реагентов. Активная форма диоксида титана и мокрое смешение способствуют образованию твердого раствора NiSb2O6 в ТЮг без посторонних примесей.
Технология титаноникелевого пигмента состоит из следующих операций: 1) приготовление шихты — смеси метатитановой кислоты, т.е. кристаллического диоксида титана (ТЮ2 Н2О), карбоната никеля и триоксида сурьмы; 2) сушка шихты при температуре 300° С; 3) термообработка смеси при 1000—1100° С в течение трех часов; 4) размол и фасовка продукта.
В ходе технологических операций исходные продукты претерпевают следующие качественные изменения: до 720° С продукты термообработки состоят из анатаза, размер монокристаллов которого колеблется в пределах 65—243А; при 740° С появляется рутил (~23%); при 760—780° С количество рутила составляет 94%. Выше 800° С образцы состоят лишь из рутила.
ГЛАВА 10
АЗОТ И ЕГО СОЕДИНЕНИЯ
10.1. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА АЗОТА
Содержание азота в земной коре составляет 1-10’2% (масс.). Наибольшая часть азота в свободном состоянии в атмосфере (азот — главная составная часть воздуха: 75,6% (масс.) 78,09% (об.)). В связанном состоянии азот встречается в воздухе, в водах рек, морей и океанов. В земной коре он образует три основных типа минералов, содержащих ионы CN~, NO3 и NH<. Промышленное значение имеет чилийская селитра (NaNCh), индийская селитра (KNO3).
Природный азот состоит из двух стабильных изотопов 14N (99,635%) и 15N (0,365%). Конфигурация внешней электронной оболочки 2г2р3; степень окисления азота в соединениях: -3, -1, +1, +3, +5, а также +2 и +4.
Молекула азота двухатомна, связь между атомами тройная (длина 0,110 нм). Энергия термической диссоциации молекулы 941,64 кДж/моль.
Азот — бесцветный газ, с температурой кипения -195,8° С; температура плавления -210° С; плотность 1,25046 кг/см3 (0°С), жидкого -0,808 г/см3 (-195,80° С); тройная точка: температура -210,00° С, давление 125,03 гПа; 4рет = -146,95° С, Ркрт = -0,304 г/см3; уравнение температурной зависимости давления пара: 1g р(мм рт. ст.) = = 7,65894-359,093/7(52-483 К); для газа Ср°= 29,125 Дж/(моль-К), 5°98 = 191,498 Дж/(моль-К); теплопроводность 0,077 (82 К), 0,243 (273 К), 0,315 (373 К), 0,725 Вт/(м-К) (1273 К).
В твердом состоянии при обычном давлении азот существует в двух модификациях: ниже 237,54° С устойчива a-форма с кубической решеткой типа СО (а = 0,5667 нм, пространственная группа 7*213; плотность 1,0265 г/см3 при -252,5° С); выше -237,54° С — 0-форма с гексагональной решеткой типа MgO (а = 0,393 нм, с = 0,650 нм, пространственная группа PG^/mmc, плотность 0,8792 г/см3 при -210° С); HJf перехода 0,231 кДж/моль. Выше 350 МПа существует третья модификация с тетрагональной кристаллической решеткой.
Растворимость азота в воде (см3 в 100 мл): 2,33 (0°С), 1,42 (40° С), 1,32 (60° С). В некоторых углеводородах (гексане, гептане и 415
др.) азот растворяется лучше, чем в воде. Растворимость в этаноле и q метаноле при 0° С примерно такая же, как в воде.
Низкая реакционная способность азота объясняется большой Ч энергией диссоциации молекулярного азота. При невысоких температурах азот реагирует с некоторыми активными металлами, например ' Li, Cs, а с другими металлами азот реагирует при высокой температуре, а иногда лишь каталитически.
Молекулярный азот может быть активирован соединениями переходных металлов, а затем превращен при обычных температуре и давлении в аммиак, гидразин или ароматические амины. К этим соединениям относятся соединения титана, ванадия, хрома, молибдена и железа, а восстановителями служат литий-, магний- или алюминий-органические соединения, гидриды металлов, щелочные металлы и их аддукты с ароматическими углеводородами и др.
Решающим моментом всех этих реакций является связывание азота в комплекс с соединениями переходного металла, при котором активизируется молекула азота.
С кислородом азот заметно взаимодействует лишь выше 2000° С с образованием NO, который уже легко окисляется до NO2. ?
С водородом азот реагирует лишь при высокой температуре и i давлении в присутствии катализатора с образованием аммиака. Кос- ' венными путями получены гидразин N2H4 и азотистоводородная кислота HN3, образующая соли — азиды. Ниже -15° С существует тетразен h2nn=nnh2.
Азот с галогенами непосредственно не реагирует. Однако косвенными путями получены соединения со связями N—Hal: галогенами-ны NHal3, NHHal2, NH2Hal и др.
С серой азот также непосредственно не реагирует. Косвенными методами получены нитриды серы: N4S4 — оранжево-желтые кристаллы, которые выше температуры плавления (179° С) или при ударе разлагаются со взрывом; весьма неустойчивый N5S5, (SN)X, S2N2, S4N2, S5N6 и др.
Азот при действии на раскаленный угольный кокс образует дициан (CN)2, при реакции с СаС2 при высокой температуре образует CaCN2. Нагревая смесь карбоната натрия с углем на воздухе, получают NaCN, а в процессе нагревания до 1500° С азот взаимодействует с ацетиленом, образуя HCN. С металлами азот дает нитриды. При высоких температурах азот взаимодействует с кремнием, а также с кальцием, стронцием, барием, титаном, вольфрамом, ванадием, хромом, марганцем, цирконием, танталом, молибденом, ураном и редкоземельными элементами.
В процессе действия электрического заряда на молекулярный азот при давлении 130—260 Па образуется активный азот — смесь 416
возбужденных молекул и атомов азота. Активный азот энергично взаимодействует при комнатной температуре с атомарными кислородом и водородом, парами серы, белым фосфором и некоторыми металлами, образуя при этом соответствующие соединения.
Азот применяется в процессе получения аммиака. Свободный азот применяют в качестве инертной среды при некоторых химических и металлургических процессах, при перекачивании горючих жидкостей. Он является консервантом в овощехранилищах и хладагентом в криостатах, вакуумных установках и др.
10.2. ОКСИДЫ АЗОТА
Азот образует с кислородом следующие оксиды: геми-, моно-, се-скви-, ди- и пентаоксид.
Гемиоксид N2O (оксид диазота, «веселящий газ») имеет слабый приятный запах и сладковатый вкус (табл. 10.1), молекула линейна: ^о71з^оп9^’ '^ЛЯ О,51-1О'30 Кл-м. Растворимость в воде (г на 100 г): 0,257 (0°С) и 0,108 (25° С), растворяется в спирте, эфире и серной кислоте. С водой, растворами кислот и щелочей, кислородом не взаимодействует.
Таблица 10.1. Свойства оксидов азота
Показатели n2o NO no2 n2o4 n2o3 N2O5
Цвет Бесцветный Бесцветный Красно-бурый Бесцветный Голубой* Бесцветный
Т °C 1 кип» -88,5 -151,6 — 20,6 -40 33**
т ° с 1 пл» -91,0 -163,7 — -11,2 -101 41
Плотность газа, г/дм3 1,9778 1,3402 1,491 (0° С) — — —
Плотность жидкого или твердого вещества, г/см3 1,226 (-88,5° С) 1,332 (-163° С) — 1,536 (-20° С) — 2,05 (15°)
t ° С *крит, 36,45 -93,0 — 158,2 — —
Ркрит» МПа 7,254 6,48 — 10,13 — —
Критический объем, ДМ/МОЛЬ 0,0973 0,0580 — 0,167 — —
С?, Дж/(моль-К) 38,63 29,86 37,18 79,2 72,7 95,3
кДж/моль 81,6 91,26 34,2 11,1 86,6 13,3
S^, Дж/(моль-К) 219,90 210,64 240,06 304,4 314,6 355,6
’Цвет кристаллов. "Температура возгонки.
14 Химическая технология неорганических веществ, кн. 1
Выше 500° С разлагается на азот и кислород с заметной скоростью и проявляет сильные окислительные свойства (в N2O, как и в кислороде, вспыхивает тлеющая лучина).
В растворе H2SOj восстанавливается до N2, а в солях Sn2+ до NH2OH, Ti3+ до аммиака. С водородом, аммиаком, оксидом углерода, а также с органическими веществами гемиоксид азота образует взрывоопасные смеси.
Гемиоксид азота получают разложением нитрата аммония при 250° С:
NH4NO3 -> N2O + 2Н2О
Процесс экзотермичен и протекает с ускорением, а при быстром нагревании возможен взрыв.
Монооксид NO парамагнитен и склонен к димеризации. Жидкий монооксид азота содержит 25% димера, а твердый полностью состоит из него. Длина связи N—О 0,115 нм, энергия связи 626,84 кДж/моль; ц 6,48-10'30 Кл-м; А//0 димеризации —11,2 кДж/моль. Растворимость в воде (г на 100 I*): 0,00988 (0°С) и 0,00348 (100° С); растворим в спирте, сероуглероде и серной кислоте.
Ниже 1000° С монооксид азота практически не разлагается. Равновесные концентрации монооксида, получающегося по реакции N2 + О2±> 2NO, составляют (в %): 0,61 (2000° С), 4,48 (3200° С), 10,00 (4200° С). С водой, разбавленными растворами кислот и щелочей не реагирует. При обычных условиях легко окисляется до диоксида азота, с повышением температуры скорость реакции уменьшается. Это связано с тем, что с кислородом взаимодействуют молекулы димера N2O2, содержание которого с повышением температуры уменьшается. Монооксид азота присоединяет галогены с образованием нитрозилгалогенидов NOHal:
NO + Hal = NOHal
С серной кислотой в присутствии воздуха образует нитрозилсер-ную кислоту:
2NO + 1/2О2 + 2H2SO4 - 2HNOSO4 + Н2О
Восстанавливается углеродом, фосфором, серой, диоксидом серы, водородом и металлами до молекулярного азота:
2NO + С = N2 + СО2
10NO + Р4 = 5N2 + 2Р2О5 418
16N0 + S8 = 8N2 + 8SO2
2N0 + 2SO2 = N2 + 2SO3
2N0 + 2H2 = N2 + 2H2O
Восстанавливается также солями Cr2* до NH3 в нейтральной среде и до NH2OH в кислой. Окисляется монооксид азота хроматами и перманганатами до азотной кислоты. С солями многих металлов образует нитрокомплексы, например нитропруссид Na2[Fe(NO)(CN)s]~, широко применяемый в аналитической химии реактив.
Монооксид азота получают каталитическим окислением аммиака как полуфабрикат в производстве азотной кислоты. Разработан плазмохимический метод получения монооксида азота. Энергия активации реакции получения NO высокая. Образование NO протекает по цепной реакции О2 + hv -> 2-О-; N2 + -О- -> NO + -N-; -N- + О2 —> NO + О. Монооксид азота применяют для получения NH2OH.
Сесквиоксид N2O3 (триоксид диазота) существует как индивидуальное соединение ниже -101° С. При повышенных температурах находится в равновесии с продуктами диссоциации моно- и диоксидов
Молекула плоская. Для N2O3 ц = 6,77-10'30 Кл-м. Растворяется в кислотах и эфире. С водой образует HNO2. Образуется из NO и NO2 в процессе охлаждения по реакции NH3 с As2O3, а также при пропускании электрической искры через жидкий воздух.
Диоксид азота NO2 парамагнитен; длина связи N—О 0,119 нм, угол О—N—О 134,5°; ц = 0,87-10’30 Кл-м. В процессе отщепления электрона (энергия ионизации около 942 кДж/моль) образуется нитроний-ион NOj, а при присоединении (сродство к электрону -228 кДж/моль) — нитрит-ион NOj.
В обычных условиях NO2 существует в равновесии с димером— тетраоксидом диазота N2O4 (ДД° димеризации —57,3 кДж/моль). При нормальном давлении и 40° С в такой смеси содержится 31% NO2, при 100° С — 88% NO2, выше 140° С N2O< полностью переходит в NO2. Смесь в жидком виде состоит в основном из и. 419
N2O4, а твердое вещество — чистый димер. Молекула димера также диамагнитна, имеет плоскую структуру.
Диоксид азота взаимодействует с водой и водными растворами щелочей:
2NO2 + Н2О 7* HNO3 + HNO2
2NO2 + 2NaOH -> NaNO3 + NaNO2 + H2O
С хлороводородной кислотой диоксид азота образует нитрозилхло-рид (NOC1), с серной кислотой — нитрозилсерную кислоту [(NO)HSO4], со фтором — нитрозилфторид NO2F. В смеси с сероуглеродом взрывается. Водородом в присутствии платины или никеля восстанавливается до аммиака. Поскольку NO2 и N2O4 являются сильными окислителями, уголь, сера, фосфор, органические соединения в них сгорают. В жидком N2O4 существует равновесие: N2O4 NO+ + NOj. По этой причине некоторые металлы, например цинк и медь, реагируя с жидким N2C>4, образуют безводные нитраты:
Zn + 2N2O4 -> Zn(NO3)2 + 2NO
Си + 2N2O4 -> Cu(NO3)2 + 2NO
Растворы N2O4 в безводной азотной кислоте — более сильные окислители, чем чистая HNO3.
Диоксид азота является полуфабрикатом в производстве азотной кислоты. Диоксид азота и тетраоксид диазота применяются в качестве окислителя в жидком ракетном топливе, смесевых взрывчатых веществ. Применяют их в процессах очистки нефтепродуктов от сераорганических соединений, в качестве катализаторов процессов окисления органических соединений, например бензола до фенола, метана до формальдегида.
Молекула газообразного оксида азота (V) N2Os (пентаоксид диазота) имеет неплоскую структуру (длины связи в нм):
420
Кристаллический N20s— ионное соединение (ЫО2)+(ЫОз)_. При комнатной температуре самопроизвольно разлагается на NO2 и 62, быстрое нагревание приводит к взрыву. Растворяется в CHCI3. С водой образует HNO3. В лабораторных условиях N2Oj получают взаимодействием азотной кислоты с P2Os, жидкого NO2 или Ы20з с озонированным кислородом.
Оксиды азота — физиологически активные вещества. N2O — средство для наркоза, в больших концентрациях вызывает удушение. Другие оксиды азота сильно ядовиты.
10.3. АММИАК
Физико-химические свойства аммиака, применение. Аммиак NH3, бесцветный газ с резким удушливым запахом. Жидкий аммиак — бесцветная жидкость, сильно преломляющая свет. Молекула имеет форму правильной пирамиды (рис. 10.1). Связи N—Н поляр-ны; ц = 4,85-1О’30 Кл-м (0—150° С); энергия связи N—Н 389,4 кДж/моль. Поляризуемость молекулы 22,6-10’25 см3. У атома азота имеется неподеленная пара электронов, которая обусловливает способность аммиака к образованию донорно-акцепторной и водородной связей. Существование водородных связей и значительная полярность молекул аммиака являются причиной сильного взаимодействия между ними, вследствие чего физические свойства аммиака во многом аномальны по сравнению со свойствами однотипных соединений, например РНз, SbH3 и AsH3.
Температура плавления аммиака -77,7° С; температура кипения -33,35° С. Плотность аммиака при -40° С составляет 0,790, а при -33,35° С — 0,681 г/см3. Изменения значения плотности жидкого аммиака от температуры приведены на рис. 10.2. Температура критическая 133° С; давление критическое 11,425 кПа; плотность критическая 0,235 г/см3. Данные по изменению теплоемкости в зависимости от температуры приведены на рис. 10.3. ДЯИСП ~ 23,27 кДж/моль, ДЯ°П = 5,86 кДж/моль; для газа С° = 35,63 Дж/(моль-К); ДЯ^р =-45,94 кДж/моль, = 192,66 Дж/(моль-К).
Твердый аммиак, бесцветные кристаллы с кубической решеткой (а - 0,515 нм, z = 4, пространственная группа Р2]3). В жидком аммиаке молекулы ассоциированы вплоть до критической температуры, электролитическая диссоциация практически отсутствует. Произведение концентраций [NH«] [NHj] составляет 10“22 (-33,4° С), р 8-106 Ом-см; s 25,4 (-77° С).
421
Рис. 10.1. Структура молекулы аммиака (длина связи в нм)
Рис 10.2. Изменение плотности жидкого аммиака в зависимости от температуры
, Дж/моль-град)
Рис 10.3. Зависимость теплоемкости аммиака от температуры
Растворимость аммиака в воде (%, масс.): 42,8 (0°С), 33,1 (20° С), 23,4 (40° С), 14,1 (60° С) (рис. 10.4). Плотность водных растворов (г/см3): 0,970 (8% по массе NH3), 0,947 (16%), 0,889 (32%), 0,832 (50%), 0,739 (75%). Для бесконечно разбавленного водного раствора Д//раств = 133,26 кДж/моль. Аммиак хорошо растворяется (но хуже, чем в воде) в спирте, ацетоне, хлороформе, бензоле и других органических растворителях. Образует гидраты с двумя (температура плавления -90° С), одной (-79° С) и 0,5 (-78,2° С) молекулами воды. В системе NH3—Н2О установлено существование эвтектик: ЫНз-Н2О (33,23% по массе NH3, температура плавления -100,3° С), NH3H2O + NH3 0,5H2O (55,11%, -83,3° С), NH3 H2O + NH3 (80,05%, -92,5° С). В водном растворе аммиака частично ионизирован на NH4 и ОН’, что обусловливает щелочную реакцию раствора (рК, 9,247).
Жидкий аммиак растворяет щелочные и щелочноземельные металлы, Al, Eu, Yb, Р, S, I, многие интерметаллиды и др. Растворы металлов в жидком аммиаке имеют металлическую проводимость,
Температура, t
Рис 10.4. Растворимость аммиака в воде при 10—70° С и давлении 0,5—4 ат
423
поскольку содержат ионы металла и сольватированные электроны. Они являются сильнейшими восстановителями. Растворенные в аммиаке соединения с полярной ковалентной или ионной связью диссоциируют на ионы.
В жидком аммиаке многие соединения способны отщеплять протон, а кислотные свойства проявляют в нем даже углеводы, амиды кислот и некоторые углеводороды.
Разложение аммиака на водород и азот становится заметным выше 1200—1300° С, а в присутствии катализаторов — выше 400° С. Аммиак весьма реакционноспособен. Для него типичны реакции присоединения, в частности протона при взаимодействии с кислотами. В результате образуются соли аммония, которые по многим своим свойствам подобны солям щелочных металлов.
Аммиак — основание Льюиса, присоединяет не только Н+, но и другие акцепторы электронов, например BF3 с образованием BF3 NH3. В процессе взаимодействия с солями образует амины. С жидким и газообразным аммиаком взаимодействуют щелочные и щелочноземельные металлы, образуя амины. В процессе нагревания в атмосфере аммиака многие металлы и неметаллы (Zn, Cd, Fe, Сг, В, Si и др.) образует нитриды. Жидкий аммиак взаимодействует с элементной серой по реакции
5Sg + I6NH3 = 24H2S + 4N4S4
Около 1000° С аммиак реагирует с углеродом, образуя HCN и частично разлагаясь на N2 и Н2. Аммиак реагирует с диоксидом углерода, образуя карбамат аммония:
2NH3 + СО2 -> NH2COONH4
Водород в аммиаке может быть заменен галогенами:
NH3 + ЗС12 = NCI3 + ЗНС1
Аммиак горит в атмосфере кислорода, образуя при этом воду и N2:
4NH3 + ЗО2 = 2N2 + 6Н2О
Каталитическим окислением аммиака получают NO — полуфабрикат в производстве азотной кислоты. Каталитическое окисление аммиака в смеси с метаном дает HCN<->H2O2. К2Сг2О7 и КМПО4 окисляют аммиак в водных растворах. Газообразный аммиак окисляется также бромом и хлором до азота. 424
Аммиак применяется в производстве азотной кислоты, карбамида, нитрата, карбоната, сульфата и фосфата аммония, уротропина, в качестве жидкого удобрения и хладагента.
Технология аммиака. Жидкий аммиак производится промышленностью двух сортов. В соответствии ГОСТ 6221—82 он должен удовлетворять следующим требованиям:
I сорт II сорт Содержание NH3, %.................................. 99,8 99,0
Содержание влаги, %................................ 0,2 1,0
Содержание примесей в жидком аммиаке не должно превышать: водород, метан и аргон (растворенные газы), %... — 1
масло, мг/л....................................... — 5—20
катализаторная пыль, мг/л......................... — 4—25
окалина, мг/л..................................... — 1—3
карбонат аммония, г/л............................. — 5
За основу промышленного способа получения аммиака положена реакция взаимодействия азота и водорода:
N2 + ЗН2 2NH3
Реакция является обратимой. Сдвигу равновесия вправо способствует повышение давления и понижение температуры, а также соотношение H2:N2. Как видно из приведенных на рис. 10.5 данных, тепловой эффект реакции при 29,4 МПа составляет 52,38 кДж/моль при температуре 500° С и 51,29 кДж/моль при 400° С (с учетом теплоты смешения газов). Процесс проводят в присутствии катализаторов (Fe), активированных К2О, А12О3, СаО и др. Для
Рис. 10.5. Зависимость равновесного содержания NH3 в газовой смеси (H2:N2=3) от давления при разных температурах
425
известных катализаторов скорость реакции описывается уравнением Темкина — Пыжева:
w= KlPt,t / р^2 у-кг(р^} / £
где W—наблюдаемая скорость процесса, равная разности скоростей образования и разложения A; A"i и К2—константы скорости образования и разложения А; > р^2 и — парциальное давление соответствующего газа, а = 0,5 для большинства промышленных катализаторов.
Блок-схема современного агрегата большой единичной мощности (450 тыс. т/г) представлена на рис. 10.6, из которой видно, что основными стадиями являются: 1) получение и очистка водорода из природного газа; 2) получение и очистка азота из воздуха; 3) получение и очистка синтез-газа и аммиака.
Физико-химические основы производства технологического газа. Основным сырьем для производства технологического газа являются природные газы, состоящие в основном из метана (33—99%). Содержание в них углеводородов С2 — С4 не превышает 7%, тяжелые же углеводороды от С5 и выше отсутствуют. Углеводородные газы на производство аммиака поступают после предварительного выделения из них газового бензина, состоящего в основном из парафиновых углеводородов С5 и выше. В
Рис. 10.6. Блок-схема получения аммиака:
1 — блок компрессии и сероочистки природного газа; 2 — блок паровой конверсии в трубчатых печах; 3— блок паровоздушной конверсии; 4 — котел-утилизатор; 5 — блок двухступенчатой конверсии оксида углерода; 6 — блок извлечения диоксида углерода; 7 — блок метанирования;
8 — компрессор синтез-газа; 9 — паровая турбина; 10 — блок синтеза аммиака; 11 — блок конденсации аммиака
426
качестве сырья в производстве аммиака могут быть использованы следующие газы: природный, попутный нефтяной, коксовый, метановая фракция коксового газа, газы гидрирования нефти и др.
Природные газы практически не содержат примесей сульфидных соединений. Однако газы при дальней транспортировке одорируют, добавляя этилмеркаптан до концентрации 10 мг/м3 (в расчете на серу). Поэтому в производстве аммиака во избежание отравления катализаторов сооружают сероочистные установки.
Сульфидные соединения вызывают коррозию аппаратуры, они являются сильнодействующими ядами для большинства катализаторов. Особенно чувствительны к сульфидным соединениям никелевые катализаторы трубчатой конверсии метана и низкотемпературной конверсии оксида углерода.
В производстве аммиака количество сульфидных соединений в исходном газе не должно превышать в сумме 56 мг S/м3, причем меркаптанной серы должно быть не более 36, сульфида водорода не более 20 мг/м3.
Содержащиеся в природном газе органические сульфидные соединения разнообразны: меркаптаны — метилмеркаптан CH3SH, этилмеркаптан C2H5SH; тяжелые меркаптаны, например СбН58Н; сульфидоксид углерода COS; сероуглерод CS2; тиофен C4H4S; тетрагидротиофен C4H8S; сульфиды R2S (например, диэтил сульфид С2Н5—S—С2Н5); дисульфиды R2S2 (например, диэтилсульфид С2Н5—S—S—С2Н5). Установлено, что чем выше молекулярная масса сульфидных соединений, тем труднее они удаляются из газов. Труднее всего удаляется тиофен, циклическое соединение с двумя двойными связями, легче других — этил- и метилмеркапта-ны и сульфид водорода.
Легко удаляющиеся из газов сульфидные соединения (меркаптаны, сульфид водорода, сульфидоксид углерода) практически полностью удаляют поглотителями на основе оксида цинка. Исходный газ от тяжелых меркаптанов, сульфидов и дисульфидов перед трубчатой конверсией подвергают двухстадийной сероочистке. Сначала сераорганические соединения гидрируют на катализаторе в сульфид водорода, который затем связывается поглотителем на основе оксида цинка.
Процесс гидрирования, протекающий при 350—400° С, описывают следующими реакциями:
C2H5SH + Н2 = H2S + С2Н6
CH3SC2H5 + 2Н2 = H2S + СН4 + С2Нб
С2Н5—S—S—С2Н5 + ЗН2 = 2H2S + 2СбНб
427
CS2 + 4H2 = 2H2S + CH4
COS + H2 = H2S + CO C4H4S + 4H2 = H2S + C4H10
В условиях процесса приведенные выше реакции являются необратимыми и достигается практически полное гидрирование.
Образующийся сульфид водорода при 380—410° С поглощается оксидом цинка:
H2S + ZnO = ZnS + Н2О
В интервале температур 200—500° С реакция необратима и обеспечивает высокую степень очистки газа.
В тех случаях, когда природный газ содержит более 80—100 MrS/м3 сульфидных соединений, применяют либо цеолитный, либо абсорбционный методы сероочистки. Адсорбционное сероудаление проводится на цеолитах или активированном угле. Цеолиты способны адсорбировать сульфид водорода, меркаптаны и другие сульфидные соединения и даже тиофен. Процесс очистки проводят при температуре окружающей среды. Некоторые марки цеолитов обеспечивают практически 100%-ную очистку газа от сульфида водорода и меркаптанов.
Абсорбционные методы сероочистки основаны на способности ряда жидких соединений или растворов сорбировать сульфидные соединения. Хорошо поглощают сульфидные соединения растворы аминов, щелочей, а также органические растворители (метилпирромедон, трибутилфосфат, керосин и др.). Схемы абсорбционной сероочистки включают стадии абсорбции и регенерации.
Наиболее широко применяемыми для процесса гидрирования являются алюмокобальтмолибденовые (АКМ) и алюмоникельмолиб-деновые (АНМ) катализаторы. Реакции гидрирования сераорганиче-ских соединений проходят через ряд промежуточных стадий при 350—400° С.
Сульфидирование катализаторов производят либо специально, либо оно проходит в процессе очистки. Катализаторы поглощают 4—6% серы от их массы. При этом могут образовываться следующие сульфиды: NiS, Ni3S2, Co9Sg, CoS, Сой89 и др. Обычно сера растворяется в сульфидах, образуя пирротины.
Давление ускоряет процесс гидрирования примерно в степени 0,5—0,6. Процесс проводят при давлениях от атмосферного до 40 105Па.
428
Для поглощения сульфида водорода, образующегося в процессе гидрирования сераорганических соединений, используют оксид-но-цинковые поглотители. При этом протекает процесс:
ZnO + H2S = ZnS + Н2О
Установлено, что количество вещества, поглощаемое в единицу времени таблеткой оксида цинка, пропорционально поверхности границы раздела между отработанной и неотработанной зонами таблетки. При проведении процесса очистки не удается полностью насытить серой все зерно поглотителя. Величина сероемкости находится в прямой связи с величиной его удельной поверхности, пористости и размерами гранул.
Разработан также поглотитель ГИАП-10. Адсорбционную очистку рекомендуется проводить в двух последовательно работающих аппаратах, что позволяет полностью использовать емкость поглотителя по сере и обеспечивает высокую степень очистки газа. Поглотитель ГИАП-10 можно регенерировать, после чего использовать повторно без выгрузки из аппарата. Процесс регенерации проводят путем окисления сульфида цинка кислородом при 500—550° С, при этом в газовую фазу выделяется диоксид серы.
Отечественный поглотитель ГИАП-10 обеспечивает высокую степень очистки и имеет высокую сероемкость при содержании сульфидных соединений в очищаемом газе до 40 мгБ/м3. В дальнейшем плани
руется производить поглотитель в формованном виде, что повышает его емкость на ~28%.
На сероочистку (рис. 10.7) природный газ из сепаратора 1 поступает после второй ступени компрессора 2, в котором сжимается до давления 43-103 Па. К сжатому газу с температурой около 150° С примешивают синтез-газ, отбираемый со всоса II ступени компрессора синтез-газа. Содержание водорода в смеси составляет 0,125 моль на моль природного газа или 10% (об.). Газовая смесь направляется в змее-
Рис. 10.7. Схема двухступенчатой сероочистки природного газа:
/ — сепаратор; 2 — компрессор; 3 — подогреватель;
4 — контактный аппарат; 5 — адсорберы
429
вики огневого подогревателя 3, в которых нагревается до 370—400° С за счет теплоты сжигания топливного газа в специальных горелках. Наличие такого подогревателя позволяет осуществить пуск и налаживание работы всей системы.
Нагретый газ поступает в контактный аппарат 4, в котором на алюмокобальтмолибденовом или алюмоникельмолибденовом катализаторе при 400° С и объемной скорости 1000—2000 ч’1 проходит гидрирование сераорганических соединений, в результате чего образуется сульфид водорода. Свежий катализатор поглощает сульфид водорода, т.е. происходит его сульфидирование. Количество поглощенной серы соответствует термодинамическому равновесию, практически это составляет 4—6% серы от массы катализатора. Содержание сульфидных соединений после катализатора в начальный период процесса равно нулю, а затем постепенно увеличивается, и в момент насыщения количество серы в газе до и после катализатора становится одинаковым. В процессе изменения условий очистки (температура, состав газа) возможно выделение части поглощенной серы или, наоборот, дополнительное поглощение.
Сульфид водорода поглощается оксидно-цинковым поглотителем в двух абсорберах 5. В схеме предусмотрено подключение адсорберов как на параллельную, так и на последовательную работу. Обычно адсорберы работают последовательно. При таком режиме процесс поглощения серы осуществляется в первом по ходу газа аппарате. После появления сульфида водорода в газе и когда его концентрация достигает определенной величины (0,5—3 мгБ/м3), его отключают и перегружают поглотитель. При этом второй аппарат подключают первым по ходу газа, во время перегрузки поглотителя он работает один, а затем вторым по ходу газа включают аппарат со свежим поглотителем. Такая система подключения аппаратов позволяет наиболее полно использовать поглотители и обеспечить надежную и глубокую очистку газа. Способ обеспечивает содержание сульфидных соединений в очищенном газе в пределах менее 0,5 мгБ/м3.
На газовых линиях после каждого адсорбера имеются свечи с вентилями для сброса, при необходимости, газа в атмосферу.
В промышленности применяется также способ цеолитной очистки природного газа от серы, основанный на их способности адсорбировать с высокой избирательностью полярные соединения, какими являются соединения серы, из неполярных — в данном случае природного газа.
Процесс сорбции осуществляют при температуре около 25° С. После насыщения цеолитов сульфидными соединениями их регенерируют при 300—400° С. Для сероочистки используют цеолит состава 430
Ыа2О-А12Оз-(2,5—3,5)SiO2 с удельной поверхностью от 800 до 1000 м2/г, диаметром пор 11-10'8м, насыпной массой (0,60—0,65)103 кг/м3. Статическая сероемкость цеолитов колеблется от 15 до 25%, емкость до проскока (динамическая) в зависимости от объемной скорости и давления составляет всего 1—5%.
Процесс очистки газа на цеолитах включает три стадии: абсорбцию, нагрев и регенерацию, охлаждение.
Очищенный от сульфидных соединений природный газ подвергают паровой и пароуглекислотной конверсии в трубчатых реакторах. Процесс может быть описан следующими стехиометрическими уравнениями реакций окисления:
СН4 + Н2О СО + ЗН2 -206,41 кДж (1)
СН4 + СО2 # 2СО + 2Н2 -248,28 кДж (2)
СО + Н2О СО2 + Н2 + 41,03 кДж (3)
Реакция окисления гомологов метана протекает аналогично. Взаимодействие их с водяным паром может быть в общем виде выражено уравнением
С„Н„ + Н2О # иСО + Й2- Q (4>
2
Состав технологического газа определяется равновесием параллельно протекающих независимых реакций. Выбор окислителей и соотношение (НгО + СОг) и метана в исходной смеси, влияющие на равновесие реакции, определяются требованиями к составу технологического газа.
Процесс получения технологического газа ведут в две стадии. В трубчатом реакторе происходит взаимодействие углеводородов с водяным паром. При этом выбирают такие условия, чтобы содержание остаточного метана в сухом конвертированном газе составляло 8—10%.
В шахтном реакторе проводят вторую стадию — конверсию метана водяным паром и кислородом воздуха. При этом весь кислород реагирует в свободном объеме аппарата с водородом, метаном и оксидом углерода.
Суммарная теплота, выделяющаяся в результате реакций, должна быть достаточной для протекания эндотермической реакции оставшегося метана с водяным паром по уравнению (1). Количество воздуха, подаваемого в шахтный реактор, подбирают из расчета получения технологического газа с отношением (СО + H2):N2 - 3,05 — 3,10 и 431
80
Рис. 10.8. Зависимость концентрации остаточного метана при каталитической конверсии метана водяным паром от соотношения Н2О:СН4 при различных давлениях (Па) и температуре 827° С:
1 — 1010s; 2 — 2010s; 3 — ЗО Ю5
Температура, °C
Рис 10.9. Зависимость состава сухого конвертированного газа при каталитической конверсии метана водяным паром от температурного при 30 105Па и соотношении Н2:СНг=4:1
Рис. 10.10. Зависимость состава сухого конвертированного газа при каталитической конверсии метана от температуры и содержания воды во влажном газе (р=30 105Па; Н2:СН4=4:1; воздух:СН4=1,43 в исходной смеси)
О 10 20 30
Давление 1(f, Па
Рис. 10.11. Зависимость концентрации остаточного метана от давления при каталитической паровоздушной конверсии метана (/=900° С) и отношении воздух:СН4=1,43 для различных соотношений Н2О:СН4: 7 — 2:1; 2 — 4:1
конечной температуры системы, при которой содержание остаточного метана равнялось бы заданному, т.е. 0,3—0,5% (об.).
На рис. 10.8 представлена зависимость равновесного содержания остаточного метана в конвертированном газе от количества водяного пара в исходной смеси при различных давлениях. При увеличении содержания водяного пара в исходной смеси и неизменных прочих условиях концентрация остаточного метана в конвертированном газе снижается пропорционально давлению.
На рис. 10.9 приведена зависимость равновесного состава конвертированного газа от температуры.
Данные рис. 10.10 отражают зависимость равновесного состава конвертированного газа от температуры на выходе из шахтного реактора паровоздушной конверсии.
На рис. 10.11 приведена зависимость равновесного содержания остаточного метана в конвертированном газе от давления системы при различных отношениях Н2О:СН4 в исходной парогазовой смеси перед трубчатой печью.
На рис. 10.12 представлена зависимость равновесной концентрации остаточного метана от температуры и давления после трубчатой печи и отношения пар:углерод в исходной смеси.
Данные рис. 10.13 отражают зависимость равновесной концентрации метана от температуры и давления после шахтной конверсии и отношения пар:углерод в исходной смеси перед трубчатой печью.
Скорость реакции метана и других углеводородов с водяным паром и диоксидом углерода ниже температуры 1100° С без катализатора очень низка. При 700° С и значительной длительности пребывания смеси газов СН4:Н2О = 1:2 в течение 3 ч в зоне нагрева степень превращения метана составляет лишь 3% от равновесной. В трубчатом реакторе (промышленном) конверсия метана до получения равновесной смеси должна проходить в течение 1,0—1,5 с. Поэтому с целью повышения скорости реакции углеводородов с водяным паром и диоксидом углерода процесс проводят в присутствии катализаторов.
Процесс каталитической конверсии углеводородов водяным паром является гетерогенным, протекает на границе раздела твердой (катализатор) и газообразной (смесь углеводородов, водяного пара, водорода и оксидов углерода) фаз. Началу же химических взаимодействий углеводородов с водяным паром предшествует концентрирование реагентов на поверхности катализатора (адсорбция).
Лучшей каталитической активностью в рассматриваемых реакциях обладают никельсодержащий катализатор. Содержание никеля в различных катализаторах колеблется от 3 до 40%. Изготовляют катализаторы двумя способами. Согласно первому способу, исходные соединения никеля и промоторов (активирующих и стабилизирующих добавок) смешивают с порошкообразным носителем и образующуюся 433
Избыточное давление, 105 Па Равновесная концентрация СН4 в сухом газе, % (об.)
Рис. 10.12. Зависимость равновесной концентрации остаточного метана от температуры и давления после трубчатой печи при различных отношениях пар:углерод в исходной смеси
однородную массу формуют экструзией или таблетированием в полусухом или сухом состоянии. По второму методу каталитически активные вещества наносят на сформированный и термообработанный инертный носитель путем двух-, трех- и четырехкратного пропитывания его растворами солей никеля и промоторов. В качестве носителей применяют чистый оксид алюминия либо с добавками оксидов других металлов. Катализатор-сырец подвергают термообработке при 400° С для перевода солей никеля в его оксиды.
Производительность реактора характеризуется линейной объемной или эффективной скоростью. Объемная скорость — это отношение объема сухого природного газа, подводимого в единицу времени, к объему загруженного в реактор катализатора [м3/(м3-ч) или ч'1]. Эффективная же скорость — это отношение объема исходного сухого природного газа, подводимого в единицу времени, к сумме геометрических
Рис 10.13. Зависимость равновесной концентрации метана от температуры и давления после шахтной конверсии остаточного метана с воздухом и соотношения пар:углерод в исходной смеси перед трубчатой печью
435
поверхностей гранул катализатора [м3/(м-ч) или м/ч]. Величина эффективной скорости дает возможность при расчете производительности реактора учитывать размеры и форму гранул катализатора.
Активность никелевых катализаторов конверсии достаточно велика при 750—830° С. При объемных скоростях порядка 4000 ч'1 на гранулах катализатора достигается степень превращения углеводородов, близкая к расчетной равновесной. Скорость химических реакций не лимитирует процесс конверсии. В трубчатых печах лимитирующими стадиями являются подвод реагентов и теплопередача.
Скорость подвода реагентов ограничена гидравлическим сопротивлением слоя катализатора, которое ограничивается цифрой (4—6)-Ю5 Па, а скорость теплопередачи — механическими свойствами сталей, из которых изготовлены реакционные трубы. Максимальная рабочая температура стенок реакционных труб печей не должна превышать 900—930° С, а количество теплоты потока — 25,121-Ю4 —4,187-Ю5 кДжДмЧ).
В процессе конверсии природного газа водяным паром и диоксидом углерода на никелевых катализаторах может образовываться твердая фаза — углерод (сажа). Это снижает активность катализатора, приводит к механическому разрушению гранул и соответственно росту гидравлического сопротивления слоя катализатора.
Процесс конверсии газов водяным паром или смесью его с диоксидом углерода на никелевом катализаторе при температуре в пределах 400—1000° С и соответствующем избытке (~ 1,5 раза от стехиометрических расчетов) протекает без образования углерода. Процесс конверсии газов при незначительном избытке водяного пара в смеси также проходит без образования сажи. В процессе с двойным избытком водяного пара конверсия углеводородных газов, содержащих до 7—8% (об.) олефинов, при температуре выше 700° С протекает без выделения свободного углерода. Это объясняется тем, что в этих условиях скорость реакции углерода с водяным паром на никелевом катализаторе больше, чем скорость процесса распада олефинов до углерода.
Отложившийся на поверхности катализатора углерод в виде кокса и сажи удаляют путем пропускания через катализатор водяного пара. Обработка никелевых катализаторов водяным паром или паровоздушной смесью при 400—800° С приводит к их частичной дезактивации в результате окисления активного никеля и образования его оксидов:
Ni + Н2О 4* NiO + Н2
2Ni + О2 т» 2NiO
436
В процессе нагревания никелевого катализатора, нанесенного на оксид алюминия, до 700° С и выше в среде, не содержащей восстановителей, в результате взаимодействия NiO с А120з образуется алюминат натрия NiAl2O4, относящийся к группе шпинелей. Это соединение не может служить катализатором конверсии углеводородов.
При температуре 800° С и выше в среде водорода или конвертированного газа с высоким содержанием водорода происходит медленный переход NiAl2O4 в NiO, а затем в металлический никель. Однако даже при длительном восстановлении от 30 до 50% никеля может оставаться в виде шпинели более сложного состава, например NiOAl2O3-NiAl2O4.
При содержании в значительном количестве NiO процесс восстановления окисленного катализатора проходит практически до конца как в среде водорода, так и в среде конвертированного газа с высоким содержанием водорода и оксида углерода. При этом протекают следующие реакции:
NiO + Н2 = Ni + Н2О
NiO + СО = Ni + СО2
Восстановление исходного окисленного катализатора (не содержащего шпинели) проводят непосредственно в трубчатой печи в период разогрева и пуска агрегата.
Никелевые катализаторы чувствительны также к действию сульфидных соединений, содержащихся в природном или другом углеводородном газе. Процесс отравления катализаторов происходит в результате образования на их поверхности сульфидов никеля.
С повышением температуры процесса конверсии и увеличением концентрации водорода в исходной смеси степень отравления никелевых катализаторов серой снижается. Паровая конверсия исходных углеводородов на никелевых катализаторах при 600—800° С возможна лишь при тщательной очистке исходного газа от соединений серы. Предельно допустимая концентрация (ПДК) сульфидных соединений в реакционной смеси в современных схемах производства аммиака с использованием паровой конверсии метана в трубчатых печах под давлением 30-105 Па не должна превышать 0,5 мг/м3 в пересчете на природный газ.
Никелевый катализатор, отравленный сульфидными соединениями, регенерируют при 800° С и выше, если содержание серы в исходном газе не превышает допустимого. При глубоком же отравлении катали-437
Природный газ
затора обрабатывают его при 800—900° С смесью водяного пара с водородом, а затем чистым водородом.
Промышленное получение технологического газа. Согласно схеме получения технологического газа (рис. 10.14), для производства аммиака природный газ под давлением около 12105 Па проходит через расходомер и делится на два потока. Один поток, идущий на конверсию, смешивают в соотношении примерно 10:1 с азотоводородной смесью, поступающей из отделения синтеза аммиака, и направляют в сепаратор 27 для отделения высших углеводородов, находящихся в капельно-жидком состоянии. Затем газ направляют на сжатие в двухступенчатый турбокомпрессор 29, между ступенями которого установлены воздушный холодильник 31 и сепаратор газового конденсата 27.
Из компрессора газ под давлением 46-105 Па при 130—140° С поступает в радиационно-конвективный огневой подогреватель 7, откуда выходит при 400° С. Далее его направляют в аппарат 2 гидрирования сульфидных соединений до сульфида водорода на алюмокобальтмолиб-деновом катализаторе. В двух, последовательно установленных адсорберах 3 (на схеме показан один) происходит очистка газа от сульфида водорода поглотителем на основе цинка до содержания серы не выше 0,5 мг/м3 газа. Затем природный газ смешивают в смесителе 4 с водяным паром в соотношении пар:газ, равном 4.
Полученную парогазовую смесь направляют в подогреватель 11, расположенный в конвективной части трубчатой печи, в которой температура ее повышается от 370 до 500—550° С за счет теплоты дымовых газов. Нагретая парогазовая смесь поступает в распределительные коллекторы 14, из которых через газоподводящие трубки при давлении 37-105 Па смесь попадает в реакционные трубы 15, установленные в радиационной камере трубчатой печи 17. В реакцион-
Рис. 10.14. Принципиальная схема получения технологического газа под давлением 30 105Па в агрегате синтеза аммиака производительностью 1360 т/сут:
I—питательная деаэрированная вода; II—насыщенный водяной пар (106 105Па); III—питательная вода; IV—пар (106 105Па)^ к турбине отделения синтеза аммиака. И—пар от котла первой ступени конверсии СО; VI—газ на конверсию СО; VII—пар (4010Т1а); VIII—пар (3,5105Па; IX—вода питательная деаэрированная; X—пар (7105Па); XI—паровой конденсат; XII—отопительный газ; XIII — сброс жидких углеводородов; XIV—азотоводородная смесь; 1— огневой подогреватель; 2 — реактор гидрирования сульфидных соединений; 3 — адсорбер; 4 — смеситель; 5 — турбины с противодавлением; 6 — дымосос; 7 — экономайзер; 8 — подогреватель отопительного газа; 9 — пароподогреватель; 10 — вспомогательный котел; II — подогреватель парогазовой смеси; 12 — подогреватель воздуха; 13 — паросборник; 14 — коллектор парогазовой смеси; 15 — реакционные трубы; 16— общий верхний коллектор конвертированного газа; 17 — трубчатая печь; 18— секционная труба для отвода конвертированного газа; 19 — нижний секционный коллектор; 20 — шахтный реактор; 21 — котел-утилизатор I ступени; 22 — котел-утилизатор 11 ступени; 23 — насосы парового конденсата; 24 — конденсационные турбины; 25 — воздушный фильтр; 26 — турбокомпрессор технологического воздуха; 27 — сепараторы; 28 — насос питательной воды; 29 — газовый турбокомпрессор; 30 — отстойник; 31 — воздушные холодильники; 32 — дегазатор
439
ных трубах на никелевом катализаторе происходит конверсия природного газа водяным паром. )
Необходимую для реакции теплоту получают путем сжигания ; природного газа в межтрубном пространстве печи. Из реакционных труб конвертированный газ при 800—830° С, содержащий около 10% остаточного метана, проходит через нижние секционные коллекторы 19 и секционные подъемные газоотводящие трубы 18, расположенные в обогреваемом пространстве печи. Далее газ направляется в верхний коллектор 16, футерованный теплоизоляционным бетоном и помещенный в водяную рубашку.
Далее газ поступает в смеситель шахтного реактора 20. Одновременно компрессором 26 под давлением 30-105 Па в реактор 20 нагнетается очищенный от механических примесей в фильтре 25 и нагретый в теплообменнике 12 до 500° С технологический газ. В свободном пространстве верхней части шахтного реактора 20 часть водорода, метана и оксида углерода, содержащихся в конвертированном газе, поступающем из трубчатой печи, сгорает с кислородом воздуха. При этом выделяется теплота, необходимая для эндотермической реакции оставшегося метана с водяным паром на никелевом катализаторе.
Процессы частичной паровой конверсии в трубчатой печи и окончательной паровоздушной конверсии в шахтном реакторе четко связаны материальными и тепловыми потоками. В шахтный реактор 20 подводят расчетное количество технологического воздуха, чтобы содержание остаточного метана не превышало заданное (0,3%) и технологический газ имел соотношение (СО + Н2): Nj, равное 3,05—3,10.
Из нижней части шахтного реактора 20 конвергированный газ под давлением 30-105 Па при 980—1000° С поступает в котлы-утилизаторы первой и второй ступеней. Первая — высокотемпературная — ступень 21 состоит из двух парогенераторов, расположенных симметрично по отношению к шахтному реактору 20. На вторую ступень 22 поступает технологический газ при температуре около 600° С. Шахтный реактор 20 и первая ступень котла-утилизатора 21, как и общий коллектор 16, снабжены водяной рубашкой.
Технологический газ из котла-утилизатора второй ступени при давлении 27-105 Па и температуре около 400° С с отношением пар: газ = 0,7 поступает на дальнейшую переработку в аппараты конверсии оксида углерода.
Второй поток природного газа, предназначенный для сжигания в трубчатой печи, огневом подогревателе и топках вспомогательного и пускового котлов, поступает в дегазатор 32. В этот аппарат сбрасывают также газовый конденсат природного газа из сепаратора 27 и промежуточных холодильников 31 и компрессора 29. Легкие фракции конденсата испаряются при нагреве дегазатора глухим паром и обогащают топливный газ.
440
Часть топливного газа подогревается в теплообменнике 8 и при температуре около 150° С подается в сводовые короткофакельные горелки для сжигания в межтрубном пространстве трубчатой печи, затем в дополнительные горелки, установленные в конвективной части трубчатой печи перед пароподогревателем 9, и во вспомогательные туннельные горелки, установленные в газоходах реакционной части трубчатой печи.
Другая часть топливного газа сжигается в огневом подогревателе 1, вспомогательном котле 10 и (при пуске агрегата) в топке пускового котла (на схеме не показан).
Дымовые газы покидают межтрубное пространство трубчатой печи 17 при температуре -1000° С. Физическая теплота горячих дымовых газов используется для нагрева реакционной парогазовой и паровоздушной смесей и для перегрева водяного пара. Далее к основному потоку дымовых газов подмешивают дымовые газы после вспомогательного котла 10. Общий поток дымовых газов с температурой около 700°С отдает свою теплоту для перегрева водяного пара в первой ступени пароподогревателя 9, для подогрева котловой воды в экономайзере 7 и нагрева топливного газа в подогревателе 8. Далее дымовые газы при температуре около 160° С выбрасываются в атмосферу двумя дымососами 6 через дымовую трубу. Питательная деаэрированная химически очищенная вода при 100° С подается насосом 28 под давлением 110-105 Па в экономайзер 7 и теплообменники, находящиеся в отделениях метанирования остаточных количеств оксида и диоксида углерода и синтеза аммиака. Здесь она подогревается до 300° С и поступает в паросборник 13, а затем во вспомогательный котел, встроенный в конвективную часть трубчатой печи, в котлы-утилизаторы 21 и 22, установленные после шахтного реактора, и в котел-утилизатор после конвертора оксида углерода первой ступени (на схеме не показан).
Насыщенный пар из котлов-утилизаторов под давлением 106-105 Па при 314° С возвращается в паросборник 13, проходит пароподогреватель 9 и под давлением 101-105 Па при 480° С поступает на основную турбину компрессора синтез-газа (на схеме не показан), работающую с противодавлением. Часть пара из основной турбины под давлением 41,5-105 Па при 370° С поступает в смеситель 4 для конверсии природного газа трубчатой печи. Остальное количество пара распределяется между конденсационными турбинами и турбинами, работающими с противодавлением.
Контроль температуры и давления в конвективной и радиационной камерах трубчатой печи агрегата осуществляют с помощью приборов, установленных на ЦПУ и рабочих площадках.
441
Радиационная и конвективные камеры трубчатой печи имеют точ- у ки контроля температуры по тракту дымовых газов от выхода из | трубчатой печи до дымососов. Кроме температуры контролируют | разрежение в радиационной камере печи, в топке вспомогательного ; котла, перед дымососами, причем датчик разрежения в радиационной камере печи через систему автоматического управления связан с регулятором оборотов паровых турбин дымососов.
Для повышения надежности работы агрегата и удобства управления им для температурного контроля оборудованы точки на стенках подогревателей пара, парогазовой смеси, питательной воды, технологического воздуха, отопительного и конвертированного газов на выходе из труб. Трубчатая печь оборудована приборами для измерения перепада давления в слое катализатора.
В табл. 10.2 приведен материальный баланс паровоздушной конверсии природного газа под давлением 35-1О5Па в агрегатах аммиака производительностью 1360 т/сут.
В табл. 10.3 приведены основные нормы технологического режима отделения конверсии метана агрегата аммиака.
Основным реакционным аппаратом в производстве конверсии исходного газа является трубчатая печь (рис. 10.15). Неотъемлемой частью печи является конвективная камера с расположенными в ней теплообменными поверхностями для нагрева технологических потоков и получения перегретого водяного пара. Конвективная камера объединяет все блоки трубчатой печи, образуя единый печной комплекс.
Конструктивной особенностью трубчатых печей является значительное число (от нескольких десятков до нескольких сотен) одинаковых трубчатых реакторов — реакционных труб, образующих трубные экраны. Трубы заполнены катализатором и объединены коллектором парогазовой смеси на входе и конвертированного газа на выходе. Каждая труба является самостоятельным реактором, в котором в присутствии катализатора углеводороды взаимодействуют с водяным паром за счет теплоты, подводимой через стенку трубы.
Прямоточная реакционная труба (рис. 10.16), работающая под давлением до 32-105 Па, снабжена верхним и нижним приварными фланцами с крышками. Фланцы уплотнены металлическими кольцами или специальными плоскими асбометаллическими прокладками. Верхняя и нижняя бобышки 8 и 5 сваркой соединены с газоподводящей и газоотводящей трубками. Установленный на нижнюю крышку опорный стакан 7 и приваренная к нему опорная коническая катали-заторная решетка из жаропрочной стали 12 предназначены для отвода конвертированного газа. Для этой же цели служит боковое окно над слоем бетона в стенке стакана. Нижняя часть опорного стакана заполнена теплоизоляционным бетоном для защиты нижней крышки трубы от воздействия высокой температуры. 442
Таблица 10.2. Материальный баланс паровоздушной конверсии природного газа под давлением 35405 Па в агрегатах производительностью 1360 т аммиака в сутки (на 1000 м3 природного газа)
Компонент газовой смеси Поступление Смешанный сухой газ на входе в трубчатую печь Выход газа
влажного сухого
м3 % м5 % м3 1 % а м3 1 % м3 1 %
СО2 При 0,8 годный газ 0,08 Первая с Азотоводо До 5” гупень — конв родная смесь ;рсия в тр1 0,8 гбчатой не» 0,07 и* 386,71 5,93 386,71 10,70
СО — — До 20 — — — 324,73 4,98 324,73 8,96
Н2 — — 74,40 74,40 74,40 6,76 2523,26 38,60 2523,26 69,80
n2 14,5 1,45 24,60 24,60 39,10 3,56 39,10 0,60 39,10 1,08
Аг — — 0,31 0,31 0,31 0,03 0,31 0,01 0,31 0,01
СН< 937,0 93,70 0,69 0,69 937,69 85,25 343,69 5,27 343,69 9,45
QHs 32 3,20 — — 32,0 2,90 — — — —
СзН, 11,4 1,14 — — 11,40 1,04 — — — —
С«Н1о 3,20 0,32 — — 3,20 0,29 — — — ——
ОД, 0,9 0,09 — — 0,90 0,08 — — — —
С<Л14 0,2 0,02 — — 0,20 0,02 — — — —
Н2О Всего 1000 1 Водяной г I 100 ар, 4000 м3 100 1 I 100 1100 100 2903,46 6521Д6 44,52 100 3617,80 100
СО2 Газ из 386,71 ] [рубчатой печи 5,93 вторая cryi В тень — юнвер >здух зия в шахт ном реактс ре*** 415,48 5,01 415,48 8,08
СО 324,73 4,98 — — — — 625,18 7,55 625,18 12,12
н2 2523,26 38,69 — — — — 2936,49 3530 2936,49 56,88
n2 39,10 0,60 1119,30 78,0 — — 1158,40 13,95 1158,40 22,44
Аг 0,31 0,01 14,35 1,0 — — 14,66 0,18 14,66 0,28
СНч 343,69 5,27 — — — — 14,47 0,18 14,47 0,28
Н2О 2903,46 44,52 — — — — 3150,00 37,83 — —
о2 Всего 6521,26 100 301,35 1435,0 21,0 100 — — 8314,68 100 5164,68 100
* Влажность газа после трубчатой печи равна 0,803м3 на 1м3 сухого газа.
♦♦Содержание, см3/м3.
♦♦♦Влажность конвергированного газа равна 0,61м3 на 1м3 сухого газа. Отношение (СО + Нг)^ = 3,07.
Рис 10.15. Трубчатая печь:
— коллектор парогазовой смеси; 2 — коллектор конвертированного газа; 3 — реакционные трубы; 4 — радиационные горелки; 5 — футеровка топочной камеры; 6 — дымоход
Таблица 10.3. Технологический режим конверсии метана в агрегате аммиака
Параметр Норма Примечание
Общий расход природного газа, в том числе на Не выше
технологию, м3/ч 38 000
Расход пара в линию природного газа, т/ч 110—120 Сигнализация по минимуму
Расход пара в линию воздуха, т/ч 4,5—7,0 При отключении воздуха— до 27
Расход природного газа на отопление, м3/ч Не выше 28 000
Топливный газ на потолочные горелки (смесь 26 000—
танковых и продувочных газов), м3/ч 27 000
Расход топливного газа на горелки вспомогательного котла, м3/ч Не выше 7000 При пуске компрессора азотоводородной сме- си— не выше 7500
Расход азотоводородной смеси на дозировку в Не выше Содержание водорода в
природный газ перед сероочисткой, м3/ч 6300 смеси до 10,7% ( об.)
Расход конденсата в рубашке конвертора мета- Не более Сигнализация по мини-
на 2-й ступени, т/ч 10 муму
Температура конвертированной парогазовой смеси после реакционных труб, °C 810—830 —
Температура конвертированной парогазовой Не выше
смеси на выходе из подъемных труб, °C 860
Температура конвертированной парогазовой 970—980 При пуске не выше 1010
смеси на выходе из конвертора метана, °C
Температура пара под давлением 40,5 Ю5Па, °C 370—380 —
Сигнализация по мини-
Температура пара после пароподогревателя, °C 480-485 муму (460) и по максимуму (490)
Температура конвертированной парогазовой смеси после котлов-утилизаторов 1-й ступени, °C 370—480
Температура конвертированного газа после воздушного холодильника, °C 40 —
Температура топливного газа после блока теплоиспользующей аппаратуры трубчатой печи, °C 100—110 —
Температура конвергированной парогазовой сме- 330—380 Сигнализация по миниму-
си на выходе из котла-утилизатора 2-й ступени, °C му 325 и по максимуму 390
Давление газовой смеси после сероочистки, К^Па 36-40 —
Давление технологического воздуха перед Не выше
шахтным реактором 1-й ступени, 105Па 35
Давление пара в паросборнике, 105Па Не выше 105,5 Сигнализация по минимуму 100 и по максимуму 108
Давление топливного газа на горелки основного подогревателя природного газа, 105Па 0,2—0,4 Блокировка при 0,1
Давление топливного газа на горелки печи ри- Не выше
форминга, 105Па 2,5
Давление топливного газа перед горелками Не выше
вспомогательного котла, 105Па 2,5
Разряжение в топочном пространстве трубчатой печи, Па 80—180 Сигнализация при 200
Разряжение в топочном пространстве вспомогательного котла, Па 100—200 Сигнализация 500
Разряжение в топке огневого подогревателя, Па До 500 —
Перепад давления в печи, Па » 50 —
Перепад давления в шахтном реакторе, Па » 10 —
445
Выше газоотводящего штуцера в наружной стенке реакционной
трубы приварены четыре планки 6 для крепления деталей (сепараторов), фиксирующих интервалы между реакционными трубами. К нижнему фланцу приварен стержень (палец) 2, которым реакционная труба свободно опирается на несущую балку. К верхнему фланцу приварены
две серьги 9 для подвески трубы с противовесом к траверсе, восприни-
Рис. 10.16. Реакционная прямоточная труба, работающая под давлением до 32Ю5Па:
1 — нижняя крышка; 2 — опорный стержень; 3 — фланец; 4 — корпус трубы; 5 — бобышка к газоотводящей трубке; 6 — планки для крепления распределителей (сепараторов) трубного экрана; 7 — опорный стакан катализаторной решетки; 8 — бобышка к газоподводящей трубке; 9 — серьги для подвески реакционной трубы; 10 — уплотняющие кольца; 11 — верхняя крышка; 12 — коническая катализаторная решетка;
13 — теплоизоляционный блок из бетона
мающей около 95% массы четырех труб, загруженных катализатором.
Температура дымовых газов, выходящих из топочного пространства, на 80—200° С выше, чем температура поверхности реакционных труб, т.е. колеблется в пределах 900—1100° С. За радиационной топочной камерой размещают конвективную часть, в которой располагают теплоиспользующую аппаратуру. К ней относятся подогреватели поступающих на конверсию природного газа и водяного пара, подогреватель воздуха для конверсии и для горения, а также котлы-утилизаторы для получения водяного пара.
Для осуществления практически полной конверсии оставшихся в конвертированном газе после трубчатой печи углеводородов и введения в его состав необходимого количества азота газы из трубчатых печей поступают в шахтный реактор.
На рис. 10.17 приведены схемы шахтных реакторов, согласно которым конвертированный газ из трубчатых печей по двум газопроводам подводится к верхней части реактора. Газ вводится в реактор радиально или танген
446
циально при 750—780° С. В начале конусной части реактора конвертированный газ смешивается с нагретым до 500—550° С воздухом, поступающим в реактор по специальному распределительному устройству (горелке, рис. 10.17, а). Нижняя часть горелки снабжена двумя концентрическими кольцами, с большим числом отверстий-сопел диаметром 10 мм на нижней и боковой поверхностях, через которые поступает воздух в реактор. Скорость воздуха в процессе смешения составляет 30—60 м/с, конвертированного газа—10—20 м/с.
Существуют конструкции (рис. 10.17, б), согласно которым воздух поступает в верхнюю часть реактора тангенциально или радиально врезанными соплами. В этом варианте конвертированный газ поступает в реактор сверху, одним потоком.
В конусной части реактора сгорает часть водорода и метана конвертированного газа с содержащимся в нем кислородом воздуха. При этом обеспечивается полное перемешивание реагирующих потоков, заканчивается процесс горения, температура газов в верхней части реактора составляет 1200—1300° С.
На рис. 10.17, в приведена схема шахтного реактора с нижней подачей реакционных потоков. При этом горячие компоненты конвертированного газа реагируют с кислородом воздуха в выносной топке, после которой равномерным потоком поступают в слой катализатора шахтного реактора. Линейная скорость газа в реакторе равняется 1—2 м/с, что значительно ниже скорости начала псевдоожижения слоя катализатора (~3 м/с), но вполне достаточно для обеспечения высокой производительности аппарата.
В слое никелевого катализатора протекает эндотермическая реакция оставшегося метана с водяным паром. Температура конвертированного газа на выходе из слоя катализатора при давлении в реакторе ~30 ат и остаточном содержании метана 0,25—0,30% составляет 950—1000° С. Теплота конвертированного газа используется для получения и перегрева пара. Для этого котлы-утилизаторы совмещают в одном корпусе с реактором (рис. 10.17, а, в) и выносные (рис. 10.17, б).
Из котла-утилизатора конвертированный газ с температурой -400° С направляется на конверсию оксида углерода. Процесс конверсии проводят по реакции
СО + Н2О = СО2 + Н2 + 41 кДж/моль
Главной целью процесса, естественно, является максимально полное получение целевого продукта — водорода. Оксид углерода служит балластом, накапливается в синтез-газе и удаляется при продувке вместе с водородом.
447
Рис 10.17. Шахтный реактор П ступени:
а — с встроенным котлом-утилизатором; б — с выносным котлом-утилизатором; в — с выносной камерой сгорания; 1— горелка; 2 — защитный слой катализатора; 3 — катализатор; 4 — высокоглиноземистая огнеупорная футеровка; 5 — теплоизоляционная футеровка; б — корпус реактора, 7 — водяная рубашка, 8— колосниковая решетка; 9— монтажный люк; 10— котел-утилизатор; 11— коллектор воздуха; 12 — коллектор конвертированного газа; 73 — камера сгорания; 14— горелка; 75 — огнеупорная насадка
В производствах аммиака осуществляется глубокая конверсия до остаточной концентрации оксида углерода 0,2—0,5%. Практика показывает, что при снижении концентрации оксида углерода на 0,1% количество продувочных газов снижается примерно на 10%, а производительность по аммиаку увеличивается на 0,7—1,0%.
Конверсия оксида углерода является обратимой и экзотермической реакцией. Она протекает тем полнее, чем ниже температура. При этом изменение давления влияния не оказывает. Степень конверсии оксида углерода можно повысить также путем увеличения содержания пара в реакционной смеси или удалением образующегося диоксида углерода из конвертированного газа.
Процесс между оксидом углерода с водяным паром протекает лишь в присутствии катализаторов, а температура процесса определяется их активностью.
Конверсию оксида углерода проводят в две стадии под давлением (20—30) 105Па. В первой стадии на железохромовом катализаторе конвертируется значительная часть оксида углерода и температура повышается от 320—380° С на входе в аппарат до 400—450° С на выходе из него. Вторую стадию осуществляют на медьсодержащем катализаторе при 200—250° С, и поскольку перерабатывается незначительное количество СО, температура в слое катализатора повышается лишь на 15—20° С.
Для первой ступени конверсии применяют железохромовые среднетемпературные катализаторы, содержащие около 90% оксида железа (III), 6—10% сесквиоксида хрома и 0,1—0,5% серы. В исходном состоянии катализаторы содержат неферромагнитный оксид железа (a-Fe2O3). Введенный хром внедряется в кристаллическую решетку сесквиоксида железа. В условиях конверсии оксида углерода a-Fe2O3 восстанавливается до оксида железа (II, III) и в нем растворяется се-сквиоксид хрома.
Повышения производительности железохромовых катализаторов достигают за счет повышения их удельной каталитической активности введением в смесь промотирующих добавок, лучшим из которых является калий. Модернизированные путем введения в промотора калия катализаторы имеют активность, превышающую в 2—3 раза по сравнению с существующими.
Для железохромовых катализаторов ядами являются соединения кремния, фосфора, бора, мышьяка, непредельные углеводороды и сульфидные соединения. Органические сульфидные соединения в присутствии водяного пара на катализаторе конвертируются в сульфид водорода, который реагирует с катализатором, образуя сульфид железа (II):
Fe3O4 + 2H2S + Н2 <2 3FeS + 4Н2О 4- 79,5 кДж/моль.
15 Химическая технология неорганических веществ, кн. 1
449
Реакция обратима, и в зависимости от соотношения пар:газ и температуры возможно как поглощение серы, так и выделение ранее поглощенной серы в парогазовую смесь. Сульфид железа также катализирует реакцию конверсии, но по сравнению с РезО4 менее активен.
На активность катализатора оказывает влияние температурный режим процесса. С достижением 530° С начинается снижение каталитической активности, а при 630° С активность падает более ускоренными темпами.
Перед эксплуатацией катализатор восстанавливают реакционной газовой смесью при объемной скорости 150—200 ч"1. Процесс восстановления железохромового катализатора начинается при температуре 170—180° С и заканчивается при 320° С. Нагрев выше 500° С не допускается, поскольку это приводит к изменению фазового состава катализатора.
Процесс восстановления сопровождается выделением теплоты:
ЗРе20з + Н2 ;2 2Ре2О4 + Н2О + 9,6 кДж/моль,
ЗРе20з + СО ^2 2РезО4 + СО2 + 50,6 кДж/моль.
При отсутствии водяного пара или при его недостатке в реакционной системе возможно перевосстановление катализатора до образования металлического железа, которое является катализатором сильно экзотермической реакции метанирования:
СО + ЗН2 ^2 СН4 + Н2О + 206,4 кДж/моль.
С целью предотвращения этого процесса в газе поддерживают не менее 15% водяного пара.
В процессе с соотношением пар:СО < 1 на железохромовом катализаторе протекает реакция:
2СО <2 С + СО2 + 172,5 кДж/моль.
Образующийся при этом углерод откладывается на поверхность катализатора, что резко снижает его активность.
При содержании оксида углерода в газе-восстановителе параллельно с восстановительным процессом протекает и процесс конверсии:
СО + Н2О ^2 Н2 + СО2 + 41 кДж/моль.
В результате выделяется теплота. Известно, что конверсия 1% СО приводит к повышению температуры в пределах 8—10° С.
450
В процессе восстановления катализатора из него выделяется сульфид водорода. Значительная часть содержащейся в катализаторе серы связана в FeSO4, реагирующей с водородом и водяным паром:
FeSO4 + 4Н2 -> FeS + 4Н2О
3FeS + 4Н2О Fe2O4 + 3H2S + Н2 — 75 кДж/моль.
Для исключения процесса отравления низкотемпературного катализатора (НТК) обессеривают железохромовый катализатор. Стадия обессеривания протекает эффективно при высокой температуре, объемной скорости и соотношении пар:газ. Более эффективной из этих операций является увеличение соотношения пар:газ. Процесс проходит с высокой скоростью при 400° С и соотношении пар:газ ~3. В этом варианте в крупном агрегате процесс обессеривания можно провести за 1—1,5 сут. В производственных условиях температуру обычно устанавливают на уровне 340—380° С, а соотношение пар:газ составляет 1,4—1,5, при этом процесс завершается за 2—2,5 сут. На НТК допускается подавать газ с содержанием не более 0,5 мгБ/м3.
Для устойчивой работы железохромового катализатора нельзя допустить попадания таких примесей, как фосфор, кремний, бор. Концентрация в газе сажи и пыли не должна быть более 1 мг/м3, соле-содержание в паре и конденсате должно быть не более 2,5 мг/л, а содержание кремния и хлора —0,1 мг/л.
Температура в слое катализатора не должна быть выше 530° С, поскольку при более высоких температурах происходит спекание катализатора и снижение его активности.
Вторая ступень конверсии оксида углерода проводится на низкотемпературном катализаторе НТК-4, химический состав которого состоит из следующих составляющих: СиО — 54±3; Сг20з —14,0±3,0; А120з —19,6±2,0; ZnO —11,0±1,5; Na2O<0,2; S<0,2; CKO,02; содержание влаги не более 37±1 и потери при прокаливании при 1000° С не должны превышать 13%. Насыпная плотность катализатора—1650—1750 кг/м3.
Ядами для низкотемпературных катализаторов конверсии оксида углерода являются соединения серы и хлора, а также соединения фосфора, мышьяка, кремния и ненасыщенные углеводороды.
Схема процесса конверсии оксида углерода приведена на рис. 10.18, согласно которой поступающий в конвертор первой ступени газ имеет следующий состав: 57% Н2, 22—23% N2, 12,0—12,5% СО, 7,5—8,0% СО2, 0,25Аг и 0,35% СН4. Соотношение пар:газ составляет (0,57—0,66): 1. Наибольшее донасыщение газа паром при необходимости осуществляют в увлажнителе 7 за 15. 451
Рис 10.18. Схема двухступенчатой конверсии оксида углерода:
1 — увлажнитель; 2—конвертор первой ступени;
3— котел-утилизатор; 4 — подогреватель газа; 5 — конвертор II ступени
счет впрыска в газ конденсата. Количество подаваемого конденсата регулирует также температуру на входе в конвертор.
В конверторе первой ступени 2 процесс проводят на железохромовом катализаторе при объемной скорости 2000—3000 ч~* и температуре на входе 320—380° С. По мере прохождения через слой катализатора температура парогазовой смеси повышается до 400—450° С. Из конвертора газ поступает в котел-утилизатор 3, в котором его теплоту используют для получения пара давлением (105—106)-105Па. Далее газ поступает в теплообменник 4, где осуществляется нагрев
газа, поступающего на процесс метанирования. При этом парогазовая смесь охлаждается до 210—220° С и направляется в конвертор второй ступени 5. Газ имеет следующий состав: 57—61% Н2, 20,5—21,0 N2, 2,5—4,0% СО, 15—17% СО2, 0,23% Аг и 0,35% СН4. На низкотемпературном катализаторе (НТК) при 210—250° С и объемной скорости 2000—3000 ч-1 происходит конверсия оксида углерода до остаточного содержания 0,2—0,55% СО.
Температуру на входе газа во вторую ступень конверсии регулируют путем байпасирования части газа мимо котла-утилизатора. Имеются также схемы, в которых регулирование температуры осуществляется впрыском конденсата в газ, поступающий в конвертор.
После конверсии оксида углерода второй ступени конвертированный газ с температурой 240—275° С проходит охладитель, в котором за счет впрыска конденсата охлаждается до 180° С и поступает в газовые кипятильники моноэтаноламинной очистки. Затем он в сепараторе отделяется от сконденсировавшейся в кипятильниках воды и охлаждается в теплоиспользующих установках от 140—180° С до 107° С, после чего снова охлаждается в подогревателе, где отдает свое тепло очищенному от диоксида углерода газу (после моноэтаноламинной очистки), идущему на метанирование. Конвертированный газ окончательно охлаждается до 35—45° С в воздушном холодильнике, после чего он про-452
ходит сепаратор-влагоотделитель, в котором освобождается от конденсата и направляется на очистку от диоксида углерода.
Для повышения выхода аммиака и улучшения эффективности процесса очистки рекомендуется на стадии охлаждения газа после НТК включать в схему установку селективного окисления оксида углерода (рис. 10.19). Процесс заключается в том, что оксид углерода, оставшийся в газе после НТК, селективно окисляется на специальном катализаторе, содержащем металлы платиновой группы. Остаточное содержание оксида углерода составляет при этом несколько сотых долей процента. Образующийся диоксид углерода удаляется. Применение селективной очистки увеличивает срок службы НТК конверсии оксида углерода.
Реакция селективного окисления протекает в интервале температур 60—150° С в присутствии незначительного количества водяных паров. Метод применяют при содержании углерода в газе от 0,1 до 1,0%.
Для проведения процесса конверсии оксида углерода применяют аппараты двух типов — с аксиальным и радиальным ходом газа. В аксиальных реакторах парогазовая смесь проходит параллельно оси аппарата, а в радиальных конверторах — перпендикулярно оси аппарата.
Основными аппаратами аксиального и радиального типов, которые используются на стадиях как сероочистки, так и метанирования, являются полочные и шахтные реакторы, конверторы оксида углерода и каталитические аппараты.
Рис 10.19. Схема установки окисления оксида углерода:
1 — теплообменник; 2 — сепаратор-холодильник; 3 — каталитический реактор
Рис 10.20. Полочный конвертор оксида углерода:
1 — входной штуцер; 2 — катализатор; 3 — решетка; 4—выходной штуцер; 5— разгрузочный люк; 6 — загрузочный люк
453
Полочный конвертор, применяемый в настоящее время в крупных агрегатах производства аммиака, приведен на рис. 10.20. В аппарате катализатор располагают слоями на полках, размещенных в цилиндрическом корпусе. Полки могут работать по газу как последовательно, так и параллельно. В аппарате на опорную балочную конструкцию укладывают колосниковую решетку 3, покрываемую металлической сеткой с размерами ячеек несколько меньшими частиц катализатора. Поверх сетки насыпают слой керамической или металлической насадки (кольца Рашига, седла Инталокс и т.п.), которая препятствует забиванию сетки частицами катализатора, а затем загружают катализатор. Сверху также укладывают металлическую сетку, под которой размещают слой насадки, который предохраняет катализатор от солей, содержащихся в паре. При этом соли откладываются преимущественно на элементах насадки. Слой насадок выравнивает также температуру и скорость потока по сечению аппарата.
Полочный конвертор, применяющийся в крупных агрегатах производства аммиака, имеет диаметр 3,8 м. Катализатор располагается на двух полках, работающих параллельно, высота слоя катализатора составляет 4 м, а объем разовой загрузки — 45,5 м3. Аппарат теплоизолирован снаружи.
Шахтные реакторы являются разновидностью полочных аппаратов (рис. 10.21), катализатор располагается в один слой, в нижней части аппарата уложены шары 6 керамические, мулитовые или корундовые диаметром около 20 мм, высота слоя шаров достигает 1,5 м. На слой шаров укладывают металлическую сетку 3, а затем катализатор 7. Шарами заполняют также патрубки для выгрузки катализатора. На выходном патрубке 5 устанавливают сетку, поверхность катализатора делают ровной, на нее укладывают металлическую сетку, а затем слой колец Рашига 8 высотой 250 мм и прижимную решетку. Аппарат снаружи имеет теплоизоляционный слой. В промышленных агрегатах производительность аммиака 1360 т/сут, конвертор шахтного типа имеет высоту 12 м, диаметр 5 м. Толщина слоя катализатора составляет 3—4 м.
В процессе работы полочных и шахтных аппаратов существенно возрастает сопротивление. Например, в шахтных реакторах исходное сопротивление составляет 0,5-105Па, а к моменту выгрузки катализатора увеличивается до 2,5—3,0, а иногда и до 4-10‘5Па. Это приводит к необходимости замены катализатора, который по показателям своей активности мог бы еще эксплуатироваться. Это объясняется наличием пыли и разрушенных гранул в исходном катализаторе. Поэтому перед загрузкой каждую партию подвергают гранулометрическому анализу.
По данным ГИАП, гидравлическое сопротивление катализатора, в котором содержалось до 7% пыли и мелочи, а также обеспы-454
ленных образцов катализаторов превышало почти в 30 раз. Поэтому при содержании пыли и мелких фракций более 1% катализатор рассеивают на специальной установке, оборудованной набором вибрирующих сит. Загрузку катализатора производят с помощью автоматически опорожняющегося бункера при высоте падения гранул не более 1,0—1,5 м.
В радиальных реакторах (рис. 10.22) катализатор находится в коаксиально расположенных центральной трубе 4 и наружной обечайке, рабочие поверхности которых перфорированы и покрыты сеткой со стороны катализатора. Между корпусом 2 реактора и наружной обечайкой перфорированной трубы 5 образуется кольцевой канал, по которому либо отводят продукты реакции, либо вводят сырье. Следовательно, в радиальном реакторе имеет место сложное движение потока одновременно в осевом направлении (по
Рис 10.21. Шахтный конвертор оксида углерода:
I — входной штуцер; 2 — термопарный карман; 3 — металлические сетки; 4 — разгрузочный люк; 5 — выходной штуцер; 6 — керамические шары; 7 — катализатор; 8 — металлические кольца Рашига; 9 — загрузочный люк
Рис 10.22. Радиальный двухкорзиночный конвертор оксида углерода:
1 — загрузочный люк; 2 — корпус; 3 — катализатор; 4 — центральная перфорированная труба; 5—наружная перфорированная труба; 6 — разгрузочный люк; 7—катализатор для компенсации усадки
455
кольцевому каналу и центральной трубе) и в радиальном — через слой катализатора 3. Радиальные реакторы могут быть двух типов: Z- и П-образные. В Z-образном реакторе газ в центральной трубе и кольцевом зазоре движется в одном направлении, в П-образ-ном — в противоположных. Число катализаторных корзин в реакторе обычно не превышает трех.
Одним из достоинств конструкции радиального реактора является малое гидравлическое сопротивление, что позволяет использовать мелкозернистые катализаторы и достичь высоких объемных скоростей газа.
Горизонтальные реакторы включают элементы как полочных, так и радиальных аппаратов (рис. 10.23) и имеют соответственно промежуточные значения гидравлических сопротивлений и степени полезного использования объема. Преимуществом горизонтального реактора является возможность использования коротких слоев катализатора, для которых меньшее влияние оказывает усадка катализатора. Однако они могут работать лишь при ограниченном перепаде температур на слое катализатора. Поэтому их применяют обычно для низкотемпературной конверсии оксида углерода.
Для горизонтальных реакторов характерны те же особенности движения и распределения потоков, что и для радиальных аппаратов.
Очистка конвертированного газа от диоксида углерода. В производстве аммиака очистку конвертированного газа от диоксида углерода осуществляют хемосорбцией водным раствором моноэтанолами-на (МЭА) или водным раствором карбоната калия, активированным диэтаноламином (ДЭА).
Рис 10.23. Многопоточная схема МЭА-очистки с двумя потоками регенерированного раствора и тремя потоками насыщенного раствора:
1 — абсорбер; 2 — холодильники; 3 — центробежные насосы; 4, 8 — теплообменники; 5 — конденсатор; 6 — регенератор; 7 — кипятильник; 9 — испаритель
456
В процессе очистки конвертированного газа 19—21%-ным раствором моноэтаноламина протекают следующие реакции:
2NH2CH2CH2OH + Н2О + СО2 т* (NH3CH2CH2OH)2CO3
(NH3CH2CH2OH)2CO3 + Н2О + СО2 2NH3CH2CH2OHHCO3
Процесс проводят при температурном интервале 35—60° С. Раствор регенерируют снижением давления и повышением температуры, в результате выделяются вода и диоксид углерода.
Реакцию между диоксидом углерода и МЭА следует рассматривать как схему, устанавливающую возможность получения в качестве конечного продукта как карбоната, так и гидрокарбоната моноэтаноламина.
В процессе поглощения диоксида углерода повышается степень карбонизации раствора (а), которая выражается в молях СО2 на моль МЭА. В производстве в процессе извлечения СО2 из газа раствором МЭА с концентрацией 2,5—3,3 моль/м3 под давлением 2,45—2,94 МПа может быть достигнута степень карбонизации (а = 0,65—0,75). Содержание СО2 в газе после очистки составляет 50—300 см3/м3. Практически установлено, что, увеличивая концентрацию МЭА, можно уменьшить количество циркулирующего раствора.
Процесс очистки газа от диоксида углерода проводится по многопоточной схеме (см. рис. 10.23), согласно которой исходный конвертированный газ (205 000 м3/ч), содержащий 16—18% СО2, подается в нижнюю часть абсорбера 1. Абсорбер разделен на две секции— нижнюю и верхнюю. Газ проходит нижнюю секцию, в которой осуществляется грубая очистка (до содержания 1—2% СО2), а затем верхнюю, после чего остаточное содержание СО2 составляет не более 0,03% (об.). В верхней части аппарата очищенный конвертированный газ проходит колпачковые тарелки, орошаемые флегмой, и сепарирующее устройство. С целью уменьшения брызгоуноса в схеме предусмотрен брызгоуловитель-сепаратор (на схеме не показан). Очищенный газ направляется на метанирование.
Верхняя секция абсорбера 1 орошается глубоко регенерированным раствором, в котором он насыщается диоксидом углерода (от 10—20 до 65 г), смешивается с грубо регенерированным раствором, идущим из регенератора.
Насыщенный раствор с содержанием СО2 80—100 г/л и температурой не выше 60° С в количестве 1100—1150 м3/ч выходит из нижней секции абсорбера и делится на три потока.
Первый поток — холодный байпас (около 10% — до 100 м3/ч) направляется непосредственно на верх регенератора 6. Второй поток (475—540 м3/ч, ~45%) проходит трубное пространство теплообменни-457
ка 4, в котором нагревается до 95—100° С за счет теплоты грубо регенерированного раствора, дросселируется и поступает в регенератор. Третий поток (475—540 м3/ч, -45%) проходит трубное пространство теплообменника 8, нагревается до ПО—115° С за счет теплоты грубо регенерированного раствора, дросселируется и поступает в трубное пространство испарителя 9, в котором за счет теплоты грубо регенерированного раствора происходит нагрев и частичная десорбция СО2 и паров воды. После испарителей насыщенный раствор поступает в регенератор.
Регенератор разделен на две секции. В верхней секции происходит регенерация (десорбция) насыщенного раствора до содержания СО2 не более 50 г/л за счет теплоты парогазовой смеси, поступающей из нижней секции регенератора. Затем раствор делится на два потока. Первый поток — грубо регенерированный раствор (500—600 м3/ч) с температурой 115—120° С — поступает в межтрубное пространство испарителя 9, в котором охлаждается до ПО—И5° С насыщенным раствором, пройдя теплообменники и воздушный холодильник 2, поступает на орошение нижней секции абсорбера. Другой поток грубо регенерированного раствора поступает в нижнюю часть регенератора для более тонкой регенерации. Окончательная десорбция происходит в процессе кипячения его в выносных кипятильниках 7 до остаточного содержания СОг в раствор 10—12 г/л. Глубоко регенерированный раствор из куба регенератора с температурой 125—130°С поступает в теплообменники 8, охлаждается в воздушном холодильнике 2, а затем с температурой около 70°С поступает на орошение верхней части абсорбера.
Если теплоты для регенерации раствора оказывается недостаточно, ее количество может быть увеличено за счет использования дополнительных газовых и паровых кипятильников.
Газы десорбции — диоксид углерода с небольшим содержанием СО, СН4 и N2 — подаются в цех переработки или выбрасываются в атмосферу. Содержание горючих веществ в газах десорбции не должно превышать 2% (об.). Парогазовая смесь охлаждается в воздушном холодильнике, а затем поступает в сборник флегмы (на схеме не показан), в котором происходит отделение газа от конденсата-флегмы, возвращаемой в цикл для поддержания баланса воды в системе.
С целью очистки раствора МЭА, постоянно находящегося в цикле, от продуктов разложения, осмоления и окисления моноэтаноламина, вызывающих эрозию аппаратуры и способствующих увеличению его вспениваемости, предусмотрена разгонка части циркулирующего раствора в кипятильнике 7 под давлением на 0,01—0,02 МПа выше, чем в регенераторе.
Разгонку осуществляют в две стадии (полунепрерывный процесс). На разгонку в межтрубное пространство кипятильника 7 из глухой 458
тарелки нижней секции регенератора 6 поступает 5—10 м3 раствора МЭА в час, туда же предварительно закачивают 1,0—1,5 м3 42%-ного раствора гидроксида натрия для нейтрализации кислых продуктов разложения моноэтаноламина. Раствор гидроксида натрия готовят в растворителе NaOH и подают в смоловыделитель центробежным насосом. В трубки смоловыделителя поступает греющий пар. Раствор кипит при температуре от 120 до 145° С. Пары МЭА и воды из смоловыделителя поступают в куб регенератора. В процессе повышения температуры раствора в смоловыделителе до 143—145° С и содержания смолистых веществ в нижней части аппарата до 280—400 г/л прекращают подачу раствора в аппарат разгонки и переходят ко второй стадии — отгонке МЭА с водяным паром. Процесс проводят до содержания МЭА в парах не более 0,5%, в кубовом остатке — 15—20% (масс.). В процессе упарки контролируют уровень раствора в аппарате и наличие смолистых веществ в парогазовой смеси. Оптимальный расход пара, подаваемого в трубное пространство, составляет около 3 т/ч. По окончании выпаривания кубовой остаток сливают, аппарат промывают и снова включают в работу. Продолжительность первой стадии разгонки зависит от скорости накопления смол в смоловыделителе и составляет 10—20 сут. Продолжительность второй стадии составляет одни сутки.
Конвертированный газ очищают от диоксида углерода раствором карбоната калия. Способ основан на взаимодействии диоксида углерода с карбонатом калия по суммарному уравнению:
К2СО3 + СО2 + Н2О 2КНСО3
Скорость взаимодействия реагентов практически полностью лимитируется скоростью медленно протекающей реакции в жидкой фазе:
СО2 + он- нсо3
Недостатками процесса являются: малая растворимость карбоната калия в воде (особенно гидрокарбоната) при обычной температуре; низкая скорость процесса абсорбции, которая требует значительной циркуляции раствора, что обусловливает увеличение габаритов аппаратов. Кроме того, равновесное парциальное давление диоксида углерода над растворами карбоната калия относительно велико, поэтому не удается достичь тонкой очистки газа (0,08—0,15% СОг в газе после очистки). Преимуществами способа являются: применение недорогостоящего карбоната калия и сравнительно низкий расход тепла на регенерацию.
Значительное распространение получили методы очистки газа горячими растворами карбоната калия, активированными различными добавками.
459
Диэтаноламин (ДЭА) — наиболее распространенный в промыш- । ленности активатор поташной очистки является одновременно хорошим абсорбентом по отношению к диоксиду углерода и ускорителем процесса. Ускоряющее действие ДЭА возрастает по мере насыщения раствора, т.е. в нижней части абсорбера.
В действующих агрегатах производства аммиака применяют две разновидности процесса поташной очистки: «Карсол» и «Бенфилд». В процессе «Карсол» для абсорбции применяют 25—28%-ный раствор карбоната калия, содержащий 1,8% ДЭА. В процессе «Бенфилд» — более концентрированные растворы: 29—30% К2СО3 и 2,9% ДЭА.
Процессы абсорбции и регенерации ведут при 65—115° С. Абсорбцию осуществляют по двухпоточной схеме. Большую часть (80%) грубо регенерированного раствора подают в среднюю часть абсорбера при 100—103°С, остальная часть при 65—70°С поступает в верхнюю часть абсорбера. Это позволяет достичь содержания в газе 0,10—0,15%, а иногда и 0,05—0,08% СОг.
Насыщенный раствор при 106—110° С сначала поступает в турбину, а затем в регенератор. Регенерацию проводят по схеме с разделением потоков регенерированного раствора.
Метанирование оксидов углерода. После стадии очистки от диоксида углерода газ содержит СО и СОг, которые в смеси с кислородом и водяным паром являются ядами для катализатора синтеза аммиака. Поэтому в процессе синтеза аммиака проводят тонкую очистку газа от оксидов углерода методом каталитического гидрирования.
После абсорбционной очистки от диоксида углерода газ содержит 0,3—0,7% СО и 0,02—0,10% СО2. В соответствии с существующими регламентами очищенный газ должен содержать СО не более 20,0%, СО2 —0,5%.
Процесс гидрирования оксидов углерода протекает по следующим реакциям:
СО + ЗН2 ^2 СНд + Н2О + 206 кДж/моль,
СОг + 4Н2 ^2 СНд + 2НгО +165 кДж/моль.
Приведенные реакции обратимы и сильно экзотермичны, протекают в присутствии катализаторов. Наиболее широко применяемыми в промышленности катализаторами являются никелевые, стабилизированные оксидом алюминия. Они могут быть активированы оксидом хрома. Никельалюминиевые катализаторы активны в интервале 350—400° С, а никельалюмохромовые — в интервале 200—250° С. Присутствующий в газе кислород на этих катализаторах гидрируется полностью.
460
В процессе метанирования применяют катализаторы НКМ-1, ТО и С-13-4. ТО в отличие от остальных катализаторов содержит в своем составе оксид хрома (10—20% Сг2О2). Его получают из основного карбоната никеля, сесквиоксида хрома и гидроксида или активного оксида алюминия.
В крупных агрегатах производства аммиака процесс метанирова-ния проходит при объемной скорости 4000 ч’1, температуре 310—350° С, под давлением (24—25)-105Па. При объемной скорости до 8000 ч"1 обеспечивается тонкая очистка. Для катализатора НКМ-1 эта величина составляет -6000 ч'1.
Ядами для никелевых катализаторов метанирования являются соединения серы, мышьяка, хлора. Катализатор, содержащий 0,1—0,2% серы, является полностью дезактивированным.
Перед началом эксплуатации катализаторы метанирования восстанавливают. Процесс восстановления происходит одновременно с разогревом метанатора перед пуском и занимает 8—10 ч. Катализатор восстанавливают технологическим газом, поступившим после НТК и очистки от СО2. Процесс начинается при 200° С и происходит по реакциям:
NiO + Н2 = Ni + Н2О - 2,55 кДж/моль,
NiO + СО = Ni + СО2 + 30,3 кДж/моль.
После восстановления катализатора начинается реакция метанирования. Процесс восстановления проводят при давлении (24—25)-105 и объемной скорости 3000—4000 ч'1. При этом давление процесса постепенно повышают при температуре около 200° С.
Катализатор выдерживают 20—24 ч при 400° С, а затем снижают температуру до рабочей. Во избежание образования карбонила никеля [Ni(CO)4] разогрев катализатора до 200° С ведут при давлении не более (3,5—4,0)-105Па. При этом исходный газ должен содержать меньше оксида углерода.
Восстановленный катализатор метанирования пирофорен, поэтому перед выгрузкой он подвергается окислению или пассивации. Единовременная загрузка катализатора в крупном агрегате производства аммиака составляет 35—40 т.
Схема процесса метанирования представлена на рис. 10.24. Установка метанирования обычно устанавливается после аппаратов очистки конвертированного газа от диоксида углерода. Согласно схеме, газ после очистки от диоксида углерода проходит через сепаратор (на схеме не показан), в котором от него отделяются капли абсорбента. После сепаратора газ подогревается в двух последовательно располо-
461
Теплоноситель
Рис 10.24. Принципиальная схема установки метанирования:
1, 2 — теплообменники; 3 — влагоотделитель; 4 — воздушный холодильник; 5, 6 — холодильники; 7 — метанатор
женных теплообменниках 7 и 2 и направляется в метанатор 7. Очистку осуществляют при 350—380° С, объемной скорости 4000—5000 ч'1 и давлении до 30-105Па. Линейная скорость газа в аппарате составляет 0,3—0,4 м/с.
Для подогрева газа до температуры реакции в теплообменниках 1, 2 в зависимости от энерготехнологической схемы всего агрегата синтеза аммиака используют различные теплоносители.
Теплоту очищенного газа после метанатора 7 используют для подогрева газа или воды (в зависимости от схемы производства аммиака). В отечественном производстве аммиака теплота реакции метанирования используется для подогрева питательной воды.
Очищенная азотоводородная смесь из метанатора с температурой 380° С проходит межтрубное пространство подогревателей воды 6, где охлаждается до 130° С, и межтрубное пространство подогревателя глубоко обессоленной воды 5, где охлаждается до 60—90° С. Окончательное охлаждение и конденсация водяных паров, образовавшихся в результате реакции метанирования, происходят в аппарате воздушного охлаждения 4. После отделения газового конденсата во влагоотделите-ле 3 азотоводородная смесь (АВС) направляется на компрессию.
Синтез аммиака. Процесс синтеза аммиака идет по реакции
N2 + ЗН2 # 2NH3, ДЯ£8 = -92,52 кДж, сопровождается уменьшением числа молекул, вследствие чего разность энтропии продуктов реакции и исходных реагентов является величиной отрицательной: S?9s = -198,6 кДж/(моль-град). Данное об-462
Концентрация NH3, %(об.) Тепловой эффект, кДж/моль NH3
Давление, МПа
Рис. 10.25. Значения теплового эффекта реакции образования аммиака при 500° С и давлениях до ЮОМПа
Рис. 10.26. Равновесная концентрация аммиака в азотоводородной смеси (N2+3H2) как функция давления и температуры
Рис 10.27. Концентрация аммиака в газе при ЗОМПа и 500° С как функция объемной скорости
стоятельство и то, что количество теплоты, выделяющееся в процессе образования аммиака (рис. 10.25), невелико (92,52 кДж на 2 моль NH3), обусловливает практическую невозможность синтеза аммиака при атмосферном давлении. Данная реакция может протекать слева направо лишь при AG<0.
Из уравнения свободной энергии процесса AG = &H-TAS определяем температуру процесса Г, при которой AG = 0:
92,52 - (-198,4)7= 0,
отсюда Т = 466 К (193° С). Следовательно, синтез аммиака при атмосферном давлении осуществим лишь с заметными степенями превращения при температуре ниже 193° С.
С целью проведения реакции в нужном направлении процесс синтеза аммиака в производстве проводится под давлением. Однако и при высоком давлении, и повышенных температурах азот и водород вступают в реакцию лишь частично. Поэтому после конденсации образовавшегося аммиака азотоводородная смесь возвращается в колонну синтеза, т.е. осуществляется циркуляция реагентов.
Равновесная концентрация аммиака в газе в зависимости от температуры и давления показана на рис. 10.26. С уменьшением продолжительности контакта газа с катализатором (с увеличением объемной скорости) концентрация аммиака в газе снижается (рис. 10.27).
Практический выход (отношение фактического содержания NH3 в газе к теоретически возможному, т. е. к равновесному содержанию аммиака при данных температуре и давлении) также уменьшается с ростом объемной скорости (рис. 10.28).
Несмотря на уменьшение содержания аммиака в газе и снижение эффективности конверсии с увеличением объемной скорости, удельная производительность катализатора (в кг NH3 на 1 л катализатора, или в т NH3 на 1 м3 катализатора) возрастает с увеличением объемной скорости (рис. 10.29). Это объясняется тем, что содержание аммиака и эффективность конверсии снижаются значительно медленнее, чем увеличивается объемная скорость.
Удельный расход газа в производстве аммиака в значительной мере зависит от содержания инертных примесей (аргона, метана) в поступающем на синтез свежем газе, а также от их концентрации в циркулирующей азотоводородной смеси (рис. 10.30).
В описании кинетики и механизма процесса синтеза аммиака наибольшее распространение получило уравнение, предложенное Темкиным и Пыжевым. Они исходили из представления о неоднородной поверхности и о том, что лимитирующей стадией является 464
Рис 10.28. Коэффициент эффективности конверсии при различных объемных скоростях
Рис 10.29. Удельная производительность катализатора синтеза аммиака при 300 ат и 500° С как функция объемной скорости
Рис 10.30. Концентрация аммиака в газе на выходе из колонны синтеза при различном содержании инертных газов в азотоводородной смеси (N2+3H2) при объемной скорости 30 000 ч~‘
адсорбция азота, причем на поверхности находится лишь азот, лимитирующей стадией процесса разложения аммиака — десорбция азота. При этом количество атомов азота на поверхности катализаторов больше, чем любых адсорбированных азотсодержащих соединений, ни водород, ни аммиак не влияют на скорость адсорбции азота, а энергии активации адсорбции Ея и десорбции Еа линейно уменьшаются с увеличением степени заполнения поверхности:
Ч
2 PNH.
^2
I
PNH3
(5)
где со — общая скорость процесса; к\, кг — константы скоростей прямой и обратной реакции; ры, Рцг, — парциальные давления во-
дорода, азота и аммиака.
Значение а характеризует степень покрытия поверхности катализатора азотом в условиях синтеза аммиака. Для железных катализаторов его принимают равным 0,5.
Уравнение (5) применимо только в области, не слишком далекой от равновесия. Из уравнения (5) следует, что максимальная скорость реакции не соответствует эквивалентному составу газовой смеси. В начальной стадии процесса, когда содержание аммиака значительно меньше равновесного и реакция разложения аммиака может не учитываться, максимальная скорость определяется максимумом произведения р^р£- Отсюда следует, что максимальная скорость в начальной стадии' процесса соответствует составу Рнг/р^ =1,5. По мере приближения выхода к равновесному оптимальный состав приближается к составу /р^ = 3, поскольку по закону действующих масс
равновесный выход достигает максимума при эквивалентном составе.
Поскольку реакция синтеза аммиака обратимая, экзотермическая, то для нее оптимальной является убывающая последовательность температуры. В начале зоны катализа высокая температура обеспечивает наибольшую скорость реакции, а по мере увеличения содержания аммиака в газовой фазе и приближения к состоянию равновесия следует снижать температуру в зоне катализа.
Для обеспечения максимальной производительности реактора необходимо реализовать оптимальные температуры по длине каталитической зоны для каждого поперечного сечения, обеспечивающие максимальную скорость процесса в данном сечении.
Основываясь на вышеуказанном и считая, что температурная зависимость констант прямой и обратной реакций следует уравне-466
нию Аррениуса, выведено уравнение для расчета оптимальной температуры:
т гр __ равн
Е2~Е\ Е\
где Е\ и Ег — энергии активации реакций синтеза и разложения аммиака, причем Е\ - Ег = ЛЯ, где кН—тепловой эффект реакции, кДж/моль.
В процессе синтеза аммиака применяются железные плавленые промотированные катализаторы. Промышленный катализатор содержит промоторы двух типов: структурообразующие —AI2O3, MgO, SiOz и модифицирующие — КгО, СаО и др. При условии оптимальных количеств и соотношений промоторов можно получить катализатор с большой и активной поверхностью.
При этом К2О повышает удельную каталитическую активность поверхности, но уменьшает поверхность, снижает термоустойчивость и устойчивость к кислотосодержащим ядам. Сесквиоксид алюминия увеличивает поверхность, термоустойчивость и устойчивость к кислородсодержащим ядам с одновременным снижением удельной каталитической активности.
Железный катализатор синтеза аммиака сначала готовят в окисленном состоянии методом плавления с добавками промотирующих соединений, после чего восстанавливают водородом или азотоводородной смесью до активного состояния. Заключительной стадией является окончательное формирование катализатора в рабочих условиях катализа в колонне синтеза. При этом в результате химического взаимодействия с компонентами реакционной среды в реальных условиях эксплуатации реакционная смесь и условия каталитической реакции оказывают влияние на химический состав и свойства катализатора.
В окисленном состоянии катализатор содержит 90—93% РезОд с некоторым избытком FeO по сравнению со стехиометрическим, 7—10% промотирующих (AI2O3, КгО, СаО) и примесных (БЮг и др.) соединений.
Катализатор синтеза аммиака производят двух марок СА-1 (окисленный) и СА-1 В (восстановленный), в гранулированном или дробленом виде.
Активность катализатора (содержание аммиака в газе, % об.) на выходе из колонны синтеза при давлении 300-105Па, объемной скорости 3000 ч’1 для обеих марок и видов в зависимости от температуры процесса должна быть не ниже:
Температура, °C.................. 550 600 475 450 400
Активность катализатора......... 15,0 19,0 20,0 20,0 14,0
467
16
в сжатой азотоводородной смеси (N2+3H2)
Рис 10.32. Равновесная концентрация аммиака в азотоводородной смеси при 300 ат, 500° С и различном отношении H2:N2 в газе
27
Катализатор марки СА-1 восстанавливают в колонне синтеза аммиака. Колонну, загруженную восстановленным вне агрегата синтеза и запассивированным катализатором СА-1 В, пускают после его до-восстановления.
Наибольшая эффективность процесса синтеза аммиака достигается при оптимизации таких параметров производства, как давление (рис. 10.31), мольное соотношение H2:N2 (рис. 10.32), температурный режим (см. рис. 10.26), объемная скорость (см. рис. 10.30), а также состав газа по основным компонентам и примесям (яды, инерты, рис. 10.30).
На рис. 10.33 приведена схема агрегата синтеза аммиака производительностью 1360 т/сут. Согласно схеме, исходная азотоводородная смесь, сжатая во II ступени центробежного компрессора до давления 320 1 05Па, после охлаждения в воздушном холодильнике (на схеме не приведена) поступает в нижнюю часть конденсационной колонны 8 для очистки от остаточных СО2 и Н2О. Очистку осуществляют барботажем газа через слой сконденсировавшегося жидкого аммиака. 468
Циркуляционный газ из турбокомпрессора
Природный газ ____________ Жид кий NH3
в горелки Свежий газ из турбокомпрессора
Рис 1033. Схема агрегата синтеза аммиака производительностью 1360 т/суг.
1 — подогреватель газа; 2—колонна синтеза аммиака; 3 — подогреватель воды; 4 — выносной теплообменник; 5—блок аппаратов воздушного охлаждения; 6 — сепаратор жидкого аммиака; 7—магнитные фильтры; 8 — конденсационная колонна; 9—испарители жидкого аммиака; 10—конденсационная колонная продувочных газов; 11— испаритель жидкого аммиака на линии продувочных газов; 12 — испаритель жидкого аммиака на линии танковых газов; 13 — сборник жидкого аммиака; 14—промежуточная дренажная емкость; 15—сепаратор
Азотоводородная смесь, проходя слой жидкого аммиака, насыщается им до 3—5% NH3 и смешивается с циркулирующим газом. Образующаяся смесь проходит по трубам теплообменника конденсационной колонны и направляется в межтрубное пространство выносного теплообменника 4, в котором нагревается до температуры не выше 195° С за счет теплоты газа, выходящего из колонны синтеза 2. Из выносного теплообменника циркуляционный газ поступает в колонну синтеза 2, проходя снизу вверх по кольцевой щели между корпусом колонны и кожухом насадки, а затем в межтрубное пространство внутреннего теплообменника, размещенного вверху в горловине корпуса колонны. В теплообменнике газ нагревается до 400—450° С за счет теплоты газа, выходящего из катализаторной коробки, последовательно проходит четыре слоя катализатора, на котором осуществляется синтез аммиака. Температурный режим перед каждой полкой поддерживают путем подачи газа между полками. Газ отбирают из потока перед колонной с температурой до 190° С.
Пройдя четвертый (нижний) слой катализатора, азотоводород-но-аммиачная смесь с содержанием аммиака около 15% и температурой 500—515° С по центральной трубе поднимается вверх, входит в трубки внутреннего теплообменника, охлаждаясь при этом до 330° С, выходит из колонны синтеза. Далее газовая смесь проходит трубное пространство подогревателя питательной воды 3, охлаждаясь при этом до 215° С. После прохождения трубного пространства выносного теплообменника 4 газовая смесь охлаждается входящим циркуляционным газом до 65° С. Далее с 65 до 40° С газ охлаждается в аппаратах воздушного охлаждения 5 (узел первичной конденсации), в которых из газа конденсируется часть аммиака. Сконденсировавшийся аммиак отделяется от газа в сепараторе 15, & газовая смесь, содержащая 10—12% NH3, поступает в азотоводородный компрессор, в котором сжимается еще раз до 320-105Па.
Циркуляционный газ с температурой 50° С поступает в систему вторичной конденсации, состоящую из конденсационной колонны 8 и испарителей жидкого аммиака 9. Газ подается в конденсационную колонну сверху, проходит межтрубное пространство теплообменника, охлаждаясь до 18° С газом, проходящим по трубкам. Газ далее поступает в испаритель жидкого аммиака, в котором, проходя по U-об-разным трубкам высокого давления, охлаждается до -5° С за счет кипения аммиака при температуре -10° С в межтрубном пространстве испарителя. Газообразный аммиак из межтрубного пространства испарителя направляется в холодильную установку для сжижения аммиака и возврата в испарители. 470
Из трубного пространства испарителя смесь охлажденного циркуляционного газа и сконденсировавшегося аммиака поступает в сепарационную часть конденсационной колонны, в которой происходит отделение жидкого аммиака от газа. При этом свежий газ смешивается с циркуляционным. Далее газовая смесь проходит корзину, заполненную полуфарфоро-выми кольцами, где отделяется от капель жидкого аммиака, поднимается по трубкам теплообменника конденсационной колонны и направляется в выносной теплообменник, а далее — в колонну синтеза аммиака. Таким образом, циркуляционный цикл замыкается.
Жидкий аммиак после первичного сепаратора с температурой 40° С, пройдя магнитные фильтры 7, очищается от катализаторной пыли, дросселируется до давления 20-105Па и поступает в сборник жидкого аммиака 13. в который также под давлением 20-105Па поступает аммиак из конденсационной колонны. В процессе дросселирования жидкого аммиака с высокого давления до давления 20-105Па происходит выделение растворенных в жидком аммиаке газов Н2, N2, Аг, СН4. Газы, называемые танковыми, содержат около 16% NH3. Утилизация аммиака из танковых га
Рис 10.34. Четырехполочная колонна синтеза аммиака мощностью 1360 т/сут:
1 — люк для выгрузки катализатора; 2 — центральная труба; 3 — корпус катализаторной коробки; 4 — корпус колонны; 5 — загрузочный люк; 6 — теплообменник; 7, 9, 11, 13— ввод байпасного газа; 8, 10, 12, 14 — катализатор
зов производится конденсацией его в испарителе 12 на линии танковых газов при температуре от минус 20 до минус 25° С.
Из испарителя танковые газы и сконденсировавшийся аммиак поступают в сепаратор 15. в котором жид
кий аммиак отделяется и направляется в сборник жидкого аммиака 13. С целью поддержания в циркуляционном газе содержания инертных
471
примесей в пределах 14—18% после первичной конденсации производят постоянный отбор части газа. Количество продувочных газов зависит от содержания инертных примесей в исходном газе, давления в системе синтеза, активности катализатора и колеблется в пределах 3—8 тыс.м3. Целевой продукт (аммиак) из продувочных газов выделяется при температуре (-25)-*-(-30)° С в конденсационной колонне 10 и испарителе 11. Смесь танковых и продувочных газов после выделения ам
миака, аргона, водорода и гелия применяют в качестве топливного газа, для чего она направляется на сжигание в блок риформинга метана.
Основным аппаратом в производстве аммиака является колонна синтеза (рис. 10.34), которая состоит из корпуса 4. Колонна снабжена люками 1 и 5 для выгрузки и загрузки катализатора соответственно. В верхней части колонны вмонтирован теплообменник 6. Внутри корпуса колонны имеются четыре слоя катализатора 8, 10, 12 и 14, которые находятся в корпусе катализаторной коробки 3. Через корпус колонны вмонтированы вводы байпасного газа 7, 9, 11 и 13. Из выносного теплообменника циркуляционный газ поступает в колонну через центральную трубу 2.
Температурный режим четырехполочной колонны синтеза аммиака (% об. — содержание NH3) приведен на рис. 10.35.
Аммиак хранят в изотермических складах (рис. 10.36), рассчитанных на поступление как холодного, так и теплого аммиака. Холодный аммиак подается в изотермический резервуар 17 снизу, а теплый охлаждается путем последовательного дросселирования в паровое пространство резервуара 17 через ресиверы 1, 2 и 3. В ре
сивере 3 аммиак охлаждается до температуры минус 33,4° С, соответствующей необходимому давлению для поступления в изотерми-
Рнс. 10.35. Температурный режим четырехполочной колонны синтеза аммиака (% об. — содержание NH3)
ческий резервуар, которое поддерживается с помощью трехступенчатого компрессора.. 24. В случае применения поршневых компрессоров перед холодильниками устанавливают маслоотделители. Газообразный аммиак из верхней части резервуара 17 через сепаратор 4 поступает в первую ступень компрессора 24. Затем через холодильник 20 поступает в ресивер 3, в котором смешивается с газообразным аммиаком, образующимся
472
Одно-двухстенный резервуар
Рис. 1036. Принципиальная схема изотермического склада для хранения жидкого аммиака под атмосферным давлением: a — изоляция; б — разбрызгиватель жидкого аммиака; в — распределительный коллектор азота; г—фундамент; 7, 2, 3, 10, 23 — ресиверы; 4, 5 — сепараторы; 6, 8 — маслоотделители; 7—промежуточный сосуд; 9, 22 — холодильники; 11, 24 — компрессоры; 12, 19 — сборники жидкого аммиака; 13 — факел; 14 — газгольдер азота; 15, 18 — насосы; 16 — фильтры; 17—изотермический резервуар; 20, 21 — холодильники-конденсаторы
в процессе дросселирования жидкого аммиака. После ресивера 3 | сжимается во второй ступени компрессора, охлаждается в холодиль- | нике 21 и поступает в ресивер 2, в котором смешивается с газо- 1 образным аммиаком, образующимся в процессе дросселирования I жидкого аммиака из ресиверов 1 и 23. Из ресивера 2 аммиак на- 1 правляется в третью ступень компрессора, после которой конден- ’ сируется в холодильнике 22 и стекает в ресивер 23.
Для регулирования режима работы компрессорной установки предусмотрены байпасы в каждой ступени компрессора и впрыск жидкого аммиака в сепаратор 4. Отделившийся в сепараторе 4 жидкий аммиак периодически сливается в сборник 19, из которого насосом 18 откачивается в резервуар 17. В сепараторе 4, обогреваемом теплоносителем, проходящим по змеевику, жидкий аммиак может испаряться.
В режиме хранения и при приеме холодного аммиака включается компрессорно-конденсационная установка меньшей мощности. Газообразный аммиак из резервуара через сепаратор 5 поступает в первую ступень двухступенчатого компрессора 11 и через маслоотделитель 6 — в промежуточный сосуд 7, в котором охлаждается впрыскиваемым жидким аммиаком из ресивера 10. После этого аммиак сжимается во второй ступени компрессора и проходит через маслоотделитель 8 в холодильник 9, в котором конденсируется, а затем стекает в ресивер 10, из которого поступает в змеевики промежуточного сосуда 7 на переохлаждение жидким аммиаком, находящимся в сосуде. Незначительная его часть отводится для впрыска в этот же промежуточный сосуд. Из змеевиков жидкий аммиак дросселируется в резервуар, а из сепаратора 5 стекает в сборник 12, из которого периодически передавливается газообразным аммиаком в ресивер 10. При остановке компрессора 11 возможно превышение рабочего давления в резервуаре. Для предотвращения этого включают в работу резервный (аварийный) компрессор (на схеме не показан), который имеет привод от дизеля или от электродвигателя, подключенного ко второму независимому источнику электроэнергии.
Расход жидкого аммиака из резервуара 17 производят насосом 15 через два поочередно работающих фильтра 16. Отвод в резервуар 17 служит для циркуляции жидкого аммиака во время охлаждения и выравнивания температуры хранения аммиака. Для исключения деформации резервуара он оснащен импульсными предохранительными клапанами и вакуумным (дыхательным) клапаном. Жидкий аммиак от предохранительных клапанов оборудования и трубопроводов сбрасывают в резервуар, а газообразный — на сжигание на факеле 13, горелки которого работают на природном или испаряемом газе (пропане).
Объемный коэффициент заполнения вертикального резервуара не должен быть больше 0,93.
474
10.4. КАРБАМИД
Физико-химические свойства карбамида. Карбамид (диамид угольной кислоты, мочевина) (NHj^CO, бесцветные кристаллы, без запаха. Кристаллическая решетка тетрагональная (а = 0,566 нм, b -= 0,4712 нм, z = 2, пространственная группа P42im.) Претерпевает полиморфные превращения, температура плавления 132,7° С; плотность 1,33 г/см3 (25° С); п2° = 1,484; С ° = 93,198 Дж/(моль-К); AG^p =-197,3 кДж/моль, ДЯ^, = -333,3 кДж/моль, ДЯ°гор = -632,5 кДж/моль, ДЯ^ = 14,53 кДж/моль; = 104,67 Дж/(моль-К); ц= 14,О-1О'зоКл-м. Для расплава плотность 1,225 кг/м3; т] = 0,00258 Па-с; у = 0,036 Н/м; теплопроводность (135° С) 0,42 Вт/(м-К); р = 2,3 Ом-м. Кристаллический карбамид обладает сильной гигроскопичностью.
Растворимость карбамида (г в 100 г растворителя): в воде—51,8(20° С), 71,7(60° С), 95,0 (120° С); в жидком аммиаке —49,2 (20° С, 709 кПа), 90 (100° С, 1267 кПа); в метаноле — 22 (20° С); в этаноле — 5,4 (20° С); в изопропаноле — 2,6 (20° С); в изобутаноле 6,2 (20° С); в этилацетате 0,08(25° С); в хлороформе не растворяется. Растворяется в аммиаке, образуя при этом соединение CO(NHz)-NH3, состоящее из 77,9% CO(NH2)2 и 22,1% NH3 и существующее лишь в растворах, содержащих избыток аммиака. Из диаграммы (рис. 10.37) видно, что кривая растворимости имеет точку перегиба, которая отвечает указанному выше составу и соответствует температуре плавления 46° С аммиаката CO(NH2)2-NH3. Участок кривой ниже точки перегиба характеризует растворимость аммиаката, а участок выше этой точки— растворимость карбамида. Справа на диаграмме указано давление р насыщенного пара аммиаката при различных температурах, которое (в мм рт. ст.) для любой температуры Т (в К) можно вычислить по формуле
i 7820 __
lgp =--------+ 9,2.
4,57Г
Рентгено- и спектроскопическими, а также нейтронографическими методами исследований установлено,- что молекула кристаллического карбамида имеет плоскую симметричную конфигурацию, имеющую форму: О = С( Следовательно, весь азот карбамида находится в амидной форме. Карбамид образует следующие комплексы: CO(NH2)2-NH3, СО(МН2)2-НзРО4, CO(NH2)2H2O2, СО(МН2)2-СНзОН, CO(NH2)2-HNO3, 4СО(ЫН2)2-Са(ЫОз)2, 4CO(NH2)rCa(H2PO4)2 и др.
В процессе нагревания до 150° С и выше карбамид последовательно превращается в NH4NCO, NH3, СО2, биурет, циануровую кислоту; в замкнутом сосуде, особенно при добавлении аммиака — про-475
Рис. 10.37. Кривая растворимости аммиака в жидком аммиаке
дукгы аминирования циануровой кислоты, например меланин. В разбавленных растворах при ~200°С возможен полный гидролиз карбамида с образованием NH3 и СО2.
В водных растворах выше 80° С карбамид переходит в изоцианат аммония, а далее в карбонат аммония:
(NH2)2CO ^ИНдНСО
NH4NCO + 2Н2О (NHthCOj
который впоследствии превращается в гидрокарбонат аммония, распадающийся затем на аммиак и диоксид углерода:
(МН4)гСОз NH4HCO3 + NH3
NH4HCO3 # NH3 + СО2 + Н2О
Карбамат аммония в разбавленных растворах практически полностью переходит в карбонат:
NH2COONH4 + Н2О = (NH4)2CO3
476
160 - 152 й
О 20 60 100
Рис. 10.38. Кривая растворимости в системе карбонат аммония — вода
Степень разложения карбамата аммония значительно понижается в присутствии аммиака. Приведенные на рис. 10.38 величины растворимости в системе NH2COONH4—Н2О показывают, что ниже 60° С из растворов карбамата аммония кристаллизуются углеаммонийные соли: от -13 до 5° С — карбонат , а от 5 до 61° С — сесквикарбонат (NH^COj-ZNHjHCOj. Температура плавления чистого карбамата аммония 152—155° С. В присутствии же углеаммонийных солей карбамида и воды эта температура понижается.
В расплавленном виде карбамид реагирует со щелочными металлами и
их амидами с образованием солей цианамида. В процессе сплавления с карбонатом натрия разлагается до NaNCO, СО2> NH3 и Н2О. При сплавлении с нитратом аммония в присутствии диоксида кремния образуется гуанидин; при взаимодействии с хлорсульфокислотами — амидосульфокислоты; с концентрированным олеумом— сульфаминовая кислота; с бромом — циануровая кислота, с NaClO — гидразин, с сероуглеродом (110° С) — NH4SCN и COS.
Карбамид устойчив к действию Н2О2 и КМпО4.
Карбамид применяют в качестве концентрированного азотного удобрения (в чистом карбамиде содержится 46,6% азота) для многих сельскохозяйственных культур на любых почвах, а также как заменитель естественного белка в кормах для жвачных животных. Карбамид является сырьем для производства карбамидных смол, меламина, циануровой кислоты и ее эфиров, цианидов калия и натрия, гидразина, гидразоформамида, карбамидных лаков, в бумажной промышленности, в фармацевтической промышленности для приготовления успокаивающих (адалин, бромурал), снотворных (веронал, люминал) и мочегонных средств, составов для заживления ран, дезинфицирующих средств, в качестве умягчителя в производстве косметических кремов. В нефтяной промышленности карбамид применяют для депарафинизации масел и моторных топлив с выделением мягкого парафина— сырья для получения белково-витаминных препаратов, жирных спиртов и кислот, моющих средств и т.п.
Согласно ГОСТ 2081—75, гранулированный и кристаллический карбамид марки А высшей и первой категории, применяемой в промышленности, должен содержать соответственно не менее 46,3 и 46,2% азота, не более 0,6 и 0,9% биурета, 0,2 и 0,3% Н2О; в предназначаемом для животных допускается до 3% биурета.
477
Карбамид марки Б используется в качестве удобрения. Он должен содержать не менее 46,0% азота и не более 0,9% биурета и 0,25% влаги; размер гранул: 1—4 мм — не менее 94%, меньше 1 мм — не более 5%.
Физико-химические основы получения карбамида из аммиака и диоксида углерода. Технология карбамида основывается на реакции аммиака и диоксида углерода, протекающей при 150 —220° С и под давлением 7—100 МПа. Процесс протекает в две стадии: первая стадия— образование карбамата аммония; вторая — дегидратация карбамата аммония с образованием карбамида:
ONH4
2NH3+ со2
NH2
ОС
zonh4 \nh2
+ H2O
Таким образом, карбамид образуется из карбамата преимущественно в жидкой фазе.
На рис. 10.39 представлена диаграмма состояния системы карбамид— карбамат аммония — аммиак, с помощью которой можно установить влияние карбамида на растворимость карбамата аммония в жидком аммиаке. Точка d соответствует раствору, который при
Рис 10.39. Диаграмма состояния системы карбамид — карбонат аммония — аммиак 478
20° С имеет состав: 40% CO(NH2)2, 25% NH4COONH2 и 35% NH3. Лежащая на изотерме 80° С точка b соответствует раствору, содержащему 40% CO(NH2)2, 40% NH4COONH2 и 20% NH3.
Кроме изотерм (тонкие линии) на диаграмме имеются кривые совместной кристаллизации (пограничные линии), которые делят ее на четыре области. Поле, примыкающее к вершине NH4COONH2, соответствует области насыщения по отношению к карбамату аммония растворов всех трех компонентов системы. К вершине CO(NH2)2 примыкает область растворов, насыщенных по отношению к карбидам. Выше нее расположена область растворов, насыщенных по отношению к комплексному соединению CO(NH2)2-NH3. Поле же, ограниченное эллипсоидальной кривой, соответствует
100 150 200 250 Температура, *С
Рис. 10.40. Давление паров над плавом карбамида, получаемым при молярном отношении NH3:CO2 = 2. Плотность загрузки, г/см3: 1 — 0,94-1,1; 2 — 0,676; 3 — 0,5; 4 — 0,3
области расслоения, в которой система содержит два раствора, каждый из которых состоит из трех компонентов. Составы этих раство-
ров отвечают точкам, полученным путем пересечения эллипсоидальной кривой с соответствующими изотермами. На пограничных линиях лежат точки, выражающие составы растворов, насыщенных по отношению к двум соответствующим солям. Точка пересечения трех пограничных линий характеризует насыщенный раствор, находящийся в равновесии с твердой фазой. Эта фаза состоит из карбамата аммония, карбамида и комплексного соединения CO(NH2)2*NH3.
Карбамат аммония обладает высоким давлением паров, в результате он при температуре 150—220° С разлагается на аммиак и диоксид углерода. Поэтому процесс получения карбамида ведут под высоким давлением (200—300 атм). На рис. 10.40 показано изменение давления паров над плавом карбамида в зависимости от температуры процесса. Величина давления зависит и от состава системы, в частности от соотношения исходных компонентов NH3:CO2. Давление резко повышается в процессе возрастания содержания диоксида углерода в исходной смеси сверх стехиометрического соотношения. Избыток аммиака не приводит к подобному сильному повышению давления. При избытке аммиака давление понижается при добавлении воды к системе. Кроме температуры и состава системы на величину равновесного давления влияет и степень заполнения реактора жидкой фазой (плотность загрузки)— чем она больше, тем больше давление (рис. 10.41).
479
Рис. 10.41. Влияние плотности загрузки на выход карбамида при 170° С
массы. Так, при со-плавления карбамата
Скорость образования карбамата аммония растет пропорционально квадрату давления. При атмосферном давлении и невысокой температуре образование карбамата аммония протекает медленно, а с повышением температуры до 150° С и давления до 10 МПа проходит практически мгновенно. С повышением же общего давления в системе возрастает также степень конверсии карбамата аммония в карбамид (рис. 10.42).
Карбамид образуется из расплавленного карбамата при температуре ниже точки его плавления (152° С). В процессе выделения воды снижается температура плавла держании в плаве 9,2% влаги температу
аммония снижается до 140° С, а при 14,7% — до 130° С. При этом увеличивается объем жидкой фазы. Под действием воды часть карбамата переходит в карбонат и затем в гидрокарбонат аммония, которые как и образующийся карбамид также понижают температуру плавления карбамата. Степень понижения температуры плавления карбамата аммония в присутствии карбамида представлена на рис. 10.43. Эвтектическая смесь, содержащая 51% карбамата аммония и 49% карбамида, плавится при 98° С.
Образующаяся в начале процесса вода увеличивает степень конверсии, поскольку в системе появилась жидкая фаза. Однако по мере накопления в системе воды реакция замедляется в ре
зультате сдвига равновесия в сторону исходных веществ выхода
Давление, МПа
Рис. 10.42. Влияние давления на степень конверсии карбамата аммония в карбамид
Рис. 10.43. Диаграмма плавкости системы карбонат аммония — карбамид
480
Рис. 10.44. Изменение выхода
(в долях от теоретического) карбамида из карбамата аммония во времени при разных температурах
Число молей воды на 1 моль карбамида аммония
Рис. 10.45. Влияние добавки воды к карбонату аммония на равновесный выход карбамида (по СО2) при 160° С
карбамида, достигнув известного предела, далее не увеличивается (рис. 10.44), а в процессе добавки воды к карбамату аммония— уменьшается (рис. 10.45).
Избыток диоксида углерода, вводимый сверх стехиометрического количества, практически не влияет на увеличение выхода карбамида (рис. 10.46). Обычно источником диоксида углерода служит
отходящий газ производства аммиака, содержащиеся в нем примеси (Н2СО3, N2, Ог и др.) снижают парциальное давление аммиака и его растворимость в жидкой фазе. Снижение содержания диок-
сида углерода в исходном газе с 98 — 99 до 85 — 86% снижает степень конверсии с 65 до 45%.
Максимальное влияние на увеличение выхода карбамида оказывает избыток аммиака в исходной смеси против стехиометрического количества, определяемого молярным отношением МНз:СОг = 2 (рис. 10.47). Избыточный аммиак уменьшает тормозящее действие
выделившейся в процессе воды — он связывает ее и ускоряет реакцию перехода карбамата аммония в карбамид. По этой же причине замедляется разложение карбамата аммония с образованием побочных продуктов. Кроме того, избыточный аммиак уменьшает
коррозию аппаратуры. При выборе соотношения исходных реагентов в смеси необходимо решить вопрос его дальнейшего использования либо возвращением аммиака в начало процесса, или организовать переработ
CO2:NH3
Рис. 10.46. Степень влияния избытка диоксида углерода на выход карбамида
ку его в другие продукты, например в нитрат аммония или в жидкие удобрения.
16 Химическая технология неорганических веществ, кн. 1
481
Молярное отношение NH3:CO2
Рис. 10.47. Влияние избытка аммиака на равновесный выход карбамида (по средним данным для 185—195° С, 18,5—32 МПа и плотности загрузки 0,72 г/см3)
Способы получения карбамида. Полученный в виде плава карбамид, содержащий в своем составе карбамид, карбамат, аммиак, карбонаты аммония и воду, подвергают дистилляции. При термообработке плава разлагают содержащийся в нем карбамат и карбонаты и извлекают аммиак и диоксид углерода, после чего водный раствор карбамида перерабатывают в твердый продукт.
Существующие способы получения карбамида различаются в основном методами улавливания и использования газов дистилляции плава — смеси аммиака с диоксидом углерода, степень превращения которых в карбамид не превышает соответственно 50 и 70%.
Современные производства карбамида большой мощности работают по замкнутым системам, по которым продукты дистилляции полностью возвращаются на синтез карбамида.
Рециркуляцию газов дистилляции осуществляют двумя способами: 1) с газовым рециклом — продукты дистилляции полностью возвращаются в газообразной форме; 2) с частичным или полным жидкостным рециклом — в цикл возвращаются жидкий аммиак или растворы (суспензии) углеаммонийных солей.
Непосредственный возврат в систему синтеза газовой смеси (аммиак и диоксид углерода), получаемой в процессе дистилляции, требует ее сжатия до давления синтеза при высокой температуре во избежание процесса образования твердого карбамата аммония. В способах с газовым рециклом предварительно разделяют газы дистилляции, обрабатывая их селективными абсорбентами для извлечения аммиака и диоксида углерода. Для этого в абсорбере газы промывают раствором нитрата карбамида и извлекают аммиак, а оставшийся диоксид углерода в газовой фазе вновь используют. В процессе регенерации поглотительного раствора в десорбере выделяют аммиак, который возвращают в цикл синтеза.
Если применяемый абсорбент моноэтаноламин NH2CH2CH2OH, то из газов дистилляции извлекают диоксид углерода, а остающийся газообразный аммиак сжижают и возвращают в цикл. В процессе нагревания вытекающего из абсорбера поглотителя из него десорбируют диоксид углерода, а регенерированный раствор вновь направляют в абсорбер.
482
Рис. 10.48. Схема получения карбамида с жидкостным рециклом (синтез и дистилляция сплава):
1 — четырехступенчатый компрессор для диоксида углерода; 2 — танк жидкого аммиака; 3 — плунжерный насос для аммиака; 4— конденсатор I ступени; 5 — колонна синтеза; 6 — смеситель; 7 — плунжерный насос; 8— промывная колонна; 9 — ректификационная колонна I ступени; 10—подогреватель I ступени; 11 — сепаратор I ступени; 12 — ректификационная колонна П ступени; 13 — подогреватель П ступени; 14 — сепаратор II ступени; 15— вакуум-испаритель; 16 — сборник раствора карбамида; 17—маслоотделитель; 18— центробежный насос; 19—конденсатор; 20—вакуум-насос; 21 — конденсатор П ступени; 22 — резервуар; 23 — центробежный насос; 24 — абсорбер; 25 — сборник раствора карбонатов аммония;
26 — десорбер; 27—холодильник; 28 — теплообменник; 29—центробежные насосы
Плав
I Реагенты
Рис. 10.49. Колонна синтеза карбамида: 1 — корпус колонны; 2 — футеровка; 3 — коллектор контроля футеровки; 4 — решетки; 5 — крышка; 6 — штуцер для термопары
В настоящее время процессы с жидкостным рециклом получили наибольшее распространение. По ним газы дистилляции абсорбируют водой, а образовавшиеся концентрированные растворы карбонатов аммония возвращают на получение карбамида. Наиболее совершенными считаются процессы, согласно которым дистилляцию плава, т.е. разложение карбамата аммония и отгонку аммиака, производят в токе аммиака или диоксида углерода при давлении синтеза карбамида.
Получение карбамида с полным жидкостным рециклом. Один из вариантов получения карбамида с двухступенчатой дистилляцией плава и жидкостным рециклом приведен на рис. 10.49.
Газообразный диоксид углерода, полученный в качестве отхода в процессе очистки азотоводородной смеси, предварительно осушенный и очищенный от механических примесей, от сульфида водорода и сульфидсодержащих соединений, сжимается в четырехступенчатом компрессоре 1 до 20 МПа и при температуре 95 — 100° С направляется в смеситель 6. Одновременно в смеситель плунжерным насосом 3 подается жидкий аммиак под давлением 20 МПа с температурой 90° С, а плунжерным насосом 7 — раствор карбонатов аммония (/»95° С), в виде которого возвращаются в систему аммиак и диоксид углерода. В процессе перемешивания компонентов в смесителе при 175° С начинается образование карбамата аммония, после чего реакционная масса [молярное отношение NH3:CO2:H2O = (3,8-ь4,5):1:(0,5-ь0,8)] поступает в колонну синтеза 5, в которой при 180—230° С и давлении 12—25 МПа заверша
ется образование карбамата аммония и его разложение до карбамида.
Колонна синтеза карбамида (рис. 10.49) представляет собой полый цилиндрический аппарат со сферическим днищем, изготовленный из углеродистой низколегированной стали. Соприкасающиеся с
плавом внутренние поверхности аппарата защищены листовой хромоникельмолибденовой сталью Х17Н16МЗТ или титаном. Многослой-
ный корпус высокого давления (25 — 30 МПа) изготовляется из листовой углеродистой стали. Для контроля за состоянием футеровки в
484
корпусе колонны имеются отверстия, соединенные с общим коллектором. Реакционная масса, состоящая из карбамида, карбамата аммония и карбонатных солей, аммиака и воды, поступает в колонну через нижний штуцер и, постепенно заполняя колонну, движется к выходному штуцеру в плоской крышке аппарата. С целью более интенсивного перемешивания образующегося плава в нижней части колонны имеются решетчатые перегородки. Габаритные размеры колонны при производительности агрегата 1250 т/сут (450 тыс. т/год): диаметр 2,0—2,5 м, высота 30—35 м (объем до 160 м3).
Образующийся в колонне синтеза плав, содержащий 30—31% карбамида, 21—22% карбамата аммония, 33—34% избыточного аммиака и 16—17% воды, направляется на двухступенчатую дистилляцию. Агрегат дистилляции каждой ступени состоит из трех аппаратов: ректификационной колонны, подогревателя и сепаратора.
Плав карбамида, выходящий из колонны синтеза 5 (см. рис. 10.48), дросселируется от 20 до 1,8—2,0 МПа и поступает в верхнюю часть ректификационной колонны 9 агрегата дистилляции I ступени. В колонне 9 при температуре 120—125° С происходит выделение в газовую фазу избыточного аммиака, после чего плав для полного разложения карбамата аммония нагревается до 158—162°С в теплообменнике 10. Образовавшаяся при этом парожидкостная смесь разделяется в сепараторе 11 — газовая фаза возвращается в нижнюю часть (барботажный слой) ректификационной колонны 9, а жидкая фаза дросселируется до давления 0,25—0,4 МПа и направляется на дистилляцию II ступени.
Газовая фаза из ректификационной колонны 9, содержащая 75—76% NH3, 21 —22% СОг и около 3% воды, направляется в нижнюю часть промывной колонны 8, в которой с помощью парового подогревателя поддерживают температуру 92—96° С. Одновременно в колонну 8 подается и раствор карбонатов аммония со II ступени дистилляции, в процессе которой поглощается основное количество диоксида углерода и конденсируется водяной пар с образованием раствора, содержащего 38—45% аммиака, 30—37% диоксида углерода и 22—27% воды. Далее раствор сжимается плунжерным насосом 7 до 20 МПа и возвращается в смеситель 6. Газообразный аммиак при 45—50° С отделяется от диоксида углерода в верхней насадочной части колонны 8, орошаемой концентрированным водным раствором аммиака (93—96% NH3), и направляется в конденсатор 4, где он сжимается и, проходя, возвращается в систему (цикл). Несконден-сировавшиеся газы (в основном азот, водород и кислород) отмываются от содержащегося аммиака в системе абсорбции, дросселируются до атмосферного давления и сбрасываются в атмосферу.
Раствор, поступающий на дистилляцию II ступени, содержит 55—61% карбамида, 4—5% карбамата аммония, 6—7% избыточного
аммиака и 28—35% воды. Процесс дистилляции во II ступени протекает так же, как и в I, т. е. раствор вначале проходит через ректификационную колонну 12, охлаждаясь до 110° С за счет испарения аммиака и разложения карбамата аммония, затем в подогревателе 13 нагревается до 140—142° G и поступает в сепаратор 14, в котором происходит разделение газообразной и жидкой фаз. Во II ступени дистилляции заканчивается разложение карбамата аммония и отгонка аммиака и диоксида углерода. Остающийся раствор, содержащий 70—72% карбамида, из сепаратора 14 дросселируется и поступает в вакуум-испаритель 75, в котором при остаточном давлении 40 кПа происходит его концентрирование до 74—76% и охлаждение до 90° С за счет самоиспарения. Раствор далее через сборник 16 и маслоотделитель 17 направляется на переработку в целевой продукт.
Газовая фаза из ректификационной колонны 12, содержащая 55—56% NH3, 24—25% СОг и 20—21% НгО, направляется в конденсатор 27. Образующийся при 40° С неконцентрированный раствор карбонатов аммония (33—50% NH3, 10—16% СОг и 35—55% Н2О) через резервуар 22 центробежным насосом 23 направляется в промывную колонну 8. Газовая фаза из конденсатора 27 и другие отходящие газы, содержащие NH3 и СОг, направляются в абсорбер 24, в котором NH3 и СОг поглощаются при 40° С раствором карбонатов аммония, циркулирующих через холодильник 27. Инертные же газы из адсорбера удаляются в атмосферу.
Образовавшийся в абсорбере 24 раствор карбонатов аммония подогревается в теплообменнике 28 до 90—95° С и направляется в десорбер 26, в котором при температуре 135—145° С и под давлением 0,3—0,4 МПа в присутствии острого пара происходит полное его разложение на NH3, СОг и Н2О. Газообразные аммиак и диоксид углерода вместе с водяными парами направляются в конденсатор II ступени 27, а оставшаяся вода сливается в отстойники.
Недостатками рассмотренной схемы получения плава карбамида являются сравнительно низкая (62—65%) степень конверсии карбамата аммония в карбамид, сложность системы регенерации и возврата непрореагировавших компонентов, отсутствие полного использования теплоты синтеза. Поэтому все более широкое распространение получают усовершенствованные процессы, так называемые стрип-пинг-процессы, основанные на отгонке и конденсации большей части непрореагировавших аммиака и диоксида углерода под давлением синтеза, что позволяет упростить схему, уменьшить объемы возвращаемой в узел синтеза воды и лучше утилизировать теплоту конденсации. Процесс дистилляции в таких процессах осуществляется при противоточной обработке плава диоксидом углерода или аммиаком, что обеспечивает возможность проведения дистилляции плава при 486
относительно низкой температуре и, главное, предотвращает гидролиз карбамида.
Один из видов стриппинг-процесса (рис. 10.50) включает синтез карбамида под давлением 13 МПа и 180—190° С в реакторе-автоклаве 6, разделенном 8—10 ситчатыми перегородками для предотвращения продольной циркуляции реакционной массы в процессе подачи в аппарат свежего жидкого аммиака и водно-аммиачного раствора карбамата аммония (/~170° С) из конденсаторов высокого давления 5 и 7. За период прохождения реакционной массы через автоклав (45—60 мин) достигается 90—95%-ная степень приближения к равновесию. Выходящий из реактора плав при том же давлении (13 МПа) подвергают дистилляции в теплообменнике-дистилляторе 3, обогреваемом паром (2,5 МПа) с помощью свежего СОг, к которому добавляют воздух для подавления коррозии аппаратуры. Газы из дистиллятора направляют в поверхностный конденсатор высокого давления 7, в котором под давлением синтеза образуется основное количество раствора карбонатов аммония, возвращаемое в колонну синтеза, и пар низкого давления (0,35 МПа). В конденсатор 7 центробежным насосом подают некоторое количество раствора карбамата из конденсатора низкого давления 12. Степень конденсации за счет подачи воды поддерживают на уровне 80% для обеспечения автотермичности
На конденсацию Инертные газы f Изконден-
Рнс 10.50. Принципиальная схема синтеза карбамида и дистилляции плава (стриппинг-процессы):
1, 11, 14 — сепараторы; 2 — компрессор; 3—теплообменник-дистиллятор; 4— насосы; 5, 7— конденсаторы высокого давления; 6 — реактор; 8— дроссельный клапан; 9 — ректификационная колонна; 10 — подогреватель; 12—конденсатор низкого давления; 13, 17—сборники; 15— десорбер; 16 — теплообменник; 18— скруббер
487
процесса синтеза. Плав из теплообменника-дистиллятора 3 дросселируют до 0,3—0,4 МПа и направляют в узел дистилляции II ступени, работающей аналогично описанной выше.
Переработка раствора карбамида в целевой продукт (рис. 10.51). Из сборника 1 фильтруют 74—75%-ный раствор на ФПАКМ 2 для очистки его от механических примесей. Отфильтрованный раствор из напорного сборника 3 поступает на двухступенчатое выпаривание. С целью предотвращения образования биурета процесс упарки ведут в выпарных аппаратах пленочного типа при минимально возможных температуре и продолжительности процесса упарки сначала при остаточном давлении 20—40 кПа и 118—125° С до концентрации 92—95%, а затем при 2,5—6,5 кПа и 135—140° С до 99,5—99,8%.
Выпарной аппарат I ступени состоит из греющей камеры 4 и сепаратора 5 сокового пара. Выпарной аппарат II ступени 11 роторного типа. В вертикальной испарительной трубе аппарата, находящейся в паровой рубашке, имеется ротор — вертикальный вал с радиально расположенными пластинами. В процессе его вращения выпариваемый раствор распределяется по поверхности нагрева в виде тонкой пленки.
В атмосферу
Рис. 10.51. Схема получения гранулированного карбамида:
1— сборник раствора карбамида; 2 — фильтр ФПАКМ; 3, 16—'напорные сборники; 4 — греющая камера выпарного аппарата 1 ступени; 5 — сепаратор; 6 — сборник упаренного раствора; 7 — поверхностный конденсатор; 8 — брызгоуловитель; 9 — вакуум-насос; 10 — барометрический бак; И — ретурный выпарной аппарат II ступени; 12 — барометрический конденсатор; 13 — пароструйный эжектор; 14—гранулирующая башня; 15 — гранулятор; 17—вентилятор; 18— ленточный транспортер; 19 — холодильник с кипящим слоем гранул; 20 — элеватор; 21 — грохот; 22 — растворитель пыли и крупных гранул; 23 — центробежный насос
488
Полученный после II ступени выпарки плав карбамида перекачивается в расположенную над грануляционной башней 14 напорную емкость 18, обогреваемую паром, откуда он поступает в гранулятор 75. Образующиеся капли плава охлаждаются встречным потоком воздуха и затвердевают в гранулы. Охлаждение гранул от 60—70 до 40—50° С производится в аппаратах с кипящим слоем, расположенных внутри башни или рядом с ней. С целью получения целевого продукта с размером гранул 1—4 мм его подвергают сортировке на двухситном грохоте 21, а частицы с размерами менее 1 мм и более 4 мм собирают в растворитель 22. Полученные от растворения мелких (менее 1 мм) и крупных (более 4 мм) частиц растворы возвращаются на выпарку. Применение в производстве вибрационных грануляторов обеспечивает получение монодис-персного целевого продукта.
Для получения 1 т карбамида по описанной схеме расходуется: 0,58 т NH3 (в пересчете на 100%-ный), 0,75 т СОг (100%-ный), 4,82 ГДж пара, 65 м3 охлаждающей воды и 480 МДж электроэнергии.
Холодильники с кипящим слоем гранул работают со значительно большей интенсивностью, чем вращающиеся барабанные. Однако в процессе охлаждения в кипящем слое гранулы карбамида раскалываются и истираются, в результате чего образуется запыленность. В результате часть мелких частиц уносится с газом, а другая часть остается в продукте и способствует его слеживанию.
Процесс гранулирования производят также на наклонном вращающемся грануляторе тарельчатого типа. Мелкие гранулы ретура, находящиеся на тарелке, опрыскивают 99,5—99,8%-ным плавом карбамида. Полученные стандартные гранулы охлаждают и рассеивают.
Перспективными являются способы получения гранулированного карбамида в аппарате кипящего слоя непосредственно из растворов, выводимых из цикла после дистилляции, содержащих до 30% воды. Раствор карбамида выпрыскивается горячим газом через форсунки в кипящий или фонтанирующий слой гранул. Отводимые из аппарата гранулы охлаждают, рассеивают, а фракцию менее 1 мм возвращают в кипящий слой или растворяют в исходных растворах.
Для получения кристаллического карбамида исходный раствор упаривают до 92—94%, а затем направляют его в кристаллизаторы шнекового типа или в вакуум-кристаллизаторы, в которых охлаждают до 40—45° С. Отделение кристаллов от маточного раствора ведут на центрифугах с одновременной подсушкой воздухом.
Незначительные добавки полифосфата натрия существенно увеличивают прочность гранул карбамида. Образующийся полифосфат карбамида затвердевает в виде пространственной сетки из переплетающихся волокон, выполняющей роль укрепляющей гранулу арматуры.
489
10.5. АЗОТНАЯ КИСЛОТА
Физико-химические свойства, применение. Азотная кислота HNO3, бесцветная жидкость, существующая также в виде моно- и тригидрата (табл. 10.4). Молекула азотной кислоты имеет плоскую структуру (длины связей в нм):
Твердая азотная кислота образует две кристаллические модификации— с моноклинной и ромбической решетками. Концентрированная кислота малоустойчива, при нагревании или под действием света частично разлагается:
4HNO3 4NO2 + 2Н2О + О2
Образующийся диоксид азота окрашивает кислоту в бурый цвет и придает ей специфический запах.
Таблица 10.4. Свойства азотной кислоты и ее кристаллогидратов
Показатель HNO, Моногидрат Тригидрат
Плотность, г/см3 1,513 — —
Т С° -41,59 (82,6)* -37,62 -18,47
ДЯ^, кДж/моль 10,47 17,50 29,09
ДЯ^, (293 К), кДж/моль 39,1 — —
ДЯ^, кДж/моль 174,1 (-134,9)** -473,5 -1056,1
С“ (300 К), Дж/(моль К) 109,9 (54,22)** 182 325
кДж/моль -33,68 -19,82 -8,92
5^, Дж/(моль К) 155,6 (266,9)** 217 344
♦Температура кипения. ♦♦Для газа при 298 К.
Азотная кис пота смешивается с водой во всех соотношениях. В водных растворах HNO3 практически полностью диссоциирует на Н+ и NOj. С водой образует азеотропную смесь (68,4%, масс. HNO3) с температурой кипения 120,7° С и плотностью </°4 = 1,41 моно- и тригидра-490
ты (рис. 10.52). Данные по плотности безводной азотной кислоты в зависимости от температуры приведены на рис. 10.53. Для водных растворов плотность при 20° С составляет 1,0543 (10%-ная кислота), 1,1150 (20%-ная), 1,3100 (50%-ная), 1,4134
(70%-ная), 1,4826 (90%-ная). На рис 10.54 приведены данные по общему давлению р паров HNO3 и Н2О над концентрированной азотной кис
Рнс. 10.52. Диаграмма кристаллизации системы NHO3 — HjO
лотой при 0—100° С, а на рис. 10.55 приведены данные по температуре кипения азотной кислоты при атмосферном давлении, а также по составу жидкой и паровой фаз. На рис. 10.56 видно, что наибольшую вязкость имеет азотная кислота при концентрации 65%.
Азотная кислота — сильнейший окислитель. Под ее действием металлы (за исключением Pt, Rh, Ir, Nb, Zr, Ta, Au) превращаются в нитраты или оксиды. Сера
энергично окисляется в H2SO4, фосфор — в Р2О5, а органические соединения окисляются и нитруются. В разбавленной азотной кислоте стойки хромоникелевые стали, титан, в концентрированной — чистый алюминий, высококремнистый чугун, хромоникелькремниевые стали. Титан в среде концентрированной кислоты, содержащей растворенные оксиды азота, приобретает пирофорные свойства. В азотной кислоте любой концентрации стойки стекло, кварц, фторопласт-4.
Для практических целей применяют 30—60%-ные водные растворы азотной кислоты или 97—99%-ные
Температура, %
Рис. 10.53. Плотность безводной азотной кислоты в зависимости от температуры
491
HNO3>% (масс.)
р, мм pm. cm.
Рис. 10.54. Общее давление паров HNO3 и Н2О над концентрированной азотной кислотой при 0—100° С
растворы (концентрированная азотная кислота). Смесь концентрированных азотной и хлороводородной кислот (соотношение по объему 1:3) называется царской водкой, растворяющей даже благородные металлы. Смесь 100%-ной HNO3 и 96%-ной H2SO4 при их соотношении по объему около 9:1 называется меланжем.
Неконцентрированная азотная кислота применяется в основном в производстве нитрата аммония и сложных минеральных удобрений, нитратов натрия, калия, кальция, бария и др., а также в гидрометаллургии. Концентрированная азотная кислота используется в производстве взрывчатых веществ, серной и фосфорной кислот, ароматических нитросоединений и красителей. Она входит в состав ракетного топлива. Азотная кислота применяется также для травления металлов, полупроводниковых материалов и др.
492
Концентрация HNO3, %(масс.)
Концентрация NH3, %(масс.)
Рис. 10.56. Вязкость азотной кислоты при 20°С
Рис. 10.55. Температура кипения азотной кислоты при 760 мм рт.ст. и состав жидкой и паровой фаз
Способы получения азотной кислоты. Химическая промышленность производит разбавленную (30—60%-ные растворы) и концентрированную азотную кислоту (97—99%-ные водные растворы).
Промышленные способы получения разбавленной азотной кислоты включают следующие стадии: получение оксида азота (И), окисление его диоксида азота (NOi), абсорбцию NO2 водой, очистку отходящих газов (содержащих в основном молекулярный азот) от оксидов азота.
Концентрированную азотную кислоту получают в основном двумя основными способами. Первый заключается в ректификации тройных смесей, содержащих азотную кислоту, воду и водоотнимающие вещества (обычно серная кислота или нитрат магния). В результате получают пары 100%-ной азотной кислоты (которые конденсируют) и водные растворы водоотнимающего агента. Последние упаривают и возвращают в производство. Второй способ основан на реакции
2^С>4(ж) + 2Н2О(Ж) + С>2(г) “ 4НЬЮз(Ж) + 78,8 кДж.
При давлении ~5МПа и использовании чистого кислорода образуется 97—98%-ная кислота, содержащая до 30% (масс.) растворенных оксидов азота. Целевой продукт получают разгонкой этого раствора.
493
Азотную кислоту особой чистоты получают ректификацией 97—98,5%-ной HNO3 в аппаратуре из силикатного или кварцевого стекла. Содержание примесей в такой кислоте менее 1-Ю"6 % (масс.).
Физико-химические основы производства азотной кислоты. Окисление аммиака. Исходное сырье—аммиак — каталитически окисляется кислородом воздуха с последующим поглощением образующихся оксидов азота водой. Процесс окисления аммиака является сложным процессом и может протекать как до целевого оксида азота (П), так и до оксидов азота (I) и молекулярного азота по реакциям:
4NH3 + 5О2 = 4NO + 6Н2О + 904,0 кДж,
4NH3 + 4О2 = 2N2O + 6Н2О + 1104,4 кДж,
4NH3 + ЗО2 = 2N2 + 6Н2О + 1268,8 кДж.
(6)
(7)
(8)
Кроме основных реакций в зависимости от условий проведения процесса могут протекать и другие, приводящие к образованию молекулярного азота, например:
2NO = N2 + О2 + 180,6 кДж,
6NO + 4NH3 = 5N2 + 6Н2О + 1810,5кДж,
2NH3 = N2 + ЗН2 - 91,6 кДж,
а образующийся водород горит с образованием паров воды:
2Н2 + О2 = 2Н2О
(9)
(Ю)
(11)
(12)
С целью получения полуфабриката — оксида азота (II), и исключения протекания других реакций процесс окисления аммиака проводят с участием активного и селективного катализатора. Общепринятым катализатором является платина, проявляющая высокую каталитическую активность в реакции (6) и обеспечивающая в температурном интервале 600—1000° С до 99% выход оксида азота (7) при продолжительности реакции около 1-Ю"4 с. При этом она имеет низкую температуру инициирования реакции (195—200° С), имеет хорошую пластичность, ковкость и тягучесть. С целью снижения разрушения платины при температурах конверсии под воздействием реакционной смеси и значительного количества «ядов» исходный катализатор изготовляют в смеси (в виде сплавов) с металлами платиновой группы. К ним относятся родий, палладий, рутений и осьмий. В настоящее время в качестве катализаторов процесса окисления аммиака служат 494
Рис 10.57. Зависимость выхода NO от температуры на различных платиноидных катализаторах: / — сплав Pt+10% Rh; 2 — Pt+2% Rh;
3— Pt+2% Ph; 4 — Pt+1% lr, 5 — чистая плагина
сплавы платины с родием, палладием, реже с рутением. Это объясняется близостью их свойств. Они обладают высокой темпера!у-рой плавления (>1500° С), легко поддаются механической обработке, а палладий и платина—и сварке. Родий, палладий и рутений образуют сплавы непрерывного ряда твердых растворов и они обладают лучшими механическими свойствами, чем платина.
При использовании в качестве катализаторов сплавов в них сохраняются свойства, присущие чистой платине: увеличение выхо
да оксида азота (II) при повышении температуры. Сплавы обладают лучшими механическими свойствами с одновременным влиянием на селективность катализатора, например увеличение содержания родия в сплаве до 4% обеспечивает в равных условиях повышение выхода оксида азота (II) на 4—5%, а палладия — на 2—3% (рис. 10.57).
В настоящее время в азотной промышленности в качестве катализатора широко применяются сплавы платины с 7—10% родия.
В производстве азотной кислоты платиноидные катализаторы применяются в виде сеток, так как эта форма удобна в эксплуатации и позволяет применять наиболее простой и удобный в обслуживании тип контактного аппарата. Для изготовления сеток применяют проволоку диаметром от 0,06 до 0,09 мм с размерами стороны ячейки 0,22 мм. Таким образом, на 1 см2 приходится 1024 ячеек.
Термодинамические расчеты изменения свободной энергии показывают, что приведенные реакции (6—8) при темпера!уре 1173 К имеют следующие значения константы равновесия:
кх = р™'р°г =ю~53, (13)
^NO^H2O
- _ Pnh^O2 _in-6i 2“1 6 1U
^N2oAi2O
K PnH3Po2 in-67
T-=1°
Pn2 Рй2о
(14)
(15)
495
Практически все они могут считаться необратимыми. При нормированных условиях наибольшую термодинамическую вероятность имеет реакция (8). Таким образом, процесс окисления аммиака может привести лишь к образованию элементного, а затем молекулярного азота. Однако направление реакции и ее конечный продукт в данном случае определяются селективностью катализатора. Поэтому, изменяя катализатор и условия проведения процесса, можно изменить и состав продуктов реакции.
Энергетическое состояние экзотермической реакции окисления аммиака характеризуется данными, приведенными на рис. 10.58.
Процесс контактного окисления аммиака начинается со стадии активированной адсорбции кислорода на поверхности катализатора К О
с образованием промежуточного соединения К—О, после чего происходит активированная адсорбция аммиака, требующая меньшей энергии активации. В процессе образуется переходный комплекс О О—NH3
К.—О—NH3 с последующей перегруппировкой его в комплекс К.—О • Вследствие перераспределения электрических связей возможны и другие промежуточные соединения: гидроксидамин NH4OH, нитроксил HNO, амид NH2, имид NH, атомарный азот N и полученные в конечном результате NO, N2 и НгО.
Учеными было предложено несколько механизмов процесса каталитического окисления аммиака. Согласно адсорбционно-химической теории катализа, процесс окисления представляется следующим образом. Кислород и аммиак диффундируют из газовой смеси к поверхности катализатора. При стехиометрическом отношении C>2:NH3 = 5:4 скорость процесса определяется диффузией кислорода и аммиака к поверхности катализатора (платины). Вследствие высокой температуры контактирования ковалентная связь между атомами этих газов ослаблена, в результате чего образуется пе-роксидный комплекс катализатор — кислород. В последующей
Рис 10.58. Энергетическое состояние экзотермической реакции окисления аммиака:
/, 2 — исходное и конечное состояния; К—катализатор; £], Е2 и £?— энергии активации исходных, промежуточных и конечных продуктов; U\ и U]— внутренняя энергия в начальном и конечном состояниях; Q — тепловой эффект реакции (кривая а—процесс гомогенного окисления аммиака, кривая б — процесс гетерогенного окисления)
496
стадии активированной адсорбции аммиака образуется новый комплекс катализатор — кислород-аммиак (рис. 10.58). Далее происходит перераспределение электронных связей, и атомы азота и водорода соединяются с кислородом:
4N3’H* + 5О°2 = 4N2+O2’ + 6Н2'О2’
Механизм процесса окисления аммиака на поверхности платины схематично представлен на рис. 10.59.
Адсорбированный кислород не входит в кристаллическую решетку платины, незначительное его количество находится в растворенном состоянии, часть адсорбированного кислорода образует с платиной непрочные связи. В результате возникает комплекс катализатор — кислород.
Молекулы аммиака также подвергаются активизированной адсорбции, причем высокое химическое сродство водорода к кислороду ориентирует аммиак к поверхности катализатора атомами водорода. При этом происходит перераспределение электронных связей, причем с кислородом комплекса реагирует как водород с образованием воды, так и азот аммиака с образованием оксида азота(11). Поскольку эти соединения обладают меньшей адсорбционной способностью, чем исходные ингредиенты, то они уносятся с поверхности катализатора потоком воздуха. В процессе повторной адсорбции кислорода снова образуются активированные комплексы платина — кислород, и процесс повторяется.
Образование элементного, а затем и молекулярного азота происходит в процессе неполного покрытия поверхности катализатора кислородом (недостаток кислорода в газе) и за счет протекания побочных реакций после контактного аппарата.
Для реакции каталитического окисления аммиака характерны зависимость скорости от линейной скорости газа, незначительная продолжительность процесса контактирования (НГ4—10’5) и небольшая энергия активации (до 2,5 кДж/моль), что свойственно диффузионным процессам. Исходя из этого предложена следующая зависимость
Рис. 10.59. Схема процесса окисления аммиака на поверхности платины (сплошными линиями обозначены ранее возникшие связи, пунктирными — вновь образующиеся связи)
497
для определения количества прореагировавшего аммиака от параметров сетки и скорости газа:
1g = 0,951 — [0,45 + 0,288(cZKo )056 ], (16)
Ст dvo
где Со и Ст — концентрации аммиака соответственно в исходной АВС (аммиачно-воздушная смесь) и в смеси за сетками, %; S — геометрическая поверхность 1 см2 сетки, см2; п — число сеток; d—диаметр проволоки, см; Ко — скорость АВС, приведенная к 0° С и 0,1013 МПа, л/(ч/см2).
Зависимость дана для температуры 850° С с оговоркой, что температура оказывает незначительное влияние на количество прореагировавшего аммиака. Уравнение (16) позволяет определять лишь общее количество прореагировавшего аммиака, но не выход оксида азота (II), поскольку последний значительно зависит от температуры. Расчеты по этому уравнению показывают, что продолжительность контактирования (0,9-1 О'4 при 900° С), оптимальная для выхода NO, количество непрореагировавшего аммиака, находящееся в нитрозном газе, составляет 0,13% от его начального объема.
Предложены также несколько кинетических уравнений для определения оптимальной продолжительности контактирования:
^т=Ла + Ва3, (17)
где a — степень конверсии исходного аммиака до NO (в пределах 90—98%); А и В — коэффициенты, существенно зависящие от температуры процесса [900 и 800° С они соответственно равны:-0,107 и -0,0857 (для А), 7,02-10'6 и 5,49-1 О’6 (для В)].
Кинетика процесса окисления аммиака до оксида азота (II) под давлением в пределах a = 95—98% описывается уравнением
dPflQ _ PnH* ~ PNHi прев (18)
dx Pno
Интегральная форма уравнения (18) имеет вид
прев111 ** >
апрев-а
где ^прев — парциальное давление аммиака, превратившегося в N2O и N2, МПа; к — константа скорости, с’1, определяемая по уравнению
498
33494 к - Ае яг ,
(20)
А =9-105еЧ25 |0".р2 -3,43-10> + 7,8-Ю5,
где 33 494 — энергия активации, Дж/моль; р — давление, МПа; R = = 8,32 Дж/(моль-К).
Реальная продолжительность контактирования может быть представлена в виде отношения свободного объема катализатора (Ксв, м3) к объемной скорости газа в условиях конверсии (К2, м3/с). После замены указанных величин на параметры, доступные для применения, уравнение для расчета продолжительности будет иметь вид
_ 80,85(1 - l,57d4n)SdmpK (21)
Х= ’
где d—диаметр проволоки сетки, см3; п — число плетений в 1 см3 площади сеток; S—площадь сетки, м2; т — число сеток в слое; Vo— скорость газа при 0°С и 0,1013 МПа, м3/(м2*с); ТК — температура конверсии, К; рк— давление газа в процессе конверсии, МПа.
Определяя из кинетических уравнений величину продолжительности контактирования и исходя из принятых условий конверсии, можно вычислить число сеток, а задаваясь продолжительностью их работы— объем вложения платиноидов.
Изучение кинетики процесса окисления аммиака до оксида азота (II) осложняется протеканием параллельных и последовательных побочных реакций, конечным продуктом значительного количества из которых является молекулярный азот. К ним относятся: 1) реакции окисления аммиака до N2O и N2; 2) разложение оксида азота; 3) термическое разложение исходного аммиака; 4) реакции (1) и (2), протекающие в основном на стенках аппарата до поступления на катализатор при перегреве АВС.
Они совершаются также при загрязнении сетки или при снижении температуры ниже оптимальной.
Реакция разложения оксида азота (II) может протекать также после появления NO в газе, но скорость этой реакции намного ниже скоростей реакций (6) — (8). Поэтому ее влияние имеет место на участке кинетической кривой после максимума (рис. 10.60).
Кинетическое уравнение разложения оксида азота (И) имеет вид
= (22)
499
Реакция (10) проходит в более благоприятных условиях в период контактирования меньше оптимального (участок кривой до максимума, рис. 10.62). Она более вероятна после появления NO при наличии в газах (в пространстве между сетками) значительных концентраций аммиака. Кинетическое уравнение
4NH,] [N^flNOr
rft [NO]
(23)
достаточно хорошо согласуется с экспериментальными данными. Имеются также сведения, что взаимодействие аммиака и оксида азота (II) сильно тормозится в присутствии кислорода.
Реакция (10) может протекать также за катализаторными сетками при проскоке аммиака через них при большой линейной скорости газа или по другим причинам. При этом в результате быстрого охлаждения при температурах ниже 600° С может протекать также реакция
8NH3 + 6NO2 = 7N2 + 12Н2О + 2734 кДж. (24)
Протекание подобных реакций во всех случаях снижает выход оксида азота (II).
На выход оксида азота наиболее существенное влияние оказывает температура. Реакция окисления аммиака на металлах начинается при относительно низких температурах, например на чистой платине при 195° С, а на платиноидных катализаторах наблюдается при температурах около 300° С. В процессе дальнейшего повышения температуры выход оксида азота (II) увеличивается как за счет уменьшения проско-
ка аммиака, так и в результате снижения содержания в продуктах реак-
Время контактирования, с-
Рис 10.60. Зависимость выхода оксида азота (II) от продолжительности процесса контактирования (t = 850° С; Р = 0,44 МПа и V = 4 м/с)
1(f
ции N2 и N2O, т.е. проявление катализатором большей селективности к реакции (1). Максимальный же выход оксида азота (II) наблюдается при 900—920° С. На чистой платине он достигает 96%, а на сплавах Pt—Rh и Pt—Rh—Pd—99%. При этих температурах вторым продуктом окисления является азот.
Поскольку при атмосферном давлении и повышении температуры от 800 до 900° С выход увеличивается незначительно, а потери платины существенно возрастают, процесс окисления аммиака в промышленных установках проводят при 800° С. При средних давлениях (0,3 — 0,5 МПа) окис-
500
Рис. 10.61. Зависимость выхода оксида азота (II) от соотношения [O2]:[NH3] при различной температуре:
7 — 680° С; 2 —760° С;
3 — 860° С
Давление, МПа
Рис. 10.62. Зависимость выхода оксида азота (II) от давления и температуры при оптимальном времени контактирования:
1 — температура 900° С; 2 —950° С; 3 — 980°С; 4—1010° С
ление проводят при 850 — 870° С, а при высоких давлениях (0,7—1,0 МПа) температура процесса возрастает до 900 — 920° С.
Существенное влияние на выход оксида азота (II) оказывает также концентрация кислорода в исходной АВС. Теоретически соотношение [O2]:[NH3] » п в АВС в соответствии с реакцией (1) должно быть равно 1,25 (по объему). При использовании воздуха максимально возможной концентрацией аммиака в АВС, когда достигаются высокие выходы оксида азота (II), является 11,0—11,5% и [С>2]:[МНз]»1,7, причем это характерно для интервала температур 870 — 900° С. При более низких температурах (750 — 800° С) содержание аммиака в АВС должно быть снижено примерно до 10% (и<1,8) (рис. 10.61).
Обычно выход оксида азота (II) при проведении процесса конверсии под давлением несколько ниже, чем в сравнимых условиях при атмосферном давлении.
Установлено, что можно достичь высокого выхода оксида азота (II) и при больших давлениях, вплоть до 3,59 МПа (рис. 10.62). Основным условием при этом является повышение температуры конверсии, например для достижения степени конверсии на уровне 98% под давлением 1,57 МПа необходимо поднять температуру до 980—1000° С.
Повышение давления в процессе конверсии увеличивает линейную скорость газа и напряженность катализатора (количество аммиака, окисляемого на 1 мг поверхности сетки или отнесенного к 1 г катализатора в единицу времени).
Однако изменение этих параметров требует увеличения числа ка-тализаторных сеток. Эти факторы взаимосвязаны, и для получения высоких выходов оксида азота (II) при осуществлении процесса под давлением они должны быть подобраны с необходимой точностью.
501
Напряженность, кг NHg/fM*cyr)
Рис. 10.63. Зависимость выхода NO от напряженности катализатора при различной температуре (С^ — 11,0—11,2%; Р = 0,101*3 МПа):
/ — 800° С; 2 — 860° С
При изменениях напряженности катализатора в процессе при атмосферном давлении в пределах от 900 до 1100 кг КНз/(м2-сут) выход оксида азота проходит через максимум (рис. 10.63). В то же время он имеет тенденцию к сниже
нию при увеличении содержания аммиака в АВС и снижению температуры контактирования (рис. 10.63) при прочих равных условиях. В узком интервале изменения напряженности 500—900 кг/(м2-сут) выход оксида азота (II) меняется незначительно.
Применение повышенного давления резко повышает скорость окисления азота до оксида азота (II). Оно позволяет повысить производительность системы и предопределяет компактность всей установки. Так, в результате применения платинородиевых сеток и перехода на многослойные компоненты (16—18 сеток) в процессе окисления аммиака под давлением 0,8 МПа, предварительном подогреве газа до 300° С и температуре конверсии около 900° С удалось добиться 96%-ного выхода оксида азота (II).
Производительность конвертора повышается пропорционально увеличению числа сеток и возрастанию давления газа. С ростом давления оптимальная температура конверсии возрастает выше 900° С.
Интенсивность образования оксида азота зависит и от продолжительности пребывания газа при высокой температуре, развиваемой раскаленными стенками конвертора. Это требует увеличения числа сеток в процессе окисления аммиака. Увеличение числа сеток сокращает, в свою очередь, продолжительность пребывания газа в конверторе.
Данные исследований, проведенных в Харьковском политехническом институте (В. И. Атрощенко и др.), приведены на рис. 10.64,
1,4 1,2 1,0 О,в 0,6 1,6 1,4 1,2 1,0 0,8 0,6
Продолжительность катализа т-ТО4, с а) б)
Рис. 10.64. Зависимость степени конверсии аммиака от времени катализатора и температуры:
а — давление I МПа на 20 сетках; б — давление 2 МПа на 30 сетках
502
Давление, МПа Рис. 10.65. Зависимость продолжительности катализа аммиака от давления и температуры при максимальном выходе NO:
1 — при 900° С; 2—при 950° С; 5 —при 980° С
Рис. 10.66. Зависимость степени конверсии аммиака от давления и температуры при оптимальной продолжительности процесса катализа:
1 — при 1010° С; 2 —при 980° С; 3 —при 950° С; 4 — при 900° С
10.65 и 10.66. Как видно из данных рисунков, переход к повышенному давлению в процессе окисления аммиака требует увеличения числа сеток, повышения температуры и сопровождается некоторым повышением продолжительности процесса конверсии аммиака.
Изучена также зависимость выхода оксида азота (II) от нагрузки по аммиаку, приходящейся на единицу поперечного сечения сетки в 1 с (рис. 10.16). Эта величина — так называемая секундная удельная напряженность (СУН) — объединяет два фактора: количество реагирующего аммиака и линейную скорость газа. Из данных на рис. 10.67 видно, что большинство промышленных установок работает при СУН, равной 0,06—0,18 кг NH3 (м2-с), что соответствует линейным скоростям газа в рабочих условиях 1,0—1,5 м/с. При этом обеспечивается получение устойчивой степени конверсии на уровне 96—97%.
Особенностью зависимости выхода оксида азота (II) от СУН является резкое его возрастание при снижении напряженности менее 0,15 кг/(м2-с), или 540 кг/(м2-сут). По мере снижения СУН выход NO стремится к 100% и, наоборот, увеличение ее более 0,2 кг МНз/(м2-с) изменяет выход NO незначительно. При этом требуемое увеличение числа сеток непропорционально возрастанию удельной напряженности.
При снижении линейной скорости газа или СУН одновременно уменьшаются удельные потери катализатора (рис. 10.67) и увеличивается продолжительность безостановочного пробега сеток.
На основании практических данных принято продолжительность работы сеток при 800° С ограничить выработкой 1500—1600 кг HNO3, а при 900—920° С— 1300—1400 кг HNO3 на 1 г массы сеток. Исходя 503
из этого продолжительность пробега сеток (в ч) может быть рассчитана по уравнению
Qgn
(25)
а расход катализатора в г на 1 т HNO3 (100%-ной), выработанной за пробег, по уравнению
(26)
Т0’
где Q — производительность контактного аппарата, т/ч; q — масса сетки, г; п — число сеток; 0 — выработка HNO3 (100%) за пробег т/ч с 1 г массы сеток.
Анализ уравнений (25) и (26) показывает, что при одной и той же выработке азотной кислоты в расчете на единицу массы сеток можно значительно изменять продолжительность пробега за счет варьирования числа сеток или диаметра их нитей.
Для комбинированных схем, в которых конверсию ведут при средних давлениях и линейных скоростях газа 1,0—1,5 м/с, пробег обычно составляет полгода (4000 ч), вложения платиноидов 0,62—0,65 г/т кислоты, а потери катализатора меньше 0,1 г/т HNO3. При высоких же давлениях вследствие увеличения напряженности катализатора потери составляют 0,72—0,85 г/т HNO3 в зависимости от давления до 0,15—0,20 г/т HNO3.
Основными причинами, вызывающими потери платиноидного катализатора, являются окисление аммиака при высоких температурах,
СУН, кг
Рис. 10.67. Зависимость выхода NO от числа сеток (d = 0,09 мм) и удельных потерь от СУН катализатора
(/ = 820—900° С, р = 0,101—0,98 МПа) 504
присутствие в газе окислителя, воздействие примесей исходных газов. При этом происходит изменение катализаторных сеток, разрыхление их поверхности, увеличение размеров кристаллов и т.д.
Установлено, что при высоких температурах в окислительной среде происходит равномерное по всей поверхности окисление металлов платиновой группы. По устойчивости к окислению металлы этой группы располагаются в ряд: родий, платина, палладий.
Окисление платины происходит по реакции
Pt<ra) + О2(г) = PtO2(r) + 172,9 кДж. (27)
Зависимость константы равновесия данной реакции от температуры имеет вид
= _^£_08861+0j55361gr_0j2557.10-37’-0,0765-1057”2. (28)
т
Исследования в производственных условиях показали, что прямые удельные потери катализатора растут с повышением температуры процесса (рис. 10.68), особенно выше 850° С. Из рисунка видно, что применение сплавов платины с родием, а также родия с палладием позволяют значительно снизить потери, которые значительно выше в системах под давлением.
Неравноценны потери отдельных сеток в комплекте, например первые по ходу газа сетки теряют в 2,6—25 раз больше массы, чем сетки, расположенные в нижней части комплекта. При этом первая по ходу газа сетка теряет несколько меньше массы, чем вторая (рис. 10.69). Одновременно с этим, чем ниже линейная скорость газа, тем больше потери первых по ходу газа сеток. Низкие потери первой сетки объясняются более низкой температурой (к ней подходит более холодная АВС).
Установлено, что в зависимости от температуры, скорости газа и других технологических факторов потери сеток за счет испарения
Температура, Ъ
Рис. 10.68. Зависимость потерь массы катализаторных сеток от температуры:
1 — сетки из платины; 2 — из Pt+10% Rh (нить диаметром 0,075 мм); 3 — сплав №1 (нить диаметром 0,09 мм, пробег 250 ч; давление — 0,1013 МПа)
Номер сетки по ходу газа
Рис. 10.69. Зависимость потерь массы сеток от износа:
1—р = 0,588 МПа, скорость газа 3,0—3,5 м/с; 2 — 0,715 МПа, 4,6 —5,0 м/с; 3 — 0,785 МПа, 5,5—6,0 м/с
505
оксидов платины составляют 40—70% общих потерь, а остальные 30—60% вызваны механическим уносом частиц катализатора.
Потери платиноидов могут быть снижены: 1) оптимизацией технологических процессов конверсии; 2) разработкой катализаторных сплавов с добавками более тугоплавких металлов; 3) заменой в сетках части нитей из платиноидных сплавов на нити из жаростойких сплавов других металлов; 4) переходом на двухступенчатое окисление аммиака с применением в качестве второй ступени недефицитных оксидных катализаторов; 5) разработкой различных способов улавливания.
Переработка оксидов азота в азотную кислоту. Полученный при каталитическом окислении аммиака оксид азота (II) — слабое реакционноспособное, не реагирующее с водой соединение. Для получения азотной кислоты его окисляют до оксидов, способных образовывать искомую кислоту.
В процессе окисления оксида азота (II) кислородом образуется диоксид:
2NO + О2 = 2NO2 + 124 кДж. (29)
Образующийся оксид азота может полимеризоваться до N2O4 и взаимодействовать с оксидом азота (II) по схеме
2NO2 = N2O4 + 56,9 кДж, (30)
NO2 + NO = N2O3 + 40,1 кДж. (31)
Равновесие приведенных реакций при низких температурах смещено вправо. Поэтому в нитрозном газе при избытке кислорода и достаточной продолжительности контактирования все приведенные оксиды азота после охлаждения могут быть превращены в тетраоксид диазота. В реальных же условиях непрерывно протекающих процессов окисления и кислотообразования равновесие не достигается, в результате чего в смеси газов присутствуют все указанные оксиды азота — NO, NO2, N2O3 и N2O4. Соотношение между их количеством определяется в основном температурными условиями. С понижением температуры равновесие реакции сдвигается в сторону образования диоксида азота. Установлено, что при атмосферном давлении в нитрозных газах, полученных конверсией аммиака воздухом, при температуре около 150° С в газе находится лишь NO2, а при 700° С — оксид азота (II). Повышение давления сдвигает равновесие в сторону образования оксида азота (IV). Процесс является двухстадийным:
NO + О2 = NO3
NO3 + NO = 2NO2 506
Первая стадия реакции протекает быстро, с повышением температуры скорость ее увеличивается, однако равновесие смещается влево. Вторая стадия медленная. Поэтому она определяет скорость суммарного процесса.
С появлением в системе NO2 при температурах ниже 150—200° С начинаются процессы по реакциям (30) и (31).
Скорость процесса полимеризации диоксида азота очень велика. Равновесие реакции (30) устанавливается практически мгновенно— за Ю^с, поэтому в расчетах принимают, что в любой взятый отрезок времени смесь NO2—N2O4 находится в равновесии.
Образованию сесквиоксида азота способствуют пониженные температуры. Равновесие реакции (31) сдвигается в сторону образования N2O3 при понижении температуры, повышении давления и увеличении концентрации оксидов азота в газе. Приближение степени окисления NO к 50% также способствует увеличению содержания в ни-трозном газе N2O3.
Поскольку содержащийся в газе диоксид азота немедленно полимеризуется (особенно при пониженных температурах), то содержание N2O3 в нитрозных газах невелико (около 7%). Обычно в нитрозных газах при 25° С и степени окисления 50% содержание N2O3 не превышает 2%. Поэтому при расчете процесса переработки нитрозных газов содержанием в них N2O3 пренебрегают, хотя и в определенных условиях кислотообразование может идти путем поглощения сесквиоксида азота.
Поскольку азотная кислота термодинамически устойчива при температуре ниже 250° С, в процессе охлаждения нитрозных газов ниже этой температуры единовременно с окислением оксида азота (II) протекают процессы кислотообразования. В газовой фазе при высоких степенях окисления оксида азота (II) оксид азота (IV) взаимодействует с водяным паром. Однако количество образовавшихся паров азотной кислоты невелико. При этом образуется также и азотистая кислота (HNO2), но и ее концентрация даже в наиболее благоприятных условиях при 50%-ной степени окисления оксида азота (II) в нитрозных газах, полученных конверсией аммиака воздухом, невелика.
Присутствующие в нитрозных газах контактного окисления аммиака ди- и сесквиоксиды азота, а также тетраоксид диазота (NO2, N2O3, N2O4) способны взаимодействовать с водой. Так, сесквиоксид образует с водой азотистую кислоту, а тетраоксид диазота — азотную и азотистую кислоты:
N2O3 + Н2О # 2HNO2 + 55,6 кДж, (32)
N2O4 + Н2О HNO3 + HNO2 + 59,2 кДж. (33)
507
Азотистая кислота — неустойчивое соединение и в растворах азотной кислоты разлагается:
4HNO2 N2O4 + 2NO + 2Н2О (34)
Суммарный процесс разложения с учетом протекания обратимых реакций (33) и (34) можно записать в виде
3HNO2 = HNO3 + 2NO + Н2О - 75,8 кДж. (35)
Образующийся при этом оксид азота (II) в растворах примерно до 60%-ной азотной кислоты частично окисляется в жидкой фазе до NO2, а в основном — газовой фазе — после выделения из раствора:
2NO + О2 = 2NO2
В общем виде процесс образования 55—60%-ной HNO3, протекающей при поглощении оксидов азота из нитрозных газов, полученных конверсией аммиака воздухом, описывается следующими реакциями:
3NO2 + Н2О = 2HNO3 + NO + 136,2 кДж, (36)
3N2O4 + 2Н2О = 4HNO3 + 2NO + 101,8 кДж, (37)
3N2O3 + Н2О = 2HNO3 + 4NO + 15,2 кДж. (38)
Механизм образования азотной кислоты путем поглощения оксидов азота считается бесспорным лишь при получении кислоты концентрацией до 60% HNO3. В процессе образования кислоты более высокой концентрации имеют место процессы гидратации, сольватации и ионизации молекул в растворе. Так, взаимодействие тетраоксида диазота с водой в жидкой фазе протекает по реакции
2N2O4 + Н2О = 2HNO3 + N2O3 (39)
При этом одновременно с азотной кислотой образуется эквивалентное по связанному азоту количество сесквиоксида азота.
В данном случае не утверждается, что реакция взаимодействия N2O4 и Н2О является первичной. Считается, что вначале идет реакция (33), а разложение азотистой кислоты протекает не по реакции (35), а с образованием сесквиоксида азота и воды или азотной кислоты и сесквиоксида азота:
HNO2 + N2O4 = HNO3 + N2O3 (40)
508
Во всех случаях суммарной реакцией является реакция (39), которая и определяет состояние равновесия в растворе. Влияние реакции (39) на процесс кислотообразования начинает проявляться лишь при достижении определенного содержания молекул недиссоцииро-ванной азотной кислоты в растворе (около 56—60% HNO3).
Важным вопросом в производстве азотной кислоты является обезвреживание отходящих нитрозных газов. В связи с высокой токсичностью оксидов азота для снижения их содержания в выхлопных газах до концентраций менее 0,01% (об.) NOX используют специальные методы очистки.
Одним из методов очистки является высокотемпературное восстановление оксидов азота до молекулярного азота. До поступления на катализатор выхлопной газ подогревают до температуры начала гетерогенной реакции и в него вводят газ-восстановитель. Температура подогрева определяется в зависимости от состава катализатора и характером газа-восстановителя и меняется в пределах от 250 до 550° С. В качестве газа-восстановителя могут быть применены водород, природный газ, оксид углерода, азотоводородная смесь, пары керосина и др.
Процесс восстановления оксидов азота газами протекает без кислорода. Поэтому первой стадией процесса является «выжигание» кислорода. В случае применения природного газа в качестве восстановителя горение протекает по реакции
СН4 + 2О2 = СО2 + 2Н2О + 804,58 кДж.
(41)
При неполном сгорании метана образуются водород и оксид углерода:
СН4 + 0,5О2 = СО + 2Н2 + 35,13 кДж.
(42)
Восстановители реагируют на катализаторе с оксидом азота:
NO2 + 2Н2 = NO + Н2О
(43)
2NO2 + 2Н2 = N2 + 2Н2О
(44)
4NO2 + СН4 = 4NO + СО2 + 2Н2О
4NO + СНд = 2N2 + СОг + 2Н2О +1160 кДж.
(45)
(46)
Обычно во всех реакциях отношение [СН4]:[О2] поддерживают на уровне 0,55—0,56.
509
В качестве катализатора высокотемпературного восстановления используют различные металлы, нанесенные на разнообразные носители, например палладиевый катализатор АПК-2, представляющий собой таблетированный оксид алюминия, на который наносят 2% Pd. В качестве восстановителя применяется природный газ. Процесс восстановления протекает при 400—800° С. Для достижения остаточной концентрации оксидов азота 0,002—0,008% в процессе поддерживают ~ 10%-ный избыток природного газа.
Разработано и применяется в различных производствах селективное восстановление оксидов азота. В качестве катализаторов селективного восстановления при 200—350° С и скорости газа 10 000 ч'1 убывает в последовательности: Р1>МпО2>У2О5>СиО>Ре2Оз>Сг2Оз>Со2Оз>МоОз> NiO>WO3>Ag2O>ZnO>Bi2O3>A12O3>SiO2>PbO.
Селективная каталитическая очистка используется в агрегатах, оборудованных низкотемпературной рекуперационной турбиной. При этом применяют алюмованадиевый катализатор типа АВК-10. На этом катализаторе (CN0 = 0,08—0,1%, СОг= 4%, СНг0 = 3%, р = 0,101 МПа) при 240—280° С степень восстановления оксидов азота близка к 100%. Катализаторы в промышленных условиях служат три года. За этот период степень очистки снижается лишь до 96%.
В качестве абсорбентов для поглощения оксидов азота различных растворителей используют водные растворы ацетата натрия, диметилсульфоксид, трибутилфосфат (ТБФ), адиподинитрил и др. Установлено, что ТБФ обладает высокой поглотительной способностью по отношению к оксиду азота (IV). Так, при 20° С и при р^0 = 1,96 кПа в 1 м3 ТБФ растворяется 17,5 м3 NO2, а при рЖг = 2,94-104Па — 460 м3 NO2. Поглощение диоксида азота (IV) сопровождается образованием сольватов ТБФ-ЫОг или 2ТБФ^гО4 и физическим растворением NO2 при содержании его в растворе сверхстехиометрического количества в соотношении 14О2:ТБФ = 1. Десорбция NO2 из ТБФ проходит достаточно легко при снижении давления или нагревании раствора до 96—105° С.
В качестве адсорбентов применяют активированные угли, силикагель, цеолиты, торфощелочные сорбенты, гидроксиды бария, стронция, магния, оксид кальция, порошкообразный карбонат натрия, фосфориты и др. Главными из них являются силикагели и цеолиты.
Установлено, что эффективность адсорбции увеличивается при понижении температуры. Так, при 17° С в течение 20 мин поглощается в два раза больше NO2, чем при 40° С. С увеличением продолжительности контакта влияние температуры возрастает. Однако равная степень насыщения силикагеля при повышенных температурах (например, при 40° С) достигается в два раза быстрее, чем при пониженных (12° С).
510
Производство азотной кислоты под давлением 0,650 — 0,716 МПа. Основным видом сырья для получения азотной кислоты является аммиак, который испаряют в специальных испарителях. Существующий ГОСТ 6221—82 на аммиак предъявляет следующие требования:
Норма для марок
Показатель А Ак Б
Содержание:
аммиака, %, не менее 99,96 99,6 99,6
азота, %, не менее — 82 82
воды, %, не более 0,04 0,2—0,4 0,2—0,4
масла, мг/дм3, не более 2 2 8
железа, мг/дм3, не более 1 1 2
общего хлора, мг/кг, не более — 0,5 —
диоксида углерода, мг/кг, не более — 30±10 —
На рис. 10.70 показано влияние примеси масла в аммиачно-воздушной смеси на степень конверсии аммиака. Из рисунка видно, что при содержании масла в смеси 0,5 мг/м3 и менее степень конверсии аммиака может достигать 97% и более. С целью повышения степени конверсии исходный аммиак пропускают через фильтр из ультратон-кого стекловолокна, что позволяет повысить степень конверсии на 1—2%. Для очистки исходного газа от железа (катализаторной пыли) применяют метод магнитной сепарации. При прохождении исходного жидкого аммиака в магнитном поле, создаваемом насадкой из постоянных магнитов, ферромагнитные частицы оседают. Степень очистки аммиака от примесей железа составляет ~90%.
При содержании диоксида серы в аммиачно-воздушной смеси более 2,3 мг/м3 (в пересчете на серу) происходит отравление платинового катализатора и снижается степень конверсии аммиака (рис. 10.71). Процесс отравления платинового катализатора серой
Содержание аэрозолей масла, мг/м3
Рис. 10.70. Зависимость степени конверсии аммиака от содержания аэрозолей масла в АВС
Рис. 10.71. Зависимость степени конверсии аммиака от содержания серосодержащих соединений (SO2 и H2S) в воздухе
511
имеет обратимый характер: работа на чистом воздухе в течение 10 ч восстанавливает активность катализатора. Поэтому в производстве используют одноступенчатую очистку воздуха на различных фильтрующих тканях (грубошерстное сукно, ткани Петрянова из синтетических волокон).
На рис. 10.72 приведена схема производства азотной кислоты под единым давлением 0,716 МПа, согласно которой атмосферный воздух проходит трехступенчатую очистку. Первые две ступени — сухая — на суконном фильтре и мокрая — на тарельчатом промывателе. Они конструктивно совмещаются в одном фильтре 7, расположенном перед входом воздуха в компрессор.
Очищенный атмосферный воздух далее поступает в компрессорное отделение 2. Повышение давления воздуха до 0,716 МПа выполнено в две ступени. Первая ступень повышения давления до 0,343 МПа происходит в осевом компрессоре а, конструктивно выполненном на одном валу и в одном корпусе с газовой турбиной в. Нагретый до 174° С за счет адиабатического сжатия воздух охлаждается в промежуточном поверхностном холодильнике е до 42° С и поступает в центробежный компрессор (нагнетатель) б, в котором давление воздуха повышается до 0,716 МПа.
Заданная (расчетная) температура воздуха на выходе из центробежного компрессора равна 135° С.
Из компрессора б поток воздуха направляется на производство азотной кислоты, на каталитическую очистку и на собственные нужды ГТУ. Поток воздуха, поступающий на производство азотной кислоты, также делится на несколько потоков. При этом основной поток после подогрева его теплом нитрозных газов в подогревателе 5 направляется в смеситель 16, где воздух смешивается с очищенным газообразным аммиаком. Количество газовой смеси автоматически регулируется в зависимости от заданной выработки азотной кислоты.
Второй поток воздуха поступает в трубопровод нитрозных газов перед абсорбционной колонной 70, которая обеспечивает полноту переработки оксидов азота в азотную кислоту. Расход этого потока также регулируется автоматически по заданной температуре газов после реактора каталитической очистки 3.
Третий поток направляется в продувочную колонну 77 для отдувки оксидов азота из полученной азотной кислоты. Поток воздуха на каталитическую очистку направляется в камеру подготовки газов (камеру сгорания реактора) 4 для сжигания горючего газа. Количественно этот поток автоматически регулируется по заданной температуре газов за камерой 4. Температура газов задается со щита управления в зависимости от активности катализатора очистки хвостовых газов от оксидов азота. Поток воздуха на собственные нужды ГТТ-3 также разветвляется на несколько потоков. Один из них, предназначенный 512
Химическая технология неорганических веществ, кн.
Рис. 10.72. Схема производства азотной кислоты под единым давлением 0,716 МПа:
1 — фильтры воздуха; 2 — газотурбинная установка ГТТ-3 (а— осевой компрессор, б—центробежный компрессор-нагнетатель, в — газовая турбина, г—электродвигатель-генератор (2 ФАЗ 800—6000), д — пусковая камера сгорания, е — промежуточный холодильник воздуха); 3 — реактор каталитической очистки; 4— камера подготовки газов (сгорания); 5, 6—подогреватели; 7, 9—сепараторы; 8 — холодильник-конденсатор; 10—абсорбционная колонна; 11— продувочная колонна; 12 — окислитель; 13 — контактный аппарат; 14, 17—котлы-утилизаторы; 15—фильтр АВС; 16—смеситель; 18, 19—экономайзеры; 20—деаэратор; 21 — питательный насос
для поддержания дежурного факела в пусковой камере горения д на номинальном режиме, проходит через пусковую камеру сгорания турбины. В процессе переменного режима это позволяет регулировать расход воздуха в химико-технологических процессах в любых пределах— от нуля до номинального.
Другие потоки воздуха на собственные нужды ГТУ направляются на охлаждение и в турбину. Количественно они зависят от состояния лабиринтных уплотнений турбины.
Смешение аммиака с воздухом происходит в смесителе 7 <5. Соотношение количеств аммиака и воздуха, подаваемых в смеситель, регулируется автоматически. Из смесителя аммиачно-воздушная смесь поступает в фильтр 75, в котором завершается процесс очистки смеси. Смеситель и фильтр совмещены в одном аппарате. Из фильтра 75 АВС поступает на катализаторные сетки контактного аппарата 13, в котором при 900° С происходит процесс окисления аммиака с образованием оксида азота (II), паров воды и азота. Образующиеся в процессе окисления аммиака горячие нитрозные газы поступают в котел-утилизатор 14, на котором установлен контактный аппарат. Конструктивно котел выполнен с учетом полного обеспечения максимально возможного окисления нитрозных газов в объеме. В котле-утилизаторе вырабатывается пар с давлением 1,67 МПа и с температурой 250° С.
Из котла-утилизатора нитрозные газы поступают в окислитель 72, представляющий собой полый сосуд, в котором продолжается процесс окисления оксида азота (II) в тетраоксид диазота с соответствующим повышением температуры нитрозных газов. Расчетная степень окисления нитрозных газов на выходе из окислителя — 80%. В верхней части окислителя установлен фильтр для улавливания платиноидов.
Из окислителя нитрозные газы поступают в подогреватели воздуха 5 и хвостовых газов 6. Охлажденные нитрозные газы направляются в скоростные холодильники 8, в которых охлаждаются оборотной водой до 45—55° С. В холодильниках 8 происходит конденсация водяных паров, окисление NO в NO2, а также образование кислоты. Из холодильников смесь газа и кислота направляются в сепаратор 9, в котором отделяется около 75% от всего количества образовавшейся кислоты.
Из сепаратора 42—47%-ная азотная кислота поступает в абсорбционную колонну 10 на тарелки с соответствующей концентрацией кислоты. Нитрозные газы, содержащие диспергированную кислоту, также передаются в абсорбционную колонну 10. Образующаяся в колонне азотная кислота самотеком направляется в продувочную колонну 77, в которой из кислоты выдуваются растворенные в ней оксиды азота. Отбеленную кислоту из продувочной колонны направляют на склад азотной кислоты. 514
Нитрозные газы после продувочной колонны возвращаются в абсорбционную колонну. Газы, выходящие из абсорбционной колонны, содержат непоглощенные оксиды азота и направляются в сепаратор для улавливания капель кислоты. После подогрева в подогревателе 6 они поступают в камеру подготовки газов 4, в которой нагреваются до 400—550° С топочными газами, полученными в процессе сжигания природного газа. Температура газов задается по активности катализатора в реакторе каталитической очистки 3. Поскольку поддерживается восстановительный режим сжигания, то в газах за камерой 4 содержится значительное количество водорода, оксида углерода и радикалов, способствующих интенсификации процесса на катализаторе в реакторе 3.
В трубопровод до или после камеры сгорания 4 вводится газ-восстановитель для каталитической очистки хвостовых газов от оксидов азота в реакторе 3. Нагретые до 705—730° С хвостовые газы, смешиваясь с относительно холодным потоком продуктов сгорания топлива и воздуха из пусковой камеры 0, охлаждаются до 700° С и направляются на расширение в газовую турбину в. Нижний предел температуры (705° С) определен из условий безопасной эксплуатации пусковой камеры сгорания д, верхний предел (730° С) — по условиям безопасной эксплуатации реактора 3 и длительности пробега катализатора. Отработанные в турбине газы под давлением 0,104 МПа и при температуре около 400° С поступают в котел-утилизатор 17, где вырабатывается пар с теми же параметрами, что и в котле-утилизаторе нитрозных газов 14. Дальнейшее охлаждение хвостовых газов до 120—130° С происходит в экономайзерах 18 и 19, конструктивно совмещенных с котлом 17.
Рассмотренная схема производства вырабатывает ~400 т/сут азотной кислоты (в пересчете на 100%).
Расходные коэффициенты на 1т HNO3 составляют: около 290 кг аммиака, НО м3 природного газа, 50 МДж электроэнергии, 150 м3 охлаждающей воды.
Степень конверсии аммиака составляет 95—96%, а степень абсорбции оксидов азота ~99,0—99,3%. Концентрация продукционной кислоты равна 58,0—58,78% (масс.). Содержание оксидов азота в продукционной кислоте в пересчете на N2O4 0,005—0,0034% (масс.). Хвостовые газы содержат 0,0034—0,005% (масс.) оксидов азота и 0,056—0,13% (масс.) оксида углерода.
Производство азотной кислоты в агрегате А К-7 2 состоит из следующих стадий производства: 1) фильтрацию воздуха от пыли, сжатие его до 0,412 МПа; 2) испарение исходного жидкого аммиака под давлением 0,588 МПа; 3) фильтрацию газообразного аммиака; 4) смешение газообразного аммиака с воздухом; 4) фильтрацию АВС; 5) окисление аммиака кислородом воздуха; 6) ох-17* 515
лаждение нитрозных газов, совмещая его с промывкой их от ни-трит-нитратов аммония и получением конденсата азотной кислоты концентрацией 40—45% HNO3; 7) сжатие нитрозных газов до 1,079 МПа; 8) охлаждение сжатых нитрозных газов; 9) абсорбцию оксидов азота с образованием 60%-ной азотной кислоты; 10) подогрев выхлопных газов до 480—500° С; 11) каталитическую очистку их от оксидов азота с одновременным подогревом их до 750—770° С; 12) расширение выхлопных газов в газовой турбине от 0,932—0,981 до 0,103 МПа и охлаждение расширенных выхлопных газов в подогревателе до 200° С.
Схема агрегата АК-72 приведена на рис. 10.73, согласно которой атмосферный воздух после очистки от механических примесей на фильтрах грубой и тонкой очистки 1 засасывается осевым воздушным компрессором 2. Сжатый в компрессоре воздух разделяется на два потока. При этом основной поток воздуха направляется в аппараты окисления аммиака 10, а другой поток (10—14% от общего расхода воздуха) направляется последовательно в подогреватель газообразного аммиака б, продувочную колонну 25 и смешивается с ни-трозными газами на линии всасывания нитрозного нагнетателя 20.
Жидкий аммиак поступает в ресивер 3, а затем в испаритель 4, в котором испаряется за счет теплоты циркулирующей воды. Увлажненный аммиак в фильтре 5 очищается от механических примесей (в основном от катализаторной пыли) и паров масла, нагревается в подогревателе 6 сжатым воздухом, а в холодное время года — дополнительно паром в теплообменнике 7. Горячий газообразный аммиак смешивается с воздухом в смесителе 8, встроенном в верхнюю часть контактного аппарата 10. АВС подвергается дополнительной тонкой очистке в фильтре 9, также встроенном в контактный аппарат.
Аммиак подвергается конверсии на катализаторных сетках из платинородиево-палладиевого сплава.
Горячие нитрозные газы охлаждаются последовательно в котле-утилизаторе 11, расположенном под катализаторными сетками, в экономайзере 12, подогревателе химически очищенной воды 13, холодильнике-конденсаторе 14 и промывателе 15. В промывателе 75 наряду с процессами охлаждения нитрозного газа и конденсации паров с образованием азотной кислоты нитрозные газы промываются от аммиака, не прореагировавшего на катализаторных сетках, и ни-
Рис. 10.73. Схема производства азотной кислоты на агрегате АК-72:
1 — фильтр воздуха; 2 — воздушный компрессор; 3 — ресивер жидкого аммиака; 4 — испаритель аммиака; 5 — фильтр газообразного аммиака; 6, 7, 13, 22, 27, 28— подогреватели; 8, 29 — смесители; 9 — фильтр АВС; 10 — контактный аппарат; 11 — котел-утилизатор; 12 — экономайзер; 14, 23 — холодильники-конденсаторы; 15 — промыватель газа; 16, 19, 21 — центробежные насосы; 17, 18 — теплообменники; 20 — нагнетатель тетрозных газов; 24 — абсорбер; 25 — продувочная колонна; 26 — ловушка; 30 — реактор каталитической очистки; 31— газовая турбина; 32 — паровая турбина
517
трит-нитратов аммония, образующихся из аммиака и оксидов азота в тракте до промывателя.
Промыватель 75 орошается азотной кислотой, циркуляция которой осуществляется насосом 16 через холодильник 77, охлаждаемый оборотной водой, и в холодильник 18, охлаждаемый циркулирующей через испаритель жидкого аммиака 4 охлажденной водой. Из промывателя 75 40—45%-ная азотная кислота центробежным насосом 21 передается в абсорбционную колонну 24, орошаемую паровым конденсатом и конденсатом сокового пара из производства нитрата аммония. Продукционная 60%-ная азотная кислота поступает в продувочную колонну 25, в которой при давлении 0,392 МПа из нее отдувают растворенные оксиды азота воздухом, и далее самотеком направляется в хранилище склада. Выхлопные газы из абсорбционной колонны направляются в ловушку 26 с встроенным теплообменником 27, в котором газы подогреваются для испарения мелких брызг, а затем в подогреватель 28. Противоточный подогрев сжатых выхлопных газов осуществляется последовательно расширенными выхлопными газами из газовой турбины и дымовыми газами, образующимися в процессе горения природного газа в горелках радиационной части подогревателя 28. Нагретые выхлопные газы проходят рубашку реактора каталитической очистки 30 и смешиваются с природным газом в смесителе 29. Смесь далее поступает в реактор каталитической очистки 30, в котором на двухступенчатом катализаторе при избытке природного газа происходит процесс восстановления оксидов азота до азота с одновременным подогревом выхлопных газов до 750—770° С. Горячие выхлопные газы направляются на ре-куперационную газовую турбину 31. Энергия расширения горячих выхлопных газов практически полностью соответствует затратам механической энергии на сжатие воздуха и нитрозных газов. При этом некоторый недостаток механической энергии восполняется работой паровой турбины 32. Расширенные выхлопные газы из турбины поступают в подогреватель 28, в котором охлаждаются, после чего выбрасываются через выхлопную трубу в атмосферу.
Технологический агрегат АК-72 обеспечивает получение 60%-ной, а в зимнее время 65%-ной HNO3. Производимая кислота содержит не более: 0,05% растворенных оксидов азота; 0,004% (масс.) прокаленного остатка; 10 мг/кг хлоридов (в пересчете на хлор-ион) и следы нитрата аммония.
На 1 т производимой азотной кислоты (100%) расходуют: 0,286 т аммиака, 0,120 г платиноидного сплава, 0,028 г палладированного катализатора АПК-2 (в пересчете на палладий), 22 г активированного оксида алюминия, 1,54 т химически очищенной (обессоленной) воды, 0,34 т конденсата водяного пара, 129 м3 производственной оборотной 518
воды, 82 м3 природного газа (теплотворная способность 37,2 МДж/м2), 14,4 кВт-ч электроэнергии.
При производстве 1т целевого продукта получают 4,731 т водяного пара (50% р = 3,92 МПа, t = 440° С; 50% р = 1,47 МПа, t = 250° С).
Производство азотной кислоты в агрегате Г И А П - Г П (рис. 10.74). Согласно схеме производства, исходное сырье — жидкий аммиак — поступает на склад под давлением до 1,57 МПа. Перед подачей в испарительную установку, работающую под давлением 0,7—0,8 МПа, он дросселируется клапаном регулятора уровня для частичного испарения. Образующаяся парожидкостная смесь аммиака поступает в ресивер низкого давления 7, в котором происходит разделение фаз. Жидкий аммиак из ресивера направляется в испаритель 2. В схеме установлены два испарителя ИТГ-630, работающие параллельно. Процесс испарения осуществляется при температуре 15° С. Постоянное давление обеспечивается подачей пара в паровой испаритель аммиака 3. Образующийся холод от испарения аммиака используется для охлаждения газа перед нитрозным компрессором 46, а также для отвода теплоты в абсорбционной колонне 29. Теплоносителем является вода (охлаждается от 28 до 23° С), для циркуляции которой используют насос.
Поступающий в цех жидкий аммиак содержит до 0,1% воды. С целью поддержания постоянной концентрации воды в жидком аммиаке часть его постоянно выводится из испарителей в дистилляционную колонну 6, снабженную кипятильником 7. В дистилляционной колонне, работающей под давлением 0,735 МПа, отгоняется газообразный аммиак, а образующийся в кубе раствор гидроксида аммония содержит 25% аммиака.
Раствор гидроксида аммония непрерывно выводится из куба дистилляционной колонны и направляется в сборник 8, в котором охлаждается и реализуется в виде товарной продукции.
Выделившийся в дистилляционной колонне газообразный аммиак, а также аммиак из испарителей очищается от механических примесей в двух параллельно работающих фильтрах 9. При этом масло удаляется фильтрами из полипропиленового волокна, а тонкая очистка производится в аэрозольном фильтре А-17.
Очищенный аммиак далее направляется в перегреватель аммиака 10, в котором он нагревается от 15 до 100° С водяным паром под давлением 0,37 МПа и температуре 141° С, вырабатываемым в котле низкого давления 22.
Фирма «Сосьете Шишик де ла Гранд Паруасс» (Франция).
519
Рис. 10.74. Схема установки ГИАП-ГП:
1 — ресивер; 2, 3 — испарители аммиака; 4— машинный агрегат (4а—воздушный компрессор, 46 — паровая турбина); 5 — центробежные насосы; 6 — дистилляционная колонна; 7—кипятильник; 8— сборник водных растворов аммиака; 9—фильтр аммиака; 10—перегреватель аммиака; И — фильтр воздуха; 12 — подогреватель воздуха; 13 — контактный аппарат; 14—холодильник продувочного воздуха; 15—продувочная колонна; 16 — пароперегреватель; 17—испаритель котла; 18 — барабан котла; 19 — среднетемпературный теплообменник; 20—котел высокого давления; 21 — низкотемпературный теплообменник; 22 — котел низкого давления; 23 — подогреватель выхлопных газов; 24 — газовый промыватель; 25, 26—холодильники кислоты; 27—экономайзер; 28—конденсатор высокого давления; 29 — абсорбционная колонна; 30 — ловушка; 31 — смеситель;
32 — реактор; 33 — выхлопная труба; 34—воздушный конденсатор; 35 — сборник конденсата; 36 — холодильник; 37—градирня
Необходимое количество воздуха для производства отбирают через воздухозаборную трубу и очищают от механических примесей в фильтре 11. В зимнее же время воздух при необходимости подогревают в подогревателе 12 для предотвращения процесса забивания фильтров инеем. Далее очищенный воздух сжимается до 0,53 МПа воздушным компрессором 4а, входящим в состав комплексного машинного агрегата 4. При этом температура воздуха повышается до 235° С. Сжатый в компрессоре воздух разделяется на два потока, первый из них поступает в два параллельно работающих контактных аппарата 12, а второй — добавочный воздух — после охлаждения в холодильнике 14 поступает в продувочную колонну 75. В продувочной колонне из продукционной азотной кислоты отделяются растворенные в ней оксиды азота, после чего он смешивается с нитрозным газом, выходящим из газового промывателя 24.
В контактных аппаратах после смешивания воздуха и перегретого аммиака образуется АВС, содержащая 9,5—10% (об.) аммиака, которая проходит фильтры тонкой очистки из базальтового волокна, огнепреградительный слой и поступает на двухступенчатый катализатор, состоящий из четырех платиноидных сеток и слоя неплатинового катализатора. Степень конверсии аммиака на этом катализаторе составляет 96,2%.
Образовавшийся в контактных аппаратах газ, температура которого составляет 880° С, проходит последовательно пароперегревательный 16 и испарительный 77 пакеты парового котла-утилизатора, расположенного непосредственно под контактными аппаратами.
Котельный агрегат включает экономайзер 27, котел низкого давления 22, котел высокого давления 20, испарительную поверхность нагрева с пароперегревателями 16 и 17 и барабан с сепарационным устройством 18. В котле за счет охлаждения нитрозного газа вырабатывается перегретый пар под давлением 3,92 МПа и температуре 450° С. Меньшая часть этого пара передается в заводской коллектор, а основная часть направляется на паровую турбину 4г, в которой расширяется до 0,01225 МПа, охлаждаясь при этом до 50° С. В паровой турбине, входящей в состав комплексного машинного агрегата 4, тепловая энергия преобразуется в механическую и используется для привода воздушного 4а и нитрозного 46 компрессоров.
Расширенный в турбине пар конденсируется в воздушных конденсаторах паровой турбины 34. Часть конденсата водяного пара направляется на орошение абсорбционной колонны 29, а другая часть после предварительного подогрева до 81° С в холодильнике продувочного воздуха 14 поступает в деаэрационную установку, после чего возвращается на питание котла-утилизатора. Подпитка котла-ути-521
лизатора осуществляется химически очищенной водой, также подаваемой в деаэрационную установку.
Вода в деаэрационной установке нагревается до температуры кипения (115° С). При этом растворенные в воде газы выделяются из нее и вместе с частью пара поступают в холодильник (охладитель выпара), в котором происходит конденсация пара. Газы выбрасываются в атмосферу.
Деаэрированная вода, подаваемая центробежным насосом 5, разделяется на два потока. Меныпий поток направляется на испарение в котел низкого давления 21, в котором за счет теплоты нитрозных газов образуется насыщенный пар с давлением 0,37 МПа и температурой 141° С, который направляется в деаэрационную установку и в теплообменники на собственные нужды. Большая часть деаэрированной воды поступает в экономайзер 27, нагревается от 115 до 208° С, а затем поступает в барабан с сепарационным устройством 18. Из барабана вода центробежным насосом непрерывно подается через испарительную поверхность котла 17 контура принудительной циркуляции. Образующийся при этом насыщенный пар под давлением 4,3 МПа собирается в барабане, освобождается от влаги и направляется в пароперегреватель котла 16, в котором пар перегревается до 450° С.
Для поддержания в воде, находящейся в котлах, содержания солей на допустимом уровне часть воды из барабана 18 и котла низкого давления 22 выводится в процессе продувки, охлаждается в теплообменниках оборотной водой, поступающей из градирни, а затем сбрасывается в канализацию. В результате охлаждения в котле-утилизаторе температура нитрозного газа снижается до 475°С, а дальнейшее его охлаждение до 370° С осуществляется выхлопным газом в среднетемпературном теплообменнике газ — газ. При этом выхлопной газ нагревается от 250 до 400° С.
Нитрозный газ поступает в трубное пространство котла высокого давления 20, в котором, охлаждаясь до 266° С, отдает свое тепло на испарение части котловой воды, поступающей из барабана 18. Затем нитрозный газ нагревается до 304° С в результате окисления NO в NOj в трубопроводе, ведущем к низкотемпературному теплообменнику газ — газ 21, в котором нитрозный газ охлаждается до 203° С выхлопным газом, нагревающимся от ПО до 245° С. Далее нитрозный газ поступает в трубное пространство котла низкого давления 22 и охлаждается до 150° С. Далее его используют для подогрева выхлопного газа от 27 до 110° С в подогревателе выхлопных газов 23. Здесь он охлаждается от 160 до 100° С и далее направляется в два работающих параллельно газовых промывателя 24, представляющих собой тарельчатые аппараты барботажного типа. 522
Нагревшийся в промывателе до 63° С конденсат азотной кислоты охлаждается в выносных холодильниках 25 и 26 вначале водой из оборотного цикла, затем захоложенной водой. Часть конденсата азотной кислоты в количестве, соответствующем образующемуся в промывателе, насосом подается в абсорбционную колонну 29 на тарелку, содержащую 42,5%-ную HNO3.
Охлажденный нитрозный газ смешивается с продувочным газом после продувочной колонны 15 и при температуре 56° С поступает в нитрозный компрессор 46, в котором сжимается до 1,96 МПа, нагреваясь при этом до 230° С. Далее он охлаждается вначале в экономайзере 27 до 139° С, а затем до 42° С в конденсаторе высокого давления 28 водой из оборотного цикла. В процессе охлаждения нитрозного газа образуется конденсат азотной кислоты (65,3% HNO3), который вместе с нитрозным газом поступает в нижнюю часть абсорбционной колонны. Абсорбционная колонна, имеющая 28 тарелок, орошается конденсатом водяного пара из сборника 35 после воздушных конденсаторов паровой турбины 34.
Паровой конденсат перед подачей в колонну охлаждается до 30° С захоложенной водой, поступающей из испарителя аммиака 2, после чего захоложенная вода направляется на охлаждение верхних тарелок (с 18-й по 25-ю) абсорбционной колонны, после чего возвращается в испаритель аммиака. Нижние тарелки (с 1-й по 17-ю) абсорбционной колонны охлаждаются водой из водооборотного цикла.
Образующаяся в процессе абсорбции 60%-ная азотная кислота поступает в продувочную колонну 75, в которой из нее выдуваются растворенные оксиды азота. Из нижней части продувочной колонны продукционная кислота направляется в хранилище.
Выхлопной газ абсорбционной колонны, содержащий 0,04% (об.) оксидов азота, при температуре 27° С проходит ловушку 30 для улавливания брызг азотной кислоты, после чего нагревается в подогревателе выхлопного газа 23 до 110° С и далее до 245° С в теплообменнике 27.
Необходимое для каталитической очистки количество аммиака отбирается из межцехового трубопровода и подается через ресивер высокого давления в испаритель аммиака и затем в пароперегреватель аммиака (на схеме не показан). Температура перегретого аммиака 100° С. Процессы испарения и перегрева аммиака осуществляются паром из котла низкого давления 22.
Выхлопной газ и перегретый аммиак поступают в смеситель 30, а далее в реактор для каталитической очистки от оксидов азота. Процесс разложения оксидов азота производится на катализаторе аммиаком при 240—250° С. Содержание оксидов азота на выходе из реактора составляет 0,005% (об.), а аммиака 0,008% (об.). В вариан-523
те отсутствия каталитической очистки проводят водно-кислотное поглощение в абсорбционной колонне с повышенным числом тарелок, остаточное содержание оксидов азота 0,01—0,02% (об.).
Очищенный газ подогревается до 400° С и направляется в реку-перационную газовую турбину 4в. Энергию, вырабатываемую газовой и паровой турбинами, используют для привода воздушного и ни-трозного компрессоров. Выхлопной газ, выходящий из газовой турбины под давлением 0,101 МПа и при температуре 90° С, выбрасывается в атмосферу через трубу 33 диаметром 2,6 м и высотой 100 м. Это отвечает требованиям санитарных норм по содержанию оксидов азота в приземном слое.
На производство 1 т азотной кислоты (в пересчете на 100% HNO3) расходуют 0,2875 т жидкого аммиака, 0,130 г платинового катализатора, 2,63 г неплатинового катализатора КН-2Т, 1,84 г селективного катализатора, 0,694 т химически очищенной воды, 98 м3 оборотной воды, 16,5 кВт-ч электроэнергии, 0,1 ГДж пара (0,59 МПа и 158° С). Производство выпускает 1,08 т пара (3,92 МПа и 450° С) на 1 т производимой HNO3 (100%-ной).
Физико-химические основы получения концентрированной азотной кислоты. Существуют два основных способа получения концентрированной азотной кислоты. Согласно первому способу, ее получают путем ректификации тройных смесей, содержащих HNO3, воду и водоотнимающее вещество (обычно серная кислота или нитрат магния). В процессе получают пары 100%-ной азотной кислоты, которые конденсируют, и водные растворы водоотнимающего агента. Второй способ основан на реакции
2И2О4(ж) + 2Н2О(ж) + О2(г) = 4ННОз(ж) + 78,8 кДж.
При проведении процесса под давлением ~5 МПа и использовании чистого кислорода образуется 97—98%-ная азотная кислота, содержащая до 30% (масс.) растворенных оксидов азота. Азотную кислоту получают разгонкой этого раствора. При 0,7—1,0 МПа и использовании воздуха образуется 80—85%-ная HNO3 и азеотропная смесь азотной кислоты с водой. Жидкий N2O4 получают путем растворения оксида азота (IV), содержащегося в нитрозном газе 80—100%-ной HNO3 с последующей разгонкой этого раствора и конденсацией жидкого N2O4.
Получение концентрированной азотной кислоты, используя водоотнимающие вещества. Лабораторные исследования показали, что в процессе перегонки разбавленных водных растворов азотной кислоты в присутствии водоотнимающих веществ давление паров воды над смесью сильно снижается.
524
HNO3
Рис. 10.75. Температура кипения тройной смеси HNO3—Н2О—H2SO4
Зависимость температуры кипения от состава тройной смеси приведена на рис. 10.75, а на рис. 10.76 — зависимость состава паров, выделяющихся в процессе кипения тройной смеси при атмосферном давлении, от состава жидкой смеси. Построение диаграмм в виде треугольника авторы основали на том, что сумма перпендикуляров, опущенных из любой точки внутри равностороннего треугольника на его стороны, равна высоте треугольника. Пользуясь этими диаграммами, можно определить состав паровой фазы, содержащей азотную кислоту и воду, а также температуру кипения в зависимости от исходного состава жидкой тройной смеси. Например, если исходная смесь содержит 40% серной кислоты, 30% азотной кислоты и 30% воды (рис. 10.76, точка А), то в парах будут находиться азотная кислота и вода, которые при конденсации образуют раствор 90%-ной HNO3. Из рис. 10.75 видно, что такая смесь кипит при температуре около 120° С (атмосферное давление). Если задаться определенной конечной концентрацией азотной кислоты, то для каждой исходной концентрации HNO3, подвергаемой концентрированию, должен существовать максимальный расход серной кислоты. Очевидно, требуемое количество серной кислоты будет наименьшим в том случае, когда в процессе перегонки применяют не разбавленную, а безводную серную кислоту (вершина треугольника).
Определяют состав тройной смеси, состоящей из 50%-ной азотной кислоты и 100%-ной серной кислоты, при условии получения в процессе перегонки 95%-ной HNO3 следующим образом. Соединяют пря-525
HNO3
Рис. 10.76. Равновесная концентрация паров азотной кислоты над тройной смесью HNO3—Н2О—H2SO4
мой линией вершину треугольника (H2SO4) на рис. 10.76 с точкой на противоположной стороне, соответствующей 50%-ной HNO3. Прямая пересекает кривую 95%-ной HNO3 в парах в точке Б, которой соответствует содержание 48% H2SO4 в исходной смеси. Следовательно, на долю азотной кислоты и воды приходится 52%, тогда начальная тройная смесь должна состоять из 48% H2SO4, 26% HNO3 и 26% Н2О. Таким образом, пользуясь диаграммами, можно определить минимальный расход серной кислоты при определенных условиях.
В процессе использования разбавленной серной кислоты для смешения ее минимальный расход изменяется. Например, определяют состав тройной смеси, содержащей 90%-ную серную кислоту и 50%-ную разбавленную азотную кислоту. Серной кислоте такого состава соответствует точка Г (рис. 10.76), лежащая на стороне треугольника Н2О—H2SO4. Эту точку соединяют с точкой, соответствующей 50%-ной азотной кислоте, на стороне Н2О—HNO3. Прямая, соединяющая обе точки, пересекает кривую 95%-ной HNO3 в парах в точке В, которой соответствует следующий состав: 55% серной кислоты, 19% азотной кислоты и 26% воды.
Тройная смесь такого же состава может быть получена прибавлением безводной серной кислоты к раствору, содержащему 40% HNO3 и 60% Н2О.
В процессе отгонки концентрированной азотной кислоты определенного состава расход безводной серной кислоты зависит от содержания HNO3 в разбавленной азотной кислоте, при этом расход 526
H2SO4 будет тем больше, чем сильнее разбавлена азотная кислота. Для одной и той же исходной разбавленной азотной кислоты расход серной кислоты обратно пропорционален ее концентрации: чем меньше концентрация серной кислоты, тем больше ее расход.
При переменных концентрациях разбавленной азотной и концентрированной серной кислот следует подобрать такую смесь, в процессе перегонки которой образуются пары нужного состава. Через точку В можно провести множество прямых, которые зафиксируют определенные точки на сторонах треугольника HjO—H2SO4 и НгО—HNO3. Помимо этого при перемещении точки В по кривой, соответствующей постоянной концентрации паров, состав исходной смеси пройдет через множество различных значений. Это доказывает, что при перегонке смеси различного состава можно получать азотную кислоту одной и той же концентрации.
Расход серной кислоты сильно уменьшается при снижении содержания воды в исходной разбавленной азотной кислоте, поэтому целесообразно проводить концентрирование разбавленных растворов азотной кислоты в две стадии: сначала отгонкой воды при нагревании до получения 60—65%-ной HNO3, а затем перегонкой раствора в присутствии серной кислоты. В процессе перегонки разбавленных растворов азотной кислоты в смеси с серной кислотой H2SO4 разбавляется водой и насыщается HNO3 и оксидами азота, образующимися вследствие частичного разложения азотной кислоты.
В процессе взаимодействия сесквиоксида азота с менее чем 75%-ной серной кислотой реакция протекает с образованием нитро-зил серной кислоты:
N2O3 + 2H2SO4 = 2HNSO5 + Н2О
Оксид азота (IV) реагирует с концентрированной серной кислотой с образованием нитрозилсерной и азотной кислот:
2NO2 + H2SO4 = HNSO5 + HNO3
В процессе разбавления серной кислоты водой происходит гидролиз нитрозилсерной кислоты:
2HNSOs + Н2О = 2H2SO4 + N2O3
в результате чего в растворах концентрацией ниже 57,5% H2SO4 нит-розилсерная кислота отсутствует:
Содержание H2SO4, % (масс.)..... 98,0 95,6 92,0 87,0 80,0 70,0 57,5
Содержание нитрозилсерной кислоты
в серной кислоте, % (мол.).... 98,0 96,6 92,7 80,6 72,3 50,2 0
527
Образующаяся при гидролизе азотистая кислота неустойчива и распадается в серной кислоте концентрацией ниже 75%:
3HNO2 = HNO3 + 2NO + Н2О
С повышением температуры степень гидролиза нитрозилсерной кислоты возрастает. Оксид азота (II), образующийся при разложении азотистой кислоты, растворяется в разбавленной серной кислоте:
Концентрация H2SO4, % (масс.).. 100 68,5 45 38 24 0
Содержание NO в растворе, 104%. 2,55 1,425 2,03 2,78 5,19 9,65
Следовательно, оксид азота (II) наименее растворим в серной кислоте, содержащей 68,5% H2SO4.
Для денитрации отработанной серной кислоты ее нагревают паром (лучше острым паром) с таким расчетом, чтобы вводимое паром тепло было достаточным для нагревания смеси до 150—160° С. Помимо этого кислород разбавляют конденсатом до конечной концентрации, равной 68—70%. В условиях подачи в колонну пара, перегретого до 250° С и поддержания при этом концентрации серной кислоты около 68%, содержание нитрозилсерной кислоты будет находиться в пределах 0,05—0,1%. В процессе же нагревания отработанной серной кислоты глухим паром содержание нитрозилсерной кислоты составит около 1—2%. При этом расход пара на денитрацию будет больше, а расход концентрированной серной кислоты и концентрирование азотной кислоты меньше, чем при нагревании острым паром.
По данным Ф.Я.Крайней, скорость процесса денитрации может быть выражена следующим уравнением:
~ ^?CN2O,£H2SO4‘t>
где G — масса выделившихся оксидов азота, кг; К—константа скорости процесса денитрации; F—поверхность контакта процесса денитрации, м2; сН2Оз и с^5О4—средние концентрации оксидов азота и серной кислоты в нитрозе, мол. доли; т— продолжительность процесса денитрации, ч.
Показатели степени: для концентрированной серной кислоты (выше 75% H2SO4) п - 2 и т = 8,5, для более разбавленных растворов серной кислоты (ниже 70% H2SO4) п = 1 и т = 10.
В процессе денитрации отработанной серной кислоты в колонне барботажного типа и разбавления ее до концентрации 68—70% продолжительность денитрации можно определить по уравнению
528
где т — продолжительность процесса денитрации, мин; К—константа скорости процесса денитрации, мин’1; а — степень денитрации, доли единицы.
Зависимость константы скорости процесса денитрации К от температуры (Г, К) выражается уравнением
. „ Л Л 3700
IgAT = 9,049--—
В промышленности широко применяется схема концентрирования разбавленной азотной кислоты (рис. 10.77). Согласно схеме, исходная разбавленная азотная кислота из напорной емкости 1 через расходомер 3 дозируется в концентрационную колонну 6. Часть азотной кислоты проходит через испаритель 5 и подается в виде смеси жидкости и пара на десятую тарелку колонны 6, другая часть (холодная) подается на вышележащую тарелку.
Серная кислота поступает из напорной емкости 2 через регулировочную коробку 4 на шестнадцатую тарелку колонны. В нижнюю часть колонны (под первую тарелку) вводится острый пар для денитрации отработанной серной кислоты. Пары азотной кислоты уходят через штуцер в крышке колонны (выше 20-й тарелки) и конденсируются в холодильнике 7. Кислый конденсат содержит большое количество растворенных оксидов азота, поэтому его возвращают на верхнюю тарелку колонны и продувают парами азотной кислоты, поступающими из колонны в конденсатор. Концентрированная кислота проходит две отбелочные тарелки, в которых освобождается от оксидов азота и выводится из колонны. После освобождения в холодильнике 8 она поступает на склад готовой продукции.
Смесь паров азотной кислоты, не сконденсировавшихся в холодильнике 7, оксидов азота и воздуха, проникающего в систему через неплотности в аппаратуре, поглощается водой в башне 13 с образованием 50%-ной азотной кислоты. Раствор циркулирует в башне насосом 10 через водяной холодильник 9. Разбавленная азотная кислота, полученная в башне 13, поступает на концентрирование, инертные газы выбрасываются в атмосферу вентилятором 14. Отработанная серная кислота по выходе из колонны 6 направляется в концентрационные аппараты или охлаждается водой в холодильнике 12, а затем направляется в промежуточные хранилища.
Из рис. 10.77 видно, что кислоты подаются на три тарелки колонны: на одну — серная кислота, на две другие — азотная. На некоторых производствах кислоты подаются на пять тарелок. Например, на 4-ю тарелку (считая сверху) подается холодная серная кислота, на 8-ю — холодная азотная; затем на 10-ю — горячая серная, на 12-ю — парообразная азотная и на 14-ю — горячая жидкая азотная кислота. Такое распреде-529
Рис 10.77. Схема концентрирования разбавленной азотной кислоты в присутствии серной кислоты:
1, 2 — напорные емкости; 3— расходомеры; 4—дозатор; 5—испаритель; 6—концентрационная колонна; 7—холодильник-конденсатор; 8— холодильник концентрированной азотной кислоты; 9 — холодильник кислоты, циркулирующей в башне; 10—центробежный насос; 11 — сборник 50%-ной азотной кислоты; 12—холодильник отработанной серной кислоты; 13 — поглотительная башня; 14 — вентилятор
ление кислот увеличивает зону выделения теплоты по высоте колонны и устанавливает режим более ровным и устойчивым.
Температура паров HNO3 на выходе из колонны 6 составляет 70—85° С, после конденсатора — 7 около 30° С. В верхней части колонны поддерживают вакуум в пределах 20—30 мм рт.ст. Отработанная 65—70%-ная серная кислота, содержащая не более 0,03% оксидов азота и азотной кислоты, выходит из колонны при 150—170° С. Абсолютное давление перегретого пара на входе в колонну не превышает 0,15 МПа, температура его около 250°С. Давление насыщенного пара, поступающего в испаритель 5, равно 0,4—0,6 МПа.
Согласно приведенной технологии, на получение 1 т азотной кислоты (в пересчете на моногидрат HNO3) расходуют 1,01—1,015 т азотной кислоты, 91—92%-ная H2SO4 (в т) при начальной концентрации:
48—50%............................... 4,0—^4,5
58—60%............................... 3,0—3,3
67—68%............................... 2,1—2,3
Охлаждающая вода, м3.................. 20—50
Пар, Гкал: перегретый............................. 0,27—0,35
насыщенный.......................... 0,2
Электроэнергия, кВт-ч.................. 8—10
Основным аппаратом в производстве является концентрационная колонна (рис. 10.78), состоящая из отдельных царг, изготовленных из ферросилицида. Каждая царга отливается вместе с днищем (тарелкой), переходящим в центре в горловину. Горловина тарелки закрывается круглым зубчатым колпаком для барботирования паров через слой жидкости. На горловине сделаны приливы, в барботажных колпаках им соответствуют пазы, которые при повороте в горизонтальной плоскости зацепляются, и таким образом фиксируют положение колпака на тарелке, не давая ему смещаться при движении паров. Жидкость перетекает с тарелки на тарелку через переточные трубки, расположенные поочередно с противоположных сторон от центра тарелки.
Обычно колонна состоит из 20—22 царг, а сами колонны изготовляют самых разнообразных типов и размеров. Иногда вместо одного колпака ставят семь барботажных колпаков или один колпак звездообразной формы, что позволяет увеличить линию барботажа и уменьшить сопротивление аппарата.
Перед пуском агрегата колонну разогревают паровоздушной смесью. Затем включают выхлопной вентилятор, создают в колонне вакуум около 15—20 мм рт.ст. и через нижний штуцер подают пар низкого давления и атмосферный воздух. Начальная температура паровоздуш-531
ной смеси не должна превышать 50° С. Дальнейшее повышение температуры смеси проводится равномерно, без рывков, со скоростью 10° за 15 мин. Через 2—3 ч нагревания в верхней царге достигается температура 80—90° С, при этом температура паровоздушной смеси должна быть около 150° С. Затем уменьшают подсос воздуха и повышают температуру смеси. В колонну для промывки парового конденсата подают концентрированную серную кислоту. Теплота, выделяющаяся в процессе разбавления H2SO4 конденсатом, а также физическая теплота пара расходуется на поддержание в верхней части колонны температуры не ниже 80° С.
В разогретую колонну постепенно вводят разбавленную азотную кислоту, доводя нагрузку агрегата до нормальной. Период пуска агрегата до установления полной нагрузки и нормального режима составляет от 4,5 до 6 ч для колонны небольшого размера (одноколпачковой) и до 10 ч для колонны большего размера (многоколпачковой). Длительность пускового периода обусловлена хрупкостью фер-росилида и повышенной его чувствительностью к изменениям температуры.
Рис. 10.78. Колонна барботажного типа для концентрирования азотной кислоты:
7, 2 — перегонные трубки
532
При остановке агрегата прекращают подачу азотной кислоты и уменьшают подачу серной кислоты, после чего через полчаса прекращают ввод пара холоднаянфя в в испаритель и колонну. За это время азотная кислота будет удалена .из колонны и испарителя, после чего можно прекратить подачу серной кислоты.
По данным исследований В.Н.Крайнова и М.И.Рябого (результаты которых представлены на рис. 10.79) температура на тарелках колонны для перегонки разбавленной
HNO3 из конденсатора 0
Продукционная2
HNO3 4 92,5-94%-ная „
Горячая HNO^
14
16
18
20
70____110 150 190 t, С
Давление, мм рт. ст. _______________-40-2040 2040 60 80 100
6 §
О
23
12$
14 п
18^ 20
50 70 90 H2SO4 в жидкой фазе, %
0 5 3 4 6 1012 14 HNO3 в жидкой фазе, %
Рис. 10.79. Изменение температуры, давления и концентрации азотной и серной кислот на тарелках концентрированной колонны (по данным В.Н.Крайнова и М.И.Рябого):
1 — температура; 2 — концентрация H2SO4 в жидкой фазе; 3 — концентрация HNO3 в жидкой фазе; 4 — давление
азотной кислоты в смеси с концентрированной серной кислотой ниже температуры кипения жидкой тройной смеси HNO3—Н2О—H2SO4, находящейся на данной тарелке. Максимальная концентрация серной кислоты в жидкой фазе наблюдается в точке ввода H2SO4. Выше и ниже точки ввода серной кислоты содержание ее в жидкой фазе уменьшается, однако минимальная концентрация H2SO4 соответствует не нижней, а примерно 13—11-й тарелке.
В нижней части колонны происходит процесс концентрирования серной кислоты за счет испарения азотной кислоты.
Острый пар подается в нижнюю часть колонны и при прохождении до 13—11-й тарелки отдает тепло перегрева, на вышележащих
тарелках подается тепло конденсации.
Температура плавно снижается по высоте концентрационной колонны: начиная от нижней тарелки до места ввода холодной кислоты. Все показатели режима в диапазоне 6—10 тарелок, т.е. в зоне ввода концентрированной серной кислоты, а также горячей и холодной азотной кислоты, имеют скачкообразный характер. Это объясняется бурным протеканием процесса, обусловленным поступлением большого количества теплоты с подогретой азотной кислотой и выделением теплоты разбавления концентрированной H2SO4 в процессе смешения ее с разбавленной азотной кислотой. Такой характер протекания процесса сказывается и на давлении, которое в этой зоне 533
резко увеличивается. С целью создания спокойного, устойчивого режима в средней части колонны азотную и серную кислоты необходимо вводить на несколько тарелок параллельно.
Физико-химические основы получения концентрированной азотной кислоты методом прямого синтеза. Механизм образования концентрированной азотной кислоты методом прямого синтеза базируется на следующих реакциях:
1Ч2О4(ж) 2NO2(r) - 56,9 кДж
2NO2(r) + Н2О(ж) 7* HNO3(p) + HNO2(p) + 74,8 кДж
3HNO2(p) HNO3(p) + Н2О(ж) + 2NO(r) - 75,87 кДж
2HNO2(p) + О2(г) 2НИО3(ж) + 8,96 кДж
2NO(r) + О2(г) 2NO2(r) ^Ы2О4(ж) + 212,7 кДж
Суммарная реакция изображается следующим образом:
2Ы2О4(ж) + 2Н2О(ж) + О2(г) 4НЫО3(ж) + 78,8 кДж.
Схема производства азотной кислоты взаимодействием жидкого тетраоксида диазота с водой состоит из стадий, изменяющихся в зависимости от вида применяемых нитрозных газов в процессе получения жидкого Ы2О4:газы, полученные окислением аммиака воздухом (или кислородом); газы, образовавшиеся при инверсии нитритных солей, или полученные в процессе динитрации нитрозы.
В варианте применения нитрозных газов, получаемых в процессе окисления аммиака воздухом, непосредственное получение концентрированной азотной кислоты состоит из следующих стадий: 1) охлаждение нитрозных газов и удаление избытка воды; 2) окисление оксида азота (И) до диоксида азота (IV); 3) поглощение диоксида азота (IV) концентрированной азотной кислотой; 4) разложение полученного раствора и выделение из него чистого диоксида азота (IV). При этом производится также отбелка полученной концентрированной азотной кислоты от оксидов азота; 5) конденсация тетраоксида диазота; 6) переработка смеси тетраоксида диазота и разбавленной азотной кислоты под давлением с участием кислорода.
В варианте применения концентрированных оксидов азота меняется последовательность проведения стадий процесса получения концентрированной азотной кислоты. При этом процесс получения кон-534
центрированной азотной кислоты состоит из следующих стадий: 1) охлаждение исходных нитрозных газов и удаление избытка воды; 2) окисление оксида азота (II) и конденсация тетраоксида диазота; 3) поглощение остатков оксидов азота концентрированной азотной кислотой; 4) переработка под давлением смеси жидкого тетраоксида диазота, конденсата и продуктов поглощения оксидов азота в концентрированную азотную кислоту; 5) отбелка полученной азотной кислоты и конденсация отогнанных оксидов азота.
В полученных контактным окислением аммиака нитрозных газах содержится значительное количество паров воды.
В соответствии с реакциями:
NH3 + 2О2 = NO + 1,5Н2О + 0,75О2 = HNO3 + Н2О
для получения 100%-ной HNO3 требуется около 1/3 реакционной воды, а остальные 2/3 воды следует удалить из нитрозных газов.
В процессе конденсации водяных паров происходит образование диоксида азота и его поглощение. Процесс окисления аммиака проводят при атмосферном давлении. При этом конденсат может содержать всего 2—3% кислоты. Для удаления из нитрозных газов 1/3 реакционной воды и водных паров (вносимых в систему воздухом) устанавливают скоростные холодильники, рассчитанные на интенсивное охлаждение смеси до температуры, соответствующей давлению насыщенных паров 1/3 воды, оставляемой в газах. Конденсат, образующийся в скоростных холодильниках, может быть использован в производстве разбавленной азотной кислоты. Конденсат, полученный при последующем охлаждении нитрозных газов в холодильнике-конденсаторе, содержит 15,25% HNO3 и поступает на производство концентрированной азотной кислоты.
Скорость окисления оксида азота по схеме
2NO + О2 = 2NO2 + 124 кДж снижается при повышении температуры и снижении концентрации NO. Продолжительность процесса окисления 50, 90, 95 и 98% оксида азота в нитрозных газах, полученных контактным окислением аммиака, растет в соотношении примерно 1:20:120:230. Степень конденсации оксидов азота, или степень их растворения в концентрированной азотной кислоте, зависит от степени окисления NO. Поэтому непрореагировавшую часть оксида азота переводят в диоксид (NO2) в процессе взаимодействия его с азотной кислотой по реакции
NO + 2HNO3 = 3NO2 + Н2О - 73,6 кДж
535
По данным В.И.Атрощенко и Е.Г.Сегашевой, стадией, определяю-щей скорость процесса гетерогенного окисления оксида азота(П), яв- 1 ляется взаимодействие NO с HNO3. Скорость реакции при 1 cHN0 = const выражается уравнением
Продолжительность, необходимая для доокисления NO концентрированной азотной кислотой, определяют по уравнению
Т= к р
где р — гидравлический радиус насадки, равный отношению свободного объема насадки к ее поверхности, см; р° и рх — парциональное давление NO в исходном и конечном газе, МПа; х— продолжительность окисления, с; к — константа скорости реакции, см/с.
Значение константы к в зависимости от концентрации азотной кислоты и температуры приведено на рис. 10.80. В пределах изменения концентрации HNO3 от 98 до 70% при линейной скорости газа в свободном сечении 0,2 м/с значения к можно определить по уравнениям:
при 15° С к = 1,43 - 0,033(100 - z),
при 25° С к = 1,76 - 0,0407(100 - z),
при 35° С к = 2,03 - 0,0467(100 - z),
где z — средняя концентрация азотной кислоты, % (масс.).
Скорость реакции гомогенного окисления NO парами азотной кислоты выражается уравнением
4^0, _ ,
_ *Чю Чою, Чю, •
Процесс окисления оксида азота (II) продолжается несколько секунд. На его осуществление расходуется часть концентрированной азотной кислоты. Несмотря на небольшое снижение скорости процесса окисления оксида азота концентрированной HNO3 со снижением температуры, процесс ведут с охлаждением газа, поскольку при этом уменьшается количество паров HNO3, уносимых нитрозными газами. 536
Технологические условия проведения окисления оксидов азота зависят от оптимального соотношения доли оксида азота, окисленного кислородом и азотной кислотой, температуры и давления процесса. Так, при атмосферном давлении степень окисления исходного оксида азота (II) кислородом составляет 92—94%, а при давлении 0,4 МПа она возрастает до 94—96%. Остальное же количество оксида азота окисляют азотной кислотой.
В промышленности применяются нитрозные газы различного состава. Примерный состав при окислении аммиака парокислородной смесью приведен в табл. 10.4.
Рис. 10.80. Зависимость константы скорости реакции окисления оксида азота азотной кислотой от концентрации кислоты и температуры (концентрация оксида азота 10 %)
Таблица 10.4. Состав газа (в %) при парокислородиом окислении аммиака
Компоненты газовой смеси Перед катализатором После катализатора После скоростного холодильника После охлаждения и окисления газа
NH3 14,2 — — —
О2 28,4 10,4 30,2 22,5
NO — 13,4 38,5 10,8
no2 — — — 48,0
n2 3,0 3,0 8,9 16,7
H2O 54,4 73,2 22,4 2,0
В зависимости от концентрации оксидов азота подбирается метод их выделения, одним из которых является процесс их конденсации. Метод основывается на обычном процессе их сжижения путем изменения давления насыщенного пара в зависимости от температуры, которое в пределах от -20 до + 20° С выражается формулой
Igp = 14,61g/’—33,17526,
которая графически представлена на рис. 10.81.
Давление паров над жидкими оксидами азота, содержащими 2—4% (масс.) воды:
Температура, °C.................. -20 -10 0 +10 +20
Давление, мм рт. ст.............. 70 192 244 396 598
Насыщенные пары над жидким тетраоксидом диазота состоят из NO2 и N2O4, находящихся в состоянии подвижного равновесия. Следовательно, величина давления насыщенных паров р является суммой парциальных давлений:
Р = Рм2 +Pn2o4-
537
Рис. 10.81. Зависимость давления паров над жидким тетраоксидом диазота от температуры
Рис. 10.82. Диаграмма растворимости системы N2O4—Н2О
Концентрация N2O4, % (масс.)
Жидкий тетраоксид диазота и вода взаимно растворяются лишь ограниченно. Диаграмма растворимости системы N2O4—Н2О приведена на рис. 10.82, из которой видно, что при 0°С два жидких слоя содержат 47 и 98% (масс.) N2O4, а при 20° С — 52 и 97,5% (масс.) N2O4.
Критическая температура растворения равна 67° С и соответствует содержанию 89% (масс.) N2O4.
Из однородных водных растворов при температуре до -50° С кристаллизуются лед и оксиды, содержащие до 33% тетраоксида диазота. Из слоя же сжиженного газа кристаллизуется N2O4 в обычной стабильной форме и неустойчивой форме, которая плавится при температуре на 11° С ниже первой.
Установлено также, что тетраоксид диазота в растворе азотной кислоты диссоциирует с образованием нитрат-иона и иона нитрозония:
N2O4 -» 2NO2 -> NO; + NO+
При пониженных температурах в растворе образуется довольно стойкое соединение HNO3 N2O4. В системе существует следующее равновесие, изменяющееся в зависимости от температуры:
2NO2 + 2HNO3 <2 NO+ + (HNO3)2NO;
Л.И.Кузнецовым-Фетисовым исследованы парциальные давления компонентов паровой фазы, состоящей из диоксида азота, тетраоксида диазота и азотной кислоты, над растворами оксидов азота в азотной кислоте и определены температуры кипения растворов. Установ-538
лено, что по мере увеличения содержания оксидов азота в растворе давление паров азотной кислоты снижается, а давление паров оксидов азота возрастает. При содержании NO2 в растворе больше 10% (мол.) парциальное давление паров азотной кислоты с уменьшением давления от 760 до 350 мм рт. ст. изменяется незначительно, в то время как общее давление паров оксидов азота снижается довольно сильно. В связи с этим повышение давления наиболее благоприятно для разделения раствора на составные части.
Рис. 10.83. Парциальные давления паров оксидов азота и азотной кислоты над растворением NOi в азотной кислоте при температуре кипения:
1 — температура кипения; 2 — давление паров NO2; 3 — давление паров N2O4; 4 — давление паров HNO3
На рис. 10.83 показаны кривые парциального давления паров над растворами диоксида азота в азотной кислоте при температуре кипения и общем давлении 760 мм рт. ст.
На рис. 10.84 представлена диаграмма кипения системы HNO3—NO2 при 760, 600 и 350 мм рт. ст.
Процесс отгонки оксидов азота из их раствора в азотной кислоте
проводят при температуре, близкой к 85° С, в колоннах, выполненных из чистого алюминия, с тарелками или с насадкой из колец. Свежий, охлажденный до 0° С раствор подают в верхнюю часть колонны. Раствор, стекая вниз, соприкасается на насадке с поднимающимися навстречу ему парами и нагревается путем теплообмена с ними, выделяя при этом оксиды азота и обогащаясь азотной кислотой. На выходе из колонны пары имеют температуру около 40°С и содержат 97—98% оксидов азота и 2—3% азотной кислоты. Кислота, стекающая из колонны в холодильник, имеет температуру 85°С и содержит около 0,1% оксидов азота. Колонну обогревают через рубаш
ку или при помощи нагревательных паровых змеевиков.
В области высоких температур скорость разложения раствора оксидов азота в азотной кислоте очень велика. Скорость удаления оксидов азота из раствора (процесс отбелки) определяется скоростью теплообмена.
Из жидких оксидов азота, смешивая их в определенной пропорции с водой и обработкой кислородом, в автоклаве под давлением получают концентрированную азотную кислоту.
В процессе производства жидких оксидов из нитрозных газов в качестве побочного продукта образуется разбавленная азотная кислота. С
539
Рис. 10.84. Диаграмма кипения растворов NO2 в азотной кислоте при различном давлении
целью предотвращения получения ела-бой кислоты при составлении сырой смеси в автоклав вместо воды вводят разбавленную HNO3. Вводимая азотная кислота в реакции не участвует, а оксиды азота, вода и кислород реагируют по следующему уравнению:
2N2O4 + О2 + 2Н2О = 4HNO3
Получение концентрированной азотной кислоты связано с гидролизом тетраоксида диазота водой и образованием, параллельно с азотной и азотистой
кислот, частично окисляющихся в концентрированных растворах непосредственно в жидкой фазе, а в разбавленных растворах — преимущественно с выделением NO. Образующийся оксид в процессе окисления кислородом образует диоксид
азота и тетраоксид диазота.
Скорость процесса зависит от давления и температуры реакций. При небольшом давлении и высокой температуре скорость определяется
окислением оксида азота или азотистой кислоты, при низкой температу-
Рис. 10.85. Изменение концентрации азотной кислоты в смеси N2O4 + Н2О + HNO3 при протекании промежуточных реакций в процессе образования HNO3:
/— область преобладания реакции окисления оксида азота кислородом в газовой фазе; II — область преобладания реакции окисления азотной кислоты; III — область преобладания реакции гидроксида тетраоксида диазота или (по данным авторов) гидролиза диоксида азота (NO2)
ре — разложением азотистой кислоты, а при большом давлении кислорода и получении концентрированных растворов азотной кислоты — скоростью гидролиза оксидов азота водой.
На рис. 10.85 приведена диаграмма, характеризующая изменение концентрации азотной кислоты в зависимости от условий протекания реакций, определяющих скорость процесса при давлении 20 атм.
Реакция тетраоксида диазота с водой ускоряется с повышением содержания N2O4 в растворе разбавленной кислоты. Поэтому и поддерживают большой избыток тетраоксида диазота для ускорения реакции образования концентрированной азотной кислоты.
Процесс разложения азотистой кислоты интенсифицируется повышением температуры и хорошим перемешиванием раствора. Увеличение скорости
540
процесса с повышением давления можно определить, пользуясь диаграммой, приведенной на рис. 10.86. Если при этом принять скорость реакции при давлении 0,5 МПа за единицу, то при 1 МПа скорость реакции увеличивается в 2 раза, при 2 МПа — в 3,8 раза и при 5 МПа — в 5,3 раза. Увеличение же давления свыше 5 МПа мало эффективно и связано с большими затратами энергии. Оптимальная
Общее давление в автоклаве, МПа
Рис. 10.86. Относительное изменение скорости в автоклаве в зависимости от общего давления в нем
температура процесса, проводимого
при 50 атм, находится в температурных интервалах 60—80° С. Теоретическое повышение температуры показано на рис. 10.87. Практически
температура в автоклаве повышается в зависимости от продолжительности операции на 10—15% меньше, чем соответствует диаграмме.
В существующих установках исходная (сырая) смесь, поступающая в автоклав, состоит из 57—70% N2O4, 18—32% HNO3 и 8—12% Н2О. По мере возрастания концентрации целевого продукта (HNO3) общая скорость процесса резко снижается. На рис. 10.88 приведены
данные о скорости поглощения кислорода в автоклавном процессе в зависимости от температуры и давления.
С повышением концентрации продукционной кислоты растет и продолжительность проведения операции процесса, например если вместо 90%-ной азотной кислоты получать 95%-ную, то продолжительность реакции увеличивается в два раза, а при получении 98%-ной кислоты — в четыре раза.
Относительная скорость реакции, выражаемая отношением AN2O4/At = к, приведена на рис. 10.89, из которых видно, что скорость процесса образования
Содержание HNOa в смеси H2O+f^O4+HNO3,4 (масс.)
Рис. 10.87. Теоретическое повышение температуры в автоклаве в зависимости от начального содержания HNO3 в смеси: 1 — при эквимолекулярном отношении N2O4 и Н2О; 2 — при 30%-ном избытке N2O4; 3— при 50%-ном избытке N2O4
кислоты в автоклаве находится в линейной зависимости от расхода кислорода (сплошные линии) и поверхности пузырьков газа (пунктирные линии). Особенно велико влияние поверхности пузырьков кислорода для смесей с небольшим избытком тетраоксида диазота. Скорость реакции кислорода с раствором N2O4—Н2О зависит также от интенсивности перемешивания. Установлено, что скорость поглощения кислорода раствором, 541
кислорода растворами оксидов азота в азотной кислоте
в зависимости от температуры и давления (NiO^HiO = 5,36)
Рис. 10.89. Зависимость скорости образования HNO3 от количества пропускаемого кислорода и содержания тетраоксида диазота в смеси
содержащим 10,5% NO2, 26,3% Н2О и 63,2% HNO3, зависит от скорости вращения мешалки:
Скорость вращения мешалки, об/мин...... 500 1000 1500 2000
Скорость реакции (dG^Jdx)............. 0,79 2,2 2,8 3,4
Повышение скорости реакции с увеличением поверхности пузырьков кислорода, пропускаемого через раствор N2O4—Н2О, связано с большей скоростью процесса окисления или разложения азотистой кислоты под действием интенсивного перемешивания реакционной массы и возможным окислением оксида азота в жидкой фазе. На возможность протекания процесса гетерогенного окисления растворенного оксида азота указывают данные о растворимости оксида азота в азотной кислоте. Процесс растворения оксида азота в азотной кислоте, по-видимо-му, сопровождается прямым восстановлением азотной кислоты до азотистой с выделением оксида азота (IV), который остается в растворе. Процесс описывают следующей реакцией:
NO(r) + HNO3(p) = HNO2(p) + NO2(p) - 20,1 кДж.
На рис. 10.90 приведены коэффициенты скорости поглощения оксида азота 74,64%-ной азотной кислотой при 20° С. Примерно такое же значение имеют коэффициенты, отнесенные к 1см2 площади соприкосновения газа и жидкости. Опыт показывает, что в результате обработки кислородом под давлением 5 МПа 69%-ной азотной кислоты, насыщенной при 20° С оксидом азота, получается 98%-ная азотная кислота. 542
Установлено, что из растворов N2O4, N2O3 и NO в азотной кислоте наибольшей скоростью образования концентрированной азотной кислоты отличается раствор оксида азота (II). Процесс образования протекает по реакциям:
HNO3xNO + 0,5хН2О + 0,75хО2 = = HNO3 + xHNO3
HNO3N2O3 + Н2О + О2 = 3HNO3
HNO3N2O4 + Н2О + 0,5О2 = 3HNO3
Рис. 10.90. Степень поглощения NO в азотной кислоте при различных давлениях (в см3 газа/см3 кислоты)
Данные по изменению концентрации азотной кислоты в автоклавной жидкости во времени и в зависимости от числа тарелок приводятся на рис. 10.91, а значения констант скорости реакции взаимодействия жидкого тетраоксида диазота с водными растворами азотной кислоты и кислородом приведены на рис. 10.92.
На рис. 10.93 показана зависимость константы скорости реакции от давления, а на рис. 10.94 — влияние соотношения N2O4 и Н2О на концентрацию получаемой азотной кислоты.
Зависимость константы скорости реакции от давления при температуре процесса 90° С и соотношении N2O4:H2O = 8—8,5 определяется уравнением
Ы03 = 0,632р + 0,8.
Рис. 10.91. Изменение концентрации азотной кислоты в автоклаве в зависимости от числа тарелок и времени при 5 МПа и 70° С: 1 — при N2O4:H2O=5,11; 2 —при N2O4:H2O=7,6;
3 — при N2O4:H2O=10,2
Рис. 10.92. Константы скорости реакции взаимодействия жидкого тетраоксида диазота с водными растворами азотной кислоты и кислородом при различных температурах и давлениях
543
Рис. 10.93. Зависимость константы скорости реакции от давления (температура 90° С, отношение N2O4:H2O = 84-8,5)
Отношение N2O4/H2O
Рис. 10.94. Зависимость концентрации азотной кислоты от соотношения N2O4:H2O (температура 85—90° С, давление 4 МПа)
Производство концентрированной азотной кислоты из нитрозных газов, полученных при атмосферном давлении. Согласно приведенной на рис. 10.95 схеме, аммиак окисляется воздухом по той же технологии, что и в производстве разбавленной кислоты. Концентрация нитрозных газов в смеси находится в пределах 10—11,5%.
Нитрозные газы отделяют от избытка реакционной воды в скоростном холодильнике 2, в котором температура их снижается с 200 до 34—40° С. С целью исключения процесса окисления нитрозных газов скоростной холодильник обычно монтируется непосредственно за паровым котлом без промежуточной соединительной коммуникации. В холодильнике отделяется 2—3%-ная азотная кислота. После холодильника слабо окисленные нитрозные газы (отношение оксидов азота к воде составляет 5,1:1) поступают в газовый холодильник 3, в котором выделяется 25—35%-ная кислота. Кислота направляется в смеситель сырой смеси 20, а нитрозные газы вентилятором 4 подаются в две последовательно включенные окислительные башни 5, работающие под давлением до 1400 мм рт. ст. Теплота реакции окисления оксида азота отводится циркулирующей через водяные холодильники б кислотой, образующейся в башне в результате частичной конденсации паров воды. Концентрация азотной кислоты в первой башне достигает 50—55%, а во второй — 60—62%. Из первой башни кислота передается во вторую, а из второй отводится в смеситель 20.
Для окисления оксида азота до 93% удельный окислительный объем составляет 11,5 м3 на 1т HNO3 в сутки.
Дальнейшее окисление оксида азота проводят 98%-ной азотной кислотой в доокислителе 8 с соблюдением количества вводимой кислоты с таким расчетом, чтобы отходящая кислота содержала не более 75% HNO3.
Азотная кислота после доокислителя также собирается в смесителе 20. Окисленные нитрозные газы охлаждаются рассолом в трубчатом холодильнике 9 до -10° С и поступают в башню 10 на поглощение 98%-ной азотной кислоты.
544
8 Химическая технология неорганических веществ, кн.
Рис. 10.95. Схема производства концентрированной азотной кислоты из нитрозных газов, полученных окислением аммиака воздухом при атмосферном давлении:
1 — паровой котел; 2 — скоростной холодильник; 3— холодильник-концентратор; 4—вентилятор; 5—окислительные башни; 6 — кислотные холодильники; 7—центробежные насосы; 8 — доокислитель; 9—газовый холодильник; 10—абсорбер; 11—рассольные холодильники; 12 — холодильник 98%-ной азотной кислоты; 13— промывная башня; 14 — отбелочная колонна; 15 — головной холодильник; 16—водяной холодильник; 17 — сборник чистой 98%-ной азотной кислоты; 18—водяной концентратор N2O4; 19—рассольный холодильник N2O4; 20—смеситель; 21 — поршневой насос; 22 — автоклав; 23 — компрессорный холодильник; 24—кислородный баллон; 25 — емкость сырой кислоты
Процесс поглощения диоксида азота проводится в три ступени при температуре около -10° С. В первой ступени колонны циркулирует кислота, содержащая до 30% диоксида азота, во второй — до 20, в третьей — до 10% NO2. В верхнюю часть колонны подается свежая, предварительно охлажденная до -10° С азотная кислота. Теплота, выделяющаяся при растворении оксидов азота, отводится в холодильниках 11.
Каждая ступень поглощения в колонне имеет два слоя насадки: верхний слой орошается перетекающим раствором, нижний — раствором, циркулируемым насосами. Насыщенный диоксидом азота раствор азотной кислоты отводится из нижней ступени в отбелочную колонну 14, а отходящие газы — в башню 13, в которой промываются поступающей из холодильника 3 разбавленной азотной кислотой. Кислота предварительно отбеливается от растворенных оксидов азота продувкой воздухом.
Промывная башня 13 работает без охлаждения. За счет поглощения паров кислоты и частично диоксида азота, кислота концентрируется до ~40%НМОз и отводится в смеситель сырой смеси. Газ промывается окончательно водой в башне 13 на верхнем слое насадки. Разбавленная кислота после промывки выводится из системы.
В процессе нагревания раствора оксидов азота в азотной кислоте в отбелочной колонне 14, снабженной паровой рубашкой, отгоняется чистый диоксид азота. Азотная кислота после охлаждения в холодильнике 16 собирается в сборник 17, откуда она частично отводится в качестве товарного продукта, а оставшееся количество возвращается в систему для поглощения диоксида азота из нитрозных газов. Диоксид азота после отбелочной колонны 14 охлаждается до 40° С в холодильнике 15. Образующийся конденсат — азотная кислота в виде флегмы поступает на орошение отделочной колонны 14.
Оксиды азота охлаждаются примерно до 20° С водой в первом холодильнике 18 и рассолом до -8° С во втором холодильнике 19. Жидкий тетраоксид диазота направляется в смеситель 20, в которую поступает и разбавленная азотная кислота. Образующаяся смесь насосом (периодически или непрерывно) подается в автоклав 22, в котором она взаимодействует с кислородом, подаваемым компрессором 23 через буферный баллон при 40—50 атм.
Полученная в автоклаве кислота содержит -25% избыточного тетраоксида диазота. Раствор собирается в емкости 25 и отбеливается в колонне 14. Все выделяющиеся из сборников нитрозные газы, а также продувочные газы присоединяются к нитрозным газам перед вентилятором 4.
75%-ная азотная кислота из доокислителя перерабатывается в автоклаве в 98%-ную. Из всего количества кислоты, получаемой в автоклаве, около 20% расходуется на окисление оксида азота.
546
На получение 1 т азотной кислоты (100% HNO3) расходуют: 0,294 т аммиака, 150 м3 кислорода, 270 кВт-ч электроэнергии, 190 м3 воды и 0,05 г платины.
Производство концентрированной азотной кислоты из нитрозных газов, полученных под давлением. Согласно приведенной на рис. 10.96 схеме, исходная аммиачно-воздушная смесь, получаемая в смесителе 1 под давлением 6—8 атм, сжигается в конверторе 2 при 880—910° С. Образующееся при этом тепло реакции окисления аммиака используется в котле-утилизаторе 3 для получения пара, а также в подогревателе отходящих газов 4. Далее нитрозные газы, с температурой 300° С, в трубчатом холодильнике 5 охлаждаются водой. В этом процессе образуется ~30%-ная азотная кислота и происходит осушка нитрозных газов. Газы далее проходят холодильник-окислитель 6, в котором основная масса оксида азота окисляется до его диоксида. В холодильнике 6 происходит процесс частичного образования кислоты и в результате выделяется 58—60%-ная HNO3. Степень окисления регулируется внесением некоторого количества воздуха. По выходе из окислителя образующаяся кислота отделяется от нитрозных газов и поступает в холодильник 6, а газы проходят последовательно два, охлаждаемые рассолом, холодильника-конденсатора 7 и 8. Охлаждение нитрозных газов в присутствии разбавленной азотной кислоты дает возможность проводить процесс при температуре до -20° С. В этих условиях отпадает процесс забивки труб твердым N2O4. При этом жидкая фаза состоит из азотной кислоты, воды, сесквиоксида азота и тетраоксида диазота. Полученная жидкая фракция собирается в смеситель 9 и передается насосом 10 в автоклав 11 на переработку в концентрированную азотную кислоту, а нитрозные газы после конденсатора 8 поступают в тарельчатые колонны, орошаемые водой (на схеме не показаны), для получения 55%-ной азотной кислоты.
Сырую смесь обрабатывают в автоклаве 11 кислородом под давлением 50 атм в присутствии избытка N2O4, 98—99%-ная азотная кислота, содержащая 25% N2O4, непрерывно собирается в емкость 12, в которой давление снижено до 1 атм. Выделяющаяся смесь газообразного диоксида азота и других газов из смесителя 9 добавляется к основному потоку из отбелочной колонны 14, конденсируется в процессе охлаждения рассолом в трубчатке 16 и возвращается в смеситель 9.
Сырая кислота, проступающая в емкость 12 с температурой 70—80° С, охлаждается рассолом примерно до 0° С и направляется в отбелочную колонну 14. Это позволяет уменьшить унос паров азотной кислоты с газообразными оксидами азота, выходящими из колонны, и сократить ее количество в цикле. Чистая 98%-ная азотная кислота выходит из нижней части колонны 14 и после охлаждения водой в трубчатке 15 собирается в емкости 17.
18*
547
На абсорбцию
Рис. 10.96. Схема производства концентрированной азотной кислоты из нитрозных газов, полученных под давлением: 1 — смеситель; 2 — конвертор; 3 — котел-утилизатор; 4— подогреватель отходящих газов; 5— скоростной холодильник; 6 — холодильник-окислитель; 7, 8— конденсаторы оксидов (I и П степени); 9 — смеситель; 10—насос для сырой смеси; 11 — автоклав; 12 — сборник автоклавной кислоты; 13— холодильник сырой кислоты; 14 — отбелочная колонна; 15 — холомыътшь чистой азотной кислоты; 16—конденсатор диоксида азота; 17—сборник товарной 98%-ной кислоты; 18 — центробежный насос
По рассмотренной схеме в концентрированную азотную кислоту перерабатывается лишь часть оксидов азота, а остальное количество служит полуфабрикатом для производства разбавленной (50—55%-ной) азотной кислоты. Однако нитрозные газы, выходящие из конденсаторов 7 и 8, можно полностью окислить до диоксида азота, с дальнейшим поглощением его концентрированной азотной кислотой в процессе охлаждения. В процессе нагревания нитрозные газы выделяются из раствора и перерабатываются в автоклаве в 98%-ную азотную кислоту. В таком варианте производства в концентрированную азотную кислоту можно перевести до 70% оксидов азота и лишь 30% выделить из скоростного холодильника 5 в виде разбавленной 30%-ной кислоты.
10.6. НИТРАТ АММОНИЯ
Физико-химические свойства. Нитрат аммония (аммонийная селитра) NH4NO3, бесцветные гигроскопичные кристаллы. Температура плавления 169,6° С, а температура кипения 235° С. При атмосферном давлении в интервале от -50° С до температуры плавления существует в пяти кристаллических формах I—V (табл. 10.5), различающихся структурой, удельным объемом и другими свойствами кристаллов.
Таблица 10.5. Свойства кристаллических модификаций нитрата аммония
Модификация Кристаллическая решетка Область существования, °C Параметры решетки, нм
а b с
I Кубическая 125,8—169,6 0,441 — —
II Тетрагональная 84,2—125,8 0,576 — 0,502
III Ромбическая 32,2—84,2 0,718 0,771 0,583
IV » от 32,2 до —16,9 0,494 0,544 0,575
V Тетрагональная Ниже —16,9 0,572 — 1,600
Д№ полиморфных переходов IV III 1,7 кДж/моль, III II 13 кДж/моль. В процессах перехода II -> III, IV -> III и IV -> V объем элементарной ячейки увеличивается.
Для модификации IV: плотность 1,725 г/см3; Cj = 139,4 Дж/(моль-К); А/7П°Л = 5,86 кДж/моль, - -365,7 кДж/моль, ДО^р = -184 кДж/моль; S2°98 = 151,1 Дж/(моль-К).
Для жидкого нитрата аммония — плотность 1,436 г/см3 (170° С), 1,414 г/см3 (220° С); Г) = 5,71 МПа-с (170° С), 3,23 МПа-с (220° С); у = 98,8 МН/м2 (179° С), 98,4 МН/м2 (187° С); р = 2,5 Ом-см (175° С). Уравнения температурной зависимости давления пара:
1g р (мм рт. ст.) = 10,708—4670/7(349—438 К),
1g р (мм рт. ст.) = 9,987—4360/7(373—513 К).
549
-20 0 20 40 60 80 100 120 140 160 Температура, °C
Рис. 10.97. Растворимость нитрата аммония в воде
Хорошо растворяется в воде (рис. 10.97). Например, при 100° С в 1 кг воды растворяется более 10 кг, а при 50° С —34,6; 25° С —21,2 и 0° С — 11,9 кг. Растворяется в жидком аммиаке — 39,1 в 100 г воды при 25° С. Растворяется также в этаноле, метаноле и пиридине.
В процессе нагревания при 200—270° С разлагается:
NH4NO3 -> N2O + 2Н2О + 36,8 кДж/моль.
Выше 270° С разлагается со взрывом:
NH4NO3 -> N2 + 05О2 + 2Н2О + 112,6 кДж/моль.
Нитрат аммония очень гигроскопичен. Приведенная на рис. 10.98 номограмма показывает, что при 30° С давление пара над его насыщенным (70,2%) раствором составляет 2,46 кПа, а гигроскопическая точка равна 60%; при относительной влажности воздуха более 60% нитрат аммония будет увлажняться. Гигроскопичность нитрата аммония и скорость процесса поглощения им влаги из воздуха, как установлено, растут при участии растворимых минеральных солей. Например, внесение 1,2% нитрата магния снижает гигроскопическую точку нитрата аммония на 8—12%, а скорость поглощения влаги при этом растет.
Значительная растворимость в воде и высокий температурный коэффициент растворимости, гигроскопичности и имеющие место полиморфные превращения являются причиной слеживаемости нитрата аммония. Это в конечном итоге затрудняет фактическое его применение.
С целью исключения вышеперечисленных причин нитрат аммония производят с минимальным содержанием влаги (не более 0,2%) и охлаждение гранул до перехода его в ромбическую бипирамидаль-ную форму кристаллов, стабильных при температуре ниже 32,3° С. Перед процессом кристаллизации в раствор вводят кондиционирующие добавки (нитрат магния, нитраты кальция и магния, сульфат алюминия или смеси его с диаммонийфосфатом) и т.д.
С этой же целью гранулы нитрата аммония обрабатывают поверхностно-активными веществами, способными образовывать гидрофобные пленки. Для этой цели применяют 40%-ный раствор диспергатора НФ, получаемого конденсацией сульфокислот нафталина с водным раствором формальдегида, а также жирные кислоты и их алкины. 550
Растворимость NH4NO3, кг соли на 100 г воды ।__1 । । । 1 । । । । । । iiiiiiiiii
О 5 10 1520 253035 40 50 60 70 80 90 100110120130140150160170
Температура, °C
Рис. 10.98. Равновесное давление водяного пара над насыщенными растворами нитрата аммония
Нитрат аммония является концентрированным азотным удобрением. Его применяют в производстве взрывчатых веществ — аммонитов и гранулитов, в качестве реагента для растворения циркониевых оболочек твэлов в процессе регенерации облученного ядерного топлива.
Согласно существующему в России ГОСТ 2—85, производят гранулированный нитрат аммония двух марок: А — высшей категории качества и Б — высшей категории качества (высший сорт) и первой категории качества (первый сорт). Нитрат аммония марки А должен содержать не менее 98% NH4NO3, а марки Б — не менее 34,4% N в высшем сорте и 34,0% N в первом сорте. В зависимости от сорта нитрат аммония должен содержать влагу в пределах 0,2—0,3%. Содержание добавок (в пересчете на сухое вещество) в продукте высшей категории качества марок А и Б: нитратов кальция и магния — 0,2—0,5% (в пересчете на СаО), фосфатов — 0,5—1,2% (в пересчете на Р2О5), сульфата аммония 0,3—0,7%, сульфата аммония + фосфата аммония 0,4-—0,6%. В продукте первого сорта марки Б содержание добавок не нормируется. Величина pH 10%-ного водного раствора для всех сортов с сульфат-551
Рис. 10.99. Теплота процесса нейтрализации азотной кислоты газообразным аммиаком при атмосферном давлении и 18° С
но-фосфатной добавкой 4,0, а с другими добавками — 5,0. Согласно действующему ГОСТу регламентируются также размеры гранул.
Технология нитрата аммония.
Процесс получения нитрата аммония базируется на реакции азотной кислоты с аммиаком:
ННО3(ж) + NH3(r) = = NH4NO3(tb) + 144,9 кДж.
В процессе нейтрализации 47—60%-ной HNO3 образуется
водный раствор нитрата аммония, который для получения целевого продукта необходимо упаривать. В процессе упарки используется теплота нейтрализации согласно приведенному выше уравнению реакции. Количество же теплоты, выделяющейся по реакции, зависит от концентрации исходной азотной кислоты. Как видно из данных рис. 10.99, чем меньше концентрация кислоты, тем меньше выделяется теплота. Данные рис. 10.100 показывают, что при соответствующей организации технологии за счет выделяемого теп
ла реакции можно выпарить значительное количество воды, вводимой в систему с азотной кислотой, и получить насыщенный рас
твор и даже плав нитрата аммония.
Процесс нейтрализации азотной кислоты аммиаком производится при температуре 180—200° С под давлением 0,35—0,6 МПа, а самоиспарение раствора — при меньшем или при атмосферном давлении. Соковый пар при этом используют для дальнейшей упарки раствора от 75—80 до 95—99% основного вещества в целевом продукте в вакуум-аппаратах. Это исключает процесс кипения раствора в зоне реакции. В существующем способе производства и нитрата аммония отвод теплоты осуществляют в самом нейтрализаторе, в котором параллельно с нейтрализацией происходят процессы кипения и упарки раствора. Поэтому реактор назван ИТН (использователь теплоты нейтрализации).
Рис. 10.100. Зависимость массовой доли нитрата аммония в получаемом растворе от содержания нитрата водорода в исходной кислоте при 70° С: 1 — при использовании теплоты реакции (учтены 3%-ные потери теплоты); 2 — без использования теплоты реакции
552
Процесс нейтрализации азотной кислоты разбавленной другими газами (при утилизации продувочных и танковых газов из цеха синтеза аммиака) проводят при температуре 90—100° С в аппаратах колонного типа скрубберных или тарельчатых, орошаемых циркулирующим раствором нитрата аммония, в который вводится азотная кислота. Образующийся при этом горячий раствор направляется в вакуум-испаритель, в котором при остаточном давлении 13—20 кПа охлаждается до температуры кипения (70—75° С), после чего часть его возвращают на процесс орошения нейтрализатора, а другую часть направляют на упарку.
В процессе применения 47—55%-ной азотной кислоты в аппаратах ИТН образуются растворы, содержащие 62—83% NH4NO3. Для получения плава целевого продукта растворы упаривают в ваку-ум-выпарных аппаратах, используя в качестве теплоносителя соковый пар из аппаратов ИТН, а при недостатке его и свежий пар. Процесс гранулирования плава, содержащего до 98,7% целевого продукта, осуществляют в потоке воздуха в футерованных кислотоупорным кирпичом железобетонных башнях.
В настоящее время в химической промышленности России применяются крупнотоннажные технологические линии нитрата аммония АС-67, АС-72 и модернизированные АС-72М, обеспечивающие объем производства 1360—1575 т /сут и обеспечивающие высокую надежность бесперебойной работы.
Аппарат ИТН (рис. 10.101) состоит из двух цилиндрических частей: нижней — реакционной и верхней — сепарационной. Внутри корпуса 1 реакционной части находится реакционный стакан 2, в нижней части которого имеются отверстия. В нижней части реакционной зоны по титановым барботерам 3 и 4 поступают аммиак и азотная кислота. Скорость аммиака в отверстиях барботера 30—50 м/с, количество отверстий (диаметром 3 мм) — 6650. Скорость поступления азотной кислоты через 2160 отверстий (диаметром 1,5 мм) — 2—3 м/с. В процессе за счет теплоты реакции из образующегося нитрата аммония испаряется часть воды, в результате чего возникает подъемная сила, и парожидкостная смесь выбрасывается из верха реакционного стакана через погруженный в раствор завихритель 5, способствующий разделению парожидкостной смеси. По цилиндрическому пространству между реакционным стаканом 2 и корпусом аппарата 1 раствор нитрата аммония движется вниз, упариваясь в процессе циркуляции за счет теплоты, передаваемой через стенку стакана, и возвращается в него через нижние отверстия. За счет кратковременности пребывания реагентов в реакционной зоне (0,5—1 с) обеспечиваются лишь незначительные потери азота за счет термического разложения азотной кислоты и нитрата аммония.
553
Рис. 10.101. Аппарат ИТН:
1 — корпус реакционной части аппарата; 2 — реакционный стакан; 3— барботер аммиака; 4 — барботер азотной кислоты; 5 — завихритель; 6 — корпус сепаратора; 7 — колпачковая тарелка; 8 — отбойник брызг
Рис. 10.102. Комбинированный выпарной аппарат:
1 — кожухотрубная часть; 2—концентрационная часть; 3 — ситчатые провальные тарелки; 4 — змеевик для подвода теплоты; 5 — очистная часть; 6 — сетчатый отбойник брызг;
7 — ситчатые переточные тарелки
Верхняя часть аппарата служит сепаратором 6, в котором соковый пар, поднимаясь со скоростью 0,6 м/с, промывается на четырех барботажных колпачковых тарелках 7. На двух нижних тарелках пар отмывается от аммиака 20—25%-ным раствором нитрата аммония, подкисленным азотной кислотой, а на двух верхних — конденсатом сокового пара улавливают пары азотной кислоты и брызги раствора нитрата аммония. Процесс окончательного освобождения от брызг происходит в отбойнике 8. Промывные растворы возвращаются в аппарат ИТН.
Процесс получения плава нитрата аммония из образующегося в аппарате ИТН концентрированного раствора (~ 90%) проводят упариванием его в комбинированном выпарном аппарате (рис. 10.102). Основная вертикальная кожухотрубная часть 1 аппарата служит для упаривания раствора нитрата аммония, стекающего по внутренним стенкам трубок. Источниками теплоты являются пар с давлением 1,3—1,5 МПа, подаваемый в межтрубное пространство, и горячий 554
В атмосферу
В атмосферу
Рис 10.103. Схема получения нитрата аммония на агрегате АС-72М:
1 — подогреватель аммиака; 2 — подогреватель азотной кислоты; 3 — аппарат ИТН; 4, 5 — доней-трализаторы; 6 — комбинированный выпарной аппарат; 7 — промыватель паровоздушной смеси; 8, 18 — скрубберы; 9 — гидрозатвор; 10 — фильтр плава; 11 — сборник для плава; 12 — погружной насос; 13 — центробежный насос; 14 — сборник; 15 — напорная емкость; 16 — акустический гранулятор; 17 — грануляционная башня; 19, 22 — вентиляторы; 20 — ленточный транспортер; 21 — холодильник с кипящим слоем гранул; 23, 24 — подогреватели воздуха; 25— нагнетатель воздуха
(180° С) воздух, движущийся внутри трубок противотоком стекающему раствору. Воздух поступает из нижней концентрированной части 2, проходя через расположенные в ней пять ситчатых провальных тарелок 3. На трех верхних тарелках смонтированы змеевики 4 для дополнительной подачи теплоты. Количество влаги в воздухе, поступающем в доупарочный аппарат, не должно превышать 20 г/кг. Из концентрационной части вытекает плав, содержащий 99,7% нитрата аммония с температурой 175—185° С. Температура плава в доупароч-ном аппарате должна быть не выше 190° С.
В верхней, очистной, части 7 аппарата расположены две ситчатые переточные тарелки 4, верхняя из которых орошается конденсатом, а нижняя — раствором нитрата аммония. Они отмывают отходящую из аппарата паровоздушую смесь и частично упаривают поступающий раствор нитрата аммония.
555
Согласно приведенной на рис. 10.103 технологической схеме, при получении нитрата аммония на агрегате А-72 М исходный газообразный аммиак, пройдя подогреватель 1, в котором нагревается до 120—160° С и 58—60%-ная азотная кислота из подогревателя 2 с температурой 80—90° С поступают в два параллельно работающих аппарата ИТН 3. В аппаратах ИТН поддерживается близкое к атмосферному давление. Поскольку при избытке азотной кислоты давление пара азотной кислоты над раствором нитрата аммония меньше, чем давление аммиака при его избытке в растворе с целью уменьшения потерь связанного азота с соковым паром в виде NH3, HNO3, NH4NO3 и NO2, процесс ведут в слабокислой среде. Концентрация азотной кислоты в отходящем из аппарата растворе (2—5 г/л) регулируется автоматически. Температура этого раствора 150—170° С, а содержание в нем нитрата аммония находится в пределах 89—92%.
Вытекающий непрерывно из аппаратов ИТН раствор нитрата аммония нейтрализуется в двух донейтрализаторах — основном 4 и контрольном 5. В донейтрализаторы же вводят кондиционирующую добавку — 30—40%-ный раствор нитрата магния, приготовленный растворением технического оксида магния в азотной кислоте. Раствор нитрата аммония с содержанием избыточного аммиака (0,1—0,5 г/л) направляется на доупаривание в комбинированный выпарной аппарат 6, откуда плав, проходя гидрозатвор-донейтрализатор 9 и фильтр 10, поступает в приемный сборник 11. Далее плав погружным насосом 12 по трубопроводу с антидетонационной насадкой перекачивается в напорную емкость 15, находящуюся над грануляционной башней 17.
Из напорной емкости плав подается в башню через три вибро-акустических гранулятора 16. Струя
Рис. 10.104. Акустический гранулятор:
1 — корпус; 2 — сопло; 3 — пластина; 4 — сетчатый фильтр; 5 — вибрирующее перфорированное днище
плава, поступающего через внутреннее сопло 2 гранулятора на пластину 3, генерирует ее колебания, передающиеся перфорированному днищу 5 (рис. 10.104).
Через отверстия в нижней части в грануляционную башню 17 засасывается наружный воздух и воздух из аппарата кипящего слоя 21, в котором гранулы досушиваются и доохлаждаются. Высота падения капель плава в башне не превышает 50 м. В потоке воздуха они затвердевают, превращаются в гранулы. Температура плава, подаваемого на процесс грануляции, на 5—7 К превышает температуру его кристаллизации. Плав нитрата ам-
556
мония с влажностью 0,2% начинает кристаллизоваться при 167° С, а полностью затвердевает при 140° С. Количество воздуха — 300—500 тыс. м3/ч в зависимости от погодных условий.
Из нижней конусной части башни гранулы поступают на ленточный транспортер 20, с помощью которого подают их в трехсекционный аппарат 21 для доохлаждения в кипящем слое с автономной подачей воздуха в каждую секцию. Температура гранул после холодильника 20—50° С. В процессе гранулирования с последующим охлаждением в потоках воздуха гранулы подсушиваются. Целевой продукт содержит более 99,8 NH4NO3. При относительной влажности атмосферного воздуха более 60% продукт пропускают через подогреватели 23 так же, как и воздух, подаваемый в доупарочный аппарат 6.
Аппараты кипящего слоя применяют не только для охлаждения гранул, но и в процессе гранулирования частиц нитрата аммония из растворов. Для этого в кипящий слой подают 80—85%-ный раствор NH4NO3. При этом вода выпаривается в потоке горячего воздуха.
Из верхней части грануляционной башни воздух поступает в шесть скрубберов 18, где отмывается от пыли нитрата аммония и аммиака циркулирующим 20%-ным раствором NH4NO3, и вентилятором 19 выбрасывается в атмосферу. Через эти же скрубберы проходят и газы из выпарного аппарата после промывателя 7 и из доней-трализатора после скруббера 8.
Прочность гранул целевого продукта зависит от технологического режима получения плава и его гранулирования. После затвердевания их при прохождении в башне происходит модификационный переход I -> II (см. рис. 10.97). В процессе дальнейшего охлаждения гранул и при последовательных превращениях модификаций II -> III -> IV изменяются удельные объемы, что предопределяет образование малопрочных гранул. Возможно осуществлять метастабильное превращение II -> IV, при котором удельные объемы изменяются незначительно, поскольку параметры кристаллических решеток модификаций II и IV близки друг к другу. В этом варианте гранулы получаются прочными. Переход II IV обеспечивается режимом работы, при котором в плаве, поступающем на гранулирование, содержатся добавки, а его влажность не превышает требуемого предела. Считается, что наиболее эффективной является добавка нитрата магния (0,3—0,6%), но при этом содержание влаги в плаве не должно быть выше 0,25%. Эти условия обеспечивают переход II -> IV при 50,8° С, т.е. в процессе охлаждения гранул в нижней части башни и в кипящем слое в холодильнике.
На производство 1 т гранулированного нитрата аммония (34,58% N) (при мощности агрегата 56,8 т/ч целевого продукта) расходуется: 557
Рис. 10.105. Барабанный кристаллизатор для получения чешуйчатого нитрата аммония:
1 — вращающийся барабан; 2 — неподвижный барабан; 3 — камера для выхода воды; 4 — полый вал; 5 — корыто; 6 — нож; 7 — электродвигатель
0,213 т аммиака (100%-ного), 0,793 т азотной кислоты (100%-ной), 0,96 ГДж пара, 28,3 кВт ч электроэнергии.
В промышленности нитрат аммония производят также в чешуи-рованном виде. Их получают в процессе кристаллизации плава (97,5—98,5% целевого продукта) на поверхности охлаждаемого изнутри водой барабана (рис. 10.105). Обычно в процессе кристаллизации плав нитрата аммония подсушивается на 0,5—0,7%, и чешуйчатый продукт имеет влажность около 2%. Его досушивают в барабанной сушилке воздухом, нагретым в калорифере до 110—120° С, поэтому сушимый продукт не нагревается выше 75° С.
Производят также водоустойчивый нитрат аммония путем добавления раствора сульфата железа (III) (600—800 г на 1 т) и кристаллизации на вальцах. Полученную массу докристаллизовывают в шнековом кристаллизаторе в процессе опрыскивания массы гидрофобизирующей смесью жирных кислот с парафином. Образующаяся кристаллическая масса охлаждается до 50° С в барабанной сушилке.
Пористый гранулированный нитрат аммония для изготовления взрывчатых веществ получают сушкой гранул с повышенным содержанием влаги при добавке в плав порообразующих легколетучих веществ.
Нитрат аммония применяют для получения известково-аммоний-ной селитры с различными соотношениями МНдЫОзгСаСОз — от 80:20 до 53:47. Выпускают плав нитрата и сульфата аммония (МНд^ЗОд • 2NH4NO3 — сульфат-нитрат аммония, а также производят комплексное удобрение — калийно-аммонийную селитру, получаемую сплавлением NH4NO3 и КС1.
Указанные продукты производят в чешуйчатом или гранулированном виде.
558
10.7. СУЛЬФАТ АММОНИЯ
Физико-химические свойства. Сульфат аммония (МН4)г8О4, бесцветные кристаллы с орторомбической решеткой (а = 0,7782 нм, Ъ = = 0,5993 нм, с = 1,0636 нм, z = 4, пространственная группа Рпта); плотность 1,766 г/см3; С° = 187,4 Дж/(моль-К); ДЯ^р =-1180,26 кДж/моль, =-901,31 кДж/моль; - 220,1 Дж/(моль-К). Выше 100° С ступенчато разлагается с выделением аммиака и образованием сначала гидросульфата аммония, а затем (NH4)2S2O7 и сульфаниловой кислоты. При 205° С давление NH3 над (ЫНд^БОд равно 0,067 кПа, а при 300° С — 6,772 кПа. Данные о растворимости сульфата аммония в воде показаны на рис. 10.106, из которых видно, что он не образует кристаллогидратов и его растворимость изменяется незначительно с повышением температуры.
Сульфат аммония под действием сильных окислителей (КМпО4) окисляется до N2. Он образует двойные соли с сульфатами других металлов, например железа, алюминия, калия, натрия и др.
Сульфат аммония применяют в качестве азотно-серного удобрения, компонента осадительной ванны в процессе формования вискозного волокна и добавки к стекольной шихте (для улучшения ее плавкости).
Согласно существующему ГОСТ 9097—82, сульфат аммония должен содержать (в %):
Высшей категории качества I категория качества
Азот (на сухое вещество), не менее Вода, не более: 21 21
в гранулированном 0,6 —
в кристаллическом Серная кислота, не более: 0,2 0,3
в гранулированном 0,5 —
в кристаллическом Фракции, не менее: 0,03 0,05
в гранулированном (1—4 мм) 90 —
в кристаллическом ( + 0,5 мм) 60 не нормируется
Рассыпчатость продукта 100%. Остаток на сите 6 мм должен отсутствовать.
Физико-химические основы получения сульфата аммония нейтрализацией серной кислоты аммиаком. Процесс нейтрализации серной кислоты газообразным аммиаком по реакции
2NH3(r) + Н28О4(ж) = (NH4)2SO4(tb) + 274 кДж протекает с выделением значительного количества теплоты, которая расходуется на испарение из системы воды, в результате чего целевой 559
Рис. 10.106. Политерма системы (NH4)2SO4—Н2О (108,9° С — температура кипения насыщенного раствора под давлением 0,101 МПа)
продукт кристаллизуется из пересыщенного раствора. В процессе кристаллизации необходимо обеспечить образование средней соли, не допуская выделения кислых солей. Содержащиеся в исходной серной кислоте примеси сульфатов металлов затрудняют процесс кристаллизации сульфата аммония.
В процессе нейтрализации кислоты осаждаются коллоидные гидрокси-
ды железа и алюминия:
Fe2(SO4)3 + 6NH3 + 6Н2О = 2Fe(OH)3 + 3(NH4)2SO4
A12(SO4)3 + 6NH3 + 6H2O = 2A1(OH)3 + 3(NH4)2SO4 которые обволакивают кристаллы сульфата аммония и препятствуют их росту. С целью исключения этого явления растворы нейтрализуют не полностью. В реакторах поддерживают кислую реакцию среды.
Как видно из данных рис. 10.107, в системе (NH4)2SO4—H2SO4—Н2О в твердой фазе существуют четыре различные кислые соли. Поле кристаллизации (NH4)2SO4 лежит в области составов систем, содержащих незначительные количества серной кислоты — а\Е\С при 10° С и а2Е2С при 90° С. Во избежание выделения кислых солей содержание серной кислоты в жидкой фазе системы должно быть меньше, чем в точках Е\ и £2>т.е. меньше 11,08% при 10° С или 19,77% при 90° С. В процессе нейтрализации реакционная масса имеет высокую температуру, но при последующем выделении кристаллов она охлаждается. Поэтому при выборе состава реакционного раствора необходимо исходить из положения поля кристаллизации сульфата аммония при невысоких температурах. В производстве кислотность раствора поддерживают на уровне 4—12% свободной серной кислоты, что исключает возможность кристаллизации кислых солей.
Получение сульфата аммония из аммиака коксового газа. В коксовом газе содержится 7—15 г/см3 аммиака. Разработаны три способа его переработки в сульфат аммония: коксовый, прямой и полупрямой. Согласно коксовому способу, коксовый газ охлаждают. При этом из него конденсируется смола и надсмольная вода, насыщенная аммиаком. Оставшийся в газе аммиак абсорбируют водой в аммиачных скрубберах. Из полученного водного аммиака и надсмольной воды в дистилляционных колоннах аммиак абсорбируют серной кислотой. Способ требует громоздкого оборудования и значительных энер-560
H2SO4
20 40 60 80 (NH4hSO4
(NH4)2SO4i %
Рис. 10.107. Изотермы растворимости в системе (NH4)2SO4—H2SO4—Н2О при 10, 30, 50, 70 и 90° С. Участки кривых соответствуют насыщению раствора:
7 —(NH^SC^; 2 —4(NH4)2SO4-4H2SO4; 3 — 3(NH4)2SO4 H2SO4; 4 — (NH4)2SO4 H2SO4; 5 —(NH4)2SO4-3H2SO4
гозатрат. Прямой способ сводится к поглощению аммиака серной кислотой с получением сульфата аммония. Процесс проводят, получая целевой продукт непосредственно из коксового газа, предварительно охлажденного до 68°С и очищенного от смолы в электрофильтрах.
Наиболее широко применяемым в промышленности способом является полупрямой способ, согласно которому исходный коксовый газ охлаждают до 25—30° С для конденсации смолы. При этом конденсат расслаивается на два слоя: нижний — смолу и верх
ний — надсмольную воду, в которой растворена часть аммиака. Надсмольную воду обрабатывают суспензией оксида-гидроксида кальция, а выделившийся аммиак абсорбируют серной кислотой вместе с аммиаком, оставшимся в доочищенном в электрофильтрах от смолы коксового газа.
Абсорбцию аммиака из коксового газа можно производить в сепараторах барботажного типа (сатураторный способ) или в скрубберах (бессатураторный способ). В сатураторном способе абсорбция аммиака из коксового газа и кристаллизация образующегося сульфата аммония совмещены в одном аппарате — сатураторе. Однако это ограничивает возможность выбора технологического режима, который был бы оптимальным одновременно для обоих процессов, т.е. обеспечивающего более полное поглощение аммиака и образование крупнокристаллического сульфата аммония. Поэтому эти процессы ведут раздельно — поглощение аммиака в скрубберах, а кристаллизацию сульфата аммония — в кристаллизаторах.
Согласно представленной на рис. 10.108 схеме получения сульфата аммония, исходный очищенный от смолы коксовый газ подогревают в подогревателе 2 до 50—60° С. Затем его направляют в сатуратор 3, примешивая пароаммиачную смесь, получаемую в процессе дистилляции надсмольной воды. Сатуратор представляет собой стальной вертикальный цилиндрический аппарат с коническим днищем, футерован кислотоупорными плитами. Он заполнен суспензией, состоящей из раствора и кристаллов сульфата аммония, уровень которой поддержи-561
Рис. 10.108. Схема получения сульфата аммония сатураторным способом:
1 — сборник серной кислоты; 2 — подогреватель коксового газа; 3 — сатуратор; 4 — барботер; 5 — циркуляционный сборник; 6 — кислотная ловушка; 7 — сборник кристаллов; 8 — сборник маточного раствора; 9 — центрифуга; 10 — сушилка с кипящим слоем; 11 — сборник для обеспири-диненного раствора; 12 — сборник подкисленного раствора; 13 — сборник маточного раствора; 14, 15 и 16 — центробежные насосы
вается стоком через циркуляционный сборник 5 с гидравлическим затвором, препятствующим прорыву газа. Температура суспензии поддерживается в пределах 55—60° С. Содержание серной кислоты в жидкой фазе 4—5%. В сатуратор непрерывно дозируется серная кислота в необходимом количестве для связывания поступающего аммиака. Коксовый газ поступает в сатуратор через центральный барботер 4 с газораспределительным зонтом, погруженным в суспензию на 0,25 м. Зонт имеет направляющие лопасти для завихрения жидкости, что способствует лучшему ее контакту с газом и выравниванию концентрации серной кислоты. Дополнительное перемешивание производится циркуляционным насосом 14 и возвратом жидкости в сатуратор насосом 15 из циркуляционного сборника 5 и запасного резервуара 13. Через сливной карман циркуляционного сборника выводится всплывающая на поверхность жидкости уловленная из газа кислая смолка. При диаметре 6,25 м и общей высоте 9,5 м производительность сатуратора по газу составляет 40—45 тыс. м3/ч.
Выходящий из сатуратора газ, содержащий водяной пар, через кислотную ловушку брызг 6 направляется на охлаждение. Концентрация аммиака в нем меньше 0,03 г/м3. Суспензия кристаллов из нижней части сатуратора насосом 16 непрерывно откачивается в сборник кристаллов 7, в котором она сгущается и направляется в центрифугу 9. Сульфат аммония для снижения его кислотности промывают водой, а полученные промывные воды направляют на раз-562
бавление подаваемой в сатуратор кислоты до 75% H2SO4. Маточные растворы из верхней части сборника кристаллов и из центрифуги возвращаются самотеком через приемник 8 в сатуратор.
Содержащиеся в коксовом газе пиридиновые основания связываются в сатураторе серной кислотой. Для извлечения образующегося сульфата пиридина часть маточного раствора из сборника кристаллов 7 отводят в пиридиновую установку, а обеспиридиненный раствор подкисляют серной кислотой и возвращают через сборники 77 и 72 в сатуратор. Из сборника 11 удаляют шлам, образующийся в процессе подкисления раствора.
Сульфат аммония, выходящий из центрифуги с влажностью 1—3%, сушат, продувая горячим воздухом в аппарате с кипящим слоем 10, а затем охлаждают холодным воздухом. Целевой продукт получается мелкокристаллическим. Поэтому его гранулируют прессованием на вальцах.
На производство 1 т сульфата аммония расходуют: 0,73—0,75 т H2SO4 (100%-ной), 0,26—0,27 т аммиака, содержащегося в 30—35 тыс. м3 коксового газа, 100—108 МДж электроэнергии, 8 м3 воды и 2,7—6 т пара.
К недостаткам сатураторного способа относятся: 1) получение мелких кристаллов; 2) большой расход электроэнергии на преодоление гидравлического сопротивления абсорберов. Этих недостатков лишены бессатураторные способы.
В бессатураторных процессах абсорбцию аммиака из коксового газа ведут в полых скрубберах кислым ненасыщенным раствором сульфата аммония с последующей вакуум-выпаркой на кристалл или кислым насыщенным раствором с выращиванием образовавшихся мелких кристаллов в кристаллизаторах под атмосферным давлением. Обычно применяют первый способ — орошение абсорбера ненасыщенным раствором предотвращает их засоление, а кристаллизация в выпарных аппаратах позволяет регулировать размеры получаемых кристаллов.
Согласно приведенной на рис. 10.109 схеме, исходный аммиак улавливается в полом скруббере 2, снабженном форсунками. Скруббер разделен на две ступени. Нижняя его часть орошается раствором, содержащим 3—4% свободной серной кислоты, верхняя — раствором, содержащим 10—12% H2SO4. Коксовый газ из скруббера проходит через брызгоуловитель 1 и направляется на дальнейшую переработку. Серная кислота и вода (необходимая для разбавления и компенсации испарения) поступают в сборник 4 раствора, циркулирующего в верхней части скруббера с помощью центробежного насоса 5. Часть раствора через смолоотделитель 3 подается в сборник 11 маточного раствора, циркулирующего в нижней части скруббера с помощью центробежного насоса 12, куда также поступает маточный раствор из центрифуги 8.
Из нижней части скруббера часть раствора, содержащего около 1 % свободной серной кислоты и 40% сульфата аммония, отбирается через смолоотделитель 3 в сборник 10 и центробежным насосом 9 перекачивается в вакуум-выпарной аппарат 6. Образующиеся при этом кристал-563
Рис. 10.109. Схема получения сульфата аммония бессатураторным способом с вакуум-выпаркой:
1— брызгоуловитель; 2 — скруббер; 3 — смо-лоотделитель; 4, 10, 11 — сборники; 5, 7, 9, 12 — центробежные насосы; 6 — выпарной аппарат; 8 — центрифуга
Рис. 10.110. Схема получения сульфата аммония бессатураторным способом с кристаллизацией под атмосферным давлением:
1—скруббер; 2 — кристаллизатор; 3 — сборник маточных растворов; < 5, б, 7 — центробежные насосы; 8—сгуститель кристаллов; 9 — центрифуга; 10—теплообменник; 11—ловушка брызг
лы отпускаются в нижнюю коническую часть аппарата, выполняющего роль кристаллорастворителя, в котором мелкие кристаллы поддерживаются во взвешенном состоянии в восходящем потоке свежеприготовлен-ного раствора. Эта операция обеспечивает рост кристаллов при небольшом пересыщении раствора и более 60% получаются с размерами, превышающими 0,5 мм. Такие же результаты достигаются при использовании выпарных аппаратов, снабженных выносными кристаллорасти-телями. Суспензия из выпарного аппарата, содержащая 50—60% кристаллов, передается для фильтрования на центрифугу 8, где кристаллы промываются горячим конденсатом (70—80° С) для удаления остатков серной кислоты. Затем целевой продукт направляется на сушку.
Бессатураторная схема с выделением сульфата аммония в кристаллизаторе под атмосферным давлением приведена на рис. 10.110. Согласно схеме, аммиак из коксового газа улавливается в скруббере 1, под которым расположен кристаллизатор (кристаллораститель) 2, из верхней части которого маточный раствор направляется в сборник 3, 564
куда поступает серная кислота и вода. Из сборника 3 жидкость центробежным насосом 4 подается на верх скруббера через распылительные форсунки. Часть маточного раствора циркулирует через подогреватель 10, что позволяет поддерживать температуру на уровне 110° С.
В кристаллизаторе происходит рост кристаллов. При этом крупные кристаллы оседают в нижней конической части, а мелкие в смеси с маточным раствором центробежными насосами 5 и 7 возвращаются форсунками в скруббер. Добавка к раствору небольшого количества сульфата железа способствует образованию кристаллов с размерами, превышающими 0,25 мм. Суспензия кристаллов из нижней части кристаллизатора непрерывно перекачивается насосом 6 в приемник 8, в котором она сгущается, и далее поступает в центрифугу 9. Маточный раствор из приемника и центрифуги возвращается в сборник 3, а кристаллы сульфата аммония направляются на сушку.
Разработан также способ гранулирования сульфата аммония из 40—45%-ных растворов в кипящем слое гранул в потоке топочных газов.
Получение сульфата аммония из гипса. По этому способу, который основывается на реакции
CaSO4 + (ЫНдЬСОз = (NH4)2SO4 + СаСОз
роль серной кислоты выполняет гипс, ангидрит или фосфогипс — отход производства экстракционной фосфорной кислоты. Процесс проводят при температуре 50—55° С, применяя 32—33%-ный раствор карбоната аммония так называемым жидкостным методом. Ход реакции определяется значительной скоростью растворения сульфата кальция в воде по сравнению с растворимостью карбоната кальция.
После реакции и отделения осадка карбоната кальция на фильтрах полученный -40%-ный раствор сульфата аммония перерабатывают в твердый продукт выпаркой, кристаллизацией, фильтрацией кристаллов в центрифуге и их сушкой.
На производство 1 т сульфата аммония расходуют: 1,13 т гипса, 0,74 т карбоната аммония, 1,4 т пара, 25 м3 воды, 65 кВт-ч электроэнергии и 71,5 кг условного топлива.
Существует также газовый метод получения сульфата аммония из гипса, применяя вместо карбоната аммония газы — аммиак и диоксид углерода:
CaSO4 + 2NH3 + СО2 + Н2О = (NH4)2SO4 + СаСОз
Недостатком газового метода является то, что по нему получают мелкие кристаллы карбоната кальция, которые фильтруются плохо. С целью получения крупных кристаллов увеличивают длительность процесса проведения реакции, чем при жидкостном способе.
ГЛАВА 11
ФОСФОР И ЕГО СОЕДИНЕНИЯ
11.1. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ФОСФОРА
Содержание фосфора в земной коре составляет 0,105% (масс.), в воде морей и океанов — 0,07 мг/л. Известно около 200 фосфорсодержащих минералов, важнейший из которых апатит является основой фосфоритов. Практическое значение имеют также монацит СеРС>4, ксенотим УРОд, амблигонит LiAlPO4(F,OH), трифилин Li(Fe,Mn)PC>4, торбернит Си(иО2)2(РО4)2’12НгО, отунит CaCUChMPC^-lOFhO, вивианит Рез(РО4^-8НгО, пироморфит РЬ5(РО4)зС1, бирюза СиА16(РО4)4(ОН)8-5Н2О.
Фосфор имеет один устойчивый нуклид 31Р. Конфигурация внешней электронной оболочки атома 3s23p3, степень окисления -3, +3 и +5. Энергия последовательной ионизации при переходе от Р°до Р5+ (эВ): 10,486, 19,76, 30,163, 51,36, 65,02. Сродство к электрону 0,6 эВ. Атомный радиус 0,134 нм, ионные радиусы (в скобках указаны координационные числа) 0,186 нм для Р3-, 0,044 нм (6) для Р3+, 0,017 нм (4), 0,029 нм (5), 0,038 нм (6) для Р5+.
Известно свыше 10 модификаций фосфора, важнейшими из которых считаются белый, красный и черный фосфор. Технический белый фосфор называют желтым фосфором. Некоторые свойства важнейших модификаций представлены в табл 11.1.
Белый фосфор (а, Р, III) образуется в процессе конденсации паров и затвердевания расплава. Дифракционным методом исследований было установлено, что молекулы Р4 имеют форму правильных тетраэдров (рис. 11.1) с атомами фосфора в вершинах. Каждый атом фосфора связан с остальными тремя атомами молекулы тремя валентностями. Таким образом, три валентности фосфора имеют пирамидальное расположение. Расстояния между двумя атомами фосфора (ребро тетраэдра) равно 2,21 А Следовательно, углы между валентностями (направленными вдоль ребер тетраэдра) равны 60°. Фосфор представляет собой белую воскообразную массу или прозрачные кристаллы, похожие на алмазы из-за высокой дисперсии света и высокого показателя преломления. В процессе охлаждения белый фосфор становится хрупким. При -76,9° С он переходит в ^-модификацию 566
a
б
Рис. 11.1. Структура молекулы белого (а) и черного (б) фосфора
белого фосфора (Р, IV) с плотностью 1,88 г/см3 с гексагональной (а = 0,734 нм) кристаллической решеткой. ДЯ перехода 0,522 кДж/моль. В структуре белого фосфора содержатся тетраэдрические молекулы Р4. В а-фосфоре они могут свободно вращаться, что отсутствует в Р-фосфоре. Такие же молекулы наблюдаются в расплаве и парах фосфора, лишь выше ~800° С начинается диссоциация на молекулы Р2. В процессе конденсации таких паров при -190° С образуется неустойчивая модификация — коричневый фосфор, состоящий из молекул Р2. Выше -100° С он превращается в смесь белого и красного фосфора (Р II).
Таблица 11.1. Свойства важнейших модификаций элементного фосфора
Показатель Белый Р, III Красный Р, II Черный Р, I
Сингония Кубическая Моноклинная Ромбическая
Параметры элементарной
ячейки:
а, нм 1,851 1,02 0,33136
Ь, нм — 0,936 0,4376
с, нм — 2,51 1,0478
Р, град — 118,8 —
Z 224 84 8
Пространственная группа J43m Р2/с Стеа
Плотность, г/см3 1,828 2,31 2,702
Т °C 1 ПЛ 5 44,14 593* 1000*
^возо °C 287” 429 453
С°р, Дж/(мольК) 23,8 21,2 21,6
ДН^, кДж/моль 0,66 25,96 —
ДЯ^.!Г, кДж/моль 13,Г” 128,6 53,75
кДж/моль 0 -17,4 -39,35
5з98, Дж/(мольК) 41,1 22,85 22,7
*Под давлением. “Температура кипения. ***&Нжп.
567
Зависимость давления пара белого фосфора от температуры: для твердого 1g р(мм рт. ст.) = 19,0925—35291,4/T—3,51g7(273—317 К); для жидкого lgp(MM рт. ст.) = 4,512—2660/Г + 1,2431g7(3 17,3—530 К). Расплав белого фосфора легко переохлаждается. Плотность расплава 1,749 г/см3.
В процессе нагревания без доступа воздуха выше 180° С разрушается система связи Р4, вследствие чего начинается процесс полимеризации, приводящий его в красный фосфор. Существует целый ряд разновидностей красного фосфора, различающихся по цвету,— от оранжевого и ярко-красного до коричневого и черно-фиолетового, по плотности, температуре плавления и т.п. В процессе нагревания до 250—300° С образуется аморфный красный фосфор (у-Р) с плотностью 2,16 г/см3. При 360—450° С образуется моноклинный красный фосфор (Р, II, 5-Р). При ~500°С — другая моноклинная модификация, также образующаяся в процессе нагревания белого фосфора со свинцом или висмутом, фиолетовый фосфор в виде кристаллов с параметрами решетки: а = 0,924 нм, b = 0,921 нм, с = 2,23 нм, р = 107,4°. В процессе длительной термообработки при ~600°С образуется кубический красный фосфор (пространственная группа РтЗт, а = 1,131 нм, z = 66, плотность 2,35 г/см3). Существует также триклинный, тетрагональный, гексагональный красный фосфор и другой переход белого фосфора в красный фосфор ускоряется при действии УФ-облучения, а также в присутствии примесей I2, S, Se и т.д. В структуре красного фосфора существуют связанные между собой бесконечные цепи из тетраэдров Р4. В этой структуре могут быть выделены группировки Pg и Р9. В процессах испарения и плавления красного фосфора (при атмосферном давлении он сублимируется и может быть расплавлен при давлении 4,5 МПа) образуются молекулы Р4. Уравнение зависимости давления пара твердого красного фосфора от температуры: 1g р (мм рт. ст.) = 14,207—6300/Г + + 3,298Т + 0,1561g:r (298—866 К).
При давлении более 1,2 ГПа белый фосфор переходит в кристаллы черного фосфора (Р1, е-Р). Для перехода красного фосфора требуется более высокое давление (2,5 ГПа), нагревание до ~200° С облегчает переход. При более низких давлениях образуется аморфный черный фосфор (i-P) с плотностью 2,25 г/см3. Кристаллический черный фосфор получают и при атмосферном давлении — длительной термообработкой красного фосфора с ртутью при ~300° С в присутствии затравки.
Черный фосфор графитоподобный, его структура (см. рис. 11.1, б) состоит из слабо связанных между собой гофрированных слоев. При атмосферном давлении черный фосфор возгоняется без плавления, уравнение зависимости давления пара от температуры Igp (мм рт. ст.) = 568
= 13,36—7560/Г. Плавится он при -1000° С под давлением 1,8 ГПа. Экстраполяция на атмосферное давление дает температуру плавления 606° С. В процессе нагревания черный фосфор при давлении собственных паров при 560—580° С переходит в красный.
При давлении 8,6 ГПа образуется фосфор V с ромбоэдрической структурой типа a-As с плотностью 3,56 г/см3 (пространственная группа Ют, z = 2, а = 0,3524 нм, a = 57,25°). При давлении 10 ГПа он переходит в модификацию VI с кубической структурой типа NaCl (a = 0,2377 нм, z = 1) с плотностью 3,83 г/см3.
Белый и красный фосфор — диэлектрики, черный фосфор — полупроводник, модификации высокого давления, возможно, обладают металлическими свойствами. Сверхпроводимость наблюдается у модификаций P(V) и P(VI) ниже соответственно 4,7—6 К и 7,5—10 К (в зависимости от давления).
Белый фосфор очень активен. В процессе перехода к красному и особенно черному фосфору химическая активность резко снижается. Белый фосфор на воздухе светится в темноте, с чем исторически связано его название (от греч. — светоносный). Свечение обусловлено окислением (с образованием низших оксидов) паров фосфора (белый фосфор обладает заметной летучестью даже при низких температурах). Мелкодисперсный белый фосфор самовоспламеняется на воздухе, для компактного — температура воспламенения 34—50° С. В воде он практически не растворим (340^ при 15° С), хорошо растворяется в сероуглероде (89,8% при 10° С), растворяется в РС1з, POCI3, жидких SO2, NH3, малорастворим в этаноле (0,31%), диэтиловом эфире (1,04%), бензоле (3,2%), ССЦ (1,27%), глицерине (0,17%), уксусной кислоте (~1%), ксилоле. Для красного и черного фосфора растворители не найдены.
Красный фосфор при комнатной температуре окисляется медленно. Однако некоторые его образцы могут самовоспламеняться при наличии следов белого фосфора, а также наличии высокоактивного аморфного фосфора по границам зерен. Температура воспламенения красного фосфора 210° С (черного фосфора около 500° С). Однако большие количества красного фосфора в процессе длительного хранения на воздухе могут загораться из-за повышения температуры вследствие окисления.
В процессе нагревания фосфора с водными растворами щелочей выделяется фосфин:
Р4 + ЗКОН + ЗН2О = Н3Р + ЗКН2РО2
2Р4 + ЗВа(ОН)2 + 6Н2О = 2Н3Р + ЗВа(РО2Н2)2
569
Белый фосфор реагирует с растворами солей электроположительных металлов (Си, Pb, Ag и др.), вытесняя их из растворов с образованием фосфидов. Красный и черный фосфор этой способностью не обладают.
С водородом фосфор в обычных условиях не реагирует. Его гидриды (фосфины) получают косвенным путем.
Пары фосфора реагируют с азотом в электрическом разряде или на накаленной вольфрамовой нити с образованием смеси аморфных нитридов P3N5 и PN. Обычно нитрид P3N5 (оранжево-коричневого цвета с плотностью 2,6 г/см3) получают взаимодействием P4S10 с NH3 при нагревании. При его термическом разложении при ~800° С может быть получен полимерный низший нитрид PNX переменного состава (х = 0,7 до 1,19). Нитриды фосфора инертны, холодная вода на них не действует, они не разлагаются при нагревании в растворах НС1, разбавленной HNO3, растворах щелочей, не реагируют с хлором, на воздухе начинают окисляться выше 250° С. Нитрид P3N5 применяют в качестве газопоглотителя в лампах накаливания и в галогенных лампах, для легирования кремния в технологии полупроводниковых материалов.
Белый фосфор в процессе сплавления с серой при температурах до -100° С образует систему эвтектического типа с ограниченными твердыми растворами, а выше 230° С компоненты реагируют с образованием серии сульфидов — P4S3 (рис. 11.2), P4S10 (P2S5), P4S5, P4S7 и моносульфид PS. Получены также Р4Без и Р2Тез. Пары фосфора реагируют лишь выше 2000° С, а с кремнием выше 1000° С образуют силицид фосфора Si2P. Фосфор с бором, а также со всеми металлами (кроме Sb и Bi) образует фосфиды.
Фосфор большей частью образует ковалентные связи как с одноименными атомами, так и с атомами других элементов. Редкое исключение— частично-ионные связи в некоторых фосфидах. Обычно наблюдаются простые 8-связи.
Применение. Около 90% всего вырабатываемого фосфора расходуют на получение термической фосфорной кислоты, применяемой в производстве различных фосфорных удобрений и фосфорных солей (фосфатов). Белый фосфор применяют в качестве компонента дымообразующих и зажигательных средств, для изготовления трассирующих боеприпасов.
Красный фосфор применяют в спичечной промышленности в качестве основного компонента обмазки зажигательной поверхности спичечных коробок, компонента термопластичных композиций и в качестве газопоглотителя в производстве ламп накаливания.
Рис. 11.2. Структура молекулы P4S3
570
Фосфор применяют в металлургии в качестве раскислителя в процессе получения некоторых сплавов, например хромаля как легирующую добавку (фосфористый чугун содержит до 0,8% Р, некоторые стали до 0,3% Р, фосфористая бронза до 1,2% Р), как компонент припоев и антифрикционных сплавов, магнитомягких сплавов, для фосфатирования поверхности стальных изделий с целью увеличения их коррозионной стойкости, для получения ферромагнитных пленок в элементах памяти вычислительных машин. Фосфор высокой чистоты применяют в процессе получения полупроводниковых фосфидов, например типа Ап‘ Bv.
11.2. ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ПОЛУЧЕНИЯ ЭЛЕМЕНТНОГО ФОСФОРА
Элементный фосфор получают восстановлением его из природных фосфатов углеродом. С целью связывания оксида кальция и смещения равновесия реакции в сторону образования элементного фосфора в исходную шихту вводят необходимое количество диоксида кремния.
Реакцию восстановления фосфора проводят по следующему уравнению:
2Са3(РО4)2 + ЮС + 4SiO2 = Р4 + ЮСО + 2Ca3Si2O7
Процесс проводят в электрических рудно-термических печах, являющихся одновременно реактором, в котором происходят процессы плавления исходного сырья и химического взаимодействия. Степень восстановления трикальцийфосфата повышается с увеличением отношения SiO2:CaO в исходной шихте. С целью облегчения выгрузки шлака из печи процесс проводят при 1400—1600° С.
Механизм восстановления фосфата кальция многостадийный, с одновременным протеканием нескольких химических реакций. Современные представления механизма сводятся к тому, что фосфат кальция поступает в расплав фосфатных минералов. В расплаве Са3(РО4)2 частично диссоциирует на ионы Са2+, РО3’, Р5+ и О2'. Допускается также возможный распад части фосфата кальция с образованием РО и РО2:
2Са3(РО4)2 = бСаО + 4РО + ЗО2
2Са3(РО4)2 = бСаО + 4РО2 + О2
Отсюда следует, что в расплаве появляются подвижные фосфорсодержащие частицы (молекулы, ионы), которые диффундируют к 571
поверхности углерода. В результате интенсивного газовыделения и значительных конвективных потоков в реакционной зоне углерод реагирует с расплавом по схеме
Са3(РО4)2 + 5С = ЗСаО + Р2 + 5СО 4РО’~ + ЮС + 6SiO2 = Р4 + ЮСО + 6SiO’’ 2Са3(РО4)2 = бСаО + 4РО + ЗО2 4РО + 4С = Р4 + 4СО
2С + О2 = 2СО
Роль диоксида кремния в процессе восстановления фосфатов заключается в связывании оксида кальция, что ускоряет диссоциацию исходного фосфора и позволяет отстранить из зоны реакции тугоплавкий оксид кальция (Zra = 2580° С) в виде относительно легкоплавких силикатов кальция, которые легко выводятся из печи (системы) в виде жидкого шлака.
Процесс восстановления фосфатов состоит из следующих стадий: 1) нагрев исходной шихты и удаление из них влаги; 2) смешение фосфата кальция и оксида кремния с расплавом; 3) разложение исходного трикальцийфосфата на более простые составляющие (молекулы и ионы); 4) диффузия составляющих к поверхности углерода; 5) диффузия силикатных частиц к поверхности углерода; 6) взаимодействие с углеродом с образованием Р4, СО и СаО; 7) удаление из зоны реакции СаО в виде силикатов кальция.
Скорость реакции между фосфатом кальция и углеродом определяется в основном температурой процесса, модулем кислотности Мк (массовым отношением SiO2:CaO), диффузией реагентов, а также качеством исходных видов сырья. Из приведенной диаграммы состояния системы SiOi—СаО (рис. 11.3) видно, что в области значений Мк от 0 до 2 существуют ортосиликат кальция 2CaO-SiO2 (fra = 2127° С), диортосиликат кальция (полуторный силикат кальция) 2SiO2-3CaO (f™, =
t, °с м*
4,0 1,5 0,66 0,25 О
2600
2200
1800
1400
в s'
А
« Я1
2СаО - S1O2+ !
+ЗСаО * SiO2 1
ж1+ж2'
SiOg
Рис
/сао+ж
-Q со
СО СО О со
2 2 +
3 3 С\1 со
20 40 60 80 100
СаО, % СаО
11.3. Диаграмма состояния системы СаО—SiO2
572
= 1475° С) и метасиликат кальция SiO2-CaO = 1547° С). Мк метасиликата кальция, содержащего 51,7% SiO2 и 48,3% СаО, равен 1,07. Минимальная же температура плавления в рассматриваемой системе соответствует Мк = 0,82 и равна 1447° С. На печных агрегатах большой мощности работают на шихте с модулем кислотности 0,7—0,9. При работе с повышенными пределами модуля кислотности увеличиваются потери фосфора, а получаемый продукт имеет низкие качественные показатели.
В процессе получения элементного фосфора основные реакции сопровождаются побочными реакциями. Так, в результате восстановления содержащихся в исходном сырье соединений железа образуются фосфиды железа:
Fe2O3 + 4С = 2Fe + ЗСО
2Fe + Р2 = 2FeP
4Fe + Р2 = 2Fe2P
Образующиеся фосфиды железа, называемые феррофосфором, содержат до 15—28% элементного фосфора. Их выгружают из печи в виде расплава.
Диоксид кремния реагирует с углеродом, образуя элементный кремний:
SiO2 + 2С = Si + 2СО
SiO2 + С = SiO + СО
SiC + 4Fe = FeSi + Fe3C
Диоксид кремния, содержащийся в шлаке, реагирует с содержащимся в природных фосфатах фторидом кальция:
3SiO2 + 2CaF2 = 2CaSiO3 + SiF4
11.3. ПОЛУЧЕНИЕ ЭЛЕМЕНТНОГО ФОСФОРА
Для получения фосфора применяют фосфориты, содержащие 21—23% Р2О5, до 10% СО2, соединения серы, щелочные металлы, глинистые сланцы и др. В качестве флюса используют кварциты, состоящие из 80—92% SiO2, 2—4% Fe2O3, до 2% P20s и другие кремнистые породы. Восстановителем служит доменный кокс, содержащий 80—85% углерода с зольностью около 12% и содержанием серосодержащих веществ до 0,7% (в пересчете на серу).
573
С целью получения качественной шихты исходное фосфатное сырье подвергают термообработке. Руду с размерами кусков 10—70 мм обжигают в шахтных печах. При этом происходит процесс декарбонизации. Образующиеся при этом мелкие частицы (менее 10 мм) дополнительно размалывают, перемешивают с глиной, окомковывают, полученные гранулы подвергают термообработке на конвейерных обжиговых машинах. Температуру спекания поддерживают в пределах 1200—1300° С за счет горения содержащегося в шихте кокса. Спек дробят, после чего фракционируют грохочением. Агломерат с размерами частиц 8—50 мм направляют на переработку. Флюсы и кокс предварительно высушивают. При расчете состава шихты соотношение исходных фосфорита (Ф) и кварцита (К) определяют по выбранному модулю кислотности и содержанию в них оксидов кальция, магния, кремния и алюминия:
К 4[CaO+MgO]t-[SiO; + А12О3]ф Ф- [SiOj + AljOJ^-A/JCaO+MgO],'
В расчетах принимают, что для связывания оксида магния служит AI2O3, который, как и SiC>2, является флюсом.
Расчет дозировки кокса ведут с учетом реакций восстановления фосфата, диоксида углерода (до СО на 80%), оксида железа на 80% и разложения воды с образованием СО и Нг (на 90%), а избыток углерода принимают равным 10%.
Процесс восстановления фосфата кальция до элементного фосфора проводят в трехфазных электрических печах (рис. 11.4), представляющих собой рудно-термические печи с круглой ванной. Печь оснащена самоспекающимися электродами 1 диаметром 1,4—1,7 м, расположенными по вершинам равностороннего треугольника. Кожух ванны 9 сваривают из углеродистой листовой стали толщиной 20—25 мм. Наружную поверхность кожуха охлаждают водой. Нижнюю зону ванны печи футеруют шамотным кирпичом. Арочный свод 14 изготовляют из жаропрочного армированного бетона. Центральная часть свода снабжена встроенными змеевиками для охлаждающей воды. Для герметизации ванны печи на свод 14 устанавливают трехсекционную крышку 7, изготовленную из немагнитной стали. Через крышку и свод проходят электроды, имеющие сухое сальниковое уплотнение 5. Электродержатели 4, расположенные над крышкой печи, перемещаются по вертикали с помощью гидроподъемников 3 с реечными синхронизаторами. Перепуск электродов дистанционный, пружиногидравлический. Самоспека-ющийся электрод представляет собой цилиндрическую трубу из стали толщиной 3 мм, расположенную вертикально и заполняемую сверху сырой электродной массой — смесью термоантрацита, пекового кокса, 574
Рис. И.4. Электропечь для получения фосфора:
1 — электрод; 2 — механизм для передвижения электрода; 3 — гидравлический подъемник; 4 — электродержатель; 5 — уплотняющие сальники; 6 — система водяного охлаждения; 7—крышка ванны; 8 — футеровка внутренней части ванны; 9 — конус ванны; 10 — летка для феррофосфора; 11 — поднос ванны; 12 — летка для шлака; 13 — загрузочный патрубок; 14 — свод ванны;
15— гибкий токопровод; 16 — короткая сеть; 17 — трансформатор для печи
графитовых добавок, каменноугольной смолы и каменноугольного пека. Углеродистый материал нижней части электрода участвует в процессе в роли восстановителя. По мере расходования электрода его опускают со скоростью 2—3 мм/ч. Сверху к электроду по мере его опускания приваривают новые звенья-обечайки и заполняют их электродной массой.
Печь оснащается тремя трансформаторами с независимыми источниками питания энергией. Энерготехнологический режим поддерживается автоматической системой управления.
575
Согласно схеме получения элементного фосфора, исходная шихта поступает в электропечь из бункеров. При поступлении в печь шихта распределяется равномерно по площади сечения печи. В бункеры шихта поступает транспортерами. Для исключения подсоса воздуха в печи и во всей системе печного отделения поддерживают давление в пределах 0,3—0,6 КПа. Бункеры должны быть постоянно заполнены шихтой и закрыты крышками, а в нижнюю их часть непрерывно подается азот, который служит буфером, не допускающим проникновения печного газа в окружающий воздух производства.
Феррофосфор, накапливающийся под шлаком, периодически сливается из печи через летку 12 в ковш, стоящий на железнодорожной колее, и отводится на разлив в изложницы, из которых извлекается после застывания в слитки.
Через две другие летки, расположенные выше уровня летки, непрерывно выпускают шлак, который сливают по желобу в разборные чугунные кокили, установленные на железнодорожной платформе.
Выходящий из электропечи газ, проходя газоотсекатель, поступает в электрофильтры, где он очищается от печной пыли. С целью исключения процесса конденсации фосфора температуру в электрофильтрах поддерживают в пределах 280—300° С. Оседающая в газоходе пыль сбрасывается шнеком в камеру, где собирается пыль, уловленная в электрофильтрах. Из камеры пыль удаляется шнеком.
Из электрофильтров газ с температурой 250—300° С поступает в конденсаторы фосфора, представляющие собой орошаемые циркулирующей водой вертикальные стальные башни. Конденсационная башня состоит из двух ступеней — «горячей» и «холодной». Газ в первом по ходу «горячем» конденсаторе движется снизу вверх по спиральной траектории и охлаждается в процессе испарения разбрызгиваемой форсунками водой, а также за счет оборотной воды, орошающей наружную поверхность конденсатора. В горячем конденсаторе конденсируется 99% фосфора, содержащегося в печном газе. Охлаждение газа и слив фосфора производятся водой, циркулирующей по замкнутому контуру: сборник фосфора — насосы — трубопроводы — форсунки — конденсатор — сборник фосфора. Оборотная вода, орошающая наружную поверхность конденсатора, направляется на охлаждение в градирни.
С целью повышения степени извлечения фосфора газы из печи после горячего конденсатора направляют в холодный, в котором циркулирует вода, охлаждаемая в паро-эжекционной установке. При этом температура газа понижается с 27 до 17° С, суммарная степень извлечения фосфора достигает 99,95%.
Циркулирующая в конденсаторах вода постепенно подкисляется вследствие гидратации содержащихся в составе газа кислотных окси-576
дов и гидролиза тетрафторида кремния с образованием H2SiF6. С целью уменьшения коррозии аппаратов и трубопроводов воду нейтрализуют карбонатом натрия или аммиаком и периодически отводят ее на станцию нейтрализации с заменой на новую.
Из конденсаторов газ удаляется газодувкой, которая поддерживает заданное избыточное давление в системе печь — газоход с помощью байпасной линии и автоматического регулятора давления. Освобожденный от фосфора газ применяется в качестве топлива в процессе термической подготовки исходного сырья или же его сжигают на «свече».
Жидкий фосфор-сырец отстаивают при 60—70° С в стальных резервуарах с метальными устройствами и рубашками. Плотность образующегося шлама ниже плотности жидкого фосфора и он всплывает. Шлам содержит твердые частицы, воду и фосфор. Над шламом находится слой воды, предохраняющий фосфор от контакта с воздухом. Фосфор погружными насосами перекачивают на склад, а шлам направляют на переработку путем дистилляции с получением чистого фосфора.
Существующий ГОСТ 8986—82 регламентирует содержание фосфора в целевом продукте марок А, В и С соответственно не менее 99,9; 99,5 и 94,5%.
На 1 т получаемого целевого продукта расходуют: 13,2 т фосфатного сырья (21,5% Р2О5), 0,8 т кварцита (95% SiO2), 2,6 т кокса (84% С), 70 кг электродной массы, 540 м3 оборотной воды, 4,44 гДж пара, 216 м3 природного газа, 150 м3 сжатого воздуха (н.у.), 500 м3 инертного газа — азота (н.у.), 14 000 кВт-ч электроэнергии (печной), 16 500 кВт-ч электроэнергии (технологической), 9,6 т шлака, 0,11 т феррофосфора, 2700 м3 печного газа (н.у.), 0,2 т пыли и 0,15 т шлама с 30% влаги.
11.4. ОКСИДЫ ФОСФОРА
В настоящее время в качестве индивидуальных веществ установлены следующие оксиды фосфора: Р4О, Р4О2 (Р2О), Р4О6 (Р20з), Р4О8, Р4Ою (Р2О5) и РО3. Теоретически показана вероятность существования (в процессе окисления паров фосфора) метастабильных оксидов фосфора ряда Р4О„, где п = 6—9, и продуктов их диссоциации — РО и РО2. Молекула РО обнаруживается лишь спектроскопически в парах. Наиболее изучены Р40б и Р4Ою (табл. 11.2).
Монооксид тетрафосфора Р4О— красно-бурые кристаллы плотностью 1,891 г/см3. В обычных условиях с достижением 270—280° С воспламеняется, а в сухом воздухе устойчив до 350° С. В процессе нагревания в инертном газе разлагается с выделением элементного фосфора и декаоксида тетрафосфора (Р4Ою). С водой не реагирует, но во влажном состоянии разлагается на воздухе выше 100° С.
577
19 Химическая технология неорганических веществ, кн. 1
Монооксид тетрафосфора получают в процессе сжигания фосфора в недостатке кислорода или в процессе взаимодействия РОС1з с металлическими алюминием, цинком при 100° С или белым фосфором при 250° С. Получают также в процессе длительной выдержки на воздухе раствора фосфора в трихлориде фосфора и диэтиловом эфире.
Таблица 11.2. Некоторые свойства оксидов фосфора
Показатель Р4О6 Р4О8 P4Q10
н О О'
Т °C 1 пл» 23,9 — 420,5 562,5 580,5
Т °C 1 КИП» 174 180** 340,5** 605,5 605,5
Плотность, г/см3 2,135 2,537 2,28 2,72 2,84
С°р, Дж/(моль К) 148,6* 169,9* 215,6 — —
кДж/моль -1606* -2302* -3010,1 — —
S^, Дж/(моль-К) 356,3* 394,7* 231 — —
АЯПЛ кДж/моль 14,1 — 27,2 74,1 63,1
АЯИСП, кДж/моль 48,0 — 67,8 78,3 78,3
*Для газа, для твердого
Р4О6 Д/7° Обр = -1668 кДж/моль. "Температура возгонки.
Диоксид тетрафосфора Р4О2 (Р2О) — желто-красные кристаллы. При нормальном давлении устойчив до 100° С, в вакууме — до 135° С, а при более высоких температурах разлагается с выделением элементного фосфора. Устойчив в сухом воздухе. С водой не реагирует, а в запаянной трубке в присутствии воздуха водный раствор Р4О2 окисляется до Н3РО3. В этаноле, ацетоне не растворяется.
Диоксид тетрафосфора получают пропусканием кислорода или воздуха через раствор белого фосфора в тетрахлориде углерода при 50—60° С. Получают также нагреванием POCI3 и Н4РВГ2 до 773° С в запаянной трубке. Является сильным восстановителем Fe3+, Cu2+ и Ag+. Это свойство РОг применяют для восстановления примесей железа в экстракционной фосфорной кислоте.
Гексаоксид тетрафосфора Р4О6 — белые хлопья или кристаллы с неприятным запахом. Структура молекулы твердого Р4О6 приведена на рис. 11.5. Р4О6 более устойчив, чем Р4О и Р4О2. Выше 200° С гексаоксид тетрафосфора разлагается с частичным отщеплением элементного (красного) фосфора и различных оксидов фосфора (Р4О8, Р4О10). Кристаллизуется вблизи точки затвердевания. Хорошо растворяется в органических растворителях (бензоле, сероуглероде), что используют для очистки его перекристаллизацией. В 100 г гексаоксида тетрафосфора растворяется до 1,7 г элементного фосфора, который может быть извлечен путем перевода в красный фосфор в процессе ультрафиолетового облучения с последующим растворением оксида в сероуглероде. 578
В обычных условиях Р40б медленно окисляется до декаоксида тетрафосфора. В атмосфере сухого разряженного кислорода он окисляется при 40° С, во влажном кислороде — при 130° С. Сухой гексаоксид тетрафосфора устойчив в сухом кислороде до 77—127° С, а в воздухе — до 450° С. При сильном нагревании в отсутствие воздуха разлагается до элементного фосфора и Р2О4. В процессе растворения в холодной воде гексаоксид тетрафосфора образует Н3РО3, а в горячей— Н3РО4, РНз и Р. С хлоридом водорода Р4О6 реагирует с образованием Н3РО3 и РС13. Энергично реагирует с хлором, бромом, иодом и серой выше 150° С.
Гексаоксид тетрафосфора получают в процессе сжигания элементного фосфора с ограниченным доступом сухого воздуха.
Октаоксид тетрафосфора Р4О8— белые хлопья или кристаллы. Молекула октаоксида тетрафосфора в газообразном состоянии соответствует формуле PgOie- В кристалле она имеет сложную структуру и включает агрегаты от мономера до тримера — Р4О8, P8Oi6, Pi2O24-
Известны две кристаллические формы. Ромбическая a-форма содержит молекулы Р4О8 и Р4О9 в различных соотношениях и имеет средний состав в пределах Р4О8]—Р4О90. Моноклинная |3-форма состоит из молекул Р4О7 и Р4О8; ее состав Р4О7 7—Р408>о. В литературе имеются данные о существовании аморфной формы Р4О8.
Октаоксид тетрафосфора устойчив в процессе нагревания до 100° С, возгоняется при 180° С. Окисляется до Р4Ою в среде чистого кислорода выше 400° С. Кристаллический Р4О8 расплывается на воздухе, хорошо растворяется в воде, но не растворяется в органических растворителях. В процессе растворения в воде образует равные количества молей Н3РО3 и НРО3. Водные же растворы трудно окисляются ДО Н3РО4.
19*
Рис И.5. Строение молекулы Р40б:
• — атом кислорода; о — атом фосфора; плоскость симметрии проходит через атомы Р(1), р(1П) q(2) 0(3)
579
Октаоксид тетрафосфора получают в процессе низкотемпературного сжигания элементного фосфора в ограниченном количестве воздуха. Получают также в процессе нагревания гексаоксида тетрафосфора (Р4О6) в течение 48 ч в запаянной трубке при температурном интервале 200—250° С.
Триоксид фосфора РО3 (по некоторым данным Р2О6 или Р4О11) — твердое вещество фиолетового цвета из-за присутствия примесей. Триоксид стабилен в течение 120—150 ч при 17—37° С в сухой атмосфере. Разлагается с выделением кислорода. В процессе гидратации РОз или его димера образуются пероксофосфорная Н3РО5 или пероксопирофосфорная Н4Р2О7 кислоты. С оксидами металлов образует пероксофосфаты.
Триоксид фосфора получают при действии на пары декаоксида тетрафосфора РдОю электрического разряда в вакууме (133,3 Па).
Декаоксид тетрафосфора РдОю — бесцветное аморфное, стекловидное (G-форма) или кристаллическое вещество. Существуют две метастабильные кристаллические модификации: гексагональная Н-форма {а = 0,744 нм, а = 87°, пространственная группа R3C) и орторомбическая О-форма (а = 0,923 нм, b = 0,718 нм, с = 0,494 нм, пространственная группа Рпат)\ одна стабильная орторомбическая О'-форма (а = 1,63 нм, b = 0,814 нм, с = 0,526 нм, пространственная группа FdcQ-, по некоторым данным, тетрагональная Т-форма). Структура молекулы Р4О10 Н-формы построена из четырех групп РО4 в виде тетраэдра, вершины которого занимают атомы фосфора; шесть атомов кислорода располагаются вдоль ребер, а четыре — по оси третьего порядка тетраэдра (рис. 11.6). Другие модификации имеют слоистую полимерную структуру, также построенную из тетраэдров РОд, которые объединены в кольца—10-членные в случае О-фор-мы и 6-членные в случае O'-формы (рис. 11.7). Н-форма переходит в О-форму при 300—360° С (процесс заканчивается при 378° С), а также при нагревании (127° С) в течение 2 ч в запаянной ампуле. В процессе нагревания О-формы в запаянной трубке (при 450° С) в течение 24 ч образуется более устойчивая O'-форма. При плавлении Н-формы получается, по-видимому, мономолекулярная жидкость, при плавлении О- и О'-модификаций — полимерная форма жидкого Р4О10. Тройные точки: для Н-формы 420,5° С, давление 478,8 кПа; для О-формы 562,5° С, 58,0 кПа; для О'-формы 580,5° С, 73,82 кПа.
Р4О10 весьма гигроскопичен (Н-форма поглощает воду со взрывом), что причисляет его к наиболее эффективным осушителям. Р4О10—сильный дегидратирующий агент, например: 4HCIO4 + Р4Ою->(НРОз)4 + 2CI2O7. Переводит также H2SO4 в SO3, HNO3 в N2O4, амиды — в нитриды и т.д. 580
РдОю реагирует как со спиртами, эфирами, так и с фенолами, кислотами и др. При этом происходит разрыв связей Р—О—Р и образуются фосфорорганические соединения. Он реагирует с аммиаком и с галогеноводородами, образуя соответствующие конденсированные фосфаты аммония и оксигалогениды фосфора. С основными оксидами образует различные фосфаты.
Декаоксид тетрафосфора является осушителем газов и жидкостей, полуфабрикатом в производстве ортофосфорной кислоты термическим способом. Он применяется в органическом синтезе (в реакциях дегидратации и конденсации), в
производстве поверхностно-активных веществ, фосфатных стекол и
других неорганических материалов.
Получение декаоксида тетрафосфора. Процесс получения декаоксида тетрафосфора состоит из следующих стадий (рис. 11.8): 1) окисление элементного фосфора предварительно осушенным воздухом; 2) осаждение РдОю; 3) очистка отходящих газов.
Окисляют исходный фосфор в камере сжигания 2. Для диспергирования исходного элементного фосфора в форсунку подается воздух под давлением 500—600 кПа. Основной объем воздуха поступает в камеру сжигания через завихритель спирального типа. Сжигание фосфора — экзотермический процесс:
+ 501 -> РдОю + 24534,6 кДж.
Около 20% теплоты реакции отводится охлаждающей водой, а остальное количество выделяется в газовую фазу. Из камеры сжигания образующийся декаоксид тетрафосфора в смеси с газами через водоохлаждаемый газоход 3 направляется в осадительные башни 4, 6. В газоходе газы охлаждаются и происходит частичная конденсация ультра-581
В атмосферу
12
Воздух
Фосфор
10
Вода
Рис. 11.8. Принципиальная схема получения Р4Ою-1— форсунка для распыления элементного фосфора; 2 — камера сжигания фосфора; 3 — газоход; 4, 6 — осадительные башни; 5 — газоход; 7 — разгрузочные Р4Ою шнеки; 8—насадочный скруббер; 9 — сборник фосфорной кислоты; 10— центробежный насос; 11 — брызгоулови-тель; 12 — гидрозатвор
Н3РО4
фосфорной кислоты, сорбирующей находящиеся в продуктах реакции примеси (оксиды тяжелых металлов и мышьяк). Конденсирующийся ниже 364° С целевой продукт осаждается в виде белого порошка на стенках в нижней части башен. В первой башне, в которой образуется более крупнокристаллический продукт, конденсируется свыше 75% от общего содержания РдОщ, а во второй — около 23%. После осадительных башен газы проходят в насадочный скруббер 8, который орошается фосфорной кислотой. Орошающая кислота циркулирует в системе насадочный скруббер 9 — брызгоуловитель 11 — сборник кислоты— центробежный насос 10. Система оснащена гидрозатвором 12. Незначительная часть (около 2—3%) РдОю в системе превращается в фосфорную кислоту и по мере необходимости кислоту центробежным насосом выводят из системы (перекачивают на склад).
11.5. КИСЛОРОДСОДЕРЖАЩИЕ КИСЛОТЫ ФОСФОРА
Кислородные кислоты фосфора в степенях окисления от +1 до +5 построены из структурных единиц XYP(O)OH, где X и Y — О, Р, Н.
Сокращенная запись формул по Блазеру и Вормсу (Р'-кислота, Р3-кислота и т.д., в индексе над атомом фосфора указывается его степень окисления) позволяет различать изомеры кислот одинакового состава, например изомеры кислоты Н4Р2О5 — кислоты Р3—О—Р3 и Р2—Р4. Примеры кислородных кислот фосфора: Р'-кислоты — фосфор-новатистая (гипофосфористая) Н3РО2 (формула I), фосфиновые кислоты RR'P(O)OH (II), фосфонистые кислоты RH2PO2 (III); Р2—Р2 — кислота — гиподифосфорная Н4Р2О4 (IV); Р3-кислоты — фосфористая Н3РО3 (V), фосфоновые кислоты RP(O)(OH)2 (VI); Р4—Р4 кислота— фосфорноватая (гипофосфорная) Н4Р2О6 (VII); Р-кислота — фосфорная кислота (ортофосфорная) Н3РО4 (VIII).
582
о
о
о
о о
о
н—р-он
R—Р-ОН
Н—Р—он
R
II
ОН III
ОН V
он он
IV
О 0 0 О О 0 0
R—р—он НО—Р—Р—ОН НО—Р—ОН НО—Р—О—ОН НО—Р—0—0—Р—ОН
он но он он он он он
VI VII VIII IX X
н
В кристаллическом состоянии выделены кислоты: одноосновные 3 3 4 4
Н3РО2, (НРО3)„; двухосновные Н4Р2О5 (Р—О—Р) Н4Р2О6 (Р—Р); тре-2 4 3 5
хосновные Н3РО4, Н4Р2О5 (Р—Р), Н4Р2О6 (Р—О—Р); четырехосновная
Н4Р2О7 (Р—О—Р); пятиосновная H5P30g (Р—Р—Р); шестиосновная
НбРбОп ( циклическая, или (Р—Р)зс) — кислота.
Анионной конденсацией Н3РО4 с отщеплением воды получают конденсированные кислоты, в молекулах которых тетраэдрические группы РО4, соединенные между собой через общий атом кислорода, образуют цепи (полифосфорные кислоты), кольца (метафосфорные кислоты), развлетвленные структуры (ультрафосфорные кислоты):
2НзР°^ЩРА ЗН3РО4—Н5Р3О10 4Н3РО4-^&-
иН3РО4 Нп+2РлО3л+1
Р2-кислота (пиро - или дифосфорная)
Р3-кислота (трифосфорная)
Р4- кислота (теграфосфорная)
Р„- кислота (полифосфорная, цепочечная)
ЗН3РО4-^
Рд,-кислота (трициклофосфорная)
4н3ро4 (нро4)4
__ —---ЛНоО V
лН3РО4-----2-*- (НРО3)„
Р*-кислота (тетрациклофосфорная) Р^.-кислота (метафосфорная, п^З)
583
При со Р„_и Р„с— кислоты неразличимы. Молярное отношение Н2О:Р2О5 = R в гомологическом ряду Рлс — кислот не меняется (R = 1), в ряду Р„ — кислот при росте п уменьшается в пределах 3>R<1. При R<3 кислоты представляют собой смеси, состав которых характеризуется молекулярно — массовым распределением анионов. По мере уменьшения R смеси обогащаются высшими кислотами, при этом вязкость кислот увеличивается вплоть до образования твердой массы. Вблизи значения R = 1 в смесях появляются Рис — кислоты, а также ультрафосфорные кислоты сетчатого строения (область их существования l,2>R<0,7). В качестве товарного продукта производят умеренно вязкую фосфорную кислоту (2,5>R>1,5).
Существуют конденсированные кислоты фосфора с различным сочетанием атома фосфора в различных степенях окисления. К ним могут быть отнесены изомеры три^осфорных кислот Н5Р3О9 (Р—О— Р—О—Р и Р—О—Р—Р), Н5Р3О8 (Р—О—Р—Р и Р—Р—Р). Известны также надфосфорные кислоты с терминальной или срединной пероксогруппой: пероксофосфорная (IX), пероксопирофосфорная (X).
Наиболее широко применяемой в народном хозяйстве кислотой из рассмотренных кислот является фосфорная кислота.
11.6. ФОСФОРНАЯ КИСЛОТА
Физико-химические свойства. Фосфорная кислота Н3РО4, бесцветные гигроскопические кристаллы моноклинной сингонии (а = 0,5762 нм, b = 0,4831 нм, с = 1,1569 нм, 0 = 95,31°, пространственная группа Р21/с) с плотностью 1,88 г/см3. Температура плавления 42,50° С; ДН^р = -1283 кДж/моль. Расплывается на воздухе. Наиболее стабильное соединение в ряду кислородсодержащих кислот фосфора. В расплавленном состоянии склонна к переохлаждению; при 15° С образует густую маслянистую жидкость, а при -121° С — стеклообразную массу.
Фосфорная кислота смешивается с водой в любых соотношениях. Из высококонцентрированных растворов кристаллизуется в виде ге-мигидрата (полугидрата) Н3РО4'0,5Н2О, представляющего собой бесцветные кристаллы моноклинной сингонии (а = 0,7922 нм, &= 1,2987 нм, с = 0,7470 нм, р = 109,9°, пространственная группа Р21/<я). Молекула безводной Н3РО4 и ее кристаллогидрата содержит тетраэдрическую группу РО4. В безводной фосфорной кислоте образуются водородные связи типа Р-0—Н...О = Р (рис. 11.9), которые удерживают в виде слоев, параллельных одной из плоскостей кристалла. Водородные связи сохраняются и в концентрированных (70—80%) растворах фосфорной кислоты. В разбавленных (до 40—50%) растворах отмечена более устойчивая водородная связь фосфат-анионов с молекулами воды, а не с другими фосфат-анионами. В водных рас-584
Рис. 11.9. Водородные связи в молекулярных структурах Н3РО4: ... — водородная связь
Рис. 11.10. Фазовая диаграмма системы Н3РО4 — Н2О (Ж — жидкая фаза)
।___i___1__।___1___।___1___1
О 20 40 60 72,4
Содержание Р2О5, %
творах фосфорной кислоты существует обмен атомами кислорода между группами РОд и водой.
Н3РО4 — сильная кислота, К\ = 7,1-10’3(рЛ'а = 2,12), /<г = 6^-10'8(рЯа = = 7,20), Кз = 5,О1О’13(рАа ~ 12,32); значения К\ и Кг зависят от температуры. Диссоциация по первой ступени экзотермична, по второй и третьей — эндотермична. Фазовая диаграмма системы Н3РО4—Н2О приведена на рис. 11.10, из которой видно, что максимум кривой кристаллизации находится при температуре 302,4 К и концентрации фосфорной кислоты 91,6%, а твердая фаза — гемигидрат.
В табл. 11.3 приведены свойства водных растворов фосфорной кислоты. Плотность 100%-ной фосфорной кислоты при 15° С составляет 18 829 г/см3.
Таблица 11.3. Свойства водных растворов фосфорной кислоты
Содержание,% (масс.) Температу-ра затвердевания, °C Температура кипения, °C с;, кДж/(ю-К) т], Па-с (25° С) Удельная электрическая проводимость, Ом/м (25° С) Давление пара, Па (25° С)
Н3РО4 Р2О5
5 3,62 0,8 100,10 4,0737 0,0010 10,0 3129,1
10 7,24 -2,10 100,20 3,9314 0,0011 18,5 3087,7
20 14,49 -6,00 100,80 3,6467 0,0016 18,3 2986,4
30 21,73 -11,80 101,80 3,3411 0,0023 14,3 2835,7
40 28,96 -21,90 103,90 3,0271 0,0035 11,0 2553,1
50 36,22 -41,90 104,00 2,7465 0,0051 8,0 2223,8
60 43,47 -76,90 114,90 2,4995 0,0092 7,2 1737,1
70 50,72 -43,00 127,10 2,3278 0,0154 6,3 1122,6
75 54,32 -17,55 135,00 2,2692 0,0200 5,8 805,2
585
», г/см
а б
Рис. 11.11. Кривые плотности (а) и вязкости (б) водных растворов Н3РО4 (при 20—180° С)
9
2* 1—
0 10 20 30 40 50 60 70 80 90 100
Температура, °C a
Рис. 11.12. Константа диссоциации фосфорной кислоты первой (а — Н3РО4 # 1Г + Н2РО;)
и второй ступени (б — Н2РО“ Н+ + НРО*-)
На рис. 11.11, а приведены кривые плотности Н3РО4 при 15 и 80° С, а на рис. 11.11, б — данные по изменению вязкости растворов Н3РО4 при 20—180° С.
В химических реакциях фосфорная кислота диссоциирует в две ступени (рис. 11.12):
Н3РО4 Н+ + Н2РО42-, уравнение константы К\ диссоциации:
7QQ 0 1
Ig/G = 4,5535--^F-l,3486-Ю-2-Т;
Н2РО4 Н+ + НРО42-, уравнение константы диссоциации К?.
lgtf2 = 5,9884-^-2,0912-10-2-Т.
Фосфорная кислота при нормальных условиях малоактивна и реагирует только с карбонатами, гидроксидами и некоторыми металлами. При этом образуются одно-, двух- и трехзамещенные фосфаты. В процессе нагревания выше 80° С реагирует даже с неактивными оксидами, кремнеземом и силикатами. При повышенных температурах фосфорная кислота является слабым окислителем для металлов. В процессе действия на поверхность металлов раствором фосфорной кислоты, содержащей цинк или марганец, образуется защитная пленка. В процессе нагревания фосфорная кислота теряет воду с образованием пиро- и метафосфорных кислот по схемам:
2НзРО4 -> Н2О + Н4Р2О7 + 71,17 кДж,
Н4Р2О7 -> Н2О + 2НРОз + 100,46 кДж/моль.
Фосфолеум (жидкий фосфорный ангидрид, суперфосфорная кислота) включает кислоты, содержащие от 72,4 до 88,6% Р2О5, и представляет собой равновесную систему, состоящую из орто-, пиро-, Триполи-, тетраполи- и других кислородсодержащих фосфорных кислот. В процессе разбавления суперфосфорной кислоты водой выделяется значительное количество теплоты, и они быстро переходят в ор-тофосфорную кислоту.
Применевие фосфорной кислоты. Фосфорная кислота применяется в основном (около 85% производимого) для получения фосфорных и сложных удобрений, кормовых фосфатов, синтетических моющих и водоумягчающих средств. В металлообрабатывающей промышленности фосфорная кислота применяется в процессе фосфатирования поверхности металлов, а в текстильной — для обработки и крашения шерсти, натуральных и синтетических волокон. В технологии органических веществ фосфорную кислоту применяют в качестве катализатора. Фосфорную кислоту и ее производные применяют в
587
процессе приготовления буровых суспензий при нефтедобыче, в производстве стекла, в фотографии (для приготовления светочувствительных эмульсий), в медицине (приготовление медикаментов, получение лекарственных средств, зубных цементов), в процессе обработки древесины (для придания огнестойкости). Производные фосфорной киодоты применяют также в пищевой промышленности (хлебопекарные порошки, приготовление плавленых сыров, в колбасном производстве и сахароварении).
Способы получения фосфорной кислоты. Фосфорную кислоту в промышленности получают двумя способами: термическим и экстракционным.
Термический способ получения фосфорной кислоты основывается на окислении элементного фосфора в избытке воздуха с последующей гидратацией и абсорбцией образующегося РдОю, конденсацией фосфорной кислоты и улавливанием тумана из газовой фазы.
РдОю получают окислением элементного фосфора в виде капель или пленки. Степень окисления фосфора в промышленных условиях определяется температурой в зоне окисления, диффузией компонентов и другими факторами. Вторую стадию получения термической фосфорной кислоты — гидратацию декаоксида тетрафосфора — осуществляют абсорбцией кислотой (водой) либо взаимодействием паров Р4О10 с парами воды. Гидратация Р4О10 идет последовательно до мета-, ди- и ортофосфорной кислоты:
иР4Ою + 2иН2О = 4(НРО3)„ (при 700° С),
4(НРОз)„ + 2иН2О = 2иН4Р2О7 (при 450° С),
2пН4Р2О7 + 2иН2О = 4иН3РО4 (ниже 230° С).
Суммарное уравнение процесса гидратации:
Р4Ою + 6Н2О = 4НзРО4
Количество теплоты, выделяющейся в процессе сжигания 1 кг элементного фосфора, равно 23 614 кДж, а в процессе гидратации— 3035 кДж. В процессе разбавления фосфорной кислоты водой до концентрации продукционной (товарной) кислоты выделяется дополнительное количество теплоты.
Термическую фосфорную кислоту можно получать двумя способами: одноступенчатым и двухступенчатым. Первый способ основан на сжигании отходящего из печи фосфорсодержащего газа (без предварительной конденсации паров фосфора) с гидратацией образующе-588
гося РдОщ. Способ не применяют из-за низкой концентрации паров фосфора в печном газе, что требует громоздкого оборудования для его переработки, а содержащиеся в газе примеси загрязняют получаемую фосфорную кислоту.
Термическую фосфорную кислоту производят двухстадийным способом. При этом конденсируют из газа фосфорной печи фосфор, а затем перерабатывают его в фосфорную кислоту.
В процессах окисления фосфора и гидратации Р4Ою выделяется значительное количество теплоты. В зависимости от принципа охлаждения газов существуют три способа производства термической фосфорной кислоты: испарительный, циркуляционно-испарительный, теплообменно-испарительный. Испарительные системы, основанные на отводе теплоты при испарении воды или разбавленной фосфорной кислоты, наиболее просты в аппаратурном оформлении. Однако из-за относительно большого объема отходящих газов считается, что использование таких систем целесообразно лишь в установках небольшой единичной мощности.
Циркуляционно-испарительные системы позволяют совместить в одном аппарате стадии сжигания элементного фосфора, охлаждения газовой фазы циркулирующей кислотой и гидратации Р4Ою. Рассматриваемые системы совмещают два способа отвода теплоты: через стенку башен сжигания и охлаждения, а также путем испарения воды из газовой фазы. Существенным преимуществом системы является отсутствие контуров циркуляции кислоты с насосно-холодильным оборудованием.
В настоящее время на заводах эксплуатируют технологические схемы с циркуляционно-испарительным способом охлаждения (двухбашенная система), отличающиеся наличием дополнительной башни для охлаждения газа.
Физико-химические основы экстракционного способа получения фосфорной кислоты. Экстракционный метод получения фосфорной кислоты основан на разложении природных фосфатов серной кислотой по реакции
Ca5(PO4)3F + 5H2SO4 + 5иН2О = 5CaSO4nH2O + ЗН3РО4 + HF
Одновременно происходит разложение и других примесей в исходном фосфатном сырье. К ним относятся: кальцит, доломит, сидерит, глауконит, силикаты железа и алюминия. Они также разлагаются серной кислотой по схеме
CaCO3 MgCO3 + 2H2SO4 + (и - 2)Н2О = CaSO4 nH2O + MgSO4 + 2СО2
K2ONa2OAl2O3-2SiO2 + 5H2SO4 =
= Na2SO4 + K2SO4 + A12(SO4)3 + 2SiO2 + 5H2O
589
Диоксид кремния реагирует с HF, образуя SiF4:
SiO2 + 4HF = SiF4 + 2Н2О
Часть тетрафторида кремния выделяется в газовую фазу, а другая часть образует гексафторкремниевую кислоту, остающуюся в растворе:
SiF4 + 2HF = H2SiF6
Разложение примесей приводит к увеличению расхода серной кислоты, а также снижает степень извлечения P2Os в целевой продукт вследствие образования нерастворимых фосфатов железа FeHj(PO4)2-2,5H2O при концентрациях P2Os выше 40% и FePO4-2H2O—при более низких концентрациях. Выделяющийся в процессе разложения карбонатов диоксид углерода образует в экстракторах стойкую пену. Растворимые фосфаты магния, железа и алюминия снижают активность исходной фосфорной кислоты, а также уменьшают содержание усвояемых форм Р2О5 в удобрениях при последующей переработке фосфорной кислоты.
С учетом влияния примесей фосфаты с повышенным содержанием соединений железа, алюминия, магния и карбонатов непригодны для производства фосфорной кислоты. Поэтому в процессах сернокислотного разложения обычно применяют руды или концентраты, характеризующиеся массовым отношением Ре2Оз:Р2О5 не более 0,08.
Основой для выбора технологических параметров процессов сернокислотного разложения является выделение сульфата кальция в виде достаточно крупных, легко отделяемых и хорошо отмываемых от фосфорной кислоты кристаллов. В системе CaSO4—Н3РО4—Н2О сульфат кальция может существовать в трех формах: одной безводной (ангидрита CaSO4) и двух кристаллогидратов (гемигидрата CaSO4-0,5H2O и дигидрата или гипса CaSO4-2H2O). Температурные и концентрационные области кристаллизации приведенных форм определяются соотношениями их растворимостей в фосфорной кислоте или соотношениями давлений паров над раствором и давлений диссоциации обратимых реакций превращения гипса в гемигидрат или ангидрит и гемигидрата в ангидрит. Из изотерм растворимости сульфата кальция при 80° С (рис. 11.13) вид-
590
Рис 11.13. Изотермы растворимости сульфата кальция в фосфорной кислоте при 80° С:
А — ангидрит; П — полугидрат; Г — гипс
но, что с увеличением содержания фосфорной кислоты растворимость всех трех модификаций сначала возрастает, достигает максимума при 16—22% Р2О5, а затем уменьшается. Минимальную растворимость при 80° С имеет ангидрит, являющийся равновесной твердой фазой. Мета-стабильный гипс в растворах, содержащих 33,3% Р2О5 (точка А пересечения изотермы метастабильных кристаллогидратов), превращается непосредственно в ангидрит. В более концентрированных растворах сначала происходит конверсия гипса в менее растворимый гемигидрат, после чего последний дегидратируется до ангидрита.
На рис. 11.14 приведена политермическая диаграмма, характеризующая направление и последовательность фазовых превращений сульфата кальция в системе CaSCU—Н3РО4—Н2О. Стабильными твердыми фазами в системе являются гипс (ниже кривой ab) и ангидрит (выше кривой ab). В области, расположенной над кривой cd, гемигидрат, который большей частью является первой кристаллизующейся фазой системы, переходит в ангидрит.
В области между кривыми cd и ab стабильной формой также является ангидрит, но здесь гемигидрат переходит в ангидрит не непосредственно, а сначала оводняется до гипса. Кривая cd является множеством точек сосуществования этих метастабильных фаз при разных температурах. Аналогично кривая ab является множеством точек сосуществования стабильных гипса и ангидрита. Согласно правилу Оствальда, ниже линии ab стабильная фаза — гипс, и последовательность превращений должна быть такой, как показано на рис. 11.14. На практике образования ангидрита не наблюдается и гемигидрат переходит в гипс. Это объясняется кинетическими причинами, меняющими маршу-ты реакций, например значительно более быстрым превращением ангидрит -> гипс, чем гемигидрат -> ангидрит, в данной области температур и концентраций Р2О5. В других условиях скорость превращения ангидрита может быть меньшей. На скорость и маршрут взаимных переходов могут влиять и ионы примесей.
Следовательно, степень гидратации сульфата кальция, отделяемого в процессе экстракции, может не соответствовать стабильным формам и зависит от конкретных условий осуществления процесса. На рис. 11.15 приведена диаграмма, показывающая практическую степень гидратации сульфата кальция в зависимости от технологического режима процесса экстракции, и в первую очередь от температуры и концентрации фосфорной кислоты. Как видно из рисунка, в области ниже кривой 2 сульфат кальция отделяется в виде гипса, выше кривой 1—в виде ангидрита, а между этими линиями — в виде гемигидрата.
В производственных условиях осадок загрязнен примесями РдОю в виде неразложенных фосфатов, неотмытой Н3РО4, сокристаллизо-ванных фосфатов различных металлов и др. Поэтому образующиеся 591
сульфаты кальция называют соответственно фосфогипс, фосфогеми-гидрат и фосфоангидрит. В зависимости от типа осаждаемого сульфата различают три способа получения экстракционной фосфорной кислоты: дигидратный, гемигидратный (полугидратный) и ангидритный, а также комбинированные: гемигидратно-дигидратный и дигид-ратно-гемигидратный. Наиболее распространен на практике дигидратный режим, который осуществляют при 65—80° С, получая при этом кислоту с содержанием до 30—32% Р2О5.
Гемигидратный метод, осуществляемый при 90—105° С, обеспечивает получение кислоты с содержанием до 50% Р2О5 (без дополнительного упаривания). Согласно способу, фосфорную кислоту, содержащую 36—38% Р2О5, можно получить из апатитового концентрата практически на том же оборудовании, что и в типовом дигидратном процессе с воздушным охлаждением суспензии. Гемигидратные процессы широкого распространения пока не получили из-за высокой температуры (80—100° С), выделения при этих температурах фторида водорода в газовую фазу, более низкого выхода Р2О5 в кислоту, чем в дигидратном методе. В усовершенствованных промышленных схемах предусмотрено предварительное смачивание апатитового сырья в скоростном смесителе, разделение зон разложения и кристаллизации и др. Проведение основной реакции при содержании серной кислоты в жидкой фазе суспензии 0,2—1,0% в первом реакторе и 2,0—3,0% во втором реакторе позволяет снизить количество растворенного CaSO4 в продукционной фосфорной кислоте, значительно уменьшить зарастание оборудования и трубопроводов, существенно интенсифицировать работу основных технологических узлов.
110 с
100
90
80
? 70
| 50 £
I 40
30
20
10
О 5 10 15 20 25 30 35 40 45 50 55
Концентрация Р2О5
Рис. 11.14. Схема превращений кристаллогидратов сульфата кальция в растворах фосфорной кислоты
Рис 11.15. Влияние температуры и содержания Р20з в растворе на практическую гидратированность отделяемого осадка сульфата кальция
592
Ангидритный способ имеет ряд преимуществ перед дигидратным и гемигидратным: 1) позволяет без упаривания получать кислоту содержащую 50% Р2О5; 2) в процессе экстракции большая часть фтора выделяется в газовую фазу; 3) получаемая кислота меньше загрязнена сульфатом кальция.
Использование способа в промышленности сдерживают: коррозионные условия (высокие температуры и концентрации фосфорной кислоты), образование мелких кристаллов и необходимость с этим большего числа ступеней противоточной промывки.
Комбинированные способы получения экстракционной фосфорной кислоты — гемигидратно-дигидратный и дигидратно-гемигидрат-ный — более технологичны и экономичны, чем одноступенчатые. Они обеспечивают повышение степени использования исходного фосфатного сырья (за счет снижения технологических потерь Р2О5), повышение концентрации целевого продукта, более чистого сульфата кальция с большими возможностями его дальнейшей переработки.
Однако комбинированные процессы усложнены двойным фильтрованием или нетехнологичны из-за значительной продолжительности стадии перекристаллизации в гемигидратно-дигидратном способе. Из усовершенствованных комбинированных технологий наибольший интерес представляет разработанный в НИУИФ дигидратно-гемигидратный процесс с промежуточным фильтрованием. Этот способ позволяет получать из апатитового концентрата фосфорную кислоту, содержащую 33—34% Р2О5, из фосфоритов — фосфорную кислоту, содержащую 28—30% Р2О5. Степень извлечения Р2О5 из исходного сырья составляет около 99%. Продолжительность процесса не превышает 6 ч. Фосфоге-мигидрат содержит менее 0,5% Р2О5 и 0,15% F и может быть применен в качестве вяжущего вещества в строительных материалах.
Получение экстракционной фосфорной кислоты дигидратным способом. На рис. 11.16 приведена принципиальная схема получения фосфорной кислоты (28—32% Р2О5) из апатитового концентрата. По этой схеме возможно получение фосфорной кислоты (20—22% P20j) из фосфоритов. Согласно схеме, в первый реактор экстрактора 3 из бункера 1 дозатором 2 непрерывно вводят апатитовый концентрат. В этот же реактор погружными насосами 5 вводят оборотную фосфорную кислоту из барометрического сборника 16 и циркуляционную суспензию после вакуум-испарительной установки 8 [кратность циркуляции (8^-12):!] и серную кислоту из сборника 4. Серную кислоту возможно частично или полностью вводить во второй реактор.
Соотношение Ж:Т в суспензии в экстракторе поддерживают равным (1,7-5-2,5): 1. Из первого реактора суспензия перетекает во второй, откуда основная часть ее погружным насосом 7 подается в вакуум-испаритель 8, представляющий собой резервуар, в котором 593
Рис. 11.16. Схема получения экстракционной фосфорной кислоты дигидратным способом:
1— бункер фосфатного сырья; 2 — дозатор; 3 — Двухступенчатый экстрактор; 4—сборник серной кислоты; 5 — погружные насосы; 6 — расходомер серной кислоты; 7 — погружной насос (циркуляционный); 8 — испаритель; 9 — брызгоуловитель; 10—конденсатор; 11 — барботажный нейтрализатор; 12 — лотки карусельного вакуум-фильтра; 13 — рессиверы (сепараторы); 14 — промежуточный сборник суспензий—после регенерации фильтрованной ткани; 75, 16, 17 — барометрические сборники: для первого (основного) фильтра (75), для оборотной фосфорной кислоты (76), для промывного фильтра (77)
вакуум-насосом поддерживают пониженное давление. Вследствие этого поступающая в него жидкость оказывается перегретой и закипает с выпаркой части воды. Это приводит к понижению температуры на 3—5° С. Газы из вакуум-испарителя через брызгоуловитель отводят в поверхностный конденсатор 10, в котором конденсируются пары воды и улавливается часть соединений фтора. Окончательную очистку газа от соединений фтора производят в барботажном нейтрализаторе 11.
Продукционная суспензия поступает на карусельный лотковый фильтр, в котором гипс отделяется от растворов, а осадок промывается по трехфильтратной системе.
Карусельный лотковый фильтр состоит из 24 отдельных лотков, на днищах которых уложена фильтровальная ткань (капрон, лавсан и т.п.). Лотки установлены на каретках с колесами, движущимися по круговым рельсам. С помощью двух шайб, образующих головку фи-594
льтра,— подвижной, вращающейся вместе с лотками, и неподвижной— фильтраты отсасываются в соответствующие вакуум-сборники (15, 16 и 17). После прохождения зон фильтрации и промывок каждый лоток с помощью направляющих автоматически опрокидывается для выгрузки лепешки фосфогипса. Фильтровальная ткань промывается водой и подсушивается воздухом. Затем лоток вновь принимает рабочее положение и перемещается в зону основного фильтрования. Воду, используемую для регенерации фильтровальной ткани, подают на последнюю или предпоследнюю зону промывки осадка, что сокращает потери Р4О10 и позволяет создать на экстракционных установках замкнутую систему водооборота. Гигроскопическая влажность фосфогипса 15—40%. Количество фосфогипса (в пересчете на сухое вещество) составляет 1,2—1,6 т на 1 т переработанного природного фосфата. В процессе переработки апатита выход гемигидрата кальция равен 1,4; гипса—1,6 т.
Газожидкостная смесь разделяется в сепараторах 13, в которых поддерживается разрежение 65—85 кПа. Первый фильтрат Ф] направляется в сборник готовой продукции, а часть его переливается в барометрический сборник оборотной кислоты 16, куда также поступает и второй фильтрат Ф2, полученный в процессе промывки осадка третьим фильтратом Фз Фильтрат Фз образуется при промывке осадка суспензией, получаемой в процессе регенерации фильтровальной ткани, и свежей горячей (60—70° С) водой. Промытый гипс передается с лотка в сборник 14, из которого в виде суспензии перекачивается в отстойник гипса. Содержание Р2О5 в фильтратах: Ф1—28—32%, Ф2 —22—25%, Ф3 —5—10%.
В процессе получения фосфорной кислоты дигидратным способом выделение фтора в атмосферу (преимущественно в виде тетрафторида кремния) невелико — 3—5% от содержащегося в исходном сырье (около 80% переходит в целевой продукт, 15—17% — в фосфогипс). Соответственно концентрация фторидов в отводимых из экстрактора газах в зависимости от способа охлаждения и вытяжки вентилятора в пересчете на фтор составляет 0,2—2,5 г/м3.
Согласно дигидратному способу, на 1 т продукционного Р2О5 расходуется 2,65—2,73 т апатита и 2,45—2,48 т 100%-ной серной кислоты. Экстракционная фосфорная кислота, полученная из апатита дигидратным способом, содержит (в %): Р2О5 25—32; SO3 — 1,8—2,8; СаО 0,1—0,4; А12О3 0,3—0,4; Fe2O3 0,3—0,5; F 1,7—2,20.
Получение экстракционной фосфорной кислоты г е м и г и д р ат н ы м и г е м и г и д р ат н о - д и г и д-ратным способами. Полугидратный способ осуществляется идентично описанному дигидратному с введением всех исходных реагентов в первую секцию реактора. Процесс осаждения полугидрата сульфата кальция происходит при содержании в жидкой фазе 35—38% 595
Р2О5 и 1,0—1,5% SO3, температуре 95—105° С. Второй вариант включает процесс предварительного разложения апатита в 3—4-кратном избытке концентрированной (45—48% Р2О5) фосфорной кислоты (смеси первого фильтрата и циркулирующей суспензии) при 95—102° С. Образовавшаяся при этом суспензия, содержащая дигидрофосфат кальция, обрабатывается 92—93%-ной серной кислотой. Аппаратурное разделение стадий разложения апатита и кристаллизации гемигидрата обеспечивает достижение высокой степени разложения сырья (97—98,5%) и получение концентрированного (45—48% Р2О5) целевого продукта, содержащего (в %): СаО 0,2—0,4; SO3 — 0,5—0,8; (Fe, А1)20з 1,0—1,2; F 1,0—1,1 или 0,2—0,3 (в процессе обесфторива-ния жидкой фазы суспензии карбонатом натрия).
В гемигидратных процессах требуется отмывать водой и вывозить из цеха нестабильный осадок сульфата кальция. Однако переход его в дигидрат, особенно при добавке в водную суспензию незначительного количества гидроксида кальция, происходит медленно, что позволяет удалять его гидротранспортом. В связи с высокой температурой процесса и концентрации Р2О5 в жидкой фазе в гемигидратных процессах степень выделения фтора в газовую фазу значительно больше, чем в дигидратных, и составляет 15—50%, что способствует его переработке. Выход Р2О5 в гемигидратных процессах на 1—2% меньше, чем в дигидратных.
Согласно гемигидратно-дигидратному процессу, исходная фосфатная руда разлагается серной кислотой с получением гемигидрата сульфата кальция, который затем перекристаллизовывается в дигидрат, процесс позволяет достичь высокого перехода Р2О5 в кислоту (98—99%) и получить гипс с 0,1%-ным содержанием водорастворимого Р2О5, пригодный для последующей переработки. Процесс позволяет разлагать исходное сырье более крупного помола, не снижая переход Р2О5 в кислоту, поскольку в процессе перекристаллизации гемигидрата в дигидрат происходит доразложение частиц, заблокированных сульфатными покрытиями.
В отличие от полугидратного способа в комбинированных процессах предусмотрено регулирование условий гидратации с получением крупнокристаллического (200—500x40—80 мкм) гипса с незначительным содержанием Р2О5. Комбинированный процесс предусматривает осаждение гемигидрата, смешением исходного фосфорита с оборотной фосфорной и серной кислотами при 90—95° С, охлаждение суспензии до 50—65° С и гидратацию гемигидрата с введением затравочных кристаллов гипса, серной кислоты и активного диоксида кремния для связывания фторид-ионов, замедляющих в сочетании с ионами алюминия обводнение. Продолжительность процесса гидратации — 5—16 ч. Осадок после отмывки содержит 1,8—1,9 моль Н2О на 1 моль СаБОд, 0,3% 596
общего Р2О5 (в дигидратном процессе 0,5—1,5%) и 0,02—0,08% водорастворимого Р2О5. В связи с проведением процесса осаждения геми-гидрата и его гидратации в идентичных растворах описанный способ позволяет получать фосфорную кислоту, содержащую не более 32% Р2О5. На получение 1 т Р2О5 расходуется 2,95 т фосфорита (1,03 т Р2О5), 2,72 т H2SO4, 0,25 т пара и 160 кВт-ч электроэнергии.
Разработан гемигидратно-дигидратный процесс с промежуточным выделением из системы продукционной кислоты. Согласно этому способу, осаждение полугидрата проводят при 90—100° С в кислоте, содержащей 45—50% Р2О5. Образующуюся суспензию фильтруют, центрифугируют или отстаивают. Осадок репульпируют в растворах, содержащих 10—25% Р2О5 и 5—10% H2SO4. Процесс гидратации ге-мигидрата проводят при 55—65° С. Образующийся гемигидрат отделяют от жидкой фазы, возвращаемой в процесс.
11.7. СУПЕРФОСФАТЫ
Суперфосфаты являются широко распространенными фосфорными удобрениями. В зависимости от усвояемого растениями фосфора различают простой, двойной и обогащенный суперфосфат. Разновидностями суперфосфата являются фосфорно-кальциевое серосодержащее удобрение (ФКСУ) и суперфос (табл. 11.4).
Таблица 11.4. Физико-механические свойства суперфосфатов и их разновидностей
Характеристика Простой суперфосфат Двойной суперфосфат ФКСУ Суперфос
Насыпная плотность, г/см3: до уплотнения после уплотнения Прочность гранул на раздавливание, МПа Угол естественного откоса, град Теплоемкость, кДж/(кг °С) Гигроскопическая точка ,% 0,90—0,95 1,00—1,05 1,50—2,0 36—39 1,173—1,215 50-60 0,90—1,05 1,00—1,15 1,80—4,0 40—42 1,215—1,257 60-65 0,95—1,00 1,00—1,08 2—3 39-42 1,180—1,210 55—65 0,95—1,05 1,00—1,15 40-42 1,210—1,240 55—60
* Равновесная влажность над насыщенным раствором при 20° С.
Простой суперфосфат. Выпускаемый промышленностью моногидрат дигидрофосфата кальция (простой суперфосфат) представляет собой порошок или зерна серого цвета. Он состоит из нескольких твердых фаз: фосфатов кальция (в основном моногидрат дигидрофосфата кальция), магния, железа, алюминия, ангидрита с примесью ге-мигидрата сульфата кальция (при переработке апатитового концентра-
та SrSO4), остатки неразложившихся минералов, входящих в состав исходного фосфорсодержащего сырья, кремнегель SiO2-nH2O и др. Содержание твердых фаз составляет 65—72%, в том числе 50—55% CaSO4 (SrSO4). Жидкая фаза состоит из водного раствора фосфорной кислоты насыщенного дигидрофосфатом кальция и содержащего ионы Mg2+, Fe3+, Al3+, F', SiF^ и др.
Порошкообразный суперфосфат гигроскопичен и сильно слеживается в результате кристаллизации из жидкой фазы моногидрата дигидрофосфата кальция. Менее слеживаемым является хорошо вызревший и охлажденный суперфосфат, что показывает завершенность процесса кристаллизации. Нейтрализованный и гранулированный суперфосфат практически не слеживается.
Физико-химические основы получения простого суперфосфата. Процесс получения простого суперфосфата состоит из двух стадий. В первой стадии фторапатит с серной кислотой образует фосфорную кислоту и гемигидрат сульфата кальция:
2Ca5(PO4)3F + 10H2SO4 + 5Н2О = 6Н3РО4 + 10CaS040,5H20 + 2HF
Реакция заканчивается за 20—40 мин в период созревания (твердения) суперфосфатной массы в реакторе. Образующийся гемигидрат в течение нескольких минут превращается в ангидрит
2CaSO4 0,5H2O -► 2CaSO4 + Н2О
что объясняется высокой температурой реакционной массы в суперфосфатном реакторе (ПО—120° С) и значительным содержанием Р2О5 в жидкой фазе (42—46% в конце первой стадии процесса).
После первого периода реакции начинается вторая стадия — разложение оставшегося количества апатита фосфорной кислотой, выделившейся в первой стадии, по реакции
Ca5(PO4)3F + 7Н3РО4 + 5Н2О = 5Са(Н2РО4)2Н2О + HF
Образующийся моногидрат дигидрофосфата кальция находится сначала в растворе и начинает кристаллизоваться в процессе его пересыщения. Из диаграммы равновесия в системе СаО—Р2О5—Н2О (рис. 11.17) видно, что при содержании Р2О3 в растворе 42—-46% и температуре выше 100°С кристаллизуется моногидрат дигидрофосфата кальция [Са(Н2РО4)2 Н2О].
При стехиометрическом соотношении реагентов согласно суммарному уравнению
2Ca5(PO4)3F + 7H2SO4 + ЗН2О = ЗСа(Н2РО4)Н2О + 7CaSO4 + 2HF 598
в первой стадии процесса реагирует 70%, а во второй — 30% исходного апатита. В первой стадии процесса образуется структурная сетка из микрокристаллов сульфата кальция, заполненная значительным количеством воды. Схватывание реакционной массы происходит до полного израсходования серной кислоты, в присутствии которой образование моногидрата дигидрофосфата кальция невозможно. Поэтому причина затвердевания массы объясняется процессом кристаллизации сульфата кальция. Вторая стадия процесса начинается в период камерного созревания и завершения при дозревании продукта на складе товарной продукции в течение 6—25 сут (в зависимости от вида сырья, технологии и условий дозревания).
В процессе получения моногидрата дигидрофосфата кальция стехиометрическая норма серной кислоты и фосфата определяется соотношением ТНгБОдЗРгО;, равным 1,61 ч. H2SO4 на 1 ч. Р2О5. Например, при содержании в апатитовом концентрате 39,4% Р2О5
Рис 11.17. Система СаО—Р2О5—Н2О. Составы растворов, насыщенных двумя солями (многовариантные равновесия):
В—при 25—132° С — Са(Н2РО4)2Н2О + СаНРО4, при 0° С — Са(Н2РО4)2Н2О + СаНРО4-2Н2О; Я, —Я5—при 40—132°С — Са(Н2РО4)гН2О + Са(Н2Ю4)2; Я и Я, —при 0 и 25°С — 2Н,РО4Н2О + + CaCHjPO^-HjO. Составы растворов, насыщенных тремя соединениями (инвариантные равновесия): U— при 30° С — Н3РО. + Ca(H2PO4)j-H2O + СаСВДЮЛ; Е— при 152° С — Са(Н2ГО4)2 Н2О + + Са(Н2РО4)2 + СаНРО4; 0 —при 100° С — 2Н}РО4 Н2О + СаСНгРО.^ НгО + лед
599
стехиометрическая норма H2SO4 составляет 39,4-1,61 = 63,4 ч. на 100 ч. сырья. С целью ускорения процесса разложения и с учетом наличия примесей практическую норму кислоты принимают более высокой— от 68 до 72 ч.
При отсутствии в исходном фосфате элементов, образующих не растворимые в кислоте сульфаты (Sr, Ва), стехиометрическую норму серной кислоты определяют по формуле
п = 98[Z/56 — (от/142 —рМ — <?/160 — г/102)], где /, т, р, q, z — содержание в кислоторастворимых минералах сырья: СаО, Р2О5, MgO, FejOj и А^Оз.Уо (масс.); 56, 142, 40, 160 и 102 — соответствующие молярные массы.
Реальная норма серной кислоты будет несколько меньше рассчитанной, поскольку часть кальций-иона связывается с ионами F" и SiF62’. Поэтому норму серной кислоты обычно устанавливают опытным путем.
Температура процесса и концентрация исходной серной кислоты заметно отражаются на структуре и физических свойствах целевого продукта. Скорость разложения фосфата зависит от концентрации серной кислоты, состава и степени пересыщения жидкой фазы суперфосфата продуктами реакции. На рис. 11.18 показан общий вид зависимости степени разложения фосфата за определенное время (изохрона) от концентрации исходной серной кислоты при периодическом перемешивании реакционной массы. С увеличением концентрации разбавленных растворов и уменьшением концентрации крепких растворов степень разложения фосфата растет. Однако, начиная с некоторых концентраций кислоты, изменяется состав выделяющегося сульфата кальция и уменьшаются размеры его кристаллов, что вызывает отложение последних на поверхности частиц фосфата и снижает Поэтому полная зависимость степени разложения от концентрации кислоты изображается кривой, которая имеет два максимума и между ними один минимум. Положение максимумов зависит от вида сырья, температуры и продолжительности процесса. Скорость и достигаемая степень разложения кислотой низкой концентрации (левый максимум) высоки.
В процессе разложения апатита раствором серной кислоты, содержащим выше 63% H2SO4, выделяются кристаллы гемигидрата сульфата кальция и ангидрита в форме мелких иголочек длиной 5—7 и шириной 1—2 мкм. Они, отлагаясь на поверхности частиц
степень их разложения.
Концентрация H2SO4
Рис. 11.18. Общий вид зависимости степени разложения фосфата от концентрации исходной серной кислоты (изохрона)
600
фосфата и образуя плотный шламовый покров, резко снижают скорость реакции. Повышение концентрации исходной кислоты наравне с уменьшением влажности продукта и возрастанием содержания в нем Р2О5 также вызывает образование корки на зернах фосфата.
Из вышеизложенного видно, что начальную концентрацию серной кислоты поддерживают около 68%. При этом оптимальная продолжительность пребывания суспензии составляет 5—1 мин, а температура суспензии на выходе из смесителя — ПО—115° С.
Фазовый состав фосфатного комплекса суперфосфата, получаемого из апатитового концентрата, может быть определен по диаграмме растворимости в системе СаО—Р2О5—Н2О (см. рис. 11.17).
На рис. 11.19, б приведены изотермы системы СаО—Р2О5—Н2О для 25, 40 и 100° С. Пучок лучей, исходящих из начала координат диаграммы, представляет шкалу степени нейтрализации. Луч 100%-ной нейтрализации заканчивается в точке N, дающей состав моногидрата дигидрофосфата кальция СаЩгРО^-НгО (56,34% Р2О5 и 22,22% СаО). На рис. 11.19, а дана зависимость степени нейтрализации от степени разложения апатитового концентрата при 200% от расчетной нормы серной кислоты (и), а на рис. 11.19, в — вспомогательный график зависимости между концентрацией Р2О5 в жидкой фазе к началу второй стадии процесса. На номограмме нанесены лучи растворения гидроксидапатита [Са5(РОд)з(ОН)] в фосфорной кислоте разных концентраций. Они исходят из точки состава гидроксидапатита (42,39% Р2О5 и 55,82% СаО), находящейся за пределами диаграммы.
Точки составов фосфатного комплекса лежат на пересечении лучей растворения и степени нейтрализации. Например, при норме серной кислоты 72 ч, степени разложения апатита 92% и влажности суперфосфата 12% состав фосфатного комплекса изобразится точкой А (46,2% Р2О5 и 8,5% СаО). Эта точка находится в поле кристаллизации Са(Н2РО4)4-Н2О при 25—100° С. Следовательно, в приведенном примере фосфатный комплекс состоит из жидкой фазы (раствор фосфорной кислоты и моногидрата дигидрофосфата кальция и дигидрофосфата кальция в твердой фазе). С помощью луча кристаллизации, проведенного из точки N через точку А, подсчитывают по правилу рычага соотношение между жидкой и твердой фазами в комплексе при различных температурах. Точки пересечения луча кристаллизации с изотермами (например, точка Р для 25° С) определяют состав жидкой фазы при соответствующей температуре.
По практическим данным при указанной норме серной кислоты (72 г) степень разложения апатита в камерном суперфосфате составляет ~87% (точка С на рис. 11.17, а), а степень нейтрализации жидкой фазы равна 30%. Луч степени нейтрализации жидкой фазы пересекает лучи растворения при влажности суперфосфата 16, 14, 12 или 601
9% соответственно в точках £>b D2, D3 или Z>4. Из положения этих точек можно заключить, что при влажности 16% фосфатный комплекс при 100° С и выше не содержит твердого дигидрофосфата кальция (точка D\ лежит в поле ненасыщенных растворов), а при влажности 14% содержит его незначительно (точка Z>2 находится близко к 100-градусной изотерме).
Рис. 11.19. Номограмма для определения фазового состава фосфатного комплекса суперфосфата из апатитового концентрата
602
Вызревание суперфосфата, полученного из апатитового концентрата, ускоряется при понижении температуры до 40—50° С, поскольку в процессе охлаждения внешнего слоя жидкость, обволакивающая частицы неразложившегося фосфата, кристаллизуется до моногидрат дигидрофосфата кальция, насыщающего раствор, увеличиваются как температурные, так и концентрационные градиенты — реакция ускоряется. Кроме того, в результате кристаллизации Са(Н2РО4)2-Н2О степень нейтрализации остающейся жидкой фазы уменьшается, поэтому увеличивается ее активность.
Процесс получения суперфосфата из фосфоритов имеет специфические особенности, поскольку они содержат примеси доломита и магнезиальных силикатов. В их присутствии процесс разложения их серной кислотой протекает в три стадии.
В первой стадии идут реакции:
Ca5(PO4)3F + 5H2SO4 = ЗН3РО4 + 4CaSO4 + HF
CaCO3MgCO3 + 2H2SO4 = CaSO4 + MgSO4 + 2CO2 + 2H2O
Mg2SiO4 + 2H2SO4 = 2MgSO4 + SiO2 + 2H2O
В результате этих реакций образуется водный раствор смеси фосфорной кислоты с сульфатом магния.
Во второй стадии фосфат разлагается фосфорной кислотой, но поскольку в системе находится сульфат магния, то образуется дигидрофосфат магния:
Ca5(PO4)3F + 7Н3РО4 + 5MgSO4 = 5Mg(H2PO4)2 + 5CaSO4 + HF
В третьей стадии оставшееся фосфатное сырье разлагается фосфорной кислотой с образованием дигидрофосфата кальция:
Ca5(PO4)3F + 7Н3РО4 = 5Са(Н2РО4)2 + HF
После второй стадии фосфатный комплекс состоит из MgO, Р2О3, Н2О. Как видно из диаграммы растворимости в системе MgO—Р2О5—Н2О (рис. 11.20), дигидрофосфат магния более растворим, чем дигидрофосфат кальция в системе СаО—P20s—Н2О (см. рис. 11.17). Хотя и моногидрофосфат и дигидрофосфат инконгруэнтны по отношению к насыщенным водным растворам, т.е. разлагаются с выделением в твердую фазу менее кислых фосфатов, степень разложения фосфатов магния относительно меньше, чем фосфатов кальция (при одинаковом соотношении между солью и водой). При 25—80° С гидрофосфат магния растворяется без разложения при массовом отношении 603
Рис. 11.20. Изотермы растворимости в системе MgO—Р2О5—Н2О при 25, 80 и 130° С. Точками обозначены
Mg(H2₽O4)2:H2O<0,287. При большем значении этого отношения дигидрофосфат магния разлагается, и в твердой фазе появляется гидрофосфат магния. Разложение же дигидрофосфата кальция происходит при отношении СаСНгРО^НгОО.О!. Как и для солей кальция, с увеличением концентрации Р2О5 растворимость MgHPO4 растет, a Mg(H2₽O4)2 уменьшается (рис. 11.20), но в отличие от дигидрофосфата кальция степень разложения водой дигидрофосфата магния почти не зависит от температуры растворов.
В результате значительной растворимости дигидрофосфата магния после второй стадии реакции жидкая фаза остается ненасыщенной
составы растворов, равновесных с им — ОН ПОЛНОСТЬЮ находится В
двумя твердыми фазами растворе, и степень нейтрализации
фосфорной кислоты оказывается ощутимо большой. Дигидрофосфат магния образует с фосфорной кислотой буферную смесь, диссоциируя на Mg*+ и Н2РО^( что подавляет диссоциацию фосфорной кислоты на Н+ и Н2РОд. Поэтому разложение сырья резко замедляется уже во второй стадии процесса, а скорость его в третьей стадии становится совсем незначительной.
Приведенные данные показывают на необходимость предварительного обогащения их для удаления части соединений магния. Содержание последнего должно быть не более 6—8% от количества Р2О5. Для суперфосфата из фосфорита оптимальная температура до-разложения более высокая (65—80°С), чем для суперфосфата из апатита или фосфоритов, не содержащих магния.
Для снятия свободной кислотности товарный суперфосфат нейтрализуют, обрабатывая его добавками, легко разлагаемыми фосфорной кислотой. Обычно нейтрализацию совмещают с гранулированием. В качестве добавок применяют фосфоритную муку, костяную муку, известь, мел, известняк и др.
Одним из способов улучшения качества суперфосфата является его аммонизация — нейтрализация свободной кислоты аммиаком. В процессе нейтрализации газообразным аммиаком свободной фосфорной кислоты сначала образуется дигидрофосфат аммония:
Н3РО4 + NH3 = NH4H2PO4
604
В результате выделения теплоты нейтрализации температура массы повышается до 80—90° С, суперфосфат подсушивается.
В процессе более сильной аммонизации образуется гидрофосфат аммония, который взаимодействует с сульфатом кальция с образованием гидрофосфата кальция:
।
Н3РО4 + 2NH3 = (ЫНдЬНРОд
(NH4)2HPO4 + CaSO4 - СаНРО4 + (NH4)2SO4
Дигидрофосфат кальция также переходит в гидрофосфат кальция:
Са(Н2РО4)2Н2О + CaSO4 + 2NH3 = 2СаНРО4 + (NH4)2SO4 + Н2О
В результате глубокой аммонизации содержание водорастворимого Р2О5 в суперфосфате значительно уменьшается, однако содержание усвояемого Р2О3 изменяется мало. В процессе дальнейшей аммонизации происходит ретроградация Р2О5 — переход гидрофосфата кальция в трудно усвояемую растениями форму — дифосфат кальция:
2СаНРО4 + CaSO4 + 2NH3 = Са3(РО4)2 + (NH4)2SO4
Поэтому в процессе нейтрализации суперфосфата аммиаком получают продукт, содержащий не более 2% азота.
Процесс аммонизации проводят во вращающемся барабане-аммо-низаторе, вводя в него непрерывно суперфосфат и газообразный аммиак прямотоком. Норма аммиака составляет 2% от массы суперфосфата, а степень его поглощения 97—99%. Образующиеся в процессе аммонизации крупные комки суперфосфата отсеиваются на сите и измельчаются.
Получение простого су п е р ф о с ф ат а (рис. 11.21). Исходный фосфат (апатитовый концентрат или фосфоритная мука) ленточным транспортером 1 передается в бункер 2, откуда шнековым питателем 3 и ковшовым элеватором 4 системой шнеков 5 и 6 загружается в бункер дозатора 7. Во избежание зависания фосфата в силосах осуществляют аэрацию — через пористые плитки, уложенные в днище силосов, подают сжатый воздух. Дозировку фосфата производят с помощью весового дозатора 8. Применяются двухступенчатые дозаторы с электромагнитными вибрационными и шнековыми питателями, имеющими незначительную инерцию и высокую точность. Фосфоритная мука обладает большой текучестью, поэтому для ее дозирования применяют снабженные электронным регулятором порционные весы.
605
Рис. 11.21. Схема получения простого суперфосфата:
1 — транспортер для апатитового концентрата (фосфорита); 2 — бункер; 3 — шнековый питатель; 4—ковшовый элеватор; 5, 6 — шнеки; 9 — обратный шнек для избыточного апатитового концентрата; 7—бункер дозатора; 8 — дозатор; 10—смеситель; 11— контрольные весы; 12—бункер контрольных весов; 13— сборник серной кислоты; 14—центробежный насос; 15 — напорный сборник для кислоты; 16—кислотный смеситель; 17—напорный сборник для воды; 18 — щелевой расходомер кислоты; 19 — конценгратомер для кислоты; 20 — суперфосфатная камера; 21 — фрезер; 22 — транспортер камерного суперфосфата; 23 — разбрасыватель суперфосфата на складе
Серная кислота центробежным насосом 14 перекачивается из резервуара серной кислоты 13 в напорный сборник 15, откуда направляется на смешение с водой из сборника 77 в кислотный смеситель 16. Далее разбавленная водой кислота проходит концентратомер 18 и щелевой расходомер кислоты 19 и поступает на смешение с фосфатным сырьем.
Исходный апатит (фосфоритная мука) смешивают с кислотой в вертикальных трех- или четырехкамерных смесителях непрерывного действия (рис. 11.22), состоящих из четырех последовательно установленных (вертикально) на одном уровне сообщающихся камер 7, имеющих переливную коробку 3 и снабженных метальными устройствами 2. Смесители снабжены входным отверстием 4, съемными
Рис. 11.22. Четырехкамерный смеситель:
1 — сообщающиеся камеры; 2 — метальные устройства; 3 — переливная коробка; 4 — входное отверстие; 5 — съемная крышка; 6 — чугунный шибер; 7 — трос ручной лебедки для подъема и опускания шибера
607
крышками 5, чугунным шибером 6 и тросом ручной лебедки 7 для подъема и опускания шибера. Объем суспензии регулируют шибером 6 так, чтобы обеспечить продолжительность ее перемешивания в течение 5—7 мин (при работе с фосфоритом — 2—3 мин). Из смесителя суспензия непрерывно перетекает в суперфосфатную камеру (рис. 11.23), представляющую собой вертикальный железобетонный цилиндрический корпус 1, имеющий стальной кожух и футеровку из диабазовых плиток. Камера опирается на 16 роликовых опор, на которых она вращается вокруг неподвижной чугунной трубы 2, проходящей через сальниковое уплотнение в днище камеры. Камера вращается электродвигателем 3 через редуктор 4. В течение 1—2 ч камера делает один оборот. Железобетонная крышка камеры 5 неподвижна— между камерой и крышкой имеется уплотнение из листовой резины. К крышке подвешена вертикальная чугунная перегородка 6, примыкающая к центральной трубе и отделяющая зону загрузки от зоны выгрузки. Около этой перегородки со стороны выгрузки находится фрезер 7, представляющий собой вращающуюся на вертикальном валу стальную конструкцию («карусель»), на которой укреплены крылья с ножами. Фрезер подвешен к крышке камеры и вращается в направлении, противоположном вращению камеры с частотой 0,13—0,17 с'1 (8—10 об/мин). *
Суспензия из смесителя непрерывно поступает в камеру по трубе, вставленной в отверстие 8 в крышке. По мере вращения камеры масса суперфосфата твердеет и подходит к фрезеру готовой для выгрузки. За один оборот фрезер срезает слой целевого продукта тол-
Рис. 11.23. Непрерывно действующая суперфосфатная камера:
7 — цилиндрический корпус камеры, 2 — центральная труба; 3— электродвигатель; 4 — редуктор; 5 — крышка камеры; 6 — перегородки; 7 — фрезер; 8 — отверстие для подачи суспензии; 9 — эксцентрик
608
щиной 5—25 мм. Срезанный суперфосфат попадает в центральную трубу 2 через имеющуюся в ней широкую щель, откуда подается на транспортер, передающий продукт на склад. Для уменьшения сопротивления вращению вследствие трения суперфосфатной массы в неподвижную центральную трубу, у ее наружной стенки в зоне загрузки установлен деревянный эксцентрик 9. Он создает свободный объем, необходимый для расширения суперфосфатной массы в процессе ее созревания. При этом ее плотность уменьшается ко времени выгрузки до 1100 кг/м3 вместо начальной 1500 кг/м3. В процессе переработки содержащих карбонаты фосфатов давление на стенки камеры меньше, поскольку суперфосфатная масса более пориста, ее плотность в пределах 800—900 кг/м3. Выделяющиеся в процессе реакции в суперфосфатной камере газы отсасываются через вентиляционную трубу, по которой попадают в абсорбционное отделение.
Полученный суперфосфат передается на склад в виде мелких частичек, охлаждается окружающим воздухом с 70—90 до 30—60° С. Целевой продукт на складе вылеживается 2—3 недели. При этом его перелопачивают грейферными кранами, т.е. пересыпают кучи с места на место, что способствует его охлаждению и дозреванию.
На производство 1 т порошкообразного суперфосфата расходуют 0,53—0,55 т апатитового концентрата и 0,37—0,38 т серной кислоты (100%-ной).
В настоящее время суперфосфат производят в гранулированном виде. Согласно схеме процесса (рис. 11.24), исходный суперфосфат и нейтрализующая добавка соответственно из бункеров 1 и 2, оборудо-
Рис. 11.24. Схема получения гранулированного суперфосфата:
1 — бункер суперфосфата; 2 — бункер нейтрализующей добавки; 3 — ленточные питатели; 4— дробилка; 5,10— элеваторы; 6 — транспортер, 7— гранулятор, 8— барабанная сушилка; 9 — транспортер ретура; 11 — грохот; 12 — транспортер целевого продукта; 13 — дробилка;
14,16 — транспортеры; 15 — бункеры целевого продукта
20 Химическая технология неорганических веществ, кн. 1
609
ванных дозаторами, по ленточным питателям 3 поступают в валковую дробилку 4, в которой измельчаются и перемешиваются. Образующаяся смесь направляется в элеватор 5, в который одновременно поступает ретур — мелкая фракция гранулированного суперфосфата, возвращаемая после рассева на грохоте И. После элеватора 5 транспортером б смесь передают в гранулятор 7, представляющий собой вращающийся барабан, в котором увлажненный порошкообразный материал закатывают в гранулы. Необходимую для увлажнения материала воду (~16%) распыляют форсунками в гранулятор, а выходящие из гранулятора влажные гранулы суперфосфата высушиваются в сушильном барабане 8 до влажности 3—5%. Во избежание перехода растворимого моногидрата дигидрофосфата кальция в нерастворимые ди- (Са2Р2О7) и метафосфаты кальция [Саз(Р3О9)2] температуру материала в процессе сушки поддерживают не выше 95° С. Поэтому в процессе сушки суперфосфат и топочные газы в барабанном грануляторе движутся прямотоком (горячий газ соприкасается с более влажным продуктом, что уменьшает опасность перегрева). Топочные газы входят в сушилку с температурой 600° С, а выходят из нее с температурой 100—120° С.
В процессе сушки улетучиваются соединения фтора в виде эквивалентной смеси 2HF + S1F4, которую перерабатывают.
Высушенный продукт элеватором 10 передается в грохот 11, в котором рассеивается на фракции. Частицы суперфосфата размером более 4 мм направляются в дробилку 13 и после измельчения транспортером 14 возвращаются в элеватор, подающий их на рассев. Фракция с размером частиц 2—4 мм транспортером 12 направляется в бункеры 15, а мелкая фракция (20—30% от общего количества) ре-турным транспортером 9 передается на смешение с суперфосфатом перед его грануляцией. Целевой продукт из бункеров 15 затаривается в пятислойные бумажные кули, которые подаются транспортером к зашивочной машине. Защитные и маркированные мешки транспортером 16 грузятся в вагоны.
Для получения 1 т гранулированного продукта расходуют 1,015—0,025 т простого суперфосфата.
Производство гранулированного суперфосфата из фосфоритов сочетают с его аммонизацией, что позволяет устранить его увлажнение и сушку, а также расход известняка на предварительную нейтрализацию.
Процесс нейтрализации полученного из фосфоритов суперфосфата аммиаком до pH 4,0:4,5 не вызывает снижения содержания усвояемого Р2О5. За счет теплоты реакций, происходящих в процессе аммонизации, влажность продукта уменьшается на 4—5%. В процессе аммонизации простого суперфосфата, полученного из фосфоритов аммиакатами нитрата аммония или карбамидом, получают сложное гранулированное удобрение, содержащее 20—21% питательных ве-610
ществ, в том числе до 5—6% азота. Удобрение получается с хорошими физическими и агрохимическими свойствами.
Аммонизацию вызревшего на складах суперфосфата газообразным аммиаком проводят в аммонизаторах-грануляторах — горизонтальных вращающихся барабанах. После грануляции и аммонизации целевой продукт в течение 3-4 сут на складе перелопачивают 3—4 раза грейферным краном до температуры 28—32° С. Охлажденный продукт рассеивают и затаривают.
Степень использования аммиака в процессе аммонизации составляет 93—96%. Выхлопной газ из аммонизатора, содержащий до 5—8 г/м3 аммиака, абсорбируют водой. Образующиеся водные растворы аммиака (2,0—2,5% NH3) применяют для разбавления серной кислоты, поступающей на производство суперфосфата.
На производство 1 т аммонизированного суперфосфата расходуют 1,02 т кислого суперфосфата и 190 кг аммиака (100%). Целевой продукт содержит (в %): РгОзобщ 16,0—16,8; Р2О5усв 15,0—15,5; Р2О5ВОД 11,5—12,0; N 1,5—2,5; РгОзсвоб не более 0,5 и влаги 3.
Двойной суперфосфат. В процессе разложения природных фосфатов фосфорной кислотой получается двойной суперфосфат, содержащий моногидрат дигидрофосфата кальция, некоторое количество свободной фосфорной кислоты. Он содержит в 2—3 раза больше Р2О5, чем простой суперфосфат: РгОбус, 42—50%; Р2О5общ 45—56%; Р2О5ВОД 38—42% и Р205СВОб 1,5—5,0%.
Двойной суперфосфат получают камерным способом с использованием концентрированной фосфорной кислоты (52—54% Р2О5) и поточными методами, применяя кислоту, содержащую 28—36% Р2О5. Поточные методы, в отличие от камерного способа, обеспечивают получение целевого продукта в гранулированном виде без складского дозревания. Известен также камерно-поточный метод, по которому камерный двойной суперфосфат, полученный из легко разлагаемой фосфоритной муки, без доработки передается на грануляцию и сушку.
Физико-химические основы получения двойного суперфосфата. Разложение содержащихся в природных фосфатах минералов протекает по основным реакциям:
Ca5(PO4)3F + 7НзРО4 + 5Н2О = 5Са(Н2РО4)2Н2О + HF
CaMg(CO3)2 + 4Н3РО4 = Са(Н2РО4)2Н2О + Mg(H2PO4)2H2O + 2СО2
М20з + 2Н3РО4 + Н2О = 2[МРО4-2Н2О]
Процесс получения двойного суперфосфата состоит из двух основных стадий. На первой стадии при непрерывном смешении исходных фосфата и фосфорной кислоты взаимодействие протекает в
подвижной суспензии, жидкая фаза которой содержит фосфорную кислоту, дигидрофосфат кальция и другие водо- и кислоторастворимые продукты реакции. Концентрация их в жидкой фазе зависит, естественно, от температуры, концентрации и нормы расхода фосфорной кислоты. Процесс постепенно замедляется в результате нейтрализации фосфорной кислоты и заканчивается при насыщении жидкой фазы фосфатами кальция. Продолжительность стадии в производственных условиях может изменяться от нескольких секунд (камерно-поточный метод) до 3—10 мин (камерный процесс) и 1,0—1,5 ч (поточные способы).
Вторая стадия процесса сопровождается кристаллизацией моногидрата дигидрофосфата кальция, в результате чего составы жидкой и твердой фаз реакционной массы постепенно изменяются. В момент насыщения жидкой фазы дигидрофосфатом и гидрофосфатом кальция
реакция разложения останавливается.
В первой стадии процесса максимум скорости растворения апатита в фосфорной кислоте практически совпадает с положением узловой точки В (рис. 11.25). Концентрация ионов кальция в насыщенном растворе, образующемся у поверхности зерен фосфата, для точки В макси
Р2О5, %
Рис. 11.25. Сопоставление изохрон растворения апатита в частично нейтрализованных растворах фосфорной кислоты (при различных степенях нейтрализации z, %) с изотермой системы СаО—Р2О5— НгО при 40° С (линия АВС)
мальная, следовательно, и движущая сила диффузии этих ионов в растворе также наибольшая. Поэтому для достижения на первой стадии процесса высокой степени разложения следует применять фосфорную кислоту такой начальной концентрации, которая приведет к получению насыщенного раствора, близкого по составу к узловым точкам, равновесным моногидрату дигидрофосфата и гидрофосфату кальция. Например, для максимального разложения апатита в момент насыщения жидкой фазы при 75° С фосфорная кислота должна была бы иметь начальное содержание Р2О5 33,6%. В этом варианте степень разложения апатита составляет 36,5% при норме кислоты 110% от стехиометрической. Отсюда видно, что определяющими в достижении высокой степени разложения исходного фосфата явля
612
ются условия осуществления второй стадии реакции. Применительно к последней установлено, что при 30—75е С наибольшая скорость реакции достигается в растворах, содержащих 46—47% Р2О5, равновесных с дигидрофосфатом кальция.
В процессе получения двойного суперфосфата из апатитового концентрата камерным способом оптимальную концентрацию исходной фосфорной кислоты выбирают таким образом, чтобы к моменту достижения коэффициента разложения около 60% жидкой фазы камерного суперфосфата содержало 46—47% Р2О5. Этому условию отвечает начальное содержание кислоты (55% Р2О5), при которой максимальный коэффициент разложения исходного сырья в конце первой стадии при 75—100° С не может превысить 10% и скорость процесса разложения относительно мала. Когда коэффициент разложения апатита достигает ~60%, суспензия затвердевает и скорость реакции резко замедляется. Дальнейший процесс разложения идет в период вылеживания продукта на складе (15—30 сут). Оптимальная температура дозревания составляет 40—60° С.
Доломитизированные фосфориты разлагают в камерном процессе кислотой, содержащей 45—50% Р2О5. Процесс разложения идет быстрее и полнее, чем у апатитового концентрата. Это объясняется менее плотной структурой разрыхления шламовых покровов на неразложен-ных частицах исходного сырья диоксидом углерода, выделяющимся из карбонатов.
В бескамерных способах исходные фосфориты обрабатываются при 50—100° С фосфорной кислотой концентрацией 28—40% Р2О5, что значительно соответствует оптимальным условиям для первой стадии процесса. Однако низкая начальная концентрация кислоты неблагоприятна для второй стадии разложения, поскольку в растворах, равновесных с Са^РОд^ЬЬО + СаНРОд (рис. 11.25, точка В) или с СаНРОд, процесс практически не идет. Процесс заканчивается лишь при сушке реакционной массы. Извлечение из нее воды благоприятствует переходу системы в область, равновесную с дигидрофосфатом кальция и соответственно дальнейшему разложению апатита. В процессе сушки из реакционной массы кристаллизуется моногидрат дигидрофосфата кальция и степень нейтрализации жидкой фазы уменьшается, хотя и активность ее увеличивается. В результате достигается высокая степень разложения сырья и при незначительной степени его разложения в первой стадии.
Получение двойного суперфосфата. Схема получения двойного суперфосфата камерным способом аналогична схеме камерного способа получения простого суперфосфата и по последовательности технологических операций, и по используемому оборудованию (рис. 11.26).
613
Рис. 11.26. Схема получения гранулированного двойного суперфосфата из апатитового концентрата камерным способом:
1— бункер; 2 — дозатор; 3 — сборник Н?РО<; 4 — расходомер; 5 — смеситель; 6—шнековый смеситель; 7— суперфосфатная камера; 8— газопровод; 9 — ленточный транспортер; 10 — разбрасыватель; 11— грейферный кран; 12 — бункер для двойного суперфосфата; 13 — ленточный транспортер; 14 — дезинтегратор; 15 — барабанный нейтрализатор; 16 — элеватор; 17—грохот; 18— бункер; 19 — ленточный питатель; 20— гранулятор; 21 — барабанная сушилка; 22 — топка;
23 — дробилка
Согласно схеме, исходный апатитовый концентрат из бункера 1 дозатором 2 подается в смеситель 5. Параллельно исходная фосфорная кислота (54% Р2О5) из сборника 3 через расходомер 4 поступает в смеситель, где они перемешиваются. К образовавшейся суспензии добавляют до 3% от массы апатита известняка. Получаемую смесь шнековым смесителем 6 непрерывно загружают в суперфосфатную камеру 7. Схватившаяся в камере суперфосфатная масса («пирог») обладает значительной рыхлостью и пористостью (вследствие выделения в процессе разложения исходного известняка диоксида углерода) и легче выгружается из камеры с помощью фрезера. Продолжительность пребывания массы в смесителе 3—6 мин, в камере 1,0—1,5 ч, температура в смесителе 70—80° С, а в камере 90—100° С. Высота «пирога» в камере 1,2—2,0 м. Возможно внесение к фосфорной кислоте концентрированной серной кислоты, что позволяет уменьшить расход фосфорной кислоты, несколько снизить содержание в ней Р2О5 (до 51—52%). Степень разложения апатитового концентрата в камере не превышает 70%. Образующиеся фторсодержащие газы по газопроводу 8 отводят в абсорбционное отделение соединений фтора.
Камерный двойной суперфосфат ленточным транспортером 9 передается на склад, где разбрасывателем 10 и грейферным краном 11 перебрасывается для перемешивания. Далее грейферным краном 11 614
двойной суперфосфат загружается в бункер 12, откуда ленточным конвейером 13 подается в дезинтегратор 14 для измельчения. Измельченный суперфосфат в барабане 15 нейтрализуется молотым известняком и элеватором 16 подается в грохот 17. Крупная часть из грохота передается в дезинтегратор, стандартный же двойной суперфосфат ленточным конвейером — в бункер 18 для нейтрализо-
ванного продукта, откуда ленточным питателем передается в гранулятор 20. Образующиеся гранулы суперфосфата поступают в сушиль-
ный барабан 21. Сухой продукт направляется на упаковку.
Полученный камерным способом двойной суперфосфат содержит 43—44% усвояемого Р2О5. На производство 1 т гранулированного двойного суперфосфата (в пересчете на усвояемый Р2О5) расходуют 320—330 кг апатита и 810—820 кг фосфорной кислоты (в пересчете на Р2О5).
В настоящее время широко применяются методы получения двойного суперфосфата на основе экстракционной фосфорной кислоты, по-
лученной из апатитового концентрата и легко разлагающегося фосфорита (рис. 11.27). Фосфорит из бункера 1 ленточным транспортером подается весовым дозатором 2 в смеситель 3, куда одновременно через дозатор 4 поступает фосфорная кислота (28—36% Р2О5). Для получе-
ния хорошо текучей суспензии исходную кислоту (52—54% Р2О5) разбавляют до 34—36% P20j
растворами, образующимися в процессе промывки отходящих из аппаратов газов от уносимой ими пыли двойного суперфосфата. Из смесителя суспензия непрерывно поступает в реакторы 5, в которых реакционная масса перемешивается в течение 60—90 мин при 70—100° С. За этот период исходный фосфорит разлагается примерно на 50%, после чего реакция резко затормаживается
Рис. И.27. Поточная схема получения гранулированного двойного суперфосфата из фосфоритной муки и неупаренной экстракционной фосфорной кислоты:
1— бункер для фосфорита; 2 — ленточный весовой дозатор; 3 — смеситель; 4 — дозатор фосфорной кислоты; 5 — реакторы; 6—центробежный насос; 7— аппарат БГС; 8— топка; 9, 14—циклоны; 10 — элеваторы; 11 — грохот;
12 — дробилка; 13 — аммонизатор; 14 — циклон
из-за насыщения жидкой фазы гидрофосфатом кальция.
Процесс завершается при переработке суспензии в гранулированный продукт: суспензия пода-
615
Рис. 11.28. Барабанный гранулятор-сушилка (БГС):
1 — корпус вращающегося барабана; 2 — опорный ролик; 3 — упорный ролик; 4 — электродвигатель; 5 — редуктор; 6 — шестерня; 7 — обратный шнек; 8— подъемно-лопастная насадка; 9 — загрузочная камера; 10 — форсунка; 11 — труба для подачи внешнего ретура; 12 — патрубок для ввода теплоносителя; 13— приемно-винтовая насадка; 14 — зубчатый венец; 15 — подпорный конус;
16 — разгрузочная камера
ется насосом 6 в аппарат БГС (барабанный гранулятор-сушилка) 7, в который из топки 8 поступает топочный газ.
Аппарат БГС работает с внутренним и внешним ретуром. БГС представляет собой (рис. 11.28) вращающийся барабан 1, установленный с уклоном 1—3° к горизонту и опирающийся бандажами на опорные ролики 2. Упорные ролики 3 предотвращают сдвиг барабана. Вращение осуществляется от электродвигателя 4 через редуктор 5, шестерню 6 и зубчатый венец 14. В переднем конце внутри барабана расположена приемно-винтовая насадка 13, а по всей длине— подъемно-лопастная насадка 8 и обратный шнек 7, который возвращает ретур от подпорного конуса 15 в зону распыла суспензии. Внешний ретур подают по трубе 11. В переднем торце загрузочной камеры 9 установлены пневматические форсунки 10 для распыления подаваемой в аппарат суспензии сжатым воздухом (0,7—0,8 МПа). Влажность суспензии около 40%.
Теплоноситель входит в загрузочную камеру через патрубок 12. Подъемно-лопастная насадка создает завесу из свободно падающих высушиваемых гранул. В эту завесу направлен факел суспензии, мелкие капли которой оседают на гранулах. На поверхности гранул происходит кристаллизация из раствора, гранулы укрупняются, а затем высушиваются. На рис. 11.29 приведен график для определения количества ретура, необходимого в процессе гранулирования на 1 т двойного суперфосфата при влажности гранул в пределах 6—14%. При построении графика содержание воды в суспензии принято 40%, а влажность ретура 2%.
В загрузочной камере установлен цилиндр с тангенциальными щелевидными окнами для завихрения газа-теплоносителя, в потоке которого вводится и внешний ретур (мелкий) после дробления, и пы-616
левидный материал. Тангенциальный ввод газа образует вихревой свод порошкообразного материала, на ядро которого поступает суспензия из форсунки, после чего она попадает на массу гранул, поднимаемых приемно-винтовой насадкой, предотвращающей завал в передней части аппарата.
Ввод греющего газа и суспензии по оси аппарата снижает локальный перегрев его конструкций и продукта в горячей зоне и дает возможность доводить тем-
Рис. 11.29. График для определения количества ретура, необходимого в процессе гранулирования суспензии двойного суперфосфата
пературу теплоносителя до 950° С. Температура отходящего из БГС газа 120—125° С. Он очищается от пыли сначала в циклоне, а затем в процессе промывки в абсорбционной установке.
Сухие гранулы, выходящие из БГС с влажностью 2—3%, имеют температуру 100—105° С. Как показано на схеме (см. рис. 11.27), они элеватором 10 передаются в грохот 11, на котором отделяется фракция, соответствующая кондициям на целевой продукт (1—4 мм). Более крупная фракция после измельчения на дробилке 12 возвращается на грохот, а мелкая фракция в смеси с уловленной пылью — в аппарат БГС в качестве внешнего ретура. Ретурное число (отношение массы ретура к массе продукта) находится в пределах 1—2.
В аппарате БГС по мере испарения воды из суспензии увеличивается концентрация свободной фосфорной кислоты, уменьшается концентрация растворенного дигидрофосфата кальция, поэтому возрастает активность ионов Н+. Это обеспечивает частичное растворение шламового покрова гидрофосфата кальция и продолжается процесс разложения фосфорита. Коэффициент его разложения в высушенном двойном суперфосфате достигает 80—85%. Для уменьшения кислотности его нейтрализуют, опудривая гранулы во вращающемся барабане мелом, или аммонизируют. В барабанный аммонизатор 13 аммиак подают через коллектор под слой гранул. С целью удаления выделяющейся в процессе аммонизации теплоты в барабан с противоположного конца поступает воздух, подаваемый в несколько зон охлаждения, находящихся между зонами аммонизации. Температура выгружаемого из аммонизатора продукта 40—45° С. Уносимая воздухом пыль улавливается в циклоне 14, а затем — в процессе промывки от соединений фтора и аммиака — в абсорбционной установке.
617
Рис. 11.30. Механический абсорбер
Пыль из циклонов 9 и 14 присоединяется к ретуру. После аммониза
ции содержание свободной фосфорной кислоты в продукте снижает-
ся на 4—5% Р2О5 (43—47% усвояемого Р2О5).
В процессе сушки суспензии в поточных способах получения двойного суперфосфата в газовую фазу выделяется (в виде смеси HF и SiF4) 50—55% фтора, содержащегося в исходном фосфорите и экстракционной фосфорной кислоте. Значительные количества выделяемых газов и высокое содержание в них пыли усложняют процесс аб-
сорбции фторидов, ухудшают качество образующейся фторидкремниевой кислоты. Система очистки состоит из циклонов (для улавливания пыли) и абсорберов. По трехступенчатой схеме абсорбции применяют механические абсорберы (рис. 11.30) и абсорберы Вентури. В процессе очистки запыленных газов, а также в условиях выделения в результате гидролиза SiF4 осадка кремнегеля применяют абсорберы с псевдоожиженным слоем шаровой насадки (рис. 11.31, а) или пен-
Рис. 11.31. Схемы пенных абсорберов с шаровой насадкой (а) и со стабилизатором (б): 1 — слой шаров; 2 — оросительное устройство (распылитель); 3 — брызгоуловитель; 4 — решетки; 5 — стабилизатор пенного слоя
618
ных абсорберов со стабилизаторами пенного слоя (рис. 11.31, б) и решетками провального типа. В качестве стабилизатора, установка которого позволяет повысить скорость газа в аппарате, применяют сотовую решетку из вертикальных пластин.
Существует также камерно-поточный способ получения двойного суперфосфата. При этом способе применяют легко разлагаемую фосфоритную муку тонкого помола (содержание частиц крупнее 0,074 мм не должно превышать 20%) и фосфорную кислоту с содержанием 47—49% Р2О5. Измельченный камерный суперфосфат смешивают с ретуром, гранулируют и сушат. В процессе сушки степень разложения фосфорита возрастает от 60—70 до 80—90%. Товарную фракцию гранул нейтрализуют аммиаком, охлаждают и получают продукт, содержащий 45—47% общего, 42,5—44,5% усвояемого, 37—38% водорастворимого, 3—5% свободного Р2О5 и 1,5—2,0% азота.
На получение 1 т Р2О5 расходуют 824—835 кг фосфорной кислоты, 271—280 кг фосфоритной муки (в пересчете на Р2О5) и 35 KrNH3.
11.8. ПРЕЦИПИТАТ
Физико-химические свойства и применение. Преципитат — фосфорное удобрение и минеральная подкормка для сельскохозяйственных животных. Удобрение — тонкодисперсный порошок от белого до серого цвета с насыпной плотностью 0,86—0,87 г/м3. Негигроскопичен, не слеживается, не растворяется в воде. В зависимости от качества исходного фосфорного сырья содержит 32—35% Р2О5 в цитратно-растворимой форме.
Основой преципитата является дигидрат гидрофосфата кальция СаНРОд ^НгО (брушит), осаждающийся из водных растворов ниже 36° С.
Выше 36° С из водных растворов кристаллизуется гидроортофосфат кальция СаНРОд. Свойства гидрофосфатов кальция приведены в табл. 11.5.
Растворимость дигидрата гидроортофосфата кальция в воде (в 100 г): 0,025 (0°С), 0,133 (60° С). Хорошо растворяется в водных растворах цитратно-кислого аммония.
На рис. 11.32 приведен график зависимости паров воды дигидрата гидрофосфата кальция от температуры. На рис. 11.33 показана растворимость метастабильной фазы СаН2РОд-2Н2О и стабильной СаНРОд при 40° С, а на рис. 11.34 — скорость полиморфного превращения СаНРОд-2НгО -> СаНРОд при этой же температуре. Метаста-бильный брушит превращается в стабильную безводную соль. Процесс превращения ускоряется с повышением концентрации Р2О5 в 619
жидкой фазе. Потеря кристаллизационной воды в процессе фазового превращения брушита в растворах снижает (до 50%) содержание усвояемого (цитратно-растворимого) Р2О5 в продукте. При очень низких концентрациях Р2О5 (pH раствора от 6,4 до 8,0 и выше) в твердой фазе системы СаО—Р2О5—Н2О появляются более основные фосфаты (рис. 11.35).
Таблица 11.5. Свойства гидрофосфаггов кальция
Показатель СаНРО4 СаНРО4-2Н2О
Сингония Параметры элементарной ячейки, нм: Триклинная Моноклинная
а 0,690 0,581
b 0,665 0,518
с 0,700 0,624
Угол, град 96,3 (а) 103,9 (р) 88,7 (у) 116,4 (р)
Число формульных единиц в ячейке 4 4
Пространственная группа И Ja
Т ° С 1 пл» >900* 109"
Плотность, г/см3 2,89 2,306 (16° С)
С°р, Дж/(моль К) 110,11 197,1
АЯ^р, кДж/моль -1815, 69 -2405,31
ДС^р, кДж/моль -1682,46 -2156,31
5°98, Дж/(моль-К) 111,45 189,56
* Температура разложения. ** Температура обезвоживания.
Физико-химические основы получения преципитата. Преципитат получают осаждением его из фосфорной кислоты или фосфорно-кислотных растворов гидроксидом или карбонатом кальция. Преципитат получают в процессе утилизации растворов, образующихся в производстве фотожелатины на костеперерабатывающих заводах.
В процессе введения в раствор фосфорной кислоты водной суспензии смеси оксида и гидроксида кальция или известняка (мела) они образуют дигидрофосфат кальция:
2Н3РО4 + Са(ОН)2 = Са(Н2РО4)2 + 2Н2О
2НзРО4 + СаСОз = Са(Н2РО4)2 + СО2 + Н2О
620
Рис. 11.32. Давление водяных паров над СаНРО4-2Н2О
Рис. 1133. Растворимость метастабильной фазы СаНРО4-2Н2О и стабильной фазы СаНРО4 в системе СаО—Р2О5 — Н2О при 40° С
Рис. 11.34. Фазовое превращение СаНРО4-2Н2О в безводную соль при 40° С в зависимости от концентрации Р2О5 в жидкой фазе, равновесной с двуводной солью. Концентрация Р2О5: / — 18%; 2—16%; 3—14%; 4—12%; 5—10%
152050 200 400 600 10001200 СаО, мг/л
Рис 11.35. Система СаО—Р2О5 — Н2О при 25° С в области низких концентраций
При постепенной нейтрализации фосфорной кислоты в слабокислой среде дигидрофосфат кальция инконгруэнтно разлагается с выделением осадка дигидрата гидрофосфата кальция. Одновременно дигидрофосфат кальция реагирует с известью или карбонатом кальция, также образуя дигидрат гидрофосфата кальция:
Са(ОН)2 + Са(Н2РО4)2 + Н2О = 2[СаНРО4-2Н2О]
СаСОз + Са(Н2РО4)2 + 2Н2О = 2[СаНРО4-2Н2О] + СО2
Избыток извести сверхстехиометрического отношения CaO:P2Os в гидрофосфате кальция и соответственное увеличение pH раствора свыше 6,3 приводят к образованию фосфата кальция:
2СаНРО4 + Са(ОН)2 = Саз(РО4)г + 2Н2О
Избыток известняка не вызывает ретроградации преципитата. Содержащиеся в исходной фосфорной кислоте примеси также реагируют с известью или известняком:
H2SO4 + Са(ОН)2 = CaSO4-2H2O
H2SiF6 + ЗСа(ОН)2 = 3CaF2 + SiO2 + 4Н2О
(Fe, Д1ХН2РО4)з + 2СаСОН)2 + 2Н2О = = (Fe, А1)РО4-2Н2О + 2[СаНРО4-2Н2О]
Образующиеся илистые осадки фосфатов железа, алюминия, фторида кальция и кремневой кислоты затрудняют процесс фильтрации преципитата.
На диаграмме системы СаО — P2Os — Н2О при 40° С (рис. 11.36) показаны условия преципитирования фосфорной кислоты концентрацией 20% P20s (точка S’) водной суспензией смеси оксида и гидроксида кальция, содержащей 12% СаО (точка 5), и кислоты концентрацией 25% P20s (точка R') суспензией карбоната кальция с отношением Ж:Т, равным 1,5:1,0, и концентрацией оксида кальция около 22% (точка R). В процессе преципитирования состав системы изменяется вдоль соединительных прямых SS' и RR’.
В равновесных условиях в процессе нейтрализации кислоты до образования насыщенного раствора в точках а и а' жидкая фаза, состоящая из свободной НзРО4 и Са(Н2РО4)2, должна стать насыщенной по отношению к гидрофосфату кальция СаНРО4. На практике насыщение наступает позже, в точках b и Ь' на метастабильной ветви ML растворимости твердой фазы СаНРО4-2Н2О. Для ее кристал-622
70
СвО, %
Рис. 11.36. Диаграмма процесса осаждения дигидрата гидрофосфата кальция из фосфорной кислоты:
ОЕ—стабильная и ML — метастабильная ветви изотермы СаО—PjOj—HjO при 40° С; КТ—ветвь растворимости СаНРО, в системе СаО—Р2О,—H2SiF6—Н2О при 60° ,С
лизации необходимо образование растворов, пересыщенных метаста-бильной фазой. Интенсивное образование гидрофосфата кальция, по-видимому, начинается лишь в момент полной нейтрализации первого иона водорода фосфорной кислоты (точки с и с'), т.е. при растворении половинного количества оксида кальция, расходуемого на осаждение преципитата.
Как указывалось выше, кристаллизация гидрофосфата кальция происходит, по-видимому, в результате разложения дигидрофосфата кальция из-за инконгруэнтности состава равновесных его растворов по отношению к составу твердых фаз (луч растворения дигидрофосфата кальция 0NW проходит вне поля кристаллизации этой соли):
Са(Н2РО4)2 + 2Н2О = СаНРО4-2Н2О + Н3РО4
В указанных точках степень разложения дигидрофосфата кальция составляет соответственно 32,6 и 36,1%. Образование свободной фосфорной кислоты в момент спонтанной кристаллизации гидрофосфата кальция и является причиной интенсивного разложения оставшегося карбоната, не прореагировавшего к этому моменту.
623
10
Количество введенной Са(0Н)2, % от стехиометрической нормы
Рис 11.37. Изменение pH раствора в процессе преципитирования фосфорной кислоты
Изменение pH раствора и концентрации в нем Р2О5 в зависимости от количества введенной в систему суспензии оксида и гидроксида кальция показано на рис. 11.37. Как видно из рисунка, процесс кристаллизации дигидрата гидрофосфата кальция начинается после внесения в систему 50% извести от стехиометрического количества, когда значение pH раствора становится равным 3,0—3,2. В процессе введения суспензии смеси оксида и гидроксида кальция значение pH постепенно возрастает примерно до 4,8, а затем при добавке даже незначительного ее количества значение pH резко увеличивается, что вызывает образование фосфата кальция.
При pH около 4,8 первый ион водорода фосфорной кислоты нейтрализуется полностью. В этой точке происходит резкий перелом кривой pH, что показывает конец процесса преципитирования.
Получение преципитата. Согласно приведенной на рис. 11.38 схеме производства кормового преципитата (дигидрата гидрофосфата кальция), термическую фосфорную кислоту (70—75% Н3РО4) из сборника кислоты 7 центробежным насосом 2 перекачивают в напорную емкость 3, в которой ее нагревают до 70—80° С, и направляют в горизонтальный двухвальный смеситель 16. Одновременно в смеситель вводят расчетное количество тонкоизмельчен-ного мела и ретура.
Ретурное число, или кратность регура, т.е. отношение частиц ретура к массе продукта, составляет (0,5+1,0):1. После трехминутного перемешивания получается масса, содержащая 80% дигидрата гидрофосфата кальция. Продукт направляют во вращающийся барабан-до-зреватель 75, в котором в течение 1,5 ч при 45—50° С завершается 624
Рис. 11.38. Схема получения кормового преципитата:
1 — сборник фосфорной кислоты; 2 — центробежный насос; 3 — напорная емкость; 4 — ротаметр; 5 — бункер для мела; 6 — бункер для ретура; 7 — транспортер; 8— сепаратор; 9 — рукавный фильтр; 10— вентилятор; 11— дезинтегратор; 12 — элеватор ковшовый; 13— циклон; 14 — сушильный барабан; 15 — дозреватель; 16 — смеситель
процесс преципитирования. Выходящий из барабанного дозревателя преципитат содержит 4—6% дигидрата гидрофосфата кальция и ~18% воды. Продукт сушат в сушильном барабане 14 топочными газами до остаточной влажности 1,5—3,0%.
Процесс сушки проводят при параллельном движении топочного газа (с температурой на входе 550—650° С, на выходе менее 110° С) и дигидрата гидрофосфата кальция, температура которого не должна превышать 90° С.
Высушенный преципитат измельчают в дезинтеграторе 11, работающем в замкнутом цикле с сепаратором 8. Из сепаратора крупная фракция размером 2 мм возвращается на повторное измельчение. Часть мелкой фракции используют в качестве ретура, а часть направляют на упаковку.
Вместо термической фосфорной кислоты применяют также упаренную и очищенную от соединений фтора, мышьяка и свинца экстракционную фосфорную кислоту.
В процессе получения преципитата регулируют соотношение фосфорной кислоты и мела с корректировкой их расхода по значению pH и содержанию РгС^своб в суспензии.
Согласно существующим ТУ, кормовой дигидрат гидрофосфата кальция, получаемый из экстракционной фосфорной кислоты, должен содержать 44,1% Р2О5, растворимого в 0,4% НС1, и не более 0,2% фтора, 0,001% мышьяка и 0,002% свинца.
21 Химическая технология 04 J
неорганических веществ, кн. 1
11.9. ТЕРМИЧЕСКИЕ ФОСФАТЫ
К термическим фосфатам можно отнести продукты высокотемпературной переработки природных фосфатов с диоксидом кремния, карбонатом кальция, силикатами магния, щелочными соединениями и т.п. Такими продуктами являются обесфторенные фосфаты, термощелочные фосфаты, плавленые магниевые фосфаты и другие богатые фосфором соединения, например основные металлургические шлаки.
В процессе термообработки природных фосфатов в присутствии добавок происходит разрушение кристаллической решетки фторапатита. В результате образуются цитратно- и лимонно-растворимые фосфатные соединения и фосфат кальция Са3(РО4)2.
Термические фосфаты негигроскопичны, не слеживаются и содержат от 20 до 42% Р2О5. В зависимости от состава исходного фосфата усвояемая часть составляет 90—98% от всего количества Р2О5.
Обесфторенные фосфаты. Обесфторенные фосфаты получают термической обработкой природных фосфатов путем выделения из них фтора в газовую фазу в виде смеси HF и SiF4. Процесс обес-фторивания фосфатов ускоряется при термическом разложении их в присутствии водяного пара. Гидротермическая обработка апатитов при 1400—1550° С приводит к замещению фтора в кристаллической решетке исходного минерала гидроксид-ионом:
Ca5(PO4)3F + Н2О = Са5(РО4)3(ОН) + HF
Образовавшийся гидроксидапатит распадается:
2Са5(РО4)3(ОН) = 2Са3(РО4)2 + 4СаОР2О5 + Н2О
Продуктами распада являются лимонно-растворимые а-трикаль-цийфосфат и тетракальцийфосфат. Трикальцийфосфат существует в двух модификациях — а и 0, точка перехода которых равна И 80° С. Ниже этой температуры стабильна неусвояемая кристаллическая 0-мо-дификация, выше — аморфная усвояемая a-модификация. Сохранять аморфную a-форму можно быстрым охлаждением (закаливанием).
В присутствии диоксида кремния температура перехода a-формы в 0-форму снижается. Процесс обесфторивания значительно ускоряется, поскольку диоксид кремния способствует разрушению кристаллической структуры апатита. При этом образуются твердые растворы фосфата и силикатов кальция разного состава, что зависит от концентрации содержания диоксида кремния в шихте. Например, диоксид кремния принимает участие в разложении гидроксидапатита по реакции
4Са5(РО4)3(ОН) + SiO2 = 6Са3(РО4)2 + Ca2SiO4 + 2Н2О 626
Суммарную реакцию гидротермического разложения апатита в присутствии диоксида кремния условно представляют так:
2nCa5(PO4)3F + tfiSiCh + «Н2О = ЮиСаОЗиРгОзтиЗЮг + 2nHF
Исходная шихта из апатитового концентрата и 20—25% кремнезема (от массы апатита) плавится при более низкой температуре. Шихту с таким составом применяют для получения обесфторенных фосфатов методом плавления. Гидротермическое обесфторивание фосфатов (муки, концентратов) спеканием проводят в присутствии обесфторенной фосфорной кислоты до молярного отношения СаО:Р2О5 = 3 (4—6% Р2О5 от массы исходного фосфата). В присутствии фосфорной кислоты температура процесса снижается до 1380—1420° С. При этом параллельно с реакцией обесфторивания идет реакция:
3Ca3(PO4)F + Н3РО4 = 5Са3(РО4)2 + 3HF
Добавка фосфорной кислоты ускоряет процесс обесфторивания с одновременным повышением содержания Р2О5 в продукте. Присутствие фосфорной кислоты низкоплавких фосфоритов повышает температуру их плавления, что дает возможность обесфторивать их термообработкой во вращающихся печах или в печах КС при 1250—1300° С. Применяется также способ обесфторивания низкоплавких фосфоритов плавкой их в циклонной или в конверторной печи при 1500—1600° С.
Обесфторивание фосфатов термообработкой проводят во вращающейся печи (рис. 11.39). Согласно схеме, исходный фосфат (апатит или фосфорит) из бункера 1 через дозатор 2 поступает в смеситель 3, в который из сборника 4 подают исходную фосфорную кислоту. Образующаяся в смесителе суспензия направляется в дисковый гранулятор 5. Гранулы сушатся в барабанной сушилке 6 и направляются во вращающуюся печь 7, оборудованную газовой форсункой 8. Выходящие из печи зерна клинкера после охлаждения в рекуператорах печи до 70—80° С транспортером 9 направляются в бункер для клинкера 10, откуда подаются для измельчения в дезинтегратор 11. Далее измельченный продукт элеватором 12 передается в бункер готового продукта 13, упаковывается в упаковочной машине 14 и направляется на склад готовой продукции. Газы из барабанной печи проходят циклоны 16. Осаждаемая в циклонах пыль и пыль из печи попадают на транспортер 15 и передаются на смешение с шихтой. Горячий воздух после циклона направляется в абсорбционные башни 17, после которых, проходя через санитарную башню 19 и выхлопную трубу 20, вентилятором 23 выбрасывается в атмосферу. Образу-21. 627
Рис. 11.39. Схема получения обесфторенного фосфата термообработкой во вращающейся печи:
1— бункер фосфата; 2 — дозатор; 3—двухвальный смеситель; 4 — емкость фосфорной кислоты; 5 — дисковый гранулятор; 6 — барабанная сушилка; 7 — вращающаяся печь; 8— газовая форсунка; 9 — транспортер; 10 — бункер для клинкера; 11 — дезинтегратор; 12 — элеватор; 13 — бункер для готового продукта; 14 — упаковочная машина; 75 — транспортер пыли; 16 — барабанный циклон; 17 — абсорбер; 18— брызгоуловитель; 19 — санитарный абсорбер; 20 — выхлопная труба;
21 — сборники; 22—центробежный насос; 23 — вентилятор
ющиеся слабые водные растворы фторидов после абсорберов перекачиваются в цех переработки фторидов, а потребляемая в санитарном абсорбере 19 вода направляется на водооборот. Степень обесфторива-ния фосфата рассмотренным способом достигает 94—96%.
Разработана технология обесфторенного фосфата циклонной плавкой (рис. 11.40). Исходная фосфоритная мука из бункера 1 элеваторами 2 передается в бункер 3, откуда тангенциально поступает в циклонную печь 4. В цилиндрическую циклонную камеру поступает природный газ и подогретый (440—480° С) воздух, которые вводятся форсунками 5 так, что поступающая фосфоритная мука, попадая на стенки, плавится и стекает вниз к выходу.
На плавление расходуется около 25% теплоты, получаемой от сжигания газа, до 70% уносится с газом из циклона. Поэтому печь совмещена с паровым котлом и является единым энерготехнологическим агрегатом. В нем генерируется 3—4 т пара на 1 т целевого продукта, и общий коэффициент использования теплоты достигает 90%.
Вытекающий из циклонной камеры расплав гранулируется водой, а образующаяся при этом смесь передается в отстойник 21. Гранулы, отделяясь от воды, через бункер 22 поступают в барабанную сушилку 23. 628
Рис. 11.40. Схема получения обесфторенного фосфата плавлением фосфорита в циклонном энерготехнологическом агрегате:
1 — бункер фосфоритной муки; 2 — элеваторы; 5, 22 — бункеры; 4 — циклонная печь; 5 — форсунки для ввода топлива; 6 — сепаратор расплава; 7 — радиационный паровой котел (камера охлаждения) с охлаждающими элементами; 8—пароперегреватель; 9 — водяной экономайзер; 10— подогреватель воздуха; 11— камера обеспыливания; 12 — электрофильтр; 13— коллектор; 14 — скруббер Вентури; 15 — промывная башня; 16 — брызгоуловитель; 17 — выхлопная труба; 18— сборник раствора карбоната натрия; 19— сгуститель; 20 — центрифуги; 21 — отстойник;
23 — барабанная сушилка; 24 — шаровая мельница; 25 — бункер готового продукта
Высушенный продукт из сушилки направляется в шаровую мельницу 24 на измельчение, а после — в бункер готового продукта 25.
Отходящий от циклонной печи газ, проходя сепаратор расплава 6, направляется в радиационный котел (камера охлаждения) с охлаждающими элементами, откуда в пароперегреватель 8, водяной экономайзер 9 и поступает в подогреватель воздуха 10. После камеры обеспыливания (циклоны) 11 воздух проходит тонкую очистку в электрофильтре 12 и направляется, проходя коллектор 13, в скруббер Вентури 14, где воздух орошается растворами карбоната натрия. Воздух дополнительно орошается водой в башне 75 и через брызгоуловитель 16 выбрасывается в атмосферу выхлопной трубой 17.
Способ обеспечивает получение продукта, отвечающего требованиям существующего ГОСТ 23999—80. Продукт содержит в своем составе не более 0,2% F, 0,0002% As и 0,002% РЬ.
В промышленности производят термощелочные фосфаты, которые получают спеканием измельченных природных фосфатов с солями щелочных металлов и минералами, например карбонаты калия и натрия, сульфаты и гидросульфаты натрия и калия, щелочные шлаки после обессеривания чугуна, нефелины и др. Температура спекания зависит от состава смеси: смесь измельченного природного фосфорита и карбоната натрия подвергают термообработке во вращающихся печах при 629
1100—1250° С, смесь апатитового концентрата, сульфата натрия и углерода в соответствии 1:0,6:0,3—при 1000—1200° С, а апатита, диоксида кремния и сульфатов кальция и магния — при 1350—1400° С.
Щелочное разложение фосфатов отличается простотой. При этом можно использовать необогащенное низкокачественное («бедное») сырье и не требуется применения кислоты.
Химизм образования лимонно-растворимого фосфат-ренанита термообработкой природных фосфатов с карбонатом натрия может быть представлен уравнением
Ca3(PO4)3F + 2Na2CO3 + SiO2 = 3CaNaPO4 + Ca2SiO4 + 2CO2 + NaF
Производство осуществляется термообработкой шихты при 1100—1250° С с последующим охлаждением, дроблением и размолом полученного клинкера до частиц размером 0,15 мм.
В виде фосфорсодержащих удобрений применяют также металлургические шлаки, содержащие фосфаты, растворимые в 2%-ной лимонной кислоте. Образующийся в процессе удаления фосфора из доменного чугуна путем добавления доломита и известняка томасш-лак содержит тетракальцийфосфат и силикофосфаты кальция и железа. Процесс описывается уравнениями:
2Fe3P + 6О2 = 4FeO + РегО3 + Р2О3
Р2О3 + 4СаО = Са4Р2О9
Затвердевшие шлаки дробят и размалывают.
Известно также получение матафосфата кальция путем обезвоживания однозамещенных ортофосфатов при 275—300° С:
Са(Н2РО4)2-Н2О = Са(РО3)2 + ЗН2О
На этом основаны другие способы получения метафосфата кальция обработкой природного фосфора серной или фосфорной кислотой или их смесью и термообработкой суспензии при 350—400° С. В процессе обработки полигалита экстракционной фосфорной кислотой с последующей упаркой и термообработкой суспензии образуется смесь метафосфатов калия, кальция и магния.
Разработан также способ получения метафосфата кальция взаимодействием фосфорного ангидрида с двух- или трехзамещенными фосфатами при 1000—1200° С:
Са3(РО4)2 + 2Р2О5 = ЗСа(РО3)2
630
11.10. ФОСФАТЫ АММОНИЯ
Физико-химические свойства, применение. К фосфатам аммония относятся дигидроортофосфат (моноаммонийфосфат) NH4H2PO4, гидроортофосфат (диаммонийфосфат) (NH^HPC^ и ортофосфат (триаммонийфосфат) (NH4)3PO4; конденсированные (дегидратированные) фосфаты аммония — поли- и циклофосфаты. Дегидратированные фосфаты обычно представляют собой смесь орто- [(ТЧН4)„Нз^РО4], диорто- [(NPLOnPU-nPiO?], Триполи- [(NH4)nH5.„P3Oio], циклофосфатов с циклическим анионом [(NH4PO3),,].
Полифосфаты аммония малогигроскопичны и обладают достаточной термической устойчивостью — при нагревании до 100° С аммиак из них не выделяется.
Полифосфаты аммония могут быть получены:
1) высокотемпературной аммонизацией ортофосфорной кислоты;
2) аммонизацией полифосфорной кислоты при атмосферном или повышенном давлении;
3) дегидратацией ортофосфатов аммония;
4) взаимодействием пентаоксида фосфора и аммиака.
Полифосфаты аммония, получаемые первым и вторым способами, состоят в основном из NH4H2PO4, (ЫНдЬНРгО? и (NH4)3P2O7 с незначительным содержанием триполифосфата аммония и других конденсированных фосфатов. В процессе длительного хранения этих продуктов протекает рекристаллизация, которая приводит к образованию моноаммонийфосфата и трехзамещенного пирофосфата аммония.
В процессе взаимодействия Р2О5 и NH3 продукт содержит около 10% орто-, 50% пиро-, Триполи-, пентаполифосфатов и 40% более высококонденсированных форм.
Процесс образования линейных полифосфорных кислот из ортофосфорной может быть представлен общим уравнением:
ИН3РО4 -> Н„ + гРиОзи +1 + (п - 1)НгО
при и = 2 образуется пирофосфорная кислота:
2Н3РО4 -> Н4Р2О7 + Н2О
при п = 3 образуется триполифосфорная кислота:
ЗН3РО4 —> Н5Р3О10 + 2НгО и т.д.
Аммонизация полифосфорной кислоты сопровождается взаимодействием входящих в ее состав кислот с аммиаком по аналогичным реакциям.
631
В процессе нагревания дигидрофосфата аммония протекают следующие реакции: при 150—160° С
NH4H2PO4 -> NH3 + Н3РО4
при 160—170° С
2NH4H2PO4 -> NH3 + NH4H3P2O7 + Н2
при 170—299° С
3NH4H2PO4 -» 2NH3 + NH4H4P3O10 + 2Н2
3NH4H2PO4 -> NH3 + 2NH4H4P3O10 + Н2
4NH4H2PO4 -» 3NH3 + NH4H5P4O13 + ЗН2О
2NH4H3P2O7 -> NH3 + NH4H5P4O13 + Н2О
2NH4H2PO4 -> (ЫНОгНгРгСЪ + Н2О
NH4H3P2O7 + NH3 (NH4)2H2P2O7
при 299—350° С
5NH4H2PO4 -► NH4H6P50i6 + 4NH3 + 4Н2О
3NH4H2PO4 -> (МН4РОз)з + ЗН2О
Дигидроортофосфат аммония — бесцветные кристаллы (табл. 11.6) с тетрагональной решеткой (а = 0,750 нм, с = 0,755 нм); = 35,6 кДж/моль; 5®98 = 152 Дж/(моль-К). При 190,5° С дигидрофосфат плавится с незначительной потерей аммиака (идет его медленная дегидратация с образованием полифосфатов аммония).
Гидроортофосфат аммония (ЫРЦ^НРС^ — бесцветные кристаллы с моноклинной решеткой (а = 1,0735 нм, b = 0,6689 нм, с = 0,8000 нм, Р = 109,72°). Разлагается с выделением аммиака, давление разложения (Па): 26,7 (50° С), 146,8 (70° С), 760 (90° С).
Ортофосфат (триаммонийфосфат) (МН4)зРО4 — бесцветное твердое вещество, при 30—40° С разлагается с выделением NH3:
(МН4)зРО4 —30~40'с..» (NH4)2HPO4 + NH3, поэтому его не производят. 632
Таблица 11.6. Свойства фосфатов аммония
Показатель NH4H2PO4 (NH4hHPO4 (NH4)3PO4
Т °C 1 ПЛ» V' 192,0 (с разл.) — —
Плотность, г/см3 1,803 1,619 —
„ 20 пд 1,479 1,530 —
С", Дж/(мольК) 142,0 182,13 230,1
кДж/моль -1445,0 -1565,7 -1671,0
pH 0,1 М раствора 4,4 8,0 9,4
Содержание азота, % 12,2 21,2 21,2
Содержание Р2О5, % 61,8 53,8 47,6
Давление паров аммиака при 100° С над NH4H2PO4 практически равно нулю, над (NHOiHPC^— 1,2 и над (МН4)зРО4— 85,7 кПа. При 125° С давление NH3 над этими солями возрастает соответственно до 0,008, 4,5 и 157 кПа.
При 20° С насыщенный водный раствор содержит: 27,2% NH4H2PO4 или 40,8% (МНд^НРОд; при температуре кипения (109,4° С) —71,8% NH4H2PO4. Изотермы растворимости в системе NH3—Н3РО4—Н2О приведены на рис. 11.41.
Из диаграммы растворимости в системе NH4H2PO4—(МРЦ^НРОд—Н2О при 25° С (рис. 11.42) видно, что с повышением концентрации дигидрофосфата растворимость гидрофосфата практически не изменяется. С повышением же концентрации гидрофосфата растворимость дигидрофосфата растет, и она достигает максимума при молярном соотношении NH3:H3PO4~1,5.
Рис. 11.41. Изотермы растворимости в системе NH3—Н3РО4—Н2О при 25 и 75° С
Рис. 11.42. Диаграмма растворимости в системе NH4H2PO4—(NH^HPO,—Н2О при 25° С
633
Значения pH 0,1М раствора равны: для дигидрофосфата аммония— 4,4, для гидрофосфата аммония — 8,0 и для ортофосфата аммония (триаммонийфосфата) — 9,4.
Моно- и дигидрофосфаты малогигроскопичны. Гигроскопическая точка дигидрофосфата при 50° С равна 88%, а при 15° С — 97%.
Фосфаты аммония широко применяются в сельском хозяйстве в качестве высококонцентрированных безбалластных удобрений, содержащих азот и фосфор. Гидрофосфат аммония, полученный из термической или очищенной экстракционной фосфорной кислоты, применяют для подкормки скота.
Дигидрофосфат аммония является основным компонентом аммофоса, а гидрофосфат — диаммофоса. Они являются составной частью флюсов в процессе пайки олова, бронзы, меди, цинка. Кристаллический дигидрофосфат аммония — пьезо- и сегнетоэлектрик, а гидрофосфат является антипиреном для древесины, бумаги, тканей. Применяют его также для приготовления пищевых дрожжей и фосфатирования металлов.
Согласно действующему ГОСТ 18918—85, промышленность выпускает аммонийно-фосфорное удобрение — аммофос (дигидрофосфат аммония с 10%-ным содержанием гидрофосфата аммония). Стандартом предусмотрен выпуск продукта двух марок: А и Б. Продукт марки А высшей и первой категорий качества и марки Б высшей и первой категорий качества соответственно должен содержать: не менее 52, 50 ± 1, 44 и 42±1% Р2О5 усв, 48, 46, 34 и 32% Р2О5 вод, 12±1, 12±1, 11±1 и 10±1% N и не более 1% Н2О. Доля гранул с размерами 1—4 мм должна быть не менее 95% для высших и 90% для первых категорий качества. Соотношение N:P2Oj в аммофосе ~1:4.
Физико-химические основы получения фосфатов аммония и аммофоса. Процесс получения фосфатов аммония основывается на следующих реакциях:
Н3РО4(ж) + NH3(r) = NH4H2PO4(tb) + 147 кДж,
Н3РО4(ж) + 2NH3(r) = (NH4)2HPO4(tb) + 215 кДж.
Наибольшее выделение в твердую фазу дигидрофосфата достигается при проведении процесса по лучу АВ (см. рис. 11.41). В процессе нейтрализации 40%-ной фосфорной кислоты аммиаком выход кристаллов невелик даже при 25° С (точка С). В процессе же нейтрализации концентрированной фосфорной кислоты (75% Н3РО4) состав системы, соответствующей точке D, и количество образующейся твердой фазы велико даже при температуре массы выше 75° С. Этому кроме концен-634
трации исходной кислоты и более высокой температуры способствует и испарение части воды за счет теплоты реакции.
В производстве аммофоса применяют экстракционную фосфорную кислоту, содержащую примеси. Поэтому в процессе нейтрализации ее аммиаком при рН>3 выделяются средние фосфаты железа и алюминия типа КРОд-гНгО, железоаммонийфосфаты — NHXFe.AlXHPO^’O.SFbO и другие, дигидрат дикальцийфосфата СаНРО^НгО, гипс, фториды и фторосиликаты, магнийаммонийфосфат NHjMgPCU-FbO, а в жидкой фазе образуется сульфат аммония.
В процессе нейтрализации фосфорной кислоты аммиаком образуются также кислые суспензии, содержащие кристаллы фосфатов аммония, свободную фосфорную кислоту, воду и осаждающиеся примеси. Количество и состав компонентов суспензии по мере абсорбции аммиака и повышения температуры непрерывно меняются, как и ее свойства — величина pH, растворимость твердых фаз, вязкость и др.
Равновесное давление аммиака над насыщенным раствором зависит от молярного отношения NH3:H3PO4 (рис. 11.43). От него же зависит и величина водородного показателя pH (рис. 11.44), по которому регулируют ход процесса. При 25° С максимальными значениями плотности и вязкости обладают насыщенные растворы с молярным отношением NH3:H3PC>4 к 1,45. Изменение состава суспензий ощутимо влияет на температуру кипения их жидких фаз, что должно конкретно учитываться при выборе оптимальных технологических режимов процессов концентрирования и обезвоживания.
Рис. 11.43. Зависимость давления NH3 над насыщенными растворами фосфатов аммония от молярного отношения NH3:H3PO4
Рис. 11.44. Зависимость величины pH насыщенных растворов фосфатов аммония от молярного отношения NH3:H3PO4 при 65—75° С
635
Получение дигидрофосфата и гидрофосфата аммония. Гидрофосфат аммония (диаммонийфосфат) — концентрированное азотно-фосфорсодержащее удобрение. Получают его, применяя ортофос-форную кислоту, полученную как из апатита, так и на базе фосфоритов. При зюм получают продукт, содержащий соответственно до 45% Р2О5 Усв, 43—45,5% Р2О5 и 18% N из апатитовой кислоты и 41% Р2О5 усв, 32,4% Р2О5 .ад и 13,5% N из кислоты, полученной из фосфоритов.
Производится также кормовой диаммонийфосфат, содержащий 52±1% Р2О5 и не менее 19% N, растворимых в 0,4%-ном растворе НС1, и вредных примесей (в %, не более): 0,006 As, 0,02 тяжелых металлов (в пересчете на РЬ) и 0,1 F.
Диаммофос получают из концентрированной ортофосфорной кислоты по схеме с применением аппарата АГ аналогично производству аммофоса и с применением вакуум-кристаллизаторов. По схеме с АГ исходную ортофосфорную кислоту с содержанием 37—40% Р2О5 используют для поглощения аммиака из отходящих газов, а затем ее нейтрализуют аммиаком в две стадии. Первую стадию нейтрализации в сатураторах завершают с достижением молярного отношения NH3:H3PO4 = 1,35—1,40 (pH 5,0—5,8, р = 1,4—1,42 г/см3). По условиям растворимости в указанном отношении образуется достаточно текущая суспензия с минимальным содержанием твердой фазы. Температуру в сатураторе поддерживают в пределах 105—117° С, а влажность — 20—26%.
Вторую стадию нейтрализации ведут в аппарате АГ, в котором молярное отношение поднимается до NH3:H3PO4 = 1,9—2,0. В процессе нейтрализации термической фосфорной кислоты добавляют небольшое количество серной кислоты для улучшения прочности гранул целевого продукта за счет пластифицирующих свойств образующегося сульфата аммония. Максимальной температурой процесса в аппарате АГ является 80° С, что диктуется нестойкостью гидрофосфата аммония. Последний, выше указанной температуры, разлагается с выделением аммиака. Обычно температуру в аппарате АГ поддерживают около 75° С. Из аппарата гранулы диаммонийфосфата влажностью 2—4% направляют в барабанную сушилку, где они сушатся топочными газами до остаточной влажности 0,8—1,0%. Температура продукта в процессе сушки 62—75° С. Целевой продукт после классификации на грохоте охлаждают до 25—27° С и направляют на упаковку.
Существует и другой вариант схемы, согласно которой исходную ортофосфорную кислоту, содержащую 47—48% Р2О5, нейтрализуют аммиаком до отношения NH3:H3PO4®0,7. Образующуюся суспензию смешивают с маточным раствором и направляют в вакуум-кристаллизатор, где аммонизируют до отношения NH3:H3PO4 = 1,7. Охлаждае-636
мую суспензию сгущают на аппарате Дорра, кристаллы гидрофосфата аммония отделяют на центрифуге и сушат в прямоточной барабанной сушилке. Образующиеся маточные растворы направляют на начало процесса для смешения с первичной суспензией.
Процесс получения гидрофосфата аммония проводят в одну стадию, согласно которой 75—85%-ную термическую фосфорную кислоту и исходный газообразный аммиак непрерывно подают в сатуратор, в который одновременно поступает маточный раствор. Температура 60—70° С поддерживается испарением воды, что достигается продувкой воздуха в реакционную массу. Образующиеся при этом кристаллы гидрофосфата аммония выводят в виде суспензии кристаллов на центрифугу, после которой кристаллы направляют на сушку, а маточный раствор — на начало процесса.
Для получения более чистых сортов фосфатов аммония, применяемых в пищевой и фармацевтической промышленности, пользуются термической фосфорной кислотой, содержащей не менее 77% основного вещества. Для получения дигидрофосфата аммония исходную кислоту смешивают с маточным раствором в реакторе с метальным устройством, и после нейтрализации до молярного соотношения МН3:НзРО4~1 образующийся водный раствор дигидрофосфата аммония охлаждают в кристаллизаторе. Образующаяся суспензия кристаллов в центрифуге разделяется на кристаллы и маточные растворы. Кристаллы сушат в барабанных сушилках, а маточный раствор возвращают в реактор.
Процесс получения гидрофосфата аммония ведут в две стадии, так как при подаче расчетного количества аммиака реакционная масса сильно разогревается и образуется слишком густая суспензия, что приводит к потерям исходного аммиака. Термическую кислоту (47—48% Р2О5) нейтрализуют аммиаком до отношения КН3:НзРО4~0,7, после чего суспензию смешивают с маточным раствором и направляют в вакуум-кристаллизатор, в котором массу аммонизируют до требуемой степени. Охлажденную суспензию сгущают, кристаллы гидрофосфата аммония отделяют от маточных растворов и сушат в прямоточной барабанной сушилке. Маточные растворы возвращают в реактор.
При получении фосфатов аммония нейтрализацией экстракционной фосфорной кислоты аммиаком выделяющиеся в осадок примеси остаются в целевом продукте, загрязняя тем самым его и снижая содержание основного вещества. Для получения более чистого продукта из экстракционной фосфорной кислоты процесс нейтрализации ведут в две стадии. В первой стадии неупаренную кислоту нейтрализуют до pH 4—4,5. При этом в осадок выделяется значительная часть примесей, которые отделяют от основного раствора путем фильтрования.
637
Раствор дигидрофосфата аммония упаривают под вакуумом до содержания 34—36% Р2О5. Кристаллы дигидрофосфата аммония получают охлаждением упаренного раствора до 18—20° С, которые отделяют на центрифуге от маточных растворов, промывают чистой водой и направляют на сушку. Сушат дигидрофосфат при 100—110° С. Маточный раствор возвращают на процесс упарки.
Для получения гидрофосфата аммония выпаренный раствор дигидрофосфата аммония дополнительно насыщают аммиаком в реакторе до рН~8. Во избежание потери аммиака процесс насыщения ведут при температуре ниже 80° С. Затем раствор гидрофосфата аммония направляют на кристаллизацию, центрифугирование и сушку. Гидрофосфат аммония сушат при 60° С для предотвращения потерь им аммиака и перехода в дигидрофосфат аммония.
Получение аммофоса. Аммофос — высококонцентрированное гранулированное азотно-фосфорное удобрение. Он состоит из дигидрофосфата аммония (не менее 80%), гидрофосфата аммония (около 5%) и примесей — (NH4)2SO4, фосфатов магния, железа, алюминия и др. В зависимости от вида сырья, из которого производят ортофос-форную кислоту (апатит, фосфориты разных месторождений), содержание питательных веществ в аммофосе составляет: 10—12% азота, 42—52% усвояемого Р2О5, в том числе 34—48% водорастворимого. Влажность готового продукта не более 1%, количество в нем гранул размерами 1—4 мм — не менее 90%. Аммофос обладает наилучшими из всех выпускаемых фосфорсодержащих удобрений физико-химическими свойствами. Широко применяется в качестве компонента сухих тукосмесей, а также входит в состав многих сложных и сложно-смешанных удобрений.
В производстве аммофоса применяют следующие схемы получения:
1) нейтрализация неупаренной (20—30% Р2О5) экстракционной фосфорной кислоты с последующим обезвоживанием суспензии в сушилках (распылительных, барабанных или с кипящим слоем);
2) нейтрализация неупаренной (20—30% Р2О5) фосфорной кислоты с последующей упаркой аммофосной суспензии, гранулированием и сушкой продукта в барабанных грануляторах-сушилках (БГС);
3) нейтрализация концентрированной (48—54% Р2О5) упаренной экстракционной фосфорной кислоты. В этом варианте нейтрализацию ведут в две стадии при атмосферном давлении — сначала в реакторах, затем в барабанных аммонизаторах-грануляторах (АГ) или в одну стадию при повышенном давлении с последующей сушкой образующейся суспензии распылением в башнях или в аппаратах БГС.
638
Рис. 11.45. Схема получения аммофоса с использованием распылительной сушилки:
1 — реактор-сатуратор, 2—-сборник суспензии; 3— центробежный насос; 4 — дозатор суспензии; 5 — распылительная сушилка; 6 — циклон; 7—вентилятор; Я —абсорбер; 9 —шнек; 10 — дробилка; 11 — элеватор; 12 — бункер; 13 — двухлопастный смеситель; 14—окаточный барабан;
15 — барабанная сушилка; 16 — двухситный грохот; 17—холодильник КС; 18 — транспортер.
Схема получения аммофоса с использованием распылительных сушилок приведена на рис. 11.45. Исходную экстракционную фосфорную кислоту (22—28% Р2О5) нейтрализуют аммиаком непрерывным способом в каскадно расположенных трех реакторах 1 при 80—115° С до молярного отношения NH3:H3PO4 не более 1,1 (рН<5). Часть образующейся при этом подвижной суспензии (70—80%) с температурой 100—105° С из третьего реактора (сборника 2) центробежным насосом 3 через дозатор 4 перекачивают в распылительную сушилку 5. Одновременно в верхнюю часть сушилки подают топочные газы, образующиеся в процессе сжигания газообразного или жидкого топлива (в схеме не приведен). Выходящие из сушилки газы с температурой 100—115° С проходят для очистки от пыли циклон 6 и абсорбер 8, после чего выбрасываются в атмосферу.
Высушенный порошкообразный аммофос с влажностью до 1% после сушилки шнеком 9 передается в дробилку 10 и элеватором 11 загружается в бункер 12. Из бункера аммофос поступает в двухлопастный смеситель 13, в который одновременно поступает мелкая фракция целевого продукта и остальная часть суспензии из реакторов. Из двухлопастного смесителя влажные (10—12% Н2О) гранулы аммофоса направляют в окаточный барабан 14, а оттуда — в барабанную сушилку 75. Процесс сушки проводят дымовыми газами с температурой 350° С.
Высушенные гранулы рассеивают. Фракцию с размером зерен более 3,2 мм измельчают и передают на рассев или растворяют в фосфорной кислоте и возвращают в процесс аммонизации. Мелкую фракцию с частицами менее 1 мм направляют на процесс грануляции, а фракцию с частицами 1—3,2 мм направляют на упаковку в качестве целевого продукта. Продукт, полученный из апатита, содержит, в%: РгС^общ 52; Р2О5 усв 51; Р2О5 Вод 50; N 12; Н2О 1; F 3,5.
Более прогрессивным методом переработки аммофосной суспензии считается, когда процесс ее сушки совмещается с гранулированием в аппаратах БГС или БСГХ — барабанных грануляторах — сушилках-холодильниках.
В настоящее время широко применяется схема получения аммофоса с промежуточной упаркой аммофосной суспензии. Процесс нейтрализации очищенной от фтора экстракционной фосфорной кислоты газообразным аммиаком ведут до pH 4,4—4,7 в аппарате САИ — скоростном аммонизаторе-испарителе (рис. 11.46), состоящем из сепаратора 1, циркуляционной трубы 2 и реакционной трубы 3 (диаметром 0,6 и высотой 6 м), снабженным патрубками для ввода кислоты и аммиака и внутренней насадкой типа Вентури. Реакционная труба тангенциально входит в сепаратор 1, в котором происходит отделение паров воды, отсасываемых через верхний штуцер. В процессе 640
Отходящие газы
Рис. 11.46. Скоростной аммонизатор-испаритель. (САИ):
1 — сепаратор; 2 — циркуляционная труба; 3 — реакционная труба
нейтрализации фосфорной кислоты аммиаком в реакционной трубе выделяется теплота, образующаяся суспензия нагревается до 100—110° С, вскипает и выбрасывается в сепаратор, из которого часть суспензии отводится на дальнейшую переработку, а остальная часть стекает по циркуляционной трубе 2. Интенсивное перемешивание и испарение влаги идет за счет теплоты реакции. Многократно циркулируя через реакционную и циркуляционные трубы, суспензия нейтрализуется. Продолжительность пребывания суспензии в аппарате 2—3 мин.
Из аппарата САИ суспензия аммофоса поступает в трехкорпусную выпарную установку 1 (рис. 11.47), в которой она упаривается от 50—65 до 23—35% влажности. Трехкорпусная выпарная установка состоит из трех выпарных аппаратов с принудительной циркуляцией и доупарива-
теля. Исходная суспензия подается в корпус III установки, в котором частично упаривается за счет самоиспарения и обогрева вторичным паром корпуса II, а также конденсатом сокового пара корпуса I и до-упаривателя, поступающего в греющую камеру корпуса III через гидрозатвор. Из корпуса III суспензия перекачивается во II, обогреваемый вторичным паром первого корпуса и доупаривателя. Из корпуса II упаренная суспензия перекачивается в корпус I выпарной батареи. Упариваясь, суспензия проходит через все корпуса и поступает в до-упариватель. При этом содержание влаги в суспензии последовательно снижается до 41—43, 33—37, 31—25 и 26—22%. Доупариватель и корпус I выпарной установки работают под избыточным давлением, II и III—под вакуумом. Греющий пар давлением 0,3—0,6 МПа подают в греющие камеры доупаривателя и корпуса III, в греющих камерах корпусов II и I используют соковый пар. Конденсат сокового пара собирают в сборнике и промывают им фосфогипс в цехе экстракционной фосфорной кислоты. Необходимое разрежение в выпарной батарее поддерживают за счет откачки неконденсирующихся в барометрическом конденсаторе газов из аппарата вакуумным насосом.
Для снижения вязкости упаренной суспензии в доупаривателе поддерживают температуру 116—119° С и давление 210—240 кПа.
Упаренная суспензия из обогреваемого сборника 9 с метальным устройством подается в аппарат БГС 21 на грануляцию и сушку. Температура топочных газов на входе в БГС 600—650° С, а на выхо-641
Рис. 11.47. Схема получения аммофоса выпариванием суспензии, сушкой и гранулированием в аппарате БГС:
1— выпарные аппараты; 2 — барометрический конденсатор; 3 — скоростной аммонизатор-испари-тель; 4 — теплообменник; 5,7 — сборники; 8 — емкость для конденсата и воды; 9 — сборник упаренной суспензии; 10 — элеваторы; 11 — грохот; 12 — циклоны; 13 — вентиляторы; 14 — абсорбер с плавающей насадкой; 15 — брызгоуловители; 16—полая башня; 17 — циркуляционный сборник; 18 — рециркуляционный сборник для фосфорной кислоты; 19 — холодильник; 20 — молотковая дробилка; 21 — аппарат БГС; 22 — топка; 23 — вибротранспортер
де 90—100° С. Температура гранулированного продукта 80—85° С, а остаточная влажность не более 1%.
Гранулы классифицируют по размерам частиц на двухситном грохоте 11. Крупная фракция с размером гранул более 4 мм направляется на молотковую дробилку 20, после которой совместно с мелкой фракцией в качестве ретура возвращается в БГС, а стандартную фракцию целевого (1—4 мм) охлаждают воздухом в аппарате КС 19 до 40—45° С и направляют на склад готовой продукции.
Отходящие газы из аппаратов САИ и БГС проходят двухступенчатую очистку от фтора и аммиака в абсорберах с плавающей насадкой (АПН) 14 и в полой башне 16. Аппарат АПН орошается фосфорной кислотой, возвращаемой из системы абсорбции на нейтрализацию в САИ. Вторая степень предназначена для очистки от 642
фтора и орошается известковым молоком или водой. Газы из аппарата КС и БГС перед мокрой очисткой обеспыливаются в циклонах 12. Вместо двухстадийной очистки газов из аппаратов КС и БГС применяют турбулентный промыватель типа «Вентури» с двумя блоками каплеуловителей, в которых происходит очистка от пыли и аммиака. Очистка от фтора производится в АПН с насадкой из вспененных полипропиленовых шаров.
Очищенные газы вентилятором 13 выбрасываются в атмосферу через выхлопную трубу.
Разработан вариант аппарата БГС, в котором осуществляется также и охлаждение гранул (БГСХ).
Разработана схема получения аммофоса с использованием аммо-низатора-гранулятора (рис. 11.48), представляющего собой полый открытый вращающийся барабан диаметром 4 м, длиной 6 м, установленный под углом 1,8° к горизонтальной оси. Частота вращения барабана 10 об/мин. Аппарат изготовлен из углеродистой стали, футерован листовой нержавеющей сталью. Аппарат имеет течку 3 для загрузки регура и трубопроводы для подачи аммофосной суспензии 4 и аммиака 5. Жидкий аммиак поступает в аппарат со стороны входа и выхода материала под слой гранулируемой массы. Ретурный продукт вводят через загрузочную течку, влажные же гранулы выводят с противоположной стороны барабана через течку 9. Для очистки внутренних стенок барабана аппарат оборудован срезающим ножом 2, расположенным вдоль всего барабана.
Согласно схеме (рис. 11.49), в каскадно расположенные емкостные нейтрализаторы 1, оборудованные трехъярусными турбинными мешалками, поступают концентрированная (50—54%-ная Р2О5) фосфорная кислота, сточные воды из цикла газоочистки и газообразный аммиак. Концентрация исходной фосфорной кислоты после смешения со стоками абсорбции составляет 47—48%. Подачу аммиака производят до нейтрализации фосфорной кислоты в первом нейтрализаторе до молярного отношения МН3:НзРО4 = = 0,35—0,50 (pH 2,5), во втором — до отношения 0,50—0,60
9 8 7
Рис. 11.48. Аммонизатор-гранулятор:
1— корпус; 2 — нож для очистки внутренней стенки; 3— течка для загрузки сыпучих компонентов; 4 — трубопроводы для подачи жидких компонентов; 5 — аммиакопроводы; 6 — распылители аммиака; 7 — привод; 8 — несущий и опорный ролики; 9 — течка для влажных гранул; 10 — подпорное кольцо; 11 — газоход; 12 — бандаж; 13 — зубчатое колесо
643
Рис. 11.49. Схема получения гранулированного аммофоса с аммонизатором-гранулятором (АГ):
1— нейтрализатор; 2, 4 — абсорберы с плавающей насадкой; 3, 5 — полые абсорберы; б, 11 — циклоны; 7 — вентиляторы; 8 — сборники; 9 — элеваторы; 10 — аппарат АГ; 12 — грохот;
13— холодильник КС; 14 — валковая дробилка; 15 — сушилка
(pH 2,5—3,5). Продолжительность пребывания суспензии на первой стадии нейтрализации составляет около одного часа. За счет теплоты реакции нейтрализации температура суспензии в первом нейтрализаторе достигает 115° С, во втором—125° С. При этом испаряется 20—25% от введенной в аппарат воды и влажность суспензии после нейтрализаторов составляет 17—18%.
Суспензия из второго нейтрализатора поступает в аммониза-тор-гранулятор 10. Одновременно в АГ поступает жидкий аммиак и ретур (4-—8 т/т целевого продукта). Происходит вторая стадия нейтрализации с одновременным гранулированием продукта. В аммони-заторе-грануляторе поддерживается температура 90—95е С и молярное отношение ЫН^НзРОд = 1,0:1,05. На этой стадии испаряется около 50% воды, введенной в систему с фосфорной кислотой и абсорбционными растворами. В выходящем из АГ гранулированном продукте содержание товарной фракции составляет 70%. Окончательная сушка аммофоса до влажности 0,5—1,0% производится в прямоточной барабанной сушилке 75. Температура топочных газов на входе в сушилку 250—300° С, а на выходе—105—115° С.
Высушенный аммофос поступает на двухстадийный грохот 12, в котором разделяется на три фракции. Выход товарной фракции со-644
ставляет 60—80%. Крупная фракция из грохота измельчается в молотковой дробилке и смешивается с мелкой фракцией, образуя ретур. Целевой продукт (товарная фракция) после охлаждения в аппарате КС 13 направляется на упаковочный узел. Отходящие из нейтрализаторов и аппарата АГ газы проходят очистку в две стадии. В первой стадии очистки улавливается аммиак кислыми растворами фосфата аммония в АПН 2, а затем в абсорбере 3 улавливается (поглощается) фтор. Абсорбер орошается водой или водной суспензией оксида и гидроксида кальция. Растворы после абсорбера возвращаются в нейтрализаторы. Очистка отходящих газов из сушильного барабана и охладителя КС производится в самостоятельной системе, включающей пылеочистку в циклонах и двухступенчатую мокрую очистку от аммиака и фтора.
По описанной схеме на 1 т аммофоса (53% Р2О5 усв, 12% N) расходуется: 0,558 т Р2О5 в виде упаренной кислоты; 0,151 т NH3; 9 м3 природного газа (34,8 МДж/м3); 67 кВт-ч электроэнергии.
Нитроаммофосфаты. Физико-химические свойства. Нитроам-мофосфаты — сложные удобрения, содержащие в качестве основных питательных элементов азот, фосфор и калий. К ним относятся: нитроаммофос, включающий нитраты и фосфаты, и нитроаммофоска, содержащая кроме нитратов и фосфатов соли калия. Обычно фосфатной составляющей является дигидроортофосфат аммония (нитроаммофос, нитроаммофоска) или гидроортофосфат аммония (нитродиаммофос, нитродиаммофоска).
Наиболее часто применяемые в народном хозяйстве виды нитро-аммофосфатов приведены в табл. 11.7.
Таблица 11.7. Состав нитроаммофоса и нитроаммофоски
Соотношение N:P2O5:K2O Соотношение в исходной смеси NH4H2PO4:NH4NO?:KC1 Состав, %
No6m р2о5 К2О Суммарное содержание действующих веществ,%
1:1:0 41,4:58,6:0 25,6 25,6 0 51,2
1:1:1 29,4 : 41,7 : 28,9 18,2 18,2 18,2 54,6
1 : 1,5: 1,5 35,5 : 29,6 : 34,4 14,7 22,2 22,2 59,1
Плав нитроаммофоса (состав NiPiOs^O = 1:1:0) при 180° С и влажности 2% имеет плотность 1,492 г/см3, динамическая вязкость 6,1 мПа-c. Плав нитроаммофоса полностью плавится при 172—174° С, теплота плавления 68,62 кДж/кг, разлагается при 225° С.
Плав нитроаммофоски (состав ЪкРгС^КгО = 1:1:1) при 180° С и влажности 0,8% имеет плотность 1,526 г/см3 и динамическую вязкость 135 мПа-c. Нитроаммофоска разлагается при 220° С с выделе-645
нием теплоты и автокаталитическим ускорением. Нитроаммофоски сильно слеживающиеся, гигроскопические удобрения (табл. 11.8), требующие кондиционирования.
В промышленности выпускается также карбоаммофос, получаемый путем введения карбамида в реакционную смесь. При одновременной добавке в реакционную смесь солей калия (КС1 или K2SO4) получают карбоаммофоску.
Таблица 11.8. Физико-химические свойства иитроаммофосфатов
Соотношение N:P2O5.K2O Насыпная плотность, г/см3 Прочность частиц по фракциям, г/гранул Угол естественного отко-са, град Гигроскопическая точка*,% Слеживае-мость, кгс/см3
1 мм 2 мм 3 мм
1:1:0 0,97 НО 400 800 56 58 0,50
1:1:1 1,00 — 3200 6500 63 54 0,22
1 : 1,5 : 1,5 1,05 820 3500 7700 60 58 0,27
♦Равновесная влажность над насыщенным раствором при 20° С.
Физико-химические основы технологии сложных удобрений. Процессы получения перечисленных видов удобрений можно выделить в три группы.
1. Процессы с двухступенчатой аммонизацией, в которых концентрированную (47—54% РгО5) фосфорную кислоту нейтрализуют до молярного отношения ЬШз:НзРС>4 = 0,6-Н),7. Из диаграммы совместной растворимости дигидрофосфата аммония (рис. 11.50) видно, что растворимость дигидрофосфата увеличивается с повышением концентрации гидрофосфата аммония. Максимальная растворимость дигидрофосфата будет при молярном отношении Н3РО4 : NH3 = 1,5. Во избежание потерь аммиака в производстве нейтрализацию ведут до отношения Н3РО4 : NH3 = 1,3—1,35.
Отношение N:P2O5 в фосфатах аммония составляет: для дигидрофосфата аммония 1:5 и для гидрофосфата 1:2,5. С целью выравнивания соотношения N^Oj в систему дополнительно вводят азот за счет добавки сульфата аммония (рис. 11.51), нитрата аммония или карбамида (рис. 11.52). Возможен одновременный ввод в систему смеси нитрата аммония и карбамида (рис. 11.53).
Из диаграмм растворимости при 25° С для систем NH4H2PO4—(NHO2SO4—Н2О (рис. 11.51, а) и (ШЬНРОд—(ИНд^ЗОл —Н2О (рис. 11.51, б) видно, что в процессе увеличения содержания сульфата аммония в растворе растворимость фосфатов аммония уменьшается. При увеличении концентрации фосфатов аммония растворимость сульфата аммония изменяется незначительно. Нитрат аммония оказывает значительное высаливающее действие на дигидрофосфат аммония. 646
Рис. 11.50. Диаграмма растворимости в системе NH4H2PO4—(NH4)2HPO4—Н2О при 25°С
a
Рис. 11.51. Диаграмма растворимости при 25° С в системе: а — NFUHjPCV-(ЫНдЬБОд—Н2О; б — (ИН^НРОд—(NH^SC^—Н2О
(NH4)2SO4, r/IOOr Н2О б
Рис. 11.52. Диаграмма растворимости в системе CO(NH2)2— NH4H2PO4—Н2О при 50° С (кривая MBL — по данным после трехсуточного перемешивания раствора, кривая МВ1!— по данным после восьмисуточного перемешивания раствора)
Система NH4H2PO4—CO(NH2)2—Н20 изучена в интервале температур от 15,3 до 50° С (рис. 11.52). В присутствии дигидрофосфата карбамид гидролизуется:
CO(NH2)2 + Н2О = 2NH3 + СО2
Однако с течением времени содержание карбамида в растворе уменьшается и скорость его разложения с появлением в системе гидрофосфата аммония, который образуется в процессе взаимодействия дигидрофосфата с выделяющимся при разложении карбамида аммиаком понижается. Поэтому в присутствии гидрофосфата аммония разложение карбамида не происходит.
Как видно из изотермы растворимости, в четырехкомпонентной системе CO(NH2)2—NH4NO3—(NH4)2HPO4—Н2О при 50° С разложение карбамида ни в одной области не имеет места (см. рис. 11.53).
В производстве сложных удобрений, содержащих три основных питательных элемента (NPK), в систему вводят соль калия в виде его хлорида. Реакция
NH4NO3 + КС1 KNO3 + NH4CI
обычно не идет до конца, и степень конверсии хлорида калия зависит от технологического режима и повышается с повышением температуры и продолжительности процесса.
Данные процессы проводят в аммонизаторе-грануляторе, в который параллельно вводят ретур. Доаммонизация осуществляется в тонком
Рис. 11.53. Изотерма растворимости в системе CO(NH2)2— NH4NO3—(NH4)2HPO4— Н2О при 50° С
слое на поверхности гранул, в связи с чем процесс ведут с большим количеством регура (3,5 —10-кратным). Операция позволяет поддерживать влажность материала на входе в сушильный барабан всего около 1,5%. Получаемые гранулы целевого продукта имеют сферическую форму и высокую прочность.
2. Малоретурные процессы, в которых получение суспензии необходимого состава осуществляется в реакторах, а процессы грануляции и сушки про-
648
дукта совмещаются в одном аппарате-сушилке типа РКСГ (с распылением и кипящим слоем материала в сушилке-грануляторе). Суспензия целевого продукта распыляется в объем, в котором происходит высушивание капель и формирование гранул. Отношение количеств регура и продукта в этих процессах составляет 0,3—1.
3. Расплавные процессы, в которых нейтрализуют смесь оргофосфор-ной и азотной кислот с использованием теплоты нейтрализации (ИТН). Затем раствор выпаривают под вакуумом до состояния плава, смешивают с хлоридом калия и гранулируют в башнях или барабанных грануляторах. Согласно способу получают прочные гранулы целевого продукта.
Получение диаммонитрофоски. Согласно схеме (рис. 11.54) производства диаммонитрофоски, исходная ортофосфорная кислота концентрацией 40—42,5% Р2О? из сборника 1 центробежным насосом 2 перекачивается в напорную емкость 3, из которой она непрерывно поступает в реактор 8. До реактора кислота проходит через промывные скрубберы 4, в которых осуществляется очистка газов, выходящих из аммонизатора-гранулятора 11 и из сушильного барабана 16. В реактор 8 параллельно с ортофосфорной кислотой поступает газообразный аммиак под давлением 1,5—2 атм и при непрерывном перемешивании происходит нейтрализация ортофосфорной кислоты. Количество поступающих аммиака и кислоты обеспечивает получение в образующейся суспензии соотношения NH3 : Н3РО4 = 1,4 : 1,0 (pH суспензии 5,6—5,7). При этом соотношении образуется смесь, состоящая из 60% дигидрофосфата и 40% гидрофосфата аммония:
Н3РО4 + 1,4NH3 = 0,6NH4H2PO4 + 0,4(NH4)2HPO4
Температура в реакторе повышается и поддерживается в пределах 115—120° С за счет теплоты реакции. В этом процессе испаряется около 20—30% внесенной с кислотой воды. Суспензия, содержащая около 25% воды, смешивается в смесителе 10 плавом нитрата аммония, содержащим 96—98% NH4NO3, газообразным аммиаком, хлоридом калия из бункера 14 и ретуром из бункера 13. Образующаяся смесь поступает в аммонизатор-гранулятор 11, в котором происходит донейтрализация дигидрофосфата аммония до гидрофосфата аммония и полное смешение всех компонентов и одновременная грануляция продукта. При этом происходит также частичное взаимодействие хлорида калия с нитратом аммония в нитрат калия и хлорид аммония.
Ретур вводится из расчета снижения содержания влаги в целевом гранулированном продукте при выходе из аммонизатора-гранулятора до 4—5%. Кратность его составляет в пределах 3—3,5. При снижении влажности до 1,3% кратность ретура возрастает до 7—10. Температуру в аммонизаторе-грануляторе поддерживают в пределах 70—75° С.
649
Рис. 11.54. Схема получения диаммонитрофоски:
1— сборник ортофосфорной кислоты; 2 — центробежный насос; 3— напорная емкость; 4— промывной скруббер; 5—ловушка; 6 — вентилятор; 7 — циклон; 8— реактор; 9 — напорная емкость для плава нитрата аммония; 10— смеситель; 11— аммонизатор-гранулятор; 12 — теплообменник для подогрева воздуха; 13 — бункер регура; 14—бункер хлорида калия; 15— ленточный транспортер; 16—сушильный барабан; 17—элеватор; 18— двухситный грохот; 19—валковая дробилка; 20 — бункер; 21 — барабан-кондиционер; 22 — насос-дозатор; 23 — бункер для припудривающей добавки
В аммонизаторе-грануляторе за счет выделяющейся теплоты нейтрализации происходит частичное испарение влаги смеси. Образующиеся пары воды и непрореагировавший аммиак удаляются в смеси с воздухом, просасываемым через аммонизатор-гранулятор для снижения избыточного тепла и направляются на очистку в промывной скруббер 4. Гранулы целевого продукта из АГ поступают в сушильный барабан 16, в котором сушатся до влажности не выше 1%. Процесс сушки осуществляется топочными газами с температурой при входе 160—180° С и на выходе ~110°С. Сухой продукт элеватором 17 передается в двухступенчатый грохот 18, в котором он рассеивается на три фракции: ретур (размер частиц меньше 2 мм), целевой продукт с размерами гранул 2—4 мм и крупная фракция с размерами частиц более 4 мм, направляющаяся в дробилку 19, в которой измельчается, и элеватором 17 подается на рассев.
Целевой продукт из бункера 20 поступает в барабанный кондиционер 21, в котором омасливается и припудривается, а затем на склад, на упаковку. В качестве омасливающего вещества применяется парафинистый мазут, который передается насосом-дозатором 22 и форсункой распиливается в первой зоне кондиционера. В качестве опуд-ривающего материала применяется каолин, кизильгур, гипс и другие химикаты, подаваемые во вторую зону кондиционера.
На получение 1 т питательных веществ в диаммонитрофоске (17,5—17,5—17,5) расходуют: 0,241 т ортофосфорной кислоты (100% Н3РОд), 0,085 т аммиака (100%), 0,310 т нитрата аммония (97% NH4NO3), 0,290 т хлорида калия (95% КС1) и 0,025 т кондиционирующих добавок.
Существует малоретурная схема получения комплексного удобрения типа диаммонитрофоски состава 18:18:18 (рис. 11.55), согласно которой процессы аммонизации, смешения компонентов, гранулирования и сушки проводятся совмещенно в одном аппарате-грануляторе барабанного типа, причем сушка продукта осуществляется за счет полного использования теплоты реакции. Азотная кислота (67—69% основного вещества) из напорной емкости 1 и ортофосфорная кислота (42—49% основного вещества) из напорной емкости 2 непрерывно поступают в гранулятор 3, в который подается также непрерывно газообразный аммиак. Гранулятор, в котором проходят все стадии образования целевого продукта, представляет собой горизонтальный барабан, внутри которого концентрически установлен второй барабан меньшего диаметра. Ортофосфорная кислота и аммиак (в расчете на образование гидрофосфата аммония) вводятся во внутренний барабан на слой гранул, поступающих из внешнего барабана. Во внутренний барабан также вводится хлорид калия из бункера 5. Во внешний барабан подается азотная кислота и аммиак согласно стехиометрии образования нитрата аммония. Они поступают также на слой гранул, 651
Рис. 11.55. Малоретурная схема получения комплексного удобрения типа диаммонитрофоски состава 18:18:18:
1 — напорный бак для HNO3; 2 — напорный бак для Н5РО4; 3 — гранулятор-аммонизатор; 4 — элеватор; 5 — бункер для КС1; 6 — охлаждающий барабан; 7 — элеватор; 8 — грохот; 9 — дробилка; 10— шнек; 11 — ленточный конвейер
которые переходят во внешний барабан из внутреннего. Такая циркуляция между внешним и внутренним барабаном осуществляется непрерывно. Гранулы перемещаются по длине внутреннего барабана и поступают во внешний барабан, в котором движутся в противоположном направлении и далее поступают в ковши и с их помощью направляются во внутренний барабан. Хлорид калия и ретур подаются шнеком 10 в конец внутреннего барабана, противоположный подаче ортофосфорной кислоты. Барабан вращается со скоростью 14 об/мин. Отношение количества внутреннего регура к готовому продукту колеблется в пределах от 1:30 до 1:60 в зависимости от влажности целевого продукта, которая составляет 0,3—0,5%. Количество внешнего ретура составляет 0,4 т на 1 т целевого продукта. Процесс грануляции продукта проводится при влажности массы менее 1%, что способствует образованию мелких гранул. При этом выход товарной фракции достигает 70%. Температура в грануляторе поддерживается в пределах 65—95° С. Испарение воды, вносимой в систему с кислотами указанной концентрации, осуществляется лишь за счет теплоты реакции. Избыток теплоты из гранулятора уносится воздухом, поступающим из охлаждающего барабана 6 и частично засасываемым через неплотности, имеющиеся в системе. Воздух проходит между барабанами и далее направляется на очистку.
Целевой продукт из гранулятора ленточным конвейером 11 и элеватором 4 направляется в охлаждающий барабан 6, в котором он охлаждается атмосферным воздухом, а затем элеватором 7 подается для 652
рассева на три фракции на двухситный грохот 8. Мелкая фракция и крупная, измельченная в дробилке 9, возвращаются в качестве ретура в гранулятор, а товарная фракция подвергается кондиционированию и направляется на склад готовой продукции.
На рис. 11.56 приведена «безретурная» схема получения нитроаммофоски из расплава с гранулированием его в башне. Ортофосфор-ная кислота (54,3% Р2О5) из сборника 1 центробежным насосом 2 перекачивается в напорную емкость 3. Азотную кислоту (47% HNO3) из сборника 4 центробежным насосом 5 перекачивают в напорную емкость 6. Из напорных емкостей 3 и 6 ортофосфорная и азотная кислоты поступают в смеситель 7, в котором с помощью мешалки тщательно перемешиваются. При этом предусмотрена линия подачи конденсата в смеситель в случае необходимости понижения концентрации смеси кислот. Смесь кислот из смесителя перетекает в сборник 8, из которого центробежным насосом 9 перекачивается в напорную емкость 10, а далее в нейтрализатор 11.
Газообразный аммиак поступает в нейтрализатор по пяти трубам, погруженным в раствор. Процесс нейтрализации ведут до значения pH 2,8—3,2, при котором в растворе будут находиться дигидрофосфат и нитрат аммония. За счет теплоты реакции температура в нейтрализаторе поддерживается 120° С. При этом часть воды испаряется и концентрация раствора повышается до 76%. Раствор из нейтрализатора перетекает в сборник 12, из которого поступает на выпарку. Процесс выпарки ведут при температуре 170° С и остаточном давлении 0,3 атм. Конечную концентрацию раствора доводят до 98%, процесс ведут в однокорпусном выпарном аппарате с выносной греющей камерой и с естественной циркуляцией. В этих условиях соли находятся в расплавленном состоянии, твердая фаза не выделяется.
Плав из выпарного аппарата стекает в сборник 14, в который подается пылевидная фракция целевого продукта. Из сборника центробежным насосом 15 плав перекачивают в напорную емкость 16, установленную на грануляционной башне. Из напорной емкости плав переливается в смеситель 17, в котором интенсивно перемешивается с загружаемым хлоридом калия. С целью предотвращения процесса конверсии хлорида калия с нитратом аммония и образования хлорида аммония, что значительно увеличило бы вязкость раствора и привело бы к загустеванию суспензии, емкость смесителя должна быть небольшой, чтобы продолжительность пребывания массы в нем не превышала 40—50 с. Исходя из этих условий, поступающий на смешение хлорид калия предварительно высушивают, подвергают размолу и подогревают в смесителе до температуры 165—170° С. Из смесителя реакционная масса самотеком поступает в распылитель 18 (~300 об/мин), откуда распиливается в грануляционную башню 19 в виде капель. Образую-653
Рис. 11.56. «Безретурная» схема получения нитроаммофоски из расплава с гранулированием его в башне:
1 — сборник Н3РО4; 2 — насос Н3РО4; 3— напорный бак Н3РО4; 4— сборник HNCh; 5 — насос HNCh; 6— напорный бак HNO3; 7—смеситель; 8— сборник смеси кислот; 9—насос для смеси кислот, 10—напорный бак; 11— нейтрализатор; 12 — сборник; 13 — выпарной аппарат с выносной греющей камерой; 14—сборник для плава; 15—насос для плава; 16 — напорный бак для плава; 17—смеситель для КС1; 18 — распылитель; 19 — грануляционная башня; 20—вентилятор; 21 — скребок; 22 — ленточный конвейер; 23 — охлаждающий барабан; 24—элеватор; 25— грохот; 26 — дробилка; 27—циклон; 28—вентилятор; 29—барабан-кондиционер; 30—бункер для припудривающей добавки; 31 — конденсатор; 32—вакуум-насос; 33 — скруббер; 34 — насос
щиеся в башне гранулы с температурой 90° С собираются в нижней части башнй и скребком 21 передаются на ленточный конвейер 22. Целевой продукт охлаждается воздухом в барабане 23, откуда элеватором 24 подается на рассев в двухступенчатый грохот 25. Мелкая фракция из грохота, пыль из циклона 27 и измельченная крупная фракция в дробилке 26 направляются на смешение с плавом в сборник 14. Целевой продукт с размером гранул 1,5—3,5 мм поступает в барабан-кондиционер 29, в котором опудривается кизельгуром или инфузорной землей и направляется на склад готовой продукции.
Получаемый по рассмотренной схеме продукт состава 17:17:17 содержит: 40% NH4NO3; 30% NH4H2PO4; 27,5% КС1 и 1% влаги. На производство 1 т целевого продукта расходуется: ортофосфорной кислоты (100% Н3РО4)— 0,235 т, азотной кислоты (100% HNO3)— 0,308 т, аммиака (100%) — 0,125 т, хлорида калия (60% К2О)— 0,228 т, кизельгура — 34 кг, пара (0,3 и 1,5 МПа) — 0,34 т, электроэнергии — 79,2 МДж.
Нитрофосфаты. Физико-химические основы процесса разложения природного фосфата азотной кислотой. В процессе разложения фосфата азотной кислотой образуется раствор (вытяжка), состоящий в основном из свободной ортофосфорной кислоты и нитрата кальция. В зависимости от последующей переработки вытяжки можно получать как азотные или фосфорные удобрения, так и двойные или тройные, также сложные удобрения с широким диапазоном соотношения питательных веществ.
Процесс разложения фосфатов азотной кислотой является сложным процессом, протекающим по следующему основному уравнению:
Ca5(PO4)3F + 10HNO3 = ЗН3РО4 + 5Ca(NO3)2 + HF
Содержащиеся в природных фосфатах примеси также взаимодействуют с азотной кислотой:
(Mg,Ca)CO3 + 2HNO3 = (Mg,Ca)(NO3)2 + СО2 + Н2О
М2О3 + 6HNO3 = 2M(NO3)3 + ЗН2О
CaF2 + 2HNO3 = Ca(NO3)2 + 2HF
При наличии в фосфатах соединений железа (И) они окисляются азотной кислотой:
FeO + 4HNO3 = Fe(NO3)3 + NO2 + 2H2O
655
Выделяющийся в процессе разложения фосфатов фторид водорода реагирует с диоксидом кремния (IV), всегда присутствующим в реакционной смеси вследствие разложения содержащихся в фосфатах примесей — нефелина и эгерина, оливина, биотита и других кремнийсодержащих минералов:
6HF + SiO2 = H2SiF6 + 2Н2О
Оксиды железа и алюминия разлагаются также выделяющейся ортофосфорной кислотой с образованием нерастворимых в воде фосфатов:
Fe2O3 + 2 Н3РО4 = 2FePO4 + ЗН2О
А12О3 + 2 Н3РО4 = 2А1РО4 + ЗН2О
Содержащиеся в исходном апатитовом концентрате нефелин и эгерин также разлагаются азотной кислотой:
KAlSiO4-4NaAlSiO4«SiO2 + 2OHNO3 =
= KNO3 + 4NaNO3 + 5А1(ЫОз)з + nSiO2 + 10Н2О
Na2OFe2O3-4SiO2 + 8HNO3 = 2NaNO3 + 2Fe(NO3)3 + 4SiO2 + 4H2O
ГЛАВА 12
САНИТАРНО-ТЕХНИЧЕСКАЯ ХАРАКТЕРИСТИКА НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ И ПРОМЫШЛЕННАЯ БЕЗОПАСНОСТЬ
В разделе приведены сведения о санитарно-технических нормах в технологии неорганических веществ, а также степени их вредного влияния на здоровье людей, работающих в этих производствах. При этом описывается общий характер действия конкретного химического соединения на организм человека, предельно допустимые концентрации (ПДК) исходных видов сырья, полуфабрикатов, готовой продукции и их возможных смесей, указаны причины, условия и дозы, вызывающие отравление (острое и хроническое). Приведены также сведения об индивидуальных средствах защиты обслуживающего производство персонала и методах оказания первой медицинской помощи пострадавшим.
12.1. СОЕДИНЕНИЯ НАТРИЯ
В природе встречается в составе минералов: галита (каменная соль), мирабилита (глауберова соль), чилийской селитры (природный нитрат натрия), буры (декагидрат тетрабората натрия) и др.
Получается электролизом расплава NaCl и NaOH.
Металлический натрий у работающих в производстве вызывает тошноту, изжогу, боли в эпигастральной области. У производственников отмечены также нарушения функции нервной и пищеварительной систем, раздражение слизистой верхних дыхательных путей. В процессе соприкосновения с влажной кожей или одеждой натрий воспламеняется и вызывает ожоги.
Исходя из перечисленного, обслуживающий персонал в производстве обеспечивается спецодеждой из нейлона, стекловолокна, а также индифферентными и гидрофобными мазями.
В производстве обеспечивается устранение контакта металлического натрия с влажным воздухом и водой.
Металлический натрий хранят под слоем керосина.
Хлорид натрия. Токсическое действие обусловлено в основном раздражающими свойствами. При этом необходимо иметь сведения о чч v 657
22 Химическая технология неорганических веществ, кн. 1
содержащихся природных примесях (соли мышьяковистых, цианидоводородной и других кислот). Природные примеси могут вызвать воспаление верхних дыхательных путей, вплоть до прободения носовой перегородки.
В процессе вдыхания распыленного в камере 10%-ного раствора хлорида натрия отмечается отложение кристаллов NaCl в легких. В условиях периодического воздействия пыли хлорида натрия с концентрацией 95—150 мг/м3 у рабочих возникает особый симптомоком-плекс отравления («синдром соляной пыли»), характеризующийся головными болями, болями в грудной и эпигастральной областях. Объективно — хроническое воспаление слизистой носа, язвочки на носовой перегородке, в гортани, трахее, а также коньюнктивиты и кератиты. Рентгенологически — признаки поражения носовых и лобных пазух, а также явления пневмосклероза.
У рабочих, занятых дроблением и упаковкой соли, засолкой рыбы,— папулезная высыпь на внутренней стороне предплечий, глубокие болезненные, не заживающие продолжительное время язвы на тыльной стороне пальцев и кисти. Поражения могут осложняться инфекцией. В производстве замечены также случаи высыпи с покраснением и отечностью лица, век и ушных раковин у рабочих, занятых очисткой хлорида натрия. Возможно также выпадение волос, изменение в ногтях и иногда их выпадение.
Основной мерой предупреждения заболеваний является снижение запыленности воздуха в процессе добычи и переработки исходного хлорида натрия и чистого NaCl.
Карбонат натрия. Карбонат натрия иногда вызывает изъязвление слизистой оболочки носа, раздражение дыхательных путей, конъюнктивит, желудочно-кишечные заболевания. В процессе погрузочно-разгрузочных работ у рабочих происходит омертвление крупных участков кожи на руках и спине, а при отпадении струпа обнажаются глубокие изъязвления. В процессе длительной работы с растворами возможны: 1) экземы; 2) разрыхление кожи; 3) дерматиты (развиваются при концентрации 1,8—2,0%, редко — при 1,5%; 4) изъязвления кожи типа «птичьих глазков». Глаза поражаются растворами карбоната натрия лишь при концентрации выше 2%.
Предельно допустимая концентрация для карбоната натрия 2 мг/м3.
Работник в производстве карбоната натрия должен носить защитные очки, резиновые перчатки, а также костюм из плотной ткани.
12.2. СОЕДИНЕНИЯ КАЛИЯ
Металлический калий получают взаимодействием расплавленного гидроксида или хлорида калия с металлическим натрием при высокой температуре.
658
Действие металлического калия идентично с действием элементного натрия.
Хлорид калия. Хлорид калия находится в природе в виде минералов: сильвина КС1, сильвинита KCl-NaCl, карналлита KCl MgCh ^HiO, каинита KCl-MgSO^SHiO и др.
В процессе производства хлорида калия у рабочих отмечаются изменения на ЭКГ и снижение выделения витаминов С и Bi с мочой. Рабочие же подземных выработок калийных руд страдают гнойничковыми болезнями кожи, заболеваниями периферической нервной системы, гипотонией, нарушениями тонуса парасимпатического отдела вегетативной нервной системы.
Пыль калийсодержащих руд резко ухудшает заживление кожных ран, вызывает мацерацию пограничных здоровых участков кожи, способствует развитию гнойной инфекции. У рабочих, контактирующих с каинитом, отмечаются дерматиты с везикулезными вспышками на фоне отека и гиперемии кожи, воспаление слизистой оболочки глаз.
Основной мерой оздоровления условий труда является ликвидация запыленности воздуха в процессе добычи и переработки минералов.
12.3. СОЕДИНЕНИЯ МЕДИ
Основными неорганическими соединениями меди являются: оксид, хлорид, оксохлорид, нитрат, гидроксокарбонаты, сульфат и гид-роксосульфат меди. Широко применяются в народном хозяйстве ацетат и оксоацетат меди.
Токсическое действие меди и ее неорганических соединений. Медь содержится в организме в основном в виде комплексных органических соединений и играет важную роль в процессах кроветворения. Во вредном действии избытка меди в организме предполагают реакцию ее с SH-группами ферментов. С колебаниями содержания меди в сыворотке и коже связывают появление депигментации кожи. Соединения меди, вступая в реакцию с белками тканей, оказывают раздражающее действие на слизистые оболочки верхних дыхательных путей и желудочно-кишечного тракта.
Попадание сульфата или ацетата меди в желудок человека вызывает тошноту, рвоту, боли в животе, понос, быстрое появление гемоглобина в плазме крови и моче. Процесс может вызвать желтуху, анемию, снижение резистентности эритроцитов. Возможна также су-льфгемоглобинемия, билирубинемия (до 2,6—7 мг%) вплоть до смерти при явлениях острой почечной недостаточности. Описан острый гастроэнтерит при потреблении воды, содержащей медь (44 мг/л). При попадании внутрь 0,2—0,5 г солей меди вызывают рвоту, 1—2 г — тяжелые, а иногда смертельные отравления.
659
22*
В процессе зачистки медных валов, шлифовке медных шайб, сварке и резке изделий из меди, прокатке медных болванок или чистой медной проволоки, дуговой плавке медного лома в воздух выделяется пыль меди и ее оксиды. В двух последних случаях концентрация меди в воздухе составляет 0,22—14 мг/м3. При этом через 1—2 ч у работающих появляется раздражение слизистых, сладкий вкус во рту, а спустя несколько часов — головная боль, слабость (особенно в ногах), покраснение зева и конъюнктивы, тошнота, боль в мышцах, иногда рвота и понос, разбитость, озноб, температура 38—39е С. Такая же картина наблюдается при вдыхании пыли карбоната меди, распылении оксида меди (концентрация по меди 0,03—0,12 мг/м3), а также в процессе чистки аппаратов от остатков соединений меди. В процессе сухой протравки зерна карбонатом меди (фунгицид содержит также сульфат меди и до 0,005% мышьяка) через несколько часов — сильный озноб, температура до 39° С и выше, проливной пот, общая разбитость, боли в мышцах, головная боль, а также раздражение слизистой глотки и гортани, кашель с зеленой мокротой не только во время лихорадки, но и после нее. Все это обычно сопровождается бронхитом. «Медно-протравная лихорадка» обычно продолжается в течение 3—4 дней. Отличительной особенностью «медно-протравной» и вообще «медной лихорадки» по сравнению с «литейной (цинковой)» является поражение желудочно-кишечного тракта.
В процессе хронической интоксикации медью и ее соединениями возможны функциональные расстройства нервной системы, нарушение функции печени и почек, изъязвление и перфорация носовой перегородки.
В производстве изделий из меди и ее сплавов (3,7—9,4 мг/м3) зарегистрированы церебральные ангионеврозы, снижение фагоцитарной активности лейкоцитов, титра лизоцима и бактерицидной способности сыворотки крови. При этом повышается содержание меди в волосах.
В процессе электролитического рафинирования меди, когда ее пыль действует в сочетании с парами серной кислоты и неблагоприятным микроклиматом, у обслуживающего персонала отмечают снижение иммунобиологической реактивности, поражение зубов и слизистой оболочки рта, язвенную болезнь желудка. У сварщиков и резчиков (стаж 1—3 года, концентрации аэрозоля 1,6—45 мг/м3) не обнаружено признаков пневмокониоза и хронической интоксикации. Среди работающих с медными порошками при стаже более пяти лет снижается жизненная емкость легких, наблюдается билирубинемия и нарушается функция печени. При этом- часть рабочих жалуется на изжогу, боли в области желудка, плохой аппетит и тошноту. У рабочих медных рудников (пыль меди 40—70 мг/м3) резко увеличено содержание гемоглобина и числа эритроцитов, содержание меди в сыворотке крови. Кроме того, у них наблюдается усиление сосудисто-бронхиального рисунка, уплотнение кор-660
ней легких, базальная эмфизема, повышается активность медьоксидазы и уровня сиаловых кислот в крови, воспаление и фиброз десен.
У рабочих, соприкасающихся с соединениями меди, кожа лица, волос и конъюнктива глаз иногда окрашены в зеленовато-желтый или зеленовато-черный цвет, на деснах темно-красная или пурпурно-красная кайма. Медь, ее соли и оксиды раздражают кожу, а пыль раздражает глаза и вызывает изъязвления роговицы. Описаны случаи аллергических дерматитов при контакте с медью и латунью, положительные кожные пробы на медь выявлены почти у половины больных или переболевших дерматитами или экземой, а также у части здоровых рабочих цеха электролиза меди и у металлистов. Известны случаи аллергического дерматита у рабочих, переносивших мешки с сульфатом меди, у работающих с телефонными проводами, а также при воздействии нитрата меди при приеме лекарственного средства, содержащего медь. Ученые-медики связывают выраженные аллергенные свойства меди с высокой способностью проникать через эпидермальный барьер в кожу. О поражении слизистых у рабочих, опрыскивающих виноградники бордосской жидкостью (концентрация аэрозоля в зоне вдыхания 1—130 мг/м3), свидетельствует увеличение в четыре раза числа лейкоцитов на слизистой оболочке носа через 2—3 ч.
С пищей человек ежедневно получает 2—5 г меди, из которых усваивается около 30%. В крови человека циркулирует медь, лабильно-связанная с аг-глобулином (церулоплазмином, образующимся в печени). До 90% меди откладывается в печени. Выделение же из организма происходит в основном через желудочно-кишечный тракт. У работающих с соединениями меди повышается ее содержание в крови, органах и выделениях. В норме кровь содержит 0,06—0,1 мг меди, а моча — 0,016—0,033 мг/л.
При «медной лихорадке» рекомендовано симптоматическое лечение. При отравлении через рот производят промывание желудка 0,1%-ным раствором K4[Fe(CN)6], внутрь — тот же раствор (1—3 столовые ложки каждые 15 мин) для осаждения и образования малотоксичного плохо растворимого комплекса. Кроме того, дают белковую воду или молоко, 30 г жженой магнезии, солевое слабительное, а при болях в животе—1 мл 0,1%-ного раствора сульфата атропина под кожу. Больного помещают в теплое помещение.
Определены следующие значения предельно допустимых концентраций: 1) медь металлическая—1 мг/м3; 2) среднесменная — 0,5 мг/м3; 3) кремнемедистый сплав — 4 мг/м3; 4) оксида меди — 0,1 мг/м3; 5) для меди с добавкой до 3% графита, 10% олова и 30% никеля— 0,5 мг/м3; 6) для пыли медно-сульфидной руды при содержании свободной SiOj менее 10% — 4 мг/м.
В качестве индивидуальной защиты в атмосфере аэрозолей меди и ее соединений применяют респираторы Ф-62, У-2К, «Астра-2», 661
«Лепесток-200», носят очки ПО-2, ПО-3 и противопылевые очки. Персонал цеха носит пылезащитную спецодежду и после работы принимает душ. При сварочных работах применяют шланговые противогазы с принудительной подачей чистого воздуха. Основным же мероприятием является предупреждение выделения аэрозолей меди и всех ее соединений.
12.4. СОЕДИНЕНИЯ СТРОНЦИЯ
В народном хозяйстве широко применяются в основном следующие соединения стронция: гидроксид, хлорид, нитрат, карбонат, сульфат и хромат.
Токсическое действие стронция и его неорганических соединений. Общий характер действия сходен с действием солей бария. Однако при приеме через рот соединения стронция малотоксичны. Основные нарушения наблюдаются со стороны минерального обмена.
При введении в желудок хлорида стронция для белых мышей ЛД50 = 1,03, для белых крыс — 2,79, для морских свинок — 2,84, для кроликов—1,51 г/кг. При введении в желудок нитрата стронция для белых мышей ЛД50 = 1,02, для белых крыс — 1,89, для морских свинок— 3,07, для кроликов—1,6 г/кг. При введении в желудок белых крыс гидроксида стронция ЛД50 = 3,16 г/кг, а при введении хромата стронция ЛД50 = 3,11 г/кг. При всех случаях признаками отравления являются заторможенность, тремор, судороги. На вскрытии: полнокровие органов, ожог слизистой оболочки желудка, перитонит (при действии гидроксида стронция).
Через 3, 6 и 9 месяцев после интратрахеального введения крысам 50 мг карбоната стронция в легких увеличивается содержание оксипролина. На вскрытии — явления пневмосклероза. Хроническое отравление пылью нитрата стронция (14,7 мг/м3, 4 ч ежедневно в течение 4 месяцев) вызывает нарушение функции печени, снижение активности холинэстеразы, повышение содержания щелочной фосфатазы, уровня а-липопротеидов, содержания кальция в крови. На вскрытии — дистрофические изменения бронхов, перибронхиальный и периваскулярный склероз, а в кистях — пролиферация остеобластов и остеокластов. Установлено, что концентрацию нитрата стронция 3,2 мг/м3 (4 ч ежедневно в течение 4 месяцев) можно считать близкой к пороговой.
При рентгенографическом обследовании 40 аппаратчиков (со стажем работы более 5 лет) у пяти из них выявлены умеренно выровненные диффузные интерстициальные изменения в легких, что связано с вдыханием пыли соединений стронция.
Экспериментально установлено, что ежедневные аппликации ланолиновой мази, содержащей до 70% Sr(NCh)2 или SrCh, на спину 662
крыс (4 ч ежедневно в течение одного месяца) вызывают сухость, гиперемию, язвочки. Порошкообразный гидроксид стронция вызывает гнойный конъюнктивит, некроз, поражение всех оболочек глаз. Карбонат и сульфат стронция раздражающим действием не обладает. Установлено также, что стронций вызывает сенсибилизацию и развитие экзематозного дерматита.
Предельно допустимая концентрация для нитрата стронция 1 мг/м3, а для карбоната и сульфата стронция 6 мг/м3.
К мерам индивидуальной защиты относятся: 1) защита органов дыхания респираторами, а в условиях присутствия в воздухе аммиака и неорганических кислот — фильтрующими противогазами марки В; 2) борьба с выделением пыли соединений стронция; 3) защита кожи от воздействия соединений стронция.
12.5. СОЕДИНЕНИЯ ЦИНКА
Металлический цинк. У работников цинкового производства преобладают явно выраженные атрофические и субатрофические катары верхних дыхательных путей. Известны также случаи «литейной лихорадки». В процессе хронического воздействия пыли цинка отмечаются желудочно-кишечные расстройства и гипохромная анемия. Рабочие обычно жалуются на раздражительность, бессонницу, снижение памяти, потливость по ночам, шум в ушах и снижение слуха. Рентгенограммы показывают усиление легочного рисунка, наблюдается эмфизема, начальные признаки пневмосклероза. Повышена заболеваемость верхних дыхательных путей, а также распространен кариес зубов.
Оксид цинка. Токсичность оксида цинка объясняют ее каталитической активностью. Попадание мельчайших частиц пыли оксида цинка вызывает заболевание «литейная лихорадка», протекающее по типу инфекционного катара верхних дыхательных путей. Задержка аэрозоли оксида цинка в дыхательных путях колеблется в пределах 41—94%. В процессе отравления оксидом цинка появляется сладковатый вкус во рту, после работы — плохой аппетит, а иногда сильная жажда. Отравленный человек чувствует усталость, стеснение и давящую боль в груди, сонливость, сухой кашель. Этот период, длящийся в зависимости от степени окисления от 1 до 4—5 ч, сменяется резким ознобом, продолжающимся 1,0—1,5 ч. Температура поднимается до 37—38° С (иногда до 40° С и выше) и держится несколько часов. При этом наблюдается расширение зрачков, гиперемия конъюнктивы, глотки и лица. В моче появляются сахар, часто гематопорфирин, уробилин, возможно увеличение содержания цинка и меди. Иногда болезненное состояние длится 2—3 дня и дольше. Это зависит от индивидуальности организма больного, а также концентрации 663
паров оксида цинка. Повторные заболевания «литейной лихорадкой» приводят к ослаблению организма и активированию туберкулезного процесса, а также повышению восприимчивости к другим заболеваниям дыхательных органов. У людей, перенесших эту болезнь многократно, появляется малокровие, повышается содержание билирубина в крови, понижается секреторная функция желудка.
Цинк является биоэлементом и входит в состав некоторых ферментов. Он является антагонистом меди: добавление к пище меди снижает токсическое действие цинка. В норме кровь человека содержит 0,46—0,67, мягкие ткани 0,68—5,41, кости 10,1—17,8, волосы и ногти 16,3—22,4 мг% цинка.
В процессе «литейной лихорадки» необходимы: щелочные ингаляции, внутривенное введение глюкозы (20 мл 40%-ного раствора) с 300 мг аскорбиновой кислоты. Внутрь — крепкий сладкий чай, кофе. По показаниям — сердечные, кислород, покой, тепло.
Предельно допустимая концентрация для оксида цинка 0,5 мг/м3.
Индивидуальной защитой служат противогаз марки БКФ, респираторы типа «Астра», «Лепесток» и др., защитные очки, спецодежда из хлориновой ткани. После работы — теплый душ.
Хлорид цинка. Кратковременное (5—30 мин) вдыхание аэрозоли хлорида цинка вызывает пароксизмальный кашель, тошноту, а через 1—24 ч — одышку, повышение температуры, воспалительные явления в легких. У обслуживающего персонала наблюдается поражение слизистой оболочки верхних дыхательных путей, трахеи, бронхов. У работников возможны прободения носовой перегородки. Имеют место желудочно-кишечные расстройства (после 1 года работы), а также язвы желудка или двенадцатиперстной кишки (после 5—20 лет работы). Описаны смертельные отравления при случайном приеме внутрь 40%-ного раствора хлорида цинка или паяльной жидкости, содержащей хлорид цинка. Процесс отравления сопровождается рвотой с примесью крови, сильными болями в области живота, возбуждением, судорогами и почечной недостаточностью.
Хлорид цинка резко раздражает и прижигает кожу и слизистые оболочки. Растворы хлорида цинка могут всасываться через кожу рук. Обладает слабовыраженными сенсибилизирующими свойствами: при длительном контакте с парами может развиться аллергический дерматит.
Предельно допустимая концентрация для аэрозоли хлорида цинка принята 1 мг/м3.
К индивидуальной защите относятся: 1) механизация технологических процессов, исключающая соприкосновение кожи с хлоридом цинка; 2) тщательное мытье рук после работы теплой водой или 2%-ным раствором гидрокарбоната натрия; 3) смазывание кожи жиром. 664
Сульфат цинка. Обслуживающий персонал в цехе электролиза, подвергающийся комбинированному воздействию сульфата цинка, паров серной кислоты, сурьмы, мышьяка и др., жалуется на сухость в носу, носовые кровотечения и охриплость. В процессе обследования обслуживающего персонала установлены атрофические риниты, фарингиты, перфорация носовой перегородки, воспаление десен, изъязвление языка. У производственников — повышенная заболеваемость органов дыхания, пищеварения, кровообращения.
Сухой сульфат цинка и концентрированные его растворы вызывают изъязвления, так называемые «птичьи глазки». Тыльная часть кистей поражается гораздо чаще, чем ладонь.
К индивидуальной защите в производстве сульфата цинка относятся мытье рук 2%-ным раствором гидрокарбоната натрия и применение ожиряющих мазей.
12.6. СОЕДИНЕНИЯ БОРА
Работающие на производстве борных удобрений из датолитового концентрата (концентрация пыли 5—58 мг/м3) худеют, жалуются на ухудшение аппетита и тошноту. Считается, что с увеличением содержания бора в пыли усиливается воспалительная реакция на фоне фиброгенного действия, вызываемого диоксидом кремния. У людей, потребляющих воду с содержанием бора 2—4 мг/л, наблюдается понижение кислотности желудочного сока и активности энтерокиназы в кале. Длительное применение лечебных препаратов, содержащих бор (в частности, тартрат бора), вызывает кахексию, дерматит, гипопластическую анемию, язву желудка, облысение.
Смертельная доза борной кислоты и сесквиоксида бора при отравлении через рот для взрослого 15—20 г, для детей 4—5 г. Обследование работающих со стажем 10 лет и более в условиях действия аэрозоля Н3ВО3 (22—35 и 10 мг/м3) выявило снижение половой активности, объема эякулята, числа сперматозоидов (в два раза), относительного числа подвижных клеток и уменьшение в целом оплодотворяющей способности спермы. В структуре заболеваемости работников, занятых в производстве сесквиоксида и борной кислоты, первые места занимают заболевания верхних дыхательных путей и пищеварительных органов, а также гнойничковые заболевания кожи.
Сесквиоксид бора и борная кислота, используемые в водных растворах или присыпках, хорошо проникают через поврежденную кожу (экземы, трещины, ожоги), вызывая при этом тяжелое (иногда смертельное) отравление, особенно у детей.
Борная кислота и растворимые бораты быстро и почти полностью всасываются из желудочно-кишечного тракта. Не исключают всасывание и сесквиоксид бора. В крови бор распределяется равномерно меж-665
ду эритроцитами и плазмой, но быстро переходит в ткани. В мягких тканях обнаруживается около 10% от дозы (преимущественно в мозгу, печени и жировой ткани). В головном мозгу бор сохраняется несколько дней после отравления борной кислотой. Имеются данные на сродство бора к серой субстанции нервной системы. В печени бор образует комплекс с углеводами. Основным депо являются кости, где находят до 50% от введенного количества борной кислоты. Установлено, что выделение соединений бора происходит главным образом через желудочно-кишечный тракт, но борная кислота выделяется почти полностью с мочой (около 85%). Нормальное содержание бора в крови составляет до 0,8 мг/л, а в моче — 0,715 мг/л. При отравлении содержание бора в крови повышается до 40 мг/л и выше.
Предельно допустимая концентрация борной кислоты 10 мг/м3, сесквиоксида бора — 5 мг/м3, датолитовой руды — 2 мг/м3, а датолитового концентрата — 4 мг/м3.
Установлено, что такие бораты, как тетрагидрат пентабората калия, метаборат кальция, тетрагидрат бората бария, моногидрат метабората свинца и декагидрат тетрабората натрия, вызывают такие болезни, как хронический бронхит и трахеит.
Рабочие, занятые на производстве фторида бора (III) и полиизобутилена, где BF3 применяется в качестве катализатора (концентрация BF3 0,035—0,61 мг/л, пыли В2О3 -320 мг/м3, KBF4 до 940 мг/м3), жалуются на раздражение, сухость и кровоточивость слизистой оболочки носа, зуд открытых участков кожи, повышенную ломкость зубов, боли в суставах. В процессе обследования выявлены частые заболевания верхних дыхательных путей и кожи, функциональные расстройства нервной системы, повышенная проницаемость кровеносных сосудов.
Предельно допустимая концентрация для фторида бора (III) 1 мг/м3.
У рабочих, контактирующих с аэрозолями боридов металлов, обнаружено заметное снижение содержания альбуминов.
Предельно допустимая концентрация для аэрозолей боридов титана и ниобия установлена 10 мг/м3, для борида кальция — не выше 4 мг/м3, для боридов титана и циркония — 2 мг/м3 и для борида хрома— 1 мг/м3.
У рабочих основных профессий цеха карбида бора (стаж 12—14 лет) выявлены острые и хронические воспалительные заболевания верхних дыхательных путей, возможно развитие пневмокониоза и пневмосклероза.
Предельно допустимая концентрация для нитрида бора кубической и гексагональной модификаций, а также для карбида бора 6 мг/м3, а для карбонитрида бора рекомендовано 10 мг/м3.
Бороводороды сильно ядовиты, токсичнее фосгена и цианида водорода. Пары их приводят к слезотечению. Подобно фосгену, диборан вызывает токсический отек легких. Пентаборан и декаборан по-666
ражают нервную систему, почки и печень. Опасность работы с бороводородами усугубляется их высокой летучестью. Они способны кумулировать, вызывая хроническое отравление. Обладают бактерицидными и фунгицидными свойствами.
Острые отравления в лабораторных условиях свидетельствуют о том, что вдыхание диборана является причиной заболевания типа «литейной лихорадки». При хронической интоксикации — астмоид-ный бронхит. При легких формах отравления пентабораном — сонливость, спутанность сознания, чувство сжатия в груди, головная боль, тремор и подергивания мышц, при тяжелых — нарушение координации движений, двоение в глазах, отвисание нижней челюсти, симптом Ромберга, невнятная речь, слюнотечение, обильный пот, приступы тяжелых судорог продолжительностью 0,5—2 мин, лейкоцитоз и эритроцитоз, тахикардия, повышение кровеносного давления и температуры тела, значительные изменения на электроэнцефало- и электрокардиограмме; может при этом развиться спазм гортани, требующий оперативного вмешательства; через пять дней возможен неврит, понижение слуха. Содержание бора в крови в острый период повышается до 0,24 мкг/мл при установленной норме 0,04—0,06 мкг/мл. По данным американских авторов, при концентрации бороводородов 0,0005 мг/л допускается работа в аварийных ситуациях в течение 1 ч, при 0,01 мг/л — 30 мин, при 0,021 мг/л—15 мин, при 0,065 мг/л — 5 мин. Минимальные действующие концентрации: для диборана— 0,002—0,004 мг/л, для пентаборана — 0,0025 мг/л, для декаборана— 0,0003 мг/л. Средние концентрации, определяемые по запаху: для диборана — 0,004 мг/л, для пентаборана — 0,007 мг/л, для декаборана — 0,002 мг/л. Большинство из 83 обследованных рабочих, соприкасавшихся с бороводородами в течение 3 лет, жаловались на головную боль, головокружение, вялость, кашель, тошноту и озноб. Часть из них была госпитализирована. При этом обнаружены поражения органов дыхания и патологические изменения в печени и почках. Для декаборана характерно действие на нервную и сердечно-сосудистую системы и на активность трансаминаз в сыворотке крови.
Пентаборан и декаборан сильно раздражают кожу и слизистые оболочки.
Предельно допустимая концентрация для диборана в США принята 0,1 мг/м3, для декаборана — 0,3 мг/м3, а для пентаборана — 0,01 мг/м3.
При работе с бороводородами применяют специальный фильтрующий противогаз, а поглотителями являются гопкалит, силикагель, смесь силикагеля с алюминием. Применяется спецодежда с длинными рукавами и перчатки. Быстрое смывание попавших на кожу продуктов разбавленным раствором аммиака. Протирание пораженных участков тела губкой, смоченной этим же раствором или 1%-ным раствором триэтаноламина, и дальнейшее врачебное наблюдение.
Тщательное наблюдение за герметичностью оборудования и коммуникаций. Содержание аппаратуры под вакуумом. Местные вытяжные устройства и вентиляция помещений. Дистанционное управление, защитные ограждения в особо опасных места. Постоянный контроль за концентрацией бероводородов в воздухе помещений.
12.7. СОЕДИНЕНИЯ АЛЮМИНИЯ
Общий характер действия алюминия заключается в механическом раздражении легочной ткани с одновременным осаждением белков с образованием необратимых белковых соединений в виде волокнистых субстанций без признаков воспаления. Экспериментально выявлено, что в ряду алюминий, гидроксид, ацетат, сульфат алюминия одновременно с фиброгенностью снижается гемолитическое действие. При экспериментально вызванной анемии соли алюминия явно задерживают восстановление содержания гемоглобина до исходного. Считается, что хлорид алюминия нарушает фосфорилирование.
При вдыхании пыли алюминия поражаются в основном легкие. Заболевание называется алюминозом легких или «алюминиевыми легкими». Описаны тяжелые заболевания у рабочих, занятых распылением алюминиевой краски и в производстве пиротехнической алюминиевой пудры при концентрации 4—50 мг/м3. После года работы отмечены похудание, сильная утомляемость, одышка, кашель, сухие и влажные хрипы в легких. Заболевание прогрессирует и после прекращения работы (алюминий долго выделялся с мокротой). При обследовании рабочих этих производств — жалобы на отсутствие аппетита, иногда расстройства пищеварения, тошноту, боли в желудке и во всем теле, одышку, сухой или влажный кашель. В крови — лимфоцитоз и эозинофилия. Уровень содержания алюминия в крови повышен в несколько раз, снижено содержание щелочной фосфатазы в кишечнике, кислой фосфатазы и АТФ в сыворотке. Рентгенологически — в более легких случаях — тонкая сетчатость, в более выраженных — нерезкие пятна на фоне сетчатости в легких, затем более резко очерченные и увеличивающиеся затемнения. Правая половина сердца увеличена. Такие легочные изменения встречались у 25—50% обследованных. На производстве пиротехнической пудры были спонтанные пневмотораксы и описаны случаи повышенной чувствительности с приступами бронхиальной астмы у рабочих, занятых сверлением листов алюминия. Легочный фиброз сопровождается неврологическими расстройствами
В производстве оксида алюминия мокрым щелочным способом в 5—10 раз повышена заболеваемость обслуживающего персонала катарами верхних дыхательных путей даже у рабочих со стажем до одного года. Наиболее резко выражены изменения слизистой оболочки у рабочих цеха кальцинации. У рабочих с большим стажем развивается алю-668
миноз. В отдельных случаях на фоне диффузного фиброза появляются и мелкие узелковые образования. У рабочих производства оксида алюминия сухим способом также распространены катары верхних дыхательных путей. В литературе описаны случаи пневмосклероза, прободения носовой перегородки, неврита слухового нерва. В процессе погрузно-разгрузочных работ глинозема обнаружены кровоточивость десен, охриплость, сухость во рту и полости носа.
Тяжелые заболевания описаны у работающих на печах в процессе плавки бокситов (в смеси с железом, кварцем, углем) в производстве искусственных абразивов. Выделяющиеся при этом пыль и дым содержат 41—62% А120з, 30—40% SiO2 и наибольшие количества оксидов других металлов. Общее количество пыли на ряде предприятий колебалось от 10 до 41 мг/м3 (15—25% Si). Частицы пыли и дыма обычно не превышают 1 мкм.
Заболевания рабочих у печей и у крановщиков (стаж работы 3—8 лет) характеризовались тяжелым течением, быстрым развитием диффузного фиброза легких, резкой краевой эмфиземой и образованием спонтанных пневмотораксов, нередко со смертельным исходом. Субъективные жалобы: кашель с мокротой или без нее, быстро развивающаяся одышка. Иногда в легких грубые и более мелкие хрипы. При обследовании группы рабочих со стажем работы 2—10 лет много жалоб на легкий кашель с темной мокротой по утрам, меньше— на плевритические и загрудинные боли.
Патологоанотомически (в тяжелых случаях) установлены утолщение плевры, стенок сосудов и бронхов, иногда участки острого воспаления легких или полного замещения легочной ткани фиброзной, утолщение альвеолярных перегородок в местах с сохранившейся тканью легких, щелевидные пространства, окруженные гигантоклеточной инфильтрацией, сплошные участки фиброзно-гиалиновой ткани или пронизывающие ткань легкого фиброзно-гиалиновые тяжи. В золе легких обычно обнаруживалось до 45% А1 и 30% Si. Быстрое развитие и тяжелое течение заболевания, возможно, связано с переходом соединений алюминия в процессе плавки в сильнореакционноспособные и растворимые соединения (у-А120з). Более негативные последствия имеет одновременное вдыхание «паров» кремния или коллоидального диоксида кремния и мелкодисперсного алюминия.
У рабочих, вдыхавших мелкую пыль синтетического корунда (пыль А12О3, вероятно, в виде у-А120з) с некоторыми примесями, выявлены раздражение слизистой оболочки верхних дыхательных путей и кровотечение из носа в начале контакта с пылью.
После попадания частиц алюминия в глаза появляются очаговые омертвления, изменения пигментации роговицы, изменения капсулы хрусталика, помутнение стекловидного тела. Пыль алюминия раздражает слизистые оболочки глаз, носа, рта, половые органы.
669
Случаи экземы и дерматита встречаются в глиноземных цехах. Рабочие, подвергающиеся действию пыли корунда, жалуются на зуд кожи стоп, голеней, кистей и предплечий. Иногда зуд сопровождается потливостью, нарушением кожной чувствительности, импотенцией. В процессе действия раствора калиево-алюминиевых квасцов кожа становится жесткой, «дубленой».
Всасывание соединений алюминия из желудочно-кишечного тракта незначительно, что может быть связано со способностью алюминия образовывать в кишечнике нерастворимые соединения с фосфором.
Предельно допустимая концентрация алюминия и его сплавов 2 мг/м3; оксида алюминия в виде аэрозоля конденсации — 2 мг/м3; оксида алюминия в виде аэрозоля дезинтеграции — 6 мг/м3; электрокорунда в смеси с легированными сталями — 6 мг/м3; гидроксида алюминия, у-А120з, корунда белого и бокситов — 6 мг/м3, нитрида алюминия — 6 мг/м3.
К индивидуальным средствам защиты относятся респираторы «Лепесток», Ф-62ш, У-2К и др. Пылезащитная спецодежда и защитные очки.
12.8. СОЕДИНЕНИЯ СВИНЦА
Свинец является ядом, действующим на все живое и вызывающим изменения особенно в нервной системе, крови и сосудах. Он активно влияет на синтез белка, является энергетическим балансом клетки и ее генетическим аппаратом. Многие факты говорят в пользу денатураци-онного механизма действия. Он подавляет ферментативные процессы превращения порфиринов и инкорпорацию железа в протопорфирин с образованием гема. Дети более чувствительны к свинцу.
Все соединения свинца действуют в общем сходно. Разница в токсичности объясняется лишь одинаковой растворимостью их в жидкостях организма, в частности в желудочном тракте. Однако и труднорастворимые соединения свинца подвергаются в кишечнике изменениям, в результате чего их растворимость и всасываемость сильно повышаются. Растворимость карбоната и оксида свинца в крови выше, чем в воде. Боросиликат свинца нерастворим в воде, однако в печени и костях отравляемых животных свинец обнаруживается. То же самое относится к сульфиду свинца, который при поступлении в организм через дыхательные пути вызывал у белых крыс и морских свинок симптомы хронического отравления, характерные для отравления другими соединениями свинца. Свинцовые белила, сульфат и оксид свинца токсичнее других его соединений.
Особым действием характеризуются лишь соединения свинца, содержащие токсический анион: арсенаты, хроматы и азид свинца. При 670
хроническом отравлении арсенатом свинца интоксикация протекает как мышьяково-свинцовая. Высокой токсичностью обладает и пыль стеарата свинца. Хромат свинца при введении в брюшину не ядовит, в то же время у обследованных 185 человек, подвергавшихся в течение двух месяцев его воздействию (концентрация 1,6—7,34 мг/м3), было выявлено повышенное количество свинца в моче и увеличенное число базофильно-зернистых эритроцитов. На основании результатов опытов на морских свинках соединения свинца располагают следующим образом по убывающей токсичности: нитрат>хло-рид>лактат>оксид>олеат>карбонат>стеарат>фосфат. При этом фосфат свинца отравления не вызывал.
Некоторые данные, полученные в опытах на животных, не согласуются с результатами наблюдений над людьми. Сплавы РЪ—Sb—Sn; Pb—Sn—Ag, содержащие от 63,89 до 97,4% Pb, не токсичны для крыс при отравлении их через рот. Однако на заводе автомобильных кузовов при работе со сплавами, содержащими свинец, часто наблюдались случаи отравления свинцом.
Ранними симптомами отравления являются: свинцовая кайма по краю десен, преимущественно у передних зубов (может полностью отсутствовать при выраженных формах отравления); «свинцовый колорит» — землисто-серая окраска кожи; ретикулоцитоз свыше 10%; появление в крови базофильно-зернистых эритроцитов в количестве не менее 15 на 10 000 эритроцитов; повышение содержания порфиринов в моче свыше 50—60 мкг/л; увеличение содержания Д-аминолевулино-вой кислоты в моче (свыше 2 мг%) и снижение активности дегидратазы ее в эритроцитах, сохраняющееся несколько лет после прекращения затравки; повышение содержания свинца в крови и моче.
В картине хронического отравления можно выделить следующие основные синдромы: 1) изменение нервной системы; 2) энцефалопатия; 3) двигательные расстройства; 4) чувствительная форма полиневрита, имеющая место при скрыто текущих и умеренно выраженных формах интоксикации; 5) поражение анализаторов на ранних этапах интоксикации: а) изменения системы крови; б) обменные и эндокринные нарушения; в) изменения желудочно-кишечного тракта; г) изменения сердечно-сосудистой системы; д) прочие проявления интоксикации.
Неотложная терапия при отравлениях производственного персонала предусматривает следующие меры. При коликах — атропин, морфин под кожу, внутривенно — серно-кислая магнезия (сульфат магния) с глюкозой, тиосульфат натрия, хлорид кальция. Теплые ванны, тепло на живот. Клизмы (солевая, капельная, масляная). Диатермия. Новокаиновая блокада. Кортизон или АКТГ. При полиневритах— внутривенное вливание новокаина, дибазол.
671
Предельно допустимая концентрация для свинца и его неорганических соединений 0,01 мг/м3, а среднесменная концентрация 0,007 мг/м3. Для цирконата — титаната свинца 0,1 мг/м3. Пыль оптического стекла, содержащего свинец, рекомендовано нормировать по свинцу, т.е. 0,01 мг/м3. Для пыли свинцово-силикатного волокна марок В-50 и В-70 и пыли трехосновного сульфата свинца — 0,01 мг/м3 в пересчете на свинец.
В качестве индивидуальной защиты в производстве соединений свинца применяют респиратор «Лепесток», который задерживает 95—97% пыли свинца. При высоких концентрациях паров применяют фильтрующий противогаз или шланговые противогазы с принудительной подачей чистого воздуха (ПШ-2 и др.). Во время работы и в рабочих помещениях запрещены прием пищи и курение. После работы обслуживающий персонал обязан мыться в душе. Удаляют свинец с рук и других загрязненных участков с помощью 1%-ного раствора уксусной кислоты или отмывочной пастой.
Показания к отстранению от работы со свинцом: при свинцовой анемии — временно, до исчезновения симптомов отравления; после свинцовой колики — на срок до 6 месяцев; при хроническом спастическом колите (свинцовом), после свинцового полиневрита, при энцефалопатии, свинцовом церебральном артериосклерозе — перевод на постоянную работу вне контакта со свинцом. Основанием для отстранения от работы со свинцом считают также повышение содержания этого металла в крови и моче сверхпринятого безопасного предела. Воспрещается использование лиц, не достигших 18 лет, в профессиях, где имеется контакт со свинцом и его соединениями. Ограничение и воспрещение труда женщин в ряде профессий, в которых имеется опасность воздействия соединений свинца. Воспрещен также труд женщин на малярных работах и на работах по очистке старой краски в труднодоступных, плохо вентилируемых местах (помещениях) на судостроительных и судоремонтных предприятиях.
12.9. СОЕДИНЕНИЯ ТИТАНА
Обслуживающий производство нерастворимых соединений титана персонал имеет некоторые изменения со стороны дыхания. Обнаружены морфологические изменения и накопление титана в легких умершего рабочего, который в течение 15 лет подвергался воздействию пыли оксида титана (IV). В процессе применения титана в порошковой металлургии известны случаи диффузного пневмосклероза, а также карнификации отдельных участков легких, астмоидных бронхитов с бронхоэктазами. В некоторых случаях у умерших были об-672
наружены в легких фиброз, эмфизема и следы ряда металлов, применяемых в производстве, но отложение титана было наибольшим.
Содержание титана в крови в норме составляет 2,5 мкг%. Содержание титана в легких и лимфатических узлах у человека в норме повышается с возрастом. Основным депо титана являются кости.
Предельно допустимая концентрация для титана и оксида титана (IV) 10 мг/м3; для нитрида титана — 4 мг/м3; для силицида титана— 4 мг/м3; для карбида титана рекомендовано 10 мг/м3.
В производствах соединений титана при высокой запыленности воздуха носят респираторы типа «Лепесток», «Астра-2» и др. Работники производства носят противопылевую спецодежду.
У работающих на производстве хлорида титана (IV) (стаж 1—10 лет) — хронические бронхиты в 21% случаев, хронические гипертрофические риниты и фарингиты 22—54%, более чем у половины — изменения функции вегетативной нервной системы. Изменения кожной чувствительности в виде гипостезии было у 54% и гиперстезии у 5% обследованных. У многих рабочих со стажем более 3 лет — усиление сухожильных рефлексов, тремор век и рук, гипергидроз.
Жидкий хлорид титана (IV) не раздражал кожи мышей. При внесении в глаз кролика — конъюнктивит и помутнение роговицы. У человека при контакте с 10%-ным раствором хлорида титана (IV) — трудно заживающие термические ожоги 2-й и 3-й степени.
Предельно допустимая концентрация хлорида титана (IV) (по содержанию НС1 в воздухе) 1мг/м3.
В производстве применяют (при наличии аэрозоли хлорида титана (IV) и хлорида водорода) фильтрующий промышленный противогаз с коробкой типа БКФ или марки В, защита глаз очками типа ПО-3. При попадании на кожу стирают сухим тампоном, а затем обильно смывают водой.
12.10. СОЕДИНЕНИЯ АЗОТА
Токсическое действие азота проявляется лишь при резком снижении давления кислорода. При давлении кислорода 100 мм рт. ст. сказываются последствия аноксии. Насыщение организма азотом происходит быстро, поскольку азот выделяется пропорционально снижению давления воздуха.
При давлении воздуха 4 кгс/см2, т.е. при давлении азота 3,2 кгс/см2, отмечались смешливость и болтливость, замедление реакции на зрительные, слуховые, обонятельные раздражения, на прикосновение, ослабление умственной деятельности. При 10 кгс/см2 (давление азота 8 кгс/см2) резкое расстройство мышечных движений. Водолаз после 5 мин пребывания на глубине 100 м (давление N2 8 кгс/см2)
673
жаловался на головокружение, цветные круги перед глазами, возбуждение наподобие опьянения, помутнение сознания, расстройство координации движений. На глубине свыше 100 м наркотическое действие азота уже не дает водолазу возможности работать. При повышенном давлении (кессонные, водолазные работы) азот воздуха растворяется в крови и тканях тела и, выделяясь из них в виде пузырьков при быстрой декомпрессии, вызывает «декомпрессионные» заболевания, или «кессонную болезнь».
При работе с жидким азотом необходимы перчатки, очки, хорошая вентиляция помещений и обеспечение безопасного транспорта. При водолазных работах вместо азота применяют гелий, смеси гелия с кислородом вместо воздуха.
Оксиды азота. Азот образует следующие оксиды — N2O (оксид диазота), NO (оксид азота), N2O3 (сесквиоксид азота), NO2 (диоксид азота) и N2O4 (тетраоксид азота). При взаимодействии с водой NO2, N2Oj и N2O4 растворяются в ней с образованием азотной и малоустойчивой азотной кислоты HNO2-
NO и NO2 содержатся в нитрогазах в различных соотношениях: от 91% NO и 9% NO2 до соответственно 48 и 52%. В смеси NO и NO2 образуется некоторое количество N2Oa— красно-бурого газа с температурой кипения 3,5° С, а в жидком состоянии — синего.
В процессе возникновения в растворе, содержащем НС1, могут образоваться еще и нитрозилхлорид C1NO и другие малоизученные и более сложные соединения. В некоторых производствах в бурых «парах» оксидов азота встречаются также СО, СО2, HCN и другие летучие вещества.
Исходя из вышеизложенного, общий характер действия меняется от содержания в газовой смеси различных оксидов азота. Процесс отравления оксидами азота в основном протекает по раздражающему или нитратному типу действия. В процессе контактирования оксидов азота с влажной поверхностью легких образуются HNO3 и HNO2, поражающие альвеолярную ткань, что приводит к отеку легких и сложным рефлекторным расстройствам. С другой стороны, в процессе отравления оксидами азота в крови образуются нитраты и нитриты. Последние, действуя непосредственно на артерии, вызывают расширение сосудов и снижение кровяного давления. Попадая в кровь, нитраты превращают оксигемоглобин в метгемоглобин. Повреждение же эритроцитов приводит к появлению метгемоглобина в мозге и (как и отек легких) к кислородной недостаточности.
В наиболее типичных случаях отравление оксидами азота начинается легким кашлем, который через некоторое время проходит. В более тяжелых случаях раздражение дыхательных путей сильнее. При этом наблюдается сильный кашель, иногда головная боль, рвота, порой пострадавший чувствует невозможность сделать глубокий вздох. 674
Но и в этих случаях на свежем воздухе явления быстро проходят, наступает ремиссия.
При менее тяжелых отравлениях в легких при рентгенологическом исследовании обнаруживаются изменения, напоминающие внешне картину милиарного туберкулеза (особая форма бронхопневмонии), исчезающие через 8—25 дней. В некоторых случаях при отравлении имеют место изменения крови. При спектроскопическом исследовании в ней определяется метгемоглобин (до 30,6%) и повышенное содержание белков. В этих случаях кровяное давление несколько снижается и замедляется пульс. Наряду с этим — симптомы поражения нервной системы: возбуждение, головокружение, обморок, потеря сознания. Возможны первичные изменения в сердечной мышце.
Предполагают, что каждый тип отравления соответствует следующему составу газовой смеси: обратимый тип — преобладание NO; тип удушающих газов — преобладание NO2, комбинированный тип — смесь NO2 и NO. Шокоподобный тип отравления вызывается вдыханием очень больших концентраций NO2 и NO.
В процессе отравления оксидами азота в организме человека в результате реакции N2O4 с влагой дыхательных путей образуются HNOj и HNO2. NO2 превращается в этих условиях в HNO3 и NO. Кислоты при слабощелочной реакции секрета дыхательных путей образуют нитраты и нитриты.
При отравлении пострадавшего переносят на свежий воздух (недопустимо, чтобы он шел сам даже при удовлетворительном общем состоянии). Проводят приемы первой помощи: максимальный покой, предотвращение охлаждения, вдыхание кислорода, искусственное дыхание (с осторожностью — лишь при угрожающей или наступившей остановке дыхания). При рефлекторных расстройствах дыхания и сердечной деятельности применяют так называемую противодымную смесь (хлороформ — 40 ч., 96°-ный этиловый спирт — 4 ч., серный эфир — 20 ч.); к этой смеси добавляют 5 капель нашатырного спирта. По показаниям: 1 мл 10%-ного кофеина, 1—2 мл 20%-ной камфары, 1 мл 10%-ного коразола, 1 мл кордиамина. Лобелии — при остановке дыхания, а в других случаях он противопоказан. Далее пострадавшего транспортируют в лежащем положении в стационар.
Предельно допустимая концентрация оксидов азота (в пересчете на NO2) 5 мг/м3. В США для диоксида азота — 9 мг/м3, оксида азота— 30 мг/м3.
В качестве индивидуальной защиты обслуживающего персонала является противогаз марки В или БКФ. Персонал имеет также изолирующий шланговый противогаз с подачей чистого воздуха, герметичные очки (ПО-1 и др.) с полумаской, перчатки резиновые кислотостойкие, бесшовные, перхлорвиниловые бесшовные, кислотозащитные рукавицы КР, перчатки, покрытые латексом, наиритом.
675
Применяют спецодежду, покрытую слоем перхлорвиниловой смолы, или из ткани, обработанной парафино-стеарино-фосфатной эмульсией и латексом СВХ-1, а также сапоги, брюки поверх сапог. Производят герметизацию производственных процессов и аппаратуры, где могут образоваться и выделяться оксиды азота, удаление образующихся оксидов азота местными вытяжными устройствами до их выделения в воздух (например, при нитровании, нитрозиро-вании, при травлении металлов и т.п.) В процессе электро- и газовой сварки внутри аппаратуры, в тесных и замкнутых пространствах обязательна подача свежего воздуха для вытеснения оксидов азота, которые могут образоваться при окислении азота воздуха при высокой температуре пламени.
Гемиоксид азота N2O в высоких концентрациях вызывает удушье вследствие вытеснения кислорода из легких. В смеси с кислородом— слабый наркотик. В опытах in vitro отмечалось митоток-сическое действие.
Газовые смеси, содержащие 20, 30 и 50% N2O, избирательно ухудшают кратковременную память у водолазов, не влияя при этом на долговременную память, психомоторную активность и зрительные функции. Примесь высших оксидов к используемому для наркоза гемиоксиду азота может вызвать поражения, характерные для этих соединений.
Оксид азота NO. Кровяной яд, переводит оксигемоглобин в метгемоглобин и оказывает, по-видимому, прямое действие на центральную нервную систему.
Начальные явления при остром отравлении оксидом азота — общая слабость, головокружение, онемение ног. При легком отравлении эти явления в течение нескольких минут исчезают при выходе на свежий воздух. При более сильном к названным симптомам присоединяются тошнота, иногда повторяющаяся рвота. Одновременно головокружение и общая слабость усиливаются, лицо бледнеет, кровяное давление снижается, и наступает полуобморочное состояние. При отравлениях средней тяжести резкая слабость и головокружение продолжаются много часов. При тяжелом отравлении — еще синюшность губ; через несколько часов указанные явления стихают. Через 1—3 дня на фоне хорошего общего самочувствия наступает столь резкая слабость, что отравленный не в состоянии держаться на ногах. Кровяное давление снижается и снова усиливается головокружение. Увеличенные печень и селезенка, болезненные при пальпации; глухие тоны сердца; замедленный пульс; повышенное выделение мочи. Сильная головная боль, онемение рук и ног, сонливость.
При концентрации 0,076 мг/л оксида азота в производственных помещениях отмечено снижение мышечной работоспособности (концентрация NO в сигаретном дыме составляет 0,32 мг/л.) 676
Диоксид азота NO2. Сравнительная токсичность NO и NO2 зависит от их концентрации и продолжительности воздействия. При 1—5 мг/л NO токсичнее NO2. При 0,2—0,7 мг/л, но длительном воздействии (6—8 ч), наоборот, NO2 токсичнее оксида азота. Диоксид азота обладает выраженным раздражающим и прижигающим действием на дыхательные пути, особенно глубокие, что приводит к развитию токсического отека легких, угнетает аэробное и стимулирует анаэробное окисление в легочной ткани.
Ощущение запаха диоксида азота и небольшое раздражение во рту и зеве наблюдаются при 0,008 мг/л, а в ряде случаев — при 0,0002 мг/л. Максимальная неощутимая концентрация — 0,00011 мг/л. При 0,014 мг/л отмечается раздражение глаз и носа, а при 0,095 мг/л раздражение через 1 мин и уменьшение диффузии СО2 в легких через 15 мин. Раздражение и одышка появляются при концентрации NO2 0,12 мг/л. При более высоких концентрациях наблюдаются тяжелые отравления, вплоть до смертельных. Вдыхание в течение 5 мин 0,51—0,76 мг/л вызывает бронхопневмонию, а 0,95 мг/л — отек легких в течение 5 мин.
У людей, работавших при 0,0008—0,005 мг/л (3—5 лет), выявлены воспалительные изменения слизистой оболочки десен, хронические бронхиты, эмфизема легких, пневмосклероз, осложненный астмоидными приступами, бронхоэкстазии, тенденция к брадикардии и гипотонии и т.д.
В верхних дыхательных путях диоксид азота не задерживается, задержка в легких кролика составляла около 80%. За счет последующего образования HNO2 и HNO3 в крови могут накапливаться нитриты. Предполагается также возможность образования в организме нитрозоаминов.
Оксид азота (V) N2O5 (пентаоксид диазота) сильно раздражает верхние дыхательные пути и легкие. Однократное отравление крыс приводило к отеку легких при 0,0097 мг/л и длительности воздействия 4 ч. Симптомы дыхательной недостаточности у крыс наблюдались при 10-дневном отравлении 0,0025 мг/л по 4 ч в день. 12-дневное отравление по 4 ч в день 0,0043 мг/л вызывало отек легких. Хроническое воздействие 0,0026 мг/л (по 5—6 ч в день, 6 дней в неделю, 105—114 дней) вызывает у крыс метгемоглобинемию, обеднение тканей витамином Be, уменьшение содержания витамина А в печени и витамина С в стенках тонкого кишечника. В опытах на животных N2O5 токсичнее NO2 и в три раза ядовитее О3. При совместном действии N2Oj и Оз (наиболее возможный вариант в производственных условиях) токсичность их складывается. Однако, по некоторым данным, N2O5 сравнительно безвреден, а отравления в подобных случаях на самом деле объясняются действием обычно присутствующего в смеси Оз.
677
Аммиак. Высокие концентрации аммиака вызывают обильное слезотечение и боль в глазах, удушье, сильные приступы кашля, головокружение, боли в желудке, рвоту, задержку мочи. Тяжелое отравление протекает на фазе резкого уменьшения легочной вентиляции, острой эмфиземы, увеличения печени, ацидоза, повышения глутаминоксалатной и глутаминпируватной активности. По литературным данным, уже через несколько минут после массивного воздействия аммиака наступает мышечная слабость с повышенной рефлекторной возбудимостью, тетанические судороги, резко снижается порог слуха, вследствие чего сильный звук вызывает новый приступ судорог. Нарушается обмен глутаминовой и а-кетоглутаровой кислот в коре головного мозга, значительно уменьшается способность тканей утилизировать кислород. Аммиак обладает также курареподобным действием. После действия очень высоких концентраций пострадавшие иногда сильно возбуждены, находятся в состоянии буйного бреда, не способны стоять. Наблюдаются резкие расстройства дыхания и кровообращения; в ближайшие часы (иногда и в первые минуты) после отравления может наступить смерть от сердечной слабости или остановки дыхания в фазе вдоха при спазме голосовой щели (рефлекс тройного нерва). В ряде случаев причиной гибели является воспаление бронхов и легких. Возможен химический ожог глаз и верхних дыхательных путей.
Последствием переменного острого отравления могут быть помутнение хрусталика, роговицы, даже ее прободение и потеря глаза; охриплость или полная потеря голоса, хронический бронхит, эмфизема легких, субатрофический фаринголарингит и т.д.
Порог обонятельного ощущения (в мг/л) 0,00050-0,00055; минимально действующая концентрация 0,00045; минимальная концентрация, вызывающая изменение биопотенциалов головного мозга, 0,00035. При концентрации 0,04—0,08 мг/л резкое раздражение глаз, верхних дыхательных путей, вплоть до рефлекторной задержки дыхания, головная боль. Вдыхание 0,003 мг/л в течение 8 ч вызывает тенденцию к уменьшению утилизации кислорода и замедлению пульса. При кратковременном вдыхании (в мг/л) 0,07—0,1 появляется раздражение в носу и в полости рта, при 0,49 — раздражение глаз, при 1,2 — кашель, возможен отек легких. Концентрацию 0,25 мг/л можно выдержать, хотя и с трудом, в течение часа.
У рабочих химических заводов выявлены (при 0,0005 — 0,024 мг/л) аносмия или гипосмия, неврастения, понижение биоэлектрической нервности головного мозга, повышение активности глутамино-пировиноградной трансаминазы и снижение уровня витамина С в крови, уменьшение выведения мочевины, увеличение потребности в витамине В|. Повышена заболеваемость катарами выроста дыхательных путей, ангинами, тонзиллитами. Заметны сдвиги в жировом и белковом обмене и учащение заболеваний катаром верхних дыхатель-678
ных путей у подростков, проходящих практику на заводе, даже при 3-часовом рабочем дне. Совместные действия NH3 и H2S вызывали у рабочих потерю обоняния.
При концентрации 1% (об.) — 7 мг/л — наблюдается легкое раздражение влажной кожи, при 2% заметное раздражение, а 3% через несколько минут могут вызвать ожог с образованием пузырей.
При попадании брызг NH4OH в глаза необходимо немедленно обильное промывание широко раскрытого глаза водой или 0,5—1,0%-ным раствором квасцов; вазелиновое или оливковое масло. При резких болях— 1—2 капли 1%-ного раствора новокаина или 8 капель 0,5%-ного раствора дикаина с адреналином (1:1000). Надеть очки-консервы. В последующем применяют закапывание 0,1%-ного раствора ZnSO4, 1%-но-го раствора Н3ВО3 или 30%-ного раствора альбуцида. При поражении кожи — обмывание чистой водой, наложение примочки из 5%-ного раствора уксусной, лимонной, винно-каменной или соляной кислот. При отравлении аммиаком через дыхательные пути — свежий воздух, вдыхание теплых водяных паров (лучше с добавлением уксуса или нескольких кристаллов лимонной кислоты), 10%-ного раствора ментола в хлороформе. Пить теплое молоко с боржомом или карбонатом натрия. Кодеин (по 0,015 г) или дионин (по 0,01 г), при удушье — кислород (вдыхать до уменьшения одышки или цианоза); при спазме голосовой щели — тепло на область шеи, теплые водные ингаляция, атропин (подкожно 1 мл 0,1%-ного раствора), при необходимости — трахеотомия. При нарушениях или остановке дыхания — искусственное дыхание. По показаниям — камфара, кофеин, кордиамин, коразол, успокаивающие средства (настойка валерьяны, бромиды).
Предельно допустимая концентрация аммиака 20 мг/м3.
В производстве аммиака индивидуальным защитным средством является промышленный противогаз КД. Период защитного действия при концентрации аммиака 2,3 мг/л — 240 мин, а для противогаза КД с фильтром — 120 мин. При отсутствии в воздухе органических веществ применяют противогаз марки М с защитным временем 90 мин. Для защиты глаз применяют защитные очки марки ПО-3 и др. Перчатки из щелочестойкой резины. Спецодежда из плотной ткани. Герметизация всей аппаратуры, коммуникаций и т.д. Местная и общая вентиляция рабочих помещений, в которых возможно образование и выделение в воздух аммиака.
Азотная кислота. Аэрозоль, содержащий диоксид азота, пентаоксид диазота и чистую азотную кислоту, раздражает дыхательные пути, может вызвать разрушение зубов, конъюнктивиты и поражения роговицы глаза. Действие паров азотной кислоты резко усиливается при одновременном присутствии в воздухе различных аэрозолей дезинтеграции— SiO2 и NaCl, моторного и минерального масел. Пары азотной кислоты приблизительно на 25% токсичнее, чем NO2.
679
В процессе тяжелых отравлений у работников происходит отек легких в течение первых суток; резкая слабость, тошнота, одышка, кашель с обильной (до 1,5 л в первые сутки) пенистой мокротой лимонно-желтого цвета, цианоз губ, лица, пальцев рук; изо рта специфический едкий запах. На вскрытии — некроз слизистой оболочки твердого неба, трахеи, бронхов; легкие полнокровны, в них чередуются участки ателектаза и эмфиземы; кровоизлияние в эпикард и эндокард; печень мраморно-пятнистая, полнокровная, с начальными явлениями жировой дистрофии. При легком отравлении бронхит и нередко выраженный бронхиолит. Длительность заболевания около пяти дней. В условиях производства — разрушение зубов, потеря ими естественного цвета.
Концентрированная азотная кислота вызывает тяжелые ожоги, а разбавленные растворы могут быть причиной экземы.
При ожогах кожи после быстрого обмывания струей воды в течение 1—2 дней повязка с 2—3%-ным раствором гидрокарбоната натрия.
Предельно допустимая концентрация — 5 мг/м3.
К индивидуальной защите относится противогаз марки В (при наличии тумана HNO3 с фильтром). Защитные очки — ПО-1, ПО-2 с полумаской. Спецодежда из специальной шерсти или хлориновой ткани. Перчатки кислотостойкие, двойные из натурального или неопренового латекса, нарукавники, фартуки. Сапоги резиновые или пластмассовые. Немедленное смывание водой кислоты, попавшей на кожу, длительное промывание глаз водой. Оборудование в кислотоопасных помещениях специальных гидрантов. Основные мероприятия — устранение непосредственного контакта азотной кислоты с работающими; механизация операций по транспортировке, наливу, сливу и т.д.
Нитрат аммония. Нитрат аммония — сильно гигроскопичен. Чистый продукт не чувствителен к ударам, но взрывается от детонатора и под каталитическим действием накапливающихся при хранении в закрытом помещении продуктов распада (NO2 и паров воды). Горение в закрытом помещении почти всегда приводит к взрыву. Влажный нитрат аммония (свыше 3% воды) не взрывается даже от детонатора. Тушить горящий нитрат аммония водой нельзя (каталитическое ускорение распада!). С целью защиты от взрыва к нитрату аммония добавляют карбамид, карбонаты кальция или магния, хлориды, уротропин и др.
Жалобы работающих в производстве нитрата аммония идентичны для действия аммиака. Кроме того, хронический гастрит, холецистит и гепатохолецистит. В литературе описан случай токсико-аллергического отека легких, миокардита и гепатита у женщины, работавшей в поле с удобрениями. Нитрат аммония оказывает раздражающее действие на кожу, что выражается в сильном зуде, покраснении вокруг фолликулов, лишаевидном утолщении кожи и покраснении ее на тыльной стороне кистей и предплечья. Попадая в мелкие ранки или трещины, вызывает жгучую боль.
680
Нитрат натрия. У рабочих, занятых добычей нитрата натрия на севере Чили, примерно через год появлялись (главным образом, на ладонях и подошвах ног) утолщения кожи, достигавшие 1,3 см в диаметре и болезненные при надавливании. В ряде случаев, даже если рабочий давно оставил работу, через 12—15 лет эти утолщения превращались в раковые опухоли.
Способность нитратов восстанавливаться в организме в нитриты часто приводит к образованию метгемоглобина. При употреблении в пищу воды, содержащей 50—100 мг/л нитратов, резко увеличивается число лиц с повышенным содержанием метгемоглобина в крови. Особенно страдают дети. Описано значительное количество смертельных отравлений грудных детей; при 20—40 мг/л содержание метгемоглобина у детей может возрастать до 5% и более.
Нитрат калия. В процессе введения в желудок белым крысам смеси нитрата калия и аммония (10 и 100 мг/л) через 10 дней отмечена метгемоглобинемия, отставание в росте, нарушение эмбриогенеза; у морских свинок способность к воспроизведению потомства нарушалась при больших дозах нитрата калия.
В производстве дымного пороха (смесь нитрата калия, серы и угля) у рабочих наблюдались изъязвления слизистой носа и даже прободение носовой перегородки уже после нескольких месяцев работы.
Нитрат кальция. Нитрат кальция имеет лишь раздражающее и прожигающее действие, выражающееся в покраснении кожи, зуде, изъязвленных, иногда глубоких и занимающих обширную поверхность, медленно заживающих и оставляющих большие рубцы. Поражаются участки кожи, на которых имеются хотя бы незначительные ранки, царапины и другие нарушения ее целостности.
К индивидуальной защите относятся: мытье после работы, применение защитных мазей типа ХИОТ и других ожиряющих смазок.
12.11. СОЕДИНЕНИЯ ФОСФОРА
Известно свыше 10 модификаций фосфора, важнейшими из которых являются белый, красный и черный.
Белый фосфор. Белый (желтый) фосфор сильноядовит, а красный— малоядовит. Это объясняется нерастворимостью красного фосфора в жидкостях организма. Однако в виде пыли красный фосфор может оказывать токсическое действие, что объясняется присутствием в нем примеси белого фосфора.
Острые отравления белым фосфором возможны при приеме его внутрь, хотя возможно отравление его парами. При отравлении бывают боли в животе, рвота светящимися в темноте массами с запахом чеснока, наблюдается вздутие живота, головокружение. При этом характерны сердечная недостаточность, симптоматика инфаркта миокарда и 681
различные нарушения деятельности сердечно-сосудистой системы, что объясняется действием белого фосфора на миокард и периферические сосуды. Иногда смерть наступает при потере сознания и сердечной слабости до появления желудочно-кишечных расстройств. С момента отравления до смерти может пройти от нескольких часов до 10 и более дней. Смертельная доза 0,05—0,15 г. Отравления отмечались при концентрации белого фосфора в воздухе 0,0002—0,0012 мг/л, а также 0,0001—0,00016 мг/л. При этом наряду с парами белого фосфора в воздухе были пары РНз, Р2О5, а иногда и НЕ
При хронических отравлениях наиболее типичны изменения в костях, особенно омертвение челюстей. Процесс начинается иногда сильной зубной болью, обычно в кариозных зубах, или воспалением надкостницы около кариозного зуба. Иногда разрушение и выпадение зубов происходит безболезненно. Даже образуются гнойные свищи. Наблюдаются также поражения слизистой рта, воспаление десен, «фосфорные полоски» серо-желтого или коричневого цвета на передних зубах.
Отмечаются также головная боль, повышенная возбудимость и чувствительность к холоду и т.д. Заболевания развиваются после нескольких лет работы с белым фосфором, хотя имеют место хронические отравления и после 7-недельной работы. Иногда омертвление челюстей развивается через несколько лет после прекращения работы с белым фосфором.
Попадая на кожу, белый фосфор воспламеняется, при этом развивается высокая температура. Кроме этого, действует и образующаяся фосфорная кислота. В организм белый фосфор поступает в основном через дыхательные пути. Отложение во внутренних органах (главным образом в печени) происходит в виде элементного и в виде окисленного фосфора. Элементный фосфор выделяется с выдыхаемым воздухом, калом и потом.
При остром отравлении через рот применяют частые повторные промывания желудка 0,2%-ным раствором КМпО4 или 1%-ным CuSO4 до исчезновения запаха белого фосфора в промывочных водах, 2—3 раза через полчаса 1%-ным CuSO4 (по 0,1 г на прием). Солевое слабительное (1 столовая ложка). Повторные очистительные клизмы. Щелочное питье (2%-ный NaHCO3, боржом). Слизистые отвары. Внутривенно 40%-ная глюкоза (20—30 мл) с аскорбиновой кислотой (300 мг), 10%-ный СаСЬ (5—10 мл). По показаниям — переливание крови, сердечные средства. Не давать больному молока, касторового масла, растительных и животных жиров (они растворяют белый фосфор).
При ожогах кожи немедленно тушить горящий белый фосфор водой или песком, землей, золой, тальком, карбонатом натрия. С целью химического связывания белого фосфора обработать обожженное место растворами CuSO4, NajCOj, NaHCO3, AgNO3 или KMnO4. Промыть глаза 2%-ным раствором NaHCO3. Одежду разрезать на месте 682
горения под током жидкости, поскольку фосфор может проникнуть через ткани и продолжать гореть на коже.
Предельно допустимая концентрация 0,03 мг/м3.
В производстве белого фосфора персонал пользуется противогазом БКФ и респираторами «Астра-2», РПГ-67.
Красный фосфор. Красный фосфор при поступлении в дыхательные пути в виде пыли вызывает явления, напоминающие хроническое отравление парами белого фосфора. Известны случаи атипичной острой пневмонии у работников, занятых возгонкой красного фосфора и работавших в среде аэрозоля красного фосфора с концентрацией до 0,04 мг/л. Описан также случай омертвления челюсти у рабочего, который имел дело только с красным фосфором. Предполагают возможность его превращения в организме в белый фосфор. Существует также мнение, что ядовитость красного фосфора объясняется примесью белого.
Описаны случаи тяжелого заболевания кожи, которое вызывалось и экспериментально.
Индивидуальная защита и меры предупреждения см. Белый фосфор.
Декаоксид тетрафосфора. Декаоксид тетрафосфора РдОю (или Р2О5, пентаоксид, устаревшее название — фосфорный ангидрид) в виде аэрозоля раздражает слизистые оболочки. Ядовитость может зависеть от примесей (в частности, фосфора).
Однократные отравления кроликов в течение 2—3 ч при концентрации 5—7 мг/л абсолютно смертельны (отек легких). При концентрациях 1—4 мг/л гибель животных наступает после ряда затравок. При 0,2—0,8 мг/л и затравках до 10 дней наблюдается ринит, фарингит, ларингит, бронхит, иногда пневмония. В результате хронических затравок при концентрациях 0,01—0,08 мг/л имеют место различные поражения дыхательных путей, отек слизистой трахеи, тромбоз сосудов легких.
Картина острой интоксикации парами оксидов фосфора в условиях производства сходна с экспериментальной. Через 6—20 ч недомогание, слабость, сухой кашель, повышение температуры. Через сутки одышка, кашель с вязкой мокротой, температура 39—39,8° С. У некоторых рабочих головная боль, головокружение и боли за грудиной, насморк и носовые кровотечения. Трахеобронхит иногда сопровождается токсической пневмонией. Лейкоцитоз с увеличением количества нейтрофилов и относительной лимфоцитопенией, высокая РОЭ, белковая диспротеинемия.
Декаоксид тетрафосфора раздражающе и прижигающе действует на кожу (процесс обезвоживания тканей).
Предельно допустимая концентрация в воздухе рабочей зоны 1 мг/м3, а в атмосферном воздухе 0,05 мг/м3.
В производстве применяют респираторы, а при наличии паров аэрозоля — противогазы марок В, БКФ.
683
Фосфорная кислота. Пары фосфорной (ортофосфорной) кислоты вызывают атрофические процессы в слизистой носа, приводящие в отдельных случаях к раздражению крыльев носа и прободению носовой перегородки. Характерны носовые кровотечения, сухость в носу и глотке, образование в носу сухих корочек, крошение зубов. Отмечается лейкоцитоз, изменение формулы крови и повышенное содержание гемоглобина.
На вскрытии отравленных животных — очаги токсической пневмонии, отек, ателектаз, увеличенная печень, иногда зернистая. Для белых мышей и белых крыс ЛД50 = 1,25 г/кг, ЛК50 = 25,5 мг/м3. В процессе длительного отравления при концентрации 10,6 мг/м3 наблюдается увеличение содержания белка в сыворотке и снижение гликогена в печени. Через месяц восстановительного периода лишь частичная нормализация сдвигов. Ингаляция 2,3 мг/м3 патологических изменений и сдвигов не вызывает.
Ортофосфорная кислота оказывает значительное прижигающее действие, вызывает воспалительные заболевания кожи. Приводит к общетоксическим явлениям.
Предельно допустимая концентрация 1 мг/м3.
Фосфорные удобрения. К фосфорным удобрениям относят: аппа-титы (исходное сырье), суперфосфат (простой и обогащенный), преципитат, фосфорную муку, аммофос (смесь моно- и диаммонийфосфатов), сульфоаммофос (смесь аммофоса с сульфатом аммония) и нитрофоску (смесь аммофоса с нитратом калия).
Поскольку анион фосфорной кислоты является «физиологическим», общее токсическое действие ее солей возможно лишь при весьма высоких дозах и в производственных условиях не опасно. Напротив, имеется довольно много наблюдений относительно раздражающего и прижигающего действия кислых солей, например простого суперфосфатата (отчасти из-за присутствия свободного Р2О5) на слизистые оболочки и кожу, особенно если они попадаю? в трещины и ранки на коже.
Фосфоритоз у работников, занятых на добыче и переработке фосфоритов, характеризуется поздним развитием, медленным течением, скудостью клинической симптоматики и незначительностью функциональных нарушений. У работников, контактирующих с суперфосфатом, в редких случаях могут развиваться дерматиты: сыпь на коже, жжение и зуд, отек кожи лица, жжение в глазах и слезотечение, быстро проходящие при отстранении от работы. Попадая в глаза, пыль суперфосфата вызывала сильное раздражение, конъюнктивы, отек век, помутнение роговицы, иногда даже прободение ее и выпадение радужной оболочки. У работников, занятых в производстве суперфосфата, описаны изменения костей предплечий, ряд неврологических расстройств, изменение порога обоняния, гипергидроз, неустойчивость артериального 684
давления, изредка встречается также лабильность сердечной деятельности. Отмечены также нарушения менструальной функции у работниц. Установлено развитие пневмокониоза (апатитоза) у работников, вдыхающих апатитовую пыль. Апатитоз характеризуется ранним возникновением (через 2—5 лет после начала работы) и медленным развитием; в III стадию не переходит. Течение благоприятное. Осложнения пневмониями и бронхоэктатической болезнью редки, присоединение туберкулеза не обнаружено.
Предельно допустимая концентрация: для аэрозоля апатита и фосфорита — 6 мг/м3; для нитрофоски фосфорной, сульфатной и бес-хлорной — 2 мг/м3; для нитрофоски азотно-сульфатной — 5 мг/м3; для нитроаммофоски — 4 мг/м3; для аммофоса — 6 мг/м3.
В качестве защитных средств в производствах применяют проти-вопылевые респираторы «Лепесток», У-2К, «Астра-2», Ф-62Ш, Р-ПК, РУ-60М и очки ПО-4. При наличии в производственных помещениях фторида водорода применяют фильтрующий противогаз марки В. В порядке профилактики пользуются защитными мазями и пастами типа силиконовых кремов, а также мазями ИЭР-2 и мазью Селисского. Для мытья применяют поверхностно-активные жидкости типа олеинсульфата и др.
Полифосфаты. Полифосфаты типа [Ме„(РОз)„], [Ме„+2РпОзп+1], [МепНгРлОзп+i] широко применяются для умягчения воды; обезжиривания волокна; в качестве компонента стиральных порошков и мыла. Они являются также ингибитором коррозии, кристаллизатором, диспергатором, стабилизатором. Они широко применяются в пищевой промышленности.
Токсичность полифосфатов при парэнтеральном введении объясняется способностью к образованию комплексов с биологически важными ионами, особенно с кальцием. Токсичный эффект различных полифосфатов неодинаков и зависит от пути введения и степени их вязкости. При повышении вязкости токсичность полифосфатов уменьшается. Наиболее чувствительны к действию полифосфатов кролики, собаки менее чувствительны, чем кролики и морские свинки. В процессе введения в желудок белым мышам и крысам разных полифосфатов ЛД50 = 2,65-5-10,3 г/кг. При этом токсическое действие является результатом нарушения осмотического действия в клетках. Проявляется оно в торможении роста животных в условиях подострого опыта. В хронических опытах при ежедневном скармливании животным до 2,5 мг/кг полифосфатов не наблюдается снижения продолжительности жизни, уменьшения плодовитости, тенденции к развитию опухолей или каких-либо иных токсических эффектов.
Циклические полифосфаты большей частью не гидролизуются в организме кроликов и выводятся с мочой, в то время как полифосфаты с линейной цепью расщепляются до ортофосфатов.
685
Рекомендуемая литература
Ахметов Т.Г., Бусыгин В.М., Гайсин Л.Г. и др. Химическая технология неорганических веществ.— М.: Химия, 1998, 488 с.
Ахметов Н.С. Общая и неорганическая химия.— М.: Высшая школа, 2002. 743 с.
Атрощенко ВЛ., Каргин С.И. Технология азотной кислоты.— М.: Химия, 1970. 494 с.
Баталин Ю.В., Урасин М.А., Шаманский ИЛ. Сульфат натрия и природная сода.— М.: Химия, 1969. 278 с.
Беленький Е.Ф., Рискни И.В. Химия и технология пигментов.— Л.: Химия, 1974. 672 с.
Бонне М., Заичко Н.Д., Караваев М.М. и др. Производство азотной кислоты в агрегатах большой единичной мощности.— М.: Химия, 1985. 400 с.
Вольнов ИЛ. Пероксидные соединения щелочных металлов.—М.: Химия, 1985. 384 с.
Вредные вещества в промышленности. Т. 3. Справочник для химиков, инженеров и врачей / Под ред. Н.В. Лазарева и И.Д. Гадаскиной.— Л.: Химия, 1977. 607 с.
Высоцкий Е.А., Желннн А.А., Здановкий А.Б. и др. Галургия: теория и практика. — Л.: Химия, 1983. 368 с.
Грабовенко В. А. Производство бесхлорных калийных удобрений.—Л.: Химия, 1980. 179 с.
Дохолова А.Н., Кармышов В.Ф., Сидорина Л.В. Производство и применение фосфатов аммония.— М.: Химия, 1979. 197 с.
Жигач А.Ф., Стасиневич Д.С. Химия гидридов.— Л.: Химия, 1969. 676 с.
Зайцев ИД., Ткач Г.А., Стоев НД. Производство соды.— М.: Химия, 1986. 312 с.
Кошкаров ОД, Соколов ИД. Технология калийных удобрений.—Л.: Химия, 1978. 403 с.
Kirk - Othmar encyclopedia, 3 ed., v. 4, N. Y, 1978, p. 151.
Корбридж Д. Фосфор. Основы химии, биохимии, технологии / Пер. с англ.— М., 1982.
Кузнецов С.И., Деревянкин В.А. Физическая химия процесса производства глинозема по способу Байера.— М.: Химия, 1964. 132 с.
Лейбуш А.Г., Семенов В.П., Казарновский Я.С., Кархов Н.В. Производство технологического газа для синтеза аммиака и метанола из углеводородных газов.— М.: Химия, 1971. 288 с.
Мальцева Н.Н., Хайн В.С. Борогидрид натрия.— М.: Химия, 1985. 367 с.
Мельиик БД., Мельников Е.Б. Краткий инженерный справочник по технологии неорганических веществ.— М.: Химия, 1968. 432 с.
Михайлов Б.М. Химия бороводородов.— М.: Химия, 1967. 698 с.
Морозова Н.К., Кузнецов В.А. Сульфид цинка. Получение и оптические свойства. М.: Химия, 1987. 304 с.
Некрич М.И., Ковалев М.П., Черняева Ю.И. Общая химическая технология. Изд-во Харьковского университета, 1969. 336 с.
Неронов В.А. Бориды алюминия. Изд-во Наука. Сибирское отделение. Новосибирск, 1966. 137 с.
Печковский В.В., Александрович Х.М., Нинаев Г.Ф. Технология калийных удобрений.— Минск, 1968. 385 с.
Позин М.Е. Технология минеральных солей.— Л.: Химия, 1974. В 2 ч. Часть I и II. 1558 с.
Позин М.Е. Физико-химические основы неорганической технологии. Л.: Химия, 1985. 384 с.
Продан Е.А., Продан Д.И., Ермоленко Н.Ф. Триполифосфаты и их применение.— Минск, 1969. 214 с.
Семенов В.П., Киселев Г.Ф., Орлов А.А. и др. Производство аммиака.— М.: Химия, 1985. 368 с.
Соколовский А.А., Унянанц Т.П. Краткий справочник по минеральным удобрениям.— М.: Химия, 1977. 376 с.
Ткачев К.В., Плышевский Ю.С. Технология неорганических соединений бора—Л.: Химия, 1983. 287 с.
Фримантл М. Химия в действии. В 2 т, Т. I, II.— М.: Мир, 1991 / пер. с англ.
Харлампович Г.Д., Залевский А.А., Новиков А.А. и др. Развитие промышленности минеральных удобрений.— М.: Химия, 1984. 352 с.
Химическая энциклопедия. Изд-во Большая Российская энциклопедия.— М.; 1988-99 гг.
Чернов В.Ф. Производство кальцинированной соды.— М.: ГХИ, 1956. 310 с.
Шихеева Л.В., Зырянов В.В. Сульфат натрия. Свойства и производство.— Л.: Химия, 1978. 289 с.
Шохин И.Н., Крашенинников С.А. Технология кальцинированной соды и очищенного бикарбоната натрия.— М.: Высшая школа, 1969. 213 с.
686
ОГЛАВЛЕНИЕ
Предисловие...........................................................3
Глава 1. Натрий и его соединения..................................5
1.1. Натрий...........................................................5
1.2. Оксиды натрия..................................................7
1.3. Хлорид натрия................................................. . 10
1.4. Сульфат натрия.................................................18
1.5. Фосфаты натрия..................................................36
1.6. Конденсированные фосфаты натрия.................................43
1.7. Гидриды и боргидриды натрия.....................................53
1.8. Карбонаты натрия................................................62
Глава 2. Калий и его соединения......................................100
2.1. Калий...........................................................100
2.2. Оксиды и гидроксид калия........................................103
2.3. Хлорид калия....................................................108
2.4. Сульфат калия...................................................121
2.5. Карбонат калия..................................................136
2.6. Нитрат калия....................................................144
2.7. Фосфаты калия...................................................151
Глава 3. Медь и ее соединения........................................154
3.1. Сульфиды меди...................................................156
3.2. Оксиды и гидроксиды меди........................................157
3.3. Хлориды меди....................................................162
3.4. Сульфаты меди...................................................165
3.5. Карбонаты меди..................................................180
3.6. Нитрат меди.....................................................181
Глава 4. Стронций и его соединения...................................183
4.1. Стронций........................................................183
4.2. Галогениды стронция.............................................186
4.3. Карбонат стронция...............................................189
4.4. Нитрат стронция.................................................192
4.5. Оксид и гидроксид стронция......................................195
4.6. Титанат стронция................................................196
Глава 5. Цинк и его соединения.......................................197
5.1. Цинк............................................................197
5.2. Оксид и гидроксид цинка.........................................203
5.3. Халькогениды цинка..............................................211
5.4. Сульфат цинка..................................................216
5.5. Галогениды цинка................................................223
5.6. Цинковые пигменты...............................................227
Глава 6. Бор и его соединения.......................................230
6.1. Бор............................................................230
6.2. Борные кислоты..................................................235
6.3. Оксиды бора....................................................247
6.4. Тригалогениды бора.............................................250
6.5. Нитрид бора....................................................254
6.6. Бораты..........................................................255
6.7. Тетраборат натрия...............................................257
6.8. Бораты кальция..................................................265
6.9. Перборат натрия................................................276
6.10. Технология боратов металлов...................................282
6.11. Бороводороды..................................................287
Глава 7. Алюминий и его соединения...................................291
7.1 . Алюминий......................................................291
7.2 . Оксид алюминия................................................294
7.3 Гидроксид алюминия..............................................297
687
7.4 Галогениды алюминия............................................299
7.5 Алюмосиликаты..................................................304
7.6 Сульфат алюминия...............................................316
7.7 Гидриды алюминия...............................................328
7.8 Нитрат алюминия................................................332
7.9 . Нитрид алюминия..............................................333
7.10 . Фосфаты алюминия............................................334
Глава 8. Свинец и его соединения....................................336
8.1. Свинец.........................................................336
8.2. Оксиды свинца..................................................339
8.3. Титанат свинца.................................................354
8.4. Хромат свинца..................................................355
8.5. Галогениды свинца..............................................359
8.6. Сульфаты свинца................................................361
8.7. Халькогениды свинца............................................362
8.8. Нитрат свинца..................................................364
Глава 9. Титан и его соединения.....................................365
9.1. Титан..........................................................365
9.2. Оксиды титана..................................................367
9.3. Диоксид титана.................................................369
9.4. Хлориды тигана.................................................403
9.5. Титанаты.......................................................412
Глава 10. Азот и его соединения.....................................415
10.1. Физико-химические свойства азота..............................415
10.2. Оксиды азота..................................................417
10.3. Аммиак........................................................421
10.4. Карбамид......................................................475
10.5. Азотная кислота...............................................490
10.6. Нитрат аммония................................................549
10.7. Сульфат аммония...............................................559
Глава 11. Фосфор и его соединения...................................566
11.1. Физико-химические свойства фосфора............................566
11.2. Физико-химические основы получения элементного фосфора........571
11.3. Получение элементного фосфора.................................573
11.4. Оксиды фосфора................................................577
11.5. Кислородсодержащие кислоты фосфора............................582
11.6. Фосфорная кислота.............................................584
11.7. Суперфосфаты..................................................597
11.8. Преципитат....................................................619
11.9. Термические фосфаты...........................................626
11.10. Фосфаты аммония..............................................631
Глава 12. Саиитарно-техническая характеристика неорганических веществ и промышленная безопасность..................657
12.1. Соединения натрия.............................................657
12.2. Соединения калия..............................................658
12.3. Соединения меди...............................................659
12.4. Соединения стронция...........................................662
12.5. Соединения цинка..............................................663
12.6. Соединения бора...............................................665
12.7. Соединения алюминия...........................................668
12.8. Соединения свинца.............................................670
12.9. Соединения титана.............................................672
12.10. Соединения азота.............................................673
12.11. Соединения фосфора...........................................681
Рекомендуемая литература............................................686