/





















































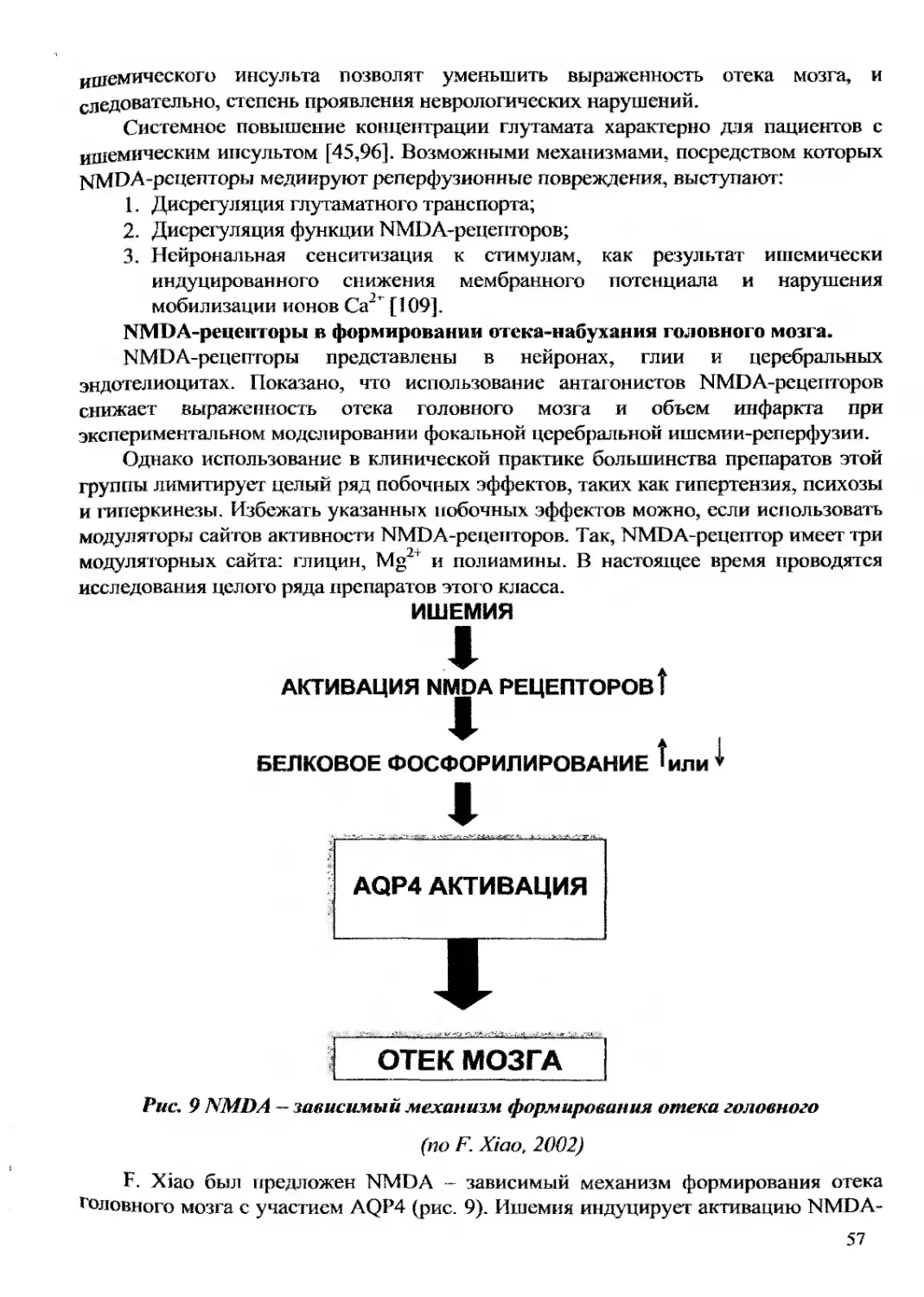



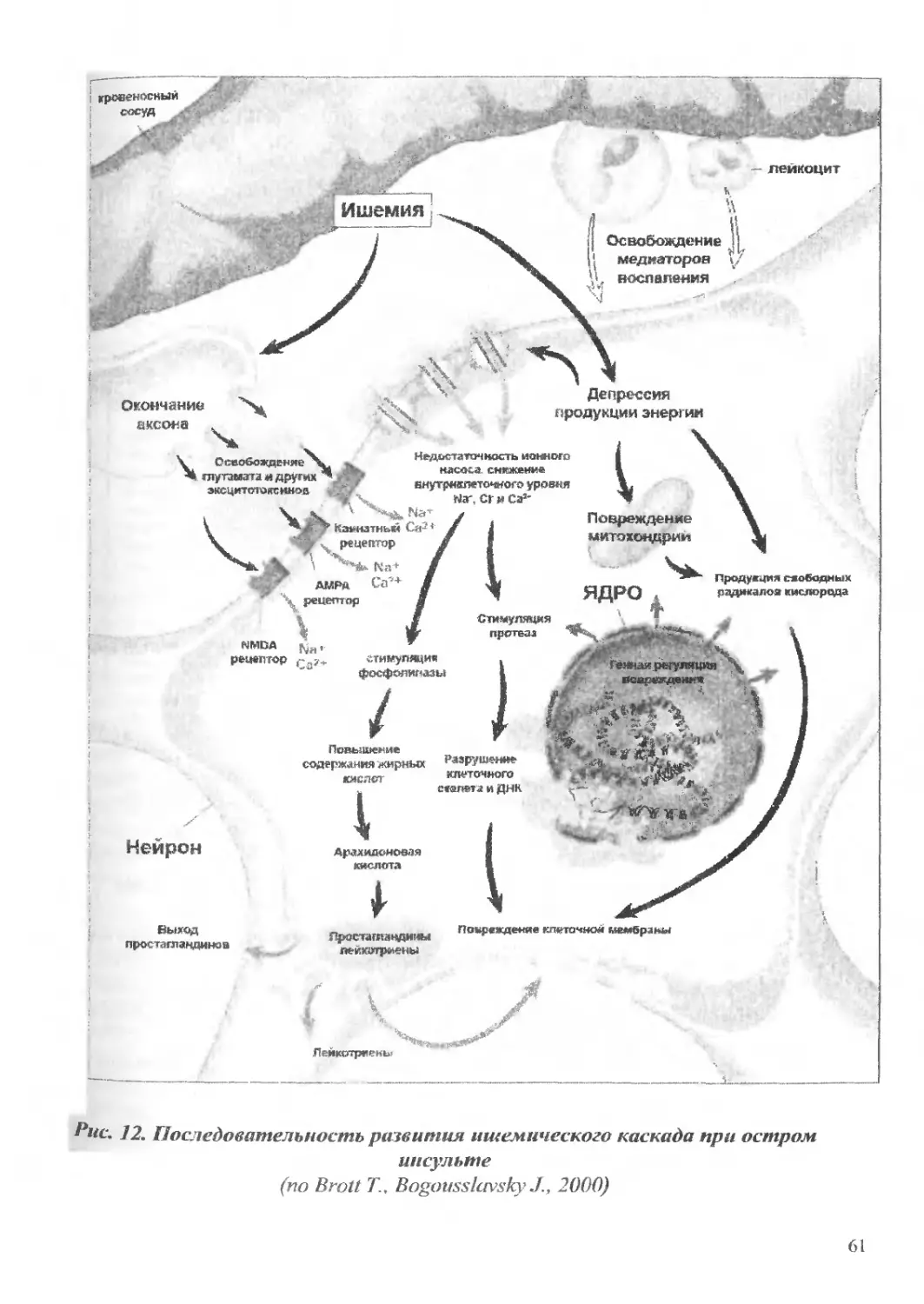















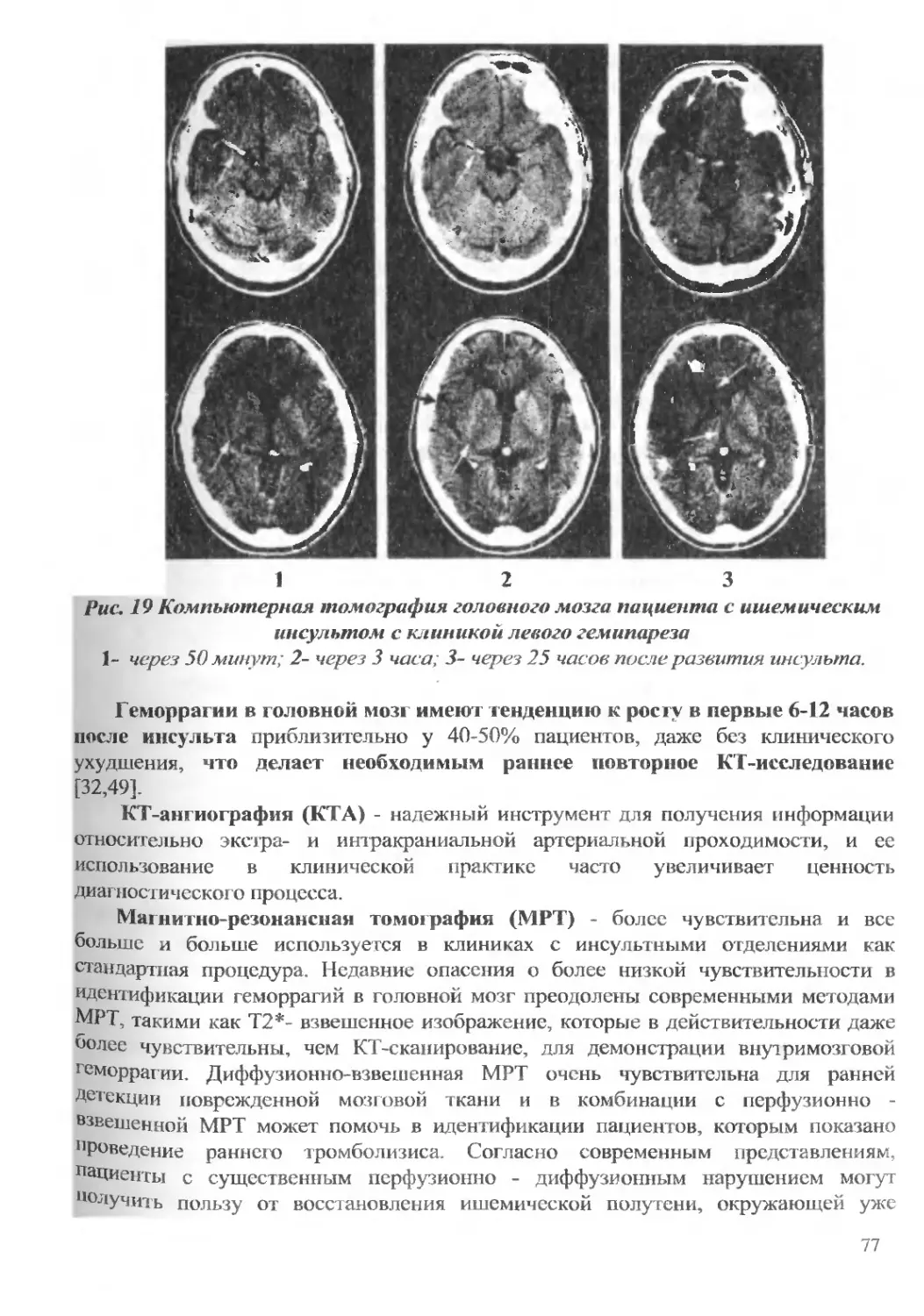















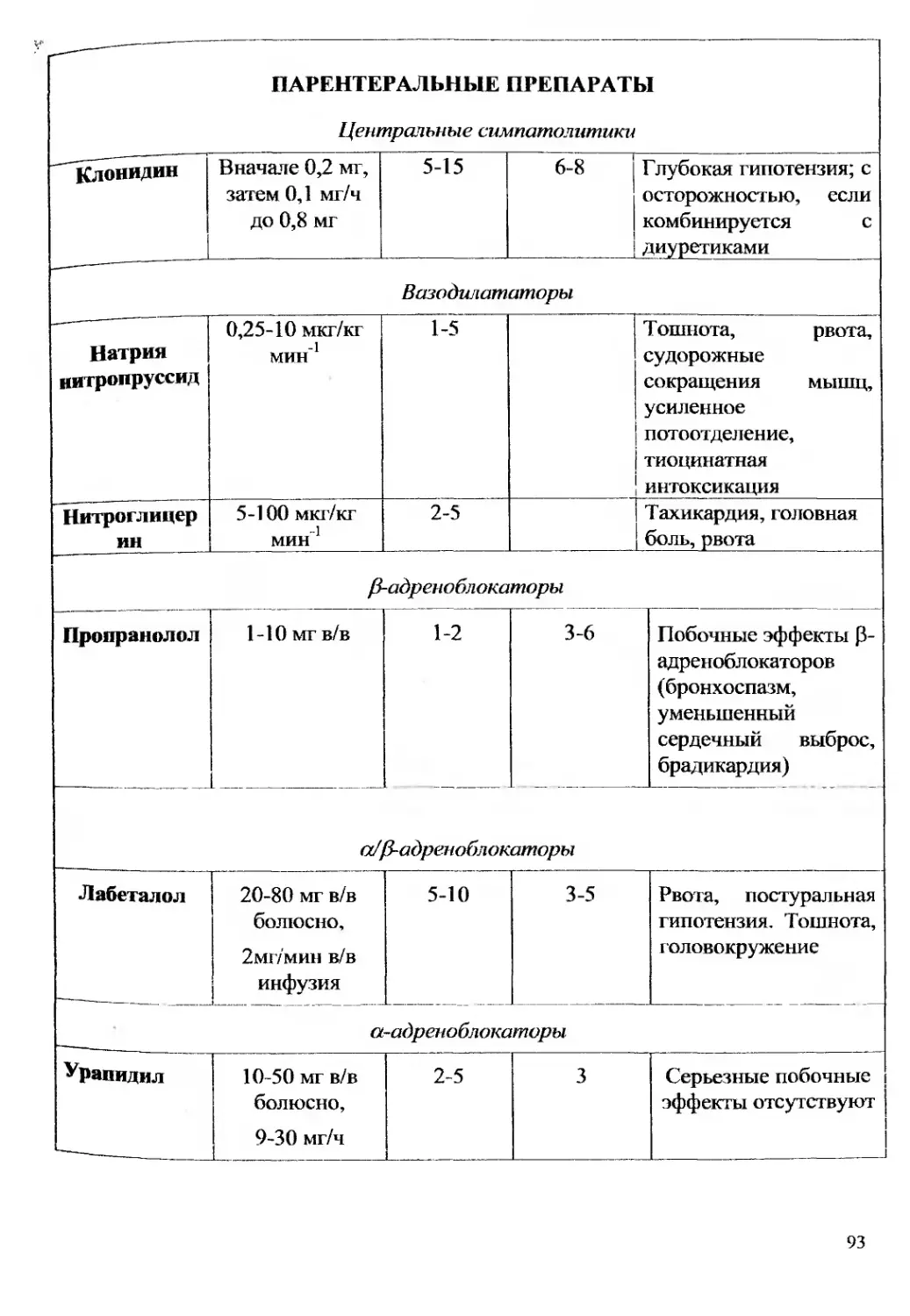

















































Текст
Л.В. Усенко, Л.А. Мальцева, А.В. Царев, В.Г. Черненко
ИШЕМИЧЕСКИЙ ИНСУЛЬТ
ГЛАЗАМИ АНЕСТЕЗИОЛОГА:
СОВРЕМЕННЫЕ ПОДХОДЫ К
ИНТЕНСИВНОЙ ТЕРАПИИ
МИНИСТЕРС ТВО ЗДРАВООХРАНЕНИЯ УКРАИНЫ
ДНЕПРОПЕТРОВСКАЯ ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ
КАФЕДРА АНЕСТЕЗИОЛОГИИ И ИНТЕНСИВНОЙ ТЕРАПИИ
Л.В. Усенко, Л.А. Мальцева, А.В. Царев, В.Г. Черненко
ИШЕМИЧЕСКИЙ ИНСУЛЬТ
ГЛАЗАМИ АНЕСТЕЗИОЛОГА:
СОВРЕМЕННЫЕ ПОДХОДЫ К
ИНТЕНСИВНОЙ ТЕРАПИИ
Днепропетровск • 2004
Ишемический инсульт глазами анестезиолога: современные подходы к
интенсивной терапии: [Монография] / Л.В. Усенко, Л.А. Мальцева, А.В. Царев,
В.Г. Черненко
Монография посвящена актуальной проблеме совремееной медицины -
интенсивной терапии острого периода ишемического инсульта, с которым, прежде
всего в своей практической деятельности сталкивается врач анестезиолог-
реаниматолог. На основании последних международных рекомендаций и научных
медицинских изданий, посвященных этой проблеме, отражены этиология,
эпидемиология и патогенез ишемического инсульта, освещены вопросы
диагностики и современные принципы интенсивной терапии острого периода
ишемического инсульта, как на догоспитальном этапе, таки и в условиях
реанимационного отделения стационара.
Для врачей анестезиологов, реаниматологов, неврологов, неотложной помощи,
терапевтов, также может быть полезной врачам-интернам и студентам медицинских
вузов.
Рецензент: профессор Л.П. Чепкий.
© Л.В. Усенко, Л.А. Мальцева, А.В. Царев, В.Г. Черненко
СОДЕРЖАНИЕ
Введение в проблему...........................................5
Глава 1
Эпидемиология ишемического инсульта......................6
Глава 2
Определение и классификация клинических форм острого нарушения
мозгового кровообращения по ишемическому типу..........7
Глава 3
Церебральный кровоток в норме и патологии
3.1 Нейрональная регуляция мозгового кровотока,.......12
3.2 Ауторегуляция мозгового кровотока....................16
3.3 Концепция порогового ишемического кровотока..........19
3.4 Концепция «ишемической полутени»..................23
Глава 4
Биохимия головного мозга в норме и патологии
4.1 Очерк биохимии головного мозга.......................24
4.2 Ликворология.................................... 35
4.3 Отек и набухание головного мозга................ 47
Глава 5
Молекулярные и клеточные механизмы повреждения головного мозга
при ишемическом инсульте
5.1 Ишемический каскад................................60
5.2 Ишемический апоптоз нейронов. Феноптоз программированная
смерть организма - миф или реальность ?..............63
5.3 Механизм ишемически-реперфузионного повреждения головного
мозга............................................... 71
Глава 6
Организация оказания медицинской помощи и вопросы диагностики при
ишемическом инсульте
6.1 «Цепочка выживания» после инсульта................73
6.2 Категории инсультных отделений................. ..75
6.3 Диагностическая визуализация.................. .76
6.4 Мониторинг витальных и неврологических функций....79
Глава 7
Интенсивная терапия ишемического инсульта
А. Поддержание экстрацеребрального гомеостаза
7.1 Респираторная терапия............................83
7.2 Коррекция циркуляторных нарушений................83
7.3 Характеристика препаратов для антигипертензивной терапии при
ишемическом инсульте.................................90
7.4 Уровень гликемии.................................109
7.5 Температура тела.................................109
7.6 Водно-электролитный баланс и осмолярность........ПО
3
Глава 8
Интенсивная терапия ишемического инсульта
В. Поддержание интрацеребрального гомеостаза
8.1 Тромболитическая терапия........................110
8.2 Антиагрегантная терапия.........................113
8.3 Антикоагулянтная терапия.........................114
8.4 Нейропротекторная терапия........................115
8.5 Г ипотермия...................................... 122
8.6 Гемодилюция......................................123
8.7 Перфторуглероды................................ 123
8.8 Профилактика и терапия огека головного мозга и повышенного
внутричерепного давления.............................123
8.9 Препараты противопоказанные в остром периоде ишемического
инсульта.............................................125
8.10 Протокол интенсивной терапии ишемического инсульта на
догоспитальном этапе.................................126
8.11 Протокол интенсивной терапии ишемического инсульта на
госпитальном этапе...............................127
Заключение......................................... 130
Литература........................................... 131
4
«Реаниматология представляет
собой науку и практику, сочетающиеся
со здравым смыслом и состраданием к
больному»
Питер Сафар (1988)
«...Если нынешнее положение не
изменится, каждый пятый житель
Украины умрет от инсульта, и еще
каждый пятый завершит свое бытие
зависящим от окружающих».
Н.Е. Полищук,
член-корр. АМН Украины (2003)
ВВЕДЕНИЕ В ПРОБЛЕМУ
За последние десятилетия вследствие углубления представлений о
формировании ишемического повреждения головного мозга существенно
изменились взгляды клиницистов на стратегию его патогенетического лечения,
подходы к применению тех или иных лекарственных средств. В связи с этим
появилась необходимость в пересмотре стратегии интенсивной терапии пациентов с
ишемическим инсультом в остром периоде, с которым прежде всего, сталкивается
в своей клинической практике врач анестезиолог-реаниматолог. При этом можно
полностью согласиться с мнением профессора Н.Е. Полищука, что на сегодняшний
день «Скорее исключением, чем правилом, является проведение простых, но
действенных мероприятий - поддержка оптимальных уровня глюкозы,
артериального давления (в частности, воздержание от резкого его снижения),
метаболическая коррекция, предупреждение пролежней, инфекционных и
тромбоэмболических осложнений, раннее начало всесторонней реабилитации, на
которых базируется ведение больных в развитых странах и которые почти ничего
не стоят (в буквальном, а не переносном смысле слова), по сравнению со
стоимостью многих лекарственных средств, широко используемых в Украине в
острый период инсульта, хотя нецелесообразность их назначения таким больным
уже доказана» [17].
В настоящем работе мы постарались, говоря словами авторов одного из
прекраснейших руководств по инсульту Ч.П. Ворлоу и соавт., «... убрать, насколько
возможно, остающиеся нейродогмы, нейрофантазии и нейроастрологию» [7] по
отношению к стратегии интенсивной терапии острого периода ишемического
инсульта.
Необходимо подчеркнуть, что залогом качества оказания медицинской
помощи, является внедрение в клиническую практику современных мировых
стандартов, базирующихся на принципах доказательной медицины. Поэтому
рассматриваемые терапевтические стратегии основываются на последних
опубликованных рекомендациях Американской ассоциации по инсульту (ASA)
2003 г. [32]; Европейской инициативной группы но инсульту (EUSI) 2003 г. [49],
Американской академии неврологии (AAN) 2002 г. [47], а также последних
литературных данных.
5
Глава 1
ЭПИДЕМИОЛОГИЯ ИШЕМИЧЕСКОГО ИНСУЛЬТА
Согласно данным ВОЗ, инсульт ежегодно поражает в мире около 20 млн.
человек, из которых 5 млн. умирают вследствие инсульта. Из 15 млн., которые
выживают, приблизительно одна треть инвалидизирована и нуждается в
постороннем уходе в повседневной жизни, и по меньшей мере 1 из 6 пациентов
переносит повторный инсульт в течение последующих 5 лет. Частота инсульта в
экономически развитых странах равняется 150 на 100 000 населения/год. В Украине
в 2001 г. этот показатель составил 307 на 100 000 населения/год [5].
В течение 3-х десятилетий популяционно частота инсульта в большинстве
стран прогрессивно возрастает; с 1985 года она увеличилась почти в 4 раза, в го
время как ишемическая болезнь сердца в 1,57 раза, а гипертоническая болезнь - в
1,96 раза. За последние три десятилетия изменилась и структура инсультов за счёт
прогрессирующего увеличения инфарктов мозга. Если до 1945 года соотношение
кровоизлияний в мозг и инфарктов мозга колебалось от 2:1 до 4:1, то во время
второй мировой войны достигло 7:1. Массивное кровоизлияние в мозг послужило
причиной смерти Т.Рузвельта, от геморрагического инсульта умер И. Сталин,
четыре инсульта перенес У. Черчиль.
Уже с конца 1950-х годов соотношение между кровоизлияниями в мозг и
инфарктами начало изменяться в противоположную сторону и в 1970-х годах в
большинстве высокоразвитых стран оно достигло современного соотношения 1:4.
На сегодняшний день среди миллионов людей, перенесших инсульт, ишемическая
форма наблюдается более чем в 80% случаев. Однако, среди выживших только у
10% неврологический дефицит почти полностью подвергается обратному развитию,
и больной возвращается к прежнему труду [ 18, 24].
По данным Н.Е. Полищука и Д.В. Гуляева [17], в Украине сложилась крайне
опасная ситуация, связанная с последствиями инсульта. В отличие от многих
других стран, где инсульт занимает среди причин смерти третье место, на Украине
он значительно опередил злокачественные новообразования и уверенно занимает
второе место. Смертность от инсульта среди мужчин в возрасте 45-74 лет
составляет 606, среди женщин - 408 человек на 100 тыс. населения. Это
соответственно в 11,2 и 12,75 раз выше по сравнению со Швейцарией и в несколько
раз, чем в других странах Европы. Даже в сравнении с Россией, от которой Украина
мало отличается по социально-экономическому развитию и структуре системы
здравоохранения, смертность от инсульта среди мужчин в 1,5 раза, а среди женщин
- почти в 2 раза выше. При этом статистика инсульта в Украине имеет тенденцию к
дальнейшему ухудшению, тогда как во многих других странах ситуация
существенно улучшается. Авторы отмечают, что смертность от инфаркта миокарда
снижается, однако это нельзя объяснить улучшением в материальной сфере,
причина скорее заключается в действенной реорганизации кардиологической
службы и внедрении в практику современных мировых стандартов.
6
Это обуславливает необходимость пересмотра существующей стратегии
интенсивной терапии острого периода ишемического инсульта, опираясь на
современные международные рекомендации, которые базируются на принципах
доказательной медицины.
Глава 2
ОПРЕДЕЛЕНИЕ И КЛАССИФИКАЦИЯ КЛИНИЧЕСКИХ ФОРМ
ОСТРОГО НАРУШЕНИЯ МОЗГОВОГО КРОВООБРАЩЕНИЯ ПО
ИШЕМИЧЕСКОМУ ТИПУ
Инсульт - острое нарушение мозгового кровообращения (ОНМК),
характеризующееся внезапным (в течение минут, реже часов) появлением очаговой
неврологической симптоматики (двигательных, речевых, чувствительных,
координаторных, зрительных и других нарушений), иногда общемозговых
нарушений (изменение сознания, головная боль, рвота и другие), подтвержденных
или нет данными компьютерной томографии, которые сохраняются более 24 часов,
либо приводят к смерти больного в более короткий промежуток времени вследствие
причины цереброваскулярного происхождения [108].
Понятие «ишемический инсульт» соответствует гипоксическому поражению
участка паренхимы головного мозга, возникающему в результате уменьшения
кровотока в определённой зоне мозга и недостаточного обеспечения
метаболических потребностей нервной ткани этого участка при дефиците
перфузионного давления. Выделяют ряд факторов риска развития ишемического
инсульта (табл. 1). Причинами ишемического инсульта выступают (табл. 2):
50 % - осложнения атеросклероза сонных, позвоночных и внутримозговых
сосудов;
22 % - стенозы и окклюзии внутримозговых артериол;
18 % - кардиогенная эмболия;
10 % - нарушения микроциркуляции.
Факторы риска ишемического инсульта
Таблица 1
Факторы Увеличение риска Меры профилактики
развития
Эндогенные
Гипертония В 6 раз Контроль артериального давления. Регулярный прием антигипертензивных препаратов
Диабет В 3 раза Контроль уровня глюкозы в крови. Регулярный прием _ антидиабетических препаратов
7
Гиперлипидемия В 2 раза Диета. Регулярный прием гиполипидемических препаратов (липанор)
—- - —————. , Консультация гематолога
Полицитемия В 2 раза
Ишемическая болезнь сердца В 6 раз при аритмии Лечение ИБС
Транзиторные ишемические атаки (ТИА) в анамнезе На 50%, в течение года после ТИА Регулярный прием антиагрегантных препаратов (тиклопедин)
Экзогенные
Курение В 3 раза Отказ от курения. _ Прием препарата никорте Применение противозачаточных средств под контролем врача
Прием оральных контрацептивов В 2-3 раза
Алкоголизм В 2 раза Лечение у нарколога. Прием препарата эспераль
Таблица 2
Причины ишемического инсульта
Сосудистые заболевания Заболевания сердца Заболевания крови
Атеросклероз Фиброзно-мышечная дисплазия Воспалительные заболевания сосудов: - гигантоклеточный артериит; - системная красная волчанка; - ревматический полиартрит; - гранулематозный ангиит; - сифилитический артериит; - СПИД. Диссекция стенки артерии Липогиалиноз мелких Ревматическое поражение сердца Бактериапьный эндокардит Небактериальный (марантический) эндокардит Миксома предсердия Внутрижелудочковый (ромб Аритмии Проляпс митрального клапана Искусственный клапан сердца Внезапная остановка сердца Парадоксальный эмболизм Тромбоцитоз Полицитемия С ерповидноклеточная анемия Лейкоцитоз Г илеркоагуляционные нарушения системы крови Коагулопатии Эритроцитоз Г емоглобинонатии Лейкоцитозы
8
артерий головного мозга Мигрень Тромбоз вен и синусов головного мозга при врожденных аномалиях сердца
Факторы ишемического инсульта подразделяют на локальные и системные
(табл. 3).
Таблица 3
Локальные и системные факторы инсульта
Локальные Системные
Морфологические изменения артерий мозга Поражение сердца Изменения шейного отдела позвоночника Нарушение центральной гемодинамики Нарушение церебральной гемодинамики Нарушение газотранспортной функции крови
По МКБ - 10 ишемический инсульт (инфаркт мозга) относится к
цереброваскулярным заболеваниям и классифицируется:
163. Инфаркт мозга
163.0 инфаркт мозга, вызванный тромбозом прецеребральных артерий;
163.1 инфаркт мозга, вызванный эмболией прецеребральных артерий;
163.2 инфаркт мозга, вызванный неуточненной закупоркой или стенозом
прецеребральных артерий;
163.3 инфаркт мозга, вызванный тромбозом мозговых артерий;
163.4 инфаркт мозга, вызванный эмболией мозговых артерий;
163.5 инфаркт мозга, вызванный неуточненной закупоркой или стенозом
мозговых артерий;
163.6 инфаркт мозга, вызванный тромбозом вен мозга, непиогенный;
163.8 другой инфаркт мозга;
163.9 инфаркт мозга неуточненный.
В зависимости от типа формирования и длительности существования
неврологического дефицита Комитет экспертов ВОЗ по сосудистой патологии
рекомендует’ выделять такие клинические формы острых ишемических нарушений
мозгового кровообращения:
> транзиторные ишемические атаки (ТИА) (регресс неврологической
симптоматики до 24 часов);
> пролонгированные ишемические атаки с обратимым развитием, или
малый инсульт (minor stroke) (регресс неврологической симптоматики в
пределах 21 дня);
> прогрессирующий ишемический инсульт (stroke-in-evolution);
> завершенный (тотальный) ишемический инсульт (major stroke);
лакунарный инфаркт.
9
Ишемический инсульт может быть:
> атеротромботическим;
кардиоэмболическим;
> лакунарным;
г гемодинамическим;
> реологическим.
Ишемический тромботический инсульт развивается в результате тромбоза,
которому способствуют патологические изменения стенки артерии (разрастание
интимы, повреждение эпителия, атероматозные бляшки, вызывающие сужение
просвета сосуда).
После 65 лет атеротромботический инсульт наблюдается в 75% случаев
данного заболевания.
Атеросклеротические бляшки поражают крупные экстракраниальные сосуды
(сонные, позвоночные артерии), интрацеребральные артерии, преимущественно в
местах их деления и извитости.
Нарушение мозгового кровообращения ишемического характера связано с
редукцией кровотока в магистральных артериях головы в результате
стенозирования сосуда, эмболизации, возникающей из-за дегенеративного распада
или тромботической закупорки сложных атеросклеротических бляшек. Спектр
клинических проявлений связан со степенью стенозирования, размером эмбола, его
состава, склонностью к распаду и состоянием коллатерального кровообращения.
Большое значение в патогенезе и течении ишемического инсульта имеет
патология сердца. Причиной 15-20% всех случаев ишемического инсульта служит
кардиогенная тромбоэмболия сосудов мозга: каждый шестой ишемический инсульт
является кардиоэмболическим. Не менее важную роль играют и расстройства
системной гемодинамики кардиального генеза, которые могут быть причиной
глобальной ишемии мозга.
Установлено, что кардиоцеребральные эмболии возникают преимущественно
при фибрилляции предсердий (ФП), остром инфаркте миокарда (ОИМ),
постинфарктных аневризмах и т ромбах в полости левого желудочка, ревматическом
поражении сердца. Так, риск развития инсульта у больных с ФП в 5 раз выше по
сравнению с лицами того же возраста без ФП.
Причиной кардиоэмболической ишемии является ОИМ. У 2,5-4% больных,
перенесших ОИМ, возникает ОНМК. Самый высокий риск ишемического инсульта
- на первой-четвертой неделе после ОИМ. Антиагрегантная терапия, начатая в
первые часы после развития ОИМ, уменьшает частоту инсульта в 2 раза.
Ревматические поражения сердца выступают причиной кардиоэмболической
ишемии мозга. Наиболее высокий эмбологенный потенциал ревматического
митрального стеноза (20%). Риск развития ишемического инсульта имеется у
больных с протезированными клапанами сердца.
Около 20-30% случаев ишемического инсульта составляют лакунарные
инфаркты (с развитием мелких очагов некроза), которые локализуются в белом
веществе полушарий большого мозга, мозжечке, стволе мозга и морфологически
представляют разновидность белого инфаркта мозга. Лакунарный инфаркт
обусловлен поражением (стенозированием, тромбозом) мелких перфорирующих
10
ветвей средней мозговой артерии, задней мозговой артерии и базилярной артерии,
которое приводит к развитию локальной ишемии и небольшого инфаркта в
бассейне пораженной артерии, что связано как с особенностями архитектоники
сосудов (отхождение от сосудов под прямым утлом, отсутствие анастомозов)
обуславливающих невозможность амортизации при резких колебаниях
артериального давления (АД), так и с поражением мелких сосудов, возрастными
изменениями, артериальной гипертензией, сахарным диабетом [4].
По МКБ-10 лакунарный инфаркт классифицируется:
G46 «Сосудистые мозговые синдромы при цереброваскулярных заболеваниях»
G46,5 моторный лакунарный синдром;
G46,6 сенсорный лакунарный синдром;
G46,7 другие лакунарные синдромы.
Лакунарный инфаркт в начале заболевания может иметь форму ТИА или
малого инсульта и возникает на фоне повышения системного артериального
давления. Общемозговая симптоматика, нарушение сознания и менингиальный
комплекс нехарактерны. Исход лакунарного инфаркта обычно благоприятный с
частичным дефицитом или полным восстановлением неврологических функций.
Гемодинамические ишемические поражения мозгового кровообращения возникают
при стенозе экстра- и интракраниальных сосудов, когда артериальное давление
Таблица 4
Периоды после развития ишемического инсульта
«Терапевтическ ое окно»
0 | 3-6 часов 24 часа 21сутки 6 месяцев 2 года
ОСТРЕЙШИЙ ПЕРИОД ОСТРЫЙ ПЕРИОД ПОЗДНИЙ ВОССТАНОВИ ТЕЛЬНЫЙ ПЕРИОД СТОЙКИЕ ОСТАТОЧ НЫЕ ЯВЛЕНИЯ
Преходящие нарушения | Малый | Инсульт со стойкими
мозгового кровообращения 1 инсульт остаточными явлениями
падает ниже границы ауторегуляции мозгового кровообращения, что ведет к
гипоперфузии головного мозга. Резкое снижение АД может возникнуть при
передозировке гипотензивных средств, при инфаркте миокарда, аритмиях,
кровотечениях и др.
Ряд гематологических нарушений, таких как полицитемия, диспротеинемия,
ДВС-синдром, тромбоцитопеническая пурпура, могут привести к гиперкоагуляции
и повышению вязкости крови, что способствует развитию тромбозов в
церебральных артериях (реологический инсульт).
Периоды после начала ишемического инсульта представлены на таблице 4.
Таким образом, первые 3-6 часов после развития инсульта, относятся к
острейшему, а от 24 часов до 21 суток к острому периоду ишемического инсульта.
Именно этот период является самым ответственным в терапии ишемического
инсульта, которая в большинстве случаев проводится в реанимационном отделении
врачами анестезиологами-реаниматологами. Последующие этапы, включающие
11
поздний восстановительный и период остаточных явлений, является прерогативной
врачей неврологов.
Глава 3
ЦЕРЕБРАЛЬНЫЙ КРОВОТОК В НОРМЕ И ПАТОЛОГИИ
ЗЛ Нейрональная регуляция церебрального кровотока
Одними из главных факторов регуляции мозгового кровотока (МК) является
нейрональная активность. Структурно-функциональное единство мозга как
целостной биологической системы зависит от постоянного поступления кислорода
и субстратов посредством МК. Имея незначительную, в сравнении с массой тела,
массу мозга (2 %), МК составляет 20 % от сердечного выброса (СВ) и
обуславливает высокий уровень метаболизма. При этом мозг не имеет в своем
распоряжении сколько-нибудь значительного энергетического пула, который бы
позволил обеспечить хотя бы временное автономное функционирование в случае
прекращения кровотока. С этих позиций мозг, по образному выражению акад. Г.А.
Рябова, «...можно уподобить высокоорганизованному промышленному
предприятию, которое в отсутствие нормативных запасов может эффективно
функционировать лишь при регулярном и бесперебойном поступлении сырьевых
ресурсов» [19]. Это обуславливает исключительную гипоксическую уязвимость
головного мозга по сравнению с другими органами и тканями. Нарушение доставки
кровью кислорода и субстратов неизбежно ведет к повреждению мозга. Увеличение
МК ведет к активации функций мозга за счет улучшения удовлетворения
энергетических потребностей и нормализации метаболизма.
Для доказательства регулирующего влияния активности отдельных нейронов
на МК выдвигают два положения:
1) Взаимосвязь между кровотоком в различных регионах мозга и
скоростью церебрального метаболизма глюкозы (СЦМ§1и) отражает
различную активность нейронов [92,73]. Поэтому области со
сравнительно низким уровнем CIJMglu (например, мозолистое тело)
имеют низкий уровень МК, а области с высоким уровнем метаболизма
глюкозы (слуховая зона коры) соответственно имеют высокий уровень
МК.
2) Повышение активности головного мозга вызывает локальное усиление
МК, пропорциональное степени активации [51,57,61]. И наоборот
снижение активности мозга вызывает пропорциональное уменьшение
МК [85,104].
В настоящее время концепция взаимосвязи между активностью нейронов и
уровнем МК является общепринятой. Напротив, соматосенсорные и визуальные
стимулы увеличивают МК и ClJMglu вне связи с повышением потребления
кислорода [52,53]. Отсутствие взаимосвязи между скоростью потребления глюкозы
и скоростью утилизации кислорода объясняется тем, что головной мозг генерирует
энергию посредством аэробного метаболизма, однако этот вопрос до конца неясен.
Так, предполагается, что энергия в мозге образуется частично путем анаэробного
метаболизма, т.е. глюкоза метаболизируется до лактата посредством гликолиза и
этот процесс, возможно, происходит в астроцитах. В подтверждение данной
12
гипотезы выступают результаты исследований, в которых было показано, что
повышение активности нейронов сопровождалось увеличением содержания лактата
в мозге [88,104].
Изучение механизмов взаимосвязи между изменениями церебрального
кровотока и нейрональной активностью позволили выдвинуть также гипотезу,
предполагающую наличие вазоактивных факторов.
Такой вазоактивный агент в идеале должен обладать следующими свойствами:
]) высвобождаться во внеклеточное пространство пропорционально интенсивности
синаптической активации; 2) обладать высокой диффузионной способностью и 3)
быстро элиминироваться. В последующем был открыт целый ряд таких агентов
(рис. 1). Рядом авторов было выдвинуто предположение, что астроциты могут
осуществлять контроль за микроциркуляцией в зависимости от активности мозга за
счет проникновения внеклеточного К+ в периваскулярное пространство [86]. Данная
концепция подверглась проверке двумя экспериментальными исследованиями, в
результате которых было показано, что нейрональная активация вызывала
повышение уровня ТС, однако недостаточное для того, чтобы вызвать повышение
МК.
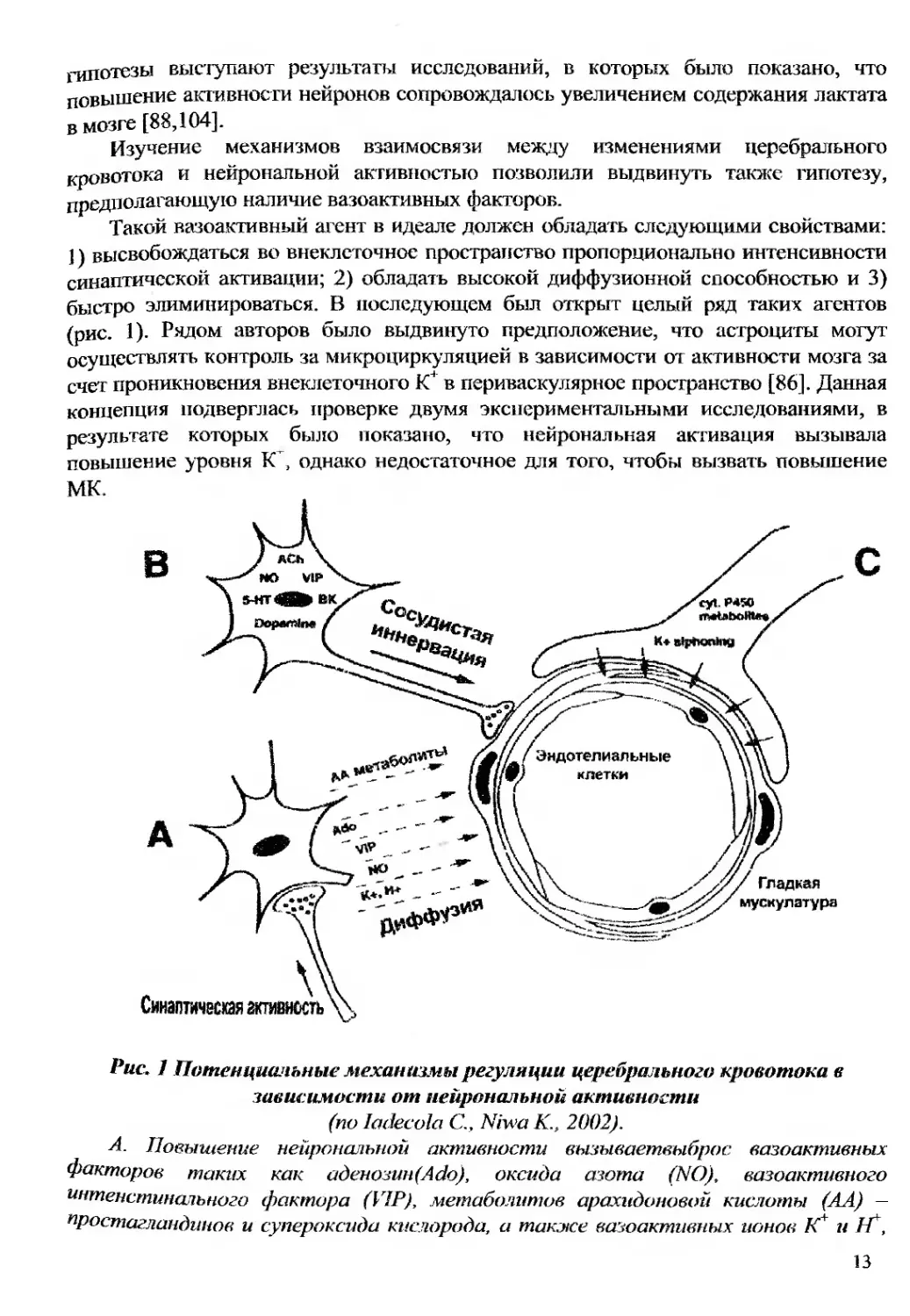
Рис. 1 Потенциальные механизмы регуляции церебрального кровотока в
зависимости от нейрональной активности
(по ladecola С., Niwa К., 2002).
А. Повышение нейрональной активности вызываетвыброс вазоактивных
факторов таких как аденозин)Ado), оксида азота (NO), вазоактивного
интенстиналъного фактора (VIP), метаболитов арахидоновой кислоты (АА) —
простагландинов и супероксида кислорода, а также вазоактивных ионов К и 1Г,
13
которые диффундируют и продуцируют релаксацию гладких мышц сосудов, что
приводит к повышению кровотока в активированном регионе мозга.
В. Церебральный кровеносный сосуд иннервируется нейронами из центральной
и периферической нервной системы, их нервные окончания содержат несколько
нейротрансмиттеров и нейропептидов: ацетилхолин (Ach), VIP, брадикинин (ВК).
катехоламины включая дофамин, как првило серотонин (5-НТ) и NO.
С. Астроцит контактирует с церебральными артериолами и капиллярами,
играя свою роль в цереброваскулярной регуляции. Все регулирующие влияния на
церебральные сосуды, астроциты оказывают посредством ионо К+ генерируемых
нейрональной активностью и ферментами системы цитохром-450.
При этом было отмечено, что К+ может действовать как медиатор
вазодилатации при нейрональной активации [44]. В качестве других вазоактивных
факторов МК выдвигаются: аденозин [71,76], катехоламины и интенстинальный
пептид (VTP) [32,33]. Также было показано, что астроциты содержа! ферменты
цитохром-450, которые являются метаболитами арахидоновой кислоты, причем
один из них - эноксиэйкозатриеновая кислота способна влиять на МК при
изменении активности нейронов [66].
Особое место занимают оксид азота (NO) и циклооксигеназа-2.
NO - самая маленькая по размерам молекула, синтезируемая в клетке и
проявляющая биологическую активность. Особый интерес к этой молекуле возник
тогда, когда была изучена природа эндотелиального фактора релаксации сосудов
(EDRF), обеспечивающего расслабление гладких мышц сосудов и было
установлено, что основным вазодилатирующим компонентом EDRFs является NO.
За открытие биологической активности NO в сердечно-сосудистой системе
R.F. Furcligott, F. Murad и L. Ignaro в 1998 г., была присуждена Нобелевская премия.
NO не является классическим трансмиттером, взаимодействующим с
мембраносвязанными рецепторами. Обладая высокой диффузионной способностью,
NO обеспечивает свою биологическую активность за счет своей реакционной
способности, поскольку молекулы NO являются свободными радикалами и легко
реагируют с другими соединениями, содержащими неспаренные электроны (О2, О2,
ионы металлов).
В организме человека NO образуется из L-аргинина и О2 посредством фермента
NO-синтазы (NOS). Существует три изоформы NOS: нейрональная, индуцируемая и
эндотелиальная. При этом нейрональная и эндотелиальная NOS являются
конститутивными ферментами, а индуцируемая NOS образуется в ряде клеток под
воздействием липополисахарида (LPS) и цитокинов.
При этом необходимо подчеркнуть, что синтез NO с участием NOS является
основным, но не единственным путем его образования. Так, в условиях тканевого
ацидоза NO генерируется путем биогрансформации нигрита (NO2-), а в условиях
гипоксии путем реакции между аргинином и перекисью водорода (Н2О2), а также
катализирования ксантиноксидазой восстановления NO2- в NO [20666]. Что, играет
определенную роль в патогенезе синдрома полиорганной недостаточности (СПОН)
при развитии критических и терминальных состояний.
Весь спектр биологических эффектов NO подразделяется на:
14
а) регуляторное влияние - на сосудистый тонус, адгезию клеток, проницаемость
сосудов, агрегацию тромбоцитов, функцию почек, систему противоопухолевого
иммунитета;
б) защитное действие - антиоксидантное, ингибирующее адгезию лейкоцитов,
блокирование токсических эффектов TNFa;
в) повреждающее действие - ингибирование ферментативной активности,
повреждение структуры ДНК, индукция процессов перекисного окисления липидов
(ПОЛ), снижение антиоксидантного потенциала клеток и повышение их
чувствительности к реперфузии, алкилирующим агентам и ионам тяжелых металлов
[20]. Т.е. биологический ответ на воздействие NO может быть разным и
определяется условиями его генерации.
Активность нейрональной NOS зависит от Са27кальмодуллина [40].
Вазодилатирующий эффект NO связан с активацией растворимой формы фермента
гуанилатциклазы, катализирующей образование цГМФ из гуанозинтрифосфата.
Следовательно, NO может действовать как внутриклеточный мессенжер и
обеспечивать дилатирующее влияние на церебральные артерии, а также, возможно,
участвовать в регуляции МК в условиях различной функциональной активности
нейронов. Однако большинство исследований не выявило взаимосвязи между NO и
вазодилатацией, вызванной нейрональной активностью. В настоящее время
считается, что в коре головного мозга NO действует больше как модулятор реакции,
нежели конечный медиатор вазодилатации [66]. Напротив, в мозжечке NO
выступает в роле облигатного медиатора вазодилатации, продуцируемый
нейрональной активностью, что указывает на различную роль, которую играет NO в
механизмах функциональной гиперемии в различных регионах мозга [33,64].
Циклооксигеназа - 2 (ЦОГ-2) - катализирует образование простагландинов и
тромбоксанов из арахидоновой кислоты. В большинстве органов ЦОГ-2 в норме
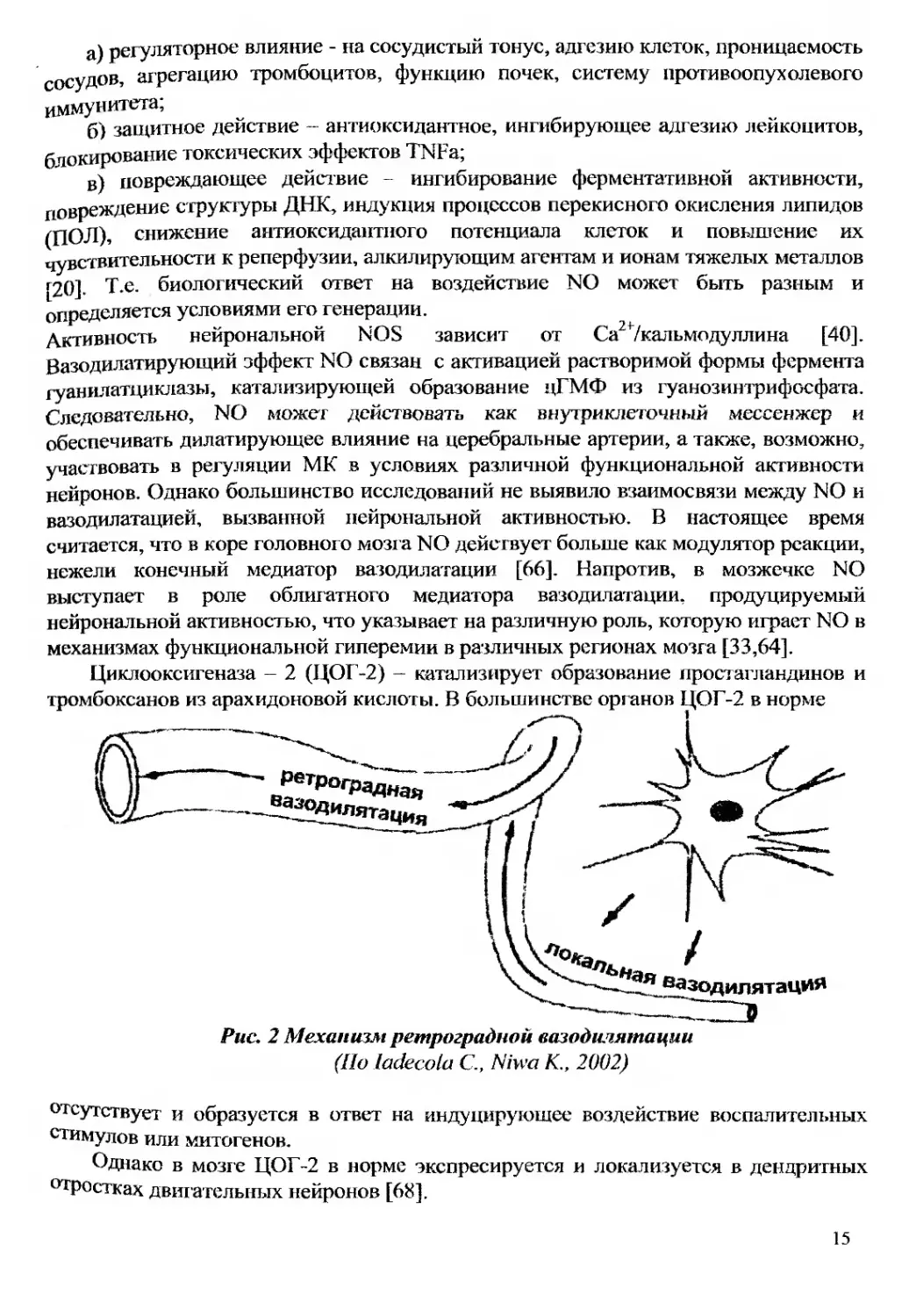
Рис. 2 Механизм ретроградной вазодилятации
(По ladecola С., Niwa К., 2002)
отсутствует и образуется в ответ на индуцирующее воздействие воспалительных
стимулов или митогенов.
Однако в мозге ЦОГ-2 в норме экспресируется и локализуется в дендритных
отростках двигательных нейронов [68].
15
Выявлено вазодилатирующее воздействие ЦОГ-2, индуцированное
нейрональной активностью посредством метаболитов, таких как простагландин Е2
(PGly) и супероксид кислорода (О2 ) [66].
Рядом исследователей было показано, что при активации коры головного мозга
или коры мозжечка вазодилатация наблюдается в артериолах, отдаленно
расположенных от места активации [61,62]. Для объяснения этого явления был
предложен механизм ретроградной вазодилатации. Предполагается, что
активированными нейронами посылается вазодилатирующий сигнал, вызывающий
локальную сосудистую реакцию, с последующим ее ретроградным
распространением в артериолы и артерии посредством внутренних сосудистых
механизмов (рис. 2)
Координация между паренхимальной нейрональной активностью и сосудистым
кровотоком осуществляется периваскулярными нервами: а) наружными,
отходящими от краниальных вегетативных ганглиев; б) внутренними нервами,
отходящими от центральных нервов. Окончания как наружных, так и внутренних
нервов содержат нейротрансмиттеры и нейропептиды, такие как NO, ацетилхолин,
VIP, нейропептид Y, катехоламины, брадикинин, субстанция Р, серотонин и
кальцитонин ген-связанный пептид [66].
Таким образом, нейрональная активность является главным фактором
регуляции МК, осуществляемой посредством разнообразных нейротрансмиттеров и
нейропептидов, при этом механизм регуляции МК зависит от нейрональной
цепочки и химической анатомии каждого региона головного мозга в отдельности.
3.2 Ауторегуляция мозгового кровотока
Головной мозг расположен в черепной коробке, которая защищает его от
внешних воздействий. Изнутри полость черепа и позвоночного канала выстлана
твердой мозговой оболочкой, которая состоит из двух спаянных между собой
фиброзных листков. В определенных местах эти листки расщепляются, формируя
венозные синусы, через которые отводится кровь из головного мозга. Стенки
синусов неподатливы и не спадаются. Поэтому при ранении синуса возникает
опасность воздушной эмболии. Впадающие в синусы вены лишены прочных стенок
и легко спадаются, что является причиной нарушения венозного оттока, например,
при повышении давления спиномозговой жидкости.
При вертикальном положении человека отток крови из полости черепа
осуществляется, в основном, по вертебробазилярной венозной системе, а в
горизонтальном положении - по яремным венам.
Между твердой мозговой оболочкой и костями черепа имеется эпидуральное
пространство. Под твердой мозговой оболочкой располагается
соединительнотканная паутинная оболочка. Между ними находится субдуральное
пространство. Третья оболочка - мягкая (сосудистая) - плотно прилегает к
поверхности головного мозга, проникая в глубину всех борозд и щелей.
Пространство между паутинной и мягкой оболочками называется
субарахноидальным пространством. Оно заполнено спинномозговой жидкостью.
Содержимое черепа - многокомпонентная среда, составные части которой занимают
16
разные объемы: спинномозговая жидкость - 10-12%, кровь - 5-10%, тканевая вода -
60-70%, твердый остаток - 10-15% от общего внутричерепного объема.
Согласно доктрине Монро - Келли, вещество головного мозга является
практически несжимаемым. В связи с этим количество крови в полости черепа
должно быть постоянным или почти постоянным. Когда вода или другое вещество
выходят или секретируются из сосудов, из черепа вытесняется количество крови,
равное по объему введенному веществу. Однако небольшие изменения в объеме
одного из компонентов содержимого черепа могут не сопровождаться
компенсаторными изменениями объемов других компонентов [29].
Резервный объем краниоспинального пространства может увеличиваться за
счет эластических свойств дурального мешка, увеличения перидурального
пространства спинного мозга, связанного косвенно с объемом плевральной полости
и «сбросом» давления в них.
Вода из интерстициальных пространств свободно проникает в клетки
головного мозга но законам осмоса.
Объем спинномозговой жидкости находится в прямой зависимости от ее
продукции и в обратной зависимости от ее резорбции, объема головного мозга и
крови в черепе.
Объем крови - наиболее быстро изменяющийся компонент содержимого
черепа, на который в полости черепа влияет ряд факторов. Чем выше тонус
мозговых сосудов, тем меньше крови содержится в них. Объем крови в полости
черепа остается постоянным при изменении системного артериального давления от
60 до 150 mm Hg за счет миогенпой и нейрогенной регуляции сосудистого тонуса.
Венозный участок сосудистого русла головного мозга называют «емкостным»,
так как в нем содержится до 50% общего внутричерепного объема крови. Объем
крови в венозном русле увеличивается в результате следующих причин:
I) увеличения венозного давления в черепе при нарушении венозного оттока;
2) увеличения центрального венозного давления (ЦВД), когда повышение
давления по системе бесклапанных полых и яремных вен свободно передается в
венозную систему черепа, что ведет к рефлюксу венозной крови.
Особенностью кровоснабжения головного мозга является существование
феномена саморегуляции мозгового кровотока, под которым понимают
механизмы, обеспечивающие постоянство ЦПД при изменениях АД или
внутричерепного давления (ВЧД), т.е. способность поддерживать свое
кровоснабжение в соответствии с метаболическими потребностями независимо от
колебаний системного АД. Так, у здоровых лиц при колебании АДсист. в диапазоне
от 50-70 до 180-200 mm Hg саморегуляция мозгового кровотока сохраняется. У
пациентов с артериальной гипертензией нижняя граница АДсист., при котором
сохраняется саморегуляция мозгового кровотока, лежит в пределах 110-120 mm Hg,
а верхняя граница достигает 240-280 mm Hg. Снижение АДсист. на 30 % от
исходного уровня вызывает уменьшение мозгового кровотока, что необходимо
Учитывать при проведении антигипертензионной терапии у таких лиц.
Механизмы ауторегуляции обеспечивают постоянство мозгового кровотока и
энергетического обмена головного мозга при изменениях среднего АД в диапазоне
60-160 mm Hg.
17
Регуляция мозгового кровотока осуществляется следующими механизмами
[29]:
метаболическим -центральным звеном регуляции является pH внеклеточной
жидкости и гладкомышечных элементов сосудистой стенки. Метаболические
сдвиги (РСО2, РО2, лактат) быстро изменяют pH и тем самым тонус и просвет
сосудов головного мозга;
нейрогенным и нейрогуморальным - симпатическая и парасимпатическая
иннервация экстра- и интракраниальных отделов сосудов головного мозга с
участием соответствующих медиаторов: ацетилхолина, норадреналина, дофамина,
адреналина;
миогенным (эффект Бейлиса-Остроумова) - сужение или расширение
сосудов головного мозга при изменении внутрисосудистого давления;
механическим типом регуляции - возрастание сосудистой резистентности (в
ответ на повышение внутрисосудистого давления), увеличение тканевого давления
вследствие экстракапиллярного пропотевания жидкости.
Основными факторами, изменяющими мозговой кровоток и энергетический
обмен, выступают:
возрастание артериального или тканевого (церебрального) напряжения
углекислого газа расширяет мозговые сосуды и увеличивает мозговой кровоток.
Сдвиг РаСО2 (в диапазоне от 20 до 60 mm Hg) изменяет мозговой кровоток в
среднем на 1 -2% на каждый 1 мм Hg;
артериальная гипокапния приводит к спазму мозговых сосудов и
уменьшению мозгового кровотока, снижению внутричерепного объема крови.
Однако падение РаСО2 ниже 15'20 mm Hg ведет к циркуляторной гипоксии
головного мозга, анаэробному обмену, нарастанию лактата, ацидозу мозговой ткани
и спинномозговой жидкости. Поэтому нецелесообразно снижать РаСО2 ниже 30-35
mm Hg, особенно во время анестезии;
колебания РаО2 меньше влияют на мозговой кровоток, чем углекислый газ.
Ингаляции 85-100% кислорода при нормальном атмосферном давлении уменьшают
мозговой кровоток на 13-15%;
искусственная вентиляция легких (ИВ Л) чистым кислородом (при
постоянном РаСО2) уменьшает кровоток в коре головного мозга соответственно на
12% и 20% при давлении 1 и 2 атм. Потребление головным мозгом кислорода
остается неизменным;
гипербарическая оксигенация (ГБО) в условиях 3,5 атм. уменьшает мозговой
кровоток на 35%;
артериальная гипоксия (при нормокапнии) расширяет сосуды и увеличивает
мозговой кровоток только при снижении РаО2 ниже 60 mm Hg;
изменения кислотно-основного состояния (КОС) метаболического характера
не влияют существенно на мозговой кровоток, так как ионы водорода или
гидрокарбоната плохо проникают через ГЭБ. Выраженные метаболические сдвиги
КОС, вероятно, изменяют тонус сосудов головного мозга, вызывая компенсаторные
изменения РаСО2;
нарушения ауторегуляции мозгового кровотока в зоне поражения приводит к
появлению феномена «внутримозгового обкрадывания» при использовании
18
зодилататоров или феномена «обратного внутримозгового обкрадывания»
/^ндром Робин Гуда) при гипервентиляции.
' При коматозных состояниях различной этиологии мозговой кровоток может
быть выше его метаболической потребности, что приводит к развитию так
называемого синдрома избыточной перфузии.
3.3 Концепция порогового ишемического кровотока
Концепция порогового ишемического кровотока определяется критически
низким уровнем мозгового кровотока (МК) и недостаточным поступлением
кислорода в ткань мозга.
Известно, что мозговой кровоток для серого и белого вещества у здорового
человека составляет 50-55 мл/ЮОг/мин1. Для мозга среднего веса (1300-1400 г)
общий мозговой кровоток (ОМК) в покое равен 750-800 мл/мин., что составляет
15-20% сердечного выброса.
При таком уровне МК общее потребление кислорода мозгом, измеряемое как
скорость церебрального метаболизма кислорода (СЦМО2) составляет 3,3-3,5
мл/Ю0г/мин 1 или 45 мл кислорода в минуту, т.е. 20% общего потребления
кислорода организмом в покое. Часть кислорода, извлеченного из крови, считается
фракцией экстракции кислорода (ФЭК), являющейся постоянной величиной
(около одной трети), потому что ОМК, МК, СЦМО2 и скорость церебрального
метаболизма глюкозы (CIJMglu) тесно связаны между собой.
Кровоток в сосудах мозга в норме определяется также церебральным
перфузионным давлением (ЦПД), которое соответствует среднему системному
АДср. в сосудах основания мозга:
ЦПД = диастолическое АД + 1/3 пульсового АД - внутричерепное венозное
давление (около 10 mm Hg)
(т.е. в норме ЦПД равняется 70-90 mm Hg)
При снижении ЦПД происходит компенсаторная вазодилатация артерий,
артериол, обеспечивающая поддержание МК на оптимальном уровне, что приводит
к увеличению объема крови в мозге и повышению ФЭК для поддержания
оксигенации тканей. При этом СЦМО2 и CUMglu поддерживается на достаточном
уровне, поэтому жалоб у пациента еще нет, т.к. снижается только перфузионный
резерв [5].
Другим показателем выступает сатурация кислородом крови оттекающей из
головного мозга по яремным венам (SvjO2), который зависит от следующих
преременных:
SvjO2 = SaO2 - СЦМО2/1,34хНЬхМКхЮ
При этом у гемодинамически стабильных и седированных пациентов,
следующие переменные, такие как SaO2, Hb и СЦМО2, являются постоянными и
таким образом SvjO2 - изменяется пропорционально изменениям МК, в связи с чем
изменил SvjO2 отражают изменения мозгового кровотока. Номальный уровень
SvjO2 составляет 55-71%. снижение этого показателя ниже 50% является
индикатором развития ишемии и нарушения между МК и метаболизмом.
При гиперемии головного мозга, SvjO2 повышается, поскольку снижается
аРгериовенозная разница по кислороду.
19
Таким образом, в неповрежденном мозге уровень МК тесно связан с
метаболическими потребностями. Эта взаимосвязь нарушается при острой
церебральной ишемии, которая сопровождается значительной диспропорцией
между МК и нейрональным метаболизмом (рис. 3).
Так, при снижении ЦПД ниже 40-50 мл/100г/мин 1 мозговой кровоток
изменяется параллельно АД, исчерпывается резерв мозговой перфузии, однако
активность метаболизма поддерживается за счет повышения ФЭК. Такое
несоответствие метаболических потребностей ткани уровню МК,
сопровождающееся увеличенной ФЭК, получило название «мизерной» перфузии
[36].
Первичные биохимические процессы в нейронах возникают в случае снижения
МК ниже 50 мл/ЮОг/мии-1 и проявляются торможением синтеза белков, но функция
нейронов ещё сохранена.
80-85
мм рт. ст.
Церебральное
j перфузионное
J давление
40-50 мм пт. ст.
750- 800 Объем крови
мл/мин ; 8 мозге
30% j- Фракция
j экстракции
! кислорода
50-55мл - Мозговой
/100г, мин ’ кровоток
850-900
! мп мин
J 46-50%
—i 20 м л
, • 00 г мин
3,3-3,5мл Скорость
/100 г,-мин ' i церебрального
j метаболизма О,
и глюкозы
Ишемии;
жалобы
-I 2,0 -2,5мл
; . 1 ПО Г МИН ‘
Ауторегулнция
сохранена;
нет жалоб
Снижение
перфузионног о
резерва;
инет жалоб
Рис. 3 Взаимосвязь между гемоциркуяяторными нарушениями и
нейрональным метаболизмом в норме и при острой церебральной ишемии в
условиях субкритической и критической («мизерной») перфузии ткани мозга
(по Виничук С.М., Черенъко Т.М., 2003)
Снижение МК ниже 35 мл/ЮОг/мин * стимулирует переключение метаболизма
глюкозы с аэробного пути на пугь анаэробного гликолиза, при этом возникают
кратковременные нарушения функции нейронов.
20
Уровень МК ниже 20 мл/ЮОг/мин1 - верхний ишемический порог (порог
ггпаты электрической функции нейронов с сохранением их мембранного
потенциала). Возникший дефицит энергообразования сопровождается повышением
тонуса сосудов мозга, нарушением реологических свойств крови и
микроциркуляции, появлением преходящих психоневрологических симптомов. При
1\4К ниже 15 мл/ЮОг/мин’1 исчезают электроэнцефалографические (ЭЭГ) и
вызванные потенциалы, но структурная организация нейронов сохраняется.
Рис. 4 Формирование инфарктного ядра и эволюция «ишемической полутени»
во времени.
Критическим порогом для необратимого повреждения клеток считают
10 мл/ЮОг/мин1 - нижний ишемический порог (порог утраты клеточного
ионного гомеостаза). На этой стадии недостаток Ог подавляет метаболизм в
митохондриях, нарушается функция энергозависимых клеточных мембран,
поддерживающих ионный гомеостаз, в результате развивается генерализованная
недостаточность функции мембран.
При снижении МК ниже 10 мл/ЮОг/мин*1 наступает абсолютная (полная)
ишемизация и в течении 6-8 мин. возникают необратимые повреждения нейронов
и клеток нейроглии - некроз, то есть формируется зона инфарктного ядра (рис. 4).
Выявление двух критических порогов кровотока - утраты функции нейронов и
Утраты клеточного ионного гомеостаза позволило сформулировать концепцию
ишемической полутени (ischemic penumbra) [34] - основной точке приложения
интенсивной терапии у пациентов с ишемическим инсультом (рис. 4).
3.4 Концепция «ишемической полутени»
Ишемическая полутень - это область ишемизированной, но жизнеспособной
ткани мозга, которая окружает зону инфарктного ядра, то есть это ткань в
21
состоянии риска, поскольку кровоток в ней находится между двумя ишемическими
порогами и соответствует «критической» перфузии, что не обеспечивает
метаболические запросы ткани мозга. Структурно-морфологическая организация
нейронов этой области сохранена, но имеется дефект её (функциональной
активности — утрата электрической функции нейронов. Вследствие того, что резерв
локальной перфузии исчерпан, нейроны в области ишемической полутени
становятся чувствительными к любому дальнейшему падению ЦПД, которое может
быть вызвано вторичной гиповолемией (после дегидратации), неадекватно
проводимой антигипертензионной терапией, быстрым вставанием больного и цц
[Ю].
К.А. Hossmann определяет ишемическую полутень, как зону с ограниченной
доставкой О2, в которой еще сохраняется энергетический метаболизм [63].
С клинической точки зрения значение ишемической полутени состоит в том,
что нарушение функции нейронов в ней в течение 1-6 часов имеют обратимый
характер. Именно за эту область мозговой ткани и ведется борьба в первые часы ц
дни заболевания, чтобы сохранить функцию нервных клеток, и, следовательно,
уменьшить выраженность неврологического дефицита.
Динамика функциональных и морфологических изменений нейронов
ишемической полутени возможна в двух направлениях: восстановление их функции
или трансформация в инфаркт.
Первичная зона инфарктного ядра формируется через 3-6 часов с момента
появления первых симптомов инсульта. Расширение очага инфаркта
продолжается в течение 24-48 часов, возможно, и позже в зависимости от степени
снижения МК и выраженности аутоиммунной реакции. После этого размеры
инфаркта практически не меняются.
Время, в течение которого с восстановлением перфузии сохраняется
возможность нормального метаболизма и функции, получило название
«терапевтического окна». В среднем продолжительность «терапевтического
окна», с момента развития инсульта для всех нейронов «ишемической полутени»
составляет 3 часа и не превышает 6 часов для нейронов самых
периферических отделов на границе с веществом мозга с нормальной перфузией и
метаболизмом кислорода [24].
В случае, если артериальная окклюзия носит временный характер или
происходит компенсаторное включение коллатеральной сети, мозговой кровоток
полностью или частично возвращается в ишемизированный участок. Однако
возвращение кровотока позже, чем через 2 минуты с момента устранения окклюзии,
на фоне протекающих уже ишемических процессов, не означает его нормализации.
Происходят поэтапные нарушения перфузии головного мозга, и начальная стадия
лостишемической гиперемии сменяется стадией постишемической гипоперфузии.
В механизме развития постишемической гиперперфузии или «роскошной
перфузии» лежит не только обильное поступление крови через коллатерали и
реканализация окклюзированной артерии, но и высвобождение из
ишемизированной ткани вазоактивных и провоспалительных метаболитов,
изменение нейрогенных механизмов вазодилатации, а также снижение вязкости
22
Причем имеющийся избыток кровотока не соответствует метаболическим
^тпебностям ткани мозга и сопровождается уменьшением ФЭК.
За стадией гиперемии происходит снижение МК ниже доишемического уровня,
является результатом отсроченных метаболических изменений в
щемизированной ткани, вызванных активацией микроглии и синтезом
большого количества провоспалительных факторов, ведущих к тяжелым
изменениям микроциркуляции и микроваскулярной обструкции. Такое
восстановление артериального кровотока, сопровождающееся неполной реперфузией
панее ишемизированной ткани, получило название «невосстановленный кровоток»
(no..reflow). Механизмами его формирования считаются: повышение вязкости и
внутрисосудистой свертываемости крови, развитие микроваскулярной окклюзии
сдавлением капилляров мозга, расположенными вокруг отечными астроцитами,
повышение внутричерепного давления, формирование отечности эндотелия
сосудистого русла, а также постишемическая гипотензия. В постишемическом
периоде механизмы саморегуляции мозгового кровотока нарушены, поэтому
функциональная активация мозга не приводит к адекватному нарастанию мозгового
кровотока.
В случае частичного восстановления кровотока компенсаторным включением
коллатералей развивается особое динамическое состояние ишемизированной ткани в
виде мозаичных изменений уровня перфузии (феномен «пространственной и
временной динамики микроциркуляции»). В зоне ишемической полутени и
инфарктного ядра возникает чередование зон относительной и/или абсолютной
гиперемии и гипоперфузии.
Согласно данных экспериментальных исследований, при острой фокальной
ишемии мозга, «терапевтическое окно» значительно более узкое, чем при глобальной
ишемии.
Исследования с использованием позитронно-эмиссионой томографии при
экспериментальном моделировании ишемического инсульта у приматов показало
значительную индивидуальную вариабельность границ «терапевтического окна» и
установило, что обратимые изменения в области ишемической полутени могут
сохраняться в течение многих часов, а иногда и нескольких дней после артериальной
окклюзии. По данным G. Marchal с соавт. (1996), через 18—24 ч после развития
инсульта в некротизированной зоне мозга была выявлена ишемизированная, но,
возможно, жизнеспособная ткань, характеризующаяся при мозговом кровотоке около
22 мл/100 г в 1 мин очень высокими значениями ФЭК и СЦМОг- Она может избежать
формирования инфаркта, и ее следует рассматривать как «подверженную риску
инфаркта». У отдельных пациентов «терапевтическое окно» оказалось значительно
Длиннее установленных ранее 3—6 ч. Такая индивидуальная вариабельность
«терапевтического окна» объясняется компенсаторными возможностями
коллатерального кровообращения, исходным состоянием метаболизма мозга,
Реактивностью нейроиммунноэндокринной системы.
В неповрежденном мозге повышение потребления глюкозы и кислорода ведет к
прямому увеличению МК, которое может достигать более чем 200% от исходного
Уровня. Напротив, в зоне ишемической полутени данный механизм нарушен из-за
снижения гемодинамической емкости коллатеральной системы обеспечивающей
23
соответствие между доставкой метаболитов кровью и потребностями ткани мозга.
Как результат это приводит к дисбалансу между высоким уровнем метаболических
потребностей мозга и низкой доставкой кислорода [10].
Исходя из вышеизложенного, построение стратегии терапевтического
воздействия на зону ишемической полугени должно базироваться на следующих
принципах: улучшение доставки кислорода за счет нормализации коллатерального
кровотока тромболизисом или реконструктивными вмешательствами на сосудах^
снижение обмена веществ головным мозгом путем применения фармакологических
препаратов или методов физической гипотермии, предотвращение
митохондриального повреждения использованием блокаторов Сал+-каналов и
ловушек свободных радикалов, что позволит сохранить зону потенциально
жизнеспособной нервной ткани и предотвратить расшерение зоны инфарктоного
ядра [63].
Глава 4
БИОХИМИЯ ГОЛОВНОГО МОЗГА В НОРМЕ И ПАТОЛОГИИ
4.1 Очерк биохимии головного мозга
Процессы, лежащие в основе таких специфических явлений, как проведение
нервных импульсов, возбудимость, способность к хранению и переработке
поступающей информации (память), а также многочисленные биохимические и
биофизические процессы, связанные с поддержанием своеобразно
пространственно-функциональной архитектоники мозга. с непрерывным
образованием функциональных ансамблей нейронов, с обновлением и
образованием синаптических структур и др., протекают со значительными
энергетическими затратами.
Эти наблюдения, а также наличие теснейшей связи между окислительными
процессами и функциональной активностью нервной ткани позволили
сформулировать принципиально важное положение о том, что интенсивность
энергетического обмена является одним из ведущих факторов, лимитирующих
деятельность мозга.
4.1.1 Потребление кислорода и глюкозы головным мозгом
Одним из важнейших показателей, характеризующих интенсивность
энергетического обмена, служит скорость дыхания. При определении артерио-
венозной разницы по кислороду установлено, что потребление кислорода мозгом
человека составляет 1,5-1,7 мкмоль/г/мин.
По интенсивности дыхания головной мозг занимаег ведущее место среди
крупных органов и тканей. Головной мозг, составляющий не более 2% массы тела,
потребляет до 20-25% всего поступающего в организм кислорода; более того, у
новорожденных животных или человека на дыхание головного мозга может
расходоваться до 50% общего количества кислорода, потребляемого организмом.
Сопоставление дыхания разных отделов мозга животных показывает общую
закономерность: снижение интенсивности дыхания по мере перехода от
эволюционно более молодых передних отделов мозга, к более старым задним
отделам. Максимальная интенсивность дыхания установлена в коре больших
24
полуШарий; далее по скорости поглощения кислорода отделы мозга можно
сположить в следующей последовательности: мозжечок и промежуточный мозг
^средний и продолговатый мозг > спинной мозг. Различия в интенсивности
дыхания отдельных зон коры больших полушарий (двигательная, слуховая,
зрительная и др.) выражены слабо. Высокая интенсивность окислительных реакций
характерна не для всей нервной ткани. Установлено, что периферические нервы
используют лишь около 3% того объема кислорода, который потребляется
эквивалентным по массе количеством ткани из центральных отделов нервной
системы.
Наряду с высокой скоростью дыхания для мозга характерно интенсивное
потребление глюкозы из крови. Ни один орган не потребляет глюкозу с такой
скоростью и в таких количествах, как мозг, и ни для одной ткани организма не
отмечено такой острой потребности в этом субстрате окисления для поддержания
нормального функционального состояния. Головным мозгом потребляется до 70%
глюкозы, образующейся в печени и выделяющейся из нее в кровь. Потребление
глюкозы мозгом взрослого человека, рассчитанное по артерио-венозной разнице,
составляет 0,25-0,30 мкмоль/г/мин [1].
Эти данные позволяют предположить, что глюкоза - основной субстрат
окисления в головном мозге. Значение дыхательного коэффициента, близкое к
единице, полностью подтверждает это и указывает на то, что основным путем
метаболизма глюкозы в головном мозге является ее окисление в реакциях
аэробного гликолиза, сопряженных с реакциями цикла трикарбоновых кислот.
Эксперименты с глюкозой показывают, что около 85-90% глюкозы,
потребляемой мозгом, полностью окисляется до СО2 и Н2О, около 5% расходуется в
реакциях гликолиза с образованием молочной кислоты и лишь 5-7% используется в
других реакциях (синтез гликогена, гликопротеидов, нейромедиаторов и др.).
Необычная зависимость функционирования головного мозга от притока
глюкозы из крови объясняется, прежде всего, тем, что собственные запасы данного
углевода в мозговой ткани чрезвычайно малы по сравнению с высокой
интенсивностью его окисления. Содержание глюкозы в мозге животных и человека
составляет в среднем 1,5-4,5 мкмоль/г. Даже при условии использования глюкозы
только для окисления запасы ее в мозге могут быть полностью исчерпаны за 3-6
минут.
При уменьшении содержания глюкозы в крови печень, почки, скелетные и
сердечная мышцы для поддержания энергетического баланса и сохранения
функциональной активности способны окислять целый ряд других субстратов
(аминокислоты, лактат, жирные кислоты, кетоновые тела и др.). Головной же мозг в
этих условиях продолжает потреблять по-прежнему большие количества глюкозы и
кислорода. И лишь при уровне глюкозы в крови ниже критического (тяжелая
гипогликемия) значительно уменьшается потребление мозговой тканью, как
глюкозы, так и кислорода и развивается коматозное состояние с потерей сознания.
Попытки компенсировать развитие комы и поддерживать энергетический
анс в головном мозге путем введения животным различных метаболитов
глюкозы (гексозофосфатов, лактата, пирувата, фруктозы, галактозы и др.) даже в
Весьма значительных количествах были неудачными; при гипогликемической коме
25
лишь внутривенные инъекции глюкозы могут нормализовать энергетически
метаболизм в мозге и вывести животное из коматозного состояния. Эти наблюдения
указывают на весьма ограниченную способность головного мозга компенсировать
уменьшенное поступление глюкозы за счет окисления других энергетических
субстратов. Основной причиной этого является низкая проницаемость
гематоэнцефалического барьера (ГЭБ) для других субстратов окисления.
Транспорт глюкозы в мозг осуществляется преимущественно с помощью
специальной системы переносчиков, активно функционирующей в широких
пределах концентраций глюкозы в крови - от 2,75 до 16,50 мкмоль/мл. На долю
пассивной диффузии приходится не более 5% общего потока глюкозы [1].
Очевидно, что в нормальных физиологических условиях поступление
глюкозы через ГЭБ не лимитирует ее метаболизм. Однако при гипогликемии или
усиленном использовании глюкозы, например, при интенсификации гликолиза в
условиях дефицита кислорода или при судорогах, скорость переноса глюкозы через
ГЭБ может ограничивать начальные этапы ее метаболизма.
Потребность мозга в кислороде и глюкозе заметно изменяется в ходе
онтогенеза, значительно повышаясь с его ростом и дифференцировкой, а также
формированием отдельных структурно - функциональных ансамблей нейронов. По
мере развития головного мозга скорость окисления глюкозы в нем возрастает, и
одновременно увеличивается зависимость функциональной активности нейронов от
интенсивности окислительных процессов как источника энергии.
4.1.2 Гликоген как возможный энергетический источник в головном
мозге.
Данные о содержании глюкозы в мозге и скорости ее потребления мозговой
тканью показывают, сколь незначительны собственные ресурсы этого метаболита в
мозге и объясняют зависимость функциональной активности головного мозга от
поступления углеводов с кровью. Возникает вопрос, в какой мере уровень глюкозы
в мозге может поддерживаться за счет гликогена.
Благодаря способности к отложению гликогена (главным образом в печени и
мышцах, и в меньшей степени в других органах и тканях) создаются условия для
накопления в норме некоторого резерва углеводов. При повышении энерготрат в
организме в результате возбуждения ЦНС обычно происходит усиление распада
гликогена и образование глюкозы. Известно, что при возбуждении ЦНС
повышаются функции ряда желез внутренней секреции (мозговое вещество
надпочечников, щитовидная железа, гипофиз и др.), гормоны которых
активизируют распад гликогена, прежде всего в печени и мышцах.
Содержание этого полисахарида в мозговой ткани разных животных
составляет 2*5-4,5 мкмоль/г (в расчете на глюкозу). Общее количество гликогена в
мозге эмбрионов и новорожденных в 2-3 раза больше, чем у взрослых. По мере
роста и дифференцировки мозга концентрация гликогена в нем быстро снижается и
далее в мозге взрослого поддерживается относительно постоянной [1].
Гликоген, как субстрат, участвует в энергетическом обмене. Расчеты
показывают, что лишь 7-10% энергетических потребностей головного мозга могут
покрываться благодаря расщеплению гликогена. Особенно важное значение этот
субстрат имеет в головном мозге при экстремальных состояниях, когда
26
енъшаетея поступление в мозг глюкозы из крови. Однако из-за небольших
У^е «пн пула гликогена в мозге полное расщепление его до глюкозы с
едуюшим окислением может произойти в течение 5-7 минут.
В Таким образом, собственные углеводные запасы в нервной ткани
сительно невелики и не могут обеспечить энергетические потребности
мально функционирующего мозга в течение длительного времени. Это
^бсгоятельство наряду с ограниченной способностью мозга использовать другие
°ч/бстоаты окисления лежит в основе характерной для нервной ткани зависимости
от постоянного поступления глюкозы из крови.
Высокая скорость потребления головным мозгом глюкозы и кислорода
сопряжена с интенсивным образованием макроэргических соединений. Среди
богатых энергией соединений в мозге основную долю составляют компоненты
адениннуклеотидной системы и креатинфосфат, в то время как доля трифосфатов,
гуанина, цитозина, уридина и других - менее 10% всех макроэргов.
Расчеты подтверждают, что в головном мозге скорость образования
макроэргических соединений значительно выше, чем в других тканях. По этому
показателю лишь сердечная мышца во время интенсивной работы приближается к
мозгу.
Интенсивность энергетического обмена тесно коррелирует с интенсивностью
функциональной деятельности головного мозга. Доказательством этого служат
данные о возрастных изменениях основных показателей энергетического
метаболизма. Многочисленными исследователями установлено, что в ходе
формирования, развития и окончательного становления функций мозга
увеличивается потребление кислорода и глюкозы и значительно повышается
скорость образования макроэргических соединений. По мере старения и
уменьшения функциональной активности головного мозга потребление кислорода,
глюкозы, а следовательно и интенсивность энергетического метаболизма
постепенно снижается.
4.1.3. Энергообеспечение специфических функций нервной ткани.
Изучение суммарных процессов окисления и образования энергопродукции в
мозге представляет собой одну сторону проблемы энергообеспечения; другая
сторона — выявление специфических процессов в нервной ткани, требующих
энергетических затрат.
Основные энергозависимые процессы, лежащие в основе специфических
функций нервной ткани: проведение нервных импульсов, образование
синаптических структур и функционирование синапсов, хранение и переработка
информации, трансмембранный перенос субстратов, поддержание определенной
пРостранственной ориентации структурных единиц нейрона, аксональный и
Ретроградный ток.
4.1.4 Свободные аминокислоты нервной системы.
Состав аминокислотного пула при нормальных физиологических условиях
Довольно стабилен и характерен для мозга. Нервная ткань обладает уникальной
способностью поддерживать содержание аминокислот на постоянном уровне при
Различных физиологических и даже некоторых патологических состояниях,
н^окислотный фонд мозга человека составляет в среднем 34 мкмоль на 1 г ткани,
27
что значительно превышает их содержание, как в плазме крови, так и в
спинномозговой жидкости.
Для головного мозга характерны высокая концентрация глутаминовой
кислоты, глутамина, аспарагиновой, N - ацетиласпарагиновой и у-аминомасляной
(ГАМК) кислот, а также их интенсивный метаболизм. Доля этих аминокислот
составляет 75% фонда его свободных аминокислот.
Постоянство качественного и количественного состава аминокислот в
метаболических фондах мозга сбалансировано благодаря слаженному
функционированию гомеостатических механизмов, гематоэнцефалическому
барьеру и мембранному транспорту.
Глутаминовая кислота (глутамат) выполняет важную функцию в
энергетическом обеспечении головного мозга, которая заключается в поддержании
метаболитов цикла Кребса на определенном и довольно высоком уровне, а также в
снабжении митохондриальных синтетических процессов восстановительными
эквивалентами.
Глутамин является важным механизмом детоксикации аммония, к которому
мозг чрезвычайно чувствителен и накопление которого губительно для ЦНС.
Глутатион выступает как нейромедиатор и нейромодулятор.
ГАМК-наиболее широко распостраненный в нервной системе тормозной
медиатор. У млекопитающих ГАМК локализована в нервных окончаниях
тормозных нейронов ЦНС.
Глицин участвует не только в биосинтезе белков, но и других
многочисленных биосинтетических процессах: в образовании пуринов,
порфиринов, креатинина, холина, глутатиона и др. Глицин функционирует также в
качестве ингибиторного медиатора. Так как потребление глицина в нервной ткани
относительно велико, а поступление из крови происходит медленно, значительная
часть его синтезируется в мозге de novo. Гюкоза и серин являются главными
источниками глицина в ЦНС.
4.1.5 Липиды ЦНС.
Липиды представляют собой не только структурные компоненты ЦНС, но и
являются важнейшими соединениями, обеспечивающими функциональную
активность. Головной мозг характеризуется высоким содержанием липидов
(приблизительно 50% сухой массы). Мозг содержит уникальные мембранные
структуры - миелиновые оболочки, которые имеют самое высокое содержание
липидов (до 80%) по сравнению с другими тканями или субклеточными
структурами. Для мембран ЦНС характерно и наибольшее структурное
разнообразие липидов по сравнению с мембранами других органов.
Липидный состав нервной ткани практически постоянен и остается неизменен
даже под влиянием внешних факторов (диета, гормоны, фармакологические
вещества, стрессы), которые меняют липидный состав висцеральных органов и
плазмы. Это следствие защищенности ЦНС от различных воздействий. Изменения
липидного состава нервной ткани рассматриваются как патология, потому что вся
сложнейшая деятельность нервной ткани опосредуется мембранами, в
формировании и функционировании которых липиды принимают непосредственное
участие.
28
Липидный состав белого вещества ближе к миелину, а серое вещество
содержит меньше типичных миелиновых липидов.
Принципы организации липидов в мембранах основываются на их
уникальных своис гвах [ 1 ].
3 1) сочетание гидрофильных и липофильных свойств, в структуре одной
молекулы, их амфифильность;
2) способность липидов четко ориентироваться на границе раздела фаз, так
что полярные группы направлены в водную среду, а неполярные экранированы от
нее;
3) способность липидов самопроизвольно упаковываться в прочные, плотные
мономолекулярные слои или пленки, устойчивые к сжатию. Такие плотные слои
создают определенный барьер для диффузии молекул;
4) способность агрегировать в хорошо упорядоченные мицеллы.
В мицеллах липиды ориентированы таким образом, чтобы максимальное
число полярных групп находилась в контакте с водной средой, а гидрофобная часть
была максимально удалена от контакта с ней.
Более 60 лет' назад высказано предположение, что основу мембран составляет
бимолекулярный слой липидов. В бимолекулярном слое гидрофобные цепочки
молекул липидов направлены друг к другу и внутренняя область бислоя
совершенно гидрофобна, а гидрофильные части образуют поверхности внутреннего
и внешнего монослоев, открытые для разнообразного рода взаимодействий.
Миелин в ЦНС.
Мозг человека содержит около 120 г миелина, что составляет 1/3 его сухой
массы. Миелин - уникальное образование, организация которого позволяет
проводить импульсы в аксоне с минимальной затратой энергии. Миелиновая
оболочка — высокоорганизованная многослойная структура, состоящая из сильно
растянутой и модифицированной плазматической мембраны олигодендроглиальной
клетки.
Плазматическая мембрана олигодендроцита образует вокруг аксона сложную
мембранную структуру - мезаксон, который является элементарной единицей
миелина, имеет пятислойную структуру: белок — липид - белок — липид - белок. Эта
структура, многократно закручиваясь вокруг аксона, конденсируется в компактную
миелиновую оболочку. На электронных микрофотографиях миелин выглядит как
серия чередующихся липидных и белковых слоев.
Благодаря особенностям своей структуры миелинизированные нервные
волокна проводят потенциалы действия чрезвычайно быстро. Только очень
короткие участки этих волокон, перехваты Ранвье, покрыты обычной клеточной
мембраной. Участки между перехватами, благодаря многослойной миелиновой
оболочке имеют значительное сопротивление мембраны. Поэтому при сдвиге
Мембранного потенциала ток, по существу, не проходит через мембрану
межперехваточных участков, и потенциал действия от одного перехвата Ранвье к
соседним перехватам распространяется через межперехваточные участки
электротонически и почти без декремента. Время проведения через
Межперехваточные участки практически равно нулю - возбуждение перескакивает
°т Одного перехвата к другому.
29
Высокая скорость проведения в миелинизированных участках обеспечивает
возможность существования у позвоночных большого количества паралелльных
быстропроводящих нервных путей.
4. 1.6 Молекулярные основы электрических процессов в нейронах.
Мембранные потенциалы.
Внутриклеточная среда существенно отличается по ионному составу от
внеклеточной. Главный внутриклеточный катион - калий, внеклеточный - натрий.
Вследствие неравномерного распределения электролитов существует разность
потенциалов между внутренней и наружной стороной плазматической мембраны
(мембранный потенциал). Когда нервная клетка находится в состоянии покоя, то
при введении в нее микроэлектрода регистрируется постоянный отрицательный
потенциал: внутренняя сторона заряжена отрицательно по отношению к наружной -
потенциал покоя (ПП) примерно -70 мВ. Электрический заряд, вносимый
входящими в клетку ионами натрия, компенсируется равным по величине
выходящим потоком калия. В результате непрерывного пассивного входа натрия и
выхода калия клетка в условиях покоя постоянно набирает натрий и теряет калий.
Если бы не активный транспорт натрия из клетки и калия в клетку, который
осуществляется зависимым от клеточного метаболизма Na+-K+ - АТФазой, то
происходило бы постепенное выравнивание концентраций ионов по обе стороны
мембраны, что в конце концов визвало бы гибель клетки. В состоянии покоя
мембранный потенциал клетки одинаков в любой точке ее поверхности.
Нерв обладает возбудимостью - способностью генерировать потенциалы
действия (нервные импульсы) в ответ на электрическое раздражение. Сома нейрона,
как правило, электрически невозбудима, в ней возникают только местные
потенциалы. Генерация импульсов осуществляется в начальном сегменте аксона.
Потенциал действия (ПД) - представляет собой кратковременную
деполяризацию, при котором мембрана разряжается до нуля, а затем (во время пика
ПД) приобретает положительный заряд на внутренней поверхности до +50 мВ.
Генерация ПД обусловлена временным повышением проницаемости мембраны для
натрия. Как только в фазу нарастания ПД мембранный потенциал приблизится к
натриевому равновесному потенциалу, входящий натриевый ток резко уменьшается
и начинается фаза спада. В это время открываются калиевые каналы. Возникающий
при открывании калиевых каналов ток возвращает потенциал к уровню
равновесного калиевого тока. Такова общая схема классической модели А.
Ходжкина и А. Хакли, описывающей генерацию нервного импульса в аксоне.
Синаптическая передача. Медиаторы.
В конце ХГХ века Ч. Шеррингтон обосновал представление об отсутствии
межклеточной непрерывности в нервной системе и ввел понятие «синапс» для
обозначения структуры, которая опосредует передачу сигнала от окончания аксона
нервной клетки к эффектору - нейрону, мышечному волокну, секреторной клетке.
В зависимости от способа передачи сигнала различают химические (наиболее
многочисленные и разнообразные) и электрические синапсы. Существуют и
смешанные синапсы.
Электрофизиологическими критериями электрической синаптической
передачи являются [1]:
30
1) отсутствие синаптической задержки;
2) проведение сигнала в обоих направлениях;
3) независимость передачи сигнала от потенциала пресинаптической
мембраиь1’ „
4) устойчивость к изменениям ионов кальция и магния в среде, к асфиксии,
нИЗКой температуре;
ж Электрические синапсы - распостраненный вид межнейронных связей у
беспозвоночных и низших позвоночных.
функциональная роль электрических синапсов состоит в осуществлении
точной (без синаптической задержки) передачи сигналов, обеспечивающих
синхронную активность группы нейронов, например, группы мотонейронов во
в емя прыжковых движений лягушки или плавательных движений рыбы.
Эволюция нервной системы сопровождается уменьшением числа
электрических синапсов в пользу другого способа передачи — химического.
В химическом синапсе нервный сигнал вызывает освобождение из
пресинаптических окончаний химического посредника - нейромедиатора, который
диффундирует через синаптическую щель и вступает во взаимодействие с белками-
рецепторами постсинаптической мембраны, в результате чего генерируется
постсинаптический потенциал. Химические синапсы преобладают в мозге
млекопитающих.
Химический механизм синаптической передачи эффективнее, чем
электрический, обеспечивает основные функции синапса:
1) одностороннее проведение сигнала;
2) усиление сигнала;
3) конвергенцию многих сигналов на одной постсинаптической клетке;
4) пластичность передачи (обучение, память).
В зависимости от- вида передаваемого сигнала химические синапсы делятся на
возбуждающие и тормозные. В возбуждающих синапсах медиатор,
высвобождаемый из пресинаптических нервных окончаний, вызывает в
постсинаптической мембране возбуждающий постсинаптический потенциал -
локальную деполяризацию, а в тормозных синапсах - тормозной постсинаптический
потенциал, как правило, гиперполяризацию.
Еще один важный принцип, лежащий в основе классификации химических
синапсов, это природа медиатора: холинергические, норадренергические,
пуринергические, пептидергические синапсы и т.д.
Квантовая теория освобождения медиатора.
Согласно этой теории процесс освобождения нейромедиатора складывается
из отдельных элементарных реакций, каждая из которых представляет собой выход
одной порции («кванта») нейромедиатора. Квантовый характер синаптической
передачи обусловлен тем, что нейромедиатор хранится в синаптических пузырьках.
ги пузырьки в нервном окончании сосредоточены в области приближенной к
синаптической щели, причем пузырьки распределяются неравномерно, группируясь
активные зоны, которые расположены напротив скоплений рецепторов
Остсинаптической мембраны.
31
Когда потенциал пресинаптической мембраны находится на уровне покоя, т.е
сигналы не поступают, кванты медиатора высвобождаются спонтанно с низкой
вероятностью. При этом в постсинаптической мембране возникают миниатюрные
постсинаптические потенциалы.
Деполяризация пресинаптической мембраны ведет к резкому повышению
вероятности высвобождения каждого кванта медиатора. Так, в нервно-мышечном
соединении во время нервного импульса практически синхронно высвобождается
до нескольких сотен таких квантов. В результате этого развивается
постсинаптический ток, обеспечивающий генерацию постсинаптического
потенциала.
Для вызванного высвобождения медиатора необходимо присутствие кальция
в окружающей среде. Основная часть синаптической задержки связана с
открыванием кальциевых каналов пресинаптической мембраны и с процессом
сближения синаптического пузырька с активной зоной, осуществляемым с участием
кальция.
Медиаторы.
До 50-х годов к медиаторам относили две группы низкомолекулярных
соединений, которые сейчас называют «классическими», «традиционными»
медиаторами, - амины (ацетилхолин, адреналин, норадреналин, дофамин,
серотонин) и аминокислоты (глутамат, аспартат, гамма-аминомасляная кислота,
глицин). В 60-е годы Дж. Бэрнсток открыл третью группу медиаторов — пуриновые
нуклеотиды. В 1953 году Ф. Лейбек выдвинул предположение о медиаторной роли
пептида - вещества Р, обнаруженного еще в 30-е годы в мозге и в стенках
кишечника в виде вещества, которое усиливало сокращения изолированной кишки
и вызывало временную гипотензию. Разработка иммуноцитохимических и
радиоиммунологических методов позволила в 70-80-е годы выявить в разных
отделах нервной системы беспозвоночных и позвоночных множество пептидов
(нейропептидов), участвующих в синаптической передаче. Нейропептиды
составляют четвертую, самую многочисленную группу медиаторов и, кроме того,
выступают как модуляторы действия других медиаторов.
Медиаторные вещества делятся па два больших класса: нейромедиаторы
(синонимы - нейротрансмиттеры, нейропередатчики), опосредующие передачу
сигнала, и нейромодуляторы, регулирующие (модифицирующие) передачу сигнала.
Одно и то же вещество в разных ситуациях может быть нейромедиатором и
нейромодулятором.
Нейромедиатор - это вещество, которое синтезируется в нейроне, содержится
в пресинаптических окончаниях, высвобождается в синаптическую щель в ответ на
нервный импульс и действует на специализированные участки постсинаптической
клетки, вызывая изменение мембранного потенциала и (или) метаболизма клетки.
Нейромодуляторы.
Понятие «модуляторные вещества» было предложено в 60-с годы Э. Флори
исходя из представлений о характере действия гормонов. В современном
понимании нейромодуляторы по сравнению с нейромедиаторами имеют следуюшйс
характеристики [1]:
32
1) нейромодуляторы не обладают самостоятельным физиологическим
йствием, а модифицируют эффект нейромедиаторов;
2) действие нейромодуляторов имеет тонический характер - медленное
развитие
и большую продолжительность действия;
нейромодуляторы не обязательно имеют синаптическое или даже
нейронное происхождение. Они могут освобождаться, например, из глии;
4) мишенью нейромодуляторов можег быть не только постсинаптическая
мембрана и мембранные рецепторы; нейромодулятор действует на разные участки
нейрона, причем его действие может быть и внутриклеточным.
Ацетилхолин.
В вегетативной нервной системе ацетилхолин служит нейромедиатором во
всех преганглионарных нервных окончаниях симпатического и парасимпатического
его отделов (в том числе и мозговом слое надпочечников) - через никотиновые
холинорецепторы; во всех постганглионарных парасимпатических нервах потовых
желез - посредством мускариновых рецепторов. В нейромышечной передаче
осуществляет роль нейромедиатора.
Холинергические системы мозга участвуют в таких функциях, как память
(кора больших полушарий, гиппокамп), регуляция движения (базальные ганглии),
уровень бодрствования (ретикулярная формация).
Установлено, что ацетилхолин в мозговой ткани накапливается в больших
величинах во время наркоза, и содержание его падает во время судорожных
припадков.
Моноамины.
Норадреналин.
Тела норадренергических нейронов в ЦНС находятся в стволе мозга, главным
образом в мосте. Нисходящие норадренергические пути спинного мозга участвуют
в регуляции сокращения мышц сгибателей и тонуса сосудов. В вегетативной
нервной системе норадреналин является медиатором постганглионарных
симпатических нервов.
В ЦНС норадреналин играет роль преимущественно тормозного
нейромедиатора, например, в коре больших полушарий, реже возбуждающую,
например, в гипоталамусе.
Адреналин.
Нейромедиаторная роль адреналина сомнительна. Тела адренергических
нейронов находятся в нижних отделах моста и продолговатого мозга.
Дофамин
Находится в среднем мозге, обонятельной луковице, гипоталамусе.
Серотонин
Играет важную роль в регуляции эмоционального поведения, двигательной
акуивпости, пищевого поведения, сна, терморегуляции, контроля
Нейроэндокринных систем. Низкая концентрация серотонина в мозгу является как
ы химической основой выраженности депрессии.
Гистамин.
тг „ Локализация и функции гистаминэргической системы весьма своеобразны.
Роны, продуцирующие гистамин, сосредоточены в очень ограниченной области
33
мозга - в заднем гипоталамусе. Эта группа нейронов посылает свои эфферентные
волокна практически во все отделы мозга. Везде обнаруживаются рецепторы
гистамина. Столь широкое распространение рецепторов гистамина позволяет
понять чрезвычайно разнообразные функции этой системы. Гистамин уменьшает
продолжительность ортодоксальной фазы сна и облегчает пробуждение. Он
стимулирует двигательную активность, половое поведение, но ослабляет
восприятие боли. Усиливая жажду, он в то же время подавляет пищедобывательное
поведение. Гистамин входит в число факторов, которые через центральные
механизмы участвуют в повышении давления крови, в терморегуляции (снижение
температуры тела) и в регуляции энергетического метаболизма мозга (стимуляция
гидролиза гликогена).
Нейромедиаторные аминокислоты.
В соответствии со своей функцией они делятся на две группы -
возбуждающие аминокислоты (глутамат, аспартат) и тормозные (ГАМК, глицин,
таурин).
L-Глутаминовая кислота.
Это главный возбуждающий нейромедиатор в мозге. Глутамат
обнаруживается во всех отделах ЦНС, очевидно, благодаря тому, что он является не
только нейромедиатором, ио и предшественником других аминокислот.
Аспарагиновая кислота.
Наиболее высокое содержание аспартата в среднем мозге. Предполагается
нейромедиаторная роль аспартата в возбуждающих интернейронах, которые
регулируют различные спинномозговые рефлексы.
Глгщин.
Выполняет нейромедиаторную роль, прежде всего в спинном мозге
млекопитающих, где она опосредует постсинасптическое торможение активности
мотонейронов. Является также тормозным нейромедиатором в интернейронах
промежуточного мозга и в ретикулярной формации.
Гамма - аминомасляная кислота (ГАМК).
Широко распостранена в ЦНС млекопитающих - она выявляется примерно в
50% нервных окончаний мозга. ГАМК — основной тормозный нейромедиатор в
ЦНС. В коре больших полушарий имеется большое количество ГАМК - эргических
тормозных интернейронов. ГАМК находится в нейронах стриатума, дающих
проекции в черную субстанцию, в нейронах мозжечка, в желатинозной субстанции
задних рогов спинного мозга.
ГАМК проявляет свой тормозной эффект на специфические нейроны,
активизируя так называемые ГАМК-рецепторы, которые контролируют поток
ионов хлора через хлоридные каналы нейронов. Гиперполяризация и повышение
резистентности нейронов к возбуждению является следствием поступления в них
иона хлора. Характерно, что при нормальном состоянии концентрация ГАМК в
мозговой ткани находится на постоянном уровне, что свидетельствует о высокой
пластичности мозгового обмена.
Наряду с медиаторной ролью ГАМК можно говорить о метаболической роли
этого биологически активного вещества. Тканевая концентрация ее значительно
выше других неаминокислотных медиаторов. Доказано, что ГАМК, как продукт
34
обМена в мозговой ткани, оказывает влияние на транспорт и утилизацию глюкозы,
раневое дыхание и окислительное фосфорилирование, участвует в осморегуляции
да уровне клеток, оказывает антигипоксическое действие.
С медиаторной функцией ГАМК связано ее участие в регуляции моторной
активности, поддержание судорожного порога в нервной системе, осуществление
рЫСщих интегративных функций головного мозга и формирование эмоционального
доведения.
Согласно современным представлениям, существуют два вида ГАМК-
ергических рецепторов: «классические» ГАМК а-рецепторы, находящиеся
преимущественно в ЦНС, и ГАМК - Р-рецепторы, широко представленные на
периферии - в предсердиях, гладкомышечных образованиях сосудов и кишечника.
Болевой стресс приводит к увеличению содержания ГАМК в коре головного
мозга. Связь ГАМК-опосредованного тормозного механизма с возрастанием
ноцицептивного потока проявляется в том, что избыток катехоламинов,
неизбежный при хирургической травме, вызывает угнетение активности ГАМК-
трансферазы, разрушающей ГАМК. Это приводит к увеличению концентрации этой
аминокислоты в мозговой ткани.
4.2 Ликворологня
Ликворная система условно делится на внешние и внутренние пространства,
которые постоянно связаны между собой (рис. 5А,Б).
Внутренние ликворные пространства (вентрикулярная система). Сюда
относятся I-1I боковые (правый и левый), III и IV желудочки.
Боковые желудочки расположены соответственно в правом и левом
полушарии головного мозга и пересекают их в продольном направлении. В каждом
боковом желудочке различают центральную часть (pars centralis) и 3 рога: передний
- cornu anterius; задний - cornu posterius; нижний - cornu inferius.
Боковые желудочки связаны с III желудочком посредством межжелудочковых
отверстий.
III желудочек представляет собой длинную узкую полоску, слегка
расширенную в конце. Он расположен между таламусом, гипоталамусом - передней
и задней спайкой большого мозга, сосцевидными телами, серым бугром,
мозолистым телом и сводом. III желудочек связан с IV желудочком через
водопровод мозга, который имеет длину около 15 мм.
IV желудочек находится под мозжечком. Он связан с подпаутинным
пространством посредством одного срединного и двух боковых отверстий IV
желудочка. Снизу он продолжается в центральный канал спинного мозга.
Все желудочки имеют сосудистые сплетения, которые играют важную роль в
образовании ликвора. Сосудистые сплетения составляют 60% внутренней
поверхности мозга. Они являются складками мягкой оболочки мозга, обильно
снабжаемыми кровью и покрыты эпителием. Ворсинчатый эпителий, в сущности,
является продолжением эпендимы, покрывающей желудочки.
Поверхность сосудистых сплетений имеет много складок, на ней образуется
большое количество отростков, проникающих в просвет желудочков. У человека
°бщая поверхность этих сплетений составляет 150-300 см2.
35
Между сплетениями
функциональные различия.
боковых, III и IV желудочков существуют
Рис. 5 А. Внешние (1) и внутренние (2) ликворные пространства головного
мозга. Б. Вид сбоку на желудочки мозга.
Сосудистые сплетения боковых желудочков снабжаются кровью через
передние ворсинчатые отверстия; III желудочка - через задние мозговые артерии, IV
желудочка - через задние спинальные и позвоночные артерии.
Венозный отток из боковых и Ш желудочков происходит в вену бугра,
полосатого тела и виуч ренние вены головного мозга; из IV желудочка - в основную
вену.
Сосудистые сплетения периваскулярно иннервируются блуждающим нервом,
внутренним сонным, позвоночным и шейным сплетениями. Иннервация
осуществляет главным образом вазомоторную функцию.
Внешние ликворные пространства (подпаутинное пространство)
Как уже указывалось выше, головной и спинной мозг человека покрыты
тремя оболочками, происходящими из мезенхимальной ткани. Твердая оболочка
мозга, состоящая из плотной фиброзной соединительной ткани, образует две
пластинки, которые в отдельных местах прочно срастаются, а в некоторых местах
отделены друг от друга (рис. 6).
36
Рис. 6 Схема внешних ликворных пространств
1- черепная кость, 2- эпидуральное пространство, твердая оболочка мозга, 4-
субдуральное пространство, 5- паутинная оболочка мозга, 6 —
субарахноидальное пространство, 7 — мягкая мозговая оболочка, 8- мозговая
паренхима.
Твердая мозговая оболочка мозга обильно снабжается кровью, содержит
лимфу и нервные волокна. С внутренней стороны она образует широкие
перегородки, разделяющие отдельные части. В складках оболочки располагаются
крупные венозные синусы. Они собирают венозную кровь из оболочки мозга,
анастамозируют между собой и обеспечивают отток через яремное отверстие в
яремную вену. Твердая оболочка спинного мозга начинается от большого
отверстия и заканчивается на уровне П-Ш крестцовых позвонков, прикрепляясь к
надкостнице крестца (рис. 7). Она состоит из двух листков, между которыми
образуется узкая щель, заполненная жировой и рыхлой соединительной тканью. В
ней размещаются большие венозные сплетения и лимфатические лакуны, которые
осуществляют механическую защиту спинного мозга. Твердая мозговая оболочка
покрывает спинной мозг, конский хвост, спинномозговые корешки и
спинномозговые ганглии.
37
Рис. 7 Схема спинального ликворного пространства.
(1) Субарахноидальное пространство.
Снабжение кровью осуществляется через позвоночные артерии и
спинномозговые вены. Иннервация происходит от ветвей оболочки мозга
спинномозговых нервов. Клетки эндотелия сосудов твердой оболочки мозга
страдают недостатком уплотненных контактов, что подкрепляет мнение, согласно
которому твердая мозговая оболочка не участвует в гематоэнцефалическом барьере.
Паутинная оболочка - рыхлая соединительная ткань, состоит из двух листков,
соединенных большим количеством трабекул. Между внутренней пластинкой
твердой оболочки мозга и наружной пластинкой паутинной оболочки находится
субдуральное пространство. Внутренняя пластинка паутинной оболочки прочно
срастается с мягкой оболочкой. Между двумя пластинками паутинной оболочки
образуется подпаутинное пространство, разделенное на большое количество ячеек и
пересекающееся трабекулами.
Подпаутинное пространство заполнено ликвором и представляет собой
внешнее ликворное пространство.
Над мозговыми извилинами это пространство узкое, а над бороздами и в
отдельных местах образует цистерны. Паутинная оболочка состоит из трех хорошо
разграниченных лептоменингеальных клеток. Это крупные клетки, которые
содержат много цитоплазмы; они являются потенциальными фагоцитами.
Паутинная оболочка лишена собственного кровоснабжения и иннервации.
Мягкая оболочка мозга (pia mater) состоит из двух пластинок: наружной,
которая прочно срастается с внутренней пластинкой паутинной оболочки и,
внутренней, которая соединена с поверхностной глиальной пограничной
мембраной.
Мягкая оболочка мозга - это тонкая нежная перепонка из соединительной
ткани, богатая кровеносными сосудами и нервами. Она плотно прилегает к
поверхности головного и спинного мозга и проникает во все борозды и углубления.
По ходу мозговых сосудов она глубоко погружается в вещество мозга. Наружная
пластинка состоит из коллагеновых волокон и в области спинного мозга образует
зубчатую связку, которая разделяет задние и передние корешки. Иннервируется
мягкая мозговая оболочка ветвями спинномозговых нервов. Ее питание зависит от
ликвора и экстрацеллюлярной жидкости.
38
Гематоэицефал и чески й барьер.
Гомеостаз ликвора поддерживается клеточными системами, которые
образуют соответсвующие барьеры.
Гематоэнцефалический барьер отождествляют с эндотелиальными клетками
мозговых капилляров, а гематоликворный барьер (ГЛБ) с эпителиальными клетками
сосудистых сплетений и арахноидальных мембран. Многие авторы предпочитают
термин «ГЭБ», включая в него ГЛБ.
ГЭБ связан с поверхностью, отделяющей мозг и ликвор от крови и
обеспечивающий двухнаправленный селективный обмен различных молекул между
кровью, ликвором и мозгом.
Морфологической базой барьера служат уплотненные контакты эндотелия
мозговых капилляров, эпителиальные клетки сосудистых сплетений и
арахноидальных мембран.
Однако в гипоталамусе, образованиях под сводом, передней спайкой такой
эндотелиальный барьер отсутствует. Эти так называемые «оконца в мозг»
позволяют составным частям плазмы непосредственно проникать к осморецепторам
и хеморецепторам.
Использование понятия «барьер» всегда требует уточнения состава обмена
(вода, электролиты, микромолекулы, аминокислоты и др.). Термин «барьер»
указывает на состояние непроницаемости для молекул определенного критического
размера.
Изучение ГЭБ началось с работ Erlich (1885) который после введения
анилиновых красок интравенозно не обнаружил окрашивания мозговой паренхимы.
Fichman (1980) характеризует гематоэнцефалический барьер следующим
образом:
ЬМорфологическая характеристика:
1. Наличие здоровых контактов между капиллярными эндотелиальными
клетками;
2. Наличие здоровых контактов между клетками и арахноидальными
мембранами;
3. Глиальные отростки, окружающие капилляры;
4. Небольшое число пиноцитозных везикул в эндотелиальных клетках;
5. Многочисленные митохондрии в эндотелиальных клетках.
И.Физиологическая характеристика:
1. Характеристика проницаемости эндотелиальных клеток: ограничение
проникновения макромолекул и липидонерастворимых веществ, в то время как
проникновение липидорастворимых веществ относительно неограниченно.
2. Осмотическое давление мозга и ликвора варьирует непосредственно с
изменениями в плазменном осмотическом давлении.
3. Существует двусторонний активный транспорт через мембраны и
эндотелиальные клетки для ионов, органических кислот, щелочей, которые
стабилизируют состав ликвора и экстрацеллюлярной мозговой жидкости.
Проницаемость плазматического вещества в ликвор зависит от:
особенностей ГЭБ;
39
липидорастворимости;
ионизированности;
молекулярной массы молекулы;
гидродинамического радиуса молекулы;
способности образования комлексов с другими протеинами;
концентрации плазмы и плазмоликворного градиента.
Низкая концентрация белков в ликворе обусловлена свойством ГЭБ не
пропускать некоторые макромолекулы. ГЭБ в отношении белков действуег как
сито.
Алкоголь и Н2О свободно проникают через ГЭБ (соответственно на 97 и
93%),. также быстро проникают через ГЭБ СО2 и (К В то же время растворимые в
воде полярные соединения с трудом проходят через него, если отсутствуют
специальные для них транспортные системы. В ликвор проникают только
ионизированные соединения, значительно ограничено проникновение в него
фракций, связанных с белками. Почти не проходит через ГЭБ билирубиново -
альбуминовый комплекс. В сосудистых сплетениях существуют специальные
транспортные системы для витаминов. Белки транспортируются с помощью
фильтрации, ультрафильтрации, везикулярного транспорта. Перенос глюкозы
противотоком происходит путем облегченной диффузии без затрат энергии.
Особенности ГЭБ определяют приблизительно одинаковое осмотическое
давление ликвора, мозга и крови.
Термины, используемые при движении раствора или раст воренных веществ
через ГЭБ:
1. Пассивная диффузия: движение растворов и веществ в направлении
химического или электрического градиента (или обоих градиентов) без затраты
энергии: вода, небольшое количество молекул белков.
2. Активный транспорт: движение растворов и растворенных веществ против
концентрационного градиента с затратой энергии. Типичный пример: движение
через насос K-Na с участием активатора К-Na АТФ-азы.
3. Везикуллярный транспорт (подобно пиноцитозу): трансцеллюлярнос
движение больших молекул. Частички вещества поглощаются цитоплазматической
мембраной и переносятся через клетки в виде пузырьков. Особенно важен для
глобулинов.
4 Облегченная диффузия: движение метаболитов через мембраны с участием
специфических мембранных переносчиков, обычно без затрат энергии. Мембрана
клетки содержит не только двухмолекулярный слой липидов, но имеет и небольшие
неэлектролитные поля и специфические молекулы переносчиков. Молекулы
переносчиков могут фиксироваться на полярных полях или свободно перемещаться
в липидных слоях и, связываясь с заданной молекулой, легко переносить ее через
мембрану.
Место образования ликвора.
Ликвор образуется в сосудистых сплетениях, эпендиме и мозговой паренхиме.
У человека сосудистые сплетения составляют 60% внутренней поверхности мозга.
40
Доказано, что основным местом образования ликвора являются сосудистые
сплетения. Морфологическое строение сосудистых сплетений можно сравнить со
строением проксимальных частей канальцев нефронов, которые выделяют и
абсорбируют различные вещества. Каждое сплетение представляет собой очень
васкуляризированную ткань, которая проникает в соответствующий желудочек.
Вне сплетений ликвор образуется в:
пиальных кровеносных сосудах;
эпендимальных клетках;
мозговой интерстициальной жидкости (здесь образуется 10-12% ликвора).
Механизм образования ликвора.
Существует несколько теорий образования ликвора, основные из них:
1. Секреционная теория: ликвор является продуктом секреции сосудистых
сплетений.
Однако этой теорией нельзя объяснить отсутствие специфического гормона и
неэффективность воздействия некоторых стимуляторов и ингибиторов желез
внутренней секреции на сплетения.
2. По фильтрационной теории ликвор является обычным диализатом или
ультрафильтратом плазмы.
Механизм ликворной секреции довольно сложный процесс. Первым этапом в
образовании ликвора является прохождение ультрафильтрата плазмы через
капиллярный эндотелий, в котором отсутствую! уплотненные контакты. Под
влиянием гидростатического давления в капиллярах, расположенных у основания
хориоидальных ворсинок. ультрафильтрат поступает в окружающую
соединительную ткань под эпителий ворсинок. Здесь определенную роль играют
пассивные процессы.
Второй этап: это трансформирование поступающего ультрафильтрата в
секрет, называемый ликвором. При этом большое значение имеют активные
метаболические процессы.
Постоянное образование ликвора говорит о существовании непрерывной
резорбции. При физиологических условиях между этими двумя процессами
существует равновесие.
Образовавшийся ликвор уходит из ликворной системы при участии
следующих структур: арахноидальных ворсинок, лимфатической системы, мозга,
капиллярного эндотелия, арахноидальной мембраны.
Арахноидальные ворсинки считают местом дренажа ликвора, поступающего
из субарахноидального пространства в синусы.
Кроме того, несомненно, что в резорбции ликвора участвуют мембраны,
соприкасающиеся с ликвором, эпителий оболочек цереброспинальной системы,
мозговая паренхима, периневральные пространства, лимфатические сосуды и
периваскулярные пространства. Участие этих дополнительных путей невелико, но
они приобретают большое значение, когда главные пути затронуты
Патологическими процессами.
41
Механизм резорбции ликвора.
Отток ликвора можна охарактеризовать как:
1. Однонаправленное просачивание через арахноидальные ворсинки
посредством клапанного механизма;
2. Резорбция, которая не является линейной, и требует определенного
давления (обычно 20-50 mm Н2О);
3. Своеобразный пассаж из ликвора в кровь, но нс наоборот;
4. Резорбция ликвора, снижающаяся при увеличении общего содержания
белка;
5. Резорбция с одинаковой скоростью для молекул различных размеров,
например, молекул маннитола, сахарозы, инсулина, декстрана.
Механизм оттока ликвора через различные структуры различен. Согласно
закрытой системе, арахноидальные ворсинки покрыты эндотелиальной мембраной,
и между клетками эндотелия находятся уплотненные контакты. Вследствие наличия
этой мембраны резорбция ликвора совершается с участием осмоса, диффузии,
фильтрации низкомолекулярных веществ, а для макромолекул - путем активного
транспорта через барьеры. Однако прохождение некоторых солей и воды остается
свободным.
В противоположность этой системе существует открытая система, согласно
которой в арахноидальных ворсинках существуют открытые каналы, связывающие
паутинную оболочку с венозной системой. Эта система предполагает пассивное
прохождение микромолекул, в результате чего абсорбция ликвора полностью
зависит от давления.
Помимо двух указанных процессов существует и динамический
трансэндотелиальный вакуолизированный процесс: в эндотелии арахноидальных
ворсинок временно образуются каналы, через которые ликвор и его составные
частицы вытекают из субарахноидального пространства в кровь.
Количество ликвора.
Объем образующегося ликвора варьирует от 0,3 до 0,8 мл/мин, от 240 до 1152
мл/сутки.
Общее количество ликвора у взрослых составляет 100-160 мл, у детей 80-92
мл.
Физиологическое значение ликвора.
Ликвор выполняет целый ряд физиологических функций:
механической защиты мозга;
экскреторную и так называемую sink функцию ликвора, т.е. выделение
некоторых метаболитов для предупреждения их скопления в мозге;
служит транспортным средством для различных веществ, особенно
биологически активных, таких как гормоны и т. д.;
контрольную функцию по отношению к мозговому окружению:
а) обеспечивает нечувствительность к быстрым изменениям состава
крови;
б) поддерживает определенную концентрацию катионов, анионов, pH, что
обеспечивает нормальную возбудимость ЦНС.
42
специфического защитного иммунобиологического барьера.
Ликворное давление (рахиметрия).
Ликворное давление значительно ниже кровяного, поэтому оно измеряется в
щгп НгО, а не в mm Hg. Оно зависит от следующих факторов:
скорости образования ликвора и ликворной резорбции;
артериального давления. Так, постоянно повышенное АД не воздействует
на давление ликвора, тогда как резкое повышение АД временно повышает его;
венозный застой и венозное давление увеличивают давление ликвора вследствие
нарушения его резорбции;
гидростатического давления ликвора, которое обусловлено различием
между вентрикулярным, субокципитальным и люмбальным давлением при прямом
положении тела;
эластичности стенок ликворной системы;
дыхания - при задержке дыхания и во время кашля давление повышается,
при глубоком дыхании понижается.
С целью измерения ликворного давления используются следующие методы:
1. на глаз;
2. при помощи стеклянной градуированной трубки диаметром 2 мм и длиной
50-60 см;
3. с помощью манометра;
4. при помощи ликворографа или ликворного тонографа;
5. электронной трансдюсерной системой.
Клиническое значение.
В норме у взрослых больных в горизонтальном положении ликворное
давление колеблется в пределах 70-200 mm Н2О, в зависимости от позы (в лежачем
положении: 150 mm Н2О, в сидячем - 400 mm Н2О); АД, а также факторов, которые
изменяют ток ликвора.
Повышенное ликворное давление встречается при опухолях головного мозга,
мозговых абсцессах, гидроцефалии, менингитах, ЧМТ, закупорке верхней полой
вены, тромбозе венозных синусов, гипотензии, расстройствах мозгового
кровообращения.
Причины повышенного ликворного давления:
увеличение образования ликвора;
снижение резорбции;
частичное или полное блокирование ликворных путей и нарушение оттока
ликвора;
увеличение объема головного мозга.
Снижение ликворного давления от 65 до 0 мм Н2О встречается при
арахноидите, разрыве связи между различными частями ликворной системы
вследствие удаления опухоли мозга, в поздиий период ЧМТ, при
Циркуляционном коллапсе, при тяжелой дегидратации, спинномозговом
блоке, гипотензии.
Причины снижения ликворного давления:
Уменьшение образования ликвора вследствие повреждения сосудистых
сплетений;
43
Артериальная гипотензия (при циркуляторном коллапсе, шоковом
состоянии).
Потеря большого количества ликвора при переломе черепных костей;
Рис. 8 Схема циркуляции ликвора.
1 - спинальные корешки, 2,3 - хориоиадльные сплетения, 4 — Ш желудочек,
5 - хориоидальное сплетение, 6 — верхняя саггитальная пазуха, 7 -
арахноидальная гранула, 8 — боковой желудочек, 9 — полушарие головного мозга,
10 - мозжечок.
Циркуляция ликвора.
Существует движение ликвора, обусловленное его непрерывным
образованием и резорбцией (рис.8).
Движение ликвора осуществляется в следующем направлении: из боковых
желудочков через межжелудочковые отверстия - в третий желудочек, из него через
водопровод большого мозга - в IV желудочек, из него через срединное и боковые
отверстия IV желудочка - в мозжечково-продолговато мозговую цистерну - вверх к
верхнебоковой поверхности мозга - вниз к конечному желудочку. Линейная
скорость циркуляции ликвора около 0,3-0,5 мм/мин.Причинами движения
выступают: сокращение сердца, дыхание, положение и движение тела, движение
ресничного эпителия сосудистых сплетений.
Ликвородинамические пробы.
Ликвородимические пробы применяются для измерения проходимости
ликворных путей и установления наличия или отсутствия спинального
44
субарахноидального блока. Однако с широким распространением контрастной
миелографии эти пробы теряют свое значение.
Проба Queckenstedt.
Измеряется ликворное давление в строго горизонтальном положении. Рукой
сдавливаются яремные вены в течение 5-10 сек. и учитывают изменение давления.
Если ликворное давление повышается в 2-3 раза - проба отрицательная, если не
изменяется - проба положительная. Так, при сдавлении яремных вен, происходит
застой в венозной системе головного мозга, увеличивается объем головного мозга,
повышается ликворное давление. В случае наличия патологических процессов,
нарушающих отток ликвора, проба положительная.
Проба Stookey.
Активно или пассивно сдавливаются мышцы живота и измеряется ликворное
давление. В норме оно повышается в 1,5-2 раза - проба отрицательная. Если
ликворное давление прежнее - проба положительная. Объяснение: сдавливание
мускулатуры вызывает застой крови в венах живота с повышением давления в
эпидуральном пространстве и в последующем в подпаутинном. При блокаде
спинномозгового канала в поясничной области, проба бывает положительной.
Синдромы ликвора.
1. Белково-клеточная диссоциация.
Характеризуется повышением ликворного белка при нормальном или
незначительном увеличении количества клеток.
Этот синдром встречается чаще всего при процессах, затрагивающих
интрарахиальное и внутричерепное пространство, реже при нейросифилисе,
менингитах, полирадикулоневрите, энцефалитах, сосудистых и дегенеративных
заболеваниях.
2. Синдром Nonne-Froin
Характеризуется ксантомией, высоким содержанием белков и спонтанной
коагуляцией ликвора, уменьшением количества альбуминов, увеличением
глобулинов и уровня фибриногена, гаптоглобинов, липопротеинов и др.
Синдром Nonne-Froin встречается при полной и неполной блокаде ликвора
вследствие опухолей спинного мозга, абсцессов, туберкулеза, арахноидитов,
костных компрессий.
3. Коллоидно-белковая диссоциация.
Характеризуется нормальным количеством белка при изолированном
повышении уровня гамма-глобулине в, иммуноглобулина-lg G и изредка остальных
Ig-
Наблюдается при пейросифилисе, рассеянном склерозе, прогрессирующем
гиперкинетическом панэнцефалите.
4. Клеточно-белковая диссоциация.
Выявляется плеоцитоз с незначительным увеличением количества ликворного
белка.
Наблюдается при быстропроходящих воспалительных процессах: вирусный
’менингоэнцефалит, полиомиелит, небактериальный менингит и др.
5. Транссудативный ликворный синдром.
45
Развивается при изменении проницаемости гематоликворного барьера и
зависит от интенсивности нарушения. Чем сильнее нарушена проницаемость, тем
чаще выявляются составные части крови в ликворном пространстве. Полное
разрушение барьера отмечается при субарахноидальном кровоизлиянии.
6. Иммунореактивный ликворный синдром.
Цитограмма характеризуется повышенным содержанием лимфоцитов и
плазматических клеток, при нормальном или слегка повышенном цитозе.
Уровень общего белка повышен. Повышается содержание глобулинов,
главным образом гамма-глобулинов. Очень важно увеличение количества
иммуноглобулинов.
Варианты:
а) Острый - с лимфоплеоцитозом и начальным повышением 1g;
б) Подострый - с лимфоплазматической клеточной картиной и последующим
повышением IgM и IgG;
в) Хронический - с лимфоплеоцитозом и повышением содержания TgG при
исчезновении IgM и нормальном содержании IgA;
г) Персистирующий - характеризуется нормальным содержанием общего
белка, повышением IgG и увеличением числа плазматических клеток.
Изменение ликвора при сосудистых заболеваниях.
При ишемическиом инсульте ликворные изменения зависят:
от размеров инфаркта мозга;
вида (белый или красный) инфаркта;
его расположения;
близости к ликворной системе;
степени выраженности общего нарушения мозгового кровообращения;
участия оболочек в патологическом процессе;
общей реактивности организма.
а) Белый инфаркт.
1. Ликворное давление повышено, но редко превышает 2,45 кПА.
2. Ликвор бесцветный, прозрачный, без фибринозной пленки, периодически
ксантохромия, преобладают билирубиновые компоненты.
При цитологическом исследовании у 80% больных нормоцитоз.
Эритроцитрахия встречается приблизительно у 1/3 больных,что можно объяснить
дисциркуляторными расстройствами и нарушениями проницаемости вблизи
некротической ткани. Клеточная реакция лимфомоноцитарная. У 30% больных
белково-клеточная диссоциация. Активность большей части ферментов слегка или
умеренно повышена.
б) Красный инфаркт.
При клиническом исследовании у 1/3-1/4 больных наблюдается кровянистый
ликвор и ксантохромия, преобладают оксигемоглобин и метгемокомпоненты, после
первой недели и билирубин. Одновременно легкий или умеренный плеоцитоз, с
преобладанием эритроциторахии, позднее (после 2-3 недели)
гемосидерофагоцитоз. У 2/3 больных гиперпротеинрахия выражена значительно со
2-го до 7-го дня, через 3 недели нормализуется. Концентрации Ь2-макроглобулинов;
В - липопротеинов, IgA, IgG, IgM значительно увеличены.
46
JJpu белом и красном инфарктах:
1. pH смещен в сторону кислых значений, наблюдается ликворная гипокапния
с гипервентиляцией. Концентрация бикарбоната снижаегся; рСО2 снижается.
Электролиты в норме.
2. Содержание адреналина, норадреналина, серотонина и их основных
метаболитов в ликворе повышается в первые дни после инфаркта. Медиаторы
усиливают ишемическое повреждение путем активирования рецепторов в стенках
сосудов, а также способствуют их спазму. В последние дни снижение содержания
катехоламинов сопровождается повышением уровня ГАМК, измененяется обмен
серотонина. Количество циклических нуклеотидов, особенно цАМФ, увеличено
вследствие ишемии мозга, при которой происходит освобождение аденозина и
калия, стимулируемое аденил атциклазой; повышение ГАМК связано с изменениями
в цикле трикарбоновых кислот.
3. Концентрация глюкозы в норме.
4.3 Отек и набухание головного мозга
По классическому определению 1. Klatzo, отек головного мозга представляет
собой патологическое накопление жидкости, ассоциированное с объемным
увеличением мозга [70]. Согласно Ю.Н. Квитницкому-Рыжову, избыточное
накопление жидкости в ткани мозга трактуется как отек - набухание головного
мозга. При отеке имеет место скопление жидкости в межклеточных пространствах,
а набухание головного мозга является самостоятельным процессом, в основе
которого лежит прочное связывание воды с внутриклеточными биоколлоидами [11].
Таким образом, отек мозга необходимо рассматривать как внеклеточную, а
набухание как внутриклеточную гипергидратацию. Чаще всего наблюдается
сочетание отека и набухания головного мозга.
В зарубежной литературе принято выделять цитотоксический и вазогенный
отек мозга, которые можно в определенной степени, отождествить соответственно с
набуханием и отеком головного мозга. Основные отличия между ними
представлены в таблице 5.
Табл. 5
Характеристики цитотоксического и вазогенного отека головного мозга
(по F. Xiao, 2002)
Цитотоксический Вазогенный
1. Вызвавшая причина Ишемический инсульт Остановка кровообрщения (аноксия) Г ипоксия Метаболические нарушения Интоксикации А. Острые состояния Черепно-мозговая травма Геморрагический инсульт Гипертензия В. Хронические состояния Опухоль головного мозга Абцесс головного мозга Инфекции ЦНС
2. Проницаемость ГЭБ тг Не изменена Повышена
41
3.Отечная жидкость Не содержит высокомолекулярных соединений плазмы Содержит компоненты плазмы
4. Локализация Внутриклеточное Интерстициальное
отека пространство: астроциты, глиальные клетки, нейроны пространство _i
Основные звенья патогенеза.
В согласии с гипотезой Старлинга отек-набухание головного мозга может быть
вызван следующими причинами, соотношение которых демонстрирует следующее
уравнение:
Jv = LpA [(Pc - Pi) - 6 (лс - m)]
1. Повышением церебрального капиллярного давления (Рс
гидростатическое давление, с постоянным интерстициальным
давлением (Pi);
2. Повышением проницаемости для воды (LpA - капиллярный
фильтрационный коэффициент);
3. Повышением проницаемости для белков (снижение 6) с незначительным
изменением градиента осмотического давления между капиллярами и
интерстицием (яс — тп). [109].
Кроме того, доказанным считается, что в формировании отека-набухания
головного мозга, свой вклад вносят нарушения функции белка аквапорина-4
(AQP4), обеспечивающего трансцеллюлярную проницаемость мембран для воды, и
повышение Na+ и Са2+ проницаемости, индуцированное глутамат — триггерной
гиперактивацией NMDA-рецепторов. Данные механизмы являются характерными
для формирования отека при фокальной ишемии мозга [78, 100].
В патогенезе отека-набухания головного мозга также принимают участие и др.
факторы: системное АД, центральное венозное давление (ЦВД),
цереброваскулярное сопротивление, объем крови в сосудах мозга, реологические
свойства и осмотическое давление крови, изменения в гематэнцефалическом
барьере (ГЭБ), которые будут рассмотрены ниже.
Роль циркуляторных факторов, участвующих в патофизиологических
механизмах развития отека головного мозга, весьма существенна, т.к. основным
источником воды, избыточно накопляющейся в мозговой ткани при отеке, является
кровь, циркулирующая в микрососудах мозга. К циркуляторным факторам в данном
случае относятся все основные параметры микроциркуляции в головном мозге, в
свою очередь связанные с центральным и периферическим (мозговым)
кровообращением. С точки зрения развития отека головного мозга, решающая роль
принадлежит с одной стороны, уровню кровяного давления в капиллярах и
прилегающих к ним мелких артериях и венах, от которого зависит разность
гидростатических давлений между кровью и тканевыми пространствами мозга, с
другой стороны, существенную роль играет осмотическое давление плазмы крови,
которое противодействует переходу воды из крови в ткань мозга. Нарушение
равновесия этих двух противодействующих сил имеет решающее значение для
48
избыточного перехода воды и других составных частей крови, в том числе белков
плазмы и даже форменных элементов в ткань мозга, что и обусловливает
дальнейшее развитие патологических изменений в ней при отеке.
Циркуляторные факторы могут играть двоякую роль в развитии отека
головного мозга. С одной стороны, нарушения мозгового кровообращения являются
одним из основных этиологических факторов в развитии отека. При этом отек
могут вызывать как гиперемия, так и ишемия в мозговой ткани. Так, известно, что
отек возникает при избыточном притоке крови в мозг в результате значительного
повышения общего артериального давления. То же самое может иметь место при
венозном застое крови, который при определенных условиях также становится
причиной развития отека. Еще более частой причиной возникновения отека мозга
является ишемия, если дефицит кровоснабжения мозговой ткани имеет
достаточную глубину и продолжительность. При этом отек мозга развивается
независимо от того, чем была вызвана ишемия — закупоркой ли просвета мозговых
артерий вследствие тромбоза или эмболии, нарушением ли реологических свойств
крови в микрососудах, или, наконец, спазмом мозговых артерий. Ишемия
становится причиной развития отека головного мозга потому', что резкий дефицит
кровоснабжения мозговой ткани вызывает ее значительные повреждения
вследствие нарушений энергетического обмена и транспортной функции клеточных
мембран. При ишемии отек мозга развивается с неодинаковой скоростью в случаях
выключения крупных или мелких сосудов разного калибра, питающих его ткань,
причем первичные нарушения кровотока в системе микроциркуляции
(микротромбы, микроэмболы) вызывают более быстрое развитие отека мозга, чем
выключение более крупных артерий.
При анализе участия параметров мозгового кровообращения в механизме
развития отека головного мозга важным представляется выделение следующих
групп явлений:
а) кровяное давление, кровоток и объем крови в кровеносном русле головного
мозга, тесно связанные с внутричерепным давлением и обусловленные основными
параметрами центрального кровообращения — системным артериальным и
венозным давлениями;
б) реологические свойства крови в системе микроциркуляции мозга;
в) осмотическое давление крови, протекающей по мозговым сосудам.
Среди параметров мозгового кровотока особо важная роль принадлежит тем,
которые непосредственно определяют переход воды сквозь стенки сосудов из крови
в ткань мозга и обратно, а именно гидростатическому и осмотическому давлениям в
сосудах мозга. Остальные параметры кровообращения, способные влиять на
развитие отека мозга, осуществляют свое действие не непосредственно, а в
основном через выше упомянутые циркуляторные факторы.
Одним из основных факторов в циркуляторной системе, определяющих
Уровень кровяного давления в сосудах мозга, является системное артериальное
Давление. Его роль чрезвычайно важна, так как уровень кровяного давления в
любой точке циркуляторного русла (в том числе в капиллярах мозга), обусловлен,
прежде всего, уровнем давления крови в аорте, но всегда ниже его, так как при
течении крови по артериальным ветвлениям происходит расход энергии на
49
преодоление сопротивления в них. Корреляция между системным артериальным
давлением и ЦПД является прямой, но пропорциональной. Она может быть лишь
при условии, когда сосудистое сопротивление на протяжении мозговых
артериальных ветвлений остается постоянным (или меняется незначительно). Из
сказанного, очевидно, что повышение системного артериального давления всегда
способствует развитию отека мозга, а понижение системного артериального
давления, наоборот, замедляет переход воды из мозговых капилляров в
окружающую ткань и таким образом, может быть использовано как лечебное
мероприятие в клинике для предупреждения или ограничения развития отека
головного мозга [28].
Центральное венозное давление является другим фактором, который может
оказывать значительное влияние на уровень кровяного давления в микрососудах
мозга. Корреляция между этими давлениями также прямая, то есть, чем выше
центральное венозное давление, тем выше кровяное давление в сосудах мозга и
наоборот. Такая зависимость связана с изменениями скорости кровотока в сосудах
мозга, обусловленными, в свою очередь, уровнем ЦВД. Однако имеются некоторые
различия между эффектами системного артериального и центрального венозного
давлений на уровень кровяного давления в микрососудах мозга. Во-первых, в
отличие от системного АД, ЦВД может только повышаться, но не понижаться. Во-
вторых, пределы изменений системного АД значительно больше, чем возможные
изменения ЦВД. Однако в то же время влияние системного венозного давления на
кровяное давление в мозге значительно больше, чем влияние системного
артериального давления. Это можно объяснить тем, что сопротивление в
артериальном сосудистом русле головного мозга сравнительно больше при условии
покоя и может значительно меняться при физиологической «ауторегуляции» и при
патологических реакциях мозговых артерий (ангиоспазме и ангиопараличе), а также
при сужении их просвега, вызванном тромбозом, эмболией и т.д. Итак, повышение
центрального венозного давления может быть важным фактором, определяющим
уровень кровяного давления в микрососудах мозга и способствующим переходу
воды из крови в ткань мозга во время развития отека.
Цереброваскулярное сопротивление является фактором, который также
влияет (наряду системным АД и ЦВД) на уровень кровяного давления в сосудах
головного мозга. При физиологических условиях изменения цереброваскулярного
сопротивления определяются главным образом изменениями диаметра мозговых
артерий, но при патологических условиях оно может меняться также в зависимости
от вязкости крови (ее реологических свойств) в микрососудах, о чем будет сказано
ниже. Кроме того, при развитии отека головного мозга может наступать сужение
просвета мозговых капилляров. В общем, кровяное давление в микрососудах мозга
находится в обратной корреляции с цереброваскулярным сопротивлением, то есть с
уменьшением последнего (преимущественно в артериях) кровяное давление в
капиллярах мозга повышается, обуславливая таким образом развитие отека мозга.
Увеличение же цереброваскулярного сопротивления в артериях должно понижать
давление в капиллярах и способствовать устранению отека мозга.
Объем крови в сосудах мозга также является фактором, влияющим на
развитие отека мозга, так как в определенной степени связан с кровяным давлением
50
в его сосудах. Но, с другой стороны, объем крови тесно связан с диаметром
мозговых сосудов и, следовательно, с площадью их сосудистых стенок, которая
является той поверхностью, через которую происходит обмен воды и других
составных частей плазмы крови между этой последней и мозговой тканью. Кроме
того, объем крови в мозге может иметь значение для развития отека еще с другой
точки зрения: ширина мозговых сосудов, особенно вен, зависит от механических
свойств ткани окружающей сосудистые стенки, и потому является показателем тех
ее изменений, которые важны для развития отека, так как определяют уровень
тканевого давления.
Как уже рассматривалось ранее, головной мозг помещен в герметически
замкнутом и неподатливом черепе, внутри которого находится три практически
несжимаемых компонента, а именно — кровь в сосудах, цереброспинальная
жидкость и мозговая ткань, изменения объема каждой из них должны вызывать,
естественно, аналогичные по степени, но противоположно направленные изменения
объема других компонентов. Поэтому, например, увеличение объема мозговой
ткани при развитии отека должно вызывать реципрокное уменьшение объема
цереброспинальной жидкости и (или) крови в мозговых сосудах. Но если такая
компенсация неполная, то повышается внутричерепное давление, и это, в свою
очередь, оказывает влияние на разные параметры мозгового кровообращения.
Однако при естественных условиях соотношения между объемами мозга,
крови и цереброспинальной жидкости внутри черепа не столь просты, как это
представляется на основании только что описанной модели. Так, давно известно,
что нет четкой корреляции между повышением внутричерепного давления и
ослаблением мозгового кровотока. Не обнаружена также четкая зависимость между
величиной отека мозга и повышением внутричерепного давления. Сложность этих
взаимоотношений при естественных условиях связана с работой физиологических
механизмов компенсации. Но эти механизмы имеют предел своей эффективности, и
как только этот предел оказывается достигнутым, влияние одного параметра на
другой становится значительно более выраженным.
Повышение внутричерепного давления может иметь самостоятельное
значение для развития отека мозга. В частности, это имеет большое значение для
его разрешения, так как удаление отечной жидкости из мозговой ткани происходит
частично в желудочки мозга благодаря градиенту давления между субовендиальной
тканью и полостью желудочков. Кроме того, когда в желудочках мозга значительно
повышается давление цереброспинальной жидкости, она начинает переходить из
них в окружающее белое вещество мозга, где возникает отек.
Реологические свойства крови связаны, прежде всего, с биомеханикой
потока эритроцитов в микрососудах с диаметром просвета до 100 мкм. При этом
явления, обусловливающие текучесть крови в микрососудах, имеющих диаметр,
соизмеримый с поперечником эритроцитов (истинных капиллярах), и в более
широких микрососудах (диаметром в 15—100 мкм) неодинаковы, и поэтому их
следует рассматривать раздельно. В первом случае решающую роль играет
Деформируемость мембран эритроцитов, без которой невозможна текучесть крови в
капиллярах, имеющих меньший просвет (примерно 5 мкм), чем диаметр
"эритроцитов (примерно 7 мкм). В более широких микрососудах главная роль в
51
текучести крови принадлежит структуре потока крови, определяемой ориентацией
эритроцитов, профилем их скоростей, а также степенью их агрегируемости в
сосудистом просвете. Последняя играет особо важную роль в нарушениях
текучести крови, так как может резко повышать вязкость крови и способствовать
замедлению кровотока в микрососудах вплоть до полного стаза. Замедление
кровотока в микрососудах может обусловливать, во-первых, дефицит
кровоснабжения (ишемию) соответствующих участков ткани мозга, способствуя
развитию отека, но, с другой стороны, это уменьшает фильтрацию воды из
микрососудов в ткань мозга и, таким образом, может ограничивать развитие отека.
Другим важным циркуляторным параметром, непосредственно влияющим на
развитие отека мозга, является осмотическое давление крови, связанное с
концентрацией разных частиц, в том числе молекул белка, в плазме крови.
Осмолярность крови находится в обратной корреляции с фильтрацией воды в ткань
мозга, то есть понижение осмолярности способствует развитию отека и наоборот.
Значительное понижение осмолярности крови может происходить в естественных
условиях, например, в результате нарушений или дисфункции желудочно-
кишечного тракта или почек и вызывать развитие отека головного мозга. Напротив
повышение осмолярности крови широко используется в медицинской практике как
лечебное мероприятие предупреждения или лечения отека мозга.
Большинство из перечисленных циркуляторных факторов (а именно, уровень
общего артериального давления, цереброваскулярное сопротивление,
реологические свойства и осмолярность крови) могут меняться в организме в двух
противоположных направлениях. Поэтому каждый из этих факторов может не
только способствовать развитию отека мозга, но и компенсировать его развитие.
Изменения проницаемости гематоэнцефалического барьера при развитии
отека головного мозга обратили на себя внимание исследователей сравнительно
давно. Основываясь на современных данных, предложено несколько механизмов,
обуславливающих повышение проницаемости ГЭБ:
1. Физическое оголение или перемещение эндотелиальной выстилки и/или
базальной мембраны;
2. Дефекты в сигнальных системах контролирующих эндотелиальное
соединение; и/или
3. Физическая деструкция или реорганизация компонентов эндотелиального
соединения [109].
При некоторых видах отека значительно повышается проницаемость ГЭБ для
веществ, которые при нормальных условиях не переходят из крови в мозговую
ткань. Поэтому одним из основных методов оценки развития отека мозга стаю
изучение избыточного перехода в мозговую ткань введенных в кровь маркеров.
Сейчас не вызывает сомнения, что переход белковых молекул и некоторых
физиологически активных веществ, сквозь ГЭБ из крови в паренхиму мозга играет
важную роль в патофизиологическом механизме развития отека головного мозга.
В процессе развития отёка отмечаются структурные изменения сосудистых
стенок мозга. Так, при вазогенном отеке выделяют следующие структурные
52
изменения сосудов микроциркуляторного русла:
I. Быстро возникающее усиление микропиноцитозного транспорта (в течение
первых 10—30 секунд);
2. Открытие тубулярно-канальцевой системы, образование аутофагических
вакуолей;
3, Открытие плотных контактов между эндотелиальными клетками, что особенно
характерно для осмотического воздействия, вызываемого введением
гиперосмолярных растворов в кровь;
4. Повреждение эндотелиальных клеток, при котором имеет место диффузное
распространение меток по клетке.
Казалось бы, что все перечисленное в настоящее время не вызывает сомнений.
Однако следует отметить, что указанные процессы еще во многом не изучены и
являются скорее схемой тех реальных процессов, которые встречаются при
исследовании конкретных и очень разных видов вазогенного отека мозга.
При цитотоксическом отеке мозга имеется не меньшее количество нерешенных
вопросов, касающихся особенностей структурных изменений сосудов мозга.
Известно, что при этом типе отека характер структурных изменений сосудов, в
общем, аналогичен таковым при вазогенном отеке мозга. Основной разницей
является липгь отсроченное время их появления. Так, ультраструктурные изменения
сосудов мозга появляются не ранее, чем через 3-4 часа. Однако остается не вполне
выясненным, можно ли говорить о какой-либо специфичности структурных
изменений стенок сосудов, характеризующих цитотоксический отек мозга вообще.
Длительность отека мозга также оказывает свое влияние на структурные
изменения сосудов мозга, в частности, на гипертрофию эндотелия сосудов. При
этом определенный вклад могут вносить изменения ткани мозга, всегда
сопровождающие развитие отека.
Отдельно стоит проблема изменений активности ферментов, определяющих
перенос разных веществ в мозговых сосудах при развитии отека. В литературе
имеется сравнительно небольшое число работ, в которых рассматривается наличие
или активность тех или иных ферментов, принимающих участие в активном
транспорте каких-либо веществ или переносе микропиноцитозных везикул, а также
в открытии системы трубочек и канальцев эндотелия. К числу таких ферментов,
прежде всего, необходимо отнести Na -K-АТФазу, обусловливающую работу
натри ево-калиевого насоса. Транспорт глюкозы, сквозь сосудистые стенки, по-
видимому, осуществляется активно, а не путем простой диффузии, как в других
органах. Показано, что стенки мозговых капилляров селективно транспортируют
глюкозу с помощью стереоспецифического переносчика гексоз и что этот транспорт
осуществляется независимо от физиологических колебаний концентрации глюкозы
в крови, не считая случаев резко выраженной гипогликемии (ниже 10 мг/100 мл),
т е. транспорт глюкозы определясгся интенсивностью ее метаболизма в мозге,
поскольку ее перенос происходит по снижающейся концентрации от плазмы крови
в мозг. Эндотелий капилляров мозга богат щелочной фосфатазой, гамма-глютамил-
транспептидазой и 6-нуклеотидазой. Предполагается, что таким же путем
осуществляется транспорт сквозь стенки мозговых сосудов не только гексоз, но и
аминокислот и их метаболитов.
53
В настоящее время весьма интересные представления существуют о регуляции
проницаемости гематоэнцефалического барьера по отношению к воде. Показано,
что капилляры мозга не являются свободно проницаемыми для воды, как это долго
предполагалось, что локальный мозговой кровоток и проницаемость барьера для
воды находятся в реципрокных взаимоотношениях и обнаруживается четкое
влияние на эту проницаемость раздражения области центральной адренергической
иннервации — голубого пятна. Согласно данным этих авторов, механизм,
ответственный за регуляцию проницаемости для воды, связан с центральными
норадренергическими терминалями, содержащими допамин-бета-гидроксилазу на
мелких внутримозговых сосудах, включая капилляры. При этом обращается
внимание на возможность перицитов и самих эндотелиальных клеток к
сокращению, а также на наличие терминалей нервных аксонов на эндотелии и на
перицитах капилляров в мозге.
Сложность влияний на проницаемость капилляров мозга связана с
комплексным воздействием на них со стороны нервных терминалей с разной
нейротрансмиттерной активностью (с наличием везикул с адреналином,
ацетилхолином и т.д.), с наличием в мозге целой системы местных источников
нейротрансмиттеров — моноаминоцитов, включая лаброциты (тучные клетки).
Последние содержат все виды моноаминов и местно влияют на сосуды мозга в
результате нейрогенных воздействий на эти моноаминоциты. В дополнение к
указанным характеристикам гематоэнцефалического барьера, как функциональной
системы, нужно добавить, что в нормальных условиях он практически не
проницаем для белков плазмы крови и белков нервной ткани. С помощью
иммуногистохимических методов с использованием радиоактивных клеток,
экзогенных электронно-микроскопических плотных белковых меток, и красок,
создающих комплексы с белками, было доказано, что в нормальных условиях в
ткань мозга проникают только следы белка. Основным путем попадания белка в
цитоплазму эндотелия капилляров является микропиноцитоз. Однако в норме его
скорость, по-видимому, позволяет метаболизировать белок, который не удается
обнаружить за пределами эндотелиальных клеток. Плотные контакты между этими
клетками также не проходимы для белка, что многократно было показано многими
авторами на разных объектах исследования. В связи с этим любые нарушения
проницаемости гематоэнцефалического барьера по отношению к белкам при
развитии отека мозга влекут за собой участие аутоимуных механизмов в патогенезе
самых различных типов отека мозга, и это надо всегда принимать во внимание при
его лечении.
Снижение или прекращение работы натриево-калиевого насоса немедленно
приводит к накоплению во внеклеточном пространстве ионов калия, накоплению
внутри клеток натрия и хлора, вместе с которыми здесь происходит и накопление
воды. Так, известно, что при ишемии мозга механизмы активного транспорта этих
ионов в мембранах быстро повреждаются, первые электронно-микроскопические
изменения можно уловить не раньше чем через 9—12 часов. Таким образом,
транспортная система указанных ионов высокочувствительна к гипоксии и эго
делает ее особенно ранимой при самых разных типах патологии центральной
нервной системы
54
Транспорт коллоидов в сосудистых стенках мозга изучен достаточно хорошо
на примере разных белков. Транспорт белков сквозь сосудистые стенки мозга
осуществляется преимущественно путем энергозависимого процесса —
микропиноцитоза. Триггером микропиноцитоза, следует считать систему
циклической АМФ и аденилатциклазы. Регуляция микропиноцитоза может
осуществляться рядом нейротрансмиттеров. К некоторым из них установлено
наличие рецепторов на эндотелии: для серотонина, гамма-аминомасляной кислоты,
вещества Р и некоторых других. Было показано, что ингибирование транспорта
микропиницитоза достоверно уменьшает выход белка из сосудов в ткань мозга при
остром подъеме артериального давления.
Повышение проницаемости гематоэнцефалического барьера для
катехоломинов, серотонина, гистамина и их предшественников при различных
видах отека мозга было показано во многих работах.
При ишемическом отеке мозга повышение проницаемости
гематоэнцефалического барьера по отношению к катехоламинам и серотонину
имеет более сложную динамику и бывает достоверной не ранее, чем через 72 часа
после реперфузии мозга кровью и через 1 час после ишемии мозга у монгольских
песчанок. Таким образом, вопрос о возможности проникновения биогенных
моноаминов в ткань мозга следует считать решенным. Однако требуют
дальнейшего анализа условия, при которых эти амины накапливаются в
соответствующих структурах (терминальных нейронах) и могут дополнительно
включаться в отдельные звенья патогенеза разных типов отёка мозга и (или)
повреждать функцию нервной системы.
Переход воды и электролитов через гематоэнцефалический барьер при
развитии отека мозга.
Предполагается, что капилляры мозга регулируют динамику проницаемости
воды и электролитов аналогично почкам, коже лягушек, кишечнику и т.д. Для
обеспечения своей регуляторной функции эндотелиальные клетки мозга, подобно
другим структурам, снабжены тесными контактами, многочисленными
митохондриями и высоким содержанием внутриклеточных ферментов, среди
которых особо важную роль играет связанная с мембраной Na+-K -АТФаза.
Выявлена роль нейрогенных и гормональных влияний, контролирующих
переход воды и электролитов в мозг. Показано, что стимуляция голубого пятна в
мозгу вызывает быстрое повышение проницаемости гематоэнцефалического
барьера для воды, одновременно с ослаблением мозгового кровотока. Несмотря на
то, что в области стенок капилляров мозга трудно обнаружить нервные окончания,
в ряде работ, было показано наличие здесь рецепторов, чувствительных к
различным нейротрансмиттерам и гормонам. Особенно хорошо было
продемонстрировано наличие бета-адренегрических рецепторов, а также
функциональной иннервации мозговых капилляров, весьма сходной с центральной
норадренергической иннервацией. В виду того, что капилляры мозга не играют роли
в изменениях общего цереброваскулярного сопротивления, можно заключить, что
эта иннервация регулирует в основном обмен веществ между кровью и мозгом. Как
стало известно, мозговые катехоламины могут способствовать освобождению
вазопрессина в ответ на изменение оемолярности крови. Возможно, что
55
ангиотензин-П также может быть причиной освобождения вазопрессина. Таким
образом, можно предполагать, что норадреналин, вазопрессин и ангиотензин- Ц
составляют часть центральной нейроэндокринной системы, которая связана с
гомеостазом воды и электролитов в мозге.
Переход белков плазмы крови через эндотелиальный барьер и их
распространение по межклеточным пространствам мозга может вызвать вазогенный
отек даже в предварительно неповрежденных тканях. В недавних исследованиях на
кроликах наблюдался прорыв гемато - энцефалического барьера при введении во
внутреннюю сонную артерию сравнительно большой порции крови того же
животного под высоким давлением, причем не было видимых повреждений
паренхимы мозга. При этом наблюдался усиленный переход воды через открытый
барьер, а затем наступало постепенное обратное развитие отека, находящееся в
прямой корреляции со скоростью устранения белков плазмы крови из
межклеточного пространства. При использовании модели холодового повреждения
мозга в различных фазах обратного развития отека мозга был выявлен параллелизм
между содержанием сывороточных белков в ткани и содержанием в ней воды.
Наконец, в экспериментах с моделированием окклюзии средней мозговой артерии
удавалось предотвратить первое открытие барьера для белков уменьшением объема
циркулирующей крови. В результате этого при взятии материала на третьем часу
после рециркуляции крови отек мозга был гораздо меньше выражен, по сравнению
с контрольной группой животных, у которых имелся значительный выход белков из
сосудов в ткань мозга. Эти данные показывают, что выход сывороточных белков в
межклеточное пространство играет первостепенную роль в динамике развития
отека головного мозга.
Роль аквапорина - 4 (AQP4) в формировании отека мозга.
Аквапорины (AQPs) представляют собой семейство белков, образующих
мембранные каналы, выступающие в качестве селективных пор, регулирующих
трансцеллюлярную проницаемость для воды. Образуемые ими каналы проницаемы
исключительно для воды и не проницаемы для ионов либо для растворимых
веществ. Представители семейства AQPs классифицируются по локализации в
определенных органах: почках, легких и т.д. AQP4 - содержится в ЦНС и
покрывает более чем 95 % поверхности церебральных капилляров. Недавние
исследования демонстрируют особую роль AQP4 в развитии отека мозга при
экспериментальном моделировании фокальной церебральной ишемии [100].
Авторы показали, что повышение экспрессии AQP4 мессенжера мРНК в
периинфарктной зоне коры на 7 день после развития фокальной церебральной
ишемии ассоциировалось с генерацией и развитием отека мозга, подтверждаемого
данными МРТ. По данным Manley G.T. et al., выраженность отека мозга была
значительно меньше у линии мышей с генетическим дефицитом AQP4 в сравнении
с контролем при моделировании острой водной интоксикации. Аналогично в линии
мышей с AQP4-дефицитом выявлен лучший неврологический исход и уменьшение
зоны инфаркта при моделировании фокальной церебральной ишемии [78].
Все эти последние данные позволяют надеяться, что разработка и последующее
включение ингибиторов AQP4 в комплекс интенсивной терапии острого
56
иП1емического инсульта позволят уменьшить выраженность отека мозга, и
следовательно, степень проявления неврологических нарушений.
Системное повышение концентрации глутамата характерно для пациентов с
ишемическим инсультом [45,96]. Возможными механизмами, посредством которых
NMDA-рецепторы медиируют реперфузионные повреждения, выступают:
1. Дисрегуляция глутаматного транспорта;
2. Дисрегуляция функции NMDA-рецепторов;
3. Нейрональная сенситизация к стимулам, как результат ишемически
индуцированного снижения мембранного потенциала и нарушения
мобилизации ионов Са"' [109].
NMDA-рецепторы в формировании отека-набухания головного мозга.
NMDA-рецепторы представлены в нейронах, глии и церебральных
эндотелиоцитах. Показано, что использование антагонистов NMDA-рецепторов
снижает выраженность отека головного мозга и объем инфаркта при
экспериментальном моделировании фокальной церебральной ишемии-реперфузии.
Однако использование в клинической практике большинства препаратов этой
группы лимитирует целый ряд побочных эффектов, таких как гипертензия, психозы
и гиперкинезы. Избежать указанных побочных эффектов можно, если использовать
модуляторы сайтов активности NMDA-рецепторов. Так, NMDA-рецептор имеет три
модуляторных сайта: глицин, Mg2T и полиамины. В настоящее время проводятся
исследования целого ряда препаратов этого класса.
ИШЕМИЯ
1
АКТИВАЦИЯ N№A РЕЦЕПТОРОВ I
БЕЛКОВОЕ ФОСФОРИЛИРОВАНИЕ ?или
1
AQP4 АКТИВАЦИЯ
1......
ОТЕК МОЗГА
Рис. 9 NMDA — зависимый механизм формирования отека головного
(по F. Xiao, 2002)
F. Xiao был предложен NMDA - зависимый механизм формирования отека
головного мозга с участием AQP4 (рис. 9). Ишемия индуцирует активацию NMDA-
57
рецепторов, регулирующих активность AQP4 посредством фосфорилирования
белков. Активация AQP4 в свою очередь ведет к формированию отека мозга путем
повышения проницаемости для воды или снижения водного клиренса.
Основной опасностью отека-набухания головного мозга является развитие
внутримозговой гипертензии с явлениями дислокации и вклинения мозга.
Для развития отека мозга характерно появление триады Кушинга: стойкой
брадикардии, повышения АД, нарушения дыхания. Данный симптомокомплекс
является классическим признаком увеличения ВЧД. Однако, триада Кушинга
проявляется только у 33% пациентов, с внутричерепной гипертезией [28].
В целом необходимо отметить, что классические проявления ВЧГ
неспецифичны. Часто развивается фокльная неврологическая симптоматика.
Наиболее частым симптомокомплексом при ВЧГ выступают головная боль,
тошнота, рвота, парез или парлич III и IV черепно-мозговых нервов. Также
характерен отек зрительного нерва, выявляемый при офтальмоскопии, но он более
характерен для длительно персистирующей ВЧГ, а на ранних стадиях этот симптом
отмечается только у незначительной части пациентов с ВЧГ.
В целом необходимо отметить, что диагностика отека-набухания головного
мозга, является трудной и подчас не решаемой задачей, в случае использования
только клинических данных.
Развитие синдрома вклинения мозга, вызывает сосудистые и окклюзионные
осложнения, приводящие к сдавлению важнейших сосудов, усилению имеющейся
ишемии, застоя и отека мозга, а также нарушению циркуляции ликвора,
обуславливающее развитие гидроцефалии. Выделяют три основных вида
супратенториальной дислокации мозга [7]:
1) Вклинение в области поясной извилины, происходящей при
дислокации увеличенного полушария поперек внутричерепной
полости, вытесняя поясную извилину под серп головного мозга,
сдавливая и смещая внутреннюю мозговую вену и переднюю мозговую
артерию, что может привести к дополнительному инфаркту в области
передней мозговой артерии (рис. 10).
Рис. 10 Вклинение поясной извилины, транстенториальное и крючка головного
мозга (по Ворлоу Ч.П. и соавт., 1998)
2) Центральное транстенториальное вклинение - промежуточного мозга,
является конечным результатом дислокации полушарий и базальных
ядер, сдавления и дислокации промежуточного мозга, а также среднего
58
мозга в рострокаудальном направлении через вырезку намета мозжечка
(рис. 11). При этом происходит сдавление большой вены мозга с
увеличением гидростатического давления из вен участка мозга
дренируемой этой веной. За счет дислокации вниз среднего мозга и
моста, происходит натяжение медиальных перфорирующих ветвей
основной артерии, что приводит к ишемии в парамедиальных зонах
ствола и кровоизлиянию, если сохраняется перфузия.
Рис. 11 Центральное транстенториальное вклинение
(по Ворлоу Ч.П. и соавт., 1998)
3) Вклинение крючка головного мозга — происходит в случае, если
увеличивающаяся в объеме зона повреждений в височной ямке или
височной доле, смещающей внутренний базальный край крючка и
гиппокампальной извилины и средней линии таким образом, что они
выпячиваются под краем вырезки намета и прижимают средний мозг к
противоположному краю вырезки намета. В тоже время III черепной
нерв и задняя мозговая артерия на стороне очага часто сдавливается
между нависающим распухшим крючком и свободным краем намета,
приводя к поражению III черепного нерва, отеку и инфаркту теменной
доли из-за ишемии бассейна задней мозговой артерии (рис.11).
Транстенториальное вклинение, является наиболее частой причиной смерти в
первую неделю после развития инфаркта мозга, составляя около 78%. Причем риск
летального исхода является максимальным на 4-5 день после развития инфаркта
мозга [7].
Клинически развитие вклинения мозга на этапе сдавления среднего мозга,
характеризуется прогрессирующим угнетением сознания, окулоцефалического и
окуловестибулярного рефлексов, развитяи контрлатерального гемипареза и
появления двухсторонних патологических стопных знаков. Следующим этапом
является стадия моста и прдолговатого мозга, на которой происходит
Двухстороннее расширение и фиксация зрачков, появление центральной
гипервентиляции и признаков децеребрационной регидности.
59
Глава 5
МОЛЕКУЛЯРНЫЕ И КЛЕТОЧНЫЕ МЕХАНИЗМЫ ПОВРЕЖДЕНИЯ
ГОЛОВНОГО МОЗГА ПРИ ИШЕМИЧЕСКОМ ИНСУЛЬТЕ
5.1 Ишемический каскад при остром инсульте
Выделяют три главных пути ишемического повреждения ткани мозга при
снижении МК до 10 мл/ЮОг/мин’1 хотя точные механизмы его ещё полностью не
установлены:
> неконтролируемое увеличение в плазме клетки концентрации ионов
Са2+;
> глутаматная эксайтотоксичность;
> образование свободных радикалов (рис. 12).
Пусковым механизмом является вызываемый недостатком кислорода переход от
аэробного к анаэробному пути утилизации глюкозы, что создает дефицит
макроэргов — АТФ и креатинфосфата. Это сопровождается, с одной стороны,
избыточным образованием лактата, закислением внутренней среды клеток,
кислотным разрыхлением их цитоплазматических мембран с последующим
набуханием и отеком клеток мозга. С другой стороны, дефицит АТФ в условиях
ишемии приводит к снижению активности мембранных АТФаз - Na+, К+ -
активируемой АТФазы цитоплазматической мембраны (Na+, К+-насос) и Са2'-
активируемой АТФазы эндоплазматического ретикулума нейронов. Недостаточная
активность Na+, К1-насоса ведет к повышению внутриклеточной концентрации Na+
и уменьшению его трансмембранного градиента. Результатом является
гипоксическая деполяризация и повышение возбудимости нейронов, а также
поступление в них Са2+ ввиду изменения вектора переноса ионов Ca2+/Na -
обменником, который при нормальном градиенге ионов Na служит инструментом
удаления Са2+ из клеток. Увеличение содержания Са2+ в нейроплазме достигаемое
этим способом дополняется затрудненным их депонированием из-за недостаточной
интенсивности Са2+-АТФазы эндоплазматического ретикулума, а также
поступлением Са2+ внутрь нейронов через потенциалзависимые Са2+-капалы.
Гипоксическая деполяризация становится механизмом, запускающим глугамат-
кальциевый каскад — последовательность процессов, ведущих к эксайтотоксической
(от excite - возбуждать) смерти нейронов. В этой цепи процессов ведущим является
деполяризация аксонных окончаний глутаматергических нейронов, широко
представленных в мозге.
Деполяризация их сопровождается долговременным и массивным высвобождением
в синаптические щели и ближайшее внеклеточное пространство эксайтотоксичных
аминокислот (глутамат, аспартат). Будучи медиаторами, аминокислоты активируют
в постсинаптических нейронах глутаматные рецепторы NMDA-типа и изоформы
АМРА-рецепторов, ионные каналы которых обладают высокой проводимостью для
наружных ионов Са2+. В результате внутриклеточная концентрация Са2+ возрастает
на 3-4 порядка.
60
Выход
простагландинов
"Г-зстатанйЖ-ъ. Пошревденне клеточной мараны
лейкотриены
Л/с. 12. Последовательность развития ишемического каскада при остром
инсульте
(по Brott Т., Bogousslavsky J, 2000)
61
Лэктат-ацидоз
Уменьшение объемной
скорости кровотока
I
Переход на анаэробный путь дыхания,
дефицит макроэргических фосфатов (АТФ)
Повреждение АТФ-зависимых
механизмов поддержания
трансмембранных градиентов
Увеличение
осмолярности 4
— нейроплазмы, набухание
и отек клеток
Увеличение внутринейронной
~ концентрации Na\ Са"
I
Гипоксическая
деполяризация нейронов
Реперфузия
О,
Устранение Мд**-блока ч.._
NMDA-рецепторов, их
активация глутаматом и
увеличение поступления
Са+‘ внутрь нейронов;
увеличение высвобождения
Са,+ из внутриклеточных
депо \
------Увеличение высвобождения
возбуждающих аминокислот
посредством активации потенциал-
зависимых Са*‘-каналов в
окончаниях глутаматергических
нейронов, затем долговременного
NO-зависимого механизма
экзоцитоза
Глутамат-Са‘* Эксайтотоксичность
Активация каспаз,
деградация цитоскелета и
белкового внутриклеточного
матрикса
Некроз клеток мозга
Запуск генетической
программы апоптоза
Активация фосфолипазы и
NO-синтазы, образование
арахидоновой кислоты.
простаноидов, оксида азота и
супероксидных свободных
радикалов (О , пероксинитрит)
I
Повреждение мембран
клеток и митохондрий,
воспаление
Инфаркт мозга
Рис. 13 Механизм повреждения клеток мозга при ишемическом инсульте
(по Комиссарову И.В., 2003)
В столь высоких концентрациях ионы Са2+ активируют многие внугриклеточные
ферменты и инициируют процессы, приводящие нервные клетки к некрозу и
апоптозу. Активация ферментов семейства каспаз (фосфолипаз, протеаз, нуклеаз),
обуславливающих образование оксида азота (NO), свободных радикалов,
разрушение внутриклеточных белков, фосфолипидов, нуклеиновых кислот.
Активация фосфолипазы А2 ведет к гидролизу фосфолипидов, составляющих
структурную основу мембран клеток и внутриклеточных органелл. При гидролизе
фосфолипидов образуется арахидоновая кислота, которая в период реперфузии
подвергается неферментативному окислению, ведущему к образованию
супероксидных форм кислорода (О2", НО', ОН ). Другим источником образования
свободных радикалов является NO, который образуется из а-аргинина и кислорода
под влиянием Ca2t-активируемой NO-синтазы нейронов. NO легко превращается в
62
пероксинитрит (OONO ) весьма агрессивную для мембран клеток и митохондрий
форму оксида азота. Резкое усиление окислительного процесса и синтеза NO при
недостаточности системы антиоксидантной защиты приводит к развитию
оксидантного стресса,
который является одним из универсальных механизмов повреждения тканей при
различных патологических состояниях.
Процессами, вызывающими реперфузионные повреждения, являются также
активация пула полиморфно-ядерных лейкоцитов, высвобождение
провоспалительных цитокинов и образование простаноидов, в совокупности
формирующих воспаление в зоне инфаркта [5,12,41,97].
Таким образом, определяющее значение в повреждающем действии на
нейроны в зоне ишемической полутени имеют последовательно возникающие
патофизиологические механизмы (рис. 13):
> фокальный дефицит мозгового кровотока;
> глутаматно-кальциевая эксайтотоксичность;
> околоинфарктная деполяризация;
> образование свободных радикалов;
> оксидантный стресс;
> местное воспаление и генетически запрограммированная смерть клеток.
5.2 Ишемический апоптоз нейронов.
Феноптоз - программированная смерть организма - миф или реальность ?
Нервная ткань в процессе своего роста и развития постоянно контролируется со
стороны генетического аппарта. Как оказалось, сама гибель клеток нервной системы
также находится под жестким генетическим контролем.
В настоящее время известно два основных механизма клеточной смерти:
описанный еще К. Фогтом (1842 г.) и Р. Вихровым (1859 г.) - некроз, и процесс
активной програмированной клеточной гибели - апоптоз, обоснованный в 1972 г.
английскими учеными J.F.R. Kerr, А.Н. Wyllie и A.R. Currie [69]. Термин «апоптоз»,
является производным от греческого и обозначает «отделение», «опадение листьев с
дерева». Причем необходимо отметить, что само сезонное опадение листьев (а также
созревших плодов) происходит именно путем запуска программы апоптоза, а само
наличие этого механизма v растений указывает на эволюционную древность апоптоза
[15].
Исторически в работе A.Gluksmann (1951 г.) было получено первое
морфологическое описание программируемой клеточной смерти (ПГС) в
эмбриональной ткани, причем автор понимал, что здесь имеет место особый вид
клеточной смерти, однако считал, что ПГС присуща только эмбриогенезу. В
последующем С. DeDuve (1964 г.) предложил концепцию «клеточного суицида»,
указывая, что в определенных условиях клетка может сама себя убить, используя
содержимое лизосом. Только в ставшей классической работе J.F.R. Kerr с соавт. [69]
представили процесс клеточной смерти как фундаментальное явление клеточной
биологии, не менее значимое чем процесс деления клеток. Авторы показали, что
наряду с процессом пассивной смерти клеток - некрозом, существует важный
физиологический механизм активной смерти клеток, запускаемый активацией т.н.
63
«генов смерти». Исследуя нормальную и патологическую ткани, они обнаружили, что
умирающие клетки делятся на 2 категории. В сильно поврежденных тканях
преобладали процессы некроза, которые затрагивали целые клеточные поля и
характеризовались пассивной дегенерацией клеток с набуханием и фрагментацией
органелл, разрушением мембран, лизисом клеток, выходом внутриклеточного
содержимого в окружающую ткань и развитием воспалительного ответа (рис. 10).
Как было показано позже, некроз всегда обусловлен трубой патологией, ею
механизмы не требуют затрат энергии и предотвратить его можно только, устранив
причину повреждения [21,10].
Апоптозные клетки в противоположность некротическим не были найдены в
соприкосновении друг с другом, а располагались по одиночке или небольшими
группами и были разбросаны по всей ткани. Они имели меньший размер,
неизмененные мембраны и органеллы наряду' с признаками сморщивания цитоплазмы
и множественными цитоплазматическими мембран-связанными выпячиваниями.
Благодаря сохранности мембран не происходило распространения литических
ферментов в окружающие клетки, в связи с чем нс наблюдалось их повреждения и
воспалительной реакции ткани, что в настоящее время признано одним из важных
морфологических отличий апоптоза от некроза [23]. И сморщенные клетки, и
апоптозные тельца содержали нетронутые массы конденсированного хроматина.
Результатом последовательной деструкции ДНК в апоптозных клетках являлась
невозможность их репликации (воспроизведения) и участия в межклеточных
взаимодействиях, так как эти процессы требуют синтеза новых протеинов.
Умирающие клетки эффективно удалялись из ткани с помощью механизма
фагоцитоза, включающегося в первые минуты-—часы после смерти. После
поглощения апоптозных тел соседними клетками, они затем сближаются, закрывая
собой дефект ткани, таким образом не вызывая изменения архитектоники ткани и в
отсутствие признаков воспаления (рис. 10).
Позже было установлено, что при ограниченной способности к фагоцитозу и
сохранении апоптозных клеток в ткани более 1—2 дней их мембраны также
дезинтегрируются и возникает «вторичный» некроз (лизис клетки, умершем
апоптозной смертью) [10,15,21].
J.F.R. Kerr с соавт. [69] обнаружили апоптозные клетки и в патологических, и в
физиологических условиях. Впоследствии выяснилось, что апоптоз является
неотъемлемой составной частью процессов развития и гомеостаза зрелой ткани. В
норме организм использует этот генетически запрограммированный механизм в
эмбриогенезе для уничтожения «избытка» клеточного материала па ранней стадии
развития тканей, в частности в нейронах, не установивших контакты с клетками-
мишенями и лишенных таким образом трофической поддержки из этих клеток. В
зрелом возрасте интенсивность апоптоза в центральной нервной системе
млекопитающих существенно снижается, хотя остается высокой в других тканях. В
основе нормальной смены клеток лежат процессы апоптоза. Выраженное подавление
апоптозной смерти приводит к развитою онкологических заболеваний. Устранение
пораженных вирусами клеток, развитие иммунного ответа также сопровождаются
необходимой апоптозной реакцией.
64
АПОПТОЗ
Рис. 14 Морфологические отличия между некрозом и апоптозом
(по В.Д. Самуилову и соавт., 2000)
Выбор пути гибели, а также жизнеспособность нейронов обеспечивается
целым рядом факторов, основными из которых являются: внутриклеточные
протеины (Р-53, Р-57, RB-I и пр.), называемые «танаканами» или «генами смерти»
(bel-1, bel-2, TRPM-2E C-fos и пр ). На выживаемость нейронов и переход к гибели
также влияет целый ряд неспецифических факторов: уровень енергетических
затрат, наличие цитотоксических ионов и радикалов. При достижении
критического уровня этих влияний происходит запуск программы гибели.
В ответ на воздействие повреждающих факторов физической, химической или
биологической природы, механизм апоптозной гибели клеток включает следующие
этапы [10]:
увеличение внутриклеточного Cai+;
активация ядерной эндонуклеазы;
фрагментация ДНК;
стимуляция активности (АДФ-рибозил) полимеразы ;
нарушения синтеза макроэргов в результате истощения пула НАВ ;
стимуляция свободнорадикальной деградации биомакромолекул;
гибель клетки.
Морфологическими маркерами апоптоза являются апоптозные тельца и
сморщенные нейроны с целостной мембраной. Биохимическим маркером, который
стал практически идентичен понятию «апоптоз», считают ДНК-фрагментацию.
Этот процесс активируется ионами Са2+ и Mg 2+, а ингибируется ионами Zn2+.
Расщепление ДНК происходит в результате действия Са2 и Mg 2+- зависимой
эндонуклеазы. Эндонуклеазы расщепляют ДНК между белками гистонами,
высвобождая фрагменты регулярной длины. ДНК первоначально делится на большие
фрагменты из 50 и 300 тыс. оснований, которые затем расщепляются на составные
части из 180 пар оснований, образующие характерную «лестницу» при сепарации
гелевым электрофорезом. Однако получены данные о том, что ДНК-фрагментация
65
не всегда коррелирует с характерной для апоптоза морфологией. Более точным
методом подтверждения апоптоза является биологически-гистохимический метод,
позволяющий не только зафиксировать факт ДНК-фрагментации, но также
наличие важного морфологического признака — апоптозных телец [10].
При фокальной ишемии мозга большинство апоптозных клеток определяются
вдоль внутренней границы ишемического ядра, что, возможно, отражает участие
апоптозного процесса в расширении зоны инфаркта. Количество апоптозных клеток
возрастает в зависимости от продолжительности фокальной ишемии мозга.
По данным М. Chopp и Y. Li, спустя 2 ч после постоянной окклюзии средней
мозговой артерии и через 24 ч от начала реперфузии в ишемизированном стриатуме
находят клетки, подвергшиеся апоптозу, среди которых 90-95 % нейронов, около 5—
10 % астроцитов и не более 1% эндотелиальных клеток. Количество апоптозных
клеток увеличивалось максимально через 24—48 ч после индуцированной ишемии,
впоследствии имело тенденцию к снижению, однако оставалось достоверно
повышенным, по сравнению с контрольными животными, около 4 нед после окклюзии.
Длительное функционирование апоптоза дополнительно подтверждает динамический
характер процесса клеточного умирания и свидетельствует о том, что мероприятия,
проводимые даже недели спустя от начала ишемического инсульта, могут защищать
ишемизированную ткань мозга [56].
В целом ишемия мозга вызывает фрагментацию нейрональной и в меньшей
степени глиальной ДНК ведущей к апоптозу клеток. Показано, что двухцепочечные
разрывы ДНК обнаруживались после 1 часа реперфузии, а одиночные разрывы - в
течение 1 минуты. Логично, что ишемия мозга одновременно вызывала и
повышенную активность ДНК-репарирующих белков. Как показало исследование
S. Love et al. [77] ДНК-репарирующие белки — поли (АДФ-рибоза)полимераза
(PARP) и Ku 80 имели высокую активность в первые часы ишемии у пациентов и
сохраняли высокий уровень экспрессии в нейронах и глие в течение нескольких
последующих дней. Было выявлено, что активация репарирующих белков и в
частности PARP способна приводить к истощению энергетического потенциала
клеток и как результат — нейрон ал ьнальной гибели после ишемического
повреждения.
Особый интерес представляют результаты исследований, в которых было
показано, что культура корковых нейронов мыши, временно лишенная кислорода
и глюкозы, погибает по типу «эксайтотоксического» некроза. Однако после
блокады эксайтотоксичности NMDA- и АМРА/каинатными антагонистами,
нейроны подвергаются апоптозу, чувствительному к действию ингибитора каспаз
z-VAD.fmk. Глутамат — одно из ключевых соединений, которое наряду с глюкозой
служит энергетическим материалом для ткани мозга, участвует в биосинтезе
углеводов, ГАМК, фолиевой кислоты, обмене ГМФ, обеззараживании аммиака
Однако длительная ативация глутаматных рецепторов, наблюдающаяся при
нарушении функций нейронов, является фактором, вызывающим клеточную смерть
[Ю].
Так, связывание глутамата NMDA-рецептором приводит к усилению потока Са7'
в нейрон. Внутриклеточное накопление Са“+ активирует фосфолипазу А2, которая в
свою очередь стимулирует освобождение глутамата, тем самым способствуя еще
66
большему повышению уровня внутриклеточного Са2+. Образуемая при этих
процессах арахидоновая кислота, ингибирует обратный захват глутамата в
астроци гы и нейроны, при ее метаболизме образуется свободный радикал, который
цовреждающе действует на нейрональный метаболизм и способствует дальнейшей
активации фосфолипазы А?, таким образом поддерживая порочный круг.
При параллельном включении в ишемизированном мозге процессов некроза
и апоптоза выбор преобладающего механизма смерти определяется несколькими
факторами, в том числе тяжестью ишемии, зрелостью нейронов, доступностью
трофического обеспечения и концентрацией свободных внутриклеточных ионов
Са24. В условиях in vitro низкие концентрации «эксайтотоксинов» индуцируют
апоптоз нейронов, в то время как более высокие — некроз [2].
60 минут 90 минут - постоянно
некроз
ИЖ апоптоз
живые нейроны
Рис. 15 Зависимость вида гибели нейронов от длительности и тяжести
ишемического поражения (по S.H. Graham, R.W. Hickey, 2002).
А. Очень кратковременная и невыраженная по тяжести ишемия, не вызывает
повреждений и изменений в экспрессии генов. Более тяжелая и длительная
ишемия вызывает сублетальное повреждение. При этом низкая выраженность
повреждения ассоциируется с экспрессией генов способствующих повышению
выживаемости нейронов, таких как hcl-2 ингибирующего апоптоз и белки
теплового шока (светло-серый цвет). Более пролонгированная или более тяжелая
ишемия наоборот индуцирует экспрессию генов, запускающих апоптоз, (темно-
серый цвет). В случае если игиемия тяжелая и длительная, нарушается
энергетический метаболизм в этих зонах с дефицитом макроэргов (АТФ) и
нарушением синтеза белков, что приводит к развитию некроза.
67
В, С, D. Изображена схема ишемии в головном мозге крыс после 30, 60 и 90
минут при моделировании окклюзии средней церебральной артерии: 30 мин. -
сублетальная ишемия, сопровождающаяся экспрессией генов ингибирующих
клеточную смерть и белки теплового шока; 60 мин. — летальная ишемия, в центре
зона инфарктного ядра, окруженная зоной апоптотических нервных клеток. Если
ишемия составляет 90 и более минут, зона некроза расширяется и апоптоз
наблюдается только в области кортикальной мантии.
Длительность и тяжесть ишемического инсульта также определяют, какие
нейроны станут на путь апоптоза после ишемии in vivo. Так, при
экспериментальном моделировании фокальной церебральной ишемии было
выявлено, что нейроны локализующиеся в тяжелой стрессовой ишемической
зоне погибают преимущественно путем некроза, а в зоне с менее выраженной
стрессовой ишемии (в зоне ишемической полутени) превалирует механизм
апоптоза (рис. 15) [56].
Состояние кальциевого гомеостаза в клетках, подвергающихся апоптозу,
еще не совсем ясно. В некоторых случаях в апоптозных клетках наблюдаются
избыточные концентрации ионов Са2', что позволило предположить их участие в
индукции механизмов апоптозного каскада (например, в активации эндонуклеаз,
расщепляющих ядерную ДНК). Вместе с тем, апоптоз может быть
приостановлен путем увеличения содержания внутриклеточных ионов Са2’ с
помощью активации потенциал-зависимых кальциевых каналов, ингибирования
секвестрации кальция или добавления малых концентраций NMDA в питательную
среду. Концентрации внутриклеточных ионов Са~+, ингибирующие апоптоз в
симпатических нейронах, значительно ниже (180—240 нмоль) его уровней,
опосредующих глутамат-кальциевую токсичность в нейронах (более 5 мкмоль).
Возможно, выживание нейронов в значительной мере зависит от оптимального
содержания внутриклеточных ионов Са-4, и на первый взгляд парадоксальная
гипотеза кальциевой нейропротекции может оказаться реальной [10].
Таким образом, кальциевый гомеостаз является мощной модулирующей
системой: при неадекватно низких внутриклеточных уровнях ионов С а" нейроны
находятся под риском апоптоза и сильно зависят от трофического обеспечения;
при промежуточных — условия оптимальны для выживания клеток и их
потребность в трофической поддержке минимальна; при повышенных —
начинаются цитотоксические процессы некроза. Логично предположить, что
избыток внутриклеточных ионов Са24 участвует в повреждении клеток
преимущественно в области центрального инфаркта в первые минуты после
начала ишемизации, в то время как их недостаток играет отрицательную роль
позже и в основном в зоне ишемической полутени. По-видимому, некоторые
клетки проходят через обе эти стадии.
Механизмы апоптоза, позволяющие клеткам погибать без признаков
выраженного воспаления и высвобождения генетического материала, включаются
позже быстрых реакций некротических каскадов — как минимум спустя 1—2 ч
после начала ишемии, начинают проявлять себя в полной мере через 12 ч,
достигая максимума активности на 2—3 сутки инсульта. Таким образом, апоптоз
наряду с другими отдаленными последствиями ишемии принимает участие в
68
«деформировании» очага инфаркта, дополнительно повреждая зону ишемической
полутени.
Необходимо отметить, что являясь крайне энергоемким и прогностически
неблагоприятным этапом клеточного цикла, апоптоз играет в случае острой
ишемии саиогеиетическую роль, выступая формой клеточной ауторегуляции и
способствуя выживанию поврежденной ткани, поскольку предотвращение
немедленной гибели (некроза) позволяет нейронам получить дополнительную
возможность структурного восстановления [15]. Механизмы апоптоза
«запускаются» тогда, когда вредное воздействие недостаточно сильно, чтобы вызвать
некроз [2, 79]. Таким образом, организм отвечает на грозящую опасность массовой
гибели клеток своеобразной защитой - самоубийством сравнительно небольшого
числа клеток, чтобы последствия патологических воздействий оказались не так
велики.
Особое место занимает предложенная В.П. Скулачевым [22] концепция
«феноптоза» или запрограммированной гибели всего организма. Механизмом
феноптоза может стать запрограммированная дисфункция одного из
жизненноважных органов. Примером феноптоза для одноклеточных организмов,
является самоубийство Е. сой, зараженной вирусом. Так, ряд штаммов Е. coli
образуют неактивную форму особой протеазы. В случае если бактерия заражается
вирусом, происходит активация этой протеазы, которая в свою очередь
инактивирует один из бактериальных белков, необходимый для белклового синтеза
(EF-Tu), выключая внутриклеточный синтез и убивая клетку, способствуя таким
образом ограничению размножения вируса и предохраняя популяцию Е. coli от
инфекции.
По мнению автора, подобно инфицированным клеткам Е. coli, также и высший
организм зараженный опасным патогенном, становится нежелательным в
сообществе себе подобных, поскольку подвергает опасности заражения. Поэтому
быстрая гибель сильно инфицированной особи могла бы решить проблему
сохранения популяции. В.П. Скулачев считает, что в частности септичесикй шок
можно рассматривать как механизм феноптоза, очищающий популяцию животных
или сообщество людей от инфицированных индивидуумов. Поскольку при
септическом шоке все характеристики указывают, что смерть больного организма
хорошо организована самим этим организмом, в то время как патогены играют в
общем пассивную роль. При этом подчеркивается, что феноптоз является
последней линей обороны популяции и в случае, если уровень патогена у
заболевшего индивидуума не так высок, чтобы стать реальной опасностью для
популяции, тогда триггерный сигнал используется только для привлечения
фагоцитов к зараженной области. И наоборот, при превышении патогеном
некоторого критического значения запускается весь каскад провоспалительных
факторов, ведущих к развитию септического шока. При этом у бактерий имеет
место феноптоз в чистом виде, а у человека феноптоз с его массовым
самоубийством клеток в жизненноважных органах развивается из исходно
полезного для организма ограниченного апоптоза.
Реперфузионное повреждение органов, вызываемое взрывом образования
активных форм кислорода (АФК) связано, в том числе с активацией программы
69
апоптоза. Так, клетка, образующая АФК не только повреждает себя, но и
становится опасной для ткани. Поэтому она и ее ближайшие соседи должны быть
как можно скорее элиминированы. Таким образом, массовая гибель клеток при
реперфузии органов будет являться следствием запуска программы апоптоза
Механизм феноптоза при ишемически-реперфузионном повреждении
предложенный автором в ответ на триггерное влияние аноксии включает в себя
(рис. 16): 1) восстановление свободных ионов железа; 2) повышение уровня
ксантиоксидазы, которая в ответ на появление кислорода в процессе реперфузии
образует активные формы кислорода (О?Н2О2), которые в свою очередьобразуют
гидроксил-радикал (ОН) при окислении Fe2+. При этом действие Fe2 и
ксантиноксидазы может быть блокировано соответственно хелаторами и
аллопуринолом. Всплеск образования АФК ведет к апоптозу АФК-генерирующих
клеток, а также соседних клеток.
Аноксия
Хелаторы ионов железа
Повышение уровня ксантиноксидазы
Аллопуринол
«Взрыв» АФК при реоксигенацми
Антиоксиданты, ингибиторы каспаз
Апоптоз клеток - супврпродулентов АФК и их соседей
Дисфункция жизне^новйжного органа из-за массового апоптоза
Смерть организма
Рис. 16 Механизм феноптоза индуцированный аноксией, при развитии
реперфузионного повреждения (по В.П Скуяачеву, 1999)
Массовый апоптоз клеток приводит к дисфункции жизненно важного органа
(мозга, сердца и т.д.), что может повлечете за собой гибель целого организма.
Исходя из этих позиций, необходимо отметить два момента. С одной стороны,
апоптоз вызванный реперфузией и реоксигенацией может быть полезен, поскольку
он устраняет опасные клетки, образующие АФК. Однако с другой стороны, апоптоз
вреден, поскольку кончают самоубийством так много клеток, что нормальное
функционирование жизненноважного органа становится невозможным
Использование блокаторов апотоза позволит в будущем значительно уменьшить
реперфузионное повреждение и выраженность полиорганной дисфункции.
Ряд экспериментальных исследований свидетельствуют о существовании
возраст - зависимой запрограммированной смерти. Так, ограничительный стресс
запускает апоптоз, контролируемый белком-шапероном Hsp70 в клетках гладких
70
МЫП1И аорты, а индукция его уменьшается у старых животных. В условиях стресса
у таких животных весь пул Hsp70 окажется связанным с денатурированными
белками (количество которых возрастает прямо пропорционально со степенью
рыраженности стресса), поскольку синтез Hsp70 не будет в достаточной мере
иНдуцир°ван. Вследствие этого, количество Hsp70, связанного с фосфатазой JNK-P,
снизится, активность фосфатазы уменьшится, в результате будет активирован
апоптоз. Данный эффект может иметь катастрофические последствия при
появлении даже сравнительно небольшой зоны ишемии в мозге, из-за массового
апоптоза клеток. И его можно рассматривать как зависящий от возраста феноптоз.
Исходя из вышеизложенного, В.П. Скулачев предполагает, что инсульт (так же, как
инфаркт миокарда и рак) может служить механизмом феноптоза, зависящим от
возраста, который освобождает популяцию от бесполезных индивидуумов,
способных к тому же ухудшать ее генофонд.
Данная концепция базируется на том факте, что в процессе эволюции жизни
возникли специфические биохимические механизмы, действие которых приводит к
ситуации, когда интересы особи приносятся в жертву ради процветания сообщества
или популяции. Однако механизмы подобного рода выглядят как опасный атавизм,
избавление от которого могло бы способствовать ликвидации определенных
человеческих недугов и прежде всего в основе которых лежит феноптоз. Исходя из
этого автором предложены направления борьбы с феноптозом [22]:
идентификация феноптических болезней человека;
выявление причин феноптоза;
устранение или сведение к минимуму причин феноптоза; описание
физиологических или биохимических механизмов реализации феиоптоза;
при гиперапоптотических феноптозах, когда апоптоз стимулируется до столь
высокого уровня, который уже может быть не совместим с жизнью человека
(инфаркт мозга) - разработка методов блокирования сигнала, запускающего
массовый апоптоз, и выключения уже начавшегося апоптоза посредством
специфических ингибиторов.
Данная интересная концепция, на наш взгляд, проливает свет на
фундаментальные представления о генезе ишемического повреждения головного
мозга и расширяет наше понимание этой проблемы, позволяя в будущем
разработать более эффективные методы как профилактики, так и лечения
ишемического инсульта, влияя на конкретные звенья патогенеза.
5.3 Механизм ишемически - реперфузионного повреждения головного
мозга
Несмотря на то, что восстановление перфузии может спасти жизнеспособные,
но функционально неполноценные клетки «ишемической полутени» и восстановить
их функционирование, оно может оказывать и пагубное действие - вызвать
реперфузионное повреждение ткани мозга. Развитие его связывают с избытком
воды, осмотически активных веществ, включением кислорода в процессы
свободнорадикального окисления, увеличением концентрации токсических
веществ, усиление асептического локального воспаления, активацию апоптоза (рис.
17).
71
D.A. Barber et aL [35] показали, что ранняя реперфузия выявляется у 61%
обследованных и состоит из 2-х компонентов: питающей (адекватной) и не
питающей (неадекватной) перфузии. Почти у 1/3 больных, перенесших
ишемический инсульт, наступает ранняя адекватная реперфузия, ешё у половины
(50%) обследованных — ранняя неадекватная реперфузия, которая затем
трансформируется в постишемическую гипоперфузию.
Усиление перфузии в зоне сформировавшегося инфаркта мозга может
увеличивать риск геморрагической трансформации, являющейся геморрагической
фазой ишемического инсульта.
Геморрагическая трансформация - распространенное осложнение тяжелого
ишемического поражения, она является маркером реперфузии ткани мозга.
По анатомо-морфологическим критериям и распространенности различают
такие типы геморрагической трансформации:
Тип 1 - петехиальный геморрагический инфаркт;
Тип 2 — геморрагический инфаркт;
Тип 3 — инфаркт-гематома или интраинфарктная гематома.
Рис. 17 Патофизиологические каскады при ишемическом повреждении
головного мозга (по Kochanek P.M. et al., 2002)
Таким образом, в процессе развития инсульта мозговой кровоток может
изменяться от значительной фокальной гипоперфузии до ранней гиперперфузии
которая может быть адекватной или неадекватной, что имеет важное значение для
выбора стратегии лечения ишемического инсульта в разные периоды его развития
[Ю].
72
Глава 6
ОРГАНИЗАЦИЯ ОКАЗАНИЯ МЕДИЦИНСКОЙ ПОМОЩИ И
ВОПРОСЫ ДИАГНОСТИКИ ПРИ ИШЕМИЧЕСКОМ ИНСУЛЬТЕ
6.1 «Цепочка выживания» после инсульта
Успешная помощь при остром инсульте начинается с признания и населением,
и специалистами в области здравоохранения того факта, что инсульт - это
неотложное состояние, подобно острому инфаркту миокарда и травме.
Большинство пациентов с инсультом не получает адекватную терапию, потому что
они не госпитализируются в достаточной степени быстро [35].
Успешная помощь при остром инсульте как неотложном состоянии зависит от
цепи из 4 шагов (рис. 18).
Рис. 18. Четыре шага е "Цепочке выживания после инсульта "
ББыстрое распознавание и реакция на клинические признаки,
предупреждающие о развитии инсульта.
2.Быстрая активация службы скорой медицинской помощи.
З.Быстрая транспортировка с уведомлением принимающей больницы.
4.Быстрая и точная диагностика и лечение в больнице.
Начало лечения ишемического инсульта в пределах «терапевтического окна»
является главным условием эффективности проводимой интенсивной терапии.
Медицинский персонал должен быть обучен распознаванию ранних признаков
ишемического инсульта, а также терапии ранних осложнений инсульта. Обучение
должно включать проведение медицинского обследования, которое должно
фокусироваться на оценке уровня сознания, наличии фокальных нарушений,
эпилептической активности и распознавании афазии и других серьезных
когнитивных нарушений [49].
Концепция ’’Время - это Мозг’’ должна быть понята всеми, кто вовлечен в
Цепочку выживания после инсульта": Не следует допускать потери времени после
поступления пациента в больницу, поскольку время - это самый важный фактор,
особенно первые минуты и часы после развития инсульта, так как патогенетически
обоснованное, в полном объеме, но начатое поздно лечение, может оказаться
практически неэффективным.
Пациент с острым инсультом, даже тот, у которого • имеется легкая
симптоматика, должен рассматриваться как неотложный пациент. После
Доставки его в приемное отделение, он должен быть осмотрел врачом в
73
приоритетном порядке, как имеющий опасное для жизни и инвалидизирующес
заболевание.
Признаки и симптомы, которые могут предсказать более поздние осложнения
такие как злокачественный инфаркт или кровотечение, повторный инсульт, а также
такие состояния как гипертонический криз, сопутствующий инфаркт миокарда
аспирационная пневмония и почечная недостаточность, должны быть рано
распознаны. Однако практика свидетельствует, что у каждого четвертого - пятого
пациента клинический диагноз характера инсульта, поставленный даже опытным
врачом, оказывается ошибочным, что в равной мере справедливо как дЛл
ишемических, так и геморрагических инсультов. Поэтому раннее определение
подтипа инсульта, основанное как на физикальном и неврологическом
обследовании, так и на квалифицированной интерпретации результатов
компьютерной томографии (КТ) и магнито-рсзонанасной томографии (МРТ), также
является существенным для предсказания высокого риска раннего рецидива. При
этом необходимо отметить, что в 80% случае КТ мозга обнаруживает зону
пониженной плотности, которая клинически соответствует инфаркту мозга в
течении первых 12-24 часов после начала заболевания [3].
Для дифференциальной диагностики ишемического и геморрагического
инсульта, можно рекомендовать использование клинической шкалы предложенной
G. Besson et al. (1995):
Шкала G. Besson для дифференциальной диагностики ишемического и
геморрагического инсульта
1. Потребление алкоголя +2
2. Подошвенные рефлексы +1,5
3. Головная боль +3
4. Артериальная гипертензия в анамнезе +3
5. Преходящий неврологический дефицит в анамнезе -5
6. Заболевание периферических сосудов -2
7. Фибрилляция предсердий при поступлении -2,1
8. Гиперлипидемия в анамнезе -1,5
Оценка проводится на основании бивариантного анализа указанных 8
показателей: 1+2+3+4-5-6-7-8. Если сумма баллов менее 1, то вероятность
ишемического инсульта >90 %.
Однако не существует клинических методов использования шкал, которые бы
позволили надежно дифференцировать ишемический инсульт от геморрагического.
Для этого необходимо проведение КТ или МРТ исследования. Так, ошибка при
использовании шкалы Besson составляет >10%.
В целом для ишемического инсульта характерны следующие диагностические
признаки [3]:
предшествующая гранзиторная ишемическая атака или транзиториая
монокулярная слепота;
выявленный ранее атеросклеротический процесс в сосудах сердив
(кардиосклероз), него (облитерирующий атеросклероз), магистральных артериях
головы; ’
74
я патология сердца (нарушение ритма сердца, чаще мерцательная аритмия,
наличие искусственных клапанов сердца, ревматизм, инфекционный эндокардит,
острый инфаркт миокарда, пролапс митрального клапана и др.);
развитие во время сна, после приема горячей ванны, физического утомления,
а также во время приступа мерцательной аритмии, в гч. на фоне острого инфаркта
миокарда, кровопотери;
постепенное развитие неврологической симптоматики, в ряде случаев ее
мерцание, т.е. нарастание, уменьшение и повторное нарастание;
возрает более 50 лет;
превалирование очаговой симптоматики над общемозговой
симптоматикой.
Одним из важнейших методов дифференциальной диагностики инфаркта мозга
и кровоизлияния в мозг, является проведение люмбальной функции. Однако
необходимо подчеркнуть, что проведение люмбальной пункции у пациентов с
явлениями нарастающего отека головного мозга может резко ухудшить их
состояние, вплоть до развития вклинения ствола мозга в большое затылочное
отверстие, о чем необходимо помнить. Поэтому люмбальная пункция без
предварительного проведения компьютерной томографии (КТ), является
потенциально опасной у пациентов с внутримозговой гематомой. При наличии
негативных данных КТ, примерно в 3% случаев есть шанс, что люмбальная пункция
обнаружит доказательства САК, по крайней мере, в течении первых трех дней.
После этого периода доля ложноотрицательных данных КТ становится все больше,
а значение люмбальной пункции возрастает, особенно в течение первых двух
недель.
Не рекомендуется проведение люмбальной пункции, в первые 12 часов после
появления у больного головной боли, для того чтобы можно было отличить кровь,
полученную вследствие травм аги чес кой пункции от кровоизлияния в
субарахноидальное пространство. Это связано с тем, что необходимо около 12
часов, для того чтобы эритроциты подверглись лизису, а гемоглобин превратился в
оксигемоглобин и после центрифугирования ликвора появляется желтовато
окрашенная осадочная жидкость (ксантохромия). Поскольку ксантохромия является
единственнвм надежным доказательством того, что геморрагически окрашенный
ликвор не является результатом травматичной люмбальной пункции, необходимо,
чтобы ликвор исследовался не ранее чем через 12 часов после начала головной
боли. Поэтому авторы рекомендуют, что предпочтение необходимо отдать методу
обнаружения ксантохромия, в отличие от традиционного метода «трех пробирок».
6.2 Категории инсультных отделений
Согласно европейской классификации инсультные отделения подразделяются
на несколько категорий [49]:
1) Острое инсультное отделение, принимающее пациентов в остром периоде и
продолжающее лечение в течение нескольких дней, но обычно менее чем 1 неделю.
) Инсультное отделение, принимающее пациентов в остром периоде, и
продолжающее лечение и реабилитацию в течение нескольких недель, если это
необходимо.
75
3) Реабилитационное инсультное отделение, принимающее пациентов спустя I или
2 недели и продолжающее лечение и реабилитацию в течение нескольких недель
или месяцев, если это необходимо.
4) Мобильная инсультная бригада - мобильная бригада, оказывающая помощь при
инсульте, а также лечение пациентов с инсультом в различных, отделениях. Такие
бригады обычно формируются в больницах, где нет специализированных
инсультных отделений.
На Украине, как и большинстве стран бывшего Советского Союза, функцию
инсультных отделений выполняют отделения реанимации и интенсивной терапии
(ОРИТ) общего профилия или специализированные отделения нейрореаиимации.
Многочисленные мультицентровые исследования и систематические обзоры
доказали преимущества специализированных «инсультных» или реанимационных
отделений, в сравнении с обычными неврологическими отделениями, в которых
было показано, что лечение в специализированных отделениях ассоциировано с
достоверным снижением летальности и инвалидности, а также значительным
улучшением качества жизни выживших пациентов в отдаленном периоде после
перенесенного инсульта [10].
6.3 Диагностическая визуализация
Краниальная компьютерная томография (КТ) широко доступна и не только
надежно дифференцирует геморрагию и ишемический инсульт или
субарахноидальную геморрагию, но также может исключить многие другие
заболевания головного мозга. Ранние признаки ишемии иногда могут быть
обнаружены уже спустя два часа после начала инсульта, но это может быть
трудным даже для обученного исследователя, особенно при очень рано
выполненных исследованиях. Ранние признаки инфаркта включают стирание
борозд, увеличение базальных ганглиев и гиперинтенсивный сигнал от средней
мозговой артерии (рис. 19). Ранние признаки обширного инфаркта со смещением
срединных структур указывают на очень серьезный случай и высокий риск, как
вторичной геморрагии, так и формирования большого злокачественного отека и
могут оправдывать повторную визуализацию после короткого интервала.
Паренхиматозная геморрагия может быть идентифицирована почти немедленно или
в глубоких структурах у пациентов с гипертензией, или в нетипичных областях у
пациентов без гипертензии, или при адекватном лечении, обычно вследствие
церебральной амилоидной ангиопатии. Инфратенториальная геморрагия или
мозжечковые инфаркты могут быть идентифицированы подобно
супратенториальным повреждениям, но более мелкие геморрагии/ишемические
инфаркты, в особенности в стволе мозга, могут легко быть пропущены. Кроме того,
КТ может детектировать субарахноидальную кровь в большинстве случаев
субарахноидальных геморрагий. Иногда геморрагии могут даже быть
интерпретированы как первичные, но в действительности быть вторичными к
ишемическим событиям. Вовлечение четко определенных сосудистых бассейнов
показательно в отношении таких состояний, которые более легко
идентифицировать при МРГ — исследованиях.
76
1 2 3
Рис. 19 Компьютерная томография головного мозга пациента с ишемическим
инсультом с клиникой левого гемипареза
1- через 50 минут; 2- через 3 часа; 3- через 25 часов после развития инсульта.
Геморрагии в головной мозг имеют тенденцию к росгу в первые 6-12 часов
после инсульта приблизительно у 40-50% пациентов, даже без клинического
ухудшения, что делает необходимым раннее повторное КТ-исследование
[32,49].
КТ-ангиография (КТА) - надежный инструмент для получения информации
относительно экстра- и интракраниальной артериальной проходимости, и ее
использование в клинической практике часто увеличивает ценность
диагностического процесса.
Магнитно-резонансная томография (МРТ) - более чувствительна и все
больше и больше используется в клиниках с инсультными отделениями как
стандартная процедура. Недавние опасения о более низкой чувствительности в
идентификации геморрагий в головной мозг преодолены современными методами
МРТ, такими как Т2*- взвешенное изображение, которые в действительности даже
оолее чувствительны, чем КТ-сканирование, для демонстрации внутримозговой
геморрагии. Диффузионно-взвешенная МРТ очень чувствительна для ранней
Детекции поврежденной мозговой ткани и в комбинации с перфузионно -
взвешенной МРТ может помочь в идентификации пациентов, которым показано
проведение раннего тромболизиса. Согласно современным представлениям,
пациенты с существенным перфузионно - диффузионным нарушением могут
получить пользу от восстановления ишемической полутени, окружающей уже
77
некротизированное ядро инфаркта и напротив у пациентов, у которых области
диффузионного и перфузионного дефицита накладываются, имеют менее
благоприятное соотношение пользы и риска. Данные методы МРТ еще не доступны
для широкого клинического применения, но представляются многообещающими
инструментами для будущего рутинного использования.
MP-ангиография может использоваться для идентификации окклюзий
крупных интракраниальных артерий, но должна тщательно интерпретироваться,
если отсутствуют исследования экстракраниальных церебральных артерий. В этом
случае ультразвуковое исследование может быть эффективно для идентификации
тяжелых гемодинамически значимых каротидных обструкций, которые, вероятно.,
продуцируют значительные перфузионные нарушения при небольших
эмболических или лакунарных инсультах и могут таким образом имитировать
значительное перфузионно - диффузионное нарушение. MP-ангиография также
играет роль в оценке венозной системы и аневризм до 3 мм в диаметре.
Главная цель состоит в том, чтобы идентифицировать крупные обструктивные
поражения эктракраниальных, а также интракраниальных базальных артерий.
Кроме того, транскраниальная допплерография может быть эффективна для
мониторирования спонтанного или медикаментозно индуцированного
тромболизиса у большинства пациентов. Приблизительно у одной четверти
пациентов невозможно или очень трудно получить адекватные сигналы через
височное окно без применения контрастных агентов. Обнаружение редких
этиологических факторов развития ишемического инсульта, таких как расслоение,
гиперплазия интимы и других менее частых причин, облегчается при
систематическом использовании ультразвуковых исследований.
Трансэзофагеальная и трансторакальная эхокардиография часто
показательна при подозрении на кардиоэмболический инсульт, но обычно не
выполняется как неотложное исследование Применение этих исследований в
течение первых 24 часов после начала инсульта может быть полезным для выбора
лучшей вторичной профилактики, особенно при наличии кардиальных источников
эмболии.
В целом, EUSI был предложен следующий стандартный алгоритм неотложных
диагностических тестов при остром ишемическом инсульте (табл. 6) [49].
Таблица 6
Неотложные диагностические тесты при остром инсульте
(* в отдельных случаях)
1. Компьютерная томография.
2. ЭКГ и рентгенологическое исследование органов грудной клетки.
3. Лабораторные исследования:
- общий анализ крови;
- протромбиновое время, АЧТВ;
- тромбоциты;
- электролиты сыворотки;
- глюкоза крови;
78
-анализ содержания газов артериальной крови (если заподозрена
гипоксия);
-печеночный и почечный комплексы.
4 Пульсоксиметрия.
5 Люмбальная пункция (только если клинически заподозрено субарахноидальное
«•повоизлияние, а компьютерная томография дала отрицательный результат ).
^дуплексное и транскраниальное ультразвуковое исследование.
7 ЭЭГ*
s. МРТ* и МРТ/КТА*.
9 ДиффУзионнь™ ^Р* и перфузионный МР*.
10 Эхокардиография (трансторакальная и трансэзофагиальиая)*.
6.4 Мониторинг витальных и неврологических функций
У всех пациентов с инсультом неврологический статус и витальные функции
(АД, частота пульса и температура) должны непрерывно и регулярно
мониторироваться
Стандартом для динамического мониторинга неврологического статуса
являются неврологические шкалы: NIH Stroke Scale (Шкала инсульта
Национального Института Здоровья США - NIHSS) (табл. 7) и Glasgow Coma Scale
(Шкала ком Глазго - GCS) [32].
В ряде случаев, когда в анамнезе были заболевания сердца и/или аритмии, а
также в случае нестабильного АД, желателен мониторинг ЭКГ в режиме online.
Пульсоксиметрия эффективна для непрерывного мониторинга в инсультных
отделениях. Она обеспечивает важную информацию о оксигенационном статусе
пациента.
Центральный венозный катетер и мониторинг центрального венозного
давления необходим у пациентов с тяжёлым инсультом, находящихся в отделениях
реанимации. Посредством центрального венозного катетера, может быть получена
косвенная информация о внутрисосудистом объёме, функции сердца и
эластичности венозной сист емы.
ШКАЛА ИНСУЛЬТА NIHSS
Таблица 7
Функция Степень нарушений Балл
не изменено 0
1А оглушение 1
Уровень сознания сопор 2
— кома 3
правильно отвечает на 2 вопроса 0
Ш Ответы на вопросы правильно отвечает на 1 вопрос 1
— не отвечает 2
правильно выполняет 2 команды 0
Реакция на команды правильно выполняет 1 команду 1
не выполняет ни одной команды 2
79
2 Парез взгляда взгляд нормальный частичный парез взгляда полный парез взгляда 0 1 1 2 1
сохранены 0 '1
3 Поля зрения частичная гемианопсия полная гемианопсия 1 1 2
билатеральная гемианопсия
отсутствует 0 ~~ 1
4 Парез мимической мускулатуры лёгкий частичный 1 | 2
полный з I
5 Двигательные функции верхней конечности: Л. Левом Б. Правой пареза нет опускается медленно, за 5 сек быстро падает, менее чем за 5 сек не может преодолеть силу притяжения движений в руке нет 0 1 1 1 2 1 3 4
6 Двигательные функции нижней конечности: А. Левой Б. Правой пареза нет опускается медленно, за 5 сек быстро падает, менее чем за 5 сек не может преодолеть силу притяжения движений в ноге нет 0 1 2 5 4
не нарушена 0
7 Чувствительность гипестезия 1
анестезия 2
нет 0
8 Атаксия в руке или ноге 1
в руке и ноге 2
нормальная 0
9 Речь лёгкая афазия выраженная афазия 1 2
тотальная афазия 3
нет 0
10 Дизартрия умеренная 1
выраженная 2
нет 0
11 Невнимательность легкая степень 1
тяжелая степень 2 _
Оценка по шкале NIHSS: 0- состояние удовлетворительное
3-8 неврологические нарушения легкой степени
9-12 неврологические нарушения средней степени
13-15 тяжелые неврологические нарушения
16-34 неврологические нарушения крайней степени
тяжести
34 кома.
Менее 10 баллов - указывает па благоприятный исход, а более 20 баллов
ассоциировано с неблагоприятным исходом.
80
для оценки исхода инсульта был предоложен индекс Бартела (табл. 9) и модифицированная шкала Rankin - Оксфордская шкала социальной дезадаптации *табЛ'8)' Таблица 8. Оксфордская шкала социальной дезадаптации.
Г~ Балл Описание
-"—"о Нет симптомов
1 Минимальная симптоматика, которая не мешает обычному стилю жизни
2 Минимальная степень дезадаптации. Жалобы, несколько ограничивающие обычный образ жизни, но позволяющие больному ухаживать за собой
3 Средняя степень дезадаптации. Жалобы, значительно ограничивающие обычный образ жизни, но больной не полностью зависит от окружающих
4 Средне - тяжелая степень дезадаптации. Жалобы, четко определяющие невозможность самостоятельного существования, хотя не требующие постоянного ухода и внимания
5 Тяжелая степень дезадаптации. Полная зависимость, требующая постоянного внимания днем и ночью
Таблица 9
Индекс Бартела, показывающий две альтернативные системы подсчета
(Mahoneyf Barthel, 1965)
Функциональные возможности Балл Балл Степень нарушения
Кишечник 0 0 Недержание или необходимость клизм
5 1 Периодическое недержание (менее одного раза в неделю)
10 2 Удержание
Мочевой пузырь 0 0 Недержание /невозможность справляться с катетером
5 1 Периодическое недержание (менее одного раза в день)
10 2 Удержание
Уход за собой 0 0 Нуждается в помощи при бритье, умывании, причесывании, чистке зубов
5 1 Самостоятельно
Пользование 0 0 Полностью несамостоятельно
туалетом 5 I Нуждается в некоторой помощи
10 2 Самостоятельно одевается, раздевается, осуществляет гигиену
-•^ормление 0 0 Полностью несамостоятельно
81
5 1 Нуждается в некоторой помощи (например, разрезании.
намазывании)
10 2 Самостоятельно, если пища находится в пределах
досягаемости
Пересаживание 0 0 Невозможно, не удерживает равновесия сидя
(например, с 5 1 Нуждается в значительной помощи
кровати на стул) 10 2 Нуждается в минимальной помощи
15 3 Самостоятельно
Передвижения 0 0 Невозможны
5 1 Самостоятельное перемещение в инвалидном кресле в помещении
10 2 Прогулки с чей-либо помощью либо под наблюдением Самостоятельно (но может использовать вспомогательные
15 з средства)
Одевание 0 0 Невозможно
5 1 Нуждается в некоторой помощи
10 2 Самостоятельно, включая застегивание
Ходьба по 0 0 Невозможна
лестнице 5 1 Нуждается в некоторой помощи или наблюдении
10 2 Самостоятельно вверх и вниз
Прием ванны 0 0 Несамостоятельно
5 1 Самостоятельно в ванной или под душем
Общий итог 0-100 0-20
Глава 7
ИНТЕНСИВНАЯ ТЕРАПИЯ ИШЕМИЧЕСКОГО ИНСУЛЬТА
А. Поддержание экстрацеребрального гомеостаза
Поддержание экстрацеребрального гомеосгаза нацелено на стабилизацию
витальных функций пациента в критическом состоянии, нарушения которых могут
отрицательно влиять на исход инсульта, а также на обеспечение оптимальной
физиологической базы, для проведения специфической терапевтической стратегии.
Поддержание экстрацеребрального гомеостаза у пациентов с инсультом
включает лечение респираторных и кардиологических нарушений, коррекцию
водно-электролитных и метаболических расстройств, контроль АД и коррекцию
повышенного внутричерепного давления. Кроме того, терапия эпилептических
приступов и профилактические меры в отношении тромбоза глубоких вен, эмболии
лёгочной артерии, дисфагии, аспирационной пневмонии, пролежней и других
инфекционных процессов являются частью комплекса интенсивной терапии
пациентов с ишемическим инсультом.
82
Необходимо подчеркнуть, что обеспечение адекватного поддержания
ьных функций составляет базовую терапию ишемического инсульта.
вИ j респираторная терапия
Для лечения инсульта требуется поддержание нормальной респираторной
шгпии и адекватной оксигенации крови, что является важным фактором для
падения метаболической функции в зоне «ишемической полутени».
С Гипоксия может быть следствием обширного инфаркта ствола мозга или
полушария, обширной церебральной геморрагии, длительной эпилептической
активности или таких осложнений, как тяжёлая пневмония (особенно у пациентов с
иском аспирации), сердечная недостаточность, эмболия лёгочной артерии или
обострение хронического обструктивного заболевания легких.
При снижении SaO2<94 % при пульсоксиметрии, необходимо проведение
оксигснотерапии через лицевую маску или назальные катетеры со скоростью 2-4
л/мин. Головной конец кровати следует приподнять на 15-30°.
Пациентам без признаков гипоксии, проведение оксигснотерапии не
рекомендуется [93].
При SaO2<90 %, несмотря на проводимую оксигенотерапию, РаО2 <55 mm Hg,
в случаях серьезного нарушения респираторного пазтерна, гиперкапнии, а также
когда неврологический дефицит составляет <8 баллов по шкале ком Глазго и у
пациентов имеющих высокий риск аспирации, необходима ранняя интубация
трахеи, а по показаниям и проведение ИВЛ.
При проведении ИВЛ необходимо подобрать такой режим, при котором
повышение ЦВД будет минимальным - использование низких величин ДО при
достаточной минутной вентиляции в режиме умеренной гипервентиляции и по
самым строгим показаниям использовать режим ИВЛ с ПДКВ.
«Борьба» пациента с респиратором не допустима [25,32,103].
Проведение кинетической терапии (только на фоне стабилизации
гемодинамики), наиболее эффективна следующая схема: положение на спине - 3
часа, на боку - 1час, на животе -3 часа (с валиком под плечевым поясом и крыльями
подвздошных костей), на другом боку -1 час [30].
7.2 Коррекция циркуляторных нарушений
При ишемическом инсульте, особенно мерцательная аритмия, встречаются
часто, в то время как могут осложнять клиническое течение сердечная
недостаточность, острый инфаркт миокарда (ИМ) или внезапная смерть.
Каждому пациенту с ишемическим инсультом, с момента поступления
необходимо проведение ЭКГ - исследования. Выше были приведены показания к
непрерывному мониторингу ЭКГ. Оптимизация сердечного выброса с
поддержанием высокого нормального АД и нормальной частоты сокращений
сеРДЦа является важной основой лечения инсульта.
Центральное венозное давление должно поддерживаться приблизительно на
Уровне 8-10 см Н2О, и его мониторинг позволит заранее предупредить развитие
сфицита или перегрузки объёма, поскольку и то, и другое отрицательно влияет на
Ребральную перфузию. Внутрисосудистый объём должен сохраняться
доильным.
83
Развитие артериальной гипотензии у лиц с ишемическим инсультом редкОе
явление, если она и наблюдается то, как правило, за счет сопутствующей патологии-
острого инфаркта миокарда, аритмии, передозировки лекарственных препаратов
сепсисе и пр.
В случае ее развития необходимо, как можно быстрее коррегировать
гипотензию, т.к. она суживает «терапевтическое окно», способствуя расширению
зоны инфаркта, и усугубляет прогноз. Путем адекватного волемического
восполнения кристаллоидами (при этом не допускается введение растворов 5%
глюкозы из-за риска увеличения отека мозга вследствие уменьшения осмолярности
плазмы) и коллоидами, лечения сердечных аритмий. При неэффективности
допустимо применение вазопрессоров.
Увеличение сердечного выброса может улучшить церебральную перфузию в
тех областях, которые утратили свою ауторегуляторную способность после
развития острой ишемии [32,49].
Среди инотропных агентов добутамин имеет преимущество, поскольку он
увеличивает сердечный выброс, не оказывая существенного влияния на частоту
сердечных сокращений и на АД. Добутамин является смесью двух изомеров: D-
изомера с [V и р2-адренергическими эффектами и L-изомера с рг и а(-
адренергическими эффектами. Начальные эффекты добутамина являются
инотропными через стимуляцию -рецепторов с различной динамикой в изменении
АД. При дозе от 2 до 28 мкг/кг/мин., увеличение сердечного индекса (СИ)
колебалось от 20% до 66%, ЧСС от 10 до 25%. При средней дозе от 5 до 12
мкг/кг/мин. индекс работы левого желудочка повышается от 23 до 37%.
Допамин является непосредственным предшественником норэпинефрина и
эпинефрина. Допамин вызывает несколько дозозависимых фармакологических
эффектов. В дозе < 5 мкг/кг/мин доминирующим эффектом является стимуляция
дофаминергических ДАГ и ДА2- рецепторов в почках, кишечнике, коронарном
ложе, приводящее к вазодилятации, повышению уровня гломерулярной
фильтрации, почечного кровотока и экскреции Na\ В дозах 5-10 мкг/кг/мин
преобладают Pi- адренергические эффекты, вызывая повышение сократительной
функции миокарда и ЧСС. В дозах выше 10 мкг/кг/мин доминируют а-
адренергические эффекты, вызывая артериальную вазоконстрикцию с повышением
АД. Таким образом, у допамина имеется большое количество перекрывающих друг
друга эффектов.
Гемодинамические эффекты допамина.
Допамин приводит к повышению САД на 24% первоначально путем
повышения СИ с минимальным эффектом на общее периферическое сопротивление
соусудов (ОПСС). Повышение сердечного выброса вызвано возрастанием ударного
объема (УО) и, в меньшей степени, за счет увеличения ЧСС. Средние дозы,
требующиеся для восстановления АД, составляют 15мкг/кг/мин. У больных с
высоким давлением заклинивания легочных капилляров (ДЗЛК), допамин, может
способствовать его возрастанию, увеличивая венозный возврат. Использование
допамина в дозах выше 20 мкг/кг/мин, вызывают повышение давления в правом
сердце и увеличение ЧСС. Допамин показан для улучшения сократительной
функции правого желудочка у больных с правожелудочковой недостаточностью.
84
Измеярцие газового состава крови.
До^ахняГпоказал стойкое возрастание легочной фракции шунта, снижение a-v
ы по Ог, повышение сатурации смешанной венозной крови со снижением
Рт И,1И без его изменений. Допамин угнетает вентиляционный ответ на
Р^-лкапнию. Повышение сердечного выброса после применения допамина
нижаст легочное сосудистое сопротивление, повышает легочный кровоток и
чтгубляет шунтирование путем закрытия сосудов в слабо вентилируемых участках
легких, что и играет ведущую роль в повышении фракции легочного шунта. Четко
станавливается тенденция к повышению содержания О2 в смешанной венозной
^рови, вопреки возрастанию шунтирования.
Доставка кислорода.
Допамин повышает рО2 выше уровня, определяемого до начала лечения или
уровня с применением конкурентных катехоламинов. Его эффекты на расчетное
или измеряемое содержание О2 носят неопределенный характер. Снижение уровня
экстракции О2 указывает на отсутствие положительной динамики в тканевой
оксигенаттии и является следствием недостаточно улучшенного
микропиркуля горного тока в витальных органах или сохраняющейся кислородной
задолженности.
Допамин является очень эффективным в повышении САД у больных с
сохраняющейся гипотонией после оптимальной жидкостной ресусцитации. САД
повышается как результат возрастания сердечного индекса, что является весьма
благоприятным фактором для больных с гипотензией со снижением сердечного
выброса. Главными побочными эффектами допамина являются тахикардия,
повышение окклюзионного давления в легочной артерии, повышение легочного
шунта, снижение рО2 и pHj.
Эпинефрин (адреналин)
Эпинефрин стимулирует а- и Р-рецепторы. В низких дозах Р-адренергические
эффекты доминируют; при его дозах от 0,1 до 0,5 мкг/кг/мин. повышается СИ на 23-
54%. Имеются данные о том, что эпинефрин усугубляет лактат-ацидоз и нарушает
перфузию кишечника.
У больных, нечувствительных к увеличению объема или инфузии других
катехоламинов, эпинефрин может повышать САД, СИ и УО с более уверенным
повышением ОПСС и ЧСС. Так как ОПСС повышается с увеличением доз
эпинефрина, эти проявления имеют непредсказуемые дозозависимые ответы. Moran
EL. et al. показали линейную зависимость между дозой эпинефрина, ЧСС, САД,
СИ, УО, DO2 и VO2. У больных с правожелудочковой недостаточностью эпинефрин
повышает функцию правого желудочка с улучшением сократимости. Препарат
снижает спланхнитический кровоток с временным повышением концентрации
лактата; артерио-венозной разницы рСО2, снижением pHj. Применение эпинефрина
ассоциировало с повышением концентрации системного и регионального лактата,
этиология данного явления пока не ясна. Несмотря на респираторную компенсацию
И Снижение артериального рСО2, повышение концентрации лактата было связано со
снижением артериальной pH и избытком оснований. К негативным эффектам
эпинефрина можно отнести повышение ЧСС.
85
Таким образом, эпинефрин повышает АД у больных, нечувствительных к
традиционным вазопрессорным агентам.
Фенилэпинефрин (мезатон).
Фенилэпинефрин - селективный а,-агонист адренорецепторов, используется
для быстрой внутривенной инфузии при лечении суправентрикулярной тахикардии
вызванной рефлекторной вагусной стимуляцией сердца вследствие быстрого
повышения кровяного давления. Фенилэпинефрин внутривенно применяется в0
время анестезии для повышения АД. Его быстрое действие, короткий период и
первичные сосудистые эффекты делают его привлекательным для лечения
гипотензии. Рекомендуется применение фенилэпинефрина коротким курсом при
гипердинамическом состоянии больных без гипотензии: во время применения
препарата в дозе 70 мкг/кг/мин. повышались САД, СИ, У О; ЧСС снижалось
незначительно; ОПСС оставалось на прежнем уровне. Фенилэпинефриа
характеризуется дозозависимым эффектом. Так, при применении его у
гипердинамических больных с нормальным АД в дозе 0,5-8 мкг/кг/мин.
повышались САД, ОПСС и УО без изменения СИ; ЧСС была достоверно ниже. Эти
исследования выявили подъем параметров транспорта О2 и не находили
статистически достоверных изменений DO2 и VO2; клинически значимое
повышение VO2 (>15%) было зарегистрировано у 8 из 10 больных при одинаковых
дозах. Введение фенилэпинефрина начинали с 0,5 мкг/кг/мин и титровали для
поддержания САД>70 мм рт.ст.; необходимость во введении препарата составляла
65 часов, при этом максимальные дозы соответствовали 3,7 мкг/кг/мин (от 0,4 до
9,1); повышались САД, ОПСС, СИ, УО, без изменений ЧСС, клинически значимо
повышался диурез, отличалась стабильностью концентрация креатинина в плазме
крови; повышались DO2 и VO2.
Следовательно, у больных после жидкостной ресусцитации, фенилэпинефрин
повышает АД, не повреждая сердечных и почечных функций. Фенилэпинефрин
может быть хорошим выбором, когда тахиаритмия ограничивает терапию с
другими вазопрессорами.
Норэпинефрин (норадреналин - НА).
Норэпинефрин, подобно эпинефрину, стимулирует1 а- и Р-рецепторы, в итоге
а-адренергический ответ является доминирующим. Эффекты норэпинефрина на СИ
весьма умеренны, ЧСС не изменяется или даже снижается па 10%.
НА является мощным сс-адренергическим агонистом. С учетом потенциально
вредного вазоконстрикторного эффекта на регионарное сосудистое ложе,
норэпинефрин традиционно не должен использоваться или должен быть в резерве,
как последняя надежда у критических больных с заведомо плохими исходными
результатами. Однако, последние наблюдения с использованием НА показали, что
этот препарат может успешно повышать давление без сопутствующего ухудшения
органных функций. Во многих исследованиях больные получали жидкость для
коррекции гиповолемии, затем титровался допамин до достижения надлежащего
АД. Если допамин не оказывал эффекта, дополнительно к его режиму применялся
НА. НА обычно используется в средней дозе 0,2-1,3 мкг/кг/мин, известной в
литературе является высшая доза 3,3 мкг/кг/мин.
86
Кяплиоваскулярные эффекты НА. Терапия с НА обычно повышает САД с
«Дпльшим изменением ЧСС и сердечного выброса, приводя к повышению ОПСС.
ЙС^как сердечный выброс увеличивается или не изменяется и САД устойчиво
Та£ьппается, индекс объема левого желудочка имеет тенденцию к возрастанию,
ом исследований установлено повышение (1-3 мм рт.ст.) окклюзионного
вления в легочной артерии, другие указывают на наличие изменений. Среднее
** яление в легочной артерии или не меняется, или слегка повышается. НА, как и
vine вазопрессоры, должен применяться только для восстановления уровней САД
и ОПСС. Титрование по САД вернее, чем по ОПСС и зависит от многих факторов,
включая возраст и тяжесть состояния. НА более мощный препарат, чем допамин и
может быть эффективнее для стабилизации гемодинамики.
Почечные эффекты НА. У больных с гипотензией и гиповолемией
вазаконстрикторные эффекты НА могут иметь выраженное вредное влияние на
почечную гемодинамику с повышением почечного сосудистого сопротивления и
почечной ишемией. НА в эксперименте вызывает ишемия-индуцированную ОПН.
Ситуация отличается при гипердинамическом септическом шоке, когда есть
надежда, что диурез снижается преимущественно как результат снижения
почечного перфузионного давления, когда НА имеет наибольшее воздействие на
эфферентное артериолярное сопротивление и повышает фильтрационную фракцию,
нормализация почечного сосудистого сопротивления может эффективно
восстанавливать диурез. Комбинированная терапия, включая вазоактивные агенты с
фазным сосудистым действием, может быть рассмотрена как путь к
максимизированному терапевтическому улучшению с минимальными побочными
эффектами. Принимая это во внимание, могут быть рекомендованы низкие,
«почечные» дозы допамина: 1-3 мкг/кг/мин. В новых исследованиях дополнение
низких доз допамина НА у здоровых добровольцев значительно повышает
почечный кровоток и экскрецию Na . Следовательно, почечное вазодилятационное
действие низких доз допамина может персистировать вопреки инфузии мощных
вазопрессоров.
Эффекты НА на концентрацию лактата.
Эффекты НА на концентрацию лактата в сыворотке крови были установлены
во многих исследованиях: изменение концентрации лактата возникало через
короткий промежуток времени (1-3 часа) и поскольку кровоток имел тенденцию к
значительному улучшению, снижение лактата было незначительным. Отсюда
вытекает вывод, что применение НА не носит повреждающего эффекта и может
Даже улучшать тканевую оксигенацию.
Все катехоламиновые вазопрессоры могут вызывать значительную
тахикардию, особенно у больных с неадекватной жидкостной ресусцитацией. У
льных с сопутствующей коронарной болезнью, вазопрессор-индуцированное
Повышение потребления кислорода миокардом может способствовать
миокардиальной ишемии и инфаркту. При наличии миокардиальной дисфункции
ыточная вазоконстрикция может снижать У О, сердечный выброс и DO2. Когда
начата инфузия вазопрессоров, дозы должны быть осторожно титрованы для
Становления САД, без снижения У О. Если УО начинает страдать, дозы
87
вазопрессоров должны быть снижены или рассмотрен вариант применения
добутамина. При наличии дисфункции правого желудочка, повышение
преднагрузки в правом предсердии может ухудшить функцию правого желудочка.
С целью профилактики данной клинической ситуации при инфузии вазопрессоров
следует удерживать легочное сопротивление на низком уровне одновременно с
восстановлением адекватной системной гемодинамики. Такие мощные
вазопрессоры, как НА, снижают кровоток, в то время как вазопрессорная терапия
должна обеспечивать адекватный уровень СИ для оптимизации почечного
кровотока. Применение вазопрессоров может повреждать кровоток в
спланхнитической системе, и это проявляется стресс-язвами, илеусом и
мальабсорбцией.
Алгоритм вазопрессорной терапии
Если соответственная жидкостная ресусцитация не приводит к восстановлению
адекватного АД и органной перфузии, должна быть начата терапия с
вазопрессорными агентами:
- добутамин и допамин является агентом первой линии для повышения АД;
катетеризация легочной артерии служит для оперделения давления в легочной
артерии, измерения сердечного выброса и SvO2;
- допамин и НА эффективны для повышения АД при условии адекватной
жидкостной ресусцитации; допамин повышает сердечный выброс больше, чем НА,
но его применение может быть лимитировано тахикардией; НА обладает
выраженным вазопрессорным эффектом;
- адреналин должен назначаться при рефрактерной гипотензии;
- рутинное применение низких доз допамина для поддержания почечной
функции не рекомендуется, но низкие дозы допамина могут повышать почечный
кровоток у некоторых больных, когда дополняются к НА.
Комбинация и одновременное применение.
Установлено, что норадреналин/добутамин или норадреналин/допамин
комбинации не лучше, чем норадреналин или добутамин, или допамин отдельно
взятые для улучшения СИ. Адреналин является веществом, наиболее активно
стимулирующим сердечную деятельность. Адреналин ассоциируется с повышением
концентрации лактата в артериальной крови и снижением гастрального pH!, которое
указывает на нарушение перфузии регионарного сосудистого ложа. Добутамин
повышает СИ и УИ в большей мере, чем норадреналин, но увеличение левого и
правого индекса ударного объема были одинаковыми в обоих случаях. При
применении норадреналин не наблюдалось достоверной тахикардии. Возможно
нарушение мезентериальной перфузии при сочетании норадреналин с добутамином.
У пациентов с ишемическим инсультом чаще, нежели гипотензия,
наблюдается артериальная гипертензия.
Коррекция артериального давления.
В июне 2003 года опубликованы новые рекомендации Европейского общества
гипертензии и Европейского общества кардиологии по лечению артериальной
гипертензии (АГ). За месяц до этого, в мае 2003 года, в США также появились
новые рекомендации, посвященные той же проблеме: они подготовлены и
88
лv6 пикованы Объединенным Национальным комитетом США по выявлению,
оценке и лечению высокого артериального давления (JNC).
Новые европейские рекомендации пришли на смену ранее опубликованным
екомендациям ВОЗ/МОГ (международное общество гипертензии) 1999 года.
Обновление этих документов обусловлено накоплением новых данных, полученных
в крупны* многоцентровых исследованиях, посвященных проблеме лечения АГ и
необходимостью их внедрения в практику.
Поражение органов-мишеней, влияющее на степень риска:
- Гипертрофия левого желудочка: ЭКГ критерии Соколова-Лайона >38 мм,
Корнельский >2440 мм/мс;
- Эхокардиографические критерии: индекс массы миокарда левого желудочка
для мужчин >125 г/м2, для женщин > 110г/м;
- УЗИ - признаки утолщения стенки сосудов: толщина интимы — меди сонной
артерии > 0,9 мм; наличие атеросклеротической бляшки;
- Небольшое повышение концентрации креатинина (у мужчин 115-
133мкмоль/л или 1,3-1,5 мг/дл, у женщин - 107-124 мкмоль/л или 1,2-1,4 мг/дл);
- Микроальбуминурия (30-300 мг/24 часа, отношение альбумин/креатинин >22
мг/дл, или > 2,5 мг/моль у мужчин и > 31 мг/г или > 3,5 мг/моль у женщин).
Рекомендации JNC 7
Одна из целей, заявленных составителями JNC 7 — это упрощение
классификации (табл. 10).
Таблица 10.
Классификация АД JNC 7 (2003).
Уровень АД, mm Hg САД, mm Hg ДАД, mm Hg
нормальное <120 <80
прегипертензия 120-139 80-89
1 стадия АГ 140-159 90-99
2 стадия АГ >160 >100
Третья стадия не выделяется в связи с тем, что терапевтические подходы к
таким больным мало отличаются от тех, которые используются при лечении
больных со второй стадией АГ.
Европейские рекомендации 2003 года предлагают рассматривать не только
ведение больных с АГ, но и лиц с «высоким нормальным» АД (130-139/85-89мм
рт.ст.). В последнем случае предлагается проводить немедекаментозную терапию, а
В случае высокого дополнительного риска (то есть, при наличии трех или более
факторов риска или поражения органов - мишеней, или сопутствующей патологии)
- начинать медикаментозное лечение, несмотря на то, что АД не достигает
гипертензивного уровня.
В соответствии с этим терапию можно начинать с низкой дозы одного
препарата или с комбинации двух антигипертензивных препаратов - также в низких
Дозах. Такой подход впервые предлагается практическому врачу: все предыдущие
Международные рекомендации предусматривали монотерапию в начале лечения,
комбинированную терапию — только при ее неэффективное™.
89
Коррекция артериальной гипертензии. Снижение АД преследует цель
уменьшения отека головного мозга и риска развития геморрагической
трансформации инфаркта. Однако агрессивное лечение артериальной гипертензии
может привести к вторичному' снижению перфузии в ишемических регионах
способствуя, таким образом, расширению зоны инфаркта. Это связано с тем, что
кровоток в зоне «ишемической полутени» пассивно зависит от среднего
артериального давления [87]. Следовательно, резкого падения АД необходимо
избегать, для поддержания адекватного церебрального перфузионного давления.
Необходимо отметить, что повышение артериального давления может быть
следствием состояний, которые могут быть корригированы (тревога, боль, реакция
на гипоксию, гипогликемию или повышение ВЧД), и обычно АД спонтанно
уменьшается в течение первых дней после начала инсульта.
Артериальная гипертензия не лечиться до тех пор, пока АД систолическое
не будет >220 mmHg, а АД диастолическое >120 mmHg.
Очевидно, что чрезвычайно высокие уровни АД недопустимы. Значения
систолического давления выше 220 mm Hg и диастолического давления выше 120
мм Hg являются показаниями для раннего, но осторожного медикаментозного
лечения, избегая резкого уменьшения артериального давления.
Целевое систолическое АД 180 mm Hg и диастолическое АД 100-105 mm Hg
рекомендуется у пациентов с гипертензией в анамнезе. В других случаях,
желательны более низкие значения артериального давления (160-180/90-100 mm
Hg).
Показаниями для ургентной антигипертензионной терапии являются: острый
инфаркт миокарда (хотя чрезмерное снижение АД у пациентов с ИМ является
вредным), гинертензионная энцефалопатия, сердечная недостаточность с развитием
отека легких, острая почечная недостаточность или расслоение дуги аорты. У
пациентов, подвергающихся тромболизису или введению гепарина следует избегать
систолического давления выше 180 mm Hg.
7.3 Характеристика препаратов для антигипертензивной терапии
при ишемическом инсульте
Лекарственные препараты для лечения артериальной гипертензии у пациентов
с ишемическим инсультом должны обладать короткодействующим эффектом и
незначительно влиять на церебральный кровоток [32,49].
Цель - снижение АД на 10-15 %
от исходного уровня
Препаратом первой линии для купирования артериальной гипертензии при
ишемическом инсульте, отвечающим указанным требованиям является лабетолол
[26].
Лабеталол является неселективным Р-адреноблокатором, который
обеспечивает одновременно селективное a i-адреноблокирующее и прямое
сосудорасширяющее действие, что обуславливав! быстрый и выраженный
антигипертензивный эффект. При этом снижение артериального давления, не
вызывает развитие тахикардии. Лакардия (лабеталол) блокирует а- и Р~
90
еНОрецепторы в соотношении 1:3. Артериальное давление снижается, главным
^азом, за счет уменьшения общего периферического сопротивления сосудов, не
или незначительно снижая сердечный выброс. Лакардия (лабеталол) также
снижает активность ренина плазмы.
фармакокинетика и особенности применения.
После введения в вену максимальная концентрация Лакардии (лабеталола)
ваблюдается через 2 минуты, но уже через 8,5 минут снижается. Период
полувыведения составляет 3,5-4,5 часа и не изменяется при почечной
недостаточности. Выводится из организма, главным образом, с мочой в виде
неактивных метаболитов.
Согласно рекомендациям ВОЗ при беременности с артериальной гипертензией
следует избегать назначения блокаторов АПФ, антагонистов ангиотензиновых
рецепторов и диуретиков. Рекомендуется для быстрого снижения АД, назначать
Лакардию (лабеталол), нифедипин, гидралазин.
Препарат следует использовать осторожно у больных с бронхиальной астмой.
.. • _ ____________ ______ . "" " --
Рекомендуемый режим дозирования лабеталола
а) 10-20 мг в/в струимо в течении 1-2 мин. Можно повторять введение
каждые 10 мин. (суточная доза 200 мг)
или
б) в/в капельно 50-200 мг/сут. в 200 мл 0,9 % раствора NaCl.
Противопоказания.
Препарат противопоказан при атрио-вентрикулярной блокаде, синдроме
слабости синусового узла, выраженной сердечной недостаточности.
Возможно применение следующих препаратов для купирования артериальной
гипертензии:
а) Никардипин 5 мг/час в/в капельно в качестве начальной дозы, затем в
зависимости от эффекта, титруя, повышают дозу до 2,5 мг/час каждые 5 минут до
максимальной дозы 15 мг/час.;
6) Клонидин (клофелин) 0,15 мг в/в;
в) Каптоприл 6,25-12,5 мг per os, однако он имеет короткую продолжительность
действия и может вызывать резкий эффект;
г) При АДдиаст. >140 mmHg возможно применение натрия нитропруссида в/в
капельно 0,5 мкг/кг/мин, в качестве начальной дозы с последующим мониторингом
АД- Однако необходимо учитывать возможные серьезные побочные эффекты при
его применении, такие как рефлекторная тахикардия и ишемия коронарных артерий
(табл. ЦД2) [32,49].
Противопоказано
Использование нифедипина сублингвально, нимодипина (нимотопа)
парентерально противопоказано из-за риска резкого уменьшения артериального
Давления, развития ишемического синдрома обкрадывания [49,58].
91
Таблица 1]
Предлагаемая схема антигипертензивного лечения при остром
ишемическом инсульте
(.модификация Brott et al. 1994, и Ringleb et al., 1998)
АД систолическое 180-220 мм Hg и или АД диастолическое 105-140 ммНд Не лечить
АД систолическое >220 мм Hg и/или АД диастолическое 120-140 мм Hg, при повторных измерениях 1) Лакардия (лабеталол) 10-20 мт в/в струйно в течении 1-2 мин. Можно повторять введение Лакардии каждые 10 мин (суточная доза 200 мг) или в/в капельно 50-200 мг/сут в 200 мл 0,9 % растворе NaCl или 5% глюкозе.; 2) Никардипин 5 мг/час в/в капельно в качестве начальной дозы, затем в зависимости от эффекта, титруя повышают дозу до 2,5 мг/час каждые 5 минут до максимальной дозы 15 мг/час., 3) Клонидин 0,15-0,3 мг в/в или п/к; 4) Каптоприл 6,25-12,5мг перорально
Диастолическое АД >140 мм Hg 1) Натрия нитропруссид в/в капельно 0,5 мкг/кг/мин; 2) Нитроглицерин 5 мг в/в, затем 1-4 мг/час в/в.
Таблица 12
Характеристика антигипертензивных препаратов, которые могут быть
использованы при остром ишемическом инсульте
(модификация Kaplan, 1990 и Ringleb et al., 1998)
Препараты Доза Начало, мин Длитель- ность, часы Неблагоприятные эффекты
ПЕРОРАЛЬНЫЕ ПРЕПАТАРЫ Ингибиторы ангиотензин-превращающего фермента
Каптоприл 6-12,5 мг сублингвально 15-30 4-6 1 Уменьшение мозгового кровотока, ортостатическая гипотензия
92
ПАРЕНТЕРАЛЬНЫЕ ПРЕПАРАТЫ
Центральные симпатолитики
Клонидин Вначале 0,2 мг, затем 0,1 мг/ч до 0,8 мг 5-15 6-8 Глубокая гипотензия; с осторожностью, если комбинируется с диуретиками
Вазодилататоры
Натрия нитропруссид 0,25-10 мкг/кг мин1 1-5 Тошнота, рвота, судорожные сокращения мышц, усиленное потоотделение, тиоцинатная интоксикация
Нитроглицер ин 5-100 мкг/кг мин 1 2-5 Тахикардия, головная боль, рвота
Р-адреноблокаторы
Пропранолол 1-10 мг в/в 1-2 3-6 Побочные эффекты 0- адреноблокаторов (бронхоспазм, уменьшенный сердечный выброс, брадикардия)
а/р~ адреноблокаторы
Лабеталол ———- 20-80 мг в/в болюсно, 2мг/мин в/в инфузия 5-10 3-5 Рвота, постуральная гипотензия. Тошнота, головокружение
а-адреноблокаторы
Урапидил 10-50 мг в/в болюсно, 9-30 мг/ч 2-5 3 Серьезные побочные эффекты отсутствуют
93
Органические нитраты.
Органические нитраты являются полиольными эфирами азотной кислоты (R-
O-NO2).
Механизм действия нитратов.
1) Внутри гладкомышечной клетки сосуда, в том числе коронарной артерии,
нитраты взаимодействуют с SH-группами (нитратными рецепторами), образуя
оксид азота (NO), который структурно соответствует физиологическому
эндотелиальному расслабляющему (релаксирующему) фактору. Под влиянием
оксида азота повышается активность гуанилатциклазы, что ведет к увеличению в
гладкомышечной клетке цГМФ и далее к снижению содержания в клетке
ионизированного кальция, расслаблению гладкомышечной клетки и вазодилятации,
включая расширение коронарных артерий.
У больных ИБС продукция эндотелиального релаксирующего фактора
(EDRF) коронарными и другими артериями значительно снижена, нитраты
восполняют этот недостаток (рис. 20).
Таким образом, нитраты устраняют коронароспазм, расширяют коронарные
артерии, улучшают коронарный коллатеральный кровоток.
RONO? Эндотелиальный
(органические нитраты) расслабляющий фактор
Рис. 20 Механизм релаксации гладкомышечной клетки под воздействием NO и
органических нитратов.
2) Оказывают дилатирующее влияние на периферические сосуды, особенно на
вены. Венозная дилатация приводит к снижению венозного возврата крови,
падению давления наполнения левого желудочка с уменьшением ударного объема.
Однако минутный объем при этом поддерживается рефлекторной тахикардией в
ответ на снижение главным образом диастолического давления. Под влиянием
нитратов происходит умеренная дилатация артериол, снижается периферическое
сопротивление, что сопровождается увеличением ударного объема. Таким образом,
нитраты вызывают снижение пред - и постнагрузки, что облегчает работу сердца и
уменьшает потребности миокарда в кислороде, снижает выраженность ишемии.
Уменьшаются также размеры сердца и напряжение стенки левого желудочка.
94
3) Перераспределяет внутримиокардиальный кровоток в пользу
ишемизированного участка, благодаря расширению коллатералей в коронарной
системе (рис. 21).
4) Уменьшают агрегацию тромбоцитов и улучшают микроциркуляцию.
[Псриф^римескоб действие
[Расшкрение
[артериол
|и&ацире:мне
| коронарных
(артерий
| Расширение
I |перйферичесзад« вен
Предотвращение
спазма
ксроиарных
артерий
|Снижоние ОПСС "1 |Уменыиамие
----- -. - ^aeHOjHO'oBosepara
т
| Снижение М€Ж|<—|7нфныиемие КДД
Т |и объема жалудочков
Снижение АД
диасо
пичеех&га налряицт-
нир стенки желудем-
яемз
________ ___ * _ ...
Г Уменьшения погребное |гм |
I миокарда в кислорода '
Г Увеличение коронарного
кроиотока в эоне ишемии
Антиангинальный эффект
Рис. 21 Механизмы антиангинального действия нитратов.
Нитроглицерин для внутривенного введения.
Нитроглицерин для внутривенного введения выпускается в следующих
лекарственных формах.
Нерлинганит - выпускается в виде ампул по 10 мл с содержанием 10 мг
нитроглицерина в стерильном изотоническом 5% растворе глюкозы либо во
флаконах по 50 мл с содержанием 50 мг нитроглицерина.
Нитро-поль - ампулы по 5 и 25 мл и флаконы по 50 мл 0,1% раствора
нитроглицерина.
Нитро 5мг/мл - является концентратом нитроглицерина для инфузий (в 1 мл
содержится 5 мг нитроглицерина, 345 мг спирта этилового, 300 мг
пропиленгликоля, до 1 мл воды для инъекций). Перед употреблением разводят в
изотоническом растворе натрия хлорида до получения 0,01% раствора
иитроглицерина.
Нитроглицерин /% спиртовый раствор - по 2 мл в ампуле.
Концентрат нитроглицерина - 1% раствор в ампулах по 1 мл.
Нитро-Бид I.V - стерильный раствор, содержащий по 5 мг в 1 мл,
выпускается в ампулах по 1, 5, 10 мл (консервант - этанол, 70% раствор).
Тридил - стерильный раствор нитроглицерина, содержащий 0,5 мг в 1 мл,
выпускается в ампулах ио 10 мл с концентрацией нитроглицерина 5 мг в 1 мл.
95
Все выпускаемые фармацевтической промышленностью растворы
нитроглицерина являются концентрированными. Перед внутривенным введением
раствор нитроглицерина следует разводить в изотоническом растворе натрия
хлорида до получения 0,01% раствора (0,1 мг -100 мкт в 1 мл, т.е. 5 мкг в 1 капле).
Например, для получения 0,01% раствора нитроглицерина следует растворить
5 мл 1% раствора нитроглицерина в 500 мл изотонического раствора.
Для внутривенного капельного введения нитроглицерина надо использовать
стеклянные емкости с полиэтиленовыми (нс полихлорвиниловыми’) трубками для
исключения абсорбции препарата на стенках системы.
Полученный 0,01% раствор нитроглицерина вводят с начальной скоростью 25
мкг/мин (т.е. 5 капель в минугу). Скорость введения в дальнейшем увеличивают
(при недостаточном эффекте) на 25 мг/мин (5 капель в минуту) каждые 15-20 мин
до оптимальной скорости, при которой происходит снижение АД на величину,
составляющую 25% от исходной. Однако необходимо следить, чтобы
систолическое АД было не ниже 90-100 mm Hg, диастолическое АД - не ниже 60
mm Hg.
Обычно количество нитроглицерина, необходимое для получения эффекта, не
превышает 100 мкг (1 мл 0,01% раствора) в минуту, но допустимая скорость может
составить 150-200 мкг/мин.
Побочные эффекты внутривенного введения нитроглицерина:
- артериальная гипотензия (у 18% больных), она зависит от скорости введения
и положения больного (чаще наблюдается при быстром введении и возвышенном
положении больного);
- головная боль;
- синусовая брадикардия вследствие активации блуждающего нерва (у 4%
больных);
- синусовая тахикардия - увеличение ЧСС на 10-20 мин1 при высокой
скорости введения (у 1% больных);
- побочный эффект пропиленгликоля (растворителя нитроглицерина в
фирменных препаратах) в виде артериальной гипотензии и выраженной
брадикардии.
Толерантность к нитратам.
Толерантность к нитратам - это постепенное снижение антиангинального и
гемодинамического эффектов нитратов при продолжающемся их применении. Под
толерантностью к нигратам можно понимать привыкание больного к этим
препаратам.
В некоторых случаях возможно развитие тахифилаксин, т. е. быстрого (после
приема всего лишь нескольких доз нитратов) появления толерантности.
Обычно толерантность к нитратам развивается при длительном, регулярном,
частом применении препаратов, особенно в высоких дозах. Надо отметить, что к
препаратам нитратов короткого действия (нитроглицерин) толерантность нс
развивается. Однако при комбинированном использовании нитроглицерина
сублингвально и нитратов внутрь может развиться перекрестная толерантность к
нитроглицерину.
Механизм развития толерантности к нитратам:
96
_ постоянно высокий уровень нитратов в крови ведет к насыщению их
едепторов, расположенных в гладкой мускулатуре сосудов, истощение запасов SH-
rnvnH, обеспечивающих превращение молекул нитратов в NO; вследствие этого
теряется реактивность сосудов и ослабляется вазодилатирующий эффект нитратов;
- при приеме нитратов внутрь на 20% снижается почечный кровоток (что
поддерживает высокую концентрацию нитратов в крови) и повышается продукция
контррегулирующих нейрогуморалытых факторов для поддержания адекватного
кровотока в почках (ренин - ангиотензиновая система), что в свою очередь
затрудняет вазодилатирующее действие нитратов;
- уменьшается активность гуанилатциклазы и содержание ц-ГМФ.
Мероприятия по предупреждению и преодолению толерантности к нитратам:
- следует обеспечить прерывистый прием нитратов внутрь в течение суток
таким образом, чтобы свободный период времени с момента поступления их в
кровь составлял 10-12 часов. Надо учитывать, что существует циркадный ритм
ишемии миокарда.
- иногда удается преодолеть толерантность назначением более высоких доз
нитратов на прием, но этот метод бывает эффективен в течение нескольких дней, а
затем толерантность к нитратам все-таки развивается снова, несмотря на большие
дозы;
- толерантность к нитратам развивается гораздо реже при лечении малыми
дозами препаратов, поэтому можно попытаться индивидуально подобрать дозу и
принимать ее с интервалом 8-12 часов. Если такая доза неэффективна, то можно
попытаться сочетать эту дозу препарата с другими антиангинальными средствами
(b-адреноблокаторами);
- можно сочетать прием органических нитратов с ингибиторами АПФ,
содержащими SH-группу, например, капотеном, периндоприлом. Эти препараты
потенцируют антиаш инальный и антиишемический эффекты изосорбида динитрата
и других нитратов;
- при развитии толерантности к нитратам необходимо отменить их на 3-5
дней, а на это время назначить другое антиангинальное средство (например,
корватон, к которому толерантность не развивается); в дальнейшем
антиангинальный эффект нитратов восстанавливается;
- можно провести лечение унитиолом (он является донатором SH-rpynn)
внутримышечно по 5 мл 5% раствора 2 раза в день в течение 5-7 дней, однако такой
метод не получил широкого распостранения, так как считается малоэффективным.
Противопоказания к назначению нитратов:
- кровоизлияние в головной мозг;
- повышение внутричерепного давления;
- выраженная артериальная гипотензия (систолическое АД ниже 100 mm Hg,
Диастолическое АД ниже 60 mm Hg);
- гиповолемия (ЦВД ниже 4-5 см вод.ст.);
- аллергическая реакция на нитраты;
- закрытоугольная форма глаукомы с высоким внугриглазным давлением.
97
8-адреноблокаторы.
Симпатоадреналовая система оказывает влияние на сердечно - сосудистую
систему через катехоламины и адренорецепторы.
Основными катехоламинами являются адренергический нейромедиатор
норадреналин и гормон мозгового слоя надпочечников - адреналин.
Адренорецепторы представляют собой участок клеточной мембраны,
взаимодействующий с катехоламинами. При взаимодействии катехоламинов с
адренорецепторами активируется аденилатциклаза с последующим повышением
содержания внутриклеточного цАМФ и развитием соответствующих эффектов
стимуляции адренорецепторов.
Различают а - (а ] иа2) и В -адренорецепторы (51 и 52).
а -Адренорецепторы локализуются преимущественно в сосудах, сфинктерах
ЖКТ, в миометрии, в трабекулах селезенки. Возбуждение а-рецепторов вызывает
сужение сосудов, сокращение сфинктеров.
Hi-Адренорецепторы локализуются постсинаптически, возбуждаются
норадреналином, а2-адренорецепторы расположены пре- и постсинаптически.
Пресинаптические а2-адренорецепторы возбуждаются преимущественно
норадреналином и участвуют в системе отрицательной обратной связи,
регулирующей высвобождение норадреналина.
Постсинаптические а2-адренорецепторы локализованы вне синапсов и
возбуждаются в основном циркулирующим в крови адреналином.
в- Адренорецепторы подразделяются на в}- и 6'2-рецсшоры, которые
располагаются практически во всех органах и тканях, однако в том или ином органе
преобладает один из двух типов Ь-адренорецепторов:
51-Адренорецепторы локализуются преимущественно в сердце, кишечнике,
адипоцитах, возбуждаются главным образом норадреналином.
52-Адренорецепторы расположены преимущественно в бронхах, матке,
сосудах, возбуждаются в основном циркулирующим в крови адреналином.
В зависимости от расположения по отношению к синапсу различают в-
адренорецепторы постсинаптические (5]-и 52-рецепторы) и пресинаптические (В2-
рецепторы). Пресинаптические В 2-адренорецепторы осуществляют положительную
обратную связь и стимулируют высвобождение норадреналина.
Классификация 8 -адреноблокаторов.
Общепринятой классификации 5-адреноблокаторов не существует. Наиболее
часто 5-адреноблокаторы классифицируются в зависимости от следующих их
свойств:
кардиоселективность (или 5(-селективность): способность 5-
адреноблокаторов избирательно блокировать Bt -адренорецепторы миокарда, и нс
влиять на 52-адренорецспторы (в частности, бронхов). Кардиоселективные 5г
адреноблокаторы мало влияют на реакции, опосредуемые 52-адренорецепторами
(расширение бронхов, секреция инсулина, мобилизация глюкозы из печени,
вазодилатация и сократительная активность матки во время беременности). В связи
с этим в[-селективные блокаторы можно применить при сочетании ИБС и
артериальной гипертензии с хроническим обструктивным бронхитом, сахарным
диабетом, снижением кровообращения в нижних конечностях. Кроме того, 5г
98
ддреноблокаторы не вызывают вазоконстрикцию в сосудах скелетной мускулатуры
0 поэтому при их применении не наблюдается резкой мышечной слабости. Однако
слеДУ61 Учесть’ что кардиоселективность - свойство - относительное и
наблюдается лишь при сравнительно небольших дозах -селективных препаратов,
при назначении этих средств, в больших дозах селективность исчезает. При
бронхиальной астме (особенно в периоде обострения) кардиоселективные в-
адреноблокаторы (даже такой высокоселективный 01-адреноблокатор, как
бисопролол) следует назначать осторожно;
- внутренняя симпатомиметическая активность (ВСА) - частичный
агонизм в отношении 0-адренорецепторов, т. е. способность не только блокировать,
но и частично стимулировать 0-адренорецепторы.
Внутренняя симпатомиметическая активность присуща вискену (пиндололу),
алпренололу, оксипренололу и др. Препараты с внутренней симпатомиметической
активностью обладают следующими свойствами по сравнению с 0-блокаторами,
которым не присуща внутренняя симпатомиметическая активность: меньше
урежают ритм сердца в покое, меньше снижают сократительную способность
миокарда, реже вызывают синдром отмены, реже оказывают отрицательное влияние
на липидный обмен. Установлено, что ВСА может быть направлена на
определенный подтип 0-адренорецепторов (селективная ВСА) или на оба подтипа
(неселективная ВСА). В связи с этим различают три группы 0-адреноблокаторов с
ВСА.
а) 0 -адреноблокаторы с ВСА в отношении 01-адренорецепторов (эпанолол);
б) 0-адреноблокаторы с ВСА преимущественно в отношении 02-
адренорецепторов (пиндолол, дилевалол, целипролол и др.);
в) 0-адреноблокаторы с неселективной ВСА, т.е. обладающие агонизмом в
отношении как 0]-, так и 02-адренорецепторов (окспренолол, буциндолол).
0-Адреноблокаторы с селективной ВСА в отношении 02-адренорсцепторов
обладают вазодилатирующими свойствами (например, пиндолол);
- наличие вазодилатирующих свойств. Имеются 0-адреноблокаторы с
вазодилатирующими свойствами. Механизм вазодилатирующих свойств 0-
адреноблокаторов:
а) выраженная ВСА в отношении 02-адренорецепторов сосудов (например,
пиндолол);
б) блокада а!- и а2-адренорецепторов сосудов (например, лабеталол);
в) прямое сосудорасширяющее действие. У большинства новых 0-
ДДреноблокаторов механизм сосудорасширяющего действия комбинированный.
Например, лабеталол одновременно блокирует а।-адренорецепторы и обладает ВСА
в отношении 02-адренорецепторов; целипролол блокирует а2-адренорецепторы,
обладает ВСА в отношении 02- адренорецепторов и непосредственно оказывает
Прямое сосудорасширяющее действие;
- продолжительность 0-адреноблокирующего действия. Длительность 0-
ДДреноблокирующего действия имеет очень большое значение, так как с ним
6Вязана продолжительность антиангинального и гипотензивного эффектов,
длительность действия 0-адреноблокаторов зависит от особенностей химического
Строения, липофильности и путей выведения.
99
Липофильные (жирорастворимые) 5-адреноблокаторы (пропранолол
метопролол, окспренолол) хорошо всасываются в ЖКТ, хорошо проникают через
гематоэнцефалический барьер, метаболизируются в печени, имеют короткий период
полувыведения (1-5 часов) и должны назначаться не реже 2-3 (а иногда 4-6) раз в
сутки.
Гидрофильные (водорастворимые) 5-адреноблокаторы (атенолол, надолол
соталол и др.) не полностью и неравномерно всасываются в ЖКТ, незначительно
метаболизируются в печени, выделяются преимущественно почками с мочой в
неизменном виде (40-70%) или в виде метаболитов, проникают через
гематоэнцефалический барьер, имеют продолжительный период полувыведения (6-
12 часов), большую длительность действия, могут назначаться 1-2-3 раза в день.
Некоторые 5-адреноблокаторы являются одновременно водо - и
жирорастворимыми (конкор, пиндолол, целипролол и др.) и имеют два пути
элиминации - печеночный метаболизм и почечную экскрецию.
Существуют также 5-адреноблокаторы, которые разрушаются эстеразами
крови, имеют очень короткий период полувыведения - 9 минут (эсмолол,
бревиблок, флестолол) и очень малую продолжительность действия - около 30
минут.
Классификация В -адреноблокаторов.
I. Некардиоселективные (5ь 53) блокаторы.
А. Без внутренней симпатомиметической активности: пропранолол, надолол
(коргард), соталол, тимолол, нипрадилол, флестролол.
В. С внутренней симпатомиметической активностью: окспренолол (тразикор),
пиндолол (вискен), алпренолол (аптин), пенбуталол, бопиндолол, буциндолол,
дилевалол, каргеолол, лабеталол.
II. Кардиоселективные (преимущественно 5() блокаторы.
А. Без внутренней симпатомиметической активности: метопролол (спесикор),
атенолол (тенормин), бетаксолол, эсмолол, бисопролол, карведилол, небивалол.
В. С внутренней симпатомиметической активностью: ацебуталол (сектраль),
талинолол (корданум), целипролол, эпанолол.
III. Ь-Адреноблокаторы с вазодилатирующими свойствами.
А. Некардиоселективные (5] + 52) блокаторы: амозулалол, буциндолол,
лабеталол, дилевалол, нипрадилол, пиндолол.
В. Кардиоселективные (преимущественно в\) блокаторы: карведилол,
небиволол, целипролол.
IV. b-Адреноблокаторы длительного действия.
А. Некардиоселективные (5j + 52) блокаторы: бопиндолол, надолол, соталол.
В. Кардиоселективные (преимущественно 5|) блокаторы: конкор, бетаксолол
атенолол, эпанолол.
V. 5-Адреноблокаторы сверхкороткого действия: эсмолол
(кардиоселективный).
Побочные реакции при лечении 5-адреноблокаторами.
Побочные реакции со стороны сердечно — сосудистой системы.
5-адреноблокаторы могут вызывать выраженную брадикардию (в связи с
угнетением функции автоматизма синусового узла). Реже брадикардия развивается
100
й назначении 6-адренолокаторов с внутренней сиспатомиметической
Живностью (пиндолол, окспренолол, эпанолол). 6-Адреноблокаторы замедляют
атриовентрикулярную проводимость и могут вызвать атриовентрикулярную
блокаду различной степени. Они также снижают сократительную способность
^окарда (отрицательное инотропное, кардиодепрессивное действие). В
наименьшей степени ухудшают сократительную способность миокарда Ь-
адреноблокаторы с вазодилатирующими свойствами (буциндолол, карведилол,
пиндолол, целипролол, лабеталол и др.).
При лечении 6 -адреноблокаторами возможно также значительное снижение
дд, особенно при назначении больших доз и использовании препаратов
парентерально. Снижение АД следует рассматривать скорее не как побочное
действие, а как фармакологический эффект, свойство 6-адреноблокаторов, но
чрезмерное снижение АД, конечно, нежелательно. Необходимо обратить внимание
на то, что 6 -адреноблокатор с 6 -1 - ВСА эпанолол не снижает АД в покое.
6 -Адреноблокаторы могут привести к ухудшению периферического кровотока
в связи с вазоконстрикцией, вызывают похолодание конечностей, ухудшают
течение болезни Рейно. Сосудистый спазм в области кисти обусловлен блокадой
внесимпатических 6-адренорецепторов, регулирующих расширение
артериовенозных шунтов. Способность ухудшать кровоток менее всего выражена у
в-адрено блокаторов с вазодилатирующими свойствами (см. классификацию 6-
адренобл о катеров).
6-Адреноблокаторы могут также уменьшить почечный кровоток вследствие
артериальной вазоконстрикции.
Побочные реакции со стороны органов дыхания.
6-Адреноблокаторы вызывают резкое ухудшение бронхиальной
проходимости, так как блокада 62-адренорецепторов способствует спазмированию
бронхов. Это влияние менее выражено у кардиоселективных 6-адреноблокаторов,
избирательно блокирующих 61-адренорецепторы, однако оптимальные дозы этих
препаратов, вызывающие оптимальный антиангинальный эффект,могут оказаться
достаточно высокими и кардиоселективность утрачивается.
Влияние на углеводный обмен.
в -Адреноблокаторы могут снижать толерантность к глюкозе, подавляя
секрецию инсулина 6- клетками островков поджелудочной железы. Секреция
инсулина опосредуется 6 Ь2-адренорецепторами, применение 6-адреноблокаторов
неселективного действия может снизить продукцию инсулина. Кардиоселективные
bi-адреноблокаторы не влияют существенно на секрецию инсулина и поэтому при
сочетании ИБС, артериальной гипертензии и сахарного диабета более рационально
применять эти средства.
В то же время 6-адреноблокаторы тормозят мобилизацию глюкозы из печени
в ответ на гипогликемию, вызванную гликемизирующими средствами (инсулином,
производными сульфанилмочевины, бигуанидами), голоданием, физическими
Нагрузками. Мобилизация глюкозы из печени опосредуется 62-адренорецепторами,
поэтому блокада их 6-адреноблокаторами нарушает поступление глюкозы из
Печени в кровь. Имеет также значение снижение секреции глюкагона под влиянием
101
б-адреноблокаторов. В связи с указанным эффектом б-адреноблокаторы могут
способствовать затяжному течению гипогликемий у больных сахарным диабетом.
Необходимо отметить и другой аспект проблемы взаимоотношений
гипогликемических состояний и б-адреноблокаторов. Известно, что в отвез на
развитие гипогликемии происходит значительный выброс в кровь адреналина
который возбуждает а-адренорецепторы и вызывает значительное повышение Ад
При лечении неселективными б-адреноблокаторами вазодилатирующие Ь,-
адренорецепторы заблокированы и, таким образом, в ответ на выброс адреналина в
связи с гипогликемией может наблюдаться выраженный подъем АД.
Артериальная гипертензивная реакция в ответ на гипогликемию бывает
значительно реже и выражена меньше при применении кардиоселективных Ь-
адреноблокаторов.
В связи с вышеизложенным в случае необходимости назначения при
сахарном диабете б-адреноблокаторов предпочтение следует отдать
кардиоселективным -адреноблокаторам.
Синдром отмены в -адреноблокаторов.
Внезапная отмена б-адреноблокаторов после длительного их применения,
особенно в больших дозах, может вызвать тяжелые проявления синдрома отмены,
характеризующегося клиникой нестабильной стенокардии, желудочковой
тахикардии, инфаркта миокарда, а иногда и развитием внезапной смерти. Синдром
отмены начинает проявляться через несколько дней (реже через 2 недели) после
прекращения приема б-адреноблокаторов.
Предполагается следующий механизм развития синдрома отмены: при
длительном применении б-адреноблокаторов на клетках-мишенях образуется
дополнительное число /?-адренорецепторов (по принципу отрицательной обратной
связи) и внезапная отмена в-адреноблокаторов (внезапное прекращение блокады 8-
адренорецепторов) приводит к чрезмерной ^-адренергической стимуляции сердца,
других органов и тканей. Не исключается также рикошетное усиление некоторых
процессов, подавляемых во время лечения б-адреноблокаторами (потребление
кислорода миокардом, агрегация тромбоцитов, секреция ренина).
Во избежание развития тяжелых последствий синдрома отмены 8-
адреноблокаторов необходимо придерживаться следующих рекомендаций:
- отменять б-адреноблокаторы медленно, в течение 2 недель, по следующей
схеме: в 1-й день суточную дозу пропранолола уменьшают не более чем на 80 мг, на
5-й день - на 40 мг, на 9-й день - на 20 мг и па 13-й день - на 10 мг;
- больным ИБС во время и после отмены б-адреноблокаторов следует
ограничить физическую активность и при необходимости увеличить дозу нитратов.
Ограничения к назначению б-адреноблокаторов.
Абсолютные противопоказания к назначению б-адреноблокаторов:
- острая сердечная недостаточность (отек легких, кардиогенный шок);
- застойная сердечная недостаточность, обусловленная систолической
дисфункцией левого желудочка и рефрактерная к стандартной медикаментозной
терапии (диуретики, сердечные гликозиды, периферические вазодилататоры,
ингибиторы АПФ);
102
_ бронхиальная астма и тяжелая степень обструктивной дыхательной
недостаточности;
- синдром слабости синусового узла (если не имплантирован искусственный
водитель ритма);
- атриовентрикулярная блокада П~Ш степени (если не имплантирован
искусственный водитель ритма);
- артериальная гипотензия (систолическое АД 100 mm Hg и ниже);
- синусовая брадикардия с ЧСС менее 50 мин'1;
- лабильный инсулинзависимый сахарный диабет - такая форма сахарного
диабета, которая характеризуется частыми и быстрыми переходами от
гипергликемии и кетоацидоза, с одной стороны, к гипогликемии, с другой, несмотря
на постоянное количество углеводов в диете и постоянную дозу инсулина в течение
суток.
Из большого числа В-адренобтюкаторов 3 препарата считаются жизненно
важными: пропранолол (некардиоселективный), атенолол (кардиоселективный,
умеренно пролонгированного действия), надолол (некардиоселективный, с
выраженным пролонгированным действием).
На сегодня приоритетное требование к в-адреноблокаторам - это высокая
селективность по отношению к В\-адренорецепторам, блокада которых и составляет
основу терапевтического эффекта от их применения.
Конкор (Nycomed) (бисопролол) - это высокоселективный б'-адреноблокатор
с клинически доказанным преимуществом высокой .^-селективности, индекс
кардиоселективности которого (1:75) и выше, чем у бетаксолола (1:35), атенолола
(1:35), метопролола (1:20) и пропранолола (1,8:1).
С этим связано отсутствие побочного действия препарата на углеводный
обмен у пациентов с сахарным диабетом 2 типа. Кроме того, клинические
исследования показали, что в терапевтических дозах, конкор (бисопролол) не
оказывает выраженного влияния на б2-рецепторы бронхов и не влияет на
функциональные параметры легких у больных с бронхообструктивными
заболеваниями. Препарат не оказывает влияния на уровень липидных фракций
плазмы при длительном лечении. Конкор оказывает надежное антиангинальное
Действие в течение суток при однократном приеме, значительно снижает
количество, продолжительность и тяжесть приступов боли при стенокардии.
Особенно ценно в клинической практике наличие сбалансированного пути
выведения — 50% препарата выводится почками, 50% - печенью. Поэтому у
пациентов как с недостаточностью печени, так и с недостаточностью почек
Коррекция дозы не является необходимой. Что касается оптимальной дозы, то опыт
показал, что т действие бисопролола усиливается при повышении дозы от 2,5 до 10
Мг.
Гемодинамика под воздействием конкора изменяется следующим образом:
Уменьшается частота сердечных сокращений (в дозах 5-10 мг он уменьшает частоту
СеРДечных сокращений на 24-25%), снижается артериальное давление (как
систолическое, так и диастолическое). Длительное лечение селективными В-
^Реноблокаторами, как правило, не вызывает существенного снижения скорости
гломерулярной фильтрации и эффективного почечного кровотока. Необходимо
103
отметить, что при однократном введении конкор снижает систолическое и
диастолическое артериальное давление. Этот эффект является дозозависимым.
Систолические интервалы левого желудочка увеличиваются, но отношение периода
предизгнания ко времени изгнания (показатель функции левого желудочка) не
изменяется. При длительном лечении таких больных изменений со стороны
фракции изгнания не происходит, уменьшается выраженность гипертрофии левого
желудочка и улучшается его диастолическое наполнение.
Конкор (5-10 мг однократно в сутки) обеспечивает и длительное дозозависимое
снижение АД (Kirsten R, et al, 1986). Даже спустя 40 ч. после приема 10 мг Конкора
на 4-й неделе лечения при мониторировании АД наблюдалось значительное
снижение АД и ЧСС (Asmar R., 1987).
Плавное снижение АД на протяжении суток, в том числе в ранние утренние
часы: коэффициент конечный эффект/пиковый эффект для Конкора составляет 91,2,
что свидетельствует о выраженном и равномерном гипотензивном действии (Keim
HJ, 1988; Метелица В.И., 1995).
Конкор может применяться длительное время без снижения эффективности: в
исследовании Giesecke HG et al (1990) 102 больных с артериальной гипертензией
наблюдались в течение 3 лет. У 85% пациентов АД адекватно контролировалось
при приеме 5-10 мг Конкора.
Антигипертензивная эффективность Конкора не зависит от возраста: в
исследовании, проведенном Hoffler D et al (1990), участвовали 2012 пациентов.
После 8 недель лечения на терапию Конкором в дозе 5-10 мг “ответили” 94,9%
пациентов моложе 60 лет и 90,6% пациентов старше 60 лет.
Конкор вызывает регрессию гипертрофии миокарда (Gosse Р., 1990)
В лечении гипертонической болезни конкор (бисопролол) зарекомендовал себя
как эффективный и надежный препарат. Однократный его прием в дозе 5-10 мг
эффективно снижает артериальное давление в состоянии покоя и при нагрузке на
протяжении 24 часов. При монотерапии гипертонической болезни конкором
(бисопрололом) показана высокая эффективность лечения во всех возрастных
группах. У больных гипертонической болезнью терапия конкором приводит к
значительном}' снижению риска развития инсульта (29%) и застойной сердечной
недостаточности (42%).
Лечение хронической сердечной недостаточности, гипертонической
болезни и ИБС являются официальными показаниями назначения препарата
Конкор.
а-Адреноблокаторы.
at-Адреноблокаторы блокируют а,-адренорецепторы (постсинаптические) на
уровне периферических артериол, что снижает периферическое сопротивление и
вызывает гипотензивный эффект.
Для лечения артериальной гипертензии используются высокоселективные
постсинаптические ai-адреноблокаторы - празозин и препараты II поколения
доксазозин, теразозин, урапидил.
Постсинантические aj-адреноблокаторы второго поколения.
104
Постсинаптические а( -адреноблокаторы второго поколения обладают
продленным действием, лучше переносятся, для них менее характерен феномен
первой Дозы-
Урапидил - обладает двумя механизмами действия:
а) блокирует постсинаптичсские рецепторы;
б) стимулирует серотониновые рецепторы подтипа IA продолговатого мозга,
ч1о снижает активность симпатической нервной системы, усиливает гипотензивный
эффект и предупреждает появление тахикардии.
ДД снижается постепенно, максимальный гипотензивный эффект наступает к
началу второй недели лечения.
Препарат выпускается в капсулах продленного действия по 30; 60 и 90 мг и
ампулах для внутривенного введения по 25 и 50 мг.
Лечение начинают с дозы 30 мг 2 раза в день. В дальнейшем можно
постепенно повышать суточную дозу до 180 мг в 2 приема. Препарат можно
комбинировать с b-адреноблокаторами, салуретиками, антагонистами кальция.
агАгоншлпы центрального действия.
а?-Агонисты центрального действия стимулируют а2-адренорецепторы в
вазомоторном центре продолговатого мозга, что приводит к торможению
симпатической импульсации из головного мозга и снижению артериального
-давления. Стимуляторы а2-адренорецепторов центрального действия вызывают
обратное развитие гипертрофии левого желудочка.
Клонидин (клофелин, катапрессин, гемитон) — выпускается в таблетках по
0,075; 0,1; 0,15; 0,2 и 0,3 мг и в ампулах по 1 мл 0,01% раствора для
парентерального введения.
Клонидин — липофильное лекарственное вещество, хорошо проникающее
через гематоэнцефалический барьер. Клонидин связывается с постсинаптическими
Яг-адренорецепторами в вазомоторном центре продолговатого мозга и
гипоталамусе, стимулируя эти рецепторы, что вызывает уменьшение
периферического сосудистого тонуса. Наряду с этим клонидин уменьшает
высвобождение норадреналина и повышает тонус блуждающего нерва. Это
приводит к уменьшению сердечного выброса и частоты сердечных сокращений, что
способствует снижению АД. При длительном применении клонидина снижается
секреция ренина.
При приеме таблеток клонидина внутрь начало гипотензивного эффекта
отмечается через 30-60 мин., максимум действия - через 2-4 ч., продолжительность
Действия составляет 6-12 ч.
При внутривенном введении клонидина гипотензивный эффект проявляется
через 3-5 мин., максимум действия - через 15-20 мин., продолжительность действия
составляет 4-8 ч.
При внутримышечном введении клонидина начало гипотензивного эффекта
отмечается через 30-60 мин., его продолжительность составляет 2-5 ч.
При сублингвальном приеме препарата эффект проявляется через 15-30 мин.,
пР°Должительность гипотензивного дейсивия составлят 6ч.
При пероральном лечении клонидином артериальной гипотензии
Первоначальная доза составляет 0,075 — 0,1 мг 2 раза в день, затем каждые2-4 дня
105
суточную дозу увеличивают на 0,075 - 1 мг и доводят до 0,3-0,45 мг (в 2-3 приема)
После достижения гипотензивного эффекта дозу можно постепенно понижать д0
поддерживающей, которая обычно составляет 0,15-0,2 мг в сутки.
При тяжелой артериальной гипертензии в ургентных случаях возможно
начинать лечение с дозы 0,15-0,2 мг, а затем через каждый час принимать по 0.075»
0,1 мг до достижения гипотензивного эффекта, однако в течение суток можно
принимать не более 0,8 клонидина. В дальнейшем следует переходить на
поддерживающие дозы.
У пожилых больных доза клонидина должна быть уменьшена из-за
возрастного снижения функции почек. Обычно назначают 0,075 мг препарата в
день.
Для потенцирования гипотензивного эффекта клонидин можно
комбинировать с диуретиками.
При использовании клонидина возможны побочные эффекты:
- выраженная сухость во рту в связи с угнетением секреции слюнных желез
(одновременный прием витамина С уменьшает сухость во рту);
- сонливость, вялость, иногда депрессия;
- у мужчин среднего возраста возможно снижение половой функции;
- задержка натрия и воды вследствие повышения их реабсорбции в почках
(повышение объема внеклеточной жидкости приводит к развитию толерантности к
препарату и снижению гипотензивного эффекта; для уменьшения этого побочного
эффекта рекомендуется сочетание приема клонидина с салуретиками);
- запоры при длительном применении;
- нарушение толерантности к углеводам, развитие утренней гипергликемии
при длительном лечении клонидином;
- значительное повышение АД (вплоть до гипертонического криза) при резкой
отмене клонидина, что обусловлено увеличением выделения норадреналина из
нервных окончаний и снижением ингибирования пресинашических аз-
адренорецепторов. В связи с этим рекомендуется медленное снижение дозы
препарата и постепенная его отмена;
- при внутривенном введении клонидина наблюдается кратковременная фаза
повышения АД, продолжающаяся не более 5 мин., за счет стимуляции
периферических а-адренорецепторов; при пероральном приеме этой фазы нет;
- угнетение секреции желудочного сока;
- одновременный прием с клонидином алкоголя приводит к резкому падению
АД, утрате сознания и последующей амнезии;
- возможно снижение клубочковой фильтрации.
Ингибиторы АПФ.
Ингибиторы АПФ имеют следующие механизмы гипотензивного действия:
- торможение превращения циркулирующего ангиотензина I в мощный
вазоконстриктор ангиотензин И;
- уменьшение секреции альдостерона, что способствует натрийурезу;
с пецифическая вазодилатация почечных сосудов, способствующая
натрийкрезу;
[06
- уменьшение инактивации вазодилататора орадикинина;
- торможение ренин - ангиотензиновой системы в тканях и сосудистой стенке;
- увеличение образования депрессорных простагландинов благодаря прямой
Стимуляции фосфолипазы мембраны клеток;
_ торможение инактивации предсердного натрийуретического фактора,
обличения выделения в кровь этого гормона,
вазодилатирующее и натрийуретическое действие;
стимуляция выхода из эндотелиальных
оказывающего мощное
клеток азота оксида
(эндотелиального расслабляющего фактора), вызывающего вазодилатацию
снижение артериального давления;
- томожение активности вазоконстрикторной симпатоадреналовой системы;
и
- подавление гипертрофии гладкой мускулатуры артерий, гиперплазии и
дролиферации гладкомышечных клеток, что снижает периферическое
сопротивление и, следовательно, АД;
- снижение продукции антидиуретического гормона (вазопрессина).
Помимо гипотензивного действия, ингибиторы АПФ оказывают также
следующие положительные эффекты:
- уменьшают гипертрофию миокарда левого желудочка;
- значительно улучшают качество жизни;
- обладают кардиопротекторным действием (уменьшают вероятность
развития повторного инфаркта и риск внезапной смерти, увеличивают коронарный
кровоток, устраняют дисбаланс между потребностями миокарда в кислороде и его
Доставкой);
йй? “ уменьшают возбудимость миокарда, тахикардию и частоту экстрасистолии,
^^ обусловлено повышением содержания в крови калия и магния, уменьшением
Гипертрофии и гипоксии миокарда;
ййу' - благоприятно влияют на углеводный обмен, повышают усвоение глюкозы
Щетками в связи с тем, что увеличение содержания брадикинина под влиянием
ингибиторов АПФ повышает проницаемость клеточных мембран для глюкозы;
- проявляют калий сберегающий эффект;
5 - повышают содержание в крови липопротеинов высокой плотности.
Ингибиторы АПФ обладают также антиишемичсским действием.
" Ингибиторы АПФ подразделяются на группы в зависимости от особенностей
химического строения:
1. содержащие SH - группу (капотен, метиолрил, алацеприл, зофеноприл);
2. содержащие карбоксильную группу (эналаприл, лизиноприл, квинаприл,
Р®Миприл, беназеприл, псриндоприл, спираприл, цилазаприл);
3. содержащие фосфонильную группу (фозиноприл);
4. содержащие гидроксамовую группу (индаприл).
Различают также ингибиторы АПФ прямого действия (каптоприл,
ЛИзинопрпл) и пролекарства, т.е. фармакологически активные только после
^^Таболических превращений в печени (остальные ингибиторы АПФ).
Некоторые авторы предлагают подразделять ингибиторы АПФ следующим
Аразом:
107
1. препараты первого поколения (капотен, каптоприл, тензиомин, каприл),
средней продолжительности действия;
2. препараты второго поколения (остальные ингибиторы АПФ), продленного
действия.
Для лечения артериальной гипертензии наиболее часто применяют
следующие ингибиторы АПФ.
Каптоприл (капотен, тензиомин) - выпускается в таблетках по 12,5; 25; 50 и
100 мг, а также в виде фиксированных комплексных препаратов капозид - 25
(каптоприл и гидрохлортиазид по 25 мг) и капозид -50 (каптоприл и
гидрохлортиазид по 50 мг). Каптоприл быстро и хорошо всасывается (75%),
концентрация в крови достигает максимума уже через 1 час после приема
препарата. Каптоприл метаболизируется в печени, образуя дисульфидный и
цистеиндисульфидный димеры. Продолжительность гипотензивного действия -
около 4-6 ч. Гипотензивное действие каптоприла начинается через 60-90 мин. после
приема препарата и длится 12 ч. При необходимости суточную дозу каптоприла
можно повысить до 200-300 мг.
Эналаприл (энап, ренитек, вазотек, ксанеф) - выпускается в таблетках по 2,5;
5; 10 и 20 мг и ампулах для внутривенного введения (1,25 мг в 1 мл). Препарат
является «пролекарством», в печени превращается в активный метаболит
эналаприлат, который, собственно, и является ингибитором АПФ. Эналаприл -
длительно действующий препарат. Начальная доза - 5 мг внутрь 1 раз в сутки. При
необходимости можно постепенно повышать дозу до 20-40 мг/сутки в I -2 приема.
Поддерживающая доза - 10 мг в сутки. Максимальный гипотензивный эффект
отмечается через 4-6 ч. после приема, длительность действия - около суток.
Выделяется эналаприл преимущественно через почки, при эзом 70% выводимого с
мочой препарата является активным метаболитом. Препарат обладает
ренопротекторным действием даже при значительной почечной недостазочности.
Лизиноприл (привинил, сипоприл, корик) - лизиновый аналог эналаприла,
препарат длительного действия. Выпускается в таблетках по 5; 10; 20 и 40 мг.
Бысгроусвояемость лизиноприла составляет 25%. В организме он не
метаболизируется и не связывается с белками, выводится почками в неизмененном
виде, при нарушении функции почек может накапливаться в организме.
Гипотензивный эффект лизиноприла отмечается через 1 ч. после приема, достигает
максимума через 6ч. и сохраняется в течение 24 ч.
Ингибиторы АПФ обладают следующими побочными действиями:
- при длительном лечении возможно угнетение кроветворения (лейкопения,
анемия, тромбоцитопения); при лечении препаратами, не содержащими SH-rpynny,
нейтропения не наблюдается;
- вызывают аллергические реакции - зуд, покраснение кожи, крапивницу,
фотосенсибилизацию;
- со стороны органов пищеварения иногда наблюдается извращение вкуса,
тошнота, рвота, неприятные ощущения в эпигастральной области, понос или запор,
возможны появления афтозных высыпаний на слизистой оболочке полости рта,
нарушение функции печени;
108
_ возможно парадоксальное повышение АД у больных с односторонним
пезким стенозом или окклюзией почечной артерии вследствие значительного
снижения кровяного давления на уровне клубочков здоровой почки
/вазодилатирующий эффект препаратов на уровне как эфферентных, так и
афферентных артериол клубочков).
Притивопоказания к лечению ингибиторами АПФ:
- индивидуальная гиперчувствительность, в том числе при наличии в
анамнезе указаний на ангионевротические отеки;
- выраженный аортальный стеноз (опасность снижения перфузии коронарных
артерии с развитием ишемии миокарда);
- артериальная гипотензия;
- беременност ь (токсичность, развитие гипотензии у плода), лактация;
- стеноз почечной артерии.
7.4 Уровень гликемии
Увеличение уровня глюкозы в сыворотке может часто обнаруживаться при
поступлении больного в больницу, что обусловлено ранее имевшимся известным
или неизвестным диабетом.
Гипергликемия вредна при инсульте. Это справедливо не только для
пациентов с диабетом, у которых метаболические расстройства могут значительно
ухудшиться в острой фазе инсульта, но также и для пациентов без диабета.
Гипергликемия ухудшает неврологический исход ишемического инсульта,
поэтому необходимо регулярно контролировать уровень глюкозы крови каждые 2
часа [42,106].
Уровень глюкозы в крови 10 ммоль/л или выше оправдывает немедленное
использование инсулина. Если уровень глюкозы в крови не известен, то пациенту
С инсультом нельзя давать растворов, содержащих глюкозу. С другой стороны,
гипогликемия в ряде случаев может имитировать клинику острого ишемического
инсульта, поэтому её следует коррегировать внутривенным введением болюса 40%
глюкозы или инфузией 10-20 % глюкозы, предпочтительно через центральный
венозный доступ [49].
7.5 Температура тела
Лихорадка ассоциирована с плохими неврологическими исходами при
ишемическом инсульте. Она является маркером тяжести состояния и высокого
риска летальности при ишемическом инсульте [60,91].
Это, по-видимому связано с увеличением метаболических потребностей ткани
мозга, повышением активности нейротрансмиттеров и свободно радикальных
процессов. Напротив купирование острой гипертермии может улучшать прогноз
Пациентов с тяжелыми инсультами.
Лихорадка часто имеет место в течение первых 48 часов после начала
инсульта. С другой стороны, необходимо помнить, что инфекция является
Фактором риска инсульта, и что у многих пациентов инфекция развивается после
инсульта. Следовательно, рекомендуется поиск возможного источника инфекции
того, чтобы начать соответствующее лечение, хотя эмпирическое лечение
109
антибиотиками, антимикотическими и антивирусными препаратами це
рекомендуется у иммунокомпетентных пациентов.
Лечить лихорадку, необходимо, при температуре тела > 37,5°С, с
применением антипиретиков и охлаждения тела [32,49].
7.6 Водно-электролитный баланс и осмолярность
Большинство пациентов необходимо вести с положительным водным
балансом, при наличии признаков отека мозга - со слегка отрицательным.
Электролитный дисбаланс может имитировать картину церебральной ишемии,
например при церебральном понтинном миелите и гипонатриемии. Коррекция
гипонатриемии, требуется, когда уровень Na+ плазмы снижается ниже 120-125
ммоль/л, поскольку этот пограничный уровень грозит развитием гипоосмолярной
комы.
Гипернатриемия сопровождается повышением осмолярности плазмы и ростом
гематокрита.
Превышение уровня осмолярности более 320 мос.моль/л, является
пргностически неблагоприятным фактором при ишемическом инсульте.
Необходимо контролировать уровень К+ поскольку гиперкалиемия может
приводить к остановке кровообращения, а гипокалиемия вызывать нарушения
сократительной функции мышц и вызывать паралич скелетной мускулатуры,
развитие сердечных аритмий и кишечной непроходимости.
Особенно важно контролировать электролиты при проведении дегидратации с
использованием диуретиков [3].
ГЛАВА 8
ИНТЕНСИВНАЯ ТЕРАПИЯ ИШЕМИЧЕСКОГО ИНСУЛЬТА
Б. Поддержание интрацеребрального гомеостаза
8.1 Тромболитическая терапия
Тромболитическая терапия рассматривается в качестве наиболее эффективного
направления в лечении ишемического инсульта, если она осуществляется в первые
3 часа и геморрагический компонент поражения исключен. В настоящее время
среди всех тромболитических агентов, рекомендуется только рекомбинантный
тканевой активатор плазминогена (rt-PA) - актилизе []18,23,105]. Препарат
обеспечивает реканализацию артериальной окклюзии, путем лизиса тромба
(эмбола), за счет превращения плазминогена в активный плазмин, который в свою
очередь разрушает фибрин образующий тромб. Эффективность при ишемическом
инсульте других громболитиков, таких как ретиплаза, урокиназа, анистреплаза и
стафилокиназа не изучена. Применение стрептокиназы при ишемическом инсульте,
согласно данным проведенных мультицентровых клинических испытаний, не
допустимо, поскольку ассоциировано с плохими неврологическими исходами и
возрастанием летальности [62,84].
rt-PA (актилизе) вводится в первые 3 часа после развития острого
ишемического инсульта, внутривенно в дозировке 0,9 мг/кг массы тела
ПО
^максимальная доза 90 мг), 10% дозы - болюсно, с последующей инфузией в
^цение 60 минут, с предварительным строгим отбором пациентов с учетом
глютивопоказаний. Пациентам, имеющим >22 баллов по шкале NIHSS проведение
омболизиса противопоказано, поскольку согласно данным мультицентровых
Следований rt-PA, было показано, что проведение тромболизиса у пациентов
реющих -20 баллов по шкале NIHSS ассоциирована с 17% случаев
усМОрРягических осложнений, а риск кровоизлияний среди пациентов имеющих <10
баллов составил только 3 % [101 ].
Проведение тромболизиса, это всегда очень серьезный шаг сделанный врачом,
поэтому необходимо тщательно взвесить риск и потенциальную пользу данного
метода у каждого конкретного пациента, исходя из высокого уронвя возможных
геморрагических осложнений.
Основные требования к проведению тромболизиса могут быть
^^формулированы в трех пунктах:
\\ 1. Точная постановка диагноза ишемического инсульта (поскольку ошибочное
Проведение тромболизиса пациенту с геморрагическим инсультом неизбежно
закончится фатально);
2. Проведение врачом имеющим опыт применения тромболитиков;
v3. Предварительный строгий отбор пациентов с учетом противопоказаний.
ASA’2003 были предложены критерии пациента, которому возможно
^ведение фомболизиса [32]:
* Диагноз ишемического инсульта с умеренной неврологической
симптоматикой;
Неврологическая симптоматика не должна быть незначительной или
изолированной;
Неврологическая симптоматика не должна спонтанно регрессировать;
я Неврологический дефицит не должен быть большим;
Должно быть исключено субарахноидальное кровоизлияние;
Начало заболевания должно составлять менее 3 часов до момента
планируемого проведение тромболизиса;
Отсутствие в анамнезе фавмы головы или инсульта, в предшествующие 3
месяца;
Отсутствие в анамнезе перенесенного инфаркта миокарда, в
предшествующие 3 месяца;
Отсутствие кровотечений из желудочно-кишечного тракта и
Мочеиспускательной системы в предидущий 21 день;
Отсутствие в анамнезе проведения больших хирургических вмешательств, в
предидущие 14 день;
• Отсутствие пункций артериальных сосудов в местах недоступных для
прижатия, в предшествующие 7 дней;
Отсутствие в анамнезе внутричерепных кровоизлияний;
Отсутствие повышения АД. АД в диапозоне: АДсист. <185 mm Hg, АДдиаст.
< И0 mm Hg.
ill
На момент обследования отсутствует ативное кровотечение или острая
травма (перелом);
Отсутствие приема антикоагулянтов;
Отсутствие проведения гепаринотерапии в предшествующие 48 часов;
Содержание тромбоцитов >100 000 мм3;
Уровень гликемии >2,7 ммоль/л;
Отсутствие по данным компьютерной томографии мультилобарного
инфаркта (очаг низкой плотности > 1/3 гемисферы мозга)и Возраст менее 80
лет;
Пациент или члены его семьи информированы и понимают потенциальный
риск и пользу от проводимого лечения.
Разработан следующий протокол проведения тромболизиса с использованием
rt-PA [32]:
1) Инфузия 0,9 мг/кг (максимально 90 мг) в течении 60 мин., при этом 10% всей
дозы вводится болюсно в течении 1 мин.
2) Перевод пациента в ОРИТ для осуществления мониторинга витальных и
неврологических функций;
3) Оценка неврологического статуса каждые 15 мин. на протяжении инфузии rt-
PA и каждые 30 мин. в последующие 6 часов, затем каждый час в пределах 24
часов после начала тромболизиса.
4) В случае если, у пациента развивается интенсивная головная боль, острая
гипертензия, тошнота или рвота - прекращают инфузию тромболитика и в
ургентном порядке проводят компьютерную томограмму головного мозга;
5) АД измеряют каждые 15 мин. в течении первых 2 часов, каждые 30 мин.
последующие 6 часов, затем каждый час в пределах 24 часов с момента начала
тромболизиса;
6) АДсист. >180 mm Hg или АДдиаст. >105 mm Hg удерживается на этом или
более низком уровне при помощи антигипертензивных препапратов;
7) При АДдиаст. 105-120 mm Hg или АДсист. >230 mm Hg, внутривенно вводят
10 мг лабеталола в течении 1-2 мин. Можно повторить введение той же (10 мг)
или удвоенной дозы (20 мг) каждые 10-20 мин. до максимальной дозы 300 мг.
Альтернативно можно ввести болюсно стартовую дозу лабеталола, с
последующей равномерной инфузией со скоростью 2-8 мг/мин.
Если применение лабетолла не позволит нормализовать АД, тогда переходят к
введению натрия нитропруссида.
8) При АДциас. >140 mm Hg, то сразу начинают инфузию натрия
нитропруссида со скоростью 0,5 мг/кг/мин;
9) Не нужно спешить с постановкой назогастрального, мочевого или
артериального катеров впервые часы после проведения тромболизиса;
10) Не допустимо применение антикоагулянтов и антитромбопитарных
препаратов, в течении 24 часов после проведения трмболизиса.
Необходимо отметить, что проведение тромболизиса максимально эффективно
в первые 90 мин. и сохраняет свою эффективность в течении 180 мин. Согласно
данным Brott [49] тромболизис действует по крайней мере до 4,5 часов, а
112
потенциально до 6 часов после начала инсульта. Однако проведение тромболизиса
более чем через 3 часа после развития инсульта не рекомендуется.
Методами ангиографии было продемонстрировано, что при внутривенном
введении rt-PA темп реканализации составляет 30%. Вероятность реканализации
снижается при более проксимальной локализации окклюзии в артериальной
циркуляции [105].
Согласно данным Т. Tomsik et al. [102] внутривенное введение rt-PA не
является эффективным у больных имеющих >10 баллов по шкале NIHSS и
окклюзией крупных сосудов средней мозговой артерии. В этих случаях
эффективной альтернативой может стать методика внутриартериального
тромболизиса. Сущность метода заключается в подведении при помощи катетера,
под ангиографическим контролем тромболитика непосредственно к месту
оккулюзии [105].
Однако сложность выполнения данной методики и недостаточная изученность,
ограничивает ее широкое применение. Также ограниченными клиническими
исследованиями, изучалась эффективность комбинированного внутривенно-
внтриартериального тромболизиса, в результате которых было выявлено, что
данная методика позволяет достич более эффективной реканализации.
В целом необходимо подчеркнуть, ято методы внутриартериального и
комбинированного тромболизиса, требуют дальнейшего изучения и в настоящее
время не являются достаточно обоснованными, с точки зрения безопасной и
эффективной альтерантивы внутривенному тромболизису rt-PA, который является
рекомендованным стандартом методом доказавшим свою высокую эффективность
и относительную безопасность, с учетом всех вышеизложенных положений по его
проведению.
8.2 Антиагрегантная терапия
Аспирин
Результаты двух очень крупных рандомизированных, мультиценгровых
исследований показали, что аспирин, использованный в течение первых 48 часов
после развития ишемического инсульта, снижает смертность и частоту повторного
инсульта минимально, но статистически значимо [32,47]. Причем необходимо
отметить, что использование аспирина более безопасно, поскольку имеется более
низкий риск развития геморрагической трансформации ишемического инсульта в
геморрагический, по сравнению с использованием как пефракционированных, так и
низкомолекулярных гепаринов.
Рекомендуется применение аспирина per os в дозе 100-300 мг в первые 24-48
часов с момента развития инсульта. Этот доступный и одновременно эффективный
метод терапии, должен обязательно быть использован и рассматриваться в качестве
минимального требования к оказанию неотложной помощи при ишемическом
инсульте. Одним из препаратов ацетилсалициловой кислоты является
кардиомагнил (NYCOMED) содержащий в одной таблетки 75 мг
ацетилсалициловой кислоты и 15,2 мг гидроксида магния, который защищает
слизистую оболочку ЖКТ от вредного воздействия ацетилсалициловой кислоты.
113
Аспирин не должен применяться в течении 24 часов после проведения
тромболизиса [49,55].
В качестве альтернативы возможно использование дипиридамола
(курантила), при противопоказаниях к применению аспирина или
неэффективности. Показано, что применение аспирина в комплексе с
дипиридамолом (200 мг/сут.) более эффективно, чем применение одного только
аспирина в профилактике повторного инсульта.
Пентоксифиллин (трентал) - в/в капельно в дозе 2-3 мг/кг/сут. Улучшает
реологию крови путем угнетения агрегации тромбоцитов и улучшения
деформируемости эритроцитов, при отсутствии вазодилатирующего эффекта (не
вызывая синдрома «обкрадывания») и активации метаболических процессов в
нейронах [12,18,23].
8.3 Антикоагулянтная терапия
Ранняя антикоагулянтная терапия с помощью нефракционированных гепаринов
(НФГ) или низкомолекулярных гепаринов (НМГ) часто применялась для лечения
острого ишемического инсульта. Но, к сожалению, ни одно из мультицентровых
клинических испытаний ранней антикоагулянтной терапии, выполненных в
последние годы, не выявило эффективности НФГ и НМГ при лечении
ишемического инсульта. Показано, что применение НФГ или НМГ несколько
улучшало исход, а также частоту повторных инсультов и риск развития венозной
тромбоэмболии, однако этот эффект всегда уравновешивалося увеличением числа
геморрагических осложнений.
Согласно современным международным рекомендациям, гепарин не должен
рутинно использоваться у пациентов с острым ишемическим инсультом
[32,47,49,55].
Гепарин может быть использован, если есть показания, такие как риск
тромбоза глубоких вен, инсульт вследствие кардиогенной эмболии с высоким
риском развития повторной эмболии (искусственные клапаны сердца, мерцательная
аритмия, инфаркт миокарда с интрамуральными тромбами, тромбоз левого
предсердия), тромбоз венозных синусов.
Рекомендуется профилактическое использование умеренных доз НФГ — 5000
ME подкожно 2 раза/сутки, или НМГ:
фраксипарин 0,3 мл подкожно 2 раза/сутки
или
эноксапарин (клексан) 0,2 мл 1 раз/сутки.
Противопоказания для лечения гепарином включают обширные инфаркты
(более 50% бассейна СМ А), неконтролируемая артериальная гипертензия. Гепарин
не должен применяться в течение 24 часов после проведения тромболизиса
[12,18,49].
114
8.4 Нейропротекторная терапия
Целью нейропротекторной терапии является удтинение периода
«терапевтического окна», расширение возможности для тромболитической терапии,
уменьшение размеров инфаркта мозга и защита от реперфузионного повреждения.
Необходимо отметить, что поддержание нормального или высокого уровня
ЦПД и обеспечение хирургической декомпрессии, являются наиболее
значительными и эффективными методами нейропротекции. Концепция
фармакологической нейропротекции включает методы увеличения МК в
ишемической зоне, снижения церебрального метаболизма и внутричерепного
давления, ингибирования аккумуляции лактата и эксайтотоксичности,
предотвращения проникновения Са , ингиоирования перикисного окисления
липидов и генерации свободных радикалов [43].
Первичная нейропротекция - направлена на прерывание быстрых реакций
глутамат-кальциевого «каскада», механизмов свободно-радикального окисления.
Она должна быть начата с первых минут ишемии и продолжаться на протяжении
первых 3 дней инсульта, особенно активно в первые 12 часов.
Вторичная нейропротекция - направлена на уменьшение выраженности
отдаленных последствий ишемии: блокаду провоспалительных цитокинов и
ферментов, молекул клеточной адгезии, апоптоза и энергокоррекцию. Она может
быть начата спустя 3-6 часов после развития инсульта и должна продолжаться по
меньшей мере 7 дней [9,107].
Практически для каждого этапа ишемического каскада был разработан
нейропротекторный препарат (табл. 13). Однако в настоящее время ни одно
мультицентровое клиническое испытание препаратов для нейропротекторной
терапии при инсульте, не продемонстрировало эффективности их применения,
поэтому международные рекомендации по лечению пациентов
нейропротекторными препаратами отсутствуют [32,43,49].
Так результаты клинических испытаний таких препаратов для первичной
нейропротекции как блокаторы Са24 (нимодипин, флунаризин, исрадипин,
цереброкраст), антагонистов глутаматных рецепторов (гавестинел, селфотел,
церестат (антиганель)\, дизолципин, десгрофан, декстрометорфан), ингибиторов
синтеза пресинаптического высвобождения глутамамата (лубелузол, фенитоин,
пропентофенилин, фос-фентоин), агонистов ГАМК (клометиазол), а также
препаратов для вторичной нейропротекции: ганглизидов, ловушек свободных
радикалов (тирилазад), нейротрофических факторов (bFGF - основной фактор роста
фибробластов), моноклональные антитела (энлимомат) оказались отрицательными
и были связаны как с их неэффективностью, так и с наличием грубых побочных
эффектов [10,32].
115
Таблица 13
Нейропротекторные препараты и их предполагаемый механизм действия
(по Виничук С.М., Черенько Т.М., 2003)
1. Средства первичной нейропротекции
Модуляторные участки NMDA- рецепторов Церестат, Декстрометорфан, Декстрорфан, Ремацемид, Дизолципин, Сульфат магния, Селфотел, AR-R15896AR
Антагонисты АМРА-рецепторов NBOX, ZK200775
Агонисты ГАМК-рецепторов Клометиазол, Мусцимол, Глицин
Агонисты 5-НТ1 А-рецепторов Урапидил, 8-OH-DPAT, Репинотан
2. Средства вторичной нейропротекции
Антиоксиданты, защита митохондрий Витамин Е, Эмоксипин, Лубелузол, Аминогуанидины, Эбселен, Тирилазад
Препараты-“ловушки” свободных радикалов Тирилазад мецилат, Солкосерил
Нейротрофические средства Актовегин, Солкосерил, Пирацетам, ! Церебролизин, Флогэнзим, Цитиколин, Семаке
Ангиопротекторы Актовегин, Солкосерил,Пирацетам, Сермион
Эта ситуация очень напоминает неудачу клинических испытаний
антицитокиновой терапии при сепсисе, первой половины 1990-х гг. [31]. В качестве
основных причин отрицательных результатов клинических испытаний препаратов
для нейропротекции выделяют: гетерогенность групп исследования, проблемы
статобработки полученных результатов и несовершенство дизайна самих
испытаний [50,59,80].
Препараты для первичной нейропротекции
Одним из нейропротекторных препаратов, к которому в последнее время
значительно возрос интерес, является сульфат магния. Сульфат магния ослабляет
развитие начальных звеньев поражающего нейроны глутаматного каскада, угнетая
высвобождение медиатора - глутамата из пресинаптических везикул. Кроме того,
сульфат магния вызывает блокаду потенциал-зависимого ионного канала NMDA-
рецепторов и в больших концентрациях действует как их неконкурентный
антагонист. В головном мозге Mg2+ превалирует в комплексе с АТР и является
важным кофактором в клеточном энергетическом метаболизме и белковом синтезе.
116
Уровень Mg2" регулируется активным транспортом через ГЭБ, при этом
концентрация его в ликворе выше чем в плазме крови (1,1 ммоль/л в сравнении с 0,8
ммоль/л соответственно). Концентрация Mg2+ в ликворе повышается на 20-25%
после ведения (в/в или в/м) обычных терапевтических доз сульфата магния,
достигая максимума концентрации через 4 часа псоле введения. При этом
концентрация Mg2+ селективно увеличивается в очаге патологии мозга у животных с
экспериментальной фокальной ишемией.
Магний выступает в качестве антагониста кальция в биологических системах
[81]. В целом ряде экспериментов на животных с моделированием фокальной
церебральной ишемии, применение сульфата магния значительно уменьшало
размеры инфаркта мозга.
Проведенное в 1995 г. клиническое испытание сульфата магния, у пациентов с
ишемическим инсультом, в дозе 8 ммоль внутривенно болюсно в 50 мл
физиологического раствора, с последующим введением 65 ммоль на 100 мл
физиологического раствора, в ивде равномерной капельной инфузии в течение 24
часов. В результатате было продемонстрирована безопасность и достоверное
снижение на 10% летальности или инвалидизации (Индекс Бартела<60) в сравнеии
с контрольной труппой [82]. В последующем было проведено клиническое
испытание позволившее определить оптимальную дозу вводимого сульфата магния.
Так было показано, что наиболее эффективной и безопастной является схема:
вначале введение 16 ммоль сульфата магния в 100 мл физиологического раствора в
течение 15 мин., с последующей равномерной инфузией 65 ммоль сульфата магния
в 100 мл физиологического раствора в течение 24 часов [83]. Однако дальнейшие
мультицентровые клинические испытания должны подтвердить
эффективностьсульфата магния в качестве нейропротекторного препарата и
установить оптимальную дозировку и длительность введения.
Более выраженный и быстрый эффект наблюдался в тех случаях, когда
применение сульфата магния начиналось в первые 3-6 часов от начала развития
мозгового инсульта. В целом суточная доза для взрослого составляет около 20 мл
25% раствора на физиологическом растворе, медленно капельно в режиме
равномерной инфузии. Во время введения магния сульфата необходим мониторинг
АД [25].
Глицин (аминоуксусная кислота) - нейромедиатор, он облегчает функцию
глутаматергических синапсов и вызывает все присущие ноотропам эффекты, но в
высоких дозах депрессирует глутаматергическую передачу и таким образом
блокирует эксайтотоксичность. Было показано, что ГАМК и глицин являются
равноценными тормозными нейротрансмитерами, обеспечивающими защитное
торможение в ЦНС, роль которого возрастает в условиях повышенного выброса
глутамата. Представляя собой, естественный метаболит мозга, глицин не проявляет
токсичность, даже в дозе более 10 г/сут. Единственным побочным эффектом может
считаться легкая седация.
Выявлено, что назначение глицина в первые 6 часов снижает показатели 30-
Дневной летальности и улучшает восстановление неврологических функций.
Отмечается преобладание эффективности глицина при тяжелом инсульте, вне
зависимости от его локализации и варианта развития.
117
Препарат назначают в первые 6 часов от момента инсульта сублингвально в
дозах 1-2 г/сутки в течение 5 дней [9].
Препараты для вторичной нейропротекции
Глиатилин - соединение, содержащее 40% холина и превращающееся в
организме в метаболически активную форму фосфорилхолин, способный проникать
через гематоэнцефалический барьер и активировать биосинтез ацетилхолина в
пресинаптических мембранах холинергических нейронов. Он предупреждает
развитие деменции, облегчает процессы обучения и запоминания за счет
увеличения синтеза и высвобождения ацетилхолина в мозговых структурах. При
внутривенном применении глиатилина в острый период тяжелого ишемического
инсульта в дозе 1г 3-4 раза в сутки в течение 5 дней был выявлен его
«пробуждающий» эффект [6].
Пирацетам (ноотропил) - структурной основой пирацетама, является
циклическая форма ГАМК (2-пирролидон), по при этом препарат не является
источником метаболически активной ГАМК. Пирацетам проходит через ГЭБ
достигая максимума концентрации в ликворе через 3 часа, а период его
полувыведепия составляет 7,5 часов.
Препарат обеспечивает улучшение окислительного метаболизма в условиях
ишемии, а также других звеньев энергетического обмена в головном мозге,
улучшает’ реологию мозгового кровотока, уменьшает выраженностьспазма
мозговых артерий нейрогенного происхождения.
Пирацетам был первым нсйрометаболическим препаратом в Советском Союзе
разрешенным к применению в клинике при остром ишемическом инсульте.
Начиная с 1970-х гг. пирацетам использовали в дозировке от 1,4 до 4 г/сут. Однако
данные дозировки не оказывали существенного влияния на исход и восстановление
неврологических функций после ишемического инсульта. Эффект пирацетама
проявляся только в случае кортикальной локализации очага инфаркта с
превалированием нарушений высших психических функций и астенического
синдрома.
Поэтому было предложено проведение клинических испытаний с
использованием высоких доз пирацетама. Пирацетам вводят болюсом или
инфузионно в дозах до 12 г/сут в 2 приема в/в или в/м. Курс парентерального
лечения 7-10 дней, после которого пирацетам можно назначать per os из расчета
130-150 мг/кг/сут в 3-4 приема. Однако проведенные рандомизированные плацебо -
контролируемые исследования (в дозе 12 г в/в болюсно при поступлении, затем в/в
капельно в течение 4 недель по 12 г/сут. и в последующие 8 недель - по 4,8 г/суг per
os) не выявили влияния пирацетама на летальность пациентов с ишемическим
инсультом. Было только показано, что в группе пациентов, лечение которых было
начато не позже 7 часов с момента развития инсульта, отмечалась тенденция к
улучшению неврологического статуса [10,23,48].
Семаке - нейропептид, аналог адренокортикотропного гормона (АКТГ 4-10).
Препарат обеспечивает нейропротекторный эффект за счет торможения механизмов
отсроченной смерти нейронов: цитокинового дисбаланса, воспаления, синтеза NO,
оксидантного стресса и трофической дисфункции. У препарата отсутствует
118
гормональная активность и он обладает в 24 раза более пролонгированным
действием нежели у природного аналога, через 4 минуты, после интраназального
введения семакс проникает через ГЭБ. Терапевтический эффект при однократном
введении продолжается около 20-24 часов, что обусловлено его последовательной
деградацией при поступлении в мозг, когда большая часть эффектов нейропептида
сохраняется у его фрагментов.
Лечение семаксом наиболее эффективно при каротидном ишемическом
инсульте, хотя положительный эффект проявляется и при вертебрально-
базиллярной локализации очага поражения. В суточной дозе 12-18 мг препарат
снижает 30-дневную летальность и улучшает степень неврологического
восстановления, что коррелирует с ранним назначением препарата, в пределах 6
часов.
Препарат назначают в первые 6 часов развития инсульта интраназально 6 мг 2
раза/сут. при средней тяжести течения и 9 мг 2 раза/сут. при тяжелом течении на
протяжении 5 дней [8].
Цито-Мак или цитохром-С - вводится в дозе 0,25-0,5 мг/кг в сутки [23].
Токоферола ацетат по 2 мл 5% раствора I -2 раза в сутки в/в или 2 капсулы 3
раза в сутки. Проведенные исследования не выявили нейропротекторного действия,
этого традиционного препарата, а была только отмечена тенденция к более
благоприятному течению заболевания [10];
Реамберин 4,5% раствор для инфузии - препарат янтарной кислоты.
Антиоксидантное действие связано с ее влиянием на транспорт медиаторных
аминокислот, а также увеличением в мозге ГАМК, за счет шунта Робертса.
Янтарная кислота нормализует содержание гистамина и серотонина, прежде всего в
мозге, не влияя существенно на АД и показатели работы сердца. Восстанавливает
активность ключевого фермента митохондриальной цепи - цитохромоксидазы.
Реамберин вводят только внутривенно капельно со скоростью не более 90
кап/мин (4-4,5 мл/мин) до 800 мл/сут. Курс 3-5 дней.
Цитофлавин - метаболический антигипоксант, представляет собой
комбинированный препарат из двух метаболитов (янтарной кислоты и рибоксина) и
двух коферментов-витаминов (витамина В2 и никотинамида - РР). Вводят
внутривенно капельно по 10 мл 2 раза/сутки, курсом 10 суток. Длительность
инфузии - 60 минут [23].
Эмоксипин - структурный аналог витамина В6 - проявляет антирадикальное и
активирующее систему антиоксидантной защиты действие, улучшает
микроциркуляцию в головном мозге, за счет ингибирования фибринообразования и
торможения агрегации тромбоцитов. Назначают 1 % раствор в дозе 15 мл в/в
капельно 10 дней, затем 5мл в/м 14 дней [10];
Мексмдол - соль эмоксипина и янтарной кислоты. Препарат тормозит ПОЛ и
свободнорадикальные процессы, активирует эндогенную антиоксидантную
систему, а также проявляет антигипоксический эффект за счет прямого окисления
сукцината, входящего в его состав с усилением эндогенного дыхания
сопровождающегося восстановлением флавинопротеидов. Улучшения
реологических свойств крови и ингибирования агрегации тромбоцитов. Вводят от
100 до 1000 мг/сутки в/в капельно [10,18].
119
Энцефабол (пиридитол) - удвоенная молекула витамина В6. Активирует
клеточный метаболизм за счет одновременного усиления мозгового кровотока и
интенсификации проникновения глюкозы через ГЭБ. Повышает метаболизм
нуклеиновых кислот и высвобождение ацетилхолина в синапсах нервных клеток,
улучшая тем самым холинергическую передачу между клетками нервной ткани.
Пиритинол способствует стабилизации структуры клеточной мембран нервных
клеток и их функций с помощью ингибирования ферментов лизосом, предотвращая
этим образование свободных радикалов. Пиритинол улучшает реологические
свойства крови, повышает пластичность эритроцитов с помощью увеличения
содержания АТФ в их мембране, что приводит к снижению вязкости крови и
улучшению кровотока. Купирует нейротоксическое действие лактатацидоза.
Назначают per os по 2-3 чайные ложки 3-4 раза/сут. суспензированной формы, либо
по 2 таблеткм 3 раза/сут. (600 мг.)
Актовегин (Nycomed) — является одним из высокоэффективных
антиоксидантов, способствующий активации церебральных и мультиорганных
реституционных механизмов. Актовегин представляет собой высокоочищенный
гемодиализат, получаемый методом ультрафильтрации крови телят. Помимо
неорганических электролитов и др. микроэлементов он содержит 30%
органических веществ, таких как олигопептиды, аминокислоты, нуклеозиды,
промежуточные продукты углеводного и жирового обмена, липиды и
олигосахариды. Молекулярный вес органических соединений составляет менее
5000 D. Исследование структурного анализа раствора актовегина выявило в нем
помимо углерода, кислорода, водорода, азота, хлора еще 6 макроэлементов, таких
как натрий, фосфор, кальций, магний и др. Магний входит в состав на правах
компонента нейропептидных фрагментов и ферментов, в качестве каталитического
центра. Это представляет особый интерес, т.к. магний является каталитическим
центром всех известных на сегодняшний день нейропептидов головного мозга и
имеет статус нейроседативного иона. Актовегин обладает выраженным
антигипоксическим действием за счет активации антиоксидантной ферментной
системы, в т.ч. таких ферментов, как пируватдегидрогеназа, активная фракция
актовегина повышает транспорт 3-0-метилглюкозы в зависимости от дозы в пять
раз. Под его влиянием значительно улучшается диффузия и утилизация кислорода
как в нейрональных структурах, так и в клетках организма в целом, прежде всего в
альвеолярной системе легких. Это значимо улучшает оксигенацию во всей
микроциркуляторной системе, в коже, слизистых ЖКТ, что позволяет уменьшить
выраженность вторичных трофических расстройств и представляется особенно
важным у пациентов с обширными ишемическими и диффузионными
гипоксическими повреждениями мозга. Под действием актовегина значительно
повышается обмен высокоэнергетических фосфатов, прежде всего АТФ,
активируются ферменты окислительного фосфорилирования, такие как
сукцинатдегидрогенеза, цитохром-С—оксидаза, ускоряется синтез углеводов и
белков, ускоряется процесс распада продуктов анаэробного гликолиза, прежде всего
лактата и гидроксибутирата. Под действием актовегина значительно повышается
устойчивость церебральных структур к гипоксии, уменьшается выраженность
диффузных постишемических повреждений.
120
Применяют в дозировке 1000-2000 мг/сут. (10-20 % раствор Актовегина по
250мл.).
Инстенон (Nycomed)
Инстенон состоит из трех компонентов: гексобендина, этамивана и
этофиллина.
Гексобендин активирует и увеличивает транспорт и потребление глюкозы и
кислорода клетками мозга за счет активации анаэробного гликолиза. Этот процесс
наиболее эффективен при ишемии и гипоксии, когда нарушается цикл аэробного
окислительного фосфорилирования.
Стимуляция анаэробного окисления дает энергетический субстрат для синтеза
и обмена медиаторов нейротрансмиттеров, а также для восстановления
синаптической передачи. Депрессия синаптической передачи, наряду с деструкцией
мембран нейронов, является ведущим патогенетическим механизмом расстройств
сознания и причиной возникновения неврологического дефицита при гипоксии и
ишемии головного мозга.
Гексобендин стимулирует нейрональный метаболизм, стабилизируя
физиологические механизмы ауторегуляции церебрального и кардиального
кровотока. Поддержание адекватной регуляции церебрального кровообращения и
блокирование вазоконстрикции осуществляются местным воздействием лактата и
пирувата (продуктами активизации цикла анаэробного окисления глюкозы) на
рецепторный аппарат интракраниальных артериол и капилляров.
Этамиван оказывает выраженное активирующее действие на лимбико-
ретикулярный комплекс. Нарушение функционального состояния
лимбикоретику л яркого комплекса (активирующей ретикулярной формации ствола
головного мозга) возникают, как правило, при различных патологических процессах
- гипоксии, ишемии, травме, интоксикации. Активация ретикулярной формации
ствола головного мозга - это основной пусковой механизм сохранения адекватного
функционирования нейронных комплексов коры и подкорково-стволовых структур.
Обычно именно этот эффект обеспечивает выход больного из тяжелого состояния
на более высокий уровень сознания при ишемическом инсульте, постгипоксической
энцефалопатии, травме мозга, интоксикации. При этом наблюдается быстрый
регресс неврологической дефицитарной симптоматики и активация вегетативной
сферы.
Этофиллин активирует метаболизм миокарда с увеличением минутного
объема сердца. Это способствует росту перфузионного давления в сосудах краевой
зоны ишемии, при чем системное артериальное давление существенно не меняется.
Активирующее действие на нервную систему проявляется также в стимуляции
Подкорковых образований, структур среднего мозга и оральных отделов ствола
(дыхательного и сосудодвигательного центров, центра вегетативной регуляции), а
также ядер черепно-мозговых нервов, прежде всего, блуждающего. Благодаря
Многокомпонентному воздействию на центральную нервную систему, Инстенон
способен улучшать функциональное состояние головного мозга тремя различными
путями, так как три компонента Инстенона действуют на различные звенья
Патогенеза ишемии и гипоксии головного мозга;
121
- гексобендин повышает энергетический статус нейрона за счет активации
утилизации глюкозы и кислорода.
- этамиван улучшает интегративную деятельность г оловного мозга за счет
активации восходящих отделов ретикулярной формации;
- этофиллин улучшает микроциркуляцию головного мозга вследствие увеличения
минутного объема сердца и повышения перфузионного давления в зоне ишемии и
гипоксии.
В силу комбинированной структуры Инстенон является достаточно мощным
агентом, способным положительно воздействовать на метаболические процессы,
протекающие в головном мозге в условиях патологии. При этом очень важно
отсутствие так называемого синдрома "обкрадывания", наблюдаемого при
назначении других вазотропных препаратов.
Применяется по следующей схеме: инстенон назначается в дозе 2-4мл
внутривенно капельно или внутримышечно с первых часов нахождения больного в
реанимационном отделении в случаях отсутствия признаков внутричерепного
кровоизлияния. Данная терапия проводится в течение 3-5 дней до стабилизации
положительной динамики. По мере улучшения состояния лечение может быть
продолжено пероральными формами препарата.
В ряде случаев при быстром внутривенном введении может незначительно
понизиться артериальное давление, поэтому продолжительность введения одной
ампулы должна составлять не менее трех часов.
На табл. 9 представлены основные группы препаратов для первичной и
вторичной нейропротекции.
8.5 Гипотермия
В настоящее время гипотермия рассматривается многими исследователями,
как наиболее многообещающий физический метод нейропротекторной защиты
мозга. В 2001 г. были проведены пилотные клинические испытания применения
гипотермии в остром периоде ишемического инсульта [72]. Гипотермия была
индуцирована в среднем через 6 часов с момента появления клинических
симптомов, охлаждение проводилось до достижения целевой температуры 32±1°С
в течении от 12 до 72 часов. В результате было получены данные,
свидетельствующие о безопасности данной методики и способности предотвращать
расширение зоны инфарктного ядра, а также улучшать неврологическое течение
ишемического инсульта.
Предполагаемый механизм действия гипотермии связан с ее способностью
снижать церебральный метаболизм и ингибировать патохимический каскад,
связанный с реперфузионным повреждением, включая реакции, связанные с
генерацией свободных радикалов, эксайтотоксичных аминокислот и кальция,
вызывающих митохондриальное повреждение и апоптоз.
Показано, что проведение мягкой гипотермии (34°С) на 20-30% снижает
мозговой кровоток, повышает в ткани РСГ и обеспечивает сохранения пула АТР в
нервной ткани, а также достоверно снижает внутричерепное давление.
122
Также выявлено, что ректальная температура при проведении гипотермии,
коррелирует с температурой мозга, которая измерялась с помощью катетера,
снабженного микротермистером при дренировании желудочков мозга.
Будущие исследования должны будут определить оптимальную
продолжительность лечебной гипотермии, уровень температуры и скорость
охлаждения и согревания тела [79,95].
8.6 Гемодилюция
Несколько крупных клинических испытаний изоволемической гемодилюции
не продемонстрировали уменьшение смертности или инвалидизации при лечении
ишемического инсульта. Гиперволемическая гемодилюция изучалась в небольших
рандомизированных исследованиях, которые дали противоречивые результаты
[67,94,98].
8.7 Перфторуглероды
Остается открытым вопрос о возможности применения в остром периоде
препарата Перфторан, однако его использование может быть перспективным,
исходя из его газотранспортных, гемореологических, противоотечных и
мембраностабилизирующих свойств, в связи с чем необходимо проведение
серьезных клинических исследований его эффективности [13]. Ранее были
проведены преклинические и начаты ограниченные клинические испытания
препарата Ревоксин (NeuronTherapeutics Corp.) в остром периоде ишемического
инсульта, представляющего собой перфторуглеродную эмульсию в комплексе с
нутриентами. Была предложена техника вентрикулостомии боковых желудочков
мозга, с последующей их перфузией ревоксином. В результате была показана
способность указанной методики уменьшать размеры инфаркта мозга и снижать
ВЧД [37,38,39].
8.8 Профилактика и терапия отека и набухания головного мозга.
Ишемический отек головного мозга развивается в течении 24-48 ч. после
начала инсульта и достигает пика на 3-5 сутки, а с 7-8 суток в случае если пациент
выживает, медленно регрессирует.
Целью противоотечной терапии является: а) снижение ВЧД; б) поддержание
адекватного ЦПД; в) предотвращение вторичного повреждения мозга вследствие
набухания [32]. Противоотечная терапия должна строится на следующих
принципах:
- ограничение объема вводимых инфузионных сред (недопустимо введение 5%
глюкозы);
исключение факторов повышающих ВЧД (гипоксия, гиперкапния,
гипертермия);
- придание возвышенного положения (20-30°) головного конца кровати
(пациентам с тяжелым инсультом не поворачивать голову в стороны в первые 24
часа);
123
- если доступен мониторинг ВЧД, то церебральное перфузионное давление
должно поддерживаться >70 мм Hg.
Для лечения отека и набухания головного мозга рекомендуются следующие
фармакологические препараты и нефармакологические методы:
Гипервентиляция -поддержание РаСО? в пределах 30-35 mm Hg. Снижение
РаСО2 на 5-10 mm Hg уменьшает внутричерепное давление на 25-30 %. При этом
снижение РаСО2 ниже 30 mm Hg опасно углублением ишемических повреждений
мозга. Применение гипервентиляции ограничено у пациентов с гиповолемией (ЦВД
ниже 30 мм вод. ст. и САД ниже 70-80 mm Hg). Управляемую гипервентиляцию
целесообразно проводить не более 2 часов в условиях медикаментозной седации.
Однако этот метод остается дискутабельным, поскольку вазоконстрикторный
эффект гипокапнии уменьшает церебральную перфузию и может усугубить
ишемическое повреждение мозга [28,32,109];
На Украине нашел широкое применение препарат L-лизина эсцинат -
комплекс водорастворимой соли сапонина эсцина из семян конского каштана и
аминокислоты L-лизина. В сыворотке крови соль L-лизина эсцината быстро
диссоциирует на ионы лизина и эсцина. Эсцин защищает от разрушения
лизосомальными гидролазами гликозаминогликаны в стенках микрососудов и
окружающей их соединительной ткани, нормализуя повышенную сосудисто-
тканевую проницаемость и оказывая антиэкссудагивное и быстрое противоотечное
действие. Препарат вводится строго внутривенно в дозе 10 мл (8,8 мг эсцина) 2 раза
в первые 3 суток, затем по 5 мл - 2 раза/сутки. Максимальная суточная доза
препарата не должна превышать 25 мл — 22 мг эсцина. Курс - до получения
стойкого клинического эффекта, как правило, 7-8 суток [27,28].
Гиперосмолярные растворы. Данные препараты мобилизируют свободную
жидкость во внутрисосудистое пространство и обеспечивают снижение
внутричерепного давления, но только при интактном ГЭБ, при его повреждении
может развиться синдром «рикошета».
а) маннитол - 25-50 г (0,25-0,5 г/кг) (1370 мосмоль/л) каждые 3-6 часов -
(осмотерапия эффективна на протяжении 48 - 72 часов), под контролем
осмолярности плазмы (не должна превышать 320 мосм/л) [32,49]. Было показано,
что противоотечный эффект достигается при использовании указанных умеренных
доз препарата, т.к. применение высоких доз маннитола (1,5 г/кг) приводит к
парадоксальному нарастанию отека мозга за счет аккумуляции осмотически
активных частиц в веществе мозга, вследствие повреждения ГЭБ или при
пролонгирования введения данного препарата более 4 дней [109] Маннитол
снижает ВЧД на 15-20 %, повышает ЦПД на 10% и в отличие от фуросемида
улучшает мозговой кровоток за счет снижения гематокрита, увеличения объемного
церебрального кровотока, пугем мобилизации внеклеточной жидкости и улучшения
реологических свойств крови за счез' снижения вязкости крови на 16% (фуросемид
напротив повышает вязкость крови на 25%);
б) реосорбилакт (900 мосмоль/л), сорбилакт (1670 мосмоль/л), в дозе 200-400
мл/сут.;
фуросемид - болюсно 40 мг внутривенно;
124
барбитураты короткого действия, такие как тиопентал натрия, используемые в
виде болюса, могут быстро и значительно уменьшить ВЧД, но эффект
кратковременный, и препараты могут быть использованы только’ для лечения
острых кризисных ситуации, например, перед операцией. Лечение барбитуратами
требует мониторинга ВЧД и ЭЭГ, а также тщательного мониторирования
гемодинамических параметров, так как может иметь место значительное падение
уровня АД [29,32].
Глюкокортикостероиды не рекомендуются для лечения ишемического
инсульта, поскольку использование как обычных, так и высоких доз неэффективно
для снижения ВЧД и ассоциировано с развитием иммуносупрессии и
последующими инфекционными осложнениями, гипергликемии и кровотечений
[90].
В случаях быстрого нарастания признаков вклинения ствола головного мозга
(снижение уровня сознания, появление децеребрационной ригидности,
двухсторонних рефлексов Бабинского), рекомендовано быстрое внутривенное
введение на физиологическом растворе 0,5 г/кг массы тела маннитола в течении 20-
25. мин., и/или навязывания больному (если он находится на ИВЛ) режима
гипервентиляции.
В случаях быстрого нарастания признаков вклинения ствола головного мозга
(снижение уровня сознания, появление децеребрационной ригидности,
двухстороннего рефлекса Бабинского) рекомендовано быстрое внутривенноке
введение маннитола в дозе 0,5 г/кг на физиологическом растворе в течение 20-25
миут и/или навязывания пациенту (если он находится на ИВЛ) режима
гипервентиляции [3].
8.9 Препараты противопоказанные в остром периоде ишемического
инсульта
1) Вазоактивные препараты:
-дибазол
-эуфиллин
-папаверин
-но-шпа
-кавинтон (винпоцетин)
-ницерголин (сермион)
Обоснование:
Целью терапии ишемического инсульта, является улучшение
ишемизированных зон. Гипоперфузия в зоне «ишемической полутени» ведет к
развитию локального тканевого ацидоза, выраженность которого зависит от степени
снижения перфузии. Т.к. ионы Н+ являются естественными химическими
факторами, обуславливающими вазо дилятацию, особенно артериол и
прекапиллярных сфинктеров, то при развитии ацидоза в зоне «ишемической
полутени» возникает застойная гиперемия. При этом максимально расширенные
сосуды стаиовяться практически нс реагирующими на церебральные
вазодилататоры. Поэтому при применении вазодилататоров в основном происходит
расширение кровеносных сосудов в интактных зонах, окружающих зону
125
«ишемической полутени». Вследствие этого, за счет снижения сопротивления
кровотоку в бассейнах сосудов, кровоснабжающих ткань мозга вокруг
«ишемической полутени», происходит падение перфузионного давления в этой
зоне, развивается синдром «обкрадывания». При этом перфузия еще больше
снижается, а зона инфаркта расширяется, увеличивая таким образом, объем
нейронов с необратимыми структурными повреждениями [5,23,32,49].
Эуфиллин, кроме того, являясь производным ксантина, в зоне «ишемической
полутени» способен генерировать свободнорадикальное (О2 ) перекисное окисление
липидов, дополнительно вызывая повреждение и гибель нейронов в этой зоне [25].
2) Блокаторы Са2'-каналов:
-нифедипин
-нимодипин (нимотоп)
Обоснование:
Использование Са2+-каналов широко практиковалось в лечении острого
ишемического инсульта и было патогенетически обосновано. Однако результаты
проведенных мультицентровых клинических испытаний перорального применения
нифедипина и парентерального применения нимодипина (нимотопа) при остром
ишемическом инсульте оказались крайне отрицательными. Из-за вызываемой этими
препаратами резкой гипотензии и развития ишемического синдрома обкрадывания.
Так у пациентов получаших нимодипин внутривенно в дозе 2 мг/час выявлено
статистически достоверное ухудшение неврологического и функционального
исхода, в сравнении с группой получавшей плацебо. При использовании дозы
нимодипина равной 1 мг/час различий в эффективности в сравнении с плацебо
выявлено не было. При этом побочный эффект нимодипина - дозозависимое
снижение АДсит. и АДдиаст. прямо коррелировало с частотой неблаг опрятных
исходов и увеличением летальности. Поэтому данные препараты были исключены
из протоколов терапии острого ишемического инсульта [10,32].
8.10 ПРОТОКОЛ
интенсивной терапии ишемического инсульта на догоспитальном этапе
1) Постановка диагноза (типичная клиника), отметка времени начала развития
инсульта.
2) Оценка витальных функций (дыхательные пути, дыхание, гемодинамика),
определение SaO2, пульсоксиметром.
3) При <94% необходимо проведения ингаляции кислорода через лицевую маску
или носовой катетер.
4) При SaO2 <90% на фоне проводимой оксигенотерапии, при уровне сознания
<8 баллов по шкале ком Глазго, серьезного нарушения паттерна дыхания
необходима интубация трахеи (или как минимум введение воздуховода
Гведела) и проведение ИВЛ по показаниям.
5) Обеспечение периферического венозного доступа катетером с медленной
инфузией 0,9% раствора хлорида натрия.
126
6) Оценка неврологического статуса по шкалам инсульта NIHSS или ком Глазго.
7) Аспирин 100-300 мг/сут per os.
8) Сульфат магния в дозе 7-10 мг/кг из расчета по магнию (в среднем 10-15 мл
25% раствора) в/в.
9) Коррекция гипотензии, нулем адекватного волемического восполнения
кристаллоидами и коллоидами, при необходимости введение вазопрессоров
(мезатон, дофамин).
Недопускается введение растворов 5% глюкозы из-за риска увеличения отека
мо зга вследствие уменьшения осмолярности плазмы.
10) Неотложная ангигипертензионная терапия не рекомендуется, за исключением
случаев чрезвычайно высоких значений АД>220/120 - лабеталол, или натрия
нитропруссид в/в, или каптоприл per os.
11) Ранняя госпитализация в инсультное отделение.
Не рекомендуется использовать:
эуфиллин, папаверин, но-шпу, дибазол, таблетированный нифедипин, нимотоп.
8.11 ПРОТОКОЛ
интенсивной терапии ишемического инсульта на госпитальном этапе
а) Неспецифическая терапия
1) Мониторирование витальных функций и мониторирование неврологического
статуса (шкала ком Глазго, шкала NIHSS).
2) Непрерывный мониторинг сердечной деятельности рекомендуется в первые 48
часов после начала инсульта, особенно у пациентов, имеющих:
• предшествующее известное заболевание сердца;
• аритмии в анамнезе;
• нестабильное артериальное давление;
» клинические признаки/симптомы сердечной недостаточности;
• отклонения от нормы в базовой ЭКГ;
• инфаркт, поражающий инсулярную кору головного мозга.
3) Купирование повышения температуры тела >37,5°С, антипиретики, охлаждение
тела.
4) В случае лихорадки рекомендуется поиск очага возможной инфекции
(локализация и этиология) дая начала антибиотикотерапии.
5) Антибактериальную, антимикотическую и антивирусную профилактику
проводить не рекомендуется у иммунокомпетентных пациентов.
6) Пациентам без признаков гипоксемии не рекомендуется проводить
оксигенотерапию.
7) Поддержание нормогликемии. (при уровне >10 ммоль/л проводится коррекция
инсулином).
8) Рекомендуется немедленная коррекция гипогликемии путём внутривенного
введения болюса 40 % и по козы или инфузии 10-20 %- глюкозы.
127
9) Коррекция водно-электролитного баланса (большинство пациентов необходимо
вести с положительным водным балансом, в случае признаков отека мозга -
слегка отрицательным балансом).
10) Поддержание ЦВД на уровне 8-10 см Н2О путем адекватного волемического
восполнения кристаллоидами и коллоидами. Рутинное снижение АД не
рекомендуется, за исключением случаев чрезвычайно высоких значений (>200-
220 АД сист. или 120 АД диаст.), подтвержденных повторными измерениями.
11) Неотложная антигипертензивная терапия при более умеренной гипертензии
рекомендуется в случае инсульта и сердечной недостаточности,
расслаивающейся аорты, острого инфаркта миокарда, острой почечной
недостаточности, тромболизиса или внутривенного введения гепарина, но её
следует применять с осторожностью.
12) Рекомендуемое целевое АД у пациентов:
с гипертензией в анамнезе: 180/100-105 мм Hg;
без гипертензии в анамнезе: 160-180/90-100 мм Hg;
при тромболизисе избегать АД сист. выше 180 мм Hg.
13) Рекомендуемые препараты для коррекции АД:
лабеталол в/в;
или
нитропруссид натрия или нитроглицирин в/в;
или
каптоприл перорально.
14) Недопустимо применение нифедипина и нимодипина и любого резкого
снижения АД.
15) Избегайте и корригируйте гипотензию, особенно у нестабильных пациентов,
посредством введения адекватных объёмов жидкости и по показаниям введение
вазопрессоров. Растворы глюкозы применять не рекомендуется вследствие
вредного действия гипергликемии.
б) Специфическая терапия
1. Тромболитическая терапия с использованием рекомбинантного тканевого
активатора плазминогена (rt-PA) — актилизе. В первые 3 часа с момента
появления первых клинических симптомов ишемического инсульта в дозе 0,9
мт/кг (максимальная доза 90 мг) в/в.
2. Антитромботическая терапия - аспирин в дозе 100-300 мг/сут. в течении 48
часов per os достоверно снижает летальность и частоту повторных инсультов.
3. Антикоагулянтная терапия - рутинное использование как
нефракционированных гепаринов, так и низкомолекулярных гепаринов для
улучшения неврологических исходов, с точки зрения доказательной медицины, не
рекомендуется. Показания для применения гепарина: риск тромбоза глубоких вен,
инсульт вследствие кардиогенной эмболии с высоким риском развития повторной
эмболии (искусственные клапаны сердца, мерцательная аритмия, инфаркт миокарда
с интрамуральными тромбами, тромбоз левого предсердия), тромбоз венозных
128
синусов. В этих случаях рекомендуется профилактическое использование
умеренных доз НФГ - 5000 ME подкожно 2 раза/сутки, или НМГ: фраксипарин 0,3
мл подкожно 2 раза/сутки или эноксапарин (клексан) 0,2 мл 1 раз/сутки.
Противопоказаниями для лечения гепарином являются: обширные инфаркты
(более 50% бассейна СМА), неконтролируемая артериальная гипертензия.
Гепарин не должен применяться в течение 24 часов после проведения
тромболизиса
4. Противоотечная терапия:
- L-лизина эсценат - в дозе 10 мл 2 раза в первые 3 суток, затем по 5 мл 2
раза/сутки строго внутривенно;
- маннитол 25-50 г (0,25-0,5 г/кг) в/в каждые 3-6 часов под контролем
осмолярности плазмы, или
- реосрбилакт, сорбилакт по 200-400 мл/сут., или
- фуросемид болюсно 40 мг в/в;
- в случае случаях быстрого нарастания признаков вклинения ствола головного
мозга рекомендовано быстрое внутривенное введение на физиологическом растворе
0,5 г/кг массы тела маннитола в течении 20-25 мин., и/или навязывания больному
(если он находится на ИВЛ) режима гипервентиляции.
5. Первичная нейропротекция:
-сульфат магния 20 мл 25% раствора, медленно капельно в режиме
равномерной инфузии, под контролем мониторинга АД;
-глицин - в первые 6 часов от момента инсульта сублингвально в дозах 1-2
г/сутки в течение 5 дней.
5. Вторичная нейропротекция:
- пирацетам - вводят болюсом или инфузионно в дозах до 12 г/сут в 2 приема
или в/м. Курс парентерального лечения 7-10 дней, после которого можно назначать
per os из расчета 130-150 мг/кг/сут в 3-4 приема;
- реамбирин - внугривенно капельно со скоростью не более 90 кап/мин (4-4,5
мл/мин) до 800 мл/сут. Курс 3-5 дней,
или
- мексидол - соль эмоксипина и янтарной кислоты от 100 до 1000 мг/сутки в/в
капельно;
129
ЗАКЛЮЧЕНИЕ
Несмотря на углубления наших знаний о генетических, молекулярных и
клеточных механизмах повреждения мозга при ишемическом инсульте, проблема
поиска оптимальной стратегии интенсивной терапии остается еще дапека от своего
решения. В нашей работе мы хотели подчеркнуть важность базовой терапии
ишемического инсульта направленной на поддержание экстрацеребрального
гомеостаза. Огромным прорывом в терапии инфаркта мозга, безусловно можно
считать появление троболизиса с использованием rt-PA, позволившего существенно
повлиять на исходы заболевания. При этом крайне противоречивыми и
неоднозначными, являются результаты исследований препаратов для
нейропротекции. Несмотря на большое количество проведенных во всем мире
клинических испытаний разнообразных групп препаратов для нейропротекции, ни
один из них не может считаться эффективным и безопастным с позиций
доказательной медицины. Необходимы дальнейшие исследования свойств
потенциальных препаратов для нейропротеции, с переосмыслением причин неудач
применения современных методов фармакологической нейропротекции. В
настоящее время открываются новые перспективы физической нейропротскции -
проведения гипотермии, относительно которой получены обнадеживающие
результаты.
В целом хотелось бы отметить, что при планировании комплекса интенсивной
терапии необходимо руководствоваться, говоря словами патриарха реаниматологии
- П. Сафара, вынесенных в заглавие книги, здравым смыслом в выборе тех или
иных методов и препаратов, опираясь на современные международные
рекомендации, которые прошли длительный путь апробации в многочисленных
мультицентровых клинических испытаниях. С учетом индивидуальных
особенностей пациета вариантата и степени тяжести ишемического инсульта.
В монографии мы обрисовали круг как отсчетсвенпых, так и зарубежных
препаратов, обладающих потенциальными нейропротекторными эффектами, при
этом мы считаем, что при их назначении, необходимо не допускать полипрогмазии.
Также необходимо подчеркнуть, что успех лечения больных с ишемическим
инсультом, зависит от хорошей организации оказания медицинской помощи,
начиная с догоспитального этапа слубы скорой медицинской помощи,
эффективного взаимодействия врачей анестезиологов-реаниматологов, неврологов
и нейрохиругов на госпитальном этапе, и последующего этапа восстановительного
лечения с актиным участием на ранних этапах заболевания врачей реабилитологов.
В заключение необходимо отметить, что в настоящей монографии мы не
претендуем на однозначность трактовки и всеобъемлимость в освещении вопросов
интенсивной терапии ишемического инсульта, а только наметили ключевые
проблемы и приглашаем к размышлению над ними. При этом важным является
понимание того факта, что лечение ишемического инсульта в пределах
«терапевтического окна», является главным условием эффективности проводимой
интенсивной терапии, поскольку патогенетически обоснованное, в полном объеме,
но начатое поздно лечение, может оказаться практически неэффективным.
130
. А „ ЛИТЕРАТУРА
1 . Ашмарин И.П., Стукшюв, П.В., Ещенко Н.Д. (Ред.) Биохимия мозга. СПб,
Санкт-Петероургский университет, 1999 328 с
2 . Бондарев А.А. Дискриминация между апоптозом и некрозом нейронов под
влиянием окислительного стресса // Биохимия, 2000. 65(7): 981 -990.
3 . Верещагин Н.В., Пирадов М.А., Суслина З.А. (Ред.) Инсульт. Принципы
диагностики, лечения и профилактики. М., Интермедика, 2002, 208 с.
4 . Виничук С.М., Довбонос Г.А. Современная диагностика и лечение острых
нарушений мозгового кровообращения. (Метод, рек.). К., 2000, 32 с.
5 . Виничук С.М., Черенько Т.М. Ишемический инсульт: эволюция взглядов на
стратегию лечения. Киев, 2003, 120 с.
6 . Вознюк А.А.,Одинак М.М., Кузнецов А.И. Применение тлиатилина у больных
с острым нарушением мозгового кровообращения // В кн.: Сосудистая
патология нервной системы. СПб, 1998. С. 167-172.
7 . Ворлоу Ч.П., Деннис М.С., ВанГейн Ж и соавт. Инсульт. Практическое
руководство для врачей. СПб, 1998, 629 с.
8 . Гусев Е.И., Скворцова В.И., Мясоедов Н.Ф. Эффективность семакса в остром
периоде полушарного ишемического инсульта (клинические и
электроэнцефалографические исследования) // Ж. невропатол. и психиатрии,
1997, 6: 26 34.
9 . Гусев Е.И., Скворцова В.И. Нейропротективная терапия ишемического
инсульта. Первичная нейропротекция // Инсульт, 2002, 5: 3-16.
10 .Гусев Е.И., Скворцова В.И. Ишемия головного мозга. М., Медицина, 2001,
328 с.
11 .Квитницкий-Рыжов Ю.Н. Отек и набухание головного мозга. К., Здоровье,
1978, 183 с.
12 .Комиссаров И.В. Средства лечения ишемического инсульта: предпосылки к
применению и эффективность /7 Лгкування та д1агностика, 2003,1: 35-40.
13 .Максимишин С.В, Мороз В.В., Семченко В.В., Степанов С.С. Перфторан как
средство нормализации микроциркуляции головного мозга в
постишемическом периоде /7 В кн.. Мороз В.В. (Ред.) Реаниматология. Ее
роль в современной медицине. Мат. конф. Москва, 13-15 мая, 2004 г. С. 144-
148.
14 Молчанова Л.В. Патохимия церебральной ишемии. В кн.: Мороз В.В. (Ред.)
Фундаментальные проблемы Р^^р0,™, ™СПП>та
реаниматологии РАМН. Том 3. N ‘ ,,
15 .Новиков В.С. (Ред.) Программированная клеточная гибель. СПб, Наука, 1996,
16 Павленко I Ф. K,TiHiKo45ioxiMi4«e обгрунтування депдратащйноГ терапй при
о.ыавленкс . Mj // Бшь, знеболювання г интенсивна терашя
ТЯЖК1И черепно-мозковш травм И<Ш1Я,
2004, 2(Д).2(В’20^ q делать? Или необходимость организационных
17 .Полищук Н.Е., Гуляев Д.В. чмд н Doctor 2003, 3 79 ™ных
изменений в борьбе с инсультом в Украг , / ч.
131
18 . Полищук Н.Е., Трещинский А.И. Интенсивная терапия при остром
ишемическом инсульте // Doctor, 2003, 3: 20-23.
19 .Рябов Г.А. Синдромы критических состояний. М., Медицина, 1994, 368 с.
20 .Рябов Г.А., Азизов Ю.М. Роль оксида азота как регулятора клеточных
процессов при формировании полиорганной недостаточности //
Анестезиология и реаниматология, 2001, 1:8-13.
21 .Самуилов В.Д., Олескин А.В., Лагунова Е.М. Программированная клеточная
смерть // Биохимия, 2000, 65 (8): 1029-1046.
22 .Скулачев В.П. Феноптоз: программированная смерть организма И Биохимия,
1999, 64(12): 1679-1688.
23 .Старченко А.А. Клиническая нейрореаниматология. СПб., Санкт-
Петербургское медицинское издательство, 2002, 672 с.
24 .Трещинский А.И., Глумчер Ф.С., Короткоручко А.А., Полищук Н.Е.
Ишемический инсульт (Эпидемиология, патогенез, клиника, интенсивная
терапия) // Бшь, знеболювання i штенсивна терашя, 1997, Г. 79-90.
25 .Трещинский А.И., Глумчер Ф.С., Короткоручко А.А., Полищук Н.Е.
Интенсивная терапия при остром ишемическом инсульте И Бшь,
знеболювання i штенсивна терашя, 1998, 1: 49-74.
26 .Усенко Л.В., Мальцева Л.А., Царев А.В., Черненко В.Г. Современный взгляд
на интенсивную терапию ишемического инсульта: место лакардии в
комплексной терапии. Днепропетровск, 2004, 47 с.
27 .Усенко Л.В., Слива В.И., Криштафор А.А., Площенко Ю.А., Хмельницкий
Э.Е. L-лизина эсцинат как средство предупреждения и купирования
локальных отеков. В ки.: Достижения и перспективы современной
анестезиологии и интенсивной терапии. Арт-Пресс, Днепропетровск, 2003.
С.151-152.
28 .Черний В.И., Городник В.А., Кардаш А.М., Дроботько В.Ф., Островой Е.Л.
Принципы и методы диагностики и интенсивной терапии отека и набухания
головного мозга. (Метод, рек.) Донецк, 2003, 47 с.
29 .Черний В.И., Городник В.А. Острая церебральная недостаточность. Киев,
Здоров’я, 2001,425 с.
ЗО .Чеченин М.Г. Кинетическая терапия в комплексе лечения респираторного
дистресс-синдрома взрослых: Дисс. канд. мед. наук. Новосибирск, 1998, 19 с.
31 .Abracham Е., Matthay М.А., Dinarello A. et al. Consensus conference definitions
for sepsis, septic shock, acute lung injury, and ARDS: Time for a reevaluation //
Crit. Care Med., 2000, 28(1): 232-235.
32 .Adams H.P., Adams K.J., Brott T. et al. American Stroke Association (ASA)
Guidelines for the early management of patients with ischemic stroke // Stroke,
2003,29: 1056-1083.
33 .Akgoren N., Fabricius M., Lauritzen M. Importance of nitric oxide for local
increases of blood flow in rat cerebellar cortex during electrical stimulation /7 Proc.
Nat. Acad. Sci. USA, 1994, 91: 5903-5907.
34 .Astrup J., Siesjo B.K., Symon L. Thresholds in cerebral ischemia - the ischemic
penumbra//Stroke, 1981, 12(6): 723-725.
132
35 fe^Ksociated whh-™ M D?V'S et a1' sP°ntaneous reperfusion after ischemic stroke
„ С Ц OU,COmfi" S,roke. 2001, 32: 2356-2358.
^rome^’bv . M G" М A' et aL Rele'sal of 6*al “misery-perfusion
;/^Х198ъatteriai bypass m h—° “rebrai ischem,a-
’ f^Zei/5!f*rock ff.O. et al. Reduction of cefbral infarction using the
n'n n'T T" Nt;uroio®- 2000,54 (Suppl. 3): A66.
3 8.Bell R. Frazer G.D., Osterholm J.L., Duckett S.W. A novel treatment for
ischemic intracranial hypertension in cats // Stroke, 1991, 22: 80-83.
^'??.Se J-L., Triolo A. Focal cerebral ischemical reduction of size of
in aict у ventriculo-subarachnoid perfusion with fluorocarbon emulsion // Brain
Research, 1985, 328: 223-231.
4O.Bredt D., Snyder S. A. Nitric oxide: a physiologic messenger molecule // Annu.
Rev. Biochem., 1994, 63: 175-195.
41.Brott T„ Bogousslavsky J. Treatment of acute ischemic stroke // N. Engl. J. Med.,
2000, 343(10): 710-722.
4 2.Bruno A., Biller J., Adams H.P. Acute blood glucose level and outcome from
ischemic stroke: Trial of ORG 10172 in acute stroke treatment (TOAST)
investigators 11 Neurology, 1999, 52: 280-284.
4 3.Buchan A.M. Neuroprotective stroke trials: a ten year dry season // In: Pinsky M.
(Ed.) Cerebral Blood Flow: mechanisms of ischemia, diagnosis and therapy. Berlin,
Springer, 2002. P. 236-251.
44 .Caesar K., Akgoren N., Mathiesen C., Lauritzen M. Modification of activity —
dependent increases in cerebral blood flow by extracellular potassium in
anaesthetized rats П J Physiol (Lend), 1999; 520 Pt 1: 281-92.
45 .Castillo J., Davalos A., Noya M. Progression of ischemic stroke and excitotoxic
aminoacids // Lancet, 1997, 349: 79-83.
46 .Corgulu A., Kins T., Cabanogly S. Reduction of edema and infarction by
Memantine and MK-801 after focal cerebral ischemia and reperfusion in rat // Acta
Neurochir. Wien., 142: 1287-1292.
47 .Coull B.M., Wiliams L.S., Goldstein L.B. Anticoagulants and antiplatelet agents in
acute ischemic stroke. Report of the Joint Stroke Guidelines Development
Committee of the American Academy of Neurology (AAN) and ASA//Neurology,
2002,59:13-22. , _ 4 t f .
48 Пр ripvn P P Reuck I P Deberdt W- et al. Treatment of acute ischemic stroke
Xnacet^. Members of the Piracetam in Acute Stroke Study (PASS) // Stroke,
49 .Euro^n StrokeMtiadve (EUSI) for stroke - Update 2003 // Cerebrovasc. Dis„
5O.f2>er M.,3Bren T.G. Emerging therapies for acute ischemic stroke. New therapy
on trial II Stroke, 2003,,34. depen(jence of regional blood flow human
a”'ss,on ™,hy"
1984,51: 1109-20.
ВЗ
52 .Fox, P.T., Raichle M.E. Focal physiological uncoupling of cerebral blood flow and
oxidative metabolism during somatosensory stimulation in human subjects // Proc.
Nat. Acad. Sei. USA, 1986, 83: 1140-4.
53 ,Fox P.T., Raichle M.E., Mintun M.A., Dence C. Nonoxidative glucose
consumption during focal physiological neural activity // Science, J 988, 241: 462-
464.
54 .Gjcdde A. Coupling and compartmentation of cerebral blood flow and metabolism
// In: Pinsky M. (Ed.) Cerebral Bloof Flow: mechanisms of ischemia, diagnosis and
therapy. Berlin, Springer, 2002 P. 72-95.
55 .Goupil A., Bourghol B., Derouet N. et al. Prise en charge de 1’accitent ishemique
cerebral de sujet jenne en urgens: aspects etiologiques et therapeutiques //
Reanimation, 2002, 11(7): 502-508-
56,Graham S.H., Hickey R.W. The genetic control of iaschemic neronal cell death //
In: Pinsky M. (Ed.) Cerebral Blood Flow: mechanisms of ischemia, diagnosis and
therapy. Berlin, Springer, 2002. P. 96-105.
57 .Greenberg J., Hand P., Sylvestro A., Reivich M. Localized metabolic flow couple
during functional activity // Acta Neurol. Scand. 1979, 72: 12-13.
58 .Grossman E., Messerli F.H., Grodzinski T., Kowey P. Should a moratorium be
placed on sublingual nifedipine capsules given for hypertensive emergencies and
pseudoemergencies? // JAMA, 1996, 276: 1328-1331.
59 .Grotta J.S. Acute stroke therapy at the millenium: Consummating the marriage
between the laboratory and bedside- The Feinberg lecture // Stroke, 1999, 30: 1722-
1728.
6O .Hajat C., Hajat S., Sharma P. Effects of poststroke purexia on stroke outcome: a
meta-analysis of studies in patients // Stroke, 2000, 31: 410-414.
61 .Hartman B., Zide D., Udenfriend S. The use of dopamine — betahydroxylase as a
marker for the central noradrenergic nervous system in rat brain И Proc. Natl. Acad.
Sci. USA. 1972, 69: 2722-2726.
62 .Hommell M., Boissel J.P., Cornu C. Termination of trial of streptokinase in severe
acute ischemic stroke: MAST Study Group // Lancet, 1995, 345: 57.
63 .Hossmann K..A. The ischemic penumbra: pathophysiology and therapeutic
implicatons // In: Pinsky M. (Ed.) Cerebral Blood Flow: mechanisms of ischemia,
diagnosis and therapy. Berlin, Springer, 2002. P. 137-148.
6 4.1adecola C., Li J., Yang G., Xu S- Neural mechanisms of blood flow regulation
during synaptic activity in cerebellar cortex // J. Neurophysiol, 1996, 75: 940-950.
6 5.1adecola M., Narakai S., Mraovitch D.A., Ruggiero L.W., Tucker D., Reis.J. Global
increase in cerebral metabolism and blood flow produced by focal electrical
stimulation of dorsal medullary reticular formation in rat // Brain Res., 1983,
272:101-114.
6 6.ladecola C., Niwa K. Neural regulation of cerebral circulation // In: Pinsky M. (Ed.)
Cerebral Blood Flow: mechanisms of ischemia, diagnosis and therapy. Berlin,
Springer, 2002. P. 7-16.
6 7.Italian Acute Stroke Study Group. Haemodilution in acute stroke: results of the
Italian haemodilution trial П Lancet, 1988,1:318-321.
134
68 .Kaufmann W.E., Worley P.F., Pcgg j„ Brcmer M-> lsakson p. COX-2, a
synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic
sites in rat cerebral cortex // Proc Natl Acad Sci USA, 1996, 93: 2317-21.
69 .Kerr J.F.R., Wyllie A.H., Currie A.R. Apoptosis: a basic biological phenomen with
wide-ranging implication in tissue kinetics // Brit. J. Cancer, 1972, 26(4): 239-257.
70 .Klatzo I. Presidental address. Neuropathological aspects of brain edema И J-
Neuropathol. Exp. Neurol., 26: 1 -] 4.
71 .Ko K. R., Ngaj A. C., Winn R. H. Role of adenosine in regulation of regional
cerebral blood flow in sensory cortex // Am J. Physiol., 1990, 259: Hl 703 -H1708.
72 .Krieger D.W., De Georgia A., Abou-Chebl A. et al. Cooling for acute ischemic
brain damage (COOL AID). An open pilot study of induced hypothermia in acute
ischemic stroke // Stroke, 2001, 32: 1847-1854.
73 .Kuschinsky W. Coupling ot function metabolism and blood flow in the brain //
NIPS, 1987, 2: 217-220.
74 .Lassen N.A., Agnoli A. The upper limit of autoregulation of cerebral blood flow in
the pathogenesis of hypertensive encephalopathy // Scand. J. Clin. Lab. Invest.,
1972, 30: 113-116.
75 .Lewandowski C.A., Frankel M., Tomsick T.A., Broderick J., Freg J., Clare W.,
Starleman S., Grotta L, Spilker J., Khoury J., Brott T. Combined intravenous and
intra-arterial therapy of r-TPA versus intra-arterial therapyof acute ischemic stroke.
EMS Bridging Trial H Stroke, 1991, 30: 2598-2605.
76 .Li J., ladecola C. Nitric oxide and adenosine mediate vasodilatation during
functional activation in cerebellar cortex II Neuropharmacology, 1994, 33: 1453-
1461.
77 .Love S., Barber R., Wilcock G.K Apoptosis and expression of DNA repair proteins
in ischemic brain injury in man //NeroR eport, 1998, 9: 955-959.
78 .Manley G.T., Fujimura M., Ma T. Aquporin-4 deletion in mice reduces brain
edema after acute water intoxication and ischemic stroke // Nat. Med., 2000,6: 159-
163.
79 .Marion D.W. Therapeutic moderate hypothermia and fever 7 In: Pinsky M. (Ed.)
Cerebral Blood Flow: mechanisms of ischemia, diagnosis and therapy. Berlin,
Springer, 2002. P. 272-276.
8O .Muir K.W. Heterogencti of Stroke pathophysiology and neuroprotective clinical
trial design // Stroke, 2002, 33: 1545-1550.
81 .Muir K.W. Magnesium in stroke tratment // Postgrad. Med. J., 2002, 78: 641-645.
82 .Muir K.W., Lees K.R. A randomized, duble-blind. placebo-controlled pilot trial of
intravenous magnesium sulfate in acute stroke // Stroke, 1995, 26. 1183-1188.
83 .Muir K.W., Lees K.R. Dose optimization of intravenous magnesium sulfate after
acute stroke // Stroke, 1998, 29: 918 923.
84 .Multicenter Acute Stroke Trial - Europe Study Group. Thrombolytic therapy with
streptokinase in acute ischemic stroke (1996) // N. Engl. J. Med., 335: 145-150.
85 -Nilsson B., Rehncrona S., Siesjo B.K. Coupling of cerebral metabolism and blood
flow in epileptic seizures, hypoxia and hypoglycaemia In: blhot, К OConnor M.
(Ed.) Cerebral Vascular Smooth Muscle and its Control. New Yoric Elsevmr, 1978,
pp. 199-218.
135
86 .Paulson O.B., Newman E.A. Does the release of potassium from astrocyte endfeet
regulate cerebral blood flow? I I Science, 1987, 237: 896-898.
87 .Powers W.J. Acute hypertension after stroke: the scientific basis for treatment
decisions // Neurology, 1993,43: 461-467.
88 .Prichard J.W., Rothman D.L., Novotny E.J., Petroff O.A.C, Kuwabara T., Avison
M., Howseman A., Hanstock C., Shulman R.G. Lactate rise detected by III NMR
in human visual cortex during physiologic stimulation // Proc. Nat. Acad. Sci. USA,
1991,88: 5829-5831.
89 .Puybasset L. Management of systemyc haemodynamics in patiets with nerological
disorders: Pathology, monithoring and tratment strategies. In: Abstracts of 13th
World Congress of Anaesthesiologists. WFSA, April 18-23, 2004, Paris, CDROM,
C027d.
90 .Qizilbash N., Lewington S.L., Lopez-Arrietta J.M. et al. Corticosteroids for acute
ischemic stroke // Cochrane Database Syst. Rev. 2002, (2): CD 000064.
91 .Reith J., Jorgensen H.S., Pedersen P.M. Body temperature in acute stroke: relation
of stroke severity, infarct size, mortality, and outcome //Lancet, 1996, 347: 422-
425.
92 .Reivich, M. Blood flow metabolism couple in brain // Res. Publ. Assoc. Res. Nerv.
Ment. Dis., 1974, 53:125-140.
93 .Ronnig O.M., Guldvog B. Should stroke victims routinely receive supplemental
oxygen? A quasi-randomized controlled trial И Stroke, 1999, 30: 2033-2037.
94 .Scandinavian Stroke Study Group. Multicenter trial of nemodilution in acute
ischemic stroke: results of subgroup analyses И Stroke, 1988,19: 464-471.
95 .Schwab S., Schwarz S., Spranger M. et al. Moderate hypothermia in the treatment
of patients with severe middle cerebral artery infarction // Stroke, 1998, 29: 2461-
2466.
96 .Serena J., Leira R., Castillo J., Pumar J.M., Castellanos M., Dvalos A. Neurological
deterioration in acute lacunar infarctioin: the role of cxicitatory and inhibitory
neurotransmitters И Stroke, 32: 1154-1161.
97 .Siesjo B.K., Kristian T., Uchino H Triggering events in ischemic brain injury // In:
Pinsky M. (Ed.) Cerebral Blood Flow: mechanisms of ischemia, diagnosis and
therapy. Berlin, Springer, 2002. P. 45-59.
98 . Strand T. Evaluation of long-term outcome and safety after hemodilution therapy in
acute ischemic stroke // Stroke, 1992, 23: 657-662.
99 .Strandgaard S. Autoregulation of cerebral circulation in hypertension // Acta
Neurol. Scand., 1978, 57: 1-82.
100 . Taniquchi M., Yamashita T., Kumura E. Induction of aquaporin -4 wwater
chahnell mRNA after focal cerebral ischemia in rat /7 Brain Res. Mol. Brain. Res.,
2000,78: 131-137.
101 . The NINDS t-PA Stroke Study Group intracerebral hemorrhage after
intravenous t-PA therapy for ischemic stroke // Stroke, 19997, 28: 2109-2118.
102 . Tomsick T., Brott T., Barsan W., Broderick J., Haley E.C., Spilker J.
Prognostic value of the hyperdense middle cerebral artery sign and stroke scale
before ultraearly thrombolytie therapy // Am. J. NeuroradioL, 1996, 17: 79-85.
136
103 . Treib J., Grauer M.T., Wossner R., Morgenthaler M. Treatment of stroke an
intensive stroke unit: a novel concept // Intensive Care Med., 2000,26: 1598-1611.
104 . Ueki M., Linn F., Hossmann K..A. Functional activation of cerebral blood
flow and metabolism before and after global ischemia of rat brain // J. Cereb. Blood
Flow Metab., 1998, 8: 486-494.
105 . Wechsler L.R. Thrombolysis for acute stroke /7 In: Pinsky M. (Ed.) Cerebral
Blood Flow: mechanisms of ischemia, diagnosis and therapy. Berlin, Springer,
2002. P. 229-235.
106 . Weir CJ., Murray G.D., Dyker A.G., Lees K.R. Is hyperglycaemia an
independent predictor of poor outcome after acute stroke? Results of a long-term
follow up study // BMJ, 1997, 314: 1303-1306
107 . Werner C. Neuronal protection. In: Abstracts of 13th World Congress of
Anaesthesiologists. WFSA, April 18-23, 2004, Paris, CDROM, L20.
108 . Whisnant J.P., Basford J.R., Benstein E.F. et al. Special report from the
National Institute of Neurological Disorders and Stroke: Classification of
Cerebrovascular Desease III // Stroke, 1990, 21: 637-676.
109 . Xiao F. Brain edema and cerebral resuscitation: The present and future. Acad.
Emerg. Med., 2002, 9(9): 933-946.
110 . Yaksh T.L., Wang J.-Y., Go V.L. Harty G. J. Cortical vasodilatation
produced by vasoactive intestinal polypeptide (VIP) and by physiological stimuli
in the cat// J. Cereb. Blood Flow Metab., 1987, 7: 315-326.
137
РЕШАЮЩИЙ ШАГ В
БОРЬБЕ С БОЛЬЮ
Эффективность эквивалентна 20 мг
морфина при снятии болевых ощущений
после хирургических операции.
Предпочтителен морфину у пациентов
после хирургических вмешательств.
Обладает существенно лучшей
переносимостью по сравнению с
наркотическими анальгетиками.
Одинаково хорошо переносится
разными категориями больных.
Показывает эффективность на уровне
максимальных доз стандартных НПВП.
Превосходит напроксен при
обезболивающей терапии
у онкологических больных.
Превосходит трамадол при
послеоперационном обезболивании.
кое окам
Лк^рноксифам
НОВЫЙ НПВП С ОБЕЗБОЛИВА
СИЛОЙ ОПИОИДОВ
NYCOMED
Lornoxicain
и.п
оболочкой
8мг
1