/























































































































































































































































































Автор: Пешкова В.М. Громова М.И.
Теги: спектральные методы анализа оптические методы анализа химия аналитическая химия
Год: 1976




Текст
В. М. Пешкова, М. И. Громова
Методы абсорбционной спектроскопии в аналитической химии
Под редакцией акад. И. П. Алимарина
Допущено Министерством высшего и среднего специального образования СССР
в качестве учебного пособия для студентов химических специальностей университетов
Москва «Высшая школа» 1976
543
П23
УДК 543.4(075)
Рецензенты: проф. К- В. Астахов (Военно-химическая академия) и кафедра аналитической химии Казанского государственного университета (зав. кафедрой проф. В. Ф. Торопова)
ВАЛЕНТИНА МОИСЕЕВНА ПЕШКОВА МАРГАРИТА ИВАНОВНА ГРОМОВА
МЕТОДЫ АБСОРБЦИОННОЙ СПЕКТРОСКОПИИ В АНАЛИТИЧЕСКОЙ ХИМИИ
Редактор Т. П. Федорова. Художественный редактор Т. М. Скворцова. Художник Ю. Д. Федичкин. Технический редактор Н. В. Яшу-кова. Корректор Р. К- Косинова
Сдано в набор 10/XI—75 г. Поди, к печати 28/V—76 г. Формат 60Х90У16. Бум. тип. № 3. Объем 17,5 печ. л. Усл. п. л. 17,5 Уч.-изд. л. 19-Изд. № ХИМ—527. Тираж 10 000 экз. Заказ 518. Цена 83 коп.
План выпуска литературы издательства «Высшая школа» (вузы и техникумы) на 1976 г. Позиция № 71. Москва, К-51, Неглинная ул., д. 29/14, издательство «Высшая школа»
Московская типография № 4 Союзполиграфпрома при Государственном комитете Совета Министров СССР по делам издательств, полиграфии и книжной торговли, Москва, И-41, Б. Переяславская, дом 46.
Пешкова В. М. и Громова М. И.
П23 Методы абсорбционной спектроскопии в аналитической химии. Под ред. И. П. Алимарина. Учеб, пособие для ун-тов. М., «Высш, школа», 1976.
280 с. с ил.
В книге даны теоретические основы метода абсорбционной спектроскопии (спектрофотометрии). Рассмотрены отдельные методы количественного абсорбционио-спектроскопического анализа: спектрофотометрического титрования, многокомпонентной системы, дифференциального, экстракциоино-спектрофотометрического и кинетического. Приведены примеры применения а бсорбционно-спектр ©фотометрического метода для изучения равновесий в растворах и ряд конкретных примеров определения констант диссоциации реагентов, констант устой чивости комплексов и их состава. Уделено большое внимание опреАс леиию отдельных элементов, в том числе редких В предлагаемых методах показано нахождение селективных условий определения элементов в присутствии сопутствующих элементов, близких по свойствам. Даны оптические схемы и принципы действия отдельных спектрофотометрических приборов. Приведены методы статической обработки полученных результатов.
20606—301
П 001(01)— 76
71—76
543
15) ИЗДАТЕЛЬСТВО .ВЫСШАЯ ШКОЛА», 1976 г
ПРЕДИСЛОВИЕ
Методы абсорбционной спектроскопии ввиду их большой чувствительности и избирательности широко применяются при решении многих задач аналитической химии. Эти методы используют при контроле производства и анализе готовой продукции ряда отраслей промышленности: химической, металлургической, металлообрабатывающей, в почвенном, биохимическом анализе, а также для определения малых и ультрамалых количеств примесей в веществах особой чистоты (IO-5—10~6 %). Для определения больших количеств веществ с точностью, не уступающей гравиметрическим и тит-риметрическим методам, а также при анализе многокомпонентных систем применяют различные варианты дифференциальной спектрофотометрии. При автоматизации контроля производства рационально использовать метод спектрофотометрического титрования. Методы абсорбционной спектроскопии остаются труднозаменимыми при анализе объектов, содержащих ядовитые летучие соединения, что делает ограниченным применение атомно-абсорбционного метода и методов эмиссионной спектроскопии. Особенно большое значение имеют методы абсорбционной спектроскопии для исследования процессов комплексообразования и получения количественных характеристик комплексных соединений.
В учебном пособии рассмотрены теоретические основы методов спектрофотометрии в современном аспекте и показаны возможности применения УФ и видимой областей спектра в этих методах. Должное внимание уделено вопросам точности спектрофотометрических методов. На большом числе примеров показана селективность спектрофотометрических методов. Для определения одного какого-либо элемента рекомендовано несколько методов, что дает возможность выбора в зависимости от природы анализируемого объекта и требуемой чувствительности. Для оценки величины поглощения рекомендуется использовать объективный способ, т. е. проводить измерения на различных приборах с той или другой степенью монохроматичности потока излучения.
Практическая часть содержит ряд методов определения элементов с использованием каталитических реакций (кинетических методов).
Для повышения точности определения ультрамалых (микрограммы) количеств вещества и достоверности получаемых результатов рекомендуется использовать градуировочные графики, построенные по
экспериментальным данным, обработанным методом наименьших квадратов, и обрабатывать полученные результаты статистически.
Описаны этапы исследования фотометрической реакции для выбора оптимальных условий ее проведения, а также ряд методов определения состава и констант устойчивости комплексных соединений и даны конкретные примеры их применения.
Авторы приносят глубокую благодарность сотрудникам лаборатории спектрофотометрии кафедры аналитической химии химического факультета МГУ Ю. А. Барбалату, И. Ф. Долмановой, Р. Д Ворониной, Е. К. Ивановой, С. О. Кобяковой, Н. В. Мельчаковой, Р. Г, Опа-совой, В. М. Савостиной, О. А. Шпигуну, а также сотрудникам лаборатории гетерополисоединений С. А. Моросановой и Е. Н. Дороховой за практическую подготовку и помощь в написании ряда работ.
Выражаем большую признательность | В. Г. Горюшиной | и ее сотрудникам по аналитической лаборатории Гиредмета за предоставление методик определения фосфора и мышьяка в ряде объектов.
ВВЕДЕНИЕ
Метод абсорбционной спектроскопии (спектрофотометрии) относится к оптическим методам анализа и основан на взаимодействии вещества с излучениями ультрафиолетовой (УФ), видимой и инфракрасной (ИК) областей электромагнитного спектра, а именно на избирательном поглощении электромагнитного излучения однородными нерассеивающими системами.
Энергетические характеристики электромагнитного излучения. Электромагнитное излучение может быть охарактеризовано следующими параметрами: длиной волны X, частотой v, или волновым числом v, и соответствующей им энергией Е излучения (табл. 1).
Электромагнитый спектр излучения (область оптических спектров)
ТАБЛИЦА I
Спектральная область Длины волн Z, нм Волновые числа V, см-1 Энергия Е, эВ Процессы, протекающие в результате поглощения (или излучения)
Ультрафиолетовая: вакуумная близкая <200 200—400 >5-10* 5-10*—2,5-10* 1 102—10 Электронные переходы
Видимая 400—700 2,5-10*—1,5-10* 10—1 То же
Инфракрасная: близкая фущамен- 700—1,5-10s 1,5-Ю8—7,5-10* 1,5-10*—6,6-10s 6,6-10s—1,3-10- 1 — 10-2 Колебания молекул То же
тальная далекая 7,5-10*—106 1,3-102—10 Вращение молекул
Длина волны есть расстояние между двумя вершинами волны. Основными единицами измерения длин волн служат в УФ и видимой области нанометры* (1 нм = 10-9 м = 10“7 см), в ИК-области — мик
* Ранее применялись единицы измерения длин волн ангстремы (I А = — 10“8 см), микроны (1 мк = 10“* см) и миллимикроны (1 ММК = 10“7 см = = 1 нм). т
рометры (1 мкм = 10s нм). Длина волны зависит от показателя преломления среды, в которой излучение распространяется, так как длина волны непосредственно связана со скоростью распространения волн. Скорость распространения излучения в различных средах различна. Поэтому для характеристики определенного участка спектра часто используют частоты или волновые числа, которые не зависят от рефракции среды.
Частота излучения v есть число колебаний в одну секунду и выражается отношением скорости распространения излучения (скорости света) с к длине волны:
с V==T'
Скорость света и длина волны должны рассматриваться для одной и той же среды, в которой распространяется излучение. Частота измеряется в обратных секундах (с-1) или герцах (Гц) (1 Гц = с-1).
Волновое число v показывает, какое число длин волн приходится на 1 см пути излучения в вакууме и определяется соотношением v — = 1 А, где л — длина волны в вакууме. Размерность волновых чисел см-1. С частотой волновое число связано соотношением v = cv, где с—скорость света в вакууме, равная ~3 • 1010 см/с*. Например, если К = 250 нм, то v = 40 000 см-1 и v = 1200 • 1012-с-1.
Энергия излучения Е непосредственно связана с частотой: Е = hv и выражается в электрон-вольтах (эВ), 1 эВ = 8,066 • 103 см"1.
Набор длин волн (или частот) представляет собой электромагнитный спектр излучения. Деление электромагнитого спектра на ряд областей (см. табл. 1) не является резким и основано главным образом на способах получения и регистрации излучений различных длин волн (или частот) и связано также с использованием различных оптических материалов.
Поглощение электромагнитного излучения однородными системами. Элементарная теория, описывающая процесс поглощения электромагнитного излучения какой-либо системой, состоящей из атомов или молекул, может быть представлена следующим образом [1] — [4].
Внутренняя энергия молекул состоит в основном из энергии вращения молекулы как целого, энергии колебания ядер друг относительно друга и энергии движения электронов, находящихся в электростатическом поле атомных ядер. Поэтому общая энергия молекулы, находящейся на определенном энергетическом уровне, может быть представлена как сумма этих энергий
£ = £эл + АоЛ + £вр-
На рис. 1 приведены энергетические уровни молекулы. Электронный энергетический уровень молекулы, находящейся в обычном, не возбужденном состоянии (£0), называют основным состоянием, а бо-
* В литературе по электронным и колебательным спектрам обычно принято вместо термина «волновое число» использовать термин «частота» с размерностью см-1, что не является правильным.
лее высокие энергетические уровни — соответственно первым (EJ, вторыми (Е2) и т. д. возбужденными состояниями. Каждому электронному уровню соответствует один основной и несколько возбужденных колебательных уровней; аналогично каждому колебательному уровню соответствует один основной и несколько возбужденных вращательных уровней. Описанная схема имеет упрощенный вид и примерно
показывает соотношение энергетических уровней двухатомных молекул. Электронная энергия более сложных молекул характеризуется потенциальной кривой (кривой Морзе) [3] — [5] и зависит прежде всего от расстояния между ядрами.
Если нет воздействия на атомы или молекулы, которое переводит их в возбужденное состояние, то они все оказываются на самом низком, основном уровне, который и является начальным уровнем всех линий поглощения. (Поэтому спектры поглощения, как правило, гораздо более бедны линиями, чем спектры излучения, так как при испускании всякий уровень, кроме самого низкого, может быть начальным.) Если молекула поглощает излучение, то ее энергия повышается и происходит переход с более низкого энергетического уровня на более высокий. Внутренняя энергия молекул не может изменяться непрерывно. Каждая молекула обладает набором дискретных квантованных состояний, которые отличаются друг от друга значениями всех видов энергий (см. рис. 1). Следовательно, для осуществления процесса поглощения необходимо, чтобы энергия излучения была равна разности энергий квантованных состояний молекулы
ДЕ = £!—Ео.
Рис. 1. Схема энергетических уровней двухатомных молекул и электронных переходов:
Е — электронные уровни; v — колебательные уровни; / — вращательные уровни
Возрастание энергии при этом равно энергии поглощенного фотона
he
EE=hv=—,
X
где h — постоянная Планка; v — частота излучения; X — длина волны излучения; с — скорость света.
Як Переход системы от одного энер гетического уровня к дру гому всегда сопряжен с частичной перестройкой электронной системы. Эта перестройка для различных переходов различна и поэтому вероятность переходов неодинакова. Интенсивность поглощения при электронном переходе для любой длины вечны определяется вероятностью перехода и размером молекулы. Для симметричных молекул вероятность пере ко-
да определяется и требованиями симметрии [31, [61 (здесь они не рассматриваются): так, разрешенными дипольными переходами являются только переходы между электронными состояниями, распределение зарядов в которых характеризуется различной симметрией; переход между состояниями с одинаковой симметрией запрещен. Интенсивность запрещенных переходов меньше разрешенных. Однако симметрия распределения зарядов может быть нарушена колебаниями ядер, так что и запрещенные переходы могут характеризоваться заметными интенсивностями [2]. i
Меньше всего энергии требуется для возбуждения вращательных уровней: разность энергий соседних вращательных уровней молекулы имеет порядок 10-2— 10-3 эВ. Следовательно, для возбуждения этих уровней достаточно энергии излучений далекой ИК-области спектра (см. табл. 1). Разность энергий соседних колебательных уровней составляет примерно 0,1—1 эВ и для их возбуждения требуются излучения средней и ближней ИК-областей спектра.
Для возбуждения электронных уровней необходимы излучения УФ-участка спектра, так как разность энергий этих уровней равна примерно 10 эВ. Если электронные уровни молекул расположены достаточно близко друг к другу, то для осуществления перехода между ними достаточно воздействия излучений видимого участка спектра. Таким образом, изменение колебательной энергии сопровождается в большинстве случаев и изменением вращательной. Изменению электронной энергии сопутствует также изменение колебательной и вращательной энергий.
Поэтому при поглощении молекулой ультрафиолетового излучения высокой энергии наблюдаемый спектр поглощения состоит из широких полос, являющихся результатом наложения большого числа узких полос, соответствующих различным переходам между близко расположенными подуровнями. Сложная природа электронных спек-ров многоатомных молекул делает очень трудным их полный анализ даже при использовании приборов высокого разрешения, т. е. высоко монохроматичных потоков излучений. Отсутствие вращательной и вращательно-колебательной структур можно наблюдать в спектрах жидких веществ и растворов, что связано с взаимодействием между соседними молекулами растворенного вещества и влиянием сольватации (большинство химических исследований относится именно к этим условиям). Полярные растворители обусловливают обычно значительно большие изменения в полосах поглощения, чем неполярные. Это объясняется тем, что оптические спектры возникают в результате поглощения или излучения света внешними электронами, наименее прочно связанными с ядром, которые требуют для возбуждения меньше энергии, чем внутренние электроны.
_В спектрах поглощения газообразных веществ наблюдаются или отдельные линии, или широкие полосы с тонкой структурой (наличие отдельных пиков на фоне общей полосы), которая соответствует отдельным переходам.
Так как внешние электроны атомов наиболее подвержены воздействию внешнего окружения (межмолекулярное, взаимодействие, взаи-
Содействие с молекулами растворителя и т. п.), которое сказывается на энергетическом состоянии поглощающей системы, спектры поглощения веществ в растворах практически полностью теряют тонкую структуру, соответствующую отдельным переходам, и имеют вид широких полос.
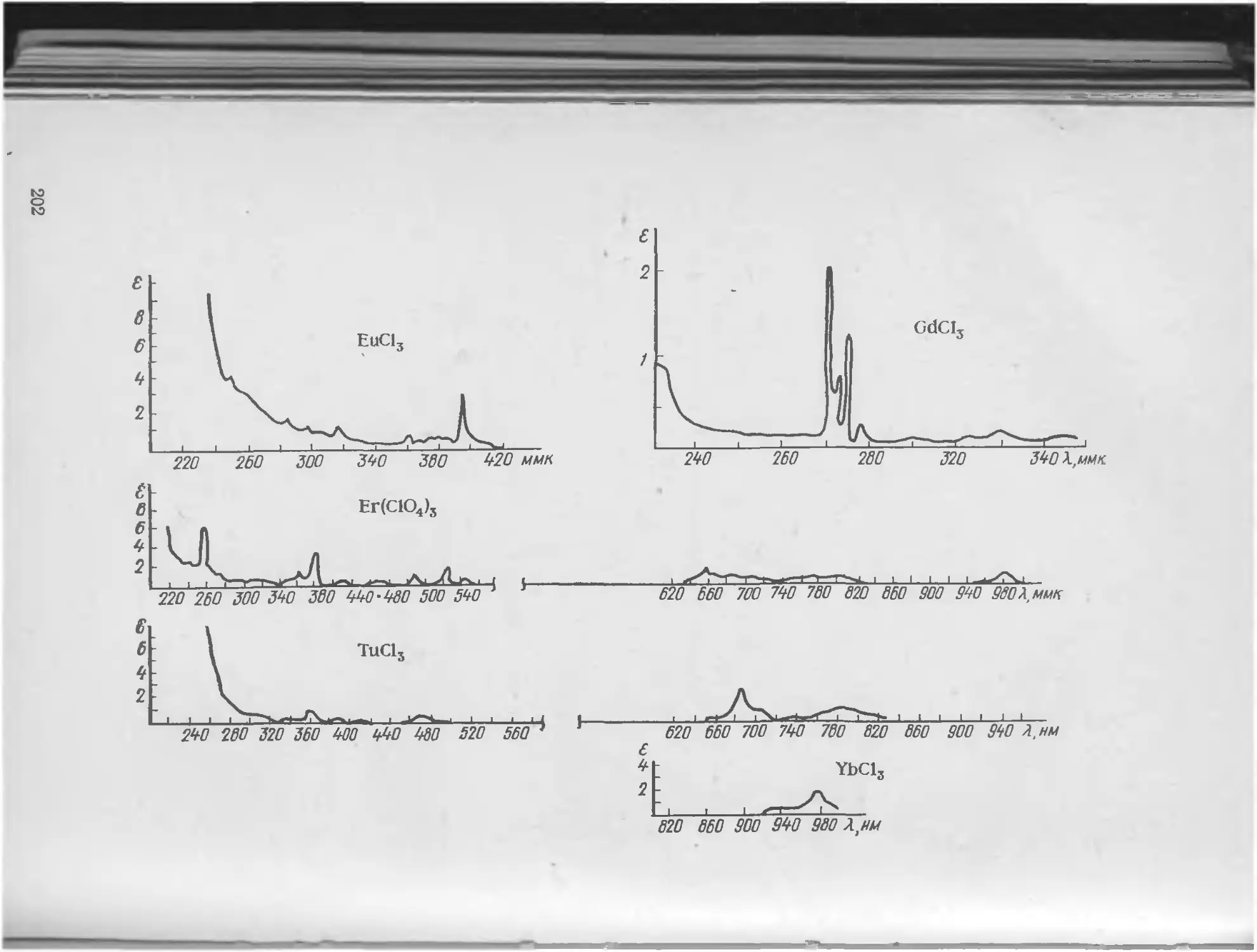
V Особый интерес представляют спектры поглощения растворов солей редкоземельных элементов и актиноидов. Они имеют почти одинаковое электронное строение внешних орбиталей. Поэтому данные элементы очень похожи друг на друга по своему химическому поведению и трудно разделимы. У редкоземельных элементов наименее прочно связанные электроны принадлежат достраивающемуся ^-подуровню, который защищен от внешних воздействий завершенными подуровнями 5s, 5р, 6s. Оптические спектры редкоземельных элементов возникают в результате возбуждения именно этих наименее прочно связанных, но достаточно экранированных электронов. Эти спектры очень сложны и резко отличаются друг от Друга. Они состоят из широких полос, расположенных в УФ-области спектра, и большого числа узких полос, отличающихся малой интенсивностью [1].
Широкие и интенсивные полосы получаются при переходе электрона с внутреннего незавершенного 4/-подуровня на внешние уровни. На этих уровнях электрон находится под сильным и нерегулярным воздействием электронных полей молекул растворителя, что ведет к образованию широкой полосы поглощения. При возникновении узких полос 4/-электрон не покидает своего подуровня, меняется лишь его взаимодействие с другими электронами. Хорошая защищенность 4/-электронов от внешних воздействий приводит к тому, что спектры поглощения растворов солей редкоземельных элементов в некоторой мере сохраняют дискретную структуру, похожую на структуру спектров этих атомов в газообразном состоянии. Различия в строении 4/-подуровней у отдельных редкоземельных элементов обусловливает индивидуальный характер их спектров поглощения.
Строгая квантовомеханическая трактовка спектров поглощения возможна только для самых простейших молекул (в основном двухатомных молекул). Существует ряд методов приближенных квантовомеханических расчетов энергетических уровней многоатомных молекул [7], [8].
Спектр поглощения можно получить, если на пути электромагнитного излучения помещено вещество, не излучающее, но поглощающее лучи определенных длин волн. В видимой части спектра воспринимаемый цвет есть результат избирательного поглощения этим веществом определенного участка сплошного спектра электромагнитного 1 с лучения (белого света). Цвет раствора всегда является дополнительным к цвету поглощенного излучения (табл. 2).
Основной характеристикой спектральной линии или участка спектра является их положение в спектре. Оно определяется длиной волны или частотой, При одноэлектронном переходе полоса поглощения характеризуется тремя основными параметрами: максимальным значением коэффициента погашения ешах, частотой v, соответствующей етят, и эффективной шириной полосы 2а (рис. 2). Чем больше значе-
Таблица 2
Наблюдаемые цвета и соответствующие им поглощенные участки спектра
Интервал длин волн поглощенного излучения X, нм Цвет поглощенного излучения Наблюдаемый цвет (дополнительный цвет) Интервал длин волн поглощенного излучения X, нм Цвет поглощенно го излучения Наблюдаемый цвет (дополнительный цвет)
380—420 Фиолетовый Желто- 520—550 Желто- Фиолетовый
зеленый зеленый
420—440 Синий Желтый 550—580 Желтый Синий
440—470 Голубой Оранжевый 580—620 Оранжевый Голубой
470—500 Голубовато- Красный 620—680 Красный Голубовато-
зеленый зеленый
500—520 Зеленый Пурпурный 680—780 Пурпурный Зеленый
ние 2сг, тем труднее анализировать смесь соединений из-за наложения полос поглощения, соответствующих отдельным компонентам смеси. Зависимость е от v выражается уравнением кривой распределения Гаусса:
где а—emax; b — vmax; о—полуширина полосы на высоте 1/2 а. Спектры поглощения таких многоэлектронных систем, как комплексные соединения, имеют сложный характер и для их расшифровки не
обходимо проводить разложение на гауссовы составляющие [6]. Чем дальше в коротковолновой области спектра лежит линия поглощения, тем на более высокий энергетический уровень переходит молекула при поглощении. Серия линий поглощения заканчивается в далекой ультрафиолетовой области сплошным поглощением. Эти участки соответствуют ионизации молекул в результате поглощения излучений больших энергий.
В практике абсорбционно-спектроскопических (спектрофотометрических) методов используются
Рис. 2. Параметры, характеризующие полосу поглощения в спектре:
а — максимальное значение е; b — волновое
число (V), соответствующее Emax; о — полуширина полосы поглощения на высоте а/2
только такие участки спектра, в которых процесс поглощения не сопровождается нарушением целостности молекул (т. е. ближняя УФ, видимая и ИК-области спектра). Это позволяет наряду с решением задач количественного анализа использовать этот метод также для изучения строения соединений и химических равновесий.
В результате протекания процессов, вызывающих изменение строения поглощающих частиц или смещение равновесий в растворах, меняются также спектральные свойства поглощающих систем. Эти из
менения в спектрах поглощения характеризуются гиперхромным аффектом— увеличением поглощения, гипохромным эффектом—уменьшением поглощения, батохромным сдвигом — смещением максимума поглощения в сторону больших длин волн, гипсохромным сдвигом — смещением максимума поглощения в сторону коротких длин волн.
Смещение полосы поглощения от фиолетового конца видимого спектра к красному (т. е. в сторону больших длин волн) дает такую последовательность воспринимаемых цветов: желтый-> оранжевый-> красный -> фиолетовый -> синий -> сине-зеленый. Смещение полосы в обратную сторону приводит к обратному изменению наблюдаемых цветов.
В настоящее время для сложных молекул предсказать точное положение максимума, а тем более оценить степень их поглощения, весьма затруднительно. Поэтому обычно экспериментальные данные о спектрах поглощения используют для выводов о строении соединений. Вид спектра поглощения обусловлен в первую очередь состоянием электронов внешних орбиталей, участвующих в образовании химической связи. Так, например, спектральные свойства органических молекул обычно систематизируют в соответствии с типом содержащихся в них следующих валентных электронов: электроны, образующие одинарную связь, называются о-электронами; образующие двойную связь — л-электронами. Кроме того, в молекулах, содержащих атомы таких элементов, как азот, кислород и т. п., имеется свободная пара электронов, или n-электронов. Различным типам электронов свойственны различные электронные переходы, обусловливающие возникновение спектров с характерными полосами поглощения в том или ином его участке. Кроме того, воздействие окружающей среды, например растворителя, вызывает различные изменения в спектре также в зависимости от типа присутствующих валентных электронов [2], [5].
Избирательное поглощение в определенной области спектра связано с наличием в молекуле определенных групп атомов. Они содержат одну или несколько кратных связей или неподеленные пары электронов. Такие группы, определяющие окраску веществ, если поглощение происходит в видимой области спектра, называют хромофорами. В табл. 3 приведены некоторые хромофоры и указаны длины волн, соответствующие положению характерных для них максимумов поглощения. Присутствие той же группы атомов в молекуле, иногда принадлежащей к совсем другому классу соединений, сопровождается появлением того же, характерного для нее поглощения, лишь в некоторой мере искаженного иным окружением этой группы. Иногда в молекуле рядом с-хромофором находится активная группа атомов, которая сама по себе не определяет поглощения, но может усиливать интенсивность поглощения, смещать максимум поглощения в длинноволновую область спектра. Такие группы (— NH2, — N(CH3)2, — ОН, —ОСН3) называются ауксохромами.
Растворы солей неорганических соединений элементов первых периодов системы Д. И. Менделеева в большинстве своем не обладают
ТАБЛИЦА 3
Характеристика некоторых хромофоров
Хромофорная группа Формула 1 Соединение Растворитель ^тах’ нм е
Карбонильная >с=о Ацетон Спирт 270,0 15,8
» >с=о Ацетальдегид » 293,4 11,8
Карбоксиль- —соон Уксусная кислота Вода 204 60
на я Этиленовая —с=с— Этилен (пары) » 193 10-Ю3
Азометиновая >C=N— Ацетоксим » 190 5 103
Нитрозо- —N=O Нитрозо-бутан (зеленый) Октилнитрит Эфир 665 20
Нитритная —ONO Гексан 230 2,2-103
Бензол N Бензол » 255 230
Нитратная —ono2 Этилнитрат Диоксан 270 12
характерным поглощением в УФ-, видимой и ИК-областях спектра. Точнее, наблюдается в этих областях только поглощение некоторых анионов, но катионы (вернее их аква-комплексы) обычно «бесцветны». Это зависит оттого, что данные катионы имеют, как правило, устойчивую замкнутую оболочку, образовавшуюся после того, как атом, переходя в катион, потерял все наименее прочно связанные электроны, и для возбуждения оставшихся электронов недостаточно даже энергии ультрафиолетового излучения. Электронная система анионов, менее стабильна. Так, например, отрицательно заряженные ионы галогенов, содержащие один лишний электрон, возбуждаются меньшими квантами излучений и полосы поглощения их попадают в доступную УФ-область спектра.
Окраска аква-комплексов катионов, имеющих незаполненные //-орбитали, обусловлена возможностью легких переходов между энергетически близкими //-орбиталями. Поэтому растворы солей, например, меди и никеля, окрашены, а цинка и кадмия бесцветны, поскольку их ионы имеют заполненный //-подуровень (табл. 4) [9] — [11].
ТАБЛИЦА 4
Заполнение d-орбиталей и окраска аква-ионов
• некоторых элементов в водном растворе
Ион Число электронов на d-орбитали Число неспаренных электронов Цвет Иои Число электронов на d-орбитали Число неспарен-иых электронов Цвет
Со2+ 7 3 Розовый Мп2+ 5 2 Розовый
Си2+ 9 1 Синий Zn2+ 10 —. Бесцветный
Ni2+ 8 2 Зеленый Cd2+ 10 — »
Появление окраски у переходных металлов объясняется расщеплением энергетических уровней центрального иона комплексообразо-вателя под влиянием поля лигандов £121.
Вообще вопрос о поглощении электромагнитных излучений комплексными соединениями представляет большую сложность, поскольку не всегда может быть достаточно четко определена роль, которую играют центральный ион и лиганд в возникновении того или иного вида спектра [13] — [16].
ЛИТЕРАТУРА
1. В. М. Чулаиовский. Введение в молекулярный спектральный анализ. М., Техтеориздат, 1951.
2. Ч. Н. Р. Ра о. Электронные спектры в химии. М., «Мир», 1964.
3. Р. Драго. Физические методы в неорганической химии. М., «Мир», 1967.
4. Дж. Гаррисон, Р. Лорд, Дж. Луфбуров. Практическая спектроскопия. М., ИЛ, 1950.
5. О. В. Свердлова. Электронные спектры в органической химии. М.—Л., «Химия», 1973.
6. Г. Джаффе, М. О р ч и н . Симметрия в химии. М., «Мир», 1967.
7. М. В. Базилевский. Метод молекулярных орбит и реакционная способность органических молекул. М.—Л., «Химия», 1969.
8. Р. Мак-Вини, Б. Сатклиф. Квантовая механика молекул. М., «Мир», 1972.
9. Т. Данн. Спектры поглощения комплексных соединений в видимой и ультрафиолетовой областях. Сб. статей «Современная химия координационных соединений». Под ред. Дж. Льюиса и Р. Уилкинса. М., ИЛ, 1963.
10. К. Дей, Д. С е л б и н. Теоретическая неорганическая химия. М.—Л., «Химия», 1969.
11. Л. О р г е л. Введение в химию переходных металлов. М., «Мир», 1964.
12. К. Бальхаузен. Введение в теорию поля лигандов. М., «Мир», 1964.
13. М. Сиенко, Р. П л е й н, Р. Хестер. Структурная неорганическая химия. М., «Мир», 1968.
14. А. К. Бабко, А. Т. Пилипенко. Фотометрический анализ. М.—Л., «Химия», 1968.
15. Л. А. Грибов, С. Б. Саввин, Э. Л. Кузин. ЖАХ, 1967, 22, 1799; ЖАХ, 1968, 23, 490.
16. С. Б. Саввин, Л. А. Грибов, В. Л. Лебедев. ЖАХ, 1971, 26, 2108.
Глава I
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДОВ
АБСОРБЦИОННОЙ СПЕКТРОСКОПИИ
1. ОСНОВНЫЕ ЗАКОНЫ ПОГЛОЩЕНИЯ ЭЛЕКТРОМАГНИТНОГО ИЗЛУЧЕНИЯ
Монохроматический поток электромагнитного излучения, падая на объект, частично поглощается, отражается и проходит через однородный слой вещества (рис. 3). Интенсивность первоначального монохроматического потока излучения после прохождения через кювету с поглощающим раствором можно представить как сумму интенсивностей излучений:
I^k+Ia+lr,
где /0 — интенсивность первоначального монохроматического излучения, падающего на объект; Ц — интенсивность монохроматического излучения, 'прошедшего через объект; 1а — интенсивность поглощенного объектом монохроматического излучения; /<Г— интенсив
ность монохроматического излучения, отраженного стенками кюветы и растворителем. Величина 1Т слагается из двух величин: 1Г1 — ин-
Рис. 3. Изменение интенсивности потока излучения при прохождении через «окрашенный» раствор
тенсивности монохроматического излучения, отраженного стенками сосуда, и 1Га — отраженного растворителем.
Интенсивность потока электромагнитного излучения, прошедшего через исследуемый раствор, всегда измеряют относительно раствора сравнения. Таким образом, величина 1Т может быть в целом исключена.
Зависимость между ослаблением интенсивности направ
ленного параллельно монохроматического потока электромагнитного излучения и толщиной поглощающего слоя, установленная Бугером в 1729 г. и подтвержденная Ламбертом в 1760 г., составляет сущность первого закона поглощения И!, [21:
относительное количество поглощенного пропускающей средой излучения не зависит от интенсивности падающего излучения. Каждый слой равной толщины поглощает равную долю падающего монохроматического потока излучения. Поглощающая способность однородного
вещества не зависит от интенсивности падающего излучения при изменении его энергии в широком пределе [3].
Представим, что слой вещества I состоит из бесконечно тонких слоев dl, и в этот тонкий слой поступает поток монохроматического электромагнитного излучения с длиной волны к. После прохождения через поглощающий слой с толщиной dl интенсивность потока излучения уменьшается в результате поглощения на величину dl
—dlldl=al. (LI)
или
dl[l=—adl, (1.2)
где a — коэффициент поглощения среды. Следовательно, наблюдается относительное ослабление потока излучения при прохождении его через каждый тонкий слой однородного раствора вещества. Интегрируя уравнение (1.2) от /0 до Ц по всей толщине слоя, получим
Ъ л, 1
С dl г „
J—=— a]dl> U-3>
'о °
1г. -1п/0=— al, (1.4)
или в экспоненциальной форме
h=Ioe~at. (1.5)
При переходе к десятичным логарифмам, уравнение (1.5) принимает вид
/z=Zo-IO-ftZ, (1.6)
а = 2,3025й, /г = 0,4303а,
где k — коэффициент поглощения — величина, обратная толщине слоя, которая необходима для ослабления в десять раз интенсивности первоначального падающего излучения. Если k = 1/ь то Л//о = Х4о.
Второй закон поглощения электромагнитного излучения установлен Бером в 1852 г. [4], [5] и выражает связь между интенсивностью монохроматического потока и концентрацией вещества в поглощающем растворе: поглощение потока электромагнитного излучения прямо пропорционально числу частиц поглощаюи^го вещества, через которое проходит поток этого излучения.
Следовательно, k = ес, где е — коэффициент пропорциональности. Объединенный закон Бугера — Ламберта — Бера выражается уравнением: ---------
' I (J.7)
I_______ )
ИЛИ ------------
'g~ = elc. (1.8)
Величину 1g ^2 называют оптической плотностью поглощающего вещества и обозначают буквой А:
A = lg-^-=sfc. С1-9)
Отношение интенсивности монохромического потока излучения, прошедшего через исследуемый объект, к интенсивности первоначаль-
Рис. 4. Зависимость оптической плотности и пропускания от длины волны
Рис. 5. Зависимость оптической плотности от концентрации вещества в растворе.
1 — закон соблюдается; 2 — закон не со- * блюдаетс"
кого потока излучения называется прозрачностью, или пропусканием раствора, и обозначается буквой Т:
Т = — = \0~ъ1с. (1.10)
Величина Т, отнесенная к толщине слоя 1 см, называется коэффи-циентом пропускания. Оптическая плотность А и пропускание (прозрачность) Т связаны уравнением
А= — IgT, (1.11)
ИЛИ
Т= 10~л=е_ 2>зЛ. (1.12)
Обычно Т выражают в процентах, тогда
A-lg-^-100 = 2—lg7. (1.13)
Величины А и Т зависят от длины волны и концентрации вещества в растворе (рис. 4 и 5). При подчинении растворов закону поглощения наблюдается прямолинейная зависимость оптической плотности от концентрации вещества в растворе при постоянном значении I (см. рис. 5). Эта пропорциональность строго соблюдается только для монохроматических излучений.
Линейная зависимость между величинами, характеризующими процесс поглощения излучения (Л, Ig Т), и концентрацией вещества в растворе или толщиной поглощающего слоя может быть получена только при постоянном значении коэффициента погашения е. Как видно из уравнения (1.9), е не зависит от с и I и характеризует степень
Рис. 6. Зависимость оптической плотности от длины волны для ряда растворов одного и того же соединения различной концентрации (С!>С2>Сз>С4>Сб)
поглощения электромагнитного излучения каким-либо веществом. Степень поглощения неодинакова при различных длинах волн и зависимость величины, характеризующей поглощение (Л, е, 1gЛ, 1gе), от длины волны (X), частоты (v) или волнового числа (v) можно изобразить спектральной кривой поглощения. Не существует единой системы построения кривых спектров поглощения.
Рабочие графики для выяснения влияния различных факторов на спектр поглощения обычно вычерчивают в координатах Л — X. Спектры поглощения в координатах Л — X имеют одну и ту же форму независимо от толщины слоя раствора или концентрации вещества в растворе (рис. 6) и характеризуются сохранением положения максимума при одной и той же длине волны.
Спектры поглощения более оправданно представлять в координатах е—X, е—V. Такого рода спектры Йе требуют дополнительных данных о концентрации с и толщине I погло
щающего раствора. При построении спектра поглощения в координа-
тах е—v полосы поглощения оказываются более симметричными.
Закон аддитивности — важное дополнение к закону Бугера — Ламберта — Бера. Сущностью закона аддитивности является независимость поглощения индивидуального вещества от наличия других веществ, обладающих собственным поглощением, или индиферентных к электромагнитному излучению. Таким образом, при данной длине волны оптическая плотность смеси компонентов, не взаимодействующих между собой, равна сумме оптических плотностей отдельных компонентов при той же длине волны:
Д=2е/ lct.
(I-14)
Уравнение закона аддитивности положено в основу метода анализа многокомпонентных систем.
В методах абсорбционной спектроскопии (спектрофотометрии) не измеряют абсолютных значений 10 и It.
Для оценки степени поглощения исследуемого раствора, содержащего какое-либо соединение, проводят сравнение интенсивности потока излучения, проходящего сквозь этот раствор с интенсчаностью по
Тока излучения, проходящего сквозь раствор, Поглощение которого принимают равным нулю — раствор сравнения. Под исследуемым подразумевают любой раствор, поглощение которого измеряют относительно раствора сравнения. Раствор, содержащий неизвестную концентрацию элемента, называют испытуемым. Для определения концентрации элемента в этом растворе используют чаще всего градуировочный график в координатах А —с. Для построения градуировочного графика готовят ряд эталонных растворов, содержащих известное количество определяемого элемента, в тех же условиях, в которых приготовлен испытуемый раствор. Необходимую концентрацию определяемого элемента в эталонных растворах создают добавлением стандартного раствора с точно известной концентрацией этого элемента. Конечные объемы всех эталонных и испытуемого растворов должны быть строго одинаковы, что необходимо для сравнения величин поглощения этих растворов. Растворы готовят в мерных колбах или в градуированных пробирках одинаковой емкости.
Стандартные растворы готовят растворением соли определяемого элемента точного состава. Если отсутствует такая соль, то готовят раствор большей концентрации, чем необходимая, и определяют его концентрацию каким-либо надежным методом (гравиметрически или титриметрически). После этого соответствующим разбавлением готовят раствор нужной концентрации. Стандартные растворы делят на исходные и рабочие. Для приготовления исходных растворов используют в некоторых случаях достаточно большую концентрацию кислоты для предотвращения гидролиза, чтобы создать условия их длительного хранения. Рабочие растворы с концентрацией 1 — 10 мкг/мл обычно готовят соответствующим разбавлением (~10 — 100-кратным) исходного раствора в день употребления, используя мерные колбы и пипетки.
Объем стандартного раствора, используемого для приготовления эталонных растворов, следует отмеривать с максимальной точностью градуированной пип еткой или бюреткой).
2. МОЛЯРНЫЙ КОЭФФИЦИЕНТ ПОГАШЕНИЯ
Основной характеристикой поглощения любой системы при данной длине волны является коэффициент погашения е. Поскольку поглощение при различных длинах волн различно, то е меняется с изменением длины волны. Если кривая зависимости е от л имеет максимум ПРИ ^-тах (рис. 7), то на кривой зависимости А от 7 также будет максимум при imai, а на кривой зависимости Т от К (см. рис. 4) при этой длине волны будет наблюдаться минимум.
Р Коэффициент погашения е называют молярным коэффициентом погашения, если концентрация вещества выражена в моль/л, а толщина слоя — в сантиметрах. Молярный коэффициент погашения представляет собой оптическую плотность 1 М. раствора, помещенного в кювету с толщиной поглощающего слоя 1 см: при с = 1 моль/л и I — 1 см в =; Л и имеет размерность [см. уравнение (1.9)1 сма/моль.
Рис. 7. Зависимость е от X при соблюдении (cj—с4) и не соблюдении (сь сз> ci) законов поглощения излучений
так как А—безразмерная величина. Однако принято приводить значения молярного коэффициента погашения без обозначения его размерности. Значение молярного коэффициента погашения характеризует два существенно важных свойства поглощающей системы.
Во-первых, постоянство значения е говорит о соблюдении закона поглощения в определенном интервале концентраций.
Во-вторых, значение молярного коэффициента погашения удобно использовать для сравнительной оценки чувствительности фотометрической реакции. Как будет показано в дальнейшем, значения А, измеряемые с достаточной точностью, находятся в пределах от ~0,1 до 1,0. Поэтому минимальная концентрация, которая обусловливает оптическую плотность, соответствующую этому интервалу, зависит от величины молярного коэффициента погашения, как следует из уравнения (1.9) : чем больше е, тем меньшую концентрацию должен иметь раствор, чтобы оптическая плотность соответствовала указанному интервалу (при постоянном Z). Таким образом, повышение чувствительности может быть достигнуто в первую очередь выбором таких реакций, в результате которых образуются соединения, обладающие большими значениями е.
Сравнивая е в максимумах погло-щенйя растворов двух различных
комплексных соединений одного и того же элемента, можно сделать вывод о том, использование какого из' этих соединений дает возможность определить меньшие концентрации данного элемента. Обычно значения е для реакций, используемых в спектрофотометрии, имеют порядок 102— 10Б. Теоретически максимально возможное значение е может быть оценено из следующих соображений [7].
Интенсивность поглощения при электронном переходе для любой длины волны определяется вероятностью этого перехода и размером поглощающей молекулы. Максимум полосы поглощения соответствует наиболее вероятному электронному переходу в данной области длин волн. Исходя из этого, молярный коэффициент погашения может быть рассчитан по формуле
c=kPa, (1.15)
где k— константа порядка 10го; Р — вероятность перехода; а — площадь поперечного сечения молекулы. Если принять вероятность электронного перехода за- единицу, то максимальное теоретическое значение коэффициента погашения средней молекулы (а ~ 10 А2) должно бьпь порядка 105. Действительно, наибольшие экспериментально найденные значения 8 имеют порядок 105.
В практической работе всегда необходимо иметь надежный способ расчета значения е для данного случая. Измерение А = е невозможно, так как оптические плотности 1 М растворов намного превышают те их значения, которые доступны измерению. Наиболее просто получить представление о значении е путем расчета по формуле
< А''
(116) '
где А—'оптическая плотность раствора с концентрацией «1 моль/л. Однако расчет по формуле (1.16) не всегда дает представление об истинном значении е, т. е. такой величине, которая характеризует поглощение излучения с длиной волны Z, одним молем вещества. Чаще всего по формуле (1.16) вычисляют лишь среднюю величину е. Это происходит потому, что для расчета истинного значения е?- по формуле (1.16) необходимо выполнение ряда условий: 1) излучение, падающее на исследуемый объект, должно быть строго монохроматичным; 2) при данной длине волны должен поглощать только один тип частиц; 3) истинная концентрация данных поглощающих частиц в растворе должна быть известна; 4) данный индивидуальный тип частиц не должен взаимодействовать ни с какими посторонними компонентами раствора, в том числе и с растворителем; 5) на поглощение данных частиц не должна влиять ионная сила раствора.
Каждое их этих условий практически трудно выполнимо. На опыте всегда имеют дело с каким-то определенным интервалом длин волн, даже если используется спектральная аппаратура высокого разрешения. Поэтому получают значение А, соответствующее суммарному поглощению в том участке спектра, который охватывает этот интервал длин волн, а следовательно, при расчете е получается также средняя величина.
Часто при данной длине волны поглощают несколько компонентов, в том числе и используемый реагент. Поэтому не выполняется второе условие.
Если имеется область спектра, где поглощает только комплекс и не поглощают остальные компоненты реакции, для получения истинной величины е необходимо, чтобы реакция практически протекала до конца, тогда исходная концентрация иона-комплексообразователя фактически будет равна концентрации комплекса. Это выполнимо только при условии образования устойчивых комплексных соединений (1g Куст > 15).
Поскольку при расчете е по формуле (1.16) используют значение с, соответствующее общему содержанию вещества во всех формах, и число поглощающих частиц определенного типа, которое моЖ^т меняться в результате смещения химического равновесия, обычно неизвестно, то трудно выполнить третье условие. Например, если известно исходное количество данного элемента, взятое для получения раствора комплексного соединения по реакции с каким-либо реагентом, то обычно бывает неизвестна доля этого иона, перешедшая в комплекс, поглощение которого измеряется.
Таким образом, расчет е по формуле (1.16) обычно не дает представления о точной величине ех как энергетической характеристике поглощающих свойств системы при данной длине волны. Величина е, близкая к истинной, может быть получена в определенных условиях: образование очень устойчивых комплексов, поглощающих в области, где отсутствует поглощение реактива, или измерение поглощения растворов, приготовленных растворением точной навески изучаемого соединения в неполярном растворителе, где диссоциация комплекса ничтожно мала.
Идеальное соблюдение закона поглощения возможно лишь при отсутствии какого-либо взаимодействия между поглощающими частицами в растворе, т. е. при ионной силе р = 0 и отсутствии каких-либо побочных химических процессов.
Под влиянием изменения ионной силы раствора меняется энергетическое состояние поглощающих частиц и, следовательно, их способность к поглощению излучений различных длин волн. При изменении концентрации реагирующих веществ в широких пределах, а также при изменении концентрации посторонних веществ в растворе (р 0) наблюдаются отклонения от законов поглощения и средний молярный коэффициент погашения не сохраняется постоянным. Иногда значение среднего молярного коэффициента погашения сохраняет постоянство в определенном интервале концентраций, т. е. зависимость А = f (с) остается прямолинейной, что очень существенно при количественном анализе. При этом постоянство е для растворов с различной концентрацией не говорит о том, что получено истинное значение молярного коэффициента погашения. В данном случае лишь соблюдается соотношение А,/А к = с,7ск; если сг = пс2, то Аг = пА2, т. е. доля исходной концентрации определяемого иона, переходящего в комплекс, всегда постоянна, нет побочных реакций и поэтому оптическая плотность пропорциональна концентрации. Совпадение кривых зависимости е от X для растворов с различными концентрациями (см. рис. 7) также не означает, что данные величины е являются истинными молярными коэффициентами погашения какого-либо компонента, а показывает, что спектрофотометрически невозможно исследовать данную систему.
С другой стороны, непостоянство средней величины молярного коэффициента погашения служит основой спектрофотометрических методов изучения состояния веществ и равновесий в растворах. Если при изменении концентраций реагирующих веществ изменяется средний молярный коэффициент погашения, то это указывает на возможность возникновения побочных процессов (изменение степени диссоциации комплекса, полимеризацию, ступенчатое образование комплексов и др.). Это позволяет спектрофотометрически исследовать состояние веществ в растворах. В дальнейшем будут даны некоторые приемы расчета истинных молярных коэффициентов погашения, которые необходимы для вычисления равновесных концентраций при получении количественных характеристик процессов комплексообразования.
3. ВЫЧИСЛЕНИЕ ИСТИННЫХ ЗНАЧЕНИЙ МОЛЯРНЫХ КОЭФФИЦИЕНТОВ ПОГАШЕНИЯ
Величина молярного коэффициента погашения, вычисленная по формуле (1.16), обычно не соответствует истинному его значению. Однако используя высоко монохроматизированные потоки излучений, проводя все опыты при постоянной ионной силе (р = const), исключив поглощение посторонних компонентов и найдя способ расчета равновесной концентрации поглощающего компонента, можно вычислить молярный коэффициент погашения при какой-то определенной длине волны. Это значение можно считать истинным. Рассмотрим несколько примеров таких расчетов.
1. Метод, предложенный Н. П. Кокарем [18], требует предварительного определения состава комплексного соединения и выяснения уравнения реакции его образования. Пусть реакция протекает по уравнению
Mn+ +9Н+ (а)
Для всех опытов, которые отличаются исходными концентрациями реагирующих компонентов, соблюдаются следующие условия: а) начальные концентрациии реагирующих компонентов cHR и см* должны сохранять постоянное соотношение, т. е. cHR = qc!A; б) величины Т, р, pH, I и Z должны быть постоянны.
Рассмотрим случай, когда «окрашенными» (т. е. обладают поглощением в исследуемой области длин волн) являются используемый реагент и образующийся комплекс. Введем обозначения:
см = с; [HR]=9c—qx — q(c —х)
[Н]=ЛЖ (используется буферный раствор);
cHR=9c; еня и емя —молярные коэффициенты погашения , 9 реагента и комплекса соответственно;
[MRg] = r;
[М] = с—х.
Для опыта i по закону действия масс
д-________xi^4_____' п .н,
(с£—л£) [9 (с£—х,]9 ’ ( °
Откуда
а« д-
Xi= (Ci“Xz)’+1- (L18)
Согласно основному закону поглощения излучений
А• = ? (Ci-xt) 4r l+x. eMRg I, (1.19)
откуда
А,~'?eHR cl
Для простоты записи заряды ионов опущены.
II
Объединяя уравнения (1.18) и (1.20), получим
Ai~‘3eHRcil I Я \ч „ ~Г7 7 — I ~7"
1 (®MR “ ?eHR) К /
Ci s№Rql~Ai
‘(EMR^~9ehr)
(1-21)
Разделив уравнение (1.21) на аналогичное уравнение для опыта k, получим:
1 ci l^Rq-Ai _ (Л.-?енк Chl^~Ah (4-?«hr^)^
Правую часть уравнения (1.22), в которую входят величины, известные или определяемые экспериментально, обозначим через В. Исходные концентрации комплексообразователя в опытах i и k можно взять в соотношении ct — nch. Тогда, решая уравнение (1.22)
относительно емяд, получим
n(Ai—BAh) ZKRq~ la (П-В)
(1-23)
Вычисленное значение bMr9 позволяет рассчитать по уравнению (1.17), если предварительно определен bHr- Следовательно, по уравнению (1.17) можно рассчитать кон-
станту равновесия реакции образования комплексного соединения.
2. В методе, предложенном Юнг-пен-Тонгом и Кингом [9], при изучении полимеризации в растворах хрома (VI), рассматривается равновесие
2HCrOf # Сг2О? - + Н2О
Исследование проводят в таком интервале pH, при котором в заметных количествах содержатся только ионы HCrQf и Cr20f_. Поэтому константа равновесия
[Сг2ОГ1
[НСгО4-р
2,4-
1 .1 , 1------. , ! ,.| , I
-V -3,7 -3,3 Lgc
Рис. 8. Зависимость 1g в от 1g с для системы НСгО^—Сг2О при различ-
ных длинах волн
(1.24)
зависит от с и не зависит от [Н+]. Для этой системы в области длин волн 380—400 нм закон поглощения не соблюдается. Это видно из кривых зависимости 1g е от 1g с (рис. 8). Удобнее всего проводить исследование в области 390—400 нм. Для среднего молярного коэффициента погашения можно записать уравнение
(1.25)
где»! молярный коэффициент погашения HCrO^; sa — молярный коэффициент погашения Сг2О^~; у —. доля Сг (VI) в димерной форме.
8=е1(1—i/) + eai/,
Соотношение между К, с и у выражается уравнением
у
п-26)
Таким образом, имеем два уравнения'с тремя неизвестными: К, еь е2, поэтому однозначно решить их невозможно. Для решения этой системы используют метод последовательных приближений с предварительным предположением одной из неизвестных величин. Предположение делается относительно величины Д’, которой задают несколько значений и на основании этого вычисляют у. Для удобства расчетов вычисляют таблицу функций
у
в интервале значений от 0,600 до 0,001. Эта таблица позволяет получить путем интерполяции значение у, соответствующее любому F (у), и, следовательно, величину 2 Кс.
Уравнения для е, число которых определяется числом растворов с различной концентрацией, решаются по методу усреднений. Мерой точности в определении величин К, еъ еа является среднее процентное отклонение вычисленной по уравнению (1.25) величины е от наблюдаемой е в опыте.
3. В методе, предложенном Циленом и Конником [10], для вычисления молярного коэффициента погашения комплекса циркония с тено-илтрифторацетоном (ТТА) используют константу распределения этого комплекса между двумя растворителями. Мак Ви [11] установил, что цирконий образует с ТТА комплекс в соотношении 1 : 1 (если отсутствуют гидролиз и полимеризация) по уравнению реакции
Zr4 + +HR;±ZrR3++H+
Комплекс имеет максимум поглощения при X 336 нм. Для вычисления молярного коэффициента погашения данного комплексного соединения при этой длине волны Цилен и Конник использовали две серии растворов:
1. Водные растворы. Серия состоит из нескольких (например, шести) пар растворов и в каждой паре концентрация ТТА одинакова, но цирконий содержит только один из растворов.
2. Водно-бензольные растворы. Двухфазная система состоит из таких же пар растворов, как и водная серия, но водная фаза во всех растворах приведена в равновесие с равным объемом бензольной фазы.
Предполагается, что поглощает только ТТА и комплекс ZrR3+. Тогда разность оптических плотностей двух растворов одной и той же пары водной серии будет равна:
A^=i(W+~eHR)[ZrR?+1’ <L28>
так как концентрация циркония намного больше концентрации реактива (ТТА) и, следовательно, [ZrR8+] = IHR]HCXOH. Уравнение (1.28) может быть написано для каждой пары растворов
Кроме того, для всех шести пар растворов отношение АД при X 366 нм к АД при любой другой длине волны Z будет величиной постоянной:
366 366
а^= (129)
АЛ1 eZrRs+~eHR
Выражение для константы распределения Kd реагента HR может быть записано
к IHR1c8h.
D IHR1h2o '
Так как [Zr4-t 1 » [ТТА], то в водной фазе
^2 = eHR^CHRH!o И ^2 = eZrR3+ ^cZrRs+,
(1.30)
НО
[Н*1Исх=[ВДЧо + [НК]с,н.
Учитывая зависимость (1.30), можно записать
[ВД1ИСХ=[ВД1Нзо+ [HR]H2o Kd=[HR1h8O (1+KD),
откуда
run]______[Hgkcx,
ihRJh2o— (1+kd) ’
но HRHCX = [ZrR3+], следовательно,
ДЛ2 — A2 — A 2 = I и для водной фазы имеем
(*д+1)4к3+-енк дл£ (^o+I)ezrR®+— eHR
(1.+ KD) eZrRs+ — eHR
(1+KD)
[ZrRs+]
(1.31)
Совместным решением уравнений (1.29) и (1.31) получим e|®R3+. Таким образом, для вычисления е по данному методу нужно знать коэффициент распределения реагента в системе бензол — вода, состав комплекса и уравнение реакции его образования.
Для применения этого метода необходим большой избыток ком-плексообразователя, чтобы весь реагент перевести в комплекс. Комплекс должен нести заряд и не переходить в слой органического растворителя. Метод непригоден для малоустойчивых комплексов.
Приведенные примеры показывают большое разнообразие в подходе к вычислению истинных значений молярных коэффициентов погашения. Это объясняется различиями в свойствах исследуемых сис
J
тем и в первую очередь особенностями их спектральных характеристик. Поэтому невозможно дать какой-то один универсальный способ расчета молярных коэффициентов погашения, пригодных для любых систем*.
4. ОТКЛОНЕНИЯ ОТ ЗАКОНА БУГЕРА — ЛАМБЕРТА — БЕРА
Отклонения от линейной зависимости оптической плотности от толщины поглощающего слоя и концентрации объясняются, с одной стороны, недостаточной монохроматичностью потока электромагнитного излучения, с другой — изменением состояния исследуемого вещества в растворе. Недостаточная монохроматичность потока электромагнитного излучения вызывает обычно отрицательное отклонение как от закона Бугера — Ламберта, так и закона Бера.
Предположим, что измерения оптической плотности проводят, используя два потока электромагнитных излучений (рис. 9, о): первый — монохроматический с длиной волны Хтах, второй — немонохроматический, охватывающий достаточно большой интервал длин волн
Рис. 9. Влияние монохроматичности излучения на соблюдение закона поглощения излучения
— кп. Измерения с первым из- этих потоков дают максимальную оптическую плотность Лтнх, со вторым среднюю А, состоящую из суммы оптических плотностей, соответствующих интервалу — ?.п:
п
А = □----- (1.32)
п
Из”рис. 9, а видно, что Лтвх > А, так как оптические плотности, соответствующие всем длинам волн, отличным от Xmai, будут иметь меньшие значения. Кроме того, при малой концентрации q кривая спектра поглощения имеет менее крутой подъем, чем кривая спектра поглощения более концентрированного раствора с2. Следовательно,
* Помимо рассмотренных методов определения s существует и ряд других (см. стр. 149, 162), [6], [12].
Рис. 10. Влияние концентрации бензилового спирта на поглощение его в растворе ССЦ прн X 2,7 н
А 3,0 нм
при малых концентрациях разность Лтнх — А будет мала, а при боль-щИХ__велика, вследствие чего наблюдается отрицательное отклоне-
ние от основного закона поглощения электромагнитного излучения (рис. 9,6).
Изменение состояния вещества в растворе приводит к несоблюдению закона Бера:
1. Под влиянием посторонних электролитов происходит деформация молекул или заряженных частиц поглощающего вещества, что приводит к изменению спектра поглощения этого вещества. Так, например, с изменением ионной силы раствора изменяется спектр комплексного соединения титана с хромотроповой кислотой [13]. Поэтому для получения воспроизводимых результатов градуировочный график для определения титана в виде соединения с хромотроповой кислотой при непостоянстве ионной силы растворов следует строить при Z, соответствующей изобестической точке.
2. При изменении концентрации раствора меняется сила взаимодействия частиц в нем и могут наблюдаться полимеризация или деполимеризация. Например, раствор бензилового спирта в СС14 при различных концентрациях СвН5СН2ОН может находиться в виде мономера или полимера:
nCeH5CH2OH^i (СвНБСН2ОН)п
мономер, >.„„„2,7 нм полимер, Х„__3,0нм
Эти^ jpopMbi поглощают излучение различных участков спектра
На отсутствие полимеризации указывает постоянство е и соблюдение линейной зависимости А от с для растворов с различной концентрацией вещества. В растворах с концентрацией более 10'8 моль/л обычно наблюдается появление полимерных частиц.
3. При изменении степени сольватации (гидратации), которая зависит от концентрации раствора, меняется поглощение раствора.
4- -Изменение концентрации водородных ионов в растворе определяемого вещества проявляется в нескольких направлениях:
а) если реагент обладает кислотными свойствами, то полнота образования окрашенного соединения зависит от pH раствора (см. уравнение (а), стр. 36). Чем менее устойчиво образующееся соединение, тем сильнее сказывается влияние pH раствора;
б) под влиянием [Н+] в растворе изменяется форма существования веществ. Например, равновесие
2СгО| - + 2Н+ Сг2О? -+Н2О
смещается в зависимости от [Н+], что изменяет характер спектра.
Из рис. 11 видно, что при L 440 нм наблюдается изобестическая точка и при этой длине волны значение А не зависит от pH раствора;
в) при изменении pH раствора меняется состав образующегося соединения. Например, в зависимости от pH раствора железо (III) с сульфосалициловой кислотой образует три комплексных соединения различного состава;
г) изме нение pH раствора (уменьшение кислотности) часто приводит к разрушению комплексного соединения или неполноте его образования вследствие склонности центрального иона — комплексообразова-
Рис. 11. Спектры поглощения ионов СгО£~ (7) и Сг2О?~ (2)
теля присоединять гидроксильные группы, давая металл—гидроксо-комплексы.
5. Изменение степени диссоциации вещества в растворе при разбавлении приводит к изменению величины поглощения.
Таким образом, во избежание ошибок при использовании фотометрических методов большое значение имеют выбор реагента и способы приготовления анализируемых растворов. Если в результате реакции образуется устойчивое комплексное соединение, то разбавление раствора не влияет практи
чески на состояние определяемого вещества и при приготовлении анализируемых растворов не требуется избытка реагента [13]. При работе с мало устойчивым комплексным соединением требуется особое внимание к способу приготовления
применяемых растворов.
5. ПРЕИМУЩЕСТВА РАБОТЫ
С МОНОХРОМАТИЧЕСКИМИ ИЗЛУЧЕНИЯМИ
В результате избирательного поглощения одной или нескольких длин волн из сплошного излучения видимого участка спектра система приобретает определенный цвет. О величине поглощения визуально судят по интенсивности этой окраски, поэтому метод, основанный на
использовании немонохроматических Излучений, называется колориметрическим. В отличие от этого метод, основанный на работе с высоко-монохроматизированными электромагнитными излучениями, называется спектрофотометрическим.
Хотя спектрофотометрический и колориметрический методы анализа основаны на одном общем законе поглощения электромагнитных излучений, спектрофотометрический метод имеет следующие преимущества по сравнению с колориметрическим:
Использование высокомонохроматизированных потоков электромагнитных излучений позволяет подробно изучать узкополосные спектры поглощения, например спектры поглощения аквакомплексов
Рис. 12. Влияние монохроматичности потока электромагнитного излучения на значение молярного коэффициента погашения
интервале концентра-
’ редкоземельных элементов и изменения в них под влиянием различных факторов (pH, концентрация и т.п.).
2. Определение концентрации ве-шесть может быть выполнено с большей точностью и чувствительностью. Избирательность методов определения повышается.
~ Выигрыш в точности обусловлен тем, что использование монохроматических излучений приводит к более строгому соблюдению законов поглощения. Как видно из рис. 5, прямолинейная зависимость обеспечивает одинаковую точность отсчета во всем используемом ций, так как А и с меняются пропорционально (сравнить с непрямолинейной).
Повышение чувствительности объясняется следующим. Предположим, что для измерения оптической плотности анализируемых растворов используют два различных потока электромагнитных излучений (рис. 9, а, стр. 26). Поскольку оптическая плотность есть разность логарифмов интенсивностей двух потоков излучений
A=lglo-lgh, (1.33)
то при монохроматическом излучении эта разность будет иметь большее значение, чем при немонохроматическом. Следовательно, прямая А = f (с) в первом случае будет иметь более крутой подъем (см. рис. 9, б), что соответствует большему значению е, который пропорционален тангенсу угла наклона прямой. Это обусловливает большую чувствительность определения. Например, при использовании потока излучения с интервалом длин волн В—Г (рис. 12) молярный коэффициент погашения равен 2,7 • 103, с интервалом Б — Д —
• Ю3, с интервалом А — Е — 1,7 • 103 [14].
Повышение избирательности метода и анализ многокомпонентных стем также возможны лишь при использовании спектральных при-ров высокого разрешения.
3. В дифференциальном спектрофотометрическом методе использование монохроматических излучений обеспечивает соблюдение законов поглощения в более широком интервале концентраций, что имеет еще большее значение, чем в обычной спектрофотометрии.
4. В методе спектрофотометрического титрования наряду с повышением чувствительности определения расширяется круг систем, для которых может быть реализован этот метод, так как использование монохроматических излучений обеспечивает правильный выбор длины волны, при которой следует измерять оптическую плотность в процессе титрования.
5. Спектрофотометрический метод дает возможность исследовать процессы комплексообразования, изучать состояние веществ в растворе: определять константы диссоциации органических реагентов, состав комплексных соединений, константы устойчивости комплексных соединений [15] — [181.
6. ПРЕДЕЛЫ ТОЧНОСТИ ИЗМЕРЕНИЯ
ВЕЛИЧИН ПОГЛОЩЕНИЙ
Изучение факторов, влияющих на точность спектрофотометрических измерений [19] — [27], показывает, что причины ошибок в спектрофотометрии могут быть весьма разнообразны и многочисленны. Ошибки возникают, например, за счет действий оператора, условий проведения реакций, недостаточной чистоты кювет, непостоянства их установки в кюветные отделения, невоспроизводимости настройки шкалы прибора на 0 и 100% пропускания, непостоянства излучения источника освещения, нестабильности работы фотоэлектрической системы [24] — [27].
Рассмотрим в первую очередь ошибки, вытекающие из самой суш ности законов поглощения излучений, и основные закономерности, установленные еще в 1937 г. Туайменом и Лотианом [19]. Найденная ими зависимость ошибки измерения Л от ее абсолютного значения является определяющей в оценке ошибог спектрофотометрических измерений.
Теоретически Л = 1g (70//г) изменяется от 0 до оо , а Т = = Zz//0—от 0 до 1 (или от 0 до 100%). Однако не все значения Л могут быть измерены с достаточной точностью. Критерий соблюдения законов поглощения — постоянство е — и построение зависимости в координатах Л — с не дают указаний относительно того, какие значения Л могут быть измерены с наибольшей точностью.
Более наглядны графики в координатах Т—с или Л — Ц. Они показывают, что интервал, в котором Т и Л меняются прямо пропорционально с, ограничен (рис. 13, 14). При выводе формулы для расчета относительной ошибки измерения следует сделать некоторые допущения:
1) ошибка при отсчете значений Л или Т (ДЛ и ДТ) на всем про тяжении шкалы прибора одинакова;
2) закон поглощения строго соблюдается во всем интервале исследуемых значений А, т. е. ЛА/А = Лс/с.
Как то, так и другое условие практически невыполнимо. Но в первом приближении, чтобы получить представление об основных закономерностях в изменении ошибок измерения, они допускаются.
Рис. 13. Зависимость пропускания Т от концентрации раствора с
Рис 14. Зависимость оптической плотности А от интенсивности It потока излучения, прошедшего через поглощающий раствор
Для нахождения относительной ошибки ЛА/А — Лс/с [14], [19] — [22] нужно продифференцировать уравнение (1.33)
dA = d (1g /0-lg li),
di di
dA =0—(lg e) — = —0,43 ——, Л II
dA 0,43dI 0,43d/ — 0,43dT
= —---------------T- (L34)
A Al, AI010-A A10~a
Заменяя дифференциалы конечными разностями, получим
Дс ДД 0.43ДТ 0.43ДТ
с ~ A AW~a ~~ АТ ’
Дифференциал второго порядка при условии, что Л1 и /0 постоянны (Д = const, так как интенсивность первоначального потока излучения во всех измерениях постоянна, Л1 = const, так как ДЛ — const), будет равен
°A3dI \ 0.43Д/ / юл 1п 10 10z\JB „ „„
Приравняв выражение (1.36) нулю, получим
10л1пЮ юл А — да -0’
10л1п10=———,
А
, 1
Amln = —- =0,4343 (1.37)
In JU '
Таким образом, построение графика зависимости ошибок измерения от А (или Т) дает кривую с минимумом при А = 0,43 (Т = 36,8%). Если ошибка не должна превышать 2Аш1п, то измеряемые величины А должны укладываться в интервал 0,2—0,8 (рис. 15).
Эта теоретическая зависимость была получена с учетом ряда ограничений. Прежде всего предполагалось постоянство ошибки отсчета АЛ на всей шкале прибора. Практически ошибка АЛ меняется на раз-
Рис. 15. Зависимость относительной ошибки от измеряемой оптической плотности:
1 — кривая Шмидта; 2 — кривая рассчитана Н. П. Комарем и В. П. Самойловым
личных участках шкалы и сильно воз-растает в области больших значений Л.
Кроме того, не учитывался также ряд факторов, существенно влияющих на точность измерений Л (см. стр. 30). Совершенно очевидно, что общая ошибка будет значительно больше, чем только одна инструментальная ошибка.
Учесть влияние некоторых факторов (см. стр. 30) на общую ошибку опреде ления концентрации спектрофотометрическим методом можно, используя прямолинейную зависимость между оптической плотностью и концентрацией, вытекающую из закона поглощения [261:
А/1—ac+b,
где а—угловой коэффициент,равный тангенсу угла наклона прямой и пропорциональный коэффи циенту погашения; b — отрезок, отсекаемый прямой на оси АН. Отсюда следует, что с = Общая
абсолютная ошибка определения с равна сумме абсолютных значений частных дифференциалов этой функции
дс дс дс дс ,
de = dA -|- dl -J- db -|- “ da. dA dl db da
(1.38)
Подставляя в выражение (1.38) соответствующие значения частных производных, получим
1 А 1 АЦ_f,
dc = — dA- — dl-—db- —-— da, (1.39)
al dl2 a • efi
где dA, dl, da и db— абсолютные ошибки в определении оптической плотности, толщины слоя кюветы и ее пропускания и оценке параметров градуировочного графика а и b соответственно. Они могут быть заменены модулями стандартных отклонений Sa, sz, sb и sa, если предполагать, что условия определения с наименее благоприятные и все ошибки складываются:
1 , А ,1 , А/1—b
Sc=^^+^S£+TSb+’^~So- (L40)
Тогда уравнение для расчета относительной ошибки будет иметь вид — = Sa + Asi 4- Sb 1 4- — (I 4В
с А — ЬГ 1(А — Ы) (А — ЬГ) а' ’
Если предположить, что градуировочный график проходит через начало координат, т. е. отсутствуют систематические ошибки, то уравнение (1.41) примет вид:
Из уравнения (1.42) следует, что ошибка будет меньше если: 1) оптическая плотность раствора близка к 0,4343 [см. уравнение (1.37)]; 2) стандартные отклонения зд, sh sb, sa малы; 3) коэффициент а велик; 4) концентрация раствора большая, так как
sb II A = sbllcal = sb/ca.
Расчет каждой из этих ошибок может быть сделан в отдельности [26]. Однако при расчете каждой из них на основании обработки экспериментальных данных методом математической статистики [28] должно быть сделано предположение о независимости одной из них от остальных или их постоянстве. Так, для расчета зд/Л по формуле (1.42) необходимо знать, как меняется вд в зависимости от абсолютного значения А. Экспериментальная оценка одной из ошибок: в определении коэффициента погашения sja, стандартного отклонения sb или ошибки sjl может быть сделана лишь в предположении, что две другие не имеют в условиях эксперимента существенного значения. Некоторые попытки [24] — [29] оценить вклад отдельных факторов в общую ошибку спектрофотометрии показывают, что она в значительной степени зависит от надежности определения, например, параметров градуировочного графика а и Ь, а не только от инструментальной ошибки АЛ/Л. В формуле (1.42) в явном виде не отражается влияние таких факторов, как постоянство работы усилительного устройства, постоянство интенсивности излучения источника освещения, воспроизводимость балансировки шкалы отсчетного устройства. Таким образом, вопрос об ошибках в спектрофотометрии весьма сложен.
В то же время экспериментальная проверка показывает, что общий ход зависимости ошибки измерения от абсолютного значения оптической плотности очень близок к теоретическому. Он практически не зависит от класса прибора и лишь отличается по абсолютному значению ошибки АЛ: чем выше класс прибора и точнее отсчет по шкале, тем меньше абсолютное значение относительной ошибки при сохранении примерно постоянным общего хода зависимости. Так, в работах Н. П. Комаря и его сотрудников, было показано [24], [25], что ошибка измерения Л на приборах разных типов (спектрофотометрах и фотоколориметрах) в 20—30 раз выше, чем это следует из теоретических расчетов Туцймена и Догнана, но интервал, в котором А<2АШ1П, может быть расширен, главным образом, в сторону больших значений до 1,8—1,9 без значительного снижения точности (см. рис. 15). Но 2 '« 518 33
более высокие значения А измерять с достаточной точностью практически невозможно.
Однако преимущества спектрофотометрического метода (его сравнительная простота и экспрессность) делают заманчивым использование его для определения не только малых, но и достаточно больших концентраций веществ взамен длительных и трудоемких классических методов анализа (гравиметрия, титриметрня). Поэтому в последнее время стали уделять большое внимание дифференциальному спектрофотометрическому методу.
ЛИТЕРАТУРА
1. Р. В о и g u е г. Essai d’optique sur la gradation de la lumieere, 1729.
2. В. M. Пешкова, M. Г. Цюрупа. Вести. Моск, ун-та, серия II, Химия, 1959, № 4, 215.
3. Методы спектрального анализа. Сб. статей под ред. В. Л. Левшина. Изд-во МГУ, 1962, с. 192.
4. A. Beer. Ann. Phys. Chem., 1952, 2, 86, 78.
5. В. M. Пешкова, М. Г Цюрупа. Вести. Моск, ун-та, серия 11, Химия, 1956, № 6, 58.
6. Сб. статей «Спектроскопические методы в химии -комплексных соединений». Под ред,. В. М. Вдовенко. М.—Л., «Химия», 1969.
7. Ч. Н. Р. Ра о. Электронные спектры в химии. М., «Мир», 1964.
8. Н. П. Комар ь. Ученые записки Харьковского ун-та, 1951, XXXVII, (8), 57.
9. I. Y i ng - р е п - Т о ng, Е. King. 1. Am. Chem. Soc., 1953, 75, 6180.
10. A. Z i e I e n, R. С о n n i k. J. Am. Chem. Soc., 1956, 78, 5786.
11. W. H. McVey. United States Atomic Energy Commission, HW-21487, June, 1951.
12. M. И. Булатов, И. П. Калинки н. Практическое руководство по фотоколориметрическим и спектрофотометрическим методам анализа. М.—Л., «Химия», 1972.
13. А. К. Бабко, А. Т. Пилипенко. Фотометрический анализ. М.—Л., «Химия», 1968.
14. Г. В. Юинг. Инструментальные методы химического анализа. М., Атомиздат, 1963.
15. И. В. Т а н а н а е в. ЖАХ, 1949, 4, 68; 1948, 3, 276; 1950, 5, 82;
16. А. К. Бабко. Физико-химический анализ комплексных -соединений в растворах. Киев, Изд-во АН УССР, 1955.
17. И. П. К о м а р ь, В. И. Толмаче в. ЖАХ, 1950, 5, 21.
18. И. П. Комарь, ЖАХ, 1950, 5, 139.
19. F. Twyman, G. F. Lothian. Proc. Phys. Soc., 1933, 45, 642.
20. T. W. Schmidt. Z. Instrumenten Kunde, 1935, 55, 336, 357
21. C. F. Hiskey. Anal. Chem., 1949, 21, 1440.
22. G. H. Ayres. Anal. Chem., 1949, 21, 652.
23. С. M. Crowford. Anal. Chem., 1959, 31, 343.
24. И. П. Комарь, В. П. Самойлов. ЖАХ, 1963, 18, 1284.
25. Н. П. Комарь, В. П. Самойлов. ЖАХ, 1967, 22, 1285.
26. G. Sv eh la, A. Pall, L. Erdev. Taianta, 1963,10,719.
27. Г. С. Терешин. ЖАХ, 1959, 13, 388.
28. В. В. Налимов. Применение математической статистики при анализе веществ. М., Физматгиз, 1960.
29. Н. П. К о м а р ь, В. П. Самойлов. ЖАХ, 1969, XXIV, в. 8, 1133.
Глава II
ПОЛУЧЕНИЕ «ОКРАШЕННЫХ СОЕДИНЕНИЙ» И ИСПОЛЬЗОВАНИЕ ИХ В КОЛИЧЕСТВЕННОМ СПЕКТРОФОТОМЕТРИЧЕСКОМ АНАЛИЗЕ*
Любое спектрофотометрическое определение состоит из трех этапов: 1) перевод анализируемой пробы в раствор; 2) получение окрашенного соединения; 3) измерение поглощения испытуемого раствора (фотометрирование).
В настоящем пособии не уделяется специального внимания способам разложения или вскрытия проб различного рода объектов. Очень редко фотометрирование проводят сразу же после переведения анализируемой пробы в раствор, так как поглощение в этом случае, как правило, бывает очень незначительно и определение малых количеств вещества становится невозможным. Поэтому определяемый компонент переводят в соединение, обладающее значительным поглощением в одном из участков спектра, т. е. проводят фотометрическую реакцию**.
К реакциям, которые используются в спектрофотометрическом методе анализа, предъявляются в основном те же требования, что и к к любым другим, применяемым в аналитической химии. Но критерии оценки того или иного свойства реакций в спектрофотометрическом методе обладают своими особенностями, поскольку метод основан на поглощении электромагнитных излучений растворами окрашенных соединений. При выборе реакций оцениваются такие свойства, как специфичность и чувствительность. Кроме того, они должны удовлетворять еще двум требованиям: хорошей воспроизводимости окраски и ее устойчивости во времени. Существенно, чтобы закон Бера для растворов изучаемого соединения соблюдался в широком интервале концентраций, хотя это требование в отдельных случаях может и не выполняться.
1. ТИПЫ ФОТОМЕТРИРУЕМЫХ СИСТЕМ
В спектрофотометрическом методе анализа могут быть использованы различные типы поглощающих систем:
мяст окРашенные соединения» взято в кавычки, так как исследуе-
™ может обладать специфическим поглощением в УФ- и ИК-об.тастях спектра, а не только в видимой.
^еа.К Ю’ л Ре3- чьтате которой образуются окращенные соединения, называют спектрофотометрической или фотометрической.
1. Растворы аква-ионов различных неорганических солей: кобальта, меди, никеля, хрома, редкоземельных элементов, обладающих поглощением в видимой области. Определение редкоземельных элементов в виде аква-комплексов существенно ввиду особой специфики их спектров поглощения, обладающих узкополосными максимумами поглощения. Однако чувствительность таких методов очень мала: значения молярных коэффициентов погашения растворов аква-комп-лексов не выше п • 102.
2. Смесь органических соединений, а также их изомерных форм. Их анализируют по спектрам поглощения в УФ- и ИК-областях, где большинство органических соединений обладает характерными полосами поглощения, которые обычно имеют тонкую структуру.
3. Некоторые элементы в определенных степенях окисления образуют ярко окрашенные соединения. Например, марганец (II) может быть окислен до марганцовой кислоты, растворы которой обладают интенсивным поглощением при X 525 нм. Ванадий в различных его степенях окисления образует ряд соединений, растворы которых обладают интенсивным поглощением в различных участках видимого спектра. Ион СгО2- (Сг2О^“) в воде или OsO4 и 12 в органическом растворителе обладают также значительным поглощением.
4. Растворы комплексных соединений:
1) соединения с неорганическими лигандами, которыми могут быть анионы неорганических кислот (хлорид- и иодид-ионы), аммиак, перекись водорода и др. Молярные коэффициенты погашения в растворах соединений с неорганическими лигандами не превышают п • 103. Поэтому чувствительность определения металлов в виде этих соединений не очень велика;
2) гетерополисоединения, которые используются для определения Si, Р, As, Се, Zr;
3) комплексные соединения с органическими лигандами, обладающие значительно большим поглощением, чем комплексы с неорганическими лигандами. Органические реагенты классифицируют различным образом; "например, по типу заряда органического лиганда [1]:
а) органический лиганд — анион—характерен для большой группы органических реагентов, общая формула которых HR; при реакциях с катионами металлов они ведут себя -как кислоты, в которых протон замещается на ион металла. Общий вид уравнения реакции может быть записан:
Mn+ +mHR^±MR^~m>+ 4-mH+ (а)
Из уравнения (а) видно, что полнота образования комплексного соединения зависит от концентрации ионов водорода. Оптимальная концентрация ионов водорода будет определяться как свойствами катиона, так и кислотно-основными свойствами реагента. Если реагент является достаточно сильной кислотой, то реакция (а) протекает полностью уже в кислой среде. Однако большинство органических реагентов обладает слабо выраженными кислотными свойствами. По
этому Для полного смещения равновесия (а) вправо требуется значительно повысить pH (чтобы увеличить концентрацию свободных анионов за счет увеличения диссоциации реагента). Но значительное повышение pH может вызвать гидролиз и привести к образованию гидро-ксокомплексов или гидроокисей металлов, при недостаточной прочности комплекса. Для органического реагента, содержащего в качестве функциональных группировок гидроксогруппы, оптимальная кислотность соответствует pH начала гидролиза катиона, образующего комплекс. Если лиганд является многозарядным анионом, могут образоваться как заряженные (положительно или отрицательно), так и нейтральные молекулы комплексов;
б) органический лиганд — незаряженная молекула. Образующийся комплекс обычно является катионом. Примером такого соединения может служить ферроин — соединение железа с 1,10-фенантроли-ном. Такие реагенты обладают основными свойствами и в водных растворах существуют в протонированной форме RH+. Реакция взаимодействия реагента с катионами может быть также выражена уравнением (а), и, следовательно, сдвиг равновесия зависит от концентрации водородных ионов;
в) органический лиганд — катион. Он образует соединения с ионами металлов, связанными в комплекс анионного типа, например хлоридный или иодидный. Образующееся соединение относится к типу ионных ассоциатов. Например, родамин Б, который существует в растворах только как катион, используется для определения сурьмы, галлия, золота в виде соединений (RH)SbCle, (RH)GaCl4, (RH)AuC14.
_Эти соединения относятся к смешанным комплексным соединениям, в которых одновременно присутствуют два лиганда.
Для окрашенных органических реагентов очень важно, какие изменения в спектрах поглощения происходят при комплексообразовании с этими реагентами; чем больше смещается положение максимума поглощения реагента при комплексообразовании (т. е. чем больше величина AZ), тем большую ценность имеет реагент.
5. Смешанные комплексные соединения — полиядерные комплексы, в состав которых входит несколько атомов металла, и моноядер-ные соединения, в состав которых входит несколько различных лигандов. Введение второго лиганда придает комплексу ряд новых свойств, например появляется окраска или увеличивается ее интенсивность, усиливается способность экстрагироваться органическими растворителями (ионные ассоциаты).
6. Соединения, получаемые в результате так называемых «усилительных реакций», позволяющих получать «эффективные» молярные коэффициенты погашения, значения которых превышают даже теоретически допустимый предел 105. Суть таких реакций заключается в том, что определяемый элемент переводят в соединение, в состав которого он входит в низком стехиометрическом соотношении. Определяют при помощи достаточно чувствительной реакции концентрацию другого элемента, входящего в состав этого же соединения в относительно высоком стехиометрическом соотношении по сравнению с определяемым. На основании данных определения второго элемента находят
Эквивалентную концентрацию первого элемента. Тем самым молярный коэффициент погашения возрастает в такое число раз, каково отношение числа атомов второго и первого элементов в данном соединении. Так, например, фосфор может быть определен [2] в виде фосфоро-молибдат-иона [РМо12О40]3- экстрагируемого смесью бутанола и хлороформа (е = 2,4 • 104). Если после реэкстрации аммиачным буферным раствором молибден определить 2-амино-4-хлорбензотиолом 13] и рассчитать соответствующую концентрацию фосфора, то, поскольку в комплексе на один атом фосфора приходится 12 атомов молибдена, эффективный молярный коэффициент погашения оказывается равным 3,6 • 105.
7. Окрашенные соединения, образующиеся в результате реакций, в которых катализатором является определяемый элемент. Основанные на этом кинетические методы [41 или кинетохроматические реакции [5] отличаются значительно большей чувствительностью, чем методы, основанные на прямом фотометрировании растворов комплексов определяемого элемента.
2. УСТРАНЕНИЕ ВЛИЯНИЯ СОПУТСТВУЮЩИХ КОМПОНЕНТОВ
Определение какого-либо элемента обычно проводят в присутствии большого числа других компонентов, содержащихся в анализируемом образце. Многие из этих сопутствующих компонентов могут внести ошибку в результаты определения, в первую очередь, за счет их собственного поглощения. Поэтому очень важно предусмотреть наличие сопутствующих компонентов, необходимо знать и учитывать свойства элементов-спутников для выявления и нахождения средств их маскирования: например, использовать различия в устойчивости комплексных соединений определяемого и сопутствующих элементов и т. п.
Для устранения влияния сопутствующих компонентов и повышения специфичности спектрофотометрических методов' анализа применяют различные способы.
1. Нахождение области спектра, в которой поглощает только соединение определяемого компонента и отсутствует поглощение всех сопутствующих компонентов, в том числе и реагента. Тогда, используя приборы с высокой монохроматичностью потока излучения, можно исключить помехи, вызванные поглощением посторонних компонентов, если максимумы их поглощения находятся в другой области спектра.
2. Нахождение специфических условий образования комплексных соединений каждого из компонентов, если реагент дает с определяемым и каким-то из сопутствующих элементов комплексные соединения, поглощающие в одной и той же области спектра. Используя, например. различия в оптимальном значении pH образования или различия в устойчивости комплексов, можно, регулируя pH и количество прибавляемого реагента, создавать такую его концентрацию, которая необходима- лишь для образования более устойчивого комплексного
единения определяемого элемента. Если сопутствующий элемент пбоазует комплекс, не поглощающий в области максимума поглощения комплекса определяемого элемента, то это также может вызвать осложнение, поскольку часть реагента расходуется на образование комплекса с мешающим элементом и его оказывается недостаточно для полного связывания в комплекс определяемого элемента. Это устраняется прибавлением большого избытка реагента.
3. Маскировка мешающего компонента путем перевода его в бес
цветное комплексное соединение, если не удается устранить помехи со стороны сопутствующих компонентов регулированием pH или концентрации реагента. Предположим, что константа устойчивости комплекса определяемого элемента (MJ с реагентом (R) значительно больше, чем с маскирующим лигандом (L), а константа устойчивости мешающего элемента (Мп) с реагентом значительно меньше, чем с мас-
кирующим лигандом, т. е.
Km[L и Kmhr ^мпь-
Комплекс мешающего элемента]/: реагентом будет’ разрушаться в результате его перехода в более устойчивый бесцветный комплекс с маскирующим лигандом Мц L. При этом компплекс определяемого элемента с реагентом разрушаться не будет./В качестве таких маскирующих веществ часто используются органические оксикислоты: лимонная, винная и некоторые другие.
4. Использование экстракционно-фотометрического метода позволяет устранить влияние посторонних элементов, а также избытка реагента подбором таких условий (pH, растворителя, концентрации реагента), когда экстрагируется только комплекс определяемого элемента. Влияние посторонних комплексов и реагента устраняется также реэкстракцией их в водную фазу. Например, удаление из органической фазы некоторого избытка диоксимов, используемых для определения малых количеств никеля.
Если ни один из приведенных способов не дает возможности устранить помехи со стороны сопутствующих компонентов, то может быть использован какой-либо из вариантов анализа многокомпонентных систем (см. стр. 72), в том числе и различные варианты дифференциальных способов измерений.
При проведении спектрофотометрических определений следует помнить о значительном поглощении ряда неорганических анионов, которые могут присутствовать в качестве примесей в объектах (табл. 5).
^Поэтому при выборе условий проведения фотометрических реакций необходимо учитывать область поглощения анионов, рекомендуя использование тех или иных вспомогательных реагентов (кислот, щелочей, компонентов буферных растворов). При применении экстракционно-спектрофотометрического метода для исследования процессов комплексообразования, разделения и определения многих элементов используются различные органические растворители. При выборе растворителей нужно учитывать их прозрачность в определенных участках спектра (табл. 6).
ТАБЛИЦА 5
Максимумы поглощения некоторых неорганических анионов [6]
Ион X, нм 8 Ион X, нм В
С1- Вг- 181 190 1-Ю4 1,1-Ю4 S2O|~ 220 4-Ю3
I- 226 1.2-104 S2O|- 254 2,2
он- SH- 187 230 5-103 8-103 1 Ol 1 П о о Z Z 354,6 302,5 2,3 7
ТАБЛИЦА 6
Граница пропускания ультрафиолетового излучения растворителями [7]
Растворитель Предельная длина волны X, нм Растворитель Предельная длина волны X, нм
Уксусная кислота 270 Этиловый спирт 215
Ацетон 330 Этиловый эфир 220
Ацетонитрил 220 Глицерин 230
Аллиловый спирт 330 Метиловый спирт 210
Амилацетат 255 - Метилформиат 260
Бензол 280 Изопропиловый спирт 210
н-Бутиловый спирт 220 Серная кислота (96%) 210
Сероуглерод 380 Тетралин 330
Четыреххлористый угле- 270 Толуол 285
род 2, 2, 4-Триметилпентан 215
Хлороформ 245 Вода 210
Циклогексан 210 Ксилол 290
Этилацетат 255 Пиридин 305
Бензиловый спирт 250
3. ИССЛЕДОВАНИЕ СПЕКТРОФОТОМЕТРИЧЕСКОЙ РЕАКЦИИ И ВЫБОР ОПТИМАЛЬНЫХ УСЛОВИЙ ЕЕ ПРОВЕДЕНИЯ
Рассмотрим этапы исследования фотометрических реакций с органическими реагентами, в результате которых образуются комплексные соединения. Оптимальные условия проведения реакции требуют возможно более полного связывания определяемого элемента в комплекс. Большинство органических реагентов обладает кислотно-основными свойствами. Если предполагается, что ион эвемента вступает в реакцию с органическим реагентом, являющимся одно-, двух- или многоосновной кислотой (т. е. реакция протекает по типу замещения протона кислоты ионом металла), то в общем виде реакцию образования комплексного соединения можно представить уравнением (а) (см. стр. 36). Следовательно, условия образования комплексного соединения будут зависеть не только от избытка реагента, но также от pH раствора. Особое значение указанные факторы приобретают, 49
комплексное соединение отличается малой прочностью и исполь-зчемый реагент является слабой кислотой.
3-> Поглощение органического реагента очень часто меняется с изменением кислотности раствора, что следует учитывать при выборе оптимальной длины волны для измерения поглощения комплекса.
Использованию фотометрической реакции для количественного
определения элемента должно предшествовать:
1) изучение ионного состояния и спектральных характеристик компонентов, вступающих в реакцию; 2) выяснение оптимальных условий полноты образования комплексного соединения; 3) предварительное изучение скорости протекания реакции.
Задача выбора оптимальных условий проведения фотометрической реакции облегчается, если уже известны некоторые физико-химические характеристики исходных компонентов и продукта реакции (Куст комплекса, ЛдиСС реагента).
Исследование органического реагента
1. Органические реагенты, растворимые в воде, часто обладают кислотно-основными функциями и, следовательно, в водном растворе протекает реакция
HR^R- + H+ (б)
равновесие которой может смещаться в зависимости от кислотности раствора. Если недиссоциированная и диссоциированная формы реагентов поглощают при различных данных волн, то при сдвиге равновесия (б) происходит изменение оптической плотности в области максимумов поглощения той и другой формы. Если обе формы поглощают при одной и той же длине волны, но каждая из форм обладает собственной величиной е, то будет происходить только изменение оптической плотности раствора в области максимума поглощения этих форм.
Если реагент представляет собой многоосновную кислоту, то изменения в спектре поглощения, с изменением кислотности раствора, могут быть весьма сложными в зависимости от характера поглощения и диссоциации кислоты.
Исследование реагента проводится в последовательности:
1) снимают спектры поглощения водного раствора реагента с постоянной его концентрацией при различных значениях pH (рис. 16). При изучении спектральных характеристик комплексных соединений сравнивают спектры поглощения комплекса и реагента и выбирают область длин волн, где различие в поглощении наибольшее;
) 2) находят зависимость А = f (pH) на основании измерения величин А в максимумах поглощения той и другой формы (Xhr и Xr-). Эга зависимость может быть использована для расчета Кднсс реагента (см. стр. 90). Участки спектра с постоянным значением А на кривых
/ (pH), которые соответствуют поглощению недиссоциированной (НК) и диссоциированной форм (R-) реагента, могут быть использованы для расчета е обеих форм. Полученные таким образом значения eHR
A
wo
050
Рис 16 Спектры поглощения водных растворов бензоилацетона при различных значениях pH
/-2,03 -6,58, 2 — 7,8; 3 — 8,4, 4 — 8,9; 5—10,02—11,08
25Z7 270 200 510 550 550^S%W\hm
и eR- являются практически истинными для данной ионной силы, если отсутствует наложение в поглощении посторонних веществ (например, компонентов буферного раствора);
3) если изучают реакцию, используемую в экстракционно-спектрофотометрическом методе, то получают спектральные характеристики растворов реагента в органическом растворителе, применяемом для экстракции комплексного соединения. В органическом растворителе (особенно неполярном) реагент обычно находится в какой-то одной форме (чаще всего в виде нейтральной молекулы), если отсутствует взаимодействие молекул реагента с молекулами растворителя и нет полимеризации. Поэтому достаточно снять спектр поглощения одного какого-либо раствора. Зная концентрацию этого раствора (если раствор приготовлен по точной навеске реагента), можно вычислить е, практически точно соответствующее истинному/ его значению в данном растворителе. Распределение реагента между
водной и органической фазой зависит от pH водной фазы. Эта зависимость может быть представлена кривой А = /ДрН) (рис. 17) для одной и той же концентрации реагента (на рис. 17 оптическая плотность А органической фазы пропорциональна проценту экстракции R) или кривой D = /"(pH), если можно вычислить коэффициент распределения D;
4) определяют, не наблюдается ли полимеризация реагента в водном растворе или в неводном растворителе. Для этого снимают спектры поглощения растворов реагента различной концентрации и рассчи
тывают е. Постоянство этой величины указывает на отсутствие полимеризации в интервале исследованных концентраций. К-такому же заключению приходят, если значение Ддисс водорастворимого реагента является постоянной величиной для растворов реагента различной концентрации.
A
Рис. 17 Зависимость процента экстракции хлороформом от значения pH: 1 — гептоксилгйта никеля; 2 — гептоксима
Исследование раствора определяемого элемента
Для исследования комплексообразования необходимы сведения о ионном состоянии элемента-комплексообразователя, который должен быть в негидролизованной форме. Данные по гидролизу большого числа соединений можно найти в соответствующих справочниках [8] а также в периодической литературе.
Необходимо также знать, при какой концентрации аква-ион элемента находится в мономерной форме. На основании большого числа исследований поведения ионов Zr, Ti, Ni в водных растворах известно, что полимеризация зависит от кислотности растворов и наблюдается при концентрации растворов выше 5 • 10-3— 1 • 10-4 моль/л. Сведения о /Сует аква-комплексов изучаемого иона элемента и их способности к замещению молекул воды на другие лиганды (их инертности) можно получить, главным образом, из периодической литературы; число таких работ пока не очень велико.
Для оценки поглощения водного раствора соли определяемого элемента снимают спектр поглощения в условиях существования негидролизованного иона элемента и рассчитывают значение е.
Исследование комплексного соединения
На полноту образования комплексных соединений, растворимых в воде или органических растворителях (экстрагируемых соединений), влияет ряд факторов: pH, избыток реагента, скорость образования соединения. Влияние pH может сказываться различно. Если в реакции в качестве лиганда участвуют анионы слабой органической кислоты, от величины pH раствора будет зависеть концентрация той формы лиганда, которая участвует в комплексообразовании. Малоустойчивые комплексные соединения при увеличении pH раствора разрушаются или меняют состав вследствие гидролиза иона комплексооб-разователя. От pH водной фазы зависит процент экстракции комплекса в органическую фазу.
Водорастворимые комплексные соединения. 1. Выясняют влияние pH на поглощение раствора исследуемого соединения: а) снимают спектры поглощения растворов, содержащих определяемый элемент и реагент (взятый без большого избытка) при различных значениях pH (рис. 18) и определяют Хтах комплекса; б) строят график в координатах А — pH при Хтак комплексного соединения. Максимумы поглощения комплекса и диссоциированной формы реагента часто совпадают. На рис. 19 приведена зависимость А = f (pH) для комплекса и реагента при А, соответствующей максимуму поглощения комплекса, и показано рНопт, при котором комплекс уже образовался в максимальной степени, а поглощение реагента сказывается на суммарной оптической плотности в наименьшей степени.
2. Выясняют влияние избытка реагента на полноту образования комплексного соединения, изучая зависимость А — LM]/[R] при ^-тах комплекса. При образовании устойчивого комплексного соединения на графике наблюдается резкий излом (рис. 20, кривая 1), ко-
Рис. 18. Спектры поглощения растворов, содержащих празеодим и арсеназо М, прн различных значениях pH
торый определяет необходимый избыток реагента и соответствует соотношению компонентов в комплексном соединении. Кривая 2 говорит или о получении малоустойчивого соединения или о более сложном процессе комплексообразования и не позволяет сделать однозначный вывод о соотношении компонентов в комплексе. В этом случае необходимо использовать большой избыток реагента, который определяют по горизонтальному участку кривой.
3. Выясняют зависимость
поглощения комплексного соединения от времени, строя график в координатах А=/:(т). Фотометрирование испытуе
мого раствора проводят в течение промежутка времени, соответствующего горизонтальному участку кривой на рис. 21.
4. Выясняют влияние разбавления раствора на диссоциацию комплекса: проводят разбавление раствора в п раз и увеличивают толщину поглощающего слоя в п раз. Если комплексное соединение устойчиво, значение А не изменяется. Однако при разбавлении возможен гидролиз данного соединения, тогда произведение cl изменяется. Для поддержания постоянного значения pH разбавление проводят буферным раствором. При этом необходимо знать, не происходит ли побочное комплексообразование с компонентами буферного раствора (конкурирующие реакции). Для этого нужно проверить, соблюдается
Рис 19. Зависимость оптической плотности растворов реагента HR и комплекса MR/, от pH
Рис 20 Кривые насыщения устойчивого комплексного соединения [М] : [R] = l -2 (/) и неj стойчивого комплексного соединения [М] : [R] =
-1:3 (2)
заКОн аддитивности спектров поглощения растворов всех компонентов, участвующих в реакции образования комплексного соедине-
15. Выясняют области соблюдения законов поглощения: а) снимают спектры поглощения растворов с различной концентрацией определяемого элемента при оптимальных значениях pH, cr и наблюдают, сохраняется ли положение максимума при одном и том же значении А (см. рис. 6); б) рассчитывают средние значения молярного коэффициента погашения для растворов с различной концентрацией при различных X, строят графики в координатах в —X и наблюдают, про-
Рис. 22. Оценка величин АЛ и Ае при сравнении спектров поглощения реагента HR и комплекса MR„
Рис. 21. Виды зависимости оптической плотности от времени образования комплексного соединения
исходит ли их наложение при всех X. Графики в координатах е—X дают
возможность сравнивать спектры поглощения независимо от концент-
рации растворов.
Для выбора наиболее выгодной длины волны при количественных определениях следует учитывать величину АХ, т. е. разницу в положении максимумов поглощения комплекса и реагента, а также разность в значениях молярных коэффициентов погашения Ае. Большое значение величин АХ и Ае повышает чувствительность и ценность рассматриваемой реакции (рис. 22). Особенно важны эти величины при сравнительном исследовании новых реагентов; в) находят зависимость А == / (с) при Х1Т)ах комплекса и выявляют, имеет ли она прямолинейный характер. Если объединенный закон поглощения не соблюда-
ется, то проверяют соблюдение закона Бугера — Ламберта, т. е. за-B1 симость А от толщины слоя раствора I. Для этого исследуемый раствор наливают в кюветы различного диаметра и измеряют А при посто-янных концентрации и длине волны. Прямолинейная зависимость между А и I позволяет полученные значения А пересчитать на одну и ту же величину / и нанести на график в координатах А— с. Если было установлено, что закон Бугера — Ламберта выполним, то необходимо изучить причины, вызывающие отклонение от закона Бера (см. стр. 15).
Комплексные соединения, экстрагируемые неводными растворителями. 1. Снимают спектр поглощения А = f (Z) экстракта изучаемого комплекса и находят %так.
2. Определяют интервал pH максимальной экстракции соединения. Обычно при экстракции комплексного соединения реагент, имеющий собственное поглощение, также переходит в слой органического растворителя и мешает выявлению поглощения комплексного соединения. Поэтому определяют различие в зависимости коэффициентов распределения реагента и исследуемого соединения от pH (см. рис. 17). Для этого изучают зависимость А (или D) экстрактов от pH водной фазы, сравнивают зависимость Лорг = f (pH), полученную для комплекса, с аналогичной кривой для реагента и выбирают значение pH, при котором наблюдается полная экстракция комплекса и минимальная реагента.
3. Изучают влияние концентрации реагента и времени на полноту экстракции (см. стр. 43, пп. 2,3).
4. Выясняют условия соблюдения закона поглощения (см стр. 45, п. 5).
В настоящее время для выяснения оптимальных условий проведения фотометрической реакции применяют метод факторного планирования [9]. Если известны константы, характеризующие процесс комплексообразования (Луст комплекса, /Сгидр элемента), то используют алгебраические методы расчета pH и избытка реагента, которые необходимы для получения оптимального выхода продукта реакции.
Рассмотрим некоторые из этих методов расчета, пригодные при отсутствии полимеризации [10],
Расчет [Н+] для оптимальных условий проведения реакции. При образовании комплексного соединения (см. уравнение (а), стр. 36) важно найти значение pH, при котором получается оптимальный выход продукта реакции. Для этого -следует располагать количественными характеристиками следующих процессов:
1) образование фотометрируемого соединения
^равн^Рл» (I)
2) протонизация реагента
R~b?H^HgR; -Кравп=огр: (II)
3) образование гидроксокомплексов металл-иона
М-НН2О^М(ОН);-НН; КРаЕн = ш. (Ш)
Предлагается ввести следующие обозначения:
см — общая концентрация металл-иона; Cr — общая концентрация реагента; [Н] = h\ [MnRml = х.
Из уравнений (II) и (III) по закону действия масс находим:
[М (ОН);] = Щ [М] ft-'; [HgR] = Og [R] h°.
Составляя уравнение материального баланса, можём записать
I Q
cM = nx-^ fM] Л'1' и cR = mx-|-[R] \ а5 h4,
i=0 ?=о
откуда
см — ПХ Cn—mx
[M] = , "7 ; [R] =----
^ih-1 hcQh4
Применяя закон действия масс к уравнению (I) и делая соответствующие замены, получим
см—пх \п( cR—тх \т
) I )
(Н. 1)
Частная производная от уравнения (II. 1), взятая по h, после приравнивания ее нулю позволяет вычислить [Н+], отвечающую максимальному выходу комплексного соединения. После преобразования получим
д dh.
(II.2)
Взяв производную и раскрыв знаки сумм в (II. 2), получим
(1 +<11 h+a2ft2+ . - .) —
" ( + hs +
(от + 2<12^+ За3й2 + . - - ) —0.
(П-3)
Подставив в уравнение (II.3) значения для п, т, т)г, о9 и решив его, получим оптимальное значение [Н+], которое не зависит ни от начальных концентраций исходных компонентов, ни от значения константы образования комплексного соединения.
Если соединение имеет состав I : 1 и отсутствуют ступенчатые процессы гидролиза иона металла и протонизации реагента, уравнение (П.3) преобразуется
<11=0,
откуда
(II 4)
Расчет количества реагента, необходимого для полноты образования соединения. Рассмотрим пример, когда отсутствуют побочные конкурирующие реакции. Уравнение (II. Н позволяет рассчитать концентрацию реагента, при которой доля иона металла, несвязанного в комплекс, не превышает некоторого заранее заданного минимального значения 6. По условию См — пх — 6см; пк = см; х = см/п-
I <2
Введем обозначения f= V ?]гЛм; s = Vgq^, i=0 g=0
fnSm fl \n/ Q \m
( S 1ъ/1-г I У I
\Z= 0 I ' <7 = 0 /
Определение ведется при оптимальном (ранее найденном) значении pH. В уравнение (II.5) подставляют значение [Н+], а также значения константР/,, 1], и о9 и вычисляют В. Тогда по уравнению (II. 1), учитывая (II.5), имеем:
= в (бсм)" -^}т
Откуда 1-п ___;
ск=“см+см,П (Вп6") Ш- (П-6)
Если п — т = I = Q -= 1, получаем
fs
CR=CM +(^4 5)-1 — См + ^- (В.7)
Следующим этапом в изучении фотометрической реакции, после того как найдены оптимальные условия образования комплексного соединения, является выбор прибора с различной степенью монохроматичности потока излучения для измерения поглощения растворов полученного соединения.
4. ЧУВСТВИТЕЛЬНОСТЬ ФОТОМЕТРИЧЕСКИХ МЕТОДОВ
Одной из основных характеристик каждого фотометрического метода является его чувствительность. Особенно важен этот критерий при выборе методики определения очень малых количеств (следов) веществ, что имеет большое значение при анализе продуктов промышленных производств, например таких, как полупроводниковые материалы.
Критерии оценки чувствительности реакции, например в визуальном колориметрическом и спектрофотометрическом цетодах, различны. Так, в визуальном колориметрическом методе критерием чувствительности может быть количество вещества, необходимое для получения заметного различия в интенсивности окраски анализируемого раствора и холостого опыта. Было установлено, что между двумя слабо окрашенными растворами можно обнаружить меньшую абсолютную разницу, чем между слабо окрашенным раствором и водой [1]. Следовательно, в этом имеется определенная аналогия с дифференциальным методом (стр. 65).
В настоящее время при анализе следов веществ испольвуется только объективный фотоэлектрический способ измерения поглощения.
При фотоэлектрической регистрации поглощения минимально улавливаемое различие в оптических плотностях, а следовательно, и минимально обнаруживаемое количество веществ определяется прежде всего воспроизводимостью результатов измерений. Сендэл [1] считает, что при измерении на современных спектрофотометрах может быть достигнут предел точности измерений порядка 0,001-оптической плотности. В действительности, такой высокий предел воспроизводимости отсчетов достигается только при таком повторении балансировки фотометрической системы прибора, которая проводится без изменения положения кюветы в кюветном отделении и смены в ней испытуемого раствора. В противном случае ошибка воспроизводимости при параллельных измерениях будет значительно выше. При повторном проведении фотометрической реакции для получения новой серии эталонных и испытуемого растворов ошибка воспроизводимости будет еще больше. Еще большую ошибку в результаты определения вносят предварительные операции растворения, отделения мешающих компонентов и т. п.
Таким образом, при оценке чувствительности фотометрического определения следует, ВЦ-первых, разлнчать^чувствительность фотометрической реакции |н чувствительность данного фотометрического метода определения того или иного компонента в соответствующих конкретных условиях (объектах). Во-вторых, правильная оценка чувствительности метода определения не может быть сделана без учета точности данного метода.
Рассмотрим некоторые способы оценки чувствительности, которые используются при изучении новых фотометрических реакций, применяемых в спектрофотометрии. Для оценки чувствительности фотометрических реакций используются следующие способы:
1. Для сравнительной оценки чувствительности двух фотометрических реакций удобно пользоваться молярным коэффициентом погашения е, вычисленным для условий, близких к получению его истинного значения (стр. 22).
К недостаткам такого способа оценки чувствительности относятся: а) невозможность оценить минимальное количество вещества, которое может быть определено; б) невозможность судить о том, какие минимальные значения оптических плотностей в данном случае можно использовать, чтобы ошибка определения не превышала допустимую, так как расчет е обычно проводится на основании оптических плотностей, измеренных в области их оптимальных значений (0,2—0,8). На практике обычно имеют дело со средней величиной е. Так как е пропорционален тангенсу угла наклона прямой на градуировочном графике, а иногда равен ему, то для сравнительной оценки чувствительности реакций можно использовать тангенсы углов наклона, которые должны определяться в одинаковых условиях.
Даким образом, на основании значения е можно лишь оценить, какой из реагентов образует соединение, обладающее наибольшим поглощением, но нельзя судить о чувствительности реакции в реальных условиях, т. е. о возможности определения реальных количеств вещества.
2. Чувствительность реакций выражают в мг/мл определяемого вещества. Этот способ имеет ряд недостатков: а) практически определение и проведение фотометрической реакции в объеме 1 мл невозможно; обычно этот' объем составляет мимимум 10—20 мл, следовательно, действительная чувствительность на порядок ниже; б) без указания воспроизводимости метода такой критерий оценки чувствительности реакций ненадежен
3. Сендэл [1] за критерий чувствительности принимает количество микрограммов вещества в слое раствора с поперечным сечением 1 см, которое дает оптическую плотность 0,001. В соответствии с законом поглощения излучений этот способ оценки чувствительности имеет следующий смысл.
Поскольку оптическая плотность А = ес/, то, подставляя в эту формулу выражение для с
т
с=----, ,
иМ
где т — масса вещества; М — молекулярный вес; v — объем, в котором проводится определение, получим
eml А =---.
Mt>
Обозначим vll = q — эффективное сечение кюветы. Тогда чувствительность (мкг/см2) может быть равна
Следовательно, при А — 0,001 чувствительность может быть вычислена как MI&, если известен молярный коэффициент погашения. Для реакций, обладающих достаточной чувствительностью, величина mlq находится в пределах 0,01—0,001 мгк/см2.
Этот способ оценки чувствительности также носит условный характер, так как предел точности измерения А, равный 0,001, недостижим практически.
Указания относительно пределов точности определения оптических плотностей различны [11] — [13] (0,01; 0,05). Однако без оценки точности определения минимальных значений А с точки зрения методов математической статистики эти указания носят условный характер.
4. Для оценки чувствительности фотометрической реакции и фотометрического метода в целом Бланк 114], [15] вводит понятие определяемый минимум (мкг), для расчета которого дает формулу
Он рассуждает аналогично Сендэлу. Но учитывая, что в состав соединения может входить несколько атомов элемента, для расчета концентрации он получил следующее выражение:
т-ю-3 с =--------,
nt’A
(11.10)
где д___атомный вес определяемого элемента; п — число атомов
в молекуле соединения, в виде которого проводится определение; у__объем раствора, мл; 10-3 — коэффициент пересчета атомного
веса в граммах на вес в миллиграммах (А • 10®) и объема в миллилитрах на объем в литрах (о • 10‘3). Подставляя выражение (11.10) в уравнение закона поглощения излучений (1.9) и решая его относительно т, получим
АиДпЮ3
т =-------
ё1
(11.11)
Если vll = q — эффективное сечение кюветы, то для расчета определяемого минимума т получим уравнение (II.9).
Чтобы вычислить т, необходимо прежде всего определить значение А, соответствующее определяемому минимуму. Минимальное значение А будет определяться погрешностью анализа (величиной стандартного отклонения). Но величина стандартного отклонения, в свою очередь, будет зависеть от абсолютной величины А (т. е. количества определяемого вещества). Следовательно, необходимо найти такую оптическую плотность, погрешность в определении которой не превышала бы ее значения, например была вдвое (критерий 2а) или втрое (критерий За) меньше измеряемой А. Так, для доверительной вероятности а 0,95 и числа опытов п = 3 (ta --= 4,3) погрешности
C'SA „ „ е“= Vn =2'55sa.
а минимальная оптическая плотность, равная удвоенной погрешности, составляет
/1т1п=5вд. (11.12)
Чтобы найти экспериментально Ат111, необходимо изучить зависимость sa — f (А) для исследуемой реакции, определяя стандартные отклонения серии значений А. Так, например, для расчета определяемого минимума меди в виде диэтилдитиокарбамината в образце иодида натрия (о. ч.) 115] было проведено несколько серий опытов с различным содержанием меди (т. е. с различными оптическими плотностями).
Число опытов в каждой серии было равно 10. Значения оптических плотностей соответствовали интервалу 0,008—0,19. Стандартные отклонения изменялись в пределах (1,79—4,34) • 10-3. Из рис. 23 следует, что зависимость = /(А) прямолинейна. Коэффициенты
а и b в уравнении этой прямой sa = а + ЬА были вычислены по методу наименьших квадратов, в результате чего получено уравнение1.
sA= 1,761 IO-3 4- 1,446 10-2 а. (11.13)
Совместное решение уравнений (11.13) и (11.12) аналитическим или графическим методами (см. рис. 23) позволяет найти Лт1п. Для рассматриваемого случая было получено: sa = 1,9 • Ю-3; Дш1п = = 9,5 • 10-3.
Если не все величины, входящие в уравнение (II.9), известны, то
для вычисления т можно
воспользоваться уравнением градуировочного графика. Так, в рассматривае-
s-Ю1
Рис. 23. Определение 4min графическим способом
мом примере для расчета определяемого минимума меди было использовано уравнение градуировочного графика, полученное при работе на фотоэлектроколориметре ФЭК-М:
Д = 2,706-10-2 т—1,77110-* тг. (11.14)
После подстановки в уравнение (II. 14) значения Лт111 = 9,5 I0-3 было получено т = 0,35 мкг Си.
Существуют еще другие подходы к оценке чувствительности фотометрической реакции [11], [131. Однако Бланк предлагает наибе лее строгий подход к оценке чувствительности спектрофотометрических методов оп
ределения.
Ошибки могут возникнуть на различных этапах определения: при разложении пробы, отделении от мешающих компонентов, проведении фотометрической реакции, измерении величины поглощения ит. п. Поэтому одна и та же фотометрическая реакция может иметь различную чувствительность в зависимости от конкретных условий ее
применения.
При определении Дт1п нельзя не учитывать флуктуаций фона (т. е. колебаний в поглощении холостого опыта). Ошибки за счет флуктуаций фона можно учесть и исключить, если проводить измерения оптической плотности по отношению к холостому опыту.
Снизить значение можно, повышая А за счет увеличения е и уменьшения эффективного сечения кюветы q (т. е. уменьшения v и увеличения /). Увеличить в можно подбором новых реагентов или использованием методов, которые позволяют получать «эффективные» молярные коэффициенты погашения (см. стр. 37). Уменьшать эффективное сечение кюветы за счет увеличения I можно не беспредельно. Поперечное сечение кюветы можно уменьшить сохраняя максимально возможной ее длину. Тогда резко сократится объем, требуемый для заполнения кювет. Можно уменьшить объем раствора, необходимый для заполнения кюветы, снижая верхнюю границу прохождения потока излучения через кювету [14].
Уменьшить Лтin, понизить определяемый минимум т и повысить увствительность определения можно за счет повышения точности сех операций и сокращения ошибок при их выполнении.
ЛИТЕРАТУРА
1 Е. С е н д э л. Колориметрические методы определения следов металлов. М «Мир», 1964.
’ 2. V. D jur kin, G. F. Kir kbr ight, T. S. Wes t. Analyst., 1966, S^G. F. К i r k b г i g h t, J. H. Yoe. Taianta, 1964, 11, 415.
4'. К. Б. Яцимирский. Кинетические методы анализа. М., «Химия», 1967.
5. Т. S. West. Chem. a. Ind. 1966, 25, 1005.
6. Р. Драго. Физические методы в неорганической химии. М., «Мир», 1967.
7. Г. В. Юинг. Инструментальные методы химического анализа. М., Госатомиздат, 1963.
8. J. Bjerrum, G. Schwarzenlach, G. S i 1 1 е n. Stability constans. The Chemical Society, Burlington House, v. 1 London, 1957.
9. Ю. В. Грановский. Основы планирования экстремального эксперимента для оптимизации многофакторных технологических процессов. Московский ин-т народного хозяйства им. Плеханова, 1971.
10. Л. П. Адамович. Рациональные приемы составления аналитических прописей. Изд-во Харьковского ун-та, 1973.
11. А. К. Бабко, А. Т. Пилипенко. Фотометрический анализ. М., «Химия», 1968.
12. М. И. Булатов., И. П. К а л и н к и н. Практическое руководство по фотоколориметрическим и спектрофотометрическим методам анализа. М.—Л., «Химия», 1968.
13. 3. Марченко. Фотометрическое определение элементов. М., «Мир», 1971.
14. А. Б. Блан к. Сб. статей «Методы анализа веществ особой чистоты и монокристаллов». ВНИИ монокристаллов. Харьков, 1962, ЖАХ, 1962, 17, 1040; 1964, 19, 363; Сб. статей «Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты». ВНИИ монокристаллов. Харьков, 1971.
15. А. Б. Бланк, А. М. Булгакова, Н. Т. С и з о н е н к о. ЖАХ, 1961, 16, 715.
Глава III
МЕТОДЫ КОЛИЧЕСТВЕННОГО АБСОРБЦИОННО-
СПЕКТРОСКОПИЧЕСКОГО АНАЛИЗА
1. КЛАССИФИКАЦИЯ МЕТОДОВ И СПОСОБЫ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИЙ ВЕЩЕСТВ В РАСТВОРАХ
Классификация фотометрических методов может быть проведена по следующим основным принципам.
1. По характеру используемого излучения: монохроматического или сплошного. На этом основано различие между спектрофотометрическими и колориметрическими методами.
2. По способу регистрации интенсивности излучений: визуальному или фотоэлектрическому; оба способа регистрации могут использоваться и в колориметрических и в спектрофотометрических методах.
3. По способу сравнения интенсивности потоков излучений до и после прохождения их через поглощающий раствор. При этом способ регистрации также может быть визуальным или фотоэлектрическим. Каждый способ сравнения интенсивности потоков излучений требует определенного метода расчета концентрации испытуемого раствора. В зависимости от того, используется или нет в этих расчетах формула основного закона поглощения, можно сделать вывод о том, требуется ли строгое соблюдение этого закона при определении концентрации соответствующим методом.
Остановимся на рассмотрении лишь отдельных, наиболее распространенных способов сравнения интенсивности потоков излучений и расчета концентраций.
Метод стандартных серий — один из классических визуальных методов. Для определения концентраций по этому методу сравнивают интенсивности окраски испытуемого раствора и ряда эталонов, которые готовят в колориметрических пробирках с притертыми пробками. Наблюдение можно проводить как в горизонтальном, так и вертикальном направлениях. Если окраска испытуемого раствора занимает промежуточное положение между окраской двух растворов эталонного ряда, то принимают за искомое среднее из значений их концентраций или готовят новый ряд эталонов в этом интервале концентраций с меньшим Ас.
Метод не требует строгого соблюдения законов поглощения излучений, в нем могут быть использованы реакции, протекающие во времени. Единственным требованием применимости метода является воспроизводимость интенсивности окраски.
Если окраска получаемых соединений сохраняется длительное время, готовят постоянный ряд эталонов в запаянных пробирках;
эТо особенно удобно для полевых лабораторий. Пробирки можно заменить набором специально подобранных цветных стекол, пленок или Устойчивыми растворами, имитирующими окраску. Возможность использовать пробирки малого диаметра и большой высоты, для заполнения которых не требуется значительного объема раствора, позволяет повысить чувствительность определения при вертикальном способе наблюдения. Кроме того, метод позволяет проводить оценку изменения оттенка окраски, что является его преимуществом даже перед фотоэлектрическим методом в тех случаях, когда не наблюдается заметного изменения абсолютного значения А. Этот метод рационально использовать в однотипных массовых анализах.
Метод диафрагмирования основан на оптическом способе компенсации различий в интенсивности двух потоков излучений. Для уравнивания интенсивности потоков излучений, проходящих через испытуемый и раствор сравнения, используются диафрагмы с переменной величиной щели. Щель диафрагмы раскрывают, чтобы увеличить интенсивность потока излучения, ослабленного после прохождения через поглощающий раствор, и сделать ее равной сравнительному потоку. Диафрагма соединена с барабаном, который имеет шкалу, проградуированную в значениях оптической плотности А и процента пропускания Т. К такому типу относится большинство визуальных и фотоэлектрических приборов отечественного производства.
В нерегистрирующих спектрофотометрах типа СФ-4 (СФ-16) осуществляется как оптическая (изменением ширины щели), так и электрическая (изменением напряжения на потенциометре в цепи усилителя фототока) компенсации.
Если есть возможность измерить величины А и Т, то используют следующие пути расчета концентрации определяемого вещества при условии соблюдения законов поглощения:
1. Концентрацию определяют по градуировочному графику А = = f (с), построенному по значениям оптических плотностей ряда эталонных растворов (см. рис. 5). Определив оптическую плотность испытуемого раствора Ах в аналогичных условиях, находят сх определяемого вещества. Для получения более точных результатов при построении градуировочного графика используют метод наименьших квадратов. Можно также пользоваться градуировочным графиком, если зависимость А = f (с) криволинейна (см. рис. 5). Однако в этом случае для точного построения градуировочного графика необходимо приготовить большее число эталонных растворов. Кроме того, как видно из рис. 5, прямолинейная зависимость повышает точность определения (пропорциональные изменения оптических плотностей соответствуют пропорциональным изменениям концентраций растворов сь с2, Сз, если зависимость прямолинейна, и непропорциональным сь с2, с3, если криволинейна).
хкюветы подбирают так, чтобы измеряемые оптические плотности У[ ладывались в интервал от 0,1 до 1,0 (о точности измерений величин бег>СМ" СТР 30)’ Если растворы подчиняются закону Бугера — Лам-рта, то выбор кюветы облегчается. Например, если оптические плот-сти, измеренные в кювете с толщиной слоя 2 см, оказываются за
пределами указанного интервала, то можно измерить оптические плотности начальных и конечных растворов эталонного ряда в кюветах с большей или меньшей толщиной слоя соответственно Пересчитав полученные оптические плотности на /-== 2 см, используя прямую пропорциональность между Ди/, наносят их на один и тот же градуировочный график.
2. Концентрацию рассчитывают, используя прямо пропорциональную зависимость между Д и с
3. Концентрацию рассчитывают по уравнению (1.9), если известен е?- определяемого вещества.
2. МЕТОД СПЕКТРОФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
В последнее время наряду с другими титриметрическими методами стали широко применять спектрофотометрическое титрование [1] — [13], которое позволяет определять значительно меньшие концентрации веществ, чем обычными титриметрическими методами.
При спектрофотометрическом титровании могут быть использованы все химические реакции, которые применяются в других титриметри-ческих методах. Особенно широко используют реакции комплексообразования. Часто в качестве реагентов применяют комплексоны и, главным образом, этилендиаминтетрауксусную кислоту (ее двунатриевую соль— комплексон III).
Метод спектрофотометрического титрования основан на последовательном измерении поглощения раствора, которое меняется в процессе титрования; и построении зависимости Д = f (и) (кривой титрования). При спектрофотометрическом титровании необходимо, чтобы один из компонентов реакции — определяемое вещество М, титрант R или продукт реакции MR — обладал характерным поглощением в доступной области спектра (безындикаторное титрование). Если ни один из компонентов реакции не поглощает, то можно использовать индикаторное титрование, для чего в титруемую систему вводят новое вещество, образующее окрашенное соединение с каким-либо из компонентов основной реакции.
Тогда с изменением концентрации поглощающего компонента в процессе титрования оптическая плотность раствора изменяется. Момент эквивалентности или конечная точка титрования фиксируются как точка пересечения двух прямых линий, выражающих зависимость поглощения титруемого раствора от количества прибавленного титранта до и после момента эквивалентности (рис. 24).
Вид кривой спектрофотометрического титрования зависит от того, какой из компонентов реакции поглощает при той длине волны, при которой измеряют оптическую плотность Д. На рис. 24 приведена кривая титрования (/), соответствующая тому случаю, когда в системе
гЛощает продукт реакции. Излом на кривой титрования может не П°егда совпадать с моментом эквивалентности по стехиометрическому равнению реакции. Тогда для определения конечной точки титрования необходимо вводить эмпирическую поправку, которую находят в процессе титрования ряда растворов с известной переменной концентрацией.
Спектрофотометрическое титрование по способу определения момента эквивалентности сходно с кондуктометрическим и особенно амперометрическим методами, в которых момент эквивалентности определяют также по излому на кривой титрования (как точку пересечения двух прямолинейных ее участков). Отсюда вытекает требование со-
Рис. 24. Определение конечной точки титрования (к. т. т.) экстраполяцией
блюдения закона Бугера — Ламберта — Бера, т. е. прямолинейной зависимости между оптической плотностью титруемого раствора и объемом (или концентрацией) титранта. Однако требование соблюдения закона может выполняться не на всех участках кривой титрования. Если вблизи от точки эквивалентности зависимость Л от и непрямолинейна, то для определения конечной точки титрования прибегают к экстраполяции.
Для экстраполяции используют такие участки кривой титрования, где в избытке находится или титруемый ион, или реагент, т. е. участки, где равновесие практически полностью сдвинуто в сторону образования продукта реакции. На этих участках кривой зависимость А = = f (с) [или А = f (о)] обычно имеет прямолинейный характер, т. е. закон Бера соблюдается. Конечную точку титрования находят, продолжая прямолинейные участки кривой титрования до их пересечения (рис. 24, кривая 2). .Благодаря этому спектрофотометрический метод определения конечной точки титрования позволяет 11роБоДить~ титрование растворов с низкими концентрациями и использовать ре- . акции, обладающие малыми константами равновесия (например, обр!ь зование малоустойчивых комплексов, титрование слабых кислот и оснований, реакции окисления — восстановления при малых значениях разности потенциалов), и имеет большие преимущества по сравнению с методами, в которых точку эквивалентности определяют по скачку титрования (например, потенциометрическое титрование). Помимо графического метода определения конечной точки титрования рекомендуется применять также алгебраический метод, основанный на использовании ^метода наименьших квадратов. Для прямолинейных участков кривой 1 (см. рис. 24) до и после момента эквивалентности удут справедливы соответственно следующие уравнения прямых:
^1=414- byv,
Аз = 4з+ 62V,
(III.2)
(III.3)
где независимая переменная v — объем титранта, а зависимая переменная А — оптическая плотность, соответствующая v в каждый момент титрования. Для определения момента эквивалентности нужно найти v, при котором Аг = Л2,
щ 4- ^2 (П1.4)
где аг, а2, Ьг и Ь2 — коэффициенты, которые рассчитывают методом наименьших квадратов: для уравнения первой прямой (III.2)
п2иД1—2Ж41 а,— reSta—(Sv)2 2 Hi Iv2 — 2ч2Лх у bl~ й2с>2 —(St')2 для уравнения второй прямой (III.3) n2vA2—2а2Д2 й2= , П2ц2— (St))2 2Л2 2с>2 — 2й2 Д2 v b&-— , nSc2—(St>)2 (III. 5) (III.6) (HI.7) (III.8)
где п — число измерений А, соответствующих определенным объемам п, добавленным в процессе титрования.
Для удобства расчетов рекомендуется все необходимые для них данные свести в таблицу следующего вида:
№ V A vz vA .
1 2 3 n 2 2o 2Д 2v2 2 vA
(III.9)
После того как будут определены коэффициенты alt а2, Ьг, Ь2, из уравнения (III.4) рассчитывают значение v:
й2—щ v=------
bi—bz
Вычисленное таким образом значение v будет соответствовать конечной точке титрования.
Рассмотрим теоретические основы безындикаторного и индикаторного вариантов спектрофотометрического титрования, в основе которых лежат реакции комплексообразования.
Безындикаторное титрование. Пусть в процессе титрования образуется комплексное соединение по уравнению
M+R^MR
т. е. компоненты реагируют в соотношении 1 : 1 (И.
Рели при выбранной длине волны поглощают все три компонента (ион-омплексообразователь, лиганд и комплекс), то согласно основному закону поглощения излучений
Л=2е* [‘11’
(III. 10)
е yq__суммарная оптическая плотность; ег — молярный коэффи-
циент погашения каждого компонента; [i] — концентрация каждого (i-ro) компонента; I — толщина поглощающего слоя. Ввиду того, что в процессе титрования толщина поглощающего слоя остается постоянной, произведение ег/ = kt есть постоянная величина. Выражая суммарную оптическую плотность через равновесные концентрации компонентов реакции, можно записать:
Л = [М] £M + [MR] Amr+[R] *r,
(III.П)
где [M], [R] и [MR]— равновесные концентрации компонентов реакции.
В каждый момент титрования должны выполняться следующие соотношения:
cM = [Ml + [MR], (III. 12)
_ cR= [R] + [MR], (III. 13)
где См — общая концентрация иона металла; cr — общая концентрация лиганда.
Для теоретических исследований удобнее вместо объема прибавляемого реагента по оси абсцисс откладывать «степень комплексообразования» р, выражаемую соотношением
p=cR/cM. (III. 14)
Комбинируя уравнения (III.II), (III.12), (III.13) и (III.14), получим
А = см Pmr + ^r (р—1)] + (^m + ^r—^mr) Iм]. (Ш-15)
Уравнение (III.15) показывает, что оптическая плотность, меняющаяся в процессе титрования, является функцией величин р и [М]. Однако [М] может быть также выражена как функция р путем комбинации (III.12), (III.13) и уравнения для кажущейся константы устойчивости комплекса (которую вычисляют с учетом реальных условий: pH, присутствия посторонних лигандов и т. д.):
к,_ [MR]
[М] [R]
Таким образом:
гмт-I&U-pI-’J + Vik'cmO-pI-iis-HK'cm
1М] “ ^77—-----------------• (Ш. 16)
Решая совместно уравнения (II 1.15) и (III. 16), получим общее уравнение кривой безынднкаторного спектрофотометрического титрования:
А=~^- (2^'С,М IAMr+ (и — 1)1 + (йм+ kR~~k!AR) Х
xk'cM(l- и) — i+VK'W1— И)— 1]2 + 4К'см]}. (III. 17)
Если существует длина волны, при которой поглощает только образующееся комплексное соединение, то уравнение (III.15) упрощается, так как = йр = 0, и принимает вид
^ = ^mr(cm-1mD. (41.18)
Из уравнений (II1.16) и (III.17) видно, что при заданных молярных коэффициентах погашения вид кривой зависит от произведения К'см. На рис. 25 приведены кривые титрования, рассчитанные для одного и того же значения kwR и различных значений К'см, (табл. 7).
ТАБЛИЦА 7
Данные для построения кривых титрования (см. рис. 25)
Кривая на рис. 25 ^уст см, моль/л AMR=EMR 1
1 ю5 10-2 1000 100
2 5-Ю3 ю-2 50 100
и Рис. 25. Вид кривых титрования при различных Куст комплексов
Е1.
При малых Кем отклонение от прямолинейной зависимости вблизи точки эквивалентности наблюдается в более широком интервале. Однако, как показывают экспериментальные исследования, при величине /<'см = 50 момент эквивалентности может быть установлен экстраполяцией прямолинейных участков, удаленных от точки эквивалентности, с достаточной точностью. Таким образом, метод без-ындикаторного спектрофометриче-ского титрования можно применять для определения малых концентраций с использованием малоустойчивых комплексов.
На рис. 25 приведены также кривые зависимости рА1 (— 1g [Ml) от р, для визуального титрования. Отсутствие скачка на обеих кривых показывает невозможность проведения количественных определений.
Из сравнения этих кривых с кривыми спектрофотометрического титрования становятся ясны преимущества последнего перед визуальным комплексонометрическим титрованием. Этот вывод справедлив в отношении всех титриметрических методов, момент эквивалентности в которых определяется по скачку на кривых титрования.
14з уравнения кривой спектрофотометрического титрования (III. 18) яКЖе следует, что чувствительность метода можно повысить увеличением толщины слоя кюветы (/) и выбором соответствующей длины волны, при которой ^mr имеет наибольшее значение, так как А пропорциональна &mr = 8mr/-
Индикаторное титрование. Если в доступной области длин волн ни
один из компонентов реакции не поглощает, используют метод индикаторного спектрофотометрического титрования. Рассмотрим несколько примеров использования различных индикаторов.
1. В качестве индикатора может быть использован ион такого металла, который дает менее устойчивое комплексное соединение, чем
определяемый элемент; причем играющего роль индикатора с титрантом, должно иметь поглощение в видимой области спектра. Так, определение тория комплексоном можно проводить в присутствии соли меди [2]. В процессе титрования тория происходит образование бесцветного комплекса (рис. 26). В момент эквивалентности образуется
комплексное соединение элемента,
окрашенное соединение меди,
поглощение которого И фикси- Рис. 26. Кривая титрования тория ком-руется прибором. Пока в раст- плексоном III в присутствии меди воре будут находиться ионы ме-
ди, не связанные в комплекс, прибавление комплексона будет вызывать увеличение оптической плотности, так как будет возрастать концентрация комплекса меди с комплексоном. Второй перегиб на кривой титрования показывает, что все ионы меди находятся в виде комплексного соединения. Таким образом, этот метод ,не только позволяет
установить момент конца титрования тория, но также позволяет провести последовательное титрование тория и меди в их смеси.
2. В качестве индикатора при титровании комплексоном III используют реагент, который дает менее устойчивое комплексное соединение с определяемым элементом, чем комплексон. Например, могут использоваться те же индикаторы, что и при визуальных комплексонометрических титрованиях (эриохром черный Т, мурексид, арсеназо I).
Теории методов индикаторного комплексонометрического спектрофотометрического и визуального комплексонометрического титрования аналогичны. Но использование спектрофотометрического контроля и графического способа определения конца титрования дает первому из этих методов ряд преимуществ.
Предположим, что определяемый элемент образует комплекс с комплексоном MR с константой устойчивости Kmr по уравнению
M-+R
MR, Kmr=-^-, MR [M][R]’
а также комплекс с индикатором Mln с константой устойчивости Kian по уравнению
, [Mln]
M + In-Mln, KMIn = ——.
Приведенные уравнения даны в упрощенном виде без учета констант диссоциации комплексона и индикатора, а также побочных реакций комплексообразования, но предполагается, что в расчетах используются кажущиеся константы
А 05
0,2-
OJ
0,7-
0,6-
0.5-
0,4-
0,3-
2? 22'ij j 7
Ы6 A35A5AA7650Q52655558B625 К,НМ
Рис. 27. Спектры поглощения комплексных соединений кислотного хром-синего К (I) и его комплексов с кальцием (2) и магнием (5) [4]
j стойчивости комплексов. Если Amr 7> Дмтп. то постепенное прибавление комплексона к раствору комплекса Mln вызовет реакцию
Mln-j-R -» MR4-In,
т. е. в процессе титрования определяемый ион постепенно переходит из комплекса с индикатором в комплекс с реагентом. Если свободный индикатор и его комплекс с определяемым элементом поглощают при разных длинах волн (рис. 27), то при этой реакции будет происходить изменение окраски раствора, что и используется для определения момента конца титрования в визуальном методе.
Если в любом участке спектра наблюдается наложение в поглоще-
индикатора и его комплекса, оптическая
НИИ свободного будет равна [5]:
плотность
/ = eIn/[In] + eMItl/[MIn] .
(III. 19)
меняется соотношение компонентов процессе титрования. Степень комплексообразования [см. (111.14)1 меняется от нуля (в начале титрования) до 1 (в момент эквивалентности). Отношение концентрации свободного индикатора к общей его концентрации выражается
[In] _ [In] С1П [Mln] + [In] ’
где Cjn и [In] — общая и равновесная концентрации индикатора. В идеальном случае в момент эквивалентности а = 1. Зависимость' между р, а, см, cjn и константами /<мк и Маш выражается уравнением [5]
Рассмотрим,
как
реакции в уравнение
(III.20)
1
^Mtn f 1
- си —:— i —
^МА ^1 —
a^MIn
1
CM
Mln
(111.21)
Если Kmr » Кмш (примерно на 4—5 порядков) и не требуется тчислять р с точностью большей чем 0,001, то вторым слагаемым ^равнении (Ш-21) можно пренебречь. Уравнение (III.21) можно ре-В ить относительно а и получить зависимость а = f (р). Чтобы полу-1“ь зависимость А = f (и), которая осуществляется практически, необходимо установить зависимость А от р и а. Из уравнения (II 1.20) рассчитываем [Mln]
[Mln] = [1п](1~-1 =с (1-К). (Ш.22)
сс
Подставляя уравнение (III.22) в (III.19), получим
/ = е1п tacin + eMln ^cIn (1 а) = ЕМ1п еМ1п) ^ac^n
Поскольку ewin^in = const, то окончательно получим
А' =А— 8MIn ZcIn = (eIn—eMjn) ZacIn. (III. 23)
Рис. 28. Зависимость оптической плотности от объема прибавленного реагента при индикаторном титровании
Из уравнения (Ш.23) видно, что если измерение проводить при длине волны, соответствующей максимальному поглощению свободного индикатора (см. рис. 27, X 588), то изменение оптической плотности в процессе титрования будет тем более заметно, чем больше разность Bin — 6М1п-
Если выразить а через р преобразованием уравнения (III.21), а р — через v и см:
са nv 1
р =----=--------,
СМ »<• СМ
где v0 — начальный объем титруемого раствора; п — нормальность титранта; v—объем титранта, прибавленный в каждый момент титрования, тогда можно рассчитать кривые зависимости А = f (и) по уравнению (III.23). На рис. 28 приведены кри
вые, рассчитанные по данным, указанным в табл. 8. Излом на кривых соответствует моменту эквивалентности.
Подробное изучение [5] влияния различных факторов: констант устойчивости комплексов с титрантом и индикатором, их соотношения, концентраций металла (см) и индикатора (С[П), констант кислотной диссоциации реагента и индикатора, приводит к следующим
ТАБЛИЦА 8
_____Даннь*е для построения кривых спектрофотометрического титрования
Кривая на рис. 28 с№ м/л с1п- м/л К Mln KMR (ещ- —eMIn)z п vDi мл
1 2 1/3-10-г 1 310-2 1-ю-‘ 0,4-10-* 1/8-10е 1/8-106 4-Ю12 4-1013 0,8-10* 0,8-10* 0,1 0,1 150 150
выводам. Излом на кривой титрования будет тем более резким, чем больше величина Дмгп, выше исходная концентрация определяемого иона см и ниже концентрация индикатора Cin, если Кмъ Кмы-Однако при использовании низкой концентрации индикатора сильно понижается абсолютное значение оптической плотности. Поэтому оптимальная концентрация индикатора определяется, с одной стороны, выбором оптимальной длины волны, для которой разность 8in — ьмш имеет максимальное значение, с другой — максимально возможной толщиной поглощающего слоя. Для получения положительных результатов величина кажущейся константы /Смш должна быть порядка 104—10®, следовательно, необходимо учитывать оптимальное значение pH образования комплекса с индикатором.
При этом совершенно очевидны преимущества работы с монохроматическими излучениями: чем более узок интервал длин волн, выделяемый монохроматором, тем легче разграничить области максимального поглощения комплекса с индикатором и самого индикатора, а следовательно, получить большую разность е1п — емш- Те же преимущества имеет использование монохроматических излучений прч безындикаторном титровании, если поглощением обладает не один из компонентов реакции, а два или все три.
Кроме того, при работе с монохроматическими излучениями более строго соблюдается закон Бера, а следовательно, прямолинейная зависимость А = f (п) сохраняется на большом участке кривой титрования.
Метод спектрофотометрического титрования имеет ряд преимуществ по сравнению с другими методами определения концентраций.
По сравнению с визуальными методами титрования метод спектрофотометрического титрования обладает более высокой чувствительностью, которая соизмерима с чувствительностью спектрофотометрических методов.
По сравнению с прямым спектрофотометрическим методом метод титрования имеет более высокую точность, так как в процессе титрования определяется не абсолютная оптическая плотность, а ее изменение. Более высокую точность обеспечивает также экстраполяционный метод определения конечной точки титрования. Повышение точности увеличивает чувствительность. К преимуществам метода относится возможность проводить определение в присутствии компонентов, частично поглощающих при выбранной длине волны, если они не взаимодействуют с титрантом, так как важно изменение оптической плотности в процессе титрования, а не абсолютное значение ее. Кроме того, метод более специфичен, так как позволяет легко проводить последовательное титрование двух компонентов. Преимущество перед методами, основанными на определении конца титрования по скачку на кривой титрования, были обсуждены ранее (см. стр. 60).
Построение кривой зависимости А = f (v) позволяет определить' молярное отношение компонентов в комплексе, которому соответствует излом на кривой при титровании раствора какого-либо элемента известной молярной концентрации раствором титранта [61.
3 ДИФФЕРЕНЦИАЛЬНЫЙ СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД
В обычном спектрофотометрическом методе анализа раствор срав-ня практически не поглощает при той длине волны, при ко-неной проводят измерение. Этот метод измерения называют методом Абсолютной» или непосредственной спектрофотометрии. Точность Апеделения концентраций при таком методе измерений невелика, что 2е позволяет проводить определение компонентов, содержащихся в пробе в высоком процентном отношении. Кроме того, интервал оптических плотностей, а следовательно, и определяемых концентраций, которые могут быть измерены методом непосредственной спектрофотометрии с достаточной точностью, ограничен (см. стр. 32). Для расширения этого интервала и повышения точности измерений может быть использован дифференциальный способ измерений [81—[22].
В настоящее время дифференциальный спектрофотометрический метод анализа развивается в трех направлениях:
1) спектрофотометрическое определение больших концентраций!
2) устранение мешающего влияния посторонних компонентов; 3) исключение поглощения реагента.
Определение больших количеств веществ можно провести спектрофотометрическим дифференциальным методом с точностью, не уступающей классическим гравиметрическим и титриметрическим методам.
Теоретические основы дифференциального спектрофотометрического метода
Для выяснения преимуществ, которые дает дифференциальный способ измерения, нужно рассматривать схему четырех основных способов балансировки фотометрических приборов, которая была дана Рейли и Кроуфордом [20].
Измерению на любом спектрофотометре (в том числе и упрощенном ФЭК-Н-57, ФЭК-56 и т. п.) предшествует балансировка шкалы прибора по двум предельным значениям пропускания 0 и 100%.
В обычной спектрофотометрии при балансировке шкалы на Т = 100/о, кювета заполняется растворителем. Другой конец шкалы соответствующий Т = 0%, балансир „ри „^^ьЛереХых потоках излучении (рис. 29, /). При такой настройке прибора при измерении А растворов, имеющих малое поглощение (Л 0, Т -> - 1 0/о), основная часть шкалы остается нерабочей. Точность измерения можно повысить, если балансировку на Т - 0% прово-няТ1рг^е ПРИ полностью перекрытом потоке излучения, а поместив бопьшр ппгИ кюветУ с Раствором, поглощение которого несколько получается зн=п ИСВЬГГУеМ0Г0 Раствора (рис. 29, II). При этом тяжения птрп ит^льныи выигрыш в точности измерения за счет рас-Этот метпп п 6711011 шкалы прибора и уменьшении цены деления.
Д олучил название метода «анализа следов».
J 18
Точно такой же выигрыш в точности получается, если при измере-нии сильно поглощающих растворов (Л -> оо, Т 0%) для настройки шкалы на Т = 100% использовать раствор, поглощение которого несколько меньше поглощения испытуемого (рис. 29, III). Этот метод балансировки шкалы называют методом «определения больших концентраций».
Самый большой выигрыш в точности будет получен, если оба конца шкалы настроить по двум растворам, близким по поглощению.
Рис. 29. Четыре способа балансировки спектрофотометров (схема Рейли и
Кроуфорда):
I — обычный метод; II—метод «анализа следов»; III — метод определения больших концентраций; IV — метод «предельной точности»
Растворы выбирают таким образом, чтобы оптическая плотность испытуемого раствора находилась между оптическими плотностями этих двух растворов (рис. 29, IV). Этот вариант балансировки называют методом «предельной точности». Обычный метод .измерения (вариант I) может считаться предельным случаем этого последнего варианта.
Однако рядом исследований показано [23]—[25], что не все эти варианты измерений могут быть осуществлены на различных приборах. Не все они дают также выиг
рыш в точности измерения величин А и Т ввиду конструктивных ограничений приборов. Так, для реализации варианта II приходится использовать для настройки конца шкалы Т = 0 % настолько концентрированный раствор, что его прозначению и ника
пускание оказывается близким к нулевому
кого растяжения шкалы не наблюдается. По той же причине вариант IV осуществляется лишь на участке шкалы, соответствующем малым значениям пропускания, так как второй раствор (для настройки на Т = 1С0%) должен быть взят возможно близким к первому, чтобы получить какой-либо эффект «растяжения» шкафы и выигрыш
в точности.
Поэтому в практике фотометрического анализа наибольшее распространение получил метод измерения больших величин А (вариант III), который обычно называют дифференциальным спектрофотометры ческам методом или методом отношения пропусканий. Его широко применяют, если в анализируемом материале определяемый компонент содержится в высоком процентном отношении. Сущность варианта III дифференциального метода состоит в том, что при измерении в качестве раствора сравнения используют один из растворов эталонного ряда, а не растворитель или раствор, содержащий все применяемые реагенты, кроме определяемого компонента. При таком измерении из большого значения Л2 испытуемого раствора как бы
чигается А раствора сравнения, поглощение которого условно зЬ'иНИмают равным нулю. Это не только позволяет измерять большие ПР чения А, но вообще повышает точность измерений [10]. Ошибка зНседеления концентрации с2 раствора при дифференциальном изме-рении С-цией Q на [22b
оптической плотности по отношению к раствору с концентра-при постоянстве ДД и соблюдении закона Бера будет рав-
Ig^OTH— Л0ТН— s/(с2 С]), /отн—А2 А1, 0,43с!Г d (lg Toth) = d [ eZ (с2 Cj)], ' отн
bide.
Откуда
dc =
о,4зат е/Т отн
и относительная ошибка равна dA2 dc2 0,ШТ
Л2 с2
Q,43dT
/ 10 отн 1П ю ю ОТН
=—0,43 АТ ------------- ——
\ А2 А\
. (III.25)
d
0,43dT
- -АЛ----• (Ш. 24)
е1с2Тотн АгТотн Л210 Л°тн
Дифференцирование уравнения (III.24)] приводит к выражению (_ °’43dT ' ' ‘ ,'>u"Х Лг ' Л2-10-Л°тн
Приравняв уравнение (III.25) нулю, получим Л2-10л°тн 1пЮ=10Лотн. Следовательно, эта функция будет иметь минимум при
1/1п 10=Л2; 0,43—Л2 = 0; 0,43— Л1~ЛОтн = 0-
Откуда получаем выражение для оптической плотности, измеряемой с наименьшей ошибкой
ЛОтн=0,43—Ль (III.26)
Таким образом, при всех значениях А! > 0,43 оптимальная величина относительной оптической плотности равна нулю. Это означает, что наименьшая ошибка при относительном измерении получается, если концентрации измеряемого раствора и раствора сравнения близки. Зависимость относительной ошибки от оптической рисТ30СТИ В диФФеРенциальн°й спектрофотометрии приведена на
А)птимальный интервал оптических плотностей, при измерении °РЫХ °Аи^ка не превышает минимальную более чем на 25—30%, плот Т В области от 0 ДО 0,2—0,3 (см. рис. 30). Чем выше оптическая Н°СТЬ РаствоРа сравнения, тем с большей точностью измеряется чувсЧеСКаЯ плотность анализируемого раствора А2 и с тем большей мтиВпТеЛЬН0СТЬ1° улавливается разница между двумя сравнивае-растворами, что видно также из следующих рассуждений [10].
Предположим, что в широких пределах меняется отношение концентраций двух растворов, один из которых (q) служит раствором сравнения при измерении оптической плотности второго (с2). Интенсивности потоков излучений, проходящих через каждый из этих растворов, соответственно равны:
Л = /() io~ec*z, /2= 10 ю~Е<Ч
при этом с2 > с19 тогда
с-> ci 4- Ас — —---------=а; Дс = с1(сх—I).
ci С1
Из рис. 31 видно, что отношение этих интенсивностей
(Q—е/(с2—с,)_ JQ—eZAc=j0—Л,(а—I)
(Ш.27)
при одном и том же значении а будет зависеть от оптической плотности раствора сравнения Аг. Чем выше Лъ тем меньше /2//х и, следовательно, тем легче различить /2 и при а I, т. е. для растворов с близкими концентрациями.
Рис. 30 Зависимость ошибки измерения Лотн от ее абсолютного значения при различных оптических плотностях раствора сравнения До
Рис. 31. Зависимость соотношения интенсивностей потоков излучения /г/Л, прошедших через испытуемый раствор и раствор сравнения, от оптической плотности раствора сравнения Д1 для различных значений а-с21с\
Таким образом, чем выше оптическая плотность раствора сравнения, тем выше и точность измерения А (максимальная точность при = 1).
Однако повышать концентрацию раствора сравнения, т. е. увеличивать его оптическую плотность, нельзя беспредельно, так как поток излучения, прошедший через сильно поглощающий раствор, будет иметь недостаточную интенсивность, чтобы вызвать заметный фототок. Следовательно, необходимо или увеличить интенсивность свечения источника излучения, или повышать чувствительность фотометрической установки, т. е. иметь достаточно мощный усилитель.
Рис. 32. Выбор оптимального раствора сравнения в дифференциальном методе при несоблюдении закона поглощения
Пуги увеличения интенсивности свечения источников излучений пгпаничены конструктивными особенностями приборов (возможно лазерные источники излучений помогут решить эту проблему). На спектрофотометрах это может быть достигнуто лишь увеличением ширины щели. Однако увеличение интенсивности потока за счет раскрытия щели может привести к большим отклонениям от закона поглощения, которые при дифференциальных измерениях вызывают большие ошибки, чем в обычном способе измерения [12].
'Итак, необходимо найти тот оптимальный раствор сравнения, который при дифференциальном измерении будет наиболее пригоден. Для этого следует учесть факторы, влияющие на ошибки дифференциального измерения при несоблюдении закона Бера [12].
Недостаточная монохроматичность потока излучения чаще всего вызывает отрицательные отклонения от закона поглощения излучений (рис. 32). Пусть для двух растворов, один из которых является
испытуемым (с2), а второй — раствором сравнения (су), справедливы соотношения (II 1.27). Тогда соотношения интенсивностей в обычном методе будут равны
y-=io~A* ю~(е^+л:); 1g А = _(е.С2+х)
где 8г = (Лг—х)/с,—текущее значение молярного коэффициента погашения (см. рис. 32); х и х' — отрезки, отсекаемые на оси ординат касательной к кривой в точках Аг и Л2. Если концентрации растворов близки, т. е. а = ~ 1, то х const, j2//i ~ 1 и 1g /2//х о.
Тогда с учетом соотношений (III.27)
• А
1 1'г
lg Z1---—l) = eic1(l—а) = ег Дс. (111.28)
Отсюда
Чтобы найти
/2 -К =----------------
ejcj
(111 29)
da/a = dc2/c2,
продифференцируем выражение (111.29) для 1g (72//г): , /2 А
d I Ig— j =d [Bj C1(l—a)] = 0—8f ci da,
0,43d
2
Следовательно,
0,43d
da =
1%
----ei ci
(I II.30)
Поделив (111.30)
на (III.29), получим
da
ei Cl
/2
— EjCi
— BiC!
^2 । . /а
~ 1g W1
(III.31)
a
Поскольку по условию I2Hi = 1, a 1g (12/0, следовательно, ошибка det/a = dc2/c2 будет тем меньше, чем больше е^. Таким образом, оптимальный раствор, честв° раствора сравнения,
который следует использовать в ка-должен удовлетворять условию
Рис. 33. Зависимость оптической плотности от концентрации в методе двусторонней дифференциальной спектрофотометрии
Зависимость относительной оптической плотности измеря-
Рис. 34. ошибки от емого раствора в методе двусторонней дифференциальной спектрофотометрии для различных значений оптической плотности растворов сравнения До
где не наблюдается значительных от-
Следовательно, в области, клонений от закона Бера, оптимальным является такой раствор сравнения, произведение концентрации которого на текущее значение ег, соответствующее этой концентрации, имеет максимальное значение.
Такой оптимальный раствор сравнения наиболее удобно исполь-ять е методе полной (двусторонней) дифференциальной спектрофо-3°метрии принцип которого был теоретически обоснован В ф Барковским и В. И. Ганопольским с сотрудниками [21], [22].
Метод позволяет при измерении получать как положительные, ак и отрицательные значения оптических плотностей (рис. 33). Причем соблюдение закона Бера в области отрицательных значений оптических плотностей наблюдается в широком интервале концентраций, несмотря на то, что теоретически ошибка в этой области возрастает быстрее, чем в области положительных значений (рис. 34).
Определение концентраций дифференциальным методом
При определении больших концентраций в данном методе в качестве раствора сравнения используют один из эталонных растворов. Этот раствор должен, во-первых, иметь достаточно высокую оптиче-' скую плотность, во-вторых, его пропускание должно быть как можно более близким к пропусканию испытуемого раствора.
При соблюдении закона поглощения излучений для определения концентраций могут быть использованы следующие варианты:
1. Графический — по градуировочному графику, построенному по серии эталонных растворов, оптические плотности которых измерены по отношению к первому или любому из растворов этого ряда. Прямая на графике отсекает на оси абсцисс отрезок, соответствующий концентрации раствора сравнения.
2. Алгебраический — по уравнению [26], [27]:
cx = A^F + cOi, (III.32)
где cOi i — концентрация вещества в растворе сравнения; Ах — оптическая плотность испытуемого раствора, измеренная по отношению к раствору сравнения; F — фактор пересчета.
Фактор в уравнении (III 32) представляет собой отношение разности концентраций двух растворов к оптической плотности более концентрированного раствора с2, измеренной по отношению к раствору с меньшей концентрацией сг.
„ Для двух растворов с известной концентрацией q и с2 и раствора с неизвестной концентрацией сх при измерении их поглощения по отношению к растворителю оптические плотности обозначим соответственно А1г А2 и Дж. При соблюдении основного закона поглощения
(А2 — Ат) / (Аж — Дх) = (с2—С1) / (сж—d).
fi Разности Аг 41 и Ах — An обозначенные через А2 и Ах, представляют ’ и оптические плотности растворов с2 и'сх, измеренные дифференциально по отношению к раствору с концентрацией сг
А'2!А'х = (сй —d) / (сх—d),
As сх—А2 d=4J( (с2—d), ,, [ с2 — d \
Сх = АЛ—+с1. (III. 33)
где c-^z£l = f
Для вычисления фактора F готовят ряд эталонных растворов в интервале концентраций, в котором соблюдается основной закон поглощения. Измеряют оптическую плотность каждого из растворов по отношению к первому из них; затем измеряют оптические плотности всех последующих растворов по отношению ко второму и г. д. По формуле
F = (Ш. 34)
/1/
на основании всех измерений вычисляют фактор F и находят среднее его значение. Оптическую плотность испытуемого раствора измеряют по отношению к одному из эталонных растворов, поглощение которого (соответственно концентрация) наиболее близко к испытуемому и вычисляют его концентрацию по формуле (III.32).
Преимущество метода расчета по формуле (II 1.32) по сравнению с графическим методом состоит в том, что концентрация раствора сравнения может быть взята максимально близкой к концентрации испытуемого раствора.
При несоблюдении закона поглощения излучений необходимо для построения градуировочного графика предварительно выбрать оптимальный раствор сравнения. Оптимальную концентрацию раствора сравнения выбирают следующим образом [9]. Готовят ряд эталонных растворов с постоянной величиной Де и интервалами ДА не более 0,2—0,4. Измеряют оптические плотности каждого последующего раствора по отношению к предыдущему и вычисляют - А,/Дс и е{с01 i, где с0,; — концентрация раствора, который в i-м измерении является раствором сравнения.
Для построения градуировочного графика измеряют оптические плотности серий растворов по отношению к раствору, для которого произведение е;с01 г имеет максимальное значение. При этом оптические плотности будут иметь отрицательные значения, если поглощение испытуемого раствора меньше поглощения раствора сравнения [21], [22]. Обычные спектрофотометрические приборы не приспособлены для измерения отрицательных значений А. Последние могут быть получены, если измерять оптическую плотность раствора сравнения по отношению к исследуемым растворам и брать их со знаком «—»•
По полученным данным строят градуировочный график в координатах А' — с. Измеряют А испытуемого раствора по отношению к тому же раствору сравнения и определяют его концентрацию по градуировочному графику, используя положительную и отрицательную ветви /'см. рис. 33).
4. АНАЛИЗ МНОГОКОМПОНЕНТНЫХ СИСТЕМ
^_Одно из преимуществ работы с монохроматическими излучениями заключается в возможности спектрофотометрического анализа многокомпонентных систем без предварительного разделения компонентов.
Взаимные помехи между отдельными компонентами смеси, анализируемой спектрофотометрическим методом, при их определении 72
Условлены, главным образом, наложением поглощений в области чин волн, соответствующих максимумам поглощения этих композитов. Наиболее часто затруднения при спектрофотометрическом Нпоеделении вызывает избыток окрашенного реагента. Чтобы исключить помехи со стороны реагента, иногда используют его в качестве Оаствора сравнения (рис. 35). Поскольку равновесная концентрация
реагента обычно бывает неизвестна, то в раствор сравнения вводят такое же его количество, которое взято для проведения фотометрической реакции [39]. Но это допустимо только, если используют большой избыток реагента и можно пренебречь той долей, которая израсходована на получение комплексного соединения, как, например, при образовании недостаточно прочных комплексов. При этом собственное поглощение реагента должно быть невелико. Однако большинство органических реагентов, с одной стороны, образует с катионами металлов достаточно устойчивые комплексные соединения, с другой — обладает значительным собственным поглощением. Вследствие этого избыток реагента обычно бывает небольшим (двух—
02
Z 0,3
VfO 560 560 600 020 6W Л,им
Рис. 35. Спектры поглощения реагента 2-(п-сульфонилазо)--1,8-диоксинафталин-3,6-ди-сульфокислота (СПАДНС) (1) и его комплекса с цирконием (2), снятые по отношению к воде, и дифференциальная кривая (3)
четырехкратный) и пренебречь долей, вступившей в реакцию комплексообразования (если комплекс образуется, например, с соотношением компонентов Me : R - 1:2), уже нельзя.
Поэтому для исключения помех со стороны реагента, а также других со-
путствующих компонентов более пра-
вильно применять какие-либо из вариантов спектрофотометрического
анализа многокомпонентных систем, в которых используются дифференциальные варианты измерений.
I. Классический метод спектрофотометрического анализа многокомпонентных си-
стем основан на решении системы уравнений, число которых должно быть равно или больше числа определяемых компонентов. При соблюдении закона аддитивности для каждой длины волны оптическая плотность раствора, состоящего из п компонентов, равна сумме оптических плотностей всех компонентов:
^=4+4+--- + а£.
В зависимости от относительного расположения максимумов полос поглощения компонентов, содержащихся в смеси, могут быть использованы различные варианты определения компонентов смеси.
1. Кривые спектров поглощения компонентов смеси накладываются по всему спектру. Измерив оптические плотности раствора при п к.
длинах волн и определив предварительно значения е.1 для п компонентов при и длинах волн, решением системы уравнений:
— 61’ ci е2* с2 Z-T... + е^1 сп I,
(Ш.35)
Рис. 36. Спектры поглощения комплексных соединений кобальта, никеля и меди с комплексоном III
А ПClZ-p ®2П С2/-р • “|-Ert7iC7i Z
находят концентрации всех п компонентов *. При выбранных длинах волн поглощение каждого из определяемых компонентов должно быть заметным. Иначе ошибка в определении е/ будет очень велика.
Этот вариант был применен для анализа смеси никеля, кобальта и меди в виде их комплексонатов (40]. Электронные спектры поглощения соединений этих элементов с комплексоном III представлены на рис. 36. Оптимальные длины волн измерения суммарной оптической плотности приведены в табл. 9. Использование большой серии измерений при длинах волн, близких к оптимальным, и решение системы уравнений, число которых значительно больше числа определяемых компонентов, существенно повышает точность результатов анализа. Однако решение системы такого большого числа уравнений требует применения ЭВМ.
2. Кривые спектров поглощения накладываются не по всему спектру, т. е. имеется участок
спектра (или длина волны), в котором поглощение одного из компонентов велико, а поглощение второго практически отсутствует. Тогда при анализе двухкомпонентной смеси определяют поглощение первого компонента при этой длине волны и вычисляют его концентрацию сг по уравнению
А^1
С1=-т—. (П1.36)
Концентрацию второго компонента с2 вычисляют, измерив оптическую плотность в области максимума поглощения этого компонента
i4i=e2* c2Z-|-ei* cjZ.
(III. 37)
ТАБЛИЦА 9
Оптимальные длины воли для измерения оптической плотности при анализе системы, состоящей из никеля, кобальта и меди, в виде их комплексонатов
Элемент з е-10 при й, (нм)
436 367 328
Медь 12,8 1,26 2,23
Кобальт 2,26 14,3 21,8
Никель 1,72 3,91 35,2
Подставив в уравнение (III.37) значение с1г вычисленное по уравнению (III.36) и решив его относительно с.2, получим:
АКг ei1 —ё12 е22 ej’* I
(III.38)
3. Кривые спектров поглощения не накладываются ни при каких длинах волн. Концентрация каждого из компонентов может быть вычислена по уравнению (1.9), если предварительно были определены значения- & .
В варианте 3 для измерения поглощения каждого из компонентов, а также в варианте 2 для измерения поглощения первого компонента длину волны выбирают таким образом, чтобы в этой области поглощение мешающих компонентов было минимальным. Рассмотренные варианты анализа многокомпонентных систем не могут быть применены, если нельзя определить предварительно молярные коэффициенты погашения с достаточной степенью точности (например, нельзя приготовить раствор поглощающего реагента точной концентрации).
II. Метод анализа многокомпонентных систем, основанный на дифференциальных измерениях, имеет несколько вариантов.
1. Выбирают две длины волны таким образом, чтобы разность молярных коэффициентов погашения (еМ — еМ для определяемого компонента (су) была достаточно велика, а для сопутствующего (с2) и реагента (с3) — минимальна, т. е. разности (е^ — е^>) и (ф — ез’) фактически стремятся к нулю. Для двух длин волн оптические плотности выражаются
= (ej-1 Су + е2* с2 е2* с3) I,
Лг12-(е^с1 + е’-2с2-Ье^Сз)/.
Тогда их разность
"= Ki I (fc'i1 — Si2)-|-c2 l (е2‘— s22)+c3Z(e31 — е32)] оказывается пропорциональной концентрации определяемого компонента: так как (ф — ф) ~ О и (ф — ф) ~ 0, то
ДА=дМ_ AK = k'ci = f (С1), (III.39)
где k' = I (e^« — e^). По зависимости ДА - f (сг) можно определить содержание компонента в смеси. Измерив поглощение анализируемого раствора при тех же двух длинах волн, при которых делались измерения при построении градуировочного графика, находят разность этих измерений ДА, соответствующую ординате градуировочного графика.
2. Используют вариант 1 в сочетании с дифференциальным способом измерения, если отсутствуют две длины волны, при которых Де для всех мешающих компонентов практически равна нулю.
Предположим, что максимальное суммарное содержание смеси анализируемых компонентов всегда постоянно и меняется только относительное содержание двух компонентов в смеси. При построении градуировочного графика по методу 2, при измерении поглощения при двух длинах волн в качестве раствора сравнения используют не растворитель, а раствор эталонного ряда с минимальной концентрацией определяемого компонента. При измерении поглощения анализируемого раствора при тех же длинах волн в качестве раствора сравнения берут раствор смеси, содержащий определяемый компонент в той же концентрации, которая была взята при измерении эталонных растворов.
3. Исключают помехи со стороны посторонних компонентов, используя для приготовления раствора сравнения и испытуемого раствор анализируемого образца [41]. Перед проведением фотометрической реакции в три мерные колбы помещают определенные объемы анализируемого раствора: в первую несколько меньший, в две следующие одинаковый, но больший, чем в первую. Кроме того, в третью колбу прибавляют еще определенное количество стандартного раствора анализируемого компонента. После этого во всех трех колбах проводят фотометрическую реакцию и измеряют оптическую плотность второго и третьего растворов по отношению к первому. Пусть с0 — содержание определяемого компонента в объеме испытуемого раствора, прибавленного в первую колбу; сх — содержание определяемого компонента в объеме v2 испытуемого раствора, прибавленного во вторую и третью колбы; А х — оптическая плотность раствора, с содержанием определяемого компонента сх, измеренная по отношению к растворителю; Ао — оптическая плотность раствора с содержанием определяемого компонента с0, измеренная по отношению к растворителю; са — количество определяемого компонента, добавленного в третью колбу; Ая — оптическая плотность раствора с содержанием определяемого компонента са, измеренная по отношению к растворителю. Согласно закону поглощения
-Ло сх—с0
Лх~Ь Ла—Л о cK-J-ca— со
После простых преобразований получим
са (Л v — Ло) = Л а (сх — С0), I-----.--I I_________I
А сх
me Л v — Ло = А*, сх — с0 = с'х. Тогда 1 A*-- v
c'x=£^L, х л
(111.40)
— оптическая плотность второго раствора с содержанием определяемого компонента сх, измеренная по отношению к первому раст-; oDV с содержанием определяемого компонента с0; с'х — содержание определяемого компонента в таком объеме испытуемого раствора, который равен разности объемов, прибавленных во вторую и первую колбы (р2 — у1)’’ /а — разность оптических плотностей третьего и второго растворов, измеренных по отношению к первому раствору.
4. Измеряют оптическую плотность раствора, содержащего определенное количество смеси двух компонентов по отношению к раствору с той же массой одного из компонентов. Строят зависимость измеренной оптической плотности от процентного содержания компонентов в смеси и используют ее для определения их содержания.
Предположим, что имеется смесь компонентов Mi и Мц, причем атомный вес компонента Mj меньше, чем компо
нента Мц. Тогда для одной и той же массы т согласно закону поглощения излучений получим:
Рис. 37. Зависимость оптических плотностей растворов соединений двух элементов М[ и
М1Г от концентрации
m
AMt
« ш.
AMj < АМц-
Зависимости оптических плотностей растворов, содержащих компоненты Mi и Мц, от концентрации представлены на рис. 37. Причем поглощение любой смеси компонентов Mi и Мц с постоянной массой т будет находиться в промежутке между поглощениями одного только компонента Mi или одного компонента Мц с той же массой т. Оптическая плотность раствора, содержащего только компонент Mi, может быть представлена как сумма двух оптических плотностей, одна из которых равна поглощению раствора компонента Мц с массой т:
тх
т
EMi I д eMi д
т.
/л, т—тх
ЛМ[ = ЕМ — + t’Mi— "Mj *'М]
или
, т tn—тх
^5=%! Г—+ ^-7----------’ (Ш.41)
1 АМП ЛМ;
-'-X
AMnEMj
Таким образом, если в качестве раствора сравнения при измерении поглощения смеси компонентов Mi и Мц взять раствор, содержащий компонент Мц в количестве т, то измеряемая оптическая плотность будет прямолинейно зависеть от относительного содержания компонента Mi в смеси суммарного веса т. Тангенс угла наклона прямой будет зависеть от соотношения оптических плотностей компонентов Mi и Мц. Это соотношение (т. е. разница в оптических плотностях) равно
еМ[ Ам„
---=----------- (если т = тмг = Ц?мТт) АМц емп Амг
и будет прямо пропорционально молярным коэффициентам погашения и обратно пропорционально атомным весам компонентов Mi и Мц. Следовательно, чем больше это соотношение, тем больше относительная чувствительность определения одного компонента в присутствии другого. Кроме того, как следует из теории дифференциальной спектрофотометрии, точность измерений будет тем выше, чем большую оптическую плотность будет иметь раствор сравнения. На рис. 37 видно, что чем выше масса т, тем больше разница в оптических плотностях растворов компонентов Mi и Мц и, следовательно, при большой массе т будут выше и чувствительность и точность определений. Процентное содержание компонента Mi определяют по градуировочному графику, измеряя оптическую плотность раствора, содержащего анализируемую смесь в количестве тпо отношению к раствору компонента Мц с той же массой т.
Этот метод был использован для анализа смеси Zr — Hf в виде их соединений с ализарином S [42] и с арсеназо III [43]. Молярные коэффициенты погашения соединений Zr и Hf с тем и другим реагентом были одинаковы, но достаточной была разница в атомных весах. Различие в величинах молярных коэффициентов погашения соединений редкоземельных элементов с такими реагентами как арсеназо М и ортаниловый А позволяет анализировать двухкомпонентные смеси элементов этой группы, несмотря на незначительные различия в атомных весах.
5. ЭКСТРАКЦИОННО-СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД
Экстракционные методы широко применяются в аналитической химии. Несмотря на развитие целого ряда инструментальных методов, отличающихся избирательностью, не всегда удается непосредственное определение отдельных элементов в объектах и сложных
месях; оно может быть выполнено после предварительного разделения методом экстракции, основанном на различной растворимости тдеЛьных соединения в системе «неводный растворитель — вода». Очень удачно сочетание экстракционного разделения с последующим спектрофотометрическим определением элементов по поглощению органической фазы. Этот метод назван экстракционно-спектрофотометрическим. Метод позволяет разделять вещества, резко отличающиеся по концентрации, поэтому особое значение он приобретает при определении микропримесей элементов в особо чистых веществах.
Метод экстракции применяют для отделения микрокомпонентов от макрокомпонентов (основы) с последующим определением их спектрофотометрическим методом. Этот метод используют и для отделения основного вещества от микрокомпонентов, остающихся в водной фазе, с последующим их определением также спектрофотометрическим методом.
/ Распределение вещества между двумя несмешивающимися жид-1&стями при установлении равновесия при постоянной температуре характеризуется термодинамической константой равновесия 7<терм = = —, где а0 и аБ — активности веществ в органической и водной фа-пв
зах соответственно.
Чаще всего пользуются концентрационными константами распределения (Ко):
V [А1° ь- _ Уо к [A]bVb-Kd
Уо
Ув
где yj — коэффициенты активности в органической и водной фазах.
Константа распределения Kd в условиях равновесия выражает отношение концентрации вещества определенного состава в органической фазе к концентрации того же самого состава его в водной фазе. Например, для 8-оксихинолина
[НОх]о D [НОх]в •
Константа распределения не зависит от pH раствора, но зависит от ионной силы раствора и температуры.
Для исследования процесса комплексообразования необходимо знать распределение вещества между двумя фазами в зависимости от побочных процессов, которые могут протекать в водных и органических средах. Распределение вещества между двумя фазами характеризуется коэффициентом распределения D, который зависит от pH, ионной силы р и температуры t раствора:
р_ [А]о сс0
[А]в ссв
где а0 и ав — коэффициенты побочных реакций в соответствующих средах.
Константу экстракционного равновесия называют константой экстракции. Например, образование 8-оксихинолината иона металла протекает по уравнению
М"+4-пНОхо5±М(Ох)По+пН+,
тогда
[М (Ох)п]о[Н+г [М'Ч ] [НО<
(Ш .42)
Для области pH, характерной для образования только одного экстрагирующегося комплексного соединения и при отсутствии в водной , [М (Ох)пр
фазе комплексов другого состава, отношение -тЛау можно заме-
T-JB
нить значением D (коэффициента распределения), тогда
[Н+]п
= lHOxjn- (III.43)
В настоящем пособии большое внимание уделяется использованию экстракционно-спектрофотометрического метода для определения малых и ультрамалых количеств отдельных элементов, как примесей в чистых веществах и естественных объектах. Вопросы механизма процесса экстракции освещены в ряде монографий и сборников [28]—[321.
Методы прямой, обменной и последовательной экстракции для определения элементов
Для аналитической химии большой интерес представляет экстракция незаряженных внутрикомплексных соединений в условиях образования мономерных частиц в органической фазе. Обычно в качестве лигандов применяют органические реагенты, обладающие кислотными функциями (А'дасс — — 10~1в), являющиеся чаще
всего бидентатными. Примером определения элементов в виде внутрикомплексных соединений является определение никеля в ряде объектов а-диоксимами (стр. 186), кобальта нитрозо-нафтолами (стр. 160), цинка дитизоном (стр. 220), алюминия 8-оксихинолином и др. Для этого определяемый элемент в виде внутрикомплексного соединения переводят в органическую фазу с последующим фотометрированием экстракта.
В последнее время все большее применение находит метод обменной экстракции с последующим определением элементов спектрофотометрическим методом. Ю. А. Золотовым [29] даны теоретические ос
новы метода и приведен ряд примеров его практического применения. В качестве реагента обычно применяют раствор внутрикомплексного соединения какого-либо элемента в органическом растворителе. Определяемый элемент (Мх) из водной фазы при перемешивании фаз переходит в органическую фазу, содержащую элемент (М2), вытесняет этот элемент, образуя более устойчивое и лучше экстрагируемое комплексное соединение. Таким путем повышается избирательность
метода- Например, при использовании в качестве экстрагента диэтил-читиокарбамииата свинца значительно сокращается число элементов, мешающих спектрофотометрическому определению меди (Ti, Bi, Ag, jjg) чем при применении диэтилдитиокарбамината натрия (Ti, V, Hg ’ Ag, Со, Ni, Au, Pb, Pt, Os). Метод пригоден для определения ме-й Б солях кобальта и никеля [33].
Д Некоторыми авторами составлены ряды элементов в порядке их вытеснительной способности: 8-меркаптохинолинатов элементов [34], дитизонатов [35], аналогов диэтилдитиокарбаминатов [33].
Существует связь между константами реакций обмена и экстракции [29]. Константы экстракции для соединений двух элементов будут соответственно равны
. {АЦАпМН+р
[М"+][НА]" ' (Ш.44)
I, lMn Am]ofH+Jm
Кех = [HA]m ’ (Ш .45)
где Mi — определяемый элемент (в); Мц — вытесняемый элемент (о).
В условиях равновесия, комбинируя уравнения (III.44) и (II 1.45), можно написать выражение для константы обмена
<u’“)
Уравнение для Кех будет
Рма Ко, I КнА
Кех =-----~п-----. (III.47)
'D
где Рмап — общая константа устойчивости комплексного соединения МАП; Ко, I — константа распределения комплексного соединения МАП; Ко — константа распределения реагента; Уна — константа диссоциации реагента.
Подставив значение Кех Для обоих комплексов в (III.46), получим
(₽МА KDii)m
Кобм= . Д ~
(III.48)
Из уравнения (III.48) видно, что полнота обмена зависит от соотношения констант устойчивости и констант распределения обоих комплексов.
Спектрофотометрические методы с использованием последовательного экстрагирования представляют практический интерес. Так, например, более устойчивое комплексное соединение — дитизонат меди— экстрагируется при pH 2, менее устойчивое — дитизонат цинка — в более щелочной среде при pH ~ 9, что позволяет последовательно определять эти элементы в их смеси или разделять их {37]. Экстракционно-спектрофотометрический метод применяется для од-
повременного определения двух элементов, образующих комплексы с общим лигандом, но имеющими области максимального поглощения в различных участках спектра; Хшах хлороформного экстракта бен-зоилфенилгидроксиламината циркония 275 нм, для аналогичного соединения титана Хтах 355 нм.
Известен большой класс соединений, называемых ионными ассоциатами, которые образуются путем ассоциации противоположно заряженных частиц. Эти соединения широко используются в экстракционно-спектрофотометрических методах определения многих элементов [36].
Большую группу ионных ассоциатов составляют соединения, в состав которых входят в виде положительно или отрицательно заряженных частиц красители основного или кислотного типа. Так, напри мер, сульфофталеиновые красители, содержащие хромофорные группы, повышают чувствительность методов. Известны методы, в которых определяемые ионы металла координируют неорганические лиганды, образуя комплексный анион, а в качестве противоиона содержат основные красители, например определение галлия (стр. 136). Аналогичного типа ионные ассоциаты образуют также соединения, у которых комплексный анион сочетается с аминами, например роданид железа с дидециламином (стр. 150). В ряде других методов в качестве противоиона используют краситель, который является отрицательно заряженной частицей. Например, при определении железа в виде ферроин-иодида, экстрагируемого хлороформом. Иодид-ион в органической фазе заменяется на бромфеноловый синий (стр. 158), что увеличивает чувствительность метода определения железа.
Экстракционно-спектрофотометрические методы применяются при исследовании процессов комплексообразования, определении констант экстракции и устойчивости комплексных соединений и констант равновесия химических реакций. При использовании метода экстракции (распределения) равновесную концентрацию иона металла — комплексообразователя в системе М—R «органический растворитель — вода» определяют в органической фазе, если поглощение образующегося соединения может быть измерено (см. стр. 126). Зная общую концентрацию металла, по разности находят его концентрацию в водной фазе. Например, при исследовании комплексообразования никеля с рядом диоксимов в интервале концентраций 4,5х X 10-5— 1,3 • 10-4 моль/л равновесную концентрацию никеля определяют в органической фазе по поглощению соответствующего диок-симата никеля.
6. КИНЕТИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ЭЛЕМЕНТОВ
СО СПЕКТРОФОТОМЕТРИЧЕСКИМ КОНТРОЛЕМ
Кинетические методы анализа основаны на использовании зависимости между скоростью реакции и концентрацией реагирующих веществ:
г=Асасв, (HI-49)
v___скорость реакции; k — константа скорости реакции; сА и св —
ГДнцентрации реагирующих веществ А и В соответственно. Для опре-К°ления малых количеств вещества обычно используют каталитические реакции, в которых катализатором служит определяемый элемент
(III. 50'
^2 — с в скат t
где ____скорость каталитической реакции; х — каталитический ко-
эффициент; скат — концентрация катализатора.
^Реакция, скорость которой определяется концентрацией катализатора, называется индикаторной, а вещества, по изменению концентрации которых судя'т о скорости реакции, — индикаторными веществами. Если индикаторное вещество — продукт реакции и концентрация его обозначается через х, то уравнение (II 1.50) принимает вид
dx
= хсдСвсКат- (III. 51)
Если по мере протекания реакции концентрации реагирующих веществ А и В существенно не меняется, то, заменив в уравнении (II 1.51) их произведение сАсв на постоянную Пс, получаем
—, = у.Пс Скат (III.52)
dt
или
dx
,, = k скат> (III.53)
dt
где k' — условная константа скорости, в величину которой входят концентрации реагирующих веществ, взятых в избытке. Тогда между концентрацией индикаторного вещества и временем протекания реакции (I) существует прямо пропорциональная зависимость (дифференциальный вариант кинетических методов анализа);
x=k Скат t. (III.54)
Если концентрация одного или нескольких реагирующих веществ во времени заметно меняется, применяется интегральный вариант кинетических методов анализа. В этом случае прямо пропорциональная зависимость наблюдается между какой-либо функцией концентрации индикаторного вещества и временем реакции. Если в ходе реакции существенно меняется концентрация одного из реагирующих веществ, например вещества А, то уравнение (III.51) можно записать в следующем виде:
dx
С’г=~т-=х(я—*)свскат, (III.55)
где а — начальная [концентрация вещества А; (а — х) — концентрация вещества А в каждый данный момент времени.
Преобразуя уравнение (III.55) и интегрируя его, получаем
— k скат/, а—х
(III. 56)
а , 1g = k скат /. а—х
(III. 57)
Если каталитическая реакция протекает с изменением окраски раствора (или ее интенсивности), то, измеряя оптическую плотность раствора, можно следить за изменением концентрации индикаторного вещества во времени.
Рис. 38. Зависимость оптической плотности от времени для различных концентраций определяемого вещества
Рис 39 Определение концентраций методом тангенсов
Для определения концентрации по данным кинетических измерений чаще всего используют метод тангенсов. По методу тангенсов оптическую плотность растворов, содержащих различное количество элемента — катализатора, измеряют через определенные промежутки времени. Затем строят графики в координатах оптическая плотность — время (дифференциальный вариант кинетического метода анализа, рис. 38) или функция оптической плотности — время (интегральный вариант). По графику определяют тангенсы угла наклона полученных прямых и строят градуировочный график в координатах tg а — концентрация катализатора (рис. 39). Для определения концентрации по данным кинетических измерений используют также способы фиксированного времени, фиксированной концентрации и др. [44].
7. ОБРАБОТКА РЕЗУЛЬТАТОВ ИЗМЕРЕНИИ
Применение метода наименьших квадратов для построения градуировочного графика
При построении градуировочного графика по эталонным растворам может быть значительный разброс результатов измерения особенно при работе с малыми концентрациями веществ, В этом случае
построение градуировочного графика следует проводить, используя метод наименьших квадратов.
Для метода наименьших квадратов справедливо утверждение: «Для лучшей кривой сумма квадратов отклонений минимальна». На рис. 40 символами О2, О3, О4, О5 обозначены точки, соответствующие экспериментально полученным данным. Если из каждой точки опустить перпендикуляр на кривую, то сумма квадратов отклонений (6) должна быть минимальной, т. е. У
У, <5f = min. (III.58)
i= 1
Рассмотрим применение метода наименьших квадратов для построения градуировочного графика. Урав-ненйе прямой можно записать в виде y=f (х)=В0 + Bix, (III.59)
Рис 40 Разброс эксперименталь ных данных относительно истинном кривой
где Во и Вг — постоянные (Во — отрезок, отсекаемый прямой на оси ординат; Bt — тангенс угла наклона прямой к оси абсцисс); х - переменная величина.
Если рассчитать Вй и Bt с учетом основного требования метода
наименьших квадратов [см. уравнение (111.58)1, то можно построить прямую, наилучшим образом удовлетворяющую экспериментальным точкам х2 ••• хп- Прямую можно построить по двум экспериментальным точкам (Xjj/x) и (х2у2). Согласно условию метода наименьших
квадратов:
^o + Sl^l-У1=&1, Bg-YByX^----1/2=62,
тогда
F=61 + 6|=(fil) + B1xi-z/1)2 4-(fi() + fiix2-z/2)2 = min. (III.60)
Значение функции минимально, если ее частные производные равны 0. Поэтому продифференцировав уравнение (III.60) по переменным Во и Вг и приравняв частные производные нулю, получим следующие уравнения:
dF
= 2(В0-рВ1х1—//j)-|-2 (Во—1/2),
О£50
dF
—— —2 (В„-| BlA’i — У’) A'i (-2 (Во— //г) х2', uBi
| В0 + В1Х1 — //i + Bo + Bi^a — {/2 — 0,
I Во X] + В! X* — У! X! + Во х2 + Brxf — х2 у2 = 0. Сгруппировав члены уравнений (III.61) по Во и Вг, ( 2B0-}-Bi(xi+x2) = f/i + i/2,
I Во (Х1 +х2) + В4 (xf + xl) = х± Ух+х2 ys.
(III.61)
получим
(III. 62)
Если градуировочный график строится по п экспериментальным точкам, то система уравнений (III.62) примет вид:
лВ04-б! (xi+x2 + . .. +хп) = У1-\-уг-\- ... -}-уп,
(xi4-x2~P • ~Pxn) (xi 4* — (III.63)
= *1 У1 + х2 У2 + • • • + ХП Уп или . i~n i=n
ябоЧ-Bl У xi= . yi,
i==1 i=1 (III. 64)
i = n i=n i = n
Bi £ 4+fio У xi= 1 xivi-i=i i=i i=t
Решая эту систему относительно Во и Въ получим
п п п
Рассчитав Во и Blt по полученным данным строят прямую, используя уравнение (II 1.59).
Пример. При определении меди кинетическим методом по способу тангенсов (стр. 178) получены следующие данные (п = 4):
х—количество Си-103, мкг/мл 4 6 8 10
у—tga 7,7 10,4 15,0 20,3
Подставляя экспериментальные данные в уравнение (111.63) или (Ш.64), получим 4В0 + (4 + 6 + 8 + 10) = 7,7 + 10,4 + 15,0 + 20,3, В„ (4 Е
+ 6 + 8 + 10) + Bi I(4)s _[_ (6)2 + (8)2 + (10)2] = 4 . 7 7 + 6 . ]0>4 + 8 Х15,010 -20,3). Откуда рассчитываем по уравнению (III.65) Вг = 2,05 и В(,= —1 и получаем уравнение для прямой: z/=2,05 х~ 1. Используя полу ченную зависимость, находим точки для построения градуировочного графика по методу наименьших квадратов-
х—количество Си-103, мкг/мл 4 6 8 10
У—Tga 7,2 11,3 15,4 19,5
Статистическая обработка экспериментальных результатов
Оценка достоверности получаемых результатов имеет первостепенное значение, особенно при работе с микроколичествами веществ. Поэтому результаты измерений обрабатывают с применением методов математической статистики [45], [46]. Методы математиче-
ТАБЛИЦА 10
Значения ta в зависимости от /г и а (таблица Сююдента)
k ta при а
0,9 | 0,95 | 0,99 0,999
1 6,314 12,706 63,657 636,619
2 2,920 4,303 9,925 31,598
3 2,353 3,182 5,841 12,941
4 2,132 2,776 4,604 8,610
5 2,015 2,571 4,032 6,859
6 1,943 2,447 3,707 5,959
7 1,895 2,365 3,499 5,405
8 1,860 2,306 3,355 5,041
9 1,833 2,262 3,250 4,781
10 1,812 2,228 3,169 4,587
И 1,796 2,201 3,106 4,487
12 1,782 2,179 3,055 4,318
13 1,771 2,160 3,012 4,221
14 1,761 2,145 2,977 4,140
15 1,753 2,131 2,947 4,073
16 1,746 2,120 2,921 4,015
17 1,740 2,110 2,898 3,965
18 1,734 2,103 2,878 3,922
19 1,729 2,093 2,861 3,883
20 1,725 2,086 2,845 3,850
21 1,721 2,080 2,831 3,819
22 1,717 2,074 2,819 3,792
23 1,714 2,069 2,807 3,767
24 1,711 2,064 2,797 3,745
25 1,708 2,060 2,787 3,725
26 1,706 2,056 2,779 3,707
27 1,703 2,052 2,771 3,690
28 1,701 2,048 2,763 3,674
29 1,699 2,045 2,756 3,659
30 1,697 9,042 2,750 3,646
40 1,684 2,021 2,704 3,551
60 1,671 2,000 2,660 3,460
120 1,658 1,980 2,617 3,373
1,645 1,960 2,576 3,291
ской статистики применяются для оценки случайных ошибок измерения. Если было сделано п измерений, то можно вычислить х — среднее арифметическое значение для выборки п наблюдаемых результатов:
- *1 + *2 + *з+• - +*п Х =------------------- ,
п
s2 — выборочную дисперсию для п наблюдений:
s *- среднее квадратичное отклонение (или среднеквадратичную ошибку) определения:
S=ys=i/
» У п — 1
Дисперсия и среднеквадратичное отклонение характеризуют воспроизводимость метода, т. е. рассеяние отдельных измерений относительно их среднеарифметического х.
При отсутствии систематических ошибок оценка точности определения проводится вычислением доверительного интервала р, внутри которого с заданной степенью надежности а лежит истинное значение определяемой величины
— ®
р = Х zt • (III.66)
У п
где ta — табличная величина распределения Стьюдента (табл. 10), которая табулирована для определенного значения степени надежности а и числа степеней свободы k (или /) = п — 1. Степень надежности задается экспериментатором (обычно а принимают равным 0,95 или 0,90). Таким образом, полученные экспериментальные данные сводят в таблицу по образцу:
X n s2 s s KT — Vs x ± r - V n
ЛИТЕРАТУРА
1. Н. F I a s с h k a. Taianta, 1961, 8, 381.
2. R. Malmst a dt, Е. Gohrbandt. Anal. Chem., 1954, 26, 442.
3. A. Ringbom, E. V a n n i n e n. Anal. Chim. Acta, 1954, 11, 153.
4. M. И. Громова, О. Д. Ларина, Т. А. Рязанова. Новые методы испытаний металлов. Сб. трудов ЦНИИЧМ. «Металлургия», вып. 73, 1969, с. ПО.
5. J. М. Н. F о г t u i n, Р. Karsten, И. Kies. Anal. Chim. Acta, 1954, 10, 356.
6. C. L i t e a n u. Taianta, 1960, 7, 18.
7. Г. Л. Ill лефер. Комплексообразование в растворах. М.—Л., «Химия», 1954.
8. G. Kortum. Angew. Chem., 1937, 50, 193.
9. R. Bastian. Anal. Chem., 1949, 21, 972; 1957, 25, 259; 1950, 22, 160; 1951, 23, 580.
10. C. F. H i s k e y. Anal. Chem., 1949, 21, 1440.
II. C. F. H i s k e у, I. Rabinowitz, I. G. Young. Anal. Chem., 1950, 22, 1469.
12. C. F. Hiskey, I. G. Young. Anal. Chem., 1951, 23,506,1196.
13. C. F. Hiskey, D. Firestone. Anal. Chem., 1952, 24, 342.
14. R. Bastian, R. Weber 1 i ng, F. P a 1 1 1 1 a. Anal. Chem., 1950, 22, 160.
15. Г. С. Терешин. ЖАХ, 1959, 14, 516.
16. G. S v e h 1 a, A. Pall, L. Erdey. Taianta, 1963,10,719.
17. A. Pall, G. S v e h 1 a, L. Erdey. Taianta, 1964, 11, 1383.
18. G. S v e h 1 a. Taianta, 1966, 13, 641.
19^ С. M. Crawford. Anal. Chem., 1959, 31, 343.
2(L С. H. R e i 1 1 e у, С. M. Crawford. Anal. Chem., 1956, 27, 716.
21. В. И. Ганопольский, В. Ф. Барковский, Т. А. Г а-но п ол ьс к а я. Зав. лаб., 1964, 30, 267.
22. В. Ф. Барковский, В. И. Ганопольский. Дифференциальный спектрофотометрический метод. М.—Л., «Химия», 1969.
23. С. Banks, J. S р о п е г, J. О. L a u g h 1 i п g. Anal. Chem., 1958, 30, 458; 1956, 28, 1894.
24. S. Poss, D. В i I s о n. Analyst., 1960, 85, 51.
25. В. Kent ni k. Chem. Anal., 1963, 18, 5, 649; 1964, 19, 5, 857; 1965, 10, 3, 329 (Polsky).
26. C. D. S и s а п о, O. Menis, С. K. Talbott. Anal. Chem., 1956, 28, Ю72.
27. Б. M. Добкина, T. M. Малютина. Зав. лаб., 1958, 24,1336, 1961, 27, 650.
28. Ю. А., 3 о л о т о в, И. П. А л и м а р и и. ДАН СССР, 1960, 138, 603.
29. Ю. А. Золотов. Экстракция внутрикомплексных соединений. М.—Л., «Наука», 1968.
30. Дж., Моррисон, М. Фрейзер. Экстракция в аналитической химии. М.—Л., Госкомиздат, 1960.
31. И. Стары. Экстракция хелатов. М.—Л., «Мир», 1966.
32. Р. М. Даймонд, А. Г. Та к. Экстракция неорганических соединений. Атомиздат, 1962.
33. G. Gottschalk. Z. anaL chem., 1963, 194, 321.
34. Ю. А. Банковский; АЛ Ф. И е в и н ь ш, 3. Э. Л и е и и-н я. ЖАХ, 1960, XV, 4.
35. Г. И в а н ч е в. Дитизон и его применение. М., ИЛ, 1961.
36. И. А. Блюм, Н. Н. Павлова. ЖАХ, 1965, 20, 898.
37. А. Б- Блан к. Методы концентрирования веществ в аналитической химии. XV. М.—Л., «Наука», 1965.
38. В. М. Пешкова, Н. В. М е л ь ч а к о в а. Е. Д. С и н и ц и-н а. Известия высших учебных заведений. Химия и хим. технология, VII, вып. I, 1960, 72.
39. G. В е п е г j е е. Anal. Chem., 1957, 29, 55.
40. Н. Fl ascii k a. Taianta, 1960, 1, 90.
41. В. М. Пешкова, М. И. Громова. Практическое руководство по спектрофотометрии и колориметрии. М., Изд-во МГУ, 1965.
42. F. Holbrook, Н. Freund. Anal. Chem., 1958, 30, 462.
43. Н. В. М е л ь ч а к о в а, Н. И. Трубецкая, В. М. Пешкова. Вестник Моск, ун-та. Серия II, Химия, 1964, №2, 45.
44. К- Б. Я Ц и м и р с к и й. Кинетические методы анализа. М.—Л., «Химия», 1967
45. В. В. Налимов. Применение математической статистики при анализе веществ. М., «Физматгиз», 1960.
. 46. К. Доерфель. Статистика в аналитической химии. М., «Мир»,
Глава IV
ПРИМЕНЕНИЕ МЕТОДА
АБСОРБЦИОННОЙ СПЕКТРОСКОПИИ
ДЛЯ ИЗУЧЕНИЯ РАВНОВЕСИЙ В РАСТВОРАХ
При поглощении электромагнитных излучений УФ, видимого и ИК диапазонов не нарушается цельность поглощающих соединений, т. е. не происходит разрыва химической связи. Это позволяет использовать метод абсорбционной спектроскопии для определения состава соединений и изучения равновесий в растворах.
Все расчеты количественных характеристик равновесий в растворах спектрофотомерическим методом основаны на одном общем принципе — на совместном решении уравнений двух законов: закона действия масс и общего закона поглощения электромагнитных излучений. Различия заключаются только в путях математической обработки экспериментальных данных при варьируемых условиях, т. е. при изменении каких-либо факторов, влияющих на сдвиг равновесия в ту или иную сторону (pH, концентрации реагирующих компонентов и т. и.).
Большое число спектрофотометрических методов, используемых для изучения равновесий в растворах, объясняется тем, что изменения в спектрах поглощения исследуемых систем отличаются большим разнообразием, поскольку на оптическую плотность могут влиять многие факторы. Поэтому для изучения каждой новой системы следует подобрать единственно пригодный для данного случая метод математической обработки экспериментальных данных.
Рассмотрим некоторые спектрофотометрические методы определения констант диссоциации органических реагентов, определения состава комплексных соединений и их констант устойчивости. Помимо чисто спектрофотометрических будут рассмотрены также и другие, в которых спектрофотометрический метод применяется для определения равновесных концентраций реагирующих компонентов.
1. РАСЧЕТ КОНСТАНТ ДИССОЦИАЦИИ
ОРГАНИЧЕСКИХ РЕАГЕНТОВ
Если положение максимумов поглощения недиссоциированной и диссоциированной форм органического реагента, являющегося слабой кислотой, отличается, то, изучая поглощение растворов этого реагента при различных значениях pH, можно спектрофотометрически определить константу его диссоциации. Пусть органический реагент
диссоциирует кик одноосновная кислота по уравнению HR^H+4-R-, (а}
тогда
где _ константа диссоциации реагента.
При увеличении pH раствора возрастает степень диссоциации реагента, т. е. концентрация его недиссоциированной формы уменьшается, а диссоциированной увеличивается'.
Оптическая плотность раствора при длине волны, соответствующей максимуму поглощения недиссоциированной формы, будет уменьшаться и достигнет минимума, а диссоциированной — возрастать и достигнет максимума в момент, когда реагент перейдет полностью в ионизированную форму. Например, максимум поглощения недиссоциированной формы бензоилацетона находится при А 250 нм, а диссоциированной формы А 320 нм (см. рис. 16), 11]. По мере увеличения pH поглощение растворов при А 250 нм будет уменьшаться, а при А 320 нм возрастать до тех пор, пока упадет до минимума и не достигнет максимума при pH 10 соответственно в момент, когда реагент перейдет полностью в диссоциированную форму. При дальнейшем возрастании pH оптическая плотность будет оставаться постоянной (обозначим ее Лк-) при отсутствии последующих стадий диссоциации, вызывающих изменение в спектре поглощения реагента.
Аналогично, при увеличении кислотности растворов наступает такой момент (pH ж 6), когда весь реагент перейдет в недиссоцииро-ванную форму и при дальнейшем увеличении кислотности не будет меняться концентрация этой формы реагента; следовательно, с этого момента оптическая плотность растворов будет также оставаться постоянной (обозначим ее Анч). Все прочие растворы с промежуточными значениями pH будут содержать в равновесии смесь диссоциированной и недиссоциированной форм реагента и иметь промежуточные значения оптической плотности (Асм).
Если концентрация реагента, ионная сила и температура растворов постоянны, то все кривые пересекутся в одной так называемой изобестической точке (см. рис. 16, А — 270 нм). При длине волны, соответствующей этой точке, обе формы реагента (недиссоциирован-ная и диссоциированная) имеют одинаковое поглощение, а растворы одной и той же концентрации независимо от pH — постоянное поглощение. Поэтому для определения общей концентрации органических реагентов следует использовать именно эту длину волны.
Рассмотрим несколько методов вычисления констант диссоциации органических реагентов, используемых в спектрофотометрическом методе. Во всех методах есть ряд общих положений. Поглощение раствора реагента, содержащего недиссоциированную и диссоциированную формы, при определенной длине волны А равно (в уравнениях опущены заряды частиц):
= +e^lcx, (IV.2)
где А см — оптическая плотность раствора, содержащего смесь R и HR; с — общая концентрация реагента (во всех опытах сохраняется постоянной); bhr и eR — молярные коэффициенты погашения не-диссоциированной и диссоциированной форм реагента при длине волны X; / — толщина поглощающего слоя раствора; х = [R] — доля диссоциированной формы; (1 — х) HR — доля недиссоциирован-ной формы;
1. Алгебраический метод. Подставив в выражение (IV. 1) величины с. х и (1 — л-), получим [2], i31
[Н] сх—Кс (I— х).
Решая уравнение (IV .3) относительно х, находим
К Х~ [Н]+.К ’
Комбинируя (IV.2) и (IV.4), получим
___W/c
[Н]+К R
(IV.3)
(IV.4)
К \
[Н]+К )’
(IV. 5)
^CM“~eHR 1с
где 6hr/c = ?4hr и eRfc = ?4r — оптические плотности раствора реагента в недиссоциированной и диссоциированной формах при длине волны 1, концентрации с и толщине поглощающего слоя I.
Решая уравнение (IV.5) относительно К, получим
дХ _дХ
----^-[Н].
R Лсм
(IV. 6)
Следовательно, для вычисления константы диссоциации реагента данным методом необходимо определить /1hr> ^r и Лем хотя бы при одном значении pH.
Чтобы определить константу диссоциации опытным путем, готовят ряд растворов, сохраняя постоянной концентрацию реагента и меняя значение pH в широких пределах. Снимают спектры поглощения всех приготовленных растворов, используя кювету с постоянной толщиной слоя. На основании полученных спектров выбирают область длин волн, в которой наблюдаются наибольшие различия в величинах Ак. При выбранной А определяют значения Лнд и Ar (см. стр. 91). При этой же длине волны измеряют Лем раствора, имеющего значение pH, промежуточное между теми, которые соответствуют Л hr и Лр. Найденные значения оптических плотностей подставляют в формулу (IV.6) и вычисляют К. Величину К вычисляют по нескольким значениям и Л см и [Н+], и обрабатывают результаты статистически.
Этот метод (его часто называют методом изобестических точек) может быть использован и для определения констант устойчивости комплексных соединений 14).
2. Графический метод. Константа диссоциации органического реагента, функционирующего как кислота, может быть найдена графически на основании изучения зависимости оптической плотности (измеренной при той длине волны, при которой происходит наибольшее изменение оптической плотности при диссоциации реагента) от pH растворов (рис. 41). На кривой этой зависимости есть участок крутого подъема. Он ограничен двумя горизонтальными прямыми с постоянными значениями А. В области низких значений pH горизон-
тальный участок соответствует интервалу, в котором не происходит
диссоциации реагента, а в области высоких значений pH — интервалу, в котором реагент существует практически в полностью диссоциированной форме. Перпендикуляр, опущенный из точки, соответствующей 50% диссоциации реагента (середина участка крутого подъема), на ось абсцисс, дает значение обратного логарифма константы диссоциации реагента. Если
^r+^hr л л _ -------
см 2
Рис. 41. Зависимость оптической плотности от pH водного раствора реагента (бензоилацетон)
(IV. 7)
то подставив (IV.7) в уравнение (IV.6), получим lgtf=lg[H] и-lgK=pH.
Этот метод применим и для многоосновных кислот при достаточно большом различии в значениях констант диссоциации. При этом необходимо, чтобы разные формы диссоциации кислоты максимально поглощали при различных длинах волн или величины е этих форм при одной и той же длине волны заметно отличались.
3. Метод Комаря. Данный метод [51 не требует предварительного определения bHr и eR (или соответствующих величин Л hr и Лк) в максимуме поглощения диссоциированной и недиссоциированной форм кислоты, поэтому он может быть применен, если нельзя полностью сдвинуть равновесие в сторону образования диссоциированной или недиссоциированной форм и определить их поглощение.
Если предполагается, что реагент диссоциирует как одноосновная кислота [см. уравнение (а) на стр. 91], то в серии растворов, в которых исходная концентрация реагента с одинакова, а концентрация водородных ионов меняется во всех опытах, для опыта i имеем
[H]=ftb [R]-Xi, [НК]=с-х4.
Тогда по закону действия масс в момент равновесия
K=-^-L-. (IV.8)
с—xt
Есл^и eHR — молярный коэффициент погашения HR; er — мо-ярный коэффициент погашения R, то согласно уравнению (IV.2)
Л^= [(с — xt) eJiR+xi е£] /, (IV.9)
шку Да
eHR Ci~
Подставляя выражение (IV. 10) в (IV.8), получим для опыта j А^К—Ker cl = hie^Rcl— (IV. Ц)
— уравнение с тремя неизвестными /<, chr и е£. Для опыта k аналогично получим
ЛАК—7Ce^c/=/zAe^RC/ — hh Л\. (IV.12)
Вычитая уравнение (IV. 12) из (IV. 11), имеем
(аЬ-А^ К = e*R cl UH-h^A^hi-A^hn), (IV. 13)
Уравнение (IV. 13) содержит уже только две неизвестных величины: Bhr и /<. Комбинируя опыт i с каким-либо опытом п, получим X ту уравнение с теми же неизвестными eHR и К:
(Л^-Л^)К=е^с/(/г1— hn)-(A^hi-A^hn). (IV. 14)
Решая два линейных уравнения (IV.13) и (IV. 14) с двумя неизвестными, получим
еь I (4~4)(л^г-4М-(л^-4)(л^;-4М ,
eHR Cl (л^-Л^^-ад-^-Л^^-ад
(hi-hk) (Abhi - A^)-(hi-hk) (A^ht-A^hk)
К—' ✓ г 1 ’ 'I* ’ 6)
(A*-4) (ht-hn)-(A^-A^ (hi-hb)
Вычисленные значения £hr и /< можно подставить в уравнение (IV. 11) и получить из него 4-
Если можно найти такой участок спектра, в котором поглощает только одна из форм (диссоциированная или недиссоциированная), то расчеты упрощаются. Пусть при данной длине волны поглощает только R, тогда Bhr = 0, А} = хг/е£ и
хг = Л^/1бк. (IV. 17)
Подставив выражение (IV. 17) в уравнение (IV.8), получим
1ц Л^=К(е^с/ — Л*). (IV. 18)
Аналогичное выражение получим для опыта k; решая два линейных уравнения для опыта i и k с двумя неизвестными, получим
^_4^К (ivj9)
4-4
I 4 4 (*-»>) (IV2O)
R cl A^hi-A^hk
Используя данные измерений оптических плотностей серии растров с различными значениями pH, получают ряд значений КдисС, которые затем обрабатывают статистически (стр. 97, 98). Уравнения nV 19) и (IV.20) можно применять и для спектрофотометрического 'поёделения pH растворов. Если известны К, е и начальная концентрация с, то на основании измерений величины А всегда можно вычислить [Н+1.
4. Метод распределения. Константу диссоциации Кдисс реагента можно определить методом экстракции, изучая его распределение между водой и органическим растворителем при различных значениях pH. Рассмотрим пример определения Кдисс реагента, нейтральные молекулы которого способны присоединять протоны.
Например, большинство органических реагентов, содержащих аминный азот, склонно к протонизации, которая в зависимости от основности азотсодержащей группы может, наблюдаться или в кислой. или уже в нейтральной среде. Кроме того, этот реагент может содержать также другие группы, способные отдавать протон. Если реагент присоединяет один протон, то первая ступень его диссоциации как кислоты соответствует отщеплению этого протона. В результате образуются нейтральные молекулы реагента [6]. Эти'молекулы затем подвергаются дальнейшей диссоциации, отщепляя один протон и давая отрицательно заряженные ионы. Уравнения рассматриваемых реакций и соответствующие им константы могут быть записаны следующим образом:
йн+ [BR] Thr
Н2 R+^± H+ + HR, К1=——-----------(IV.21)
[Н2 R+] уН2 R+
°н+ IR~] ”Vr-
HR^H+ + R-, К2 = [HR]?hr (1V.22)
или для постоянной ионной силы
йн+ [HR] _ Тн2 R+
[ [Н2 R+] Yhr
йн+ тг Thr „
гтттм —Кг —Ай-
(IV. 23)
(IV.24)
Если используется распределение в системе Н2О — неполярный растворитель, то в органическую фазу могут переходить только нейтральные молекулы HR. В дальнейшем для простоты записи опустим заряды частиц, помня что все остальные частицы заряжены. Коэффициент распределения выражается как
B=-ffRlopr , (IV.25)
сн,о
ch3o = [HR] + [H2R+] + [RL (IV. 26)
а константа распределения HR — как
л- _ tHR]opr [HR]HaO •
Выразив концентрации [R+] и [H2R+] через IHR], используя
нения (IV.23) и (IV.24) и подставив их в (IV.26), получим
[HR] fHR] йн /
<4o=IHR]+ -J,2 + ——=IHR]^1 +
(IV.27)
урав-
—J-йн )
Объединяя уравнения (IV.25), (IV.27) и (IV.28), получим
Впер 60 5,0
В/пах 30 20
йн
(IV .28)
Kd hr
(1V.29)
б б Ю pH
ИЛИ
! + —~+ V" Ь °н *i /
(IV. 30)
Уравнение (IV.30) выражает зависимость коэффициента распределения D, который может быть определен экспериментально, от pH, Ki и К2 (рис. 42). Уравнения асимптот, проведенных из начального и конечного участков этой кривой, будут иметь вид:
lgD=lg*Oi HR+lsKi + pH, (IV.31
lgP=lg*DiHR-lg*2-pH. (IV.32)
(D =- £>пер) справедливо соотношение lg*i*2= — 2pH. (IV.33)
Этот же результат может быть получен дифференцированием уравнения (IV.30) по pH и приравнивания производной нулю.) Подставив в уравнение (IV.30) выражение (IV.33), получим для коэффициента распределения в максимальной точке
10
00
-1,0 >__।___i—
0 2 И
Рис. 42. Зависимость lg D органического реагента от pH водной фазы
Для точки их пересечения
18 Дпах — 1S *о, hr — 1S I Id-
Откуда
*2
У*Г*^
У *1*2 )
*1 /’
(IV. 34)
1/ Jk.= _L(—p’ HR —I j.
|/ Ki 2 ( Umax /
С другой стороны, из уравнений (IV.31) и (IV.32) следует
. /~*T Кр- нк .
V К1 °пер
(IV.35)
(IV. 36)
решая совместно уравнение (IV.35) и (IV.36), получим
%d. hr = J ~ (IV.37)
Dmax -Опер
Если можно экспериментально определить £>max и £>пер, то Kd.hr может быть вычислена. Тогда по уравнениям (IV.31) и (IV.32) можно вычислить /<1 и К2.
Зависимость D от pH можно получить, определяя спектрофотометрически равновесные концентрации реагентов в водной и органической фазах. При этом концентрация в органической фазе определяется по градуировочному графику, построенному по эталонным растворам, приготовленным растворением точной навески реагента в органическом растворителе. В водной фазе поглощение испытуемого и эталонных растворов, приготовленных также по точной навеске, целесообразнее всего измерять при X, соответствующей изобестической точке (см. стр. ООО), так как при этой длине волны поглощение водных растворов реагентов не зависит от pH. Если измерить поглощение реагента в органической фазе невозможно, то, определив концентрацию его в водной фазе, вычисляют концентрацию реагента в органической фазе по разности между общей исходной концентрацией и найденной в одной фазе. Если коэффициент распределения мал, практически весь реагент находится в водной фазе, то для определения его концентрации в органической фазе после разделения фаз реагент ре-экстрагируют в водную фазу и в ней определяют его концентрацию. В этом случае концентрация реагента в водной фазе определяется по разности.
2. ОПРЕДЕЛЕНИЕ СОСТАВА КОМПЛЕКСНЫХ СОЕДИНЕНИИ
Состав комплексного соединения часто меняется при переходе его из твердого состояния в раствор, а многие комплексные соединения существуют только в растворе.
Для определения стехиометрических коэффициентов в уравнениях реакций образования комплексов, находящихся в растворе, широко применяется метод физико-химического анализа, разработанный И- В. Тананаевым, А. К. Бабко, Н. П. Комарем и другими учеными i'i — 128]. Метод основан на построении диаграмм состав — свойство- В качестве свойства изучаемой системы при спектрофотометрических исследованиях используют оптическую плотность А. Этот метод позволяет определять и состав комплексных соединений, если известно ионное состояние компонентов, участвующих в образовании комплексного соединения.
Метод изомолярных серий
ре ^ЛЯ- опРеДеления стехиометрических коэффициентов в уравнении ^ЛР ЦИИ комплексо°бразования (или соотношения компонентов в ком-ксном соединении) часто применяют метод Остромысленского —
Жоба [9], [10], называемый методом изомолярных серий или методов непрерывных изменений [8], [13]. Пусть образование комплексное соединения происходит по уравнению
M+«R^±MRn, (а)
которому соответствует константа равновесия
[MJ [RJ” [MR]n
(IV.38)
С возрастанием концентрации компонента R количество образовавшегося комплекса MRn будет увеличиваться. Если приготовить серию растворов, в которых относительное количество компонентов М. и R различно, но общее количество одинаково, то один из растворов згой серии будет содержать комплекс MRn в максимальной концентрации. Состав такого раствора будет зависеть от исходных концентраций М и R а также от 7Сравн. Если приготовить серию растворов исходя из растворов М и R равной молярной концентрации, в которых сумма молярных концентраций М + R постоянна, но меняются относительные количества этих компонентов (изомолярные растворы), то зависимость от исходной концентрации и /Спавн отпадает. Максимальное количество комплекса будет находиться в том растворе, в котором соотношение компонентов М и R равно их соотношению в комплексе. Предположим, что растворы обоих компонентов одинаковой молярной концентрации смешивают в следующих соотношениях (мл):
раствор компонента R 123456789 раствор компонента М 987654321
Введем следующие обозначения: т — исходная концентрация компонентов; х — доля компонента R в каждом из приготовленных растворов; (1 — х) — доля компонента М в тех же растворах; сг = = [М] — равновесная концентрация компонента М; с2 = [R] — равновесная концентрация компонента R; с3 — [MRJ — равновесная концентрация комплекса MRn. Тогда можно записать с учетом уравнений (IV.38) и (а):
с1=лг(1—х) — с3, ci — f(x), (IV.39)
с2 = тх—пса, c2 = f(x), (IV.40)
<73./C=crc" c3=/(x). (IV.41)
Для раствора, в котором комплекс MRn содержится в максимальной концентрации, необходимо выполнение условия dejdx = 0. Производные от (IV.39), (IV.40) и (IV.41) по х будут соответственно равны
, (IV.42)
dx dx
=т_п , (IV.43)
dx dx
^ + пе1сп2-1^. (IV’44)
ах ах цх
Подставляя (IV.42) и (1V.43) в (1V.44) и учитывая, что dc-Jdx =• О, получим
0= m(—Cz + nci).
Так как с2 и т не могут быть равны нулю, то их можно сократить. В результате получим
с2=пс1. (IV.45)
Умножив (1V.39) на п, получим
nci = mn (1— х) — пс3. (IV.46)
и вычитая (IV.40) из (IV.46), имеем
пс!—с2 = т[п(1—х)—х]. (IV.47
Учтя (IV.45) и решая уравнение (IV.47) относительно п, получим
(IV.48)
Следовательно, для вычисления п по уравнению (IV.48) необходимо установить, для какого раствора с3 имеет максимальное значение. Если в результате реакции образуется «окрашенный» комплекс,
то, измеряя оптические плотности приготовленной серии, можно выбрать раствор, имеющий наибольшую оптическую плотность, который и будет соответствовать максимальной концентрации комплекса с3. Если при выбранной длине волны поглощают помимо комплекса также исходные компоненты, то вместо величины А используют ее отклонение от аддитивности ДА = А — Ао, где А — измеренная оптическая плотность, а Ао — сумма оптических плотностей, соответствующих концентрации всех посторонних компонентов в испытуемом растворе.
При построении графической'зависимости А от соотношения ком-
/ 2 3 4 5 6 7 8 9Уи,мл 9 8 7 6 5 4-321
Рис. 43. Определение соотношения компонентов в комплексном соединении методом изомолярных серий (непрерывных изменений)
понентов М и R в растворе максимум на кривой состав — свойство (рис. 43) определяет стехиометрические коэффициенты в уравнении образования комплексного соединения (см. уравнение (а) на стр. 98). При образовании малоустойчивых комплексных соединений на кривых не будет наблюдаться резкого излома. Максимум определяют экстраполяцией участков кривой, соответствующих избытку одного из компонентов в растворе, что способствует сдвигу равновесия (а) В СТ?Р°НУ образования комплекса (см. рис. 43).
Использование рассматриваемого метода для определения соот-ошения компонентов в комплексе справедливо только для образова
ния в системе одного комплекса, причем в условиях опыта компоненты М и R не диссоциируют, не гидролизуются, не полимеризуются Если на графике состав — свойство положение максимумов совпадают для растворов различной концентрации, то это говорит о постоянстве состава комплексного соединения. Смещение максимума на кривых обычно происходит при образовании нескольких комплексных соединений. При этом условия применения метода и математическое его доказательство более сложны [11]—[19]. Если нельзя найти область длин волн, в которой поглощает только комплекс, то для надежного определения положения максимума на графике зависимости свойства от состава необходимо внести поправку на поглощение избытка исходных компонентов. Поскольку первоначально состав не известен и невозможно рассчитать этот избыток исходных компонентов и соответствующую ему оптическую плотность, то используют последовательное приближение. В начале вычитают оптическую плотность, соответствующую поглощению всех присутствующих в растворе компонентов. Затем, определив ориентировочно состав, уточняют, какой их избыток содержится в каждом растворе.
Существенное осложнение наблюдается при определении экстремумов на кривых состав — свойство при образовании высококоординационных соединений типа MR4, MR5, MR6. Для таких соединений п равно 0,80; 0,83; 0,86 соответственно, т. е. мало различимо. То же самое относится к многоядерным комплексным соединениям (MpR5) с дробным соотношением стехиометрических коэффициентов 9 Р-
При определении состава образующих комплексных соединений методом изомолярных серий следует иметь в виду, что если происходят различные побочные процессы, то точная величина п может быть получена лишь в отдельных случаях. Таким образом, рассматриваемый метод не являегся универсальным и имеет определенные границы применяемости [12], [14] — [19].
Чтобы избежать ошибок при использовании этого метода, рекомендуется готовить несколько серий растворов (две-три), отличающихся исходной концентрацией, и снимать поглощение приготовленных растворов при нескольких длинах волн (не менее двух). Кроме того, полнота образования комплексного соединения часто зависит от pH раствора. Оптимальное значение pH образования комплекса должно быть предварительно установлено (см. стр. 43,46) и его следует соблюдать при приготовлении серии растворов. Обычно для этого используют буферные растворы. При выборе буферного раствора необходимо, чтобы комплексообразование иона металла с компонентами буферного раствора отсутствовало. Для получения воспроизводимых результатов ионная сила раствора также должна быть постоянной.
К экстракционным системам метод изомолярных серий впервые применили Бабко и Пилипенко [20]. Теоретическая возможность применения данного метода к экстракционным системам освещена в ряде работ Комаря [14], [15], Ирвинга и Пирса [21].
Для экстракционных систем, как показал Комарь, соотношение q ; р может быть определено, если даже: 1) в водной фазе протекают
процессы ступенчатого комплексообразования; 2) кроме комплексного соединения, экстрагируется реагент HR, поглощающий при той }ке длине волны X, что и экстрагируемое комплексное соединение; 3) реагент димеризуется в неводном растворителе.
Методы, основанные на использовании кривой насыщения
При исследовании какой-либо новой реакции образования окрашенного комплексного соединения практически всегда проводят построение кривой насыщения — зависимости А (или ее отклонения от аддитивности — АЛ) от соотношения М : R, например при определении необходимого избытка реагента для проведения фотометрической реакции (см. рис. 20). Она может быть использована для определения состава комплексного соединения, если оказывается непригодным метод изомолярных серий.
При построении кривой насыщения обычно сохраняют постоянной концентрацию одного из реагирующих компонентов и меняют концентрацию другого. При образовании в системе достаточно прочного комплекса на кривой насыщения получается резкий излом в точке, абсцисса которой соответствует молярному соотношению иона-ком-плексообразователя и лиганда в комплексе. При получении кривой насыщения, не имеющей резкого излома, о составе или соотношении компонентов в комплексном соединении можно иногда судить на основании экстраполяции прямолинейных участков на кривой насыщения. Однако при образовании малопрочных .комплексов, когда получаются кривые насыщения с плавным переходом, бывает трудно определить состав или соотношение компонентов. Тогда следует использовать специальные способы обработки экспериментальных данных. Некоторые из них рассмотрены ниже.
Метод Адамовича [221. Если комплекс образуется по уравнению
mM4-nR^±MmRn, (а)
то по закону действия масс
(х-ту)т(^—пу)п=Ку, (IV. 49)
где сц — начальная (постоянная) концентрация компонента R; х — переменная концентрация компонента М; у — равновесная концентрация комплекса MmRn; /<нест — константа нестойкости комплекса.
При Клест -> 0 (для устойчивого комплекса) уравнение (IV.49)-вырождается в две прямые
1 1
У = — X и у = — Со. т п
Первая из них представляет прямую, проходящую через начало ко-°РДинат под углом, тангенс которого равен 1/т. Вторая не зависит °т х и параллельна оси 0 — х. Абсцисса их точки пересечения отвечает равенству х/сд = т/п и позволяет определить т/п в реакции омплексообр азования.
Следовательно, при всех значениях тип кривые насыщения имеют асимптоту, параллельную оси абсцисс, уравнение которой у -= (1/n) Cr_ При т — 1 (моноядерный комплекс) форма всех кривых насыщения в первом квадранте весьма сходна с гиперболической.
При различных соотношениях т и п и значениях К, когда К О, характер кривой насыщения зависит от типа химической реакции.
Следовательно, уравнение кривой насыщения заранее неизвестно и вывести его можно только на основе аппроксимации. В качестве такой аппроксимирующей функции удобно взять гиперболу второго порядка. Допустимость такой аппроксимации может быть решена путем сопоставления экспериментальных величин с данными расчета по аппроксимирующему уравнению.
В общем виде уравнение кривой второго порядка
х2+2щ ху а23 у2 -\-2а^3 х 2с23 у -j- с33=0,
коэффициенты а33 и аи обращаются в нуль, так как должны выполняться условия: кривая проходит через начало координат и ома из ее асимптот параллельна оси абсцисс. Тогда оставшиеся члены могут быть представлены в виде уравнения
ау2—2 (b +х) у + 2сх — 0 (IV. 50)
или
ф/(^+л:)2—2ссх
У=—------- -------------- (IV.51)
Исследование кривой, описываемой уравнениями (IV.50) и (IV.51), показывает, что: 1) при х = 0 у = 0, т. е. кривая действительно проходит через начало координат; 2) при х -> оо у -> с, т. е. параметр с есть расстояние горизонтально расположенной асимптоты кривой от оси абсцисс; 3) параметр b является абсциссой точки пересечения касательной к кривой, проведенной в начале координат, с горизонтальной асимптотой.
Для решения вопроса о составе комплекса необходимо найти параметр Ь. При образовании моноядерного комплекса (т = 1) (см. уравнение (а) на стр. 98) согласно закону действия масс имеем
(х—у) (cR—ny)n -------------’—=К. (IV.52)
У
В начальном участке хода этой кривой, к которой проводится касательная, определяющая параметр Ь, получаем
lim -----— = lim —:; lim —- = -. (IV.53)
х-о у г/-о (cR—ny)n х-*о у
’ SI-+0 R
Если KjcR 0,02, можно считать, что lim —si, т. е. при х->0 х->0 У
и у -> 0 х = у. Тогда b равен концентрации компонента М, стехио-метрически соответствующей cr, откуда cR/y = п = cr/Ь, т. е. иско
мому коэффициенту п уравнения реакции. Чем больше будет слагаемое K/cr в выражении (IV.53), тем больше угол наклона касательной к кривой насыщения будет отличаться от ее предельного значения и тем меньше оснований использовать параметр b для определения состава комплекса.
Для расчета параметров b поступают следующим образом. Готовят серию растворов, сохраняя концентрацию одного компонента, например лиганда cr, постоянной и меняя концентрацию металла См. При этом соблюдают те условия, которые были указаны для приготовления серии изомолярных растворов (см. стр. 98). Измеряют оптическую плотность А (или ее отклонение от аддитивного значения ДЛ). На основании этих измерений вычисляют параметры гиперболы по уравнениям, которые были получены применением метода наименьших квадратов к основному аппроксимирующему уравнению (IV.50):
aS«/4—2с£х«у2 = 2£х «у3;
dZy3—Ib'S.y2+2с2ед = 2Sx#2, aLxyV—2feSx^-j-2cSx2 = 2Sx2^,
где x — переменная концентрация металла см; у — оптическая плотность А (или АЛ). Определив параметры гиперболы, вычисляют по уравнению (IV.50) для всех концентраций второго компонента (х) оптические плотности (у) и сопоставляют рассчитанные величины с опытными. Если расхождение между ними укладывается в пределы точности измерений на данном приборе, вид кривой насыщения может быть описан уравнением гиперболы (IV.50).
Аппроксимирующее уравнение гиперболы (IV.50) может быть использовано также для определения молярного коэффициента погашения е и константы нестойкости комплекса. Для оценки е может быть использован параметр с, так как он характеризует удаленность асимптоты от оси абсцисс и соответствует предельной оптической плотности при полном связывании реагента в комплекс. Если такая предельная оптическая плотность не может быть определена опытным путем, то вычисляется, как параметр с; тогда
e = cn/lcRj (IV.54)
где cR — концентрация реагента; I — толщина слоя кюветы; п — число лигандов, входящих в молекулу комплекса. Параметр с может быть использован для расчета е только в том случае, если исходные компоненты реакции при выбранной X не поглощают и для построения гиперболы используются величины Л, а не АЛ.
Применяя уравнение гиперболы (IV.50) при расчете Кнест, для пждого опыта находят значение
--------
1 (eMRn~eM—8r)
где emr, ем и sr — молярные коэффициенты погашения комплекса и компонентов М и R соответственно (если ем = eR = 0, то в числителе вместо ЛЛ будет Л). Для каждого опыта вычисляют серию значений Кнест
(.х—у) (cR—ny)n
Кнест= ’
У
(IV. 55)
Логарифмируя
(IV. 56)
где п — число одним ионом
(IV. 57)
постоянство которых подтверждает правильность определения п.
Метод сдвига^равновесия. Рассмотрим применение этого метода для условий, когда при построении кривой насыщения постоянной сохраняют концентрацию комплексообразователя М., а переменной является концентрация лиганда R. Образование моноядерного комплекса протекает по уравнению (а) (см. стр. 98) [11], [19]. Общая константа устойчивости комплекса выражается
[MR„J Рп — и •
[М] [R]
уравнение (IV.55), получаем
lg₽n=lg [MRnJ-lg [М]~п lg [R],
лигандов, участвующих в комплексообразовании с металла. Преобразуя уравнение (IV.56), получаем ig^^igpn+nigiRj.
Если за свойство комплекса взять его оптическую плотность и выбрать такую длину волны, при которой поглощает только один комплекс MRn, то [MRn] пропорциональна оптической плотности А, а концентрация металла [MI, не связанного в комплекс, пропорциональна Ло — At, где Ао — оптическая плотность раствора, в котором практически весь металл связан в комплекс MRn. На кривой насыщения Ai соответствует участку, где происходит возрастание оптической плотности, а Ао — горизонтальному участку этой кривой (см. рис. 20).
Тогда уравнение (IV.57) может быть преобразовано:
lg .Ai. =lgPn+nlg[Ri].
A0—Ai
A-
На графике lg-^—= f (IR1), n — тангенс угла наклона пря-Ао —
мой к оси абсцисс, a lg (Зп — отрезок, отсекаемый этой прямой на оси ординат.
Если комплекс MRn экстрагируется несмешивающимся с водой растворителем, уравнение (IV.58) несколько видоизменяется. Константа распределения MRn между водной и органической фазами Kd, п описывается уравнением
(IV.58)
[MR„]0
Kd’ п ~ [MRn]B
(IV. 59)
Комбинируя (1V.59) и (1V.55), получаем
₽n %D, п —
[MRnlo
[М]в [RJS
(IV. 60)
После логарифмирования уравнения (IV.60), заменив [MRI0 и ГМ]В на пропорциональные им Ai 0 и Ао 0 — Ait0, получаем выражение, аналогичное (IV.58), из которого легко определить п и логарифм двухфазной константы 1g РпКо, п
1g 'л Лг’°л =lg₽*^D.n+п 1g[RjJb. (IV.61)
О—О
Для определения п можно использовать другой метод. Как видно из уравнения (IV.60), для 50%-ной экстракции =1,
тогда справедливо соотношение
₽nKD.„ = l/IRln.
(IV. 62)
Таким образом, определив по кривой насыщения концентрацию реагента [R1, соответствующую той точке, для которой At = 1/2 Ло, и зная п, по уравнению (IV.62) можно вычислить ₽n/<D, «
Метод предельного логарифмирования. Если устойчивость образующегося комплекса очень мала и исследования проводят при большом разведении, т. е. соблюдаются условия, в которых концентрация исходных компонентов много больше концентрации образовавшегося комплекса (см смяп и cr смкп), тогда справедливы соотношения ГМ] = см = const и [R] =cR. В этом случае уравнение (IV. 57) может быть записано в виде
lg[MRn] = lgcM+nlgcR+lg₽n (IV. 63)
и при построении зависимости в координатах 1g Л — lg cr получают прямую , тангенс угла наклона которой равен п. Такой вариант метода сдвига равновесия для определения соотношения компонентов в комплекс называется также методом Бента и Френча [25].
3. РАСЧЕТ КОНСТАНТ УСТОЙЧИВОСТИ
КОМПЛЕКСНЫХ СОЕДИНЕНИЙ
При обсуждении методов вычисления истинных молярных коэффициентов погашения (стр. 22) и определения состава комплексных соединений (стр. 97) одновременно были приведены некоторые методы вычисления констант равновесия реакций комплексообразования, так как эти задачи часто решаются совместно. Рассмотрим некоторые методы расчета констант устойчивости комплексных соедине-нии, котсфые не требуют предварительного определения состава этих соединений каким-либо методом.
Расчет констант устойчивости
при ступенчатом комплексообразовании
При ступенчатом образовании моноядерных комплексов наблюдаются равновесия (для простоты в уравнениях опущены заряды частиц):
(IV. 64)
MRn MRn — i+R 7-^ MRn_2 + 2R M+ + nR. ^6)
При большой и постоянной ионной силе для расчета констант вместо активностей можно пользоваться концентрациями. Тогда ступенчатые (последовательные) константы устойчивости kt запишутся как k [MR! k [MRal k [MRn] 1 [MJ [R] ’ 2 [MR] [R] n [MRn_J [R] "
Общие константы устойчивости будут иметь вид: [MR] [MR2] [MRn]
[M][R] P1’ [M][R]2 P2... [M][R]« Pn’
Суммарная концентрация металла См будет равна см= IM]+[MR]+[MR2[ +... + [MRn],
(IV. 65)
(IV. 66)
суммарная
(IV. 67)
концентрация лиганда cR
cR= [R] + [MR]+2 [MR]2 +... + n [MRn],
[M], [R1 — равновесные концентрации комплекса, иона лиганда соответственно; см и cr — общие концентрации лиганда.
где [MRn], металла и металла и
Метод Яцимирского. Если предполагать [24], что все комплексы, образующиеся ступенчато и находящиеся в равновесии в соответствии с уравнением (б), окрашены, а лиганд бесцветен, то оптическая плотность раствора, содержащего все компоненты, будет
А= ев [М] I + £1 [MR] I + е2 [MR2] /+...+ en [MRn] /.
Средний молярный коэффициент погашения равен
- _ А ! ei fa [Rl-r-eq р2 [Rja -| . 4-en pn [R]n
е“см/ “ l + Pi[RJ+P2[R]2+...+Pn[RJn
(IV. 65)
(IV. 69)
вычитая е0, получим
-=-_ AeiPi[R]+Ae2p2[R]2-|-...-|-A8npn[Rr
е-е 60 l + Pi[Rl+P2 [RP+. • +Pn [RR
Уравнение (IV.70) справедливо не только для е, но и для все измерения производились в кювете с одной и той же толщиной слоя. В результате выполнения серийных определений можно получить большое число значений Ае и, следовательно, большое число уравнений типа (IV.70). Для расчета констант устойчивости необходимо найти коэффициенты этих уравнений. Предполагаемый метод решения сводится к построению ряда вспомогательных функций
(IV. 70)
А, если
экстраполяции их на нулевое значение. Рассмотрим простую вспомогательную функцию
/£=Лё/[К]. (IV. 71)
ц3 уравнения (IV.70) следует
Деу Pi4~Aea р2 [R]+As3 р3 [R]2-}- .. +Аеп рп [R]n~1
l + PllRJ+PalRP+.-.+PnlRl" ’ ( ’
Экстраполяция на нулевую концентрацию лиганда дает lim fi=ai=AeiPi.
[R]-*o
Эту экстраполяцию можно осуществить графическим методом, если по оси абсцисс откладывать равновесную концентрацию лиганда, а по оси ординат — значение /\. На оси ординат при этом отсекается отрезок, равный Леъ 0у. Дифференцирование функции fi и экстраполяция производной на нулевую концентрацию лиганда дает:
lim /р =а2 = Ле2р2— ЛеуР^.
[RJ-*o d [RJ
Значение а2 можно определить построением новой вспомогательной функции /2:
/2 = (/1—«13/IR1- (IV.73)
Функция (IV.73) при экстраполяции на нулевую концентрацию лиганда принимает следующее значение:
lim /2 = а2 = Ае2р2—АеуР?.
[R]-o
Аналогично строится третья вспомогательная функция f3:
fa= (/2—°2) / [R] •
При экстраполяции [3 на нулевую концентрацию лиганда получим lim fs = а3 = Дез ₽3 —Деу Р|.
[RJ-0
Этот же результат может быть получен при дифференцировании /2 и экстраполяции производной на нулевую концентрацию лиганда. Совершенно аналогично могут быть построены функции f3, fi, f5, ft и т. д.
A = (/i-i-«i-i)/[R]. (IV-74)
Экстраполяция такой функции на нулевую концентрацию лиганда дает
lim /£ = aj=Ae£ р£—Деу^-
[R]-0
Общее число экстраполяционных уравнений оказывается в два Раза меньше, чем число подлежащих определению величин 0 и Ае. в связи с этим целесообразно ввести новую переменную величину у, связанную с концентрацией лиганда соотношением
J/=1/[RJ- (IV.75)
После деления числителя и знаменателя уравнения [RIn и учета соотношения (IV.75) получим
- AenPn+Aen-iPn-i y+'-.+AeiP^»-1
6~ Pn + Pn-1 У + - +Р1^П_1 + ^П
(IV.70) на
(IV. 76)
Экстраполяция значения Ае на нулевое значение у дает
lim Ae=fe1 = Aen. (IV. 77)
</-*0
Дифференцирование Ае по у и экстраполяция производной на нулевое значение у приводит к пределу
йАе Рп-1
1 im —г— = (Леп _! —Деп) .
0-0 ду рп
(IV. 78)
Ф1=
Аналогичный результат может быть получен и при построении вспомогательной функции
Ле—Ьг-
У
Как видно из уравнений (IV.76) и (IV.77), при экстраполяции этой функции на нулевое значение у имеем
1- , . Рп —1
lim <[!= h2= (Ae^_j — Ае) —— . 0—0 Рп
По этой же схеме строят функции <р2, <р3, ..., (рп_т и экстраполируют их на нулевое значение у.
При сочетании значений, полученных экстраполяцией функций Ае, <рх, <р2, • <Pn-i. со значениями, полученными экстраполяцией функций Д, f2, fs....fn, можно определить все коэффициенты урав-
нения (IV.70). Так, например, при наличии в растворе только двух комплексов MR и MR2 получим:
а1 = Де1р1, с2=Де2 р2—Aei р^,
Pi
Ь1=Де2, Ь2 = (Де!—Де2)-—- .
Рг
Отсюда находим константы устойчивости рх и р2 и их оптические характеристики Аег и Ае2:
gi bi—д2 bj
gi~|-g2 bi
P2~ + '
g?b2+gife?
Дб1=-----------
gl bi — G% b2
Для устойчивых комплексов затруднительно вычисление равней весных концентраций лиганда. Если комплексы не обладают высокой устойчивостью, то общая концентрация лиганда не будет существенно отличаться от равновесной [24].
(IV. 79)
Метод Бьеррума. Для расчета констант устойчивости комплексов в системах, содержащих ряд ступенчато образующихся моноядер-ных комплексных соединений, можно использовать функцию [25]
- cfe-[R] п=---------
см
(IV. 80)
Изучение зависимости функции п от равновесной концентрации лиганда позволяет рассчитать ступенчатые константы комплексообразования. Функция (IV.80) представляет собой отношение концентрации лиганда, связанного в комплексы, к общей концентрации металла-комплексообразователя. По смыслу п является средним координатным числом. Среднее число лигандов п, приходящееся на каждый ион металла в растворе, можно выразить через концентрации комплексов следующим образом:
n = N
V и FMRln
_ [MR]+2 [MR2]+ ••+# [ММ nfi
n=[M] + [MR] + [MR2] + ...+[MRw]“ n=N ’ (IV‘81)
[M]+ 2 IMR]
n= 1
где N — максимальное число лигандов.
Подставляя уравнения (IV.65) в (IV.81) и деля на [М], получают функциональную зависимость п от общих констант образования отдельных комплексных соединений и концентрации свободного лиганда:
n=N
_ Pi [R]+2pa [RF + ЗРз [RF+ Щ [R]N „S ”MR]n
1+ PiIRl+Рг[RJ2 + Ps [R]3+• ••+Pw [K]N ~ , , "v'b row
1 + 2j Р» [RJ n=l
(IV. 82)
Функция ti называется функцией образования. Кривая, выражающая зависимость п от p[Rl, называется кривой образования соответствующей системы, где p[R] = — lg [R] — показатель концентрации лиганда.
Функцию, или кривую образования, системы можно рассчитать, если концентрация свободных, не связанных в комплекс лигандов, может быть определена экспериментально. Если концентрация лиганда известна, то функция образования может быть вычислена по уравнению (IV.80). Уравнение (IV.80) применимо для расчета п только в тех случаях, если равновесная концентрация лиганда заметно отличается от его общей концентрации и можно достаточно точно определить их разность cR — [R1.
Форма кривой образования зависит от соотношения последовательных констант комплексообразования систем. Если константы комплексообразования достаточно различаются по величине, то кри
вая образования имеет волнообразную форму (рис. 44, кривая ]). Минимум наклона кривой наблюдается вблизи всех целых значений п максимум наклона — вблизи всех половинных значений п. Если константы образования мало отличаются одна от другой, то получается S-образная кривая с одной точкой перегиба посредине (см. рис. 44 кривая 2).
При построении кривой образования для расчета равновесной кон-
центрацииишганда и п Бьеррум
Рис. 44. Два вида кривых образования:
1 — волнообразная; 2 — s-образная
предложил использовать метод соот-ветственных растворов. Этот метод применим, если соблюдаются следующие условия:
1. Поглощение лиганда R должно быть настолько мало по сравнению с поглощением образующихся комплексных соединений, что им можно пренебречь.
2. Реакция взаимодействия иона металла с лигандом должна проводиться в среде нейтральной соли с постоянной и высокой ионной силой. При этом условии закон действия масс может применяться в его классической форме.
3. Растворы всех комплексных соединений должны подчиняться
объединенному закону поглощения изучений Бугера — Ламберта — Бера.
4. Аддитивность оптической плотности для смеси растворов всех комплексных соединений должна соблюдаться:
Лсм = ^ У 8n[MRnJ.
1
5. Компоненты, участвующие в ступенчатых равновесиях, не диссоциируют и не полимеризуются.
Среднее число лигандов п, связанных с центральным ионом, рассчитывается по уравнению (IV.82). Заменив в уравнении (IV.82) общие константы на ступенчатые, получим
- П?1 П n R Ai[RJ+2fe1fe2[RJ2+...4-wnfen[Rl" /ivcq.
« =-----^Г~п---------=--------------------------n--------’ <IV'83)
1+ S n*n[Rl« 1+^ilRI [Rp+... +ШП [R]n n=i
где [R] — концентрация свободных лигандов; N — максимальное координационное число; kt — ступенчатые константы комплексообразования системы, состоящей из п комплексов. Общая константа вы-
зислйется на основании последовательных констант kt по уравнению
[MRnl u t t L
₽« = [M] [R]" =kik2ks-'*- fyv-fpn-
Равновесная концентрация лиганда [R1 и функция образования могут быть определены спектрофотометрическим методом. Для этого выби-пают два раствора с различными общими концентрациями cr, см и cr, Р" которые имеют одинаковые оптические плотности, т. е. одинаковое Процентное содержание комплексом (соответственные растворы). Для
_ — -ЛГГПГЧЮП ПТ7ПТ Т Г'ГЛГ'ИТЧУГЛ-
таких растворов справедливы соотношения:
_ Cr —[R] C£-[R]
П =
си
CR Cr «= ’
СМ Cr
СМ CR сМ Сд
[Rl =
CM.
(IV. 84)
(IV.85)
8
6
4
2
c" c'"
см~см
Для выбора соответственных растворов поступают следующим образом. Готовят две серии растворов, в которых концентрация металла различна, но постоянна в каждой серии, меняется, постепенно возрастая. Оптические плотности всех растворов этцх серий измеряют при определенной длине волны. Рассчитывают значения средних молярных коэффициентов погашения е и строят график их зависимости от концентрации лиганда (рис. 45). По этим графикам для одинакового значения молярного коэффициента пога-
Cft
Рис. 45. Зависимость е от концентрации лиганда, используемая для выбора соответственных растворов
а концентрация лиганда из-
шения находят соответствующие ему концентрации металла и лиганда, которые используют для расчета п и [R] — концентрации свободных, не связанных в комплекс лигандов по уравнениям (IV.84) и (IV.85).
После построения кривой образования п = f [R] находят ступенчатые ki или общие константы комплексообразования р,. Бьеррум предложил метод, который позволяет рассчитать ориентировочно константы комплексообразования. Путем последовательного приближения можно вычислить окончательные значения констант комплексообразования [26]. Дляуориентировочного определения констант можно использовать величины, обратные концентрациям лиганда при всех половинных значениях п [1/2, 3/2, 5/2... — 1/2),
kn~ [R]_ ’ ! '
n=n—~
~ Это соотношение получают, учитывая, что раствор, в котором п ~ п — М2, содержит приблизительно равные количества комплексов MRn-1 и MRn. Тогда при образовании двух последователь-
ных моноядерных комплексов общая концентрация металла будет равна
cM=[MRnl + [MRn_1]. (IV.86)
Если предположить, что [MRnl = [MRn-il, то из уравнения (IV.86) получим
Cjt=2 [MRn], а концентрация лиганда, который связан с металлом, равна cR - [R] = 2 (п -1) [MRn _!] + [MRn] = (2n -1) [MR„].
Тогда
_ (2n —I)[MRn] I
п=------------= п ——
2 [MRn] 2
С другой стороны, при [MRJ = [MRn-il k [MRn] 1 n [MRn-d IR] [R]
(IV. 87)
(IV. 88)
Если кривая образования волнообразна, то уравнение (IV.88) дает хорошее приближение при расчете kn. Но если кривая образования имеет S-образный характер, то лучше использовать другие методы расчета констант устойчивости, например использовать фактор рассеяния [131, 121], [27], [28].
Метод пересечения кривых. Метод пересечения кривых применяют для определения состава и прочности моноядерных комплексов [27]. Пусть комплекс образуется по уравнению
M + nR#MRn.
Каждой концентрации металла см и реагента cr в растворе соответствует определенная концентрация комплекса ск. Тогда согласно закону действия масс для КуС1 комплекса запишем
(см~ ск) (Cr — пск)п
(IV. 89)
Если общая концентрация реагента будет значительно больше, чем металла, то cr ск, тогда уравнение (IV.89) примет вид
₽=-------(IV. 90)
(СМ Ск) cr
Логарифмируя выражения (IV.90), получим уравнение прямой
ig ₽K=ig ——д- п1еск, (См Ск)
(IV.91)
тангенс угла наклона которой равен lg cR, если независимой переменной является п, а зависимой 1g 0К. Л1ожно получить серию прямых lg рк = f (п), для каждой из которых см, cr и ск постоянны. Абсцисса точки их пересечения соответствует соотношению п компонентов в ком-
плексе. При построении этих прямых равновесные концентрации комплекса MRn определяют по кривой насыщения (см. рис. 46) по уравнению
• _ ( Al
ск — СМ ( « I >
3 n
Рис 46. Определение n методом пересечения кривых
%
%
где д — предельное значение оптической плотности раствора при см = const; At — измеряемые оптические плотности.
Зная равновесную концентрацию комплекса и задаваясь различ
ной величиной п (п = 1, 2, 3, 4), рассчитывают по уравнению (IV.91) значения 1g Р для каждого из растворов, отличающихся cm/cr. По полученным данным строят графики зависимости Ig 0К от п (рис. 46). Координаты точки пересечения этих двух прямых соответствуют числу присоединенных лигандов п и 1g рк.
4. ПРИМЕРЫ ПРИМЕНЕНИЯ СПЕКТРОФОТОМЕТРИЧЕСКОГО МЕТОДА ДЛЯ ИЗУЧЕНИЯ РАВНОВЕСИЙ В РАСТВОРАХ
Определение /СДИсс органических реагентов
При определении констант диссоциации органических реагентов соблюдаются следующие этапы:
1) готовят серию растворов с одинаковой концентрацией реагента и различными значениями pH. Значение pH растворов после их приготовления измеряют на ламповом потенциометре;
2) записывают на регистрирующем спектрофотометре (СФ-10, СФ-14, Unicam, Hitachi) или измеряют на нерегистрирующем спектрофотометре спектры поглощения трех растворов реагента, значительно отличающихся значениями pH. Из серии растворов целесообразно выбрать Два, имеющих граничные значения pH, и один с промежуточным;
3) выбирают длину волны на основании полученных кривых спектров поглощения, при которой наблюдаются наибольшие изменения в оптических плотностях растворов с изменением pH;
4) измеряют при выбранной длине волны оптические плотности остальных растворов приготовленной серии и вычисляют Лдисс, ис-пользуя алгебраические и графические методы обработки результатов фор^ЭПИСЬ РезУльтатов измерений может быть сделана по следующей
’'max. и» — А
pH 1,23 pH 3,12 | pH 4,50 | pH 5,30 pH 7,12
I
Определение константы кислотной диссоциации нитрозо-Р-соли. Нитрозо-Р-соль способна к кислотной диссоциации за счет водорода оксимной группы.
Максимум поглощения недиссоциированной формы лежит в области 260—265 нм, диссоциированной — 420—430 нм. Серия изучаемых растворов должна иметь значения pH в интервале 3—12 и ионную силу, равную ~0,5.
Реагенты
1. Нитрозо-Р-соль, водный раствор, содержащий 0,2 г/л соли.
2. Буферный раствор, pH 6. Берут 500 мл 0,1 М бифталата калия, 454,5 мл 0,1 н. щелочи и доводят объем раствора до 1 л.
3. Буферный раствор, оН 7. Берут 400 мл 0,15 М NaH,PO4 и 600 мл 0,15 М Na2HPO4.
4. Буферный раствор, pH 8. Берут 50 мл 0,15 М NaH2PO4 и 950 мл 0,15 М Na2HPO4.
5. Тетраборат натрия, 0,05 М раствор.
6. Перхлорат натрия, 1 М раствор.
7. Едкий натр, 0,1 н. раствор.
8. Хлорная кислота, 0,1 н. раствор.
Приготовление растворов
Серию растворов готовят в мерных колбах емкостью 50 мл.
1. Раствор pH < 3: берут 5 мл раствора нитрозо-Р-соли, 3 мл хлорной кислоты и 9,7 мл раствора перхлората натрия.
2. Раствор pH 3: берут 5 мл раствора нитрозо-Р-соли, 1 мл хлорной кислоты и 9,9 мл раствора перхлората натрия.
3. Раствор pH 6: берут 5 мл раствора нитрозо-Р-соли, 20 мл буферного раствора с pH 6 и 6,4 мл раствора перхлората натрия.
4. Раствор pH 7: берут 5 мл раствора нитрозо-Р-соли, 20 мл буферного раствора с pH 7 и 3,4 мл раствора перхлората натрия.
5. Раствор pH 8: берут 5 мл раствора нитрозо-Р-соли, 20 мл буферного раствора с pH 8 и 1,35 мл раствора перхлората натрия.
6. Раствор pH 9: берут 5 мл раствора нитрозо-Р-соли, 20 мл 0,05 М раствора тетрабората натрия и 8 мл раствора перхлората натрия.
7. Раствор pH 12: берут 5 млраствора нитрозо-И-соли, 5 мл щелочи и 9,5 мл раствора перхлората натрия.
8. Раствор pH > 12; берут 5 мл раствора нитрозо-Р-соли, 10 мл щелочи и 9 мл раствора перхлората натрия. После добавления необходимого количества перхлората натрия объем раствора доводят водой до метки.
Дальнейшие этапы-работы и запись результатов см. на стр. 113.
Определение констант кислотной диссоциации тимолсульфофталеина (тимолового синего). Кислотно-основной индикатор тимолсуль-
фофталеин является двухосновной кислотой. При изменении кислотности его водных растворов в интервале pH от 1 до 10 их окраска меняется дважды: из красной в желтую при pH ~ 2, что соответствует диссоциации его по первой ступени; из желтой в синюю при pH ~ 8,8 — диссоциации по второй ступени:
H2R^HR- + H+^:R2-+2H+ красная желтая синяя
Максимумы поглощения различных форм: H2R, HR-, R2- находятся при Z, 555, 425 и 600 нм соответственно. Изучая спектры поглощения растворов тимолового синего в интервале pH 1—7, можно определить первую константу его диссоциации Klt а вторую /С2 в интервале pH 7—10. Кроме указанных на стр. 114 необходимы реагенты:
1. Тимоловый синий, 0,5%-ный раствор.
2. Гликоколь, 0,1 и. раствор.
3. Соляная кислота, 0,1 н. раствор (рН1).
4. Раствор, pH 1. Соляная кислота, 0,1 и. раствор.
5. Раствор, pH 1,2. Берут 100 мл гликоколя и 400 мл соляной кислоты.
6. Раствор, pH 1,6. Берут 200 мл гликоколя и 300 мл соляной кислоты.
7. Раствор, pH 1,9. Берут 300 мл гликоколя и 300 мл соляной кислоты.
8. Раствор, pH 2,3. Берут 300 мл гликоколя и 200 мл соляной кислоты.
9. Раствор, pH 2,9. Берут 400 мл гликоколя и 100 'мл соляной кислоты.
Ю. Раствор, pH 4,5. Берут 233,8 мл двузамещенного фосфата натрия и ^66,2 мл 0,1 М лимонной кислоты.
11. Раствор pH 7; приготовление см. стр. 114
!? Раствор, pH 7,6. Берут 425 мл 0,2 М борной кислоты и 75 мл 0,05 М буры.
13. Раствор, pH 8. Берут 350 мл 0,2 М борной кислоты и 150 мл 0,05 Л! буры.
14. Раствор, pH 8,8. Берут 450 мл гликоля и 50 мл щелочи.
1с РаствоР’ pH 9,3. Берут 400 мл гликоля и 100 мл щелочи.
16. Раствор, pH 9,6 Берут 350 мл гликоля и 150 мл щелочи.
17. „створ, pH 10 Бер^т 300 мл гликоля и 200 мл щелочи.
18- Раствор, pH 12. Берут 100 мл Na2HPO4 и 150 мл 0,1 н. NaOH
Приготовление растворов
Готовят две серии (соответственно двум константам диссоциации) растворов, имеющих следующие значения pH:
1 серия 1,0; 1,2; 1,6; 1,9; 2,3; 2,9; 4,5; 7,6
2 серия 7,0; 7,6; 8,0; 8,8; 9,3; 9,6; 10,0; 12,0
В мерные колбы емкостью 25 мл помещают по 0,4 мл раствора тимолового синего, доводят объем раствора до метки соответствующими буферными растворами и тщательно перемешивают. Дальнейшие этапы работы и запись результатов см. стр. 113.
Определение первой константы кислотной диссоциации диметилдиоксима. Первая константа диссоциации диметилдиоксима
Н3С—С—С—СН3
II II
НО—N N—ОН
(сокращенно ЩО)
соответствует уравнению H2D HD- + Н+ и может быть определена спектрофотометрически на основании изучения спектров поглощения растворов для интервала pH 3—12. Максимум поглощения недиссоциированной формы лежит в области 220—230 нм, диссоциированной — в области 260—270 нм. По второй ступени диметилдиоксим диссоциирует в более щелочной среде (pH > 12).
Реагенты
1. Диметилглиоксим, насыщенный водный раствор, разбавленный в 100 раз.
2. Хлорная кислота, 0,1 н. раствор.
3. Едкий натр, 0,1 н. раствор.
4. Буферный раствор, pH 9,24 (0,05 М раствор тетрабората натрия).
5. Перхлорат натрия, 1 М раствор.
Приготовление растворов
Серию растворов готовят в мерных колбах емкостью 100 мл.
1. Раствор pH 2: берут 10 мл раствора диметилдиоксима, 3 мл хлорной кислоты, 1 мл раствора перхлората натрия.
2. Раствор pH 3: берут 10 мл раствора диметилдиоксима, 1 мл хлорной кислоты, 10 мл раствора перхлората натрия.
3. Раствор pH 6: берут 10 мл раствора диметилдиоксима, Ю мл раствора перхлората натрия.
4. Раствор pH 9: берут 10 мл раствора диметилдиоксима, 66 мл раствора тетрабората натрия.
5. Раствор pH 10: берут 10 мл раствора диметилдиоксима, 1 мл 0 01 н. раствора щелочи (10 мл 0,1 н. щелочи разбавляют водой до 100 мл), Ю мл раствора перхлората натрия.
6. Раствор pH 11: берут 10 мл раствора диметилдиоксима, 1 мл раствора щелочи, 10 мл раствора перхлората натрия.
7. Раствор pH 12: берут 10 мл раствора диметилдиоксима, 10 мл раствора щелочи, 9 мл раствора перхлората натрия.
8. Раствор pH > 12: берут 10 мл раствора диметилдиоксима, 20 мл раствора щелочи, 8 мл раствора перхлората натрия.
После добавления необходимого количества реагентов во всех колбах объем раствора доводят водой до метки. Все растворы имеют постоянную ионную силу 0,1. Дальнейшие этапы работы и запись результатов см. стр. 113.
Определение константы диссоциации бензоилацетона (НБА).
Длины волн, соответствующие максимальному поглощению недиссоциированной и диссоциированной форм реагента
соответственно равны 250 и 320 нм (см. рис. 16). Серия изучаемых растворов должна иметь переменные значения pH в интервале 4—11 и ионную силу, равную 0,1. Константа диссоциации НБА определяется по методу изобестических точек (см. стр. 91) при 25 + 0,5°.
Реагенты
1. Бензоилацетон, насыщенный водный раствор (~ 1,5 • 10~4 М).
2. Хлорная кислота, 0,1 н. раствор.
3. Едкий натр, 0,1 н. раствор, не содержащий карбонатов.
4. Перхлорат натрия, 0,2 н. раствор.
вносят по 1 мл раствора НБА, 5 мл воды и по каплям раствор
В 8 стаканов емкостью 25—30 мл 1Д5 мл раствора перхлората натрия, хлорной кислоты или раствор едкого натра для создания pH 4,5,6,7,8, Ю,11; pH растворов измеряют на ламповом потенциометре. Растворы переносят в мерные колбы емкостью 25 мл и объем растворов доводят метки водой с соответствующим значением pH. Далее поступают, Как указано на стр. 113.
Исследование равновесий в системе Ti (IV) — тайрон в водных растворах
ОН
I
Титан (IV) образует с тайроном ОН окрашенное
HO3S—IJ-SOsH
комплексное соединение, растворимое в воде и не растворимое в органических растворителях. Реакцию применяют для спектрофотометрического определения титана. Спектрофотометрическое определение /Сдисс тайрона затруднено из-за малого различия Zmax диссоциированной и недиссоциированной форм реагента. Поэтому при изучении равновесий в системе Ti-тайрон используют значения Кдисс, определенные методом потенциометрического титрования: рКх = 7,7; р/С = = 11,0 [28].
Изучение спектра поглощения комплекса. Изучая спектры поглощения растворов с различными значениями pH, находят область спектра, в которой наблюдается максимальное поглощение комплекса. Одновременно выбирают pH, при котором комплекс образуется наиболее полно.
Реагенты
1. Хлорид титана (IV), 1 • 10-3 М раствор. Готовят растворением точной навески металлического титана в концентрированной соляной кислоте. После растворения добавляют несколько капель концентрированной азотной кислоты для окисления Ti (III) в Ti (IV) до исчезновения фиолетовой окраски. Избыток азотной кислоты удаляют кипячением. Разбавляют раствор до определенного объема.
2. Тайрон, 1 - 10-2 М раствор. Готовят по точной навеске реагента. 3. Ацетатные буферные растворы с pH: 2,8; 3,0; 3,5; 4,0; 4,5; 5,0; 6,0; 7,0.
Готовят восемь растворов в мерных колбах емкостью 50 мл. В каждую колбу вводят по 2 мл раствора соли Ti, 10 мл раствора тайрона и 10 мл ацетатного буферного раствора с соответствующим значением pH. Объем раствора доводят до метки водой. Записывают спектры поглощения приготовленных растворов на регистрирующем спектрофотометре в интервале длин волн от 350 до 500 нм (Z = 1 см). Раствором сравнения служит раствор тайрона той же концентрации, что и испытуемых. По полученным спектрам поглощения определяют Z, и pH, соответствующие наибольшим значениям оптической плотности.
Определение состава и констант устойчивости. Определение состава комплекса титана с тайроном проводится по методу сдвига равновесия (см. стр. 104). Для этого в десять мерных колб емкостью 50 мл вводят 2 мл раствора титана, последовательно 1; 1,2; 1,5; 1,7; 2,0; 3,0; 4,0; 5,6; 7,0; 8,0 мл раствора тайрона и 10 мл ацетатного буферного раствора с оптимальным значением pH, доводят объем раствора до метки водой, оставляют стоять 5 мин. Измеряют оптические плотности растворов на фотоэлектроколориметре ФЭК-60 при X, соответствующей максимальному поглощению комплекса (Z = 1см) по отношению к воде.
По полученным данным строят кривую насыщения. Так как координирующим ионом в комплексе титана с тайроном является анион HR3-[29], ионизированный по двум сульфо- и одной ОН-группам, кривую насыщения строят как зависимость А от [HR3-]. Концентрацию [HR3-] находят по значению константы диссоциации тайрона Ki = 7,7, соответствующей равновесию
К,
H2R2-^HR3- + H+, м
учитывая оптимальное значение pH образования комплекса. Пользуясь данными кривой насыщения, строят график зависимости А 1(А0 — Лг) от 1g [HR3-], где Ао — оптическая плотность раствора, соответствующая горизонтальному участку кривой насыщения; Лг — оптические плотности, соответствующие участку кривой насыщения.
Тангенс угла наклона прямой соответствует числу молекул тайрона, координированных титаном, при этом происходит возрастание А (см. уравнение (IV.58), рис. 20). В точке, где А0/(А0 — А) = 0,
lg [HR3-] = — lg ₽n. n
Определение состава комплексных соединений Си (II) и Ni (II) с нитрозой-солью методом изомолярных серий
Медь (II) и никель (II) образуют с нитрозо-R-солью растворимые в воде комплексные соединения состава MeR, MeR2, MeR3. В кислой среде существуют преимущественно комплексные соединения состава 1:1, в нейтральной и слабокислой — состава 1:2, в щелочной— 1:3. Образование того или иного комплексного соединения в растворе зависит от концентрации нитрозо-R-соли. Соединение MeR3 образуется при большом избытке реагента. До сих пор нет определенной ясности в структуре образующихся соединений.
А. ОПРЕДЕЛЕНИЕ СОСТАВА СОЕДИНЕНИЙ Си (II)
С НИТРОЗО-Р-СОЛЬЮ,
ОБРАЗУЮЩИХСЯ ПРИ РАЗЛИЧНЫХ ЗНАЧЕНИЯХ pH
Реагенты
1. Раствор CuSO4, содержащий 1 - 10~3 г-ион/л Си.
2- Нитрозо-Р-соль, I • 10_3 М раствор.
3. Сульфат натрия, 0,5 М раствор.
4. Серная кислота, 0,1 М раствор.
°- Едкий натр, 0,1 и. раствор.
В стаканы емкостью 25—50 мл вводят растворы соли металла и Реагента в следующих соотношениях [32]:
раствор реагента, мл 5,0; 4,8; 4,5; 4,0; 3,0; 2,0; 1,5; 1,2; 1,0»
раствор соли меди, мл 1,0; 1,2; 1,5; 2,0; 3,0; 4,0; 4;5; 4,8; 5,0
Добавляют 5 мл раствора сульфата натрия и затем растворами серной кислоты и едкого натра доводят значения pH растворов до 4,0 ± 4 i Измерения pH проводят на ламповом потенциометре. После этого растворы из стаканов переносят в мерные колбы емкостью 25 мл, доводят объемы растворов до меток водой, имеющей pH 4,0 ± 0,1, и перемешивают. pH проверяют на потенциометре.
Для введения поправок на поглощение реагента готовят серию растворов, содержащих реагент в тех же количествах и условиях, как была приготовлена изомолярная серия растворов, не содержащих меди. Из приготовленной изомолярной серии растворов выбирают средний (4-й или 5-й) и снимают его спектр поглощения относительно раствора, содержащего один реагент концентрации, соответствующей выбранному раствору. Выбирают длину волны, при которой наблюдается максимальное поглощение и проводят измерение оптической плотности каждого из растворов приготовленной серии по отношению к раствору, содержащему соответствующее количество реагента.
Строят график в координатах ЛА — сц/см- Максимальное значение оптической плотности соответствует составу комплексного соединения меди с нитрозо-Я-солью, образующегося в кислой среде.
Для определения состава комплексного соединения меди с нитро-зо-R-солью, образующегося в слабокислой и нейтральной средах, поступают аналогично, но значение pH растворов поддерживают 6,0±0,1.
Б. ОПРЕДЕЛЕНИЕ СОСТАВА СОЕДИНЕНИЯ Ni (II)
С НИТРОЗО-И-СОЛЬЮ,
ОБРАЗУЮЩИХСЯ ПРИ РАЗЛИЧНЫХ ЗНАЧЕНИЯХ pH
Кроме указанных на стр. 119 необходим реагент: раствор сульфата никеля, содержащий 1 • 10-3 г-ион/л Ni.
В стаканы емкостью 25—50 мл вводят растворы соли никеля и реагента в следующих соотношениях:
раствор, реагента, мл 5,0; 4,8; 4,5; 4,0; 3,0; 2,0; 1,5; 1,2; 1,0
раствор соли никеля, мл 1,0; 1,2; 1,5; 2,0; 3,0; 4,0; 4,5; 4,8; 5,0.
Добавляют 5 мл раствора сульфата натрия и затем растворами серной кислоты и едкого натра доводят pH растворов до 4,0 ±0,1 и перемешивают. Измерения pH проводят на ламповом потенциометре. После этого растворы из стаканов переносят в мерные колбы емкостью 25 мл, доводят объем раствора до метки водой, имеющей pH 4,0 ±0,1, и перемешивают. pH проверяют на ламповом потенциометре.
Для введения поправок на поглощение реагента готовят серию растворов, содержащих реагент в тех же количествах и условиях, как была приготовлена изомолярная серия растворов, не содержащих никеля. Из приготовленной изомолярной серии растворов выбирают средний (4-й или 5-й) и снимают его спектр поглощения относительно раствора, содержащего один реагент концентрации, соответствующей выбранному раствору. Выбирают длину волны, при которой наблюдается максимальное поглощение и проводят измерение оптической плотности
каждого из растворов приготовленной серии по отношению к раствору, содержащему соответствующее количество реагента. Строят график в координатах ДЛ — Cr/cm- Максимальное значение оптической плотности соответствует составу комплексного соединения никеля с нитро-зо-R-солью, образующегося в кислой среде (см. стр. 119).
Для определения состава комплексного соединения никеля с нитро-зо-И-солью, образующегося в слабокислой и нейтральной средах, поступают аналогичным образом, но значение pH растворов поддерживают 6,5 ±0,1.
Определение состава и константы устойчивости ассоциата феороина с иодид-ионами
Ферроин — довольно прочный комплексный катион [Fe (Rhen)3]2+, который образует с 1“, СЮ4 и некоторыми другими анионами комплексные соединения (ассоциаты), экстрагируемые органическими растворителями. Эти ассоциаты обладают невысокой прочностью. Для определения их состава и констант устойчивости применяют метод, описанный на стр. 104. Комплекс [Fe(Rhen)312+ принимают как практически недиссоциированную частицу Mz+ (301.
Реагенты
1. Стандартный раствор соли Мора, содержащий 1 мг/мл Fe3+ (приготовление см. на стр. 156).
2. Гидроксиламин, хлоргидрат, 10%-нын водный раствор.
3. 1,10-Фенантролин, 1%-ный водный раствор.
4. Ферроин, 3 • 10~4 М раствор: Берут 16,8 мл раствора соли Мора, 90 мл раствора фенантролина, 20 мл раствора гидроксиламина — хлоргндрата и разбавляют водой до 1 л.
5. Хлороформ.
6. Ацетат натрия, 1 М раствор.
7. Иодид калия, 0,1 М и 0,05 М растворы.
В девять делительных воронок с притертыми пробками помещают по 1 мл раствора ферроина и растворы ацетата натрия и иодида калия в следующих количествах:
Номер воронки 1 2 3 4 5 6 7 8 9
Раствор ацетата натрия, мл 8 7 6 5 4 3 5 4 3
Раствор иодида калия, мл 1 2 3 4 5 6 4 5 6
Для приготовления растворов в первых шести воронках используют 6>05 М растворы KI, а в воронках с 7-й по 9-ю — 0,1 714 KI.
После перемешивания добавляют во все растворы по 10 мл хлоро-Ф°Рма и встряхивают их содержимое 2—3 мин. Через 15 мин после Расслаивания жидкостей хлороформные экстракты отделяют и снимают спектр поглощения одного из последних в этом ряду хлороформных Растворов в области 400—600 нм. Находят 7тах, измеряют при этой ллине волны оптическую плотность остальных растворов на спектро-
фотометре. Строят график зависимости ^орг = f ([I-]). По участку кривой насыщения, на котором достигается постоянство оптической плотности, определяют Л°Рг и строят график зависимости
•^орг
lg77-у ) =№i)~ \Лорг ^oprj
Состав комплекса определяют по этому графику как тангенс угла наклона полученной прямой к оси абсцисс. Из точки на кривой, где А‘ = 1/2Л0 (50%-ная экстракция), опускают перпендикуляр на ось II-] и вычисляют двухфазную константу $пКог = 1/11-К
Определение константы устойчивости диметилдиоксимата никеля методом экстракции (распределения)
Методом экстракции (распределения) удобно определять константы устойчивости комплексных соединений, малорастворимых в воде, но хорошо растворимых в неполярных органических растворителях, когда лиганд является слабой кислотой, а комплексное соединение заметно растворяется в кислых средах [31]. Если металл в степени окисления +2 образует со слабой органической кислотой HR комплексное соединение типа MR2:
M2+ + 2HR MR2 Орг+2Н+, (a)
то константа равновесия реакции (а) называется константой экстракции:
[MR2] [H+J2 1\ р у •
]М2+] [HR]2
MR2 OprZiMR2H о ; KD = I j E , (IV.92)
Lmk2Jh2o
где Kd2 — константа распределения комплекса MR2;
HRopr^±HRH O ; [»R1°pr , (IV.93)
IHR2Jh2o
где Kd — константа распределения реагента.
Общая константа устойчивости |32 комплекса MR2 равна
р2_____[MR2] (IV.94)
[М2+] [RJ2
К экстракции диметилдиоксимата никеля применимо уравнение (а)' Коэффициент распределения D для этой системы равен (последовательным комплексообразованием пренебрегаем):
Р________[NiRglopr____ (j v. 95)
~ ([Ni2+] + [NiR2])H2O
ИЛИ, Преобразуя (IV.95) с учетом уравнения (IV.94), получим
KD р2[R-р 1 + ₽2 [R-]2
(IV. 96)
равновесную концентрацию [R] вычисляют, используя константы диссоциации реагента (Л^исс) и его константы распределения Ко (количеством реагента, израсходованным на образование NiR2, пренебрегают, так как применяют большой избыток HR)
lg[R] = lg^jmcc+pH-|-lgcHR—Ig^p+i), (IV.97)
при различных значе-
1g И
где chr — общая (исходная) концентрация реагента. Экспериментально определяют коэффициент распределения D ниях pH. Строят на основании полученных данных график зависимости lg D от lg IR1 (рис. 47). К полученной кривой распределения [см. уравнение (IV.96)] строят две асимптоты, уравнения которых
lgP=lgKD2; [R] -> оо, прямая а;
и
lg D= 1g + lg Р2—2рКдисс +
+21g cHR-|-2pH, [R]->0 прямая b.
В точке пересечения асимптот 0 имеем
2 1g Р2 = Р^СдисС — lgCHR pH = lg [RJ.
(IV. 98)
Опуская перпендикуляр из точки 0 на ось абсцисс (точка с), находим
ig 1R1=—~igP2-
(IV. 99)
Значение lg О в точке пересечения асимптоты а с осью ординат численно равно Ко, — константе распределения незаряженного комплекса-
Реагенты
1. Диметилдиоксим, 3,0- 10-3 М раствор. Готовят растворением реагента в 0,075 М перхлорате натрия.
2. Буферные растворы (ацетатные), pH 3,0; 3,5; 4,0; 4,3; 4,7; 5,0; 5,5; 6—7.
3- Соль никеля, 1,7 • 10-2 М раствор (исходный 1 мг/мл).
Для приготовления 0,85 - 10~2 М и 2,125 • 10~3 М растворов никеля бе-РУт25,0 мл и 6,35 мл исходного раствора, помещают соответственно в мерные колбы емкостью 50 мл, добавляют воду до объема ~ 45 мл, устанавливают pH Д° значения 4—5 и доводят объем раствора водой до метки).
4. Едкий натр, 2 и. раствор.
В восемь сосудов для экстракции емкостью 100 мл помещают 15 мл ХлоРоформа, 10 мл водного раствора диметилдиоксима, 4 мл соот-Ветствующего буферного раствора; в сосуды с 1-го по 4-й добавляют по
1 мл 2,125 • 10~3 М раствора соли никеля, а в сосуды с 5-го по 8-й_
0,85 • 10-2 М раствор. Для создания определенного значения pH в сосуды добавляют буферные растворы: 1 — pH 3,0; 2 — pH 3 5-3 — pH 4,0; 4 — pH 4,3; 5 ~ pH 4,7; 6 — pH 5,0; 7 — pH 5,5; 8 pH 6—7. После приготовления растворов сосуды встряхивают на механическом вибраторе в течение 30 мин для установления равновесия. После расслоения фаз при помощи делительных воронок отделяют водную фазу в стаканы емкостью 50 мл. В полученных растворах проверяют pH и проводят определение никеля. Для этого в колбы емкостью 25 мл отбирают пипетками следующие аликвотные части: из 1-го раствора 3 мл, из 2, 3 и 4-го по 5 мл из 5, 6, 7 и 8-го по 10 мл. Определение никеля проводят в условиях, описанных на стр. 184. Содержание никеля в эталонных растворах (мкг): 1,0; 3,0; 5,0; 7,0; 10, которые готовят в колбах емкостью 25 мл.
Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов. Используя полученные значения, вычисляют г-ион/л никеля в водной фазе при различных значениях pH. Концентрацию никеля в органической фазе определяют по разности между исходной и концентрацией никеля в водной фазе. Далее вычисляют коэффициент распределения D для всех растворов по уравнению
NiOpr /С^*Н2О
и строят кривую распределения [зависимость lg D от lg [R], которую рассчитывают по формуле (IV. 97)].
Общую константу устойчивости р2 и константу распределения незаряженного комплекса ЛЪ2 вычисляют по данным кривой распределения графически (см. рис. 47) или по уравнениям (IV.98) и (IV.99), используя значение константы распределения 1g До = —1,03 и Кдисс
Исследование комплексообразования в системе
V (¥)-1-фенил-3-метил-4-бензоилпиразолон-5 (НФМБП) — н-бутиловый спирт — хлороформ — вода методом экстракции
Ванадий (V) с р-дикетонами (бидентатными реагентами) образует комплексное соединение состава VO(OH)R2 [32], [33]. Метод экстракции (распределения) пригоден для определения Муст этого соединения, хорошо растворимого в неполярных растворителях. Равновесие реакции можно выразить уравнением
Кех
VO (OH)2++2HRopr?z± VO (ОН) R2Opr+2H+
Константа равновесия реакции называется константой экстракции
Кех—
[VO (ОН) R2]opr[H+]2
tVO (0H)2+J [HR ]o2pr
(IV. 100)
Пля системы V (V) — НФМБП— СНС13— Н20 было установлено, что lg Кех = I,03 ± °>16- Равновесие при экстракции этого комплексного соединения устанавливается медленно (~5 ч), но при наличии тпстьего компонента — н-бутилового спирта (синер гента S) этот процесс протекает в течение 15—30 мин. Изучение смешанного комплексного соединения проводят при ионной силе р, = 0,1 и 20 ± 0,5°. В присутствии н-бутилового спирта экстракция ванадия (V) хлороформным раствором НФМБП описывается уравнением
кех. s
VO (OH)2++nHRopr+mSopr^z^ VO (ОН) R„.Sm Орг+пН+ (б)
к _ [VO (ОН) Rn-Sm]opr [Н+]га _ [Н+Р
ех- S [VO (OH)a+J [HR]"pr [S]-pr ~D [HRJ2pr [S]-Pr ’ (IV-101)
где Kex — константа образования смешанного комплексного соеди-‘ [VO (ОН) Rnsm]opr , ,
нения: D -=~- -—(oh)2+] — коэффициент распределения вана-
дия IV].
Для определения состава экстрагируемого смешанного комплексного соединения в системе V(V) — НФМБП — н-С4Н9ОН — СНС13— — Н2О изучают зависимость коэффициента распределения ванадия (V) от pH водной фазы. Используя уравнение (IV. 101) можно написать
lg D = lg КеХ> s 4- n lg [HR]opr4-m lg [S]opr4- npH. (IV. 102)
Зависимость IgZ) от pH должна выражаться прямой линией с углом наклона п (при постоянной концентрации HR и S в органической фазе). Аналогично из зависимостей lg D от 1g [НФМБП] (при постоянных pH и концентрации S в органической фазе) и lg D от 1g [н-С4Н9ОН] (при постоянном значении pH и концентрации HR в органической фазе) устанавливают количество молекул НФМБП и н-С4Н9ОН, которые входят в состав комплексного соединения.
Образование смешанного комплексного соединения ванадия (V) с НФМБП и н-бутиловым спиртом происходит в органической фазе:
VO (ОН) Rn opr“bm*^opr (ОН) Rn-Sm орг >
_ [VO (ОН) Rn-Sm]opr
Рп, т— _ • (IV.103)
[VO (OH)Rn]oPr [5]^рг
Используя уравнения (IV. 100) и (IV. 101), находят зависимость рп т ^ех И Aex,S
А с ₽n,m=-^-- (IV. 104)
^ех
Таким образом, для определения рп, т необходимо знать Дех и экспериментально определить Мех, s-
Реагенты
1. Раствор ванадия, содержащий 1 мг/мл V (V). Навеску 2,44 г метавайада-та натрия помещают в мерную колбу емкостью 1 л, растворяют в 0,2 н. соляной кислоте и доводят объем раствора той же кислотой до метки. Концентрацию раствора проверяют титриметрическим методом по соли Мора. Перед употреблением проводят разбавчение, чтобы получить раствор, содержащий 50 мгк/мл V (V) и 0,1 н. по соляной кислоте.
2. 1-Фенил-3-метил-4-бензоилпиразолон-5 (НФМБП), 2 • 10“3 М хлороформный раствор готовят растворением точной навески реагента (0,112 г в 200 мл хлороформа).
3. Соляная кислота, 0,4 и., 0,2 н., 0,1 и. растворы.
4. Буферные растворы: pH 1,0; 1,2; 1,4; 1,6; 2,0; 0,2 и. (хлорид калия + 0,2 и. соляная кислота); 2,6; 3,8 (0,1 и. бифталат калия + 0,1 н. соляная ки слота).
5. н-Бутиловый спирт (пл. 0,8098 г/см3).
6. Хлороформ очищенный.
Для определения количественных характеристик необходимо:
1. Определение времени установления равновесия. В пять сосудов для экстракции емкостью 50 мл вносят 0,8 мл соли V (V) (40 мкг) и 9,2 мл буферного раствора с pH 1,6. Органическую фазу готовят, вводя в каждый сосуд 5 мл раствора НФМБП, 1 мл н-бутилового спирта и 4 мл хлороформа. Встряхивают сосуды на механическом вибраторе 5,10,15,20,30 мин. Снимают спектр поглощения одного из приготовленных экстрактов и измеряют оптическую плотность экстрактов при Z,max. Раствором сравнения служит раствор органической фазы. Строят график в координатах А — т и по горизонтальному участку кривой находят время достижения равновесия.
2. Построение градуировочного графика. В шесть сосудов для экстракции вводят раствор, содержащий V (V) (мкг): 10; 20; 30; 40; 50; 60. Затем добавляют буферный раствор с pH 3,8 до объема 10 мл (в интервале pH 3—4 коэффициент распределения V (V) постоянен). Состав органической фазы тот же, что и в п. 1. Встряхивают сосуды на механическом вибраторе в течение того времени, которое определено в п.1. После наступления равновесия фазы разделяют в делительных воронках и измеряют оптическую плотность хлороформных экстрактов, как указано в п. 1. Строят градуировочный график в координатах А—с.
3. Определение состава смешанного комплексного соединения.
Определение зависимости lg D от pH. В девять сосудов для экстракции вносят по 40 мкг V (V), добавляют в первые три сосуда 0,4 н., 0,2 н., 0,1 н. соляную кислоту соответственно, в остальные — буферные растворы с pH 1,0; 1,2; 1,4; 1,6; 2,0; 2,6 до объема 10 мл соответственно. Состав органической фазы см. в п. 1. Сосуды встряхивают в течение времени, необходимого для достижения равновесия (см. п. 1). фазы разделяют. Содержание ванадия в органической фазе определяют по градуировочному графику (см. п. 2), в водной фазе — по разности. На потенциометре проверяют pH водной фазы. Рассчитывают коэффициент распределения ванадия и строят график в координатах lg D pH (см. стр. 125). Таким образом определяют заряд комплексной частицы, участвующей в комплексообразовании.
Определение зависимости lg D — lg [НФМБП}. В четыре сосуда дЛя экстракции вводят по 40 мкг V(V) и буферный раствор с pH 1,6 до объема 10 мл. Затем добавляют в каждый сосуд по 1 мл «-бутилового спирта; 0,5; 1; 3; 5 мл раствора НФМБП и 8,5; 8; 6 и 4 мл хлороформа соответственно. Сосуды встряхивают на механическом вибраторе до достижения равновесия (см. п. 1) и измеряют оптическую плотность экстрактов. Рассчитывают коэффициент распределения ванадия и строят график в координатах lg D — 1g [НФМБП] и находят число молей реагента, входящих в состав комплексного соединения (см. стр. 125).
Определение зависимости IgD от В четыре сосуда
для экстракции вносят по 10 мл водной фазы (см. п. 3), добавляют по 5 мл раствора НФМБП; 0,25; 0,5; 0,75; 1 мл н-бутилового спирта и соответственно 4,75; 4,5; 4,25; 4,0 мл хлороформа. Строят график в координатах lg О — 1g [н-С4Н9ОН] и находят число молей спирта, входящих в состав комплексного соединения (см. стр. 125). Определив состав комплекса, подставляют полученные коэффициенты пит в уравнение (б) и вычисляют Лех, s по формуле (IV. 101). По уравнению (IV. 104) рассчитывают константу образования смешанного комплексного соединения т.
ЛИТЕРАТУРА
1. А. П. Зозуля, Н. Н. Мезенцева, В. М. Пешкова, Ю. К. Юрьев. ЖАХ, 1959, 14, вып. I, 17.
2. W. St enst гоп, N. Goldsmith..!. Phys. Chem., 30, 1683 (1926).
3. С. V. Vanks, A. В. Carlson, Anal. Chim. Acta, 1952, 7, 293.
4. L. V a r e i 1 1. Bull. Soc. Chim. France, 1955, № 6, 870.
5. H. П. Кома рь. Ученые записки ХГУ, 1957, XXXVII. (8), 57.
6. М. Г. Панова, В. И. Левин. Радиохимия, 1960, 5, 568.
7. И. В. Т а н а и а е в. ЖАХ, 1949 4, 68, 136,; 1948, 3, 276; 1950, 5, 82.
8. А. К. Бабко. Физико-химический анализ комплексных соединений в растворах. Киев, Изд. АН УССР, 1955.
9. Л. И. Остром ыс лен с кий. Вег, 1910, 43, 197; 1910; 44, 268.
10. Р. Job. Ann. Chim. Phys., 1928, 9, 113; Compt. rend. 1925, 180, 928.
И. Г. Л. HI лефер. Комплексообразование в растворах. М.—Л., «Химия, 1964.
12. L. К a t z i n, Е. Gerber. J. Am. Chem. Soc., 1950, 72, 5455-
13. А. К. Бабко. ЖОХ, 1946, 16, 1549; 1947, 17, 642; 1948, 18, 816.
14. H. П. К о м а р ь. Труды НИИхимии ХГУ, 1954, 12, 1513, 31, 53.
15. Н. П. Комарь. ЖНХ, 1956, 1, 1243, 1957, 2, 1015.
16. Л. П. Адамович. ЖНХ, 1959, 4, 1552.
17. В. Н. Толмачев. Труды НИИхимии ХГУ, 1954, 12, 7795.
18. В Н. Толмачев. Укр. хим. ж., 1961, 27, 5.
19. Сб. статей «Спектроскопические методы в химии комплексных соединений». Под ред. В. М. Вдовенко. М.—Л., «Химия», 1964.
20. А. К. Бабко, А. Т. Пилипенко. ЖАХ, 1947, 2, 33.
21 Н. Irving, Т. В. Pierce. J. Am. Chem. Soc. 2656 (1959).
22. Л. П. Адамович. ЖНХ, 1960, 5, 782.
23. Н. Bent, С- French. J. Am, Chem. Soc., 1941, 63, 568.
24. К. Б. Я Ц и м и р с к и й. ЖНХ, 1956, 1, 2306.
м -Я. Б ь е р р у м. Образование амминов металлов в водном растворе.
Ф. и
Ф.
R.
Россотти, X. Россотти. Определение констант устой-других констант равновесия в растворах. М., «Мир», 1965.
Д. Шевченко. L. Gustafson, , 1960, 82, 1526.
26. чивости
27.
28.
Chem. Soc,
29. В. М. Пешкова, ЖАХ, 1974, 29, 592.
30. С. О. Кобякова, В. М. С а ЖНХ, 1974, 19, 3085.
31. В. М. Савостина, Е. ЖНХ, 1964, 9, 80; 1964, 9, 817.
32. Н. А. Краснянская, к о в а. ЖАХ, 1972, 27, 1620.
33. С. А. Николаева, И.
в а. ЖАХ, 1974, 27, 2055.
Укр. хим. ж., 1965, 31, С. Richard, А. Е.
229
Martell.
J • Am.
В. M. С
а в
о
в о
стина,
О. А. Ш п
и
г
У
и.
с
тина,
В. М.
П е ш
к
о
в
а.
К.
Н.
В.
А
а
х о в а,
В. М.
П е ш
к
о
в
а.
В. Мельча
кова,
В. М.
Пеш-
Мельчакова, В.
М. П е ш к о-
Глава V
ОПРЕДЕЛЕНИЕ ОТДЕЛЬНЫХ ЭЛЕМЕНТОВ
В этой главе изложены методы определения элементов, которые являются примесями в различных объектах. Рассматриваются элементы, в том числе редкие, которые дают соединения (главным образом комплексные), близкие по свойствам. Предлагается несколько методов, чтобы можно было сравнить их селективность и чувствительность. Для определения некоторых элементов рекомендованы весьма чувствительные кинетичесеие методы, в которых каталитические реакции протекают с изменением окраски растворов или ее интенсивности.
1. РАСТВОРЫ СРАВНЕНИЯ ПРИ ИЗМЕРЕНИИ ВЕЛИЧИН
ПОГЛОЩЕНИЯ
Фотометрические измерения, независимо от конструкции прибора, всегда проводят относительно раствора сравнения, поглощение которого принимается равным нулю. При выборе раствора сравнения необходимо учитывать область максимального поглощения раствора определяемого элемента и влияние поглощения сопутствующих компонентов. Растворами сравнения могут быть:
1) растворитель — вода, хлороформ, толуол и др. Границы прозрачности и ряда растворителей даны в табл. 6;
2) раствор реагента при наличии наложения в спектрах поглощения растворов реагента и комплексного соединения. Если комплексное соединение неустойчиво и требуется большой избыток реагента, то в растворе сравнения соблюдают ту же концентрацию его, которая взята для приготовления исследуемого раствора. Если комплексное соединение устойчиво и берется малый избыток реагента, но поглощение его значительно, то следует использовать один из вариантов дифференциального метода (стр. 75);
3) раствор «холостого опыта», который готовят в условиях приготовления эталонных растворов, но не вводят при этом определяемый элемент. При использовании «холостого опыта» исключается влияние загрязнений, имеющихся в применяемых реагентах. «Холостой опыт» Рекомендуется при определении ультрамалых количеств элементов, являющихся примесями;
4) раствор анализируемого объекта, если другие элементы, при-утствующие в этом растворе, не дают соединений с применяемым реа-5 Зак. 518 129
гентом, но поглощают при той же длине волны. Например, определение никеля в стали (стр. 184).
Измерение оптической плотности испытуемого и эталонных растворов необходимо проводить соблюдая требования, предъявляемые к чистоте и установке кювет.
2. СРАВНИТЕЛЬНОЕ ИЗУЧЕНИЕ СПЕКТРАЛЬНЫХ ХАРАКТЕРИСТИК ДВУХ РАЗЛИЧНЫХ СИСТЕМ НА ПРИБОРАХ РАЗНОГО ТИПА
Одной из основных характеристик любого фотометрического прибора, определяющей его возможности, является степень монохроматичности потока излучения.
Однако в зависимости от характера спектров поглощения изучаемых систем (широкополосных, узколинейчатых или с тонкой структурой спектральных полос) при работе на приборах, имеющих различную монохроматичность потоков излучений, могут быть получены или совершенно идентичные, или резко отличающиеся спектральные характеристики.
В работе предлагается изучение спектрофотометрических характеристик двух систем, обладающих различными спектрами поглощения. Измерения проводят на приборах двух типов, у которых моно-хроматизация потоков излучения, с одной стороны, достигается при помощи светофильтров, с другой стороны, — диспергирующей призмы или диффракционной решетки. Ширина спектрального интервала, пропускаемого светофильтром, у различных марок приборов составляет от 80 —100 (ФЭК-М) до 30—40 нм (ФЭК-56 и ФЭК-60). Приборы второго типа — спектрофотометры дают ноток излучения со спектральным интервалом 1—2 нм.
Порядок выполнения работы
1. Приготовить растворы'.
а) 5 эталонных и один испытуемый растворы, пользуясь методикой фотометрического определения марганца, никеля, железа в водном растворе (стр. 151, 172, 184);
б) раствор нитрата или хлорида редкоземельного элемента с концентрацией порядка 0,05 моль/л.
2. Измерить поглощение но фотоэлектроколориметре ФЭК-М'.
а) оптические плотности одного из приготовленных растворов эталонного ряда [п. 1(a)] с тремя имеющимися в этом приборе светофильтрами и выбрать светофильтр, для которого оптическая плотность имеет наибольшее значение;
б) оптические плотности всех пяти растворов эталонного ряда с выбранным светофильтром: построить градуировочный график в координатах А — с; вычислить средние молярные коэффициенты погашения растворов и оценить соблюдение законов поглощения;
в) оптическую плотность испытуемого раствора с тем же светофильтром и определить его концентрацию по градуировочному графику [п. 2(6)1;
г) измерить оптическую плотность раствора редкоземельного элемента с каждым из светофильтров; вычислить молярный коэффициент погашения этого элемента для светофильтра, с которым получено наибольшее значение оптической плотности.
3. Измерить на фотоэлектроколориметре ФЭК-Н-57 (ФЭК-56 или ФЭК-60):
а) оптические плотности приготовленных 1-го, 3-го, 5-го растворов эталонного ряда [п. 1(a)] со всеми светофильтрами, имеющимися в указанных приборах; вычертить кривые спектров поглощения этих растворов в координатах А — к и оценить, сохраняется ли постоянство положения А. максимума поглощения для растворов различных концентраций;
б) оптические плотности остальных двух растворов эталонного ряда со светофильтром, соответствующим максимуму поглощения; построить градуировочный график в координатах А — с по данным измерения оптических плотностей всех растворов эталонного ряда и, вычислив средние молярные коэффициенты погашения этих растворов в максимуме поглощения, оценить соблюдение законов поглощения;
в) оптическую плотность испытуемого раствора со светофильтром, соответствующим максимуму поглощения, и определить его концентрацию по градуировочному графику [см. п. 3(6)1;
г) оптическую плотность раствора редкоземельного элемента со всеми светофильтрами; вычертить кривую спектра поглощения в координатах А — к и вычислить молярный коэффициент погашения в максимуме поглощения.
4. Измерить на спектрофотометре СФ-4 (СФ-4А,СФ-5, СФ-16)-.
оптические плотности одного из растворов эталонного ряда [п.1(а)1 и раствора соли редкоземельного элемента [п. 1(6)1 в области 400— 600 нм с интервалом 10 нм; и, установив область длин волн, в которой расположены полосы поглощения этих растворов, измерить в этой области оптические плотности раствора редкоземельного элемента через 1 нм, а оптические плотности раствора, приготовленного согласно п. 1 (а), через 2—5 нм. Вычертить кривые спектров поглощения изучаемых растворов и вычислить молярные коэффициенты погашения в максимумах поглощения этих растворов.
з. оценка преимуществ при измерениях поглощения
С РТУТНОЙ ЛАМПОЙ НА ФОТОЭЛЕКТРОКОЛОРИМЕТРЕ ФЭК-56
Фотоэлектроколориметр ФЭК-56 имеет два источника излучений: лампу накаливания и ртутную лампу. Производить измерения можно как с той, так и с другой лампой. Однако ввиду того, что соответствующий светофильтр выделяет из спектра испускания ртутной лампы отдельные линии, а из спектра испускания лампы накаливания определенный интервал длины волн, то монохроматичность падающего ка измеряемый объект излучения будет в первом случае выше.
1. Снять спектры поглощения исследуемого раствора, пользуясь ртутной лампой и лампой накаливания, делая измерения при соответствующих светофильтрах и длинах волн.
2. Измерить, используя длину волны, соответствующую полосе максимального поглощения, оптические плотности эталонных растворов с той и другой лампой и построить соответствующие градуировочные графики.
3. Определить, в каком из графиков (см. п. 2) tg угла наклона прямой будет больше.
4. Вычислить значения е из результатов измерений (см. п.1). Данные обработать статистически и определить погрешности.
Пр и выполнении п. 1 можно использовать растворы соединений:
а) кобальта с нитрозо-R-солью (стр. 166);
б) трисульфосалицилата железа (стр. 154);
в) перманганата калия (стр. 169);
г) диоксимата никеля в присутствии окислителей (стр. 184);
д) сульфата церия.
Примечание. 1. Пять эталонных растворов сульфата церия готовят с содержанием церия от 0,5 до 5 мг в объеме 50 мл.
2. При приготовлении эталонных растворов кобальта с нитрозо-R-солью берут 1,5 мл реагента вместо 3 мл, указанных на стр. 162.
3. Для приготовления растворов диоксимата никеля при наличии окислителей следует добавить в каждый эталонный раствор вместо щелочи (стр. 184) 3 мл концентрированного раствсГра аммиака.
4. МЕТОДЫ ОПРЕДЕЛЕНИЯ ЭЛЕМЕНТОВ
Определение алюминия
Более распространенными для определения алюминия являются методы с применением 8-оксихинолина [1, 2] и эриохромцианина R [1].
Образование соединений алюминия с указанными выше реагентами в большой мере зависит от pH раствора, поэтому очень важно строго соблюдать рекомендуемые условия определения [2].
«I
ОПРЕДЕЛЕНИЕ АЛЮМИНИЯ 8-ОКСИХИНОЛИНОМ
(кислая среда)
ККИсл = 2-1О-1о
Косн= 1-10-ю
(щелочная среда)
8-Оксихинолин —- амфотерное соединение и в зависимости от условий реагирует или как слабая кислота, или как слабое основание [3], [4]. Реагируя как основание, 8-оксихинолин дает продукты присоеди-
нения, например, с гетерополикислотами фосфора, кремния, германия.
8-Оксихинолин проявляет свойства слабой кислоты, вступая в реакцию с ионами металлов. В результате реакции образуется внутрикомплексное соединение состава Al (C9H6ON)3 мало растворимое в воде и хорошо растворимое в неводных растворителях (бензол, хлороформ) [4], [1]. Растворы в хлороформе окрашены в желтый цвет J'-max 395 нм, е = 7,3-103. Поскольку есть некоторое наложение в спектрах комплексного соединения алюминия и
350 ^00
Рис. 48. Спектры поглощения в СНОз:
1 — оксихинолинат алюминия; 2 — оксихинолин
раствора реагента в хлороформе (Хтах 320 нм), то это следует учитывать при приготовлении раствора сравнения (рис. 48) [1].
А. Определение алюминия в растворе его соли
Реагенты
1. Стандартный раствор соли алюминия, содержащий 1 мг/мл А1. Для его приготовления 1,00 г металлического алюминия (проволока или тонкая пластинка) помещают в мерную колбу емкостью 1 л, растворяют в 20 мл соляной кислоты (пл. 1, 12), доводят объем до 1 л — раствор А. Затем 10 мл раствора А разбавляют водой до 1 л с предварительным добавлением в него 5 мл соляной кислоты той же концентрации — раствор Б : 1 мл раствора Б содержит 0,01 мг А1; раствор пригоден в день его приготовления.
2. 8-Оксихинолин, 1%-ный раствор в хлороформе.
3. Хлороформ.
4. Соляная кислота (х. ч.), 2 н. раствор.
5. Буферный раствор, pH 5. Для его приготовления берут 70 мл 0,2 М раствора ацетата натрия и 30 мл 0,2 н. уксусной кислоты.
6. Тайрон, 1%-ный водный раствор.
Для приготовления эталонных растворов в пять делительных воронок емкостью 100 мл вводят 40 мл воды, стандартный раствор, содержащий алюминий (мкг) 0,0; 0,2; 0,4; 0,6 и 0,8 соответственно, добавляют 5 мл буферного раствора и 2 мл раствора тайрона. Затем приливают 10 мл раствора 8-оксихинолина и встряхивают в течение 3 мин на механическом вибраторе. Экстрагирование повторяют дважды 5 мл раствора 8-оксихинолина в течение того же времени. Объединенные экстракты доводят хлороформом до метки колбы и измеряют оптическую плотность хлороформного раствора при к 395 нм на спектрофотометрах различных марок. В качестве раствора сравнения используют раствор эталонного ряда (1-я воронка) .Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов. Для определения алюминия в испытуемом растворе из общего объема 100 мл берут аликвотную часть 40 мл, проводят все операции, указанные при приготовлении эталонных растворов, и находят содержание алюминия по градуировочному графику.
Б. Определение алюминия в образце бериллия
При определении алюминия в бериллии используют свойство ок-сихинолината алюминия экстрагироваться в более кислой среде по сравнению с оксихинолин атом бериллия. Этот метод позволяет определять 50 мкг алюминия в 0,1 г бериллия при pH 5; соединение бериллия экстрагируется при pH 9. В предлагаемом методе имеющееся железо связывают в комплексное соединение тайроном [85].
Примечание. Метод Сендела [3] видоизменен в лаборатории спектрофотометрии кафедры аналитической химии МГУ [85].
Для приготовления эталонных растворов используют условия, указанные при определении алюминия (см. стр. 133).
Для определения алюминия в металлическом бериллии берут две навески по 0,5 г металла, помещают каждую в коническую колбу емкостью 300 мл и растворяют в 20—25 мл 2 н. соляной кислоты при осторожном нагревании. Охлажденные растворы переводят в мерные колбы емкостью 100 мл и доводят объем раствора водой до метки. Для определения 10 мл этого раствора переводят в делительную воронку и проводят определение в условиях, указанных для приготовления эталонных растворов. Содержание алюминия находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают с применением метода математической статистики.
ОПРЕДЕЛЕНИЕ АЛЮМИНИЯ ЭРИОХРОМЦИАНИНОМ
Эриохромцианин R относится к группе трифенилметановых красителей:
Растворы реагента в зависимости от pH раствора имеют различную окраску, что соответствует различным его формам [1]:
ER+ ER ER- ER2- ER3-рН —0 ~ 1 ~ 3 ~ 7 — 12
цвет розовый оранжево- розовато- желтый желтый красный
в литературе имеются разноречивые сведения о составе образующегося соединения [1]. В оптимальных условиях образования комплекса в интервале pH 6,2—6,5 [1] значение в 6,5е 10*, 7тах 535 нм.
Рис. 49. Спектры поглощения растворов:
1 — эрнохромцианин R; 2 — комплексное соединение с алюминием
ОЛОВО И
В спектрах комплекса и реагента есть некоторое наложение, что необходимо учитывать при приготовлении раствора сравнения (рис. 49). Определению алюминия мешает ряд элементов: железо (III) восстанавливают аскорбиновой кислотой до Fe(II); Си (II) переводят в бесцветное
комплексное соединение тиосульфатом. Свинец, никель, марганец не мешают определению алюминия.
А. Определение алюминия в растворе его соли
Реагенты
1. Стандартный раствор алюминия, содержащий 1 мг/мл А1; приготовление (см. стр. 133).
2. Аскорбиновая кислота, 1%-ный раствор, свежеприготовленный.
3. Ацетат аммония, 50%-ный раствор (pH 7).
4. Эрнохромцианин R, 0,04%-ный раствор. В мерную колбу емкостью 250 мл вносят 0,1 г реагента, добавляют 1 мл 1 н. раствора соляной кислоты и доводят объем раствора водой до метки.
5. Соляная кислота, 1 н. раствор.
6. Раствор NaOII, 1 н. раствор.
Для приготовления эталонных, растворов берут пять мерных колб емкостью 50 мл, вводят по 25 мл воды и стандартный раствор соли алюминия в количестве (мкг): 0,0; 5,0; 7,5; 10,0; 15,0 соответственно, добавляют в каждую 2 мл раствора аскорбиновой кислоты, доводят раствор добавлением НС1 или NaOH до pH 2 по индикаторной бумажке и через 5 мин добавляют 5 мл раствораэриохромцианина R, 5 мл ацетата аммония и раствор перемешивают, доводя водой его объем до метки. Через 5 мин измеряют оптическую плотность раствора при 7535 нм на фотоэлектроколориметрах марок ФЭК-56, ФЭК-57 и ФЭК-60 или любых спектрофотометрах. В качестве раствора сравнения используют 1-й раствор эталонного ряда. Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
. „^ля определения алюминия в испытуемом растворе из общего объема W мл берут аликвотную часть 25 мл, проводят все операции, указанные при приготовлении эталонных растворов, и находят содержание алю-иния по градуировочному графику.
Б. Определение алюминия в медно-цинковых сплавах дифференциальным методом
Кроме указанных на стр. 133, необходимы реагенты:
1. Тиосульфат натрия, 15%-ный раствор.
2. Соляная кислота (х. ч.), раствор 1:1.
3. Азотная кислота (х. ч.), раствор 1:1.
Определение алюминия проводят одним из вариантов дифференциального метода, см. стр. 76.
Для определения алюминия в образце сплава (латуни, бронзы) берут две навески по 0,5 г, помещают каждую в коническую колбу емкостью 50 мл и растворяют при нагревании, добавляя 5 мл раствора азотной кислоты. После растворения переносят раствор в мерную колбу емкостью 100 мл, доводят объем раствора водой до метки. Берут три мерные колбы емкостью 100 мл, в 1-ю вводят 20 мл приготовленного раствора, во 2-ю колбу —30 мл того же раствора и в 3-ю — также 30 мл приготовленного раствора и добавляют 0,1 мг стандартного раствора алюминия. Во все колбы добавляют по 2 мл аскорбиновой кислоты, 0,25 мл раствора тиосульфата натрия, тщательно растворы перемешивают, доводят раствор до pH 2 по индикаторной бумажке добавлением раствора NaOH или НС1, приливают 5 мл реагента, 20 мл ацетата натрия и доводят объем раствора водой до метки. Измеряют оптическую плотность второго раствора А* и третьего Ах+а по отношению к 1-му раствору при X 535 нм и находят содержание алюминия в 10 мл контрольного раствора, чго соответствует разности объемов, прибавленных во второй и первый растворы. Содержание алюминия рассчитывается по формуле
сх~^ cal Аа-
По полученному результату вычисляют процентное содержание алюминия в анализируемом образце сплава.
Определение галлия
Для определения галлия большее распространение получили методы, в которых предусматривается образование ионных ассоциатов хлоргалловой кислоты с основными красителями. Галлию всегда сопутствует алюминий, близкий по ряду свойств этому элементу. В методах 'определения галлия это необходимо учитывать.
В предлагаемом экстракционно-спектрофотометрическом методе определения галлия используется образование ионного ассоциата HGaCl4 с родамином С (тетраэтилродамин) [6]
^страгируемого смесью толуола с бутилацетатом (2:1) из 6н. раствора соляной кислоты. Соотношение компонентов в образующемся ионном ассоциате 1:1; е = 6- 104, Хп1ах 540 нм [61. При этом отпадает необходимость предварительного отделения галлия от алюминия. Данным методом возможно определение галлия при соотношении Ga : А1 = = 1 : 5 • 106.
Реагенты
1. Стандартный раствор соединения галлия; 0,1 г металлического галлия растворяют в 6 н. НС1, опустив в раствор платиновый шпатель или проволоку. Раствор переводят в мерную колбу емкостью 100 мл и доводят объем раствора до метки 6 н. НС1. Прн этом 1 мл раствора содержит 1 мг Ga — раствор А. Растворы меньшей концентрации готовят в день употребления разбавлением раствора А в 1000 раз; 1 мл этого раствора содержит 1 мкг Ga.
2. Соляная кислота (х. ч.), пл. 1,19, 6 н. раствор.
3. Хлорид титана (III), 20%-ный раствор. Для приготовления его 6 г металлического титана растворяют в 100 мл 6 н. соляной кислоты. Раствор сохраняют в склянке из темного стекла.
4. Толуол (х. ч.).
5. Бутилацетат. Используют смесь толуола и бутилацетата в соотношении 2 : 1.
6. Родамин С, 0,5%-ный раствор: растворяют 0,25 г реагента в 50 мл 6 и. НС1, раствор фильтруют и сохраняют в склянке из темного стекла.
А. Определение галлия в гидроокиси алюминия
Для приготовления эталонных растворов берут шесть делительных воронок емкостью 50 мл, вводят в пять из них стандартный раствор, содержащий галлий в количестве (мкг): 0,0; 0,5; 1,0; 2,0; 3,0; 4,0 соответственно; в 1-ю воронку стандартный раствор не вводят. Во все воронки добавляют по 0,3 мл раствора хлорида титана (III) ибн. соляной кислоты до объема 5 мл. По истечении 5 мин добавляют 1,0 мл раствора родамина С и вновь перемешивают растворы. Затем добавляют 8 мл смеси толуола и бутилацетата и экстрагируют в течение 0,5 —1,0 мин образовавшийся ионный ассоциат галлия с родамином. После разделения фаз органический слой фильтруют через ватный тампон в сухой • цилиндр или пробирку с притертой пробкой, промывают тампон смесью растворителей и доводят объем до 10 мл этой же смесью и перемешивают. Оптическую плотность растворов измеряют на фотоколориметрах марок ФЭК-56, ФЭК-57, ФЭК-60 или спектрофотометрах любой марки при 7 540 нм. Раствором сравнения служит экстракт, полученный обработкой раствора, в который не вводился стандартный раствор соединения галлия. Градуировочный график строят по экспериментальным данным, обработанным методом наименьших Квадратов.
Для определения галлия в гидроокиси алюминия берут две навески по 0,5 г, помещают в конические колбы емкостью 50 мл, растворяют каждую при нагревании до кипения в 30—40 мл 6 н. НС1, раствор переводят в мерную колбу емкостью 50 мл и доводят объем раствора до метки той же кислотой. Аликвотную часть раствора 5 мл переносят в Делительную воронку емкостью 50 мл, добавляют 0,3 мл раствора хлорида титана (Ш) и перемешивают. По истечении 5 мин добавляют
1 мл раствора родамина С и вновь перемешивают, добавляют 8 мл смеси толуола и бутилацетата и экстрагируют в течение 0,5—1,0 мин образовавшийся ионный ассоциат галлия с родамином. В дальнейшем поступают так же, как указано при приготовлении эталонных растворов (см. стр. 136). Содержание галлия находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики.
Б. Определение галлия в карбонате кальция
Приготовление эталонных растворов см. на стр. 136.
Для определения галлия в карбонате кальция берут две навески по 1 г, растворяют каждую при нагревании в 15—20 мл 6 н. HCL Раствор переводят в мерную колбу емкостью 25 мл и доводят объем раствора до метки той же кислотой. Атиквотную часть раствора 5 мл переносят в делительную воронку емкостью 50 мл, добавляют 0,3 мл раствора хлорида титана (III) и перемешивают. Через 5 мин добавляют 1 мл раствора родамина С и вновь перемешивают, добавляют 8 мл смеси толуола и бутилацетата и экстрагируют в течение 0,5—1,0 мин образовавшийся ионный ассоциат. В дальнейшем поступают так же, как указано при приготовлении эталонных растворов (см. стр. 136). Содержание галлия находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики.
Определения фосфора, кремния и мышьяка в виде гетерополисоединений
В основе фотометрических методов определения кремния, фосфора и мышьяка лежит реакция образования молибденовых гетерополикислот состава: H4[SiMO12O40] • пН2О; Н3 [РМо12О40ЬпН2О; Н3 [AsMo12O4J • иН2О.
Гетерополикислоты (ГПК) кремния, фосфора, мышьяка (так называемые желтые формы) образуются в кислой среде при избытке молибдена (VI) в растворе.
Кислотность среды и концентрация молибдена (VI) влияют совместно на образование ГПК. Взаимную связь кислотности и концентрации Mo (VI) при образовании гетерополииона (ГА) подчеркивали многие исследователи: 3. Ф. Шахова, М. Л. Цап, Л Дюваль и др. Последние исследования Е. Пунгора с сотрудниками [7] показали, что ГА образуется в водных растворах количественно при определенном соотношении кислотности и концентрации молибдата. Авторы [7] ввели условную величину — кислотное число z, равное числу протонов, участвующих в реакции с одним молем молибдена (VI). По стехиометрическому уравнению для 12 молибдат-ионов
POJ- + 12MoOJ - + 24Н+ = [Р (Мо3 Ою)4] + 12Н2О
г = 24: 12=2.
Для фосфора и мышьяка ГА образуется количественно при г = 1,60. Для кремния z = 1,50.
Молибденофосфорная ГПК получается в 0,85 н. растворе минеральной кислоты, молибденомышьяковая в 0,6—0,9н. растворе, молибдено-кремниевая кислота в слабокислом растворе (pH 1,5—2,0 и pH 3—4). Различная устойчивость указанных комплексных соединений широко используется при определении кремния, фосфора’ и мышьяка в их смеси. При фотометрическом определении этих элементов по желтым формам следует учитывать различные модификации а- и |3- форм, природа которых не совсем ясна. По-видимому, решающим в образовании этих форм является степень полимеризации молибдата; возможно различия заложены в структуре ГА.
Молибденомышьяковая кислота всегда образуется в ct-форме, которая при pH 1 медленно переходит в [1-форму. Все молибденовые ГПК могут быть получены в [1-форме в водно-органических средах [8], чем обусловлено проведение реакции образования гетерополикислот фосфора, кремния в смешанных средах [9]. Этот метод [9], не уступая по простоте выполнения обычному методу фотометрического определения фосфора в водных растворах, несколько превосходит его по чувствительности. В последнее время для получения синих форм ГПК в качестве восстановителей используют преимущественно более мягкие восстановители [11]: аскорбиновую кислоту, аскорбиновую кислоту + + антимонилтартрат и аскорбиновую кислоту с солью висмута, что предотвращает восстановление молибдена из молибдата аммония, который берут в избытке; [10] применяют также соль Мора, хлорид олова (II), сульфит натрия, гидрохинон, аминонафтолсульфат (эйконоген) [11].
При определении кремния следует иметь в виду, что реакционноспособной является лишь мономерная форма, образующая молибдено-кремниевую кислоту в течение 75 с. Обычно для полного развития окраски необходимо 10—15 мин, в течение которых происходит деполимеризация димерной формы в мономерную. Для молибденокремниевой кислоты лучшими восстановителями являются аскорбиновая кислота, аминонафтолсульфат натрия, оксалат олова, смесь Mo (VI) и Mo (V), что приводит к непосредственному образованию синей формы ГПК без стадии образования желтой формы.
При растворении железосодержащих материалов в серной кислоте также наблюдается образование синей формы без предварительного получения желтой формы [12], [13].
Восстановленные и невосстановленные формы гетерополикислот экстрагируются органическими растворителями, обладающими электронно-донорными атомами кислорода или азота (спиртами, кетонами, эфирами, высокомолекулярными аминами), что позволяет определять Микрограммовые количества фосфора, кремния и мышьяка.
ОПРЕДЕЛЕНИЕ ФОСФОРА
Определение фосфора проводят в виде синей формы фосфорно-молибденового гетерополианиона с применением различных восстано-
А. Определение фосфора в стали
При определении фосфора в стали [14] в качестве восстановителя для получения синей формы гетерополикислоты используют сернокислый гидразоний.
Реагенты
1. Стандартный раствор, содержащий 0,1 мг/мл Р, готовят растворением в воде 0,439 г (х. ч.) фосфата калия КН2РО4, предварительно высушенного при 110° С, в мерной колбе емкостью 1 л и доводят объем раствора водой до метки более разбавленный раствор с содержанием 1 мкг/мл готовят соответствующим разбавлением в день употребления.
2. Азотная кислота, разбавленный раствор: 380 мл концентрированной азотной кислоты добавляют к 620 мл воды.
3. Бромистоводородная кислота, разбавленный раствор" 20 мл 48%-ной НВг добавляют к 80 мл воды.
4. Сульфит натрия, растворяют 100 г безводного сульфита натрия в 500 мл воды, разбавляют раствор до 1 л и фильтруют.
5. Молибдат аммония, растворяют 20 г молибдата аммония (ЫН4)в Мо7О24-•4Н2О в 1 л 10 н. раствора серной кислоты.
6. Сульфат гидразиния, растворяют 1,5 г сульфата гидразиния в мерной колбе емкостью 1 л и доводят объем раствора водой до метки.
7. Смесь молибдата аммония и сульфата гидразиния. Берут 25 мл раствора молибдата аммония разбавляют до 80 мл водой, добавляют 10 мл раствора сульфата гидразиния и разбавляют водой до объема 100 мл. Этот раствор неустойчив, поэтому его следует готовить непосредственно перед употреблением
8. Хлорная кислота, 2 и. раствор.
Для приготовления эталонных, растворов в пять мерных колб емкостью 50 мл вводят в каждую 25 мл воды и стандартный раствор, содержащий фосфор (мг): 1,0; 2,0; 4,0; 6,0 соответственно, добавляют 5 мл раствора молибдата и 2 мл раствора сульфата гидразиния, доводят объем раствора водой до метки и перемешивают. Погружают колбу в кипящую водяную баню на 10 мин, вынимают и затем быстро охлаждают. Снова перемешивают содержимое .колбы и, если необходимо, доводят объем до метки добавлением нескольких капель воды. Измеряют оптическую плотность раствора при К 830 нм относительно раствора, не содержащего фосфора (1-й эталонный раствор), и строят градуировочный график.
Для определения фосфора в образце стали берут две навески по 0,15 мг углеродистой или низколегированной стали, помещают каждую в коническую колбу емкостью 100—125 мл, добавляют 5 мл разбавленного раствора азотной кислоты и нагревают до полного растворения пробы. (При анализе некоторых хромовых сталей, кроме азотной кислоты, добавляют еще 3 мл соляной кислоты 1:1.) Пипеткой добавляют 5 мл раствора хлорной кислоты. Осторожно выпаривают до появления паров и после этого продолжают нагревание 3—5 мин до удаления азотной кислоты. Охлаждают, и, если сталь содержит более 0,05% мышьяка, добавляют 5 мл разбавленного раствора бромистоводороднои кислоты и осторожно выпаривают до удаления НВг, переводят в мерные колбы емкостью 25 мл и доводят объем раствора водой до метки. Берут две порции по 10 мл из каждой колбы, помещают растворы в конические колбы емкостью 50 мл, добавляют в каждую по 15 мл раство
ра сульфита. Растворы нагревают до кипения и продолжают кипятить около 30 с, охлаждают и переносят в мерные колбы емкостью 50 мл, применяя минимальное количество воды для споласкивания. Добавляют 20 мл реагента — смесь молибдата аммония с сульфатом гидрази-ния. Доводят объем раствора до метки водой и перемешивают его. Погружают колбу на 10 мин в кипящую водяную баню, вынимают и быстро охлаждают. Если необходимо, прибавляют несколько капель воды до метки. Измеряют оптическую плотность при X 830 нм в кювете I = 1 см относительно раствора, приготовленного из одинаковой навески и тех же количеств реагентов, но с прибавлением 5 мл 10 н. раствора серной кислоты вместе с 5 мл раствора молибдата.
Содержание фосфора находят по градуировочному графику. Данные из параллельных (не менее четырех) опытов обрабатывают методами математической статистики.
Для определения фосфора в чугуне берут две навески по 100 мг, помещают каждую в коническую колбу емкостью 100 —125 мл, добавляют 25 мл разбавленного раствора азотной кислоты, удаляют азотную кислоту длительным кипячением, разбавляют водой раствор, переводят в мерную колбу емкостью 100 мл, доводят объем раствора до метки водой, дают осесть графиту и отбираю! 5 мл полученного раствора для анализа. Далее определение производят, как описано выше, начиная с добавления раствора хлорной кислоты (см. стр. 140). Содержание фосфора находят по градуировочному графику. Данные из параллельных (не мейее четырех) опытов обрабатывают методами математической статистики.
Б. Определение фосфора в мышьяке, трехокиси мышьяка
Метод основан на реакции образования фосфорномолибденовой гетерополикислоты с последующим восстановлением ее в среде соляной кислоты аскорбиновой кислотой в присутствии тартрата калия антимонила.
При анализе мышьяка и трехокиси его мышьяк отделяют отгонкой в виде галогенида на стадии разложения материалов и последующей экстракцией четыреххлористым углеродом из 9 н. соляной кислоты, содержащей иодид калия.
При анализе хлорида мышьяка первоначально проводят его экстракцию четыреххлористым углеродом, а остаточные количества отделяют отгонкой в виде галогенида.
Кроме указанных на стр. 140, необходимы реагенты:
1. Соляная кислота (о. ч.), 9 н., 6 н. и 5 н. растворы.
2. Азотная кислота (х. ч.), пл. 1,4, очищенная перегонкой.
3. Смесь соляной (пл. 1,19) и азотной (пл. 1,4) кислот, взятых в соотношении 5 : 1 (свежеприготовленная).
4. Кислота бромистоводородная (х. ч.), очищенная перегонкой.
о. Бром, очищенный перегонкой.
6- Иодид калия (ч. д. а.), 1,7%- и 3,4%-ные растворы в 9 н. НС1. Для их риготовления 1,7 г или 3,4 г KI соответственно растворяют в 15 мл воды
и доводят концентрированной соляной кислотой до объема 100 мл; готовят в день употребления.
7. Едкий натр (о. я.), 40%-ный раствор.
8. Четыреххлористый углерод, перегнанный.
9. Эфир диэтиловый, перегнанный.
10. Молибдат аммония (х. я.), 4%-ный раствор.
11. Аскорбиновая кислота (я. д. а), 0,1 М раствор.
12. Хлорид калия (о. я.), 1%-ный раствор.
13. Раствор тартрата калня антимонила, готовят растворением 0,27 г соли в 100 мл воды.
Для приготовления эталонных растворов берут пять мерных колб емкостью 25 мл, вводят в четыре из них стандартный раствор, содержащий фосфор (мкг): 0,0; 0,05; 0,10; 0,30; 0,50 соответственно; приливают воду до объема 20 мл (в первую колбу не вводят стандартный раствор), добавляют во все колбы по 2 мл соляной кислоты, 0 6 мл раствора молибдата аммония, 1,2 мл раствора аскорбиновой кислоты и 0,2 мл раствора тартрата калия антимонила. Доводят объем раствора водой до метки и перемешивают. Через 3—5 мин измеряют оптическую плотность растворов на спектрофотометрах различных марок при X 830 нм, строят градуировочный график по экспериментальным данным, обработанным методом наименьших квадратов. Раствором сравнения служит содержимое первой колбы.
Примечание. Этот метод приготовления эталонных растворов используют при анализе всех объектов (мышьяка и его соединений, сурьмы, галлия и его соединений), в которых для получения синей формы фосфорномолибденовой гетерополикислоты применяют аскорбиновую кислоту с тартратом калием антимонилом.
Для определения фосфора в мышьяке и трехокиси мышьяка две навески по 2,5 г тонкорастертого мышьяка или окиси мышьяка помещают каждую в коническую колбу емкостью 50 мл, приливают 9 мл 6 н. соляной кислоты. При анализе мышьяка добавляют по каплям через воронку 6 мл брома и растворяют на холоду.
Раствор переводят в мерную колбу емкостью 25 мл, доводят объем раствора тем же раствором кислоты до метки. Отбирают 10 мл, переносят в колбу для отгонки и отгоняют мышьяк при нагревании до тех пор, пока в колбе останется 1 мл жидкости; добавляют еще 3 мл 6 и. соляной кислоты и отгонку продолжают. Эту операцию повторяют. (Летучие соединения мышьяка поглощают раствором едкого натра.) Оставшийся раствор (~1 мл) переводят в делительную воронку емкостью 25 мл, колбу споласкивают 5 мл 1,7%-ного раствора иодида калия в соляной кислоте и присоединяют к содержимому в воронке. Затем приливают 10 мл четыреххлористого углерода и содержимое воронки встряхивают в течение 2 мин. По расслаивании жидкостей органический слой сливают, а водный переводят в кварцевую чашку и упаривают досуха на плитке со слабым нагревом.
Для удаления иода сухой остаток тщательно обрабатывают 20— 25 каплями азотной кислоты, приливая ее по стенкам чашки и упаривают досуха. Затем остаток обрабатывают 20—25 каплями 6 н. соляной кислоты и вновь упаривают досуха. (Остаток в чашке должн быть бе
лым.) Остаток растворяют в 2 мл 5 н. соляной кислоты и проводят оп-ределение фосфора, как указано при приготовлении эталонных растворов. Содержание фосфора находят по градуировочному графику. Данные (не менее четырех) параллельных определений обрабатывают методом математической статистики.
Примечание. 1. Для измерения оптической плотности при определении п • Ю-во/о фосфора рекомендуется использовать кюветы / = 10 см. При отсутствии кювет указанной длины применяют экстракцию фосфорномолибде-новой гетерополи кислоты 5 мл изобутилового спирта. Измерение оптической плотности следует проводить при X 830 нм и / = 1 см.
2. Во всех объектах, содержащих мышьяк, сурьму и галлий, раствор сравнения готовят аналогично приготовлению эталонных растворов.
В. Определение фосфора в сурьме особой чистоты
Метод основан на реакции образования фосфорномолибденовой гетерополикислоты с последующим восстановлением ее в среде соляной кислоты аскорбиновой кислотой с добавлением тартрата калия антимонила.
При анализе сурьмы ее отделяют отгонкой в виде галогенида на стадии растворения металла, а при анализе хлорида сурьмы первоначально проводят его экстракцию эфиром из 11 н. соляной кислоты, а остаточные количества сурьмы отгоняют в виде галогенида.
При определении фосфора в сурьме берут две навески по 1г тонко-растертого препарата, помещают каждую в кварцевую чашку емкостью 50 мл, приливают 5 мл 6 н. соляной кислоты и по каплям при охлаждении 2 мл брома. Чашку помещают на электроплитку, находящуюся в боксе из оргстекла, накрытую кварцевой или графитовой пластиной, и упаривают содержимое чашки досуха. Сухой остаток растворяют в 2 мл 6 н. соляной кислоты и 1 мл брома и вновь упаривают досуха. Эту операцию повторяют два раза (в последний раз бром не добавляют). Остаток растворяют в 2 мл 5 н. соляной кислоты и проводят определение фосфора, как указано при приготовлении эталонных растворов (стр.142 ). Содержание фосфора находят по градуировочному графику. Данные (не менее четырех) параллельных определений обрабатывают методами математической статистики.
Г. Определение фосфора в галлии
Метод основан на реакции образования фосфорномолибденовой гетерополикислоты с последующим восстановлением ее аскорбиновой кислотой в среде соляной кислоты с добавлением тартрата калия антимонила. Метод пригоден для определения фосфора в образцах хлорида, арсенида и антимонида галлия.
Галлий и сурьму в основном удаляют из раствора однократной экстракцией диэтиловым эфиром; мышьяк удаляют в виде галогенида отгонкой. Полное отделение остаточных количеств мышьяка и сурьмы, мешающих определению, достигается упариванием раствора с бромистоводородной кислотой досуха.
Приготовление эталонных растворов см. на стр. 142.
Для определения фосфора в галлии берут три навески металла noir помещают каждую в кварцевый стакан или чашку емкостью 50 мл' приливают 12 мл смеси соляной и азотной кислот, накрывают часовым стеклом и проводят растворение при умеренном нагревании. Полученный раствор переводят в делительную воронку емкостью 50 мл, смывая стакан 2—3 мл 6 н. соляной кислоты, приливают 15 мл диэтилового эфира и экстрагируют галлий, встрахивая содержимое воронки в течение 1 мин. По расслаивании жидкостей водную фазу отделяют в кварцевую чашку и упаривают при умеренном нагревании досуха. К остатку добавляют 2 мл 5 н. соляной кислоты, смывают его водой в мерную колбу емкостью 25 мл, добавляют 0,6 мл раствора молибдата аммония, 1,2 мл раствора аскорбиновой кислоты и 0,2 мл раствора тартрата калия антимонила. Доводят объем раствора до метки водой и перемешивают. Измерение оптической плотности см. на стр. 142. Содержание фосфора находят по градуировочному графику. Данные параллельных (не менее четырех) определений обрабатывают методами математической статистики.
Методы определения фосфора в других объектах, содержащих галлий, различаются только условиями предварительной подготовки образца.
Д. Определение фосфора в растворе его соли в водноорганических средах
Метод основан на образовании ^-формы молибденофосфорной кислоты в водноорганических средах [9].
Кроме указанных на стр. 141, 142, необходимы реагенты:
1. Молибдат натрия Na2MoO4 • 2Н2О, 1%-ный раствор.
2. Хлорная кислота 2,5 н. раствор. Готовится разбавлением 129 мл хлорной кислоты (пл. 1,28) до 250 мл водой.
3. Ацетон.
При приготовлении эталонных растворов берут шесть мерных колб емкостью 25 мл, вводят в пять из них стандартный раствор, содержащий фосфор в количестве (мг) 0,0; 0,01; ,0,03; 0,05; 0,075; 0,1 соответственно, добавляют во все шесть колб по 2,5 мл раствора хлорной кислоты, 12,5 мл ацетона и 1 мл раствора молибдата натрия, доводят объем раствора до метки водой. Измеряют оптическую плотность через 60 мин в кварцевых кюветах I = 1 см при X 310 нм на спектрофотометре относительно раствора, содержащего все компоненты, кромё фосфора (1-я колба). Измерение оптической плотности можно провести на фотоэлектроколориметре марки ФЭК-60 со светофильтрами Лаф 340 нм. Строят градуировочный график.
Для определения фосфора в испытуемом растворе из общего объема 150 мл берут аликвотную часть 10 мл, проводят все операции, указанные при приготовлении эталонных растворов, и находят содержание фосфора по градуировочному графику.
ОПРЕДЕЛЕНИЕ КРЕМНИЯ
А. Определение кремния в металлическом бериллии
Кремний в металлическом бериллии определяют в виде кремнемолибденовой сини. В качестве восстановителя используют смесь растворов соли Мора и аскорбиновой кислоты.
Реагенты
1. Стандартный раствор, содержащий 0,1 мг/мл Si. В платиновом тигле сплавляют 0,2140 г прокаленной и тонкоизмельченной двуокиси кремния с 2 г карбоната натрия. Плав растворяют в воде, количественно переносят в мерную колбу емкостью 1 л, добавляют до 900 мл воды, подкисляют 2 н. серной кислотой до рН~1,5, доводят раствор водой до метки. Раствор содержит 0,1. мг/мл Si. Более разбавленный раствор готовят в день употребления.
2. Молибдат аммония (х. ч.), 5%-ный раствор.
3. Соляная кислота (о. ч.), раствор 1 : 1 и 0,5%-ный раствор.
4. Соль Мора (х. ч.), 4%-ный раствор.
5. Аскорбиновая кислота, 5%-ный раствор.
6. Едкий натр (х. ч.), 20%- и 10%-ный растворы.
7. Смесь восстановителей. Готовят перед употреблением равные объемы растворов аскорбиновой кислоты и соли Мора.
Для приготовления эталонных растворов берут шесть мерных колб емкостью 50 мл, вносят в пять из них стандартный раствор, содержащий кремний в (мкг): 0,0; 5,0; 10,0; 15,0; 20,0; 25,0 соответственно, прибавляют во все колбы 10 мл воды, 5 мл 0,5%-ного раствора соляной кислоты, 2 мл раствора молибдата аммония, растворы перемешивают и выдерживают при комнатной температуре 15 мин. Затем приливают 10 мл соляной кислоты (1:1), 1 мл восстановителя, доводят раствор водой до метки, содержимое тщательно перемешивают и выдерживают 40 мин. Измеряют оптическую плотность эталонных растворов на спектрофотометрах при X 800 нм или фотоэлектроколориметрах ФЭК-56 и ФЭК-57 при X 660 нм и ФЭК-60 X 750 нм. В качестве раствора сравнения используют содержимое 1-й колбы. По экспериментальным данным, обработанным методом наименьших квадратов, строят градуировочный график.
Для определения кремния в бериллии берут три навески металла по 0.5 г в конические колбы емкостью 300 мл, растворяют каждую в 20 мл соляной кислоты (1:1), по окончании растворения раствор упаривают До 15 мл, охлаждают и осторожно нейтрализуют раствором едкого натра до pH ~ 2, затем раствор переносят в мерную колбу емкостью о0 мл, далее ведут анализ в условиях приготовления эталонных растворов и по градуировочному графику, находят содержание кремния. Результаты не менее трех определений обрабатывают методом математической статистики.
„„.Примечание. Данный метод разработан в лаборатории спектрофотометрии кафедры аналитической химии МГУ.
Б. Определение кремния в сталях
Описываемой метод пригоден для определения кремния при наличии фосфора в сталях.
Для приготовления эталонных растворов 0,1г стали, не содержащей кремния (если такая отсутствует, то берут сталь с известным содержанием кремния, которое учитывают при приготовлении эталонных растворов), помещают в коническую колбу емкостью 80—100 мл, добавляют 10 мл 2 н. серной кислоты и нагревают на водяной бане при 85° С до прекращения выделения пузырьков. После этого приливают 5 мл азотной кислоты и продолжают нагревание раствора в течение 2 мин. Небольшой осадок графита дальнейшему ходу анализа не мешает. Охладив раствор, переносят его в мерную колбу емкостью 250 мл, доводят объем раствора водой до метки, перемешивают и дают ему отстояться. Для приготовления эталонных растворов в шесть мерных колб емкостью 100 мл берут по 5 мл приготовленного раствора, вводят кроме 1-й колбы в каждую соответственно стандартный раствор, содержащий кремний в количестве (мг): 0,005; 0,01; 0,015; 0,02; 0,025 соответственно, приливают 15 мл 0,15 и. серной кислоты, 5 мл раствора молибдата аммония и оставляют стоять 5 мин. Прибавляют 25 мл 8 н. серной кислоты и после перемешивания добавляют 4 мл раствора хлорида олова. Измерение оптической плотности эталонных растворов проводят на фотоэлектроколориметре при X 800 нм на ФЭК-60 или спектрофотометрах различных марок и строят градуировочный график. В качестве раствора сравнения берут содержимое 1-й колбы.
Для определения кремния в стали берут две навески по 0,1 г стали, растворяют каждую при тех же условиях, которые были указаны для приготовления эталонных растворов. После соответ :твующего разбавления для определения берут 5 мл раствора и проводят операции в условиях, аналогичных приготовлению эталонных растворов. Содержание кремния находят по градуировочному графику. Результаты параллельных (не менее четырех) опытов, обрабатывают методами математической статистики.
В. Определение микроколнчеств кремния в водах
Метод основан на восстановлении Р-молибденокремниевой кислоты мягким восстановителем 1-амино-2-нафтол-4-сульфокислотой («эйконо-геном») [151. Метод позволяет определять от 2 мкг до 75 мг кремния в котловых, питьевой и деионизованной водах. Для приготовления раст-' вора сравнения используется оригинальный способ: искусственное торможение основной реакции в растворе «холостой пробы».
Реагенты
1. Стандартный раствор, содержащий 10 мкг/мл, Si (см. стр. 145).
2. Подкисленный раствор молибдата аммония: 4,85 г парамолибдата аммония (х. ч.) растворяют в 400 мл воды, добавляют 100 мл 9 н. H2SO4 и разбавляют водой до 500 мл.
3. 1-Амино-2-нафтол-4-сульфокислота («эйконоген»): 0,2 г эйконогена и 2,4 г сульфита натрия растворяют в 70 мл воды, добавляют 14 г метагидросуль
фита калия и встряхивают до полного растворения, разбавляют до 1 00 мл во-Д0И’4. Винная кислота, 2,8%-ный раствор.
Для приготовления эталонных растворов берут шесть мерных колб емкостью 100 мл, вводят в каждую 90 мл деионизованной воды, стандартный раствор, содержащий кремний (мкг): 0,0; 2,0; 5,0; 10,0; 15,0; 20,0 соответственно, добавляют 2,5 мл раствора молибдата аммония, через 10 мин добавляют 2,5 мл раствора винной кислоты и через 3—5 мин 2 мл раствора эйконогена. Измеряют оптическую плотность через 20__30 мин в кюветах длиной 1—5 см при X 810 нм. Раствором сравне-
ния служит содержимое 1-й колбы, в которую не введен стандартный раствор. Поданным измерения оптической плотности эталонных растворов, обработанным методом наименьших квадратов, строят градуировочный график.
Для on ределения кремния в испытуемой воде в полиэтиленовую колбу помещают 100 мл исследуемой воды, содержащей 2—100 мкг кремния, добавляют 2,5 мл раствора молибдата аммония, через 10 мин добавляют 2,5 мл раствора винной кислоты и через 3—5 мин 2 мл раствора эйконогена. Измеряют оптическую плотность через 20—30 мин в кюветах длиной 1—5 см при Z, 810 нм относительно раствора сравнения.
Для приготовления раствора сравнения «холостой пробы» к 5 мл исследуемой воды добавляют 2,5 мл раствора молибдата аммония, разбавляют до 100 мл водой и добавляют те же реактивы в условиях, указанных при приготовлении эталонных растворов.
Содержание кремния находят по градуировочному графику.
ОПРЕДЕЛЕНИЕ МЫШЬЯКА
Определение мышьяка производят по восстановленной форме мо-либденовомышьяковой кислоты, используя в качестве восстановителя аскорбиновую кислоту, содержащую ионы висмута (III) и тартрат калия антимонила.
Реагенты
1. Стандартный раствор соли мышьяка г.отовят растворением в воде 0,416 г Na2HAsO4 7Н2О в мерной колбе емкостью 100 мл; 1 мл раствора содержит 1 мг As; более разбавленный раствор с содержанием 1 мкг/мл готовят соответствующим разбавлением в день употребления.
2. Молибдат аммония, 5 • 10-2 М раствор, 0,83 г соли растворяют в 100 мл воды.
3. Нитрат висмута, 2,5 • 10-2 М раствор, 0,125 г соли растворяют в 100 мл 5 н. серной кислоты и разбавляют аликвотную порцию раствора в 10 раз 2 н. H2SO4 Разбавление осуществляют в день употребления.
4. Аскорбиновая кислота, свежеприготовленный 0,7%-ный раствор.
5. Серная кислота, 2 н. раствор.
А. Определение мышьяка в растворе его соли
Для приготовления эталонных растворов берут шесть мерных колб емкостью по 25 мл, в пять из них вводят стандартный раствор, содержащий мышьяк в количестве (мкг): 0,0; 0,5; 1,0; 2,0; 3,5; 5,0 соответ-
ственно, во все колбы добавляют по 3 мл раствора молибдата аммония 1 мл раствора нитрата висмута, 4 мл серной кислоты, 10 мл раствора аскорбиновой кислоты, доводят объем раствора водой до метки. Измерение оптической плотности эталонных растворов производят при Я 840 нм на фотоэлектроколориметре марки ФЭК-60 или спектрофотометрах любых марок. Раствором сравнения служит раствор 1-й колбы в которую не введен стандартный раствор, содержащий мышьяк. Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения мышьяка в испытуемом растворе из общего объема 100 мл берут аликвотную часть 25 мл, проводят все операции, указанные при приготовлении эталонных растворов, и находят содержание мышьяка по градуировочному графику.
Б. Определение мышьяка в галлии
В качестве восстановителя для получения синей формы ГПК применяют раствор аскорбиновой кислоты с добавлением раствора тартрата калия антимонила.
Кроме указанных на стр. 147, необходимы реагенты:
1. Серная кислота (х. ч.), пл. 1,84, 9 н , 6 н., 5 н., 1 н. растворы.
2. Сульфат аммония (о. ч.).
3. Азотная кислота (х. ч.), раствор 1 : 1.
4. Раствор диэтилдитиокарбамината цинка в хлороформе: 0,4 г ацетата цинка растворяют в 40 мл воды и отдельно растворяют 0,4 г диэтилдитиокарбамината натрия в 40 мл воды. Оба раствора переводят в делительную воронку емкостью 500 мл, приливают 250 мл хлороформа и встряхивают содержимое воронки в течение 1 мин. После расслаивания фаз слой хлороформа сливают через сухой фильтр в сухую склянку с притертой пробкой нз темного стекла. Раствор устойчив длительное время.
5. Едкий натр (о. ч.), 0,5 н. раствор.
6. 5%-ный водный раствор молибдата аммония и 1%-ный раствор 6 н. серной кислоты; хранят в полиэтиленовой посуде, в темноте.
7. Тартрат калия антимонила растворяют 0,27 г соли в 100 мл воды.
8. Аскорбиновая кислота 0,1 и. раствор; 0,76 г растворяют в 100 мл воды, употребляют свежеприготовленный раствор.
9. Смесь реагентов (готовят перед употреблением): сливают 10 мл 5 н. серной кислоты, 3 мл 5%-ного раствора молибдата аммония, 6 мл раствора аскорбиновой кислоты, разбавляют до объема 25 мл водой и перемешивают.
10. Смесь растворителей—изоамилового спирта и этилового эфира в соотношении 1 : 2.
Для приготовления эталонных растворов берут четыре делительные воронки емкостью 20 или 25 мл, вводят стандартный раствор, содержащий мышьяк (мкг): 1,0; 2,0; 3,0; 5,0 соответственно, добавляют воду до объема 3 мл, приливают 1 мл смеси реагентов, 1 мл воды и оставляют стоять. Через 25—30 мин приливают 5 мл смеси растворителей, перемешивают содержимое воронок в течение 1—2 мин и экстракты переводят в градуированные цилиндры или пробирки емкостью 5 мл или 10 мл, добавляя, если нужно, несколько капель смеси растворителей до объема 5 или 10 мл. Измерение оптической плотности эталонных растворов проводят на спектрофотометрах различных марок при 840 нм. Раствором сравнения служит смесь растворителей. Градуиро-148
вечный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения мышьяка в галлии берут три навески металла по 0,5 г, помещают каждую в кварцевый стакан емкостью 100 мл, добавляют 5 г сульфата аммония; приливают 10 мл серной кислоты (пл.1,84), накрывают стакан часовым стеклом и проводят растворение при умеренном нагревании на плитке. По окончании растворения металла содержимое стакана охлаждают и образовавшиеся сульфаты растворяют в 25 мл воды при нагревании. Охлажденный раствор переводят в делительную воронку емкостью 100 мл, смывая стенки стакана 5 мл 9 н. серной кислоты. В воронку приливают 5 мл диэтилдитиокарбамината цинка и экстрагируют соединения мышьяка, встряхивая содержимое воронки в течение 1 мин. После расслаивания слой хлороформа сливают в другую делительную воронку емкостью 50 мл и проводят реэкстракцию мышьяка 5 мл азотной кислоты при встряхивании воронки в течение 0,5 мин. Эту операцию повторяют дважды. Объединенные азотнокислые растворы, содержащие мышьяк, помещают в делительную воронку, промывают 5 мл хлороформа, перевертывая воронку 5—6 раз.Отстоявшийся слой хлороформа тщательно отделяют (не захватывая водной фазы) и отбрасывают, а водный слой переводят в кварцевую чашку и упаривают досуха на плитке с умеренным нагревом, избегая прокаливания сухого остатка. По охлаждении в чашку приливают 3 мл воды, нейтрализуют раствором едкого натра по индикаторной бумаге до pH 6—7 и переводят в делительную воронку, приливают 1 мл смеси реагентов, 1 мл воды и оставляют стоять. В дальнейшем проводят все операции, указанные при приготовлении эталонных растворов.
Содержание мышьяка находят по градуировочному графику. Данные параллельных (не менее четырех) опытов обрабатывают методом математической статистики.
Определение железа
Для определения железа существует ряд методов, позволяющих определять этот элемент в степени окисления (+'2) и (+3). Наиболее известными из них являются методы определения” железа (III) в виде роданида и соединения Fe (II, III) с сульфосалициловой кислотой. Интерес представляют методы определения железа (II) с 1,10-фенант-ролином, 4,7_-дифенил-1-10-фенантролином (батофенантролином), которые в настоящее время получают все большее растпространение.
ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА (III) В ВИДЕ РОДАНИДА
Определение железа (III) в виде роданида. Железо (III) в кислой среде образует с роданид-ионом в зависимости от его концентрации и кислотности среды ряд комплексных соединений различного состава, отличающихся сравнительно малой устойчивостью (табл. 11) [15].
В растворе могут сосуществовать комплексы состава [Fe(SCN)n]3—", где n = 1, 6. Комплексный ион [Fe (SCN)]2+ в растворе образуется
при концентрации роданид-ионов 5 • 10-5 моль/л. При увеличении
ТАБЛИЦА II
Состав и устойчивость комплексов железа с роданид-ионами
Состав комплекса рК ступенчатых констант диссоциации [16] Состав комплекса рК ступенчатых констант диссоциации [16]
[Fe(SCN)]2+ [Fe(SCN)o]+ [Fe(SCN)3] OS ГО 1-1 II II II КЗ Ф» Ф» КЗ [Fe(SCN)4]-[F =(SCN)BF-[Fe(SCN)6]3- 1 F- СП О о COQO -11 II II II 41 Ю (О Q.Q.Q.
концентрации реагента до 1,2 • 10-2 моль7л в значительном количестве образуется [Fe (SCN)21+.
В водном растворе всегда содержится смесь комплексных соединений (рис. 50). Поэтому для получения воспроизводимых результатов важно соблюдать точную концентрацию роданид-ионов в испытуемом и эталонных растворах. При одинаковом составе комплексных частиц соответственно получается одинаковая интенсивность окраски растворов. Рекомендуется всегда добавлять большой избыток роданида. При соблюдении указанных условий растворы подчиняются закону Бугера—Ламберта—Бера в большом интервале концентраций железа. В качестве реагентов можно применять роданиды аммония или калия.
При определении железа в виде роданида важно создавать определенную кислотность раствора. При увеличении кислотности и соответственно увеличении концентрации сульфат- и хлорид-ионов возникает опасность образования комплексов [Fe (SO4) 3]3— и HFeCl4. Оптимальной кислотностью считают 0,05 н. — 0,2 н.; подкисление можно проводить кроме серной, соляной, азотной и хлорной кислотами.
При проведении реакции в вод-но-ацетоновой среде чувствительность реакции повышается. Молярный коэффициент погашения (е) увеличивается от 8,5 • 103 в водном растворе до 1,8 • 104 в растворе, содержащем 50% ацетона [16].
Соединения роданида железа хорошо экстрагируются кислород-
содержащими растворителями, высшими спирами (изоамиловым, 6yj типовым, изобутиловым), эфирами, кетонами, смесью растворителей (трибутилфосфат + СС14). В зависимости от характера растворителя экстрагируются комплексы железа различного состава. Соединение при соотношении [Fe3+] : [SCN~] =1:4 экстрагируется эфирами, при соотношении [Fe3+] : [SCN-] = 1 : 3 — трибутилфосфатом. Чувствительность родановой реакции увеличивается с использованием в качестве экстрагента смеси трибутиламмонпя и амилового спирта (е — = 2,4 • 104).
Рис. 50. Образование железороданидных комплексов (%) в зависимости от концентрации роданида аммония-I— Fe(3f^2Q) ; II—VI комплексы с п—1—5
При проведении реакции в присутствии аминов, например дигек-силамина, образуется соединение состава tFe(SCN)e]Am3, которое в виде ионного ассоциага экстрагируется амилацетатом; е = 3 • Ю4 [17].
Реакцию с роданидом применяют 'только для определения железа (III); железо (II) не вступают в реакцию с,роданидами.
Для окисления Fe (II) в Fe (III) используют азотную кислоту, а также другие окислители в зависимости от природы анализируемого объекта: пероксидисульфат аммония, перманганат калия. Проведению реакции мешает ряд веществ. Прежде всего должны отсутствовать анионы кислот, которые дают более прочные комплексные соединения, чем роданиды железа: фосфаты, ацетаты, арсенаты, фгориды, бораты, а также значительные количества хлоридов и сульфатов. Также должны отсутствовать элементы, ионы которых дают комплексные соединения с роданидом: кобальт, хром, висмут, медь молибден, вольфрам, титан (III, IV), ниобий, палладий, кадмий, цинк, ртуть.
А. Определение железа (III) в растворе его соли
Реагенты
1. Стандартный раствор соли железа (III). Навеску 0,864 г железоаммо -паевых квасцов (х. ч.) (отбирают прозрачные кристаллы'' помещают в мерную колбу емкостью 1 л, растворяют в воде, подкисленной 5 мл серной кислоты (пл. 1,84), доводят объем раствора водой до метки и тщательно перемешивают — раствор А. 1 мл раствора А содержит 0,1 мг железа; 10 мл раствора А разбавляют водой в мерной колбе на 1000 мл — раствор Б: 1 мл раствора Б содержит 0,01 мг железа. Раствор Б готовят в день его употребления.
2. Роданид аммоний, 4 н. раствор.
3. Серная кислота (х. ч.), 4 н. раствор.
4. Азотная кислота (х. ч.), раствор 1:1.
5. Изоамиловый спирт (бутиловый спирт).
Для приготовления эталонных растворов в делительные воронки на 50 или 100 мл последовательно в каждую вводят 15—20 мл воды, стандартный раствор, содержащий железо (мг): 0,01; 0,02; 0,04; 0,06 и 0,1 соответственно, добавляют по 0,5 мл азотной кислоты, роданида аммония, серной кислоты и 10 мл изоамилового (бутилового) спирта. После энергичного перемешивания содержимого воронок и расслоения фаз отделяют органические фазы, вливая их в мерные колбы емкостью в 25 мл. Повторяют экстракцию 10 мл растворителя, экстракты объединяют, доводят объем раствора взятым растворителем до метки. Измерение оптической плотности экстрактов производят на фотоэлектроколориметрах различных марок при X 480 нм и строят градуировочный график. В качестве раствора сравнения берут применяемый растворитель.
Для определения железа в испытуемом растворе из общего объема 100 мл берут аликвотную часть 15—20 мл, помещают в коническую колбу, добавляют 0,5 мл азотной кислоты, нагревают раствор до начала кипения и после охлаждения переносят его в делительную воронку емкостью 50 или 100 мл, добавляют те же количества реагентов в той же последовательности, как и при приготовлении эталонных растворов, не вводя стандартного раствора железа; экстракцию и измерение оптической плотности экстракта см. в приготовлении эталонных растворов
Б. Определение железа в пятокиси тантала
Железо в Та2О5 определяют в виде ионного ассоциата [17].
Кроме указанных на стр. 151, необходимы реагенты:
1. Роданид аммония, 50%-ный раствор.
2. Дигексиламин.
3. Амилацетат.
4. Винная кислота, 25%-ный раствор.
5. Бисульфат калия, х. ч.
6. Серная кислота (о. ч.) (пл. 1,34).
7. Соляная кислота (о. ч.), концентрированная.
Для приготовления эталонных растворов в четыре делительные воронки емкостью 50 мл вводят в каждую 10 мл воды, стандартный раствор, содержащий железо (мкг): 0,4; 0,6; 0,8; 1,0 соответственно, добавляют 3—4 капли НО до pH 1—2, 2 мл раствора роданида аммония, перемешивают, прибавляют 0,5 мл дигексиламина и 5 мл амилацетата. Встряхивают содержимое воронок в течение 1—2 мин. Экстракты сливают в мерные градуированные цилиндры или пробирки и доводят раствор амилацетатом до объема 5 мл. Затем растворы помещают в кювету и измеряют оптическую плотность при X 480 нм относительно амилацетата в качестве раствора сравнения. Измерение оптической плотности эталонных растворов проводят на спектрофотометрах или фотоэлектроколориметре ФЭК-60. По экспериментальным данным, обработанным методом наименьших квадратов, строят градуировочный график.
Для определения железа в Та2ОБ три навески образца по 0,1 г сплавляют каждую в кварцевом тигле с 2,5 г бисульфата калия, сначала на Горелке, а затем в муфельной печи при 760° С. Сплав растворяют в Змл H2SO4 конц. (пл. 1,84), охлаждают и прибавляют 3 мл раствора винной кислоты. Затем раствор количественно переносят в делительную воронку и прибавляют 2 мл раствора роданида аммония, 0,5 мл дигексиламина и 5 мл амилацетата. Комплексное соединение железа экстрагируют в течение 1—2 мин и после разделения фаз переносят в градуированный цилиндр, доводят амилацетатом до объема 5 мл и фотометрируют органическую фазу при X 480 нм.
Содержание железа определяют по градуировочному графику (см. стр. 151). Результаты трех определений обрабатывают методом математической статистики.
ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА СУЛЬФОСАЛИЦИЛОВОЙ (2-ОКСИ-5-СУЛЬФОБЕНЗОИНОИ) КИСЛОТОЙ
Железо с сульфосалициловой кислотой
/ч/ОН / J-cooh HO3SZ х
образует ряд комплексных соединений в зависимости от кислотности определяемого раствора. В кислой среде в интервале pH 1,8—2,5 об-
разуется комплексный катион моносульфосалицилата железа. В интервале pH 4—8 образуется комплексный анион дисульфосалицилата железа. В щелочной среде в интервале pH 8—11 образуется также комплексный анион, в состав которого входят три остатка сульфосалици-ловой кислоты — трисульфосалицилат [18], [19].
Для соединений, образующихся при pH 1,8—2,5 и pH 8—11, спектры поглощения (рис. 51) имеют области максимального поглощения при
Рис. 51. Спектры поглощения моно-(/) и трисульфосалицилата железа (2)
510 и 416 нм и молярными коэффициентами погашения 1,8- 103 и 5,8 • Ю3 соответственно.
Об устойчивости комплексных соединений железа с сульфосалициловой кислотой имеются несколько разноречивые све. дения. По данным работы [20] константы устойчивости равны.
р1= 1,13-1014 (pH 1,8—2,5)
р2 = 2,14-1024 (pH 4,0-8,0),
ps = 1,25-1033(pH 8,0—11,0).
В работе Никольского [20] несколько иначе рассматривается комплексообразование железа с сульфосалициловой кислотой.
Авторы предполагают, что в растворах при pH < 2 образуется моносульфосалицилат железа состава [Fe(COO)(OH)C6 H3HSO3]1 2 3+, Куст — = (2 + 0,1)-104; при pH 4—8 образуется дисульфосалицилат {Fe[(COO)(OH)C6H8(SOa)]2}1-, Куст (2,25 + 0,5) • 102. Для соединения, образующегося в щелочной области, приписывают состав {Fe[(COO)(O)C6H3(SO3)]2}2-.
Образование сульфосалицилатов железа может быть использовано для определения железа (III) в кислой среде и суммы железа (II) и (III) в щелочной среде. Комплексные соединения сульфосалицилата железа являются более устойчивыми, чем роданиды железа, что позволяет применить рассматриваемый метод для определения железа в присутствии фосфатов, ацетатов, боратов.
Определению железа при проведении реакции в кислой среде не мешают медь и алюминий, так как соответствующие комплексные соединения этих элементов менее устойчивы, чем комплексное соединение сульфосалицилата железа (III). Но соединение моносульфосалицилата железа менее устойчиво, чем трисульфосалицилат железа. В кислой среде исключается возможность определения железа при наличии фтор-иона, в то время как фториды не мешают определению железа в виде трисульфосалицилата в щелочной среде.
Кроме указанных на стр. 152, необходимы реагенты:
1. Серная кислота (х. ч.), 2 н. раствор.
2. Сульфосалициловая кислота, 10%-ный раствор.
3. Аммиак, 10%-ный раствор.
А. Определение железа в виде моносульфосалицилата в растворе его соли
Для приготовления эталонных растворов в мерные колбы емкостью 50 мл вводят по 10 мл воды, стандартный раствор соли железа от 0,05 ди 0,3 мг, увеличивая содержание железа в каждом эталонном растворе на 0,05 мг. Добавляют во все растворы 1 мл азотной кислоты, 1 мл серной кислоты, 5 мл сульфосалициловой кислоты и объем раствора доводят водой до метки. Строят градуировочный график по данным измерения оптических плотностей эталонных растворов на фотоэлектроколориметрах различных марок при X 510 нм. В качестве раствора сравнения берут воду.
Для определения железа в испытуемом растворе берут из общего объема 100 мл аликвотную часть 10—20 мл этого раствора, помещают в коническую колбу емкостью 50 мл, прибавляют 1 мл азотной кислоты, осторожно нагревают раствор в течение 1—2 мин, охлаждают его и переводят в мерную колбу емкостью 50 мл. Добавляют в эту колбу 1 мл серной кислоты, 5 мл сульфосалициловой кислоты и доводят объем раствора водой до метки. Измерение оптической плотности раствора см. в условиях приготовления эталонных растворов. По градуировочному графику находят содержание железа.
Б. Определение железа в виде трисульфосалицилата в растворе его соли
Для приготовления эталонных растворов в мерные колбы емкостью 50 мл вводят по 10 мл воды, стандартный раствор соли железа от 0,05 до 0,3 мг, увеличивая содержание железа в каждом эталонном растворе на 0,05 мг, добавляют по 5 мл растворов сульфосалициловой кислоты и аммиака и объем раствора доводят водой до метки. Строят градуировочный график, измеряя оптическую плотность эталонных растворов при X 416 нм на фотоэлектроколориметрах различных марок. Раствором сравнения служит вода.
Для определения железа в испытуемом "растворе берут из общего объема 100 мл аликвотную часть 10—20 мл раствора, помещают в мерную колбу емкостью 50 мл, прибавляют по 5 мл растворов сульфосалициловой кислоты, аммиака и объем раствора доводят водой до метки. Измерение оптической плотности раствора см. в условиях приготовления эталонных растворов. Содержание железа находят по градуировочному графику.
В. Определение железа в виде трисульфосалицилата в фосфорнокислых солях
Две навески анализируемого препарата по 2,5 г (в зависимости от содержания железа) помещают каждую в коническую колбу емкостью 50 мл, добавляют 30 мл воды, 15 мл раствора сульфосалициловой кислоты и растворяют при слабом подогревании (60—70°С) [21]. При неполном растворении для получения прозрачного раствора добавляют несколько капель азотной или соляной кислот. После охлаждения раствор
J51
переводят в мерную колбу емкостью 50 мл. Берут из каждой колбы две порции по 20 мл, переносят в другие мерные колбы емкостью 50 мл, добавляют 5 мл аммиака и доводят объем раствора водой до метки. Измерение оптической плотности растворов см. в условиях приготовления эталонных растворов. Результаты определений (не менее четырех) обрабатывают, пользуясь методом математической статистики.
ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА 1ДО-ФЕНАНТРОЛПНОМ И БАТОФЕНАНТРОЛИНОМ
Для определения Fe (II) используют 1,10-фена нтро л ин и его производное 4,7-дифенил-1,10-фенантролин (батофенантролин). Данные
1,10 - фенантролин
батофенантролин
реагенты образуют окрашенные комплексные соединения Fe, Со, Ni, Си и бесцветные с Cd, Zn, Ag, Bi и др. Указанные реагенты бидентатны и их устойчивые комплексы образуются при соотношении компонентов Ме2+ : R = 1:3. 1,10-Фенантролин — слабое основание и присоединяет в кислой среде один протон Ph + Н PhH+. Протонированный реагент (Кцр1,1 • 10-5) имеет две характеристические полосы при X 226 и X 265 нм. Наиболее изучено соединение Fe (II) с 1,10-фенантролином, образующееся в интервале pH 2—9. Соединение с батофенантролином образуется в более узком интервале pH 4—7.
Для восстановления Fe (III) до Fe (II) обычно используют гидроксиламин хлоргидрат или аскорбиновую кислоту. Комплексное соединение железа (II) с 1,10-фенатролином состава [FePh 312+ отличается сравнительно большой устойчивостью, 1g р3 21,3; 21,5; оно получило название ферроина. Устойчивости ферроина способствует образование трех пятичленных циклов в молекуле комплекса:
Устойчивость ферроина позволяет применять ряд маскирующих веществ, что повышает селективность метода определения железа. Фосфаты, фториды, хлориды и сульфаты, а также ацетаты, цитраты и тартраты не влияют на результаты определения железа (II). При
наличии иодидов и перхлоратов ферроин образует ионные ассоциаты FePh3X2 (X = I-, или СЮ?), экстрагируемые хлороформом. Значение молярного коэффициента погашения для водного раствора ферроина и ионного ассоциата ферроин-иодида очень близко. Для водного раствора е = (11,1 ± 0,2) • 103, X 515 нм и для хлороформного е = = (П,5 ± 0,3) • 103, X 520 нм. Для двухфазной константы ферроин-иодида 1g Рр2 = 4,2 ± 0,2 [23].
Соли никеля, кобальта и меди также дают ионные ассоциаты с 1,10-фенантролином при наличии иодид-иона, которые экстрагируются хлороформом, но экстракты слабо окрашены: для никеля е = 25 (X 528 нм), для кобальта е — 28 (А 428 нм); для меди е = 63 (А 770 нм). Определение 1—5 мкг железа (II) в виде ферроин-иодида возможно при указанных элементах в соотношении Fe : Me = 1 : 10, если проводить определение, вводя комплексон III.
Железо (III) также взаимодействует с 1,10-фенантролином, образуя растворы зеленовато-синего цвета (Ашах = 585 нм), которые при стоянии переходят в более устойчивые во времени растворы желтого цвета (А 396 нм). Метод определения железа (III) с 1,10-фенантролином менее чувствителен и его используют для определения сравнительно больших количеств железа [22].
При определении ультрамалых количеств железа необходимо применять реагенты особой чистоты или при отсутствии таких подвергать их дополнительной очистке. Воду нужно брать деионизованную.
ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА В ВИДЕ ФЕРРОИН-ИОДИДА
Реагенты
1. Стандартный раствор соли железа (II). Навеску 0,702 г соли Мора (х. ч.) помещают в мерную колбу емкостью 1 л, растворяют в воде, подкисляют 4 мл серной кислоты (пл. 1, И), доводят водой объем раствора до 1 л и тщательно перемешивают, 1 мл раствора содержит 0,1 мг железа.
2. Гидроксиламин хлоргидрат (х. ч.), 10%-ный раствор.
3. 1, 10-Фенантролин, 0,5%-ный водный раствор.
4. Иодид калия (х. ч.), 20%-ный раствор.
5. Комплексон III, 10%-ный раствор; 50 г комплексона III помещают в мерную колбу емкостью 0,5 л, добавляют 200 мл воды, 20 мл раствора аммиака (пл. 0,92), растворяют соль и доводят объем раствора водой до метки.
6. Хлороформ перед употреблением промывают дважды водой.
7. Аммиак, раствор (1 : 1).
8. Соляная кислота, раствор 1 : 10.
А. Определение железа (II) в растворе его соли
Для приготовления эталонных растворов в пять делительных воронок емкостью 50 мл вводят по 10 мл воды, 5 мл раствора трилона III, 1 мл гидроксиламина хлоргидрата 1 мл, 1,10-фенантролина, 1млиодида калия и вводят стандартный раствор, содержащий железо (II) (мкг): 0,3; 0,6; 1,0; 2,0; 3,0 соответственно. Устанавливают pH 3—4 , добавляя NH з или НС1 по универсальной индикаторной бумаге и оставляют растворы стоять 40 мин. Далее добавляют 10 мл хлороформа и экстрагируют образовавшееся соединение ферроин-иодида в течение 1 мин.
Хлороформные экстракты помещают в градуированные пробирки емкостью 10 мл и добавляют, если нужно несколько капель хлороформа до указанного объема. Измеряют оптическую плотность экстрактов при X 520 нм на спектрофотометрах или фотоэлектроколориметре ФЭК-60. В качестве раствора сравнения используют хлороформ, которым обработан раствор «холостого опыта». Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения железа в испытуемом растворе из общего объема 50 мл берут 5 или 10 мл этого раствора (в зависимости от содержания железа), помещают в делительную воронку емкостью 50 мл и в дальнейшем проводят все операции, указанные при приготовлении эталонных растворов и раствора сравнения, используя те же количества реагентов. По градуировочному графику находят содержание железа в испытуемом растворе [23].
Б. Определение железа в гипофосфите натрия
Три навески соли по 1 г переносят каждую в коническую колбу емкостью 50 мл, растворяют в 5 мл раствора комплексона III, прибавляют 5 мл воды, по 1 мл растворов гидроксиламина, 1,10-фенантро-лина и иодида калия. Создают кислотность раствора, добавляя растворы аммиака или соляной кислоты до pH 3—4 по универсальной индикаторной бумаге и доводят водой объем раствора до 15 мл. Через 40 мин раствор переносят в делительную воронку, приливают 10 мл хлороформа и экстрагируют образовавшееся соединение ферроин-иоди-да в течение 1 мин/ Приготовление эталонных растворов и раствора сравнения проводят в условиях, указанных на стр. 156. Содержание железа находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистикки.
Примечание. В аналогичных условиях определяют железо в фосфате натрия.
В. Определение железа в соляной кислоте
К трем пробам по 8,4 мл (10 г) испытуемой кислоты (пл. 1,19), помещенным каждая в коническую колбу емкостью 50—100 мл, прибавляют 10 мл воды, нейтрализуют раствором аммиака до pH 3—4 по универсальной индикаторной бумаге, добавляют 5 мл комплексона 111 и по 1 мл растворов гидроксиламина хлоргидрата, 1,10-фенантролина и иодида калия. Устанавливают раствор рН~3—4 добавлением растворов соляной кислоты или аммиака. Через 40 мин раствор переносят в делительную воронку, прибавляют 10 мл хлороформа и экстрагируют соединение ферроин-иодида в течение 1 мин. Приготовление эталонных растворов и раствора сравнения проводят в условиях, описанных на стр. 156. Содержание железа в соляной кислоте находят по градуировочному графику. Результаты параллельных определений (не менее трех) обрабатывают методом математической статистики.
Примечания. 1. В аналогичных условиях в 7,1 мл (10 г) определяют железо в азотной кислоте.
2. Метод рекомендз ется при анализе соляной и азотной кислот марки х. ч. и о. ч.
Г. Определение железа в солях кобальта
в виде ассоциата ферроина с бромфеноловым синим
В основу метода положено предварительное выделение железа экстракцией дибутиловым эфиром в виде HFeCl4, реэкстракцией этого соединения в водную фазу с последующим определением железа в видеферроин-иодида. Для повышения чувствительности метода можно вместо иодид-иона использовать сульфофталеиновые красители, например бромфеноловый синий. При этом образуется ионный ассоциат (Хтах 610 нм, е = 5,9 • 104). Но этот последний метод при непосредственном определении железа в солях кобальта имеет два недостатка: 1) очень узкий интервал значений pH при экстракции ассоциата (pH 8,7—8,9); 2) малую избирательность, так как следы никеля, кобальта и меди при замене иодида на бромфеноловый синий дают интенсивно окрашенные, экстрагирующиеся ионные ассоциаты.
Для устранения влияния железа, кобальта и меди предлагается следующий метод. Следы железа экстрагируют хлороформом в виде ферроин-иодидного ассоциата в интервале pH от 2 до 8, отделяют слой хлороформа, содержащий комплексное соединение железа и встряхивают его с концентрированным водным раствором бромфеноловым синим при pH 8,7—8,9 (ацетатный буфер). При этом иодид замещается на бромфеноловый синий; окрашенный экстракт фото-метрируют.
Кроме указанных на стр. 156, необходимы реагенты:
1. Ацетатный буферный раствор, готовят из 1 М раствора ацетата натрия с добавлением 1 и. раствора NaOH до pH 8,8.
2. Бромфеноловый синий, 0,1%-ный водно-щелочной раствор.
Для приготовления эталонных растворов в пять делительных воронок вносят 2, 4, 6, 8, 10 мл раствора, содержащего 0,1 мкг/мл железа, прибавляют 5 мл раствора комплексона III, по 1 мл растворов гидроксиламина хлор гидрата, 1,10-фенетролина и иодида калия и выдерживают в течение 20 мин. Далее добавляют 10 мл хлороформа и встряхивают содержимое воронок в течение 1 мин. К экстракту добавляют 1 мл раствора бромфенолового синего, 10 мл ацетатного буфера с pH 8,7— 8,9 и продолжают экстракцию в течение 1 мин. После разделения фаз фотометрируют органическую фазу на СФ-4А, СФ-16 в кюветах с I = = 1 см при X 610 нм. В качестве раствора сравнения используют хлороформ, которым обработан раствор «холостого опыта». Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения Fe в нитрате кобальта навеску образца 1 г растворяют в 10 мл соляной кислоты и помещают в делительную воронку, куда добавляют 10 мл дибутилового эфира, и экстрагируют образовавшееся комплексное соединение железа HFeCl4 в течение 3 мин. Орга
ническую фазу промывают последовательно двумя-тремя порциями соляной кислоты (по 10 мл каждая) так, чтобы последняя порция была бесцветной. Из органической фазы железо реэкстрагируют в водную фазу добавлением 5 мл воды и 5 мл раствора комплексона III. Водную фазу собирают в делительную воронку, добавляют по 1 мл растворов гидроксиламина хлоргидрата и 1,10-фенантролина, устанавливают pH 3—7 и добавляют 1 мл иодида калия.
Через 20—30 мин экстрагируют образовавшийся ферроин-иодид двумя порциями хлороформа (по 5 мл каждая) в течение 1 мин. После разделения фаз сливают органическую фазу в чистую воронку, добавляют 1 мл бромфенолового синего и 10 мл ацетатного буфера с pH 8,8 и встряхивают содержимое воронки в течение 1 мин. При этом иодид замещается на бромфеноловый синий. После разделения фаз сливают органическую фазу в градуированную пробирку на 10 мл, добавляют хлороформ, если нужно, до объема 10 мл и проводят фотометрирование на спектрофотометре при X 610 нм в кюветах с / = 1 см относительно раствора хлороформа (см. приготовление эталонных растворов). Содержание железа определяют по градуированному графику (см. стр. 156).
Д. Определение железа (II) 4,7-дифенил-1,10-фенантролином (батофенантролином) в растворе его соли
Кроме указанных на стр. 156, необходимы реагенты.
1. Батофенантролин, 0,02%-ный раствор в этиловом спирте.
2. Ацетат натрия, 50%-ный раствор.
3. Этиловый спирт.
Для приготовления эталонных, растворов в пять конических колб емкостью 50 мл вводят по 10 мл воды, стандартный раствор, содержащий железо в количестве (мкг): 0,5:1,0; 2,0; 3,0; 5,0 соответственно. Добавляют растворы 1 мл гидроксиламина хлоргидрата, 2 мл ацетата натрия, нагревают растворы до начала кипения, охлаждают их, переносят в делительные воронки, добавляют 10 мл раствора батофенан-тролина и экстрагируют соединение железа двумя порциями хлороформа по 5 мл каждая, энергично перемешивая содержание воронки в течение 1—2 мин. После расслаивания органические фазы переносят в мерные колбы емкостью 25 мл или градуированные пробирки емкостью 15 мл, доводят объем раствора до метки колбы или пробирки этиловым спиртом. Измерение оптической плотности эталонных растворов проводят на спектрофотометрах различных марок или фотоэлектроколориметрах ФЭК-56, ФЭК-57, ФЭК-60 при X 533 нм и строят градуировочный график.
Для определения железа в испытуемом растворе из общего объема 50 мл берут аликвотную часть 10 мл этого раствора, помещают в делительную воронку, добавляют те же количества реагентов и проводят те же операции, что и при приготовлении эталонных растворов. Удержание железа находят по градуировочному графику [11, [25].
Определение кобальта
Для определения кобальта наибольшее распространение получили методы, в которых используются органические реагенты—производные нитрозо-нафтолов [24]—[26], пиридиновые азосоединения, из которых большее распространение получил 4-(2-пиридилазо)-резорцин (ПАР),
ОПРЕДЕЛЕНИЕ КОБАЛЬТА НИТРОЗО-НАФТОЛАМИ
М. А. Ильинский впервые наблюдал образование окрашенных соединений кобальта с 1,2- и 2,1-изомерами нитрозо-нафтола [24]:
1 - нитрозо-2-- нафтол
нитрозо-R-соль
Спектрофотометрическое исследование реагентов этого типа показало, что преимущественно они находятся в хиноноксимной форме. Нитрозонафтолы обладают малой избирательностью и помимо кобальта взаимодействуют с рядом элементов: Fe, Си, Ni, Pd, Ru, Rh, Os, U, Zr, Сг и др.
При’образовании комплексных соединений возможно, что атомы одних металлов связываются с молекулами нитрозо-нафтолов через атомы азота и кислорода, а других металлов—через атомы кислорода. Это приводит к образованию пятичленных и шестичленных хелатных циклов.
Кобальт с указанными реагентами в зависимости от условий проведения реакций дает два типа соединений. В щелочной среде образуется соединение, легко растворимое в воде, состава CoR2, в кислой среде — состава CoR3, которое мало растворимо в воде и хорошо растворимо в неводных органических растворителях. Плохая растворимость в воде комплексных соединений типа CoR2 обусловлена тем, что из шести координатных мест кобальта заполненными являются четыре, а два других заняты молекулами воды. Комплексы состава CoR 3 кинетически инертны и не разрушаются даже в сильнокислых средах, в то время как комплексы Ni, Си, Fe состава MeR2 с аналогичными лигандами разрушаются в кислых средах, что и позволяет проводить определение кобальта в присутствии больших количеств данных элементов.
Преимущество 2,1-изомера нитрозонафтола видно из сравнения спектров поглощения комплексных соединений обоих изомеров с кобальтом (рис. 52). В ближней УФ-области тля соединения кобальта с 2,1- изомером X 307 нм, е = 5,3 • 104; для соединения кобальта с 1,2-изомером X 460 нм, 8 = 3,0 • 104. Реакция определения кобальта с 2,1-изомером более чувствительна. Преимуществом 2,1-изомера является также образование комплекса CoR3 без дополнительного окисления кобальта (II), что требуется при использовании 1,2-изомера.
Соединения кобальта с 1,2- и 2,1-изомерами, получаемые в кислой среде, обладают преимуществом перед соединениями, выделяемыми в щелочной среде, так как первые экстрагируются неводными растворителями. Сам реагент также экстрагируется, но может быть легко переведен в водную фазу обработкой 2 н. раствором щелочи. Таким путем при измерении оптической плотности испытуемых растворов избавляются от влияния избытка реагента.
Медь, железо и никель не мешают определению кобальта, так как они, дают менее устойчивые комплексные соединения с 1,2- и 2,1-изомерами нитрозонафтолов, легко разрушаемые 2 н. раствором соляной кислоты.
Кроме 1,2- и 2,1-изомеров нитрозонафтолов для определения кобальта широко используют растворимое в
Рис. 52. Спектры поглощения соединений кобальта:
1 — с 1-нитрозо-2-нафтолом; 2 — с 2-нитрозо-!-нафтолом; /' — 1-нитрозо-2-наф-тол: 2' — 2-нитрозо-1-нафтол
воде сульфопроизводное 1-нит-
розо-2-нафтола—нитрозо-К-соль. В кислой среде нитрозо-И-соль образует с кобальтом водорастворимое соединение состава CoR3.
Реагенты
1. Стандартный раствор соли кобальта, содержащий 0,1 мг/мл Со. Навеску сульфата кобальта без никеля (х. ч. или ч. д. а.) с известным содержанием кобальта (0,1 г Со) помещают в мерную колбу емкостью 1 л, растворяют в воде, добавляют 4 мл серной кислоты (пл. 1,84), доводят объем раствора водой до метки. Более разбавленные растворы с содержанием 0,01 и 0,001 мг/мл готовят соответствующим разбавлением; эти растворы пригодны только в день его приготовления.
2. Нитрозонафтолы (1,2- и 2,1-изомеры), 0,5%-ный раствор в ледяной уксусной кислоте.
3. Едкий натр, 2 н. раствор.
4. Соляная кислота, раствор 1 : 4.
5. Хлороформ.
6. Перекись водорода, 30%-ный раствор.
7. Ацетатный буферный раствор, pH 5,2.
8. Смесь концентрированных соляной и азотной кислот (х. ч.) 3 : 1.
9. Уксусная кислота (х. ч.), концентрированная.
А. Определение кобальта 2-нитрозо-1-нафтолом в растворе его соли
Для приготовления эталонных растворов в пять стаканов емкостью 50 мл вносят по 5 мл ацетатного буферного раствора, стандартный раствор, кобальта соответственно указанным ниже 1-му, 2-му или 3-му ряду в зависимости от содержания кобальта в испытуемом растворе, прибавляют 1 мл раствора реагента, нагревают растворы почти до
кипения, охлаждают и переносят в делительные воронки емкостью 50 мл, добавляют 5 мл буферного раствора и через 15 мин экстрагируют образовавшееся соединение кобальта в течение 10 мин двумя порциями хлороформа по 5 мл каждая, используя механический вибратор. Обе порции хлороформа объединяют в другой делительной воронке и промывают в течение 20 мин 10 мл раствора щелочи и один раз 10 мл воды. Хлороформный слой через сухой фильтр переносят в градуированную пробирку емкостью 10 мл. Строят градуировочный график по данным измерения оптической плотности хлороформных растворов на фотоэлектроколориметрах ФЭК-56, ФЭК-57, ФЭК-60 при X 365 нм или спектрофотометрах при к 307 нм.
В качестве раствора сравнения используют хлороформ, которым обработан раствор «холостого опыта». В зависимости от содержания кобальта (мкг) готовят три ряда эталонных растворов:
1-й ряд
2-й ряд
3-й ряд
0,01 0,05
0,5
0,02 0,03 0,05
0,07 0,10 0,20 0,50
1,5 3,0 6,0 10,0
Для определения кобальта в испытуемом растворе из общего объема 25 мл берут 3—5 мл этого раствора в зависимости от содержания кобальта и переносят в делительную воронку емкостью 25—30 мл, добавляют 1 мл реагента и проводят определение кобальта в условиях см. приготовление эталонных растворов. Содержание кобальта находят по градуировочному графику.
Грглечаьие. При работе с количестиом кобальта, соответствующим 1-му и 2 mv рядам, градуировочрый график строят по экспериментальным данным, сбрабстагтым методом наименьших квадратов.
Б. Определение кобальта 2-нитрозо-1-нафтолом в присутствии солей железа и никеля
Эталонные растворы кобальта готовят в условиях, указанных на стр. 161 соответственно 3-му ряду.
Для определения кобальта в растворе, содержащем никель и железо, после экстракции соединения кобальта с 2-нитрозо-1-нафтолом хлороформом (см. стр. 161) хлороформный экстракт промывают последовательно раствором соляной кислоты двумя порциями по 20 мл каждая, один раз 10 мл воды и затем двумя порциями раствора щелочи по 5 мл каждая и 5 мл воды. Так как при этой операции освобождается некоторое количество реагента, которое входило в комплексное соединение железа и никеля, то раствор хлороформа еще паз последовательно промывают раствором щелочи и водой. Хлороформный слой через сухой фильтр переносят в градуированную пробирку емкостью 10 мл. Измерение оптической плотности растворов проводят на фотоэлектроколориметрах ФЭК-56, ФЭК-57 или ФЭК-60 при X = 365 нм или спектрофотометрах при Z 307 нм. Содержание кобальта находят по градуировочному графику (см. стр. 162)
Ь. Определение кобальта 2-нитрозо-1 -нафтолом в стали
Две навески стали по 0,1 г переносят каждую в жаростойкий стакан емкостью 100 мл, добавляют 10 мл раствора смеси соляной и азотной кислот и нагревают на песчаной бане. Раствор упаривают почти досуха; остаток растворяют в воде и переносят раствор в мерную колбу емкостью 250 мл. Для определения берут 2—5 мл раствора, в зависимости от содержания кобальта в стали. В дальнейшем определение проводят, как при определении кобальта, в присутствии никеля и железа. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики.
Г. Определение кобальта-2-нитрозо-1-нафтолом в азотной кислоте (о. ч.)
Кроме указанных на стр. 161, необходимы реагенты:
1. 2-Нитрозо-1-нафтол, 0,02%-ный водный раствор. •
2. Фосфатный буферный раствор, pH 6,1.
Для приготовления эталонных растворов в пять стаканов емкостью 50 мл вносят по 5 мл фосфатного буферного раствора, стандартный раствор кобальта, соответственно 1-му или 2-му ряду (см. стр. 162) в зависимости от содержания кобальта прибавляют 2 мл раствора реагента и в остальном соблюдают условия, указанные на стр. 161.
Для определения кобальта в азотной кислоте (о.ч.) три пробы по 10 г (7,1 мл) азотной кислоты упаривают каждую в кварцевой или тефлоновой чашке на водяной бане почти досуха. К остатку добавляют 5 мл буферного раствора и 2 мл раствора 2-нитрозо- 1-нафтола. Раствор нагревают почти до кипения, охлаждают и переносят в делительную воронку емкостью 50 мл. К полученному раствору приливают 5 мл хлороформа, оставляют стоять на 15 мин и экстрагируют соединение кобальта в течение 10 мин на механическом вибраторе. Избыток реагента удаляют из хлороформа в условиях, описанных при приготовлении эталонных растворов. Если в кислоте содержится никель, то хлороформный раствор обрабатывают в условиях, описанных на стр. 162.
Примечание. Для опр еделения кобальта в азотной кислоте о. ч. берут деионизованную воду.
Д. Определение кобальта 2-нитрозо-1 -нафтолом в щелочи (о. ч.)
Эталонные растворы готовят в условиях, указанных на стр, 161.
Для определения кобальта в щелочи три навески по 10 г растворяют каждую в 25 мл воды, растворы нейтрализуют до pH ~7 по индикаторной бумаге, добавляют небольшой избыток азотной кислоты (2—3 мл) и растворы выпаривают. Сухой остаток растворяют в 20 мл воды, добавляют 5 мл буферного раствора и далее поступают, как описано при определении кобальта в азотной кислоте.
Примечание. Если в азотной кислоте, используемой для нейтрализации щелочи, содержится кобальт, то должно быть известно его содержание и сделана соответствующая поправка.
Е. Определение кобальта 2-нитрозо-1-нафтолом в чистом алюминии
Кроме указанных на стр. 161, необходимы реагенты:
1. Едкий натр, 4 н. растзор.
2. Лимонная кислота, 25%-ный водный раствор.
Для приготовления эталонных растворов точно соблюдают условия, указанные при определении кобальта в азотной кислоте (стр. 163).
Для определения кобальта в алюминии берут две навески металла по 1 г, растворяют каждую в 20 мл едкого натра, прибавляют постепенно раствор лимонной кислоты до pH 8. Раствор переносят в мерную колбу емкостью 50 мл и доводят объем раствора водой до метки. В стакан емкостью 50 мл переносят 10 мл приготовленного раствора, добавляют 2 мл раствора 2-нитрозо- 1-нафтола, нагревают почти до кипения, охлаждают и переносят раствор в делительную воронку емкостью 50 мл. К этому раствору приливают 5 мл хлороформа, оставляют стоять 15 мин и экстрагируют соединение кобальта в течение 20 мин на механическом вибраторе. Bi >дный слой отбирают пипеткой (используя резиновую грушу). Для удаления избытка реагента хлороформный слой обрабатывают 5 мл щелочи в течение 20 мин, используя механический вибратор, затем промывают водой. Если имеется примесь железа, то его комплексное соединение разрушается раствором щелочи при удалении избытка реагента из хлороформа. Для разрушения комплексных соединений никеля и меди, которые могут также содержаться в качестве примесей, раствор хлороформа промывают 5 мл соляной кислоты в течение 5 мин и снова водой, используя механический вибратор. Так как при этой операции освобождается некоторое количество реагента, которое входило в комплексные соединения меди и никеля, то еще раз раствор хлороформа промывают последовательно раствором щелочи (1 мл) и водой (5 мл). Раствор хлороформа переводят в мерный цилиндр или градуированную пробирку, добавляют хлороформ до 5 мл и измеряют оптическую плотность раствора на спектрофотометрах при "к 307 нм. Раствор сравнения готовят в условиях, указанных на стр. 162.
Содержание кобальта находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики.
Ж. Определение кобальта 2-нитрозо-1-нафтолом в гипофосфите натрия
Для приготовления эталонных растворов в конические колбы емкостью 50—100 мл вводят в каждую 15 мл фосфатного буферного раствора, стандартный раствор, содержащий кобальт (мкг): 0,01; 0,025; 0,05; 0,1; 0,2 соответственно, прибавляют 2 мл раствора реаген
та, растворы нагревают до начала кипения, охлаждают и переносят в делительные воронки емкостью 50 мл. К полученному раствору добавляют 5 мл хлороформа, оставляют стоять 15 мин и экстрагируют полученное соединение кобальта в течение 10 мин на механическом вибраторе. Хлороформный слой отделяют, отбирая для этого водный слой пипеткой с резиновой грушей. Для удаления избытка реагента хлороформный слой обрабатывают раствором щелочи последоательно двумя порциями по 5 мл в течение 10 мин на механическом вибраторе. В качестве раствора сравнения используют хлороформ (см. стр. 162). Измерение интенсивности окраски экстрактов проводят на спектрофотометрах при 7,307 нм. Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения кобальтов гипофосфите берут две навески по 2,5 г, переводят каждую в мерную колбу емкостью 50 мл, растворяют в воде и доводят объем раствора до метки водой. Затем 20 мл этого раствора помещают в воронку емкостью 50 мл, устанавливают кислотность раствора, добавляя соляную кислоту или раствор щелочи до pH 6,0— 6,5, прибавляют 2 мл раствора реагента, экстрагируют соединения кобальта и измеряют оптическую плотность в условиях, указанных при приготовлении эталонных растворов. Для определения содержания кобальта данные из параллельных опытов (не менее четырех) обрабатывают методом математической статистики.
Примечания. 1. Определение кобальта в фосфате натрия аналогично его определению в гипофосфате натрия.
2. Методики определения кобальта в HNO3 (о. ч.), щелочи (о. ч.) и чистом алюминии разработаны в лаборатории спектрофотометрии кафедры аналитической химии МГУ.
3. Определение кобальта 1-нитрозо-2-нафтола в растворе его соли
Для приготовления эталонных растворов в пять стаканов емкостью по 25—30 мл вносят 5 мл ацетатного буферного раствора, стандартный раствор соли кобальта, соответственному 2-му или 3-му ряду (см. стр. 162) в зависимости от содержания кобальта в испытуемом растворе добавляют 0,2 мл раствора перекиси водорода и раствора щелочи (постепенно) до полного образования осадка (~1 мл). Выпавший осадок гидроокиси растворяют при слабом подогревании в 2 мл уксусной кислоты. К горячему раствору добавляют 1 мл раствора 1-нитрозо-2-нафтола, переносят содержимое стаканчиков в пять делительных воронок, добавляют воды примерно до 20 мл, осталяют стоять 15 мин и экстрагируют образовавшееся соединение кобальта в течение 5 мин двумя порциями хлороформа по 5 мл каждая. Водный слой отбирают пипеткой, используя резиновую грушу, слой хлороформа промывают двумя порциями раствора щелочи по 5 мл каждая. Строят градуировочный график по данным, измерения оптической плотности растворов на фотоэлектроколориметрах ФЭК-56, ФЭК-57 и ФЭК-60 или спектрофотометрах при 7, 460 нм. В качестве раствора сравнения используют хлороформ.
Для определения кобальта в испытуемом растворе из общего ооъема 25 мл берут 5 мл этого раствора и помещают в стакан емкостью 30— 40 мл, добавляют 0,2 мл раствора перекиси водорода и постепенно раствора щелочи до образования осадка (~1 мл). Выпавший осадэк гидроокиси растворяют при слабом подогревании в 2 мл концентрированной уксусной кислоты. К горячему раствору добавляют 1 мл раствора 1-нитрозо-2-нафтола, переносят содержимое стакана в делительную воронку, добавляют воды примерно до объема 20 мл, оставляют стоять на 15 мин и экстрагируют соединение кобальта в течение 5 мин двумя порциями хлороформа по 5 мл каждая. В дальнейшем проводят все операции, указанные при приготовлении эталонных растворов. Содержимое кобальта находят по градуировочному графику.
И. Определение кобальта нитрозо-И-солью в растворе его соли
Кроме указанных на стр. 161, необходимы реагенты:
1. Нитрозо-И-соль, 0,1%-ный водный раствор.
2. Соляная кислота (х. ч.), раствор 1:1.
3. Азотная кислота (х. ч.), раствор 1:1.
4. Ацетат натрия, 40%-ный раствор.
Для приготовления эталонных растворов в конические колбы емкостью 50—100 мл вводят в каждую по 15 мл воды, стандартный раствор, содержащий кобальт в количестве (мг): 0,005; 0,0075; 0,01; 0,02 соответственно, добавляют 0,5 мл соляной кислоты и несколько (3—5) капель азотной кислоты, прибавляют 2 мл раствора нитрозо-R-соли и 5 мл раствора ацетата натрия. Растворы нагревают до кипения, прибавляют 2 мл азотной кислоты и осторожно кипятят их в течение 1—2 мин. После этого растворы охлаждают и переводят в мерные колбы емкостью 50 мл, доводят объем раствора водой до метки, перемешивают, определяют оптические плотности растворов при "к 460 нм , используя фотоэлектроколориметры различных марок и строят график. Раствор сравнения готовят в тех же условиях, что и эталонные растворы, не вводя стандартного раствора кобальта.
Для определения кобальта в испытуемом растворе берут 10—20 мл этого раствора (в зависимости от содержания кобальта), добавляют все реагенты и проводят операции, указанные при приготовлении эталонных растворов, не вводя стандартного раствора соли кобальта. Содержание кобальта находят по градуировочному графику.
ОПРЕДЕЛЕНИЕ МИКРОКОЛИЧЕСТВ КОБАЛЬТА
КИНЕТИЧЕСКИМ МЕТОДОМ
Для определения микроколичеств кобальта используют катализируемую им реакцию окисления ализарина перекисью водорода в боратном буферном растворе при pH 12,4. Определение проводят способом тангенсов интегрального варианта кинетических методов анализа (см. стр. 82) 127].
Реагенты
1. Стандартный раствор соли кобальта (см. стр. 161).
2. Ализарин (1,2-диоксиантрахинон), 4 10~* М водный раствор. Навеску 0,0247 г ализарина, очищенного двойной перекристаллизацией из воды, растьоряют в 8 мл 0,01 н. раствора едкого натра и доводят объем раствора водой до 250 мл.
3. Перекись водорода (о. ч.), 3,3 М раствор.
4. Боратный буферный раствор (pH 12,37). Для приготовления 1 л буферного раствора смешивают 400 мл 0,05 М раствора тетрабората натрия и 600 мл 0,1 н. раствора едкого натра. Используют едкий натр (о. ч.) и тетраборат натрия (х. ч.), дважды перекристаллизованный из воды. Все растворы готовят на деионизованной воде.
При построении градуировочного графика способом тангенсов в кварцевый смеситель с тремя отростками вводят последовательно: в первый отросток 0,5 мл перекиси водорода, во второй — раствор, содержащий соль кобальта, в третий — 0,5 мл раствора ализарина и такое количество боратно-щелочного буферного раствора, чтобы общий объем раствора во всех трех смесителях был равен 10 мл. Растворы тер-мостатируютв течение25—30 мин при 25° С, в момент смешения реагирующих веществ включают секундомер и измеряют оптическую плотность раствора каждую минуту, начиная с первой, в течение 10 мин на приборе ФЭК-Н-52 с термостатируемым корпусом или ФЭК-М, ФЭК-54 или ФЭК-57 с термостатируемой камерой для кювет {1 = 3 см; Хэфф 530 нм).
По полученным данным строят графики в коодинатах 1g оптической плотности — время. Углы наклона (tg а) этих прямых пропорциональны концентрации кобальта. Затем строят график в координатах tg а — концентрация кобальта. Содержание кобальта в эталонных растворах может быть от 5 • 10~3—5- 10'2 мкг в 10 мл раствора.
Для определения кобальта в испытуемом растворе берут 1 мл этого раствопа и помещают в один из отростков смесителя, в другие два отростка добавляют указанные реагенты и после термостатирования и смешения, как указано выше, фиксируют изменение оптической плотности во времени, строят график в координатах 1g А — время и находят тангенс угла наклона этой прямой. По градуировочному графику определяют содержание кобальта, соответствующее полученному значению tg а [27].
Определение марганца
ОПРЕДЕЛЕНИЕ МАРГАНЦА В ВИДЕ ПЕРА1АНГАНАТ-ИОНА
Для определения марганца используют ряд методов. Из них большее распространение получили методы, основанные на окислении марганца (II) в марганец (VII) (марганцовую кислоту). Эти методы обычно применяют для определения марганца в сталях, чугунах, горных породах.
В качестве окислителей наиболее часто используют перйодат калия и персульфат аммония, последний в присутствии катализаторов нитрата серебра или солей кобальта. Применяют также другие окис-
Рис 53 Спектры поглощения марганцовой кислоты (7) и формалъдоксимата марганца (2)
лители: висмутат натрия и нитрат аммония-церия (IV). Реакцию окисления проводят в кислой среде. Растворы марганцовой кислоты, полученные при окислении марганца (II) перйодатом калия, более устойчивы во времени по сравнению с растворами, в которых окислителем был персульфат аммония. Определению марганца мешают восстановители и особенно хлорид-ионы. Если в анализируемом объекте содержатся хлориды, то их предварительно удаляют выпариванием с азотной или серной кислотами.
При окислении персульфатом аммония могут образоваться соединения марганца (III), окрашивающие раствор в винно-красный цвет.
Во избежание образования этих соединений необходимо: 1) проводить окисление марганца в растворе, имеющем достаточно высокую кислотность; 2) вводить фосфат-ион в виде фосфорной кислоты или ее солей для образования промежуточного комплексного соединения марганца (III). Если образуется раствор винно-красного цвета, рекомендуют прибавить несколько кристаллов формиата натрия для восстановления марганца (III) до марганца (II), который затем снова окисляют до марганцовой кислоты. Однако не следует брать избыток формиата натрия во избежании восстанов
ления самой марганцовой кислоты. Для восстановления марганца (III) можно применять также перекись водорода и нитрит натрия.
При определении сравнительно больших количеств марганца полнота окисления достигается перйодатом калия, который добавляют после окисления персульфатом. При успешном течении реакции окисления растворы должны иметь малиново-розовый цвет. Спектр поглощения марганцовой кислоты приведен на рис. 53. В спектре поглощения наблюдается два максимума: X 528 нм, 8 = 2,9 • 103 и 8 = 2,1 • 103.
Для определения больших количеств марганца в виде марганцовой кислоты используют дифференциальный метод. Большей чувствительностью обладают методы, в которых используют образование ионных ассоциатов НМпО4 • амин. В качестве аминов применяют тетра-фениларсоний ди-н-дициламин или ди-н-гексиламин.
Реагенты
1. Стандартный раствор соли марганца (II), содержащий 0,1 мг/мл Мп, готовят двумя способами: а) в мерную колбу емкостью 1 л переносят навеску 0,273 г сульфата марганца (х. ч. или ч. д. а.), предварительно прокаленного до постоянного веса в воздушной бане, растворяют в воде, добавляют 4 мл сер
ной кислоты (пл. 1,84) и водой доводят объем раствора до метки; б) 0,287 г перманганата калия (ч. д. а.) помещают в мерную колбу емкостью 1 л, растворяют в воде, перегнанной в присутствии перманганата, добавляют 4 мл серной кислоты (пл. 1,84), доводят объем раствора водой до метки.
2. Серная кислота (х. ч.), 6 н. раствор.
3. Нитрат серебра 0,5 н. раствор.
4. Персульфат аммония, 20%-ный раствор, свежеприготовленный.
5. Перйодат калия, кристаллический.
6. Фосфорнокислый натрий двузамещенный, 10%-ный раствор.
7. Раствор, содержащий смесь кислот (серной, фосфорной, азотной). Для приготовления указанной смеси кислот берут 500 мл воды, вводят туда при постоянном перемешивании 100 мл серной кислоты (пл. 1,84), 125 мл форсфорной кислоты (пл. 1,7), 250 мл азотной кислоты (пл. 1,4) и разбавляют водой до объема 1 л.
8. Перекись водорода, 1 н. раствор.
9. Фосфорная кислота (х. ч.), концентрированная.
А. Определение марганца в сталях окислением его персульфатом
Для растворения стали применяют смесь серной, фосфорной, азотной кислот. Серную и азотную кислоту используют в качестве растворителей; фосфорную кислоту добавляют для связывания железа (III), образующегося в процессе растворения стали в слабо окрашенный комплекс.
Для приготовления эталонных растворов 0,5 г стали b известным содержанием марганца помещают в коническую колбу и растворяют при нагревании на песчаной бане в 50 мл смеси кислот. Раствор нагревают до прекращения выделения окислов азота, охлаждают его, переносят в мерную колбу емкостью 100 мл, доводят объем раствора водой до метки — раствор А. Затем для приготовления эталонных растворов отбирают пипеткой по 20 мл раствора А и переносят в мерные колбы емкостью 100 мл. Приливают стандартный раствор перманганата калия с таким расчетом, чтобы общее содержание марганца в каждом из эталонных растворов было соответственно равно (мг):
1-й ряд 0,5 0,7 0,9 1,1
2-й ряд 1,1 1,3 1,5 1,7
К полученным растворам для обесцвечивания окраски перманганата добавляют 3—4 капли перекиси водорода, избыток которой разрушают кипячением. Содержимое колб охлаждают и добавляют в каждую по 0,5 мл нитрата серебра и 10 мл раствора персульфата аммония. Растворы нагревают на водяной бане (60—70° С), пока окраска их не перестанет усиливаться, затем охлаждают, доводят объем раствора до метки дистиллированной водой, тщательно перемешивают и измеряют оптическую плотность, пользуясь фотоэлектроколориметрами различных марок при к 530—550 нм. По полученным данным строят градуировочный график. В качестве раствора сравнения берут воду.
Примечание. В зависимости от содержания марганца в стали используют 1-й или 2-й ряд эталонных растворов.
Для определения марганца в стали берут две навески по 0,25 г, расгворяют каждую в 25 мл смеси кислот в конической колбе емкостью 100 мл, нагревают раствор до прекращения выделения окислов азота, охлаждают и переносят в мерную колбу емкостью 50 мл. Отбирают из обеих колб две порции по 20 мл, переносят в конические колбы емкостью 50—60 мл и приливают к каждой порции 0,5 мл раствора нитрата серебра и 10 мл раствора персульфата аммония. Раствор нагревают на водяной бане, пока интенсивность окраски не перестанет увеличиваться, охлаждают, доводят водой объем раствора до метки и измеряют оптическую плотность растворов.
Примечание. Если содержание марганца в стали превышает 1%, то в раствор после окисления персульфатом и охлаждения его добавляют 0,1—0,2 г перйодата калия, снова нагревают почти до кипения, выдерживают при этой температуре 5 мин, охлаждают и доводят объем раствора до метки.
По градуировочному графику находят содержание марганца. Результаты параллельных определений (не менее четырех) обрабатывают, пользуясь методом математической статистики.
Б. Определение марганца в сталях окислением его перйодатом
При окислении марганца (II) перйодатом добавляется фосфорная кислота для связывания железа (III) в комплексное соединение и предотвращения осаждения перйодата или йодата марганца.
Для приготовления эталонных растворов вначале соблюдают условия, указанные на стр. 169, кончая операцией разрушения избытка перекиси водорода, добавляют в каждую колбу после охлаждения растворов 6 мл серной кислоты, 2 мл фосфорной кислоты и 0,3 г перйодата калия. Растворы нагревают до кипения и выдерживают при температуре, близкой к кипению, 5 мин, затем, охладив, переносят их в мерные колбы емкостью 50 мл и доводят объем расгвора дэ метки. Строят градуировочный график по измерениям оптической плотности эталонных растворов на фотоэлектроколориметрах различных марок при К 530—550 нм; в качестве раствора сравнения берут воду.
Для определения марганца в стали берут две навески по 0,25 г, растворяют каждую в 25 м п смеси кислот в конической колбе емкостью 100 мл, нагревают раствор до прекращения выделения окислов азота, охлаждают и переносят в мерную колбу емкостью 50 мл. Отбирают из обеих колб 2 порции по 20 мл, переносят в конические колбы емкостью 50—60 мл и приливают к каждой порции 6 мл серной кислоты и 2 мл фосфорной кислоты, 0,3 г перйодата калия. Производят окисление и измерение оптической плотности в условиях, указанных для приготовления эталонных растворов с применением перйодата калия. Результаты параллельных определений (не менее четырех) обрабатывают, пользуясь методом математической статистики.
В. Определение больших количеств марганца дифференциальным методом в растворе его соли
Для приготовления эталонных растворов в шесть конических колб емкостью 100 мл вводят в каждую 30 мл воды и стандартный раствор, содержащий марганец в количестве (мг): 2,0; 4,0; 6,0; 8,0; 10,0; 12,6 соответственно. Окисление марганца (II) проводят в условиях, указанных при окислении перйодатом в качестве окислителя (см. стр. 170), увеличив количество перйодата калия до 0,6 г. После получения окрашенных растворов их охлаждают, переносят в мерные колбы емкостью 100 мл и доводят объем раствора водой до метки. Измерение оптических плотностей этих растворов производят на любом фотоэлектроколориметре при X 525 нм, I = 0,5 см в том порядке, который указан на стр. 65, 71, начиная с выбора оптимального раствора сравнения. По полученным данным строят градуировочный график.
Для определения марганца в испытуемом растворе из общего объема его 100 мл берут 20 мл этого раствора, переносят в коническую колбу емкостью 100 мл, добавляют 10 мл воды, проводят реакцию окисления марганца в условиях приготовления эталонных растворов. После получения окрашенного раствора его переводят в мерную колбу емкостью 100 мл и доводят объем раствора водой до метки. Измеряют оптическую плотность и находят содержание марганца по градуировочному графику.
ОПРЕДЕЛЕНИЕ МАРГАНЦА ФОРМАЛЬДОКСИМОМ
Для определения марганца в воде, золе растений, если присутствует железо в небольших количествах, можно использовать в качестве реагента формальдоксим [28], [29]. Марганец (II) реагирует с фор-мальдоксимом в щелочной среде с образованием окрашенного в красно-бурый цвет соединения, X 455 нм, е = 1,1 • 104 (см. рис. 52). В растворе образуется комплексное соединение марганца с соотношением компонентов 1 : 6 и состава [Мп (CH2NO)e]2_ [29].
Формальдоксим пригоден и для определения марганца, находящегося в виде перманганат-иона. Здесь формальдоксим вначале играет роль восстановителя. В дальнейшем в растворе образуется комплексное соединение указанного состава. Формальдоксим дает окрашенные соединения, кроме марганца также с Се, Fe, Си, V, Ni и Со. Комплексные соединения большинства этих элементов не устойчивы в растворе при нагревании до 70—90° С [29].
Реагенты
1. Стандартный раствор соли марганца (П), содержащий 1 мг/мл Мп (стр. 167).
2. Формальдоксим, 1 /И раствор. К 7,9 мл 38%-кого раствора формалина добавляют 7,0 г хлоргидрата гидроксиламина и разбавляют водой до 100 мл.
3. Едкий натр, 1 нЛ раствор.
А. Определение марганца в растворе его соли, не содержащей хлоридов
Для приготовления эталонных растворов в пять мерных колб емкостью 50 мл вводят по 10 мл дистиллированной воды, стандартный раствор, содержащий марганец в количестве (мг): 0,025; 0,05, 0,075, 0,1 и 0,125 соответственно, 0,5 мл раствора формальдоксима и 1—3 мл раствора щелочи. Растворы перемешивают и через 5 мин добавляют еще 4—5 капель формальдоксима, 0,5 мл раствора щелочи и наблюдают, не увеличивается ли интенсивность окраски растворов. Если окраска их постоянна, то объем раствора доводят водой до метки. Строят градуировочный график по оптической плотности эталонных растворов, измеренной на фотоэлектроколориметрах различных марок при А 455 нм. В качестве раствора сравнения берут воду.
Для определения марганца в испытуемом растворе из общего объема 100 мл берут 10—15 мл этого раствора, помещают в мерную колбу емкостью 50 мл, добавляют то же количество реагентов, в той же последовательности. как это было указано для приготовления эталонных растворов. По градуировочному графику находят содержание марганца.
Б. Определение больших количеств марганца
дифференциальным методом в отсутствие посторонних ионов
Берут 10—15 мл испытуемого раствора, содержащего 0,2—0,7 мг марганца, помещают в мерную колбу емкостью 50 мл, прибавляют 1,5 мл свежеприготовленного раствора формальдоксима, 10 мл раствора щелочи, оставляют стоять 5 мин, вновь прибавляют такое же количество формальдоксима и щелочи, после чего доводят объем раствора водой до метки. '
В тех же условиях готовят 6—7 эталонных растворов с содержанием в них марганца в указанных пределах. Измерение оптической плотности растворов производят на любом фотоэлектроколориметре, используя светофильтр с А 455 нм и кювету с толщиной слоя 0,3—0,5 см, в том порядке, который указан на стр. 65, 71, начиная с выбора оптимального раствора сравнения.
В. Определение больших количеств марганца дифференциальным методом в присутствии железа
Для определения марганца в присутствии железа может быть использован вариант дифференциального спектрофотометрического метода (см. стр. 71). Раствор анализируемого образца, содержащий марганец и железо в количестве 0,5—3 мг, помещают в мерную колбу емкостью 100 мл и доводят объем раствора водой до метки. Готовят три раствора:
1-й раствор —• в мерную колбу емкостью 100 мл помещают 10 мл испытуемого раствора;
2-й раствор — в мерную колбу емкостью 100 мл помещают 20 мл того же испытуемого раствора;
3-й раствор — в мерную колбу емкостью 100 мл помещают 20 мл испытуемого раствора и прибавляют 0,1 мг марганца, используя его стандартный раствор.
Все растворы готовят по методике, приведенной на стр. 172. Измеряют оптические плотности 2-го А'х и 3-го Ах_^а растворов, используя в качестве раствора сравнения 1-й раствор и кювету с толщиной слоя 0,3—0,5 см. Вычисляют содержание марганца в 10 мл испытуемого раствора (что соответствует разности объемов прибавленных во 2-й и 3-й растворы) по формуле, приведенной на стр. 136.
ОПРЕДЕЛЕНИЕ МАРГАНЦА И ХРОМА В СТАЛЯХ
Марганец и хром в стали можно определять одновременно, окисляя соответственно до бихромата и перманганата персульфатом аммония. Растворы фотометрируют при А 440 нм, соответствующей максимуму поглощения бихромата, и А 545 нм, соответствующей максимуму поглощения перманганата (см. рис. 11 и 53). Определение содержания марганца и хрома при совместном присутствии облегчается тем, что при А 545 нм поглощает только перманганат. Для расчета процентного содержания марганца и хрома в стали могут быть использованы два метода.
Метод градуировочных графиков. Для марганца стро^ два градуировочных графика, измеряя оптическую плотность эталонных растворов при двух длинах волн: 440 и 545 нм; для хрома строят один градуировочный график при А 440 нм. Измеряют оптическую плотность испытуемого раствора при указанных длинах волн. По оптической плотности при А 545 нм находят содержание марганца, используя градуировочный график. По второму градуировочному графику для марганца при А 440 нм определяют оптическую плотность, соответствующую найденному количеству марганца, и вычитают это значение из оптической плотности испытуемого раствора при А 440 нм. Эта разность оптических плотностей будет соответствовать содержанию хрома, которое и определяют по градуировочному графику, построенному для хрома при А 440 нм.
Содержание хрома или марганца (в процентах) рассчитывают по формуле
, а 100
Сг(Мл)‘-"ГЙ7г
где а — найденное количество марганца или хрома; мг; w — навеска анализируемого образца стали.
Алгебраический метод. Решая систему уравнений (III.35) относительно концентраций [МпОГ 1 и [Сг2О7-1 при условии, что есг2о|~ =0 и I = 1 см, получим:
А545 [МпО4-]=—------,
Мп®.
4 g 545 /440_о440 /546
МпОА МпО.
[Сг2 О? - ] =-----------4----
1271 ₽545 г 4 40
еМ»О, есг20,2-
где Л440 и Л545 — поглощение растворов образца стали при длинах волн 440 и 545 нм. Значения е для каждого из компонентов при указанных длинах волн находят по градуировочным графикам (как тангенс угла наклона прямой). Подставляя в уравнения (III.35) полученные значения е и оптической плотности испытуемого раствора, измеренные при А, 440 и А 545 нм, находят содержание бихромата и перманганата (моль/л). Процентное содержание каждого из компонентов в образцах стали рассчитывают по уравнениям
Мпо/о = 54,9-^- Ю-4[МпОгЬ
Сг% = 52,0-2— Ю-4[Сг2О2-],
где v — объем мерной колбы, в которой проводят определение; w — навеска образца стали, г; 54,9 и 52,0 — атомные веса марганца и хрома соответственно.
Реагенты
1. Стандартный раствор Cr (Ilk), содержащий 1 мг/мл Сг. Навеску 9,197 г квасцов CrNH4 (SO4)2 • 12Н2О помещают в мерную колбу емкостью 1 л, растворяют в воде, добавляют 2 мл H2SO4 (пл. 1,84) и доводят объем раствора водой до метки.
2. Стандартный раствор Мп (II) , содержащий 1 мг/мл Мп (приготовление см. стр. 168).
3. Смесь кислот серной и фосфорной. В мерную колбу емкостью 1 л вводят 500 мл воды, добавляют постепенно, при помешивании 150 мл H2SO4 (пл. 1,84), 125 мл концентрированной Н3РО4, перемешивают раствор и доводят объем раствора водой до метки.
4. Смесь кислот серной, форсфорной н азотной: в мерную колбу емкостью 1 л вводят 500 мл воды, добавляют постепенно, при помешивании 100 мл H2SO4 (пл. 1,84), 125 мл концентрированной Н3РО4, 200 мл концентрированной HNO3 и доводят объем раствора водой до метки.
5. Нитрат серебра, 1%-ный раствор.
6. Персульфат аммония, 20%-ный водный раствор, свежеприготовленный.
7. Перйодат калия, кристаллический.
Для определения марганца и хрома в сталях три параллельные навески стали по 0,1г растворяют в конической колбе объемом 50 мл в 10 мл смеси азотной, форсфорной и серной кислот, нагревая на песчаной бане. После полного растворения образца, растворы упаривают до появления белых паров SO3. Затем растворы охлаждают, добавляют по 10 мл смеси серной и фосфорной кислот, 2 мл раствора нитрата серебра, 10 мл персульфата аммония и нагревают растворы до прекращения выделения пузырьков. Если окисление марганца произошло не полностью (разные оттенки растворов), то добавляют по 0,1 г перйодата калия и нагревают до тех пор, пока интенсивность окраски растворов не станет одинаковой. Затем растворы охлаждают и количественно переносят в мерные колбы емкостью 50 мл. Доводят объем растворов до метки водой, перемешивают и измеряют оптическую плотность их на спектрофотометре типа СФ-4А в кюветах с / = 1 см при А, 440 и 545 нм.
Дальнейший расчет содержания марганца и хрома в образце стали проводят, как описано выше, по градуировочным грфикам или алгебраическим методом.
Определение меди
Для определения меди известен ряд методов с использованием комплексных соединений этого элемента, главным образом, с органическими лигандами. Наиболее распространенными являются методы определения меди с дитизоном и диэтилдитиокарбаминатом свинца [31]. Ранее широко применялся метод определения меди в виде аммиачного комплексного соединения. Но чувствительность его мала и он используется в настоящее время, главным образом, для определения больших количеств меди дифференциальным методом.
ОПРЕДЕЛЕНИЕ МЕДИ ДИЭТИЛДИТИОКАРБАМИНАТОМ СВИНЦА
Диэтилдитиокарбаминат
/N (С2НБ)2 натрия S=C< r xSNa
образует с медью
комплексное соединение, растворы которого в органических растворителях окрашены в зеленовато-коричневый цвет, 436 нм, е = 1,4 • 104. В качестве растворителей применяют четыреххлористый углерод, хлороформ или изоамиловый спирт. Для повышения селективности метода применяют диэтилдитиокарбаминат свинца. Все элементы, образующие менее устойчивые комплексы, чем диэтилдитиокарбаминат свинца, не мешают определению меди. Кроме того, растворы диэтилдитиокарбамината свинца более устойчивы при хранении, чем диэтилдитиокарбаминат натрия.
Реагенты
1. Стандартный раствор соли меди, содержащий 2 мг/мл Си. Навеску сульфата меди (х. ч.), соответствующую 0,5 г металла, помещают в мерную колбу емкостью 250 мл, растворяют в воде, подкисленной 1 мл серной кислоты (пл. 1,84), доводят объем раствора водой до метки. Более разбавленные растворы готовят соответствующим разбавлением, раствор с содержанием 1 мкг/мл готовят ежедневно.
2. Раствор диэтилдитиокарбамината свинца. Готовят из диэтилдитиокарбамината натрия, предварительно очищенного выделением эфиром из насыщенного спиртового раствора. Для этого 10 г диэтилдитиокарбамината натрия растворяют в 50 мл этилового спирта и фильтруют через сухой фильтр. Полученный раствор вливают в 300 мл эфира. Выделившиеся кристаллы отсасывают на воронке Бюхнера, промывают 2—3 раза эфиром, высушивают на воздухе и сохраняют в темной склянке. Берут 0,1 г очищенной соли, растворяют в воде, добавляют 0,1 г нитрата свинца (х. ч ), 0,5 г сегнетовой соли, нейтрализуют раствором аммиака до pH 9 по индикаторной бумаге и встряхивают раствор последовательно с двумя порциями хлороформа по 50 мл каждая. Экстракты собирают, промывают двумя порциями воды и, пропуская через сухой фильтр, переводят в мерную колбу емкостью 0,5 л, доводят объем раствора до метки хлороформом.
3. Хлороформ очищенный.
4. Аммиак, раствор 1 : 5.
5. Соляная кислота х. ч., 2 н. раствор.
6. Цитратный буферный раствор, pH <— 9.
А. Определение меди в растворе ее соли
Для приготовления эталонных растворов берут пять делительных воронок емкостью 50—60 мл, вводят в каждую 20 мл воды, стандартный раствор, содержащий медь в количестве (мкг): 0,1; 0,5; 1,0; 3,0; 5,0 соответственно, устанавливают pH раствора до 3—5 по индикаторной бумаге,, добавляя, если необходимо, раствор аммиака или соляной кислоты. Экстрагируют комплексное соединение меди двумя порциями по 5 мл каждая хлороформного раствора реагента, перемешивают в течение 3 мин каждую порцию. Объединенные экстракты переводят в мерную колбу емкостью 25 мл и доводят объем раствора хлороформом до метки. Для измерения оптической плотности эталонных растворов при А 436 нм используют спектрофотометры различных марок или фотоэлектроколориметр ФЭК-60. Строят градуировочный график по экспериментальным данным, обработанным методом наименьших квадратов.
Раствор, содержащий все реагенты, кроме стандартного раствора меди и прошедший через все стадии, указанные выше, обрабатывают хлороформом, который и служит раствором сравнения.
Для определения меди в испытуемом растворе из общего объема 100 мл берут 10—20 мл этого раствора (в зависимости от содержания меди) и проводят определение ее в условиях см. приготовление эталонных растворов. Содержание меди находят по градуировочному графику-
Б. Определение меди в гипофосфите натрия
Приготовление эталонных растворов см. на стр. 175.
Для определения меди в гипофосфите натрия берут две навески по 2,5 Г', помещают каждую в мерную колбу емкостью 50 мл, растворяют в воде, доводят объем раствора водой до метки. Отбирают из обеих колб по две порции по 20 мл, помещают каждую в делительную воронку емкостью 50—100 мл, добавляют 20 мл воды и проводят все операци, указанные при приготовлении эталонных растворов. Полученные данные (не меньше четырех из параллельных опытов обрабатывают методом математической статистики.
Примечания. 1. Метод определения меди в гипофосфите натрия пригоден для определения меди в фосфате натрия.
2. Метод определения меди в гипофосфите натрия разработан в лаборатории спектрофотометрии кафедры аналитической химии МГУ.
В. Определение меди в нитрате кобальта
Для приготовления эталонных растворов в пять делительных воронок емкостью 60—70 мл вводят по 20 мл воды, стандартный раствор, содержащий медь в количестве (мкг): 1,0; 3,0; 5,0; 7,0; 9,0 соответственно. В остальном проводят все операции, указанные при приготовлении эталонных растворов (см. стр. 175).
Для определения меди в нитрате кобальта берут две навески пр 1 г, переводят каждую в мерную колбу емкостью 100 мл, доводят раствор
водой до метки. Для определения 20 мл раствора помещают в делительную воронку емкостью 60—70 мл, устанавливают pH раствора до 3—5 по индикаторной бумаге, добавляя, если это необходимо, раствор аммиака или соляной кислоты. Экстрагируют соединение меди последовательно двумя порциями (по 5 мл каждая) хлороформного раствора реаганта, перемешивая на механическом вибраторе в течение 3 мин каждую порцию. Объединенные экстракты переводят в мерную колбу емкостью 25 мл и добавляют хлороформ до метки. Измерение оптической плотности проводят при X 436 нм. Содержание меди находят по градуировочному графику. Данные из (не менее четырех) параллельных опытов обрабатывают методом математической статистики [31].
ОПРЕДЕЛЕНИЕ БОЛЬШИХ КОЛИЧЕСТВ МЕДИ ДИФФЕРЕНЦИАЛЬНЫМ МЕТОДОМ
Определение меди производят в виде аммиачного комплексного соединения, е = 1 • 102, X 610 нм. Работа может быть выполнена на фотоэлектроколориметрах ФЭК-56, ФЭК-Н-57, ФЭК-60.
Реагенты
1. Стандартный раствор меди (см. стр. 175).
2. Аммиак, концентрированный водный раствор.
3. Азотная кислота (х. ч.), раствор 1:1.
А. Определение меди в растворе ее соли
Для приготовления эталонных растворов в мерные колбы емкостью по 50 мл вводят по 10 мл воды в каждую, стандартный раствор, содержащий медь в количестве (мг): 5,0; 7,0; 9,0; 11,0; 13,0; 15,0 или 10,0; 12,0; 14,0; 16,0; 18,0; 20,0 соответственно, добавляют 5 мл раствора аммиака и доводят объем раствора водой до метки. Измерение оптической плотности этих растворов производят на любом фотоэлектроколориметре при К 610 нм, I = 0,5 см в том порядке, который указан на стр. 65, 71, начиная с выбора оптимального раствора сравнения.
Для определения меди в испытуемом растворе из общего объема 50 мл берут 10—20 мл этого раствора в зависимости от содержания меди в нем, переносят в мерную колбу емкостью 50 мл, добавляют 5 мл раствора аммиака и доводят объем раствора водой до метки. Измеряют оптическую плотность раствора и находят содержание меди (см. стр. 65, 71).
Б. Определение меди в бронзе, в томпаке
Две навески по 0,5 г исследуемого образца бронзы или томпака растворяют каждую при легком нагревании в 25 мл азотной кислоты, раствор упаривают до небольшого объема, переносят в мерную колбу емкостью 50 мл и разбавляют водой до метки. Затем 1—2 мл этого раствора (в зависимости от содержания меди) переносят в мерную
колбу емкостью 50 мл, добавляют 5 мл раствора аммиака, разбавляют водой до метки и тщательно перемешивают [32].
Содержание меди в контрольном образце определяют по градуировочному графику или по формуле (III.32), вычислив предварительно фактор F. Результаты (не менее четырех) определений обрабатывают методом математической статистики.
ОПРЕДЕЛЕНИЕ МЕДИ МЕТОДОМ
СПЕКТРОФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Медь с комплексоном III образует устойчивое комплексное соединение (1g К = 18,3) с соотношением компонентов 1:1, растворы которого имеют максимум поглощения при X 745 нм [30].
Комплексонат 'железа в этой области не поглощает, поэтому возможно последовательное титрование железа и меди [331. Вначале титруется железо, образующее с комплексоном более прочное соединение, чем медь, при этом изменения оптической плотности не наблюдается. После того как будет оттитровано железо, начинает оттитровываться медь, в результате чего наблюдается постепенное возрастание оптической плотности до ее постоянного значения. На кривой титрования наблюдаются два излома, первый из которых соответствует объему комплексона III, израсходованному на титрование железа, второй — на титрование меди.
Реагенты
1. Стандартный раствор солн меди, содержащий 2 мг/мл Си.
2. Комплексон III, 1 М раствор. Раствор готовят из препарата (х. ч.) по точной навеске. Концентрацию его проверяют методом спектрофотометрического титрования по соли меди известной концентрации.
3. Ацетатный буферный раствор, pH 3. К 900 мл 0,2 н. раствора уксусной кислоты добавляют 100 мл 0,2 н. раствора ацетата натрия.
Для определения меди в испытуемом растворе в отсутствие железа в мерную колбу емкостью 50 мл помещают раствор, содержащий от 10 до 20 мг меди, и доводят объем ацетатным буферным раствором до метки. После перемешивания отбирают пипеткой 20 мл этого раствора и переносят в кювету для титрования. Раствор комплексона III прибавляют из микробюретки порциями по 0,1 мл, перемешивают и измеряют оптическую плотность при А 745 нм после прибавления каждой порции реагента. По полученным данным строят кривую титрования.
Конечную точку титрования находят графическим путем или рассчитывают алгебраическим методом (см. стр. 57).
ОПРЕДЕЛЕНИЕ МИКРОКОЛИЧЕСТВ МЕДИ
КИНЕТИЧЕСКИМ МЕТОДОМ
Для определения микроколичеств меди применяют катализируемую медью реакцию окисления гидрохинона перекисью водорода. Реакцию проводят в присутствии пиридина в боратном буферном растворе при pH 7,8. Содержание меди рассчитывают по способу тангенсов дифференциального варианта кинетических методов анализа [341.
Реагенты
1. Стандартный раствор, содержащий 2 мг/мл Си (см. стр. 175). Растворы с меньшим содержанием меди (0,1 и 0,01 мкг/мл) готовят ежедневно с разбавлением исходных растворов
2. Гидрохинон, 5,4 - 10-2 М водный раствор. Гидрохинон очищают возгонкой. Свежие растворы готовят каждые 2—3 дня.
3. Пиридин высушивают над едким калн и перегоняют в кварцевом приборе; готовят 3 • 10-2 М водный раствор.
4. Перекись водорода (о. ч.), 1,6 М раствор. Точную концентрацию перекиси водорода устанавливают титриметрически каждые 5—7 дней.
5. Боратный буферный раствор, pH 7,8. Для приготовления 1 л буферного раствора смешивают 800 мл 0,2 н. раствора борной кислоты и 200 мл 0,5 М раствора тетрабората натрия. Тетраборат натрия (х. ч.) и борную кислоту (х. ч.) дважды перекристаллизовывают из воды.
Для построения градуировочного графика в кварцевый смеситель с тремя отростками вводят: в первый отросток 0,5 мл раствора гидрохинона, во второй — 1 мл раствора перекиси водорода, а в третий — последовательно 1 мл раствора пиридина, раствор соли меди и такое количество боратного буферного раствора, чтобы общий объем раствора во всех смесителях составлял 10 мл. Растворы термостати-руют 25—30 мин при 25° С. В момент смешения растворов включают секундомер и измеряют оптическую плотность раствора через 1 мин в течение 10 мин на приборах ФЭКЧМ, ФЭК-54, ФЭК-57 с термоста-тируемой камерой для кювет или ФЭК-Н-52 с термостатируемым корпусом (1 = 3 см; А 420 нм). По данным измерений строят график в координатах оптическая плотность — время; тангенс угла наклона прямых (tg а) соответствует концентрации меди.
Содержание меди в эталонных растворах 5 • 10-2—1 • 10-1 мкг в 10 мл раствора.
А. Определение меди в растворе ее соли
Для определения меди 1 мл испытуемого раствора из общего объема 25 мл помещают в один из отростков смесителя, в другие два отростка добавляют реагенты. После термостатирования и смешения фиксируют изменение оптической плотности во времени, строят график в координатах оптическая плотность — время и определяют тангенс угла наклона полученной прямой. По градуировочному графику находят содержание меди.
Б. Определение меди в азотной и соляной кислотах
Кроме указанных на стр. 177, необходимы реагенты:
1. Едкий натр, 2 н. раствор.
2. Серная кислота (о. ч.), (пл. 1,8).
В тефлоновые чашки помещают 10—15 мл испытуемой кислоты, раствор упаривают до объема 5 мл в боксе на плитке, покрытой асбестом, добавляют 1—2 капли серной кислоты и далее упаривают раствор до 0,5 мл, добавляя 1 мл деионизованной воды. Раствор нейтрализуют рствором едкого натра до pH 7 по универсальной индикаторной
бумаге, переносят в отросток кварцевого смесителя, споласкивают тефлоновую чашку буферным раствором, добавляют в другие два отростка кварцевого смесителя реагенты (см. стр. 179), чтобы общий объем во всех смесителях был равен 10 мл. Содержание меди находят по градуировочному графику.
Определение молибдена
Для определения молибдена большое распространение до последнего времени имели роданидный и пероксидный метод. В настоящее время большое внимание уделяют методам определения молибдена в виде ионных ассоциатов, к которым относятся триоксифлуорон— антипириновые комплексные соединения [35].
ОПРЕДЕЛЕНИЕ МОЛИБДЕНА В БЕРИЛЛИИ И ЕГО СОЛЯХ В ВИДЕ РОДАНИДА
Реагенты
1. Стандартный раствор, содержащий 0,1 мг/мл Мо; 1,500 г МоОз растворяют в 25 мл 2 н. раствора NaOH , раствор подкисляют соляной кислотой и разбавляют водой в мерной колбе до 1 л; 100 мл раствора перед использованием разбавляют водой до 1 л.
2. Роданид аммония 10%-ный раствор.
3. Аскорбиновая кислота, 10%-ный раствор.
4. Серная кислота, 10%-ный и 2 н. растворы.
5. Тетра-н-бутиламмонийхлорид (ТБА), 0,001 М раствор.
6. Аммиак, раствор водный 1:1.
7. Хлороформ.
Для приготовления эталонных растворов берут пять делительных воронок емкостью 100 мл, вводят по 5 мл воды, стандартного раствора, содержащего молибден (мкг): 0,0; 2,0; 4,0; 6,0; 8,0 соответственно, приливают в каждую 3 мл раствора роданида аммония, 2 мл аскорбиновой кислоты, перемешивают и выдерживают растворы в течение 2 ч, после чего приливают 1 мл ТБА, 3 мл серной кислоты, 10 мл хлороформа и встряхивают содержимое воронок в течение 10 мин на механическом вибраторе. После расслаивания отделяют органическую фазу и измеряют оптическую плотность хлороформных растворов при А 465 нм относительно раствора, не содержащего молибдена (1-я воронка). Измерение производят на спектрофотометрах различных марок и строят градуировочный график по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения молибдена в бериллии берут две навески металла по 1,5 г (или соли бериллия, соответствующей 1,5 г металла).
Навеску металла растворяют в 2 н серной кислоте при осторожном нагревании до получения прозрачного раствора, который переводят в мерный цилиндр на 150 мл и добавляют той же кислоты до метки. Для определения берут аликвотную часть раствора (0,5 г), осторожно добавляют раствор аммиака до pH ~ 4 (по индикаторной бумаге),
переносят в делительную воронку и проводят определение в условиях см. приготовление эталонных растворов. Результаты (не менее четырех) определений обрабатывают методом математической статистики.
ОПРЕДЕЛЕНИЕ МОЛИБДЕНА САЛИЦИЛФЛУОРОНОМ
Молибден с триоксифлуороном и его производными образует комплексное соединение при соотношении компонентов Mo : R 1:1 и 1 : 2 в зависимости от кислотности раствора. Эти соединения не экстрагируются органическими растворителями. В присутствии антипирина и его производных образуются комплексы катионного характера, которые при добавлении анионов сильных кислот хорошо экстрагируются хлороформом в виде ионных ассоциатов, предполагаемого состава [МоО2ФАнт2]+ А-, где Ф — триоксифлуорон; АнТ*— антипирин; А — анион кислоты (НВт), Хшах 515 нм, г = 1,3 • 105.
Для повышения селективности метода предлагается предварительное выделение молибдена в виде диэтилдитиокарбамината с последующим определением его салицилфлуороном [36].
Реагенты
1. Серная кислота (х. ч.), пл. 1,84 и 2 н. раствор.
2. Хлороформный раствор диэтилдитиокарбаминовой кислоты. В делительной воронке к 100 мл охлажденного до 5—10° С диэтилдитиокарбамината натрия приливают 8 мл 6 н. соляной кислоты и немедленно взбалтывают с 100 мл охлажденного хлороформа до растворения белой мути. Хлороформный раствор устойчив не более 6 ч при 5—10° С и до 3 суток при 0° С.
3. Лимонная кислота, 2%-ный раствор в 2 н. растворе H2SO4.
4. Сульфаты меди, 2%-ный раствор.
5. Комплексон III, 5%-ный раствор.
6. Аскорбиновая кислота, 10%-ный раствор.
7. Бромид калия, 2,5 М раствор.
8. Салицилфлуорон, 0,001 М (0,034%-ный) раствор в 96%-ном этаноле (0,03 н. по НС1).
9. Антипирин, 10%-ный раствор.
10. Хлороформ.
11. Соляная кислота (х. ч.), 6 н., 1 н. растворы.
12. Аммиак (х. ч), 12,5 н. раствор.
13. Перманганат калия, 1%-ный раствор.
14. Метиловый оранжевый, 0,01 %-ный раствор.
А. Определение молибдена в растворе его соли
Для приготовления эталонных растворов берут пять делительных воронок емкостью 200 мл, вводят стандартный раствор, содержащий молибден в количестве (мкг): 0,0; 0,5; 1,0; 2,5; 5,0 соответственно, приливают по 4 мл 2 н. серной кислоты, разбавляют водой до 75 мл, затем вводят 3 мл бромида калия, 3 мл салицилфлуорона и 15 мл антипирина, доводят объем водой до 100 мл. Через 5 мин экстрагируют 10 мл хлороформа, производя энергичное перемешивание в течение 1 мин. Органические фазы переводят в мерные пробирки емкостью 10 мл, добавляют, если нужно, 2—3 капли хлороформа до объема 10 мл. Измерение оптической плотности экстрактов проводят на спектрофотометрах
различных марок или фотоэлектроколориметре ФЭК-60 при Z 515 нм относительно раствора сравнения, которым служит хлороформный раствор 1-й пробирки, в которую не введен стандартный раствор молибдена. Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения молибдена в испытуемом растворе из общего объема 25 мл берут 5 или 10 мл и проводят определение см. приготовление эталонных растворов. Содержание молибдена находят по градуировочному графику.
Б. Определение молибдена в едких щелочах
В щелочи обычно содержатся элементы, мешающие определению молибдена указанным методом. Поэтому рекомендуется проводить предварительное его выделение диэтилдитилкарбаминовой кислотой [36].
Приготовление эталонных растворов проводят в условиях, указанных на стр. 181.
Для определения молибдена в едких щелочах (NaOH, КОН, L1OH) берут две навески по 15—30 г (в зависимости от содержания в них молибдена), помещают каждую в мерную колбу емкостью 0,5 л, добавляют 200—300 мл воды и после растворения щелочи объем раствора доводят водой до метки. Берут две аликвотные части по 150 мл каждая в коническую колбу емкостью 250—300 мл, нейтрализуют 6 н. соляной кислотой до слабокислой реакции по индикаторной бумаге, добавляют 12,5 мл серной кислоты (пл. 1,84), 2 г лимонной кислоты, разбавляют водой до 200 мл и охлаждают раствор до 2—5° С. Переводят раствор в делительную воронку емкостью 250—300 мл и экстрагируют двумя порциями (по 5 мл каждая) хлороформного раствора диэтилдитиокар-баминовой кислоты, перемешивая в течение 2 мин, 3-ю экстракцию производят 5 мл хлороформа. Объединенные хлороформные экстракты переводят в делительную воронку емкостью 150 мл и промывают их 20 мл 2%-ного раствора лимонной кислоты. Водный слой отделяют, к экстрактам приливают 25 мл воды, 3,0 мл 12,5 н. раствора аммиака и 3 мл раствора сульфата меди. Производят энергичное перемешивание в течение 2 мин. Через 5 мин отделяют и отбрасывают хлороформный слой, а водный слой промывают хлороформом до получения бесцветного экстракта. К ярко-синему водному раствору приливают 2 капли метилового оранжевого и (около 6 мл) 2 н. серной кислоты до розовой окраски. Затем прибавляют еще 4 мл той же кислоты, разбавляют водой до 75 мл, прибавляют 1 каплю раствора КМпО4, через 2 мин приливают 1 мл раствора комплксона III и оставляют стоять до исчезновения розовой окраски. После этого приливают 2 мл раствора аскорбиновой кислоты (в отсутствие железа эта операция опускается), добавляют 3 мл раствора бромида калия, 3 мл раствора салицилфлуоро-на, 15 мл раствора антипирина и добавляют воду до 100 мл. Через 5 мин добавляют 10 мл хлороформа, перемешивают фазы энергично в течение 1—2 мин, экстракт фильтруют через сухой фильтр и изме
ряют его оптическую плотность (см. стр. 181). Содержание молибдена находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают методами математической статистики.
Определение никеля
Наиболее распространенными реагентами для определения никеля являются а-диоксимы. Впервые предложенные Л. А. Чугаевым [37] для открытия и определения никеля они не потеряли своего значения до настоящего времени.
а-Диоксимы, являясь бидентатными реагентами, образуют с никелем комплексные соединения по схеме
Ni2+ + 2H2D^±Ni (HD)2+2H+
Распространение получили две группы методов определения. В первой используются окрашенные соединения никеля с а-диоксимами, которые образуются при действии ряда окислителей в щелочной среде, во второй — соединения, образующиеся при растворении в неводных растворителях диоксиматов никеля, которые поглощают излучение в УФ-области; исключение составляют соединения никеля с а-фурилдиоксимом (А, 435 нм).
ОПРЕДЕЛЕНИЕ НИКЕЛЯ ДИМЕТИЛ ГЛИОКСИМОМ
В ПРИСУТСТВИИ ОКИСЛИТЕЛЯ
Соединения никеля в присутствии окислителей образуют растворы, окрашенные в бурый цвет. Максимальное поглощение наблюдается при Z, 470 нм, 8 = 1,3 • 103. В настоящее время известно, что в условиях
проведения реакции происходит окисление никеля. В присутствии аммиака и щелочи образуются два раз-
ных комплексных соединений, отличающихся по спектрам их поглощения [38], [40] (рис. 54), соотношение Ni : H2D = = 1:3. В настоящее время установлено, что степень окисления никеля в этих соединениях (III) или (IV). В присутствии окислителя персульфата аммония образуется соединение, в котором степень окисления никеля +4 [39], в присутствии иода +3 [40]. Более устойчивые по времени соединения образуются в йрисутствии щелочи. В качестве окислителя лучше использовать раствор иода, так как в его присутствии не окисляется избыток а-диоксима.
Рис. 54 Спектры поглощения:
1 — диметилдиоксим; 2 — диоксим ат никеля в присутствии ЫНз; 3 — ди-оксимат никеля в присутствии NaOH
Никель может быть определен данным методом в сталях, содержащих кобальт, ванадий, молибден; вольфрам,
хром и титан могут содержаться до 18%. Элементы, которые дают осадки гидроокисей при действии растворов аммиака и едкой щелочи, и медь, которая дает окрашенные комплексные соединения с а-дио-ксимами, мешают определению.
При определении никеля в сталях, содержащих медь, никель предварительно отделяют в виде диметилдиоксимата, экстрагируя соединение хлороформом. Влияние железа и хрома устраняют добавлением винной кислоты или ее соли (сегнетовой). После реэкстракции в водную фазу при подкислении определяют никель диметилглиоксимом в присутствии окислителя.
Реагенты
1. Стандартный раствор соли никеля, содержащий 0,1 мг/мл Ni. Навеску сульфата никеля (х. ч.) (с известным содержанием никеля), соответствующую 0,1 г металла, помещают в мерную колбу емкостью 1 л, растворяют в воде, подкисляют 2 мл серной кислоты (х. ч.) (пл. 1,84), доводят объем раствора водой до метки и тщательно перемешивают — раствор А; 1 мл раствора А содержит 0,1 мг Ni. Раствор А разбавляют в 10 раз — раствор Б: 1 мл раствора Б содержит 0,01 мг Ni; раствор Б и более разбавленные растворы пригодны в день их"приготовления.
2. Иод, 0,05 М раствор.
3. Диметилглиоксим, 0,05 М спиртовой раствор.
3. Едкий натр, (х. ч.) 1 н. раствор.
5. Азотная кислота (х. ч.), раствор 1 : 5.
6. Сегнетова соль, 20%-ный раствор.
7. Соляная кислота (х. ч.), 1 н. раствор.
А. Определение никеля в сталях, не содержащих меди
Для приготовления эталонных, растворов навеску 0,1 г стали, не содержащей никеля, переносят в коническую колбу емкостью 50 мл, растворяют в 10 мл азотной кислоты, нагревают на песчаной бане до удаления окислов азота, раствор охлаждают, переносят его в мерную колбу емкостью 100 мл и доводят объем раствора водой до метки — раствор А. В мерные колбы емкостью 50 мл отбирают по 2,5 мл раствора А и вводят стандартный раствор соли никеля в таких количествах, чтобы содержание никеля (мг) в эталонных растворах соответствовало:
1-й ряд 0.005; 0,01; 0,02; 0,03; 0,04; 0,05
2-й ряд 0,06; 0,07; 0,08; 0,09; 0,1
К растворам добавляют по 5 мл раствора сегнетовой соли, объем их доводят водой до 20—25 мл, после этого добавляют 0,5 мл раствора иода, 0,5 мл раствора диметил глиоксима, 3 мл раствора едкого натра и доводят водой объем раствора до метки. По приготовленным эталонным растворам строят градуировочный график, беря в качестве раствора сравнения воду.
Для определения никеля в стали две навески по 0,1 г исследуемого объекта переносят каждую в коническую колбу емкостью 50 мл, добавляют 10 мл азотной кислоты, нагревают раствор на песчаной бане до удаления окислов азота, охлаждают, переносят в мерную колбу
емкостью 100 мл и доводят водой объем раствора до метки — раствор А. В мерные колбы емкостью по 50 мл отбирают пипеткой по 2,5 мл раствора А, добавляют те же количества реагентов и в той же последовательности, как это указано для приготовления эталонных растворов, не вводя стандартных растворов соли никеля. Измерение оптической плотности растворов проводят в условиях, указанных на стр. 184. Содержание никеля находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают, применяя метод математической статистики.
Примечания. 1. При содержании никеля в стали около 0,20% и меньше берут для определения 5 или 10 мл раствора А.
2. При определении никеля в стали можно использовать эталонные растворы, не содержащие стали. Но при измерении оптической плотности испытуемого раствора в качестве раствора сравнения следует использовать раствор, в котором не проведена фотометрическая реакция (без добавления к нему диметилдиоксима, щелочи и окислителя).
Б. Определение никеля в сталях, содержащих медь
Примесь никеля предварительно рекомендуется отделить экстракцией в виде диметилдиоксимата никеля хлороформом.
Для опредеения никеля в сталях, содержащих медь, сохраняют условия растворения навески и получение раствора А, описанные на на стр. 184.
Берут 5 или 10 мл раствора А, переносят в делительную воронку, добавляют 10 мл раствора сегнетовой соли, 2 мл раствора диметилглиок-сима, 10 мл раствора фосфата натрия и доводят pH раствора до 8~ 9,5 по универсальной индикаторной бумаге, добавляя раствор щелочи или уксусной кислоты, если pH > 9,5. После стояния через 10 мин проводят экстракцию последовательно двумя порциями хлороформа, первая 10 мл, вторая 5 мл, перемешивая содержимое воронки в течение 5 мин (каждую порцию). После расслаивания отделяют органические фазы и объединяют их в делительной воронке: никель из экстракта переводят в водный раствор встряхиванием содержимого воронки в течение 1мин последовательно с двумя порциями 1 н. соляной кислоты по 5 мл каждая. В дальнейшем определение никеля проводят в условиях, описанных на стр. 184.
В. Определение никеля в металлическом бериллии
Кроме указанных на стр. 184, необходимы реагенты:
1. Цитрат натрия трехзамещенный (х. ч.), 0,7 М раствор.
2. Едкий натр (х. ч.), 50%- и 4%-ные растворы.
3. Соляная кислота (х. ч.), пл. Ц19, 5%-ный раствор.
4. Раствор аммиака (х. ч.), концентрированный.
5. Хлороформ.
6. Серная кислота (х. ч.), 25% ный раствор.
Для приготовления эталонных растворов строго соблюдают условия, указанные на стр. 184, не вводя в водные растворы сталь.
Для определения никеля в металлическом бериллии берут две навески по 0,25 г, растворяют каждую при слабом подогревании в 20 мл
серной кислоты, переносят раствор в делительную воронку емкостью 50—60 мл, добавляют 5 мл раствора диметил глиоксима, 10 мл раствора цитрата натрия и доводят pH раствора до 8—10 по универсальной индикаторной бумаге. После стояния через 10 мин экстрагируют диметил-диоксимат никеля двумя порциями хлороформа по 5 мл каждая встряхиванием на механическом вибраторе в течение 5 мин. После расслаивания отделяют органические фазы и объединяют их в делительной воронке; соединение никеля из экстракта переводят в водный раствор встряхиванием содержимого воронки в течение 1 мин последовательно двумя порциями 1 н. соляной кислоты каждая по 5 мл. Переносят водную фазу в мерную колбу емкостью 50 мл, добавляют постепенно около 1 мл 50%-ного раствора NaOH до pH 3 по универсальной индикаторной бумаге и затем все реагенты в той же последовательности, как это указано для приготовления эталонных растворов. Содержание никеля находят по градуировочному графику. Результаты параллельных определений Сне менее четырех) обрабатывают методом математической статистики.
Примечание. Метод разработан в лаборатории спектрофотометрии кафедры аналитической химии МГУ.
ЭКСТРАКЦИОННО СПЕКТРОФОТОМЕТРИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ НИКЕЛЯ а-ДИОКСИМАМИ
Известен ряд а-диоксимов, кроме диметилглиоксима, которые широко используются для фотометрического определения никеля с применением метода экстракции. В экстракт переходит соединение никеля состава
Устойчивость образующихся диоксиматов никеля объясняется образованием четырех пятичленных циклов. Применение других а-диоксимов позволяет определять ультрамалые количества никеля в ряде объектов. Применяют а-бензилдиоксим [41], а-фурилдиоксим, [42], диоксим 1,2-циклогександиона (ниоксим) [43], [411 и диоксим 1,2-цик-логекптандиона (гептоксим) [45].
Комплексные соединения никеля со всеми а-диоксимами в растворе хлороформа имеют три полосы поглсщения: более интенсивная из них лежит в ультрафиолетовой области, в которой поглощают и сами реагенты (табл. 12). Кроме того, последними работами установлено наличие плеча в интервале 430—440 нм. Более устойчиво соединение никеля с а-бензилдиоксимом; близкими по устойчивости являются соединения никеля с гептокспмом и ниоксимом. Комплексные соединения никеля
Нб
ТАБЛИЦА 12
Характеристики комплексных соединений (к. с.) диоксиматов никеля
Реагент pK dh2 lg ₽2 ^та? он2 | с. НМ к. с. е-ю-3 Рн50%
СНз—С—с—сн3 II II HON NOH диметилглиоксим pKi=10,55 pK2= 12,60 17,40* 240 262 327 374 24,5 4,58 3,43 4,69
СБНБ-С-С-СвНБ II II HON NOH а-бензилдиоксим pKl-10,12 рК2=11,73 25,32** 260 273 361 406 50,0 9,9 10,9 2,46
Н2С —СН2 —C=NOH Н2С—СН2—CH^C=NOH диоксим 1,2-циклогептандиона (гептоксим) pK1=10,65 рК2=12,16 21,80* 220 263 330 374 24,9 5,05 4,05 2,67
СН2 — C=NOH Н2с/ \c=NOH СН2-СН2 диоксим 1,2-циклогександиона (ниоксим) рК]=9,80 рК2=12,16 21,59* 220 265 334 383 24,8 5,02 3,57 2,90
НС—СН НС—СН 1 Lc_ с-1 1 II || хк HON NOH а-фурилдиоксим рК1= 9,80 рК2-11,25 14,90* 265 293 384 438 51,0 10,4 19,0 5,20
* Значение 1g ₽2 получено методом экстракции.
** Значение 1g получено методом растворимости
с этими тремя реагентами, а также комплекс никеля с диметилглиокси-мом не разрушаются при обработке экстрактов раствором щелочи, применяемой для разрушения соответствующих комплексных соединений кобальта и меди, часто сопутствующих никелю. Действием щелочи изб] jtok этих реагентов удаляется из слоя органического растворителя и можно измерять поглощение только комплексного соединения в ультрафиолетовой области спектра, где 8 имеет более высокое значение.
Комплексное соединение никеля с а-фурилдиоксимом менее устойчиво; оно разрушается при удалении избытка реагента щелочью, поэтому в ультрафиолетовой области проводить измерение поглощения а-фурилдиоксимата никеля невозможно. Тем не менее указанный реагент является ценным для фотометрического определения никеля, так как имеется возможность проводить измерение оптической плотности в видимой области спектра, К 438 нм, 8 = 1,9 • 104, реагент в этой об
ласти спектра не поглощает. Предварительное отделение никеля в виде диметилдиоксимата методом экстракции позволяет отделять его от больших количеств Си, Со, Fe, Сг, Мп и А1. Для устранения мешающего влияния легко гидролизующихся элементов экстракцию проводят в присутствии солей лимонной и винной кислот.
А. Определение никеля в сталях а-фурилдиоксимом
Реагенты
1. Стандартный раствор соли никеля, содержащий 0,001 мг/мл Ni. Готовят разбавлением 10 мл раствора А (стр. 184) до 1 л. Раствор пригоден в день его приготовления.
2. а-Фурилдиоксим, 0,2%-ный водный раствор (пригоден в течение недели).
3. Фосфатный буферный раствор, pH 9,6.
4. Хлороформ.
5. Азотная кислота, раствор (х. ч.), (пл. 1,4), раствор 1 : 5.
6. Едкий натр, 10%-ный раствор.
7. Аммиак, раствор 1 : 50.
8. Уксусная кислота, 2 н. раствор.
9. Винная кислота, 25%-ный водный раствор.
10. Соляная кислота (х. ч.), пл. 1,19.
Для приготовления эталонных растворов берут пять делительных воронок, вводят в каждую стандартный раствор, чтобы содержание никеля (мг) соответствовало:
1-й ряд 0,005 0,007 0,01 0,025 0,05 2-й ряд 0,025 0,05 0,075 0,10 0,20
Добавляют 5 мл раствора а-фурилдиоксима, 10 мл буферной смеси и доводят pH раствора до 8,5—9,5 по универсальной индикаторной бумаге, прибавляя по каплям раствор NaOH; если раствор будет иметь щелочную реакцию >9,5, добавляют несколько капель уксусной кислоты. Дают раствору постоять 15 мин и образовавшееся соединение диоксимата никеля экстрагируют в течение 10 мин на механическом вибраторе двумя порциями хлороформа по 5 мл каждая. Перед второй экстракцией добавляют в воронку еще 5 мл раствора а-фурилдиокси-ма. Обе порции хлороформного раствора сливают в градуированную пробирку емкостью 10 мл или мерную колбу емкостью 25 мл и доливают до метки хлороформом. Измерение оптической плотности эталонных растворов производят на фотоэлектроколориметрах ФЭК-56, ФЭК-60 или спектрофотометрах при К 438 нм (рис. 55) и строят градуировочный график. В качестве раствора сравнения используют хлороформ, которым обработан раствор «холостого опыта».
Примечание. При содержании никеля от 0,05 мг и выше экстракцию проводят из 15—20 мл водного раствора, беря первую порцию растворителя 10 мл, и делая повторную обработку водного раствора еще двумя порциями (по 5 мл) растворителя. Все порции растворителя сливают в мерную колбу емкостью 25 мл и доводят хлороформом объем раствора до метки.
Для определения никеля в стали берут две навески по 0,1 г, переносят каждую в коническую колбу емкостью 50 мл, растворяют в 10 мл азотной кислоты, нагревают раствор на песчанной бане до удаления окислов азота, выпаривают его до объема 2—3 мл, переводят в мерную колбу емкостью 50 мл, доводят раствор водой до метки. К 1—2 мл приготовленного раствора (в зависимости от содержания никеля), помещенного в делительную воронку, добавляют 10 мл раствора а-фурилдиоксима, 10 мл раствора фосфата натрия и доводят pH раствора до 8,5—9,5 по универсальной индикаторной бумаге, добавляя по каплям раствор NaOH; если pH > 9,5, нужно добавить несколько капель уксусной кислоты. Раствор оставляют стоять 15 мин.
Рис. 55. Спектр поглощения соединения — Ni с а-фурил-диоксимом в СНС1з
Экстрагирование образовавшегося соединения диоксимата никеля см. приготовление эталонных растворов стр. 188.
Содержание никеля находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики.
Б. Определение никеля а-фурилдиоксимом в чистом алюминии
Приготовление эталонных растворов см. на стр. 188. Содержание никеля в эталонных рядах (мкг):
1-й ряд 0,05 0,1 0,2 0,3 0,5
2-й ряд 0,5 0,7 0,9 1,1 1,5
Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения никеля в алюминии берут две навески по 2,5 г, помещают каждую в тефлоновую или кварцевую чашку емкостью 100 мл и растворяют в 20—25 мл концентрированной соляной кислоты, добавляя ее небольшими порциями, прикрывая чашку часовым стеклом. Раствор выпаривают почти досуха, добавляют 40 мл 25%-но-го раствора винной кислоты и переносят в мерную колбу емкостью 50 мл, смывая чашку двумя порциями воды по 4 мл и доводят объем раствора водой до метки. Переносят в две делительные воронки по 20 мл этого раствора, добавляют в каждую 10 мл буферной смеси, 5 мл раствора а-фурилдиоксима. После этого измеряют pH раствора по универсальному индикатору. Если pH раствора будет меньше или больше 8,5—9,5, то добавляют щелочь или уксусную кислоту. Через 15 мин полученное соединение никеля экстрагируют двумя порциями (по 2,5 мл) хлороформа. Перед прибавлением второй порции еще добавляют 5 мл раствора а-фурилдиоксима. Содер-
Жймое боронки встряхиваю!4 в течение 15 мин после прибавлений каждой порции хлороформа на механическом вибраторе. Объединенные экстракты промывают 10 мл раствора аммиака (1 : 40) и после этого промывают 10 мл воды, перемешивая содержимое воронки в течение 1—2 мин. Далее поступают согласно описанию определения никеля в стали (стр. 188). В зависимости от содержания никеля используют тот или другой эталонный ряд, измеряя оптическую плотность растворов, см. приготовление эталонных растворов (стр. 188). Содержание никеля находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики [46].
В. Определение никеля а-фурилдиоксимом в галлии
Приготовление эталонных растворов см. стр. 188.
Примечание. При использовании 1-го ряда для экстракции берут две порции хлороформа по 2,5 мл каждая; применяют кювету с толщиной поглощающего слоя 5 или 10 см. Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения никеля в галлии берут две навески по 2,5 г металлического галлия, помещают в тефлоновую или кварцевую чашку емкостью 100 мл и растворяют осторожно в смеси 5 мл соляной и 15 мл азотной кислот, прикрывая чашку часовым стеклом. После растворения полученный раствор упаривают почти досуха на водяной бане. Остаток растворяют в 20—40 мл 25%-ного раствора винной кислоты и переносят в мерную колбу емкостью 50 мл, смывая чашку двумя порциями воды по 4 мл и доводят объем раствора водой до метки. Переносят в две делительные воронки по 20 мл этого раствора. К раствору, в каждую делительную воронку добавляют 10 мл буферной смеси, 5 мл раствора а-фурилдиоксима. После этого проводят все операции, указанные при определении никеля в алюминии (стр. 189). Содержание никеля находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают с применением метода математической статистики.
Г. Определение никеля а-фурилдиоксимом в двуокиси кремния
При определении никеля в двуокиси кремния основная масса кремния удаляется плавиковой кислотой в виде H2SiF6 и SiF4.
Кроме указанных на стр. 188, необходимы реагенты:
1. Плавиковая кислота (ч. д. а.), концентрированная, очищенная перегонкой в тефлоновом приборе.
2. Серная кислота (х. ч.), раствор 1:1.
Приготовление эталонных растворвв (см. стр. 188).
Для определения никелявдвуокиси кремния берут три навески по 2 г SiO2, помещают каждую в тефлоновую чашку и обрабатывают плави
ковой кислотой (около 25 мл) с несколькими каплями серной кислоты. Затем раствор выпаривают примерно до объема одной капли на воздушной бане при 80° С. После выпаривания остаток растворяют в 5 мл винной кислоты и раствор нейтрализуют в чашке раствором щелочи до pH 8,5—9,5 по индикаторной бумаге. Если pH > 9,5, добавляют несколько капель уксусной кислоты. Полученный раствор переносят в делительную воронку емкостью 50 мл, промывая чашку 5—10 мл воды. Затем добавляют 5 мл раствора а-фурилдиоксима и оставляют стоять 15 мин. После этого экстрагируют комплексное соединение никеля двумя порциями (по 2,5 мл каждая) хлороформа. Содержимое воронки встряхивают в течение 15 мин после прибавления каждой порции хлороформа, используя механический вибратор. Хлороформные экстракты объединяют и промывают их 10 мл раствора аммиака.
Полученные хлороформные растворы сливают в мерные цилиндры емкостью 5 мл и доводят хлороформом объем до 5 мл. Полученный раствор помещают в кювету и измеряют его оптическую плотность при X 438 нм. В качестве раствора сравнения используют хлороформ, которым был обработан раствор «холостого опыта».
Содержание никеля находят по градуировочному графику. Результаты трех параллельных определений обрабатывают с применением метода математической статистики.
Д. Определение никеля гептоксимом в индии
Гептоксим имеет преимущество перед другими а-диоксимами, так как позволяет проводить определение никеля в большом интервале pH 4—12.
Кроме указанных на стр. 188, необходимы реагенты:
1. Азотная кислота (х. ч.1, пл. 1.4.
2. Гептоксим, 0,2%-ный раствор.
3. Едкий натр, 6 н. и 1 н. растворы.
4. Ацетатный буферный раствор, pH 4; в мерную колбу емкостью 250 мл берут 50 мл 1 в. раствора ацетата натрия, 40 мл 1 н. НС1 и доводят объем раствора водой до метки.
5. НС1 (о. ч.) 3 н. раствор.
6. H2F2 (о. ч.), концентрированная.
7. Тетраборат натрия, насыщенный раствор.
8. Фторид аммония, 10%-ный раствор.
9. Ацетат аммония, 50-%-ный раствор.
10. Серная кислота, 2 н. раствоп.
Для приготовления эталонных растворов в делительные воронки емкостью 50 мл помещают 5 мл стандартного раствора никеля в количестве (мкг):
1-й ряд 0,05 0,1 0,2 0,3 0.5
2-й ряд 0,5 0,7 0,9 1,1 1,5
Добавляют 3 мл раствора гептоксима, доводят pH до 8,2—8,5 6 н. раствором щелочи и выдерживают 30 мин. После этого экстрагируют двумя порциями (по 2,5 мл каждая) хлороформа. Содержимое воронок встряхивают в течение 15 мин на механическом вибраторе после при
бавления каждой порции хлороформа. После отделения объединяют хлороформные экстракты, промывают раствор хлороформа 10 мл 1 н. раствора щелочи в течение 15 мин и водой (3—5 мин), используя механический вибратор. Сливают слой хлороформа в сухую градуированную пробирку емкостью 10 мл, доводя объем раствора до 10 мл, и измеряют оптическую плотность экстракта при X 263 нм в тефлоновых кюветах (/ = 10 см). В качестве раствора сравнения используют хлороформ, которым обрабатывают раствор «холостого опыта». Строят градуировочный график по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения никеля в индии берут три навески по 0,5 г металла, помещают каждую в тефлоновую или кварцевую чашку емкостью 70— 100 мл и растворяют в 5 мл азотной кислоты. Раствор выпаривают почти досуха, добавляют к остатку 10 мл раствора винной кислоты при нагревании. После охлаждения добавляют 3 мл раствора гептоксима и доводят pH раствора до 8,2—8,5 6 н. раствором щелочи. Полученный раствор переносят в делительную воронку емкостью 50 мл и оставляют стоять в течение часа. После этого производят экстракцию тремя порциями (по 2,5 мл каждая) хлороформа. Содержимое воронки встряхивают на механическом вибраторе в течение 15 мин после прибавления каждой порции хлороформа. После отделения хлороформные экстракты объединяют и промывают 10 мл 1 н. раствора щелочи в течение 15 мин и водой (3—5 мин), используя механический вибратор. Сливают слой хлороформа в сухую пробирку емкостью 10 мл и измеряют поглощение органического экстракта при X 263 нм в тефлоновых кюветах (/ = 10 см). Приготовление раствора сравнения см. выше. Содержание никеля находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают с применением метода математической статистики [46].
Е. Определение никеля гептоксимом в карбонате кадмия
Для приготовления эталонных растворов берут пять делительных воронок емкостью 50 мл, помещают в каждую 5 мл воды стандартный раствор никеля в количестве (мкг): 0,5; 0,7; 0,9; 1,1; 1,5, добавляют 3 мл гептоксима, 10 мл ацетатного буферного раствора и оставляют стоять. Через 30 мин добавляют 5 мл хлороформа и содержимое воронки встряхивают в течение 15 мин на механическом вибраторе; водную фазу отбрасывают. Экстракт промывают 10 мл 1 н. раствора едкого натра в течение 15 мин и один раз водой при встряхивании на механическом вибраторе, после этого переводят его в сухую градуированную пробирку. Оптическую плотность экстракта измеряют в тефлоновой кювете (/ = 10 см) при Х263 нм на спектрофотометрах различных марок. Раствором сравнения служит хлороформ, которым обработан раствор «холостого опыта». Строят градуировочный график по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения никеля в карбонате кадмия две. навески по (2,5 г) растворяют каждую в конической колбе емкостью 100 мл в 15 мл 3 н. НО, раствор переносят в мерную колбу емкостью 50 мл, смывая
колбу двумя порциями воды по 4 мл и доводят объем раствора водой до метки. Берут две порции по 10 мл, переносят каждую в делительную воронку, нейтрализуют 6 н. раствором NaOH до pH 3—4 по индикаторной бумаге, добавляют 10 мл ацетатного буферного раствора и оставляют стоять 30 мин. В дальнейшем проводят определение никеля в условиях см. приготовление эталонных растворов (стр. 192). Содержание никеля находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики [47].
Ж. Определение никеля гептоксимом в цирконилнитрате
Приготовление эталонных растворов см. на стр. 192.
Для определения никеля в анализируемом образце в тефлоновые чашки берут три навески по 1 г, растворяют каждую при нагревании на плитке, закрытой асбестом, в 4—5 мл H2Fa и выпаривают раствор досуха. Сухой остаток переносят в делительную воронку при помощи 10 мл 2 н. H2SO4 и 10 мл раствора тетрабората натрия, затем добавляют 10 мл раствора фторида аммония, 50 мл раствора ацетата аммония, 1 мл раствора гептоксима и оставляют стоять 30 мин. После этого производят экстракцию 5 мл хлороформа, встряхивая содержимое воронки на механическом вибраторе в течение 15 мин. Органическую фазу промывают 10 мл 1 н. раствора щелочи в течение 15 мин при встряхивании на механическом вибраторе и 1 раз водой в тех же условиях. Оптическую плотность экстракта измеряют при А 263 нм. Содержание никеля находят по градуировочному графику (см. стр. 192). Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики [47].
3. Определение никеля ииоксимом в нитрате кобальта
Содержание больших количеств кобальта осложняет непосредственное определение никеля а-диоксимами, поэтому необходимо предварительно разделение этих элементов. Для этого используют метод отделения больших количеств кобальта в виде роданида экстракцией этилацетатом. Никель определяют ниоксимом турбидиметрическим методом или фотометрически, используя хинолин для растворения ниоксимата никеля [481. Метод был доработан в лаборатории спектрофотометрии кафедры аналитической химии МГУ.
Кроме указанных ранее, необходимы реагенты:
1. Роданид аммония, 50%-ный раствор.
2. Соляная кислота (х. ч.), 5 н. раствор.
3. Едкий натр (х. ч.), 2 н. раствор.
4. Диоксим 1,2-циклогексадион (ниоксим), перекристаллизованный из воды, 0,05 М водный раствор.
5. Этилацетат, перегнанный.
6. Аммиак, (х. ч.), водный, концентрированный.
7. Толуол.
Для приготовления эталонных растворов в делительные воронки емкостью по 100 мл вводят в каждую 20 мл воды, стандартный раствор, содержащий никель в количестве (мкг): 0,0; 1,0; 2,5; 5,0; 7,0; 10,0; доводят pH раствора до 8—10 по универсальной индикаторной бумаге добавлением раствора едкого натра, вносят 1 мл раствора ниоксима и оставляют стоять на 15 мин. Затем добавляют 5 мл хлороформа и встряхивают содержимое воронок в течение 10 мин на механическом вибраторе. Хлороформный слой отделяют и фотометрируют при X 263 нм на спектрофотометрах различных марок. Для получения раствора сравнения используют хлороформ (первая воронка). Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения никеля в нитрате кобальта берут две навески соли по 2,5 г, переносят их в мерные колбы емкостью 50 мл и добавляют 40 мл соляной кислоты, растворяют соль и доводят объем раствора до метки той же кислотой. Две порции по 20 мл переносят каждую в делительную воронку емкостью 100 мл, прибавляют 10 мл раствора роданида аммония и экстрагируют роданид кобальта 50 мл этилацетата в течение 5 мин на механическом вибраторе. Значение pH водной фазы не должно быть >2 по универсальной индикаторной бумаге, в противном случае добавляют по каплям соляную кислоту. После отделения экстракта добавляют к водному раствору 25 мл этилацетата и вновь проводят экстракцию. Эту операцию повторяют трижды, добавляя по 2 мл раствора роданида аммония перед каждой экстракцией, следя, чтобы значение pH водной фазы не превышало 2. После экстракции к водному раствору добавляют раствор щелочи до pH 8—10 по униьерсальной индикаторной бумаге, затем добавляют 5 мл раствора ниоксима. Через 10 мин добавляют 10 мл толуола и встряхивают содержимое воронки на механическом вибраторе в течение 5 мин. Водный слой отбрасывают, органическую фазу промывают трижды 5 мл раствора аммиака и последовательно двумя порциями воды по 5 мл каждая. После этого к органическому слою добавляют 5 мл соляной кислоты и встряхивают содержимое воронки на механическом вибраторе в течение 10 мин. Водный слой сливают в другую делительную воронку, а оставшуюся органическую фазу промывают дважды 5 мл воды. Промывание воды объединяют с водной фазой и добавляют раствор щелочи до pH 8—10, вносят 1 мл раствора ниоксима и образовавшееся соединение никеля экстрагируют хлороформом в условиях (см. приготовление эталонных растворов). Для определения содержания никеля берут данные (не менее четырех) из параллельных опытов и обрабатывают их методом математической статистики.
И. Определение никеля кинетическим методом
Для определения микроколичеств никеля используют катализируемую им реакцию окисления дифенилкарбазона перекисью водорода в боратном буферном растворе при pH 8,9 [49].
Содержание никеля определяют по способу тангенсов дифференциального варианта кинетического метода.
Реагенты
1. Стандартный раствор соли никеля с содержанием 1 мкг/мл Ni. Готовят разбавлением исходного раствора (см. стр. 184).
2. Дифенилкарбазон (ч. д. а.), 4 • 10-4 М спиртовой раствор. Препарат перекристаллизовывают из этанола до получения раствора с постоянным значением оптической плотности.
3. Перекись водорода (о. ч.), 0,9 М раствор. Точную концентрацию перекиси водорода устанавливают титриметрически каждые 5—7 дней.
4. Боратный буферный раствор, pH 8,84. Для приготовления 1 л буферного раствора смешивают 300 мл 0,2 М раствора борной кислоты и 700 мл 0,05 М раствора тетрабората натрия. Тетраборат натрия (х. ч.) и борную кислоту (х. ч.) дважды перекристаллизовывают из воды. Все растворы готовят на деионизованной воде.
Для построения градуировочного графика в кварцевый смеситель с тремя отростками вводят: в первый отросток 1 мл перекиси водорода, во второй — 1 мл дифенилкарбазона и стандартный раствор никеля в объеме не более 1 мл с содержанием (мкг) от 0,01 до 0,1, в третий такое количество буферного раствора, чтобы общий объем раствора во всех смесителях был равен 10 мл.
Растворы термостатируют при 25 ± 0,1° С в течение 20—30 мин В момент смешения растворов включают секундомер и измеряют оптическую плотность растворов через 1 мин в течение 10 мин на приборах ФЭК-М, ФЭК-54, ФЭК-57 с термостатируемой камерой для кювет или ФЭК-Н-52 с термостатируемым корпусом X 420 нм. По результатам измерений строят график в координатах оптическая плотность — время (см. стр. 82).
Тангенсы углов наклона полученных прямых (tg а) пропорциональны концентрации никеля в интервале 0,1—1 мкг в 10 мл. По градуировочному графику в координатах тангенс угла накона прямой — концентрация никеля находят его содержание.
Для определения никеля в растворе его соли 1 мл испытуемого раствора помещают в один из отростков смесителя. В два другие отростка добавляют реагенты, указанные для построения градуированного графика, и после термостатирования и смешения растворов измеряют оптическую плотность раствора во времени и строят график оптическая плотность — время. Находят тангенс угла наклона полученной прямой. По градуировочному графику определяют концентрацию никеля, соответствующую полученному значению tg а [49].
Определение рения
Для определения рения известен ряд спектрофотометрических методов, в которых используют образование комплексных соединений с а-диоксимами, преимущественно с а-фурилдиоксимом [50] мочевиной и ее производными и роданидами. Заслуживают внимания методы определения рения в виде ионных ассоциатов перренат — основные красители [51].
ОПРЕДЕЛЕНИЕ РЕНИЯ а-ФУРИЛДИОКСИМОМ
Метод определения рения а-фурилдиоксимом отличается большой чувствительностью и избирательностью. Молибден, вольфрам и ванадий, обычно сопутствующие рению в природных соединениях и сплавах, в соответствующих условиях не мешают определению малых количеств рения а-фурилдиоксимом. Соединение рения с а-фурилдиоксимом, полученное в присутствии хлорида олова (II) и ацетона (24--26 об. %), при кислотности 0,6—1,0 н. НО поглощает при Хтах 530 нм; е = 4,3 • 104. Раствор реагента в ацетоне поглощает в УФ-области спектра (220—330 нм) и не мешает измерению оптической плотности комплексного соединения рения.
Оксикислоты образуют комплексные соединения с рением, имеющим степень окисления ниже семи. При изучении влияния оксикислот на реакцию образования комплексного соединения рения с а-фурил-диоксимом было установлено [52], [53], что максимальная окраска в их присутствии развивается в течение 15 мин; без добавления оксикислот необходимо 45 мин. Видимо, оксикислоты (щавелевая или винная), фиксируя определенную степень окисления рения (IV), ускоряют образование соединения рения с а-фурилдиоксимом. При определении рения в присутствии больших количеств молибдена с добавлением винной кислоты рекомендуется использовать вариант дифференциального метода (стр. 71), при котором большие количества молибдена не мешают определению рения.
Реагенты
1. Стандартный раствор соли рения; 0,155 г. перрената калия KReO4 (х. ч.) растворяют в воде в мерной колбе емкостью 250 мл — раствор А; 1 мл раствора А содержит 0,1 мг/мл Re; 10 мл раствора А разбавляют водой до 100 мл —раствор Б: 1 мл раствора Б содержит 0,01 мг Re; годен к употреблению в течение 1—2 дней.
2. а-Фурилдиоксим, 0,015 А1 раствор в ацетоне.
3. Соляная кислота, 5 н. раствор.
4. Изоамиловый спирт.
5. Ацетон, свежеперегнанный.
6. Хлороформ, свежеперегнанный.
7. Хлорид олова (II), 10%-ный раствор в 5 н. соляной кислоте.
8. Винная кислота, 20%-ный водный раствор.
9. Хлорид бария, 10%-ный водный раствор.
А. Определение рения в растворе его соли
Для приготовления эталонных растворов берут четыре делительных воронки емкостью 50—60 мл, вводят в каждую 5 мл воды, стандартный раствор рения в количестве (мг): 0,005; 0,01; 0,02; 0,03 соответственно, добавляют 3 мл соляной кислоты, 10 мл винной кислоты, 6,5 мл раствора а-фурилдиоксима в ацетоне, 2,5 мл хлорида олова (И), разбавляют раствор водой до объема 25 мл и через 45 мин экстрагируют комплексное соединение рения двумя порциями хлороформа (по 6 мл), встряхивая в течение 1 мин каждую порцию раствора.
После разделения слоев слой хлороформа переносят в мерную колбу емкостью 25 мл, в которую предварительно добавлено 10 мл изоамилового спирта, доводят объем раствора до метки изоамиловым спиртом и фотометрируют при X 580 нм; раствором сравнения является смесь изоамилового спирта и хлороформа (1 : 1). Измерение оптической плотности растворов производят на фотоэлектроколориметрах ФЭК-56, ФЭК-57, ФЭК-60 или спектрофотометрах любой марки. По данным измерения оптической плотности А строят градуировочный график.
Для определения рения в испытуемом растворе из общего объема 25 мл берут 5 мл этого раствора, помещают в делительную воронку и проводят все операции, указанные при приготовлении эталонных растворов; содержание рения находят по градуировочному графику.
Б. Определение рения в присутствии молибдена
Рений в природных соединениях обычно сопутствует молибдену. Описанный метод может быть применен для определения рения в присутствии молибдена. Молибден в малых концентрациях не мешает определению рения. Для соотношений Re : Мо, равных 1 : 1, 1 : 5, 1 : 10 : 1 : 15, рений определяется количествено в интервале концентраций 6—30 мкг.
Приготовление эталонных растворов см. на стр. 196. В эталонные растворы вводят рений в количествах (мг):
1-й ряд 0,005 0,0075 0,01 0,015
2-й ряд 0,015 0,02 0,025 0,03
Измерение оптической плотности эталонных растворов проводят в условиях см. стр. 196.
Для определения рения в присутствии молибдена из общего объема 25 мл берут 5—7 мл водного раствора, содержащего рений (VII) и молибден (VI) и помещенного в делительную воронку, добавляют 3 мл соляной кислоты, 10 мл винной кислоты, 6,5 мл раствора а-фурилдиоксима в ацетоне, 2,5 мл раствора хлорида олова, разбавляют раствор до объема 25 мл водой, экстрагируют комплексное соединение рения двумя порциями хлороформа (по 6 мл), добавляют изоамиловый спирт и фотометрируют в условиях, указанных на стр. 196.
Концентрацию рения в растворе находят графическим методом [53].
В. Определение рения в присутствии больших количеств молибдена дифференциальным спектрофотометрическим методом
Для определения рения в присутствии больших количеств молибдена следует использовать вариант дифференциального метода (стр. 71).
Для определения рения в молибденовой руде берут две навески по 2г руды (молибденового или медного концентрата), помещают каждую в фарфоровый тигель, добавляют 0,2 г КС1О3, 3 г СаО и содержимое тигля осторожно перемешивают. Тигель помещают в тигельную печь,
температуру которой постепенно доводят до 650' С и нагревают 2 ч при 650 ± 10° С. Затем тигель вынимают и охлаждают. При охлаждении спек выщелачивают 150 мл горячей воды в стакане емкостью 300 мл при постепенном и тщательном перемешивании раствора. Затем содержимое стакана упаривают на песчаной бане до объема 80 мл, осадок отфильтровывают в горячем состоянии на небольшой воронке Бюхнера. Осадок молибдата кальция промывают двумя небольшими порциями воды. Фильтрат переносят в стакан емкостью 150 мл, приливают 5 мл 10%-ного раствора хлорида бария для осаждения ионов сульфата, которые могут присутствовать в растворе. Раствор упаривают на песчаной бане до объема 20—30 мл, фильтруют через фильтр с синей лентой в мерную колбу емкостью 50 мл, охлаждают и доводят объем раствора водой до метки. При обработке руды большая часть молибдена отделяется [54].
Для определения рения берут две порции по 10 мл (цж) приготовленного раствора, одна из них имеет концентрацию сх и вторая с той же концентрацией искомого элемента, но к ней добавляют известное количество стандартного раствора Re (Са), например 0,01—0,02 мг Re. Для приготовления раствора сравнения берут 9 мл (ц0) анализируемого раствора. Ко всем трем растворам добавляют по 10 мл винной, кислоты, 6,5 мл раствора а-фурилдиоксима в ацетоне, 4 мл раствора SnCl2 и воды (из бюретки) до объема 25 мл. Через 15 мин проводят экстракцию окрашенного комплексного соединения двумя порциями хлороформа (3+4 мл) в течение 1—2 мин. Хлороформные экстракты собирают в мерные колбы объемом 25 мл и доводят объем раствора до метки изоамиловым спиртом (небольшое количество изоамилового спирта следует прилить в колбы перед сливанием хлороформного слоя). Измеряют оптические плотности двух растворов Аж и Аж + а относительно раствора сравнения при X 530 нм. Содержание рения определяют по формуле сх = (AxcjAa) (vx/vx — v0). Данные не менее четырех определений обрабатывают методом математической статистики.
Г. Определение рения в сплавах рений-вольфрам
Кроме указанных на стр. 196, необходимы реагенты:
1. Азотная кислота (х. ч.), пл. 1,4.
2. Плавиковая кислота (х. ч.) концентрированная.
3. Едкий натр (х. ч.), 50%-ный раствор.
Две навески сплава по 0,2 г (содержание рения около 20%) или 0,8 г (при содержании около 5%) растворяют каждую в тефлоновой чашке в смеси 10 мл HF и 5 мл HNO3 (азотную кислоту добавляют по каплям). После растворения раствор упаривают на водяной бане досуха, осадок растворяют в воде с добавлением 2,5 мл щелочи, 10 мл винной кислоты (из расчета 1 мл на 20 мл W) и разбавляют раствор водой до объема 200 мл. Этот раствор дополнительно разбавляют в 10 раз и в нем уже проводят определение рения. Для этого берут две порции по 3 мл испытуемого раствора, к одной из них добавляют 0,01 мг рения. Для приготовления раствора сравнения берут 2 мл испытуемо
го раствора. Далее определение ведут так же, как указано при определении рения в присутствии молибдена, с тем лишь различием, что экстрагируют соединение рения через 20—30 мин после прибавления всех реагентов.
Д. Определение рения при наличии ванадия
Наличие 10 000-кратного избытка ванадия не мешает прямому определению рения. Поэтому приготовление эталонных растворов и определение рения проводят в условиях, указанных на стр. 196 [55].
ОПРЕДЕЛЕНИЕ РЕНИЯ ТИОМОЧЕВИНОИ
В РАСТВОРЕ ЕГО СОЛИ
Комплексное соединение рения с тиомочевиной образуется в водном растворе при соотношении компонентов: Re(IV): Thio= 1 :4 и ему нужно предположительно приписать формулу [ReO (ThiO)4] Cl2 [55]: В зависимости от условий образования могут существовать комплексы. [ReO (ОН) (Thio)4] Cl и [Re (ОН)2 (Thio)4] С12. В качестве восстановителя используют хлорид олова (II). В спектре поглощения комплексного соединения наблюдается два максимума: X 390 нм, е = 10,5 X X 103 и А 450 нм, е = 6,9 • 103.
Реагенты
1. Стандартный раствор соли рения, содержащий 0,1 мг/мл Re. Более разбавленные растворы готовят в день употребления.
2. Соляная кислота (х. ч.), 10 н. раствор.
3. Тиомочевина, 5%-иый водный раствор.
4. Хлорид олова, 30%-ный раствор в 10 н. соляной кислоте.
Для приготовления эталонных, растворов в пять мерных колб емкостью 25 мл вводят стандартный раствор соли рения в количестве (мг): 0,1; 0,25; 0,5; 0,75; 1,0 соответственно, добавляют в каждую 6 мл раствора соляной кислоты, 3 мл раствора тиомочевины, 1,5 мл раствора хлорида олова и доводят объем раствора водой до метки. Измерение оптической плотности проводят через 45 мин при X 390 нм на фотоэлектроколориметрах ФЭК-60, ФЭК-57, ФЭК-56 и строят градуировочный график. В качестве раствора сравнения используют воду.
Для определения рения в испытуемом растворе из общего объема 25 мл берут 5 или 10 мл его и проводят все операции в условиях, указанных при приготовлении эталонных растворов. Содержание рения находят по градуировочному графику.
ОПРЕДЕЛЕНИЕ РЕНИЯ С 1-ФЕНИ Л ТИОСЕМИКАР БАЗ ИД ОМ
В РАСТВОРЕ ЕГО СОЛИ
1-Фенилтиосемикарбазид служит реагентом для образования комплексного соединения с рением и является восстановителем для получения Re (IV). Оптимальные условия для образования комплексного Соединения — 8 и. соляная кислота. В спектре поглощения комплекс-
него соединения наблюдается один максимум при % 365 нм, е = = 1,59 • 104. Определению рения не мешают V, Ni, Со; при наличии Мо и W возможно определение при соотношении Re : Mo (W) = 1 : 10.
Кроме указанных на стр. 199, необходим реагент—1-фенилтио-семикарбазид, 4 • 10~3 714 водко-спиртовой раствор (10 : 1).
Для приготовления эталонных растворов в пять мерных колб емкостью 25 мл вводят стандартный раствор, содержащий рений в количестве (мкг): 0,5; 1,0; 2,5; 5,0; 7,5; 10,0 соответственно, добавляют в каждую 20 мл НС1, 3 мл раствора реагента и доводят объем раствора водой до метки. Через 40 мин измеряют оптическую плотность эталонных растворов при X 365 нм на фотоэлектрокилориметрах ФЭК-56, ФЭК-57, ФЭК-60 или спектрофотометрах и строят градуировочный график.
Для определения рения е испытуемом растворе из общего объема 25 мл берут 1—2 мл его (в зависимости от содержания рения) и проводят все операции в условиях, указанных для приготовления эталонных растворов.' Содержание рения находят по градуировочному графику.
Определение редкоземельных элементов
Редкоземельные элементы обладают весьма близкими химическими свойствами и при отделении их от других элементов практически всегда выделяются в виде суммы соединений всех редкоземельных элементов (например, оксалатов или фторидов). Для разделения и выделения отдельных элементов этой группы используют различные химические и физико-химические методы. Для определения отдельных редкоземельных элементов в их смеси наряду с некоторыми физическими методами используют спектрофотометрические методы.
Все спектрофотометрические методы анализа редкоземельных элементов можно разделить на две группы:
1. Методы определения суммы редкоземельных элементов. Для этого используют реакции, в которых эти элементы проявляют одни и те же химические свойства. Растворы образующихся соединений имеют одинаковые характеристики спектров поглощения (Хтах, етах).
2. Методы определения индивидуальных редкоземельных элементов в смеси. В этих методах используют различия в свойствах следующих соединений:
а) общий вид спектров растворов аква-комплексов различных редкоземельных элементов и расположение в них максимумов поглощения имеют строго индивидуальный характер (рис. 56) и узкополосную структуру (они приближаются по своему виду к спектрам поглощения газообразных веществ). По этим спектрам можно провести качественный анализ и количественное определение ряда элементов этой группы, используя максимумы поглощения, свободные от наложения максимумов других редкоземельных элементов [561—[591. Для этого применяют спектрофотометры с достаточно высокой монохроматизацией потока излучения, иначе невозможно разрешить очень близко расположенные (на расстоянии 5--10 нм) узкие полосы поглощения.
Недостатком метода является его малая чувствительность (е не превышает 10) и отсутствие характерных полос поглощения в УФ, видимой и ближней ИК-областях спектра у некоторых элементов этой группы (La, Lu), в результате чего они не могут быть определены. В присутствии церия, имеющего широкие и интенсивные полосы поглощения в УФ-области, невозможно определить Eu, Gd, Dy, Tb, полосы поглощения которых также лежат в УФ-области, что требует предварительного отделения церия;
б) если при образовании комплексных соединений с неорганическими и органическими лигандами сохраняется индивидуальность полос поглощения, то в растворах этих комплексных соединений можно определять отдельные редкоземельные элементы. При этом может наблюдаться некоторое смещение Хтах и увеличение интенсивности индивидуальных полос поглощения; последнее приводит к увеличению чувствительности определения этих элементов. Такие лиганды должны быть «бесцветны»* и их комплексы с редкоземельными элементами могут быть растворимы или в воде, или в органических растворителях. Для повышения чувствитепьности определения редкоземельных элементов используют этилендиаминтетр ауксусную кислоту (ЭДТА) [60] в воде, теноилтрифторацетон (ТТА1 [59] в бензоле, трибутилфосфат (ТБФ) [60] и 5,7-дихлор-8-оксихинолин [60] в хлороформе; 7-диод-8-оксихинолин-5-сульфокислоту [60] в водно-спиртовой среде; тайрон в воде [59].
Эти методы также имеют ряд недостатков: Интенсивность поглощения в максимумах не у всех редкоземельных элементов возрастает в одинаковой степени; напротив, иногда наблюдается ее снижение при комплексообразовании; ввиду значительного поглощения большинства этих реагентов в УФ-области спектра (за исключением ЭДТА) затрудняется или вообще исключается возможность определения ряда элементов, полосы поглощения которых находятся в УФ-области (Се, Gd, Eu, Sm). Следовательно, ни один из этих методов не может быть использован для анализа смеси, содержащей все элементы этой группы. Выбор реагента определяется качественным составом смеси редкоземельных элементов; непригодны такие лиганды, как лимонная, триоксиглутаровая, сульфосалициловая и винная кислоты, образующие смешанные комплексы, одна молекула которых может содержать несколько элементов этой группы, при этом один из них влияют на форму и интенсивность полос поглощения других;
" |в) при образовании комплексов редкоземельных элементов с органическими реагентами, обладающими широкими и интенсивными полосами поглощения в той области спектра, где расположены характерные максимумы поглощения аква-ионов редкоземельных элементов, исключается возможность их наблюдения. В результате комплексообразования максимум полосы поглощения такого реагента обычно претерпевает батохромный или гипсохромный сдвиг и определение проводят по поглощению в этом максимуме; чувствительность определения воз-
* «Бесцветными» называют реагенты, не поглощающее в области максимумов поглощения редкоземельных элементов.
£
YbCl3
820 860 900 940 980 Л,нм
Рис 56. Спектры поглощения растворов солей редкоземельных элементов
растает в 1 • 103—1 • 104 раз. Однако изменение в спектре поглощения реагента, которые происходят под влиянием комплексообразования, мало отличимо для различных редкоземельных элементов, поэтому специфичность определения отдельных элементов этой группы часто теряется, так как величины е и положения максимумов у различных элементов этой группы близки.
В настоящее время намечаются три основных пути использования «окрашенных» органических реагентов для спектрофотометрического определения индивидуальных редкоземельных элементов в их смеси.
Первая группа методов основана на изменении устойчивости комплексных соединений в ряду редкоземельных элементов как с реагентом, с которым проводят определение, так и с реагентом, который используют для маскировки сопутствующих элементов этой группы [611—164].
Вторая группа методов основана на различиях в спектрофотоме^ рических характеристиках соединений редкоземельных элементов с одним и тем же реагентом. Эти различия чаще всего проявляются в значениях молярных коэффициентов погашения. В этом случае для определения индивидуальных редкоземельных элементов могут быть использованы некоторые варианты дифференциальной спектрофотометрии (стр. 65—71).
Сравнительно недавно было обнаружено [64], что при реакциях некоторых производных арсеназо III с элементами иттриевой и цериевой подгрупп редкоземельных элементов образуются соединения, сильно отличающиеся по своим спектральным характеристикам: у элементов цериевой подгруппы наблюдается длинноволновый максимум поглощения в области X 720 нм, который отсутствует в спектрах поглощения элементов иттриевой подгруппы. Были сделаны попытки использовать эти различия для определения редкоземельных элементов одной подгруппы в присутствии другой подгруппы [64].
Третья группа методов основана на использовании различий в скорости образования соединений отдельных редкоземельных элементов с одним и тем же реагентом [65].
Все эти методы пригодны для определения какого-либо одного редкоземельного элемента при наличии другого элемента этой группы (иногда нескольких), расположенного достаточно далеко от определяемого в ряду редкоземельных элементов. Это объясняется тем, что используемые различия в свойствах соединений редкоземельных элементов достаточно ощутимы лишь при значительной удаленности по порядковому номеру элемента друг от друга;
г) ряд редкоземельных элементов склонен проявлять так называемую аномальную степень окисления +2 (европий, самарий, иттербий) и +4 (церий, празеодим, тербий). Более изучены соединения Се (IV), которые устойчивы в растворах. Поэтому церий является наиболее легко определяемым элементом и известно большое число спектрофотометрических методов его определения. Соединения других редкоземельных элементов в аномальных степенях окисления устойчивы лишь в твердом состоянии, поэтому число спектрофотометрических методов их определения весьма ограничено.
Известны методы определения европия [66] по спектрам поглощения аква-иона Eu (II) в присутствии самария и гадолиния. Чувствительность определения европия в виде аква-иона Eu (И) значительно выше, чем в виде аква-иона Eu (III): молярные коэффициенты погашения аква-иона Eu (II) при X 248 и X 320 нм равны 1,21 • 103 и 4,29 • 102 соответственно; молярный коэффициент погашения акваиона Eu (III) при Z, 394 равен 2,7. Кроме того, могут использоваться восстановительные свойства Eu (II) в реакциях с «окрашенными» реагентами, обладающими окислителными свойствами (например, пиридилазонафтолом или пиридилазорезорцином) [67], что тоже приводит к значительному повышению чувствительности определения европия.
Однако получение европия (II) и изучение его спектра поглощения [66] требует специального аппаратурного оснащения.
Спектрофотометрическому анализу (качественному и количественному) смеси редкоземельных элементов обязательно предшествует выделение суммы этих элементов из анализируемого объекта в виде окислов [57], [58], [60].
Методы выделения суммы окислов редкоземельных элементов зависят от их природы [57]. Многие минералы, содержащие редкоземельные элементы, разлагаются только фтористоводородной кислотой и переходят в осадок в виде фторидов; в дальнейшем их переводят в -сульфаты, из растворов которых осаждают их или в виде гидроокисей аммиаком, или оксалатов. Тот и другой осадок после прокаливания дают сумму окислов редкоземельных элементов.
СПЕКТРОФОТОМЕТРИЧЕСКИЙ АНАЛИЗ СМЕСИ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ НА СОДЕРЖАНИЕ ОТДЕЛЬНЫХ КОМПОНЕНТОВ ПО ИХ ИНДИВИДУАЛЬНЫМ СПЕКТРАМ ПОГЛОЩЕНИЯ
А. Качественный анализ смеси
редкоземельных элементов в растворах их аква-комплексов
Спектрофотометрическое определение индивидуальных редкоземельных элементов возможно в растворах, содержащих (в качестве посторонних ионов) в основном только неорганические анионы, поглощающие лишь в далекой УФ-области спектра, так как молярные коэффициенты погашения аква-комплексов редкоземельных элементов очень незначительны (табл. 13, 14) и присутствие посторонних компонентов может исказить результаты определения. Значения молярных коэффициентов погашения несколько меняются в зависимости от природы аниона (табл. 13). После выделения суммы редкоземельных элементов в виде окислов их переводят чаще всего в хлориды, так как хлорид-ион поглощает в далекой УФ-области. Избыток соляной кислоты легко удаляется выпариванием раствора, при этом исключается возможность образования комплексов.
Для проведения качественного анализа в смеси редкоземельных элементов навеску свежепрокаленной смеси их окислов помещают в стакан емкостью 50 мл, смачивают небольшим количеством воды, прибавляют рассчитанное с 10%-ным избытком по отношению к на-
Молярные коэффициенты погашения растворов солей редкоземельных элементов (по Моллеру и Бринтли [58])
Соль ^max* HM e Ширина щели, мм Минимальная концентрация, г/л
РгС13 444,5 9,85 0,025 0,143
Pr(NO3)3 444,5 9,20 0,025 0,153
NdCl3 521,8 4,33 0,013 0,333
Nd(C104)3 521,8 4,30 0,013 0,336
Nd(NO3)3 521,8 3,88 0,013 0,372
Nd(C2H3O2)3 52^,0 3,80 0,013 0,380
SmCl3 402,0 3,14 0,044 0,480
Sm(NO3)3 402,0 3,28 0,044 0,458
Sm(C2H 302)3 402,5 3,68 0,044 0,410
EuCl3 393,9 2,92 0,029 0,520
Eu(NO3)3 394,5 2,05 0,053 0,743
Eu(C2H 302)3 394,7 1,92 0,053 0,793
GdCl3 272,8 2,34 0,205 0,640
Gd(C2HgO2)3 272,9 3,28 0,205 0,479
Er(C104)3 379,2 4,00 0,030 0,419
TmCl3 682,5 2,58 0,018 0,659
Tm(C104)3 682,5 2,49 0,018 0,680
YbCl3 975,0 1,94 0,026 0,901
Yb(NO3)3 975,0 1,91 0,026 0,912
Yb(C104)3 975,0 1,96 0,026 0,885
Yb(C2H3O2)3 972,0 1,99 0,026 0,864
ТАБЛИЦА 14
Молярные коэффициенты погашения солей редкоземельных элементов (по Холлеку и Хартингеру [69])
Соль ^max» HM e Минимальная концентрация, г/л Мешающие элементы
РгС13 444,0 10,50 0,78
NdCl3 575,2 7,11 0,78 Рг
NdCl3 521,9 4,51 1,56 —
SmCl3 401,6 3,49 1,56 Ег
EuCl3 394,3 2,7 1,56 Sm
GdCl3 272,9 3,53 2,9 Sm, Dy, Но, Er
DyCl3 907,5 2,15 3,13 Но, Er, Yb
DyCl3 350,0 2,44 6,25 Но
Hocig 536,8 4,26 0,78 —
HoCl3 640,7 2,81 3,13 Er
ErCl3 523,0 3,04 3,13 Но
ErCl3 487,1 1,92 3,13 Но
TmCl3 682,5 2,52 2,70 Er
TmCl3 777,5 1,07 2,70 —
YbCl3 972,5 1,90 1,56 Er
веске количество соляной кислоты (1:1) и нагревают на водяной бане до полного растворения. Если соляной кислоты окажется недостаточно для полного растворения, то еще прибавляют некоторое ее количество небольшими порциями до тех пор, пока все окислы не растворятся. Этот метод применяют для растворения окислов в отсутствии церия (СеО2 в данных условиях растворяться не будет). После полного растворения упаривают раствор на водной бане до образования влажных солей. Остаток после выпаривания растворяют в воде при слабом нагревании, переносят в мерную колбу емкостью 25 мл и доводят объем раствора до метки водой.
Примечание. В табл. 13 и 14 приведены данные нли для разных соединений одного элемента, или для одного и того же соединения при разных Z.
Снимают спектр поглощения этого раствора по отношению к дистиллированной воде в интервале длин волн от 220 до 1100 нм; первоначально измерения производят через 5—10 нм и в области намечающихся максимумов повторяют измерение оптической плотности через 0,5— 1 нм. Используя спектры поглощения аква-комплексов редкоземельных элементов (см. рис. 56), устанавливают присутствие отдельных элементов в смеси.
Б. Количественное определение отдельных редкоземельных элементов в их смеси в виде аква-комйлексов
В табл. 13 и 14 приведены длины волн и соответствующие им молярные коэффициенты погашения, которые могут быть рекомендованы для количественногого определения редкоземельных элементов в их смеси. При этих длинах волн помехи со стороны других редкоземельных элементов проявляются в наименьшей степени, а молярные коэффициенты погашения имеют наибольшую величину.
Испытуемый раствор соли редкоземельного элемента или смеси элементов этой группы, приготовленный, как указано на стр. 205, и качественный состав которого известен, помещают в предварительно высушенную кювету спектрофотометра. Пользуясь кривыми спектров поглощения растворов аква-комплексов редкоземельных элементов (см. рис. 55) и данными табл. 13 и 14, определяют область длин волн, в которой есть характерные для данного элемента полосы поглощения. Снимают спектр поглощения испытуемого раствора в этой области длин волн по отношению к дистиллированной воде, производя измерения через 0,5—1 нм. Пользуясь данными табл. 13, 14, по формуле (1.9) вычисляют концентрацию исследуемого раствора на основании измеренной оптической плотности в соответствующем максимуме поглощения.
В. Определение неодима 5,7-дихлор-8-оксихинолнном в смесях Рг—Nd и Nd—Sm
Для повышения чувствительности определения индивидуальных редкоземельных элементов по их характерным максимумам поглощения применяют галогенопроизводные 8-оксихинолина, например 5, 7-дихлор-8-оксихинолин (дихлороксин).
Оптимальное значение pH, при котором следует проводить экстракцию дихлороксинатов, 9,2—9,7. Поскольку максимальная растворимость дихлороксинатов редкоземельных элементов в хлороформе равна 1 • 10-3 моль/л, навеска анализируемой смеси должна соответствовать предельной растворимости их в том объеме хлороформа, который используется при выполнении анализа. Для полноты экстракции необходим 100—200-кратный избыток реагента. Для определения концентраций используют классический вариант метода анализа многокомпонентных систем уравнения (III.35). Длины волн, при которых следует проводить измерения и соответствующие им значения е, приведены в табл. 15. При наличии больших количеств неодима ошибка определения празеодима и самария велика, первого из-за близости значений е дихлороксинатов неодима и празеодима (X 640 нм), второго из-за малого значения е его дихлороксината при X 1085 нм.
ТАБЛИЦА 15
Молярные коэффициенты погашения дихлороксинатов редкоземельных элементов
Элемент \пах» нм 8
Неодим 581 640 1085 63,0 7,8 1,8
Празеодим 581 640 10,34 7,85
Самарий 1085 581 5,0 3,6
Реагенты
1. 5,7-Дйхлор-8-зэксихинолин, 0.1%-ный раствор в 3 н. соляной кислоте. 2. Раствор аммиака, 1%-ный раствор.
3. Хлороформ, очищенный.
4. Метиловый спирт.
Для анализа смеси редкоземельных элементов из общего объема 25 мл берут 5 мл, содержащих 3,5—3,6 мг смеси редкоземельных элементов, помещают в стакан емкостью 30—40 мл, прибавляют 3 мл раствора дихлороксина по каплям, раствор NH3 до pH 9,2—9,7 (измерения pH проводят на потенциометре) и переносят в делительную воронку. Образовавшуюся суспензию экстрагируют последовательно двумя порциями хлороформа (по 10 мл) в течение 3—5 мин. Экстракты сливают в сухие стаканы и затем переносят в сухую мерную колбу емкостью 25 мл и доводят объем раствора до метки метиловым спиртом. Измеряют на спектрофотометре поглощение полученных хлороформных растворов (/ = 5 см) при длинах волн (нм): смесь Рг—Nd 581 и 640, смесь Nd—Sm 581 и 1085. В качестве раствора сравнения используют хлороформный раствор реагента той же концентрации, которая была использована для приготовления испытуемых растворов. Концентрации вычисляют так, как указано на стр. 74.
Г. Определение индивидуальных редкоземельных элементов в их смеси 7-иод-8-оксихинолин-5-сульфокислотой
В отличие от дигалогенпроизводных 8-оксихинолина, 7-иод-8-оксихинолин-5-сульфокислота и ее комплексы растворимы в воде. Комплексные соединения оптимально образуются в щелочной среде (pH 8-11). В этих условиях полоса поглощения иод-оксин-сульфо-кислоты претерпевает сильный гипсохромный сдвиг, в результате чего поглощение реагента оказывается незначительным в области максимумов поглощения комплексов редкоземельных элементов, расположенных в видимой области спектра. Для достижения максимальной чувствительности следует использовать такое количество окислов редкоземельных элементов, которое соответствует предельной растворимости комплексов в объеме воды, необходимом для проведения анализа. Предельная растворимость реагента в воде равна 1,4 • 10-2 моль/л, а поскольку для полного связывания иона редкоземельного элемента необходим четырехкратный избыток реагента, то максимальная кон-цен'т’рация суммы редкоземельных элементов ие должна превышать 3 • 10~3 моль/л. Длл повышения растворимости используют водноспиртовую среду [60].
Реагенты
1. Стандартные растворы солей Nd, Pr, Sm, Er, Но, Tm, Yb, Lu, содержащие 1 мг/мл элемента (приготовление растворов см. на стр. 205).
2. 7-Иод-8-оксихинолин-5-сульфокислота, 0,5%-ный водно-спиртовой раствор (3 : 2).
3. Раствор вода — гпирт (3 : 2).
4. Аммиак, 1%-ный водный раствор.
1. Анализ смесей Nd—Pr, Nd—Sm, Er—Tm. В области максимумов поглощения комплексов неодима и эрбия с иод-оксин-сульфокисло-той комплексы празеодима, самария и тулия с этим реагентом, а также сам реагент не имеют характерных максимумов, но поглощение их в этой области заметно (рис. 57, 58). Чтобы исключить это поглощение при анализе данных смесей, можно использовать вариант дифференциального метода (стр. 75). В табл. 16 приведены значения X, при которых разность величин е для определяемого элемента велика, а для сопутствующих мала, а также все данные, необходимые для проведения анализа конкретных смесей.
Эталонные растворы готовят в соответствии с данными табл. 16. В градуированные пробирки емкостью 10 мл вносят растворы солей неодима или эрбия, прибавляют по 2,5 мл иод-оксин-сульфокислоты и раствора аммиака до соответствующего значения pH (объем раствора аммиака определяется предварительно добавлением его к отдельной аликвотной части испытуемого раствора и измерением pH на потенциометре) и доводят объем раствора до 6 мл водно-спиртовой смесью. После перемешивания измеряют поглощение каждого раствора при двух длинах волн, указанных в табл. 16 (/ = 10 см). Раствором сравнения служит водно-спиртовая смесь. Строят градуировочный график в координатах = Д’1» = f (с), используя метод наименьших квадратов (стр. 84).
ТАБЛИЦА 16
Условия определения редкоземельных элементов в смесях
Nd — Pr, Nd—Sm, Er—Tm, Er—Ho, Yb — Lu (2р.3э.—1.5 мг)
Диализируемая смесь элементов Пределы концентраций определяемого элемента в эталонных растворах, мг Интервалы между эталонными растворами, мг Длины волн, им pH Раствор сравнения для измерения
А, ^2 эталонных растворов испытуемого раствора
Nd—Рг 0,02—0,22 0,04 581 590 9—10 Смесь вода — спирт Смесь вода— спирт
Nd—Sm 0,02—0,22 0,04 581 590 9—10 То же io же
Er—Tm 0,08-0,23 0,05 519 525 10,5±0,2 » »
Er—Ho 0,08—0,23 0,05 519 525 Ю,5±0,2 0,08 мг Ег смесь Ег —Но сЕг = =0,08 мг
Yb—Lu 0,18—1,05 0,08 960 980 7,3±0,5 0,18 мг Yb смесь Yb—Lu cYb = =0,18 мг
Для определения неодима или эрбия берут 2 мл испытуемого раствора, содержащие ~1,5 мг суммы редкоземельных элементов и проводят все операции, как указано при приготовлении эталонных растворов. Находят разность оптических плотностей и определяют содержание неодима (эрбия) по градуировочному графику.
2. Анализ пар Ег—Но и Yb—Lu. При анализе этих пар приведенный метод (см. стр. 209) не_дает удовлетворительных результатов, так как разница в величинах е при двух выбранных длинах волн для
Рис. 57. Спектры поглощения соединений Nd, Pr и Sm с 7-иод-8-оксихинолин-5-сульфо-кислотой и реагента (R)
Рис. 58. Спектры поглощения соединений Ег, Но и Тгп с 7-иод-8-оксининолин-5-сульфо-кислотой и реагента (R)
Но и Lu довольно велика в сравнении с разностью е для Ег и Yb при тех же длинах волн. Положительный результат в этом случае дает сочетание метода, приведенного выше, с дифференциальным способом измерений (стр. 76). Основным условием применения этого варианта является постоянство содержания суммы редкоземельных элементов в аликвотной части анализируемой смеси.
Для приготовления эталонных и испытуемого растворов используют методику, приведенную на стр. 210. Растворами сравнения служат соответственно первый раствор эталонного ряда и раствор смеси анализируемых компонентов с той же концентрацией определяемого элемента, что и в первом эталонном растворе (см. табл. 16). Дальнейшие измерения при длинах волн, указанных в табл. 16, построение градуировочных графиков и расчет концентрации определяемых элементов в их смеси проводят в условиях, указанных на стр. 210.
ОПРЕДЕЛЕНИЕ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
В ВИДЕ КОМПЛЕКСОВ «ОКРАШЕННЫМИ» РЕАГЕНТАМИ
«Окрашенные» реагенты применяются как для определения суммы редкоземельных элементов, так и индивидуальных элементов в смеси. При этом можно использовать прямые спектрофотометрические методы, методы спектрофотометрического титрования, применяя в качест-
Рис. 59 Спектры поглощения арсена- Рис 60. Спектры поглощения арсеназо III (/) и его комплексных соедине- зо I (1), ортанилового А (2) и их
ний с редкоземельными элемента- комплексов с неодимом (Г) и (2')
ми (2) соответственно
ве индикаторов эти реагенты, а также некоторые варианты дифференциального спектрофотометрического метода. К «окрашенным» реагентам относятся азо-производные хромотроповой кислоты и в первую очередь о-арсеназо-о-оксиазосоединения, которые имеют большое значение для спектрофотометрического анализа редкоземельных элементов. Первый из реагентов этой группы, арсеназо I был предложен В. И. Кузнецовым. Позже С. Б. Саввин синтезировал аналог арсеназо I — арсеназо III [68], а затем С. Б. Саввин, Ю. М. Дедков и их сотрудники [69], [70] синтезировали большую группу аналогов арсеназо
III. У большинства комплексных соединений редкоземельных элементов с данными реагентами в спектрах поглощения имеются два максимума (рис. 18, 59, 60), положение которых незначительно меняется в зависимости от структуры реагента (АХ~ ПО—120 нм для реагента и комплекса). У некоторых нитрозопроизводных АХ, ~ 170 нм. Имеются также комплексные соединения, в спектре которых есть только один максимум. Изучение [701 ряда аналогов арсеназо III показало, что наиболее ч) вствительным из этой группы реагентов является арсеназо М. Однако при анализе смеси на содержание отдельных редкоземельных элементов иногда более важна не абсолютная, а относительная чувствительность, т. е. возможность определения малых количеств отдельных элементов этой группы в их смеси.
А. Определение суммы редкоземельных элементов арсеназо III
Арсеназо III (бензол-2'-арсоновая кислота-(Г-азот-2-)-бензол-2"-арсоновая кислота-(1"-азо-7-)-1,8-диоксинафталин-3,6-дисульфокисло-
та широко используется для определения редкоземельных элементов.
Комплексные соединения редкоземельных элементов с арсеназо III образуются в интервале pH 2—4 и имеют два максимума поглощения: при X, 600 и 650 нм (см. рис. 59). При pH > 3 максимумы поглощения при этих же длинах волн имеет реагент. Для реагента оба эти максимума поглощения являются характерными, в то время как для комплексного соединения более характерен максимум поглощения при X, 650 нм. Поэтому определение рекоземельных элементов арсеназо III проводят, измеряя оптическую плотность растворов, с pH 2,5—3 при X, 650 нм. (В данном методе не рекомендуется использовать буферные растворы для создания необходимого значения pH, так как это снижает оптическую плотность растворов.)
Реагенты
1. Стандартный раствор соли редкоземельного элемента, содержащий 0,1 мг/мл элемента (приготовление см. на стр. 205). Раствор с содержанием 100 мкг/мл элемента готовят в день употребления.
2. Арсеназо III, 0,015%-ный водный раствор.
3. Хлорная кислота, 0,08 н. раствор.
Для приготовления эталонных растворов в пять мерных колб, емкостью по 25 мл вводят раствор, содержащий редкоземельный элемент (мкг): 5. 10, 20, 30, 40, доводят объемы во всех колбах до 5 мл, прибавляют 6—7 мл раствора арсеназо III, 1 мл хлорной кислоты и доводят объем раствора до метки водой. Измеряют оптическую плот
ность полученного раствора по отношению к воде при X 650 нм, используя спектрофотометр. По полученным данным строят градуировочный график, используя при этом метод наименьших квадратов (стр. 84).
Для определения редкоземельного элемента в испытуемом растворе из общего объема берут 5 мл этого раствора, содержащего 4- -40 мкг элемента, помещают в колбу емкостью 25 мл и прибавляют те же количества реагентов, которые указаны для приготовления эталонных растворов. После доведения объема раствора до метки водой измеряют его оптическую плотность на спектрофотометре при X 650 нм и определяют содержание в нем редкоземельного элемента по градуировочному графику. Это определение повторяют три-четыре раза и обрабатывают результаты, используя метод математической статистики (стр. 86).
Б. Спектрофотометрическое титрование редкоземельных элементов комплексоном III с арсеназо I*
Возможность спектрофотометрического титрования редкоземельных элементов комплексоном III с арсеназо I в качестве индикатора обусловлена меньшей устойчивостью комплексных соединений этих элементов с индикатором, чем с комплексоном III.Комплексное соединение редкоземельного элемента с арсеназо I имеет максимум поглощения в области 570—575 нм, в то время как сам реагент поглощает при X 510 нм (см. рис. 59). Титруя растворы редкоземельных элементов с арсеназо I комплексоном III, наблюдают постепенное уменьшение оптической плотности при X 575 нм в результате перехода редкоземельных элементов из менее устойчивого комплексного соединения в более устойчивое, которое в данной области длин волн не поглощает. Таким образом, в процесе титрования получают ход кривой, представленной на рис. 24.
Арсеназо I как индикатор имеет ряд преимуществ, поскольку его растворы в воде устойчивы довольно длительное время, не подвергаются окислению кислородом воздуха, как это наблюдается с индикатором эриохромом черным Т. Комплексы редкоземельных элементов с арсеназо I более устойчивы, чем с ксиленоловым оранжевым, что также имеет преимущества при индикаторном комплексонометрическом титровании.
Примечания. 1. При титровании суммы редкоземельных элементов при наличии церия необходимо добавлять 3 мл аскорбиновой кислоты.
2. Для получения надежных результатов концентрация реагента (комплексона III) не должна превышать концентрацию определяемого элемента более чем в 100 раз, а концентрация индикатора должна быть одного и того же порядка или в крайнем случае ниже на попядок, чем концентрация определяемого элемента.
3. При определении суммы резкоземельных элементов расчет производят по среднему атомному весу.
* Арсеназо I —• бензол-2-арсоновая кислота (I -азо-2)-1,8-диоксинафталин-3,6-дисульфокислота.
Кроме указанных на стр. 212, необходимы реагенты:
1. Комплексон III, 1 - 10-4 М раствор. Раствор готовят по точной навеске. Комплексон III можно очистить перекристаллизацией.
2. Арсеназо I, 1 • 10“в Л1 раствор. Реагент очищают сливанием насыщенного раствора с равным объемом HCI конц., с последующим промыванием осадка спиртом.
3. Уротропин, 25%-ный раствор.
4. Соляная кислота, 0,1 н. раствор.
5. Аскорбиновая кислота, 1%-ный раствор (свежеприготовленный).
Определение редкоземельных элементов в отсутствие посторонних ионов.
К испытуемому раствору соли редкоземельного элемента (отдельному или смеси), помещенному в мерную колбу емкостью 50 мл и содержащему редкоземельный элемент в количестве от 5 мкг до 0,5 мг, прибавляют 5 мл раствора арсеназо I, 0,5 мл соляной кислоты и 1,5 мл уротропина (для создания pH ~ 6,5) и доводят объем раствора водой до метки. После перемешивания отбирают пипеткой 20 мл этого раствора и переносят в кюветку для титрования. Раствор комплексона III прибавляют из микробюретки отдельными порциями по ~0,1 мл, перемешивают и измеряют оптическую плотность при Л 575 нм после прибавления каждой порции реагента. По полученным данным строят график в координатах А—v, графически определяют конечную точку титрования и вычисляют количество редкоземельного элемента, учитывая, что редкоземельные элементы образуют с комплексоном III соединение в соотношении 1:1.
Определение редкоземельных элементов в присутствии тория. Возможность последовательного титрования тория и редкоземельных элементов обусловлена тем, что торий образует с комплексоном III более устойчивое комплексное соединение, его соединение с арсеназо I образуется в кислой среде при pH ~ 2, а соединение редкоземельного элемента при pH 6,5. Соединения тория и редкоземельного элемента поглощают при одной и той же длине волны К 575 нм. Таким образом, имеется возможность провести последовательное титрование сначала тория при pH ~ 2, а затем редкоземельного элемента при при pH ~ 6,5. При этом добавления индикатора перед титрованием редкоземельного элемента не требуется, так как индикатор регенерируется в процесс тирования тория. Однако этот метод дает хорошие результаты при титровании смесей, содержащих элементы при соотношении Th: редкоземельный элемент =1:1; 1 : 10; 1 : 100.
Если содержание тория превосходит содержание редкоземельного элемента более чем в 10 раз, то совместное титрование также возможно. Но при этом содержание редкоземельного элемента вычисляется по разности результатов титрования двух отдельных проб, проведенных при pH 2 и 6 5
Для определения редкоземельного элементов испытуемом растворе, содержащем 5 мкг — 0,5 мг редкоземельного элемента и около 5 мкг тория и помещенном в мерную колбу емкостью 50 мл, прибавляют 5 мл арсеназо I, 1 мл соляной кислоты для создания pH ~2 и доводят объем раствора до метки водой. После перемешивания отбирают пн-
петкой 20 мл этого раствора, переносят в кювету и титруют раствором комплексона III, прибавляя его по 0,1 мл из микробюретки и измеряя оптическую плотность после каждой прибавленной порции реагента. Когда оптическая плотность установится постоянной, создают в растворе pH 6,5 добавлением 1 мл уротропина и продолжают титрование. Кривая зависимость А от v будет иметь два излома: один соответствует количеству тория, второй — количеству редкоземельного элемента. Графически определяют конечные точки титрования, соответствующие тому или другому излому, и вычисляют содержание тория и редкоземельного элемента [71].
В. Применение арсеназо М для анализа двухкомпонентных смесей Рг—Er, Y—La •
дифференциальным спектрофотометрическим методом
Арсеназо М бензол-2'-арсоновая кислота (Г-азо-2)-3'-сульфо-бензол (1"-азо-7)-1,8 -диоксинафталин-3,6-дисульфокислота) являемся
наиболее чувствительным реагентом для определения редкоземельных элементов1 (см. рис. 18). Кроме того, молярные коэффициенты погашения соединений с этим реагентом в ряду редкоземельных элементов заметно различаются. Вариант дифференциального спектрофотометрического метода (см. стр. 77) при анализе смеси двух компонентов дает тем лучший результат, чем больше отношение молярных коэффициентов погашения соединений анализируемых компонентов. Поэтому Арсеназо М имеет преимущества при анализе смеси Рг — Ег перед арсеназо I не только в том, что позволяет повысить чувствительность определения на один порядок, но также дает возможность определения меньших относительных содержаний компонентов в смеси (см. ниже).
Реагент Арсеназо 1 Рг ; Ег Арсеназо М Ортаннловый А Рг : Ег
Рг : Ег La : Y
Л 'лМц 1,19 1,30 1,40 1,83
Метод может быть применен к анализу смеси La с Y, так как обратное соотношение в величинах е (у Y меньше, у La больше) перекрывается большой разницей в атомных весах.
Реагенты
1. Стандартные растворы солей празеодима, эрбия, Лантана и иттрия, содержащие 0,1 мг/мл металла (приготовление см. на стр. 205). Перед употреблением раствор разбавляют в 10 раз (10 мкг/мл).
2. Арсеназо М, 1 - 10-3 М водный раствор.
3. Ацетатный буферный раствор, pH 3.
Выбор оптимальной концентрации суммы редкоземельных элементов в эталонных растворах (а следовательно, концентрации элемента с большим атомным весом в растворе сравнения) проводят по методу Бастиана (см. стр. 72). Выяснять пределы соблюдения закона поглощения (выбирать оптимальную концентрацию суммы редкоземельных элементов) следует по растворам элемента с меньшим атомным весом (Pr, Y). В мерные колбы емкостью 25 мл вводят раствор соли элемента с меньшим атомным весом (Рг или Y) в количествах (мкг): 8, 16, 24, 32, 40, 48, 56 (интервал в концентрациях должен быть постоянным, Ас = = 8мкг), прибавляют 1 мл арсеназо М, 15 мл ацетатного буферного раствора и доводят объем раствора до метки водой. Измеряют на спектрофотометре при X 640 нм оптическую плотность каждого последующего раствора по отношению к предыдущему (Д',)- Далее поступают, как указано на стр. 205.
Для построения градуировочного графика (условия приведены выше) готовят серию растворов с постоянной суммой редкоземельных элементов, соответствующей содержанию в выбранном оптимальном растворе, но содержание элемента в которых с меньшим атомным весом (Рг или Y) меняется от 10 до 90%. Измеряют при Z. 640 нм оптическую плотность этих растворов по отношению к раствору элемента с большим атомным весом (Ег или La), имеющему концентрацию, равную сумме редкоземельных элементов в эталонных растворах. По данным этих измерений строят градуировочный график в координатах А — процентное содержание элемента с меньшим атомным весом (см. стр. 84), используя метод наименьших квадратов.
Для определения содержания редкоземельных элементов в смеси аликвотную часть испытуемого раствора с таким количеством суммы редкоземельных элементов, которое соответствует содержанию их в оптимальном растворе сравнения, обрабатывают как указано при выборе этого раствора. Измеряют его оптическую плотность при Л 640 нм по отношению к тому же раствору сравнения, который использовали при прстроении градуировочного графика. По градуировочному графику находят процентное содержание компонентов в смеси. Это определение повторяют три-четыре раза и обрабатывают результаты, используя метод математической статистики (см. стр. 86).
Г. Применение ортаиилового А для анализа двухкомпонентных смесей
Рг—Ег, Nd—Ег дифференциальным спектрометрическим методом
- Несмотря на то, что абсолютная чувствительность определения редкоземельных элементов с ортаниловым А (бензол-2'-арсоновая кислота (Г-азо-2)-2"-сульфобензол-(-Г-азо-7-)-1,8-диоксинафталин-3,
6-дисульфокислота) (см. рис. 60)
ниже, чем с арсеназо М, соотношение Л₽г/Лег для соединений с этим реагентом больше, чем с арсеназо М (стр. 215). Это позволяет определять меньшие относительные количества редкоземельных элементов в смеси. Кроме указанных на стр. 214, необходимы реагенты:
1. Ортаниловый А, 1 • 10-3 М водный раствор.
2. NHaOH, 0,1 н. раствор.
Выбор оптимальной концентрации суммы редкоземельных элементов в эталонных растворах следует проводить по растворм празеодима (неодима), так как закон Бера для растворов комплексного соединения эрбия соблюдается в более широком интервале концентраций, чем для празеодима и неодима.
В мерные колбы емкостью 25 мл помещают раствор празеодима, содержащий от 0,02 до 0,14 мг Рг (с постоянным интервалом Ас= 0,02), прибавляют 2 мл ортанилового А, несколько капель NH40H до pH 4,5 и доводят объем раствора до метки водой.
Дальнейшие измерения при X 650 нм и расчеты производят, как указано при работе с арсеназо М (см. стр. 216), с той лишь разницей, что относительное содержание Pr(Nd) в Ег при одном и том же оптимальном содержании суммы редкоземельных элементов может меняться не только от 10 до 90%, но также от 1 до 9% и от 10 до 100%. Таким образом относительная чувствительность в этом методе на порядок выше, чем с арсеназо М.
Примечание. Методы В и Г разработаны в лаборатории спектрофотометрии на кафедре аналитической химии МГУ.
Определение титана
Большое распространение получили методы определения титана в виде его пероксидного соединения, образующегося в сернокислой среде, и комплексных соединений с хромотроповой кислотой. Очень большой интерес представляет определение титана тайроном 172].
ОПРЕДЕЛЕНИЕ ТИТАНА ПЕРОКСИДНЫМ МЕТОДОМ
В РАСТВОРЕ ЕГО СОЛИ
Соединению титана, образующемуся в сернокислой среде в присутствии Н2О2, приписывают состав [TiOH2O2P+, оно мало устойчиво Синеет — 9 • 10-3) [73], [74]. Растворы комплексного соединения окрашены в желтый цвет, ?.тах 410 нм, е = 7,0 • 10а. Метод широко применяется для определения титана в сталях и сплавах несмотря на его
малую селективность. Ряд элементов: V, Mo, U, Nb, Сг дают также окрашенные растворы с Н2О2 в условиях определения титана. Данным методом можно раздельно определять V, Мо и Ti.
Реагенты
1. Стандартный раствор соли титана, содержащий 1 мг/мл Ti. Для этого 0,4175 г. двуокиси титана, предварительно прокаленной в платиновом тигле, сплавляют с 3—4 г пиросульфата калия. Плав выщелачивают 70—80 мл серной кислоты (1 • 4), слегка нагревают и после полного растворения переносят в мерную колбу емкостью 250 мл, раствор доводят до метки серной кислотой (1 : 4).
2. Перекись водорода, 3%-ный раствор.
3. Серная кислота, 5%-ный раствор.
Для приготовления эталонных растворов берут пять мерных колб емкостью 50 мл, вводят в каждую 10 мл дистиллированной воды, стандартный раствор, содержащий титан (мг) : 0,5; 1,0; 1,5; 2,0; 2,5 соответственно, добавляют 5 мл раствора серной кислоты и 3 мл раствора перекиси водорода, объем раствора доводят водой до метки. Растворы фотометрируют При X 410 нм, используя фотоколориметры различных марок. В качестве раствора сравнения берут воду и строят градуировочный график.
Для определения титана в испытуемом растворе из общего объема 50 мл берут 10 или 20 мл этого раствора в зависимости от содержания титана и помещают в мерную колбу емкостью 50 мл. Проводят те же операции с добавлением тех же количеств реагентов, которые указаны при приготовлении эталонных растворов. Содержание титана определяют по градуировочному графику.
ОПРЕДЕЛЕНИЕ ТИТАНА ХРОМОТРОПОВОЙ КИСЛОТОЙ
В РАСТВОРЕ ЕГО СОЛИ
Титан образует с хромотроповой кислотой в зависимости от pH
НО ОН
два комплексных соединения, растворимых в воде. Соединения различаются по интенсивности полос максимального поглощения: при pH 1,2—1,5 ?-тах 470 нм, е = 1,1 • 104, при pH 4,5—5,0 Хтах 430 нм, е = 2,7 • 104. Состав соединений также различен. В интервале pH 1,2— 1,5 образуется соединение с соотношением Ti : R = 1 : 2, в интервале pH 4—5 с соотношением Ti : R 1 : 3. При определении титана указанным реагентом необходимо точно соблюдать идентичность в условиях приготовления испытуемого и эталонных растворов.
В присутствии третичного амина—трибутиламина комплексные соединения титана экстрагируются хлороформом [72].
Кроме указанных на стр. 218, необходимы реагенты:
1. Хромотроповая кислота, 2%-ный раствор.
2. Трибутиламин (перегнанный).
3 едкий натр, 1 н. раствор.
4. Серная кислота, 1 и. раствор.
5. Сульфат натрия, 1 и. раствор.
6. Хлороформ.
Для приготовления эталонных растворов берут шесть делительных воронок емкостью 50 мл, вводят в пять из них стандартный раствор, содержащий титан (мг): 0,0; 0,005; 0,025; 0,05; 0,075; 0,1 соответственно, добавляют во все колбы по 2 мл раствора хромотроповой кислоты, 1 мл трибутиламина, H2SO4 илиИаОН до pH 1,2—1,5, доводят раствор до объема 10 мл раствором сульфата натрия. После этого прибавляют 10 мл хлороформа, встряхивают содержимое воронок в течение 3 мин, отделяют органическую фазу в градуированные пробирки емкостью 10 мл и добавляют несколько капель хлороформа до метки. Измерение оптической плотности хлороформных растворов проводят при X 470 нм на спектрофотометре относительно раствора сравнения, которым служит хлороформный раствор первой воронки. По данным измерения строят градуировочный график.
Для определения титана в испытуемом растворе из общего объема 50 мл берут 10 или 20 мл этого раствора и проводят с ним все операции, указанные при приготовлении эталонного раствора. Содержимое титана находят по градуировочному графику.
ОПРЕДЕЛЕНИЕ ТИТАНА ТАЙРОНОМ В БЕРИЛЛИИ
Титан (IV) образует с тайроном
ОН ООН so3-
комплексное соединение, растворы которого окрашены в лимонножелтый цвет (X 380 нм, е = 1,5 - 104). Наибольшая интенсивность окраски растворов наблюдается в интервале pH 4,0 — 5,5. В присутствии трибутиламина комплексное соединение экстрагируется хлороформом, образуя ионный ассоциат состава 1:3:5 (е = 1,8 • 104), при том же значении X 380 нм [72].
Кроме указанных на стр. 218, необходим 0,5%-ный раствор тайрона.
Для приготовления эталонных растворов берут шесть делительных воронок емкостью 50 мл, в пять из них вводят стандартный раствор, содержащий титан (мг): 0,0; 0,05; 0,10; 0,15; 0,20; 0,25 соответственно, прибавляют во все воронки по 1 мл триб^тиламмония, H2SO4 или NaOH до pH 4,5—5,5 по индикаторной бумаге, затем раствором сульфата натрия доводят объем до 10 мл. Приливают 10 мл хороформа и встряхивают содержимое воронок в течение 3 мин. Отделяют органические фазы в мерные пробирки емкостью 10 мл и добавляют несколько капель хлороформа до объема 10 мл. Измерение интенсивности окраски растворов производят на спектрофотометре при 'К 380 нм. Раствором сравнения служит хлороформный раствор первой воронки. По данным измерения строят градуировочный график.
Для определения титана в бериллии берут две Навески металла по 1 г, переносят каждую в коническую колбу или стакан емкостью 50— 100 мл, растворяют в 10 мл серной кислоты (1 : 1) при слабом подогревании. После получения прозрачного раствора переводят его в мерную колбу емкостью 100 мл и доводят объем раствора водой до метки. В делительную воронку емкостью 60—70 мл берут аликвотную часть 20 мл, прибавляют 4 мл тайрона, 4 мл трибутиламина, 5 мл сульфата гидразина и устанавливают pH раствора 4,5—5,0 добавлением 1 и. раствора едкого натра. После этого приливают 10 мл хлороформа и экстрагируют соединение титана, производят перемешивание в течение 3 мин. Измерение оптической плотности проводят в условиях см. приготовление эталонных растворов (стр. 219). Содержание титана находят по градуировочному графику. Результаты (не менее четырех) определений обрабатывают методом математической статистики.
Определение цинка
Для определения цинка применяют ряд органических реагентов. Из их числа большого внимания заслуживает дитизон,
,NH—NH—свНв s=c/
XN=N-CeH6
который в зависимости от кислотности раствора может реагировать в кето- или енольной формах.
Дитизон—один из распространенных реагентов, применяемых для определения тяжелых металлов. Реакции эти отличаются большой чувствительностью; молярный коэффициент погашения в ~ 104. Ди-
тизонаты металлов хорошо экстрагируются неводными растворителями [761. Найдены условия раздельного определения многих элементов (Zn, Cd, Си, Pb, Hg и др.), которые проводят при различной
кислотности раствора и применении ряда маскирующих веществ
(тиосульфат натрия, диэтилдитиокарбаминат натрия, тиомочевина). При экстракции из 0,1 н. раствора минеральной кислоты можно предварительно выделить соединения Си, Ag, Hg, а понижая кислотность до pH 5,0—5,5—соединения цинка. Раствор дитизоната цинка в СС14 поглощает при 538 нм, в = 9,2 • 104 (рис. 61).
Реагенты
1. Стандартный раствор цинка: 0,1 г металлического цинка растворяют в мерной колбе емкостью 100 мл в НС1 и доводят
Риг. 61. Спектры поглощения объем раствора водой до метки — раствор в ССЦ дитизона (/) и дитизоната А: 1 мл раствора А содержит 1 мг цинка.; цинка (2) 1 мл раствора А разбавляют в мерной
колбе емкостью 200 мл водой — раствор Ё: 1 мл раствора Б содержит 0,005 мКГ цинка; готовят перед употреблением.
2. Азотная кислота (х. ч.), концентрированная, разбавленная 1 : 3, 0,01 н.
3. Борная кислота, 5%-ный раствор.
4. Винная кислота, 15%-ный раствор.
5. Плавиковая кислота (1 : 5).
6. Аммиак, водный раствор.
7. Этилацетат, перегнанный.
8. Диэтилдитиокарбаминат натрия, 2%-ный раствор.
9. Ацетатный буферный раствор, pH 4,7.
10. Ацетат натрия, 50%-ный раствор.
11. Тиосульфат натрия, 50%-ный раствор.
12. Четыреххлористый углерод, перегнанный, бесцветный.
13. Дитизон, 0,02%-ный раствор в четыреххлористом углероде (запасный). Навеску дитизона 0,020 г помещают в делительную воронку емкостью 500 мл и растворяют в 50 мл четыреххлористого углерода; дитизон, растворяясь, окрашивает раствор в зеленый цвет. По растворении дитизона добавляют в воронку 100 мл аммиака (1 : 100) и воронку энергично встряхивают; при этом дитизон переходит в аммиачный раствор, окрашивая его в оранжевый цвет, а продукты окисления дитизона остаются в слое органического растворителя. Последний сливают, в воронку добавляют 100 мл чистого четыреххлористого углерода, подкисляют аммиачный раствор несколькими каплями разбавленной серной кислоты (1: 1) и при перемешивании переводят дитизон обратно в четыреххлористый углерод. Отделив раствор дитизона от водного слоя дважды промывают его водой встряхиванием в делительной воронке, а затем фильтруют (для просушки) через бумажный фильтр и хранят в склянке из темного стекла, под слоем 2 н. серной кислоты.
Для получения 0,005%-ного раствора дитизона к 25 мл 0,02%-него раствора добавляют 75 мл. Раствор готовят в день употребления.
А. Определение цинка в металлическом титане
Для приготовления эталонных растворов берут пять делительных воронок емкостью 50—70 мл, вводят в каждую 10 мл 0,01 н. азотной кислоты и стандартный раствор, содержащий цинк (мкг): 0; 3,0; 6,0; 10,0; 20,0 соответственно, приливают 1 мл ацетата натрия, после перемешивания добавляют 15 мл 0,005%-ного раствора дитизона в четыреххлористом углероде. В качестве раствора сравнения используют содержимое первой воронки.
Измерение оптической плотности при X 538 нм производят на спектрофотометре. Градуировочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Примечание. Раствор ацетата натрия предварительно очищают обработкой его раствором дитизона.
Для определения цинка в титане берут две навески по 0,3 г металла, помещают каждую в платиновую чашку и обрабатывают на холоду 10 мл плавиковой кислоты, прибавляя ее небольшими порциями; очередную порцию плавиковой кислоты прибавляют по прекращении бурной реакции. Помешиваю! раствор легким покачиванием платиновой чашки, растворение заканчивают при слабом нагревании на водяной бане. После полного растворения добавляют 5—6 капель концентрированной азотной кислоты и выпаривают раствор на водяной бане до объема 1—1,5 мл, не допуская выпаривания до меньшего объема. Приливают 3 мл винной кислоты, 10 мл борной кислоты и разбавляют водой до объема 25 мл. Раствор нейтрализуют аммиаком до переходной
ЬКраски по бумаге конго, охлаждают и переводят в делительную Воронку емкостью 100 мл. Платиновую чашку дважды промывают небольшими порциями воды из промывалки, сливая ее в воронку. Туда же приливают 5—8 мл этилацетата, 1 мл раствора диэтилдитиокарба-мината натрия и энергично встряхивают воронку в течение 10—20 с.
После отстаивания тщательно отделяют слой этилацетата от водного слоя, переводя последний во вторую делительную воронку. К водному слою снова приливают этилацетат и диэтилдитиокарбаминат натрия в тех же количествах и воронку встряхивают. Экстракцию повторяют еще два-три раза, пока окраска слоя этилацетага сохраняется без изменения. Объединенный экстракт, собранный в одну делительную воронку, отделяют от отстоявшихся капелек водного слоя, промывают его два раза по 5 мл ацетатным буферным раствором и водный слой отбрасывают. Экстракт фильтруют через плотный сухой складчатый фильтр, собирая его в другую делительную воронку емкостью 50 мл. Туда же приливают 10 мл азотной кислоты (1 : 3) и воронку энергично встряхивают для извлечения цинка в водный слой. После отстаивания тщательно отделяют водный слой, собирая его в стакан емкостью 50 мл. Обработку экстракта той же кислотой повторяют еще два раза. Объединенную азотнокислую вытяжку упаривают на песчаной бане досуха.
Сухой остаток смачивают несколькими каплями концентрированной азотной кислоты и слегка подсушивают на водяной бане. Затем остаток растворяют в 0,01 н. азотной кислоте, раствор переводят в мерную колбу емкостью 50 мл и доводят объем раствора до метки той же кислотой. Отбирают пипеткой 5—10 мл раствора и переносят в воронку для экстрагирования ( в растворе не должно быть более 10 мкг цинка), приливают 1 мл раствора ацетата натрия и 1 мл тиосульфата натрия (растворы перед употреблением очищают взбалтыванием в делительной воронке с дитизоном до тех пор, пока вновь добавленная порция дитизона не будет оставаться зеленой) и после перемешивания 15 мл 0,005 %-ного раствора дитизона в четыреххлористом углероде. Полученные растворы фотометрируют в условиях, указанных для приготовления эталонных растворов. Данные из параллельных опытов (не менее четырех) обрабатывают методом математической статистики.
Примечание. Необходимо проверять чистоту применяемых реагентов, остерегаться применения химической посуды, содержащей цинк, и работать с перегнанными (желательно в кварцевой посуде) кислотами и водой.
Б. Определение цинка в металлическом бериллии
Кроме указанных на стр. 221, необходимы реагенты:
1. Соляная кислота, 2 н. раствор.
2. Тиосульфат натрия, 25%-ный раствор.
3. Аммиак, 10%-ный раствор.
4. Буферный ацетатный раствор, pH 4,7.
Для приготовления эталонных растворов в шесть делительных воронок емкостью 100 мл помещают 50 мл соляной кислоты, прибавляют стандартный раствор, содержащий цинк (мкг). 0, 0; 1,0; 2,0; 3,0; 4,0; 5,0 соответственно, добавляют раствор аммиака по универсальной ин-
д^каторной бумаге до pH 5, затем 1 мл раствора тиосульфата натрия и 5 мл буферного раствора. Растворы перемешивают, прибавляют 5 мл раствора дитизона и энергично встряхивают содержимое воронки в течение 5 мин, органический слой отделяют, сливая его в мерную пробирку емкостью 15 мл. К водному слою в делительную воронку прибав- ляют еще 5 мл дитизона и вновь производят перемешивание содержимого воронки в течение 5 мин и сливают органический слой, объединяя его с первым. Объединенные органические фазы доводят четыреххлористым углеродом в мерной пробирке до метки. Измерение оптической плотности растворов дитизоната цинка производят при X 538 нм на фотоэлектроколориметре ФЭК-60 или спектрофотометрах любого типа и строят градуировочный график по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения цинка в металлическом бериллии берут три навески металла по 0,5 г, растворяют каждую в 50 мл соляной кислоты, охлаждают, добавляют аммиак по универсальной индикаторной бумаге до pH 5, прибавляют 1 мл раствора тиосульфата натрия, 5 мл ацетатного буферного раствора и переносят в делительную воронку. В дальнейшем определение цинка проводят в условиях, см. приготовление эталонных растворов. Результаты трех параллельных определений обрабатывают методом математической статистики.
Определение циркония
При использовании всех спектрофотометрических методов определения циркония решающую роль играет его ионное состояние в вод-
ных растворах.
Исследование ионного состояния циркония в растворах начали проводить Конник и Мак Ви [77]. Соловкин [78] вычислил общие константы комплексообразования и константы гидролиза; рассчитано процентное распределение циркония между гидроксокомплексами [79]. В результате расчета показано, что цирконий в одном растворе дает ряд оксикатионов, которые образуют соединения различного состава (рис. 62).
Для определения циркония
применяется ряд органических реагентов: арсеназо (III) (80), ксиленоловый оранжевый [81] и о, о'- диоксиазосоединения [82]. Из последней группы соединений представляет интерес пикрамин-эпсилон. При использовании отдельных реагентов можно применить дифференциальный спектрофотометрический метод, позволяющий определять цирконий и гафний [83] в их смеси. Для определения циркония известен также ряд методов спектрофотометрического
62 Распределение гидроксокомплек-сов циркония
титрования. Все указанные методы не являются селективными, они в одинаковой мере пригодны и для определения гафния.
ОПРЕДЕЛЕНИЕ ЦИРКОНИЯ КСИЛЕНОЛ» )ВЫМ ОРАНЖЕВЫМ
Ксиленоловый оранжевый
в кислой среде образует с цирконием комплексное соединение (рис. 63), растворы которого окрашены в интенсивный красный цвет, с соотношением Zr : R 1:1; ?vmax 535 нм, 8 = 3,5 • 104 (для реагента Хюах 440 нм). Определению циркония мешают фториды, фосфаты и оксалаты.
Реагенты
1. Стандартный раствор, го цирконила ZrOCl2 8Н2О, кислоте. Содержание циркония $ определяют гравиметрическим методом в виде
содержащий 1 мг/мл Zr. Берут 3,9 г хлористо-растворяют в 2 н. соляной или 2 н. хлорной
Рис. 63. Спектр поглощения ксиленолового оранжевого (/) и его комплекса с Zr (2)
ZrO2. Если концентрация Zr в растворе больше или меньше 1 мг/мл. то соответствующим разбавлением одной из кислот получают раствор с содержанием 1 мг/мл Zr. Более разбавленные растворы готовят добавлением соответствующей кислоты до определенного объема.
2. Ксиленоловый оранжевый, 0,1 %-ный водный раствор.
3. Серная кислота, 1 н. раствор и концентрированная.
4. Перекись водорода, 30%-ный раствор.
5. Сульфат аммония, кристаллический.
6. Комплексон III, 0,05 М раствор.
А. Определение циркония в растворе его соли
Для приготовления эталонных растворов в шесть мерных колб емкостью 50 мл вводят стандартный раствор, со
держащий цирконий (мкг): 0; 5,0; 10,0; 20,0; 30,0; 50,0 соответственно, добавляют 1 н. серную кислоту до 30 мл, прибавляют 1 мл раствора реагента и доводят объем раствора водой до метки. Измеряют оптическую плотность эталонных растворов при X 535 нм на фотоэлектроколориметре ФЭК-60 или спектрофотометрах различных марок относительно первого раствора, в котовый не вводили цирконий. Градуи
ровочный график строят по экспериментальным данным, обработанным методом наименьших квадратов.
Для определения циркония в испытуемом растворе из общего о бъема 50 мл берут 10—15 мл в зависимости от его содержания и проводят анализ, соблюдая условия, указанные при приготовлении эталонных растворов. Содержание циркония находят по градуировочному графику.
Б. Опеределение циркония в сплавах на основе ниобия и тантала
Метод [81] позволяет производить определение Zr в присутствии ниобия в ряде легирующих металлов. Ниобий в определенных условиях взаимодействует с ксиленоловым оранжевым. Однако проведение реакции при соответствующей кислотности раствора и способ приготовления раствора сравнения делают метод селективным для определения циркония.
Приготовление эталонных растворов производят в условиях, указанных для определения циркония в растворе его соли (см. стр. 224).
Для определения циркония в сплавах берут две навески его по 0,1 г, растворяют каждую в стакане из жаропрочного стекла емкостью 150— 200 мл, добавляют 0,3 г сульфата аммония и 3 мл H.2SO4 (пл. 1,84), нагревая содержание стакана на электрической плитке. После разложения сплава добавляют0,1—0,2 мл перекиси водорода, раствор переводят в мерную колбу емкостью 100 мл и объем раствора доводят водой до метки. В две мерные колбы емкостью 50 мл отбирают в каждую аликвотные части по 5—10 мл, содержащие не более 50 мкг Zr, и добавляют 1 н. H2SO4 до объема 20 мл. В одну из колб добавляют 0,2 мл раствора комплексона, тщательно перемешивают, затем в обе колбы вводят по 1 мл раствора ксиленолового оранжевого и доводят объем раствора водой до метки; кислотность раствора должна быть 0,4 н. по серной кислоте. Оптическую плотность этого раствора измеряют на фотоэлектроколориметрах ФЭК-56, ФЭК-60 или спектрофотометрах различных марок при X 535 нм относительно раствора, в который не вводится комплексон. Содержание циркония находят по градуировочному графику. Результаты параллельных определений ( не менее четырех) обрабатывают методом математической статистики.
ОПРЕДЕЛЕНИЕ ЦИРКОНИЯ ПИКРАМИНОЧ-ЭПСИЛОН
2,4- Динитрофенол-<6 азо 2>-1 нафтол, 3,8-дисульфокислота (пик-рамин-эпсилон)
отличается большой избирательностью и чувствительностью в отношении циркония. В определенных условиях образуется комплексное соединение с соотношением Zr : R 1 : 2. В спектрах поглощения реа-
Рис. 64. Спектры поглощения пикрамин-эпсилона (/) и его комплекса с Zr (2)
гента и комплексного соединения циркония есть некоторое наложение, что следует принимать во внимание при приготовлении раствора сравнения, для HR Zmax 496 нм, е = 3,0 • 104, для ZrR2Xmas 540 нм, е = 3,7 104 (рис. 64). При использовании ряда комплексующих веществ: тиомочевины, винной и аскорбиновой кислот, возможно определение циркония в присутствии меди, алюминия, титана, ванадия, железа и других элементов при соотношении 1: 1 102 (Си); 1 :3 • 104 [Fe(II)]; 1 : 10Б (редкоземельные элементы); 1 : 4 • 102 [Ti (IV)]; 1:2,5 • 103 (А1) [83].
Примечание. Пикрамин-эпсилон служит реагентом и для определения гафния. Для HfR2 Чпах 520 нм, е = 4,0 104.
Кроме указанных на стр. 224, необходимы реагенты:
1. Стандартный раствор, содержащий 1 мг/мл Zr. Приготовление см. на стр. 224. Для растворения хлористого цирконила берут соляную кислоту.
2. Пикрамин-эпсилон, 0,1%-ный водный раствор.
3. Соляная кислота, 1 н. и 5 н. растворы.
А. Определение циркония в растворе его соли
Для приготовления эталонных растворов в шесть мерных колб емкостью 25 мл вводят стандартный раствор, содержащий цирконий (мкг): 0,0; 2,5; 5,0; 7,6; 10,0; 15,0 соответственно; добавляют в каждую колбу 1 мл раствора реагента и доводят объем раствора 1 н. соляной кислотой до метки. Через 10 мин измеряют оптическую плотность растворов при Z 540 нм, / = 3 см на фотоэлектроколориметре ФЭК-60 относительно 1-го раствора. Строят градуировочный график по экспериментальным данным, обработанным методом наименьших квадратов. ,
Для определения циркония в испытуемом растворе из общего объема 50 мл берут 5 или 10 мл этого раствора (в зависимости от содержания циркония) и проводят все операции, указанные при приготовлении эталонных растворов. Содержание циркония находят по градуировочному графику.
Б. Определение циркония в сплавах на основе железа
Для определения циркония в сплавах берут две навески образца по 0,1 г, помещают каждую в коническую колбу емкостью 50 мл, растворяют в 20 мл 5 н. НС1 при нагревании. После растворения переносят
в мерную колбу емкостью 100 мл и доводят объем раствором 2 н. соляной кислотой до метки. Из каждой колбы берут по две аликвотные части (5 или 10 мл) в мерные колбы емкостью 25 мл и проводят все операции см. приготовление эталонных растворов (стр. 226). Содержание циркония находят по градуировочному графику. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики.
ОПРЕДЕЛЕНИЕ ЦИРКОНИЯ СПЕКТРОФОТОМЕТРИЧЕСКИМ
ТИТРОВАНИЕМ РАСТВОРОМ АРСЕНАЗО III
Реагент арсеназо III (см. стр. 212), предложенный С. Б. Саввиным 168] и успешно примененный для прямого спектрофотометрического определения циркония, может быть использован также и в методе спектрофотометрического титрования. Реагент максимально поглощает при Z 535 нм. У комплексного соединения циркония с арсеназо III имеются две полосы максимального поглощения при 7 620 и 670 нм. Чувствительность и селективность метода увеличивается, если титрование проводить при К 670 нм и кислотности 6 н. по НС1О4.
Кроме указанных на стр. 224, необходимы реагенты:
1. Арсеназо III, 10-3 М водный раствор. К 100 мл раствора арсеназо III добавляют 1—2 мл 2 н. НСЮ4. Титр раствора арсеназо ill определяют титрованием соли циркония известной концентрации.
2. Хлорная кислота, 6 н. раствор.
При использовании арсеназо III для титрования циркония необходимо предварительно выяснить степень частоты реагента, т. е. установить, есть ли примеси, имеющие поглощение при данной длине волны. Для этого готовят раствор, содержащий 100 мкг циркония в 25 мл 6 н. НСЮ4. В кювету спектрофотометра вводят последовательно по 0,5; 1,0; 1,5; 2,0 мл раствора циркония, добавляют 6 н. НС1О4 до закрытия окошечка кюветы и титруют каждую порцию из бюретки арсе
назо III, добавляя по 0,1 мл (X 670 нм), строят кривые титрования в координатах А — объем реагента и находят экстраполяцией точку эквивалентности. Полученные значения наносят на график в координатах объем реагента концентрация циркония (рис. 65). Все точки должны лежать на прямой; при экстраполяции на ось ординат получают отрезок, который дает поправку, учитывающую объем арсеназо III, израсходованный на титрование посторонних веществ. Эту поправку вносят при титровании раствора с неизвестным содержанием циркония. Данный способ можно рекомендовать и для титрования с различными органическими реагентами, если не по
По экспериментальным данным
Рис. 65. Зависимость объема раствора арсеназо III, израсходованного на титрование, от концентрации Zr в 25 мл
лучают пропорциональной зависимости в координатах А — v [84].
Конечную точку титрования находят графическим путем, а также рассчитывают алгебраическим методом.
А. Определение циркония в растворе его соли
В мерной колбе емкостью 25 мл готовят испытуемый раствор соли перхлората циркония с содержанием от 0,1 до 1 мг. В кювету для титрования переносят 0,5 мл испытуемого раствора, добавляют раствор хлорной кислоты до 18—20 мл и помещают в кювету спектрофотометра. Раствор арсеназо III добавляют из микробюретки отдельными порциями по 0,1 мл, перемешивают и измеряют оптическую плотность при X 670 нм после прибавления каждой порции реагента. По полученным данным строят кривую титрования и находят точку эквивалентности.
Б. Определение циркония в сплавах на основе магния
Навеску сплава (0,1—0,15 г) растворяют в 10 мл соляной кислоты и раствор упаривают до небольшого объема (1 мл), добавляют 30 мл 6 н. хлорной кислоты и вновь упаривают на водяной бане до удаления паров соляной кислоты. Раствор переносят в мерную колбу емкостью 50 мл и доводят объем раствора до метки хлорной кислотой. Переносят в кювету 3—5 мл этого раствора и проводят титрование см. определение циркония в растворе его соли. Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики.
ОПРЕДЕЛЕНИЯ ЦИРКОНИЯ
КОМПЛ ЕКСОНОМЕТРИЧЕС КИМ ТИТРОВАНИЕМ
В качестве индикатора при комплексонометрическом титровании используют п-нитробензолазопирокатехин.
Цирконий с комплексоном III и п-нитробензолазопирокатехином образует комплексные соединения различной устойчивости, обладающие поглощением в разных областях спектра. Область максимального поглощения комплексного соединения циркония с п-нитробензолазопирокатехином лежит при 7 510 нм, комплексного соединения циркония с комплексоном III — при 7 375 нм. Если индикатор добавлен в количестве, недостаточном для связывания всего циркония в комплексное соединение, то при титровании раствором комплексона сначала титруются свободные ионы циркония, а затем связанные в комплексное соединение с индикатором (см. рис. 24)
Кроме указанных на стр. 226, необходимы реагенты:
1. Комплексон III, 10~1 2 3 М раствор. Раствор готовят из препарата (х. ч.) по точной иавеске. Определение концентрации его проводят методом спектрофотометрическою титрования по соли циркония известной концентрации.
2. /1-Нитробензолазопирокатехин, 0,01 %-ный спиртовой раствор.
3. Соляная кислота, раствор 1:1.
к. Определение циркония в растворе его соли
В мерной колбе емкостью 25 мл готовят раствор соли перхлората циркония, содержащий от 0,1 до 5 мг Zr. В кювету для титрования переносят 5 мл испытуемого раствора, добавляют раствор хлорной кислоты до 18—19 мл, 0,2 мл раствора индикатора и помещают кювету в спектрофотометры СФ-4, СФ-5. Раствор комплексона прибавляют из микробюретки отдельными порциями по 0,1 мл, перемешивают и измеряют оптическую плотность при Z 510 нм после прибавления каждой порции реагента. По полученным данным строят кривую титрования, находят точку эквивалентности графическим и алгебраическим методами и вычисляют содержание циркония.
Б. Определение циркония в сплаве на основе магния
Две навески сплава по 0,1—0,25 г растворяют каждую в 10 мл соляной кислоты и раствор упаривают до небольшого объема (~1 мл), затем добавляют 30 мл хлорной кислоты и вновь упаривают на водяной бане до удаления паров соляной кислоты. Раствор переносят в мерную колбу емкостью 50 мл и доводят объем раствора до метки хлорной кислотой. Переносят в кювету аликвотную часть (15—20 мл) и проводят титрование точно в условиях, указанных для определения циркония в растворе его чистой соли.
Результаты параллельных определений (не менее четырех) обрабатывают методом математической статистики.
ЛИТЕРАТУРА
1.3. Марченко. Фотометрическое определение элементов. М., «Мир», 1971.
2. U. Т. НИ 1. Anal. Chem., 1956, 26, 1419.
3. Е. Б. Сендэлл. Колориметрические методы определения следов металлов. М., «Мир», 1964.
4. D. D у г s s о п, V. Dahlberg. Acta Chem. Scand., 1953, 7, 1186.
5. Ф. В. 3 а й к о в с к и й. Бюллетень научно-технической информации Мин-ва геологии и охраны недр СССР. 1957, № 7 (12) 64.
6. Т. В. Черкашина. Материалы совещания по вопросам производства и применения индия, галлия и таллия. Информация 2 (13), ч. 1. Гиред-мет, 1960.
7. А. Н а 1 a h, Е. Р u ng er. Taianta, 1971, 18, 557, 569, 577.
8. Р. A. Chalmers, A. G. S i с 1 a i г. Anal. Chim. Acta, 1965, 33, 384, 1966, 34, 312-
9. Ф. П. Судаков, Л. А. Обухова, Т. И. Ц е и с к а я. ЖАХ, 1970, XXV, 78, 5.
10. А. В. Т у. м у р о в а и др. ЖАХ, 1971, XXV, 2125.
11. Н. Е. Виноградова, Л. В. Д у б о в с к а я, О. Г. Жуковский. ЖАХ, 1964. XIX, 8, 6, 997.
12. 3. Ф. Шахова, Е. Н. Дорохова. Вести. Моск, ун-та, вып. 2, 1965, 72, 77.
13. С. Н. L и е с, D. F. Boltz. Anal. Chem., 1956, 28, 1168.
14. V. R. Morrison, A. L. Wilson. Analyst. 1963, 88, 100.
15. А. К. Бабко, A. T. Пилипенко. Фотометрический анализ. M., «Химия». М.—Л., 1968.
16. М. Morin, М. R. Paris, J. К. S с h а г f t. Anal. chim. acta, 1971, 57, № 1, 12B.
17. Е. К. Иванова, В. А. Парамонова, В. М. Пешкова. Вести. Моск, ун-та, 1968, серия 2, вып. 4, 69.
18. К. O“g a w a, N. Т о b е. Bull. Chem. Japan, 1966, 39, 223, 227.
19. J. Bjerriim, G. Schwarzenbach. Stability Constants, Chem. Soc., London, part I (1956).
20. Б. M. Никольской др. Доклады АН СССР, 1:970, 194, № 5, 1100.
21. В. М. Пешкова, А. Д. Егоров. Зав. лаб., 1936, 4, 885.
22. Н. L. Watts. Anal. Chem., 1964, 36, 364.
23. С. О. Кобякова, В. М. Савостина, Н. П. Добычи-
и а. ЖАХ, 1970, XXV, 1348.
24. М. I 1 i n s к у, G. К п о г г е. Вег., 1885, 118, 699.
25. В. М. Пешкова, В. М. Бочкова. Труды комиссии по аналитической химии, VIII (XI). Изд-во АН СССР, 1958.
26. В. М. Пешкова, Ю. А. Б а р б а л а т. Вести. Моск, ун-та, 1972, серия 2. № 1, 78.
27. В. М. Пешкова, И. Ф. Д о л м а и о в а, Н. М. Семене-в а. ЖАХ, 1963, XVIII, вып. 10, 1228.
28. В. М. Пешкова, А. А. Овсянникова. Зав. лаб., 1937, 6, 800.
29. Marczenko. Z. Anal. Chem., 1961, 6, 477; Anal. Chim. Acta, 1964, 31, 224.
30. G. Gottschalk. Z. anal. Chem., 1963, 194, 321.
31. И. П. К а л и и к и h, Г. С. С e м и к а з о в. Зав. лаб., 1961, 27,17.
32. Т. Г. Малкина, В. Н. Подчайнова. ЖАХ, 1964, XIX, вып. 6, 668.
33. Г. Шарле. Методы аналитической химии, т. 1. М.—Л., «Химия», 1969.
34. И. Ф. Д о л м а и о в а, В. П. П о д д у б и е и к о, В. М. Пешкова. ЖАХ, 1973, XXVIII, вып. 13, 592.
35. В. А. Назаренко, М. Б. Шустова, Е. И. Шеилихи-и а, ЖАХ, 1970, XXV, вып. И, 2139.
36. Г. Я- Я г и я т и и с к а я, Е. Н. П о л у э
к т о в а, Е. И. Ше-ли хи н а. Труды химии и химической технологии. Йзд-во Горьковского ун-та, 1969, вып. 3 (24), 129.
37. Л. А. ” 38. В. М.
анализа редких “ Ю. А.
И. н в. м.
1961, XVI, 596.
42. В. М. Пешкова 43. В. М. Пешкова 1948, III, 368.
R. С. Ferguson, В. М. Пешкова, В. М. Пешков 1960, XV, 610.
47. В. М. Савости ЖАХ, 1968, XXIII, 938.
48. W. С. Jonson,
49. И. Ф. Д о л м а н о в а, Г. А. Вести. Моск, ун-та, серия 2, 1964, § 2, 50. В. М. П е ш к о в а, 1952, № 10, 85.
39.
40.
41.
ЖАХ,
ЖАХ, 44. 45. 46.
ЖАХ,
Чугаев. Избранные труды, т. 1. Изд-во АН СССР, 1951. Пешкова, Н. В. Мельчакова. Сб. статей «Методы и цветных металлов». Изд-во МГУ, 1956.
Золотов, Г. Е. Власова. ЖАХ, 1973, 28, Маров и др. ЖНХ, 1975, 20, вып. 1, 135. Пешкова, В. ' ’ ~
М. Бочкова, Е. К.
а,
вып. 8, 1540.
А
стахова.
и др М.
ЖАХ, 1953, VIII, 114.
И. Ведерникова, Н. Н.
С. V.
Н. Г.
, В. М. Б о ч
Г
о и т а е в а.
Banks. Anal. Chem., 1951, 23, 448, 1486.
Игнат
ь к
на, С. О. К о б я
е в а. ЖАХ, 1962, XVII, 1086. Лазарева.
о в а, Л. И.
к о в а, В. М.
Analj4, 1946.
Пешкова.
71, 554. Пешкова.
М. S i m m о n s.
Золотова, В. M. 50.
м о в а. Вести. Моск, ун-та, Химия,
М. И. Г р
о
51. В. М. Т а р а я н, Е. В.
---- ---- ------- XIVH, 214:
В. М. Пешкова, Н. Г. Иг совещания по рению. М.
В. М. Пешкова, Н. Г. 1963, XVIII, 496.
ССР, 1967, XLV, 121; 1968,
52. “ юзного
53. ЖАХ,
В а
Р
т а и я н. Доклады АН Армянской
н а т ь е в а. Труды Второго всесо-«Наука», 1974.
- Игнатьева, Г. П. Озерова.
54. Б. Н. Райский. Зав. лаб , 1958, 24, 803.
55. Л. В. Борисова. ЖАХ, 1969, XXIV, вып. 9, 1361.
56. К. Б. Я Ц и м и р с к и й и др. Химия комплексных соединений редкоземельных элементов, Киев, «Наукова думка», 1966.
57. Д. И. Рябчиков, В. А. Рябухин. Аналитическая химия редкоземельных элементов и иттрия. М.—Л., «Наука», 1966.
58. М о е 1 1 е г Th., В г а п t 1 а у J. Anal. Chem., 1950, 22, 433, 5447.
59. Н. С. Полуэктов, Л. Н. Кононенко. Спектрофотометрические методы определения индивидуальных РЗЭ. Киев, «Наукова думка», 1968.
60. М. И. Громова, Т. И. Романцева, В. М. Пешкова.
Вести. Моск, ун-та, серия П, Химия, 1964, № 4, 57; 1966, № 4, 73; 1965, Ns 6, 74.
61. Н. С. Полуэктов Р. С. Лауэр, М. А. Санд}. ЖАХ,
1970, 25, 899, 2118; 1971, 26, 496.
62. Н. С. Полуэктов и др. Зав. лаб. 1971, 37, 895.
63. М. К- Спицин, В. С. Ш в а р е в, Т. П. П о п ы в а н о в а.
ЖАХ, 1971, 26, 86.
64. С. Б. Саввин, Т. В. Петрова, П. Н. Романов. ЖАХ, 1971, 26, 297; 1973, 28, 272.
65. К. Б. Я Ц и м и р с к и й, Л. И. Бударин, А. Г. Хачатрян. Доклады АН СССР, 1970, 4, 898; ЖАХ, 1971, 26, 1499.
66. М. И. Громова, М. Н. Литвина, В. М. Пешкова.
Вести. Моск, ун-та, серия II, Химия, 1969, № 1, 30; 1968, № 5, 98.
67. М. И. Громова, М. Н. Литвина, В. М. Пешкова.
ЖАХ, 1969, 24, 1598.
68. С. Б. Саввин. Арсеназо III. М., Атомиздат, 1963.
69. С. Б. Саввин, Ю. М. Дедков. ЖАХ, 1964, 19, 21.
70. С. Б. Саввин, Р. Ф. П р о п и с ц о в а, Р. В. Стрельникова. ЖАХ, 1969, 24, 31.
71. В. М. Пешкова, М. И. Громова, Н. М. Александр о-в а. ЖАХ, 1962, 17, 218.
72. В. А. Пешкова, В. М. Савостина, О. А. Шпигун ЖАХ, 1974, 29, вып. 3, 592.
73. А. К- Бабко, А. И. Волкова, С. Л. Л и с и ч е н о к. ЖНХ, 1966, 11, 478.
74. Г. В. Юинг Инструментальные методы химического анализа. М., Госатомиздат, 1963.
75. L. Sommer. Taianta, 1962, 9, 439.
76. Н. Irving, С. F. Bell, R. L. Williams. J. Chem. Soc., 1952, 356.
77. -R. Connik, Ney W. Me, Vey. J. Amer. Chem. Soc., 1949, 71, 3182.
78. А. С. С о л о в к и н. ЖНХ, 1957, 2, 611.
79. В. М. Пешкова, Н. В. М е л ь ч а к о в а, С. Г. Ж е м ч у -ж и н. ЖНХ, 1961, 6, 1233.
80. В. Г. Горюшииа, Е. В. Романова, Т. А. Арчакова. Зав. лаб., 1960, 26, 415.
81. С. В. Елинсон, К. И. Петров. Аналитическая химия циркония н гафния. М , «Наука», 1965.
82. Ю. М. Дедков, А. Н. Ермаков, Н. В. Корсакова. ЖАХ, 1970, XXV, вып. 10, 1912.
83. Н. В. Мильчакова, Н. И. Трубецкая, В. М. Пешко-в а. Вести. МГУ, 1964, № 2, 45.
84. Н. В. М е л ь ч а к о в а, М. Н. Станиславская, В. М. Пешкова. ЖАХ, 1964, XIX, вып. 6, 701.
85. О. А. Шпигун и др., ЖАХ, 1974 29, 597.
Глава VI
АППАРАТУРА В СПЕКТРОФОТОМЕТРИИ
1. ПРИНЦИПЫ КОНСТРУИРОВАНИЯ ПРИБОРОВ
ДЛЯ СПЕКТРОФОТОМЕТРИЧЕСКОГО АНАЛИЗА
4
Фотометрическое измерение заключается в оценке различий в интенсивности двух потоков излучений: падающего /0 на испытуемый объем и прошедшего /г через этот объект.
Общая схема фотометрической установки представлена на рис. 66. Схемы однолучевых и двулучевых приборов различаются только способом оценки соотношения /0 : /г. При измерениях на однолучевых приборах на пути потока излучения поочередно устанавливают «нулевой» образец и детектор фиксирует соответствующую интенсивность излучения /0- Затем на пути потока излучения устанавливают испытуемый образец и детектор показывает изменение в интенсивности потока излучения. При измерениях на двулучевых приборах два одинаковых по интенсивности потока излучений одновременно проходят «нулевой» и испытуемый образцы и на детектор попадает уже суммированный (при помощи соответствующего интегрирующего устройства) поток излучений.
Возможность использования какого-либо прибора для измерений в той или иной области спектра определяется прежде всего оптическими свойствами материала, из которого выполнены элементы разрешающей
Рис 66 Принципиальная схема фотометрической установки
1 — монохроматор; 2 — кювета; 3 — анализатор; 4 — детектор; 5 — усилитель, 6 — прибор--индикатор
и фокусирующей систем прибора, и в первую очередь его прозрачностью. На рис. 67 и в табл. 17 приведены данные о прозрачности некоторых оптических материалов в основных спектральных областях, в которых они используются.
Использование того или иного оптического материала определяется не только его прозрачностью, но также преломляющими свойствами.
Кюветы для работы в определенной области спекгра должны быть изготовлены из соответствующего материала.
ТАБЛИЦА 17
Прозрачность оптических материалов и спектральные области их использования [1]
Материал Область прозрачности Спектральная область использования
Различные сорта стекла Кварц кристаллический Фторид лития LiF Флуорит CaF2 Каменная соль NaCl Сильвин КС1 Бромид калия КВг 300 нм —2,5 мкм 175 йм—3,5 мкм 108 нм—7 мкм 125 нм —9 мкм 200 нм—17 мкм 220 нм—21 мкм 220 нм—28 мкм 300 нм—2,5 мкм 185 нм—400 нм 700 нм—3,5 мкм 2,5—6,0 мкм 125—200 нм 3—7 мкм 6—15 мкм 10—20 мкм 15—25 мкм
Приборы со стеклянной оптикой пригодны для работы только в видимой и ближней ИК-области спектра от 400 нм до 2,5 мкм. Для работы в УФ-области спектра необходимы приборы с кварцевой оптикой, которые менее пригодны для работы в видимой области, чем стеклянные, хотя кварц полностью прозрачен в этой области спектра.
Рис. 67. Прозрачность оптических материалов: 1 — стекло, 2 — кварц, 3 — УФ кварц: 4 — ИК-кварц
Однако в области длин волн короче 200 нм кварц непрозрачен. Кроме того, в этой области непрозрачным становится также воздух. Поэтому для измерений при X < 200 нм используют вакуумные приборы или на всем пути излучения воздух вытесняют азотом, который не имеет характерных полос поглощения в данной области. В качестве материала для преломляющей оптики в этой области используется флу-орит, прозрачный до 125 нм.
В ближней ИК-области помимо стеклянных можно использовать приборы с кварцевой оптикой (до 3,5 мкм), так как в этой области диспергирующие свойства кварца более выгодны, чем в видимой. В более далекой ИК-области спектра используют специальные оптические материалы (см. табл. 17): каменную соль, бромид калия, а также бро
мид цезия в области И—35 мкм. Флуорит и фторид лития пригодны для работы, как в УФ-, так и ИК-области.
Спектральные приборы классифицируют по следующим основным принципам:
1. По спектральным областям, в которых работают данные приборы. Это определяется, в первую очередь, прозрачностью оптических материалов и их диспергирующими свойствами.
2. По способу монохроматизации потока излучений. Для получения монохроматических излучений используют светофильтры, призмы, дифракционные решетки.
3. По типу регистрации интенсивности излучения, т. е. по характеру приемника (детектора), применяемого в данном приборе. Приемником может служить глаз, в этом случае приборы относят к типу визуальных фотометров или спектроскопов. Приборы с фотографической регистрацией называются спектрографами. Наиболее удобны в фотометрическом анализе приборы с фотоэлектрической регистрацией — фотоэлектроколориметры и спектрофотометры.
2. ХАРАКТЕРИСТИКА ОСНОВНЫХ УЗЛОВ СПЕКТРАЛЬНЫХ ПРИБОРОВ
Характеристики узлов схемы, приведенной на оис. 66, определяют возможности приборов различных конструкций, их пригодность для измерений в определенной области спектра и их принадлежность к тому или иному типу [1] — [9].
Источники излучений __
Для работы в определенной области спектра прибор должен быть снабжен источником излучений достаточной интенсивности с соответствующим спектральным интервалом.
При работе в УФ-области спектра в качестве таких источников применяют водородную и дейтериевую лампы, которые дают сплошной спектр излучения в этой области и пригодны для измерений в области 200—350 нм. Кроме того, дейтериевая лампа обеспечивает работу также в УФ вакуумной области. Ртутная лампа также дает излучения в УФ-области, но ее спектр имеет линейчатый характер, что позволяет проводить измерения только при определенных длинах волн, соответствующих линиям эмиссионного спектра ртути. Иногда это при измерении затрудняет выбор оптимальной длины волны. Тем не менее эта лампа обеспечивает определенные преимущества при работе на фотоэлектроколориметрах, в которых монохроматорами служат светофильтры.
При работе в видимой и ближней ИК-областях спектра источником излучений служит обычная вольфрамовая лампа накаливания, дающая сплошной спектр.
В ИК-области, соответствующей основным фундаментальным частотам, и далекой ИК-области применяются специальные источники тепловых излучений (глобар, лампа Нернста).
Монохроматоры
Рис. 68. Кривые пропускания светофильтров в фотоэлектроколориметре ФЭК-М (/) и фотоэлектроколориметрах ФЭК-56 и ФЭК-60 (2)
действию идеально-монохромати-
Монохроматор — основная часть спектрального прибора. Одной из главных характеристик прибора, определяющей его возможности, будет степень монохроматичности потока излучения, используемого при измерениях.
Рассмотрим основные способы получения монохроматических потоков излучений, которые используются в современных спектральных приборах.
Светофильтры. Светофильтры представляют собой твердые (стеклянные, желатиновые, целлофановые и т. п.) или жидкие среды, обладающие избирательным пропусканием излучения с достаточно узким интервалом длин волн. Каждый светофильтр характеризуется кривой пропускания с Хтах (рис. 68) и ее полушириной ДМ72, которая соответствует спектральному интервалу, ограниченному кривой пропускания светофильтра на высоте х/2 максимального.
Светофильтры в визуальных приборах характеризуются Хэфф, т. е. эффективной длиной волны, которая вычисляется по специальным формулам, учитывающим чувствительность глаза. Величина %Эфф указывает, что данный светофильтр примерно эквивалентен по своему
ческому светофильтру, пропускающему только излучение с длиной волны, к которой наиболее чувствителен глаз в области пропускания светофильтра. Для желтых и зеленых светофильтров Хэфф приблизительно совпадает с максимумом пропускания. Для красных светофильтров такое совпадение отсутствует. Например, для красного светофильтра максимум пропускания лежит в области 700—680 нм, а 2.0фф — 659 нм. Такое расхождение между максимумом пропускания и Хдфф является результатом уменьшения чувствительности глаза на краю красной части спектра.
Полуширина максимума пропускания для различных светофильтров колеблется от 100 до 30—40 нм (см. рис. 68). При измерениях на приборах, снабженных светофильтрами с ДХ ~ 100 нм, нельзя получить какое-либо представление о спектральной характеристике исследуемого объекта. В этом случае светофильтр может лишь способствовать некоторому повышению точности и чувствительности количественных определений. Такого рода светофильтрами снабжены фотоэлектрокалориметры типа ФЭК-М.
Фотоэлектроколориметры, снабженные светофильтрами с полушириной максимума пропускания 30—40 нм (ФЭК-Н-57, ФЭК-56, ФЭК-60 и т. п.), позволяют получать достаточно точную спектральную харак-
теристику большинства систем, обладающих широкими полосами поглощения. Однако аппаратура такого типа не дает возможности изучать спектры поглощения систем с узкими полосами (20—30 нм) и систем, полосы поглощения которых имеют тонкую структуру, как, например, растворы аква-комплексов редкоземельных элементов.
Рис. 69. Схема хода лучей в диспергирующей призме: к —красный конец спектра; ф— фиолетовый конец спектра
В фотоэлектроколориметрах последних марок (ФЭК-60) имеется ряд светофильтров, монохроматизирующее действие которых основано на явлении интерференции, что уменьшает полуширину максимума
пропускания этих светофильтров.
В спектральных приборах более совершенных конструкцй для мо-
нохроматизации излучений в
1, ^0---1-----1--------1___I_____1 ।
0Л 0,8 1,2 1,6 2U & 26
Я Ю'^нм
Рис. 70. Зависимость коэффициента преломления п от X для различных материалов:
1 — тяжелый флинт; 2 — кристаллический кварц; 3 — каменная соль NaCl;
4 — кронглас; 5 — сильвин КС1; 6 — плавленный кварц; 7 — флуорит CaF2
п
качестве диспер ги р ующи х элементов используются призмы и дифракционные решетки.
Призмы. Проходя призму, параллельный пучок лучей претерпевает двойное преломление и отклоняется к основанию призмы, так как коэффициент преломления материала [1], из которого сделаны призмы, всегда больше единицы (рис. 69).
Коэффициент преломления зависит не только от материала призмы (рис. 70), но также и от длины волны падающего излучения (лучи с малыми значениями X отклоняются сильнее). Поэтому лучи с различной длиной волны выходят из призмы под разными углами. Это позволяет получить в плоскости, перпендикулярной направлению хода лучей, вышедших из призмы, серию излучений различной монохроматичности.
Одной из основных характеристик призмы является обратная Линейная дисперсия — dUdl, которая показывает, сколько нанометров укладывается на расстоянии 1 мм в фокальной плоскости собирающей линзы. Обратная линейная дисперсия дает определенное представление о разрешающей способности спектральных приборов.
Обратной линейной дисперсией призмы определяется разрешающая сила R прибора, которая представляет собой отношение средней длины волны двух самых близких линий, разрешаемых данным прибором, к разности их длин волн. Две спектральные линии равной интенсивности считаются разрешенными, если дифракционный максимум одной из них находится против минимума другой линии (рис. 71).
7? = У/ДУ, ^k=pdkldl, R = Ullpdf., где р — наименьшее расстояние между двумя линиями, раздельно регистрируемыми прибором. Чем меньше обратная линейная дисперсия, тем выше разрешающая сила прибора. Обратная линейная дисперсия
Рис. 71. Распределение интенсивности в дифракционном изображении спектральной линии за щелью монохроматора (вид двух разрешаемых спектральным прибором линий)
Рис 72. Зависимость коэффициента преломления материала от длины волны в области его максимального поглощения
спектральных приборов низкого разрешения равна — 50 нм/мм, среднего— ~1 нм/мм и высокого—~0,1 нм/мм. Соответственно разрешающая сила приборов меняется от 103 до 10s.
Обратная линейная дисперсия зависит как от материала призмы, так и от конца спектра: d’Kidl для данной призмы различна для длин волн в ИК- и УФ-областях. Поэтому выбор оптического материала для работы в той или иной части спектра определяется не только его прозрачностью, но также его преломляющими свойствами. По мере приближения к области максимального поглощения материала, из которого сделана призма, показатель преломления возрастает (рис. 72), а следовательно, уменьшается обратная линейная дисперсия призмы и увеличивается разрешающая способность прибора, но при этом падает его светосила. Поэтому приборы с кварцевой оптикой пригодны для работы не выше 7600 нм, так как при больших длинах волн сильно возрастает обратная линейная дисперсия, хотя кварц прозрачен не только в ультрафиолетовой части спектра, но также в видимой и ИК-области до 3,5 мкм.
Для области спектра длин волн больше 45 нм призменные приборы непригодны и используются только приборы с дифракционными решетками.
Рис 73. Ход лучей после прохождения через дифракционную решетку
Дифракционная решетка. Дифракционная решетка представляет собой большое число щелей, расположенных близко друг к другу на одинаковых расстояниях (рис. 73). Претерпевая дифракцию от каждой из этих щелей, параллельный пучок излучения превращается в ряд точечных источников излучений, число которых равняется числу щелей. Излучения от этих точечных источников распространяются по всем направлениям. Если направление плоской волны, падающей на решетку, перпендикулярно ее поверхности, то предполагают, что фаза колебаний вторичных волн, излучаемых точечными источниками, одинакова.
Расстояние от начала одной щели до начала другой называют постоянной дифракционной решетки. Обозначим ее d. Пусть разность хода лучей от двух соседних щелей будет равна й; она будет одинакова для всех соседних щелей. Разность хода лучей будет зависеть от угла а. Если на пути лучей, выходящих из решетки, поставить линзу так, чтобы ее главная ось совпадала с их направлением, то в главном фокусе линзы соберутся все лучи, исходящие от всех щелей под углом а. В направлениях, где геометрическая разность хода двух лучей окажется равной целому числу длин волн, будут наблюдаться максимумы интенсивности (дифракционные максинаправлении все вторичные волны, наклады
ваясь, усиливают друг друга. Поэтому условие получения дифракционного максимума выражается уравнением
a=d sin « = Л/Х, которое называется уравнением дифракционной решетки. Поскольку N может равняться любому целому числу, то должно наблюдаться соответствующее число максимумов интенсивности, или порядков. Во всех направлениях, не соответствующих целочисленным значениям 7V, будет наблюдаться ослабление интенсивности вплоть до минимума. Таким образом, для монохроматического излучения (ZJ в фокальной плоскости линзы получим зависимость интенсивности от угла а, пред-сталенную на рис. 71. Для каждой другой длины волны (Z2) получается аналогичная картина. Следовательно, в результате прохождения излучений через диффракционную решетку получают целую группу спектров различных порядков.
Чем больше число щелей и меньше постоянная дифракционной решетки, тем уже и более интенсивными становятся дифракционные максимумы. При этом расстояние между дифракционными максимумами, соответствующими излучениям с различными длинами волн, увеличивается с возрастанием порядка спектра (рис. 74). Следовательно, в приборах высокого разрешения должны использоваться решетки с большим числом щелей, работающие в достаточно высоких порядках.
, так как в этом
Длина волны
Л 2 4 у ^-2 ^2 1 4; ^2 4; ^-2^1 ^2
Порядок спектра
Рис. 74. Распределение спектров различных порядков, получающихся при помощи дифракционной решетки для двух ДЛИН ВОЛН /.1 И /-2
Прозрачные дифракционные решетки изготовляют нанесением на какой-либо прозрачный материал большого числа штрихов (600____
1200 штрихов на 1 мм), промежутки между которыми и представляют собой щели. В настоящее время они почти полностью вытесняются отражательными решетками, которые представляют собой отражающую поверхность, на которой резцом выдавлено большое число параллельных друг другу канавок одинакового профиля и на одинаковом расстоянии. Дифракционные решетки, работающие на отражение, имеют то преимущество, что спектральная область их применения не зависит от прозрачности материала, использованного для их изготовления. Поэтому отражательные решетки пригодны для работы в довольно широких спектральных областях.
Основным преимуществом, приборов с дифракционными решетка
ми по сравнению с призменными приборами является то, что дисперсия их не зависит от длины волны и они обладают одинаковой разрешающей способностью во всех участках спектра.
Детекторы
2 5
80V Д0\-20
900 500 600 700 800 900 Л,нм
Рис. 75. Кривые спектральной чувствительности фотоэлементов:
/— сурьмяио-цезиевый; 2 — селеновый;
3 — глаз, 4 — кислородно цезиевый
Визуальный детектор (глаз). Приборы с визуальной регистрацией излучений, в которых детектором служит глаз, пригодны для работы только в видимой области спектра: глаз человека чувствителен к излучениям с длинами волн от 400 до 700 нм (рис. 75) с максимумом чувствительности в зеленой области (Z 550 нм). Кроме того, при работе на визуальных приборах неизбежны субъективные ошибки, свойственные наблюдателю.
В прецизионных приборах используется объективный способ оценки интенсивности’ потоков излучений, основанный на применении светочувствительных датчиков или фотографических пластинок.
Фотографическая пластинка. Приборы с фотографической регистра
цией излучений более удобно использовать в эмиссионном спектральном анализе. Хотя приборы такого типа могут быть использованы и для спектрофотометрического анализа. Для этого следует заменить дугу или искру каким-либо более стабильным источником излучения. Для получения зависимости поглощения от длины волны необходимо
измерять на фотоэлектрическом микрофотометре интенсивность почернений на фотопластинке при соответствующих длинах волн.
Фотоэлементы. Фотоэлементы как приемники излучений наиболее часто используют в современных спектральных приборах, применяемых для количественного фотометрического анализа—фотоэлектроколориметрах, а также в нерегистирующих спектрофотометрах.
Явление фотоэффекта, открытое А. Г. Столетовым в 1883 г., заключается в том, что под действием света с поверхности различных тел вырываются электроны, вследствие чего данное тело приобретает заряд. Причем это явление наблюдается только при условии, если энергия светового кванта больше работы, необходимой для отрыва электрона с поверхности данного вещества, и сообщения ему некоторой кинетической энергии. Энергия светового кванта определяется длиной волны света Л или частотой его колебаний у;
E—hv=h с/'К.
Таким образом, для каждого фоточувствительного вещества существует определенная длина волны (или соответственно частота колебания) света, называемая порогом фотоэффекта, при которой начинает наблюдаться фотоэффект.
Фотоэлементы характеризуются спектральной чувствительностью, т. е. чувствительностью к определенным длинам волн электромагнитного излучения и интегральной чувствительностью — чувствительностью к суммарному потоку излучения сложного спектра. Спектральное распределение чувствительности для различных фотоэлементов зависит от природы фоточувствительного слоя (см. рис. 75). Кроме того, спектральная чувствительность фотоэлементов сильно зависит от температуры. А. Г. Столетовым было установлено, что сила фототока прямо пропорциональна интенсивности падающего на фотоэлемент излучения, хотя строгая пропорциональность существует только для монохроматических излучений.
При работе с фотоэлементами на получение точных и воспроизводимых результатов влияет ряд факторов.
{. «Старение» фотоэлементов, так как спектральная и интегральная (равная силе тока, возникающего при освещении поверхности элемента световым потоком в 1 лм) чувствительность их может со временем меняться.
2. «Утомление» (уменьшение силы фототока со временем) фотоэлементов, которое наблюдается при длительном непрерывном освещении фотоэлемента достаточно ярким светом. Поэтому во время работы необходимо временное прекращение облучения фотоэлемента.
3. Неодинаковость чувствительности разных участков поверхности фотоэлемента. Поэтому следует осветитель настраивать так, чтобы при параллельных измерениях всегда освещался один и тот же участок поверхности фотоэлемента. Диаметр освещаемого пятна должен быть равен примерно 1 см. Иногда равномерная освещенность фотоэлементов достигается применением матовых рассеивателей.
По принципу действия фотоэлементы подразделяются на: 1) фотоэлементы с запирающим слоем (вентильные), 2) фотоэлементы с внеш
ним фотоэфектом и 3) фотоэлементы с внутренним фотоэффектом [3,5] (фотосопротивления).
1. Действие фотоэлементов с запирающим слоем заключается в том, что световой поток, падающий на поверхность полупроводника, нанесенного на железную пластинку и обладающего односторонней проводимостью, возбуждает на ней движение электронов, которые не могут проникнуть в нижний слой (фронтальный фотоэффект). Если соединить верхний и нижний слои каким-либо проводником через гальванометр, можно измерить фототок, появляющийся во внешней цепи. Вентильные фотоэлементы обладают некоторым преимуществом перед фотоэлементами с внешним фотоэффектом, так как не требует дополнительных источников питания и имеют невысокое внутреннеее сопротивление, что позволяет непосредственно подключать к ним измерительный прибор. При непосредственном включении в цепь вентильного фотоэлемента измерительного прибора необходимо, чтобы последний обладал малым внутренним сопротивлением. Из вентильных фотоэлементов наибольшее распространение получил селеновый фотоэлемент [4].
Спектральная чувствительность селенового фотоэлемента и глаза очень близки (см. рис. 75), поэтому приборы с селеновыми фотоэлементами пригодны для работы только в видимой области спектра. Селеновые фотоэлементы получили широкое распространение, так как имеют ряд положительных свойств. Интегральная чувствительность их достаточно велика (350—500 лм), что позволяет использовать гальванометры с чувствительностью 10-6—10-7 А. Селеновые фотоэлементы обладают инертностью и после включения источника освещения ток стабилен. Чувствительность данных фотоэлементов уменьшается по прошествии года не более чем на 1 %.
Кроме селеновых фотоэлементов с фронтальным фотоэффектом, существуют селеновые фотоэлементы с тыловым фотоэффектом, у которых электроны выбиваются под действием света на границе между селеном и железом. Эти фотоэлементы обладают большим внутренним сопротивлением, вследствие чего сила тока во внешней цепи мала. Они обладают более постоянными характеристиками. К фотоэлементам с запирающим слоем относятся и меднозакисные [запирающий слой окись меди (I)], серносеребряные (запирающий слой сульфид серебра) и серноталлиевые (запирающий слой сульфид таллия). Меднозакисный фотоэлемент не выдерживает сравнения с селеновым из-за малой интегральной чувствительности большой зависимости его фототока от температуры. Серносеребряный и серноталлиевый фотоэлементы обладают более высокой интегральной чувствительностью и могут быть использованы в более широкой спектральной области, чем селеновый фотоэлемент. Их широко используют для измерений в ближней инфракрасной области спектра.
2. Фотоэлементы с внешним фотоэффектом представляют собой эвакуированный или газонаполненный баллон с двумя электродами. При этом катод является светочувствительным. Выбитые из светочувствительного катода электроны устремляются к аноду, в результате чего во нешней цепи возникает электрический ток. Спектральная
Рис 76 Схема многокаскадного фотоумножителя:
К — катод; А — анод; /—4 — диноды
чувствительность такого фотоэлемента зависит от материала катода. Вакуумные и газонаполненные фотоэлементы имеют различные вольт-амперные характеристики [5]. Характеристика вакуумного фотоэлемента имеет вид кривой с насыщением. Зависимость фототока насыщения от интенсивности падающего излучения линейная, что и используется для измерения интенсивности светового потока. Вакуумные фотоэлементы обладают малой инерционностью и могут применяться в быстро действующих схемах.
В газонаполненных фотоэлементах происходит некоторое усиление фототока за счет вторичных электронов, выбиваемых из молекул газа. Однако газонаполненные фотоэлементы менее устойчивы в работе, так как при большом освещении может наступить газовый разряд фотоэлемент перестанет быть управляемым. Кроме того, эти фотоэлементы обладают большой инерционностью и не пригодны для работы с прерывистым освещением, а также они имеют более высокий уровень шумов.
Характеристики фотоэлементов с внешним фотоэффектом мало зависят от температуры, поэтому они могут быть исполь
зованы без термостатирования. К недостатку этих фотоэлементов относится их утомляемость, выражающаяся в уменьшении чувствительности при длительной работе. Величина фототока в данных фотоэлементах может составлять доли микроампера, поэтому требуется дополнительное его усиление. При этом усилитель фототока должен обладать большим входным сопротивлением, так как внутреннее сопротивление фотоэлементов составляет несколько сотен мегомов.
Из фотоэлементов с внешним фотоэффектом наиболее распространены сурьмяно-цезиевые и кислородно-цезиевые. Первые имеют максимум чувствительности в области 430 нм и используются для работы в ультрафиолетовой и видимой областях спектра. Вторые являются газонаполненными и имеют два максимума чувствительности (см. рис. 75): один в УФ-области, около 350 нм, второй в близкой ПК-области, около 800 нм. Минимум чувствительности лежит в области 500 нм. Поэтому кислородно-цезиевые фотоэлементы используют обычно для работы в ближней ПК-области.
По принципу действия вакуумным фотоэлементам аналогичны фотоэлектронные умножители (ФЭУ) [1,5]. Электроны, вылетающие из катода под влиянием падающего (рис. 76) излучения, попадают на промежуточный электрод—динод и выбивают из него дополнительное количество электронов, которые, попадая на следующий динод, вызывают появление новых электронов. Усиленный таким образом ноток электронов достигает анода. При десяти каскадах в таком умножителе можно достигнуть усиления порядка миллиона. Чувствительность ФЭУ и темновой ток сильно зависят от напряжения на динодах. По
этому при работе с фотоумножителями необходимо устанавливать оптимальный режим питания, который обычно указывают в паспорте ФЭУ. Зависимость анодного тока ФЭУ от интенсивности падающего излучения линейна только до определенного значения. Спектральная чувствительность ФЭУ также определяется материалом катода, которые чаще всего бываю! цезиевыми или сурьямно-цезиевыми. Одним из достоинств ФЭУ является также их малая инерционность. ФЭУ заключают в светонепроницаемый экран, который одновременно служит защитой от электростатических и электромагнитных полей.
3. Действие фотосопротивлений основано на внутреннем фотоэффекте, при котором сопротивление полупроводника зависит от его освещения. Под действием света электроны из кристаллической решетки полупроводника переходят в свободное состояние (в зону проводимости). Изменение сопротивления обнаруживается по изменению тока в слое проводника.
В фотосопротивлениях используют таллофид (смесь таллия, серы и кислорода) с максимумом чувствительности в области около 1 мкм, теллурид свинца с максимумом в области 4,5 мкм, селенид кадмия с максимумом в области 0,75 мкм, сульфид свинца с максимумом в области 2,4 мкм, сульфид висмута с максимумом в области 0,7 мкм. Таким образом, большинство фотосопротивлений пригодно для работы в ИК-области спектра. Только фотосопротивления из сульфида кадмия имеют максимум чувствительности в видимой области и пригодны для работы в этом участке спектра.
К недостаткам фотосопротивлений относится их большая инерционность и чувствительность к изменениям температуры. Однако некоторые фотосопротивления обладают высокой чувствительностью, сравнимой даже с чувствительностью фотоумножителей (например, фотосопротивления из сульфида кадмия ФС-КМ) или выше чувствительности фотоэлементов (например фотосопротивления из селенида кадмия: ФС-ДО, ФС-Д1, ФСД-П) [5].
В качестве детекторов могут быть использованы фотодиоды и фототриоды, максимум чувствительности которых лежит так же, как у большинства фотосопротивлений, в ИК-области. Для работы в инфракрасной области спектра, соответствующей основным фундаментальным частотам, применяют специальные тепловые приемники излучений — термоэлементы и балометры.
Кюветы
Жидкие образцы перед измерением должны быть помещены в соответствующие кюветы.
Выбор кювет. Объем кюветы выбирают в зависимости от количества раствора. При незначительном количестве слабопоглощающего раствора следует использовать узкие кюветы, но с достаточной толщиной слоя (/)•
Основным критерием выбора кювет является необходимость выдерживать оптимальным интервал измеряемых оптических плотностей (см. гл. I). При построении градуировочного графика концентра
ции эталонных растворов и толщина слоя кюветы должны быть выбраны такими, чтобы измеренные оптические плотности находились в интервале ~ 0,1 — 1,0. При определении очень малых концентраций измеряемая оптическая плотность может быть ниже 0,1, но при этом для обработки экспериментальных результатов необходимо применять методы математической статистики.
Типы кювет. В зависимости от конструкции прибора кюветы имеют различную форму. Наиболее распространены прямоугольные кюветы (рис. 77). Наборами таких кювет снабжено большинство фотоэлектроколориметров и спектрофотометров. В каждом из наборов
Рис. 77. Кюветы:
1 — прямоугольные; 2 — цилиндрические; 3 — тефлоновые
имеется несколько пар кювет с толщиной слоя от 1 до 5 см. Парные кюветы должны иметь одинаковую пропускаемость при заполнении их одинаковыми растворами. В комплекте спектрофтометров СФ-4, СФ-4А, СФ-5, СФ-16 имеются два вида кювет: прямоугольные кварцевые и цилиндрические со сменными кварцевыми и стеклянными крышками.
Для измерения поглощения растворов малых концентраций и малых объемов для повышения чувствительности определения удобно использовать цилиндрические кюветы г большой толщиной слоя, но малым диаметром. Представленные на рис. 77 [10] кюветы с толщиной слоя 10 см сделаны из тефлона и имеют кварцевые окна диаметром 5 мм. Для их заполнения требуется лишь около 2,5 мл испытуемого раствора. Однако следует уделять особое внимание тщательности их установки в кюветном отделении в положении, строго параллельном световому потоку. Это достигается при помощи специального держателя (рис. 78).
Обращение с кюветами. Большое значение в спектрофотометрии имеет чистота рабочих стенок кювет. Многие вещества обладают способностью адсорбироваться на поверхности и загрязнять стенки кювет. Это может вызвать большие ошибки при измерении оптических
плотностей исследуемых растворов. Поэтому не следует надолго оставлять кюветы с испытуемыми растворами. По окончании работы их необходимо сразу же вымыть, ополоснуть дистиллированной водой и вытереть снаружи мягким материалом. Во время мытья или вытирания нельзя скоблить стенки кювет чем-либо жестким, чтобы не повредить их поверхность. Не следует допускать вытекания раствора из кюветы на внешнюю стенку или в кюветное отделение, так как это сильно меняет прозрачность стенок кювет.
Для очистки кювет от загрязнений их необходимо на некоторое время опустить в концентрированную соляную кислоту, после чего
Рис. 78. Держатели кювет к спектрофотометрам типа СФ-4:
1 — для прямоугольных кювет; 2 — для цилиндрических кювет;
3 — для твердых образцов; 4 — для тефлоновых кювет
тщательно промыть водой. Не следует для мытья кювет использовать бихромат калия в серной кислоте, так как он содержит компоненты, имеющие интенсивную окраску и способные адсорбироваться на стенках кювет. От огранических веществ кюветы следует очищать органическими растворителями. Пропускание кювет, наполненных водой, следует периодически сверять с данными аттестата.
Заполнение кювет растворами. Чисто вымытые кюветы насухо вытирают снаружи. Не следует высушивать кюветы изнутри при работе с водными растворами.
Перед заполнением кюветы ее следует ополоснуть небольшой порцией исследуемого раствора, чтобы его концентрация не изменилась вследствие разбавления водой, оставшейся в кювете. Если измеряют плотность ряда эталонных растворов, то мыть кювету перед заполнением ее очередным раствором, отличающимся только по концентрации, не следует. Перед заполнением кюветы очередным раствором следует только ополоснуть ее этим же раствором. Вначале измеряют оптическую плотность менее концентрированных растворов, затем более концентрированных.
При работе с растворами в органических растворителях, несмеши-вающихся с водой, кювету следует высушить изнутри. Нельзя приме
нять воздушную сушилку, так как это вызывает загрязнение кювет, а кроме того, кювета может дать трещины. Для ускорения процесса высушивания можно после дистиллированной воды последовательно ополоснуть кювету спиртом и эфиром. Работа с органическими растворителями во избежание их испарения требует более тщательного закрывания кювет пробками; иногда для этого используют кюветы с тефлоновыми крышками. Заполняют кювету до такого уровня, чтобы поток излучения попадал целиком в слой раствора.
Установка кювет в кюветное отделение. Кюветы устанавливают в кюветное отделение всегда в строго определенном положении, чтобы избежать ошибок, связанных с различиями в отражении или рассеянии излучений. При длительной работе происходит некоторый нагрев прибора. Температура в кюветном отделении повышается, в результате чего на внутренних стенках кювет появляются пузырьки воздуха, иногда раствор в кювете мутнеет, что сильно меняет поглощение. Поэтому требуется заново наполнить кювету.
Для твердых образцов в некоторых приборах имеются специальные держатели (см. рис. 78), конструкция которых определяет минимальные размеры испытуемого образца.
3. ОПТИЧЕСКИЕ СХЕМЫ И ПРИНЦИПЫ ДЕЙСТВИЯ
РАЗЛИЧНЫХ ФОТОМЕТРИЧЕСКИХ ПРИБОРОВ
В зависимости от характеристик каждого узла (см. рис. 66) прибор будет обладать теми или иными возможностями, что определяет целесообразность использования его при решении конкретных задач спектрофотометрии.
В зависимости от характера спектров поглощения изучаемых систем (широкополосных, узколинейчатых или имеющих тонкую структуру спектральных полос) при работе на приборах, имеющих различную монохроматичность потоков излучений, могут быть получены или совершенно идентичные, или резко отличающиеся спектральные характеристики. Изучение спектров поглощения систем, обладающих тонкой структурой спектров, требует использования приборов с высоко монохроматизированным потоком излучения (призменные приборы или приборы с дифракционными решетками). Для проведения количественного анализа в большинстве случаев достаточно иметь прибор, в котором монохроматизация осуществляется при помощи светофильтров.
Однако недостаточная монохроматичность потока излучения может вызвать несоблюдение закона поглощения излучений и снижение чувствительности фотометрической реакции, а также не позволит проводить фотометрический анализ многокомпонентных систем.
Чтобы получить спектр поглощения при работе на нерегистрирующих приборах, необходимо произвести измерения оптической плотности при ряде длин волн, которые входят в изучаемую область спектра, и затем построить графическую зависимость величины А от длины
волны X. При работе на приборах* со светофильтрами следует соответственно произвести измерения со всеми имеющимися в приборе светофильтрами и построить зависимость А или Т от Хтах, его пропускания. Регистрирующий прибор позволяет сразу получить спектр поглощения в виде записи на бланке. Значения А (Г), соответствующие какой-то определенной длине волны X (или v), могут быть получены путем отсчета по бланку.
Перед началом измерений на приборе необходимо провести проверку правильности работы всех его узлов. Тесты, которые при этом необходимы, указаны в описании соответствующего прибора. Для получения надежных результатов при измерении величин А и Т отсчет по шкале прибора следует делать несколько раз, повторяя балансировку полностью до получения воспроизводимых результатов. Иногда полезно также повторно наполнить кювету и сделать измерение вновь. Отсчет по шкале производят с точностью, указанной в аттестате соответствующего прибора. Для спектрофотометров она будет равна 0,001, а для фотоэлектроколориметров — 0,01 А.
Фотоэлектрический колориметр ФЭК-М
Фотоэлектроколориметр ФЭК-М относится к типу объективных приборов, в которых детекторами служат селеновые фотоэлементы.
Фотоэлектроколориметр ФЭК-М имеет стеклянную оптику, прозрачную только для лучей видимого участка спектра. В качестве источника излучений служит лампа накаливания (вольфрамовая лампа), дающая излучение в видимой части спектра. Селеновые фотоэлементы чувствительны только к излучениям видимого участка спектра. Следовательно, данный прибор пригоден для измерений в интервале 400— 700 нм. Кроме того, для работы в этом интервале прибор снабжен тремя светофильтрами с полушириной пропускания 80—100 нм (см. рис. 68) и поэтому его используют только при определении концентрации. Он непригоден для изучения спектров поглощения.
Принципиальная схема прибора ФЭК-М представлена на рис. 79, внешний вид—на рис. 80. Конструкция прибора основана на принципе уравнивания интенсивности двух световых потоков при помощи щелевой диафрагмы. От источника света 1 (см. рис. 79) световые лучи отражаются двумя зеркалами 2, 2', проходят светофильтры 3,3', кюветы 4,4' с растворами и попадают на селеновые фотоэлементы 5,5'. Фотоэлементы включены через гальванометр 10 (см. рис. 80) по дифференциальной схеме: при одинаковой интенсивности потоков лучей стрелка гальванометра стоит на нуле. На пути левого пучка света, падающего на фотоэлемент 5, расположены нейтральные клинья 6 и 7; на пути правого пучка света, падающего на фотоэлемент 5', щелевая диафрагма 8, связанная с двумя отсчетными барабанами 9,9' (см. рис. 80). Барабаны имеют две шкалы: красную— оптическая плотность, черную — процент пропускания.
* При опис ании прибора главное внимание уделяется его оптической схеме и лишь незнач ите.:ъно затронуты данные об электрических схемах.
Рис 79. Принципиальная оптическая схема фотоэлектроколориметра ФЭК-М
Рис 80. Общий вид фотоэлектроколориметра ФЭК-М
Порядок работы. Работу на приборе следует начинать с проверки установки осветителя. Лампу осветителя включают через стабилизатор за 15—20 мин до начала работы, чтобы она успела приобрести устойчивую эмиссию. Чтобы световой поток был равномерным и полностью заполнял отверстие измерительной дифрагмы и нейтральные клинья, нить лампы осветителя должна изображаться на поверхности одного из светофильтров в виде резкой и отчетливой спирали, расположенной в центре. Изображение нити наблюдают, прикрыв светофильтры папиросной бумагой. Устанавливают осветитель при помощи регулировочных винтов.
После проверки установки осветителя можно приступать к измерениям. Включают гальванометр при помощи рукоятки 11, которая имеет три положения: «0» — гальванометр выключен, «1» малая чувствительность, «2» большая чувствительность. Начинать работу всегда следует при положении рукоятки на малой чувствительности «1». Большая чувствительность включается после того, как произведено уравнивание световых потоков при малой чувствительности. Ни в коем случае не следует перемещать рукоятку 11 вправо за положение «2», так как при обратном переключении гальванометр сразу включается на большую чувствительность, что приводит к его порче.
На фотоэлектроколориметре ФЭК-М оптическую плотность (или коэффициент пропускания) можно измерять двумя способами: по правому и левому барабану. Шкала правого барабана имеет значения А от 0 до 0,52; Т — от 100 до 30%. Шкала левого барабана имеет значения А от 0 до 2,5; Т — от 100 до 0%.
Измерение оптической плотности (или процента пропускания) по правому барабану производят следующим образом:
а) устанавливают по шкале правого барабана 9’ (см. рис. 80) оптическую плотность на «0» (при этом щель диафрагмы имеет минимальную ширину);
б) вводят кюветы с растворителем или раствором, по отношению к которому производится измерение оптической плотности испытуемого раствора в правый и левый пучок света. Кюветы устанавливаются в кюветодержатели 12, каждый из которых имеет три гнезда для установки кювет и вращается на оси, имеющей три фиксированных положения. Правая и левая кюветы должны быть установлены в кювето-держателях на одинаковом расстоянии от входного отверстия;
в) устанавливают определенные светофильтры поворотом рукоятки 13 на пути потоков излучения;
г) компенсируют фототоки, устанавливая стрелку гальванометра на «0», пользуясь рукоятками нейтральных клиньев: 14 — грубая настройка, 15 — плавная, при помощи которых меняется освещенность левого фотоэлемента. Компенсация производится сначала при положении «I» выключателя II, а затем — «2»;
д) вводят в правый световой поток кювету с испытуемым раствором, поставив выключатель 11 в положение «0», поворотом кювето-держателя. При этом стрелка гальванометра отклоняется; ее возвращают в нулевое положение при помощи правого барабана 9', по шкале которого производят отсчет оптической плотности или процента пропускания. Поставив поворотом кюветодержателя в правый световой поток кювету с раствором другой концентрации, можно, не меняя компенсации фототоков нейтральными клиньями, произвести измерение плотности второго раствора и т. д. Смена кювет производится при положении руксятки И на «0». Оптическую плотность концентрированных растворов измеряют по левому барабану в следующем порядке:
а) устанавливают кювету с растворителем на пути левого светового потока, на пути правого — с исследуемым раствором;
б) устанавливают левый барабан в положение, соответствующее «0» оптической плотности (или 100% пропускания). При этом диафрагма оказывается полностью открытой;
в) подводят стрелку гальванометра к 0 при помощи нейтральных клиньев;
г) ставят кювету с растворителем на пути правого светового потока, при этом освещенность правого фотоэлемента увеличивается и необходимо закрывать правую диафрагму до тех пор, пока стрелка гальванометра не придет в нулевое положение. Отсчет производят по шкале левого барабана.
Работа с правым барабаном имеет ряд преимуществ: во-первых, точность отсчета по шкале правого барабана значительно выше, так как на полный оборот его приходится меньший интервал значений, например, оптической плотности от 0 до 0,52, в то время как на тот же оборот левого барабана — от 0 до 2,0; во-вторых, если светофильтр не меняют, по правому барабану можно измерить плотность всего ряда
9 Зак. 518
249
эталонных растворов при однократной компенсации фототоков нейтральными клиньями.
Определение концентрации испытуемого раствора начинают с выбора светофильтра. Фотоэлектроколориметр ФЭК-М снабжен всего тремя широкополосными светофильтрами: зеленым, красным и синим (спектральные характеристики светофильтров представлены на рис. 68). Светофильтры устанавливаются попарно на пути световых потоков при помощи рукоятки 13. С каждым из светофильтров измеряют оптическую плотность А или пропускание Т одного из растворов эталонного ряда и выбирают светофильтр согласно общим принципам.
Фотоэлектрический колориметр-нефелометр ФЭК-56
Фотоэлектроколориметр ФЭК-56 относится к типу объективных приборов, в основу конструкции которого положен принцип уравнивания интенсивности двух световых потоков, падающих на два фотоэлемента, при помощи диафрагмы с переменной шириной. Фотоэлементы включены по дифференциальной схеме: равенство световых потоков (а следовательно, и фототоков) соответствует нулевому показанию прибора-индикатора.
Помимо оптических плотностей и процентов пропускания, прибор позволяет измерять турбидиметрически (в проходящем свете) светорассеяние, вызываемое взвесями, эмульсиями и коллоидными частицами. Светорассеяние растворов со слабой мутностью на данном приборе измерить невозможно.
Прибор имеет оптику, которая позволяет работать в ближайшей УФ-области спектра от 300 нм. Приемниками световой энергии служат сурьмяно-цезиевые фотоэлементы.
В качестве источников света в приборе используют две лампы: лампу накаливания, дающую сплошной спектр испускания в видимой области; ртутно-кварцевую лампу, дающую линейчатый спектр испускания в ультрафиолетовой и видимой областях. В качестве монохроматоров служат светофильтры с узкими полосами пропускания 30— 40 нм. Прибор может быть использован как упрощенный спектрофотометр при изучении спектров систем, обладающих широкими полосами поглощения, для измерений в области 300—700 нм. Максимумы пропускания большинства светофильтров практически совпадают с рядом линий в эмиссионном спектре ртути (табл. 18). Поэтому с ртутно-кварцевой лампой можно производить измерения в ультрафиолетовой и видимой областях спектра с очень узкими монохроматическими пучками при следующих длинах волн (нм): 577,9; 546; 436; 405,8; 365 313.
Оптическая схема прибора дана на рис. 81, общий вид—на рис. 82. Свет от источника 1 (см. рис. 81), пройдя через светофильтр 2, попадает на призму 3, которая делит пучок на два: левый и правый. Далее параллельные пучки света проходят кюветы 4,4', диафрагмы 5 и 5' и попадаю" на фотоэлементы 6,6', включенные по дифференциальной схеме через усилитель на индикаторную лампу 7.
ТАБЛИЦА 18
Характеристики светофильтров фотоэлектроколориметра ФЭК-56
Номер светофильтра Длина волны, соответствующая максимуму пропускания, нм Длина волны, соответствующая линии испускания ртутной лампы, нм Номер светофильтра Длина волны, соответствующая максимуму пропускания, нм Длина волны, соответствующая линии испускания ртутной лампы, нм
1 315 313 5 490
2 364 365 6 540 546
3 400 405, 408 7 582 577
4 440 436 8 610 —
Неодинаковая освещенность фотоэлементов вызывает размыкание сектора индикаторной лампы, которая служит прибором-индикатором. Интенсивности световых потоков, проходящих через правую 4' и левую 4 кюветы, уравнивают при помощи диафрагм «кошачий глаз»: левая 5 является компенсационной и служит для балансировки нуля отсчета на шкале барабана правой диафрагмы 5'; правая диафрагма является измерительной; отсчет величин поглощения производят всегда по шкале правого барабана. На пути левого светового потока
Рис. 81. Принципиальная оптическая схема фотоэлектроколориметра ФЭК-56
постоянно находится кювета с нулевым раствором (например, с растворителем). На пути правого светового потока могут быть последовательно установлены кюветы с испытуемым и нулевым растворами.
Лампа накаливания и ртутно-кварцевая лампа имеют каждая собственную панель 8 (см. рис. 82), при помощи которой их поочередно устанавливают в осветителе 9, вдвигая по пазам 10 и закрепляя винтом 11. Лампу накаливания 12 включают в телефонные гнезда 13, ртутно-кварцевую лампу 14 — в телефонные гнезда 15. В диск, на-
9*
251
холящийся на задней стенке корпуса прибора, вмонтированы девять светофильтров. Усилитель и источник света получают питание от сети переменного тока через специальное питающее устройство.
Порядок работы. Питающее устройство соединяют с прибором и
включают в сеть переменного тока напряжением 220 В. Переключателем ламп на панели стабилизатора включают соответствующую лам-
пу. Включают стабилизатор и дают усилителю и лампе прогреться 20—30 мин. При работе с ртутно-кварцевой лампой усилитель прогревают при выключенной лампе, включая ее за 5 мин до начала измерений.
Устанавливают лампу при помощи юстировочных винтов 16 (см. рис. 82, б); в трубки измерительных диафрагм вставляют металлические пробки, имеющиеся в комплекте прибора. При помощи юстировочных вин
Рис. 82. Общий вид фотоэлектроколориметра-нефелометра ФЭК-56: а — вид спереди; б — задняя панель с осветителем
тов 16 добиваются положения, когда пучки света будут располагаться концентрически относительно центров металлических пробок.
После включения прибора в сеть переменного тока следует: 1) установить «электрический нуль» прибора: пучки света перекрыть шторкой, нажав рукоятку 17 (см. рис. 82) внутрь вдоль оси, после чего повернув ее в положение «3». Рукояткой 18 сомкнуть края сектора индикаторной лампы 7. Затем вновь открыть шторку рукояткой 17 (положение «0»); 2) поставить в левый кюветодержатель кювету с раствором сравнения и оставить ее там на все время работы. В правый кювето-
держатель поставить кювету с испытуемым раствором; 3) установить правый барабан на «100%» пропускания или «0 оптической плотности»; 4) установить рукояткой 19 на пути потока излучения определенный светофильтр; 5) добиться смыкания краев сектора индикаторной лампы вращением левого, компенсационного, барабана 5; 6) заменить на пути правого пучка света кювету с исследуемым раствором на кювету с раствором сравнения поворотом рукоятки 20\ при этом края сектора индикаторной лампы расходятся или перекрываются; 7) добиться сомкнутого положения индикаторной лампы вращением правого, из-'мерительного, барабана 5'; по шкале 21 этого барабана снять показания оптической плотности А (красная шкала) или процента пропускания Т (черная шкала).
Примечание. Большое значение для получения надежных результатов измерений имеет чувствительность прибора, которая определяется числом делений шкалы, на которое нужно повернуть барабан для полного размыкания сектора индикаторной лампы (чем число делений меньше, тем выше чувствительность прибора). Чувствительность проверяют следующим образом: 1) устанавливают электрический нуль прибора и открывают шторку; 2) включают светофильтр № 6; 3) по шкале правого барабана устанавливают деление Т= 80%; 4) поворотом левого барабана приводят сектор индикаторной лампы в сомкнутое положение; 5) меняя щель правой диафрагмы рукояткой барабана 5', приводят сектор в полностью разомкнутое состояние. Отсчет по шкале правого барабана должен при этом быть в пределах 73—87%. Если полученные значения выходят за пределы данного интервала, то это указывает на неисправность одного из электроузлов: индикаторной лампы, фотоэлемента или лампы 6НС усилителя.
Новая модель фотоэлектроколориметра данного типа—ФЭК-56М отличается от предшествующей модели следующим:
а) рукоятка, при помощи которой опускаются шторки, перекрывающие потоки света, расположена за кюветным отделением перед осветителем в верхней части корпуса прибора;
б) вместо индикаторной лампы в качестве прибора-индикатора используется миллиамперметр, он снабжен потенциометром чувствительности: чувствительность можно повысить, если изменение ширины щели диафрагмы вызывает лишь незначительные отклонения стрелки миллиамперметра, меняя положение рукоятки, расположенной рядом с рукояткой настройки электрического нуля.
Фотоэлектрический колориметр-нефелометр ФЭК-60
Данный прибор относится к типу объективных приборов, в основу которых положен принцип уравнивания интенсивности двух световых модулированных потоков при помощи переменной щелевой диафрагмы. Помимо измерения оптических плотностей и процентов пропускания прибор используют для косвенного определения светопропуска-ния мутных растворов по отношению к прозрачному растворителю или воде. Светорассеяние растворов со слабой мутностью, светопропуска-ние которых незначительно отличается от светопропускания растворителя, на данном приборе определить нельзя.
Прибор имеет оптику, позволяющую работать в видимой, ближней ПК- и УФ-областях. Помимо сурьмяно-цезиевого фотоэлемента, ра
ботающего в УФ и видимой областях спектра, прибор снабжен также кислородно-цезиевым фотоэлементом, что позволяет проводить измерения в ближней ИК-области до 1000 нм. Однако в отличие от фотоэлектроколориметра ФЭК-56 в этом приборе отсутствует ртутная лампа, поэтому нет возможности проводить измерения при длинах волн короче 360 нм.
Фотоэлектроколориметр ФЭК-60 — однофотоэлементный прибор. На фотоэлемент попадает суммированный поток, который является результатом сложения двух потоков излучений, проходящих через исследуемый и сравнительный растворы и модулированных в противофазе. Это исключает ошибки, возникающие в результате неодинаковой спектральной чувствительности фотоэлементов. Большая чувствительность прибора позволяет измерять пропускание растворов высоких концентраций (с оптической плотностью > 3) методом дифференциальной спектрофотометрии.
Прибор снабжен девятью парами светофильтров: одна пара для измерений в ближней УФ-области, пять — в видимой и три — в ближней ИК-области (табл. 19).
ТАБЛИЦА 19
Характеристики светофильтров фотоэлектроколориметра ФЭК-60
Номер светофильтра Длина волны, соответствующая максимуму пропускания, нм Номер светофильтра Длина волны, соответствующая максимуму пропускания, нм
1* 364 9 980
2* 390 10** 420
3 450 11 480
4 520 12 560
5 590 13 620
6 670 14 720
7 750 I 15 800
8 870 j 16 930
* Светофильтры Ns 1 —2— стеклянные, остальные интерференционные.
•* Светофильтры № 10—16 входят в дополнительный комплект.
Принципиальная оптическая схема прибора приведена на рис. 83, общий вид прибора—на рис. 84. Нить накала лампы Л при помощи двух конденсаторов /<1 и К2 и двух зеркал Зг и 32 изображается на линзах (\ и 02. Эти изображения проектируются линзами Оэ и 04 и сводятся зеркалами З3, 3^ и призмой П в плоскость фотокатода фотоэлемента Ф. Модулятор Л4, помещенный за конденсаторами, модулирует световые потоки, правый и левый, в противофазе. Модулятор представляет собой полый цилиндр с семью окнами, вращающийся от синхронного электродвигателя. Частота модуляции светового потока 350 Гц. Модулированные световые потоки, пройдя светофильтры Сг и С2 и кюветы Рг и Р2, попадают на фотоэлемент и возбуждают переменный электрический ток, пропорциональный разности правого и левого световых потоков. В правый световой поток последовательно может быть введена или кювета с раствором сравнения, или с исследуемым.
Переключение кювет производится поворотом рукоятки 3 (см. рис. 84) до упора. Левый световой поток оставляют свободным или
помещают на его пути раствор сравнения.
Щелевая диафрагма Дг (см. рис. 83), расположенная на пути правого светового потока, является измерительной и связана с отсчетным
барабаном 1 (см. рис. 84), проградуированным в значениях А и Т.
Шкала на отсчетном барабане нанесена таким образом, что максималь-
ное раскрытие щелевой диафрагмы соответствует 100% пропускания, а полное ее закрытие—0%. Шкала пропускания равномерная, так как мощность пучка лучей, проходящего через диафрагму, пропорциональна ширине щели. Щелевая диафрагма Д2, расположенная на пути левого светового потока, является компенсационной и шкалы не имеет. При вращении рукоятки 2 по стрелке происходит раскрытие щелевой диафрагмы Д2.
Сурьмяно-цезиевый и кислородно-цезиевый фотоэлементы устанавливаются поочередно в одни и те же гнезда. Для замены фотоэлемента нужно снять крышку 8. Питание прибора осуществляется от сети переменного тока 220 В, 50 Гц через питающее устройство, которое обеспечивает постоянное напряжение питания фотометрической лампы и усили
теля. На вилке, посредством которой п о„ „
_ r г Рис. 83. Принципиальная опти-
питающее устройство включается в сеть, веская схема фотоэлектроколо-выведен контакт с гайкой. Перед нача- риметра-нефелометра ФЭК-60 лом работы к этому контакту подсоеди-
няется заземляющий провод. Питание на прибор подается от питаю-
щего устройства через разъем, включение высокого напряжения производится выключателем, расположенным на передней стенке пи-тающегоу стройства.
Светофильтры располагаются в двух барабанах, вращение которых производится рукояткой 6. В каждом барабане имеется девять светофильтров, которые поочередно устанавливаются на пути потоков излучений. Рабочее положение каждого светофильтра фиксируется и указывается цифрой на рукоятке 6. Для замены светофильтра открывают крышку 12 и при помощи ключа, прилагаемого к прибору, вынимают светофильтр. Если в процессе работы светофильтр, с которым производилось измерение, меняется на другой, то измерение рекомендуется производить не ранее чем через 1 мин после смены светофильтра. Для уменьшения чувствительности прибора в оба плеча включаются нейтральные светофильтры из прилагаемого комплекта. Нейтральный светофильтр надевается на оправу линз Ог и О2.
Порядок работы. Измерения на приборе можно начинать спустя
t
15—20 мин после включения прибора. В течение этого времени электросхема будет прогрета и наступает стабильный режим ее работы.
Установку лампы в осветителе осуществляют при помощи юстировочных винтов 9 и 10 (см. рис. 84). Для правильной работы прибора необходимо, чтобы нить лампы резко изображалась на линзах Ог и О2 (см. рис. 83); для удобства наблюдения изображения нити лампы к поверхности этих линз прикладывают папиросную бумагу. После регулировки осветитель закрепляется двумя винтами 11.
После этого следует:
1) закрыть измерительную диафрагму Дг, поставив указатель ее шкалы на значение 0% поворотом рукоятки 4\ 2) закрыть компенсационную диафрагму Д2, повернув рукоятку 2 против часовой стрелки до упора;
Рис 84 Общий вид фотоэлектроколориметра-нефелометра ФЭК-60 а —вид спереди: б —задняя панель с осветителем
3) добиться нулевого положения стрелки микроамперметра, вращая рукоятку потенциометра 5; 4) установить правый барабан рукояткой 4 на значение Т = 100% (или А = 0); 5) установить рукояткой 6 на пути излучения соответствующий светофильтр; 6) установить в кюветное отделение 7 на пути правого потока кювету с испытуемым раствором, левый поток остается свободным; 7) установить вращением левого барабана 2 стрелку микроамперметра на нуль; 8) ввести в правый поток кювету с раствором сравнения поворотом рукоятки 3; 9) восстановить вращением барабана 4 нарушенное равновесие и привести стрелку микроамперметра к нулю; отсчет оптической плотности или процента пропускания производить по шкале 1 барабана 4.
Можно использовать также второй способ измерения: операции 1—5 те же, что и в первом способе; 6) установить в правый и левый потоки света кюветы с раствором сравнения (растворителем); 7) установить вращением левого барабана 2 стрелку микроамперметра на нуль; 8) заменить в левом потоке кювету с раствором сравнения на кювету с испытуемым; 9) возвратить вращением правого барабана 4 стрелку микроамперметра в нулевое положение ; по шкале 1 этого же барабана произвести отсчет А или Т.
Преимуществом первого способа является то, что измерение и настройка начала отсчета проводится по о том} и тому же бланку (воз-256
дух). Второй способ имеет то преимущество, что настройка начала отсчета шкалы производится только один раз, после чего измеряется поглощение всей серии растворов (способ более производителен).
Нерегистрирующие спектрофотометры СФ-4, СФ-4А, СФ-16, СФ-5*
Спектрофотометры данного типа имеют одинаковую оптическую схему и несколько различаются электрическими схемами, методикой измерений и спектральной областью, в которой их используют.
Основной частью монохроматора в этих приборах является диспергирующая призма, которая разлагает сплошное излучение в спектр, в результате чего через выходную щель монохроматора, осуществляющую дополнительную монохроматизацию, проходит излучение той или иной монохроматичности в зависимости от дисперсии призмы и рабочей ширины щели в данном спектральном интервале.
Спектрофотометры СФ-4, СФ-4А, СФ-16 имеют кварцевую оптику, что позволяет производить измерения, помимо видимой и ближней ИК-области, также в УФ-области спектра. В качестве источников сплошных излучений в них используются водородная лампа в УФ-области (200—350 нм) и вольфрамовая лампа в видимой и ближней ИК-областях (320—1100 нм). Кроме того, в спектрофотометре СФ-16 имеется дейтериевая лампа для работы в области 185—200 нм, что требует полной эвакуации или вытеснение воздуха азотом на всем оптическом пути. Для измерений в широком спектральном интервале используют в качестве детекторов два фотоэлемента сурьмяно-цезиевый в области 186—650 и кислородно-цезиевый—в области 600—1100 нм. Длина волны, при которой следует переходить от измерений с одним фотоэлементом к измерениям с другим, указана в аттестате прибора.
Спектрофотометр СФ-5 имеет стеклянную оптику и поэтому работает только в видимой и ближней ИК-областях спектра. В качестве источника излучений в нем используется только вольфрамовая лампа, а в качестве детекторов—те же фотоэлементы. Ртутная лампа, имеющаяся в комплекте каждого из этих приборов, дает линейчатый спектр и используется для проверки градуировки шкалы длин волн. Для уменьшения рассеянного излучения на пути луча, выходящего из монохроматора, устанавливают светофильтры: из стекла УФС-2 — при работе в области 320—380 нм, из стекла ОС-14 — при работе в области 590—700 нм. Таким образом, эти светофильтры не играют роли монохроматоров, как это осуществляется в фотоэлектроколориметрах.
Принципиальная оптическая схема спектрофотометров приведена на рис. 85.
Свет от источника 1 падает на зеркало-конденсор 2, которое собирает и направляет пучок лучей на плоское зеркало 3, поворачивающее лучи на 90° и направляющее их через защитную кварцевую пластинку 4 на входную щель монохроматора 5. Зеркальный объектив—колли
* Здесь дана характеристика спектрофотометров всех указанных моделей. Подробное описание методики измерений дается только для спектрофотометров последних моделей СФ-4А и СФ-16.
матор 6, в фокусе которого расположена щель, направляет параллельный пучок лучей на кварцевую диспергирующую призму 7 с отражающей задней гранью. Призма разлагает свет в спектр и направляет его обратно на объектив. Луч, прошедший призму под углом, близким к углу наименьшего отклонения, попадает на выходную щель 8, расположенную под входной Щелью. Поворачивая призму вокруг оси, можно получить на выходе монохроматора лучи различных длин волн. Выходящий монохроматический пучок света проходит кварцевую линзу 9, фильтр 10, измеряемый образец 11, линзу 12 и падает на свето-
Рис. 85. Принципиальная оптическая схема спектрофотометров СФ-4, СФ-5, СФ-4А, СФ-16
чувствительный слой фотоэлемента 13. Фототок, возникающий в фотоэлементе, передается на усилитель постоянного тока. Усиленный ток попадает на прибор - индикатор (миллиамперметр).
Данные спектрофотометры относятся к типу однолучевых приборов, поэтому в процессе измерений на пути потока излучения устанавливается поочередно «нулевой» и испытуемый образцы. Происходящее при этом изменение интенсивности излучения, падающего на фотоэлемент, вызывает изменение напряжения в системе усилителя, которое компенсируется изменением напряжения на потенциометре, связанном с отсчетным устройством.
Спектрофотометр состоит из четырех частей (рис. 86): монохроматора с фотометрической частью, заключенного в основном корпусе прибора 15, кюветного отделения 17, камеры с детекторами и усилителем 20 и осветителя с источниками освещения 31. Кроме того, каждый спектрофотометр снабжен стабилизатором (рис. 87), через который осуществляется питание источников освещения, а также усилителя (СФ-4А, СФ-16). Осветители жестко крепятся к корпусу прибора и имеют держатели источников освещения с соответствующими механизмами юстировки, которые несколько отличны у различных моделей.
Поворот призмы и направление на выходную щель излучения определенной длины волны осуществляется рукояткой 19 и фиксируется на шкале 29. Входная и выходная щели монохроматора конструктивно составляют одно целое, расположены одна под другой и раскрытие их
Рис. 86. Внешний вид спектрофотометра СФ-16: а—вид спереди; б — задняя панель с осветителем
осуществляется одновременно при помощи рукоятки 22 (в СФ-16 — микрометрический винт) в пределах от 0 до 2 мм. Ширина раскрытия щели фиксируется по шкале 21.
Кюветная камера 17 предназначена для установки испытуемых образцов и образцов сравнения и имеет подвижную каретку, перемещаемую рукояткой 18. Каретка имеет четыре фиксированных поло
жения, отмеченных цифрами на рукоятке 18. Измерение образцов производится при плотно закрытой крышке кюветного отделения.
Фотоэлементы и усилитель размещены в герметической камере, на стенке которой имеется рукоятка 16 для перемещения фотоэлементов. В камере имеется патрон с осушителем—силикагелем, пропитанным солями кобальта (си-
стабилизатор
Рис. 87. Электронный < к спектрофотометрам СФ-16 и СФ-4А
ний цвет осушителя указывает на нормальную влажность, розовый — на повышенную)*. Излучение попадает на фотоэлемент через окно в кюветной камере, закрываемое шторкой, механически связанной с переключателем 24.
При помощи рукоятки 10 или диска (СФ-16) на пути потока излучения можно установить поочередно фильтры УФС-2, ОС-14 или свободное отверстие, в которое можно вставить любой другой фильтр. На передней панели корпуса прибора расположены также миллиамперметр 14 (нуль—инструмент), рукоятка 27 и шкала 28 отсчетного
* Для регенерации осушителя его следует просушить при температуре не выше 100° С, иначе силикагель теряет способность поглощать влагу.
потенциометра, а также рукоятки регулировки темнового тока 25 и чувствительности 26.
Порядок включения приборов различных марок в электрическую сеть, включение источников освещения, конструкция осветителя, установка в нем соответствующих ламп и порядок измерений (последовательность операций при установке темнового тока, регулировка чувствительности, балансировка нуля отсчета и т. п.) указаны при описании спектрофотометров каждой модели. Прежде чем приступить к измерению оптических плотностей исследуемого объекта, необходимо проверить работу всех узлов прибора: 1) правильность установки ламп в осветителе; 2) правильность градуировки шкалы длин волн прибора; 3) правильность показаний шкалы оптических плотностей.
1. Проверяют правильность установки ламп в осветителе.
Правильность установки ртутной лампы проверяют визуально: поворотом рукоятки 22 (см. рис. 86) открывают щель на полную ширину (2 мм). Указатель шкалы длин волн 29 устанавливают на X 546,1 нм (зеленая линия ртути). В кюветное отделение со стороны, противоположной щели, вставляют лист белой бумаги и наблюдают освещенность прямоугольника, являющегося изображением щели. При правильной установке лампы этот прямоугольник должен равномерно освещаться зеленым светом. Если освещенность прямоугольника неравномерна или наблюдаются затемненные углы, то следует, пользуясь специальным ключом и часовой отверткой, настроить лампу при помощи винтов и конденсорного зеркала до получения равномерной освещенности прямоугольника зеленым светом.
Проверку правильности установки водородной лампы и лампы накаливания проводят как визуально, так и фотоэлектрически.
Визуальную проверку установки водородной лампы проводят так же, как ртутной лампы, но по красной линии водорода 656 нм. Фотоэлектрическую проверку правильности, установку водородной лампы проводят по следующим критериям.
Оптимальная установка водородной лампы должна обеспечить возможность работы в достаточно далекой ультрафиолетовой области (213—215 нм). Это может быть достигнуто при полностью открытой щели (2 мм) и минимальной чувствительности прибора, т. е. в условиях, в которых интенсивность излучения водородной лампы обеспечивает достаточную величину фототока. Поэтому, проверяя установку этой лампы, раскрывают ширину щели 2 мм (максимальное раскрытие) и потенциометр чувствительности, ставят в положение минимальной чувствительности: в крайнее левое положение (СФ-4, СФ-5) или в положение «1» (СФ-16, СФ-4А). Регулируют темновой ток, открывают шторку фотоэлемента и вращают шкалу длин волн при помощи рукоятки 19 в сторону меньших длин волн. Значение длины волны, при котором стрелка миллиамперметра приходит к нулю, сравнивают с паспортом данной лампы, в котором указана предельная граница излучения лампы в ультрафиолетовой области. Если показания шкалы длин волн и данные паспорта отличаются более чем на 2—3 нм, считается, что водородная лампа установлена плохо и ее устанавливают при помощи юстировочных винтов и конденсорного зеркала.
Визуальная проверка установки лампы накаливания проводится аналогично установке ртутной лампы по зеленой линии 546,1 ммк. При фотоэлектрической проверке установки этой лампы учитывается, что интенсивность излучения лампы накаливания и чувствительность фотоэлементов неодинакова при различных длинах волн. Максимум приходится на область 520—550 нм. Следовательно, в этой области имеется возможность для работы с минимальной щелью. Поэтому при проверке установки лампы накаливания, скомпенсировав, как обычно, темновой ток при закрытом фотоэлементе, устанавливают по шкале длин волн значение 546,1 нм (находящееся как раз в области максимальной интенсивности излучения этой лампы). Выбирают среднюю чувствительность прибора. Для этого рукоятку 26 на СФ-4 и СФ-5 поворачивают на 4—5 оборотов от одного из крайних положений; на СФ-16 ставят рукоятку потенциометра чувствительности 26 в положение «2» или «3». Открыв шторку фотоэлемента, приводят стрелку миллиамперметра к нулю, уменьшая щель. Если стрелка миллиамперметра приводится к нулю при раскрытии щели не более чем на 0,02— 0,03 мм, то установку лампы накаливания можно считать удовлетворительной. Если для приведения стрелки к нулю требуется большее раскрытие щели, то это вызывает необходимость работать при иных длинах волн с еще более широкой щелью и снижает монохроматичность потока излучения. Поэтому плохо установленную лампу следует настроить при помощи юстировочных винтов.
2. Проверяют градуировку шкалы длин волн 29 по линиям ртутного спектра. Длины волн эмиссионного спектра ртути помимо атласа спектральных линий можно найти в аттестате каждого прибора, где имеется таблица длин волн линий ртутной лампы, по которым проверяют градуировку шкалы при выпуске прибора. Сущность данной проверки заключается в том, что, наблюдая визуально или фотоэлектрически прохождение через щель определенной линии ртути при вращении рукоятки 19 (см. рис. 86) шкалы длин волн, останавливают это вращение как раз в тот мсмент, когда через щель проходит максимум линии, и сравнивают полученное показание шкалы с данными атласа или аттестата.
Визуальную проверку градуировки целесообразно проводить только при первоначальной установке прибора или переносе его на новое место, т. е. когда может произойти сдвиг шкалы. Визуальную градуировку проводят по зеленой линии ртути 546,1 нм. Щель уменьшают настолько, чтобы можно было видеть только резкую линию (0,02— 0,04 мм). Передвигая шкалу длин волн при помощи рукоятки 19 и подводя линию 546,1 нм со стороны коротких длин волн (от ~530 нм), наблюдают момент наиболее яркого освещения щели, что соответствует прохождению через щель максимума интенсивности этой линии. В этот момент останавливают вращение шкалы длин волн и отмечают, насколько точно показание шкалы 29 совпадает с данными аттестата. Если отклонение невелико (не более 3—5 нм), то можно перейти к фо' о-электрической проверке градуировки. Если же отклонение значительное, то следует его уменьшить поворотом зеркального объектива 30, Для этого крышку с надписью «регулировка зеркала» снимают 261
и винт зеркального объектива поворачивают при помощи специального ключа; регулировочный винт следует вращать по часовой стрелке, если линия на шкале устанавливается на отсчете больше табличного значения, и против часовой стрелки, если меньше табличного значения.
Фотоэлектрическую, более точную, проверку градуировки проводят систематически через 1—2 месяца, так как шкала длин волн может сместиться в процессе длительной работы прибора. Для этого следует:
1) поставить рукояткой 16 в рабочее положение фотоэлемент, соответствующий проверяемой области спектра; 2) поставить переключатель 23 в положение «I» (или «выкл.» — СФ-16 и СФ-4А) и при закрытой шторке-переключателе 24 (опущена вниз) компенсируют темновой ток; 3) установить потенциометр чувствительности в рабочее положение: на СФ-4 сделать 4—5 оборотов рукоятки 26 от крайнего положения (правого или левого); на СФ-16 и СФ-4А переключатель 26 поставить в положение «3»; 4) поставить отсчетный потенциометр 28 рукояткой 27 на 3—5% пропускания; 5) открыть шторку фотоэлемента (рукоятка 24 поднята вверх до отказа); 6) привести стрелку миллиамперметра 14 в пределы шкалы, регулируя щель рукояткой 22; 7) подвести линию 546, 1 нм, медленно вращая шкалу длин волн рукояткой 19; при этом стрелка миллиамперметра двигается влево. Регулируя щель, следует удерживать стрелку в пределах шкалы. В момент прохождения через щель максимума интенсивности линии наблюдается также максимальное отклонение стрелки миллиамперметра влево. Вращение шкалы длин волн следует остановить как раз в этот момент, до начала обратного движения стрелки вправо. Отсчет по шкале 29 должен быть равным 546,1 нм с отклонением, не превышающим указанного в аттестате для данной длины волны (~ ±0,2 нм). Таким же образом нужно проверить градуировку во всем диапазоне от 220 до 1100 нм по длинам волн, указанным в аттестате или инструкции. Если не наблюдается достаточно хорошего совпадения в показаниях, то необходимо этого добиться поворотом зеркального объектива, как указано выше.
3. Проверяют правильность показаний шкалы отсчетного потенциометра. Это можно сделать по так называемым нейтральным светофильтрам, представляющим собой стеклянные стандарты пропускания. В комплектах спектрофотометров имеются четыре светофильтра: НС-6 толщиной 1 мм и НС-8 толщиной 1,0; 2, 3 и 4,0 мм, данные о пропускании которых при различных длинах волн приведены в паспорте каждого прибора. Однако эти данные следует периодически проверять, так как со временем пропускание стекол меняется. Эта проверка осуществляется по дискам, имеющим определенное число прорезей, при вращении которых может быть получена соответствующая оптическая плотность.
Примечание. При отсутствии этих дисков в комплекте прибора проверка пропускания светофильтров может быть сделана на предприятии, выпускающем эти стандарты пропускания.
В качестве стандартов можно использовать растворы некоторых веществ, спектральное пропускание которых при различных длинах 262
волн, концентрациях и толщинах слоев было измерено с достаточной ' точностью (с ошибкой в отсчете не выше 0,001) на приборах различных моделей 16]. Кроме того, оценку правильности измерений оптических плотностей можно сделать по молярным коэффициентам погашения (табл. 20) растворов: 0,3 М раствора KNO3 (30,36 г KNO3 растворяют в 1 л воды); 0,0001 М раствора К2СгО4 в 0,05 М КОН (0,01942 г К2СгО4 растворяют в 1 л 0,05 М КОН).
ТАБЛИЦА 20
Молярные коэффициенты погашения для спектральных стандартов пропускания
X, нм В X, нм С
К,СгО4 | KNO, К.СгО4 | KNOa
254 2570 3,59 313 193,5 5,26
265 3160 1,56 334 985 0,45
280 3290 3,68 366 4410 —
289 2086 5,61 405 1330 —
297 927 6,84 436 310 —
303 498 6,93
В комплекте спектрофотометров имеются два вида кювет', прямоугольные с толщиной слоя в 1 см и цилиндрические с различной толщиной слоя (см. рис. 77). Для прямоугольных кювет имеется металлический держатель (см. рис. 78) с четырьмя гнездами. В одно из этих гнезд устанавливают кювету с раствором, поглощение которого принимается равным нулю (чаще всего это растворитель.) В три остальных гнезда вставляют кюветы с исследуемыми растворами.
Таким образом, можно одновременно измерять поглощение трех растворов, если оценивают их поглощение по отношению к одному и тому же раствору сравнения. Металлический держатель с кюветами помещают в кюветное отделение (см. рис. 86, 88) таким образом, чтобы белая точка на каретке в кюветном отделении соответствовала такой же точке на держателе кювет. На это следует обращать внимание, так как каретка имеет четыре фиксированных положения (соответственно числу установленных в держателе кювет), обозначенных на ее рукоятке и соответствующих только определенному держателю при правильной его установке в каретку. Нельзя пользоваться держателем из другого спектрофотометра.
Цилиндрические кюветы комплектуются попарно: по две кюветы с толщиной слоя (мм): 4,05; 4,10; 4,20; 4,50; 5,0; 10; 20; 50 и 100. Для измерения пропускания тонких слоев жидкостей 0,05; 0,1; 0,2; 0,5 и 1 мм в соответствующие стаканы следует помещать вкладыши, представляющие собой пластины из кварцевого стекла толщиной 4 мм. Соответственно имеются по два металлических держателя (см. рис. 78) для каждой из пяти пар цилиндрических кювет, кюветы с толщиной слоя от 4,0 до 4,5 мм вставляются в держатели для кювет с толщиной 5,0 мм. Точные данные относительно толщины слоя кювет и их пропускаемое™ приведены в аттестате соответствующего прибора. Цилиндрические кюветы (см. рис. 77) представляют собой стеклянные
стаканы, закрывающиеся с обеих сторон крышками, кварцевыми или стеклянными в зависимости от того, в какой спектральной области предполагается вести работу. Стеклянные крышки, обозначенные буквой «С», используют только для работы в области 400—1000 нм.
При заполнении кюветы одну из крышек помещают на чистую подкладку и сбоку на нее надвигают стакан кюветы. Затем наполняют кювету раствором до образования выпуклого мениска. Вторую крышку надвигают сверху так, чтобы в стакане не оставалось пузырьков воздуха. После этого осторожно снимают кювету с подкладки, придерживая крышки, и вставляют ее в отвинченную крышку металлического держателя, в которую предварительно помещают каучуковую прокладку. Сверху помещают вторую каучуковую прокладку и навинчивают металлический стакан вместе со второй металлической крышкой.
В кюветное отделение устанавливают одновременно два держателя с кюветами с исследуемым раствором и раствором сравнения. При работе используют два фиксированных положения «1» и «4» на рукоятке 18 (см. рис. 86).
Для крепления плоских твердых образцов в кюветной камере служит держатель 3 (см. рис. 78) с четырьмя окнами и пружинами, позволяющий устанавливать и измерять одновременно пропускание образцов разной толщины от 0,5 до 2 мм. При его установке также следует соблюдать правильное положение белой точки указателя.
Для фиксации положения твердых образцов и прямоугольных кювет в каретке используется рамка, вставляемая в одну из прорезей в стенках каретки. Для фиксации положения держателей с цилиндрическими кюветами используют пружинящие прижимы, вставляемые в горизонтальные прорези стенок каретки.
СПЕКТРОФОТОМЕТР СФ-16
Прибор данной модели (см. рис. 86) имеет четыре источника излучений: дейтериевую, водородную, вольфрамовую и ртутную лампы Их рабочий диапазон указан на стр. 257.
Лампы помещают в одном и том же осветителе 31, имеющем два цоколя. Водородная, дейтериевая и ртутная лампы устанавливаются поочередно в один и тот же цоколь. Для лампы накаливания имеется отдельный цоколь. Излучение определенного интервала длин волн от той или иной лампы направляется на входную щель монохроматора зеркалом-конденсором, помещенным между лампами, и поворот которого осуществляется рукояткой 34. Держатель каждой лампы имеет свой механизм юстировки. Держатели лампы и стойка с конденсором крепятся на отдельном кронштейне, жестко связанном с основанием прибора. Сверху лампы закрываются кожухом, в котором есть отверстие с подвесной крышкой для доступа к рукоятке 34 переключения конденсора на ту или другую лампу.
Питание всех ламп и усилителя осуществляется от одного и того же блока питания (см.рис. 87), в котором осуществляется стабилизация тока. Стабилизатор питается от сети переменного тока 220 В.
Для обеспечения работы в области 185—200 нм необходимо вытеснить воздух азотом на всем пути излучения, так как отдельные компоненты воздуха имеют характерные полосы поглощения в этой области. Газ (обычно N2) от баллона, пропущенный через манометр и ротаметр (тип РС-3 с пределами измерений 4—15 л/мин), по которому определяется скорость продувки монохроматора газом, вводится внутрь корпуса прибора через штуцер 40 (см. рис. 86). Продувать прибор следует в течение 10—15 мин со скоростью 10 л/мин. Измерения рекомендуется производить при постоянной продувке со скоростью 5 л/мин.
Для расширения границ точности измерения потенциометры чувствительности 26 и темнового тока 25 (см. рис. 86) имеют переключатели на четыре рабочих интервала. Положение «1» рукоятки потенциометра чувствительности соответствует самой высокой точности измерения, но при этом приходится работать с широкой щелью; положение «4» соответствует обратной зависимости. Так как в соответствии с электрической схемой прибора напряжение на этих потенциометрах взаимно связано, то вначале выбирается необходимая чувствительность, а затем подбирается напряжение на потенциометре темнового тока.
Спектрофотометр СФ-16 имеет несколько пределов измерений процентов пропускания. Переключение пределов измерения осуществляется рукояти >й 23: положение «1» соответствует пределу измерений от 0 до 100%, положение «2» — от 0 до 10%, положение «3» — от 90 до 100%.
Приспособление для спектрофотометрического титрования к нерегистрирующим спектрофотометрам рассматриваемого типа может быть выполнено довольно просто. Металлическую крышку кюветного отделения заменяют эбонитовой, проделывают в ней два отверстия: одно для Микробюретки, второе для механической мешалки. Кювета из оптического стекла должна иметь объем около 25 мл (для титрования в видимой области спектра можно использовать, например, кювету от фотоэлектрокалориметра ФЭК-М с I = 4 см). Стенки кюветы покрывают черным лаком, оставляя отверстие диаметром в 1 см на пути светового потока. Мешалка вводится через отверстия в крышке кюветного отделения так, чтобы ее конец находился против затемненного участка кюветы. Раствор реагента прибавляют из микробюретки, чтобы избежать значительного разбавления титруемого раствора.
Порядок работы. Усилитель подключается к стабилизатору в момент установки выключателя 30 (см. рис. 87) в положение «вкл.». При этом выключатель 23 (см. рис. 86) в начале работы должен находиться в положении «выкл.».
Включение ламп осуществляется через стабилизатор в следующем порядке: 1) подключить при помощи шлангов усилитель и лампы к стабилизатору; 2) вынуть из гнезд 35 и 36 на стабилизаторе короткозамкнутые вилки и подключить амперметр переменного тока на 2,5 — 2 А к гнездам «ток накала», а миллиамперметр на 500—700 мА к гнездам «разрядный ток». Далее в зависимости от используемой лампы соблюдают следующий порядок.
Для лампы накаливания: а) проверить, находится ли выключатель 30 в положении «выкл.»; б) повернуть рукоятку 32 влево до упора; в) тумблером 38 включить в цепь требуемую лампу; г) включить стабилизатор в сеть 220 В шлангом 37; д) поставить рукоятку 30 в положение «накал»; е) нажать кнопку 39; ж) подождать, пока на миллиамперметре установится показание 300 мА; з) поставить рукоятку 30 в положение «лампа накал». После этого можно приступить к измерениям.
Для водородной, ртутной и дейтериевой лампы: пп. а — д см. для лампы накаливания; е) установить, вращая рукоятку 32, движок потенциометра в положение, при котором пусковой ток лампы соответствует указанному в паспорте лампы; ж) выждать 2—5 мин; з) нажать кнопку 39; и) снизить ток до рабочего значения, указанного в паспорте лампы, вращая рукоятку 32. После этого можно приступать к измерениям, предварительно прогрев прибор 10 мин.
Примечания: 1. Если после лампы накаливания требуется включить водородную лампу, необходимо полностью выключить стабилизатор, а затем включить снова.
2. Для включения лампы накаливания после водородной нужно рукоятку 30 поставить в положение «лампа накал», а тумблер 38 в положение «лампа накаливания».
После включения лампы и усилителя в электрическую сеть следует: 1) установить в кюветодержателе кюветы с раствором сравнения и испытуемыми образцами, поместить его в кюветное отделение 17 (см. рис. 86) таким образом, чтобы на пути потока излучения находился раствор сравнения (кюветодержатель должен быть повернут точкой к оператору); закрепить его прижимом; закрыть крышку кюветного отделения; 2) установить рукояткой 16 в рабочее положение сурьмя-но-цезиевый-Ф (СФ-4А — рукоятка вдвинута) или кислородно-цезиевый-К (СФ-4 — рукоятка выдвинута) фотоэлемент; 3) поставить переключатель 23 в положение «выкл.» и закрыть фотоэлемент, поставив шторку 24 в положение «закр.»; 4) установить, вращая рукоятку 19, по шкале 29 требуемую длину волны, подводя ее со стороны малых значений. Если при этом случайно будет пройдено нужное значение, то следует возвратить шкалу к значению на 3—5 нм меньше требуемого и снова установить на соответствующее деление; 5) установить рукоятку (диск) 10 держателя светофильтров на указатель нужного светофильтра (см. стр. 257) или «воздух» (СФ-4А—рукоятка вдвинута); 6) поставить рукоятку 26 в одно из положений—4. Если требуется производить измерения с большой чувствительностью и можно пренебречь снижением монохроматичности, работая с широкой щелью, то следует поставить рукоятку 26 в положение «1». Если требуется работать с узкой щелью (измерение узкополосных максимумов), то следует проводить измерения при'положении «4»; 7) скомпенсировать темновой ток рукояткой грубой 25 и плавной 25' регулировки, подводя стрелку миллиамперметра 14 к нулю; 8) открыть фотоэлемент, поставив рукоятку 24 в положение «откр.»; 9) установить стрелку миллиамперметра вновь на нулевое значение, меняя ширину щели вращением рукоятки 22,
более плавно это может быть сделано поворотом рукоятки потенциометра чувствительности 26'; 10) установить на пути излучения испытуемый образец, перемещая рукояткой 18 каретку с кюветодержателем; 11) установить рукоятку 23 в положение «1» и, поворачивая отсчетный потенциометр рукояткой 27, привести стрелку миллиамперметра к условному нулю; снять отсчет по шкале 28 оптических плотностей (верхняя шкала) или процентов пропускания (нижняя шкала). Отсчет рекомендуется производить 3—5 раз, повторяя весь порядок компенсации; за истинное значение принимается среднее из этих отсчетов.
Примечания. 1. Если при установке рукоятки 24 в положение «закр.» стрелка миллиамперметра постоянно возвращается к условному нулю, то можно производить измерения, не проверяя компенсацию темнового тока перед каждым измерением.
2. Если требуется с большой точностью произвести измерение пропускания очень плотных растворов (Т < 10%), то рукоятку 23 устанавливают в положение «2» и снятый по шкале пропусканий отсчет умножают на 0,1, а к показаниям оптической плотности прибавляют 1
3. Если требуется с высокой точностью измерить коэффициент пропускания (больше 90%), то рукоятку 23 устанавливают в положение «3» и снятый по шкале пропускания отсчет умножают на 0,1 и к нему прибавляют 90.
При проведении спектрофотометрического титрования следует: 1) настроить темновой ток; 2) наполнить кювету для титрования водой; 3) открыть шторку 24 (см. рис. 86); 4) настроить нуль шкалы отсчетного потенциометра (потенциометром чувствительности или меняя ширину щели); 5) закрыть шторку; 6) наполнить кювету титруемым раствором; 7) открыть шторку и измерить начальную оптическую плотность титруемого раствора; 8) прибавлять титрант небольшими порциями (~0,1 мл) и компенсировать отклонение стрелки миллиамперметра в процессе титрования, приводя ее к условному нулю только отсчетным потенциометром (сохраняя постоянной компенсацию темнового тока и нуля отсчетной шкалы).
Примечание. Необязательно прибавлять порции титранта точно по 0,1 мл, но егр объем, прибавляемый в каждый момент титрования, следует точно отсчитывать по бюретке, так как это необходимо для выявления правильного хода кривой титрования.
Рис 88. Внешний вид спектрофотометра СФ-4А
СПЕКТРОФОТОМЕТР СФ-4А
Данный прибор имеет в принципе ту же электрическую схему, что и СФ-16, поэтому порядок измерений аналогичен. Однако эта модель имеет несколько иное расположение рукояток для настройки. Оно показано на рис. 88 с сохранением нумерации, данной при описании модели СФ-16. В спектрофотометре СФ-4А рукоятка 23 не имеет положения, соответствующего измерению больших значений процентов пропускания. В приборе отсутствует дейтериевая лампа и приспособление для вытеснения воздуха азотом и, следовательно, он не может работать в области 185—200 нм.
Спектрофотометр фирмы Hitachi
(Япония), модель 124
Спектрофотометр модели 124 фирмы Hitachi — двухлучевой регистрирующий прибор, работающий в области спектра 190—800 нм. Диспергирующим элементом в нем служит дифракционная решетка, позволяющая работать с излучениями трех различных спектральных интервалов: 0,5; 1,0; 2,0 нм. Прибор позволяет измерять проценты пропускания и оптические плотности жидкостей и газов по отношению к выбранному сравнительному образцу. Измерения могут быть сделаны непосредственно по шкале прибора или зарегистрированы на линейном самописце.
Пропускание измеряют в интервале 0—100% (±0,5%), оптические плотности — в интервалах 0—1 (+0,005 — + 0,5), 0 — 2,0 (+0,01 — — + 0,5) (возможно также измерять А в интервале от 0,5 до ±0,5 и от —0,5 до ±1,5 перемещением нуля отсчета). Запись на бланке можно проводить со скоростью 30, 60, 120 и 240 нм/мин.
В качестве источников освещения используются вольфрамовая в видимой области (370 — 800 нм) и дейтериевая в УФ-области (190— 370 нм) лампы. Детектором служит фотоумножитель. Кюветное отделение имеет достаточно большие размеры, чтобы можно было установить кювету с толщиной слоя I = 10 см.
Прибор может работать как однолучевой при измерении спектральной интенсивности или энергии излучения каких-либо источников. Основная схема прибора приведена на рис. 89, его общий вид — на рис. 90. Прежде чем приступить к измерениям, необходимо внимательно ознакомиться с инструкцией о порядке работы на приборе. Подготовив испытуемый раствор, наполнить им кювету и установить ее в держатель, предварительно вынув его из кюветного отделения. В кюветном отделении нельзя ничего проливать. По окончании работы, прежде чем покрыть прибор пленкой, защищающей его от пыли, проверить, выключен ли блок питания и прибор (тумблеры «Operation Swith» 1 и «Power Swith» 2 должны быть в положении «off»). Перед началом работы рукоятки управления должны-находиться в следующих положениях (см. рис, 90);
Рукоятка управления Положение
Power Swith (выключатель блока питания) ........................ . Off (выключено)
Operation Swith (переключатель рода работ)....................... .... Off
Energy— ABS/% Т . .Oft
ABS—% T (переключатель вида измеряемых величин) .................ABS
D2 Lamp, (дейтериевая лампа) . Off
W. Lamp (вольфрамовая лампа) . .Off
Cell Compartmant Cover (крышка кюветного отделения)..............Closed (закрыто)
Slit Selector (выбор ширины щели) 1,0
Для установки механического нуля снять магнитную крышку 3
с отверстия и, повернув винт отверткой, установить стрелку на нуль
шкалы 6. Для установки ламп необходимо снять крышку осветителя 10.
Измерения без записи на бланке. Для этого следует: 1) проверить, стоят ли все рукоятки управления прибором в тех позициях, которые указаны выше; 2) выбрать нужную лампу для работы в соответствующей области спектра (см. стр. 268), поставив рукоятку «Visible — ultraviolet» («видимая — ультрафиолетовая») 9 в нужное положение; 3) установить рукояткой 18 по шкале длин волн 17 требуемую длину волны; 4) поставить рукоятку «Power» 2 в положение «ON», (т. е. включить блок питания 20 в электрическую сеть). Индикаторная лампа 19 должна загореться; 5) поставить рукоятку 4 или 5, соответствующую выбранной лампе, на панели блока питания 20 в положение «ON»; при включении водородной лампы 5, прежде чем поставить переключатель этой лампы на «ON», следует подождать около 10 мин после включения блока питания в сеть; порядок переключения ламп описан на стр. 272; 6) поставить «Operation swith» (переклю-чате, 1ь рода работ) 1 в положение
Рис. 89. Принципиальная схема регистрирующего спектрофотометра Hitachi, модель 124:
1 — осветитель; 2 — вторичный светофильтр, 3 — входная щель, 4 — монохроматор, 5 — выходная щель; 6 — делитель световых потоков; 7 — кюветное отделение; 8 — соединитель потоков; 9 — фотоумножитель, 10 — усилитель, // — анализатор сигналов, 12 — цепь сравнительного канала; 13 — питание фотоумножителя, 14 — цепь канала ис пытуемого образца, 15 — отсчетная шкала: 16— самописец
«ON». Мотор-прерыватель начинает вращаться и стрелка на шкале 6 находится около значения 0,0. Дать прогреться прибору около 10 мин; 7) поставить рукоятку «Skale selektor» (выбор шкалы) 7 в положение «0 — 1» А; 8) установить стрелку на шкале оптических плотностей 6 точно в положение 0,0, пользуясь рукояткой «Zero adjust» (регулировка нуля) 8, расположенной на передней панели
прибора; 9) повернуть переключатель «АВ — % Ту>, расположенный на правой стороне прибора, в положение «%Т». На шкале6 стрелка должна находиться на значении 1,0; 10) поставить стрелку на шкале 6 точно на 100% (1,0), регулируя рукояткой «% Т Span» (протяженность шкалы %7), расположенной на правой стороне прибора; 11) открыть крышку кюветного отделения 11 и вставить пустой кюветодержатель; 12) вставить металлическую пластину (непрозрачное тело), имеющуюся в комплекте прибора, на пути потока излучения, проходящего через испытуемый образец (s), и закрыть крышку кюветного отделения; 13) установить стрелку на 0% шкалы
Рис. 90 Внешний вид регистрирующего спектрофотометра Hitachi, модель 124
6, вращая рукоятку «Zero check» (контроль нуля), расположенную на правой стороне прибора; 14) удалить металлическую пластину и восстановить положение стоелки шкалы 6 на «100 % 7», если это необходимо;
Примечание. Иногда необходимо проверить протяженность обеих шкал оптических плотностей перед измерением.
15) вынуть кюветодержатель и вставить в него кюветы с испытуемым раствором и раствором сравнения. Держатель в кюветном отделении должен быть поставлен так, чтобы штырек, имеющийся на дне кюветного отделения, вошел в отверстие в дне кюветодержателя. Закрыть крышку кюветного отделения; 16) отсчитать по шкале б показания оптической плотности или процента пропускания. (Если А > > 1,0, то переключатель «Scale selector» 7 должен стоять в положении, соответствующем интервалу 0 — 2.)
Выключают прибор в следующем порядке: 1) поставить переключатели «Operation swith» 1 и «Power svxith» 2 в положение «Off»; 2) вынуть кюветодержатель и убедиться, что в кюветном отделении ничто не пролито; 3) закрыть прибор чехлом.
Запись спектров поглощения (пропускания) на бланке при помоши самописца. Самописец подключают к прибору в соответствии с прилагаемой инструкцией.
Перед началом записи прибор подготовить к работе согласно указаниям, данным на стр. 269, и пп. 1—15 инструкции для измерений А и Т без записи за исключением того, что длина волны (п. 3) устанавливается несколько большей, чем требуется для начала записи. Сканирование происходит от больших длин волн к меньшим.
Далее нужно подготовить самописец 22 (см. рис. 90) для записи: 1) установить рулон бумаги, для чего следует: а) вставить стержень 27 в отверстие рулона 21 и соединить рулон с пластиковыми концами стержня; б) поднять прижим 13 (он должен удерживаться в этом положении защелкивающейся пружиной); в) обрезать конец бланка в виде острого треугольника с высотой ~ 150 мм; г) поместить треугольный конец бумаги между двумя ведущими пластинами 23; д) укрепить концы стержня в прорези нижней ведущей пластины; е) вращать постепенно рулон так, чтобы бумага двигалась внутрь самописца между ведущими пластинами и появилась в промежутке между управляющим барабаном 12 и прижимом 13; после того как конец бумаги выйдет из отверстия на 100 мм, установить перфорацию на штырьки барабана слева и справа и опустить прижим 13, повернуть вручную барабан рукояткой 24 против часовой стрелки и убедиться, что бланк хорошо закреплен и передвигается (после окончания записи бланк должен свободно свисать вниз; отрыв бланка проводят у переднего края столика самописца); ж) чтобы сдвинуть бланк в обратную сторону, следует повернуть ручку 24 барабана по часовой стрелке до желаемого положения;
2) настроить самописец, для чего следует: а) проверить, стоит ли рукоятка «Power selector swith» 14 в положении «Off»; б) поставить указатель скорости движения бланка 25 на требуемую скорость (она может иметь значения 5, 10, 20, 60, 100 и 240 мм/мин); в) повернуть’ рукоятку 14 в положение «Ашр», что соответствует подключению усилителя; индикаторная лампа должна загореться; дать прогреться прибору 30 мин; г) опустить перо на бланк рукояткой 15; д) повернуть рукоятку 14 в положение «Servo»; е) поставить переключатель «Zero check swith» (контроль нуля) 16 в положение «Zero» (нуль) и рукояткой «Zero set knob» (установка нуля) 26 помести перо к выбранному положению нуля оптической плотности на бланке (это положение может находиться на любом участке бланка); отпустить переключатель 16, при этом он самопроизвольно переходит в положение «Meas».
Примечание. Переключатель «Polarity chang over swith» (переключатель полярности) позволяет установить нуль начала отсчета А (Т) на любой (левой или правой) стороне бланка.
Запись спектра проводить следующим образом: 1) установить рукоятку «X speed» (скорость длин волн), расположенную на задней панели прибора, в нужное положение. Для большинства операций рекомендуется пользоваться положением «Fast» (быстро) 60 нм/мин. Если изучаемая система обладает узкополосными острыми максимумами, то запись следует вести на малой скорости при положении переключателя «Slow»; 2) повернуть рукоятку «Operation swith» 1 в положение «Scan» (сканирование). Когда указатель длин волн подойдет точно
к значению, соответствующему началу записи, поставить переключатель «Operation swith» 1 в положение «ON»; 3) включить движение бланка на самописце, переключив рукоятку 14 в положение «Chart» (бланк) и остановить его, когда перо подойдет к нужной точке, поставив рукоятку 14 в положение «ОН» (это можно сделать вручную рукояткой 24)\ 4) повернуть рукоятку «Operation swith» 1 в положение «Scan». Движение рукоятки длин волн и самописца должно быть начато одновременно. По окончании записи поставить рукоятку «Operation swith» в положение «Off».
Переключение ламп. 1. Если переходят от работы в видимой области спектра к УФ, то соблюдают следующий порядок операций и переключения с вольфрамовой на дейтериевую лампу: а) если дейтериевая лампа не горит, поставить переключатель «Operation swith» в положение «Off», установить шкалу длин волн примерно на значение 370 нм, поставить переключатель «Visible-ultrav» в положение «Ultrav» л переключатель «W. Lamp» в положение «Off» и подождать 5 мин; поставить выключатель «D2Lamp» в положение «ON», поставить переключатель «Operation swith» в положение «ON»; б) если дейтериевая лампа горит, подвести шкалу длин волн примерно к 370 нм. Поставить рукоятку «Visible-ultraviolet» в положение «Ultraviolet». Поставить выключатель «W. Lamp» в положение «Off» (если не требуется, чтобы «W. Lamp» горела далее).
2. Если переходят от УФ к видимой области спектра, то соблюдают следующий порядок операций переключения с дейтериевой на вольфрамовую лампу:
а) если требуется, чтобы дейтериевая лампа горела в то время, когда ведется работа с вольфрамовой лампой, подвести шкалу длин волн к 7> 370 нм; поставить выключатель «W. Lamp» в положение «ON»; поставить рукоятку «Visible-ultrav» в положение «Visible»;
б) если не требуется, чтобы дейтериевая лампа горела во время работы с вольфрамовой лампой, то установить по шкале длин волн значение 7, >» 370 нм, поставить выключатель «W. Lamp» в положение «ON», рукоятку Visible-ultrav.» — в положение «Visible», выключатель «D2Lamp» — в положение «Off».
Запись по времени. Запись поглощения (или пропускания) может проводиться при фиксированной длине волны как функция времени. Для этого нужно выполнить все операции, указанные на стр. 269— 270, за исключением пп. 16. Самописец регистрирует сигнал изменения оптической плотности или процента пропускания.
Измерение в далекой УФ-области спектра. Если необходимо проводить измерения при длинах волн короче 196 нм, из монохроматора и на всем пути излучения должен быть удален кислород продуванием током азота. Используемый азот должен быть чистым и сухим, чтобы никакие частицы не попали в монохроматор. Трубки, подводящие газ, должны быть из полихлорвинила; резиновые и т. п. не рекомендуются. Если газ содержит какие-либо загрязнения в виде твердых частиц, то используются фильтры. Запись спектра проводят в следующем порядке:
1. Удалить пробки из отверстий «N2Gas inlet» (ввод газа) и «Outlet» (вывод) и подвести сжатый, чистый и сухой газ.
2. Поставить рукоятку «Visible — ultraviolet» в положение «Ultraviolet». Поставить переключатель «ABS—% Т/Energy» в положение «Energy».
3. Поставить шкалу длин волн на значение 190 нм.
4. Поставить давление азота на 2 lb/sq. in (2 psi ~0,136 атм).
5. Поставить тумблер «Power swith» в положение «ON».
6. Поставить тумблер «D2Lamp» в положение «ON».
7. Повернуть переключатель «Operation swith» в положение «ON».
8. Повернуть рукоятку контроля «ABS — % Т/Energy» в положение «Energy» и установить шкалу примерно на 50% пропускания.
9. Открыть вентиль баллона с азотом и пустить газ в прибор, так как кислород будет вытесняться из прибора, его поглощение при Я, 190 нм будет падать, а следовательно, энергия, попадающая на детектор, возрастать. Через 30—40 мин показание на шкале достигнет максимального значения. После этого интенсивное продувание можно прекратить, оставив только слабый ток газа, и далее использовать прибор для измерений.
Регистрирующие спектрофотометры СФ-10, СФ-14
Регистрирующие спектрофотометры указанных моделей позволяют записывать спектры поглощения и пропускания, а так же измерять коэффициенты диффузного отражения различных твердых и порошкообразных веществ. Запись по всей длине видимого спектра может быть произведена в несравненно более короткое время (2—12 мин), чем измерение поглощений в этом же участке спектра на нерегистрирующем спектрофотометре типа СФ-4, СФ-16. Прибор имеет двойной монохроматор, поэтому монохроматизация света здесь достаточно высокая. Ширина входной и выходной щелей монохроматора изменяется во время работы прибора автоматически соответственно дисперсии призм. Таким образом, при достаточно высокой монохроматизации вырезается спектральный участок постоянного спектрального интервала. Источником освещения служит кинопроекционная лампа К-30, приемником энергии—мультищелочной фотоэлемент Ф-10. Рабочий диапазон прибора охватывает только видимую часть спектра от 400 до 700 нм, и, следовательно, он обладает в этом отношении меньшими возможностями, чем нерегистрирующие кварцевые спектрофотометры типа СФ-4 (СФ-16).
Спектрофотометры моделей СФ-10 и СФ-14 в принципе ничем не отличаются друг от друга ни по оптической схеме, ни по порядку измерений, за исключением того, что запись спектров на спектрофотометре СФ-10 можно проводить лишь в интервале оптической плотности от 0 до 2,5 и в интервале процента пропускания от 1 до 100%, в то время как на СФ-14 имеется четыре предела измерений: оптическую плотность измеряют от 0 до 2,5 и от 0 до 1,0; проценты пропускания—от 1 до 100рб и от 0 до 10%.
Спектрофотометры СФ-10, СФ-14 состоят из осветителя, двойного призменного монохроматора, фотометра поляризационного типа, приемно-усилительной части и записывающего механизма. Монохроматический пучок света делится призмой Рошона на два плоскополя-ризованных пучка. Один пучок диафрагмируется, другой проходит через призму Волластона и снова делится на два пучка, поляризованных во взаимно перпендикулярных плоскостях. Так как на призму Волластона падает плоскогюляризованный пучок света, интенсивность пучков света за призмой Волластона определяется угловым положением по отношению к ней призмы Рошона. Далее пучки перекрываются вращающимся барабаном прерывателя таким образом, что интенсивность световых потоков в каждом пучке изменяется по форме трапеции и началу открытия одного пучка соответствует начало закрытия другого. Конструкция барабана прерывателя и скорость его вращения выбраны так, что световой поток меняется с частотой 50 Гц.
Свет, отраженный от образца и эталона, после многократного отражения от стенок шара освещает фотоэлемент, расположенный за окном шара, закрытым молочным стеклом. Освещенность фотоэлемента в каждый момент времени определяется суммой мгновенных потоков, отраженных от образца и эталона. Если световые потоки, отраженные образцом и эталоном, равны, освещенность фотоэлемента будет постоянна в любой момент времени и переменный сигнал на входе усилительной системы будет отсутствовать. Если испытуемый образец заметно поглощает, то суммарный световой поток на фотоэлементе будет изменяться с частотой 50 Гц и на входе усилителя появится сигнал такой же частоты. Напряжение сигнала усиливается и подается на обмотку якоря электродвигателя отработки, который при помощи фотометрического кулачка поворачивает призму Рошонд до тех пор, пока не исчезнет сигнал на входе усилителя, т. е. пока не исчезнет разность световых потоков. Одновременно с поворотом призмы происходит перемещение пера, фиксирующего на бланке пропускание, отражение или оптическую плотность образца. Изменение длины волны света, выходящего из монохроматора, производится перемещением вдоль спектра средней щели прибора. Перемещение щели осуществляется от электродвигателя одновременно с поворотом барабана записывающего механизма. Таким образом, на бланке, закрепленном на барабане записывающего механизма, записывается кривая спектрального пропускания, отражения или оптической плотности.
Оптическая схема приборов приведена на рис. 91, их общий вид — на рис. 92. Оптическая система приборов состоит из спектральной (двойного монохроматора) и фотометрической частей. Нить лампы 1 (см. рис. 91) изображается конденсором 2 через входную щель 3 в плоскости объектива 4 коллиматора. Входная щель расположена в фокальной плоскости объектива. Выходящий из него параллельный пучок света проходит диспергирующую призму 5 и разлагается в спектр. Объектив 6 первого монохроматора дает спектральное изображение входной щели в плоскости средней щели по линии А — А. Средняя щель двойного монохроматора, образованная зеркалом 7 и ножом 8,
вырезает участок спектра, Который проходит второй монохроматор 5, и проектируется в плоскость выходной щели 9.
По выходе из монохроматора пучок света попадает в фотометрическую часть прибора. Сначала пучок проходит через линзу 10 и двояко-преломляющую призму Рошона //. Линза 10 дает изображение объектива выходного коллиматора вблизи диафрагмы 12, призма 11 разделяет это изображение на два, поляризованных во взаимноперпендикулярных плоскостях: одно, расположенное симметрично относительно оси, проходит через призму Волластона 13 и линзу 14, другое, сме-
Ркс. 91. Принципиальная оптическая схема регистрирующих спектрофотометров СФ-10 и СФ-14
щенное, срезается диафрагмой 12. Линза 14 дает изображение выходной щели в плоскости полулинз 15. Вследствие двойного лучепреломления призмы Волластона в плоскости полулинз получаются два изображения выходной щели. Пройдя полулинзы 15, установленные внутри барабана прерывателя 16, оба пучка отклоняются на 90° призмой 17, проходят через входные окна интегрирующей сферы 18 и падают на окна, к которым прижимаются образец и эталон при измерении коэффициента отражения или два эталона—при измерении коэффициента пропускания. Свет, отраженный от образца и эталона, суммируется шаром и падает на фотоэлемент за выходным окном шара.
Питание осветителя, электромеханической системы и усилителя осуществляется от сети переменного тока напряжением 127 В; сетевой шланг подключают к разъему 35 (см. рис. 92); заземляют прибор через клемму 36. Кюветы с растворами и твердые образцы укрепляются в специальных держателях.
Порядок работы. 1. Для подготовки прибора к измерениям следует: а) провеоить, выключены ли выключатели на щитке управления 26\
б) установить ширину входной и выходной щели. Для этого отодвигают шторку правого 25 и левого 25' окон в кожухе прибора и, ослабив винты, устанавливают требуемую ширину спектрального интервала согласно цифрам на барабанчиках; в) установить рукояткой 27, оттянув ее на себя и повернув в нужную сторону, в рабочее положение кулачок 19: для записи по оптическим плотностям против рукоятки 19 должна появиться буква «Д», коэффициентам отражения или пропускания — соответственно буква «У»; г) установить перо в каретку 33 и приподнять его, оттянув кнопку 20 на себя; д) установить по шкале
Рис 92. Внешний вид регистрир^ющик спектрофотометров СФ-10 и СФ 14
длин волн 22 значение 400 нм при помощи рукоятки 23. Вращать шкалу длин волн рукояткой 23 можно как против часовой стрелки в сторону длин волн, так и по часовой стрелке. Однако это вращение вручную ограничено пределами шкалы; подводить какое-либо значение шкалы следует вращая ее всегда в сторону больших Если при этом шкала случайно провернется дальше, нужно отвести ее назад и снова подвести требуемое деление; е) установить бланк для записи на барабане 24. Для этого предварительно обрезают бланк, имеющийся в комплекте прибора так, чтобы линия отреза прошла через два штриха, нанесенные ниже шкалы длин волн на расстоянии 15 ± 0,2 мм. Поднять прижим 34 и плотно навернуть бланк на барабан, вводя его снизу так, чтобы линия отреза вплотную прилегла к правому бортику барабана. После этого прижать бланк прижимом 34, опустить перо, нажав кнопку 20, рукой повернуть барабан так, чтобы перо при опускании его на бланк стояло на абсциссе, соответствующей 400 нм.
2. Далее включают прибор и проверяют правильность его работы. Для этого необходимо: а) включить прибор в электросеть с напряжением 127 В; б) включить выключатель «сеть» на щитке 26 и дать прибору прогреться в течение 5 мин; в) включить выключатель «лампа» на щитке 26. Произвести юстировку лампы при помощи рукояток 29. Юстировка лампы контролируется по резкому изображению нити лампы в центре поверхности коллиматорной линзы (для наблюдения следует отодвинуть шторку левого окна 25' в кожухе прибора). Если необ
ходимо сменить лампу, то снимают кожух осветителя 30. По окончании юстировки лампу выключить; г) включить тумблер «модулятор» (прерыватель) на щитке 26; д) включить мотор «отработка» выключателем на щитке 26; е) повернуть рукоятку «усиление» 28 в крайнее правое положение (максимальное усиление); ж) включить лампу. Если в кюветном отделении отсутствуют какие-либо поглощающие объекты и в окнах интегрирующей сферы установлены два одинаковых эталона, то при положении «Д» рукоятки 27 перо должно двигаться вправо, т. е. к нулю оптической плот ности, и соответственно влево при положении «Т», т. е. в сторону 100% пропускания; з) переключить рукоятку «изменение направления» на щитке 26, если перо движется не в том направлении, в котором следует; и) установить перо винтом 19 на линию бланка, соответствующую примерно 95% пропускания; к) установить рукояткой 23 на шкале длин волн значение 420 нм, при этом должны наблюдаться колебания пера. Вращать рукоятку 28 влево до тех пор, пока не прекратятся колебания пера. Для проверки правильности установки усиления следует коснуться правой клеммы «контроль» пальцем, при этом перо должно отойти в сторону. Затем отнять палец, при этом перо должно возвратиться в первоначальное положение с одним-двумя колебаниями. Если наблюдается несколько колебаний, усиление нужно уменьшить. Если перо медленно подходит к первоначальному положению, усиление следует увеличить. Проверку проводят при незазем-ленном приборе. После проверки прибор обязательно заземлить.
3. После этого необходимо произвести запись прямой, соответствующей Т = 100%: а) установить по шкале длин волн 22 значение 400 нм рукояткой 23; б) установить максимальную скорость записи, поставив переключатель «скорость развертки спектра» 32 в положение «4»; в) включить последовательно выключатели «отработка» и «развертка спектра» на щитке 26 и записать кривую 100%-ного пропускания; г) по окончании записи (при 750 нм) выключить выключатель «развертка спектра»; д) отрегулировать положение пера поворотом винта 19 через правое окно 25 в кожухе прибора, если оно не идет по прямой, соответствующей Т — 100% при отсутствии измеряемых образцов на пути светового потока, или стремится выйти за ее пределы; при этом в окне 21 должно наблюдаться совмещение индекса со значением 100%. Если этого нельзя добиться винтом 19, то освободив винт, крепящий каретку с держателем пера к капроновому шнуру, добиться такого положения, когда острие пера будет совпадать с линией 100% на бланке, а отметка «100%», наблюдаемая в окно 21, совпадает с индексом (регулировку производят отверткой, придерживая каретку во избежание растяжения капронового шнура); е) выключить по окончании регулировки линии 100%-ного пропускания тумблер «отработка» на щитке 26.
4. Проверка правильности градуировки шкалы длин волн и записи оптических плотностей и процентов пропускания проводится следующим образом: а) установить на шкале длин волн 22 отсчет 400 нм ру- -кояткой 23; б) установить переключатель «скорость развертки спектра» в положение «3»; в) укрепить в держателе светофильтр № 1 («дидимо-вое стекло» — стекло с добавками солей неодима и празеодима) и уста-
повить его в кюветное отделение 31 на пути правого (по ходу лучей) пучка света; закрыть крышку кюветного отделения; г) включить выключатель «отработка» и подождать, пока перо займет положение, соответствующее пропусканию светофильтра № 1 при К 400 нм; д) включить тумблер «развертка спектра» и записать кривую пропускания светофильтра № 1; по окончании записи выключить последовательно выключатели «развертка спектра» и «отработка». Сравнить полученную запись с записью, приложенной к аттестату прибора (сравнить 1 максимумов поглощения); смещение кривой по длинам волн должно быть не более±0,5 нм; е) установить в кюветное отделение держатель со светофильтром № 2 и записать его кривую пропускания таким же образом, как и для светофильтра № 1 (пп. а — д); отклонения в пропускании от контрольной записи, прилагаемой к аттестату, не должны превышать 0,3% по всему спектру; ж) установить по шкале длин волн значение 400 нм; з) установить рукояткой 27 в рабочее положение кулачок «Д»; и) включить выключатель «отработка»; переключить рукоятку «изменение направления», если перо начнет двигаться влево, т. е. от нуля оптической плотности; к) установить перо винтом 19 на линию А = 0; выключить тумблер «отработка»; л) установить в кюветное отделение держатель со светофильтром № 3, закрыть крышку кюветного отделения; м) включить тумблер «отработка» и подождать, пока перо не займет положение, соответствующее оптической плотности светофильтра № 3 при X 400 нм; н) включить тумблер «развертка спектра» и записать кривую спектра поглощения светофильтра № 3; после этого выключить последовательно выключатели «развертка спектра» и «отработка»; при сравнении полученной записи с контрольной, прилагаемой к аттестату прибора, отклонение не должно превышать ±0,0075 единиц оптической плотности по всему спектру.
Примечания: 1. При переходе от кулачка «Т» к кулачку «Д» необходимо всегда проверять установку нулевой оптической плотности, а при переходе от «Д» к «Г»— установку 100%-ного пропускания.
1 ^"чись с кулачком «7\» производится в масштабе, увеличенном в 10 раз, поэтом^ получения истинных результатов необходимо значения, отсчитанные по шкале пропусканий бланка, разделить на 10.
3. Запись с кулачком «Д[» производится в масштабе, увеличенном в 2,5 раза, поэтому для получения истинных результатов измерений необходимо значения, снятые по шкале оптических плотностей, разделить на 2,5.
4. Проверка правильности записи значений Д и Т при работе на спектрофотометре СФ-14 может быть сделана со светофильтром №2 при установке в рабочее положение кулачка «Др>, соответствующее интервалу в оптической плотности 0—1, 0, и со светофильтром № 3 при установке в рабочее положение кулачка «Т1» (соответствующее интервалу 0—10% Т). Критерии точности те же, что и при записи с кулачками «Д» и «7».
5. Во избежание поломки пера следует держать его в приподнятом положении до тех пор, пока прибор не будет полностью подготовлен к записи. После установки и снятия бланка прижим записывающего механизма необходимо опустить, неопущенный прижим может привести к поломке пера. Чтобы перо не чертило на бланке ненужных линий, его также следует поднимать при возвращении шкалы длин волн к началу записи.
6. Скорость записи по спектру регулируется рукояткой «скорость развертки спектра». Спектры поглощения веществ, обладающих широкими и мало интенсивными максимумами, можно записывать на большой скорости (при положении рукоятки «3»—«4»), Спектры поглощения таких объектов, как аква-
комплексы редкоземельных элементов, следует регистрировать при минимальной скорости записи, иначе при большой скорости поворота барабана перо не успевает пройти вдоль барабана и выписать резкий и узкий максимум.
7. Проверка градуировки шкалы длин волн и запись спектров контрольных фильтров проводится только периодически после прохождения некоторого периода работы прибора — через 1—2 месяца.
5. Запись спектра поглощения испытуемого образца проводят аналогично тому, как проводили запись спектров светофильтров в следующем порядке: а) проделать все операции, указанные в пп. 1—3 (а также в пп. 4, з—к, если запись будет проводиться в величинах оптических плотностей); б) возвратить шкалу длин волн к значению 400 нм рукояткой 23, подняв перо и выключив мотор длин волн («развертка спектра»), а затем мотор «отработка»; в) поднять кожух 31 кюветного отделения, установить на пути светового потока кювето-держатели с испытуемым раствором и раствором сравнения. Раствор сравнения устанавливают всегда в правом световом потоке, исследуемый — в левом. Закрывают кюветное отделение кожухом; г) включить мотор «отработка» и дать перу занять положение, соответствующее поглощению или пропусканию образца при 2.400 нм; д) включить мотор длин волн («развертка спектра») и произвести запись по спектру.
Останавливать запись и выключать прибор рекомендуется в следующем порядке: 1) выключить мотор «развертка спектра»; 2) выключить мотор «отработка». Если нужно записать спектр нового образца, то именно в этот момент можно поставить его в кюветное отделение и, установив шкалу длин волн, как обычно, на требуемое значение, произвести запись, включив сначала мотор «отработка» и затем мотор «раз-вептка спектра». Чтобы выключить прибор полностью, выключают отдельные его узлы в следующем порядке; 3) мотор-модулятор («прерыватель»); 4) «лампа»; 5) «сеть». Выключают прибор из сети.
ЛИТЕРАТУРА
1. В. М. Ч у л а н о в с к и й. Введение в молекулярный спектральный анализ. М.—Л., Гостехиздат, 1951.
2. Дж. Гаррисон, Р. Лорд, Дж. Луфбуров. Прак, еская спектроскопия. М., ИЛ, 1950.
3. В. С. А с а т и а н и. Биохимическая фотометрия. М.—Л., Изд-во АН СССР, 1957.
4. И. П. А л и м а р и н. Фотоэлектрические колориметры с селеновыми фотоэлементами и применение их в химическом анализе. М., Госгеолтехиздат, 1944.
5. Н. Г. Алексеев, В. А. Прохоров, К. В. Ч м у т о в. Современные электронные приборы и схемы в физико-химическом исследовании. М .—Л., «Химия», 1971.
6. «Абсорбционная спектроскопия». Под ред. Э. В. Шпольского. ИЛ, 1953.
7. Г. В. Ю и н г. Инструментальные методы химического анализа. М., Атомиздат, 1963.
8. М. И. Корсунский. Оптика, строение атома, атомное ядро. М.—Л., «Наука», 1964.
9. В. М. Т а т е в с к и й. Спектроскопия. М., Изд-во МГУ, 1951.
10. В. М. Пешкова, В. М. Бочкова, Е. К- Астахова. ЖАХ, 1961, 16, 596.
т-, Стр.
Пре дисловие . . ................ .... 3
Вве дение . ....... . . . . 5
Гла ва I. Теоретические основы методов абсорбционной спектроскопии 14
1. Основные законы поглощения электромагнитного излучения . . . 14
2. Молярный коэффициент погашения . ...................... 18
3. Вычисление истинных значений молярных коэффициентов погашения 22
4. Отклонения от закона Бугера — Ламберта — Бера . . 26
5. Преимущества работы с монохроматическими излучениями 28
6. Пределы точности измерения величин поглощений 30
\/ Г л а’в а II. Получение «окрашенных соединений» и использование их в количественном спектрофотометрическом анализе * 35
1. Типы фотометр ируёмых систем............ 35
2. Устранение влияния сопутствующих компонентов . . 38
3. Исследование спектрофотометрической реакции и выбор оптимальных условий ее проведения ................................... .... 40
4. Чувствительность фотометрических методов . . 48
Глава III. Методы количественного абсорбционно-спектроскопического /анализа . . . . .... 54
'/ 1. Классификация методов и способы определения концентрации веществ в растворах.........................................................54
Метод спектрофотометрического титрования . . . 56
(.71 Дифференциальный спектрофотометрический метод . 65
4. Анализ многокомпонентных систем .... 72
5. Экстракционно-спектрофотометрический метод........................78
6. Кинетические методы определения элементов со спектрофотометрическим контролем . ... . .82
7. Обработка результатов измерений . . 84
Глава IV. Применение метода абсорбционной спектроскопии для изучения равновесий в растворах ... 90
1. Расчет констант диссоциации органических реагентов . .90
2. Определение состава комплексных соединений .... .97
3. Расчет констант устойчивости комплексных соединений . . . .105
4. Примеры применения спектрофотометрического метода для изучения равновесий в растворах .113
Гл ава V. Определение отдельных элементов ... 129
1. Растворы сравнения при измерении величин поглощения . .129
2. Сравнительное изучение спектральных характеристик двух различных систем на приборах разного типа......................................130
3. Оценка преимуществ при измерениях поглощения с ртутной лампой на фотоэлектроколориметре ФЭК-56 - .131
4. Методы определения элементов . - 132
Гл ава VI. Аппаратура в спектрофотометрии . - 232
1 Принципы конструирования приборов для спектрофотометрического ме-
\\ / тода анализа............... . .... 232
V 2. Характеристика основных узлов спектральных приборов . ... 234
3. Оптические схемы и принципы действия различных фотометрических приборов . ............. 246