/











































































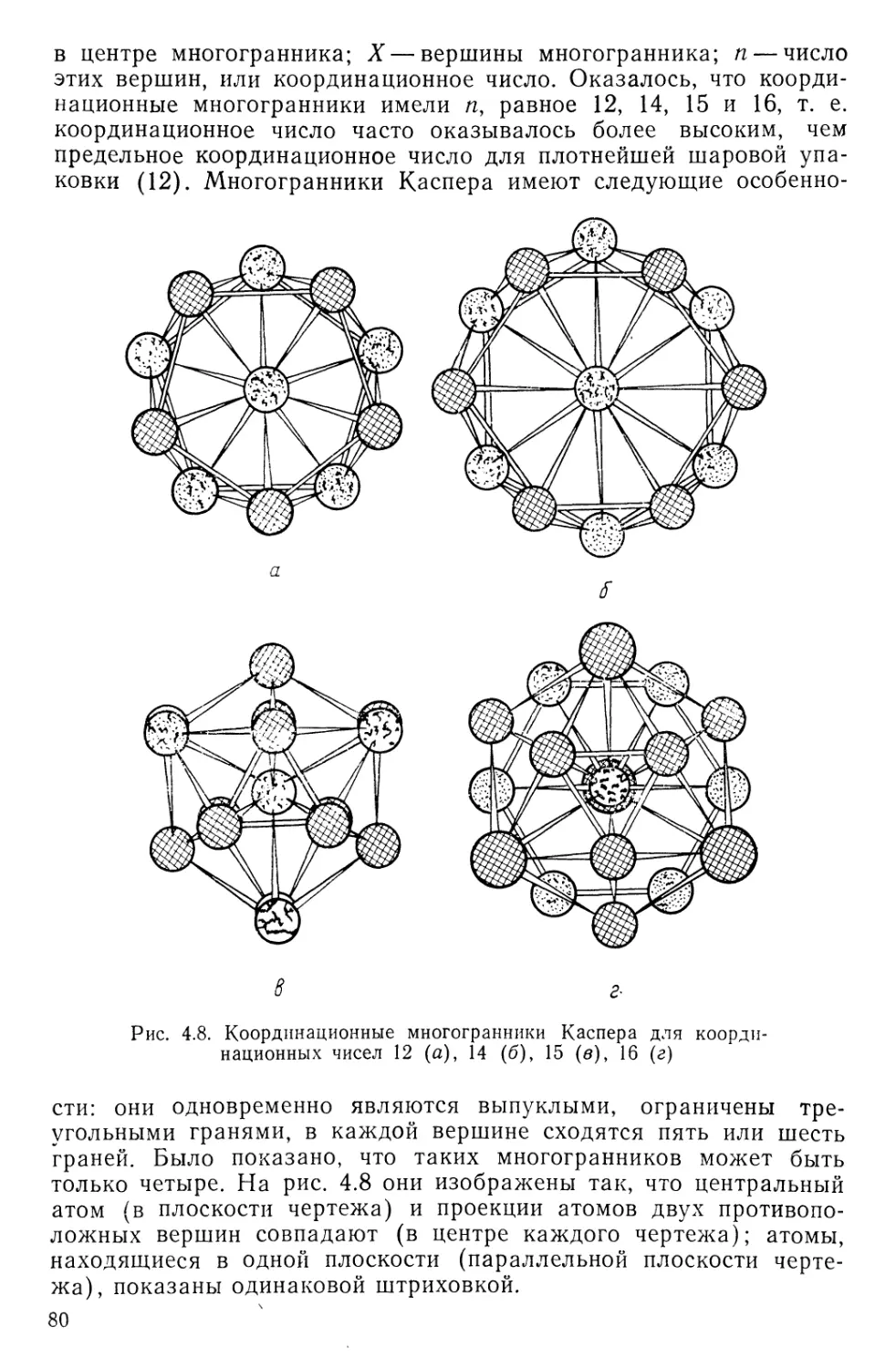










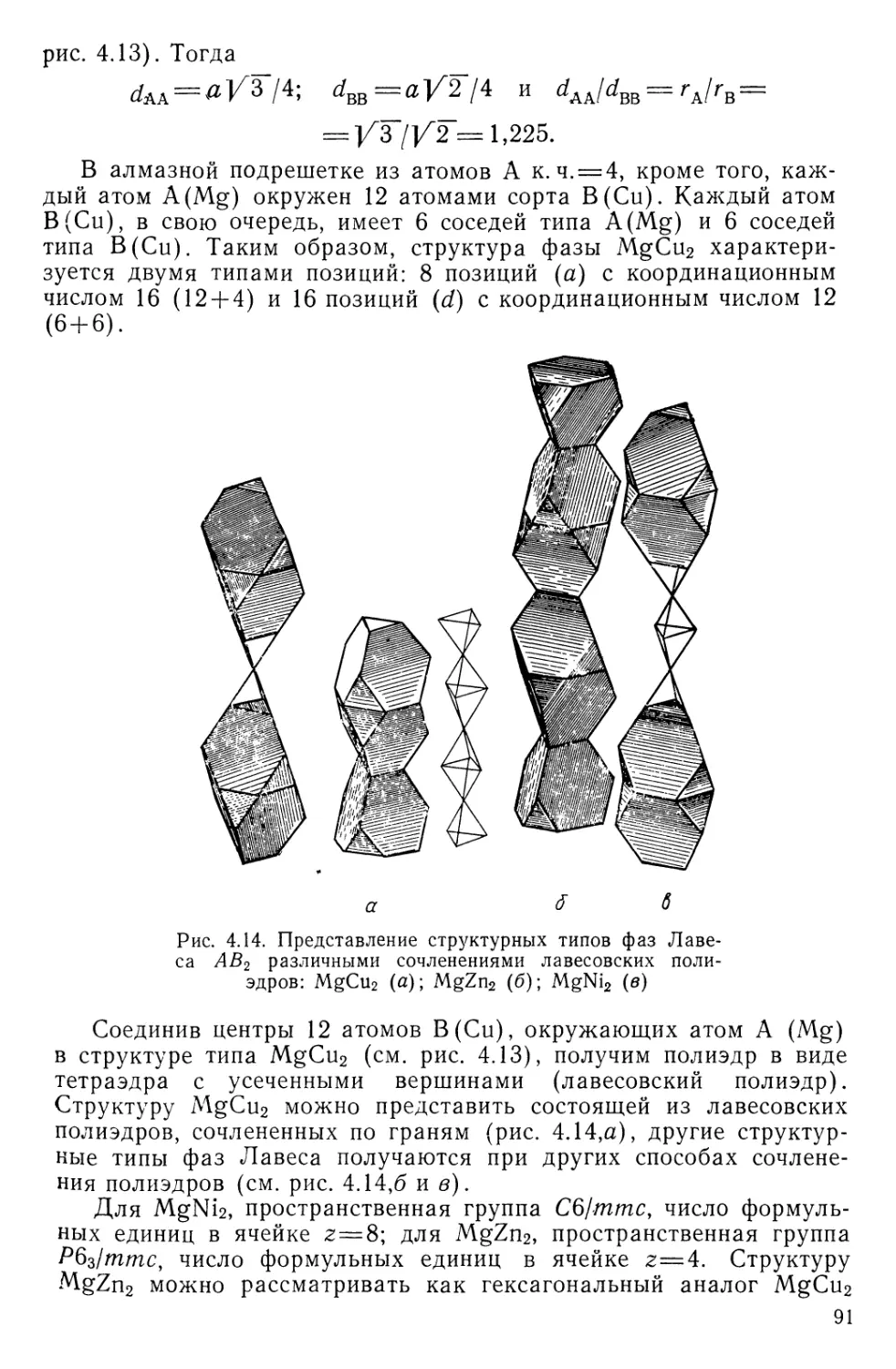


















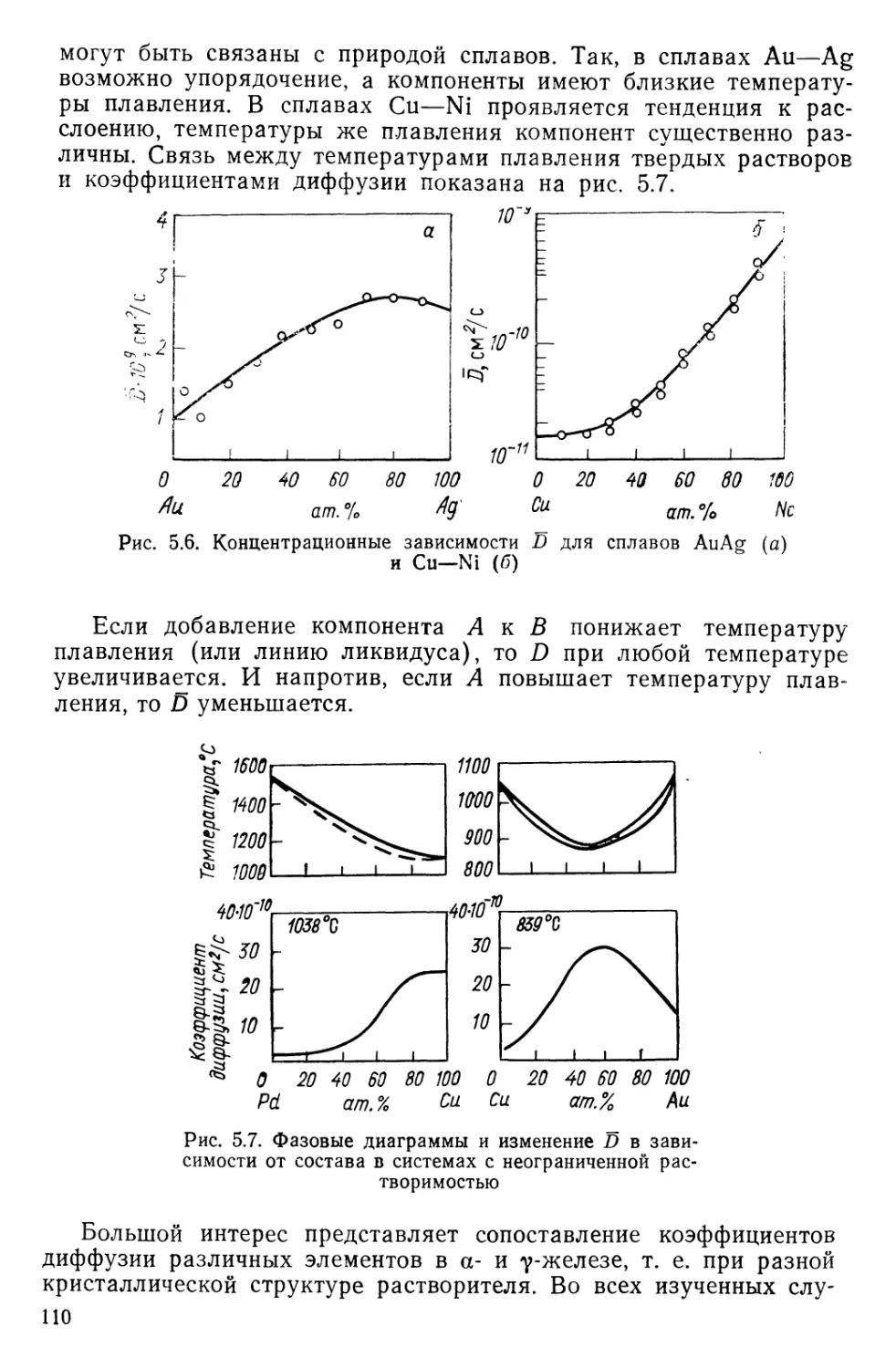




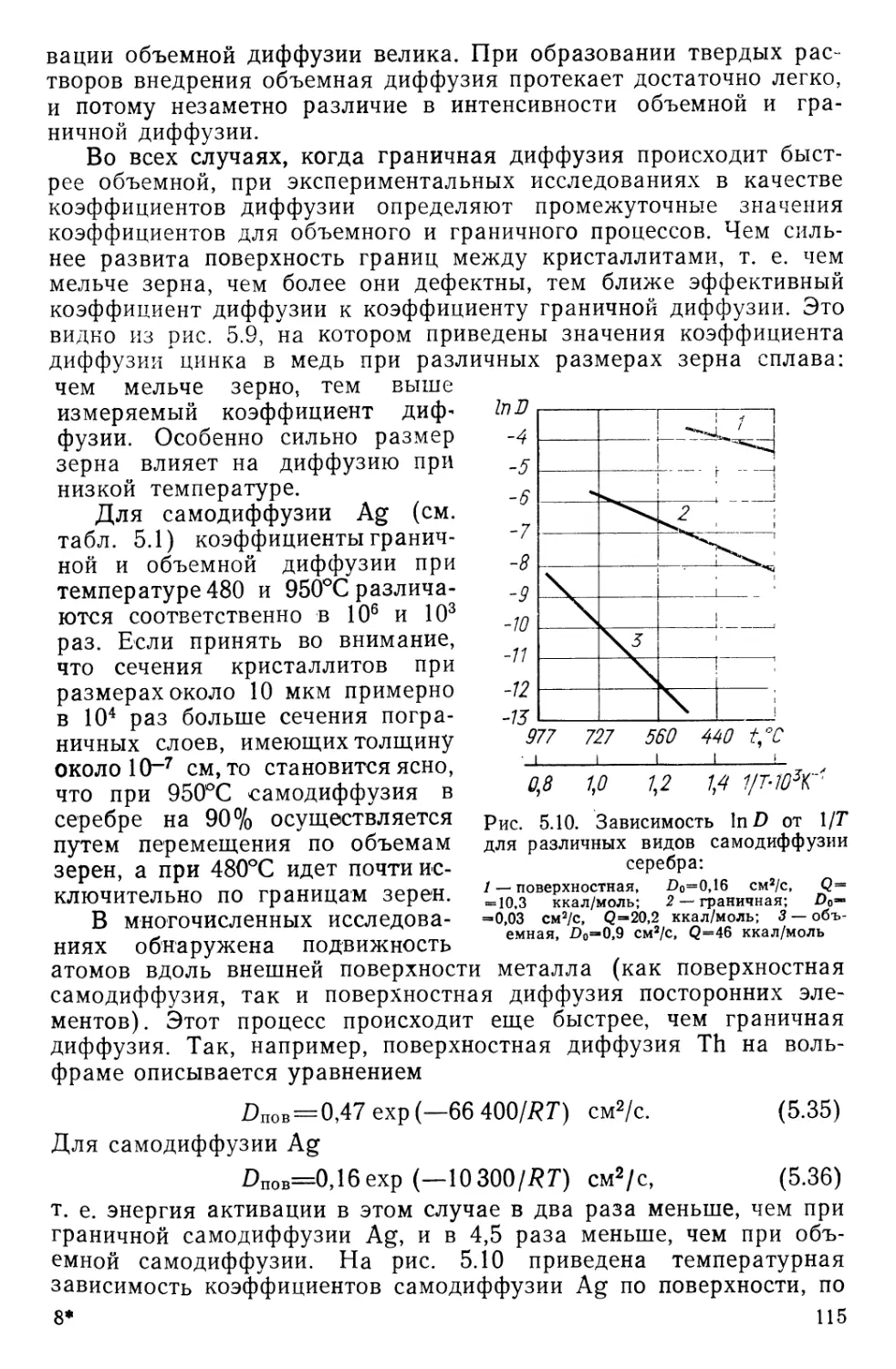



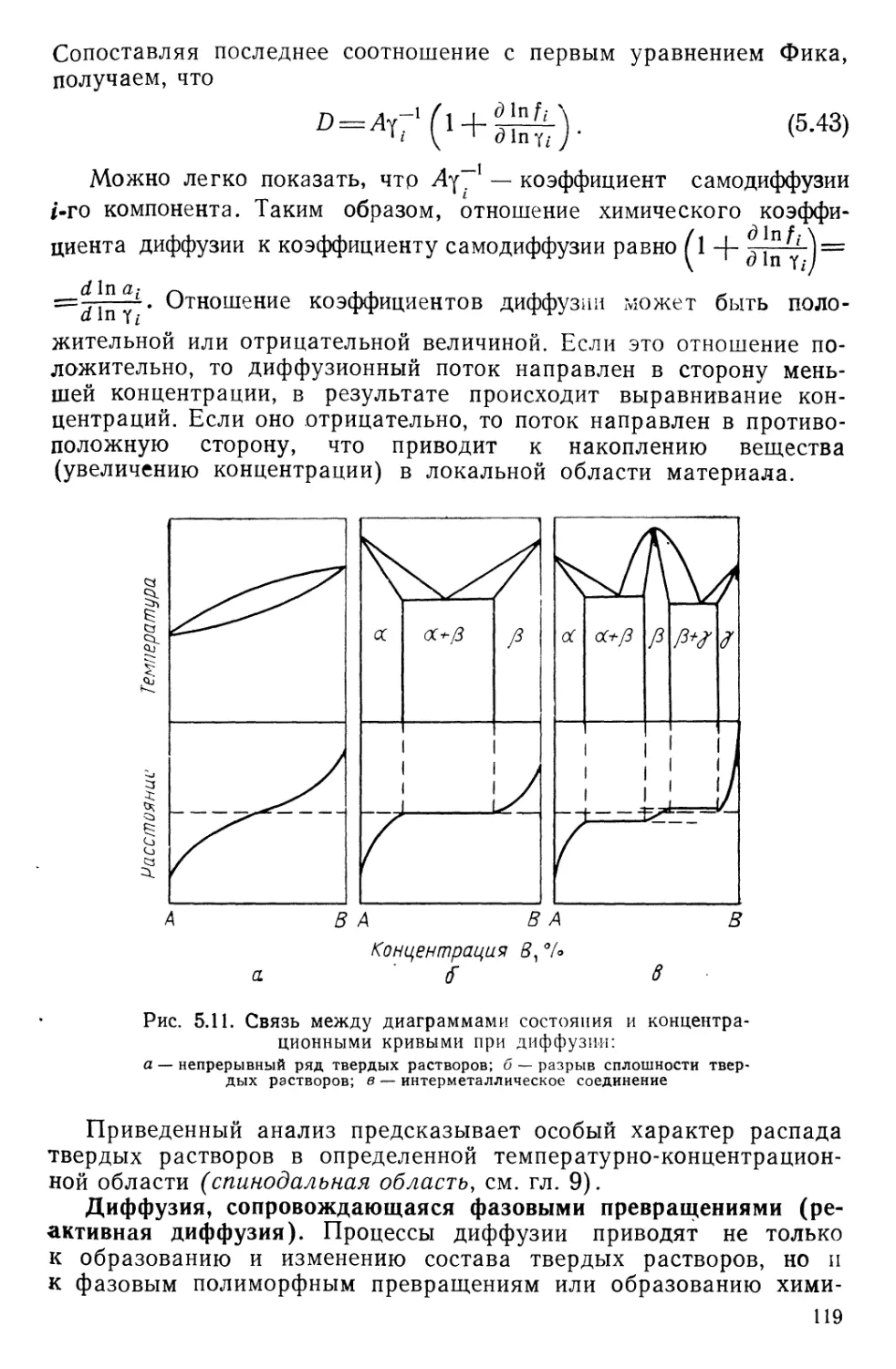

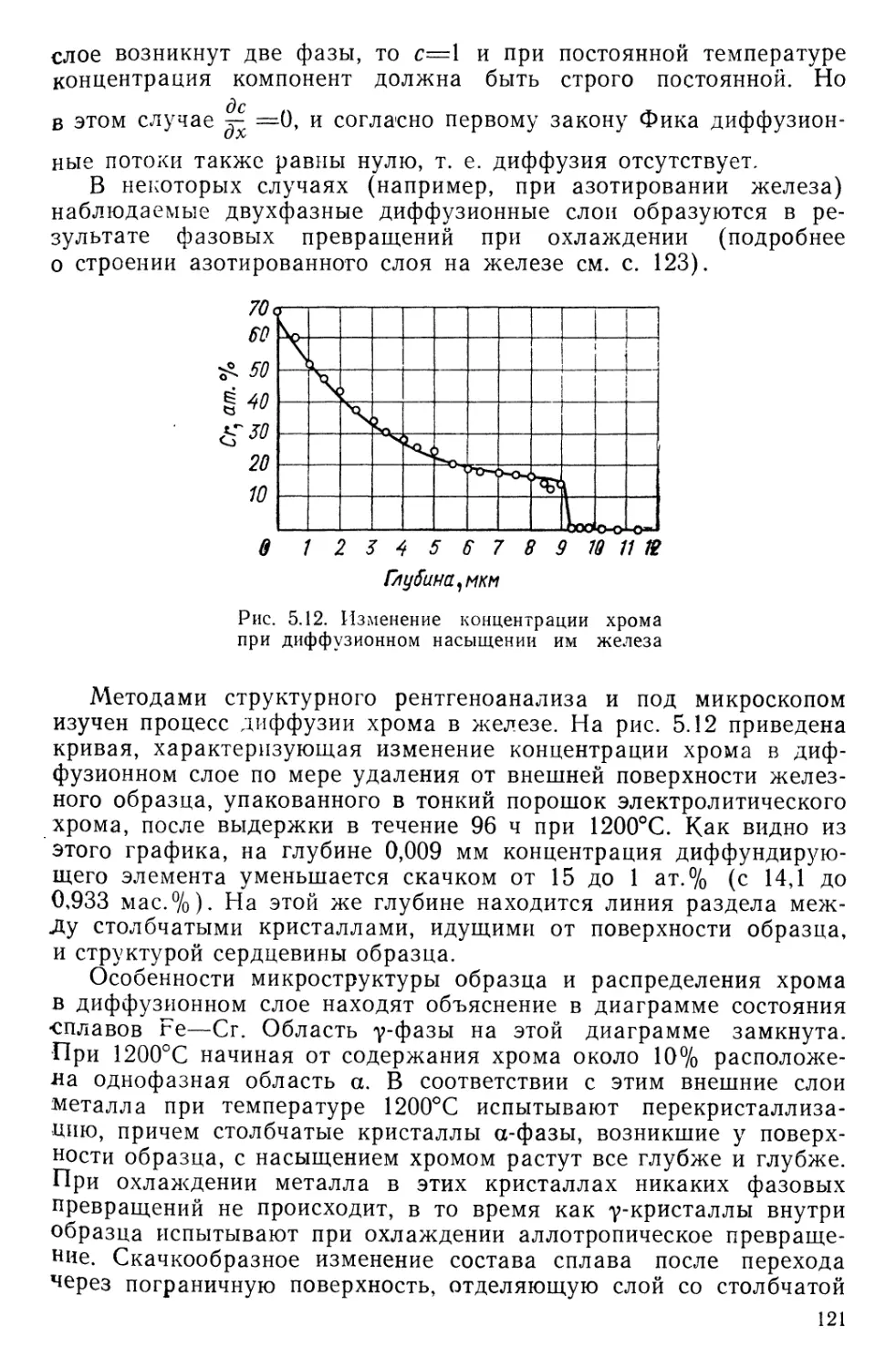























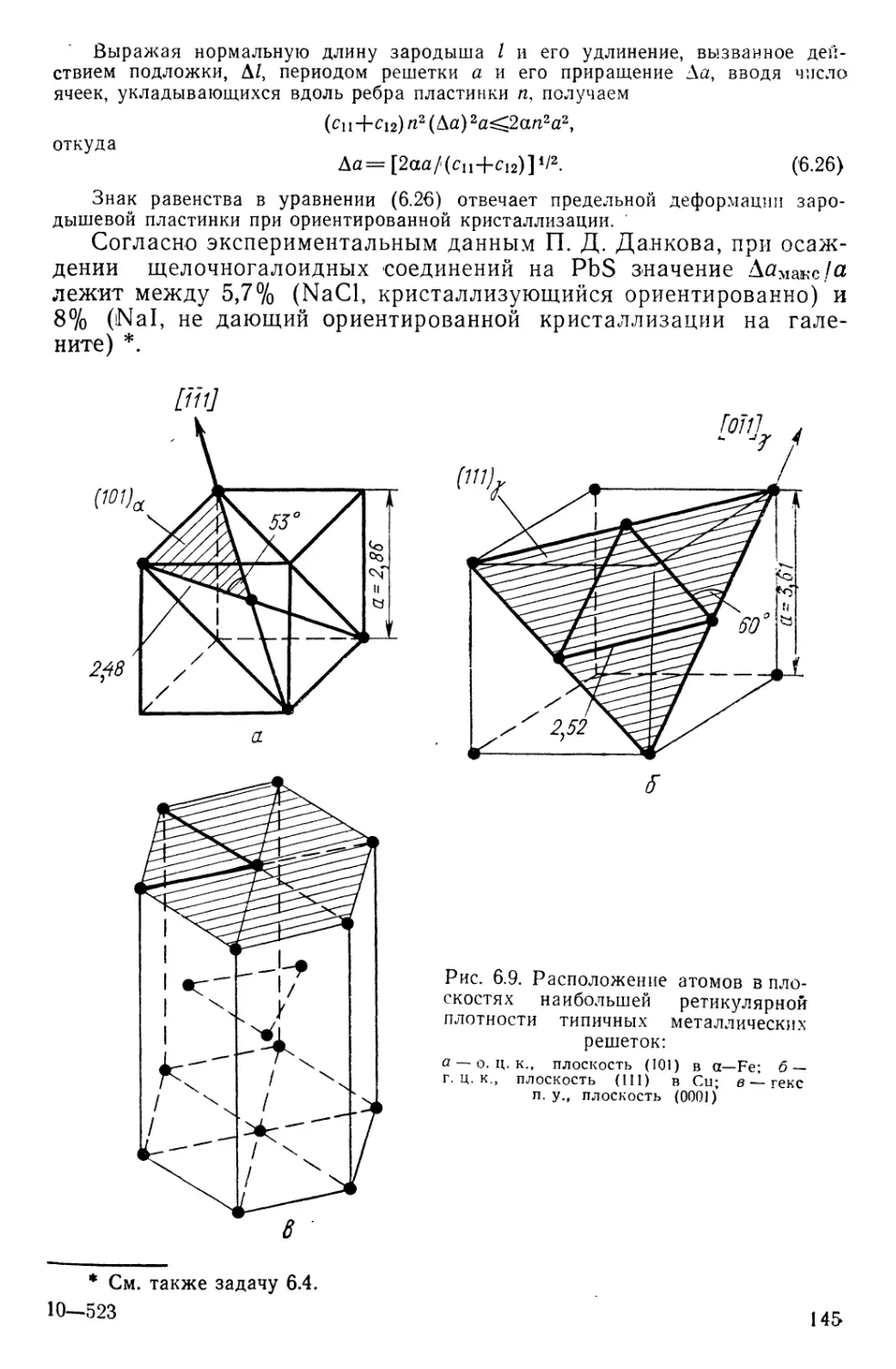


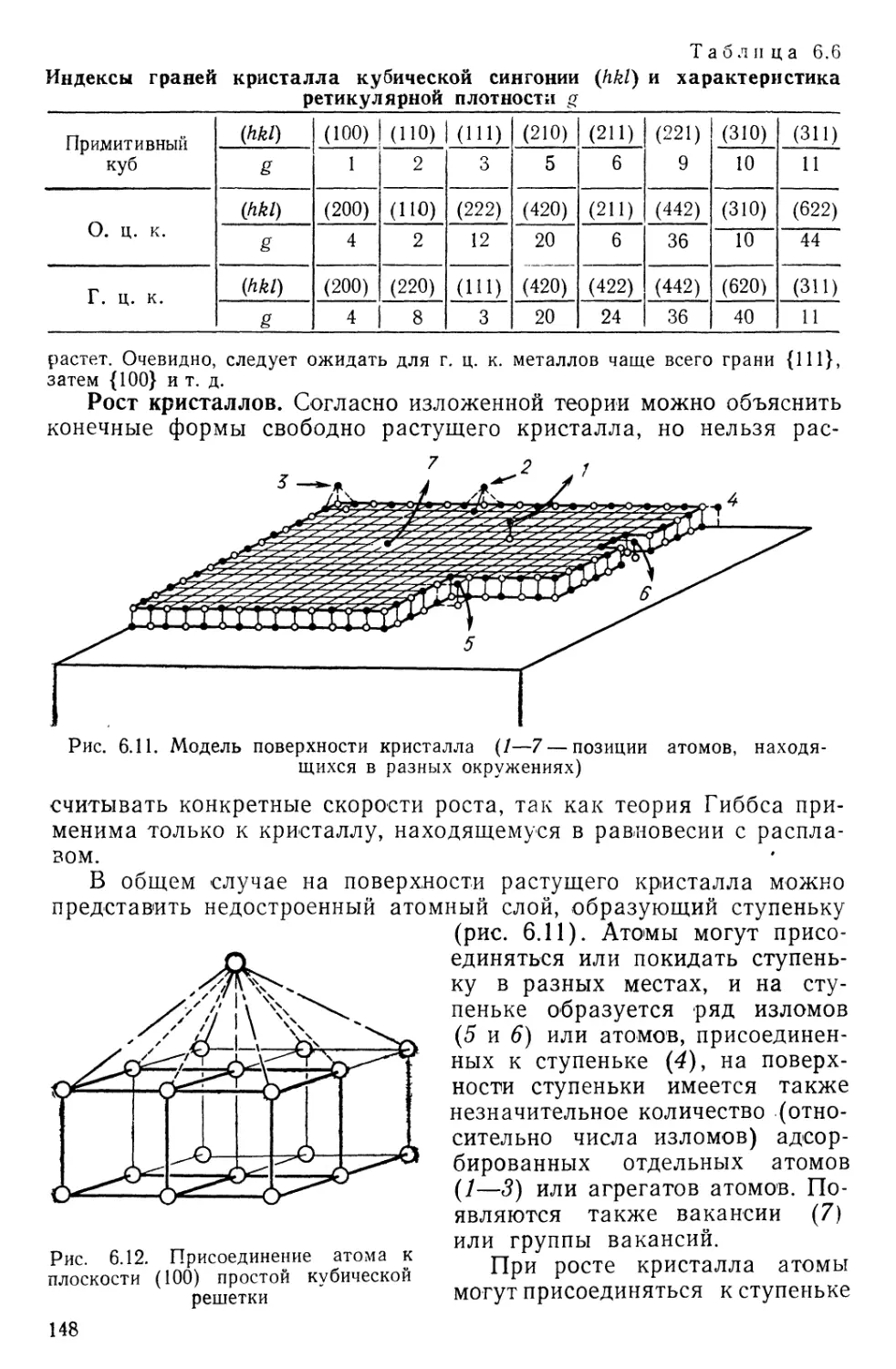

































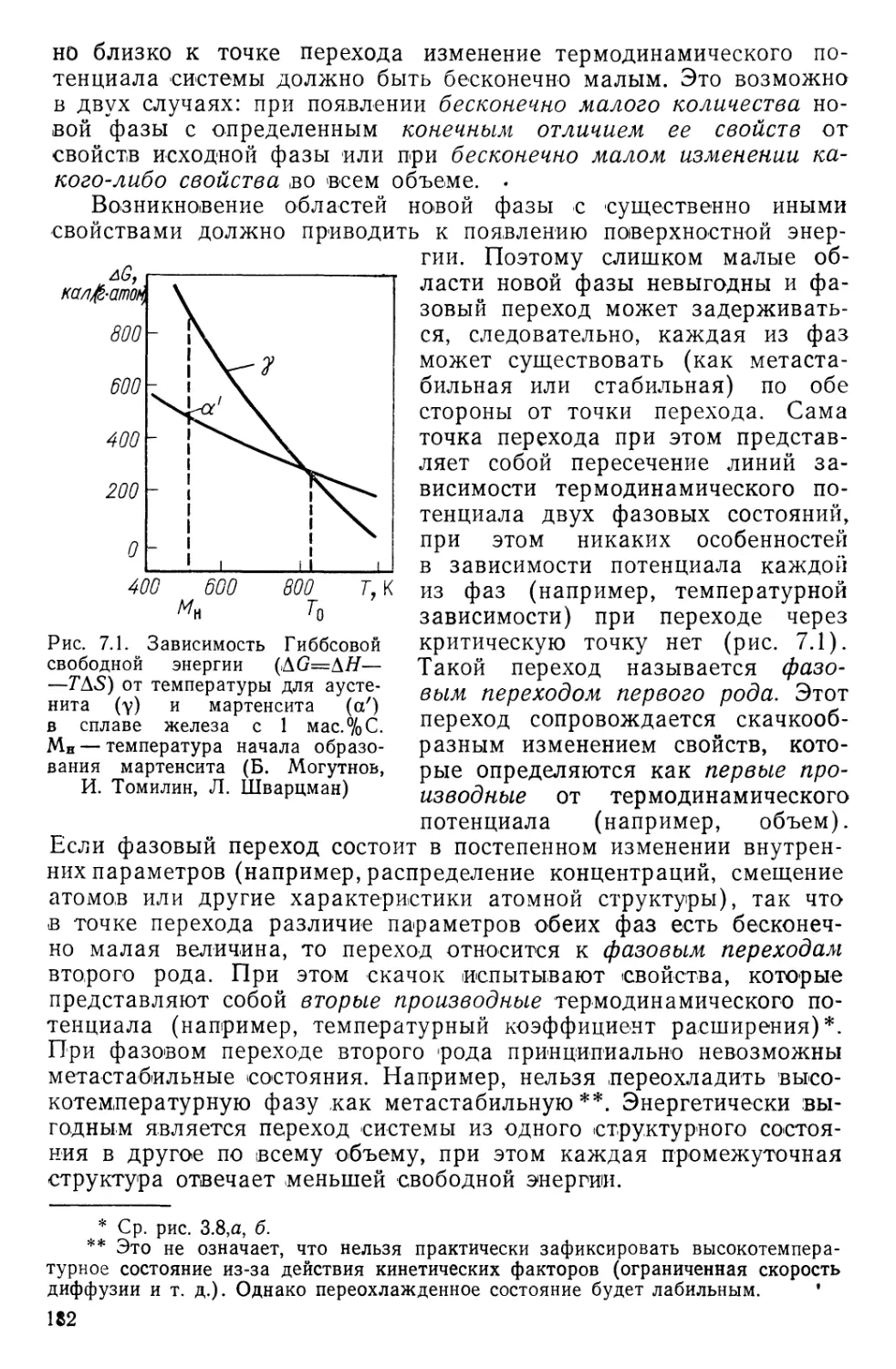













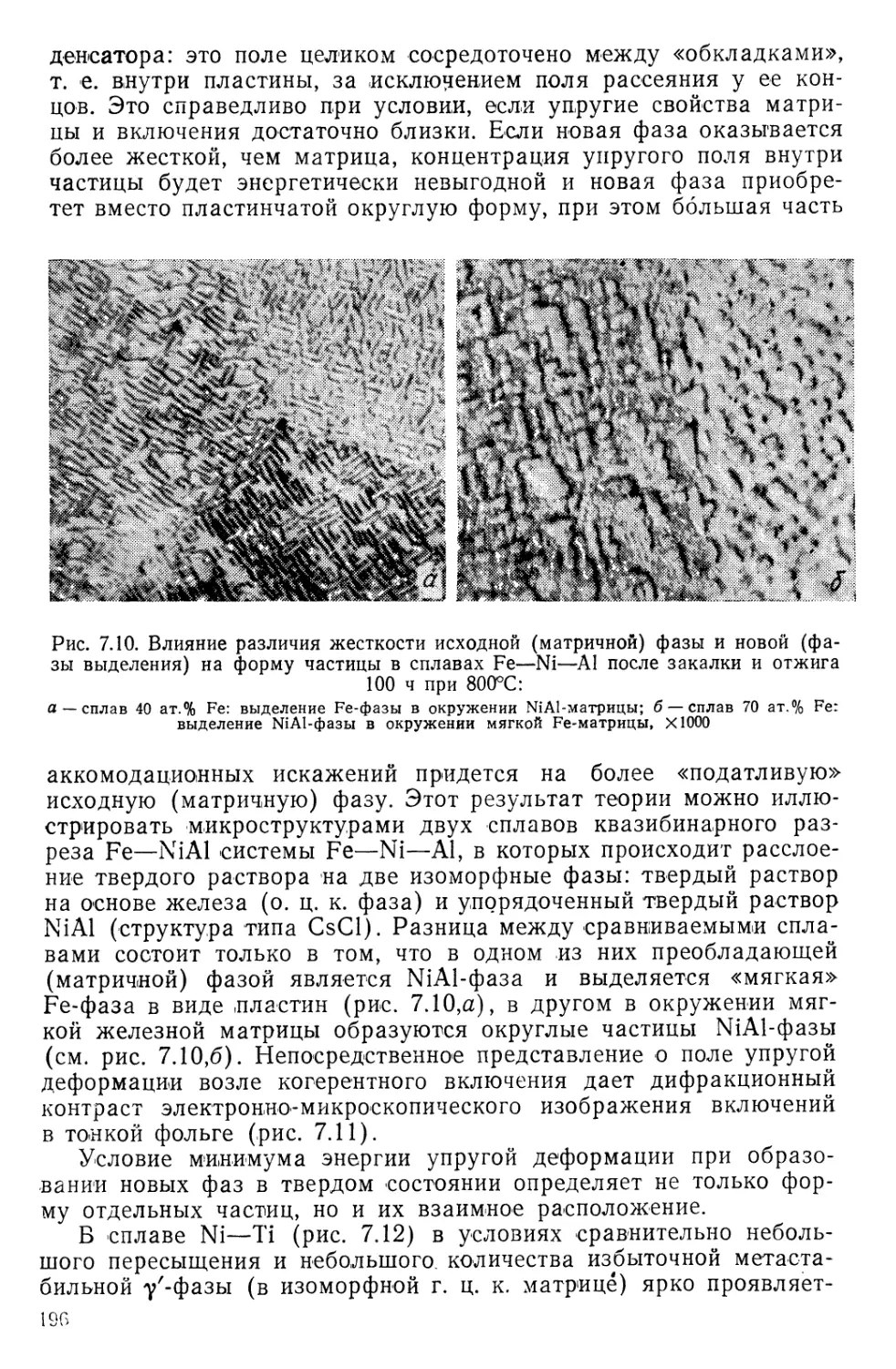
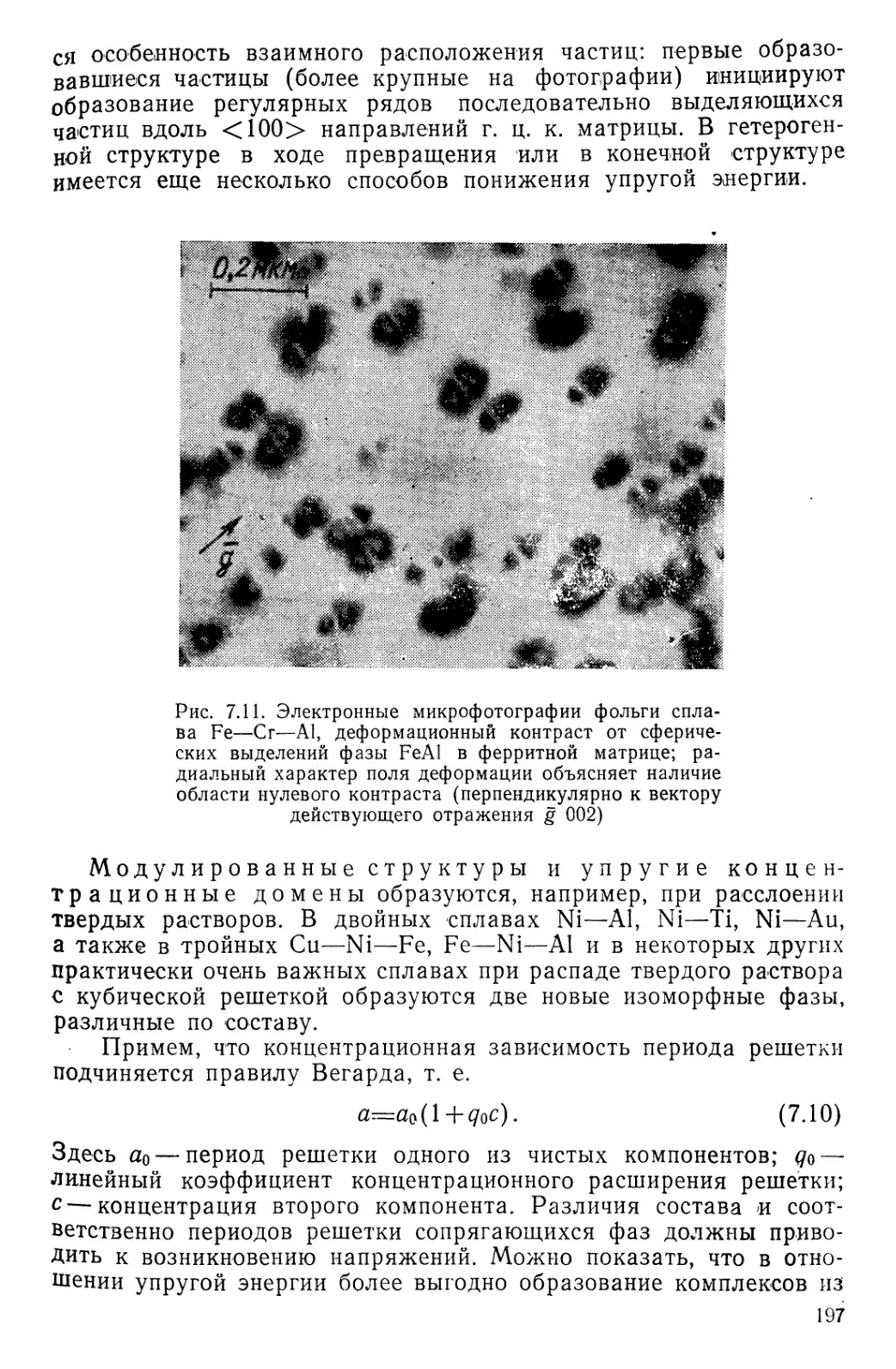












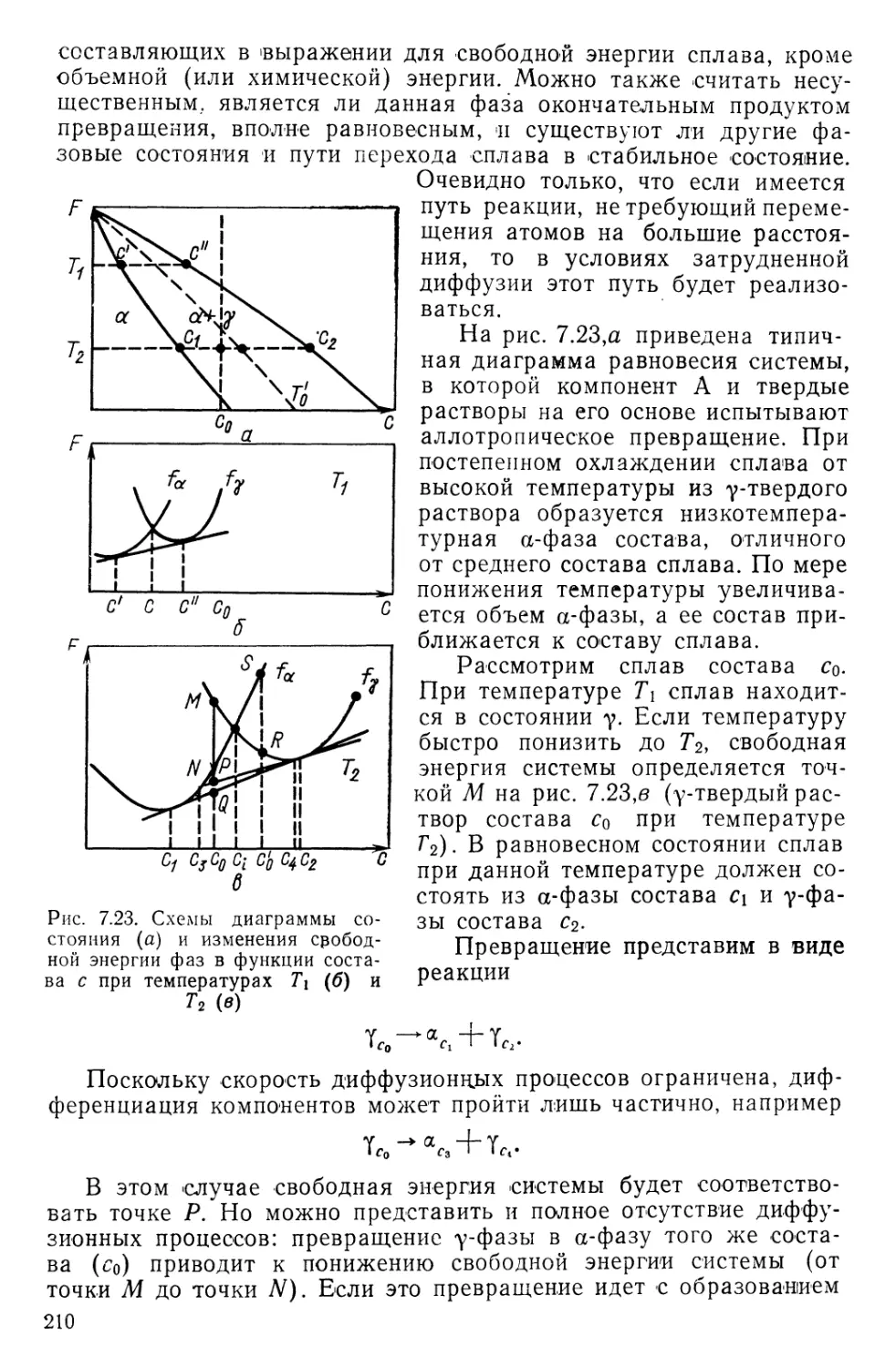


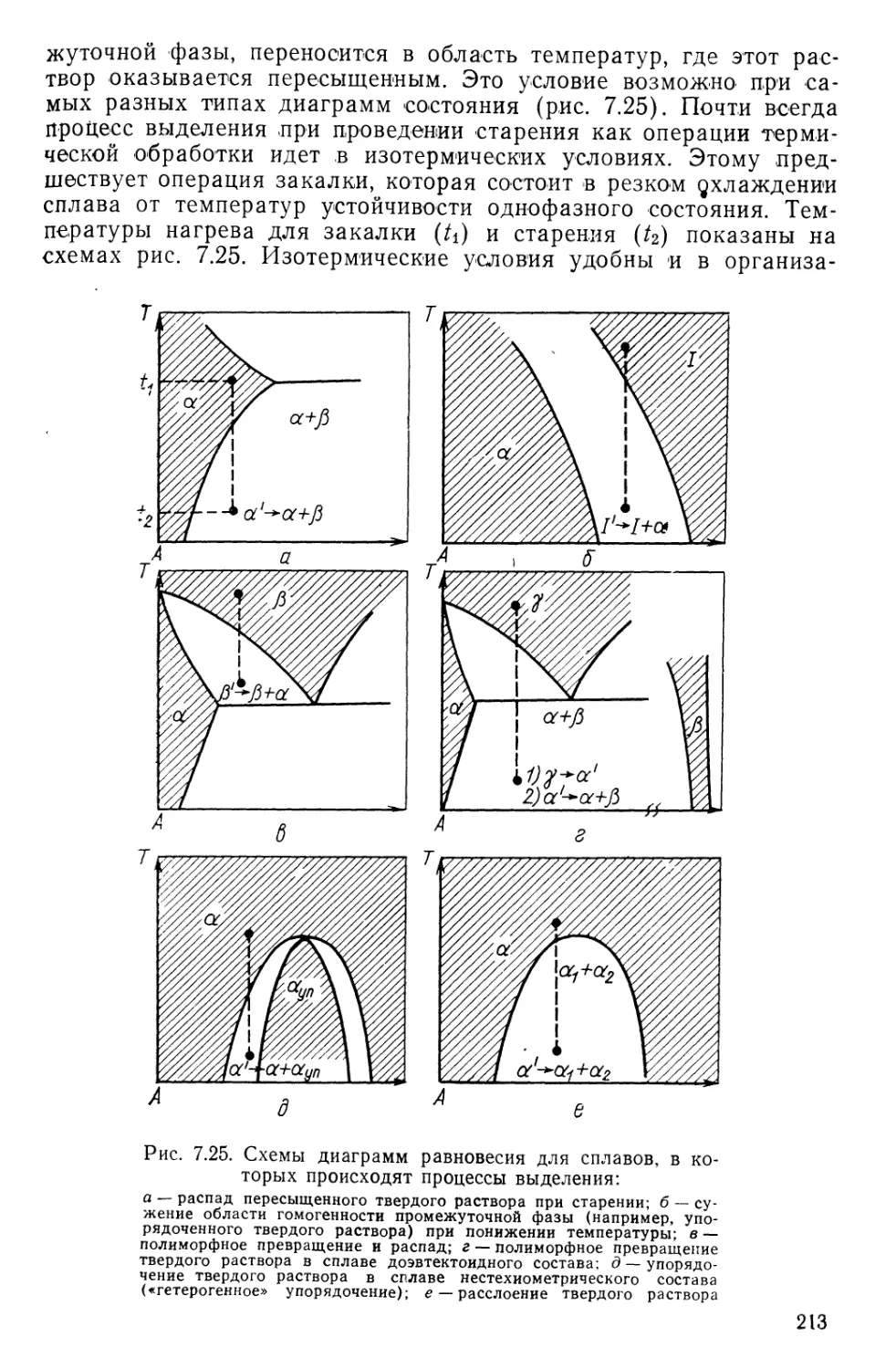









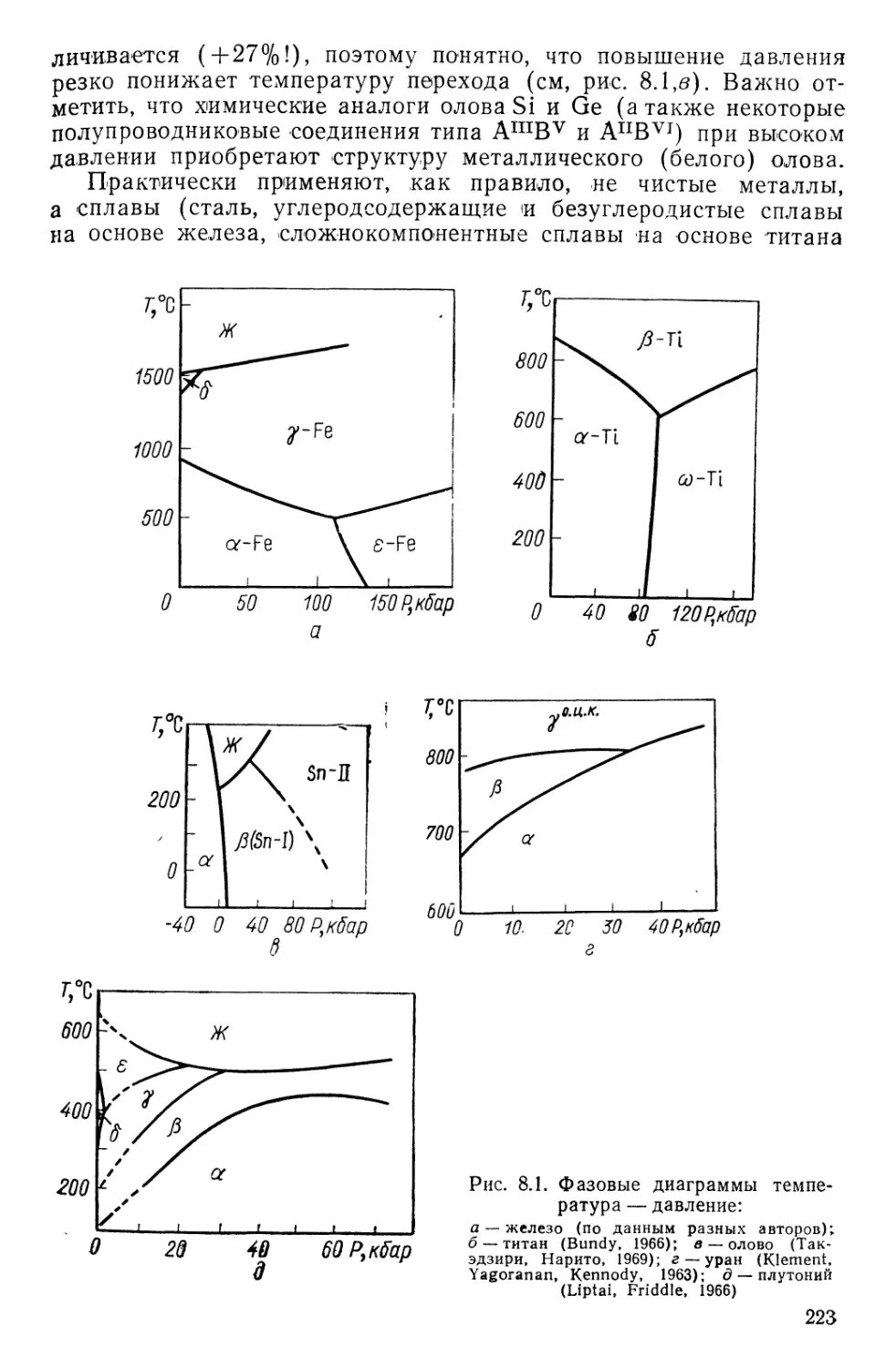
































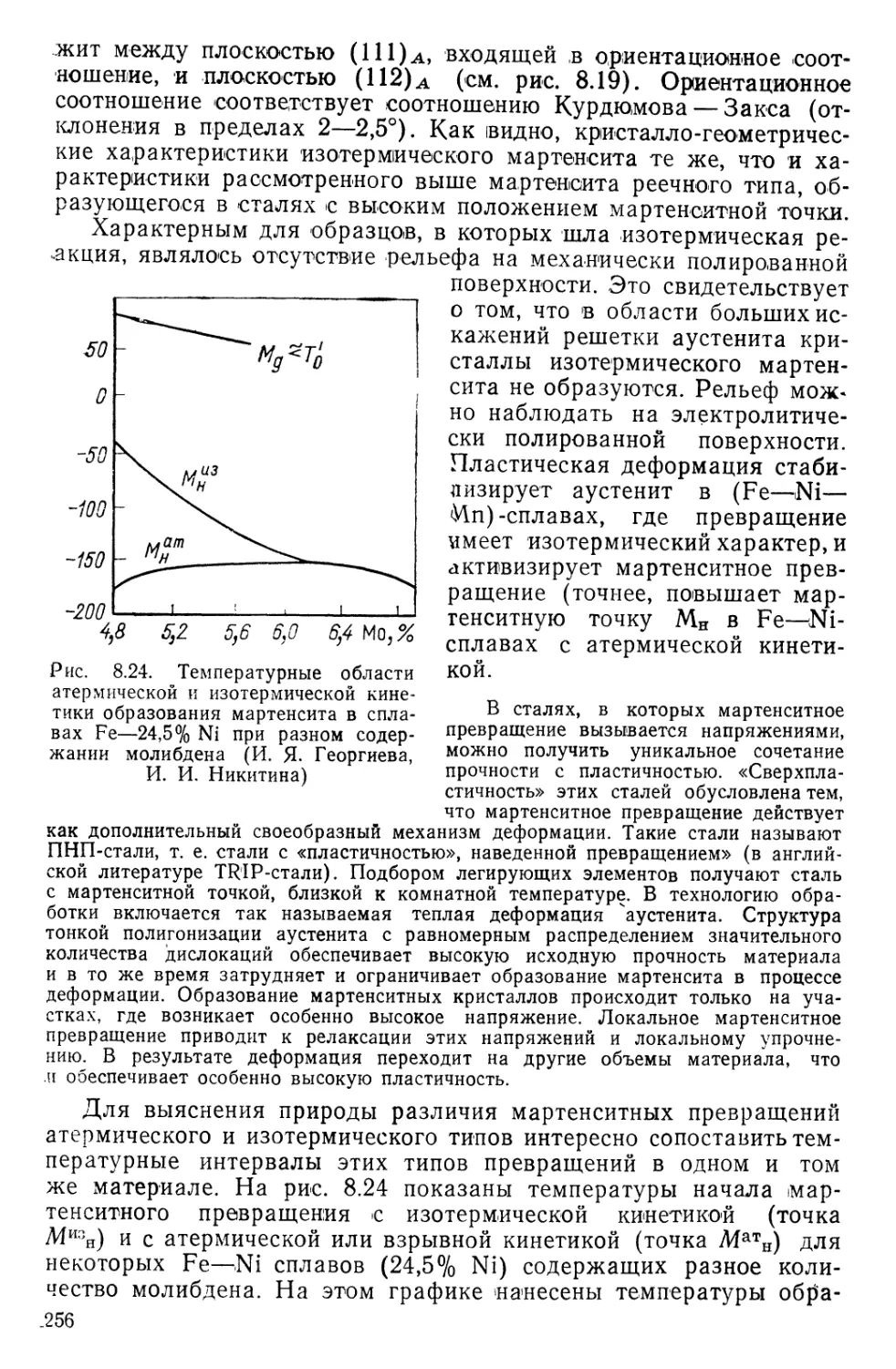



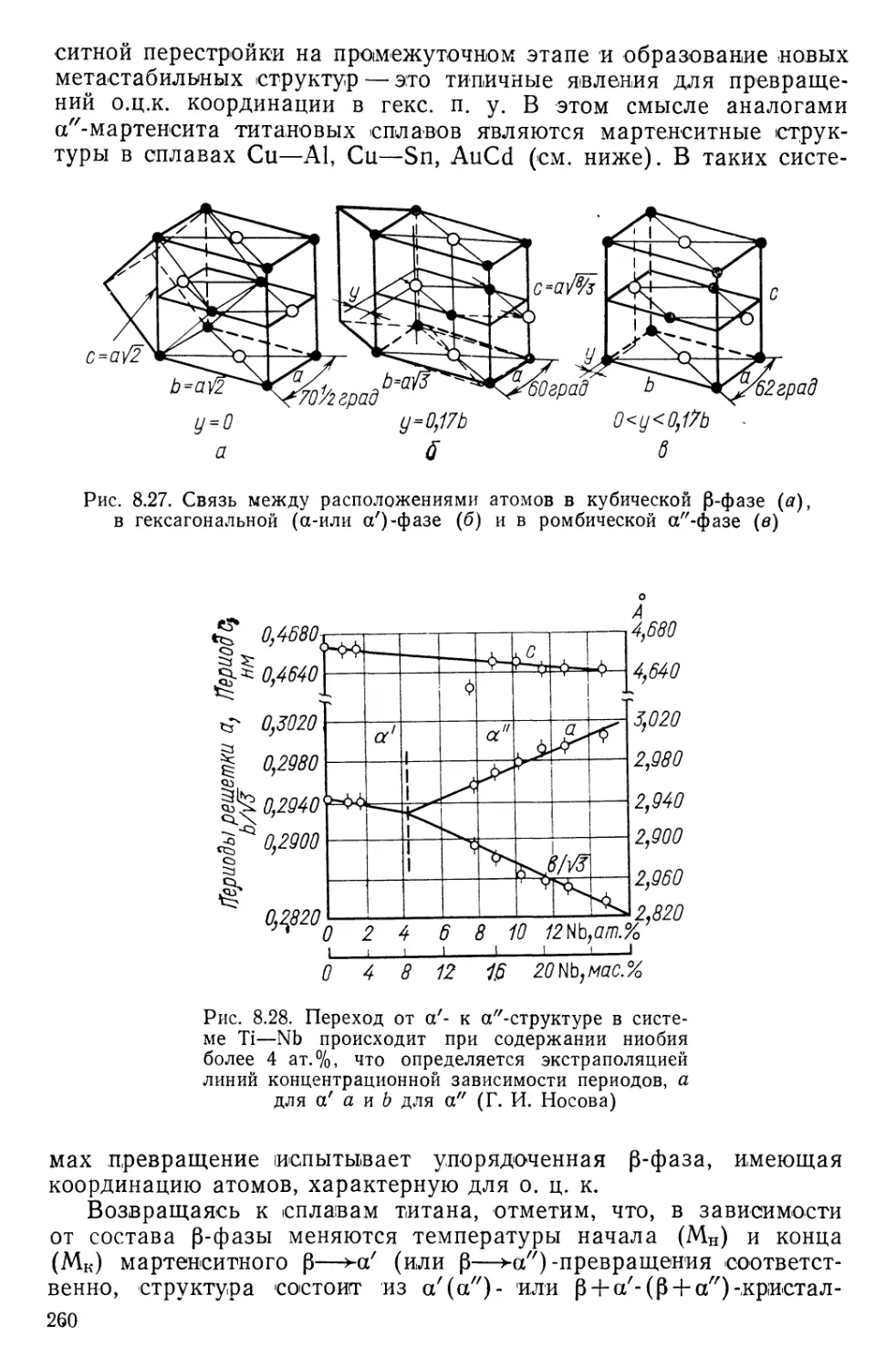




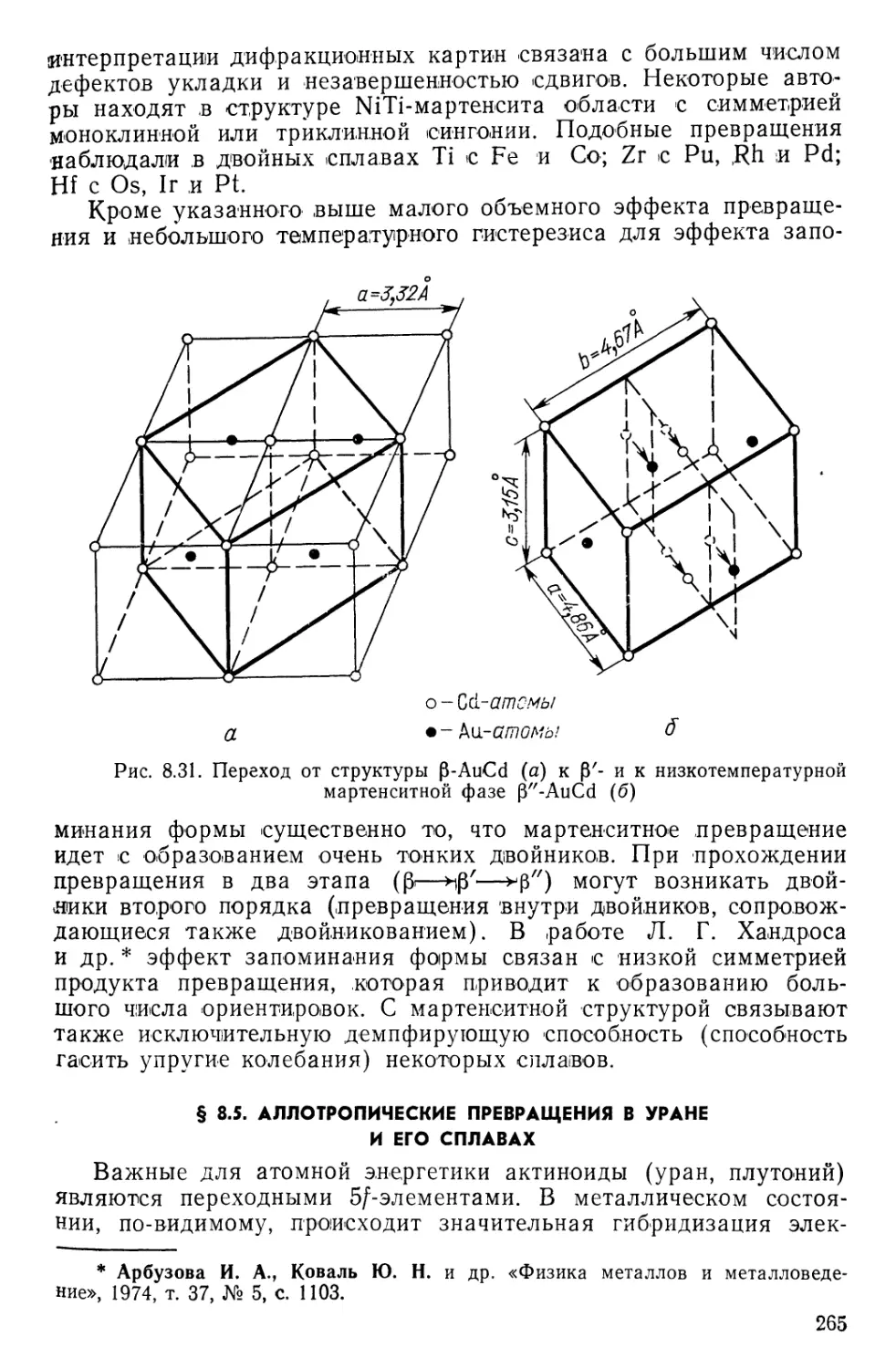
































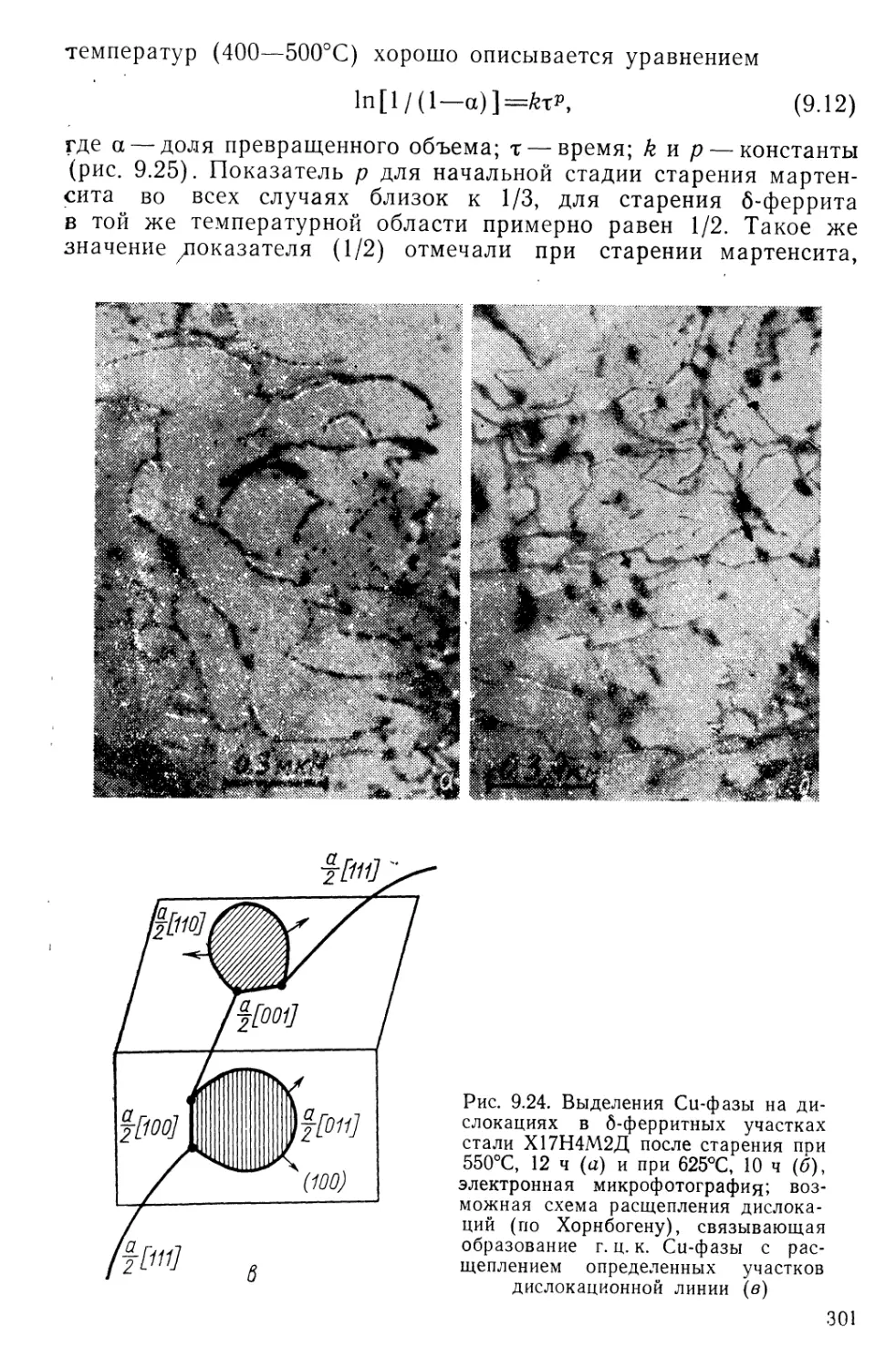
















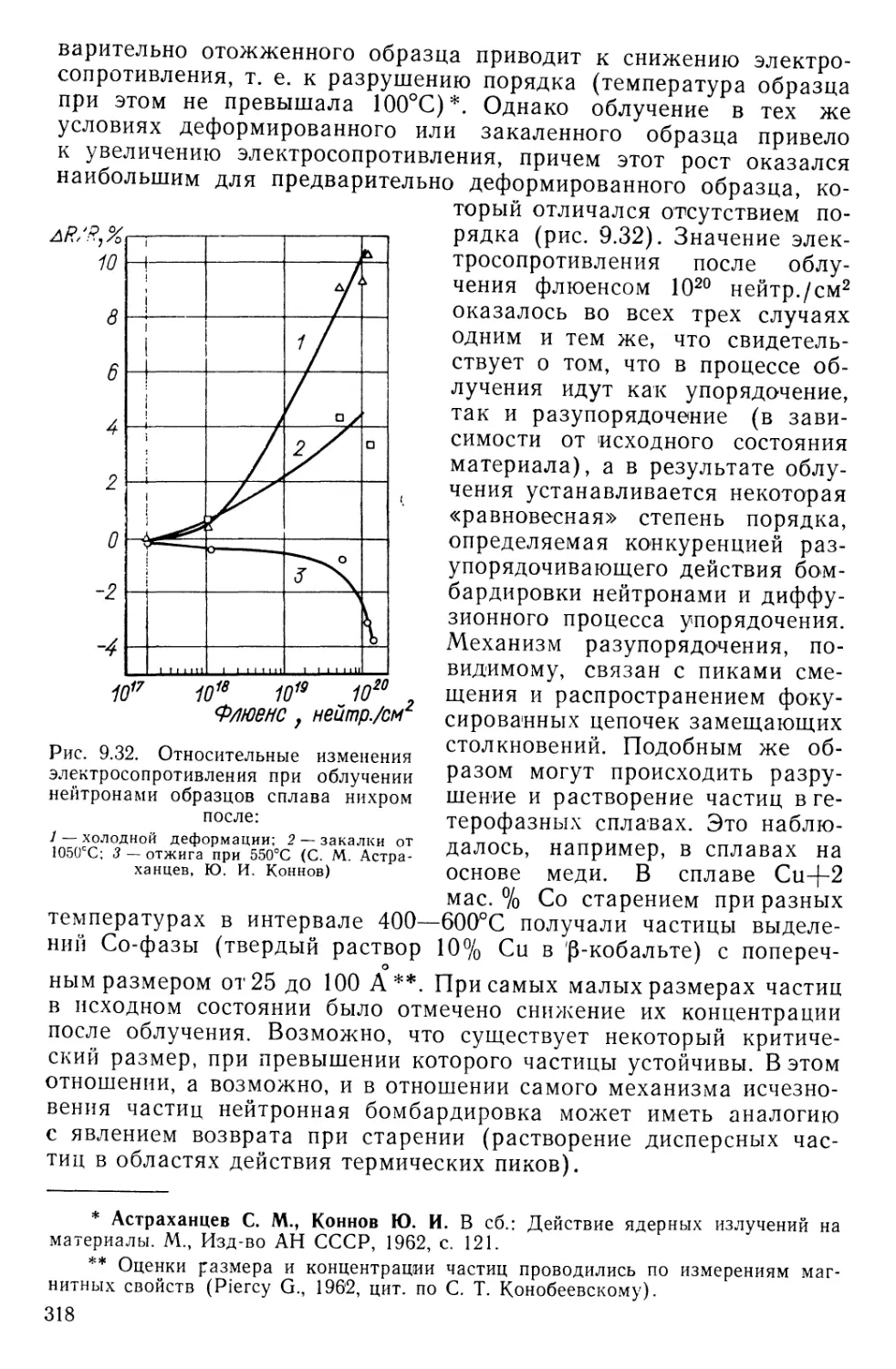

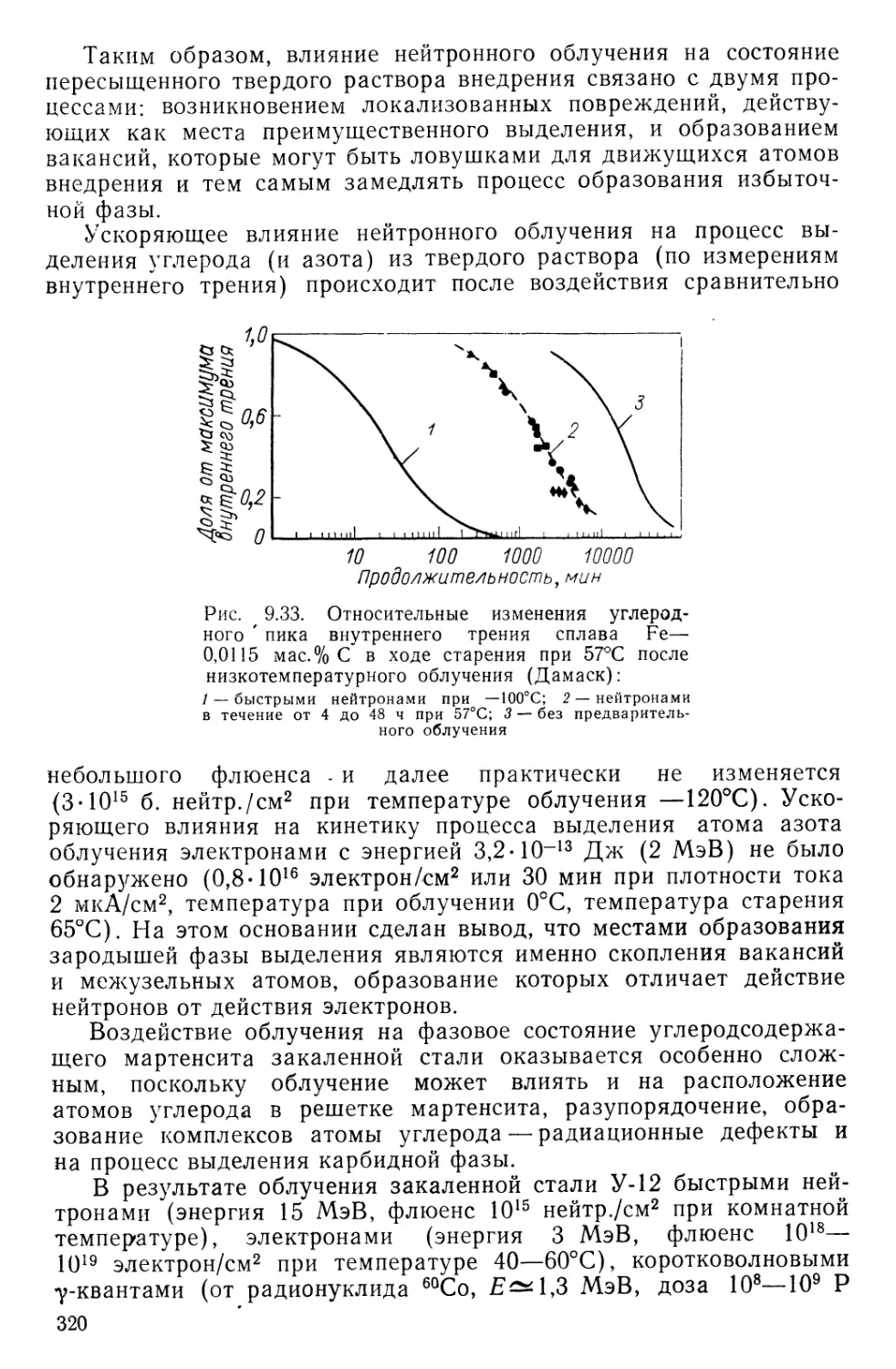




































Текст
Я. С. УМАНСКИЙ, Ю. А. СКАКОВ
ФИЗИКА
МЕТАЛЛОВ
АТОМНОЕ СТРОЕНИЕ МЕТАЛЛОВ И СПЛАВОВ
Допущено Министерством высшего и среднего
специального образования СССР
в качестве учебника для студентов
высших учебных заведений,
обучающихся по специальности «Физика металлов»
МОСКВА АТОМИЗДАТ 1978
УДК 669.539.2
Уманский Я. С., С к а к о в Ю. А. Физика металлов.
Атомное строение металлов и сплавов: Учебник для вузов. —
М.: Атомиздат, 1978. — с. 352.
Рассмотрены атомная и электронная структура металлов
и фаз металлических сплавов, феноменология и атомные меха-
низмы фазовых превращений (затвердевание расплавов, алло-
тропические превращения металлов и сплавов, процессы упоря-
дочения и распада твердых растворов), а также теории диф-
фузии и роль атомной структуры, дефектов решетки и микро-
структуры в явлениях диффузии. Особое внимание уделено
закономерностям возникновения кристаллических структур и
микроструктур. В числе рассматриваемых фаз находятся ме-
таллические соединения с замечательными магнитными свой-
ствами, фазы, перспективные в отношении сверхпроводимости
и т. д. Среди фазовых превращений особенно подробно рас-
смотрены мартенситные превращения в металлах и сплавах
на основе железа, кобальта, титана, урана и др. и процессы
распада пересыщенных твердых растворов.
Книга является учебником для студентов металлургиче-
ских, политехнических и инженерно-физических вузов по спе-
циальности «Физика металлов», а также может служить учеб-
ным пособием по специальности «Металловедение и термиче-
ская обработка».
Рис. 194. Табл. 27. Список литературы 117 наименований.
Рецензенты:
Кафедра физики твердого тела физического фа-
культета МГУ, зав. кафедрой доктор физ.-мат. наук
Г. С. ЖДАНОВ
Доктор химических наук О. С. ИВАНОВ
„ 20403, 31102-020 „„
У 034(01)—78 20 78
© Атомиздат, 1978
ПРЕДИСЛОВИЕ
Физика металлов весьма обширная область в физике твердого
тела и в учебных планах высших учебных заведений для материа-
ловедческих специальностей имеется несколько дисциплин, которые
можно было бы объединить общим названием физика металлов
или физика твердого тела. К ним относятся кристаллография и
теория дефектов, электронная теория металлов, теория фаз и фазо-
вых превращений, физические и механические свойства. Известны
книги, в которых охвачены все или почти все эти вопросы, напри-
мер, книги Г. Шульца «Металлофизика» (Пер. с нем. М., «Мир»,
1971) и Л. Ван Флека «Теоретическое и прикладное материалове-
дение» (Пер. с англ. М., Атомиздат, 1975). Первая книга отлича-
ется чрезвычайной конспективностью большинства разделов, вто-
рая— упрощенностью изложения, поскольку в добавлении к пере-
численным выше вопросам она содержит вопросы технического
материаловедения.
Цикл металлофизических курсов начинается с кристаллографии
и теории дефектов кристаллического строения. Эти курсы иногда
составляют часть более широкого курса структурного анализа и
освещаются в соответствующих главах учебников по структурному
анализу и рентгенографии, однако имеются вполне современные
специальные учебники: М. П. Шаскольской «Кристаллография»,
(М., «Высшая школа», 1972), Д. М. Васильева «Физическаякри-
сталлография» (М., «Металлургия», 1972) и И. И. Новикова
«Дефекты кристаллического строения» (М., «Металлургия», 1975).
Теория фаз и фазовых превращений вместе с теорией металли-
ческого состояния посвящены изучению структуры — электронного
строения и атомнокристаллического строения металлов и сплавов.
Эти разделы физики металлов являются теоретической основой
других специальных курсов.
В начале учебника изложены основы электронной теории метал-
лов. Это позволяет использовать учебник в тех случаях, когда от-
дельный курс по теории металлического состояния отсутствует. Со-
держание остальных глав полностью соответствует программе кур-
са «Физика металлов, атомное строение металлов и сплавов» для
металлофизиков.
Программы металлофизических курсов других специальностей
ограничены меньшим объемом и меньшим числом разделов. Для
3
металловедов-технологов это ограничение прежде всего связано
с более узкой специализацией в отношении круга материалов (ме-
талловедение стали, металловедение цветных металлов, реакторное
металловедение и т. д.). Дальнейшее ограничение объема курса
для металловедов связано с исключением некоторых теоретических
вопросов (частично теория упорядочения, специальные вопросы
геометрической теории структур, диффузия, математический анализ
кинетики процессов роста). При написании учебника предусмотре-
на возможность выделения таких вопросов для курсов разного
объема.
В конце каждой главы учебника приведены задачи, а в конце
учебника — ответы или решения наиболее сложных задач.
Главы 1—4 написаны Я. С. Уманским, гл. 5 — Я. С. Уманским
и А. А. Бабад-Захряпиным, гл. 6 — Я. С. Уманским, гл. 7—10,
а также § 3.4, 4.3—4.5 — Ю. А. Скаковым, § 5 гл. 7 написан
Л. И. Гомозовым.
В написании § 3.3 принимал участие А. А. Бабад-Захряпин,
гл. 6 — Н. В. Чириков, § 9.2 — Я. С. Уманский, гл. 3 и 6 —
Ю. А. Скаков, § 9.4 — Н. В. Еднерал.
ГЛАВА 1
ХИМИЧЕСКАЯ СВЯЗЬ И МЕТАЛЛИЧЕСКОЕ СОСТОЯНИЕ
ВЕЩЕСТВА
При определенных температуре и давлении атомы металл — не-
металл, неметалл — неметалл и металл — металл соединяются друг
с другом в молекулы и кристаллы. Свойства этих сложных образо-
ваний при данных внешних условиях и их строение определяются
природой составляющих их атомов, которая и обусловливает ха-
рактер связи между ними.
Основные два типа химической связи: ионная или гетерополяр-
ная и гомеополярная или ковалентная (атомная).
Химическая связь в металлах не представляет принципиально
нового вида связи, она приближается по типу к ковалентной свя-
зи, но в некоторых отношениях сходна с ионной.
В настоящее время эта классификация типов химической связи
представляется несколько условной. Она соответствует предельным
случаям, в реальных веществах связь может иметь смешанный ха-
рактер. К ионным или гетерополярным относятся галоидно-водо-
родные соединения и галоидные соединения щелочных элементов:
НС1, HBr, NaCl, NaBr и др. В этих, как и во всех других случаях
ионной связи, каждому из атомов (или группе атомов) можно при-
писать определенное электронное строение, практически не зави-
сящее от присутствия других атомов или групп атомов. Молекула
NaCl, как и кристалл, состоит из ионов Na+ и С1~, обладающих
электронным строением соответственно атомов Ne и Аг. Взаимо-
действие этих двух противоположно заряженных составных частей
молекулы или кристалла сводится в основном к электростатиче-
скому их притяжению. К ионным кристаллам относятся окислы,
например рутил (ТЮ2), корунд (А12О3) и многие другие соеди-
нения.
Рассмотрение гомеополярной химической связи начнем с про-
стейшей молекулярной системы — положительного иона водород-
ной молекулы Н*. Эта система состоит из двух одинаковых
ядер — протонов, находящихся на расстоянии R друг от друга, и
одного электрона, движущегося в электрическом поле обоих про-
тонов. Можно показать, что такая система устойчива: состоянию
равновесия отвечает значение расстояния между ядрами 7?=1,0 6 А.
Энергия диссоциации молекулярного иона Н* на атом Н и ион
Н+ составляет 61 ккал/моль.
5
В водородной молекуле в электрическом поле двух протонов на-
ходятся два электрона. Это типичный пример ковалентной связи,
осуществляемой двумя электронами с антипараллельными спи-
нами.
Первая из рассмотренных систем — положительный ион водо-
родной молекулы Н* — показывает, что и одного электрона доста-
точно для связи между двумя протонами. Однако парная связь,
осуществляемая двумя электронами с антипараллельными спина-
ми, оказывается более прочной; энергия диссоциации водородной
молекулы достигает 102 ккал/моль.
Не любые два атома образуют при своем сближении молекулу.
Для образования последней необходимо, чтобы при сближении со-
ответствующих атомов происходило занятие электронами наиболее
низких из тех энергетических уровней, которые возникают вместо
уровней изолированных атомов. Только при этом условии уменьше-
ние энергии электронов компенсирует происходящее при таком
сближении возрастание энергии кулоновского отталкивания поло-
жительно заряженных ядер.
Ковалентная связь наблюдается в кристаллах неметаллических
элементов.
Из всех известных в настоящее время химических элементов
более половины — металлы. Сопоставление опытных данных, отно-
сящихся к металлам, показывает, что все особые физические свой-
ства последних в большей или меньшей степени связаны с присут-
ствием электронов проводимости.
Металл в твердом состоянии представляет собой совокупность
ионов, образующих кристаллическую решетку, и электронов прово-
димости, движущихся внутри решетки и взаимодействующих как
с ионами, расположенными в узлах решетки, так и друг с другом.
В отсутствие внешнего электрического поля это движение электро-
нов проводимости носит неупорядоченный характер и не дает ре-
зультирующего электрического тока. При наложении внешнего
электрического поля в металле возникает макроскопический элек-
трический ток, который остается неизменным при данном значении
напряженности внешнего поля.
Другим важным признаком металлов, отличающим их от неме-
таллических проводников, является особый характер температур-
ной зависимости электропроводности при низких температурах, до-
статочно близких к нулю градусов Кельвина. В то время как элек-
тропроводность неметаллов с приближением к 0 К уменьшается,
у металлов она возрастает.
Следует отметить, что правильнее говорить о металлическом
состоянии вещества, так как некоторые вещества обладают метал-
лическими свойствами только в определенной аллотропической
модификации или при некоторых определенных температурах и
давлениях.
В поведении электронов проводимости обнаружены закономер-
ности, до некоторой степени аналогичные тем, которые характер-
ны для сочленения двух электронов в молекуле водорода.
6
Молекула водорода представляет собой систему, качественно от-
личную от атомов, соединением которых она образовалась. В моле-
куле водорода каждый из электронов нельзя считать принадлежа-
щим одному из ядер: при образовании молекулы водорода проис-
ходит коллективизация электронов. В изолированном атоме водо-
рода электрон находится в силовом поле одного ядра. В молекуле
водорода каждый электрон движется в поле двух ядер. Металл
можно рассматривать как своего рода гигантскую молекулу, со-
стоящую из ионов и электронов проводимости; образование таких
электронов при сближении атомов приводит к уменьшению энер-
гии.
Теория свободных электронов в металлах. Со-
гласно этой теории электроны проводимости образуют идеальный
электронный газ, подчиняющийся обычным газовым законам.
В 1900—1905 гг. эта теория была успешно применена к вычисле-
нию отношения электро- и теплопроводности металлов. Однако
возникли два затруднения: предсказываемая этой теорией темпе-
ратурная зависимость электропроводности противоречила наблю-
даемой на опыте и теория приводила к слишком большому вкладу
электронов проводимости в теплоемкость металла, что такж^7 ока-
зывалось в противоречии с опытными данными.
Лишь с открытием законов квантовой статистики (1927 г.) элек-
тронная теория металлов получила дальнейшее развитие.
Рассмотрим свойства электронного газа при абсолютном нуле
температуры. Предположим, что N свободных электронов, взаимо-
действием которых друг с другом мы пренебрегаем, находятся
в объеме у, и, следовательно, объемная плотность электронов, т. е.
число их в единице объема, равно
n^=N/v. (1.1)
Частицы, подчиняющиеся законам обычной механики, при абсо-
лютном нуле температуры должны находиться в состоянии с наи-
меньшей энергией 8=0. Однако такой результат противоречит
принципу Паули, не допускающему одновременного пребывания
в одном состоянии более чем двух электронов с противоположны-
ми спинами. Состояние одной частицы с энергией 8 можно харак-
теризовать значением импульса или количества движения р, свя-
занного с энергией 8 и массой частицы иц соотношением
е = р2/2т,, (1.2)
ИЛИ
р = У2тое.
Введем понятие о пространстве импульсов, каждую точку ко-
торого можно характеризовать радиусом-вектором р, проведенным
из начала координат. Таким образом, геометрическим местом то-
чек, которым соответствует одно и то же значение импульса р,
является сфера радиуса р. Но согласно соотношению (1.2) всем
точкам этой поверхности можно приписать одно и то же значение
энергии.
7
Следовательно, сферическая поверхность радиуса р — это вме-
сте с тем поверхность равных энергий. Пространство импульсов
в отсутствие внешнего силового поля обладает сферической сим-
метрией, являющейся выражением изотропности пространства им-
пульсов для свободно движущихся частиц. Если обозначить ро зна-
чение импульса, соответствующее максимальному значению энер-
гии электрона при абсолютном нуле температуры, то объем
в пространстве импульсов, внутри которого расположатся все N
электронов, равен
Объемом V фазового пространства назовем произведение объе-
ма и обыкновенного пространства и объема 4/злр3о в пространстве
импульсов. Этот объем имеет следующую размерность:
(г<омЖ=[-Ч^]'=^ с’ = (эрг-с)’.
Согласно квантовой теории наименьший объем фазового про-
странства, в котором могут находиться не более двух электронов
с противоположными спинами, равен Л3 (Л— постоянная Планка).
Для размещения в фазовом пространстве при абсолютном нуле N
электронов необходимо tV/2 таких элементарных объемов. Отсюда
получаем соотношение
-~^h3 = v^r.p\, (1.3)
или
A- h3= v A it (2m0s0)3/2,
откуда
г0 =/г2/2m0 (3/V/8tot)2/3 , (1.4)
где Ео — граничная энергия электрона при абсолютном нуле, или
энергия Ферми.
Формулу (1.4) после перестановки входящих в нее констант
можно представить в виде
80=36,1 (Сэл/^ат) 2/3 эВ,
(1-5)
где ^ат — атомный объем, А3*; сэл— число свободных электронов
на атом.
В табл. 1.1 приведены вычисленные по формуле (1.5) значения
энергии Ферми для некоторых одновалентных элементов (сэл=1).
Как видно, энергия Ферми составляет несколько электронвольт и
значительно превосходит среднюю энергию теплового движения,
которая (на атом) при обычной температуре (Г^ЗОО К) равна
приблизительно 0,03 эВ.
При абсолютном нуле температуры полностью заняты все со-
стояния электронами с энергией, не превышающей ео. Функция
распределения частиц электронного газа по энергиям при Т=0 К
* 1 Аа= 10-24 см3.
8
Таблица 1.1
Энергия Ферми при О К и температура вырождения
для некоторых элементов
Элемент Li Na к Rb Cs Си Au
е0, эВ 4,74 3,16 2,06 1,79 1,53 7,10 5,56
9фЮ-‘, К 5,40 3,60 2,35 2,04 1,74 8,09 6,34
представляет собой ступенчатую линию на рис. 1.1 (kT=Q). Для
других значений температуры вероятность для частицы, подчиняю-
щейся принципу Паули, обладать энергией е определяется функ-
цией Ферми:
/(8)=1/[еХр(е-еф)/^4-1], (1.6)
где еф —уровень Ферми, который для металлов мало зависит от
температуры и остается близким к ео-
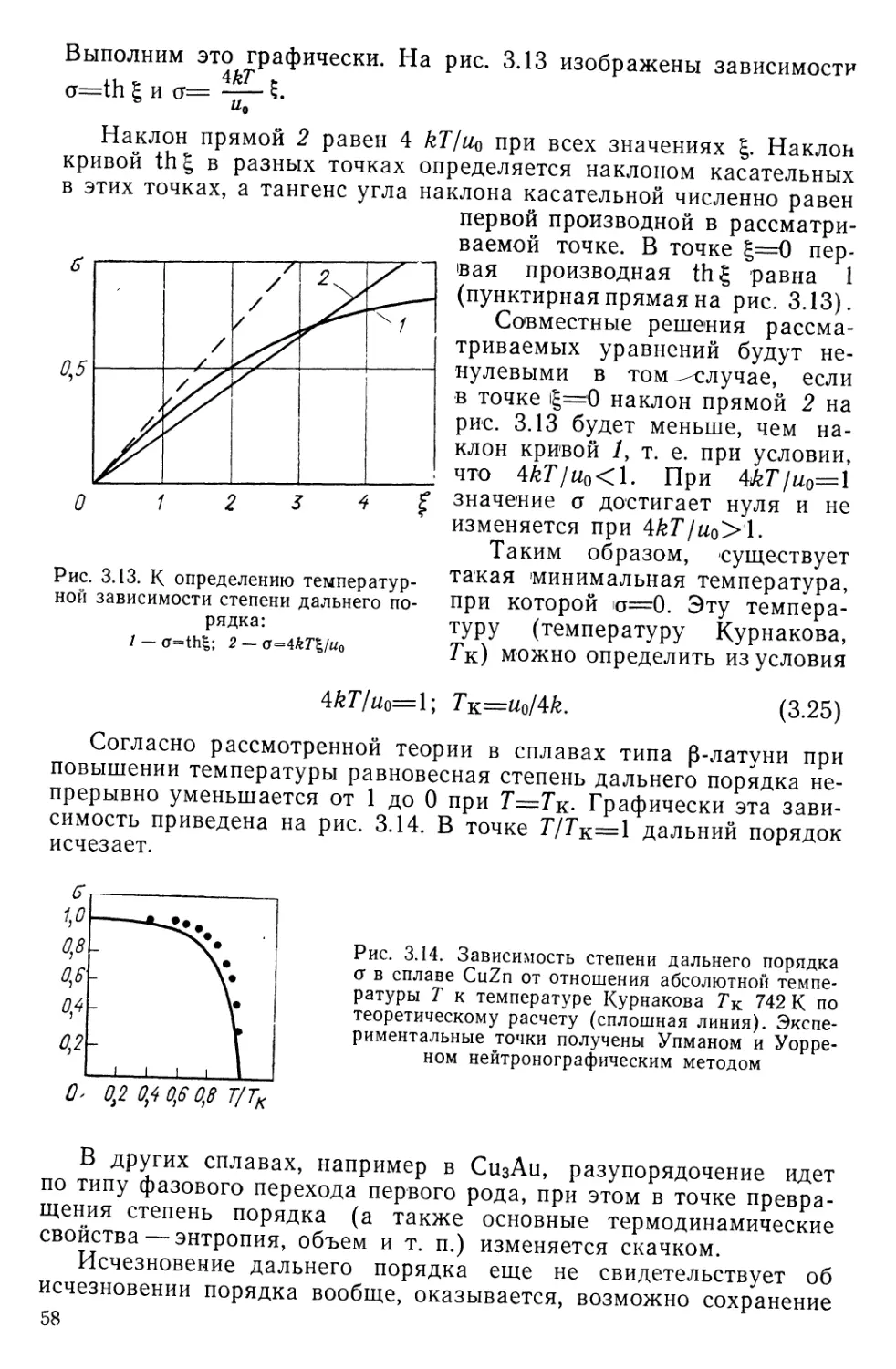
Рис. 1.1. Функция распределения Ферми (для Си):
/ — kT=--0 эВ; 2 — 0,1 эВ (Т«1200 К); 3—1 эВ
(Т«12 000 К); 4 — 2,5 эВ (Т«30 000 К)
Появление какого-либо значительного числа электронов на
энергетических уровнях, лежащих выше граничного значения е0,
может произойти только при такой температуре Г, для которой
средняя энергия теплового движения kT соизмерима с энергией е&
(см. кривые для kT— 1 и 2,5 эВ на рис. 1.1). Температуру, при ко-
торой распределение электронов по состояниям существенно изме-
нится по сравнению с распределением при абсолютном нуле, мож-
но определить из выражения
£0ф = е<>, (1.7)
откуда
Температура вф, названная темпе partly рой вырождения элек-
тронного газа или температурой Ферми, значительно превышает
£
температуру плавления соответствующих металлов. Отсюда следу-
ет, что во всем температурном интервале существования твердого
металла свойства электронного газа практически не должны зави-
сеть от температуры и определяются значениями соответствующих
величин при абсолютном нуле.
Вычислим полную кинетическую энергию £0 всех N электронов
в объеме v при абсолютном нуле, равную
£0 = 2 J sn(s)ds. (1.8)
и
Здесь n(e)de— число электронных состояний в объеме vy приходящихся на
интервал энергий, лежащих между значениями е и е+^8. Эта величина имеет
важное значение для теоретического изучения многих свойств металлов (тепло-
вых, магнитных и пр.). Для ее вычисления возьмем объем шарового слоя в фа-
зовом пространстве, лежащий между значениями импульса р и p-\-dp, равный
v4np2dр = ц2л (2щ0)2 Ke de.
Разделив его на /z3, получим число состояний, приходящихся на интервал
энергии de:
п (е) de — 2nv (2m9/h2)3^ Иe de. (1.9)
Величину n(e) можно назвать плотностью числа состояний.
Если изобразить зависимость этой величины от энергии 8, откладывая по
оси абсцисс 8, а по оси ординат п(е), то получится парабола.
Максимальное число электронов, приходящихся на этот интервал энергий,
равно удвоенному числу состояний, т. е. 2n(e)t/e. Таким образом,
ЕЛ = 2 J sn (е) de = 4то (2т,/Л2)3/2 j e3/2de =
О о
о //2\3/2 5/2 8т: /2т0 \3/2 3/2
— 8тсу/5 (2mo//z) Sq 5 ( а2 ) so so*
В то же время из выражения (1.4) следует
е3/2 = (/г2/2и0)3/2Зпэл/8те.
Подставив эту величину в предыдущее равенство, получим
£» = Т-у^Ге« = 3^/5- 0-Ю)
Отсюда для средней энергии е, приходящейся при абсолютном нуле на один
электрон, имеем
r=:E0/^ = 3s0/5. (1.11)
Благодаря почти полной независимости внутренней энергии электронного га-
за от температуры теплоемкость, определяемая производной ст энергии по тем-
пературе, оказывается практически равной нулю, и, таким образом, отпадает
указанная выше главная трудность первоначальной электронной теории метал-
лов. Состояние системы, в котором ее свойства слабо зависят от температуры,
называется вырожденным. Электронный газ полностью вырожден не только
вблизи абсолютного нуля, но и при более высоких температурах, малых по
сравнению с температурой вырождения 9Ф . Теплоемкость электронного газа
можно представить формулой
1 R Т
С'„ = —(Ы2)
10
или
= 3,26* 10~5"Ипо у Т кал/(град-моль),
где А — атомная масса; р — плотность металла.
Для сравнения приведем формулу для теплоемкости, обусловленной тепло-
выми колебаниями решетки в области низких температур:
C"v = 468(Г/0d)3 кал/(град • моль),
где 0d — характеристическая (дебаевская) температура.
При очень низкой температуре вклад в теплоемкость от электронов прово-
димости сравним с теплоемкостью, обусловленной колебаниями решетки. Так,
например, для серебра при 2 К: С\=0,00031 кал/(град-моль); C"v=
=0,00037 кал/(град*моль).
Таким образом, влияние электронов проводимости на теплоемкость металла
может проявиться вблизи абсолютного нуля, где происходит резкое падение
(пропорциональное Г3) той части теплоемкости, которая связана с колебаниями
решетки.
Следующий шаг в развитии электронной теории металлов дол-
жен состоять в учете силового поля, в котором движутся электро-
ны проводимости, а также в учете их взаимодействия друг с дру-
гом. Этот шаг делается в так называемой одноэлектронной теории,
представляющей следующее приближение к действительности.
В этой теории рассматривается движение отдельных электронов
проводимости в периодическом поле кристаллической решетки.
Начнем рассмотрение с простейшего случая свободного движе-
ния электрона.
Уравнение Шредингера для движения электрона в отсутствие
внешнего силового поля имеет вид
= (1.13)
где Н — оператор, соответствующий функции Н — — ; Ч* —
волновая функция, характеризующая вероятность местоположения
электрона; Е — энергия электрона.
Следовательно, волновая функция Ч* (х, у, г) удовлетворяет
дифференциальному уравнению
— дх2 ‘ ду2 dz* h2
Решение этого уравнения можно представить в виде
Ч(х, у, z)=expi (kxx-\-kyy-\-kzz),
где kx, ky, kz — составляющие некоторого постоянного вектора к,
физический смысл которого выясним несколько позже. Дифферен-
цирование волновой функции дает
= i'kx exp i (kxx + kyy + kzz) = i ,
и, следовательно,
+ k\) ЧГ = - k2W,
где k2=k2x-\-k2y-\-k2z.
11
Подставляя этот результат в левую часть уравнения (1.14), по-
лучаем
>2__87T2/Z7o р
72 — Л2 с
или
E=h2k2/8n2mQ. (1.15)
Из сравнения соотношения (1.15) с выражением (1.2) для ки-
нетической энергии свободной частицы массы ш0, обладающей им-
пульсом р, видно, что введенный выше вектор к связан с импуль-
сом частицы р соотношением
Р = 2Гк- <Ы6)
Используя выражение для длины электронной волны, получаем
^=hlp\
£=2лД. (1.17)
Таким образом, величина k дает число длин волн X, укладыва-
ющихся на отрезке, длина которого равна 2л. В соответствии с этим
величину k назвали волновым числом, а вектор к — волновым век-
тором.
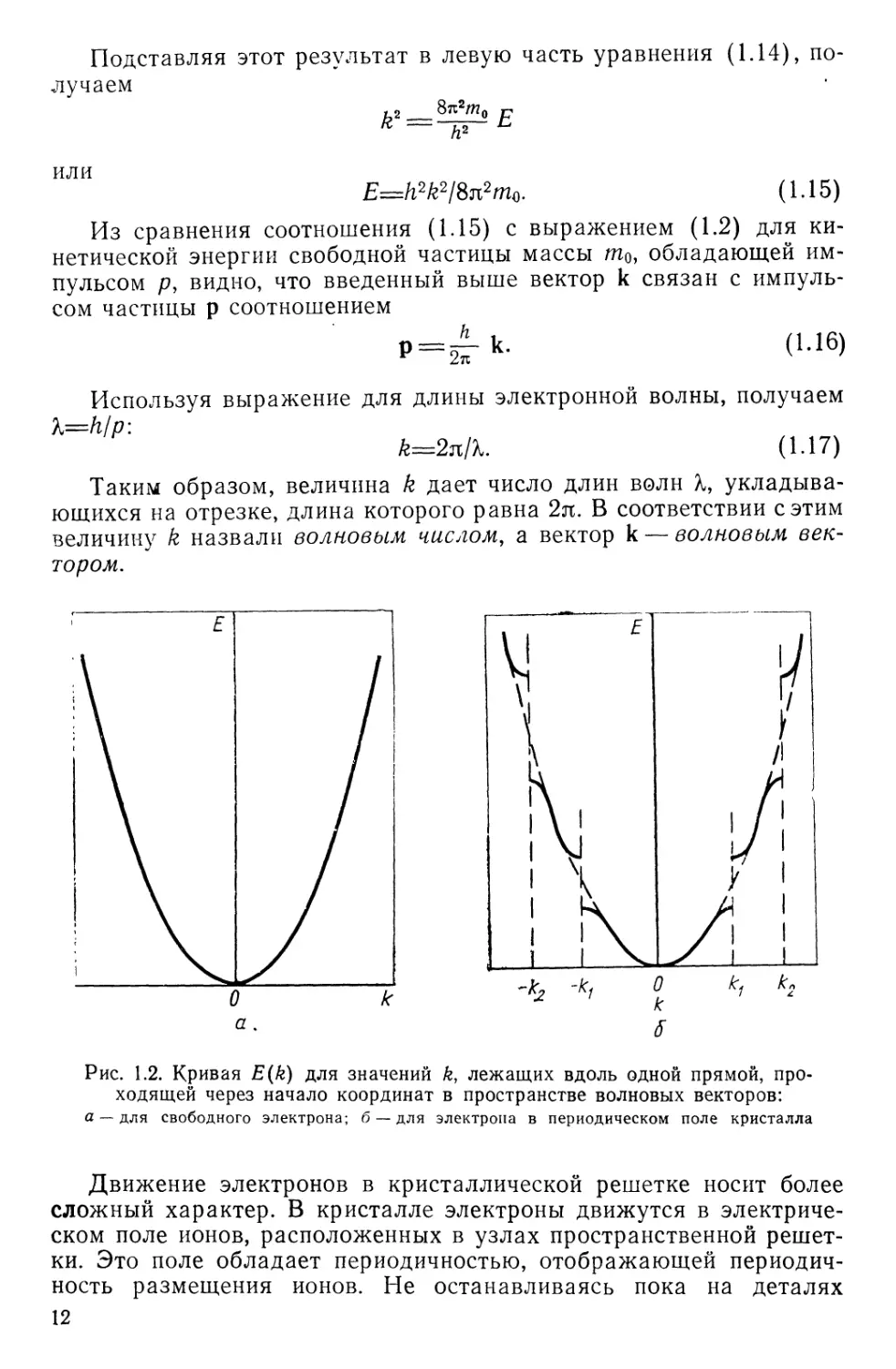
Рис. 1.2. Кривая E(k) для значений k, лежащих вдоль одной прямой, про-
ходящей через начало координат в пространстве волновых векторов:
а — для свободного электрона; б — для электрона в периодическом поле кристалла
Движение электронов в кристаллической решетке носит более
сложный характер. В кристалле электроны движутся в электриче-
ском поле ионов, расположенных в узлах пространственной решет-
ки. Это поле обладает периодичностью, отображающей периодич-
ность размещения ионов. Не останавливаясь пока на деталях
12
решения задачи о движении электрона в периодическом силовом
поле, укажем следующий общий результат.
Для свободного движения электрона энергия является непре-
рывной функцией волнового вектора, с которым она связана соот-
ношением (1.15), и, следовательно, в этом случае возможны любые
движения с импульсами, имеющими всевозможные направления и
величины (рис. 1.2,а).
Такой простой результат получается только для электрона, свободно движу-
щегося в бесконечном пространстве. Если же область, внутри которой движется
электрон, ограничена («электрон в ящике»), то дозволенные значения энергии
электрона образуют не непрерывную, а дискретную последовательность. В том
случае, когда линейные размеры «ящика», в котором движется электрон, велики
по сравнению с атомными размерами, высота «порога» между соседними энер-
гетическими уровнями оказывается настолько мала, что дискретностью уровней
можно пренебречь и пользоваться формулой (1.17). Действительно, уже для
«ящика» с линейными размерами L порядка 1 см разница между первыми двумя
энергетическими уровнями электрона имеет порядок 10~14 эВ.
Ограничимся случаем движения электрона в одномерной решет-
ке. Уравнение Шредингера принимает вид
[Е _ v (Л)] т = 0 (1 18)
Здесь V — потенциальная энергия.
Предположим, что период одномерной решетки равен а, т. е.
V(x+a)=V(x). (1.19)
В этом случае уравнение Шредингера имеет решения типа
e/xTz(x); e-Zx’Fz(%);] €~ikxWk(x),
где I и k — вещественные параметры, зависящие от энергии.
Решения, содержащие только действительный параметр /, дол-
жны быть отброшены, так как функция не может неограниченно
возрастать *.
Этот результат означает, что имеются «запрещенные» интерва-
лы значений энергии, для которых не существует стационарных со-
стояний электронов. Движение электрона в этом случае модулиру-
ется силовым полем таким образом, что энергетический спектр
электрона распадается на зоны дозволенных энергий — зоны Брил-
люэна, разделенные конечными интервалами значений энергии, за-
прещенными для электронов. Одна зона от другой отделена «энер-
гетической щелью» ДЕ, ширина которой определяется свойствами
симметрии кристаллической решетки и различна для разных на-
правлений импульса электрона (рис. 1.2,6).
Волновая функция свободного электрона
W=expi(kr), ’ (1.20)
* Эти решения не удовлетворяют граничным условиям: ^-функция должна
обращаться в нуль при х—0 и x = L (на поверхности «ящика»).
13
описывающая движение электрона с постоянным импульсом р,
соответствует плоской волне с волновым вектором:
В трехмерной кристаллической решетке электроны движутся
в трехмерном периодическом силовом поле; потенциальная энер-
гия V (х, у, z) также является периодической функцией координат.
Волновая функция электрона должна удовлетворять уравнению
vT + 8j^[E-V(x,i/,z)]iF = 0. (1.21)
Можно показать, что в рассматриваемом случае волновая
функция имеет вид
Т(х, у, z)=expi (kr)4f/l(x, у, г), (1.22)
где ЧД(х, у, z)—периодическая функция координат, обладающая
теми же периодами, что и V(x, у, z), кроме того, функция
ЧД(х, у, z) зависит от к.
Таким образом, решение волнового уравнения для заданного
V (х, у, z) по-прежнему представляет плоскую волну с волновым
вектором к, но модулированную периодическим полем решетки.
Величину р=(й/2л)к назвали квазиимпульсом электрона в пе-
риодическом поле. В отличие от свободно движущегося электрона,
для которого имеет место сохранение импульса, в случае электро-
на, движущегося во внешнем поле, являющемся функцией коорди-
нат, импульс не сохраняет постоянного значения. Энергия электро-
на Е представляет функцию волнового вектора, или квазиимпуль-
са; определение этой функции требует решения уравнения Шредин-
гера при заданной потенциальной энергии. Некоторые важные
свойства волновой функции в зависимости от симметрии кристал-
лической решетки можно получить с помощью методов теории
групп.
Для свободного движения электронов пространство импульсов
(соответственно, волнового вектора) изотропно, поверхности рав-
ных энергий (поверхности Ферми) в этом пространстве представ-
ляют собой сферы. Особо интересна сфера поверхности для макси-
мальной энергии электронов при Т=0.
Для электронов, движущихся в периодическом поле решетки,
пространство импульсов утрачивает свой радиально-симметричный
характер вблизи границ зон, где форма поверхностей Ферми иска-
жается, значительно отклоняясь от сферической. Определение точ-
ной формы поверхностей Ферми как для чистых металлов, так и
для сплавов представляет большие математические трудности, пре-
одолеть которые полностью не удается; поэтому приведенные ре-
зультаты являются приближенными. Схематически зависимость
энергии Е от волнового числа представлена на рис. 1.2,6. Кривая
построена для значений k, лежащих в пространстве импульсов на
прямой, проходящей через начало координат и, следовательно, от-
вечающей определенному направлению импульса или волнового
14
вектора. Разрывы на кривой происходят в точках k2 и т. д.,
в которых эта прямая пересекает поверхности, являющиеся грани-
цами зон. Эти точки различны для разных направлений. Пунктир-
ная парабола представляет зависимость для совершенно свобод-
ных электронов.
Рис. 1.4. Простейшая двух-
мерная решетка.
Рис. 1.3. Перекрытие смежных зон
Переходы электрона из одной зоны в соседнюю требуют скач-
кообразных изменений энергии на величины \Е2 и т. д., раз-
личные для разных направлений волнового вектора или импульса.
Однако возможно частичное перекрытие смежных зон как для
отдельных направлений импульса электрона, так и (в некоторых
случаях) для всех направлений. Схематически оба случая пред-
ставлены на рис. 1.3.
Построение зон дозволенных энергий (зон
Бриллюэна). Электроны, обладающие импульсом p=(/i/2n)k,
ведут себя аналогично рентгеновским лучам с длиной волны Л=
=h/p или K=2n/k, распространяющимся в направлении вектора к.
Из теории дифракции рентгеновских лучей известно, что если дли-
на волны излучения Z, и угол падения а удовлетворяют соотноше-
нию Вульфа — Брэгга:
nX=2d cos а,
(1-23)
где п — произвольное целое число; d — расстояние между двумя
соседними атомными плоскостями, то лучи отражаются, не прони-
кая в глубь кристалла.
В применении к электронам это значит, что в данном кристалле
отсутствуют электроны с «запрещенными» энергиями, удовлетво-
ряющими указанному соотношению.
Для различных направлений волнового вектора к, т. е. для раз-
личных направлений в кристалле, расстояние d различно. На
рис. 1.4 изображена двухмерная решетка, представляющая собой
сечение простой кубической решетки плоскостью, параллельной
грани куба.
15
Подставляя в уравнение (1.23) соотношение (1.17), получаем
2nnlk=2d cos а,
откуда
k cos QL—nnld.
Но &cosa=x — проекция вектора к на нормаль к рассматри-
ваемой плоскости и, следовательно,
^=nn!d.
(1.24)
Формула (1.24) представляет собой в пространстве волнового
вектора уравнение плоскости, проходящей на расстоянии пл/d от
начала координат. Зная расположение в кристалле всех «отража-
ющих» атомных плоскостей для
различных порядков отражения (со-
ответствующих всевозможным зна-
чениям целого числа п), можно по-
строить зоны Бриллюэна. Для этого
необходимо воспользоваться струк-
турным фактором интенсивности от-
ражения электронов (или рентге-
новских лучей) от определенного
Рис. 1.5. Первая (/) и вторая (II)
зоны Бриллюэна для простой ку-
бической решетки
семейства атомных плоскостей в
кристалле.
Рассмотрим более подробно по-
строение этих зон для простой
кубической решетки. Формула (1.24) показывает, что наименьшему
значению х соответствует наибольшее значение межплоскостного
расстояния d. В простой кубической решетке такими плоскостями
являются грани ячейки; при этом d=a (а — период решетки).
Направляющие косинусы нормали к этим плоскостям равны соот-
ветственно: (±1, 0, 0), (0±, 1,0), (0,0, ±1). Первая зона пред-
ставляет куб, ограниченный плоскостями: kx=±nja, ky=±nfa, kz=
= ~t~ л/n.
Следующая совокупность отражающих плоскостей характери-
зуется межплоскостным расстоянием d = a/]/r2 , т. е. плоскости
совокупности {НО}, направляющие косинусы нормалей к этим пло-
скостям, равны: (zt 1,± 1,0), (±1,0, ±1), (0, ± 1, ± 1). Наружными
границами второй зоны являются плоскости: ± kx ± ку — У~2 те/а;
± kx ± kz = У 2 к/a; ±ky± кг = У2 id а, образующие додекаэдр
(рис. 1.5).
Для г. ц. к. решетки первая зона Бриллюэна (рис. 1.6,я) обра-
зуется плоскостями, параллельными плоскостям кристалла {111} и
{100}; структурный множитель отличен от нуля для отражений пло-
скостями {100} второго порядка [в уравнении (1.24) п=2]. Для
о. ц. к. решетки (см. рис. 1.6,6) граница первой зоны образуется
плоскостями, параллельными плоскостям {110} кристалла. Случай
гекс. п. у. (см. рис. 1.6,в) решетки рассматривается в задаче 1.4.
16
Таким образом, форма зон дозволенных энергий определяется
структурой кристалла.
Физические свойства кристалла существенно зависят от распре-
деления электронов по зонам, формы поверхностей Ферми и от
энергетических скачков границ между зонами.
Связь
тронов и
между энергетическим спектром элек-
свойствами кристаллов. На рис. 1.7 схемати-
чески изображены кривые, полу-
чающиеся при пересечении по-
Рис. 1.6. Первые зоны Бриллюэна
для г. ц. к. (а), о. ц. к. (б) и гекс. п. у.
(в) решеток
Рис. 1.7. Поверхности равных энер-
гий в плоскости /?2=0 для простой
кубической решетки
верхностей равных энергий плоскостью Az=0 для простой кубиче-
ской решетки. В первой зоне показаны кривые, соответствующие
различным уровням энергии вплоть до границы зоны, во второй
k>±2n/a — только несколько наиболее низких уровней.
Для самых низких уровней первой зоны форма поверхностей
равных энергий не отличается от сферической, свойственной сво-
бодному движению электронов, когда связь между энергией элек-
трона и волновым числом выражается простой квадратичной зави-
симостью (1.15).
Предположим, что электрон с энергией, которая далека от границы зоны,
находится в постоянном внешнем электрическом поле F. Если заряд электро-
на —е, скорость движения v, то работа электрических сил за единицу времени
—eFv равна скорости изменения кинетической энергии
dE
dt ’
т. е. —eFv
~ dt Е “ dk dt ’
В то же время на основании соотношения (1.15)
dE —
dk 4п2т0 ’
(1.25)
hk 2п dE h dk
но mQv — р = hk/2n или v = ~~~dk * 0ТКУДа — #Fv = и и, сле-
2—523
17
довательно,
dk
ИГ
(— 2л 'h) eF.
(1.26)
Вычислим ускорение dv/dt, приобретаемое электроном во внешнем поле F
dv
~dt
d / 2л dE \ 2л d dE 2л d2E dk
-dr[~-dk) = ~-dF-dk~~^lF> или’ согласно ФОРМУЛ (1.26)
dv 4л2 d2E
~dt hT'd^'eF’
(1.27)
ИЛИ
[(4л2/А2) (d2E/dk2)] -1 = —eF.
В правой части этого уравнения стоит сила, действующая на частицу, следо-
74712 d2E X-1
вательно, множитель I 1 при уск фении dv/dt в левой части должен
представлять массу частицы.
d2E h2
Для свооодного электрона из соотношения (1.15) получаем
г dv — г
Следовательно, пг0 — — ег.
Получили обычный закон движения отрицательного электрического заряда
в электрическом поле напряженностью F. В рассмотренном случае свободного
движения электрона вторая производная d2E/dk2, характеризующая кривизну
кривой зависимости энергии электрона от его волнового числа (или импульса),
всегда положительна. Иначе обстоит дело в случае электрона, движение которо-
го модулировано периодическим силовым полем кристаллической решетки.
Как показано на рис. 1.2,6, с приближением энергии к крити-
ческому значению у границы зоны кривизна кривой E(k) изменяет
свой знак на противоположный и, следовательно, величина
d2E/dk2 становится отрицательной.
Таким образом, для электрона, движущегося в периодическом
поле кристаллической решетки, величина m*=/z2/[4n2(d2E/dk2)],
названная эффективной массой электрона, может быть и положи-
тельной, и отрицательной величиной. Кроме того, эффективная мас-
са может зависеть и от направления движения электрона в кри-
сталлической решетке. В общем случае эта величина носит не ска-
лярный, а тензорный характер.
Эффективная масса электрона характеризует его взаимодейст-
вие с кристаллической решеткой. У свободного электрона эффек-
тивная масса /п* совпадает с его обычной массой т0. Назовем иде-
альным металлом кристалл, обладающий совершенно свободными
электронами проводимости (т*=т0). Отклонение реального ме-
талла от идеального (в указанном смысле) можно характеризовать
отношением m0/m* для электронов проводимости. Так, например,
у щелочных металлов Li, Na, К это отношение составляет соответ-
ственно 0,653, 1,069 и 1,72. У натрия эффективная масса электрона
проводимости наиболее близка к его обычной массе. Таким обра-
зом, по своим свойствам натрий должен быть ближе к идеальному
металлу, чем литий или калий.
Уже отмечалось, что для понимания многих свойств кристаллов
необходимо знать распределение электронов по уровням энергии,
т. е. величину n(e)de, представляющую собой число электронов
в единице объема, энергии которых лежат в интервале 8 и e-[-de.
Для свободных электронов n(8)d8=(2m0//i2)3/2 ]/s de.
Выражая энергию через волновое число, получаем
n(s) ds = -^-2-k2dk.
В то же время элемент объема в пространстве волнового векто-
ра равен ^nk2dk и, следовательно, n(e)de=-g^r dkxdkydkz.
Если это выражение проинтегрировать по объему куба с реб-
ром 2л/а (первой зоны Бриллюэна), то в правой части получим
1/8л3(2л/а)3==1/а3. В левой части получим число электронов в 1 см3
металла. Следовательно, число электронов, заполняющих первую
зону Бриллюэна, для простой кубической решетки в расчете на
1 см3 равно числу ячеек в 1 см3, в расчете на одну ячейку получа-
ется один электрон.
Периодическое поле кристалла, модулируя движение электро-
нов, усложняет зависимость п(е). Ограничимся только качествен-
ным рассмотрением. Для малых значений k (далеких от границы
зоны) поверхности равных энергий мало отличаются от сфер. По-
этому в области малых энергий сохраняется пропорциональность п
величине У Но мере отклонения формы поверхностей от сфери-
ческой п(е) начинает расти быстрее, чем У е, до тех пор, пока по-
верхность не коснется плоско-,
стей, ограничивающих зону.
С этого момента п(б) начинает
уменьшаться. На рис. 1.8 при-
ведена зависимость л (в). Рез-
кий спад кривой в точке А
(или D) обусловлен «запреще-
нием» состояний с этими энер-
гиями для некоторых направ-
лений импульса. Этот спад
продолжается до точки, соот-
ветствующей заполнению пер-
вой зоны.
Если в данном кристалле
электроны проводимости зай-
мут все энергетические уровни
первой зоны, то приложенное
Рис. 1.8. Кривые распределения электро-
нов по энергиям (в непереходных ме-
таллах) :
-------- для свободных электронов; ------
для г. ц. к. решетки (ЛВС);----для
о. ц. к. решетки (DEF)
к кристаллу внешнее поле не создаст электрического тока. Дей-
ствительно, электроны, разгоняясь полем, должны переходить на
более высокие энергетические уровни, но свободные уровни име-
ются только в соседней зоне. Если вторая зона отделена от первой
энергетической щелью конечной ширины, то для перевода электро-
нов на свободные уровни потребуется внешнее электрическое поле
2*
19
очень большой напряженности, в обычных же полях в таком кри-
сталле электронная проводимость будет отсутствовать, т. е. он
окажется диэлектриком. Для большинства металлов энергетиче-
ский спектр электронов характеризуется перекрытием зон (см.
рис. 1.3).
На рис. 1.8 изображены кривые распределения электронов по
энергиям для непереходных металлов с г. ц. к. и о. ц. к. решетками
(у переходных металлов картина сложнее из-за большого числа
электронов — см. ниже).
Тип решетки, в которой кристаллизуется тот или иной металл,
должен быть таков, чтобы энергия электронного газа имела мини-
мальное значение. При этом электроны, как правило, стремятся
разместиться на уровнях внутри первой зоны.
Рассмотрим два вещества с различным числом валентных элек-
тронов, приходящихся на один атом (это число называют электрон-
ной концентрацией). Может случиться, что в первой зоне решетки,
характерной для вещества с меньшей электронной концентрацией,
не будет свободных энергетических уровней для большего числа
электронов. Тогда для второго вещества энергетически более вы-
годной окажется другая решетка, в которой все валентные элек-
троны смогут разместиться на уровнях первой зоны.
На поверхности Ферми, касающейся границы первой зоны, зна-
чение энергии
впРед =-о^Лред- (Ь28)
Здесь рПред=^/2б/ — длина перпендикуляра, опущенного в простран-
стве импульсов из начала координат на плоскость, ограничиваю-
щую зону. Для г. ц. к. решетки d\\\=aV 3. Период г. ц. к. решетки
а связан с числом атомов AfaT в объеме v соотношением AfaT/u=
=4/а3 (в элементарную ячейку г. ц. к. решетки входят четыре ато-
ма). Следовательно,
Рпр^ = НШ = УЗ Нуа=УЗ h/2 (^)1/3 . (1.29)
В то же время из формул (1.4) и (1.28) получаем
Ap.« = (W'!C = 2(»..),b n/2«. (gi)1'1 , (1.30)
где N3„ — число валентных электронов в объеме v. Сравнив отно-
шения (1.29) и (1.30), получим
33/2Уат/ 32о = ЗЛГэл/8™ или ЛГэл/ЛГет = УЗ it/4 = 1,362.
Найдем общее выражение для расчета электронной концентра-
ции (сэл), при которой поверхность Ферми касается границ первой
зоны Бриллюэна. В случае кубического кристалла расстояния меж-
ду атомными плоскостями
Gnhl=al(h2+k2+l2y2. (1.31)
20
Если число атомов на элементарную ячейку пат, то для куби-
ческого кристалла с периодом a NSLTiv=nSirr/a3. Тогда формула
(1.29) примет следующий вид:
Рпред=^/2 (N^/n^v) V3 (Л24-^+/2) 1/2. (1.32)
Из формул (1.32)’И (1.30) получим
Сэл=Л/’эл/^аТ=я/ЗПат (Й2+^2+/2) 3'2. (1.33)
Расчет для о. ц. к. решетки дает сэл= 1,480.
Для атомов нормальных элементов (с целиком заполненными
внутренними электронными уровнями) под числом валентных элек-
тронов па атом подразумевается число электронов, находящихся
вне застроенной оболочки.
Для сплавов (твердых растворов или промежуточных фаз) рас-
считывают электронную концентрацию как среднее число электро-
нов на атом, исходя из валентности компонентов (например, для
сплава Си — 50 ат.% Zn это число составляет 1 /2-1+1/2-2=3/2).
При анализе электронных соединений (фаз Юм-Розери) ва-
лентность элементов VIII группы периодической системы Д. И. Мен-
делеева (железо, кобальт, никель и др.) принимают равной нулю,
иногда—равной 1.
Энергетические уровни электронов проводимости в кристалли-
ческой решетке здесь рассмотрены в приближении одноэлектронной
модели металла, т. е. изучением поведения валентного электрона
в периодическое поле решетки, образованной ионами.
Возможен, однако, и другой способ решения этого вопроса. По-
добно тому как в простейшей молекулярной системе Н2 происходит
раздвоение каждого энергетического s-уровня электрона, в кристал-
ле, представляющем собой гигантскую молекулу из N атомов, каж-
дый s-уровень валентного электрона расщепляется на N уровней.
Благодаря большему числу N они настолько близко лежат друг
к другу, что их совокупность с достаточно хорошим приближением
можно считать сплошной полосой, имеющей конечную ширину.
Рассмотрим образование металлической связи на примере ли-
тия. В нормальном атоме лития энергетической уровень 1s целиком
заполнен, так как на нем находятся два электрона, а уровень 2s
заполнен частично: на нем находится один валентный электрон.
При сочленении атомов лития в кристаллическую решетку электро-
ны в зоне 2s, возникающей расщеплением уровней 2s изолирован-
ных атомов, которые заполнены лишь частично, разместятся на
нижних уровнях зоны, благодаря чему средняя энергия валентного
электрона окажется в металле меньше, чем в изолированном атоме.
Этот выигрыш в энергии с избытком компенсирует как возрастание
энергии электростатического отталкивания ионов лития, которое
произойдет при их сближении, так и те силы отталкивания, кото-
рые обусловлены кинетической энергией валентных электронов.
Если же уровень в изолированном атоме заполнен целиком, то за-
полненной окажется и зона, образующаяся при расщеплении этого
уровня. При этом не произойдет уменьшения энергии валентных
не только валентных электронов, но
7/г
Рис. 1.9. Размытие 3d и 4s уровней и их
перекрытие при образовании кристалла '
переходного металла (г — межатомное
расстояние)
электронов в металле по сравнению с энергией в изолированных
атомах. Так обстоит дело в случае атомов гелия, где уровень Is
целиком заполнен.
В переходных металлах происходит размытие в полосу уровней
и энергетических уровней внут-
ренних незаполненных под-
групп.
На рис. 1.9 показано отно-
сительное расположение уров-
ней 4s и 3d в изолированных
атомах никеля и их размытие
в полосы при сближении ато-
мов и их сочленении в кристалл
в зависимости от обратного
значения межатомного рас-
стояния.
На рис. 1.10 приведены кри-
вые распределения электронов
по энергиям в переходных ме-
таллах с о. ц. к. и г. ц. к. ре-
шетками. Для о. ц. к. решетки
характерно наличие резко выраженного минимума в средней ее
части, для г. ц. к. — наличие крутого пика в конце.
При образовании кристаллов нивелируется различие электрон-
ного строения переходных металлов одной и той же группы пе-
Рис. 1.10. Кривые распределения элек-
тронов по энергиям в переходном
металле с о. ц. к. (Сг) (а) и г. ц. к.
(Pd) (б) решетками (точки на а —
значения n(s), полученные из элек-
тронной теплоемкости чистых метал-
лов)
рпсдической системы. Так, если изолированные атомы Ni имеют
8 d-электронов, a Pd—10 d-электронов, то кристаллы Ni и Pd —
примерно по 9,5 d-электрона.
Рассмотренное приближение (не взаимодействующие между собой электро-
ны в периодическом поле положительно заряженных ионов) позволяет вычислить
такие важные характеристики, как энергия сублимации металла и упругие
спектры лишь для самых «рыхлых» структур, таких, как у щелочных металлов,
у которых суммарный объем ионов очень мал по сравнению с объемом кристал-
ла и малы потенциалы поля решетки. Гаррисон, Коэн и др. предложили для
непереходных металлов учитывать при расчетах лишь слабый потенциал, суще-
ствующий вне атомных остовов («псевдопотенциал»). Допустимость использо-
вания псевдопотенциала мотивируется «экранирующим» действием электронов
проводимости, вероятность пребывания которых вблизи атомных остовов велика.
Метод псевдопотенциала позволил с удовлетворительной точностью вычислить
энергии связи, сжимаемость и атомные радиусы металлов II—IV групп и неко-
торых полупроводников и рассчитать спектры тепловых колебаний металлов.
Кроме того, удалось оценить влияние точечных дефектов кристаллической решет-
ки (примесных атомов, вакансий и т. д.) на свойства металлов и некоторых
твердых растворов.
Задачи к гл. 1
Задача 1.1. Определить энергию кристалла алмаза массой Л4=0,5 г, если
энергия каждой связи С — Св алмазе равна 85,4 ккал/(г-моль).
Указание: принять во внимание особенности структуры алмаза (с. 25).
Задача 1.2. Найти функцию распределения Ферми для: 1. е^8Ф и Т=0 К;
2. е—£ф=0, Г>0 К; 3. е—еф —kT\ 4. е——4kT.
Задача 1.3. Найти индексы плоскостей, ограничивающих первую зону Брил-
люэна для кристалла, имеющего гекс. п. у. структуру.
Указание: использовать плоское сечение обратной решетки для плоскостей
зоны [00,1] и [10,0]; первая зона Бриллюэна образуется плоскостями, соответ-
ствующими ближайшим к началу координат узлам обратной решетки (проводят-
ся через середины радиус-векторов обратной решетки).
ГЛАВА 2
КРИСТАЛЛИЧЕСКАЯ СТРУКТУРА ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
§ 2.1. СТРУКТУРА КОВАЛЕНТНЫХ КРИСТАЛЛОВ
Как известно, резкую границу между металлами и неметаллами
в периодической системе элементов провести невозможно.
Если все элементы первых трех групп периодической системы,
за исключением водорода, гелия и бора, а также все элементы пе-
реходных групп безусловно являются металлами, а все галогены
и благородные газы — неметаллами, то в IV—VI группах имеются
как неметаллические элементы — диэлектрики (углерод в алмаз-
ной модификации, N, Р, As, О, S), так и элементы, по свойствам
промежуточные между металлами и неметаллами (Si, Ge, a-Sn, Se,
Те, Sb, Bi), и, наконец, несомненно металлические элементы (p-Sn
и особенно РЬ).
Рис. 2.1. Схема распределения валентных электронов в атомах углерода
по состояниям: в изолированном атоме (а) и в кристалле алмаза (б).
Кристаллические структуры неметаллических и полуметалличе-
ских элементов IV—VII групп периодической системы удовлетво-
ряют простому правилу: координационные числа равны валентно-
сти соответствующих элементов по водороду Z—8—р, где р — число
валентных электронов атома, равное номеру группы периодической
системы.
Это правило показывает, что в кристаллах таких элементов
основную роль играет ковалентная (гомеополярная) связь, осуще-
ствляемая парами электронов, одновременно принадлежащими
к двум соседним атомам, причем каждый атом стремится иметь
максимальное число соседей, отдавая для связи с каждым из них
один из своих неспаренных электронов. Валентные электроны
в атоме, образующем молекулу, стремятся занять наибольшее воз-
24
можное число ячеек фазового пространства. Так, например, если
в изолированном атоме углерода (рис. 2.1,а) имеется два s-элек-
трона, так что на долю р-подгруппы приходится также лишь два
электрона и одна из р орбит свободна, то в результате возмущаю-
щего действия соседних атомов один из двух s-электронов перехо-
дит в свободную p-ячейку (см. рис. 2.1,6). При этом число неспа-
ренных (т. е. способных к взаимодействию с соседними атомами)
электронов увеличивается с двух до четырех.
а
Рис. 2.2. Кристаллические
структуры: алмаза (а) и гра-
фита[б)
В кристалле алмаза (рис. 2.2,а) каждый атом окружен четырь-
мя соседями, находящимися в вершинах правильного тетраэдра.
Связь с каждым соседом осуществляется парой электронов, общих
для обоих атомов. За счет общих электронов происходит заполне-
ние всех фазовых ячеек валентной оболочки каждого атома.
Представления о наличии общих пар электронов у соседних ато-
мов наиболее четко подтверждаются данными рентгенографиче-
ского исследования распределения электронной плотности в кри-
сталле алмаза.
Фактически кристалл алмаза представляет собой гигантскую
гомеополярную молекулу CN, где N — общее число атомов углеро-
да в кристалле. То же координационное число четыре имеют и кри-
сталлы кремния, германия и а-олова (модификация, устойчивая при
температуре ниже 18°С и часто называемая «серым оловом»).
В кристалле графита каждый атом углерода связан ковалентными
связями с тремя соседями, принадлежащими одному слою (рис.
2.2,6). Четвертый электрон каждого атома при образовании кри-
сталла делается общим для всего кристалла, обусловливая высо-
кую электропроводность графита. У графита весь кристалл не
является уже единой гигантской молекулой. Такими молекулами
можно считать лишь отдельные атомные слои кристалла. Эти слои-
молекулы связаны между собой силами металлического взаимо-
действия. Если теплота испарения углерода в алмазной модифи-
кации равна 150 ккал/(г-атом), то для отделения атомных слоев
в графите нужно затратить лишь 2—3 ккал/(г-атом).
25
Сложную ромбоэдрическую структуру имеют кристаллы эле-
ментов V группы: мышьяка, сурьмы и висмута (рис. 2.3,а). В соот-
ветствии с общим правилом координационное число в этих кри-
сталлах равно трем (2=8—р=8—5).
Каждый атом имеет трех ближайших соседей в соседнем слое —
с ними он связан ковалентной связью, это определяет наличие оси
симметрии третьего по-
рядка. Соседние двуслой-
ные пластинки связаны
между собой вандерва-
альсовскими силами.
Каждая из таких пла-
Рис. 2.3. Характерные кристаллические структуры: элементов V (As) (а) и эле-
ментов VI (Se) (б) групп
стинок аналогична молекуле (так же, как при алмазной струк-
туре одной молекулой является по существу весь кристалл). Кова-
лентные связи не пронизывают непрерывной связью кристаллы
As, Sb и Bi — они охватывают лишь отдельные группировки ато-
мов кристалла. Поэтому As, Sb и Bi более пластичны, чем алмаз,
Si, Ge или a-Sn и менее прочны.
Таблица 2.1
Степень компактности некоторых кристаллических решеток*
Тип решетки Координационное число Коэффициент запол- нения кристалла атомами Объем межатомных промежутков, % сбще- го объема кристал- лов
Гекс. п. у. 12 0,74 26
Г. ц. к. 12 0,74 26
О. ц. к. 8 0,68 32
Простая кубическая 6 0,52 48
Решетка алмаза 4 0,34 66
Решетка теллура 2 0,23 77
* Степень компактности кристаллов подсчитана в предположении, что распределение электронного
заряда в атоме обладает сферической симметрией.
26
Кристаллы элементов VI группы (Se и Те) состоят из спираль-
ных атомных цепочек, связанных между собой вандерваальсовы-
ми силами (см. рис. 2.3,6). Каждый атом имеет две ковалентные
связи (под углом 90°) с соседями, принадлежащими той же цепоч-
ке. Роль молекул играют здесь атомные цепочки. Эти кристаллы
еще менее прочны, чем кристаллы As, Sb и Bi.
Наконец, кристалл иода (VII группа) состоит из двухатомных
молекул (координационное число равно единице), которые связаны
между собой вандерваальсовыми силами. Молекулярная связь
в кристаллах иода обусловливает высокую летучесть этого веще-
ства.
Чем меньше координационное число структуры, тем меньше сте-
пень компактности кристаллической решетки, тем больше места
в решетке приходится на долю межатомных промежутков. Доля
пространства, занятого атомами в некоторых кристаллических ре-
шетках, указана в табл. 2.1.
Таблица 2.2
Атомные радиусы и объемы некоторых элементов
третьего периода таблицы Д. И. Менделеева
Порядковый номер п 12 13 14 15 16
Элемент Атомный объем, 1024 см3/г-атом Na 23,7 Mg 14,1 Al 10 Si 12 P 13,2 s 15,5
Атомный радиус, А 1,92 1,60 1,43 1,17 1,10 1,04
С увеличением номера группы периодической системы (начиная
с четвертой) компактность кристаллической решетки уменьшается.
Этим объясняется расхождение между кривыми, построенными для
атомных радиусов, и кривыми атомных объемов соответствующих
элементов. Атомные радиусы в пределах одного периода периоди-
ческой системы с увеличением порядкового номера элементов
уменьшаются, а атомные объемы начиная с середины каждого пе-
риода всегда возрастают (табл. 2.2).
Причина этого расхождения заключается в том, что в ковалент-
ных кристаллах влияние уменьшения атомных радиусов на атом-
ные объемы не только компенсируется, но даже перекрывается про-
тивоположным влиянием уменьшения компактности, связанного
с уменьшением координационного числа.
§ 2.2. СТРУКТУРА МЕТАЛЛИЧЕСКИХ ЭЛЕМЕНТОВ
Валентные электроны в металле являются общими не для пары
соседних атомов, а для всего кристалла. Рентгенографические ис-
следования распределения электронной плотности в кристаллах
магния и алюминия показали, что вероятность нахождения валент-
27
ного электрона в любой точке вне иона одна и та же*, что свиде-
тельствует об отсутствии направленных связей в металлических
кристаллах. Отсутствие направленных связей определяют сфериче-
скую симметрию силовых полей в металлических кристаллах, вы-
сокую симметрию и компактность структур большинства металли-
ческих элементов.
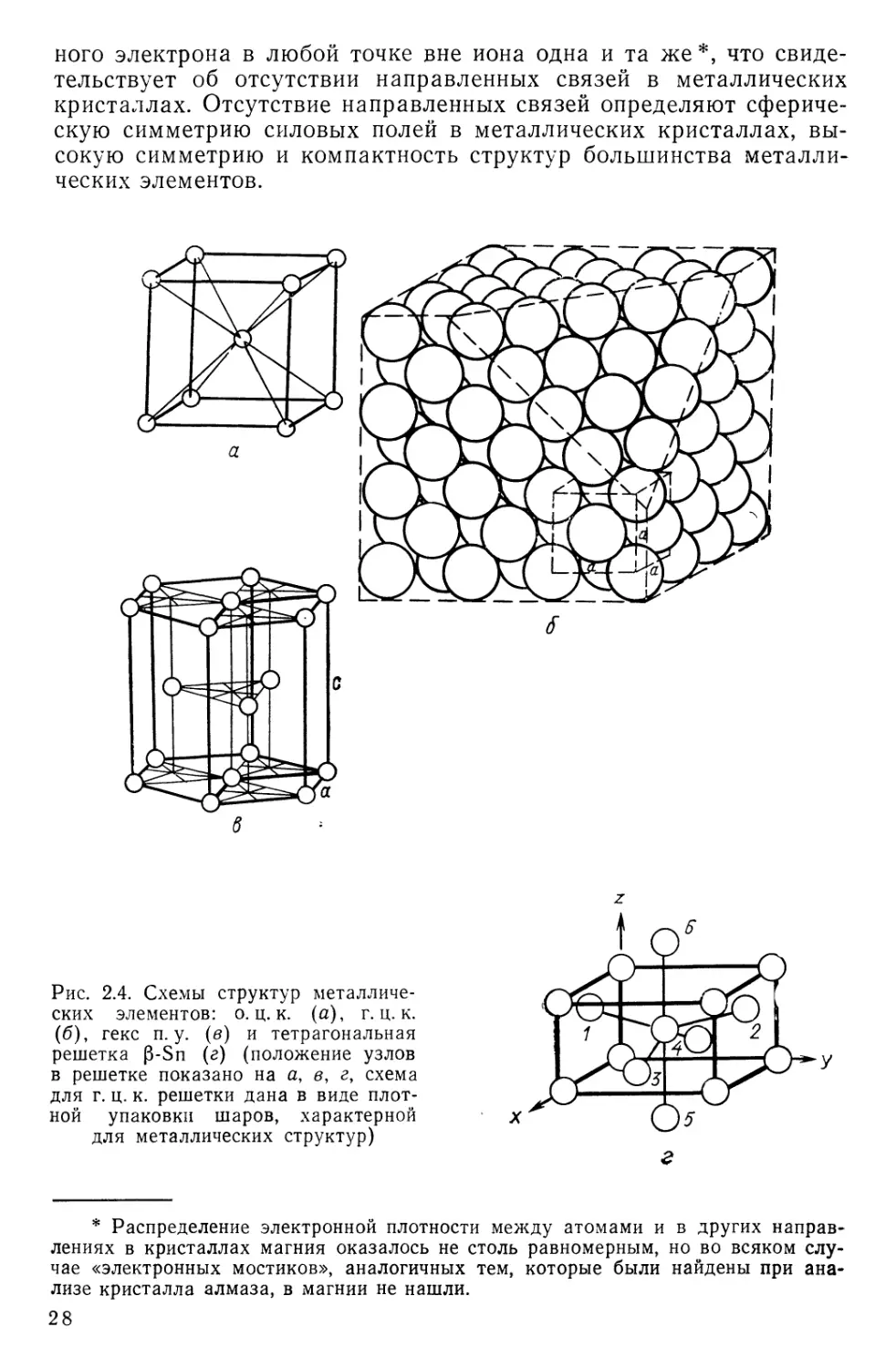
Рис. 2.4. Схемы структур металличе-
ских элементов: о. ц. к. (а), г. ц. к.
(б), гекс п.у. (в) и тетрагональная
решетка P-Sn (г) (положение узлов
в решетке показано на а, в, г, схема
для г. ц. к. решетки дана в виде плот-
ной упаковки шаров, характерной
для металлических структур)
* Распределение электронной плотности между атомами и в других направ-
лениях в кристаллах магния оказалось не столь равномерным, но во всяком слу-
чае «электронных мостиков», аналогичных тем, которые были найдены при ана-
лизе кристалла алмаза, в магнии не нашли.
28
Наиболее распространены среди металлов гекс. и. у. и г. ц. к.
решетки с координационным числом 12 и о. ц. к. решетка с коорди-
национным числом 8 (рис. 2.4).
Коэффициент заполнения у о. ц. к. решетки лишь на 10% ниже,
чем у г. ц. к. и гекс. п. у. решеток. Столь высокая компактность
о. ц. к. решетки обусловлена тем, что кроме восьми ближайших
соседей, находящихся на расстоянии 0,867 а, каждый атом имеет
еще шесть соседей на расстоянии а.
Периодический закон проявляется и в строении металлических
кристаллов. Все щелочные металлы имеют о. ц. к. решетку (по
имеющимся данным, о. ц. к. решетка цезия при давлении 22 000 атм
превращается в более компактную гекс. п. у. решетку*). Все три
•металла подгруппы меди (Си, Ag и Au) имеют г. ц. к. решетку.
Металлы II группы с достроенными внутренними электронными
оболочками (Be, Mg, Zn и Cd) имеют гекс. п. у. решетку.
Следует отметить, что в то время как в кристаллической ячейке
Mg отношение осей с/а=1,624 близко к числу 1,633, отвечающему
плотной упаковке шаров, у Be оно заметно уменьшено (с/а=1,568),
так что ячейка этого металла сплюснута в направлении оси с,
а у Zn и Cd это отношение превышает 1,8, их элементарные ячейки
вытянуты в вертикальном направлении. Поэтому из 12 соседей, ко-
торыми окружеды каждый атом в кристаллах Zn и Cd, шесть ле-
жащих в той же плоскости (001) являются ближайшими, а шесть
(по три в двух параллельных атомных слоях) расположены при-
мерно на 10% дальше ближайших соседей, у Be же ближайшими
соседями являются шесть атомов, лежащих в соседних слоях.
У Hg, наиболее тяжелого элемента II группы с заполненными
внутренними электронными оболочками, простая ромбоэдрическая
решетка с координационным числом 6.
Два из металлов второй группы, у которых внутренняя d-обо-
лочка не заполнена, —Са и Sr — имеют г. ц. к. решетку, но каль-
ций при температуре 450°С превращается в р-модификацию
с гекс. п. у. решеткой, а в присутствии некоторых примесей имеет
о. ц. к. решетку.
Кроме трех, наиболее характерных решеток есть несколько спе-
цифических решеток у металлов III группы Ga, In, p-Sn (белое
олово — металл с тетрагональной решеткой, четыре атома на
ячейку, к. ч.б, см. рис. 2.4,г); у Мп кроме о. ц. к. (6-Мп) встреча-
ются решетки, присущие металлическим соединениям, у а-Мп —
решетка х-фазы, у р-Мп — решетка одного из типов р-электронных
соединений, у у-Мп — решетка, присущая упорядоченному твердо-
му раствору АВ слоистого типа, сжатая по оси с. Сложные решет-
ки встречаются у актинидов (p-U имеет ту же решетку, что и о-
фаза системы Fe—Сг, — тетрагональную, с 30 атомами на элемен-
тарную ячейку). У некоторых редкоземельных металлов (Nd, Рг)
компактные гексагональные решетки со сложным чередованием
слоев: ABAC..., у Sm чередование еще сложнее.
* Литий при температуре ниже 77 К и Na при 36 К переходят в модифика-
цию с гекс. п. у. решеткой.
29
У полиморфных металлов, имеющих о. ц. к. модификации, по-
следние устойчивы при более высоких температурах (единственное
исключение a-Fe).
Температура полиморфного превращения сильно меняется при
введении примесей и изменении давления. Изменение давления от
атмосферного до 50 кбар понижает температуру перехода a-Fe—>
—>y-Fe от 910 до 600°С и при 150 кбар до комнатной, а темпера-
туру перехода а-Со—>-р-Со повышает от 450 (при атмосферном
давлении) до 600°С при 50 кбар. Воздействие высокого давления
в некоторых случаях приводит к возникновению новых полиморф-
ных модификаций, которые не обнаруживаются при атмосферном
давлении (например, при давлении около 130 кбар и комнатной
температуре возникает гексагональное e-Fe).
В соответствии с принципом ле Шателье высокое давление сни-
жает температуру перехода, когда низкотемпературная модифика-
ция менее компактна, чегл высокотемпературная (y-Fe—>a-Fe-ne-
реход), и повышает в противоположном случае. Если фазовый
переход не сопровождается изменением плотности, то температура
перехода не зависит от давления (магнитные превращения).
Введение примесей также оказывает сильное влияние на тем-
пературу полиморфных превращений. Например, при введении
в iFe 20% Ni температура у—^a-перехода снижается до комнатной.
Особое положение занимают металлы IA подгруппы (Си, Ag, Аи).
У меди, когда она двухвалентная, ЗбЛоболочка не полностью за-
полнена. Таким образом, в СиО ион Си ведет себя как ион пере-
ходного металла. Поэтому Си, Ag и Au можно отнести условно
к переходным металлам.
Переходные металлы разбиваются на несколько групп:
I группа (Зй-металлы) переходных металлов начинается с Sc
до Ni.
II группа (4б/-металлы) —с Y до Pd. Особая переходная груп-
па— редкие земли (4/-металлы) от Се до Lu, заполняется 4/-обо-
лочка.
III группа (5d) —переходная группа от Hf до Pt.
Ac (Z=89)—элемент, начинающий ряд актиноидов (биметал-
лы). У Th (Z=90) и последующих элементов заполняется 5/-обо-
лочка. Все актиноиды радиоактивны.
Области образования определенных металлических структур
расположены почти вертикально в периодической системе, т. е.
определенная кристаллическая структура соответствует примерно
одинаковому числу s-f-d-электронов на 1 атом:
Гекс. п. у.
О. ц. к. О. ц. к. Гекс. п. у. Г. ц. к.
Zr Nb, Mo Тс, Ru Rh, Pd
Hf Ta, W Re, Os Ir, Pt
В конце первой переходной группы (Мп, Fe, Со) это правило не
вполне соблюдается (видимо, в связи с антиферромагнетизмом и
ферромагнетизмом этих элементов).
30
Т аб лица 2.3
Теплота сублимации, температура плавления и модуль упругости металлических элементов
Металл К Са Ti V Cr Mn Fe Co Ni Cu Zn
Q, ккал/моль 21,5 42 113 128 95 \67 99,5 101 103 81 31,2
Ts, к 336 1140 1950 2190 2176 1567 1809 1768 1725 1357 792
£-10~3, кгс/мм2 I-3 — 10,5 15 19 20,1 21,6 20,4 19,7 12,5 8,0
Металл Rb Sr Zr Nb Mo Tc Ru Rh Pd Ag Cd
Q, ккал/моль 19,5 39 146 173 157 •— 155 133 91 68 26,7
л, к 312 1040 2170 2740 2890 — 2773 2239 1823 1234 594
£-10”3, кгс/мм2 — — 8,5 16 33,6 — — 44 12,6 8,0 5,0
Металл Cs Ba Hf Ta w Re Os Ir Pt An Hg
Q, ккал/моль 18,5 42 160 187 200 187 187 155 135 87 15,3
Л, К 303 990 2500 3270 3650 3450 2970 2727 2043 1336 234
Е-10“3, кгс/мм2 — — 8,0 18,8 41,5 47,0 57,0 53 17 8,0 2
Чем больше электронов принимает участие в связи, тем сильнее
взаимодействие атомов в решетке. Мерой связи можно считать
упругие константы, теплоту сублимации, температуру плавления.
Если рассматривать нормальные металлы, то в пределах группы
периодической системы с увеличением атомного номера все харак-
теристики прочности связи уменьшаются (табл. 2.3), например Q
для К, Rb, Cs.
Но при приближении к переходной группе четкой картины уже
нет, см., например Q и Ts для Са, Sr, Ва (см. табл. 2.3).
У переходных металлов сначала не все характеристики прочно-
сти связи изменяются одинаково (Q, Ts и Е для Ti, Zr, Hf
в табл. 2.3).
В V группе все характеристики уже растут с увеличением атом-
ного номера (см. табл. 2.3). Дальше эта закономерность всюду вы-
полняется, т. е. в то время как у нормальных металлов все показа-
тели связи падают, у переходных металлов они возрастают. В груп-
пе Си, Ag, Аи характеристики связей практически неизменны, но
в следующей группе Cd, Hg, Zn все показатели уменьшаются с рос-
том атомного радиуса (см. табл. 2.3).
Итак, в пределах группы периодической системы прочность
связи у нормальных металлов с увеличением Z.n гат падает, у пе-
реходных металлов начиная с V группы прочность связи возрастает
с увеличением Z, несмотря на увеличение г. Как уже отмечалось,
дело здесь в участии d-электронов в межатомном взаимодействии,
которое максимально в пределах каждого длинного периода не-
сколько правее их середин, вплоть до VIII группы. Самый большой
модуль упругости Е у Os. У элементов же IV группы Ti, Zr и осо-
бенно у Hf модуль даже меньше, чем у Си.
Характеристики упругости металлов имеют очень важное значе-
ние в технике высоких давлений. При давлении в несколько сот
килобар, упругая деформация может существенно менять форму
деталей. В связи с этим применяют W и другие элементы с высо-
ким модулем упругости Е. Модуль упругости увеличивается при
введении углерода. Самое большое значение модуля имеет моно-
карбид вольфрама WC (Е=71 • 103 кгс/мм2), что в 3,5 раза боль-
ше, чем у Fe.
Модуль сдвига G входит в предэкспоненциальный множитель
в выражении коэффициента диффузии и влияет на энергию акти-
вации. Там, где вреден высокий коэффициент диффузии (например,
для жаропрочных материалов), надо иметь дело с основой, имею-
щей большое значение G.
Задачи к гл. 2
Задача 2.1. Найти плотность кремния, если длина связи между атомами
равна 2,351 А.
Задача 2.2. Найти наименьший радиус одновалентного положительного
иона, который может соседствовать в ионном кристалле с шестью одновалент-
ными ионами фтора.
Задача 2.3. Начертить структуру белого олова. Найти расстояние до его
первых и вторых ближайших соседей и отношение этих расстояний.
ГЛАВА 3
ТВЕРДЫЕ РАСТВОРЫ
§ 3.1. КРИСТАЛЛИЧЕСКАЯ СТРУКТУРА ТВЕРДЫХ РАСТВОРОВ
Твердыми растворами называют такие твердые фазы, в кото-
рых соотношения между компонентами могут изменяться. Сущест-
вуют три структурных типа твердых растворов: замещения, внед-
рения, вычитания.
В твердых растворах замещения, возникших на основе химиче-
ского элемента, атомы растворенного вещества замещают в кри-
сталлической решетке атомы растворителя, распределяясь среди
них хаотически (рис. 3.1).
ж<
ж<
а
®§Wo®
о2о5£с£о
оеееоео
•оеоеее ° <
оео»о ео ^~в
• О • О •
оеоеоео
6
О О О О
О О О 0*0 *о»о о-^
°о2о°оо
ГЛ гл гл гл • О • О • О • Вакант-
о о О О 0«0«0«0<Ж
О О О* ‘OOOZ позиции
О О О О ox<rio»o
д
Q-Fe •<
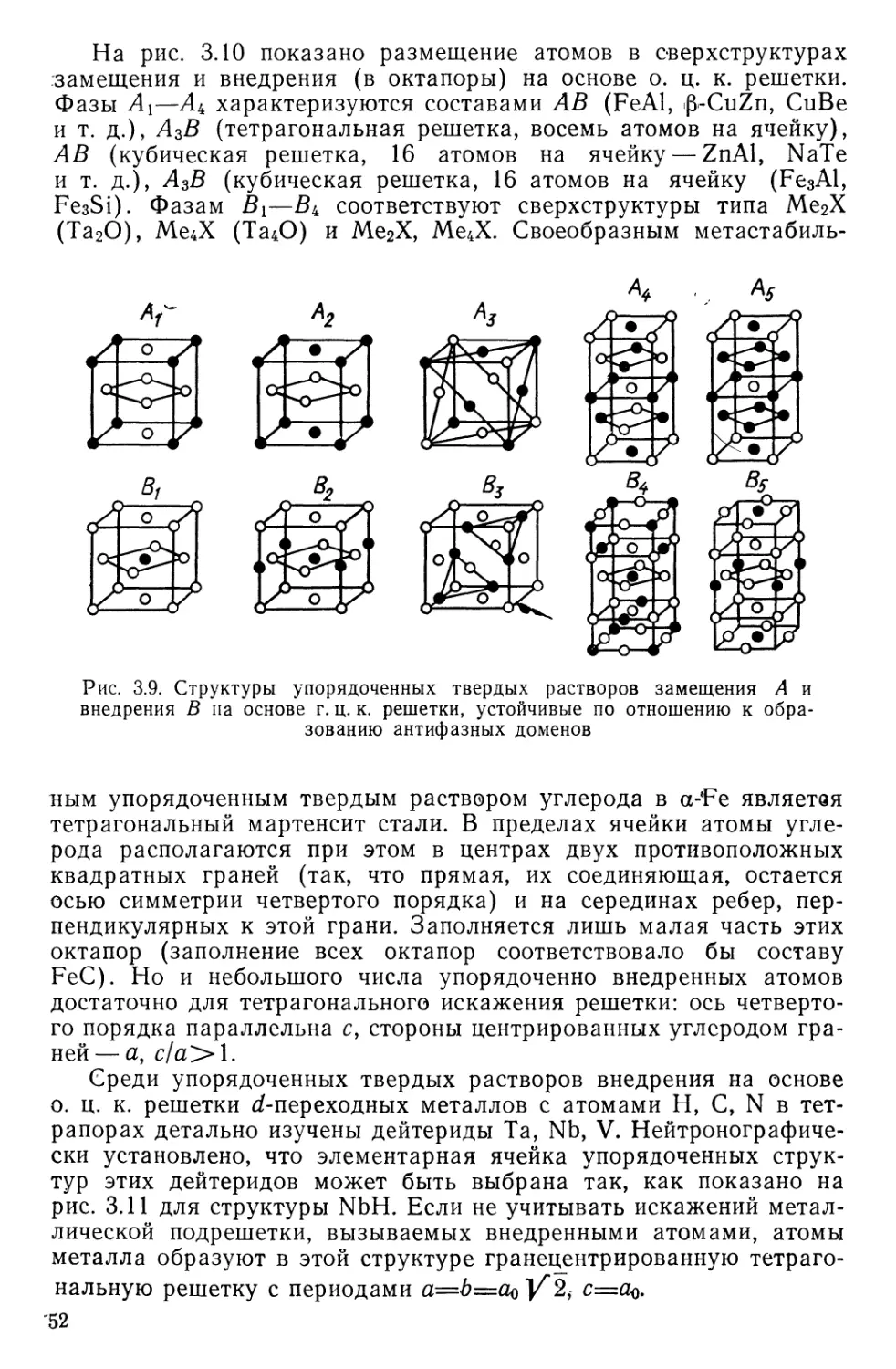
Рис. 3.1. Схемы расположения атомов в твердых растворах [плоскость (100)
в г. ц. к. решетке]:
а — металл-растворитель; б — твердый раствор замещения (Си—Zn); в — твердый раствор
замещения между двумя химическими соединениями (Ti, V)C; г — твердый раствор
внедрения (аустенит); д — твердый раствор вычитания (вюстит)
В твердых растворах между химическими соединениями АВ и
ВС, например NaCl — КС1 или TiC — TiN, построенных по типу
замещения, атомы (или ионы) компонентов размещаются в соот-
ветствии со схемой, приведенной на рис. 3.1,в.
В кристаллах твердых растворов внедрения атомы растворен-
ного элемента располагаются в межатомных промежутках крис-
3—523 33
таллической решетки (рис. 3.2). Положение внедрения атома (Н;
С, N, О, В) в межузлии устойчиво, если радиус внедренного атома
достаточно мал, чтобы не слишком смещать окружающие его ато-
мы металла (обычно переходного), и достаточно велик, чтобы
с ними соприкасаться.
Известны также и растворы внедрения на основе химических
соединений (например, как показал Е. С. Макаров, раствор никеля
в соединении NiSb).
В значительных количествах растворяются по типу внедрения
указанные металлоиды только в переходных металлах. Большая
часть других металлов образует с С, N, В, Н соединения ионного
типа («солеобразные» карбиды, нитриды и бориды).
Металлические атомы в твердом растворе внедрения образуют
кристаллическую решетку, характерную для металлов. Присутст-
вие внедренных атомов приводит к местным искажениям кристал-
лической структуры растворителя и увеличению среднего периода
решетки. Искажения, вносимые в матрицу атомами внедрения, ве-
лики из-за малых размеров пор, поэтому увеличение объема ячей-
ки при внедрении атомов металлоида значительно. В связи с этим,
а также из-за того, что непрерывный ряд твердых растворов внед-
рения запрещен топологически, предельная растворимость по отно-
шению к общему числу позиций внедрения обычно мала (но иногда
атомная доля металлоида достигает 0,25—0,30 (N в Ti и О в Zr),
0,5(Н в Pd) и даже 0,75 (Н в Се).
В г. ц. к. и о. ц. к. решетках существуют октаэдрические и тет-
раэдрические типы позиций, удобных для размещения внедренных
атомов (см. рис. 3.2) В г. ц. к. решетке октаэдрические позиции
образуют одну г. ц. к. подрешетку, а тетраэдрические—две г. ц. к.
подрешетки. В о. ц. к. решетке имеются три о. ц. к. подрешетки
октаэдрических позиций внедрения и шесть о. ц. к. подрешеток тет-
раэдрических пор.
В неупорядоченном состоянии с равной вероятностью заполня-
ются все позиции определенного сорта — октаэдрические или тет-
раэдрические. Конкретная конфигурация неметаллической подре-
шетки характеризуется тем, что одни поры заняты, а другие сво-
бодны. Поэтому раствор внедрения можно рассматривать как трех-
компонентную систему из металлических и внедренных атомов и
пустых пор.
Очень важно знать, какой сорт позиции внедрения (октаэдриче-
ские или тетраэдрические) заполняется в данной системе внедре-
ния. Было показано теоретически *, что атомы углерода и кисло-
рода в о. ц. к. решетке Fe, Та, Nb и V занимают октаэдрические
поры, водород в Та и Nb — тетраэдрические, а в V — те и другие.
Экспериментально определить тип занятых позиций внедрения
можно в большинстве случаев только с помощью методов нейтро-
нографии, так как рентгеноструктурный и даже электронографиче-
* Хачатурян А. Г., Шаталов Г. И. «Физика металлов и металловедение»,
1968, т. 25, с. 687.
34
ский методы часто непригодны из-за малой, по сравнению с атома-
ми металлической матрицы, рассеивающей способности атомов ме-
таллоидов.
Твердые растворы вычитания (растворы с дефектной решеткой)
образуются лишь на основе химических соединений. В таких твер-
дых растворах некоторые позиции атомов (ионов) второго недоста-
чу?
а
Рис. 3.2. Возможные позиции внедренных атомов в твердых раство-
рах внедрения:
а, б — тетрапоры и октапоры в г. ц. к. решетке; в, г — то же в гекс. п. у. ре-
шетке; д, е — то же в о. ц. к. решетке
3*
35
точного компонента оказываются пустыми (см. рис. 3.1,5). Так, на-
пример, в кристалле вюстита, являющемся твердым раствором
кислорода в FeO, все позиции решетки, в которых должны нахо-
диться кислородные ионы, заполнены, часть же позиций, в которых
должны расположиться ионы железа, вакантна.
Такие же «пустые места» в решетке имеют растворы Sb
в NbSb со структурой никель-арсенида, а также растворы S в CoS,
Se в CoSe и Те в СоТе и FeTe. Установлено, что дефектные решет-
ки образуются в сплавах Ni — Al, Со — Al, Си — Al.
В нитридах циркония (ZrN1+x) и гафния (HfN1+x) с избытком
азота имеются вакантные места в позициях, которые должны быть
заняты атомами Zr и Hf.
Замечательную дефектную структуру на основе решетки типа
NaCl имеют окислы титана, содержащие от 38 до 56 ат. % кислоро-
да. Здесь вакантными могут быть как позиции металлического ком-
понента, так и позиции, в идеальной решетке занимаемые ионами
кислорода. В кристаллах с максимальным содержанием титана
(ТЮо,б) все дефекты (20% общего числа позиций) находятся в кис-
лородной части решетки (не хватает 40% кислородных ионов).
С уменьшением содержания титана уменьшается и общее число
вакантных мест, причем они уже распределяются между обеими
частями решетки, кислордной и титановой, так что в окисле, отве-
чающем формуле TiO, и в кислородной, и в титановой части решет-
ки имеется по 15% вакантных мест. Наконец, у окисла, наиболее
богатого кислородом, имеется 20% вакантных мест в одной лишь
титановой части решетки.
Дефектные решетки с ионной связью характеризуются наличи-
ем металлических ионов разной валентности. Так, например, в кри-
сталлах вюстита при избытке кислорода имеются кроме двухва-
лентных также и трехвалентные ионы железа, в кристаллах окисла
титана с избытком титана кроме двухвалентных присутствуют и
одновалентные ионы титана, а при дефиците титана этот окисел
содержит двух- и трехвалентные ионы. Видимо, эта особенность
является следствием перекрытия 3d- и 4$-энергетических уровней
в атомах, присущего металлам переходных групп. Именно среди
химических соединений, в которых участвуют металлы переходных
групп, и встречаются чаще всего фазы с переменным числом ато-
мов в элементарной ячейке, к которым относятся твердые раство-
ры внедрения и вычитания.
Следует отметить, что независимо от типа металлического твер-
дого раствора все они имеют атомарный характер: в кристалличе-
ской решетке твердого раствора можно обнаружить лишь атомы
(или ионы), но не молекулы растворенного вещества. Например,
в решетке аустенита, который находится в равновесии с цементи-
том, внедрены атомы (ионы) углерода, а не молекулы РезС.
Поскольку атомы растворителя и растворенного элемента име-
ют различные радиусы, кристаллическая решетка твердого раство-
ра замещения будет искажена. Еще более резкие искажения возни-
36
кают при внедрении атомов и образовании пустых мест. Это под-
тверждается следующими важными фактами.
1. Интенсивность интерференционных линий (особенно с боль-
шими индексами) на рентгенограммах твердых растворов меньше,
чем на рентгенограмме металла-растворителя, а интенсивность
сплошного фона увеличена — эффект, аналогичный эффекту тепло-
вых колебаний атомов.
По данным В. И. Ивероновой, средние значения статических от-
клонений атомов от правильных положений в узлах решетки
в сплавах Ni— Си и Fe — Со в несколько раз превышают разности
атомных радиусов этих металлов. Следовательно, эти искажения
нельзя рассматривать только как результат несовпадения атомных
размеров — здесь несомненно проявляется и перераспределение
электронной плотности в решетке растворителя под действием ато-
мов растворенного металла.
2. Образование твердых растворов всегда сопровождается зна-
чительным увеличением электрического сопротивления и умень-
шением температурного коэффициента электросопротивления.
3. Твердые растворы обычно менее пластичны и более тверды,
чем чистые металлы.
По существу нельзя говорить о каком-то определенном значении
размера элементарной ячейки кристаллической решетки твердого
раствора: размер этот в разных участках решетки различен, поэто-
му можно говорить лишь о среднем значении периода (его и изме-
ряют при рентгенографическом исследовании). В дальнейшем та-
кую среднюю величину будем называть периодом кристаллической
решетки твердого раствора.
Определение периодов решетки твердого раствора в сочетании
с точным измерением плотности позволяет решить вопрос о том,
как построен изучаемый твердый раствор. Для этого необходимо
на основании экспериментальных данных определить общее число
атомов п, приходящихся на элементарную ячейку кристаллической
решетки твердого раствора. Если п совпадает с nQ (nQ — нормаль-
ное число атомов в ячейке данной кристаллической структуры), то,
значит, раствор построен по типу замещения; при п>п$— по типу
внедрения и при — по типу вычитания.
Во всех исследованных случаях внедряются при растворении
атомы меньших размеров, чем атомы растворителя (например,
атомы С в Fe, Н в Pd, Ni в химическом соединении NiSb). Объем
элементарной ячейки при образовании раствора внедрения всегда
возрастает.
По способу вычитания растворяется тот из компонентов хими-
ческого соединения, атомы (или ионы — в зависимости от типа хи-
мического соединения) которого имеют большие размеры (напри-
мер, металлоиды VI группы О, S, Se и Те в соединениях FeO, FeS,
FeSe, CoS, CoSe, CoTe, сурьма в NiSb, металлические элементы
в нитриде TiN и карбидах TiC, ТаС, ZrC, NbC), «дырки» образу-
37
ются в позициях, занятых ранее малыми атомами (Fe в FeO, Ni —
в NJSb, N — в TiN, С — в карбиде TiC и т. д.) *.
При образовании твердых растворов замещения, как правило,
растворение элементов с меньшим атомным радиусом, чем атом-
ный радиус растворителя, вызывает уменьшение периода решетки,
при растворении же элементов с большим атомным радиусом пери-
од решетки возрастает. На рис. 3.3 показано изменение периода
кристаллической решетки с концентрацией для нескольких двой-
ных систем, образующих непрерывные ряды твердых растворов.
Рис. 3.3. Периоды решетки твердых
растворов в бинарных сплавах
с медью (а) и железом (б)
Первоначальные исследования привели к заключению, что
в твердых растворах металлов с одинаковой структурой период
кристаллической решетки изменяется по линейному закону:
а=ах-\- (а2—ах) с, (3.1)
где и а2— периоды решетки растворителя и растворенного ком-
понента соответственно; с — концентрация твердого раствора
(атомная доля второго компонента).
Позднейшие исследования показали, что уравнение (3.1) (на-
зываемое законом Вегарда) является лишь первым, иногда весьма
грубым, приближением. На самом деле зависимость a=f(c) выра-
жается кривыми линиями или несколько провисающими, но без
минимума по сравнению с прямыми линиями или выпуклыми, но
без максимума (см. рис. 3.3,а).
* Исключение составляют рассмотренные выше окисел Ti и нитриды Zr, Hf,
Nb и Та с избытком неметалла по сравнению с формулой МеХ, где X — неме-
таллический компонент.
38
Лишь для сплавов Ti — Nb, Ti — Та, Ag — Au зависимость a=
выражается кривой с минимумом, а для сплавов 'Ni — y-Fe-—
с максимумом.
При растворении 10% Na в РЬ, имеющем меньший атомный
радиус, период решетки уменьшается с 4,94 до 4,92 А.
Известны случаи противоположной зависимости, например при
растворении Ni, атомный радиус которого равен 1,244 А, в W
с атомным радиусом 1,408 А период кристаллической решетки не
уменьшается, как следовало бы ожидать, а увеличивается.
У сплавов ia-Fe с Сг период решетки меняется по сложной кри-
вой, имеющей точку перегиба. Аналогичная кривая для сплавов
Сг с Ti имеет три точки перегиба.
Отклонения от закона Вегарда объясняли прежде всего упру-
гим взаимодействием компонентов, обусловленным различием их
коэффициентов сжимаемости xi и х2, и значением модуля сдвига
растворителя 6ь По Б. Я. Пинесу, величина поправки составляет
- а,, f g' ~ п. (3.2)
Приведенную на рис. 3.4 зависимость размеров атомов от кон-
центрации твердого раствора Авербах объяснил перераспределени-
ем электронов. Таким образом,
на период решетки твердого рас-
твора может влиять изменение
электронного состояния атомов,
обусловленное различием валент-
ности fi и f2 (или заряда ионов).
Для металлической системы
можно принять *, что атомные
объемы компонентов зависят от
среднего значения валентности
fcp=Cif 1+^2- При этом средний
атомный объем для твердого рас-
твора будет равен:
Wifi +
Clf 1 4" С2^2
Подсчитанные с помощью форму-
лы (3.3) периоды решетки твер-
дых растворов на основе А1, Си,
Ag и Au отличались от измерен-
ных не более чем на 0,004 А.
При образовании твердых
растворов с гексагональной или
тетрагональной кристаллической
решеткой изменяются не только
размеры, но и форма элементар-
(3.3)
^Т.р
Рис. 3.4. Размеры атомов Au и Ni
в твердых растворах, рассчитанные
по интенсивности диффузного рассея-
ния рентгеновских лучей; г — среднее
значение межатомного расстояния,
определяемое по периоду решетки;
Го, Ni и г0, au — атомные радиусы Ni
и Au
* Кузнецов Г. М., Барсуков А. Д. «Изв. вузов. Сер. Черная металлургия».
1969, № 5. с. 118.
39
ной ячейки. Например, в твердых растворах Си в Zn (трфаза)
с гекс. п. у. решеткой соотношение осей с/а изменяется от 1,86 для
Zn до 1,81 для сплава с 2% Си.
§ 3.2. ВЗАИМНАЯ РАСТВОРИМОСТЬ МЕТАЛЛОВ
До настоящего времени не удалось установить совокупность
условий, которые были бы не только необходимы, но и достаточны
для неограниченной взаимной растворимости двух металлов. На
основании имеющегося экспериментального материала были опре-
делены следующие необходимые для этого условия.
1. Непрерывные ряды твердых растворов по типу замещения
образуют металлы с одинаковой кристаллической решеткой. Если
сплавляемые металлы полиморфны, то неограниченная взаимная
растворимость может наблюдаться лишь между изоморфными мо-
дификациями (например, в системе Fe — Со между y-Fe и р-Со
с г. ц. к. решеткой).
2. Атомные радиусы и, следовательно, периоды кристалличе-
ской решетки сплавляемых элементов не должны различаться бо-
лее чем на 10—15%.
3. Электрохимические свойства (сродство к электрону) сплав-
ляемых элементов не должны сильно различаться, так как при
значительном различии электрохимических свойств проявляется
тенденция к образованию металлических соединений. Очевидно,
для выполнения этого условия сплавляемые элементы должны при-
надлежать к одной и той же группе периодической системы эле-
ментов или к смежным, родственным группам.
Необходимость выполнения первого условия очевидна. Для
разъяснения второго и третьего условий вспомним, что из двух кон-
курирующих между собой состояний конденсированной системы
устойчиво при заданной температуре то, которое обладает мень-
шей свободной энергией F—U—TS (U — внутренняя энергия; S —
энтропия). При высокой температуре устойчиво состояние с боль-
шей энтропией (S^^S7), температура равновесия TQ определяется
из условия F'—F"\
TQ= (U"— U') I ; (3.4)
она тем выше, чем больше тепловой эффект превращения.
Энтропия однофазного состояния — твердого раствора — S"
всегда выше, чем энтропия S' того же сплава, распавшегося на два
твердых раствора разной концентрации*. Поэтому если образова-
ние твердого раствора сопровождается выделением тепла (U'>
>U"), то при отсутствии склонности к возникновению химических
соединений или упорядоченных твердых растворов (см. с. 48) си-
стема должна представлять собой непрерывный ряд твердых рас-
творов, не распадающихся на два твердых раствора разной кон-
центрации при понижении температуры.
* В курсе физической химии дано понятие энтропии смешения (или конфигу-
рационной энтропии). Читателю предлагается самостоятельно вывести зависи-
мость конфигурационной энтропии твердого раствора замещения от состава (см.
задачу 3.3).
40
Разрывы непрерывности областей однофазных твердых раство-
ров наблюдаются тогда, когда при образовании твердого раствора
происходит поглощение тепла (т. е. при
Внутренняя энергия каждой фазы U может быть выражена
следующим образом:
U=UQ+jt (3.5)
где Uo — потенциальная энергия кристаллической решетки при тем-
пературе абсолютного нуля; /— энергия тепловых колебаний ато-
мов при заданной температуре.
Энергия тепловых колебаний в первом приближении не зависит
от состояния сплава, поэтому при анализе вопроса о равновесии
в сплавах ее можно не учитывать и вместо величины U исполь-
зовать Uq.
Учитывая взаимодействия между одними лишь ближайшими со-
седями в кристаллической решетке* и считая значения энергии та-
ких парных взаимодействий (иАА, ивв и иАВ) не зависящими от
концентрации, т. е. используя приближение регулярных растворов
для молярной потенциальной энергии раствора концентрации х
можно закисать следующее:
= -^)2 + ubb^ + 2u/bx(1 -*)]> (3-6)
где иАА и иВв — взаимная потенциальная энергия двух одноимен-
ных атомов А и В соответственно; иАВ— то же для разноименных
атомов А и В; z— координационное число; У — число Авогадро.
Эта величина отличается от энергии обособившихся кристаллов
компонентов Л и В на энергию смешения, равную
—(uaa-\-Ubb) /2. (3.7)
Знак величины определяет устойчивость твердого раствора.
Если энергия смешения отрицательна \иАВ< у (^аа+^вв)] и,
следовательно, взаимодействие разнородных атомов сильнее, чем
взаимодействие атомов одного и того же элемента, то компоненты
А и В образуют непрерывный ряд твердых растворов**. Если же
энергия смешения положительна Arz>0 [«ав> у (^аа+^вв)], то
при определенной температуре происходит распад на два твердых
раствора разной концентрации в соответствии с изменениями сво-
бодной энергии*** от состава и температуры (рис. 3.5,а). Схема
соответствующей диаграммы состояния для такого случая (ниже
* На самом, деле взаимодействуют и атомы, находящиеся на большом рас-
стоянии, но силы взаимодействия резко убывают с увеличением межатомных
расстояний.
** Как будет показано ниже, в таких сплавах при понижении температуры
происходит особый процесс — образование упорядоченного твердого раствора.
*** Вывод выражения для свободной энергии двухкомпонентного твердого
раствора замещения предлагается читателю выполнить самостоятельно (см. за-
дачу 3.4).
41
0 .Ctf 0,5 Cj3 1,0 0 Cg 0,5 Ср 1,0
А Концентрация, % В А Концентрация,°/<> В
Рис. 3.5. Диаграммы равновесия (схемы) при iAu>0:
а — зависимость свободной энергии от состава твердого раствора при раз-
ных значениях параметра температуры p=fc77(zAu); б — кривая распада
твердого раствора, построенная в соответствии с расчетами для свободней
энергии; в — несимметричная кривая распада при резкой разнице темпе-
ратур плавления компонентов; г — появление эвтектики в системе с боль-
шой энергией смешения (Azz>0)
линии солидуса) приведена на рис. 3.5,6. Из жидкой фазы сплавы
любой концентрации кристаллизуются как однофазные в виде твер-
дых растворов, распадающихся с понижением температуры. При
приближении к абсолютному нулю взаимная растворимость компо-
нентов стремится к нулю. Чем больше положительная энергия сме-
шения, тем выше температура распада.
42
Если энергия смешения достаточно велика, температура распа-
да сплавов, лежащих в средней части диаграммы, может оказаться
выше температуры их плавления. Такие сплавы и из жидкой фазы
кристаллизуются двухфазными (см. рис. 3.5,а).
Если искажения кристаллической решетки твердого раствора,
вызываемые различием атомных радиусов компонентов А и В, не
учитываются, то кривая расслоения должна быть симметричной и
иметь максимум при с=0,5 (см. рис. 3.5,6). Температура этого ма-
ксимума
Тмакс=гАи/2&*. (3.8)
Если энергия смешения А«>0, то взаимная растворимость ком-
понентов при абсолютном нуле равна нулю, поскольку по теореме
Нернста при Т—>0 5—>0.
Изменение потенциальной энергии при образовании твердого
раствора всегда больше энергии смешения, вычисленной по фор-
муле (3.7), так как из-за неравенства атомных радиусов сплавляе-
мых металлов в твердом растворе возникают искажения кристал-
лической решетки.
При равенстве упругих констант компонентов уравнение кривой
распада сохраняет свой вид, но вместо надо взять сумму «хими-
ческой» энергии смешения Ди и упругой энергии 8 (всегда поло-
жительной), т. е. величину
киг = Дн-|- е, (3.9)
так что
Тмакс=г(Д«-Н)/2£. (3.10)
В соответствии с этим вся кривая распада, сохраняя симмет-
ричную форму, поднимается кверху. Если упругая энергия доста-
точно велика, кривая распада пересекается с линией солидуса при
малых и даже отрицательных значениях энергии смешения.
При сплавлении элементов, принадлежащих к одной и той же
группе периодической системы, химическая составляющая энергии
смешения должна быть мала. Очевидно, в этих случаях взаимная
растворимость металлов определяется упругой энергией искажений
решетки твердого раствора 8, зависящей от трех величин: относи-
тельной разности атомных радиусов &R/R, коэффициента сжимае-
мости и модуля сдвига. Очевидно, при сопоставлении сплавов ме-
таллов с одними и теми же упругими константами основное значе-
ние имеет относительное различие атомных радиусов — чем сильнее
различаются по размерам атомы, тем больше упругая энергия
искажений решетки, чем выше идет кривая распада.
Обычно упругие константы сплавляемых элементов бывают раз-
личны. При этом, как было показано Б. Я. Пинесом, кривая рас-
пада должна быть несимметричной: при равных концентрациях
’твердого раствора фаза, основой которой является компонент, име-
ющий большую сжимаемость, будет обладать меньшей упругой
См. также задачи 3.4 и 3.5.
43
энергией по сравнению с фазой, основной компонент которой имеет
меньшую сжимаемость. Поэтому температурный максимум на кри-
вой распада смещается в сторону того металла, сжимаемость кото-
рого меньше, и растворимость в этом компоненте при понижении
температуры остается ниже, чем во втором компоненте. Это можно
иллюстрировать схемой рис. 3.5,в и кривой распада сплавов Au —
Ni (см. рис. 3.6,а), у Ni коэффициент сжимаемости при комнатной
температуре и небольшом давлении равен 0,53-10-6 см2/кг, у Au —
0,59-10~6 см2/кг, в этом элементе растворимость больше.
Металлы с высокими модулями упругости являются одновре-
менно и тугоплавкими металлами. Поэтому максимум на кривой
распада должен быть смещен в сторону более тугоплавкого компо-
Ni, мае. °/о
Aq Си, ат. °/<> Си
пента (75 ат.% Ni в системе Au—Ni). При заданной температуре
растворимость больше в том металле, температура плавления ко-
торого ниже.
Разрывы сплошности области твердых растворов в сплавах
с меньшей сжимаемостью наблюдают при таких различиях в атом-
ных радиусах, при которых сплавы с большей сжимаемостью пред-
ставляют собой непрерывный ряд твердых растворов.
Максимальная разница атомных радиусов при неограниченной
взаимной растворимости у элементов V и VI групп (As — Sb,
и Se — Те, б/?//?=0,17): эти элементы легкоплавки и об-
ладают большой сжимаемостью.4
Если сплавляемые элементы принадлежат к разным группам
периодической системы, взаимодействие между разнородными ато-
мами будет резко отличаться от взаимодействия между атомами
одного и того же элемента; энергия смешения велика и взаимная
растворимость ограниченна. В случае отрицательной энергии сме-
шения при этом возникают промежуточные фазы, в случае поло-
жительной— смесь твердых растворов компонентов А в В и В в А
(при этом возможно даже расслоение в жидком состоянии).
Разительные примеры, показывающие недостаточность перечис-
ленных выше условий неограниченной взаимной растворимости ме-
Содержание Аи,ат.Ч>
Рис. 3.6. Диаграммы равновесия сплавов Au—Ni
(а), Си—Ag (б) и Си—Au (в)
45
таллов, можно найти среди сплавов металлов железной группы
с Си, Ag и Au.
Медь, образующая непрерывный ряд твердых растворов с Ni,
не полностью смешивается даже в жидком состоянии с Fe и Со,
имеющими те же атомные радиусы, что и Ni, и принадлежащими
к той же группе периодической системы. Золото образует непре-
рывный ряд твердых растворов с Ni и ограниченные области твер-
дых растворов с Со, атомный радиус которого ближе к атомному
радиусу Au. Серебро не смешивается с Со и Ni ни в твердом, ни
в жидком состоянии.
Еще менее ясен вопрос о растворимости металлов в системах
с ограниченной растворимостью. В большинстве случаев второе и
третье условия образования непрерывных рядов твердых растворов
сохраняют силу и при ограниченной растворимости: чем благопри-
ятнее объемный и электрохимический факторы, тем выше бывает
взаимная растворимость металлов.
Число нарушений этой закономерности велико. Достаточно ука-
зать, например, что Fe, растворяющее в твердом состоянии не более
10% близкой к нему в периодической системе Си, атомный радиус
которой отличается от атомного радиуса Fe менее чем на 1%, рас-
творяет 50 ат. % А1.
Обычно металл с большим числом валентных электронов слабее
растворяет в себе металл с малым числом валентных электронов,
чем сам в нем растворяется. Так, например, Ag растворяет до 30%
Mg, a Mg — до 3,9 Ag; Си — до 6,5% Mg, но Mg — не более 0,01%
Си и т. д.
о All • Си
в
Рис. 3.7. Кристаллические структуры упорядоченных твердых растворов в си-
стеме Си—Аи: сверхструктуры СизАи (а); СиАи—I (сплошными линиями
обозначены ребра г. ц. тетрагональных ячеек, пунктиром — о. ц. тетрагональ-
ной ячейки (б) и CuAu—II (в)
46
Особо стоит вопрос о растворимости элементов II—V групп
периодической системы в металлах группы меди. Юм-Розери от-
мечает, что максимальная растворимость в этих случаях приблизи-
тельно обратно пропорциональна валентности растворяемого эле-
мента р, уменьшенной на единицу так, что произведение СмаксХ
Х(р-1)=40% (табл. 3.1).
Заметно уменьшенная растворимость наблюдается только при
растворении металлов, атомные радиусы которых сильно отлича-
ются от атомного радиуса растворителя (Sn и As в сплавах
с медью).
Таблица 3.1
Растворимость элементов II—V групп в Си
Растворимый элемент Номер груп- пы p ^макс» ат' O//° C (p-1) Максимальная электронная концентрация б R/R-100%
Zn 2 38,4 38,1 1,38 +8
Al 3 20,4 40,8 1,41 + 12
Ga 3 19,3 38,6 1,39 +6
Si 4 14,0 42,0 1,42 +5
Ge 4 11,4 34,2 1,34 +9
Sn 4 9,3 27,9 1,28 4-20
As 5 6,8 27,2 1,27 +15
Количественная теория растворимости многовалентных элемен-
тов в металлах группы меди основана на представлении о заполне-
нии первой зоны Бриллюэна для электронов в г. ц. к. структуре
кристалла. В соответствии с принципом Паули энергетические зоны
в кристаллах различных структур заполняются при наличии опре-
деленного количества валентных электронов, приходящихся на
каждую элементарную ячейку (а при неизменности числа атомов
в ячейке — на каждый атом).
Сопоставим две различные кристаллические структуры А и В,
отличающиеся друг от друга тем, что в кристалле со структурой А
«емкость» первой зоны меньше, чем в кристалле В. Соответственно
у кристалла А ниже критическое значение импульса (Ркрл<Ркрв)
и ниже расположена полоса «запрещенных» энергий (см. рис. 1.8).
Если концентрация электронного газа такова, что первые зоны
двух конкурирующих между собой структур А и В не заполнены,
то устойчива структура А, у которой первая зона заполняется рань-
ше, чем у В. После же заполнения первой зоны кристаллической
решетки А делается более устойчивой структура В.
Условия заполнения первой зоны дозволенных энергий выведе-
ны в гл. 1 [см. формулу (1.33)].
В случае г. ц. к. решетки при увеличении концентрации элек-
тронов раньше всего поверхность Ферми касается границ первой
зоны Бриллюэна в-направлениях <111> (см. рис. 1.6); это долж-
47
но быть при концентрации электронов
сэл — 4^з'33/2 = L36 электрон/атом.
В случае о. ц. к. решетки границы первой зоны Бриллюэна об-
разуют плоскости hkl (110), поэтому
с9л — -23/2 = 1,48 электрон/атом.
В соответствии с этим предельная растворимость многовалент-
ных металлов в одновалентных Си и Ag, имеющих г. ц. к. решетку,
должна соответствовать электронной концентрации 1,36 — после
перехода через эту концентрацию должна появиться новая фаЗа
с более широкой первой зоной дозволенных энергий (например,
о. ц. к.). Эта теория хорошо согласуется с опытными данными, при-
веденными в табл. 3.1.
§ 3.3. УПОРЯДОЧЕННЫЕ ТВЕРДЫЕ РАСТВОРЫ (СВЕРХСТРУКТУРЫ)
В 1914 г., изучая золотомедные сплавы, образующие при высо-
кой температуре непрерывный ряд твердых растворов, Н. С. Курна-
ков, С. Ф. Жемчужный и А. М. Заседателев заметили, что отжиг
закаленных сплавов, состав которых отвечает формулам Cu3Au,
CuAu, приводит к уменьшению их электрического сопротивления
в два-три раза. В соответствии с этими данными Н. С. Курнаков
с сотр. отметили на диаграмме состояния Au — Си области суще-
ствования новых фаз (см. рис. 3.6,в).
Известные в то время методы физико-химического анализа
сплавов не позволяли установить природу превращений, обнару-
женных в золотомедных сплавах. Решающим в данном случае ока-
залось применение структурного рентгеноанализа. Появление так
называемых сверхструктурных линий можно объяснить, предполо-
жив, что отжиг твердых растворов, по составу отвечающих форму-
лам Cu3Au и CuAu, приводит к упорядоченному размещению ато-
мов кристаллической решетки.
Как известно, г. ц. к. решетку можно представить как совокуп-
ность четырех простых решеток, вставленных одна в другую. В не-
упорядоченном твердом растворе, по составу отвечающем)/ форму-
ле Cu3Au, атомы Au равномерно распределены между всеми че-
тырьмя простыми решетками, так что в каждой содержится по
25% атомов. При идеальном упорядочении одна из простых реше-
ток целиком состоит из атомов Au, а три остальные — из атомов
Си. При упорядочении сплава состава CuAu атомы Au занимают
решетки I и II, а атомы Си — решетки III и IV, так что общая
решетка становится слоистой. При упорядочении фаза Cu3Au со-
храняет кубическую решетку (рис. 3.7,а), упорядоченная фаза
CuAu имеет тетрагональную решетку с несколько укороченной вер-
тикальной осью с, так что с/а=0,935. Это изменение формы эле-
ментарной ячейки легко объяснить: в то время как в горизонталь-
48
ных ее гранях «сидят» атомы Au, в центрах вертикальных граней
«сидят» меньшие по размеру (на 12%) атомы Си (см. 3.7,6).
Тетрагональную структуру имеют сплавы, содержащие от 47
до 53 ат. % Au, после отжига при температуре 200—400°С. Те же
сплавы, закаленные после отжига при температурах 410—420°С,
а также отожженные при температуре 200—400°С сплавы с 36—
47 и 53—65% Au, обладают более сложной ромбической решеткой
(см. рис. 3.7,в). Элементарная ячейка этой решетки состоит из
десяти расположенных вдоль одной прямой г. ц. к. ячеек и содер-
жит 20 атомов Си и 20 Au. Такое взаимное расположение атомов
можно представить, если в тетрагональной решетке CuAu произво-
дить сдвиги вдоль направления [010] на расстояние а/2 через каж-
дые пять ячеек.
В последние годы были открыты подобные «длиннопериодные»
сверхструктуры и в других системах. Их образование связывают
с энергией электронного газа и изменением конфигурации зон
Бриллюэна.
При повышении температуры упорядоченные твердые растворы
превращаются в неупорядоченные, например в золотомедных спла-
вах (см. диаграмму состояния на рис. 3.6,в), или распадаются на
две фазы (например, Co3W).
Температуру перехода упорядоченного твердого раствора в не-
упорядоченное состояние называют «точкой Кюри упорядочения»
или точкой Курнакова.
У твердых растворов, обладающих очень высокой начальной магнитной про-
ницаемостью (например, у сплава альсифер, содержащего 85% Fe, 9,5% Si и
5,5% Al), при переходе в упорядоченное состояние начальная магнитная прони-
цаемость уменьшается в несколько раз. Неупорядоченные сплавы Ni3Mn парамаг-
нитны, упорядоченные (со структурой типа Cu3Au) — ферромагнитны. Коэрцитив-
ная сила в них мала при низких температурах и имеет максимум при температу-
ре перехода в неупорядоченное состояние. Не содержащий ферромагнитных ком-
понентов сплав Cu^AlMn парамагнитен при неупорядоченном расположении ато-
мов, в результате отжига, приводящего к образованию сверхструктуры, он при-
обретает ферромагнитные свойства.
При нагревании упорядоченного твердого раствора увеличива-
ется его теплоемкость (рис. 3.8), так же как растет при приближе-
нии к точке Кюри теплоемкость ферромагнитных металлов. Уве-
личение теплоемкости упорядоченного твердого раствора при
приближении к температуре превращения обусловлено частичным
Уничтожением порядка в расположении атомов. Однако есть раз-
ница в поведении сплавов возле самой критической температуры.
В сплаве CuZn (0-латунь) имеется скачок в изменении теплоемко-
сти (или разрыв непрерывности в изменении второй производной
от внутренней энергии по температуре), что характерно для фазо-
вого перехода второго рода.
Теплоемкость сплава Cu3Au в точке упорядочения обращается
в бесконечность, так как этот сплав в отличие от 0-латуни имеет
возле точки полного разупорядочения конечную степень порядка
(см. стр. 55) и конечную (около 2 кал/моль) теплоту превращения.
Это характерно для фазового перехода первого рода.
4—523 49
Структура упорядоченных твердых растворов замещения и внед-
рения. Упорядочение твердых растворов всегда сопровождается
уменьшением числа элементов симметрии. Л. Д. Ландау и
Е. М. Лифшиц* установили жесткие условия, которым должна
удовлетворять симметрия неупорядоченного и устойчивого в одно-
родном состоянии упорядоченного твердого раствора замещения,
чтобы превращение могло идти как фазовый переход второго рода.
а
Рис. 3.8. Теплоемкость упорядоченно-
го твердого раствора Cu3Au в зави-
симости от температуры при нагреве
(падение теплоемкости происходит
в точке Курнакова):
а — СизАи; б — CuZn
Не приводя этих условий, укажем, что из числа фаз, которые
можно представить при упорядочении твердых растворов с г. ц. к.
решеткой, по типу фазового перехода второго рода могут возни-
кать лишь фазы со структурами CuPt (ромбоэдрическая решетка)
* Ландау Л. Д. «Sow. Phys.», 1937, v. 11, N 26, p. 545.
Лифшиц E. M. «Журн. эксперим. и теор. физ.», 1941, т. 11, с. 255, 269.
50
и CuPt7. Среди сверхструктур, возникающих в результате фазового
перехода второго рода твердых растворов с о. ц. к. решеткой, ре-
шетки типа CsCl (FeAl, р-фаза CuZn) и Fe3Al.
Л. Д. Ландау и Е. М. Лифшиц не рассматривали условия упорядочения,
протекающего как превращение первого рода. Однако Е. М. Лифшиц сформули-
ровал критерий для всех возможных сверхструктур, в которых возникающие
в начале процесса антифазные домены не обладают термодинамической устой-
чивостью и к концу процесса упорядочения фаза делается однородной. Этот кри-
терий сохранил свое значение и в развитой А. Г. Хачатуряном общей для упо-
рядоченных твердых растворов (как замещения, так и внедрения) теории, осно-
ванной на представлениях современной статистической физики. Для простейшего
случая, когда атомы размещены в узлах одной только решетки Бравэ, вероят-
ность нахождения атома в позиции R определяется уравнением
n(/?) = <[eXp[-^-+y]-£E^lLn(/?,) +1}"*, (3.11>
где Цг — неопределенный множитель Лагранжа, играющий роль химического по-
тенциала; /?, R'— радиус-векторы, определяющие положения узлов решетки
Бравэ. u(R—R') —потенциал парного взаимодействия атомов в позициях R и /?'.
А. Г. Хачатурян решил уравнение (3.11) методом статических концентра-
ционных волн для самого общего случая взаимодействия атома с любым числом
окружающих его координационных сфер. Это особенно существенно для сплавов
переходных металлов, характеризующихся взаимодействием с далекими коорди-
национными сферами, и для растворов и фаз внедрения на их основе.
Решение уравнения (3.11) неоднозначно. Очень существенно, что каждому
решению соответствует как сверхструктура упорядоченного твердого раствора
замещения, так и выводимая из нее с помощью простой процедуры структура
фазы внедрения. Атомы внедрения располагаются в октапорах металлической
подрешетки, причем если состав упорядоченного твердого раствора замещения
описывается формулой ЛтВп, то соответствующая фаза внедрения имеет состав.
Мет+пХщ или Mem+nXn Атомы неметалла X и незаполненные октапоры
являются компонентами упорядоченной неметаллической подрешетки, и соотно-
шение их концентраций совпадает с соотношением металлов А и В в прототи-
пе — упорядоченном растворе замещения.
Было показано, что общее число сверхструктур, устойчивых
по отношению к образованию антифазных доменов, невелико. Для
исходной г. ц. к. решетки Бравэ возможны семь сверхструктур за-
мещения А и семь упорядоченных растворов внедрения В. Пять
таких структур приведено на рис. 3.9. Сверхструктуры Ai и В[
соответствуют фазам типа Cu3Au (кубическая решетка, четыре
атома в ячейке) и Ме4Х (или антиизоморфной Ме4Х3). Фазам А2 и
В2 соответствуют соединения типа CuAu—I и Ме2Х (тетрагональ-
ная решетка), А3 и В3 — это CuPt и Ме2Х (ромбоэдрическая ре-
шетка), соединения типа А^ и В4, как и В3, пока не обнаружены;
А5 и В5 — это Ni3V (тетрагональная решетка, восемь атомов на
ячейку) и Ме4Х (или антиизоморфная Ме4Х3). Большинство рас-
считанных сверхструктур внедрения обнаружено экспериментально:
Bi —Fe4N, Nb4C3, Та4С3; В2— (Ni, Fe)2N; В5 — Ni4N, Nb4N3. Кроме
изображенных на рис. 3.9 сверхструктур решение уравнения (3.11)
дает еще две сложные фазы кубической сингонии с удвоенным пе-
риодом решетки и составами А7В (PtCu7), Ме8Х (Fe8N) и Ме»Х7
(V8C7), а также фазу АВ и Ме2Х (структура с 32 атомами металла
и 16 атомами неметалла — фазы Ti2C, Zr2C и, по-видимому, Hf2C).
4* 51
На рис. 3.10 показано размещение атомов в сверхструктурах
замещения и внедрения (в октапоры) на основе о. ц. к. решетки.
Фазы Л!—Л4 характеризуются составами АВ (FeAl, p-CuZn, CuBe
и т. д.), А3В (тетрагональная решетка, восемь атомов на ячейку),
АВ (кубическая решетка, 16 атомов на ячейку — ZnAl, NaTe
и т. д.), А3В (кубическая решетка, 16 атомов на ячейку (Fe3Al,
Fe3Si). Фазам В{—В± соответствуют сверхструктуры типа Ме2Х
(Та2О), Ме4Х (Та4О) и Ме2Х, Ме4Х. Своеобразным метастабиль-
Рис. 3.9. Структуры упорядоченных твердых растворов замещения А и
внедрения В на основе г. ц. к. решетки, устойчивые по отношению к обра-
зованию антифазных доменов
ным упорядоченным твердым раствором углерода в a-'Fe являетея
тетрагональный мартенсит стали. В пределах ячейки атомы угле-
рода располагаются при этом в центрах двух противоположных
квадратных граней (так, что прямая, их соединяющая, остается
осью симметрии четвертого порядка) и на серединах ребер, пер-
пендикулярных к этой грани. Заполняется лишь малая часть этих
октапор (заполнение всех октапор соответствовало бы составу
FeC). Но и небольшого числа упорядоченно внедренных атомов
достаточно для тетрагонального искажения решетки: ось четверто-
го порядка параллельна с, стороны центрированных углеродом гра-
ней— a, da>\.
Среди упорядоченных твердых растворов внедрения на основе
о. ц. к. решетки d-переходных металлов с атомами Н, С, N в тет-
рапорах детально изучены дейтериды Та, Nb, V. Нейтронографиче-
ски установлено, что элементарная ячейка упорядоченных струк-
тур этих дейтеридов может быть выбрана так, как показано на
рис. 3.11 для структуры NbH. Если не учитывать искажений метал-
лической подрешетки, вызываемых внедренными атомами, атомы
металла образуют в этой структуре гранецентрированную тетраго-
нальную решетку с периодами a=b=aQ 2, с=а$.
'52
Атомы дейтерия занимают часть тетраэдрических позиций внед-
рения, указанных на рис. 3.11 (нейтронографически обычно иссле-
дуют дейтериды, а не гидриды).
В сплаве с концентрацией D, меньшей эквиатомной, например
NbD0,75, при некоторой температуре происходит упорядочение внед-
ренных атомов по типу МеХ. Дополнительные «пустые» межузлия,
присутствующие из-за того, что не выполняется соотношение NbD,
размещены хаотически, т. е. тип решетки (и характер нейтроно-
граммы) остается тем же, что и у NbD.
При дальнейшем понижении температуры (до 100 К и ниже)
размещение этих пустых межузлий упорядочивается, элементарная
Рис. 3.10. Структуры упорядоченных твердых растворов замещения А и
внедрения В на основе о. ц. к. решетки (внедрение в октапоры)
Рис. 3.11. Структура гидрида (дейтерида) Nb (Та) МеХ:
а — исходная о. ц. к. ячейка с центрами октапор и тетрапор на грани; б —
тетрагональная ячейка сверхструктуры; в — проекция ячейки сверхструктуры
на грань (001)
53
ячейка увеличивается и на нейтронограмме появляются новые
сверхструктурные рефлексы. Таким образом, упорядочение в дан-
ном случае идет ступенчато. Хорошо известным аналогом такого
процесса в растворах замещения является последовательное упо-
рядочение в Fe3Al.
Общая теория, позволяющая предсказать структуры упорядо-
ченных твердых растворов замещения и внедрения на основе
гекс. п. у. решетки, не построена. Экспериментально установлено
несколько типов таких упорядоченных фаз. Например, при упоря-
Рис. 3.12. Структуры упорядоченных твердых растворов Mg3Cd (а) и
NbN (б)
дочении твердых растворов замещения характерны две сверхструк-
туры, возникающие в сплавах MgCd и Mg3Cd (или MgCd3). У обе-
их период с тот же, что и неупорядоченного твердого раствора
(если пренебречь влиянием различия атомных радиусов). У упоря-
доченной фазы Mg3Cd (Ni3Sn, Co3W, MgCd3) гексагональная ре-
шетка с удвоенным (по сравнению с неупорядоченным твердым
раствором) периодом а (рис. 3.12). В упорядоченном твердом рас-
творе MgCd чередуются плотноупакованные слои, построенные из
атомов Mg или Cd. Неравенство соответствующих атомных радиу-
сов приводит к тому, что шестиугольники, из которых построены
плотноупакованные слои, перестают быть равносторонними. Решет-
ку можно описать как ромбическую а —а0 ]/"2, c~cQ, где aQ
и с0 — периоды решетки неупорядоченного твердого раствора.
Стехиометрический состав почти всех известных упорядоченных
растворов внедрения на основе гекс. п. у. подрешетки переходного
металла — Ме2Х. Внедренные в октапорах атомы неметалла С,
N образуют частично заполненную простую гексагональную подре-
шетку с периодом с0/2 (где с0 — период гекс. п. у. металлической
подрешетки).
В упорядоченном NbN (рис. 3.12,6) атомами азота заполнены
все октапоры — структура антиизоморфна широко распространен-
ным фазам типа NiAs.
54
Термодинамика процесса упорядочения *. Пусть рассматривае-
мый сплав состоит из металлов А и В, Полностью неупорядоченное
состояние характеризуется тем, что любой узел решетки может
быть занят этими атомами с вероятностью, пропорциональной их
концентрациям сА и св> а полностью упорядоченное состояние —
тем, что имеются две подрешетки, образуемые А и В. Узлы под-
решетки А обозначим а, а подрешетки В — Ь.
Рассмотрим какое-то промежуточное между этими двумя со-
стояние. Пусть ра — вероятность того, что А находится в узле а,
a wa — вероятность того, что А находится в узле Ь. Соответственно
ръ и Wb — вероятности того, что В находится в узле b или а.
Если сплав полностью упорядочен, то ра=р6=1, a pb—wb. При
полном беспорядке pa==wa—ca, Рь = ^ь = сь.
Введем понятие степени дальнего порядка в расположении ато-
мов
о=(ра—С а) 1(1—С а) • (3.12)
Из такого определения следует, что для полного беспорядка
ра=сА, рь=св, о=0, для полного порядка ауа = ^& = 0, о=1.
Пусть произошло малое увеличение степени дальнего порядка
на величину do. При этом некоторое число атомов А перешло из
узлов b в узлы а. В результате относительное число атомов Л,
находящихся в узлах а, возросло на dpA. Число атомов, перешед-
ших в узлы а, равно NAdpa, так как cA=NA]N, где N—общее
число атомов в сплаве, то NAdpA=cANdpA,
Однако da=dpa/(l—cA)=:dpalcB, т. е. dpA=cBdo. Следова-
тельно,
cANdpa=cAcBNdo. (3.13)
Дальнейшая задача состоит в том, чтобы получить выражение сво-
бодной энергии
F=U—TS (3.14)
в зависимости от о. Для этого следует вычислить U=U(о) и S=
=5(<т).
Вычисление [/=(7(о). Изменение характера расположения
атомов в решетке связано с последовательными перескоками ато-
мов из одного узла решетки в другой. Если перескоки не изменяют
общего характера расположения атомов, то, следовательно, не из-
меняется и состояние сплава, а тем самым и его энергия U. Если
же в результате перескоков все большее число атомов А оказыва-
ется в положении а, а атомов В — в положении &, то изменяется
общий характер расположения атомов, а тем самым и состояние
сплава. Возникающее при каждой температуре равновесное значе-
ние от по отношению к предыдущему значению приводит к изме-
нению полной энергии сплава, так как при переходе атома из
неправильного (чужого) положения в правильное изменяется его по-
* Здесь рассмотрена теория образования дальнего порядка по типу превра-
щения второго рода, разработанная в Англии Брэггом и Вильямсом и независи-
мо от них В. С. Горским.
55
тенциальная энергия. При малых сг, когда относительная доля пра-
вильно занятых мест невелика, изменение потенциальной энергии
атома не зависит от положения других атомов. С ростом <у начи-
нает сказываться характер расположения всех атомов сплава.
Пусть при о=1, т. е. при полном упорядочении, изменение энергии
перескочившего атома равно Uo*.
Таким образом, при изменении о от 0 до 1 энергия растет от О
до Uq. Естественно поэтому в первом приближении допустить, что
u=uoo. (3.15)
С этим предположением найдем значение U в выражении для
свободной энергии. Для этого используем выражение (3.13), пра-
вую часть которого умножим на обе части равенства (3.15). По-
лучим
ucAcBNd(3——иоСАСвМв(1о. (3.16)
Знак минус показывает, что при увеличении сг(д!а>0) энергия
сплава уменьшается. Интегрируя обе части этого равенства, полу-
чаем
= + (3.17)
где с — постоянная интегрирования, которую можно определить из
условия, что при о=1 дальнейшего изменения U происходить не
будет, т. е. dU (о)3=1 =0. Окончательно получим
t/(o)=4-^u0(l-a2). (3.18)
Вычисление S(o). Рассматриваемое состояние сплава ха-
рактеризуется тем, что в нем NA атомов находятся: раМл в своих
и waNA в чужих узлах; NB: рьМв атомов находятся в своих и
WbNB — в чужих узлах.
Вероятность реализации такого состояния пропорциональна про-
изведению чисел способов размещения NA и NB атомов по своим
и чужим узлам (Ра и Рь), но
Pa=NA4(PaNA)l(WaNA)l;
Pb=Nb4 (PbNB) l(wbNB)! (3.19)
Подставив эти выражения в формулу энтропии и, учитывая, что
х\=х In х—х, получим
S = k In Р = — kN [сА (ра In ра + wa In wa) + св (р In pb + оЦп W(,)].
(3.20)
Преобразуем полученное выражение таким образом, чтобы по-
лучить S(o). Для простоты рассмотрим сплав эквиатомного соста-
* Читателю предлагается самостоятельно вывести связь изменения энергии
Uq с энергией смешения (uQ^2zAu).
56
ва, т. е. Са=Св=*/2. В этом случае ст=2ра—1 = 1—2wa=2pb—1 =
= 1—2wb и
Pa = Pt> = 4~(1 +0); ®а=“’б = (1 — °)/2- (3.21)
В окончательном виде имеем
5(о) = --^ЛЛ/[(1 4-а)1п(1+а) + (1 — о)1п(1 -а)-21п2]. (3.22)
Рассмотрим предельные случаи. Пусть <т=1. Тогда S (1) =0, т. е.
полное упорядочение можно осуществить одним способом.
ПуСТЬ 0=0. Тогда *$(0)=*5цеупор.тв.раств*
Вычисление F=F(g). Подставив найденные выражения
U(о) и S(o) в (3.14), получим F(o)=t/(o) — TS(o). Для каждой
температуры сплава имеется равновесное значение о, определяю-
щее минимум свободной энергии сплава.
Определение температурной зависимости сг.
Найдем зависимость о от температуры из условия минимума сво-
бодной энергии:
dF (o)/do = 0
или
-2«ec + -^^T[ln(l+a)-ln(l -а)]=0.
Отсюда
1п (т~;)=и»о/2^т- (3.23)
Запишем это выражение в виде (1+ст)/(1—ст)=ехр (аст/2), где
Ч—UQlkT.
Выполним следующие преобразования:
(1 —ст)/(1—ст)—1 =2сг/ (1—ст)=ехр (аст/2)—1;
(1+ст)/(1—<т)+1=2/(1—ст)=ехр (аст/2)+1.
Разделив почленно оба уравнения, получим
ст=[ехр (аст/2)— 1]/[ехр (аст/2)+1].
Умножим числитель и знаменатель на ехр (—ао/4), а=[ехр (аа/4)—
—ехр(—аа/4)]/[ехр(аа/4)-|-ехр(—ao/4)] = th(aa/4), где th — гипер-
болический тангенс.
Положив аа/4 = £, получим
a = th£ (3.24а)
и
o = 45/a = (4AT/u.)£. (3.246)
Для установления характера зависимости ст=ст(7') следует
определить область совместного решения двух уравнений (3.24).
57
Выполним это графически. На рис. 3.13 изображены зависимости
4kT j.
o=th о, и в= --— t
Uq
Наклон прямой 2 равен 4 kTlu$ при всех значениях Наклон
кривой thg в разных точках определяется наклоном касательных
в этих точках, а тангенс угла наклона касательной численно равен
первой производной в рассматри-
ваемой точке. В точке £=0 пер-
Рис. 3.13. К определению температур-
ной зависимости степени дальнего по-
рядка:
1 — or=thfe; 2 — a=4feT^/«o
вая производная thg равна 1
(пунктирная прямая на рис. 3.13).
Совместные решения рассма-
триваемых уравнений будут не-
нулевыми в том случае, если
в точке ig=0 наклон прямой 2 на
рис. 3.13 будет меньше, чем на-
клон кривой /, т. е. при условии,
что 4^Г/п0<1. При 4^Г/и0=1
значение о достигает нуля и не
изменяется при 4йТ/ио>1.
Таким образом, существует
такая минимальная температура,
при которой о=0. Эту темпера-
туру (температуру Курнакова,
Тк) можно определить из условия
4йТМо=1; TK=u0/4fe. (3.25)
Согласно рассмотренной теории в сплавах типа [3-латуни при
повышении температуры равновесная степень дальнего порядка не-
прерывно уменьшается от 1 до 0 при Т=ТК. Графически эта зави-
симость приведена на рис. 3.14. В точке Т/Тк=1 дальний порядок
исчезает.
5*
0,8
0,6
0,2
О' 0.2 0,4 0,6 0,8 Т/Тк
Рис. 3.14. Зависимость степени дальнего порядка
а в сплаве CuZn от отношения абсолютной темпе-
ратуры Т к температуре Курнакова Гк 742 К по
теоретическому расчету (сплошная линия). Экспе-
риментальные точки получены Упманом и Уорре-
ном нейтронографическим методом
В других сплавах, например в Cu3Au, разупорядочение идет
по типу фазового перехода первого рода, при этом в точке превра-
щения степень порядка (а также основные термодинамические
свойства — энтропия, объем и т. п.) изменяется скачком.
Исчезновение дальнего порядка еще не свидетельствует об
исчезновении порядка вообще, оказывается, возможно сохранение
58
ближнего порядка, т. е. состояния, в котором имеется порядок
в ближайшем окружении каждого атома. Для ближнего, как и для
дальнего порядка, характерно уменьшение степени порядка с рос-
том температуры, но ближний порядок не исчезает при определен-
ной температуре.
Если Ди<0, наблюдается «ближнее упорядочение», когда вы-
годны разноименные соседства (Л — В). Если Ди>0, имеет место
«ближнее расслоение», когда преимущественно возникают соседст-
ва одноименных атомов А — А и В — В.
§ 3.4. БЛИЖНИЙ ПОРЯДОК В ТВЕРДЫХ РАСТВОРАХ
Для характеристики преимущественных соседств атомов (Д — В
или А—А и В — В) используют параметр ближнего порядка, ко-
торый получают из распределения интенсивности диффузного рас-
сеяния рентгеновских лучей (или нейтронов):
а; = 1 — ti^cBZi, (3.26)
где число атомов В на z-й координационной сфере, окружа-
ющей атом Л; Zi— общее число атомов на r-й координационной
сфере; cBZi— число атомов В на i-й координационной сфере, ко-
торое могло бы быть при статистически равномерном распределе-
нии атомов (св — атомная доля В).
При = а=0. Если п^<с5г(1), а, > 0, т. е.
имеется тенденция к группировкам атомов одного сорта (ближнее
расслоение; <0 (Яд2?> сBz(^) означает наличие преимущественных
соседств чужеродных атомов — ближнее упорядочение). Отношение
ct2/cti характеризует особенности атомной структуры сплава и изме-
нение энергии упорядочения в различных координационных сферах.
За последние 10—20 лет накоплен большой фактический мате-
риал по диффузному рассеянию рентгеновских лучей, показавший,
что практически нет в природе металлических твердых растворов
со статистически однородным хаотическим распределением атомов
компонент. Выяснение действительной атомной структуры твердых
растворов осложняется неоднозначностью интерпретации опытных
данных. Максимумы интенсивности рассеяния рентгеновских лучей
могут быть истолкованы как результат диффузного рассеяния для
статистического (или гомогенного) ближнего порядка, при котором
число атомов каждого сорта с любым образом выделенными под-
решетками должно отвечать атомной доле каждого компонента.
Однако те же максимумы можно принять за селективные сверх-
структурные отражения от областей с дальним порядком, при этом
размытие максимумов легко объяснить дисперсностью областей
дальнего порядка. Для твердых растворов вне области концентра-
ций, в которой может возникать состояние дальнего порядка, пред-
ложена модель «локального» упорядочения, согласно которой дис-
59
Таблица 3.2
Опытные данные о знаке ближнего порядка*
Номер
группы
раств ори-
те ля
Номер группы растворяемого элемента
2
3
4
5
6
8
2
3
4
5
6
8
Ag—Au <0
Cu—Au <0
Ag—Zn <0
Li—Mg <0
Cu—Zn <0
Ag—Al <0
Cu-Al <0
Au—Fe
Cu-Pt
Cu—Ni
Cu-Pd
Au—Ni
Au—Pd
Al—Ag >0
Mg-Cd <0
Al—Zn >0
Mg—In <0
Nb—U <0
Ni—Au >0
Pd—Au <0
Fe—Al <0
Ni-Al <0
Si—Ge
Nb—Ti
Mo—Ti
Ni-Si
>0
>0
Ti—Nb >0
Nb—Ta >0
Ni—Ta <0
Ti—Mo <0
Mo—W >0
Fe—Mo >0
Ni—Cr <0
Ni—Mo <0
Ni—W <0
Pd—W <0
co-Pt <0
Ni—Fe <0
Ni—Pt <0
Pd—Co <0
Pd—Pt <0
* Иверонова В. И., Кацнельсон А. А. „Изв. вузов. Сер. Физ.“. 1976, № 8, с. 43.
персные области дальнего порядка находятся в совершенно неупо-
рядоченной матрице. Важным для выяснения возможных локаль-
ных распределений при известных из рентгеновских исследований
значениях параметров ближнего порядка является машинное кон-
струирование атомной структуры твердого раствора, а также ма-
шинное моделирование установления равновесного состояния спла-
ва при заданных значениях энергии взаимодействия (метод Монте-
Карло). Результаты машинного моделирования для Си — Аи
(с помощью метода Монте-Карло) * свидетельствуют о том, что
выше Тк в твердом растворе стехиометрического состава можно
выделить упорядоченные домены в окружении неупорядоченной
матрицы.
В. И. Иверонова и А. А. Кацнельсон предложили следующую
классификацию типов ближнего порядка:
1) «жидкоподобный ближний порядок», при котором атомное
распределение одно и то же около всех узлов, т. е. каждый узел
можно принять за центр координационной сферы;
2) «локальный дальний порядок», когда имеются субмикрооб-
ласти, различающиеся по концентрациям, степени и виду упорядо-
чения, степень порядка максимальна в центре области и уменьша-
ется к периферии;
3) «микродоменный дальний порядок», когда кристалл состоит
из очень мелких антифазных доменов; благодаря высокой дисперс-
ности доменов распределение атомов по подрешеткам остается
однородным. Из-за сильного размытия сверхструктурных рефлек-
* Голосов Н. С., Смирнов В. А., Дудка Б. В. «Изв. вузов. Серия Физика»,
1973, № 2, с. 76.
60
сов распределение интенсивности качественно мало отличается от
картины диффузного рассеяния в случае ближнего порядка пер-
вого типа, но отношение параметров ближнего порядка для разных
координационных сфер должно соответствовать структуре даль-
него порядка.
Первый тип должен соответствовать термодинамическому равно-
весию при высокой температуре. Третий тип (микродоменная
структура) может возникать, например, в процессе закалки от тем-
ператур более высоких, чем критическая температура упорядоче-
ния, т. е. в условиях ограниченной диффузионной подвижности для
упорядочивающегося сплава; второй тип реализуется в процессе
низкотемпературного отжига сплава, имеющего дефекты кристал-
лической структуры (деформационные, закалочные или радиаци-
онные).
Согласно Фриделю, равновесный ближний порядок типа рас-
слоения (си >0) возможен в случае, если валентность растворителя
больше валентности растворяемого элемента; если же в металле
низкой валентности растворяется металл с более высокой валент-
ностью, то следует ожидать ближнего упорядочения (щСО). Экс-
периментальные данные (табл. 3.2) соответствуют этому правилу
при небольшом различии валентности компонентов и достаточно
низкой валентности растворителя. Очевидно, нельзя искать простой
закономерности для сплавов с участием металлов переходных
групп, хотя энергетический спектр электронов в сплаве и электрон-
ная часть энергии упорядочения имеют важнейшее значение в опре-
делении структуры дальнего порядка и типа ближнего порядка.
Значительная разница атомных радиусов компонентов может быть
причиной ближнего упорядочения (cti<0), однако роль этого
фактора не очень велика (например, для Ni — Au oti>0).
В некоторых сплавах отжиг после закалки приводит к так на-
зываемому /(-состоянию, которое характеризуется аномально вы-
соким электросопротивлением (например, Fe — Al, Ni — Mo, Ni —
Cr). Б. Г. Лившиц предположил, что это состояние аналогично со-
стоянию зонного распада пересыщенных твердых растворов. Рент-
генографические и другие структурные исследования действитель-
но показали устойчивую субмикронеоднородность или высокодис-
персную структуру в системах, где вообще имеет место упорядоче-
ние в области составов, примыкающих к границе области неупоря-
доченного твердого раствора. Ю. А. Скаков дал простое объясне-
ние устойчивости такой структуры. Межфазовую границу упоря-
доченной фазы в неупорядоченной матрице (обедненной, поскольку
исходный твердый раствор далек от стехиометрии) надо рассма-
тривать как соседства чужеродных атомов. В этом случае поверх-
ностная энергия (точнее, ее химическая составляющая) должна
быть очень небольшой или даже иметь отрицательный знак, так
как энергия смешения Az/<0. Только потеря когерентности дает
стимул к росту частиц, однако когерентность сохраняется благода-
ря изоморфности фаз. Поэтому рассматриваемое состояние можно
определить как локальный дальний порядок.
61
Важное значение изучение типа ближнего порядка имеет для сплавов, кото-
рые на существующих диаграммах равновесия характеризуются неограниченной
взаимной растворимостью при любых температурах (например, Мо—W). Эти
диаграммы противоречат теореме Нернста, по которой энтропия твердых и
жидких тел стремится к нулю при приближении к О К. Поэтому при достаточно
низких температурах такие сплавы должны были бы испытать распад на почти
чистые компоненты, или на упорядоченные твердые растворы стехиометрических
составов. Превращениям при низких температурах в этих сплавах препятствует
невозможность диффузии — сплав остается «замороженным». Знание знака энер-
гии смешения позволяет раскрыть характер неосуществимых в обычных условиях
превращений. Очень длительные выдержки при эксплуатации деталей из такого
сплава, облучение их нейтронами могут привести к реализации превращений
и нежелательному изменению свойств (например охрупчиванию).
Следует отметить, что число таких диаграмм постепенно уменьшается. Так
например, установлен распад сплавов Сг—'Мо (при 700°С), в сплавах Se—Те
обнаружено упорядочение после того, как был выявлен ближний порядок.
Особый случай представляет система Fr—Сг. В области a-твердого раство-
ра можно было предполагать отрицательное значение энергии смешения, посколь-
ку в системе имеется химическое соединение — о-фазы (ниже 800°). Однако
сейчас известно, что при низких температурах (<530°) о-фаза не образуется
и равновесной является гетерогенная смесь почти чистых компонент. Это состоя-
ние экспериментально обнаруживается, например, методом рассеяния нейтронов
под малыми углами (Е. 3. Винтайкин, В. И. Колонцов) или методом дифракци-
онной электронной микроскопии после очень длительных выдержек. Этот пример
свидетельствует о недостаточности приближения регулярных растворов.
Задачи к гл. 3
Задача 3.1. Найти изменение среднего атомного объема при образовании
эквиатомного твердого раствора MgCd.
Задача 3.2. Найти энергию связи иАА из энергии сублимации для Си.
Задача 3.3. Определить изменения энтропии системы при образовании рас-
твора в двухкомпонентной системе А—В (определяется так называемая энтро-
пия смешения или конфигурационная энтропия, изменения «вибрационной» энтро-
пии при сравнении конденсированной системы из смеси фаз — чистых компонен-
тов и системы в однофазном состоянии не учитываются).
Дать анализ зависимости энтропии смешения от состава твердого раствора.
Задача 3.4. Вывести уравнение кривой растворимости в бинарной системе
(Ди>0), упругая энергия не учитывается, для упрощения расчета можно при-
нять
Задача 3.5. Определить зависимость максимальной температуры существо-
вания двух твердых растворов от энергии смешения при отсутствии эффекта
упругой энергии. Максимуму кривой расслоения на диаграмме состояния соответ-
ствует концентрация с=0,5.
Задача 3.6. Найти энергию смешения для сплава Au—Мп по температуре
максимума кривой расслоения твердого раствора (Тмакс —1100 К).
Задача 3.7. Определить степень дальнего порядка о сплава AuCu3, если
известно, что вероятность нахождения атома В в узле a Wb равна 0,06 и 0,12.
Задача 3.8. Для задачи 3.7 определить число правильно (ра и рь) и непра-
вильно (wa и Wb) занятых мест кристаллической решетки при степени дальнего
порядка а=0,25.
Задача 3.9. Решить задачу 3.8 для дальнего порядка в (3-латуни состава
Си — 45 ат. % Zn и степени дальнего порядка о=0,67.
ГЛАВА 4
МЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ
Упорядоченные твердые растворы можно рассматривать как
металлические соединения, хотя по своей структуре и условиям
образования они тесно связаны с твердыми растворами. В тех
случаях, когда упорядочение происходит как переход первого ро-
да, области существования упорядоченных твердых растворов на
диаграммах фазового равновесия отделены от областей неупо-
рядоченных твердых растворов двухфазными областями, и есть
все основания относить упорядоченные твердые растворы к так
называемым промежуточным фазам, отличая их, таким образом,
от твердых растворов на основе элементов. Особую группу метал-
лических соединений образуют электронные соединения (фазы
Юм-Розери) и фазы внедрения, структуры которых хотя и не
совпадают со структурами образующих их металлов, но представ-
ляют собой те же характерные для металлических элементов
структуры о. ц. к., гекс. п. у., г. ц. к. или близкие к ним по коорди-
нации атомов.
В отдельную группу металлических соединений выделяют ме-
таллические фазы, образуемые только переходными металлами и
имеющие сложные кристаллические структуры (icr и родственные
ей фазы). Поскольку подобные структуры обнаружены и для
некоторых металлических элементов (p-U, а-Мп) и состав не опи-
сывается простыми атомными соотношениями (как и в случае
фаз Юм-Розери), эти фазы можно рассматривать как своеобраз-
ные электронные соединения.
В следующую группу металлических соединений входят фазы,
для которых характерны более или менее строгие стехиометриче-
ские атомные соотношения и, как следствие этого, более или ме-
нее узкие области гомогенности (например, р-, Х-фазы или фазы
Лавеса).
§ 4.1. ЭЛЕКТРОННЫЕ СОЕДИНЕНИЯ
Кристаллическая структура и химический состав электронных
соединений. При установлении закономерностей, определяющих
взаимную растворимость металлов (см. гл. 3), было обращено
внимание на то, что предельная концентрация твердых растворов
металлов II—V групп периодической системы элементов в Си, Ag
и Au соответствует определенной электронной концентрации, т. е.
63
определенному отношению числа валентных электронов к числ}
атомов, равному 1,4. Еще до обнаружения этого факта было
замечено, что электронная концентрация определяет структуру
многочисленных промежуточных фаз в сплавах металлов переход-
ных групп и I группы периодической системы (так называемые
металлы I класса) с металлами II—V (основных) групп периоди-
ческой системы (металлы II класса).
К металлам I класса относятся: Мп, Fe, Со, Ni, Rh, Pd, Pt,
Ce, La, Pr, Cu, Ag, Au, Li, Na. К металлам II класса: Be, Mg, Zn,
Cd, Hg, Al, Ga, In, Si, Ge, Sn, Pb, As, Sb.
Рис. 4.1. Структура типа у-латуни:
а — без учета смещений атомов между вакансиями; б — реальная структура
Найдены четыре типа таких электронных соединений или «фаз
Юм-Розери». Три типа фаз по кристаллической структуре анало-
гичны фазам р, у и 8 системы Си—Zn. Фазы четвертого типа (так
называемые фазы |У) имеют сложную кубическую решетку
с 20 атомами в элементарной ячейке, аналогичную решетке р-Мп.
Фазы р в неупорядоченном состоянии имеют о. ц. к. решетку. Фазы
у имеют кубическую решетку с 52 атомами на элементарную ячей-
ку (рис. 4.1). Гигантскую элементарную ячейку у-фазы можно
представить следующим образом: нужно сложить 27 элементар-
ных ячеек о. ц. к. решетки (по три вдоль каждой кристаллогра-
фической оси) и удалить атомы, сидящие в вершинах и в центре
построенного таким образом куба (очевидно, атомы, окружающие
освободившиеся места, должны несколько сблизиться)*. Наконец,
Е-фазы имеют гексагональную компактную решетку**. Большинст-
во электронных соединений — вещества переменного состава.
* Обозначения электронных соединений на диаграммах состояния не всегда
совпадают с приведенными здесь обозначениями типов этих фаз: так, например,
к типу у-фаз относятся фазы системы Си—Sn и Си—А1, обозначаемые на диа-
граммах Е.
** При наблюдаемом иногда упорядочении 8-фаз уменьшается число элемен-
тов симметрии в результате незначительного искажения решетки (так, например,
последняя может сделаться ромбической).
*64
Юм-Розери установил, что область гомогенности фаз с решеткой
0-латуни и 0-Мп лежит вблизи составов, для которых отношение
числа валентных электронов к числу атомов равно 3/2. В фазах
с решеткой у-латуни электронная концентрация близка к 21/13
(предельная концентрация достигает 22/13), в фазах типа 8-ла-
туни — 7/4.
Число валентных электронов берется равным номеру группы
таблицы Д. И. Менделеева как для металлов группы меди, так
и для непереходных элементов II—V групп.
Металлам переходных групп приписывается в различных элек-
тронных соединениях различная валентность: 0, 1 или 2. Это
связано с перекрытием их nd- и (и-|-1) s-энергетических уровней,
что приводит к переменной валентности. Чаще всего металлы
переходных групп являются в электронных соединениях нуль-
валентными элементами не отдающими своих валентных электро-
нов в первую зону Бриллюэна.
В табл. 4.1 приведены составы некоторых электронных соеди-
нений. Необходимо отметить, что чаще всего состав, отвечающий
«идеальной» химической формуле фазы, лежит у той границы
области гомогенности этой фазы, которая соответствует макси-
мальному содержанию двух- или многовалентного металла, одна-
ко имеются и отступления от этого правила.
Среди фаз с электронной концентрацией около 1,5 имеется
несколько веществ с гекс. п.у. решеткой, присущей 8-фазам при
концентрации 1,75 (в табл. 4.1 приведены данные об этих фазах).
Квантовая теория электронных соединений. Теория С. Т. Ко-
нобеевского и Джонса, объясняющая закономерности образования
электронных соединений, основана на том же представлении о зо-
нах дозволенной энергии электронов, которое было использовано
в теории растворимости (см. гл. 3).
Разберем простейший пример — электронную концентрацию
0-фаз, имеющих о. ц. к. решетку. В кристаллах с такой решеткой
плоскости ромбического додекаэдра наиболее густо усажены ато-
мами; отражением электронных волн от этих плоскостей и опре-
деляется разрыв на кривой, выражающей зависимость энергии
электронов от их импульсов, т. е. заполнение первой зоны Брил-
люэна.
С помощью уравнения (1.33) для о. ц. к. кристалла найдем
электронную концентрацию, при которой поверхность Ферми под-
ходит к границе первой зоны Бриллюэна (или удовлетворяется
условие, отражения электронных волн плоскостями {НО}):
сэл = 3Т2 (1 + 1)3/2 — 1 ’48 электрон/атом.
Уточненная теория, учитывающая искажение поверхности Фер-
ми при ее приближении к зоне Бриллюэна, показывает, что на
самом деле разрыв на кривой, выражающей зависимость энергии
электрона от импульса 8=/(р), происходит при концентрации,
несколько превышающей числа, полученные по формуле (1.33).
5— 523 65
Таблица 4.1
Состав некоторых электронных соединений
Обозначение фазы Формула электронного соединения, отвечаю- щая правилу Юм-Ро- зери, и соответствую- щее ей содержание металла II класса, ат. % Границы гомогенной области электронного соединения на диаграм- ме состояния (содер- жание металла II класса), ат. % Электронная концен- трация на границах гомогенной области
Фазы со структурой [1-латуни
Теоретическое значение предельной электронной концентрации 1,5
P-Ag—Mg AgMg (50) 39,9—65,0 1,40—1,66
p-Ag—Zn AgZn(50) 39,1—57,5 1,39—1,57
y-Al—Co AlCo (50) 24,6—52,7 0,74—1,58
P- Al—Cu AlCu3(25) 18,9—31,8 1,38—1,64
p-Al—Fe AlFe (50) 50 1,50
p-Al—Ni AlNi (50) 33,8—54,9 1,01—1,64
P-Au—Mg AuMg (50) 34,1—58,9 1,34—1,58
P-Au—Zn AuZn(50) 36,5—57 1,36—1,57
BeCo BeCo(50) 50,0 1,50
y-Be—Cu BeCu (50) 47,3—49,7 1,47—1,50
Р-Be—Cu BeCu(50) 20—47 1,20—1,47
Be—Ni BeNi (50) 50,0 1,50
P-Cu—In Cu3In(25) 18,2—23,8 1,36—1,48
P-Cu—Sn Cu3Sn(16,7) 13,5—22 1,40—1,66
P-Cu—Zn CuSn (50) 36,4—55,8 1,36—1,55
Фазы со структурой Р-Мп
p-Ag-Al Ag3Al(25) 20,3—80,8 1,41 — 1,62
AlAu3 AlAu3(25) 25 1,50
CoZn3 CoZn3(75) 75 1,50
у-Cu—Si Cu6Si(16,7) 18,0 1,54
Фазы со структурой у-латуни
у-Ag—Cd Ag5Cd8(61,5) 57,5—62,9 1,53—1,68
o-Al—Au Au9A14(30,8) 33,6—35 1,67—1,70
y-Al—Cr Al8Cr5(61,5) 65 1,65
y-Al—Cu Cu9Al4 1
y'-Al—Cu y2-Al—Cu Cu17A119(30,8) Сц3гА119 30,8—40 1 1,61—1,80
e-Al—CU Cu3A12 J
y'-Au—Zn Au5Zn8 (61,5) 64,9—78,7 1,65—1,79
Be2Ni5~ Be2Ni5(80,8) 81,9—82,5 1,64—1,65
6-Cd—Cu Cd8Cu5(61,5) 52,3—64,2 1,52—1,64
6-Cd—Ni Cd2INi5(80,8) 80,8 1,62
6-Cu—Ca Cu9Ca4(30,8) 29,7—37,8 1,50—1,76
a-Cu— Si Cu3]Sl8 (30,8) 28,9—30,8 1,58—1,62
d-CuSn Cu31Sn8 (20,2) 18,0 1,54
y-Cu—Zn Cu5Zn8(61,5) 57,1—69,9 1,57—1,73
Na3Pb8 Na3,Pb8 (20,2) 20,2 1,62
Г-Fe—Zn Fe5Zn21 (80,8) 72—79 1,44—1,58
Г-Ni— Zn Ni5Zn21 (80,8) 78—84,9 1,66—1,70
Pt5Zn21 Pt5Zn21 (80,8) 80,8 1,62
66
Продолжение табл. 4.1
Обозначение фазы Формула электронного соединения, отвечаю- щая правилу Юм-Ро- зери, и соответствую- щее ей содержание металла II класса, ат. % Границы гомогенной области электронного соединения на диаграм- ме состояния (содер- жание металла II класса), ат. % Электронная концен- трация на границах гомогенной области
Фазы с гекс. п. у. структурой (е- и f-фазы)
Y-Ag—А1 Ag5Al3(37,5) 27,1—40,0 1,54—1,80
( frAg—Cd* AgCd(50) 50—53 1,50—1,53
1 e-Ag-Cd AgCd3(75) 64,6—81,4 1,65—1,81
J p-Ag—Sn* — 12,4—2,1 1,37—1,63
l Y'-Ag—Sn** Ag3Sn(25) — 1,75
Y'-Au—Cd AuCd3(75) 67,8—64,6 1,62—1,65
e-AgSb* Ag„Sbs (18,75) 9,9—14,4 1,39—1,58
e-Cll—Zn CuZn3(75) 78—87,2 1,78—1,87
e-Cu—Sn Cu3Sn(25) 24,5—25,1 1,73—1,75
p-Cu—Si* — 14,5 1,44
T]-Fe—Zn FeZn, (87,5) 87—91 1,74—1,82
e-Ni—Sn Ni3Sn(25) 25 1,75
* По электронной концентрации не соответствует е-фазам.
** Решетка ромбически искажена.
Для о. ц. к. решетки «коэффициент заполнения» первой зоны ра-
вен 1,02, таким образом, можно принять сэл = 3/2.
После перехода через критическую концентрацию электронного
газа, равную 3/2, правильная о. ц. к. решетка делается неустойчи-
вой. Дальнейшее растворение многовалентных элементов возмож-
но при условии такой пере-
стройки решетки, которая
приведет к уменьшению
среднего числа атомов, при-
ходящегося на элементар-
ную ячейку. При этом число
электронов ’в ячейке, равное
произведению электронной
концентрации с на среднее
число атомов в ячейке п, мо-
жет остаться неизменным:
Сэл/яч=Сэл • ^ат/яч =const.
Как показал С. Т. Коно-
беевский, один из способов
такой перестройки — обра-
зование твердых растворов
вычитания. Так растворяет-
ся А1 в фазах СоА1 и NiAl,
имеющих кубические ре-
шетки типа CsCl.
Рис. 4.2. Зависимость периода кристалличе-
ской решетки (/) и плотности (2) фазы
NiAl от состава сплава
5*
67
На рис. 4.2 показана зависимость периода кристаллической
решетки фазы NiAl от химического состава. При содержании алю-
миния, отвечающем стехиометрическому составу NiAl, период
решетки максимален. При растворении в NiAl никеля, ко-
торый замещает отдельные алюминиевые атомы, измеряемый
рентгенографически период решетки уменьшается. Это объясняет-
ся тем, что радиус атома Ni меньше радиуса А1. Резкое же
уменьшение периода решетки этой фазы при растворении алюми-
ния объясняется тем, что при этом образуется твердый раствор
вычитания: атомы А1 не становятся на место атомов Ni, а нехват-
ка последних приводит к образованию пустых мест в кристалли-
ческой решетке. Сколько бы А1 ни растворялось в фазе СоА1 или
NiAl, количество валентных электронов, приходящихся на каж-
дую элементарную ячейку решетки твердого раствора, остается
равным 3.
Пустые места в решетках твердых растворов алюминия в СоА1
и NiAl распределены статистически так, что симметрия решетки
при образовании этих твердых растворов не изменяется. Но при
определенной концентрации пустых мест в таких дефектных ре-
шетках эти пустые места могут разместиться упорядоченным об-
разом, что должно привести к образованию своеобразной сверх-
структуры. Такими сверхструктурными решетками являются
описанные выше сложные кубические решетки типа у-латуни.
В большой ячейке решетки
у-латуни, содержащей 27 дефор-
мированных о. ц. к. ячеек, за-
полненных 52 атомами, в соот-
ветствии с правилом Юм-Розери
для у-фаз при насыщении эле-
ментом II класса содержится
88 валентных электронов, так
что на каждую из малых о. ц. к.
ячеек со средним числом атомов,
равным 1,927, приходится
88/27=3,26 валентных электро-
нов. Исследования показали, что
за счет деформации у-решетки
в системе Си—А1, сопровождаю-
щейся дальнейшим уменьшением
числа атомов в элементарной ячейке до 49, возможно дальнейшее
увеличение содержания А1 в у-фазе.
И в этом случае уменьшение среднего числа атомов v в каж-
дой малой ячейке компенсирует увеличение электронной концен-
трации сэл так, что количество электронов в малой ячейке при
всех описанных выше трансформациях у-решетки остается неиз-
менным и равным примерно 3,25 (рис. 4.3).
В сплавах Си—А1 электронное соединение со структурой
е-латуни не возникает. В результате деформации кубической ре-
шетки, связанной с уменьшением среднего числа атомов на эле-
Рис. 4.3. Зависимость среднего числа
(ггат) атомов в малой элементарной
ячейке фаз у, у' и т. д. в сплавах
Си—А1 в зависимости от электронной
концентрации сэл
68
Рис. 4.4. Диамагнитная восприимчи-
вость сплавов Си—Zn
ментарную ячейку до 1,74, координация атомов, характерная для
о. ц. к. решетки, сохраняет устойчивость вплоть до электронной
концентрации, равной 1,85, что даже превышает электронную
концентрацию, присущую фазам со структурой &-латуни.
Квантовая теория электронных соединений основывается на
Представлении о почти свободных электронах проводимости и не
может ввиду этого учитывать индивидуальные особенности тех
атомов, которые образуют кристаллы указанных фаз. Она яв-
ляется первым приближением, схемой, которую приходится при
анализе конкретных фактов дополнять специальными соображе-
ниями (часто не имеющими еще серьезных теоретических обосно-
ваний). Так, например, для фаз,
содержащих никелевые атомы,
правило Юм-Розери соблюдается
в одних случаях, если приписать
Ni нулевую валентность (напри-
мер, NiAl с |3-решеткой), в других,
если считать Ni одновалентным
элементом (Ni3Sn с решеткой е-
латуни). Редкоземельные метал-
лы ведут себя в электронных со-
единениях типа |3-латуни то
как нуль-валентные элементы
(LaMg3, PrMg3), то как однова-
лентные (CeMg, LaMg, PrMg).
Таким образом, переменная
валентность, характерная для ме-
таллов переходных групп при
взаимодействии с неметаллами,
проявляется и в металлических
сплавах.
В табл. 4.1 имеется большое число относительно незначитель-
ных отступлений от соотношений Юм-Розери, которые объясняют-
ся индивидуальными особенностями атомов разных элементов, не
учитываемыми в приведенных расчетах, обезличивающих атомы
в соответствии с теорией почти свободных электронов.
Насыщению зоны Бриллюэна какой-либо фазы всегда соот-
ветствует граница области гомогенности этой фазы со стороны
элемента II класса, т. е. двух- или многовалентного. Это утвер-
ждение иллюстрируется следующим фактом. Согласно теории
магнитных свойств сплавов насыщению первой зоны кристалла
соответствует пик диамагнитной восприимчивости. На рис. 4.4
приведена кривая диамагнитной восприимчивости сплавов Си—Zn.
Максимум на этой кривой соответствует 70 ат.% Zn, т. е. границе
у-области в системе Си—Zn (58—70 ат.% Zn). Между тем элек-
тронной концентрации 21/13=1,615 отвечает сплав с 61,6 ат.% Zn,
лежащий внутри области гомогенности. Очевидно, в кристаллах
Y-латуни насыщение первой зоны происходит при повышенной
электронной концентрации.
69
Существование фаз со структурой гекс. п. у. при электронной
концентрации, присущей p-фазам (около 1,5), пока объяснения
не имеет. Несомненно, однако, что эти вещества нельзя относить
к s-фазам, руководствуясь одними лишь данными об их структуре.
Интересно отметить, что в системах Ag—Cd и Ag—Sn, где имеют-
ся p-фазы с гексагональной структурой, также и нормальные
8-структуры с электронной концентрацией 1,75. В системе Ag—Cd
область фазы |3 с гекс. п. у. решеткой, устойчивая при температуре
210—440°С, вклинивается между областями фаз почти того же
состава, но с о. ц. к. решеткой (p-фаза с неупорядоченным распо-
ложением атомов, устойчивая при температуре выше 440°С, и
P-фаза с упорядоченным расположением атомов, т. е. с решеткой
типа CsCl, устойчивой при температуре ниже 210°С).
По-видимому, целесообразно ввести представление о третьем
типе структур р с гексагональной компактной решеткой. Очевид-
но, концентрация 3/2 соответствует при каких-то условиях запол-
нения зоны дозволенной энергии гекс. п. у. решетки.
Массальский показал, в частности, что такое заполнение воз-
можно при малых различиях атомных радиусов (до 2—4%), и
установил, что фазы с гекс. п. у. решеткой и разными значениями
с/а могут возникать при электронных концентрациях от 1,32
до 1,89.
Многие p-фазы при высоких температурах имеют неупорядо-
ченное расположение атомов, например в системе Си—Zn. Фазы
СоА1 и NiAl обладают упорядоченной структурой вплоть до тем-
пературы плавления. Эти фазы — устойчивые, как указывалось
выше, в широком интервале концентраций — обладают резко вы-
раженными максимумами на линиях плавкости, отвечающие со-
ставам СоА1 и NiAl.
р-Фазы систем Au—Zn и Ag—Mg также сохраняют упорядо-
ченность вплоть до плавления.
Неупорядоченные p-фазы при низких температурах обычно
переходят в упорядоченное состояние (p-фазы систем Си—Zn,
Ag—Cd) с решеткой типа CsCl или испытывают эвтектоидный
распад (системы Си—А1 и Си—Sn).
В сплавах Си—Be высокотемпературная p-фаза с неупорядо-
ченным расположением атомов испытывает при 576°С эвтектоид-
ный распад, причем один из продуктов распада, у-фаза, имеет
ту же о. ц. к. решетку, что и p-фаза, но с упорядоченным распо-
ложением атомов (решетка типа CsCl).
В большинстве случаев фазы с решетками типа р-Mn имеют
неупорядоченное расположение атомов. Фазы со структурой
у-латуни имеют упорядоченные структуры (иногда не вполне),
у большинства гексагональных фаз структуры неупорядоченные.
Электронные соединения в трехкомпонентных сплавах. В большинстве иссле-
дованных трехкомпонентных сплавов однотипные электронные соединения обра-
зуют непрерывные ряды твердых растворов. Например, в системе Си—Zn—Sn
при температуре, превышающей 520°С (температура эвтектоидного распада
P-фазы системы Си—Sn), (3- и е-фазы системы Си—Zn, Си—Sn и у-фаза системы
70
Cu—Zn с имеющей ту же структуру 6-фазой системы Си—Sn образуют непре
рывные ряды однофазных сплавов. Этого и следовало ожидать, если исходить
из представлений о почти свободных электронах: любой сплав на двойном раз-
резе между однотипными (т. е. имеющими одно и то же число валентных элек
тронов в каждой элементарной ячейке) электронными соединениями должен
иметь то же число электронов на ячейку.
В сплавах Fe—Ni—Al при высокой температуре (свыше 1300°С) широкая
область гомогенных (3-фаз простирается между двойными фазами NiAl и FeAl.
Распад (3-твердых растворов некоторых сплавов этой системы при пониже-
нии температуры с образованием высокодисперсной смеси неупорядоченной фер-
ромагнитной фазы (на основе Fe) и упорядоченной немагнитной (на основе
NiAl) широко используется для получения магнитожестких материалов.
Среди тройных (3-фаз также встречаются «дырчатые» структуры (сплавы,
в которых на ячейку приходится менее двух атомов). Образование дырчатых
структур и здесь в соответствии с теорией С. Т. Конобеевского и Джонса обес-
печивает сохранение трех электронов на элементарную ячейку при электронной
концентрации, превышающей 1,5.
В заключение необходимо отметить, что в трехкомпонентных сплавах, содер-
жащих Ni, обнаружены следующие фазы с упорядоченной (на основе о. ц. к.)
решеткой типа Fe3Al: (CuNi)3Sn; (CuNi)3Sb, Ni2MgSn, Ni2MgSb.
По-видимому, все эти вещества можно считать электронными соединениями
типа (3-латуни, но электронную концентрацию, равную 3/2, имеет лишь фаза
Ni2MgSn.
Электронные соединения между переходными металлами. При
сплавлении переходных металлов VI группы (о. ц. к. решетка),
с Ir, Pd, Rh и Pt (г. ц. к. решетка) в некоторых случаях (в сплавах
1г—Мо, 1г—W, Rh—Mo, Rh—W, Pt—Мо) возникают промежуточ-
ные фазы с гекс. п. у. решеткой. Их, очевидно, следует считать
электронными соединениями, образующимися в результате запол-
нения s +d-электронами не только первой, но и второй (а может
быть, и третьей) зоны Бриллюэна о. ц. к. решетки; о заполнении
зон приходится судить косвенно по кристаллическим структурам
и, наконец, пользоваться таким критерием, как число s + d-элек-
тронов, приходящихся на атом.
Весьма существенно для суждения о природе этих фаз то, что значения сво-
бодной энергии для каждого из переходных металлов с «виртуальными» структу-
рами а (г. ц. к), |3 (о. ц. к) и 8 (гекс. п. у.) чрезвычайно мало отличаются друг
от друга. Кауфман * систематизировал данные о термодинамических характери-
стиках ДН и AS как наблюдающихся на опыте, так и виртуальных переходов
Р—и а—>8 для всех ^-переходных металлов второго и третьего длинных
периодов. Значение е лишь для Мо и W равно 2000 кал/(г-атом), а для
всех остальных металлов — меньше.
Предсказания возникновения 8-фаз в сплавах с участием переходных эле-
ментов, сделанные на основе этих расчетов для многих систем, показали хоро-
шее совпадение с опытом.
Последовательное образование 8-, а затем a-фаз в твердых растворах заме-
щения при увеличении числа nd-, (гг+1)5р-электронов, приходящихся на один
атом, аналогично чередованию структур при образовании обычных фаз Юм-Ро-
зери, только с перестановкой первого звена цепи а—>-(3—>8 на последнее место
₽—^8—э-а.
Области существования 8-фаз на диаграммах равновесия, приведенных
Кауфманом, включают состав с таким же числом dsp-электронов как у Ru и Os,
т- е. элементов с 8-решеткой. Это же относится к 8-фазе системы Сг—1г. Оче-
* Кауфман Л. В сб.: Устойчивость фаз в металлах и сплавах. Пер. с англ.
Под ред. Д. С. Каменецкой. М., «Мир», 1970, с. 134.
71
видно, эти фазы следует рассматривать как е-электронные соединения. Современ-
ное состояние теории переходных металлов не позволяет еще связать их возник-
новение с заполнением определенной (как указывалось, не первой) зоны Брил-
люэна о. ц. к. решетки; приходится ограничиваться приблизительными данными
об их электронной концентрации. Такую же стадию переживало учение об элек-
тронных соединениях непереходных металлов после установления соотношений
Юм-Розери для их электронных концентраций, пока эти соотношения не были
вычислены методами теории зон Бриллюэна.
К числу е-электронных соединений переходных металлов следует отнести и
фазы, обычно описываемые формулой АзВ, где А— переходный металл VIII груп-
пы, а В — IV—VI групп. Электронная концентрация у некоторых из этих фаз
доходит до 9 электрон/атом.
§ 4.2. ФАЗЫ ВНЕДРЕНИЯ
Схемы структур упорядоченных растворов внедрения (см.
рис. 3.2) и рассмотрение диаграмм состояния сплавов, в которых
концентрация атомов внедрения достигает 0,35—0,5 (например,
приведенных на рис. 4.5), показывают, что металлическая подре-
шетка в них испытывает концентрационные полиморфные превра-
Рис. 4.5. Диаграммы равно-
весия сплавов Nb—С (и);
Ti—С (б); W—С (в)
щения. Если металл-растворитель имеет о. ц. к. решетку, то уже
при введении долей процента неметалла (в рассматриваемых слу-
чаях— углерода) наряду с о. ц. к. решеткой появляется фаза
с гекс. п. у. решеткой. Максимум температуры плавления этой
фазы соответствует примерно составу Ме2Х, область гомогенности
при высокой температуре — около 10 ат.%, с понижением темпе-
ратуры атомы неметалла размещаются в решетке упорядоченно
(в NbC примерно при 2000°С, в Мо2С — при 1200°С), в обоих слу-
чаях при упорядочении область гомогенности сужается.
72
Дальнейший рост концентрации неметалла приводит к появ-
лению фаз с г. ц. к. решеткой металлических атомов. У металлов
IV группы (Zr, Ti, Hf) концентрации углерода в г. ц. к. фазах от
33 до 50%, у карбидов Nb и Та — примерно от состава МеС0,б ДО
МеС, у карбидов Мо и W — примерно от МеС0,б до МеС0)75. Куби-
ческие карбиды Мо и W устойчивы лишь при высокой темпера-
туре. При комнатной температуре устойчив лишь карбид WC
с простой гексагональной металлической подрешеткой.
Хэгг впервые систематизировал многочисленные фазы в спла-
вах Me—X, содержащие большие концентрации неметалла * *. Он
установил, что структуры внедрения с большой концентрацией
неметалла и металлическими подрешетками, отличными от реше-
ток образующих их металлов (но относящимися к числу типичных
металлических о.ц. к., г. и. к., гекс. п. у*2), образуются лишь при
соблюдении следующего соотношения атомных радиусов: гхЛме^
^'0,59. Это условие не удалось вывести из соображения плотней-
шей упаковки атомов, тем не менее оно почти всегда соблюдается.
Фазы с упорядоченными (по неметаллу) структурами внедре-
ния мы будем называть «фазами внедрения» *3. Как твердые
растворы внедрения, так и фазы внедрения образуются при взаи-
модействии переходных металлов с Н, С, N и О.
При рассмотрении свойств фаз внедрения следует всегда учи-
тывать возможность их загрязнения [например, карбиды титана
TiC или урана UC на самом деле почти всегда следует описывать
формулами Ti(C, О, N) или U(C, О, N)]. Особенно склонны
к загрязнениям карбиды и нитриды металлов IV группы, менее —
V группы. Только в последние годы научились синтезировать доста-
точно чистые материалы и даже монокристаллы, пригодные для
надежного измерения их механических и физических свойств.
Электронное строение фаз внедрения. Хэгг отметил, что струк-
тура и свойства этих фаз, несмотря на высокое содержание не-
металла, определяются сохранением в них металлической связи.
В дальнейшем высказывались предположения о ковалентной связи
Me—X и даже ионной связи между положительно заряженными
ионами металла и отрицательно заряженными ионами неме-
талла.
Уббелоде *4 применил представления зонной теории к твердому
раствору внедрения — гидриду палладия. Предположив, что водо-
род отдает валентный электрон в имеющую примерно 0,5 элек-
тронных вакансий на атом d-полосу металла, Уббелоде объяснил
интересные особенности сплавов Pd—Н: уменьшение их магнитной
восприимчивости (вплоть до нуля) с увеличением концентрации Н
(подобное влияние Ag), уменьшение растворимости Н в Pd при
легировании его Ag и т. д.
* Hagg G. «Z. Phys. Chem.», 1931, Bd 12, S. 33.
*2 Иногда слегка искаженными.
*3 Андриевский P. Я., Уманский Я. С. Фазы внедрения. М., «Наука», 1977,
‘4 Ubbelohde A. R. «Ргос. Roy. Soc.», 1937, А. 159, р. 295.
73
Я. С. Уманский*, используя структурные аналогии и тот факт,
что карбиды и нитриды со структурами внедрения обладают
электропроводностью, близкой к электропроводности металлов, и
положительным температурным коэффициентом электросопротив-
ления, предположил, что углерод, водород и азот в этих фазах
также отдают часть своих валентных электронов в перекрываю-
щиеся nd- и (п+ 1)5-полосы материала. При этом в узлах решетки
должны располагаться положительно заряженные ионы металла,
в части межузлий (а в таких фазах, как TiC, — во всех октапо-
рах)— положительно заряженные ионы неметалла, а электроны
проводимости движутся по всему объему образца, заполняя его
с осциллирующей плотностью.
Эта модель позволила объяснить широкие области гомогенно-
сти фаз со структурами внедрения, не присущие обычным хими-
ческим соединениям с ионной (например, NaCl) и ковалентной
(например, SiC) связями и несоблюдение законов валентности
(например, NbC). Высокий коэффициент диффузии С и N, рас-
творенных в у-железе (по сравнению, например, с кислородом,
имеющим меньший атомный радиус), и направления электропере-
носа водорода в гидриде Pd и С в аустените согласуются с этой
гипотезой об электронном строении фаз со структурами внедрения.
Однако большую часть этих фактов можно объяснить, предпо-
ложив, что атомы металла и металлоида взаимодействуют между
собой непосредственно по законам ковалентной связи в твердом
теле, сформулированным в несколько модифицированной (по
сравнению с химической связью в молекулах) теории Полинга.
В последние годы появились дополнительные важные доводы
в пользу представления о металлической природе фаз внедрения.
Один из этих доводов связан с теорией А. Г. Хачатуряна,
предсказывающей возможность существования разнообразных фаз
внедрения — карбидов, нитридов, гидридов и низших юкислов,
структуры которых однозначно выводятся из полученных ранее
в тех же работах сверхструктур замещения**. Теория А. Г. Хача-
туряна не содержит каких-либо предположений о характере свя-
зей в упорядоченных твердых растворах замещения, но все они —
вещества металлические. Естественно поэтому предположить, что
в фазах внедрения также доминирует металлическая связь.
Об отсутствии направленных связей Me—X свидетельствуют
данные анализа мягких эмиссионных рентгеновских спектров.
Было установлено, что преимущественной системой скольжения
в карбидных растворах и фазах внедрения с г. ц. к. металлической
подрешеткой является система {111} <110>—та же, что у чис-
* Уманский Я. С. «Изв. сектора физ.-хим. анализа», 1943, т. 16, с. 127.
** Сверхструктура внедрения, выводящаяся из сверхструктуры замещения
АтВп, по А. Г. Хачатуряну имеет состав Mew+nXw или Мет+пХп. Отсюда сле-
дует, что при внедрении в октапоры не может быть неупорядоченной фазы со
структурой NaCl или NiAs; такие фазы являются твердыми растворами, октапо-
ры которых заполнены до предела.
74
тых металлов с г. ц. к. решеткой. Точно так же в карбиде М02С
скольжение происходит вдоль наиболее плотноупакованной пло-
скости гекс. п. у. подрешетки металла (0001) в направлении
<2Н0>, как и у большинства чистых металлов с гекс. п.у^_ре-
шеткой (Be, Zr, Cd и т. д.). Система скольжения {111} <110>
исключена для ионных соединений. Она маловероятна и при на-
личии сильных локализованных связей Me—X, поскольку при
скольжении по этим плоскостям в кристаллах перерезалось бы
максимальное число связей (на 1 см2 плоскости сдвига).
Разрез Re—0-W2C системы Re—W—С или Re—Мо2С (рис. 4.6)
представляет собой непрерывный ряд твердых растворов на основе
С
Рис. 4.6. Диаграмма равновесия сплавов Мо—Re—С
гекс. п. у. металлической подрешетки. Предположение об уходе
в полосу проводимости р-электронов углерода приводит к выводу
о том, что вдоль всего разреза концентрация электронов на один
атом равна семи.
Чередования структур металлических подрешеток у карбидов
металлов V и VI групп при увеличении концентрации углерода
совпадает с чередованием структур в сплавах металлов с о.ц. к.
и г. ц. к. решетками (см. § 2.2). Это чередование также прекрасно
согласуется с представлением о переходе валентных электронов
углерода в полосу проводимости металла, причем для количест-
венного совпадения значений сэл электрон/атом приходится пред-
положить, что углерод отдает все четыре валентных электрона.
При этом, так как у W и Мо в dsp-полосе на один электрон
больше, чем у металлов VA группы, необходимая для образования
г. ц. к. решетки электронная концентрация достигается при мень-
шей, чем у металлов V группы, концентрации углерода, что и
реализуется в фазах МоСо>7 и WCo,7 (см. рис. 4.5).
75
Переход в полосу проводимости всех валентных электронов
неметалла маловероятен. Однако, поскольку форма ферми-по-
верхности у переходных металлов сложна, касание этой поверх-
ности и граней зоны Бриллюэна может происходить при электрон-
ных концентрациях, существенно отличающихся от величин,
вычисленных в предположении о сферической форме ферми-по-
верхности. Сложна даже форма ферми-поверхности меди — про-
стейшего переходного металла. От почти сферической ее части
отходят восемь почти цилиндрических отростков, оси которых
перпендикулярны к граням {111} зоны Бриллюэна г. ц. к. решетки,
пересекаясь с этими гранями. Даже и в этом простейшем (для
переходных металлов) случае поверхность Ферми многосвязна.
Из расчетов следует, что поверхность Ферми у сплавов благород-
ных металлов с двух- и многовалентными металлами касается
указанных выше граней при электронной концентрации 1,36.
Несомненно, еще более сложными могут быть ферми-поверх-
ности твердых растворов и фаз внедрения.
Однако качественно описанная здесь модель электронного
строения вполне объясняет последовательность чередования струк-
тур при растворении по крайней мере углерода — структуры ме-
няются так, как изменяются они при продвижении вдоль второго
или третьего большого периода, т. е. при увеличении концентра-
ции электронов проводимости: о. ц. к., гекс. п. у., г. ц. к. Металлы
IVA группы ведут себя так же, как металлы с о. ц. к. решеткой,
которая у них стабильна при высокой температуре и стабилизи-
руется при комнатной температуре незначительными (около
10 ат.%) добавками металлов VA группы. Эти металлы не обра-
зуют растворов и фаз внедрения с гекс. п. у. металлической под-
решеткой, но и в металлических сплавах, образующих фазы
Юм-Розери, нередко отсутствуют электронные соединения с 8-ре-
шеткой (сплавы Си—А1, Со—Al, Ni—Al).
Очень характерно сравнение изменений свойств при образова-
нии фаз внедрения с изменениями свойств металлов переходных
групп вдоль периода. При этом становится понятным резкое
(в 2,5—4 раза) увеличение упругих модулей (табл. 4.2). Модули
карбидов Zr и Nb превосходят модуль Ru, самого «жесткого»
металла второго d-периода; модули карбидов Hf и Та близки
к модулям металлических Re и 1г, но ниже, чем у Os. Вся сово-
купность особенностей структур и свойств твердых растворов
углерода в d-металлах и образующихся фаз внедрения позволяет
считать, что растворяющийся углерод отдает в зону проводимо-
сти кристалла часть своих валентных электронов. Это вызывает
увеличение прочности связей в решетке и, подобно фазам Юм-Ро-
зери, «концентрационные» полиморфные превращения. Надежных
данных о количестве отдаваемых атомами углерода электронов
пока нет.
Ясно, что электронные концентрации, соответствующие у твер-
дых растворов и фаз внедрения переходу р->8-^а, ниже числа spd-
электронов у переходных металлов с 8 и а-решетками.
76
Электронное строение нитридных и оксидных растворов и фаз
внедрения, несомненно, сложнее, чем у карбидов. У большинства
этих фаз при концентрациях неметалла, приближающихся к МеХ,
температурный коэффициент электросопротивления если положи-
тельный, то близок к нулю или может быть отрицательным. По-
Таблица 4.2
Теплоты образования ккал/r-моль), максимальная
температура плавления (7\, К) и модуль упругости
(£, кгс/мм210~3) карбидных и нитридных фаз со структурами
внедрения
Фаза —A/faee Фаза (состав) Фаза (состав) Модуль упругости
фазы металла
TiC 44 TiC0>8 3340 TiC0>9 45,5 10,5
ZrC 49,5 ZrC<,>8 3700 ZrC0>9 41 10,0
HIC 55 Hfc0>9 4200 HfC0>97 47 8,0
vc0>5 16,5 ?-VC0,5 2440 — — —
VC0>88 24,5 VC0>85 3000 VC0,88 43 —
₽-Nb2C 23 pbco>54 3300 — — —
NbC 32,2 NbC0>8 3870 NbC0>97 52 16
TaC0>6 25 ?"TaC0>55 3700 — — —
TaC 34 TaC0>9 4250 TaC0>99 54,5 19
MoC 5,5 f-MoC0> 5 2750 Mo2C 23 29
wc0>5 6 WC0>46 3050 WCO>99 73,5 41
TiN 81 TiN0>8 3100 — — —
ZrN 88 — — — —-
HfN 88 — — — —- —.
NbN 56,5 — — — — —
NbN0>5 30 — — — — —
TaN 60 — •— — —
TaN0>5 32 — — .L — —
этому не следует считать, что в этих фазах заметная доля валент-
ных электронов неметалла уходит в полосу проводимости.
Описанная здесь модель электронного строения, вероятно, спра-
ведлива для фаз системы Fe—N(/, 8 и a"-Fe8N), а также Nb4O,
Та4О, Ta8N, Tai6O.
§ 4.3. ПРИНЦИП ПЛОТНОЙ УПАКОВКИ И ЕГО РЕАЛИЗАЦИЯ
В СТРУКТУРАХ ИНТЕРМЕТАЛЛИЧЕСКИХ СОЕДИНЕНИИ
Особенности металлической связи объясняют тенденцию
к плотнейшей упаковке атомов в структурах металлических
кристаллов. Важнейшим фактором существования «электронных»
металлических соединений оказалась электронная концентрация;
при этом устойчивость той или иной структуры соединения опре-
деляется минимумом кинетической энергии электронов связи. Са-
ми кристаллические структуры во всех рассмотренных типичных
случаях (структуры а-, 0-, у- и 8-фаз) оказались основанными на
77
тех же атомных координациях, которые вообще характерны для
металлических элементов (К12, К8, Г12). Существование таких
структур, как и в случае металлических элементов, легко объяс-
нить с точки зрения минимума потенциальной энергии атомов
(или ионов) в плотнейших упаковках, поскольку атомные радиусы
компонентов в электронных соединениях должны быть близки
между собой и характер связи остается тот же, что и в чистых
металлах. Смену кристаллических структур при изменении состава
сплава элемента группы Си с многовалентным элементом (и фаз
внедрения), в общем, можно считать подобной смене структур при
переходе от простых металлических элементов более низкой ва-
лентности к элементам с более высокой валентностью в таблице
Д. И. Менделеева. Перед описанием других металлических фаз
необходимо отметить значительное разнообразие их кристалличе-
ских структур и существенное отличие от рассматривавшихся вы-
ше структур плотнейших упаковок.
Прежде чем описывать особенности каждого из этих типов
химических соединений и искать причины их образования, попы-
таемся найти общие признаки между разными типами соединений,
а также между структурами этих соединений и уже рассмотренных
классов металлических фаз — металлов и электронных соединений,
которые могли казаться аномальными. Во-первых, напомним, что
среди структур металлических элементов, кроме о. ц. к., г. ц. к. и
гекс. п. у., а также близких к ним по координации и симметрии
имеют весьма сложные структуры, например, а- и 0-Мп, 0-U.
Известны случаи подобных сложных структур и среди фаз
металлических сплавов. Выше было указано, что некоторые элек-
тронные соединения при электронной концентрации 3/2 имели
структуру типа (3-Мп. Эта структура близка к кубической плот-
нейшей упаковке с координационным числом 12. Структура элек-
тронных соединений типа у-латуни близка к структуре а-Мп. Обе
структуры имеют координацию атомов, близкую к о. ц. к.
Отмечая отклонения в кристаллических структурах от коорди-
наций плотнейшей упаковки (к. ч.= 12 для упаковки из шаров оди-
накового размера), следует указать, что и широко распространен-
ная среди металлических фаз структура о. ц. к. решетки представ-
ляется в этом смысле аномальной. Предлагались различные объ-
яснения причин этой аномалии (изменения энергии электронного
газа, выигрыш в энтропии при повышенных температурах, дейст-
вие направленных химических связей). Однако при этом важно
отметить, что о. ц. к. решетка не очень сильно уступает плотней-
шим упаковкам г. ц. к. и гекс. п. у. в отношении компактности.
Коэффициент заполнения пространства немного меньше (0,68
вместо 0,74), а координационное число, если пренебречь неболь-
шими различиями радиусов первой и второй координационных
сфер (гх=ЕА=Ес=0,87 а и r<2=EF=d), можно принять рав-
ным 14 (8 + 6).
Н. В. Белов отметил интересную особенность структуры о. ц. к.
В отличие от гекс. п. у. и г. ц. к. в о. ц. к. решетке все пустоты
78
можно представить тетраэдрическими (рис. 4.7, см. также
рис. 3.2). Взяв в качестве «кирпича» полиэдры, образованные при
соединении центров соседних атомов, можно наглядно представить
заполнение пространства кристалла. Для заполнения пространства
о. ц. к. кристалла можно использовать «кирпич» одного типа:
неправильные тетраэдры (AB = EF=a, AF=BF=AE = BE —
= а ]ЛЗ/2=0,87 а). В гекс. п.у. или г. ц. к. решетке для такого
представления структуры надо взять два типа полиэдров — пра-
вильные октаэдры и тетраэдры.
Это приводит к большей ком-
пактности структуры и к значи-
тельным флуктуациям плотно-
сти (в тетраэдрических полиэд-
рах плотность выше, в октаэдри-
ческих— ниже, чем в о. ц. к.).
Таким образом, рассмотрение
типичных кристаллических струк-
тур металлических элементов
приводит к выводу, что наиболее,
плотное заполнение пространства
может обеспечиваться двумя спо-
собами:
1. Плотнейшей упаковкой
сфер, которая при одинаковом
радиусе сфер дает координацион-
ное число 12 и характеризуется
двумя типами пустой (тетраэдри-
ческие и октаэдрические).
2. Идеальной упаковкой (т. е.
Рис. 4.7. Координационный много-
гранник для о. ц. к. решетки. Решетка
может быть представлена плотной
упаковкой неправильных тетраэдров
без промежутков) тетраэдров,
которая в случае о. ц. к. решетки дает координационное число
около 14 и характеризуется наличием только одного типа пор
(тетраэдрические).
Координационный многогранник вокруг одного атома в о. ц. к.
решетке должен состоять из 24 плотноуложенных тетраэдров, из
14 вершин 8 должны быть общими вершинами шести соседних
граней, а 6 — четырех граней. Однако в случае о. ц. к. решетки
треугольные грани тетраэдров в многограннике вырождаются
в четырехугольные грани и сам многогранник имеет вид ромбо-
додэкаэдра (см. рис. 4.7). Более общие представления о плотных
упаковках из тетраэдров были развиты Каспером* в результате
анализа сложных структур в соединениях на основе переходных
элементов (сг-, ц- и некоторые другие фазы). Особенность многих
из этих соединений состоит в том, что атомные радиусы их ком-
понентов мало различаются. Если не учитывать различий в хими-
ческой природе атомов, то во всех этих структурах можно вы-
делить координационные многогранники YXV, где Y — атом
* Каспер Д. С. В сб.: Теория фаз в сплавах. Пер. с англ. Под ред.
Я. С. Уманского. М., «Металлургия», 1961, с. 320.
79
в центре многогранника; X — вершины многогранника; п— число
этих вершин, или координационное число. Оказалось, что коорди-
национные многогранники имели п, равное 12, 14, 15 и 16, т. е.
координационное число часто оказывалось более высоким, чем
предельное координационное число для плотнейшей шаровой упа-
ковки (12). Многогранники Каспера имеют следующие особенно-
Рис. 4.8. Координационные многогранники Каспера для коорди-
национных чисел 12 (а), 14 (б), 15 (в), 16 (г)
г-
сти: они одновременно являются выпуклыми, ограничены тре-
угольными гранями, в каждой вершине сходятся пять или шесть
граней. Было показано, что таких многогранников может быть
только четыре. На рис. 4.8 они изображены так, что центральный
атом (в плоскости чертежа) и проекции атомов двух противопо-
ложных вершин совпадают (в центре каждого чертежа); атомы,
находящиеся в одной плоскости (параллельной плоскости черте-
жа), показаны одинаковой штриховкой.
80
Если соединить вершины многогранника Каспера с централь-
ным атомом, то он окажется разделенным на тетраэдры. Таким
образом, в этих случаях, так же как и в случае о. ц. к. решетки,
пространство кристалла можно построить из тетраэдров (непра-
вильных, упаковка правильных тетраэдров невозможна без про-
межутков).
Описанные представления позволили показать, что и в струк-
турах металлических фаз, которые трудно или невозможно опи-
сать как плотнейшие упаковки, действует характерная для метал-
лической связи тенденция к наибольшему заполнению простран-
ства. Разумеется, при выборе конкретных структур, так же как и
при плотнейших шаровых упаковках, должны иметь значение
конкретные точные характеристики химической связи, соотношение
атомных радиусов и другие конкурирующие в определении крис-
таллической структуры факторы. В икосаэдрической упаковке (см.
рис. 4.8,а) даже для сфер равного радиуса может быть достигнута
большая локальная плотность, компактность упаковки, чем при
обычной упаковке по типу г. ц. к. или гекс. п. у. Особенно выгод-
ными рассмотренные координации должны быть при несколько
различающихся размерах атомов. Эта разница естественна в хи-
мических соединениях. Но она может возникнуть и при кристал-
лизации химического элемента, если атомы этого элемента могут
быть в различных электронных состояниях, как, например, в слу-
чае а-Мп или p-U и в случае других переходных элементов. Лавес
отмечает, что наилучшие способы заполнения пространства —
плотнейшая упаковка шаров с к. ч.=12 и плотная упаковка непра-
вильных тетраэдров, к.ч.^12 можно реализовать в пределах одной
решетки (например, а-Мп).
§ 4.4. ИНТЕРМЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ ТИПА o-FeCr
И РОДСТВЕННЫЕ ИМ ФАЗЫ
Соединения типа cr-фаз встречаются во многих практически
важных системах.
Впервые предположение о новом типе металлического соеди-
нения возникло при выяснении причин охрупчивания сплавов
системы Fe—Сг*. Бейн и Гриффитс (1927 г.) назвали предпола-
гаемую фазу cr-фазой. Эти фазы представляют собой интерес для
кристаллохимии как особый тип.
Сигма-фазы образуются только при взаимодействии элементов
переходных групп, существуют в очень широкой области концен-
траций и имеют характерную кристаллическую структуру, подоб-
ную структуре чистого металла — элемента переходной группы —
урана (p-U).
* Сейчас известно несколько видов структурных превращений, способных
вызвать охрупчивание промышленных сплавов на основе Fe—Сг металлических
фаз в связи с особенностями химического состава и кристаллической структуры.
(Меженный Ю. О., Скаков Ю. А.—«Изв. вузов. Сер. Черная металлургия», 1967,
№ 11, с. 120—125.)
6—523 81
Достоверные данные о структуре cr-фазы получили только
к середине 50-годов. Рентгенографически исследовали (1951 г.)
монокристалл сплава Fe—Сг, в котором при отжиге образовалась
о-фаза с ограниченным числом кристаллографических ориентиро-
вок. По этим данным, a-фаза имеет тетрагональную структуру,
пространственная группа Р4/т/гт, элементарная ячейка состоит
из 30 атомов, размер ячейки фазы a-FeCr: а=8,790 А, с=4,559 А
(рис. 4.9,а).
Рис. 4.9. Элементарная ячейка a-фазы (а); представление структуры о-фазы
слоями (сетки Кагомэ) .(светлые и черные кружки относятся к гексагональ-
ным сеткам, координаты которых по оси с z=x/^ и 3/4; заштрихованные круж-
ки — позиции атомов, расположенных друг над другом по вертикали, с коор-
динатами по оси с z=Q и !/2 (б)
Расположение атомов в структуре а-фазы удобно представить в виде гекса-
гональных сеток (типа Кагомэ), повернутых относительно друг друга на 90°,
между которыми располагаются еще атомы, проектирующиеся в плоскости ху
в центры гексагональных ячеек сеток Кагомэ (позиция типа £), с координатами
по вертикали z, равным 0 и 1/2 (см. рис. 4.9,6). Из рис. 4.9,6 видно, что для
атомов в позициях типа Е и С координационное число составляет 14, в позициях
А и D оно равно 12, а в позиции В— 15. Этим координационным числам соот-
ветствуют многогранники, рассмотренные выше '(см. рис. 4.8). Число атомов
в каждой кристаллографической позиции следующее: А — 2, В — 4, С, D и Е—
по 8 атомов. Однако физически неэквивалентных позиций, различающихся по
значениям координационных чисел и по межатомным расстояниям, можно выде-
Таблица 4.3
Характеристики позиций в решетке д-фазы в соединении Со14Сг16
Тип позиций Число узлов в каждой позиции Координационное число Среднее межатсм- ное расстояние, А Сорт дтомов
А 2 12 2,49 1
D 8 12 2,48 J СО
с 8 14 2,65 1 Со и Сг
Е 8 14 2,63 /
в 4 15 2,70 Сг
82
лить только три. В табл. 4.3 указано распределение атомов между этими пози-
циями в соединении СонСг^; подобное распределение было предложено и для
фазы o-FeCr.
Поскольку рассеивающие способности атомов Сг и Fe или Со для рентгенов-
ских лучей мало различаются, непосредственные экспериментальные данные об
упорядоченном расположении атомов в о-фазах получены только из нейтроногра-
фических исследований.
В табл. 4.3 приведены результаты анализа a-фаз Со—Сг и других систем
А—В, где А — элементы таблицы Д. И. Менделеева, располагающиеся левее
Мп, а В — правее Мп (марганец может быть и в той, и в другой группе) *.
Таблица 4.4
Области существования a-фаз в бинарных системах с ванадием
Система (А—В) V, ат. % 4s-|-3r7, электрон/атом
V—Мп 14—25 6,7—6,51 1,03
V—Fe 37—57 7,3—6,9 1,06
V—Со 40—55 7,4—6,8 1,08
V—Ni 55—65 7,2—6,7 1,08
Сигма-фазы образуются переходными металлами V—VI и
VII—VIII групп** при не очень большой разнице атомных радиусов
(обычно до 8%). Область гомогенности cr-фаз в бинарных систе-
Таблица 4.5
Области существования a-фаз в системах с хромом
Система (А—В) Сг, ат. % Температурная область существования а-фаз rairb
Сг—Мп 19—24 (800°С) До температуры плав- ления 0,98
Сг—Fe 44—49 (600°С) Устойчивы в твердом состоянии 1,01
Сг—Со 57—61 — 1,02
Сг—Ni — Нет о-фазы 1,08
мах может быть довольно широкой, но положение этой области
закономерно связано с положением компонентов в таблице
Д. И. Менделеева. Например, в ряду систем Мп, Fe, Со и Ni с V
область существования cr-фазы сдвигается от состава, приближен-
но описываемого как АВ3 для системы V—Мп, до состава А2В
* Это легко понять, если учесть, что атомы Мп могут быть в различных
электронных состояниях, как, например, при образовании сложной структуры
металлического а-Мп.
** Обнаружены о-фазы с участием Al (Nb—А1 и Та—А1) и отмечено стабили-
зирующее действие Si по отношению к о-фазам в ряде тройных систем; А1 и Si
занимают позиции, относящиеся к компоненту типа А (т. е. позиции, занимаемые
обычно Мп, Fe, Со и т. д.).
6* 83
84
для системы V—Ni. Важно отметить, что сохраняется примерно
постоянным среднее количество 4$+Зб/-электронов на атом: около
7 электрон/атом (табл. 4.4, 4.5). Температурная область существо-
вания о-фаз также находится в связи с положением компонентов
в таблице Д. И. Менделеева: чем дальше друг от друга компо-
ненты, тем меньше устойчивость о-фазы (см. табл. 4.5 и рис. 4.10).
В двойной системе Сг—Ni * о-фаза не обнаружена, но неболь-
шие (6%) добавки Мо или Si стабилизируют <г-фазу (рис. 4.11,6).
При значительной разнице атомных радиусов образование о-фазы
возможно только при высокой температуре (рис. 4.10,в). На
рис. 4.11 показаны концентрационные области существования
о-фазы в тройных системах.
Как в двойных (см. табл. 4.4), так и в тройных системах (см.
рис. 4.11) отчетливо видна связь между составом о-фаз и поло-
жением компонентов в периодической системе элементов. Как и
* В двойной системе Сг—Ni or-фаза наблюдалась только в тонких пленках,
полученных конденсацией из пара.
в
Рис. 4.10. Диаграммы состояния сплавов: Сг—Мп (а), Сг—Fe (б) и Fe—Мо (в)
85
в случае электронных соединений типа фаз Юм-Розери, о-фазам
нельзя приписать определенный состав в атомных соотношениях.
Естественно поэтому связать концентрационные области сущест-
вования о-фаз с определенными значениями электронной концен-
трации. Для атомов переходных элементов в металлическом
состоянии точное число электронов, участвующих в связи, неиз-
вестно, поэтому для ориентировочных расчетов можно взять за
основу сумму s+d-электронов. Эта сумма для среднего состава
о-фаз во всех случаях составляет 6,5—7 электронов на атом (см.
табл. 4.4). В ряде двойных сг-фаз была обнаружена сверхпроводи-
мость, дричем максимальная критическая температура оказалась
при составах фаз, соответствующих сумме s+d равно 6,5-электро-
нов на атом. Предполагается, что именно при такой электронной
концентрации плотность состояний на поверхности Ферми дости-
Рис. 4.11. Изотермические сечения
фазовых областей тройных систем
Fe—Сг—V при 600° (а); Сг—Ni—
Мо при 1000° (б); Мп—Со—Мо
при 1240° С (в)
86
гает максимума. Эти факты лежат в основе представления
о сг-фазе как о своеобразном электронном соединении.
Такое представление подтверждается также тем, что о-фаза—
это одна из фаз в последовательности о-, Р-, ц-, R-, /-фаз, доволь-
но строго определенной значениями электронных концентраций.
Это видно на примере тройной системы Мп—Со—Мо, где обра-
зуются все указанные типы фаз (см. рис. 4.11,в). В виде общей
схемы эта смена фаз при изменении электронной концентрации
показана на рис. 4.12.
/-Фаза имеет сложную кубическую ячейку, которая так же, как
и в случае a-фазы, изоморфна структуре металлического элемента,
в данном случае а-Мп (58 атом/ячейка). х-Фазу наблюдали в ряде
двойных систем с участием переходных элементов из групп Sc,
Ti, V или Сг, с одной стороны, и Fe или Re и Os — с другой, а так-
же в некоторых тройных системах с участием уже указанных
элементов в качестве компонентов А и Fe, Со, Ni вместе с Сг, Si,
Ge* в качестве компонентов В.
P-фаза обнаружена в тройных системах (Мо—Fe—Ni,
Со—Мо—Ni, Мо—Мп—Со). Решетка P-фазы ромбическая, про-
странственная группа РЬпт, 56 атом/ячейка. Можно описать
ячейку P-фазы как удвоенную по оси с-ячейку о-фазы. Р-фазу
наблюдали в системе Ti — Мп, а также в ряде тройных систем
с участием переходных металлов, расположенных слева от Мп
в качестве компонента А, и в качестве компонента В—Мп и ме-
* Предполагается, что Si и Ge уменьшают число электронов, участвующих
в связи, т. е. играют роль акцепторов электронов.
87
таллы, расположенные справа от Мп, а также Si. Ячейка 7?-фазы
ромбоэдрическая, пространственная группа /?3 содержит
53 атом/ячейка.
ц-Фаза известна в ряде двойных систем Nb, Та, Мо, W, с од-
ной стороны, и Fe, Со, Ni — с другой. Во всех этих случаях имеется
значительная разница атомных радиусов, и область гомогенности
оказывается довольно узкой. В то же время компоненты принад-
Число электронов вне заполненных оболочек
Рис. 4.12. Схема последовательности фазовых об-
ластей or-, Р-, jx-, R- и %-фаз в зависимости от
электронной концентрации
лежат к ограниченному числу групп таблицы Д. И. Менделеева,
поэтому в отличие от предыдущих фаз для ц-фазы можно указать
атомное соотношение: Fe7Mo6, Co7W6 и т. д. Ячейка ц-фазы ром-
боэдрическая, пространственная группа R3m, 13 атом/ячейка.
Для всех этих фаз характерны большие координационные
числа (12, 14, 15, 16) и высокая степень компактности. Упорядо-
ченное (или частично упорядоченное) расположение атомов, ха-
рактерное для всех этих фаз, в известной степени связано с тем,
что более крупные атомы занимают позиции с большим координа-
ционным числом; кроме того, образование этих фаз и ширина
областей гомогенности ограничиваются соотношением атомных
радиусов, однако определяющим в образовании рассмотренных
фаз является фактор электронной концентрации.
§ 4.5. НЕКОТОРЫЕ ИНТЕРМЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ
ПРИ ПРОСТЫХ СТЕХИОМЕТРИЧЕСКИХ (АТОМНЫХ) СООТНОШЕНИЯХ
Одна из характерных для металлических фаз особенностей со-
стоит в том, что их области гомогенности имеют значительную
ширину. Эта особенность характерна для всех рассмотренных вы-
ше фаз. При этом часто область существования фаз данного типа
нельзя привязать к определенным простым атомным соотноше-
88
ниям. Простые стехиометрические атомные соотношения характер-
ны для упорядоченных твердых растворов. Однако среди интер-
металлических соединений встречаются и другие «промежуточные
фазы» *, для которых характерны простые атомные соотношения.
В общем случае для металлических фаз эти простые соотношения
не соответствуют соотношениям валентности (как для соединений
с ионным или ковалентным типом связи), но обязательно соответ-
ствуют соотношению определенного сорта позиций в кристалличе-
ской решетке при упорядоченном расположении атомов.
Известно около 200 различных структурных типов среди изу-
ченных 1300 интерметаллических бинарных соединений. Однако
примерно 2/3 соединений принадлежат к одному из 13 типов,
приведенных в табл. 4.6. Значительное число интерметаллических
соединений имеет кристаллические структуры с теми же коорди-
нациями атомов, которые типичны вообще для металлических
кристаллов с к. ч.= 12 и к. ч. = 8. Как видно из табл. 4.6, соеди-
Таблица 4.6
Распределение бинарных интерметаллических соединений
по структурным типам (Лавес, 1967 г.)
Структурный тип Число соединений Характер координации атомов
MgCu2 MgZn2 AuCu. Mg AuCu CsCl (р-латунь) W CaZn5 f-U(a) у-латунь Cr3Si A1B2 CuAi2 158 1 оол 76 J 234 108 1 45 | 182 29 J ‘Ji } >5' 58 49 45 34 29 25 Фазы Лавеса,. к.ч. = 12,16 Фазы на основе плотней- ших упаковок (гекс. п.у. и г.ц.к.), к.ч.=12 Фазы на основе о.ц.к., к.ч.=8
нения на основе плотнейшей шаровой упаковки с к. ч.= 12 или
на основе о. ц. к. упаковки с к. ч. = 8 в большинстве случаев имеют
упорядоченные структуры AuCu и AuCu3, CuZn(CsCl) и Fe3Al,
и поэтому их состав должен описываться формулами АВ и АВ3.
АВ. Из металлических фаз бинарных систем равноатомного
состава было рассмотрено два структурных типа (CuAu и (3-ла-
тунь или CsCl). Всего найдено 11 разных структурных типов
интерметаллических соединений такого состава, но более 2/3 всех
* Термин «промежуточные» фазы обычно употребляют для обозначения фаз,
образующихся не на основе структуры компонентов, т. е. занимающих на диа-
граммах состояния «промежуточное» положение.
89
изученных соединений АВ имеют структуру типа CsCl. К ним от-
носятся многие фазы Юм-Розери с электронной концентрацией
3/2 (тип fJ-CuZn) и ряд соединений переходных элементов (NiTi,
FeTi и др.).
АВ2. Для всех рассмотренных выше фаз (кроме фаз внедрения)
характерна относительно небольшая разница атомных радиусов
компонентов. Это обстоятельство часто рассматривают как усло-
вие образования указанных фаз. Отметим также, что при одина-
ковых размерах шаров максимальное к. ч.= 12. При некотором
различии атомных радиусов компонентов становится возможным
появление в решетке позиций с более высокими координационны-
ми числами. В a-фазах это различие должно быть небольшим.
При более значительном различии атомных радиусов, а именно
когда Га/гв—1,225, при упорядоченном расположении атомов и
стехиометрическом соотношении АВ2 возникают фазы с компакт-
ными структурами, характеризующимися координационными
числами 12 и 16,— фазы Лавеса.
самое большое число изученных
в бинарных системах и много
соединений в тройных системах.
Кроме перечисленных в табл. 4.6
структурных типов MgCu2 и
MgZn2 имеется еще тип MgNi2,
не включенный в эту таблицу, по-
скольку число соединений этого
типа в бинарных системах очень
невелико. Число фаз типа Лаве-
са (MgCu2, MgZn2 и MgNi2) из
интерметаллических соединений
состава АВ2 достигает 2/3, при-
чем большинство соединений
АВ2 относится к структурному
типу MgCu2.
ственная группа Fd3tn) легко
представить, выделив подрешет-
ку больших атомов (Mg) типа
решетки алмаза (рис. 4.13), меньшие по размеру атомы Си зани-
мают тетраэдрические поры, располагаясь в них группами из
четырех атомов в виде тетраэдра.
Таким образом, элементарная ячейка MgCu2 содержит 8 ато-
мов Mg и 16 атомов Си (т. е. 8 формульных единиц).
Идеальное соотношение атомных радиусов компонентов гА1гв,
одинаковое для фаз Лавеса всех трех структурных типов, удобно
определить на примере кубической структуры MgCu2. Для иде-
альной плотной шаровой упаковки будем считать, что происходит
соприкосновение больших атомов (А) вдоль пространственной
диагонали алмазной решетки и одновременно имеет место сопри-
косновение меньших атомов (В) вдоль плоской диагонали (см.
90
К фазам Лавеса относятся
интерметаллических соединений
Рис. 4.13. Элементарная ячейка фазы
типа MgCti2:
А — большие атомы (Mg); В— меньшие
атомы (Си)
Структуру MgCu2 (простран-
рис. 4.13). Тогда
= /4; dBB = а/2 /4 и ^дд/^вв = га/гв =
= /3 //2 = 1,225.
В алмазной подрешетке из атомов А к. ч. = 4, кроме того, каж-
дый атом A (Mg) окружен 12 атомами сорта В (Си). Каждый атом
В (Си), в свою очередь, имеет 6 соседей типа A (Mg) и 6 соседей
типа В (Си). Таким образом, структура фазы MgCu2 характери-
зуется двумя типами позиций: 8 позиций (а) с координационным
числом 16 (12 + 4) и 16 позиций (d) с координационным числом 12
(6 + 6).
а
б 6
Рис. 4.14. Представление структурных типов фаз Лаве-
са ДВ2 различными сочленениями лавесовских поли-
эдров: MgCu2 (a); MgZn2 (б); MgNi2 (в)
Соединив центры 12 атомов В (Си), окружающих атом A (Mg)
в структуре типа MgCu2 (см. рис. 4.13), получим полиэдр в виде
тетраэдра с усеченными вершинами (лавесовский полиэдр).
Структуру MgCu2 можно представить состоящей из лавесовских
полиэдров, сочлененных по граням (рис. 4.14,а), другие структур-
ные типы фаз Лавеса получаются при других способах сочлене-
ния полиэдров (см. рис. 4.14,6 и в).
Для MgNi2, пространственная группа С§1ттс, число формуль-
ных единиц в ячейке г=8; для MgZn2, пространственная группа
Ptymmc, число формульных единиц в ячейке г=4. Структуру
MgZn2 можно рассматривать как гексагональный аналог MgCu2
91
(подобно аналогии гексагональной и кубической модификации
ZnS — вюрцита и сфалерита). Если MgZn2 характеризуется двух-
слойной (гексагональной) упаковкой лавесовских полиэдров, то
MgCu2 — трехслойной (кубической), a MgNi2 — четырехслойной
упаковками.
Размерное соотношение является определяющим фактором об-
разования фаз Лавеса, поэтому даже в тех случаях, когда соот-
ношение радиусов металлических элементов не совпадает с иде-
альным*, можно предполагать такую деформацию электронных
оболочек, которая устанавливает соответствие размеров атомов
Mg-Cu-Al
Mq-Cu-Zn
Mg-Ag-Zn
Mq-Cu-Si
Mq-Co-Zn
Mg-Zn-AI
Mq-Cu-Ag
Mg-Ag-Al
1,33 1,6 1,6 1,8 2,0 электрон/атом
ГПТ11111 Tuna Mg Си2 e Tuna MqZn
4--Ы-++ Tuna Mq Ni2
Рис. 4.15. Области существования фаз Лавеса разного
типа в некоторых тройных сплавах на основе Mg (кон-
центрация многовалентного элемента отсчитывается как
электронная концентрация)
плотной упаковке. Однако может быть существенным фактор
электронной концентрации. Об этом свидетельствует закономерная
смена фаз от MgCu2 до MgZn2 в тройных сплавах на основе
MgCu2—MgZn2, MgCu2—MgAl2 или MgCu2—MgSi2 (рис. 4.15).
Это же доказывает анализ изменения некоторых свойств
(растворимость водорода, магнитная проницаемость) в зави-
симости от содержания многовалентного компонента в тех же
системах. Наконец, представление о существовании предель-
ной электронной концентрации согласуется с тем, что при взаимо-
действии переходных металлов образование фаз Лавеса невоз-
можно при s + d=8. Однако известны фазы Лавеса, которые,
несмотря на очевидное различие электронных концентраций,
имеют одну и ту же структуру (тип MgCu2—Na2K, Mg2Ba и
А12Ва).
АВ5. Фаза Лавеса типа MgCu2 образуется при взаимодейст-
вии Со и Ni с редкоземельными элементами, однако при7 другом
соотношении тех же компонентов (например, SmCo5) образуется
* В известных фазах Лавеса Га/гв может изменяться от 1,1 до 1,3 (и даже
от 1,05 до 1,68).
92
фаза с гексагональной структурой, похожей на структуру MgZn2
(рис. 4.16). В других случаях соединений типа АВ5 (например,
PdBe5 и UNi5) структура оказывается похожей на структуру
MgCu2. Эти аналогии можно понять, если представить формулу,
например, UNi5 в виде UNiNi4 и сопоставить ее с удвоенной фор-
мулой Mg2Cu4. Очевидно, при U'Nis половина положений атомов
Mg в структуре MgCu2 занята большими атомами U, а все осталь-
ные положения Mg и Си заняты атомами Ni (см. рис. 4.13).
Рис. 4.16. Структура SmCos (тип CaZn5)
А3В. Структура типа Cr3Si*. Своеобразную структуру имеют
некоторые соединения состава А3В, в которых атомы А — один из
переходных элементов IV—VI групп, а атомы В — один из пере-
ходных элементов VIII группы таблицы Д. И. Менделеева либо
Ga, Si, Ge, Sn, As, Sb, а также Hg и металлы группы Cu.
Ячейка Cr3Si кубическая (рис. 4.17), а=5,04 А, пространствен-
ная группа РтЗп, число формульных единиц на ячейку г=2.
Атомы Si (А) образуют о. ц. к. подрешетку, атомы Сг (В) зани-
мают центры тетраэдров, располагаясь попарно на каждой грани
куба. Позиции первого типа характеризуются координационным
числом 12 (для Cr3Si di = 2,82 А), в позициях второго типа коор-
динационное число 14 (2—d2=2,52, 4—d3 = 2,82, 8—d4 = 3,09 А),
что дает координационный полиэдр в виде икосаэдра (см.
рис. 4.8,а).
Можно назвать сплавы, свойства которых в значительной сте-
пени определяются рассмотренными здесь фазами. Фазы равно-
атомного состава со структурой типа CsCl (например, NiAl) или
фазы типа Fe3Al позволяют получить тонкодисперсную гетероген-
ную микроструктуру, если матрица имеет о. ц. к. структуру. Такая
* Ранее ошибочно обозначили этот структурный тип (3-W. В действительно-
сти ^-модификации W не существует; структура (3-W представляет собой низший
окисел W, отличающийся от подобного окисла Сг3О и интерметаллических фаз
типа Cr3Si не вполне упорядоченным расположением атомов W и О. В литера-
туре часто используется обозначение А15 (как для чистого элемента).
93
структура создается, в частности, в высокопрочных мартенситно-
стареющих высоконикелевых или хромоникелевых сталях. Подоб-
ная структура обеспечивает высокую коэрцитивную силу в сплавах
типа алии (Fe—Ni—Al)
или магнико (Fe—Со—
Ni—Al—Ti). Фазы на
основе компактной г. ц. к.
решетки (АВ или А3В)
также дают высокоди-
сперсные микроструктуры
в г. ц. к. матрице. К таким
Рис. 4.17. Структура типа Cr3Si: элементарная ячейка (а), проекция на
плоскость (001) (б)
сплавам относятся многие дисперсионно-твердеющие сплавы, на-
пример жаропрочные сплавы на основе Ni—Сг—Ti—Al.
Фаза Лавеса оказывается упрочняющей фазой в некоторых
теплоустойчивых сталях.
На основе соединения CosSm в последние годы удалось полу-
чить постоянные магниты с рекордной магнитной энергией, а на
основе некоторых фаз со структурой типа Cr3Si — сверхпроводя-
щие материалы с высокой критической температурой (например,
Nb3Sn).
Задачи к гл. 4
Задача 4.1. Найти эффективный радиус углерода в карбиде тантала ТаС0)7.
Задача 4.2. Кристалл вюстита состава FeO содержит 76,08% Fe. Определить
тип твердого раствора, если плотность кристалла равна 5,613 г-см-3, период
решетки 4,2816 А.
Задача 4.3. Объяснить, почему область существования высокотемпературно-
го карбида вольфрама с г. ц. к. металлической подрешеткой со стороны углерода
ограничена меньшей концентрацией (WCo,?), чем для карбида тантала или
ниобия (ТаС, NbC).
о
Задача 4.4. Атомные радиусы Си—1,28; Ag—1,44 и Mg — 1,60 А. Почему
в сплавах Ag—Mg возникают фазы Юм-Розери, а в сплавах Си—Mg их нет?
ГЛАВА 5
ДИФФУЗИЯ В МЕТАЛЛАХ
Изучение диффузии в металлах и сплавах необходимо, во-пер-
вых, для понимания изменений, происходящих в твердых телах
при высоких температурах, так как процессы диффузии во многом
определяют кинетику процессов выделения фаз, окисления, пол-
зучести и т. д., во-вторых, для получения сведений о поведении
точечных дефектов в кристаллах.
Проблему изучения диффузии можно условно разделить на
две части. В первой — феноменологической — металл рассматри-
вают как континуум, т. е. пренебрегают его атомной структурой.
При этом может быть рассмотрена большая часть структурных
изменений, происходящих в результате диффузионных процессов.
Кинетику изменений обычно описывают системой дифференциаль-
ных уравнений. При составлении уравнений используют предпо-
ложения, впервые сделанные еще в 1855 г. Фиком. Во второй
части — микроскопической — учитывают атомное строение и атом-
ные процессы, связанные с диффузией.
§ 5.1. ФЕНОМЕНОЛОГИЧЕСКАЯ ТЕОРИЯ ДИФФУЗИИ
Уравнения диффузии (первый и второй законы Фика). Если
градиент концентрации одного из компонентов направлен по оси
х, то поток этого компонента направлен в сторону уменьшения
градиента концентрации и равен
J = - D
дх *
(5-1)
Отношение = —
ас/dx
D называют коэффициентом диффузии.
Уравнение (5.1)—первый закон Фика — неудобно для практи-
ческого использования. Более удобным оказывается другое диф-
ференциальное уравнение, которое можно получить из уравнения
(5.1) и из условия, что концентрация в рассматриваемом объеме
изменяется во времени (условие баланса вещества). В оконча-
тельном виде это уравнение — второй закон Фика имеет вид
дс____д I г\ дс \
dt дх дх J'
(5-2)
95
Для трех измерений уравнение (5.2) запишем в виде —^~ =
=—div 7. Это уравнение выражает закон сохранения вещества
в форме уравнения непрерывности и остается справедливым и
в тех случаях, когда уравнением (5.1) пользоваться нельзя (на-
пример, при наличии добавочных градиентов, кроме концентраци-
онного).
Коэффициент диффузии — это параметр, характеризующий
скорость диффузии. С увеличением температуры для металлов
коэффициент диффузии резко возрастает, изменяясь по экспонен-
циальному закону:
D(T)=Doexp(-Q//?T). (5.3)
Здесь Do — предэкспоненциальный или частотный множитель; Q и
Do связаны с физико-химическими свойствами металла, а также
диффундирующих частиц; Q — энергия активации; R — газовая
постоянная; Т — абсолютная температура.
Экспоненциальный характер изменения коэффициента диффу-
зии в зависимости от температуры был установлен эмпирически
(закон Аррениуса), а затем обоснован теоретически.
Размерность коэффициента диффузии можно определить из
уравнения (5.1). Обычно используют размерность сантиметр квад-
ратный на секунду [см2/с], которая означает, что 1 моль вещества
проходит сквозь площадку в 1 см2 за 1 с при градиенте концентра-
ции dcfdx, равном молю на сантиметр. Иногда коэффициенты диф-
фузии относят не к секунде, а к суткам. В этом случае размер-
ность имеет вид [см2/сут]. Значения констант, входящих в урав-
нение (5.3) для некоторых диффузионных пар, приведены
в табл. 5.1.
В практике физических исследований обычно встречаются две
группы диффузионных задач. В первой из них неизвестны коэф-
фициент диффузии D, а также предэкспоненциальный множитель
DQ и энергия активации Q. Требуется их определить из известных
диффузионных характеристик. Во второй группе задач по заранее
известному коэффициенту диффузии необходимо определить те
или иные диффузионные характеристики: распределение концен-
трации диффундирующего вещества, поток вещества через какую-
либо поверхность, количество вещества, проникшего в материал
за известное время или же вышедшее из него.
Для решения указанных диффузионных задач необходимо
.знать аналитические зависимости между диффузионными харак-
теристиками в интегральном виде. Эти зависимости могут быть
получены интегрированием основного уравнения (5.2) при соот-
ветствующем выборе начальных и граничных условий, характери-
зующих конкретные физические условия рассматриваемой задачи.
Примеры решения основного уравнения диффузии. Предположим, что на-
чальное распределение концентрации в теле задано в виде
с(х, O)=f(x) (5.4)
96
Таблица 5.1
Константы диффузии, входящие в уравнение D=D0 exp (—Q/RT)
Растворитель Диффундирующий элемент | Do, см2/с Q. кал /г-атом
f-Fe Fe (самодиффузия) | 0,6 68 000
с I 0,04+0,08% (C) 31 400+800
N 3,3-10-* 34 600
А1 — 44 000
р — 46 100
S — 26 700
Сг — 80 000
Мп 0,486+0,011 (Mn)% 66 000
Ni 0,344+0,012 (Ni)% 67 500 ,
Мо 6,8.10-2 59 000
W 62 500
a-Fe Fe (самодиффузия) 5,8 59 700
N 4,6-10-* 17 950
С 0,02 20 000
a-Co Со(самодиффузия) 0,367 67 000
₽-Co Со(самодиффузия) 0,18 46 500
А1* l,2-10-2 37 500
Zn* 8-10-1 38 000
Sn* 1,0 45 000
Si* 5,2-10-2 39 900
Be* 4,5-10“5 27 900
Cd* 3,5-10-9 8200
Ni Ni (самодиффузия) 1,3 66 800
Mo 0,013 50 800
Cu Си (самодиффузия) 0,20 47 100
Zn Zn (самодиффузия | ] осис) 0,13 21 800
„ 1 оси c Al (самодиффузия) 0,58 24 300
Al 1,71 34 000
Al Cu 2,3 34 900
Mg 15-10-2 38 500
Si (при 0,5% Si) 0,58 31 400
Zn (при 0,84% Zn) 5,5-10-2 25 900
Pb Pb (самодиффузия) 4,0 27 500
TI 3,6-10-2 21000
» Sn 3,4-10“2 24 000
Au 4,9-Ю-1 13 000
Ag 7,5-10-2 15 200
Bi 7,7-lC-3 18 600
Hg 3,6-Ю-1 19 000
Cd 2,1 - IO"2 18 000
Au Au (самодиффузия) 0,157 53 000
Au Cu 5,8-10-4 27 400
Pt 1,2-Ю-3 39 000
Ag Ag (самодиффузия 0,9 45 900
объемная)' 5,9-Ю-5
Au 24 800
Sn 7,9-10-5 21 400
» Cd 4,9-10-5 22 350
w "(большие зер- W (самодиффузия) Th 11,5 4,1-Ю"3 142 000 94 400
на) w(малые зерна) Th 7,5-Ю"1 94 000
w (объемная _ диффузия) Th 1,0 120 000
7—523
97
Продолжение табл. 5.1
Растворитель Диффундирующий элемент Do, см2/с Q. кал/г-атом
W Zr 1,0 78 000
Мо 5-10-3 80 500
TiC0,68 С НО 11 400
Bi Bi (самодиффузия || оси с) io-3 30 000
9 Bi (самодиффузия J_ оси с) 2,4-Ю46 137 000
* Значения констант Q и £>0 даны для концентраций диффундирующего элемента, равных нулю.
Значения эти получены экстраполяцией данных для весьма малых концентраций.
и коэффициент диффузии не зависит от концентрации. Решение уравнения (5.2)
в общем случае для неограниченного тела получают в виде
с(х, t)=T(t)X(x), (5.5)
где функция T(t) зависит только от времени, а Х(х)—только от координаты
(для простоты ограничиваемся одномерной диффузией).
Общее решение уравнения диффузии практически используется редко, так
как имеется мало практических задач, в которых требуется рассчитать измене-
ние со временем распределения концентрации в телах достаточно больших раз-
меров. Однако оно используется для получения частных решений.
Решение для диффузии из бесконечно тонкого слоя.
В этом случае распределение концентрации имеет вид
а
с (x,t) = iVlzDt ехр х2/4£>4 • (5-6)
Здесь q — количество вещества (в тех же единицах, что и концентрация),
приходящееся на 1 см2 слоя.
Функция ехр (—x2/4Dt) четная, поэтому распределение концентрации, созда-
ваемое бесконечно тонким слоем в обоих полупространствах, симметрично отно-
сительно начала координат или середины слоя. Со временем начальное распре-
деление постепенно расплывается, оставаясь симметричным относительно х = 0.
Величина максимума кривой распределения в точке х = 0 сМакс = ^/2К nDt
уменьшается обратно пропорционально корню квадратному из времени
(рис. 5.1,а).
Решение для диффузии из слоя конечной толщины. Рас-
пределение концентрации при толщине слоя 2h имеет вид
с (х, t) = е0/2 [erf (Л + х)/2 VDt + erf (h — х) /2 VDt], (5.7)
где erf — функция ошибок (error function) Гаусса:
у
еНг/ = 2/Ктс ехр (—z2) dz.
о
Из графика функции с(х, t) (см. рис. 5.1,6) видно, что при t—>оо концентрация
всюду обращается в нуль (erf 0 = 0), так как конечное количество вещества q =
=2c0h распределяется по бесконечной области.
Решение для диффузии из полубесконечного простран-
ства. Начальное распределение концентрации задано так, что при всех х<0
с(х, О)=со, а при всех х>0 с(х, 0)=0. Для этого случая решение имеет вид
с (х, t) = Cfi/2(l — erf (х/2/D?))- (5-8)
98
Следует отметить, что для всех />0 концентрация в плоскости раздела (х =
=0) постоянна и равна с0/2 (см. рис. 5.1,в).
Некоторые общие закономерности. Выше отмечено, что значе-
ние максимума на кривой распределения концентрации при диф-
фузии из бесконечно тонкого слоя уменьшается обратно пропор-
ционально корню квадратному из времени. При диффузии из слоя
конечной толщины наблюдается тот же эффект, но он становится
заметным через значительно большее время. Теперь определим,
по какому закону изменяется расстояние до плоскости, в которой
Рис. 5.1. Распределение концентра-
ции при диффузии из бесконечно тон-
кого слоя в тело неограниченных раз-
меров (а); из слоя конечной толщи-
ны в тело неограниченных размеров
(б); из полубесконечного простран-
ства (в)
концентрация в е раз меньше, чем в плоскости х=0. При х=0
с(0, t) = q/2 ехр(О). В плоскости, где концентрация в е раз
меньше, с(х, /)=с(0, /)/е. Приравнивая концентрации в обеих
рассматриваемых плоскостях, получаем
с (0, f)/e = (q[2 У itDt )ехр ( — xt1/4D0-
Решая это уравнение относительно получаем
(5.9)
т. е. расстояние между этими плоскостями изменяется пропорцио-
нально корню квадратному из времени.
При решении уравнения диффузии предполагалось, что мате-
риал в направлении диффузионного потока имеет бесконечную
протяженность.
Произведем оценку размера тела, который может считаться
бесконечно большим. Для этого зададимся количеством продиф-
фундировавшего вещества, которым можно пренебречь начиная
с какого-то размера тела Х[.
Тогда общее количество вещества, продиффундировавшее за
00
время t, равно qt = J exp (— x^/^Dt)dx. Количество же вещества,
—00
которым условились пренебречь,
q2 — J exp (— x*]Wt) dx. (5.10)
Xi
Искомую величину хг можно найти из заданного отношения qjqv
Если 92/д1 = 10“3, т. е. составляет 0,1%, то хх = 4УDt. Други-
ми словами, при протяженности тела большей 4 ]/DZ условие бес-
конечности выполняется. Следует иметь в виду, что Xi зависит от
D и от времени. Поэтому для разного времени требуемая протя-
женность материала разная.
Здесь рассмотрены лишь некоторые решения уравнения диф-
фузии, наиболее часто встречающиеся и на практике.
Решение уравнения диффузии при переменном значении коэф-
фициента диффузии. Рассмотренные примеры решения диффузи-
онного уравнения справедливы только для постоянного значе-
ния D. В реальных экспериментах D может зависеть от состава,
и, поскольку есть градиент концентрации, должен меняться от
точки к точке. Второе уравнение Фика для случая, когда
D=D[c(x)];—D(x), имеет вид
дс dD дс . ~ д2с ,с 1 п
-дГ^дг+я-д^-- <5-Н)
Введем новую переменную i]=x/Z1/2 и будем считать, что с(х, t)
определяется теперь только новой переменной, т. е. c=c(ij).
В этом случае второе уравнение Фика примет вид
de __ d In de
2 dri di\ y-
Применительно к случаю диффузии в бесконечной паре, началь-
ные условия для которой в переменных х и t записывались в виде
с = Со при х<0, t=0 и с=0 при х>0, /=0, переход к новой пере-
менной rj можно осуществить следующим образом. Точка х=0
исключается из рассмотрения при /=0, и исходная концентрация
не зависит от расстояния, поэтому, если отбросить разрыв при
х=0, начальные условия для переменной г] будут иметь вид с—с^
при т] = —оо и с=0 при т] = оо. Так как уравнение для переменно?!
т] содержит только полные дифференциалы, можно произвести
сокращение на 1/dr] и провести интегрирование от с=0 до с=с',
где с' — любая концентрация в интервале 0<с'<с0’.
100
Значения с(х) можно получить для любого фиксированного зна-
чения времени, поэтому, переходя опять к переменным х и t, имеем
с'
---J Xdc = dcjdx]^ = Dt (dcldx)c~c,. (5.13)
О
Последнее равенство отражает тот факт, что на бесконечности
dcldx—Q при c—G. Учитывая, что dc/dx—О при c—Cq, получаем
условие
Со
j xdc = 0.
о
(5.14)
Это условие определяет плоскость, на которой х=0, т. е. плос-
кость, не совпадающую с исходной плоскостью раздела и распо-
ложенную между диффузионными зонами. Эту плоскость назы-
вают поверхностью Матано,
Теперь окончательно запишем выражение для коэффициента
диффузии для любого значения концентрации:
(5.15)
о
Вычисления по этому соотношению можно производить графиче-
ским интегрированием и дифференцированием экспериментально
полученной зависимости с(х).
При массовых расчетах целесо-
образно использовать электронные
вычислительные машины (ЭВМ).
Для этого экспериментальная кри-
вая с(х) по точкам с интервалом
Дх вносится в память ЭВМ. Вели-
чина Ах определяется необходимой
точностью конечного результата.
Алгоритм вычислений следующий.
Весь заданный интервал по х де-
лится пополам и вычисляются пло-
распределения
поверхность М.а-
Рис. 5.2. Кривая
концентрации и
тано (площади заштрихованных
участков справа
центрационной
быть равны)
и слева пт кон-
кривой должны
щади Si и S2 (рис. 5.2). Затем от
большей площади вычитается ма-
лая часть и прибавляется к мень-
шей. Эта процедура продолжается
до тех пор, пока Si и S2 не станут
равны в пределах заданной точности.
В результате этой операции определяют положение плоскости
Матано, т. е. находят ХМ-
Затем для каждого значения х, в котором вычисляют коэффи-
циент диффузии, определяют dx[dc = Axl(cn — сп_х) и по формуле
101
трапеций вычисляют xdc. Полученные значения перемножают,
о
а результат умножают на 1/2Л
Расчет всей экспериментальной кривой с(х) на машине М-20
занимает около 2 мин.
Диффузия в поле напряжений. Рассмотренные решения урав-
нения диффузии справедливы лишь в случае, когда перемещение
диффундирующего вещества определяется исключительно гради-
ентом концентрации. В общем случае поток вещества будет пред-
ставлять собой сумму потоков, возникающих под действием гра-
диента концентрации и добавочного силового поля. Частным
случаем такого поля является поле неоднородных напряжений
(деформаций).
Причины возникновения поля напряжений в твердом теле сле-
дующие: его неоднородная деформация, фазовые превращения,
неравномерный нагрев и т. д. Микроскопические деформации мо-
гут быть вызваны внедренными атомами или вакансиями. Поле
напряжений, возникающее вокруг внедренного атома в твердом
растворе, способствует притяжению атома к дислокации. В ре-
зультате может оказаться, что суммарный диффузионный поток
будет направлен в сторону не меньшей, а большей концентрации.
Это так называемый случай восходящей диффузии.
Второе уравнение диффузии с учетом влияния поля напряжений
имеет вид
(5.16)
dt дх2 дх2’ ' '
гдё е — упругая деформация. Коэффициент диффузии D" харак-
теризует поток, связанный только с влиянием градиента напряже-
ний (деформаций). По вычислениям С. Т. Конобеевского величи-
на D" пропорциональна относительной разнице атомных радиусов
компонентов сплава (гв—Га)/га> Атомы с большим радиусом при
наличии деформации стремятся переместиться в растянутые слои
сплава, а атомы с меньшим радиусом — в сжатые. Если перво-
начально компоненты сплава были распределены в кристалличе-
ской решетке сплава статистически равномерно, так как градиент
концентрации был равен нулю, то при наличии неоднородного
напряженного состояния за счет второго члена уравнения в твер-
дом растворе будет протекать диффузия, приводящая не к вы-
равниванию концентраций, а к разделению компонентов. Создаю-
щаяся в результате восходящей диффузии химическая неоднород-
ность, характеризуемая в каждом участке градиентом концентра-
ции, приводит к возникновению встречного диффузионного потока,
обусловливаемого первым членом правой части уравнения (5.16)
и стремящегося восстановить первоначальное равномерное рас-
пределение компонентов в кристаллической решетке твердого
раствора. Равновесное распределение компонентов при неизмен-
102
ной деформации решетки наступит при равенстве обоих потоков,
т. е. при условии
С эффектом восходящей диффузии С. Т. Конобеевский связы-
вает упрочнение деформированных твердых растворов при на-
греве.
Для объяснения зависимости коэффициента диффузии от
концентрации диффундирующего элемента Б. Я. Любов и
Н. С. Фастов* применили представление о влиянии неоднородно
напряженного состояния на диффузионные явления. Переменная
концентрация твердого раствора всегда связана с неоднородно
напряженным состоянием, причем градиент концентрационных на-
пряжений определяется упругими константами твердого раствора,
зависимостью периода решетки твердого раствора от концентра-
ции последнего и, наконец, градиентом концентрации.
Влияние поля напряжений на интенсивность протекания диф-
фузии может оказаться весьма значительным. Я. С. Уманский**
установил, что толщина карбидного слоя, образовавшегося на
поверхности недеформированной танталовой пластинки, находив-
шейся в карбонизующей среде в течение 12 ч при температуре
1900°С, была меньше 0,01 мм, а та же пластинка после холодной
прокатки с обжатием на 75% покрывалась за 1 ч карбидным
слоем толщиной 0,5 мм.
При электронографическом исследовании структуры тонких
слоев цинка, последовательно нанесенных конденсацией на поли-
рованную поверхность медной пластинки, обнаружено, что первые
слои цинка при комнатной температуре целиком растворились
в меди (интерференционные линии цинкового слоя исчезли через
'10 с после конденсации). Полированная поверхность приобрела
характерный оттенок латуни. Такая интенсивная диффузия при
комнатной температуре обусловлена чрезвычайно большими ис-
кажениями кристаллической решетки, вызванными полировкой.
Очевидно, искаженность кристаллической решетки увеличивает
коэффициент диффузии. Но не следует представлять этот эффект
объемным, равномерно охватывающим всю массу деформирован-
ного металла. Это видно из анализа приведенных в литературе
экспериментальных данных об энергии остаточных упругих напря-
жений после деформирования металла (т. е. об энергии искаже-
ний кристаллической решетки). Оказалось, что ее запас не превы-
шает 100—200 кал/(г-атом), составляя лишь ничтожную долю
теплоты диффузии, измеряемой несколькими десятками тысяч
калорий. Рентгенографические исследования показали, что энергия
эта распределена по объему деформированного металла очень
неравномерно: около 90% остаточной энергии деформирования
*Любов Б. Я., Фастов Н. С. «Докл. АН СССР», 1952, т. 84, с. 947.
** Уманский Я. С. Карбиды твердых сплавов. М., Металлургиздат. 1947.
103
сосредоточено в тончайших слоях у плоскостей сдвига, охватыва-
ющих 2—3% общего числа атомов. Здесь искаженность кристал-
лической решетки значительна. В этих слоях энергия деформации
достигает 10 ккал/(г-атом). Очевидно, вдоль плоскостей сдвига и
происходит в основном диффузия в пластически дефррмированном
металле.
Необходимо отметить, что диффузия в реальных технологиче-
ских процессах происходит при температурах, достаточных для
возврата и даже рекристаллизации деформированных металлов,
снижающих или устраняющих совсем искаженность кристалличе-
ской решетки*.
Кроме поля напряжений (деформаций) на диффузионную по-
движность атомов в твердых телах могут существенно влиять и
другие поля, в частности электрические.
При изучении диффузии Си в Ge обнаружено, что при темпе-
ратуре выше 800°С наложение электрического поля приводит
к смещению концентрационной кривой в сторону отрицательного
электрода. При температуре ниже 800°С такого смещения не
наблюдали. Обнаруженная закономерность позволяет считать, что
при высокой температуре Си присутствует в Ge в виде положи-
тельных ионов.
Зависимость коэффициента диффузии от симметрии кристаллической решет-
ки. Металлы и сплавы представляют собой совокупность кристаллов разных
размеров, которые могут быть расположены относительно друг друга различным
образом. Если размеры этих кристаллов малы и расположены они так, что не
образуют текстуры, то предположение об изотропности при диффузии в таком
материале справедливо. При рассмотрении же диффузии в монокристалле сле-
дует иметь в виду его симметрию (пространство, где происходит диффузия, уже
не изотропно). В общем случае суммарные диффузионный поток и градиент кон-
центрации могут оказаться направленными не параллельно. В этом случае надо
считать, что каждая из компонент потока зависит от всех компонент градиента
концентрации:
дс__ дс
Jx=—Dlld>: Dl2 ду D13 dz;
(5.18)
дс__ de _
= — ду. D23 D33 dz .
Рассмотрим монокристалл с кубической решеткой. Систему координат выбе-
рем так, чтобы она совпадала с ребрами элементарной ячейки. Благодаря сим-
метрии кубической решетки оси х, у и z неразличимы. Отсюда сразу следует,
что 2)ii = D22=^33. Свойством кубической решетки является также то, что при
i#=j Z)fj=O. Таким образом, используя только свойства симметрии кубических
решеток, можно показать, что они являются изотропными для процессов диффу-
зии. Решетки с другой симметрией не изотропны. Для гексагональных кристаллов
Dii = Z)22¥=^зз, т. е. имеется изотропность только в плоскости базиса. Аналогич-
ная картина наблюдается и в тетрагональных решетках. В ромбических решет-
ках все три коэффициента диффузии (1)ц, D22 и D33) различны.
Таким образом, в случае текстуры металла с некубической симметрией ре-
шетки значения коэффициентов диффузии следует привязывать к типу и направ-
* Самый факт диффузии обусловливает по существу возможность рекристал-
лизации.
104
лению оси текстуры. Для металлов с кубическими решетками анизотропию диф-
фузии можно наблюдать при возникновении в материале одноосного напряжен*
ного состояния *, на что обратил внимание М. А. Кривоглаз **.
§ 5.2. АТОМНАЯ ТЕОРИЯ ДИФФУЗИИ
Рис. 5.3. Схема перескоков атомов
при их случайных блужданиях
До сих пор мы рассматривали диффузию как статистический
эффект, в результате которого происходит макроскопическое из-
менение концентрации в какой-то части тела.
Предположим, что диффузия — это результат периодических
перескоков атомов из одного узла решетки в другой. Если это
действительно так, то, используя математическую обработку
модели процесса диффузии в виде атомных случайных перескоков,
можно найти связь между измеряемыми макроскопическими ко-
эффициентами диффузии, частотой и длиной перескоков диффун-
дирующих атомов. Таким образом,
диффузионный опыт можно исполь-
зовать для изучения атомных про-
цессов и межатомных взаимодей-
ствий.
Случайные перемещения и коэф-
фициент диффузии. Рассмотрим
для простоты линейную модель
диффузии. Отметим, что перемеще-
ния атомов в этом процессе подчи-
няются тем же законам, что и броу-
новское движение частиц, взвешен-
ных в жидкости. Проблема броу-
новского движения, или, как ее ино-
гда называют, проблема случайных
блужданий, впервые была рассмо-
трена в 1905 г. Смолуховским и
Эйнштейном.
Пусть атомы растворенного компонента В сплава в начальный
момент находятся на плоскости РР (рис. 5.3) и далее через рав-
ные промежутки времени то перескакивают в направлении, пер-
пендикулярном к этой плоскости, на расстояние 6 с равной
вероятностью как вправо, так и влево.
Рассмотрим размещение атомов после Z-го и (/+ 1)-го скачков.
Обозначим расстояние атомов от исходной плоскости РР после
ьго скачка Xi,i, x2>i, . . ., Xk,i. После следующего скачка расстояние
изменится следующим образом:
-Ч,-+±8; х2Д+1=х2Д ±8; ... xk'i+i = xkti ± 8.
* Герт Л. М., Бабад-Захряпин А. А. «Физика металлов и металловедение»,
1969, 28, № 2, с. 375.
** Кривоглаз М. А. «Физика металлов и металловедение», 1964, 17, с. 161.
105
Знаки плюс и минус указывают, что возможны скачки как
вправо ( + ), так и влево (—). Очевидно,
, i+1 = А, i =t . z8 4- 82.
Найдем среднее значение квадрата смещения атома после
(г+1)-го скачка. Если п — общее число атомов, то
’г\-+1 = (л;21,/+1 + ^22,/+1 + ••• + Л%,/+1 + ••• + л2п,/+1)/п =
In п \ / п
= n~^x2k,i~\r П^1Л —(5.19)
п
поскольку 2—i8=0 [числа отрицательных и положительных чле-
нов суммы равны между собой, так как скачки вправо и влево
при переходе от f-ro к (г + 1)-му положению равновероятны].
Если принять во внимание, чго после первого скачка Xk,i =
= ±6, так что x2i = 62 и далее x22=x2i + 62=262, то
x2i = i62. (5.20)
Если обозначить время, в течение которого протекал процесс,
т, то /=т/то, так что х2—т62/то=с2т, где константа c=62/xq.
Такой же результат можно получить для трехмерного движе-
ния атомов в кристалле, т. е. для реальной модели диффузион-
ного процесса (величину х2 нужно будет только в этом случае
заменить s2, где s — смещение атома в произвольном направ-
лении) .
Вводя обозначение с2 = 2О, получаем
x2 = 2Dx. (5.21)
Для трехмерного случая диффузии имеем
s2=6Dt. (5.21а)
Таким образом, среднеквадратическое значение смещений ато-
мов при диффузии пропорционально квадратному корню из дли-
тельности процесса диффузии, а коэффициент диффузии равен
среднему значению квадрата смещения атомов, деленному на
удвоенную длительность процесса диффузии.
Естественно считать, что значения перескоков атомов того же
порядка, что и межатомные расстояния в решетке, т. е. поряд-
ка 1 А. Зная коэффициент диффузии, можно определить частоту
перескоков. Для углерода в a-железе при 900°С D~10-6 см2/с.
Если 6~10-8 см, то 1/то составит 1010 1/с, т. е. каждый атом угле-
рода меняет свое положение примерно 1010 раз в 1 с.
Аналогичный расчет для самодиффузии в металлах с г. ц. к. и
гекс. п. у. решетками для температуры, близкой к точке плавления,
показывает, что каждый атом в этих условиях меняет место
108 раз в 1 с. Если эта цифра кажется очень большой, то следует
106
вспомнить, что частота колебаний (дебаевская частота) таких
атомов составляет 1012—1013 с-1. Таким образом, атом меняет
положение только один раз на 104—105 колебаний. Даже вблизи
точки плавления большую часть времени атом колеблется около
равновесного положения.
Атомные механизмы диффузии и вычисления коэффициентов
диффузии. Теоретическое и экспериментальное исследование
диффузионных процессов непосредственно связано с теорией и
методами изучения дефектов в кристаллических телах. По совре-
менным представлениям, именно дефекты определяют механизм и
скорость диффузионного перемещения вещества. Для диффузии
в металлах при высокой температуре основное значение имеют
Рис. 5.4. Схемы элементарных актов при диффу-
зии в твердых растворах замещения:
1 — простой обмен; 2 — циклический обмен; 3 — переме-
щение в вакансию; 4 — перемещение по межузлиям; 5 —
перемещение по межузлиям с вытеснением; 6 — переме-
щение кроудионное
атомные дефекты, а среди них — точечные и их ассоциации: бива-
кансии и тривакансии, спаренные межузлия, комплексы вакан-
сия— атом примеси и т. п. При относительно низкой температуре
увеличивается роль диффузии по дислокациям и двумерным де-
фектам.
Наибольшая роль, по современным воззрениям, принадлежит
вакансиям. Впервые идею о доминирующей роли вакансий выска-
зал Я. И. Френкель более 40 лет назад.
На рис. 5.4 изображены схемы элементарных актов при диф-
фузионном перемещении атомов в растворах замещения. Атомные
перемещения могут происходить в результате простого и цикли-
ческого обмена мест, обмена мест с вакансией и движения по
107
межузлиям, перемещения, связанного с возникновением кроудио-
нов*.
При обсуждении механизма диффузии и оценках величин
коэффициентов диффузии в первую очередь встает вопрос, какие
дефекты ответственны за перенос и можно ли рассчитать кон-
центрацию этих дефектов и их подвижность.
Относительно просто вычислить коэффициент диффузии для
случая самодиффузии** в чистых металлах или для диффузии
внедренных атомов в сильно разбавленных бинарных сплавах.
Для этого можно использовать выражение для коэффициента диф-
фузии, полученное при рассмотрении случайных блужданий
в твердом теле:
D=l /6($2/т) . (5.22)
Будем считать, что рассматривается самодиффузия меченого
компонента в чистом г. ц. к. металле по вакансионному механизму.
При этом предположении 1/то=6рщ, где р — вероятность того, что
соседнее место свободно или, что то же самое, концентрация ва-
кансий; со — вероятность перескока атома в единицу времени;
числовой коэффициент показывает число ближайших соседей.
Если предположить, что диффузионный поток возникает в резуль-
тате перескоков атомов с одной плоскости на другую, то этот
коэффициент для г. ц. к. решетки равен 6, так как координацион-
ное число для этого типа решетки равно 12. Длина перескока
в рассматриваемом случае совпадает с расстоянием а между эти-
ми плоскостями. Таким образом, выражение для коэффициента
диффузии можно записать в виде
D = a2pa. (5.23)
В равновесных условиях величину р или концентрацию вакан-
сий можно найти.из соотношения
p = exp(-AGP//?T), (5.24)
где AGp = \Hp—TASp — изменение свободной гиббсовой энергии
бесконечного кристалла на один моль вакансий за вычетом энтро-
пии смешения.
Для вычисления со необходимо рассмотреть модель перескока
атома из одного положения в другое (рис. 5.5). Предположим, что
диффузия происходит вдоль оси х в одной плоскости. Изменение
свободной энергии решетки при изменении конфигурации атомов
от 1 до 3 показано на этом же рисунке. Среднюю частоту пере-
скоков на атом можно записать в виде щ=ЛД, где N — молярная
доля активированных комплексов типа 2 на рис. 5.5, a v — часто-
та, с которой атомы в перевальной точке, соответствующей кон-
* Кроудион — группа атомов, сж,атая (обычно вдоль направления плотной
упаковки) за счет наличия в ряду одного или нескольких лишних атомов
(crowd — скопление, толпа).
** Напомним, что самодиффузия — это перемещение атомов в чистом метал-
ле, «гетеродиффузия» — перемещение атомов растворенного элемента.
108
фигурации 2, переходят на новое место. Молярная доля активи-
рованных комплексов может быть выражена значением свободной
гиббсовой энергии в перевальной точке 7V=exp(—\GmIRT), где
Дбт— изменение свободной энергии в области перескока атома.
Окончательно имеем
= vexp (—AGm/RT). (5.25)
Частота ухода атомов из перевального положения v порядка
величины средней частоты колебаний атомов около положения
равновесия. Обычно v принимают равной дебаевской частоте. Та-
Рис. 5.5. Схемы последовательно возникающих
конфигураций (1—3) при перескоке атома из
одного равновесного положения (/) в другое
(3) и характер изменения свободной энергии
решетки при таком перескоке
ким образом, коэффициент диффузии можно записать в следую-
щем виде:
D = [a2v ехр (Д5₽ + &Sn/R)] exp (- ДЯР + &Hm/RT). (5.26)
Из сравнения выражений (5.3) и (5.26) следует, что
Do=a2vexp (ASp + AS™) /R (5.27)
F
Q=AHp + AH™. (5.28)
В настоящее время разработаны приемы расчета величин, вхо-
дящих в эти выражения. Однако неопределенность значений неко-
торых параметров, и в первую очередь v, позволяет при вычисле-
ниях коэффициентов диффузии получать значения, совпадающие
с экспериментальными с точностью до порядка величины.
Факторы, влияющие на коэффициент диффузии. Концент-
рационная зависимость коэффициентов диффу-
зии. На рис. 5.6 приведены концентрационные зависимости коэф-
фициентов взаимной диффузии D для сплавов Au—Ag и Си—Ni.
Из рисунка видна существенная зависимость коэффициента взаи-
модиффузии от состава обоих сплавов. Однако отклонения от
линейной зависимости различное: для сплава Au—Ag — положи-
тельные, а для сплава Си—Ni — отрицательные. Эти отклонения
109
могут быть связаны с природой сплавов. Так, в сплавах Au—Ag
возможно упорядочение, а компоненты имеют близкие температу-
ры плавления. В сплавах Си—Ni проявляется тенденция к рас-
слоению, температуры же плавления компонент существенно раз-
личны. Связь между температурами плавления твердых растворов
и коэффициентами диффузии показана на рис. 5.7.
Рис. 5.6. Концентрационные зависимости D для сплавов AuAg (а)
и Си—Ni (б)
Если добавление компонента А к В понижает температуру
плавления (или линию ликвидуса), то D при любой температуре
увеличивается. И напротив, если А повышает температуру плав-
ления, то D уменьшается.
Рис. 5.7. Фазовые диаграммы и изменение D в зави-
симости от состава в системах с неограниченной рас-
творимостью
Большой интерес представляет сопоставление коэффициентов
диффузии различных элементов в а- и у-железе, т. е. при разной
кристаллической структуре растворителя. Во всех изученных слу-
110
чаях диффузия в a-железе протекает быстрее, чем в -у-железе.
Прежде всего это относится к самодиффузии. Коэффициенты
самодиффузии Fe выражаются следующим образом:
Da = 5,8 exp (- 59700//??) (5.29)
и
Пт = 0,58 ехр (- 67900//??), (5.30)
так что
Da/DT = 10.exp(4100/?)> 1.
Так, например, в точке превращения (? = 1183К) DjD^ — 280.
По данным Ю. М. Лахтина, при температуре 600°С коэффици-
ент диффузии W в a-фазе в 2000 раз выше коэффициента диффузии
в у-фазе (Da = 1400-10"11 см2/с: D; = 0,7-10’11 см2/с).
Коэффициенты диффузии Мо, образующего с Fe твердые рас-
творы замещения, в a-фазе также значительно выше, чем в у-фазе:
Da = 0,34 ехр (^-57 700//??) см2/с. (5.31)
DT = 6,8 • 10~2 ехр (- 59 000//??);
Da/DT = 5exp(- 1300//??). (5.32)
Аналогично для циркония при 825°GDp/Da 10. Обычно боль-
шее значение коэффициента диффузии в о. ц. к. решетке (по срав-
нению с г. ц. к. и гекс. п. у. решетками) связывают с меньшей ее
компактностью.
При выяснении влияния разных факторов на скорость диффу-
зии необходимо обращать внимание на характер изменения не
только D, но и Do и Q.
Очевидно, увеличение коэффициентов диффузии при одновре-
менном увеличении энергии активации возможно лишь в резуль-
тате значительного увеличения предэкспоненциального множителя.
Действительно, как видно из табл. 5.2, при повышении концентра-
та б л и ц а 5.2
Характеристики диффузии Al, Si и Sn в меди и их изменения
при изменении содержания диффундирующего элемента
Диффундирую- щий элемент и его содержание в твердом ра- Q, ккал/г-атом Do, см2/с D при 800° С, см2/с 1010 AS, кал/(градХ Хг-атом) кал/(г-атом) 1 6Q/T, кал/(градХ Хг-атом) б (AS), кал/(градХ Хг-атом)
створе ( ат. %)
А1 2 16 39 54 0,03 17,1 3,9 24,2 —2,8 +8,25 + ’5 + 14 + 11,05
Si 2 8 44 53 0,32 160 3,4 20 +0,75 +12,8 +9 + 11,2 +12,05
Sn 2 4 40,5 31 0,151 0,04 9,5 19 +0,66 —7,5 —9,5 —8,8 —8,16
111
ции твердого раствора предэкспоненциальный множитель воз-
растает для диффузии А1 и Si в Си на несколько порядков.
При увеличении содержания в твердом растворе Sn значение
Do уменьшается, но не так резко, чтобы скомпенсировать одно-
временное уменьшение энергии активации Q: коэффициент диф-
фузии несколько увеличивается.
П. Л. Грузин, Ю. В. Корнев и Г. В. Курдюмов исследовали
самодиффузию железа в аустените с разным содержанием угле-
рода. Оказалось, что увеличение содержания углерода вызывает
ускорение самодиффузии Fe, особенно при низкой температуре:
в то время как в безуглеродистом y-Fe коэффициент самодиффу-
зии при 860°С равен 0,55-10~12 см2/с, в стали с 1,Г% С (4,7 ат.%)
он достигает 9-10~12 см2/с, т. е. в 16 раз больше.
Рис. 5.8. Зависимость коэффициентов диффузии различных
элементов в меди при 800°С от их концентрации
С увеличением температуры влияние содержания С на коэф-
фициент диффузии уменьшается: при 1100°С для безуглеродистого
y-Fe £)=12-10~12 см2/с, а для стали с 1,1 %С .0=38-10~12 см2/с,
т. е. только в три раза выше.
Коэффициент самодиффузии Fe в аустените можно выразить
следующим соотношением:
~ / 69 000 —- 7000С \ ,, /г ппч
D=lOcexp (----------------- см2/с, (5.33)
где с — концентрация углерода, ат.%.
На рис. 5.8 приведена зависимость коэффициента диффузии от
концентрации при диффузии различных металлов в медь. Влияние
концентрации становится заметным, когда она превышает 1/4—1/5
концентрации насыщения: вблизи границы растворимости при
данной температуре влияние концентрации особенно резко.
112
Для диффузии углерода в y-Fe установлена следующая зави-
симость константы Do, (см2/с) от концентрации углерода С,
(мае. %): /)о=О,О4 + О,О8 С. Значение Q в этом случае остается
(в пределах погрешности измерений) постоянным.
Влияние третьего компонента. Переход от диффу-
зии в чистых металлах к бинарным системам, как видно, значи-
тельно усложняет задачу. Переход же к многокомпонентным
системам приводит к еще большим усложнениям.
Рассмотрим это на примере перехода к тройным системам.
Было проведено сравнительное изучение плотно соединенных об-
разцов сплавов Fe+0,4% С и Fe+0,4% С +4% Si при темпера-
туре 1050°С. При этой температуре сплавы однофазны (аустенит).
Так как между сплавами отсутствует градиент концентрации
углерода, то по первому закону Фика не должно быть потока
углерода. Однако после отжига в течение 13 дней в образце был
обнаружен градиент концентрации в области стыка. Таким обра-
зом, диффузия С привела к возникновению градиента концентра-
ции. Этот эффект связан с возрастанием химического потенциала
С при добавлении к сплаву Si. В результате происходит перерас-
пределение С в области стыка таким образом, чтобы обеспечива-
лось локальное равновесие. В этой области имелось резкое изме-
нение концентрации Si, поэтому для локального равновесия необ-
ходимо столь же резкое изменение концентрации С.
Приведенный пример показывает, что при анализе диффузии
первое уравнение Фика следует использовать в виде
J=-Ddg-, (5.34)
где ц — химический потенциал диффундирующего элемента.
При этом значения D представляют собой эмпирические кон-
станты, зависящие от градиента концентрации и от состава сплава.
Природа диффундирующего металла. Чем силь-
нее природа диффундирующего элемента отличается от природы
растворителя, тем больше значение коэффициента диффузии. Воз-
можно, это связано со степенью искажения силовых полей вблизи
растворенного атома. В то же время искажение силовых полей
снижает предельную растворимость диффундирующего элемента.
Подобная зависимость была, например, установлена при диффузии
Sn, Si, Al, Zn в меди. Оказалось, что при постоянной температуре
коэффициенты диффузии тем больше, чем дальше диффундирую-
щий элемент от меди в периодической системе элементов. Для
самодиффузии в металлах характерны малые значения коэффи-
циентов.
Влияние типа твердого раствора. Энергия актива-
ции при диффузии элемента, растворяющегося по типу внедрения,
меньше, чем при диффузии элемента, растворяющегося по типу
замещения в том же растворителе (см. табл. 5.1).
В твердых растворах вычитания диффузия того компонента,
часть узлов которого в кристаллической решетке вакантна, осу-
8—523 113
ществляется тем легче, чем больше «пустых мест» в решетке. Это
подтверждается данными В. И. Архарова, установившего, что Fe
легко диффундирует сквозь вюстит FeO, имеющий значительный
дефицит железных ионов, в то время как сквозь окись никеля и
кобальта с очень незначительным избытком кислорода металли-
ческие ионы диффундируют весьма медленно.
Аналогичный результат получен при исследовании диффузии
Со в электронном соединении СоА1, способном растворять Со по
типу замещения и А1 по типу вычитания.
Граничная и поверхностная диффузия; влия-
ние размера зерна на коэффициент диффузии. По
данным многочисленных работ, диффузия в поликристаллических
металлах значительно интенсивнее, чем в монокристальных. Так,
например, при 1600°С коэффициент диффузии Мо в поликристал-
лическом W со средним размером зерна 20 мкм в 10 раз выше,
чем в монокристалле W. Различие в скоростях диффузии для
монокристаллов и поликристаллических веществ обусловлено тем,
что диффузия по границам зерен происходит быстрее, чем по
объему кристалла.
Влиянием граничной диффузии следует объяснить различия
диффузионных констант для диффузии Th в поликристаллическом
Рис. 5.9. Зависимость InD от 1/Т при диф-
фузии цинка в меди (цифры указывают
средний размер зерна)
вольфраме с разной величи-
ной зерна и в монокристал-
ле (см. табл. 5.1). При
2100°С коэффициент гранич-
ной диффузии Th в W при-
мерно в 100 раз больше,
чем объемной.
Граничная диффузия ха-
рактеризуется меньшими
значениями энергии актива-
ции, чем объемная. Причи-
ной такого различия явля-
ется скопление дефектов на
границах зерен. В тех слу-
чаях, когда коэффициент
диффузии вдоль границ зе-
рен намного превышает
коэффициент объемной диффузии, диффузионный слой по мере
удаления от источника диффузии делается все более неодно-
родным по химическому составу. Атомы диффундирующего эле-
мента, перемещаясь вдоль границ зерен, быстро насыщают по-
верхностные слои кристаллов, далеко расположенных от источ-
ника диффузии. Внутрь глубоко расположенных кристаллов диф-
фундирующий элемент проникает в гораздо меньшем количестве.
Диффузия углерода в y-Fe при обычных условиях цементации
происходит с одинаковой скоростью по толщине объема и по гра-
ницам зерен. Диффузия по границам зерен идет преимущественно
при низких температурах и особенно тогда, когда энергия акти-
114
вации объемной диффузии велика. При образовании твердых рас-
творов внедрения объемная диффузия протекает достаточно легко,
и потому незаметно различие в интенсивности объемной и гра-
ничной диффузии.
Во всех случаях, когда граничная диффузия происходит быст-
рее объемной, при экспериментальных исследованиях в качестве
коэффициентов диффузии определяют промежуточные значения
коэффициентов для объемного и граничного процессов. Чем силь-
нее развита поверхность границ между кристаллитами, т. е. чем
мельче зерна, чем более они дефектны, тем ближе эффективный
коэффициент диффузии к коэффициенту граничной диффузии. Это
видно из рис. 5.9, на котором приведены значения коэффициента
диффузии цинка в медь при различных размерах зерна сплава:
чем мельче зерно, тем выше
измеряемый коэффициент диф^
фузии. Особенно сильно размер
зерна влияет на диффузию при
низкой температуре.
Для самодиффузии Ag (см.
табл. 5.1) коэффициенты гранич-
ной и объемной диффузии при
температуре 480 и 950°С различа-
ются соответственно в 106 и 103
раз. Если принять во внимание,
что сечения кристаллитов при
размерах около 10 мкм примерно
в 104 раз больше сечения погра-
ничных слоев, имеющих толщину
около 10-7 см, то становится ясно,
что при 950°С самодиффузия в
серебре на 90% осуществляется
0,8 1,0 1,2 1,4
Рис. 5.10. Зависимость 1п£> от 1/Т
путем перемещения по объемам
зерен, а при 480°С идет почти ис-
ключительно по границам зерен.
В многочисленных исследова-
ниях обнаружена подвижность
атомов вдоль внешней поверхности металла (как поверхностная
для различных видов самодиффузии
серебра:
1 — поверхностная, Do=O,16 см2/с, Q=
= 10,3 ккал/моль; 2 — граничная; Ро—
=0,03 см2/с, Q=20,2 ккал/моль; 3 — объ-
емная, В0=0,9 см2/с, Q=46 ккал/моль
самодиффузия, так и поверхностная диффузия посторонних эле-
ментов). Этот процесс происходит еще быстрее, чем граничная
диффузия. Так, например, поверхностная диффузия Th на воль-
фраме описывается уравнением
ЛПОВ = 0,47 ехр (—66 400/7?Т) см2/с. (5.35)
Для самодиффузии Ag
DnoB=0,16 ехр (—10 300/7?Т) см2/с, (5.36)
т. е. энергия активации в этом случае в два раза меньше, чем при
граничной самодиффузии Ag, и в 4,5 раза меньше, чем при объ-
емной самодиффузии. На рис. 5.10 приведена температурная
зависимость коэффициентов самодиффузии Ag по поверхности, по
8* 115
границам зерен и по объему зерен. Хорошо известно, что Th, ло-
кально адсорбированный на вольфрамовой нити, при нагревании
быстро покрывает всю ее поверхность, не проникая в заметном
количестве внутрь металла.
§ 5.3. ДИФФУЗИЯ В ПОЛЕ ГРАДИЕНТА КОНЦЕНТРАЦИЙ
Коэффициенты самодиффузии элементов, образующих раство-
ры замещения, различны. Если соединить два полубесконечных
образца, содержащих два элемента в различных количествах, то
можно определить один коэффициент диффузии 5(c), который
полностью описывает процесс гомогенизации. В связи с этим воз-
никает задача нахождения связи между 5(c), отвечающего дан-
ному составу, и коэффициентами самодиффузии при том же
составе. Величину 5 часто называют химическим коэффициентом
диффузии.
Впервые различия потоков диффундирующих навстречу друг
другу компонентов были обнаружены в 1947 г. Смигельскасом и
Киркендалом.
Эффект Киркендала. Прямоугольный пруток латуни обматы-
вали тонкой молибденовой проволокой (молибден нерастворим
в меди и латуни), а затем покрывали слоем меди. Такой образец
последовательно отжигали. После каждого отжига от прутика
отрезали кусок, в котором определяли расстояние между молиб-
деновыми проволочками. Оказалось, что расстояние между ними
уменьшается со временем. Расстояние между проволочками может
измениться только в том случае, если поток атомов цинка из
латуни заметно превосходит поток атомов меди внутрь через
ту же плоскость. Аналогичный эффект впоследствии был обнару-
жен для различных металлов. Таким образом, эффект Киркендала
имеет совершенно общий характер.
Анализ Даркена. В основу теории эффекта Киркендала Даркен
(1948 г.) положил представление о перемещении границы раздела
между латунью и медью. Если скорость перемещения с, то пере-
нос масс компонентов через границу составит
_ ^СА
дс
&mB = DB-^~ (5.37)
тт дс НО Т Q IZ VQ V ** ' dJ то могут быть получены следующие
А Ю 9 1 dib. IXdxY -ч . ’ dt дх *
соотношения: дсА _ д ( г> дСА Д
dt ~ дх ( DA дх CAVJ
и дсв д 1 дсз \
dt дх Da-% CrV г \ в дх в )
116
Складывая их, получаем
(СА + СВ> = + DB — 1СА + Св1 °) •
Если выполняется условие сА + св=с и с сохраняет в каждой точ-
ке постоянное значение, то имеем
дсА &CR
-----= const.
А дх ' в дх
На бесконечности градиенты концентраций и v обращаются в нуль,
поэтому const —0 и
дсА $св
Поскольку
дсА 1 СА
(Da — Db) или, вводя молярные доли —уд, получаем
(Da-DJ^-=V. (5.38)
Из (5.38) следует, что v пропорционально DA—DB. Подставляя
выражение для v в одно из исходных уравнений и дифференцируя
€го по х, имеем
д^пА дсА д Гп дсА сА
дх — dt ~~ дх [ л дх с ив> ‘
После упрощений получаем
<5-39>
Из сопоставления (5.39) со вторым законом Фика (5.2) следует,
что коэффициент взаимной диффузии равен
Д=ПА(1-?А) +£>в?а (5.40)
или
D=DAyB-\-DByA,
так как 1—уА = ув. Из эксперимента определяют значение D. Для
нахождения DA и DB необходимо второе уравнение, связывающее
их с какой-либо экспериментально определяемой величиной. На-
пример, со скоростью перемещения проволочек или другой
какой-либо метки, т. е.
” = (04-О») (5.41)
Многочисленными работами установлено, что расстояние,
пройденное метками от исходного положения, выражается квад-
ратичной зависимостью от времени. В диффузионной паре такого
типа область любого заданного состава сдвигается как Д2, по-
этому состав области, окружающей метки, не меняется. Значение
117
v находят дифференцированием зависимости хт от t. Таким обра-
зом, эксперимент позволяет определить DA и DB для изотермиче-
ской диффузии в бесконечном образце.
Используя эти соображения, Даркен вычислил, что при содер-
жании в латуни 22,5% Zn Dcu=2,2-10“9 см2/с, DZn=5,lX
Х10~9 см2/с. Следует отметить, что эти значения коэффициентов
диффузии нельзя приравнивать к коэффициентам диффузии, полу-
ченным в результате эксперимента с мечеными атомами в отсут-
ствие градиента концентрации.
Со времени работы Даркена выполнено большое число иссле-
дований эффекта Киркендала. Большинство из них показало
согласие эксперимента с теорией. Однако слабость этой теории
состоит в возможности появления отрицательных значений пар-
циальных коэффициентов диффузии при достаточно больших ско-
ростях перемещения меток, что лишено физического смысла.
В связи с этим было предложено учесть осмотическое давление,
которое может возникнуть благодаря различию в подвижности
компонентов в связи со стремлением к выравниванию химических
потенциалов компонентов раствора*. Результаты расчетов по
первоначальной теории Даркена и с учетом осмотического давле-
ния при Da~Db совпадают. Однако различие становится весьма
существенным при возрастании различия в значениях DA и DB.
Связь между химическим коэффициентом диффузии и коэффи-
циентом самодиффузии. Химические потенциалы компонент сплава
можно записать в виде
In aiy (5.42)
где ц°г- — химический потенциал чистого f-го компонента; ai— ак-
тивность f-ro компонента в сплаве. Если рассматривать в качестве
движущей силы при диффузии не градиент концентрации, а гра-
диент химического потенциала, то перенос массы можно предста-
вить в виде Д/пг-= Ад (где А — коэффициент пропорцио-
нальности). Приведем это выражение к виду первого уравнения
Фика. Для этого введем коэффициент активности (?г—
молярная доля компонента):
— Л^ • дх у>
♦ Бокштейн Б. С., Бокштейн С. 3., Жуховицкий А. А. Термодинамика и ки-
нетика диффузии в твердых телах. М., «Металлургия», 1974.
118
Сопоставляя последнее соотношение с первым уравнением Фика,
получаем, что
D=-V <5-«)
Можно легко показать, чтр Ду. 1 — коэффициент самодиффузии
/-го компонента. Таким образом, отношение химического коэффи-
циента диффузии к коэффициенту самодиффузии равно ^1
Отношение коэффициентов диффузии может быть поло-
жительной или отрицательной величиной. Если это отношение по-
ложительно, то диффузионный поток направлен в сторону мень-
шей концентрации, в результате происходит выравнивание кон-
центраций. Если оно отрицательно, то поток направлен в противо-
положную сторону, что приводит к накоплению вещества
(увеличению концентрации) в локальной области материала.
Рис. 5.11. Связь между диаграммами состояния и концентра-
ционными кривыми при диффузии:
а — непрерывный ряд твердых растворов; б — разрыв сплошности твер-
дых растворов; в — интерметаллическое соединение
Приведенный анализ предсказывает особый характер распада
твердых растворов в определенной температурно-концентрацион-
ной области (спинодальная область, см. гл. 9).
Диффузия, сопровождающаяся фазовыми превращениями (ре-
активная диффузия). Процессы диффузии приводят не только
к образованию и изменению состава твердых растворов, но и
к фазовым полиморфным превращениям или образованию хими-
119
ческих соединений. Большое практическое значение имеет анализ
роста кристаллов новой фазы, контролируемого диффузией. При
рассмотрении реактивной диффузии необходимо учитывать законы
гетерогенного равновесия: если фазы находятся в равновесии,
химический потенциал любого компонента одинаков в каждой из
фаз, т. е. он сохраняет непрерывность при переходе через поверх-
ность их раздела. Если равновесие установилось, то концентрация
на границах каждой фазы может быть найдена по диаграмме
фазового равновесия.
На рис. 5.11 показана связь между кривыми распределения
концентрации и диаграммами состояния. При образовании в диф-
фузионном слое интерметаллического соединения (см. рис. 5.11,в)
на концентрационной кривой появляется ступенька, высота кото-
рой соответствует ширине области гомогенности этого соединения.
Изменение толщины слоя интерметаллического соединения, по
данным многочисленных исследований, описывается параболиче-
ским уравнением х=2& У Dt. Решение диффузионной задачи для
многофазной диффузии проводят с учетом скачков концентрации
на концентрационной кривой и закона изменения толщины слоев
растущих фаз.
На границе раздела фаз а и £ в диффузионной зоне потоки
должны быть равны
D = D
а \ А $ \ дх
Отсюда следует, что ’
D /(-¥-}, (5.44)
а' $ \ А/ \ А 7
т. е. значения коэффициентов диффузии обратно пропорциональны
градиентам концентрации диффундирующего элемента в фазах.
Из отношения толщины слоев двух сопредельных фаз в предпо-
ложении параболического закона изменения их толщины
следует, что оно определяется отношением коэффициентов диффу-
зии. При значительном различии коэффициентов диффузии толщи-
на одной из фаз может быть меньше толщины другой. Это необхо-
димо иметь в виду при сопоставлении фаз в диффузионной зоне
с наличием фаз на диаграмме состояния (из-за малости толщины
фазы в диффузионной зоне можно ошибочно считать, что она пол-
ностью отсутствует).
Следует иметь в виду, что при диффузионном насыщении
чистого металла другим элементом образующийся диффузионный
слой не может состоять более чем из одной фазы. Это следует из
правила фаз: c=k+\—ft где k — число компонентов; f — число
фаз. В нашем случае с=3—f. Если в сопредельном с металлом
120
слое возникнут две фазы, то и при постоянной температуре
концентрация компонент должна быть строго постоянной. Но
в этом случае ~ =0, и согласно первому закону Фика диффузион-
(/X
ные потоки также равны нулю, т. е. диффузия отсутствует.
В некоторых случаях (например, при азотировании железа)
наблюдаемые двухфазные диффузионные слои образуются в ре-
зультате фазовых превращений при охлаждении (подробнее
о строении азотированного слоя на железе см. с. 123).
Рис. 5.12. Изменение концентрации хрома
при диффузионном насыщении им железа
Методами структурного рентгеноанализа и под микроскопом
изучен процесс диффузии хрома в железе. На рис. 5.12 приведена
кривая, характеризующая изменение концентрации хрома в диф-
фузионном слое по мере удаления от внешней поверхности желез-
ного образца, упакованного в тонкий порошок электролитического
хрома, после выдержки в течение 96 ч при 1200°С. Как видно из
этого графика, на глубине 0,009 мм концентрация диффундирую-
щего элемента уменьшается скачком от 15 до 1 ат. % (с 14,1 до
0,933 мае.%). На этой же глубине находится линия раздела меж-
ду столбчатыми кристаллами, идущими от поверхности образца,
и структурой сердцевины образца.
Особенности микроструктуры образца и распределения хрома
в диффузионном слое находят объяснение в диаграмме состояния
•сплавов Fe—Сг. Область у-фазы на этой диаграмме замкнута.
При 1200°С начиная от содержания хрома около 10% расположе-
на однофазная область а. В соответствии с этим внешние слои
металла при температуре 1200°С испытывают перекристаллиза-
цию, причем столбчатые кристаллы a-фазы, возникшие у поверх-
ности образца, с насыщением хромом растут все глубже и глубже.
При охлаждении металла в этих кристаллах никаких фазовых
превращений не происходит, в то время как у-кристаллы внутри
образца испытывают при охлаждении аллотропическое превраще-
ние. Скачкообразное изменение состава сплава после перехода
через пограничную поверхность, отделяющую слой со столбчатой
121
структурой от сердцевины образца, вызвано отсутствием двух-
фазной области а + у при протекании диффузии.
Если металл насыщается вторым компонентом из газообразной
или жидкой среды, то параболический закон в начальных стадиях
процесса насыщения при небольшой скорости адсорбции диффун-
дирующего элемента и большой скорости диффузии нарушается:
концентрация диффундирующего элемента во внешнем погранич-
ном слое не остается вначале постоянной, а увеличивается посте-
пенно до предельного значения. Толщина слоя новой фазы при
этом растет в начале процесса не по параболическому закону.
Зависимость от толщины слоя и длительности процесса имеет бо-
лее сложный вид, приближенно описываемый при непродолжи-
тельных выдержках формулой
x?=k%, (5.45)
где р— 14-2.
Концентрация диффундирующего элемента в поверхностном
слое изменяется (например, при цементации стали углеродом), это
также вызывает нарушение параболического закона в цементаци-
онном процессе.
Предельная концентрация углерода в поверхностном слое це-
ментуемого изделия зависит от соотношения скоростей адсорбции
и диффузии. Если при неизменной скорости адсорбции скорость
диффузии возрастает, адсорбированный слой не успевает достав-
лять атомы диффундирующего элемента в необходимом количест-
ве, и концентрация этого элемента в поверхностном слое падает,
но зато глубина проникновения увеличивается.
Такое явление наблюдается при цементации технически чистого
железа в атмосфере СО в результате повышения температуры
свыше 900°С. Предельная концентрация углерода в поверхностном
слое при всех температурах ниже концентрации насыщения.
Типичным примером реактивной диффузии является диффузия при окислении
металлов. Процесс окисления железа начинается с образования на поверхности
металла тонкой пленки высшего окисла Ре20з (в модификации у при низкой тем-
пературе и а при высокой). При температурах, превышающих 100—150°С вслед
за Fe2O3 во внутренних слоях окалины образуется магнитная окись Fe3O4,
а выше 570°С — также закись железа FeO (вюстит). С течением времени каждая
из трех зон слоя окалины утолщается (как и весь слой).
При окислении железа сквозь слой окалины диффундируют навстречу друг
другу кислород и железо. Коэффициенты диффузии железа в различных окислах
различны — самый большой коэффициент диффузии железа в закиси FeO, в ко-
торой имеются вакантные узлы на месте недостающих ионов железа.
Поскольку реакция окисления железа идет быстрее диффузии сквозь нарас-
тающий слой окалины, скорость образования окалины определяется скоростью
лимитирующего процесса диффузии. Поэтому нарастание слоя окалины на по-
верхности железа в сухом воздухе резко ускоряется при переходе через темпе-
ратуру образования закиси железа FeO, равную 575°С.
Присутствие некоторых легирующих элементов (например, Мп и Сг) повы-
шает температуру образования вюстита и тем самым повышает жаростойкость
соответствующих сплавов по сравнению с технически чистым железом и угле-
родистой сталью.
Как считает В. И. Архаров, одной из важнейших причин повышения стойко-
сти стали при легировании Ni и Со является то, что в сложных окислах
(Fe, Со)0 и (Fe, Ni)O число вакантных узлов в кристаллической решетке значи-
122
тельно меньше, чем в вюстите, а это затрудняет диффузию металла сквозь слой
окалины.
Процессы реактивной диффузии происходят при азотировании
стали. На рис. 5.13 приведены построенные Ю. М. Лахтиным кри-
вые распределения азота в азотированном слое технически чистого
На кривой отчетливо видны скачки концентрации азота при
переходе из одной фазовой области в другую.
При температуре 700°С, которой соответствуют эти графи-
ки, в системе Fe—N устойчивы три фазы: а) 8-фаза — нитрид
Рис. 5.14. Микроструктура азотированного слоя
технически чистого железа в закаленном состоя-
нии (а) и после медленного охлаждения (6),
Х450
123
переменного состава с гекс. п. у. решеткой металлических атомов,
часто описываемый формулой Fe2N, которая соответствует макси-
мальному содержанию азота; б) твердый раствор азота в у-же-
лезе с г. ц. к.; в) твердый раствор азота в a-железе с о. ц. к. ре-
шеткой. На микрофотографии (рис. 5.14,а) слои 8- и у-фаз
обладают резко выраженной столбчатой структурой, что объяс-
няется условиями фазовых превращений при диффузионном на-
сыщении.
При медленном охлаждении образца (см. рис. 5.14,6) у-фаза
испытывает эвтектоидный распад, превращаясь в смесь а- и у'-фаз
(нитрид Fe4N). В связи со снижением растворимости азота в а-фа-
зе при понижении температуры медленное охлаждение азотиро-
ванного слоя сопровождается выделением у'-кристаллов в слое
a-фазы там, где она всегда сильнее насыщена азотом (т. е. вблизи
поверхности раздела между фазами а и у).
Микроскопическое исследование и наблюдение за изменением
концентрации азота в поверхностном слое азотируемого образца
с увеличением выдержки от 5 до 45 мин при 520°С привели
Ю. М. Лахтина к заключению, что нитриды не сразу образуются
на поверхности железа в результате химической реакции между
железом и азотом. По-видимому, сначала азот растворяется
в a-Fe, причем концентрация его в поверхностном слое постепенно
повышается. Когда a-фаза насыщается до предела, в ней возни-
кают кристаллы устойчивой при этой температуре у'-фазы, кото-
рая образует затем сплошной слой. В этом слое при дальнейшем
насыщении возникают кристаллы е-фазы.
Чередование фаз в диффузионном слое при реактивной диф-
фузии обычно соответствует диаграмме состояния взаимодейству-
ющих элементов.
Задачи к гл. 5
Задача 5.1. Сталь, содержащая 0,1% С, должна быть подвергнута цемента-
ции при 930°С. При этом концентрация углерода на глубине 0,05 мм должна до-
стигнуть 0,45%. Карбюризатор обеспечиваем постоянную концентрацию углерода
1 % на поверхности насыщения в течение всего времени насыщения. Определите
потребное время цементации, если Z) = l,4-10~7 см2/с.
Задача 5.2. Определить время, в течение которого при отжиге можно уда-
лить 90% водорода из железной пластинки толщиной 0,175 см. На поверхности
пластинки предполагается нулевая концентрация водорода, D— 10“4 см2/с.
Задача 5.3. Медный прут соединен с прутом из сплава 29,4% Zn+70,6% Си.
После отжига в течение 360 ч данные о зависимости содержания Zn на опреде-
ленном расстоянии от плоскости стыка обработаны по методу наименьших квад-
ратов.
Zn, ат. %
0,3
1,5
4,4
8,8
14,7
20,6
х, 10"2 см
50,05
48,15
46,45
44,95
43,15
39,65
Zn, ат, %
23,5
25,0
26,5
27,9
28,8
29,1
х, IO’2 см
36,55
34,05
30,75
25,15
18,95
14,95
124
Найти положение плоскости Матано и рассчитать D (с) через каждые
5% Zn.
Задача 5.4. Сто шариков, которые могут перескакивать с места на место со
средней длиной прыжка 0,25 см, размещены вдоль прямой линии с интервалом
15 см. Через 12 ч измерено расстояние от каждого шарика до линии. Сумма
квадратов расстояний, деленная на 100, равна 90 см2. Определить коэффициент
диффузии шариков и среднюю частоту перескоков.
Задача 5.5. Две метки помещены внутрь образца, который состоит из двух
полубесконечных стержней А и В, — одна на исходную плоскость раздела, дру-
гая рядом. Покажите качественно, как меняется положение каждой из них в за-
висимости от времени, если DA>DB- Изобразите результат графически, отложив
jV(x) и dN [dx (N— относительная концентрация) против х, пользуясь уравне-
dN л
нием / = (Da—Db) -з—
дх
Задача 5.6. Хром диффундирует в железо при 1000°С. Его концентрация на
поверхности железа 50% • Система Fe—Сг имеет замкнутую у-область. При
1000°С максимальное содержание хрома в у-области 12%, а минимальное его
содержание в а-фазе 13%• Коэффициент диффузии хрома в a-фазе больше, чем
в у-фазе. Показать схематически зависимость концентрации хрома от глубины
его проникновения.
ГЛАВА 6
СТРУКТУРА РАСПЛАВОВ И КРИСТАЛЛИЗАЦИЯ
§ 6.1. СТРУКТУРА РАСПЛАВЛЕННЫХ МЕТАЛЛОВ
Природа жидкостей ближе к природе твердых тел, чем газов.
Частицы соприкасающихся между собой твердых тел также спо-
собны перемешиваться в результате диффузии, как и частицы
двух жидкостей. Было открыто вязкое течение кристаллических
тел при достаточно высокой температуре, происходящее под дей-
ствием очень малых сил.
Прочность твердых металлов при испытании на разрыв и тем-
пературе, близкой к температуре плавления, совершенно ничтож-
на. Удавалось без разрыва растянуть жидкую ртуть на 1,4%, что
соответствует отрицательному давлению около 100 кгс/мм2, а воду
на 1,5% (опыты проводили в условиях, исключающих возможность
течения жидкости) .
Таблица 6.1
Теплоемкость [кал/(моль-град)]
некоторых веществ в твердом, жидком
и газообразном состоянии вблизи
температуры плавления
Элемент Твердое состояние Жидкое состояние Г азообразное состояние
Na 7,6 8 3
К 7,1 7,6 3
Zn 7,2 7,9 3
Теплоемкость веществ с простой кристаллической структурой
меняется не очень сильно (обычно несколько возрастает) в ре-
зультате плавления и значительно отличается от теплоемкости
соответствующих газов (табл. 6.1).
Электрическое сопротивление большинства металлов при плав-
лении увеличивается примерно вдвое и при дальнейшем нагреве
продолжает возрастать (у висмута, сурьмы, галлия плавление
уменьшает электрическое сопротивление в полтора-два раза), ме-
таллическая природа вещества после плавления сохраняется.
Следовательно, при плавлении характер сил взаимодействия меж-
ду частицами не изменяется, мало изменяется и величина сил
взаимодействия, по крайней мере у металлов, поскольку теплота
126
плавления металлов не превышает 5—10% теплоты испарения.
Как указал впервые Я. И. Френкель, в жидкости, как и в твердом
теле, частицы не движутся свободно, как в газе, а колеблются
около положения равновесия. В отличие от твердых тел, в которых
длительность пребывания частиц около положения равновесия
в миллионы раз превышает периоды колебаний, в жидкостях час-
тицы переходят в новое положение, совершив несколько тысяч
колебаний около положения равновесия. Спектр тепловых колеба-
ний в жидкости сходен со спектром тепловых колебаний в соот-
ветствующих кристаллах.
Дифракционная картина, получающаяся в результате рассея-
ния рентгеновских лучей и тепловых нейтронов жидкостями вблизи
температуры плавления, во многом сходна с дифракционной кар-
тиной, характерной для рассеяния лучей теми же веществами
в кристаллическом состоянии, существенно отличаясь от картины,
которую дают пары тех же веществ.
Дифракционные максимумы при рассеянии рентгеновских лу-
чей или нейтронов жидкостью обусловлены наличием определен-
ного порядка во взаимном расположении молекул (или атомов —
в жидкостях атомарной структуры).
В качестве характеристики «порядка» в расположении частиц
жидкости принимается функция атомного размещения, определяе-
мая из соотношения
4nr2drp(r), (6.1)
где г — расстояние от центра какого-либо фиксированного атома;
nr,T+dr — число центров атомов, находящихся в шаровом слое меж-
ду сферами с радиусами г и r + dr.
Очевидно, р(г) представляет собой плотность жидкости на
расстоянии г от центра фиксированного атома, выраженную в чис-
лах атомов на единицу объема. На достаточно большом расстоя-
нии от центра атома величина р(г) должна стремиться
к постоянному значению, совпадающему со средней плотностью
жидкостей р0.
Функция атомного размещения вычисляется из кривой интен-
сивности рассеяния рентгеновских лучей в зависимости от угла
рассеяния ф (для удобства расчета по оси абсцисс обычно откла-
дывают величину sin 0/Z, где А,— длина волны).
В идеальном кристалле при отсутствии теплового движения
функция атомного размещения должна иметь дискретный харак-
тер: все атомы кристалла должны находиться на некоторых опре-
деленных расстояниях от одного, центр которого принят за начало
координат.
На одном и том же расстоянии от начального атома на-
ходится несколько атомов Ni. Центры всех атомов с одним и
тем же значением лежат на одной и той же координационной
сфере (I— номер сферы).
Для характеристики размещения атомов в идеальной решетке
вводят «поверхностную плотность» рпов, равную числу атомов,
127
центры которых лежат на единице площади поверхности сферы
радиусом гг-, т. е.
рпов — N i/4лГ2ц
На рис. 6.1 показан характер распределения центров атомов
в г. ц. к. решетке [за единицу измерения длины принят атомный
диаметр (2г0), длины вертикальных прямых дают значения A/’J.
Тепловое движение в кристалле изменяет размещение атомов —
их центры отклоняются от идеальных положений на соответствую-
щих сферах.
Рис. 6.1. Размещение центров атомов по координационным сферам
г. ц. к. решетки без учета тепловых смещений (а) и с учетом (б)
Вероятность нахождения атома вне соответствующей сферы
оказывается отличной от нуля, она плавно спадает по обе стороны
от Гц для которого вероятность максимальна, причем форма кри-
вых размещения для всех одна и та же: вероятность определен-
ного изменения расстояния Дг одна и та же для всех значений гг-.
Если принять, например, простейшее (на самом деле неточное)
предположение о том, что вероятность нахождения атома линей-
но спадает по мере удаления от основной сферы, то вертикали на
рис. 6.1,а нужно заменить треугольниками с одинаковыми осно-
ваниями (см. рис. 6.1,6).
Такое размещение атомов характеризует наличие дальнего по-
рядка в решетке. Средние смещения атомов по отношению к сфе-
ре, в которой они располагались бы в отсутствие тепловых коле-
баний, не зависят при наличии дальнего порядка от гг-.
Анализ кривых рассеяния рентгеновских лучей жидкостями
привел к представлению о том, что в жидкости сохраняется лишь
ближний порядок расположения частиц. Ближний порядок харак-
теризуется тем, что средние смещения атомов по отношению к иде-
альным сферам растут с увеличением гу, относительное смещение
двух далеко расположенных атомов А и В оказывается при на-
личии одного лишь ближнего порядка векторной суммой взаимных
смещений промежуточных атомов. .Применение к этому статисти-
ческому суммированию теории броуновского движения приводит
128
к выводу, что это смещение пропорционально корню квадратному
из расстояния между атомами А и В. Различие в распределении
атомов при ближнем и дальнем порядках на примере плоской сет-
ки из плотноупакованных шаров показано на рис. 6.2.
а
Рис. 6.2. Однослойная укладка равновеликих ша-
ров:
а — дальний порядок; б — ближний порядок
Представление о ближнем порядке связано с представлением
об исходной идеальной структуре, которая «размывается» при
плавлении. На рис. 6.3,а показа-
на функция атомного размещения,
Рис. 6.3. Радиальное распределение
плотности в расплавленном свинце
(а) и расплавленном натрии (б). Вер-
тикальные линии характеризуют ко-
ординационные сферы, соответствен-
но кристаллического свинца (г. ц. к.)
и кристаллического натрия (о. ц. к.);
монотонные кривые характеризуют
равномерное распределение плотно-
сти. На графиках для Na видно хо-
рошее согласие результатов исследо-
вания различных авторов (кружки и
точки)
9—523
129
построенная по данным о рассеянии рентгеновских лучей для свин-
ца. Эта кривая весьма сходна с линией, построенной теоретически
для размытой г. ц. к. решетки. Площадь кривой под первым мак-
симумом определяет среднее число ближайших соседей у каждого
атома жидкости, местоположение первого максимума — наиболее
вероятное расстояние между соседними атомами.
Точно также сходна со своим кристаллическим «прототипом»
кривая распределения атомной плотности у расплавленного натрия
Рис. 6.4. Рассеяние нейтронов
расплавленным рубидием при
различной температуре (темпера-
тура плавления 38,8°С)
(см. рис. 6.3,6, кривая а).
У металлов с к. ч.=12 (в твер-
дом состоянии) при плавлении, как
видно из рис. 6.2, усредненное к. ч.
бывает несколько меньше 12, у ме-
таллов с «рыхлыми» решетками
плавление вызывает вместе с об-
щим размытием структуры и неко-
торое изменение упаковки атомов,
направленное в сторону повыше-
ния плотности упаковки. Это об-
стоятельство вызывает уменьше-
ние удельного объема Bi при плав-
лении.
Как видно из рис. 6.3, по мере
увеличения расстояния г от началь-
ного атома кривая, вычисленная по
экспериментальным данным, все
меньше отличается от кривой, по-
строенной для равномерного распределения, порядок ощущается
все слабее, поэтому он и называется ближним.
С повышением температуры расплавленного металла интерфе-
ренционные максимумы на кривой рассеяния рентгеновских лучей
или нейтронов (рис. 6.4) размываются, что свидетельствует об уве-
личении размытия структуры.
В. И. Данилов показал, что различные типы идеальных реше-
ток размываются при плавлении по-разному: более четко сохраня-
ют ближний порядок после расплавления металлы с компактными
решетками (например, свинец), сильнее сглаживаются интерфе-
ренционные максимумы на кривых рассеяния расплавленных ме-
таллов с рыхлыми упаковками, характеризующимися небольшими
координационными числами (Bi).
Резкое ослабление ближнего порядка после плавления метал-
лов с рыхлыми упаковками объясняется тем, что тепловое дви-
жение частиц стремится сделать размещение атомов наиболее ком-
пактным, т. е. приблизить упаковку атомов к плотнейшей.
Исследуя рассеяние рентгеновских лучей в расплавленных эв-
тектиках РЬ—Bi, Pb—>Sn, Bi—Sn, В. И. Данилов и И. В. Радченко
показали, что интерференционные картины, наблюдающиеся при
этом, можно объяснить как результат наложения интерференцион-
ных картин, характерных для расплавов компонентов, входящих
130
в эти эвтектики. Таким образом, в эвтектических сплавах имеют*
ся области, ближний порядок в которых соответствует кристалли-
ческой структуре компонента А, и области с ближним порядком,
характерным для компонента В.
В последние годы проведены нейтронографические и рентгено-
графические исследования структуры расплавов с относительно
высокой температурой плавления (1000°С). Для жидкого железа
получены значения координационного числа от 11,5 до 13,5. Было
высказано * предположение, что упаковка атомов в жидком желе-
зе близка к структуре его высокотемпературной модификации
б-Fe. При исследовании расплавов Fe—С показано **, что введе-
ние углерода в жидкое железо приводит к изменению дифракцион-
ной картины: первый максимум интенсивности сдвигается в сто-
рону меньших углов рассеяния.
§ 6.2. ГОМОГЕННАЯ КРИСТАЛЛИЗАЦИЯ
Кристаллизация расплава. Рассмотрим самопроизвольное обра-
зование зародышей кристаллитов в расплаве чистого металла.
Практически осуществить гомогенное зарождение очень сложно,
так как надо исключить влияние любой готовой поверхности ча-
стиц, примесей, стенок. При охлаждении расплава степень ближ-
него порядка возрастает. Из-за
интенсивного теплового движе-
ния малые области с располо-
жением атомов, подобным их §
расположению в кристалличе- Й,
ском состоянии, то разрушаются, |
то возникают снова. Однако
при дальнейшем охлаждении §
расплава вероятность возникно-
вения областей с таким же ко-
ординационным числом, как у
твердого металла, будет возра-
стать. Эти объемы можно рассма-
тривать как зародыши кристал-
лизации, если при их дальнейшем
росте свободная энергия систе-
мы Дф уменьшается. Рассчитаем
Рис. 6.5. Изменение объемной свобод-
ной энергии металла в жидком (Гж)
и твердом (FTB) состояниях с темпе-
ратурой
ДФ как изменение свободной энергии при затвердевании объема
с линейным размером г. Учитывая изменение свободной энергии,
связанное с переходом из жидкого состояния в твердое AF—
=Ftb—Fm, и энергию поверхности раздела площадью S=02r2 меж-
ду расплавом и кристаллическим зародышем и предполагая, что
* Клименков Е. А., Гельд П. В. и др. «Докл. АН СССР». 1976, т. 230,
№ 1, с. 71.
** Ватолин Н. А., Веселова С. И. и др. «Физика металлов и металловед.»
1974, т. 37, № 1, с. 181.
9* 131
зародыш сохраняет неизменную форму для общего изменения сво-
бодной энергии, получаем
ДФ=₽1Д^г3+₽2аг2, (6.2)
где Pi и — коэффициенты формы для расчета объема и поверх-
ности зародыша; а — поверхностное натяжение между жидкой и
твердой фазами.
Поверхностное натяжение * и, значит, второе слагаемое в урав-
нении (6.2) всегда положительны. Свободная энергия единицы
объема металла в зависимости от температуры изменяется для
твердой и жидкой фаз, как показано на рис. 6.5.
Твердая и жидкая фазы находятся в равновесии при температу-
ре плавления Го(Д/?=О). С изменением температуры Д/7 меняет
знак, так как при температуре выше TQ (AF>0) стабильна жид-
кая фаза, а при переохлаждении расплава Д/7<0. Изменение сво-
бодной энергии ДФ(г) для различной температуры показано на
рис. 6.6.
Из анализа хода ДФ(г) по рис. 6.6,а ясно, что при Т>Т0 ДФ
всегда увеличивается, и кристаллический зародыш, случайно об-
* При образовании зародыша на инородной подкладке в некоторых случаях
поверхностная энергия а может быть отрицательна.
132
разующийся при тепловом движении, нестабилен. При Г<70 функ-
ция ДФ имеет максимум, соответствующий определенному крити-
ческому значению размера зародыша гк. Зародыш новой фазы,
имеющий критический размер, находится, как видно из рис. 6.6,6
и в, в состоянии неустойчивого равновесия с окружающей его сре-
дой: если он немного уменьшится, то последует дальнейшее его
самопроизвольное уменьшение вплоть до полного расплавления.
При г>Гк зародыш будет расти, так как это ведет к уменьшению
ДФ. В соответствии с этим зародыш критической величины назы-
вается равновесным.
Критический размер гк получим из условия максимума функции
ДФ(г) =0:
гк„2р2а/ЗР1ДЕ (6.3)
Например, для зародыша, имеющего форму сферы радиуса
г(Р1=4/Зл, р2=4л) или куба с ребром 2r (£1=8, р2=24), имеем
rK=2a/|AF|. (6.4)
Из выражения (6.3) или (6.4) для Т—имеем гк—>оо. Поэто-
му, очевидно, образование кристаллов возможно лишь при неко-
тором переохлаждении.
Разность свободных энергий А/7 в функции переохлаждения А Г можно опре-
делить для небольших переохлаждений из следующих соображений. При темпе-
ратуре плавления /7тв = ^ж. В соответствии с этим
F^(T)-F^(T)=[F^(T)-F^(To)]-[F^(T)~F^To)].
Каждое выражение, стоящее в квадратных скобках, может быть разложено
в степенной ряд, причем при небольших переохлаждениях ДТ = Г0—Т можно
ограничиться только первыми членами этих рядов, так что:
fTB (7'о) = 4г^г~7’<>);
dF
F* -То);
f dFr3 дРж \
^тв (O - F ж (Г) = j (Г - T„) = (S;K - STB) (T - Tt),
поскольку dF/dT——S, где S — энтропия системы.
Вводя скрытую теплоту плавления на единицу объема Q=7\AS,
получаем
Гтв(Т)-Гж(Г)=-^-(Г-Т0)
1 о
ИЛИ
ДГ = —ATQ/T,. (6.5)
Из уравнения (6.3) имеем
гк = 2р2аГ0/Зр^дГ (6.6)
Работа образования зародыша размером гк равна
Дф"=4т^. <67а>
133
или
дф — —__________ zg
а * 27 ^Q2 (ДТ)2 ’ (о-/и»
ИЛИ
ДФк=-^р2аг2к=~5ка- (6.8а)
где sK —общая поверхность зародыша.
Эта формула выведена в предположении, что поверхностная
энергия изотропна. Однако для кристаллов различные кристал-
лографические плоскости (hkl) имеют разные значения щкм), по-
этому формулу (6.8а) следует преобразовать следующим образом:
п
А®к з“ $i (hkl) (hkl)i (6.86)
x=l
где Sickly — площадь f-й грани типа (htkili) кристалла, имеющего
п граней.
Таким образом, работа образования равновесного зародыша
новой фазы равна 1 /3 его поверхностной энергии.
Критическая величина равновесного зародыша пропорциональ-
на коэффициенту поверхностного натяжения или удельной поверх-
ностной энергии а на поверхности раздела кристалл — расплав.
О поверхностном натяжении вещества можно судить по теплоте испарения,
температуре плавления, твердости: чем больше теплота испарения, температура
плавления, твердость, тем выше при прочих равных условиях силы в кристалле,
тем больше поверхностное натяжение (табл. 6.2).
Таблица 6.2
Удельная поверхностная энергия некоторых металлов и воды
Металл a0 (расплав- пар), эрг/см3 a (расплав- кристалл), эрг/см'1 a/a0 Металл a0 (расплав— пар), эрг/см3 a(расплав— кристалл), эрг/см3 a/a0
Sn 610 59 0,09 Ag 880 126 0,14
Bi 465 54 0,12 Au 1030 132 0,13
Pb 450 33 0,07 Fe 1800 204 0,12
Sb 320 101 0,33 Pt 1820 240 0,14
Al 900 93 0,10 H2O 73 32 0,44
Ge 900 181 0,20
Атомы (ионы) поверхностных слоев двух соприкасающихся конденсирован-
ных фаз (твердых или жидких) находятся в иных условиях, чем атомы на по-
верхности твердой или жидкой фазы, соприкасающейся с газовой фазой. Поверх-
ностное межфазовое натяжение на границе раздела конденсированных фаз на-
много ниже, чем поверхностное натяжение любой из этих фаз по отношению
к газовой фазе. Это уменьшение поверхностного натяжения, очевидно, опреде-
ляется близостью типов связи в соприкасающихся фазах и сходством во взаим-
ном расположении атомов (ионов) на соприкасающихся поверхностях фаз. При
соприкосновении веществ, в которых действуют силы химической связи разного
типа, поверхностное натяжение должно быть выше, чем при соприкосновении фаз
с одним и тем же типом связей. Так, на поверхности неметаллических включе-
134
ний в расплаве поверхностное натяжение выше, чем на поверхности раздела
двух металлических фаз.
Наконец, поверхностное натяжение кристаллического вещества по отноше-
нию к расплаву того же вещества намного ниже, чем натяжение на поверхности
соприкосновения этого вещества в твердом или жидком состоянии с газовой
фазой. Поверхностное натяжение особенно сильно должно быть снижено при
соприкосновении расплава с твердой фазой в тех случаях, когда она имеет ком-
пактную кристаллическую решетку (г. ц. к. или гекс. п. у.)
Анализ гомогенного зарождения при кристаллизации позволяет
оценить поверхностную энергию на границе кристалл — расплав.
Из табл. 6.2 видно, что удельная поверхностная энергия на по-
верхности раздела кристалл — расплав (а) составляет 7—33%
удельной поверхностной энергии расплавленного металла по от-
ношению к газовой фазе (а0). Особенно сильно снижается по-
верхностное натяжение при соприкосновении расплава с твердой
фазой для некоторых металлов с компактными (или близкими
к компактным) кристаллическими структурами (для РЬ а/ао—
=0,07, для А1 0,10, для Au 0,13). Для сурьмы, кристаллическая
решетка которой весьма далека от компактной, значение «/«о
остается достаточно большим (0,33).
Для твердых тел можно теоретически рассчитать энергию свя-
зи и затем энергию поверхностного натяжения. Такие расчеты
дают для граней (100) NaCl «=150 эрг/см2, а для (111) алмаза
ct=l 1400 эрг/см2.
Расчеты показывают, что даже при значительном переохлаж-
дении равновесный зародыш в однородной жидкости много боль-
ше элементарной ячейки.
Конденсация пересыщенного пара, кристаллизация из пересы-
щенного жидкого или твердого раствора.
С пересыщением пара Р/Роо (Р<х> — равновесное давление) свя-
зана свободная избыточная энергия (на 1 г-моль)
In (Р/Роо). (6.9)
Подставляя это выражение для АР в формулу для критической
сферической капли, получаем
rK=2Ma/RTp In (Р/Роо), (6.10)
(здесь р — плотность; М — молекулярная масса; Р — газовая по-
стоянная).
Приведенное уравнение можно считать условием метастабиль-
ного равновесия капли радиусом г и упругости пара над нею Рг:
In (Рг/Роо) =2Л4а/РТрг. (6.11)
В этом уравнении (Томсона — Кельвина) Роо— равновесная
упругость пара над плоской (г->оо) поверхностью жидкой фазы.
Формула, выражающая зависимость растворимости кристаллов
в жидкости от размеров кристаллов, отличается от уравнения
(6.11) лишь тем, что в ней упругости пара Рг и Роо заменены рас-
творимостями ст и Соо, т. е.
1 п (сг I сж) = 2М a/RTpr. (6.12)
135
Критическая величина зародыша при кристаллизации из пере-
сыщенного раствора выражается так:
гЕ=2М а / [ 7? Тр I п (сг / . (6.13)
Из уравнения (6.11) -следует, что пар, насыщающий простран-
ство над плоской -поверхностью жидкости, будет ненасыщенным
по отношению к капле жидкости, причем тем в большей степени,
чем меньше капля. Капля, находящаяся в замкнутом пространст-
ве по соседству с плоской поверхностью жидкости, должна поэто-
му испаряться, а испарившаяся с ее поверхности масса должна'
конденсироваться на плоской поверхности. В результате капля
полностью перетопится на плоскую поверхность жидкости (или на
каплю большого размера). Точно так же мелкие кристаллы в на-
сыщенном растворе, где имеются кристаллы больших размеров,
должны растворяться и в конце концов перекристаллизоваться на
более крупные1 кристаллы.
Таблица 6.3
К расчету равновесного размера капель ртути и воды при конденсации
из пара (Р/Р^ — 1,1, 7 —300° К)
Вещество р, г/см3 а, эрг/см2 г, см Объем капли, (М3 Число молекул (атомов) в капле
Н2О Hg 1 13,6 72 470 10-6 5,5-Ю-6 4.2-10-18 6,7-10-16 1,4.10s 2,7-107
Приведем несколько численных примеров.
Определим прежде всего равновесные при комнатной темпера-
туре (Т=300 К) размеры капель воды и ртути при конденсации
из пара, пересыщенного на 10% (Р/Р ОО-1 , 1) (табл. 6.3).
Как видно из табл. 6.3, равновесный размер капли, конденси-
рующийся из пересыщенного пара при небольшом пересыщении,
весьма значителен—устойчивы лишь капли, содержащие несколь-
ко сот тысяч и даже миллионы молекул (атомов).
В табл. 6.4 приведены данные о влиянии размеров кристаллов
гипса (CaSO4) на его растворимость в воде при 0°С (Т—273°С).
Размер кристаллов влияет на их растворимость, если кристалл
меньше 10 мкм (особенно если г^ОД мкм).
Таблица 6.4
Растворимость гипса в воде при различных размерах кристаллов
г, мкм In (с /с ) Г ОО с /с г ОО с , ммоль/л
ОО 0 1 12,93
10 0,0033 1,0033 12,97
1 0,033 1,034 13,37
0,1 0,33 1,39 18,0
0,03 1 2,72 35,2
136
Из формулы Томсона — Кельвина следует, что упругость пара
над вогнутой поверхностью (например, над мениском жидкости,
смачивающей стенки в капилляре) при г<0 должна быть меньше
упругости пара над плоской поверхностью (1п(Рг/Роо) <0; Рг/Роо<
<1). Поэтому в капиллярах пар начинает конденсироваться, когда
он еще не насыщен по отношению к плоской поверхности
жидкости (явление капиллярной конденсации). Аналогично в ка-
пиллярах могут зарождаться и кристаллы из раствора. Как будет
показано ниже, это обстоятельство объясняет некоторые особен-
ности процесса кристаллизации, объединяемые общим названием
«наследственность». Учитывая связь между уравнениями (6.6) и
(6.10), следует отметить, что кристалл с вогнутой поверхностью
(т. е. находящийся в капилляре) имеет повышенную температуру
плавления.
Работу образования равновесного зародыша новой фазы из
пересыщенного пара или пересыщенного раствора можно выразить
так:
ДФК у-asK з ^Пр2 1П2 ’ (6.14)
ДФк 3 In2 (cr/Coo) • (6.15)
Вероятность образования равновесных зародышей новой фазы.
В соответствии с уравнением для вероятности W энергетической
флуктуации АФ W=C ехр{—АФ/йГ}, определим вероятность об-
разования любого зародыша новой фазы:
Ц73=Сехр{—а$к/3&Л- (6.16>
В развернутом виде уравнение (6.16) имеет следующий вид:
кристаллизация из переохлажденного расплава:
W —Сехо ___________4рз2а370Л42/У ) J
^3 Ь ехр | 27p\QYRT (ДГ)2 (
конденсация пересыщенного пара:
1V7 ( 16тса3Л42Л^ ) /р 1О>
— С exp зрзтз ]п2 р2 |: (6.18)
выделение из пересыщенного раствора:
1V7 Г» ( 16тга3Л42.У ) /р
Г3 = С exp J----QD3T3 2. 2 ,—;—-— I. (6.19)
3 г | 3/<3Г3р2 In2 (Сг/с^) ( v 7
Равновесный зародыш жидкой фазы, возникший в пересыщен-
ном паре, не обязательно будет центром конденсации. В соответст-
вии с зависимостью АФ=/(г), изображенной на рис. 6.6,6, в,
этот зародыш, состоящий из п молекул, находится в состоянии не-
устойчивого равновесия с окружающим его пересыщенным паром:
присоединив одну молекулу (п-+п+1), он дальше будет самопро-
извольно расти; потеряв молекулу —1), он будет далее испа-
ряться. Очевидно, центрами конденсации являются лишь капли,
выросшие по сравнению с равновесными размерами, т. е. такие,
137
число молекул в которых увеличивается от п до п+1. Вероятность
образования такой капли должна быть равна произведению двух
величин: вероятности W3 образования равновесного зародыша, со-
держащего п молекул, и вероятности W' того, что он присоединит
к себе еще одну молекулу.
Расчеты показывают *, что при небольших пересыщениях ве-
роятность образования равновесной капли -исчезающе мала; пар
должен конденсироваться -с заметной скоростью лишь при 5—10-
кратном пересыщении. Действительно, в очень тщательно постав-
ленных опытах самопроизвольная конденсация пара идет быстро
лишь при 5—8-кратном пересыщении (т. е. 5-кратное пересыщение
соответствует при таких условиях границе метастабильности).
Однако в обычных условиях пар начинает конденсироваться при
самых малых пересыщениях, когда вероятность гомогенного обра-
зования капли исчезающе мала (при малых пересыщениях приня-
тое в расчетах С=1 не играет никакой роли: по сравнению со зна-
чением ехр{—cusK/3feT}, равным, например, для двукратного пере-
сыщения 10~82, любое значение С заметно изменить картину не
может). Этот факт связан с влиянием готовых поверхностей раз-
дела (например, поверхностей взвешенных мельчайших твердых
частиц, находящихся в атмосфере, поверхностей стенок сосуда,
в котором происходит конденсация, и т. д.).
Влияние растворимых примесей на процессы кристаллизации.
Поверхностное натяжение расплавленного вещества по отношению
к его кристаллам изменяется при введении растворимых приме-
сей. Растворимые примеси, снижающие поверхностное натяжение,
называют поверхностно-активными. Такие примеси концентриру-
ются у поверхности растворителя, адсорбируются ею. Поверхност-
ная плотность такого вещества Г, т. е. выраженное в молях его ко-
личество, приходящееся на 1 см2 поверхности раздела, определяет-
ся уравнением Гиббса:
Г=—(сраст//?Т) (За/дс), (6.20)
где Сраст — концентрация поверхностно-активного вещества в рас-
творе; а — коэффициент поверхностного натяжения.
Такое действие поверхностно-активных примесей увеличивает
скорость образования центров кристаллизации и приближает гра-
ницу метастабильности к температуре плавления. Если при этом
линейная скорость кристаллизации не возрастает или увеличива-
ется незначительно, поверхностно-активные добавки должны из-
мельчать зерно закристаллизовавшегося вещества. Установлено**,
что малые добавки (до 0,05 ат. %) К уменьшают переохлаждение,
необходимое для начала кристаллизации ртути, с 18—19 до 7°С и
делают порог метастабильности более резким.
* См. задачу 6.3.
** Данилов В. И., Каменецкая Д. С. В сб.: Проблемы металловедения и фи-
зики металлов. Вып. 2. ЦНИИчермет, 1951, с. 3.
138
Таким образом, изменяя поверхностное натяжение металлов
введением малых поверхностно-активных добавок, можно управ-
лять микроструктурой слитка, вызывать «модифицирование»
сплава.
Растворимые примеси, повышающие поверхностное натяжение,
не только не концентрируются у поверхностей возникающих кри-
сталлитов, но и стремятся удалиться от этих поверхностей. Поэто-
му влияние их на кристаллизацию должно быть очень незна-
чительно, если только они в достаточной степени растворимы
в жидкой и твердой фазах.
Примеси, растворимые в жидкой фазе и практически нераство-
римые в твердой фазе, всегда тормозят процесс кристаллизации,
концентрируясь у поверхности кристаллов и нарушая контакт
между кристаллами и питающей их жидкой фазой.
§ 6.3. ГЕТЕРОГЕННОЕ ОБРАЗОВАНИЕ ЗАРОДЫШЕЙ
Общие положения. Большое значение в процессах образования
фаз имеют готовые поверхности раздела: стенки сосуда, взвешен-
ные частицы и т. д.
Рассмотрим образование капли на плоской стенке при конден-
сации пара. Если обозначить 7?к радиус кривизны равновесной
капли, 0 — краевой угол смачивания капли на стенке (рис. 6.7),
Я1,о, си,2, ct2,o — поверхностные натяжения между каплей и ее насы-
щенным паром, между каплей и стенкой и между стенкой и на-
сыщенным паром соответственно, то изменение свободной энергии
системы при образовании равновесного зародыша жидкой фазы
ДФК выразим согласно уравнению Гиббса в виде
Дфк=4" f2lc7?2K (1 — cos 0) а, „ + itfl2K sin2 0 (а,>г — аг ,)]. (6.21)
В этом уравнении внешняя поверхность
капли — шарового сегмента, отвечающего
углу 0, равна sKi=2n7?2K(l—cos 9), пло-
щадь соприкосновения капли и стенки
=nJ?2Ksin2 0 и работа образования 1 см2
поверхности соприкосновения капли и стен-
ки С0],2—Ct2,0«
По второй теореме Лапласа из теории
капиллярности имеем <ц,о—ai,2=ai,ocos 0, и
окончательно
ДФк=-Т-тг7?2к (2 — 2cos0 — sin 6 cos0)a,
О *
(6.22)
При заданном пересыщении ра-
диус кривизны равновесной капли 7?к дол-
жен быть один и тот же для сферического
зародыша, возникшего в свободном про-
странстве, и для зародыша, образовавше-
Рис. 6.7. Образование
равновесной капли на
плоской стенке при кон-
денсации пара
139
гося на стенке. Но для сферической капли
л
Дф'к = ^-
так что
ЛФк/ЛФ,к:=(2—2 cos 0—sin20 cos 0) /4. (6.23)
При полном несмачивании стенки жидкостью (0=180°, cos0=
=—ЦДФ'к—АФ. При любой же степени смачивания (0<
< 180°) АФ/к<АФк, т. е. работа образования капли на стенке мень-
ше работы образования сферической капли, причем с уменьшением
краевого угла работа уменьшается. При полном смачивании (0=0,
cos 0=1) работа образования капли на стенке равна нулю.
Следовательно, вероятность образования капли на стенке дол-
жна превышать вероятность образования капли в свободном про-
странстве. Стимулирующее конденсацию действие стенок возра-
стает с уменьшением краевого угла.
Как уже отмечалось, самопроизвольная конденсация водяного
пара в свободном пространстве в отсутствие стенок становится за-
метной лишь при 5—8-кратном пересыщении. На стенках, смачи-
ваемых конденсирующейся жидкостью, конденсация должна на-
чинаться при меньших пересыщениях, полное смачивание (0=0,
Дф—0) обеспечивает конденсацию уже при ничтожно малых пе-
ресыщениях. Поэтому практически конденсация пара всегда начи-
нается на готовых поверхностях раздела—на стенках, взвешен-
ных пылинках и т. д.
Заключения о роли готовых поверхностей раздела между фаза-
ми в процессе конденсации пара справедливы применительно к об-
разованию любой новой фазы.
Существование готовой поверхности при плавлении кристаллов приводит
к тому, что в обычных условиях перегреть твердое тело выше температуры плав-
ления невозможно. Зародыши жидкой фазы возникают на поверхности плавяще-
гося кристаллического вещества при сколь угодно малых перегревах, потому что
поверхностное натяжение жидкой фазы по отношению к кристаллу («1,2) значи-
тельно меньше, чем поверхностное натяжение кристаллического тела (а2,о) или
расплава (oti,o) по отношению к насыщенному пару, расплав вполне смачивает
плавящееся твердое тело. При этих условиях сохранить твердую фазу в мета-
стабильном состоянии выше температуры плавления невозможно. Однако удалось
сохранить олово в твердом состоянии при температуре, на 2°С превышающей
температуру плавления, прогревая кристалл электрическим током и охлаждая
его поверхность (С. Э. Хайкин, Н. П. Бене).
Влияние перегрева расплавленного металла на структуру слитка.
Известно, что слиток, полученный из сильно перегретого металла,
имеет обычно более крупное зерно, чем слиток из слабо перегре-
того металла.
В. И. Данилов и В. Е. Неймарк* исследовали кристаллизацию
воды, Hg, Bi, Sn и Al, структура которых в жидком состоянии бы-
* Данилов В. И. Строение и кристаллизация жидкостей. Киев, изд. АН
УССР, 1956.
140
ла изучена рентгенографически. Кристаллизация первых четырех
веществ характеризуется достаточно четкой границей метастабиль-
ности. Все жидкости подвергали обработке, после которой в них
оставалось очень небольшое количество нерастворимых примесей.
На рис. 6.8 показана зависимость переохлаждения при кристалли-
зации воды от температуры, до которой нагревали воду перед ох-
лаждением (каждая точка соответствует среднему значению, полу-
ченному из нескольких опытов). Из рис. 6.8 видно, что переохлаж-
дение при кристаллизации растет с увеличением перегрева вплоть
Рис. 6.8. Зависимость переохлаждения при кри-
сталлизации воды от температуры предваритель-
ного нагрева
до 25—ЗО°С. Дальнейшее увеличение перегрева на переохлажде-
ние не влияло. Если бы зародышами кристаллизации после малых
перегревов являлись анизотропные молекулы (неустойчивые кри-
сталлические образования), то увеличение перегрева влияло бы
до температуры 90—1ОО’°С, так как ближний порядок с четверной
координацией молекул Н2О сохраняется вплоть до кипения, а при
температуре 25—30°С резких изменений в структуре воды не про-
исходит.
Аналогичные результаты получены и при исследовании Sn и Bi,
здесь увеличение перегрева влияет на переохлаждение лишь до пе-
регрева на 10—15°С. При кристаллизации ртути заметного влия-
ния перегрева жидкости на переохлаждение не обнаружено. Алю-
миний вообще переохладить не удалось.
Искусственно вводя нерастворимые примеси (РЬО и WOs)
в расплавленные и профильтрованные металлы, добились того, что
при малых перегревах расплава склонность его к переохлажде-
нию резко снижалась, но большие перегревы вновь повышали пе-
реохлаждение при кристаллизации.
В. И. Данилов объясняет влияние предварительного перегрева
на переохлаждение яри кристаллизации и на число центров кри-
сталлизации постепенным растворением и дезактивацией твердых
частиц примесей, на которых образуются после небольших пере-
охлаждений зародыши кристаллизации. Температура плавления
141
кристалла, имеющего вогнутую поверхность (например, кристалла,
образовавшегося в капиллярной поре или трещине тугоплавкой
частицы), выше, чем нормальная температура плавления. Поэто-
му при небольшом перегреве металл, находящийся в порах и тре-
щинах тугоплавких посторонних частиц, не плавится — такие
нерасплавившиеся металлические кристаллы и являются при кри-
сталлизации наиболее активными центрами. При достаточно боль-
ших перегревах плавится и металл, находящийся в порах и тре-
щинах частиц примесей, и частицы в значительной степени утра-
чивают свою активность. Этот процесс может быть необратимым,
если при перегреве расплава поверхность частиц благодаря по-
верхностной миграции атомов делается более гладкой, поры и
трещины исчезают. Все нерастворимые примеси, способствующие
зарождению кристаллов в переохлажденной жидкости, можно раз-
делить на две группы. Примеси первой группы (они и были рас-
смотрены выше) становятся существенно активными в отношении
того или иного вещества после того, как побывают в контакте
с кристаллами последнего. Активность таких примесей может быть
резко снижена прогревом выше точки плавления кристаллизую-
щегося вещества. Такие примеси называют активирующимися,
а приобретаемую ими активность — наведенной.
Существуют примеси, активность которых нельзя устранить пе-
регревом. Такую активность проявляют по отношению ко многим
расплавленным металлам их собственные окислы; поведение ча-
стиц изучено на примерах Na и К, Pb и Bi, а также каменной со-
ли в расплаве а-салола и кальцита ,в расплаве гидрохинона. Та-
кие примеси В.И.Данилов назвал активными, а их активность—
естественной. Естественная активность — это следствие определен-
ного сходства во взаимном расположении атомов на соприкасаю-
щихся между собой гранях кристаллизующегося вещества и ча-
стиц примесей.
Как показали исследования Тарнбалл и сотр. *, влияние нерас-
творимых примесей на процесс кристаллизации удается устранить
почти полностью лишь при кристаллизации мельчайших капелек
(диаметр несколько микрон). Если число частиц нерастворимых
примесей в 1 см3 тщательно очищенной жидкости равно п, а сред-
ний объем капель, полученных в результате разбрызгивания жид-
кости, а<1/п, то, очевидно, некоторые мелкие капли не будут со-
держать ни одной частицы примеси. Такие капли должны кристал-
лизоваться по-другому, чем капли, содержащие хотя бы по одной
частице примеси. Действительно, некоторые мельчайшие капли
тщательно очищенной жидкости удается переохладить намного
сильнее (вплоть до О,2ТоК), чем переохлаждаются большие массы
жидкости. Это видно из сопоставления данных В. И. Данилова
и В. Е. Неймарка, полученных для значительных объемов жидко-
сти, с данными Тарнбалла (табл. 6.5).
* Холломон Д., Тарнбалл Д. В сб.: Успехи физики металлов. Т. 1. Пер.
с англ. Под ред. Я- С. Уманского и Б. И. Финкельштейна. М., «Металлургия»,.
1956, с. 330.
142
Таблица 6.5
Переохлаждение при кристаллизации некоторых веществ для больших
объемов (1 см3) и для микроскопических капель
Вещество Переохлаждение, °C Вещество Переохлаждение, °C
для смз ДЛЯ V~10 MKM3 для v~l смз | для V~10 MKM3
Н2О 11,6 39 Fe 295
Bi 32,2 90 Сц — 235
Sn 30,6 118 Ge — 227
Hg 4,5 77 Pd — 332
Al 0 130 Pt — 370
Ni 319 Au — 230
Co — 330 Pb — 80
Данные, полученные при изучении кристаллизации дисперсных
капель, позволили подсчитать по формулам теории гомогенного
образования фаз значение удельной поверхностной энергии на гра-
нице раздела кристалла с его собственным расплавом (см.
табл. 6.2).
Кристаллизация на анизотропной подкладке. Если взаимное
расположение атомов или ионов на стенке, у которой возникает
кристаллический зародыш, такое же, как на соприкасающейся
с ним грани этого зародыша, и расстояния между атомами или
ионами по обе стороны от поверхности раздела одни и те же,
то коэффициент поверхностного натяжения должен быть очень
мал (особенно если и валентности ионов в обоих кристаллических
телах одни и те же). Очевидно, в таких случаях размеры кристал-
лического зародыша должны быть очень малы и зародыш должен
иметь плоскую форму.
В предельном случае при ai,2->0 зародыш, как и при конденса-
ции пара, может представлять собой одноатомный слой. Новый
кристалл как бы продолжает в процессе своего роста тот кри-
сталл, на котором он образуется. Кристалл новой фазы должен
быть совершенно определенно сориентирован по отношению к тому
кристаллу, на грани которого он образуется. Это явление называ-
ют эпитаксией.
Приведем предложенную С. Т. Конобеевским общую формули-
ровку основного закона структурного и размерного соответствия
при ориентированной кристаллизации, которая дополняет извест-
ный закон Гиббса — Кюри — Вульфа: «Форма и ориентировка за-
родышей новой фазы при кристаллизации в анизотропной среде
должны соответствовать минимуму поверхностной энергии при дан-
ном объеме, а минимум поверхностной энергии обеспечивается
при максимальном сходстве в расположении атомов на соприка-
сающихся гранях старой и новой фаз».
При равенстве периодов кристаллической решетки обеих сопри-
касающихся фаз коэффициент поверхностного натяжения между
ними, очевидно, будет минимален (близок к нулю) для взаимной
142
ориентировки, при которой кристаллическая решетка одной фазы
служит продолжением решетки другой. Любые другие ориенти-
ровки невыгодны, так как направленные внутрь кристалла силы,
действующие на атомы поверхностных слоев, не будут компенси-
роваться силами, направленными в сторону поверхности соприка-
сающегося кристалла. Компенсация из-за притяжения к атомам
соседнего кристалла не происходит даже при правильной взаимной
ориентировке кристаллов, если расстояние между атомами на со-
прикасающихся плоскостях не совладает. При небольшом разли-
чии периодов (Да) атомы размещаются в поверхностном слое
зародыша на измененных расстояниях, равных расстояниям меж-
ду атомами кристалла подложки (а). При этом объемная свобод-
ная энергия зародыша несколько завышена из-за упругой дефор-
мации (Да/а), но зато поверхностная энергия на плоскости разде-
ла почти равна нулю.
Вероятность образования плоского закономерно ориентированного зароды-
ша имеет вид
WQ=C, ехр {—(А+В)/кТ}, (6.24)
где А — работа образования плоского ориентированного зародыша без учета
упругой деформации; Е — энергия упругой деформации.
Вероятность образования зародыша произвольной ориентировки определится
уравнением
IF = С2 ехр (6.25)
где ДФК — работа образования зародыша.
Предэкспоненциальные множители Cj и Сг мало влияют на величину W и
не могут 'значительно отличаться друг от друга. Поэтому, чтобы закономерно
ориентированная кристаллизация была более вероятна, чем неориентированная,
т. е. Wo>№, необходимо соблюдение условия Д4-Е^АФК-
П. Д. Данков вычислил предельную энергию деформирования, при затрате
которой еще выгодна ориентированная кристаллизация, введя два упрощающих
предположения:
1) работа (ДФК образования закономерно не ориентированного по отноше-
нию к кристаллу-подложке зародыша равна работе ДФК образования нормально-
го трехмерного зародыша в пересыщенном растворе при отсутствии готовой по-
верхности раздела ДФК= ~у
2) закономерно ориентированный зародыш представляет собой квадратную
пластинку, длина ребра которой I равна длине ребра нормального кубического
трехмерного зародыша, а толщина — периоду элементарной ячейки а, причем
работа образования его поверхности Д = 0.
Из приближенных вычислений, основанных на нестрогом применении обоб-
щенного закона Гука для плосконапряженного состояния, П. Д. Данков нашел,
что энергия Е деформации зародышевой пластинки, длина и ширина которой
благодаря деформации изменились от / до 1-[Л1, выражается следующей фор-
мулой:
£=(01+02) (ДОЧ
где Си и Ог— упругие постоянные растущего кристалла.
Образование ориентированного зародыша энергетически выгодно при усло-
вии, что Е^ДФК, или, принимая во внимание, что для кубического зародыша
1 1
ДФк= — asK = -ya 6/2='2a/2, то (O1+O2) (M)2a^2al2.
144
Выражая нормальную длину зародыша I и его удлинение, вызванное дей-
ствием подложки, Д/, периодом решетки а и его приращение Да, вводя число
ячеек, укладывающихся вдоль ребра пластинки п, получаем
(С11+С12) п2 (Да) 2а^2а/г2а2,
откуда
Да-^ааДсп+^г)]1/2. (6.26)
Знак равенства в уравнении (6.26) отвечает предельной деформации заро-
дышевой пластинки при ориентированной кристаллизации.
Согласно экспериментальным данным П. Д. Данкова, при осаж-
дении щелочногалоидных соединений на PbS значение Аймаке/а
лежит между 5,7% (NaCl, кристаллизующийся ориентированно) и
8% (iNal, не дающий ориентированной кристаллизации на гале-
ните) *.
Рис. 6.9. Расположение атомов в пло-
скостях наибольшей ретикулярной
плотности типичных металлических
решеток:
а — о. ц. к., плоскость (101) в a—Fe; б—
г. ц. к., плоскость (111) В Си; в —гекс
п. у., плоскость (0001)
* См. также задачу 6.4.
10—523
145
Экспериментально неоднократно наблюдали деформацию по-
верхностного слоя новой фазы, снижающую до минимума поверх-
ностное натяжение ai,2.
Ориентированная кристаллизация наблюдается на анизотроп-
ной «подложке» и при неполном совпадении структур, если в этих
кристаллах имеются «сопрягающиеся» атомные комплексы
(рис. 6.9).
Ориентированная кристаллизация из жидкой и парообразной
фаз на анизотропной подложке распространена широко. Еще бо-
лее распространена ориентированная кристаллизация из твердой
фазы в результате фазовых превращений и химических реакций
в твердом состоянии.
§ 6.4. АТОМНАЯ ТЕОРИЯ РОСТА КРИСТАЛЛОВ
Форма кристалла. Рассмотрим идеальный случай бездефектного
зародыша. Его минимальный линейный размер определяется
формулой (6.6), а форма растущего монокристалла при совершен-
ной кристаллической решетке определяется из условия равнове-
сия кристалла с расплавом. Согласно теории Гиббса это условие
сводится к тому, что свободная энергия для данного объема дол-
жна быть минимальной. Принимая, что объемная свободная энер-
гия постоянна, имеем
2 az (hkl) si (hkl) =min. (6.27)
z=i
Здесь обозначения те же, что для уравнения (6.86).
Если общая свободная энергия постоянна, то из (6.27) полу-
чаем
2 as (hkl) dst (hkl) =0. (6.28)
1=1
Развиваться будут те грани, которые уменьшают общую сво-
бодную поверхностную энергию. Для оценки скорости роста раз-
ных граней \hikili) с поверхностью Si(hkl) рассмотрим кристалл,
ограненный п гранями (/=1, 2, ..., п), для которого рг— рас-
стояние от центра кристалла до грани (hikili).
Объем кристалла v равен
п
v^-^-^PiS^hkl), (6.29)
i=l
и изменение объема, когда одни грани растут за счет других,
= 2 s^hktydpi- (6.30)
z=i
146
В то же время из (6.29) имеем
dv = ~- [sz (hkl) dpt + {hkl)\. (6.31)
z=i
Если при этом объем не меняется, то dv=Q и из (6.30) и (6.31)
2 pidsi(hkl) = Q. (6.32)
;=1
Из сравнения уравнений (6.28) и (6.32) следует, что
pi^a.i (hkl).
(6.33)
Так как линейная скорость роста Сг
определяется для перемещения грани
(hikih) в перпендикулярном направле-
нии, из уравнения (6.33), следует, что cL
пропорциональна удельной поверхност-
ной свободной энергии грани. Развивая
идею Бравэ, предположили, что грани,
обладающие наибольшими ретикулярны-
ми плотностями, имеют минимальные
поверхностные энергии, т. е. для них
Ci=min.
Тогда если ретикулярная плотность
плоскостей (1.00) выше, чем плоскостей
(101), то Сюо<Сю1 (рис. 6Л0). Более
быстро растущие плоскости, как видно
из рис. 6.1'0, зарастают и исчезают сов-
сем. Форма кристалла при этом будет
определяться медленно растущими пло-
скостями с наибольшими ретикулярными
ПЛОТНОСТЯМИ 2" hkl-
с(ооп
Рис. 6.10. Зависимость конечной фор-
мы кристалла от скоростей роста с^йг
граней
Для грани (hkl) (hkl) = $0 1 (hkl), где s0’(hkl) —площадь параллелограмма >
вершины которого образованы четырьмя ближайшими атомами этой плоскости.
Объем элементарной ячейки равен v0=s0 (hkl)dhki (dhki— межплоскостное
расстояние), поэтому
&(hkl)=dhhi/v0, (6.34)
Или для кубической сингонии
^(/i&)=ao/(vog), (6.35)
где g2=h2+k2+l2.
Ретикулярная плотность обратно пропорциональна g и для примитивной ку-
бической решетки уменьшается с увеличением индексов hkl (табл. 6.6). Это
объясняет конечную форму кристалла, имеющего примитивную кубическую ре-
шетку (куб).
Для о. ц. к. и г. ц. к. решеток надо учесть, что действительные расстояния
между атомными плоскостями уменьшаются вдвое при определенных ориента-
циях плоскостей (для о. ц. к. при h-\-k+l^2n и для г. ц. к. при неодинаковой
четности /г, k и /), в этих случаях для вычисления g надо брать удвоенные
индексы (’200) вместо (100) и т. д.
Из табл. 6.6 следует, что для о. ц. к. металлов ретикулярная плотность по-
нижается в ряду: (ПО), (100), (2.11), (310), (111).
По табл. 6.6 2? для г. ц. к. решеток уменьшается в ряду (111), (100), (ПО),
(311), (331), (210) и т. д.» а скорость роста этих граней соответственно теории
10* 147
Таблица 6.6
Индексы граней кристалла кубической сингонии (hkl) и характеристика
ретикулярной плотности g
Примитивный куб (hkl) (100) (110) (111) (210) (211) (221) 9 (310) (311)
g 1 2 3 5 6 10 11
О. ц. к. (hkl) (200) (HO) (222) (420) (211) (442) (310) (622)
g 4 2 12 20 6 36 10 44
Г. ц. к. (hkl) (200) (220) (111) (420) (422) (442) (620) (311)
g 4 8 3 20 24 36 40 11
растет. Очевидно, следует ожидать для г. ц. к. металлов чаще всего грани {111},
затем {100} и т. д.
Рост кристаллов. Согласно изложенной теории можно объяснить
конечные формы свободно растущего кристалла, но нельзя рас-
Рис. 6.11. Модель поверхности кристалла (7—7 — позиции атомов, находя-
щихся в разных окружениях)
Рис. 6.12. Присоединение атома к
плоскости (100) простой кубической
считывать конкретные скорости роста, так как теория Гиббса при-
менима только к кристаллу, находящемуся в равновесии с распла-
вом.
В общем случае на поверхности растущего кристалла можно
представить недостроенный атомный слой, образующий ступеньку
(рис. 6.11). Атомы могут присо-
единяться или покидать ступень-
ку в разных местах, и на сту-
пеньке образуется ряд изломов
(5 и 6) или атомов, присоединен-
ных к ступеньке (4), на поверх-
ности ступеньки имеется также
незначительное количество (отно-
сительно числа изломов) адсор-
бированных отдельных атомов
(/—3) или агрегатов атомов. По-
являются также вакансии (7)
или группы вакансий.
При росте кристалла атомы
могут присоединяться к ступеньке
148
в разных позициях 1—7, но энергетически эти позиции неравно-
ценны. Исследуя количество связей, реализующихся в результате
присоединения атомов в разных позициях, можно оценить силу
связи, что позволит определить места, где присоединение атомов
энергетически наиболее выгодно. При этом для металла достаточ-
но рассматривать первые три координационные сферы (/=1, 2, 3).
Например, при присоединении атома к плоскости (100) (рис. 6.12)
примитивной кубической структуры для первой координационной
сферы на расстоянии (период решетки) расположен один
атом, для второй сферы на расстоянии Г2=аУ 2 — четыре и для
третьей сферы на расстоянии г3=а]/г 3 — также четыре. Запишем
это в следующем виде: Ф"'=1 /4/4.
При присоединении атома к двумерной сетке плоскости (100)
для введенных выше первых трех координационных сфер число со-
седей запишем в виде Ф"— 1 /2/0.
При присоединении атома к концу цепочки атомов [100] име-
ем: Ф'=1/0/0. Здесь Ф', ф" и Ф'"— константы, которые для ион-
ного кристалла аналогичны константам Маделунга, определяющим
энергию связи. При росте кристалла присоединение атома в по-
зиции 1 (рис. 6.11) дает выигрыш энергии, пропорциональный
Ф"'=1/4/4, в позиции 4 атом находится одновременно над кри-
сталлической плоскостью (100) и на ребре, т. е. выигрыш энергии
будет пропорционален сумме Ф'" -\-ф"=2/6/4. Максимальный
энергетический выигрыш будем иметь, очевидно, для позиции 5,
так как при этом Ф1/2=Ф' + Ф" + Ф"'—3/6/4.
Индекс 1 /2 здесь означает, что число соседей у присоединенно-
го атома в позиции 5 составляет половину максимально возмож-
ного для данной структуры.
В процессе роста ступенька двигается к краю кристалла и
в конце концов исчезает, оставив завершенную кристаллическую
поверхность. Дальнейший рост происходит в том случае, если воз-
никают новые ступеньки. Вероятность образования ступеньки опре-
деляет линейную скорость роста. Предположим, что образование
ступенек происходит по механизму зарождения способных к даль-
нейшему росту двухмерных зародышей на поверхности кристалла.
Если радиус зародыша р, то при его образовании изменение сво-
бодной энергии:
ДФ=~лр2ДЕ+2лра', (6.36)
где Д/7— свободная энергия на единицу площади, приобретенная
при присоединении зародыша к кристаллу; а' —свободная энергия
на единицу длины торцевой поверхности монослоя.
уравнение (6.
=;0. При этом
pK=a'/bF. (6.37)
Используя (6.37) и ZK=2jrpK из уравнения (6.36), получаем
ДФ,к=а7к/2. (6.38а)
149
36), определим рк из условия —
Рк
Уравнение (6.37) аналогично уравнению (6.8) для равновесно-
го трехмерного зародыша, только вместо поверхности SK вводится
периметр /к двухмерного зародыша, а поверхностное натяжение а
заменяется удельной периферийной энергией (а'). В функции пе-
реохлаждения для свободной энергии двухмерного зародыша кри-
тической величины получим
АФК= [л (а') 2Л] / Q'AZ, (6.386)
где Q' — скрытая теплота плавления на единицу поверхности мо-
нослоя.
Радиус критического зародыша определяется выражением
pK=aTs/Q'A7\ (6.39)
Вероятность образования равновесного двухмерного зародыша
в соответствии с формулой для энергетических флуктуаций
имеет вид
W2=C ехр{—а'1к/2кТ}. (6.40)
Таким образом, в соответствии с описанным механизмом за-
рождения ступенек двухмерный зародыш быстро растет, последо-
вательно присоединяя к себе ряды атомов, так что одноатомный
слой, образовавшийся в результате его роста, покрывает грань
кристалла. После заполнения грани наступает пауза, длительность
которой т обратно пропорциональна вероятности W2 образования
нового двухмерного зародыша:
-^ехр (6.41)
затем наращивается новый атомный слой и т. д.
В заключение отметим, что рост реальных кристаллов обычно
осложняется множеством побочных факторов, из которых важней-
шим является влияние силы тяжести, выделяющегося при кри-
сталлизации тепла и примесей. Все эти факторы часто искажают
внешнюю форму реальных кристаллов по сравнению с равновесной
и приводят к дендритной кристаллизации расплавленных метал-
лов.
Атомная структура поверхности раздела расплав — кристалл и
механизмы роста кристаллов. Структуру поверхности растущего из
расплава кристалла исследовали экспериментально с применением
светового микроскопа для прозрачных органических и неорганиче-
ских кристаллов, в случае металлических кристаллов использовали
технику декантации — исследовали поверхность фронта роста кри-
сталла после его извлечения из расплава. Иногда наблюдали от-
клонения от плоской формы фронта кристаллизации — образова-
ние выпуклостей. Их возникновение при кристаллизации чистого
вещества можно объяснить только существованием отрицатель-
ного градиента температуры в расплаве перед фронтом растущего
кристалла. При положительном градиенте (см. рис. 6.13,а) любой
выступ попадает в область температур, превышающих Го, и дол-
150
жен расплавляться. Отрицательный градиент температур в рас-
плаве непосредственно перед фронтом кристаллизации может воз-
никать благодаря выделению скрытой теплоты кристаллизации.
В предельных случаях, когда существует сильная анизотропия ско-
рости роста в разных кристаллографических направлениях или
различие поверхностной (межфазовой) энергии для разных граней,
выступы приобретают форму дендритов *. Сложный характер гео-
Рис. 6.13. Морфология поверхности раздела
при кристаллизации чистого вещества в зави-
симости от знака градиента температуры
в жидкой фазе перед фронтом кристалли-
зации:
а — плоский фронт; б — ячейки в дендритные ячейки
метрии поверхности раздела благодаря структурным дефектам,
связанным с действием силы тяжести и явлениями усадки, а так-
же наличию даже самого малого количества посторонних приме-
сей проявляется в фрагментации всего объема кристалла {ячеи-
стая структура}.
Экспериментальных данных об атомной структуре поверхности
растущего кристалла нет, однако имеются теоретические сообра-
жения, которые уже получили некоторое косвенное подтвержде-
ние.
Рассмотрим два крайних случая строения границы: 1) атомно-
гладкая или кристаллографически совершенная плоскость, когда
на N узлов на плоскости идеальной решетки приходится очень
мало «лишних», т. е. выступающих, или недостающих атомов
(Л^а); 2) шероховатая поверхность, когда из N позиций атомного
* Каждую ветвь дендрита второго, третьего и т. д. порядков можно рассма-
тривать, в свою очередь, как «выступ».
151
слоя беспорядочным образом заполнено атомами около 50%. Сво-
бодная энергия слоя в зависимости от энтропии плавления (изме-
нения энтропии ASw^L/Tq, где L — скрытая теплота кристалли-
зации; То — равновесная температура плавления) и вероятностной
функции от N и Na имеет один или два минимума, соответствую-
щих указанным структурам. При рассмотрении модели границы
раздела, представляющей собой два слоя (два уровня), шерохова-
тая поверхность должна быть при £/&7о<2: при L/kTo>2 грани-
ца раздела должна быть атомно-гладкой. В предположении мно-
гоуровневой границы (т. е. более постепенного перехода от жид-
кого состояния к кристаллическому) оказывается выгодной шеро-
ховатая поверхность раздела при Л/£То<3,5*. Оценки энтропии
плавления позволяют сделать вывод, что для металлов должна
быть характерной шероховатая поверхность, тогда как для полу-
металлов и полупроводников, а также для большинства органиче-
ских соединений энтропия плавления велика и в контакте с рас-
плавом выгодны гладкая поверхность и кристаллографически пра-
вильная огранка.
Для атомно-гладкой поверхности рост кристалла возможен
только образованием двухмерного зародыша и его бокового роста.
При наличии ступеньки от винтовой дислокации также возможен
боковой рост (см. ниже). В случае шероховатой поверхности рост
кристалла происходит присоединением одиночных атомов — нор-
мальный рост (термин связан с направлением элементарного про-
цесса— вдоль нормали к поверхности).
Теория позволяет выделить крайние случаи. В действительно-
сти большинство неорганических и органических веществ характе-
ризуются промежуточными значениями энтропии, а переход от
гладкой к шероховатой поверхности должен зависеть от конкрет-
ной координации атомов, зависящей от типа структуры кристал-
ла и от условий кристаллизации (прежде всего от переохлажде-
ния). Скорость для нормального роста должна увеличиваться про-
порционально переохлаждению сНорм=^нормА7’. Здесь, кинетический
коэффициент £норм=(£>повД5пл) I&RT, Опов — коэффициент диффу-
зии вдоль поверхности раздела, а — размер скачка для атомов.
Кинетический коэффициент для металлов равен 1 см • c~J • К”*1, сле-
довательно, скорость роста должна быть большой уже при незна-
чительном переохлаждении. Однако измерения давали порядок
10~3 см-с-1. Следовательно, можно предположить, что действи-
тельное переохлаждение на фронте кристаллизации из-за выделе-
ния скрытой теплоты кристаллизации мало и существенно отлича-
ется от измеряемых в макрообъемах. В общем случае, по-видимо-
му, нормальный рост возможен при достаточно большой движущей
силе кристаллизации (AF).
* Темкин Д. Е. В кн.: «Механизм и кинетика кристаллизации». Минск,
«Наука и техника», 1964, с. 86.
152
Экспериментальные результаты * подтверждают предсказание
теории о послойном (т. е. боковом) росте кристаллов с большой
энтропией плавления. Устойчивые формы роста (огранка) для этих
веществ сохраняются при значительно большем переохлаждении
расплава (висмут, пиперонал при ДТ^5^-10°), чем для вещества
с низкой энтропией плавления (олово, циклогексанал при
ДТ^Доли градуса).
§ 6.5. КИНЕТИКА КРИСТАЛЛИЗАЦИИ
Число центров кристаллизации. Скорость образования зароды-
шей пропорциональна произведению вероятности образования
.трехмерного зародыша на вероятность того, что зародыш этот
растет.
Рассмотрим случай, когда трехмерный кристаллический заро-
дыш растет не за счет присоединения единичных молекул (атомов)
из окружающей среды, а в результате образования и роста на его
плоских гранях двухмерных равновесных зародышей. При этом
число центров п, образующихся в единице объема за 1 с (или
^скорость образования центров),
п = К (г2к ехр {—a/lJ2kT}Y exp {—asK/3£T}. (6.43)
Выражение, стоящее в скобках, дает вероятность образования
^двухмерного зародыша на одной из граней трехмерного зародыша,
длина ребра которого обозначена гк. Очевидно, вероятность эта
должна быть пропорциональна поверхности грани >г2к. Поскольку
равновесная форма кристалла, зарождение которого анализирова-
лось,— куб, образование двухмерного зародыша на одной лишь
грани не завершает первого этапа роста трехмерного зародыша:
последний должен нарастить не менее одного атомного слоя на
каждой из трех взаимно перпендикулярных граней. Перемножение
вероятностей образования двухмерных зародышей на трех гранях
и приводит к тому, что выражение, стоящее в скобках, возводится
в куб.
Принимая во внимание зависимость размеров равновесных
трехмерных и двухмерных зародышей от коэффициента поверх-
ностного натяжения а и удельной периферийной энергии а', а так-
же от переохлаждения АТ, уравнение (6.43) можно записать
в следующем виде:
п = К0 ехр {—2[ТКГ} ехр {—К2а’/Т (ДТ)2},
где Kq, Ki и Кг— константы.
В тех случаях, когда удельная периферийная энергия при об-
разовании двухмерного зародыша мала, а переход атомов или мо-
лекул от жидкости к растущему кристаллу связан с преодолением
* Овсиенко Д. Е., Алфинцев Г. А. В кн.: «Механизм и кинетика кристалли-
зации. Минск, «Наука и техника», 1973, с. 144.
153
высокого потенциального барьера (большая энергия активации),
в формулу (6.43) для п вводится кинетический фактор:
^-1 ехр J
п — К0 ехр
-^2а3 ]
Т (ДГ)2 j
(6.44)
(Ks—qfR, где q — энергия активации при переходе атома или мо-
лекулы от жидкой фазы к твердой). Первому члену (кинетический
фактор) на рис. 6.14 соответствует кривая /, второму — кривая 2.
Уравнение (6.44) справедливо для кристаллизации жидкостей
высокой вязкости. Очевидно, величина q имеет решающее значение^
в процессах кристаллизации жидкостей при большом переохлаж-
дении. Чем больше энергия акти-
вации при переходе частиц от жид-
кой фазы к твердой и чем мень-
ше коэффициент поверхностного
натяжения а, тем меньше переох-
лаждение, отвечающее макси-
мальной скорости образования
центров кристаллизации. Нали-
чие готовых поверхностей разде-
ла (стенок изложницы, взвешен-
ных твердых частиц, или собст-
венно активных или активирован-
ных) смещает максимум скоро-
сти образования центров в сторо-
ну еще меньших переохлаждений.
Линейная скорость роста кри-
сталлов. Рассмотрим процесс рос-
та кристаллов. Как уже отмеча-
лось, процесс этот носит скачко-
образный характер, наращивание
каждого атомного слоя начинает-
то # гмет лтгр
Рис. 6.14. Зависимость линейной ско-
рости с и числа центров кристалли-
зации п от переохлаждения:
1 — кинетический фактор; 2 — термодина-
мический фактор в определении п
ся с образования «двухмерного зародыша». Однако равновесный
двухмерный зародыш может расти, покрывая всю грань, или рас-
творяться (плавиться). Поэтому при вычислении линейной скоро-
сти роста кристалла необходимо учитывать не только вероятность
образования двухмерного зародыша, но и вероятность присоедине-
ния к его торцу одной молекулы или целого атомного ряда. Моле-
кула, присоединяющаяся к двухмерному зародышу, должна отде-
литься от жидкой фазы, совершив определенную работу против
сил сцепления и, поэтому вероятность такого присоединения дол-
жна быть пропорциональна величине ехр{—u/kT}. Формула для
линейной скорости кристаллизации с имеет следующий вид:
с=Ко ехр{—К/ТДТ}ехр{—К3/Л-
(6.45)
В этой формуле первая экспоненциальная функция пропорцио-
нальна вероятности образования двухмерного зародыша, вторая—
вероятности присоединения к нему одной молекулы, т. е.
154
Как следует из формул (6.43) и (6.45), линейная скорость кри-
сталлизации в зависимости от температуры имеет максимум при
меньших переохлаждениях, чем скорость образования зародышей
(см. рис. 6.14).
При анализе формулы (6.45), так же как и при рассмотрении
влияния переохлаждения на скорость образования центров кри-
сталлизации, следует иметь в виду, что упрощающие предположе-
ния, лежащие в основе вывода этих формул, не приводят к сущест-
венным ошибкам лишь при малых переохлаждениях. При больших
переохлаждениях, когда жидкость приближается к стеклообразно-
му состоянию, изменяется по существу механизм роста кристаллов.
Справедливость представления о росте кристаллов при опреде-
ленных условиях образованием двухмерных зародышей и правиль-
ность выведенной на основании этого представления формулы
(6.45) были подтверждены опытами по кристаллизации салола
В. И. Даниловым и В. И. Малкиным.
Кинетика процесса. При малых переохлаждениях и температуре
выше начала подъема кривой п (ДТ) вещество может не кристал-
лизоваться даже при длительных выдержках. В этом случае сплав
находится в метастабильной области (Тмет<Г<Г0, см. рис. 6.14).
Однако с увеличением переохлаждения п растет, и после перехо-
да через границу метастабильности расплав начинает кристалли-
зоваться. Ход кристаллизации, а также размеры зерен в литом
металле зависят от соотношения скоростей образования зароды-
шей и роста кристаллов.
Представления о кристаллизационных параметрах были при-
менены не только к анализу процессов образования кристаллов
из жидкой фазы, но и к анализу процессов, протекающих в твер-
дой фазе (рекристаллизация, фазовые превращения). В связи
с изучением явлений рекристаллизации и фазовых превращений
сделаны первые расчеты кинетики Б. В. Старком и И. Л. Мирки-
ным *. В самой общей форме задача была решена А. Н. Колмо-
горовым **.
В дальнейшем приведен расчет по А. Н. Колмогорову приме-
нительно к простейшему случаю, когда скорость зарождения цен-
тров и линейная скорость роста кристаллов остаются все время
постоянными. Причем кристаллы растут с одной и той же скоро-
стью по всем направлениям, сохраняя до момента соприкоснове-
ния с другими кристаллами сферическую форму.
А. Н. Колмогоров определял вероятность P(t) того, что какая-либо точка
объема заполненного кристаллизующимся веществом, спустя t с от начала
процесса кристаллизации попадает внутрь закристаллизовавшейся массы. Если
начальный объем маточной фазы и0 намного больше размеров отдельных кри-
сталлов, то закристаллизовавшийся за 1 с объем v(t) равен произведению пер-
воначального объема vQ на вероятность P(t), т. е,
u(t)=P(t)vQ (6.46)
в начальный момент времени при /=0 и и(/)=0.
* Старк Б. В., Миркин И. Л. Тр. Моск, ин-та стали. М., Металлургиздат,
1935.
** Колмогоров А. Н. «Изв. АН СССР», 1937, № 3, с. 355.
155
Для того чтобы определить, по какому закону растет объем закристаллизо-
вавшейся массы v (/), рассмотрим, что происходит за небольшой промежуток
времени At между t и t4~At.
Прежде всего в некристаллизовавшемся объеме vQ—v возникают новые цент-
ры кристаллизации. Вероятность образования одного центра кристаллизации
в некоторой части незакристаллизовавшегося объема —v за промежуток
времени между t и t-\-At равна nv'At (п скорость образования центров кристал-
лизации). Центры кристаллизации, возникшие за время At, как и кристаллы, воз-
никшие ранее, растут с постоянной линейной скоростью с. Рассмотрим произволь-
ную точку О, находящуюся на расстоянии, превышающем ct, от границ объема
t’o. Эта точка к моменту t окажется внутри закристаллизовавшейся массы, если
в какой-то момент времени ti<t в какой-либо точке О', находящейся на рас-
стоянии г^о(/—ti), возникает центр кристаллизации. Объем v'(ti), внутри ко-
торого должен возникнуть центр кристаллизации, чтобы точка О оказалась
внутри закристаллизовавшейся массы, определим так:
г/(/1.)==4/Злг3макс-=4/Злс3(/—/г)3. (6.47)
Вероятность Pi того, что за промежуток времени в объеме, равном v'(ti),
образуется центр кристаллизации, равна
Pi = 7\v'(ti)Ati = 4/3nc3n(t— tiYAt. (6.48)
Вероятность qt того, что в объеме v'(tP) за t с не возникнет ни одного цент-
ра кристаллизации, равна
qi = \-Pi. (6.49)
Разделим промежуток времени t от начала кристаллизации до данного мо-
мента на т равных промежутков At — t/m. Для того чтобы точка О оказалась
к моменту времени t вне закристаллизовавшегося объема, необходимо, чтобы ни
в одном из этих промежутков времени не зародился такой центр кристаллиза-
ции, который за время до момента t мог бы дорасти до точки О. Поскольку ве-
роятность совпадения нескольких независимых событий равна произведению ве-
роятностей каждого из них, вероятность q(t) того, что точка О останется до
момента t вне закристаллизовавшегося объема, выразится так:
i—m i—m
q(t)= П = П (б.50>
1=1 1=1
где ii — iAt.
Для очень малого промежутка времени At вероятность Pi образования цент-
ра кристаллизации в объеме Vi также чрезвычайно мала, поэтому можно при-
нять
ехр{—Pi} = 1—Pi.
В соответствии с этим преобразуем уравнение (6.50):
i=m i—m
q(t) = J] (1 - Л) = П ех₽ (6.51>
i—\ Z=1
Логарифмируя уравнение (6.51) и используя (6.48), получим
т т
In <7 (0 = — Pi = -у- лс3п V (ti — О’ ~t;,
1=1 1=1
и, переходя от малых конечных промежутков времени At к бесконечным малым
промежуткам di:
t
In q (t) — -jj-rcc3n J (t— t)3 di = (— Tic3n/4/3).
0
156
Учитывая уравнения (6.46) и (6.49), найдем окончательное вы-
ражение для объема v (/), закристаллизовавшегося за t с:
v(t) = vQ (1 — ехр {—• (6.52)
На рис. 6.15 приведена кинетическая кривая v(t)/vo, построен-
ная для значения параметра лс3п/3=1,5-10~6. Как видно из этого
графика, закристаллизовавший-
ся объем сначала растет очень
медленно (небольшое число рас-
тущих кристалликов и малая по-
верхность каждого из них), затем
рост резко ускоряется, кривая
круто поднимается вверх, и, на-
конец, когда незакристаллизовав-
шийся объем уменьшается до
10—20% первоначального, кривая
вновь становится пологой.
Формула (6.52) является при-
ближенной. Она была бы точна
лишь в том случае, если бы пер-
воначальный объем был бесконеч-
но велик. Действительно, при ко-
нечном значении первоначального
объема Vq он должен был бы пол-
Рис. 6.15. Кинетическая кривая и/уои
кривая общей скорости кристаллиза-
ции Q/Qmhkc
ностью закристаллизоваться за
конечный промежуток времени, между тем как по формуле (6.52)
процесс кристаллизации длится бесконечно долго. Формула (6.52)
делается заметно неточной, когда незакристаллизовавшийся объем
делается очень малым (меньше 1 —10% у0), причем неточность ее
проявляется тем сильнее, чем меньше кристаллитов содержит за-
кристаллизовавшееся тело, т. е. чем крупнее зерно.
По формуле (6.52) можно вычислить промежуток времени, в течение которо-
го кристаллизуется любая определенная доля кристаллизующейся массы В —
==^(^)М-
При Р = 0,5
/о,5*=0,901 (с3п) ~4/4. (6.53}
При 0 = 0,99 можно считать, что кристаллизация практически заканчивается:
tK = 1,45 (пс3)—1/4. (6.54}
Число кристаллов /V(/), образовавшихся за промежуток времени /, можно
определить по формуле
t
N (/) = уоп J q (т) dt, (6.55}
о
Для полностью закристаллизованного объема
W=iO,9t>o(n3c~3)1/4. (6.56)
При анализе процесса кристаллизации важно знать величину
= которую можно назвать общей (объемной) скоростью
кристаллизации (превращения). Общая (объемная) скорость кри-
157
сталлизации равна объему, кристаллизующемуся в 1 см3 за 1 с.
Общая скорость кристаллизации достигает максимума, когда
закристаллизовавшийся объем составит половину первоначального
объема (см. рис. 6.15).
Закристаллизовавшаяся масса в начале процесса очень мала и
медленно увеличивается. Это приводит иногда к предположению,
что процесс превращения при изотермической выдержке начина-
ется не сразу, а спустя некоторый промежуток времени, называе-
мый инкубационным периодом. В большинстве случаев термин
«инкубационный период» следует понимать условно: на самом де-
ле, и за этот период обычно возникают и растут центры кристал-
лизации, новые кристаллы можно обнаружить лишь в результате
большого числа опытов *. При кристаллизации жидкости процесс
роста кристаллов усложняется и тормозится из-за выделения скры-
той теплоты плавления. Скорость роста кристалла в жидкой фазе
с течением времени обычно уменьшается.
Дислокации и рост несовершенных кристаллов. Реальные кри-
сталлы могут расти с гораздо большей скоростью, чем это дает
расчет для механизма роста путем образования двухмерных заро-
дышей. При нормальном росте, когда присоединение атомов к по-
верхности растущего кристалла происходит повсеместно, скорость
роста должна быть пропорциональной переохлаждению (для не-
больших переохлаждений).
Результаты экспериментов дают зависимость с^ДТ1’75*0’5
(табл. 6.7). Чтобы объяснить это, приходится допустить, что при
Таблица 6.7
Скорость роста некоторых кристаллов*
Вещество Скорость роста, см-с"1
наблюдаемая вычисленная по (6.60)
Вода Сб.Ю-'ДГ1’7 1,3.10-2Д72
Глицерин З-Ю^ДГ1'7 7-10~6ДГ2
Салол ЧИО-МГ1’7 10“3Д72
Олово 7.10"1Д7'1»8 1,4- 10-3Д72 ‘
Фосфор 102ДГ’7 4’10“*ДГ2
* Хиллиг У., Тернбалл Д. В кн.: Элементарные процессы роста кристаллов. Пер. с англ. Под
ред. Г. Г. Леммлейна и А. А. Чернова, М., Изд-во иностр, лит., 1959.
данном переохлаждении присоединение атомов или молекул про-
исходит не повсеместно, а только в отдельных узлах, число кото-
рых составляет часть у от полного числа ячеек на поверхности,
причем у — функция ДТ. Тогда
с=#гу(ДТ)ДТ. (6.57)
Таким образом, снова возвращаемся к идее существования
на поверхности кристалла ступенек, к которым присоединяются
атомы. Важная особенность обсуждаемых экспериментов состоит
* Неизбежным является инкубационный период длительностью То = и-1уТ1
158
в том, что поверхность кристалла должна все время сохранять сту-
пени роста.
Если винтовая дислокация выходит на поверхность кристалла,
как показано на рис. 6.16, появляется ступенька, к которой будут
присоединяться атомы в процессе роста. Грань зарастает, но так
как поверхность является геликоидальной, ступенька все время
сохраняется, поворачиваясь вокруг точки выхода дислокации (см.,
рис. 6.16,6).
Рис. 6.16. Ступенька, обусловленная винтовой дислокацией (а). В процессе
роста она сохраняется, передвигаясь вращением вокруг фиксированной
точки (б—д)
Если линейная скорость роста <с постоянна по длине ступеньки,,
она, поворачиваясь, искривляется, так как более удаленные от
центра вращения участки перемещаются с меньшей угловой ско-
ростью 0. Здесь 0=сг (г — радиус-вектор атома на спирали;
0 — его угловая координата). Этим объясняется экспериментально'
наблюдаемый факт развития на зарастающей грани спирали, одно-
образно вращающейся вокруг дислокации (см. рис. 6. 16,6—6).
Для квазистационарных условий роста можно принять, что г=
=2рс0, т. е. это будет спираль Архимеда. Вблизи выхода линии
159*
дислокации радиус кривизны рс не может быть меньше критиче-
ского двухмерного зародыша для данного ДТ, иначе атомы будут
самопроизвольно испаряться до тех пор, пока радиус не увеличит-
ся до равновесной величины, определяемой по формуле (6.37), т.е.
/АТ. (6.58)
Скорость роста с при этом по сравнению с (6.42) увеличивается
в у раз:
Т=//(4лрс). (6.59)
Здесь I — линейный размер области у моноатомной ступени,
где, по предположению, присоединяются атомы в процессе роста.
Тогда по (6.57) — (6.59) получим
с=7<(ДТ)2, (6.60)
где 1\ — новая постоянная.
Описанный механизм роста, как видно из табл. 6.7, хорошо
объясняет зависимость с от АТ.
Если на поверхность кристалла выходит группа близко рас-
положенных винтовых дислокаций одного знака, образуется много-
слойная ступенька.
Установлено, что скорость роста спирали зависит от кристалло-
графического направления ее различных участков на поверхности
растущего кристалла.
Кристаллизация из пара через жидкую фазу. В определенных
условиях осуществляется кристаллизация из пара через жидкую
фазу по так называемому ПЖТ-механизму (пар->жидкость->
-^твердое). При этом кристаллы получаются почти совершенны-
ми и имеют разнообразную форму: от усов или нитевидных кри-
сталлов до эпитаксиальных пленок.
Например, для выращивания нитевидных монокристаллов крем-
ния по ПЖТ-механизму на кристалл-подложку кремния помещают
мельчайшие частички золота размером 10”1—10~3 см. При нагрева-
нии подложки золото плавится, и образовавшаяся капелька насы-
щается кремнием. При пропускании над капелькой водорода и
тетрахлорида кремния в газовой фазе образуется атомарный крем-
ний, который переходит через границу раздела газ — жидкая фа-
за, растворяясь в золоте. Пересыщение жидкой фазы (золота
кремнием) происходит быстрее, чем кристаллизация по механизму
зарождения и роста слоев, поэтому обогащение продолжается до
тех пор, пока движущая сила кристаллизации не возрастет на-
столько, что начнет действовать механизм нормального роста, т. е.
присоединение атомов по всей поверхности твердой фазы.
§ 6.6. ОСОБЕННОСТИ КРИСТАЛЛИЗАЦИИ И СТРУКТУРЫ СПЛАВОВ
Затруднение процесса кристаллизации при несовпадении соста-
ва кристаллов и состава жидкой фазы. При кристаллизации из пе-
ресыщенного раствора и при эвтектической кристаллизации воз-
никновение равновесного зародыша связано не только с образо-
ванием поверхности раздела, но и с необходимостью скопления
160
молекул или атомов кристаллизующегося вещества в объеме это-
го зародыша, т. е. с необходимостью «концентрационной» флук-
туации. При кристаллизации твердого раствора концентрации с
гк=2аЛ1 /{р [ (1 —с) Дца + сД 4цв]}, (6.61)
где Дрл 'И Дрв — разности химических потенциалов для компонен-
тов в расплаве и в твердом растворе.
При достаточно сильном переохлаждении образование и рост
зародышей могут происходить без перераспределения концентра-
ции. Возможность такой бездиффузионной кристаллизации следу-
ет из сопоставления значений химической свободной энергии для
исходной жидкой фазы в сплаве твердой фазы того же состава
и смеси жидкой и твердой фаз, равновесных при данной темпе-
ратуре Т' (рис. 6.17). Свободная
энергия твердой фазы (точка 61
оказывается меньше свободной
энергии жидкой фазы (точка
что определяет термодинамиче-
скую возможность образования
твердой фазы того же соста-
ва (С1).
Как видно из схемы, приведен-
ной на рис. 6.17, возможность без-
диффузионной кристаллизации
зависит от состава сплава: если
состав сплава лежит правее точ-
ки с0, то при данной температуре
Т' термодинамически невыгодно
образование твердой фазы, совпа-
дающей по составу с жидкой фа-
зой (точка а2 ниже точки б2), и
должны возникать кристаллы
твердой фазы, обогащенные ком-
понентом А (в соответствии с диа-
граммой на рис. 6.17,6). Если на
диаграмму равновесия нанести
температуры, при которых значе-
ния свободной энергии твердой и
жидкой фаз одного и того же со-
става равны (т. е. зависимость
состава с0, показанного на схеме
рис. 6.17,а, от температуры), то
положение фигуративной точки
сплава по отношению к этой ли-
нии (Т'о, штрих-пунктир на
рис. 6.17,6) определяет возмож-
ность бездиффузионной кристал-
лизации. Для сплава с2 центры
кристаллизации могут иметь тот
1523
Рис. 6.17. Изменение свободной энер-
гии жидкого (/7ж) и твердого (/’’тв)
растворов в зависимости от состава
при заданной температуре Т' (а);
для той же системы линии ликвидус
(7\), солидус (Тs) и зависимость
температура — состав для условия
7?тв=77ж (например, Т'^—Т' при
С=Съ) (б)
161
же состав, что и исходная жидкая фаза, например, при температуре
Т". Расчеты показали, что для многих металлических систем (на-
пример, Ag — Си) бездиффузионная кристаллизация может идти
при переохлаждениях около 100—ЗОО’°С *.
Перераспределение примесей и морфология поверхности раздела
кристалл — расплав. При кристаллизации сплавов, как правило,
примеси в кристалле распределяются неоднородно. Из-за особен-
Рис. 6.18. Части диаграмм равновесия для случаев, когда примесь
понижает (а) и повышает (б) температуру плавления сплава
костей атомного механизма роста, когда грань растет за счет
присоединения и зарастания отдельных ступеней, может проис-
ходить слоистое распределение примесей. Неоднородности обра-
зуются и из-за химической адсорбции молекул на поверхности за-
растающей грани. Еще большие неоднородности в масштабе всего
слитка возникают, если примеси имеют разную растворимость
в твердой фазе и в расплаве. Пусть в сплаве с концентрацией
примеси со при температуре начала кристаллизации Т' выпадает
фаза с концентрацией примеси ств (рис. 6.18). Разную раствори-
мость примеси в твердой и жидкой фазах выразим через коэффи-
циент распределения k^:
ко=Ст!в I бо- (6.62)
Рассмотрим подробнее случай &о<1. Из диаграммы равнове-
сия следует (см. рис. 6.18,а), что при этом примесь менее раство-
рима в твердой фазе, чем в расплаве. Поэтому в процессе кристал-
лизации она оттесняется от поверхности раздела в расплав. Если
скорость роста достаточно нелика и нет полного выравнивания
концентрации и температуры в жидкой фазе, примесь скапливается
* Аптекарь И. Л., Каменецкая Д. С. В кн.: Проблемы металловедения и
физики металлов. Вып. 8. М., «Металлургия», 1964, с. 204.
162
перед фронтом кристаллизации (см. рис. 6.19,6) и концентрация
примесей у границы повышается до сж0. Концентрационная неод-
нородность распространяется в расплаве на величину 3 от фронта
кристаллизации.
Обогащение жидкости примесью вызывает понижение темпе-
ратуры начала кристаллизации (что следует из диаграммы рав-
новесия на рис. 6.18ц). В области кон-
центрационной неоднородности (на рис.
6.19,6 от 0 до 6) температура ликвидуса
понижается от Т' до температур, обозна-
ченных сплошной линией (см. рис. 6.19,в).
По мере затвердевания общая концен-
трация сж в расплаве растет, и поэтому
концентрация ств в твердой фазе тоже
увеличивается, так как ств=$сж.
Для случая частичного перемеши-
вания коэффициент распределения k не
достигает равновесного значения k0
(полное перемешивание, см. рис. 6.19ц),
а определяется соотношением
k/k0=l/[k0—(\ —
—kQ) ехр{ (—'dc/D) (ротв/рж)}], (6.63)
где с — скорость роста; D — коэффици-
ент диффузии примеси в расплаве; ртв и
рж— плотности жидкой и твердой фаз
соответственно.
В результате оказывается, что обла-
сти слитка, где кристаллизация прошла
в первую очередь, будут обеднены при-
месями, а в участках, затвердевших позд-
нее, концентрация примесей возрастет.
Таким образом, изменяя условия кри-
сталлизации, можно контролировать рас-
пределение примесей по слитку, доби-
ваясь заданного распределения по кон-
центрациям или же очищая сплав от
примесей. Наиболее перспективна мето-
дика зонной плавки.
Форма поверхности раздела растущего кристалла и жидкой
фазы при кристаллизации сплавов в еще большей степени может
быть неустойчивой, чем при кристаллизации чистого вещества.
В случае кристаллизации чистого вещества форма поверхности
зависит от температурного поля, ,в случае кристаллизации сплава
добавляется фактор распределения концентрации. Только при до-
статочно большом (положительном) градиенте температуры перед
фронтом кристаллизации поверхность раздела сохраняется пло-
ской. С уменьшением градиента и соответствующим увеличением
и* 163
Рис. 6.19. Концентрация при-
меси ств в твердой и сж
в жидкой фазе; сж —в жид-
кой фазе перед поверх-
ностью раздела для полно-
го (а) и частичного (б) пе-
ремешивания; в — распреде-
ление температуры в окрест-
ности фронта затвердевания
(/ и температуры ликвиду-
са— II)
зоны концентрационного переохлаждения * возникает ячеистая,
а затем дендритная структура (рис. 6.20).
Дефекты роста. Структура слитка может быть слоистой или
ячеистой в связи с механизмом (роста кристаллов и морфологи-
ей поверхности раздела твердая фаза — расплав. Сегрегация при-
Тв. Ж ид к.
Рис. 6.20. Морфология поверхности раздела при
кристаллизации сплава в зависимости от градиен-
та температуры в жидкой фазе при наличии кон-
центрационного переохлаждения (/ — распределе-
ние температуры перед фронтом затвердевания;
II — температуры ликвидуса):
а — плоский фронт; б — ячейка; в — дендритные ячейки;
г — свободно растущие дендриты; д — образование кри-
сталлов перед фронтом затвердевания
I
Расстояние от k
Фронта затвердевания
месей способствует закреплению дефектов роста в объеме кристал-
лов. Если атомные размеры примеси и растворителя различны,
изменения концентрации примеси приводят к вариации периода
решетки и возникают напряжения, величина которых дается вы-
ражением
б—(&аДсЕ') /а, (6.64)
* Это явление было теоретически предсказано Г. П. Иванцовым («Докл. АН
СССР». 1951, № 81, с. 179).
164
где Аа— разность периода решетки а растворителя и растворен-
ного компонента (при изоморфных решетках); Ас — разница кон-
центрации в атомных долях по сечению области сегрегации. При
образовании ячеистой структуры дислокации располагаются по
периметру ячеек (рис. 6.21,а), плотность дислокаций 106—1010 см-2,
при периодическом росте слоев (рис. 6.21,6) плотность дислокаций
106— Ю7 см"2.
Образуются дислокации и из-за термических напряжений
в сплаве при наличии градиентов температуры в процессе роста
или в результате быстрого охлаждения.
Рис. 6.21. Схема распределения дислокаций в кристалле,
когда области сегрегации примеси образуют ячеистую
структуру при поперечном (а) и продольном (б) сече-
ниях ячеек
а
При резком охлаждении кристалла в нем сохраняется повы-
шенная концентрация вакансий (до 1017-^—102а см-3). Они имеют
тенденцию конденсироваться, создавая полости, которые затем мо-
гут превращаться в дислокационные петли. Из вакансий для отме-
ченной выше концентрации, по расчетам, может возникнуть
106—109 см-2 дислокаций.
Если дислокации заканчиваются на растущей поверхности кри-
сталла (см. рис. 6.16), то, как было показано, они сохраняются,
способствуя росту, и могут проходить через весь кристалл. Можно
свести к минимуму вероятность выхода дислокации на поверхность
раздела кристалл — расплав и таким образом уменьшить возмож-
ность распространения дислокаций, если выращивать кристалл
в определенном кристаллографическом направлении *.
Участки металлического кристаллита с ненарушенной кристалли-
ческой решеткой, отделенные от соседних, правильно построенных
участков малоугловыми границами, называются блоками мозаич-
ной структуры. Блоки мозаики бывают повернуты по отношению
друг к другу на малые углы — порядка нескольких минут. Разме-
ры блоков в литых металлах весьма различны: от нескольких до-
* См. также задачу 6.7.
165
лей до десятков м-икрон. Границы с углом разориентации порядка
1 град разделяют монокристалл на фрагменты (размер — от не-
скольких долей до нескольких миллиметров).
Мозаичная структура литых сплавов весьма устойчива. Она ча-
сто не устраняется при гомогенизирующем отжиге и ,в значитель-
ной степени сохраняется после обработки давлением.
Эвтектическая кристаллизация. Сплавы эвтектического состава
имеют техническое значение как материалы для отливок и при-
пои, так как они имеют выгодные технологические свойства (низ-
кую температуру плавления). Однако в настоящее время эти спла-
вы получили применение как ценный конструкционный материал—
естественный композиционный материал, в котором выгодное со-
четание механических свойств обеспечивается вязкой матрицей и
прочными включениями второй фазы в виде волокон пли пластин.
Наилучшие свойства обеспечивают выбором сплава, т. е. свойства-
ми фаз эвтектики, и выбором условий кристаллизации, приводя-
щих к выгодной микроструктуре.
Как и для выращивания монокристаллов, для приготовления
изделий из эвтектических сплавов применяют направленную кри->
сталлизацию.
Формирование микроструктуры при затвердевании идет по пу-
ти, обеспечивающем возможно более низкую поверхностную (меж-
фазовую) энергию, эта микроструктура может быть весьма ста-
бильной в процессе службы материала при повышенных темпе-
ратурах.
Механизм эвтектической кристаллизации.
Экспериментальному исследованию кристаллизации эвтектик и
анализу этого процесса посвящены классические работы акад.
А. А. Бочвара *. Были исследованы сплавы прозрачных органиче-
ских веществ — азобензола (С12Нк№) и пиперонала (СвНбОз). Эти
сплавы оказались наиболее подходящими для исследования по
многим причинам: удобная для работы область температур кри-
сталлизации (равновесные температуры плавления азобензола
68°С, пиперонала 37°С, эвтектики 26°С; кристаллы компонентов
резко отличаются по окраске: оранжево-красный азобензол и бес-
цветный пиперонал); линейные скорости кристаллизации фаз-ком-
понентов в эвтектической жидкости и чистого пиперонала порядка
1 мм/мин, что позволяет легко их измерять и делать микрофото-
графии на разных стадиях процесса при 10—50-кратном увеличе-
нии без применения киноаппаратуры; состав эвтектики: 26% азо-
бензола и 74% пиперонала.
Основные результаты наблюдений сводятся к следующему.
1. Скорость роста первичного кристалла каждого компонента
в сплавах разного состава уменьшается с увеличением содержа-
ния второго компонента (рис. 6.22). Состав жидкости влияет на
линейную скорость роста кристаллов азобензола, в чистом виде
* Бочвар А. А. Исследование механизма и кинетики кристаллизации спла-
вов эвтектического типа. М., ОНТИ, 1936.
166
растущего необычайно быстро, значительно сильнее, чем на мед*
ленный рост кристаллов пиперонала.
2. Даже в переохлажденной жидкости точно эвтектического со-
става самопроизвольно зарождаются и раздельно растут кристал-
лы каждой из фаз.
3. Слой жидкости, окружающей растущий кристалл, резко обед-
няется молекулами данного компонента. Об этом можно судить по
изменению окраски слоя: жидкость, окружающая оранжевый кри-
сталл азобензола, была заметно обесцвечена, вокруг же бесцвет-
ного кристалла пиперонала ок-
раска жидкости была особенно
интенсивна.
4. При сближении растущих
кристаллов скорость роста кри-
сталлов азобензола увеличивает-
ся. В момент соприкосновенияпер-
вичных кристаллов кристалл азо-
бензола выбрасывает иглы, ко-
торые быстро прорастают жид-
кость, окружающую кристалл пи-
перонала.
5. Собственно эвтектическая
кристаллизация начинается после
соприкосновения кристалликов
различных фаз. Эвтектическая
кристаллизация совершается с го-
раздо большей скоростью, чем
кристаллизация отдельных фаз в
Рис. 6.22. Зависимость линейной ско-
рости роста первичных кристаллов
азобензола и пиперонала от состава
расплава при температуре 17°С (пе-
реохлаждение 9°С):
1 — азобензол; 2 — пиперонал; 3 — скорость
роста эвтектической колонии а«обензол —
пиперонал
жидкости эвтектического состава.
6. На границе твердая эвтектика — жидкость происходит пре-
имущественное выделение одной из фаз, которое сопровождается
обогащением пограничного слоя жидкости веществом, составляю-
щим основу второй фазы. Вследствие этого одна из фаз ведет
кристаллизацию, создавая скелет эвтектического кристаллита, вто-
рая же фаза отлагается в междуосных ,пространствах этого ске-
лета.
Причина влияния состава жидкости на скорость роста кристал-
лов очевидна: это уменьшение количества молекул какого-либо
компонента, приходящих в соприкосновение с поверхностью расту-
щего кристалла этого компонента, из-за снижения содержания по-
следнего в расплаве и уменьшение количества молекул, присоеди-
няющихся к кристаллу за 1 с.
Обеднение слоя, окружающего растущий кристалл какого-либо
компонента молекулами этого компонента в ходе процесса, еще
более затрудняет рост кристалла в растворе. При кристаллизации
эвтектического образования картина существенно меняется: кри-
сталл компонента А в процессе роста очищает жидкость в слое,
прилегающем к растущему зерну, от молекул А и этим облегчает
рост кристалла В, последний же оказывает аналогичную помощь
167
кристаллу А. Такое диффузионное разделение жидкости на ком-
поненты приводит к тому, что при любых переохлаждениях ско-
рость роста эвтектики в жидкости эвтектического состава выше,
чем скорость роста первичных зерен обоих компонентов.
Как видно из рис. 6.22, скорость роста зерен эвтектики не за-
висит от состава жидкости; при этом эвтектика растет быстрее,
чем первичные кристаллы в жидкости не только эвтектического
состава, но и составов, несколько отличных от эвтектического,
причем интервал концентраций, выгодных для роста эвтектики,
возрастает с увеличением переохлаждения. А. А. Бочвар отмечал,
что кривые линейной скорости обоих компонентов пересекаются
вблизи от эвтектического состава, и предположил, что если бы
было возможно получить данные о кинетике кристаллизации при
бесконечно малых переохлаждениях, то оказалось бы, что скоро-
сти роста первичных кристаллов обоих компонентов и скорость
роста эвтектики в жидкости строго эвтектического состава совпа-
ли бы.
Сплавы составов, для которых скорость роста эвтектического
образования превышает скорость роста первичных кристаллов
обоих компонентов, должны иметь эвтектическую (или «квазиэв-
тектическую») структуру.
Азобензол часто образует ободки вокруг кристаллов пиперона-
ла, в то время как ободки пиперонала вокруг кристаллов азобен-
зола никогда не наблюдаются. Это объясняется большим влия-
нием состава жидкости на линейную скорость кристаллизации азо-
бензола. Поэтому кристалл азобензола в момент соприкосновения
с кристаллом пиперонала чрезвычайно быстро прорастает в обед-
ненный пипероналом слой жидкости вокруг кристалла последнего.
Тот факт, что в слое, окружающем кристалл одного' из .компонен-
тов, не зарождаются, несмотря на благоприятную концентрацию,
новые кристаллы второго компонента вплоть до момента соприкос-
новения двух независимо образовавшихся первичных кристаллов,
А. А. Бочвар объясняет влиянием нагрева жидкости в слое за счет
выделения скрытой теплоты кристаллизации. Как известно, при
малых переохлаждениях вероятность образования центров кри-
сталлизации очень мала даже в однокомпонентном расплаве. На
скорость роста кристаллов уменьшение переохлаждения влияет го-
раздо меньше — это и делает возможным образование ободков.
Такие явления наблюдали и в металлических эвтектических спла-
вах.
При рентгенографическом исследовании эвтектик* было вы-
яснено, что по крайней мере одна из фаз эвтектической колонии
является монокристальной. В эвтектиках Bi—Sn и Cd—Sn моно-
кристалл образует олово. В общем случае эвтектическую колонию
можно рассматривать как бикристальное образование.
При анализе различных эвтектических систем замечено, что
для ведущей фазы часто характерны более сложная кристалличе-
* Авакян С. В., Лашко Н. Ф. «Жури. физ. химии», 1949, № 6, с. 729.
168
ская «структура, большая доля ковалентных межатомных связей
Например, в системах твердый раствор — химическое соединение
(Ре—Fe3C, AI—CuAI2) ведущей фазой являются именно соедине-
ния; в сплавах Ре—С, А1—Si, Sn—Bi эвтектические колонии воз-
никают соответственно на кристаллах графита, кремния, висмута.
Более прямая характеристика ведущей фазы при эвтектической
кристаллизации может быть получена из рассмотрения изменений
поверхностной энергии при зарождении одной фазы на другой.
Такой анализ проведен при исследовании эвтектической кристал-
лизации в системе Ag—Си, компоненты которой являются химиче-
Рис. 6.23. Структура медленно охлажденных сплавов Ag—Си, содержащих
28,6% Си (а) и 34,8% Си (б), Х200
скими аналогами, фазы — твердые растворы соответственно на ос-
нове Ag и Си — имеют одну и ту же типичную для металлических
кристаллов г. ц. к. решетку. Ведущей при образовании эвтектики
оказалась Cu-фаза (рис. 6.23), поверхностная энергия меди на гра-
нице с расплавом больше, чем поверхностная энергия серебра на
границе с расплавом (17,7 и 12,6 Дж/м2 соответственно). Цоэто-
му более выгодным является образование кристалла серебра на
поверхности кристалла меди, чем наоборот* (см. рис. 6.7).
Количественные характеристики микрострук-
туры при эвтектической кристаллизации метал-
лических сплавов. Состав сплава и условия кристаллизации
должны обеспечивать одновременное выпадение обеих фаз при
плоском (или почти плоском) фронте кристаллизации («нормаль-
ная» эвтектика). При этом существенны как выбор самой системы
(состав эвтектики и объемное соотношение фаз), так и наличие
примесей или дополнительное легирование.
В условиях направленной кристаллизации получают правиль-
но ориентированные и регулярно расположенные пластины или
* Таран Ю. Н., Мирошниченко И. С., Галушко И. М. «Докл. АН СССР».
1970, т. 193, № 1, с. 143.
169
стержни, растущие из расплава перпендикулярно к фронту кри-
сталлизации. Приближенный анализ условий дифференциации ком-
понентов на фронте кристаллизации приводит к простой зависимо-
сти расстояния между частицами X от скорости движения фронта
кристаллизации с:
%^c=const. (6.65)
Эта зависимость хорошо видна на рис. 6.24.
Морфология эвтектической структуры зависит от объемной до-
ли каждой из фаз. Если поверхностная межфазовая энергия для
фаз эвтектики изотропна, то из условия минимума поверхностной
энергии при объемной доле второй фазы <0,28 должна быть вы-
Рис. 6.24. Зависимость межпластиночного расстояния
в эвтектическом сплаве Sn—Pb от скорости роста (по
данным разных авторов)
годной структура в виде округленных стержней, а при объемной
доле >0,28 — в виде пластин. Если анизотропия поверхностной
энергии создает одну плоскость габитуса с низкой поверхностной
энергией, то проявляется тенденция к образованию пластинчатой
структуры; если может быть несколько плоскостей сопряжения
фаз эвтектики с низкой поверхностной энергией, то возникают
стержни с огранкой.
Возможность образования когерентных межфазных границ за-
висит от небольших изменений межплоскостных расстояний в кри-
сталлах сопрягающихся фаз, которые могут происходить под дей-
ствием легирования. Эвтектика в сплаве 34 ат. % Сг + 66 ат. %
NiAl* состоит из двух изоморфных фаз—твердого раствора на
основе хрома (о. ц. к., а=2,88 А) и интерметаллида NiAl (тип
CsCl, п=2,89А), кристаллы этих фаз в эвтектике имеют совпа-
дающие ориентировки. При добавлении около 0,6—1 ат. % Мо пе-
риод решетки Cr-фазы становится почти равным периоду NiAl-
фазы при температуре плавления эвтектики (на рис. 6.25 приве-
дены значения периодов, измеренные при комнатной температуре).
При дальнейшем увеличении содержания Мо стержневая структу-
ра сменяется пластинчатой, по-видимому, этот переход связан
с нарушением когерентности на границах раздела; одновременно
* Cline Н. Е., Walter J. L. «Metallurg. Trans.», 1970, v. 1, p. 2907.
170
2,91
2,90
2,89
2,94
2,95
292
а, А
2,97
2,95
2,95
Рис. 6.25. Зависимость периода ре-
шеток твердого раствора на осно-
ве хрома (/) и NiAl-фазы (2)
от содержания молибдена в эвтек-
тическом сплаве 34 ат. % Сг+Мо
и 66 ат. % NiAl
0 1 2 3 4 5 6 7 8 9 10
Содержание Мо, ат. ° о
изменяются кристаллографические характеристики структуры:
для стержневой структуры ориентация стержней и направ-
ление роста [100]niaiII [100]сг-фаза; для _ пластинчатой струк-
туры: плоскости габитуса (112)niaiII (112)сг-фаза,’ направления
роста [11 1]niaiII [111] Сг+фаза-
Переход от стержневого роста
к пластинчатому может происхо-
дить также при изменении скорости
роста. Указанное выше критическое
значение объемных долей фаз эв-
тектики для смены морфологии
роста выведено в предположении
одинакового расстояния между ча-
стицами— стержнями или пласти-
нами. Если межпластиночное рас-
стояние принять в 1,5 раза боль-
шим, чем расстояние между стерж-
нями, то критическое значение объ-
емной доли должно составлять 0,10,
а не 0,28. С увеличением скорости
роста при кристаллизации эвтекти-
ки Ni—W уменьшение расстояния
между стержнями происходит быст-
рее, чем уменьшение расстояния
между пластинами (в области ско-
ростей роста 10~4 см/с существуют
как стержни, так и пластины фазы,
поэтому при больших скоростях ро-
ста более устойчивой оказывается
пластинчатая структура*).
Изменения скорости роста могут
изменения структуры, если имеются примеси или легирующие до-
бавки (рис. 6.26). Чтобы .предотвратить ячеистый (или дендрит-
ный) рост, когда поверхность раздела перестает быть плоской,
сплавы должны быть достаточно чистыми. При некотором крити-
ческом значении G/c (G— градиент температуры в расплаве пе-
ред фронтом кристаллизации) могут возникать ячейки с веерооб-
t разной структурой из-за концентрационного переохлаждения.
Затвердевание металлических расплавов при очень быстром ох-
лаждении («закалка из жидкого состояния»). Известно несколько
способов достижения сверхвысоких скоростей охлаждения при за-
твердевании металлических расплавов. В основе этих способов
лежит общий принцип: небольшое количество жидкого металла
вводится в соприкосновение с хорошо отводящей тепло средой. Это
осуществляется распылением жидкого металла в потоке газа, «вы-
стреливанием» капли металла на массивную медную подложку,
захлопыванием капли между пластинами, при падении капли на
вызывать особенно сильные
* Kurz W., Lux В. «Metallurg. Trans.», 1971, v. 2, p. 329.
171
Содержание Мо,ат.7о
Рис. 6.26. Влияние добавок Мо и
скорости роста при направленной
кристаллизации эвтектики в спла-
ве Сг—А1:
/ — стержневая структура; 2 — пластин-
чатая структура; 3 — ячеистая струк-
тура; 4 — дендритная структура
, град/с
Рис. 6.27. Зависимость переохлажде-
ния А1 от скорости охлаждения при
различных перегревах расплава
поверхность вращающегося диска
или цилиндра, при затвердевании
тонкой струи металла между вра-
щающимися прокатными валками
и пр. Скорости охлаждения распла-
ва достигают 105—108 и даже
1010 град/с.
Уже при скорости охлаждения
около 105—106 град/с затвердевание
технических металлов и сплавов
происходит при таких переохлажде-
ниях, которые в случае обычных
условий охлаждения достигаются
лишь после тщательной очистки
расплавов, высокого перегрева или
при охлаждении очень мелких ка-
пель (ср. данные, приведенные на
рис. 6.27, с данными табл. 6.5). За-
твердевание расплавов при таких
(и еще больших) переохлаждениях
может привести к кристаллизации
пересыщенных твердых растворов
вплоть до образования твердого
раствора, состав которого совпадает с составом исходной жидко-
сти, к образованию метастабильных промежуточных фаз и аморф-
ного состояния.
Образование однофазного состояния пересыщенного твердого
раствора можно рассматривать
как результат бездиффузионной
кристаллизации в соответствии со
схемой, приведенной на рис. 6.17.
Это подтверждается следующи-
ми наблюдениями. Образование
сильно пересыщенных твердых
растворов с увеличением скорости
охлаждения происходит не по-
степенным повышением пересы-
щения и устранением признаков
перераспределения концентрации
(в сплаве заданного состава), а
внезапно после достижения не-
которой критической скорости ох-
лаждения. Отмечалась также
исключительная четкость рентге-
новских линий, свидетельствую-
щая о высокой степени однородности состава твердого раствора *.
Однако возможность достижения topi или иной растворимости, по-
Мирошниченко И. С. Рост кристаллов, т. XI. Ереван, 1975, с. 337—348.
172
Таблица 6.8
Метастабильные состояния при закалке из жидкого состояния
Расширение области некоторых твердых растворов
Система Растворимость, ат. % Система Растворимость, ат. %
равновесная достигнутая равновесная достигнутая
Ag—Си 8,8 Полная раство- Си—Fe 4,5 20,0
римость Fe—Си 7,2 15,0
А1—Си 2,5 18,0 Fe—Ti 9,8 16,0
Al—N1 0,023 7,7 Ni—С 2,6 8,2
Al—Si 1,59 11,0 Ni—W 16,0 32,0
12,0 25,0 Pb-Sb 5,9 17,0
Си—Со 5,5 15,5
Аморфные твердые фазы
Система Состав или интервал состава, ат. % второго компонента Система Состав или интервал состава, ат. % второго компонента
Au—Si 15—40 Pd—Si 15—23
Au—Sn 29—31 Pt—Ge 17—30
Cu—Ti 30—35 Pt-Sb 33—37
Fe-P-C 13 Р; 7 С Те—Ag 33—40
Nb—Ni 33—78 Zr—Co 28
Pb—Au 25 Zr—Cu 40—75
Fe—Pd—P 10,5 Pd; 20 Р Zr—Ni 20—40
Zr—Pd 20—35
видимому, зависит от некоторых факторов, определяющих макси-
мальную растворимость в кристаллическом состоянии. Экспери-
ментальные данные (табл. 6.8) показывают, что хотя равновесная
предельная растворимость германия и меди в серебре примерно
одинаковая (около 9 ат. %), предельная концентрация твердого
раствора германия в серебре оказалась равной 13%, тогда как
в системе Ag—Си при закалке из расплава достигается неограни-
ченная растворимость при менее благоприятном объемном факто-
ре, чем для Ag—Ge. Определяющее значение здесь имеет ограни-
чение растворимости по правилу Юм-Розери [см. с. 47—48].
В некоторых исследованных системах расширение растворимости
ограничивалось составом эвтектики, что иногда объясняют необ-
ходимостью более резкого охлаждения при таком составе. Важно
отметить, что именно для сплавов эвтектического состава легче
всего (т. е. при меньших скоростях охлаждения) достигается
аморфное состояние (рис. 6.28).
Если кристаллизация идет с образованием новых промежуточ-
ных фаз (металлических соединений), то можно найти аналоги
этих фаз в других подобных системах или эти же фазы в той же
системе при других особых условиях кристаллизации: при конден-
сации из пара, воздействии высокого давления и пр. Например,
173
в ^системе Au—Ge при обычных условиях кристаллизации не обра-
зуются какие-либо промежуточные фазы, но после закалки из рас-
плава наблюдались фазы типа £- и у-латуни, образования которых
вообще можно было ожидать, исходя из положения этих элементов
в таблице Д. И. Менделеева. Закалка расплава Fe—С—Si (чугун)
приводит к образованию фазы с гекс. п. у. структурой (8-фаза) и
с ромбической структурой (Х-фаза) *, близких к 8-модификации
железа, обнаруживаемой при высоких давлениях. Однако- отмеча-
лось также появление совершенно новых структур с особенно боль-
шими ячейками (например, длиннопериодная 24-слойная плотно-
упакованная структура в системе Au—Ge) и фаз с примитивной
Рис. 6.28. Диаграммы фазовых состояний Au—Ge:
а — диаграмма равновесия; б — фазовые состояния в зависимости от степени пере-
охлаждения при затвердевании расплава ДТ (или скорости охлаждения)
кубической ячейкой (например, в системах Au—Sb и Au—Те).
Иногда связывают появление новых метастабильных состояний при
сверхбыстром охлаждении расплавов с фиксированием высокотем-
пературных состояний, придавая определяющее значение тем ко-
ординациям (или группировкам) атомов, которые могут быть в ис-
ходной жидкости. В подтверждение этой точки зрения указывают
на изменения структуры твердой фазы при изменениях степени
перегрева расплава, наблюдавшихся в некоторых системах (на-
пример, в Au—Ge).
Однако следует иметь в виду, что перегрев расплава может
приводить к увеличению переохлаждения. Возможно представле-
ние о новых фазах как о фазах, связанных условиями фазового
равновесия (метастабильного) с жидкой фазой при тех переохлаж-
дениях, которые достигаются сверхбыстрым охлаждением. Среди
* Еднерал Н. В., Лякишев В. А., Ревякин А. В., Скаков Ю. А. В сб. Струк-
турный механизм фазовых превращений сплавов. М., «Наука», 1976, с. 183—186.
174
этих фаз могут быть и так называемые аморфные фазы (или «ме-
таллические стекла»). Если условием возникновения мета-стабиль-
ных кристаллических фаз (пересыщенных твердых растворов, но-
вых промежуточных фаз) является невозможность диффузионных
процессов, то дополнительным кинетическим условием возникно-
вения аморфных фаз должно быть уменьшение до нуля скорости
образования центров кристаллизации (см. рис. 6.14).
Аморфное состояние легче всего получается при затвердевании
S, Se, С, т. е. элементов, для которых в конденсированных состоя-
ниях характерна ковалентная химическая связь. Среди металли-
ческих элементов аморфное состояние при закалке расплавов из-
вестно для никеля *. Более общим является получение аморфного
состояния в сплавах на основе переходных металлов и металлов
группы Си, содержащих более электроположительные элементы
и металлоиды, такие, как В, Те, С, Si, Ge, Sn, Р. Можно предпо-
Рис. 6.29. Функции радиального распределения g (г)
для аморфного железа, полученного конденсацией или
«закалкой из паровой фазы» (/), и для расплавлен-
ного железа при температуре 1560°С (2)
лагать и в этих системах наличие значительных ковалентных свя-
зей. Аморфные состояния получали закалкой из расплавов в таких
системах, как Mg—Cu, Gd—Со, La—Au, Ag—Си, Ag—Мп, А1—Си.
Многие из систем отличаются низкой- температурой эвтектики.
В отношении обычных (например, силикатных) стекол распро-
странено мнение, что они имеют «замороженную» структуру жид-
ких расплавов, неустойчивы по отношению к конфигурационным
изменениям, что неизбежно должно вызывать некоторую неопре-
деленность свойств; от жидкости это состояние отличается резко
увеличенным временем релаксации. Аморфные металлы в самом
деле дают картины рассеяния рентгеновских лучей, близкие к кар-
* Аморфное состояние чистых металлов особенно часто наблюдали при «за-
калке из паровой фазы», т. е. при конденсации на охлажденные и высокотепло-
проводные подложки.
175
Рис. 6.30. Конфигурации атомов
в плотных упаковках по Берналу:
а — тетраэдр; б — октаэдр; в — тригональ-
ная призма с тремя полуоктаэдрами; г —
архимедова антипризма с двумя полуокта-
эдрами; д — тетрагондодекаэдр
тинам для металлических расплавов (см. с. 130). Однако для неко-
торых аморфных металлических систем отмечались большая интен-
сивность и острота главного (первого) максимума и усложнение
второго диффузионного максимума. Это можно связать с более
высокой степенью ближнего порядка в твердом аморфном состоя-
нии, чем в расплавах. Более сложный вид может иметь функция
радиального распределения для аморфного' состояния (рис. 6.29).
Представление о ближнем порядке не является общепринятым,
некоторые авторы рассматривают «рентгеновски аморфные» со-
стояния (т. е. не дающие четких дифракционных максимумов) как
мелкокристаллические с размером частиц менее 50—20 А, иногда
находят и истинно аморфные, и
мелкокристаллические структуры
в закаленных сплавах разного
состава одной и той же системы.
Однако имеются некоторые осно-
вания выделять аморфное состоя-
ние металлов как метастабильное
твердое состояние, которое отли-
чается от жидкого состояния не
только по кинетическим свой-
ствам, но и по степени и харак-
теру порядка в расположении
атомов. Координационное число,
определяемое по площади перво-
го пика функции распределения
радиальной плотности (см. рис.
6.29), во всех изученных твер-
дых аморфных металлических
системах оказалось более высо-
ким, чем для жидких металлов,
и близким к 12±1.
Атомные структуры могут быть
слишком различны и то же вре-
мя достаточно неопределенны,
чтобы их описывать, пользуясь
только моделью ближнего поряд-
ка (как и металлических распла-
вов) или микрокристаллической
моделью (г. ц. к., о. ц. к. и т. д.).
Наиболее подходящей оказалась модель плотной неупорядоченной
упаковки жестких сфер, предложенная ранее Берналом * для опи-
сания некоторых одноатомных жидкостей. Сочетания приведенных
на рис. 6.30 типов атомных конфигураций могут быть самыми раз-
ными. При сочетании всех типов в пропорции 86,1; 5, 9; 3,8; 0,5 и
3,7% достигается коэффициент заполнения объема р=0,64 (как
известно из кристаллографии, для плотных упаковок в кристаллах
* Bernal J. D. Proc. Roy., Soc., 1964, A280, p. 299.
176
•с г. ц. к. и гекс. л. у. решетками г]—0,74, а в о. ц. к. кристаллах
г]=0,68). Можно предположить, что устойчивость плотных упако-
вок по Берналу в сплавах металл — металлоид повышается, если
металлоидные атомы ионизируются и занимают определенную
часть пор.
Изменения свойств при переходе жидкость — аморфное состоя-
ние подробно изучали для неметаллических систем, где это пре-
вращение наблюдали в удобной для
вания области температур и при
умеренных скоростях охлаждения.
Как видно из рис. 6.31, переход
жидкость — аморфное состояние но-
сит характер фазового перехода
второго рода (отсутствие скачка
в изменении объема при резком из-
менении температурного коэффици-
ента). Для металлов изменение
удельного объема в точке превра-
щения расплав — кристалл для ком-
пактных решеток достигает 6%.
Различия удельных объемов в
аморфном и кристаллическом со-
стояниях металлических систем при
нормальных условиях (после охла-
ждения) составляют всего 1—2%.
Металлические сплавы в аморф-
ном состоянии имеют высокую проч-
ность (например, для сплава на
основе железа получены предел
прочности при растяжении более
300 кг/мм2 и удлинение 1—2%) и
большую коррозионную стойкость.
Замечательное сочетание высокой
экспериментального исследо-
Рис. 6.31. Схема изменений объ-
ема жидкой фазы (/), кристалла
(2) и стеклообразной фазы (5)
в зависимости от температуры и
при фазовых переходах жид-
кость—кристалл (Ткр). переох-
лажденная жидкость — стеклооб-
разное состояние (ТСт)
прочности и магнитной проницаемости получено в сплавах на
основе Fe и Ni. В некоторых случаях уже удается получить аморф-
ные состояния при толщине ленты 0,1 — 1 мм. Во многих сплавах
аморфное состояние сохраняется при нагреве до 300—400°С и
выше.
Задачи к гл. 6
Задача 6.1. Рассчитать критическую величину кубического зародыша, кри-
сталлизующегося из расплава железа, при переохлаждениях на 10 и Ю0°С, если
скрытая теплота плавления Q=15-1010 эрг «см-3, а поверхностная энергия а=
==204 эрг-см-2.
Задача 6.2. Как изменится энергия в процессе сфероидизации металлическо-
го стержня длиной 10 см и диаметром 0,02 мм, если удельная поверхностная
энергия равна 300 эрг-см-2.
Задача 6.3. Рассчитать вероятности образования равновесных зародышей
Жидкой фазы из водяного пара при разных степенях его пересыщения и 0°С,
принимая постоянную С—1, для воды удельная поверхностная энергия равна
72 эрг-см-2, Л4=Г8, р = 1 г-см-3.
12—523 177
Задача 6.4. Определить предельную деформацию зародыша, когда еще воз-
можна ориентированная кристаллизация для NaCl при кристаллизации на гале-
ните: Сц=4,84-1011 дин/см-2, с12=1,37*1011 дин-см-2, а=150 эрг-см~2, а=5,6 А.
Задача 6.5. Предсказать, какие грани наиболее вероятны у алмазных кри-
сталлов.
Задача 6.6. Через сколько времени закристаллизуется 30% жидкого метал-
ла, если при данном переохлаждении линейная скорость роста кристаллизации
равна 0,5 мм-с-1, а скорость зародышеобразования равна 103 с-1.
Задача 6.7. Показать, что в кристаллах с г. ц. к. решеткой дислокации
в плоскости скольжения {111} в процессе кристаллизации «зарастают» при на-
правлении роста, например, < 100> или могут «прорастать» во вновь кристал-
лизующиеся области для направления роста, например, <110>.
ГЛАВА 7
ОБЩИЕ ЗАКОНОМЕРНОСТИ ФАЗОВЫХ ПРЕВРАЩЕНИЙ
В ТВЕРДОМ СОСТОЯНИИ,
КЛАССИФИКАЦИЯ ПРЕВРАЩЕНИЙ
Фазовые превращения, в частности превращения атомно-кри-
сталлической структуры, происходят при изменении температуры,
давления, а также при изменении вида, числа или соотношения
компонентов системы, т. е. химического состава сплава. Все эти
факторы используются в современной технологии для получения
металлических материалов с заданными свойствами. Наибольшее
значение имеют фазовые превращения, обусловленные изменением
температуры: температурный полиморфизм, изменение раствори-
мости, упорядочение в твердых растворах. При этом уделяют осо-
бое внимание превращениям, которые происходят при непрерыв-
ном или ступенчатом охлаждении. Фазовое превращение доходит
до конца или прерывается на некоторой промежуточной стадии,
обеспечивающей оптимальные свойства материала. Эти свойства
определяются не только фазовым составом и свойствами фаз, но
и микроструктурой сплава, т. е. дисперсностью и распределением
фаз и нарушениями атомно-кристаллической структуры основы
сплава или субструктурой зерен. Значение некоторых операций
термической обработки состоит именно в том, чтобы сформировать
подходящую субструктуру.
Как правило, температура заключительной операции термиче-
ской обработки выше температуры службы материала, поэтому
материал длительное время (иногда неограниченное) сохраняет
свою структуру и, следовательно, приданные ему свойства.
Проблема изучения фазовых превращений усложняется тем,
что механизм и кинетика фазового превращения одного и того же
типа (полиморфное превращение, упорядочение твердого раствора
и т. д.) могут быть существенно' различными. Тем не менее су-
ществуют общие для фазовых превращений в твердом состоянии
закономерности, а также системы признаков, позволяющих разли-
чать механизмы превращений (по морфологии конечной структу-
ры, временной и температурной зависимостям хода превращения).
Возвращаясь к анализу фазовых превращений в конкретных спла-
вах, приходится снова рассматривать комбинации разных меха-
низмов превращений, поскольку они могут действовать последо-
вательно (сменять друг друга) или одновременно в структурно
различающихся частях материала (у поверхности изделия и в объе-
ме или у границ зерен и внутри зерен). Таким образом, в рас-
12* 179
смотрении фазовых превращений должно быть два взаимосвязан-
ных и одинаково важных аспекта: выделение и исследование от-
дельных типов (или механизмов) фазовых превращений и
исследование фазовых превращений во всей их сложности в кон-
кретных сплавах.
Теория фазовых превращений в твердом состоянии использует
закономерности, выявленные и хорошо изученные при исследова-
нии процессов конденсации и кристаллизации из расплавов или
жидких растворов. Кинетика многих превращений в твердом со-
стоянии описывается, так же как и кинетика процесса кристалли-
зации, известным уравнением Колмогорова; во многих случаях
сохраняет силу представление о параметрах превращения — скоро-
сти образования центров и линейной скорости роста; одинаково
обосновывается положение о необходимости переохлаждения (или
пересыщения). Важное место занимает представление о «критиче-
ском» зародыше; в твердом состоянии, как и в жидком, образо-
вание новой фазы может начинаться от готовых центров и т. д.
Вместе с тем необходимо учесть, что изменение агрегатного со-
стояния (конденсация из пара, кристаллизация из расплава или
раствора) всегда представляет собой фазовый переход первого
рода, однако среди изучаемых превращений в твердом состоянии
имеются и превращения второго рода, для которых характерна
особая кинетика.
Отметим еще два специфических фактора превращений в кри-
сталлической среде.
1. Фактор упругой энергии. Изменение свободной энер-
гии ДФ при образовании новой фазы в кристаллической среде кро-
ме изменения объемной химической свободной энергии (F2—F\)v
и энергии межфазовых границ (2аг5г-(^)) должно обязательно
включать энергию поля упругих напряжений (dE7Trp/dv)v:
дЕ
дФ==^2_Р1)о + 2яА(0 + —^v. (7.1)
Фактор упругой энергии вместе с граничной межфазовой энергией
определяет морфологию, ориентацию и взаимное расположение
частиц новой фазы в ходе их образования и роста.
2. Фактор ограниченной диффузионной подвиж-
ности атомов в кристалле и возможность коопера-
тивного движения совокупности атомов. Ограничен-
ная подвижность атомов позволяет получать мета-стабильные или
лабильные (абсолютно неустойчивые) состояния с помощью обыч-
ных для современной технологии средств (имеются в виду пределы
изменения внешних параметров равновесия — температуры и дав-
ления, градиенты и скорости их изменения). Резкое изменение дав-
ления в широком диапазоне (от нескольких десятков или сот ки-
лобар до атмосферного) при низкой температуре или большая
скорость охлаждения в условиях ограниченной подвижности ато-
мов позволяет достигать очень большого* переохлаждения (или
180
пересыщения), что часто совершенно изменяет ход фазового пре-
вращения.
Кооперативный характер движения атомов в отсутствие (или
при слабом развитии) релаксационных процессов при низкой тем-
пературе дает особый механизм превращений кристаллической
структуры — мартенситный. Кинетика мартенситных превращений
?ак сильно отличается от кинетики других фазовых превращений,
что до сих пор обсуждается вопрос о применимости к ним обыч-
ных представлений об образовании и росте зародышей.
§ 7.1. ОБЩАЯ КЛАССИФИКАЦИЯ ФАЗОВЫХ ПРЕВРАЩЕНИЙ
И ПРОБЛЕМА ЗАРОЖДЕНИЯ НОВОЙ ФАЗЫ
В основу классификации фазовых превращений можно поло-
жить сравнение атомно-кристаллической структуры и химического
состава фаз в исходном состоянии системы и фаз — продуктов
превращения. В этом отношении превращение состоит в образова-
нии новой фазы (или нескольких новых фаз), отличающейся:
— кристаллической структурой, т. е. координацией атомов в ре-
шетке (аллотропические превращения в металлах и сплавах, упо-
рядочение) ;
— химическим составом при сохранении координации атомов
в решетке (расслоение твердого раствора);
— структурой и составом (аллотропическое превращение
в твердых растворах, выделение избыточных фаз из твердых рас-
творов, эвтектоидный распад).
Эта классификация не включает превращений, связанных толь-
ко с электронной структурой (многие случаи магнитных переходов,
переход в сверхпроводящее состояние).
Наиболее общее представление о конденсированной системе да-
ется с помощью элементов вращательной и трансляционной сим-
метрии в расположении и свойствах частиц, т. е. определением
эквивалентности некоторых направлений и точек пространства в си-
стеме. С этой точки зрения фазовый переход состоит в изменении
симметрии. Например, при затвердевании расплава полная транс-
ляционная симметрия однородной жидкости переходит в более
низкую симметрию кристаллической решетки. Понижение симме-
трии системы происходит при упорядочении и распаде твердых
растворов, при переходе вещества из парамагнитного состояния
в ферромагнитное (или антиферромагнитное) и т. д. В связи
с этим наиболее общая классификация фазовых переходов может
быть дана по группам симметрии *.
В зависимости состояния термодинамической системы от внеш-
них параметров равновесия особенно интересны точки, в которых
происходит качественное изменение — скачкообразное изменение
тех или иных свойств, что и означает фазовый переход. Если иметь
в виду требование термодинамической устойчивости, то бесконеч-
Браут Р. Фазовые переходы. Пер. с англ. М., «Мир», 1967, с. 286.
181
но близко к точке перехода изменение термодинамического по-
тенциала системы должно быть бесконечно малым. Это возможно
в двух случаях: при появлении бесконечно малого количества но-
вой фазы с определенным конечным отличием ее свойств от
свойств исходной фазы или при бесконечно малом изменении ка-
кого-либо свойства во всем объеме. *
Возникновение областей новой фазы с существенно иными
свойствами должно приводить к появлению поверхностной энер-
гии. Поэтому слишком малые об-
ласти новой фазы невыгодны и фа-
зовый переход может задерживать-
ся, следовательно, каждая из фаз
может существовать (как метаста-
бильная или стабильная) по обе
стороны от точки перехода. Сама
точка перехода при этом представ-
ляет собой пересечение линий за-
висимости термодинамического по-
тенциала двух фазовых состояний,
при этом никаких особенностей
в зависимости потенциала каждой
400 600 800 Т. К
г0
Рис. 7.1. Зависимость Гиббсовой
свободной энергии (AG—AH—
—TAS) от температуры для аусте-
нита (у) и мартенсита (а')
в сплаве железа с 1 мае. % С.
Мн — температура начала образо-
вания мартенсита (Б. Могутнов,
И. Томилин, Л. Шварцман)
из фаз (например, температурной
зависимости) при переходе через
критическую точку нет (рис. 7.1).
Такой переход называется фазо-
вым переходом первого рода. Этот
переход сопровождается скачкооб-
разным изменением свойств, кото-
рые определяются как первые про-
изводные от термодинамического
потенциала (например, объем).
Если фазовый переход состоит в постепенном изменении внутрен-
них параметров (например, распределение концентраций, смещение
атомов или другие характеристики атомной структуры), так что
в точке перехода различие параметров обеих фаз есть бесконеч-
но малая величина, то переход относится к фазовым переходам
второго рода. При этом скачок испытывают свойства, которые
представляют собой вторые производные термодинамического по-
тенциала (например, температурный коэффициент расширения)*.
При фазовом переходе второго рода принципиально невозможны
метастабильные состояния. Например, нельзя переохладить высо-
котемпературную фазу как метастабильную **. Энергетически вы-
годным является переход системы из одного структурного состоя-
ния в другое по всему объему, при этом каждая промежуточная
структура отвечает меньшей свободной энергии.
* Ср. рис. 3.8,а, б.
* * Это не означает, что нельзя практически зафиксировать высокотемпера-
турное состояние из-за действия кинетических факторов (ограниченная скорость
диффузии и т. д.). Однако переохлажденное состояние будет лабильным. •
182
На рис. 7.2,а представлена зависимость свободной энергии О
системы в функции некоторого термодинамического внутреннего
параметра при фиксированных температурах. Таким параметром
может быть, в частности, степень порядка для процесса упорядо-
чения, развивающегося по типу перехода второго рода.
Феноменологическая теория фазовых переходов второго рода
л. Д. Ландау и Е. М. Лифшица применима к таким превращени-
ям, в которых пространственная группа симметрии одной фазы
является подгруппой симметрии другой фазы. Теория предсказала
возможность протекания процессов упорядочения в некоторых
Рис. 7.2. Зависимости Гиббсовой свободной энергии G от некоторого тер-
модинамического параметра для гипотетической системы, в которой имеет-
ся фазовый переход второго рода (а) и для гипотетической системы,
в которой имеется фазовый переход первого рода (б)
сплавах по типу фазового перехода второго рода (см. гл. 3).
Для кристаллизации из расплава и для фазовых превращений
первого рода в твердом состоянии высокотемпературная фаза су-
ществует при температуре ниже температуры равновесия фаз, по-
скольку для образования центра новой фазы необходимо переох-
лаждение.
Необходимость переохлаждения (пересыщения и т. д.) для об-
разования новой фазы обусловлена не только затратой энергии на
-образование поверхности раздела новой и старой фаз и на преодо-
ление упругого сопротивления среды. Более детальный анализ
фазового перехода первого рода показывает, что при определен-
ных условиях прохождение системы через промежуточные состоя-
ния на пути к равновесному (например, непрерывное изменение
степени дифференциации компонентов или гомогенный сдвиг при
деформации решетки) может приводить к повышению свободной
183
энергии. Преодоление такого энергетического барьера можно пред-
ставить через флуктуационное образование критических зароды-
шей. На рис. 7.2,6 представлена зависимость свободной энергии
системы в функции от некоторого термодинамического параметра
для превращения .первого рода. За начало отсчета принят уровень
свободной энергии исходной фазы р. Схема, приведенная на
рис. 7.2,6, подходит, например, для термодинамического анализа
процесса распада твердого раствора, тогда по оси ординат следует
отсчитывать изменение средней свободной энергии системы, а по
оси абсцисс — состав раствора. В области стабильного существова-
ния (например, при температурах 7\ и Т2 для высокотемператур-
ной p-фазы на рис. 7.2,6) фаза устойчива по отношению к малым
и большим флуктуациям параметра структуры или состава (рас-
пределения концентраций, смещений атомов и пр.). В точке фазо-
вого' перехода (температура То) равновесие фаз означает равенст-
во свободных энергий (или термодинамических потенциалов) фаз
при определенной (конечной) разнице параметра структуры (а/
и х"). Ниже температуры Т3_TQ исходная фаза неустойчива по
отношению к большим изменениям параметра х. Переохлаждение
приводит к понижению энергетического барьера на пути тех изме-
нений в атомной структуре, которые переводят фазу р в фазу а.
Вплоть до переохлаждений, соответствующих температуре Г>Т5,
система остается устойчивой по отношению к малым флуктуациям
параметра х. При Т-.кр=Т5 происходит потеря устойчивости исход-
ной системы по отношению к бесконечно малым изменениям па-
раметра х. Температура 7]кр называется температурой абсолютной
потери устойчивости исходной фазы. Область метастабильных со-
стояний переохлажденных или перегретых фаз при фазовом пере-
ходе первого рода ограничена температурами абсолютной потери
устойчивости соответственно при прямом и обратном переходах.
В случае фазовых переходов второго рода температура абсолют-
ной потери устойчивости совпадает с температурой фазового пе-
рехода.
В области температур Tq>T>T^ постепенное изменение па-
раметра х должно приводить к возрастанию энергии системы, по-
этому фазовый переход осуществляется не сразу по всему объему
материала, а флуктуационным образованием малых участков за-
родышей новой фазы и их ростом (например, при распаде пересы-
щенного раствора возникают флуктуации состава). Однако при
достаточно большом переохлаждении (или пересыщении) возмо-
жен так называемый безбарьерный, беззародышевый или гомоген-
ный переход: при свободная энергия всего ряда промежу-
точных состояний меньше свободной энергии исходной фазы. Имен-
но так идет процесс дифференциации компонентов раствора при
спинодальном распаде. Для превращения, которое состоит в пере-
стройке кристаллической решетки, на схеме (см. рис. 7.2,6) вдоль
оси абсцисс откладывают параметр, характеризующий промежу-
точные структуры (сдвиг атомов или промежуточные координации
атомов). Не исключено, что температура начала мартенситной ре-
184
акции при аллотропических превращениях связана с температурой
абсолютной потери устойчивости старой решетки (точка AfH на
рис. 7.1). Если спинодальный распад при расслоении твердых
растворов теоретически и экспериментально хорошо изучен (под-
робно это будет рассмотрено' в гл. 9), то применение понятия аб-
солютной неустойчивости к мартенситным превращениям пока
является только гипотезой.
В наших представлениях о фазовых превращениях образование зародышей
новой фазы является до сих пор наименее ясным. Экспериментальные исследо-
вания затруднены из-за высокой дисперсности тех частиц, которые должны рас-
сматриваться как «критические зародыши», или из-за большой скорости их роста
в первые моменты после образования. Теоретическое решение вопроса затруд-
няется тем, что при очень малом размере частиц новой фазы, по-видимому, нель-
зя пользоваться обычным понятием поверхностной (или граничной) энергии и
во всяком случае нельзя использовать количественные оценки граничной энергии,
полученные в измерениях макроскопических объектов. Вероятно, нельзя полно-
стью относить к зародышам — дисперсным новообразованиям — те представле-
ния об атомной структуре, которые получаем из исследований равновесной но-
вой фазы.
Как и в анализе процесса кристаллизации из расплава, при
•превращениях в твердом состоянии различают два типа зародыше-
образования: гомогенное и гетерогенное зарождения. Под гомо-
генным зарождением понимают образование зародышей в резуль-
тате флуктуаций— случайных и локальных (местных) отклонений
концентрации избыточного компонента или концентрации точеч-
ных дефектов (вакансий, межузельных атомов), образование ком-
плексов из этих дефектов или малых группировок атомов в коор-
динациях, совпадающих с координацией атомов в решетке ста-
бильной фазы. Под гетерогенным зарождением понимают
преимущественное образование зародышей в местах с дефектной
структурой, которые из-за тех или иных особенностей новой фазы
понижают работу образования зародыша и потому могут рассма-
триваться как потенциальные места зарождения: поверхность кри-
сталла, границы зерен, некогерентные границы двойников, границы
антифазных доменов, когерентные границы двойников, дефекты
-упаковки и дислокации.
Группировки атомов, соответствующие структуре новой фазы,
или некоторые промежуточные координации могут, очевидно, воз-
никать в результате флуктуаций в тепловом движении атомов, но
могут и присутствовать в исходном материале в виде тех или иных
дефектов кристаллической решетки. Примером таких дефектов
являются расщепленные дислокации в г. ц. к. решетке, представ-
ляющие собой прослойку гекс. п. у. структуры (и наоборот). По-
добные дефекты вследствие различий в мощности или разного
приближения к равновесной структуре могут давать спектр темпе-
ратур начала аллотропического превращения — температур, ниже
которых они оказываются способными к росту. С понижением тем-
пературы происходит как бы «спуск курка», вступают в действие
все новые и новые центры роста новой фазы. Такой механизм был
предложен для объяснения мартенситных превращений, которые
идут при очень низких температурах, когда трудно ожидать появ-
185
ления зародышей в результате флуктуации. Гипотеза «спускового»
механизма перестройки решетки близка к представлению о посте-
пенной потере устойчивости решетки исходной фазы ко все мень-
шим и меньшим смещениям атомов при увеличении переохлажде-
ния (в соответствии со схемой, приведенной на рис. 7.2,6) .вплоть
до состояния потери устойчивости к самым малым смещениям.
Остановка роста мартенситных кристаллов и сохранение исходной
фазы легко объясняются накоплением упругой энергии в системе
(см. с. 180, 202).
Одно и то же фазовое превращение может происходить путем
гомогенного или гетерогенного зарождения, иногда эти механизмы
действуют одновременно, иногда—только один из этих механиз-
мов в определенных температурно-концентрационных условиях.
Например, при распаде пересыщенных твердых растворов гете-
рогенное зарождение на дефектах имеет место при небольшом
пересыщении или при низкой температуре, когда затруднена диф-
фузия.
Неправильно считать, что всегда дефекты облегчают процесс
образования новой фазы. Пластическая деформация может затор-
мозить мартенситное превращение (тормозится рост мартенситных
кристаллов). Большая концентрация дефектов при сильном взаи-
модействии избыточных атомов с этими дефектами может настоль-
ко понизить пересыщение в матрице, что распад твердого раствора
окажется возможным только после отжига этих дефектов.
§ 7.2. РОЛЬ ЭНЕРГИИ УПРУГОЙ ДЕФОРМАЦИИ
И МЕЖФАЗОВОЙ (ПОВЕРХНОСТНОЙ) ЭНЕРГИИ
В процессе фазового превращения первого рода возникает ге-
терогенное состояние, даже если начальное и конечное состояния
являются однофазными. После образования центра новой фазы
фазовое превращение состоит в движении границы раздела новой
и исходной фаз. Уровень энергии межфазовой границы и ее свой-
ства определяются атомным строением границы (степень неупоря-
доченности) и различиями в химическом составе граничащих
фаз. Очевидно, что чем больше различия в кристаллической струк-
туре фаз (различия межатомных расстояний и координации ато-
мов) и природы атомов, тем выше должна быть энергия межфазо-
вой границы.
В граничащих кристаллах разных фаз могут быть опреде-
ленные кристаллографические направления, вдоль которых меж-
атомные расстояния совпадают (или почти совпадают). Примеры
полного (трехмерного) совпадения расположения атомов можно
найти в тех случаях, когда образуются домены упорядочения или
когда из твердого раствора выделяется фаза, изоморфная матри-
це, но отличающаяся от нее химическим составом. В таких слу-
чаях можно говорить о полной когерентности решеток новой и
исходной фаз (рис. 7.3,<з). Различия удельных объемов (атомных
радиусов и соответственно межатомных расстояний) приводят
186
к упругой деформации. Несоответствие решеток в процессе роста
кристалла новой фазы может привести к появлению дислокаций,
снимающих эту упругую деформацию (см. рис. 7.3,6). Возмож-
ность образования когерентной межфазовой границы определяется
выигрышем энергии, которая зависит от степени несоответствия
сопрягающихся решеток е=(«0—ам) /ам (рис. 7.4). Разумеется, при
Рис. 7.3. Схемы структуры межфазовых границ:
а — когерентная граница выделения упорядоченной фазы
на основе структуры, изоморфной структуре матричного
твердого раствора при различии периодов решетки выде-
ления ав и матрицы ам, вызывающем относительную
деформацию 8= (ав—ам)/ам; б — частично когерентная
(дислокационная) граница кристаллов тех же фаз, что
и на рис. 7.3,а; в — некогерентная граница зерен раз-
ных фаз
этом имеют значение также упругие характеристики сопрягающих-
ся фаз. Значение упругой энергии Еулр при полной когерентности
решеток можно выразить через коэффициент сжимаемости х, мо-
дуль сдвига G и объем v, в котором заключена частица новой фа-
зы при несоответствии решеток г:
£’ynp=6Gye2/ (1 +4/3Gx). (7.2)
187
Рис. 7.4. Зависимость энергии
системы от степени несоответ-
ствия решеток 8 (энергия не-
когерентной границы принята
равной 1):
1 — упругая энергия системы при
одномерном сопряжении решеток;
2 — энергия дислокационной гра-
ницы
Это уравнение выведено с учетом
только эффекта дилатации (сокраще-
ния или расширения) объема без из-
менения его формы: сферическая ча-
стица радиусом г0(1+е) заключена
в полом сферическом объеме матри-
цы и=4/3лг3о; упругие характеристики
матрицы и включения приняты оди-
наковыми.
При достижении достаточно боль-
шого размера частицы когерентность
решеток нарушается: уровень упру-
гой энергии понижается благодаря
образованию на межфазовых грани-
цах дислокаций несоответствия, или
эпитаксиальных дислокаций. Полная
релаксация упругих напряжений на-
ступает при образовании такого коли-
чества дислокаций, при котором рас-
стояние между дислокациями
/=|Ь|/е, (7.3)
где b — вектор Бюргерса эпитаксиальных дислокаций. Механизм
появления дислокаций на границе раздела фаз может быть раз-
ным: образование призматических петель вокруг частицы в матри-
це, зарождение дислокационных петель внутри выделяющейся ча-
стицы конденсацией точечных дефектов, зарождение дислокаций
несоответствия на самой межфазовой границе, наконец, к поверх-
ности раздела могут притягиваться дислокации, ранее образован-
ные в самой матрице. Энергетический барьер образования меж-
фазовых дислокаций при 8<0,005 очень велик (ЛЕ на рис. 7.4).
Даже при большом несоответствии когерентность существует (на-
пример, между частицами Co-фазы и Cu-матрицей в сплаве
Си—Со при 8^0,015).
Когерентность (или частичная когерентность) решеток новой
и исходной фаз может быть не только в случае изоморфности ре-
шеток. Легко найти в гекс. п. у. и г. ц. к. структурах совпадающие
по расположению атомов плоскости (см. рис. 6.9,6 и в). Действи-
тельно, превращение в Со идет при полной когерентности а- и
фаз, сопрягающихся по (0001)tt [| (111)р. Близкими по расположе-
нию атомов являются наиболее плотноупакованные плоскости
(111) в г. ц. к. и (ПО) о. ц. к. решетках (см-, рис. 6.9,а, 6), что
имеет важное значение для понимания атомного механизма поли-
морфных превращений в железе и его сплавах. Определенные
ориентационные соотношения кристаллических решеток фаз ха-
рактерны для превращений в твердом состоянии, во многих слу-
чаях эти соотношения проявляются в микроструктуре — в форме
и закономерном расположении частиц относительно матрицы
(видманштеттовы структуры, рис. 7.5) и в закономерных кристал-
188
Рис. 7.5. Видманштеттова структура
феррита (светлые пластины) в стали
45 после высокотемпературного от-
жига, Х200
Рис. 7.6. Зерна d-феррита (темные),
оставшегося после д—^'-превращения
при высокой температуре в хромо-
никелевой стали, Х200
лографических индексах плоскостей габитуса частиц; эти индексы
обычно задаются в кристаллографической координатной системе
матричной фазы (табл. 7.1).
При отсутствии когерентности (см. рис. 7.3,в) и преобладаю-
щем действии фактора поверхностной энергии конфигурация гра-
ниц стремится к равновесной (рис. 7.6), которой при равенстве
энергий границ зерен и межфазовых границ соответствуют 120°-
ные стыки (рис. 7.7).
Рост мартенситного кристалла
прекращается при нарушении коге-
рентности вследствие релаксации
упругих напряжений на границе
раздела фаз.
Частицы новой фазы, образую-
щиеся в результате превращений
в твердом состоянии, могут иметь
весьма различные формы — дендри-
ты, многогранники, сферы, пласти-
ны, диски, иглы.
^7
Рис. 7.7. Равновесная форма час-
тицы при полностью неупорядочен-
Плоскости габитуса частиц,
имеющих правильную огранку, па-
раллельны определенным кристал-
лографическим плоскостям исход-
ной фазы; кристаллическая решет-
ка новой фазы закономерно ориен-
тирована относительно решетки ис-
ходной фазы; наконец, сами частицы
ных границах раздела: — энер-
гия некогерентной границы зерен
матрицы; а5 —энергия межфазовой
границы
обычно имеют закономерное
взаимное расположение, часто образуя одно-, двух- или трехмер-
189
to
о
Таблица 7.1
Характеристики фаз, образующихся при распаде твердых растворов некоторых технических спчавов
№ п/п Сплав или система, мае. % Фаза выделения, ее структура о и периоды решетки, А Ориентационное соотношение Форма частиц Примечание
1 Fe—8Ni—ITi NiTi, типа CsCl, <2=2,99 1Ы1] II [hkl]a Равноосная
2 Fa—16Ni—IT! Tj—Ni3Ti, гекс. п. у. упо- рядоченная <2=5,1, с=8,3 (00.1) || {110}а 1100],, II <111>а Игольчатая
3 Fe—16Ni—5Mo (FeNi)2Mo, типа MgZn2 <1=4,74, с =7,75 (112) || {110}а 1- [243] || <111>а 2- [823] || <111>а Линзообразная
4 Fe—18N1—lOCo— —5Mo—0,5Ti, <2=2,869 1. Ni3Mo, орторомбическая, Л=5,064, 6=4,224, с=4,448 Для неупорядоченной фазы Ni3Mo, гекс. п. у. 2. 7)=Ni3Ti, гекс. п. у. «=5,101, с=8,307 (ОЮ) || (011)а 1- [ЮО] || [ 111]а 2- [102] || (]Ц]а (42') (0001) ||(011)а ' F1120] 1] [П1]а См. № 2 Стержни <111>а
5 Fe—16Cr—4Ni— —2Mo—1,3Cu Си—твердый раствор, г. ц. к. (HI)CU II (И0)а [Н0]Си II [1Ю|а Пластинчатая На дислокациях старение при 550—800°С
6 Fe—13Cr—0,4N Cr2N, гекс. п. у. <2=2,772, с=4,478 (00.1) II (110)а [10.0] II [П3]а Игольчатая по <311>а
7 Fe—С (сталь) Бейнит нижний 6-Fe3C, орторомбическая, «=4,525, 6=5,090, с—6,744 (100)о II (011)а (ОЮ)0 II (111)а (001)е II (211)а Пластины по {110}а или {211}а Рост вдоль <111>а
№ п/п Сплав или система, мае. % Фаза выделения, ее структура о и периоды решетки, А
8 Fe—С—N (техни- ческое железо), Fe—N a"-Fe16(N, С)2, тетраго- нальная, <7=5,72 £=6,29 V-Fe4(N, С), г. ц. к., я=3,78
9 a) Fe—V—С б) Fe—V—Ti— С (0,18V, 0,13Ti, 0,08С) Старение при 700®С 9-Fe3C V4C8, г. ц. к., <7=4,16 (Ti, V)x Cj-д;, типа NaCl
10 Fe—Мо—С Мо2С, гекс. п. у., <1=3,00, с=4,77
11 Fe—W—С М6С, сложная кубическая
12 а) Аустенитная сталь Fe—14Cr—14N1— —0,4 С, а=3,585 б) Fe—14Cr—14Ni— —2.7W—0,45Мо М2аС5> сложная кубическая, «=10,57
13 Аустенитная сталь Fe—24N1—21Сг—4Мо м28с6 e-фаза, сложная тетрагональная, <7=8,80, с=4,58
Продолжение табл. 7.1
Ориентационное соотношение Форма частиц Примечание
lhkl]a„ || [hkl]a Пластины по {100}а Пластины по {012}а | | {112}т, В матрице и на дислокациях
(Ю0)к |1 (ЮО)а (ОЮ) к || (011)а (001)к || (011)а Дендрит Пластины по {100}а
[10.0] II [100]» (00,1)к II (011)а (01.0)к II (011)а Иглы <100>а
(1Н)к II (01|)а [0111k II [ill]»
a. (001)к || (111) (111)к П (001) (ИО)к II (1Ю)7 б. [Ш]к || |^/]т Пластин ы по {111}7
См. № 12 (001). II (111) (НО), II (110)т (111), II (1Ю)Т Неправильная форма о зарождается на частицах кар- бида М23С6
ю
to
№ п/н Сплав или система, мае. % Фаза выделения, ее структура о и периоды решетки, А
14 Ni—Ли, а-г. Ц. К., arAu—твердый раствор, г. ц. к., «=3,97 a2-Ni—твердый раствор, г. uv. к., «=3,56
15 Ni—2% Be лм=3,48 y'-NiBe, тетрагональная искаженная типа CsCl
16 Жаропрочный сплав Ni—20 Сг— —2 Ti—1,5 Al Y'-Ni3 (TiAl), кубическая, типа Cu3Au
17 Al—4Cu 0-CuA12, тетрагональная, искаженная, типа флюорита, <7=5,71, с=5,80
0-СиА12, тетрагональная, «=6,066, е=4,87
18 Al—20 Ag у', гекс. п. у., <1=2,858, с=4,602 у, гекс. п. у., <2=2,879 с=4,573
19 Al—Cu—Mg 5—Al2CuMg, ромбическая
Продолжение табл. 7.1
Орие нта цисшиое соо гношение Форма частиц Примечание
[AWJal II [hkl]^ (Ч0)а1в2 || (П_1)а [°°Чал II 1Н0]а Пластины В колониях пре- рывистого распада
а) (110) z || (111)а [001J , II [110]а б) (120) , II (112)а [001]т, || [Н0]а
Ihkl]^ || [Ш]т Равноосная Ь=я/2 [110]
(001)е, II (001)а [100]9, II [110]и Разные, нет коп Пластины фентности Цислокации несо- ответствия, Ь—а [001] Ь=а [010]
(00.1)т II (111)а [11.0]т II [И0]а Пластины по {111} Дислокации несоответствия, Ь=а [001] Ь=а/2 [112]
[100]. II [Ю0]а [010]s II [021]а [001]. II [012]а Иглы Дислокации несоответствия, Ь=а/2 (101)
ные сетки*. Такое расположение при распаде твердых растворов
бывает настолько строгим, что в дифракционной картине (рентге-
новской или электронной) рядом с определенными вульф-брэггов-
скими отражениями могут возникать сателлиты — признак квази-
периодической или «модулированной» структуры.
Морфология** гетерогенной структуры, а также дисперсность
этой структуры определяются совместным действием упругой и
поверхностной (межфазовой) энергией. А. Л. Ройтбурд и А. Г. Ха-
чатурян дали наиболее общее решение задачи об упругой энергии
а
Рис. 7.8. Пластина гыделения проходящая через
весь кристалл исходной фазы (а), и образование
кристалла внутри объема исходной фазы (6):
у края пластины внутри исходного кристалла
условно показано поле упругой деформации
частицы, рассматриваемой как изолированное когерентное вклю-
чение в упруго-анизотропной матрице. Минимальная упругая энер-
гия получается для включения пластинчатой формы. Поверхность
пластины совпадает с той плоскостью кристалла, которая меньше
всего искажается при превращении. Фактор поверхностной энергии
препятствует превращению включения в бесконечно тонкую (и бес-
конечно протяженную) пластину, поскольку поверхностная энергия
минимальна для включения равноосной формы.
Выражение для упругой энергии кристалла, содержащего ко-
герентное ,пластинчатое включение (или систему включений), со-
стоит из двух членов: первый пропорционален объему включения
и поэтому складывается с объемной химической свободной энер-
гией, второй связан с деформацией у торцов пластины и формаль-
но может быть интерпретирован как энергия дислокационной пет-
ли, охватывающей включение по .его периметру. Первый член ра-
вен нулю, если деформация при превращении есть деформация
с инвариантной плоскостью и плоскость габитуса параллельна
инвариантной плоскости. Деформация с инвариантной плоскостью
означает, что в процессе превращения, которое при изменении
кристаллической структуры можно рассматривать как «структур-
ную» деформацию, имеются плоскости, в которых расположение
атомов не изменяется и которые сами не поворачиваются. Про-
* Гаврилов А. В., Тяпкин Ю. Д., Усиков М. П. «Докл. АН СССР», 1967,
1. 176, с. 1052.
** Балли Д., Захарова М. И. «Докл. АН СССР», 1954, т. 96, с. 453.
13—523 193
стейшей моделью деформации с инвариантной плоскостью может
служить движение игральных карт в колоде (скольжение). Де-
формацией с инвариантной плоскостью является двойникование
в кристаллах, а также подробно рассматриваемое ниже мартен-
ситное превращение. Второй член должен быть равен нулю, если
пластина включения проходит через весь кристалл (рис. 7.8,а).
Если пластинчатое выделение оказывается внутри кристалла, то
поле упругих искажений возникает у торцов пластины (см.
рис. . 7.8,6) и величина упругой энергии будет пропорциональной
периметру пластинчатого выделения.
Для пластины в виде диска радиусом R и толщиной D имеем
BD2 [ D ИМ
£упр = [- 1п "R" + 2 1п 2 - ~),2^’ (7.4)
здесь (и — модуль упругости; е0 — деформация, характеризующая пере-
ход от исходной к кристаллической структуре новой фазы). Энергию Еупр мож-
но представить как энергию линейного натяжения струны длиной 2jtR с коэф-
фициентом линейного натяжения
. Р2/ В 1 \
8 = 17 (~1ПТГ+21П2~—) ~~(хЬ2’
(7.5)
где b = De0 имеет смысл вектора Бюргерса.
Для того чтобы выяснить пределы применимости предположения о том, что
форма включения (пластина толщиной D и длиной L) определяется условием
минимума упругой энергии, оценим порядок величины Еупр из уравнения (7.4)
Eynp^He?‘oLD2. (7.6)
Энергия поверхностного натяжения пропорциональна поверхности включения и
без учета влияния торцов пластины имеет порядок aL2, где a — коэффициент
поверхностного натяжения.
Примем, что когда сумма поверхностной и упругой энергии минимальна, по-
рядок величины для упругой и для поверхностной энергий один и тот же:
p,e2oD2L^aL2.
(77)
При этом условие получения пластинчатой формы включения, т. е. D/L<C1, опре-
деляется неравенством
D/L~a/p,e20D<Cl. (7.8)
Это условие получается более наглядным, если ввести некоторую характерную
длину г0^а/це20:
(ro/Ll^Cl. (7.9)
Если принять а^10 эрг/см2, ц~1012 эрг/см3, е2о~1О_4, то получим го^10 А.
Итак, выводы теории, принимающей во внимание только фак-
тор упругой энергии, справедливы для включений, протяженность
которых существенно больше 10 А. В приведенной выше оценке
была принята деформация около 1%, очевидно, при 8»—>0 эти со-
отношения теряют свою силу, и при изменении знака неравенства
(7.9) возможна равноосная форма включения. В связи с этим при
некоторых значениях 8о можно ожидать изменения формы частиц
по мере их роста. Наглядным подтверждением теории являются,
например, экспериментальные данные для сплавов на никелевой
194
основе (г. ц. к. структура), в которых выделяется изоморфная
матрице фаза типа /-Nis(Al, Ti) (упорядоченная фаза типа Сп3Ап).
В сплавах системы Ni—Ti при относительной разнице периодов
решетки в ~ 0,01 выделения, видимые в электронном микроскопе
(Lo^lOO А), имеют пластинчатую форму; в сплавах Ni—Al при
£~0,005 частицы сначала имеют равноосную форму, а при уве-
личении длительности старения приобретают пластинчатую форму;
в сплаве Ni—Сг—Ti—Al относительная разница периодов решет-
ки матрицы и выделения уменьшается до 0,008, поэтому частицы
выделений /-фазы сохраняют равноосную форму. При дальней-
шем уменьшении различия периодов решетки форма выделений
Рис. 7.9. Влияние степени несоответствия е решеток фазы выделения и матрицы
на форму частиц в сплавах Ni—Сг—Ti—Al, после отжига 1000 ч при 850°С.
Электронные микрофотографии углеродных реплик с фиксированными частицами:
а —сплав 75 Ni—15,5 Сг-0,5 Ti—9 А1 ат.%; 8=0,0035; б — сплав 75 Ni—18 Сг—4 Ti—3 А1 ат.%;
8=0,0080
изменяется от полиэдрической до правильной сферической
(рис. 7.9). В данном случае кроме различий в периодах решетки
сказывается разное влияние титана и алюминия на поверхностную
энергию. Подобный переход от пластинчатых форм к равноосным
может быть также следствием потери когерентности решеток ча-
стицы новой фазы и матрицы и релаксации упругих напряжений
при образовании на границах раздела дислокаций. Эффект этих
изменений эквивалентен уменьшению деформации е и увеличению
поверхностной энергии.
Поле деформации в неограниченной матрице от одиночной плос-
кой частицы надо рассматривать как суперпозицию полей двух
параллельных межфазовых границ, в 'Случае очень тонкой пластины
эти поля в любой точке кристалла вне пластины одинаковы по
величине, но противоположны по знакам и взаимно уничтожаются
везде, кроме самой пластины. А. Л. Ройтбурд уподобляет упругое
поле такого кристалла электростатическому полю плоского кон-
13* 195
денсатора: это поле целиком сосредоточено между «обкладками»,
т. е. внутри пластины, за исключением поля рассеяния у ее кон-
цов. Это справедливо при условии, если упругие свойства матри-
цы и включения достаточно близки. Если новая фаза оказывается
более жесткой, чем матрица, концентрация упругого поля внутри
частицы будет энергетически невыгодной и новая фаза приобре-
тет вместо пластинчатой округлую форму, при этом большая часть
Рис. 7.10. Влияние различия жесткости исходной (матричной) фазы и новой (фа-
зы выделения) на форму частицы в сплавах Fe—Ni—А! после закалки и отжига
100 ч при 800°С:
а — сплав 40 ат. % Fe: выделение Fe-фазы в окружении NiAl-матрицы; б — сплав 70 ат.°/о Fe:
выделение NiAl-фазы в окружении мягкой Fe-матрицы, Х1000
аккомодационных искажений придется на более «податливую»
исходную (матричную) фазу. Этот результат теории можно иллю-
стрировать микроструктурами двух сплавов квазибинарного раз-
реза Fe—NiAl системы Fe—Ni—Al, в которых происходит расслое-
ние твердого раствора на две изоморфные фазы: твердый раствор
на основе железа (о. ц. к. фаза) и упорядоченный твердый раствор
NiAl (структура типа CsCl). Разница между сравниваемыми спла-
вами состоит только в том, что в одном из них преобладающей
(матричной) фазой является NiAl-фаза и выделяется «мягкая»
Fe-фаза в виде пластин (рис. 7.10,а), в другом в окружении мяг-
кой железной матрицы образуются округлые частицы NiAl-фазы
(см. рис. 7.10,6). Непосредственное представление о поле упругой
деформации возле когерентного включения дает дифракционный
контраст электронно-микроскопического изображения включений
в тонкой фольге (рис. 7.11).
Условие минимума энергии упругой деформации при образо-
вании новых фаз в твердом состоянии определяет не только фор-
му отдельных частиц, но и их взаимное расположение.
Б сплаве Ni—Ti (рис. 7.12) в условиях сравнительно неболь-
шого пересыщения и небольшого, количества избыточной метаста-
бильной у'-фазы (в изоморфной г. ц. к. матрице) ярко проявляет-
196
ся особенность взаимного расположения частиц: первые образо-
вавшиеся частицы (более крупные на фотографии) инициируют
образование регулярных рядов последовательно выделяющихся
частиц вдоль < 100> направлений г. ц. к. матрицы. В гетероген-
ной структуре в ходе превращения или в конечной структуре
имеется еще несколько способов понижения упругой энергии.
Рис. 7.11. Электронные микрофотографии фольги спла-
ва Fe—Сг—А1, деформационный контраст от сфериче-
ских выделений фазы FeAl в ферритной матрице; ра-
диальный характер поля деформации объясняет наличие
области нулевого контраста (перпендикулярно к вектору
действующего отражения g 002)
Модулированные структуры и упругие концен-
трационные домены образуются, например, при расслоении
твердых растворов. В двойных сплавах Ni—Al, Ni—Ti, Ni—Au,
а также в тройных Cu—Ni—Fe, Fe—Ni—Al и в некоторых других
практически очень важных сплавах при распаде твердого раствора
с кубической решеткой образуются две новые изоморфные фазы,
различные по составу.
Примем, что концентрационная зависимость периода решетки
подчиняется правилу Вегарда, т. е.
a=a^(l+qoc). (7.10)
Здесь а0 — период решетки одного из чистых компонентов; qo—
линейный коэффициент концентрационного расширения решетки;
с — концентрация второго компонента. Различия состава и соот-
ветственно периодов решетки сопрягающихся фаз должны приво-
дить к возникновению напряжений. Можно показать, что в отно-
шении упругой энергии более выгодно образование комплексов из
197
перемежающихся пластин обогащенной и обедненной фаз
(рис. 7.13), чем независимое образование пластин тех же фаз.
Соотношение толщин этих пластин в комплексе определяется диа-
граммой состояния (правило рычага).
Если справедливо правило Вегарда, то любое перераспределе-
ние атомов компонентов, включая перераспределение, приводящее
Рис. 7.12. Электронная микрофотография реплики
выделений у'-фазы в сплаве Ni—Ti после старе-
ния при 800°С в течение 5 ч (стрелками отмечены
первые образовавшиеся частицы). X10 000
(С. Б. Масленков)
к образованию двух изоморфных твердых растворов, не вызывает
изменения заданного объема (объема комплекса). Поэтому на
достаточно большом расстоянии от комплекса (больше, чем пе-
Рис. 7.13. Упругие концентрацион-
ные домены при расслоении твер-
дого раствора (А. Г. Хачатурян)
риод внутренней неоднородности
комплекса D) поле упругих напря-
жений в матрице равно нулю. Если
пластины комплекса не выходят на
поверхность кристалла, то упругие
напряжения должны быть в слое,
прилегающим к торцам пластин.
Толщина этого слоя порядка тол-
щины элементов комплекса D, объ-
ем слоя около DL2 (L — линейный
размер комплекса), величина де-
формации —qokc (Ас— разность
концентраций в фазах — слоях ком-
плекса); плотность упругой энергии
—ри?2оАс2 и пблная энергия
(7.11)
198
Из соотношения (7.11) следует, что упругая энергия тем мень-
ше, чем тоньше концентрационные домены комплекса (ЕУпр—>0
при D—>0), однако дробление в комплексах должно вызывать по-
вышение поверхностной энергии. Порядок этой величины
£пов—aL2L/D. (7.12)
Здесь L2 — площадь одной межфазовой границы в комплексе;
L/D — число границ в комплексе. Выгодная величина периодич-
ности доменов Do определяется из условия минимума суммы энер-
гии Eyirp И £*пов"
De~)/rJL. (7.13)
Величина го^а/м2о\с2 характеризует соотношение удельных зна-
чений поверхностной энергии и энергии упругой деформации. Вы-
воды данной теории сохраняют свое значение и при росте ком-
плекса, если элементы его структуры много меньше размера ком-
плекса (DCL), так как при таком соотношении релаксация
в структуре комплекса будет идти быстрее, чем относительное уве-
личение размера комплекса.
Полидоменные пластины. Если фазовое превращение
состоит в изменении кристаллической структуры, то обычно имеет-
ся несколько равноценных вариантов деформации .решетки. На-
пример, при образовании тетрагональной фазы из кубической ось
с может быть параллельной одному из трех направлений ребра
куба, т. е. образующиеся в пределах одного исходного кубическо-
го кристалла области тетрагональной структуры могут иметь три
разных ориентировки. Деформация решетки при превращении ку-
бической структуры в тетрагональную, например при образовании
мартенсита в углеродистой стали, не является деформацией с ин-
вариантной плоскостью, поэтому образование пластины тетраго-
нальной фазы конечной толщины должно приводить к возникно-
вению поля деформации. На рис. 7.17,а показана пластина,
состоящая из чередующихся пластин — доменов тетрагональной
фазы с различными ориентациями тетрагональной оси. При под-
ходящей ориентации полидоменной пластины и определенном со-
отношении толщины доменов двух типов получается суммарная
макроскопически плоская деформация, которая уже может быть
деформацией с инвариантной плоскостью. В этом случае сопряже-
ние фаз не приводит к возникновению поля напряжения в объеме
пластины. Определить оптимальную структуру полидоменной плас-
тины (толщина одного из типов доменов и расстояние между эти-
ми доменами) можно минимизацией суммы поверхностной энер-
гии и упругой энергии, связанной с торцевой (некогерентной) по-
верхностью полидоменной пластины *.
Упругое взаимодействие кристаллов новой
фазы. Поскольку дальнодействующее поле когерентного пластин-
* Ройтбурд А. Л. В кн.: Несовершенства кристаллического строения и мар-
тенситные превращения. М., «Наука», 1972, с. 15.
199
чатого кристалла новой фазы внутри исходного кристалла можно
уподобить полю дислокационной петли с вектором Бюргерса
|b|=eD, то по аналогии с обычными дислокациями можно пред-
ставить такие взаимные расположения этих кристаллов, которые
приводят к понижению уровня суммарных напряжений. Во-первых,
это достигается образованием стыков пластинчатых кристаллов,
Рис. 7.14. Микроструктуры при образовании тет-
рагональной фазы (мартенсит в стали):
а — полидоменная пластина, составленная из доменов
с разной ориентацией тетрагональной оси; границы раз-
дела параллельны плоскостям (ПО) и (ОН); б, в—устой-
чивые расположения монодоменных пластинчатых кри-
сталлов («фермы», параллельные ряды) (А. Л. Ройтбурд)
если энергия результирующего поля будет меньше суммы энергий
упругой деформации у торцов независимо взятых кристаллов:
(28гОг)2<2(8гОг)2. (7.14)
Во-вторых, уменьшения упругой энергии надо ожидать при обра-
зовании таких расположений пластинчатых кристаллов, которые
аналогичны расположению дислокаций в вертикальных стенках
(при полигонизации). Такое расположение показано на рис. 7.14,в.
Стыки кристаллов (см. рис. 7.14,6) при подходящем соотношении
толщин могут дать результирующий вектор Бдоргерса, перпенди-
кулярный к линии стыка кристаллов, а изменение длины стыкую-
щихся кристаллов от стыка к стыку может создать расположение
стыков («дислокаций») по типу вертикальной стенки. Следова-
тельно, в этом расположении реализуются оба интерференционных
эффекта, уменьшающих дальнодействующее поле упругой дефор-
мации от торцов пластинчатых кристаллов. Фермы из пластин и
параллельные расположения пластинчатых кристаллов характер-
ны, например, для микроструктур мартенсита в сплавах на же-
лезной основе (рис. 7.15).
Рис. 7.15. Взаимные расположения
кристаллов новой фазы, обусловлен-
ные влиянием упругой энергии (элек-
тронные микрофотографии тонких
фольг):
а — упругие тетрагональные домены при
упорядочении А и В в сплаве Со—Pt
(О. Д. Шашков); б — двойниковые ориен-
тации соседних мартенситных кристаллов
в хромоникелевой стали (В. И. Изотов);
в — разные ориентации пластинчатых ча-
стиц фазы СиВе (структура типа CsCl)
в чередующихся областях тетрагональной
деформации (г. ц. к. матрица + включения)
сплав Си—1 мае.% Be (В. Г. Холодова-
III а рш а ткин а)
Рассмотренные особенности структурообразования, связанные
с энергией упругой деформации, проявляются в той или иной сте-
пени при всех превращениях в кристаллах, сопровождающихся
изменением (деформацией) решетки при условии сохранения
сплошности кристалла. К этим превращениям относятся кроме
превращений с изменением координации атомов и упорядочение
в твердых растворах, часто сопровождающееся изменением класса
симметрии, и расслоение твердых растворов, поскольку перерас-
пределение концентраций вызывает локальные изменения периода
решетки (см. рис. 7.15 и рис. 7.16). Доменные структуры в фер-
ромагнетиках и сегнетоэлектриках во многом аналогичны струк-
турам гетерофазных систем, поскольку во всех этих случаях струк
турообразование определяется минимумом свободной энергии, ко-
торая в качестве существенной составляющей содержит энергию
дальнодействующего поля.
20Г
Внутренние напряжения могут контролировать развитие фазо-
вого превращения, если это превращение идет в ограниченном
объеме исходной фазы, 'сохраняется контакт между фазами (т. е.
сохраняется сплошность) и отсутствуют или проходят в ограни-
ченной степени релаксационные процессы. Наиболее ярко прояв-
ляется роль упругой энергии (и отсутствие релаксационных про-
цессов) в опыте Г. В. Курдюмова и Л. Г. Хандроса с термоупру-
гим мартенситным кристаллом в сплаве на Cu-основе: при пони-
Рис. 7.16. Упругие домены вдоль <011> на ранних стадиях распада твер-
дого раствора в сплаве Си—1,6 мес.% Be (20(У*С, 1 ч):
а — электронная микрофотография фольги, Х80 ООО; б — схема расположения частиц —
плоских зон, создающих тетрагональные искажения внутри доменов
жении температуры выигрыш объёмной химической энергии обес-
печивал работу против сил упругости и кристалл мартенсита под-
растал; наоборот, повышение температуры приводило к обрати-
мому уменьшению размера кристалла. Благодаря удачному под-
бору состава сплава это явление удалось легко наблюдать с по-
мощью светового микроскопа при небольшом изменении темпера-
туры вблизи комнатной температуры; теоретический анализ этого
явления упрощался тем, что мартенситный кристалл сохранял
когерентность с матрицей, и поэтому можно было пренебречь фак-
тором энергии межфазовой границы в уравнении (7.1). Равнове-
сие при понижении температуры наступало при 6(ДФ)=0, т. е.
когда
(3£ynp/cto) 8v—&Fbv. (7.15)
Дальнейшее понижение температуры приводило к увеличению
движущей силы превращения — разности свободных энергий Д£=
=£2(—£i, что обеспечивало подрастание кристалла.
При определенных условиях в ходе фазового превращения про-
исходят генерация дислокаций и пластическая деформация путем
202
сдвига или двойникования. Деформация превращенного объема
с инвариантной решеткой является составным элементом мартен-
ситного превращения в стали. Под деформацией с инвариантной
решеткой понимают деформацию, при которой не изменяется кри-
сталлическая -структура, это может быть в тех случаях, когда ато-
мы перемещаются относительно друг друга на полную трансля-
цию. Примером такой деформации является сдвиг, осуществляе-
мый движением дислокаций с целочисленным вектором Бюргерса;
двойникование относят к такого рода деформации только условно.
Релаксация упругих напряжений может идти или непрерывно
в ходе -самого превращения, или представлять собой определенный
этап фазового превращения.
§ 7.3. МЕХАНИЗМЫ РОСТА КРИСТАЛЛОВ
ПРИ БЕЗДИФФУЗИОННЫХ ПРЕВРАЩЕНИЯХ.
ТЕРМОДИНАМИЧЕСКАЯ ВОЗМОЖНОСТЬ БЕЗДИФФУЗИОННОГО
ПРЕВРАЩЕНИЯ ТВЕРДЫХ РАСТВОРОВ
Если скорость превращения определяется скоростью переме-
щения границы раздела фаз, различающихся только своей кри-
сталлической -структурой, превращение называется бездиффузион-
цым. К бездиффузионным превращениям относятся полиморфные
превращения в однокомпонентных системах, а также в некоторых
сложных по составу фазах — химических соединениях стехиоме-
трического состава и так называемое массивное превращение
в твердых растворах. Кинетически при этом различают нормаль-
ное полиморфное и мартенситное превращения.
Нормальное превращение. Кинетика некоторых превращений
в твердом состоянии названа «нормальной», поскольку как вре-
менная, так и температурная зависимости скорости превращения
оказались подобными тем, которые ранее были установлены при
исследовании процесса кристаллизации. При понижении темпера-
туры (для реакций, идущих при охлаждении) скорость превра-
щения сначала увеличивается, а затем уменьшается. Возрастание
скорости превращения связано с облегчением образования заро-
дышей и ускорением их роста при увеличении переохлаждения,
поскольку при этом возрастает термодинамический стимул пре-
вращения; уменьшение скорости превращения при дальнейшем
увеличении переохлаждения объясняется уменьшением подвиж-
ности атомов при понижении температуры.
Термодинамический стимул превращения определяется полным
изменением свободной энергии системы при образовании новой
фазы: АФ(и) для области новой фазы, состоящей из п атомов.
Движущей силой процесса роста области новой фазы называют
величину с?АФ(и)/б/и; рост области новой фазы происходит, если
при увеличении п АФ (и) уменьшается, т. е. ^АФ(и) /dn<S}. Из
термодинамики неравновесных процессов получим * следующее
* Полный вывод по Б. Я. Любову приведен в гл. 10.
203
общее выражение для скорости роста:
dn/dt = - (К/Т) п* d^n(n) . (7.16)
Здесь п*— число атомов, составляющих поверхность раздела об-
ласти новой фазы и матрицы; К — кинетический фактор, который
определяется механизмом процесса роста.
Рост области новой фазы при бездиффузионном превращении
состоит в присоединении к новой фазе атомов, находящихся непо-
средственно у поверхности раздела фаз. Нормальная кинетика
предполагает, что присоединяются (и, наоборот, отрываются) от-
дельные атомы и эти акты
присоединения (или от-
рыва) никак не связаны.
П п+1^
Координата реакции
а
Рис. 7.17. Энергетический барьер у поверхности частицы новой фазы, ха-
рактеризующий состояние «активированного комплекса» при присоединении
к новой фазе атома (а); центр новой фазы, состоящий из п атомов (б)
В процессе перехода атома от одной фазы к другой возникают
некоторые промежуточные конфигурации, обладающие повышенной
свободной энергией по сравнению с исходным состоянием. Такая
конфигурация называется активированным комплексом. На
рис. 7.17,61 состоянию активированного комплекса соответствует
максимум, высота которого обозначена [АФ (п)-j-^АФ1 (п-{-1) ]
величина q, характеризующая уровень активированного комплек-
са, имеет порядок энергии активации процесса самодиффузии.
Значения qt и q2 определяют энергетические барьеры переходов
атомов соответственно при увеличении и уменьшении объема час-
тицы новой фазы.
При небольших переохлаждениях, когда
ddn(n) ^kT’ (7.17)
для скорости изменения радиуса R сферической частицы было по-
лучено (без учета упругой энергии) следующее выражение:
dR /dt= (K/T)exp(-qlkT) &Fq (1 —RKp / R). (7.18)
204
1/Т; ы^-q/kT); aF0; dK/dt
Рис. 7.18. Температурные зависи-
мости кинетических коэффициен-
тов 1/Т и Д^ехр(—q!kT), термо-
динамического стимула AFo и ско-
рости роста dR/dT центра новой
фазы для «нормального» превра-
щения
В этом выражении AF0 — изменение химической свободной
энергии на единицу объема; f/?«p=2a/AFo — радиус критиче-
ского зародыша, зависящий от поверхностного натяжения а, ко-
эффициент К включает параметр, характеризующий частоту коле-
баний атомов (например, температуру Дебая). Как видно из урав-
нения (7.18), в соответствии с общей теорией образования фаз
рост частицы возможен при /?>ь/?кр.
Температурная зависимость скоро- Т
сти роста, схематически представ-
ленная на рис. 7.18, в общем согла-
суется с экспериментальными дан-
ными для разных систем.
Мартенситное превращение. Ки-
нетика превращения характеризу-
ется обычно очень большой ско-
ростью роста отдельных кристаллов
и максимальной объемной ско-
ростью в начальный момент превра-
щения в изотермических условиях.
На предварительно полированной
поверхности металлографического
шлифа, появляется рельеф, связан-
ный с образованием единичных кри-
сталлов. Важнейшая особенность
мартенситного механизма превра-
щения — это слабая зависимость
подвижности границы фаз от тем-
пературы: мартенситное превраще-
ние может идти при весьма низких температурах. Во многих слу-
чаях развитие превращения в изотермических условиях не наблю-
дается и превращение идет только по мере охлаждения; такую ки-
нетику иногда называют «атермической» или «взрывной».
Формальное объяснение слабой температурной зависимости скорости мартен-
ситного роста следует из анализа уравнения бездиффузионного превращения
(7.16), если вместо условия (7.17) принять условие
^АФ (n)/dn^kT.
(7-19)
Очень большие значения термодинамической движущей силы характерны
для мартенситных превращений, идущих при больших переохлаждениях. Усло-
вие (7.19) дает
dn/dt=—ri4 ехр {—7-[-1/2 [б/ДФ (п)/б?л] }/^Т. (7.20)
Как видно из уравнения (7.20), эффективная энергия активации процесса роста
меньше величины q на 1 [2dАФ (n) /dn (^ДФ(п) /dn<$).
О скорости движения фронта превращения можно судить по
времени образования отдельного кристалла. Время роста мартен-
ситного кристалла из аустенита в стали около 10~7 с при конечном
размере кристалла приблизительно 10-2 см, что соответствует ско-
рости движения межфазовой границы порядка 105 см/с, что близ-
205
ко к скорости звука. Такая большая скорость перестройки решет-
ки обеспечивается согласованным (кооперативным) движением
атомов.
Перемещения атомов в кристаллической решетке не могут быть
независимыми. Только при достаточно высокой температуре, когда
тепловые колебания приводят к значительным нерегулярным сме-
щениям атомов в кристаллической решетке, движение атомов мо-
жет иметь более или менее автономный характер. С понижением
температуры влияние межатомного взаимодействия становится все
сильнее и перемещение атомов приобретает все более организо-
ванный, кооперативный характер. Обязательным условием коопе-
ративного движения фронта реакции превращения — границы раз-
Рис. 7.19. Пример деформации с инвариант-
ной плоскостью. Стрелки показывают на-
правления деформации s и ее величину:
а — сложная однородная деформация объема;
б — дилатация (растяжение); в — сдвиг; г —
сдвиг и дилатация
дела новой и исходной фаз — должна быть строгая регулярность
расположения атомов на границе. Это достигается при когерент-
ности решеток, когда атомные плоскости в решетке новой и исход-
ной фаз, параллельные границе раздела фаз, имеют одинаковое
расположение атомов.
Если перестройку >решетки при образовании новой фазы пред-
ставлять как деформацию решетки с инвариантной плоскостью, то
инвариантной плоскостью является граница между когерентными
областями новой и исходной фаз.
На межфазовой границе имеет место скачок деформации Де, отличающей
новую фазу от исходной:
Де= l/2(s0n+ns0) *. (7.21)
Здесь вектор s — направление смещения атомов; и — нормаль к инвариант-
ной плоскости; sn соответствует деформации с инвариантной плоскостью, когда
величина смещений атомов пропорциональна расстоянию от некоторой инва-
риантной плоскости, обозначенной направлением нормали к ней п (рис. 7.19).
* См.: Несовершенства кристаллического строения и мартенситные превра-
щения. М., «Наука», 1972, с. 26.
206
На рис. 7.20,а показана зависимость свободной энергии решетки от параметра
деформации 8. Первый относительный минимум соответствует структуре исход-
ной фазы, второй (абсолютный) минимум при деформации s0 соответствует
структуре новой фазы. AF0— термодинамический стимул превращения.
Локальная свободная энергия зависит не только от величины деформации,
но и от ее градиента. Поэтому на рис. 7.20,6 межфазовая граница представлена
как переходный слой, в котором вектор деформации s, характеризующий поло-
жение атомов, постепенно меняется по направлению и по величине. Такая гра-
ница подобна стенке в доменной структуре ферромагнетика *. Равновесную
Рис. 7.20. Схемы изменения свободной энергии при одно-
родной деформации (а) и энергии межфазовой границы
в виде переходного слоя (б) (А. Л. Ройтбурд)
структуру, толщину границы 1п и ее энергию Г(п) определяют из условия ми-
нимума свободной энергии. В процессе превращения, идущего как деформация
решетки с инвариантной плоскостью, межфазовая граница движется параллель-
но самой себе. Это движение сопровождается периодическим увеличением энер-
гии границы в соответствии с периодичностью в структуре в направлении п. Если
толщина границы равна периоду решетки, то приращение энергии границы
ДГ = ыАа. (7.22а)
Толщина границы может быть на порядок больше периода решетки, так как чем
сильнее размыта граница, тем меньше активационный барьер. Поскольку менее
резко изменяется структура (деформация), то изменение энергии при движении
границы:
ДГ = цАаф(/п), (7.226)
здесь ф(/„) —убывающая при увеличении I функция; при 1п — а ф = 1; кинетика
движения границы должна зависеть от соотношения между барьером для пере-
мещения АГ и термодинамическим стимулом превращения АЕ0. Величина AF0
возрастает с увеличением отклонения системы от состояния равновесия (для
превращений при охлаждении максимум AFoMaKc должен быть при Т — 0 К).
Если АГ>АЕмакс, то движение границы во всем температурном интервале пре-
вращения требует термической активации. Скорость роста кристалла изменяется
от нуля при температуре возле точки равновесия до максимума при некотором
переохлаждении и затем снова уменьшается до нуля при Т—>0 К в связи
* Разница в том, что магнитные моменты атомов меняют только направ-
ление.
207
с уменьшением вероятности необходимых для превращения тепловых флуктуа-
ций (кривая 1 на рис. 7.21).
Если Аммане>ДГ, то при достаточном отклонении от равновесия (т. е., на-
пример, при достаточном переохлаждении) движение границы не требует терми-
ческих флуктуаций и превращение идет «безбарьерным», или, лучше сказать,
«надбарьерным» путем. Скорость такого надбарьерного роста должна быть близ-
кой к скорости звука. С повышением температуры растет динамическое трение,
поэтому температурная зависимость скорости надбарьерного роста (кривая 2 на
рис. 7.21) при низкой температуре противоположна зависимости для термически
активируемого роста. Среди исследованных мартенситных превращений в боль-
К 7”ра6н 7~
1 .. 7.21. Зависимость скорости дви-
щия границы от температуры при
1ермически-активируемом (/) и «над-
барьерном» (2) росте
шинстве случаев наблюдалась надбарь-
ерная кинетика роста. В связи с очень
большой скоростью роста мартенситных
кристаллов в определении температур-
ной зависимости скорости превращения
(или объема превращения) существенна
температурная зависимость зарождения
мартенситных кристаллов.
В случае нормального превра-
щения движение границы осуще-
ствляется через термические
флуктуации, так как некоторые
промежуточные конфигурации
атомов у границы раздела фаз
отличаются повышенной свобод-
ной энергией. Преодоление этих
энергетических барьеров при
очень низкой температуре оказы-
вается маловероятным в связи с уменьшением вероятности соот-
ветствующих энергетических флуктуаций. Аналогичный механизм
перемещения границ зерен действует при рекристаллизации. Тем-
пературная зависимость этих процессов определяется энергией
активации, близкой к энергии активации самодиффузии. Если
представить такую границу состоящей из некоторой сетки дисло-
каций, способных только к неконсервативному движению, и коге-
рентных участков, то превращение (или движение границ) должно
быть связано с установлением равновесия раствора точечных де-
фектов вакансий и межузельных атомов, 'испускаемых или погло-
щаемых дислокациями. Между кристаллами новой фазы возникает
«самодиффузионное» взаимодействие посредством обмена вакан-
сиями через исходную фазу, при этом кинетика фазового превра-
щения контролируется релаксацией средней концентрации вакан-
сий за счет диффузии к сторонним источникам или стокам (напри-
мер, к внешней поверхности). Протекание релаксационных про-
цессов у границы будет тем полнее, чем меньше движущая сила
роста новой фазы. При малых отклонениях системы от равнове-
сия более вероятен рост кристаллов, контролируемый самодиффу-
зией, при больших — кооперативный рост. Таким образом, один и
тот же фазовый переход в однокомпонентной системе при разных
внешних условиях может протекать независящей (или слабо зави-
сящей) от температуры скоростью роста (мартенситная кинети-
ка) и со скоростью, экспоненциально зависящей от температуры
208
т
То№
12 3
' Тоу/а"
7
4
при энергии активации, близкой к энергии активации самодиффу-
зии (нормальная кинетика). Хотя мартенситное превращение мо-
жет идти как термически активируемый процесс, энергия актива-
ции этого превращения значительно меньше энергии активации
нормального превращения или энергии активации самодиффузии.
Например, для мартенситного превращения в стали была получе-
на энергия активации около 1 ккал/моль, тогда как энергия акти-
вации самодиффузии составляет более 60 ккал/моль.
Массивные превращения. Мартенситный механизм предполагает
отсутствие какого-либо перераспределения атомов, т. е. бездиффу-
зионный характер превращения в сложнокомпонентных сплавах
(в твердых растворах). Однако нор-
мальное превращение в сложноком-
понентных сплавах также может
идти без перераспределения компо-
нентов между фазами в ходе самого
превращения. Этот тип превраще-
ния называется массивным. Непо-
средственным результатом такого
Превращения является однофазное
состояние, причем в классических
случаях при нормальной кинетике
зерна имеют вид бесформенных
массивов, без какой-либо простой
линии границ. Отсюда название
превращения — массивное. Общим
для массивных превращений (рис.
7.22) является отсутствие каких-
либо перераспределений концентра-
ций компонентов. Если в ходе
превращения происходят диффузи-
онные процессы, кинетика услож-
няется и превращение тормозится. Массивное превращение может
происходить и при непрерывном охлаждении с достаточно большой
скоростью, предупреждающей заметное развитие диффузионных
процессов, и в изотермических условиях (в одно- и двухфазной
областях) при достаточно большом переохлаждении.
Превращения, не требующие перераспределения концентраций,
происходят в чистых компонентах, в химических соединениях сте-
хиометрического состава и в монотектоидных сплавах; однако
можно показать термодинамическую ( т. е. не связанную с затруд-
ненностью диффузии и т. д.) возможность бездиффузионного пре-
вращения твердых растворов.
Термодинамическое обоснование возможности
бездиффузионного образования новой ф а з ы долж-
но означать только одно: рост новой фазы, состав которой соот-
ветствует исходной (маточной) фазе, происходит с понижением
свободной энергии. При решении этого вопроса в принципе мож-
но не рассматривать процесс зарождения и не учитывать других
14—523 20$
А В
Рис. 7.22. Возможные случаи мас-
сивного превращения:
/ — полиморфное превращение чистого
металла; 2 — полиморфное превращение
твердого раствора V—астаб (например,
в Cu—Zn); 3 — полиморфное прев-
ращение твердого раствора “Y—амет
(например, аустенит -* мартенсит в
Fe—С); 4 — превращение твердого рас-
твора в интерметаллидную фазу (на-
пример, упорядочение a—(Zi в Cu—Au)
составляющих в выражении для свободной энергии сплава, кроме
объемной (или химической) энергии. Можно также считать несу-
щественным. является ли данная фаза окончательным продуктом
превращения, вполне равновесным, и существуют ли другие фа-
зовые состояния и пути перехода сплава в стабильное состояние.
Очевидно только, что если имеется
путь реакции, не требующий переме-
щения атомов на большие расстоя-
ния, то в условиях затрудненной
диффузии этот путь будет реализо-
ваться.
Рис. 7.23. Схемы диаграммы со-
стояния (а) и изменения сробод-
ной энергии фаз в функции соста-
ва с при температурах Л (б) и
Г2 (в)
На рис. 7.23,а приведена типич-
ная диаграмма равновесия системы,
в которой компонент А и твердые
растворы на его основе испытывают
аллотропическое превращение. При
постепенном охлаждении сплава от
высокой температуры из у-твердого
раствора образуется низкотемпера-
турная сс-фаза состава, отличного
от среднего состава сплава. По мере
понижения температуры увеличива-
ется объем сс-фазы, а ее состав при-
ближается к составу сплава.
Рассмотрим сплав состава с0.
При температуре сплав находит-
ся в состоянии у. Если температуру
быстро понизить до Т2, свободная
энергия системы определяется точ-
кой М на рис. 7.23,в (у-твердый рас-
твор состава с0 при температуре
Г2). В равновесном состоянии сплав
при данной температуре должен со-
стоять из a-фазы состава С\ и у-фа-
зы состава с2.
Превращение представим в виде
реакции
Поскольку скорость диффузионных процессов ограничена, диф-
ференциация компонентов может пройти лишь частично, например
В этом 'случае свободная энергия системы будет соответство-
вать точке Р. Но можно представить и полное отсутствие диффу-
зионных процессов: превращение у-фазы в сс-фазу того же соста-
ва (с0) приводит к понижению свободной энергии системы (от
точки М до точки N). Если это превращение идет с образованием
210
зародышей и нормальным ростом (без участия кооперативных пе-
ремещений атомов), то оно называется массивным и может быть
записано реакцией
Yr ~.
• Со СО*
Как видно из рис. 7.23, такое превращение возможно лишь
в определенной температурно-концентрационной области.
При температуре Т2 в сплаве состава с' однофазное состоя-
ние у тоже нестабильно. Однако переход yfQ -> af,Q должен сопро-
вождаться возрастанием свободной энергии (от точки R до S
см. рис. 7.23,в), поэтому превращение у-фазы в этом сплаве при
данной температуре возможно только диффузионное. Очевидно,
так же происходит превращение при температуре Т2 и в других
сплавах системы правее точки Только в сплавах левее точки Сг
однофазное состояние а при температуре Т2 более стабильно, чем
однофазное состояние у, хотя только при составах левее точки Ct
оно является 'вполне стабильным (в остальных сплавах ci—Ci оно
метастабильно).
Состав сплава c-L имеет простой физический смысл: при данной
температуре значения свободной энергии фаз и yf одинаковы.
На схеме диаграммы состояния (см. рис. 7.23,а) пунктирной ли-
нией показано изменение состава cL с температурой. В сплавах
данной области концентраций массивное превращение у-фазы уСо -►
-► а возможно при температуре ниже линии Ро и обратное прев-
ращение a-фазы ПРИ нагреве выше линии Г'о.
Хотя первоначальное значение термина «массивный» относи-
лось к микроструктуре, во многих случаях структура после пре-
вращения массивного характера может не иметь характерных
черт. Это объясняется процессами полигонизации и рекристалли-
зации, меняющими вид микроструктуры, а также развитием диф-
фузионных процессов в ходе превращения. Развитие диффузион-
ных процессов может привести к остановке массивного превраще-
ния. у-Фаза в сплаве cQ при температуре Т2 может полностью
перейти в a-фазу. Однако если в ходе превращения успевает про-
изойти перераспределение концентраций и состав непревращенной
части у-фазы приблизится к сг-, то кинетика превращения резко
изменит свой характер, поскольку дальнейшее превращение воз-
можно только с участием диффузионного перераспределения ком-
понентов между а- и у-фазами.
§ 7.4. ТИПЫ ДИФФУЗИОННЫХ ПРЕВРАЩЕНИЙ
В двойных и многокомпонентных сплавах рост центров новой
фазы, состав которой отличается от состава исходной фазы, мо-
жет ограничиваться скоростью подвода атомов «избыточного»
компонента к растущему кристаллу, т. е. скоростью диффузии.
14* 211
Можно выделить два типа превращений, контролируемых диф-
фузией: непрерывное и прерывистое (или ячеистое) превращения.
В первом случае в ходе процесса сосуществуют две фазы: исход-
ная, «состав которой непрерывно (во времени и по объему) изме-
няется, и новая фаза. На рис. 7.24,а показано распределение кон-
центраций у границы частицы растущей фазы. Так могут проис-
ходить процессы полиморфного превращения (например, у—
превращение в стали) и распада пересыщенного твердого раство-
ра (например, AI—Si). Второй тип превращений, связанный с из-
Рис. 7.24. Схемы распределения концентрации у границы
частицы, растущей при непрерывном выделении (а) и
при образовании колонии ячеистого (или прерывистого)
распада (б)
менением состава фаз приведен на рис. 7.24,6. Так идет, например,
образование перлита в эвтектоидной стали. Подобный механизм
может осуществляться при распаде пересыщенных твердых рас-
творов (например, в сплавах А1—Ag или Ni—Au). В обоих слу-
чаях конечное (стабильное) состояние двухфазное, однако в ходе
прерывистого превращения (в изотермических условиях) сосу-
ществуют три фазы: исходный (закаленный) и обедненный (рав-
новесный) твердые растворы и новая фаза выделения в составе
колонии. Эти три фазы могут иметь различные кристаллические
структуры (как, например, при эвтектоидном распаде аустенита
в сплавах Fe—С на феррит и цементит), но могут быть и все изо-
морфными (например, распад пересыщенного твердого раствора
в сплавах Ni—Au или в промышленном сплаве 36НХТЮ).
Условие процесса выделения состоит в том, что сплав, представ-
ляющий собой твердый раствор на основе компонента или проме-
212
жуточной фазы, переносится в область температур, где этот рас-
твор оказывается пересыщенным. Это условие возможно при са-
мых разных типах диаграмм состояния (рис. 7.25). Почти всегда
процесс выделения при проведении старения как операции терми-
ческой обработки идет в изотермических условиях. Этому пред-
шествует операция закалки, которая состоит в резком охлаждении
сплава от температур устойчивости однофазного состояния. Тем-
пературы нагрева для закалки (/i) и старения (t2) показаны на
схемах рис. 7.25. Изотермические условия удобны и в организа-
Рис. 7.25. Схемы диаграмм равновесия для сплавов, в ко-
торых происходят процессы выделения:
а — распад пересыщенного твердого раствора при старении; б — су-
жение области гомогенности промежуточной фазы (например, упо-
рядоченного твердого раствора) при понижении температуры; в —
полиморфное превращение и распад; г — полиморфное превращение
твердого раствора в сплаве доэвтектоидного состава; д — упорядо-
чение твердого раствора в сплаве нестехиометрического состава
(«гетерогенное» упорядочение); е — расслоение твердого раствора
213
ции технологии обработки и .в теоретическом анализе превраще-
ния, хотя в действительности процессы выделения могут частично
идти при закалке и может применяться неизотермическое (комби-
нированное или ступенчатое) старение. Можно выделить четыре
последовательные стадии выделения: 1) зарождение частиц новой
фазы; 2) рост частиц до установления термодинамического равно-
весия компонентов на границе раздела частицы новой фазы с ма-
точной фазой; 3) рост частиц в условиях равновесия на границе
раздела фаз, когда скорость процесса определяется диффузией —
скоростью подвода (или отвода) вещества; 4) коалесценция
частиц.
Непрерывное выделение. При распаде пересыщенного твердого
раствора (см. рис. 7.25,а и б), так же ка.к и при полиморфном
превращении твердого раствора (см. рис. 7.25,в), выделяющаяся
фаза отличается от маточной по составу и по структуре. Поэтому
диффузионное перераспределение атомов компонентов твердого
раствора должно сочетаться с перестройкой кристаллической ре-
шетки. Такое превращение в бинарной системе характеризуется
шестью кинетическими коэффициентами: двумя определяющими
подвижность первого компонента в исходной и новой фазах; дву-
мя аналогичными для атомов второго компонента; двумя опреде-
ляющими скорости перехода атомов каждого из компонентов че-
рез границу раздела фаз.
При теоретическом рассмотрении обычно делают два допуще-
ния: 1) число атомов растворенного вещества п мало по сравне-
нию с полным числом атомов N, т. е. 2) подвижности
атомов растворителя и растворенного вещества существенно раз-
личаются, так что можно рассматривать диффундирующие атомы
растворенного компонента как движущиеся в решетке неподвиж-
ных атомов растворителя. Это полностью справедливо для твер-
дых растворов внедрения. Далее можно принять, что кинетический
коэффициент, характеризующий скорость перехода атомов раство-
ренного вещества через границу раздела фаз, также много больше
коэффициента, который определяет скорость перестройки решетки
растворителя. Наконец, можно не учитывать диффузию внутри
частицы новой фазы. Таким образом, из названных шести кине-
тических коэффициентов остаются только два: коэффициент диф-
фузии атомов растворенного вещества в маточной фазе (Ом) и
коэффициент, определяющий перестройку решетки у границы раз-
дела фаз (хо) *. Задачу можно еще упростить, если принять, что
только один из этих процессов — переход атомов основного ком-
понента через границу раздела или подвод (отвод) атомов раство-
ренного вещества к растущему центру — ограничивает скорость
роста центров. Однако полный теоретический анализ должен учи-
тывать оба фактора и определять те крайние случаи (или стадии
роста), когда процесс контролируется одним из этих факторов.
Любов Б. я. Кинетическая теория фазовых превращений. М., «Металлур-
гия», 1969, с. 73.
214
На начальных -стадиях роста частиц можно не учитывать их
диффузионное взаимодействие, т. е. считать, что со-став маточной
фазы вдали от растущего центра не изменяется. Когда частица
мала, состав маточной фазы у ее поверхности мало отличается от
исходного, следовательно, градиенты концентрации растворенного
вещества малы и рост ча-стицы не должен лимитироваться достав-
кой атомов (или их отводом) выделяющегося компонента из вну-
тренних областей маточной фазы.
Скорость перестройки решетки, т. е. перехода атомов раство-
рителя А через границу раздела фаз, пропорциональна разности хи-
мических потенциалов этих атомов у границы раздела фаз со сто-
роны маточной фазы <рмл(с”ов) и со стороны выделения новой фазы
?Вд(Св°В^ Расчет Дает при /-*0 для скорости увеличения радиуса R
сферического центра
.^=х (ср“н_с"°в). (7.23)
Здесь — равновесная концентрация в маточной фазе; свов — со-
став маточной фазы у поверхности центра, который при /->0 можно
принять равным исходной концентрации с”сх. Значение х0 опреде-
ляется конкретными характеристиками атомного механизма про-
цесса и включает работу образования активированного комплекса
при присоединении нового атома к растущей частице q, имеющей
порядок величины энергии активации самодиффузии атомов основ-
ного компонента системы (см. § 7.3).
По мере роста частицы и приближения составов фаз у поверх
ности раздела к равновесным (с^авн и ^авн) должна уменьшаться
разность химических потенциалов атомов по обе стороны границы
и возрастать роль диффузии. Подвод атомов избыточного компо-
нента к границе, обусловленный градиентом концентрации в ок-
рестности частицы, вызывает нарушение локальных условий равно-
весия и соответствующее увеличение размера частицы.
Скорость роста контролируется диффузией, если xoR^^m:
</R/d/=(Z)M/R) [(С™в - си“)/(<,ов - <™)]. (7.24)
Таким образом,
R^x0(^BH-cy)Z при >0; (7.25)
Г рравн_____ исх
R= V t при (7.26)
Этот анализ неприменим в области R, близких к размеру кри-
тического зародыша (RKp), поскольку не учитывалось влияние по-
верхностного натяжения а на концентрацию растворенного компо-
нента у поверхности частицы. Л. Н. Александров и Б. Я. Любов
рассчитали концентрации растворенного элемента в. маточной фазе
215
и скорость роста частицы в зависимости от ее размера для выде-
ления доэвтектоидного феррита из аустенита в сплаве Fe—С. Как
видно из рис. 7.26,6, при малых размерах частицы состав матри-
цы у ее поверхности почти не изменяется; при этом скорость ее
роста определяется как скорость перестройки решетки (кривая 1
на рис. 7.26,в). При больших размерах частицы скорость ее роста
определяется диффузией углерода в аустените (кривая 2). Кри-
вая 3 построена с учетом обоих факторов.
Рис. 7.26. Изменение кон-
центрации углерода у по-
верхности растущего
ферритного центра со
стороны аустенита (а);
зависимость (с"ов) от раз-
мера центра R (б) и за-
висимость скорости роста
центра и его размера при
разных условиях (в):
1 — при определяющей роли
перестройки решетки; 2 —
при определяющей роли
диффузии углерода; 3 — при
одновременном учете обоих
факторов (Л. И. Александ-
ров, Б. Я. Любов)
Развитие процесса выделения приводит к изменению состава
маточной фазы — твердого раствора между растущими центрами.
До сих пор рассматривали рост частиц при выделении избы-
точного компонента непосредственно из твердого раствора. Однако
структурные изменения при распаде твердых растворов на этом не
заканчиваются. Когда частицы имеют достаточно большой размер,
а пересыщение становится очень малым, структура изменяется бла-
годаря процессу коалесценции — росту более крупных частиц за
счет растворения мелких. Анализ этой последней стадии структур-
ных изменений при распаде твердых растворов имеет важное
практическое значение. Уже при термической обработке в тепло-
устойчивых сплавах должен в известных пределах идти процесс
коалесценции, стабилизирующий структуру; явление коалесценции
следует рассматривать при анализе структурных изменений в про-
цессе службы жаропрочных и теплоустойчивых сплавов. Анализ
процесса коалесценции проведен И. М. Лифшицем и В. В. Слезо-
вым (см. гл. 9, 10).
216
В основе всех рассмотренных выше случаев анализа процесса
выделения взяты уравнения нормальной химической диффузии,
в которых поток атомов принимается пропорциональным гради-
енту концентрации. Однако в реальных системах должны возни-
кать еще потоки атомов под действием градиента напряжений,
вызванных дислокациями и другими дефектами матричного кри-
сталла или дефектами, развивающимися в процессе самого выде-
ления (различия в удельных объемах фаз, изменение формы пре-
вращенного объема и особенности строения поверхности границы
раздела фаз). Поскольку само по
себе образование раствора связано с
с изменением объема, градиент кон-
центрации твердого раствора должен по6
давать в свою очередь градиент упру-
гих напряжений. Таким образом, про-
исходит наложение структурных и с0
концентрационных напряжений. Среди
эффектов взаимодействия примесей и
выделений с дислокациями широко ^поб
известны как в отношении теории, так
и в экспериментальных исследованиях сн.ф
два эффекта — образование атмосфер
Коттрелла возле дислокаций и заро- распределение кон-
ждение новых фаз на дислокациях. цеНТрации растворенного ком-
Первый из этих эффектов относит- понента у границы растущего
ся в большей степени к непересыщен- кристалла (/) в случае опреде-
ным растворам в связи со свойством ляющей роли нормальной диф-
«насыщения» атмосфер, второй пре- ’ауми доказаны направления
жде всего относится к некогерентным нормальных диффузионных по-
выделениям, так как стимулом выде- токов; и* —сложная безраз-
ления на дислокациях считается ре- мерная текущая координата,
лаксация напряжении от дислокации пластины (Б. Я. Любов,
благодаря потере когерентности. л. И. Александров)
Однако имеются основания считать,
что атмосферы возле дислокаций в пересыщенном твердом раство-
ре могут быть устойчивыми центрами его расслоения, а возле дис-
локаций могут преимущественно возникать и когерентные части-
цы, и частицы изоморфных матрице фаз.
Кинетика роста выделения с учетом анизотропии суммарных
потоков атомов рассмотрена Л. А. Александровым и Б. Я. Лю-
бовым.
Для толщины пластинчатого выделения в условиях преобла-
дающего влияния дрейфа получена зависимость от времени
(7.27)
При преобладающей роли нормальной диффузии
У (0 V
(7.28)
217
Очевидно, что при низкой температуре, когда релаксация струк-
турных напряжений идет медленно, их .роль в кинетике процесса
выделения может быть определяющей. Они придают реакции вы-
деления автокаталитический характер, а нормальная диффузия
может оказаться фактором, тормозящим процесс (рис. 7.27).
При спинодальном распаде диффузионные потоки в отличие от
рассмотренных процессов роста частиц направлены в сторону соз-
дания и усиления градиентов концентрации.
Рис. 7.28. Ячеистый (прерывистый) распад в сплаве Ni—2 мае.% Be (старение
при 500°С, 10 мин):
а — микроструктура, Х200; б — схема микроструктуры при большом увеличении
Прерывистый распад (образование ячеек). Колонии или ячейки,
содержащие две фазы (стабильные или метастабильные), зарож-
даются на границах зерен или на некогерентных границах двойни-
ков и растут внутрь одного из примыкающих зерен (рис. 7.28,а).
Фронт реакции обычно имеет приблизительно сферическую фор-
му, частицы одной из фаз обычно имеют пластинчатую (или во-
локнистую) форму и регулярно расположены в матрице. В неко-
торых подробно исследованных системах было установлено, что
кристаллографическая ориентировка всех этих частиц в составе
одной колонии (в ячейке) одна и та же, так что можно их рассма-
тривать как разветвленный монокристалл. Кристаллографическая
ориентировка матрицы в ячейке в случае распада пересыщенного
раствора (когда одна из образующихся фаз обязательно изо-
морфна исходной фазе) совпадает (или очень близка) с ориенти-
ровкой соседнего матричного зерна, от которого начинается рост
ячейки (рис. 7.28,6). Последнее обстоятельство позволяет предста-
вить превращение как движение большеугловой границы зерен —
фронта реакции, подобно движению границ при рекристаллизации.
В процессе роста ячейки расстояние между частицами сохраняет-
ся примерно постоянным; при условии сохранения сферической
формы фронта реакции это означает, что по мере роста ячейки
218
должно происходить зарождение новых частиц p-фазы или ветвле-
ние ,р-фазы внутри колоний. Следует отметить преимущественное
развитие ячеистого распада пересыщенных твердых растворов
именно при низких температурах. Не касаясь вопроса о механиз-
мах зарождения -центров новой фазы .внутри зерен и ячеек рас-
пада по границам, можно объяснить преимущественное развитие
ячеек наличием коротких и облегченных путей диффузии при диф-
ференциации компонентов пересыщенного твердого раствора вдоль
границы зерен — по фронту реакции. Межпластиночные расстоя-
ния при изотермических условиях реакции постоянны и уменьша-
ются с понижением температуры. Ячеистый распад с заметной
скоростью идет при очень низкой температуре: ,в системе РЬ—Sn
при —78°С, в системе тугоплавких металлов, таких как Ni—Au,—
при комнатной температуре. Вопрос о механизме ячеистого рас-
пада еще остается предметом теоретических и экспериментальных
исследований.
Скорость роста ячеек может определяться диффузией в объеме
исходной фазы Добыли граничной диффузией, т. е. диффузией вдоль
фронта реакции по границе растущей ячейки 7)гр. В обоих случаях
пути диффузии тем меньше, чем меньше расстояние между пласти-
нами в колонии d. Однако измельчение структуры должно приво-
дить к увеличению поверхностной энергии ЕПов и, 'Следовательно,
к уменьшению термодинамического стимула превращения. Конку-
ренция между фактором сокращения путей диффузии при умень-
шении межпластиночного расстояния и фактором уменьшения тер-
модинамического стимула АФ^=—АЕ(у)4-Епов устанавливает такое
значение d, которое обеспечивает максимальную скорость движе-
ния фронта реакции.
Зная коэффициент поверхностного натяжения для фаз, составляющих коло-
нию, можно определить межпластиночное расстояние d0, при котором термоди-
намический стимул, а следовательно, и скорость реакции, становятся равными
нулю. По Зинеру, который впервые провел такой анализ для эвтектоидного рас-
пада, межпластиночное расстояние при максимальной скорости роста колонии
•^макс=2б/0, если определяющей является объемная диффузия, и ^макс=1,5^о,
«ели определяющей является диффузия вдоль границы. При увеличении пере-
охлаждения растет выигрыш объемной свободной энергии и должно уменьшаться
и, следовательно, tZMaKc, т. е. должно происходить измельчение внутренней
структуры ячеек. По Тарнбаллу, для прерывистого распада пересыщенного твер-
дого раствора скорость роста ячеек в случае действия объемной диффузии
V—[(Смех—Сравн) /Сисх/] (2/)об/^) • (7.29)
Здесь сИСх—состав исходной (маточной) фазы; сравн— равновесный состав
твердого раствора *.
При определяющем действии граничной диффузии имеем
V=l[(cMCx—Сравн) Дисх] (7)rpZ/t/2) (7.30)
(Z—толщина границы раздела ячейка—маточная фаза; обычно в диффузион-
ных расчетах она принимается равной 5-Ю-8 см**).
* Непрерывный распад твердого раствора внутри зерен (перед фронтом
ячеек) снижает сЖСх- Это уменьшает скорость ячеистого распада.
** Из диффузионных опытов определяют величину Drpl.
219
Для решения вопроса о действующих путях диффузии при росте ячеек кро-
ме оценки d/dQ измеряли скорость движения фронта реакции v и температурную
зависимость этой скорости (для сопоставления энергии активации роста ячеек
и значений энергии активации граничной и объемной диффузии). Определяли
также значения скорости роста и межпластиночные расстояния. Согласно су-
ществующим моделям, если процесс контролируется объемной диффузией,
если диффузией по границе,
vd2.=const,
vd3=const.
(7-31)
(7.32) *
Исследовали *2 эвтектоидный распад в системе Cu—In: превращение £—>
—^a-j-6-Cu9In4. Эвтектоидный распад в исходном монокристальном образце
проводили при нагреве ограниченной области образца токами высокой частоты
Рис. 7.29. Зависимость скорости
роста ячеек в сплаве А1—28 ат.%
Zn от температуры:
точки — экспериментальные определе-
ния; 1, 2 —расчетные данные для объ-
емной диффузии (Qo6=24,5 ккал/моль,
Do=0,13 • 10-8 см2/с) без учета (/) и
с учетом (2) непрерывного распада
в матрице; 3 — теоретический расчет
для механизма граничной диффузии
(Qrp=9 ккал/моль, £>о=1О-4 см2/с) с
учетом распада в матрице; 4 — темпе-
ратурная граница однофазной области
с заданным градиентом температуры, что
обеспечивало направленный рост колоний и
отсутствие огрубления структуры за фрон-
том реакции распада. Малый объемный эф-
фект превращений (0,6%, сжатие) и ис-
пользование сравнительно узкого темпера-
турного интервала превращения (30°С) по-
зволяли не учитывать влияния температуры
и фактора кинетического переохлаждения.
При малых скоростях роста (<2,5 мм/ч)
действует закон vJ1>80±0’2=const, что свиде-
тельствует о контролирующем действии
объемной диффузии; при увеличении скоро-
сти роста (от 2,5 до 80 мм/ч) показатель
степени постепенно растет до 3,35±0,25, что
соответствует действию граничной диффу-
зии. При исследовании образования перлита
в эвтектоидной стали получили значение
показателя степени в уравнении vdP=const
в пределах р=2,3-г-2,7, что не позволило ре-
шить вопрос о действующих путях диффу-
зии *3.
При исследовании ячеистого рас-
пада пересыщенных твердых рас-
творов в сплавах Ni—Ti и Со—Та
сделаны выводы о действии меха-
низма объемной диффузии, хотя
большинство исследований кинети-
ки говорит о контролирующем дей-
ствии граничной диффузии. При
анализе кинетики распада твердых
растворов важно учитывать влияние
возможного непрерывного распада
перед фронтом ячеистого распада.Поэтому особенно интересно ис-
следование роста ячеек начиная с их зарождения непосредственно
в просвечивающем электронном микроскопе, поскольку при этом
возможен непосредственный контроль состояния маточной фазы пе-
ред фронтом распада. С помощью высоковольтного электронного
* На величину показателя степени в уравнениях (7.31) и (7.32) могут
влиять температурная зависимость коэффициента диффузии и другие факторы.
*2 Mellor В., Chadwick G. «Metal. Science», 1974, v. 8, p. 65—72.
*3 Bolling G., Richman R. «Met. Trans.», 1970, v. 1, p. 2095.
220
микроскопа (106 В) в процессе старения сплава А1 — 28 ат. % Zn
непосредственно в электронном микроскопе при разных температу-
рах измеряли межпластиночное расстояние и скорость движения
фронта ячеек*. Сопоставление рассчитанной температурной зави-
симости скорости роста с экспериментальными данными (рис. 7.29)
приводит к выводу, что ячеистый распад контролируется гранич-
ной диффузией.
Задачи к гл. 7
Задача 7.1. Показать схематически изменение объема V, электросопротивле-
ния 7? и температурного коэффициента электросопротивления а* в зависимости
от температуры в области полиморфного перехода в однокомпонентной системе.
Задача 7.2. а. Определить разницу в значениях периода и степень несоответ-
ствия решеток Fe—Cr-матрицы и когерентных частиц выделений фазы ЕезА1 по
картине дифракционного контраста типа муара в электронно-микроскопическом
Изображении фольги: расстояние между полосами в картине муара 150 А, дей-
ствующее отражение g (200) о. ц. к. решетки матрицы, период решетки матрицы
о
> принять 2,87 А.
б. Оценить энергию упругой деформации когерентной частицы Fe3Al
й Fe—Сг-матрице, если форму частицы аппроксимировать сферой диаметром
1 мкм, упругие характеристики матрицы и включения принять одинаковыми.
в. Определить расстояние между дислокациями при нарушении когерентно-
сти на границе частиц Fe3Al и Fe—Сг-матрицы.
Задача 7.3. Определить степень несоответствия решеток а-мартенсита (а=
= 2,869 А) мартенситно-стареющей стали и фазы выделения Ni3Mo вдоль на-
правлений матрицы, параллельных направлениям [100], [010] и [001] фазы вы-
деления с ромбической решеткой.
Задача 7.4. Объяснить особенности формы частиц выделений, используя дан-
ные об ориентационных соотношениях, степени несоответствия решеток матрицы
и фазы выделения в разных направлениях для фаз V4C3 (г. ц. к. решетка, а =
о о
=4,16 А), М02С (гекс. п. у. решетка, а=3,00; с=4,72 А) в сплавах на основе
о о
a-Fe (о. ц. к., а = 2,86 А) и фазы y'-Ni3Ti (г. ц. к., а=3,60 А) в аустенитном
°
Fe-Ni-сплаве (г. ц. к., а=3,58 А). Данные об ориентационных соотношениях
взять из табл. 7.1.
* Butler Е., Ramaswamy V., Swann Р. «Acta Metalurgica», 1973, v. 21, p. 517.
ГЛАВА 8
АЛЛОТРОПИЧЕСКИЕ ПРЕВРАЩЕНИЯ
В МЕТАЛЛАХ И СПЛАВАХ
Аллотропические превращения состоят в изменении кристалли-
ческой структуры элементов, твердых растворов или химических
соединений (промежуточных 'фаз) при изменении внешних пара-
метров равновесия — температуры или давления * (рис. 8.1). Для
металлов эти изменения состоят в изменении координации и ком-
пактности упаковки атомов (например, г. ц. к.^о. ц. к. в Fe,
о. ц. к.^гекс. п. у. в Ti), в изменении последовательности упаков-
ки плотноупакованных атомных слоев [например, при охлаждении
Со трехслойная структура АВСА ... (г. ц. к.) переходит в двух-
слойную структуру АВА ... (гекс. п. у.)] или в резком изменении
межатомных расстояний при сохранении координации атомов в ре-
шетке (например, переход Се при повышении давления из г. ц. к.
с периодом а =5,16 А ,в г. ц. к. с периодом а=4,84 А).
Фазовый переход в Се не меняет типа атомной структуры, но
сопровождается очень большим изменением объема (16%). Этот
переход рассматривается как проявление изменений в электрон-
ной структуре: действием давления (7 кбар при комнатной тем-
пературе) 45-электрон «вжимается» в 5й-состояние.
Перечисленные -случаи аллотропических превращений относят-
ся к взаимным переходам типичных металлических структур, де-
тально рассмотренных в гл. 1. Имея в виду проблемы общей тео-
рии фазовых превращений, а также .практическое значение ряда
систем, назовем еще несколько примеров полиморфных превра-
щений.
С изменением состояния электронов, осуществляющих меж-
атомную связь, обусловлен аллотропический переход в Sn: при
понижении температуры металлическая модификация р—Sn
(к. ч.=6, см. рис. 2.4) переходит в модификацию а—Sn со струк-
турой типа алмаза (к. ч.=4, см. рис. 2.2,а), характеризующуюся
ковалентными связями. Удельный объем при переходе р—а уве-
* Термин «аллотропия» чаще применяется для обозначения изменений атом-
но-кристаллической структуры простых веществ, термин «полиморфизм» имеет
более широкое значение. Основа металлических материалов (сплавов), как пра-
вило, представляет собой фазу со структурой, характерной для металлических
элементов. Поэтому анализ превращений в сплавах основан на рассмотрении
аллотропических превращений в металлах.
222
личивается (+27%!), поэтому понятно, что повышение давления
резко понижает температуру перехода (см, рис. 8.1,в). Важно от-
метить, что химические аналоги олова Si и Ge (а также некоторые
полупроводниковые соединения типа AinBv и AnBVI) при высоком
давлении приобретают структуру металлического (белого) олова.
Практически применяют, как правило, не чистые металлы,
а сплавы (сталь, углеродсодержащие и безуглеродистые сплавы
на основе железа, сложнокомпонентные сплавы на основе титана
-40 0 40 30 Ргкбар
8
Рис. 8.1. Фазовые диаграммы темпе-
ратура — давление:
а — железо (по данным разных авторов);
б — титан (Bundy, 1966); в — олово (Так-
эдзири, Нарито, 1969); г — уран (Klement,
Yagoranan, Kennedy, 1963); д — плутоний
(Liptai, Friddle, 1966)
223
и т. д.), целесообразно рассматривать превращения .в этих метал-
лах вместе с теми особенностями, которые накладываются легиро-
ванием. Прежде всего следует отметить изменения механизма пре-
вращения. Например, в чистом железе у—>а-превращение только
в особых условиях (очень большая скорость охлаждения) идет
мартенситным путем, но при достаточном содержании Ni превра-
щение может идти только мартенситным путем из-за того, что
температура превращения оказывается очень низкой. В некоторых
сплавах на основе Ti высокотемпературная p-фаза (о. ц. к. струк-
тура) при охлаждении превращается не в a-фазу (гекс. п. у. струк-
тура), а в совершенно новую модификацию — со (гексагональная,
4?/6Z=0,613, 3 атом/ячейка).
Последующие рентгеноструктурные исследования (1963 г.) Ti
и Zr при высоком давлении показали, что со-модификация являет-
ся фазой высокого давления в этих металлах: при давлении более
60 кбар (см. рис. 8.1,6) идет превращение а—но, причем со-фаза
сохраняется и после снятия высокого давления при комнатной
температуре.
Подобное положение имеет место и для Fe: гексагональная
компактная упаковка атомов Fe, .ранее известная в структуре
8-нитрида (и метастабильного 8-карбида), найдена при давлении
больше 130 кбар как новая модификация чистого железа — фаза
высокого давления (s-Fe, см. рис. 8.1,а). Эти примеры показыва-
ют, насколько широкое значение могут иметь фазовые Р—Т- (тем-
пература— давление)-диаграммы в объяснении появления разных
фазовых состояний в технических материалах и при использова-
нии современных технологий обработки этих материалов.
Образование новых кристаллических модификаций (метаста-
бильных при нормальных условиях) наблюдали после «закалки»
из жидкой или паровой фазы (т. е. при сверхбыстром охлаждении
жидких расплавов или при конденсации из пара при низкой тем-
пературе подложки). В частности, в технических сплавах железа
с углеродом и кремнием (чугун) при быстром затвердевании так-
же обнаруживается гекс. п. у. 8-фаза (см. гл. 6). Современные
технологические способы обработки, такие, как деформация взры-
вом или обработка поверхности лучом лазера, могут создать усло-
вия для образования метастабильных состояний, которые ранее
получали только в лабораторных исследованиях. Следует также
иметь в виду, что некоторые из метастабильных модификаций мо-
гут быть промежуточными состояниями при образовании стабиль-
ных фаз в процессе самого превращения. Например, имеется мне-
ние, что образование гекс. п. у. модификации железа в некоторых
сплавах является промежуточным • этапом у—>а-превращения.
Среди полиморфных превращений в сплавах надо выделить
очень специфические по характеру и важные в развитии новых
материалов превращения p-фаз Ю^-Розери (AuCd, NiTi и др.).
Полиморфное превращение в' железе и стали, открытое
Д. К. Черновым более 100 лет назад, исследовано особенно под-
робно. В настоящее время имеются надежные количественные дан-
224
ные о кинетике полиморфного у—>а- (и а—>у) -превращения,
о морфологии, субструктуре и атомной структуре продуктов пре-
вращения как в железе и разбавленных растворах на основе же-
леза, так ;и во многих (сложных по химическому составу сплавах
железа. Термин «мартенситное превращение» происходит от на-
званной в честь известного металлурга Мартенса основной струк-
турной составляющей закаленной стали. Сейчас можно считать
общепринятым, что мартенситный механизм аллотропических пре-
вращений является самым характерным не только для металлов
и сплавов, но и для минералов и органических кристаллов.
§ 8.1. АЛЛОТРОПИЧЕСКОЕ у—^-ПРЕВРАЩЕНИЕ ЖЕЛЕЗА
И ТВЕРДЫХ РАСТВОРОВ НА ОСНОВЕ ЖЕЛЕЗА
Рис. 8.2. Зависимость разности свободных
энергийлДба~7 = — Ga для Fe от темпе-
ратуры (по данным Б. М. Могутнова, И. А.
Томилина и Л. А. Шварцмана)
в a-состоянии: структура массивного
В области температур ниже 1183 К (910°С) свободная энергия
железа со структурой у выше, чем свободная энергия a-Fe, y-Fe
нестабильно и должно переходить в состояние со структурой а.
С помощью измерений теплоемкости а- и y-Fe при различных тем-
пературах вычислены значения разности ДО=С7 — Ga (рис. 8.2),
которая собственно является термодинамическим стимулом пре-
вращения. Объемный эффект превращения y-Fe в a-Fe порядка 1%
(уменьшение компактности решетки в о. н. к. модификации в зна-
чительной степени компенсируется уменьшением атомного радиу-
са, с повышением температуры разница удельных атомных объе-
мов у- и a-фаз уменьшается) *. Большая скорость уч^а-превра-
щений в чистом железе за-
трудняет изучение его кине-
тики. Уже небольшое содер-
жание примесей (например,
0,01—0,001 мае. % углерода)
может существенно изменить
кинетику превращения. По-
этому необходимо при рас-
смотрении эксперименталь-
ных данных о кинетике ал-
лотропического превраще-
ния в железе учитывать со-
держание в нем примесей.
При исследовании желе-
за и сплавов на его основе
отмечались различия микро-
структуры, связанные с осо-
бенностью механизма у—>а-
превращения в различных
условиях. Обычно отмеча-
ют два типа микроструктуры
феррита, которая характеризуется более или менее извилистыми
границами, вплоть до правильной полиэдрической формы зерен,
* См. задачу 8.1.
15—523
225
соответствующей минимальной поверхностной (эернограничной)
энергии, границы феррита могут пересекать границы исходных зе-
рен аустенита, структура мартенсита отличается еще большим раз-
нообразием морфологии и субструктуры кристаллов. Эту смену
структур можно наблюдать при увеличении скорости охлаждения.
Например, для железа, содержащего около 0,003 мае.% С, при
переходе от скорости охлаждения порядка 103 град/с к скорости
порядка 105 град/с (рис. 8.3) или при постоянной скорости охлаж-
Рис. 8.3. Микроструктуры железа после охлаждения со скоростью 103 град/с
(а) и 5-Ю4 град/с (б) (Д. А. Мирзаев, М. М. Штейнберг)
# 1000
600'—1——1—1—1—1—1—d
О 10 20 50 40 50 60 70 80
Скорость охлаждения, 10^град/с
а
Рис. 8.4. Температура начала превра-
щения в сплаве Fe — 0,0015 мас.% С
при различной скорости охлажде-
ния (а), зависимость температуры
массивного (/) и начала мартенсит-
ного (2) превращения (Мн) от содер-
жания углерода (б); зависимость
температуры Мн от содержания угле-
рода при значительном его содер-
жании (в)
800
700
600
500
^400
1*300
I 200
*- 100
0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6
Содержание углерода, мас.%>
226
дения с повышением содержания углерода (например, при ско-
рости охлаждения 104 град/с микроструктура существенно изме-
няется при переходе от сплава Fe — 0,003 мае. % С к сплаву Fe —
0,03 мае. % С). Эта смена микроструктур и, следовательно, меха-
низмов превращения обусловлена изменением степени переохлаж-
дения или температурных интервалов превращения. На рис. 8.4,а
показана зависимость температуры начала превращения для весь-
ма чистого железа (0,0015 мае. % С) от скорости охлаждения.
Увеличение скорости охлаждения понижает температуру массивно-
го превращения с нормальной кинетикой, но не меняет темпера-
туру начала мартенситного превращения* *. На рис. 8.4,6 показана
зависимость температуры массивного и мартенситного превраще-
ний от содержания углерода, на рис. 8.4,в приведены данные для
сплавов со сравнительно высоким содержанием углерода.
Кинетику изотермического превращения у—нх
удалось детально исследовать в сплавах железа с никелем и хро-
мом, которые сильно замедляют массивное превращение за счет
существенного понижения температурного интервала превращения
(и соответственно уменьшения линейной скорости роста центров
новой фазы). Легирование вносит новую особенность в превраще-
ние: превращение в сплавах при определенных условиях контро-
лируется скоростью диффузионного перераспределения компонен-
тов между исходной и новой фазами, т. е. оно может быть диффу-
зионным. Механизм собственно массивного превращения в сплаве
принципиально не должен отличаться от превращения в чистом
железе.
Магнитометрическим методом изучали *2 кинетику аллотропиче-
ского' превращения в сплаве железа с 0,025 С; 5,06 Сг; 2,18 Ni,
0,25 Si и 0,30 мае. % Мп (03Х5Н2). Аустенитизацию проводили
при 950°С.
С помощью металлографического высокотемпературного микро-
скопа микроструктуру мартенсита (рельеф) наблюдали на поверх-
ности шлифа в процессе изотермического превращения ниже475°С.
Кинетические кривые для температуры 450 и 400°С (рис. 8.5,а)
показывают характерные признаки мартенситной кинетики: отсут-
ствие инкубационного периода, большую скорость превращения,
незавершенность превращения (см. изотерму для 450°С), с пони-
жением температуры увеличение объема превращения (ср. изотер-
мы для 450 и 400°С).
В интервале температур 575—625°С превращение начинается
после инкубационного периода *3 с небольшой скоростью, макси-
* При еще более высокой скорости охлаждения (до 3,4-105 град/с) возмож-
ны дальнейшая смена механизма превращения и снижение температуры превра-
щения до 545°С и даже до 420°С (Морозов О. П., Мирзаев Д. А., Штейн-
берг М. М. «Физика металлов и металловедение», 1972, т. 36, № 4, с. 795—800).
*2 Коган Л. И., Энтин Р. И. «Физика металлов и металловедение», 1971,
т- 31, вып. 2, с. 379.
*3 Как и в анализе процесса кристаллизации (см. с. 158), это не истинный
инкубационный период, т. е. не время «созревания» зародыша, а время «ожида-
ния» первого зародыша.
15* 227
мальная скорость наступает в момент, когда объем превращения
составляет 50%. Такая кинетика характерна для нормального
аллотропического превращения с неупорядоченным ростом центров
новой фазы. Как будет видно из сопоставления с кинетикой пре-
вращения при более высоких температурах, превращение в интер-
вале 575—625°С следует считать бездиффузионным массивным
превращением (подобным превращению в однокомпонентной си-
Рис. 8.5. Превращения в системе Fe— (Cr+Ni):
а — кинетика изотермических превращений в сплаве 03X5H2; б — диаграмма темпера-
тура — время — объем превращения переохлажденного аустенита; в — температуры
Го, при которых свободные энергии а- и у-фаз одинакового состава равны
стеме у—>a-Fe). Первая особенность кинетических кривых для
температуры 650—670°С — наибольшая скорость превращения
в начальный момент, затем превращение замедляется и прекра-
щается при сохранении некоторого количества аустенита. После
закалки в воде в микроструктуре наблюдали полиэдрические зер-
228
на феррита, образовавшиеся в процессе изотермической выдержки,
и участки мартенсита, образовавшиеся в процессе последующего
резкого охлаждения в аустенитных участках.
Очевидно, эти температуры соответствуют двухфазному состоя-
нию сплава на диаграмме фазового равновесия. При этом следо-
вало бы ожидать диффузионного перераспределения компонентов
между исходной (у) и образующейся (а) фазами. О составе
остающейся у-фазы можно довольно определенно судить по темпе-
ратуре начала мартенситного превращения при охлаждении (точ-
ка Л4Н). Эксперимент не показал каких-либо изменений.
Таблица 8.1
Кинетика изотермического превращения сплава 03Х5Н2
а->а + у(у) (при нагреве из а-состояния)
Температура изотермичес- кой выдержки, °C Время выдержки Содержание, % Ма, °C
а-фазы 7-фазы
900 (однофазная Без выдержки в 100,0 475
область) двухфазной области
660 4 ч 95,0 5,0 —
680 2 ч 15 мин 94,3 5,7 —
700 2 ч 30 мин 91,0 9,0 —
720 5 мин 87,0 13,0 —
15 мин 82,0 18,0 390
40 мин 71,0 29,0 365
1 ч 30 мин 56,5 43,5 340
770 2 ч 1,5 98,5 465
Превращение а—>у ПРИ нагреве из ферритной области наблю-
дали при температуре 660°С и выше; при 770°С превращение идет
практически до конца: после 2 ч выдержки остается 2% a-фазы и
мартенситная точка образовавшегося аустенита почти совпадает
с точкой Мн после аустенитизации при 900°С. При более низкой
температуре в результате изотермической выдержки остается двух-
фазное состояние, а в процессе выдержки меняются количествен-
ное соотношение фаз и их химический состав, как показывают
определения точки Л1н для остающейся у-фазы (табл. 8.1).
Обращает на себя внимание существенное расхождение коли-
чественных соотношений фаз в результате изотермических превра-
щений у—>а (см. рис. 8.5,6) и а—>у (см. табл. 8.1). Например,
при 720°С превращение из а- в у-(нагрев) идет до образования
43,5% ^у-фазы после 1,5—2 ч выдержки; при topi же температуре
превращение из у-состояния в а (охлаждение) практически сов-
сем не происходит, в то же время при более низкой температуре
(660°С) из у-состояния превращение идет до образования 50%
a-фазы, а из a-состояния образуется всего 5% у-фазы. Существен-
ным является разница коэффициентов диффузии в а- и в у-со-
стояниях. При одной и той же температуре в более рыхлой о. ц. к.
решетке a-фазы диффузия идет много быстрее (т. е. быстрее под-
водится избыточный компонент, например Ni в системе Fe—Ni,
2^9
к растущему центру у-фазы, чем отводится тот же компонент при
росте центра a-фазы в у-матрице). Это важно в том случае, когда
превращение контролируется диффузией — перераспределением
компонентов между 'исходной и образующейся фазами. Однако
различие коэффициентов диффузии не может объяснить резуль-
таты изотермических превращений у—и а—>у ПРИ 660°С. Оче-
видно, нельзя считать, что превращение может контролироваться
только процессом диффузии. Существенным является соотношение
значений свободной энергии (химической) у- и a-фаз одинакового
состава (состава сплава) при температуре превращения (см.
рис. 7.23). На схеме участка диаграммы в квазибинарном сечении
Fe—(Cr + Ni) (см. рис. 8.5,в) эти составы в функции температуры
обозначены штрих-пунктиром (Т'о), проведенным внутри двухфаз-
ной области а+у так> чтобы объяснялись полученные кинетические
закономерности. При 660°С для сплава данного состава (7 мае. %
Cr + Ni) свободная энергия у-фазы больше, чем a-фазы, поэтому
при нагреве из a-состояния нельзя ожидать образования у-фазы
без перераспределения концентраций — превращение должно кон-
тролироваться процессом диффузии; напротив, при превращении
у—>а (при охлаждении), поскольку свободная энергия сплава
в состоянии a-фазы меньше свободной энергии сплава в состоянии
у-фаз, сплав может полностью перейти в a-состояние. Это будет
метастабильным состоянием (стабильно двухфазное состояние), и
оно может реализоваться (и сохраниться), если заторможены
диффузионные процессы. Обратная картина должна быть при тем-
пературе превращения 720°С — выше температуры равенства сво-
бодных энергий а- и у-фаз состава сплава. При 720°С свободная
энергия данного сплава в у-состоянии меньше свободной энергии
сплава в состоянии а, поэтому возможно превращение а—>у, без
перераспределения концентраций в процессе роста у-центров. Од-
нако при такой высокой температуре в процессе изотермической
выдержки происходит перераспределение примесей, которое при-
водит к уменьшению содержания Cr + Ni в a-фазе. Когда состав
a-фазы приблизится к концентрации, соответствующей точке Т'о
при данной температуре, дальнейшее превращение ее в у-фазу
простой перестройкой решетки (как массивное превращение без
перераспределения концентрации) становится невозможным. Кон-
тролируемое диффузией превращение, по-видимому, идет с замет-
ной скоростью при 720°С (а—нх+у); фазовый состав сплава
0Х5Н2 после выдержки 1,5 ч при 720°С (нагрев из а-состояния)
взят как близкий к .равновесному при построении схемы участка
диаграммы, приведенной на рис. 8.5,в. При охлаждении из у-со-
стояния превращение у—>а+у практически совсем не происходит,
так как диффузионные процессы в г. ц. к. решетке у-фазы слишком
медленны, а массивное превращение невозможно. Остановка пре-
вращения у—>а+у при изотермической выдержке 670—660°С пос-
ле образования некоторого количества a-фазы, если эта темпера-
тура ниже Т'о, .может быть также связана с некоторым перерас-
пределением компонентов и приближением состава у-фазы
230
к концентрации, соответствующей точке Т'о. При этом массивное
бездиффузионное превращение становится 'невозможным, а диффу-
зионное иде1 очень медленно при такой температуре.
Как у;ке отмечалось, мартенситная точка для сплава 03Х5Н2
(0,03 мае. % С) находится около 475°С. Понижение содержания
углерода до 0,034 мае. % привело к повышению точки Л4Н до
640—650°С. Высокое положение точки Мн позволило наблюдать
наложение мартенситного и нормального превращений: при 620°С
после выдержки 30 мин практически завершается мартенситная
реакция — образование мартенситного рельефа. При последующем
охлаждении оставшийся аустенит претерпевает мартенситное пре-
вращение. Если продолжительность изотермической выдержки
увеличить до 6 ч, этот аустенит нормальным образом превращал-
ся в феррит (после охлаждения наблюдали кристаллы мартен-
сита и полиэдрические зерна феррита).
Образование видманштеттовой структуры. На-
ложение нормального и мартенситного превращений возможно не
только при температуре ниже точки Л4Н для сплава, но и при бо-
лее высокой температуре. Ферритные кристаллы, образующиеся
«сдвиговым» или мартенситным путем при сравнительно высокой
температуре, называют видманштеттовой структурой. Особенно
ярко эта структура проявляется в стали, где кроме избыточного
феррита, образующегося в виде крупных закономерно ориентиро-
ванных пластин, имеется перлит (см. рис. 7.5). Образование вид-
манштеттовой структуры избыточного феррита происходит при
достаточно крупном зерне аустенита и высокой степени совершен-
ства его внутризеренной структуры. Эти условия предполагают вы-
сокую температуру аустенитизации *. Б. А. Леонтьев **' и др. по-
казали, что образование видманштеттова феррита сопровождается
появлением характерного рельефа на полированной поверхности
шлифа.
Очевидно, есть определенная зависимость скорости образования центров но-
вой фазы от температуры (степени переохлаждения) и для аллотропического
превращения с нормальной кинетикой, и для мартенситного превращения. Извест-
но, что в превращении с нормальной кинетикой местами преимущественного
образования центров являются границы зерен. В рассмотренных случаях в струк-
туре кроме пластин видманштеттова феррита наблюдались равноосные зерна
феррита, расположение которых свидетельствовало об их образовании возле гра-
ниц аустенитных зерен.
Анализ кинетики образования избыточного феррита, проведенный *** на спла-
вах железа с 0,15 мае. % С и 2—4 мае. % Сг с разделения кинетики образо-
ния центров и кинетики роста центров, показал очень резкую зависимость скоро-
сти зарождения от температуры (рис. 8.6). При температурах, близких к Аз
скорости образования зародышей (см. рис. 8.6,6) и их нормального роста (см.
рис. 8.6,в) могут быть очень незначительными. Вместе с тем при высокой темпе-
ратуре можно предполагать образование полукогерентных (дислокационных)
границ растущего кристалла из-за низкой упругости среды. Для компенсации
* В связи с этим появление видманштеттовой структуры рассматривается
как признак перегрева стали.
** «Физика металлов и металловедение», 1963, т. 16, с. 516.
*** Swinden D., Woodhed J. «J. Iron. St. Inst.», Nov. 1971, p. 883—899.
231
энергии сдвига, необходимой для преодоления сопротивления консервативному
движению поверхностных дислокаций, может хватить запаса объемной свобод-
ной энергии уже при относительно небольших переохлаждениях. По оценке
Б. А. Леонтьева, если принять предел текучести аустенита 1 кгс/мм2 и сдвиго-
вую деформацию усдв=0,2, удельная энергия сдвига составит еСдв~
~2 кал/(г-атом) *. При небольшой движущей силе превращения препятствия
Рис. 8.6. Кинетика образования
избыточного феррита в доэв-
тектоидной стали, содержащей
0,17 мае. % С и 3,16 мае. % Сг:
а — диаграмма температура — вре-
мя — степень превращения (Ф —
ферритное, П— перлитное превра-
щение); б—скорость зарождения
феррита при разном объеме прев-
ращения (1—15 об.% выделившего-
ся феррита); в—линейная скорость
роста в начале процесса (1 об. %
феррита)
на пути консервативного движения полукогерентной границы (дислокации, пе-
ресекающие плоскость скольжения, атомы примесей, сопротивление самой решет-
ки или силы Пайррлса — Набарро) могут преодолеваться только термической
активацией (см. рис. 7.21). Это объясняет относительно медленный рост кристал-
лов и остановку их роста внутри (а не на границе) исходного аустенитного зер-
на. Увеличение степени переохлаждения может привести к преобладанию конку-
рирующего механизма аллотропического превращения неконсервативным движе-
* Работа перемещения единицы длины дислокации xb (где х— напряжение
сдвига), перемещающийся при сдвиговой деформации -усдв объем равен е/уСдв;
таким образом, удельная энергия сдвига £сдв==тусдв.
232
нием границ раздела. Этот механизм единственный, если имеется большое число
препятствий к кооперативному перемещению атомов (т. е. при недостаточно со-
вершенной внутризеренной структуре) при ограниченном числе реализуемых за-
родышей мартенситных кристаллов, а также развитая поверхность границ зерен,
на которых происходит зарождение центров нормального роста кристаллов фер-
рита. Только при очень большом переохлаждении, когда движущая сила дости-
гает около 100 кал/(г-атом), барьеры на пути консервативного движения границ
кристаллов мартенсита могут преодолеваться без термической активации и про-
исходит «атермический» рост мартенситных кристаллов при большом числе
реализуемых зародышей. Верхняя температура образования видманштеттова
феррита в стали с 0,1 мае. % С равна 800—810°С; с увеличением содержания
углерода до 0,4% эта температура понижается до 690—7О0°С; при содержании
углерода 0,8—0,9 мае. % самостоятельная реакция образования феррита исклю-
чается и происходит бейнитное превращение (ниже 540—550°С), в котором так-
же действует мартенситный механизм у—>а-превращения наряду с диффузион-
ным процессом образования карбидных частиц (из аустенита).
Видманштеттов феррит в процессе образования не пересыщен
углеродом, поскольку, по электронно-микроскопическим данным
(В. И. Изотов, Б. Н. Леонтьев, 1971 г.), отсутствуют карбидные
частицы. Следовательно, рост кристаллов контролируется ско-
ростью диффузионного отвода углерода, что объясняет сравни-
тельно малую скорость роста (10~3—10“4 см/с).
* Особенность внутреннего строения видманштеттовых кристал-
лов— это большая плотность дислокации (значительно большая,
чем в зернистом феррите), что согласуется с представлением
о сдвиговом характере превращения. Габитусные плоскости плас-
тин видманштеттова феррита близки к плоскостям {111} исходного
аустенита, такой габитус наблюдали в мартенсите малоуглеродис-
той стали, в верхнем бейните, а также при образовании мартен-
сита в тонких пленках, т. е. в условиях, когда уровень энергии
упругих искажений понижен (из-за высокой температуры превра-
щения, малого содержания углерода или возможности легкой ре-
лаксации напряжений в одном направлении в тонкой фольге).
Значительный разброс ориентации габитусной плоскости для от-
дельных кристаллов объясняется различиями объема и формы
этих кристаллов, влияющими на энергию упругих искажений.
§ 8.2. ТЕОРИЯ МАРТЕНСИТНЫХ ПРЕВРАЩЕНИЙ
Кристаллографическая теория мартенситно-
го превращения. Следствием и одновременно условием коопе-
ративного движения атомов является закономерная связь или «ко-
герентность» решеток исходной и растущей фаз на фронте превра-
щения— границе раздела. На этом положении, впервые сформу-
лированном Г. В. Курдюмовым, основана современная теория
мартенситных превращений. В классической работе Г. В. Кур-
дюмова и Г. Закса (1930 г.) при исследовании закаленной стали
была установлена закономерная связь между ориентировками
кристаллической решетки мартенсита и решеткой исходной фа-
зы— аустенита. Это позволило рассматривать превращение как
деформацию решетки аустенита. В дальнейшем (в 50-х годах)
233
была разработана кристаллографическая (феноменологическая)
теория мартенситных превращений, на основании которой, зная
кристаллические структуры исходной и конечной фаз, можно
предсказать ориентационные соотношения между решетками обеих
фаз, габитусные плоскости мартенситных кристаллов и макроско-
пическую деформацию превращенного объема. Эта теория рассма-
тривает изменение кристаллической структуры как результат де-
формации решетки с инвариантной плоскостью.
Рис. 8.7. Характер рельефа, воз-
никающего на поверхности поли-
рованного шлифа в результате об-
разования кристалла мартенсита:
1 — исходная фаза; 2 — новая фаза
(мартенсит); 3 — линия, проведенная
на плоской поверхности образца, в ис-
ходном состоянии она остается непре-
рывной и после образования мартен-
ситной фазы
превращения, в результате
Из анализа рельефа на поверх-
ности предварительно полированно-
го шлифа в месте образования ново-
го кристалла можно определить ха-
рактер макроскопической деформа-
ции, которая является результатом
смещений атомов или деформации
решетки в макроскопическом объе-
ме. Такое исследование было прове-
дено Гренингероми Трояно (1949 г.)
на образцах сплава Fe — 22 мае.%
Ni — 0,8 мае.% С, в котором превра-
щение аустенита в мартенсит проис-
ходило только при охлаждении ниже
комнатной температуры. Установле-
но, что поверхность превращенной
области материала наклоняется к
плоскости шлифа, но остается пло-
ской (рис. 8.7); прямые риски, про-
веденные на поверхности шлифа до
образования кристалла мартенсита
претерпевают излохМ на выходе поверхности габитуса и меняют
свое направление, но остаются прямыми; в целом сохраняется не-
прерывность этих линий. Это свидетельствует о том, что рельеф
является результатом однородной деформации с инвариантной
плоскостью, т. е. имеется плоскость (плоскость габитуса кристал-
ла мартенсита), которая не искажается и не поворачивается; все
смещения материала направлены в одну сторону и пропорциональ-
ны расстоянию от инвариантной плоскости.
Примером деформации с инвариантной плоскостью может слу-
жить одноосное растяжение или сжатие (вектор смещения пер-
пендикулярен к инвариантной плоскости (см. рис. 7.19) или сдвиг
(вектор смещения параллелен инвариантной плоскости); любую
деформацию с инвариантной плоскостью можно представить ком-
бинацией этих двух простых деформаций. Все смещения при де-
формации с инвариантной плоскостью происходят в одной плос-
кости, в которой лежат нормаль к инвариантной плоскости и век-
тор смещения; таким образом, деформация оказывается плоской.
Следует отметить, что не всякая плоская однородная деформация
происходит при наличии инвариантной плоскости.
234
В этом легко убедиться, рассматривая деформацию сферического объема
кристалла (рис. 8.8). Однородная деформация превращает этот объем в эллип-
соид. Деформация с инвариантной плоскостью имеет место, если по одной из
главных осей нет деформации (например, по оси %i = l), а по двум другим
главным осям деформации различны по знаку: по одной (например, по оси х2)
деформация Л2>1, а по другой (%з) деформация Х3<1 (см. рис. 8.8,а). Если
%2 и Хз одновременно больше или меньше 1, эллипсоид не пересекает исходную
сферу (см. рис. 8.8,6), так что ни одно из плоских сечений эллипса не сохраняет
длины исходного диаметра; при Xi = l происходит только касание сферы и эллип-
соида вдоль одной линии (оси *1). При условии Х2>1, Х3<1 (или, наоборот,
Рис. 8.8. Плоская деформация сфе-
рического объема кристалла с инва-
риантной ПЛОСКОСТЬЮ (Л1=1, Л2>1,
Л3<1) (я) и без инвариантной пло-
скости (^<1, Х3<1) (б)
^2<1, Х3>1) легко найти плоское сечение, которое сохраняется инвариантным.
На рис. 8.9,а показаны сечения исходной сферы и эллипсоида деформации
в координатной плоскости х2х3. Пересечения окружности и эллипса в точках А
и В, Р и Q означают сохранение длины исходного диаметра. Поскольку М = 1,
то не меняет своей длины и диаметр по оси Xt. Плоскость, проведенная через
диаметр А'В' (или P'Q') и ось X], остается неискаженной, все линии в этой пло-
скости не изменяют своей длины при деформации, хотя она может поворачи-
ваться.
235
Для определения положения инвариантной плоскости до деформации необхо-
димо построить так называемый взаимный эллипсоид, т. е. выделить в кристал-
ле такой эллипсоид, который в результате деформации превратился бы в сферу
единичного радиуса (см. рис. 8.9,6). Пересечение этой сферы и взаимного эллип-
соида дает положение инвариантной плоскости до деформации (сечения АВ и
PQ на рис. 8.9,6).
Чтобы представить характер деформации при сосуществовании двух фаз
(т. е. в ходе превращения), рассмотрим превращение в эллипсоид половины сфе-
ры, «отрезанной» по инвариантной плоскости в ее исходном положении (АВ на
рис. 8.10). В результате деформации решетки полусфера превращается в полу-
эллипсоид, при этом изменяется положение границы превращенной области (А'В'
на схеме рис. 8.10).
Рис. 8.9. Определение инва-
риантной плоскости сфериче-
ского объема и эллипсоида де-
формации (а) и взаимного эл-
липсоида деформации (6)
Для сохранения контакта с непревращенной частью кристалла
необходимо вращение, поэтому деформация решетки с инвариант-
ной плоскостью включает чистую деформацию (растяжение, сжа-
тие, сдвиг) и вращение.
Если превращение идет с одной поверхностью раздела, т. е.
начинается с одного конца исходного монокристалла, фронт пре-
вращения занимает всю толщину кристалла и продвигается вдоль
кристалла, то сама поверхность раздела является инвариантной
плоскостью (в соответствии со схемой рис. 7.19 и аналогично не-
подвижной карте на границе сдвигаемой части колоды). Не ока-
зывает стесняющего действия на непревращенный объем исход-
ной фазы мартенситная пластина, проходящая сквозь весь кристалл
(см. рис. 7.8). В этом случае габитусная поверхность совпа-
дает с инвариантной плоскостью. Для образования отдельного
кристалла мартенсита внутри исходной фазы (см. рис. 7.8) теоре-
тически было показано (А. Л. Ройтбурд, 1967 г.), что минимум
236
энергии искажений системы достигается при отклонении габитуса
х>т инвариантной плоскости; это отклонение тем больше, чем боль-
ше толщина пластины (точнее, отношение толщины пластины к ее
размеру *в плоскости). При незначительной толщине пластины ее
габитусные поверхности остаются параллельными инвариантной
плоскости.
Кристаллографическая теория мартенситного превращения
усложняется тем, что в общем -случае при чистой деформации ре-
шетки условие инвариантности плоскости не выполняется. Эле-
ментарный кристаллографический анализ решеток, испытывающих
взаимное превращение, для большинства известных -случаев не
обнаруживает каких-либо кристаллографических рациональных
Рис. 8.10. Превращение половины объема сферы с инвариантной плоскостью:
и — положение инвариантной плоскости до деформации (АВ) (штриховкой выделена
половина сферы, в которой предполагаем отсутствие деформации решетки); б — резуль-
тат чистой деформации (т. е. деформация без какого-либо вращения); граница превра-
щенной части объема должна занять положение А'В'; в — вращение превращенного
объема, необходимое для сохранения контакта с непревращающейся частью
плоскостей, по которым возможно было бы точное сопряжение
решеток, и деформация решетки являлась бы плоской. Это, в част-
ности, относится к практически очень важному у—мх-превраще-
41ию в стали. Исключение составляют превращения в компактных
.упаковках (например, превращение г. ц. к.^гекс. п. у. в кобаль-
те), в которых деформация решетки является плоской (простой
сдвиг) с инвариантной плоскостью. Экспериментальное исследо-
вание Гренингера и Трояно показало, что при превращении ау-сте-
яит—^мартенсит фактическая макроскопическая деформация пре-
вращенного объема (рельеф на поверхности полированного шли-
фа) является плоской, но отлична от деформации решетки. Сле-
довательно, в процессе превращения на деформацию решетки на-
кладывается некоторая дополнительная деформация, так что ре-
зультирующая макроскопическая деформация становится плоской.
Эта дополнительная деформация не должна менять решетку (де-
формация при инвариантной решетке), это может быть скольже-
ние или двойникование* (ср. рис. 8.11,а с рис. 8.11,в; рис. 8.11,6
* Двойникование, строго говоря, не является деформацией при инвариант-
ной решетке, однако, при двойниковании изменяется только ориентировка, но
не тип кристаллической структуры (см. с. 241).
237
с рис. 8.11,г и д). Чтобы результирующая деформация оставалась
макроскопически однородной, расстояния между плоскостями
скольжения и величины этого скольжения должны быть постоян-
ными в пределах всей испытывающей превращение области кри-
сталла; между этими плоскостями скольжения происходит дефор-
мация решетки (см. рис. 8.11,а). Возможна также комбинация
различающихся по направлению смещений атомов деформаций
решетки (см. рис. 8.11,(5), эквивалентная двойникованию в исход-
ной или конечной фазе (до или после деформации решетки).
В общем случае нет точных оснований для выбора исходных
положений деформации решетки и дополнительной деформации.
Правильность этого выбора определяется только сопоставлением
рассчитанных и экспериментально найденных ориентационных со-
отношений решеток и ориентации плоскости габитуса.
§ г д
Рис. 8.11. Деформация решетки и деформация при
инвариантной решетке:
а, б — образование новой фазы (б) как результат однород-
ной деформации решетки исходной фазы (а); в—д — изме-
нение формы кристалла в результате деформации при инва-
риантной решетке (пластическая деформация по механизму
скольжения или двойникования)
Однородная деформация в случае, когда ни одно из значений Х=#1 (объем-
ный случай) не дает инвариантной плоскости, но сохраняет инвариантность ли-
ний вдоль образующих конуса (рис. 8.12). Задача теоретического анализа со-
стоит в том, чтобы при известных параметрах исходной и образующейся фаз,
а следовательно, при заданной чистой деформации решетки выбрать дополни-
тельную деформацию, не меняющую решетку (сдвиг, двойникование), которая
давала бы в сумме с заданной деформацией решетки результирующую плоскую
деформацию с инвариантной плоскостью. Дополнительный сдвиг должен для
этого удовлетворять следующим условиям: 1) плоскость сдвига Р должна пере-
секать конус инвариантных направлений; 2) плоскость, перпендикулярная к век-
тору направления сдвига 1, также должна пересекать конус инвариантных на-
правлений.
Выбор деформации решетки для описания превращений. При-
меры превращений, осуществляемых деформацией решетки без до-
полнительной деформации. Кооперативный характер перестройки
решетки состоит в согласованном движении атомов, когда атомы
не обмениваются местами, а лишь смещаются относительно своих
соседей на расстояния, не превышающие межатомных. При этом.
238
хотя каждый атом по отношению к своему соседу смещается на
расстояние меньше межатомного, смещение атома, отстоящего от
места начала роста кристалла новой фазы на макроскопическую
величину, будет также макроскопическим. Если вектор смещения
(сдвига) равен полной трансляции решетки, то в результате лю-
Рис. 8.12. Деформация решетки (объемный случай):
1 — единичная сфера в исходной решетке; 2 — эллипсоид деформации;
3 — конус инвариантных линий
бого числа таких смещений кристаллическая структура не изме-
нится. Примером таких смещений является пластическая дефор-
мация, осуществляемая при движении полных дислокаций (де-
формация при инвариантной решетке, см. рис. 8.11,в, г). Хорошо
известным примером кооперативного
мов, приводящего к перестройке
кристаллической решетки, является
механическое двойникование. Чтобы
описать результат фазового превра-
щения как деформацию решетки,
необходимо принять условие соот-
ветствия всех узлов исходной решет-
ки и решетки новой фазы. Это усло-
вие связывает положения узлов эле-
ментарных ячеек решеток новой и
исходной фаз, которые выбирают
так, чтобы они содержали одинако-
вое число атомов. В случае однород-
ной деформации решетки должны
быть выбраны главные оси и значе-
ния главных деформаций для пере-
хода исходной решетки в новую. Вы-
бор условий соответствия, эквива-
лентных по числу узлов ячеек, и,
следовательно, величин деформаций
процесса перемещения ато-
Рис. 8.13. Многозначность выбо-
ра деформаций решетки
239
может иметь много вариантов (рис. 8.13). Очевидно, наиболее ре-
альным является предположение, что атом узла исходной решетки
занимает ближайший к нему узел новой решетки, т. е. деформация
выбирается минимальной. Условие деформации решетки с инвари-
антной плоскостью (без какой-либо дополнительной деформации)
выполняется лишь в исключительных случаях. Деформация ре-
шетки является плоской и идет с инвариантной плоскостью при
превращении гекс, п.у^г.ц.к. (например, а-Со^р-Со), а также
при механическом двойниковании. В последнем случае определить
Рис. 8.14. Двойникование в о. ц. к. решетке:
а — схема движения атомов в плоскости (110) при двой-
никовании; б — положение плоскости двойникования
(112) в элементарной ячейке, заштрихована плоскость,
изображенная на схеме а; в —деформация сферической
области в результате двойникования
деформацию решетки очень просто, поскольку форма и размер
элементарной ячейки не изменяются. В обоих случаях деформации
решетки представляют собой простой сдвиг, матрица и новый
кристалл сохраняют когерентность по границе раздела без каких-
либо искажений. Граница раздела фаз (поверхность габитуса)
плоская и параллельная инвариантной плоскости.
Образование двойника в о. ц. к. кристалле. Про-
цесс двойникования можно рассматривать как превращение, не
связанное с действием химической движущей силы. Здесь этот про-
цесс рассматривается как наглядный пример кооперативного дви-
жения атомов. В свою очередь, мартенситное превращение, по-
скольку оно сопровождается изменением формы превращенного
24 0
объема, может быть своеобразным механизмом пластической де-
формации в определенного типа сплавах. Образование мартенси-
та во время пластической деформации дает эффект чрезвычай-
но высокой пластичности и одновременно приводит к упроч-
нению.
На рис. 8.14 приведена схема кооперативного движения атомов
в плоскости (110) при образовании двойников в о. ц. к. решетке
с плоскостью двойникования (112) и направлением скольжения
fill], Положение атомов в результате двойникового сдвига от-
мечено светлыми кружками. Плоскость, выделенная толстыми
пунктирными линиями, остается инвариантной, т. е. не изменяю-
щейся в процессе двойникования, общей для исходного кристалла
(матрицы) и двойника и представляет собой одну из когерентных
границ раздела. Величину сдвига атомов при двойниковании легко
определить из условия, что в двойнике сохраняется тип кристал-
лической структуры, но его решетка оказывается зеркально-сим-
метричной по отношению к исходной решетке. Плоскость, от ко-
торой начинается рост двойника, является плоскостью симметрии.
Величина сдвига атомов в плоскости, соседней с этой плоско-
стью, в данном случае составляет s1 = 6zj/3/6 (или 1/3 полной
трансляции в направлении сдвига [111]). Атомы следующей плоско-
сти, находящейся на расстоянии 2d(112), смещаются на то же рас-
стояние по отношению к атомам только что рассмотренной первой
плоскости, но общее (абсолютное) смещение составляет величину,
вдвое большую: s2 = 2d)/3/6; для n-плоскости имеем sn=na]/3/6.
Поскольку расстояние от этой плоскости до исходной границы
двойника равно rff112) и = па/)/б, то рассматриваемая деформация
характеризуется сдвигом постоянной величины: 4S — я/с1=УЗ/У 6 =
= 0,707.
Двойникование сопровождается изменением формы объема,
в котором оно прошло: сферический объем исходного кристалла
становится эллипсоидальным (см. рис. 8.14,в).
Дислокационная модель полиморфного мар-
тенситного превращения в кобальте. Для кристалло-
графического анализа наиболее простым примером аллотропичес-
кого превращения, протекающего мартенситным путем, является
Р^а-превращение в кобальте. При охлаждении ниже 400°С высо-
котемпературная модификация [3-Со с г. ц. к. решеткой переходит
в модификацию а-Со с гекс. п. у. решеткой. Эти решетки различа-
ются только последовательностью укладки плотноупакованных
слоев: решетка г. ц. к. — трехслойная (АВСАВСА ...), решетка
гекс. п. у. — двухслойная (ABABA ...). Рост кристалла низкотем-
пературной фазы начинается от некоторой плоскости исходного
кубического кристалла типа (111), которая остается инвариант-
ной (межфазовая граница). Перестройку решетки из трехслойной
упаковки в двухслойную можно описать как сдвиг на 7б трансля-
ции в направлении <211> по каждой второй из последователь-
16—523 241
ностей плотноупакованных плоскостей. Как видно из рис. 8.15,
сдвиг плоскости 3 (С) переводит этот слой атомов в положе-
ние А(С—>А), а следующий слой (4) при этом оказывается в по-
ложении В (А—>В). Описанный механизм перестройки решетки
может реализоваться при расщеплении дислокации а/2 [ПО]
в плоскости (111) и движении частичных дислокаций а/6 [12Г] и
.«/6 [211] (/ и Г на рис. 8.15,6). Дефект упаковки, образующий-
ся при таком расщеплении, уже представляет собой слой гексаго-
Рис. 8.15. Превращение (3-Со—^а-Со: структуры плотнейшей упаковки {а) и
дислокационный механизм превращения в кобальте (б):
О, ф, X — узлы однотипных плоскостей последовательности АВСА... (г. ц. к.); О, ф —
то же для последовательности АВА... (гекс. п. у.); стрелки показывают величину и на-
правление сдвига
нальной упаковки. Расстояние, на которое расходятся частичные
дислокации, вообще определяется энергией дефекта упаковки.
При температуре равновесия а- и p-фаз эта энергия .равна нулю.
Движение частичных дислокаций может перейти в другую плос-
кость, если перпендикулярно к той же последовательности плос-
костей в кристалле имеется винтовая дислокация. Представим
точку Д' на рис. 8.15,6 как дислокационный узел. Если сверху
В42
к ней подходит дислокация 2 с вектором Бюргерса а/2 [211], то
для точки К можно записать реакцию а/2 [211]=2/За [111]+
+ а/6 [21 Г]. Аналогично для дислокации 2', подходящей к точке
К снизу, имеем а/2 [121]=2/За [111]+а/6 [121].
Таким образом, в точке К можно выделить винтовые компо-
ненты указанных дислокаций с вектором Бюргерса, вдвое большим
межплоскостного расстояния dm (2а/]/*3), что обеспечивает пере-
ход частичной дислокации (например, а/6 [121]) в каждую вто-
рую плоскость последовательности плотноупакованных плоскос-
тей (111). Очевидно, по мере развития превращения частичные
дислокации а/6 <2П>> будут удаляться друг от друга по высоте:
между ними остается винтовая дислокация 2 а [111]. Эта схема,
предложенная Зегером, представляет собой один из возможных
механизмов превращения с участием дислокаций. По существу
этот механизм аналогичен механизму роста кристалла из жидкой
или газовой фазы присоединением атомов к ступеньке, обуслов-
ленной винтовой дислокацией.
В плоскости (111) имеются три эквивалентных направления
типа < 112>. При статистическом распределении смещений атом-
ных плоскостей между этими направлениями в результате пре-
вращения не возникла бы заметная макроскопическая деформа-
ция. Однако опыт показывает, что направление смещения остается
общим для многих сотен или тысяч атомных плоскостей, так что
возникает макроскопический сдвиг. Это служит аргументом в поль-
зу рассмотренного механизма роста.
§ 8.3. МАРТЕНСИТНОЕ ПРЕВРАЩЕНИЕ В СПЛАВАХ
НА ОСНОВЕ ЖЕЛЕЗА
Деформация решетки при у—>а-превращении по Бейну. Для
записи условия соответствия решеток аустенита и мартенсита их
следует представить элементарными ячейками, содержащими оди-
наковое число атомов и требующими минимальной деформации *
для перехода одной решетки в другую, т. е. ячейка, выделенная
в решетке аустенита, должна быть возможно более близкой к ко-
нечной ячейке мартенсита. На рис. 8.16 приведена сдвоенная
г. ц. к. ячейка у-фазы (аустенита). Такую решетку (оси z?)
можно описать как о. ц. т. решетку (оси ла, уа, га) с размерами
ячейки: ао.ц.т.=&о.ц.т=аг.ц.к. /2 и Со.ц.т.=аг.ц.к; с/а=1,43**. Та-
кое представление целесообразно для анализа деформации решет-
ки при полиморфном превращении и означает, что именно узлы,
выделенные на схеме (рис. 8.16,6), в результате деформации ре-
шетки при превращении примут конфигурацию элементарной
* Минимальная деформация определяется как минимальное значение суммы
квадратов деформаций по главным осям.
** Для описания других свойств кристалла ячейка должна обладать симмет-
рией самого кристалла.
243
ячейки решетки мартенсита (о. ц. к. — в безуглеродистых сплавах,
например Fe—Ni, тетрагональная — при наличии углерода).
Соответствие решеток аустенита и мартенсита удобно записать
в матричной форме. Такая запись позволяет определить кристал-
лографические индексы направлений при изменении системы ко-
ординат.
Рис. 8.16. Деформация Бейна при перестройке решетки г. ц. к.о. ц. к.:
а — сдвоенная обычная элементарная ячейка в г. ц. к. решетке; б — выделенная о. ц.,
тетрагональная ячейка, с/а=1,43, описывающая структуру г. ц к. решетки исходной фазы
(аустенита); в — о. ц. (кубическая при отсутствии углерода) ячейка, возникающая в ре-
зультате деформации решетки исходной фазы (сжатие по оси z и растяжение по осям
ос
х и у ); X — обозначение одной из систем октаэдрических позиций — позиции внедре-
а ос
ния для атома углерода
Как известно, кристаллографическое направление определяется как направ-
ление вектора из начала координат, координаты конца вектора, упирающегося
в узел решетки, являются кристаллографическими индексами данного направле-
ния [iH7O>]a. Можно определить индексы этого же направления в другой коорди-
латной системе если известна матрица переход
1
Для получения матрицы перехода от системы координат решетки мартенси-
та к системе координат решетки аустенита запишем координатные направления
решетки мартенсита (ха, Уа, za) или направления ребер элементарной ячей-
ки— [Ю0]др [010]M и [°011м —в координатной системе решетки аустенита.
Если векторы ребер элементарной ячейки мартенсита 1'ь 1'г и 1'з, а единичные
векторы координатной системы аустенита 1Ь Ь и 13, то из рис. ЗЛб,^ следует:
244
Коэффициенты правых частей этих уравнений образуют матрицу перехода
от системы координат аустенита к системе координат мартенсита:
(8.1)
Матрица обратного перехода т* получается из матрицы т заменой столбцов
на строки:
/1 1 0\
т* = | Г1 О (8.?)
\0 0 1/
Х1 П р« '
Уу =Т* Ул .
А _
Матрицы (8.1) и (8.2) позволяют найти соответствующие векторы решеток
исходной и новой фаз («соответствующие», т. е. переходящие один в другой
при превращении). Чтобы теперь описать изменения длины этих векторов (или
координат узлов) решетки при превращении, необходимо найти главные дефор-
мации.
Выделенная на рис. 8.16,6 о. ц. т. ячейка характеризуется сте-
пенью тетрагональное™ с/а=1,43. В углеродистом а'-мартенсите,
как известно, имеется тетрагональность, но гораздо меньшая и
зависящая от содержания углерода; безуглеродистый мартенсит
(в хромоникелевой или высоконикелевой стали) имеет кубичес-
кую ячейку. Поэтому превращение г. ц. к.->о. ц. к. требует дефор-
мации, которую в координатной системе исходной фазы можно
Записать в виде матрицы:
(7] 0 0 \
о т] о I, (8’3)
о о —731 /
где т] = (]Л2аа — ат)/ау; ?ь = (аа — а7)/аг Эта деформация пере-
водит атомы из узлов г. ц. к. решетки в ближайшие узлы о. ц. к.
решетки, объемноцентрированная ячейка, выделенная на рис. 8.16,6
сжимается по оси га (ось с) и несколько расширяется по осям ха
й уа. Для любого вектора гт решетки у можно найти соответст-
вующий ему вектор решетки:
( 7] о о \
г«= о 71 о г =ОБг (8.4)
\ 0 0 -7), /
С помощью дифракционных методов (рентгенографии и элек-
тронографии) всегда удается найти близкие или практически сов-
падающие (параллельные) кристаллографические направления и
245
плоскости решеток новой и исходной-фаз. Из рис. 8.16,6? видно,
что при отсутствии другой деформации (кроме ЬБ) ориентацион-
ную связь у и а решеток можно записать в виде
(001) || (001)в;
(8-5)
|Ю0]т || [110]а.
Такое ориентационное соотношение известно как соотношение
Бейна. Оно наблюдалось при превращении в очень тонких плен-
ках.
Мартенситные пластины в обычных (массивных) образцах ста-
ли с содержанием 0,5—1,4 мае. % С находятся в следующем ори-
ентационном соотношении с аустенитом:
II (Ю1)в;
(8.6)
Ж. II [ШЦ.
Это соотношение известно как соотношение Курдюмова — Закса.
В сплаве Fe—30 мае. % Ni установлено соотношение вида
(1Т1)т II (101)а;
(8.7)
(1121, II [10IL
Это соотношение, известное как соотношение Нишиямы, так же
как и соотношение (8.6), связано с деформацией Бейна
Рис. 8.17. Ориентационные соотношения
Бейна, Курдюмова — Закса (К—3) и
Нишиямы (Н):
Слева темный кружок показывает проекции
атомов выше- и нижележащих слоев, что
означает двухслойную упаковку плоскостей
типа (101) в о. ц. к. решетке; справа — проек-
ции атомов выше- и нижележащих слоев не
совпадают, что означает трехслойную упаков-
ку плоскостей типа (111) в г. ц. к. решетке
246
(см. рис. 8.16) и отличается от
соотношения Курдюмова —
Закса не очень сильно: напри-
мер, угол между направления-
ми [101]асоставляет около 5Э
(рис. 8.17). Кроме указанных
соотношений известно соотно-
шение Гренингера и Трояно
(найдено в сплаве Fe — 22
мае.% Ni—0,8 мае.% С). Это.
промежуточное соотношение
между (8.6) и (8.7) и не выде-
ляет каких-либо других совпа-
дающих рациональных кри-
сталлографических направле-
ний в тех же сопрягающихся
плоскостях (1Н)Т и (101)а.
Кроме перечисленных ориен-
тационных соотношений, сле-
дующих из схемы Бейна или по
крайней мере не противореча-
щих ей, существует еще один
весомый аргумент в пользу реальности этой схемы деформации ре-
шетки. Тетрагональность решетки мартенсита углеродсодержащей
стали объясняется упорядоченным расположением атомов углеро-
да в одной из трех подсистем октаэдрических позиций решетки
«-железа: вдоль оси [001]. Как видно из рис. 8.16, деформация
Бейна автоматически переводит все октаэдрические поры г. ц. к.
решетки аустенита в одну подсистему октаэдрических пор о. ц. к.
решетки мартенсита.
Рис. 8.18. Деформация решетки при перестройке решетки г. ц. к.->о. ц. к.:
а~плоскосгь (Ш)^ и (101)а, пунктирной линией показан след плоскости сдвига (121)^, нап-
равление сдвига [101 ^(„второй* сдвиг Курдюмова—Закса); б—плоскость (110)^ и (010)а>
плоскость сдвига (П1)^—верхняя линия чертежа; направление сдвига [П2]у („первый* сдвиг
Курдюмова—Закса)
Убедившись в реальности деформации Бейна, следует поискать
дополнительную к ней деформацию, которая приведет к .извест-
ному из эксперимента ориентационному соотношению.
Из рис. 8.18 видно, что для сохранения первоначальной ориен-
тации плоскости (111)т после ее превращения в результате бей-
новской деформации в плоскость (Ю1)а необходимо вращение вок-
руг направления [110]^. Такая сложная деформация приведет к реа-
лизации соотношения Нишиямы. Эту деформацию можно записать
как произведение матриц
— ^[И0]7
(8.8)
247
Наиболее простой вид полная деформация решетки приобретает, если вместо
обычных координатных осей решетки аустенита взять систему (ортогональных}
осей [Т12]г [1Г1]Г [1 Ю]т:
(а 0 0 \ /1 у 0\
Of 0 || 0 1 О =РГР (8.9}
о о т), у \0 0 1/
Выражение (8.9) показывает, что полная деформация решетки может быть
представлена как простой сдвиг вдоль [112]^ по плоскости (111)^ с последую-
щим изменением размеров вдоль выбранных координатных осей (Р).
Как видно из рис. 8.18,6 сдвиг Г1 приводит к замене чередования плотно-
упакованных плоскостей типа АВСА ..характерного для г. ц. к. решетки, на
чередование типа АВ'А ..., характерное для о. ц. к. решетки в последовательно-
сти плоскостей {ПО}. Сдвиг каждой плоскости (111) относительно рядом рас-
положенной происходит на 1/12 а [112] (1/12 доля трансляции в направлении
[И 2]); поскольку эти плоскости отстоят друг от друга_ на расстояние 1/За
[111], то величина сдвига у=1/12 а [112]/1/3 а [111]= И*2/4«0,35.
Деформация Р относится к изменению межплоскостного
элемент матрицы f) и к изменению расположения атомов в
расстояния
плотноупакованных
слоях путем растяжения вдоль [112]^ и сжатия по [110JT (для сплава Fe — 30%.
Ni а= + 13 %, = — 7,5 %, f близка к нулю).
Ориентировка Курдюмова—Закса может быть получена при до-
бавлении поворота вокруг оси [111]7 на угол 5° (см. рис. 8.17).
Преобразования матрицы деформации, подобные тем, которые были
описаны выше, приводят к тому, что перестройка решетки описы-
вается рассмотренным выше сдвигом (111)7 [Т12]т (fj, вторым-
сдвигом (Г2) по плоскости (121 )т в направлении [101]^ (или (121)а
[111]а) и небольшими изменениями межатомных расстояний вдоль
[Г12]т и [110Ц Схема сдвига Г2 приведена на рис. 8.18,а. Направ-
ление этого^ сдвига [101]7 должно быть [инвариантным, поскольку
оно входит в ориентационное соотношение Курдюмова—Закса. Ве-
личина сдвига у2 = 1/3 ]^3 0,19.
Из приведенного анализа, исходными данными для которого
были только представления о чистой (т. е. без вращения) дефор-
мации решетки по Бейну и экспериментальная характеристика
конечного состояния— ориентационное соотношение, следует, что:
1) перестройка решетки описывается деформацией Бейна; учет
конечного ориентационного соотношения добавляет сравнительно
небольшие деформации;
2) один и тот же конечный результат (полная деформация)
может быть описан различными последовательностями деформа-
ций, в частности, повороты вместе с дилатационными изменениями
могут быть представлены как однородные сдвиги и соответствую-
щие (другие) дилатации по главным осям. Таким образом, рас-
248
смотренные микроскопические сдвиги (например, .по Курдюмову—
Заксу) являются лишь .вероятными, но не обязательными меха-
низмами перестройки;
3) в данном анализе инвариантной считалась плоскость плотной
упаковки аустенита однако только в определенных случаях
кристаллы мартенсита имеют такой габитус. Наиболее часто встре-
чается габитус (252)7, а также (259)^. Теория должна объяснить
появление таких габитусов.
Морфология и субструктура кристаллов мартенсита. Кристалло-
графическая теория, описывающая мартенситное превращение как
деформацию решетки с инвариантной плоскостью, предъявляет
определенные требования к параметрам собственно деформации
решетки (деформация Бейна): 1) i]i и т]2 не должны быть одно-
временно больше или меньше 1; 2) деформации должны быть до-
статочно малыми: t]2i + t]?2^2. Только при этих условиях можно
подобрать дополнительную деформацию при инвариантной решет-
ке, чтобы суммарная деформация обеспечила инвариантность
некоторой плоскости. Для практического анализа превращения
в стали удобно задавать деформацию решетки степенью тетраго-
нальное™ и относительным изменением объема ячейки при пре-
вращении (Ду/ц=г]2т]1). Для очень широкой области составов
сплавов на железной основе эти параметры деформации изменя-
ются в .следующих пределах: Ди/у=1,02-^-1,06; см/аЛг=1,00-г-1,08.
В работе авторов одного из вариантов кристаллографической
теории Векслера, Либермана и Рида (1953 г.) проведен расчет
для превращения в сплаве Fe—Ni—С. Из экспериментальных дан-
ных .в расчете использованы параметры собственно деформации
решетки Ди/и=1,0384 и См^м—1,04. В качестве свободного па-
раметра теории для дополнительного сдвига была выбрана си-
стема (112)а [Ш]а. Сопоставление экспериментальных и полу-
.ченных в теоретических расчетах характеристик превращения
(табл. 8.2) показывает очень хорошее совпадение.
Таблица 8.2
Экспериментальные и расчетные определения кристалло-геометрических
характеристик мертенсита в сплаве Fe—Ni—С
Кристалло-геометрическая характеристика Эксперимент Теория
Габитус Ориентационное соот- ношение Угол макросдвига % /10\ /0,5472 \ I 3 ) = | 0,1642 | \15/ \0,8208/ (111)а II (101)М с точностью 1° [П0]л Д [Ш]м =2,5° 10,66е /0,5691\ 1 0,1783 ) У0,8027у (111)аЛ(1°1)л!= 15' Л [И1]м = 3* 10,33*
249
В высокоуглеродистых нелегированных (>1,5 мае. °/0С) сталях
наблюдается габитус {259}^, близкий к габитусу {3, 10, 15}^ в только
что рассмотренном примере. Этот габитус также получается в пред-
положении дополнительной деформации в виде простого (однократ-
ного) сдвига. Во многих случаях, в частности в нелегированных
сталях с содержанием углерода 0,5— 1,4 мае. °/0, отмечается га-
битус {225}7 (рис. 8.19). Уменьшение содержания углерода в неле-
гированных сталях приводит к изменению не только габитуса кри-
сталлов мартенсита, но и морфологии и их субструктуры: при не-
высоком содержании углерода вместо внутреннедвойникованных
пластин, характерных для высоконикелевых или высокоуглеродис-
тых сплавов, наблюдаются рейки со сложной дислокационной
структурой. Хотя изменение габитуса от {259}у к {225}7 проис-
ходит раньше, чем становятся
Рис. 8.19. Стереографические проек-
ции выходов полюсов габитусных пло-
скостей пластин мартенсита в спла-
вах железо — никель [(31510)^ или
(295)у], в сплавах железо — углерод
[от (295)т до (252)7] и в нержавею-
щих хромоникелевых сталях [(121)^ —
(252)^] (схема)
явными изменения внутренней
структуры и формы кристаллов,
надо полагать, что при этом уже
меняется характер дополнитель-
ной деформации (появление но-
вой системы сдвига и дру-
гие, более сложные деформа-
ции).
Для большинства промышлен-
ных конструкционных углеродис-
тых (до 0,6 мае.% С) и легиро-
ванных сталей, для двойных спла-
вов железа с содержанием никеля
до 28—29%, а также для безуг-
леродистых высоконикелевых ста-
лей (18—25 мае. % Ni+Mo, Ti, Al
и другие легирующие добавки)
характерна форма кристаллов в
виде реек, образующих пакеты
(рис. 8.20) и разделенных мало-
угловыми границами (типа кру-
чения). По данным В. И. Изото-
ва *, полученным при исследова-
нии серии сплавов Fe—Ni, плос-
кость границы близка к {110}м>
которая составляет угол 1,5°
с плоскостью {П1}а, входящей ib ориентационное соотношение.
Длинное ребро реек идет вдоль направления [111] аг, которое со-
ставляет угол 2° с направлением [110]л, входящим в ориента-
ционное соотношение. Таким образом, в данном случае плоскость
габитуса, инвариантная в условиях деформации решетки, в пре-
делах точности данного эксперимента совпадает с плоскостью
наилучшего соответствия решеток исходной и конечной фаз.
* Изотов В. И. «Физика металлов и металловедение», 1972, т. 34, вып. 1,
с. 123.
250
Важной особенностью субструктуры мартенситных кристаллов
в сплавах, содержащих до 28% Ni, является значительная плот-
ность дислокаций (около 1010 см~2), принадлежащих к разным
системам скольжения (см. рис. 8.20,а). При увеличении содержа-
ния никеля (и углерода) наблюдается «мидриб» — закономерно
расположенное скопление тонких двойников превращения (в све-
товом микроскопе эти скопления видны просто как темные линии,
см. рис. 8.21,я), одновременно отмечается переход от реечной фор-
мы кристаллов к пластинчатой.
Рис. 8.20. Структура «реечного» мартенсита:
а, б — форма и расположение кристаллов мартенсита в сплаве Fe—24% Ni—3% Мп
(схема и электронная микрофотография фольги); в — субструктура кристаллов рееч-
ного мартенсита (сплав Fe—5% .Ni, электронная микрофотография при большом уве-
личении)
251
Описанные здесь типы микроструктуры мартенсита связывали с различиями
химического состава стали. Однако такая связь еще не объясняет причину по-
явления разных типов структуры мартенсита. Поскольку уменьшение содержания
никеля (и углерода) приводит к повышению температуры начала мартенситного
превращения (точка Мн), можно именно этим объяснять изменения габитуса,
субструктуры и морфологии кристаллов мартенсита, имея в виду самые общие
температурные зависимости, важные для фазовых превращений в твердом со-
Рис. 8.21. Структура «атермического» в сплаве Fe—31% Ni (а, б) и «изо-
термического» в сплаве Fe—24% Ni—3% Мп (в, е) мартенсита:
а, в — световые микрофотографии (О. П. Максимова); б, г — электронные микрофо-
тографии (В. И. Изотов)
252
стоянии — термодинамического стимула превращения (степени переохлаждения)
и упругих характеристик исходной и новой фаз. Однако сопоставление темпера-
туры Мн разных сплавов не позволяет говорить определенно о различиях тер-
модинамического стимула из-за изменений температуры (метастабильного) рав-
новесия T'q и не исключает зависимости от химического состава других свойств
(например, энергии дефекта упаковки, размера зерен, флуктуаций состава и пр.).
Поэтому было важным получить типы мартенсита разной морфологии в одном
и том же сплаве и при этом знать температурные интервалы их образования.
Такой эксперимент возможен с хромоникелевой нержавеющей сталью переход-
ного аустенитно-мартенситного класса, которая переходит в мартенситное со-'
Рис. 8.22. Кинетические характеристики мартенситного превра-
щения в сплавах Fe—Ni—С:
а — изменение объема мартенсита при непрерывном охлаждении
(10 град/мин); б — изменение объема мартенсита в изотермических
условиях [при температуре, близкой к точке Мд для трех типов спла-
вов (кривые 1—3), различающихся по области температур мартенсит-
ного превращения]; в — температурно-концентрационные области (1—3)
мартенситного у-^а-превращения (положение температуры начала прев-
ращения — точка Мд) в зависимости от содержания никеля при раз-
ном содержании углерода (И. Я. Георгиева, О. П. Максимова)
стояние в процессе холодной деформации (при комнатной температуре) или
в условиях глубокого охлаждения (погружением в жидкий азот). В первом слу-
чае получен реечный (с большой плотностью дислокаций) мартенсит (или по
терминологии, использованной в оригинальной публикации*, — «игольчатый»).
При охлаждении в жидком азоте получен пластинчатый, внутреннедвойникован-
ный мартенсит. Таким образом, морфология мартенситных кристаллов (и, следо-
вательно, механизм превращения) определяется температурой превращения.
Образование реечного мартенсита связывают с таким механиз-
мом превращения, в котором происходят релаксационные процес-
* Иорданский В. Н., Скаков Ю. А., Спиридонов В. Б. «Физика металлов и
металловедение», 1964, т. 18, вып. 6, с. 929.
25&
сы. Пластинчатый мартенсит с внутренними двойниками, напро-
тив, по-видимому, образуется в условиях, когда протекание релак-
сационных процессов затруднено.
Кинетика мартенситного превращения и микроструктура мар-
тенсита в сплавах на железной основе. Кинетика мартенситного
превращения даже в сплавах одной системы (например, Fe—С,
Fe—Ni, Fe—Ni—С или Fe—Ni—Mo) имеет большое разнообразие.
Именно это разнообразие (а не просто «большая скорость») от-
личает мартенситные превращения от нормальных. Действитель-
но, в одних случаях наблюдали мартенситное превращение как
взрывную реакцию (кривые 2 и 3 на рис. 8.22,я), а в других —
Рис. 8.23. Диаграмма температура — время —
объем превращения (0,5; 1; 2 и 4%) для образо-
вания мартенсита в сплаве Fe—22,7% Ni—
3,5% Мп (т — время, мин; Мн.и— предельная или
«истинная» температуры начала мартенситного
превращения) (Г. В. Курдюмов, О. П. Максимова)
постепенное развитие процесса по мере охлаждения (кривая 1 на
рис. 8.22,а) и медленное увеличение превращенного объема в изо-
термических условиях (кривые 1 и 2 на рис. 8.22,6) и даже подав-
ление этого превращения при быстром охлаждении в область тем-
ператур жидкого азота.
При исследовании сплавов системы Fe—Ni—С самый сложный
вид кинетики наблюдался в сплавах с температурой начала мар-
тенситного превращения в области 2 (см. рис. 8.22,в) (примерно
от комнатной до —100° С): в ходе охлаждения превращение отме-
чалось скачкообразным увеличением намагниченности и звуковым
эффектом. От такого «(взрыва» могло возникнуть по объему до
40—50% мартенситной фазы (кривая 2 на рис. 8.22,а). Если про-
цесс охлаждения прервать, то непосредственно после взрывной
реакции происходит еще некоторое увеличение объема мартенсит-
ной фазы (кривая 2 на рис. 8.22,6).
254
При меньшем содержании углерода и (или) никеля (область
1 на рис. 8.22,в) в ходе превращения при охлаждении нет призна-
ков взрывной реакции (нет и звуковых эффектов), а при посто-
янной температуре ниже точки тИн во времени идет образование
мартенсита (до 20—30% по объему, кривая 1 на рис. 8.22,6). За-
висимость скорости этого превращения о;т температуры (рис. 8.23)
характеризует этот процесс как термически активируемый.
Наконец, в высоконикелевых сплавах с самым низким поло-
жением мартенситной точки (область 3 на рис. 8.22,в) превраще-
ние в ходе охлаждения идет как серия мелких взрывов
(см. рис. 8.22,а, кривая 3) и нет подрастания количества мартен-
сита при постоянной температуре (линия 3 на рис. 8.22,6). По-
нижение температуры не замедляет этого превращения и увеличе-
ние скорости охлаждения соответственно, не позволяет задержать
образования мартенсита (как это можно было сделать в сплавах
с точкой Ми в области /). Поэтому такой тип превращения был
назван «атермическим». В тех сплавах, где мартенсит может воз-
никать при постоянной температуре, тип мартенситной реакции
называют «изотермическим». Эти же термины используют для
обозначения морфологического типа мартенситных кристаллов
независимо от конкретных условий их образования (см. рис. 8.21).
Атермический мартенсит (см. рис. 8.21,а, 6) имеет вид правиль-
ных пластин с четко очерченными ровными гранями. Длина кри-
сталлов может быть равной поперечнику аустенитного зерна, на
границах зерен (или двойников) можно наблюдать как бы «пре-
ломленное» продолжение кристаллов — результат инициирующего
действия растущего кристалла на зарождение нового кристалла
в соседнем зерне, в промежутках между ранее образовавшимися
кристаллами возникают ряды более мелких кристаллов в виде
«ферм» или «молний». Эти ряды также свидетельствуют об ини-
циирующем действии растущих кристаллов с «отражением» упру-
гих волн. При самой низкой температуре быстрый рост мартенсит-
ных кристаллов приводит к их взаимной деформации «вторже-
нием» и пересечением. Внутри кристаллов в световом микроскопе
можно видеть четкие линии—мидрибы, представляющие собой
скопления очень тонких двойников; в остальной части кристаллов,
атермического мартенсита методом дифракционной микроскопии
также выявляются двойники, но более широкие и редкие. В объеме
кристалла область мидриба имеет плоскую форму, и ее ориентация
в атермическом мартенсите практически совпадает с ориентацией
плоскости габитуса (для рассматриваемых сплавов {259}а>
см. рис. 8.19). Ориентационное соотношение для атермического
мартенсита близко к соотношению Нишиямы.
Кристаллы изотермического мартенсита имеют неправильные
очертания; часто они выстраиваются с некоторым смещением
вдоль общего направления — линии мидриба (см. рис. 8.21,в) и
образуют тупоугольные стыки. Плоскость мидриба в мартенсите
рассматриваемых сплавов близка к {259Ц, габитусная плоскость
(при спрямлении контуров поверхности границ и усреднении) ле-
255
жит между плоскостью (111) а, входящей ,в ориентационное соот-
ношение, и плоскостью (112) А (см. рис. 8.19). Ориентационное
соотношение соответствует соотношению Курдюмова — Закса (от-
клонения в пределах 2—2,5°). Как видно, кристалло-геометричес-
кие характеристики изотермического мартенсита те же, что и ха-
рактеристики рассмотренного выше мартенсита реечного типа, об-
разующегося в сталях с высоким положением мартенситной точки.
Характерным для образцов, в которых шла изотермическая ре-
акция, являлось отсутствие рельефа на механически полированной
Рис. 8.24. Температурные области
атермической и изотермической кине-
тики образования мартенсита в спла-
вах Fe—24,5% Ni при разном содер-
жании молибдена (И. Я. Георгиева,
И. И. Никитина)
поверхности. Это свидетельствует
о том, что в области больших ис-
кажений решетки аустенита кри-
сталлы изотермического мартен-
сита не образуются. Рельеф мож-
но наблюдать на электролитиче-
ски полированной поверхности.
Пластическая деформация стаби-
лизирует аустенит в (Fe—Ni—
Wn)-сплавах, где превращение
имеет изотермический характер, и
активизирует мартенситное прев-
ращение (точнее, повышает мар-
тенситную точку Мн в Fe—Ni-
сплавах с атермической кинети-
кой.
В сталях, в которых мартенситное
превращение вызывается напряжениями,
можно получить уникальное сочетание
прочности с пластичностью. «Сверхпла-
стичность» этих сталей обусловлена тем,
что мартенситное превращение действует
как дополнительный своеобразный механизм деформации. Такие стали называют
ПНП-стали, т. е. стали с «пластичностью», наведенной превращением» (в англий-
ской литературе TRIP-стали). Подбором легирующих элементов получают сталь
с мартенситной точкой, близкой к комнатной температуре. В технологию обра-
ботки включается так называемая теплая деформация аустенита. Структура
тонкой полигонизации аустенита с равномерным распределением значительного
количества дислокаций обеспечивает высокую исходную прочность материала
и в то же время затрудняет и ограничивает образование мартенсита в процессе
деформации. Образование мартенситных кристаллов происходит только на уча-
стках, где возникает особенно высокое напряжение. Локальное мартенситное
превращение приводит к релаксации этих напряжений и локальному упрочне-
нию. В результате деформация переходит на другие объемы материала, что
.и обеспечивает особенно высокую пластичность.
Для выяснения природы различия мартенситных превращений
атермического и изотермического типов интересно сопоставить тем-
пературные интервалы этих типов превращений в одном и том
же материале. На рис. 8.24 показаны температуры начала мар-
тенситного превращения с изотермической кинетикой (точка
Мизн) И с атермической или взрывной кинетикой (точка А4атн) для
некоторых Fe—Ni сплавов (24,5% Ni) содержащих разное коли-
чество молибдена. На этом графике нанесены температуры обра-
.256
зования мартенсита, вызванного -пластической деформацией — точ-
ка Л4д. Температура Л4Д должна быть близкой (чуть ниже) к тем-
пературе термодинамического равновесия аустенита и мартенсита
(Т'о). Из приведенных данных следует, что в сплавах, где есть и
изотермическое, и атермическое превращение, последнее начинает-
ся при большем переохлаждении; переохлаждение при изотерми-
ческом превращении может быть в два раза меньше, чем при
атермическом. Таким образом, можно считать, что термодинами-
ческий стимул для осуществления изотермического типа превра-
щения существенно меньше, чем для атермического.
В общей классификации (см. с. 181) было принято, что для мар-
тенситных превращений характерно отсутствие релаксационных
процессов. Однако это представление следует считать просто край-
ним случаем, удобным при общей грубой классификации превра-
щений. В реальных случаях мартенситный рост кристаллов, оче-
видно, может быть и при некоторой степени релаксации напря-
жений, осуществляющейся образованием на межфазовой границе
некоторого числа дислокаций, способных к движению. Релаксация
напряжений образованием дислокаций облегчает образование но-
вой фазы, поскольку уменьшается требуемая движущая сила пре-
вращения или термодинамический стимул (уменьшается работа
против сил упругости). Представление о релаксации напряжений
объясняет тот факт, что мартенситное превращение с так назы-
ваемой изотермической кинетикой является термически активи-
руемым процессом.
Анализ образования зародышей, когерентных и частично коге-
рентных с матрицей, проведенный А. Л. Ройтбурдом в терминах
термодинамической теории зарождения (см. с. 207), показал, что
при малых переохлаждениях большей оказывается скорость обра-
зования частично когерентных зародышей, при значительном тер-
модинамическом стимуле превращения (большие переохлаждения)
особенно велика скорость образования когерентных кристаллов.
§ 8.4. АЛЛОТРОПИЧЕСКИЕ ПРЕВРАЩЕНИЯ ТИТАНА, ТВЕРДЫХ РАСТВОРОВ
НА ОСНОВЕ Р-Ti И НЕКОТОРЫХ ИНТЕРМЕТАЛЛИДОВ
При нормальном давлении металлический титан может быть
в двух модификациях: ниже 883°С устойчива гекс. ,п. у. модифика-
ция a-Ti с периодами а=2,951 А, с—4,684 А*, выше 883°С устой-
чива о. ц. к. модификация р, период решетки p-Ti при комнатной
температуре можно принять равным 3,28 А**. Объемный эффект
превращения невелик (около 0,2%), поэтому превращение р—
идет при довольно небольших переохлаждениях. Увеличение ско-
рости охлаждения от 4 до 10 000 град/с приводит к понижению
* Отношение с/а=1,587, что несколько меньше обычного значения с/а для
компактной решетки.
** Получено экстраполяцией к чистому Ti зависимости периода решетки от
состава в сплавах, в которых P-фаза сохраняется до комнатной температуры.
Методом высокотемпературной рентгеновской съемки при 900±5°С получено а —
= 3,3065 А.
17—523 257
температуры превращения от 882 до 850°С. При скорости охлаж-
дения более 200 град/,с на поверхности шлифов наблюдали рель-
еф и игольчатую форму кристаллов a-фазы, характерные для мар-
тенситного превращения, при меньшей скорости охлаждения (и
соответственно при более высокой температуре) нет характерного
проявления мартенситного механизма из-за интенсивного проте-
кания релаксационных явлений (во время или после превраще-
ния). Во всех случаях наблюдали* строгое ориентационное соот-
ношение (с отклонениями до 1 град)
{0001}, И{210};
(8.10)
<1120>,||<111>э.
Связь решеток а- и [3-модификаций показана на рис. 8.25 (на схеме указа-
ны размеры ячеек для Zr). Слева приведена о. ц. к. ячейка (3-фазы; далее в о. ц. к.
решетке выделена о. ц. т. ячейка, ограниченная плоскостями {110}^ (нижняя
и верхняя плоскости тетрагона) и {211}^ (боковые плоскости). Справа внизу по-
казана гексагональная ячейка а = Zr, которая получается из тетрагональной ячей-
ки в результате сдвигов: [в плоскостях (112)^ и (112)^ соответственно^в напра-
влениях [111]^ и [llljg, кроме*того, происходит расширение [ячейки в одном на-
правлении и сжатие в другом.
При указанном ориентационном соотношении возможно в р-кри-
сталле образование 12 кристаллографических ориентировок а-фа-
* Это ориентационное соотношение впервые было рентгенографически полу-
чено Бюргерсом в 1934 г. для превращения аналога титана — циркония.
[111]
[111]
0}361 нм (3,61А)
122рад(70°52') /?\ 0,540нм (5,40А) 4а 4 0,312нм(3,12А)
’ Л r^j — —?'
(110)
0,51 Они (5,10А)
1/2aV3-
0,31?нм(3,12А)
j3-Zr-o(.-Zr :
0,510нм(5,10])~
"о^16нм(5,1б\]
0,340нм (3]2А4
' 0,524 нм (3,24А)
^[1010] J
[1120] W о
0,512нм(3,12 АН 0№hm(3,24A)
Рис. 8.25. Схема перестройки о. ц. к. решетки в гекс. п. у. (P-Zr->a-Zr) (по Бур-
герсу)
258
зы, что и наблюдалось экспериментально. Обратное превращение
приводит к восстановлению при нагреве исходного 0-кристалла.
Зафиксировать 0-фазу в чистом титане не удавалось при самой
высокой скорости охлаждения.
Новые метастабил ьные состояния возникают
Рис. 8.26. Схема диаграммы состоя-
ния титан — переходный металл, на
которой показаны температуры нача-
ла (Л4Н) и конца (Мк) мартенситного
массивного превращения при охлаж-
дении 0->а и температура начала
превращения 0-но (Т Вдоль оси
концентрации указан фазовый состав
охлажденных сплавов; сКр — «крити-
ческая» концентрация легирующего
элемента, при которой наблюдается
появление о-фазы (Г. И. Носова)
йка решетки 0-фазы в новую
в сплавах титана. Особенно характерны сплавы титана с переход-
ными элементами, повышающими устойчивость 0-модификации
(бета-стабилизаторы). Схематически Ti-угол диаграммы состоя-
ния такой системы приведен на рис. 8.26. В связи со сравнительно
малой растворимостью этих элементов в a-Ti в широкой области
составов возникает метастабильная а'-фаза — пересыщенный твер-
дый раствор на основе a-Ti — как результат массивного (мартен-
ситного) превращения. В более концентрированных сплавах титана
с переходными элементами при благоприятном объемном факторе
(разница атомных радиусов небо-,
лее 7—7,5%) может возникать
новая по структуре метастабиль- *
ная а"-фаза, близкая к a-Ti (или 7/
а'), но с более низкой симметри- 7^
ей. Структура а"-фазы описыва-
ется в ромбической системе, связь
между структурами 0-, а' (или
а)- и а"-фаз показана на рис.
8.27. Для всех трех структур вы-
делена ромбическая ячейка с пе-
риодами а, Ь, с\ различие между
структурами выражается параме-
тром сдвига (кроме очевидных
изменений в соотношении перио-
дов и углов, показанных на схе-
мах). Связь между а'(а)- и а"-
фазами иллюстрируется концен-
трационной зависимостью перио-
дов решетки (рис. 8.28). Таким
образом, можно считать *, что а"-
фаза занимает промежуточное по-
ложение между 0' и а'(а)-фаза-
ми. При небольшом содержании
легирующего компонента nepecTj
структуру идет до конца с образованием гекс. п. у. а(а'); при зна-
чительном содержании легирующего компонента перестройка
(сдвиг) останавливается на некотором промежуточном этале; этот
этап характеризуется параметром у [от у=0 для 0-фазы до у—
= 0,17^ для а (а')-фазы (см. рис. 8.27)], степень приближения
к равновесной структуре а может характеризоваться также отно-
шением периодов решетки в ромбической системе (при завершении
перестройки а/Ь—>1, см. рис. 8.28). Остановка сдвиговой мартен-
* Багаряцкий Ю. А. и др. В кн.: Труды ЦНИИчермета. М., Металлургиздат,
1958, № 4, с. 61.
17* 2 59
ситной перестройки на промежуточном этапе и образование новых
метастабильных структур — это типичные явления для превраще-
ний о.ц.к. координации в гекс. п. у. В этом смысле аналогами
а"-мартенсита титановых сплавов являются мартенситные струк-
туры в сплавах Си—А1, Си—Sn, AuCd (см. ниже). В таких систе-
Рис. 8.27. Связь между расположениями атомов в кубической 0-фазе (я),
в гексагональной (a-или а')-фазе (б) и в ромбической а"-фазе (в)
Рис. 8.28. Переход от а'- к (/'-структуре в систе-
ме Ti—Nb происходит при содержании ниобия
более 4 ат.%, что определяется экстраполяцией
линий концентрационной зависимости периодов, а
для а' а и b для а" (Г. И. Носова)
мах превращение испытывает упорядоченная 0-фаза, имеющая
координацию атомов, характерную для о. ц. к.
Возвращаясь к сплавам титана, отметим, что, в зависимости
от состава 0-фазы меняются температуры начала (Мн) и конца
(Мк) мартенситного 0—>ах (или 0—их")-превращения соответст-
венно, структура состоит из (/(а")- или 04-а'-(04-а")-кристал-
260
лов* (см. рис. 8.26). При дальнейшем повышении концентрации
второго компонента (обязательно переходного) возникает новая
метастабильная фаза со. Кроме того, отмечают еще два условия
образования co-фазы из р-фазы: 1) количество второго компонен-
та должно соответствовать электронной концентрации 4,14—•
4,16 электрон/атом (для переходных элементов берется сумма
s+\d электронов или номер группы); 2) состав p-твердого раство-
ра, соответствующий «критической» концентрации, отвечает при-
мерно постоянному значению периода решетки (около 3,25А).
«Атермическое» ** мартенситное превращение р—но не сопровож-
дается заметным изменением микроструктуры, видимой в свето-
вом микроскопе. Практический интерес к выяснению природы это-
го фазового перехода сначала был вызван сильным его влиянием
на механические свойства. В настоящее время с образованием
co-фазы связывается существенное улучшение свойств сверхпро-
водящих материалов на основе Ti.
Рентгеновское исследование co-фазы в образцах, приготовлен-
ных как монокристаллы p-фазы, позволило однозначно опреде-
Рис. 8.29. Элементарная ячей-
ка co-фазы (а) и ее связь
с о. ц. к. решеткой р-фазы,
представленной в гексагональ-
ной системе (б); расположение
атомов в плоскости (011)^—,
О, Н----то же в соседней пло-
скости, X — положение атомов
внутри ячейки <о(в)
* Одновременно а'- и а"-фазы не наблюдались, это подтверждает мнение
о непрерывности перехода одной из них в другую.
** Возможно изотермическое диффузионное образование co-фазы при отпуске
(или изотермической обработке) переохлажденной Р-фазы; при этом превраще-
ние контролируется диффузией и сопровождается изменением состава р-фазы.
261
лить ее структуру *. со-Фаза имеет гексагональную ячейку
(3 атом/ячейка, рис. 8.29) с периодами а~4,60 А, с=2,82 А, от-
ношение с/а=0,613; базис структуры (ООО, ±1/3 2/Зг), простран-
ственная группа P3ml; значение z несколько различается в раз-
ных сплавах, но остается близким к 1 /2. Ориентационное соот-
ношение .о- и р-фаз:
(0001)ф ||(111)?;
(8.П)
[ИЖ || [1Т0]р.
Из-за близости структур со- и p-фаз возможно сопряжение фаз
по всей поверхности частицы. Ю. А. Багаряцкий показал, что
о. д. к. решетка p-фазы, представленная в гексагональной системе
координат, когда за ось с выбрано одно из направлений <111>
кубической решетки (см. рис. 8.29,6), оказывается очень близкой
к решетке со-фазы по форме ячейки (с/я=0,6126), по величине
периодов, если период ~3,25 А, и по числу атомов на ячейку
(3 атома); различие только в положении внутренних атомов
(см. рис. 8.29,в). Таким образом, для перестройки решетки р-фазы
в решетку со-фазы необходимы лишь смещения атомов внутри
ячеек вдоль оси [111]^ на расстояние, составляющее долю меж-
атомного расстояния. Эти смещения можно представить как сдвиг
соседних атомных рядов [111]^ навстречу друг другу (рис. 8.30,а);
можно также эту перестройку представить как движение навстре-
чу друг другу соседних атомных плоскостей (Ш)^ до их совме-
щения, тогда как каждая третья плоскость остается неподвижной.
Удобной моделью перестройки р—ив является действие в о. ц. к.
решетке продольной волны смещений с волновым, вектором к=
=(2/а)^31, где 1—единичный вектор в направлении [111]^ (см.
рис. 8.30, 6).
Совершенно ясно, что описанные движения атомов при р—><о-
превращении не приводят к накоплению какой-либо макроскопи-
ческой деформации, и никакого поверхностного рельефа, харак-
терного для описанных ранее мартенситных превращений, в дан-
ном случае быть не должно. Однако имеются другие особенности
превращения, которые позволяют предполагать кооперативный
характер движения атомов, и по этому главному признаку счи-
тать р—)-(о-превращение мартенситным: превращение идет при
такой низкой температуре как 90 К, его не удается подавить са-
мым быстрым охлаждением (например, 11 000 град/с), оно пол-
ностью обратимо.
Термодинамический анализ условий появления co-фазы при закалке ^атерми-
ческое бездиффузионное превращение) и при изотермических условиях (при от-
пуске) был проведен Вильямсом с сотр. (4972 г.) на примере системы Ti—Мо.
* Багаряцкий Ю. А. и др. «Докл. АН СССР», 1955, т. 105, № 6, с. 1225.
262
б
Рис. 8.30. Расположение атомов в плоскости (011)^ и их
перемещение при перестройке (3->со:
а — атомные структуры представлены в виде плотной упаковки
шаров: слева — для 0-фазы, справа — для co-фазы; сплошные
линии выделяют сечения элементарных ячеек; стрелками пока-
заны смещения атомных рядов [111]^ в противоположных на-
правлениях; б — частица to-фазы внутри 0-кристалла, стрелкой
показаны плоскости (Ш)^; в — образование to-фазы как дей-
ствие продольной волны смещений атомных плоскостей (Ш)р
’Было показано, что прямой переход р—без концентрационных изменений
(массивное превращение) возможен в довольно узкой области. При меньшей
концентрации образуется а' (или а")-фаза, при слишком высокой концентрации
Мо образование со-фазы возможно только при изменении состава (невозможно
массивное превращение). Кинетика изотермического образования co-фазы отли-
чается от других диффузионных процессов выделения своей быстротой. Например,
в сплаве Ti — 25% Мо при 350°С доля превращения уже через 10 мин дости-
гает 0,7. Это объясняется образованием высокодисперсных частиц co-фазы при
очень низком уровне поверхностной межфазовой и упругой энергии.
Особый интерес представляет превращения в интерме-
таллических р-ф а з а х Юм-Розери (AuCd, CuAuZn2,
NiTi, RuNb и др.). В объяснении полиморфного превращения этих
интерметаллидов считается существенным упорядочение атомной
структуры в исходной p-фазе (CsCl).
Связь между исходной структурой р-фазы и низкотемператур-
ной ромбической структурой р"-фазы для AuCd показана на
рис. 8.31. В четырех соседних ячейках р-фазы структуры типа
CsCl выделяется тетрагональная ячейка типа CuAu (р'-фаза).
263
Ромбическая базоцентрированная ячейка р" (4 атом/ячейка) по-
лучается сдвигом 1/6 [110]^ в плоскости (1Ю)ре Ориентационное со-
отношение
(110>? ||(100)?„;
(8.12)
[НОЦ [010]₽„.
Превращение идет как мартенситное с очень малым температур-
ным гистерезисом (AT=AH—Мн) порядка нескольких градусов* **,
тогда как, например, в сплаве Fe—30% Ni Д7^400 С. движущая
сила превращения 3—5 кал/моль вместо нескольких сот для спла-
вов на железной основе. Эти особенности объясняются очень ма-
лой деформацией решетки (для AuCd^2%, в Ре-сплавах^20%).
Именно в сплавах такого типа Г. В. Курдюмов «и Л. Г. Хандрос
впервые наблюдали описанное выше (см. с. 202) термоупругое мар-
тенситное превращение. На этой основе в настоящее время раз-
работаны сплавы, «запоминающие» форму. Эффект запоминания
формы состоит в том, что' если при температуре ниже температуры
начала превращения (Мн) проволоку согнуть в спираль, то при
нагреве выше температуры превращения (Ан) спираль полностью
разогнется. Обратный эффект—если проволоку свернуть в спи-
раль при Т>ЛН, затем охладить и выпрямить при 7<МН (т. е.
в мартенситном состоянии), то при повторном нагреве выше Лк
проволока сама свернется в спираль. Техническое применение (тем-
пературные датчики) получил сплав нитинол — сплав никеля с ти-
таном примерно равноатомного состава. Изменяя состав сплава,
удается изменять температуру полиморфного превращения от —160
до -РЗЗО°С *. Общее объяснение эффекта запоминания формы со-
стоит в том, что полиморфное превращение, которое идет мартен-
ситным (т. е. сдвиговым) способом, подобно скольжению и двойни-
кованию, является механизмом формоизменения.
В сплавах типа NiTi структура мартенсита точно не установ-
лена. Описанная выше ромбическая структура р", возможно,
является промежуточной. Имеются данные о том, что превращение
состоит в кооперативных, но негомогенных сдвигах атомов в на-
правлении < 111 > в плоскости (НО) исходной p-фазы. Движение
атомов начинается при температуре начала превращения (Мн),
заканчивается при температуре конца превращения (Мк), так что
ход превращения напоминает превращение второго рода. Эти сдви-
ги приводят к структурам, образованным определенными последо-
вательностями укладки плотноупакованных слоев гексагональной
или ромбоэдрической сингонии (2//, 477, 12/?, 18/?). Трудность
* Ан— температура превращения при нагреве.
** См., например: Апаев Б. А., Вороненко Б. И. «Металловедение и терми-
ческая обработка металлов», 1973, № 1, с. 23—28; 1975, № 5, с. 28—33.
264
интерпретации дифракционных картин связана с большим числом
дефектов укладки и незавершенностью сдвигов. Некоторые авто-
ры находят в структуре NiTi-мартенсита области с симметрией
моноклинной или триклинной сингонии. Подобные превращения
наблюдали в двойных сплавах Ti с Fe и Со; Zr с Pu, Rh и Pd;
Hf с Os, Ir ,и Pt.
Кроме указанного выше малого объемного эффекта превраще-
ния и небольшого температурного гистерезиса для эффекта запо-
о - №-атсмы
а •- ки-атомы &
Рис. 8.31. Переход от структуры 0-AuCd (а) к 0'- и к низкотемпературной
мартенситной фазе 0"-AuCd (б)
минания формы существенно то, что мартенситное превращение
идет с образованием очень тонких двойников. При прохождении
превращения в два этапа (0,—>10'—*0") могут возникать двой-
ники второго порядка (превращения внутри двойников, сопровож-
дающиеся также двойникованием). В работе Л. Г. Хандроса
и др. * эффект запоминания формы связан с низкой симметрией
продукта превращения, которая приводит к образованию боль-
шого числа ориентировок. С мартенситной структурой связывают
также исключительную демпфирующую способность (способность
гасить упругие колебания) некоторых сплавов.
§ 8.5. АЛЛОТРОПИЧЕСКИЕ ПРЕВРАЩЕНИЯ В УРАНЕ
И ЕГО СПЛАВАХ
Важные для атомной энергетики актиноиды (уран, плутоний)
являются переходными б/^-элементами. В металлическом состоя-
нии, по-видимому, происходит значительная гибридизация элек-
* Арбузова И. А., Коваль Ю. Н. и др. «Физика металлов и металловеде-
ние», 1974, т. 37, № 5, с. 1103.
265
тронов 5f- и 6б/-подуровней и одного электрона 75-уровня. Связи,
возникающие при этом в низкотемпературных модификациях ура-
на и плутония, можно .считать частично ковалентными. Степень
локализации f-электронов сильно зависит от межатомных расстоя-
ний и, следовательно, от температуры и давления. Поэтому уран
имеет три, а плутоний — шесть аллотропических модификаций.
В низкотемпературных модификациях возникают сложные, «уни-
кальные» кристаллические структуры, свойства которых резко
Рис. 8.32. Кристаллическая структура a-U (а) и проекция его
решетки на плоскость (001) (б)
анизотропны. Такие структуры можно описать как плотную упа-
ковку несферических атомов. Теплота превращений и изменение
атомных объемов при аллотропических переходах часто очень вы-
сокие. Так, для р—^у’пРевРаЩення Урана изменение энтропии со-
ставляет 1,11 кал / (град-г-атом), а увеличение объема 0,71%. По-
этому превращения обнаруживают значительный температурный
гистерезис: при охлаждении превращения даже в чистом металле
происходят при температурах на 50—80°С более низких, чем при
нагреве. Резкая анизотропия сил связи приводит к необычным
физико-механическим свойствам и отражается на поведении урана
и плутония под облучением. Так, 6- и трфазы плутония сжимаются
при нагреве; a-U подвержен радиационному «росту» за счет ани-
зотропной конденсации возникающих при облучении вакансий.
Кристаллические структуры урана. Низкотемпе-
ратурная a-модификация, стабильная до 667°С, имеет ромбичес-
кую решетку с четырьмя атомами в элементарной ячейке
(рис. 8.32,я). Решетка образована «гофрированными» слоями ато-
мов, параллельными плоскости (010). Среднее координационное
число равно 12, однако четыре атома (входящие в слои) распо-
ложены на значительно меньшем расстоянии друг от друга. Та-
кую решетку можно рассматривать как искаженную гексагональ-
ную, вытянутую в направлении оси b (см. рис. 8.32,6). Атомы
266
промежуточных слоев, обозначенные на этом рисунке светлыми
кружками, смещены из положений (точки р), в которых они на-
ходились бы в компактной гексагональной решетке. Отношения
b : а и с\а для a-U равны соответственно 2,06 и 1,77, в то время
как для гекс. п. у. решетки 1,73 и 1,63. Н. Т. Чеботарев* устано-
вил, что при нагреве или легировании, например молибденом,
сходство решетки a-U с гексагональной увеличивается. Это про-
исходит потому, что кратчайшие расстояния между атомами в сло-
ях при нагреве от 0 до 600°С возрастают почти на 2%,, а между
слоями практически не изменяются. В результате системы сколь-
жения в a-U изменяются с (010) [100] при комнатной темпера-
туре на (НО) [ИО] и (001) [100] при повышенной. Эти особен-
ности наряду с наличием высокотемпературной о. ц. к. модифика-
ции урана сближают его с ^-переходными металлами IV группы.
Ниже 43К периоды а и Ь решетки a-U скачком возрастают, а пе-
риод с начинает ускоренно уменьшаться.
0-U, существующий до температуры 772°С, имеет сложную те-
трагональную ячейку с 30 атомами. Она оказалась идентичной
структуре так называемых cr-фаз, возникающих в сплавах некото-
рых переходных металлов (см. гл. 4). Лишь плоскости {110} явля-
ются плотноупакованными и «плоскими». Поэтому при комнатной
температуре 0-U (его структура может быть зафиксирована за-
калкой малолегированных сплавов) деформируется лишь в систе-
ме (НО) <001 > и имеет ограниченную пластичность. С увеличе-
нием давления должна происходить делокализация /-электронов,
что приводит к переходу к «обычным» металлическим связям и
выклиниванию области 0-U на Р—Т диаграмме (см. рис. 8.1,г).
y-U стабилен до температуры плавления (1132°С), имеет, как
и высокотемпературные модификации многих металлов, о. ц. ю
структуру. Экстраполированное на комнатную температуру значе-
ние периода решетки равно 3,474 А. Особенности y-U—очень низ-
кие модули упругости и соответственно малая механическая проч-
ность.
Некоторые особенности взаимодействия у р а-
на с легирующими элементами. Равновесные диаграммы
состояния урана с различными элементами имеют некоторое сход-
ство с диаграммами на основе металлов IV группы. Растворимость
в низкотемпературных (0- и особенно a-модификациях) очень низ-
ка. Для a-фазы она не превышает 1 ат. % даже при 650°С. Было
показано **, что величина парциальной энтальпии растворения
в a-фазе лишь частично определяется факторами размерного не-
соответствия урана и легирующего элемента и в значительной сте-
пени искажением специфичных электронных связей. Только плуто-
ний, для которого вклад /-электронов в связь также существен,
* Чеботарев Н. Т. «Атомная энергия», 1961, т. 10, № 1, с. 43.
** Гомозов Л. И., Лютина Э. М., Иванов О. С. «Изв. АН СССР», 1970, №2,
с. 210.
267
широко растворим в a-U. В о. ц. к. (у) модификации урана, ха-
рактеризующейся «обычной» металлической связью, растворимость
элементов, имеющих благоприятный размерный фактор и близкую
электроотрицательность, весьма высока. Так, непрерывный ряд
твердых растворов с y-U образуют Ti, Zr, Hf, Nb. Большое прак-
тическое значение имеют у-сплавы U—Мо (рис. 8.33).
Особенность урана — очень низкая (на уровне 0,1 ат.%) рас-
творимость в нем примесей внедрения С, N, О.
Превращения в нелегированном уране. Для вы-
сокочистого урана закалкой не удается зафиксировать высоко-
температурные фазы. Механизм у—^-превращения эксперимен-
тально не изучен. Модель, разработанная для этого превращения,
предсказывает*, что структурное соотношение должно иметь вид
(11Т)т || (001)?, [1Т0]т II [Т4Ь]?. (8.13)
Низкие модули упругости у-фазы позволяют релаксировать на-
пряжения, и в результате y^p-переходов удается выращивать со-
Рис. 8.33. Диаграмма состояния
U—Мо
вершенные кристаллы р-урана.
Диаграмма изотермического
р—^«-превращения урана высо-
кой чистоты показана на рис. 8.34.
Верхняя С-кривая описывает пре-
вращение нормального типа (за-
рождение и рост зерен а-фазы).
С увеличением переохлаждения
превращение идет по бейнитному
Рис. 8.34. Диаграмма изотермиче-
ского р-нх-превращения для ура-
на высокой чистоты (цифры у кри-
вых— степень превращения, %)
или мартенситному типу и возникают структуры, в которых зерна
a-фазы имеют неправильную форму, их границы «изрезаны».
При непрерывном охлаждении со скоростями выше 65 град/с
мартенситная точка для (3—>а-превращения равна 590°. Установ-
* Kitchingman W. J. «Acta Cryst.», 1968, т. А-24, № 2, р. 282.
268
лено следующее ориентационное соотношение:
(Ю0)о II (101) ;
(8-14)
[001]в || [212]₽.
При термообработке урановых стержней (сердечников тепло-
выделяющих элементов атомных реакторов) большое внимание
уделяют устранению текстур, развивающихся под влиянием тер-
мических градиентов, а также механических напряжений, возни-
кающих при закалке. Обратное а—>р-превращение идет нормаль-
но при любых скоростях нагрева.
При многократной циклической обработке урана с переходом
через температуру а—^-превращения происходит сильное изме-
нение формы образцов, обусловленное анизотропией термического
расширения, объемным эффектом превращения, а также, как по-
казал А. А. Бочвар *, градиентами температуры в образцах.
Превращения в сплавах урана. В сплавах кроме за-
фиксированной закалкой p-фазы наблюдается целый ряд морфо-
логически связанных метастабильных состояний. Среди них а'-пе-
ресыщенная легирующим элементом фаза, ромбическая решетка
который имеет уменьшенный пе-
риод b при мало изменяющихся
периодах а и с по сравнению со
стабильной структурой а (см.
рис. 8.32). При дальнейшем уве-
личении содержания легирующе-
го элемента в пересыщенном (за-
фиксированном закалкой) твер-
дом растворе обнаруживается
а"-фаза с моноклинным искаже-
нием ромбической решетки, что
проявляется в расщеплении ряда
линий на дебаеграммах. Угол мо-
ноклинности плавно увеличивает-
ся с ростом концентрации. Такая
картина наблюдается, в частно-
сти, для подробно изученных
сплавов урана с молибденом и
ниобием. В сплаве с 15 ат.% Nb
при —150°С наблюдали обрати-
мое превращение а"^а'.
Закалка еще более легирован-
ных сплавов от температур суще-
ствования у-твердого раствора
приводит к возникновению тетра-
Рис. 8.35. Температуры мартенсит-
ных превращений (Мн) при охлажде-
нии сплавов урана с молибденом.
Скорости охлаждения, град/с
* Бочвар А. А. и др. В кн.: Труды Второй международной конференции по
мирному использованию атомной энергии. Женева, 1958. Доклады советских
ученых. Т. 3. Ядерное горючее и реакторные материалы. М., Атомиздат, 1959,
с. 554.
269
гональной у°-фазы. Эта структура возникает из о. ц. к. решетки
в результате удвоения периодов а и b и сжатия по оси с. Возмож-
но, что при образовании у°-фазы в системе U—Мо происходит час-
тичное упорядочение атомов различного сорта, однако для у°-фа-
зы системы U—Zr—Nb такое упорядочение не обнаружено.
Наконец, для наиболее легированных сплавов, например
с 13—20 ат.% Мо, .при закалке образуется метастабильная фаза
ys. Ее структура образуется из решетки y-U путем удвоения пе-
риодов; структура частично упорядочена: атомы U расположены
в центрах кубических ячеек, атомы Мо и оставшиеся атомы U
статистически размещены в остальных узлах.
Концентрационные области, в которых фиксируется вышеопи-
санный набор метастабильных фаз, зависят и от условий охлаж-
дения образцов. Поэтому считают, что yQ- и «"-фазы являются
промежуточными ступенями в процессе сдвиговой деформации,
трансформирующей структуры в последовательности у—>у°—>
—>а"—их'. Величина этой деформации у-структуры уменьшается
при введении молибдена (по сравнению с ниобием) и увеличивает-
ся с ростом стимулирующих сдвиговые процессы термических на-
пряжений, т. е. с увеличением скорости охлаждения образцов. При
охлаждении сдвиговая деформация происходит последовательно,
т. е. наблюдаются две мартенситные точки (рис. 8.35).
Прямое -превращение у—>а наблюдается в сплавах урана с та-
кими элементами, как Мо, Nb и пр., при невысокой их концентра-
ции. При температуре немного выше точки Мн механизм является
промежуточным между чисто сдвиговым и диффузионным.
Первый этап сдвигового превращения можно представить как
неоднородную деформацию, состоящую в сдвиге каждого второго
слоя типа (110)7 в направлении [223J? (рис. 8.36, а, б). Второй этап
представляет собой гомогенный сдвиг плоскостей (112)? в направ-
лении (111)? (см. рис. 8.36, в), который превращает моноклинную
решетку в ромбическую. Кроме того, необходима дополнительная
деформация с целью „подгонки" параметров: сжатие вдоль [Ю0]а
и расширение вдоль [010]а и [001]а. Один и тот же механизм опи-
сывает мартенситное превращение и первые стадии диффузионно-
го. О. С. Иванов, Р. М. Софронова и др. показали, что такое рас-
смотрение объясняет и кристаллографические механизмы последо-
вательных у—>у°—^«"-превращений в сплавах урана.
Имеется аналогия превращений у-твердого раствора в сплавах
урана и |3—>«'(«")-превращения в сплавах металлов IV группы
(Ti, Zr, Hf). Прямые переходы у—>а в сплавах U и |3—>а
в сплавах Ti или Zr описываются подобными кристаллографичес-
кими соотношениями. В сплавах урана с Ti, Zr, Hf имеются про-
межуточные фазы с решеткой типа С32, близкой к о. ц. к-фазе и
co-фазе, возникающей при закалке сплавов на основе металлов
IV группы. Отличие состоит в том, что в решетке промежуточных
фаз U происходит упорядочение атомов различного сорта, в то
время как co-фаза не упорядочена.
270
Рис. 8.36. Кристаллографический ме-
ханизм прямого у—нх-превращения
в сплавах урана:
а, б — неоднородная деформация при прев-
ращении, общий вид переходной ячейки и
проекция на плоскость (ПО) у возможных
путей смещений атомов соответственно;
в — однородная деформация переходной
ячейки, следующая за неоднородной де-
формацией
В закаленном состоянии сплав имеет полосчатую структуру (так называе-
мая а'ь-фаза), характерную и для сплавов на основе титана или циркония (а").
Оказывается, что каждая наблюдаемая с помощью светового микроскопа полоса
состоит из тонких параллельных двойников. Электронная микроскопия на про-
свет обнаруживает, что эти двойники состоят, в свою очередь, из еще более
тонких двойников второго и даже третьего порядка. Деформация таких структур
происходит в основном миграцией границ двойников в поле напряжений. Дей-
ствие этого механизма подтверждается проявлением «эффекта памяти» для неко-
торых сплавов урана со структурами а'ъ и а"&. Для сплава с 5 ат. % Мо ста-
рение на максимальную прочность не приводит к появлению электронно-микро-
скопически различимых выделений. Считают, что упрочнение в этом случае
обусловлено закреплением границ двойников скоплениями атомов легирующего
элемента или примесей.
Задачи к гл. 8
Задача 8.1. а. Вычислить относительное изменение объема при переходе же-
леза из у-модификации (г. ц. к.) в a-модификацию (о. ц. к.).
б. То же при переходе в кобальте.
в. Полагая, что объемные эффекты изменяются незначительно при изменении
температуры перехода, сделать вывод о соотношении температурного гистерезиса
обратных переходов в железе и кобальте.
Для расчета: периоды решетки для a-Fe при 911°С а~ 2,8985 А,
• о
для y-Fe при 911 °C а — 3,6394 А; периоды решетки для a-Со при 20°С а = 2,514 А,
с = 4,105 А, для {I-Со при 20°С, а = 3,554 А.
2 71
Задача 8.2. а. Определить число возможных ориентировок кристаллов а-Со
при мартенситном превращении монокристалла P-Со; то же для превращения
у—в стали.
б. Определить число возможных ориентировок при обратном мартенситном
превращении а-Со—>£-Со.
Задача 8.3. а. Какое число различных векторов о. ц. к. решетки может пре-
вращаться в ось с гексагональной ячейки при мартенситном превращении £-Ti
в a-Ti?
-б. Равно ли это число числу возможных различных ориентировок кристал-
лов P-Ti, получающихся из монокристалла a-Ti?
в. Что ограничивает образование всех возможных ориентировок новой фазы
в пределах одного монокристалла (или зерна) при мартенситном превращении?
Задача 8.4. Вывести матричное уравнение преобразования индексов пло-
скостей для ориентационного соответствия, приведенного на схеме рис. 8.25 (для
Zr или Ti).
Задача 8.5. Записать чистую деформацию решетки 5 в матричной форме для
мартенситного перехода в соответствии со схемой, приведенной на рис. 8.25
(для Zr).
ГЛАВА 9
РАСПАД ПЕРЕСЫЩЕННЫХ ТВЕРДЫХ РАСТВОРОВ
§ 9.1. ИЗМЕНЕНИЯ СВОЙСТВ И СТРУКТУРЫ
ПРИ СТАРЕНИИ СПЛАВОВ
Процессы распада пересыщенных твердых растворов и выде-
ления избыточных фаз занимают особое место среди других фа-
зовых превращений, поскольку именно эти процессы происходят
во время заключительных операций термической обработки (ста-
рение, отпуск), формирующих свойства сплавов — конструкцион-
ных высокопрочных, сплавов с особыми магнитными свойствами
и пр. Состояние пересыщенного твердого раствора достигается
обычно быстрым охлаждением (закалкой) сплава от температур,
при которых компоненты сплава образуют твердый раствор (на-
пример, Т[ на рис. 7.25,а). Состояние пересыщенного твердого
раствора можно получить при аллотропическом превращении по
мартенситной кинетике, .когда высоко- и низкотемпературная мо-
дификации сильно различаются по предельной растворимости и
мартенситная кинетика превращения позволяет получить особенно
большое пересыщение. Так обстоит дело в случае сплавов на же-
лезной основе (углеродистых и безуглеродистых) (см. рис. 7.25,г).
Очень большое пересыщение удается получать в сплавах, форми-
рующихся при гальваническом осаждении, а также при закалке
сплавов из жидкого состояния. Наконец, можно перевести сплав
из гетерогенного в гомогенное состояние при введении большого
числа структурных дефектов облучением или пластической дефор-
мацией в условиях сильного взаимодействия этих дефектов с ато-
мами компонента, образующего избыточную фазу.
Распад твердого раствора происходит в области температур
устойчивого гетерогенного состояния (например, Т2 для сплава на
рис. 7.25,а), одновременно обеспечивающей достаточную диффу-
зионную подвижность атомов компонентов сплава. Это может про-
исходить в определенных сплавах при атмосферной («естествен-
ное» старение) или повышенной («искусственное» старение)
температуре. Процессы распада дают состояния сплавов с замеча-
тельным разнообразием физических и механических свойств. Осо-
бый интерес представляет процесс упрочнения — «дисперсионное
твердение» (рис. 9.1). Можно назвать стареющие или так назы-
ваемые дисперсионно-твердеющие сплавы, которые в результате
старения приобретают прочность, сопоставимую с прочностью тер-
мообработанной углеродистой стали: сплавы на основе Си—Be,
сплавы на основе Со, стареющие мартенситные безуглеродистые
18-523 273
стали (предел прочности более 200 кгс/мм2). Дисперсионно-твер-
деющимк являются большинство современных металлических жа-
ропрочных материалов. В этих сплавах сохранение высокопрочного
состояния в течение длительного времени при высокой температу-
ре обеспечивается не только замедлением диффузионных процес-
Рис. 9.1. Зависимость твердости (HV) сплава
Си—1,68% Be от времени выдержки при разной
температуре старения:
1 — 100; 2—150; 3 — 200; 4 — 250; 5 — 280; 6 — 300; 7 — 330;
8 — 360°С
сов (без которых не могут происходить изменения структуры, при-
водящие к разупрочнению), но и подбором таких композиций, ко-
торые обеспечивают наибольшую -стабильность структурно-фазо-
вого состояния высокой прочности.
Существенное значение в формировании свойств стареющих
сплавов имеет обязательный третий компонент сплава — дефекты
15% А1 от времени выдержки при
разной температуре старения после
закалки:
1 — 400; 2 — 500; 3 — 700; 4 — 750; 5 — 800;
6 — 850; 7 — 900°С (Б. Г. Лившиц)
кристаллического строения осно-
вы (матрицы) сплава (вакансии,
дислокации, дефекты упаковки и
границы зерен). Вид и концен-
трации этих дефектов задаются
предысторией сплава и пластиче-
ской деформацией, предшествую-
щей операции старения, или об-
работкой, создающей в матрице
наклеп при фазовом превраще-
нии (например, аустенит-мартен-
ситное превращение предшеству-
ет операции старения в сталях
типа мартенситно-стареющих).
Кроме высокой прочности ста-
реющие сплавы имеют и другие
274
ценные физические свойства, например высокую коэрцитивную си-
лу (рис. 9.2). В сплавах для постоянных магнитов типа алии и
магнико состояние с высокой коэрцитивной силой возникает в ре-
зультате расслоения пересыщенного твердого раствора, причем
наилучшие магнитные свойства достигаются при наложении ориен-
тированного магнитного поля во время термической обработки.
В сверхпроводящих стареющих сплавах (например, Ti—Nb) мак-
симальные значения критической плотности тока достигаются при
определенной дисперсности выделений.
Рис. 9.3. Зоны Гинье — Престона:
а схема плоской зоны в твердом растворе А1—Си; б — схема сферической зоны в твердом
растворе Al—Ag; в — зонная структура в техническом железе (2 месяца при 20°С)
18*
275
Процессы распада могут приводить и к нежелательным изме-
нениям свойств сплавов, например к ухудшению пластичности и
вязкости малоуглеродистой стали или к увеличению коэрцитив-
ной силы и потерь на перемагничивание электротехнического же-
леза.
Особенность процессов распада твердых растворов во многих
сплавах—разнообразие продуктов распада и структурных состоя-
ний. На разных стадиях распада превращение может включать и
чисто диффузионные перераспределения компонентов без измене-
ний в кристаллической структуре, и перераспределения атомов,
связанные с упорядочением, и перестройки кристаллической струк-
туры по разным механизмам, в том числе и по мартенситному.
Характеристику структурного состояния сплава на ранних стадиях
распада пересыщенного твердого раствора (до максимума проч-
ности) впервые дал анализ эффектов аномального диффузного
рассеяния рентгеновских лучей. Эти эффекты для сплава А1—Си
(типа дуралюмина) Гинье и Престон (1938 г.) объяснили образо-
ванием неоднородностей в распределении атомов компонентов,
различающихся по рассеивающей способности для рентгеновских
лучей. В сплаве А1—Си образуются плоские скопления атомов Си
(рис. 9.3,а). Неоднородности структуры подобного типа назвали
зонами Гинье—Престона (зоны Г—П). В последнее время новые
данные о структурных изменениях в течение всего процесса рас-
пада— от образования зон Г—П до выделения стабильных фаз и
их коагуляции — получены методом дифракционной электронной
микроскопии.
Для количественного описания временной и температурной за-
висимостей процесса распада пересыщенного твердого раствора
используют измерения различных свойств. Если старение при раз-
ной температуре приводит к одному и тому же структурному со-
стоянию, то зависимость времени достижения этого состояния от
температуры термической обработки по измерению структурно-
чувствительного свойства (например, твердости или коэрцитивной
силы) дает характеристику температурной зависимости скорости
процесса — энергию активации процесса старения. Почти всегда
это некоторая эффективная величина, прежде всего потому, что
процесс распада сложный и состоит из нескольких элементарных
процессов: например, образования зародышей новых фаз (гомо-
генное и гетерогенное), диффузии (при разных видах и концентра-
циях дефектов), роста частиц. Для анализа временной зависимос-
ти лучше использовать структурно-нечувствительные свойства,
отражающие изменение состава и, следовательно, степень пересы-
щения твердого раствора. Такие исследования проводили по рент-
геновским прецизионным измерениям периода решетки твердого
раствора, по измерениям электросопротивления и намагниченнос-
ти насыщения. Эти методы имеют свои ограничения: процесс вы-
деления избыточного компонента из твердого раствора должен
приводить к снижению электросопротивления, однако зонный рас-
пад сопровождается ростом электросопротивления.
276
Рис. 9.4. Зависимости электросопротивления
(&R/R, %) от времени старения:
а — сталь с 0,6% С и 23% Ni (Мн=30°С), старение при
разной температуре; измерения при температуре — 196°С;
б — сталь с 1,5 мае.% С, вылеживание при комнатной
температуре
Эффект роста электросопротивления на начальной стадии рас-
пада затрудняет количественную характеристику кинетики, но сам
эффект имеет большое значение в диагностике превращения: мно-
гие авторы рассматривают рост электросопротивления как признак
того, что распад твердого раствора идет через образование зон
Гинье — Престона. Рост электросопротивления наблюдали и при
распаде твердых растворов внедрения, в частности при выделении
углерода (и азота) из феррита в техническом железе и <при низ-
Рис. 9.5. Изменения периода
решетки твердого раствора
и твердости в процессе ста-
рения сплава А1—5% Си
для образцов разного сече-
ния (пунктирная линия 1,6
и сплошная 12,7 мм), пере-
несенных непосредственно в
соляную ванну 275°С (Хар-
ди, Хилл)
котемпературном отпуске мартенсита углеродсодержащей стали
(рис. 9.4*). Эти эффекты электросопротивления, а также элек-
тронно-микроскопические наблюдения распада феррита в техни-
ческом железе (см. рис. 9.3,в) и эффекты аномального рассеяния
электронов при отпуске мартенсита позволяют считать, что обра-
зование зон «меет общее значение ,при распаде твердых растворов
замещения и В1недрения.
Обычно не наблюдают изменений рентгенографически измерен-
ного периода решетки твердого раствора на зонной стадии распа-
да (рис. 9.5). Это один из аргументов в пользу представления
о данном состоянии .как о состоянии неоднородного твердого рас-
твора (по Гинье) и об отсутствии «истинного» выделения. С истин-
ным выделением связывают момент, когда начинается изменение
периода решетки твердого раствора (см. рис. 9.5) **. Устойчивость
* Еднерал Н. В., Скаков Ю. А. «Физика металлов и металловедение», т. 26
(1968), вып. 5, с. 850—856. Следует иметь в виду сложный характер изменения
при старении не только электросопротивления /?, но и температурного коэффи-
циента. При распаде феррита в техническом железе и мартенсита в высокоугле-
родистой стали повышение 7? было замечено только в измерениях при комнатной
температуре (см. рис. 9.4,6). В случае стали — достаточно низкая температура
образования мартенсита, чтобы не происходило распада мартенсита сразу после
его образования при закалке.
** Некоторые важные особенности кривых, приведенных на рис. 9.5, анали-
зируются в задаче 9.1.
278
зонной структуры, длительность зиншои стадии в изотермических
условиях и возможность истинного выделения зависят от темпе-
ратурно-концентрационных условий равновесия и от уровня диф-
фузионной подвижности.
Осложнение в анализе процесса выделения по измерениям пе-
Рис. 9.6. Период решетки
твердых растворов в про-
цессе старения при
250°С сплавов Ag—
7,5 мае. % Си (ф, X) и
Ag—6,3 мае.% Си (О,
+ ). Состав (и период
решетки) твердого рас-
твора, образующегося
при выделении в обоих
сплавах, один и то же
(X, +) (Н. В. Агеев,
Хансен, Закс)
риода решетки твердого раствора возникает при так называемом
двухфазном распаде. В классических 'Случаях двухфазного распа-
да (например, в сплавах Ag—Си) пе-
риод решетки исходного твердого рас-
твора не изменяется, но на рентгено-
грамме обнаруживается вторая систе-
ма линий от твердого раствора равно-
весной концентрации (рис. 9.6). Интен-
сивность этих линий (т. е. объем обед-
ненного твердого раствора) со време-
нем старения увеличивается. О степе-
ни распада в этом случае судят не по
изменению периода решетки, а по
изменению соотношения интенсивности
рентгеновских отражений от матрич-
ной фазы — пересыщенного твердого
раствора, мало или совсем не меняю-
щего периода своей решетки, и отра-
жений от участков матричной фазы,
претерпевшей распад (часто практиче-
ски до равновесного состава). Таким
образом, рентгеновское изменение пе-
риода решетки твердого раствора вы-
являют два типа распада: двух- и
однофазный. Эта терминология со-
ответствуют не числу фаз в сплаве,
а числу фаз, выявленных обычным
методом рентгеновского анализа поликристаллического мате-
риала на тех стадиях распада, когда существенно изменяют-
ся свойства материала. При этом дисперсность образующейся
новой фазы (см. рис. 7.28,6) слишком высока, чтобы для нее полу-
чилась система четких рентгеновских отражений. Двухфазный рас-
пад часто называют гетерогенным в отличие от однофазного — го-
могенного. Гетерогенный распад предполагает преимущественное
зарождение новой фазы на разного рода микроскопически выяв-
ляемых дефектах — границах зерен и блоков, на дефектах упаков-
ки и дислокациях. Рентгенографически выявленный двухфазный
распад — это частный случай гетерогенного распада и по микро-
скопическим данным состоит в образовании у границ зерен ячеек—
колоний из равновесных фаз распада и в росте этих колоний пе-
ремещением границы—фронта распада* (см. рис. 7.28). Более
* Это не относится к двухфазному распаду углеродсодержащего мартенсита,
механизм которого пока не выяснен (см« § 9.4).
279
определенным для обозначения двухфазного распада является тер-
мин ячеистый распад.
Параллельно с образованием колоний или ячеек у границ зе-
рен может происходить гомогенный распад внутри зерен. Это об-
наруживается при электронно-микроскопическом исследовании и
в измерениях микротвердости (рис. 9.7).
Поме закалки.
Рис. 9.7. Зависимость микротвердости HV сплава
Ni—2% Be от времени старения при 500°С:
1 — в ячейках прерывистого распада; 2 — в области гомо-
генного распада (внутри зерен)
Для конструкционных сплавов ячеистый распад считали вред-
ным, и его старались предотвратить (например, с помощью спе-
циального легирования в бериллиевой бронзе, используемой для
изготовления пружин). Однако в современной технологии ячеис-
тый распад при выделении наряду с эвтектоидным распадом и
эвтектической кристаллизацией используют для получения так на-
зываемых естественных композиционных материалов.
§ 9.2. НЕКОТОРЫЕ ВОПРОСЫ ТЕРМОДИНАМИКИ И КИНЕТИКИ
ПРОЦЕССОВ РАСПАДА ПЕРЕСЫЩЕННЫХ ТВЕРДЫХ РАСТВОРОВ
Стадии распада. Процесс распада пересыщенного твер-
дого раствора часто многостадиен. Число стадий выделения для
сплавов одной и той же системы зависит от состава сплава и от
температуры. Чем выше степень пересыщения, тем больше стадий
распада, различающихся прежде всего по природе продукта рас-
пада: зоны Г—П, метастабильные фазы, изоморфные матрице,
неупорядоченные или упорядоченные. Это легко показать с по-
мощью схематической диаграммы: свободная энергия фазы (а
или а', аУц, iff, Р)—состав (рис. 9.8). Взята достаточно сложная
гипотетическая система, которая предполагает четыре стадии про-
цесса выделения при большом пересыщении (как наблюдалось,
например, в известных сплавах системы А1—Си, рис. 9.9,а).
Схема предполагает расслоение твердого раствора (а—«1) в спла-
ве состава правее х4. Далее должно быть упорядочение в участках
280
ai(a), затем образование промежуточной фазы и, наконец,
образование стабильной фазы р. Последний переход pz->p может
быть аллотропическим превращением. В равновесии (метастабиль-
ном) с фазами аь а1! и р' находится твердый раствор соответст-
венно состава с4, с3, с^. В равновесии (стабильном) с фазой р
находится раствор состава Ср Твердые растворы состава правее С\
при данной температуре являются нестабильными (или метаста-
бильными). При составе (исходном)
твердого раствора между С\ и с<>
сразу должна возникать стабильная
фаза Р; при составе между с2 и с3
распад должен быть двухстадий-
ным, на первой стадии выделяется
Р'; при составе исходного твердого
раствора между с3 и с4 первый про-
дукт распада — это упорядоченная
фаза а'уп, и только при достаточно
большом пересыщении (при исход-
ном составе правее с±) первым про-
дуктом распада могут быть неупо-
рядоченные области (или зоны)
az— твердого раствора. Если нане-
сти зависимость состава твердого
раствора, находящегося в метаста-
бильном равновесии с фазами си,
a'i, pz от температуры, то диаграм-
ма состояния примет вид, подобный
рис. 9.9,а.
Рис. 9.8. Схематическая диаграм-
ма свободная энергия — состав
(температура задана) системы,
в которой образуются фазы:
1 — неупорядоченный твердый раствор
(а или а'); 2 — упорядоченный твердый
раствор (а'уп); 3 ~ метастабильная
(промежуточная) фаза 4 — стабиль-
ная (конечная) фаза 3
и экспериментальные данные
На рис. 9.9,6 приведены реаль-
ная диаграмма состояния сплавов
А1—А§для областей» и »+у, а так-
же теоретически рассчитанная гра-
ница существования метастабиль-
ных группировок (зоны) атомов Ag
о температурно-концентрационной области сплавов, обнаруживаю-
щих зонную стадию распада. Теоретический расчет (Делингер и
Кнапп, 1952) включал предположение о том, что повышение энтро-
пии системы — это единственный фактор появления относительно-
го минимума свободной энергии при образовании атомных группи-
ровок в твердом растворе.
Коллоидное равновесие и коалесценция час-
тиц при выделении. Следует отметить одну условность
диаграмм метастабильных состояний, связанную с тем, что устой-
чивость дисперсных частиц тех или иных фаз в равновесии с твер-
дым раствором должна зависеть от дисперсности этих частиц.
Устойчивость гетерогенной системы частиц ограниченного размера
определяет не минимум объемной (химической) свободной энер-
гии, но минимум общей свободной энергии, включающей гранич-
ную (поверхностную) межфазовую энергию, которая обусловлена
281
структурой границ раздела, различиями кристаллических структур
или просто химического состава граничащих фаз. В соответствии
с этим образование и рост частицы новой фазы возможны при
определенном «критическом» размере зародыша гКр [см. уравне-
ние (6.97)].
С. Т. Конобеевский* впервые применил к твердым растворам
законы, связывающие растворимость кристаллического вещества
в жидкости с размером кристалликов (см. гл. 6). Это позволило
ему объяснить прекращение (или торможение) распада, связанное
с наступлением коллоидного равновесия. Если отвлечься от ани-
зотропии поверхностного натяжения кристаллов и принять, что
частицы имеют сферическую форму, то ранее выведенное уравне-
ние (6.96) можно использовать как условие метастабильного рав-
новесия этих частиц с твердым раствором.
Уравнение (6.96) запишем в виде
In с(г) = 2aAf/(/?7pr) +1п с^. (9.1)
Условие равновесия между частицами новой фазы и окружающим
их твердым раствором имеет вид гиперболы, асимптотически при-
а
ближающеися с увеличением г к
горизонтальной прямой, соответ-
ствующей In Соо (рис. 9.10). Об-
ласть, расположенная между ги-
перболой и координатными ося-
ми, отвечает ненасыщенным твер-
Рис. 9.9. Диаграмма состояния А1—Си, стабильная (о)/и)4-0) и метастабильные
(co/cdH-O') и ((о/со + Э") —кривые растворимости. Объяснения для линии тп даны
на с. 291 (а); диаграмма состояния А1—Ag: линия предельной растворимости
Ag в А1 или зависимость концентрации твердого раствора а от температуры
в условиях равновесия а- и у-стабильных фаз (б):
1 — зависимость концентрации a-твердого раствора от температуры при его метастабильном
равновесии с группировками — зонами Ag-атомов; 2 — температурный предел существования
первой стадии старения по измерениям твердости; 3 — по данным термического анализа
дым растворам, выше лежит область пересыщенных растворов.
Анализируя данные о различных стадиях распада твердых раство-
* «Изв. АН СССР. Сер. Хим.», 1937, № 5, с. 1209.
282
ров, С. Т. Конобеевский исходит из следующей упрощенной модели
процесса. В некоторый момент времени по всему объему одно-
временно возникают частицы новой фазы (Л^ частиц в единице
массы вещества). Все эти частицы растут с одной и той же по-
стоянной линейной скоростью, так что в любой момент размеры
всех частиц одни и те же. С увеличением размеров выделяющихся
частиц происходит обеднение твердого раствора вторым компонен-
том. Если начальная концентрация пересыщенного твердого рас-
твора с0, концентрация в фазе выделения сВыд, то данному общему
объему частиц 4/3лг3?/1 с плотностью р в массе материала Р будет
соответствовать концентрация твердого раствора с, определяемая
по уравнению
cNt = С„ - wW,cBbIAр/Р =с„ — КУ/. (9.2)
На рис. 9.10,а точка с0 на оси ординат изображает начальную
концентрацию распадающегося твердого раствора и пунктирная
кривая 1 показывает уменьшение концентрации с ростом кристал-
ликов новой фазы Ni. Эта кривая пересекает линию равновесия
в точках М' и U. Обозначим г0 радиус равновесного зародыша
новой фазы; зародыш с радиусом, превышающим г0, будет увели-
Рис. 9.10. Графическое изображение условий равновесия при выделении высоко-
диспеосной фазы из раствора, начальный состав которого с0 (С. Т. Конобеев-
ский) (а), и равновесная граница растворимости (г->оо) и линии метастабильно-
го «коллоидного» равновесия (Г1<У2) (б)
чиваться. Однако одновременный рост AG кристаллов не продол-
жается до достижения устойчивого равновесия, чему соответство-
вало бы пересечение пунктирной кривой с линией 0 = 0^ (вдоль
этой линии концентрация твердого раствора с равна концентрации
насыщения для кристалликов бесконечно большого радиуса).
Рост кристаллов второй фазы прекращается по достижении ими
радиуса гь что отвечает пересечению кривой изменения реальной
концентрации твердого раствора с гиперболой, соответствующей
уравнению (9.1). Наступает метастабильное «коллоидное» равнове-
сие кристалликов радиуса Г\ с твердым раствором концентрации
й>Соо (см. также рис. 9.10,6, где одна из пунктирных кривых
изображает границу растворимости для кристалликов радиуса гь
283
а вторая — несколько большего радиуса г2). Если бы число
выделившихся частиц #1 оставалось в дальнейшем неизменным,
коллоидное равновесие было бы устойчивым. Когда распад про-
текает при таких низких температурах, что диффузия на сколько-
нибудь значительные расстояния практически исключается (на-
пример, при низкотемпературном отпуске закаленной бериллиевой
бронзы или закаленной стали), коллоидное равновесие действи-
тельно затягивается на неопределенно долгое время. Однако при
более высокой температуре состояние коллоидного равновесия на-
рушается благодаря коалесценции выделений. Описанная идеаль-
ная схема распада с равновеликими частицами выделений на самом
деле никогда не реализуется: в момент наступления коллоидного
равновесия в сплаве имеются частицы разного размера. Если кон-
центрация твердого раствора по всему объему сплава достаточно
постоянна, то по отношению к крупным частицам раствор в соот-
ветствии с уравнением (9.1) будет несколько пересыщен, и части-
цы эти медленно растут, обедняя твердый раствор и делая его
ненасыщенным по отношению к мелким частицам—последние
растворяются вплоть до полного их исчезновения. В результате
коалесценции крупные кристаллики выделившейся фазы увеличи-
ваются не только за счет поглощения меньших выделившихся
частиц (см. рис. 9.10), одновременно происходит дополнительное
понижение концентрации твердого раствора. Пунктирная кривая 2,
показывающая изменение состава твердого раствора при росте N2
кристалликов (A^Cl/Vi), проходит более полого, чем кривая Л и
пересекает линию коллоидного равновесия в точке, соответствую-
щей большему равновесному радиусу частицы г2 и меньшей кон-
центрации твердого раствора c2<^i (см. рис. 9.10). По мере
растворения все новых и новых малых частиц и укрупнения боль-
ших кристаллов второй фазы сплав постепенно приближается
к устойчивому равновесию, отвечающему концентрации твердого
раствора с^. Как отмечалось выше, при анализе формулы (9.1)
применительно к жидким растворам уже при достижении кристал-
ликами размеров в несколько микрон величина сг практически
перестает отличаться от Соо (см. табл. 6.4). Влияние размеров
частиц на их растворимость тем сильнее, чем больше коэффи-
циент поверхностного натяжения а. Поскольку поверхностное на-
тяжение между частицами, выделившимися из твердого раствора,
и матрицей существенно меньше (по крайней мере не превышает
нескольких десятков эрг на квадратный сантиметр), то можно
считать, что равновесная концентрация Соо твердого раствора уста-
навливается уже тогда, когда размеры выделившихся частиц
приближаются к 1 мкм.
Однако процесс приближения к равновесию должен быть очень
медленным, потому что он связан с необходимостью растворения
малых частиц выделившейся фазы и с диффузией на большие
расстояния, а время диффузии, как известно, пропорционально
квадрату пути диффузии.
Большая поверхностная (межфазовая) энергия при выделении
284
из твердого раствора возможна при отсутствии когерентности, что
чаще всего соответствует поздней стадии выделения, обычно за
максимумом твердости при проведении старения или в процессе
эксплуатации материала при повышенных температурах.
На стадии коалесценции средний размер частиц изменяется по
закону
R(*W/3> (9.3)
а число частиц
ЛЦ/)~1/./. (9.4)*
Метастабильное и лабильное состояние пере-
сыщенного твердого раствора. Спинодальный
распад. При определенных температурно-концентрационных
условиях состояние пересыщенного раствора оказывается абсолют-
но нестабильным (лабильным). Эти о
условия можно выяснить с помощью
равновесной диаграммы состояния,
отражающей только значения объ-
емной (химической) свободной энер-
гии. Это оправдано, так как при
анализе образования зон или коге-
рентных выделений можно прене-
бречь поверхностной энергией и рас-
сматривать процесс распада с об-
разованием «сегрегатов» флуктуа-
цией состава. Этот подход был
впервые развит С. Т. Конобеев-
ским.
Типичная зависимость свободной
энергии твердого раствора от соста-
ва приведена на рис. 9.11. Для спла-
ва состава Xi при образовании сег-
регатов отклонением их состава от
среднего (x7i и x"i) свободная энер-
гия возрастает по сравнению со сво-
бодной энергией однородного твер-
дого раствора; термодинамически
выгодными могут быть только сегре-
гаты, состав которых приближает-
ся к составу равновесной фазы с2
(например, x"'i). Для твердого рас-
твора состава х2, напротив, самое
небольшое отклонение состава от
среднего приводит к понижению
свободной энергии, причем увеличе-
ние разности концентраций матри-
цы и сегрегатов непрерывно пони-
жает энергию системы — происхо-
дит самопроизвольное расслоение
О II I и i и, 11
А х^х] х2х^ В
с2
Рис. 9.11. Равновесная диаграмма
состояния сплавов, образующих
при затвердевании непрерывный
ряд твердых растворов (а); зави-
симость свободной энергии от со-
става при температуре (6); точ-
ки Qj и Q2—составы метастабиль-
ных твердых растворов при тем-
пературе 1\, которым соответст-
вуют точки перегиба qr и q2 на
кривых изменения свободной энер-
гии F
* Вывод приведен в гл. 10.
285
твердого раствора на стабильные фазы. Очевидно, понятие «за-
родыш выделения» в данном случае не имеет смысла. Спла-
вы, в которых распад происходит сегрегацией или расслое-
нием без образования зародышей, характеризуются кривой зави-
симости свободная энергия — состав, обращенной выпуклостью
вверх, т. е. d2F/dc2<S} (см. рис. 9.11,6 между точками q{ и q2).
Если за движущую силу диффузии принять градиент свободной
энергии, то знак в уравнении диффузии определяется знаком
d2F/dc2\ разница концентраций должна увеличиваться при отрица-
тельном d2F!dc2 (эффект «восходящей» диффузии).
Рис. 9.12. Схемы распределения концентрации вдоль некото-
рого направления при распаде твердого раствора: путем обра-
зования частиц и их роста (а) и развития флуктуаций состава
твердого раствора (б); Ci — конечный состав твердого раство-
ра (равновесный при температуре старения), с2— состав вто-
рой фазы (равновесный); А—начальная стадия распада; Б —
некоторый промежуточный момент; В — конечное состояние
Температурная зависимость концентрации твердых растворов,
при которой d2Fldc2 меняет свой знак, показана на рис. 9.11,а
(спинодалъ или спинодальная линия). Распад в области темпера-
тур и составов внутри спинодали не связан, как при обычном вы-
делении, с преодолением энергетического барьера для создания
зародыша и определяется только диффузионными процессами.
В более детальной теории критический зародыш характеризуется
как локальная, но размытая группировка атомов в однородной
матрице. Размер этой группировки соответствует размеру устой-
чивого зародыша в классической теории и стремится к бесконеч-
ности, если состав сплава приближается к пределу растворимости
или к спинодальной кривой. Минимальный размер должен быть
при некотором промежуточном составе сплава. Различие состава
зародыша (в центре группировки) и матрицы также зависит от
состава сплава и становится равным нулю, если состав сплава
приближается к спинодальному. Это должно приводить к исчезно-
вению энергетического барьера при зарождении, несмотря на то,
286
что размер зародыша растет. При спинодальном распаде в твер-
дом растворе возникают флуктуационные волны концентрации —
модулированная структура, длина волны или период такой моду-
лированной структуры определяется температурой старения (мо-
жет быть, порядка нескольких десятков и сот ангстрем), ампли-
туда (или разница концентраций) может возрастать вплоть до
полного распада твердого раствора и потери когерентности.
Распределение концентрации при распаде твердого раствора
с образованием и ростом зародышей схематически показано на
рис. 9.12,а, при беззародышевом или спинодальном распаде — на
рис. 9.12,6. Конечная структура может быть одинаковой (ста-
дия В). Принципиальное различие в морфологии распада свойст-
венно начальным стадиям А, Б. Зародыши растут в результате
диффузии, направленной в сторону уменьшения градиентов кон-
центрации, при спинодальном распаде, напротив, идет процесс
«восходящей» диффузии — градиенты концентрации в твердом
растворе усиливаются.
Начальная стадия спинодального распада состоит в образова-
нии синусоидальных флуктуаций состава с очень малой амплиту-
дой (см. А на рис. 9.12,6). Границы раздела в процессе незавер-
шенного распада следует представлять весьма размытыми, с не-
прерывным изменением состава. С таким характером границ
связаны два дополнительных члена в выражении для свободной
энергии системы: градиентная энергия как аналог поверхностной
энергии и энергия упругих искажений, которые стабилизируют
твердый раствор по отношению к малым флуктуациям состава
r = J {f (с) + &(ус)2 + 712[£/(1 - Vn)](c-co)}tfo. (9.5)
V
Здесь f(c)—свободная энергия на единицу объема гомогенного
твердого раствора состава с; £(Vc)2— градиентная энергия; по-
следний член представляет собой упругую энергию; г] — линейное
расширение решетки твердого раствора при изменении его состава;
Е — модуль упругости; vn—коэффициент Пуассона; cf,— средний
состав сплава.
Разница значений свободной энергии на единицу объема ДБ/е
между исходным гомогенным раствором и неоднородным твердьш
раствором
= 0/4] [-gt-|-2т)2У + 2£02 j. (9.6)
Здесь Y=E/(1—vn); Л(0, /) и 0 = 2лД характеризуют амплитуду
и волновой вектор синусоидальных флуктуаций* состава в момент
времени t\ г — вектор положения, т. е.
с—со=Д(0, 0cos(0г). (9.7)
Очевидно, состояние однородного твердого раствора неустой-
чиво, если в уравнении (9.6) правая часть оказывается отрица-
тельной, т. е. если
2>+2?У<0, (9.8)
287
и волновое число окажется меньше критической величины, опре-
деляемой
i [да-+2’>’4 <9-9)
Область метастабильности отделяется от области лабильного
твердого раствора линией, для которой
d2f/dc2 + 2vfY=0. (9.10)
Эта линия называется когерентной спинодалъю (рис. 9.13,а).
Степень понижения температуры максимума спинодали может
быть оценена в приближении регулярных растворов
ДТ=2т]2Ус(1—с)/Ж. (9.11)
Здесь k — постоянная Больцмана и Nv — число атомов на единицу
объема. В табл. 9.1 для нескольких систем с расслоением в твер-
Таблица 9.1
Степень переохлаждения ДТ для достижения
максимума когерентной спинодали*
Система 4 T° , c макс Д7”, c
Al—Zn 0,026 350 40
Au—Pt 0,038 1260 200
Au—Ni 0,150 810 2000
* Cahn J. Chem. Phys.“, 1965, v. 42, № 1, p. 93—99.
дом состоянии, имеющих диаграммы состояния типа рис. 9.11,
приведены значения максимальной температуры расслоения Т°макс
и положение максимума когерентной спинодали (ДТ) по отноше-
нию к этой температуре. Согласно этим данным спинодальный
распад можно ожидать в сплавах Au—Pt при температуре ниже
1000°С (1260—200°С). В сплавах системы Au—Ni, где могут воз-
никать очень большие напряжения из-за различий в периодах
решетки Au и Ni (и соответственно твердых растворов на основе
компонентов), спинодальный распад вообще не должен реализо-
ваться.
На рис. 9.13,а для системы Au—Ni нанесена «когерентная» спинодаль (кри-
вая 3), полученная в более поздней работе*. Экспериментальная проверка для
этой системы затруднительна, поскольку вообще для нее характерен гетероген-
ный ячеистый распад. Однако в области довольно низких температур (ниже
250—200°С), несмотря на значительное уменьшение уровня диффузионной по-
движности, становится заметным непрерывный распад. Эта область близка к по-
ложению когерентной спинодали. Приведенная на рис. 9.13,6 спинодальная кри-
вая для сплавов Au—Pt вычислена ** без учета упругой энергии («химическая»
* Golding В., Moss S. «Acta Met.», 1967, v. 15, № 7, p. 1239—1241.
** Borelius G. «Phys. Scr.», 1971, v. 4, p. 127—131.
288
спинодаль). Из анализа изохронных кривых изменения электросопротивления,
характеризующих скорость распада при разных температурах, Борелиус опреде-
лил температуры резкого замедления или ускорения процесса распада. Как вид-
но из графика, эти точки хорошо ложатся на химическую спинодаль, что под-
тверждает небольшое различие химической и когерентной спинодалей для спла-
вов данной системы в отличие от Ап—Ni.
Из уравнения (9:9) следует, что в твердых растворах внутри концентрацион-
но-температурной области, называемой когерентной спинодалью, будут устойчи-
выми и станут усиливаться (т. е. увеличиваться амплитуда) все концентрацион-
ные волны флуктуаций с волновым числом Р<рКр.
Рис. 9.13. Положение спинодальных линий на диаграммах состояния Au—Ni (а)
и Au—Pt (б):
/—линия расслоения твердого раствора ограничивает область стабильного равновесия двух
фаз (а' + а"); 2 — линия «химической» спинодали ограничивает область метастабильного со-
стояния пересыщенного твердого раствора (выше 2) от области его лабильного состояния
(ниже 2) без учета упругой энергии; 3 — «когерентная» спинодаль, т. е. то же, что 2, но
с учетом упругой энергии. Спинодаль для Al—Pt построена по температурам резкого за-
медления (О) или ускорения (X) процесса распада
Как видно из рис. 9.14*, наибольшей скоростью увеличения амплитуды
характеризуются волны в очень узкой области значений волнового числа возле
₽М. Следовательно, наблюдаемое расстояние между частицами соответствует (Зм,
которое, по расчетам Кана, равно PKp/F 2.
При увеличении степени переохлаждения (в пределах когерентной спинода-
ли) (Зм возрастает, а длина волны концентрационных флуктуаций (Хм —2л/(Зм)
становится меньше, поскольку при большой степени переохлаждения величина
d^f/dc2 по абсолютному значению растет, оставаясь отрицательной, и соответст-
венно увеличивается (3Кр [см. уравнение (9.9)]. Амплитуда флуктуаций не зави-
сит от переохлаждения и определяется условием потери когерентности, которая
происходит, если различия в периодах приводят к когерентным деформациям
порядка 1% (см. с. 188).
В связи с упругой анизотропией решетки развитие флуктуаци-
онной волны идет в направлениях, соответствующих наименьшим
значениям упругих констант. При (сп—с12)/2с44<1 наименьший
модуль упругости оказывается вдоль <100>, следовательно, мо-
* См. также § 10.6.
19—523
289
Рис. 9.14. Зависимость факто-
ра скорости развития модули-
рованной структуры при спино-
дальном распаде от волнового
числа (Р=2л/Х), характери-
зующего период модуляции или
длину волны Л
дуляции химического состава (и периода решетки) твердого рас-
твора будут именно вдоль направлений куба. При (сц—Ci2)/2с44> 1
возможны модуляции вдоль направлений < 111 >.
Модуляция рассеивающей способности атомов в решетке (или
модуляция межплоскостных расстояний) дает характерный эф-
фект на рентгенограммах — сателлиты возле определенных реф-
лексов.
Многие сплавы в начальной стадии старения дают интенсивное
рассеяние рентгеновских лучей в виде сателлитов возле основных
линий твердых растворов. Интенсив-
ность этих сателлитов позволяет заме-
тить их при съемках поликристалли-
ческих образцов. Впервые это наблю-
дали при исследовании сплавов Си—
Ni—Fe. Пересыщенный твердый рас-
твор в этом сплаве имеет г.ц.к. решет-
ку; в равновесном состоянии при низ-
кой температуре сплав состоит из
двух изоморфных фаз (г.ц.к. с разни-
цей в периоде около 2%). Рентгенов-
ские данные о модулированной струк-
туре подтверждены электронно-микро-
скопическими исследованиями. Однако
наблюдение «модулированной» струк-
туры еще не позволяет считать распад
спинодальным, поскольку периодиче-
ское расположение частиц благодаря
упругому взаимодействию возникает и
при обычном механизме распада образованием и ростом зароды-
шей (см. с. 196). Исследование, проведенное с помощью анализа
эффекта Мёссбауэра, показало, что состав выделяющейся фазы
в сплаве Си—27Fe—9Ni при температуре старения ниже 400°С
непрерывно изменяется со временем. При более высоких темпера-
турах состав выделения не меняется. Это говорит о том, что спи-
нодальный механизм старения действует только при низких темпе-
ратурах (<400°) *.
Роль вакансий при зонном или низкотемпе-
ратурном распаде. Максимальную скорость зонного распа-
да наблюдают в самом начале процесса.
Особое значение при зонном распаде имеет большая концентра-
ция избыточных вакансий, которая возникает из-за очень боль-
шой разницы между температурами гомогенизации (закалки) и
старения.
Скорость старения сплавов резко уменьшается после кратко-
временного нагрева (возврата), закалки с замедленным охлаж-
дением, слабой пластической деформации, когда движение дисло-
каций только одной системы скольжения «смывает» избыточные
(«замороженные» закалкой) вакансии. Пример влияния прерыва-
* Данные Б. С. Бокштейна, А. Л. Мирского, Ю. А. Скакова.
290
ния охлаждения при закалке (ступенчатая закалка) на развитие
зонного распада (по эффекту подъема электросопротивления) по-
казан на рис. 9.15. Большую скорость образования зон часто объ-
ясняют только очень большим коэффициентом диффузии, обуслов-
ленным избыточными вакансиями. Однако отмечалось несоответ-
ствие между временем образования зон и временем существования
избыточных вакансий. Поэтому не-
правильно считать, что вакансии
влияют только на кинетику процес-
са через коэффициент диффузии.
Может быть существенным участие
вакансий в зонном распаде как
«третьего» компонента, способст-
вующего уменьшению критического
размера зародышей и увеличению
их стабильности или спинодальному
механизму распада*. О термодина-
мическом характере (в том смысле,
дчто влияние не сводится к простому
'Изменению кинетики, т. е. скорости
^процесса) влияния избыточных ва-
кансий на процесс старения свиде-
тельствуют данные, полученные при
наблюдении за так называемыми
свободными от выделений пригра-
ничными областями. Такие зоны
(в Al-сплавах шириной обычно по-
рядка 0,1 мкм) иногда связывали
Рис. 9.15. Зависимость относи-
тельного изменения удельного
электросопротивления сплавов
А1 + 1,9%Си от времени старения
при 0°С (Де Сорбо и др.):
1 — после прямой закалки до 0°С; 2 —
после ступенчатой закалки (выдержка
при 200°С в течение 10 с, затем охлаж-
дение также до 0°С и пластической де-
формации); 3 — то же, что 2, но без
пластической деформации
с уходом атомов избыточного компонента к границе зерен. Однако
существует резкая граница области, свободной от выделения (при
непрерывном изменении концентрации избыточного компонента
должно бы быть постепенное изменение количества выделений),
а при понижении температуры старения процесс распада идет
в этих же областях. Сделан вывод, что при данной температуре
старения должна быть критическая концентрация вакансий, при
которой возможны зарождение и рост частиц. Представление
о том, что нельзя сводить влияние избыточных вакансий к про-
стому ускорению диффузии, подкрепляется тем, что, несмотря на
различные механизмы диффузии в растворах замещения и вне-
дрения и принципиально различную роль вакансий в этих меха-
низмах, при низкотемпературном распаде растворов внедрения
образуются такие же приграничные области, свободные от вы-
делений или зон (см. рис. 9.3,в).
Возврат при старении. В процессе старения при до-
статочно низкой температуре сплав существенно изменяет свойст-
ва: растет прочность, понижается пластичность, часто повышается
электросопротивление и т. д. Если после достижения некоторого
* Вакансии могут, например, приблизить упругую (или реальную) спино-
даль к химической.
19* 291
эффекта старения сплав нагреть до более высокой температуры,
чем температура, при которой проводилось старение, то проис-
ходит возврат свойств: твердость, прочность, электросопротивле-
ние и другие свойства сплава «возвращаются» к уровню исходного
состояния, т. е. к уровню свойств закаленного состояния. При этом
температура нагрева, при котором может происходить возврат,
существенно ниже температуры нагрева для закалки и находится
ниже равновесной температуры перехода сплава в однофазное
состояние. Тем не менее если
Рис. 9.16. Изменение прочности техни-
ческого железа в процессе старения при
150°С (1 ч), в результате возврата при
250°С (70 с), а также в процессе повтор-
ного старения при 250°С (до 20 ч)
(Б. Г. Лившиц, Л. И. Цупрун)
уровень свойств оказывается
характерным для однофазного
состояния, то следует считать,
что при таком нагреве возни-
кает именно однофазное со-
стояние. Прямым доказатель-
ством того, что температура,
при которой происходит воз-
врат, соответствует гетероген-
ному равновесному состоянию,
служит развитие процесса ста-
рения при продолжении вы-
держки. На рис. 9.16 показа-
но изменение прочности тех-
нического железа (измеряли
число оборотов шпинделя машины при скручивании образца
до разрушения) в процессе старения при 150°С (до 1 ч), воз-
врат прочности (разупрочнение) в результате кратковременно-
го (70 с) нагрева при 250°С (участок ab дан не в масштабе) и
последующее старение (т. е. снова упрочнение) в процессе вы-
держки при 250°С. В данных условиях возврат свойств практиче-
ски полный.
Полнота возврата зависит от длительности старения, т. е. от
того, как далеко зашел процесс распада при старении, и от тем-
пературы нагрева для возврата; в свою очередь, температура
полного возврата зависит от состава сплава.
Зависимость температуры, при которой наступает полный воз-
врат, от содержания меди, по данным Д. А. Петрова, была при-
ведена на рис. 9.9,а (линия шп). Для объяснения явления воз-
врата гетерогенное состояние, возникающее после старения при
температуре ниже линии тп (см. рис. 9.9,а), следует считать тер-
модинамически нестабильным при температуре выше линии тп.
Эта нестабильность может быть связана с тем, что линия тп пред-
ставляет собой метастабильное равновесие твердый раствор — ме-
тастабильный продукт выделения (метастабильная промежуточ-
ная фаза — 0"), либо с высокой дисперсностью частиц выделения.
В последнем случае линия тп соответствует метастабильному
равновесию твердого раствора и дисперсных частиц фазы вы-
деления определенного размера. Обоснование этого представления
292
было дано С. Т. Конобеевским в термодинамическом анализе
процесса выделения.
Как следует из общей теории образования фаз, равновесные
зародыши фазы выделения, возникшие при низкотемпературном
старении (т. е. при большем пересыщении), должны быть меньше
кристалликов, устойчивых при более высокой температуре
(Г2>Г1). Поэтому если частицы новой фазы, возникшие при
низкотемпературном ста-
рении, успевают за время
выдержки при низкой
температуре 7\ (см. рис.
9.10,6) вырасти до разме-
ров Г1 <г2, то в результа-
Рис. 9.17. Изменение твердости HV и электросопротивления R/Ro стали 0X24,
содержащей 0,05 мае.% N, в процессе старения при 475СС после закалки в воду
(?) и в растворе щелочи (2) (а); микроструктура после закалки в воду и старе-
ния 3 мин при 475СС (б); то же, но закалка в растворе щелочи (в) (Ю. О. Ме-
женный, Ю. А. Скаков, Р. С. Ярославцева)
те нагрева до Т2 они окажутся меньше критических размеров и
почти все растворяются — произойдет возврат. Однако после неко-
торой выдержки при Т2 появятся новые центры с размерами г2, они
будут расти — разовьется процесс высокотемпературного старения.
Следует отметить еще одно явление, связанное с возвратом. В начальном
периоде старения иногда место повышения прочности, которое всегда должно
быть при переходе от действительно гомогенного к гетерогенному состоянию, на-
блюдается понижение прочности. Как видно из рис. 9.17 (см. также рис. 9.5),
после короткого периода разупрочнения прочность увеличивается, достигает
максимума и снижается в соответствии с общим представлением о связи процес-
293
сов распада и упрочнения. В рассматриваемом случае (сплав Х24 с азотом) мож-
но считать, что в процессе охлаждения при закалке в воду идет процесс распада
пересыщенного азотом железо-хромистого феррита (по-видимому, возникает гете-
рогенность типа зонной структуры) *. Максимальная степень протекания этого
процесса, очевидно, соответствует температурам ниже 500°С, которые сплав про-
ходит достаточно медленно. Поэтому нагрев до 475' С в течение очень короткого
времени (порядка нескольких десятков секунд) приводит к возврату, при про-
должении выдержки при 475°С идет процесс распада в последовательности, свой-
ственной именно температуре старения 475°С. Закалка в растворе щелочи
фиксирует однородный твердый раствор, об этом свидетельствует низкая твер-
дость после закалки. Задержка в изменении электросопротивления при кратко-
временном старении после закалки в щелочном растворе объясняется образова-
нием структуры типа зонной.
Изотермические и комбинированные обработ-
ки. Изменения скорости охлаждения при закалке приводят к из-
менениям кинетики и эффектов старения при выделении когерент-
ных и частично когерентных фаз, с которыми обычно связывают
процессы упрочнения при технологическом старении. Эти измене-
ния можно связывать как с изменениями состояния кристалличе-
ской решетки матрицы (концентраций и типа дефектов, образую-
щихся в процессе закалки), так и с распадом в процессе замед-
ленного охлаждения. Установление точных механизмов влияния
подобных вариаций режимов термической обработки имеет важ-
ное значение для рационализации технологии обработки старею-
щих сплавов.
Существенное различие кинетики и эффектов твердения при
высокотемпературном старении сплава А1—4 мае. % Си (выделе-
ние 6"- и 9'-фаз) после резкой закалки в воду и охлаждения в спо-
койном воздухе (кривые 1 и 3 на рис. 9.18) нивелируются, если
материал подвергается пластической деформации. На этом осно-
вании авторы считают, что замедление процесса старения и умень-
шение эффекта твердения после замедленного охлаждения свя-
заны с уменьшением числа закалочных дефектов, способствующих
выделению упрочняющей фазы. Восстановление эффекта тверде-
ния после дополнительной деформации (кривые 2 и 4, см.
рис. 9.18) свидетельствует о несостоятельности предположения
о распаде твердого раствора в процессе замедленной закалки и
одновременно о том, что пластическая деформация создает в ма-
териале такие конфигурации дефектов, которые близки к обра-
зующимся после резкой закалки. Однако должна существовать
некоторая критическая скорость охлаждения, ниже которой в про-
цессе охлаждения идет распад твердого раствора.
Эту скорость можно определить методами, разработанными при исследова-
нии термической обработки стали, например с помощью диаграмм изотермическо-
го превращения. В последнем случае развитие превращения во времени легко
количественно характеризовать как изменение количества превращенного твердо-
го раствора только для двухфазного или ячеистого распада. Обычно ограничи-
ваются определением времени начала превращения. Поскольку начальная стадия
распада может не сопровождаться заметными изменениями механических свойств
* Меженный Ю. О., Скаков Ю. А. «Изв. ®узов. Черная металлургия», 1967,
№ 11, с. 120—125.
294
.(электросопротивления и т. д.), но существенно влияет на кинетику последующе-
го превращения, то после фиксирования состояния заданной изотермической вы-
держки быстрым охлаждением образец подвергается контрольному, или «диагно-
стическому», старению. Режим этого старения выбирают близким к обычному
технологическому, т. е. таким, чтобы обеспечивалось максимальное изменение
свойств. Например, для бериллиевой бронзы берут температуру 300°С и выдерж-
ку 2 ч. Опыт проводят по следующей схеме: обрабатывают на твердый раствор
(нагревают до температуры закалки), быстро переносят в изотермическую ван-
ну, выдерживают в ванне заданное время, резко охлаждают (например, зака-
ливают в воду), нагревают до температуры контрольного старения и после про-
Рис. 9.18. Влияние скорости охлаждения при за-
калке на кинетику твердения сплава А1—
4 мае.% Си при температуре 200°С непосред-
ственно после закалки (/, 3) и после холодной
прокатки с обжатием 10% (2, 4):
1,2- охлаждение в воде; 3, 4 — охлаждение на возду-
хе (Тим, Боршерс)
показаны значения предела прочности сплава Си—Be после контрольного старе-
ния для состояний (точки на графике), соответствующих заданным температуре
и времени изотермической обработки. Точки с заданным уровнем свойства, на-
пример (Тв== 140 кге/мм2, примерно соответствующим уровню свойства при ста-
рении после обычной закалки, принимают соответствующим состоянию нераспав-
шегося твердого раствора. Как видно из рис. 9.19, таким образом можно отме-
( тить время начала распада при разной температуре. Зависимость времени начала
превращения от температуры имеет С-образный характер, что подтверждается
и измерениями других свойств <(см. рис. 9.19). Эта зависимость следует из теории
фазовых превращений, идущих через образование зародышей и их рост. В связи
с отмеченной ролью при старении дефектов решетки, формирующихся в процессе
'закалки, имеется некоторая условность в истолковании С-образных кривых;
кроме того, в определении свойств после контрольного старения может оказать
влияние наложение процессов старения в разных температурных интервалах. По-
следнее очень важно в теоретическом и практическом отношениях.
Для магнитных сплавов типа алии (на основе системы
Fe—Ni—Al) известно, что наибольшая коэрцитивная сила полу-
чается не при обычной обработке (закалка плюс старение),
а в процессе охлаждения с определенной (критической) скоростью
или при комбинированном старении в двух температурных интер-
валах: высокотемпературное старение плюс низкотемпературное
старение (ВТС + НТС). Подобное явление было отмечено в от-
ношении прочностных свойств жаропрочных сплавов на
Ni—Сг-основе.
295
Существенные изменения кинетики и эффектов упрочнения
недавно обнаружены при сложном старении некоторых сплавов
в комбинации НТС + ВТС. Например, длительное пребывание
(вылеживание) сплава А1—Mg—Si при комнатной температуре
5 // 50 60 90 500 600 900
Ьремя изотермической выдержки, с
а
Рис. 9.19. Диаграммы температура — время — превращение для сплава
Си—2,46 мае. % Be—0,3 мае.% Со:
а — предел прочности после изотермической выдержки и контрольного старения
при 300°С, 2 ч; б — С-кривые, построенные по измерениям разных свойств; 1 —-
ав = 140 кг/мм2; 2 — HV = 390 ед.; 3 — повышение электропроводности (23%), 4 —
HV=400 ед. для эвтектоидного превращения; 5 — критическая скорость охлаж-
дения (60 град/с) (Тода)
после закалки приводит к задержке твердения при последующем
искусственном старении и снижению уровня прочности на 10%,
а для сплава Ni—Be обработка НТС + ВТС приводит к дополни-
тельному упрочнению (рис. 9.20). Эти результаты свидетельствуют
о том, что нагрев после низкотемпературного старения нельзя
296
сводить только к явлению возврата, т. е. к растворению дисперс-
ных образований, возникающих при низкотемпературном старе-
нии. Эти образования в зависимости от размера и степени остав-
шегося пересыщения твердого раствора при высокотемпературном
Рис. 9.20. Зависимость твердости сплава Ni—
1,3 мае. % Be после высокотемпературного старе-
ния при 550°С, 1 ч от времени предварительного
низкотемпературного старения при различной
температуре
Рис. 9.21. Влияние времени старения при 90°С,
предшествующего старению при 175°С, 16 ч, на
твердость сплавов А1—Mg—Si с разным со-
держанием Mg2Si (Баба, Такашпма)
старении могут развиваться, определяя тем самым число и дис-
персность частиц упрочняющей фазы.
Эффект дополнительного упрочнения при комбинированном
старении изменяется при изменении содержания основных компо-
нентов и при дополнительном легировании. Например, этот эф-
фект практически отсутствовал в двойном сплаве А1—Си, но был
обнаружен в том же сплаве при добавке 0,5% Мп. В системе
297
Al—Mg—Si отрицательный эффект (или отсутствие эффекта)
имеет место при значительном пересыщении (1,2% Mg2Si и более),
но при небольших пересыщениях обнаруживается существенное
дополнительное упрочнение (рис. 9.21).
§ 9.3. ВЛИЯНИЕ ПЛАСТИЧЕСКОЙ ДЕФОРМАЦИИ НА РАСПАД
ПЕРЕСЫЩЕННЫХ ТВЕРДЫХ РАСТВОРОВ
И РОЛЬ ДЕФЕКТОВ КРИСТАЛЛИЧЕСКОГО СТРОЕНИЯ
Влияние дефектов кристаллического строения, возникающих при
пластической деформации, на процесс старения часто сводилось
к ускорению процессов выделения. В действительности, влияние
на скорость распада может не иметь однозначного решения, так
как процесс распада обычно состоит из нескольких стадий и
влияние структурных дефектов на протекание этих стадий раз-
лично.
При анализе процессов распада в сплаве с высокой плотностью
дефектов необходимо рассматривать не только изменение энергии
активации обычных реакций распада, но и возможность возникно-
вения под действием структурных дефектов новых состояний с от-
носительным минимумом свободной энергии.
Высокая плотность деформационных дефектов решетки может
привести к задержке процесса выделения. В сплаве К40НХМ
(40% Со, по 15—20% Fe, Ni и Сг, 7% Мо, около 0,1% С) из г. ц. к.
твердого раствора возможно выделение карбида типа СобМо6С.
После сильной пластической деформации (холодная прокатка
с обжатием около 70%) старение при 500°С приводит к значи-
тельному упрочнению, однако методом дифракционной электрон-
ной микроскопии выделение не обнаружено. Выделение наблюдали
при более высокой температуре старения только на тех участках,
где прошла рекристаллизация (участок Р на рис. 9.22). При не-
большом пересыщении и значительной плотности дефекта распад
можно представить как образование атмосфер избыточных атомов
(например, С и N в a-Fe)’возле дислокаций вследствие упругого
(коттрелловского) взаимодействия и дальнейшее развитие объем-
ных сегрегатов из-за химического пересыщения матрицы. Таким об-
разом, процесс гетерогенизации возможен непрерывным понижени-
ем свободной энергии: повышение химической свободной энергии,
связанное с началом дифференциации компонентов твердого раство-
ра неспинодального состава, компенсируется уменьшением упругой
энергии при образовании атмосфер. Образование и рост сегрега-
тов, последующее упорядочение и образование промежуточной
фазы — это важнейшие элементы процесса распада твердого рас-
твора при низкотемпературном старении после деформации.
Образование выделений на дислокациях показано на рис. 9.23
(распад твердого раствора типа внедрения — углерод в феррите)
и на рис. 9.24 (распад твердого раствора замещения — медь
в феррите).
298
В последние годы получили распространение стареющие высо-
коникелевые и хромоникелевые стали, в которых мартенситное
превращение создает высокую концентрацию дефектов.
Значительная растворимость легирующих элементов (Ti, Al,
Мо, Си и др.) в аустените позволяет получить большие пересыще-
ния в результате бездиффузионного превращения в мартенсите.
После старения, приводящего к некоторому перестарению (при
Рис. 9.22. Микроструктура сплава К40НХМ после закал-
ки, холодной прокатки и старения при 700°С, 1 ч (элек-
тронная микрофотография фольги), X70000
500—550°С), методом дифракционной электронной микроскопии
в некоторых мартенситно-стареющих сталях удается определить
природу выделяющихся фаз (см. табл. 7.1). Среди этих фаз есть
фазы, изоморфные о. ц. к. матрице (NiAl, NiTi), фаза на основе
почти чистой меди (г. ц. к.), Ni3Al и фазы с более сложными ре-
шетками (№зМо, Fe2Mo, /?-фаза и др.). Однако, несмотря на раз-
личное легирование и природу выделяющихся фаз, для всех этих
сталей имеются одни и те же особенности кинетики старения,
обусловленные, очевидно, структурными особенностями мартенсит-
ной матрицы.
Старение мартенситных сталей обычно проводят при темпера-
туре от 450 до 500°С (при температуре выше 500—550°С возможно
обратное превращение мартенсита в аустенит). При исследовании
старения большого числа хромоникелевых и высоконикелевых
мартенситно-стареющих сталей было показано, что временная за-
висимость объема превращения по всей исследованной области
299
(001)
IIIIIIIIIIIIIIIIIIIH!
мяйв
направлений/
роста /
I частиц /
К^ПГНаправление о
X неконсерватид-
X ного движения
\,дислокации
г
Рис. 9.23. Выделения на дислокациях в сплавах на основе a-железа (о. ц. к. ре-
шетка) :
а — когерентная промежуточная карбидная (или карбонитридная) фаза в техническом же-
лезе (0,04% С), в результате старения при 200°С (1 ч) образуются на участках дислокацион-
ной линии с большой краевой компонентой — А (на винтовых дислокациях участки Б н Б'
выделения отсутствуют); б — некогерентные частицы карбидной фазы NirnTinC (упорядо-
ченная решетка на о. ц. к. основе, а=4а0) в стали 1X21Н5Т после старения при 620°С (2 ч);
частицы выстраиваются вдоль <110>, что соответствует направлению линий дислокации
краевого типа; при отклонении линии дислокации от этого направления размер частиц
уменьшается (электронная микрофотография фольги), схемы расположения выделения и
линий дислокации в случае, когда существенно упругое взаимодействие атомов избыточного
компонента и дислокации и образование атмосферы по Коттреллу (в) и в случае, когда
взаимодействие поля растущей частицы и поля дислокаций приводит к «подтягиванию» ли-
нии дислокации к плоскости роста частицы (г)
температур (400—500°С) хорошо описывается уравнением
In[ 1 / (1—a)]— kxp,
(9.12)
где а — доля превращенного объема; т — время; k и р — константы
(рис. 9.25). Показатель р для начальной стадии старения мартен-
сита во всех случаях близок к 1/3, для старения 6-феррита
в той же температурной области примерно равен 1/2. Такое же
значение показателя (1/2) отмечали при старении мартенсита,
Рис. 9.24. Выделения Cu-фазы на ди-
слокациях в d-ферритных участках
стали Х17Н4М2Д после старения при
550°С, 12 ч (а) и при 625°С, 10 ч (б),
электронная микрофотография; воз-
можная схема расщепления дислока-
ций (по Хорнбогену), связывающая
образование г. ц. к. Cu-фазы с рас-
щеплением определенных участков
дислокационной линии (в)
301
когда по электронно-оптическим данным выделяли избыточные
фазы, т. е. при достаточно большом содержании легирующего ком-
понента в стали и при достаточно высокой температуре старения
(>500°С). Если р=1/2 характерно для выделения новой фазы,
то /3 характерно для начальной стадии старения мартен-
сита независимо от природы продукта распада. Поэтому есте-
ственно предположить связь такого значения показателя р
с процессом старения в условиях высокой плотности дефектов.
Рис. 9.25. Анализ кинетики превращения (по 'эффекту
электросопротивления) при старении мартенситной ста-
ли Н13М5 после закалки при различной температуре
(Ю. А. Скаков, В. Б. Спиридонов)
В стали Н13М5 в результате старения при 500—550°С вы-
деляется фаза Ni3Mo; в соответствии с этим обнаруживаются две
стадии старения (см. рис. 9.25,6): в начальной стадии (до 10—
20 мин при 550°С, т. е. до достижения состояния максимального
упрочнения) р~1/3, во второй стадии (т. е. на стадии выделения
и разупрочнения) р—1/2. Если старение стали Н13М5 проводили
при очень низкой температуре (475°С), то вторая стадия (р~ 1/2)
не обнаруживалась (выдержка 1 ч), в противоположность старе-
нию при 500—550°С значение р уменьшалось; в этом случае,
по-видимому, можно говорить об относительной устойчивости (ме-
тастабильности) состояния, возникающего в начальной стадии
распада.
Вывод об устойчивости состояния, возникающего после старе-
ния при температуре ниже 500°С, подтверждается измерениями
твердости, предела упругости и ширины рентгеновской линии
(рис. 9.26).
Некоторое уменьшение физической ширины линии (Впо) после
кратковременного старения, по-видимому, обусловлено перерас-
302
пределением дислокации, которые в исходном мартенсите могли
не занимать самых выгодных с точки зрения общей упругой энер-
гии взаимных положений. Затем, после некоторого увеличения
ширины в результате старения, при 450°С ширина линии не изме-
няется, тогда как при 500°С идет ее непрерывное уменьшение,
которое может быть связано не только с уменьшением плотности
дислокаций, но и с компенсацией упругих полей дислокаций и
частиц в ходе их образования и роста, сопровождающегося по-
Рис. 9.26. Изменение твердости, физической ширины линии и
предела упругости в результате старения стали Х17И7Т
(К. В. Варли, Л. И. Никитина, Ю. А. Скаков)
терей когерентности. Сравнительно быстрое и значительное по
общему эффекту изменение электросопротивления в первый пе-
риод старения сопровождается основным эффектом упрочнения.
На следующей (медленной) стадии изменения электросопротивле-
ния распад приводит к дополнительному упрочнению.
При дополнительном упрочнении стали значения твердости,
которые достигаются за первые 30—60 мин старения при 450 и
500сС, сильно различаются. Изменение предела упругости связано
только с начальным процессом распада. Об этом свидетельствуют
почти не изменяющиеся во времени значение предела упругости
после начального возрастания и небольшая зависимость этой ве-
личины от температуры старения при 400—500°С. При 450°С вы-
держка 200 мин дает близкое к максимальному значение предела
текучести, однако даже выдержка в течение 1000 ч не вызывает
перестарения (см. рис. 9.26). Таким образом, структура, форми-
рующаяся в интервале температур максимального упрочнения,
оказывается весьма устойчивой.
Температура 500°С находится выше границы устойчивости со-
стояния высокой прочности, и уже выдержка в течение 1 ч при
303
этой температуре приводит к разупрочнению. Если бы не было
принципиальной разницы в условиях равновесия при 450 и 500°С
и степень развития процесса старения контролировалась бы диф-
фузией с энергией активации (около 60 ккал/моль), то состояние
разупрочнения, наступающее при 500°С после нескольких часов,
при 450°С должно было бы наступать не позднее 100 ч.
Этот анализ указывает на существование температурной гра-
ницы относительной устойчивости структуры, отвечающей состоя-
нию высокой прочности, аналогичной температурной границе ме-
тастабильного существования неоднородного твердого раствора
(зонной структуры) при старении после закалки сплавов типа
л41—Си (см. рис. 9.9,а).
Кинетическое уравнение с показателем 1/3 получено эмпири-
чески при исследовании процесса выделения углерода из мартен-
сита. В работах Б. Я. Любова и др. этот процесс рассмотрен
в связи с влиянием на диффузию поля напряжений от дислокаций
на полукогерентной границе карбид — мартенсит при одновремен-
ном ее перемещении*. Этот вывод имеет общее значение для вы-
деления частиц при наличии дислокаций, поскольку учитывает
кроме нормальной восходящую диффузию в результате дрейфа
примесных атомов в поле напряжений.
В настоящее время еще нет достаточных структурных данных
для установления точного механизма выделения на дислокациях
в о. ц. к. решетке, хотя выделения на дислокациях в сплавах на
основе a-железа в случае раствора внедрения (углерод, азот) и
растворов замещения (медь) совершенно бесспорны (см. рис. 9.24
и рис. 9.25).
Роль дислокаций в процессе выделения обычно обсуждают
в связи с фактом возрастания упругой или поверхностной энергии
при образовании частиц новой фазы и компенсации этого возрас-
тания при образовании частиц на (или около) дислокации. При
этом обращают внимание на облегчение зарождения фаз со струк-
турой, отличной от структуры матрицы, подчеркивают роль
«несоответствия» решеток матрицы и значения вектора Бюргерса
дислокации. Чаще всего обсуждают выделение на дислокациях
некогерентных частиц и придают значение дислокациям только
в тех системах, где новая фаза имеет структуру, отличающуюся
от структуры матрицы (некогерентное выделение). Все эти сооб-
ражения бесспорно имеют значение при действительном выделении
новой фазы. Однако в связи с выяснением механизма упрочнения
при старении после деформации, как уже отмечалось, очень важно
состояние, предшествующее образованию частиц фазы с новой
структурой.
При низкотемпературном старении в условиях высокой плот-
ности дефектов может бЬтть устойчивым (точнее, метастабильным)
состояние, подобное зонной структуре в недеформированных спла-
* Рассмотрение дрейфа атомов внедрения в поле одиночных дислокаций
привело Коттрелла и Бейлби к уравнению, которое имеет вид обычной времен-
ной зависимости выделения с показателем р = 2/3.
304
вах. Распределение, состав и размеры группировок избыточных
атомов или сегрегатов в деформированном материале обусловлены
распределением и типом дефектов, с которыми взаимодействуют
атомы избыточного компонента. Эти сегрегаты не следует
отождествлять с «атмосферами» примесных атомов возле дефек-
тов в ненасыщенных твердых растворах. В пересыщенных твердых
растворах «атмосферы» могут явиться устойчивыми центрами рас-
слоения. В связи с тем, что состояние неоднородного (или рассло-
ившегося) твердого раствора более стабильно, чем состояние од-
нородного (закаленного) твердого раствора, само расслоение
может не ускорять появления стабильной фазы.
Таким образом, дислокации способствуют распаду твердого
раствора по типу расслоения, но тем самым создают более устой-
чивое состояние системы, чем однородный твердый раствор, и при
не слишком большом пересыщении затрудняют выделение частиц
стабильной фазы (с новой структурой). При низкой температуре
это состояние можно считать метастабильным.
Стадии распада, которые анализируются в связи с основным
эффектом упрочнения при старении, можно рассматривать как
единый процесс расслоения твердого раствора. Только при доста-
точной концентрации примесей и при температуре старения, пре-
вышающей предельную температуру устойчивости состояния неод-
нородного твердого раствора, возможен дополнительный эффект
упрочнения, проявляющийся в повышении сопротивления большим
деформациям (твердость, предел текучести). Такое упрочнение
наблюдается в сталях, в которых процесс старения завершался
образованием избыточной фазы с новой структурой.
§ 9.4. ОТПУСК СТАЛИ (РАСПАД МАРТЕНСИТА)
Атомная структура мартенсита углеродистой
стали. Мартенсит — главная структурная составляющая зака-
ленной стали, образование которой происходит в результате без-
диффузионного сдвигового превращения аустенита. Такое превра-
щение обусловливает передачу мартенситу всего углерода,
содержащегося в аустените. В о. ц. к. решетке a-Fe можно выде-
лить три подрешетки октаэдрических межузлий (см. рис. 3.2,д).
Схема расположения октаэдрических позиций г. ц. к. решетки
аустенита и образующейся деформацией Бейна о. ц. к. решетки
приведена на рис. 8.16. Каждая из указанных трех подрешеток
представляет собой о. ц. к. решетку со своим направлением оси
тетрагональной деформации Бейна: [100]^, [010]т или [001]7.
Деформация Бейна задает направление оси тетрагональности с,
вдоль которой октаэдрические позиции о. ц. к. решетки совпадают
с октаэдрическими позициями исходной г. ц. к. решетки (см.
рис. 8.16,а, в, следовательно, распределение атомов углерода, ко-
торое возникает в результате деформации Бейна, является пол-
ностью упорядоченным.
20—523 305
Упорядоченное распределение атомов углерода метастабильно
не только потому, что мартенсит представляет собой пересыщен-
ный твердый раствор углерода в a-Fe, но и потому, что темпера-
тура перехода в неупорядоченное состояние (точка Курнакова)
лежит ниже комнатной температуры при малых концентрациях
углерода и быстро повышается с увеличением его концентрации* *.
Непосредственно после образования мартенсита в нем происходят
релаксационные и диффузионные процессы, которые сопровож-
даются снижением свободной энергии. При рассмотрении распада
мартенсита необходимо учитывать процессы, связанные с изме-
нением состояния дефектов решетки мартенсита и поведением
углерода в твердом растворе.
Прежде всего необходимо рассмотреть процессы перераспреде-
ления атомов углерода в пределах одной подрешетки октаэдриче-
ческих межузлий. В закаленном состоянии атомы углерода
заполняют эту подрешетку октаэдрических межузлий неупоря-
доченным образом, занимая только часть ее узлов, поскольку
число атомов углерода много меньше числа разрешенных позиций
внедрения. Такое состояние можно рассматривать как двухкомпо-
нентный раствор замещения, в котором одним из компонентов
служит углерод, а вторым — оставшиеся незанятыми (вакантные)
узлы данной подрешетки октаэдрических межузлий. В таком рас-
творе возможны все те явления, которые происходят в обычных
бинарных растворах замещения: расслоение, образование ближ-
него и дальнего порядков и т. д. Как ближний порядок, так и
спинодальный распад должны приводить к диффузному рассеянию
электронов вблизи дифракционных максимумов, т. е. к размытию
узлов обратной решетки мартенсита. Впервые такие эффекты
диффузного рассеяния наблюдались при электронно-микроскопи-
ческом исследовании свежезакаленного мартенсита стали с содер-
жанием углерода 1,5% *2.
Экспериментальные электронно-микроскопические данные для
свежезакаленного мартенсита хорошо согласуются с теоретически-
ми выводами (рис. 9.27). Интенсивность наблюдаемых диффузных
эффектов пропорциональна содержанию углерода в твердом рас-
творе. Наличие этих эффектов при исследовании закаленного
состояния стали 60Н20 *3 свидетельствует о том, что этот процесс
характерен не только для высокоуглеродистых, но и для средне-
углеродистых сталей. Эффект диффузного рассеяния не наблю-
дается в сталях с высоким положением мартенситной точки
* Сохранению упорядоченного состояния, как показали теоретические расче-
ты, способствует упруго-напряженное состояние, в котором находятся кристаллы
мартенсита после своего образования. (А. Г. Хачатурян. В кн.: Несовершенства
кристаллического строения и мартенситные превращения. М., «Наука», 1972,
с. 34).
*2 Изотов В. И., Утевский Л. М. «Физика металлов и металловедение»,
1968. т. 25, с. 98.
*3 Еднерал Н. В., Скаков Ю. А. «Физика металлов и металловедение»,
1968, т. 26, с. 850.
306
(Л4Н>15О°С), в которых заметно протекают процессы распада во
время закалки.
Взаимодействие атомов углерода с дефектами
кристаллического строения. В решетке мартенсита
из-за относительной малости размера позиций для внедрения
атомы углерода создают значительные упругие смещения соседних
атомов железа. Наличие полей упругих искажений, окружающих
внедренный атом углерода, в основном определяет природу их
взаимодействия с такими дефектами кристаллического строения,
Рис. 9.27. Пространственная форма узла обратной решетки мартен-
сита при возникновении корреляции в расположении атомов углеро-
да (а); вид сечения обратного пространства в сечении (110)* (схе-
ма дифракционной картины) для свежезакаленного мартенсита ста-
ли (6)
как дислокации и вакансии. Взаимодействие внедренных атомов
с дислокациями является движущей силой процесса, приводящего
к образованию около дислокаций атмосфер примесных атомов:
атмосфер Сноека или Коттрелла.
Рассмотрим одну особенность взаимодействия углерода с дис-
локациями в мартенсите закаленной стали. Упорядоченное рас-
положение атомов углерода определяет весьма устойчивое
существование в пределах кристалла углеродистого мартенсита
единой оси тетрагональности. Неравнозначность позиций внедре-
ния существенно влияет на распределение атомов углерода в атмо-
сферах Коттрелла и Сноека*. В атмосфере около краевой дисло-
кации все атомы могут располагаться в системе октаэдрических
пор, соответствующей направлению тетрагональности. Около же
винтовой дислокации позиции внедрения, занятые атомами угле-
рода, попеременно расположены в каждой из трех подрешеток
* Тавадзе Ф. П., Зоидзе Н. А., Бадзошвили В. И. В кн.: Взаимодействие
между дислокациями и атомами примесей в металлах и сплавах. Тула, 1969,
с. 72.
20* 307
октапор. Вследствие этого образование атмосфер около винтовых
дислокаций невыгодно в высокоуглеродистом мартенсите и долж-
но приводить к разупорядочению в малоуглеродистом мартенсите.
Методом внутреннего трения было установлено, что закрепление
дислокаций атмосферами углерода имеет место уже при темпе-
ратуре —40 или —50°С и происходит весьма интенсивно при
комнатной температуре. При этом из твердого раствора уходит
примерно 0,2% С. Это количество углерода достаточно для эффек-
тивного блокирования дислокаций, к которому чувствительна из-
меряемая амплитудная зависимость внутреннего трения. Однако,
по-видимому, процесс выхода углерода из твердого раствора
к дислокациям этим не ограничивается. Наличие подъема электро-
сопротивления на ранних стадиях распада закаленного углероди-
стого мартенсита также может свидетельствовать об образовании
сегрегаций углеродных атомов (см. рис. 9.5). Причем время до-
стижения максимума электросопротивления несколько больше
времени основного уменьшения амплитудной зависимости внутрен-
него трения, что свидетельствует о дальнейшем прохождении
процесса развития сегрегаций возле дефектов кристаллического
строения дислокационного типа.
При облучении мартенсита высокоэнергетическими частицами
(«-частицами, нейтронами, электронами)* возникают значитель-
ные количества вакансий, которые служат «ловушками» для ато-
мов углерода. Энергия взаимодействия атомов углерода с вакан-
сиями (в отсутствие взаимодействия атомов углерода между
собой) равна 0,41 эВ. Однако при достаточно высоком содержании
углерода возникает тенденция атомов углерода к упорядоченному
расположению в октаэдрических позициях внедрения. Поэтому
эффективная энергия связи углерода с вакансией уменьшается
с ростом содержания углерода в твердом растворе и при концен-
трации 1,5% С составляет всего 0,03 эВ. Наличие радиационных
дефектов приводит к тому, что атомы углерода могут находиться
в октаэдрических межузлиях одной подрешетки и в вакансионных
«ловушках». Согласно теоретическим расчетам** при концентра-
ции углерода 0,8—1 мае. % и примерно равном количестве позиций
возле вакансий процесс перехода атомов углерода в позиции возле
вакансий носит характер фазового превращения первого рода
с четко выраженной температурной границей, разделяющей ука-
занные два состояния твердого раствора. При этом охлаждение
облученного мартенсита ниже критической температуры приводит
к спонтанной конденсации атомов углерода в вакансионных ло-
вушках. Уход атомов углерода из нормальных позиций внедрения
сопровождается падением тетрагональности, пропорциональному
доле диффундирующих атомов углерода. Такой процесс наблюда-
ли в стали Fe—1% С, облученной электронами с энергией
* См. § 9.5.
** Хачатурян А. Г., Шаталов Г. А. «Физика твердого тела», 1970, т. 12,
с. 1969.
. 308
2,5 МэВ*. Охлаждение мартенсита до температуры —40°С умень-
шало степень тетрагональности. Последующий отогрев увеличивал
степень тетрагональности до исходного уровня. Таким образом,
процессы перераспределения атомов углерода происходят значи-
тельно раньше возникновения карбидных фаз.
Процесс двухфазного распада. Рентгенографические
исследования стали после отпуска в температурном интервале до
100—150° (первая стадия распада мартенсита по Г. В. Курдю-
мову) показала уменьшение тетрагональности решетки мартенсита
вследствие выделения углерода из твердого раствора. Распад мар-
тенсита имеет двухфазный характер, т. е. наряду с раствором
с исходной концентрацией появляется «мартенсит отпуска» — твер-
дый раствор, содержащий 0,2—0,3% С, что соответствует степени
тетрагональности, равной 1,012—1,013. По мере протекания про-
цесса отпуска количество обедненного углеродом раствора уве-
личивается. На прохождение двухфазного распада мало влияют
такие легирующие элементы как Сг, Мо, W. Увеличение содержа-
ния углерода тормозит процесс распада, легирование никелем—
ускоряет, а кремнием — замедляет его.
Кинетика процесса двухфазного распада мартенсита описы-
вается экспоненциальным уравнением типа (9.12) при р=1/3.
Такое значение показателя р в кинетическом уравнении соответст-
вует одномерному росту частицы за счет дрейфа атомов углерода
в поле напряжений.
На стадии двухфазного распада, по-видимому, отсутствует нор-
мальная диффузия С на значительные расстояния, которая мог-
ла бы обеспечить формирование частиц карбидной фазы достаточ-
но крупных и в достаточном количестве для их выявления
с помощью современной методики структурного анализа (напри-
мер, электронографически).
Процесс к арб идо о бразо в ани я при отпуске
стали. Изучение структуры карбидных выделений, их размеров,
формы и распределения — один из важных вопросов отпуска ста-
лей, поскольку процесс карбидообразования во многом определяет
физические и механические свойства сталей.
При распаде мартенсита малоуглеродистых сталей, содержа-
щих менее 0,2—0,3% С, образуется цементит, при более высоком
пересыщении углеродом возможно образование метастабильных
карбидов.
Взаимодействие атомов углерода с дефектами кристаллической
решетки может быть термодинамически и кинетически более вы-
годным, чем образование карбида. Энергия связи углерода
в 8-карбиде составляет 0,27 эВ, т. е. меньше, чем энергия взаимо-
действия углерода с дислокациями и вакансиями. При увеличении
температуры отпуска усиливается тепловое движение атомов угле-
рода, вследствие чего уменьшается взаимодействие атомов угле-
* Крицкая В. К., Нархов А. В. «Физика металлов и металловедение», 1970,
т. 29, в. 6, с. 1'293.
309
рода с дефектами, и при температуре отпуска выше 200°С в этих
сталях идет выделение равновесного 0-карбида-цементита.
На этой стадии распада (вторая стадия по Г. В. Курдюмову)
происходит непрерывное уменьшение тетрагональности мартенси-
та, что дает рентгеновский эффект «однофазного» распада. Прак-
Рис. 9.28. Кристаллические струк-
туры (тонкие линии) е-карбида
типа £=Fe3N (толстые линии)
ориентационных соотношения
матрицей:
тически в стали с не очень высоким
содержанием углерода эти стадии
распада бывает трудно различить
и в значительной мере распад мар-
тенсита успевает пройти уже в ходе
охлаждения при закалке.
В средне- и высокоуглеродистых
сталях первой выделяющейся кар-
бидной фазой является метаста-
бильный 8-карбид, который имеет
гекс.п.у. решетку с периодами а=
=2,73 А и с=4,36 А (см. рис. 9.28) *.
Выделение 8-карбида наблюдали
возле дислокаций в виде тонких
длинных пластин. При этом реали-
зуются два близких между собой
между 8-карбидом и мартенситной
(101), II (101)M; (001), || (011)м; [110], || [ 100]м; (9.13а)
(001), || (011)м; [100], || [111]м. (9.136)
При дальнейшем увеличении температуры (~300°С) наблю-
дается выделение стабильного цементита. Цементит имеет орто-
ромбическую решетку с 12 атомами железа и четырьмя атомами
углерода на элементарную ячейку со следующими значениями
периодов решетки: а = 4,525, 6 = 5,090, с=6,744 А. Образование
цементита может происходить перестройкой решетки 8-карбида
в цементит или без предварительного выделения 8-карбида в двой-
никованной части мартенситного кристалла на границе раздела
двойник — матрица. Расположение атомов Fe на такой границе
близко к расположению атомов железа в решетке цементита
(рис. 9.29).
* Впервые гексагональная структура карбида низкоотпущенной стали была
определена Я. С. Уманским и Е. И. Онищиком (см. «Свойства стали». М., Ме-
таллу рг из дат, 1949, с. 160—179). Трудности расшифровки рентгенограмм были
связаны с сильным размытием линий из-за высокой дисперсности частиц карби-
да. Поэтому важное значение имело электроннографическое исследование {Ска-
ков Ю. А. и др. «Докл. АН СССР», 1958, т. 118, № 2, с. 284—287).
Некоторые исследователи находят после отпуска при низких температурах
(например, Г20°С) т]’каРбид, который весьма близок к е-карбиду. Различие свя-
зано г упорядоченным расположением атомов С, приводящим к ромбическим
искажениям гексагональной решетки (см. рис. 9.28).
310
При выделении цементита соблюдаются два ориентационных
соотношения:
[100]ц II [011]ф; [010]ц II [111]ф; [001 ]ц || (211)ф (9.14а)
И _
(103),t II (011)м; [110]ц II [1П]Ы. . (9.146)
Различие между соотношениями Ю. А. Багаряцкого (9.14а) и
И. В. Исайчева (9.146) состоит в повороте на 4° вокруг оси
[010]ц||'<111>м. В двойникованной части кристалла мартенсита
строго соблюдается соотношение Исайчева, габитус пластин це-
ментита (101)ц|| (112)м, где (112)м — плоскость двойникования
(рис. 9.29,6). В других случаях соблюдается соотношение Бага-
Рис. 9.29. Расположение
атомов Fe в сопрягающихся
плоскостях феррита и це-
ментита при разных ориен-
тационных соотношениях:
а - (100ц|| (110) ф;
б - (101)ц|| (112)ф;
в- (001)ц|| (112)ф.
черные кружки — атомы Fe в
решетке a-Fe, светлые — атомы
Fe в карбиде, цифра — номер
слоя
®—с
—
6
ряцкого, при этом габитус (110)ц||{110}м, пластины вытянуты
вдоль направления < 111 >м|| [010]ц, где имеется хорошее совпа-
дение положений атомов железа в мартенсите и в цементите
(рис. 9.29,а). Структуры 8-карбида и цементита имеют отдельные
весьма сходные элементы. Это приводит к тому, что в некоторых
случаях на микроэлектронограммах практически трудно различить
е-карбид и цементит*. Анализ рентгенограмм** и кольцевых элек-
* Еднерал А. Ф., Еднерал Н. В., Скаков Ю. А. «Физика металлов и ме-
талловедение», 1972, т. 33, с. 614.
** Арбузов М. П., Бушуев Ю. Е. «Физика металлов и металловедение»,
1968, т. 25, с. 882.
311
тронограмм от поликристаллических образцов дает более одно-
значную трактовку типов выделяющейся фазы.
Количество углерода в стали и легирование влияют на темпе-
ратурные интервалы устойчивости карбидных фаз. Увеличение
содержания углерода несколько повышает устойчивость 8-карбида.
В легированных сталях в отличие от углеродистых распад
мартенсита связан с процессами перераспределения углерода и
легирующих элементов. Процессы распада в сталях, легирован-
ных Сг, Мп, W, Мо, протекают по той же схеме, что и в углеро-
дистых сталях (если не образуются специальные карбиды).
Различие заключается лишь в некоторых сдвигах температурных
интервалов устойчивости различных карбидов. Кремний в отличие
от других легирующих элементов расширяет температурный интер-
вал устойчивости 8-карбида, это связано, вероятно, с тем, что Si
может входить в состав 8-карбида, но нерастворим в цементите.
Так, в стали с 1 мае.% С и 2 мае. % Si е-карбид сохраняется при
нагреве до 400°С.
§ 9.5. ВЛИЯНИЕ ИЗЛУЧЕНИЯ ВЫСОКОЭНЕРГЕТИЧЕСКИХ ЧАСТИЦ,
РЕНТГЕНОВСКОГО И у-ИЗЛУЧЕНИЯ НА ФАЗОВЫЕ ПРЕВРАЩЕНИЯ
Радиационные дефекты в металлах. Бомбардировка металлов
ионами и элементарными частицами (протонами, нейтронами,
электронами) при достаточно высокой энергии этих частиц, а так-
же воздействие у-квантов большой энергии приводят к образова-
нию дефектов кристаллического строения, которые названы
радиационными повреждениями.
Простейшие точечные дефекты — смещенные (промежуточные
или межузельные) атомы и вакансии. Их количество, распределе-
ние и вид возможных комплексов из этих дефектов зависят от вида
и энергии частиц, а также от природы материала, температуры и
других условий облучения.
Смещения атомов из нормальных положений внутри решетки требуют за грач
энергии Ео, которая как по разным теоретическим оценкам, так и по эксперимен-
тальным определениям для металлов порядка нескольких десятков электронвольт
(что в несколько раз превышает энергию сублимации). Согласно классической
механике движущаяся частица массой Mi может сообщить покоящейся частице
массой М2 энергию Ед, если обладает энергией не меньше чем
(М, + М2)2ЕД
^мин— 4MiM2
Для нейтронов и протонов, не вызывающих ядерных реакций, эта энергия
изменяется от 0,004 до 0,007 МэВ, если атомная масса материала увеличивается
от 50 до 100*. В случае электронов ЕМин слишком велика, чтобы можно было
* Если нейтроны вызывают радиоактивные превращения, сопровождающиеся
возникновением новых высокоэнергетических частиц, то радиационные поврежде-
ния возможны даже при очень малых энергиях первичных частиц.
Облучение некоторых металлов и полупроводников нейтронами, вызывающи-
ми радиоактивные превращения, приводит к появлению в решетке точечных де-
фектов типа раствора замещения; это явление используется для дозированного
легирования практически нерастворимыми примесями (например, А1 в Si, As в Ge
и т. д.).
312
(9.15)
использовать классическую механику, и применение релятивистской механики
дает для того же интервала атомных масс А от 50 до 100 энергию на два по-
рядка больше, чем для нейтронов (Емин—0,5 МэВ) у Рентгеновское и у-излучения
непосредственно не вызывают смещения атомов, но передают энергию электро-
нам (комптоновские электроны, фотоэлектроны), движение которых может при-
вести к смещению атомов. Очевидно, что кинетическая энергия этих электронов
сравнима с энергией у-квантов, поэтому значение £Мин для у- или рентгеновско-
го излучения имеет тот же порядок, что и приведенное выше значение для облу-
чающих электронов.
Энергия электрона поглощается уже в тонких слоях вещества
и тратится не только на смещение атомов, но и на взаимодействие
с электронами внутренних оболочек атомов, сопровождающееся
излучением, поэтому повреждения атомной структуры сравнитель-
но невелики и охватывают поверхностные слои материала. Еще
меньшая степень радиационного повреждения характерна для об-
лучения у- и рентгеновским излучениями. Кванты этих лучей
способны передать атому лишь малую часть своей энергии, и то
не непосредственно, а через возбуждение электронов вещества.
Основная доля энергии у-излучения тратится на ионизацию. Для
металлов это не приводит к дефектам кристаллической структуры
и выражается в выделении тепла.
Протоны и другие заряженные частицы (а-частицы ядра гелия,
осколки деления) с большой массой имеют малую длину свобод-
ного пробега в веществе, и их влияние ограничивается приповерх-
ностными слоями материала. Например, для а-частиц при энергии
от 2 до 130 МэВ длина их пробега составляет 2—6 мкм.
Нейтроны вызывают смещения атомов в результате непосредст-
венного столкновения с ядром или в результате ядерных реакций,
когда смещения атомов производят продукты этих реакций. В слу-
чае непосредственного упругого или неупругого* столкновения
нейтрона с ядром атом почти всегда получает энергию достаточно
большую, чтобы вызвать ряд смещений других атомов. Поврежде-
ния от быстрых нейтронов распространяются на большую толщу
материала, поскольку свободный пробег нейтрона между столкно-
вениями обычно составляет несколько сантиметров. Вместе с тем
повреждение, которое вызывает каждый из первично смещенных
атомов, очень концентрированно. Степень локализации этих по-
вреждений возрастает с увеличением атомной массы вещества.
В предельном случае каскад смещений, вызванных первично вы-
битым атомом, образует некоторый объем сложной перестройки
атомов в решетке, который называют пиком смещений. Если пер-
вичный атом получает энергию меньшую Ед, то эффектов смеще-
ния нет, но окрестный объем получает значительную тепловую
энергию (например, около 1 эВ на 20 атомов). Такую область
называют тепловым пиком.
* При неупругом рассеянии часть энергии нейтрона поглощается ядром. Воз-
бужденные ядра распадаются (возвращаются в исходное состояние) с испуска-
нием у-квантов.
313
В веществе, построенном из легких атомов, пиков смещения не бывает и
смещения, производимые первично выбитыми, вторичными и т. д. атомами, мож-
но рассматривать ка случайно расположенные и независимые точечные дефекты.
При движении тяжелого атома его столкновения с другими атомами про-
исходят все более часто по мере растрачивания кинетической энергии, и расстоя-
ния между столкновениями становятся соизмеримыми с межатомными расстоя-
ниями в кристаллической решетке. При столь частых актах взаимодействия
трудно говорить о столкновениях, и кажется более приемлемой модель локально-
го расплавления. Относительно строения пика смещения высказывали разные
предположения: пик смещения — область высокой плотности вакансий, окружен-
ная плотной оболочкой смещенных атомов, внутренняя часть пика — правильная
решетка, образованная в результате кристаллизации расплавленного объема, на
периферии отдельные скопления межузельных атомов и вакансий; пик смещения
после охлаждения — область с высокой концентрацией вакансий.
В более детальных рассмотрениях учитывали кристаллическую структуру
облучаемого вещества. Выла высказана гипотеза, что передача энергии в кристалле
идет преимущественно вдоль направлений плотной упаковки атомов.
В г. ц. к. решетке такими направлениями являются направле-
ния < 110>, но при более низких энергиях можно ожидать сме-
щения вдоль других направлений, например <100>. Передача
части энергии каскадного смещения от первично выбитых атомов
в определенные кристаллографические направления решетки на-
зывается явлением фокусировки. При передаче смещений вдоль
ряда плотной упаковки атомов могут возникать сгущения ато-
мов— динамические краудионы (см. рис. 5.4). При высокой тем-
пературе существенное влияние на распределение повреждений
в решетке оказывают тепловые колебания атомов. Эти колеба-
ния должны приводить к появлению компоненты импульса, пер-
пендикулярной к направлению фокусировки. Эта компонента
может привести к образованию побочного следа из смешанных
атомов и вакансий в начале сфокусированного ряда смещений.
В этом состоит один из механизмов влияния температуры на число
и распределение дефектов. Для решеток, не принадлежащих кплот-
ноупакованным, возможен эффект каналирования, когда выбитый
атом движется, не вызывая смещений, вдоль ряда пустот, которые
могут быть в веществах преимущественно неметаллического типа.
Кроме одиночных межузельных атомов и вакансий (или пар
Френкеля) в облученном материале возможны и более сложные
дефекты как в связи с механизмом самого радиационного повреж-
дения, так и в связи с последующим движением и взаимодейст-
вием точечных дефектов. После облучения электронами, рентге-
новскими и у-лучами, а также после облучения быстрыми
нейтронами вещества, состоящего из легких атомов, возникают
одиночные межузельные атомы и вакансии; при достаточно вы-
сокой температуре (более 0,3—0,4 ТПл) может оказаться значи-
тельной подвижность одного из этих типов дефектов (обычно
более подвижны межузельные атомы) и происходит образование
комплексов (бивакансии, тривакансии, гантели из межузельных
атомов и т. д.) и скоплений этих дефектов, или рекомбинация.
Образование скоплений происходит по объему кристалла (гомо-
генно) или на уже имеющихся дефектах решетки (гетерогенное
зарождение), на дислокациях, границах и других дефектах. При
314
наличии пиков смещения надо различать гомогенное зарождение
скоплений и зарождение внутри пиков смещения.
В компактных гекс. п. у. и г. ц. к. решетках скопления вакансий
и 'межузельных атомов приводят соответственно к образованию
дефектов упаковки вычитания или внедрения и дислокационных
петель. Такие петли, а также тетраэдры из дефектов упаковки по
плоскостям {111} многие исследователи наблюдали в г. ц. к. метал-
Рис. 9.30. Изменение электрического со-
противления W, Мо, Zr и Pt под дей-
ствием облучения быстрыми нейтронами
при 30сС. Крайние точки для W соответ-
ствуют флюенсу 2-'1О19 нейтр./см2 (Том-
сон — для W и Мо, А. Н. Иванов,
Н. Ф. Правдюк — для W, Мо, Zr и Pt)
Рис. 9.31. Увеличение длины образца
А/ (/) и периода кристаллической ре-
шетки Да (2) при нейтронном облу-
чении молибденовой проволоки (Том-
сон)
лах с помощью просвечивающего электронного микроскопа.
В о. ц. к. металлах подобных характерных дефектов не наблю-
дали. В некоторых случаях отмечалось образование дефектов типа
пор и газовых пузырей*.
Движение и взаимодействие точечных дефектов происходят и
при облучении, если температура достаточно высока. Даже при
низкой окружающей температуре эти процессы развиваются в об-
ластях торможения тяжелых частиц — в термических пиках. Это
объясняет уменьшение эффекта радиационных повреждений (на-
пример, эффекта роста электросопротивления) при очень большой
дозе облучения («радиационный отжиг»). Взаимодействие первич-
* При облучении а-частицами (или при их образовании в результате ядер-
ных реакций в материале) атомы гелия служат местами скопления вакансий.
315
ных радиационных дефектов с примесями является еще одним
фактором, существенно усложняющим характер дефектов. При-
мером такого взаимодействия является картина дефектов, наблю-
давшихся при дифракционной электронной микроскопии в облу-
ченном быстрыми нейтронами (1 МэВ) железе. Уже после
сравнительно малого флюенса около 1015 нейтр./см* 2 были замечены
области нарушения около 100 А с концентрацией 1014 см~3 4, ко-
торые можно было связать с наличием примеси углерода и азота
и процессом их выделения. Количественная оценка степени
радиационного повреждения производится по эффекту повышения
электросопротивления. Как видно из рис. 9.30, эффект возрастания
электросопротивления различен для разных металлов и зависит
от атомной массы и типа кристаллической решетки (в случае
компактных структур эффект меньше). О соотношении между
концентрациями вакансий и межузельных атомов можно судить
по изменениям периода решетки и удлинению образцов: более
быстрое увеличение длины проволочного образца, чем периода
решетки (рис. 9.31), свидетельствует о большей концентрации
вакансий. Расчет показывает, что флюенс 1018 нейтр./см2 создает
одну вакансию на 105 атомов облучаемого металла (для о. ц. к.
решетки было принято, что объем вакансии составляет 0,6—0,8
атомного объема). Представление о различии радиационных
дефектов после облучения нейтронами и электронами дает изме-
нение ширины и интенсивности рентгеновских интерференций.
Исследование* проводили на монокристаллах Мо, что позволило
выявить эффекты анизотропии воздействия радиации. Облучение
электронами привело к ослаблению интенсивности отражений
пропорционально е~2лг (при М—т. е. пропорциональном сум-
ме квадратов индексов интерференции); ширина линий не измени-
лась. Это означает, что облучение электронами создает только
точечные дефекты (вакансии и межузельные атомы) и не вызывает
заметных изменений субструктуры Мо. Наименьшими (практиче-
ски отсутствовали) ^ыли рентгеновские эффекты для направления
в кристалле [ИО], наибольшими — для [111}.
Основные рентгеновские эффекты нейтронного облучения:
размытие интерференций и относительное повышение интенсивно-
сти интерференций с малыми индексами. Оба эффекта можно
истолковать как измельчение блоков мозаики, так как эффект
уширения линий пропорционален функции тангенса угла Вуль-
фа— Брэгга, а рост интенсивности связан с уменьшением экстинк-
ции. Таким образом, характерными для радиационного поврежде-
ния от нейтронного облучения являются дефекты дислокацион-
ного типа (наряду с точечными дефектами).
* Крицкая В. К., Ильина В. А. и др. «Физика металлов и металловедение».
1974, т. 37, вып. 2, с. 307.
Облучение электронами проводили на линейном ускорителе, энергия 3—
4 МэВ, флюенс 1018 эл/см2, температура 50°С; облучение нейтронами велось
в реакторе при 80°С, доза облучения (по быстрым нейтронам) ГО19 н/см2.
316
Точечные дефекты, возникающие при облучении электронами;,
а также квантами при подходящих условиях могут образовывать
более или менее сложные комплексы, взаимодействуя между собой
или с примесными атомами. Был сделан вывод* о появлении упо-
рядоченных группировок точечных дефектов в молибдене непо-
средственно после облучения у-квантами (энергия 1,3 МэВ при
35±5°С): на рентгенограммах кроме возрастания диффузного
фона наблюдали резкий пик, соответствующий индексам отраже-
ния (100). Этот пик исчезал при нагреве в области 200—400°С,
диффузный фон от хаотически расположенных точечных дефектов
исчезал при нагреве до 600—800°С.
2. Фазовые превращения под действием облучения. Превра-
щения в сплавах, связанные с изменениями рас-
творимости или степени порядка твердого
раствора под действием облучения возможны как в направ-
лении приближения к стабильному состоянию, если исходное
состояние материала было неравновесным, так и, наоборот, могут
создавать новое относительно устойчивое или метастабильное
состояние. Такое сложное влияние облучения на фазовое состояние
сплавов обусловлено тем, что точечные дефекты в кристаллической
решетке способствуют развитию диффузионных процессов в рас-
творах замещения, приводящих систему в состояние равновесия:
сами смещения атомов могут быть важными кинетическими фак-
торами, если облучению подвергается сплав, находящийся в не-
равновесном состоянии. Как кинетический фактор установления
фазового равновесия можно рассматривать образование под дейст-
вием облучения мест, где облегчается образование частиц новой
фазы (скопления точечных дефектов, дефекты упаковки, дислока-
ции, пороги на дислокациях и т. д.). Однако сами дефекты могут
быть термодинамическим фактором равновесия (метастабильно-
го), и в их присутствии относительно устойчивы новые фазовые
состояния системы.
В некоторых твердых растворах нестехиометрического состава
процесс упорядочения происходит в малых объемах (/(-состояние,
локальное упорядочение или особый случай ближнего порядка,
см. также § 3.3) и сопровождается повышением электросопротив-
ления. Разрушение порядка связано с малыми перемещениями
атомов и легко обнаруживается в таких сплавах по весьма значи-
тельному снижению электросопротивления. Поэтому такие сплавы
очень удобны для выявления радиационных эффектов. В сплаве
типа нихром (никель — 20% хрома) максимальное электросопро-
тивление и, следовательно, максимальное развитие локального
порядка достигается после отжига при 550°С, сильная холодная
деформация разрушает это состояние (минимальное электросо-
противление), а закалка от высокой температуры дает некоторое
промежуточное состояние. Облучение быстрыми нейтронами пред-
Малиненко И. А., Шиврин О. Н. Докл. на IV Всесоюз. совещании по упо-
рядочению атомов и его влиянию на свойство сплавов. Т. 2. Томск, Томск, ун-т,
1974, с. 121 — 126.
317
варительно отожженного образца приводит к снижению электро-
сопротивления, т. е. к разрушению порядка (температура образца
при этом не превышала 100°С)*. Однако облучение в тех же
условиях деформированного или закаленного образца привело
к увеличению электросопротивления, причем этот рост оказался
наибольшим для предварительно деформированного образца, ко-
Рис. 9.32. Относительные изменения
электросопротивления при облучении
нейтронами образцов сплава нихром
после:
1 — холодной деформации; 2 — закалки от
1050сС; 3 — отжига при 550°С (С. М. Астра-
ханцев, Ю. И. Коннов)
температурах в интервале 400-
ний Co-фазы (твердый раствор
торыи отличался отсутствием по-
рядка (рис. 9.32). Значение элек-
тросопротивления после облу-
чения флюенсом 1020 нейтр./см2
оказалось во всех трех случаях
одним и тем же, что свидетель-
ствует о том, что в процессе об-
лучения идут как упорядочение,
так и разупорядочение (в зави-
симости от исходного состояния
материала), а в результате облу-
чения устанавливается некоторая
«равновесная» степень порядка,
определяемая конкуренцией раз-
упорядочивающего действия бом-
бардировки нейтронами и диффу-
зионного процесса упорядочения.
Механизм разупорядочения, по-
видимому, связан с пиками сме-
щения и распространением фоку-
сированных цепочек замещающих
столкновений. Подобным же об-
разом могут происходить разру-
шение и растворение частиц в ге-
терофазных сплавах. Это наблю-
далось, например, в сплавах на
основе меди. В сплаве СиЦ-2
мае. % Со старением при разных
-600°С получали частицы выделе-
10% Си в р-кобальте) с попереч-
ным размером от 25 до 100 А**. При самых малых размерах частиц
в исходном состоянии было отмечено снижение их концентрации
после облучения. Возможно, что существует некоторый критиче-
ский размер, при превышении которого частицы устойчивы. В этом
отношении, а возможно, и в отношении самого механизма исчезно-
вения частиц нейтронная бомбардировка может иметь аналогию
с явлением возврата при старении (растворение дисперсных час-
тиц в областях действия термических пиков).
* Астраханцев С. М., Коннов Ю. И. В сб.: Действие ядерных излучений на
материалы. М., Изд-во АН СССР, 1962, с. 121.
** Оценки размера и концентрации частиц проводились по измерениям маг-
нитных свойств (Piercy G., 1962, цит. по С. Т. Конобеевскому).
318
Особенности влияния облучения на фазовое состояние сплавов
с участием элементов, растворяющихся по типу внедрения, свя-
заны прежде всего с особенно сильным взаимодействием радиа-
ционных дефектов с атомами внедрения. В отличие от растворов
замещения точечные дефекты не ускоряют диффузию атомов
внедрения, напротив, происходит образование сравнительно мало-
подвижных комплексов. В сплаве a-Fe—С энергия связи атом
углерода — вакансия оценивается в 0,66-10~19 Дж (0,41 эВ), при
этом атом углерода несколько смещается из центра октаэдриче-
ской позиции к вакансии (см. рис. 3.2); энергия связи атом угле-
рода— межузельный атом железа 0,8-10-19 Дж (0,5 эВ) для
расположения атома С в октаэдрической позиции типа
[[1 1/2 1/2]] и пары атомов железа с общим центром тяжести
в позиции [[1/2 1/2 1/2]];. В одном из первых исследований влия-
ния облучения на сплавы Fe—С изучали поведение атомов угле-
рода в закаленных образцах Fe — 0,0115 мае.% С, подвергавшихся
старению (при 35—57°С) после облучения в реакторе (при —100
и +57°С разной длительности) методом измерения пика внутрен-
него трения Сноека. Высота этого пика зависит от содержания
углерода в феррите, атомы углерода, захваченные радиационными
дефектами, не участвуют во внутреннем трении (как и при вы-
делении карбидной фазы). Измерения относительных изменений
электросопротивления при последующих изохронных отжигах (до
350°С) позволили проследить за другими процессами (освобожде-
ние углерода из ловушек, уменьшение концентрации точечных
дефектов, выделение карбидов). Пришли к выводу, что межузель-
ные атомы Fe подвижны при —196°С, пары межузельный атом
железа — атом углерода стабильны до —30°С. Взаимодействие
атомов углерода и вакансий является реакцией второго порядка,
это означает взаимодействие одного атома углерода с одной ва-
кансией. Явление вакансионного захвата не всегда удается за-
метить, поскольку в ходе облучения атомы углерода успевают
дойти до мест выделения прежде, чем образуется заметное коли-
чество вакансионных ловушек. При температуре старения 57°С
уменьшение высоты пика Сноека происходит после облучения го-
раздо быстрее, чем при старении необлученных образцов
(рис. 9.33, кривые 2 и 3), увеличение времени облучения от 4 до
48 ч заметно не влияет на скорость процесса, понижение темпе-
ратуры облучения (до —100°С, кривая /) еще более ускоряет
процесс. Однако электронно-микроскопическое исследование по-
казало, что выделение карбидных частиц после облучения задер-
живается. Образование частиц цементита наблюдали при нагреве
выше 230°С, когда атомы углерода уже освобождаются из вакан-
сионных ловушек*.
* При низкотемпературном старении технического железа (около 0,01 мас.°/о
С) после закалки как на дислокациях, так и гомогенно сначала образуются час-
тицы карбидной фазы изоморфной матрицы; гомогенное зарождение этой фазы
идет при участии вакансий или их группировок (Скаков Ю. А. «Докл. АН
СССР», 1967, т. 175, № 6, с. 401).
319
Таким образом, влияние нейтронного облучения на состояние
пересыщенного твердого раствора внедрения связано с двумя про-
цессами: возникновением локализованных повреждений, действу-
ющих как места преимущественного выделения, и образованием
вакансий, которые могут быть ловушками для движущихся атомов
внедрения и тем самым замедлять процесс образования избыточ-
ной фазы.
Ускоряющее влияние нейтронного облучения на процесс вы-
деления углерода (и азота) из твердого раствора (по измерениям
внутреннего трения) происходит после воздействия сравнительно
Рис. ' 9.33. Относительные изменения углерод-
ного ' пика внутреннего трения сплава Fe—
0,0115 iMac.% С в ходе старения при 57°С после
низкотемпературного облучения (Дамаск):
1 — быстрыми нейтронами при —100°С; 2— нейтронами
в течение от 4 до 48 ч при 57°С; 3—без предваритель-
ного облучения
небольшого флюенса - и далее практически не изменяется
(3-1015 б. нейтр./см2 при температуре облучения —120°С). Уско-
ряющего влияния на кинетику процесса выделения атома азота
облучения электронами с энергией 3,2-10-13 Дж (2 МэВ) не было
обнаружено (0,8-1016 электрон/см2 или 30 мин при плотности тока
2 мкА/см2, температура при облучении 0°С, температура старения
65°С). На этом основании сделан вывод, что местами образования
зародышей фазы выделения являются именно скопления вакансий
и межузельных атомов, образование которых отличает действие
нейтронов от действия электронов.
Воздействие облучения на фазовое состояние углеродсодержа-
щего мартенсита закаленной стали оказывается особенно слож-
ным, поскольку облучение может влиять и на расположение
атомов углерода в решетке мартенсита, разупорядочение, обра-
зование комплексов атомы углерода — радиационные дефекты и
на процесс выделения карбидной фазы.
В результате облучения закаленной стали У-12 быстрыми ней-
тронами (энергия 15 МэВ, флюенс 1015 нейтр./см2 при комнатной
температуре), электронами (энергия 3 МэВ, флюенс 1018—
К)19 электрон/см2 при температуре 40—60°С), коротковолновыми
у-квантами (от радионуклида 60Со, Е—1,3 МэВ, доза 108—109 Р
320
при температуре 40°С) наблюдалось размытие рентгеновских дуб-
летов и уменьшение тетрагональное™*. Можно было бы предпо-
ложить, что это связано с выделением углерода, однако в отличие
от процесса отпуска при облучении изменение с/а происходит не
только за счет уменьшения с. Такой характер изменения с/а и а
свидетельствует об изменении положения атомов углерода в ре-
шетке мартенсита: углерод выходит из октаэдрических позиций,
соответствующих направлению оси тетрагональности [001], В дру-
гие позиции. Этот вывод подтверждается еще тем, что.уменьше-
ние с/а, вызванное облучением, не
является необратимым, как при от-
пуске мартенсита. Вылеживание при
комнатной температуре образца
после облучения в течение несколь-
ких месяцев приводит к повышению
тетрагональности. При более высо-
кой температуре и длительном вы-
леживании тетрагональное™ снова
уменьшается уже за счет процессов
отпуска (рис. 9.34).
При охлаждении стали с содер-
жанием 1 мае. % С после облучения
в интервале температур от —40 до
—50°С наблюдалось резкое умень-
шение с/а, которое имело характер
фазового перехода. Этот эффект
уменьшался с увеличением времени
вылеживания образцов при комнат-
Рис. 9.34. Изменения междублет-
ного расстояния (AL, отн.ед.), ха-
рактеризующего степень тетраго-
нальности мартенсита:
1 — после облучения и в результате
нагрева; 2 — необлученный образец
(В. К. Крицкая)
ной температуре и уменьшении дозы
облучения. Поскольку все эти эффекты наблюдали после облуче-
ния электронами, их возникновение следует связывать с точечны-
ми дефектами.
В теории ** рассматриваются два типа позиций в облученном
мартенсите для атомов внедрения: октаэдрические межузлия,
расположенные вдоль оси тетрагональности, и позиции, связан-
ные с вакансиями (радиационными дефектами). При понижении
температуры атомы углерода переходят в эти «ловушки», что
соответствует разупорядочению и снижению тетрагональности ре-
шетки мартенсита. При нагреве эти комплексы разрушаются, и
тетрагональное™ восстанавливается. Теоретический анализ опре-
деляет эти превращения как своеобразный фазовый переход пер-
вого рода.
Изменения кристаллической структуры под
действием облучения могут происходить не только в сплавах (как
в рассмотренном выше случае углеродсодержащего мартенсита
* Крицкая В. К. В кн.: Несовершенства кристаллического строения и мар-
тенситные превращения. М., «Наука», 1972, с. 94—100.
** Хачатурян А. Г., Шаталов Г. А. «Физика металлов и металловедение»,
1970, т. 12, № 12, с. 2969.
21—523 321
стали), но и в однокомпонентных системах, в частности в метал-
лах.
Изменения периодов решетки, регистрируемые рентгенографи-
ческим методом и не связанные с изменениями химического
состава, вызываются образованием точечных дефектов. В случае
кубической структуры изменение периода решетки Ла/а опреде-
ляется через изменение объема Aviv, которым сопровождается
появление вакансий и межузельных атомов. Если на одну пару
дефектов прирост объема равен (ссг-рагО^а (здесь иа— атомный
объем; аг^а и аг^а— изменения объема соответственно при пере-
ходе атома в межузельное положение и при образовании вакансии),
то
Да/а=(1 /3) (Av/v)=(1/3) (с^+аДг),
или Aala= (1/3) (ai + av)c, если cv=Ci = c.
Величина щ не может быть меньше 1, а ее наибольшее значе-
ние должно зависеть от положения атома. Если представить
образование межузельного атома как образование гантельной
пары в г. ц. к. решетке, то соседние ячейки должны получить
тетрагональную деформацию и относительное увеличение объема
составит 04=1,71. Вклад вакансии оценивается или нулем, или
отрицательной величиной, около —0,1 или —0,2. Таким образом,
по С. Т. Конобеевскому, получаем l,7>av + ai>0,8.
В случае о. ц. к. решетки или других, менее компактных реше-
ток можно ожидать меньшей величины для верхнего предела, чем
для плотноупакованных структур, в связи с большей способностью
рыхлых структур к релаксации. Образование группировок дефек-
тов должно уменьшать их влияние. При увеличении дозы облуче-
ния скорость/прироста а уменьшается (и наблюдается даже
уменьшение Абсолютного эффекта) за счет образования группи-
ровок и аннйгиляции точечных дефектов. Например, в результате
облучения Мо быстрыми нейтронами при температуре 50°С значе-
ние периода достигало максимума (увеличение на 0,048%) при
5-Ю19 нейтр./см2, а при флюенсе 102° нейтр./см2 снова уменьша-
лось.
Смещение атомов при облучении может привести к закономер-
ной перестройке решетки или ее разрушению и аморфизации. На
схеме рис. 9.35 показано смещение атомов (Е и D) решетки
алмаза, приводящее к разрушению тетраэдрической координации
атомов углерода в алмазе и образованию характерной слоистой
структуры графита. Возможность такой перестройки следует из
того, что при низких давлениях стабильной является именно моди-
фикация графита, а не алмаза. Перестройке решетки в значи-
тельных объемах препятствует очень большое напряжение, раз-
вивающееся из-за резкого увеличения объема (атомный объем
о •
углерода в алмазе 5,63 А, а в графите 8,80 А). Это объясняет как
очень значительное возрастание Aafa алмаза при облучении, так
и возврат радиационных изменений в результате отжига.
322
Облучение облегчает фазовый переход в олове (при 12°С), где
(в противоположность рассмотренному выше случаю алмаза)
возникает модификация с решеткой типа алмаза. В железных
сплавах облучение нейтронами не только не способствует пре-
вращению, но, напротив, может стабилизировать аустенитную
структуру*.
Фазовое состояние вещества может изменяться под действием
бомбардировки ускоренными ионами. Внедрение (имплантация)
ионов в кристаллическую решетку приводит к образованию новых
фазовых состояний как химическим взаимодействием (образова-
ние. 9.35. Схема перестройки ячейки кубической решетки
алмаза в ячейку графита (С. Т. Конобеевский)
нием химических соединений), так и независимо от химической
природы иона. Под действием бомбардировки ионами Не+
В. Н. Быков и др. наблюдали переход железа (о. ц. к.) и никеля
(г. ц. к.) (в виде тонких пленок) в гекс. п. у. структуру. Такой же
результат получен в более поздней работе при облучении ионами
Аг+ энергией 40 кэВ (флюенс 1015—1017 ион/см2, нагрев образцов
не превышал 100—150°С), для молибдена наблюдали переход
в г. ц. к. структуру. Те же переходы наблюдали** при использова-
нии ионов N+ и С+. Независимость превращения от химической
природы ионов позволяет считать, что определяющим фактором
при ионной бомбардировке в рассматриваемых условиях является
чисто радиационное воздействие. В цитируемой работе наблюдав-
шиеся перестройки атомных структур объясняются возбуждением
электронных систем при внедрении ионов в решетку металлов и
отмечается, что новые структуры могут быть характерны для фаз
высокого давления или структур, возникающих под действием
примесей внедрения. Перестройке решетки при ионной бомбарди-
ровке способствуют локальные напряжения, связанные с накоп-
лением радиационных дефектов, и термические пики. Проявления
* По данным А. И. Захарова и О. П. Максимовой, для сплава Fe
с 22 мае. % Ni и 3 мае. % Мп.
** Павлов В. П. и др. «Докл. АН СССР», 1974, г. 217, № 2, с. 330.
21*
323
«химического» действия внедряемых ионов следует ожидать для
ионов элементов, образующих между собой и с атомами мишени
более прочные связи (фосфор, кислород и пр.).
Задачи к гл. 9
Задача 9.1. Дать анализ следующих особенностей в изменениях периода ре-
шетки твердого расплава и твердости сплава А1 — 5% Си (см. рис. 9.5):
а. Периоды решетки твердого раствора после закалки образцов разного се-
чения различаются, хотя температура нагрева под закалку была одинаковой и
выше температуры перехода сплава в однофазное состояние.
б. В процессе старения образцов большого диаметра период решетки матри-
цы достигает больших значений, чем период решетки чистого алюминия 4,041 А.
в. Твердость в начальный период старения понижается.
Указание: при решении задачи следует принять во внимание условия прове-
дения термической обработки: закалка осуществлялась предельно резким охлаж-
дением, образцы имели разное сечение, известно, что сплавы на основе А1 могут
испытывать старение при комнатной температуре.
Задача 9.2. Оценить эффективную энергию активации процесса старения бе-
риллиевой бронзы, используя данные о изменении твердости в зависимости от
времени старения при разной температуре (см. рис. 9.1).
Сопоставить результат с известными диффузионными характеристиками. Для
самодиффузии в Си энергия активации 2,03—2,13 эВ, энергия образования вакан-
сий 0,95—1,17 эВ, энергия миграции вакансий 0,80—1,10 эВ (см. также данные
для системы Си—Be на рис. 5.8).
Задача 9.3. Обсудить роль вакансий, дислокаций и границ зерен в процессе
низкотемпературного распада твердых растворов внедрения на примере картины
естественного старения технического железа (Fe—С—N), используя электронную
микрофотографию фольги (см. рис. 9.3,в).
а. Определить концентрацию частиц (зон), образующихся при закалочном
старении технического железа (Fe — '0,04% С), используя электронную микрофо-
тографию фольги (см. рис. 9.3,в, увеличение задано масштабом на фотографии,
обычная толщина фольги 0,2—0,4 мкм).
б. Оценить по порядку величины концентрацию вакансий, полагая, что за-
калкой они фиксируются полностью (в достаточном удалении от стоков, в част-
ности от границ зерен). Температура закалки 700°С (973 К), для энергии обра-
зования вакансий принять £^ = 1,5 эВ.
в. Оценить по порядку величины коэффициент диффузии D для процесса,
определяющего образование свободной от выделений области вдоль границ зе-
рен, предполагая, что рассматриваемый процесс идет при закалке.
Сопоставить полученное значение D с коэффициентом диффузии для атомов
внедрения Dc (уход С и N в границу) и с коэффициентом самодиффузии D?e
(сток вакансий в границу).
Учитывая особенности режима охлаждения при закалке в воде, принять
в оценочном расчете температуру около 100°С и время порядка 1 с, ширина
области ~0,1 мкм (см. рис. 9.3,в); данные для расчета коэффициентов диффу-
зии (Dq, Q) взять из табл. 5.1.
При обсуждении результатов принять во внимание, что характерная для экс-
периментов по старению избыточная концентрация вакансий может повышать
коэффициент самодиффузии Dpe до значений, соответствующих температуре за-
калки (700°С).
ГЛАВА 10
АНАЛИЗ КИНЕТИКИ ПРОЦЕССОВ
РОСТА ЧАСТИЦ НОВОЙ ФАЗЫ
ПРИ НОРМАЛЬНЫХ ПРЕВРАЩЕНИЯХ
§ 10.1. КИНЕТИКА РОСТА ЦЕНТРА НОВОЙ ФАЗЫ
ПРИ БЕЗДИФФУЗИОННОМ ПРЕВРАЩЕНИИ НОРМАЛЬНОГО ТИПА *
Движущей силой процесса роста центра новой фазы является
изменение свободной энергии системы при изменении числа атомов
п в превращенном объеме dAF (n)/dn\ термодинамический стимул
роста имеется, если с увеличением nAF(n) уменьшается. С точки
зрения термодинамики неравновесных состояний все изменения
системы при постоянных внешних условиях можно описать как
процессы возрастания энтропии, скорость изменения энтропии
представляется как сумма произведения потоков I и соответству-
ющих сил X для всех направлений k:
{dS/dt)^^IkXk. (10.1)
k
Поток в направлении k может быть результатом суперпози-
ции полей (сил), поэтому представим
. (10-2)
i
тогда
(10.3)
ik
где Kik — скалярные величины, характеризующие подвижность
атомов, постоянные при постоянных температуре и давлении.
Необратимое изменение энтропии dSHeo6p равно сумме измене-
ний энтропии в системе и в окружающей среде:
dSueo6p== dS -\~dSo'C.
В изотермических условиях, когда выделяющееся тепло погло-
щается окружающей средой, а температура остается постоянной:
dS0C^= — dQ/T;
dQ = dU^-\-PdV,
* Материал § 10.1 и 10.2 излагается по книге: Любов Б. Я. «Кинетическая
теория фазовых превращений» (М., Металлургия, 1069).
325
тогда
rfSBeo5P = dS - (dUt 4- PdV)/T = (TdS- dUt - PdV)/T,
следовательно,
dUQ + PdV— TdS=dG,
dSHeo6p=—dGjT^—dF/T,
если учесть малую сжимаемость тел в конденсированном состоя-
нии и относительно небольшие давления
idSIdt)^^
\_dF_
Т dt ’
Применительно к процессу роста центра новой фазы удобно
представить изменение свободной энергии на один атом
из числа атомов и*, составляющих поверхность раздела фаз,
тогда
Л* d^F (Л) Л* d£dF ап /10 4Л
(rfS/^)fleo5p=- у--at---Г ~dR---------- dP (10Л)
В соответствии с (10.1) движущая сила X = I = dn/dt,
тогда в соответствии с (10.2)
Величина К может быть выяснена только из определенных
представлений о механизме процесса. С точки зрения теории абсо-
лютных скоростей реакций переходу атома в новое равновесное
состояние предшествует образование некоторой промежуточной
конфигурации («активированный комплекс»), обладающей повы-
шенной свободной энергией по сравнению с исходным состоянием
(центр из n-атомов, свободная энергия на один атом AFn) и с ко-
нечным состоянием (в центре всего п+1 атомов, свободная энер-
гия на один атом AFn+i). На рис. 7.17 эти состояния представлены
как «координаты» реакции.
При нормальном (немартенситном) превращении отдельные
акты присоединения атомов к центру считают независимыми, по-
рядок величины энергетического барьера q — энергия активации
процесса самодиффузии.
Очевидно, скорость роста центра новой фазы определяется
разностью потоков атомов, переходящих от центра в окружающую
среду и из среды к центру:
Л-4; (Ю.6)
Л = ехр (— qJkT)\ /2 = n2v2 ехр (— qJkT),
326
где П\ и п2, vi и V2 — число атомов и частота их колеоании возле
контактной поверхности с внутренней и внешней сторон, т. е. со
стороны новой фазы и со стороны окружающей среды. Примем, что
ni = n2==n*, vi=V2=v и AFn+i—&Fn~d\F(ri)ldn\
<71 = (ДГл + Д77„+’1)/2 + ^— =
q2 = (kFn 4- ДГ„+1)/2 + q - ДГ„+1 = — (ДГ„+1 - ДГ„)/2 + q,
dn/dt = n*v [exp (—qJkT) — exp (—qJkT)\ = n*v exp (—qlkT)y^
X {exp [(1/2АГ)с?ДГ(п)/с/п] — exp \—(l/'2kT)d^F(n)/dn]} —
=n*v exp(—q[k7’)sh [(1 !2kT) d\F (n)jdn\... (10.7)
При небольших переохлаждениях можно допустить, что
rfAf(n) (10.8)
dn ' '
тогда, учитывая, что при малых х shx^x, можно записать
dn
dt
n*v ехр (—qfkT) rfA„(n)
(10.9)
Теперь, сравнивая (10.5) и (10.9), можно записать выражение
для Д в уравнении (10.5):
Д= (y/k) ехр (—qlkT).
(10.10)
Если подставить вместо v значение максимальной частоты
колебаний атомов через характеристическую температуру Дебая
(vMaKc=^0nM), а число атомов на поверхности я* и в объеме
сферического центра п выразить через его радиус R и радиус
атома га, то для скорости роста получим
dR __
dt “ 9/zF
RkpM
(10.11)
Здесь AFo — изменение свободной энергии на единицу объема.
При выводе формулы (10.11) не учитывали изменения упругой
энергии, т. е. принимали, что AF(R) =—4/3^R^AFo+4^R2a, RKp=
=2a/AF0 — радиус критического зародыша.
Как видно из уравнения (10.11), при R<RKp скорость роста
dRJdt приобретает отрицательное значение и должно происходить
растворение частйцы новой фазы. Возникновение центров новой
фазы, способных к росту (R^RKp), рассматривается как флуктуа-
ционный процесс (см. с. 137).
При очень больших переохлаждениях можно принять
d\F(п)/dn^>kT, тогда из уравнения (10.7) получим
dnfdt = — n*vexp
t 1 dAF (n)
' 2 dn
kT
(10.12)
327
Температурная зависимость скорости роста при большом пере-
охлаждении ослабляется тем сильнее, чем больше термодинами-
ческая движущая сила превращения \dAF(ti)ldn 0]. Слабая
температурная зависимость характерна для мартенситных превра-
щений.
§ 10.2. АНАЛИЗ ПРОЦЕССА РОСТА ФЕРРИТНОГО ЦЕНТРА
В АУСТЕНИТНОЙ МАТРИЦЕ СПЛАВА ЖЕЛЕЗО — УГЛЕРОД
Примем, что благодаря большой подвижности атомов углерода
при выделении феррита из аустенита сразу устанавливается ра-
венство химических потенциалов углерода в а- и в у-фазах
(10.13)
В приближении разбавленных растворов
(10.14)
kT 1пс7.
В уравнениях (10.14) ф* и ф^-—теплота растворения углерода
в а- и в у-фазах, отнесенная к одному атому; сл и с — концентра-
ции углерода в феррите и аустените; числовой коэффициент / вво-
дится для учета разницы в числе позиций для внедрения атома
углерода в решетках а- и у-железа. В г. ц. к. решетке y-Fe число
октаэдрических пор (возможных позиций внедрения при растворе-
нии углерода) равно числу атомов железа; в о. ц. к. решетке число
возможных позиций внедрения в три раза больше (/ = 3). Боль-
шая подвижность атомов углерода создает стационарные условия
процесса, что еще не означает создания условий равновесия.
Начальная концентрация углерода в образующемся центре фер-
рита свач, так же как и концентрация углерода у границы раздела
феррит — аустенит с™в, может отличаться от с*авн, соответ-
ственно и концентрация углерода у поверхности раздела со сторо-
ны аустенитной матрицы буд^т неравновесной (свов). Тогда из урав-
нений типа (10.14) следует
нов
Г = 4г ехр W^),
(10.15)
здесь AFC = (фса — фс) NA (Na — число Авогадро).
Характер распределения концентрации углерода в системе
ферритный центр —«аустенитная матрица можно представить ре-
шением второго уравнения Фика. Для сферической симметрии
328
перемещений атомов (рост сферического центра феррита) это
уравнение запишем в виде
(10.16)
При очень медленном изменении распределения концентрации
можно принять, как для стационарного состояния,
dcldt~Q.
Тогда остается решить уравнение
д2с/дг2 + (2/г)дс/дг=0. (10.17)
Простой подстановкой легко показать, что
с=А+В/г, (10.18)
где А и В — постоянные интегрирования. Для области системы,
которая включает точку г=0, надо считать В = 0, чтобы не было
расходимости в значении с. Тогда для внутренней области сферы
(область ферритного центра)
са = А= const. (10.19)
За пределами ферритного центра, т. е. в аустенитной матрице,
с7=А7+В/г. (10.20)
Полученные распределения схематически приведены на
рис. 7.26,а.
Постоянные А и В? определяют из дополнительных условий
задачи. Очевидно, вдали от центра (г-юо)г =с^ач (исходный со-
став аустенита); таким образом,
Л7 = сн7ач.‘ (10.21)
У поверхности ферритового центра в аустените с7 |R=c”0B, в то же
время |R = ^ач 4“ “Б" > отсюда
В7 = (с*ов - ^ач) R. (10.22)
Составим уравнение баланса масс на поверхности растущего
центра, исходя из того, что количество углерода, освобождающе-
гося при росте центра за время dt, должно равняться количеству
углерода, отводимого в аустенит диффузией:
(с
пов
7
люв\ dR
дг
(10.23)
329
Здесь D — коэффициент диффузии углерода в у-фазе. Поскольку
/ дса \
концентрация углерода в феррите принята постоянной =
то в правой части уравнения баланса содержится только выраже-
ние (в соответствии с первым законом Фика) для массы угле-
рода, диффундирующего от границы раздела фаз в аустенит.
Подставим (10.18) в (10.23):
-т* ***
с •
Подртавляя значение
скорости роста центра:
c™B)(dR/dt) = DBJR*.
(10.24)
В из (10.22), получаем выражение для
dR/dt = (DIR) (с"ов - снД/(сП0В - <ов). (10.25)
II II а
Уравнение (10.25) содержит только условие отвода углерода
от поверхности растущего центра, т. е. оно не задает значение
скорости роста, а ограничивает ее скоростью отвода углерода.
Величины концентрации с^ов и с"ов пока остаются неизвестными
функциями времени. Термодинамической движущей силой роста
ферритного центра является изменение свободной энергии системы
при увеличении размера этого центра (см. § 10.1).
В приближении разбавленных растворов химические потенциалы
атомов железа в феррите [концентрация железа (1—cj] и аусте-
ните [концентрация железа (1 — с)] можно записать*:
(10.26)
kTc^
Здесь ф* и <{ле — значения свободной энергии на атом в a-и у-
модификациях чистого железа.
Сделанное допущение о том, что в процессе роста ферритного
центра быстро, т. е., не ограничивая скорость реакции, устанав-
ливается равенство химических потенциалов атомов углерода
у поверхности центра со стороны феррита и аустенита, означает
определенное соотношение между этими концентрациями, которые
сами по себе могут быть долями от равновесных [см. уравнение
(10.15)]. Скачок химического потенциала железа на границе раз-
дела феррита и аустенита зависит именно от этих (изменяющихся
в ходе процесса) концентраций углерода:
(О = (10.27)
* Из-за малости сп и и близости 1 —с„ и 1 —к 1 вместо In (1 —"с) за-
Qu J (A J > м г
— с. ♦
писано
330
В условиях равновесия Асрре=0 и Асрс=О и уравнения (10.14)
и (10.26) позволяют определить равновесные концентрации ком-
понентов. Соотношение равновесных концентраций углерода
в феррите и аустените записывается по типу уравнения (10.15),
а из уравнения (10.26) получается разность равновесных кон-
центраций
' ^авН “ <авн = ^еД/7оЖ- (10-28)
I
Здесь AFo — изменение свободной энергии железа при фазовом
превращении на единицу объема; — объем грамм-атома железа
[vF(AF0=Na (Ф^е —ф«е)]-
Выражение для скорости роста ферритного центра можно за-
писать по типу уравнения, выведенного в § 10.1 с учетом влияния
поверхностной энергии [уравнение (10.11)]:
dR[dt — KjRT [Na^ (c)/vFe] (1 - RKp/R). (10.29)
Здесь 7(1 = (16w4a0DR/9A) exp(—(?/И'); Д <pFe (c)/t»Fe=Д F, — раз-
ность свободных энергий для атомов железа на единицу объема.
Раскрывая значение А<рре(с) из уравнений (10.27) и (10.26),
учитывая RKr=2cc/AF (см. с. 326) и используя выражение (10.28),
получаем
б/R ^Fe А-F» / п°в /1П0В\ 2сг
------<СТ -Са WFe
(10.30)
Из не определенных пока функций времени в уравнениях (10.25)
и (10.30) легко решить вопрос о концентрации на поверхности
ферритного центра с”ов. Учитывая, что с самого начала процесса
свов<с"ов [см. уравнение (10.15)], изменение абсолютного значения
свов не может влиять на кинетику реакции, поэтому можно при-
нять, что с™в = const, свов= с*авн. В уравнении (10.30) вместо
v„&FJRT можно записать разность концентраций ^авн—^авн
" С I **
[см. уравнение (Ю.28)]; окончательно общие выражения для ско-
рости реакции под действием термодинамической силы и для ско-
рости реакции, контролируемой диффузией углерода в аустените
в условиях стационарного процесса, получаем
dR/dt = и (^авн - с™ - 2olvfJRTR).
Здесь % = 7<1/Пре;
dR/dt = (£>T/R) (С;св - ^ач)/(сптов - Свн).
Из уравнения (10.31а) можно получить
с„ J_ ж _ 2avrJRr
(10.31а)
(10.316)
(10.32)
331
Подстановка полученного значения с (R) в уравнение
баланса масс (10.316) позволяет для скорости роста (с некото-
рыми упрощениями*) получить
dR/dt и {(^авн - с;ач - 2*vFJRTR)l [(^аан - <авн
2xvpJRTR)
(10.33)
На рис. 7.26,6 и в приведены результаты расчетов по формулам
(10.32) и (10.33), выполненных Б. Я. Любовым и И. П. Гаркушей,
для конкретного случая роста ферритного зерна при 700°С (973 К)
из аустенита, содержащего 0,5% С, коэффициент диффузии угле-
рода в аустените принят равным D—1,42 -10-8 см2/с, энергия
активации перестройки решетки у->а 35 ккал/моль; vf&^Fq/RT=
= 0,04, поверхностное натяжение для межфазной границы
феррит — аустенит а=40 эрг/см2.
Как видно из рис. 7.26 б, в, при небольших размерах центра
при этом можно рассматривать процесс как аллотропи-
ческое превращение в однокомпонентной системе, и именно пере-
стройка структуры определяет скорость процесса (R<10~5 см).
Когда происходят значительные изменения концентрации углерода
у границы в аустените (область размеров центра от 10-5 до
10-3 см), в реакции постепенно приобретает доминирующую роль
отвод углерода от движущегося фронта растущего ферритного
центра. Процесс диффузии полностью контролирует реакцию при
больших размерах частиц (больше Rrp). С повышением темпера-
туры Rrp уменьшается, так что в верхней области субкритических
температур значение перестройки решетки в определении скорости
аллотропического превращения может стать незначительным уже
вскоре после начала роста ферритного центра.
§ 10.3. АНАЛИЗ РОСТА СФЕРИЧЕСКИХ ЧАСТИЦ
С УЧЕТОМ ПЕРЕКРЫТИЯ ДИФФУЗИОННЫХ ПОЛЕЙ
Рассмотренное выше решение задачи диффузии относилось
к случаю, когда состав матрицы вдали от растущего центра мож-
но считать неменяющимся. Это справедливо только для началь-
ных стадий распада, развитие распада приводит к изменению
состава матрицы — твердого раствора между растущими центра-
ми, что необходимо учитывать в расчете.
Взаимное влияние отдельных частиц в процессе их роста
Верт и Зенер учитывали, введя в кинетическое уравнение множи-
теля 1—а, где а — доля выделившегося компонента.
♦Допустимыми при условии, что Цавн — с™)/(с^авн — с^И) 1, т. е. при очень
большом различии равновесных концентраций углерода в аустените и феррите,
когда вообще процесс диффузии можно рассматривать как стационарный.
332
В работе И. П. Гаркуши и Б. Я. Любова* приведено решение
задачи о диффузионном росте сферических частиц новой фазы
при распаде пересыщенного твердого раствора с учетом неста-
ционарное™ процесса диффузии.
Объем кристалла делят на области («сферы влияния») вокруг
5?нтра каждой растущей частицы новой фазы. Для конкретности
асчета принято расположение этих
етки. Многогранники, образованные
и (делят пополам расстояние меж-
У соседними частицами), можно
ппроксимировать сферами того же
объема. На рис. 10.1 радиус этих
эквивалентных сфер обозначен
^о(О—радиус частицы новой фа-
зы; г — текущая координата. Здесь
же схематически показано распре-
деление концентраций при выделе-
нии фазы, обедненной растворен-
ным веществом (например, выделе-
ние феррита из аустенита в сплаве
Fe—С). Распределение концентра-
ций в пределах эквивалентной сфе-
ры описывается уравнением для не-
стационарных условий
центров по типу г. ц. к. ре-
эквидистантными плоскостя-
Рис. 10.1. Распределение концен-
трации при росте сферической ча-
стицы новой фазы с учетом нало-
жения диффузионных полей
Спов
dr2
г dr
При использовании безразмерных переменных (относительный
радиус частицы р0 = г0/Кэ, обобщенная текущая координата
p=r/R3, обобщенное время T=Df/R23) уравнение принимает вид
(10.34)
Концентрация в начальный момент с(р, О)=Со, состав матрицы
у поверхности растущей частицы сохраняется постоянным:
нов
Состав новой фазы сн.ф также постоянный.
Уравнение массового баланса на движущейся границе раздела
фаз
(10.35)
f
* Гаркуша И. П., Любов Б. Я. «Физика металлов и металловедение», 1970,
т. 29, вып. 3, с. 449.
333
Взаимное влияние растущих частиц выражается в том, что
суммарный диффузионный поток через эквидистантную плоскость
(или через поверхность эквивалентной сферы) отсутствует:
р:
(10 36)
Таким образом, задача сводится к вычислению концентрации
с(р, т) и радиуса частицы ро(т) только для одной ячейки. Анали-
тические выражения для указанных зависимостей слишком гро-
Рис. 10.2. Кинетика выделения при учете перекрытия диффузионных полей:
а — зависимость относительной концентрации ^(т)==с1,Г“сПов/сн ф— спов в эквидистантной
точке (на поверхности эквидистантной сферы) от безразмерного параметра времени т при
разной исходной концентрации р0; б — относительный радиус частицы р0 новой фазы в за-
висимости от безразмерного параметра времени х при разной исходной избыточной концен-
трации Ро=-(с1>0-спов)/(сн ф-спов)
моздки, поэтому авторы расчета дают их графическое представле-
ние (рис. 10.2). Кривые на рис. 10.2,а позволяют судить, в част-
ности, о моменте времени начала эвтектоидного превращения по
достижении концентрации х ==сЭвтект (например, при выделе-
нии цементита из аустенита стали эвтектоидного состава); для
этого надо знать состав эвтектоида, коэффициент диффузии и
радиус эквивалентной сферы (или число центров в единице
объема).
Из рис. 10.2,6 видно, что учет диффузионного взаимодействия
растущих частиц приводит к меньшим размерам частиц по срав-
нению с размерами частиц при отсутствии взаимодействия.
§ 10.4. КОАЛЕСЦЕНЦИЯ ЧАСТИЦ
В работе И. М. Лифшица и В. В. Слезова*, так же как и
в других рассмотренных выше теоретических исследованиях, при-
нята сферическая форма частиц. Равновесная концентрация твер-
дого раствора у поверхности частиц Cr выделяющейся фазы
* Лифшиц И. М., Слезова В. В. «Журн. эксперим. и теор. физ.», 1958, т; 35,
вып. 2(8), с. 479.
334
связана с радиусом кривизны частицы R* этой поверхности, т. е.
с радиусом
с1<=‘;«+т. <‘°'37>
гДе а включает межфазовое поверхностное натяжение; — «ис-
тинная» концентрация насыщения (плоская поверхность раздела,
т. е. R = oo).
Истинное пересыщение А = с—(с — действительная концен-
трация твердого раствора).
Если пересыщение А (и начальное пересыщение До) очень мало
(А<С1) и можно не учитывать прямое диффузионное «взаимо-
действие» между частицами выделяющегося вещества (т. е. отно-
шение размера частиц к среднему расстоянию между ними очень
мало), то диффузионный поток растворенного вещества на еди-
ницу площади поверхности раздела можно записать в следующем
виде:
dj = D^- ^dt=^(c-cR)dt = ^(b--^dt. (10.38)
Предположим, что это вещество выделяется в чистом виде
(сы.ф=1), тогда этот поток дает приращение радиуса частицы,
скорость которого
dRldt=(D/R)^ (Ю-39)
Это уравнение показывает, что каждому пересыщению Л соответ-
ствует критический радиус частицы RKp=a/A, при котором новая
фаза находится в равновесии с раствором (dR/d/=0), при R>RKp
частица растет, а при R<RKp растворяется. В последующих вы-
кладках авторы использовали безразмерные величины:
p=R/KP;
где До — начальное пересыщение и RKPo— начальный критический
радиус);
^W = RKp/RKPo=Ao/A
— безразмерный критический радиус [х(0) = 1].
В этих переменных уравнение (10.39) примет вид
dp3/dr=3(p/x—1). (Ю.40)
Далее вводится функция распределения частиц по размерам
/ (р, т) и рассматривается v =dpld% как скорость «перемещения»
частицы в пространстве размеров. Искомые функции f(p, т)
* Это упрощенный вид известной формулы Томсона In сл/сто=а//?.
335
и х(т); для их определения можно составить два уравнения: усло-
вие непрерывности в пространстве размеров
<+т(м=° <1ОЛ1>
и закон сохранения вещества
М=Ао+шо=Л+т,
(10.42)
где М — полное количество избыточного растворенного вещества,
включая количество этого вещества в частицах (ш0 — в начальный
момент, т=0, m — в ходе процесса) и пересыщение раствора
(начальное До или А — в ходе процесса):
ОО
m=41t7?3KPo ?
р=о
Начальное условие состоит в том, что имеется некоторое исходное
распределение f (р, O)=fo(p)« Задача решается в асимптотическом
случае (т->оо). Направление изменения размера частицы R при
коалесценции, как показывает анализ уравнения (10.39), связано
со значением критического радиуса RKp. Уравнение (10.40) вклю-
чает критический радиус и поэтому оказывается более удобным
для анализа. Действительно, если р<х, скорость изменения раз-
мера частицы или скорость «движения» частицы в пространстве
размеров оказывается отрицательной.
! х(ъ)
Рис. 10.3. Схема к анализу уравнения (10.40)
Если представить ось размеров (рис. 10.3), то точки р по этой
оси, находящиеся левее х(п), должны ускоренно двигаться влево
(скорость этого движения возрастает с уменьшением р), будут до-
стигать начала координат и «выходить из игры» (р->0, полное
растворение частицы). Точки р, находящиеся справа от х, должны
двигаться направо (рост частиц), однако по мере роста частиц
пересыщение раствора А уменьшается и критический размер
(х=А0/А) возрастает. Поэтому точка х(т) на оси размеров дого-
няет некоторые точки, движущиеся направо, после чего эти точки
ускоренно начинают движение в обратном направлении и «исче-
зают» в начале координат (частицы растворяются). Все эти дви-
жения контролируются общим уравнением, и первоначальный
336
порядок взаимного расположения точек на оси размеров сохра-
няется и также упорядоченно происходит исчезновение ряда точек
(растворение ряда частиц).
Движение точки х(т)=Ао/Л происходит со временем монотон-
ным образом: при т->оо х(т)->оо и пересыщение Д->0.
Рассмотрим результаты математического анализа. Функция
л*(т) получена в следующем виде:
х* 9 х 4 (1m)2 +•’•)]•
(10.43)
Если (1пт)2^>1, можно принять, что критический размер частиц
имеет зависимость от времени:
х= (4/9 т)1/3.
(10.44)
Количество частиц в единице объема
N (т) = (3/2)2/3 Л/т,
где Л = 0,22M/R’KPo.
Средний размер частиц
(10.45)
R-3=4/9Dctt (10.46)
т. е. пропорционален .
Степень пересыщения
L L
где Л = (3/2)3 (а2/£))3 .
Все эти асимптотические решения
при условии
9<lnR/4J!> 1 или R>RKPo.
Приведенный анализ вообще спра-
ведлив, если средний начальный раз-
мер частицы Ro~RKPo, т- е- Достиг-
нут такой размер частиц, такое пе-
ресыщение, что дальнейший рост мо-
жет быть связан с растворением не-
которой доли частиц (см. рис. 9.10).
Если же ==-£-. то дЪлжен
продолжаться рост частиц непосред-
ственно за счет растворенного ве-
щества в матрице до тех пор,
пока пересыщение твердого рас-
твора не станет достаточно малым
22—523
_ 1
, (10.47)
(10.44) — (10.47) справедливы
Рис. 10.4. Зависимость размера
частиц от времени: до момента
рост за счет выделения избыточ-
ного компонента, после f — коа-
лесценция
337
(соответственно критический радиус поднимается до уровня
R ~JR0) (рис. 10.4). Если исходное пересыщение Д', исходное
число частиц N', то рост частиц непосредственно из раствора дол-
жен продолжаться до размера
(10.48)
Продолжительность этой стадии (предшествующей коалесцен-
ции)
f ^R20/ZM'. (10.49)
§ 10.5. ВЛИЯНИЕ НАПРЯЖЕНИЙ НА ПРОЦЕСС РОСТА ВЫДЕЛЕНИЙ
Дифференциальное уравнение диффузии, описывающее и диф-
фузию, и дрейф атомов растворенного компонента в поле от струк-
турных дефектов с учетом концентрационных напряжений*:
dc[dt=^ (Рэфус) — ?cv<e; (10.50)
Озф = Д f 1 4- -- 2Ev{-^ со2 - 2га/А. (10.51)
Здесь (ysee — сумма диагональных компонент тензора структурных
напряжений; v — объем на 1 гратом; со — параметр, характеризу-
ющий зависимость периода решетки твердого раствора от кон-
центрации; Е — модуль упругости; vn —коэффициент Пуассона,
последний член в уравнении учитывает концентрационную зави-
симость коэффициента диффузии, обусловленную химическим
взаимодействием при энергии смешения Ди#=0; z— координаци-
онное число; а1=|Ди/-йТ; для разбавленных растворов можно счи-
тать, что D не зависит от концентрации.
Расчеты, проведенные Б. Я. Любовым, показали, что, напри-
мер, в случае диффузии углерода в аустените зависимость ОЭф(с)
обусловлена главным образом концентрационными напряжениями,
в отличие от диффузии цинка в меди, где основное значение в от-
клонении поведения твердого раствора от идеального имеет хими-
ческое взаимодействие.
Если считать щ = 0 и ввести энергию взаимодействия диффун-
дирующих атомов с полем структурных напряжений цп=—
(здесь Уо — объем атома), то уравнение (10.50) примет вид
de [ р. . D
dt — V (D3$yc —|
(10.52)
Будем считать, что при возникновении когерентной частицы упру-
гое поле дислокации сохраняется и влияет на кинетику роста.
* Любов Б. Я. Кинетическая теория фазовых превращений. М., «Металлур-
гия», 1968, с. 150.
338
Тогда первоначально возникшее выделение, например, в форме
цилиндра возле краевой дислокации постепенно обволакивает ее,
приобретая асимметричную форму в поперечном сечении и сохра-
няя асимметричное расположение относительно линии дислокации.
В случае выделения цементита из аустенита при высокой тем-
пературе дрейф влияет на форму выделения только в самом нача-
ле процесса (по расчету при 973 К до 10~2 с), далее преобладаю-
щее влияние имеет нормальная диффузия.
Следующая задача, которая была решена в работах Б. Я. Лю-
бова,— одномерный рост пластины с полукогерентной границей,
состоящей из горизонтального ряда дислокаций эпитаксиального
типа (например, рост пластины 8-карбида при выделении углерода
из мартенсита в стали).
Если принять преобладающую роль дрейфа (10.52), уравнения
диффузии (10.50) — (10.52) примут вид
Здесь у — текущая координата; у0— координата границы раз-
дела;
coy P-b (1 + уп)
RT ти(1 —vn)
Ь — вектор Бюргерса; ц — модуль сдвига.
Уравнение баланса масс
Пусть вдали от дислокационной границы концентрация рас-
твора остается постоянной с(оо, /)=с0, — состав фазы выде-
ления; состав твердого раствора у границы в условиях пре-
обладающего влияния дрейфа с[у0(/), /]1=с2=0, т. е. все коли-
чество подводимого к границе растворенного компонента сразу
отбирается. Функция уо(О имеет вид
(10.55)
При преобладающей роли нормальной диффузии
у,(/) = 2рг/Д^.
(10.56)
§ 10.6. КИНЕТИКА СПИНОДАЛЬНОГО РАСПАДА
Применение зависимостей, приведенных выше (см. § 10.1 —
10.4), к анализу распада твердых растворов замещения при из-
менении состава в широких пределах возможно, если везде вместо
коэффициента диффузии растворенного компонента подставить
22*
339
в формулы коэффициент гетеродиффузии, зависящий от концентра-
ции. Учет этой зависимости может дать не просто уточнение чис-
ловых параметров кинетических уравнений, но и качественное
изменение процесса распада, изменение его механизма. Взаимо-
действие атомов компонентов твердого раствора приводит к тому,
что диффузионный поток определяется не градиентом концентра-
ции, а градиентом химического потенциала и при определенных
условиях вызывает не устранение, а усиление градиентов кон-
центрации.
Дифференциальное уравнение диффузии, выведенное на основе
рассмотрения градиента химического потенциала с учетом кон-
центрационных напряжений (в связи с градиентом концентрации)
и структурных напряжений (в связи с когерентностью обогащен-
ных и обедненных областей), имеет вид (без нелинейных членов)
=М g v2e - 2AfTj2 t - 2Mky*c. (10.57)
Здесь M — подвижность [см. также уравнение (9.5)].
Решение этого уравнения можно представить в форме сину-
соидальных волн концентрации:
c=Co + exp[R(p)/]cos(0r), (10.58)
.со — исходная концентрация гомогенного твердого раствора; 0=
= 2лД— волновой вектор; R(₽) —фактор роста:
R Ф) = - М2? (S-+2T)2 2^2) • (10-59)
Учет влияния напряжений приводит к сужению] области спино*
дального распада (см. рис. 9.13), так как] условие для химической
d2f Е
спинодали df2/dc2 =0 заменяется -^—-4-27] "f_z—— 0 (когерент-
ная спинодаль). В начальной стадии распада распределения кон-
центраций можно описать суммой синусоидальных волн. Ампли-
туда каждой из этих волн будет во времени увеличиваться или
уменьшаться в зависимости от знака R(0).
Знак и величина фактора роста R ф) зависят от знака и вели-
чины d2f/dc2 и 1/Л. При-^-4-2т]2 >0, т. е. вне спино-
дальной области, R((3) отрицательны для всех р, следовательно,
небольшие флуктуации концентрации должны со временем исчезать.
Для системы внутри спинодальной области существует крити-
ческое значение 0кр (и соответственно ХКр), для которого R(p)=O.
Было показано также, что некоторому значению периода % соот-
ветствует максимальный фактор роста (см. рис. 9.14); дифферен-
циация компонентов будет наиболее быстро происходить в тех
340
волнах неоднородности, которые имеют периоды, близкие к ХМакс-
В результате такого отбора устанавливается определенный, при-
мерно постоянный по объему кристалла период неоднородностей.
Даже если амплитудные значения концентрации близки к соста-
вам равновесных фаз, образованием модулированной структуры
процесс не завершается: должен идти процесс, подобный процессу
коалесценции.
Одно из затруднений теоретического анализа огрубления мо-
дулированной структуры связано с размытостью границ между
обогащенными и обедненными областями. Приведенное диффу-
зионное уравнение, в котором опущены нелинейные члены, при-
годно только для анализа начальных стадий процесса. Тем не
менее имеются попытки теоретического анализа поздних стадий
спинодального распада — процесса огрубления модулированной
структуры. Для одномерной модуляции (пластинчатые области
вдоль одного направления) получена * логарифмическая временная
зависимость для периода модуляции
I (/) = Z. + -|-In (1 + ? exp-M (10.60)
iде l(t) и Zo— безразмерные величины периода (Zo к моменту
/=0); Z=i£X (X — период модуляции; g — характеристическая дли-
на, зависящая от температуры).
Для модели трехмерной периодичности (рассматриваются сфе-
рические области, образующие трехмерную сетку) получено
dldt(Ws) =е/3то, (10.61)
где Rs — радиус сферических областей.
Этот результат подобен закону Д/3, выведенному И. М. Лиф-
шицем и В. В. Слезовым.
Если изменения концентрации в модулированной структуре
находятся в экспоненциальной зависимости от времени, а рост
частиц (или периода) происходит пропорционально то можно
считать, что концентрационные изменения заканчиваются на-
столько быстро, что растут частицы, состав которых близок к со-
ставу конечных фаз.
* Langer L S. «Anna!. Phys.», 1971, v. 65, p. 53—86.
СПИСОК ЛИТЕРАТУРЫ
Общая литература
Beficc Р. Физика твердого тела. Пер. с англ. М., Атомиздат, 1968.
Жданов Г. С. Физика твердого тела. М., Изд-во МГУ, 1961.
Физические основы металловедения. М., Металлургиздат, 1955. Авт.: Я. С. Уман-
ский, Б. Н. Финкельштейн, М. Е. Блантер и др.
Шульце Г. Металлофизика. Пер. с нем. М., «Мир», 1971.
К гл. 1
Абрикосов А. А. Введение в теорию нормальных металлов. М., «Наука», 1972.
Физика металлов. Ч. I. Электроны. Под ред. Д. Займана. Пер. с англ., М.,
«Мир», 1972 (гл. 1, 2, 8).
К гл. 2
Устойчивость фаз в металлах и сплавах. Пер. с англ. Под ред. Д. С. Каменец-
кой. М., «Мир», 1970 (статьи 5, 8).
Электронная структура переходных металлов и химия их сплавов. Пер. с англ.
Под ред. Я. С. Уманского и Р. А. Суриса. М., Металлургиздат, 1966.
К гл. 3
Гольдшмидт X. Дж. Сплавы внедрения. Пер. с англ. М., «Мир», 1971, вып. 1,
гл. 1—3.
Иверонова В. И., Кацнельсон А. А. Ближний порядок в твердых растворах. М.,
«Наука», 1977.
Кривоглаз М. А., Смирнов А. А. Теория упорядочивающихся сплавов. М., Физ-
матгиз, 1958.
Скаков Ю. А., Глезер А. М. Упорядочение и внутрифазовые превращения. —
В сб.: Итоги науки и техники. Сер. «Металловедение и техническая обра-
ботка». Т. 9, М., ВИНИТИ, 1975, с. 5—72.
Строение металлических твердых растворов. Пер. с франц. Под ред. Л. Н. Лари-
кова. М., «Металлургия», 1966.
Тот Л. Карбиды и нитриды переходных металлов. Пер. с англ. М., «Мир», 1974.
Хачатурян А. Г. Теория фазовых превращений и структура твердых растворов.
М., «Наука», 1974.
К гл. 4
Интерметаллические соединения. Пер. с англ. Под ред. И. И. Корнилова. М.,
«Металлургия», 1970.
Устойчивость фаз в металлах и сплавах. Пер. с англ. Под ред. Д. С. Каменец-
кой. М., «Мир», 1970 (статьи 10, 13).
Шуберт К. Кристаллические структуры двухкомпонентных фаз. Пер. с нем. М.,
«Металлургия», 1970 (гл. 2, 3).
К гл. 5
Бокштейн Б. С., Бокштейн С. 3., Жуховицкий А. А. Термодинамика и кинетика
диффузии в твердых телах. М., «Металлургия», 1974.
Болтакс Б. И. Диффузия в полупроводниках. М., Физматгиз, 1961.
342
Герцрикен С. Д., Дехтяр И. Я. Диффузия в металлах и сплавах. М., Физмат-
гиз, 1961.
Томпсон Г. Дефекты и радиационные повреждения в металлах. Пер. с англ. М.,
«Мир», 1971.
Федоров Г. Б., Смирнов Е. А. Диффузия в металлах и сплавах. — В сб.: Итоги
науки и техники. Сер. «Металловедение и термическая обработка». Т. 8.
М., ВИНИТИ, 1974, с. 5—63.
К гл. 6
Варма А. Рост кристаллов и дислокаций. Пер. с англ. М., Изд-во иностр, лит.,
1958.
Данилов В. И. Строение и кристаллизация жидкостей. Избр. статьи. Киев, Изд-во
АН УССР, 1950.
Скрышевский А. Ф. Структурный анализ жидкостей. М., «Высшая школа», 1971
(гл. 1, 5).
К гл. 7
Любов Б. Я. Кинетическая теория фазовых превращений. М., «Металлургия»,
1969 (гл. 1—3).
Хачатурян А. Г. Теория фазовых превращений и структура твердых растворов.
М., «Наука», 1974 (гл. 1, 2, 4, 5).
К гл. 8
Иванов О. С., Бадаева Т. А., Софронова Р. М. и др. Диаграммы состояния и
фазовые превращения сплавов урана. М., «Наука», 1972.
Курдюмов Г. В., Утевский Л. М., Энтин Р. И. Превращения в железе и стали.
М., «Наука», 1977.
Лысак Л. И., Николин Б. И. Физические основы термической обработки стали.
Киев, «Техника», 1975.
Носова Г. И. Фазовые превращения в сплавах титана. М., «Металлургия», 1968.
Ройтбурд А. Л., Эстрин Э. И. Мартенситные превращения. — В сб.: Итоги нау-
ки и техники. Сер. «Металловедение и термическая обработка». Т. 4. М.,
ВИНИТИ, 1970, с. 5—70.
Сокурский Ю. Н., Стерлин Я. М., Федорченко В. А. Уран и его сплавы. М.,
Атомиздат, 1971.
К гл. 9
Гинье А. Неоднородные металлические твердые растворы. Пер. с англ. М., Изд-
во иностр, лит., 1962.
Захарова М. И. Атомно-кристаллическая структура и свойства металлов и
сплавов. М., Изд-во МГУ, 1972.
Конобеевский С. Т. Действие облучения на материалы. М., Атомиздат, 1967.
Курдюмов Г. В. Явления закалки и отпуска стали. М., Металлургиздат, 1960.
Чуистов К. В. Модулированные структуры в стареющих сплавах. Киев, «Нау-
кова думка», 1975.
Тяпкин Ю. Д., Гаврилова А. В. Старение металлов. — В сб.: Итоги науки и
техники. Сер. «Металловедение и термическая обработка». Т. 8. М.,
ВИНИТИ, с. 64—124.
К гл. 10
Кристиан Дж. У. Фазовые превращения.—В кн.: Физическое металловедение. Под
ред. Р. Кана. Пер. с англ. Вып. II. М., «Мир», 1968, с. 227—340.
Любов В. Я. Кинетическая теория фазовых превращений. М., «Металлургия»,
1969.
РЕШЕНИЯ И ОТВЕТЫ К ЗАДАЧАМ
1.1. Решение. Координационное число для кристаллической решетки типа
алмаза равно 4, а каждая связь одновременно принадлежит двум атомам угле-
рода, поэтому общее число связей для N атомов равно 4А/2 = 2А=2уИАа/Ас>
где Na—число Авогадро; Ас—атомный вес углерода. Имеем: E=2MNA'X
ХЕсв/Ас — Ъ,]. ккал.
Ответ: Энергия кристалла алмаза равна 6,1 ккал.
1.2. Решение. Используя уравнение f (е) = 1/1-|-е8 гф^Т,
Ответ: 1. f(<F)=l; 2. f (<Г)=0,5; 3. 0,269 и 0,731; 4. 0,018 и 0,982.
Замечание. Для комнатных температур kT—0,025 эВ, т. е., учитывая сим*
метричность распределения Ферми, отклонение больше ±0,1 эВ от еф будет
у 3,6% электронов, а &f имеет порядок несколько электронвольт.
1.3. Ответ. Первая зона Бриллюэна для кристалла с гекс. п. у. структурой
ограничена 20 плоскостями, образованными тремя совокупностями кристаллогра-
фических плоскостей (или плоскостей интерференции): шестью гранями совокуп-
ности {10.0}, двумя— {00.2} и двенадцатью — {10.1}.
2.1. Решение. Длина связи равна 0,25 пространственной диагонали решетки
с периодом а. р=латАт/а3=2,33 г-см~3, где пат— число атомов на ячейку;
А — атомная масса; пг— масса атома водорода.
2.2. Решение. Радиус г октаэдрической поры, образованной атомами радиу-
сом 7?а, равен г=0,414; /?а ионный радиус фтора, /?а=1,33 А, г=0,414-1,33=
е о
=0,55 А. Это может быть ион лития Li+ (г=0,59 А).
2.3. Ответ. Отношение равно 0,95.
3.2. Энергия сублимации Q для металла определяет изменение внутренней
энергии системы при полном разрыве связей между атомами (переход кри-
сталл—>пар). Не учитывая изменения при этом энергии теплового движения
атомов и взаимодействия других атомов, кроме ближайших соседей, из уравне-
ния (3.6) при х=0 (т. е. для чистого компонента А) получим
1 2La
UА “ ~ NZUAA> иАА = fife' '
Из табл. 2.3 для меди Q = 81 ккал/моль; z— 12 (г. ц. к. структура), в расчете
2-81 13,5
на 1 моль берем N—\\ ^Cu_Cl1 = = 13,5 ккал/моль — эВ^0,6 эВ.
3.3. Энтропию смешения для двухкомпонентного твердого раствора опреде-
ляем через число способов W размещения атомов компонентов А и В по узлам
кристаллической решетки твердого раствора:
Scm= k In IT, (3.3.1)
344
пусть NA—число атомов компонента A; NB— число атомов В; если N— полное
число узлов, то Na=cN, Nb=(1—c)N, где с — концентрация компонента А.
Число способов размещения одного атома А среди W узлов — число способов
размещения двух атомов А среди N узлов — W (У—1)/2 [для второго атома А
остается АГ—1 узлов, делим произведение У (У—1) на два, так как взаимная
перестановка первого и второго -атомов не дает нового состояния системы, по-
скольку эти атомы неразличимы]. Число способов размещения трех атомов А
среди N узлов
У (У — 1) (У —2)
1-2-3 ’
число способов размещения Na атомов среди У узлов
У (У —1) (N — 2). . . ,(N — (Na — 1))
1-2-3. . . .ftд
Умножая числитель и знаменатель последнего выражения на (У—Nа)! = Ув!»
получаем
у»
<3-3-2»
Легко видеть, что каждому (одному) распределению атомов сорта А отве-
чает одно распределение атомов сорта В, т. е.
У!
/V!
*$см = д/ I дг | •
° А* В’
Заменяя Na и Nb значением N и с и используя приближение Стирлинга
(In N\ —N In N—N), получаем окончательно
Scm==—kN[c In c+(l— c) In (1—c)]. (3.3.3)
3.4. Исходным уравнением является выражение для свободной энергии твер-
дого раствора ДГ=Д£/—ТДЗ, которое получается из выражений для внутренней
энергии сплава [см. уравнение (3.6)] и энтропии [уравнение (3.3.3), выведенное
в задаче 3.3].
Учитывая только члены, зависящие от фазового состояния сплава и его кон-
центрации, и полагая Uaa^Ubb, в расчете на атом получаем
р
f _ —В-= const + гДц {с (1—с) + р [с In с + (1 — с) In (1—с)]}, (3.4.1)
'Р у
здесь переменный параметр p=kT/zbu.
Легко видеть, что зависимость f(c) при р=0 (т. е. при О К) имеет вид мо-
нотонной кривой с максимумом при с=1/2, что соответствует неустойчивости
состояния твердого раствора во всей области концентраций;
гДп
при р^1/2, т. е. при температурах, превышающих ТМакс= “?£" > зависимость
f(c) имеет вид монотонной кривой с минимумом также при с=1/2, что соответ-
ствует полной взаимной растворимости компонентов. При промежуточных зна-
чениях параметра 0<р<1/2, т. е. в области температур от 0 до ТМакс, зависи-
мость f(c) имеет сложный вид кривой с двумя минимумами. При принятых выше
допущениях эта зависимость симметрична относительно вертикали с=0,5. При
этом составы твердых растворов, находящихся в равновесии в двухфазной
области (0<р<1/2), определяются общей касательной к кривой f(c) в точках
минимумов; таким образом, состав твердого раствора определяется условием
dj/dc — b
1—2с+р[1п с—In (1—с)] =0. (3.4.2)
345
Отсюда
JTZ7 = exP[---£Г~(1— 2с) j - (3.4.3)
При малых концентрациях (1—с=е1) кривая растворимости должна иметь вид
экспоненты:
с~ехр(—гДи/kT). (3.4.4)
3.5. Температуру, соответствующую максимуму кривой растворимости (или
расслоения), можно получить, решая уравнение (3.4.2), выведенное в задаче
3.4, относительно параметра р:
1___
р = kT/zbu = ln[(j _С) /С] - (3.5.1)
Максимум кривой расслоения в отсутствие влияния фактора упругой энергии
имеет место при концентрации с—1/2, однако подстановка с=1/2 в уравнение
(3.5.1) приводит к неопределенности.
Для раскрытия неопределенности в уравнении (3.5.1) находим отношение
производных числителя и знаменателя:
__Ы макс
/^макс —
(3.5.2)
(3.5.3)
3.7. Решение.
_ Pg—С А _ Pb~cB X—Wb — CB
1 — сА 1 — св 1 — св
для <у=0,76; для w6=0,12 0=0,5,
3.8. Решение.
р а = а (1—Са )= 0,44.
Рв — & (1—Св) +св = 0,81.
мА = 1—рА=0,56, ^в = 1—рв = 0,19.
3.9. Ответ. рА = 0,82, Ша=0,18, рБ = 0,85, шв = 0,15.
4.1. Твердый раствор внедрения ТаСод имеет г. ц. к. решетку с периодом
о
4,41 А, а атомы углерода размещаются в октаэдрических пустотах. Атомный
о 4,41—2,92
радиус Та гТа=И,46 А, гс эф=----%----- =0,745 А.
4.2. Плотность вакансий по железу равна 4,6 *1021 см~3.
6.1. Для кубического зародыша с ребром 2г для ДГ=10° 2г=9.6-10~6 см.
При ДТ=100° 2г=9,6-10~7 см. Если принять период решетки железа вблизи
температуры плавления а = 3,05 А, то в первом случае зародыш состоит более
чем из 107 ячеек, а во втором — из 104.
6.2. Объем стержня равен объему шара, имеем ДЕ——а2ш7+а4л/?2=
=—17,3 Эрг. В ходе самопроизвольной сфероидизации энергия системы умень-
шается.
6.3. Задаваясь разными Рт/Р^, получаем
Рг/Р^ 1,15 2 5 10
Wr/P«) 0,0453 0,301 0,699 1
ехр { — а5к/З^Г} |Q- 300® Ю-83 Ю-is Ю-’л
346
6.4. Максимальное Да=0,5-10~8 см, Да/а=0,09.
6.7. Дислокации в растущих кристаллах с г. ц. к. решеткой, перемещаясь
в плоскости скольжения (111), могут'выходить, если направление роста совпс
дает с осью [100], на боковую поверхность, т. е. «зарастают». При направлении
роста [НО] дислокации выходят на растущую торцевую поверхность, создавая
там ступеньки роста, и «прорастают» во вновь кристаллизующиеся области.
7.2. а. Указание. Решетки Fe—Сг-матрицы и фазы ЕезА1 можно рассматри-
вать как изоморфные. При параллельной ориентации решеток расстояние между
полосами картины муара D определяется формулой l/Z^l/d'—\/d"=(d"—
—df)/d''d”, где d' и d" — межплоскостные расстояния матрицы и включения для
действующих отражений соответственно;
Д^=d”—d' = d'd" fD^d'2/D.
Для отражения (200)
в. Вектор Бюргерса эпитаксиальной дислокации в о. ц. к. решетке Ь =
— а <100>, т. е. Ь=2,87 А, 7=550 А.
7.3. Ответ. еюо = —1,86% = +5,35% (сжатие). 7.4. а. Ответ. (расширение); 80ia=+3,93% (сжатие); 8Ooi =
а V.c. е, %
(100) II (ЮО) —27,2!
(011) II (ОЮ) +2,8
(ОН) II (001) + 2,8
Выделения имеют вид пластин габитус (100)а, ограниченный рост в направ-
лении [Ю0]ж V£3.
а Мо2С *>•/.
( 100) II (10.0) + 4,7!
(011) II (00.1) + 16,5
(ОН) II (01.0) + 28,7
Выделения имеют вид игл вдоль [100]а, рост в других направлениях ограни-
чен из-за большого несоответствия межатомных расстояний.
В. f V е, %
(100) II (100) — 0,5
(010) II (010) — 0,5
(001) II (001) — 0,5
нет направлений преимущественного роста, выделения равноосны.
8.1. а.
2 55,85
Ра-Fe— 2,8985’- 10_” ’ 6,023-10” ~7,62 г-см~’,
4 55,85
Рт-Fe — 3,6394’-Ю-24 6,023-10” —7>73 г-см-’
Дц /1 1 \ / 1
~’Ю0= (7,62 —7,7з) / Л62 = 1153
8.2. а. 4 ориентировки а-Со.
б. 2 (двойниковые) ориентировки р-Со.
347
8.3. a. 12 векторов <110>.
б. Да.
(10 0 \
—1 /2 1/2—1/21.
0 1 1 /
8.5.*
—0,10
о
о
0
0,10
О
О
О
0,2
9.1. а. б. В массивных образцах первой серии (диаметр 12,6 мм) при рез-
ком охлаждении (закалке) возникают значительные термические напряжения зо-
нального характера (на периферии цилиндра — деформация растяжения).
Разница периодов образцов первой и второй серий со временем старения
уменьшается за счет частичной релаксации этих напряжений.
в. Понижение твердости в начальной стадии старения, очевидно, связано
с тем, что твердость исходного («закаленного») состояния может заметно воз-
расти за время пребывания этого образца при комнатной температуре (интервал
времени между закалкой и моментом измерения). Этот эффект «естественного»
старения снимается при последующем кратковременном нагреве («возврат»).
9.2. Принимая температурную зависимость скорости процесса старения типа
Аррениуса, надо построить график в координатах In (1/тмакс)—1/Т, где Тмакс—
время достижения максимума твердости; Т — температура старения (К).
9.3. а. Число частиц (зон) около 1015 см~3.
б. Равновесное количество вакансий на 1 моль: пВак = Na X
Хехр (-^вак/7?Т).
Для сопоставления с картиной распада следует найти объемную концентра-
цию вакансий, используя данные о плотности a-Fe (7,8 г/см3) или об атомной
о
структуре (о. ц. к., а=2,86 А).
в. Д^КРт, отсюда 7)^Д2/т^ 10~10 см2/с.
* См. также: Келли А., Гровс Г. Кристаллография и дефекты в кристаллах.
М., «Мир», 1974, с. 373.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аллотропия 30, 222
— концентрационная 76
Аморфные металлы и сплавы 173
Атмосферы примесных атомов 304,
307, 319, 339
Атомного размещения функция 127,
175
Блоки мозаичной структуры 165
Вакансии при старении 290, 319
Видманштеттова структура 188, 231
Возврат при старении 291
Волновая функция 11, 13
Волновое число (для электронов) 12
Габитус частиц новой фазы 146, 153,
189, 249
Гетерогенное зарождение 139, 185,279
Гомогенное зарождение 131, 142, 185,
279
Границы
— межфазовые 150, 186, 207
— зерен при зарождении 218, 231,
291
Даркена анализ 116, 118
Двойники
— деформации 240
— превращения 237, 255, 265
Двухфазный распад 218, 279, 309
Деформация
— с инвариантной плоскостью 206,
234
— при инвариантной решетке 237
— решетки в кобальте 242
— решетки ОЦК по Бейну 243
— эллипсоид 235
Дислокации
— при выделении 188, 298, 339
— при мартенситных превращениях
208, 251, 256
Дисперсионное твердение 273
Диффузия, атомные механизмы 107
— восходящая химическая 102, 286
— граничная 114
— в поле напряжений 102, 103
— теория 105
— и фазовые превращения 119, 16ГГ
180, 203, 211, 216, 329
— в электрическом поле 104
Домены
— антифазные 51
— упругие 198, 202
Зонная плавка 163
Зоны Бриллюэна 15, 65
Зоны Гинье — Престона 275
Интерметаллические соединения 63,.
77, 81, 88, 120
Карбидные фазы при распаде мар-
тенсита 309
Кинетика кристаллизации 153
— коалесценция частиц 284, 334
— роста частиц 205, 216, 232, 325
— спинодального распада 289, 339
— старения мартенситных сталей
277, 301
— ячеистого (двухфазного) распада
219
Коллоидное равновесие (при старе-
нии) 282
Кристаллизация
— зародыши (центры) 131
----двумерные 144, 150, 153
----критический размер 133
---- работа образования 134
----активные 142
----поверхностно-активные 138
---- дезактивация 141
----естественная активность 142
— скорость
---- роста (линейная) 149, 154
----зародышеобразования 153, 178
----объемная (общая) 157
— ступени роста 158, 159
Коэффициент диффузии
— величина зерна 114, 115
— влияние третьего компонента 113
— и искаженность кристаллической
решетки 102
349
— концентрационная зависимость 112
— и тип твердого раствора 113
— химической связь с коэффициен-
том самодиффузии 118
Мартенсит
— морфология кристаллов 250
— отпуска 309
— субструктура 251
Мартенситное превращение
— атермическое 255
— кинетика 254
— кристаллографическая теория 233
— изотермическое 255
— термоупругое 202, 264
Мартенситно-стареющие стали 273,
294
Матано, метод 101
Многогранники
— Каспера 80
— координационные 79
Модулированная структура 197, 289
Ориентационные соотношения
---в железе и стали 246, 310
--- в титане и его сплавах 258,
261
--- в уране и его сплавах 268
— в процессе выделения 190
Плотность состояний 10
Поверхность Ферми 14, 44, 65, 76
Поверхностное натяжение 132, 134
Превращения
— бездиффузионные 161, 203, 325
— массивные 209, 228
— мартенситные 181, 205, 233, 276
— с нормальной кинетикой 202
Принцип
— размерного и структурного соот-
ветствия 143
— плотного заполнения пространства
(в кристаллах) 77
Пространство импульсов 7
Распад твердых растворов
— при комбинированном старении
294
— непрерывный 212, 214, 278
— прерывистый (ячеистый) 212, 218,
279
— спинодальный 119, 185, 285, 339
— стадии выделения 214, 280, 302,
309
— и упругая анизотропия 197, 289
Ретикулярная плотность (кристалла)
145, 147
Спинодаль
— когерентная (упругая) 288
— химическая 119, 285
350
Твердые растворы
— замещения 33
— внедрения 23, 34
— вычитания 33, 37
— упорядоченные 48, 51
Температура
— абсолютной потери устойчивости
(фазы) 184
Уравнения
— диффузии, решения 98, 99
— для бесконечно тонкого слоя 98
— для неограниченного тела 99
— общие закономерности 95, 96
— при переменном значении коэффи-
циента диффузии 100
— для полубесконечного простран-
ства 98
— Шредингера И, 13
— Фика 95
Фазовое пространство 8
Фазовые переходы первого и второго
рода 49, 182, 264
Фазы
— внедрения 72
----- электронная теория 74
— свойства 73, 77
— Лавеса 63, 90
— «промежуточные» 63, 173
— сигма 63, 81
Химическая связь 5, 6
Элементы
— металлические 24, 27
— радиоактивные 30
— редких земель 30
— переходных групп 30, 31
Электрон
— вырождение 10
— квазиимпульс 18
— эффективная масса 18
Электронные соединения
— фазы Юм-Розери 63, 263
— между переходными металлами
71, 263
Энергия
— активации
-----при диффузии 96
----- при старении 276
— градиентная 287
— поверхностная 134, 189, 194
— смешения 41, 62
— Ферми 8
— электрона 8, 14
Эпитаксия 143
— дислокации 188
Эффект
— Киркендала 117
— «памяти» 264, 271
ОГЛАВЛЕНИЕ
Предисловие............................................................ 3
Глава 1. Химическая связь и металлическое состояние вещества 5
Задачи к гл. 1.......................................................23
Глава 2. Кристаллическая структура химических элементов .... 24
§ 2.1. Структура ковалентных кристаллов..............................24
§ 2.2. Структура металлических элементов.............................27
Задачи к гл. 2......................................................32
Глава 3. Твердые растворы...............................................33
§ 3.1. Кристаллическая структура твердых растворов ..... 33
§ 3.2. Взаимная растворимость металлов...............................40
§ 3.3. Упорядоченные твердые растворы (сверхструктуры) .... 48
§ 3.4. Ближний порядок в твердых растворах..........................59
Задачи к гл. 3......................................................62
Глава 4. Металлические соединения......................................63
§ 4.1. Электронные соединения.......................................63
§ 4.2. Фазы внедрения...............................................72
§ 4.3. Принцип плотной упаковки и его реализация в структурах
интерметаллических соединений.......................................77
§ 4.4. Металлические соединения типа o-FeCr и родственные им фазы 81
§ 4.5. Некоторые интерметаллические соединения при простых стехио-
метрических (атомных) соотношениях..................................88
Задачи к гл. 4.....................................................94
Глава 5. Диффузия в металлах...........................................95
§ 5.1. Феноменологическая теория диффузии...........................95
§ 5.2. Атомная теория диффузии......................................105
§ 5.3. Диффузия в поле градиента концентраций......................116
Задачи к гл. 5......................................................124
Глава 6. Структура расплавов и кристаллизация.........................126
§ 6.1. Структура расплавленных металлов............................126
§ 6.2. Гомогенная кристаллизация...................................131
§ 6.3. Гетерогенное образование зародышей..........................139
§ 6.4. Атомная теория роста кристаллов.............................146
§ 6.5. Кинетика кристаллизации.....................................152
§ 6.6. Особенности кристаллизации и структуры сплавов .... 160
Задачи к гл. 6.....................................................177
Глава 7. Общие закономерности фазовых превращений в твердом со-
стоянии. Классификация превращений..................................179
§ 7.1. Общая классификация фазовых превращений и проблема за-
рождения новой фазы................................................181
§ 7.2. Роль энергии упругой деформации и межфазовой (поверхност-
ной) энергии.......................................................186
§ 7.3. Механизмы роста кристаллов при бездиффузионных превраще-
ниях. Термодинамическая возможность бездиффузионного пре-
вращения твердых растворов.........................................203
§ 7.4. Типы диффузионных превращений...............................211
Задачи к гл. 7.....................................................221
351
Глава 8. Аллотропические превращения в металлах и сплавах . . 222
§ 8.1. Аллотропическое у—>а-превращение железа и твердых раство-
ров на основе железа .............................................225
§ 8.2. Теория мартенситных превращений ..........................233
§ 8.3. Мартенситное превращение в сплавах на основе железа . 243
§ 8.4. Аллотропические превращения титана, твердых растворов на
основе Р—Ti и некоторых интерметаллидов......................... 257
§ 8.5. Аллотропические превращения в уране и его сплавах . . . 265
Задачи к гл. 8...................................................271
Глава 9. Распад пересыщенных твердых растворов.......................273
§ 9.1. Изменения свойств и структуры при старении сплавов . . 273
§ 9.2. Некоторые вопросы термодинамики и кинетики процессов рас-
пада пересыщенных твердых растворов...............................280
§ 9.3. Влияние пластической деформации на распад пересыщенных
твердых растворов и роль дефектов кристаллического строения 2.98
§ 9.4. Отпуск стали (распад мартенсита)..........................305
§ 9.5. Влияние излучения высокоэнергетических частиц, рентгеновского
и у-излучения на фазовые превращения “............................312
Задачи к гл. 9...................................................324
Глава 10. Анализ кинетики процессов роста частиц новой фазы при
нормальных превращениях........................................ 325
§ 10.1. Кинетика роста центра новой фазы при бездиффузионном пре-
вращении нормального типа..........................................325
§ 10.2. Анализ процесса роста ферритного центра в аустенитной мат-
рице сплава железо — углерод.......................................328
§ 10.3. Анализ роста сферических частиц с учетом перекрытия диффу-
зионных полей .....................................................332
§ 10.4. Коалесценция частиц.......................................334
§ 10.5. Влияние напряжений на процесс роста выделений .... 338
§ 10.6. Кинетика спинодального распада............................339
•Список литературы...................................................342
Решения и ответы к задачам...........................................344
Предметный указатель................................................ 349
ИБ № 477
Яков Семенович Уманский, Юрий Александрович Скаков
ФИЗИКА МЕТАЛЛОВ
Атомное строение металлов и сплавов
Редактор В. К. Мелешко
Художественный редактор А. Т. Кирьянов
Переплет художника В. А. Бондарева
Технический редактор Н. А. Власова
Корректор Н. М. Загудаева
Сдано в набор 6.1.1978 г. Подписано к печати 22.VI.1978 г. Т-12903
Формат 60Х90!/1в Бумага тип. № 2 Усл. печ. л .$22,0
Уч.-изд. л. 25,07 Тираж 11 500 экз. Зак. изд. 75218 Зак. тип. 523 Пена 1 р. 20 к.
Атомиздат, 103031, Москва, К-31, ул. Жданова, 5
Московская типография № 10 Союзполиграфпрома при Государственном комитете Совета
Министров СССР по делам издательств, полиграфии и книжной торговли.
Москва, М-114, Шлюзовая наб., 10.