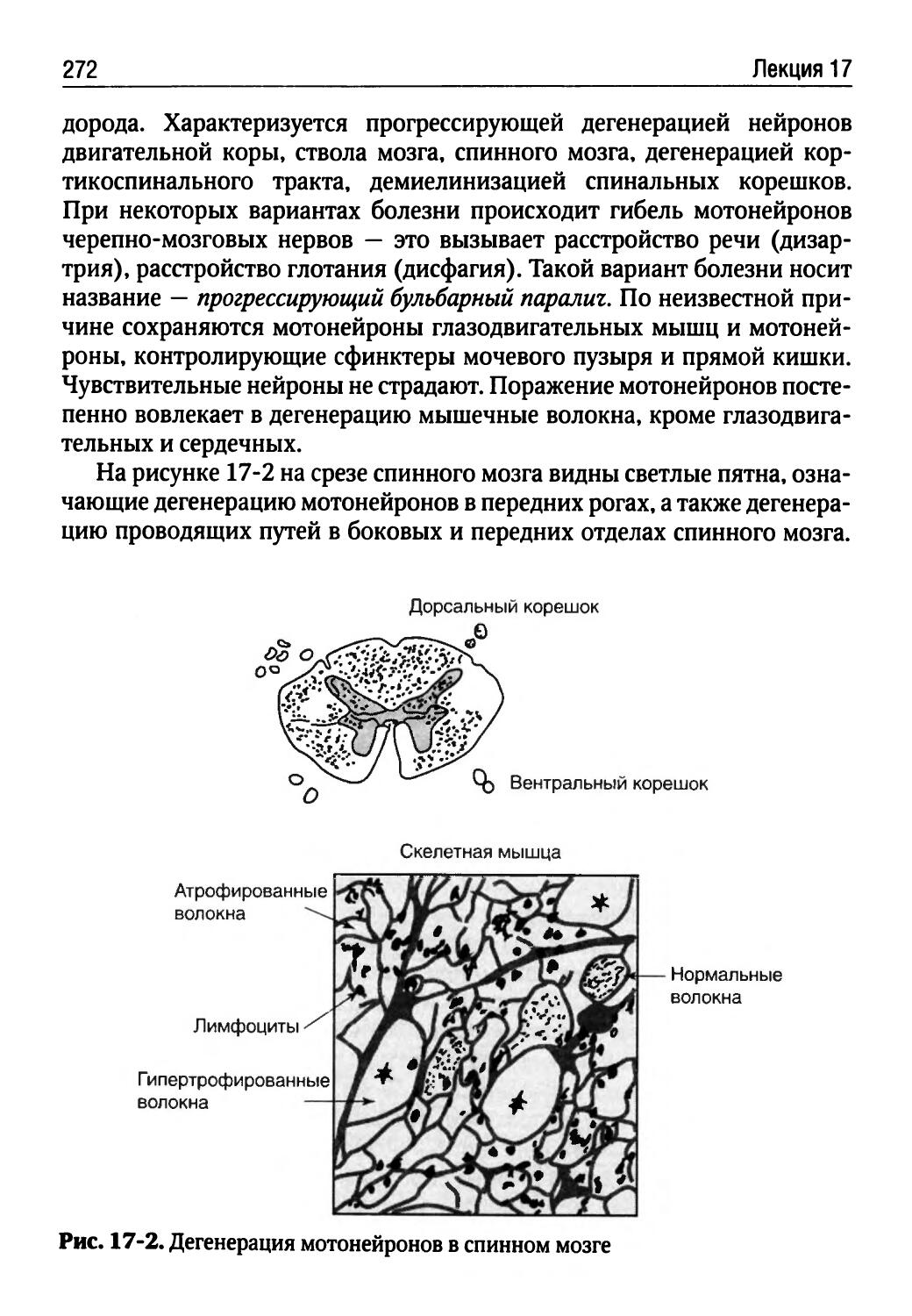
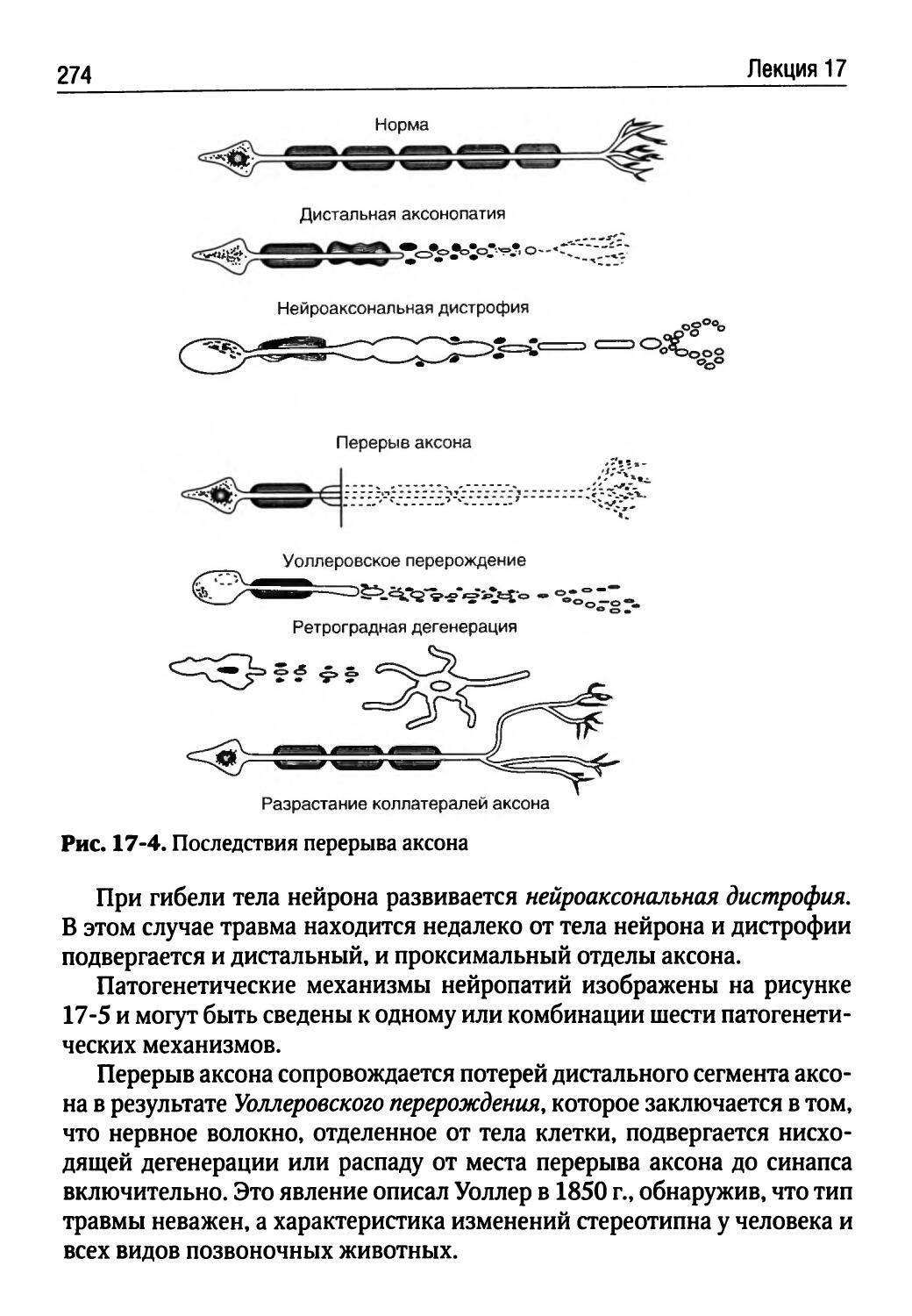
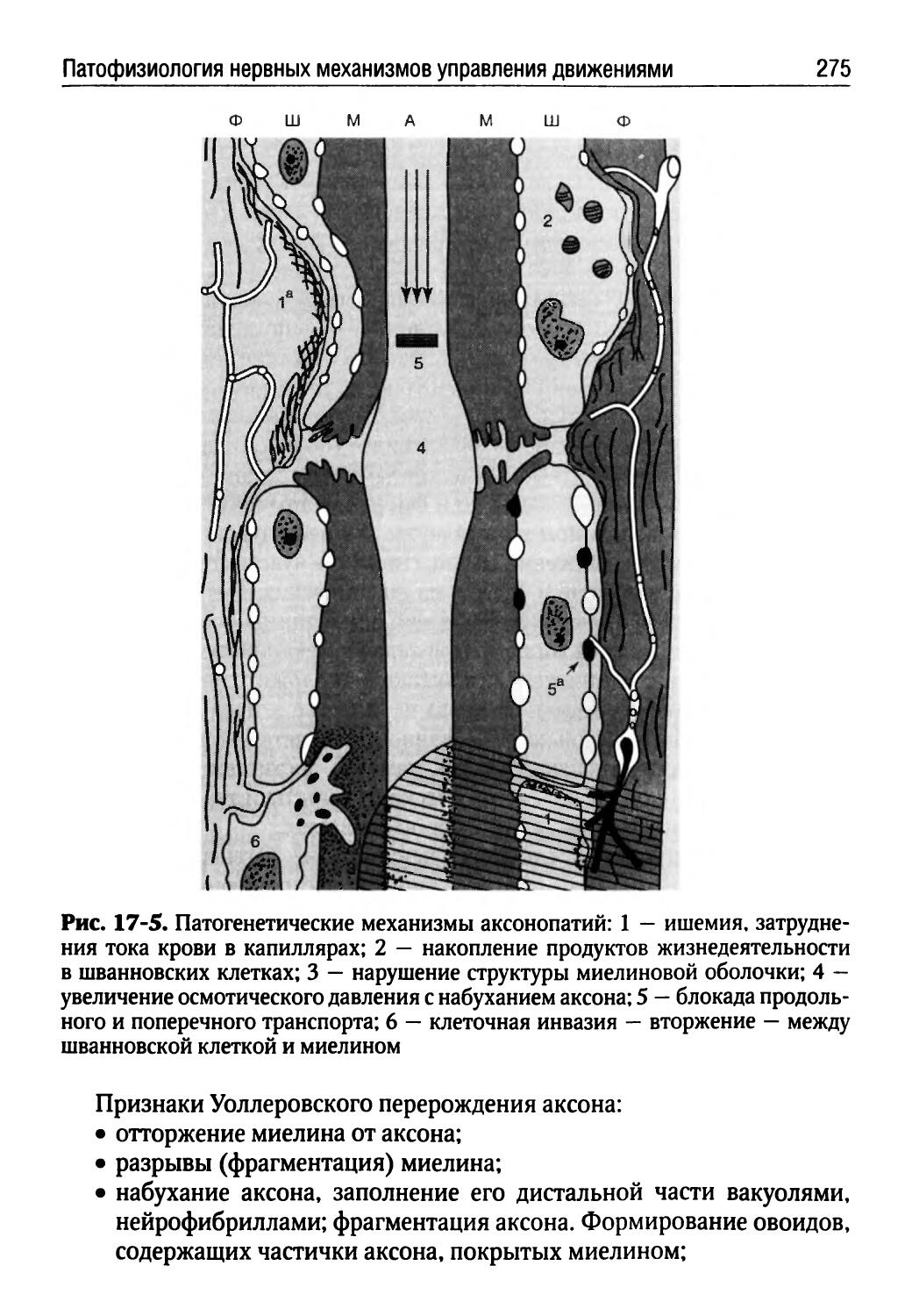
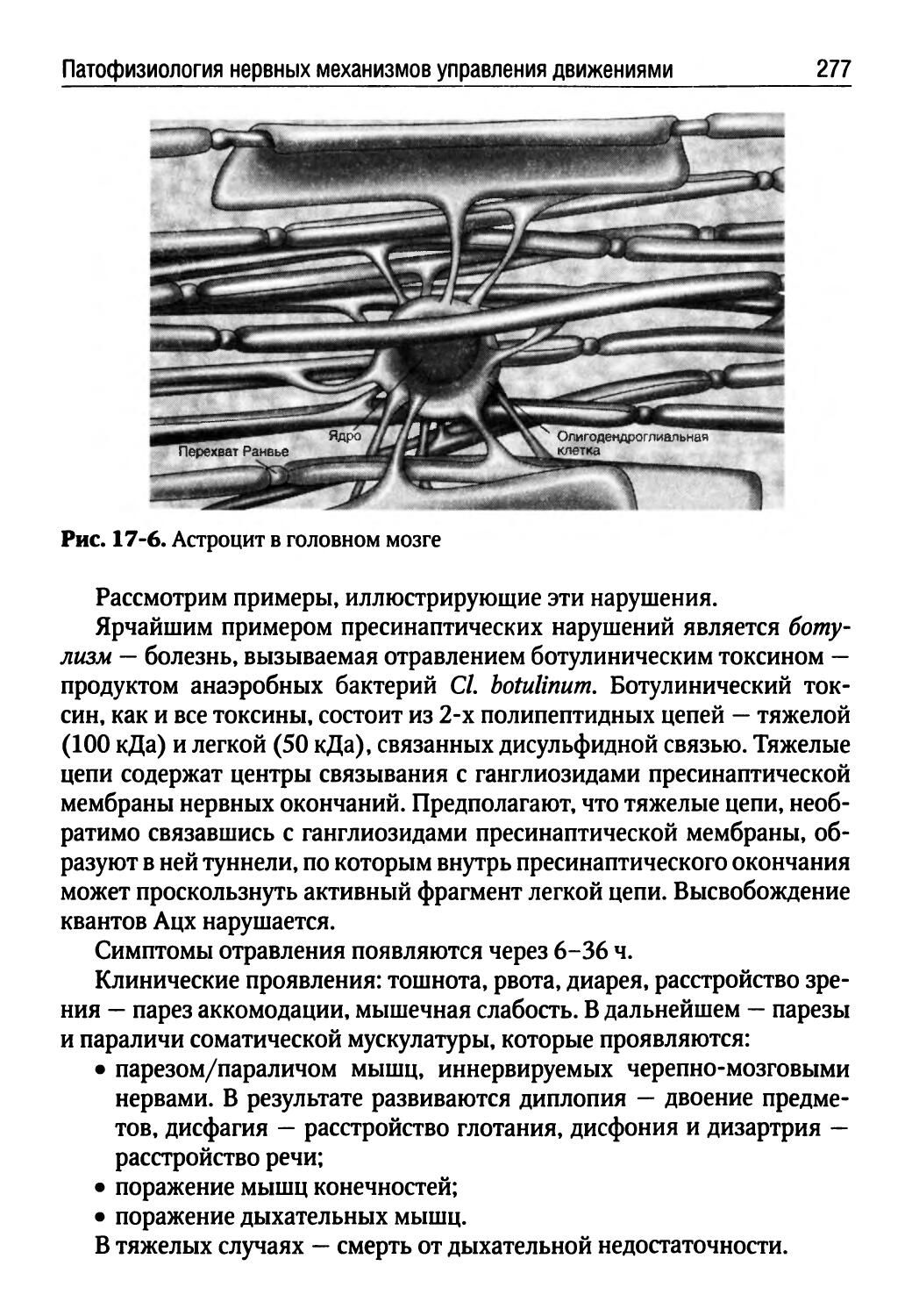
/


















































































































































































































































































































































































































































































































































































Автор: Порядин Г.В.
Теги: патологическая физиология формы развития заболеваний патогенез учение о происхождении заболеваний общая патология патофизиология
ISBN: 978-5-9704-2903-7
Год: 2014




Текст
ПАТОФИЗИОЛОГИЯ
КУРС ЛЕКЦИЙ
УЧЕБНОЕ ПОСОБИЕ
Под редакцией члена-корреспондента РАМН, профессора Г.В. Порядина
ИЗДАТЕЛЬСКАЯ ГРУППА «ГЭОТАР-Медиа»
ГЭОТАР
МЕДИЦИНСКИЕ УЧЕБНЫЕ ПОСОБИЯ
подразделение крупнейшей в России и странах СНГ компании, работающей в сфере медицинского и фармацевтического образования
КОМПЛЕКСНОЕ ОСНАЩЕНИЕ ЦЕНТРОВ ПРАКТИЧЕСКИХ УМЕНИЙ
УЧЕБНОЕ ОБОРУДОВАНИЕ
Анатомические и биологические модели
Модели патологий
Модели для обучения пациентов
Тренажеры, манекены и симуляционные модели для отработки практических умений (врачебных и сестринских):
• сердечно-легочная реанимация
• первая помощь при травмах и кровотечениях
• физикальное обследование
• хирургические манипуляции
• инвазивные процедуры
• родовспоможение
• уход за больными
• ультразвуковая диагностика
Расходные материалы и медицинские инструменты для симуляционного оборудования
УЧЕБНЫЕ МАТЕРИАЛЫ
Наглядные пособия (плакаты и атласы)
Мультимедийные материалы
Виртуальные пациенты
МЕТОДИЧЕСКОЕ ОБЕСПЕЧЕНИЕ
КОМПАНИИ-ПРОИЗВОДИТЕЛИ
Limbs & Things
PAY Laerdal
AdamJtouiUy ( otoSim
SynDaver™ Labs -4*1-J- f
Synthetic Human Tissues <ЗаГСНоЩС8
кокни
tru corp
Г KYOTO KAGAKU
Перечень компаний постоянно расширяется
ЗАКАЗ МОДЕЛЕЙ И КОНСУЛЬТАЦИИ
Телефакс: (495) 921-39-07 (доб. 237,267), (916)876-98-03.
E-mail: info@geotar-med.ru
Полный каталог наглядных учебных пособий и интернет-магазин
www.geotar-med.ru
ЭЛЕКТРОННО-БИБЛИОТЕЧНАЯ СИСТЕМА ДЛЯ МЕДИЦИНСКОГО И ФАРМАЦЕВТИЧЕСКОГО ОБРАЗОВАНИЯ
КОНСУЛЬТАНТ
СТУДЕНТА
ЭЛЕКТРОННАЯ БИБЛИОТЕКА МЕДИЦИНСКОГО ВУЗА
www.studmedlib.ru
СТРУКТУРА
Учебники,учебные пособия Руководства к практическим занятиям
Атласы
Тестовые вопросы и ситуационные задачи_______
Лекции ведущих преподавателей
Практические умения_________
Учебные программы по дисциплинам Мультимедийные материалы
• Быстрый и удобный доступ для студентов и преподавателей к высококачественной медицинской информации
• Расширение возможностей для самостоятельной работы студентов
• Обеспечение контроля преподавателями за самостоятельной работой студентов в режиме реального времени
• Ускорение перехода на модульное, проблемно-ориентированное и дистанционное обучение
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ:
тел.: (495) 921-39-07 (доб. 266), (917) 550-49-19 e-mail: chmarov@geotar.ru
ИЗДАТЕЛЬСКАЯ ГРУППА «ГЭОТАР-Медиа»
ПАТОФИЗИОЛОГИЯ
КУРС ЛЕКЦИЙ
Под редакцией члена-корреспондента РАМН, профессора Г.В. Порядина
УЧЕБНОЕ ПОСОБИЕ
Министерство образования и науки РФ
Рекомендовано ГОУ ВПО «Первый Московский государственный медицинский университет имени И.М. Сеченова» в качестве учебного пособия для студентов учреждений высшего профессионального образования, обучающихся по специальностям 31.05.01 «Лечебное дело», 31.05.01 «Медико-профилактическое дело», 31.05.03 «Стоматология», 31.05.02 «Педиатрия» по дисциплине «Патофизиология. Клиническая патофизиология»
Регистрационный номер рецензии 122 от 21 апреля 2011 года ФГУ «Федеральный институт развития образования»
Москва ИЗДАТЕЛЬСКАЯ ГРУППА « ГЭОТАР-Медиа» 2014
УДК 616-092(075.8)
ББК 52.5я73-1
П20
Авторский коллектив - сотрудники кафедры патофизиологии ГБОУ ВПО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России: Порядин Геннадий Васильевич, зав. кафедрой патофизиологии, д-р мед. наук, проф., чл.-кор. РАМН; Салмаси Жеан Мустафаевич, проф.; Шарпань Юрий Владимирович, доцент; Осколок Лариса Николаевна, доцент; Богуш Нина Львовна, доцент; Бережкова Неля Ивановна, доцент; Щелкунова Галина Петровна, доцент; Зеличенко Людмила Иосифовна, доцент; Ручинская Татьяна Юрьевна, доцент; Семенова Людмила Юрьевна, доцент.
П20 Патофизиология : курс лекций : учеб, пособие / под ред. Г. В. Порядина. - М.: ГЭОТАР-Медиа, 2014. - 592 с.: ил.
ISBN 978-5-9704-2903-7
Учебное пособие соответствует требованиям действующего Федерального государственного образовательного стандарта высшего профессионального образования. Подготовлено сотрудниками кафедры патофизиологии Российского национального исследовательского медицинского университета имени Н.И. Пирогова Минздрава России.
Пособие состоит из 38 лекций, в которых последовательно разбираются вопросы общей и частной патофизиологии. Теоретический материал иллюстрирован таблицами и рисунками.
Предназначено студентам и аспирантам всех факультетов медицинских вузов.
УДК 616-092(075.8)
ББК 52.5я73-1
Права на данное издание принадлежат ООО Издательская группа «ГЭОТАР-Медиа». Воспроизведение и распространение в каком бы то ни было виде гасти или целого издания не могут быть осуществлены без письменного разрешения ООО Издательская группа «ГЭОТАР-Медиа».
ISBN 978-5-9704-2903-7
© Коллектив авторов, 2014
© ООО Издательская группа «ГЭОТАР-Медиа», 2014 © ООО Издательская группа «ГЭОТАР-Медиа»,
оформление, 2014
ОГЛАВЛЕНИЕ
Предисловие...............................................5
Сокращения и условные обозначения.........................6
Лекция 1. Введение в патофизиологию (Г.В. Порядин)........9
Лекция 2. Патофизиология алкоголизма, наркоманий и токсикоманий (Ю.В. Шарпань).........................................22
Лекция 3. Молекулярные механизмы повреждения клетки (Л.Н. Осколок).........................................37
Лекция 4. Воспаление I (сосудистые реакции) (НЛ. Богуш).....50
Лекция 5. Воспаление II (клеточные реакции) (НЛ. Богуш).....63
Лекция 6. Ответ острой фазы (Ж.М. Салмаси)..................75
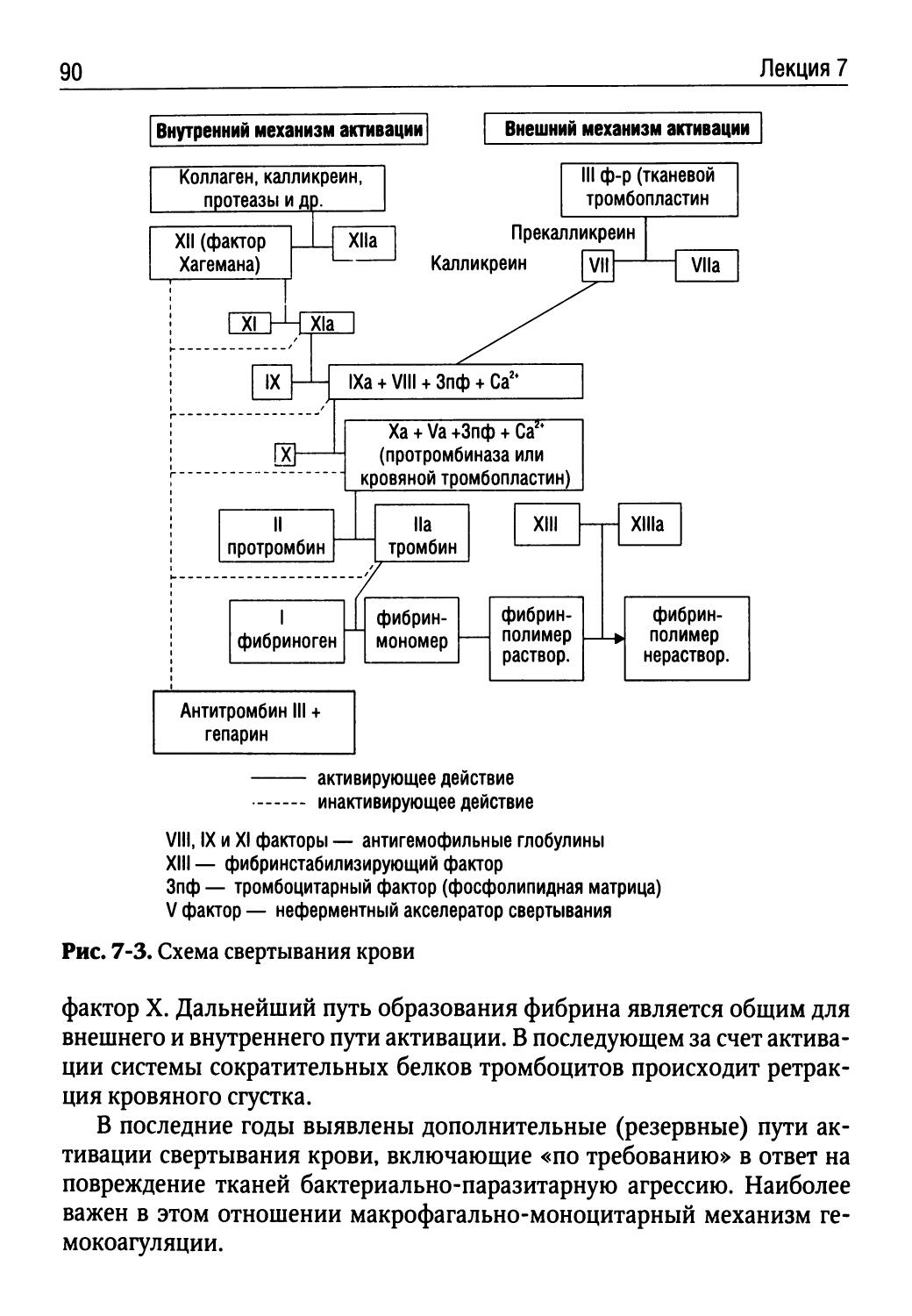
Лекция 7. Патофизиология системы гемостаза (Н.И. Бережнова).85
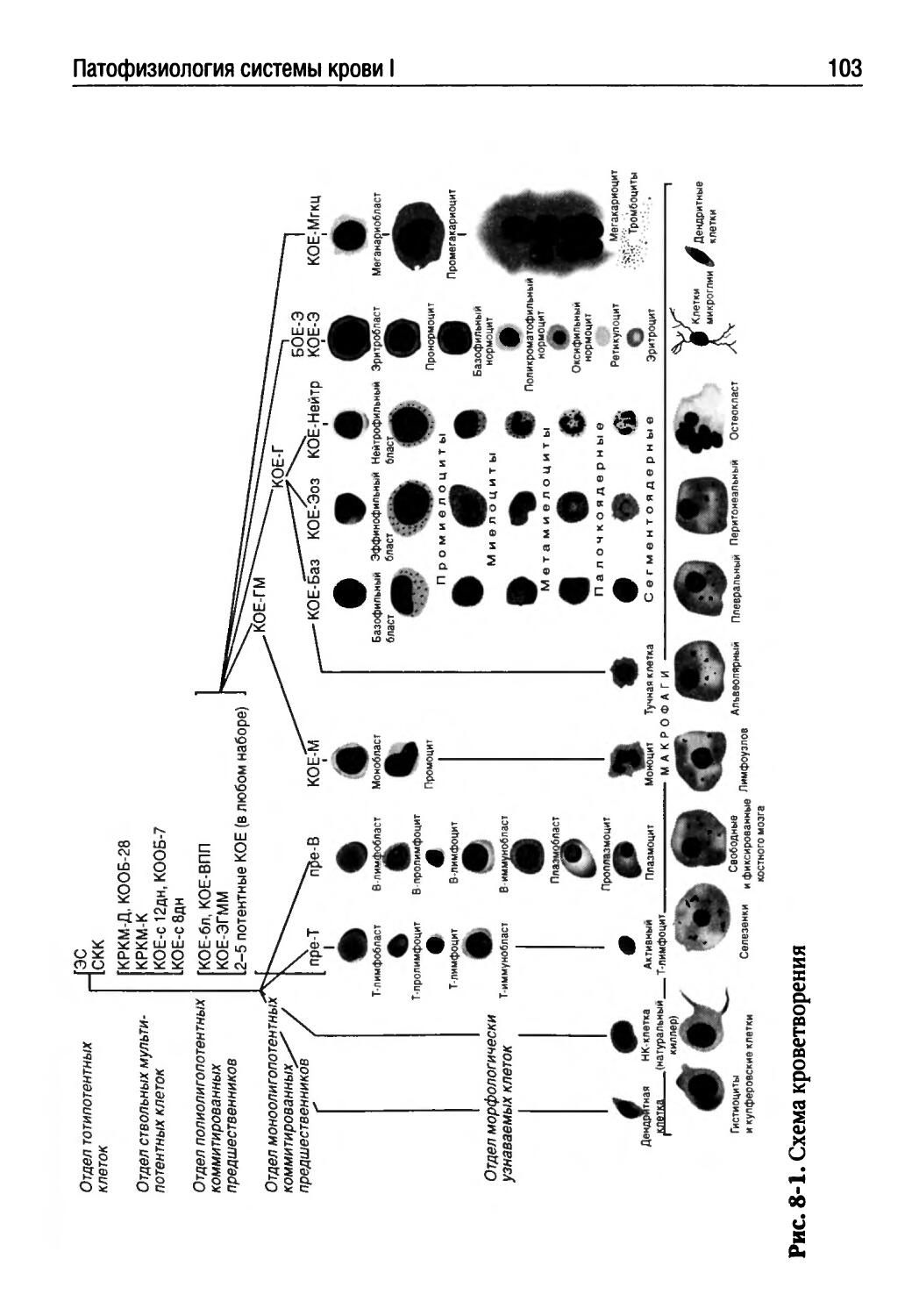
Лекция 8. Патофизиология системы крови I (Г.П. Щелкунова)...99
Лекция 9. Патофизиология системы крови II (Г.П. Щелкунова)..129
Лекция 10. Патофизиология белкового обмена (Г.В. Порядин)...145
Лекция 11. Патофизиология углеводного обмена (Г.В. Порядин).158
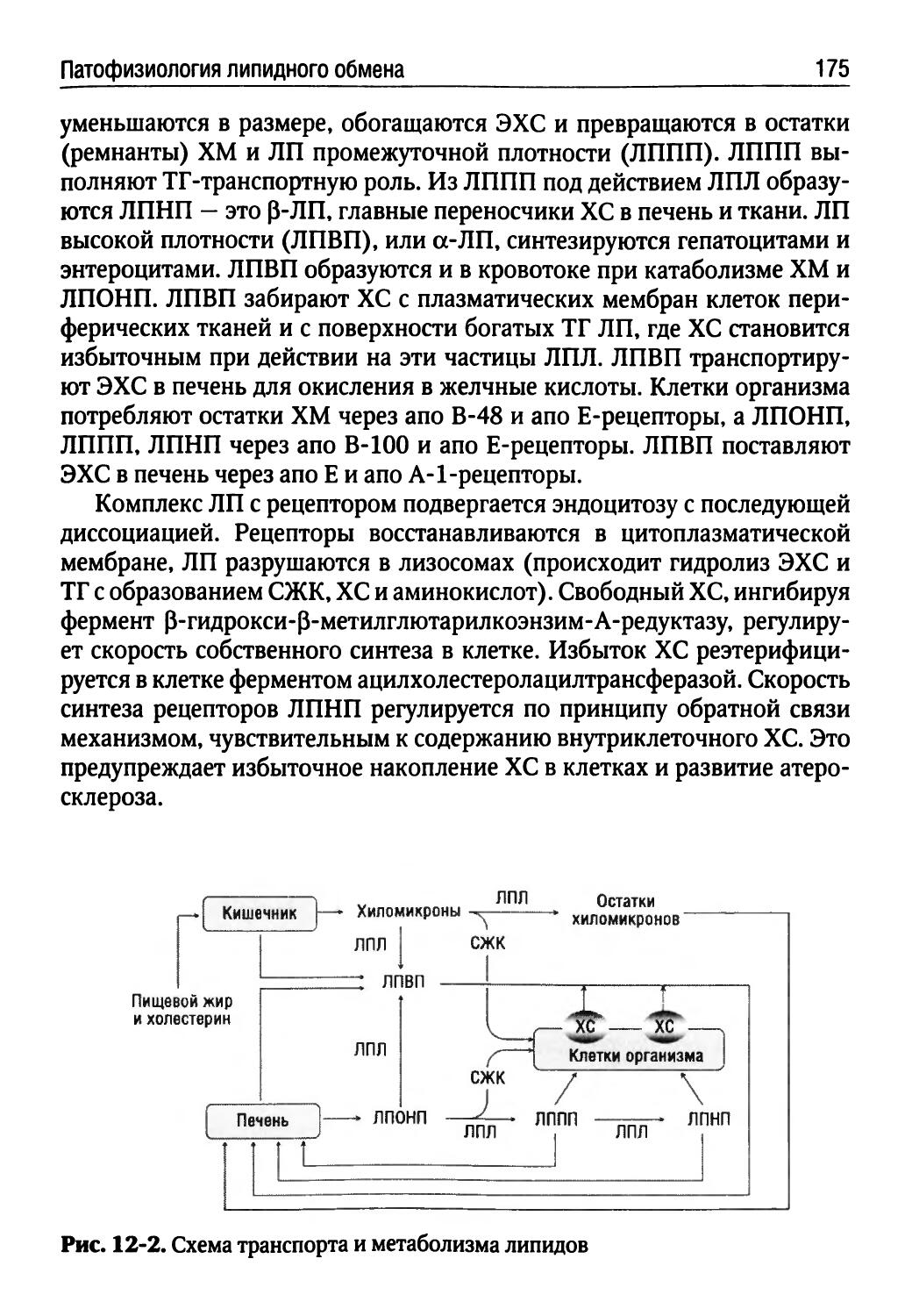
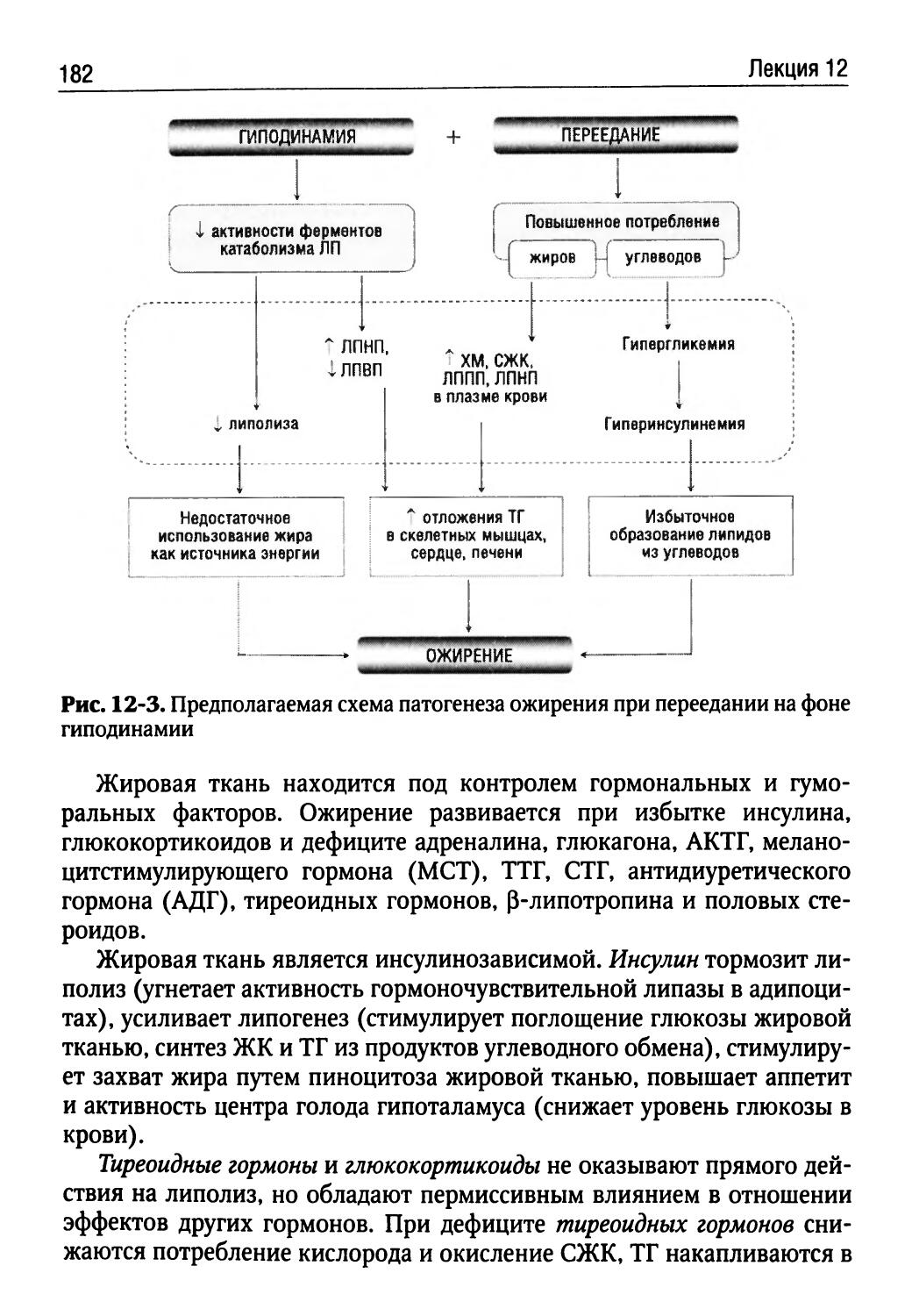
Лекция 12. Патофизиология липидного обмена (Л.Н. Осколок)...173
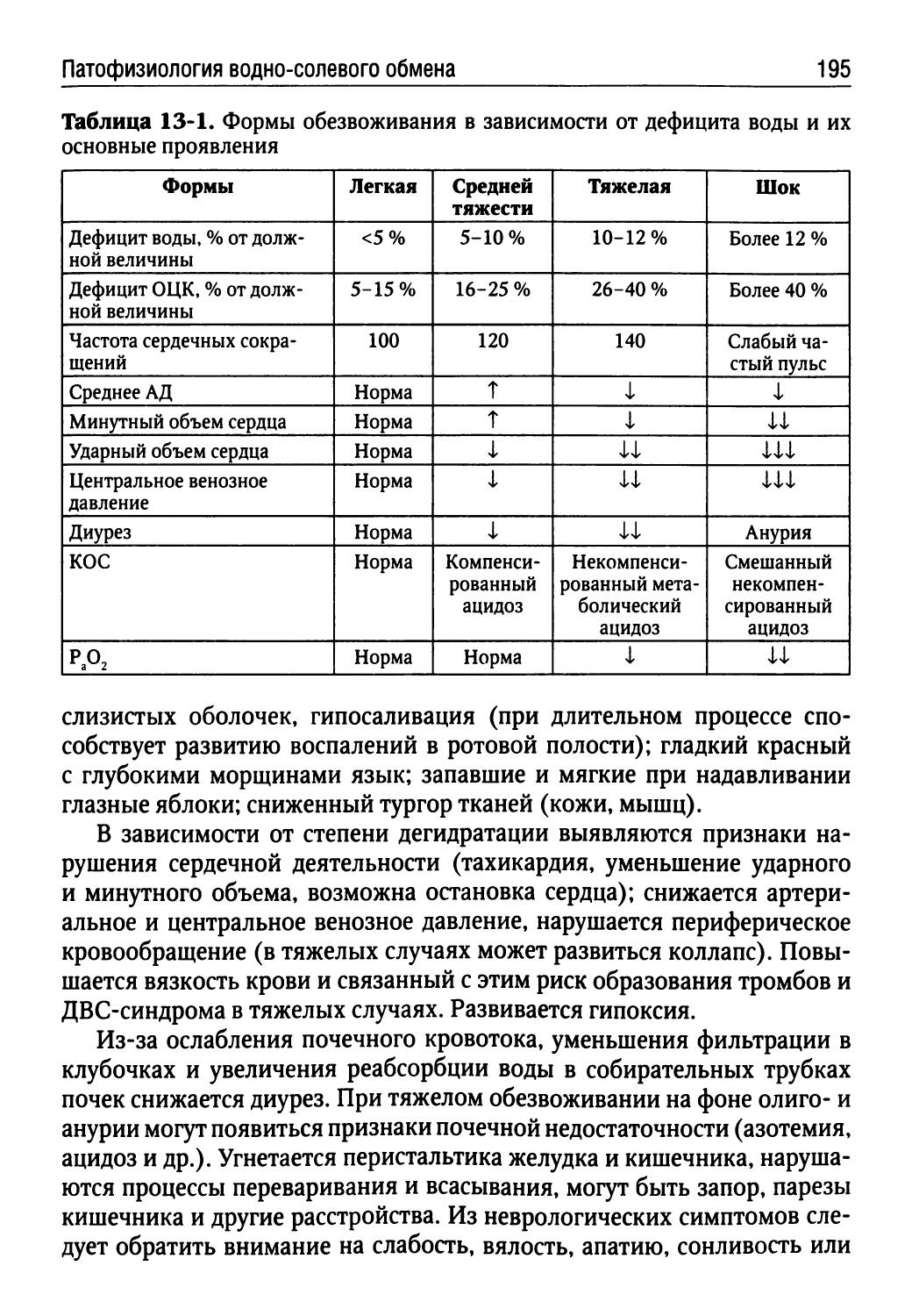
Лекция 13. Патофизиология водно-солевого обмена (Г.П. Щелкунова)......................................185
Лекция 14. Патофизиология кислотно-основного состояния (Г.П. Щелкунова)......................................205
Лекция 15. Патология опухолевого роста. Канцерогенез (Л.И. Зелигенко)......................................236
Лекция 16. Патофизиология нервной системы (Г.В. Порядин)........................................254
Лекция 17. Патофизиология нервных механизмов управления движениями (Т.Ю. Ругинская)................268
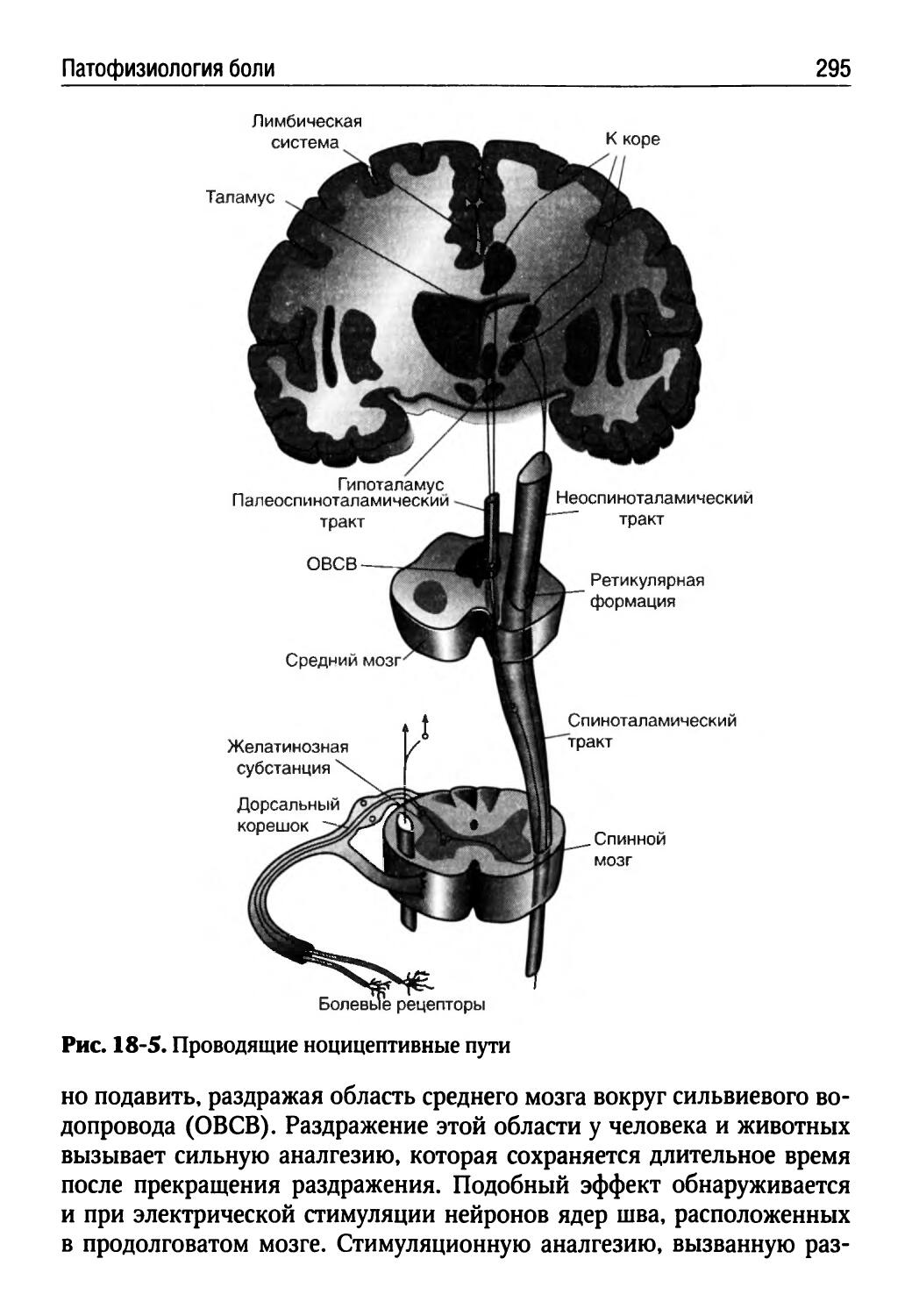
Лекция 18. Патофизиология боли (Т.Ю. Ругинская).........288
Лекция 19. Патофизиология эндокринной системы
(общая этиология и патогенез эндокринных нарушений) (Г.В. Порядин).......................................305
Лекция 20. Патофизиология эндокринной системы (эндокринопатии —
патологические процессы в самой железе) (Г.В. Порядин)...319
4
Оглавление
Лекция 21. Нарушение гормональной регуляции основных физиологических процессов организма (Г.В. Порядин)....334
Лекция 22. Стресс и общий адаптационный синдром. Их роль в патологии (Л.И. Зелитенко)..................348
Лекция 23. Патофизиология сердечно-сосудистой системы (Н.Л. Богуш)..........................................364
Лекция 24. Патофизиология сердечной недостаточности (Н.Л. Богуш)..........................................375
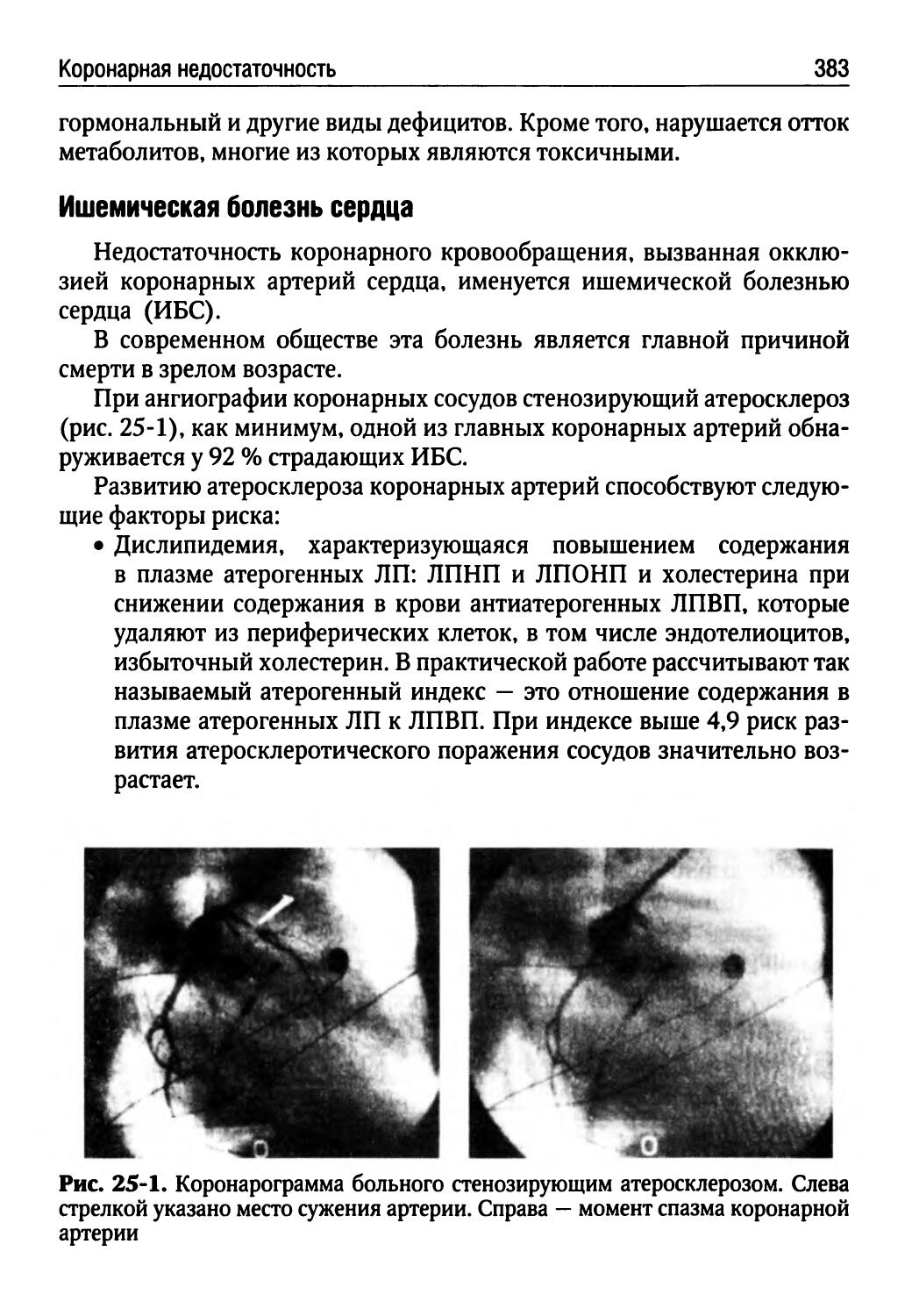
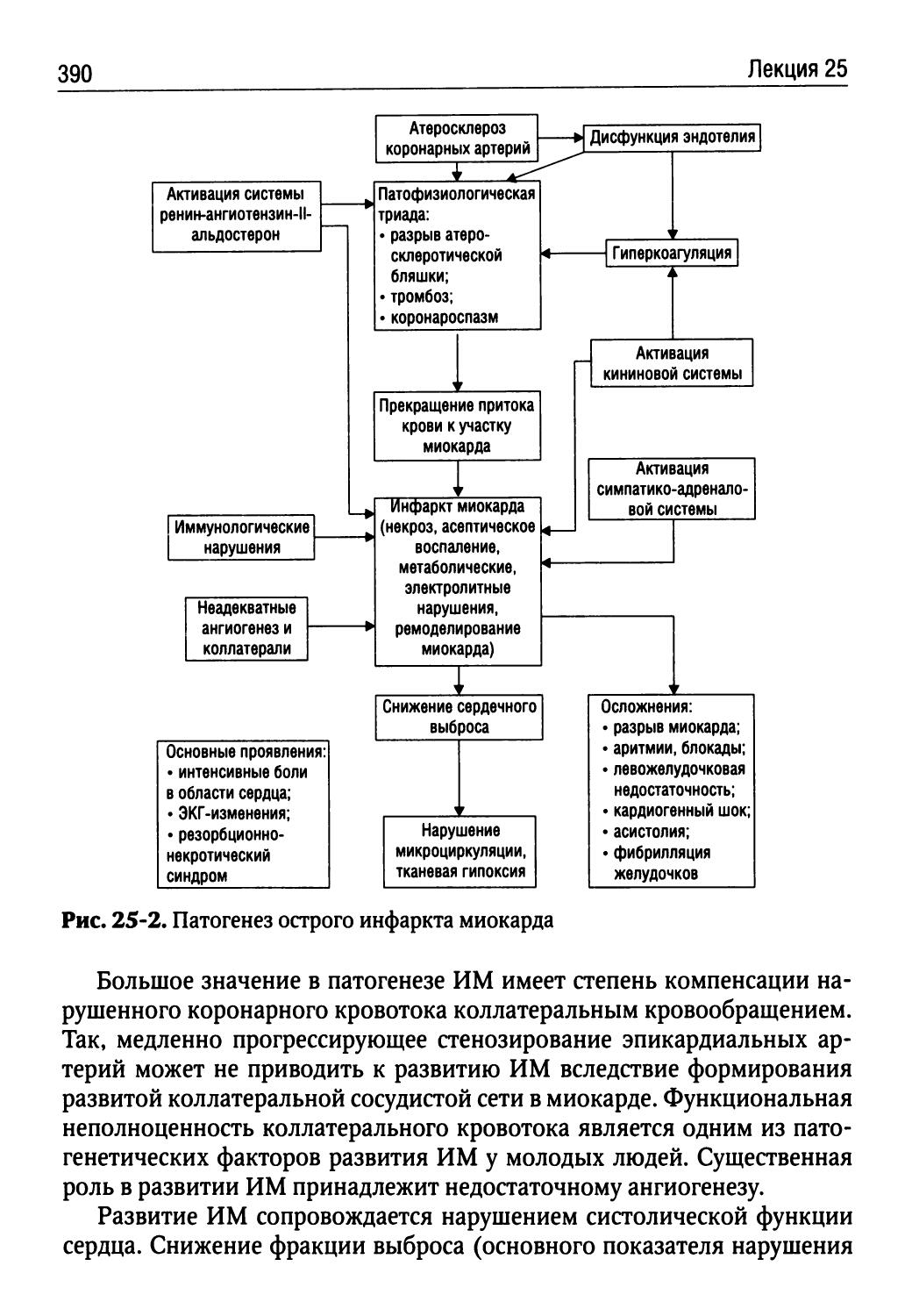
Лекция 25. Коронарная недостаточность (Н.И. Бережнова)...382
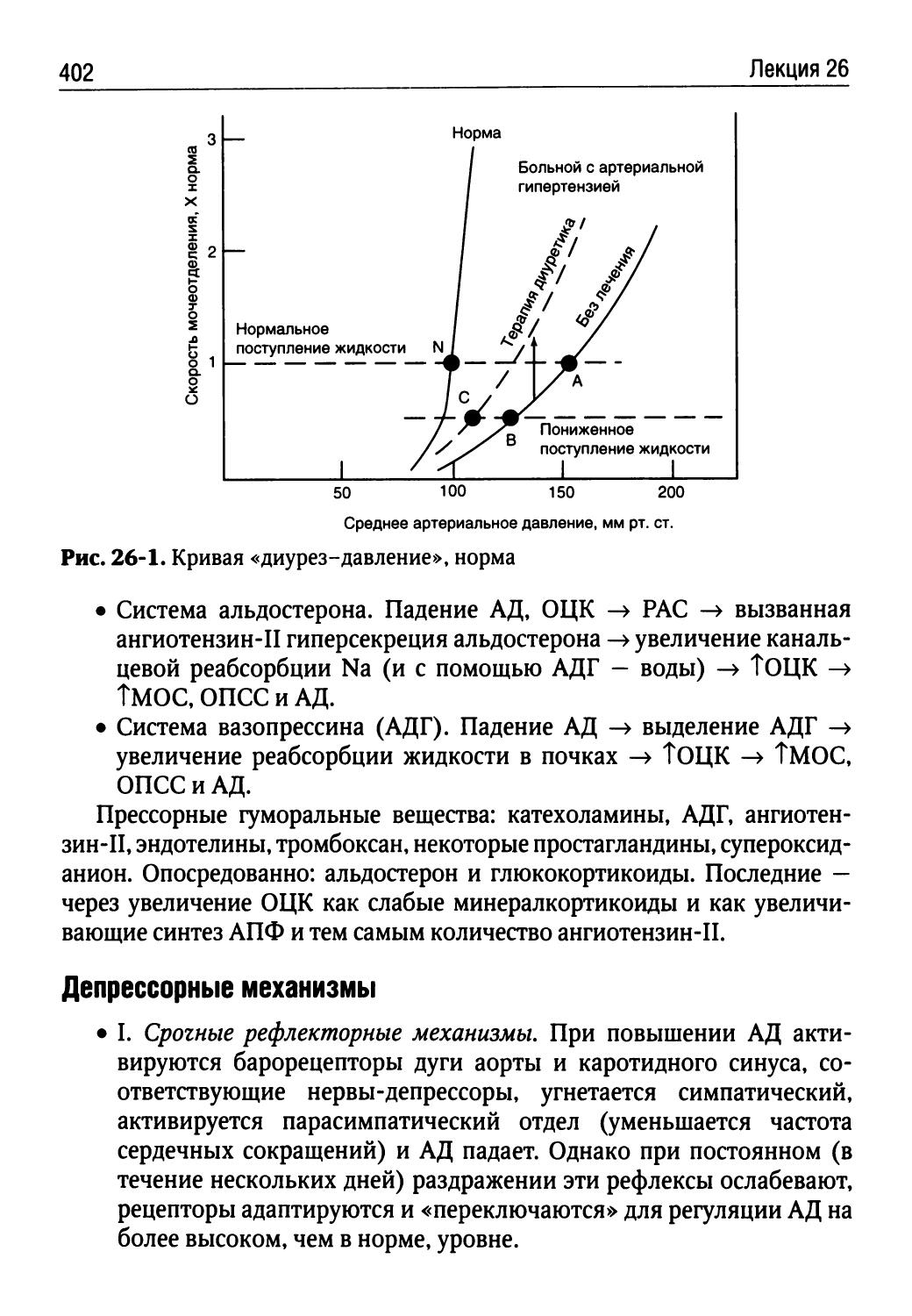
Лекция 26. Патофизиология артериальной гипертензии (НЛ. Богуш)...396
Лекция 27. Роль реактивности организма в патологии (Г.В. Порядин) ...415
Лекция 28. Аллергия. Понятие и общая характеристика.
Актуальные проблемы аллергии (Г.В. Порядин)...........431
Лекция 29. Аутоиммунные болезни (НЛ. Богуш)..............443
Лекция 30. Патофизиология иммунодефицитных состояний (Ж.М. Салмаси)........................................455
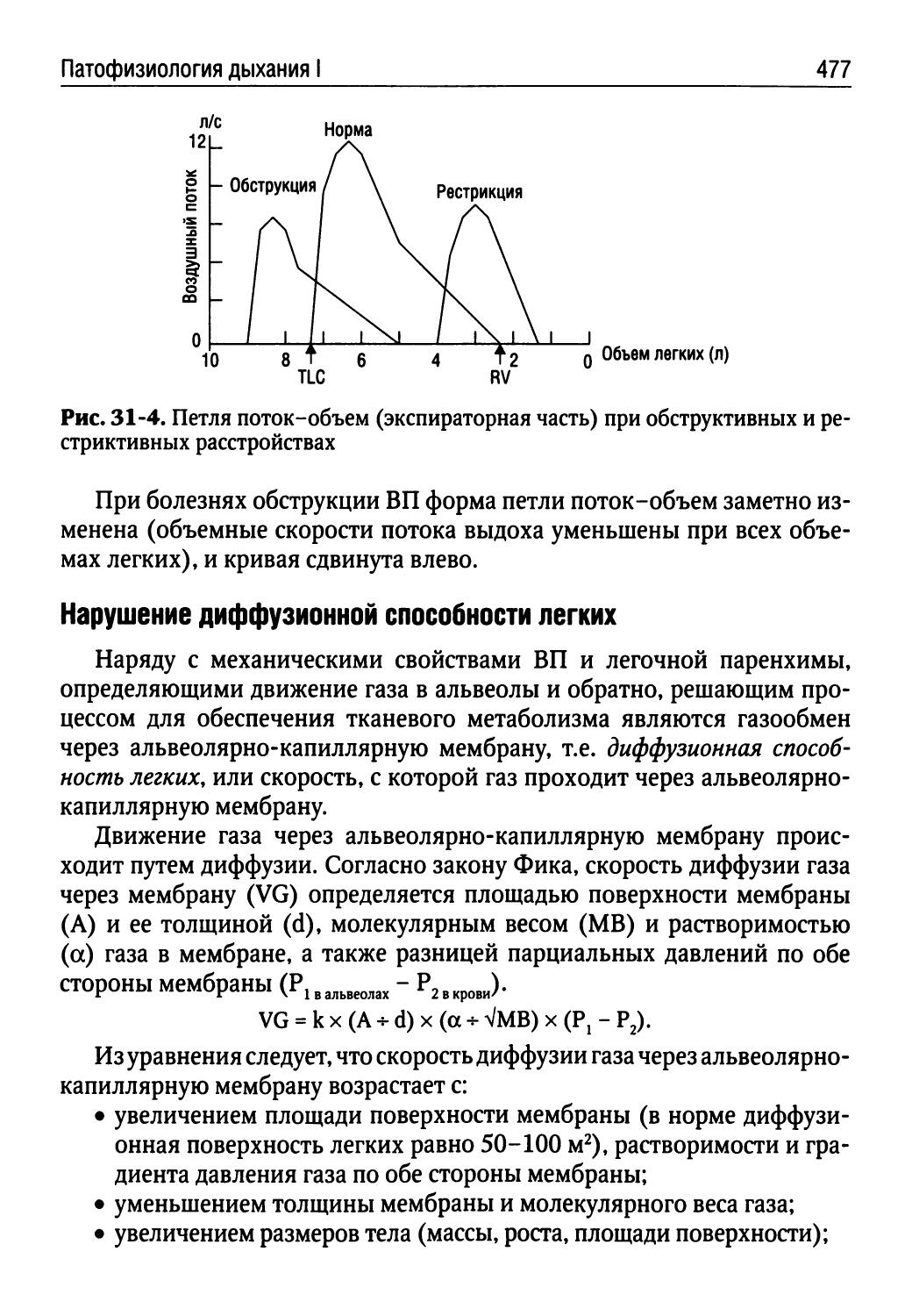
Лекция 31. Патофизиология дыхания I (Г.В. Порядин).......469
Лекция 32. Патофизиология дыхания II (Г.В. Порядин)......484
Лекция 33. Патофизиология кишечного пищеварения (Л.Ю. Семенова).......................................494
Лекция 34. Патофизиология язвенной болезни желудка и двенадцатиперстной кишки (Ж.М. Салмаси).............509
Лекция 35. Патофизиология печени (Н.И. Бережнова)........523
Лекция 36. Патофизиология почек I (Г.П. Щелкунова).......539
Лекция 37. Патофизиология почек II (Г.П. Щелкунова)......553
Лекция 38. Патофизиология шоковых состояний (Т.Ю. Путинская)......................................569
Предметный указатель.....................................584
ПРЕДИСЛОВИЕ
Значение фундаментальной подготовки в общем образовании и становлении врача невозможно переоценить. Это положение особенно усиливается в свете новой концепции подготовки специалистов-медиков, регламентированной последними государственными образовательными стандартами по медицинским специальностям и внедряемой в рамках нового учебного плана медицинскими вузами России. Патофизиология является одним из важных компонентов успешной и глубокой подготовки врача.
Вместе с тем динамичное развитие патофизиологии, «проникновение» патофизиологии в клинику значительно усложняет как преподавание этой дисциплины, так и важного процесса написания учебника для студентов медицинских вузов и его оперативного издания.
В связи с этим коллектив кафедры патофизиологии Российского национального исследовательского медицинского университета им. Н.И. Пирогова подготовил курс лекций по патофизиологии, который соответствует указанным целям и предназначен, в первую очередь, для самостоятельной работы студентов.
Вниманию читателя представлены лекции, читаемые на кафедре патофизиологии РНИМУ им. Н.И. Пирогова студентам III курса лечебного и педиатрического факультетов, И, III, IV курсов стоматологического факультета. В материалах лекций представлены как классические данные, так и новейшие результаты фундаментальных и прикладных исследований в области патофизиологии.
Цикл лекций, публикуемый кафедрой, содержит современный взгляд на изложенные проблемы. Он призван облегчить усвоение патофизиологии и помочь студенту разобраться в сложных вопросах, знание которых необходимо любому практикующему врачу.
Учитывая, что патофизиология не только предваряет клинический этап подготовки врача, но и постоянно востребована студентами при изучении клинических дисциплин, в материалах лекций приводятся сведения не только о типовых формах патологии систем и органов, но и о патофизиологии синдромов, наиболее часто встречающихся в клинической практике.
КОЛЛЕКТИВ АВТОРОВ
СОКРАЩЕНИЯ И УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
• — торговое наименование препарата
АГ — антиген
АД — артериальное давление
АДГ — антидиуретический гормон
АИБ — аутоиммунные болезни
АКТГ — адренокортикотропный гормон
АПК — антигенпредставляющая клетка
АПФ — ангиотензинпревращающий фермент
АТ — антитело
БА — бронхиальная астма
БАБ — биологически активные вещества
ВИП — вазоинтестинальный пептид
ВОЗ — Всемирная организация здравоохранения
ГАМК — у-аминомасляная кислота
ГД — гидростатическое давление крови
Г-КСФ — гранулоцито-колониестимулирующий фактор
ГМ-КСФ — гранулоцито-моноцито-колониестимулирующий фактор
ГН — гломерулонефрит
ГПУВ — генератор патологически усиленного возбуждения
ГЭБ — гематоэнцефалический барьер
ДВС-синдром —синдром диссеминированного внутрисосудистого свертывания крови
ДМП — давление в мочевыводящих путях
ДН — дыхательная недостаточность
ДНК — дезоксирибонуклеиновая кислота
ДО — дыхательный объем
ДЦ — дыхательный центр
ЖЕЛ — жизненная емкость легких
ЖК — желчные кислоты
ЖКБ — желчнокаменная болезнь
ЖКТ — желудочно-кишечный тракт
ИБС — ишемическая болезнь сердца
ИДС — иммунодефицитное состояние
ИЛ — интерлейкин
КБ — конъюгированный (связанный или прямой) билирубин
ККС — калликреин-кининовая система
КОС — кислотно-основное состояние
Сокращения и условные обозначения 7
КСФ — колониестимулирующий фактор
ЛП — липопротеин
ЛПВП — липопротеины высокой плотности
ЛПЛ — липопротеинлипаза
ЛПНП — липопротеины низкой плотности
ЛПОНП — липопротеины очень низкой плотности
ЛПС — липополисахариды
ЛППП — липопротеины промежуточной плотности
МАВ — минутная альвеолярная вентиляция
МВЛ — максимальная вентиляция легких
MX — митохондрии
МОД — минутный объем дыхания
МОС — минутный объем сердца
МОС50%вд — максимальная объемная скорость вдоха
НКБ — неконъюгированный (несвязанный, прямой) билирубин
НЭЖК — неэстерифицированные жирные кислоты
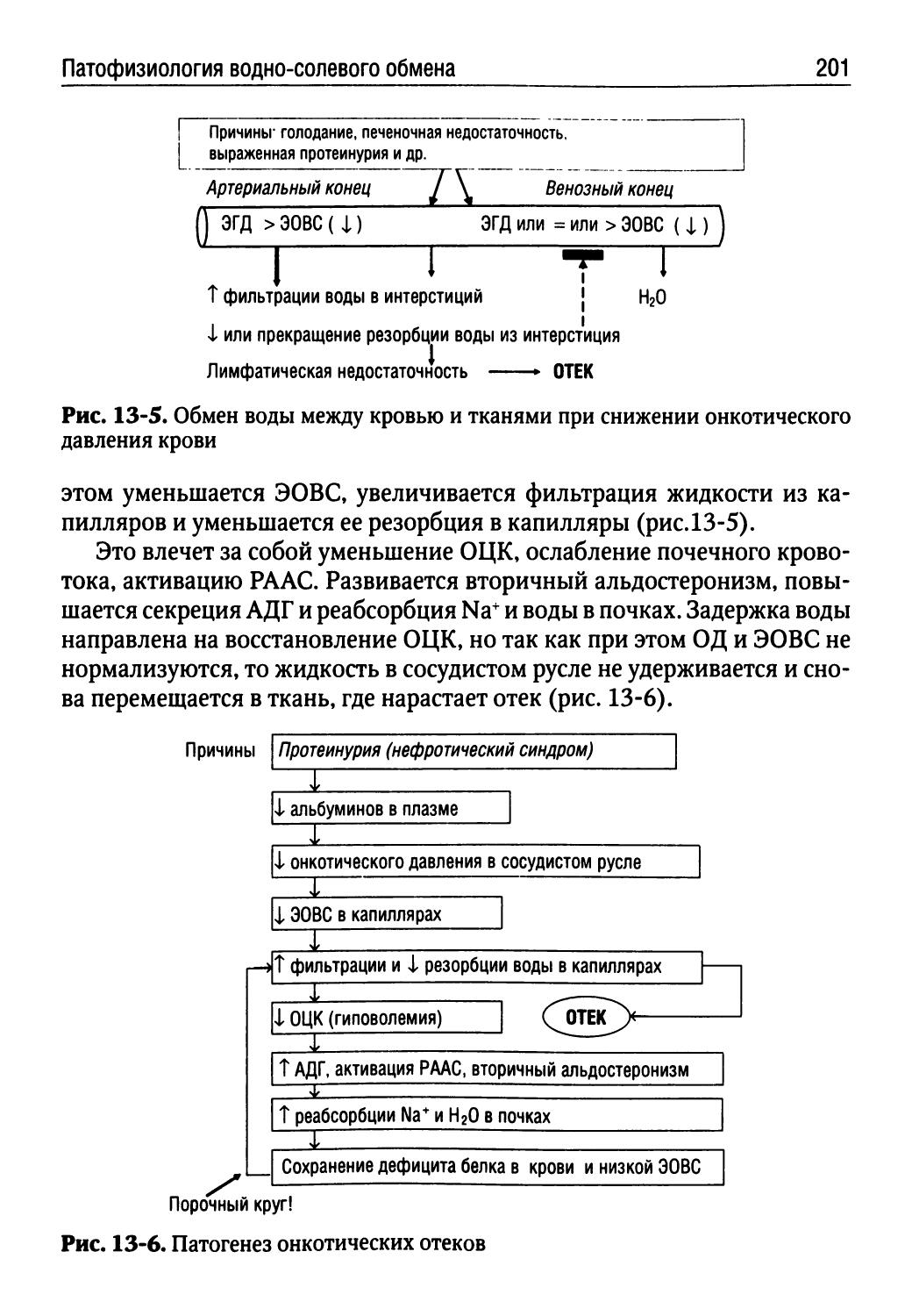
ОД — онкотическое давление крови
ОЕЛ — общая емкость легких
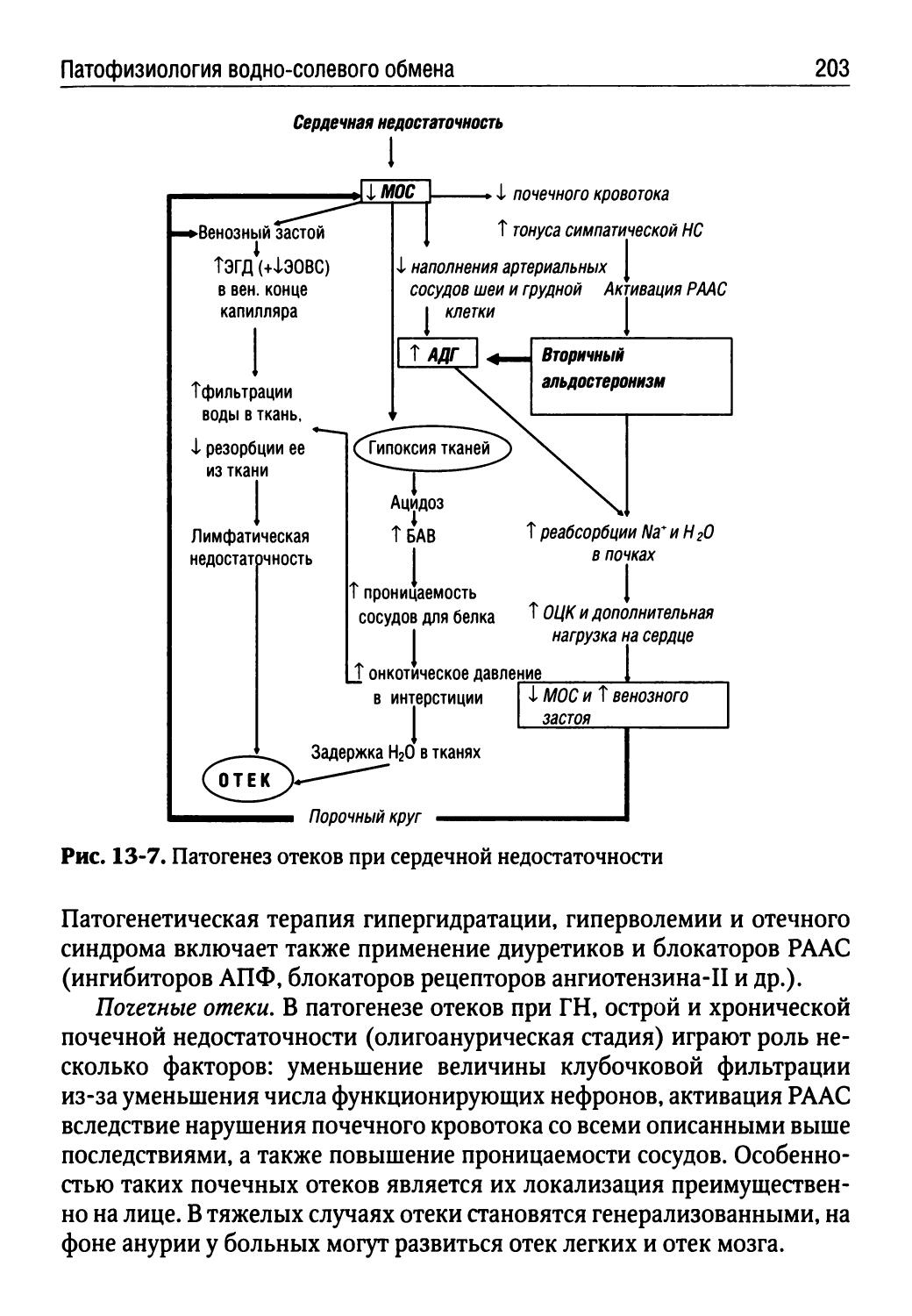
ОЛЛ — острый лимфобластный лейкоз 00 — остаточный объем
ООФ — ответ острой фазы
ОПН — острая почечная недостаточность
ОПСС — общее периферическое сопротивление сосудистого русла
ОФВ] — объем форсированного выдоха за 1 секунду
ОЦК — объем циркулирующей крови
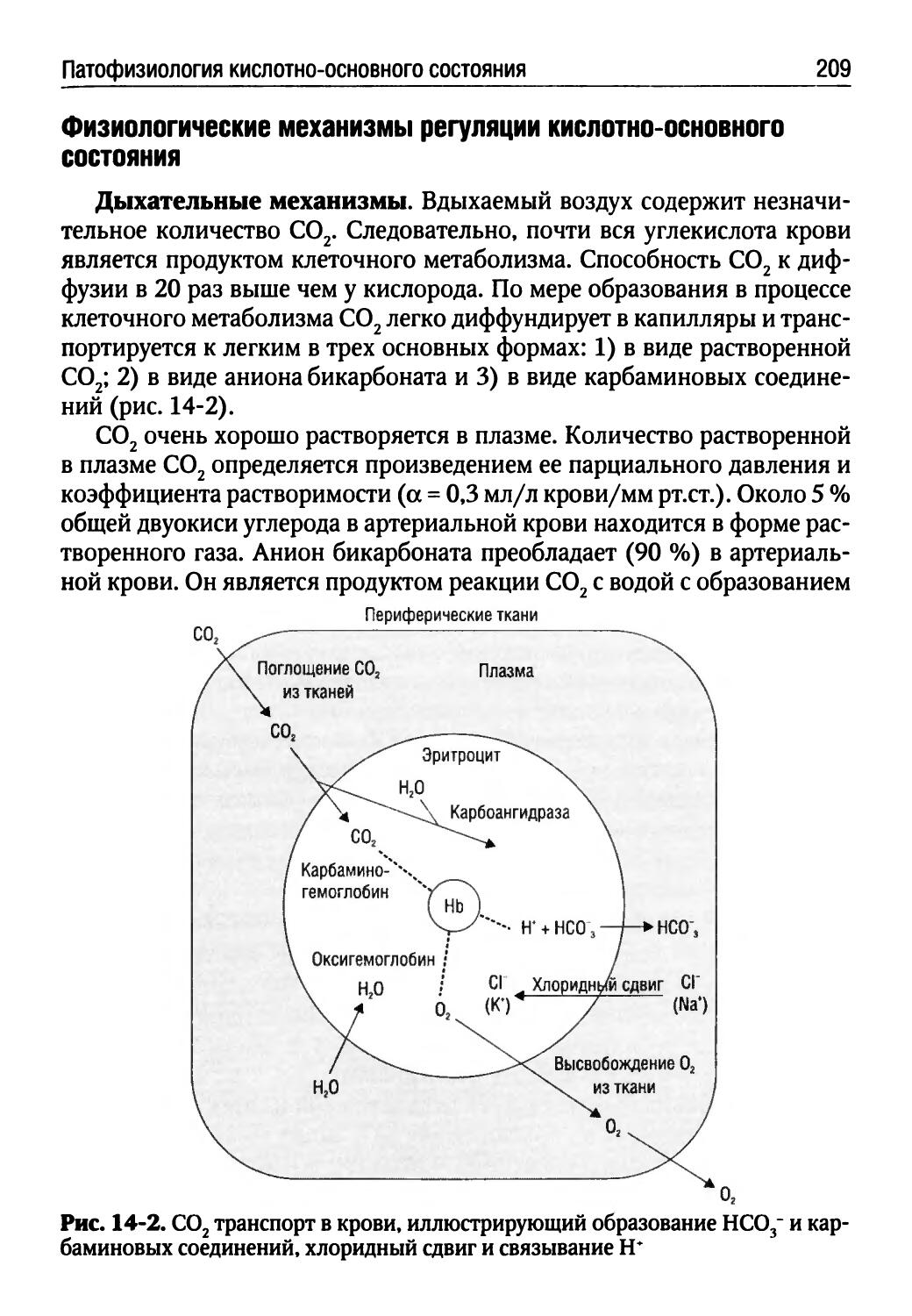
ПГ — простагландин
ПН — пиелонефрит
ПНЖК — полиненасыщенные жирные кислоты
ПНУФ — предсердный натрийуретический фактор
ПНФ — предсердный натрийуретический пептид
ПОЛ — перекисное окисление липидов
ПОС — пиковый экспираторный поток
ПТВ — психотропное вещество
РААС — ренин-ангиотензин-альдостероновая система
РДСВ — респираторный дистресс-синдром взрослых
СБ — стеркобилиноген
СБА — сидеробластная анемия
СЖК — свободные жирные кислоты
СИ — сердечный индекс
8
Сокращения и условные обозначения
СН — сердечная недостаточность
СР — сердечный резерв
СТГ — соматотропный гормон
СТТ — спиноталамический тракт
ТГ — триглицериды
ТТГ — тиреотропный гормон
УБ — уробилиноген
УОС — ударный объем сердца
ФАТ — фактор активации тромбоцитов
ФЖЕЛ — форсированная жизненная емкость легких
ФЛ — фосфолипиды
ФНО — фактор некроза опухолей
ФОБ — функциональная остаточная емкость
ФЭП 25-75 % — форсированный экспираторный поток между 25 и 75 % форсированной жизненной емкости легких
ХЛЛ — хронический лимфолейкоз
ХМ — хиломикрон
ХМЛ — хронический миелолейкоз
ХПН — хроническая почечная недостаточность
ХС — холестерин
ЦИК — циркулирующие иммунные комплексы
ЦНС — центральная нервная система
ЦП — цветовой показатель
ЦПМ — цитоплазматическая мембрана
ЦТЛ — цитотоксический лимфоцит
ЧД — частота дыхания
ЭАГ — эссенциальная артериальная гипертония
ЭГД — эффективное гидростатическое давление
ЭЗДП — экспираторное закрытие дыхательных путей
ЭОВС — эффективная онкотическая всасывающая сила
ЭПО — почечные эритропоэтины
ЭР — эндоплазматический ретикулум
ЭФД — эффективное фильтрационное давление
ЭХС — эфиры холестерина
ЭЦМ — экстрацеллюлярный матрикс
ЮГА — юкстагломерулярный аппарат
ЯБ — язвенная болезнь желудка и двенадцатиперстной кишки
Ht — гематокрит
НЬ — гемоглобин
Лекция 1
ВВЕДЕНИЕ В ПАТОФИЗИОЛОГИЮ
Г.В. Порядин
Предмет и задачи патофизиологии
В общих чертах патофизиологию можно определить как учение о болезненных явлениях или науку о жизнедеятельности больного организма. Патофизиология выясняет причины заболевания, условия, в которых оно возникает, механизмы развития и исходы болезни.
Как при решении научных задач, так и в клинической практике часто возникает необходимость решения трех взаимосвязанных вопросов.
• Почему возникает та или иная болезнь, развивается тот или иной патологический процесс, т.е. где причина и каковы условия, порождающие данное заболевание? Это первый вопрос, который возникает при столкновении с болезнью. Найти причину — означает найти и правильный способ профилактики и лечения болезни. Древние врачи говорили: «Sublata causa tollitur morbus — устраняя причину, устраняешь болезнь». Этот вопрос труден, он составляет суть важного раздела патофизиологии: общей этиологии — учения о причинах и условиях возникновения, а также развития болезней.
• Как действует фактор, ставший причиной заболевания, как он функционирует в определенных условиях, т.е. каков механизм возникновения и развития данной болезни, данного патологического процесса?
Если этиология изучает причину возникновения болезни, то раздел патофизиологии, отвечающий на вопрос как развивается болезнь, носит название общий патогенез.
Такие вопросы патогенеза, как соотношение функций регулирующих и исполнительных систем, смена ведущих звеньев болезни, кольцевые связи патологического характера (порочные круги) и ряд других, занимают центральное место в патофизиологии.
10
Лекция 1
• Наконец, что получается в результате действия причинного фактора, каков возможный результат (исход) болезни, т.е. чем завершится болезнь (патологический процесс): выздоровлением, затягиванием (переход в хроническую форму) болезни, гибелью.
Ответ на все эти вопросы — путь к пониманию сути болезни, а также к подбору адекватной терапии.
Патофизиология как наука официально существует около 250 лет. Свое развитие она получила со времени опубликования выдающимся итальянским ученым, анатомом и врачом Морганьи Джованни Батиста классического 6-томного руководства «De sedibus et causa morborum» — «О местонахождении и причинах болезни» (1761).
Последующие 100 лет патофизиология (ее первое название «патология») развивалась во всем мире по преимуществу как морфологическая наука.
Вместе с тем расширялось внедрение экспериментального метода (подхода) в изучение патологических процессов и заболеваний. С1835 г. в российской высшей школе начинается формирование двух самостоятельных направлений фундаментальной подготовки студентов-медиков — морфологического и экспериментального.
Видный русский ученый А.И. Полунин выделил из общей патологии экспериментальный раздел, организовав на медицинском факультете Московского императорского университета самостоятельную кафедру экспериментальной патологии (патофизиологии), которая затем была создана и в других российских университетах (Казанском, Томском, Петербургской медико-хирургической академии, Московских высших женских курсах).
Этот акт символизировал чрезвычайно важный период для отечественной медицинской науки — переход от морфологического пути к позициям экспериментальной (функциональной) патологии.
Название «общая и экспериментальная патология» за этой наукой и учебной дисциплиной сохранялось до 1924-1925 гг.
В 1925 г. по инициативе академика А.А. Богомольца данное направление медицины стало называться «патологическая физиология». Следует сказать, что исторически впервые термин «патологическая физиология» был предложен профессором Эрфуртского университета А.Ф. Геккером, издавшем в 1791 г. учебник «Основы патологической физиологии» (через 30 лет после выхода 6-томного руководства Морганьи Джованни Батиста).
Почти через 30 лет (в 1819 г.) другой ученый Л. Галлиот повторил этот термин в учебнике «Pathologic generale etphyziologie pathologique».
Введение в патофизиологию
11
Структурно-системная характеристика патофизиологии как фундаментальной науки и учебной дисциплины
В соответствии с задачами, которые призвана решать патофизиология как фундаментальная и учебная наука, она, будучи важнейшим разделом биологии и медицины, имеет три составные части.
• Первая часть: общее угение о болезни — изучение наиболее общих закономерностей возникновения, течения и исхода патологических процессов и болезни в целом. Два вопроса: почему возникла болезнь и как она развивается, а иными словами, этиология и патогенез, — главные вопросы патофизиологии, рассматриваются как раз в этой части предмета.
• Вторая часть — типовые (типигескиё) патологигеские процессы (воспаление, повреждение клетки, лихорадка, гипоксия, опухолевый рост, голодание и др.), встречающиеся в виде постоянных сочетаний или комбинаций.
Например, одни клинические изменения наблюдаются при холецистите, другие — при пневмонии. Иная клиническая картина характеризует артрит или гепатит и т.д.
Однако все эти заболевания объединяет общий процесс — воспаление, для которого характерны свои закономерности как в развитии и течении, так и в прогрессировании и исходе. Знание этих закономерностей необходимо врачу любой специальности для выбора правильной тактики ведения больного.
• Третья составляющая патофизиологии — частная патофизиология или патофизиология органов и систем — включает общий анализ нарушений отдельных, но взаимосвязанных систем организма: кровообращения, дыхания, эндокринной, нервной и т.д.
И хотя эта часть условно называется частной патофизиологией, в ней главным образом рассматриваются общепатологические аспекты.
Например, при рассмотрении патологии почек следует выяснить общие закономерности нарушения их функции, компенсаторные возможности системы выделения, которые окажутся необходимыми в дальнейшей клинической (нефрология, урология) практике, в ходе анализа отдельных форм патологии, связанной с функцией системы выделения.
Этот последний (третий) важнейший раздел патофизиологии по существу является клинигеской патофизиологией.
Клиническая патофизиология — вершина сравнительной патологии, так как описывает именно патологию человека. Патофизиологи
12
Лекция 1
ческий подход в решении различных задач клинической медицины все более и более внедряется в самые различные области практической медицины.
Сегодня трудно представить, например, работу квалифицированного кардиологического отделения без фазового анализа сердечной деятельности больных, механо-фоно-вектор-кардиографии.
В практике кардиологического отделения постоянно назначают определенные лекарства, производят ритмовождение или кардиостимуляцию, дефибриляцию. Применение этого невозможно без знания и учета патофизиологии кровообращения.
Патофизиологические критерии лежат в основе современных определений и классификаций недостаточности кровообращения и дыхания. Правильной диагностике и лечению детских болезней способствуют сведения о механизмах наследования той или иной патологии.
Место патофизиологии в высшем медицинском образовании
В 1835 г. уставом российских университетов было введено преподавание этой дисциплины. С этого момента патофизиология как фундаментальная дисциплина в системе высшего медицинского образования занимает особое место. Находясь на стыке теоретических и клинических дисциплин, она способствует развитию врагебного мышления.
С одной стороны, патофизиология опирается на те дисциплины, которые предшествуют ей в программе обучения практического врача. Особенно много общего у патофизиологии (в плане преемственности) с нормальной физиологией, биологией, биохимией, микробиологией, иммунологией. Но в то же время патофизиология опирается так же на такие морфологические дисциплины, как анатомия и гистология.
Конечная цель патофизиологии в подготовке врача — познание процессов, общих для всех болезней или для их групп, получение фундаментальных сведений о сущности болезни и законах ее развития; формирование и развитие в конечном счете у студента — будущего врача клинического мышления.
Эта задача тесно связывает патофизиологию с клиническими дисциплинами, поскольку конечная цель едина.
Однако ближайшие задачи, методы и объекты различны. Объект изучения клинических наук — конкретный больной человек с конкретными проявлениями болезни.
Например, онкология изучает конкретные формы проявления доброкачественных и злокачественных опухолей. Чтобы поставить диаг
Введение в патофизиологию
13
ноз «рак желудка», «саркома бедра», «миома матки», врачу необходимо знать, чем они отличаются друг от друга, их особенности.
В то же время углубленное понимание этих болезней требует раскрытия того общего, что их объединяет, и ответа на следующие вопросы:
• как нормальная клетка превращается в опухолевую:
• как меняется ее наследственность;
• откуда она черпает энергию для своего безудержного роста:
• какие звенья ее метаболизма наиболее чувствительны к внешним воздействиям и, прежде всего, действию лекарственных средств.
Изучить общие неспецифические механизмы развития болезней — задача патофизиологии.
Основные понятия общей нозологии
Итак, патофизиология — это наука, изучающая жизнедеятельность больного организма.
Необходимо сразу оговориться, что патофизиология, как и медицина в целом, имеет дело с двумя обобщенными категориями: болезнь и здоровье. Обе эти категории являются особыми формами жизни.
Коренной особенностью существования живых существ является приспособление (адаптация) организма к постоянно изменяющимся условиям внешней среды.
При этом реакции организма направлены на сохранение в нем постоянства внутренней среды или гомеостаза.
Чем же обеспечивается включение адаптационных (приспособительных) механизмов, обеспечивающих гомеостаз? Только одним — разнообразными факторами внешней среды, по силе воздействия они могут быть трех видов.
• Постоянные, обычные воздействия на организм человека, в подавляющем своем большинстве оптимальные, вызывающие многочисленные отклонения в организме, которые вполне корригируются соответствующими процессами ауторегуляции.
• Сверхоптимальные воздействия, которые в современной жизни человека встречаются все чаще, — немедленно включают более высокие уровни адаптивной регуляции.
• Чрезвыгайные (экстремальные) воздействия, доля которых тоже растет, — имеет место мобилизация нейроэндокринных механизмов, которые выражаются в стресс-реакции.
14
Лекция 1
Все названные реакции — реакции здорового организма. Но, поскольку они имеют особое значение для обеспечения нормальной жизнедеятельности и при возникновении патологических процессов, знание их, учет и правильная оценка необходимы для выбора тактики ведения больного.
Поэтому наряду с больным организмом патофизиология изучает и такие понятия, как стресс, предболезнь, резистентность, конституция, относящиеся к здоровым индивидам.
Понятие «здоровье» — довольно сложное понятие. Этот вопрос настолько сложен, что специально обсуждался в ВОЗ. После оживленной дискуссии эксперты ВОЗ приняли следующее определение.
Под здоровьем следует понимать полное физигеское, психигеское и социальное благополугие геловека, допускающее наиболее полноценное его угасшие в общественной трудовой деятельности, а не только отсутствие у него болезни или физигеских дефектов.
Другое понятие, с которым медицина имеет дело, — «норма». «Норма» — термин, близкий к понятию «здоровье», но не исчерпывающий его вполне (А.Д. Адо). Понятия «здоровье» и «норма» не совпадают.
Норма — присущее большинству популяций, наиболее типичное значение того или иного параметра: средний рост, вес, артериальное давление. При этом может иметь место нарушение (расстройство) на молекулярном, клеточном и даже органном уровнях, но до определенного времени не проявляющееся, и человек будет практически здоров (компенсированный порок сердца, анемия, связанная с недостаточностью фермента глюкозо-6-фосфатдегидрогеназа вне дополнительных нагрузок).
С другой стороны, можно быть здоровым, но отличаться от общепринятых эталонов нормы массы, роста, интеллекта.
При любом патологическом процессе (или болезни) в организме неизбежно возникают и чисто патологигеские явления, т.е. нарушения нормальной структуры и функции, и компенсаторные процессы, представляющие проявление деятельности нормальных ре1уляторных механизмов.
Эти два вида явлений находятся обычно в сложном взаимодействии. Врачу необходимо разобраться, какие проявления болезни представляют нарушения функций, а какие из них — компенсаторные процессы, так как лечебные мероприятия должны быть подобраны таким образом, чтобы они устраняли патологические реакции и способствовали компенсации нарушенных функций. Сказанное можно продемонстрировать на примере острой кровопотери (рис. 1-1).
Введение в патофизиологию
15
Кровотечение
Уменьшение массы крови (гипоксия) I
Снижение возбудимости дыхательного центра и синокаротидного узла
I I
Патологические явления Компенсаторные процессы
Нарушение регуляции функции сердечно-сосудистой системы (снижение АД, ослабление сердечных сокращений) Возбуждение дыхательного центра (тахипноэ)
Нарушение регуляции дыхания (дыхательная недостаточность, гипоксия) Возбуждение сердечнососудистого центра (спазм сосудов, повышение АД)
Нарушение обмена веществ (углубление гипоксии) Спазм сосудов (выброс крови из депо)
Рис. 1-1. Патогенез кровопотери
Наголо нарушения оптимального взаимодействия организма и внешней среды получило название предболезнь или предпо-вреждение (например, преморбидное состояние при инфекционном заболевании).
Болезнь — это жизнь поврежденного организма при участии процессов компенсации нарушенных функций.
Болезнь — качественно новое состояние организма.
Тем не менее при определении болезни мы должны учитывать, что человек — существо социальное. Поэтому мы добавляем, болезнь снижает трудоспособность человека.
Поскольку речь идет о популяции людей, которые, в отличие от популяции животных, в своем поведении подгоняются социальным законам, необходимо всегда иметь в виду, что в формировании и болезни, и здоровья не только внешние (средовые) причинные факторы, но и условия, их формирующие, социально детерминированы.
Это обстоятельство выдвигает чрезвычайно важную проблему в учении о болезни — соотношение биологического и социального.
Безусловно, особенности организма человека обусловливают своеобразие его заболеваний. Например, существенную роль в формировании заболеваний человека играют такие биологические свойства человека, как факторы антропогенеза, популяционной генетики (инбридинг — степень родства брачных партнеров; смешение, дрейф генов — слу
16
Лекция 1
чайные колебания концентраций отдельных аллелей; неоднородность генетических групп), особенности конституции, в том числе иммунологической и т.д.
Однако эти особенности, в свою очередь, определяются социальными условиями жизни.
Социальные факторы опосредуют начало болезней питания людей (голодание, недоедание, авитаминозы, ожирение); имеют большое значение в возникновении и распространении эпидемий.
Социальные факторы создают у людей особые, свойственные только человеку болезни, почти не встречающиеся в природе у животных (инфаркт миокарда, гипертоническая болезнь, язвенная болезнь желудка и двенадцатиперстной кишки, бронхиальная астма, психические болезни и ряд других).
Научно-технический прогресс сопровождается появлением многих новых, ранее неизвестных, болезнетворных факторов, таких как ионизирующее излучение, токсичные, канцерогенные и аллергенные продукты промышленного производства.
Процессы профилактики, лечения болезней и ухода за больными (деонтология) у людей полностью социально опосредованы.
Патологическая реакция, патологический процесс (синдром). Патологическое состояние
Патологическая реакция — кратковременная необычная реакция организма на какое-либо воздействие (патофизиологические рефлексы: симптом Бабинского, фагоцитоз при воспалении, централизация кровообращения при шоке).
Патологический процесс (синдром) — сочетание патологических и защитно-приспособительных реакций, развивающихся под действием патогенного фактора (фурункул, тромбоз, гипоксия, алкалоз, ацидоз).
Типовой (типический) патологический процесс — патологический процесс, встречающийся в виде постоянных сочетаний или комбинаций (независимо от биологического вида животного и причины, вызвавшей процесс: воспаление, опухоль, лихорадка и др.).
Патологическое состояние — это медленно (вяло) текущий патологический процесс (рубец, родимое пятно, состояние после ампутации, удаления зуба).
Введение в патофизиологию
17
Роль причин и условий в возникновении болезни.
Их диалектическая взаимосвязь
Конкретное проявление болезни — следствие действия причинного фактора и его функционирования в определенных условиях.
Определение притин и условий возникновения болезней необходимо для практической деятельности врача, для выбора рациональной терапии, для профилактики болезней.
Что же понимается под причиной болезни?
Причиной болезни называют тот фактор, который вызывает заболевание и сообщает ему специфические черты.
Например, причиной лучевой болезни служит ионизирующая радиация, причиной инфекционного заболевания — патогенные микроорганизмы.
При этом следует подчеркнуть, что причиной болезни служит не какой-либо изолированный фактор внешней среды, а взаимодействие внешнего (или внутреннего) патогенного фактора и организма (туберкулез).
Причина болезни действует на организм не изолированно от окружающей среды, а непременно в каких-либо конкретныхусловиях, имеющих чрезвычайно большое значение. Они могут способствовать действию фактора-причины, а могут и противодействовать ему.
Причинно-следственные отношения в патогенезе
Общая этиология тесным образом связана с проблемами общего патогенеза болезней — учения о механизмах возникновения и течения болезней.
Выяснение патогенеза представляет большой интерес для практической медицины. Более того, учение о патогенезе болезней составляет ее основу.
Патогенез болезни или механизм развития болезни — это совокупность взаимосвязанных и взаимообусловленных непрерывно развивающихся функциональных (физиологических), структурных, биохимических и биофизических изменений.
Чтобы в полном объеме понять патогенез болезни и на этой основе целенаправленно управлять им, врач должен решить ряд следующих задач.
1. Определить патогенетическую значимость внешнего (внутреннего) этиологического фактора. Такой фактор может участвовать лишь в первигном повреждении, выполняя пусковую роль, либо детерминировать патогенез болезни на отдельных этапах или на всем протяжении.
18
Лекция 1
Следует подчеркнуть, что в редких случаях фактор внешней среды, ставший причиной патологического состояния, исчезает сразу после своего воздействия, т.е. совершает роль толчка (как, например, механическая сила, тепло, радиация).
Чаще фактор продолжает свое действие (носит постоянный характер), определяя различные этапы или звенья патогенеза болезни, которые связаны между собой причинно-следственными отношениями (инфекции, гипертензия).
Знание динамики взаимодействия этиологического фактора с организмом — важная задача для выбора и своевременной смены способов и средств этиотропной терапии.
2. Выявить основное звено патогенеза. В ходе развития болезни нарушаются функции многих систем и органов, меняется их структура, появляются разнообразные симптомы, которые позволяют врачу поставить правильный диагноз.
Среди различных проявлений болезни имеются главные (основные) и второстепенные, нередко случайные изменения. Для применения рациональной терапии необходимо оценить значение этих изменений для развития и течения болезни.
То явление или процесс, который совершенно необходим для развертывания всех звеньев патогенеза и предшествует им, называется основным звеном патогенеза.
Например, основным звеном кровопотери служат уменьшение объема циркулирующей крови и связанная с ним гипоксия (гипоксемия).
Отмеченное основное звено обусловливает развитие как приспособительных реакций со стороны крови (эритроцитоз), дыхания (одышка), сердегно-сосудистой системы (повышение тонуса сосудов) и других систем, так и патологических изменений при кровопотере: нарушение регуляции дыхания (дыхательная недостаточность — ДН), работы сердегно-сосудистой системы [снижение артериального давления (АД), ослабление работы сердца], обмена веществ и др.
Восстановление объема циркулирующей крови после переливания ее или кровезамещающих жидкостей приводит к устранению всех изменений, характерных для кровопотери.
3. Наряду с установлением основного звена патогенеза болезни весьма важно определить последовательную цепь причинно-следственных отношений, без чего невозможно рациональное применение средств симптоматической или патогенетической терапии.
Введение в патофизиологию
19
Патогенный агент
Повреждение (альтерация) раздражение рецепторов
Сосудистая реакция (сужение, расширение сосудов)
Пролиферация
Нарушение тканевого обмена Образование медиаторов воспаления (ФНО, гистамин, интерлейкины), накопление ионов калия, некротических гормонов — Образование факторов, стимулирующих регенерацию и пролиферацию (стимуляторы роста и др.)___________________
Повышение проницаемости стенок сосудов, экссудация с эмиграцией лейкоцитов, развитие отека, сдавление корней мелких вен — венозная гиперемия, дальнейшее развитие отека________
Образование противовоспалительных медиаторов, гормонов, интерлейкинов
Рис. 1-2. Причинно-следственные связи при воспалении
Например, при воспалении вслед за повреждением закономерно возникает сосудистая реакция с экссудацией, а затем — пролиферация. Все эти явления тесно связаны причинно-следственными связями (рис. 1-2).
Сосудистая реакция (сужение, расширение сосудов) возникает в результате раздражения рецепторов. Наряду с этим в тканях образуются сосудорасширяющие вещества, понижается pH среды, что также поддерживает вазодилатацию.
Под влиянием продуктов тканевого распада и образующихся гуморальных факторов в очаге воспаления повышается проницаемость стенок сосудов, начинается экссудация с миграцией лейкоцитов, возникает отек, следствием которого становятся сдавление корней мелких вен и венозная гиперемия, которая, в свою очередь, способствует развитию отека.
Но уже в стадии альтерации образуются факторы (некротические гормоны и др.), стимулирующие регенерацию и пролиферацию.
Из вышесказанного видна тесная связь всех основных процессов (альтерация, сосудистая реакция, экссудация, регенерация) воспалительной реакции.
Знание причинно-следственных отношений в патогенезе болезней позволяет целенаправленно влиять на механизмы течения болезней, применять средства патогенетитеской терапии.
Нередко цепь явлений в ходе развития болезни замыкается в поротный круг, когда событие, возникшее под воздействием предыдущего эта
20
Лекция 1
па, становится определяющим и еще более нарушающим ход развития патологии. В результате образования такого порочного круга организм не может самостоятельно (без посторонней помощи) выйти из этого состояния.
Например, при травматическом шоке развиваются гипотония и нарушение внешнего дыхания, что приводит к кислородному голоданию тканей, прежде всего сказывающемуся на функции ЦНС, которое ведет к расстройству обмена веществ, еще больше усиливающему гипоксию (гипоксемию) и углублению нарушений функции нервной системы. Разорвать указанный порочный круг удается лишь проведением комплексной терапии, направленной на основные патогенетические звенья шока, разрывающие возникший порочный круг (переливание крови, восстановление дыхания, регуляция обмена веществ).
4. Большая практическая задача любого врача — оценить состояние механизмов выздоровления и компенсации, или саногенетические механизмы.
Саногенез (от греч. sanus — здоровье и genesis — развитие) буквально означает «развитие здоровья» (С.М. Павленко).
Поскольку саногенез — это динамигеский комплекс механизмов, направленный на восстановление нарушенной саморегуляции организма, от правильного и своевременного их выявления зависят и степень точности прогноза развития заболевания, и выбор терапевтических средств.
Главная задага врага — устранить патологигеские процессы и способствовать компенсации нарушенных функций.
Моделирование
Важнейшей проблемой современной клинической и экспериментальной медицины является моделирование заболеваний — основной метод патофизиологии.
С помощью различных моделей раскрываются механизмы патогенеза болезней.
1. Физическое или материальное (на биологических моделях-объектах) моделирование.
Например, модель алиментарной дистрофии у крыс и собак уточняет значение возраста и обменных процессов в развитии дистрофии.
Иммунизация обезьян гомологичными антигенами (АГ) из латерального коленчатого тела мозга позволяет представить механизмы развития некоторых форм эпилепсии.
Введение в патофизиологию 21
Известны методы воспроизведения биологических моделей по наиболее актуальным вопросам патологии, таким как аллергия, лихорадка, гипертрофия сердца, атеросклероз, гипертензии и т.д.
2. Компьютерное (математическое) — нематериальное (формализованное) моделирование.
Несомненно, моделирование различных заболеваний и отдельных звеньев патогенеза на разных уровнях интеграции организма помогает решать важные вопросы этиологии, диагностики и терапии многих болезней.
Однако всегда необходимо помнить, что в полном объеме смоделировать болезнь человека невозможно.
Лекция 2
ПАТОФИЗИОЛОГИЯ АЛКОГОЛИЗМА, НАРКОМАНИЙ И ТОКСИКОМАНИЙ
Ю.В. Шарпань
Медико-социальное значение алкоголизма, наркоманий и токсикоманий определяется их значительной распространенностью, представляющей серьезную угрозу для здоровья населения в целом. За последние три десятилетия употребление алкоголя в промышленно развитых странах возросло в среднем в 15 раз и привело, по данным ВОЗ, к поражению алкоголизмом от 1 до 10 % взрослого населения. Алкоголизм характеризуется высокой летальностью, что связано не только с большим травматизмом, сопровождающим опьянение, но и с такими его осложнениями, как цирроз печени, язвенная болезнь, кардиомиопатия, а также множество других хронических заболеваний, в той или иной мере поражающих практически все жизненно важные системы (табл. 2-1).
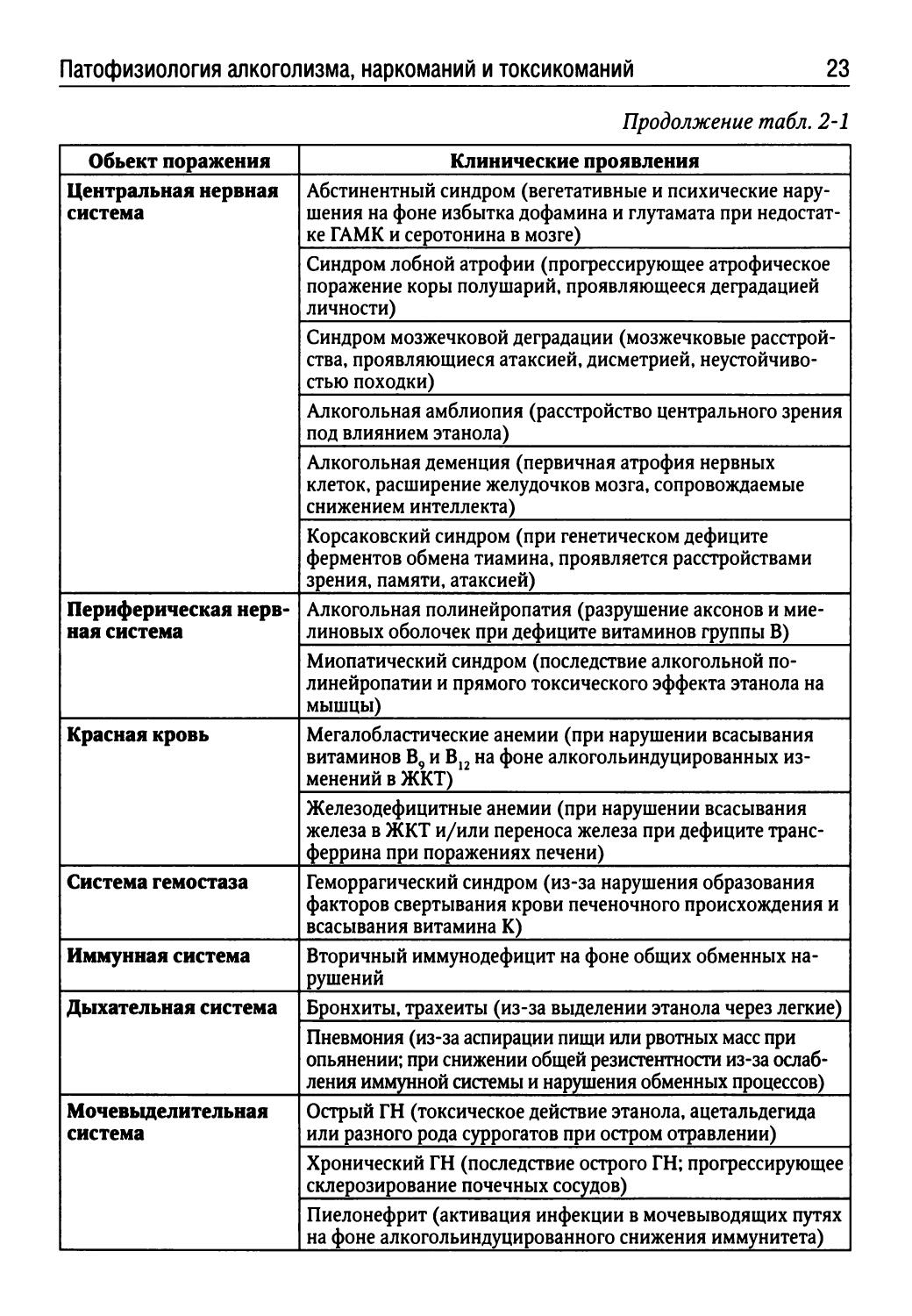
Таблица 2-1. Заболевания, провоцируемые хроническим алкоголизмом
Объект поражения Клинические проявления
ЖКТ Печень Гепатомегалия; алкогольный гепатит; алкогольный цирроз
Поджелудочная железа Острый и хронический панкреатит
Пищевод Алкогольный эзофагит
Желудок Гастрит атрофический, геморрагический; увеличенный риск развития язвенной болезни желудка
Кишечник Энтерит
Сердце Алкогольная миокардиодистрофия (гипертрофия и дистрофия кардиомиоцитов, кардиомегалия, нарушения ритма и проводимости сердца)
Ишемическая болезнь сердца
Сосудистая система Артериальная гипертензия
Потенцирование атеросклеротических поражения артерий головного мозга и магистральных артерий
Повышение давления в портальной системе при циррозе (частое последствие — развитие кровотечений из расширенных вен желудка и пищевода)
Патофизиология алкоголизма, наркоманий и токсикоманий
23
Продолжение табл. 2-1
Объект поражения Клинические проявления
Центральная нервная система Абстинентный синдром (вегетативные и психические нарушения на фоне избытка дофамина и глутамата при недостатке ГАМК и серотонина в мозге)
Синдром лобной атрофии (прогрессирующее атрофическое поражение коры полушарий, проявляющееся деградацией личности)
Синдром мозжечковой деградации (мозжечковые расстройства, проявляющиеся атаксией, дисметрией, неустойчивостью походки)
Алкогольная амблиопия (расстройство центрального зрения под влиянием этанола)
Алкогольная деменция (первичная атрофия нервных клеток, расширение желудочков мозга, сопровождаемые снижением интеллекта)
Корсаковский синдром (при генетическом дефиците ферментов обмена тиамина, проявляется расстройствами зрения, памяти, атаксией)
Периферическая нервная система Алкогольная полинейропатия (разрушение аксонов и миелиновых оболочек при дефиците витаминов группы В)
Миопатический синдром (последствие алкогольной полинейропатии и прямого токсического эффекта этанола на мышцы)
Красная кровь Мегалобластические анемии (при нарушении всасывания витаминов В9 и В12 на фоне алкогольиндуцированных изменений в ЖКТ)
Железодефицитные анемии (при нарушении всасывания железа в ЖКТ и/или переноса железа при дефиците трансферрина при поражениях печени)
Система гемостаза Геморрагический синдром (из-за нарушения образования факторов свертывания крови печеночного происхождения и всасывания витамина К)
Иммунная система Вторичный иммунодефицит на фоне общих обменных нарушений
Дыхательная система Бронхиты, трахеиты (из-за выделении этанола через легкие)
Пневмония (из-за аспирации пищи или рвотных масс при опьянении; при снижении общей резистентности из-за ослабления иммунной системы и нарушения обменных процессов)
Мочевыделительная система Острый ГН (токсическое действие этанола, ацетальдегида или разного рода суррогатов при остром отравлении)
Хронический ГН (последствие острого ГН; прогрессирующее склерозирование почечных сосудов)
Пиелонефрит (активация инфекции в мочевыводящих путях на фоне алкогольиндуцированного снижения иммунитета)
24
Лекция 2
Оконгание табл. 2-1
Объект поражения Клинические проявления
Половая система У мужчин: снижение половой функции (импотенция)
У женщин: нарушение течения беременности (увеличение частоты выкидышей и мертворождений)
Алкогольный синдром плода (пороки развития, умственная отсталость, повышенный риск развития алкогольной болезни в постнатальном периоде)
Для конца XX - начала XXI века характерно увеличение потребления наркотических и токсических веществ. Актуальной проблемой, связанной с внутривенным потреблением наркотиков, стало распространение заболеваний, передающихся с кровью (СПИД, гепатиты). Согласно статистическим отчетам последних лет, на долю ВИЧ-инфицированных наркоманов приходится примерно 70 % от числа всех зарегистрированных ВИЧ-инфицированных больных.
Высокая частота хронического алкоголизма, наркомании и токсикомании создает не только медицинскую проблему, связанную с лечением пациентов с такими заболеваниями, но и сопровождается тяжелыми социальными, криминальными и экономическими последствиями. Хорошо известно, что значительная часть лиц, страдающих пристрастием к алкоголю, наркотическим и токсическим веществам, неизбежно выключается из экономических отношений, пополняя асоциальный и криминальный слой общества. Это способствует как росту общей преступности, совершаемой в состоянии алкогольного или наркотического опьянения, так и росту преступлений, связанных с изготовлением и сбытом наркотиков и нелегально производимого алкоголя.
Механизм токсического действия этанола
Эндогенный этанол относится к незаменимым метаболическим факторам. В норме его концентрация в крови составляет 0,01-0,03 % (0,Ol-О.03 г/л). Окисление эндогенного этанола обеспечивает до 10 % всех энергетических потребностей организма (1 г этанола дает 7,1 ккал).
В организме этанол подвергается быстрому и эффективному окислению: в печени метаболизируется до 80 % этанола, в других тканях — до 10 % и столько же выделяется с мочой и легкими.
В метаболизме этанола участвуют три системы (рис. 2-1).
1. Система алкогольдегидрогеназы.
2. Микросомальная этанолокисляющая система (МЭОС).
3. Система каталазы пероксисом.
Патофизиология алкоголизма, наркоманий и токсикоманий
25
Рис. 2-1. Метаболизм этанола
На долю этих систем приходится соответственно 75-90 %, 10-20 % и 1-2 % метаболизируемого этанола. При посредстве трех указанных систем этанол превращается в ацетальдегид, который, в свою очередь, под влиянием фермента ацетальдегиддегидрогеназы трансформируется в угольную кислоту, распадающуюся в конечном итоге на Н2О и СО2.
В организме этанол способен действовать на различные органы и системы. Это связано с его физико-химическими особенностями. Этанол способен смешиваться с водой в любых соотношениях. Также легко растворяется он в липидах, беспрепятственно проходит через гематоэнцефалический (ГЭБ) и плацентарный барьеры. В результате взаимодействия этанола с гидрофобными участками липопротеидов повышается текучесть мембран, что сопровождается нарушением работы мембранных насосов (К’/Na*- и Са2’-АТФаз) и других мембранных ферментных систем. В связи с повышенным сродством этанола к липидам при отравлении этиловым спиртом прежде всего страдают органы, богатые ими. При введении этанола в кровоток, если его концентрацию в крови принять за единицу, то в печени она составит 1,5, а в головном мозге — 1,75.
Реакция ЦНС на этанол при его однократном приеме у здоровых лиц зависит от дозы и индивидуальной переносимости этилового спирта, связанной с активностью алкогольдегидрогеназы и ацетальдегиддегидрогеназы (рис. 2-2).
26
Лекция 2
Рис. 2-2. Последствия употребления алкоголя
При концентрации этанола в крови менее 0,3 г/л не обнаруживается каких-либо отклонений со стороны ЦНС.
Концентрация этанола от 0,3 до 3,0 г/л вызывает различный по выраженности эффект опьянения, для которого характерен симптомоком-плекс, связанный с ослаблением центрального торможения и развитием эйфории. В основе эйфории лежат биохимические изменения, обусловленные повышенными концентрациями ацетальдегида. Ослабление центрального торможения связано с начинающимися нарушениями в работе мембранной K’/Na -АТФазы в тормозных нейронах ЦНС. Кроме того, при таких концентрациях этанола в крови начинает повышаться проницаемость ГЭБ, увеличивается выделение ацетилхолина, уменьшается выделение адреномедиаторов. Именно активацией парасимпатического отдела вегетативной нервной системы объясняются наблюдаемые при опьянении усиление рвотного рефлекса, слюнотечение, усиление перистальтики, ослабление гладкомышечных сфинктеров желудочно-кишечного тракта (ЖКТ) и мочеполовой системы. Со временем при постоянном употреблении алкоголя снижается скорость окисления глюкозы, утилизации АТФ, синтеза белков и липидов нейроглии, что становится причиной органических дегенеративных изменений в ЦНС при хроническом алкоголизме.
Концентрация этанола в крови от 3,0 до 6,0 г/л вызывает острое отравление независимо от индивидуальной переносимости этилового спирта. Происходит глубокое угнетение ЦНС, клинически выражающееся в ступорозном состоянии, сменяющемся потерей сознания. В ЦНС отмечается ряд биохимических изменений: угнетение тканевого
Патофизиология алкоголизма, наркоманий и токсикоманий
27
дыхания, снижение активности окисления глюкозы и утилизации АТФ, резкое повышение проницаемости ГЭБ, еще более возрастающее выделение ацетилхолина, приводящее к падению артериального давления, резкой брадикардии; происходит увеличение выделения у-аминомасляной кислоты (ГАМК) в синапсах тормозных нейронов, значительно падает активность K'/Na'- и Са2-АТФазы.
Концентрация этанола более 6,0 г/л несовместима с жизнью. В таких концентрациях этанол вызывает в ЦНС биохимические сдвиги, угнетающие ферменты, отвечающие за тканевое дыхание и фосфорилирование. Это сопровождается глубокими нарушениями работы жизненно важных органов вследствие практически полного прекращения их регуляции со стороны ЦНС, что, собственно, и становится причиной смерти.
Патофизиологическая характеристика хронического алкоголизма
Постоянный прием алкоголя, создающий концентрацию этанола в крови в пределах 0,3-3,0 г/л, со временем может привести к развитию хронического алкоголизма. В ряду событий от первого знакомства со спиртными напитками до конечной стадии хронического алкоголизма можно выделить несколько этапов:
1) умеренное (эпизодическое) употребление алкоголя;
2) злоупотребление спиртными напитками (бытовое пьянство);
3) хронический алкоголизм, в проявлении которого выделяют три стадии.
Бытовое пьянство по отношению к хронигескому алкоголизму рассматривается как предболезнь. Переход постепенно усугубляющегося бытового пьянства (предболезни) в хронический алкоголизм (болезнь) констатируется при наличии трех симптомов:
1) нарастающий при бытовом пьянстве симптом желательности алкоголизации трансформируется в неподконтрольное воле пациента влечение к регулярным приемам алкоголя (больные перестают искать повод для приема спиртных напитков и потребляют их без каких-либо ссылок на внешние обстоятельства);
2) постепенно утрачиваемое при усугублении бытового пьянства чувство меры при потреблении алкоголя переходит в полную утрату количественного контроля из-за потери ощущения насыщения алкоголем (больные стремятся к потреблению всего имеющегося запаса алкоголя, остановить на время их может лишь исчерпание таких запасов или состояние глубокого опьянения, лишающее возможности продолжать прием алкоголя);
28
Лекция 2
3) утрачивается воля к отказу от приема алкоголя (алкоголь употребляется даже тогда, когда пациент осознает, что ситуация этого категорически не позволяет).
При прогрессировании хронигеского алкоголизма (после его формирования как болезни) выделяют 3 стадии (табл. 2-2).
Таблица 2-2. Клинические проявления хронического алкоголизма
Стадии хронического алкоголизма Признаки
1-я стадия Наличие психической зависимости от алкоголя; нарастание толерантности к этанолу (постоянно увеличивается объем разового потребления алкоголя для достижения прежнего эффекта); проявления физической зависимости (абстинентного синдрома) отсутствуют
2-я стадия Появляется абстинентный синдром, отражающий развитие физической зависимости от алкоголя; толерантность к этанолу достигает максимума; вне алкоголизации (в периоды «случайной» трезвости) отмечаются стойкие эмоционально-психические нарушения в виде депрессии и угнетения настроения (дисфория); прогрессируют признаки алкогольной деградации (нарушения памяти, утрата ценностных ориентиров, ослабление интеллекта); могут возникать алкогольные психозы; соматические расстройства становятся труднообратимыми; усугубляется социальная дезадаптация
3-я стадия Отмечается снижение толерантности к алкоголю (амнезии опьянения наступают даже после приема незначительных доз этанола); проявления абстинентного синдрома становятся максимально тяжелыми; алкогольная деградация сопровождается органическими поражениями ЦНС; учащаются алкогольные психозы; соматические расстройства характеризуются необратимостью; отмечается глубокая социальная дезадаптация, трудоспособность практически утрачивается
Главным признаком первой стадии хронического алкоголизма является формирование алкогольного стиля жизни, при котором ведущей смыслообразующей потребностью становится употребление спиртного. Пьянство становится главной мотивирующей силой, основным содержанием жизни. Круг интересов резко сужается (что особенно контрастно проявляется у людей достаточно образованных), начинается личностная и профессиональная деградация, оказывающая все более заметное негативное влияние на трудоспособность и социальное функционирование. Это характеризует психигескую зависимость от алкоголя, появление
Патофизиология алкоголизма, наркоманий и токсикоманий 29
которой и позволяет поставить диагноз первой стадии хронического алкоголизма. В этой стадии абстинентного синдрома еще не наблюдается, хотя появляются стертые вегетососудистые реакции, субдепрессивное настроение, которые, однако, еще купируются неспецифическими средствами (крепким чаем, холодным душем и др.).
Для второй стадии хронического алкоголизма характерно выдвижение на первый план биологических механизмов заболевания. Происходят окончательное уплощение и деформация личности алкоголика. Влечение к алкоголю в этой стадии носит навязчивый характер, отражающий возникновение физической зависимости, выражающейся в появлении абстинентного синдрома — выраженных соматовегетативных и психических нарушений в периоде протрезвления, снимаемых только приемом очередной дозы алкоголя (опохмелением).
В третьей стадии хронического алкоголизма зависимость от алкоголя принимает витальные черты, потребность в спиртном становится в один ряд с потребностью в пище, сне и т.д. Нормальная жизнедеятельность вне алкоголизации просто невозможна. Состояние опьянения лишено индивидуальных черт, нет повышения настроения и стимулирующего эффекта. Интоксикация алкоголем лишь стабилизирует психосоматические функции. Абстинентный синдром сопровождается наиболее выраженными соматовегетативными и неврологическими расстройствами.
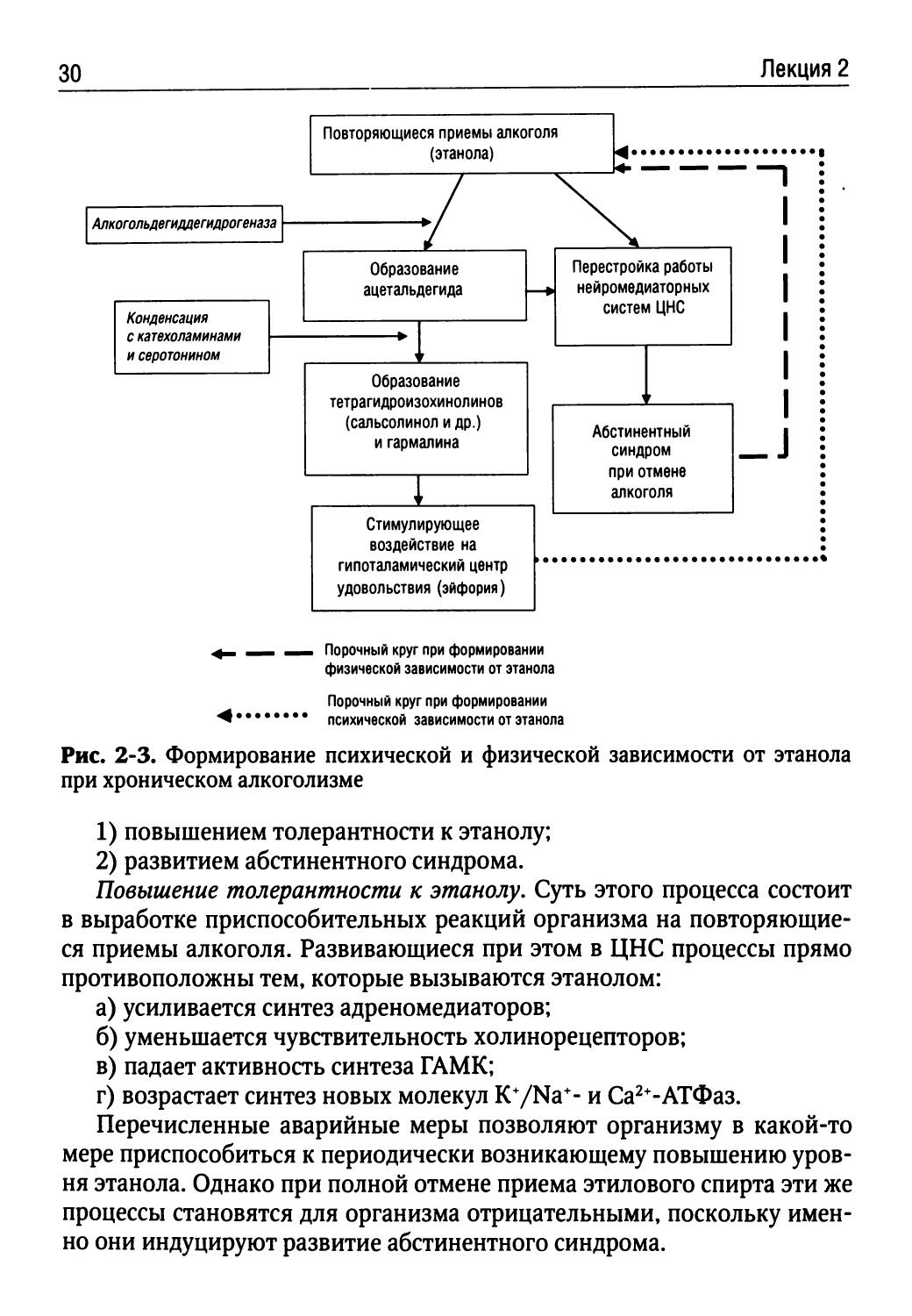
С патофизиологической точки зрения развитие феномена психиге-ской зависимости от алкоголя связано с нарушениями биохимических процессов в ЦНС, вызванными избытком ацетальдегида, уровень которого кратковременно повышается в крови после каждого очередного приема экзогенного этанола. Ацетальдегид путем конденсации с дофамином способен образовывать сальсолинол и тетрагидропапаверолин, а при взаимодействии с серотонином — гармалин, которые обладают морфиноподобной активностью. Эти вещества воздействуют на опиатные рецепторы гипоталамического центра удовольствия, индуцируя эйфорию при приеме алкоголя, и формируют патологическую привязанность к нему, обозначаемую как психическая зависимость от этанола. Возникает порочный круг (рис. 2-3):
• прием алкоголя -> образование повышенных концентраций ацетальдегида образование сальсолинола, тетрагидропапаверолина, гармалина —> стимуляция гипоталамического центра удовольствия (эйфория) -> побуждение к новым приемам алкоголя.
Развитие физигеской зависимости от этанола обусловлено прогрессированием двух тесно взаимосвязанных процессов:
30
Лекция 2
4— _ _ Порочный круг при формировании физической зависимости от этанола
Порочный круг при формировании ...... психической зависимости от этанола
Рис. 2-3. Формирование психической и физической зависимости от этанола при хроническом алкоголизме
1) повышением толерантности к этанолу;
2) развитием абстинентного синдрома.
Повышение толерантности к этанолу. Суть этого процесса состоит в выработке приспособительных реакций организма на повторяющиеся приемы алкоголя. Развивающиеся при этом в ЦНС процессы прямо противоположны тем, которые вызываются этанолом:
а) усиливается синтез адреномедиаторов;
б) уменьшается чувствительность холинорецепторов;
в) падает активность синтеза ГАМК;
г) возрастает синтез новых молекул K*/Na*- и Са2+-АТФаз.
Перечисленные аварийные меры позволяют организму в какой-то мере приспособиться к периодически возникающему повышению уровня этанола. Однако при полной отмене приема этилового спирта эти же процессы становятся для организма отрицательными, поскольку именно они индуцируют развитие абстинентного синдрома.
Патофизиология алкоголизма, наркоманий и токсикоманий
31
Абстинентный синдром. Механизм абстинентного синдрома состоит в том, что перестроившиеся в период формирования повышенной то-лератности к этанолу биохимические процессы продолжают протекать по-прежнему, в то время как противостоящие им эффекты повышенных концентраций этанола исчезают при отмене его приема. В результате происходит резкое нарушение работы вегетативной нервной системы, проявляющееся в резкой активации ее симпатического отдела при одновременном ослаблении холинергических эффектов. В ЦНС, кроме того, ослабляется центральное торможение, активизируется работа К ‘/Na*- и Са2‘-АТФаз. Клинически это выражается в развитии тахикардии, гипертензии, ангиоспазмов (вызывающих сильнейшие головные боли, боли в сердце), резком возбуждении, сопровождающемся агрессивностью и субъективным чувством тревоги. Все эти нарушения могут быть купированы лишь очередным приемом алкоголя.
Таким образом, индукция физической зависимости от алкоголя связана с образованием еще одного порочного круга (см. рис. 2-3):
• повторяющиеся приемы алкоголя -» перестройка работы нейромедиаторных систем ЦНС (повышение толерантности к этанолу) -> возникновение абстинентного синдрома при отмене очередного приема алкоголя -> побуждение к повторным приемам алкоголя с целью прекращения/недопущения проявлений алкогольной абстиненции.
Принципы патогенетической терапии острого и хронического отравления этанолом
При остром отравлении этанолом (концентрация в крови более 3 г/л) проводят общую дезинтоксикационную терапию, применяемую при многих других отравлениях. Суть ее сводится к активации выведения токсического вещества и к проведению симптоматического лечения.
Для активации выведения этанола — введение большого количества жидкости (изотонический раствор хлорида натрия, гемодез*, полиглю-кин* и др.) при одновременном форсировании диуреза (введение фуросемида).
При атонии мочевого пузыря — его катетеризация. Промывание желудка.
Интенсивная симптоматическая терапия: при падении артериального давления — адреномиметики, холинолитики, большие дозы глюкокортикоидов: для улучшения газообмена — дыхательные смеси с повышенным содержанием кислорода (карбоген); при угнетении ды
32
Лекция 2
хания — аналептики (лобелии), при необходимости перевод больного на управляемое дыхание. Для уменьшения центрального торможения — бемегрид, кофеин, для стимуляции сердечной деятельности — кордиамин, камфора.
Лечение хронического алкоголизма преследует одновременно достижение двух целей: подавление патологического пристрастия к этанолу (психической зависимости от алкоголя) и устранение абстинентного синдрома (физической зависимости от алкоголя).
Патогенетические механизмы наркоманий и токсикомании
Наркомания и токсикомания, как и хронический алкоголизм, относятся к болезням патологической зависимости, которые имеют три ведущих клинических признака:
• нарастание пристрастия к повторяющимся приемам психотропного вещества (ПТВ);
• повышение толерантности к принимаемому ПТВ, что обусловливает необходимость увеличения принимаемой дозы для достижения прежнего эффекта:
• возникновение абстинентного синдрома при прекращении приемов ПТВ.
Первый признак ассоциируется с формированием психической зависимости от ПТВ, второй и третий — с физической зависимостью от ПТВ.
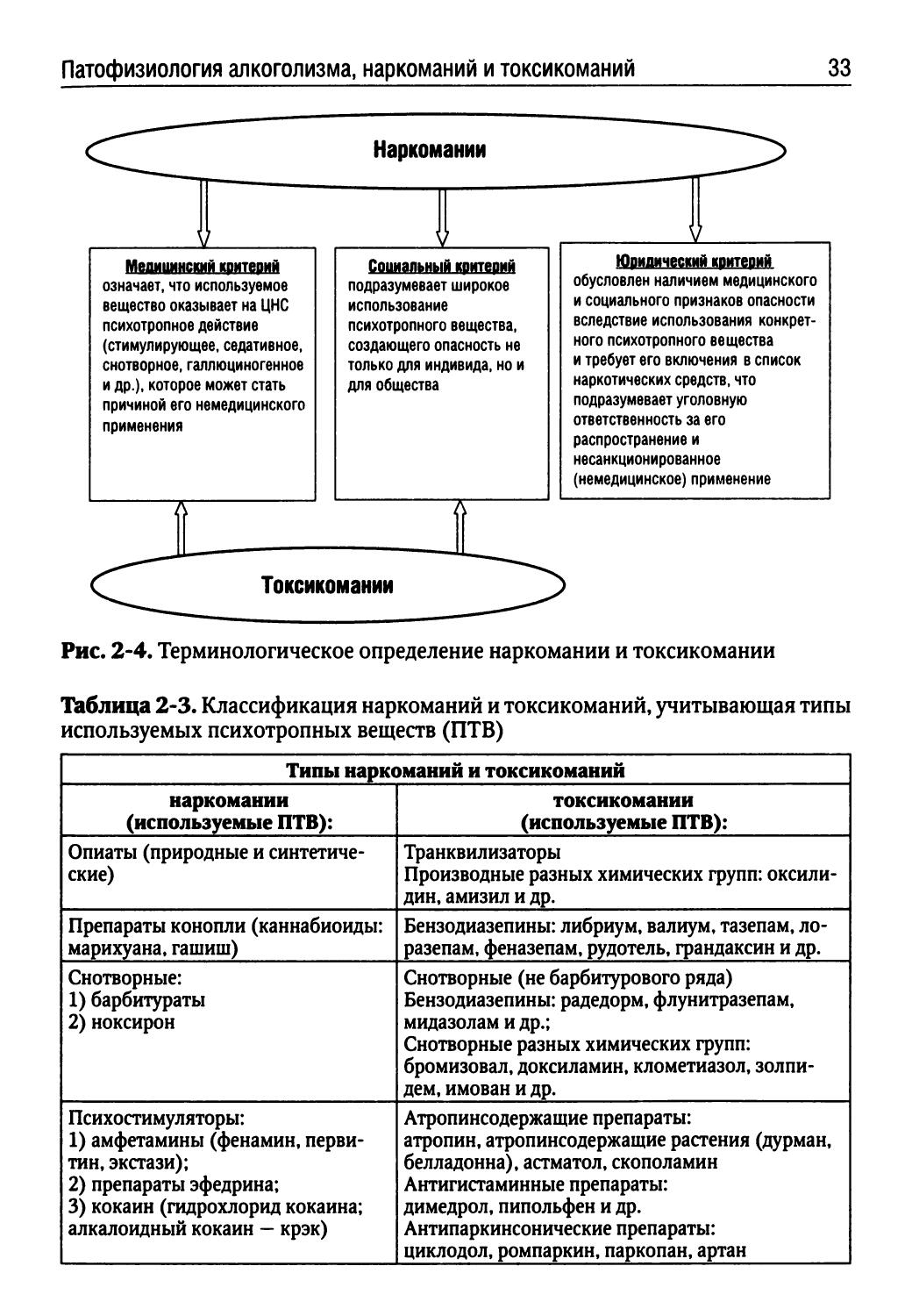
В отечественной наркологии злоупотребление веществом, официально внесенным в список наркотиков, трактуется как наркомания. Если употребляемое ПТВ не внесено в этот список, то злоупотребление им обозначается как токсикомания. С клинико-динамических позиций принципиальных различий между наркоманиями и токсикоманиями нет. Таким образом, различия между наркоманиями и токсикоманиями определяется лишь юридическим критерием (рис. 2-4).
Наиболее распространенные наркомании и токсикомании (табл. 2-3):
1. Наркомания производными опия и морфина, конопли (марихуаны), психостимуляторами (кокаином, эфедрином, амфетаминами), снотворными (ноксироном, барбитуратами), галлюциногенами (фенциклидином, препаратами лизергиновой кислоты).
2. Токсикомания транквилизаторами, снотворными небарбитурового ряда, антипаркинсоническими и антигистаминными средствами, атропинсодержащими препаратами, летучими растворителями, средствами для ингаляционного наркоза.
Патофизиология алкоголизма, наркоманий и токсикоманий
33
Рис. 2-4. Терминологическое определение наркомании и токсикомании
Таблица 2-3. Классификация наркоманий и токсикоманий, учитывающая типы используемых психотропных веществ (ПТВ)
Типы наркоманий и токсикоманий
наркомании (используемые ПТВ): токсикомании (используемые ПТВ):
Опиаты (природные и синтетические) Транквилизаторы Производные разных химических групп: оксили-дин, амизил и др.
Препараты конопли (каннабиоиды: марихуана, гашиш) Бензодиазепины: либриум, валиум, тазепам, лоразепам, феназепам, рудотель, грандаксин и др.
Снотворные: 1) барбитураты 2) ноксирон Снотворные (не барбитурового ряда) Бензодиазепины: радедорм, флунитразепам, мидазолам и др.; Снотворные разных химических групп: бромизовал, доксиламин, клометиазол, золпидем, имован и др.
Психостимуляторы: 1) амфетамины (фенамин, первитин, экстази); 2) препараты эфедрина; 3) кокаин (гидрохлорид кокаина; алкалоидный кокаин - крэк) Атропинсодержащие препараты: атропин, атропинсодержащие растения (дурман, белладонна), астматол, скополамин Антигистаминные препараты: димедрол, пипольфен и др. Антипаркинсонические препараты: циклодол, ромпаркин, паркопан, артан
34
Лекция 2
Оконгание табл. 2-3
Галлюциногены: 1) мескалин, псилоцибин; 2) препараты лизергиновой кислоты (ЛСД); 3) препараты фенциклидина (кетамин) Летучие ингалянты: алифатические, ароматические и галогенезиро-ванные углеводороды; эфиры, кетоны, входящие в состав средств бытовой химии Кофеиновая токсикомания
Полинаркомания — зависимость от 2-х наркотических веществ и более, применяемых одновременно или в разной последовательности Никотинизм (табачная зависимость) Политоксикомания — зависимость от 2-х ПТВ и более (не включенных в список наркотиков), применяемых одновременно или в разной последовательности
Осложненная наркомания - зависимость от наркотического средства, сочетаемого с приемом ПТВ, не относимого к наркотикам, или с алкоголем
Существует два объяснения наблюдаемого при наркомании и токсикомании механизма формирования психической зависимости от ПТВ.
Согласно первой точке зрения, в организме некоторых людей может иметь место дефицит эндогенных адаптогенов (морфиноподобных веществ), выполняющих в норме роль антистрессоров, анальгетиков и эйфоригенов. Это может быть связано либо с индивидуальными генетическими особенностями, проявляющимися в виде ослабленного синтеза таких соединений, либо с частыми и длительными эмоционально-стрессовыми нагрузками, приводящими к перерасходу эндогенных адаптогенов. Таким образом, прием наркотических веществ можно рассматривать как неосознаваемую попытку искусственного восполнения эндогенных адаптогенов соединениями, способными имитировать их действие. Эта гипотеза хорошо объясняет эффекты морфиноподобных соединений и алкоголя, но не других наркотических и токсических веществ.
Согласно второй точке зрения, наркотические и токсические вещества, взаимодействуя с липопротеидными компонентами клеточных мембран, вызывают их конформационные изменения, что нарушает нормальное функционирование нейронов. Воздействуя в дозах, не вызывающих тяжелых поражений нейронов, эти вещества прежде всего нарушают самые сложные нервные процессы, протекающие с одновременным и взаимосвязанным участием многих нервных клеток. К таковым относятся многие процессы активного коркового торможения, процессы тонкой регуляции работы отдельных корковых зон и подкорковых центров. При таких нарушениях возможны растормаживание центра удовольствия (индукция эйфории), несбалансированность
Патофизиология алкоголизма, наркоманий и токсикоманий
35
работы чувствительных центров (индукция галлюцинаций). В более высоких концентрациях наркотики и токсические вещества подавляют корковую активность в целом, что клинически проявляется состоянием оглушенности. Наконец, еще более высокие концентрации этих соединений угнетают работу подкорковых жизненно важных центров (дыхательного и сосудодвигательного), что часто является причиной смерти при передозировке наркотиков и токсических веществ. Данная теория хорошо объясняет эффекты барбитуратов, летучих растворителей (бензина, ацетона, нитролаков), средств для ингаляционного наркоза, а также этанола (учитывая его липотропность).
Патогенез формирования психической зависимости от психотропных веществ сложен, его следует рассматривать по видам отдельных наркоманий и токсикоманий. Наркоманы проходят через фазу привыкания к наркотику к собственно наркотическому синдрому, т.е. от психической зависимости к физической. Вначале, в зависимости от типа высшей нервной деятельности, вида наркотика, дозы и пути его поступления в организм, а также мотивационной психологической установки наркотик вызывает желаемый эффект — эйфорический, анальгетический, антистрессовый и т.д. Формируется предпочтительность наркотика, его начинают употреблять регулярно. Организм автоматически отвечает повышением толерантности к этому веществу (за счет активации метаболизирующих систем и снижения чувствительности рецепторов к этим веществам, а также путем синтеза медиаторов, проявляющих конкурентный или функциональный антагонизм к ним). В результате наркоманы и токсикоманы вынуждены увеличивать потребляемые дозы наркотиков. Резко возросшая толерантность к наркотику и становится основой физической зависимости от него, так как именно она порождает клинические проявления абстинентного синдрома при нарушении регулярного поступления наркотика. В конце концов наступает истощение всех защитных систем, происходят изменения в психической сфере в виде невротизации и психопатизации, появляются признаки органического поражения ЦНС.
Принципы терапии наркоманий
Лечение наркоманий предусматривает обязательную госпитализацию.
Первый этап — борьба с абстинентным синдромом, когда необходимо купировать симптомы физической зависимости. Используются те же средства, что и при подавлении абстинентного синдрома в процессе
36 Лекция 2
лечения хронического алкоголизма. Большое значение имеет нормализация сна.
На втором этапе проводят борьбу с психической зависимостью от наркотиков. Здесь важно значение психотерапии, гипноза, выработки отрицательных условных рефлексов на наркотик, а также трудотерапии.
На третьем этапе (уже вне стационара) проводится поддерживающая терапия, которая в основном сводится к психотерапии и активному наблюдению за больным.
Лекция 3
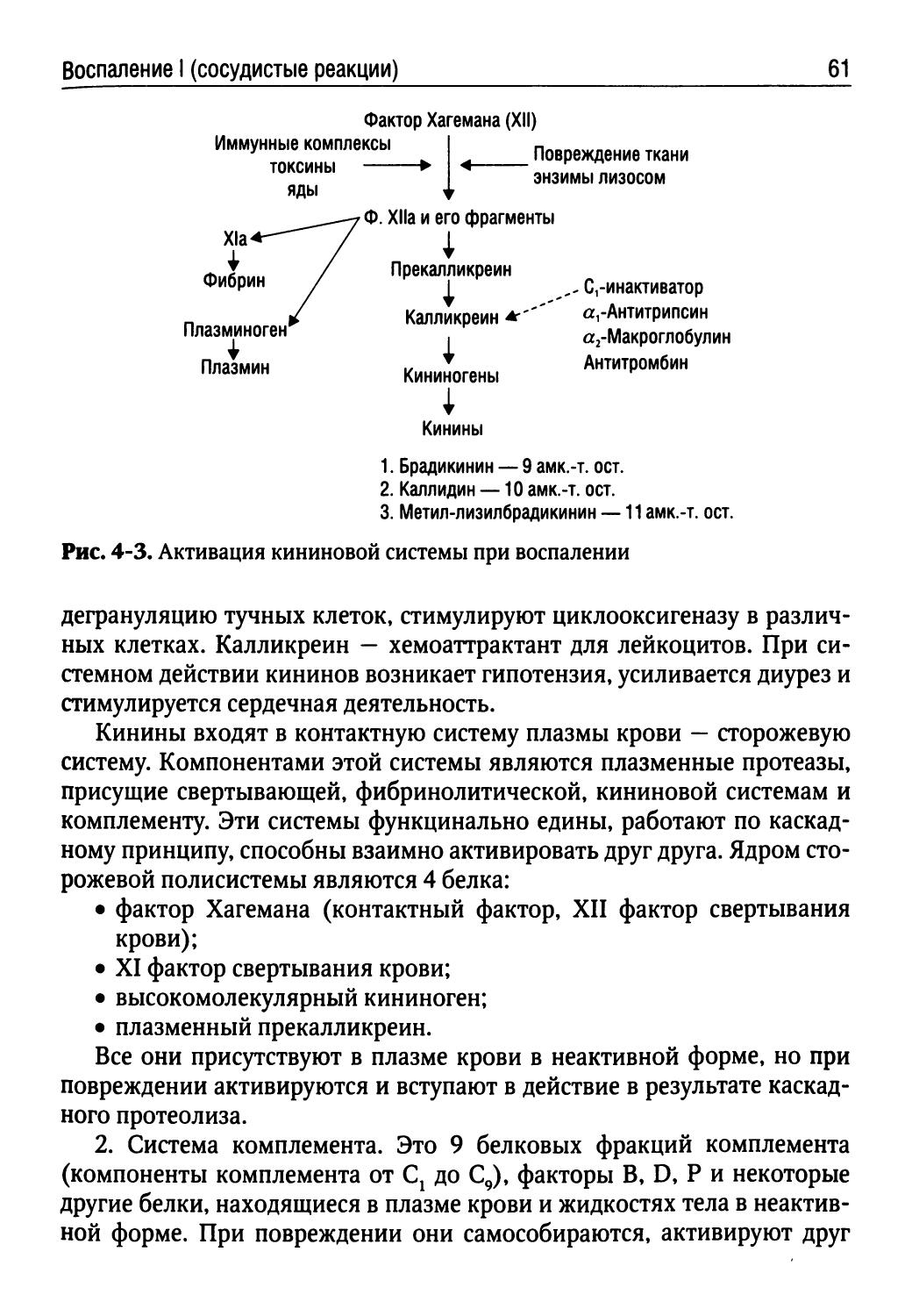
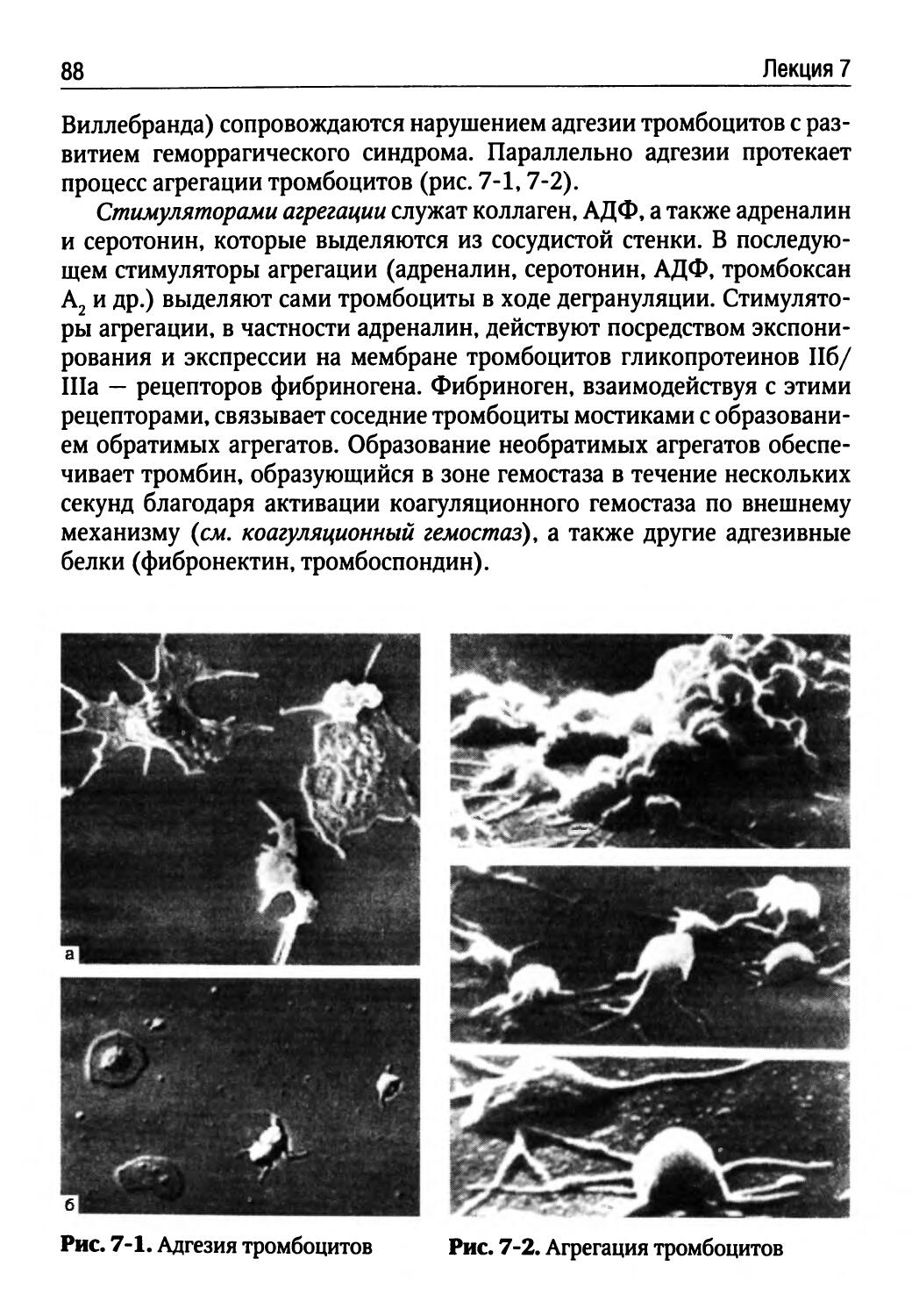
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ ПОВРЕЖДЕНИЯ КЛЕТКИ
Л.Н. Осколок
Любая болезнь человека включает в себя повреждение клеток, характеризующееся таким нарушением внутриклеточного гомеостаза, которое ограничивает функциональные возможности клеток, угрожает их жизни или сокращает ее продолжительность. В понятие гомеостаза клетки входит постоянство концентрации Н\ содержания О2, электролитов, воды, субстратов для энергетического и пластического обеспечения клетки, ферментов, нуклеотидов и других веществ.
По этиологии повреждение клеток может быть механическим, электрическим, термическим, химическим, радиационным, инфекционным и др. По времени развития повреждение клеток может быть острым и хроническим. При механической травме возникает острое повреждение клеток. При длительной гипергликемии развивается хроническое повреждение макрофагов, которые, поглощая в избытке гликозилированные липопротеины низкой плотности (ЛПНП), расщепляя их и накапливая эфиры холестерина, превращаются в пенистые клетки. Повреждение клетки может быть обратимым и необратимым. Если последствия повреждения могут быть устранены самой клеткой, повреждение имеет обратимый характер, например, при кратковременной ишемии миокарда. Если защитные механизмы не в состоянии устранить последствия повреждения, клетка либо сразу гибнет, либо значительно сокращается срок ее жизни. Немедленную смерть клетки может вызвать повреждение цитоплазматической мембраны (ЦПМ) и лизосом. Накопление гликогена в лизосомах клеток при гликогенозах приводит к постепенному разрушению клеток.
При повреждении клетки могут быть выявлены специфические и неспецифические признаки. Специфические признаки определяются особенностями патогенного фактора, вызвавшего это повреждение. Неспецифические признаки не зависят от вида патогенного фактора и выявляются при повреждениях любых клеток.
38
Лекция 3
Специфигеские проявления повреждения клетки:
• образование радиотоксинов при радиационном повреждении;
• появление кавитации при повреждении ультразвуком;
• подавление активности цитохромоксидазы при отравлении цианидами;
• расширение синаптической щели, сглаживание складок постсинаптической мембраны нервно-мышечных синапсов вследствие аутоиммунного повреждения при тяжелой миастении.
Различают морфологические, функциональные и биохимические не-специфигеские проявления повреждения клетки.
Морфологигеские проявления повреждения клетки:
• набухание клетки, нарушение контакта с соседними клетками;
• набухание митохондрий (MX), просветление их матрикса, вакуолизация;
• расширение канальцев эндоплазматического ретикулума (ЭР), разрыв и фрагментация их мембран;
• уменьшение числа рибосом и нарушение их связи с мембранами;
• изменение размера и формы ядра, разрывы ядерной оболочки;
• повреждение и фрагментация плазматической мембраны клетки. Функциональные признаки повреждения клетки:
• нарушение барьерной функции клеточных мембран: увеличение их проницаемости для ионов, микро- и макромолекулярных структур (белков, коллоидных красителей, аминокислот, нуклеотидов);
• нарушение специализированной функции клетки, например снижение сократительной функции кардиомиоцитов; белково-синте-тигеской функции, например снижение синтеза альбуминов гепатоцитами; энергетигеской функции клетки (снижение синтеза АТФ); секреторной функции, например снижение синтеза и секреции стероидных гормонов клетками коры надпочечников;
• повышение возбудимости клетки (снижение мембранного потенциала);
• активация синтеза медиаторов воспаления и ответа острой фазы (лейкотриенов, простагландинов, тромбоксанов, фактора активации тромбоцитов, интерлейкинов, фактора некроза опухолей).
Биохимигеские проявления повреждения клетки:
• повышение содержания в крови внутриклеточных белков и ферментов;
• повышение содержания в крови пуриновых оснований;
Молекулярные механизмы повреждения клетки
39
• уменьшение внутриклеточного содержания АТФ и увеличение внутриклеточного содержания АДФ, АМФ и неорганического фосфора;
• повышение внутриклеточного содержания Na\ Н\ Са2+, Н2О и внеклеточного содержания К?;
• денатурация молекул белка.
Признаки необратимого повреждения клетки:
• стойкое набухание митохондрий и выпадение солей кальция в осадок внутри MX;
• выход интегральных и высокомолекулярных белков из ЦПМ;
• потеря внутриклеточных пуриновых оснований;
• сморщивание и дегенерация ядра;
• разрывы мембран лизосом и аутолиз клетки.
Живая клетка находится в неравновесном состоянии с окружающей средой. В частности, в здоровых клетках содержание Са2‘ и Na‘ существенно меньше, а содержание К‘ и белков больше, чем во внеклеточном пространстве. На поверхности мембран возникает электрохимический градиент (диффузионный потенциал), происходит постоянная пассивная диффузия ионов по градиенту концентрации внутрь клетки и из нее, из цитоплазмы в органеллы и обратно в цитоплазму через мембранные поры и гидрофильные белковые каналы. Пассивная диффузия ограничивается исходно низкой проницаемостью гидрофобного слоя липидов для водорастворимых веществ. Для поддержания ионной асимметрии в мембранах клетки функционируют системы ионных насосов — АТФ-зависимого транспорта против градиента концентрации с помощью ферментов (Ыа‘/К*-АТФаза, Са2*-АТФаза и др.), Na‘/Ca2‘-и Н‘/Са2*-ионообменных механизмов. Существуют липопротеиновые и другие комплексы, обладающие сродством к электролитам и выполняющие роль внутриклеточных депо. Благодаря мембранам клетки изменение показателей гомеостаза происходит в сравнительно узких пределах, обеспечивающих оптимальные условия для жизнедеятельности клетки.
Структуру мембран клетки можно представить по модели жидкокристаллической мембраны Сингера-Никольсона (рис. 3-1). Основными компонентами мембран являются липиды и белки, которые находятся в постоянном движении. Наиболее подвижны жидкие липиды бислоя. Аннулярные липиды менее подвижны и входят в состав монопротеи-новых комплексов. К мембранным липидзависимым белкам относятся Ыа*/К*-АТФаза, аденилатциклаза, цитохромоксидаза, ацил-СоА и
40
Лекция 3
канал
белок
Рис. 3-1. Модель жидкокристаллической мембраны Сингера-Никольсона
другие, главным образом, интегральные белки. Физико-химическое состояние липидов влияет на конформационные свойства, доступность активных центров для субстрата, подвижность, возможность образования полиферментных комплексов связанных с ними белков. Состав и конформационные свойства белков влияют на состояние мембранных липидов; в частности, экранируя липиды, белки снижают действие на них фосфолипаз, радикалов и других патологических факторов.
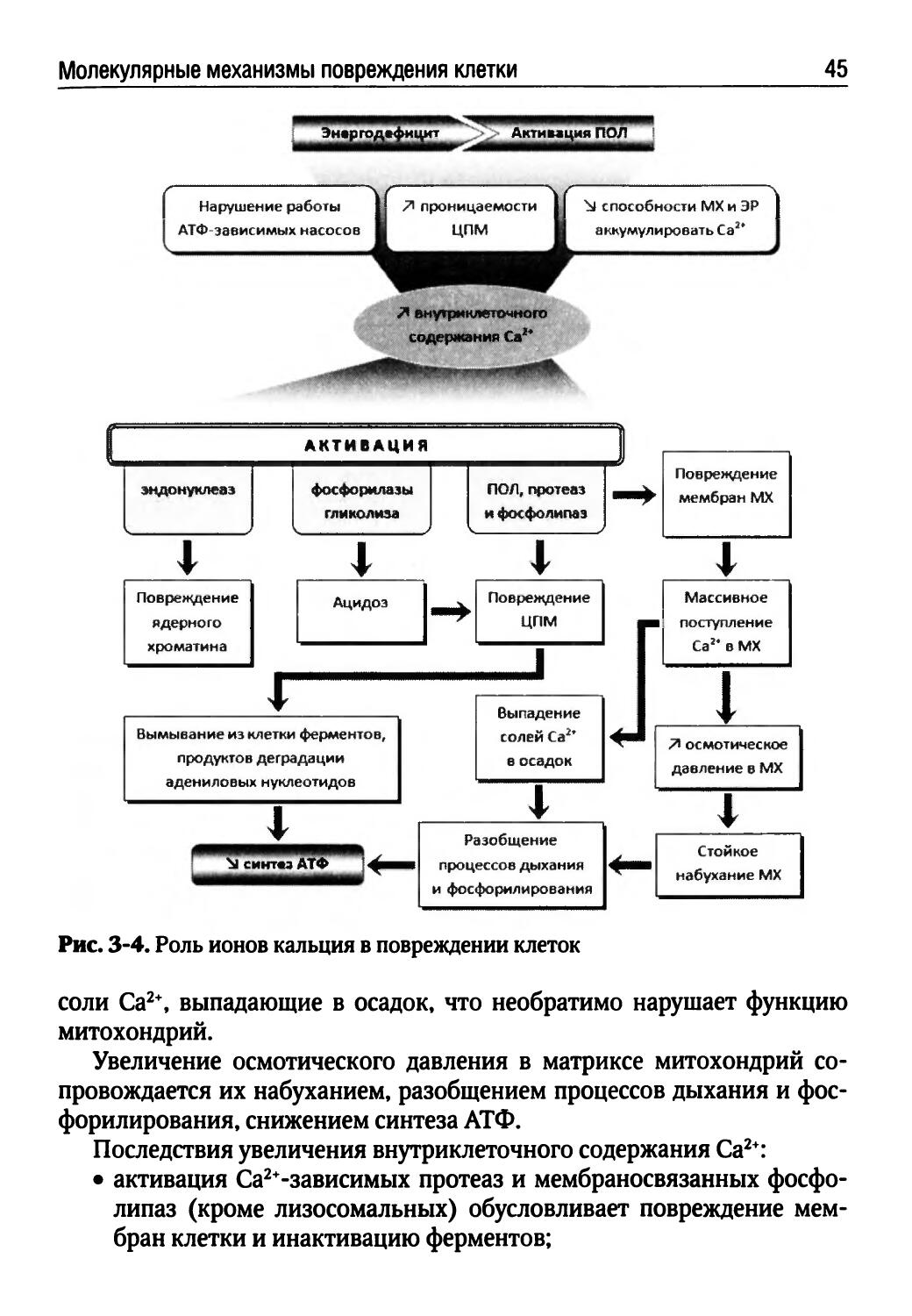
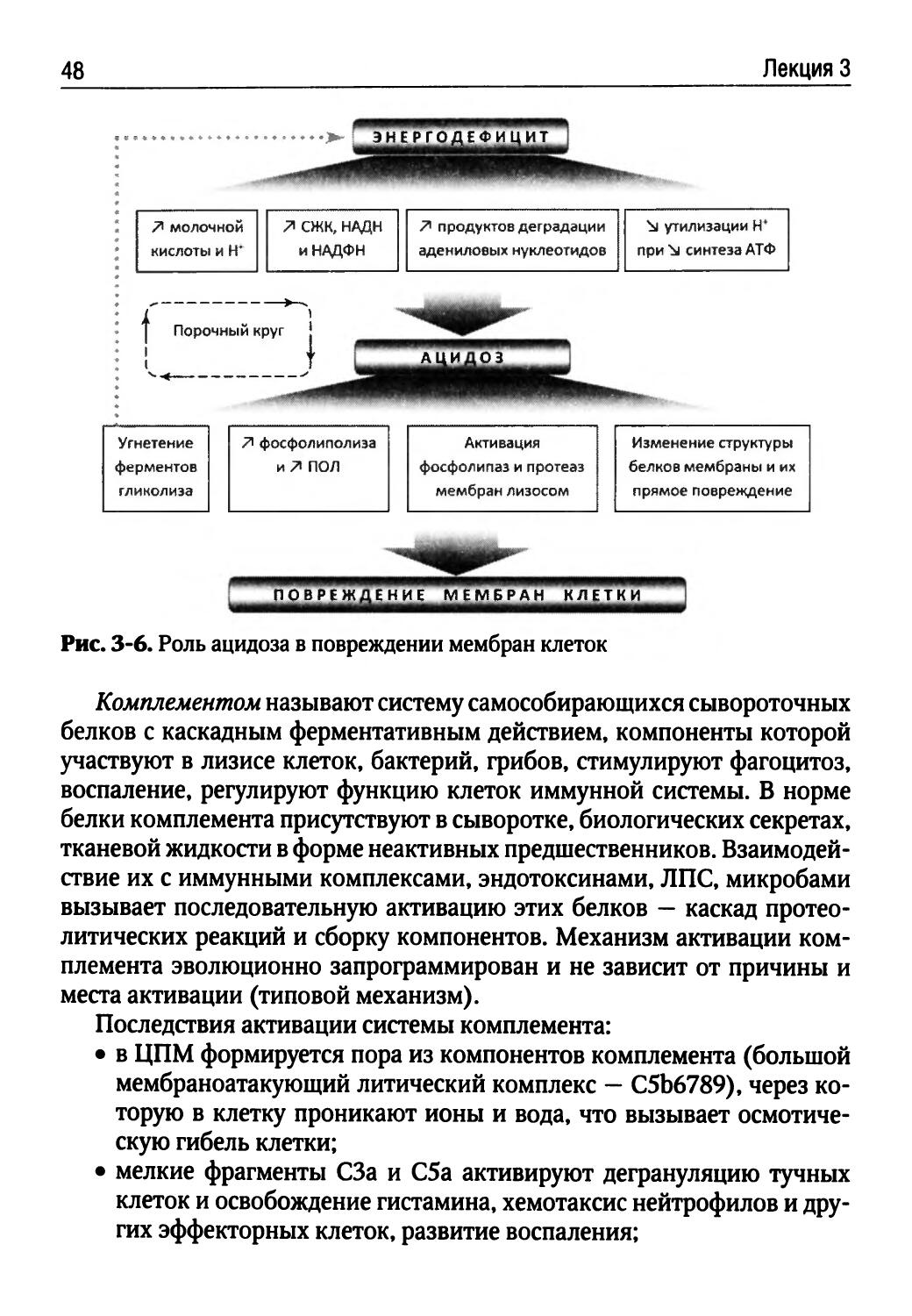
При нарушении структурной целостности биологических мембран нарушается гомеостаз клетки, и она повреждается. Эволюционно сформированы типовые механизмы повреждения биологических мембран: развитие энергодефицита; активация перекисного окисления липидов (ПОЛ), фосфолиполиза, липолиза и протеолиза; встраивание мицелл из амфифильных соединений в мембрану; внутриклеточный ацидоз; растяжение и микроразрывы мембран при набухании клеток и их органелл; активация системы комплемента и повреждение мембран макромолекулами и иммунными комплексами.
Типовые механизмы не зависят от вида патогенного агента, вызвавшего повреждение, и присущи всем клеткам. Они взаимно обусловливают друг друга. К важнейшим факторам повреждения клетки относят: увеличение внутриклеточного содержания Са2+, Na+, Н+ и Н2О; накопление недоокисленных продуктов метаболизма углеводов, белков и липидов; увеличение проницаемости ЦПМ и потерю клеткой пуриновых оснований и ферментов.
Развитие энергодефицита характерно для любого повреждения клетки, поскольку нарушаются гомеостаз, активность ферментов, целостность ЦПМ и мембран митохондрий. На дефицит О2 и субстратов
Молекулярные механизмы повреждения клетки 41
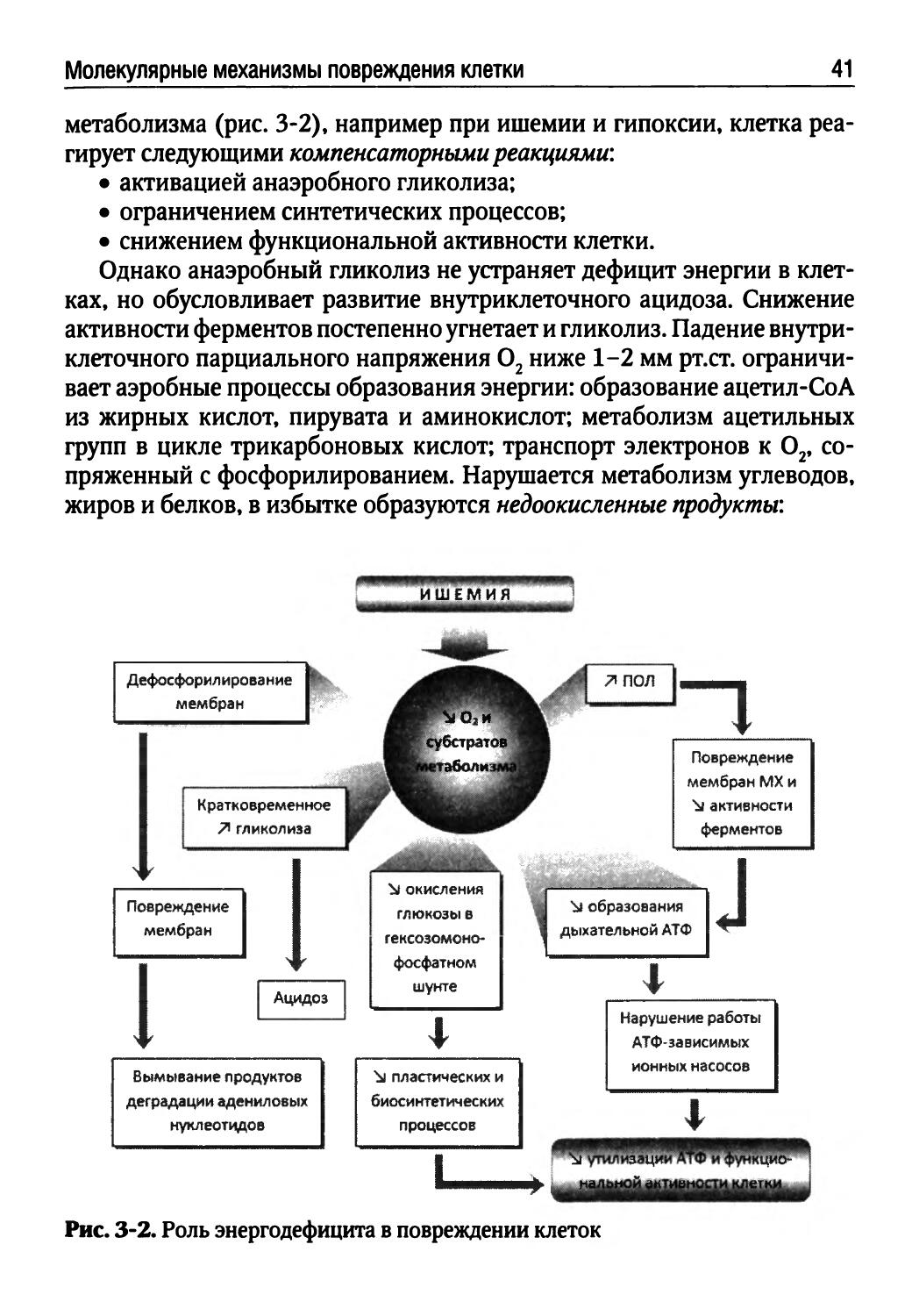
метаболизма (рис. 3-2), например при ишемии и гипоксии, клетка реагирует следующими компенсаторными реакциями:
• активацией анаэробного гликолиза;
• ограничением синтетических процессов;
• снижением функциональной активности клетки.
Однако анаэробный гликолиз не устраняет дефицит энергии в клетках, но обусловливает развитие внутриклеточного ацидоза. Снижение активности ферментов постепенно угнетает и гликолиз. Падение внутриклеточного парциального напряжения О2 ниже 1-2 мм рт.ст. ограничивает аэробные процессы образования энергии: образование ацетил-СоА из жирных кислот, пирувата и аминокислот; метаболизм ацетильных групп в цикле трикарбоновых кислот; транспорт электронов к О2, сопряженный с фосфорилированием. Нарушается метаболизм углеводов, жиров и белков, в избытке образуются недоокисленные продукты:
Повреждение мембран
Кратковременное Z гликолиза
Дефосфорилирование мембран
'Si окисления
глюкозы в
Ы образования дыхательной АТФ
Повреждение мембран MX и \ активности ферментов
гексозомоно-фосфатном шунте
Вымывание продуктов деградации адениловых нуклеотидов
Ацидоз
процессов
\ пластических и биосинтетических
Нарушение работы АТФ-зависимых ионных насосов
Рис. 3-2. Роль энергодефицита в повреждении клеток
42
Лекция 3
• СЖК, ацилкарнитин и ацил-СоА (СЖК, соединенная с СоА), которые, внедряясь в липидный компонент мембран, повреждают мембраны и ферменты, в том числе участвующие в синтезе АТФ, транспорте АТФ от места продукции к эффекторным структурам клеток и утилизации энергии АТФ;
• токсичные средне- и низкомолекулярные продукты деградации белков;
• продукты катаболизма адениловых нуклеотидов, которые, связываясь с Fe2*, усиливают его прооксидантную роль и активируют ПОЛ. При повреждении ЦПМ они, а также ферменты вымываются из клетки, что затрудняет ресинтез АТФ и способствует энергодефициту;
• лактат и пируват, продукты гидролиза АТФ, СЖК и их производные, НАДН, НАДФН, ФАДН обусловливают внутриклеточный ацидоз.
Как следствие, нарушается работа АТФ-зависимых ионных насосов, пластических процессов, восстанавливающих поврежденные мембраны, активность мембранных белков при их дефосфорилировании.
Дефицит энергии и снижение активности ферментов, в частности ци-тохромоксидазы, нарушает процесс полного четырехэлектронного восстановления О2 до Н2О. В месте повреждения накапливаются продукты его неполного восстановления — кислородные радикалы: при присоединении одного электрона образуется супероксидный анион-радикал; при присоединении двух электронов — перекись водорода, которая не является радикалом, но легко образует радикалы; при присоединении трех электронов — гидроксильный радикал. Свободные радикалы — это молекулярные частицы, имеющие неспаренный электрон на внешней орбитали и обладающие высокой реакционной способностью.
Кислородные радикалы инициируют перекисное окисление липидов — цепную реакцию последовательного окисления жирных кислот (ЖК) или их остатков в составе других липидов (рис. 3-3). Субстратами процессов ПОЛ в клеточных мембранах являются полиненасыщенные жирные кислоты (например, арахидоновая кислота). Кислородные радикалы отнимают водород из групп -СН2- полиненасыщенной жирной кислоты (ПНЖК), находящихся рядом с двойной связью. Это для них энергетически более выгодно, поскольку происходит делокализация неспаренного электрона между тремя атомами углерода. Образуется радикал ЖК, который легко присоединяет О2 и превращается в пероксидный радикал жирной кислоты. Этот радикал может отнимать водород от
Молекулярные механизмы повреждения клетки
43
- НО X
Разрыв связей
------------------1
[ — сн = сн-сн |сн2-] I;
O-J-OH
[-СН = СН-СН-СН;-] I 0-0-
Продолжение цепной реакции
[-СН = СН-СН =О] + [НО-СН,-]
Продукты окисления
Рис. 3-3. Механизм перекисного окисления липидов
другой молекулы жирной кислоты. Возникает цепная реакция. Пероксиды легко распадаются с образованием альдегидов путем разрыва в ЖК углерод-углеродной связи, соседствующей с пероксидной группой. Это энергетически более выгодно, поскольку альдегиды имеют систему сопряженных двойных связей (С=С, С=0).
Интенсивность ПОЛ регулируется соотношением факторов, активирующих (прооксиданты) и подавляющих (антиоксиданты) этот процесс.
К прооксидантам относятся витамин D, нафтохиноны, НАДФН, НАДН, липоевая кислота, низкие концентрации аскорбиновой кислоты, ионы металлов с переменной валентностью (железо, медь, марганец). Антиоксидантные факторы бывают двух видов — ферментные и неферментные. К неферментным факторам антиоксидантной защиты клеток относятся токоферол (витамин Е), витамин С, церулоплазмин, ретинолы, убихиноны, стероидные гормоны.
Ферменты антиоксидантного действия:
• супероксиддисмутаза, локализующаяся в цитоплазме, MX и ядре, одновалентно восстанавливает супероксидные радикалы до перекиси водорода;
• каталаза, преимущественно находящаяся внутри пероксисом, восстанавливает перекись водорода до воды;
44
Лекция 3
• глутатионпероксидаза, содержащая селен, восстанавливает перекись водорода до воды, а глутатионпероксидаза, не содержащая селен, разлагает гидроперекиси липидов нерадикальным способом.
При повреждении клеток активность прооксидантов усиливается, а активность антиоксидантной системы резко снижается. По-видимому, антиоксидантную систему повреждают продукты ПОЛ, ацидоз, липидные перекиси. Ионизирующая радиация и у-излучение ингибируют антиоксидантные ферменты.
Последствия активации ПОЛ:
• раннее увеличение пассивной проницаемости липидного слоя мембраны для ионов и воды из-за формирования сквозных каналов;
• увеличение в клетке содержания Na*, Са2* и Н2О, потеря клеткой К*;
• нарушение структурной целостности ЦПМ — изменение пространственной формы молекул, образование между ними ковалентных сшивок, нарушение свойств рецепторных белков и гликопротеинов (цАМФ-зависимой протеинкиназы), активности мембраносвязанных ферментов, липидзависимых белков, выход их из мембраны.
Повышение внутриклетогной концентрации Са2* — важнейший фактор повреждения клетки. В норме концентрация Са2* во внеклеточной среде примерно в 10 000 раз больше, чем внутри клетки. Через потен-циал-зависимые и лиганд-зависимые каналы ЦПМ Са2* входит в клетку, связывается с мембранами, белками-переносчиками и депонируется в митохондриях (Са2* является кофактором ферментов дыхательной цепи митохондрий) и эндоплазматическом ретикулуме. Са2* — внутриклеточный вторичный посредник: как положительно заряженный ион деполяризует мембрану и как химический агент влияет на работу ионных каналов, ферментов, рецепторов. При возбуждении клетки Са2* выходит из депо в цитоплазму, связывается с белками-переносчиками и участвует в фосфорилировании белков, в том числе насосов, удаляющих избыток Са2* из клетки.
При повреждении клетки увеличивается содержание Са2* в цитоплазме и матриксе митохондрий (рис. 3-4). Вначале это обусловлено дефицитом энергии, активацией ПОЛ, нарушением барьерной функции ЦПМ. При падении электрического трансмембранного потенциала снижается способность депо аккумулировать Са2*, его уровень в цитоплазме резко повышается. Активируются Са2*-зависимые протеазы и фосфолипазы мембран митохондрий, повышается проницаемость мембран для Са2*. При перенасыщении митохондрий Са2* образуются фосфорные
Молекулярные механизмы повреждения клетки
45
Нарушение работы АТФ-зависимых насосов
Л внутриклеточного содержания Са2*
Рис. 3-4. Роль ионов кальция в повреждении клеток
соли Са2+, выпадающие в осадок, что необратимо нарушает функцию митохондрий.
Увеличение осмотического давления в матриксе митохондрий сопровождается их набуханием, разобщением процессов дыхания и фосфорилирования, снижением синтеза АТФ.
Последствия увеличения внутриклеточного содержания Са2+:
• активация Са2*-зависимых протеаз и мембраносвязанных фосфолипаз (кроме лизосомальных) обусловливает повреждение мембран клетки и инактивацию ферментов;
46
Лекция 3
• увеличение образования кислородных радикалов, субстратов ПОЛ (ПНЖК), снижение активности антиоксидантной системы обусловливает интенсификацию ПОЛ;
• повреждение MX, разобщение процессов окисления и фосфорилирования уменьшает синтез АТФ;
• развитие мышечной контрактуры уменьшает запасы АТФ в клетке;
• повышение осмотического давления в клетке, набухание органелл и клетки, растяжение и микроразрывы необратимо повреждают мембраны;
• активация фосфорилазы гликолиза усиливает внутриклеточный ацидоз;
• активация эндонуклеаз повреждает ядерный хроматин.
Дефицит энергии, накопление продуктов деградации адениловых нуклеотидов и активация ПОЛ нарушают структурную организацию мембран, что облегчает взаимодействие фосфолипаз с фосфолипидами мембран. Фосфолипазы ЦПМ и внутриклеточных мембран (кроме лизосомальных) активируются при повышении внутриклеточной концентрации Са2\ но работают фосфолипазы только при закислении среды.
По своей форме молекулы фосфолипидов похожи на сплющенный цилиндр, */4 которого по длине приходится на гидрофильную головку, а 3/4 — на гидрофобные жирнокислотные цепи. Продукты гидролиза фосфолипидов и триацилглицеридов (лизофосфолипиды, СЖК, их не-доокисленные метаболиты и карнитинпроизводные) являются водорастворимыми амфифилами, которые тоже имеют в своей структуре гидрофильную и гидрофобную группы. Однако форма их молекулы напоминает конус, поскольку размер их гидрофильной головки в плоскости мембраны превышает размер гидрофобной жирнокислотной цепи. Это объясняется окислением жирнокислотных цепей фосфолипидов при активации ПОЛ или отщеплением от фосфолипидов под действием фосфолипаз продуктов гидролиза, например арахидоновой кислоты. Такие дефектные молекулы, собираясь вместе, образуют не бислой, а сферические мицеллы (рис. 3-5). В низкой концентрации амфифилы, встраиваясь в гидрофобный слой мембраны, стабилизируют ее и изменяют функции мембранных белков. В высокой концентрации амфифилы повреждают мембрану, образовывая мицеллы, выталкивая кальций, отдельные фосфолипиды и белки из мембраны. Повышается проницаемость мембраны, повреждаются функции белков (Ыа*/К+-АТФазы, Са2+-АТФазы и АТФ/АДФ-транслоказы).
Молекулярные механизмы повреждения клетки
47
Рис. 3-5. Образование мицелл при активации перекисного окисления липидов и фосфолиполиза
Несколько мицелл могут формировать пору, внутренняя часть которой образована гидрофильными головками. Повышается неселективная проницаемость ЦПМ и мембран органелл, увеличивается внутриклеточное содержание Na* и Са2*. Повышение осмотического давления в клетке обусловливает избыточное поступление в клетку воды из внеклеточного пространства. При растяжении и микроразрывах мембран нарушается барьерная функция мембран (осмотигеская гибель клетки). Фосфолипазы мембран лизосом активируются Н* (pH = 5,0-3,5). Нарушается структура мембран, повышается их проницаемость, ферменты лизосом выходят в цитозоль, развивается аутолиз клетки. Это происходит на стадии перехода обратимых изменений в необратимые или даже уже после гибели клетки.
Внутриклеточный ацидоз при повреждении (рис. 3-6) возникает вследствие дефицита энергии, гидролиза макроэргических соединений, увеличения внутриклеточного содержания Са2*, активации анаэробного гликолиза, накопления недоокисленных продуктов метаболизма жиров и углеводов, снижения утилизации Н* из-за уменьшения синтеза АТФ, восстановления НАД и НАДФ до НАДН и НАДФН.
Умеренный ацидоз (до pH = 7,2-6,8) оказывает защитный эффект, ингибируя фосфолипазы, увеличивая активность Са2*-АТФазы на фоне высокого содержания Са2* в клетке.
Выраженный ацидоз (pH < 6,8-5,7) повреждает мембраны клетки и внутриклеточные процессы, ингибируя ферменты гликолиза, повышая гидролитический эффект ряда фосфолипаз, активируя ПОЛ, изменяя конформационные свойства мембранных белков, угнетая ферменты (креатинкиназную систему, АТФазу миозина), активируя лизосомальные фосфолипазы и протеазы, прямо повреждая белки мембран.
48
Лекция 3
Изменение структуры белков мембраны и их прямое повреждение
Угнетение ферментов гликолиза
фосфолиполиза и Л ПОЛ
Активация фосфолипаз и протеаз мембран лизосом
ПОВРЕЖДЕНИЕ МЕМБРАН КЛЕТКИ
Рис. 3-6. Роль ацидоза в повреждении мембран клеток
Комплементом называют систему самособирающихся сывороточных белков с каскадным ферментативным действием, компоненты которой участвуют в лизисе клеток, бактерий, грибов, стимулируют фагоцитоз, воспаление, регулируют функцию клеток иммунной системы. В норме белки комплемента присутствуют в сыворотке, биологических секретах, тканевой жидкости в форме неактивных предшественников. Взаимодействие их с иммунными комплексами, эндотоксинами, ЛПС, микробами вызывает последовательную активацию этих белков — каскад протеолитических реакций и сборку компонентов. Механизм активации комплемента эволюционно запрограммирован и не зависит от причины и места активации (типовой механизм).
Последствия активации системы комплемента:
• в ЦПМ формируется пора из компонентов комплемента (большой мембраноатакующий литический комплекс — С5Ь6789), через которую в клетку проникают ионы и вода, что вызывает осмотическую гибель клетки;
• мелкие фрагменты СЗа и С5а активируют дегрануляцию тучных клеток и освобождение гистамина, хемотаксис нейтрофилов и других эффекторных клеток, развитие воспаления;
Молекулярные механизмы повреждения клетки
49
• фрагменты СЗЬ и С4Ь (опсонины) усиливают фагоцитоз и экзоци-тоз гранул нейтрофилов.
Когда нарушения гомеостаза поврежденной клетки достигают критического уровня, наступает ее гибель. Существует два механизма гибели клетки — апоптоз и некроз.
Апоптоз (программированное самоуничтожение клетки) реализуется в норме и патологии, обусловливает гибель и удаление единичных клеток, продуцирующихся в избытке, неправильно развивающихся или имеющих поврежденную дезоксирибонуклеиновую кислоту (ДНК). Это активный процесс, требующий затрат энергии и сопровождающийся синтезом специфических белков. Апоптоз инициируется активацией внутриклеточной суицидной программы, рецепторов внеклеточными факторами (фактором некроза опухолей, интерлейкинами-1 и 10, глюкокортикоидами, у-интерфероном), свободными радикалами, этанолом.
Последовательность событий при апоптозе:
• сигналы от рецепторов — ядро — активация летальных генов (интеграция информации, запуск и регулирование программы апоптоза);
• экспрессия генов, контролирующих внутриклеточную концентрацию Ca2t, которая модулирует активность ядерных эндонуклеаз;
• распад ДНК на нуклеосомальные фрагменты (по величине кратные 185-200 парам оснований);
• упорядоченная активация протеолитических процессов в клетке.
Морфологические изменения клетки при апоптозе:
• сморщивание и фрагментация клетки на апоптозные тельца, содержащие фрагменты ядра и органеллы и ограниченные мембраной;
• фагоцитоз апоптозных телец соседними клетками или макрофагами;
• сближение окружающих клеток без изменений архитектоники ткани, сохранение целостности ЦПМ;
• содержимое клетки не попадает в окружающую ткань и не образуется воспалительный очаг.
Некроз возникает при грубом химическом или физическом воздействии на клетки (ишемии, гипотермии, гипертермии, бактериальной и вирусной инфекции и др.). Повреждение обычно затрагивает множество клеток, и образуются некротические зоны. Поврежденные клетки набухают, нарушается целостность их мембран, содержимое клеток попадает в окружающее тканевое пространство и вызывает воспалительную реакцию. Некроз — пассивный неуправляемый распад клетки, заканчивающийся ее аутолизом. Ткань очищается от клеточных остатков и репарирует.
Лекция 4
ВОСПАЛЕНИЕ I (СОСУДИСТЫЕ РЕАКЦИИ)
НЛ. Богуш
Воспаление — типовой патологический процесс, развивающийся в ответ на повреждение и заключающийся в реакциях микроциркулятор-ных сосудов, нервной системы, крови и соединительной ткани. В процессе воспаления в тканях последовательно развиваются альтерация, экссудация и пролиферация. Целью воспаления являются отграничение повреждающего агента от здоровых тканей, уничтожение повреждающего агента и создание условий для восстановления поврежденной ткани.
Воспаление — местная реакция. В месте повреждения возникает боль, развиваются покраснение, припухлость, кожа становится горячей на ощупь, функция воспаленного органа нарушается.
Древние греки называли воспалениеphlogosis, а римляне — inflammatio (воспламенение, поджег). 4 классических признака воспаления — «rubor, tumor cum calore etdolore» описаны Цельсом 2000 лет назад, 5-й признак — «functio laesa» добавлен Галеном (129-200 год н.э.).
Причиной воспаления может стать любой флогогенный агент: механический, химический, физический, биологический, даже психический (в эксперименте можно вызвать воспалительную гиперемию путем внушения). Воспаление вызывают как экзогенные, так и эндогенные факторы (например, камень при мочекаменной или желчнокаменной болезни).
Патогенез воспаления и его симптомов
Флогогенный агент вызывает первичную альтерацию — раздражение и повреждение тканей. Одной из первых реакций организма является чувство боли. Оно характерно для острого повреждения, вызванного механическими, химическими или термическими факторами. Однако даже если повреждающий фактор перестал действовать (например, палец отдернули от горячей поверхности), то боль может продолжаться
Воспаление I (сосудистые реакции)
51
все то время, пока не заживет рана. Следовательно, в ходе самого воспаления появляются факторы, поддерживающие болевую реакцию.
Боль — это результат возбуждения болевых рецепторов (ноцицепто-ров). Факторы, которые могут раздражать ноцицепторы:
• в ряде случаев — сам повреждающий агент;
• повышение осмотического давления в очаге воспаления, вызванное усиленным распадом тканей и изменением обмена веществ (известно, что введение растворов с повышенным осмотическим давлением под кожу или внутрикожно вызывает боль, как и попадание соли на царапину);
• повышение концентрации ионов калия и коэффициента К/Са2' — результат повреждения и распада клеток;
• повышение концентрации ионов водорода — результат нарушения обмена веществ (введение под кожу растворов калия или буферных растворов с pH ниже 7,2 вызывает боль);
• механическое растяжение тканей отечной жидкостью;
• действие медиаторов воспаления (следует отметить роль простагландина Е2, который участвует в болевой реакции, повышая чувствительность ноцицепторов к раздражителям);
• накопление метаболитов: молочной кислоты, АДФ, аденозина и др.
Очень быстро в зоне повреждения появляется покраснение — развиваются сосудистые воспалительные реакции. Основная заслуга в открытии и описании их принадлежит Ю. Конгейму (1867). Сосудистые реакции протекают в следующей последовательности:
• кратковременный спазм микрососудов -»артериальная гиперемия
-> венозная гиперемия -> стаз.
Одновременно во всем очаге воспаления мы можем найти все 3 последних вида сосудистых реакций.
Начинаются сосудистые реакции с кратковременного сосудистого спазма. Он наблюдается не всегда, а только при остром механическом, термическом, химическом, лазерном повреждении и длится от нескольких секунд до 5 мин. Повреждающий агент раздражает гладкомышечные клетки сосудистой стенки, сосудодвигательные окончания, выделяющие норадреналин, эндотелиоциты, синтезирующие эндотелии. В ответ происходит спазм артериол. Однако норадреналин быстро разрушается, а поврежденные гладкомышечные клетки теряют способность к сокращению — спазм сменяется расслаблением, развивается артериальная гиперемия.
52
Лекция 4
Патогенез ее сложен, это не просто пассивное расслабление. Вспомним, что в основе изменения микроциркуляции при этом типе расстройств лежит увеличение притока артериальной крови. Следовательно, необходимо расширить приводящие артериолы (увеличение притока) и расслабить прекапиллярные сфинктеры, чтобы заполнить капиллярную сеть. Этому способствует несколько факторов.
• Развивается аксон-рефлекс, расширяющий артериолы (рис. 4-1). Начинается он с возбуждения ноцицепторов различными агентами очага повреждения (см. выше). Потенциалы действия распространяются по дендриту к нейронам спинального ганглия, а также по ответвлениям — в обратном направлении, к месту повреждения. Там их окончания, являющиеся пресинаптическими, контактируют непосредственно со стенкой артериолы или с тучной клеткой. Выделяются нейромедиаторы — вещество Р, нейрокинин А, нейропептид Y, которые вызывают расслабление гладких мышц артериол и дегрануляцию тучных клеток с высвобождением медиаторов, в первую очередь гистамина. Предполагают, что и нейромедиаторы, и гистамин вызывают расслабление гладких мышц опосредованно, через оксид азота (NO), синтезируемый эндотелиоцитами.
• Начинается действие медиаторов сначала преформированных, а затем и вновь синтезированных. К медиаторам, расслабляющим артериолы, относятся гистамин, серотонин, брадикинин, простагландины Е2, D2,12, лейкотриены С4, D4, Е4. Продукты активации комплемента С3а, С4а и С5а — анафилатоксины также участвуют в
Рис. 4-1. Аксон-рефлекс при воспалении
Воспаление I (сосудистые реакции)
53
расширении сосудов, способствуя высвобождению гистамина из тучных клеток и серотонина из тромбоцитов.
• Накопление в очаге воспаления сосудорасширяющих и расслабляющих сфинктеры метаболитов и ионов: СО2, Н*. К\ продуктов деградации АТФ, аденозина, молочной кислоты.
• Развивается необратимое повреждение вазомоторных окончаний и гладких мышц микрососудов.
Следовательно, артериальная гиперемия при воспалении развивается по всем 3-м механизмам: нейротоническому (аксон-рефлекс), нейропаралитическому и миопаралитическому.
При воспалительной артериальной гиперемии в первую очередь расширяются артериолы (почти в 2 раза), венулы и затем капилляры. Капилляры расширяются особенно сильно по сравнению с другими вариантами артериальных гиперемий, причем как активные, функционирующие, так и пассивные, плазменные или закрытые, и их расширение носит диффузный тотальный характер. Капилляры находятся в межклеточной среде, в норме представляющей гель. Окружающая среда обеспечивает 99 % жесткости капиллярной стенки. Флогогенные факторы и экссудация увеличивают растяжимость соединительной ткани, что приводит к снижению упругости капиллярной стенки. В дальнейшем под действием протеаз разрушение соединительного каркаса продолжается. В таких условиях усиленный приток крови при открытых сфинктерах растягивает капилляры максимально. В результате паралича гладких мышц исчезает «игра» сосудов (спонтанное чередование спазма и расширения), нет сокращения в ответ на воздействие катехоламинов. В расширенных сосудах возрастает гидродинамическое давление, например, в артериолах диаметром до 100 мкм — на 35 см водного столба, в капиллярах — на 7, в венулах — на 25 см, гидродинамическое давление начинает превышать онкотическое, что способствует экссудации. Значительно возрастает объемная скорость кровотока (Q), а вот линейная (V) может даже уменьшаться, учитывая площадь (S) расширенных сосудов (согласно формуле V = Q/S). Адекватно возрастает лимфоотток. В ткани повышается парциальное давление кислорода, увеличивается приток питательных веществ, естественных антител.
Артериальная гиперемия, таким образом, становится барьером между поврежденными тканями и здоровыми, повышая их защитный потенциал. Внешне артериальная гиперемия обусловливает появление красноты (увеличение количества и диаметра функционирующих сосудов, увеличение оксигемоглобина в венозной крови) и локальное
54
Лекция 4
повышение температуры (увеличенный приток теплой артериальной крови). Однако дополнительное тепло усиленно образуется в самих воспаленных тканях из-за бурных обменных процессов, что образно названо «пожаром обмена веществ». Наиболее чувствительна к повышению температуры кожа, в других органах разница температур не улавливается.
Через 30-60 мин кровоток в эпицентре начинает решительно замедляться, развивается венозная гиперемия. В основе ее лежит нарушение оттока, следовательно, уже в стадию артериальной гиперемии появляются факторы, постепенно все более и более затрудняющие отток. Их можно разделить на внутрисосудистые, суживающие венулы и мелкие вены изнутри, и внесосудистые.
Внутрисосудистые факторы
Просвет венул становится меньше, так как изнутри к эндотелиоци-там начинают прикрепляться лейкоциты (развивается краевое стояние лейкоцитов), сами эндотелиоциты набухают и выступают в просвет сосудов, появляются «пробочки» из агрегатов эритроцитов и нитей фибрина. Нарушению оттока способствует также сгущение крови из-за выхода жидкой части крови из сосудов в ткань (экссудации).
Внесосудистые факторы
Вначале венулы могут суживаться под действием серотонина тромбоцитов. Затем с развитием экссудации венулы и мелкие вены сдавливаются механически экссудатом. Имеет значение и разрушение соединительнотканного поддерживающего каркаса вен и десмосом протеазами.
Ю. Конгейм утверждал, что без сосудов нет воспаления. Действительно, замедление тока крови способствует дальнейшему развитию воспаления. Без замедления кровотока и ухудшения оттока невозможно было бы на коротком участке сконцентрировать медиаторы и стимуляторы воспаления, прокоагулянты и проагреганты, направить поток жидкости из сосудов в ткани, обеспечить эмиграцию лейкоцитов в очаг воспаления.
Венозная гиперемия сменяется стазом. Ему предшествуют особые характерные изменения кровотока: сначала толчкообразное, а потом маятникообразное движение крови по сосудам, или престаз. Эти изменения обусловлены продолжающимся затруднением оттока
Воспаление I (сосудистые реакции)
55
и возрастанием гидростатического давления (ГД) на венозном конце микрососудов, с одной стороны, и параличом артериол — с другой стороны. Паралич артериол приводит к тому, что в капиллярах появляется пульсовая волна, она больше не гасится паралитически расширенными артериолами (субъективно появляется дергающая, пульсирующая боль). Возрастание ГД на венозном конце до диастолического рождает толчкообразное движение крови, кровь движется от артериолы к венуле только в систолу. При превышении ГД над диастолическим давлением появляется маятникообразное движение крови, кровь движется в систолу от артериолы к венуле, а в диастолу — от венулы к артериоле. Когда ГД на венозном конце становится равным ГД на артериальном конце, движение крови прекращается, возникает стаз. Все факторы, вызывающие стаз, сформировались в ходе предыдущих сосудистых реакций. В основе истинного капиллярного стаза, который наблюдается при воспалении, лежит значительное ухудшение реологии. Факторы, ухудшающие реологию:
• сгущение крови в результате экссудации;
• увеличение вязкости крови из-за снижения скорости кровотока, вызванного нарушением оттока;
• повреждение эритроцитов в очаге воспаления, снижение их дефор-мабильности, образование ими «монетных столбиков» — агрегатов, препятствующих движению крови.
При стазе создаются условия для образования тромбов — это и приближение тромбоцитов к стенке поврежденного сосуда, и несмываемость током крови прокоагулянтов и проагрегантов, отсутствие притока антикоагулянтов, и повреждение эндотелиоцитов движениями тока крови в престаз. Появление тромбов в венуле — причина развития дополнительно к истинному стазу застойного стаза.
С течением времени вначале обратимый стаз превращается в необратимый. Характерно появление сладж-феномена (от англ, sludge — тина, ил, грязь). Эритроциты слипаются в комки, опутанные нитями фибрина, теряют границы между собой, гемолизируются. Стаз развивается в течение нескольких часов, однако при очень тяжелом повреждении стаз может развиться почти мгновенно, без предварительного престатиче-ского замедления кровотока.
Итак, при воспалении в эксперименте, например, через 24 ч после ожога, вокруг центральной зоны, состоящей из некротической ткани, находилась зона стаза и кровоизлияния; вокруг нее располагалась зона выраженной вазодилатации, гиперемии и клеток экссудата; наружная
56
Лекция 4
зона была нормальной, но сосуды расширены. Воспалительная гиперемия ограничена очагом поражения. Однако гиперемический центр (в котором обычно имеется припухлость, т.е. отек) окружен красной каймой или ярким ободком. Льюис (1927) показал, что механизм, обусловливающий наличие ободка, по-видимому, вызван аксон-рефлексом.
В патогенезе воспаления большая роль принадлежит медиаторам воспаления, в этом смысле их даже называют дирижерами воспаления.
Медиаторы — гуморальные эндогенные вещества, чей уровень возрастает в очаге воспаления в сочетании с возникновением по крайней мере одной тканевой реакции или структурного изменения. Медиаторы действуют в пределах очага воспаления, поддерживая и развивая там воспалительный процесс.
Источники медиаторов воспаления разнообразны — это и жидкостные среды (плазма, межклеточная жидкость), и клетки (крови и тканей), и внеклеточные элементы соединительной ткани.
По химической природе медиаторы также разнообразны и являются моноаминами, белками, пептидами, нуклеозидами и нуклеотидами, протеогликанами, липидами.
Все медиаторы можно разделить на 2 группы.
I. Преформированные, т.е. уже синтезированные, готовые немедленно вступить в реакцию и хранящиеся в гранулах в клетках. Такие клетки отвечают на повреждение или стимуляторы дегрануляцией — секрецией гранул с готовыми медиаторами воспаления.
II. Вновь образующиеся медиаторы. Они синтезируются в ответ на повреждение и вступают в реакции воспаления с некоторым запозданием, так как на их синтез необходимо время.
Преформированные медиаторы (клеточного происхождения)
1. Гистамин (моноамин) образуется при декарбоксилировании гистидина, содержится у человека главным образом в гранулах тучных клеток и базофильных лейкоцитах крови. В гранулах, окруженных мембраной, гистамин связан ионной связью с кислыми группами гепарина (тучные клетки) или хондроитин-сульфата (базофилы). После выхода из клетки вытесняется из связи ионами Na*.
Дегрануляция возникает в результате повреждения тучной клетки внешними факторами (механическими, температурными), в результате воздействия на тучную клетку анафилатоксинов (фрагментов комплемента С3а, С5а), нейропептидов (вещества Р), АТФ, комплекса антиген-антитело при аллергии (АТ-иммуноглобулины IgE-класса) и многочис
Воспаление I (сосудистые реакции)
57
ленных факторов, высвобождаемых клетками при воспалении (нейтрофилами, эндотелиоцитами, тромбоцитами и др.). Большая часть гистамина инактивируется N-метилтрансферазой, остальная — диаминоксидазой, т.е. гистаминазой (содержится, в частности, в эозинофилах).
Гистамин действует на клетки-мишени, имеющие к нему рецепторы. Последние делятся на 2 типа: Н,- и Н2-. При действии гистамина на клетки с ^-рецепторами он вызывает:
• расслабление гладких мышц артериол — расширение артериол:
• сокращение эндотелиоцитов, в первую очередь посткапиллярных венул, — увеличение сосудистой проницаемости;
• раздражение ноцицепторов — зуд, боль;
• сокращение гладких мышц бронхиол и ЖКТ;
• раздражение чувствительных окончаний блуждающего нерва в воздухоносных путях;
• стимуляцию образования простагландинов.
При действии на клетки с Н2-рецепторами гистамин увеличивает:
• секрецию слизи эпителием воздухоносных путей;
• содержание цАМФ в клетках-мишенях;
• секрецию соляной кислоты в желудке.
Следовательно, гистамин принимает участие в развитии всех признаков воспаления и так как он преформированный, то появляется в очаге воспаления почти одновременно с повреждением, можно сказать, запускает острый воспалительный ответ.
2. Факторы хемотаксиса для эозинофилов (ФХЭ-А) и нейтрофилов (ФХН-А) также содержатся в гранулах тучных клеток. Стимулируют хемотаксис эозинофилов и нейтрофилов.
3. Протеазы — триптаза и химаза; разрушают базальную мембрану кровеносных сосудов и увеличивают сосудистую проницаемость, расщепляют протеогликаны соединительной ткани и способствуют эмиграции лейкоцитов, способствуют заживлению очага повреждения, расщепляя продукты распада тканей и активируя факторы роста.
4. Серотонин содержится в гранулах тромбоцитов, эозинофилов, энтерохромаффинных клетках. Вызывает расширение артериол, спазм венул, повышение проницаемости, агрегацию тромбоцитов, бронхоспазм.
Вновь образующиеся медиаторы
Они могут быть клеточными, происходящими из фосфолипидов клеточных мембран (ФАТ, простагландины и лейкотриены), или син
58
Лекция 4
тезированными пептидами (цитокины ИЛ-1, ФНО-а , ИЛ-6), а также плазменными (кинины, фрагменты комплемента).
Вновь образующиеся клеточные медиаторы
1. Простагландины. Относятся к эйкозаноидам — производным двадцати-(эйкоза-)углеродных полиненасыщенных жирных кислот. При повреждении клетки активируется фосфолипаза А2, которая высвобождает из фосфолипидов мембраны арахидоновую кислоту (содержит 20 углеродных атомов и 4 двойные связи). Под действием циклооксигеназы (ЦОГ) и О2 арахидоновая кислота окисляется до промежуточного продукта простагландина G2, который превращается в большинстве клеток в простагландины (ПГ) Е2 или простагландины F2a, в тучных клетках — в простагландины D2 (рис. 4-2).
• Эффекты ПГЕ2: расширяет микрососуды, увеличивает их проницаемость, повышает чувствительность (сенситизация) окончаний ноцицепторов, расширяет бронхи, стимулирует секрецию синовиальной жидкости в суставах и протеолиз в мышцах, стимулирует дегрануляцию тучных клеток, подавляет функцию лимфоцитов и гранулоцитов.
• Эффекты ПГО2: как и ПГЕ2 расширяет микрососуды и повышает их проницаемость, стимулирует дегрануляцию тучных клеток, но в отличие от него вызывает спазм бронхов.
• Эффекты ПГР2а: спазм микрососудов, подавление эмиграции лейкоцитов и бронхоспазм.
Хспюкокортикоиды
Фосфолипиды
фосфолипаза А,
Липоксигеназа Циклоксигеназа
I I аспирин
ЛТ „ ♦ „
А., В., С., D„ Е, + ПГЕ’ D’ +
МРС-А простациклин- тромбоксан-
синтетаза синтетаза
(ЭЦ) 4 ЛГУ, (простациклин)
(Тр)
ТД
Рис. 4-2. Образование эйкозаноидов из мембраны клетки
Воспаление I (сосудистые реакции)
59
2. Тромбоксаны. Образуются из ПГН2 в тромбоцитах под влиянием тромбоксансинтетазы (см. рис. 4-2). Вызывает спазм сосудов, бронхов, хемотаксис и адгезию нейтрофилов, адгезию, агрегацию и дегрануляцию тромбоцитов.
3. Простациклин ПП2. Образуется из ПГН2 в эндотелиоцитах под влиянием простациклинсинтетазы (см. рис. 4-2). Вызывает расширение сосудов и бронхов, оказывает антиадгезивное и антикоагуляционное действие, является антиагрегантом, стимулирует коллатеральный кровоток и фибринолиз.
4. Лейкотриены. Образуются из арахидоновой кислоты, но при действии фермента 5-липоксигеназы (ЛОГ), который содержится в гранулоцитах, макрофагах, моноцитах и в тучных клетках. Промежуточным продуктом является нестабильный ЛТА„, который быстро трансформируется либо в ЛТВ4, либо в ЛТС4 (см. рис. 4-2). Последний последовательно превращается в JITD4 и затем в ЛТЕ4. Смесь ЛТС4, JITD4 и ЛТЕ4 определяли раньше как медленно реагирующую субстанцию анафилаксии (МРС-А).
ЛТВ4 — мощный хемоаттрактант для нейтрофилов и эозинофилов. ЛТС4, JITD4 и ЛТЕ4 увеличивают сосудистую проницаемость, поддерживают гиперемию при воспалении, способствуя высвобождению вазодилататоров эндотелиоцитами. Они вызывают сокращение гладкой мускулатуры бронхов и ЖКТ. Лейкотриены играют большую роль в патологии легких, особенно при бронхиальной астме. Под их влиянием в легких возникают бронхоспазм, усиление секреции слизи бронхиальным эпителием, спазм легочных сосудов.
5. ФАТ. Из фосфолипидов клеточных мембран образуется еще один важный медиатор — ФАТ. Его образуют и секретируют активированные нейтрофилы, эозинофилы, базофилы, моноциты, макрофаги, тучные клетки, эндотелиоциты и др. Синтез начинается с гидролиза мембранных фосфолипидов фосфолипазой А2, затем получившийся неактивный лизо-ФАТ ацетилируется ацетилтрансферазой до активного ФАТ, который является смесью 1-алкил-2-ацетил-глицеро-3-фосфохолинов. ФАТ вызывает активацию и дегрануляцию тромбоцитов, гранулоцитов, увеличивает проницаемость микрососудов (в 10 000 раз активнее гистамина), способствует продукции активных кислородных радикалов и эйкозаноидов, стимулирует эмиграцию нейтрофилов и базофилов, сокращает гладкую мускулатуру бронхов и сосудов (вызывает спазм коронарных и легочных сосудов), вызывает гиперемию и отек кожи.
60
Лекция 4
6. Интерлейкин-1 (ИЛ-1). Пептид, к синтезу которого способны все ядросодержащие клетки. При воспалении синтез основных количеств ИЛ-1 происходит в макрофагах/моноцитах, нейтрофилах, эндотелио-цитах. ИЛ-1 местно вызывает изменения сосудистой стенки, способствующей адгезии лейкоцитов, стимулирует синтез ПГЕ2, ПП2 и ФАТ, усиливает прокоагулянтную активность эндотелия и одновременно вызывает усиление синтеза активатора плазминогена.
7. Фактор некроза опухолей а (ФНО-а). Полипептид. При воспалении продуцируется макрофагами/моноцитами и лимфоцитами. Местно способствует адгезии лейкоцитов к эндотелию, стимулирует цитолитическую активность Т-киллеров, вызывает пролиферацию фибробластов и стимулирует синтез ими коллагена.
8. Интерлейкин-6 (ИЛ-6). Пептид. Его синтезируют активированные макрофаги/моноциты, эндотелиоциты, фибробласты. ИЛ-6 способствует синтезу антител и увеличивает активность Т-клеток.
Местные эффекты ИЛ-1, ФНО-а и ИЛ-6 связаны с их низкими концентрациями; при значительном повреждении тканей количество цитокинов увеличивается, они распространяются вместе с кровью по организму, оказывая системное действие — ответ острой фазы (ООФ).
Вновь образующиеся плазменные медиаторы
1. Кинины (т.е. «действующие»). Это система пептидных медиаторов, активируемая после контакта фактора Хагемана с отрицательно заряженными поверхностями (контактная активация). Такими «контактными» активаторами могут быть кристаллы мочевой кислоты, коллаген, хрящ, липополисахариды бактерий, кристаллы фосфатов кальция, ДНК, стекло и др. После контакта от фактора Хагемана отщепляется короткий пептид ХПа, который активирует путем протеолиза прекалликреин (рис. 4-3). Неактивный прекалликреин превращается, таким образом, в активный фермент калликреин. Последний расщепляет плазменный высокомолекулярный кининоген а2-гликопротеид с образованием главного кинина крови — брадикинина (состоит из 9 аминокислот). Из тканевого предшественника под действием калликреинов почек, поджелудочной, слюнной и других желез в тканевой жидкости образуется другой кинин — каллидин (лизил-брадикинин, состоит из 10 аминокислот). В норме кинины служат медиаторами рабочей гиперемии, при воспалении количество их значительно возрастает. Они расширяют микрососуды, увеличивают сосудистую проницаемость, раздражают ноцицепторы, вызывают спазм гладких мышц бронхов и ЖКТ,
Воспаление I (сосудистые реакции)
61
Фактор Хагемана (XII) Иммунные комплексы токсины — яды
Повреждение ткани энзимы лизосом
Плазминоген
Плазмин
Xia
Фибрин
Прекалликреин
Калликреин *
Ф. ХПа и его фрагменты
Кининогены
(^-инактиватор с^-Антитрипсин а2-Макроглобулин Антитромбин
Кинины
1. Брадикинин — 9 амк.-т. ост.
2. Каллидин —10 амк.-т. ост.
3. Метил-лизилбрадикинин — 11 амк.-т. ост.
Рис. 4-3. Активация кининовой системы при воспалении
дегрануляцию тучных клеток, стимулируют циклооксигеназу в различных клетках. Калликреин — хемоаттрактант для лейкоцитов. При системном действии кининов возникает гипотензия, усиливается диурез и стимулируется сердечная деятельность.
Кинины входят в контактную систему плазмы крови — сторожевую систему. Компонентами этой системы являются плазменные протеазы, присущие свертывающей, фибринолитической, кининовой системам и комплементу. Эти системы функцинально едины, работают по каскадному принципу, способны взаимно активировать друг друга. Ядром сторожевой полисистемы являются 4 белка:
• фактор Хагемана (контактный фактор, XII фактор свертывания крови);
• XI фактор свертывания крови;
• высокомолекулярный кининоген;
• плазменный прекалликреин.
Все они присутствуют в плазме крови в неактивной форме, но при повреждении активируются и вступают в действие в результате каскадного протеолиза.
2. Система комплемента. Это 9 белковых фракций комплемента (компоненты комплемента от Сх до С9), факторы В, D, Р и некоторые другие белки, находящиеся в плазме крови и жидкостях тела в неактивной форме. При повреждении они самособираются, активируют друг
62
Лекция 4
друга в результате ограниченного протеолиза и выступают медиаторами воспаления.
Комплемент может быть активирован 3-мя путями: классическим, альтернативным и маннозным или лектиновым.
Классигеский путь является специфическим, так как активация компонента С\ начинается под воздействием комплекса АГ-АТ (АТ — иммуноглобулины класса G или М).
Альтернативный или неспецифигеский путь активации комплемента возникает в ответ на липополисахариды бактерий, грибки, гельминты, некоторые вирусы, гетерологичные эритроциты, продукты некроза.
Маннозный путь (неспецифический) приводит к активации комплемента после контакта с бактериальными маннозилами маннансвязы-вающего лектина плазмы и лейкоцитов и активации им сывороточных протеаз. Промежуточным общим продуктом всех путей является образование С3-конвертазы: С4Ь2а — при классическом и маннозном пути, СЗЬВ — при альтернативном пути. Большие продукты протеолиза обозначаются буквой «Ь», малые — «а».
СЗЬ фиксируется на поверхности атакуемой клетки, и в результате последующих каскадных активаций комплемента в мембране клетки формируется пора или трансмембранный канал. Этот канал составляют компоненты комплемента С5Ь6789, или мембраноатакующий, мембранолизирующий комплекс комплемента. Через канал в клетку поступают NaCl и вода, в результате чего клетка лизируется. Помимо этого фрагменты комплемента выполняют функцию медиаторов воспаления. Это компоненты С2а, СЗа, СЗЬ, С4а, С5а и др.
Образующиеся в ходе активации комплемента фрагменты СЗа, С4а и С5а называют анафилатоксинами. Самый сильный из них — С5а, самый слабый — С4а. Анафилатоксины высвобождают медиаторы из тучных клеток и базофилов, повышают проницаемость микрососудов, участвуют в формировании боли, являются хемоаттрактантами для нейтрофилов, эозинофилов, базофилов, макрофагов, стимулируют липоксигеназу фагоцитов, лейкоцитарную адгезию, вызывают спазм гладких мышц, увеличивают продукцию ИЛ-1 фагоцитами и образование ФАТ.
С2а обладает кининоподобной активностью, увеличивает сосудистую проницаемость, сокращает гладкие мышцы. СЗЬ и С4Ь — опсонины. С5а — хемоаттрактант для гранулоцитов и моноцитов/ макрофагов.
Лекция 5
ВОСПАЛЕНИЕ II (КЛЕТОЧНЫЕ РЕАКЦИИ)
НЛ. Богуш
Сосудистые реакции создают условия для осуществления экссудации. Экссудация — это выход жидкой части крови из сосудов в очаг воспаления. Наиболее тонкая стенка, являющаяся барьером между кровью и окружающей тканью, состоит из одного слоя эндотелиоци-тов, лежащих на базальной мембране. Именно она может стать проницаемой в условиях воспаления. Такое строение стенки характерно для посткапиллярных венул I порядка и капилляров. В них начинается экссудация.
I фактор экссудации — повышение сосудистой проницаемости
Развивается очень быстро. Медиаторы воспаления раздражают эн-дотелиоциты, они становятся округлыми, втягивают отростки, между ними появляются поры, через которые выходит жидкая часть крови, в том числе белки. На поверхности эндотелиоцитов скапливаются анионы, которые способствуют притягиванию катионов плазмы крови. Развивается трансцитоз — перенос плазмы крови через стенку сосуда при помощи пиноцитотических везикул. Последние есть и в норме, но при воспалении их количество и размеры значительно возрастают. Наконец, лизосомальные ферменты лейкоцитов отслаивают эндотелиоциты от базальной мембраны и разрушают, лизируют базальную мембрану и основное вещество (белки и гликозаминогликаны).
Можно наблюдать две фазы повышения проницаемости.
Ранняя фаза, достигающая максимума через 4-10 мин и длящаяся около 30 мин, возникает в посткапиллярных, малых и средних венулах и является результатом действия прежде всего гистамина (реакция эндотелия). В этой фазе принимают участие также серотонин, вещество Р, брадикинин, лейкотриены и простагландины.
64
Лекция 5
Поздняя фаза начинается через 1-2 ч, становится максимально выраженной через 4-6 ч и может длиться до 24 ч и даже нескольких суток. В ней дополнительно уже к названным медиаторам присоединяются цитокины лейкоцитарного происхождения (ИЛ-1, ФНО-а,у-интерферон). Усиливается трансцитоз, стенка сосудов повреждается лейкоцитами.
При тяжелом первичном повреждении тканей сосудистая проницаемость повышается в течение первых 30-45 мин до максимальной и остается такой на протяжении нескольких часов. Снижается повышенная проницаемость очень медленно, в течение нескольких суток. Затрагиваются не только венулы и капилляры, но и артериолы, часть из них некротизируется. Восстановление связано с новым сосудообразованием, но пролиферирующий эндотелий может продлевать экссудацию, так как он намного более проницаем для плазмы, чем зрелый.
II фактор экссудации - повышение онкотического (ОнД) и осмотического давления (ОсД) в очаге воспаления
ОД формируется мелкими белковыми молекулами, альбуминами, которые появляются и накапливаются в очаге воспаления в результате их выхода из сосудов с повышенной проницаемостью.
ОД, давление ионов, солей, молекул является результатом альтерации и изменения обмена веществ в очаге воспаления.
Воспаленные ткани начинают больше потреблять кислорода на единицу веса, при этом выделение угольной кислоты значительно отстает от потребления кислорода, вследствие чего дыхательный коэффициент СО2/О2 значительно уменьшается. Это свидетельствует о том, что окисление продуктов идет не до конца. Количество глюкозы в воспаленной ткани увеличивается не только за счет уменьшения потребления сахара поврежденными тканями, но и за счет усиленного распада сложных углеводов. Количество лактата возрастает. Увеличивается также распад жиров; из-за их неполного окисления в очаге воспаления накапливаются ацетон и фосфаты. Нарушение белкового обмена характеризуется усиленным распадом белка, нарастанием количества полипептидов, появлением альбумоз, пептонов и различных аминокислот, например лейцина, тирозина, триптофана. Меняется минеральный обмен: в 10-20 раз повышается количество калия из-за повреждения клеточных элементов и изменения их физико-химического состояния, повышается коэффициент К+/Са2\ а также возрастает концентрация ионов водорода, развивается ацидоз. pH может меняться от 7,0-7,09 в серозном экссудате до 5,96-6,44 в тканях при остром воспалении. Избыток водородных ионов
Воспаление II (клеточные реакции)
65
и одновалентных катионов (К+) повышает гидрофильность белков, они связывают больше воды, и ее количество в очаге воспаления увеличивается.
Таким образом, некроз и повреждение клеток, усиление и извращение обмена веществ и накопление продуктов неполного распада приводят к возрастанию осмотического давления.
Итак, в очаге воспаления увеличивается коллоидно-осмотическое давление. В результате не только усиливается выход воды из сосудов в ткань, но и она там удерживается, накапливается, образуя экссудат.
Ill фактор экссудации — повышение гидростатического давления в посткапиллярной венуле или на венозном конце капилляра
Причиной повышения этого давления является нарушение оттока, которое формируется теми же самыми процессами, которые были перечислены для объяснения венозной гиперемии. Венозный конец капилляра в норме реабсорбирует большую часть тканевой жидкости, образующейся на артериальном конце капилляра, так как гидростатическое давление на артериальном конце — около 30 мм рт.ст., а на венозном — 10 мм рт.ст. Если гидростатическое давление на венозном конце возрастает, приближаясь к гидростатическому давлению на артериальном конце, то всасывательная функция венозного конца падает, жидкость скапливается вокруг такого сосуда.
IV фактор - нарушение лимфатического опока в очаге воспаления
В среднем около 3-х л из 17 л межтканевой жидкости удаляется из тканей в норме через лимфатические сосуды. При воспалении поток жидкости из сосудов в ткани значительно возрастает, примерно в 5 раз превышая дренажную способность лимфатических сосудов. Кроме того, их пропускная способность может ограничиваться ближайшим лимфатическим узлом. В очаге воспаления устья лимфососудов иногда закрываются выпавшими нитями фибрина, телами погибших клеток.
Жидкость, скапливающаяся в очаге воспаления, называется экссудатом. Экссудаты классифицируются в зависимости от своего состава и проливают свет на этиологию и патогенез данного воспаления.
Различают следующие виды экссудатов:
• серозный; он беден форменными элементами крови, содержит единичные лимфоциты, количество белка в нем — от 0,5 до 8,0 %,
66
Лекция 5
часто — 2,0-2,5 %, удельный вес — 1000-1026, pH — 7,0-7,1. Характерен для ожогов или мозолей (бесцветная жидкость в пузырях на коже);
• катаральный', образуется на слизистых оболочках, содержит слизь, лизоцим, IgA;
• гнойный', в этом экссудате много лейкоцитов, находящихся в различных стадиях распада (гнойные тельца), и неповрежденных лейкоцитов. Большая часть лейкоцитов представлена нейтрофилами. Зеленовато-желтая окраска экссудата связана с присутствием там фермента нейтрофилов миелопероксидазы. Кроме того, на окраску может влиять пигмент возбудителя. Например, синегнойная палочка выделяет синий пигмент, а «чудесная палочка» — красный. В экссудате много белка (до 8-10 %), глобулинов больше, чем альбуминов, много жира, удельный вес — 1020-1040, pH — 6,9-5,6. В гнойном экссудате может находиться гноеродная микрофлора, если только это не асептическое воспаление;
• фибринозный; содержит фибрин, самый крупный белок плазмы (фибриноген). Выход в ткань этого белка свидетельствует о значительном повышении проницаемости сосудистой стенки или даже о ее серьезном поражении. Вышедший фибриноген превращается после соприкосновения с окружающей тканью в нерастворимый фибрин. Если нити фибрина образуют ворсинки, то это придает особый вид органу, например описывается «волосатое сердце». Фибрин, лежащий на поверхности органов и клеток, образует легко снимающуюся пленку, воспаление при этом называется крупозным. Иногда фибрин свертывается в предварительно некротизированных клетках многослойного эпителия, в таком случае фибринная пленка снимается с трудом, обнаженная поверхность кровоточит. Такое воспаление называется дифтеритическим;
• геморрагигеский; в экссудате находятся эритроциты, вышедшие из сосудов при их повреждении. В ткани они распадаются, продукты их распада изменяются и придают бурую окраску ткани, где развилось геморрагическое воспаление. Может наблюдаться при гриппе, сибирской язве и др;
• гнилостный; содержит анаэробную флору, встречается при анаэробной инфекции.
Экссудаты осуществляют и повреждающую, и защитную, барьерную функцию. Повреждающая функция связана с механическим растяжением и сдавлением тканей, сосудов, нервных стволов. Если экссудат
Воспаление II (клеточные реакции)
67
содержит много гидролитических ферментов лейкоцитов, то они будут «расплавлять» ткани и образовывать канал для выхода экссудата. Экссудат вызывает припухлость и боль в пораженной ткани и, тем самым, нарушение ее функции.
Защитная роль состоит в том, что экссудат — это прослойка, отодвигающая очаг повреждения от нормальных, здоровых тканей. Экссудат нарушает отток продуктов разрушения клеток, токсинов, медиаторов, метаболитов, распространение инфекционных возбудителей по кровеносным и лимфатическим сосудам, так как он вносит вклад в образование венозной гиперемии и стаза, блокирует лимфоотток. Он снижает концентрацию опасных веществ, накапливает антитела, связывающие инфекционные и неинфекционные АГ.
В ходе воспаления развивается эмиграция — выход лейкоцитов из микрососудов в очаг воспаления. Главными сосудами являются посткапиллярные венулы I порядка.
По закону И.И. Мечникова, первыми в течение 2 ч выходят нейтрофильные лейкоциты (максимально накапливаются через 4-6 ч), вторыми — моноциты (накапливаются через 16-24 ч и начинают преобладать в инфильтрате), третьими — лимфоциты. Лейкоциты проходят во время эмиграции последовательно 3 стадии:
• краевого стояния;
• прохождения через стенку;
• блуждания в очаге воспаления.
Лейкоциты и в норме мигрируют из сосудов в ткани, где они осуществляют контроль за постоянством внутренней среды, но при повреждении этот выход становится массовым и именно в той области микроциркуляции, которая находится в поврежденной и прилегающей к ней зонам.
I стадия — краевого стояния. Развивается в течение 1-2 ч. Во время венозной гиперемии и престаза лейкоциты появляются в краевой плазматической зоне, что получило название «маргинация» — краевое стояние. Этому явлению способствует замедление скорости кровотока (роль сосудистых реакций!): частицы с более легким удельным весом отклоняются от своего центрального положения и занимают периферическое. Некоторое значение придают и притяжению анионов лейкоцитов к катионам водорода (катафорез) и кальция. Однако главное заключается в появлении молекул адгезии (прилипания), индуцированных хемоаттрактантами и медиаторами, на лейкоцитах и эндотелии и лиганд-рецепторные взаимодействия между ними.
68
Лекция 5
К хемоаттрактантам относят:
• микроорганизмы и продукты их жизнедеятельности, например пептиды, содержащие N-формил-метионин, липополисахариды;
• медиаторы воспаления: компоненты комплемента — С5а, С5 des Arg, СЗа; компоненты кининовой системы — калликреин, каллидин; липидные медиаторы — ФАТ, лейкотриен В4, тромбоксан А2; ФХН-А, ФХЭ-А из тучных клеток;
• цитокины ООФ — ИЛ-1, ФНО-а;
• иммунные комплексы, особенно содержащие иммуноглобулины классов М и G;
• хемокины (представители 4-х главных семейств хемотаксических белков), например ИЛ-8, гранулоцитарный хемотаксический белок-2, лимфотактин (для лимфоцитов), эотаксин (для эозинофилов), макрофагальные воспалительные белки, моноцитарные хемоаттрактивные белки;
• продукты разрушенных тканей (обеспечивают некротаксис) — ДНК, АТФ, АДФ, фибрин-пептид В, фибронектин, коллаген, тромбин и др.
Под влиянием вышеперечисленных хемоаттрактантов и в условиях замедленного кровотока лейкоциты краевой зоны начинают останавливаться и прикрепляться к клеткам эндотелия. Сначала такое сцепление продолжается недолго, клетки уносятся потоком крови или перекатываются по поверхности эндотелия (роллинг); впервые отмечено Ю. Кон-геймом в 1873 г.
В первоначальной непрочной связи участвуют адгезивные молекулы лейкоцитов — селектины. L-селектин, трансмембранный белок, имеющийся в норме на лейкоцитах, связывается с появившимся на активированном эндотелии лигандом (рецептором) гликолипидом Р-селектином, а затем и с более поздним Е-селектином. Активация лейкоцита повышает его сродство к лигандам. Затем L-селектин сбрасывается и заменяется появлением интегринов. Для взаимодействия селектинов и интегринов необходимы ионы кальция.
Интегрины, трансмембранные белки, состоят из а- и p-цепей. При активации лейкоцита они собираются в фокус адгезии и связываются с лигандами на эндотелии. Это белки суперсемейства иммуноглобулинов, называются ICAM-1 (межклеточная адгезивная молекула) и VCAM-1 (сосудистая клеточная адгезивная молекула). Взаимодействие интегринов с лигандами на эндотелии обеспечивает прочную связь. Когда большое количество лейкоцитов прикрепляется к клеткам эндотелия, то сосуд
Воспаление II (клеточные реакции)69 изнутри принимает вид «булыжной мостовой». Внутриклеточные концы интегринов связаны с цитоскелетом лейкоцита. Под влиянием активации интегринов активируются белки цитоскелета, лейкоцит, в частности нейтрофил или моноцит, прочно связывается с поверхностью эндотелия, распластывается, образует псевдоподии и мигрирует через межэндотелиальный контакт исключительно в посткапиллярных венулах.
Происходит 2-я стадия эмиграции — прохождение герез стенку сосуда. Прямыми наблюдениями установлено, что от момента остановки лейкоцита у стенки сосуда до выхода его в окружающие ткани проходит около 4 мин.
3-я стадия — блуждание лейкоцита в огаге воспаления (скорость лейкоцита — приблизительно 20 мкм/мин) или положительный хемотаксис. Это направленное движение клеток по нарастающему градиенту концентрации распознаваемого ими вещества. Такими веществами при воспалении являются перечисленные выше хемоаттрактанты. Рецепторы лейкоцита определяют разницу концентрации в 0,1 %. Большинство рецепторов перемещается на тот полюс, где более 20 % рецепторов занято хемоаттрактантами. После активации рецепторов происходят увеличение количества внутриклеточного кальция, активация сократительных белков актина, миозина, кальмодулина, гельзолина, превращение на этом полюсе цитоплазмы в гель и подтягивание лейкоцита к переднему полюсу. При накатывании цитоплазмы на передний полюс зона желатинизации остается сзади, кальций высвобождается и сокращение заканчивается. Цитоплазма на переднем полюсе теперь находится в состоянии золя, но на мембрану начинают действовать хемоаттрактанты, и все повторяется, т.е. лейкоцит как бы вращается вокруг зоны желатинизации, постепенно перемещаясь, пусть и зигзагообразно, в сторону раздражителя. В этом ему помогают различные белки внеклеточного матрикса, за которые он цепляется интегринами. Наконец лейкоцит достигает раздражителя.
Начинается фагоцитоз — поглощение клеткой инородных частиц (от греч. phagein). Напомним, что основная цель эмиграции нейтрофилов и моноцитов — фагоцитоз, который был открыт И.И. Мечниковым как общебиологическое явление в 1883 г.
В фагоцитозе различают несколько стадий:
• I стадия фагоцитоза — приближение или положительный хемотаксис;
• II стадия — прилипание. В этой стадии главную роль играют опсонины, содержащиеся в сыворотке крови. К ним относятся антитела —
70
Лекция 5
иммуноглобулины класса IgG, IgM, СЗв компонент комплемента, С-реактивный белок, сывороточный амилоид Р, лизоцим. Опсонины одним концом легко прикрепляются к объектам фагоцитоза, а к другому концу опсонинов прикрепляются лейкоциты, у которых для этого есть специальные рецепторы (этим профессиональные фагоциты — нейтрофилы и макрофаги/моноциты отличаются от непрофессиональных). Таким образом, лейкоцит, во-первых, узнает объект фагоцитоза и, во-вторых, крепко к нему прикрепляется через опсониновый «мостик». Вот почему уголь, хорошо абсорбирующий сыворотку, во много раз лучше фагоцитируется, чем кварц, не абсорбирующий сыворотки;
• после прилипания фагоцит активируется и образует фагосому, развивается III стадия фагоцитоза — погружение.
Псевдоподии лейкоцита вытягиваются, окружают инородный объект и смыкаются. Складка мембраны фагоцита образует пузырек с заключенным в нем объектом — фагосому. На ее образование затрачивается энергия гликолиза. В цитоплазме клетки фагосома сливается с лизосомами и специфическими гранулами, образуя фаголизосому. При этом часть содержимого фаголизосомы — содержимое гранул и медиаторы — выделяется наружу в форме экзоцитоза. Это явление получило название «отрыжка при питании». Экзоцитоз может происходить и без фагоцитоза, например, если объект фагоцитоза очень большого размера. Поступление бактерицидного материала в окружающую среду, в экссудат усиливает защитные свойства экссудата, и еще до фагоцитоза микробы подвергаются повреждающему воздействию.
Последняя IV стадия — деградация или переваривание объекта. Она состоит из двух процессов: обезвреживания микроорганизма и разрушения погибшего или значительно ослабленного микроба гидролазами лейкоцита.
В цитоплазме нейтрофила помимо обычных органелл содержатся от 50 до 200 гранул, гликоген и липиды. Гранулы подразделяются на 3 типа: а, Р и у. В гранулах находятся гидролазы и бактерицидные вещества.
Бактерицидные вещества:
• лизоцим (мураминидаза); он гидролизует мурамилглюкозамин мембраны грамположительных бактерий;
• лактоферрин связывает железо, и гемсодержащие бактерии теряют способность к размножению;
• катионные белки. Встраиваясь в мембрану бактериальной клетки, они образуют в ней пору, через которую вытекает цитоплазма ми
Воспаление II (клеточные реакции)
71
кроба. Активны против грамотрицательных и некоторых грампо-ложительных бактерий. Начинают действовать уже в нейтральной среде, не дожидаясь ее закисления. К семейству катионных белков относятся и дефензины, активные в щелочной среде;
• миелопероксидаза нейтрофилов способствует галогенизации белков бактерий и их гибели, так как превращает перекись водорода и ионы хлора (брома, иода) в гипохлорид-анион.
Помимо готовых бактерицидных веществ фагоцит после активации и, особенно, после образования фагосомы начинает производить большое количество активных кислородсодержащих радикалов (АКР). При этом в лейкоците так сильно изменяется метаболизм, что его назвали даже респираторным взрывом. В спокойном состоянии основным способом получения энергии в лейкоците является анаэробный тип обмена веществ. Фагоцитоз сопровождается повышением потребления кислорода, увеличением аэробного и анаэробного гликолиза, гликогенолиза, увеличением расщепления глюкозы по гексозомонофосфатному пути и др. При образовании фагосомы на ее внутренней мембране находится часть НАДФ-оксидазной ферментной системы, которая полностью собирается в результате дополнения белками сливающихся с фагосомой гранул и цитоплазматического материала. Происходит активация НАДФ-оксидазы, резко возрастает потребление лейкоцитом кислорода и превращение его НАДФ-оксидазой в супероксидный радикал — О2 , что дает начало появлению большого количества и других АКР, в их числе перекиси водорода, синглетного кислорода, гидроксильных радикалов, оксида азота, оксийодидов и др. В результате повреждается мембрана бактерий, нарушается их размножение, и они успешно подвергаются действию лизосомальных гидролаз лейкоцита. Это нейтральные протеазы (коллагеназа, эластаза, желатиназа, катепсины), кислые протеазы (разрушают гликопротеиды и протеогликаны), кислые дезрибонуклеазы, липазы, фосфолипазы, гликозидазы (разрушают гликозаминогликаны основного вещества соединительной ткани).
В фагоцитирующих лейкоцитах (фагосома — «мешок самоубийцы») происходит все большая и большая дегрануляция вплоть до исчезновения гранул, образуется большое количество перерабатывающих вакуолей. Лейкоцит погибает, цитоплазма его становится водянистой, почти лишенной гликогена, ядро гомогенизируется и сморщивается. Такой лейкоцит называется гнойным тельцем. Продолжительность жизни гранулоцитов небольшая (в тканях — 2-4 дня), а в очаге воспаления она становится еще меньше. Остатки фагоцитов, поврежденные, некротизи
72
Лекция 5
рованные ткани и бактерии (при асептическом воспалении их может не быть), а также медиаторы, гидролазы, бактерицидные и цитотоксические факторы лейкоцитов формируют гной.
При воспалении выделяют первичную и вторичную альтерацию.
• Первигная альтерация возникает под влиянием повреждающего фактора, который может не только сам разрушать клетки и ткани, но прежде всего нарушать их функционирование, их обмен веществ, вызывать в них синтез медиаторов повреждения.
• Вторигная альтерация развивается в ходе самого воспаления. Дополнительное повреждение тканей обусловлено сосудистыми реакциями (венозной гиперемией, стазом), тромбозом, экссудацией (механическое сдавление тканей, сосудов, нервов), «расплавлением» тканей гидролазами экссудата и фагоцитирующих лейкоцитов, а также АКР.
Из-за незавершенного фагоцитоза возможно персистирование микробов и вирусов внутри лейкоцита, возникновение хронического и рецидивирующего течения инфекций, а также генерализации инфекций. Незавершенный фагоцитоз является следствием патологии лейкоцитов (наследственной или приобретенной), а также особенностями некоторых микроорганизмов, научившихся в ходе эволюции защищаться от лейкоцитов. Это характерно для возбудителей таких инфекций, как туберкулез, сифилис, бруцеллез, коклюш, сальмонеллез, гонорея, малярия и др.
Тем не менее в эволюции воспаление сохранилось, несмотря на некоторые отрицательные его стороны: боль, нарушение функции, вторичную альтерацию, потерю паренхиматозной ткани и замещение ее рубцом. В подавляющем большинстве случаев воспаление выполняет свою задачу отграничения повреждающего агента или поврежденных тканей от здоровых, а затем и ликвидации очага повреждения.
После создания некоего «протеолитического котла», в котором все уничтожается — и повреждающий агент, и поврежденные ткани (вместе с прилегающими), и фагоциты — развивается регенерация. Пролиферируют фибробласты, синтезируется соединительная ткань, заделывающая брешь. При сохранении паренхиматозных клеток, способных к делению, развивается их пролиферация. Полное заживление небольшой неинфицированной раны без нагноения получило название заживления первигным натяжением. Большое повреждение с нагноением заживает вторигным натяжением: полностью не восстанавливается паренхима, рана стягивается при помощи соединительной ткани.
Воспаление II (клеточные реакции)
73
В некоторых случаях развивается хронигеское воспаление.
• Первигно-хронигеское персистирующее воспаление — результат вялого течения воспаления и регенерации. Подобное состояние может наблюдаться в результате голодания, гиповитаминозов, недостатка микроэлементов, иммунодефицитов, местных тканевых нарушений (диабетические или атеросклеротические ангиопатии).
• Вторигное хронигеское воспаление — результат неадекватности механизмов острого воспаления. Условием хронического воспаления в данном случае является длительное существование повреждающего агента. Этим агентом может быть микроорганизм, приспособленный к персистенции в тканях (микобактерии туберкулеза и др.), неметаболизируемое вещество (кристаллы кремния, бериллия, кусочки костной ткани), аутоантигены (при аутоиммунных заболеваниях антигенами становятся ткани сердца, почек, суставов и т.д.). Тогда возможно только отграничение, в результате происходит мононуклеарная (моноцитами/макрофагами и лимфоцитами) инфильтрация пораженных тканей, образуется мононуклеарный инфильтрат. Макрофаги пролиферируют, превращаются в эпителиоидные клетки и гигантские многоядерные клетки. На периферии располагаются лимфоциты. Формируются гранулемы — главный морфологический признак хронического воспаления. Выделяемые активированными макрофагами и лимфоцитами цитокины поддерживают приток новых мононуклеаров в очаг воспаления и хро-низацию процесса.
Следовательно, итогами воспаления являются полное заживление, неполное заживление (образование рубца) и переход в хроническую форму.
При воспалении мы наблюдаем активное участие 3-х видов лейкоцитов.
• Нейтрофильные гранулоциты. Профессиональные фагоциты (микрофагоциты) . Первыми выходят в очаг воспаления (сильнейшими хемоаттрактантами для них являются гноеродные кокки и продукты их жизнедеятельности). Имеют менее широкий ассортимент фагоцитируемых объектов, чем макрофаги. В очаге воспаления живут гораздо меньше, чем макрофаги. Содержат миелопероксидазу и естественные пептидные антибиотики — катионные белки и дефензины. Не способны к презентации антигена и, таким образом, к инициации специфического иммунного ответа.
74
Лекция 5
• Макрофаги/моноциты. Профессиональные фагоциты (макрофагоциты). Являются долгоживущими клетками и могут многократно участвовать в воспалении. Способны к синтезу de novo медиаторов воспаления. Синтезируют цитокины ответа острой фазы, т.е. подсоединяют системный ответ, ответ целого организма, к местному ответу. Синтезируют факторы комплемента, свертывания крови, плазминоген и предшественники кининов. Источники факторов пролиферации, антиоксидантов и ингибиторов протеаз (например антитрипсина). Способны презентировать клеткам иммунной системы антиген и тем самым подключать к местной реакции на повреждение иммунный ответ. Участники хронического воспаления.
• Лимфоциты. Не способны к фагоцитозу. Синтезируют цитокины ответа острой фазы. Участвуют в иммунных реакциях. Синтезируют опсонины (IgG, IgM) и другие антитела, участвующие в воспалении. Осуществляют киллерную функцию по отношению к поврежденным клеткам. Единичные лимфоциты могут содержаться в транссудате и серозном экссудате. Клетки хронического воспаления.
Лекция 6
ОТВЕТ ОСТРОЙ ФАЗЫ
Ж.М. Салмаси
В процессе эволюции у высших животных и человека сформировалась способность развивать неспецифическую реакцию на повреждение, направленную на связывание и удаление патогенного фактора из организма. Местная реакция на повреждение получила название воспаления, общая реакция организма на повреждение носит название ответа острой фазы. При незначительном повреждении ткани возникает только местная воспалительная реакция, при значительном повреждении возникновение воспаления в месте повреждения сочетается с развитием ответа острой фазы (ООФ).
Ответ острой фазы влияет на функционирование практически всех систем организма, с одной стороны, повышая резистентность организма к действию повреждающих факторов внешней среды, а с другой стороны, он может стать причиной развития нарушений функционирования различных органов и организма в целом, особенно при длительном течении.
Наиболее характерные проявления ответа острой фазы:
• развитие лихорадки;
• развитие вялости, сонливости, снижения работоспособности;
• синтез белков острой фазы в печени;
• активация гипофизарно-надпочечниковой системы;
• развитие нейтрофилии;
• активация макрофагов и нейтрофилов;
• усиление пролиферации и дифференцирования фибробластов и, как следствие, активация заживления ран;
• активация клеток иммунной системы;
• снижение массы тела.
На сегодняшний день доказано, что развитие ООФ, как и развитие воспаления, связано с образованием медиаторов. Роль медиаторов ООФ
76
Лекция 6
играют цитокины, выделяемые при повреждении, в первую очередь монокины и лимфокины (цитокины, выделяемые макрофагами и лимфоцитами).
В развитии ответа острой фазы принимает участие большое количество цитокинов, которые вовлекаются в процесс по механизму каскадной реакции, т.е. один цитокин способен запускать синтез нескольких других цитокинов. Однако только 3 цитокина способны при введении в организм вызывать большинство эффектов ответа острой фазы: ИЛ-1, ИЛ-6 и фактор некроза опухоли а. Именно эти цитокины и относят к основным медиаторам ООФ.
Медиаторы ответа острой фазы
ИЛ-1 синтезируется в организме различными клетками макрофагального и лимфоидного ряда, фибробластами, эндотелиоцитами, нейтрофилами и рядом других клеток. Основным источником ИЛ-1 в организме являются макрофаги и эндотелиоциты.
Покоящиеся клетки не продуцируют ИЛ-1. Выделение синтезированного ИЛ-1 начинается через 2-4 ч после стимуляции и достигает максимума через 1-2 сут.
В качестве индуктора синтеза ИЛ-1 и других медиаторов ООФ могут выступать:
• компоненты бактериальной стенки (липополисахариды, пептидогликаны, белок А и др.);
• стимуляторы фагоцитоза;
• АГ и иммунные комплексы;
• физические факторы (УФ-облучение, термические факторы).
Биологическое действие ИЛ-1 представлено в табл. 6-1.
Таблица 6-1. Основные биологические эффекты ИЛ-1 (по С.А. Кетлинскому)
Объекты воздействия Биологические эффекты
Макрофаги Стимуляция фагоцитоза, хемотаксиса, генерации супероксид-радикала, продукции ФНО, ИЛ-1, ИЛ-6, ПГЕ2, увеличение экспрессии 1а АГ
Нейтрофилы Опосредованное усиление хемотаксиса, дегрануляции и индукции миелопероксидазы, нейтрофильный лейкоцитоз
Т-лимфоциты Усиление экспрессии ИЛ-2, стимуляция хемотаксиса, увеличение продукции ИЛ-2, ИЛ-4, у-ИФН
В-лимфоциты Усиление экспрессии рецептора ИЛ-2,1а АГ, стимуляция пролиферации преактивированных клеток
Ответ острой фазы
77
Оконгание табл. 6-1
Объекты воздействия Биологические эффекты
NK-клетки Усиление цитотоксичности, продукции у-ИФН и ИЛ-2
Фибробласты Усиление пролиферации, индукция продукции ИЛ-6, КСФ, ИФН-р, ПГЕ,
Базофилы и тучные клетки Индукция выброса гистамина
Эндотелий Стимуляция пролиферации, усиление адгезивных свойств, индукция прокоагулянтной активности
Красный костный мозг Усиление пролиферации полипотентных клеток
Костная и хрящевая ткань Стимуляция продукции коллагеназы и ПГЕ2, усиление пролиферации синовиальных фибробластов и хондроцитов, резорбция хряща и кости
Мышцы Протеолиз мышц, стимуляция продукции ПГЕ?
Печень Снижение синтеза альбумина, усиление синтеза компонентов комплемента
ЦНС Усиление продукции ПГЕ2 и АКТЕ снижение аппетита, сонливость, лихорадка
Концентрация ИЛ-1 в крови у больных с инфекционным процессом коррелирует с подъемом температуры тела.
Следует отметить, что целый ряд биологических эффектов ИЛ-1 (и других медиаторов ООФ) связан с его способностью стимулировать синтез ПГЕ2 в тканях. В частности, через синтез ПГЕ2 реализуются механизмы лихорадки, снижения массы тела, повреждение хрящевой и костной ткани и ряд других (см. табл. 6-1). Под влиянием ИЛ-1 синтез ПГЕ2 в фибробластах увеличивается в 20 раз. Подавление синтеза ПГЕ2 блокаторами фермента циклооксигеназы (ацетилсалициловая кислота, индометацин и т.п.) отменяет ряд эффектов ООФ.
Фактор некроза опухоли (ФИО). Фактор некроза опухоли первоначально был открыт как вещество, обладающее способностью вызывать некроз опухолевого узла in vivo и лизис опухолевых клеток in vitro. Фибробласты выделяют ФНО-а. При исследовании выделяемого цитотоксическими лимфоцитами медиатора лимфотоксина была показана его гомология с ФНО, что позволило присвоить ему наименования ФНО-р. Таким образом, исторически сложившееся имя не отражает спектр биологического действия этого цитокина.
Основным источником ФНО в организме служат макрофаги и лимфоциты. Следует подчеркнуть, что продукция ФНО, как и других медиато
78
Лекция 6
ров ООФ, начинается только под действием индукторов. Биологические эффекты ФНО близки к действию ИЛ-1, хотя и не идентичны. ФИО в большей степени отвечает за вторичную альтерацию при ООФ, в частности он в большей степени индуцирует снижение массы тела, активно индуцирует апоптоз в различных тканях организма. В то же время ФНО значительно слабее влияет на синтез белков острой фазы в печени.
ИЛ-6 представляет типичный индуцибельный белок и может секретироваться Т-лимфоцитами, макрофагами, фибробластами, эндотелио-цитами и другими клетками, участвующими в развитии воспаления или иммунного ответа. Наиболее выраженный биологический эффект ИЛ-6 — синтез белков острой фазы в печени.
Механизмы развития проявлений ответа острой фазы
Повышение резистентности организма к инфекционным агентам. Существуют прямые доказательства, что ИЛ-1 и ФНО, введенные животным по профилактической схеме, вызывают защитный эффект от летальных доз бактерий. Так, введение ФНО мышам за 2 ч до заражения летальной дозой К. pneumoniae снижало летальность в 2 раза. При сочетанном применении ИЛ-1 и ФНО по той же схеме удавалось уменьшить летальность при заражении патогенным штаммом Е. coli в 20 раз. Если же эти медиаторы ООФ вводили в течение 4-х дней до заражения S. aureus, то летальность снижалась в 200 раз. Поскольку ни ИЛ-1, ни ФНО не обладают антибактериальной активностью, их эффект связан с действием на различные системы организма.
На сегодняшний день важнейшим механизмом действия медиаторов ответа острой фазы считают усиление экспрессии Toll-подобных рецепторов (Toll-like receptors, TLRs).
7Ь//-рецепторы рассматривают в качестве древнего механизма узнавания «чужого» и сигнализации о проникновении в организм «чужого», представляющего опасность для этого организма. Как правило, в качестве «чужого» рассматривают патогенные и условно-патогенные микроорганизмы. Однако в последние годы стало очевидно, что TLRs могут распознавать ряд эндогенных продуктов, появление которых свидетельствует о присутствии иной (неинфекционной) опасности. К эндогенным активаторам врожденного иммунитета относят белки теплового шока и мочевую кислоту, а также продукты некроза и апоптоза.
TLRs описаны у всех многоклеточных организмов, включая растения, беспозвоночных и позвоночных животных. У человека выявлено 11 TLRs. Экспрессированы, главным образом, на клетках системы
Ответ острой фазы
79
врожденного иммунитета — макрофагах, дендритных клетках, ТК, нейтрофилах, моноцитах, а также на В- и Т-лимфоцитах. Опубликованы сообщения о выявлении TLRs на поверхности эпителиальных клеток кишечника, кератоцитов кожи и клетках микроглии.
Уровень экспрессии TLRs зависит от природы клетки-носителя, стадии ее развития (дифференцировка, созревание), характеристики действующих цитокинов и свойств распознаваемых бактериальных лигандов.
Лигандами для TLR служат повторяющиеся молекулярные карбо-гидратные и липидные структуры, однонитевая (sRNA) и двунитевая (ssRNA) РНК, cytosine phosphate guanosine (CpG) мотив ДНК.
Один рецептор может распознавать лиганды, выявляемые у разных микроорганизмов. Для TLR2 описано взаимодействие с лигандами бактерий, вирусов, несовершенных дрожжей и простейших. TLR4 взаимодействует с ЛПС бактерий, F-протеином (белком слияния) респираторно-синцитиального вируса и оболочным белком ММТ вируса (mouse mammary tumor virus).
Лихорадка — компонент ООФ, характеризующийся повышением температуры тела за счет смещения «установочной точки» на более высокий уровень. По аналогии с лабораторным термостатом, переключенным на более высокую температуру, организм поддерживает более высокую температуру за счет активного усиления теплопродукции и снижения теплоотдачи. Терморегуляция при этом, в отличие от гипертермии, не нарушена. Образовавшиеся под влиянием индукторов медиаторы ООФ играют роль эндопирогенов. Действуя на центр терморегуляции в гипоталамусе, медиаторы ООФ стимулируют синтез ПГЕ2, который, в свою очередь, повышает чувствительность холодовых нейронов, вызывая смещение «установочной точки» и повышение температуры тела. Снижение теплоотдачи происходит за счет торможения потоотделения и спазма сосудов кожи, а усиление теплопродукции связано в первую очередь с развитием мышечной дрожи. Поскольку лихорадка развивается при участии ПГЕ2, ингибиторы циклооксигеназы ее эффективно подавляют.
Синтез белков острой фазы происходит в основном в печени. В наибольшей степени усиливается синтез С-реактивного белка и сывороточного амилоида А — более чем в 100 раз. Концентрация а-антитрипсина и других протеинах возрастает в 2-10 раз, церулоплазмина — в 2 раза, а синтез сывороточного альбумина и трансферрина снижается.
Белки острой фазы обладают широким спектром биологических эффектов.
80
Лекция 6
С-реактивный белок и сывороточный амилоид А стимулируют иммунную систему и повышают неспецифическую резистентность организма к инфекциям. С-реактивный белок (СРБ) начинает интенсивно синтезироваться в гепатоцитах через 4-6 ч после повреждения. СРБ способствует активному фагоцитозу, активации комплемента по классическому пути, усиливает ответ лимфоцитов на стимулирующие факторы. Повышение концентрации СРБ в крови используют как диагностический тест. В то же время длительное повышение концентрации в крови сывороточного амилоида А может привести к развитию серьезного осложнения — амилоидоза тканей.
Вторая функциональная группа белков острой фазы — антипротеиназы. Классические представители — а-антитрипсин, а-антихимотрипсин, орозомукоид, а2-макроглобулин. Эти белки снижают активность тканевых протеаз, активно поступающих в воспаленную ткань. При их недостатке возможен запуск реакций ограниченного протеолиза: активация свертывающей системы, фибринолиза, активации комплемента и активации калликреин-кининовой системы (ККС). Это может привести к развитию различных осложнений, вплоть до развития тромбогеморрагического синдрома.
Группа транспортных белков представлена церулоплазмином, транспортирующим ионы меди, и трансферрином, обеспечивающим транспорт ионов железа.
Кроме того, при ООФ существенно возрастает синтез белков системы свертывания крови, в первую очередь протромбина и фибриногена, а также компонентов комплемента.
Влияние ООФ на гемостаз. В процессе ООФ наблюдается резкое усиление гемостаза. В зависимости от интенсивности повреждения в очаге может наблюдаться тромбоз либо капилляров в очаге воспаления, либо крупных сосудов, в особо тяжелых случаях может развиваться синдром диссеминированного внутрисосудистого свертывания крови (ДВС-син-дром). Механизмы усиления гемостаза:
• под влиянием ИЛ-1 и ФНО усиливается синтез тромбоксанов и фактора активации тромбоцитов (ФАТ) клетками эндотелия — веществ, резко усиливающих агрегацию клеток крови, повышающих проницаемость стенки сосудов и, таким образом, способствующих замедлению кровотока и развитию стаза;
• ИЛ-1 и ФНО способны значительно повышать прокоагулянтную активность плазмы крови за счет стимуляции синтеза ряда факторов свертывания крови гепатоцитами;
Ответ острой фазы
81
• ИЛ-1 и ФНО способны значительно повышать экспрессию рецепторов адгезии эндотелиоцитами сосудистой стенки.
Механизм развития нейтрофилии при ответе острой фазы
Хорошо известно, что ООФ в большинстве случаев сопровождается повышением содержания нейтрофилов в периферической крови, причем интенсивность лейкоцитоза связана с выраженностью реакции. Нейтрофилия при ООФ вызывается как прямым стимулирующим действием ФНО и ЙЛ-1 на таксис гранулоцитов, так и способностью медиаторов ООФ стимулировать продукцию КСФ. Имеются экспериментальные данные, доказывающие, что ИЛ-1, ИЛ-6 и ФНО индуцируют выработку гранулоцитарного и гранулоцитомакрофагального КСФ.
Механизм резорбции кости и хряща при ответе острой фазы
В экспериментальных работах показано, что ИЛ-1 и ФНО способствуют резорбции костной ткани. Этот процесс реализуется 3-мя путями:
• ИЛ-1 и ФНО подавляют активность остеобластов;
• ИЛ-1 и ФНО повышают активность остеокластов;
• ФНО блокирует синтез коллагена и неколлагеновых белков остео-цитами.
Аналогичным образом эти медиаторы действуют и на хрящевую ткань. Под влиянием ИЛ-1 и ФНО происходит активация коллагеназы и протеингликандеградирующих протеаз хондроцитов с нарушением структуры хрящевой ткани.
Механизмы развития кахексии при ответе острой фазы
Известно, что длительное течение ООФ сопровождается значительным снижением массы тела за счет мышечной и жировой тканей. Механизмы развития кахексии при ООФ связаны с биологическими эффектами медиаторов ООФ:
• усиленным протеолизом мышц;
• снижением аппетита;
• блокадой липопротеинлипазы (ЛПЛ).
Давно известно, что у больных с тяжелой инфекцией или значительным повреждением тканей резко увеличивается протеолитическая активность плазмы крови, при этом в крови растет концентрация свободных аминокислот и усиливается синтез белка гепатоцитами. Позднее было выявлено, что роль фактора, активирующего протеолиз мышц
82
Лекция 6
и усиливающего синтез белка в печени, играет один из фрагментов, образующихся при расщеплении ИЛ-1. Этот фрагмент имеет молекулярную массу 4 Кд (ИЛ-1 имеет ММ 16 Кд). Для врачебной практики важно знать, что протеолитический эффект фрагмента ИЛ-1 реализуется через синтез ПГЕ2.
Второй механизм снижения массы тела при ООФ связан с прямым действием ИЛ-1 и ФИО на ЦНС. В эксперименте введение этих медиаторов в третий желудочек головного мозга вызывало резкое подавление аппетита у крыс. Внутривенное введение препаратов подобной реакции не вызывало.
Третий механизм снижения массы при ООФ связан со способностью ФНО блокировать фермент ЛПЛ, что приводит к нарушению поступления жирных кислот в клетки и нарушению образования нейтральных жиров в адипоцитах. При этом происходит накопление триглицеридов в сыворотке крови. Кроме того, ФНО усиливает процесс липолиза.
Развитие стрессовой реакции при ответе острой фазы
Со времен Ганса Селье, автора теории общего адаптационного синдрома, известно, что инфекции, массивные повреждения ткани вызывают стрессовую реакцию, характеризующуюся активацией симпато-адре-наловой системы. Однако механизмы развития стрессовой реакции при ООФ в то время были неизвестны. Только в конце 1990-х годов было доказано, что медиаторы ООФ ИЛ-1, ИЛ-6, ФНО при введении в организм могут играть роль кортикотропин-рилизинг-фактора. В отношении ИЛ-1 на сегодня доказана специфичность действия на гипофиз. Он вызывает усиление продукции только адренокортикотропного гормона (АКТГ), не влияя на синтез других гормонов аденогипофиза. Вырабатывающиеся в коре надпочечников под влиянием АКТГ глюкортикоидные гормоны существенно подавляют иммунологические и воспалительные реакции, индуцируемые медиаторами ООФ, препятствуя экспрессии генов, регулирующих синтез этих медиаторов. Таким образом, включается отрицательная обратная связь между ООФ и активацией симпатоадреналовой системы, приводящая к ограничению интенсивности ООФ в организме.
Влияние ответа острой фазы на функции фибробластов
Фибробласты являются основными клетками соединительной ткани, образующими все ее неклеточные компоненты: коллаген, эластин и про
Ответ острой фазы
83
теогликаны. Скорость пролиферации фибробластов в решающей степени обеспечивает процесс заживления ран. Доказано, что ИЛ-1 способен усиливать пролиферацию фибробластов в 5-6 раз, с другой стороны, и ИЛ-1, и ФНО значительно усиливают продукцию фермента коллагеназы, что может привести к деструкции соединительной ткани. Коллагеназная активность медиаторов ООФ не связана с продукцией ПГЕ2.
Роль медиаторов ответа острой фазы в развитии септического или эндотоксического шока
Септический и эндотоксический шок можно воспроизвести на экспериментальных животных введением значительного количества патогенных микроорганизмов или их эндотоксина. При этом наблюдаются прогрессирующее повышение проницаемости стенки сосудов, приводящее к развитию гиповолемии, активация свертывающей системы крови, возникают краевое стояние лейкоцитов, отек тканей. В дальнейшем развивается классическая картина шока со снижением артериального давления и включением компенсаторных механизмов. В экспериментах на животных удалось получить состояние, аналогичное эндотоксическому шоку у человека при внутривенном введении высоких доз ФНО и ИЛ-1 одновременно. Характерно, что интенсивность нарушений гемодинамики при шоке, вызванном введением ФНО и ИЛ-1, значительно снижалась при одновременном введении ингибиторов циклооксигеназы, т.е. при блокаде синтеза ПГЕ под влиянием медиаторов ООФ.
Роль медиаторов ответа острой фазы в противоопухолевой защите
Медиаторы ООФ, будучи одновременно медиаторами иммунной системы, играют важнейшую роль в формировании противоопухолевой защиты организма. Они, с одной стороны, участвуют в развитии иммунного ответа на опухолевые АГ, а с другой — усиливают неспецифическую резистентность организма к опухоли. В рамках настоящей лекции основное внимание обращено на повышение неспецифической противоопухолевой резистентности под влиянием медиаторов ООФ.
Основные механизмы повышения неспецифической резистентности к опухолям под влиянием медиаторов ООФ:
• активация цитотоксичности макрофагов;
• активация естественных цитотоксических клеток;
• индукция лимфокин-активируемых киллеров;
84
Лекция 6
• индукция терминальной дифференцировки опухолевых клеток;
• усиление антигенности опухолевых клеток путем экспрессии антигенов гистосовместимости I и II классов;
• некроз опухолевого узла за счет тромбоза внутриопухолевых сосудов.
Роль медиаторов ответа острой фазы в защите от ионизирующего излучения
Как известно, при действии несмертельных доз ионизирующей радиации на организм человека в первую очередь страдает кроветворная ткань. Именно поэтому факторы, поддерживающие гемопоэз, должны обладать радиопротективным эффектом. Экспериментальная проверка этого предположения показала, что ИЛ-1 и ФИО обладают дозозависимым защитным эффектом. При введении мышам через 3 ч после летального облучения ИЛ-1 в дозе 2 мкг выживали 100 % животных; ФНО в дозе 5 мкг обеспечивал выживание 50 % мышей.
В заключение хотелось еще раз подчеркнуть, что современная концепция формирования ООФ сложилась только в конце XX века. Тем не менее уже сегодня полученные знания используются в клинической практике для диагностики различных патологических состояний. Предпринимают попытки использования медиаторов ООФ и их индукторов для лечения отдельных заболеваний, в частности онкологических. К сожалению, широкое применение этих препаратов невозможно из-за очень широкого спектра действия — рецепторы к этим медиаторам имеются на клетках многих тканей организма. Однако поиск препаратов, стимулирующих определенные TLR-рецепторы, представляет несомненный практический интерес.
На сегодняшний день показана важная роль избыточного образования медиаторов ООФ в патогенезе ряда заболеваний, таких как ревматоидный артрит. Попытка клинического применения препаратов на основе моноклональных антител против медиаторов ООФ, в первую очередь ФНО-а, показала, с одной стороны, их высокую эффективность при РА, а с другой — выявила высокий риск развития септических осложнений при повышении дозы препарата, что препятствует внедрению этих препаратов в широкую клиническую практику.
Лекция 7
ПАТОФИЗИОЛОГИЯ СИСТЕМЫ ГЕМОСТАЗА
Н.И. Бережнова
Система гемостаза — биологическая система, обеспечивающая сохранение жидкого состояния крови, поддержание целостности стенок кровеносных сосудов, предупреждение и остановку кровотечений при их повреждении.
Гемостаз реализуется в основном тремя взаимодействующими функционально-структурными компонентами: стенками кровеносных сосудов (в первую очередь интимой), клетками крови (тромбоцитами, эритроцитами и др.) и плазменными ферментными системами (свертывающей, фибринолитической, ККС и др.). На повреждение сосудов первыми реагируют сами сосуды (спазм, открытие шунтов выше места повреждения) и клетки крови (тромбоциты и др.). Именно тромбоцитам, а не свертыванию крови принадлежит ведущая роль в первичной остановке кровотечений из микроциркуляторных сосудов, наиболее ранимых и чаще всего бывающих источником геморрагий. На основании этого условно можно выделить сосудисто-тромбоцитарный (первичный) гемостаз и коагуляционный (вторичный) гемостаз. Чаще эти механизмы функционируют одновременно и сопряженно.
Сосудисто-тромбоцитарный (первичный) гемостаз
Неповрежденный эндотелий сосудов обладает тромборезистентно-стью, обусловленной следующими его свойствами:
• предупреждением агрегации тромбоцитов благодаря отрицательному поверхностному заряду и синтезу и секреции антиагреганта простациклина;
• подавлением коагуляционного гемостаза вследствие связывания тромбина тромбомодулином и инактивации других прокоагулянтов (V, VIII, IX и X плазменных факторов);
86
Лекция 7
• активацией антикоагулянтов:
— комплексом тромбин-тромбомодулин системы протеина С, инактивирующей V и VIII факторы;
— синтезом протеина S — кофактора протеина С;
— синтезом гепариноподобных протеогликанов (гепарин-сульфата и др.), которые активируют антитромбин III, инактивирующий, в свою очередь, все ферментные плазменные факторы свертывания крови;
• активацией фибринолитической системы благодаря синтезу тканевого активатора плазминогена (ТПА);
• способностью метаболизировать биологически активные вещества, влияющие на гемостаз (биогенные амины, плазменные кинины, ФАТ и др.);
• продукцией эндотелий-расслабляющего фактора (оксида азота);
• наличием АДФазы, расщепляющей АДФ, — одним из основных стимуляторов агрегации тромбоцитов.
Поврежденный эндотелий трансформируется в мощную прокоагулянтную поверхность вследствие:
• выделения адреналина и секреции эндотелина-1, вызывающих спазм сосуда в месте повреждения, который замедляет кровоток и улучшает взаимодействие между тромбоцитами, факторами свертывания и поврежденным эндотелием;
• понижения продукции антиагреганта простациклина (ПП2) и повышения выделения стимуляторов адгезии и агрегации тромбоцитов: адреналина, АДФ, фактора Виллебранда, тромбоксана А2, фактора агрегации тромбоцитов (ФАТ) и др.;
• ослабления антикоагулянтной активности эндотелия вследствие снижения синтеза тромбомодулина, понижения активации антитромбина III и синтеза ингибитора пути тканевого фактора;
• увеличения прокоагулянтного потенциала выделением тканевого фактора (ф. III) и активацией ф. XII (фактор Хагемана);
• ослабления активации фибринолитической системы в результате снижения синтеза тканевого активатора плазминогена (ТПА) и увеличения продукции его ингибиторов;
• обнажения субэндотелия, компоненты которого (коллаген и др.) активируют сосудисто-тромбоцитарный и коагуляционный гемостаз.
Участие тромбоцитов в гемостазе определяется выполнением следующих функций:
Патофизиология системы гемостаза
87
• ангиотрофической (ежедневно на роль физиологических «кормильцев» эндотелия расходуется около 15 % циркулирующих тромбоцитов);
• способностью поддерживать спазм поврежденных сосудов путем секреции вазоактивных веществ (адреналина, серотонина);
• участием в активации вторичного коагуляционного гемостаза за счет пластиночного фактора 3 (ф. 3) — компонента мембраны тромбоцитов, играющего роль фосфолипидной матрицы, на которой идут реакции коагуляционного гемостаза;
• способностью закупоривать поврежденный сосуд первичным тромбоцитарным тромбом, образующимся вследствие реализации адгезивно-агрегационной функции тромбоцитов;
• осуществлением репаративной функции благодаря выделению фактора роста, вызывающего миграцию к месту повреждения фибробластов, макрофагов и гладкомышечных клеток.
Процесс гемостаза при повреждении сосудистой стенки можно условно разделить на несколько взаимосвязанных между собой этапов.
• Локальная вазоконстрикция (рефлекторная и метаболическая, обусловленная действием вазоконстрикторов, выделяемых тромбоцитами и эндотелием — серотонином, ТхА2, норадреналином).
• Адгезия и агрегация активированных тромбоцитов к участку повреждения с образованием первичного тромбоцитарного тромба.
• Активация коагуляционного гемостаза, локализованного системой антикоагулянтов местом повреждения сосуда; стабилизация тромбоцитарного тромба образующимися нитями фибрина.
• Возможная реканализация сосуда вследствие активации фибринолитической системы (системы плазминогена).
Адгезивно-агрегационная функция тромбоцитов
Для полноценной адгезии тромбоцитов к поврежденному эндотелию необходимы следующие условия:
• контакт с главным стимулятором адгезии коллагеном субэндотелия в присутствии плазменного кофактора — ионов кальция;
• изменение формы тромбоцитов из дисковидной в шарообразную с псевдоподиями;
• экспрессия на мембране тромбоцитов рецепторов фактора Виллебранда — гликопротеинов 1b (ГП 1b).
Отсутствие этих рецепторов на мембране тромбоцитов (тромбоцитопатия Бернара-Сулье) и дефицит фактора Виллебранда (болезнь
88
Лекция 7
Виллебранда) сопровождаются нарушением адгезии тромбоцитов с развитием геморрагического синдрома. Параллельно адгезии протекает процесс агрегации тромбоцитов (рис. 7-1,7-2).
Стимуляторами агрегации служат коллаген, АДФ, а также адреналин и серотонин, которые выделяются из сосудистой стенки. В последующем стимуляторы агрегации (адреналин, серотонин, АДФ, тромбоксан А2 и др.) выделяют сами тромбоциты в ходе дегрануляции. Стимуляторы агрегации, в частности адреналин, действуют посредством экспонирования и экспрессии на мембране тромбоцитов гликопротеинов Пб/ Ша — рецепторов фибриногена. Фибриноген, взаимодействуя с этими рецепторами, связывает соседние тромбоциты мостиками с образованием обратимых агрегатов. Образование необратимых агрегатов обеспечивает тромбин, образующийся в зоне гемостаза в течение нескольких секунд благодаря активации коагуляционного гемостаза по внешнему механизму (см. коагуляционный гемостаз), а также другие адгезивные белки (фибронектин, тромбоспондин).
Рис. 7-1. Адгезия тромбоцитов
Рис. 7-2. Агрегация тромбоцитов
Патофизиология системы гемостаза 89
Способностью активировать агрегацию обладают также такие агенты, продуцируемые другими клетками (лейкоцитами и др.), как фактор активации тромбоцитов (ФАТ), фактор некроза опухолей (ФНО), интерлейкины, лейкотриены и др. В конечном итоге образование гемостатической тромбоцитарной пробки и остановка кровотечения из микроциркуляторных сосудов завершаются за 2-4 мин (проба Дьюка или время кровотечения).
Чрезвычайно важную роль в регуляции сосудисто-тромбоцитарного гемостаза играют такие производные арахидоновой кислоты, как синтезируемый в тромбоцитах проагрегант — тромбоксан А2, а в сосудистой стенке — основной ингибитор агрегации простациклин (простагландин 12). Мощным ингибитором агрегации является также оксид азота (NO).
Коагуляционный (вторичный) гемостаз
Коагуляционный гемостаз представляет собой многоэтапный процесс (рис. 7-3), в ходе которого можно выделить три фазы.
Во внутреннем механизме пусковым фактором служит фактор XII, активация которого происходит вследствие контакта крови с базальной мембраной сосудов при их повреждении, а также при его ферментативном расщеплении (калликреином, плазмином).
Фактор ХПа последовательно активирует XI и IX факторы. Последние два активируют фактор X, при этом действие фактора IX на фактор X усиливается в несколько тысяч раз фактором VIII. Фактор V и пластиночный фактор 3, играющий роль фосфолипидной матрицы, в совокупности повышают суммарную активность кровяного тромбопластина или протромбиназы (Ха + ф.З + ф.У + Ca2t) по сравнению с изолированным фактором Ха в 300 000 раз. Образованием кровяного тромбопластина (протромбиназы) заканчивается 1-я фаза коагуляционного гемостаза.
Фактор Ха последовательно отщепляет от фактора II (протромбина) два фрагмента, превращая его в фактор Па (итог 2-й фазы), который, в свою очередь, отщепляет от молекулы фактора I (фибриногена) четыре пептида (два пептида А и два — В), превращая его в мономеры фибрина, которые полимеризуются с образованием вначале растворимого, а затем нерастворимого фибрин-полимера под влиянием фактора XIII, активируемого тромбином (3-я фаза коагуляционного гемостаза).
Во внешнем механизме коагуляционный гемостаз стимулируется поступлением в плазму из поврежденных тканей тканевого тромбопластина (фактора III), превращающего фактор VII в Vila, который активирует
90
Лекция 7
| Внутренний механизм активации |
| Внешний механизм активации |
-----активирующее действие
----- инактивирующее действие
VIII, IX и XI факторы — антигемофильные глобулины
XIII — фибринстабилизирующий фактор
Зпф — тромбоцитарный фактор (фосфолипидная матрица)
V фактор — неферментный акселератор свертывания
Рис. 7-3. Схема свертывания крови
фактор X. Дальнейший путь образования фибрина является общим для внешнего и внутреннего пути активации. В последующем за счет активации системы сократительных белков тромбоцитов происходит ретракция кровяного сгустка.
В последние годы выявлены дополнительные (резервные) пути активации свертывания крови, включающие «по требованию» в ответ на повреждение тканей бактериально-паразитарную агрессию. Наиболее важен в этом отношении макрофагально-моноцитарный механизм гемокоагуляции.
Патофизиология системы гемостаза
91
Для оценки состояния коагуляционного гемостаза используют: время свертывания крови (5-11 мин в норме); активированное частичное тромбопластиновое время — АЧТВ (25-38 с в норме); протромбиновый индекс (95-105 % в норме); протромбиновое время — ПВ (12-15 с в норме), на основе которого рассчитываются протромбиновое отношение — ПО (отношение ПВ исследуемой плазмы к ПВ нормальной плазмы; в норме 0,7-1,1) и международное нормализованное отношение — МНО (ПО в степени МИЧ — международного индекса чувствительности тромбопластина; в норме от 1,0 до 1,4): содержание фибриногена в плазме крови (в норме составляет 2,0-4,0 г/л)
Противосвертывающие механизмы и система фибринолиза
Свертыванию крови противодействует противосвертывающая система, которая объединяет первичные и вторичные антикоагулянты.
Первичные антикоагулянты:
• гепарин — активатор антитромбина III, в комплексе с которым тормозит все три фазы ферментативной коагуляции и улучшает реологические свойства крови;
• антитромбин III — универсальный ингибитор почти всех ферментных факторов свертывания, в первую очередь тромбина и фактора Ха и 1Ха. На его долю приходится более 75 % всей антикоагулянтной активности плазмы, при этом он является основным кофактором гепарина;
• протеин С с кофактором протеином S — ингибитор неферментных факторов свертывания VIII и V;
• ингибитор пути тканевого фактора — ингибитор внешнего пути коагуляционного гемостаза;
• а2-макроглобулин — ингибитор комплекса «ф.Ш + Vila», тромбина, калликреина, плазмина;
• оц-антитрипсин — ингибитор тромбина, калликреина.
Вторигные антикоагулянты образуются в процессе свертывания крови и фибринолиза. Так, фибрин адсорбирует и инактивирует большое количество тромбина, т.е. является как фактором свертывания, так и антикоагулянтом (обозначается как антитромбин I). Продукты, образующиеся в результате фибринолиза — продукты деградации фибриногена/фибрина (ПДФ), ингибируют агрегацию тромбоцитов и полимеризацию мономеров фибрина (т.е. действуют как антиагреганты и антикоагулянты).
Возможен синтез в организме патологических антикоагулянтов, которые отсутствуют в крови в норме. К ним относятся антитела против
92
Лекция 7
ВМК калликреин **• ф. ХПа----
а2-Глобулин ---
| ПЛАЗМИНОГЕН"|
—и-----------Урокиназа, ТАП <- PAI
ПЛАЗМИН Антиплазмины
(а2-антиплазмин и др.)
| ФИБРИН
♦I ПДФ I
Неферментативный путь | ГЕПАРИН + ФИБРИНОГЕН |
Рис. 7-4. Фибринолитическая система крови
факторов свертывания (чаще к факторам VIII и IX), а также к фосфолипидам мембран (ФЛМ).
Фибринолитическая система. Ферментная система, вызывающая расщепление фибрина/фибриногена на мелкие фрагменты, обозначается как фибринолитическая или плазминовая система (рис. 7-4).
Главным компонентом этой системы служит фермент плазмин (фибринолизин), содержащийся в плазме в виде профермента — плазминогена. Активный плазмин быстро блокируется антиплазминами (главный из них относится к а2-глобулинам) и элиминируется из кровотока. В организме активация фибринолиза может осуществляться двумя путями. Внутренняя активация фибринолиза осуществляется фактором ХПа в комплексе с калликреином и ВМК. Внешняя активация фибринолиза осуществляется в основном синтезируемым в сосудистом эндотелии тканевым активатором плазминогена (ТПА), а также урокиназой, которая вырабатывается в почках. При патологии в роли активаторов фибринолиза могут выступать лейкоцитарные протеазы и бактериальные ферменты (стрептокиназа и др.).
Нарушения гемостаза можно разделить на три группы.
• Развитие тромботических состояний со склонностью к тромбозам.
• Геморрагические синдромы.
• Нарушение гемостаза смешанного характера — тромбогеморрагический синдром, развивающийся при диссеминированном внутрисосудистом свертывании (ДВС-синдром).
Тромбозы. Тромбофилии
Тромбоз — это прижизненный процесс образования в просвете кровеносных сосудов плотных масс, состоящих из форменных элементов
Патофизиология системы гемостаза
93
крови и фибрина, фиксированных к эндотелию и в той или иной мере препятствующих движению крови по сосудам.
Тромбообразование лежит в основе ряда заболеваний сердечнососудистой системы (ишемическая болезнь сердца — ИБС, атеросклероз, инсульт головного мозга, сосудистые поражения конечностей, почек и т.д.) и является в настоящее время основной причиной смерти в промышленно развитых странах. Смертность от ИБС и патологии сосудов головного мозга составляет 40-45 % общей смертности.
К факторам риска тромбообразования относят хронические стрессы, ожирение, гиподинамию, неконтролируемый прием лекарственных препаратов, в частности гормональных контрацептивов, курение, инфекции и другие повреждающие сосуды факторы.
Выделяют «тромботическую болезнь» (тромбофилию) — патологическое состояние организма, которое характеризуется повышенной склонностью к тромбообразованию, обусловленному нарушением регуляции системы гемостаза или изменением функционирования и свойств отдельных ее звеньев. Тромботические заболевания могут быть врожденными и приобретенными. Они могут протекать скрыто и проявляться в определенной ситуации тромбозами.
Факторы, способствующие тромбообразованию
• Основной фактор — повреждение сосудистой стенки (структурное или функциональное), приводящее к снижению ее тромборези-стентности.
• Нарушение гемодинамики (снижение скорости кровотока и локальный ангиоспазм).
• Активация адгезивно-агрегационной функции тромбоцитов как первичной (при болезни Вакеза), так и вторичной (при массивных травмах тканей, хирургических операциях и т.д.).
• Активация коагуляционного (вторигного) гемостаза, что наблюдается при действии различных повреждающих агентов.
• Ослабление противосвертывающей системы крови. Так, возможно истощение запасов гепарина, идущего на активацию липопротеиновой липазы при гиперлипидемии; снижение синтеза активаторов плазминогена тканевого (ТПА) и урокиназного типа, а также увеличение продукции ингибиторов этих активаторов.
При ишемической болезни сердца и других сердечно-сосудистых заболеваниях происходят сдвиг соотношения между простациклином и тромбоксаном А2 в сторону преобладания последнего, а также пони-
94
Лекция 7
жение чувствительности тромбоцитов к антиагрегационному действию простациклина.
Показана способность продуктов перекисного окисления липидов (ПОЛ), ЛПНП и никотина тормозить активность простациклина.
При острых и хронических стрессорных воздействиях в крови повышается содержание адреналина, оказывающего проагрегантное действие за счет повышения мобилизации внутриклеточного кальция и активации связывания экстрацеллярного фибриногена со специфическими фибриногеновыми рецепторами тромбоцитов (ГП Пв/Ша).
Различают тромбы: белый, который состоит главным образом из тромбоцитов и лейкоцитов; красный, образующийся в результате активации свертывающей системы с образованием нитей фибрина и задержкой в них большого количества эритроцитов, а также смешанный (встречается наиболее часто).
Возможны следующие исходы тромбоза: организация тромба, т.е. прорастание соединительной тканью, в результате чего он прочно закрепляется в сосуде; отрыв тромба и превращение его в эмбол; канализация тромба; гнойное расплавление его при инфицировании; рассасывание тромба с восстановлением кровотока в сосуде.
Принципы патогенетигеской терапии тромбозов: назначение ан-тиагрегантов, антикоагулянтов, фибринолитических препаратов; повышение тромборезисгентности сосудистой стенки, нормализация реологических свойств крови и гемодинамики.
Геморрагические синдромы
Этиопатогенетическая классификация геморрагических синдромов
• Нарушение механизмов первичного гемостаза (тромбоцитарнососудистой реакции):
— изменение количества тромбоцитов (тромбоцитопении, тром-боцитемии);
— нарушение адгезии тромбоцитов (болезнь Виллебранда, тромбоцитопатия Бернара-Сулье);
— нарушение агрегации тромбоцитов (дефицит рецепторов ПЬ/Ша — тромбастения Гланцманна, передозировка антиагрегантов);
— аномалии сосудистой стенки: врожденные (телеангиэктазии) и приобретенные — инфекционного или инфекционно-иммунного генеза (болезнь Шенлейна-Геноха).
Патофизиология системы гемостаза
95
• Нарушение механизмов вторичного (коагуляционного) гемостаза: — дефицит прокоагулянтов врожденный (нарушение синтеза анти-гемофильных глобулинов при гемофилиях; синтез аномального фибриногена) и приобретенный (нарушение синтеза прокоагулянтов группы протромбина при патологии печени);
• Преобладание эффектов антикоагулянтов и фибринолитической системы.
Тромбоцитопении
Группа заболеваний, при которых колигество тромбоцитов крови составляет 150х10ч/л и ниже {при норме 180-ЗООх.КР/л).
Снижение количества тромбоцитов может быть обусловлено повышенным их разрушением или потреблением, а также недостаточным образованием. Наиболее часто встречаются аутоиммунные тромбоцитопении (аутоиммунная тромбоцитемическая пурпура), при которых происходит разрушение тромбоцитов вследствие воздействия на них антитромбоцитарных антител. В основе патогенеза лежит срыв иммунологической толерантности. Во всех случаях тромбоцитопенической пурпуры отмечается резкое укорочение продолжительности жизни тромбоцитов до нескольких часов, вместо 7-10 сут. Число тромбоцитов, образующихся в единицу времени, увеличивается по сравнению с нормой в 2-6 раз, что обусловлено повышением синтеза тромбоцито-поэтинов в ответ на снижение количества тромбоцитов. Основным местом выработки антитромбоцитарных тел, относящихся к классу IgG3, является селезенка. Идиопатическая (врожденная) форма болезни развивается без явной связи с каким-либо предшествующим заболеванием, симптоматические формы наблюдаются при хроническом лейкозе, миеломной болезни, хроническом гепатите, системной красной волчанке (СКВ), ревматоидном артрите. Геморрагический синдром характеризуется петехиально-синячковым типом кровоточивости. Возможны кровоизлияния в мозг, реже наблюдаются кровотечения из ЖКТ, гематурия, кровохарканье и др. Кровотечения при удалении зубов начинаются сразу после вмешательства и продолжаются несколько часов или дней, чем отличаются от рецидивирующих кровотечений при гемофилии. Пробы на ломкость капилляров чаще положительны. Увеличение селезенки и печени не характеры.
При лабораторном исследовании периферической крови регистрируется снижение числа тромбоцитов (иногда вплоть до их полного исчезновения) при нормальном или повышенном содержании плазмен-
96
Лекция 7
них факторов свертывания. Признаки геморрагического диатеза появляются при снижении количества тромбоцитов до 50х109/л и ниже. Время кровотечения чаще бывает удлиненным. Ретракция кровяного сгустка уменьшена. Свертываемость крови у большинства больных нормальная. Нередко при аутоиммунной тромбоцитопении наблюдаются функциональные нарушения тромбоцитов.
Патогенетическая терапия аутоиммунных тромбоцитопений складывается из применения кортикостероидных гормонов, лечения иммунодепрессантами и спленэктомии. Симптоматическое лечение включает местные и общие гемостатические средства.
Гемофилия
Гемофилия — классигеское наследственное заболевание, характеризующееся периодигески повторяющимися, трудно останавливаемыми кровотегениями, обусловленными недостатками факторов свертывания крови-, при гемофилии А — фактора VII; при гемофилии В — фактора IX; при гемофилии С — фактора XI.
Гемофилия А
В норме фактор VIII циркулирует в крови в форме крупномолекулярного белкового полимера. При дефиците фактора VIII нарушается первая стадия коагуляционного гемостаза — образование протромбиназы (или кровяного тромбопластина). Следствием этого являются нарушение образования тромбина и развитие геморрагического синдрома. При этом тяжесть синдрома строго коррелируется со степенью дефицита фактора VIII в плазме крови. Для гемофилии характерен гематомный тип крово-тогивости. Образующиеся гематомы могут сдавливать периферические нервные стволы и крупные сосуды, что сопровождается выраженным болевым синдромом и может привести к развитию параличей и гангрен. Гемофилии свойственны продолжительные кровотечения из слизистых оболочек носа, десен, опасны любые медицинские манипуляции (экстракция зуба, внутримышечные инъекции, тонзилэктомии и т.п.). Возможны кровоизлияния в головной мозг и мозговые оболочки. Типичным симптомом являются кровоизлияния в суставы (чаще крупные) — гемартрозы, при повторении которых возможно развитие анкилозов. Особенностью геморрагического синдрома при гемофилии является отсроченный характер кровотечений (через 6-12 ч и более после травмы) вследствие того, что механизмы сосудисто-тромбоцитарного гемостаза не нарушены.
Для гемофилии характерны следующие данные лабораторных исследований:
Патофизиология системы гемостаза
97
• удлинение времени свертывания крови;
• замедление времени рекальцификации;
• нарушение образования протромбиназы;
• снижение потребления протромбина;
• уменьшение содержания фактора VIII;
• время кровотечения по Дьюку соответствует норме;
• резистентность сосудистой стенки не изменена.
Основной метод лечения — заместительная терапия, для которой пригодны только трансфузия свежеполученной крови, а также концентраты фактора VIII, антигемофильная плазма.
Синдром диссеминированного внутрисосудистого свертывания крови
Термином «синдром диссеминированного внутрисосудистого свертывания крови» (ДВС-синдром, тромбогеморрагигеский синдром) обознага-ют неспецифигеский патологигеский процесс, связанный с поступлением в кровь активаторов ее свертывания и агрегации тромбоцитов, образованием тромбина, активацией и последующим истощением плазменных ферментных систем (свертывающей, ККС, фибринолитигеской и др.), образованием в крови множественных микросгустков и агрегатов клеток, блокирующих микроциркуляцию в органах. Указанные изменения приводят к развитию тромбогеморрагий, гипоксии, ацидоза, дистрофии и глубокой дисфункции органов, интоксикации организма продуктами белкового распада и другими метаболитами.
Чаще всего возникновение ДВС-синдрома обусловливают следующие патологические процессы и воздействия: тяжелые септические состояния, массивный цитолиз, тяжелые аллергические реакции, все виды шока, обширные хирургические вмешательства, все терминальные состояния.
В патогенезе ДВС-синдрома центральное место занимают образование в сосудистом русле тромбина (тромбинемия) и истощение как гемокоагуляционного потенциала, так и механизмов, препятствующих свертыванию крови и агрегации тромбоцитов.
В течении тромбогеморрагического или ДВС-синдрома различают четыре стадии:
• 1 стадия — гиперкоагуляторная, характеризуется активацией свертывания крови, внутрисосудистой агрегацией клеток, блокадой микроциркуляции в органах и тканях;
98
Лекция 7
• 2 стадия — коагулопатия потребления, в которую происходят уменьшение количества тромбоцитов, снижение содержания фибриногена с возможным развитием гипокоагуляции;
• 3 стадия — гипокоагуляция и активация фибринолиза. В эту стадию возможно развитие тяжелых профузных кровотечений вследствие генерализации фибринолиза, повреждения при этом фибриногена и других факторов свертывания, блокады части оставшегося фибриногена РФМК (растворимыми фибрин-мономерными комплексами), блокады оставшихся тромбоцитов ПДФ (продуктами деградации фибрина);
• 4 стадия — восстановительная (или остаточных проявлений), для которой характерно постепенное восстановление механизмов гемостаза, дистрофические и некротические изменения в тканях и развитие органной недостаточности в органах-мишенях (легких, почках).
Летальность при острых формах ДВС-синдрома достигает 30-50 %.
Принципы терапии ДВС-синдрома
• Этиотропное лечение — немедленное принятие мер по устранению всех причинных факторов развития ДВС-синдрома.
• Поддержание необходимого объема и состава крови. С этой целью проводят струйное введение криоплазмы и переливание крови (только свежей).
• Коррекция нарушений гемостаза. В фазе повышенной свертываемости крови немедленно следует начинать внутривенное введение гепарина (в последующем он вводится в малых дозах для прикрытия трансфузионной терапии). Поддержанию антитромботическо-го потенциала крови (антитромбина III, протеина С, плазминогена и физиологических антиагрегантов) способствует трансфузионная терапия.
• Лечение органной патологии (шокового легкого, острой почечной недостаточности).
Лекция 8
ПАТОФИЗИОЛОГИЯ СИСТЕМЫ КРОВИ I
Г.П. Щелкунова
Изменения объема циркулирующей крови
Кровь — важнейшая составная часть организма, обеспечивающая его гомеостаз. Она переносит к тканям кислород из легких и удаляет из тканей углекислоту (дыхательная функция), доставляет клеткам необходимые для жизнедеятельности вещества (транспортная функция), участвует в терморегуляции, в поддержании водного баланса и выведении токсических веществ (дезинтоксикационная функция), в регуляции кислотно-основного состояния (КОС). От количества крови зависят величина артериального давления и работа сердца, функция почек и других органов и систем. Лейкоциты обеспечивают клеточный и гуморальный иммунитет. Тромбоциты вместе с плазменными факторами свертывания останавливают кровотечение.
Кровь состоит из плазмы и форменных элементов — эритроцитов, лейкоцитов и тромбоцитов. В 1 л крови на долю форменных элементов (главным образом эритроцитов) приходится у мужчин 0,41-0,53 л [гематокрит (Ht) 41-53 %], а у женщин — 0,36-0,48 л (гематокрит 36-48 %). Количество крови у человека составляет 7-8 % массы его тела, т.е. у человека массой около 70 кг • 5 л.
При анемии количество эритроцитов в крови снижается (Ht ниже нормы), но объем циркулирующей крови (ОЦК) сохраняется нормальным за счет плазмы. Такое состояние называется олигоцитемигеская нормоволемия. В этом случае из-за дефицита гемоглобина (НЬ) уменьшается кислородная емкость крови и развивается гипоксия гемического (кровяного) типа.
При увеличении в крови числа эритроцитов (эритроцитоз) на фоне нормального ОЦК развивается полицитемигеская нормоволемия (Ht выше нормы). В большинстве случаев эритроцитоз, исключая некото-
100
Лекция 8
рые патологические формы (см. ниже), компенсирует гипоксии различного генеза благодаря повышению кислородной емкости крови. При значительных увеличениях Ht могут повышаться вязкость крови и нарушаться микроциркуляция.
Гиповолемия
Гиповолемия — уменьшение ОЦК.
Различают 3 формы гиповолемий: простая, олигоцитемическая и по-лицитемическая.
Простая гиповолемия возникает в первые минуты (часы) после массивной острой кровопотери, когда на фоне уменьшения ОЦК Ht остается нормальным (скрытая анемия). При этом в зависимости от степени уменьшения ОЦК могут наблюдаться падение АД, уменьшение сердечного выброса (УОС, МОС), тахикардия, перераспределение кровотока, выброс депонированной крови, уменьшение диуреза, нарушения мозгового кровообращения вплоть до потери сознания и другие последствия. Из-за ослабления микроциркуляции и уменьшения общего количества НЬ развивается гипоксия циркуляторного и гемического (смешанного) типа.
Олигоцитемигеская гиповолемия характеризуется уменьшением ОЦК и снижением Ht. Такое состояние может развиться у больных, страдающих тяжелой анемией, осложненной острым кровотечением или обезвоживанием, например, при лейкозах, апластических анемиях, лучевой болезни, злокачественных опухолях, некоторых болезнях почек и т.п. При этом развивается очень тяжелая гипоксия смешанного типа, обусловленная как дефицитом НЬ, так и нарушением центрального и периферического кровообращения.
Лучшим способом коррекции простой и олигоцитемической гиповолемии является переливание крови или кровезаменителей.
Полицитемигеская гиповолемия характеризуется уменьшением ОЦК и увеличением Ht. Ее причиной главным образом служит гипогидратация, при которой из-за дефицита воды в организме уменьшается объем плазмы крови. И хотя при этом кислородная емкость крови остается нормальной (НЬ в норме), развивается гипоксия циркуляторного типа, так как в зависимости от степени обезвоживания уменьшение ОЦК приводит к падению АД, уменьшению сердечного выброса, нарушению центрального и периферического кровообращения, уменьшению фильтрации в клубочках почек, развитию ацидоза. При этом происходит увеличение вязкости крови, затрудняющее и без того ослабленную микроциркуляцию, повышающее риск образования тромбов.
Патофизиология системы крови I
101
Для восстановления ОЦК необходимо вливать жидкости и применять препараты, снижающие вязкость крови и улучшающие ее реологические свойства, а также дезагреганты и антикоагулянты для профилактики тромбоза.
Гиперволемия
Гиперволемия — увеличение ОЦК. Различают также 3 формы гиперволемий: простую, олигоцитемическую и полицитемическую.
Простая гиперволемия может наблюдаться после массивных гемотрансфузий и сопровождаться увеличением АД и МОС. Обычно носит временный характер, так как благодаря включению регуляторных механизмов ОЦК возвращается к норме.
Олигоцитемигеская гиперволемия характеризуется увеличением ОЦК и снижением Ht. Развивается обычно на фоне гипергидратаций, когда увеличение воды в организме сопровождается увеличением объема плазмы крови. Особенно опасно такое состояние у больных с почечной недостаточностью и хронической застойной сердечной недостаточностью, так как при этом повышается АД, развивается перегрузка сердца и его гипертрофия, возникают отеки, в том числе опасные для жизни. Гиперволемия и гипергидратация у этих больных обычно поддерживаются активацией ренин-ангиотензинальдостероновой системы (РААС) и развитием вторичного альдостеронизма.
Для восстановления ОЦК следует использовать диуретики, блокаторы РААС [главным образом блокаторы ангиотензинпревращаю-щего фермента (АПФ) — см. патофизиологию водно-электролитного обмена].
На фоне почечной недостаточности у больных обычно развивается и анемия, которая, в свою очередь, еще больше уменьшает Ht, а состояние больного усугубляется развитием гипоксии гемического типа.
Полицитемигеская гиперволемия характеризуется увеличением ОЦК и увеличением Ht. Классическим примером такого состояния является хроническое миелопролиферативное заболевание (см. ниже) — эритремия (болезнь Вакеза). У больных в крови резко увеличено содержание всех форменных элементов, особенно эритроцитов, а также тромбоцитов и лейкоцитов. Заболевание сопровождается артериальной гипертензией, перегрузкой сердца и его гипертрофией, нарушениями микроциркуляции и высоким риском тромбообразования. Больные часто умирают от инфарктов и инсультов.
102
Лекция 8
Патофизиология эритрона
Эритрон — это совокупность зрелых и незрелых клеток красной крови: эритроцитов. Эритроциты рождаются в красном костном мозге из стволовой клетки, как и все другие форменные элементы (рис. 8-1). Монопотентными клетками, из которых могут развиваться только эритроциты, являются бурстобразующие единицы эритроидные (БОЕэр), которые под влиянием почечных эритропоэтинов (ЭПО), ИЛ-3 и КСФ превращаются в колониеобразующие единицы эритроидные (КОЕэр), также реагирующие на ЭПО, и затем — в эритробласты. Эритробласты, одновременно пролиферируя, дифференцируются в пронормоциты, далее — нормоциты базофильные, нормоциты полихроматофильные и нормоциты оксифильные. Нормоциты (по классификации Кассирского, нормобласты) — это класс созревающих ядерных предшественников эритроцитов. Последней клеткой, способной к делению, является полихроматофильный нормоцит. На стадии нормоцитов происходит синтез НЬ. Оксифильные нормоциты, теряя ядра, через стадию ретикулоцита превращаются в зрелые безъядерные оксифильные эритроциты. 10-15 % предшественников эритроцитов гибнет еще в костном мозге (так называемый неэффективный эритропоэз).
В периферической крови здорового человека ядерных предшественников эритроцитов быть не должно. Из незрелых клеток красного ростка в крови в норме встречаются только ретикулоциты (или полихроматофильные эритроциты) от двух до десяти на тысячу эритроцитов (2-10 %о) или 0,2-1,0 %. Ретикулоциты (клетки, содержащие в цитоплазме сетчатую зернистость — остатки полирибосом) выявляются только при специальной суправитальной окраске красителем бриллианткрезил-блау. Эти же клетки при окраске по Райту или по Романовскому-Гимзе, воспринимая и кислые, и основные красители, имеют сиреневый цвет цитоплазмы без зернистости — полихроматофильные эритроциты.
Основную массу клеток периферической крови составляют зрелые безъядерные оксифильные эритроциты (RBC— red blood cells). Их количество у мужчин — 4-5х1012/л, у женщин — 3,7-4,7х1012/л. Поэтому Ht у мужчин — 41-53 %, а у женщин — 36-48 %.
Состояние крови в организме здорового взрослого человека характеризуют следующие показатели: общее содержание НЬ — 130-160 г/л у мужчин и 120-140 г/л у женщин. Среднее содержание НЬ в эритроците (МСН — mean contains hemoglobin) — 25-36 пг/кл (pg). Средняя концентрация НЬ в эритроците (МСНС — mean corpuscular hemoglobin
Отдел тотипотентных клеток
Отдел ствольных мульти-потентных клеток
Отдел полиолигопотентных коммитированных предшественников
Отдел моноолигопотентных коммитированных^ предшественников
Т-лимфобласт
Lckk
Т-пролимфоцит
КОЕ-Г
Монобласт
Промегакариоцит
Т-лимфоцит
Базофильный Эффинофильныи бласт С ласт
Неитрофильныи Эритробласт бласг
В-ли^^област В-пролимфоцит
Патофизиология системы крови I
’КРКМ-Д, КООБ-28 КРКМ-К
КОЕ-с 12дн, КООБ-7
ХОЕ-с 8дн
’КОЕ-бл, КОЕ-ВПП КОЕ-ЭГММ
£-5 потентные КОЕ (в любом наборе)
пре-Т
□Е-ГМ
пЗе-В
КОЕ-М
-КОЕ-Баз КОЕ-Эоз
\ БОЕ-Э
КОЕ-Нейтр КОЕ-Э
КОЕ-Мгкц
Мегана()иобласт
Промоцит
Пронормоцит
В-иммунобласт
Проплазмоцит
Ретикулоцит
Плазмоцит
Эритроцит
Базофильный нормоцит
Оксифильный нормоцит
Плазмрбласт
Моноцит Тучная клетка
Дендритные клетки
Свободные Альвеолярный Плевральный Перитонеальный Остеокласт
Селезенки и фиксированные Лимфоузлов костного мозга
Отдел морфологически узнаваемых клеток т-иммунобласт
В-лимфоцит
Дендритная НК-хлетха Активный JMltTtt .(натуральный________Т-лимфс
киллер)
Гистиоциты и купферовские клетки
Рис. 8-1. Схема кроветворения
Полихроматофильный нормоцит
Мегакариоцит Тромбоциты
GO
104
Лекция 8
concentration) — 310-370 г/л. Средний диаметр эритроцитов 6-8 мкм, а средний объем клетки (СОК или MCV — mean corpuscular volume) — 80-100 мкм3 (fl). Скорость оседания эритроцитов (СОЭ) у мужчин — 1-10 мм/ч, а у женщин — 2-15 мм/ч. Осмотическая резистентность эритроцитов (ОРЭ), т.е. их устойчивость к гипотоническим растворам NaCl: минимальная — 0,48-0,44 %, а максимальная — 0,32-0,28 % NaCl. Благодаря своей двояковогнутой форме нормальные эритроциты имеют резерв прочности при попадании в гипотоническую среду. Их гемолизу предшествуют перемещение воды в клетки и превращение их в легко разрушающиеся сфероциты.
Максимальная продолжительность жизни эритроцитов в крови — 100-120 сут. Разрушаются отжившие эритроциты в ретикулоэндотелиальной системе, главным образом в селезенке («кладбище эритроцитов»). При разрушении эритроцитов путем последовательных превращений образуется пигмент билирубин.
Патология эритрона может выражаться как в изменении количества эритроцитов, так и в изменении их морфологических и функциональных свойств. Нарушения могут происходить на этапе их рождения в костном мозге, на этапе их циркуляции в периферической крови и на этапе их гибели в ретикулоэндотелиальной системе.
Эритроцитозы
Эритроцитоз — увеличение содержания эритроцитов в единице объема крови, сопровождающееся повышением Ht и НЬ.
Эритроцитоз может быть относительным и абсолютным, приобретенным и наследственным.
Относительный эритроцитоз является следствием уменьшения объема плазмы крови, главным образом на фоне гипогидратации. Из-за уменьшения объема плазмы в единице объема крови увеличивается содержание эритроцитов, НЬ и растет Ht, повышается вязкость крови и нарушается микроциркуляция. И хотя кислородная емкость крови не изменяется, ткани могут испытывать кислородное голодание по причине нарушения кровообращения.
Абсолютные эритроцитозы приобретенные (вторигные) обычно являются адекватной реакцией организма на гипоксию тканей. При дефиците кислорода в воздухе (например, у жителей высокогорья), при хронической дыхательной и сердечной недостаточности, при увеличении сродства НЬ к О2 и ослаблении диссоциации оксигемоглобина в тканях, при угнетении тканевого дыхания и тому подобных ситуациях
Патофизиология системы крови I
105
включается универсальный компенсаторный механизм: в почках (главным образом) вырабатываются ЭПО, под влиянием которых чувствительные к ним клетки усиливают свою пролиферацию, и в кровь из костного мозга поступает большее число эритроцитов (так называемый физиологигеский, гипоксигеский, компенсаторный эритроцитоз). Это сопровождается увеличением кислородной емкости крови и усилением ее дыхательной функции.
Абсолютные эритроцитозы наследственные (первичные) могут быть нескольких видов:
• аутосомно-рецессивный дефект в аминокислотных участках НЬ, ответственных за его деоксигенацию, приводит к увеличению сродства НЬ к кислороду и затрудняет диссоциацию оксигемоглобина в тканях, которые получают меньше кислорода. В ответ на гипоксию развивается эритроцитоз:
• понижение в эритроцитах 2,3-дифосфоглицерата (может снижаться на 70 %) также приводит к увеличению сродства НЬ к кислороду и затруднению диссоциации оксигемоглобина. Результат аналогичный — в ответ на гипоксию вырабатываются ЭПО и усиливается эритропоэз:
• постоянная повышенная продукция эритропоэтинов почками, которые по причине аутосомно-рецессивного генетического дефекта перестают адекватно реагировать на уровень оксигенации тканей;
• генетически обусловленная усиленная пролиферация эритроидных клеток в костном мозге без увеличения ЭПО.
Наследственные эритроцитозы являются патологическими, характеризуются увеличением Ht, вязкости крови и нарушением микроциркуляции, гипоксией тканей (особенно при увеличении сродства НЬ к О2), увеличением селезенки (рабочая гипертрофия), могут сопровождаться головными болями, повышенной утомляемостью, варикозным расширением сосудов, тромбозами и другими осложнениями.
Анемии
Общая характеристика и классификация
Анемия (дословно — бескровие, или общее малокровие) — клиникогематологический синдром, характеризующийся уменьшением содержания НЬ и (за редким исключением) числа эритроцитов в единице объема крови.
В результате уменьшения количества эритроцитов снижается и показатель Ht.
106
Лекция 8
Поскольку для всех анемий характерен низкий уровень НЬ, а значит, снижена кислородная емкость крови и нарушена ее дыхательная функция, то у всех больных, страдающих анемией, развивается гипоксигеский синдром гемигеского типа.
Клинигеские проявления-, бледность кожных покровов и слизистых оболочек, слабость, повышенная утомляемость, головокружение, могут быть головная боль, одышка, сердцебиение с тахикардией или аритмией, боли в сердце, иногда изменения на ЭКГ.
Так как на фоне низкого Ht снижается вязкость крови, то следствием этого обычно служит увелигение СОЭ (чем меньше эритроцитов, тем быстрее они оседают), а также такие симптомы, как шум в ушах, систолический шум на верхушке сердца и шум «волчка» на яремных венах.
Все анемии принято классифицировать: по патогенезу, типу эритропоэза, цветовому показателю (ЦП) и МСН, диаметру эритроцитов и СОК, функциональному состоянию костного мозга (его регенераторной способности).
По патогенезу и этиологии выделяют следующие виды анемий.
• Анемии вследствие нарушенного кровообразования (гемопоэза). В эту группу входят все дефицитные анемии: железодефицитные (ЖДА), В12- и фолиеводефицитные анемии (В12-ФДА), сидеробластные анемии (СБА), анемии при дефиците белка, микроэлементов и других витаминов, а также анемии, обусловленные нарушениями самого костного мозга — гипо- и апластические анемии. В последние годы отдельно рассматривают анемии при хронических заболеваниях (АХЗ).
• Анемии вследствие кровопотери — это острые и хронические постгеморрагические анемии. Следует, однако, заметить, что хронические кровопотери обычно сопровождаются дефицитом железа и поэтому имеют патогенез и клинические проявления ЖДА.
• Анемии вследствие повышенного кроворазрушения — это все гемолитические анемии, как приобретенные, так и наследственные.
По типу эритропоэза анемии подразделяют на две группы.
• Анемии мегалобластические. Такой тип эритропоэза является главным признаком (патогномоничным) для В12-ФДА и ФДА.
• Анемии нормобластические. Нормальный тип эритропоэза характерен для всех анемий, кроме В12-ФДА и ФДА.
По цветовому показателю (ЦП) анемии подразделяют на гипо-, ги-пер- и нормохромные. В норме ЦП равен 0,85-1,05 (рассчитывается по формуле — НЬ г/лхЗ: три первые цифры числа эритроцитов). Следует
Патофизиология системы крови I
107
заметить, что по ЦП гиперхромными являются мегалобластические В12-ФДА и ФДА и некоторые гемолитические анемии на фоне гемолитических кризов. Гипохромными — ЖДА, СБА, некоторые гемолитические анемии и анемии при хронических заболеваниях. Однако с внедрением в клиническую практику современных цитометрических методов оценки крови (МСН и МСНС) определение ЦП все больше утрачивает свое значение.
По среднему объему клеток (СОК или MCV) выделяют следующие анемии:
• Анемии микроцитарные — MCV меньше 80 мкм3. Таковыми являются ЖДА, СБА, талассемия и некоторые АХЗ. Диаметр большинства эритроцитов при этих заболеваниях также меньше 6 мкм (кривая Прайс-Джонса сдвинута влево).
• Анемии макроцитарные — MCV больше 100 мкм3 (обычно — 110— 120 мкм3). В эту группу входят прежде всего мегалобластные анемии (В12-ФДА и ФДА), а также немегалобластные — анемии при хронических заболеваниях печени, алкоголизме, микседеме и др. Диаметр большинства эритроцитов в этих случаях колеблется от 8 до 12 и даже 14 мкм (кривая Прайс-Джонса сдвинута вправо).
• Анемии нормоцитарные — MCV от 80 до 95-100 мкм3. Нормальный объем эритроцитов характерен для гипо- и апластических анемий, острой постгеморрагической анемии, для сфероцитоза Минковского-Шоффара и некоторых АХЗ. Следует подчеркнуть, что по диаметру эритроцитов анемия Минковского-Шоффара является микроцитарной, а по MCV — нормоцитарной, так как эритроциты сохраняют нормальный объем за счет сферической формы.
По регенераторной способности костного мозга выделяют следующие виды анемий.
• Арегенераторные анемии — это апластические анемии, при которых костный мозг практически не функционирует.
• Гйпорегенераторные анемии, к которым относятся все дефицитные анемии, гипо- и метапластические анемии, анемии при хронических заболеваниях. В этих случаях регенерация недостающих эритроцитов в костном мозге происходит, но не адекватно степени анемии.
• Регенераторные анемии — обычно это острые постгеморрагические анемии.
• Пшеррегенераторные анемии — все гемолитические анемии, так как под влиянием продуктов гемолиза и ЭПО неповрежденный
108
Лекция 8
костный мозг усиленно функционирует, и нет дефицита веществ, необходимых для построения клеток. Продукция эритропоэтинов в почках повышена при всех анемиях в ответ на гипоксию, однако при гипо- и апластических анемиях поврежден костный мозг, а в других случаях имеется дефицит железа или витаминов либо нарушена их утилизация костным мозгом.
Критериями функциональной (регенераторной) активности костного мозга служат количество ретикулоцитов в крови и появление ядерных предшественников — нормоцитов, а также состояние других ростков крови (лейкоцитарного, тромбоцитарного). В особых случаях оценивают лейкоцитарно-эритроцитарный (Л/Э) индекс костного мозга (норма — 4/1).
При повышении регенераторной активности костного мозга в крови увеличивается количество ретикулоцитов (от умеренного при острой кровопотере до значительного при гемолитических анемиях). В мазке крови много полихроматофильных эритроцитов, появляются нормоциты, характерен нейтрофильный лейкоцитоз со сдвигом влево. В костном мозге Л/Э-индекс = 2/1-1/1.
При гипорегенераторных анемиях число ретикулоцитов остается нормальным или снижено, полихроматофилии нет, нормоцитов в крови нет, лейкоцитоза нет. При апластических анемиях ретикулоцитов нет, а главное — мало всех клеток крови, продуцируемых костным мозгом, — эритроцитов, лейкоцитов (гранулоцитов) и тромбоцитов, что называется панцитопенией. В костном мозге — опустошение (панмиелофтиз), кроветворная ткань замещается жиром.
В последнее время для оценки адекватности регенераторной активности костного мозга считают или нормализованное гисло ретикулоцитов, или индекс ретикулоцитов (ИР). Если ИР меньше 2 %, то продукция эритроцитов в костном мозге неадекватна степени анемии, и ее считают гипорегенераторной, даже если при этом число ретикулоцитов в крови повышено.
ИР = число ретикулоцитов (%) больного х Ht больного (%) нормальный Ht (%) х 2.
При всех анемиях, исключая только острую кровопотерю, происходит не только уменьшение числа эритроцитов, но и изменение их размеров и формы. Если в крови обнаруживаются эритроциты разных размеров (от микро- до макроцитов иногда с тенденцией в ту или другую сторону), то это явление называется анизоцитозом. Если в крови встречаются эритроциты различной неправильной формы — это назы
Патофизиология системы крови I 109
вается пойкилоцитозом. Пойкилоциты могут иметь специфический вид для некоторых наследственных гемолитических анемий. Анизоцитоз и пойкилоцитоз характерны для всех анемий, кроме острых постгеморрагических!
Анемии арегенераторные и гипорегенераторные (гипопролиферативные)
Гипо- и апластигеские анемии — это самые прогностически неблагоприятные заболевания, так как их патогенез связан с резким угнетением кроветворения в костном мозге.
Этиология
Этиология гипопластических анемий в более 50 % случаев остается невыясненной. Встречаются врожденные гипопластигеские анемии, например анемия Фанкони, при которой, кроме нарушения кроветворения, имеются пороки развития почек, сердца, скелета.
Причинами приобретенных гипо- и апластигеских анемий могут быть: действие на организм ионизирующего излучения, которое в первую очередь поражает лимфоидные органы, а также костный мозг: повреждение кроветворных клеток костного мозга химическими веществами (пары ртути, бензол, толуол, анилиновые красители, химические вещества, используемые в промышленности и бытовой химии). Особую актуальность это приобрело в связи с распространением токсикоманий. Длительный прием многих лекарственных препаратов, прежде всего цитостатических, используемых в лечении онкологических заболеваний, а также самых банальных лекарств (антибиотиков, сульфаниламидов, противовоспалительных и противотуберкулезных), может вызвать угнетение кроветворения. Нередки случаи развития апластических анемий после перенесенных вирусных инфекций. Метастазы злокачественных опухолей в костный мозг также подавляют кроветворение, но анемию в этом случае обычно называют метапластигеской (замещение нормальных ростков кроветворения опухолевыми клетками).
Патогенез
Может быть связан как с повреждением и уменьшением числа стволовых клеток под влиянием внешних факторов (радиация, химические вещества, вирусы) или иммунных механизмов (до 40 % апластических анемий — аутоиммунные), так и с нарушением процессов регуляции кроветворения (например, дефицит или нарушение передачи сигналов
110
Лекция 8
на клетки различных КСФ, цитокинов, ЭПО, гормонов, регулирующих гемопоэз). Особую роль может играть нарушение микроокружения в системе кроветворения. Например, под влиянием вирусов может происходить изменение генетической программы в стромальных клетках костного мозга и приводить к нарушению процессов самоподдержания стволовых клеток.
Во всех случаях количество кроветворных клеток в костном мозге уменьшается, их место занимают адипоциты — красный костный мозг замещается жиром вплоть до полного опустошения (панмиелофтиз). Иногда в некоторых участках костного мозга сохраняются очаги кроветворения — так называемые «горячие карманы» (hot pockets), которые выпускают в периферическую кровь молодые клетки, маскирующие арегенераторный характер анемии. Усиливается неэффективный эритропоэз, ускоряется апоптоз клеток крови, возникает раннее старение эритроцитов и других клеток на периферии (прогерия) и уменьшение продолжительности их жизни.
Главным следствием всех этих нарушений является абсолютная панцитопения — уменьшение в периферической крови эритроцитов, тромбоцитов, лейкоцитов (гранулоцитопения в сочетании с относительным лимфоцитозом). При лучевой болезни наблюдается и абсолютная лим-фоцитопения. Молодых клеток (ретикулоцитов, нормоцитов, юных нейтрофилов) в крови нет или очень мало (ИР всегда значительно меньше 2 %). Встречаются анизоциты и пойкилоциты. В крови повышено содержание железа, так как неработающий костный мозг его не утилизирует. Может развиться сидероз внутренних органов. СОЭ увеличена как по причине уменьшения Ht и вязкости крови, так и в связи с изменением белковых фракций крови на фоне обычно присоединяющихся вторичных инфекций.
Клиническая картина
Проявляется сочетанием трех ведущих синдромов: гипоксигеского — из-за снижения НЬ и эритроцитов, геморрагигеского — из-за дефицита тромбоцитов и нарушения гемостаза и иммунодепрессивного — из-за дефицита гранулоцитов, обеспечивающих антимикробную защиту (фагоцитоз). При лучевой болезни абсолютная лимфоцитопения приводит к резкому снижению специфического (клеточного и гуморального) иммунного ответа.
Для уточнения диагноза проводят исследования пунктата костного мозга. Заболевание дифференцируют с гемобластозами.
Патофизиология системы крови I
111
Принципы патогентической терапии сводятся к переливанию эритроцитарной и тромбоцитарной массы, стимуляции гемопоэза с помощью эритропоэтинов, цитокинов, гормонов, к профилактике инфекционных осложнений. В ряде случаев необходима трансплантация костного мозга.
Необходимо запомнить: гипоапластигеские анемии в типичных случаях по типу эритропоэза — нормобластигеские, по функциональному состоянию костного мозга — арегенераторные, по среднему диаметру и MCV — нормоцитарные, по ЦП нормохромные (редко гипохромные), МСН и МСНС — в норме.
Железодефицитные анемии
Это наиболее распространенные анемии среди населения земного шара — они составляют более 70 % от числа всех анемий.
В крови взрослого человека в норме содержится 12-32 мкмоль/л железа, которое связано с транспортным белком плазмы — трансферрином (коэффициент насыщения трансферрина железом равен 30-40 %, т.е. 2/3 молекул трансферрина могут дополнительно связывать железо). Общая железосвязывающая способность крови (ОЖСС) — 30,6-80,6 мкмоль/л, латентная железосвязывающая способность (ЛЖСС) — 20-50 мкмоль/л.
Общее количество железа в организме — 4-5 г (у мужчин 50 мг/кг, а у женщин — 40 мг/кг), из которого большая часть (около 60 %) содержится в эритроцитах в составе НЬ. Остальное железо входит в состав миоглобина, железосодержащих ферментов, трансферрина, ферритина, гемосидерина. Трансферрин — транспортный белок, синтезируемый в печени. Ферритин — внутриклеточный белок, депонирующий железо в нетоксичной форме (1 молекула ферритина связывает 4500 атомов железа). Часть ферритина превращается в гемосидерин — нерастворимое соединение, содержащее железо в менее доступной форме. Органы — депо железа: печень (гепатоциты, макрофаги), костный мозг, селезенка, мышцы.
Железо поступает в организм с пищей и лучше всего усваивается гемовое железо, содержащееся в мясе (гемового железа всасывается 20-30 %, а негемового — менее 5 %). В других продуктах, в том числе растительного происхождения, железо содержится в окисной (трехвалентной) форме и для его всасывания необходимо превращение в закисное (двухвалентное) железо. Этому способствует кислая среда желудка, где, кроме соляной кислоты, могут содержаться поступающие с пищей органические кислоты (аскорбиновая, яблочная, молочная и др.). Фос
112
Лекция 8
фаты, бикарбонаты, содержащийся в чае танин ухудшают всасывание железа, так как образуют его нерастворимые осадки.
Всасывание железа происходит в проксимальной части тонкого кишечника. В стенке кишечника содержится белок апоферритин, который, образуя железопротеиновый комплекс, превращается в ферритин. Система апоферритин-ферритин в определенной степени влияет на количество и скорость всасывания железа, не допуская его избыточного поступления. При уменьшении железа в крови его всасывание, как правило, увеличивается. В виде ферритина железо депонируется в печени и в костном мозге в клетках-предшественниках эритроцитов. Незрелые эритроидные клетки, в цитоплазме которых содержатся гранулы железа в виде ферритина, называются сидеробластами. Количество сидеробластов в норме колеблется от 20 до 80 %, но главным является не столько их число, сколько число глыбок железа (ферритина) вокруг ядра (в норме 1-2). Часть железа находится в макрофагах, которые доставляют его эритроидным предшественникам в костном мозге, а также используют железо в бактерицидных механизмах (лактоферрин). Транспортируется железо в костный мозг кровью с помощью трансферрина. Около 80 % железа, освобождающегося из разрушенных эритроцитов, снова используется в эритропоэзе, около 20 % — депонируется.
При истощении запасов железа в организме увеличиваются количество и скорость его всасывания (ОЖСС и ЛЖСС при этом повышаются). Исключением являются некоторые анемии при хронических заболеваниях, когда на фоне дефицита железа одновременно нарушено и его всасывание, или, наоборот, при талассемии на фоне высокого содержания железа в крови, но резко повышенного неэффективного эритропоэза всасывание железа увеличивается.
Этиология
Этиология железодефицитных анемий многообразна.
• Может наблюдаться врожденный дефицит железа у недоношенного ребенка или ребенка, рожденного от много рожавшей матери и имеющей ЖДА.
• Алиментарная недостатогность железа встречается у детей, находящихся на искусственном неполноценном вскармливании, а также у взрослых, не получающих полноценное питание, или у строгих вегетарианцев, не использующих специальные пищевые добавки.
• Повышенное расходование железа наблюдается при беременности и грудном вскармливании ребенка, что особенно важно при повтор
Патофизиология системы крови I
113
ных беременностях, а также при частых инфекционных заболеваниях, когда железо усиленно расходуется макрофагами, а на фоне ответа острой фазы в печени снижаются продукция трансферрина и количество железа в крови. ЖДА часто встречается у быстро растущих подростков, когда поступление железа не соответствует повышенным потребностям организма.
• Нарушение всасывания железа в кишечнике обычно является следствием резекции желудка или больших отрезков тонкого кишечника (синдром короткой кишки), а также развития воспалительных процессов (дуодениты, энтероколиты, в том числе СПРУ, глютеновая энтеропатия, болезнь Крона). Могут повреждаться рецепторы кишечника к трансферрину или апоферритин-ферритиновые системы. Долгое время считалось, что дефицит соляной кислоты в желудке на фоне атрофического гастрита (гипоацидное состояние, ахилия) приводит к нарушению всасывания железа, так как оно не превращается в закисную форму. Это оказалось справедливым только по отношению к вегетарианцам, поскольку у людей, питающихся мясной пищей, железо всасывается в достаточном количестве из мяса, и кислая среда здесь роли не играет. Однако на фоне дефицита железа снижается регенераторный потенциал слизистой оболочки ЖКТ и может развиться атрофический гастрит с пониженной продукцией желудочного сока, а также колит, сопровождающийся снижением всасывания железа. Иными словами, ахилия является скорее не причиной, а следствием ЖДА. Возможно также формирование порочного круга: дефицит железа -> атрофия слизистой обологки желудка и кишегника нарушение всасывания железа -» дефицит железа.
• Повышенная потеря железа наблюдается при хронических кровопотерях — маточных, геморроидальных, желудочно-кишечных (язвенная болезнь, язвенные колиты, варикозные расширения вен при портальной гипертензии, опухоли и т.п.), кровотечениях из десен при пародонтозе, различных геморрагических синдромах. Кровотечения — настолько частая причина ЖДА, что во многих клиниках у больных с низким содержанием НЬ сразу же при поступлении исследуют кал на скрытую кровь. В 10 мл потерянной крови содержится около 5 мг железа, что равно суточной потребности организма. Маточные кровотечения, особенно ювенильные (у девочек) или у женщин на фоне маточных опухолей и в климактерическом периоде, усугубляются гормональным дисбалан
114
Лекция 8
сом. Женские половые гормоны, в отличие от мужских, тормозят эритропоэз, ослабляют процессы включения железа в гем. При этом гемоглобинизация эритроцитов нарушается настолько, что цветовой показатель снижается до 0,5 и даже 0,4. Цвет кожи у таких больных не просто очень бледный, а бледный с зеленоватым (цвета алебастра) оттенком, что послужило поводом назвать такие ЖДА хлороз (от греч. «хлорос» — зеленый). Ранний хлороз -у девочек и поздний хлороз — у женщин. Встречается так называемый полиглобулический хлороз, характеризующийся нормальным количеством эритроцитов в крови, но очень низким содержанием в них НЬ.
• Нарушение транспорта железа может быть результатом наследственно обусловленного отсутствия трансферрина (атрансферри-немия — аутосомно-рецессивное заболевание) или нарушением синтеза этого белка при патологии печени и при ответе острой фазы (ООФ), или его повышенной потерей с мочой (протеинурия при нефропатиях).
Опухолевый процесс в организме, как правило, сопровождается железодефицитным синдромом (в том числе ЖДА).
Патогенез
Сводится к уменьшению гемоглобинизации эритроцитов. Синтез НЬ страдает больше, чем пролиферация клеток, и поэтому содержание НЬ снижается в большей степени, чем число эритроцитов. Ht снижается только при тяжелых анемиях. Это приводит к тому, что одним из ведущих признаков анемии является ее выраженная гипохромия (ЦП снижен до 0,5-0,6; МСН и МСНС резко снижены). На 1-й и 2-й стадиях ЖДА может быть нормохромная.
Железо входит в состав 72 ферментов, поэтому наряду с гипоксическим синдромом, обусловленным низким уровнем НЬ, дефицит железа проявляется многочисленными расстройствами (сидеропенигеский синдром).
Резкая мышечная слабость обусловлена не только гипоксией, но и дефицитом а-глицерофосфатоксидазы, нарушением обмена миоглобина. У девушек возможны ночное недержание мочи (энурез), частые позывы к мочеиспусканию, неспособность удерживать мочу при кашле и смехе, недоразвитие матки и различные нарушения течения беременности.
Нарушены процессы обновления кожи и слизистых оболочек. Типичные проявления сидеропенического синдрома: сухость кожи, ран
Патофизиология системы крови I
115
нее поседение и выпадение волос, повышенная ломкость и искривление ногтей, их выраженная поперечная исчерченность, воспаление око-лоногтевого валика, койлонихии — ложкообразные ногти, что можно объяснить дефицитом пролиноксидаз. Возможно развитие глоссита, эзофагита, охриплости голоса, дисфагии. Из-за нарушения образования коллагена происходит отслойка эпителия от базальной мембраны, образуются паутиноподобные наслоения и пленки в глотке, гортани и пищеводе, гиперемия небных дужек (симптом Пламмера-Уинсона), повышен риск развития опухолей пищевода. Атрофические процессы затрагивают и другие отделы ЖКТ (атрофический гастрит, энтерит). Снижение активности ферропротеинов вызывает снижение продукции соляной кислоты вплоть до ахилии, что, в свою очередь, нарушает не только всасывание железа, но и других веществ.
У многих больных отмечена склонность к кариесу зубов, наблюдаются извращения вкуса: больные едят глину, мел, лед, уголь, сырое мясо, тесто и т.п., с удовольствием вдыхают запахи керосина, ацетона, гуталина, известки и др. Данный синдром получил название pica chlorotica. Механизм его развития, возможно, связан как с атрофическими процессами на уровне рецепторного аппарата, так и с изменениями центральных анализаторов вкуса и обоняния. При восстановлении уровня железа в крови проявления данного синдрома исчезают.
На фоне сниженной антибактериальной и антигрибковой защиты у больных часто развиваются ангулярный стоматит (заеды), грибковый стоматит (молочница), угри, фурункулы и другие инфекционные заболевания.
В мазках периферической крови больных ЖДА встречаются кольцевидные эритроциты (анулоциты) с расширенной зоной просветления в центре из-за малого количества НЬ, выражена анизохромия (эритроциты разной степени окраски). На фоне выраженного пойки-лоцитоза встречаются мишеневидные и карандашеподобные клетки. Анизоцитоз характеризуется преобладанием микроцитов (диаметр клеток меньше 6 мкм, кривая Прайс-Джонса сдвинута влево, MCV меньше 80 fl). На начальных стадиях и при легких формах MCV сохраняется нормальным.
В крови снижены содержание железа (менее 12 мкмоль/л), коэффициент насыщения трансферрина, повышены ЛЖСС и ОЖСС. Индекс ретикулоцитов обычно меньше 2 %. Несмотря на нормальное или несколько повышенное число ретикулоцитов, анемия рассматривается как гипорегенераторная, так как продукция эритроцитов не адекватна сте
116
Лекция 8
пени анемии, а вновь образованные эритроциты содержат мало НЬ и не выполняют свою основную функцию — дыхательную. СОЭ увеличена, ОРЭ в норме (иногда повышена максимальная ОРЭ, так как у анулоци-тов больше резерва для набухания). В некоторых случаях бывает реактивный тромбоцитоз.
В костном мозге отсутствуют сидеробласты и усилен неэффективный эритропоэз.
Следует иметь в виду, что ЖДА развивается постепенно:
• в 1-й стадии может быть скрытый дефицит железа, который не приводит к характерным изменениям крови, но вызывает увеличение всасывания железа в кишечнике;
• во 2-й стадии — на фоне также нормальной картины крови уже снижается содержание железа в плазме крови, уменьшается насыщение трансферрина и увеличивается ЛЖСС. Обычно на 1-й и 2-й стадиях болезни больные не обращаются к врачу;
• 3-я стадия характеризуется постоянным дефицитом железа и всеми характерными признаками заболевания, регенераторная способность костного мозга снижена умеренно;
• в 4-й стадии тяжелый дефицит железа приводит к большему угнетению эритропоэза, нарушению регенерации различных тканей (особенно кожи и слизистых оболочек), ослаблению функции многих ферментов (например, цитохромоксидазы), нарушению тканевого дыхания, ослаблению функции макрофагов, к гипоксическому повреждению различных органов с явлениями дистрофии (например, миокарда) и другим последствиям.
Клиническая картина
Клиническая картина (в 3-й и 4-й стадиях) характеризуется гипоксическим синдромом, проявления которого зависят от степени снижения НЬ, а также различными проявлениями вышеописанного сидеропенического синдрома.
Принципы патогенетической терапии
Сводятся к назначению диеты (повышенное потребление мяса в сочетании с витаминами), восстановлению содержания железа в крови с помощью специальных железосодержащих препаратов, применяемых или внутрь, или парентерально, если нарушено всасывание железа в кишечнике. Важно установить и устранить причину дефицита железа.
Патофизиология системы крови I
117
Следует запомнить: ЖДА (в 3-й и 4-й стадиях) по типу эритропоэза — нормобластигеские, по регенераторной способности костного мозга — гипорегенераторные, по MCV и среднему диаметру эритроцитов — микроцитарные, по ЦП — гипохромные, МСН и МСНС — ниже нормы. В крови снижено содержание железа, ОЖСС и ЛЖСС повышены.
Сидеробластные анемии
Встречаются наследственные (сцепленные с Х-хромосомой) сидероахрестические (ахрезия — неусвоение) анемии и приобретенные сидеробластные анемии (СБА) (обычно на фоне отравления свинцом или при гиповитаминозе В,).
О'
Патогенез
Сводится или к наследственному, или к приобретенному дефициту (или блокированию) ферментов, участвующих в синтезе протопорфирина и порфирина, составляющих основу для построения железосодержащей части молекулы НЬ — гема. Железо не встраивается в молекулу НЬ и так же, как при ЖДА, нарушается гемоглобинизация эритроцитов. Поэтому общим с ЖДА признаком будет гипохромия эритроцитов (ЦП низкий, МСН и МСНС — снижены), тенденция к микроцитозу (средний диаметр и MCV меньше нормы).
Главным отличием от ЖДА являются высокий уровень железа в крови (более 32 мкмоль/л), высокий уровень насыщения трансферрина и сниженные ЛЖСС и ОЖСС.
В мазке крови — такие же изменения, как и при ЖДА: много гипохромных эритроцитов (анулоцитов), анизохромия, пойкилоцитоз и анизоцитоз с преобладанием микроцитов. Молодых клеток мало, так как анемия гипорегенераторная (исключая отравление свинцом).
В костном мозге повышено содержание сидеробластов с большим количеством глыбок железа (ферритина), которое часто располагается вокруг ядра в виде кольца (ринг-формы сидеробластов). Встречаются макрофаги, переполненные железом, — сидерофаги.
В виде ферритина, гемосидерина железо в избытке откладывается во внутренних органах.
Клиническая картина
Характеризуется гипоксическим синдромом и нарушением функции тех органов, в которых развиваются гемосидероз и дистрофия.
118 Лекция 8
Принципы патогенетической терапии
Сводятся к выведению лишнего железа из крови, например, с помощью десферала, который связывает железо и выводит его с мочой. В случаях приобретенных СБА важно устранить причину заболевания.
Следует запомнить: СБА являются нормобластигескими, гипореге-нераторными, микроцитарными, гипохромными (ЦП, МСН и МСНС — ниже нормы) с высоким содержанием железа в крови и наличием ринг-форм сидеробластов в костном мозге. ОЖСС и ЛЖСС снижены. При СБА, вызванной отравлением свинцом, как правило, наблюдаются ре-тикулоцитоз и увеличение ИР >2 %, что можно объяснить повышенным гемолизом эритроцитов (прямое действие свинца), увеличением продукции стимулирующих гемопоэз цитокинов активированными макрофагами.
В12-фолиеводефицитные и фолиеводефицитные (мегалобластные) анемии
Витамин В12 (кобаламин, внешний антианемический фактор) состоит из кобальта, связанного с 4 пиррольными кольцами и нуклеотидом: продуцируется микроорганизмами, обитателями бобовых и корнеплодов; присутствует в мышцах и паренхиматозных органах животных, питающихся этими растениями: поступает в организм человека с животной пищей.
В желудке кобаламин с помощью пепсина отщепляется от животных белков и образует комплекс с R-белками (транскобаламин-1 и -III), который поступает в двенадцатиперстную кишку, где панкреатические ферменты (трипсин, хемотрипсин) отщепляют R-белки и освобожденный В12 образует комплекс с гастромукопротеином (внутренним антиа-немическим фактором Касла, поступающим из желудка). Фактор Касла, предохраняя кобаламин от разрушения, доставляет его в дистальные отделы подвздошной кишки, где при участии специальных мембранносвязанных рецепторов происходит всасывание витамина В12 в кровь. Кровь транспортирует витамин в виде его комплекса с транскобаламином II к клеткам-мишеням, в которых происходит эндоцитоз.
В митохондриях клеток-мишеней витамин В12 (в форме аденозинко-баламина) необходим для активации метилмалонил-КоА-мутазы, обеспечивающей превращение метилмалонил-КоА в сукцинил-КоА.
В (аденозинкобаламин)
4.
Метилмалонил-КоА -» сукцинил-КоА
Патофизиологиясистемы крови I
119
В цитоплазме клеток-мишеней витамин В|2 (в форме метилкобала-мина) катализирует метаболизм метилтетрагидрофолата (метил-ТГФ) в тетрагидрофолат (ТГФ), который подвергается полиглутаминированию (что сохраняет фолиевую кислоту в клетке).
Тетрагидрофолат, участвующий в синтезе пуриновых и пиримидиновых оснований, используется в синтезе ДНК. Кроме того, метилко-баламин активирует метионинсинтазу, катализирующую перемещение метильных групп от N-метил ТГФ к гомоцистеину с образованием метионина. Гомоцистеин — очень токсичная аминокислота, способная ускорять развитие атеросклероза.
Фолиевая кислота (фолат) поступает в организм в основном с пищей растительного и животного происхождения, но, в отличие от кобаламина, всасывается в тощей кишке, а в цитоплазме клеток восстанавливается в 5-метилтетрагидрофолат, отдающий метиловую группу кобаламину в процессе образования метионина из гомоцистеина. Фолиевая кислота играет роль донора 1 атома углерода при превращении дезоксиуридина в дезокситимидин в процессе синтеза ДНК.
В12 (метилкобаламин)
фолат —> метил ТГФ —> ТГФ —> синтез ДНК
Этиология
Этиология В12-фолиеводефицитных анемий многообразна.
Алиментарные формы заболевания развиваются у лиц, получающих неполноценное питание, у строгих вегетарианцев, в течение многих лет не употребляющих мясной пищи. Собственных депонированных запасов кобаламина (всего 2-5 мг) при его небольших физиологических потерях обычно человеку хватает на несколько лет. Однако если витамин В12 перестает поступать с пищей, то его дефицит может проявиться через 2-3 года (по мнению некоторых авторов, через 10-12 лет).
Нарушение всасывания кобаламина может быть следствием резекции желудка или подвздошной кишки, атрофического гастрита или колита, рака желудка или кишечника, болезни Крона, врожденного дефицита в кишечнике В12-связывающих рецепторов (синдром Иммерслунда-Грасбека), дефицита транскобаламинов-I и -III, заболеваний поджелудочной железы, синдрома Золлингера-Эллисона и т.п. Однако наиболее частой причиной является иммунное повреждение (IgG) париетальных клеток желудка и связанный с этим дефицит внутреннего фактора Касла (пернициозная анемия Аддисона-Бирмера). Диагноз пернициозная
120
Лекция 8
(злокачественная) анемия был впервые поставлен Бирмером в 1872 г., а патогенез заболевания был раскрыт только в 1920 г., после открытия Каслом гастромукопротеина. Позже был открыт и сам витамин В12.
При дефиците транскобаламина II нарушается транспорт кобаламина к клеткам-мишеням.
Повышенное расходование витамина наблюдается при дифиллоботрио-зе (паразитировании широкого лентеца), при дивертикулезе тонкой кишки (в дивертикулах скапливается много микрофлоры, усиленно поглощающей витамин), при беременности и лактации. При миелодиспластических синдромах возможно неусвоение витамина В12 костным мозгом.
Дефицит фолиевой кислоты может быть алиментарным и развивается уже через 3 нед после прекращения поступления фолатов с пищей — животных белков, свежих овощей и фруктов (фолаты разрушаются при варке). Причиной дефицита может стать повышенная потребность в данном витамине при беременности и лактации, эксфолиативном дерматите, гемолизе, злокачественной опухоли, гемодиализе, алкоголизме (алкоголь разрушает фолаты). В эксперименте доказано, что дефицит 50 % фолиевой кислоты на фоне беременности приводит к нарушению развития нервной трубки плода. Искусственное дренирование желчи в эксперименте вызывает дефицит фолиевой кислоты уже через несколько часов.
Нарушается всасывание фолиевой кислоты после резекции тощей кишки, а также на фоне заболеваний тонкой кишки (СПРУ, глютеновая энтеропатия, болезнь Крона, синдром короткого кишечника, амилоидоз и др.). Некоторые лекарства являются антагонистами фолиевой кислоты (например, метатрексат).
Патогенез
Патогенез В12-ФДА и ФДА имеет как общие, так и отличительные особенности.
При дефиците фолиевой кислоты в цитоплазме клеток имеется дефицит метилтетрагидрофолиевой кислоты (метил-ТГФ), поэтому мало образуется ТГФ и нарушается синтез ДНК при нормальном содержании витамина В12:
J-фолата —> -1метил-ТГФ -»Г ТГФ -> нарушение синтеза ДНК.
При дефиците витамина В12 (метилкобаламина) блокируется метаболизм метил-ТГФ в ТГФ, фолат расходуется на синтез дезоксиуридина, уменьшается полиглутаминирование, увеличивается утечка фолиевой кислоты из клетки:
Патофизиология системы крови I
121
J. В., (метилкобаламин)
1
Фолат (норма) -»метил-ТГФ —> X ТГФ —> нарушение синтеза ДНК.
В обоих случаях из-за дефицита тетрагидрофолата нарушается синтез ДНК и страдают прежде всего интенсивно регенерирующие ткани — это органы кроветворения (костный мозг) и слизистые оболочки (особенно ЖКТ).
Главным следствием дефицита ТГФ является переход нормобласти-ческого типа эритропоэза на мегалобластический, который характеризуется нарушением процессов деления и дифференцировки клеток. Клетки прекращают развиваться в S-фазе клеточного цикла при частичной репликации ДНК, но не могут завершить процесс деления. В результате образуется очень мало зрелых эритроцитов большого диаметра до 12-14 мкм (макроциты, мегалоциты) измененной формы (тонкие, плоские, часто овальные, без двояковогнутости). Предшественниками этих клеток являются мегалобласты (базофильные, полихроматофильные, оксифильные), которые отличаются от нормоцитов своим большим размером и крупным ядром, характерным для незрелых клеток (ядерно-цитоплазматическая диссоциация). В цитоплазме мегалоцитов и мегалобластов часто встречаются патологические включения — тельца Жолли, кольца Кебота, базофильная зернистость. Мегалоцитам приходится сильно деформироваться, чтобы продвигаться по капиллярам, они легко подвергаются гемолизу как в периферических сосудах, так и в синусах селезенки.
Выраженный дефицит ТГФ может ослабить регенерацию и других ростков крови: возможно развитие тромбоцитопении и лейкопении. Очень характерно появление в крови крупных нейтрофилов с полисег-ментированными ядрами (сдвиг вправо) на фоне дефицита молодых клеток.
В мазке крови резко выражены анизохромия, пойкилоцитоз и анизоцитоз с преобладанием гиперхромных (без зоны просветления в центре) макроовалоцитов и мегалоцитов, содержащих патологические включения. На фоне обострения анемии появляются мегалобласты. Нейтрофилы полисегментированы.
В крови умеренно или незначительно повышено содержание железа и билирубина — следствие усиленного разрушения эритроцитов. Индекс ретикулоцитов (ИР) меньше 2 %. Средний диаметр и MCV больше нормы. МСНС нормальная, но ЦП больше 1,05. Иногда встречаются лимфоцитопения, эозинофилия. СОЭ увеличена, ОРЭ снижена.
122
Лекция 8
В костном мозге усилен неэффективный эритропоэз. При исследовании пунктата костного мозга (миелограммы) выявляются признаки мегалобластического кроветворения, а обилие базофильных (с яркосиней цитоплазмой) мегалобластов позволило выделить характерный симптом — синий костный мозг.
Кроме специфических гематологических изменений при дефиците кобаламина и фолатов нарушаются процессы регенерации слизистой обо-логки ЖКТ: атрофируются сосочки языка с изменением вкусовой чувствительности, развиваются атрофический глоссит, эзофагит, гастрит, колит.
При дефиците витамина В12 (аденозинкобаламина) из-за нарушения превращения метилмалонил-КоА в сукцинил-КоА накапливается метилмалоновая кислота, которая вызывает повреждение миелиновых обо-логек боковых и задних столбов спинного мозга {фуникулярный миелоз), что сопровождается нарушением чувствительности и вторичными двигательными расстройствами.
Клинические проявления
Клинические проявления В12-ФДА включают гипоксигеский синдром, диспептигеский синдром, расстройства вкуса в сочетании с характерным лакированным малиновым языком (язык Хантера), нарушения пищеварения, а также неврологигеский синдром {кроме ФДА) (гипер- и парастезии в виде ощущения ползания мурашек, зуда кожи, покалывания, жжения), шаткость походки, симптом «ватных ног» и т.п. Следует подчеркнуть, что при избирательном дефиците только фолатов (ФДА) неврологических нарушений нет, так как в метаболизме метилмалонил-КоА фолаты не участвуют.
У некоторых больных отмечаются легкая желтушность кожи, иктеричность склер.
Принципы патогенетической терапии
Сводятся к назначению полноценного витаминизированного питания и парентеральному введению препаратов витамина В12. После нескольких инъекций витамина в крови появляются нормоциты и ретикулоциты, отражающие переход мегалобластического кроветворения в нормобластический. Мегалобласты в крови не обнаруживаются.
Следует запомнить: В12-ФДА и ФДА — мегалобластигеские, макро- и мегалоцитарные, гиперхромные по ЦП, МСН — больше нормы, МСНС — норма, гипорегенераторные.
Патофизиология системы крови I
123
Мегалобластическими могут быть также анемии при миелодиспла-стическом синдроме, эритролейкозе и на фоне применения некоторых лекарств (противосудорожных, ингибиторов синтеза ДНК, ингибиторов дигидрофолатредуктазы).
Макроцитарные немегалобластные анемии могут развиваться у больных с патологией печени, при микседеме, алкоголизме; они связаны с нарушением липидных компонентов мембран эритроцитов.
Анемии регенераторные и гиперрегенераторные (гиперпролиферативные)
Острые постгеморрагигеские анемии. Острые кровопотери довольно часто приводят к развитию анемий, тяжесть которых зависит от количества потерянной крови. Причиной массивной кровопотери могут быть травма, маточное или язвенное кровотечение, тяжелое оперативное вмешательство, кровотечение у больного, страдающего гемофилией или другим геморрагическим синдромом и т.д.
1-я стадия — скрытая анемия, так как в первые часы и даже сутки после массивной кровопотери, несмотря на уменьшение общего количества эритроцитов и НЬ, в анализе крови нет никаких изменений (НЬ, Ht, число эритроцитов — в норме). Развивается простая гиповолемия, которая характеризуется уменьшением ОЦК и нарушением кровообращения, развитием гипоксии. Происходят выброс в сосуды крови из депо, активация симпато-адреналовой системы в целях повышения тонуса сосудов и усиления работы сердца. Если кровопотеря очень тяжелая (ОЦК <30 % от должного), развивается картина гиповолемического шока: падает АД и ЦВД, из-за уменьшения венозного возврата к сердцу уменьшается сердечный выброс, развивается гипоксическая энцефалопатия вплоть до потери сознания, уменьшается диурез, пульс становится слабым, нитевидным.
2-я стадия — гидремигеская, характеризуется восстановлением ОЦК за счет объема плазмы (олигоцитемическая нормоволемия) путем перемещения воды из тканей в сосудистое русло и включения универсального компенсаторного механизма: активации РААС — уменьшении диуреза — задержки воды в организме. Кроме того, развивающаяся гипогидратация стимулирует центр жажды, и человек потребляет больше воды.
В анализе крови имеются все признаки анемии: низкие НЬ и Ht, мало эритроцитов и ускорено их оседание, но количество молодых клеток крови не увеличено. Содержание железа в крови нормальное или незначительно снижено.
124
Лекция 8
Несмотря на восстановление ОЦК и нормализацию центрального и периферического кровообращения, у больного имеются проявления гипоксического синдрома.
3-я стадия — регенераторная (или стадия костно-мозговой регенерации). В ответ на гипоксию почки выделяют эритропоэтины в первые часы после кровопотери, но признаки регенераторной активности костного мозга обнаруживаются в периферической крови только после 4-5 сут. Hb, Ht и число эритроцитов остаются пониженными, но в крови увеличивается количество ретикулоцитов (полихроматофильных эритроцитов), появляются нормоциты, наблюдаются нейтрофильный лейкоцитоз со сдвигом влево и тромбоцитоз. Продолжительность стадии зависит от тяжести анемии.
Принципы лечения
При легких формах острой постгеморрагической анемии никакого вмешательства не требуется. При анемии средней тяжести вливают изотонические растворы, растворы глюкозы и используют кровезаменители для быстрого восстановления ОЦК. При тяжелых анемиях требуется переливание крови. Важно устранить причину кровопотери.
Следует запомнить: острая постгеморрагическая анемия — нор-мобластическая, нормохромная (ЦП, МСН и МСНС — норма), нормоцитарная (по диаметру и объему эритроцитов, MCV — норма), регенераторная (ретикулоцитоз, ИР >2 %, нейтрофильный лейкоцитоз со сдвигом влево, тромбоцитоз — но в 3-й стадии!).
Гемолитические анемии
Анемии вследствие повышенного кроворазрушения или гемолитические анемии могут быть приобретенными и наследственными.
Этиология и патогенез
Разнообразны: эритроциты могут разрушаться под действием ядов (змеиного, грибного), химических соединений (фенилгидразин), токсинов микробов (при сепсисе); при тяжелых интоксикациях на фоне ожоговой болезни или уремии; под влиянием механических воздействий, например, после трансплантации искусственных клапанов сердца или протезировании сосудов, на фоне сильной вибрации и сотрясения организма (например, маршевая гемоглобинурия у солдат).
Патофизиология системы крови I 125
Частой причиной гемолиза являются переливание несовместимой по группе крови или резус-конфликт при гемолитической болезни новорожденного, а также иммунное повреждение эритроцитов на фоне длительного применения некоторых лекарств, которые могут выступать в роли гаптенов и изменять антигенные характеристики клеток. Такие анемии называются иммунными гемолитигескими анемиями. Механизм этих анемий связан с действием на эритроциты цитотоксических антител (чаще IgG, IgM, реже IgA). Опсонизированные антителами эритроциты или фагоцитируются макрофагами селезенки, вызывая ее увеличение и болезненность, или подвергаются комплементзависимому лизису в сосудистом русле (II тип иммунного повреждения по Джеллу и Кумбсу). Иммунные гемолитические анемии по патогенезу разнообразны — с участием тепловых гемолизинов или холодовых агглютининов, ночная пароксизмальная гемоглобинурия и др.
Повышенное разрушение эритроцитов приводит к резкому снижению их количества в крови, уменьшению НЬ и Ht, развитию гипоксии, стимуляции продукции почками эритропоэтинов и усилению гемопоэза в неповрежденном костном мозге. Продуктов для построения новых клеток хватает (содержание железа в крови даже повышено из-за гемолиза), некоторые компоненты разрушенных эритроцитов сами стимулируют кроветворение. Именно поэтому анемия имеет гиперрегенераторный характер', число ретикулоцитов в крови может увеличиваться до нескольких сот промилей (десятков процентов), наблюдается нейтрофильный лейкоцитоз с гиперрегенеративным сдвигом влево. Средний диаметр эритроцитов и MCV в пределах нормы, МСНС чаще нормальная, ЦП бывает разный (на фоне гемолитического криза возможно его увеличение, но это ложная гиперхромия). Резко увеличена СОЭ, повышено количество общего билирубина (главным образом неконъ-югированного). Наличие на поверхности эритроцитов фиксированных антител и активированных компонентов комплемента выявляет прямой положительный тест Кумбса, который подтверждает иммунный характер гемолиза.
В мазке крови резко выражена полихроматофилия, встречается множество нормоцитов, наблюдаются анизоцитоз и пойкилоцитоз (при иммунном гемолизе можно встретить типичные «пузырчатые клетки»), много палогкоядерных нейтрофилов и метамиелоцитов, встречаются миелоциты.
В костном мозге лейкоцитарно-эритроцитарное соотношение1/гЧ3 (вместо Vj), что отражает резкую активацию эритропоэза.
126
Лекция 8
Клиническая картина
В клинической картине ведущими являются тяжелый гипоксический синдром и гемолитическая желтуха.
Различают три группы наследственных гемолитических анемий: гемоглобинопатии, энзимопатии и цитопатии (эритроцитопатии). При гемоглобинопатиях в эритроцитах содержится НЬ аномальной структуры, при энзимопатиях имеется дефицит или аномалия фермента, при эритроцитопатиях нарушена структура клеток.
Г емоглобинопатии (гемоглобинозы)
Серповидно-клетогная анемия часто встречается в странах Средиземноморья, Закавказья, в Средней Азии. Генетический дефект выражается в том, что в p-цепях глобина аминокислота глютамин замещается на валин. Окисленная форма такого аномального НЬ обладает меньшей растворимостью по сравнению с нормальным, и в условиях гипоксии такой НЬ кристаллизуется и выпадает в осадок, изменяя форму эритроцитов в виде серпа. Тип наследования заболевания ко-доминантный, т.е. у гетерозиготного носителя патологического гена проявляют себя оба гена и в крови такого больного большая часть эритроцитов имеет нормальный НЬ, а меньшая часть — Hb-S. Клинически такая аномалия почти не проявляется и обнаруживается только специальными тестами. Анемия может проявиться в легкой форме при экстремальных ситуациях, сопровождающихся гипоксией.
У гомозиготного носителя патологического гена большая часть эритроцитов содержит Hb-S, и заболевание проявляется всеми признаками гемолитической анемии с гипоксическим и желтушным синдромами. В мазке крови преобладают серповидные эритроциты. У больных нарушена микроциркуляция с явлениями стаза и микротромбозов по причине скопления аномальных эритроцитов в микроциркуляторном русле и нарушения реологии крови. Наблюдается спленомегалия и гепатомегалия, а также происходит гиперплазия костного мозга вследствие гиперрегенерации эритроцитов. У детей часто нарушается развитие скелета — изменяется форма черепа, грудины и трубчатых костей. Из-за постоянно повышенного уровня железа в крови могут развиться гемохроматоз и гемосидероз внутренних органов.
Талассемия (болезнь Кули, средиземноморская болезнь) характеризуется тем, что в структуре НЬ, имеющего в норме две a-цепи и две p-цепи глобина, нарушается это соотношение. При а-талассемии нару
Патофизиология системы крови I
127
шен синтез a-цепей, при р-талассемии — синтез p-цепей глобина. В обоих случаях из-за нестабильности НЬ он выпадает в осадок и приводит к появлению в крови эритроцитов-пойкилоцитов мишеневидной формы, повышенный гемолиз которых влечет за собой развитие типичной гиперрегенераторной микроцитарной гипохромной гемолитической анемии.
Так как при наследственных гемолитических анемиях эритроциты в основном разрушаются в селезенке, вызывая ее увеличение, то спленэктомия является одним из методов лечения заболевания.
Энзимопатии
Гемолитическая анемия, обусловленная дефицитом в эритроцитах фермента глюкозо-6-фосфатдегидрогеназы. Наследственный дефект сцеплен с полом (передается с Х-хромосомой). При этом заболевании нарушается пентозофосфатный цикл, обеспечивающий ликвидацию активных метаболитов кислорода (перекисных соединений, свободных радикалов), образующихся на поверхности эритроцитов под влиянием сильных окислителей, в том числе некоторых лекарств (сульфаниламидов, левомицетина, фурациллина, противомалярийных препаратов, фенацитина, метиленового синего и др.). При дефиците глюкозо-6-фосфатдегидрогеназы нарушено восстановление НАДФ, являющегося донором ионов водорода, необходимых для восстановления глютатио-на, который является сильным антиоксидантом, защищающим клетку от свободных радикалов. У лиц с данной энзимопатией после приема вышеназванных лекарств может развиться гемолитическая анемия: нормохромная, нормоцитарная, гиперрегенераторная, с повышенным содержанием в крови железа и билирубина. В эритроцитах часто обнаруживаются характерные включения — тельца Гейнца-Эрлиха.
Эритроцитопатии
Семейный наследственный микросфероцитоз (болезнь Минковского-Шоффара). Наследственная гемолитическая анемия, которая наследуется по аутосомно-доминантному типу и встречается довольно часто (200-300 на 1 млн населения).
В результате генетического нарушения в цитоплазматической мембране (ЦПМ) эритроцитов содержатся дефектные структурные белки — спектрин, анкирин и другие, вследствие чего эритроциты теряют свою эластичность и деформируемость, становятся мелкими и сферическими.
128
Лекция 8
Недостатогная АТФазная активность в ЦПМ приводит к большему поступлению в клетки натрия и воды. Продолжительность жизни сфероцитов очень мала (6-14 сут), гибнут они главным образом в селезенке, вызывая ее увеличение и болезненность. Есть мнение, что на поверхности таких аномальных сфероцитов рано экспрессируются АГ стареющих клеток, что позволяет селезеночным макрофагам распознавать такие эритроциты, как уже отжившие свой срок. Такая укороченная жизнь эритроцитов приводит к развитию гиперрегенераторной гемолитигеской анемии и гемолитигеской желтухи. В крови у больных на фоне снижения Hb, Ht, числа эритроцитов резко повышено содержание ретикулоцитов (полихроматофилов), нормоцитов, нейтрофильных лейкоцитов (см. выше), повышено содержание железа и билирубина, увеличена СОЭ.
По среднему диаметру эритроцитов анемия характеризуется как микроцитарная (кривая Прайс-Джонса, отражающая процентное соотношение в крови эритроцитов разного диаметра, сдвинута влево, так как преобладают эритроциты диаметром 4-5 мкм). По среднему объему клеток (MCV) из-за их сферичности данная анемия является нормоцитарной. МСНС при анемии Минковского-Шоффара — выше нормы, а ЦП — чаще нормальный, но на фоне гемолитического криза повышается.
Важной характеристикой данной анемии является уменьшение минимальной и максимальной осмотической резистентности эритроцитов (ОРЭ). Не имеющие резерва для набухания сферические эритроциты начинают гемолизироваться в растворах NaCl с концентрацией 0,6-0,7 % (вместо 0,48-0,44 %).
Клинические проявления
Зависят от тяжести заболевания и частоты гемолитических кризов, которые могут сопровождаться лихорадкой, болями в пояснице, животе и конечностях, а также гемоглобинурией. На фоне гепатомегалии часто развивается калькулезный холецистит с образованием камней, состоящих из билирубината кальция. Ведущими являются гипоксический синдром и гемолитическая желтуха.
Следует запомнить: все гемолитигеские анемии по типу эритропоэза — нормобластигеские, по функциональному состоянию костного мозга — гиперрегенераторные, сопровождаются повышением в крови содержания железа и билирубина, сочетаются с гемолитигеской желтухой. Другие показатели зависят от конкретной формы заболевания.
Лекция 9
ПАТОФИЗИОЛОГИЯ СИСТЕМЫ КРОВИ II
Г.П. Щелкунова
Патофизиология лейкона
Лейкон — это совокупность зрелых и незрелых клеток белой крови — лейкоцитов. Все лейкоциты подразделяют на два вида: гранулоциты — нейтрофилы, эозинофилы, базофилы, и агранулоциты — лимфоциты и моноциты. В мазках крови, окрашенных по Райту, в цитоплазме нейтрофилов выявляется мелкая нежная фиолетового цвета зернистость, в эозинофилах — крупная зернистость красноватого цвета, в базофилах — крупная зернистость темно-синего цвета. В цитоплазме лимфоцитов и моноцитов зернистости нет (исключая редкие азурофильные гранулы).
Общим предшественником всех лейкоцитов является стволовая клетка в костном мозге. Гранулоциты, моноциты и В-лимфоциты все этапы своего развития проходят в костном мозге, а предшественники Т-лимфоцитов из костного мозга очень быстро переселяются в тимус, где дифференцируются до зрелых специализированных клеток. Поэтому в норме в костном мозге преобладают предшественники гранулоцитов, представлены предшественники эритроцитов (Л/Э-индекс = 4/1), тромбоцитов и моноцитов. Лимфоидных клеток в костном мозге мало (около 10 %).
Монопотентными клетками для гранулоцитов являются колониеобразующие единицы (КОЕ-Г, КОЕ-баз, КОЕ-эоз), из которых под влиянием цитокинов (интерлейкинов, КСФ) формируются последовательно: миелобласты — промиелоциты — миелоциты — метамиелоциты (юные) — палочкоядерные и, наконец, зрелые сегментоядерные гранулоциты (нейтрофилы, эозинофилы и базофилы), которые также называют полинуклеарами из-за сегментированности ядра. У каждого вида гранулоцитов свой росток. Выйдя из костного мозга, гранулоциты циркулируют в крови и эмигрируют в ткани. Главной функцией этих
130
Лекция 9
клеток является фагоцитоз, они играют важную роль в развитии воспаления, аллергии, аутоиммунных заболеваний.
Монопотентными предшественниками моноцитов являются КОЕ-М, которые под влиянием М-КСФ дифференцируются в монобласты, далее — в промоноциты и в зрелые моноциты (мононуклеары). После циркуляции в сосудистом русле моноциты эмигрируют в ткани, где превращаются в макрофаги. Макрофаги выполняют не только роль фагоцитов, но и являются антигенпредставляющими клетками (АПК), выделяют большое количество медиаторов воспаления и ответа острой фазы, участвуют в процессах заживления, регенерации.
Лимфопоэз представлен двумя ростками — Т- и В-лимфоцитов. Из монопотентных Пре-Т-клеток (образовавшихся из мультипотентных стволовых) при участии цитокинов (ИЛ-2, ИЛ-7, ИЛ-9, ИЛ-12) в тимусе формируются Т-лимфобласты, затем — Т-пролимфоциты и зрелые Т-лимфоциты. Т-лимфоциты также циркулируют в крови, в лимфатической системе и перемещаются в ткани. После антигенной стимуляции эти зрелые клетки способны, вернувшись в лимфоидные органы, подвергнуться бласттрансформации — превратиться в Т-иммунобласты, которые будут создавать клон активированных Т-лимфоцитов, участвующих в элиминации АГ. Часть Т-лимфоцитов (CD4t) выполняет функцию хелперных клеток, регулирующих гуморальный иммунный ответ (Тх-2) и клеточный иммунный ответ (Тх-1), а также иммуномодулирующую роль (Тх-3). Т-лимфоциты (CDg ) превращаются в цитотоксические лимфоциты (ЦТЛ). Еще часть Т-лимфоцитов является естественными киллерными клетками (NK-клетки).
Аналогичный процесс развития присущ и В-лимфоцитам. Из монопотентных Пре-В-клеток при участии ИЛ (4, 5, 6, 7 и 11) образуются В-лимфобласты, затем В-пролимфоциты и зрелые В-лимфоциты, которые являются главными клетками гуморального иммунного ответа. В-лимфоциты после антигенной стимуляции и при участии Тх-2 превращаются в способные к делению плазмобласты, создающие затем клон плазматических клеток (плазмоцитов), синтезирующих специфические антитела (IgA, IgE, IgG, IgM), обеспечивающие специфический иммунитет организма, участвующие в развитии воспаления, аллергии и аутоиммунных заболеваний.
Патология лейкона может выражаться как в изменении морфологии и функции лейкоцитов, так и в изменении их количества.
Для оценки количественных изменений лейкоцитов необходимо знать общее содержание лейкоцитов в единице объема крови (WBC —
Патофизиология системы крови II
131
white blood cells) и лейкоцитарную формулу — процентное соотношение различных видов лейкоцитов.
У взрослого человека в норме содержится 4-9х109 лейкоцитов в 1 л крови (4-9 тыс./мкл). Лейкоцитарная формула выглядит следующим образом: из 100 лейкоцитов 47-72 % составляют сегментоядерные нейтрофилы, 1-6 % — палочкоядерные нейтрофилы, 0,5-5 % — эозинофилы, 0-1 % — базофилы, 19-37 % — лимфоциты и 3-11 % — моноциты. Зная общее число лейкоцитов и лейкоцитарную формулу, легко рассчитать абсолютное количество любого вида лейкоцитов в единице объема крови. Морфологические особенности клеток изучают в мазках крови, окрашенных по Райту или Романовскому-Гймзе.
Лейкоцитозы, лейкемоидные реакции
Лейкоцитозом называется увеличение общего количества лейкоцитов в единице объема периферической крови (в 1 л, 1 мкл).
Классификация. По механизму развития выделяют следующие виды лейкоцитозов:
• перераспределительный или нейрогуморальный лейкоцитоз — может возникнуть в результате перераспределения лейкоцитов в различных сосудистых областях, мобилизации их из депо. Перераспределительные лейкоцитозы большей частью физиологического происхождения, скоропреходящие и наблюдаются:
— при беременности (особенно в поздние сроки), во время родов, при мышечном напряжении (у спортсменов, у новорожденных при крике) — миогенный лейкоцитоз;
— при быстром переходе из вертикального положения в горизонтальное — статигеский лейкоцитоз;
— через 2-3 ч после приема пищи, особенно белковой — пищеварительный лейкоцитоз;
— при психическом возбуждении (связан с выбросом адреналина и прямым его действием на депо) — эмоциональный лейкоцитоз;
• патологигеский или истинный (абсолютный) лейкоцитоз, возникающий при усилении лейкопоэза в костном мозге, характеризуется появлением в крови молодых форм лейкоцитов. Количество лейкоцитов при нем может увеличиться от 10 до 40х109/л и более.
В зависимости от причины выделяют следующие виды лейкоцитозов:
• Инфекционный лейкоцитоз — наблюдается при многих инфекционных заболеваниях, воспалительных (особенно гнойных) процессах.
132
Лекция 9
• Травматигеский лейкоцитоз — при шоках, после операций, ранений черепа, сотрясении мозга.
• Токсигеский лейкоцитоз — при отравлении мышьяком, ртутью, угарным газом, при тканевом распаде, некрозе.
• Медикаментозный лейкоцитоз — в результате приема некоторых лекарственных препаратов (глюкокортикоиды, препараты серебра, жаропонижающие, болеутоляющие средства).
• Постгеморрагигеский лейкоцитоз — после обильных кровотечений (гипоксия — усиление гемопоэза).
Усиленный выход лейкоцитов из костного мозга связываютсдействи-ем КСФ, в первую очередь гранулоцитарного КСФ и гранулоцитарномакрофагального КСФ.
В зависимости от того, каких лейкоцитов в крови становится больше, различают: нейтрофильный лейкоцитоз (нейтрофилия, нейтрофилез), эозинофильный (эозинофилия), базофильный (базофилия), моноцитарный (моноцитоз) и лимфоцитарный (лимфоцитоз).
Любой из этих лейкоцитозов может быть абсолютным, если число данных лейкоцитов увеличивается в единице объема крови, и относительным, если на фоне увеличения каких-либо лейкоцитов в процентном соотношении их число в единице объема крови сохраняется нормальным или даже уменьшается.
Нейтрофильный лейкоцитоз может быть физиологическим: на фоне физической нагрузки, стресса, пищеварения, действия ультрафиолетовых лучей, и патологическим, сопровождающим самые различные заболевания. Он наблюдается при острых асептических (ожоги, инфаркты) и септических воспалениях, вызванных бактериальной инфекцией (ангина, пневмония, аппендицит, острый холецистит, острый пиелонефрит, менингит и т.п.). Характерен для злокачественных новообразований, хронических миелопролиферативных заболеваний, для метаболических нарушений (кетоацидоз, уремия). Сопровождает гемолитические анемии, обострения хронических воспалительных заболеваний (подагра, ревматизм).
Механизм нейтрофилии в большинстве случаев связан с увеличением в костном мозге гранулоцитоколониестимулирующего фактора (Г-КСФ), гранулоцитомоноцитоколониестимулирующего фактора (ГМ-КСФ), например, под влиянием медиаторов ответа острой фазы (ИЛ-1, ИЛ-6 и ФНОа), и усилением пролиферации предшественников нейтрофилов. Молодые недозревшие нейтрофилы могут выходить в периферическую кровь, и тогда говорят о сдвиге ядра нейтрофилов влево.
Патофизиология системы крови II
133
Различают: регенеративный сдвиг влево, когда в крови увеличивается количество палочкоядерных нейтрофилов и появляются метамиелоциты (юные нейтрофилы), гиперрегенеративный сдвиг, если кроме метамиелоцитов в крови появляются миелоциты и лейкемоидный сдвиг, если кроме вышеназванных клеток в периферической крови встречаются промиелоциты и даже миелобласты. Лейкемоидный сдвиг характерен для лейкемоидных реакций и хронического миелолейкоза.
Значение нейтрофильного лейкоцитоза (исключая лейкозы) в большинстве случаев приспособительное, так как усиливаются механизмы неспецифицеской (фагоцитоз) и специфической (иммунные механизмы) резистентности организма. Если на фоне острой бактериальной инфекции нейтрофильного лейкоцитоза нет, то либо заболевание протекает в легкой форме, либо снижены реактивность и резистентность организма. Нейтрофилия не характерена для вирусных заболеваний, малярии и многих форм туберкулеза.
Эозинофильный лейкоцитоз. Такой лейкоцитоз характерен для паразитарных (аскаридоз, трихинеллез, описторхоз, эхинококкоз и т.п.) и аллергических заболеваний (поллинозы, бронхиальная астма и др.); наблюдается при хроническом миелолейкозе и на фоне злокачественных новообразований (в 60-80 % случаев), при хронических системных заболеваниях соединительной ткани и при болезни Аддисона (хронической недостаточности надпочечников). Гиперэозинофильный синдром является частым предвестником лейкоза. Кратковременная эозинофилия наблюдается при инфекционных заболеваниях в стадии выздоровления («эозинофильная заря выздоровления»).
Значение эозинофилии зависит от того, какие биологическиактив-ные вещества (медиаторы) и в каком количестве эти лейкоциты высвобождают в тканях. Это могут быть ингибиторы медиаторов аллергии (гистаминаза, арилсульфатаза, фосфолипаза-D) или сильные повреждающие агенты: катионные белки, главный белок эозинофилов (ГБЭ), нейротоксины, активные метаболиты кислорода, которые полезны в борьбе с паразитами, но могут вызывать значительные повреждения тканей организма. Очень тяжело протекают так называемые эозинофильные пневмонии, эозинофильные миокардиты.
Базофильный лейкоцитоз. Данный вид лейкоцитоза встречается нечасто: при некоторых аллергических и иммунных заболеваниях, при хроническом миелолейкозе и редко на фоне хронических воспалений. Базофилы крови наряду с тучными клетками тканей являются главными источниками медиаторов воспаления и аллергии (см. соответствующий раздел).
134
Лекция 9
Лимфоцитоз. Он развивается на фоне таких заболеваний, как инфекционный мононуклеоз, вирусный гепатит, краснуха, коклюш, сифилис, бруцеллез, туберкулез, тиреотоксикоз, болезнь Аддисона, хронический лимфолейкоз. Против клеток, пораженных вирусом, или против некоторых названных выше микробов нейтрофилы оказываются бессильными (не могут фагоцитировать объект или завершить фагоцитоз), в этом случае включаются специфические гуморальные и клеточные иммунные защитные механизмы с участием лимфоцитов (см. выше).
Моноцитоз. Этот лейкоцитоз характерен для многих инфекционных заболеваний: корь, краснуха, скарлатина, свинка (инфекционный паротит), инфекционный мононуклеоз, лейшманиоз, оспа, бруцеллез, риккетсиоз, лямблиоз, малярия, острая фаза туберкулеза, бактериальный эндокардит и др. Он сопровождает гранулематозные заболевания (саркоидоз, болезнь Крона), диффузные болезни соединительной ткани (ревматоидный артрит, системная красная волчанка, узелковый полиартериит, полимиозит и т.п.), гематологические заболевания (лейкозы, лимфомы, миелопролиферативные и миелодиспластические синдромы, гемолитические анемии и др.), а также злокачественные опухоли. Моноциты — главные клетки хронического воспаления; превращаясь в тканях в макрофаги, они участвуют в специфических и неспецифических механизмах защиты (см. выше), сливаясь друг с другом могут образовывать инфильтраты из эпителиоидных клеток.
Лейкемоидная реакция. Это реактивный гиперлейкоцитоз (50-100х109/л) с лейкемоидным сдвигом влево. Она может быть нейтрофильная, эозинофильная, лимфоцитарная.
Лейкемоидная реакция может развиваться на фоне тяжелой пневмонии, сепсиса, милиарного туберкулеза, инфекционного мононуклеоза, сочетанной инфекции у детей, некоторых паразитарных заболеваний, метастазах рака, а также при шоках, комах (уремическая, диабетическая), при отравлении угарным газом и в других ситуациях.
Гйперлейкоцитоз и резкое омоложение лейкоцитов в периферической крови вызывают необходимость дифференцировать лейкемоидную реакцию с лейкемией (лейкозом). В отличие от лейкоза при лейкемоид-ной реакции гиперплазия лейкоцитарного ростка имеет не опухолевый, а реактивный характер, является вторичной по отношению к основному заболеванию и исчезает после его излечения. При лейкемоидных реакциях в костном мозге не происходит угнетения других ростков крови (эритроцитарного, тромбоцитарного и др.), нет лейкемических инфильтратов в других органах, но в самих лейкоцитах встречаются очень
Патофизиология системы крови II
135
характерные изменения: токсогенная зернистость, тельца Князькова-Деле, пикноз ядер, вакуолизация цитоплазмы, анизоцитоз и др.
Лейкопении, агранулоцитозы
Лейкопенией называется уменьшение общего числа лейкоцитов в единице объема периферической крови.
Классификация. По механизму развития выделяют следующие группы лейкопений:
• временные или перераспределительные (лейкоциты собираются в депо: легкие, селезенка);
• постоянные или истинные, происхождение которых связано либо с понижением продукции лейкоцитов в связи с нарушением процесса их дифференциации и созревания, либо с повышенным разрушением лейкоцитов и выведением их из организма.
В зависимости от причины, вызвавшей развитие лейкопении, выделяют:
• инфекционно-токсигеские лейкопении — в результате токсического воздействия промышленных ядов (бензола, тетраэтилсвинца и др.); в результете применения ряда медикаментозных препаратов (сульфаниламиды, барбитураты, мышьяк и др.); при инфекционных заболеваниях (чаще всего стрептококковый сепсис, малярия, тяжелые формы брюшного тифа, бруцеллеза, туберкулеза), острых вирусных инфекциях (грипп, ветряная оспа);
• лейкопении в результате воздействия ионизирующей радиации (рентгеновые лучи, радиоактивные изотопы);
• лейкопении при системных поражениях кроветворного аппарата {см. причины гипо(а)пластигеских анемий, болезнь Аддисона-Бирмера и др.);
• дефицитные лейкопении, которые могут возникать при недостаточном поступлении в организм белков, аминокислот, витаминов группы В (при кахексии, голодании, длительных энтеритах и колитах).
По своему течению лейкопении могут быть острыми и хроническими.
Лейкопения может быть обусловлена уменьшением в крови нейтрофилов — нейтропения, или эозинофилов — эозинопения, или лимфоцитов — лимфопения, или моноцитов — моноцитопения.
Нейтропения
Эта лейкопения наблюдается на фоне острой лучевой болезни, вирусных заболеваний (грипп, гепатит, инфекционный мононуклеоз,
136
Лекция 9
ВИЧ), бактериальных инфекций (брюшной тиф, бруцеллез, милиарный туберкулез, молниеносный сепсис), малярии, а также на фоне аутоиммунных заболеваний, гиперспленизма, приема цитостатических препаратов. Она характерна для таких заболеваний крови, как апластическая анемия, В12- и фолиеводефицитная анемия, лейкоз, миелодиспластиче-ский синдром и др.
Нейтропения может сопровождаться дегенеративным сдвигом влево, который характеризуется уменьшением в крови сегментоядерных на фоне увеличения палочкоядерных нейтрофилов при отсутствии более молодых предшественников и наличием дегенеративных изменений в клетках (токсогенная зернистость, вакуолизация цитопалазмы, аномалии строения ядра и др.). Может наблюдаться сдвиг нейтрофилов вправо — на фоне отсутствия палочкоядерных и уменьшения сегментоядерных нейтрофилов в крови встречаются крупные нейтрофилы (макроцитоз) с полисегментированными ядрами и вакуолями в цитоплазме, что наиболее характерно для мегалобластических анемий.
Агранулоцитоз
Агранулоцитозом называется синдром, характеризующийся грану-лоцитопенией (нейтропенией менее 0,75х109/л (750/мкл) на фоне выраженной лейкопении (1-Зх109/л).
Термин «агранулоцитоз» («нет гранулоцитов») отражает крайнюю степень дефицита нейтрофилов в периферической крови, приводящую к резкому ослаблению резистентности организма и развитию инфекции. Именно инфекционное заболевание чаще всего является поводом обращения больного к врачу: тяжело протекающая некротическая ангина, пневмония, язвенно-некротический стоматит бактериальной или грибковой природы, кандидозы (микозы) другой локализации.
Обычно бактериальная инфекция сопровождается нейтрофильным лейкоцитозом, который является вторичным по отношению к заболеванию. При агранулоцитозе инфекция является следствием недостатка нейтрофилов. Ряд авторов считает, что агранулоцитозом следует считать нейтропению менее 500/мкл, так как только в этом случае инфекционные процессы проявляются у 100 % больных.
По патогенезу агранулоцитоз может быть иммунным или миелоток-сигеским. Редко встречается синдром «ленивых лейкоцитов», при котором дефицит нейтрофилов в крови обусловлен нарушением их выхода из костного мозга в периферическую кровь.
Иммунный агранулоцитоз, в том числе лекарственный, является следствием разрушения нейтрофилов (часто и других гранулоцитов)
Патофизиология системы крови II
137
при участии цитотоксических антител (IgG, IgM), активированных компонентов системы комплемента, макрофагов (II тип гиперчувствительности). Другие клетки крови при этом не страдают: нет анемии, тромбоцитопении, наблюдается относительный лимфоцитоз.
Миелотоксигеский агранулоцитоз развивается на фоне повреждения костного мозга. При этом повреждаются все ростки крови. Лейкопения (прежде всего гранулоцитопения) сочетается с моноцитопенией, тромбоцитопенией и гипо(а)пластической анемией. Причины миелоток-сического агранулоцитоза те же, что и гипо- и апластических анемий. Также характерен относительный лимфоцитоз (повышено процентное содержание лимфоцитов). Абсолютное число лимфоцитов в единице объема крови или нормальное или пониженное (например, при лучевой болезни).
Клинические проявления миелотоксического агранулоцитоза, кроме картины инфекционного заболевания (главное проявление), включают также гипоксический синдром, тяжесть которого зависит от степени снижения в крови эритроцитов и НЬ, а также возможное развитие геморрагического синдрома в том случае, когда число тромбоцитов в крови снижается до критических цифр (менее 40x109/л).
Зозинопения
Эта форма лейкопении может наблюдаться на фоне стрессов, глюкокортикоидной терапии, ответа острой фазы, миелотоксического агранулоцитоза, злокачественных опухолей, при болезни и синдроме Иценко-Кушинга, при некоторых вирусных заболеваниях, возможно из-за дефицита ИЛ-5.
Лимфопения
Данная форма лейкопении развивается под влиянием избытка в организме глюкокортикоидов (стрессы, гиперкортизолизм, длительная стероидная терапия), а также при лучевой болезни и лучевой терапии, при лимфомах (болезнь Ходжкина) и иммунодефицитных синдромах (Луи-Барр, Вискотта-Олдрича, Ди Джорджи). Она характерна для многих хронических заболеваний (застойная сердечная недостаточность, системная красная волчанка, туберкулез лимфатических узлов, хроническая почечная недостаточность в стадии уремии, милиарный туберкулез, метастазы опухоли и др.). Лимфопению вызывает иммуносупрессивная терапия антилимфоцитарным глобулином или циклофосфамидом.
Моиоцитопения
Этот вид лейкопении может быть результатом избытка в организме глюкокортикоидов, а также следствием повреждения костного мозга
138
Лекция 9
(лейкоз, апластическая анемия, анемия Аддисона-Бирмера, лучевая болезнь, метастазы рака, химиотерапия и т.п.). Уменьшение моноцитов в крови может наблюдаться на фоне разгара острого инфекционного заболевания (в сочетании с нейтрофилией), обострения туберкулезного процесса, ревмокардита (в сочетании с лимфоцитозом).
Гемобластозы
Гемобластозы — системные заболевания кроветворной ткани опухолевого происхождения.
Классификация. По международной классификации все гемобластозы подразделяют на три большие группы.
I. Острые лейкозы (лейкемии).
А. Острые миелолейкозы (ОМЛ) или нелимфобластные:
— недифференцированный (МО);
- миелобластный без дифференцировки (Ml);
— миелобластный с формированием некоторых гранул (М2);
— промиелоцитарный (М3);
— миеломонобластный (М4);
— моноцитарный (М5);
— эритролейкоз (Мб);
— мегакариобластный (М7).
Б. Острые лимфобластные лейкозы (ОЛЛ):
- В-ОЛЛ, Т-ОЛЛ, пре-В-ОЛЛ.
II. Хронигеские миелопролиферативные заболевания: хронические миелолейкозы (ХМЛ), эритремия (болезнь Вакеза), эссенциальная тромбоци-темия, миелоидная метаплазия.
III. Хронигеские лимфопролиферативные заболевания: хронический лимфолейкоз (ХЛЛ), лимфомы (болезнь Ходжкина, Беркитта и др.). Этиология и патогенез
Так же как и при всех других опухолевых заболеваниях, этиология и патогенез гемобластозов рассматриваются с позиций вирусного, радиационного или химического канцерогенеза. Предполагается, что опухолевый процесс начинается с появления в красном костном мозге или в лимфоидной ткани одной клетки-мутанта, которая приобретает способность к неограниченному безудержному делению и либо полностью (при острых лейкозах), либо частично (при хронических пролиферативных заболеваниях) утрачивает способность к дифференцировке. В результате вначале появляется один клон опухолевых функционально неполноценных клеток, затем в процессе опухолевой прогрессии могут
Патофизиология системы крови II
139
появиться новые мутанты и новые клоны клеток с более злокачественными свойствами.
В процессе роста опухоли происходит подавление нормальных ростков кроветворения, появляются лейкемические инфильтраты (аналог метастазов) в различных органах. В некоторых случаях, например при ХМЛ, в опухолевых клетках утрачивается способность к апоптозу, в результате чего вместо короткой жизни, свойственной нормальным лейкоцитам, опухолевые функционально неполноценные клетки становятся почти «бессмертными».
Острые лейкозы
Независимо от морфологического варианта острые лейкозы имеют много общих проявлений. Общее количество лейкоцитов в периферической крови при острых лейкозах может быть разным — нормальным, пониженным, умеренно повышенным и чрезмерно повышенным, в связи с чем различают алейкемические, сублейкемические и лейкемические варианты.
Главными признаками острого лейкоза являются преобладание в периферической крови бластных клеток (40-90 %) и дефицит всех других форменных элементов — эритроцитов, тромбоцитов, лейкоцитов. Исключение составляют только алейкемические варианты, при которых на фоне лейкопении, анемии, тромбоцитопении бластные клетки в периферической крови практически отсутствуют, но их огромное количество находится в костном мозге.
Бластные клетки, полностью потеряв способность к дифференцировке, не дозревают до конечной стадии, поэтому в лейкоцитарной формуле можно видеть так называемый «лейкемигеский провал» — отсутствие промежуточных клеток (например, между миелобластами и сегментоядерными лейкоцитами при ОМЛ, или между лимфобластами и зрелыми лимфоцитами при ОЛЛ). Зрелые клетки являются продуктами сохранившихся нормальных ростков крови.
В костном мозге и лимфоидных органах выявляются признаки опухолевой гиперплазии, анаплазии и метаплазии. Лейкозные бластные клетки, создавая значительную массу, вытесняют нормальные клетки, а с помощью гуморальных факторов угнетают пролиферацию, дифференцировку и ускоряют их апоптоз. Нарастающая масса бластных клеток по принципу обратной связи тормозит деление стволовых и по-липотентных клеток — предшественников гемопоэза. Развиваются анемия, тромбоцитопения, гранулоцитопения (вплоть до агранулоцитоза).
140 Лекция 9 лимфопения. При ОМЛ опухолевые бластные клетки обнаруживаются в лимфоидных органах (миелоидная метаплазия лимфоидной ткани), при ОЛЛ, наоборот, опухолевые лимфобласты преобладают в костном мозге (лимфоидная метаплазия костного мозга). Появляются очаги опухолевого кроветворения (лейкемические инфильтраты) в печени, селезенке, лимфатических узлах, легких, почках, сердце, ЖКТ, эндокринных железах, стенках сосудов, головном мозге, коже (лейкемиды) и в других местах. При лейкоцитозе более 100х109/л может развиться лейкостаз в сосудах легких и мозга.
Клинические проявления острых лейкозов в развернутой стадии сводятся к сочетанию гипоксического синдрома из-за развивающейся анемии, геморрагического синдрома, обусловленного как дефицитом тромбоцитов, так и повреждением лейкемическими инфильтратами печени и сосудов, а также иммунодепрессивного синдрома. Резкое снижение в крови гранулоцитов и агранулоцитов приводит к ослаблению и клеточного, и гуморального иммунитета и присоединению инфекционных осложнений (чаще бактериальной и грибковой природы). Развитие ответа острой фазы и появление больших количеств ФНО-а могут привести к развитию кахексии. У больных умеренно увеличены лимфатические узлы (безболезненные), печень и селезенка; возникают боли в костях (оссалгии) и могут появиться признаки недостаточности функции того органа, где появились лейкемические инфильтраты (например, неврологические симптомы, гастроэнтерологические и др).
Прогноз и тактика лечения острого лейкоза во многом зависят от морфологического варианта заболевания. Поскольку в периферической крови все бластные клетки практически не распознаваемы по морфологическим признакам, в современной гематологической практике для дифференциальной диагностики острых лейкозов применяют гистохимические методы исследования, иммунное фенотипирование и генетические цитологические исследования, выявляющие аномалии хромосом.
Например, при ОМЛ-МО-форме все гистохимические реакции отрицательные, а иммунофенотип характеризуется маркерами CD13, CD33, CD34. При Ml- и М2-формах ОМЛ гистохимические исследования выявляют положительные реакции на миелопероксидазу и на липиды с су-даном черным, маркеры — CD13, CD33; обнаруживается транслокация хромосом соответственно t (9; 22) и t (8; 21). При МЗ-форме гистохимические реакции и иммунофенотип аналогичен, но генетический маркер — t (15; 17). При М4-форме положительны реакции на миелопероксидазу и неспецифическую эстеразу, иммунофенотип — CD13, CD14, CD33,
Патофизиология системы крови II
141
CDIIb, а хромосомная аномалия выявляет инверсию в 16-й паре. Наличие вышеназванных хромосомных маркеров улучшает прогноз заболевания. При Мб-форме ОМЛ положительна PAS-реакция в диффузной форме (реакция на полисахариды), иммунофенотип — CD13, CD33, а генетическим маркером является деления 5-й и 7-й хромосом (5q-7q), что серьезно ухудшает прогноз.
При ОЛЛ гистохимические реакции на миелопероксидазу и липиды отрицательные, а полисахариды (PAS-реакция) выявляются в бластных клетках в гранулярной форме, у 95 % больных ОЛЛ обнаруживается терминальная деоксинуклеотидилтрансфераза (TdT) и только у 15 % больных ОМЛ. У больных с Т-ОЛЛ иммунофенотип — CD3, CD5, CD7, а у больных с пре-В-ОЛЛ (составляющих 75 % от всех ОЛЛ) — CD10, CD19. У 5 % детей и 30 % взрослых при пре-В-ОЛЛ выявляется так называемая филадельфийская хромосома — t (9; 22) — это химерный ген «аЫ/bcr», управляющий синтезом аномальных протеинкиназ. Острый лимфобластный лейкоз — это наиболее часто встречающаяся форма лейкемии у детей, она достаточно успешно лечится. Однако наличие филадельфийской хромосомы при ОЛЛ резко ухудшает прогноз.
Хронические миелопролиферативные заболевания
Можно рассмотреть на примере наиболее часто встречающихся заболеваний — хронического миелолейкоза и эритремии.
Хронигеский миелолейкоз известен в двух формах — с филадельфийской хромосомой (95 % больных) и без нее. В отличие от острых лейкозов развивается в течение нескольких лет и проходит несколько стадий. Ранее гематологи выделяли 3 стадии: хронигескую (3-7 лет), стадию прогрессирующей акселерации (1-1,5 года) и финальную стадию бластных кризов (3-6 мес).
Долгое время считалось, что острые лейкозы не могут переходить в хронические формы (возможны только ремиссии при успешной терапии), а хронические миелолейкозы не могут переходить в острые. Последнее утверждение недавно было опровергнуто. И в настоящее время в развитии ХМЛ выделяют хронигескую стадию (при успешном лечении до 7 лет) и стадию бластной трансформации, при которой 2/3 случаев ХМЛ переходит в ОМЛ и '/3 - в ОЛЛ. Это доказано с помощью современных иммунофенотипических и генетических маркеров. Объяснить данный феномен можно только тем, что, по-видимому, опухолевый клон клеток возник из стволовых полипотентных клеток, на стадии опухолевой прогрессии из-за генетической нестабильности в одних случаях
142
Лекция 9
появляется клетка-мутант миелоидного типа, а в других случаях — лимфоидного типа.
При хронических лейкозах (в отличие от острых) опухолевые клетки не только безудержно размножаются, но и дифференцируются до зрелых клеток. Поэтому в лейкоцитарной формуле наблюдается лейкемо-идный сдвиг: в периферической крови обнаруживаются миелобласты (в хронической стадии их мало), промиелоциты, миелоциты, метамиелоциты, палочкоядерные и сегментоядерные гранулоциты («лейкемического провала» нет). Морфологическим субстратом ХМЛ чаще являются нейтрофилы, но в крови увеличивается количество эозинофилов и базофилов (эозинофильно-базофильная ассоциация). Иногда встречаются базофильные или эозинофильные варианты ХМЛ. В опухолевых клетках выключен механизм апоптоза, поэтому при ХМЛ общее число лейкоцитов в крови всегда очень высокое (выше 100х109/л). Количество тромбоцитов на ранних этапах заболевания может быть повышено (реактивный тромбоцитоз), но постепенно развивается тромбоцитопения. У всех больных выражены анемия, тяжесть которой нарастает к финалу, а также лимфопения, моноцитопения.
В костном мозге наблюдаются признаки опухолевой гиперплазии миелоидной ткани, в лимфоидных органах — признаки миелоидной метаплазии, в печени и других органах — лейкемические инфильтраты. Поскольку процесс длительный, то лимфатические узлы, селезенка, печень увеличиваются до больших размеров. Также характерны лейкемиды в коже, оссалгии, на фоне тромбоцитопении — геморрагический синдром. Гипоксический синдром нарастает параллельно тяжести анемии.
Важными следствиями процесса являются ослабление клеточного и гуморального иммунитета и развитие инфекционных заболеваний (лейкоцитов — гранулоцитов в крови очень много, но это опухолевые функционально неполноценные клетки, а моноцитов и лимфоцитов очень мало). Развивается и ответ острой фазы, со временем приводящий к развитию кахексии. Больной может погибнуть задолго до финальной стадии из-за развившегося осложнения. Во всех случаях прогноз заболевания неблагоприятный.
Истинная полицитемия (эритремия, болезнь Вакеза) отличается от ХМЛ более длительным и доброкачественным течением с характерными 3-мя стадиями:
• плеторическая, сопровождающаяся значительным увеличением массы крови (полицитемическая гиперволемия), продолжающаяся несколько лет;
Патофизиология системы крови II 143
• миелоидная метаплазия селезенки;
• анемическая.
Различают главные и малые критерии заболевания (в 1-й стадии). К главным критериям относят резкое увеличение массы эритроцитов в крови (число эритроцитов, НЬ и Ht резко повышены), спленомегалию и нормальное насыщение крови кислородом. Малые критерии', тромбоцитоз (более 400х109/л), лейкоцитоз (более 12х109/л), гранулоцитоз, базофилия, увеличение в лейкоцитах щелочной фосфатазы, увеличение в крови кобаламина и транскобаламина.
Основными клиническими проявлениями заболевания являются эритроцианоз, зуд кожи и жгучие боли в кончиках пальцев с их отеком (возможно, связанные с повышенным выделением гистамина из базофилов крови), увеличение ОЦК, АД, гипертрофия миокарда, увеличение селезенки (сначала в результате рабочей гипертрофии, затем вследствие метаплазии). Часто развиваются подагра, нефропатия, фиброз печени, калькулезный холецистит. Из-за высокого риска развития тромбоза (высокая вязкость крови, тромбоцитоз) наиболее частой причиной гибели больных являются инфаркты, инсульты и другие осложнения. Если больной доживает до финала, то эритроцитоз сменяется анемией, так как либо развивается миелофиброз, либо эритремия переходит в хронический миелолейкоз.
Хронический лимфолейкоз
Эта форма лейкоза относится к хроническим лимфопролиферативным заболеваниям, является неизлечимой патологией лиц пожилого возраста. Выделяют 5 стадий развития хронического лимфолейкоза (ХЛЛ):
• 0-я стадия характеризуется только лимфоцитозом более 15х109/л (если случайно в этой стадии больному ставят диагноз ХЛЛ, то он может прожить более 14 лет);
• 1-я стадия — к лимфоцитозу присоединяется лимфаденопатия (с этого момента продолжительность жизни может составить около 8 лет);
• 2-я стадия — присоединяется спленомегалия (продолжительность жизни может составить около 6 лет);
• 3-я стадия — развивается анемия;
• 4-я стадия — появляется тромбоцитопения (больному остается жить не более 19 мес).
144
Лекция 9
Общее количество лейкоцитов в крови в финальных стадиях заболевания в большинстве случаев около 50-100х109/л (возможны исключения). 60-80 % клеток периферической крови представлены лимфоцитами, небольшим количеством пролимфоцитов.
В мазках крови встречаются характерные тельца (тени) Боткина-Гумпрехта — это размазанные на стекле во время приготовления мазка лимфобласты (остатки их ядер). Всех других клеток крови мало (эритроцитов, тромбоцитов, гранулоцитов, моноцитов).
В костном мозге лимфоидные клетки значительно превышают 10 % (метаплазия). Характерны значительное увеличение лимфатических узлов, селезенки, наличие лейкемических инфильтратов в других органах. В 50 % случаев больные ХЛЛ погибают от инфекции из-за резко сниженного иммунитета: лимфоцитов в крови много, но они функционально неполноценные и не дифференцируются в плазматические клетки (т.е. нарушен синтез антител), не обеспечивают хелперные функции и механизмы клеточного иммунитета. Из-за метаплазии костного мозга имеется дефицит гранулоцитов и моноцитов. У некоторых больных на фоне опухолевой прогрессии могут появиться «запретные клоны» лимфоцитов, вызывающие тяжелые аутоиммунные заболевания, в том числе аутоиммунную гемолитическую анемию и тромбоцитопению (причиной смерти могут стать массивный гемолиз или кровотечение).
Лекция 10
ПАТОФИЗИОЛОГИЯ БЕЛКОВОГО ОБМЕНА
Г.В. Порядин
Белки — полимеры, состоящие из аминокислот. Они играют решающую роль во всех процессах, происходящих в живом организме.
Различают простые и сложные белки.
Простые белки — протеины (от греч. protos — первый, простой), молекулы которых содержат только белковые компоненты и при полном гидролизе распадаются только на аминокислоты.
Сложные белки — протеиды (протеины + греч. eidos — вид), в молекулы которых, кроме белкового компонента, входят низкомолекулярные компоненты небелковой природы.
Суточная потребность в белке зависит от возраста, характера трудовой деятельности, климатических условий, функционального состояния организма (для детей в возрасте до 3 мес — 2,3 г/кг массы тела, от 1 до 3 лет — 0,88 г/кг, от 7 до 12 лет — 0,77 г/кг, от 16 до 19 лет — 0,64 г/кг; для взрослых — 0,59 г/кг).
Биологическое значение белков для нормальной жизнедеятельности организма определяется теми функциями, которые они выполняют:
• в форме специфических структур обладают ферментативной активностью. Ферменты (трипсин, пепсин, цитохром С, рибонуклеаза и др.) — самая большая и наиболее важная по своему биологическому значению группа. Известно более 1000 различных ферментов;
• в форме транспортных белков (в первую очередь р-глобулины, альбумины, миоглобин, НЬ, цирулоплазмин, трансферрин) обеспечивают обмен небелковых соединений (жиров, липидов, углеводов, солей, воды), обладая способностью связывать и переносить различные вещества (образуя комплексы с липидами, витаминами, гормонами и т.д.);
146
Лекция 10
• составляют структурную основу всех тканевых элементов — структурные белки (коллаген, мукопротеиды, гликопротеиды, а-кератин) — вторая по численности группа белков;
• являются обязательными компонентами сократительных и двигательных систем — сократительные белки (актин, миозин);
• служат источником образования ряда гормонов (инсулин, тироксин, соматотропин, адреналин, норадреналин, АКТГ), медиаторов;
• выполняют защитную функцию — защитные белки крови — иммуноглобулины (антитела), система комплемента, лизоцим, интерферон;
• поддерживают гемостаз, участвуя в гемокоагуляции и фибринолизе (фибриноген, протромбин, тромбин, антитромбин, тромбопластин, плазминоген и др.).
Обеспечение организма белками из нескольких источников определяет полиэтиологичность нарушений белкового обмена.
Нарушения обмена белков могут носить первигный или вторигный характер.
Нарушения обмена белков первичного (экзогенного) происхождения
Наиболее частыми причинами общих нарушений белкового обмена являются колигественные или кагественные изменения поступающих в организм белков первичного (экзогенного) происхождения.
В этом случае дефекты и изменения в организме связаны с:
• несоответствием поступления белка потребностям организма:
— увелигение колигества поступающих извне белков;
— огранигение поступления экзогенных белков (полное или частичное голодание);
• низкой биологической ценностью пищевых белков;
• дефицитом незаменимых аминокислот (валин, лейцин, изолейцин, лизин, метионин, треонин, триптофан, фенилаланин, гистидин, аргинин).
Основные показатели патологии белкового обмена
Главным показателем белкового обмена является азотистый баланс — разность между количеством азота, который поступает в организм, и количеством азота, выводимого из организма. У взрослого человека азотистый баланс в норме близок к нулю — азотистое равновесие или нулевой азотистый баланс.
Патофизиология белкового обмена
147
Азотистый баланс может быть положительным и отрицательным (табл. 10-1).
Таблица 10-1. Показатели патологии белкового обмена
Патология Норма
Диспротеинемия 60-80 г/л
белки плазмы:
альбумины — 60 %
а-глобулины — 13 %
р-глобулины - 12 %
у-глобулины - 15 %
Патологические белки:
парапротеины
криоглобулины
макроглобулины
белки Бенс-Джонса
Гиперазотемия: увеличение количества азотистых шлаков крови
Увеличение остаточного азота (азот мочевины, креатинина, мочевой кислоты, аминокислот) 25 ммоль/л
Увеличение мочевины 1,67-4,16 ммоль/л
Увеличение мочевой кислоты 0,12-0,35 ммоль/л
Увеличение аммиака 29-86 мкмоль/л
Аминоацидемия 5 ммоль/л
Увеличение биологически активных аминов (гистамин, серотонин)
Протеинурия (альбуминурия)
Аминоацидурия
Аммиак в моче (уремия)
Положительный азотистый баланс — состояние азотистого обмена, при котором вводимое с пищей количество азота превышает количество азота, выводимого из организма (в норме — у растущих организмов и беременных; при патологии — после длительного голодания, при избыточной секреции инсулина, андрогенов, соматотропина, при недостатке тироксина).
148
Лекция 10
Отрицательный азотистый баланс — состояние азотистого обмена, при котором количество азота, выводимого из организма, превышает количество азота, вводимого с пищей. Отрицательный азотистый баланс является следствием потери организмом части собственных белков (наблюдается при недостаточном поступлении с пищей белка, голодании, стресс-реакциях, гиперпродукции глюкокортикоидов (гиперкорти-цизм), тироксина, повышении активности лизосомальных ферментов: химотрипсин, катепсины, нефротическом синдроме — протеинурии).
Увеличенное (избыточное) поступление белков
• При неправильно организованной диете, так называемое белковое перекармливание. Вызывает большую нагрузку на пищеварительные ферменты, изменяет соотношение в системе фермент-субстрат, снижает эффективность переваривания поступающих белков. Избыток белка ведет к задержке в организме аминокислот — к аминоацидемии.
• При некоторых патологических состояниях (поражение гипоталамуса — сахарный диабет, гигантизм, акромегалия и др.).
Патогенез
Выпадение тормозных импульсов из вентромедиальных ядер заднего гипоталамуса («центры сытости») вызывает пролонгированное пищевое возбуждение и резко выраженную гиперфагию.
Повышение реактивности парасимпатической системы (растормаживание) ведет к повышению функции инсулярного аппарата, развитию гипогликемии и, следовательно, усилению аппетита.
Пшерпродукция гормонов гипофиза (СТГ) усиливает выделение секретина, который активирует продукцию инсулина 0-клетками.
Основные проявления избыточного поступления белка в организм:
• положительный азотистый баланс;
• отвращение к белковой пище;
• диспептические расстройства;
• дисбактериоз;
• кишечная аутоинтоксикация.
Избыточное поступление аминокислот
• Избыток триптофана в диете. Большая часть триптофана превращается в никотиновую кислоту (витамин РР). На этом этапе образуется ряд промежуточных продуктов: ксантуреновая кислота, оксиантраниловая кислота и другие, повышение концентрации которых в крови оказывает общетоксическое действие.
Патофизиология белкового обмена 149
В частности, ксантуреновая кислота нарушает образование инсулина. Кроме того, избыток триптофана ведет к повышению синтеза и содержания серотонина в серотонинергических отделах нервной системы, изменению функции головного мозга, развитию нервно-психических заболеваний. А накопление в организме метаболитов триптофана ведет к нарушению функции эндокринной, сердечно-сосудистой, дыхательной, кроветворной и выделительной систем.
• Избыток в диете метионина ведет к усилению синтеза белков и образованию цистина (что облегчает образование камней в почках), адреналина и холина. Более тяжелое нарушение обмена цистина — цистиноз — сопровождается общей аминоацидурией, отложением кристаллов цистина в тканях, характеризуется ранним летальным исходом.
Недостаточное поступление белков
Белковая недостаточность первичного (экзогенного) происхождения, связанная с ограничением поступления белков (при голодании). Проблема века — 30 % населения земли голодает.
Голодание — состояние, которое формируется в результате недостаточного поступления в организм питательных веществ, необходимых для нормального течения обмена веществ и покрытия энергетических трат.
Голодание может стать результатом стихийных бедствий, например землетрясения, наводнения.
Голодание часто встречается в клинической практике как осложняющий болезнь фактор, особенно при таких болезнях, как инфекционные заболевания, поражения ЖКТ, которые голодание может сопровождать.
Иногда ограничение приема пищи используется в лечебных целях (например, при лечении ожирения, заболеваний пищеварительного тракта, аллергии и др.).
Особую опасность голодание представляет для детей (особенно грудного возраста). Голодание может развиться в силу особенностей их организма (пороки развития ЖКТ) либо организма матери (недостаток молока, плоские соски и др.).
Выделяют следующие виды голодания.
• Абсолютное, или полное прекращение поступления в организм пищи и воды. Эта форма голодания встречается крайне редко, в основном в связи с какими-либо экстремальными (чрезвычайными) обстоятельствами, в которых оказывается человек.
150
Лекция 10
По своему патогенезу абсолютное голодание связано с тем, что организм человека в этом случае лишен как пищи, так и воды. Отсутствие воды (обезвоживание) усиливает распад белков и общую интоксикацию организма собственными продуктами обмена. Состояние человека при абсолютном голодании усугубляется неизбежным при этом нарушением коллоидной структуры белков, что в конечном счете и приводит его к гибели на 5-7-е сутки голодания.
• Полное — полное прекращение приема пищи при сохранении приема воды.
Причинами полного голодания могут быть отсутствие пищи (внешние) либо заболевания пищеварительного тракта — полости рта, пищевода, желудка, кишечника (внутренние).
Полное голодание — это состояние, связанное с переходом организма на эндогенное питание (за счет расходования веществ своих тканей), когда организм для сохранения необходимого уровня энергетического обмена вынужден использовать собственные запасы питательных веществ (в основном жиров).
Утилизация жировых запасов и атрофия тканей организма в ходе полного голодания приводят к постепенной потере массы тела. При этом более всего атрофируются жировая и мышечная ткань, селезенка, печень, почки, кожа, легкие, и практически атрофия не распространяется на такие жизненно важные органы, как нервная система и сердечная мышца, масса которых остается неизменной вплоть до гибели человека.
В зависимости от изменения обмена веществ и энергии в развитии полного голодания можно выделить 3 периода.
1. Натальный (приспособительный) — длится 1-2 сут. Первые сутки энергетические потребности еще обеспечиваются за счет окисления углеводов. Дыхательный коэффициент около 1,0. На вторые сутки энергозатраты обеспечиваются уже на 84 % окислением жиров. Синтез белка снижается.
2. Стационарный (наибольшего приспособления) — длится 55-70 сут (2-3 мес) и более, наиболее продолжительный период. Организм адаптируется к голоданию и полностью перестраивается на эндогенное питание. Энергозатраты практически на 90 % обеспечиваются за счет распада жира. Дыхательный коэффициент снижается и становится равен 0,7. Возрастает кетонемия. Постепенно развивается ацидоз.
3. Терминальный (заключительный) период — длится 2-3 сут. Характеризуется резким усилением распада белков. Энергозатраты
Патофизиология белкового обмена
151
осуществляются за счет расходования белков. Снижается синтез многих ферментов (развивается недостаточность ферментной системы). Нарастает ацидоз. Развивается гипопротеинемия. Снижается онкотическое давление крови (ОД), что ведет к образованию голодных отеков. Гибель наступает в результате интоксикации организма.
• Неполное — или хроническое недоедание, когда калорийность принимаемой пищи (полноценной по составу) не покрывает всех энергетических затрат и пластических нужд организма. Неполное голодание (хроническое недоедание или несбалансированная диета) отличается от полного тем, что организм способен длительное время поддерживать свою жизнедеятельность в условиях недостаточного питания.
Длительность существования человека в условиях неполного голодания зависит от величины дефицита между калориями, затрачиваемыми на поддержание жизни и получаемыми с пищей, а также от количества резервных веществ самого организма.
Чаще всего причиной неполного голодания являются различные хронические заболевания, особенно ЖКТ, либо отсутствие в пище тех или иных веществ (недостаток белков, жиров, углеводов, витаминов и т.д.).
Так как необходимые питательные вещества, хотя и в ограниченном количестве, но все-таки поступают в организм, патогенез и исход неполного голодания в основном зависят от того, насколько велик дефицит калорийности пищи.
При длительном неполном голодании у человека может развиться алиментарное истощение, которое характеризуется прогрессирующим исхуданием (нередко маскируемым отеками), мышечными болями, изменением реактивности организма (повышенная восприимчивость к инфекциям), общей заторможенностью вплоть до депрессии.
Другим частым проявлением неполного голодания является белковокалорийная недостатогность (рис. 10-1).
Отмечаются задержка роста, общего развития, снижение иммунитета, анемия.
В основе перечисленных нарушений лежат снижение активности ферментов, угнетение синтеза белка, нарушение всасывания в ЖКТ железа, витаминов и других продуктов.
Тяжелая белково-калорийная недостаточность проявляется в 2-х формах:
• алиментарный маразм (кахексия) — одинаково часто поражает детей и взрослых. Характеризуется тяжелым общим истощением, по-
152
Лекция 10
Рис. 10-1. Патогенез белково-калорийной недостаточности
терей подкожного жира, атрофией мышц и большинства органов, нарушением обмена веществ: отрицательный азотистый баланс, гипогликемия, кетонемия, гиперкалиемия;
• белковая дистрофия (голодные отеки, квашиоркор — впервые описана в Индии). Заболевание характерно для детей раннего возраста. Характеризуется задержкой роста, отставанием в весе, депигментацией кожи, отеками [уменьшение количества глобулинов, увеличение продукции антидиуретического гормона (АДГ)], анемией (нарушение всасывания железа), повышенной восприимчивостью к инфекции (снижение реактивности), диареей. Развиваются жировая дистрофия печени вплоть до цирроза, атрофия ЖКТ, эндокринных желез.
Белковая недостаточность первичного происхождения, связанная с дефицитом незаменимых аминокислот
Для нормального течения в печени процессов биосинтеза белка необходимо наличие в ней всех аминокислот, входящих в состав синтезируемых белков (особенно незаменимых — не могут быть синтезированы).
Общие проявления недостатка незаменимых аминокислот:
• появляются быстрая утомляемость и атрофия мышц;
• развивается отрицательный азотистый баланс;
• в крови развивается гипопротеинемия, и прежде всего за счет уменьшения альбуминовой фракции. Замедляется синтез белков плазмы;
Патофизиология белкового обмена
153
• повышается риск возникновения инфекций и интоксикаций, в связи с повреждением иммунных механизмов (задерживается образование антител, ослабевает фагоцитоз);
• замедляются процессы физиологической регенерации: образование эритроцитов, эпителизация, рост волос и т.д.
Специфические проявления недостатка незаменимых аминокислот:
• недостаток в организме лизина — ведет к задержке роста и развития организма, понижению содержания в крови НЬ и эритроцитов (в связи с уменьшением очагов кроветворения и ослаблением пролиферации клеток красного ростка);
• для недостаточности валина характерно развитие мышечной слабости;
• при недостатке триптофана возникает гипохромная анемия, понижается содержание белков в плазме крови. Длительная недостаточность триптофана в организме сопровождается развитием катаракты;
• дефицит в организме гистидина ведет к развитию экземы, отставанию в росте, угнетению синтеза НЬ;
• при недостатке метионина ускоряется атерогенез, развиваются ожирение, гипокортицизм, гипокатехоламинемия.
Неправильные соотношения аминокислот в белках пищи и изменение нормальных отношений аминокислот при всасывании из желудочно-кишечного тракта
Для биосинтеза белка необходимо не только наличие всех аминокислот, но и определенные их соотношения.
Например, дефицит даже одной аминокислоты лимитирует весь процесс биосинтеза белков и резко отражается на обмене не только этой аминокислоты, но и всех остальных. Например, в тканевом белке триптофан, лизин и валин содержатся в равных соотношениях (1:1:1). Если с пищевым белком эти аминокислоты поступают в соотношении 1:1:0,5, то синтез тканевого белка будет обеспечиваться только наполовину.
Неправильные соотношения аминокислот могут возникать при снижении желудочного расщепления белков (после резекции желудка), отщепление тирозина и триптофана запаздывает, и их концентрация во всасывающейся смеси становится ниже, чем в норме.
Последствия: в связи с относительной недостаточностью указанных аминокислот большая, чем в норме, часть других аминокислот не может быть использована для биосинтеза белков. Образовавшийся избыток
154
Лекция 10
аминокислот подвергается действию ферментов печени, катализирующих окислительное дезаминирование, что приведет к накоплению в организме ряда промежуточных продуктов обмена, причем некоторые из них токсичны для организма, и их накопление в тканях может вызвать патологические явления (в частности развитие гипохромной анемии).
С другой стороны, полное использование тирозина (поступившего в малом количестве) в печени для биосинтеза белков ведет к уменьшению образования в печени и других органах биологически важных соединений (таких как гормон щитовидной железы-тироксин, надпочечников-адреналин, гипофиза-меланин).
Нарушения обмена белков вторичного (эндогенного) происхождения
Общая белковая недостаточность (вторигная или эндогенная) может развиться и при достаточном поступлении белков с пищей — в результате нарушения белкового обмена в самом организме вследствие:
• нарушения переваривания и всасывания белковых продуктов (гастроэнтериты, язвенный колит);
• повышенного распада белков в тканях (стресс, инфекционные болезни);
• усиленной потери эндогенных белков (кровотечения, травмы, нефроз);
• нарушения синтеза белка (гепатиты).
Это так называемая вторичная (или эндогенная) белковая недостаточность.
Как следует из сказанного, обмен веществ является важным условием нормальной жизнедеятельности организма.
Обмен веществ — это комплекс физиологических и биохимических процессов, обеспечивающих жизнедеятельность организма в условиях влияния факторов внешней среды.
Обмен веществ обеспечивает поступление энергии для постоянного синтеза и распада всех компонентов живого организма.
Сущность энергетических превращений в организме состоит в том, что энергия, заключенная в структуре белков, жиров и углеводов в конечном счете используется на осуществление специфических форм работы: мышечные сокращения, секреция, синтез соответствующих структур ит.д.
Следует отметить, что ряд болезней (так называемые молекулярные), с которыми врач сталкивается в клинической практике, непосредствен
Патофизиология белкового обмена
155
но связан с наследственным нарушением того или иного вида обмена веществ.
Например, появление патологических вариантов белковых молекул имеет особое значение для патогенеза многих болезней крови. Примером могут служить:
• гемоглобинопатии — болезни, связанные с присутствием (образованием) патологических молекул Hb (HbS);
• ферментопатии — составляющие основу ряда анемий (гемолитическая анемия при недостатке активности глюкозо-6-фосфат-дегидрогеназы);
• фибриногенопатии — патология структуры фибриногена и связанные с этим нарушения гемостаза (тромбоцитопении, гемофилии, коагулопатии).
Известен ряд нарушений сердечно-сосудистой системы, источником которых являются молекулярные дефекты белкового обмена:
• генетически обусловленная патология клапанов сердца (синдром Элерса-Данло):
• семейная миокардиопатия, характеризующаяся аритмиями, недостаточностью кровообращения, гиперхолестеринемией;
• мышечные дистрофии (типа Дюшенна) — миокардиодистрофия с гипертрофией миокарда;
• семейная синусовая тахикардия, связанная с избыточной активностью фермента аденилатциклазы и накоплением катехоламинов.
Нарушение обмена нуклеиновых кислот (пуриновых и пиримидиновых оснований)
Пуриновые (аденин, гуанин) и пиримидиновые (цитозин, урацил, тимин) основания входят в состав нуклеиновых кислот — РНК и ДНК и являются поставщиками мочевой кислоты. Нарушение их обмена приводит к повышению уровня мочевой кислоты и развитию подагры — заболевания известного еще со времен Гиппократа.
Подагра (от греч. podagra — капкан, ломота, слабость в ногах) — хроническое заболевание, обусловленное нарушением обмена пуринов. Характеризуется отложением солей мочевой кислоты в тканях, органах, суставах с развитием в них вначале воспалительных, а затем деструктивно-склеротических изменений.
Заболевание относится к наследственным, сцепленным с Х-хромо-сомой. Это болезнь пожилых мужчин.
156
Лекция 10
В основе заболевания лежат недостаточность гипоксантин-гуанин-фосфорибозил-трансферазы и гиперактивность 5-фосфорибозил-1-пирофосфат-синтетазы.
Проявляется гиперурикемией, рецидивирующим артритом, образованием подкожных узелков, симптомами мочекаменной болезни.
Гиперурикемия (концентрация урата в сыворотке более 70 мг/л). Гйперурикемия — необходимое условие развития подагры. Мочевая кислота образуется при окислении пуриновых оснований. 2/3 мочевой кислоты выводится с мочой (300-600 мг/сут), а */3 — через ЖКТ, в котором она разрушается бактериями.
Гйперурикемия может быть обусловлена:
• повышенной скоростью продукции мочевой кислоты;
• сниженной секрецией мочевой кислоты почками.
В связи с этим подагру и гиперурикемию делят на метаболическую и почечную.
I. Метаболигеская гиперурикемия и подагра обусловлены повышенной продукцией мочевой кислоты, о чем можно судить по повышенной экскреции (более 600 мг/сут) мочевой кислоты. На долю этого типа подагры приходится менее 10 % всех случаев заболевания.
Избыточная продукция мочевой кислоты может быть первичной и вторичной.
• Первигная гиперурикемия обусловлена наследственной недостаточностью гипоксантин-гуанин-фосфорибозил-трансферазы или повышенной активностью 5-фосфорибозил-1-пирофосфат-син-тетазы.
• Вторигная гиперурикемия обусловлена гиперпродукцией мочевой кислоты либо в связи с ускорением биосинтеза пуринов, либо в связи с ускорением распада пуриновых нуклеотидов, либо в связи с недостаточностью глюкозо-6-фосфатазы (гликогеноз I типа), при которой отмечается повышенная продукция мочевой кислоты.
II. Погегная гиперурикемия и подагра обусловлены снижением экскреции мочевой кислоты почками. На долю этого типа подагры приходится до 90 % всех случаев заболевания. Почечный тип гиперурикемии и подагры может быть первичным и вторичным.
• Первигная почечная подагра (гиперурикемия) встречается у больных с патологией почек (поликистоз, свинцовая нефропатия).
• Вторигная почечная подагра (гиперурикемия) может наблюдаться при приеме диуретиков, уменьшающих объем циркулирующей плазмы, что сопровождается снижением фильтрации мочевой кис
Патофизиология белкового обмена 157
лоты, усилением ее канальцевой реабсорбции и уменьшением секреции мочевой кислоты.
Патогенез подагры обусловлен:
• механизмами накопления уратов в хрящевой ткани и почках;
• механизмами провоцируемого уратами воспаления.
Нарушение обмена пиримидиновых оснований
Пиримидиновые (цитозин, урацил, тимин) основания, как и пурины, входят в состав нуклеиновых кислот — РНК и ДНК и формируют нуклеотидтрифосфаты и нуклеотидпирофосфаты — макроэргические донаторы.
Известна наследственная оротацидурия (дефект оротат-фосфорибо-зил-трансферазы и оротидин-5-фосфат-декарбоксилазы).
Аутосомно-рецессивное заболевание, при котором нарушен синтез уридина. Характеризуется замедлением роста, тяжелой мегало-пластической анемией, иммунодефицитом с поражением в основном Т-клеточного звена иммунитета, образованием кристаллов оротовой кислоты в моче.
Лекция 11
ПАТОФИЗИОЛОГИЯ УГЛЕВОДНОГО ОБМЕНА
Г.В. Порядин
Основные функции углеводов
• Являются легко утилизируемыми истогниками энергии. И в этом отношении углеводы, в частности глюкоза крови, играют особую роль в энергетике ЦНС. Дело в том, что ЦНС не имеет собственных запасов углеводов. Поэтому ею используется до 70-75 % углеводов крови или около 500 г/сут (2/3 всей энергии). Запасы же углеводов в организме относительно невелики: в печени человека содержится около 45-75 г гликогена, в мышцах около 280 г гликогена. Вот почему организм нуждается в постоянном поступлении углеводов с пищей.
• Образующийся в процессе пентозного цикла рибозо-5-фосфат входит в состав многих нуклеотидных соединений (АТФ, НАД, НАДФ, НАДФ-Н2, ацетил-КоА и т.д.). Последние имеют большое значение в клеточном обмене (протоплазмы, клеточного ядра — НАДФ-Н2 — восстановленный никотинамид-аденин-динуклеотид-фосфат).
• В виде мукополисахаридов углеводы являются важным структурным элементом соединительной ткани.
• Образующаяся из глюкозы и уридинтрифосфата уридиндифосфорно-глюкуроновая кислота принимает участие в обезвреживающих процессах в печени.
Основные патогенетические пути нарушения углеводного обмена
1. Нарушение всасывания.
2. Нарушение межуточного метаболизма.
3. Нарушение процессов регуляции углеводного обмена.
Патофизиология углеводного обмена 159
Нарушение всасывания углеводов
Как и для всех остальных компонентов пищи (белки, жиры), для углеводов первым этапом превращения являются расщепление их в ЖКТ и последующее всасывание.
В процессе последовательного ферментативного расщепления полисахариды (крахмал, гликоген) и дисахариды распадаются в двенадцатиперстной кишке до стадии моносахаров (глюкозы, фруктозы, галактозы).
В этом заложен большой биологический смысл: устраняется видовая специфичность поступающих в организм веществ: в результате пищеварительного распада все сложные высокомолекулярные соединения превращаются в низкомолекулярные элементарные структуры; поставляется основная масса энергии.
Последовательное расщепление углеводов происходит под влиянием специфического фермента амилазы или диастазы поджелудочной железы.
Нарушение всасывания углеводов может возникнуть в результате 2-х причин: расстройства ферментативного расщепления полисахаридов в кишечнике: нарушения самого процесса всасывания моносахаридов (т.е. процесса их фосфорилирования в слизистой оболочке кишечника):
• расстройство ферментативного расщепления полисахаридов, в свою очередь, может наблюдаться при поражении ацинозной ткани поджелудочной железы (диффузный панкреатит) и закупорке ее выводного протока (камнем, опухолью) — непоступление ферментов:
• нарушения процесса всасывания моносахаридов (понижение скорости всасывания) в связи с нарушением образования в слизистой оболочке кишечника фосфорных эфиров моносахаридов.
Причины нарушения фосфорилирования:
• воспаление слизистой оболочки кишечника;
• отравление ядами, угнетающими процессы фосфорилирования (монойодацетат, флоридзин). Во всех этих случаях происходит блокада гексокиназы кишечника.
Нарушение межуточного метаболизма (включения глюкозы в тканевой обмен)
Превращение углеводов (глюкозы, фруктозы, галактозы) с момента их поступления в организм до образования конечных продуктов принято называть промежуточным (или межуточным) обменом или метаболизмом.
160
Лекция 11
Нарушение межуточного углеводного обмена может проявляться следующими изменениями:
• усилением синтеза гликогена (главным образом в печени и мышцах), приводящим к его избыточному отложению: гликогенная болезнь, или гликогеноз (12 типов);
• патологическим ослаблением синтеза гликогена: уменьшение отложения:
• нарушением процесса глюконеогенеза:
• нарушением превращений глюкозы в тканях.
Патологигеское ослабление синтеза гликогена — уменьшение отложения (главным образом в печени и мышцах) — может наблюдаться в результате его усиленного расщепления (при недостаточном ресинтезе) либо при ослаблении его образования.
Усиление распада гликогена (гликогенолиз) обычно связано с повышением энергозатрат организма (например, усиленная мышечная работа, сильное эмоциональное возбуждение, боль, охлаждение и др.).
При этом возбуждение центральной нервной системы (коры головного мозга, подкорки, межуточного, продолговатого мозга) приводит к усилению импульсации по симпатическим нервным путям с последующим усилением гликогенолиза в органах и тканях.
Помимо непосредственной передачи нервных импульсов к эффекторным органам и тканям, при возбуждении ЦНС повышается функция ряда желез внутренней секреции (щитовидная, мозговой слой надпочечников, гипофиз), гормоны которых активируют фосфорилазу печени, тем самым усиливая распад гликогена.
Усиление распада гликогена может быть обусловлено и первичным возбуждением функции этих желез.
Патологигеское уменьшение отложения гликогена вследствие нарушения процессов его образования из моносахаридов (угнетение глико-генообразования) может быть результатом:
• нарушения нервной регуляции синтеза гликогена. Так, при псевдо-паралитигеской тяжелой миастении (myastenia gravis) в результате нарушения передачи нервно-трофических импульсов по парасимпатической нервной системе в скелетной мускулатуре, вследствие угнетения энергетики тканевых элементов содержание гликогена значительно снижается:
• некоторых заболеваний желез внутренней секреции (тиреотоксикоз, гормонально активные опухоли коры надпочечников), гормоны которых приводят к усилению активности фосфорилазы;
Патофизиология углеводного обмена
161
• всякого рода гипоксических состояний (синтез гликогена сопровождается довольно значительным потреблением энергии), приводящим к уменьшению образования гликогена (например, понижение парциального давления кислорода в атмосфере, анемии, заболевания органов дыхания, кровообращения, отравление ядами, действующими на тканевые окислительные ферменты, такие как СС14, дихлорэтан, фосфор, различные интоксикации):
• гипо- и авитаминозов.
Нарушение глюконеогенеза (образования глюкозы и гликогена из аминокислот и жиров). Нарушение глюконеогенеза в сторону чрезмерного усиления или ослабления может сказаться на течении межуточного обмена углеводов (более подробно при разборе патогенеза сахарного диабета).
Нарушение использования глюкозы в тканях. В основном связано с механизмами ее окисления и превращения. Из курса биохимии должно быть известно, что расщепление углеводов в тканях организма может происходить как анаэробно — без участия кислорода (гликолиз), который завершается образованием 2-х молекул молочной кислоты: (С6Н12О6->2С3Н6О3), так и аэробно (по пути пентозного цикла): С6Н12О6 + 6О2 -> 6СО2 + 6Н2О.
В условиях недостатогного снабжения организма кислородом (понижение парциального давления кислорода в атмосфере — горная болезнь, нарушение кровообращения, анемии, интоксикации) соотношение между первой и второй фазами может нарушаться, и в клетках немедленно развиваются процессы анаэробного расщепления углеводов. Это ведет к избыточному накоплению пировиноградной и молочной кислот (ги-перлактацидемия). Содержание последней в крови может доходить до 10 ммоль/л (норма — 5,5 ммоль/л). Нарушение обмена углеводов с преобладанием анаэробной фазы ведет к менее эффективному использованию их энергии: на каждую молекулу глюкозы образуется 2-3 молекулы АТФ (что дает освобождение только 32,6 ккал). Тогда как полное окисление (через цикл трикарбоновых кислот) сопровождается образованием до 38 молекул АТФ на каждую молекулу глюкозы (освобождает 680 ккал). Вместе с тем накопление молочной кислоты уменьшает щелочные резервы, а это приводит к изменению КОС.
Содержание мологной кислоты в крови может повышаться и при нарушении функции пегени, так как часть молочной кислоты, образующаяся в процессе обмена углеводов (главным образом в мышцах) и поступающая в кровь, превращается (ресинтезируется) в глюкозу и гликоген именно в печени.
162
Лекция 11
Нарушение межуточного обмена глюкозы, проявляющееся в накоплении пировиноградной кислоты, может наступать при недостаточности витамина В] (авитаминоз). Дело в том, что для окислительного превращения пировиноградной кислоты необходимо наличие фермента липотиамин-пирофосфата, представляющего собой комплекс липоевой кислоты и тиаминпирофосфата. Последний является пирофосфорным эфиром витамина В, (тиамина). Поэтому при авитаминозе Bt нарушается декарбоксилирование пировиноградной кислоты и происходит ее накопление. Одновременно повышается и количество молочной кислоты, так как часть избытка пировиноградной кислоты восстанавливается в молочную. Все это вызывает интоксикацию, и в первую очередь элементов нервной ткани.
Кроме того, с окислением пировиноградной кислоты тесно связан синтез важнейшего медиатора нервной системы — ацетилхолина. Образование его затрудняется. Поэтому при гипо- и авитаминозе В! прежде всего наступают нарушения в центральной и периферической нервной системе, которые выражаются в потере чувствительности, невритах, параличах и судорогах. Введение витамина В1 устраняет описанные нарушения.
Расстройства углеводного обмена — нарушения процессов его регуляции
Высшим нервным регуляторным аппаратом углеводного обмена является «сахарный» центр, который нужно рассматривать как функциональное объединение нервных центров, расположенных в различных отделах центральной нервной системы — коре головного мозга, подкорке (чечевичное ядро полосатого тела), подбугровой (гипоталамической) области, продолговатом мозгу (область писчего пера).
Кроме того, поглощение глюкозы мышцами и жировой тканью в соответствии с количеством и качеством пищи регулируется несколькими гормонами: инсулином, глюкагоном, адреналином, АКТГ, глюкокортикоидами (рис. 11-1).
Третий важный фактор поддержания постоянного уровня глюкозы в крови — утилизация ее различными тканями, т.е. механизм клеточной регуляции (и в первую очередь печенью).
Все эти звенья тесно связаны друг с другом. Выпадение или нарушение любого звена в этой сложной системе регуляции углеводного обмена ведет к его патологии.
Одним из показателей состояния регуляторных (адаптивных) механизмов углеводного обмена является изменение содержания глюкозы в крови.
Патофизиология углеводного обмена
163
Пентозный цикл Гликолиз
Уровень глюкозы в крови
Рис. 11-1. Обмен глюкозы в печени и влияние на него гормонов
В норме оно колеблется от 3,3 до 5,5 ммоль/л и является относительно постоянной величиной.
Выделяют две формы патологии — гипогликемию, гипергликемию.
Гипогликемия — падение уровня сахара крови ниже 3,3 ммоль/л.
Различают физиологическую и патологическую гипогликемию.
Физиологигеская гипогликемия (быстро проходящая) — встречается у здоровых людей при усиленной мышечной работе, приводящей к значительному потреблению глюкозы как источника энергии, если эти затраты не покрываются поступлением в организм легко усвояемых углеводов.
Механизм. Уменьшение глюкозы в крови возбуждает гипоталамус, его ядра, связанные с симпатическими нервами. Образование инсулина подавляется, и поток глюкозы в мышечную и жировую ткань прекращается. Одновременно усиливается гликогенолиз в печени.
При отсутствии инсулина активируются контринсулярные гормоны. Возрастает секреция мозговой частью надпочечников адреналина, который повышает содержание сахара в крови за счет активации фосфорилазы, расщепляющей гликоген. Усиливается секреция гипофизом АКТГ, который, в свою очередь, возбуждает секрецию корой надпочечников глюкокортикоидов, усиливающих глюконеогенез в печени. Увеличивается выделение а-клетками поджелудочной железы глюкагона, обладающего свойством повышать в печени гликогенолиз и глюконеогенез.
164
Лекция 11
Усиливается выделение СТГ, способствующего развитию гипергликемии. В результате увеличивается количество циркулирующей в крови глюкозы, одновременно повышается и потребление ее тканями.
Однако чаще гипогликемия связана с выпадением того или иного звена регуляции углеводного обмена и является патологигеской.
Различают:
• гипогликемию центрально-нервного характера (или нейрогенную). Ряд заболеваний нервной системы (энцефалит, диэнцефалиты, травмы мозга, прямое повреждение гипоталамуса опухолью или воспалением) часто сопровождается появлением выраженной гипогликемии. У таких больных гипогликемия обусловлена рефлекторным раздражением парасимпатической нервной системы, которое приводит к усилению продукции инсулина и повышению ассимиляторных процессов в углеводном обмене. При этом происходят усиленное образование гликогена в печени и мышцах из глюкозы и превращение ее в жир;
• гипогликемию при выпадении гормонального звена в системе регуляции углеводного обмена (или гормональную); в результате атрофии гипофиза — при гипофизарной кахексии; недостаточности надпочечников — при болезни Аддисона (бронзовая болезнь); при избыточной продукции инсулина (гиперинсулинизм абсолютный, панкреатический). Встречается при различных заболеваниях поджелудочной железы, вовлекающих 0-клетки в патологический процесс и возбуждающих их функцию (на ранних стадиях панкреатита, при аденоме и раке поджелудочной железы, инсулиноме — инсулинопродуцирующей опухоли поджелудочной железы);
• гипогликемию при тяжелых поражениях пегени (гепатит, цирроз печени, отравления хлороформом, фосфором, мышьяком, четыреххлористым углеродом и другими ядами, при алкоголизме), сопровождающихся гибелью части печеночной ткани и ослаблением гомеостатических свойств печени.
Во всех перечисленных случаях преобладает действие инсулина над контринсулярными гормонами.
Гипергликемия — повышение глюкозы крови выше 5,5 ммоль/л.
Кратковременная гипергликемия является реакцией приспособительной, физиологической, обеспечивающей доставку тканям легко утилизируемого энергетического материала при всех ситуациях, когда в этом имеется потребность:
Патофизиология углеводного обмена
165
• временная {физиологигеская) гипергликемия может наблюдаться у здоровых людей после приема большого количества сахара (глюкозы, свекловичного сахара, меда). Это так называемая пищевая, или алиментарная, гипергликемия;
• временная {физиологигеская) гипергликемия может стать следствием усиления нервной регуляции углеводного обмена. Примером подобной гипергликемии может служить эмоциональная гипергликемия, возникающая при волнениях, эмоциональных переживаниях, душевных потрясениях и сильных болях. Резкое преобладание процесса возбуждения через гипоталамус по симпатическим путям усиливает функцию мозгового слоя надпочечников, что ведет к усилению продукции адреналина, поддерживает и усиливает распад гликогена, активирует глюконеогенез в печени;
• гипергликемия при некоторых видах наркоза (эфирный, морфинный, хлороформный) — гипергликемия растормаживания. Гипергликемия наступает потому, что симпатические центры освобождаются от тормозных влияний коры головного мозга, вследствие чего усиливается продукция адреналина.
При стойком нарушении того или иного звена регуляции углеводного обмена развиваются патологигеские гипергликемии.
• Нейрогенная — при нарушении нервной регуляции (если в ЦНС создается патологический очаг возбуждения). Стойкая гипергликемия развивается при сильной и продолжительной боли, асфиксии и других состояниях, характеризующихся рефлекторным возбуждением симпатической нервной системы и раздражением «сахарного» центра.
• Эндокринная — при нарушении эндокринной регуляции. Стойкая гипергликемия может возникнуть при повышенной функции одного из звеньев эндокринной системы, ведущего к распаду гликогена и усилению глюконеогенеза:
— при избытке тиреоидных гормонов {тиреотоксикоз), опухоли мозгового слоя надпочечников (феохромоцитома — при которой в кровь поступает большое количество адреналина), при некоторых заболеваниях гипофиза {акромегалия, болезнь Иценко-Кушинга), сопровождающихся повышенной продукцией его гормонов (АКТГ, СТГ) и гормонов коры надпочечников;
— при инсулиновой недостаточности панкреатического или вне-панкреатического происхождения.
166
Лекция 11
Во всех этих случаях лежит избыточная активность контринсулярных гормонов.
Особое место среди патологических гипергликемий занимает гипергликемия при инсулиновой недостаточности.
Заболевание, в основе которого лежит панкреатическая и/или вне-панкреатическая инсулиновая недостаточность, получило название сахарный диабет (Diabetes Mellitus).
Сахарный диабет — заболевание, которое характеризуется гипергликемией, глюкозурией и обусловлено панкреатической (абсолютной) или внепанкреатической (относительной) инсулиновой недостаточностью, приводящей к нарушению обмена веществ.
Около 1-2 % населения земного шара больны сахарным диабетом (СД).
В 2000 г. численность пациентов с СД в мире составляла более 160 млн человек, к 2025 г., по прогнозам экспертов ВОЗ, предполагается увеличение числа больных до 300 млн.
Согласно классификации ВОЗ, выделяют две формы СД:
• инсулинзависимый сахарный диабет I типа. Для инсулиночувствительного варианта СД в первую очередь характерно прекращение синтеза и секреции инсулина вследствие аутоиммунного повреждения р-клеток, при сохраненном действии инсулина на периферии. Характеризуется выраженной инсулинопенией, развитием кетоацидоза, чаще встречается у детей (ранее назывался — юношеский диабет). Имеет рецессивную наследственную предрасположенность.
• инсулиннезависимый (ИНЗД) сахарный диабет II типа — инсулинорезистентный вариант. Сопровождается выраженной гиперин-сулинемией, инсулинорезистентностью периферических тканей. Характеризуется минимальными обменными нарушениями. Кетоацидоз не развивается. Инсулинопения наблюдается редко (чаще гиперинсулинемия), чаще встречается после 40 лет (ранее назывался — диабет взрослых, пожилых). Имеет выраженную генетическую основу — аутосомно-доминантный тип наследования.
При сахарном диабете нарушаются все виды обмена: углеводный, жировой, белковый.
Патофизиология углеводного обмена
167
Острые осложнения сахарного диабета
Гиперкетонемическая диабетическая кома
Наиболее грозным осложнением сахарного диабета I типа является гиперкетонемигеская (кетоацидотигеская) диабетигеская кома (табл. 11-1). Частота диабетической гиперкетонемической комы среди больных СД составляет 1-6 %.
Таблица 11-1. Острые осложнения сахарного диабета
Виды осложнений Показатели
Глюкоза крови Кетоновые тела Лактат pH, осмолярность плазмы
Кетоацидотическая кома 19-33 ммоль/л 17 ммоль/л Менее 7,3
Гйперосмолярная кома 39-56 ммоль/л Около нормы 350 мосмоль/л
Гйперлактаци-демическая кома Изменяется незначительно 2-8 ммоль/л Менее 7,3
Гипогликемическая кома 0,5-1,1 ммоль/л
НОРМА 3,3-5,5 ммоль/л 1,5-1,7 ммоль/л 0,4-1,4 ммоль/л 7,4 285 мосмоль/л
Диабетическая кома обусловлена резким увеличением в крови концентрации кетоновых тел, развитием кетоза.
Этиология
• Прекращение (недостаточное) введения больному сахарным диабетом инсулина.
• Острые гнойные инфекции.
• Стресс-реакции (травмы, операции).
• Нарушения диеты (избыток углеводов, жиров, голодание).
• Интоксикации (пищевые, алкогольные).
• Нарушения в ЖКТ (длительная рвота, диарея).
• Беременность.
Патогенез
Основу патогенеза диабетической кетоацидотической комы составляет нарастающая инсулиновая недостаточность, которая связана с:
168
Лекция 11
• нарушением утилизации глюкозы тканями в результате снижения проницаемости клеточной мембраны для глюкозы, ее окисления и энергетического использования клеткой;
• нарушением синтеза гликогена и усилением его распада;
• новообразованием глюкозы из неуглеводов (белков, жиров) — глюконеогенез;
• гиперпродукцией контринсулярных гормонов (СТГ, АКТГ, катехоламины, глюкагон и др.) и усилением липолиза.
В результате всех перечисленных изменений в углеводном обмене отмечается избыточное поступление глюкозы из печени в кровь — значительное (до 19-33 ммоль/л) повышение ее уровня — гипергликемия.
Высокая гипергликемия, характерная для диабетического кетоацидоза, ведет к повышению осмотического давления во внеклеточной жидкости, вследствие чего вода устремляется из клеток во внеклеточное пространство, — развивается внутриклетогная дегидратация.
Гиперпродукция контринсулярных гормонов (гормона роста, АКТГ, катехоламинов, глюкокортикоидов) способствует увеличению липолиза, который сопровождается накоплением в крови свободных жирных кислот, триглицеридов, фосфолипидов, холестерина, концентрация которых в крови резко возрастает.
Уменьшение окисления ацетил-КоА в цикле трикарбоновых кислот ведет к его избыточному накоплению и усиленному образованию из него в печени ацетоуксусной и р-оксимасляной кислот, что ведет к развитию кетоза. Кетонемия достигает 17 ммоль/л и более (в норме кетоновых тел —1,5-1,72 ммоль/л).
Накопление кетоновых тел сопровождается истощением щелочных резервов крови и декомпенсацией КОС с развитием метаболического ацидоза.
Сильный кетоз вызывает усиленное выделение с мочой фосфора, кальция, азота, аммиака, аминокислот, мочевины, магния, натрия, хлоридов, что способствует усилению диуреза (полиурия).
Накопление в организме кетоновых тел оказывает токсическое действие на ткани, особенно на клетки ЦНС (угнетает ферментные системы, уменьшает захват мозгом глюкозы), обусловливая потерю сознания.
Развивающаяся гиповолемия ведет к тяжелым нарушениям гемодинамики: падению АД, уменьшению ударного объема вплоть до развития коллаптоидного состояния.
Патофизиология углеводного обмена
169
Гиперосмолярная кома
Гиперосмолярная (некетонемигеская, неацидотигеская) кома — особый вид комы, специфической для СД II типа. Протекает с высокой гипергликемией (более 39-55,5 ммоль/л), но без кетоацидоза, с резким обезвоживанием, гиперхлоремией, гипернатриемией, азотемией.
Гиперосмолярная кома встречается в 0,2-0,5 %, преимущественно у лиц старше 50 лет.
Этиология
• Избыточное употребление углеводов.
• Инфекции.
• Ожоги, травмы.
• Длительные рвота, понос, прием мочегонных препаратов.
• Гемодиализ.
• Введение больших количеств гипертонических растворов.
Патогенез
Основу патогенеза гиперосмолярной комы составляют высокая гипергликемия и гиперосмолярность крови.
Гиперосмолярность обусловлена прежде всего высокой гипергликемией и задержкой электролитов (особенно гипернатриемией), которая развивается в ответ на повышенную секрецию альдостерона (как результат дегидратационной гиповолемии), а также на уменьшение выведения натрия почками в связи со снижением почечного кровотока и поражением канальцев (как результат уменьшения секреции АДГ гипофизом).
Повышение осмотического давления крови усиливает приток жидкости из клеток во внеклеточные пространства, тем самым ведет к кле-тогной дегидратации.
Высокий осмотический диурез, обусловленный примерно равной глюкозурией и выделением электролитов, ведет к быстрому развитию гиповолемии, внутри- и внеклетогной дегидратации, сосудистому коллапсу и смерти.
Смертность от гиперосмолярной комы очень высока (30-40 %), что объясняется тромбозами (из-за сгущения крови), почечной недостаточностью, дегидратацией головного мозга (что ведет к развитию внутримозговых кровоизлияний).
Отсутствие кетоза при гиперосмолярной коме обусловлено:
• ингибированием липолиза и кетонообразования в результате нарушения функции (дегидратация) клеток печени, жировой ткани, поджелудочной железы;
170
Лекция 11
• некоторой способностью р-клеток выделять инсулин, который тормозит липолиз, препятствует кетоацидозу;
• высокой концентрацией глюкозы в крови, что тормозит кетогенез.
Гиперлактацидемическая кома
Гиперлактацидемигеская (мологнокислая, лактат-ацидотигеская) кома развивается в результате метаболического ацидоза, обусловленного накоплением в организме больного сахарным диабетом молочной кислоты (более 2 ммоль/л, при норме — 0,4-1,4 ммоль/л).
Данный вид комы встречается в основном у пожилых больных, страдающих тяжелыми сопутствующими заболеваниями печени, почек, сердца.
Этиология
• Чрезмерная физическая нагрузка.
• Интоксикации с гипоксией.
• Острые инфекции, переходящие в сепсис.
• Кровотечения.
Патогенез
Связан с накоплением в организме молочной кислоты и увеличением отношения лактат/пируват (в норме 12/1).
Для СД характерны диабетические ангиопатии: сосудистые осложнения в виде капилляропатий (нефропатии, ретинопатии сетчатки глаз, поражения сосудов нижних конечностей и т.д.).
В условиях недостатка кислорода в превращениях гликогена преобладает анаэробная фаза, ведущая к избыточному образованию молочной кислоты.
Гйпоксия и функциональная недостаточность печени тормозят ресинтез лактата в гликоген, переход молочной кислоты в пировиноградную.
Недостаток инсулина создает предпосылку для повышенного образования пирувата и лактата из белков.
Гипогликемическая кома
Острое состояние, развивающееся при быстром понижении концентрации сахара в крови и резком падении утилизации глюкозы мозговой тканью.
Этиология
Гипогликемическая кома чаще всего развивается у больных сахарным диабетом:
Патофизиология углеводного обмена
171
• при избытке вводимого инсулина и неадекватном приеме углеводной пищи (инсулиногенная);
• несоответствии дозы инсулина степени утилизации глюкозы (интенсивная мышечная работа, эмоциональные состояния, психические травмы, инфекции);
• нарушении функции печени, кишечника, почек, надпочечников, гипофиза, щитовидной железы;
• приеме сахароснижающих препаратов (сульфаниламиды, салицилаты);
• ряде синдромов, сопровождающихся усиленной секрецией инсулина — гиперинсулинизм (инсулинома).
Патогенез
В патогенезе гипогликемической комы основное значение имеет снижение утилизации глюкозы клетками головного мозга. Вслед за недостатком глюкозы в клетках мозга наступает гипоксия с последующим нарушением обмена веществ. Длительное углеводное и кислородное голодание сопровождается функциональными и структурными изменениями вплоть до отека и некроза отдельных участков мозга.
Поздние осложнения сахарного диабета
Диабетитеские микро- и макроангиопатии (ДМА). Патогенез ДМ А весьма сложен и включает метаболические, гемодинамические, гормональные, генетические, иммунологические факторы.
Главными среди них являются метаболические нарушения — хро-нигеская гипергликемия. Активация обмена глюкозы при СД ведет к нарушению гемодинамики и проницаемости сосудистой стенки, что способствует утолщению эндотелия, изменению структуры базальной мембраны капилляров и нарушению клеточного метаболизма.
Одним из важнейших последствий гипергликемии является неферментативное гликозилирование белков (как внутриклеточных, так и внеклеточных), что способствует изменению их физико-химических свойств и биологической активности, приводящей к образованию избыточного количества свободных кислородных радикалов и повреждению кровеносных сосудов, повышению тонуса сосудистой стенки и снижению кровотока.
Среди механизмов, способных вызвать нарушение микроциркуляции при СД, важная роль принадлежит вазоактивному веществу эндотелия — оксиду азота (NO), мощному сосудорасширяющему фактору.
172
Лекция 11
Определенная роль в патогенезе диабетических микро- и макроангиопатий принадлежит иммунным механизмам. У больных СД обнаруживается дефицит клеточного звена иммунитета — снижение активности преимущественно Т-супрессоров, что ведет к активации аутоиммунных процессов, увеличению образования иммунных комплексов и повреждению базальной мембраны капилляров. У 5 % больных СД отмечается высокая степень предрасположенности к развитию сосудистых осложнений.
Неврологигеские осложнения сахарного диабета. Поражение периферической нервной системы (диабетическая нейропатия) относится к наиболее частым осложнениям СД (в среднем около 25 % среди пациентов с СД). Ключевую роль в развитии диабетической нейропатии играет гипергликемия.
Различают варианты диабетической нейропатии:
• острые (диабетическая радикулоплексопатия, мононейропатия) — чаще у пациентов с СД II типа;
• хронигеские (дистальная сенсомоторная и вегетативная полинейропатии) — одинаково часто у пациентов с СД I и II типов.
Лекция 12
ПАТОФИЗИОЛОГИЯ ЛИПИДНОГО ОБМЕНА
Л.Н. Осколок
Патологические изменения в обмене липидов являются следствием нарушения процессов переваривания и всасывания жиров, транспорта липидов и перехода их в ткани, окисления липидов в тканях, промежуточного липидного обмена, обмена липидов в жировой ткани.
Нарушение процесса переваривания и всасывания жиров возникает:
• при недостатке панкреатической липазы (например, при панкреатите); при этом нарушается расщепление жиров в верхних отделах тонкой кишки до свободных жирных кислот (СЖК) и р-моногли-церидов;
• дефиците желчных кислот (например, при закупорке желчного протока). Нарушается эмульгирование жира, активация панкреатической липазы и образование наружной оболочки смешанных мицелл, в составе которых высшие жирные кислоты и моноглицериды переносятся с места гидролиза жиров к всасывающей поверхности кишечного эпителия;
• усиленной перистальтике тонкой кишки и поражениях ее эпителия инфекционными и токсическими агентами, при этом не активируется и панкреатическая липаза;
• избытке в пище Са2* и Mg2*, когда образуются нерастворимые в воде соли желчных кислот — мыла;
• авитаминозах А и В, недостатке холина, нарушении процесса фосфорилирования (тормозится всасывание жира).
Вследствие нарушения переваривания и всасывания жиров развивается стеаторея: кал содержит много нерасщепленного жира, жирных кислот (ЖК) или их солей, вместе с жиром теряется и кальций.
Нарушение транспорта липидов и перехода их в ткани
В крови представлены все фракции липидов, которые содержатся в тканях человека. Холестерин (ХС), его эфиры (ЭХС), фосфолипиды
174
Лекция 12
(ФЛ) и триглицериды (ТГ) транспортируются в форме микромицелляр-ных комплексов, называемых липопротеинами (ЛП). Каждый ЛП содержит особый набор поверхностных апопротеинов, которые:
• связываются с рецепторами на клеточной поверхности;
• регулируют реакции с ферментами (например, с липазой);
• участвуют в синтезе и транспорте ЛП, регулируют взаимоотношения и связи липидов внутри частицы (рис. 12-1).
Циркулируя в крови, ЛП обмениваются ХС и апопротеинами.
На рисунке 12-2 представлена схема транспорта и метаболизма липидов. В стенке тонкой кишки после приема жирной пищи образуются хиломикроны (ХМ), транспортирующие экзогенные ТГ и ХС к местам утилизации (сердечная и скелетные мышцы, молочные железы) и депонирования (жировая ткань). ЛП очень низкой плотности (ЛПОНП), или пре-р-ЛП, синтезируются гепатоцитами и энтероцитами. Они транспортируют эндогенные ТГ. Клетки тканей, печени и других органов синтезируют и секретируют липопротеинлипазу (ЛПЛ). Она функционирует как мембраносвязанная гидролаза на поверхности эндотелия. Для проявления ее максимальной активности необходим кофактор — апопротеин С-П, компонент ХМ и ЛПОНП, субстратов для ЛПЛ. ЛПЛ высвобождает из них ТГ, происходит гидролиз ТГ до СЖК и глицерола. Часть СЖК связывается с альбуминами крови. Остальные СЖК поступают в клетку, где они либо окисляются, либо из них синтезируются ТГ. ХМ и ЛПОНП
Рис. 12-1. Схема строения липопротеина
Патофизиология липидного обмена
175
уменьшаются в размере, обогащаются ЭХС и превращаются в остатки (ремнанты) ХМ и ЛП промежуточной плотности (ЛППП). ЛППП выполняют ТГ-транспортную роль. Из ЛППП под действием ЛПЛ образуются ЛПНП — это р-ЛП, главные переносчики ХС в печень и ткани. ЛП высокой плотности (ЛПВП), или а-ЛП, синтезируются гепатоцитами и энтероцитами. ЛПВП образуются и в кровотоке при катаболизме ХМ и ЛПОНП. ЛПВП забирают ХС с плазматических мембран клеток периферических тканей и с поверхности богатых ТГ ЛП, где ХС становится избыточным при действии на эти частицы ЛПЛ. ЛПВП транспортируют ЭХС в печень для окисления в желчные кислоты. Клетки организма потребляют остатки ХМ через апо В-48 и апо Е-рецепторы, а ЛПОНП, ЛППП, ЛПНП через апо В-100 и апо Е-рецепторы. ЛПВП поставляют ЭХС в печень через апо Е и апо А-1-рецепторы.
Комплекс ЛП с рецептором подвергается эндоцитозу с последующей диссоциацией. Рецепторы восстанавливаются в цитоплазматической мембране, ЛП разрушаются в лизосомах (происходит гидролиз ЭХС и ТГ с образованием СЖК, ХС и аминокислот). Свободный ХС, ингибируя фермент p-гидрокси-р-метилглютарилкоэнзим-А-редуктазу, регулирует скорость собственного синтеза в клетке. Избыток ХС реэтерифици-руется в клетке ферментом ацилхолестеролацилтрансферазой. Скорость синтеза рецепторов ЛПНП регулируется по принципу обратной связи механизмом, чувствительным к содержанию внутриклеточного ХС. Это предупреждает избыточное накопление ХС в клетках и развитие атеросклероза.
Рис. 12-2. Схема транспорта и метаболизма липидов
176
Лекция 12
Клетки печени используют ХС и липиды для поддержания целостности мембран и синтеза желчных кислот. Часть ХС из печени экскретируется в желчь. В кровь ХС поступает в форме ЛП. В плазме, коре надпочечников (здесь ХС используется для синтеза стероидных гормонов), в атеросклеротической бляшке в основном содержится ЭХС.
Увеличение общих липидов в сыворотке крови свыше 2 ммоль/л носит название гиперлипемии. Она может быть алиментарной, транспортной и ретенционной.
Алиментарная гиперлипемия выявляется через 2-3 ч после употребления жирной пищи, в крови повышается содержание ХМ (хилоз). Через 9 ч уровень жира в крови возвращается к норме. Функциональные нарушения печени, блокада ретикулоэндотелиальной системы (РЭС), спленэктомия, недостаточность ЛПЛ, атеросклероз сопровождаются более выраженной алиментарной гиперлипемией.
Транспортная гиперлипемия обусловлена повышением тонуса симпатической нервной системы, избытком глюкагона, адреналина соматотропного гормона (СТГ), адренокортикотропного гормона (АКТГ), тиреотропного гормона (ПТ) и тироксина. Активируется гормончувствительная липаза ТГ, стимулируются липолиз и мобилизация СЖК из депо в печень. В печени повышается синтез ЛПОНП, возникает вторигная гипертриглицеридемия, возможно развитие стеатоза. При дефиците инсулина угнетается его антилиполитический эффект. Длительный избыток СТГ вызывает развитие инсулинорезистентности и активирует синтез белка, что повышает чувствительность адипоцитов к тоническим липолитическим стимулам.
Ретенционная гиперлипемия возникает при задержке перехода нейтральных липидов из крови в ткани. Это обусловлено уменьшением содержания альбуминов в крови (например, при заболеваниях почек и печени), которые транспортируют СЖК, а также снижением активности ЛПЛ, что наблюдается при:
• холемии (избыток желчных кислот в крови ингибирует ЛПЛ);
• сахарном диабете 1-го типа (дефицит инсулина снижает активность ЛПЛ, а избыток глюкагона тормозит секрецию ЛПЛ);
• увеличении осмоляльности плазмы крови (избыток Na' в крови ингибирует ЛПЛ);
• избытке СТГ (СТГ тормозит секрецию ЛПЛ);
• атеросклерозе (нарушается высвобождение гепарина, который стимулирует синтез ЛПЛ и активирует ее);
• избытке СЖК и моноглицеридов в крови (СЖК связываются с ЛПЛ, ослабляют ее взаимодействие с субстратом, уменьшают влияние апо С-П).
Патофизиология липидного обмена
177
Для ретенционной гиперлипемии характерна fi-липопротеинемия.
Увеличение содержания в плазме ЛП одного или нескольких классов носит название гиперлипопротеинемии. Различают 5 типов гипер-липопротеинемии по Фредриксону (1967). Уменьшение содержания в плазме ЛП разных видов носит название гиполипопротеинемии. Рассмотрим некоторые варианты нарушения нормального соотношения разных транспортных форм липидов и клинические последствия этого (табл. 12-1).
Таблица 12-1. Классификация гиперлипопротеинемий
Тип ХС плазмы ТГ плазмы Нарушения ЛП в крови Особенность гиперлипопротеинемии
I Повышен Повышен Избыток ХМ Недостаточность ЛПЛ
II Повышен Норма или повышен Избыток ЛПНП Дефект ЛПНП-рецепторов (апо В-100)
III Повышен Повышен Избыток ХМ и ремнантов ЛПОНП (р-ЛПОНП) Дефектная аллель апопротеина Е -> патологические ЛП (р-ЛПОНП)
IV Норма Повышен Избыток ЛПОНП Увеличен размер ЛПОНП и соотношение ТГ/апо-протеин В
V Повышен Повышен Избыток ХМ и ЛПОНП Нет апопротеина С-П
Блокирование синтеза белка (пуромицином) тормозит образование ХМ. Нарушается всасывание жиров, и они накапливаются в эпителиальных клетках кишечника. То же самое происходит при наследственном заболевании а-$-липопротеинемией, когда репрессирован синтез главного белка ХМ — апопротеина В-48.
Снижение активности ЛПЛ возможно при генетических мутациях вследствие образования неактивного белка. Это характерно для гиперхиломикронемии (I тип гиперлипопротеинемии), наследуемой по аутосомно-рецессивному типу. Комбинированная липемия (V тип гиперлипопротеинемии), вызванная нарушением обмена жиров и углеводов, наследуется полигенно. При этом заболевании отсутствует апопротеин С-П. Для обоих случаев характерно повышение уровня ТГ, ХМ, ХС плазмы. У них сходные клинические симптомы: эруптивные ксантомы (отложение ТГ в коже), гепатоспленомегалия (перегрузка клеток РЭС ХМ), абдоминальные колики (микроэмболия в сосудах, инфаркт селезенки), появление в костном мозге перегруженных липидами макрофагов, острый панкреатит (в капиллярах много ХМ -> замедляется кровоток секреция липазы в кишечник и кровоток капилляров ->
178
Лекция 12
расщепление ТГ -» СЖК, медленно перемещаясь, активируют перекисное окисление липидов (ПОЛ) -» возникает воспаление).
При II типе гиперлипопротеинемии (семейная гиперхолестеринемия, множественная бугоргатая ксантома), наследуемой по аутосомнодоминантному типу, обнаружен дефект рецепторов ЛПНП (апо В-100). Практически с рождения в плазме накапливаются ЛПНП. Уровень общего ХС в 2-4 раза выше нормального. Характерны бугорчатые ксантомы (отложения ХС) и липоидная дуга роговицы. Специфический регулируемый эндоцитоз невозможен. Развиваются атеросклероз, коронарная болезнь, инфаркт миокарда у детей.
III тип гиперлипопротеинемии («флотирующая» $-гиперлипемия, ремнантная гиперлипидемия) наследуется по рецессивному типу. В плазме накапливаются ТГ, ХМ и ремнанты ЛПОНП (р-ЛПОНП), поскольку их катаболизм в печени по рецептор-опосредованному пути нарушен. Это связано с появлением в их составе изоформы апо Е-2, которая слабо взаимодействует с рецепторами, р-ЛПОНП — это патологические ЛП. Они отличаются от ЛППП большей насыщенностью ХС, более высоким содержанием апопротеинов Е-1 и Е-2 при дефиците Е-3. р-ЛПОНП не способны превращаться в ЛПНП. Характерны липоидная дуга роговицы, локтевые и коленные ксантомы, отложения липидов в коже, атеросклероз.
IV тип (семейная гипертриглицеридемия) наследуется по аутосомнодоминантному типу. Природа генетического дефекта пока не выяснена. Характерны избыток ЛПОНП, по размеру превышающих нормальные частицы, и более высокое значение соотношения ТГ/апопротеин В при нормальном содержании сывороточного ХС. Избыточное отложение ТГ в органах приводит к ожирению, развитию инсулинорезистентности, гиперлипемии, кардиомиопатии, стеатоза, хронических заболеваний почек.
Наследственные нарушения обмена ЛПВП обусловлены в первую очередь дефектом гена, кодирующего синтез апопротеина А-1, локализованного в полигенном локусе хромосомы 11. При низком содержании в плазме ЛПВП снижается доставка ими избытка пищевых ТГ и ХС через рецепторы в подкожную жировую клетчатку (депо). Нарушение процессов этерификации ХС в ЛПВП и транспорта его между ЛП отдельных классов при наследственном дефиците лецитин-холестерин-ацилтрансферазы снижает способность ЛПВП удалять ХС из тканей. Изменяются состав и размер частиц ЛПНП, появляются патологические ЛП-Х («икс»), ЛП-Х отличаются высоким содержанием ФЛ и ХС, плот
Патофизиология липидного обмена 179
ностью, как у ЛПНП. Их наружная оболочка похожа на плазматическую мембрану клеток. Повышенная жесткость ЛП-Х увеличивает вязкость крови. В тяжелых случаях развиваются гемолитическая анемия, почечная недостаточность, катаракта, атеросклероз.
Роль нарушений липидного обмена в патогенезе атеросклероза
Развитие атеросклероза тесно связано с процессами транспорта ХС в артериальную стенку в составе ЛППП и ЛПНП и с процессами удаления ХС из артериальной стенки с помощью ЛПВП. В клинической практике используют холестериновый коэффициент атерогенности, который получают на основании определения концентрации общего ХС (Со) и ХС ЛПВП(Сллвп):
тг _ '-Q '—ЛПВП КХС ~ г '-лпвп
Это отношение идеально у новорожденных (не более 1).
У лиц 20-30 лет Кхс = 2,0-2,8, старше 30 лет без клинических признаков атеросклероза Кхс = 3-3,5, а у лиц с ишемической болезнью сердца Кхс >4-
На ранних стадиях развития атеросклероза возникают местные и системные нарушения обмена ХС и ЛП — дислипопротеинемии с преобладанием в крови ХС (больше 6,21 ммоль/л) и ЛПНП. Избыток ХС оказывает прямое повреждающее действие на эндотелий, увеличивая его проницаемость для ЛПНП и ЛППП. ЛПНП, окисляясь, повреждают как эндотелий, так и интиму сосуда. Захват макрофагами модифицированных ЛП не регулируется по механизму отрицательной обратной связи. Содержание ЭХС в клетке нарастает линейно в зависимости от концентрации ХС в окружающей среде. Избыточное накопление ЭХС приводит к трансформации макрофага в пенистую клетку. Высокая атерогенность характерна для ЛППП и ЛПНП, поскольку эти ЛП легко модифицируются в кровотоке и при контакте с клетками организма. Причиной их модификации могут быть свободные радикалы, продукты ПОЛ, различные ферменты или гипергликемия. На одну частицу ЛПНП человека приходится в среднем 1300 молекул полиненасыщенных ЖК и только 6-12 молекул а-токоферола, основного антиоксиданта этих ЛП, поэтому ПОЛ в первую очередь затрагивает ЛПНП. Постоянно возникающие в организме свободные кислородные радикалы инициируют образование гидроперекисей ненасыщенных ЖК, входящих в состав ФЛ, ТГ и ЭХС ЛПНП. Изменение структуры вызывает появление новых свойств ЛПНП.
180
Лекция 12
Модифицированные ЛПНП способны:
• повреждать эндотелий артерий и увеличивать его проницаемость;
• стимулировать экспрессию молекул адгезии и хемотаксис моноцитов и макрофагов в интиму;
• задерживать миграцию макрофагов из интимы;
• стимулировать синтез макрофагами лейкотриенов, хемоаттрактантов и митогенных факторов, активирующих миграцию и пролиферацию гладкомышечных клеток;
• тормозить зависящую от окиси азота релаксацию сосудов;
• инициировать синтез антител и развитие аутоиммунного процесса.
Перекисное окисление ЛПВП ослабляет их ХС-акцепторную функцию.
При гипергликемии происходит гликозилирование белков — необратимое ковалентное присоединение глюкозы к е-аминогруппе белка без участия ферментов. Гликозилированные ЛПНП не способны взаимодействовать с апо р-, Е-рецепторами клетки, замедляется их катаболизм, развиваются гиперлипопротеинемия и гиперхолестеринемия. Активируется нерегулируемое поглощение макрофагами гликозилированных ЛПНП, как следствие развивается атеросклероз. Гликозилирование ЛПВП ускоряет их катаболизм, возникает гипо-а-липопротеинемия.
Нарушение обмена липидов в жировой ткани
Ожирением называется избыточное отложение жира в организме. Критерием величины избытка массы тела является индекс Кетле — индекс массы тела (ИМТ), который вычисляют путем деления массы тела, выраженной в килограммах, на квадрат роста в метрах. По данным ВОЗ (1997), при:
• идеальной массе тела ИМТ = 18,5-24,9;
• избыточной массе тела ИМТ = 25-29,9;
• ожирении 1 степени ИМТ = 30,0-34,9;
• ожирении 2 степени ИМТ = 35,0-39,9;
• ожирении 3 степени ИМТ больше 39,9.
С помощью индекса Брока можно рассчитать идеальный вес.
• Идеальный вес = а-10%а, где а — рост (в сантиметрах) - 100.
По этиологии различают три вида ожирения — церебральное, гормональное и алиментарно-конституциональное.
Основные патогенетические факторы ожирения:
• повышенное потребление углеводов и жиров, превышающее энергетическое расходование жира;
Патофизиология липидного обмена
181
• недостаточное использование жира депо как источника энергии;
• избыточное образование липидов из углеводов.
Выделяют две формы ожирения — первичное и вторичное.
Первигное ожирение бывает двух видов — женский тип или ягодичнобедренное ожирение (форма груши), и мужской тип или абдоминальновисцеральное ожирение (форма яблока).
В первичном ожирении важная роль принадлежит наследственности. Обнаружены предполагаемые гены ожирения — гены, кодирующие образование Р2- и Р3-адренорецепторов, влияющих на обмен липидов в жировой и мышечной ткани, ген ЛПЛ и другие гены. Однако у большинства людей отсутствует моногенное наследование ожирения. Причины избыточной массы тела прежде всего кроются в особенностях стиля жизни и пищевого поведения. Переедание может быть следствием повышенного аппетита из-за низкой возбудимости центра насыщения и/или повышенной возбудимости центра голода. При первичном ожирении нарушены взаимоотношения между жировой тканью и пищевым центром в гипоталамусе. Адипоциты секретируют пептидный гормон лептин, который подавляет аппетит, пищевое поведение, липогенез и активирует липолиз. При увеличении размеров адипоцитов повышается секреция лептина, но развивается лептинорезистентность, которая сопровождается повышенным аппетитом и пищевым поведением, превышающем энергозатраты.
На рисунке 12-3 представлен предполагаемый механизм ожирения при переедании на фоне гиподинамии. Для него характерны повышенное потребление жиров и углеводов, превышающее энергетическое расходование жира, избыточное образование липидов из углеводов, увеличенное отложение ТГ в скелетных мышцах, печени, мышце сердца, недостаточное использование жира как источника энергии.
Вторигное ожирение — это синдром при первичных эндокринных заболеваниях, поражениях центральной нервной системы и приеме ряда лекарственных препаратов (ятрогенное ожирение). Гипоталамическое и диэнцефальное ожирение могут развиться у больных с травмой головного мозга, после перенесенного менингита или энцефалита, при внутричерепной гипертензии и опухолях головного мозга. При нормальной (соответствующей энергетическим тратам) функции центра голода причиной ожирения может быть недостаточное использование жира из жировых депо в качестве источника энергии. Такое ожирение развивается при снижении тонуса симпатической и/или повышении тонуса парасимпатической нервной системы, при тормозящем влиянии коры головного мозга на центры симпатического отдела диэнцефальной области.
182
Лекция 12
ОЖИРЕНИЕ
Рис. 12-3. Предполагаемая схема патогенеза ожирения при переедании на фоне гиподинамии
Жировая ткань находится под контролем гормональных и гуморальных факторов. Ожирение развивается при избытке инсулина, глюкокортикоидов и дефиците адреналина, глюкагона, АКТГ, мелано-цитсгимулирующего гормона (МСТ), ТТГ, СТГ, антидиуретического гормона (АДГ), тиреоидных гормонов, p-липотропина и половых стероидов.
Жировая ткань является инсулинозависимой. Инсулин тормозит липолиз (угнетает активность гормоночувствительной липазы в адипоцитах), усиливает липогенез (стимулирует поглощение глюкозы жировой тканью, синтез ЖК и ТГ из продуктов углеводного обмена), стимулирует захват жира путем пиноцитоза жировой тканью, повышает аппетит и активность центра голода гипоталамуса (снижает уровень глюкозы в крови).
Тиреоидные гормоны и глюкокортикоиды не оказывают прямого действия на липолиз, но обладают пермиссивным влиянием в отношении эффектов других гормонов. При дефиците тиреоидных гормонов снижаются потребление кислорода и окисление СЖК, ТГ накапливаются в
Патофизиология липидного обмена 183 жировых депо; снижается активность симпатической нервной системы и адреналина, угнетается липолиз.
При избытке глюкокортикоидов:
• жировая клетчатка перераспределяется в висцеральную область: здесь много глюкокортикоидных и андрогенных рецепторов, при стимуляции которых активируется адипогенез;
• развивается лептинорезистентность;
• возникает гипергликемия, снижается чувствительность центра насыщения в гипоталамусе, повышаются аппетит и потребление пищи;
• угнетаются действие СТГ, окисление и мобилизация жира из депо.
При дефиците адреналина, глюкагона, АКТГ, МСГ, СТГ и АДГ снижается активность гормоночувствительной липазы и скорость липолиза ТГ.
При дефиците ^-липотропина, ТТГ, СТГ и половых стероидов отмечается увеличение отложения жира в висцеральных депо.
При ожирении нарушается функция адипоцитов. Они в избытке секретируют фактор некроза опухолей-а, СЖК, резистин, интерлейкин-6, ангиотензин-П, ингибитор активатора плазминогена-1 и снижают секрецию адипонектина, что способствует развитию метабо-лигеского синдрома, основными клиническими проявлениями которого являются дислипидемия, ранний атеросклероз и ИБС, абдоминальновисцеральное ожирение, артериальная гипертензия, гиперинсулинемия, инсулинорезистентность, сахарный диабет 2-го типа, стеатоз печени, нарушение гемостаза, синдром обструктивных апноэ во сне, гиперури-кемия и подагра, микроальбуминурия.
Стеатоз пегени — жировой гепатоз, проявляющийся в накоплении капель жира не менее чем в 50 % печеночных клеток. Размеры капель достигают величины ядра гепатоцитов или превышают его.
Основу патогенеза стеатоза составляют:
• увеличенное поступление СЖК в печень;
• активация синтеза, ускорение этерификации, замедление окисления ЖК;
• снижение синтеза апопротеинов в печени и нарушение нормальной сборки ЛПОНП;
• затруднение экскреции ЛПОНП.
Основные этиологические факторы стеатоза печени:
• токсические воздействия на печень (алкоголь, мышьяк, хлороформ);
184 Лекция 12
• влияние лекарственных препаратов (кортикостероиды, цитостатики, антибиотики тетрациклинового ряда):
• дисбаланс пищевых факторов;
• эндокринно-метаболические нарушения (синдром Иценко-Ку-шинга, сахарный диабет);
• гипоксия (сердечно-сосудистая недостаточность, легочные заболевания).
При алкоголизме активируется поступление СЖК в печень, повышается этерификация ЖК за счет избытка глицерофосфата, снижается окисление ЖК, нарушаются сборка, внутриклеточный транспорт и секреция ЛПОНП. При активации ПОЛ и развитии аутоиммунных процессов повреждаются гепатоциты и нарушается структура ЛПОНП.
Лекция 13
ПАТОФИЗИОЛОГИЯ ВОДНО-СОЛЕВОГО ОБМЕНА
Г.П. Щелкунова
Содержание воды в организме и механизмы регуляции водного баланса
Вода — один из важнейших компонентов живого организма. У новорожденных детей она составляет 75-80 % массы тела, у взрослых мужчин массой около 70 кг — 53-55 %, а у женщин — 43-45 %. Из этого количества на внутриклеточную воду приходится 25-30 % у мужчин и 18-20 % у женщин, а на внеклеточную у тех и других — около 25 %. В свою очередь, внеклеточная вода складывается из интерстициальной (15-17 %), плазменной (4-5 %) и трансцеллюлярной (1-2 %). К транс-целлюлярной жидкости относится камерная влага глаз, спинномозговая, суставная и синовиальная жидкость, содержимое почечных канальцев, пищеварительные соки. По мере старения организма общее количество воды уменьшается до 50 % массы тела у мужчин и 42-44 % у женщин. При этом количество внутриклеточной воды уменьшается, а внеклеточной увеличивается.
В течение суток у новорожденного обменивается около 50 % объема внеклеточной воды, у взрослого около 15 %. За сутки в организм взрослого человека должно поступать около 2,5-2,7 л воды (около 1,2-1,5 л с питьем, около 1 л с пищей, 0,2-0,4 л воды образуется эндогенно в результате метаболизма). Для сохранения водного баланса и гомеостаза суточное выделение воды из организма должно соответствовать поступлению, т.е. равняться 2,5-2,7 л воды. При этом около 1,4-1,6 л выделяется с мочой, 0,5-0,7 л выводится через легкие; 0,5-0,7 л — через кожу и около 0,005-0,1 л — с калом. Поступление воды регулируется из центра жажды, главными стимуляторами которого являются увеличение осмоляльности плазмы, дегидратация клеток и ангиотензин-П.
186
Лекция 13
У здорового человека в оптимальных условиях (температура, влажность и т.д.) через легкие, кожу и кишечник выделяется постоянное количество воды, а сохранение водного баланса обеспечивается главным образом выделительной функцией почек, в регуляции которой важнейшую роль играют антидиуретический гормон гипофиза (АДГ, вазопрессин), ренин-ангиотензин-альдостероновая система (РААС), предсердный натрийуретический фактор (ПНФ), простагландины, катехоламины, глюкокортикоиды и др. Афферентным звеном регуляторных систем являются осмо-, баро- и волюморецепторы сосудистой стенки.
Роль антидиуретического гормона. АДГ секретируется в супраоптических и паравентрикулярных ядрах гипоталамуса (рис. 13-1), накапливается в задней доле гипофиза и выделяется из нее при увеличении осмоляльности плазмы крови (главным образом при увеличении в крови
| Супраоптические и паравентрикулярные ядра гипоталамуса |
| Задняя доля гипофиза |+~-----------------1 ддр |
Стимулируют выделение: ----► АДГ *— Тормозят выделение:
- t осмоляльности плазмы;
- 4 наполнения артериальных сосудов шеи и грудной клетки, предсердий, легочных вен;
- ангиотензин II (АТ-11);
- тошнота, рвота;
- некоторые стрессоры
(боль, волнение);
-^-адреномиметики;
- ацетилхолин;
- никотин, эфир;
- фенобарбитал
- 4 осмоляльности плазмы;
- t наполнения артериальных сосудов шеи и грудной клетки, предсердий, легочных вен;
- холод;
-а-адреномиметики;
- этанол;
- морфин;
- резерпин;
- глюкокортикоиды
(но t чувствительность почечных канальцев к АДГ)
Эффекты АДГ
Собирательные трубки почек: раздражение У2-рецепторов на базальной мембране эпителиальных клеток -» активация аденилатциклазы -* t цАМФ -* активация протеинкиназы А -* встраивание водных каналов в апикальную мембрану, появление в клетке аквапереносящих белков (аквапорин G) -* f реабсорбции воды -» 4 диуреза.
Гладкие мышцы сосудов: раздражение V, -рецепторов -» активация -» фосфолипазы С -* образование инозитолтрифосфата (ИТФ) и диацилглицерола (ДАГ) -* активация протеинкиназы С -»t Са2* в клетке -* сокращение гладких мышц -* спазм сосудов -> t АД.
Рис. 13-1. Регуляция секреции антидиуретического гормона и его основные эффекты
Патофизиология водно-солевого обмена
187
натрия, глюкозы, мочевины и т.п.), уменьшении наполнения предсердий, легочных вен, артериальных сосудов шеи и грудной клетки (например, при уменьшении объема циркулирующей крови и длительном вертикальном положении человека, при уменьшении сердечного выброса), а также при некоторых стрессовых ситуациях (боль, волнение), тошноте, рвоте, под влиянием ангиотензина-П, p-адреномиметиков, ацетилхолина, никотина, фенобарбитала, эфира.
Раздражая У2-рецепторы эпителия дистальных канальцев и собирательных трубок почек, АДГ увеличивает реабсорбцию воды, задерживая ее в организме. Кроме того, АДГ (вазопрессин) участвует в поддержании сосудистого тонуса и регуляции артериального давления. Повышенное его выделение вызывает спазм сосудов путем стимуляции У^рецепторов гладких мышц, что приводит к увеличению в них ионов Са2’.
Выделение АДГ может тормозиться: при уменьшении осмоляльности плазмы крови, увеличении наполнения предсердий, артериальных сосудов шеи и грудной клетки (например, при гиперволемии, длительном горизонтальном положении человека, в состоянии невесомости и др.), а также при охлаждении организма или под влиянием а-адреномиметиков, этанола, морфина, резерпина, глюкокортикоидов (при одновременном повышении чувствительности рецепторов почечных канальцев к АДГ). При уменьшении выделения АДГ диурез увеличивается и больше воды выводится из организма.
При врожденном или приобретенном дефиците АДГ развивается синдром несахарногомогеизнурения (диабета), характеризующегося полиурией до 10-20 л в сутки с низкой плотностью мочи и компенсаторной жаждой и полидипсией. Возможно также развитие нефрогенного несахарного могеизнурения, обусловленого врожденной или приобретенной нечувствительностью рецепторов почек к АДГ (например, при гипокалиемии или гиперкальциемии).
Синдром неадекватной секрециии АДГ может наблюдаться при опухолях задней доли гипофиза, нарушениях нейроэндокринной регуляции, под влиянием химических веществ (литий), лекарственных препаратов (диметилхлортетрациклин и др.), а также при введении регуляторных механизмов «в заблуждение». Примеры ошибочного регулирования водно-электролитного баланса будут рассмотрены ниже.
Роль РААС. Из субстрата, вырабатываемого печенью, в юкстагломерулярном аппарате (ЮГА) почек образуется и накапливается в виде гранул ренин (гликопротеин, мол. масса — 40 000), который как гормон выделяется в кровь: при уменьшении почечного кровотока, обусловлен
188
Лекция 13
ного как заболеванием самих почек, так и уменьшением ОЦК, снижением АД: увеличении в моче натрия и хлора; влиянии адреномиметиков, простациклина и др. (рис. 13-2).
В плазме крови под влиянием ренина из а2-глобулина образуется декапептид ангиотензин-l, который при участии АПФ, преимуществено в легких, а также в стенках сосудов, переходит в октапептид ангиотензин-Н и, далее, в гептапептид ангиотензин-lll. Последнего в пять раз меньше, чем ангиотензина-П.
Стимулируют выделение ренина:
| почечного кровотока, | ОЦК, [АД ППг, f Na в моче, раздражение Д-адренорецепторов
1 Печень -j- Субстрат ренина ЮГА почек -- 1 РЕНИН 1 1— Плазма крови — Из а2-глобулина АТ-1 (декапептид) | *- АПФ Легкие, сосуды -- АТ-11 (октапептид) 1 * А Т-Ш (гептапептид Надпочечники — АЛЬДОСТЕРОН (клубочковая зона) Почки -- f реабсорбции Na* <••••• Кишечник t секреции К* и Н* Слюнные 1 и потовые 1 железы t осмоляльности плазмы Задняя доля - - f АДГ гипофиза Почки - - t реабсорбции воды 1 1 диуреза Задержка воды Тормозят РААС: /?-адреноблокаторы, индометацин, АДГ, альдостерон, ОЦК, Na* в плазме Блокаторы АПФ Саралазин
ПНФ, блокаторы рецепторов АТ-11 t Na*, j К* в плазме, t ОЦК ••••• Спиронолактоны (альдактон)
Рис. 13-2. Регуляция ренин-ангиотензин-альдостероновой системы и ее роль в поддержании водно-электролитного баланса
Патофизиология водно-солевого обмена
189
Ангиотензин-П стимулирует центр жажды, повышает активность симпатических нервов, обладает прессорными свойствами (вызывает спазм сосудов) и уменьшает скорость гломерулярной фильтрации, стимулирует выделение АДГ, что способствует задержке воды в организме. Кроме того, ангиотензин-П и ангиотензин-Ш являются мощными стимуляторами клубочковой зоны коры надпочечников и, раздражая специальные рецепторы, усиливают выделение минералкортикоида альдостерона (вторигный альдостеронизм). Выделение альдостерона может усиливаться также под влиянием гипонатриемии, гиперкалиемии, ПГЕ, АКТГ.
Альдостерон через специальные рецепторы в дистальных канальцах и собирательных трубках почек, в дистальных отделах толстой кишки, а также в слюнных и потовых железах вызывает усиление синтеза различных специализированных внутриклеточных белков, которые увеличивают реабсорбцию из мочи в кровь Na+ и секрецию из крови в мочу К+ и Н+. Задержка Na+ в плазме крови сопровождается увеличением ее осмоляльности, что по описанному выше механизму приводит к выделению АДГ и усилению реабсорбции воды, уменьшению диуреза, задержке воды в организме и нормализации осмоляльности плазмы {осморегулирующий рефлекс).
Необходимо отметить, что помимо стимуляторов РААС у нее существуют естественные и искусственные ингибиторы. Например, выделение ренина из ЮГА почек тормозят АДГ, альдостерон (по принципу обратной связи), увеличение ОЦК, гипернатриемия, р-адреноблокаторы, индометацин и др. Блокаторы АПФ, например эналаприл, препятствуют превращению ангиотензин-I в ангиотензин-П и ангиотензин-Ш. ПНФ, ингибиторы рецепторов ангиотензин-П, блокируя рецепторы ангиотензин-П и ангиотензин-Ш в клубочковой зоне надпочечников, тормозят выделение альдостерона. Его выделениие уменьшается также при увеличении ОЦК, гипернатриемии, гипокалиемии, под влиянием дофамина. Блокировать рецепторы альдостерона в почках, кишечнике, слюнных и потовых железах могут альдактон, спиронолактон и др.
Мощным естественным ингибитором РААС является ПНФ (другое название ПНУФ — предсердный натрийуретический фактор), образующийся в кардиомиоцитах предсердий. ПНФ оказывает диуретическое и натрийуретическое действие через усиление почечной фильтрации путем уменьшения тонуса приводящих и увеличения тонуса отводящих почечных артериол, снижения секреции ренина и ингибирования
190
Лекция 13
синтеза альдостерона (ПНФ блокирует рецепторы ангиотензин-П и ангиотензин-111 на клетках клубочковой зоны надпочечников).
РААС в здоровом организме включается быстро и достаточно надежно поддерживает водно-электролитный баланс, от которого зависят ОЦК, величина АД, деятельность сердца, почек и других систем организма.
Однако в ряде случаев при поражении надпочечников продукция альдостерона может быть пониженной (например, при болезни Аддисона) или повышенной (гиперкортицизм, первигный альдостеронизм). Избыточное выделение альдостерона может быть результатом «ошибочного регулирования» (вторигный альдостеронизм). В этих случаях развиваются выраженные нарушения водно-электролитного обмена, примеры которого будут описаны ниже.
Рассмотренные выше механизмы регулируют прежде всего содержание общей воды на уровне целого организма и величину ОЦК. Обмен воды между сосудистым руслом и тканями осуществляется по известному механизму Э. Старлинга (1896), суть которого в современной интерпретации сводится к следующему. Через стенки капилляров достаточно легко перемещается вода, электролиты, некоторые органические соединения, но труднее транспортируются белки. Концентрация белка в плазме крови — 60-80 г/л, а в тканевой жидкости колеблется от 10 до 30 г/л. При этом величина ОД в крови равна 25-28 мм рт.ст., а в интерстициальном пространстве — около 5 мм рт.ст. Разность этих давлений (19-22 мм рт.ст.) есть эффективная онкотическая всасывательная сила (ЭОВС), под действием которой вода всасывается в капилляры из интерстициального пространства. Но кровь движется в сосудах с определенной скоростью и под определенным давлением. Гидростатическое давление крови в капиллярах неодинаковое и колеблется от 30-32 мм рт.ст. в артериальном конце капилляра до 8-10 мм рт.ст. в венозном конце. Величина давления тканевой жидкости отрицательна (на 6-7 мм рт.ст. ниже величины атмосферного давления) и обладает присасывающим эффектом. Разность между гидростатическим давлением крови и гидростатическим давлением интерстициальной жидкости называется эффективным гидроапатигеским давлением (ЭГД), которое колеблется от 36-38 мм рт.ст. в артериальном конце капилляра до 14-16 мм рт.ст. в венозном конце.
В тех капиллярах, где ЭГД выше ЭОВС, происходит фильтрация жидкости из сосудов в интерстициальное пространство, а в тех капиллярах, где ЭГД меньше ЭОВС, — резорбция (всасывание) жидкости из ткани в
Патофизиология водно-солевого обмена
191
сосудистое русло. Следует отметить, что один и тот же капилляр в зависимости от интенсивности кровообращения (покой или нагрузка) может или фильтровать жидкость, или ее всасывать. У здорового человека за сутки из крови в ткань фильтруется до 20 л жидкости, 17 л всасывается обратно в капилляры и около 3 л оттекает из ткани по лимфатическим капиллярам и через лимфатическую систему возвращается в сосудистое русло (рис. 13-3).
Нарушение обмена воды между кровью и интерстициальным пространством может наблюдаться при:
• изменениях гидростатического давления в капиллярах;
• изменениях ОД в крови и интерстиции;
• повышении проницаемости сосудистой стенки для белков;
• нарушениях лимфооттока.
Обмен жидкости между внутри- и внеклеточным пространством осуществляется по закону изоосмоляльности, согласно которому вода перемещается через биологические мембраны в сторону большего осмотического давления до тех пор, пока осмоляльность по обе стороны мембраны не уравняется. В норме осмоляльность внутри и вне клеток равна 285 мОсм/кг Н2О и зависит главным образом от содержания Na+, глюкозы, мочевины и других осмотически активных веществ, содержание которых может изменяться при патологии почек, надпочечников, при сахарном диабете, повреждении клеток и т.д. Это может повлечь за собой или обезвоживание клеток (при увеличении осмоляльности вне-
Капилляр
Артериальный конец Венозный конец Q
эгд > эовс
эгд < эовс
Клетка 285 мОсм/кг Н20
Вне клетки
* , Н20 285 мОсм/кг Н20
Рис. 13-3. Обмен воды между кровью и тканями
192
Лекция 13
клеточной жидкости) или их гипергидратацию (при уменьшении осмоляльности внеклеточной жидкости).
Следует отметить также, что и внутри, и вне клеток часть воды находится в связанном, а часть в свободном (мобильном) состоянии. Количественное соотношение этих фракций воды в различных органах неодинаковое и может изменяться при патологии.
Виды нарушений водно-электролитного обмена
Все нарушения водного обмена в организме (дисгидрии) принято делить на два основных вида: гипогидратацию и гипергидратацию, которые, в свою очередь, в зависимости от величины осмоляльности внеклеточной жидкости подразделяют на изоосмоляльную (изотоническую), гипо-осмоляльную (гипотоническую), гиперосмолялъную (гипертоническую)
Выделяют также ассоциированные формы нарушений: внеклеточную гипогидратацию с внутриклеточной гипергидратацией и наоборот, а также гипо- и гиперосмолярные синдромы.
Причины, патогенез, виды и последствия гипо-и гипергидратации для организма
Обезвоживание (гипогидратация, дегидратация, эксикоз) развивается в тех случаях, когда потери воды превышают поступление ее в организм и возникает отрицательный водный баланс.
Недостатогное поступление воды в организм (истинная гипертоническая дегидратация) наблюдается при водном голодании, повреждении центра жажды, при неврологических и психических заболеваниях, коматозных состояниях, при затруднении глотания, непроходимости пищевода и др. При полном прекращении поступления воды в организм продолжительность жизни взрослого человека — 6-8 сут, а ребенка массой около 7 кг — в два раза меньше. Маленькие дети больше воды теряют через кожу и легкие (перспирация), а также с мочой, так как окончательное формирование всех функций почек завершается только в возрасте 11 лет. У детей интенсивнее происходит обмен внеклеточной воды и значительно выше чувствительность к ее дефициту.
Повышенная потеря воды из организма может происходить в результате следующих причин: массивной кровопотери, полиурии, неукротимой рвоты, профузного поноса, желудочно-кишечных свищей, гипервентиляции легких, усиленном потоотделении (перспирация воды может достигать 10-14 л/сут), лихорадки (повышение температуры
Патофизиология водно-солевого обмена
193
тела на 1 °C сопровождается потерей воды 0,5 л/сут), обширных ожогов с ликвореей, диабетических ком (кетоацидотическая и, особенно, гипер-осмоляльная), первичного дефицита в организме электролитов (главным образом натрия).
При эквивалентной потере воды и электролитов, т.е. в тех случаях, когда теряется жидкость, по электролитному составу близкая к плазме крови и интерстициальной жидкости, развивается изоосмолялъная гипогидратация. Ее можно наблюдать у больных в первой стадии после массивной кровопотери, у больных с кишечным токсикозом (дизентерия, энтерит, холера, язвенный колит), при стенозе привратника, высокой непроходимости тонкого кишечника, т.е. при состояниях, сопровождающихся обильной рвотой или диареей, а также при массивных ожогах, кишечных свищах, длительном применении некоторых диуретиков и в других случаях.
При этом виде обезвоживания осмоляльность внутри и вне клеток не изменяется. Уменьшается главным образом объем внеклеточной воды, и ведущие симптомы обусловлены нарушением кровообращения: уменьшением минутного объема сердца (МОС), АД, центрального венозного давления (ЦВД) и др.
Если потеря электролитов преобладает над потерей воды, то развивается гипоосмоляльная гипогидратация. Так как при этом осмотическое давление внутри клеток оказывается выше, чем во внеклеточном пространстве, то вода по закону изоосмоляльности перемещается в клетки, вызывая их отек и набухание.
Причинами гипоосмоляльного обезвоживания могут стать обильное и длительное потоотделение, дефицит минералкортикоидов при недостаточности надпочечников (болезнь Аддисона), рвота, понос, коррекция изоосмоляльного обезвоживания водой без электролитов и др.
При неправильном проведении гемодиализа или перитонеального диализа быстрое снижение осмоляльности плазмы может вызвать отек нервных клеток, судороги, кому.
Последствия обезвоживания такого вида обусловлены нарушениями кровообращения, значительным уменьшением ОЦК, увеличением вязкости крови, нарушениями сердечной деятельности (уменьшением УОС, МОС), нарушением КОС (при неукротимой рвоте — алкалоз, при профузном поносе — ацидоз), выраженной гипоксией.
Важно отметить, что при гипоосмоляльной гипогидратации у больных может отсутствовать жажда (так как осмоляльность плазмы низкая, а в клетках много воды).
194
Лекция 13
Если потеря воды преобладает над потерей солей, то развивается ги-перосмоляльная гипогидратация. Она наблюдается при недостаточном поступлении воды в организм, лихорадке с обильным потоотделением, искусственной вентиляции легких без увлажнения дыхательной смеси, несахарном мочеизнурении, инфузиях гипертонических растворов и средств парентерального питания, питье морской воды на фоне обезвоживания, сахарном диабете (глюкозурия сопровождается полиурией, а высокая осмоляльность плазмы крови обусловлена гипергликемией), почечной недостаточности (сочетание полиурии и азотемии — увеличения содержания в крови мочевины и других осмотически активных веществ, т.е. при осмотическом диурезе).
На фоне общего обезвоживания воду главным образом теряют клетки, так как она будет перемещаться во внеклеточное пространство, т.е. в сторону большего осмотического давления. Для таких больных характерны мучительная жажда, неврологические симптомы: возбуждение, беспокойство, спутанность сознания, повышение температуры тела и др.
Компенсаторные изменения при обезвоживании сводятся к рефлекторному возбуждению центра жажды (исключая гипоосмоляльную форму) под влиянием гиперосмоляльности плазмы, уменьшения объема внутриклеточной воды и ангиотензина-П. Полидипсия (повышенное потребление воды) приводит к увеличению поступления воды (если это возможно) и восстановлению водного баланса.
Жажда может появиться даже при небольшом дефиците воды, сопровождающемся гипернатриемией. Дефицит 3-4 л воды вызывает мучительную жажду.
Активируется РААС (по описанному выше механизму), увеличивается секреция АДГ гипофизом, диурез уменьшается, и вода задерживается в организме. Компенсаторные изменения оказываются достаточными только при легкой форме обезвоживания, когда дефицит воды составляет меньше 5 % от должной величины. При этом у больных могут отсутствовать типичные симптомы обезвоживания, не считая тахикардии. При дефиците воды 5-10 % развивается обезвоживание средней тяжести, дефиците более 10 % — тяжелая дегидратация. Дефицит более 12 % воды приводит к развитию гиповолемического шока и почечной недостаточности (табл. 13-1).
Последствия дегидратации зависят главным образом от ее тяжести, но для всех форм обезвоживания характерны следующие симптомы: сухость кожи (особенно в подмышечной и паховой области), сухость
Патофизиология водно-солевого обмена
195
Таблица 13-1. Формы обезвоживания в зависимости от дефицита воды и их основные проявления
Формы Легкая Средней тяжести Тяжелая Шок
Дефицит воды, % от должной величины <5% 5-10% 10-12% Более 12 %
Дефицит ОЦК, % от должной величины 5-15% 16-25 % 26-40 % Более 40 %
Частота сердечных сокращений 100 120 140 Слабый частый пульс
Среднее АД Норма ? 1 1
Минутный объем сердца Норма т 1 и
Ударный объем сердца Норма 1 и 'L'L'L
Центральное венозное давление Норма I U XXX
Диурез Норма 1 и Анурия
КОС Норма Компенсированный ацидоз Некомпенсированный метаболический ацидоз Смешанный некомпенсированный ацидоз
Ра02 Норма Норма X и
слизистых оболочек, гипосаливация (при длительном процессе способствует развитию воспалений в ротовой полости); гладкий красный с глубокими морщинами язык; запавшие и мягкие при надавливании глазные яблоки; сниженный тургор тканей (кожи, мышц).
В зависимости от степени дегидратации выявляются признаки нарушения сердечной деятельности (тахикардия, уменьшение ударного и минутного объема, возможна остановка сердца); снижается артериальное и центральное венозное давление, нарушается периферическое кровообращение (в тяжелых случаях может развиться коллапс). Повышается вязкость крови и связанный с этим риск образования тромбов и ДВС-синдрома в тяжелых случаях. Развивается гипоксия.
Из-за ослабления почечного кровотока, уменьшения фильтрации в клубочках и увеличения реабсорбции воды в собирательных трубках почек снижается диурез. При тяжелом обезвоживании на фоне олиго- и анурии могут появиться признаки почечной недостаточности (азотемия, ацидоз и др.). Угнетается перистальтика желудка и кишечника, нарушаются процессы переваривания и всасывания, могут быть запор, парезы кишечника и другие расстройства. Из неврологических симптомов следует обратить внимание на слабость, вялость, апатию, сонливость или
196
Лекция 13
возбуждение, повышение температуры тела, в тяжелых случаях помрачение сознания, галлюцинации, бред.
Принципы терапии обезвоживания сводятся к устранению вызвавшей его причины, введению изотонических или гипертонических растворов в зависимости от вида обезвоживания. Коррекция проводится обязательно под контролем осмометрии, pH-метрии и других показателей гомеостаза. Применяют средства, улучшающие микроциркуляцию и реологические свойства крови, проводят симптоматические мероприятия.
Гипергидратация. Задержка воды в организме {гипергидратация, ги-пергидрия) наблюдается или при чрезмерном введении воды (водная интоксикация), или при недостаточном ее выведении из организма. В обоих случаях развивается положительный водный баланс. Аналогично обезвоживанию в зависимости от величины осмоляльности внеклеточной жидкости гипергидратация может быть изоосмоляльной, гиперосмоляльной и гипоосмолялъной.
Изоосмоляльная гипергидратация развивается при вливании больших количеств изотонических растворов, сердечно-сосудистой недостаточности, токсикозах беременности, болезни Иценко-Кушинга (избыточной продукции АКТГ гипофизом), вторичном альдостеронизме, гипопротеинемиях (нефротический синдром, голодание, печеночная недостаточность), нарушениях лимфооттока.
Последствия такой гипергидратации сводятся к увеличению ОЦК (олигоцитемическая гиперволемия), повышению АД, перегрузке сердца с риском развития его недостаточности, а также к выраженному отечному синдрому с угрозой развития в тяжелых случаях отека легких, отека мозга.
Гиперосмоляльная гипергидратация развивается в том случае, когда задержка солей преобладает над задержкой воды и осмоляльность внеклеточной жидкости оказывается больше, чем внутри клеток. Это может наблюдаться при введении больших количеств гипертонических растворов, вынужденном неограниченном питье морской воды, осмоляльность которой значительно превышает осмоляльность плазмы крови; вторичном альдостеронизме; патологии почек, характеризующихся преимущественным нарушением выведения солей (тубулопатии, фер-ментопатии), а также мочевины (сочетание олигурии и азотемии при почечной недостаточности).
При этом на фоне увеличения объема и осмоляльности внеклеточной жидкости происходит обезвоживание клеток. Возникает внутриклеточная дегидратация на фоне внеклеточной гипергидратации
Патофизиология водно-солевого обмена
197
(ассоциированное нарушение). Несмотря на избыток воды в организме, больных беспокоит жажда, а дополнительное поступление воды только усугубляет процесс. У больных резко возрастают ОЦК, артериальное и центральное венозное давление, развивается перегрузка сердца (возможна его остановка), выражены неврологические симптомы, признаки повышения внутричерепного давления, могут развиться отек мозга, отек легких. На фоне почечной недостаточности осмоляльность обычно повышается как вне, так и внутри клеток, так как задерживающаяся в организме мочевина легко проникает через все биологические мембраны. Происходят отек и набухание клеток.
Гипоосмоляльная гипергидратация развивается в том случае, когда задержка воды в организме сопровождается снижением осмоляльности внеклеточной жидкости. Такие нарушения могут возникать при чрезмерном введении бессолевых растворов, превышающем выделительную способность почек; повышенной неадекватной секреции АДГ, циррозе печени, почечной недостаточности, недостаточности кровообращения с отечным синдромом и др.
У больных увеличивается объем внеклеточной воды, гиперволемия выражена слабее, чем при вышеописанных формах гипергидрии, так как значительная часть воды перемещается в сторону большего осмотического давления, т.е. внутрь клеток, вызывая их осмотическое повреждение и развитие синдрома водной интоксикации (водного отравления). Происходит гемолиз эритроцитов, сопровождающийся гемоглобинурией; полиурия сменяется анурией с признаками почечной недостаточности. Характерны рвота, понос. Рано появляются неврологические симптомы: вялость, апатия, судороги, помрачение сознания.
Следует оговориться, что уменьшение осмоляльности плазмы крови менее 280 мОсм/кг Н2О, независимо от общего количества воды в организме (гипо- или гипергидратация), называется гипоосмоляльным синдромом. Он всегда сопровождается гипергидратацией клеток и признаками водной интоксикации (даже на фоне обезвоживания). Снижение осмоляльности плазмы ниже 250-230 мОсм/кг Н2О приводит к смерти. Наиболее частыми причинами развития этого синдрома являются потеря натрия с мочой при дефиците альдостерона или нечувствительности к нему канальцев почек, интенсивная потеря натрия с потом, рвотными массами, поносом, а также чрезмерное разведение его концентрации на фоне гипергидратации.
При увеличении осмоляльности плазмы крови более 300 мОсм/кг Н2О (на фоне как гипо-, так и гипергидратации) развивается гиперос-
198
Лекция 13
моляльный синдром, сопровождающийся клеточной дегидратацией. Он наблюдается у больных на фоне гипернатриемии (более ISOZOO ммоль/л) алиментарного происхождения, при гломерулонефрите, белковом голодании, первичном альдостеронизме, лихорадке, длительном приеме глюкокортикоидов и др. Гиперосмоляльность плазмы и внеклеточной жидкости возникает у больных сахарным диабетом за счет увеличения в крови глюкозы, у больных с почечной недостаточностью за счет мочевины и других осмотически активных веществ. Оба синдрома (гипо- и гиперосмоляльный) схожи по клиническим проявлениям, так как в обоих случаях ведущими являются неврологические нарушения (рвота, судороги, кома), но тактика врача должна быть разной. Коррекция проводится строго под контролем осмометрии и количества воды в организме.
Компенсаторные изменения в организме при гипергидратации зависят от вызвавшей ее причины. При избыточном поступлении воды в организм увеличивается ОЦК и тормозится выделение АДГ, повышается диурез. Положительный водный баланс развивается только в том случае, если поступление воды превышает выделительную функцию почек, а также на фоне гипернатриемии (включается осморегулирующий рефлекс — увеличивается выделение АДГ). Задержка воды, обусловленная олигурией (анурией) при почечной недостаточности, по понятным причинам не имеет эффективных механизмов компенсации, быстро приводит к развитию гипергидратации, отечных синдромов с угрозой для жизни больного и нуждается в срочной коррекции (например, путем гемодиализа).
При застойной форме сердечной недостаточности, некоторых заболеваниях печени, почек и других состояниях, сопровождающихся уменьшением наполнения артериальных сосудов, ослаблением почечного кровотока, стимуляцией адренергической системы, как правило, активируется РААС, развивается вторичный альдостеронизм, повышается продукция АДГ и, несмотря на уже имеющуюся гипергидратацию, происходит задержка воды в организме (ошибочное регулирование). Вместо компенсации регуляторные системы только усиливают положительный водный баланс и играют важную роль в патогенезе возникающих при этом отеков.
Отек. Виды, патогенез, принципы коррекции
Отек (oedema) — это избыточное накопление жидкости в тканях. Накопление жидкости в полостях называется водянкой (hydrops). Жид
Патофизиология водно-солевого обмена
199
кость может накапливаться в брюшной полости (ascites), плевральной полости (hydrothorax), желудочках мозга (hydrocephalus), околосердечной сумке (hydropericardium). Скопившаяся в тканях или полостях воспалительная жидкость называется экссудатом, а невоспалительная — транссудатом. Он содержит мало белка и клеток.
Механигеские отеки. Если ведущую роль в патогенезе отеков играет увеличение эффективного гидростатического давления (ЭГД) в капиллярах, например, при местной венозной гиперемии, то такие отеки называются механигескими. При этом жидкость задерживается в тканях по причине уменьшения или полного прекращения ее резорбции в капилляры, в которых ЭГД становится равным или даже большим ЭОВС, а также из-за перегрузки лимфооттока (динамическая лимфатическая недостаточность) и сдавления лимфатических сосудов отечной жидкостью (механическая лимфатическая недостаточность). Коррекция этих отеков сводится к устранению причины венозного застоя (рис. 13-4).
Мембраногенные отеки. Отеки, в патогенезе которых главную роль играет повышение проницаемости сосудистой стенки для белка, например: при воспалении, аллергии, укусе насекомых, называются мембраногенными. В этих случаях под влиянием токсинов насекомых или медиаторов воспаления, аллергии (гистамина, серотонина, брадикинина, анафилатоксинов, протеаз и др.) повышается проницаемость капилляров. Белок, вышедший в интерстициальное пространство, повышает ОД вне сосудов, ЭОВС в сосудах уменьшается, и жидкость задерживается в тканях.
Эти отеки являются местными и существенно не влияют на водный баланс в целом организме. Для лечения таких отеков используются противовоспалительные, противоаллергические препараты.
Причины: сдавление или обтурация вен, венозный застой при сердечной недостаточности
Капилляр
Артериальный конец Венозный конец
() ЭГД > ЭОВС ЭГД(Т) = или >ЭОВС )
I т I
? фильтрации воды в интерстиций [ Н20
। * 1 или прекращение резорбции воды из интерстиция
Лимфатическая недостаточность -- ОТЕК
Рис. 13-4. Патогенез механических отеков
200
Лекция 13
Нейрогенные отеки. Повышение проницаемости сосудов играет ведущую роль и в механизме нейрогенных отеков, которые наблюдаются при сирингомиелии (образовании полостей в сером веществе спинного мозга), спинной сухотке (поражении задних столбов и рогов спинного мозга), контузиях, истерии. Развитием отека лица часто сопровождается невралгия тройничного нерва.
Отек Квинке по механизму также является нейрогенным и мембраногенным. Чаще он локализуется на лице, но может возникать и в других местах на коже и во внутренних органах (пищевод, желудок, кишечник, дыхательные пути, матка). Появление его в глотке, в дыхательных путях может вызвать острую асфиксию и привести к смерти. Отек Квинке может быть аллергический и псевдоаллергический (наследственный ангионевротический отек Квинке, обусловленный дефицитом ингибиторов протеаз, в частности ингибитора Clq компонента комлемента). Развитие такого отека может быть спровоцировано действиями стоматолога: надавливание на губу, вмешательства в ротовой полости. Отек развивается за несколько минут, холодный и плотный на ощупь, чаще бледный, держится до нескольких часов и исчезает сам.
Лимфатигеские отеки могут быть местными и распространенными. Их развитие может быть связано с механической лимфатической недостаточностью, обусловленной сдавливанием лимфатических сосудов (рубцами, опухолью), их спазмом (при повышении давления в правом сердце), а также их закупоркой (паразитами или тромбами). В последнем случае развивается задержка тканевой жидкости (лимфостаз) и тяжелый неподдающийся лечению «синдром слоновости» {elephantiasis). Динамическая лимфатическая недостаточность развивается вследствие перегрузки лимфооттока и участвует в патогенезе отеков различного происхождения, например, играет важную роль в механизме развития отека легких, застойных сердечных отеков и др.
При заболеваниях почек, печени, сердечной недостаточности, голодании отеки имеют генерализованный характер, их патогенез сложен и включает многие факторы.
Онкотигеские (гипоальбуминемигеские) отеки. Такие отеки развиваются при голодании (кахектические), печеночной недостаточности из-за снижения синтеза альбуминов, заболеваниях почек, сопровождающихся протеинурией, особенно при нефротическом синдроме, когда потеря белка в сутки превышает Зги может достигать 80 г. Ведущим звеном патогенеза таких отеков является снижение ОД в сосудистом русле. При
Патофизиология водно-солевого обмена
201
| Причины голодание, печеночная недостаточность, выраженная протеинурия и др.
Артериальный конец _____Венозный конец
П ЭГД > ЭОВС ( J,) ЭГД или = или > ЭОВС (1)
ГТ I
Т фильтрации воды в интерстиций [ Н20
i или прекращение резорбции воды из интерстиция Лимфатическая недостаточность --------► ОТЕК
Рис. 13-5. Обмен воды между кровью и тканями при снижении онкотического давления крови
этом уменьшается ЭОВС, увеличивается фильтрация жидкости из капилляров и уменьшается ее резорбция в капилляры (рис.13-5).
Это влечет за собой уменьшение ОЦК, ослабление почечного кровотока, активацию РААС. Развивается вторичный альдостеронизм, повышается секреция АДГ и реабсорбция Na+ и воды в почках. Задержка воды направлена на восстановление ОЦК, но так как при этом ОД и ЭОВС не нормализуются, то жидкость в сосудистом русле не удерживается и снова перемещается в ткань, где нарастает отек (рис. 13-6).
Рис. 13-6. Патогенез онкотических отеков
202
Лекция 13
Сохраняющаяся гиповолемия, несмотря на избыток воды в организме, будет продолжать стимулировать РААС. Развивается порочный круг в патогенезе онкотического отека, который может быть удален путем нормализации ОД и устранения причин гипоальбуминемии. Следует заметить, что по мере развития онкотических отеков присоединяется лимфатическая недостаточность (сдавление лимфатических сосудов отечной жидкостью, перегрузка лимфооттока), которая усугубляет картину. При этом отеки распространяются по всему телу, отекают все органы, развиваются водянки, значительно увеличивается масса тела. Такой генерализованный отек носит название анасарка.
Сердегные, или застойные, отеки. Эти отеки развиваются у больных с сердечной недостаточностью. При левожелудочковой недостаточности может развиваться кардиогенный отек легких, при правожелудочковой недостаточности отекают, как правило, нижние конечности, туловище, может наблюдаться асцит. Патогенез таких отеков, с одной стороны, включает общий и местный венозный застой, увеличение ЭГД, повышение фильтрации воды из капилляров и снижение резорбции воды в капиллярах (см. рис. 13-4), развитие динамической лимфатической недостаточности из-за перегрузки лимфооттока и спазма лимфатических сосудов; с другой стороны — уменьшение МОС (начальное и ведущее звено патогенеза сердечных отеков), ослабление кровенаполнения артериальных сосудов шеи и грудной клетки, уменьшение почечного кровотока, повышение тонуса симпатической нервной системы и активацию РААС, увеличение выделения альдостерона (вторичный альдостеронизм) и АДГ. Уменьшение диуреза приводит к задержке воды и увеличению ОЦК (рис. 13-7).
Но из-за ослабления насосной функции сердца задержка воды лишь усиливает венозный застой, является дополнительной нагрузкой для сердца и может привести к еще большему ослаблению сократительной способности. Как и в случае с онкотическими отеками, возникает порочный круг по механизму ошибочного регулирования.
В патогенез застойных отеков может включиться и мембранный фактор, так как на фоне циркуляторной гипоксии тканей могут появиться биологически активные вещества, повышающие проницаемость сосудов, а также онкотический фактор, так как снижается синтез альбуминов в печени на фоне венозного застоя в большом круге кровообращения.
Коррекция застойных отеков у больных с сердечной недостаточностью должна быть направлена на устранение ведущего звена, т.е. восстановление сократительной способности сердца и нормализацию МОС.
Патофизиология водно-солевого обмена
203
Сердечная недостаточность
Тфильтрации воды в ткань, резорбции ее из ткани
Цмос I.
| X АДГ | 4— Вторичный
альдостеронизм
1 почечного кровотока
? тонуса симпатической НС
1 наполнения артериальных |
сосудов шеи и грудной Активация РААС
I клетки I
—►Венозный застой
Тэгд (+4ЛЭ0ВС) в вен. конце капилляра
Гипоксия тканей
Лимфатическая недостаточность
Ацидоз Т БАВ
? реабсорбции № и Н20 в почках
? проницаемость сосудов для белка
? ОЦК и дополнительная нагрузка на сердце
_? онкотическое давление в интерстиции
X МОС и ? венозного застоя
ОТЕК
Порочный круг
Задержка Н20 в тканях
Рис. 13-7. Патогенез отеков при сердечной недостаточности
Патогенетическая терапия гипергидратации, гиперволемии и отечного синдрома включает также применение диуретиков и блокаторов РААС (ингибиторов АПФ, блокаторов рецепторов ангиотензина-П и др.).
Погегные отеки. В патогенезе отеков при ГН, острой и хронической почечной недостаточности (олигоанурическая стадия) играют роль несколько факторов: уменьшение величины клубочковой фильтрации из-за уменьшения числа функционирующих нефронов, активация РААС вследствие нарушения почечного кровотока со всеми описанными выше последствиями, а также повышение проницаемости сосудов. Особенностью таких почечных отеков является их локализация преимущественно на лице. В тяжелых случаях отеки становятся генерализованными, на фоне анурии у больных могут развиться отек легких и отек мозга.
204
Лекция 13
Патогенез отеков и асцита при циррозе пегени включает повышение гидростатического давления в системе vena porta, а зачастую и в системе нижней полой вены, что приводит к увеличению фильтрации жидкости в ткани (эффективное гидростатическое давление больше эффективной онкотической всасывающей силы); снижение альбуминов в крови (цирроз сопровождается печеночной недостаточностью), а значит, уменьшение эффективной онкотической силы всасывания воды; вторичный альдостеронизм, обусловленный уменьшением ОЦК и нарушением инактивации альдостерона в печени.
Для лечения гипергидратаций с отечным синдромом при застойной сердечной недостаточности, заболеваниях почек и печени наряду с диуретиками с успехом применяются препараты, блокирующие РААС: 0-адреноблокаторы, тормозящие секрецию ренина; спиронолактоны, блокирующие рецепторы альдостерона в почках; но наибольшей популярностью пользуются ингибиторы АПФ или блокаторы рецепторов ангиотензина-П.
Лекция 14
ПАТОФИЗИОЛОГИЯ КИСЛОТНООСНОВНОГО состояния
Г.П. Щелкунова
Нарушения КОС (синонимы — кислотно-основной баланс или кислотно-щелочное равновесие) часто осложняют течение основного заболевания, являясь следствием изменений газового состава крови, метаболических расстройств, которые возникают, например, при недостаточности дыхания, кровообращения, печени, почек, при эндокринных заболеваниях, патологии ЖКТ, системы крови и др. При сдвигах КОС нарушается гомеостаз, изменяются функции органов и систем, нарушаются ферментативные процессы, часто снижается или даже извращается эффект проводимой терапии. Изменения КОС неизбежно ведут к нарушению физиологических процессов. Тяжелые формы нарушений КОС могут стать непосредственной причиной смерти больного.
В сутки в организме образуется 10 000-20 000 ммоль летучей угольной кислоты (при физических нагрузках в 20 раз больше) и около 1 ммоль/кг массы тела нелетучих кислот (молочной, пировиноградной, ацетоуксусной, р-оксимасляной, хлористоводородной, фосфорной и др.). Несмотря на огромное количество выделяющихся при этом во внеклеточную жидкость Н\ концентрация их в крови поддерживается около 40±5 нМ. Для поддержания концентрации ионов водорода в таком узком диапазоне в организме существуют специальные системы — это буферы и физиологические системы, главным образом легкие и почки. Важную роль играют также печень, поджелудочная железа и желудочно-кишечный тракт.
Роль буферных систем в регуляции кислотно-основного состояния
Буфер — это любая система, которая стремится противостоять изменению pH после добавления либо кислоты, либо щелочи. Буферные системы организма играют первостепенную роль в поддержании относительно узкого диапазона pH, в котором протекают физиологические
206
Лекция 14
клеточные и внеклеточные процессы. Главной буферной системой является система СО2-бикарбонат, которая действует во внеклеточной и внутриклеточной жидкостях организма. Кроме того, внутриклеточные белки, НЬ, белки плазмы и составные элементы костей (например, карбонаты и коллаген) также играют роль буферов.
Главные буферные системы крови: гидрокарбонатная (NaHCOyH^O,), гемоглобиновая (HHb/HbO2), протеиновая (Pt-COONa/Pt-COOH) и фосфатная (Na2HPO4/NaH2PO4). Буферные системы внеклеточной жидкости: гидрокарбонатная и фосфатная, а внутриклеточной жидкости — протеиновая, фосфатная и гидрокарбонатная. Буферные системы мочи — аммонийная (NH3/NH/) и фосфатная.
Большой запас буферов, которые участвуют в нейтрализации нагрузок кислотами или основаниями, представлен солями угольной кислоты в костях. При острой нагрузке кислотой кость может обеспечить до 40 % буферной емкости, а при хронических нагрузках и больше (например, при хронической почечной недостаточности). Нейтрализация ионов водорода костным карбонатом сопровождается высвобождением кальция из кости во внеклеточную жидкость.
Анион бикарбоната присутствует в большинстве жидкостей организма и является его главным щелочным резервом. Бикарбонат реагирует с ионом водорода, образуя угольную кислоту, которая существует в равновесии с СО2:
(Н- + нсо3- «—> Н2СО3 <—> СО2 + Н2О).
Превращение Н2СО3 в Н2О и СО2 катализируется ферментом карбоангидразой. По мере того как в ходе метаболизма нарабатываются Н , бикарбонатный буфер превращает их в Н2СО3 и, наконец, в СО2 и Н2О. Углекислый газ выводится через легкие. И, наоборот, когда СО2 образуется в процессе клеточного метаболизма, угольная кислота диссоциирует на Н* и НСО3~.
Водородный показатель pH (отрицательный десятичный логарифм концентрации протонов -lg[H+]) системы, в которой протекают эти реакции, рассчитывается на основе уравнения Гендерсона-Хассельбаха. Оно выводится из уравнения для константы диссоциации (Ка) угольной кислоты:
Ка= [Н‘]х[НСОЗ'] + [Н2СО3].
Или в логарифмической форме:
lgKa = lg[H ] + lg([HCO3 ] - [Н2СО3]).
Патофизиология кислотно-основного состояния
207
Поскольку pH — это отрицательный логарифм [HJ, уравнение можно записать как:
pH = рКа + lg([HCO(] + 0,03 X рСО2).
Величина рКа для буферной системы СО2 — бикарбонат равна 6,1. Концентрация бикарбоната в плазме артериальной крови здорового человека — 24 ммоль/л, а нормальное РаСО2 — 40 мм рт.ст Следовательно, нормальное pH артериальной крови равно 7,4 (pH = 6,1. lg24*0,03x40).
Величина рКадля буферов-белков = 7,4 (Н белок <—> Н*+белок_). Величина рКа для буфера-фосфата — 6,8 (Н2РО4 <—> Н +НРО/2). Белки и фосфаты являются главными внутриклеточными буферами.
В принципе, наибольшей эффективностью в качестве внеклеточного буфера должны обладать белки, так как среднее значение их рКа близко к значению pH плазмы крови (7,4). В действительности основным буфером организма является бикарбонат, несмотря на то что его рКа более чем на одну единицу ниже, чем pH плазмы. Это объясняется тем, что концентрация бикарбоната в плазме очень высока (24 ммоль/л) и бикарбонатная система — не закрытая система. СО2, образующийся в результате диссоциации угольной кислоты, быстро удаляется при дыхании. Иными словами, СО2 находится в динамическом равновесии с Н\
Концентрацию Н2СО3, Н* и НСО3 можно подставить в уравнение, описывающее главную буферную систему в плазме крови: pH = pK+lg[HCO3->[H2CO3].
Концентрация СО2, растворенного в крови в 800 раз и более выше, чем концентрация угольной кислоты (Н2СО3). Поэтому Н' в норме непосредственно связан с парциальным давлением СО2 при следующей форме уравнения: pH = pK+lg[HCO3-]-HxpCO2, где а — константа растворимости СО2в воде (0,03). Это уравнение можно переписать как простое уравнение действующих масс: [Н‘] = 24 (рСО2)/[НСО3 ]. В таком виде уравнение удобно для применения, поскольку концентрацию бикарбоната и напряжение СО2 измеряют в обычной лабораторной практике.
Если концентрация бикарбоната снижается, а РаСО2 остается постоянным, то снижается и pH. Напротив, если концентрация бикарбоната растет, а РаСО2 остается без изменения, то pH повышается. Если же РаСО2 изменяется так, что отношение бикарбоната и угольной кислоты остается на уровне 20:1, то pH сохранит значение 7,4 (рис. 14-1).
В норме оно равно 20/1, что соответствует нормальному pH. Увеличение или уменьшение соотношения приведет к отклонению стрелки в сторону алкалоза или ацидоза.
208
Лекция 14
Рис. 14-1. Изображение соотношения основание/кислота уравнения Гендер-сона-Хассельбаха в форме весов
В различных жидкостях организма концентрация ионов водорода различная, и поэтому pH желудочного сока колеблется от 1 до 3, pH мочи — 5-6, панкреатического сока — 8, pH артериальной крови в норме колеблется от 7,35 до 7,45.
В медицинской практике для оценки гомеостаза организма принято оценивать концентрацию Н* артериальной крови. Увелигение pH более 7,45 свидетельствует о защелагивании (алкалемии), уменьшение pH менее 7,35 — о закислении (ацидемии).
При существенных сдвигах pH в ту или иную сторону нарушаются функции клеток, и прежде всего многочисленных ферментных систем, изменяются направленность и интенсивность окислительно-восстановительных процессов, способность НЬ связывать и отдавать кислород, изменяются все виды обмена веществ и в первую очередь водного и электролитного, нарушаются чувствительность клеточных рецепторов, проницаемость мембран, возбудимость, проводимость и другие процессы. При значительных сдвигах pH клетки гибнут.
Следует помнить, что уменьшение pH крови менее 6,8 и увеличение более 7,7 несовместимы с жизнью!
Патофизиология кислотно-основного состояния
209
Физиологические механизмы регуляции кислотно-основного состояния
Дыхательные механизмы. Вдыхаемый воздух содержит незначительное количество СО2. Следовательно, почти вся углекислота крови является продуктом клеточного метаболизма. Способность СО2 к диффузии в 20 раз выше чем у кислорода. По мере образования в процессе клеточного метаболизма СО2 легко диффундирует в капилляры и транспортируется к легким в трех основных формах: 1) в виде растворенной СО2: 2) в виде аниона бикарбоната и 3) в виде карбаминовых соединений (рис. 14-2).
СО2 очень хорошо растворяется в плазме. Количество растворенной в плазме СО2 определяется произведением ее парциального давления и коэффициента растворимости (а = 0,3 мл/л крови/мм рт.ст.). Около 5 % общей двуокиси углерода в артериальной крови находится в форме растворенного газа. Анион бикарбоната преобладает (90 %) в артериальной крови. Он является продуктом реакции СО2 с водой с образованием
Периферические ткани
Рис. 14-2. СО2 транспорт в крови, иллюстрирующий образование НСО3‘ и карбаминовых соединений, хлоридный сдвиг и связывание Н*
210
Лекция 14
Н2СО3 и ее диссоциации на водород и ион бикарбоната: СО2 + Н2О <—> Н2СО3 <—> Н + НСО3. Реакция между СО2 и Н2О протекает медленно в плазме и очень быстро в эритроцитах, где присутствует внутриклеточный фермент карбоангидраза. Она облегчает реакцию между СО2 и Н2О с образованием Н2СО3; вторая фаза уравнения протекает очень быстро без катализатора.
По мере накопления НСО3 внутри эритроцита анион диффундирует через клеточную мембрану в плазму. Мембрана эритроцита относительно непроницаема для Н‘, как и вообще для катионов, поэтому ионы водорода остаются внутри клетки. Электрическая нейтральность клетки в процессе диффузии НСО3‘ в плазму обеспечивается притоком ионов хлора из плазмы в эритроцит, что формирует так называемый хлорид-ный сдвиг.
Часть Н\ остающихся в эритроцитах, забуферивается, соединяясь с НЬ. В периферических тканях, где концентрация СО2 высока и значительное количество Н накапливается эритроцитами, связывание Н* облегчается деоксигенацией НЬ. Восстановленный НЬ лучше связывается с протонами, чем оксигенированный. Таким образом, деоксигенация артериальной крови в периферических тканях способствует связыванию Н+ посредством образования восстановленного НЬ. Это увеличение связывания СО2 с НЬ известно как эффект Холдейна.
При поглощении О2 и высвобождении СО2 в легочных капиллярах реакции, представленные на схеме, протекают в обратном направлении.
Третьей формой транспорта СО2 являются карбаминовые соединения, образованные в реакции СО2 с концевыми аминогруппами белков крови. Основным белком крови, связывающим СО2, является НЬ. Этот процесс описывается следующей реакцией:
НЬ - NH2 + СО2 <—>НЬ - NH СООН <—> НЬ - NHCOO- + Н‘.
Реакция СО2 с аминогруппами протекает быстро. Как и в случае более легкого связывания СО2 с восстановленным НЬ, образование карбаминовых соединений легче протекает с восстановленной формой НЬ. Карбаминовые соединения составляют около 5 % общего количества СО2, транспортируемого артериальной кровью.
С точки зрения сравнительного вклада каждой из этих форм в артериовенозную разницу по концентрации СО2 около 60 % принадлежит НСО3 , карбаминовым соединениям — около 30 % и растворенной СО2 — около 10 %. Присутствие в крови всех трех форм СО2 создает равновесие между растворенным СО2 и СО2, химически связанным с другими веществами.
Патофизиология кислотно-основного состояния
211
Высокая растворимость и способность СО2 к диффузии в воде делают СО2 особенно удобным средством удаления кислоты из тканей в кровь. К способности эритроцитов переносить СО2 добавляются два феномена: 1) карбоангидраза в эритроцитах превращает значительную часть СО2 в НСО3_, который быстро переносится из эритроцитов в плазму крови в обмен на ионы Ch 2) СО2 образует карбаминовое соединение с НЬ, причем в большей степени с восстановленным НЬ, чем с оксигемоглобином. Это облегчает связывание углекислого газа в периферических тканях, где уровень О2 низкий. В легких же оба процесса идут в противоположных направлениях, и СО2быстро выводится из организма (см. рис. 14-2).
Регуляция выделения СО2 достигается изменением скорости и объема легочной вентиляции (т.е. зависит от величины минутной альвеолярной вентиляции — МАВ). Повышение МАВ приводит к снижению артериального рСО2; уменьшение МАВ приводит к увеличению артериального рСО2. Афферентные сигналы, изменяющие минутную альвеолярную вентиляцию, связаны с хеморецепторами, которые регулируют дыхательный центр. Эти рецепторы находятся в продолговатом мозгу, аортальном и каротидном тельцах и реагируют на изменения рСО2 и Н’.
Легкие — это первая линия защиты в поддержании КОС, поскольку они обеспегивают механизм погти немедленной регуляции выделения кислоты. С другой стороны, любые нарушения дыхания, сопровождающиеся увелигением или уменьшением минутной альвеолярной вентиляции, могут стать пригиной развития нарушений КОС.
Почечные механизмы. Хотя количество нелетучих кислот, образующихся в процессе метаболизма белков и других веществ, гораздо меньше, чем летучих, почки выделяют от 50 до 100 ммоль нелетучих кислот в сутки. Их выделение происходит в проксимальных канальцах и собирательных трубках почек, где секретируются протоны, а в качестве буферных систем участвуют фосфаты и сульфаты (т.е. титруемые кислоты) и аммиак. Однако, до того как может произойти экскреция всех кислот, почки должны реабсорбировать НСО3_, профильтровавшийся в клубочках.
Угольная кислота образуется в клетке из воды и СО2 под действием карбоангидразы, Н* активно переносятся через люминальную мембрану Ыа /Н*-обменником. Затем НСО3_ транспортируется через базолатеральную мембрану. Секретируемый Н+ быстро соединяется с фильтруемым НСО3‘, образуя угольную кислоту (Н2СО3). Угольная кислота превращается в воду и углекислый газ с помощью карбоангидразы (КА) на люминальной стороне щеточной каемки проксимального ка-
212
Лекция 14
Рис. 14-3. Реабсорбция бикарбоната в клетках проксимального канальца (Шейман Д.А., 1997): КА — карбоангидраза
нальца. СО2 диффундирует обратно в клетку проксимального канальца, где соединяется с Н2О и образует угольную кислоту, завершая тем самым этот цикл (рис. 14-3).
Некарбоновые кислоты секретируются вставочными клетками собирательных трубок коры и наружного мозгового слоя почек. Секрецию Н’ в просвет канальцев осуществляет Н*-АТФаза, тогда как в реабсорбции НСО3~ через базолатеральную мембрану участвует обменник С1-/ НСО3- (рис. 14-4).
Главным фактором, от которого зависит количество выделяемых кислот, является присутствие буферов в моче. Максимальный pH жидкости в просвете собирательной трубки — 4,0 (Н+ = 0,1 ммоль/л). Поэтому только 0,1-0,2 % суточной нагрузки кислот (50-100 ммоль) могут быть выведены в форме незабуференных ионов Н\ Остальная часть Н‘ в моче должна быть выведена в форме буферов, обычно таких, как фосфаты и аммоний. Концентрация аммония регулируется преимущественно
Базолатеральная
Просвет
Рис. 14-4. Секреция Н+ вставочными а-клетками собирательной трубки (Шейман Д.А., 1997): АДФ — аденозиндифосфат; АТФ — аденозинтрифосфат
Патофизиология кислотно-основного состояния
213
почками и колеблется в зависимости от КОС (рис. 14-5). Объем суточной секреции кислот в наибольшей степени зависит от количества выделяемого аммония.
NH/ образуется и секретируется клетками проксимального канальца, а затем реабсорбируется в восходящем отделе петли Генле и концентрируется в мозговом слое почки. Небольшое количество NH4’ диссоциирует на NH3 и Н\ последний реабсорбируется. NH3 может диффундировать в собирательную трубку, где служит буфером для ионов Н*. секретируемых вставочными клетками.
Факторами, регулирующими транспорт Н' и НСО3’ в проксимальных канальцах почек, являются рСО2, фильтруемая нагрузка НСО3~, карбоангидраза, паратиреоидный гормон, концентрация К* и
Рис. 14-5. Транспорт NH, и NH; в почке
4
НРО4-2 в сыворотке. В собирательных трубках регуляция транспорта катионов и бикарбоната обеспечивается градиентом pH, разностью
электрических потенциалов (электрохимический градиент), рСО2, экскрецией NH4 и альдостероном. Альдостерон способствует секреции Н посредством двух механизмов: 1) стимулирует реабсорбцию Na* и повышает отрицательный заряд просвета, что облегчает секрецию Н+; 2) непосредственно стимулирует Н’-АТФазу.
Соотношение между фильтруемым количеством бикарбоната и выделением кислот почками представлено в табл. 14-1.
Таблица 14-1. Соотношение между фильтруемым количеством бикарбоната и выделением кислот почками
Кислота или основание ммоль/сут
Фильтруемый НСО/ 4150
Канальцевая реабсорбция НСОЧ~ 4145
Выделение НСО,- с мочой 5
Выделение с мочой титруемых кислот 55
Выделение аммония с мочой 30
Общая экскреция кислот 80
Таким образом, угасшие погек в регуляции КОС сводится к удалению из крови сульфатов и фосфатов, секреции Н' эпителием канальцев и
214
Лекция 14
удалении их в виде NH4Cl (аммониогенез) и в виде NaH2PO4 (ацидогенез), а также к реабсорбции бикарбоната для восстановления щелогного резерва.
Реакция почек на изменение КОС организма более медленная, чем соответствующая реакция легких. Стимуляция канальцевой секреции Н‘ из-за изменений рСО2 начинается через несколько минут. Однако стимуляция секреции Н* в дистальных канальцах альдостероном протекает в течение нескольких часов. Реализация эффектов других факторов, влияющих на выведение Н* почками, может потребовать 2-3-х дней. Кроме того, происходит компенсаторная адаптация почек к ацидозу или алкалозу, и эти изменения носят длительный характер.
Печень, в которой синтезируются все альбумины, служит поставщиком протеинового буфера крови: синтезируя мочевину, она использует ее для нейтрализации кислот в гепатоцитах, а поддерживая нормальный уровень аммиака в крови, предотвращает защелачивание внеклеточной жидкости. Печень способствует снижению в крови и тканевой жидкости органических кислот, вовлекая их в синтез глюкозы (использование в глюконеогенезе лактата, пирувата, аминокислот). В печени 80 % лактата превращается в СО2 и Н2О, а 20 % в глюкозу. Этот процесс сопровождается регенерацией истраченного главного буферного основания — гидрокарбоната. В печени осуществляются синтез кетоновых тел и инактивация стероидных гормонов, в том числе альдостерона. С желчью из печени в кишечник выделяется ряд кислых и основных продуктов.
Желудок — главный поставщик соляной кислоты, а поджелудочная железа и кишечник — бикарбоната. В кишечнике происходит всасывание воды и электролитов.
Взаимосвязь кислотно-основного состояния с обменом воды и электролитов
Законы электронейтральности и изоосмоляльности
КОС тесно связано с обменом электролитов и воды в организме. Их объединяет два физико-химических закона: закон электронейтральности и закон изоосмоляльности.
Согласно закону электронейтральности, во всех жидкостных средах организма сумма отрицательных зарядов всех анионов должна быть равна сумме всех положительных зарядов катионов. Из ионограммы ГЬмбла (рис. 14-6) видно, что в плазме крови содержится 154 мэкв/л
Патофизиология кислотно-основного состояния
215
Рис. 14-6. Ионограмма Гэмбла. Зависимость концентрации ионов НСО,- от количества остаточных анионов (ОА)
катионов и столько же анионов. Самые подвижные анионы — НСО3‘, которые в большом количестве образуются в организме и легко выводятся или задерживаются почками.
Если количество НСО3~ в плазме крови по какой-либо причине уменьшается (ацидоз), то для сохранения электронейтральности должно увеличиться содержание ионов С1‘ и/или остаточных анионов (сульфатов, фосфатов, лактата и других анионов органических кислот) и уменьшиться количество катионов. При увеличении содержания в плазме анионов бикарбоната (алкалоз), наоборот, количество ионов СГ должно уменьшиться, а количество катионов пропорционально увеличиться. Содержание неизмеряемых остаточных анионов никогда не уменьшается, оно может только увеличиваться.
Так как сдвиги КОС, как правило, сопровождаются изменениями в ту или другую сторону количества бикарбоната, то при этом всегда меняется содержание в плазме и клетках ионов хлора, натрия, калия, магния, кальция и др. Анионы НСО3~ — главное связующее звено между электролитным и КОС.
По закону изоосмоляльности во всех жидкостных системах организма. между которыми свободно обменивается вода, устанавливается одинаковое осмотическое давление. В норме осмоляльность плазмы, межклеточной и внутриклеточной жидкости составляет 285 мОсм/кг воды. Если в каком-то месте количество осмотически активных веществ
216
Лекция 14
нарастает (например, натрия, глюкозы), то туда переходит вода, пока не установится новое равновесие. Поскольку при нарушениях КОС внутри и вне клеток изменяется содержание электролитов, то изменяется в них и содержание воды. Возможно развитие как гипо-, так и гиперосмо-ляльных синдромов. С другой стороны, первичные нарушения водного и электролитного балансов могут повлечь за собой изменения КОС.
Основные показатели (компоненты) КОС
1. Актуальный pH (Actual pH) или активная реакция крови (в норме 7,35-7,45).
2. рСО2 (Actual рСО2) — напряжение СО2 — дыхательный компонент КОС характеризует функциональное состояние дыхательной системы (в норме рСО2 артериальной крови — 35-45 мм рт.ст. (4,7-6,0 кПа), рСО2 в венозной крови — около 46 мм рт.ст. (6,1 кПа).
3. ВВ (Buffer Base) — буферные основания крови (сумма оснований всех буферных систем крови — гидрокарбонатной, белковой и гемоглобиновой), характеризует метаболигеский компонент КОС (у здоровых лиц — 48 ммоль/л, допустимые колебания — 42-52 ммоль/л).
4. BE (Base Excess) — сдвиг буферных оснований, главный критерий метаболигеского компонента КОС, характеризует изменение содержания буферных оснований по отношению к должным величинам для данной крови. В норме BE = ±2,0 ммоль/л. Отрицательное значение BE указывает на дефицит оснований или избыток кислот, положительное значение BE — на избыток оснований или дефицит кислот.
5. SB (Standart Bicarbonat) — стандартный бикарбонат, т.е. концентрация бикарбоната у данного человека, приведенная к стандартным условиям (температура тела 37 °C, рСО2 — 40 мм рт.ст. и полное насыщение крови кислородом). У здоровых лиц SB равен 25-28 ммоль/л.
6. АВ — истинный бикарбонат крови или содержание НСО3 в крови у конкретного человека (в норме 24 ммоль/л, допустимые колебания — 19-25 ммоль/л).
7. ТСО2 (Total С02) — общее содержание СО2 в крови во всех буферах и физически растворенного (в норме 19-25 ммоль/л).
Зная значения pH и рСО2 в крови у больного, с помощью специальных номограмм или карт Зиггаарда-Андерсена можно определить остальные показатели КОС (ВВ, BE, SB, АВ, ТСО2), установить форму нарушения КОС и степень его компенсации (рис. 14-7,14-8).
В обычной врачебной практике для оценки КОС у больного достаточно определять pH артериальной крови, характеризующий кон-
Патофизиология кислотно-основного состояния
217
Рис. 14-7. Виды нарушения кислотно-основного состояния крови и их компенсация: 1 — некомпенсированный респираторный ацидоз; 2 — метаболическая компенсация респираторного ацидоза; 3 — метаболический некомпенсированный ацидоз; 4 — дыхательная компенсация метаболического ацидоза; 5 — некомпенсированный дыхательный алкалоз; 6 — метаболическая компенсация дыхательного алкалоза; 7 — некомпенсированный метаболический алкалоз; 8 — дыхательная компенсация метаболического алкалоза
центрацию протонов во внеклеточной жидкости; содержание в крови бикарбонатного аниона (НСО3_); напряжение углекислого газа в артериальной крови (РаСО2), а также концентрацию в ней К+, Na+ и Ch Можно также определить у больного pH и РаСО2 и, пользуясь уравнением Гендерсона-Хассельбаха, рассчитать концентрацию в крови гидрокарбоната.
218
Лекция 14
о-------1-----1----1-----1----1-----1-----1----1-----1-----
10 20 30 40 50 60 70 80 90 100
Рис. 14-8. Номограмма, которая позволяет по двум значениям — pH и РаСО2 определить у больного содержание бикарбоната (главного щелочного резерва крови) и установить вид нарушения КОС
Весьма полезным иногда бывает определение так называемого анионного пробела плазмы (АПП) (англ. — «anion gap») или анионной разницы (АР). Ее определяют по разнице между количеством измеренных катионов Na* и суммой измеренных анионов (С1_ + НСО3").
В основе внедрения показателя АР в клиническую практику лежит предположение, что для создания электрически нейтральной среды количество отрицательно заряженных анионов и положительно заряженных катионов в плазме крови должно быть одинаковым (закон электронейтральности). Если это так, то концентрацию неизмеренных анионов и катионов можно определить, используя данные о содержании хлоридов, бикарбоната и натрия в плазме крови (они исследуются повседневной лабораторной техникой). Разница между неизмеренным колигеством анионов и катионов и будет АР. Нормальное значение АР составляет 12 мэкв/л (табл. 14-2).
Патофизиология кислотно-основного состояния
219
Таблица 14-2. Анионная разница плазмы крови
Концентрация неизмеренных анионов (НА), мэкв/л Концентрация неизмеренных катионов (НК), мэкв/л
Белки 15 РО4 2 SO4 1 Органические кислоты 5 К+ 4,5 Са2 5 Mg2* 1,5
Всего 23 Всего 11
АР = НА - НК = 12 мэкв/л НА + (Cl- + НСО3) = Na* + НК НА - НК = Na* - (Cl +НСО,)
Наибольшую часть неизмеренного пула анионов в плазме крови составляют белки, поэтому даже небольшое уменьшение концентрации альбуминов может понизить АР (уменьшение в плазме альбуминов на 50 % снижает АР на 5-6 мэкв/л). К другим причинам, приводящим к сдвигу АР, относят изменение содержания парапротеинов (аномальные белки плазмы), имеющих суммарный положительный заряд; повышение количества неизмеренных катионов (К\ Mg2* и Са2*); снижение уровня натрия в плазме крови, а также увеличение в крови органических кислот (лактата, кетокислот и др.), приводящее к увеличению АР.
Основные формы нарушений кислотно-основного состояния
Из уравнения pH = pK+IgBHCOMIHCO3 видно, что нарушения реакции крови (pH) могут наступить при первичных изменениях метаболического компонента pH, т.е. в содержании бикарбонатов (ВНСО3), либо в сдвигах респираторного компонента pH, т.е. в содержании угольной кислоты (ННСО3). Первые приводят к метаболическим нарушениям КОС, а вторые — к респираторным.
Первигное снижение содержания бикарбоната менее 22 ммоль/л вызывает метаболигеский (негазовый) ацидоз, а его увелигение более 25 ммоль/л — метаболигеский (негазовый) алкалоз.
Первигное снижение напряженияугольнойкислотыменее 35 ммрт.ст., например при гипервентиляции легких, повышает pH и вызывает респираторный (газовый) алкалоз, первигное увелигение напряжения угольной кислоты более 44 мм рт.ст. является пригиной развития респираторного (газового) ацидоза. Отношение рСО2/НСО3_ указывает на то, что содержание Н* в плазме крови прямо пропорционально уровню рСО2 и обратно пропорционально концентрации НСО3_. Из приведенного уравнения ясно также, что величина pH зависит не от абсолютных значений,
220
Лекция 14
а от отношения буферной пары (содержания бикарбоната и угольной кислоты). Поэтому величина pH может сохраниться нормальной даже в тех случаях, когда патологические процессы приведут к изменениям абсолютных концентраций бикарбоната, если, конечно, содержание угольной кислоты претерпит компенсаторные изменения. Таким образом, первичные дыхательные нарушения могут быть компенсированы вторичными изменениями метаболического характера, а первичные метаболические нарушения — изменениями дыхания (табл. 14-3).
Таблица 14-3. Классификация нарушений кислотно-основного состояния
Первичное нарушение Компенсаторная реакция
Респираторный ацидоз (увеличение рСО2) Респираторный алкалоз (снижение рСО2) Метаболический ацидоз (снижение содержания НСО3) Метаболический алкалоз (увеличение содержания НСО3) Увеличение содержания НСО3 (метаболический алкалоз) Снижение содержания НСО3_ (метаболический ацидоз) Снижение рСО2 (респираторный алкалоз) Увеличение рСО2 (респираторный ацидоз)
При респираторном ацидозе снижение pH, вызванное задержкой СО2 в крови, может частично или полностью компенсироваться увеличением содержания бикарбонатов. Снижение pH при метаболическом ацидозе, вызванное первичным снижением содержания бикарбонатов, может быть компенсировано уменьшением концентрации угольной кислоты путем гипервентиляции легких.
Важно подчеркнуть, что компенсаторные механизмы приводят только к ограничению сдвигов pH плазмы крови, но не предотвращают полностью их развития.
Нередко в клинической практике встречаются однонаправленные изменения КОС: смешанный ацидоз (увеличение рСО2 + снижение содержания НСО3 ) или смешанный алкалоз (снижение рСО2 + увеличение содержания НСО3~).
Ацидоз респираторный
Наиболее частыми причинами респираторного ацидоза являются угнетение дыхательного центра при травмах, воспалении, отеке головного мозга, передозировке наркотических препаратов; повреждение, слабость дыхательной мускулатуры; уменьшение дыхательной поверхности легких (пневмония, ателектаз, опухоли, эмфизема, пневмоторакс и др.); нарушение проходимости дыхательных путей (аспирация ино
Патофизиология кислотно-основного состояния
221
родных тел, астматический статус, спазм или отек голосовых связок и т.п.), т.е. все случаи некомпенсированной дыхательной недостаточности. Респираторный ацидоз является одним из ведущих звеньев патогенеза асфиксии новорожденных, респираторного дистресс-синдрома. Во всех этих случаях в результате гиповентиляции легких или значительного несоответствия вентиляции и перфузии возникает гиперкапния (РаСО2 больше 45 мм рт.ст.).
При некомпенсированном дыхательном ацидозе увеличение напряжения СО2 в крови приводит к снижению pH. Отсутствие метаболической компенсации выражается в том, что щелочные резервы крови остаются неизменными (содержание НСО3_ не увеличивается, показатели BE, ВВ, АВ, SB — в пределах нормы), или, несмотря на некоторое увеличение НСО3~ за счет диссоциации Н2СО3 на Н+ и НСО3 , отношение [НСО3~] к (ах РаСО2) тем не менее падает, что и снижает pH.
Если же респираторный ацидоз компенсируется метаболическими сдвигами, а именно увеличением в крови бикарбоната (BE — больше +2,0 ммоль/л, ВВ, АВ, SB — выше нормы), то при этом, несмотря на избыток в крови СО2, pH или приближается к норме (субкомпенсирован-ный ацидоз), или остается нормальным (компенсированный ацидоз). В этом случае говорят о том, что респираторный ацидоз компенсируется метаболическим алкалозом (разнонаправленные сдвиги, см. рис. 14-7).
Компенсаторные изменения направлены на связывание или выведение избытка Н+-ионов и освобождение или задержку в организме анионов НСО3‘. Сначала в этот процесс включаются буферные системы, затем физиологические. Основные изменения концентрации НСО3' обусловлены буферным действием НЬ и тканевых буферов. Хлорид обменивается на внутриклеточный НСО3_ (хлоридный сдвиг). При остром дыхательном ацидозе концентрация НСО3‘ увеличивается на 0,1 ммоль/л на каждый 1 мм рт.ст. повышения величины артериального рСО2. Если дыхательный ацидоз носит хронический характер, то в канальцах почек активнее секретируются ионы водорода, с мочой больше выделяется титруемых кислот и аммония хлорида (реакция мочи кислая), с другой стороны — увеличивается реабсорбция бикарбоната и натрия. При длительном течении периода компенсации НСО3~ возрастает на 0,3 ммоль/л на каждый 1 мм рт.ст. увеличения рСО2 в артериальной крови. Увеличение концентрации бикарбоната в плазме приводит к соответствующему снижению концентрации ионов С1‘ (для сохранения электронейтральности), которые усиленно выводятся с мочой и перемещаются в эритроциты (хлоридный сдвиг). Белки, связывая ионы во-
222
Лекция 14
дорода, освобождают катионы натрия и калия. Костная ткань, усиленно связывая Н+-ионы, освобождает в кровь ионы Са2*. Увеличение концентрации ионов натрия в плазме обусловлено также выделением их из клеток, из костной ткани (превращение двуосновного фосфата в одноосновной) и усиленной реабсорбции в канальцах почек. Гипернатриемия влечет за собой повышение осмоляльности плазмы крови (если нет сопутствующей гипергидратации) и выход воды из клеток (эритроциты сморщиваются). Полностью заменить функцию легких метаболическая компенсация не может, она эффективна только при умеренных нарушениях вентиляции, хронической гиперкапнии.
Если компенсаторные реакции при дыхательном ацидозе недостаточны, то развиваются различные патологические явления:
• гиперкапния приводит к расширению сосудов головного мозга, усилению образования ликвора, повышению внутричерепного давления (гиперкапнигеская энцефалопатия);
• спазм периферических артериол сопровождается повышением артериального давления, уменьшением фильтрации мочи в почечных клубочках, повышением давления в малом кругу кровообращения, нарушением микроциркуляции;
• возможно повышение температуры тела из-за уменьшения теплоотдачи;
• сердечная деятельность сначала усиливается от прямого действия СО2 на миокард, а затем ухудшается как в результате перегрузки «правого» сердца высоким давлением в легочной артерии, так и в результате повреждения миокардиоцитов (ишемия, дисбаланс натрия и калия); МОС уменьшается, частота сердечных сокращений увеличивается; при тяжелом ацидозе возникает аритмия с тенденцией к брадикардии;
• артериальное давление резко падает вследствие сердечной декомпенсации;
• происходит спазм гладкой мускулатуры бронхиол и гиперсекреция слизи, что ухудшает бронхиальную проходимость и усугубляет дыхательную недостаточность;
• уменьшается сродство НЬ к кислороду, кривая диссоциации НЬ смещается вправо, что в какой-то степени компенсирует гипоксию, так как облегчается диссоциация оксигемоглобина в тканях;
• повышается активность симпатоадреналовой системы, в крови увеличивается содержание катехоламинов, усиливающих спазм периферических сосудов.
Патофизиология кислотно-основного состояния
223
В результате недостаточности дыхания, вторичных нарушений центрального и периферического кровообращения развивается гипоксия смешанного типа (дыхательная и циркуляторная), которая сопровождается накоплением недоокисленных продуктов, т.е. избыточным образованием водородных ионов, а это означает, что метаболический алкалоз, компенсировавший вначале гиперкапнию, сменяется метаболическим ацидозом. Развиваются однонаправленные сдвиги КОС — респираторный и метаболический ацидоз.
При выраженном ацидозе возникают характерные клинические проявления: одышка, адинамия, головная боль, анорексия; бледность кожи и слизистых оболочек, акроцианоз. Кожа холодная и влажная, при тяжелом ацидозе становится «мраморной», резко выражен симптом «белого пятна» (результат сочетания резкого спазма артериол и паралича капилляров и вен); олигурия, повышение температуры тела; сначала повышение, а затем падение артериального давления; беспокойство, раздражительность, галлюцинации, переходящие в психоз; возбуждение, сменяющиеся в стадии декомпенсации арефлексией, потерей сознания.
Принципы коррекции: единственным мероприятием при дыхательном ацидозе является восстановление функции дыхания (устранение причины, искусственная вентиляция, бронходилататоры, отхаркивающие и т.п.). Дополнительно применяются оксигенотерапия и другие симптоматические средства.
Ацидоз метаболический
Это самая тяжелая и наиболее часто встречающаяся форма нарушения КОС. Причинами метаболического ацидоза могут быть гипоксия (особенно циркуляторная), сахарный диабет, голодание, лихорадка, почечная недостаточность, длительный понос, обширные воспалительные процессы (перитонит), отравления, передозировка кальция хлорида, аммония хлорида и др.
При некомпенсированном ацидозе pH крови снижается в результате имеющегося дефицита НСО3~ менее 22 ммоль/л (BE меньше 2,0 ммоль/л, АВ, ВВ, SB — ниже нормы), а рСО2 — остается нормальным.
Если же метаболический ацидоз компенсируется гипервентиляцией легких и усиленным выведением из организма СО2, то напряжение СО2 в крови снижается менее 35 мм рт.ст. (гипокапния), а pH приближается к норме, несмотря на сохраняющийся дефицит бикарбонатов. В этом случае говорят о том, что метаболический ацидоз компенсируется дыхательным алкалозом (см. рис. 14-7). При этом возникает очень характер
224
Лекция 14
ный симптом — глубокое, шумное дыхание Куссмауля, обусловленное чрезмерным раздражением дыхательного центра ионами водорода.
К компенсации ацидоза подключаются почки и печень. Активируется ацидо- и аммониогенез в почечных канальцах (если метаболический ацидоз не является следствием почечной недостаточности), а с другой стороны, усиливается реабсорбция бикарбоната в почках и выделение его печенью. При низкой концентрации в крови НСО3’ из эритроцитов в плазму поступают ионы С? и выводятся с мочой в виде аммония хлорида. Если процесс аммониогенеза в почках не нарушен, то концентрация ионов С1_ в плазме остается нормальной, а для сохранения электронейтральности пропорционально увеличивается содержание остаточных анионов.
Концентрация ионов К+ в плазме, как правило, увеличивается вследствие вытеснения их из клеток ионами водорода. (Следует помнить, что при диабетической коме под влиянием инсулинотерапии гиперкалиемия довольно быстро сменяется гипокалиемией, так как К* будет активно перемещаться в клетки.) Белки, связывая Н*-ионы, высвобождают в плазму ионы калия и натрия. Повышение осмотического давления плазмы за счет гипернатриемии способствует перемещению воды из клеток и развитию ги-перосмоляльного синдрома. Следует, однако, оговориться, что концентрация в крови и общее количество в организме ионов натрия, калия, кальция, магния, хлора и других в конечном итоге зависят от характера основного заболевания, количества внеклеточной воды в организме (гипер- или гиповолемия), наличия у больного неукротимой рвоты или профузного поноса, сопровождающихся потерей электролитов, функционального состояния почек, печени и других органов. Поэтому у каждого конкретного больного врач держит под постоянным контролем содержание в крови вышеназванных ионов и коррекцию проводит строго индивидуально.
При метаболигеском ацидозе возникают следующие патологические изменения:
• сосуды, как правило, расширяются при умеренном снижении pH и сужаются при выраженном ацидозе;
• при уменьшении сосудистого тонуса артериальное и венозное давление снижается, уменьшается венозный возврат крови к сердцу, в результате чего уменьшаются ударный и минутный объемы сердца. Если же преобладает спазм периферических сосудов, возникают изменения, описанные в разделе «Респираторный ацидоз»-,
• изменяется чувствительность миокардиоцитов к ионам кальция и к адреналину, что сопровождается снижением сократительной способности миокарда;
Патофизиология кислотно-основного состояния
225
• гиперкалиемия более 5,2 ммоль/л влечет за собой нарушение нервно-мышечной возбудимости и проводимости и обусловливает такие симптомы, как повышение тонуса поперечно-полосатой мускулатуры, рвоту, понос, психические расстройства, нарушение чувствительности, брадиаритмию, экстрасистолию. При повышении концентрации К+ в плазме выше 7,5 ммоль/л возможно развитие мерцания желудочков сердца и остановка его в диастоле, а также паралич скелетной мускулатуры;
• усиливается агрегация тромбоцитов, возникающие микротромбы еще больше нарушают микроциркуляцию, усугубляя гипоксию, нарушения метаболизма и ацидоз;
• в результате нарушения деятельности сердца, периферического кровообращения вторично нарушается функция почек, печени, центральной нервной системы. В тяжелых случаях развивается кома, может наступить остановка дыхания, блокирование многих важнейших ферментов;
• при чрезмерном снижении pH (менее 6,8) разрушаются лизосомы, и клетки подвергаются аутолизу под влиянием лизосомальных ферментов.
Клинические проявления метаболического ацидоза мало чем отличаются от симптомов дыхательного ацидоза, поэтому для правильной оценки сдвига КОС следует ориентироваться на объективные показатели и патогенез основного заболевания, вызвавшего нарушения.
В последнее время все случаи метаболического ацидоза принято условно разделять на две группы по величине анионной разницы (АР).
Высокие знагения АР — больше 12 мэкв/л указывают на ацидоз, вызванный повышением содержания органических кислот (например, молочной, кетокислот). К метаболическому ацидозу с высокой анионной разницей относятся: лактат-ацидоз, кетоацидоз, ацидоз при погегной недостатогности и отравлении салицилатами, метанолом, этиленгликолем, ацидоз при усиленном всасывании органигеских кислот из просвета кишегника. В случае отдачи в плазму крови Н*-ионов в количестве 1 мэкв/л связанными кислотами (например, молочной кислотой), содержание бикарбоната в ней снижается на 1 мэкв/л, а АР будет соответственно возрастать на аналогичную величину.
Нормальная велигина АР свидетельствует об ацидозе, возникшем в результате истощения бикарбонатного буфера из-за усиленного выведения НСО3’ из организма (например, при диарее).
226
Лекция 14
Виды метаболического ацидоза без увеличения АР: ацидоз при диарее, потегной недостатогности средней тяжести', при введении жидкостей, содержащих большое колигество хлоридов; при компенсации респираторного алкалоза; погегный канальцевый ацидоз. При потере бикарбоната с мочой или калом компенсаторное повышение концентрации хлоридов в плазме крови поддерживает баланс анионов, и АР не изменяется.
Мологнокислый ацидоз {лактат-ацидоз). Молочная кислота, конечный продукт анаэробного гликолиза и гликогенолиза, вырабатывается в организме в количестве 1 мэкв на килограмм в 1 час в норме при мышечном покое. Нормальный уровень лактата в сыворотке крови составляет 2 мэкв/л и менее, но при больших физических нагрузках содержание молочной кислоты в крови может достигать 4 мэкв/л. Большая часть лактата метаболизируется печенью (субстрат глюконеогенеза) и поглощается сердечной мышцей, где используется как энергетический материал. Печень способна переработать в 10 раз больше лактата, чем его образуется в норме. Пригинами развития лактат-ацидоза могут быть кардиогенный шок, сепсис, полиорганная недостаточность, кислородное голодание, введение адреналина или натрия нитропруссида, инфаркт кишечника, большие судорожные припадки, дефицит тиамина, D-лактат-ацидоз. С меньшей вероятностью лактат-ацидоз может развиться при гипоксемии, анемии и заболеваниях печени.
Гиперлактацидемия при шоке (особенно кардиогенном и септическом) возникает из-за интенсивного образования молочной кислоты и снижения способности печени к ее переработке, т.е. способности превращать лактат в глюкозу и гликоген. Накопление в крови молочной кислоты при любом виде шока считается плохим прогностическим признаком. Лактат-ацидоз при заболеваниях печени развивается, как правило, только при нарушениях печеночного кровообращения.
Дефицит тиамина {витамина BJ приводит к блокированию превращения пирувата в ацетилированную форму кофермента (ацетил-КоА) и направляет метаболизм пировиноградной кислоты по пути образования лактата.
В мышцах человека и животных найдена L-молочная кислота (левовращающий изомер), а ее правовращающий изомер образуется в результате действия ферментов микроорганизмов, расщепляющих глюкозу в кишечнике. D-молочную кислоту могут вырабатывать несколько видов бактерий (например, Bacteroides fragilis, Escherichia coli и др.). D-лактат-ацидоз встречается у больных с обширной резекцией тонкой кишки, кишечными анастомозами, а также у чрезмерно тучных лиц и
Патофизиология кислотно-основного состояния
227
лиц с дисбактериозом. Это нарушение более распространено, чем кажется на первый взгляд.
Адреналин ускоряет распад гликогена в скелетных мышцах и усиливает выработку лактата. Кроме того, из-за констрикции мелких артерий и артериол нарушается периферическое кровообращение и усиливается анаэробный гликолиз в тканях. Натрия нитропруссид быстро метаболизируется, вызывая высвобождение цианидов, способных нарушать процессы окислительного фосфорилирования (они ингибируют клеточное дыхание, оказывая токсическое действие на цитохромоксидазу). Способность цианидов вызывать лактат-ацидоз выявлена относительно недавно.
Лактат-ацидоз можно заподозрить при любой форме метаболигеского ацидоза, сопровождающейся повышением АР (особенно более 30 мэкв/л).
При лечении лактат-ацидоза необходимо не только проводить коррекцию нарушений КОС, но и улучшать доставку кислорода тканям, воздействуя на сердечно-сосудистую и дыхательную систему. Для поддержания pH артериальной крови у больных выше 7,2 можно использовать натрия гидрокарбонат. Однако применение натрия бикарбоната при лактат-ацидозе бывает неэффективным, так как вызывает ряд побочных эффектов, включая гиперосмоляльность плазмы, артериальную гипотензию, снижение сердечного выброса и увеличение лактата в крови. Гипотензия и снижение сердечного выброса могут быть результатом связывания ионов кальция анионами бикарбоната. Неэффективность натрия бикарбоната во многих случаях побудила к разработке новых препаратов (карбикарб, натрия дихлорацетат и др.). Больные сердечными заболеваниями по-разному реагируют на «кислотные» нагрузки, что требует индивидуального подхода к каждому больному.
Кетоацидоз. Кетоновые тела — группа органических соединений, являющихся промежуточными продуктами обмена жиров, белков и углеводов. Кетогенез происходит главным образом в печени (конденсация под действием тиолазы двух молекул ацетил-КоА, образованного при Р-окислении высших жирных кислот или окислительном декарбоксилировании пирувата). Кетоновые тела используются организмом в качестве источника энергии при ограниченном питании (их энергетическая ценность выше, чем у углеводов). Основные кетоновые тела — это ацетон, ацетоуксусная (АУК) и р-оксимасляная (БОМК) кислоты. При диабетическом кетоацидозе в крови больных увеличивается содержание всех кетоновых тел, при алкогольном — главным образом БОМК, что стандартными методами определения кетоновых тел не выявляется.
228
Лекция 14
В отличие от высокой АР при лактат-ацидозе (часто более 30 мэкв/л), АР при кетоацидозе повышена незнагительно (15-20 мэкв/л) или даже остается нормальной. АР значительно ниже у больных с нормальной функцией почек, так как кетоновые тела выводятся с мочой, а хлориды реабсорбируются в почечных канальцах для поддержания электронейтральности среды. Типичными проявлениями диабетического кетоацидоза являются высокая гипергликемия, гиперкетонемия и кетонурия, увеличение АР, гипогидратация (дефицит жидкости может достигать 10 % массы тела). Однако встречаются атипичные случаи диабета: кетоацидоз при уровне сахара в крови менее 19,4 ммоль/л, кетоацидоз с нормальной АР, сдвиг pH в щелочную сторону.
Легение диабетигеского кетоацидоза включает:
• устранение дефицита жидкости с помощью изотонического раствора натрия хлорида или 5 % раствора альбумина (последний предпочтительнее, так как лучше удерживает воду в сосудах, способствует быстрому восстановлению ОЦК, препятствует развитию отека мозга или легких):
• введение инсулина:
• восстановление нормального содержания калия в крови, который у больных может быть как повышенным, так и пониженным (иногда и нормальным).
Следует заметить, что больные диабетическим кетоацидозом часто переносят падение pH ниже 7,2 без всяких серьезных последствий (даже при pH ниже 7,0 не развивается коллапс). Натрия гидрокарбонат в данном слугае неэффективен и даже может усилить образование кетоновых тел. Конечной целью лечебных мероприятий является нормализация содержания НСО3_ в сыворотке крови.
Алкогольный кетоацидоз (в отличие от вызванного этиловым спиртом лактат-ацидоза на фоне неумеренного пьянства) обычно развивается через 1-3 дня после чрезмерного потребления спиртных напитков на фоне небольшой концентрации этанола в крови. Его развитию способствуют частичное голодание, сопровождающееся усилением кетогенеза, и усиленное образование НАДН в печени в процессе окисления этанола до ацетальдегида (НАД Н превращает ацетат в БОМК). Из-за того, что в крови у таких больных увеличивается БОМК (а не АУК), диагностика алкогольного кетоацидоза затруднена. Величина анионной разницы при этом значительно варьирует. Этот вид нарушения КОС обычно поддается коррекции в течение 24 ч путем внутривенного вливания 5 % раствора глюкозы, приготовленного на изотоническом растворе натрия
Патофизиология кислотно-основного состояния
229
хлорида. Необходимость введения натрия гидрокарбоната обычно отсутствует.
Гиперхлоремигеский ацидоз {ацидоз без роста АР). Многие из причин гиперхлоремического ацидоза очевидны: например, инфузия кислого раствора, содержащего ионы С1" (соляной кислоты, хлорида аммония, хлорида аргинина): потеря бикарбоната ЖКТ (тяжелая диарея, свищ тонкой кишки, фистула поджелудочной железы); значительное снижение бикарбоната в крови при компенсации хронического дыхательного алкалоза; внезапное увеличение объема внеклеточной жидкости; уменьшение секреции кислот и реабсорбции НСО3~ почками на фоне гипоальдостеронизма или при почечном канальцевом ацидозе.
При гиперхлоремигеском ацидозе накапливается НС1, и каждое снижение концентрации НСО~ уравновешивается повышением концентрации СР, поэтому разность анионов остается нормальной (в отличие от ацидоза с ростом АР, когда вместо НС1 накапливается другая кислота — лактат, кетокислота, сульфат и НСО3~ замещается иным анионом (А-), который обычными способами не измеряется). У большинства больных с таким ацидозом развивается гиперкалиемия.
Коррекция гиперхлоремического ацидоза осуществляется введением натрия гидрокарбоната или трисамина. Трисамин связывает большое количество Н*-ионов и выводит их с мочой (его применяют только при нормальной функции почек), а с другой стороны, он способствует увеличению НСО3_ в крови. Проводят также мероприятия, направленные на нормализацию гемодинамики и газообмена, улучшение микроциркуляции и обменных процессов в организме, коррекцию электролитного дисбаланса, а главное — на устранение причины, вызвавшей сдвиг КОС.
Алкалоз респираторный
Респираторный алкалоз возникает вследствие избыточного выведения СО2 из организма при гипервентиляции легких.
Наиболее частые причины его развития — неправильно проводимая искусственная вентиляция легких, невротическая одышка, поражение ЦНС (травма, энцефалит, инсульт, опухоль и др.), анемия, горная и высотная болезнь, перевозбуждение дыхательного центра после применения лекарственных препаратов (центральных аналептиков, катехоламинов, эстрогенов и др.), сепсис, тиреотоксикоз, перитонит и др.
Для некомпенсированного дыхательного алкалоза характерны снижение рСО2менее 35 мм рт.ст. (гипокапния) и увеличение pH, содержание
230
Лекция 14
НСО3_ и другие метаболические компоненты КОС (BE, ВВ и SB) — в пределах нормы.
Метаболическая компенсация респираторного алкалоза сводится к уменьшению щелочных резервов крови (BE смещается в сторону дефицита оснований, ВВ, АВ, SB — меньше нормы) и накоплению Н -ионов, что нормализует pH на фоне дефицита углекислоты. Респираторный алкалоз компенсируется метаболическим ацидозом (см. рис. 14-7). При этом в почках уменьшается секреция водородных ионов в канальцах и усиливается выведение НСО3‘ (реакция мочи щелочная). В среднем НСО3 плазмы падает на 0,4 ммоль/л при снижении рСО2 на каждый 1 мм рт.ст. Уменьшение в плазме анионов НСО3_ вызывает выход ионов хлора из эритроцитов (хлоридный сдвиг) и гиперхлоремию. Ионы водорода высвобождаются из клеток в обмен на ионы калия, из белков и костной ткани в обмен на ионы натрия и кальция. Развиваются гипокалиемия, гипонатриемия, гипокальциемия. Осмоляльность плазмы уменьшается, вода перемещается в клетки (гипоосмоляльный синдром).
Если при ацидозе (кроме метаболического ацидоза с ростом АР) всегда существует угроза развития гиперкалиемии, то при алкалозе — гипокалиемии. Установлена закономерность: если рСО2 в крови снижается на 10 мм рт.ст., то концентрация калия уменьшается на 0,5 ммоль/л: если pH крови увеличивается на 0,1 ед., то концентрация ионов калия в плазме уменьшается на 0,4 ммоль/л.
В патогенезе респираторного алкалоза можно выделить характерные патологические изменения:
• при значительном снижении СО2 в крови церебральные сосуды суживаются (развивается гипоксия мозга), а сосуды системного кровообращения расширяются, что влечет за собой уменьшение артериального и венозного давления, депонирование крови в расширенных сосудах и уменьшение ОЦК, уменьшение венозного возврата крови к сердцу, уменьшение ударного и минутного объема сердца, снижение кровотока в тканях, что постепенно ведет к развитию гипоксии и метаболическому ацидозу:
• кривая диссоциации НЬ смещается влево (эффект Бора), сродство НЬ к кислороду повышается, и оксигенация тканей затрудняется на фоне увеличения потребности клеток в кислороде; из тканей плохо выводится СО2;
• нарушается углеводный обмен, усиливается образование лактата и кетоновых тел; при снижении рСО2 менее 20 мм рт.ст. происходят торможение гликолиза и необратимые повреждения клеток;
Патофизиология кислотно-основного состояния
231
• гипокалиемия менее 3,8 ммоль/л обусловливает развитие таких симптомов, как адинамия, мышечная слабость (доходящая до параличей), парез кишечника, судороги, нарушения сердечного ритма; при уменьшении концентрации калия менее 2,0 ммоль/л возможна остановка сердца в систоле в результате гиперполяризации мембран, остановка дыхания; дефицит ионов калия в крови приводит к повышению нервно-мышечной возбудимости, головокружению, пароксизмальной тахикардии, выраженным тоническим судорогам.
Принципы коррекции: восстановление нормального дыхания, вдыхание карбогена, противосудорожная терапия, нормализация водноэлектролитного баланса.
Алкалоз метаболический
Метаболический алкалоз характеризуется повышением НСО/ более 25 ммоль/л и снижением Н’ и СР во внеклеточной жидкости. В основе этих нарушений лежит или потеря Н+, или нагрузка экзогенным НСО/.
Метаболигеский алкалоз развивается при потерях нелетучих кислот вследствие частой или неукротимой рвоты (при кишечной непроходимости, пищевой токсикоинфекции и т.п.), частых промываниях желудка, непрерывном зондировании желудка; выраженной первичной гипокалиемии (стероидная терапия, альдостеронизм, печеночная недостаточность, длительный прием мочегонных препаратов, инсулинотерапия). Мочегонные средства могут вызвать алкалоз, опосредованный потерей электролитов и жидкости (хлориды выводятся с мочой, пропорционально выделению натрия, для сохранения электронейтральности ионы хлора замещаются анионами бикарбоната, возросшая реабсорбция бикарбоната поддерживает алкалоз). Истощение запасов калия на фоне усиленной реабсорбции натрия поддерживает алкалоз путем стимулирования секреции Н* в дистальных канальцах. Магний под влиянием диуретиков тоже выводится с мочой, и его дефицит способствует потере катионов калия и развитию алкалоза. Причиной метаболического алкалоза могут быть введение в организм избыточного количества натрия гидрокарбоната (при лечении метаболического ацидоза с высокой АР), массивные гемотрансфузии, а также молочно-щелочной синдром. Метаболический алкалоз может быть вызван уменьшением объема внеклеточной жидкости. Во-первых, потеря свободной воды приводит к концентрированию бикарбоната и повышению НСО3~ в сыворотке крови. Во-вторых, уменьшение ОЦК стимулирует высвобождение альдо
232
Лекция 14
стерона, который усиливает секрецию К* и Н* в дистальных почечных канальцах, т.е. выведение их с мочой.
Компенсация метаболигеского алкалоза дыхательным ацидозом, как правило, не может быть длительной, так как в ответ на задержку СО2 тут же развивается гипервентиляция, и напряжение углекислоты нормализуется.
Показатели некомпенсированного метаболического алкалоза: BE — больше +2,0 ммоль/л (избыток оснований); НСО3_, ВВ, SB — выше нормы, pH — более 7,45, рСО2 — в пределах нормы (см. рис. 14-7).
Компенсаторные изменения направлены на выведение и связывание избытка оснований и задержку и освобождение от связей ионов водорода. Возникает ионный дисбаланс, аналогичный таковому при респираторном алкалозе: в плазме крови снижается концентрация К+, Са2+, С1\ Содержание натрия в плазме сначала уменьшается, возникает гипоос-моляльный синдром с гиповолемией. Затем под влиянием альдостерона в канальцах почек усиливается реабсорбция натрия и потеря с мочой калия. Если при этом чувствительность канальцевого эпителия к АДГ не уменьшается, что бывает при выраженной гипокалиемии, то повышается реабсорбция воды. На фоне выраженной гипокалиемии ионы водорода усиленно выводятся с мочой и перемещаются в клетки вместе с ионами натрия, что может привести к сложным нарушениям КОС — внутриклеточному ацидозу и отеку на фоне внеклеточного алкалоза. Гйпохлоремия может сопровождаться азотемией, так как уменьшается выделение с мочой аммония хлорида. Истощение запасов хлоридов ограничивает экскрецию бикарбоната почками путем усиления реабсорбции и угнетения его секреции в почечных канальцах, при этом алкалоз как бы поддерживает сам себя.
Принципы коррекции: возмещение дефицита ионов водорода, удаление избытка бикарбоната; коррекция гипохлоремии, гипокалиемии; применение антагонистов альдостерона; симптоматическая терапия. Причины метаболического алкалоза в результате потери Н+ могут быть дифференцированы в зависимости от того, сопровождается ли алкалоз истощением запасов СГ во внеклеточном пространстве или вызывается первичной гипохлоремией.
Хлоридзависимый метаболигеский алкалоз (концентрация в моче С1' <10 ммоль/л) сочетается с гиповолемией (на фоне гипогидратации) и развивается после приема диуретиков, в период восстановления после дыхательного ацидоза, кетоацидоза или лактатацидоза. Он корректируется после введения больному NaCl или КС1. Хлористоводо
Патофизиология кислотно-основного состояния
233
родную кислоту (НС1) обычно вводят лишь в тяжелых случаях алкалоза (pH >7,5). когда восполнение потерь жидкостей и ионов К не дает эффекта.
При алкалозе, не связанном с первичной гипохлоремией (концентрация в моче С1*>20 ммоль/л), пациент не реагирует на введение хлоридов.
Хлориднезависимый алкалоз сочетается с гиперволемией (на фоне гипергидратации). Он наблюдается при первичном альдостеронизме, чрезмерной активации РААС, синдроме Кушинга и стероидной терапии. Хороший лечебный эффект при этом дают восполнение дефицита калия и применение антагонистов альдостерона, например спиронолактонов, блокирующих рецепторы альдостерона в почках, или блокаторов АПФ (см. водно-солевой обмен).
В редких случаях для коррекции метаболического алкалоза (при достаточном объеме внеклеточной жидкости в организме) используют ингибитор карбоангидразы — диакарб, который подавляет реабсорбцию бикарбоната в проксимальных канальцах почек. Однако на фоне его применения трудно устранить потерю хлоридов и дефицит калия.
Смешанные нарушения кислотно-основного состояния
Смешанный метаболигеский ацидоз и дыхательный ацидоз (сочетание дефицита бикарбоната и гиперкапнии) могут снижать pH до очень низкого и опасного уровня. Это наблюдается у больных с остановкой сердца и дыхания, при сочетании недостаточности дыхания и кровообращения.
Смешанный метаболигеский алкалоз и дыхательный алкалоз (сочетание избытка бикарбоната и гипокапнии) часто наблюдаются при токсикозах первой половины беременности у женщин, страдающих тошнотой и частой рвотой.
Смешанный метаболигеский алкалоз и дыхательный ацидоз (более выраженный дефицит бикарбоната, чем того требует компенсация дыхательного ацидоза) могут развиться у больного с гиповентиляцией легких (например, при обструктивном заболевании) при назначении ему кортикостероидов или диуретиков, вызывающих хлоридзависимый метаболический алкалоз. В этих условиях задержка СО2 может стать более выраженной для компенсации метаболического алкалоза.
Смешанный метаболигеский ацидоз и дыхательный алкалоз наблюдаются довольно часто у больных в критических состояниях, и это указывает на плохой прогноз. Такая комбинация может наблюдаться при
234
Лекция 14
печеночной недостаточности, а также когда к первичному дыхательному алкалозу присоединяется метаболический ацидоз в результате сепсиса и лактат-ацидоза или алкогольного кетоацидоза.
Смешанный метаболигеский ацидоз и метаболигеский алкалоз явно противоположны друг другу, и их диагностика может быть трудной. Такая ситуация может возникнуть у больных с инсулинзависимым сахарным диабетом при развитии кетоацидотической комы, которой предшествовала многократная рвота. Рвота вызывает хлоридзависимый алкалоз с уменьшением объема внеклеточной жидкости, а гиперкето-немия вызывает ацидоз с высоким дефицитом анионов. При таком сочетании могут наблюдаться относительно нормальные величины pH, и диагностика нарушения КОС требует особой квалификации врача. Еще труднее диагностировать нарушение КОС у больных с тяжелым гастроэнтеритом, который сопровождается рвотой и поносом, что вызывает соответственно алкалоз и ацидоз с нормальным значением анионной разницы.
Может встречаться и тройной вариант нарушения КОС — согетание метаболигеского ацидоза, метаболигеского алкалоза и нарушений дыхания.
Приложение
Пользуясь предложенной на рисунке 14-8 номограммой, можно определить вид нарушения КОС у данного больного, зная только два показателя: pH и РаСО2, а также с помощью буферной линии оценить другие показатели: ВВ, BE и SB.
Предположим, у больного А. pH = 7,25 (ацидоз) и РаСО2 = 70 мм рт.ст. (гиперкапния). Перпендикуляры, восстановленные из точек, соответствующих этим значениям, пересекаются в точке А. Буферная линия «а» пересекает показатели ВВ, SB и BE в значениях нормы, что свидетельствует об отсутствии метаболической компенсации респираторного (газового) ацидоза — колигество оснований не увелигилось. (Все точки, попадающие на отрезок 1, указывают на наличие гиперкапнии и респираторного некомпенсированного ацидоза.)
У больного Б. pH = 7,4 (норма), а РаСО2 = 70 мм рт.ст. (гиперкапния). Перпендикуляры пересекаются в точке Б, а буферная линия «б» пересекает показатели ВВ, SB и BE в значениях, указывающих на наличие у больного избытка оснований. Это означает, что, несмотря на выраженную гиперкапнию, благодаря метаболической компенсации (колигество оснований увелигилось) pH остается нормальным. (Все точки, попадаю
Патофизиология кислотно-основного состояния
235
щие на отрезок 2, указывают на наличие частично или полностью компенсированного респираторного ацидоза.)
У больного С. pH = 7,25 (ацидоз), РаСО2 = 40 мм рт.ст. (норма). Перпендикуляры пересекаются в точке С, а буферная линия «с» пересекает показатели ВВ, SB и BE в значениях дефицита оснований. Это говорит о том, что у больного из-за дефицита оснований развился метаболический ацидоз без дыхательной компенсации. (Все точки, попадающие на отрезок 3, указывают на наличие некомпенсированного метаболигеского ацидоза.) Если метаболический ацидоз будет компенсироваться гипервентиляцией легких и РаСО2 начнет снижаться (отрезок 4), то pH может сохраняться нормальным, несмотря на имеющийся дефицит оснований. Это компенсированный метаболигеский ацидоз.
Аналогичным способом можно рассмотреть случаи с алкалозом респираторным (отрезки 5 и 6) и алкалозом метаболическим (отрезки 7 и 8).
Лекция 15
ПАТОЛОГИЯ ОПУХОЛЕВОГО РОСТА. КАНЦЕРОГЕНЕЗ
Л.И. Зелигенко, О.Д. Мишнев
Рост и созревание клеток являются нормальными явлениями для развивающегося организма во время эмбриогенеза, в периоды его становления после рождения, а также во время физиологической регенерации. Нарушение же регуляции клеточного обновления может привести к патологии клеточного роста и формированию в организме опухоли.
Опухоль или новообразование — это избыточная ненормальная масса ткани, которая возникает вследствие чрезмерного неконтролируемого клеточного роста, сохраняющегося даже после прекращения влияния факторов, вызвавших этот рост.
Злокагественная опухоль — это патологическое разрастание атипичных, неполноценно дифференцированных клеток, обладающих автономным прогрессирующим необратимым ростом, инфильтрирующим и разрушающим прилегающие ткани.
Терминология и классификация опухолей.
Сравнительная характеристика доброкачественных и злокачественных опухолей
Широко распространена клинико-анатомическая классификация органоспецифических опухолей, которой пользуются клиницисты. В соответствии с этой классификацией различают разнообразные опухоли различных органов (табл. 15-1).
По степени зрелости опухолевых клеток, характеру роста, клиническому течению и степени вреда для организма различают доброкачественные и злокачественные опухоли. Как правило, опухоли из зрелых тканей являются доброкачественными, однако иногда и зрелые опухоли могут привести к гибели организма, если их хирургическое удаление не произведено вовремя. Такую опасность могут представлять внутричерепные локализации опухолей.
Патология опухолевого роста. Канцерогенез
237
Таблица 15-1. Сравнительная характеристика доброкачественных и злокачественных опухолей
Доброкачественные опухоли Злокачественные опухоли
Темпы роста Медленный рост Быстрый рост
Характер роста по отношению к прилегающим тканям Экспансивный рост Инфильтрирующий (инвазивный рост)
Вид атипизма Тканевой атипизм Клеточный и тканевой атипизм
Степень зрелости Зрелые, хорошо дифференцированные клетки Незрелые клетки, имеющие различную степень дифференцировки клеток (анаплазии (недостаточной клеточной дифференцировки)
Некрозы опухолевой ткани Встречаются редко (в крупных и длительно существующих опухолях) Характерный признак, более выраженный в продвинутых стадиях
Метастазы Как правило, не метастазируют Лимфогенные, гематогенные, периневральные, имплантационные метастазы
Рецидивы после удаления После полного хирургического удаления, как правило, не рецидивируют После полного хирургического удаления нередко рецидивируют
Прогноз Обычно благоприятный Нередко (часто) неблагоприятный
Опухоли принято классифицировать и обозначать в соответствии с той тканью, из которой они берут свое происхождение (гистогенетиге-ская классификация). Так, злокачественные опухоли из эпителиальной ткани называют раками или карциномами. Злокачественные опухоли из соединительной, мышечной, костной, жировой ткани, а также из кровеносных и лимфатических сосудов называют саркомами. Злокачественные опухоли из кроветворных клеток — лейкозами, а из лимфоидных — лимфомами. Около 90 % всех опухолей человека составляют карциномы или рак. Термин «канцер» (рак) может быть использован только в случае определения опухолей из эпителиальных тканей.
Свойства злокачественных опухолей
Наиболее важное свойство злокачественных опухолей — их способность к опухолевой прогрессии. Учение об опухолевой прогрессии было разработано Фулдсом в 60-х годах XX века. Основное его положение может быть сформулировано следующим образом: по мере своей про
238
Лекция 15
грессии (развития) опухоль приобретает ряд новых свойств, которые необратимо за ней закрепляются и придают ей новые все более злокачественные черты.
Важнейшими проявлениями опухолевой прогрессии являются:
• морфологический или клеточный атипизм;
• биохимический атипизм:
• инвазия тканей и метастазирование;
• автономность роста.
Клетогный атипизм. Для злокачественных опухолей свойствены тканевой и клеточный атипизм. Общей чертой новообразований или неоплазий является неоднородность составляющих ее клеток (плеоморфизм). Важнейшей отличительной особенностью злокачественного роста является нарушение дифференцировки клеток, из которых она произошла, однако степень утраты клеточной дифференцировки может быть различной. В соответствии с этим различают высоко-, умеренно- и низкодифференцированные опухоли, а также абсолютно недифференцированные опухоли (анаплазированные опухоли).
Анаплазия — основная черта опухолевой ткани, в ее основе лежит замена специализированных клеток с высоким уровнем дифференцировки мало- и недифференцированными клетками. Считается, что с уменьшением степени дифференцировки опухоли возрастает угроза для жизни больного. В то же время такие опухоли, обладающие клетками с высоким митотическим потенциалом, как правило, более чувствительны к лучевой терапии и химиотерапии.
Признаки клеточного атипизма включают изменение ядра, цитоплазмы, ультраструктур и клеточных мембран.
Ядро опухолевой клетки имеет больший размер по сравнению с ядром нормальной клетки, и ядерно-цитоплазматическое соотношение «сдвинуто в пользу ядра», вместо 1:4 или даже 1:6 оно может составить 1:1. Для опухолевых клеток характерны активное ядро и «ленивая» цитоплазма. Число митозов увеличено, и нередко в опухолевых клетках обнаруживают разнообразные хромосомные аберрации. Некоторые из них достаточно специфичны для определенных типов злокачественных опухолей. Их цитогенетические характеристики отражают наследственные механизмы возникновения и в настоящее время используются для диагностики (табл. 15-2).
Цитоплазма опухолевых клеток часто характеризуется базофилией, свидетельствующей об активном синтезе клетками белка в условиях их активной пролиферации. Часто обнаруживается неупорядоченное
Патология опухолевого роста. Канцерогенез
239
Таблица 15-2. Цитогенетические особенности некоторых опухолей
Тип хромосомой аберрации 1 Опухолевое заболевание
Реципрокные транслокации:
t(8:14) Лимфома Беркетта
t(9;22) Хронический миелолейкоз
Делеция 13-й хромосомы Ретинобластома
Делеция 11-й хромосомы Опухоль почки Вильмса
расположение микротрубочек и микрофиламентов, а также появление промежуточных микрофиламентов, таких как десмин в опухолях из соединительной и мышечной ткани, кератин в опухолях из эпителия, виментин — в лимфомах, саркомах и некоторых карциномах, белки нейрофиламентов в опухолях симпатической нервной системы и глиальные филаменты в глиомах ЦНС.
К аномалиям ультраструктур можно отнести появление круглых, гигантских и уродливых митохондрий, свидетельствующих о нарушении энергообразования в клетках опухолей. Дело в том, что в опухолях не тормозится эффект Пастера, сутью которого является прекращение анаэробного гликолиза в присутствии кислорода.
Поверхность опухолевых клеток характеризуется повышенной склад-частью, появлением микровыростов, пузырьков и микроворсинок. Последние увеличивают площадь клеточной поверхности, что предполагает активное взаимодействие опухолевой клетки с окружающей тканью — «перекройке ее под себя». Изменение рецепторного аппарата опухолевых клеток является ключевой проблемой злокачественного роста.
Биохимический атипизм
Биохимический атипизм опухоли проявляется прежде всего в нарушении ею синтеза многих белков, в частности МНС I класса, что затрудняет распознавание опухоли клетками иммунной системы. Другой вариант проявления клеточного атипизма — синтез опухолью веществ, в норме ею не синтезируемых.
Эти вещества могут быть использованы в качестве маркеров при диагностике опухолей. Такими маркерами могут служить некоторые гормоны и а-фетопротеин, гликопротеид, синтезируемый в норме желточным мешком и печенью плода, но не свойственный тканям взрослого человека.
Паранеопластитеский синдром. Некоторые опухоли, не обладая явным эндокринным происхождением, способны синтезировать и продуцировать гормоны. К таким гормонам относятся АКТ, АДГ, инсулин.
240
Лекция 15
паратгормон и некоторые другие. Эктопическая выработка гормона в неподходящих тканях ведет к возникновению так называемого паранео-пластического синдрома. Название этого синдрома говорит о том, что он не связан напрямую с опухолевым ростом (нарушением пролиферации и дифференцировки клеток), а относится к его осложнениям в связи с дерепрессией определенных генов (принимающих участие в синтезе гормонов) из-за нарушений в клеточном геноме. Паранеопластический синдром чаще возникает при мелкоклеточном раке легких и достаточно часто при карциномах молочной железы и толстой кишки.
К паранеопластическим синдромам относят такие аутоиммунные болезни, как дерматомиозит, миастения гравис и синдром Итона-Ламберта.
Различные факторы, продуцируемые опухолевыми клетками, могут, провоцировать возникновение как анемии, так и эритроцитоза, тромбоза, тромбоэндокардита, нефротического и других форм паране-опластических синдромов. Этот синдром встречается при опухолях приблизительно в 10 % случаев. Он может оказаться первым проявлением еще недиагностируемой опухоли, а иногда даже стать причиной гибели больного. С другой стороны, паранеопластический синдром способен замаскировать симптомы опухоли и помешать как ее ранней диагностике, так и своевременному лечению (чаще всего ее удалению).
Инвазия тканей и метастазирование
Инвазия тканей и метастазирование — кардинальные признаки злокачественной опухоли. Они являются основной причиной гибели больных раком.
Способность злокачественной опухоли к метастазированию основана на:
• инфильтрирующем росте с инвазией в кровеносные и лимфатические сосуды:
• слабой связи между опухолевыми клетками:
• на благоприятной ситуации для их приживления в тканях.
Каскад метастазирования включает в себя четыре стадии:
1. Отделение опухолевых клеток от основного опухолевого узла и внедрение в экстрацеллюлярный матрикс (ЭЦМ). Инвазия ткани.
2. Проникновение опухолевых клеток в просвет сосудов (пермеация) и агрегация с другими метастазирующими клетками. Их распространение по лимфатическим и кровеносным путям (раковая эмболия).
3. Фиксация опухолевых клеток на интиме сосудов капиллярного типа с образованием тромба.
Патология опухолевого роста. Канцерогенез
241
4. Переход из кровеносных и лимфатических сосудов в прилегающую ткань. Приживление и размножение клеток (экстраваскулярная пролиферация), индукция ангиогенеза. Формирование метастаза (вторичного опухолевого узла).
Отделение клеток от основного опухолевого узла связано с отсутствием феномена контактного торможения, который хорошо изучен при их росте в культуре ткани. В культуре ткани здоровые клетки растут в форме монослоя и прекращают рост при заполнении монослоя. Опухолевые же клетки выявляют «асоциальное» поведение, поскольку растут, наползая друг на друга и формируют «бородавки» в культуре.
В лабораторных условиях они легко отделяются друг от друга с помощью стеклянной палочки, так как силы сцепления между ними малы. Это явление объясняется утратой опухолевыми клетками некоторых форм адгезивных молекул, называемых Е-кадгерины (Е-эпителиальные). Однако плотность других рецепторов, обеспечивающих прикрепление опухолевых клеток к молекулам ЭЦМ, возрастает, и эта рецептор-зависимая связь позволяет клеткам опухоли передвигаться и прикрепляться к молекулам ЭЦМ. Дело в том, что мобильность в ткани позволена лишь клеткам-фагоцитам, в меньшей мере — лимфоцитам. Это свойство записано в их геноме, и оно связано с выполнением этими клетками определенных функций, требующих передвижения по ткани. Однако опухолевые клетки, обладая измененным геномом и принадлежа к типам, не обладающим локомоцией (мобильностью) — эпителий, мышечные и другие клетки, способны передвигаться по ткани, экспрессируя молекулы адгезии к ЭЦМ. После прикрепления к ЭЦМ опухолевые клетки должны расчистить себе дорогу на пути своего передвижения. Для этой цели они продуцируют плазмин, расщепляющий фибрин, а также вырабатывают и секретируют различные протеазы, в частности коллагеназу 4-го типа, облегчая свой переход через стенку мелких сосудов.
В кровотоке и стенке сосуда агрегация опухолевых клеток облегчает их выживаемость и дальнейшую имплантацию в ткани. Попав в кровоток, клетки опухоли становятся особо уязвимыми по отношению к клеткам иммунного надзора, особенно опасны для них нативные киллеры. Однако снижение иммунологической реактивности (пожилой возраст больного и иммунодефициты наследственные и приобретенные), а также увеличивающаяся по мере прогрессии масса метастазирующих клеток, действуя вместе, легко преодолевают иммунологический барьер и сводят на нет элиминацию опухолевых клеток из организма.
242 Лекция 15
Автономность опухолевого роста
По мере прогрессирования опухоли снижается ее чувствительность к химиотерапии, а также чувствительность гормонально-зависимых опухолей ктерапии гормонами противоположного пола. Снижается чувствительность организма к лучевой терапии, и она становится менее эффективной. В значительной мере это объясняется тем, что опухоль, возникшая из одного клона клеток (моноклоновая), превращается в поликлоновую, так как в ходе опухолевой прогрессии в опухолевых клетках возникают дополнительные спонтанные мутации, и таким образом возникают новые дополнительные клоны клеток с более злокачественными свойствами. Затем идет селекция клеток с высокой стратегией выживания.
Опухолевые клетки приобретают автономность благодаря следующим их качествам:
• продукцией изнутри факторов роста, рецепторов к факторам роста и других ростовых факторов;
• уменьшению, вплоть до полного исчезновения, кейлонов-ингибиторов роста;
• способности опухоли организовать свое собственное микроокружение. Сама опухоль и ее преобразованное микроокружение синтезируют факторы ангиогенеза (роста сети сосудов);
• слабовыраженная антигенность опухолевых клеток из-за утери клеточных дифференцировочных антигенов (молекул МНС I класса) делает их малодоступными для опознания иммунокомпетентными клетками.
Системное действие опухоли на организм и симптомы опухолевой болезни
Для всех злокачественных заболеваний характерны общие симптомы, которые связаны как с местным, так и с системным действием опухоли на организм. Последние во многом перекликаются с симптомами ответа острой фазы повреждения. Однако наиболее часто встречаются следующие осложнения:
• сдавление прилежащих тканей и нарушение функции органов;
• некрозы в опухолях, резорбция продуктов тканевого распада и развитие эндотоксикоза;
• продукция опухолевыми клетками различных биологически активных веществ, в том числе гормонов, с последующим развитием паранеопластического синдрома;
Патология опухолевого роста. Канцерогенез
243
• разрушение прилегающих тканей и достаточно крупных сосудов может привести к угрожающему жизни больного кровотечению;
• резкое снижение иммунологической защиты опасно развитием инфекции, которая нередко становится причиной гибели больного;
• злокачественные опухоли в силу массивного повреждения тканей часто приводят к развитию ДВС-синдрома. Наиболее часто ДВС-синдром осложняет течение острого промиелоцитарного лейкоза и карциномы простаты.
Раковая кахексия
Больные раком, даже не страдающие непроходимостью и другими симптомами, нарушающими нормальное пищеварение, как правило, страдают от резчайшего исхудания за счет уменьшения массы сначала жировой, а затем и мышечной ткани. Это состояние называется раковой кахексией. Обычно оно сопровождается выраженной слабостью и отказом от еды. В настоящее время имеются четкие доказательства того, что кахексия при раке является результатом действия на организм цитокинов, которые продуцируются не только самой опухолью, но и клетками организма, вступившими в борьбу с ней.
Важнейшим цитокином, вызывающим раковую кахексию, является фактор некроза опухолей (ФНО-а) (отсюда его синоним — кахектин).
Теории канцерогенеза и общие механизмы опухолевой трансформации клетки
На сегодня существует четыре теории происхождения рака или теории канцерогенеза:
• вирусная;
• теория химического канцерогенеза;
• лучевого канцерогенеза;
• наследственная теория.
Наследственные опухоли, наиболее часто встречающиеся у детей, составляют относительно небольшую группу заболеваний.
Экспериментально доказано, что формирование опухоли является многостадийным процессом, начинающимся с того, что канцероген вирусной, химической или лучевой природы (отсюда название теорий возникновения опухолей) вызывает неблагоприятные мутации в геноме определенной соматической клетке. В случае же наследственных опухолей генетические повреждения передаются через половые клетки. Затем
244
Лекция 15
мутантная клетка размножается, и ее потомки являются уже семейством одного клона.
Мишенями для действия канцерогенных факторов являются следующие типы генов.
• Гены, усиливающие рост, или протоонкогены. Это нормальные гены, включающиеся в периоды роста организма, а также во время процессов обновления и регенерации тканей. Под влиянием канцерогенов протоонкогены превращаются в онкогены, ответственные за синтез онкобелков. Необходимо добавить, что протоонкогены являются доминантными генами, поэтому для клональной опухолевой пролиферации достаточно появления одного мутировавшего протоонкогена.
• Гены, тормозящие рост, или туморосупрессирующие гены. Злокачественная опухоль возникает только в случае инактивации или наследственной утраты обоих этих генов. К туморосупрессирующим относят Re-ген. Утрата двух копий этого гена приводит к злокачественной опухоли глаза — ретинобластоме. Опухоль развивается из ретинобластов и преимущественно у детей. Если же дефектным бывает один аллель, то приобретенная мутация другого аллеля в случае, когда исчезает генное прикрытие здоровым аллелем, с высокой степенью вероятности может привести к ретинобластоме.
• Гены, регулирующие апопотоз, — могут быть как доминантными, так и рецессивными. К таким генам относят bd-2, Ьах и р-53. Нарушение баланса экспрессии этих генов может привести к ослаблению апоптоза и усилению амплификации протоонкогенов. Это обеспечивает бессмертие опухолевым клеткам.
• Гены репарации ДНК обеспечивают клеточному геному стабильность, и поэтому мутации в системе этих генов увеличивают риск возникновения протоонкогенов.
Механизмы вирусного канцерогенеза
Теория вирусного канцерогенеза в основном разработана в экспериментах на животных. Существуют многочисленные доказательства онкогенного влияния некоторых вирусов на животных самого разного уровня их биологической организации. Однако что касается человека, здесь мы должны согласиться с тем, что нам известны лишь немногие опухоли вирусного происхождения.
Классификация онковирусов и опухоли, ими вызываемые
Все онковирусы делятся на ДНК- и РНК-содержащие. ДНК-содер-жащие вирусы животных, такие как аденовирусы и вирус полиомы, мо
Патология опухолевого роста. Канцерогенез
245
гут вызывать опухоли только в лабораторных условиях. Другие вирусы, например бычий вирус папилломы, вызывают как доброкачественные, так и злокачественные опухоли у их природных хозяев.
ДНК-содержащие вирусы человека участвуют в формировании злокачественных опухолей у человека; их можно разделить на 4 группы:
• вирусы, вызывающие папиллому (папилломавирусы);
• вирус Эпштейна-Барр вызывает лимфому Беркитта;
• вирус гепатита В способен вызывать рак печени;
• вирус герпеса — саркому Капоши.
К РНК-содержащим вирусам животных относят вирус куриной саркомы Роуса, а также вирусы лейкозов (Молони, Граффи) у мышей. Эти модели наиболее часто используются для изучения патогенеза лейкозов в эксперименте.
Из РНК-содержащих онковирусов человека нам известен вирус Т-лейкоза человека (HTLV-1).
Наследственные механизмы вирусного онкогенеза
Перед описанием механизмов вирусного канцерогенеза (онкогенеза) необходимо определиться в ныне существующей в онкологии терминологии:
• протоонкоген обозначается как р-onc. Это ген, как уже было сказано, кодирующий белки, определяющие нормальную пролиферацию клеток;
• клеточный онкоген с-опс (от английского слова cell — клетка). Это продукт активации протоонкогена клетки, кодирующий онкобелки, обеспечивающие высокую митотическую активность клеток;
• v-onc (от слова virus) — онкоген, присутствующий внутри вируса, аналогичный с-опс.
В настоящее время общепризнанны две основные теории превращения клеточного протоонкогена в онкоген. Первый механизм связан с интеграцией вирусного генома с протоонкогенной последовательностью. Это так называемый вставочный, или инсертный, механизм. Так, про-вирусная ДНК оказывается вставлена в клеточный протоонкоген, после чего мутировавший протоонкоген становится онкогеном. В случае с РНК-содержащими вирусами последние должны синтезировать ДНК на матричной РНК, используя для этого энзимобратную транскриптазу.
Второй механизм реализуется через вставку или прикрепление сильного вирусного промотора (он может быть использован и для собственной репликации) к протоонкогену, усиливая амплификацию последнего. Эти промоторные вирусные области называют концевыми областями
246
Лекция 15
длительного повтора (long terminal repeat units). Так, например, такой промоторный вирусный ген, прикрепляясь к гену синтеза тяжелых цепей иммуноглобулина, может привести к опухолевой патологии в форме плазмацитомы. Присоединяясь же к протоонкогену, ответственному за синтез КСФ-Г, этот промотор может привести к неконтролируемой пролиферации миелоидных клеток гранулоцитарного ряда и возникновению миелоидного лейкоза.
Химический канцерогенез
(характеристика химических канцерогенов)
Открытие химических канцерогенов началось с исследования на животных влияния полициклических углеводородов — продуктов переработки каменноугольной смолы. Первым был 3,4-бензпирен, более сильным в этом отношении оказался его синтетический аналог — ди-метилбензантрацен, однако в настоящее время более широко в эксперименте на животных используется метилхолантрен. Эти вещеста обладают как выраженными местными эффектами (при втираниии в кожу и слизистые оболочки), так и общерезорбтивными эффектами, вызывая рак в областях, удаленных от места их нанесения.
Другая группа — ароматические амины и некоторые красители. Это вещества с органотропным эффектом, т.е они вызывают рак определенных органов. Так, аминоацетофлоурен в эксперименте вызывает рак печени у крыс, а 0-нафтиламин вызывал рак мочевого пузыря у лиц, занятых в его производстве (Германия, 1920-е гг.). К природным канцерогенам, потенциально вызывающим рак печени у человека, относят афлотоксин В-1, продуцируемый некоторыми штаммами грибка аспергилус флавус. Его содержат некоторые виды земляных орехов. Нитрозамины, асбест, винилхлорид, мышьяк, хром и никель — профессиональные вредности, зарегистрированы так же, как потенциальные канцерогены.
Другую группу веществ составляют эндогенные канцерогены — вещества, являющиеся нашими естественными метаболитами и природными стероидами. К первой группе веществ относят производные триптофана (5-ОИК), оксииндолуксусную кислоту. Так, инъекции высоколейкозным линиям мышей экстракта мочи больных лейкозами, содержащими высокую концентрацию 5-ОИК, практически в 100 % случаев вызывали у мышей лейкоз — опухоль кроветворной системы. Подобными свойствами обладает и 3-оксиантраниловая кислота (производное тирозина). Что касается стероидов, то канцерогенными свойствами в опытах на животных обладали облученный холестерин, желчные кислоты (ЖК) и
Патология опухолевого роста. Канцерогенез
247
эстрогены. Эстрогены после длительной аппликации на слизистую оболочку матки мышей-самок, находящихся в определенной фазе цикла овуляции, вызывали у них рак матки.
Стадии химического канцерогенеза
Экспериментально доказано, что химический канцерогенез протекает в две стадии: инициации и промоции. Инициация является результатом прямого контакта экзогенного канцерогена с клеточным геномом. Мишенью для канцерогена являются ДНК-клетки, и считается, что химические канцерогены чаще обладают не канцерогенными, а мутагенными свойствами, и порой мутагенные и канцерогенные свойства у них не совпадают. Однако для воспроизведения в эксперименте химической опухоли одного контакта с канцерогеном недостаточно. Для увеличения числа опухолевых клеток необходимо дальнейшее влияние веществ-промоторов, которые усиливают пролиферацию генетически измененных клеток. Промоторы играют роль факторов-толкачей в плане их влияния на клеточный рост. Промоторным влиянием в эксперименте обладает кротоновое масло. На роль промоторов, участвующих в формировании злокачественных опухолей у человека, претендуют избыточный уровень жиров в пище (рак толстой кишки и молочной железы), высокий уровень эстрогенов в период начинающейся менопаузы, наконец, хроническое воспаление, требующее усиленных пролиферативных процессов и вовлекающее клетки в развернутую фазу митоза, когда возрастает опасность мутаций. При этом увеличивается потребность организма в местных ростовых факторах, которые могут сыграть промоторную роль в плане опухолевого роста (рис.15-1).
№ опыта Инициация (ДМБА) Используемое вещество (кротоновое масло или скипидар в различной последовательности) Результат
1. ДМБА Кротоновое масло Опухоль
2. ДМБА Скипидар Нет опухоли
3. ДМБА Кротоновое масло, затем скипидар Опухоль
4. ДМБА Скипидар, затем кротоновое масло Нет опухоли
ДМБА — диметилбензантрацен, полициклический углеводород: используется как инициатор канцерогенеза.
Кротоновое масло является промотором канцерогенеза.
Скипидар вызывает гибель клеток.
Рис. 15-1. Схема двустадийного химического канцерогенеза
В схеме опытов, иллюстрирующих двухстадийный канцерогенез, только последовательности, в которых за инициатором (ДМБА) следует про
248
Лекция 15
мотор (кротоновое масло), могут привести к возникновению опухолей. Если же в промежутке между ними на кожу животного апплицировать скипидар, то такая последовательность не сработает, так как скипидар уничтожит эффект инициации, т.е., другими словами, уничтожит все, в том числе и малигнизированные под влиянием ДМБА клетки.
Лучевой канцерогенез
Любые формы лучевой энергии, такие как ультрафиолетовая часть солнечного спектра, ионизирующее излучение в форме а-, р- или у-частиц, рентгеновское излучение, действие протонов или нейтронов, обладают способностью вызывать опухолевую трансформацию различных видов клеток in vitro, а также вызывать опухоли как у различных видов животных, так и у человека.
Прослеживается связь между ультрафиолетовым облучением (УФО) и раком кожи, описаны профессиональные раки у врачей-рентгенологов и лиц, занятых в атомной промышленности. Трагедии в Японии и Чернобыле также стали печальным опытом человечества, подтвердившим связь между облучением и возникновением злокачественных опухолей. Лучевыми опухолями принято считать лейкозы, опухоли щитовидной железы и молочных желез, если речь идет об общем облучении, местное же воздействие больших доз ионизирующего излучения может вызвать рак кожи и костей.
Патогенное действие ультрафиолетового спектра солнечных лучей
УФО является фактором риска в плане возникновения различных злокачественных опухолей кожи: плоскоклеточного рака, базальноклеточной карциномы и меланомы. Степень риска зависит от длины волны ультрафиолетовых лучей, времени экспозиции, площади облучения, а также от конституциональных особенностей облученного организма. Особенно чувствительны к ультрафиолету люди со светлой кожей и рыжеволосые, не обладающие должной адаптацией кожи к длительному его воздействию.
Ультрафиолетовые лучи обладают множеством патогенных эффектов в отношении клеток, включая торможение клеточного деления, инактивацию внутриклеточных энзимов, а также вызывают хромосомные и генные мутации, а в соответствующей дозе даже убивают клетки. В основе же их мутагенной активности лежит свойство формирования пиримидиновых димеров ДНК клетки. Этот тип повреждения ДНК в норме восстанавливается нуклеотидной эксцизией (вырезкой нуклеотидов) специальным ферментом — эксцизионной репаразой с последую
Патология опухолевого роста. Канцерогенез
249
щим включением репаративных энзимов. Этот процесс требует участия около 20 генов, ответственных за репарацию ДНК. Интенсивная же солнечная экспозиция может превысить репарационные возможности ДНК, и поэтому после частых и интенсивных УФО-облучений лиц с наследственными и приобретенными дефектами репарации ДНК последняя полностью не восстанавливается. Такие люди подвержены возникновению злокачественных опухолей кожи.
Патогенное действие ионизирующей радиации
В настоящее время установлены определенные иерархические последовательности лучевых раков. Первое место по частоте возникновения занимают лейкозы. У молодых лиц первое место принадлежит раку щитовидной железы. Средней частотой отмечены раки молочной железы, легких и слюнных желез.
В противоположность этому в коже, костях и в желудочно-кишечном тракте (ЖКТ) имеется относительная устойчивость к общему облучению, несмотря на то что клетки слизистой оболочки ЖКТ чувствительны к радиационной гибели. Однако любой врач, исследуя анамнез больного, в своих дальнейших умозаключениях должен придерживаться следующего правила: «воздействие ионизирующей ионизации может превратить любую нормальную клетку в опухолевую».
Молекулярные основы всех лучевых раков тесно связаны с различными мутагенными эффектами любых видов ионизирующей радиации. При этом, как и в случае эффекта УФО, важную роль играет возникающий дисбаланс между повреждением и репарацией ДНК. Последняя не выдерживает мощного напора повреждения. Помимо возможности ма-лигнизации клеток, лучевое повреждение обладает такими возможностями, как сокращение продолжительности жизни человека, его раннее старение и появление различных врожденных уродств и пороков развития внутренних органов у его потомков.
Онкобелки и их свойства
Трансформация нормального генома клетки в опухолевый приводит к синтезу такой клеткой онкобелков. Онкобелки — это продукты онкогенов.
Онкобелки играют роль ростовых факторов. С одной стороны, это могут быть белки с измененной функцией, но чаще это нормальные белки, отвечающие за процессы роста. В соответствии с их функциями онкобелки могут играть роли:
• факторов роста;
• рецепторов к факторам роста;
250
Лекция 15
• мембранных G-белков, вовлекаемых в передачу ростовых сигналов с клеточной мембраны на цитоплазму;
• вторичных мессенджеров, передающих сигналы с цитоплазмы на ядро и обладающих протеинкиназной активностью;
• ДНК-связывающих белков, вовлекающих ДНК в репликацию с последующим входом клетки в митотический цикл;
• циклинов и циклинзависимых киназ, наделенных функцией «контрольно-пропускных ворот» (check point) при входе клетки в митотический цикл.
Механизмы антибластомной резистентности организма
Механизмы антибластомной резистентности представлены в организме целым рядом биологических феноменов, приобретенных человеком и животными в процессе их биологической эволюции. Они включают неспецифические и специфические реакции организма, предупреждающие возникновение опухоли и противодействующие ее развитию и прогрессированию.
Первой линией обороны против опухолей, несомненно, являются антионкогены или туморосупрессирующие гены, данные нам нашей эволюцией как контрфактор, работающий против онкогенов. Утрата обоих антионкогенов, как это было упомянуто выше, происходит при наследственной детской опухоли — ретинобластоме. Если же утрачен только один ген, возникает высокий риск по этому заболеванию после рождения. Утрата этих генов теоретически может быть спровоцирована любыми канцерогенными факторами. Вторым важнейшим механизмом противоопухолевой устойчивости является система энзимов репарации ДНК. Клинически это доказано на примере нескольких синдромов. Один из них, синдром Фанкони, сопровождается множественными врожденными пороками развития, важнейшим из которых является аплазия костного мозга. Дело в том, что средний срок продолжительности жизни больных с этим синдромом не превышает 21-22 лет, а причиной их гибели чаще являются именно злокачественные опухоли различных органов. К такому же типу патологии, связанной с нарушением репарации ДНК, могут быть отнесены пигментная ксеро-дерма и синдром Блума.
К превентивным в отношении злокачественных опухолей относят и механизмы иммунной защиты организма, как специфические, так и неспецифические. Доказательством важной роли иммунного надзора за опухолевыми клетками служит факт повышенной заболеваемости зло
Патология опухолевого роста. Канцерогенез
251
качественными опухолями лиц с иммунодефицитами. У этих больных наиболее часто возникают лимфомы.
К лицам со сниженной иммунологической реактивностью относятся:
• больные ВИЧ-инфекцией;
• больные, подвергавшиеся достаточно длительной химиотерапии;
• больные, длительное время получавшие иммуносупрессивные препараты;
• лица после трансплантации опухолей, предварительно получавшие курс облучения;
• наконец, дети с врожденными иммунодефицитами.
Опухоль и иммунитет
Необходимо назвать несколько позиций, которые помогли бы понять проблему взаимодействия опухоли с иммунной системой организма. Это понимание, несомненно, в будущем приведет к эффективной иммунотерапии злокачественных опухолей, начало которой уже положено в наши дни.
1. В ходе опухолевой прогрессии идет селекция АГ-негативных субклонов, т.е. опухолевых клеток с низкой антигенной активностью, плохо распознаваемых системой иммунного надзора. В то же время клоны клеток с выраженной иммуногенностью элиминирутся гораздо легче.
Это связано с тем, что по мере опухолевой прогрессии опухолевые клетки, будучи менее зрелыми, не приобретают МНС I класса АГ, наличие которых так необходимо для иммунного распознавания при встрече с Т-лимфоцитами. Последние специализируются на иммунном распознавании тумороспецифических — новых АГ опухолевых клеток, которые не всегда экспрессируются должным образом. Трудности распознавания связаны с тем, что опухолевые АГ включают в себя «свое» и «чужое», другими словами, эти клетки и свои, и чужие.
2. Некоторые опухоли, такие как меланома и гепатоцеллюлярная карцинома, способны вызывать апоптоз цитотоксических СД8‘-лимфоцитов, с которыми связана важнейшая функция при уничтожении опухолевых клеток.
3. Наконец, опухолевые клетки не вырабатывают костимулирующие иммунный ответ молекулы, необходимые для активации Т-клеток.
Принимая во внимание все сказанное, можно заключить, что опухоль, с одной стороны, чаще возникает у лиц с иммунодефицитами, а с другой стороны, она сама вызывает иммуносупрессию. К этому можно добавить, что опухолевый процесс, являясь одним из вариантов острофазового от
252
Лекция 15
вета, сопровождается гиперкортизолизмом. Однако известно, что высокие концентрации кортизола в крови также способствуют иммуносупрессии.
Роль цитотоксических лимфоцитов в противоопухолевом иммунитете
Противоопухолевые защитные механизмы включают реакции клеточного и гуморального иммунитета, однако первым принадлежит более важная роль. В иммунизированном опухолевыми клетками организме работают как СД4* (хелперы 1-го типа), так и СД8* (цитотоксические лимфоциты).
Иммунологическая специфичность цитотоксических лимфоцитов направлена и против тумороспецифических, и против туморосвязанных АГ.
Роль цитокинов. Такие медиаторы ответа острой фазы, как ФНО-а и ФНО-0, усиливают местные разрушающие опухоль реакции. В свою очередь, ИЛ-2, являясь сильным митогеном, обеспечивает усиленную пролиферацию лимфоцитов. Учитывая первейшую и наиважнейшую роль лимфоцитов в борьбе с опухолями, при лечении некоторых опухолей (гипернефроидный рак почки и меланома) используется метод адоптивной терапии (от англ, adopt — усыновлять). Суть этого метода заключается в создании банка Т-лимфоцитов больного, активированных (in vitro) ИЛ-2. После такой стимуляции лимфоциты называются лейкин-активированные. После введения в организм больного обработанные подобным методом клетки более активно разрушают опухоль.
Роль натуральных киллеров
Естественные киллеры (NK*) клетки — это лимфоциты, способные разрушать опухолевые клетки без предварительной иммунизации их опухолевыми АГ. Считается, что именно этим клеткам принадлежит первая роль в противоопухолевом неспецифическом иммунитете. Наибольшая эффективность натуральных киллеров объясняется тем, что при снижении экспрессии HLA опухолевыми клетками затрудняется их распознавание рецепторным аппаратом иммунизированных против опухолевых АГ цитотоксическими лимфоцитами, а естественные киллеры исправляют это положение. Не обладая свойством распознавания АГ, они распознают лектиновые рецепторы на любых интенсивно размножающихся клетках. Опухолевые клетки относятся именно к такой категории клеток. При контакте NK* клеток с опухолевыми формируются белки-перфорины, образующие сквозные каналы в ее мембране
Патология опухолевого роста. Канцерогенез
253
и гранзимы, активирующие апоптоз клетки. Этот цитолиз и апоптоз тормозятся в случае встречи натуральных киллеров с молекулами МНС I класса на нормальных клетках.
Антитела при опухолевом иммунитете играют вспомогательную роль. Они участвуют в формировании антителозависимой клеточной цитотоксичности (АЗКЦ), антителозависимом фагоцитозе и компле-ментзависимом лизисе опухолевых клеток.
Фактор некроза опухолей и его роль в противоопухолевой защите
Роль ФНО в опухолевом процессе неоднозначна, поскольку этот медиатор ответа острой фазы оказывает системное влияние на организм (больше ФНО-а), что приводит к кахексии больного злокачественной опухолью. В то же время местный эффект больше выражен у ФНО-р, называемого лимфотоксином, что выражается высоким цитопатоген-ным влиянием на опухолевые клетки, вплоть до их полного лизиса и гибели.
Различают следующие механизмы, лежащие в основе антибластом-ного эффекта ФНО.
• Вызывает геморрагический некроз опухоли, что и отражено в его названии. Геморрагический некроз связывают с повышенным синтезом опухолевыми клетками фактора VII, участвующего в синтезе протромбиназы. Таким образом, местная активация коагуляционного гемостаза приводит к тромбозу питающих опухоль сосудов и дальнейшему ее некрозу.
• Будучи медиатором ответа острой фазы повреждения вместе с ИЛ-1 играет важную роль в активации различных феноменов иммунологической защиты: увеличивает число всех видов лимфоцитов, активирует фагоциты, вооружая их оксидантами и увеличивая мобильность.
• Способствует увеличению экспрессии МНС I класса на опухолевых клетках и МНС II класса на антигенраспознающих клетках, тем самым облегчая иммунное распознавание и дальнейшую элиминацию из организма опухолевых клеток.
• Тормозит репарацию ДНК поврежденных опухолевых клеток, препятствуя сохранению их жизнеспособности.
Лекция 16
ПАТОФИЗИОЛОГИЯ НЕРВНОЙ СИСТЕМЫ
Г.В. Порядин
Какова природа нарушений системных механизмов нервной деятельности, каковы возникающие системные патологические механизмы, какие нервные образования, начиная с нейронов и кончая высшими отделами мозга, составляют патогенетическую организацию нервных расстройств, как возникают эти расстройства в виде соответствующих нейропатологических синдромов, как они развиваются и ликвидируются — вопросы, которые составляют предмет патофизиологии нервной системы и главную задачу общей патофизиологии нервной системы — установление и изучение типовых патологических процессов в нервной системе.
Г.Н. Крыжановский, 1997
Выяснение механизмов патогенеза отдельных форм патологии нервной системы составляет частную нейропатофизиологию, которая будет изучаться в курсе неврологии и психиатрии.
Общая этиология и патогенез нервных расстройств.
Причины возникновения повреждений нервной системы
Любой патологический процесс в нервной системе (НС) начинается с ее повреждения. Повреждения могут заключаться в разрушениях, морфологических альтерациях нервных образований, молекулярных изменениях и изменениях физико-химических процессов.
Патогенные факторы, вызывающие повреждение нервной системы, могут быть экзогенной и эндогенной природы. Их можно разделить на первичные и вторичные.
Первигные экзогенные факторы, вызывающие повреждение нервной системы:
Патофизиология нервной системы
255
• биологические возбудители (вирусы бешенства, полиомиелита; микробы — возбудители лепры; микробные токсины ботулинический, столбнячный; токсины растительного происхождения — стрихнин, кураре);
• синтетические и другие яды (этиловый и метиловый спирты);
• ядохимикаты (хлорофос, отравляющие вещества);
• фармакологические препараты;
• интенсивные стрессорные воздействия;
• перегрузка информацией, требующей быстрой обработки и принятия важных решений.
Первигные эндогенные факторы, вызывающие повреждение нервной системы:
• изменения внутренней среды организма (нарушения кровообращения в ЦНС, гипоксия, ишемия);
• эндокринные нарушения (при диабете, гипо- и гипертиреозе);
• опухоли и воспалительные процессы в ЦНС;
• генетические факторы, обусловливающие наследственные формы патологии НС.
Вторигные эндогенные факторы, вызывающие повреждение нервной системы:
• поражения в самой нервной системе (изменения нейронов, секреции медиаторов, межнейрональных и системных отношений, образование генераторов патологически усиленного возбуждения, патологических систем, антител к нервной ткани).
Общие реакции нервной системы на повреждение
Патологические изменения в НС проявляются:
• альтерацией структур, нарушением и перерывом функциональных связей, повреждением и дезинтеграцией существующих физиологических систем. С них начинается любой патологический процесс. При некоторых формах патологии (повреждение мозговых структур и выпадение их функций при инсультах мозга) деструктивные изменения являются ведущими:
• возникновением новых (патологических по характеру и результатам) интеграций из поврежденных и вторично измененных образований нервной системы. При многих формах патологии НС образование и деятельность патологических интеграций являются ведущими патогенетическими механизмами.
256
Лекция 16
Примером такой патологической интеграции на уровне межнейрональных отношений является агрегат взаимодействующих гиперактивных нейронов, обладающий свойствами генератора патологически усиленного возбуждения (ГПУВ).
Примером такой патологической интеграции на уровне системных отношений является новая организация, охватывающая различные первично и вторично измененные отделы нервной системы — патологическая система.
ГПУВ и патологические системы относятся к типовым патологическим процессам в нервной системе и служат патофизиологической основой нервных расстройств.
ГПУВ — это агрегат гиперактивных нейронов, продуцирующий чрезмерный неконтролируемый поток импульсов. ГПУВ образуется в поврежденной нервной системе из первично и вторично измененных нейронов и представляет собой новую, необычную для деятельности нормальной НС патологическую интеграцию, возникающую на уровне межнейрональных отношений. ГПУВ может образовываться практически во всех отделах ЦНС.
Особенностью ГПУВ является его способность развивать самопод-держивающуюся активность.
Возникновение ГПУВ служит эндогенным механизмом развития патологического процесса в НС и нервных расстройств.
В основе формирования ГПУВ лежит недостаточность тормозных механизмов, которая возникает либо при первигной гиперактивности нейронов (хроническое раздражение рецепторов в тканях под влиянием усиленных и длительных возбуждающих воздействий; невринома), либо при первигном нарушении тормозных механизмов (действие столбнячного токсина, нарушающего выделение тормозных медиаторов, стрихнина, блокирующего рецепторы на постсинаптических нейронах).
Основное патогенетическое значение ГПУВ как эндогенного механизма развития патологического процесса в нервной системе заключается в том, что с его деятельностью связано формирование в ЦНС патологической системы, лежащей в основе соответствующего нейропа-тологического синдрома.
Патологическая система — это новая патологическая интеграция, возникающая из первично и вторично измененных образований ЦНС, деятельность которой имеет дезадаптивное или прямое патогенное значение для организма. Образование и деятельность патологической системы являются, с одной стороны, выражением и результатом воз
Патофизиология нервной системы
257
никновения патологического процесса, с другой, — механизмом дальнейшего его развития.
Активность патологической системы на уровне ЦНС служит системным патофизиологическим механизмом нейропатологических синдромов, которые, в свою очередь, представляют собой клиническое выражение деятельности соответствующей патологической системы.
Патологические системы клинически проявляются в виде мышечного гипертонуса при местном столбняке, патологических рефлексов, акинезии (брадикинезии), ригидности и тремора при паркинсонизме.
Функционально эффекты патологических систем выражаются либо в виде чрезмерного усиления, либо подавления функции того отдела ЦНС, который является конечным звеном патологической системы.
Патология НС не сводится к чисто количественным изменениям деятельности нервных образований и физиологических систем. Важную роль играют также качественно новые патологические механизмы, возникающие на разных уровнях структурно-функциональной организации НС в условиях патологии.
Выражением качественно новых патологических процессов является извращение нормальных физиологических, биохимических и биофизических механизмов.
• Измененная деятельность Na/Ca2’-насоса в условиях патологии: вместо выхода Са2’ из нейрона его вхождение в нейрон, что способствует развитию патологии в нейроне.
• Извращение деятельности переностика глутамата: вместо транспорта глютамата из синаптической щели в терминаль и в глиальную клетку усиление выхода глютамата в синаптическую щель, что приводит к патогенному действию на нейрон избытка глютамата. Трансформационные изменения ферментов — изменение деятельности моноаминооксидазы (МАО). В условиях перекисного окисления липидов (ПОЛ) МАО дезаминирует гамма-аминомасляную кислоту (ГАМК), вследствие чего исчезает тормозной эффект ГАМК, — это приводит не к торможению, а к растормаживанию нейрона.
Барьерные механизмы нервной системы
Все нейроны как периферической, так и центральной нервной системы ограждены барьерными образованиями.
• Аксоны нейронов имеют специальную миелиновую оболочку: пучки нервных волокон и нервные стволы окружены эндо- и периневральными оболочками.
258
Лекция 16
• Нейроны ЦНС окружены глиальными клетками (специализированные барьеры), которые обеспечивают трофику нейрона, удаляют продукты его метаболизма и предохраняют нейрон от различных токсических воздействий.
• Помимо поверхностных оболочек ЦНС имеет специализированный барьер — гематоэнцефалический (ГЭБ), образующийся сосудистой стенкой и глиальными элементами (астроцитами). Важнейшая функция ГЭБ — защита мозга от действия различных токсических агентов экзо- и эндогенной природы.
Пути поступления патогенных агентов в нервную систему
Патологическая проницаемость (прорыв) ГЭБ. Может возникать при действии разнообразных патогенных факторов — физических (травмы, радиация), химических (токсические жирорастворяющие вещества), инфекционно-токсических, аллергических, при шоке, патологическом стрессе, судорожных состояниях, острой артериальной гипертензии (кризовые состояния), воспалении оболочек и сосудов мозга, отеке мозга, опухолях.
Нервные стволы обладают специальными барьерами. При их повреждении нервы могут служить путями поступления патогенных агентов в ЦНС. Невральным путем осуществляется поступление в ЦНС столбнячного токсина, вирусов полиомиелита, бешенства и других патогенных агентов. Нервные стволы являются также путем распространения многих органических и неорганических ядов, что приводит к возникновению невритов, токсических поражений нервных волокон и нейронов.
Патофизиология нервной системы
Нарушения деятельности нервной системы могут быть обусловлены наследственными нарушениями обмена веществ, врожденными дефектами развития НС, патогенным действием химических и физических факторов, расстройствами мозгового кровообращения, аутоиммунными процессами, инфекциями, неопластическими образованиями, нарушением питания.
Повреждения нервной системы, обусловленные наследственными нарушениями обмена веществ
Нарушения деятельности НС при наследственных нарушениях обмена веществ могут быть обусловлены:
Патофизиология нервной системы
259
• прямым повреждением нервных клеток вследствие недостаточности какого-либо фермента;
• накоплением тех или иных нерасщепленных продуктов обмена во внеклеточной жидкости;
• повреждением других органов;
• повреждением мозговых сосудов.
К числу наследственных болезней, обусловленных прямым повреждением нервных клеток, относятся так называемые болезни накопления, возникающие в связи с дефектом лизосомальных ферментов.
Дефект фермента нарушает внутриклеточный метаболизм определенных макромолекул и приводит к накоплению их внутри клетки, что, в свою очередь, нарушает функцию клетки.
К болезням накопления относятся липидозы, мукополисахаридозы, гликогенозы.
Примером липидозов может служить болезнь Ниманна-Пика, характеризующаяся накоплением сфингомиелина в печени, селезенке, сером и белом веществе головного мозга. Накопление молекул сфингомиелина обусловлено недостаточностью фермента сфингомиелиназы. Болезнь наследуется по аутосомно-рецессивному типу. Неврологические расстройства обнаруживаются уже на первом году жизни, они характеризуются прогрессирующей деменцией, расстройствами функций пирамидного тракта, нарушением слуха. Смерть наступает обычно в возрасте 5 лет.
Мукополисахаридозы — болезни накопления мукополисахаридов обусловлены дефектом фермента, участвующего в катаболизме мукополисахаридов (глюкозоаминогликанов). Нерасщепленные молекулы мукополисахаридов (дерматан сульфата, гепаран сульфата) накапливаются в клетках кожи, хрящей, роговицы, кровеносных сосудов, коры головного мозга. Болезни наследуются по аутосомно-рецессивному типу. Характеризуются прогрессирующими расстройствами психики, связанными с дегенеративными изменениями клеток коры головного мозга, мозговых оболочек и сосудов, и нарушением развития скелета.
Гликогенозы — болезни, связанные с нарушением функций ферментов, ответственных за синтез и расщепление гликогена. Болезни наследуются по аутосомно-рецессивному типу. Характеризуются расстройствами функций печени, сердца и других органов, в том числе нервной и мышечной систем. Повреждения нервной системы в наибольшей степени выражены при болезни Помпе (гликогеноз II типа) и генерализованном гликогенезе (обусловленном дефицитом а-1,4-глюкозидазы). Частички нерасщепленного гликогена обнаруживаются в клетках печени, по
260
Лекция 16
чек, скелетных мышцах, мышце сердца, ЦНС. Значительное количество гликогена накапливается в лизосомах нейронов ганглиев дорсальных корешков, в мотонейронах спинного мозга, нейронах ствола мозга, в клетках глии и эндотелии мозговых сосудов. Неврологические симптомы болезни — нарушения моторики и прогрессирующая мышечная слабость.
Нарушения функции нервной системы, вызванные аноксическим и ишемическим повреждением мозга
Мозг человека обладает высоким уровнем окислительного метаболизма, в связи с чем на его долю приходится около 20-25 % всего потребляемого организмом кислорода. Поскольку нервная ткань не имеет собственных запасов кислорода, метаболическая активность мозга целиком зависит от кислорода, приносимого кровью. Недостаточное поступление кислорода в мозг — самая частая причина метаболигеских энцефалопатий, сопровождающихся немедленными расстройствами функции и гибелью нервных клеток. Нарушение психики обнаруживаются при снижении напряжения кислорода в артериальной крови (РаО2) до 40-50 мм рт.ст. При падении РаО2 ниже 30 мм рт.ст. наступает потеря сознания и исчезает электрическая активность мозга.
Аноксигеское повреждение мозга может быть обусловлено различными причинами, однако самой частой ее формой является ишемигеская аноксия, обусловленная нарушениями мозгового кровотока вследствие расстройства системного кровообращения или увеличения внутричерепного давления.
Ишемигеское повреждение мозга обычно приводит к более тяжелым последствиям, чем аноксическое, поскольку нарушения кровотока не только снижают поступление кислорода и питательных веществ к тканям, но и вызывают задержку в гипоперфузируемых тканях продуктов метаболизма, способных оказывать токсическое действие.
Одним из ранних последствий ишемии мозга является нарушение синтеза нейромедиаторов (катехоламинов, ацетилхолина, аминокислот), ответственных за ранние клинические проявления ишемии.
Следом за этими изменениями развиваются более тяжелые расстройства, обусловленные развитием отека мозга, накоплением в мозге молочной кислоты, свободных жирных кислот, образованием свободных радикалов, увеличением внеклеточного калия, поступлением в клетку избыточных количеств ионов кальция, образованием окиси азота (NO).
Патофизиология нервной системы
261
Особую роль в ишемическом повреждении мозга играют возбуждающие дикарбоновые аминокислоты (глутаминовая, аспарагиновая), получившие специальное название — экситотоксины (от англ, oxcitation — возбуждение). Уже в самом начале ишемии концентрация глутамата в зоне ишемии резко возрастает. Глутамат взаимодействует со своими рецепторами на мембране нейрона, что сопровождается увеличением проницаемости цитоплазматической мембраны нейронов для ионов кальция и натрия, избыточным поступлением натрия в клетку, набуханием клетки и осмотическим лизисом клетки.
Избыточное поступление в клетку ионов кальция сопровождается активацией фосфолипаз, в частности фосфолипазы А2, и высвобождением свободной арахидоновой кислоты из фосфолипидов мембраны. Дальнейшие ферментативные превращения арахидоновой кислоты сопровождаются накоплением вазоактивных продуктов (простагландины и лейкотриены), способствующих нарушению микроциркуляции в зоне ишемии. Кроме того, ферментативные превращения арахидоновой кислоты стимулируют образование свободных радикалов и активацию перекисного окисления липидов, что может вызвать необратимое повреждение и смерть клетки.
В очаге ишемии увеличивается концентрация внеклеточного калия. Это вызывает деполяризацию мембраны нейронов, набухание клеток астроглии, что усугубляет нарушения мозгового кровотока и способствует возникновению судорог.
Повреждение мозга при гипогликемии
В нормальных условиях весь потребляемый мозгом кислород расходуется на окисление глюкозы. Запасы глюкозы и гликогена в мозге минимальны. Поэтому содержание глюкозы в клетках мозга постоянно зависит от его содержания в крови. Когда концентрация глюкозы в крови снижается до 1,7-2,2 ммоль/л, обнаруживаются нарушения функций сначала коры головного мозга, а затем и ствола мозга.
Гипогликемические расстройства нервной системы обнаруживаются при:
• первичном гиперинсулинизме (аденома островков поджелудочной железы);
• передозировке инсулина;
• надпочечниковой недостаточности;
• болезнях печени.
262
Лекция 16
Неврологические симптомы выражаются в появлении головной боли, раздражительности, расстройствах координации движений, сонливости. В тяжелых случаях возможно развитие судорог и комы. Длительная гипогликемия вызывает необратимые повреждения нервной системы.
Нарушения функции нервной системы, вызванные нарушением кислотно-основного состояния
pH мозга и спинномозговой жидкости относительно не зависит от pH крови и поддерживается на постоянном уровне при колебаниях pH крови от 6,8 до 7,6 благодаря свойствам ГЭБ, клетки которого способны ограничивать поступление ионов водорода и НСО3~ из крови внутрь мозга, а также продуцировать и секретировать внутрь спинномозговой жидкости ионы НСО3_ и ионы аммония.
Нарушения деятельности нервной системы возникают при нарушениях КОС, связанных с изменениями парциального напряжения СО2 в крови (респираторный ацидоз, респираторный алкалоз), поскольку ГЭБ не является барьером для СО2.
Повышение парциального напряжения СО2 в крови оказывает угнетающее и анестезирующее действие, что можно связать со снижением содержания в мозге возбуждающих аминокислот (аспартата и глутамата) и увеличением содержания тормозящих аминокислот (ГАМК). Умеренный респираторный алкалоз вызывает парестезию, головную боль. Более выраженный алкалоз (pH 7,52-7,65), особенно связанный с гипоксией, может вызвать точечные кровоизлияния в мозге, миоклонус мышц, судороги, кому и смерть.
Нарушения функции нервной системы, вызванные изменением электролитного состава крови
Наиболее частыми нарушениями электролитного состава крови являются гипер- и гипонатриемия, сопровождающиеся неконтролируемыми изменениями объема мозга.
Гипернатриемия возникает в результате потери жидкости или, иногда, в результате избыточного приема соли.
Неврологические симптомы (беспокойство, раздражительность, спутанность сознания) возникают при уровне натрия в крови, превышающем 150 мэкв/л. Увеличение натрия до 180 мэкв/л вызывает судороги, кому и может привести к смерти.
Патофизиология нервной системы
263
В основе неврологических расстройств лежит дегидратация нервных клеток, вызванная перемещением воды из клеток во внеклеточное гиперосмолярное пространство.
Гипонатриемия возникает при значительных потерях натрия, вызванных болезнями ЖКТ, обильным потоотделением, недостаточностью надпочечников или болезнями почек.
Неврологические расстройства, вызываемые гипонатриемией, обнаруживаются при падении содержания натрия в сыворотке крови до 120-125 мэкв/л. Более значительные падения содержания натрия приводят к ступору, генерализованным судорогам и коме.
В основе неврологических расстройств лежат отек мозга, связанное с ним повышение внутричерепного давления и нарушения мозгового кровотока.
Гипокальциемия. Ионы кальция участвуют практически во всех основных процессах жизнедеятельности нейрона. Са2’ оказывает влияние на возбудимость мембраны нервных клеток, стабилизирует цитоплазматическую и внутриклеточные мембраны, участвует в регуляции ионных каналов, в высвобождении нейромедиаторов и в других процессах. Изменение содержания внутриклеточного Са2*, его чрезмерное снижение приводят к патологии нервной системы. Изменение деятельности нервной системы при гипокальциемии возникает при понижении концентрации ионов кальция в крови до 1,5-1,75 ммоль/л (при почечной недостаточности, гипопаратиреозе, недостатке витамина D). Нарушения деятельности нервной системы при гипокальциемии проявляются периферическими и центральными расстройствами.
К периферическим расстройствам относятся парестезии, повышенная возбудимость двигательных нервов.
К центральным — высокая раздражительность, психозы, судороги, бред, ступор, кома.
Гиперкальциемия — увеличение содержания кальция в крови более 4,5 ммоль/л. Сопровождается летаргией, спутанностью сознания, ступором, комой.
Нарушения функции нервной системы, вызванные нарушением мозгового кровотока
Нарушения мозгового кровотока — одна из самых частых причин неврологических расстройств у взрослых, возникающие либо в связи с расстройствами системного кровообращения {глобальные нарушения мозгового кровотока), либо в связи с повреждением сосудов мозга (местные нарушения мозгового кровотока).
264
Лекция 16
В норме мозговой кровоток поддерживается на постоянном уровне (50-60 мл/мин на 100 г массы) независимо от колебаний среднего АД в пределах от 45 до 170 мм рт.ст.
Глобальные изменения мозгового кровотока возникают либо при падении АД ниже 45 мм рт.ст., либо при отеке мозга. Тяжесть возникающего ишемического повреждения мозга определяется степенью уменьшения мозгового кровотока и его продолжительностью. Ткани мозга сохраняют жизнеспособность при снижении кровотока до */3 от нормального (до 15-20 мл/мин на 100 г массы) в течение 30-60 мин, и только после этого возникают необратимые изменения в мозге.
Неврологические проявления при глобальных тяжелых нарушениях мозгового кровообращения — амнезия, корковая слепота, атаксия, признаки локальных повреждений мозга. Локальные повреждения обусловлены различной чувствительностью клеток мозга к гипоксии и особенностями развития сосудистой сети мозга. Наибольшей чувствительностью к ишемии обладают пирамидные клетки гиппокампа, клетки Пуркинье мозжечка.
Местные (региональные) нарушения мозгового кровотока могут быть обусловлены:
• повреждением (врожденным или приобретенным) стенок мозговых сосудов:
• нарушением системы свертывания крови.
В обоих случаях создаются условия, способствующие внутрисосудистому тромбообразованию или внутримозговым кровотечениям (геморрагиям).
Причиной приобретенных повреждений относительно крупных сосудов мозга является атеросклероз.
Атеросклеротические изменения возникают чаще всего в начальном отделе внутренней сонной артерии, во внутримозговых сегментах позвоночных артерий, в средней части основной артерии, в начальном сегменте средней мозговой артерии.
Наиболее частая причина повреждений относительно мелких сосудов мозга — хроническая артериальная гипертензия, приводящая к замещению гладких мышц артериол (сосуды с наружным диаметром около 50 мкм) коллагеном и образованию микроаневризм (со средним диаметром — 200 мкм). Разрывы небольших аневризм обычно приводят к относительно небольшим кровотечениям, разрывы крупных аневризм — к массивным внутримозговым кровотечениям.
Патофизиология нервной системы
265
Региональная ишемия редко бывает полной ввиду богатой сети анастомозов в сосудистой системе мозга, что обусловливает возможность восстановления кровоснабжения пораженных участков мозга.
Нарушения функции нервной системы, обусловленные повреждением миелина
Миелин периферических нервов образован мембраной шванновских клеток, тогда как миелин центральных аксонов — мембраной олигодендроцитов. Оба миелина выполняют сходные функции, хотя и имеют некоторые биохимические и морфологические различия. И центральный, и периферический миелин содержат основной белок (18 кДа), обладающий высокой антигенностью, в силу чего он становится частой мишенью аутоиммунных реакций.
Очаговые разрушения миелина приводят вначале к замедлению, а затем и к полной блокаде проведения нервных импульсов по соответствующим нервным волокнам. Длительно сохраняющаяся демиелинизация сопровождается гибелью соответствующих аксонов.
Повреждение миелиновой оболочки аксонов нейронов центральной и периферической нервной системы лежит в основе так называемых демиелинизирующих болезней — разнородной группы болезней, общим признаком которых являются очаговые повреждения миелина при относительной сохранности аксонов.
Распространенным примером демиелинизирующих болезней является рассеянный склероз.
Болезнь характеризуется возникновением очагов демиелинизации в различных участках ЦНС с наиболее частым поражением зрительного нерва, ствола мозга, задних столбов спинного мозга, околожелудочко-вых участков белого вещества головного мозга. В соответствии с этим у больных обнаруживаются расстройства зрения, проприоцептивной чувствительности, нервных механизмов управления движениями. Рассеянный склероз имеет прогрессирующее течение, прерываемое, особенно в начальном периоде, более или менее продолжительными периодами ремиссии.
Существуют убедительные доказательства того, что возникающая при рассеянном склерозе демиелинизация — результат аутоиммунного воспаления, опосредуемого Т-лимфоцитами, проникающими в ЦНС. Взаимодействие Т-лимфоцитов с АПК приводит к Т-клеточной пролиферации, активирует Т-хелперные клетки, вызывает секрецию ими цитокинов, активацию В-лимфоцитов и макрофагов. Развивается острое
266
Лекция 16
иммунное воспаление, сопровождающееся разрушением гематоэнцефалического барьера, гибелью олигодендроцитов и демиелинизацией.
В качестве АГ, вызывающего аутоиммунное воспаление в ЦНС, могут выступать вирусы, бактерии, белки миелиновой оболочки или специфические белки, экспонированные на поверхности олигодендроцитов.
Периферигеская нейропатия — другой пример демиелинизирующих болезней, связанных с демиелинизацией периферических нервов. Заболевание может быть обусловлено аутоиммунными процессами, запускаемыми вирусами, бактериальными токсинами (дифтерийная интоксикация), метаболическими расстройствами (недостаточность витамина В12). Поражение двигательных, чувствительных и вегетативных волокон вызывает появление разнообразных симптомов: мышечную слабость, угнетение сухожильных рефлексов, утрату кожной и проприоцептивной чувствительности, появление парестезий, расстройства сердечного ритма.
Причиной неврологических расстройств могут быть так называемые дегенеративные болезни нервной системы, обусловленные гибелью определенных групп центральных нейронов в результате ускоренного процесса естественной гибели нейронов.
Характерным примером дегенеративных болезней является болезнь Альцгеймера — частая форма старческого слабоумия. Заболевание обычно возникает около 60 лет, характеризуется вначале расстройствами памяти, особенно на ближайшие события, утратой способности сосредоточить внимание. Затем присоединяются нарушения речи, абстрактного мышления, зрительно-пространственной ориентации, параноидальные симптомы, расстройства сознания, нарушения движения.
На гистологических срезах в пораженных областях коры (височные доли, гиппокамп, подкорковые ядра мозга) обнаруживаются:
• рассеянные очаги внеклеточных отложений амилоида, содержащие многочисленные измененные отростки нервных клеток — сенильные бляшки;
• внутриклеточные нейрофибриллярные клубки, состоящие из плотно скрученных друг с другом нейрофиламентов, занимающих значительную часть цитоплазмы нейронов;
• аморфные отложения амилоида, располагающиеся внутри стенки и вокруг мозговых сосудов.
Главная роль в повреждении нейронов при болезни Альцгеймера принадлежит, по-видимому, р-амилоидному белку, составляющему основу сенильных бляшек и обладающему нейротоксическим действи
Патофизиология нервной системы
267
ем. Он образуется в результате протеолитического расщепления крупного трансмембранного белка — предшественника амилоида (БПА), ген которого локализован на длинном плече 21-й хромосомы.
В обычных условиях ферментативное расщепление внеклеточного домена БПА ведет к образованию продуктов, участвующих в межклеточном взаимодействии, и не сопровождается образованием сенильных бляшек. Избыточное отложение амилоида, вызывающее повреждение нейронов, возникает либо в результате его избыточной продукции, либо вследствие нарушения катаболизма внеклеточных отложений амилоида.
При болезни Дауна (наличие трех 21-х хромосом и, следовательно, дополнительной копии гена БПА) также обнаруживается избыточная продукция амилоида. Избыточное отложение р-амилоида в мозге и связанные с этим неврологические расстройства достаточно сильно выражены у этих больных уже к 40 годам жизни.
Несмотря на то что болезнь Альцгеймера связана с дегенерацией многих нейронов, наиболее сильно поражаются холинергические нейроны ядер основания мозга (ядра Мейнерта), которые посылают аксоны в кору гиппокампа и в различные отделы коры больших полушарий. Предполагают, что причиной высокой ранимости этих нейронов может быть тот факт, что холинергические нейроны используют холин не только для синтеза главного фосфолипида клеточной мембраны — фосфатидилхолина, но и для синтеза нейромедиатора — ацетилхолина. В условиях повышенной активности значительная часть холина расходуется на образование ацетилхолина. При этом синтез фосфатидилхолина замедляется, а его содержание в мембране падает, что способствует разрушению клеток.
Лекция 17
ПАТОФИЗИОЛОГИЯ НЕРВНЫХ МЕХАНИЗМОВ УПРАВЛЕНИЯ ДВИЖЕНИЯМИ
Т.Ю. Ругинская
Нарушения движений — это тяжелые болезни, которые приводят к тяжелым нарушениям социальной адаптации человека. Относительно высокая частота этих заболеваний, тяжелая инвалидизация при большинстве из них, поражение в детском и зрелом возрасте делают эти заболевания не только важной проблемой клинической неврологии, но и социальной проблемой. Ранняя диагностика этих заболеваний особенно важна, так как она обеспечивает проведение патогенетически обоснованной терапии, способной изменить течение и прогноз болезни.
Болезни нервной системы, в том числе болезни, приводящие к нарушению движения, могут быть обусловлены внешними и внутренними причинами: действием физических, химических, биологических факторов, болезнями обмена веществ, витаминной недостаточностью, расстройствами кровообращения, аутоиммунной патологией и др. Многие нарушения движений возникают в результате болезней или повреждения тех отделов нервной системы, которые объединяют в двигательную систему — систему управления движениями.
Различают 4 главных компонента двигательной системы:
• премоторная кора и дополнительные зоны коры (поля: 9, 22, 24, 7,8):
• моторная (двигательная) кора (поля: 4,6);
• ствол мозга:
• спинной мозг.
Деятельность низших отделов, управляющих наиболее стандартными, автоматическими движениями, постоянно контролируется высшими отделами, управляющими сложными нестандартными двигательными актами, — в этом проявляется иерархическая организация двигательной системы.
Мозжечок и базальные ганглии — структуры, контролирующие взаимодействие 4-х главных компонентов двигательной системы.
Патофизиология нервных механизмов управления движениями
269
Клетками двигательной системы являются мотонейроны. Выделяют высшие (центральные) и низшие (периферические) мотонейроны.
Высшие мотонейроны не выходят за пределы ЦНС. Их тела располагаются в коре головного мозга: в двигательной коре, премоторной коре, дополнительных двигательных зонах коры — соматосенсорной и теменной. Аксоны высших мотонейронов направляются в спинной мозг, образуя кортикоспинальный (пирамидный) тракт. Начинаясь в передней центральной извилине, тракт проходит через ножку мозга, варолиев мост, продолговатый мозг. В нижнем отделе продолговатого мозга происходит неполный перекрест пирамидных волокон (рис. 17-1). Перекрещенные аксоны идут в боковые рога спинного мозга, где так же, как и в коре, существует соматотопическое распределение и оканчиваются на вставочных нейронах серого вещества. Неперекрещенные аксоны образуют синаптические контакты с клетками переднего рога контра-латерально, переходя через белую спайку спинного мозга на противоположную сторону. Очень небольшая часть кортикоспинальных волокон у человека образует моносинаптические контакты с мотонейронами дистальных мышц верхних конечностей.
Тела низших мотонейронов располагаются в вентральных рогах спинного мозга и в ядрах черепно-мозговых нервов. Аксоны этих мотонейронов непосредственно контактируют с мышечными волокнами.
Функциональным элементом двигательной системы является двигательная (моторная) единица. Двигательная единица состоит из низшего мотонейрона и группы мышечных волокон, которые этот мотонейрон иннервирует. Чем меньше число волокон в двигательной единице, тем более тонкий контроль осуществляется за движением со стороны нервной системы. Так, моторная единица, которая контролирует глазные мышцы или малые мышцы кисти, содержит от 3 до 6 мышечных волокон. В моторной единице, контролирующей икроножную мышцу, содержится около 2000 мышечных волокон.
Болезни моторных единиц возникают в результате:
• повреждения тел мотонейронов;
• повреждения аксонов мотонейронов, составляющих периферический нерв;
• нарушения передачи возбуждения с окончаний двигательных нервов на мышцу;
• повреждения самой мышцы.
Главные признаки болезней моторных единиц:
• мышечная слабость;
270
Лекция 17
Рис. 17-1. Кортикоспинальный путь
• ослабление рефлексов;
• снижение мышечного тонуса;
• атрофия мышц (без признаков болезней мышц);
• фасцикуляция (мышечные сокращения, обнаруживаемые визуально);
• фибрилляции (асинхронные возбуждения отдельных мышечных волокон, обнаруживаемые с помощью электромиографии).
Патофизиология нервных механизмов управления движениями
271
Одним из примеров болезней моторных единиц является полиомиелит. Это острое повреждение низших мотонейронов, вызываемое вирусом полиомиелита — возбудителем детского спинального паралича. Болеют дети от 2 до 7 лет. Основные пути передачи инфекции — личные контакты и фекальное загрязнение пищи. Инкубационный период — 7-14 дней. У человека наиболее распространенный путь заражения — через пищеварительный тракт. Вирус достигает тел мотонейронов по вегетативным волокнам, распространяясь вдоль осевых цилиндров в периферическом нерве и ЦНС. Считают возможным распространение через кровь и лимфу.
Механизм избирательного поражения низших мотонейронов вирусом полиомиелита не выяснен. Предполагают, что вирус проникает внутрь клеток, соединяясь со специфическими рецепторами, имеющимися только на мембране мотонейрона. Гистологические изменения при полиомиелите наиболее выражены в сером веществе спинного мозга и продолговатом мозге. В мотонейронах передних рогов спинного мозга обнаруживается различная степень дегенерации: от хроматолиза до полной деструкции. Происходит инфильтрация серого вещества лимфоцитами и нейроглией. В пораженных мышцах находят различную степень атрофии с увеличением соединительной и жировой ткани, снижение мышечного тонуса, непроизвольные фибриллярные подергивания — признаки поражения низших мотонейронов. Часты развития вялых параличей с арефлексией.
Для профилактики полиомиелита применяют инъекции инактивированной вакцины или пероральный прием живой ослабленной по-ливирусной вакцины Себина. Живая вакцина дает более длительный иммунитет. Вакцинация превратила полиомиелит из некогда опасной нейроинфекции в спорадическое заболевание.
Лечение: полный покой, так как высок риск параличей, применение сыворотки переболевших, внутримышечное введение рибонуклеазы, анальгетики, седативные препараты, глюкокортикоиды, плазмаферез.
Амиотрофигеский боковой склероз — хроническое прогрессирующее заболевание, приводящее к гибели не только низших, но и высших мотонейронов. Возникает у мужчин после 50 лет.
Этиология не ясна, предполагают, что некоторые вирусы могут быть причиной возникновения болезни, возможны аутоиммунные нарушения, а также генетический дефект 21-й хромосомы, которая кодирует супероксиддисмутазу, защищающую от разрушающего действия свободных радикалов. Нарушается метаболизм глутамата и перекиси во
272
Лекция 17
дорода. Характеризуется прогрессирующей дегенерацией нейронов двигательной коры, ствола мозга, спинного мозга, дегенерацией кортикоспинального тракта, демиелинизацией спинальных корешков. При некоторых вариантах болезни происходит гибель мотонейронов черепно-мозговых нервов — это вызывает расстройство речи (дизартрия), расстройство глотания (дисфагия). Такой вариант болезни носит название — прогрессирующий бульбарный паралиг. По неизвестной причине сохраняются мотонейроны глазодвигательных мышц и мотонейроны, контролирующие сфинктеры мочевого пузыря и прямой кишки. Чувствительные нейроны не страдают. Поражение мотонейронов постепенно вовлекает в дегенерацию мышечные волокна, кроме глазодвигательных и сердечных.
На рисунке 17-2 на срезе спинного мозга видны светлые пятна, означающие дегенерацию мотонейронов в передних рогах, а также дегенерацию проводящих путей в боковых и передних отделах спинного мозга.
Дорсальный корешок
% Вентральный корешок
Скелетная мышца
Атрофированные волокна \
Нормальные волокна
Лимфоциты
Гипертрофированные волокна
Рис. 17-2. Дегенерация мотонейронов в спинном мозге
Патофизиология нервных механизмов управления движениями
273
Рис. 17-3. Вызывание рефлекса Бабинского
Симптом повреждения кортикоспинального тракта
Дегенерации подвергаются вентральные корешки и мышечные волокна. Поскольку при амиотрофическом боковом склерозе погибают и высшие, и низшие мотонейроны, то симптоматика не ограничивается только признаками поражения моторных единиц. Первый симптом болезни — слабость в конечностях — обусловлена атрофией пучков мышечных волокон рук и ног. Денервированные мышцы обнаруживают фас-цикулляцию и фибрилляцию — это признаки повреждения низших мотонейронов. Но наряду с этим обнаруживается гиперрефлексия — повышение сухожильных рефлексов, усиление мышечного тонуса, появление патологических рефлексов. Все это — признаки повреждения высших мотонейронов. Примером патологического рефлекса может служить рефлекс Бабинского (рис. 17-3).
В начале болезни, когда погибает малое число мотонейронов, — возможна компенсация: восстановление иннервации мышц за счет разрастания аксонов переживающих мотонейронов. Лечение — симптоматическое, прогноз — неблагоприятный.
Другим примером нарушений, приводящим к болезням моторных единиц, являются повреждения двигательных аксонов. Эти нарушения могут быть вызваны травмой периферических нервов и сопровождаться утратой произвольных и рефлекторных движений, снижением мышечного тонуса, мышечной атрофией. Поскольку периферические нервы, как правило, смешанные, нарушения движений комбинируются с нарушениями чувствительности (анестезиями).
На рисунке 17-4 представлены последствия перерыва аксона. Если травма далеко от тела нейрона, наблюдается дистальная аксонопатия — дегенерация дистальной части аксона.
274
Лекция 17
Дистальная аксонопатия
Перерыв аксона
Уоллеровское перерождение
Ретроградная дегенерация
Разрастание коллатералей аксона
Рис. 17-4. Последствия перерыва аксона
При гибели тела нейрона развивается нейроаксональная дистрофия. В этом случае травма находится недалеко от тела нейрона и дистрофии подвергается и дистальный, и проксимальный отделы аксона.
Патогенетические механизмы нейропатий изображены на рисунке 17-5 и могут быть сведены к одному или комбинации шести патогенетических механизмов.
Перерыв аксона сопровождается потерей дистального сегмента аксона в результате Уоллеровского перерождения, которое заключается в том, что нервное волокно, отделенное от тела клетки, подвергается нисходящей дегенерации или распаду от места перерыва аксона до синапса включительно. Это явление описал Уоллер в 1850 г., обнаружив, что тип травмы неважен, а характеристика изменений стереотипна у человека и всех видов позвоночных животных.
Патофизиология нервных механизмов управления движениями
275
Ф Ш М А М Ш Ф
Рис. 17-5. Патогенетические механизмы аксонопатий: 1 — ишемия, затруднения тока крови в капиллярах; 2 — накопление продуктов жизнедеятельности в шванновских клетках; 3 — нарушение структуры миелиновой оболочки; 4 -увеличение осмотического давления с набуханием аксона; 5 — блокада продольного и поперечного транспорта; 6 — клеточная инвазия — вторжение — между шванновской клеткой и миелином
Признаки Уоллеровского перерождения аксона:
• отторжение миелина от аксона;
• разрывы (фрагментация) миелина;
• набухание аксона, заполнение его дистальной части вакуолями, нейрофибриллами; фрагментация аксона. Формирование овоидов, содержащих частички аксона, покрытых миелином;
276
Лекция 17
• овоиды фагоцитируются шванновскими клетками, которые активно пролиферируют.
Первые признаки дегенерации наблюдаются уже через 24-48 ч. Полная дегенерация аксона происходит в течение 1-2 мес. После дегенерации аксона соединительнотканная оболочка, окружающая аксон, может сохраниться. На месте волокна остаются пустые шванновские футляры, в которые прорастают регенерирующие аксоны. Регенерация аксона наступает, если тело мотонейрона сохранено и не нарушена связь между телом нейрона и его центральным обрубком. Из проксимального обрубка аксона немиелинизированные отростки растут наугад, формируя нейрому, подобную спруту с щупальцами. Эти фибриллярные отростки растут по периферии миелиновых элипсоидов (овоидов), начиная с 8 сут после травмы. Брешь между обоими концами перерезанного аксона закрывается шванновскими клетками и фибробластами. Когда аксон растет в ошибочно выбранном направлении, пациент чувствует слабость, неконтролируемость движения мышц, снижение чувствительности. Аксоны, случайно попавшие в футляр из шванновских клеток, растут по колее со скоростью 1-4 мм/сут. Растущий демиелинизированный конец регенерирующего аксона гиперчувствителен к механическим стимулам. Это используют в клинической практике — положительный синдром Тиннеля (перкуссия срединного нерва на запястье — парестезия — онемение кисти). Когда аксон растет, шванновские клетки образуют новый миелин, и волокна утолщяются до их диаметра, достигают области концевой пластинки, образуют синаптические контакты и теряют свою гиперчувствительность. Движение восстанавливается.
Аксоны центральных (высших) нейронов не регенерируют (рис. 17-6). В зоне травмы ЦНС аксоны и миелин быстро дегенерируют, накапливаются макрофаги, роль которых выполняет глия. Астроциты и микроглия активно делятся, фагоцитируя продукты распада. Пролиферирующие астроциты создают глиальный рубец, который препятствует регенерации аксона.
Нарушения функции двигательных единиц могут быть обусловлены нарушением синаптигеской передаги между нервом и мышцей и связаны с:
• нарушением высвобождения ацетилхолина (Ацх) из пресинапти-ческих мембран;
• блокадой холинэстеразы — фермента, разрушающего Ацх;
• нарушением взаимодействия Ацх с постсинаптической мембраной.
Патофизиология нервных механизмов управления движениями
277
Рис. 17-6. Астроцит в головном мозге
Рассмотрим примеры, иллюстрирующие эти нарушения.
Ярчайшим примером пресинаптических нарушений является ботулизм — болезнь, вызываемая отравлением ботулиническим токсином — продуктом анаэробных бактерий Cl. botulinum. Ботулинический токсин, как и все токсины, состоит из 2-х полипептидных цепей — тяжелой (100 кДа) и легкой (50 кДа), связанных дисульфидной связью. Тяжелые цепи содержат центры связывания с ганглиозидами пресинаптической мембраны нервных окончаний. Предполагают, что тяжелые цепи, необратимо связавшись с ганглиозидами пресинаптической мембраны, образуют в ней туннели, по которым внутрь пресинаптического окончания может проскользнуть активный фрагмент легкой цепи. Высвобождение квантов Ацх нарушается.
Симптомы отравления появляются через 6-36 ч.
Клинические проявления: тошнота, рвота, диарея, расстройство зрения — парез аккомодации, мышечная слабость. В дальнейшем — парезы и параличи соматической мускулатуры, которые проявляются:
• парезом/параличом мышц, иннервируемых черепно-мозговыми нервами. В результате развиваются диплопия — двоение предметов, дисфагия — расстройство глотания, дисфония и дизартрия — расстройство речи;
• поражение мышц конечностей;
• поражение дыхательных мышц.
В тяжелых случаях — смерть от дыхательной недостаточности.
278
Лекция 17
Эффективное лечение отсутствует, но можно уменьшить выраженность симптомов и оказать поддерживающий эффект. Рекомендуется введение антитоксина, антиботулинической сыворотки, антибиотиков, десенсибилизирующая терапия, кислород.
Другой пример блокады пресинаптического выброса медиатора Ацх — синдром Ламберта-Итона - псевдомиостенический синдром, относящийся к аутоиммунным заболеваниям. Это паранеопластический синдром — в 80 % случаев наблюдается связь с опухолями, чаще всего с овсяноклеточной карциномой легкого. Характеризуется патологическим мышечным утомлением при жевании, глотании, счете, ходьбе, подъеме по лестнице. Установлено, что в крови больных синдромом Ламберта-Итона имеются антитела (IgG) к структурам потенциалзависимых кальциевых каналов на пресинаптической мембране двигательных аксонов. Результатом взаимодействия антител с белками кальциевых каналов является снижение выброса Ацх в ответ на нервный импульс. Считают возможным механизм перекрестной аллергиза-ции, вызванной сходством АГ опухоли и белком кальциевых каналов. Постсинаптические структуры не изменяются (в отличие от миастении). При повторных движениях мышечный ответ увеличивается за счет накопления Ацх в синаптической щели.
Лечение при синдроме Ламберта-Итона включает плазмаферез, иммунодепрессанты, в том числе глюкокортикоиды, ингибиторы холинэстеразы — эзерин, неостигмин, гуанидин гидрохлорид (препарат, увеличивающий число квантов Ацх на одиночный стимул).
Болезни моторных единиц могут быть вызваны нарушениями нервно-мышечной передачи, связанными с блокадой ферментативного расщепления Ацх. Блокада холинэстеразы может быть обусловлена действием фосфорорганических соединений, к числу которых относятся некоторые нервно-паралитические газы (табун, зарин, зоман) и пестициды (хлорофос, тиофос). Все указанные вещества необратимо связываются с холинэстеразой, блокируя ферментативное разрушение Ацх. В результате Ацх накапливается в синаптической щели и появляются симптомы, связанные с чрезмерным возбуждением мускариновых и никотиновых холинорецепторов.
Симптомы, вызываемые возбуждением М-холинорецепторов: миоз (сужение зрачка), бронхоспазм, кардиоспазм, тошнота и рвота, увеличение продукции слизи эпителием бронхов.
Симптомы, вызываемые возбуждением Н-холинорецепторов: фибрилляции — подергивание скелетной мускулатуры и мышц лица,
Патофизиология нервных механизмов управления движениями
279
повышение артериального давления, вызванное возбуждением постганглионарных нейронов.
Фосфорорганические соединения быстро проходят через гематоэнцефалический барьер и вызывают расстройство функций М- и Н-холинорецепторов головного мозга, что проявляется расстройством психики, потерей сознания, судорогами.
Лечение предполагает введение препаратов, разрушающих связь ядов с ацетилхолинэсгеразой (пралидоксин); конкурентно тормозящих мускариновые эффекты нервных ядов. Для профилактики применяют обратимые блокаторы ацетилхолинэстеразы. На рисунке 14-7 представлена схема действия подобных препаратов, защищающих холинэстеразу.
1 2 3
ФОС
Пиридостигминобратимый блокатор холинэстеразы
Пралидоксин разрушает связь ядов с холинэстеразой
Рис. 17-7. Схема действия препаратов, защищающих холинэстеразу
280
Лекция 17
Болезни моторных единиц могут быть обусловлены нарушением взаимодействия Ацх с его рецепторами на постсинаптической мембране.
Механизмы подобных нарушений:
• необратимое связывание с Н-холинорецепторами. Примером может служить действие нейротоксинов яда змей (кобры, крайта).
• конкурентная блокада рецепторов для Ацх. Таков механизм действия кураре и курареподобных веществ. Мышечная клетка становится нечувствительной к импульсам, приходящим от мотонейронов, и к прямой аппликации Ацх, но отвечает на прямую электрическую стимуляцию. D-тубокурарин был первым натуральным агентом, который использовали в хирургической практике. В настоящее время заменен на синтетические аналоги — курареподобные вещества, имеющие большую силу, но меньшую продолжительность действия:
• аутоиммунное разрушение ацетилхолиновых рецепторов, что характерно для тяжелой миастении.
Тяжелая миастения — аутоиммунная приобретенная болезнь, которая обнаруживается у людей разного возраста. Связана с появлением иммуноглобулинов (IgG) к рецепторам Ацх и, возможно, к Т-лимфоцитам. В 75 % случаев у больных обнаруживается тимома. Выявлено, что тимус содержит миоидные, или мышцеподобные, клетки, которые обладают гистологическими характеристиками скелетных мышц и несут Ацх-рецепторы. Предполагается, что эти клетки играют роль АГ, которые сенсибилизируют лимфоциты к Ацх-рецепторам. Взаимодействие антител с ацетилхолиновыми рецепторами приводит к образованию крупных структур, которые втягиваются внутрь мышечной клетки через постсинаптическую мембрану путем эндоцитоза и разрушаются внутри клетки с помощью лизосомальных ферментов. Снижение числа Ацх-рецепторов на постсинаптической мембране скелетных мышц сочетается со снижением числа крипт на постсинаптической мембране и расширением синаптической щели (рис. 17-8,17-9).
Клинически это проявляется патологической мышечной слабостью, затруднениями при глотании, жевании. Слабость мышц, поднимающих веки, приводит к развитию птоза, слабость экстраокулярных мышц — к диплопии.
Лечение: удаление тимуса дает длительную ремиссию. Облегчения мышечной слабости достигается применением блокаторов ацетилхо-линэстеразы (эзерин, неостигмин, пиридостигмин). Так же как при лечении других аутоиммунных болезней, используют плазмаферез, иммунодепрессанты, кортикостероиды.
Патофизиология нервных механизмов управления движениями
281
Норма
Тяжелая миастения
Рис. 17-8. Нервно-мышечный синапс при тяжелой миастении
складки
Норма
Миограмма (МГ)
МГ после неостигмина
Рис. 17-9. Электромиограмма больного тяжелой миастенией
Патология моторных единиц может быть обусловлена собственно болезнями мышц — миопатиями, которые классифицируют как врожденные и приобретенные.
Примером наследственной патологии может служить мышегная дистрофия Дюшенна. Болезнь обнаруживается у мальчиков в возрасте от 2-х до 5 лет. Наследуется по рецессивному типу, сцеплена с Х-хромосомой. Существует несколько теорий происхождения болезни: нейрогенная, сосудистая, мембранная. Наиболее распространено мнение о генетической патологии мышечной мембраны — отсутствие белка — дистрофина. Проявляется мышечной слабостью, которая поражает прежде всего мышцы обеих ног, а затем и другие мышцы скелета. Появляются затруднения в беге, прыжках, подъеме по лестнице. Мышечная ткань замещается жировой, наблюдается псевдогипертрофия мышц. Постепенно процесс поднимается вверх, захватывая плечевой пояс, сердечную мышцу и дыхательные мышцы. Специфического лечения мышечных дистрофий нет.
282
Лекция 17
Примером приобретенной миопатии может служить дерматомиозит. Полимиозит, встречающийся у взрослых и подростков, сопровождается воспалительными изменениями в коже — покраснением кожи верхней части туловища, лица, кожи над суставами.
Причинным фактором могут быть вирусная инфекция, вакцинация, лечение определенными препаратами, опухоли — все, что может инициировать иммунный ответ.
Патогенез неизвестен, но полагают, что повреждение мышц вызвано иммунными процессами. Гистологически обнаруживают различную степень мышечной дистрофии и лимфоцитарную инфильтрацию мышц. Дерматомиозит — одно из немногих вылечиваемых заболеваний мышц.
При лечении применяют иммуносупрессоры, плазмаферез, противовоспалительные препараты.
Повреждение спинного мозга — одного из компонентов двигательной системы — приводит к тяжелым расстройствам движений, поскольку в спинном мозге располагаются все периферические (низшие) мотонейроны, управляющие активностью мышц туловища и конечностей. Спинной мозг является центром сбора и обработки сенсорной информации, поступающей от мышц, сухожилий, поверхности тела и внутренних органов, поэтому нарушения движений, вызванные болезнями спинного мозга, часто сочетаются с расстройствами чувствительности и вегетативными расстройствами.
Наиболее частыми причинами спинальных травм являются дорожные аварии, падения с высоты, спортивные травмы.
Неврологический синдром, возникающий немедленно после полной поперечной перерезки (разрыва) спинного мозга, называют спинальным шоком. Спинальный шок — полная утрата рефлекторной активности сегментов, расположенных каудальнее места травмы. Отсутствуют рефлексы соматических мышц и вегетативные рефлексы. Глубина и длительность спинального шока зависят от степени преобладания головного мозга над спинным. У человека стадия арефлексии продолжается 6-7 дней. Арефлексия — результат утраты возбуждающих влияний на мотонейроны вышележащих отделов мозга, в том числе и головного, но не результат травмы спинальных рефлекторных путей. Стадия арефлексии спинального шока сменяется постепенным восстановлением рефлексов, а затем и их усилением. Усиление рефлексов — гиперрефлексия — может быть результатом разрастания окончаний первичных афферентных волокон внутри спинного мозга.
Патофизиология нервных механизмов управлениядвижениями
283
Повреждение низших мотонейронов на различных уровнях может привести к периферигеским паралигам.
Симптомы периферического паралича: полная или частичная потеря произвольных движений, мышечная слабость, атрофия мышц, атония мышц, арефлексия.
Поражение высшего (центрального) мотонейрона — нейрона моторной коры головного мозга и кортикоспинального тракта, составленного аксонами центральных мотонейронов, на любом уровне приводит к развитию центрального паралига.
Симптомы центрального паралича: гиперрефлексия на стороне паралича, усиление сухожильных рефлексов, появление патологических рефлексов, гипертонус мышц на стороне паралича, снижение кожных и брюшных рефлексов на стороне паралича.
Роль базальных ганглиев и мозжечка, контролирующих взаимодействие 4 главных компонентов двигательной системы
Базальные ганглии входят в экстрапирамидную систему ЦНС и получают афферентные сигналы от различных отделов коры головного мозга. Вся приходящая к базальным ганглиям информация поступает в стриатум, все исходящие из базальных ганглиев сигналы передаются к ядрам таламуса через внутреннюю часть бледного шара и черную субстанцию. Связи базальных ганглиев и мозжечка с компонентами двигательной системы представлены на рисунке 17-10.
Контроль за двигательной функцией организма осуществляется благодаря наличию обратной связи между компонентами двигательной системы и базальными ядрами.
Существуют 2 пути передачи информации от стриатума к спинному мозгу:
• нигроретикулоспинальный путь;
• стриопалидоруброспинальный путь.
Нарушение 1-го пути приводит к появлению ригидности, акинезий, тремора в покое.
Нарушения 2-го пути приводит к гиперкинезам и мышегной дистонии. Повреждения базальных ганглиев и расстройства экстрапирамидной системы приводят к нарушению движений, которые проявляются следующими симптомами: появлением акинезий — обеднением движений, нарушениями позы, равновесия, появлением непроизвольных движений (тремор, атетоз, баллизм, хорея).
284
Лекция 17
Двигательная область коры поля 4, 6
Экстрапирамидная система
Средний мозг
Лобная область коры
Корково-паллидарные волокна
Мозжечок
Олива
К спинному мозгу
Зубчатое ядро
Рис. 17-10. Экстрапирамидная система центральной нервной системы
Бледный шар Скорлупа
Хвостатое ядро Таламус
Субталамическое ядро Красное ядро
Черное вещество
Характерными примерами патологии базальных ганглиев являются хорея Гентингтона и болезнь/синдром Паркинсона.
Хорея Гентингтона — неуклонно прогрессирующее дегенеративное наследственное заболевание (аутосомно-доминантный тип наследования). Клинические проявления: начало заболевания в зрелом возрасте. Проявляется хореическим гиперкинезом, баллизмом, атетозом, слабоумием. В основе болезни Гентингтона лежит дегенерация внутристриарных и корковых холинергических нейронов, а также ГАМК-эргических нейронов. Снижается содержание в стриатуме ацетилхолинтрансферазы и тормозного медиатора — у-аминомасляной кислоты (ГАМК), динорфина, энкефалина, вещества Р вместе с рецепторами. Уменьшение содержания ГАМК приводит к активации дофаминергической — нигростриарной системы — снимается контроль за содружественными движениями. Специфического лечения нет. Прогноз неблагоприятный. Для подавления гиперкинеза назначают антагонисты дофамина в сочетании с транквилизаторами.
Патофизиология нервных механизмов управления движениями
285
Болезнь Паркинсона — ажиотированный паралич — характеризуется ритмическим тремором в покое рук, головы, затем туловища. Усиление мышечного тонуса — ригидность мышц — приводит к появлению своеобразной позы просителя: голова, туловище наклонены вперед, пальцы, руки, ноги — в согнутом положении. Невозможность быстро начать и быстро закончить начатое движение, медленное выполнение самих движений — это проявления акинезий и брадикинезий. Исследования базальных ганглиев больных болезнью Паркинсона показали снижение содержания дофамина в стриатуме, что обусловлено гибелью дофаминергических нейронов, лежащих в компактной части черной субстанции. Окончания аксонов этих нейронов, высвобождающих дофамин, располагаются на холинергических нейронах в стриатуме. Конечным результатом гибели этих нейронов является ослабление тормозного влияния на холинергические нейроны стриатума, что вызывает тремор. Одновременно происходит усиление тормозящих влияний на релейные нейроны таламуса, передающие информацию к коре головного мозга. Постоянное торможение таламокортикальных нейронов снижает активность моторных полей, участвующих в управлении движениями.
Лечение заключается во введении L-ДОФА (левовращающий изомер дигидрооксифенилаланина), предшественника дофамина, который легко проходит через гематоэнцефалический барьер, улавливается сохранившимися нейронами черного вещества, что усиливает продукцию этими нейронами дофамина и восстанавливает нигростриарные связи. Делаются лабораторные и клинические попытки хирургического лечения болезни Паркинсона путем пересадки мозгового вещества надпочечников и даже кусочков черного вещества плода в головной мозг (рис. 17-11).
Рис. 17-11. Схема взаимодействия базальных ганглиев
286
Лекция 17
Другой структурой, контролирующей двигательную систему, является мозжегок. Последствия расстройства функций мозжечка зависят от локализации повреждения.
Вестибулоцеребеллум — самая древняя часть мозжечка, повреждение которой ведет к нарушению равновесия при стоянии — астазия, при ходьбе — абазия.
Сигнал к началу
Норма
Патология
Рис. 17-12. Нарушения при патологии мозжечка
Дисдиадохокинез
Патофизиология нервных механизмов управления движениями 287
Спиноцеребеллум получает информацию от спинного мозга и сенсорной зоны коры, имеет топографическую карту тела. При его повреждении нарушается координация в работе антагонистических мышц — асинергия, возникает дисметрия — отсутствие меры в движениях, адиа-дохокинез — невозможность быстро выполнять чередующиеся противоположно направленные движения.
Повреждения срединных отделов мозжечка — червя и прилегающих к нему областей коры — вызывают нарушения походки, связанные с нарушением координации сокращений мышц нижних конечностей (рис. 17-12). Такие повреждения встречаются у лиц, злоупотребляющих алкоголем.
Цереброцеребеллум — 2 полушария, каждое из которых имеет по 4 ядра, имеющих выходы к таламусу, моторной и премоторной коре головного мозга. Специфической функцией являются планирование и начало движения, контролирует прямохождение.
Лекция 18
ПАТОФИЗИОЛОГИЯ БОЛИ
Т.Ю. Ругинская
Боль — социальная проблема. Поданным ВОЗ, ежедневно до 3,5 млн человек страдают от боли, причем у 50 % — боль умеренная, у 30 % — непереносимая, 50-80 % больных онкологическими заболеваниями не получают удовлетворительного облегчения боли. Проблема боли волновала врачей с древности. На известном рисунке Декарта (1596-1650) изображен коленопреклоненный человек, ощущающий от ожога жгучую боль, поднимающуюся к мозгу (рис. 18-1). К настоящему времени существует несколько точек зрения на проблему боли. Прежде всего обсуждается вопрос: боль — это физиологическая или патологическая реакция организма? Большинство исследователей считают — патологическая, так как она связана с болезнями; другие считают боль нормальным физиологическим защитным механизмом. Известно, что люди.
Рис. 18-1. Коленопреклоненный человек, испытывающий боль от ожога (Декарт)
Патофизиология боли
289
лишенные чувства боли в результате травмы, инфекционного поражения или врожденной патологии, имеют более короткую продолжительность жизни, по сравнению с людьми, ощущающими боль.
Международная ассоциация по изучению боли предложила следующее определение.
Боль — это неприятное сенсорное переживание, эмоциональное сигнальное чувство и переживание, связанное с повреждением ткани либо с воздействием потенциально повреждающих факторов или описываемых в терминах повреждения.
Боль — это сигнальное чувство, дающее информацию о повреждении и о моментах, способных вызвать повреждение. Боль может возникнуть до того, как возникнет повреждение. Например: при исследовании температурной чувствительности кожи человека порог болевой чувствительности для большинства обследуемых пациентов составлял 45 °C, повреждения кожи не было. Но если длительно воздействовать такой температурой, то можно вызвать ожог. Повреждение будет связано с денатурацией белка, которая начинается при этой температуре.
Чувство боли отличается ©т других чувств тем, что не несет.информа-ции об окружающем мире и всегда имеет отрицательную эмоциональную окраску.
Ощущение боли описывается пациентом в терминах повреждения. Больной жалуется на боль: жжет, колет, режет, разрывает, стреляет, но врач не видит повреждений, соответствующих этим описаниям. Ощущения боли — субъективные переживания, которые зависят от степени тренировки в восприятии болевых раздражений. Однако реакция на боль приводит к объективным изменениям в организме. При этом повышается артериальное давление, увеличивается частота дыхания и сердечных сокращений, усиливается потоотделение, расширяются зрачки, увеличивается концентрация глюкозы в крови и снижается секреция желез желудочно-кишечного тракта (ЖКТ).
Классификация боли
Выделяют физиологическую и патологическую боль.
Физиологигеская боль возникает как адекватная реакция нервной системы на опасные для организма ситуации. В этих случаях боль выступает как фактор, предупреждающий о процессах, потенциально опасных для организма. Патологическая боль возникает при повреждении нервной системы, воспалениях различных органов, переломах костей, травмах.
290
Лекция 18
По временному аспекту различают транзиторную, острую и хроническую боль.
Транзиторная боль провоцируется активацией болевых рецепторов кожи или других тканей при отсутствии значимого повреждения. Предполагают, что функция такой боли заключается в защите человека от угрозы физического повреждения факторами внешней среды в форме своеобразного обучения, т.е. это физиологическая боль.
Острая боль длится менее 6 мес. Она часто совмещается с вегетативными реакциями «борьбы или бегства», вызванными активацией симпатической нервной системы и сигнализирует о повреждении ткани. Острую боль подразделяют на первичную острую локализованную боль и вторичную нелокализованную боль. Например: при уколе, порезе, действии тока человеком или животным определяется вначале место повреждения и боли (локализованная первичная боль). В последующем образуется большое количество медиаторов воспаления, что приводит к формированию жгучей нелокализованной боли. По локализации, этиологии и патогенезу острую боль разделяют на поверхностную, глубокую, висцеральную и отраженную.
Хронигеская боль рассматривается Международной ассоциацией по изучению боли как «боль, которая продолжается сверх нормального периода заживления». В клинике хронической считают боль, продолжительность которой свыше 6 мес.
По патофизиологической классификации боли, отражающей механизмы ее формирования, выделяют ноцицептивную боль, нейрогенную (периферическую и центральную) и психогенную.
Ноцицептивная боль возникает при любом повреждении ткани, вызывающем возбуждение периферических болевых рецепторов и соматических или висцеральных афферентных волокон.
Нейрогенная боль обусловлена повреждением или изменением состояния периферических и/или центральных отделов нервной системы.
Психогенные боли возникают при отсутствии какого-либо органического повреждения. Любое хроническое заболевание или недомогание, сопровождающееся болью, влияет на эмоции и поведение человека, ведет к появлению тревожности и напряженности, которые сами усиливают восприятие боли. Психофизиологические (психосоматические) механизмы, воздействуя через кору, изменяют состояние внутренних органов, стимулируют выделение вызывающих боль веществ и активацию болевых рецепторов. Возникающая боль, в свою очередь, усиливает эмоциональные нарушения, замыкая порочный круг.
Патофизиология боли
291
Боль — это рефлекторный процесс, который включает все основные звенья рефлекторной дуги: рецепторный аппарат, болевые проводники, образования спинного и головного мозга, а также медиаторы, осуществляющие передачу болевых импульсов.
Болевые рецепторы. Английский физиолог Ч. Шеррингтон (1906) впервые высказал предположение о наличии специальных рецепторов для восприятия боли. Болевые рецепторы (ноцицепторы) — свободно расположенные окончания наиболее тонких миелинизированных афферентных волокон группы А-дельта и немиелинизированных афферентных волокон группы С.
Ноцицептивные окончания афферентных волокон группы А-дельта активируются сильными механическими и термическими стимулами (выше +45 °C и ниже -10 °C). Окончания афферентных волокон группы С возбуждаются сильными механическими, термическими стимулами, а также химическими веществами, образующимися в очаге повреждения. Эти окончания называются полимодальными ноцицепторами. Химические вещества, возбуждающие ноцицепторы, называют алгезирующими агентами. К ним относятся гистамин, брадикинин, соматокинин, холецистокинин, серотонин, субстанция Р, ионы калия и водорода, аденозин. Простагландины алгезирующим действием не обладают, но повышают чувствительность ноцицепторов к другим воздействиям, в частности к брадикинину.
Ацетилсалициловая кислота (аспирин) — блокатор циклооксигеназы — фермента, участвующего в синтезе простагландинов, — оказывает слабое аналгезирующее действие. Характерным свойством ноцицепторов является их способность сенситизироваться, т.е. повышать свою чувствительность при длительном действии алгезирующего агента. Другое свойство — наличие «спящих», молчащих ноцицепторов, которые обнаруживают себя при хроническом воспалении. Это явление объясняют увеличением числа рецепторов по мере развития воспаления вследствие разрастания аксонов, дающих новые коллатерали, в окончаниях которых выделяется алгезирующее вещество Р (рис. 18-2).
Периферические проводники болевой чувствительности — афферентные волокна группы А-дельта и группы С — являются периферическими отростками нейронов, тела которых располагаются в спинальных ганглиях. Афферентные волокна группы А-дельта имеют диаметр от 1 до 4 мкм и проводят импульсы со скоростью 5-30 м/с. Центральные аксоны этих нейронов входят в спинной мозг в составе дорсальных корешков и образуют синаптические контакты с нейронами дорсального рога, расположенными в I и II пластинах по Рекседу. Активация волокон
292
Лекция 18
Рис. 18-2. Адгезирующие агенты, вызывающие раздражение болевых рецепторов
группы А-дельта сопровождается ощущением острой, хорошо локализованной боли. Афферентные волокна группы С имеют диаметр 0,2-1,0 мкм и проводят импульсы со скоростью 0,5-2,0 м/с. Центральные аксоны этих нейронов оканчиваются на нейронах основания дорсального рога в V-VII пластинах по Рекседу. Возбуждение этих волокон сопровождается возникновением плохо локализованной жгучей боли. В окончаниях С-волокон, как на периферии, так и в спинном мозге, выделяются нейромедиаторы двух типов: возбуждающие аминокислоты (глютаминовая, аспарагиновая) и нейропептиды (вещество Р и др.). С помощью этих медиаторов передается болевая информация к нейронам дорзального рога, которые являются вторичными нейронами в пути болевой чувствительности. Например, при уколе или щипке ладони на первые болевые стимулы выделяется глютаминовая кислота и регистрируется вспышка импульсов в шейном отделе спинного мозга. Если развивается воспаление (флегмона), то выделяется вещество Р, и в нейронах дорсального рога регистрируется длительная импульсация. Вещество Р — возбуждающий медиатор, полипептид, состоящий из 11 аминокислот, является важнейшим медиатором боли. Вещество Р обнаружено в задних рогах спинного мозга, преоптической области гипоталамуса, коре головного мозга, т.е. на всех участках релейной передачи болевой информации. Субстанция Р выделяется в окончаниях С-волокон и на артериолах и в коже, вызывая дегрануляцию тучных клеток и участвуя в аксон-рефлексе при развитии воспалительной гиперемии. Эффекты
Патофизиология боли
293
вещества Р могут быть заблокированы морфином. Кроме вещества Р и глютаминовой кислоты, вторичные нейроны в пути болевой чувствительности возбуждают холецистокинины и нейротензин, тормозящий эффект оказывают ГАМК, дофамин, энкефалин, серотонин и др.
Пути проведения болевой чувствительности. Вторичные ноцицептивные нейроны, расположенные в 1-2-й и 5-7-й пластинах по Рекседу, имеют очень длинные аксоны, которые переходят на противоположную сторону в составе передней комиссуры и поднимаются к таламусу, формируя спиноталамический тракт (СТТ). СТТ проходит в переднебоковом канатике белого вещества спинного мозга. Внутри СТТ разделяется на две части: новый — неоспиноталамический тракт, расположенный латерально, и старый — па-леоспиноталамический тракт, расположенный медиально.
Неоспиноталамигеский тракт начинается от 1-й пластины по Рекседу. Нервные волокна этого пути поступают в вентропастеролатераль-ное ядро таламуса, нейроны которого проецируются в соматосенсорные зоны коры (S-1 и S-2). Так как в этом вентробазальном комплексе таламуса сходится афферентация тактильная, суставная, мышечная и болевая, формируется локализованная болевая чувствительность, т.е. определяется локализация ноцицептивного раздражителя. Это подтверждается тем, что повреждение вентропастеролатерального ядра приводит к нарушению чувствительности и одновременно наблюдается кратковременная аналгезия (обезболивание) (рис. 18-3). Электрическая стимуляция этой зоны вызывает аналгезию и локальную парестезию.
Соматосенсорные
Неоспино-
Спинальный ганглий таламический
тракт
Рис. 18-3. Схема пути проведения острой локализованной боли
294 Лекция 18
Палеоспиноталамигеский тракт начинается от 5-й пластины по Рекседу. Большая часть волокон первоначально оканчивается на нейронах ретикулярной формации ствола мозга, которые посылают свои аксоны в задние ядра таламуса, а также в лимбическую кору — центр эмоций. Другие волокна палеоспиноталамического тракта оканчиваются на нейронах, расположенных в гигантоклеточном ядре, в ядрах шва, околоводопроводном сером веществе, клиновидном ядре, а также в тех нейронах таламуса, которые имеют связь с лимбическими образованиями и соматосенсорной корой больших полушарий (рис. 18-4,18-5). Этот путь принимает участие в передаче информации и формировании ощущения тупой плохо локализованной боли с участием реакций со стороны эндокринной, сердечно-сосудистой и дыхательной систем. Активация этого пути ответственна за мотивационные компоненты боли и защитные реакции. Повреждение заднего таламуса у человека и животных уменьшает болевую чувствительность. Электрическая стимуляция этих ядер вызывает ощущение жгучей боли.
Модуляция боли. Восприятие боли может изменяться под действием лекарств, болевых и неболевых раздражений, эмоциональных переживаний, стрессов. Такая изменчивость обусловлена существованием специальных модулирующих боль механизмов, которые действуют на всех уровнях переключения в путях передачи ноцицептивной чувствительности. В 1970-х годах было сформулировано положение об антиноцицептивной системе в ЦНС. Было обнаружено, что боль мож-
Рис. 18-4. Схема пути плохо локализованной боли
Патофизиология боли
295
Рис. 18-5. Проводящие ноцицептивные пути
но подавить, раздражая область среднего мозга вокруг сильвиевого водопровода (ОВСВ). Раздражение этой области у человека и животных вызывает сильную аналгезию, которая сохраняется длительное время после прекращения раздражения. Подобный эффект обнаруживается и при электрической стимуляции нейронов ядер шва, расположенных в продолговатом мозге. Стимуляционную аналгезию, вызванную раз
296
Лекция 18
дражением околоводопроводного серого вещества, можно заменить, вводя в эту область морфин, т.е. были выявлены нейроны, имеющие рецепторы к морфию (эти рецепторы назвали опиатными рецепторами). Наибольшее количество опиатных рецепторов найдено в желатинозной субстанции спинного мозга, ретикулярной формации, ОВСВ, лимбических структурах, гипоталамусе, коре мозга и мембране периферических окончаний ноцицептивных миелинизированных и немиелинизиро-ванных волокон. Рецепторы на мембране периферических окончаний синтезируются внутри тел чувствительных нейронов, расположенных в спинальных ганглиях, транспортируются с током аксоплазмы в окончания периферических и центральных отростков этих нейронов, где и экспрессируются на мембране. Распределение опиатных рецепторов соответствует местам, стимуляция которых вызывает аналгезию. Налоксон — специфический антагонист морфина — блокирует не только морфинную, но и стимуляционную аналгезию. В настоящее время выделяют следующие субклассы опиатных рецепторов: мю-рецепторы, К-рецепторы, эпсилон-рецепторы, дельта-рецепторы, причем мю-рецепторы преобладают в местах, связанных с ноцицепцией, а дельтарецепторы превалируют в лимбической системе. Сходный эффект между стимуляционной аналгезией и аналгезией, вызванной морфином, объясняется тем, что при раздражении ОВСВ высвобождаются эндогенные вещества, воздействующие через опиатные рецепторы на те же нейроны, что и экзогенный морфий. Эти вещества — эндогенные опиаты или опиоидные пептиды — взаимодействуют с опиатными рецепторами. Известно три семейства эндогенных опиоидных пептидов: энкефалины, эндорфины и динорфины, каждое из которых образуется из более крупных белков-предшественников. Из проэнкефалина синтезируются такие олигопептиды, как метэнкефалин и лейэнкефалин, состоящие из 5 аминокислот: тирозин-глицин-глицин-фенилаланин-метионин и тирозин-глицин-глицин-фенилаланин-лейцин соответственно. В ЦНС содержание метэнкефалина превышает концентрацию лейэнкефали-на приблизительно в 10 раз. Предшественником эндорфинов является проопиомеланокортин — большой линейный белок, который выделяется в кровь из гипофиза в ответ на стресс. Из проопиомеланокортина образуются АКТГ, меланостимулирующий гормон и бета-липотропин, из которого образуется 0-эндорфин. 0-Эндорфин в 48 раз сильнее морфина при введении в желудочки мозга и 3 раза сильнее морфина при внутривенном введении. При стрессовых ситуациях (страх, психический стресс, роды, переохлаждение) одновремено с АКТГ образуются
Патофизиология боли
297
эндорфины. Таким образом происходит инъекция опиатов, для того чтобы боль не отвлекала от адаптивной реакции. Динорфин образуется из продинорфина, который может образовываться из проэнкефалина и состоит из 13 аминокислот: тирозин-глицин-глицин-фенилаланин-лейцин-аргинин-аргинин-изолейцин-аргинин-пролин-лизин-лейцин-лизин. Динорфин в 200 раз сильнее морфина. Эндогенные опиаты синтезируются в дорсальных рогах спинного мозга, продолговатом мозге, ОВСВ, гипоталамусе, гипофизе, надпочечниках, клетках плаценты. Все эндогенные опиаты меняют восприятие боли в результате влияния и на эмоциональную кору. Действие этих веществ кратковременное.
Патогенетические механизмы антиноцицептивных влияний
Опиатные механизмы обезболивания (рис. 18-6). Тормозящее действие ОВСВ на нейроны спинного мозга опосредуется нейронами ядер продолговатого мозга — большим ядром шва (nucleus raphe magnus) и голубоватым пятном (lokus ceruleus). В ядре шва и в ОВСВ располагаются серотонинергические нейроны, аксоны которых направляются в дорсальные рога спинного мозга, где оканчиваются на короткоаксон-
Околоводопроводное
серое вещество (ОВСВ)
Ядра шва
Спинной мозг
Рис. 18-6. Эндогенные механизмы обезболивания
298
Лекция 18
Рис. 18-7. Механизмы пресинаптического торможения при активации антино-цицептивной опиатной системы
ных нейронах желатинозной субстанции и возбуждают их. Эти нейроны являются энкефалинергическими, т.е. из их окончаний выделяется эндогенный опиат — энкефалин, вызывающий деполяризацию преси-наптических окончаний центральных волокон первичных ноцицептивных нейронов. Возникает пресинаптическое торможение в пути болевой чувствительности, и болевой стимул не поступает к вторичным ноцицептивным нейронам (рис. 18-7).
Адренергигеские механизмы обезболивания. Нисходящие аксоны норадренергических нейронов голубоватого пятна образуют синаптические контакты с вторичными ноцицептивными нейронами и вызывают постсинаптическое торможение этих нейронов и блокаду пути распространения болевой чувствительности.
Холинергигеские механизмы обезболивания связаны с опиатной ан-тиноцицептивной системой. Введение прозерина (холиномиметик) в ОВСВ усиливает обезболивающий эффект, что является результатом вовлечения ацетилхолина в реакцию обезболивания на уровне среднего мозга. Предполагают, что ацетилхолин связывается с центральными мускариновыми рецепторами и стимулирует высвобождение эндогенных опиоидных пептидов.
ГАМК-эргигеские механизмы обезболивания. ГАМК — тормозной медиатор, регулирует болевую чувствительность, подавляя поведенческие реакции на боль. Обнаружено, что болевое воздействие сопровождается повышением концентрации ГАМК и снижением концентрации ГАМК-трансферазы в структурах переднего мозга. Это приводит к усилению
Патофизиология боли 299
процессов торможения в головном мозге и действует как защитный механизм.
Нарушения болевой чувствительности
Снижение болевой чувствительности, или гипалгезия (аналгезия), обнаруживается при нарушении путей проведения болевой чувствительности. Например: при перерыве периферического нерва повреждаются аксоны первичных ноцицептивных нейронов — болевая информация не поступает в спинной мозг. Причиной избирательной аналгезии могут быть травмы переднебоковых отделов спинного мозга, где располагаются волокна спиноталамического тракта, проводящего болевую чувствительность.
Другим примером гипалгезии является сирингомиелия - заболевание, которое характеризуется образованием полостей (кист) и разрастанием глии в сером веществе шейного и верхнегрудного отделах спинного мозга. При этом гибнут волокна белой комиссуры, где перекрещиваются аксоны вторичных температурных и ноцицептивных нейронов, что приводит к двусторонней потере болевой и температурной чувствительности. Тактильная и проприоцептивная чувствительность сохраняется, так как проводники этих видов чувствительности располагаются в белом веществе дорсальных канатиков спинного мозга и не повреждаются. Этиология: травмы боковых столбов спинного мозга, опухоли, пороки эмбрионального развития спинного мозга с дефектом заращения шва в месте смыкания двух половин медуллярной трубки, т.е. незаращение центрального канала, грыжи межпозвоночных дисков и гидродинамические нарушения, связанные с отсутствием или обтурацией отверстий Мажанди. В настоящее время ведущей патогенетической концепцией является теория W.J. Gardner (1950), экспериментально подтвержденная В. Williams (1969). Эта теория объясняет, что затруднение оттока ликвора из большой затылочной цистерны в спинальное субарахноидальное пространство приводит к гидродинамическим ударам ликворной волны из 4-го желудочка в стенки центрального канала спинного мозга с последующим его расширением и образованием сирингомиелических полостей. Патоморфологически установлено, что эти полости неправильной формы, располагаются вокруг центрального канала спинного мозга и окружены глиальным валиком. Заболевание начинается в 18-40 лет снижением или утратой болевой и температурной чувствительности на руках, что ведет к травмам и ожогам. В руке, плечевом поясе отмечаются жжение, парестезии, боль. Двигательные расстройства про
300
Лекция 18
являются атрофическими парезами мышц верхних конечностей, прежде всего мышц кисти (кисть приобретает вид когтистой лапы), вследствие повреждения передних рогов спинного мозга. Прогрессирование болезни ведет к сдавлению боковых и задних канатиков, т.е. к сегментарным расстройствам присоединяются проводниковые.
Усиление болевой чувствительности, или гипералгезия, является усиленным ответом на нормальный болевой стимул. Пшералгезию разделяют на первичную и вторичную.
Первигная гипералгезия обнаруживается в зоне гиперемии, окружающей место повреждения тканей, т.е. имеет четкую локализацию и характеризуется повышением возбудимости ноцицепторов (порог возбудимости снижается) в результате появления биологически активных веществ в очаге воспаления. Эти вещества включают гистамин, серотонин, субстанцию Р, брадикинин, продукты метаболизма арахидоновой кислоты, цитокины и др. Повышение возбудимости ноцицепторов объясняют появлением «спящих» ноцицепторов и повышением их возбудимости, т.е. периферической сенситизацией. Примером может быть первичная гипералгезия кожи после солнечного загара — даже легкое прикосновение вызывает боль.
Вторигная гипералгезия обнаруживается за пределами зоны гиперемии, в местах, удаленных от очага повреждения. Она отождествляется с отраженными болями и не связана с повышением возбудимости ноцицепторов. В ЦНС увеличивается афферентная импульсация от сенси-тизированных и вновь активированных «спящих» ноцицепторов, что ведет к избыточному высвобождению активирующих аминокислот и пептидов в дорсальных рогах спинного мозга и повышению возбудимости вторичных ноцицептивных нейронов. В результате происходит расширение периферической зоны гипералгезии. Первоначально неэффективная, подпороговая афферентация из тканей становится надпороговой из-за увеличения возбудимости вторичных ноцицептивных нейронов — это относится к понятию «центральная сенситизация». Периферическая и центральная сенситизация при хронических болевых состояниях сосуществуют независимо друг от друга и могут быть блокированы отдельно одна от другой.
Некоторые болевые синдромы
Патологическая боль периферического происхождения
Боль в регенерирующем нерве. Постоянные мучительные боли обнаруживаются при регенерации нерва и врастании центральной культи
Патофизиология боли
301
аксона к мышечному волокну, которое нерв иннервировал до пересечения (травмы). К регенерации способны только те волокна, которые сохранили связь с телом нейрона. На конце центральной культи регенерирующего нерва возникает неврома — патологически ветвящиеся и переплетающиеся между собой веточки регенерирующих чувствительных, двигательных и симпатических волокон, обладающих повышенной возбудимостью. Поэтому невромы становятся источником интенсивной импульсации ноцицептивных волокон, которая возникает под действием гистамина, брадикинина, адреналина, а также под влиянием механических стимулов. Гистамин является сильным раздражителем для регенерирующего аксона. Это свойство используется в медицинской практике для определения скорости прорастания и места расположения культи аксона. Пациент чувствует боль при легком поколачивании по проекции нерва на коже (симптом Тинеля). Патогенез этого явления: поколачивание -» дегрануляция тучных клеток -» выделение гистамина -»раздражение регенерирующих волокон -»боль. Повышенная возбудимость регенерирующих волокон может исчезать при образовании синаптического контакта с мышцей.
Каузалгия — интенсивная, жгучая, непереносимая боль, возникающая при воздействии неболевых раздражителей, которые могут быть контактными (тактильными или температурными) или дистанционными (свет, звук). Ощущения сильной боли сохраняются после прекращения болевой стимуляции. Каузалгия возникает после ранений крупных нервов (большеберцового, срединного), богатых симпатическими волокнами, и после ранения ткани рядом с нервным стволом. При этом возникает деформация нерва, связанная с его частичной демиелинизацией. Демиелинизированный участок нерва образует подобие синаптического контакта с эфферентным симпатическим волокном — эфакс. Возникает подобие короткого замыкания. В месте эфаптического контакта из симпатических волокон выделяются адреналин и/или норадреналин, которые способны возбуждать ноцицептивные волокна прямо или через вазомоторную систему, вызывая сокращения гладких мышц, путем высвобождения адгезирующих веществ.
Для исключения других патологических состояний, вызывающих боль, используют следующий прием: при каузалгии боль усиливается при погружении пораженной конечности в холодную воду.
Лечат каузалгию хирургически: иссекая симпатические волокна, иногда — симпатическую цепочку, фармакологически: блокадой паравертебральных симпатических ганглиев, кортикостероидами, клони-
302
Лекция 18
дином, который, снижая активность симпатической нервной системы, облегчает боль.
Фантомные боли — боли в несуществующих органах тела, появляющиеся почти у всех людей, перенесших ампутацию органа. Фантом — ощущение присутствия отсутствующего органа. В возникновении фантомных болей участвуют два механизма: периферический и центральный.
Периферический связан с повышенной возбудимостью регенерирующего нерва. Переплетения регенерирующих аксонов, которым некуда прорастать, формируют неврому и вызывают ощущение боли.
Центральный механизм — память предыдущего жизненного опыта. В ЦНС существуют схема тела и соматотопический принцип регуляции деятельности органов. Это невозможно изменить в связи с ампутацией органа или конечности. У детей при врожденной патологии опорнодвигательного аппарата — дефектах конечностей — существует схема возможного применения отсутствующей конечности. Одним из центральных механизмов может быть увеличение возбудимости первичных афферентных нейронов после их деафферентации, а также чрезмерное возбуждение вторичных ноцицептивных нейронов в дорсальных рогах спинного мозга, вызванное катехоламинами, концентрация которых повышается в крови при болевых синдромах.
Лечат фантомные боли чаще нейрохирургическими операциями: иссечением невромы, стереотаксическим разрушением задних вентрота-ламических ядер, хордотомией, таламотомией (редко).
Патологическая боль центрального происхождения
Согласно определению Международной ассоциации по изучению боли, центральная боль — это боль, возникающая в результате поражения спиноталамокорковых путей ЦНС.
Из этого определения следует, что на любом уровне в ЦНС любой процесс, приводящий к повреждению соматосенсорных структур, которые участвуют в проведении афферентной импульсации, а также повреждение образований головного мозга, которые контролируют поступающую сенсорную информацию, могут стать источником центральной боли. Практически все виды поражения головного и спинного мозга потенциально могут вызывать центральную боль.
Самой частой причиной развития центральной боли при поражении головного мозга является острое нарушение мозгового кровообращения, составляющее 90-92 % всех этиологических случаев. Главной причиной
Патофизиология боли
303
центральной боли, связанной с повреждением спинного мозга, являются его травмы.
Патогенез центральных болей окончательно неясен. Среди различных гипотез наиболее обоснованная связывает центральную боль с изменением рисунка импульсации деафферентированных центральных чувствительных ядер и усилением их спонтанной активности. Полагают также, что возникновение центральной боли контролируется взаимоотношением между глутаматергической и ГАМК-эргической нейропередачами и в основе лежит ГАМК-эргическая гипофункция. При этом возникает гиперактивация вторичных ноцицептивных нейронов на спинальном и надспинальных уровнях ноцицептивной системы. Такие нейроны составляют группы, которые генерируют возбуждающие импульсы, — генераторы патологически усиленного возбуждения (ГПУВ, по Т.Н. Крыжановскому). Эти ГПУВ могут образовываться в разных отделах ноцицептивной системы, обусловливая развитие болевых синдромов: ГПУВ в спинном мозге — спинальный болевой синдром, ГПУВ в ядрах тройничного нерва — тригеминальная невралгия, ГПУВ в ядрах таламуса — таламический болевой синдром.
Таламигеский болевой синдром — феномен центральной боли — проявляется при повреждении таламуса, тромбозе сосудов зрительного бугра в тяжелых жгучих, непереносимых болях, которые локализуются на контралатеральной стороне тела и лица. Вся афферентация: прикосновения, холод, термические стимулы, тепло, болевые стимулы — воспринимаются пациентом как неприятные на стороне, противоположной той, на которую воздействуют. Одновременно с извращением всех видов соматической чувствительности возникали и двигательные нарушения. Предполагают, что боль связана с гибелью неноцицептивных волокон, в результате чего повышается возбудимость ноцицептивных волокон. При этом обнаруживается также нарушение ГАМК-эргического торможения.
Патогенетические принципы терапии боли
Хирургические методы: симпатэктомия, перерезка симпатических волокон, перерезка периферического нерва, дорсального корешка, половинная хордотомия, хирургическое стереотаксическое разрушение восходящих путей и ядер таламуса.
Фармакологические методы: применение анестетиков, местных и общих анальгетиков, введение опиатов, препаратов, воздействующих на ГАМК-эргические структуры (баклофен, сирдалуд), применение проти
304
Лекция 18
восудорожных препаратов для лечения центральных болей, использование ганглиоблокаторов и блокаторов обратного захвата серотонина, применение психотропных препаратов (антидепрессантов, транквилизаторов).
Физиотерапевтические методы: акупунктура, электропунктура, чрескожная электростимуляция, ультразвук, электрофорез, диодинамиче-ские токи, массаж и другие направлены на различные способы активации антиноцицептивной системы. Стимуляционная аналгезия — стереотаксические методы раздражения ОВСВ и ядер шва.
Психологические методы направлены на снятие напряжения, чувства страха и повышение болевого порога.
Лекция 19
ПАТОФИЗИОЛОГИЯ ЭНДОКРИННОЙ СИСТЕМЫ (ОБЩАЯ ЭТИОЛОГИЯ И ПАТОГЕНЕЗ ЭНДОКРИННЫХ НАРУШЕНИЙ)
Г.В. Порядин
Наряду с нервной системой эндокринные железы являются важнейшей регулирующей системой, представляющей собой специальный аппарат межклеточной регуляции.
Если нервная система обеспечивает в основном быстрый и относительно кратковременный способ регуляции, то эндокринная система осуществляет преимущественно более медленный и длительный контроль.
Функция эндокринной системы осуществляется через выработку специфических веществ — гормонов.
Термин «гормон» применяют для обозначения специфических биологически активных веществ, вырабатываемых специальными эндокринными органами во внутреннюю среду организма (кровь, лимфа) и оказывающих регулирующее действие на строение и функцию клеток-мишеней вне места своего образования.
По химическому строению гормоны можно разделить на 3 основные группы.
1. Белки и пептиды — АКТГ (олигопептид), СТГ (соматотропин — гормон роста — простой белок), лютропин (ЛТГ — гликопротеид), тиреотропин (ТТГ — гликопротеид) — главный регулятор морфогенеза фолликулярного аппарата щитовидной железы, биосинтеза и секреции тиреоидных гормонов, инсулин, глюкагон, вазопрессин, окситоцин.
2. Производные аминокислот — тирозина: трийодтиронин, тироксин — гормоны щитовидной железы; адреналин, норадреналин — гормоны мозгового слоя надпочечников.
3. Стероиды имеют липидную природу. В основе всех стероидов лежит циклопентанопергидрофенантреновое кольцо. Это гормоны коры надпочечников: кортикостерон, кортизол, альдостерон; половых желез: эстрадиол, эстрон, тестостерон, прогестерон.
306
Лекция 19
Можно выделить 5 типов воздействия гормонов на ткани-мишени.
• Метаболигеское — вызывающее изменения обмена веществ.
Например, инсулин, глюкагон, адреналин регулируют углеводный обмен; минералокортикоиды влияют на содержание натрия и калия в организме; паратгормон регулирует обмен кальция и фосфора; СТГ гипофиза стимулирует синтез белка в организме.
• Влияние гормонов на морфогенетигеские процессы — стимуляция роста, дифференциации тканей и органов.
Например, соматотропин влияет на рост тела и внутренних органов; гонадотропные гормоны стимулируют рост и созревание половых желез; половые гормоны влияют на развитие полового аппарата, вторичных половых признаков.
• Кинетигеское или пусковое (способность гормонов запускать реализацию определенной функции). Например, тропные гормоны гипофиза необходимы для выработки и секреции гормонов щитовидной железы, половых желез, надпочечников (глюкокортикоиды); окситоцин вызывает сокращение мускулатуры матки; адреналин запускает механизм распада гликогена в печени и выход глюкозы в кровь; вазопрессин включает механизм обратного всасывания воды в собирательных трубочках нефронов.
• Корригирующее — изменяющее интенсивность функций организма или его органов, которые происходят и при отсутствии гормонов.
Например, адреналин учащает ритм и усиливает сокращения сердца, тормозит моторику ЖКТ, повышает тонус сосудов. Тироксин активирует окислительные процессы. Альдостерон уменьшает обратное всасывание ионов калия в почках. Нормализующий эффект гормонов, направленный на восстановление измененного (нарушенного) процесса. Так, глюкокортикоиды вызывают катаболический эффект при усилении анаболических процессов белкового обмена, но стимулируют синтез белков при преобладании распада белков.
• Реакпюгенное — способность гормона менять реактивность ткани. Например, тиреоидные гормоны, глюкокортикоиды усиливают эффект катехоламинов (пермиссивное действие), инсулин — соматотропина.
Пути регуляции функций эндокринных желез
Нормальной инкреторной функцией эндокринных желез следует считать такую, которая обеспечивает необходимый уровень гормонов в соответствии с потребностями организма.
Патофизиология эндокринной системы...
307
Это нормальное, стабильное функционирование эндокринной системы осуществляется различными сложными и многообразными путями регуляции, причем нарушение каждого из них может стать основой возникновения патологии функций организма.
Известно несколько физиологических механизмов специфического контроля функций эндокринных желез.
• нервный: нервно-проводниковый, нервно-эндокринный;
• эндокринный;
• неэндокринный (гуморальный);
• периферический (внежелезистый).
Ведущее место в регуляции эндокринных желез принадлежит ЦНС, которая осуществляет свои регуляторные влияния двумя путями и с помощью различных механизмов.
Нервный (нервно-проводниковый или рефлекторный) механизм регуляции — парагипофизарный механизм регуляции эндокринных желез, независимых от гипофиза.
Так, функция эпифиза, нейроэндокринных зон гипоталамуса, мозгового слоя надпочечников и паращитовидных желез находится под прямым влиянием нервно-проводниковой регуляции, причем нервные влияния в этих случаях играют важную или даже определяющую роль.
В ответ на различные воздействия (эмоциональные возбуждения, болевые сигналы, мышечная нагрузка, охлаждение, гипоксия и т.д.) осуществляется быстрый рефлекторный выброс катехоламинов надпочечниками.
Важнейшим регулятором (стимулятором) биосинтеза и секреции катехоламинов в надпочечниковой железе является симпатический большой нерв (большой чревный нерв). Возбуждение его запускается симпатическими центрами спинного мозга и вегетативными центрами гипоталамуса. Ветви нерва подходят к надпочечникам, выделяют ацетилхолин и вызывают усиление синтеза и секреции адреналина и норадреналина железой.
Функции мозгового слоя надпочечников и симпатической нервной системы настолько тесно связаны друг с другом, что обычно обозначаются единой симпатоадреналовой системой. Наиболее ярко видно повреждение функции симпатоадреналовой системы при феохромоцитоме.
Феохромоцитома — катехоламинопродуцирующая опухоль хромаффинной ткани, локализующаяся в мозговом веществе надпочечников.
Продукция катехоламинов при феохромоцитоме повышена в десятки раз. Ведущим физиологическим механизмом нарушений при фео
308
Лекция 19
хромоцитоме является артериальная гипертензия (возрастает уровень норадреналина).
Нервные окончания, подходящие к другим эндокринным железам, вступают в синаптические контакты с кровеносными сосудами, которые обильно оплетают гормонпродуцирующие клетки. В этих случаях перерезка нервов или их раздражение изменяет в основном кровоснабжение желез и тем самым, опосредованно, изменяет их функцию.
Нейроэндокринная (гипоталамическая) регуляция функций гипо-физ-зависимых эндокринных желез (трансгипофизарный механизм регуляции) — щитовидная железа, гонады, кора надпочечников.
В этом случае регулирующее влияние ЦНС на физиологическую активность желез внутренней секреции реализуется через гипоталамус, который является конечным морфологическим образованием, обеспечивающим функциональную связь головного мозга с эндокринной системой.
Это исключительное назначение гипоталамуса связано с тем, что гипоталамическая область имеет богатые афферентные связи с продолговатым и средним мозгом, таламусом, базальными ганглиями, гиппокампом, обонятельным мозгом, различными полями коры больших полушарий и другими мозговыми структурами, через которые гипоталамус может получать информацию от любой части организма.
Основной механизм деятельности гипоталамических нейронов — это трансформация нервного импульса в специфический эндокринный процесс, который сводится к биосинтезу гормона в теле нейрона и сбрасыванию образовавшегося секрета из окончаний аксона в кровь. При этом осуществляется два типа нейроэндокринных реакций:
• один из них связан с образованием и секрецией рилизинг-факторов в гипоталамусе — главных регуляторов секреции гормонов аденогипофиза;
• другой — с образованием нейрогипофизарных гормонов.
В первом случае гипоталамические гормоны образуются в ядрах среднего и заднего отделов подбугровой области, поступают по аксонам их нейронов в область среднего возвышения, где могут накапливаться и далее проникать в специальную систему портальной циркуляции аденогипофиза. Эти высокоактивные вещества (нейросекреты, нейрогормоны) — рилизинг-факторы — избирательно регулируют гормонообразовательные процессы аденогипофиза.
Гипоталамигеские рилизинг-факторы (по направленности эффекта: гипофизарные либерины и статины). В настоящее время из
Патофизиология эндокринной системы...
309
вестно 10 рилизинг-факторов, участвующих в регуляции секреции гормонов аденогипофиза: тиреотропин-рилизинг-фактор (тиролибе-рин); гонадотропин-рилизинг-фактор (гонадолиберин); АКТГ-рилизинг-фактор; соматостатин-ингибирующий фактор и др.
Во втором случае сами гормоны образуются в ядрах переднего гипоталамуса, спускаются по аксонам в заднюю долю гипофиза, где депонируются, а оттуда могут поступать в системную циркуляцию и действовать на периферические органы (вазопрессин-АДГ и окситоцин).
Эндокринная регуляция связана с непосредственным влиянием одних гормонов на биосинтез и секрецию других. Гормональную регуляцию эндокринных функций осуществляет несколько групп гормонов.
Особую роль в гормональной регуляции многих эндокринных функций играет передняя доля гипофиза. В различных ее клетках образуется ряд так называемых тропных гормонов, основное значение которых сводится к направленной стимуляции функций и трофики некоторых периферических эндокринных желез (кора надпочечников, щитовидная железа, гонады).
АКТГ вызывает разрастание пучковой и сетчатой коры надпочечников и усиление биосинтеза глюкокортикоидов и аналогов половых гормонов (половые стероиды).
ТТГ — главный регулятор морфогенеза фолликулярного аппарата щитовидной железы, биосинтеза и секреции тиреоидных гормонов.
Лютеинизирующий гормон (лютропин) — основной стимулятор овуляции и образования желтого тела в яичниках, биосинтеза эстрогенов и андрогенов.
СТГ — главный регулятор роста организма и анаболических процессов.
После хирургического удаления гипофиза, в эксперименте, гипофиз-зависимые периферические железы подвергаются гипотрофии, в них резко подавляются гормональный биосинтез, а также процессы, регулируемые соответствующими периферическими железами.
Аналогичная картина наблюдается у человека при полной недостаточности функции гипофиза — болезнь Симмондса-Шиена (пангипопитуитаризм).
Введение тропных гормонов гипофизэктомированным животным постепенно восстанавливает структуру и функцию зависимых от гипофиза эндокринных желез.
Среди негипофизарных гормонов, непосредственно регулирующих периферические эндокринные железы, можно назвать:
310
Лекция 19
• глюкагон — гормон а-клеток поджелудочной железы. Наряду с влиянием на углеводный и липидный обмен в периферических тканях может оказать прямое стимулируюшее действие на р-клетки той же железы, вырабатывающие инсулин;
• инсулин — непосредственно контролирует секрецию катехоламинов надпочечниками и СТГ гипофизом.
В механизмах регуляции гормон-гормон существует сложная система регуляторных взаимосвязей как прямых (нисходящих), так и обратных (восходящих), которые получили название механизма обратной связи.
Разберем их на примере системы гипоталамус-гипофиз-перифе-рические железы (рис. 19-1).
Прямые связи (нисходящие) начинаются в гипофизотропных областях гипоталамуса, которые получают по афферентным путям мозга внешние сигналы к пуску системы.
Гипоталамический стимул в форме определенного рилизинг-фактора передается в переднюю долю гипофиза, где усиливает или ослабляет секрецию соответствующего тропного гормона. Повышенные или сниженные концентрации последнего через системную циркуляцию поступают к регулируемой им периферической эндокринной железе и изменяют ее
Обратные связи (восходящие) могут исходить как от периферической железы (длинная наружная обратная связь), так и от гипофиза (короткая внутренняя обратная связь).
Восходящие наружные связи заканчиваются в гипоталамусе и гипофизе. Так, половые гормоны, кортикоиды или тиреоидные гормоны могут оказывать через кровь обратное влияние и на регулирующие их области гипоталамуса, и на соответствующие тропные функции гипофиза.
Большое значение в процессах саморегуляции имеют также внутренние короткие обратные связи, идущие от гипофиза к соответствующим гипоталамическим центрам.
секреторную функцию.
Кора головного мозга *--------
11
Гипоталамус 4-------------
(нейросекреты)
t i
Гипофиз ◄---------------
(тропные гормоны)
t i
Кровь (гормоны, метаболиты)
t i
Железы внутренней секреции ------
(собственные гормоны)
11
Периферические ткани и органы------
Рис. 19-1. Механизм обратной
Патофизиология эндокринной системы...
311
Таким образом, гипоталамус:
• с одной стороны, принимает сигналы извне и посылает приказы по линии прямой связи к регулируемым эндокринным железам:
• с другой — реагирует на сигналы изнутри системы от регулируемых желез по принципу обратной связи.
По направленности физиологического действия обратные связи могут быть отрицательными и положительными.
Первые как бы самоограничивают, самокомпенсируют работу системы, вторые — ее самозапускают.
При удалении периферической железы, регулируемой гипофизом, или ослаблении ее функции секреция соответствующего тропного гормона возрастает. И наоборот, усиление ее функции приводит к торможению секреции тропного гормона.
Примером значения механизма обратной связи для организма является викарная гипертрофия одного из надпочечников после хирургического удаления второго (односторонняя адреналэктомия). Такая операция вызывает быстрое падение содержания кортикостероидов в крови, что усиливает через гипоталамус адренокортикотропную функцию гипофиза и вызывает повышение концентрации АКТГ в крови. Повышение же концентрации АКТГ вызывает компенсаторную гипертрофию оставшегося надпочечника.
Длительный прием тиреостатиков (или антитиреоидных веществ), подавляющих биосинтез гормонов щитовидной железы (метилура-цил, мерказолил, сульфаниламиды), вызывает усиление секреции тиреотропного гормона, а это, в свою очередь, из-за блокады синтеза обусловливает зобогенный эффект (разрастание железы) и развитие зоба.
Другой пример необходимости помнить о механизме обратной связи — легение гормонами.
В этих случаях вводимый извне гормон по механизму обратной связи тормозит функцию соответствующей железы и при длительном введении приводит к ее атрофии.
Наиболее широкое применение с лечебной целью получили кортикостероидные гормоны, в первую очередь глюкокортикоидные гормоны. Объясняется это тем, что глюкокортикоиды (кортикостерон, кортизол и их аналоги) являются:
• мощными регуляторами углеводного и белкового обмена. Они вызывают повышение количества глюкозы в крови, тормозят синтез белка в мышцах, соединительной ткани и в лимфоидной ткани (ка
312
Лекция 19
таболический эффект), но стимулируют образование белка в печени (анаболический эффект);
• повышают резистентность организма к различным раздражителям (адаптивный эффект), обладают противовоспалительным и десенсибилизирующим действием (в больших дозах);
• являются одним из факторов, поддерживающих артериальное давление, количество циркулирующей крови и нормальную проницаемость капилляров.
Указанные эффекты глюкокортикоидов и обусловили их широкое клиническое применение при заболеваниях, в основе патогенеза которых лежат воспаление либо аллергические процессы.
Однако длительное введение глюкокортикоидов по механизму обратной связи приводит к атрофии коры надпочечников. В этой связи больные, прекратившие лечение препаратами глюкокортикоидных гормонов, попадая в ситуацию, когда под влиянием повреждающих факторов (как то: операция, бытовая травма, интоксикация) у них разовьется стрессовое состояние, не ответят адекватным усилением секреции собственных кортикостероидов.
В результате у них может развиться острая надпогегниковая недоста-тогность, которая сопровождается сосудистым коллапсом, судорогами, развитием комы. Смерть у таких больных может наступить через 48 ч при явлениях глубокой комы и сосудистого коллапса. Аналогичная ситуация может развиться при двустороннем кровоизлиянии в кору надпочечников (синдром Уотерхауса-Фридериксена).
Этиология:
• массивные кровоизлияния в надпочечники, вызванные травмой;
• септицемия (менингококковая, стрептококковая, стафилококковая);
• грипп, дифтерия, пневмония, гемофилия;
• опухоли надпочечников, тромбоз сосудов надпочечников.
Проявления: резкое снижение АД; сердечно-сосудистая недостаточность — коллапс; обезвоживание организма. Заболевание обычно имеет молниеносное течение, заканчивающееся гибелью в течение 24 ч.
Неэндокринная (гуморальная) регуляция
Регулирующее действие на эндокринные железы могут оказывать и некоторые негормональные метаболиты.
Этот способ регуляции в большинстве случаев является, по существу, саморегуляцией, самонастройкой эндокринной функции.
Патофизиология эндокринной системы...
313
Так, глюкоза, гуморально действуя на эндокринные клетки, изменяет интенсивность продукции инсулина и глюкагона поджелудочной железой, адреналина мозговым слоем надпочечников, СТГ аденогипофизом.
Уровень секреции паратгормона околощитовидными железами и кальцитонина щитовидной железой, контролирующих кальциевый обмен, в свою очередь, регулируется концентрацией ионов кальция в крови. Интенсивность биосинтеза альдостерона корой надпочечников обусловлена уровнем ионов натрия и калия в крови.
Рассмотренная неэндокринная регуляция эндокринных процессов представляет один из важнейших способов поддержания метаболического гомеостаза.
Для ряда желез (а- и 0-клетки островкового аппарата поджелудочной железы, околощитовидные железы) гуморальная регуляция негормональными агентами по принципу самонастройки имеет первостепенное физиологическое значение.
Особый интерес приобретает образование негормональных факторов стимуляции деятельности эндокринных желез в условиях патологии.
Так, при некоторых формах тиреотоксикоза и воспаления щитовидной железы (тиреоидит) в крови больных появляется длительно действующий тиреоидный стимулятор (long acting thyroid stimulator — названный LATS).
LATS представляет гормонально активные аутоантитела (иммуноглобулины IgG), вырабатываемые к патологическим компонентам (аутоантигенам) клеток щитовидной железы.
Аутоантитела, избирательно связываясь с клетками щитовидной железы, специфически стимулируют в ней процессы секреции тиреоидных гормонов, приводя к развитию патологической гиперфункции железы. Эти антитела действуют аналогично ТТГ, усиливая процессы синтеза и секреции щитовидной железой тироксина и трийодтироксина. Не исключено, что аналогичные метаболиты могут образовываться и к специфическим белкам других эндокринных желез, вызывая повреждение их функции.
Периферический (внежелезистый) путь регуляции эндокринных желез
Наряду с указанными механизмами регуляции, функция той или иной эндокринной железы зависит от:
• концентрации гормонов в крови;
• уровня их резервирования комплексообразующими (связывающими) системами крови;
• скорости их захвата периферигескими тканями.
314
Лекция 19
В развитии многих эндокринных заболеваний (эндокринопатий) эти механизмы могут играть весьма значительную роль.
Нарушение связывания гормонов белками-переносчиками
Выделившиеся из желез гормоны циркулируют в крови в нескольких физико-химических формах:
• в свободном виде;
• в форме комплексов со специфическими белками плазмы;
• в форме комплексов с форменными элементами крови.
Из плазменных белков, вступающих в комплексы с гормонами, наиболее хорошо известны транскортин, взаимодействующий с глюкокортикоидами и прогестинами; секс-стероидсвязывающий (тесто-сгерон-эстроген) глобулин, взаимодействующий только с андрогенами и эстрадиолом; тироксинсвязывающий глобулин, специфически взаимодействующий с тиреоидными гормонами; инсулинсвязывающий белок; соматотропинсвязывающий белок.
Сывороточный альбумин, характеризующийся широким диапазоном сродства не только к гормонам, но и к аминокислотам, сульфаниламидам, антибиотикам, витаминам и т.д„ связывает главным образом стероидные гормоны.
Физиологический смысл этого связывания состоит в том, что гормоны, связанные с комплексообразующими белками, физиологически не активны и не способны подвергаться метаболическим превращениям, т.е. связывание играет буферно-резервирующую роль по отношению к гормонам.
Некоторые формы патологии эндокринных функций (эндокринопатий) могут быть первично обусловлены нарушениями в связывании гормонов транспортными белками.
Например, некоторые формы гиперкортицизма (избыток свободных глюкокортикоидов вследствие понижения концентрации транскорти-на), сахарного диабета (повышенное связывание инсулина белками), гипо- и гипертиреоза (нарушение связывания тиреоидных гормонов).
Нарушение метаболизма гормонов
Важную роль в периферических превращениях гормонов играют процессы их катаболизма. Основной физиологический смысл катаболических процессов — это необратимая инактивация гормонов и обеспечение гормонального баланса. Вместе с тем в процессе метаболических
Патофизиология эндокринной системы...
315
превращений гормонов могут протекать обменные процессы, приводящие к активации, реактивации, взаимопревращениям гормонов и возникновению новой гормональной активности.
Так, отщепление атома йода в молекуле тироксина приводит к образованию трийодтиронина, обладающего значительно большей активностью (в 10 раз).
Примером реактивации может послужить переход кортизона в кортизол.
Примером взаимопревращений гормонов разного типа является превращение андрогенов в эстрогены в гипоталамусе, печени, переход 17-оксикортикостероидов в андрогены.
Изменение интенсивности катаболизма секретируемых железами гормональных соединений может косвенно оказывать влияние на функциональное состояние эндокринных желез.
Помимо указанных превращений, может иметь замедление метаболизма гормонов, что приводит к их задержке в организме.
Избыток кортизола по механизму обратной связи угнетает функцию пучковой зоны коры надпочечников, вызывая ее атрофию.
Снижение инактивации в печени эстрадиола у мужчин также приводит к включению механизма обратной связи, в результате угнетается образование гонадотропных гормонов в гипофизе, и, как следствие этого, происходит снижение функции тестикулов, что приводит к развитию импотенции.
Патология гормональных рецепторов
В последнее время накопилось много данных о том, что в основе ряда эндокринных и неэндокринных заболеваний может лежать не нарушение деятельности той или иной железы внутренней секреции, гормонального транспорта и метаболизма, а нарушение чувствительности клеток-мишеней к гормонам.
Чаще всего гормоны обладают достаточно выраженной тропностью (направленностью) своего физиологического действия. Так, эффекты эстрогенов преимущественно направлены на органы женской половой сферы, а андрогенов на органы мужской; АКТГ — на кору надпочечников и т.д.
Органы, ткани и клетки, на которые направлено преимущественное действие гормона, принято называть соответственно', орган-мишень, ткань-мишень, клетки-мишени.
Специфичность ответа клетки на тот или иной гормон определяется специфичностью рецептора, который связывается только со своим гор
316
Лекция 19
моном, выбирая его из большого количества соединений, а также природой специфических для клетки протеинкиназ и белковых субстратов.
Некоторые формы патологии, как оказалось, обусловлены недостаточностью рецепторов к гормонам в клетках.
Нарушение взаимодействия гормона с его рецептором может быть обусловлено:
• изменением числа рецепторов гормона;
• отклонением от нормального соотношения высоко- и низкоаффинных рецепторов;
• образованием антирецепторных антител;
• блокадой рецепторов негормональными лигандами, имеющими сходные с молекулой гормона структуры;
• перекрестным эффектом гормонов (СТГ может активировать рецепторы пролактина).
Одна из наиболее изученных форм патологии рецепторного аппарата — ожирение, связанное с резистентностью к инсулину. Такие формы ожирения человека могут быть наследственными или приобретенными.
Основной дефект при данной форме патологии обусловлен недостаточностью мембранных рецепторов инсулина, приводящей к ареактив-ности клеток-мишеней к гормону.
Снижение концентрации рецепторов может играть решающую роль в патогенезе некоторых инсулинрезистентных форм диабета у человека.
В качестве еще одного примера патологии гормональных рецепторов можно привести синдром тестикулярной феминизации. Это генетически обусловленные заболевания человека, проявляющиеся в форме врожденного мужского псевдогермафродитизма.
При данной патологии в мужском организме имеют место нормальная секреция тестостерона семенниками и чаще всего нормальный его метаболизм. Введение андрогенных препаратов таким больным оказывает слабый лечебный эффект. При синдроме тестикулярной феминизации ткани обладают пониженной чувствительностью к андрогенам, так как в клетках ряда тканей этих больных резко снижено содержание андрогенных рецепторов, вплоть до полного их отсутствия.
Описаны случаи вазопрессинрезистентных форм несахарного диабета, сопровождающиеся значительным увеличением уровня антидиу-ретического гормона в крови и отсутствием эффекта на его введение извне.
В ряде случаев карликового роста концентрация СТГ в крови нормальная, и больные не отвечают на введение экзогенного СТГ.
Патофизиология эндокринной системы...
317
Аналогичная ситуация отмечается и при псевдогипопаратиреозе — синдроме, сходном с гипопаратиреозом, сопровождающимся гипокальциемией, гиперфосфатемией и даже развитием тетании. Такие больные не реагируют на введение экзогенного паратгормона.
Показано, что при некоторых опухолях (лимфолейкозы, рак молочной железы) также имеет место недостаточное или избыточное содержание рецепторов ряда гормонов. Это делает опухолевые клетки гормоннезависимыми либо высокочувствительными к гормональным влияниям.
Знание существующих соотношений разных рецепторов в опухолевой ткани может создать основу для рациональной гормональной терапии опухолей.
Регуляторные пептиды
Наряду с гипоталамическими рилизинг-факторами, нейрогипофизарными гормонами и гормонами аденогипофиза в регуляции различных функций организма участвуют биологически активные вещества (регуляторные пептиды), которые секретируются не клетками ЦНС и эндокринными железами, а клетками других органов и тканей.
Их источниками служат одиночные гормонпродуцирующие клетки. Эти клетки способны декарбоксилировать ароматические кислоты — предшественники нейроаминов, что позволило объединить их в единую систему, получившую название APUD-система (по первым буквам английских слов Amine Precursor Uptake and Decaboxylating system — система захвата и декарбоксилирования предшественников аминов).
Апудоциты встречаются в секреторных элементах гастроинтестинального тракта (вазоактивный интестинальный пептид — ВИП, холецистокинин, гастрин); в нервной ткани (субстанция Р, нейротензин); в почках, сердце, костном мозге, легких.
Функции регуляторных пептидов
• Влияние на формирование боли (вещество Р, ВИП, холецистокинин).
• Влияние на эмоциональные реакции (ВИП).
• Влияние на вегетативные функции (ВИП, атриопептид).
• Являются антистрессорами, ограничивая развитие стрессорных реакций.
• Модулируют иммунный ответ (ВИП, субстанция Р).
318
Лекция 19
Участие регуляторных пептидов в развитии патологии
Поскольку пептидные гормоны (APUD-гормоны) участвуют в регуляции многих функций в организме, они неизбежно вовлекаются в патогенез различных заболеваний.
На сегодня выделяется особая группа заболеваний, обусловленная патологией этой системы, — апудопатии.
Апудопатии — заболевания, связанные с нарушением структуры и функции апудоцитов и выражающиеся в определенных клинических синдромах.
Различают первигные апудопатии, обусловленные патологией самих апудоцитов, и вторигные апудопатии, возникающие как реакция апудоцитов на нарушение гомеостаза организма.
Первичные апудопатии могут проявляться в гиперфункции, гипофункции, дисфункции, в образовании апудом — опухолей из клеток APUD-системы. Например, следующие.
• Гастринома — апудома из клеток, продуцирующих гастрин, который, как известно, стимулирует выделение большого количества желудочного сока с высокой кислотностью и переваривающей силой. Поэтому клинически гастринома проявляется развитием уль-церогенного синдрома Золлингера-Эллисона.
• Випома — опухоль из клеток, секретирующих вазоактивный интестинальный пептид. Может локализоваться в двенадцатиперстной кишке или поджелудочной железе. Проявляется развитием водной диареи и обезвоживанием, а также расстройством обмена электролитов.
Вторигные апудопатии. Нарушения концентрации пептидов мозга при дегенеративных неврологических заболеваниях (болезнь Альцгеймера). Недостаточность пептидов мозга при шизофрении. Увеличение концентрации атриопептида при застойных явлениях в системе кровообращения.
Лекция 20
ПАТОФИЗИОЛОГИЯ ЭНДОКРИННОЙ СИСТЕМЫ (ЭНДОКРИНОПАТИИ — ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ В САМОЙ ЖЕЛЕЗЕ)
Г.В. Порядин
Наряду с механизмами, лежащими в основе нарушения регуляторных процессов, серьезную проблему в нарушении нормальных взаимодействий в эндокринной системе представляют патологические процессы в самих эндокринных железах, вследствие прямого поражения одной (моногландулярные эндокринопатии) или нескольких (полигландуляр-ные эндокринопатии) эндокринных желез.
В патологических условиях по характеру изменения функции нарушения деятельности эндокринных желез (рис. 20-1) могут проявляться:
• неадекватной потребностям организма чрезмерно высокой инкре-цией — гиперфункцией;
• неадекватной потребностям организма чрезмерно низкой инкре-цией — гипофункцией;
• качественным нарушением гормонообразования в железе, качественным нарушением инкреции — дисфункцией;
• эндокринными кризами — острыми проявлениями эндокринной патологии (тиреотоксический криз, гипотиреоидная кома).
Рис. 20-1. Нарушение функции эндокринных желез
320
Лекция 20
По происхождению эндокринопатии могут быть:
• первичными (развивающимися в результате первичного повреждения ткани железы);
• вторичными (развивающимися в результате первичного повреждения аденогипофиза);
• третичными (развивающимися в результате первичного повреждения гипоталамуса).
Пригины поражения эндокринных желез (эндокринопатий) могут быть различными:
• инфекционные процессы и интоксикации;
• опухолевые процессы;
• аутоиммунные процессы;
• генетические дефекты синтеза гормонов.
Инфекционные процессы и интоксикации (острые, хронические)
Одной из причин, приводящей к нарушению функции желез внутренней секреции, может стать острое или хроническое инфекционное заболевание. Например, менингококковая инфекция может сопровождаться кровоизлиянием в надпочечники, что приводит к разрушению ткани железы и развитию острой надпочечниковой недостаточности. Подобная недостаточность может возникать при дифтерии в связи с коагуляционными некрозами в надпочечниках.
Эпидемитеский паротит у взрослых мужчин нередко вызывает орхит, который в 50-70 % случаев заканчивается одно- или двусторонней атрофией яичек. Тестикулы могут поражаться и при гонорее в связи с восходящей инфекцией уретры.
Такие хронически протекающие инфекционные заболевания, как туберкулез и сифилис, также поражают различные железы.
При туберкулезе идет постепенное разрушение ткани железы в связи с творожистым некрозом туберкулезных бугорков, а при сифилисе — в связи с некрозом сифилитической гранулемы (гуммы).
При локализации процесса в надпочечных железах развивается хроническая надпочечниковая недостаточность (гипокортицизм), которая называется болезнь Аддисона — по имени врача Аддисона, впервые описавшего это заболевание (или бронзовой болезнью).
Опухолевые процессы в железах
Одним из частых патологических процессов в самой железе внутренней секреции может стать опухоль.
Патофизиология эндокринной системы...
321
Если опухоль не секретирует гормон, а только сдавливает ткань железы, она вызывает атрофию нормальных участков ткани железы.
Клинически это выражается в гипофункции соответствующей железы, как, например, при хромофобных аденомах гипофиза.
Чаще же развитие опухоли сопровождается избыточным образованием гормона и клинической картиной гиперфункции.
Например, базофильная аденома передней доли гипофиза продуцируется избыточным количеством адренокортикотропного гормона (АКТГ). Это приводит к увеличению секреции кортизола надпочечными железами и развитию синдрома Иценко-Кушинга (гиперкорти-цизм).
Аутоиммунные процессы
Функция желез внутренней секреции может нарушаться в связи с повреждением их иммунными механизмами организма. Общим для всех поражений иммунного механизма является выявление в крови циркулирующих аутоантител к ткани соответствующей железы (например, надпочечников при идиопатической форме аддисоновой болезни, щитовидной железы при тиреоидите Хашимото и т.д.).
Генетически обусловленные дефекты биосинтеза гормонов
Биосинтез любого гормона представляет собой сложный и многозвеньевой процесс, в котором принимают участие многие ферменты. Образование же любого фермента, точнее его апофермента, определяется активностью соответствующего гена.
Мутация гена может привести к выпадению образования апофермента или такому его изменению, при котором образующийся фермент теряет свою активность. В таком случае будет нарушен последовательный ход биосинтеза соответствующего гормона, что приведет либо к:
• развитию гипофункции железы;
• накоплению в железе промежуточных продуктов биосинтеза, образующихся до места блокады;
• нарушению работы механизма обратной связи.
В качестве примера этому положению может служить схема биосинтеза кортизола (рис. 20-2).
При дефиците 21-гидроксилазы процесс биосинтеза заканчивается образованием прогестерона и 17а-оксипрогестерона. Кортизол не образуется.
322
Лекция 20
КОРТИКОСТЕРОН
Рис. 20-2. Схема биосинтеза кортизола
Это по механизму обратной связи растормаживает секрецию корти-котропин-рилизинг-фактора в гипоталамусе, что, в свою очередь, ведет к усилению образования АКТЕ АКТГ стимулирует стероидогенез в надпочечниках до места блокады, а так как кортизол не образуется, вся эта стимуляция переключается на образование 4-андростен-3,17-диона, обладающего андрогенными свойствами. Его поступление в кровь значительно увеличивается, что по механизму обратной связи приводит к выключению функций половых желез с последующей их атрофией как у мальчиков, так и у девочек. Ферментный дефект выявляется уже в периоде эмбрионального развития.
Избыток андрогенов у девочек вызывает развитие вирилизма и маскулинизации, которые продолжаются и после рождения. У мальчиков же появляются признаки преждевременного полового созревания.
Подобный механизм включается и при дефекте фермента 11-р-гид-роксилазы. В этом случае кортизол также не образуется, но в отличие от предыдущего синдрома здесь накапливается избыточное количество 11-дезоксикортикостерона и 17-а-окси-11-дезоксикортикостерона; первый из них обладает выраженными минералокортикоидными свойствами, что ведет к стойкому повышению кровяного давления.
Всю эту патогенетическую цепь можно разорвать введением глюкокортикоидов, которые тормозят образование АКТГ и тем самым уменьшают образование андрогенов.
Патофизиология щитовидной железы
Щитовидная железа, расположенная впереди трахеи, — самая крупная из желез внутренней секреции. Ее вес у взрослого человека состав-
Патофизиология эндокринной системы...
323
Рис. 20-3. Патология функции щитовидной железы
ляет около 20 г (для сравнения, вес надпочечника — 5 г, а гипофиза — 0,5 г).
В случае патологии функция щитовидной железы может меняться как в сторону чрезмерного снижения, так и усиления (рис. 20-3).
Расстройства, связанные с недостаточностью функции щитовидной железы (гипотиреозы)
Гипотиреоз — состояние, обусловленное недостаточностью функции щитовидной железы. Выраженные формы этого заболевания у взрослых обозначаются как микседема.
Понижение функции щитовидной железы может быть связано с воздействием внешних неблагоприятных условий:
• недостаточное питание;
• пребывание при очень высокой внешней температуре;
• действие ряда токсических веществ;
• различные инфекции и т.д.
Этот тип торможения функции щитовидной железы непрямой. Он осуществляется опосредованно через переднюю долю гипофиза и приводит к развитию вторигного гипотиреоза, возникающего в результате прекращения продукции тиреотропного гормона (гипофизарная микседема).
Помимо разрушения передней доли гипофиза, тот же эффект дает перерыв гипофизарной ножки, а также некоторые опухоли и поражения гипоталамуса (так называемый третигный гипотиреоз).
324
Лекция 20
Функция щитовидной железы может быть подавлена и прямым путем, в результате непосредственного поражения или разрушения щитовидной железы, это приводит к развитию первигного гипотиреоза.
Его причинами могут быть:
• врожденная аплазия или недоразвитие щитовидной железы (чаще наблюдается в очагах эндемического зоба (эндемигеский кретинизм)',
• полное или частичное выключение щитовидной железы (тиреоидэктомия, рентгеновское облучение):
• ослабление гормонообразовательной деятельности щитовидной железы в результате действия тиреостатических агентов, как природных, так и синтетических;
• ослабление гормонообразовательной деятельности щитовидной железы в результате воспалительных процессов, дегенерации тиреоидной паренхимы под влиянием некоторых аутоиммунных процессов:
• генетически обусловленные нарушения биосинтеза тиреоидных гормонов.
В результате воздействия указанных факторов развивается тиреоидная недостатогность — неспособность щитовидной железы вырабатывать такое количество гормона, которое необходимо для обеспечения нормальных функций организма и поддержания нормального уровня тиреоидных гормонов в крови.
Различают две формы:
• Приобретенная форма гипотиреоза:
— «спонтанная» тиреоидная недостаточность, связанная с полной аплазией или агнезией щитовидной железы (клиническая картина гипотиреоза во многом зависит от того, в каком возрасте началось заболевание):
— тиреоидная недостатогность в детстве или юности (приобретенная форма гипотиреоза), обычно называется ювенильный гипотиреоз или детская и юношеская микседема (к причинным факторам этой формы гипотиреоза относят главным образом воспалительные или дегенеративные изменения щитовидной железы, возникающие в результате инфекционных заболеваний детского возраста).
• Врожденная форма гипотиреоза: врожденная тиреоидная недостаточность (врожденная микседема), сопровождающаяся синдромом полной функциональной недостаточности щитовидной железы у
Патофизиология эндокринной системы...
325
новорожденного — кретинизм, в основе которого лежит врожденная аплазия железы.
Недостаточность щитовидной железы может быть воспроизведена экспериментальной тиреоидэктомией. Результаты тиреоидэктомии различны в зависимости от того, в каком возрасте животное подвергается удалению щитовидной железы.
После удаления щитовидной железы у молодых животных нарушения сильнее всего проявляются в росте скелета. Преждевременно прекращается рост всего тела, особенно отстают в росте длинные трубчатые кости.
Наблюдается ослабление энхондрального окостенения, незараста-ние эпифизарных швов, запаздывает развитие зубов. Явно замедлено половое развитие.
В результате тиреоидэктомированное животное приобретает характерный вид карлика: тело шарообразной формы, лапы и хвост короткие.
Удаление щитовидной железы у взрослых животных вызывает через несколько недель тиреопривную кахексию, для которой характерно преимущественное нарушение обменных процессов:
• понижение основного обмена:
• ослабление окислительных процессов в тканях;
• уменьшение выделения азота с мочой;
• задержка в организме воды и хлористого натрия;
• увеличение резистентности организма к сахару;
• падение температуры тела до низшей границы нормы;
• трофические изменения кожи и слизистых оболочек.
Шерсть выпадает, животные становятся апатичными: они больше лежат, с трудом передвигаются, становятся неуклюжими, утрачивают приобретенные ранее условные рефлексы.
В клинической практике состояние, близкое к тиреоидэктомии, может иметь место в результате удаления ткани щитовидной железы при операции, разрушении тиреоидной ткани, остром гнойном тиреоидите, метастазировании рака, туберкулезе и других заболеваниях щитовидной железы, приводящих к ее острой недостаточности.
Наиболее постоянным и характерным для тиреоидной недостатог-ности является замедление всех процессов обмена веществ.
Чтобы была понятна патология, вспомним влияние тиреоидных гормонов на организм в физиологических условиях:
326
Лекция 20
• усиливают синтез белка;
• увеличивают задержку азота в организме;
• усиливают всасывание глюкозы в кишечнике;
• форсируют гликогенолиз;
• усиливают всасывание жиров из кишечника;
• усиливают мобилизацию жиров из депо;
• усиливают окисление жиров в тканях.
Основной эффект тиреоидных гормонов проявляется стимулированием тканевого дыхания, особенно катаболической фазы обмена.
Основным последствием удаления щитовидной железы или ее разрушения с последующим понижением или отсутствием ее функции у человека является понижение поглощения кислорода при полном покое натощак, т.е. понижение основного клеточного обмена, который может уменьшаться на 40-50 % и более.
Понижено и специфически динамическое действие белка. Скорость синтеза белка у больных обычно снижается.
Наряду с нарушениями образования тканевых белков нарушается и их распад:
• увеличивается содержание мочевины и мочевой кислоты в крови;
• значительно снижено выделение азота.
Гипотиреоз приводит к уменьшению выделения креатинина и стимулирует синтез его в ткани печени. Креатин и креатинфосфат накапливаются в большом количестве в сердечной и в скелетной мускулатуре.
Ряд изменений обнаруживается и со стороны белков крови — повышение общего белка сыворотки крови за счет у-глобулинов.
Изменения углеводного обмена
При тиреоидной недостаточности наблюдаются умеренное снижение уровня сахара в крови и повышение чувствительности к инсулину.
В значительной степени ослаблены процессы гликогенолиза в печени.
Жиролипоидный обмен
Понижение окисления жиров приводит к ожирению. В связи с замедленным усвоением жира тканями при тиреоидной недостаточности уровень липидов крови имеет тенденцию к повышению: содержание нейтрального жира, жирных кислот и фосфолипидов в крови увеличивается.
Тиреоидная недостаточность сопровождается и гиперхолестеринемией, достигающей высоких показателей, что способствует развитию атеросклероза.
Патофизиология эндокринной системы...
327
Минеральный и водный обмен
При экспериментальном и клиническом гипотиреозе происходит задержка воды и хлористого натрия.
Это, а также накопление в соединительной ткани мукопротеидов, обладающих выраженными гидрофильными свойствами, вызывает развитие так называемого слизистого отека (микседема).
При гипотиреозе количество выделяемого с мочой и калом кальция и фосфора понижается, что сопровождается задержкой этих ионов в организме.
Расстройства, связанные с повышенной функцией щитовидной железы (гипертиреоз)
Диффузный токсический зоб (синонимы: базедова болезнь, тиреотоксикоз, первичный тиреотоксический диффузный зоб).
Одно из распространенных эндокринных заболеваний. Характеризуется длительным поступлением в организм избытка йодированных тиреоидных гормонов в результате стойкого усиления интратиреоидного гормоногенеза.
Впервые описано в 1840 году мерзебургским врачом Базедом у 4-х больных с клинической симптоматикой, получившей в дальнейшем наименование мерзебургской триады, зоб, пучеглазие, тахикардия.
Основным этиологическим фактором гипертиреоза служит психическая травма. Возникающий под влиянием психической травмы очаг застойного возбуждения в гипоталамусе может вызвать стойкое повышение функции щитовидной железы (третигный гипертиреоз).
Импульсы со стороны центральной нервной системы на щитовидную железу могут передаваться как через гипофиз — его ТТГ (трансгипофи-зарно), так и минуя его, непосредственно по нервным проводникам (па-рагипофизарно).
Значительное место в этиологии тиреотоксикоза занимают инфекции (они являются причиной этого заболевания у 17 % больных). Наиболее частые инфекции, вызывающие тиреотоксикоз:
• грипп и ангина;
• тиреотоксикозу могут предшествовать и другие инфекционные заболевания: ревматизм, туберкулез, скарлатина, тиф, коклюш и пр.
Гипертиреоз и тиреотоксикоз могут быть и внетиреоидного генеза, т.е. развиваться при нормально функционирующей щитовидной железе.
328
Лекция 20
Это может быть обусловлено образованием более рыхлого комплекса тироксина с белком. В связи с этим большее количество тироксина поступает в ткань.
Возможно, что в развитии тиреотоксикоза может играть роль нарушение метаболизма тиреоидных гормонов в периферических тканях и т.д.
Точно так же, как главными проявлениями тиреоидной недостаточности являются нарушения обмена веществ, гипертиреоз оказывает выраженное влияние главным образом на те же процессы.
При тиреотоксикозе у человека основной обмен повышается (на 200-400 %).
Азотистый обмен
Параллельно повышению основного обмена выраженный тиреотоксикоз сопровождается отрицательным азотистым балансом. В связи с тем, что при гипертиреозе повышается выделение аммиака и мочевой кислоты, увеличивается в крови содержание остаточного азота и азота аминокислот.
Характерной особенностью изменений азотистого обмена при тиреотоксикозе являются креатинурия и связанное с ней выделение креатинина. У больных тиреотоксикозом количество выделяемого креатинина с мочой достигает 200 мг/сут. Содержание креатина в плазме повышается (в крови N = 1 мг%).
При тиреотоксикозе особенно интенсивно теряют креатин поперечно-полосатые мышцы и миокард. У больных тиреотоксикозом резко уменьшается объем мышечных масс, напоминая картину прогрессивной мышечной дистрофии.
Углеводный обмен
Повышение функции щитовидной железы оказывает значительное влияние и на углеводный обмен. Тиреотоксикоз характеризуется усилением катаболизма углеводов. Как известно, тиреоидные гормоны являются одним из физиологических регуляторов уровня сахара в крови. Они участвуют в пополнении использованных запасов глюкозы путем стимуляции гликонеогенеза.
При гипертиреозе наряду с повышением потребности сахара усиливает гликонеогенез.
Избыток тиреоидных гормонов при тиреотоксикозе разобщает процессы окисления от одновременно и сопряженно с ним протекающих процессов фосфорилирования.
Разобщение процессов окислительного фосфорилирования снижает коэффициент энергетического полезного действия и не возмещается по-
Патофизиология эндокринной системы...
329
вишенной скоростью окисления. Это является одним из факторов, вызывающих мышечную слабость при тиреотоксикозе, а также большее выделение тепла. Последнее выражается в субфебрилитете.
Жиролипоидный обмен
Понижая содержание гликогена в печени, тиреоидные гормоны стимулируют мобилизацию жира из его депо. Активация мобилизации жира выражается в повышении содержания неэстерифицированных (свободных) жирных кислот в крови.
В то же время тироксин тормозит переход углеводов в жиры. Общим результатом такого воздействия повышенной активности щитовидной железы является исхудание больных.
Усиление окисления жира в печени приводит к избыточному образованию кетоновых тел - ацетоуксусной, р-оксимасляной кислот, а при дефиците в организме углеводов — к нарушению окисления кетоновых тел в тканях и к гиперкетонемии.
Чрезмерные количества гормона вызывают усиление перистальтики, прохождение пищи в кишечнике ускоряется, и жир не успевает всасываться. В результате наступает стеаторея.
У больных с тяжелым тиреотоксикозом обычно встречаются низкие показатели холестерина крови.
Минеральный и водный обмен
При гипертиреозе повышается выделение воды и хлористого натрия, понижается содержание воды в коже.
В связи с распадом белка, гликогена и жира происходит потеря и связанной с ними интрацеллюлярной воды. Тиреотоксикоз сопровождается повышенным выделением кальция и фосфора.
Зобная болезнь (эндемический зоб, спорадический зоб)
Если гипертиреоз и гипотиреозы определяются расстройством механизмов, регулирующих деятельность щитовидной железы, то в основе зобной болезни на первый план выступает нарушение факторов, контролирующих рост тиреоидной паренхимы.
Основным патогенетическим фактором эндемического зоба является йодный дефицит в природе, что приводит к недостаточному поступлению йода с водой и пищей и нарушению образования тиреоидных гормонов.
По механизму обратной связи пониженное образование тиреоидных гормонов ведет к растормаживанию секреции ТТГ, а увеличение последнего — к гиперплазии щитовидной железы.
330
Лекция 20
На первых этапах развития зоба или при относительно небольшом дефиците йода эта гиперплазия имеет характер компенсаторной реакции и может обеспечивать нормальное образование и поступление в кровь тиреоидных гормонов.
Однако при длительной йодной недостаточности или значительном дефиците йода гиперплазия эпителия щитовидной железы, образование новых фолликулов оказываются недостаточными для образования нормального количества гормонов, и тогда развивается гипотиреоидный зоб, в далеко зашедших случаях переходящий в микседему и кретинизм.
Патофизиология околощитовидных желез
Паращитовидные железы представлены двумя парами (верхней и нижней) небольших желез, тесно прилежащих к задней поверхности щитовидной железы, образуя с последней тиреопаратиреоидный комплекс с общим кровоснабжением и иннервацией.
Околощитовидные железы вырабатывают паратиреоидный гормон (паратгормон), под влиянием которого концентрация кальция в крови повышается (гиперкальциемический эффект).
В щитовидной железе одновременно продуцируется тиреокальцитонин (антагонист паратгормона), который, наоборот, способствует понижению кальциемии (гипокальциемический фактор). В норме паратгормон и тиреокальцитонин находятся в динамическом равновесии.
Наряду с регуляцией кальциевого метаболизма, околощитовидные железы оказывают сильное влияние на обмен фосфора (снижают концентрацию фосфора в крови).
Механизм действия паратгормона и кальцитонина
Паратгормон:
• в кости стимулирует мобилизацию и выход в кровь Са2*;
• в кишечнике усиливает всасывание Са2* в кровь;
• в почках усиливает реабсорбцию Са2* и тормозит реабсорбцию фосфатных ионов.
Кальцитонин:
• в костях усиливает отложение Са2*;
• в кишечнике тормозит всасывание Са2* и фосфатов из кишечника;
• в почках усиливает экскрецию Са2*.
Адекватным регулятором функции околощитовидных желез служит изменение уровня кальция сыворотки: понижение его активирует, увеличение тормозит секрецию паратиреоидного гормона.
Патофизиология эндокринной системы...
331
Гипопаратиреоз
Заболевание развивается как следствие недостаточности функции околощитовидных желез. Уменьшение продукции паратгормона может обусловливаться:
• удалением части околощитовидных желез;
• ослаблением их деятельности при инфекциях и интоксикациях.
Соответственно различают 2 формы недостаточности:
• острую паратиреоидную недостаточность;
• хроническую паратиреоидную недостаточность.
Причиной возникновения острой паратиреоидной недостаточности (паратиреопривной тетании) чаще всего является случайное удаление околощитовидных желез при операциях на щитовидной железе или травматическое их повреждение.
Уже в первые сутки развивается депрессивное состояние, возбудимость нервов и мышц возрастает, затем начинаются подергивания отдельных мышц головы и туловища, постепенно переходящие в тонические судороги.
Содержание кальция в крови во время тетанического припадка резко снижается. При снижении концентрации кальция в крови нарушается равновесие между моновалентными и двухвалентными катионами (натрия и калия к кальцию и магнию), в результате чего и повышается нервно-мышечная возбудимость.
Содержание калия в крови больных гипопаратиреозом нередко повышено, содержание фосфора в крови во время приступа достигает высоких цифр. При тетании наблюдается сдвиг pH крови в кислую сторону. Удаление всех паращитовидных желез, безусловно, смертельно.
Хроническая форма околощитовидной недостаточности характеризуется подавлением аппетита, прогрессирующей кахексией и рядом трофических расстройств (гиперемия, кровоизлияния в области привратника желудка и двенадцатиперстной кишки с образованием язв, выпадение волос, замедление роста — в молодом возрасте).
Болезнь Олбрайта (врожденное заболевание) — псевдогипопаратиреоз. От истинного гипопаратиреоза, обусловленного первичной недостаточностью околощитовидных желез, следует отличать псевдогипопаратиреоз. Это заболевание характеризуется нанизмом (низким ростом), аномалиями в развитии скелета, дистрофическими изменениями зубов и костей и отставанием в интеллектуальном развитии. Наблюдается тетания понижения содержания кальция и увеличения фосфора в крови. Пшофосфатурия.
332
Лекция 20
В противоположность истинному гипопаратиреозу отмечается резистентность к паратгормону.
Гиперпаратиреоз
Гиперпаратиреоз (генерализованная фиброзная остеодистрофия, болезнь Реклингаузена). В 1876 г. английский врач Педжет описал случай заболевания, проявившегося резкой деформацией костей. На секции кости оказались утолщенными и вместе с тем легкими, порозными и настолько мягкими, что их можно было свободно резать ножом.
Гиперпаратиреоз — сложный и многообразный симптомокомплекс, клинические проявления которого обусловлены избыточной продукцией паратиреоидного гормона. Патологические изменения при гиперпаратиреозе проявляются в основном в костях и почках (рис. 20-4).
Костная ткань подвергается рассасыванию вследствие усиленной остеокластической реакции и деминерализации.
Нормальное пластинчатое костное вещество замещается свободной от извести молодой остеоидной тканью. Кости делаются мягкими, происходят разнообразные, иногда причудливые, деформации скелета.
Происходят резкие изменения в минеральном обмене. Паратиреоидный гормон понижает почечный порог в отношении фосфора, ослабляя реабсорбцию в нефронах и усиливая экскрецию фосфатов с мочой.
В условиях гиперпаратиреоза потеря фосфора из организма приводит к мобилизации неорганических соединений фосфора из костей. Так как
Гиперпродукция
Активация деятельности остеокластов
Вымывание
паратгормона
Угнетение реабсорбции фосфатов I Усиленное
кальциево- -фосфорных солей из костей
Гиперкальциемия
Порозность и
выделение фосфатов с мочой
I
Выделение
размягчение костей
Развитие
с фосфатами солей кальция
Отложение
в костях фиброзной ткани
солей кальция в мочевых канальцах
Рис. 20-4. Патогенез основных нарушений при гиперпаратиреозе
Патофизиология эндокринной системы...333 эти соединения являются солями кальция, то, освобождаясь из костей, кальций в избытке поступает в кровь и развивается гиперкальциемия.
Кроме того, паратгормон стимулирует всасывание кальция из кишечника, функционально перекрываясь в этом качестве с другим «гормоном» — витамином D, а также имеет место обратное всасывание кальция из клубочкового фильтрата.
Различают первичный и вторичный гиперпаратиреоз.
Первигный гиперпаратиреоз — обусловлен развитием аденомы или гиперплазией околощитовидных желез.
Вторигный гиперпаратиреоз — гиперплазия и гиперфункция желез возникает в результате первичного изменения кальциево-фосфорного обмена (потери кальция и накопления фосфора в крови).
Как уже говорилось выше, понижение содержания кальция в крови является адекватным раздражителем околощитовидных желез, вследствие чего развивается их гиперплазия и гиперфункция.
Вторичный гиперпаратиреоз имеет компенсаторный характер.
Чаще всего вторичный гиперпаратиреоз бывает при недостаточности почек (погегная остеодистрофия), когда нарушается экскреция фосфорнокислых солей в результате клубочковой недостаточности или же вследствие нарушения обмена ионов в дистальных канальцах.
Лекция 21
НАРУШЕНИЕ ГОРМОНАЛЬНОЙ РЕГУЛЯЦИИ ОСНОВНЫХ ФИЗИОЛОГИЧЕСКИХ ПРОЦЕССОВ ОРГАНИЗМА
Г.В. Порядин
Нарушение гормональной регуляции роста
Важнейшей стороной онтогенеза организма являются процессы его роста, обусловленные в конечном счете интенсивностью синтеза нуклеиновых кислот и белка в тканях, т.е. анаболическими процессами.
Особое место в системе регуляции роста и анаболических процессов среди гормонов принадлежит СТГ. Одна из главных функций этого гипофизарного гормона — стимулирующее влияние на рост, общие размеры тела, его массу, размер и массу отдельных органов.
Во взрослом организме чисто ростовые функции гормона в значительной мере утрачиваются, а анаболические почти полностью сохраняются, играя важную роль в регуляции белкового обмена разных клеток.
Стимулирующий эффект СТГ на белковый синтез обусловлен несколькими механизмами:
• стимулирующим действием на транспорт аминокислот и глюкозы через клеточные мембраны (инсулиноподобный эффект):
• усилением процессов трансляции в рибосомах.
Секреция СТГ передней доли гипофиза находится под двойным контролем гипоталамуса: его секреция усиливается соматолиберином и тормозится соматостатином.
Гипофизэктомия у животных на ранних этапах их онтогенеза почти полностью приостанавливает рост и увеличение массы тела. Введение гипофизэктомированным животным препаратов СТГ стимулирует и полностью восстанавливает интенсивность этих процессов.
При возникновении СТГ-продуцирующих опухолей гипофиза (опухоль из эозинофильных клеток) у человека в раннем детском возрасте развивается заболевание, известное под названием гигантизм.
Нарушение гормональной регуляции основных физиологических...
335
Для этого заболевания характерно значительное пропорциональное увеличение роста и массы тела за счет увеличения массы костей, мышц, внутренних органов. При гигантизме рост может достигать 2 м 70 см, масса тела — 220 кг.
Если опухоли гипофиза, продуцирующие СТГ, возникают у человека в более позднем возрасте, когда зоны роста многих костей уже закрыты, развивается другой тип патологии — акромегалия. Она также связана с гиперпродукцией СТГ, но проявляется в неравномерном разрастании скелета, непропорциональном увеличении некоторых костей черепа, нижней челюсти, кистей рук, стоп, хрящей (нос, ушные раковины).
В случае недостаточности СТГ в организме ребенка развивается обратная форма патологии ростовых процессов — гипофизарная карликовость (лилипутизм, гипофизарный нанизм). Рост и масса тела у таких больных могут быть почти вдвое меньше, чем у здоровых людей.
Регулирующее влияние СТГ на ростовые и анаболические процессы взаимосвязаны с эффектами ряда других гормонов.
Важное место в регуляции процессов роста и прежде всего синтеза белка принадлежит инсулину.
В плане регуляции белкового обмена и ростовых процессов СТГ и инсулин являются синергистами, периферические эффекты и механизмы контроля секреции которых взаимосвязаны.
Инсулин, как и СТГ, ускоряет транспорт аминокислот через мембраны, стимулирует процессы трансляции в рибосомах.
Значительное влияние на процессы роста и синтеза белка оказывают глюкокортикоиды, являющиеся в целом антагонистами СТГ.
В основе катаболического действия глюкокортикоидов лежит прежде всего торможение синтеза белка, а не стимуляция его распада.
Одновременное введение больших доз глюкокортикоидов с СТГ может почти полностью затормозить ростовой эффект последнего.
Существенное влияние на интенсивность роста могут оказывать также тиреоидные гормоны и регулирующие их секрецию ТРФ и ТТГ.
Так, при разных формах недостаточности щитовидной железы у людей, возникшей в детстве, закономерно отмечаются карликовый рост и снижение интенсивности синтеза белка.
Введение тиреоидных гормонов при недостаточности щитовидной железы или полном выпадении ее функций вызывает восстановление скорости этих процессов.
Влияние тиреоидных гормонов на синтез белка в органах и рост тела зависят от их концентрации: в низких концентрациях данные гормоны
336
Лекция 21
стимулируют эти процессы (анаболический эффект), а в высоких — тормозят их (катаболический эффект).
Катаболическое действие тиреоидных гормонов отчетливо проявляется при тяжелых формах тиреотоксикоза.
Мощным анаболическим действием обладают андрогенные гормоны — синергисты СТГ.
Нарушения гормональной регуляции общего развития
Особенно тяжелые изменения в развитии организма наступают при тотальной недостаточности гипофиза.
Общее развитие организма определяется функцией гипофиза и секрецией его гормонов — СТГ, ТТГ, АКТГ, ГТГ.
При снижении секреции всех гормонов аденогипофиза развивается заболевание, известное под названием пангипопитуитаризм, или синдром Симмондса-Шиена.
Причины
Полная недостаточность функции гипофиза может наступить в результате врожденной аплазии и гипоплазии гипофиза, воспалительного повреждения гипофиза, сосудистых нарушений в гипофизе и гипоталамусе, травмы основания черепа, опухоли гипофиза и гипоталамуса.
Снижение секреции гормонов аденогипофиза может быть следствием нарушений образования и секреции рилизинг-факторов в гипоталамусе.
Синдром проявляется нарушением половой функции, уменьшением половых органов, выпадением волос на лобке и в подмышечных впадинах, мышечной слабостью, утомляемостью.
При развитии синдрома в детском возрасте наблюдается отставание в росте, физическом (недостаток СТГ, ТТГ, АКТГ), психическом (недостаток ТТГ), половом (недостаток ГТГ) развитии.
У мужчин, страдающих от сочетанной недостаточности секреции гормонов аденогипофиза, падает половое влечение, возникает импотенция, уменьшаются яички и исчезают некоторые из вторичных половых признаков, в частности волосы на лобке. Одновременно с падением либидо и инволюцией яичек появляются патологические изменения кожи (кожа белеет, истончается, становится сухой и похожей на пергамент). Это снижает резистентность организма к инфекциям, хирургическому и травматическому стрессу.
Нарушение гормональной регуляции основных физиологических...337
Нарушение гормональной регуляции углеводного и жирового обмена
Углеводы и жиры наряду с белками участвуют в обеспечении нормальной жизнедеятельности организма, играя роль важных поставщиков строительных, пластических материалов, входящих в структуры клетки: нуклеиновых кислот, простых белков, гликопротеидов, ряда липидов и т.д.
Синтезируемая благодаря распаду углеводов и жиров АТФ обеспечивает клетки необходимой для работы энергией и является источником образования цАМФ, участвует в регуляции активности многих ферментов.
Первостепенное значение в контроле метаболических процессов углеводного и жирового обмена (как и белкового) принадлежит эндокринной системе. Среди гормонов, участвующих в регуляции жирового и углеводного обмена, центральное место занимают гормоны ЖКТ, инсулин, глюкагон, СТГ, глюкокортикоиды, АКТГ и адреналин.
Основным показателем интенсивности обмена углеводов является концентрация глюкозы в крови (3,5-5,7 ммоль/л), которая зависит от:
• притока моносахаридов в кровь (из кишечника, печени, почек);
• активности фосфорилазной реакции;
• гликонеогенеза;
• оттока глюкозы из крови в ткани (транспорт через клеточные мембраны, скорость утилизации глюкозы, превращения ее в гликоген).
Все эти процессы непосредственно контролируются комплексом гормональных факторов (рис. 21-1).
Окисление глюкозы
инсулин:
г в тканях э (фосфорилирование)
АКТГ СТГ ГЛЮКОКОР-Переход глюкозы в жир ТИКОИДЫ
Синтез гликогена
* Гликонеогенез
* Липолиз
Гликогенолиза
АДРЕНАЛИН ГЛЮКАГОН
Рис. 21-1. Взаимодействие инсулина и контринсулярных гормонов
338
Лекция 21
Например, инсулин стимулирует утилизацию глюкозы тканями и ее депонирование в форме гликогена и в виде жира, тормозит глюконеогенез и, следовательно, снижает концентрацию глюкозы в крови, т.е. является гипогликемическим гормоном.
Такие же гормоны, как глюкагон, адреналин, СТГ, наоборот, стимулируют распад гликогена и глюконеогенез и, следовательно, вызывают повышение содержания глюкозы в крови (гипергликемическое действие).
Они являются антагонистами инсулина — контринсулярными гормонами, диабетогенными гормонами.
Важным показателем интенсивности жирового обмена служит концентрация неэстерифицированных жирных кислот (НЭЖК) (в покое — 500-600 мкмоль/л).
Регуляторами липидного обмена служат в основном те же гормоны, которые регулируют белковый обмен. Это — глюкагон, катехоламины, СТГ, АКТГ, глюкокортикоиды, липотропин, Т3, Т4. В основном они стимулируют липолиз в жировой ткани печени.
Инсулин — единственный гормональный стимулятор синтеза липидов и ингибитор липолиза. Уровень секреции комплекса гормонов, регулирующих углеводный и жировой обмен, находится в зависимости от потребностей организма в энергетических ресурсах.
При голодании, большой мышечной и нервной нагрузке, когда возрастает потребность в использовании углеводов и жиров, в здоровом организме происходит повышение скорости секреции тех гормонов, которые повышают уровень глюкозы и липидов в крови. Одновременно тормозится секреция инсулина.
И наоборот, прием пищи стимулирует преимущественно секрецию инсулина, который способствует синтезу гликогена в печени и мышцах, триглицеридов в жировой ткани и печени, а также синтезу белка. При этом секреция гормонов мобилизации тормозится.
Роль гормонов в нарушении водно-солевого обмена
Наряду с центральной нервной системой специальная группа гормонов ведает осморегуляцией (уровень осмотического давления крови) и поддержанием определенного соотношения ионов натрия и калия.
Главные гормоны этой группы — вазопрессин и альдостерон.
При заболевании гипоталамо-нейрогипофизарной системы, связанной с недостаточностью продукции АДГ, развивается несахарный диабет.
Несахарный диабет — заболевание, характеризующееся полидипсией, полиурией с низкой относительной плотностью мочи.
Нарушение гормональной регуляции основных физиологических...
339
Задняя доля ГИПОФИЗА
Уменьшение продукции АДГ
Нарушение реабсорбции воды в почечных канальцах
ПОЛИУРИЯ
Потеря ВОДЫ (и ЭЛЕКТРОЛИТОВ)
Уменьшение ОЦК
Рефлекторное и непосредственное раздражение «ПИТЬЕВОГО ЦЕНТРА»
ПОЛИДИПСИЯ Q Q
Рис. 21-2. Патогенез несахарного диабета
Этиологические факторы несахарного диабета весьма разнообразны: заболеванию могут предшествовать травмы черепа, многочисленные инфекции (малярия, скарлатина, бруцеллез и др.). Особенно часто развитие несахарного диабета связывается с перенесенным гриппом.
Патогенез несахарного диабета. В норме АДГ действует в дистальных отделах почечных канальцев, активируя реабсорбцию воды, уменьшая диурез. При недостаточности АДГ (рис. 21-2) уменьшается концентрационная способность почек, увеличивается диурез, снижается относительная плотность мочи. Большая потеря жидкости и изменение
физико-химического состава крови приводит к раздражению «питьевого» центра и компенсаторной жажде, обеспечивая пополнение водных ресурсов организма. Величина суточного диуреза резко возрастает (до 30 л мочи за сутки).
Другим гормоном, регулирующим водно-солевой обмен, является гормон коры надпочечников — альдостерон.
При повышении активности клубочковой зоны коры надпочечников развивается синдром альдостеронизма (гиперальдостеронизма). Впервые клиническую картину синдрома описал Конн (в 1955 г.).
Синдром Конна (первичный альдостеронизм) — относительно редкое заболевание, встречающееся преимущественно у женщин, чаще в возрасте 30-45 лет.
Ведущую роль в возникновении синдрома играет повышенная секреция альдостерона клубочковой зоной коры надпочечников.
В 85 % случаев причиной этого заболевания является гормональноактивная опухоль клубочковой зоны — альдостерома.
При длительном повышенном выделении альдостерона развиваются симптомы, связанные с нарушением количества и соотношения электролитов в организме:
• перемежающая тетания;
• периодическая сильная мышечная слабость;
• парестезии;
• полиурия и полидипсия;
• гипертензия и нарушение функции почек.
340
Лекция 21
Условно перечисленные симптомы первичного альдостеронизма могут быть разделены на три группы:
• нервно-мышегные (тетания, мышечная слабость, парестезии, спазмы мышц, судороги);
• погегные (полиурия, полидипсия, ночное мочеиспускание — никтурия);
• связанные с гипертензией (ослабление зрения, головные боли, головокружение).
Основным и первичным проявлением воздействия избытка альдостерона при синдроме Конна являются потери калия с мочой и развитие гипокалиемии.
Для восполнения потери калия во внеклеточной жидкости и крови последний выходит из клеток. Взамен потерянного клетками калия внутрь клеток поступают ионы натрия, хлора и водорода. Это ведет к развитию внутриклеточного ацидоза, а потеря калия с мочой и поступление хлора внутрь клеток — к гипокалиемигескому, гипохлоремигескому алкалозу, что ведет к развитию тетании, при нормальной концентрации кальция и фосфора в крови.
Длительная гиперкалийурия ведет к дегенерации эпителия почечных канальцев, в связи с чем уменьшается чувствительность последних к АДГ. В результате появляется полиурия, которая сопровождается выраженной жаждой (полидипсией).
Полиурией объясняется тот факт, что при синдроме Конна возникновение отеков — явление довольно редкое.
При потере значительного количества калия (10-30 %) наступают мышечная слабость, апатия, потеря аппетита.
Для первичного альдостеронизма характерно возникновение артериальной гипертонии (протекающей иногда в тяжелой форме), которая объясняется повышением тонуса артериол.
Повышение тонуса артериол связано с перераспределением электролитов и задержкой натрия в сосудистой стенке с последующим набуханием стенки сосуда и сужением просвета сосуда.
К тому же задержка натрия в интрацеллюлярной жидкости повышает реактивность симпатической нервной системы в связи с потенцииро-ванием действия норадреналина. У больных обнаруживается усиленное выделение с мочой альдостерона.
В отличие от первичного гиперальдостеронизма ряд неэндокринных заболеваний (такие как гипертоническая болезнь, заболевание почек, сердечная недостаточность различного генеза, цирроз печени и т.д.) мо
Нарушение гормональной регуляции основных физиологических...
341
гут сопровождаться явлением так называемого вторигного альдостеронизма.
Повышенное выделение альдостерона, например, у больных гипер-тонигеской болезнью происходит в том случае, когда в патогенетическую цепь гипертонической болезни включается почечное звено:
• при развитии ишемии почек происходит выделение юкстагломерулярным аппаратом ренина, который способствует образованию ангиотензина I.
Образующийся из него ангиотезин II стимулирует секрецию альдостерона.
Повышенная секреция альдостерона влечет за собой изменение соотношения в тканях натрия и калия, а это потенциирует эффекты катехоламинов, усиливает тонус и набухание гладкомышечных волокон артериол и таким образом способствует прогрессированию артериальной гипертензии.
При сердегной недостатогности, нефрозах, циррозах печени вторичный альдостеронизм имеет определенное значение в патогенезе развития отеков. Однако вторичный альдостеронизм при этих состояниях является только дополнительным фактором возникновения отеков в связи с вызываемым им усилением задержки натрия. Он не первопричина отеков. Задержка натрия в тканях при альдостеронизме только тогда может сопровождаться отеками, если имеется предотечное состояние.
Нарушения гормональной регуляции артериального давления
Уровень артериального давления контролируется и поддерживается нейрогуморальными факторами при участии определенных вазомоторных центров, расположенных в продолговатом мозге. В регуляции тонуса сосудов принимают участие и многие гормоны:
• глюкокортикоидные гормоны коры надпочечников;
• минералокортикоидные гормоны коры надпочечников (выработка и выделение альдостерона в норме стимулируются ионами калия и ангиотензином). Минералокортикоиды увеличивают резорбцию натрия в дистальной части нефрона. Если они присутствуют в избыточном или неконтролируемом объеме, повышение общего содержания натрия в организме ведет к увеличению объема жидкости, возрастанию объема крови и подъему артериального давления. Кроме того, прямое воздействие минералокортикоидов на гладкие мышцы повышает сопротивление периферических сосудов;
342
Лекция 21
• катехоламины, вырабатываемые мозговым веществом надпочечников (катехоламины оказывают множество эффектов на функцию сердечно-сосудистой системы и метаболизм, обеспечивая доставку крови и питательных веществ в условиях выраженного стресса);
• ренин-ангиотензин;
• вазопрессин.
Роль гормонов, вовлеченных в регуляцию артериального давления, наиболее отчетливо проявляется при их избытке или недостатке.
Избыточная продукция гормона роста, глюкокортикоидных и, особенно, минералокортикоидных гормонов корой надпочечников, катехоламинов, гипертиреоз, гиперпаратиреоз, повышенная продукция ренина-ангиотензина способны вызывать значительное повышение артериального давления.
Артериальная гипертензия может быть проявлением самостоятельного заболевания — гипертонигеской болезни (эссенциальная гипертензия, первичная артериальная гипертензия).
Термин «эссенциальная гипертензия» используется для описания состояния пациентов, у которых не известна причина повышения артериального давления. У большинства пациентов с гипертензией отсутствуют какие-либо симптомы, а данные лабораторных исследований остаются в пределах нормальных значений. При обследовании обычно выявляются только высокое артериальное давление и иногда его последствия — заболевания сердца и сосудов головного мозга, нарушение функции почек.
У многих больных прослеживается отчетливая связь повышения артериального давления с генетическими факторами и образом жизни. Стресс, чрезмерное потребление алкоголя, ожирение и содержание в рационе большого количества соли коррелируют с развитием гипертензии. Если эссенциальная гипертензия трудно контролируется или сочетается с гипокалиемией, необходимо исключить эндокринную причину ее возникновения.
Вторигная артериальная гипертензия (эндокринная). Артериальная гипертензия — ведущий компонент многих эндокринных расстройств: при поражении гипофиза (АКТГ-продуцирующая опухоль), адреналовых желез (феохромоцитома, первичный альдостеронизм).
Гипертензия адреналового происхождения (минералокортикоидная — альдостероновая гипертензия). Минералокортикоидные гормоны вызывают гипертензию различными механизмами: за счет увеличения плазмы, объема внеклеточной жидкости.
Нарушение гормональнойрегуляции основных физиологических...
343
При недостаточности гормонов коры надпочечников развивается недо-статогность надпогегников (болезнь Аддисона, бронзовая болезнь). При низком содержании кортизола в плазме нарушается реактивность гладких мышц сосудов, что вносит определенный вклад в развитие гипотензии.
Механизм гипотензии при адреналовой недостаточности отчасти связан с уменьшением объема жидкости, обусловленным дефицитом альдостерона (расстройства функции сердечно-сосудистой системы, нарушение обмена веществ, гиперпигментация кожных покровов и слизистой оболочки).
Эндокринная система и стресс
Эндокринная система занимает серьезный удельный вес в ответе организма на воздействия факторов среды и адаптации его к меняющимся внешним условиям — развитии стресс-реакции.
Термин «стресс» (давление, нажим, напряжение)был введен в медицину канадским патологом Гансом Селье. В своей жизни практически каждый человек испытывал физический и эмоциональный стресс. Без стресса невозможна жизнь. «Стресс, — как писал Селье, — аромат и вкус жизни». Однако избыточный стресс — дистресс, может стать патогенетической основой психических заболеваний, эндокринных расстройств, гипертонической болезни, ишемической болезни сердца, язвенной болезни желудка. Стресс снижает иммунную противоопухолевую защиту организма.
Под стрессом понимают экстремальные условия жизнедеятельности, возникающие после действия чрезвычайных раздражителей на организм человека и животных. Сами же раздражители называются стрессорами.
Стрессорами могут служить самые разнообразные внешние и внутренние факторы, воздействующие на организм: травмы, ожоги, инфекции, интоксикации, наркоз, кровопотеря, действие высоких и низких температур, электротравма, действие ионизирующей радиации, эмоциональные воздействия.
Системы противодействия организма чрезвычайным раздражителям способствуют сохранению гомеостаза.
Стресс-лимитирующие системы. Их роль в развитии стресса
Бурный ответ организма, направленный на ограничение действия стрессора и борьбу с теми неблагоприятными последствиями, которые он может вызвать, несет в себе потенциальную опасность. Она связана
344
Лекция 21
и с возможным истощением запасов адаптивных гормонов глюкокортикоидов, и с формированием в связи с этим острой надпочечниковой недостаточности.
• Классической стресс-лимитирующей системой является опиатная система организма (пропиомеланокортин-гипофиза, р-эндорфин, энкефалины).
• Серотонинергигеская и ГАМК-эргигеская системы, оказывающие тормозящее влияние на реакции ЦНС при стрессе.
• На периферии ограничение стресса связывают с деятельностью системы адениннуклеотидов, простагландинов, антиоксидантных систем, сдерживающих разрушительные процессы в тканях.
• NO-эргигеская система — нейроны стриатума, среднего мозга, гипоталамуса, гипофиза, чувствительные к оксиду азота, ограничивающие активацию симпатоадреналовой системы и периферических звеньев, ответственных за развитие стресс-реакции.
Роль стресса в патогенезе некоторых заболеваний
В настоящее время узаконены термины «острый» и «хронический» стресс. В возникновении и развитии болезней человека как один из важнейших причинных факторов выступает хронический стресс.
Психологический стресс, связанный с неразрешимостью жизненных ситуаций, может провоцировать возникновение не только психических болезней (в этом случае врачи говорят о большой вероятности премор-бидного состояния — готовности к болезни), но и психосоматических заболеваний (ИБС, гипертоническая болезнь, язвенная болезнь желудка и двенадцатиперстной кишки).
Описаны случаи аффективных эндокринных расстройств: нарушение функции щитовидной железы, лактации, течения беременности и климакса у женщин.
Роль стресса в патогенезе язвенной болезни желудка и двенадцатиперстной кишки
Патогенез возникновения язвенной болезни желудка при стрессе связан с чрезмерной активацией как парасимпатического, так и симпатического отделов вегетативной нервной системы, а также с повышением тонуса пептидергических волокон, иннервирующих клетки АПУД-сис-темы ЖКТ. Повышение тонуса п. vagus влечет за собой усиление секреции желудочного сока (повышается его кислотность, количество и активность пепсина), уменьшается образование слизи, происходит за
Нарушение гормональной регуляции основных физиологических...
345
брос дуоденального содержимого в пилорическую часть желудка с его дальнейшим повреждением.
Активация симпатического отдела вегетативной нервной системы приводит к ишемизации стенки желудка, нарушению ее трофики и процессов репарации слизистой оболочки. Усиленная функция G-клеток, продуцирующих гастрин, повышает агрессивные свойства желудочного сока при стрессе. Глюкокортикоиды тормозят местную выработку простагландинов Е2, что ведет к ухудшению проницаемости гистогематиче-ского барьера стенки желудка, кроме того, глюкокортикоиды тормозят процессы регенерации слизистой оболочки желудка.
Существует понятие «глюкокортикоидные язвы». Оно основано на результатах экспериментов на животных, получавших большие дозы глюкокортикоидов, и клинических наблюдениях за больными, леченными большими дозами этих препаратов.
Роль стресса в патогенезе гипертонической болезни
Длительно действующие эмоциональные стрессоры играют значительную роль в становлении гипертонической болезни. Одним из важнейших факторов патогенеза гипертонии является нарушение процессов саморегуляции артериального давления. Возбуждение симпатоадреналовой системы повышает тонус резистивных сосудов, что проявляется в периодически возникающей гипертензии. Если приступы гипертензии повторяются достаточно часто, то подъемы артериального давления уже плохо купируются рефлексами с барорецепторов крупных сосудов, так как понижается их чувствительность к колебаниям АД, и гипертензия закрепляется. Другое звено патогенеза гипертонии реализуется через повышение в крови содержания натрия. Величина артериального давления определяется уровнем ОЦК и тонусом артериол. Натрий увеличивает объем циркулирующей крови и повышает тонус гладких мышц артериол, в частности он повышает чувствительность гладкой мышцы сосудов к катехоламинам. Кроме того, накопление натрия в сосудистой стенке ведет к ее отеку и сужению просвета сосуда, что также увеличивает периферическое сопротивление в артериолах. Описано и возбуждающее влияние глюкокортикоидов на сосудодвигательный центр. Совокупность перечисленных механизмов и составляет основу патогенеза гипертонической болезни.
Стресс и ишемическая болезнь сердца
Анамнез больных инфарктом миокарда свидетельствует в пользу того, что в среднем за полгода до заболевания в жизни этих больных
346
Лекция 21
возникали стрессорные ситуации. Родилась концепция, в соответствии с которой стресс может служить причиной ИБС и ее грозного осложнения в виде инфаркта миокарда. Особенно активно этот фактор работает у лиц с избыточной массой тела, высоким уровнем холестерина в крови, курящих, живущих в условиях гиподинамии.
Высокий уровень катехоламинов при стрессе реализует свой отрицательный эффект на сердечную деятельность через следующие патогенетические звенья.
Катехоламины вызывают длительный или часто повторяющийся ко-ронароспазм, ведущий к ишемическому повреждению миокарда. В основе такого повреждения лежат процессы нарушения энергообразования в миокарде, нарушение работы ионных насосов, накопление ионизированного калия в саркоплазме миокардиоцитов, активация процессов перекисного окисления липидов с образованием биологически активных веществ, отрицательно влияющих на сократительную функцию миокарда и обладающих аритмогенным влиянием.
Прямое некоронарогенное влияние катехоламинов на сердце принято связывать с резким увеличением потребности сердца в кислороде, в связи с активацией процессов возбуждения в сердечной мышце. Катехоламины создают «ножницы» между должным и истинным потреблением кислорода сердечной мышцей — возникает дефицит в энергообеспечении миокарда, результатом которого могут стать метаболические нарушения в сердце.
Катехоламины активируют процессы тромбообразования, в частности увеличивают агрегацию тромбоцитов. Стресс способствует становлению гиперлипидемии и гиперхолестеринемии, роль которых связана с патогенезом атеросклеротического поражения сердца.
Остро возникающий психоэмоциональный стресс может создавать дефицит кислорода в сердце и другими, дополнительными путями:
• за счет резкого снижения тонуса периферических сосудов (коллап-тоидное состояние) и депонирования крови на периферии. Возникающее при этом снижение венозного возврата в сердце может привести к снижению его кровоснабжения;
• во время острой стресс-реакции может возникнуть гипервентиляция легких, которая может вызвать гипокапнический алкалоз, повышающий тонус коронарных сосудов.
Нарушение нервной регуляции деятельности сердца при стрессе, а также ишемия миокарда могут провоцировать нарушения ритма сердца: синусовую тахикардию, реже синусовую брадикардию, появление мно-
Нарушение гормональной регуляции основных физиологических...347 жественных экстрасистол и приступы пароксизмальной тахикардии. Аритмии отрицательно влияют на функциональные возможности сердца и общую гемодинамику.
Гансом Селье в эксперименте было изучено влияние больших доз минералокортикоидов на морфологию и функцию миокарда животных. Он доказал, что большие дозы минералокортикоидов в сочетании с повышенным содержанием натрия в рационе животных вызывают микронекрозы миокарда. Этот вид поражения миокарда принято считать некоронарогенным. Хронический стресс у человека сопровождается активацией ренин-ангиотензин-альдостероновой системы, что предполагает усиление гипертензивных эффектов и накопление натрия в организме. Оба эти фактора являются факторами риска в плане развития повреждений миокарда.
Лекция 22
СТРЕСС И ОБЩИЙ АДАПТАЦИОННЫЙ СИНДРОМ. ИХ РОЛЬ В ПАТОЛОГИИ
Л.И. Зелигенко
В настоящее время в медицине под термином «стресс» подразумевается состояние организма после воздействия на него различных экстремальных факторов физической, биологической и психогенной природы.
Эти факторы принято называть стрессорами. В качестве таких этиологических факторов могут быть названы тяжелые травмы организма, включая хирургические операции, электро- и баротравмы, облучение, ожоги и массивные кровотечения, а также тяжелые инфекции, опухолевые и аутоиммунные заболевания. Различают острый и хронический стресс, а также стресс психогенной природы. Стрессу можно дать следующее определение: это неспецифический набор психофизиологических реакций организма на серьезное повреждение. К этому можно было бы добавить, что если есть возможность увидеть хоть какую-то специфичность в этом «наборе», то она целиком и полностью связана с индивидуальными особенностями организма.
Экспериментальное изучение стресса
Изучая стресс на животных, Ганс Селье пришел к заключению, что стресс у животных протекает в три стадии.
Первую стадию, продолжительностью 24-48 часов, он назвал реакцией тревоги. В эту стадию происходит мобилизация всех резервных возможностей организма в целях противостояния патогенному агенту.
Вторая стадия была обозначена им как стадия резистентности, когда, мобилизуя все свои защитные компенсаторные реакции, организм адаптируется к стрессу, и в случае, когда действие стрессора прекращается или его эффекты уравновешиваются реакциями защиты, организм выходит из стрессового состояния.
Стресс и общий адаптационный синдром. Их роль в патологии
349
Название третьей стадии, обозначенной им как стадия истощения, говорит само за себя. Повреждение в эту фазу уже не может быть компенсировано защитой, а порой возникает ситуация, когда реакции самого организма на стрессорные факторы по своей разрушительной силе превышают эффект самого стрессора. В результате перехода стадии резистентности в стадию истощения нередко возникает гибель организма.
Анализируя различные ткани и органы крыс, подвергавшихся различным видам стресса (инъекции больших доз формалина, звонковый и иммобилизационный шок), Селье обнаружил заинтересованность определенных органов в ходе стрессорной реакции. Он описал ее в форме триады.
Эта триада состоит из следующих изменений в органах:
• гипертрофия коры надпочечников;
• инволюция тимуса и лимфатических узлов;
• не постоянно, в 30-40 % случаев стресса появлялись язвы по ходу ЖКТ.
В первую стадию Селье наблюдал обогащение коры надпочечников секреторными гранулами, сопровождающееся гиперсекрецией кортизола. Во вторую стадию возникала устойчивая гипертрофия коры надпочечников с признаками ее гиперплазии, подтвержденной увеличением числа митозов в клетках пучковой зоны с соответственно высокой продукцией глюкокортикоидов. Если же стресс был либо чрезвычайно интенсивным, либо очень длительным, то кора надпочечников обеднялась липидами и часто «напоминала папиросную бумагу». Эти признаки свидетельствовали о практическом истощении коры надпочечников и прекращении секреции кортикостероидов, т.е. развивалось состояние острой надпочечниковой недостаточности. Необходимо подчеркнуть, что понимание учения Ганса Селье о стрессе привело врачей к использованию в клинике медикаментозной терапии большими дозами кортизола в угрожающих жизни больного ситуациях. Данный метод заместительной терапии позволяет восполнить функции коры надпочечников организма, подвергшегося стрессу.
Это служит прекрасной иллюстрацией того, как экспериментальное исследование может разрешить определенные проблемы клинической медицины.
Инволюция лимфоидных органов, следующий признак триады Селье, может быть правильно интерпретирована, если учесть иммуносупрессивный эффект кортизола. С тех же позиций гиперкортизолизма
350
Лекция 22
можно объяснить и формирование желудочно-кишечных язв. Итак, согласно наблюдениям Селье, стресс можно было охарактеризовать как состояние гиперкортизолизма. Выделим важность этого положения, к которому будем неоднократно возвращаться.
Общий адаптационный синдром
Это понятие, также предложенное Селье, рассматривается как неспецифическая реакция целого организма на действие стрессорных агентов. По современным понятиям общий адаптационный синдром (ОАС) является частью ответа острой фазы, однако если острофазовый ответ протекает при участии четырех жизненно важных систем: ЦНС, эндокринной, иммунной и системы гемопоэза, то ОАС, по Селье, — реакция преимущественно эндокринной системы, а именно системы гипофиз-надпочечники. Кроме того, небходимо добавить, что психогенные факторы не провоцируют острофазовый ответ, в то время как стресс может иметь и психогенную природу.
Пути реализации ОАС после воздействия на организм стрессоров можно представить следующим образом:
• стрессоры активируют различные рецепторы организма: экстеро-рецепторы-ноцицепторы, располагающиеся в коже и слизистых оболочках, а также интерорецепторы, находящиеся в стенке кровеносных сосудов и в глубоких тканях («спящие» рецепторы), а также зрительные, слуховые и другие анализаторы;
• следующим этапом является активация моноаминергических нейронов, под контролем которых находятся пептидергические нейроны гипоталамуса, секретирующие рилизинг- и статин-факторы. Следует отметить, что медиаторами в моноаминергических синапсах ЦНС являются катехоламины (норадреналин, адреналин и допамин), а также такие моноамины, как серотонин и ГАМК. Они определяют баланс секреции статинов и рилизинг-факторов гипоталамусом, что, в свою очередь, решает вопрос о секреторной функции клеток аденогипофиза, продуцирующих тропные гормоны. Секреция гормонов аденогипофиза в соответствии с определенным регуляторным моноамином через либерины и статины может повышаться или снижаться;
• активация моноаминергических нейронов, сопровождающаяся ги-перкатехоламинемией, приводит к усиленному синтезу и секреции кортикотропин-рилизинг-фактора нейросекреторными клетками гипоталамуса;
Стресс и общий адаптационный синдром. Их роль в патологии
351
• КРФ-фактор, в свою очередь, усиливает синтез и секрецию АКТГ базофильными клетками гипофиза;
• последним событием в этой цепи событий, обозначаемой как стрес-сорная ось, является стимуляция синтеза и секреции глюкокортикоидов корой надпочечников, находящихся под контролем АКТГ.
Такая активация коры надпочечников во время стресса «наводняет кровь» глюкокортикоидами. Таким образом стрессорную ось, по Селье, можно представить себе следующим образом: стрессор -> активация гипоталамуса и секреция КРФ -» секреция АКТГ аденогипофизом -» увеличение синтеза и продукции глюкокортикоидов корой надпочечников.
Несомненно, что симпатическая нервная система включается при формировании ОАС первой, однако Селье не занимался изучением роли нервной системы в развитии ОАС. Предметом его изучения была именно роль гипофиз-надпочечниковой системы в механизмах стрессорных реакций. Позже, в 1960-е годы его учение было расширено и дополнено доказательствами регулирущей роли гипоталамуса в этих реакциях.
Итак, гормонами стресса являются катехоламины и глюкокортикоиды, однако эффект катехоламинов весьма кратковременный, в то время как глюкокортикоиды обеспечивают долговременную адаптацию к стрессу. Они усиливают и продлевают эффекты симпатической нервной системы, обеспечивая метаболизм глюкозой, и таким образом поддерживают долговременную энергетику. За счет глюконеогенеза глюкокортикоиды создают постоянный приток глюкозы в кровь, кроме того, глюкокортикоиды поддерживают и системный кровоток.
В настоящее время ни одна большая травматическая операция или другое серьезное повреждение организма (массивные ожоги, значительная кровопотеря) не обходятся без лечения большими дозами глюкокортикоидов. Они также используются при лечении аутоиммунных заболеваний и купирования астматического статуса у больных бронхиальной астмой. О протективной роли катехоламинов и глюкокортикоидов во время стресса речь пойдет ниже.
Протективная роль гормонов стресса
Катехоламины. В начале XX века французский физиолог Клод Бернар, изучая в экстремальных ситуациях эффект катехоламинов на организм животных, назвал их «гормонами борьбы и бегства». Это выражение подчеркивает ту неоценимую помощь, которую эти гормоны оказывают организму во время экстремальных для него ситуаций, т.е.
352
Лекция 22
во время стресса. Катехоламины, являясь первой линией защиты организма, обладают хотя и временной, но высокой способностью мобилизации всех человеческих резервов, позитивно влияя на метаболизм тканей и органов. Модулируя же психические реакции, они позволяют правильно оценить создавшуюся ситуацию и путем изменения поведенческих реакций уменьшить губительное влияние стрессорных факторов на организм.
Механизмы протективного влияния катехоламинов
• Обеспечение должного (в соответствии с изменившимся метаболизмом) уровня глюкозы и жирных кислот в крови. Известно, что катехоламины способствуют гликогенолизу в печени и, являясь контринсулярными гормонами, тормозят эффект последнего на периферии — в инсулинзависимых тканях (мышечная и жировая). Результатом этих метаболических эффектов является повышение уровня глюкозы в крови.
• Жировая ткань — одна из самых существенных мишеней для катехоламинов. Адреналин и норадреналин, через 0,-адренорецепторы стимулируют в ней липолиз, активируя гормончувствительную липазу. Свободные жирные кислоты и триацилглицерол высвобождаются в кровь. Этому эффекту способствует и одновременное ингибирование секреции инсулина, осуществляющееся через те же а-адренорецепторы. Свободные жирные кислоты могут стать весьма ценным субстратом для усиленного энергетического обмена, что особенно важно для метаболизма в сердце и мышцах. Таким образом, на должном уровне поддерживается работа сердца и скелетных мышц, что так необходимо организму во время «борьбы и бегства».
• Влияние на сердечно-сосудистую систему. Обладая положительным хроно- и инотропным влиянием на сердце, катехоламины (в большей степени адреналин) соответственно вызывают тахикардию и увеличение ударного объема, повышая таким образом МОС. Это, в свою очередь, ведет к адекватной перфузии органов в условиях их повышенного метаболического запроса. Регуляция же кровяного давления, небходимого для поддержания системного кровотока в основном осуществляется норадреналином.
• Происходит централизация кровотока за счет его перераспределения в различных органах. Адреналин оказывает сложное влияние на коронарное кровообращение, но преимущественно расширяет коронарные сосуды (стимуляция 0-рецепторов гладких мышц
Стресс и общий адаптационныйсиндром. Их роль в патологии
353
коронарных сосудов). Этот эффект может быть как прямым на гладкие мышцы коронарных сосудов, так и опосредованным, через локальное высвобождение аденозина вследствие стимуляции метаболизма во время активации сердечной деятельности. Через а-рецепторы катехоламины вызывают периферическую вазоконстрикцию в определенных участках сосудистого периферического ложа, особенно в коже, слизистых оболочках, почках, а также в венозном русле. Централизация кровотока достигается изъятием крови с периферии, что позволяет поддерживать на должном уровне центральное венозное давление и диастолическое наполнение сердца.
• Увеличение частоты и глубины дыхания. Адреналин, действуя через Р2-адренорецепторы, является мощным бронхолитическим фактором. Дыхание становится более глубоким и частым, что улучшает газовый обмен между внешней средой и легкими (минутная альвеолярная вентиляция легких повышается).
• Мобилизация психической активности, мотивационный поиск выхода из создавшейся критической ситуации, формирование новых условных рефлексов, стереотипов поведения, а также притупление боли.
Ярким примером адаптивной роли стресса является катехоламиновая адаптация при рождении. Причиной стресса при рождении, во-первых, служит неизбежная гипоксия новорожденного, которую он испытывает во время потуг матери в момент прохождения по родовым путям. При этом происходят сжатие плаценты и пережатие пуповины, что ведет к временному нарушению кровообращения ребенка во время родов. Однако, по мнению педиатров, испытываемый ребенком при рождении стресс полезен для него. При сравнительном анализе содержания катехоламинов в крови у мужчин во время интенсивной физической нагрузки, у них же в сауне, в крови женщин во время родов, в крови больных с феохромоцитомой (опухолью мозгового слоя надпочечников, продуцирующей катехоламины в высокой концентрации) и, наконец, у новорожденного оказалось, что концентрация катехоламинов крови в пикограммах последовательно нарастает в этой цепи и максимальна именно у новорожденных. Катехоламины защищают новорожденного во время рождения и адаптируют его к новой жизни. Оказалось, что в первые месяцы жизни дети, рожденные с помощью кесарева сечения, были менее устойчивы к изменившимся условиями, чем те, что рождались естественным путем. Защитные эффекты катехоламинов, помимо
354
Лекция 22
описанных выше, включают также улучшение всасывания воды с поверхности легких в первые часы после рождения, а также увеличение продукции сурфактанта, обеспечивающего оптимальное расправление альвеол во время акта дыхания.
Активация симпатической нервной системы ведет также к расширению зрачков ребенка и, возможно, участвует в становлении привязанности ребенка к матери. Последнее утверждение, однако, остается лишь предположением, но то, что рождение здоровых детей сопровождается их душевным подъемом, иллюстрируется многочисленными фотографиями новорожденных, на лицах которых запечатлены весьма положительные эмоции.
Глюкокортикоиды. Однако основными гормонами, обеспечивающими долговременную защиту организма во время стресса, необходимо считать глюкокортикоиды.
Роль гормонов коры надпочечников может быть проиллюстрирована их следующим влиянием на системный метаболизм и функции различных органов и систем.
• Активируя глюконеогенез, они обеспечивают долговременное должное поддержание уровня глюкозы в крови.
• Подобно катехоламинам крови, усиливают липолитические процессы.
• Поддерживая системный кровоток и ОЦК крови, работают как минералокортикоиды, усиливая реабсорбцию натрия в почках. Обладают гипертензивным эффектом, помогая обеспечению системного кровотока в экстремальных ситуациях.
• По последним научным данным, их считают косвенными модуляторами функции ЦНС, поскольку высокая концентрация глюкокортикоидов в крови напрямую связана с увеличением продукции кортикотропин-рилизинг фактора (КРФ). Наше поведение и, более того, наш иммунитет определенным образом связаны с продукцией этого фактора гипоталамусом.
• Они обладают мощным противовоспалительным и иммуносупрессивным влиянием. Последнее обстоятельство позволяет организму избавляться от ряда негативных явлений, возникающих во время стресса в форме воспаления и появления чужеродных субстанций, как результат интенсивного повреждения.
• Во время стресса вызванные гиперкортизолизмом изменения в крови носят следующий характер: нейтрофильный лейкоцитоз, эозинопения и лимфопения.
Стресс и общий адаптационный синдром. Их роль в патологии
355
Стресс-лимитирующие системы. Роль этих систем в ограничении стресс-реакций
Как мы уже выяснили, реакция на стрессоры в форме ОАС состоит из двух важнейших этапов:
• активация симпатоадреналовой системы, работающей как аварийная система. Эта реакция сопровождается гиперсекрецией катехоламинов;
• активация гипоталамо-гипофиз-надпочечниковой системы, ведущая к временному гиперкортизолизму, обеспечивающему более долговременную адаптацию организма к той ситуации, которая, с одной стороны, связана с теми поломами, которые вызывает сам повреждающий агент, а с другой стороны — с бурной реакцией организма на это повреждение. Как уже было сказано, последняя по своей силе может превышать негативные последствия фактора, послужившего причиной стресса. В связи с этим в процессе эволюции организма человека и, по-видимому, животных с высокоразвитой ЦНС сформировались механизмы, ограничивающие стресс-реакции.
Стресс-лимитирующие системы ограничивают как центральные механизмы активации нервной и эндокринной систем, так и те, что определяют повреждение органов-мишеней для стресса. Важнейшими центральными ограничителями стресса являются опиоидные пептиды, серотонин, ГАМК.
Возможно, что стресс-лимитирующие системы тренируют организм в отношении повторяющихся стрессорных ситуаций. К периферическим стресс-ограничивающим системам относят антиоксидантную, простагландиновую и систему адениннуклеотидов. Доказано, что центральные системы особенно важны для ограничения бурных катехоламиновых эффектов.
Классической, быстро включающейся в стресс-ограничительную реакцию организма является опиатная система, нейротрансмиттерами которой являются эндорфины и энкефалины, которые вырабатываются в различных образованиях ЦНС и мозговом веществе надпочечников. Интересен тот факт, что стрессор одномоментно с увеличением продукции АКТГ гипофизом стимулирует и образование р-эндорфина этой железой. Это объясняется тем, что оба этих вещества имеют один и тот же предшественник, белок пропиомеланокортин, синтез которого во время стресса увеличивается в гипофизе. В головном мозге син
356
Лекция 22
тезируются и другие опиоидные пептиды из семейства эндорфинов и энкефалинов. В то же время стимуляция симпатической нервной системы приводит к усиленной продукции р-энкефалина мозговым слоем надпочечников.
Опиоидные пептиды способствуют повышению порога болевой чувствительности, что приводит к притуплению чувства боли, а иногда и к полной аналгезии. Механизмы торможения боли объясняются тем фактом, что р-энкефалины, освобождающиеся во время вызванного болью включения нисходящей серотонинергической антиноцицептивной системы, тормозят выделение болевого передатчика — субстанции Р во вторичных ноцицептивных нейронах, находящихся в дорсальных рогах спинного мозга (вариант пресинаптического торможения). Если учесть, что болевые реакции резко снижают физические и психические возможности в плане ответных реакции организма, становится понятна значимость этого эволюционно выработанного механизма.
Снижение активности симпатической нервной системы опиатами связывают как с уменьшением высвобождения норадреналина из пре-синаптических терминалей в соответствующих синапсах, так и с торможением в этих синапсах пострецепторных механизмов передачи импульсов.
К стресс-лимитирующим относят и серотонинергигескую систему. Под влиянием стресса усиливается образование этого медиатора в гипоталамусе, голубом пятне (локус церулеус) и в областях, тесно связанных с лимбической системой. Несомненно, что серотонин влияет на поведенческие реакции человека во время стресса. Доказано, что он, как и опиаты, тормозит адренергические эффекты и обладает модулирующим эффектом в отношении боли (ослабляет боль).
Стресс-реакция, вызванная самыми различными факторами, также сопряжена с активацией ГАМК-эргической системы в полушариях головного мозга и прежде всего с увеличением биосинтеза глутамата и самой ГАМК. Доказано, что активация ГАМК-эргической системы предупреждает в эксперименте образование стрессорных язв желудка и нарушение электрической стабильности сердца в форме фибрилляции, а также остановку сердца при экспериментальной ишемии сердца у животных.
Интересными в отношении стресса являются соображения Мунка (1984) о том, что глюкокортикоиды требуются организму не столько для преодоления начальных эффектов стресса, сколько для предотвращения избыточности реакций на стрессоры. Они защищают организм от из-
Стресс и общий адаптационный синдром. Их роль в патологии
357
быточносги повреждения. Если к этому добавить открытые в 1949 году Хенгом и Кендаллом противовоспалительные эффекты кортизола, то становится ясно, что кортизол можно причислить и к факторам, лимитирующим стресс. Этот многоликий кортизол, с одной стороны, особенно на ранних стадиях стресса, потенцирует эффекты активации симпатической нервной системы и вносит свой вклад в перераспределение метаболитов в целях поддержания на должном уровне энергетического обмена организма с сохранением функции жизненно важных органов. С другой стороны, он лимитирует стрессорные реакции, что происходит гораздо позже, когда начинается разборка последствий повреждения. Кортизол ингибирует образование медиаторов повреждения и их эффекты на органы-мишени, что и определяет его противовоспалительные эффекты. Затянувшееся во времени повреждение вызывает нарушение тканевого гомеостаза и формирование в организме «чужого» с ответом в виде непрекращающихся иммунных реакций; последние могут спровоцировать повреждение. Кортизол гасит их, причем в наибольшей степени он ингибирует клеточные реакции, связанные с активацией Т-лимфоцитов. Именно иммунные реакции клеточного типа ведут к поражению и гибели тканей, и с этих позиций оправдана иммуносупрессорная роль кортизола в ОАС. Добавлением к сказанному может служить и тот факт, что кортизол усиливает апоптоз Т-лимфоцитов.
От стресса к заболеваниям
Эта проблема тесно связана с органами, который стресс выбирает своей мишенью для поражения. Можно предположить существование определенных факторов, от которых это зависит. К ним в настоящее время причисляют следующие.
• Генетический фактор или биологическая предрасположенность органа к повреждению (в частности к отрицательным психосоциальным воздействиям). Условия внешней среды: питание, инфекции, физическое напряжение и различного рода предсуществующие повреждения органа.
• Частота, интенсивность и продолжительность активации органа.
На сегодня очень популярна теория гипокинетического заболевания, в соответствии с которой стрессовая ситуация в сочетании с подавленной физической активностью приводит к возникновению психосоматического заболевания, первичным звеном которого являются серьезные нарушения психического статуса больного, в то время как нарушения в органах (соме) носят вторичный характер.
358
Лекция 22
Как уже было сказано выше, в настоящее время в медицине и психологии узаконены термины «острый» и «хронический» стресс, при возникновении и развитии болезней человека в роли причинного фактора чаще выступает хронический стресс. Психологический стресс, связанный с неразрешимостью неблагоприятных жизненных ситуаций, может провоцировать возникновение не только психических болезней, но и заболевания внутренних органов. Доказано, что органами-мишенями при них могут служить сердце и кровеносные сосуды, ЖКТ, под подозрением находятся эндокринная и иммунная системы, а также кожные покровы. Возникающий как ответ на действие стрессора адаптационный синдром не срабатывает, и поэтому такие болезни еще называют и болезнями адаптации.
Стресс и сердечно-сосудистые заболевания
Сердечно-сосудистая система считается основной мишенью стрессовой реакции. К болезням нарушения кровообращения в первую очередь относят гипертоническую болезнь, ишемическую болезнь сердца и ее угрожающее осложнение в форме инфаркта миокарда, а также аритмии. Есть данные, свидетельствующие о том, что головные боли по типу мигрени и болезнь Рейно с облитерацией сосудов конечностей, чаще нижних (перемежающаяся хромота), могут провоцироваться стрессорными ситуациями.
Роль стресса в патогенезе гипертонической болезни
Следующие нейрогуморальные факторы известны как определяющие при формировании этой патологии.
• Сосудосуживающий эффект катехоламинов.
• Закрепление симпатических влияний при их частой повторяемости. В основе этого явления лежит развитие тенденции барорецепторов аорты и каротидного синуса к перенастройке рефлекторного ответа на более высокое артериальное давление. В норме благодаря депрессорным влияниям подъем артериального давления сдерживается рефлексами с этих зон, однако при перенастройке их ответа на поддержание базового артериального давления требуются все более высокие его значения.
• Высокий базовый уровень кортизола в крови вызывает альдостероновый эффект увеличения содержания в крови натрия и ОЦК, увеличивается и содержание натрия в стенке резистивных сосудов.
Стресс и общий адаптационный синдром. Их роль в патологии
359
Это, в свою очередь, приводит к сужению этих сосудов и увеличению периферического сопротивления кровотоку, а кроме того, усиливает симпатомиметические эффекты гладкой мышцы мелких сосудов.
• Активация ренин-ангиотензин-альдостероновой системы (РААС) через симпатические влияния.
Стресс, ишемическая болезнь сердца и инфаркт миокарда
Анализ условий жизни людей, пострадавших от инфаркта миокарда, показал, что примерно за полгода до инфаркта они пережили стрессор-ную ситуацию. Отсюда логично было заключить, что, возможно, именно стресс стал причастным к возникновению у этих лиц инфаркта миокарда. Несомненно, что наследственность играет значительную роль в возникновении ишемической болезни сердца (ИБС), однако стресс значительно повышает роль факторов риска и часто выступает как пусковой фактор болезни.
Перечислим в высокой степени важные звенья патогенеза ИБС, возможно, связанные со стрессорной ситуацией.
Длительная и частая стимуляция симпатической нервной системы, присущая стрессу, приводит к стойкому спазму коронарных сосудов с последующим ишемическим повреждением миокарда. Важная роль в ишемическом повреждении отводится накоплению в цитоплазме кардиомиоцитов кальция. Кальциевые механизмы повреждения принято рассматривать в соответствии со следующими возникающими при этом феноменами:
• активаций различных клеточных фосфолипаз;
• увеличением активности перекисного окисления липидов;
• ослаблением мощности саркоплазматического ретикулума (СПР) с последующей остаточной контрактурой сердечной мышцы;
• ухудшением работы митохондрий в силу их перегрузки кальцием.
Все перечисленные факторы ведут к нарушению нормального формирования систолической и диастолической функций сердца, что приводит и к нарушению кровоснабжения миокарда.
• Катехоламины обладают способностью активации процесса свертывания крови и могут спровоцировать тромбозы коронарных артерий.
• Перераспределение крови во время стресса вызывает снижение перфузионного давления в сердце, что также нарушает коронарный кровоток.
• Стимуляция симпатической системы способствует различным тахиаритмиям, во время которых укорачивается суммарное диасто
360
Лекция 22
лическое время. Вспомним, что кровоснабжение сердечной мышцы происходит в основном во время диастолы.
• Ухудшению коронарного кровотока способствует характерная для хронического стресса гиперлипидемия. Она является важнейшим звеном патогенеза атеросклероза, роль которого нельзя переоценить в патогенезе ишемической болезни сердца.
Мигрень и болезнь Рейно
Оба этих варианта нарушения периферического кровообращения носят вазоспастический характер. Мигрень считается семейным заболеванием, чаще встречающимся у женщин. Основными ее симптомами являются гемикрания — боль, которую пациент ощущает только на одной стороне головы и лица, нарушение зрения, а также тошнота и рвота. В основе приступов мигрени лежат сосудистые расстройства. При мигрени болевому приступу предшествует спазм гладких мышц мелких внутричерепных сосудов, возникающий вследствие дисбаланса между вазоконстрикторными и вазодилатирующими влияниями на мозговой кровоток с преобладанием первых. В настоящее время при мигрени доказана роль усиленной агрегации тромбоцитов с выделением серотонина, вызывающего спазм венул. Возможно, что приступы головной боли при мигрени могут стать последствием временного нарушения микроциркуляции, ведущей к изменению внутричерепного давления, а также высвобождения медиаторов боли, одним из которых является серотонин. Факторами, провоцирующими мигрень, могут служить аллергены, а также те, что влияют на мозговой кровоток (кофе, курение, стресс). Доказательством участия вазоспастических реакций, связанных с симпатической стимуляции мозговых сосудов, является успешное применение адреноблокатора пропранолола для купирования приступа мигрени.
В случае болезни Рейно холод или хронический эмоциональный дистресс («дурной стресс», по Селье) могут вызывать сужение сосудов кистей, стоп, пальцев верхних и нижних конечностей. Как и при мигрени, при болезни Рейно предполагается наличие у больных повышенного симпатического тонуса.
Роль стресса в патогенезе желудочно-кишечных заболеваний
Стресс соотносят с такими заболеваниям ЖКТ, как язвенная болезнь желудка или двенадцатиперстной кишки, реже его считают повинным в возникновении некоторых форм язвенного колита.
Стресс и общий адаптационный синдром. Их роль в патологии
361
Доказано, что кратковременная стрессовая ситуация, вызванная агрессией, активирует симпатоадреналовую систему, тогда как при депрессии и аутизации (уход в себя), продолжающейся более длительный период, активируется ось гипоталамус-гипофиз-кора надпочечников. Во время эмоций гнева и ярости возрастает содержание пепсина и соляной кислоты в желудке, при депрессии продукция их снижается. Оба фактора ведут к дисбалансу секреторной функции желудка. Хорошо известно, что язвы тела желудка редко сопровождаются повышенной кислотностью желудочного сока, в то время как агрессивные свойства желудочного сока при этом повышены. Описанные нарушения, связанные с изменением секреторной функцией желудка, относят на счет активации парасимпатического отдела вегетативной нервной системы. Повышенная же активность симпатического отдела может повлечь за собой спазм сосудов, питающих стенку желудка или кишечника с последствиями в форме их необратимого ишемического повреждения с выходом в некрозы.
Помимо этого оказывается весьма неблаговидной и роль кортизола в формировании язвенных процессов в ЖКТ. С одной стороны, это было экспериментально показано Селье при обнаружении им триады клинических признаков, характеризующих стресс у животных (язвы по ходу ЖКТ). И в то же время многочисленные клинические наблюдения за состояниями гиперкортизолизма у больных, включающими опухоли, продуцирующие кортизол или АКТГ, а также лечение больных большими дозами кортизола показывают возможность осложнения этих вариантов гиперкортизолизма образованием язвенных процессов в ЖКТ. Глюкокортикоиды снижают репаративные процессы в эпителии, понижают секрецию слизи, делают ее менее вязкой, а именно густая и вязкая слизь необходима для защиты стенки желудка от агрессивных факторов. Известно также, что глюкокортикоиды уменьшают продукцию простагландинов, усиливающих местный кровоток, и таким образом ухудшают питание стенки желудка. В дополнение необходимо отметить негативное влияние кортизола на лимфоидный аппарат ЖКТ, защищающий последний от различных местных патогенных влияний, в том числе и инфекции.
Расстройства скелетно-мышечной системы
Именно эта система отвечает за подвижность тела и, следовательно, играет одну из наиболее важных ролей в реакциях типа «борьбы и бегства». В такой ситуации мышцы напрягаются, приток крови к ним воз
362
Лекция 22
растает, и само слово «напряжение» в этот момент относится именно к состоянию скелетной мускулатуры. Это мышечное напряжение необходимо для того, чтобы держать тело выпрямленным и готовым для спасения бегством. Если сокращение мышц продолжается, а действие в виде физической активности не происходит, мышечный кровоток ослабевает, и в них повышается содержание кислых метаболитов, вызывающих боль. Длительное сокращение мышц головы и шеи приводит к болевому синдрому посредством аналогичного механизма.
Стресс и приступы бронхиальной астмы
Возникающая во время приступа бронхиальной астмы неспособность дышать неизбежно вызывает у больных чувство тревоги, которое создает дополнительный психоэмоциональный стресс. Не вызывает сомнения зависимость частоты приступов бронхиальной астмы от психосоциальных факторов. Особенно ярко это проявляется в детском возрасте. Неблагоприятная психологическая обстановка в семьях таких детей делает их подверженными достаточно частым приступам болезни, и, соответственно, хороший психологический климат в семьях действует на детей в этом отношении благотворно.
Стресс и иммунная система
У хронического стресса обнаружен иммунодепрессивный эффект, который включает в себя как центральные, так и периферические механизмы. Что касается менее изученных центральных механизмов, то важнейшая роль в них отводится кортикотропин-рилизинг-фактору, тесно связанному с иммуномодулирующим эффектом. Дело в том, что при хроническом стрессе из-за повышения базового уровня кортизола по принципу обратной связи уменьшается число рецепторов к кортизолу в гипоталамусе, и секреция КРФ уменьшается. Доказано, что КРФ активирует иммунную систему, и, естественно, снижение его содержания в крови может привести к иммуносупрессии. Имеются интересные данные о том, что КРФ позитивно влияет на психический статус больного, изменение которого не в лучшую сторону влияет на течение хронического стресса.
Глюкокортикоидные эффекты на периферии включают подавление тимико-лимфатической системы (экспериментально выявленная Селье инволюция тимуса и лимфатических узлов при остром стрессе у крыс). Нарушения в системе иммунитета во время стресса дают возможность
Стресс и общий адаптационный синдром. Их роль в патологии 363
предположить его связь с опухолевыми процессами, а возможно, и острыми инфекциями типа гриппа.
Таковы клинические проявления стресса и их предполагаемые механизмы. На самом же деле гамма психосоматических явлений гораздо шире и разнообразнее описанных выше. Такие кожные заболевания, как экзема, псориаз и нейродермиты, а возможно, и угри, часто возникающие в юношеском возрасте, вероятно, тем или иным образом связаны с психологическим стрессом. В заключение можно отметить, что патогенетические механизмы этих патологических состояний нам пока не ясны, и в настоящее время их изучение остается прерогативой клиницистов. Изучение этих актуальных медицинских проблем уже начато сегодня, и залогом успеха лечения этих и других заболеваний, вызванных или спровоцированных стрессом, является умение врача докопаться до причин, вызвавших стресс, а также правильно оценить состояние органов-мишеней, вовлеченных в стресс-реакцию. Чем раньше врачом будут решены эти проблемы, тем более успешным окажется лечение больного. Не вызывает сомнения, что мы находимся на пороге того времени, когда правильная оценка психологического статуса больного с возможной последующей коррекцией (психотерапия) станет одним из патогенетических методов лечения психосоматических заболеваний.
Лекция 23
ПАТОФИЗИОЛОГИЯ СЕРДЕЧНОСОСУДИСТОЙ СИСТЕМЫ
НЛ. Богуш
Недостаточным называется такое кровообращение, которое не обеспечивает органы и ткани кровью соответственно их метаболическим потребностям.
Доля сердечно-сосудистых заболеваний в структуре общей смертности составляет около 53 %.
Многочисленные пригины недостатогности кровообращения объединяются тремя общими механизмами:
• недостаточность кровообращения развивается в результате уменьшения объема крови;
• недостаточность кровообращения развивается в результате нарушения работы сердца как насоса;
• недостаточность кровообращения развивается в результате нарушения работы сосудов.
Выделяют следующие формы и виды недостаточности кровообращения:
• острая и хроническая недостаточность кровообращения;
• компенсированная (скрытая) и декомпенсированная (явная) недостаточность кровообращения;
• сердечная, сосудистая или общая, смешанная формы недостаточности кровообращения.
Сердечная недостаточность
Сердечная недостаточность (СН) возникает в результате повреждения насосной функции сердца, когда оно не способно выбрасывать в кровоток необходимое количество крови против данного давления в сосудах.
В России насчитывается около 3,5 млн больных СН.
Патофизиология сердечно-сосудистой системы
365
Основные механизмы, приводящие к СН, делятся на 3 группы.
• Первичное повреждение сократительного миокарда, прямое повреждение мышцы. К этой группе можно отнести идиопатическую кардиомиопатию, а также инфекционные, аутоиммунные, аллергические, токсические миокардиты, постишемический кардиосклероз, кардиомиопатии при авитаминозах, амилоидозе и др.
• Вторичное повреждение миокарда в результате изнашивания от чрезмерной работы — перегрузочная форма СН.
• СН, возникающая в результате тяжелых аритмий.
СН бывает острой и хронической, компенсированной и декомпенсированной, левожелудочковой, правожелудочковой и смешанной или тотальной. Кроме этого выделяют систолическую, диастолическую и смешанную формы СН.
«Левое сердце» принимает кровь из малого круга кровообращения, которую левый желудочек выбрасывает в аорту и, следовательно, обеспечивает кровообращение в большом круге. Острое повреждение левого желудочка может привести к отеку легких, а в более тяжелых случаях — к падению артериального давления и смерти. В некоторых случаях развивается острый тубулярный некроз в почках, приводящий к азотемии. Изменения в ЦНС в результате уменьшения кровоснабжения мозга варьируют от гипоксической энцефалопатии до комы.
Наиболее частые пригины острой недостатогности левого желудочка:
• острая кровопотеря и шок;
• инфаркт миокарда;
• формирование или декомпенсация некоторых клапанных пороков сердца;
• токсические или инфекционные миокардиты;
• тяжелые аритмии (например, фибрилляция желудочков);
• гипертонический криз при гипертонической болезни, феохромоцитоме, синдроме Конна.
Пригины острой недостатогности правого желудогка:
• массивная тромбоэмболия легочной артерии или ее ветвей тромбами из вен большого круга кровообращения или «правого» сердца;
• респираторный дистресс-синдром взрослых.
Следствием является застой в большом круге кровообращения, проявляющийся быстро нарастающей одышкой, цианозом, падением артериального давления и уменьшением наполнения пульса (из малого круга кровообращения кровь слабо поступает в «левое» сердце), набуханием и пульсацией шейных вен.
366
Лекция 23
Наиболее частыми причинами хронической недостаточности левого желудочка являются:
• гипертоническая болезнь:
• недостаточность клапанов «левого сердца»:
• стеноз аортального клапана.
Более редкие причины — гипертензии при синдроме Иценко-Ку-шинга, Конна, гипертиреозе, феохромоцитоме, гипертензия и гиперволемия при болезни Вакеза.
Причины хронической недостаточности правого желудочка:
• левожелудочковая недостаточность (стеноз митрального клапана, стеноз устья аорты, инфаркт);
• хронические обструктивные заболевания легких;
• заболевания сосудов легких (первичная легочная гипертензия);
• хроническая гипоксемия (падение напряжения кислорода во вдыхаемом воздухе, например высоко в горах).
Все причины, приводящие к перегрузке сердца, можно разделить на преднагрузку и постнагрузку.
Под преднагрузкой понимается перегрузка сердца объемом, когда желудочки сердца перерастягиваются в диастолу и должны выбросить больший объем крови. Например, при недостаточности митрального клапана в систолу часть крови будет поступать парадоксально в левое предсердие (регургитация), а затем в диастолу снова возвращаться в левый желудочек, увеличивая таким образом конечный диастолический объем.
Постнагрузка объединяет 2 группы причин — перегрузку давлением, когда желудочек должен выбросить кровь в систему сосудов, где имеется гипертензия, и перегрузку сопротивлением к изгнанию, когда клапанное отверстие в аорте или легочном стволе сужено.
Всякая сверхнагрузка вызывает увеличение работы сердца, при этом будет возрастать потребление миокардом кислорода. В норме миокард потребляет около 8-10 мл кислорода в 1 мин на каждые 100 г массы, из них 6-8 мл тратится на мышечное сокращение. Наибольшее потребление кислорода характерно для преодоления постнагрузки, меньшее — для преодоления преднагрузки. Определены 4 важнейшие детерминанты потребления кислорода сердцем: 1 — давление в полости желудочков, постнагрузка; 2 — ЧСС; 3 — величина конечного диастолического объема, преднагрузка; 4 — скорость укорочения мышцы (зависит от тонуса симпатических нервов и содержания катехоламинов в крови). В качестве энергетических субстратов сердце использует свободные жирные кислоты (60 % субстратов), глюкозу (30 %) и лактат (около 10 %).
Патофизиология сердечно-сосудистой системы
367
Сердце обладает возможностью быстро увеличить силу сердечных сокращений срогными механизмами адаптации-.
• соотношение длины и силы или гетерометрический механизм, закон Франка-Старлинга: сердце сокращается более сильно (возрастает ударный объем) во время систолы, если оно наполняется в большей степени во время диастолы (желудочки сильнее растягиваются). Количество крови, перекачиваемой сердцем каждую минуту, практически полностью зависит от поступления крови в сердце из вен, обозначаемого термином «венозный возврат». Механизм Франка-Старлинга обеспечивает приспособление сердца к изменениям объемов притекающей крови;
• гомеометрический механизм — он состоит в том, что увеличение силы сокращения возникает при неизменной исходной длине волокон в ответ на увеличенную нагрузку, например, при гипертензии;
• инотропный эффект высокой частоты сокращений;
• инотропный эффект катехоламинов.
Вспомним, что сокращение кардиомиоцита происходит благодаря работе актомиозиновых мостиков, которые продвигают актиновые волокна вдоль миозиновых. Для образования мостиков необходимо поступление ионов кальция из саркоплазматического ретикулума и внеклеточного пространства (последнее обеспечивает до 40 % напряжения, развиваемого кардиомиоцитом при сокращении), активация кальцием тропонина, образование открытых актиновых центров, АТФ и хорошая АТФазная способность миозина. Для повторного сокращения кальций должен быть удален из цитоплазмы, мостики разрушены, а запасы АТФ пополнены. АТФ образуется в процессе цикла трикарбоновых кислот и окислительного фосфорилирования в митохондриях и доставляется к работающим миофибриллам креатинфосфатной транспортной системой. Чем больше при возбуждении кальция в цитоплазме (до физиологического предела), тем больше может быть образовано мостиков и тем больше будет сила сокращения.
Закон Старлинга обеспечивает увеличение развиваемого напряжения или амплитуды сокращения в ответ на увеличение исходной длины клеток сердечной мышцы и заложенных в них саркомеров. Так, сила сокращения при длине саркомера 2,2 мкм больше, чем при длине 1,6 мкм. В последнем случае между концами тонких актиновых протофибрилл, которые отходят от противоположных концов саркомера навстречу друг другу, отсутствует какая-либо дистанция и сразу же после начала сокра
368
Лекция 23
щения развивается эффект взаимозахождения тонких протофибрилл, который ограничивает зону их контакта с миозиновыми протофибриллами и тем самым количество актомиозиновых мостиков. При длине саркомера 2,2 мкм между концами тонких протофибрилл появляется больший зазор, поэтому на определенном этапе сокращения взаимозахождения тонких нитей не происходит, зона же контакта между тонкими и толстыми протофибриллами оказывается максимальной. Это обеспечивает максимальное количество актомиозиновых связей и увеличение силы сокращения. Еще одно объяснение этого феномена основано на зависящем от длины изменении чувствительности миофиламентов к кальцию: при большей длине активируется большее количество поперечных мостиков, что ведет к повышению активного напряжения и силы сокращения. Следовательно, закон Старлинга говорит о том, что по мере увеличения исходной длины сердечной мышцы в физиологических пределах зона образования актомиозиновых мостиков увеличивается, и, как результат, возрастают сила, амплитуда и скорость сокращения. Этот механизм помогает справиться сердцу при преднагрузке (перегрузке объемом).
Гомеометригеский механизм. Увеличение сопротивления изгнанию крови и укорочению сердечной мышцы приводит к уменьшению скорости укорочения. Такой фактор действует при гипертензии в сосудах, в которые желудочек должен выбросить кровь, или при стенозе отверстия, через которое кровь должна быть перемещена в сосуды. Это означает, что актомиозиновые мостики совершают свои «гребущие движения» в замедленном темпе и актиновые протофибриллы более медленно скользят вдоль миозиновых. В результате возрастают время экспозиции и процент актиновых центров, к которым успевают присоединиться головки миозина. Количество актомиозиновых мостиков увеличивается, и сила сокращения тоже увеличивается. Этот механизм помогает преодолеть сердцу постнагрузку.
Вышеприведенные инотропные эффекты не зависят от существенного трансмембранного транспорта кальция. Некоторое возрастание количества кальция в цитоплазме через несколько минут после удлинения мышцы может быть результатом активации чувствительных к растяжению кальциевых каналов в клеточных мембранах. Два других механизма, регулирующих силу и скорость сокращений сердца, увеличивают транспорт кальция через мембраны внутрь клеток (возможна перегрузка клеток ионизированным кальцием).
Инотропный эффект высокой гастоты сокращений. При физической работе может происходить 2-3-кратное увеличение частоты сердечных
Патофизиология сердечно-сосудистой системы
369
сокращений. При этом после некоторого латентного периода происходит увеличение силы сердечных сокращений (лестничный феномен Боудича). Основой увеличения силы и скорости сокращений является увеличение поступления кальция в клетку во время плато потенциала действия. Увеличение кальция -> увеличение кальциевотропониновых комплексов увеличение актомиозиновых мостиков увеличение силы сокращений.
Инотропный положительный эффект катехоламинов — усиление силы сокращения миокарда также под действием симпатической нервной системы — опосредован ^-адренорецепторами и цАМФ. В результате увеличиваются приток в клетку кальция и депонирование его, что позволяет использовать данный ион в большем количестве при следующей деполяризации. Норадреналин увеличивает также интенсивность расслабления. В целом сокращение получается более мощное и более краткое по времени.
В результате непрерывной компенсаторной гиперфункции сердца, возникающей как вариант адаптации при патологии, развивается пато-логигеская гипертрофия сердегной мышцы. Масса миокарда становится в среднем больше 500 г. Так, масса «легочного сердца» увеличивается до 350-600 г, масса сердца при гипертонической болезни, аортальном стенозе и митральной недостаточности — до 400-800 г, при аортальной недостаточности — до 600-1000 г. При пороках сердца, гипертонической болезни сердце вынуждено работать в условиях сверхнагрузки без отдыха. При диффузном атеросклерозе, авитаминозах, инфаркте миокарда поврежденная мышечная ткань не в состоянии функционировать с нормальной интенсивностью, и только благодаря гипертрофии (увеличению массы этой ткани) сердце в целом продолжает поддерживать нормальный кровоток. Следовательно, 5-й механизм адаптации сердца, возникающий при хронической перегрузке, — это компенсаторная па-тологигеская гипертрофия или ремоделирование миокарда.
Физиологическая гипертрофия развивается в результате дозированных тренировок, распространяется на все камеры сердца и характеризуется возрастанием потенциальных возможностей гипертрофированной мышцы. Напротив, установлено, что максимальная сила, скорость и энергетическая эффективность сокращения патологически гипертрофированной мышечной ткани снижаются и ее функциональный резерв уменьшается сразу же после достижения большой гипертрофии. Считается, что в основе этого явления лежит несбалансированная форма роста мышечной ткани.
370
Лекция 23
• От массы миокарда отстает рост иннервирующих его аксонов симпатических нейронов, снижается плотность симпатической иннервации органа, трата норадреналина превышает его ресинтез, и концентрация этого медиатора в миокарде падает в 3-5 раз. Снижается управляемость сердца, уменьшается положительный инотропный и расслабляющий эффект симпатической нервной системы.
• Отстает рост артериол и капилляров — уменьшается коронарный резерв сердца. При возрастании нагрузки на такое сердце доставка большего количества кислорода нарушена, уменьшается выработка энергии, могут возникать зоны ишемии. Патологическая гипертрофия — фактор риска ишемической болезни сердца и инфаркта миокарда.
• Масса клетки увеличивается в значительно большей степени, чем ее поверхность, — падает относительная или удельная поверхность миокарда. Особенно страдает функционирование ионных насосов, что способствует накоплению в клетке ионизированного кальция.
• Относительно возросшей массы мышцы снижается масса митохондрий — падает мощность энергообеспечения клеток.
• Синтезируется миозин с низкой АТФазной способностью, что снижает скорость сокращения сердечной мышцы.
Патологическая гипертрофия сердца, согласно Ф.З. Меерсону, проходит 3 стадии.
I. Аварийная или стадия острой гемодинамигеской нагрузки. Начинается сразу после воздействия повреждающего фактора на сердце. Для поддержания в этих условиях должного минутного объема включаются срочные механизмы адаптации сердца:
• увеличивается частота сердечных сокращений;
• активируется симпатическая нервная система — реализуется положительный инотропный эффект катехоламинов, при перегрузке объемом;
• реализуется механизм Франка-Старлинга;
• активируется ренин-ангиотензин-альдосгероновая система для увеличения объема крови и поддержания механизма Франка-Старлинга.
В клетках миокарда происходит синтез белков теплового шока, которые защищают миокард от повреждения. Они увеличивают мощность антиоксидантных систем, связывают СЖК и избыток ионов Са2\ уменьшают ПОЛ, связываются с белками и экранируют их от протеаз, а также способствуют восстановлению поврежденных рибосом. Структуры, участвующие в срочной адаптации, живут от 2 до 12 сут, поэтому рано на
Патофизиология сердечно-сосудистой системы 371
чинают синтезироваться сократительные белки, и уже через 30-60 мин появляются первые тонкофибриллярные структуры, которые представляют новообразованные миофиламенты. Аварийная стадия продолжается обычно 1-2 нед. В эксперименте на кроликах было показано, что после сужения устья аорты в 4 раза масса сердца возрастает на 80 % уже через 5 сут.
II. Стадия устойгивой гипертрофии сменяет аварийную. Масса сердца увеличивается в 1,5-3 раза и больше не растет. Увеличенная функция органа распределяется в его возросшей массе. Согласно формуле Т = Р х г -5- 2S, напряжение (Т), развиваемое сердечной мышцей, равно произведению давления в желудочке Р на радиус желудочка (г), деленному на удвоенную толщину стенки желудочка 2S. При увеличении толщины стенки систолическое напряжение снижается. Таким образом, гиперфункция вызывает гипертрофию, а возникшая гипертрофия устраняет эту гиперфункцию. Эта стадия может существовать многие годы, обеспечивая работу сердца в условиях его повреждения. Однако сниженный функциональный резерв патологически гипертрофированного сердца постепенно приводит к последней стадии.
III. Стадия прогрессирующего кардиосклероза и изнашивания структур или стадия недостатогности гипертрофированного сердца. Нарушение энергетического обмена приводит не только к уменьшению сократительной функции сердца, но и к уменьшению синтеза белка, восстановлению сократительных структур. Уменьшение сердечного выброса обусловливает дилатацию камер сердца (Тг), гибель кардиомиоцитов снижает толщину стенки (TS ), следовательно, напряжение (Т) в стенке увеличивается. Растет давление крови в полости сердца на каждую растянутую миофибриллу, она повреждается, погибает и замещается соединительной тканью. Развивается эффект соскальзывания или транслокации миофибрилл. Стенка становится все тоньше, полость — все больше, сила сокращения — все меньше. Развивается хроническая сердечная недостаточность. В собственном смысле слова, это состояние, при котором нагрузка, падающая на сердце, превышает его способность совершать работу.
Причины патологической гипертрофии, ее пусковые факторы
• Возрастание гемодинамической нагрузки — увеличение частоты сердечных сокращений, пред- и постнагрузки.
• Наследственные факторы. Показано, что у молодых людей с отягощенной семейной наследственностью по гипертонической болезни гипертрофия миокарда может предшествовать увеличению АД.
372
Лекция 23
• Повышение активности нейрогуморальных систем. Увеличение норадреналина, гормона роста, тромбоцитарных факторов роста приводит к развитию гипертрофии. Ангиотензин-П, который образуется в ткани сердца из ангиотензина-I под действием химазы (АПФ), приводит к гипертрофии сердца, воздействуя на рецепторы АТ2-1. Альдостерон влияет на рост стромы миокарда, способствуя появлению жесткого соединительнотканного каркаса, т.е. развитию фиброза миокарда.
• Появление цитокинов — ИЛ-1, ФНО-а и интерферона-у. Они вызывают продукцию индуцированной NO-синтазы, которая оказывает токсическое действие на миокард, стимулируя апоптоз кардиомиоцитов и развитие фиброза. ФНО-а увеличивает образование токсических радикалов кислорода, вызывая оксидативный стресс, перегрузку кардиомиоцитов ионизированным кальцием, что уменьшает выработку АТФ и сократительную способность миокарда, а также способствует эффекту соскальзывания миофибрилл.
Механизм гипертрофии. Вышеперечисленные пусковые факторы активируют клеточные протоонкогены. Кардиомиоциты начинают синтезировать факторы роста, рецепторы для них и других факторов роста, внутриклеточные переносчики и белки, регулирующие трансдукцию генов. Для патологической гипертрофии характерен изогенный сдвиг — запускаются фетальные (эмбриональные) генетические программы: миофибриллы располагаются беспорядочно как у эмбриона, цепи миозина с высокой АТФазной способностью заменяются их эмбриональными формами, тяжелые цепи меромиозина переходят в его медленно сокращающуюся 0-форму, тропонин Т — в протропонин TN и др. На крысах со спонтанной артериальной гипертензией показано, что в антенатальном периоде апоптоз отстает от пролиферации, что в итоге приводит к первичной гипертрофии миокарда.
Показатели внутрисердечной гемодинамики и функциональных свойств миокарда
Охарактеризовать состояние сердца можно с помощью показателей внутрисердечной гемодинамики и функциональных свойств миокарда:
• КДО — конечный диастолический объем — объем крови, находящийся в полости левого или правого желудочка в конце диастолы, непосредственно перед систолой. В среднем равен 120 мл крови. При сердечной недостаточности увеличивается. При преднагрузке (перегрузке объемом) КДО возрастает прежде всего (вместе с КСО).
Патофизиология сердечно-сосудистой системы
373
• КСО — конечный систолический объем — объем крови, находящийся в полости левого или правого желудочка после систолы, непосредственно перед диастолой. В среднем равен 40 мл крови. При сердечной недостаточности увеличивается.
• УО — ударный или систолический объем — объем крови, который левый или правый желудочек выбрасывает в аорту или, соответственно, в легочный ствол. В среднем равен 80 мл крови. При сердечной недостаточности УО уменьшается: УО = КДО - КСО.
• ИФ — изгоняемая фракция крови или фракция выброса — отношение УО к КДО. ИФ = УО/КДО = 0,67. При сердечной недостаточности ИФ уменьшается.
• КДД — конечное диастолическое давление — величина давления крови в желудочках в конце диастолы. КДД для левого желудочка — 5-12 мм рт.ст., для правого----2+5 мм рт.ст. При СН увеличива-
ются. Для определения величины КДД левого желудочка может использоваться такой показатель, как давление заклинивания легочных капилляров (5-13 мм рт.ст. в норме). Чем оно выше, тем больше вероятность развития отека легких. При постнагрузке КДД возрастает прежде всего.
Характеристика функциональных свойств миокарда
• Сократимость желудогков. Оценивается по максимальной скорости увеличения давления в полости желудочка во время изоволумиче-ской фазы сокращения (продолжительность этой фазы — 0,032 с). Максимальная скорость приращения давления — максимальная dP/dt — первая производная внутрижелудочкового давления — есть мера сократимости сердца. При сердечной недостаточности она уменьшается (менее 2000 мм рт.ст./с).
• Кривая функции желудогков или зависимость УО (или «работы за удар») от величины КДО (или КДД). Кривая смещается вверх и влево при увеличении сократимости сердца, физической активности, тренированности, активации симпатической нервной системы и гиперкатехоламинемии, увеличении ЧСС, возрастании кальция в клетке, действии сердечных гликозидов. При этом ИФ увеличивается. При СН кривая смещается вниз и вправо, ИФ уменьшается. Способствуют такому смещению кривой гипоксия, гиперкапния, ацидоз, перегрузка кальцием, повреждение миокарда (рис. 23-1).
• Растяжимость желудогков (compliance). Растяжимость желудочков важна для наполнения желудочков кровью в диастолу и для формирования УО. Определяется как отношение скорости изменения
374
Лекция 23
Одышка Отек легких
Растяжение / миокарда
Рис. 23-1. Кривая функции желудочков
объема желудочка (dV) к скорости изменения давления в полости желудочка (dP) — dV/dP. При толстой или тугой мышце желудочка для достижения в диастолу нормального объема крови — УО, КДО, необходимо приложить большее усилие для его растяжения, увеличения его полости. Растяжимость желудочков уменьшается при ишемии, гипертрофии, фиброзе миокарда, увеличении в желудочках КДД, дилатации желудочков и др. Снижение растяжимости может приводить к уменьшению УО, росту в желудочке КДД, а затем и росту соответствующего давления в предсердии и впадающих в него венах, возрастанию роли систолы предсердий в наполнении желудочков.
Лекция 24
ПАТОФИЗИОЛОГИЯ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
НЛ. Богуш
В больном организме при компенсаторной гипертрофии сердца из-за снижения максимальных возможностей образующей его мышечной ткани появляются начальные сдвиги гемодинамики. Они изменяют нейроэндокринную регуляцию функций организма, которая, с одной стороны, тормозит основной обмен, двигательную активность, т.е. уменьшает требования организма к системе кровообращения, а с другой стороны, активирует внесердечные механизмы компенсации, которые сдерживают нагрузку на сердце, стимулируют работу ослабевшего сердца и тормозят развитие сердечной недостаточности.
При этом показатели общей гемодинамики могут оставаться нормальными долгое время:
• МОС = 5 л/мин в покое, увеличивается до 25-30 л/мин (МОС макс) при максимальной физической нагрузке. МОС = ЧСС х УО. По методу Фика: МОС = количество поглощенного кислорода в мл/мин, деленное на артериовенозную разницу по кислороду в мл/л крови. Внимание: 1 об.% кислорода = 10 мл кислорода/л крови. При сердечной недостаточности МОС падает.
• СР — сердечный резерв = 20-25 л/мин. СР = МОС максимальный — МОС в покое. При сердечной недостаточности уменьшается.
• СИ — сердечный индекс = 3,1-3,2 л/мин/м2. СИ = МОС/площадь тела. При сердечной недостаточности уменьшается.
• АВ (артериовенозная) разница по кислороду = 6 об.%. А02 - В02 =
19 об.% - 13 об.%. Обмен кислорода происходит в капиллярах. При ослаблении сердечной деятельности МОС и скорость тока в капиллярах падают, время контакта крови с тканями в капиллярах увеличивается, ткани забирают больше кислорода, следовательно, содержание кислорода в венозной крови падает менее 13 об.%. В итоге АВ разница по кислороду увеличивается.
376
Лекция 24
• КУ02 — коэффициент утилизации кислорода = 0,3. КУ02 = (А02 -В02)/Ао2 При сердечной недостаточности (СН) увеличивается.
• САД — среднее артериальное давление = 100 мм рт.ст. При СН падает.
• Давление в крупных венах — 12-15 мм рт.ст. При СН увеличивается.
Механизмы внесердечной компенсации
1. Повышение тонуса симпатической нервной системы — первое средство защиты от опасности развития нарушений кровообращения. Происходит по механизму рефлекторной реакции с участием барорецепторов. Барорецепторы дуги аорты (находятся в стенке сосуда, являются рецепторами растяжения) и каротидного синуса не реагируют на низкое АД (до 60-80 мм рт.ст.). Их импульсация постепенно возрастает с ростом АД и достигает максимума при АД = 180 мм рт.ст. При СН снижение УО, МОС и АД инактивирует эти рецепторы, и они перестают оказывать тормозящее влияние на сосудодвигательный центр. Возбуждение симпатического отдела сосудодвигательного центра приводит к увеличению частоты и силы сердечных сокращений, перемещает сниженную кривую функции желудочков вверх и влево по направлению к нормальному положению, вызывает сужение вен и увеличение венозного возврата (включается механизм Франка-Старлинга), а также повышение периферического сосудистого сопротивления и нормализацию АД. С прогрессированием болезни симпатический тонус нарастает, происходит сужение артериальных сосудов (централизация кровотока), что внешне проявляется бледными и холодными кожными покровами.
Отрицательными сторонами этой реакции являются возрастание нагрузки на сердце (как преднагрузки, так и постнагрузки), увеличение потребности сердца в кислороде, активация РААС.
2. Гиперкатехоламинемия. Активация симпатической нервной системы вызывает выделение катехоламинов из мозгового вещества надпочечников и из вазомоторных окончаний. Концентрация их в крови возрастает с 200 до 600 пг/мл. Катехоламины поддерживают эффекты симпатической нервной системы на сердце и сосуды.
3. Активация РААС (вторичный альдостеронизм) происходит в результате активации симпатической нервной системы (клетки ЮГА почек имеют p-адренергические рецепторы) и в результате уменьшения кровотока через кору почек. Ренин + ангиотензиноген -> ангиотензин-1
Патофизиология сердечной недостаточности
377
+ АПФ ангиотензин-П секреция альдостерона задержка Na* почками секреция АДГ—> задержка воды почками -> ТОЦК. В результате: а) увеличивается венозный возврат к сердцу -> стимулируется механизм Франка-Старлинга -> увеличивается УД -> Т МОС Т АД; б) ТОПСС в ответ на возросший ОЦК и воздействия на сосуды Na*, ангиотензина-П и АДГ= вазопрессина -> Т АД. Повышенная задержка жидкости вызывает увеличение МОС до нормального уровня и одновременно позволяет снизиться активности симпатических нервов до нормы.
Натрий накапливается в организме (на 30-40 % больше нормы) в межклеточной жидкости и в стенке сосудов. В крови происходит раз-ведение натрия, гипонатриемия и его концентрация становятся ниже нормы — менее 135 ммоль/л (норма — 140-148 ммоль/л). Вследствие задержки Na происходит потеря К* через почки с развитием гипокалиемии. Это приводит к угнетению сократительной функции сердца. При значительном дефиците К* возникают некрозы миофибрилл.
4. Активация секреции предсердного натрийуретического фактора — атриопептина (ПНУФ) происходит в результате дилатации предсердий, в основном правого. Он способствует выведению натрия и воды и разгружает сердце.
5. Одышка возникает вначале в ответ на возбуждение сосудодвигательного центра. При прогрессировании болезни развиваются застойные явления в легких. Ухудшается газообмен, и одышка возникает в ответ на раздражение сосудистых хеморецепторов недостаточным количеством кислорода, избыточным — углекислого, а также снижением pH крови. Увеличение минутного объема дыхания способствует поддержанию нормального газового состава крови. Кроме того, частое и поверхностное дыхание усиливает венозный возврат к сердцу в результате деятельности дыхательного насоса.
6. Развитие абсолютного эритроцитоза из-за гипоксемии и ответного усиления синтеза эритропоэтина почками. Уменьшает гипоксию.
Эти механизмы поддерживают работу сердца и гемодинамику в организме. Но они же создают и дополнительную нагрузку на сердце, в конце концов способствуя его дальнейшему повреждению.
Так, ангиотензин-П, оказывая положительный инотропный эффект на миокард, вызывает спазм коронарных сосудов, апоптоз кардиомиоцитов, способствует развитию гипертрофии как сердца, так и мышечной стенки сосудов, поддерживая постнагрузку. Он вызывает высвобождение катехоламинов из мозгового вещества надпочечников, в почках сокращает сосуды, участвует в гибели клубочков. В головном мозге сти
378
Лекция 24
мулирует «питьевой» центр, усиливает выделение АДГ, АКТГ, активирует симпатическую нервную систему.
Противоборствующий эффект ПНУФ со временем прекращается.
В результате компенсаторная гипертрофия сердца переходит в следующую стадию — прогрессирующего кардиосклероза и развитию хронической застойной сердечной недостаточности.
Хронигеская застойная сердегная недостатогность — комплексный клинический синдром, характеризующийся патологическим изменением функции левого желудочка и нейрогуморальной регуляции. Сопровождается низкой толерантностью к физической нагрузке, явлениями задержки жидкости в органах и тканях и значительным уменьшением продолжительности жизни.
Симптомы хронической застойной СН:
• бледные и холодные кожные покровы;
• цианоз, акроцианоз;
• отеки, может быть асцит, увеличение размеров застойных печени и селезенки;
• одышка;
• тахикардия;
• уменьшение толерантности к физическим нагрузкам;
• ремоделирование миокарда, что означает при хронической СН:
— патологическое расширение полостей сердца и изменение геометрии его полостей (они становятся шаровидными);
— патологическую гипертрофию;
— резкое увеличение напряжения стенок сердца;
— выраженные изменения систолической и диастолической функции.
Характеристики ухудшенной систолической функции: снижение ИФ (менее 0,4), СИ (менее 2,5 л/мин/м2), увеличение КСО и КДО: для левого желудочка — увеличение КДД и давления заклинивания легочных капилляров.
Характеристики ухудшенной диастолической функции: ухудшение расслабления и податливости желудочков (compliance).
Механизмы, обусловливающие симптомы хронической застойной сердечной недостаточности
Бледные и холодные кожные покровы — проявление спазма сосудов кожи (периферических сосудов) и централизации кровотока. Происходит в результате активации симпатической нервной системы. Кожные
Патофизиология сердечной недостаточности
379
покровы становятся влажными, так как потоотделение — единственный механизм выведения излишков тепла в условиях спазма сосудов кожи.
Цианоз, акроцианоз — результат замедления тока крови и возрастания времени поглощения кислорода тканями. Ухудшение оксигенации крови в застойных легких также способствует развитию цианоза. В венозной крови увеличивается содержание восстановленного НЬ более 50 г/л, что придает характерный темно-вишневый цвет крови. Затруднение оттока и увеличение ОЦК приводят к расширению кожной венозной сети. Максимальное замедление скорости кровотока наблюдается в капиллярах в отдаленных от сердца частях тела. Если в этих местах кожа тонкая, эта синюшность наиболее выражена и описывается как акроцианоз.
Отеки. Пусковым фактором является повышение гидростатического давления на венозном конце капилляров в результате повышения давления в крупных венах, предсердиях и желудочках. Дополнительно способствует транссудации снижение ОД, так как синтез альбуминов застойной печенью снижается, кроме того, альбумины могут теряться с мочой при застойной почке. Накопление жидкости (положительный водный баланс) обусловливает активация РААС.
Одышка. Застойные явления в легких приводят к увеличению толщины альвеолярно-капиллярного барьера и нарушают диффузию газов, в первую очередь кислорода. Гипоксемия, гиперкапния и ацидоз раздражают хеморецепторы сосудов. Кроме того, транссудат раздражает юкстакапиллярные рецепторы в легких. Развивается частое и поверхностное дыхание. Вытеснение альвеолярного воздуха жидкостью уменьшает жизненную емкость легких (ЖЕЛ) (по рестриктивному типу нарушения вентиляции). Ригидные легкие увеличивают нагрузку для дыхательной мускулатуры, возникает чувство затруднения дыхания. Ухудшается соотношение вентиляции и перфузии, увеличивается величина мертвого пространства, усиливается гипоксемия. Отек стенок бронхов вызывает небольшую обструкцию дыхательных путей, появляется «сердечная астма» (обструктивная ДН). Особенно затруднено дыхание в лежачем положении, так как кровь из нижних конечностей и брюшной полости притекает к сердцу, и нагрузка на него возрастает. Поскольку сердце работает на плоской части кривой функции желудочка, любое увеличение притока крови приводит к росту КДД, следовательно, к росту давления в легочных капиллярах. Известно, что больные застойной сердечной недостаточностью спят практически сидя, используя 2 подушки и более. Приступ ночной сердечной астмы можно объяснить увеличенным притоком крови от конечностей, снижением активности симпатической нервной системы (выпадает ее положительный
380
Лекция 24
инотропный эффект), повышением активности парасимпатический системы, а также снижением возбудимости дыхательного центра.
Тахикардия. Развивается рефлекторно. Снижение УО, МОС и АД приводит к возбуждению латеральных отделов сосудодвигательного центра через барорецепторы дуги аорты и каротидного синуса (рецепторы высокого давления). Симпатические нервы проводят возбуждение к сердцу, и ЧСС увеличивается. Раздражение хеморецепторов этих же зон при падении парциального давления кислорода, pH и возрастания рСО2 также через симпатические нервы увеличивает ЧСС. Увеличение давления в правом предсердии из-за уменьшения насосной функции правого желудочка также вызывает тахикардию. Она возникает в соответствии с рефлексом Бейнбриджа: растяжение правого предсердия кровью стимулирует рецепторы растяжения предсердия (барорецепторы низкого давления), импульсы поступают по блуждающему нерву в продолговатый мозг, где тормозят заднее ядро п. vagus, возбуждаются латеральные отделы сосудодвигательного центра и через симпатические сердечные нервы увеличивается ЧСС. Рефлекс Бейнбриджа препятствует переполнению кровью вен, предсердий и легких.
Уменьшение толерантности к физигеским нагрузкам. Больные жалуются на быструю утомляемость, снижение физической активности. В основе этого симптомокомплекса лежит несколько механизмов. Прежде всего это недостаточный прирост МОС и перфузии работающих мышц. Неадекватна и легочная вентиляция. Происходят изменения в самих мышцах: уменьшается плотность капилляров на единицу мышечной ткани, содержание митохондрий в клетках, падает активность ферментов аэробного окисления, обмен в мышцах рано переключаются на анаэробный тип с уменьшением выработки АТФ, увеличивается доля миозина с медленной АТФазной способностью.
Принципы терапии хронической застойной сердечной недостаточности
• Разгрузка поврежденного сердца:
— уменьшение механической нагрузки — покой;
— уменьшение перегрузки объемом — ограничение соли и воды в диете, назначение диуретиков;
— уменьшение перегрузки давлением — назначение периферических вазодилататоров.
• Понижение активности нейрогуморальных систем — назначение ингибиторов АПФ, блокаторов ангиотензиновых рецепторов, блокаторов Р1-адренергических рецепторов.
Патофизиология сердечной недостаточности
381
• Разгрузка кардиомиоцитов — назначение блокаторов р^адре-нергических рецепторов.
• Инотропная стимуляция. Назначение сердечных гликозидов.
Осложнения хронической застойной сердечной недостаточности
Опасными осложнениями ХЗСН являются аритмии и отек легких.
Аритмии возникают в результате электрической негомогенности миокарда. Дилатация полостей сердца, перегрузка клеток кальцием, недостаточность в них калия изменяют возбудимость и рефрактерность кардиомиоцитов. Нарушаются автоматизм, проводимость, возникают эктопические очаги. Помимо развития тахикардии характерно появление мерцательной аритмии предсердий (резкая дилатация предсердий), экстрасистолий, блокад, вплоть до фибрилляции желудочков.
Отек легких. Механизм отека легких как проявление недостаточности левого желудочка. Начальным звеном отека являются резкое падение насосной функции левого желудочка и увеличение в нем давления (КДД). Последовательно возрастает давление в левом предсердии, в легочных венах и на венозном конце легочных капилляров (давление заклинивания капилляров легких становится выше 15 мм рт.ст.). Развиваются транссудация, отек легких. Когда давление в легочных капиллярах превысит ОнД (25-30 мм рт.ст.), отек становится максимальным. Развиваются приступ удушья, кашель, появляются клокочущее дыхание и обильная пенистая мокрота с примесью крови.
Принципы терапии отека легких при хронической застойной сердечной недостаточности
• Применение веществ (например, нитроглицерина), расслабляющих вены большого круга и таким образом уменьшающих венозный возврат к «захлебывающемуся» сердцу.
• Для уменьшения венозного возврата к сердцу можно наложить умеренные жгуты на нижние конечности.
• Для уменьшения ОЦК произвести кровопускание или ввести мощные диуретики.
• Для улучшения диффузии кислорода в легких применить дыхательную смесь с повышенным содержанием кислорода.
• Назначить бронхолитики и пеногасители.
• В этой критической ситуации показаны сердечные гликозиды. При подозрении на инфаркт миокарда и развитие отека легких именно по этой причине назначение сердечных гликозидов может привести к разрыву миокарда!
Лекция 25
КОРОНАРНАЯ НЕДОСТАТОЧНОСТЬ
Н.И. Бережнова
Коронарная недостаточность — типовая форма патологии сердца, обусловленная несоответствием поступления кислорода к миокарду его метаболическим потребностям.
Различают 2 формы коронарной недостаточности:
• абсолютная (коронарогенная), обусловленная действием коро-нарогенных факторов. Она возникает в случае первичного нарушения коронарного кровотока вследствие уменьшения просвета коронарных артерий. К коронарогенным факторам относятся: стенозирующий атеросклероз: тромбоз коронарных артерий, чаще всего являющийся следствием атеросклероза: спазм коронарных сосудов, причинами которого могут служить: возбуждение oCj-адренорецепторов на фоне блокады Р2-адренорецепторов, а также увеличение синтеза вазоконстрикторов (эндотелина-1, ангиотензина-П, тромбоксана А2, серотонина и др.) при понижении количества вазодилататоров (оксида азота — NO, простациклина, брадикинина и др.). При этом развивается ишемия миокарда;
• относительная, возникающая в случае действия некоронароген-ных факторов, значительно повышающих энергетические потребности миокарда.
К некоронарогенным факторам относятся увеличение ЧСС, которое наблюдается при повышении содержания в крови и миокарде катехоламинов, повышение напряжения внутри стенок желудочков сердца, сократимости миокарда. При относительной коронарной недостаточности интенсивность коронарного кровотока может возрастать, но все же оказывается недостаточной для возросшей потребности миокарда в кислороде.
При ишемии наблюдается уменьшение доставки к миокарду не только кислорода, но и субстратов окисления, возникают антиоксидантный,
Коронарная недостаточность
383
гормональный и другие виды дефицитов. Кроме того, нарушается отток метаболитов, многие из которых являются токсичными.
Ишемическая болезнь сердца
Недостаточность коронарного кровообращения, вызванная окклюзией коронарных артерий сердца, именуется ишемической болезнью сердца (ИБС).
В современном обществе эта болезнь является главной причиной смерти в зрелом возрасте.
При ангиографии коронарных сосудов стенозирующий атеросклероз (рис. 25-1), как минимум, одной из главных коронарных артерий обнаруживается у 92 % страдающих ИБС.
Развитию атеросклероза коронарных артерий способствуют следующие факторы риска:
• Дислипидемия, характеризующаяся повышением содержания в плазме атерогенных ЛП: ЛПНП и ЛПОНП и холестерина при снижении содержания в крови антиатерогенных ЛПВП, которые удаляют из периферических клеток, в том числе эндотелиоцитов, избыточный холестерин. В практической работе рассчитывают так называемый атерогенный индекс — это отношение содержания в плазме атерогенных ЛП к ЛПВП. При индексе выше 4,9 риск развития атеросклеротического поражения сосудов значительно возрастает.
Рис. 25-1. Коронарограмма больного стенозирующим атеросклерозом. Слева стрелкой указано место сужения артерии. Справа — момент спазма коронарной артерии
384
Лекция 25
• Артериальная гипертензия является важным фактором риска коронарного атеросклероза. Повышение АД выше 160/90 мм рт.ст. увеличивает риск развития атеросклероза у мужчин в возрасте SO-59 лет в 3 раза, а у женщин того же возраста — в 6 раз.
• Курение в 2-6 раз увеличивает риск смерти от ИБС.
• Избыточное питание, обычно сочетающееся с малоподвижным образом жизни, увеличивает риск возникновения коронарного склероза на 40 %.
• Сахарный диабет часто сопровождается развитием ИБС, к тому же в более раннем возрасте. У половины больных, страдающих при жизни сахарным диабетом, на аутопсии обнаружен тяжелый коронарный атеросклероз.
• Стрессовые состояния, вызывающие выброс катехоламинов, которые спазмируют сосуды, повышают давление и стимулируют липолиз с развитием гиперлипидемии.
• Наследственность. При отягощенном ИБС семейном анамнезе вероятность развития коронарного атеросклероза в 9 раз выше.
• Длительное применение некоторых синтетических прогестинов с контрацептивными целями у женщин.
Патогенетические факторы ишемической болезни сердца
1. Органическая обструкция коронарной артерии атеросклеротическим процессом.
При этом резкое ограничение коронарного кровотока обусловлено:
• формированием атеросклеротической бляшки, воспалительного и фиброзного процессов в ней и последующим стенозированием артерий (уровень критического стеноза составляет 75 %);
• формированием агрегатов тромбоцитов и затем тромба в области атеросклеротической бляшки.
2. Динамическая обструкция атеросклеротически измененных коронарных артерий вследствие развития эндотелиальной дисфункции. У больных ИБС регуляторные механизмы, обеспечивающие коронарную вазодилатацию, нарушаются, а вазоконстрикторные реакции начинают преобладать. Главной причиной нарушения вазодилатации является уменьшение количества оксида азота (NO), так как большинство химических факторов, синтезируемых в эндотелии или циркулирующих с кровью, реализуют свое действие через экспрессию синтеза NO. Снижение количества оксида азота происходит в результате как снижения его продукции поврежденным эндотелием, так и ускоренной
Коронарная недостаточность
385
его деградации под действием свободных радикалов кислорода. Существенную роль в повреждении эндотелия может играть образующийся при реакции NO с супероксидным радикалом высокореактивный оксидант пероксинитрит, вызывающий пероксидацию липидов мембран эндотелиальных клеток. Нарушение продукции NO способствует прогрессированию атеросклеротического процесса. NO является мульти-потентной молекулой, которая ингибирует проникновение моноцитов в субэндотелиальное пространство, дифференциацию их в макрофаги с последующим преобразованием в «пенистые клетки»: тормозит продукцию провоспалительных цитокинов, уменьшает экспрессию молекул адгезии лейкоцитов, тормозит адгезию и агрегацию тромбоцитов и высвобождение из них ростовых факторов, ингибирует пролиферацию и миграцию в интиму гладкомышечных клеток сосудов. Важную роль в ослаблении способности сосудов к вазодилатации играет также снижение продукции эндотелием простациклина (ПП2) — фактора, расширяющего сосуды и тормозящего агрегацию тромбоцитов.
Преобладание вазоконстрикции коронарных сосудов в первую очередь обусловлено повышением синтеза эндотелиоцитами эндотелина-1, которому присущи следующие свойства: сосудосуживающее действие (вазоконстрикторный эффект превышает таковой у ангиотензина-П в 10 раз); стимуляция пролиферации гладкомышечных клеток; стимуляция адгезии лейкоцитов к поверхности эндотелия: ингибирование фибринолиза. Продукцию эндотелинов повышают катехоламины, ангиотензин-11, серотонин, ИЛ-1, гипоксия, ишемия, трансформирующий фактор роста.
Вазоконстрикции коронарных сосудов способствует также активация местной ренин-ангиотензин-альдостероновой системы (РААС), так как эндотелий обладает собственным ангиотензинпревращающим ферментом — химазой, повышающим уровень ангиотензина-П и снижающим количество вазодилататора брадикинина. Вазоконстрикцию могут вызывать высвобождаемые активированными тромбоцитами серотонин и тромбоксан А2.
3. Активация прокоагулянтных свойств эндотелия.
Этому способствуют следующие обстоятельства: снижение синтеза эндотелием физиологического антиагреганта простациклина, а также увеличение синтеза мощного проагреганта тромбоксана А2; активация коагуляционного гемостаза с повышенным образованием нитей фибрина: понижение активности фибринолитической системы в результате уменьшения продукции эндотелиоцитами тканевого активатора плаз
386
Лекция 25
миногена (ТПА). При этом увеличивается синтез ингибитора активатора плазминогена под влиянием цитокинов ИЛ-1 и ФНО, продуцируемых макрофагами в зоне атеросклеротического поражения артерий.
Активация механизмов гемостаза с образованием и отложением тромботических масс на поверхности атеросклеротической бляшки приводит к более значительному сужению коронарных артерий, вплоть до критического, или к полной обтурации коронарного сосуда.
Механизмы ишемического повреждения миокарда
1. Нарушается энергетическое обеспечение миокарда. Ухудшается снабжение миокарда кислородом, что приводит к нарушению образования энергии в митохондриях в связи с падением активности как окисления глюкозы, так и 0-окисления жирных кислот. Накапливаются жирные кислоты, оказывающие детергентное действие на миокард. Неполностью метаболизированные продукты 0-окисления жирных кислот усиливают разобщение процессов гликолиза и окислительного декарбоксилирования, что еще больше нарушает образование АТФ. В результате содержание АТФ и КФ резко снижается и развивается основной метаболический признак ишемии миокарда — отставание скорости синтеза макроэргических соединений от потребности в них миокарда. Известно, что спустя несколько секунд после развития тотальной ишемии миокарда запасы КФ истощаются полностью, а через 40 мин запасы АТФ уменьшаются на 90 %. Вследствие потери кардиомиоцитами изоэнзимов КФК нарушается транспорт АТФ к местам ее утилизации.
2. Нарушается электролитный баланс в кардиомиоцитах, что обусловлено нарушением энергетического обеспечения работы ионных насосов. В клетках миокарда накапливаются ионы натрия, вызывающие набухание клеток, а также ионы кальция, что приводит к развитию контрактуры сократительных клеток, понижению растяжимости миокарда, повышению напряжения в стенке миокарда и, следовательно, повышению потребности миокарда в кислороде (замыкается порочный круг).
3. Повышенное содержание кальция в кардиомиоцитах активирует перекисное окисление липидов (ПОЛ) и фосфолипазы, повреждающие митохондрии и цитоплазматическую мембрану кардиомиоцитов.
4. Вследствие накопления лактата развивается метаболический ацидоз, активирующий лизосомальные гидролазы, повреждающие клетки миокарда. Кроме того, ионы водорода начинают конкурировать с кальцием за места связывания на тропонине, что угнетает сократительную функцию сердечной мышцы.
Коронарная недостаточность
387
5. После восстановления коронарного кровотока (снятие спазма коронарных сосудов, лизирование тромба и т.д.) может развиться реперфузионное повреждение миокарда, которое обусловлено более значительным накоплением в клетках миокарда ионов кальция с аккумуляцией их в митохондриях, ведущей к необратимому нарушению функции последних; большей интенсивностью ПОЛ мембран кардиомиоцитов вследствие недостаточности антиоксидантной защиты и притока кислорода в зону предшествующей ишемии, а также развитием феномена «по-reflow» (неполного восстановления кровотока). Основными причинами развития этого феномена являются повышение тонуса резистивных сосудов, затруднение кровотока вследствие внутрисосудистой агрегации эритроцитов и тромбоцитов, отек эндотелия, перикапиллярный отек и ишемическая контрактура.
Классификация ишемической болезни сердца
1. Внезапная коронарная смерть (первичная остановка сердца).
2. Стенокардия:
• стабильная стенокардия напряжения;
• спонтанная стенокардия;
• нестабильная стенокардия.
3. Безболевая ишемия миокарда.
4. Инфаркт миокарда.
5. Постинфарктный кардиосклероз.
6. Нарушение ритма и проводимости как единственное проявление ИБС.
7. Сердечная недостаточность.
Внезапная коронарная смерть — это смерть, возникшая мгновенно или в пределах 1 ч после появления первых симптомов коронарной недостаточности. Внезапная сердечная смерть может развиться при постинфарктном кардиосклерозе, спазме коронарных артерий, кардиомиопатии вследствие фибрилляции желудочков и желудочковой тахикардии либо асистолии или выраженной брадикардии.
Стенокардия напряжения характеризуется возникновением болевого синдрома (с локализацией боли сжимающего характера в загрудинной области, иррадиирующей в левую руку) во время физических нагрузок, причиной которого является неспособность суженных коронарных артерий обеспечить повышенную потребность миокарда в кислороде.
Нестабильная стенокардия — это стенокардия с нестабильным прогрессирующим течением с учащением приступов ангинозных болей,
388
Лекция 25
часто заканчивающихся внезапной сердечной смертью или инфарктом миокарда.
Безболевая ишемия миокарда характеризуется эпизодами транзитор-ной ишемии миокарда, клинически ничем не проявляющейся. Вследствие того что эта форма ИБС прижизненно не выявляется, она может осложняться инфарктом миокарда и быть причиной внезапной коронарной смерти.
В настоящее время выделяют такие новые ишемические синдромы, как гибернация миокарда, оглушение миокарда.
Гибернирующий («спящий») миокард — это локальное снижение сократительной способности миокарда левого желудочка, вызванное его длительной гипоперфузией, которое полностью или частично ликвидируется после улучшения коронарного кровотока. Гибернация миокарда является приспособительной реакцией в ответ на хроническое снижение коронарного кровотока. Функция миокарда снижается, благодаря чему достигается равновесие между потребностью миокарда в кислороде и доставкой его с кровью. Радикальный метод лечения этого состояния — хирургическая реваскуляризация.
Оглушенный миокард (станнинг) — это обратимая постишемическая дисфункция миокарда после восстановления коронарного кровотока. Этот феномен может развиться при раннем применении тромболитиков в остром периоде инфаркта миокарда; при хранении донорского сердца перед его трансплантацией; при наложении дистальных анастомозов во время аортокоронарного шунтирования. Патогенез этого состояния до конца не выяснен. Постишемическую дисфункцию миокарда пытаются объяснить образованием свободных кислородных радикалов в процессе реперфузии, а также перегрузкой кардиомиоцитов кальцием.
Инфаркт миокарда — одна из клинигеских форм ишемигеской болезни сердца, характеризующаяся развитием локального (огранигенного) некроза миокарда вследствие остро возникшего несоответствия коронарного кровотока потребностям миокарда.
Острый инфаркт миокарда
Инфаркт миокарда (ИМ) является одним из наиболее частых проявлений ИБС и одной из наиболее частых причин смерти в развитых странах, причем около половины смертельных исходов приходится на первый час от начала заболевания. Заболеваемость ИМ значительно увеличивается с возрастом. В молодом и среднем возрасте мужчины болеют им значительно чаще женщин. У женщин в возрасте до 60 лет ИМ
Коронарная недостаточность
389
встречается в 4 раза реже, чем у мужчин. После наступления менопаузы различия в заболеваемости ИМ среди женщин и мужчин постепенно уменьшаются, а с 70-летнего возраста исчезают.
Факторы риска развития ИМ полностью идентичны факторам риска ИБС. Уменьшение частоты развития ИМ обусловлено в первую очередь борьбой с такими факторами риска, как гиперхолестеринемия, артериальная гипертензия, курение.
Основной причиной ИМ является атеросклероз коронарных артерий и развивающийся на этом фоне тромбоз артерии с развитием острой ишемии участка миокарда. Выраженный атеросклероз коронарных артерий обнаруживается у 95 % больных, умерших от ИМ. У большинства больных (50-70 %) отмечается стенозирующий атеросклероз 2-3 магистральных коронарных артерий, при этом просвет артерий уменьшается более чем на 75 %.
Патогенез инфаркта миокарда
Основой развития ИМ является патофизиологическая триада (рис. 25-2), включающая разрыв (надрыв) атеросклеротической бляшки, тромбоз и вазоконстрикцию.
В большинстве случаев ИМ развивается при внезапно наступившем резком уменьшении коронарного кровотока вследствие тромботической окклюзии коронарной артерии, просвет которой значительно сужен предшествующим атеросклеротическим процессом. При внезапном полном закрытии просвета коронароной артерии тромбом в условиях отсутствия или недостаточного развития коллатералей развивается трансмуральный ИМ. При интермиттирующей тромботической окклюзии коронарной артерии (вследствие спонтанного или терапевтического тромболизиса) и существовавших ранее коллатералях формируется нетрансмуральный ИМ. В этом случае некроз располагается чаще всего в субэндокардиальных отделах или в толще миокарда, не достигая эпикарда.
В окклюзии коронарной артерии значительную роль играет коро-нароспазм, обусловленный развитием эндотелиальной дисфункции, ведущей к преобладанию синтеза и эффектов вазоконстрикторов при снижении синтеза вазодилататоров. ИМ является стрессовой реакцией, активирующей симпатическую нервную систему. Выброс в кровь катехоламинов повышает потребность миокарда в кислороде, способствует агрегации тромбоцитов и выделению ими вазоконстриктора тромбоксана А2.
390
Лекция 25
Рис. 25-2. Патогенез острого инфаркта миокарда
Большое значение в патогенезе ИМ имеет степень компенсации нарушенного коронарного кровотока коллатеральным кровообращением. Так, медленно прогрессирующее стенозирование эпикардиальных артерий может не приводить к развитию ИМ вследствие формирования развитой коллатеральной сосудистой сети в миокарде. Функциональная неполноценность коллатерального кровотока является одним из патогенетических факторов развития ИМ у молодых людей. Существенная роль в развитии ИМ принадлежит недостаточному ангиогенезу.
Развитие ИМ сопровождается нарушением систолической функции сердца. Снижение фракции выброса (основного показателя нарушения
Коронарная недостаточность
391
систолической функции) происходит при нарушении сократимости более 10 % массы миокарда. При нарушении сократимости более 15 % массы миокарда наблюдается повышение конечного диастолического объема (КДО) и давления (КДД) левого желудочка. При некрозе более 25 % массы миокарда развивается левожелудочковая недостаточность, а при некрозе более 40 % массы левого желудочка — кардиогенный шок. Нарушение диастолической функции сердца обусловлено снижением эластичности и растяжимости миокарда. Диастола становится неполноценной вследствие недостаточной релаксации миокарда, что ведет к повышению КДД и ухудшению коронарного кровотока. Нарушение диастолической функции наблюдается при поражении менее 10 % массы миокарда левого желудочка.
Происходит ремоделирование левого желудочка: развиваются дилатация миокарда, истончение миокарда в зоне некроза, снижение тонуса миокарда в области некроза и в периинфарктной зоне, гибернация в периинфарктной зоне, активация циркулирующей и местной РААС, активация САС, гиперпродукция эндотелием эндотелина. Указанные ней-рогуморальные стимуляторы активируют факторы роста, повышают внутриклеточный синтез протоонкогенов, факторов ядерной транскрипции, что приводит к развитию гипертрофии миокарда.
Нарушается системная гемодинамика: понижается систолическое и пульсовое АД; растет ЧСС; снижается скорость кровотока; повышается ОПСС.
При ИМ развивается болевой синдром, развитие которого связывают с действием следующих факторов: понижением порога болевой чувствительности; острой дилатацией сердца; увеличением концентрации внеклеточного калия из-за потери его кардиомиоцитами; повышением концентрации таких медиаторов, как брадикинин, субстанция Р, серотонин, аденозин и др.; развитием метаболического ацидоза.
Характерным для ИМ является развитие резорбционно-некротического синдрома со следующими признаками:
• повышением температуры тела;
• нейтрофильным лейкоцитозом со сдвигом формулы влево;
• увеличением СОЭ;
• обнаружением биохимических признаков воспаления;
• появлением в крови биохимических маркеров гибели кардиомиоцитов;
• развитием ООФ, сопровождающимся повышением содержания в крови медиаторов и белков ООФ.
392
Лекция 25
Лабораторная диагностика ИМ
1. Данные лабораторного исследования периферической крови: нейтрофильный лейкоцитоз со сдвигом лейкоцитарной формулы влево, эозинопения, лимфопения, повышение СОЭ.
2. Данные биохимического исследования крови: повышается содержание С-реактивного белка, гаптоглобина, ИЛ-1, ФНО; возрастает содержание глюкозы; развивается метаболический ацидоз: повышается содержание фибриногена: наблюдается гиперкалиемия.
3. Определение содержания в крови биохимических маркеров гибели кардиомиоцитов. При ИМ из очага некроза в кровь поступает ряд таких белковых молекул - компонентов мышечных волокон, как миоглобин, тропонин (рис. 25-3).
Рис. 25-3. Динамика ги-перферментемии при инфаркте миокарда
Инструментальные исследования
ЭКГ-диагностика ИМ. ЭКГ при ИМ формируется под влиянием трех зон, образующихся в миокарде: зоны некроза, зоны ишемического повреждения и зоны ишемии (рис. 25-4). Зона некроза характеризуется появлением патологического зубца Q. (ширина его превышает 0,03 с; а глубина больше 1/4 амплитуды зубца R в том же отведении) либо комплекса QS (при трансмуральном ИМ). Зона ишемического повреждения отражается на ЭКГ смещением интервала ST от изолинии: при субэндокардиальной локализации наблюдается депрессия ST, при субэпикардиальной либо трансмуральной — элевация ST.
Зона ишемии вследствие замедления процесса реполяризации отражается на ЭКГ появлением «коронарного» зубца Т: положительного, высокого, симметричного, с заостренной вершиной — при субэндокардиальной ишемии и отрицательного, глубокого, симметричного — при
Коронарная недостаточность
393
Рис. 25-4. Типовые измения ЭКГ при инфаркте миокарда
субэпикардиальной или трансмуральной ишемии. Локализация ИМ устанавливается по обнаружению указанных ЭКГ-признаков в соответствующих отведениях.
Дополнительные инструментальные методы диагностики
Неинвазивные методы:
• компьютерная томография;
• эхокардиография;
• фонокардиография;
• радиоизотопные методы с использованием меченых ионов таллия-201, технеция-99 и др.;
• ядерно-магнитный резонанс (ЯМР) и др.
Инвазивные:
• катетеризация полостей сердца;
• ангиография с введением рентгеноконтрастных веществ и др.
Осложнения инфаркта миокарда
Ранние осложнения
• Аритмии.
• Острая левожелудочковая недостаточность с развитием отека легких.
• Кардиогенный шок.
394
Лекция 25
Кардиогенный шок — крайняя степень левожелудочковой недостаточности, характеризующаяся резким снижением сократительной функции миокарда. Кардиогенный шок развивается примерно у 20 % больных с ИМ. Выделяют следующие формы кардиогенного шока: рефлекторный; истинный кардиогенный; ареактивный; аритмический; из-за разрыва миокарда.
Острая левожелудогковая недостатогность. Основным патогенетическим фактором является снижение сократительной способности (систолическая дисфункция) и уменьшение податливости (диастолическая дисфункция) миокарда левого желудочка, что приводит к повышению гидростатического давления в левом предсердии, а затем в сосудах малого круга.
Аритмии. Нарушения сердечного ритма и проводимости являются практически постоянным осложнением крупноочагового ИМ.
Основные механизмы развития аритмий у больных ИМ:
• изменение электрофизиологических свойств миокарда в области поражения;
• электрическая негомогенность миокарда;
• появление эктопических очагов, потеря электрической стабильности;
• электролитный дисбаланс в миокарде (потеря кардиомиоцитами калия, магния, повышение уровня калия во внеклеточной среде);
• гиперкатехоламинемия;
• острая дилатация миокарда;
• развитие феномена re-entry и высокая спонтанная диастолическая поляризация.
Единичные циклы эктопического возбуждения или круговой волны приводят к экстрасистолии; длительный период деятельности эктопического очага или циркуляции круговой волны по миокарду приводят к развитию пароксизмальной тахикардии, трепетанию и мерцанию предсердий.
Наиболее опасна ранняя фибрилляция желудочков (хаотическое подергивание отдельных волокон миокарда, отсутствие координированного сокращения желудочков), которая является основной причиной смерти больных ИМ.
Поздние осложнения
• Формирование аневризмы сердца (происходит в 12-15 % трансмурального ИМ).
• Разрывы сердца: наружные, межжелудочковой перегородки; отрыв сосочковой мышцы с развитием недостаточности митрального клапана.
Коронарная недостаточность
395
• Тромбоэмболии, которые развиваются примерно у */ш перенесших ИМ (первое место занимает тромбоэмболия легочной артерии — ТЭЛА).
• Постинфарктный аутоиммунный синдром Дресслера. Некроз миокарда, а также изменения в периинфарктной зоне приводят к появлению кардиальных аутоантигенов с последующим образованием аутоантител, усиливающих повреждение миокарда. Этот синдром развивается приблизительно у 3-4 % больных на 2-8 нед от начала ИМ.
Принципы патогенетической терапии инфаркта миокарда
• Купирование болевого синдрома.
• Назначение нитратов, прежде всего нитроглицерина, с целью расширения коронарных сосудов, что способствует ослаблению болевого синдрома, а также снижению преднагрузки благодаря венодилатации.
• Оксигенотерапия.
• Тромболитическая терапия с целью восстановления магистрального коронарного кровотока, предупреждение тромбообразования.
• Понижение потребности миокарда в кислороде.
• Ограничение размеров ИМ, что достигается путем ранней реваскуляризации с помощью тромболитической терапии и назначения периферических вазодилататоров, p-адреноблокаторов, антагонистов кальция, а также хирургических методов (чрескожной транслюминальной коронарной ангиопластики, аортокоронарного шунтирования).
• Предупреждение опасных для жизни аритмий.
• Лечение метаболическими кардиопротекторами.
Лекция 26
ПАТОФИЗИОЛОГИЯ АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИИ
Н.Л. Богуш
Артериальная гипертония — одно из самых распространенных хронических заболеваний среди взрослого населения. В мире среди мужчин и женщин в возрасте 50-60 лет около 20 % страдает гипертонией, причем с постарением населения (старше 65 лет) этот процент значительно возрастает (50 % и более). АГ не только ухудшает качество жизни больных, но и повышает риск тяжелых сердечно-сосудистых заболеваний (инфаркт, инсульт, хроническая сердечная недостаточность), приводит к инвалидности и сокращению продолжительности жизни.
Основной признак артериальной гипертензии — повышение АД.
В сосудах кровь движется под давлением в результате работы сердца. В норме в желудочках давление крови колеблется от 120 в левом и 25 в правом в систолу до 0 мм рт.ст. в диастолу. При измерении АД в сосудах (например, на уровне плечевой артерии) систолическое АД (САД) соответствует таковому в сердце, а диастолическое АД (ДАД) не бывает равным 0. В среднем ДАД равно 70-80 мм рт.ст. Следовательно, в артериях АД колеблется в значительно меньшей степени, чем в желудочках сердца. Такое отличие в колебании АД и сохранение высокого ДАД (по сравнению с аналогичными в полостях желудочков) обусловлено прежде всего структурно-функциональными особенностями сосудов-буферов — аорты и крупных артериальных сосудов. Стенки последних состоят из приблизительно равных количеств коллагеновых, эластических волокон и гладкомышечных клеток. Эластический компонент растягивается, обусловливая депонирование систолического объема крови, а коллагеновые волокна и сокращающиеся гладкомышечные клетки препятствуют перерастяжению и разрыву стенок. После захлопывания аортального клапана эластическая аорта и ее крупные ветви сокращаются, поддерживая этим градиент давления (т.е. сохраняя высокое ДАД) и делая поступление крови на периферию более равномерным. Но вклад
Патофизиология артериальной гипертензии 397
этих крупных сосудов-буферов в общее периферическое сопротивление сосудистого русла (ОПСС) небольшой, всего 19 %.
В другую группу сосудов входят сосуды-распределители (мелкие артерии и артериолы), шунты, капилляры, венулы и мелкие вены. Именно мелким артериям и артериолам (из всех сосудов) принадлежит главный вклад в регуляцию АД. Они могут, сокращаясь и расслабляясь, значительно изменять просвет сосудов, поэтому на их долю приходится около 50 % всего ОПСС. Этим сосудам свойственна высокая степень базального тонуса, который постоянно меняется под влиянием местных физических и химических факторов. Кроме того, величина просвета сосудов регулируется вегетативной нервной системой. Сосудодвигательные нервы относятся преимущественно к ее симпатическому отделу (медиатор — норадреналин). Сосудистый тонус покоя поддерживается благодаря постоянному поступлению по сосудодвигательным нервам импульсов с частотой 1-3 в секунду. При частоте импульсов, равной всего около 10 в секунду, наблюдается максимальное сужение сосудов. При уменьшении частоты импульсации развивается вазодилатация, причем последняя ограничена базальным тонусом сосудов (т.е. тем тонусом, который наблюдается при отсутствии импульсации в сосудосуживающих нервах либо при их перерезке). Норадреналин и адреналин связываются на мембране гладкомышечных клеток адренергическими рецепторами — а или |3.
Норадреналин {адреналин) действует преимущественно на а-адрено-рецепторы, что сопровождается сокращением мускулатуры сосудов (в большинстве периферических сосудов, кроме сосудов сердца, преобладают а-адренорецепторы; с возрастом количество (3-рецепторов понижается). Адреналин действует и на а-, и на p-рецепторы (возбуждение p-рецепторов приводит к расслаблению гладких мышц сосудов). Если в сосудах преобладают а-рецепторы, то адреналин вызывает их сужение, а если p-рецепторы — расширение. Порог возбуждения р-рецепторов ниже, чем а-рецепторов, хотя при возбуждении и тех, и других преобладают эффекты а-рецепторов.
Таким образом, в низких (физиологических) концентрациях адреналин вызывает расширение сосудов (при физической нагрузке, эмоциональном возбуждении), а в высоких — сужение (при кровотечении, стрессе).
Следовательно, в результате действия сосудов-буферов и сосудов-распределителей АД имеет двухфазный характер — САД и ДАД. Среднее АД будет всегда находиться между САД и ДАД, определяясь прежде всего величиной ДАД.
398
Лекция 26
СрАД вычисляем по формуле:
СрАД = ДАД + 1/3 АДпульсо|10е;
АДпульсовое - САД ~
Какие условия влияют на СрАД и определяют его величину? Рассмотрим формулу определения величины системного кровотока (F) или минутного объема сердца (МОС):
F = МОС = (АД - АД ) 4- ОПСС.
\ ^в аорте правом предсердии'
Учитывая, что АД в аорте — это СрАД, а АД в правом предсердии -около 0 и им можно пренебречь, получим, что F = МОС = СрАД/ОПСС, отсюда
СрАД = МОС х ОПСС.
В норме и покое МОС = 5 л/мин, ОПСС = 20 мм рт.ст./(лхмин).
Все изменения СрАД непосредственно определяются изменениями минутного объема сердца или общего периферического сосудистого сопротивления.
Можно заметить, что в организме АД зависит от 3-х главных условий:
• работы сердца;
• общего периферического сосудистого сопротивления (ОПСС);
• объема циркулирующей крови (отражается на величине МОС и ОПСС).
ОЦК будет увеличивать МОС, так как:
• увеличится приток крови к сердцу, что приведет к увеличению ударного объема по механизму Франка-Старлинга;
• приток крови будет растягивать устье полых вен, стимулировать рецепторы растяжения или рецепторы низкого давления этой рефлексогенной зоны и вызывать рефлекторную тахикардию.
ОПСС будет увеличиваться по механизму ауторегуляции и рефлекторной вазоконстрикции.
Врач, назначая лекарственную терапию больному гипертонией, сталкивается с тем, что он не всегда знает, какой тонкий механизм регуляции АД нарушен в данном случае. Однако, контролируя основные показатели гемодинамики (МОС, ОПСС и ОЦК), он может контролировать и АД.
Для уменьшения МОС, тахикардии назначаются блокаторы [3-адренорецепторов ([3-адреноблокаторы), уменьшения ОПСС — блокаторы кальциевых каналов, ОЦК — диуретики.
Для патогенеза большинства гипертоний ведущим механизмом является именно повышение ОПСС, следовательно, необходимо вспомнить, от каких факторов зависит величина ОПСС.
Патофизиология артериальной гипертензии
399
По закону Пуазейля-Хагена величина ОПСС = 8т|Е*-лг4, где и — вязкость крови, L — длина сосуда, г — радиус сосуда.
Если не иметь в виду особые случаи увеличения вязкости крови (при болезни Вакеза, дегидратациях и др.), то определяющим величину ОПСС окажется радиус (просвет) сосуда.
Просвет мелких артерий и артериол (а значит, и ОПСС) зависит от ряда факторов.
Прежде всего он зависит от тонуса гладких мышц сосудов, который представляет непрерывное тоническое сокращение этих мышц.
Тонус сосудистой стенки складывается из двух основных компонентов — базального и вазомоторного тонуса, кроме этого тонус зависит от структурных особенностей сосуда.
Структурные особенности
Стенка резистивных сосудов состоит на 35 % из гладкомышечных клеток, соотношение толщины стенки к диаметру сосуда (индекс Керно-гана) значительное, больше, чем у сосудов-буферов. Чем толще стенка, тем большая масса перемещается при сокращении гладких мышц сосуда от наружной поверхности сосуда в центр и может значительно сузить просвет, иногда до его полного закрытия (пример — кожные шунты). Для артериальной гипертонии характерны развитие гипертрофии гладких мышц артериальных сосудов и повышение индекса Керногана, что вносит дополнительный вклад в повышение ОПСС и морфологически «закрепляет» высокое АД.
Базальный тонус
Резистивным сосудам свойственна высокая степень внутреннего (миогенного) базального тонуса, который постоянно изменяется под действием местных химических и физических факторов.
Его величину определяют:
• структура сосудистой стенки;
• механизм миогенной (механогенной) ауторегуляции — способность гладких мышц сокращаться при их растяжении, например, при повышении давления (эффект Бейлиса). Чем выше внутрисосудистое давление, тем сильнее сокращаются гладкие мышцы;
• обмен веществ, в частности катионов (Na+, К*, Са2+, Mg2*), в мышечной клетке, так как тем самым определяется способность мышечной клетки реагировать сокращением различной силы на одну и ту же степень растяжения.
400
Лекция 26
Вазомоторный тонус
Вазомоторный тонус создается прямым влиянием вазоконстрикторной симпатической импульсации, причем мелкие артерии и артериолы богато снабжены симпатическими вазоконстрикторными волокнами.
Увеличение базального тонуса, например, миогенного компонента должно привести не только к общему увеличению сосудистого тонуса и АД, но и к увеличению вазоконстрикторного тонуса — к увеличению всех прессорных реакций на констрикторные импульсы и норадреналин. Увеличение вазоконстрикторной импульсации при неизменной величине миогенного компонента должно также привести к увеличению сосудистого тонуса и гипертонии за счет увеличения вазомоторного компонента.
Также ОПСС будет зависеть от соотношения действующих на сосуды прессорных и депрессорных механизмов. Они могут быть срочными и кратковременными, а также медленными, но длительно действующими. Последние играют главную роль в развитии хронической гипертонии.
Прессорные механизмы
I. Срогные механизмы, рефлекторные. Возникают с рецепторов дуги аорты и каротидного синуса (главных сосудистых рефлекторных полей).
• Уменьшение АД, или МОС, или ОЦК -> снижение возбуждения барорецепторов (рецепторов растяжения или рецепторов высокого давления, заложенных в стенке сосуда и возбуждающихся при ее растяжении) -> уменьшение импульсации по депрессорным нервам -> возбуждение сердечно-сосудистого центра -> возбуждение симпатических центров -> увеличение работы сердца (ТМОС -> ?АД) + сокращение вен-емкостей (ТОЦК -> ?АД) и возврата крови к сердцу ->(?УО -»ТМОС ->ТАД) + сокращение артериальных сосудов (ТОПСС-ЛАД).
• Гипоксемия, гиперкапния, ацидоз -» возбуждение хеморецепторов тех же рефлексогенных зон -> те же рефлекторные реакции, приводящие в итоге к увеличению АД.
• Ишемическая реакция ЦНС. При значительном падении АД (до 40 мм рт.ст.) развивается опасная ишемия головного мозга. Происходят активация сосудодвигательного центра, Т тонуса симпатического отдела вегетативной нервной системы, вазоконстрикция и подъем АД.
Эти рефлекторные механизмы включаются быстро и достигают максимальной активности через 10-30 с от начала возбуждения.
Патофизиология артериальной гипертензии
401
И. Механизмы, занимающие по длительности промежутогное положение. Для их развития требуются минуты, а для достижения максимума — часы.
• Механизм ауторегуляции: растяжение резистивных сосудов (например, из-за увеличения ОЦК) приводит к их сокращению и подъему АД.
• Активация ренин-ангиотензиновой системы (РАС): падение АД (СрАД ниже 65-90 мм рт.ст.) —> выброс ренина -> превращение ангиотензиногена в ангиотензин-1, ангиотензин-I при помощи АПФ — в ангиотензин-П спазм сосудов —> повышение ОПСС и повышение АД.
• Усиление секреции АДГ = вазопрессина гипоталамусом и задней долей гипофиза -> повышение ОПСС и повышение АД. Кроме того, АДГ усиливает реабсорбцию воды почками, что увеличивает ОЦК и, следовательно, АД.
• Механизм перемещения межтканевой жидкости в капилляры: при падении АД из-за кровопотери межтканевая жидкость по закону Старлинга переходит в сосуды. Увеличение ОЦК увеличение АД.
III. Поздние и длительно действующие механизмы.
• Почечная система контроля за объемом жидкости (почечный объемно-прессорный механизм). Существует тесная связь между кровяным давлением и выведением жидкости почками. На рисунке 26-1 (кривая «диурез-давление», норма) показаны результаты опытов, в которых потребление воды и электролитов менялось по отношению к исходному равновесному уровню (точка N). Крутой подъем кривой выделения мочи от точки N, соответствующей нормальному СрАД около 100 мм рт.ст., свидетельствует о том, что даже очень небольшое повышение АД сопровождается существенным увеличением выделения жидкости почками. При возрастании давления на 1 мм рт.ст. выделение воды почками повышается на 100 %. Это означает, что при увеличении давления примерно на 10 мм рт.ст. почечная экскреция должна возрасти почти в 6 раз. Из рисунка также видно, что даже при существенных колебаниях потребления жидкости СрАД меняется незначительно. Как расположение, так и форма кривой выделения мочи могут существенно различаться у разных индивидов. При этом кривая может смещаться параллельно горизонтальной оси, и равновесие между поступлением и выделением жидкости (точка N) передвинется в область больших значений АД, что и происходит при гипертонии.
402
Лекция 26
Среднее артериальное давление, мм рт. ст.
Рис. 26-1. Кривая «диурез-давление», норма
• Система альдостерона. Падение АД, ОЦК РАС -> вызванная ангиотензин-П гиперсекреция альдостерона -> увеличение канальцевой реабсорбции Na (и с помощью АДГ — воды) ТОЦК -> ТМОС, ОПСС и АД.
• Система вазопрессина (АДГ). Падение АД -> выделение АДГ -> увеличение реабсорбции жидкости в почках -> ТОЦК -> ТМОС, ОПСС и АД.
Прессорные гуморальные вещества: катехоламины, АДГ, ангиотензин-П, эндотелины, тромбоксан, некоторые простагландины, супероксид-анион. Опосредованно: альдостерон и глюкокортикоиды. Последние — через увеличение ОЦК как слабые минералкортикоиды и как увеличивающие синтез АПФ и тем самым количество ангиотензин-П.
Депрессорные механизмы
• I. Срогные рефлекторные механизмы. При повышении АД активируются барорецепторы дуги аорты и каротидного синуса, соответствующие нервы-депрессоры, угнетается симпатический, активируется парасимпатический отдел (уменьшается частота сердечных сокращений) и АД падает. Однако при постоянном (в течение нескольких дней) раздражении эти рефлексы ослабевают, рецепторы адаптируются и «переключаются» для регуляции АД на более высоком, чем в норме, уровне.
Патофизиология артериальной гипертензии
403
• II. Механизмы, занимающие по длительности промежутогное положение. Выделение предсердных натрийуретических пептидов -ПНУП. Повышение АД -> увеличение напряжения стенки миокарда и внутриполостного давления -> растяжение предсердий -> секреция ПНУП увеличение выведения почками натрия и воды -э уменьшение ОЦК, МОС, ОПСС и АД.
• III. Длительно действующие механизмы. Почечная система контроля за объемом жидкости, а также регуляции ОЦК, а следовательно, МОС, ОПСС и АД при участии альдостерона и АДГ.
Депрессорные гуморальные вещества: NO, кинины (брадикинин), простациклин, простагландин Е, ПНУП, местно — метаболиты (углекислый газ, лактат, аденозин и др.), медиаторы повреждения (гистамин, брадикинин, вещество Р), ацетилхолин.
Таким образом, против нарушений АД (и ОЦК) постоянно действуют 3 «линии обороны», каждая в свое время. При кратковременных колебаниях АД включаются сердечные и сосудистые реакции, при длительных же сдвигах преобладают компенсаторные изменения объема крови. В последнем случае сначала меняется содержание в крови воды и электролитов, а при необходимости происходят и сдвиги в содержании белков плазмы и клеточных элементов.
По определению ВОЗ, артериальная гипертония — стойкое хроническое повышение систолического и/или диастолического давления.
Таблица 26-1. Нормальные величины артериального давления
Возраст САД, мм рт.ст. ДАД, мм рт.ст.
ДЕТИ 2-6 лет 7-10 лет 11-16 лет 105-110 110-115 120-139 70-75 Около 75 75-85
ВЗРОСЛЫЕ оптимальное нормальное высокое нормальное минимальное Ниже 120 Ниже 130 130-139 105 Ниже 80 Ниже 85 85-89 60
АД — это величина непостоянная, хотя она и колеблется в довольно узких пределах (табл. 26-1). Изменение АД в течение суток характеризуется двухфазной периодикой — день/ночь с отчетливым снижением АД ночью во время сна. В дневное время АД образует плато с двумя пиками: I — от 9 до И ч, II — от 16 до 20 ч со снижением АД в вечернее вре
404
Лекция 26
мя и достижением минимального значения в ночное время в интервале от 0 до 4 ч. У большинства обследованных АД начинает подниматься в предутренние часы, за 2-3 ч до пробуждения.
Суточный ритм АД модулируется физической и психической активностью, подчиненной циклу бодрствование-сон. Он реализуется и закрепляется участием нейрогуморальных систем. В ранние утренние часы в крови увеличиваются кортизол, адреналин, норадреналин и ренин. В ночное время активность симпатоадреналовой системы и РАС снижается, уменьшается МОС и ОПСС.
Заболеваемость артериальной гипертонией нарастает с возрастом. У женщин до 45 лет показатели АД ниже, чем у мужчин. С началом климактерического периода у женщин максимальное АД выше, чем у мужчин. В пожилом возрасте уровень АД у мужчин вновь превосходит показатели у женщин.
Факторы риска (по данным ВОЗ, 1999)
• Повышенное АД — САД >140 мм рт.ст., ДАД >90 мм рт.ст.
• Возраст и пол — мужчины старше 55 лет, женщины — старше 65 лет.
• Курение.
• Холестерин (содержание в крови >6,5 ммоль/л).
• Сахарный диабет.
• Наследственная предрасположенность — семейный анамнез ранних сердечно-сосудистых заболеваний.
Другие факторы, неблагоприятно влияющие на прогноз:
• дислипидемия — снижение уровня холестерина ЛП высокой плотности (ЛПВП);
• повышение ЛП низкой плотности (ЛПНП);
• ожирение;
• сидячий образ жизни.
Классификация артериальной гипертензии
Течение артериальной гипертензии классифицируют по нескольким основаниям.
Выделяют стадии артериальной гипертензии:
• I стадия — нет объективных признаков поражения органов-мишеней. Повышение АД не достигает высоких цифр и выявляется иногда случайно, например при диспансеризации.
• II стадия — появляется один или несколько признаков поражения органов-мишеней: гипертрофия левого желудочка, сужение
Патофизиология артериальной гипертензии
405
сосудов сетчатки, альбуминурия, признаки атеросклеротического поражения крупных сосудов (аорты, сонных, подвздошных, бедренных артерий).
• III стадия — характеризуется развернутой клинической картиной поражения органов-мишеней:
— сердце — ИБС, инфаркт, хроническая сердечная недостаточность;
— мозг — нарушение мозгового кровообращения, гипертензивная (ишемическая) энцефалопатия, инсульт;
— почки — почечная недостаточность, азотемия;
— сосуды — расслаивающаяся аневризма аорты, окклюзивные поражения артерий с соответствующей клинической картиной;
— сетчатка — ретинопатия с кровоизлияниями в сетчатку.
По уровню САД и ДАД выделяют степени тяжести и формы артериальной гипертензии (табл. 26-2).
Таблица 26-2. Классификация степени тяжести или формы артериальной гипертензии
Степень тяжести Форма гипертонии САД, мм рт.ст. ДАД, мм рт.ст.
I степень Мягкая гипертензия 140-159 90-99
II степень Умеренная гипертензия 160-179 100-109
III степень Тяжелая гипертензия >180 >110
Комитет экспертов ВОЗ принял решение считать синонимами термины «эссенциальная артериальная гипертензия» и «гипертоническая болезнь».
I. Эссенциальная или первигная гипертония (~80 % случаев артериальной гипертензии).
II. Симптоматигеские или вторигные гипертонии:
• почечные (-14 %) развиваются при заболевании сосудов почек (вазоренальные) или паренхимы почек (ренопривные); обусловлены, в основном, активацией и преобладанием активности РААС;
• эндокринные (-3 %) — сопутствуют тиреотоксикозу, синдрому Конна, синдрому Иценко-Кушинга, феохромоцитоме; обусловлены различными механизмами, возникающими при гиперпродукции гормонов;
• кардиоваскулярные (-1,5 %) — при недостаточности клапана аорты, гиперкинетическом варианте работы сердца с увеличенным МОС, коарктации аорты;
406
Лекция 26
• нейрогенные (-0,8 %) - при органическом поражении структур мозга, участвующих в регуляции уровня АД (опухоль, воспаление, травма, сотрясение мозга, кровоизлияние в мозг).
Причиной симптоматических гипертоний является первичное поражение какого-либо органа, приводящее впоследствии к гипертонии.
Причина эссенциальной гипертензии неизвестна.
Эссенциальная артериальная гипертензия
Эссенциальная артериальная гипертензия (ЭАГ) — хронически протекающее заболевание неизвестной этиологии с наследственной предрасположенностью, возникающее вследствие взаимодействия генетических факторов и факторов внешней среды, характеризующееся стабильным повышением АД при отсутствии органического поражения регулирующих его органов и систем.
Большинство ученых склоняются к принятию «мозаичной» теории ЭАГ или болезни регуляции АД. По этой теории в основе ЭАГ лежат различные нарушения в частных системах регуляции АД. Это приводит к такому соотношению функций нервного, гормонального и гуморального факторов адаптации к предъявляемым организму условиям, что в итоге развиваются стойкое повышение сосудистого тонуса и увеличение АД.
Этиология
Роль факторов внутренней среды
• Генетигеские факторы в значительной степени определяют развитие гипертонии. Хорошо документирована семейная предрасположенность к артериальной гипертензии. К генам-кандидатам ЭАГ относят гены ренина, ангиотензиногена, рецепторов ангиотен-зин-П, АПФ, рецепторов глюкокортикоидов, инсулина, регуляции синтеза альдостерона, эндотелиальной NO-синтетазы, синтетазы простациклина, эндотелина-I и его рецепторов и др.
• Нарушения трансмембранного транспорта ионов. Согласно теории Ю.В. Постнова и С.Н. Орлова, ЭАГ является патологией клеточных мембран, имеющей генетическое происхождение. Вследствие мембранного дефекта в цитоплазме клеток, в том числе гладкомышечных, создается высокая концентрация ионов Na+ и Са2*, что значительно повышает чувствительность сосудистой стенки к сосудосуживающему действию ангиотензин-П и катехоламинов, способствует гипертрофии сосудистой стенки, активации симпа
Патофизиология артериальной гипертензии
407
тической нервной системы. Важная роль в этой теории отводится адаптационному почечному механизму — «переключению» почки. В условиях мембранной патологии почка может осуществлять достаточную экскрецию солей и воды лишь при более высоком, чем в норме, АД. В связи с этим происходит «переключение» почки на новый режим функционирования. На ранних стадиях ЭАГ этот процесс обратим, однако в последующем становится необратимым за счет развития структурно-морфологических изменений — гипертрофии и склероза артериол и артерий почек.
• Врожденный дефект погегной регуляции экскреции натрия. Существует предположение о том, что в развитии ЭАГ может иметь значение врожденная способность почек к повышенной задержке натрия (снижению его экскреции), что связано с врожденным дефицитом количества нефронов и нарушением клубочковой фильтрации натрия. В одной из работ было установлено, что почки больных ЭАГ содержат значительно меньшее число клубочков — 702 000 против 1 429 000 в контрольной группе. Среди причин, вызывающих врожденное уменьшение числа нефронов, обсуждаются дефекты генов, контролирующих развитие почек, действие на плод токсических веществ, блокирующих РАС (блокаторы АПФ), а также недостаточное потребление калорий и белка матерью во время беременности.
• Нарушения в деятельности почек вносят существенный вклад в развитие и поддержание первичной гипертонии. Напомним, что скорость и объем мочеотделения зависит от АД. Если посмотреть на рисунок 26-1, то можно заметить, что у нелеченого больного артериальной гипертензией отмечается очень низкая скорость мочеотделения при близком к нормальному уровню СрАД около 100 мм рт.ст. (точка В). Но если скорость поступления жидкости в организм превышает скорость мочеотделения, то объем жидкости в организме должен увеличиваться и как следствие увеличиваться МОС и СрАД. На фоне нормальной скорости поступления жидкости в организм данный больной с нелеченой гипертонией в конечном итоге стабилизируется в точке А (СрАД = 150 мм рт.ст.). Барорецепторы адаптируются на протяжении нескольких дней, так что у них отмечается нормальный уровень импульсации при господствующем уровне АД. Таким образом, поскольку больной артериальной гипертензией находился в точке А в течение недели или более, то даже барорецепторный механизм начнет противодействовать резким отклонениям от уровня 150 мм рт.ст.
408
Лекция 26
• Ожирение и гиподинамия. При ожирении отмечается активации РААС и симпатоадреналовой системы, у большинства больных имеется повышенная чувствительность к соли. Гиподинамия приводит к дезадаптации по отношению к физическим и психическим нагрузкам и характеризуется преобладанием прессорных механизмов над депрессорными в ответных реакциях организма.
Роль факторов внешней среды
Значение этих факторов наиболее существенно у лиц с генетической предрасположенностью.
Избытогное употребление поваренной соли. Считается, что адекватное количество соли для взрослого человека — это 3,5 г (60 мЭкв натрия) в сутки. В настоящее время не подвергается сомнению положение, что избыточное потребление соли является важным фактором риска развития гипертонии.
Развитие артериальной гипертензии обусловлено следующими механизмами:
• Т Na*-^ ТОЦК -> ТАД;
• на фоне генетического снижения активности Na'/K -АТФазы происходит задержка Na* в стенке артерий и артериол;
• повышение чувствительности сосудистой стенки к вазопрессорам;
• отечность сосудистой стенки, сужение просвета и рост ОПСС.
Недостатогное поступление с пищей и водой кальция и магния. Предполагается, что при дефиците кальция гладкомышечные клетки избирательно накапливают его, что повышает их активность и приводит к увеличению ОПСС. Дефицит магния повышает АД, так как:
• активируются РААС и симпатоадреналовая система;
• снижается активность Са2*- и Ыа*/К*-АТФаз; накапливается Са2*;
• снижаются эластические свойства аорты.
Курение. Предполагают, что курение, в частности никотин, повышает АД из-за угнетения синтеза простациклина эндотелием и повышения выделения норадреналина из окончаний симпатических нервов.
Алкоголь. Многими исследованиями подтверждается связь между повышением АД и приемом алкоголя, в том числе пива. Под влиянием алкоголя угнетаются барорецепторные рефлексы, активируется симпатоадреналовая система, в ЦНС повышается уровень ангиотензина-П, в гладкомышечных клетках накапливается Са2+- и Na+, повреждаются почки.
Психоэмоциональные стрессовые ситуации.
Плохое социальное и экономигеское положение.
Патофизиология артериальной гипертензии
409
Патогенез
Повышенная активность симпатоадреналовой системы. Тонус симпатической нервной системы регулируется центрами, расположенными в стволе головного мозга, в частности в рострально-вентролатеральной области продолговатого мозга. Там находятся а2-адренорецепторы и открытые в 1992 г. имидазолиновые рецепторы I и II типа (Ij- и 12-рецепторы). При возбуждении а2-адренорецепторов и ^-рецепторов (12-рецепторы не участвуют в регуляции АД) снижается тонус симпатической нервной системы, и АД уменьшается. Возбуждение 1,-рецепторов также приводит к уменьшению реабсорбции натрия и воды в проксимальных канальцах почек.
У 30-40 % больных ЭАГ повышено содержание катехоламинов в плазме в ранней стадии заболевания. У части больных с нормальным уровнем катехоламинов в крови выявили снижение количества и/или чувствительности [3-адренорецепторов и увеличение постсинаптических влияний а-адренорецепторов, что в итоге приводило к усилению вазоконстрикторных влияний катехоламинов. Дополнительный вклад в повышение АД вносит гиперсекреция ренина и активация РААС в ответ на рост симпатической активности. Причина повышенной активности симпатоадреналовой системы может быть связана с нарушением центральной регуляции, с увеличением ангиотензина-П, с постоянным использованием высококалорийной диеты, действием стрессовых факторов и малоподвижным образом жизни.
Высокая активность ренин-ангиотензин-альдостероновой системы (РААС). Различают циркулирующую (системную) РААС (цРААС) и тканевую (местную) РААС (тРААС). В цРААС входят почечный ренин, ангиотензиноген плазмы, АПФ — карбоксипептидаза или кининаза-П, ангиотензины — I, II, III, IV, ферменты, их инактивирующие, и рецепторы для ангиотензинов. Наиболее сильное вазоконстрикторное действие, а также стимуляция секреции альдостерона и ряд других важных эффектов принадлежат ангиотензину-П. Доказано существование тРААС. Она содержит все компоненты РААС и обнаружена во многих тканях, в том числе в кровеносных сосудах, миокарде, надпочечниках, почках, мозге.
тРААС имеет свои отличительные от цРААС особенности. Так, в тканях существуют другие пути образования ангиотензина-П, что делает неэффективным применение ингибиторов АПФ для снижения синтеза ангиотензина-П. Независимо от АПФ в сердце 80 % ангиотензина-П образуется из ангиотензина-I с помощью фермента химазы, в стенке арте
410
Лекция 26
рий — под влиянием химазоподобного фермента, а также под влиянием катепсина, тканевого активатора плазминогена. В других тканях ренин заменяют катепсин G, эластаза, тканевой активатор плазминогена, причем ангиотензиноген сразу превращается в ангиотензин-И.
тРААС осуществляет длительный контроль за АД, так как она способствует гипертрофии сосудистой стенки (ТОПСС) и сердца, повышению локального синтеза норадреналина, а в почках — развитию внутриклу-бочковой гипертензии, ангиосклероза и последующей гибели клубочков. Показано, что при нормо- или гипоренинной формах артериальной гипертонии отмечается высокая активность тРААС.
Все основные эффекты активации РААС опосредуются рецепторами преимущественно 1-го типа, которые подразделяются на АТ1а и АТ1В. В регуляции системного АД ведущую роль играют АТ1а. Именно через них ангиотензин-И вызывает гипертрофию и фиброз миокарда, вазоконстрикцию и повышение АД, гипертрофию (ремоделирование) артерий, стимуляцию секреции альдостерона и адреналина надпочечниками, развитие нефросклероза, стимуляцию центра жажды и выделение АДГ, повышение активности центрального звена симпатической нервной системы и др.
Ремоделированию сосудов способствует оксидантный стресс. Повышенный уровень ангиотензина-П и АД активируют НАДН- и НАДФН-оксидазы в эндотелиоцитах (ЭЦ), гладких мышцах сосудов, макрофагах и других клетках, что приводит к образованию перекиси водорода и су-пероксиданионрадикала. Они разрушают NO с образованием пероксинитрита (OONO ). Пероксинитрит инициирует ПОЛ, повреждает ДНК, подавляет выработку энергии в митохондриях.
ККС. Существуют циркулирующая плазменная ККС (цККС) и тканевая локальная ККС (тККС), из последней наиболее изучена почечная ККС. В ККС входят прекалликреин и калликреины (плазменный и тканевой), кининогены, брадикинин и другие кинины, ферменты, их расщепляющие, и рецепторы для брадикинина на мембранах клеток.
Брадикинин — главный эффекторный пептид ККС, вызывает:
• сосудорасширяющее действие. Он действует непосредственно на стенку сосудов, а также через стимуляцию эндотелия к выделению NO, простациклина и простагландина Е;
• обладает натрийуретическим действием, так как стимулирует синтез в собирательных трубках и интерстициальных клетках почек простагландина Е, способствующего выраженному натрийурезу;
• уменьшает антидиуретическое действие АДГ;
Патофизиология артериальной гипертензии
411
• уменьшает высвобождение норадреналина из окончаний симпатических волокон и секрецию катехоламинов надпочечниками;
• снижает АД.
Показано снижение активности ККС у больных гипертонией. Возможно, это связано с высокой активностью АПФ-кининазы И, который превращает брадикинин в неактивные пептиды. Не исключена и наследственная роль низкой активности ККС. В результате превалирует ответ антагонистических гипертензивных факторов (РААС, симпатоадреналовой системы и др.).
Эндотелиальная дисфункция. Эндотелиоциты участвуют в регуляции тонуса сосудов, поскольку синтезируют наряду со многими важными веществами вазодилатирующие и вазоконстрикторные факторы. На состояние и функцию ЭЦ влияют 3 основные группы факторов:
• изменение скорости кровотока (увеличение напряжения сдвига);
• медиаторы тромбоцитов (серотонин, тромбин, АДФ);
• биоактивные вещества, циркулирующие в крови и продуцируемые самими ЭЦ (катехоламины, АДГ, гистамин, брадикинин и др.).
Основным вазодилататором, продуцируемым ЭЦ, является оксид азота — NO, кроме которого ЭЦ вырабатывают еще простациклин (простагландин 12) и другие вазодилататоры. NO образуется из L-аргинина с помощью эндотелиальной NO-синтетазы. Продукция NO бывает базальной и стимулированной. Базальная, или постоянная, продукция NO играет очень важную роль. Она обеспечивает оптимальную степень дилатации сосудов и препятствует вазоконстрикции. Основными стимуляторами секреции NO являются напряжение сдвига в сосудах, брадикинин и ацетилхолин (синтез NO увеличивают еще норадреналин, АДГ, гистамин, серотонин, тромбин, эндотелии). NO вызывает в гладкомышечной сосудистой клетке активацию гуанилатциклазы, рост цГМФ (циклического гуанозинмонофосфата), уменьшение Са2’ и вазодилатацию.
При ЭАГ продукция NO понижена. Возможно, это связано с уменьшением активности NO-синтетазы (у больных выявлен полиморфизм гена NO-синтетазы), с повышением активности АПФ, разрушающего брадикинин, — стимулятор синтеза NO, или с повышенной инактивацией NO свободными радикалами, возникающими при перекисном окислении липидов.
Простациклин. Синтезируется ЭЦ из арахидоновой кислоты. Синтез стимулируется напряжением сдвига, брадикинином, веществом Р, тромбином, ИЛ-1, ангиотензином-П, гипоксией. Является сильным
412
Лекция 26
антиагрегантом в отношении тромбоцитов. Расширяет сосуды за счет увеличения синтеза цАМФ (циклического аденозинмонофосфата) в гладкомышечных клетках и снижения их чувствительности к Са2’. При ЭАГ продукция простациклина понижена.
ЭЦ вырабатывают вазоконстрикторы — эндотелины (ЭТ-1, 2, 3, 4), ангиотензин-П (превращение ангиотензина-I под влиянием АПФ), тромбоксан, простагландин Н2.
Наиболее сильный вазоконстриктор — ЭТ-1. Он образуется из своего предшественника — «большого эндотелина» — под влиянием эндотелин-превращающего фермента, обнаруженного внутри и на поверхности ЭЦ в разных органах и тканях. ЭТ разрушаются под действием различных протеаз, наибольшее значение имеют деамидаза и энкефалиназа. Синтез ЭТ-1 стимулируется ангиотензином-П, АДГ, тромбином, модифицированными ЛПНП, гиперхолестеринемией, свободными радикалами, глюкозой, некоторыми факторами роста, гипоксией и напряжением сдвига.
ЭТ-1 вызывает спазм сосудов, активируя в гладкомышечной клетке G-белки, фосфолипазу С, что приводит к повышению в клетке Са2* и сокращению клеток. ЭТ-1 стимулирует пролиферацию гладких мышц сосудов, оказывает положительное инотропное влияние на миокард и усиливает активность РАС и симпатоадреналовой системы. При ЭАГ обнаружен повышенный уровень ЭТ-1 в крови.
Таким образом, указанная эндотелиальная дисфункция — сниженная продукция вазодилататоров и повышенная — вазоконстрикторов, приводит к ремоделированию артерий (гипертрофии, фиброзу), снижению эластичности стенок артерий, к повышению ОПСС и АД.
Нарушение депрессорной функции потек. Почки вырабатывают из каллидина брадикинин и гипотензивные простагландины, главным из которых является простагландин Е2 (PgE2). Он расширяет сосуды почек, улучшает почечный кровоток, повышает экскрецию натрия и воды, ингибирует синтез ренина и ангиотензина-П. При гипертонии депрессорная система почек вначале активируется, затем истощается. Уменьшается продукция кининов, PgE2 что способствует проявлению действия вазопрессорных механизмов и стабилизации высокого АД.
Патогенез поражения органов-мишеней при эссенциальной артериальной гипертензии
Высокое АД постепенно приводит к поражению органов-мишеней.
Гипертрофия сердца, прежде всего левого желудочка, является следствием перегрузки сердца давлением (постнагрузки) и воздействия на
Патофизиология артериальной гипертензии
413
миокард нейрогуморальных факторов (катехоламинов, ангиотензин-П, альдостерона и др.).
Поражение периферигеских артерий заключается в их ремоделировании и протекает в две стадии: стадию функциональных изменений сосудов, обусловленную сосудосуживающими реакциями в ответ на трансмуральное давление и нейрогормональную стимуляцию, и морфологическую стадию, которая характеризуется уменьшением просвета сосудов вследствие утолщения их медиального слоя. Сосуды становятся ригидными, так как в них увеличивается содержание внеклеточного коллагена. В крупных артериях развивается атеросклероз. Мелкие сосуды, артериолы и капилляры, редуцируются, в тканях развивается разряжение сосудистого русла с уменьшением объемного кровотока и развитием венозной гиперемии и стаза.
Поражение потек — гипертоническая нефропатия или гипертонический артериолосклеротический нефросклероз (первично сморщенная почка) проходит ряд стадий. Вначале наблюдаются функциональные изменения сосудов почек. Повышение АД приводит к увеличению ультрафильтрации и выведения избытка натрия, способствуя нормализации АД. Для защиты от повышенного АД в почке возникает спазм приводящих афферентных артериол, постепенно приводящий к их ремоделированию. В последующих стадиях развивается гипертонус эфферентных сосудов. Артериосклеротические изменения сосудов, функциональная перегрузка клубочков и канальцев приводят постепенно к атрофии и диффузному склерозу почек.
В сосудах сеттатки при артериальной гипертензии развиваются те же механизмы поражения, как и в артериолах и капиллярах.
Головной мозг поражается в результате ремоделирования как крупных, так и мелких сосудов.
Вторичные артериальные гипертензии
Из вторичных или симптоматических артериальных гипертензий рассмотрим патогенез почечных гипертоний.
Первичные заболевания паренхимы почек, как правило, приводят к развитию стойкого повышения АД, развитию ренопривных гипертензий.
Патогенез их представляется следующим:
• активируется РААС и симпатоадреналовая система;
• задерживаются натрий и вода в результате уменьшения массы действующих нефронов;
414
Лекция 26
• снижается активность депрессорной системы почек, уменьшается синтез кининов, PgE2, NO;
• в почках активируются ПОЛ; продукты ПОЛ, свободные радикалы способствуют выделению вазопрессорных медиаторов из арахидоновой кислоты внутри клубочковых мембран, угнетают синтез эндотелием NO. В норме NO вырабатывается ЭЦ почечных артериол, капилляров клубочка, областью плотного пятна. При уменьшении выработки NO активируется синтез ренина, усиливается задержка натрия, возрастает протеинурия, прогрессирует гломерулосклероз, стабилизируется высокое АД. Кроме этого увеличивается продукция эндотелина.
Заболевания почечной артерии (атеросклероз, тромбоз, эмболия и др.) приводят к развитию реноваскулярной гипертензии.
Повышение АД развивается в результате:
• активации РААС из-за ишемии почки на стороне поражения;
• развития оксидативного стресса.
Принципы терапии (основные группы препаратов)
• Диуретики.
• Ингибиторы АПФ.
• Антагонисты кальция.
• р-адреноблокаторы.
Препараты последних поколений других групп часто назначаются дополнительно к какому-то препарату из групп 1-2. Например, агонисты имидазолиновых рецепторов, периферические вазодилататоры, открывающие калиевые каналы, а,-адреноблокаторы.
Рекомендуется ограничение в диете поваренной соли.
Лекция 27
РОЛЬ РЕАКТИВНОСТИ ОРГАНИЗМА В ПАТОЛОГИИ
Г.В. Порядин
Воздействие на организм человека и животных экзогенных и эндогенных факторов может стать результатом развития болезни или патологического процесса. Однако не всякое воздействие факторов среды завершается развитием болезни. Например, наличие в легких микобактерий Коха не всегда сопровождается развитием туберкулеза, а наличие в миндалинах патогенных микроорганизмов обходится без возникновения ангины.
Следовательно, возникновение болезни и патологического процесса определяется не только патогенным агентом, но и организмом, в который он попал, и условиями, в которых происходит взаимодействие организма и причинного фактора.
Важным в этой цепочке является свойство организма — его реактивность.
Реактивность организма — это способность организма как целого отвечать определенным образом (качественно и количественно) изменениями жизнедеятельности на воздействие факторов окружающей (внутренней и внешней) среды, приспосабливаясь (адаптируясь) к ним.
Реактивность присуща всем живым организмам наряду с такими качествами, как размножение, рост, обмен веществ, наследственность и др.
От реактивности в большой степени зависят поддержание гомеостаза, возникновение или невозникновение болезни при встрече с болезнетворным фактором, характер течения, исход болезни.
Вот почему изучение реактивности, ее механизмов имеет большое значение как для понимания патогенеза заболеваний, так и для целенаправленной их профилактики и лечения.
Формирование реактивности тесно связано с филогенетическим усложнением живых существ.
Из курса биологии вам известно, что все живые объекты обладают свойством изменять свое состояние или деятельность, т.е. реагировать
416
Лекция 27
на воздействия факторов внешней среды. Причем, как правило, генерализованной, малодифференцированной реакцией. Например, изменением формы, размеров и др.
Это свойство принято называть раздражимость.
Но все живые организмы реагируют неодинаково на одно и то же воздействие, определенным образом — это реактивность.
Одни виды животных изменяют жизнедеятельность на внешние воздействия не так, как другие виды; одни группы людей (или животных) реагируют на одно и то же воздействие не так, как другие группы; и каждый индивидуум в отдельности имеет свои особенности реагирования.
В связи с этим различают несколько видов реактивности.
1. Реактивность, которая определяется наследственными анатомофизиологическими особенностями представителей данного вида, получила название видовая (или биологигеская}. Это наиболее общий вид реактивности организма.
Видовая (биологическая) реактивность возникает под влиянием обычных (адекватных физиологическим возможностям представителей вида) воздействий окружающей среды. Эту реактивность еще называют первичной (физиологигеской) — она направлена на сохранение вида в целом и имеет адаптивный характер.
2. Кроме общих (видовых свойств реактивности) имеются особенности реактивности у отдельных групп людей (или животных), объединенных каким-то общим признаком, от которого зависят особенности реагирования всех представителей данной группы на воздействия внешних факторов, — это групповая реактивность.
К таким признакам относятся возраст, пол, конституциональный тип, группа крови, тип высшей нервной деятельности, группа людей с одним и тем же заболеванием и др.
Так, проявления реактивности организма связаны с возрастом.
Например, у детей грудного возраста в силу недостаточности функциональной зрелости больших полушарий головного мозга, недоразвития барьерных систем организма отмечается своеобразие реактивности, проявляющееся склонностью к расстройствам пищеварения и обмена веществ.
В возрасте 1-3 лет в силу функциональной незрелости иммунной системы (неспособность к выработке антител) ребенок особо восприимчив к различным инфекциям.
Наиболее оптимально выражена физиологическая реактивность организма в онтогенезе в зрелом возрасте, когда все системы сформированы и функционируют.
Роль реактивности организма в патологии
417
В старости отмечается снижение физиологической реактивности, чему, по-видимому, способствуют перемена в гормональной системе, понижение реактивности нервной системы, ослабление функции барьерных систем. Отсюда повышение восприимчивости к кокковым инфекциям, вирусным (грипп) инфекциям.
Особенность реактивности детей и стариков привело к необходимости выделения особых разделов медицины — педиатрии и гериатрии.
Большое значение в реактивности организма придается особенностям его конституции (особенности строения, характеру реакций на внешние и внутренние раздражители).
Конституция — это единый комплекс достаточно устойчивых морфологических, функциональных, психических особенностей организма, сложившийся на основе генотипа под влиянием факторов внешней среды.
3. Кроме общих (видовых) и групповых свойств реактивности имеются и индивидуальные особенности реактивности у каждого индивида в отдельности.
Например, воздействие какого-либо фактора (например, инфекционного агента) на группу людей или животных никогда не вызывает у всех индивидов этой группы совершенно одинаковые изменения жизнедеятельности.
Например, при эпидемии гриппа некоторые люди болеют тяжело, другие — легко, а третьи не болеют вовсе, хотя возбудитель и находится в их организме (вирусоносительство) или окружающей среде. Объясняется это индивидуальными особенностями реактивности каждого организма.
Биологическая значимость ответа организма на действия факторов среды определяет физиологическую или патологическую реактивность.
• Физиологическая реактивность — это изменения жизнедеятельности организма в ответ на действие факторов окружающей среды, не нарушающие его гомеостаза, это реактивность здорового человека (или животного) на непатогенные раздражители.
• Под воздействием на организм чрезвычайных болезнетворных факторов возникает патологическая реактивность. Эта реактивность характеризуется понижением приспособляемости болеющего организма. Ее еще называют вторигной (или измененной) реактивностью.
Патологическая реактивность проявляется при действии на организм болезнетворных факторов, вызывающих в организме повреждение и нарушение его гомеостаза.
418
Лекция 27
Способность организма сопротивляться воздействиям окружающей его среды, сохраняя при этом постоянство гомеостаза, тесно связана с функционированием как механизмов неспецифической защиты, так и механизмов специфической защиты.
Неспецифические механизмы реактивности
Неспецифические факторы защиты формируются в фило- и онтогенезе раньше, чем специфические или иммунологические механизмы. При возникновении заболевания неспецифическая реактивность осуществляет первую, раннюю защиту организма, давая ему время для формирования полноценного иммунного ответа.
Неспецифические механизмы реактивности разделяются на клеточные и гуморальные.
Клеточные механизмы реактивности
К их числу относится фагоцитарная активность микро- и макрофагов.
Фагоцитоз представляет филогенетически наиболее древнюю и стабильную защитно-приспособительную клеточную реакцию организма. Фагоцитирующими клетками являются макрофаги (альвеолярные, перитонеальные, макрофаги печени и т.д.), моноциты (предшественники макрофагов), нейтрофильные гранулоциты или микрофаги.
Фагоцитоз — основная функция макрофагов и нейтрофилов (микрофагов), которые поглощают частицы размером 0,5-5,0 мкм.
Фагоцитоз рассматривается как сложный многоступенчатый процесс, начинающийся с захвата чужеродного агента фагоцитом и кончающийся его перевариванием.
Кроме защитных реакций против различных инфекций, макрофаги участвуют в противоопухолевой защите, переработке и представлении антигена (АГ), регуляции иммунных процессов.
Нейтрофилы обеспечивают основную защиту от гноеродных бактерий. Нейтрофилы несут на своей поверхности рецепторы к Fc-фрагменту IgG, IgM и к С3, с помощью которых осуществляется прикрепление микробов к поверхности фагоцита.
Поглощение патогенного фактора фагоцитом является активным энергозависимым процессом, стимулирующим выработку АТФ и ее распад, гликолиз в нейтрофилах и перитонеальных макрофагах, а также окислительное фосфорилирование в альвеолярных макрофагах.
Роль реактивности организма в патологии
419
Препараты адренергического ряда, повышающие внутриклеточный уровень цАМФ в фагоцитах, ингибируют, а цГМФ и холинергические препараты, наоборот, стимулируют фагоцитоз.
Для нейтрофилов (но не макрофагов) основной является бактерицидная система: миелопероксидаза — перекись водорода — галогены (МПО-Н2О2-Г). Галогены повышают бактерицидность Н2О2. В качестве галогенов выступают ионы Г, Вг, С1_, указанные в порядке убывания их бактерицидной активности.
Ярко выраженные бактерицидные свойства имеют другие кислородные метаболиты: супероксиданионрадикал (О2~), синглетный кислород (Ю2) и ион гидроксила (ОН ), которые являются короткоживущими, реакционноспособными, они мгновенно окисляют и повреждают поверхностные структуры и содержимое микробной клетки.
Активация мембраны фагоцитов опсонизированными микробами или частицами и иммунными комплексами приводит к серии метаболических изменений, получивших название «респираторный взрыв», в результате которого значительно повышается поглощение кислорода фагоцитирующими клетками.
В нейтрофиле существуют и другие бактерицидные системы, не зависящие от кислорода. Бактерицидное и бактериостатическое действие на многие микроорганизмы оказывает кислотность фагоцитарной вакуоли (pH 3,5-4,0), миелопероксидаза гранулоцитов, эластаза, а также внутриклеточный лизоцим, щелочной и катионный белки, расщепляющие мукопептиды клеточной стенки некоторых видов бактерий.
Антибактериальными свойствами обладают лизосомальные ферменты, поступающие после дегрануляции в фагосому и способные гидролизовать белки, жиры и углеводы поглощенных частиц, осуществляя последнюю стадию фагоцитоза — переваривание микробной клетки.
Мононуклеарные фагоциты имеют сходные бактерицидные механизмы: кислотность, лизоцим, щелочной белок, Н2О2, О2, ОН-, однако они не имеют катионных белков и лактоферрина, содержат значительно меньше миелопероксидазы или вовсе не содержат ее в гранулах. Роль отсутствующей миелопероксидазы, особенно в альвеолярных макрофагах, выполняет каталаза. Малонилдиальдегид, продукт липидного пере-окисления, обладает выраженной бактерицидной активностью.
Перитонеальные макрофаги одинаково эффективно фагоцитируют и убивают ряд микробов в аэробных и анаэробных условиях, в то время как соответствующая активность нейтрофилов резко снижается при полном отсутствии кислорода.
420
Лекция 27
Реакция мононуклеарных фагоцитов, заключающаяся в выработке бактерицидных кислородных радикалов, развивается медленнее и выражена слабее, чем у нейтрофилов. Макрофаги в реактивном состоянии в отличие от нейтрофилов часто не до конца переваривают поглощенный антигенный материал. Таким образом, нейтрофилы при фагоцитозе работают более эффективно, чем макрофаги, которым необходим дополнительный активирующий сигнал для усиления их бактерицидной функции.
Гуморальные механизмы реактивности
1. Ингибиторы вирусной активности представляют первый гуморальный барьер, препятствующий контакту вируса с клеткой.
• Термолабильные ингибиторы инактивируют инфекционные, гемаг-глютинирующие и токсические свойства ингибиторочувствительных штаммов вируса.
• Термостабильные ингибиторы блокируют соединение вируса с рецепторами клеток.
Люди с высоким содержанием ингибиторов вирусной активности отличаются высокой устойчивостью к вирусной инфекции и слабой при-живляемостью вакцинных штаммов вирусов.
2. Система комплемента — термолабильная ферментная система, состоит из 20 белков сыворотки крови, составляющих 9 компонентов комплемента (Cj-C9).
Для этой системы характерно формирование быстрого многократного усиленного ответа на первый сигнал за счет каскадного процесса. В результате такой активации продукт одной реакции служит катализатором последующей.
Известны 2 основных пути активации системы комплемента: классический и альтернативный, но при определенных условиях комплемент может быть активирован фибринолитической, ККС, свертывающей системой, С-реактивным белком, лизосомальными ферментами нейтрофилов и т.д.
Система комплемента обладает рядом биологических функций:
• участвует в процессах опсонизации. Фагоцитирующие клетки имеют рецепторы для С3в и С3вГ которые участвуют в прикреплении микроорганизмов;
• образует биологически активные фрагменты С3а и С5а, которые являются хемотаксинами для нейтрофилов и анафилотоксинами для тучных клеток, вызывая их активацию и дегрануляцию;
• образует мембраноатакующий комплекс.
Роль реактивности организма в патологии
421
Система комплемента состоит из активных компонентов и их ингибиторов и для нормального ее функционирования важен баланс всех составляющих компонентов.
3. Лизоцим (мурамидаза) является ферментом лизосомальных структур клеток и относится к основным факторам неспецифической защиты. Лизоцим содержится у человека и животных во многих биологических жидкостях и тканях, синтезируясь в основном в макро- и микрофагах.
Основным биологическим эффектом этого фермента является его бактериологическое и бактериостатическое влияние на некоторые микроорганизмы.
Наиболее чувствительны к лизоциму грамположительные бактерии, у которых фермент катализирует гидролиз гликозаминогликанов, растворяет клеточную оболочку и вызывает распад всей бактериальной клетки. Грамотрицательные бактерии более устойчивы к лизоциму ввиду того, что пептидогликаны их клеточной оболочки находятся под слоем ЛП и липополисахаридов. Поэтому лизоцим оказывает менее выраженное влияние на грамотрицательные микроорганизмы.
4. Пропердин — высокомолекулярный белок, обнаруживаемый в р-глобулиновой, а-глобулиновой или одновременно во всех трех фракциях сыворотки крови.
В основе действия пропердина лежит его способность соединяться с полисахаридными структурами микробных клеток с образованием комплекса пропердин + полисахарид, который необратимо связывает третий компонент комплемента. Пропердин обеспечивает бактерицидное, гемолитическое, вируснейтрализующее свойство сыворотки крови (в совокупности с другими гуморальными факторами и в присутствии ионов магния).
5. Лейкины — термостабильные бактерицидные факторы, образующиеся при распаде лейкоцитов. Аналогичные вещества, называемые плакинами, образуются при распаде тромбоцитов. Лейкины и плакины способны инактивировать стафилококки и другие грамположительные микробы.
6. р-Лизины — термостабильные гуморальные факторы, активные в отношении некоторых аэробов и анаэробов.
7. Интерфероны — группа термостабильных, низкомолекулярных неспецифических белков, синтезируемых лимфоцитами и моноцитами и обладающих противовирусной активностью. Существуют различные типы интерферонов: а, р, у.
422
Лекция 27
Механизм действия интерферона состоит в подавлении соединения вирусной РНК с рибосомами клетки. Соединяясь с соседними незаряженными клетками, интерферон оказывает наиболее активное противовирусное действие до заражения или в самом начале репродукции вируса.
Интерферон широко применяется в клинической практике не только для профилактики и лечения вирусных заболеваний, но и как иммунокорригирующий препарат в онкологической практике и при лечении системных заболеваний, в основе которых лежит поражение иммунной системы (коллагенозы, системная красная волчанка и т.д.). В результате лечения интерфероном усиливаются функция нейтрофильных лейкоцитов, фагоцитарная активность макрофагов, повышаются титр комплемента, активность лизоцима, модулируются функции Т- и В-лимфоцитов.
В таблице 27-1 отражены наиболее типичные виды нарушения неспецифических механизмов реактивности (резистентности).
Таблица 27-1. Нарушения неспецифических механизмов реактивности (резистентности)
Виды Проявления
Недостаточность фагоцитарной активности Повышение чувствительности к инфекциям Склонность к гнойно-септическим заболеваниям, развитию хронического воспаления и болезням накопления
Недостаточность комплемента Повышение чувствительности к инфекциям, склонность к возникновению злокачественных новообразований и аутоиммунных заболеваний
Недостаточность лизоцима Повышение чувствительности к инфекциям (особенно слизистых оболочек)
Недостаточность интерферона Снижение устойчивости к вирусам
Снижение пропердина Снижение бактерицидных свойств крови
Недостаточность синтеза р-лизинов, лейкинов, плакинов Снижение бактерицидных свойств крови по отношению к грамположительным микробам
1. Недостаточность фагоцитарной активности может быть приобретенной и врожденной.
Нарушения фагоцитарных функций довольно часто сопровождают рецидивирующие гнойно-септические заболевания, такие как хроническая пневмония, пиодермия, остеомиелит, кожно-слизистый кандидоз и так далее, которые протекают с массивной деструкцией тканей и не поддаются обычным методам лечения, включая интенсивную комбинированную антибиотикотерапию.
Роль реактивности организма в патологии
423
Подобные клинические проявления, как правило, обусловлены нарушением бактерицидное™ фагоцитов.
Например, хронигеская гранулематозная болезнь возникает в результате отсутствия ферментативной активности, ответственной за превращение поглощенного кислорода в бактерицидные продукты О2, Н2О2, ОН ".
Заболевание проявляется с раннего возраста и протекает в виде экзематозных поражений кожи, пиодермий, гнойных аденитов, остеомиелитов, инфильтративной пневмонии. Быстро развиваются стафилококковые абсцессы кожи, легких и печени, несмотря на хирургическое вмешательство и массивную антибиотикотерапию.
Синдром Чедиака-Хигаси также обусловлен нарушением фагоцитарной функции. Это аутосомно-рецессивная болезнь, которая сопровождается рецидивирующими инфекциями, частичным кожно-глазным альбинизмом, фотофобией, нистагмом и появлением нейтрофилов с гигантскими цитоплазматическими гранулами. Заболевание проявляется в раннем детстве, больные часто погибают от инфекций или опухолей, не достигая 10-летнего возраста.
Синдром «ленивых» лейкоцитов характеризуется нейтропенией со сниженной спонтанной подвижностью клеток и нарушением реакции на хемотаксический стимул.
Врожденная миелопероксидазная недостатогность нейтрофилов проявляется хроническим кожно-слизистым кандидозом, поскольку такие клетки не способны убивать этот вид грибов. Потребление кислорода, окислительный метаболизм нормальны или даже повышены.
Нарушения глутатионовой системы проявляются резкими нейтропениями во время инфекций. Это обусловлено тем, что глутатионовая система, необходимая для обезвреживания агрессивных кислородных метаболитов, не справляется с нагрузкой во время фагоцитоза и происходит «нейтрофильное самоубийство».
Отсутствие щелогной фосфатазы в лейкоцитах больных вызывает недостаточность хемотаксиса и внутриклеточного киллинга. Больные с синдромом Дауна более чувствительны к инфекциям, чем здоровые, из-за снижения хемотаксиса, поглощения и бактерицидное™ фагоцитов.
При ряде инфекций возникают различные приобретенные нарушения фагоцитоза. Например, микобактерии, находясь в фагосоме, препятствуют их слиянию с лизосомами. Стафилококк тормозит нормальную опсонизацию и поглощение.
424
Лекция 27
Встречаются также формы нарушений, при которых снижено образование хемотаксических веществ, либо имеется недостаточный клеточный ответ на нормальный хемотаксический стимул. Это сочетается с нарушением поглощения инфекционного фактора фагоцитом и сопровождается тяжелыми рецидивирующими инфекциями.
Нарушения фагоцитарной функции приводят также к развитию хронического воспаления и к болезням накопления, например амилоидозу, когда поглощенный макрофагом материал не может быть катаболизи-рован в результате наследственной недостаточности ферментов.
В процессе роста опухоли, эмбрионального роста, регенерации ткани, например после травмы, в организме возникает антивоспалительная реакция, направленная в большей степени против полиморфно-ядерных клеток и опосредованная растворенными антивоспалительными факторами. Считается, что пролиферация клеток при опухолевом росте в определенной степени обусловлена подавлением функции моноцитов.
Фагоцитоз угнетается глюкокортикоидами и медиаторами: ацетилхолином, нарушениями белкового, витаминного, водно-электролитного баланса, при злокачественных новообразованиях.
Незавершенному фагоцитозу способствуют некоторые микробные яды и ингибиторы гликолиза, приводящие к дефициту энергии в клетках.
Нарушения образования и дифференцировки фагоцитов возникают при лейкопениях, микозах; встречаются также наследственный дефект образования лизосом, недостаточность ферментов. В конечном счете все это приводит к незавершенному фагоцитозу.
2. Дефицит компонентов системы комплемента способствует повышенной чувствительности к инфекциям возникновению злокачественных новообразований и аутоиммунных заболеваний.
• Недостатогность С2 вызывает снижение бактерицидности плазмы крови и способствует частому развитию инфекционных заболеваний верхних дыхательных путей, хронического гломерулонефрита, артрита, отита и др.
• Недостатогность С2 способствует инфекциям, заболеваниям почек, тромбоцитопении.
• Дефицит С3 компонента значительно снижает ферментативную и регуляторную активность системы комплемента, наблюдается высокая смертность.
Особое значение в осуществлении инфекционной защиты имеет С3а, который связывается с поверхностью бактериальной клетки, опсонизи
Роль реактивности организма в патологии
425
рует ее, что приводит к образованию терминального мембраноатакующего комплекса, который перфорирует оболочки микробов, приводит к их лизису.
• При наследственной недостатогности С5 компонента встречаются нарушение развития ребенка, дерматиты и диарея.
• Специфический артрит и нарушение свертываемости крови наблюдаются при дефиците С6.
• Диффузные поражения соединительной ткани возникают при снижении концентрации Сг
Кроме недостаточности встречается также и активация системы комплемента, например, снижение содержания ингибитора Ц компонента приводит к развитию наследственного ангионевротического отека.
Активация потребления комплемента происходит при термическом ожоге, когда создается дефицит комплемента, что может оказаться критическим фактором, определяющим исход термической травмы.
3. Уменьшение содержания пропердина наблюдается при ряде патологических состояний: при острой кровопотере, хирургическом шоке, тяжелых ожогах, заболеваниях крови, хронических инфекциях и т.д.
4. Содержание р-лизинов, лейкинов и других антимикробных гуморальных факторов снижается при хронических, затяжных инфекциях, сопровождающихся угнетением неспецифических механизмов реактивности.
Как видно, неспецифические механизмы реактивности (резистентности), клеточные и гуморальные, возникли в филогенезе для сохранения относительного постоянства внутренней среды путем уничтожения чужеродных факторов различной природы.
Эти механизмы направлены на любые органические и неорганические объекты, отличные от клеток ткани, и они достаточны для обеспечения жизнедеятельности низкоорганизованных организмов.
Появление существ с высокой организацией привело к необходимости формирования более тонкого механизма защиты, который взял бы на себя функцию прецезионного прицельного различения мутированных клеток, выявляя генетически родственные, но в чем-то отличающиеся клетки.
В результате возникла специфигеская (иммунологигеская) реактивность — высокоспециализированная форма реакции.
С учетом этих двух важнейших механизмов, определяющих формирование реактивности организма, реактивность может быть:
• неспецифической;
• специфической.
426
Лекция 27
Резистентность организма — это его устойчивость к действию патогенных факторов.
Различают следующие формы резистентности: первихную и вториг-ную; пассивную и активную.
Первичная (наследственная) резистентность может быть абсолютной и относительной.
• Абсолютная. Например, человек абсолютно невосприимчив к чуме рогатого скота, к собственным тканевым АГ. Гонорея — сугубо человеческая болезнь, и в эксперименте ни при каких условиях не удается заразить гонококком животных.
• Относительная резистентность. Человек невосприимчив к чуме верблюда, однако, будучи сильно утомлен, может заразиться.
Вторичная (приобретенная) резистентность. Например, иммунитет (невосприимчивость) после перенесенных инфекционных заболеваний, после введения вакцин и сывороток. Резистентность к неинфекционным воздействиям приобретается путем тренировок, например, к физическим нагрузкам, к действию ускорений и перегрузок, к гипоксии, к низким и высоким температурам и т.д.
Резистентность может быть:
• активной — возникает в результате активной адаптации (активного включения механизмов защиты) к действию повреждающего фактора (например, фагоцитарная активность, образование антител при инфекции);
• пассивной — не связанной с активным функционированием механизмов защиты (возникает при передаче антител от матери к ребенку, при заместительном переливании крови).
Резистентность, как и реактивность, может быть:
• специфигеской — к действию какого-либо одного определенного патогенного агента (например, устойчивость к определенной инфекции);
• неспецифигеской — по отношению к самым различным воздействиям.
Способность организма поддерживать постоянство гомеостаза тесно связана с функционированием механизмов неспецифической и специфической защиты.
Более того, иммунные (специфические) механизмы являются центральным биологическим механизмом реактивности, основной биологический смысл которого заключается в поддержании антигенного гомеостаза.
Роль реактивности организма в патологии
427
По этой причине проблема иммунологической реактивности как важнейшей составляющей реактивности организма в целом приобретает большое значение для практической медицины.
Иммунологическая реактивность — это способность организма отвечать на действие АГ, выработкой гуморальных антител и комплексом клеточных реакций, специфичных по отношению к данному АГ.
Иммунопатология — область медицины (медицинских знаний), которая изучает патологические процессы и заболевания, основу которых составляют повреждения иммунологической реактивности и резистентности.
Иммунопатология охватывает практически все разделы медицины.
• Во внутренних болезнях — выделяют большую группу самостоятельных заболеваний, получивших общее название аллергические (анафилаксия, атопия, лекарственная аллергия).
• Иммунопатологические поражения соединительной ткани составляют основу коллагеновых болезней (ревматизм, СКВ и др.).
• Широкое внедрение в медицинскую практику серотерапии, вакцинотерапии (наряду с несомненным прогрессом) ознаменовалось ростом таких осложнений, как сыворотогная болезнь.
• Гематология все чаще имеет дело с разновидностями анемий, тромбоцитопений, лейкопений (гемолитическая анемия новорожденных, ядерная желтуха, резус-несовместимость), которые в своей основе имеют иммунную природу.
• В развитии многих видов патологии (острый нефрит, ревмокардит, ревматоидный артрит, иммунный тиреоидит, ИБС) в качестве ведущих механизмов рассматриваются процессы аутоиммунизации.
Патогенетическая классификация иммунопатологических заболеваний
Условно (с учетом механизмов, лежащих в их основе) можно выделить три большие группы заболеваний, имеющих иммунную природу.
• Болезни, обусловленные иммунологической недостаточностью или повреждением иммунологической реактивности в отношении чужеродных (посторонних) АГ.
• Болезни, обусловленные срывом иммунологической резистентности (толерантности) в отношении собственных антигенных структур.
• Болезни, связанные с нарушением пролиферации иммунокомпетентных клеток.
428
Лекция 27
Болезни, обусловленные иммунологической недостаточностью (нарушение формирования иммунного ответа)
Оптимальное функционирование иммунной системы обеспечивается взаимодействием специфических клеточных элементов (лимфоцитов) друг с другом, а также с нелимфоидными элементами (нейтрофильные, эозинофильные, моноцитарные лейкоциты и др.).
Отавными из них являются АПК. В основном это мононуклеарные фагоциты, эндотелиальные и глиальные клетки.
Пусковым моментом для включения иммунного ответа является взаимодействие Т-хелпера с АПК, на поверхности которой присутствует антигенный пептид, связанный с молекулой МНС.
Схематически осуществление первичного иммунного ответа можно представить в следующем виде (рис. 27-1).
Попадание в организм чужеродного агента — АГ — связывание АГ + АПК — презентация АГ Т-хелперам.
Т-хелперы дифференцируются в две основные разновидности — Th1 и Th2, которые отличаются набором секретируемых цитокинов. Ключевыми цитокинами Tht являются — ИЛ-2 и ИНФу. Th2 - ИЛ-4, ИЛ-5, ИЛ-6.
Дифференцировка Т-хелперов контролируется цитокинами:
• ИЛ-12 — способствует развитию Thp
• ИЛ-4 — способствует развитию Th2.
Между Th1 и Th2 существуют отношения антагонизма, реализуемые их ключевыми цитокинами ИНФуи ИЛ-4 (ИЛ-10).
Возникающее доминирование одного типа хелперов над другим в дальнейшем закрепляется, что определяет преобладающую форму иммунного ответа (гуморальный или клеточный).
Развитие гуморального иммунного ответа приводит к образованию антител, способных специфически связывать АГ, для чего необходимо контактное взаимодействие Т-хелперов и В-клеток.
Стимулированные АГ и Т-хелперами В-лимфоциты начинают интенсивно делиться, дифференцироваться в плазматические клетки, в них происходит соответствующее переключение С-генов иммуноглобулинов, в результате чего на поверхности клеток появляются иммуно
^тк' АГ-.МФ-
2 Эффекторные клетки
То - Th, ПК
IgA igE igM igD
Фагоциты^ 4
Н Э М АГ+АТ-’Удаление
Рис. 27-1. Формирование иммунного ответа в норме и патогенетические расстройства иммунного ответа
3
Роль реактивности организма в патологии
429
глобулины класса IgG, IgA, IgE. Связывание антител с АГ — удаление иммунных комплексов.
Следовательно, АГ запускает последовательность реакций, которые завершаются образованием комплексов АГ + АТ, удаляющихся затем с помощью фагоцитирующих клеток.
Основываясь на этой схеме, патогенетические расстройства иммунного ответа можно классифицировать следующим образом.
• Расстройства иммунного ответа в связи с нарушением функции иммунокомпетентных клеток (А-Т-В-клеток) в результате прямого поражения иммунокомпетентных клеток.
Поражение макрофагов — при хронических инфекционных заболеваниях: лепра, туберкулез, кандидоз: острых вирусных инфекциях: корь, краснуха, грипп, гепатит.
Поражение Т-В-лимфоцитов — при длительном воздействии патогенных факторов: облучение, прием кортикостероидов, введение антилимфоцитарной сыворотки, цитостатиков).
• Расстройства иммунного ответа в связи с нарушением кооперации между иммунокомпетентными клетками и фагоцитами (дисфункция медиаторов иммунного ответа: цитокинов [ИЛ], лимфокинов, системы комплемента и др.)
• Расстройства иммунного ответа в связи с нарушением образования иммуноглобулинов различных классов (появление белков Бенс-Джонса при миеломной болезни; IgE-гиперсекреция и др.).
• Расстройства иммунного ответа в связи с нарушением образования иммунных комплексов (АГ + АТ). Известно, что удаление из организма возможно только растворимых иммунных комплексов (при определенном соотношении АГ и АТ). Нарушение соотношения в комплексообразовании АГ и АТ в сторону избытка АГ делает иммунные комплексы нерастворимыми.
Ряд заболеваний (ревматизм, нефрит, ревматоидный артрит и др.) сопровождается накоплением иммунных комплексов (АГ + АТ) и осаждением их в тканях. В результате активации системы комплемента происходит выделение медиаторов воспаления с развитием типичных симптомов заболевания.
Болезни, обусловленные иммунологической недостаточностью, могут иметь врожденный либо приобретенный характер.
I. Во всех случаях расстройства иммунного ответа приводят к повышению восприимчивости к инфекции, развитию так называемых иммунодефицитных состояний.
430 Лекция 27
И. Вторую группу иммунопатологических заболеваний составляют болезни, обусловленные срывом иммунологической резистентности (толерантности), или аутоимммунные заболевания.
III. Третью группу иммунопатологических заболеваний составляют болезни, связанные с нарушением пролиферации иммунокомпетентных клеток (иммунопролиферативные заболевания).
Лекция 28
АЛЛЕРГИЯ. ПОНЯТИЕ И ОБЩАЯ ХАРАКТЕРИСТИКА. АКТУАЛЬНЫЕ
ПРОБЛЕМЫ АЛЛЕРГИИ
Г.В. Порядин
Контакт человека (животного) с разнообразными инфекционными и токсическими агентами ведет к образованию антител, которые защищают его организм посредством лизиса, нейтрализации или элиминации (с помощью фагоцитов) чужеродных веществ, сохраняя при этом постоянство внутренней среды.
Однако результатом иммунных реакций может быть не только защита организма, но и явное повреждение.
В этом случае развивается тот или иной вид иммунопатологии — патологический процесс или заболевание, основу которого составляет повреждение иммунного ответа (иммунологической реактивности).
Основываясь на особенностях механизма иммунного повреждения, в 1969 году Джелл и Кумбс выделили четыре основных типа иммунного повреждения (иммунопатологических реакций) (рис. 28-1).
I тип (IgE-зависимый, анафилактический) связан с образованием особого типа антител, имеющих высокое сродство (аффинность) к определенным клеткам (тучным, базофилам), так называемые цитотропные антитела. АГ, вступая во взаимодействие с фиксированными на клетках антителами, приводит к секреции предсуществующих и вновь образующихся БАВ (медиаторов), которые вызывают повышение проницаемости сосудов, отек ткани, гиперсекрецию слизи из слизистых желез, сокращение гладкой мускулатуры, т.е. повреждение.
Типичным примером этого типа повреждения являются аллергиге-ские реакции (атопическая бронхиальная астма, поллиноз, аллергические риниты, конъюнктивиты, анафилактический шок, аллергическая крапивница, отек Квинке и др.).
II тип (цитотоксический или цитолитический). АГ служат компоненты естественных клеточных мембран или вещества, сорбированные на клеточной поверхности, к которым образуются антитела. Образую-
432
Лекция 28
I тип (IgE-зависимый, анафилактический) Антитела-реагины фиксированы на клетке. Свободный антиген (аллерген) циркулирует в крови
II тип (цитотоксический или цитолитический). Антитела, циркулируют в крови.
Антиген (первичный или вторичный) фиксирован на поверхности клетки
III тип (иммунокомплексный).
Антитела преципитирующие.
Антиген в избытке. Образование ИК.
Реакция протекает без предварительного связывания АГ или АТ на клетке
IV тип (клеточно-опосредованный). Сенсибилизированные лимфоциты. Реакция не связана с действием АТ
Рис. 28-1. Типы иммунного повреждения
щийся на поверхности клеток комплекс АГ-АТ активирует систему комплемента, в результате чего возникают повреждение и лизис клеток.
Примерами такого типа повреждения являются аутоиммунные заболевания (нефротоксический нефрит — аутоантигеном является базальная мембрана почечных клубочков, внеклеточные структуры; постинфарктный миокардит, миастения гравис); лекарственная тромбоцитопеническая пурпура, лекарственный агранулоцитоз (антигенами является лекарственный препарат или продукт его метаболизма, включенный в состав клеточной поверхности); гемотрансфузионные реакции, возникающие вследствие несовместимости групп крови (антигенами являются естественные клеточные структуры).
III тип связан с образованием токсических иммунных комплексов (АГ-АТ).
Примером являются аутоиммунные заболевания (ревматоидный артрит, ГН, иридоциклит, сывороточный гепатит, СКВ, васкулит, рассеянный склероз, тиреоидит Хашимото, РТПХ); сывороточная болезнь, аллергическая крапивница, феномен Артюса.
Аллергия. Понятие и общая характеристика. Актуальные проблемы аллергии 433
IV тип — клеточно-опосредованный. К этому типу принадлежат иммунные реакции, формирующиеся при некоторых инфекционных заболеваниях (туберкулез, проказа, лепра, бруцеллез, сифилис), реакция отторжения трансплантата.
Среди болезней, основу которых составляет повреждение иммунного ответа (иммунологической реактивности), важное место занимают аллергические заболевания.
В большинстве стран мира отмечается неуклонный рост числа аллергических заболеваний, значительно превышающий в ряде случаев заболеваемость злокачественными опухолями, сердечно-сосудистыми заболеваниями.
Аллергия в наши дни становится по существу национальным бедствием для многих стран мира, включая и Россию.
Высокий уровень заболеваемости аллергией — обратная сторона прогресса, своего рода расплата за цивилизацию.
Одним из тяжелейших аллергических заболеваний является бронхиальная астма, занимающая первое место среди болезней органов дыхания.
Ежегодно растет число вызовов по скорой медицинской помощи по поводу аллергических реакций, возникающих на введение вакцин, сывороток, лекарственных препаратов.
Загрязнение биосферы токсичными, раздражающими и сенсибилизирующими веществами, эмоциональные стрессы, выраженная химизация условий труда и быта, злоупотребление фармакологическими средствами способствуют постоянному напряжению гомеостатических механизмов с вовлечением резервных механизмов жизнедеятельности, создает погву для срыва механизмов адаптации, развития различных заболеваний, в том числе и аллергических.
А учитывая, что все неблагоприятные факторы действуют на человека, год из года наращивая свой эффект и то обстоятельство, что предрасположенность к аллергии имеет наследственную основу, в ближайшее время не приходится рассчитывать на снижение числа больных аллергией, более того, в ближайшие десятилетия следует ожидать сохранение неблагоприятной тенденции к росту, утяжелению и многообразию проявлений аллергических заболеваний.
Что же такое аллергия?
Аллергия — это форма иммунного ответа организма на экзогенные вещества антигенной или гаптенной природы, сопровождающаяся повреждением структуры и функции собственных клеток, тканей и органов.
434
Лекция 28
Аллергическая реактивность (аллергическая конституция) в значительной мере определяется наследственными особенностями организма.
Свыше 80 % детей с аллергией в возрасте до 10 лет имеют отягощенный семейный анамнез.
В родословных больных бронхиальной астмой наследственное предрасположение к аллергии составляет более 50 %.
Однако аллергические заболевания не относятся к группе наследственных болезней, передаваемых непосредственно от родителей к потомству. Передается только предрасположенность к аллергии.
Исследования аллергических проявлений у однояйцовых близнецов показали, что реализация аллергической предрасположенности к заболеванию строго зависит от факторов внешней среды и их природы. Особенно чувствителен к развитию аллергического процесса организм в детском возрасте.
К факторам внешней среды (или социальным факторам), способствующим аллергизации населения, относятся:
• широкая обязательная вакцинация населения против многих инфекционных заболеваний (оспа, дифтерия, коклюш и т.д.). Известно, что коклюшная вакцина повышает чувствительность тканей к гистамину, вызывает блокаду 0-адренергических рецепторов в бронхиальной ткани, играет роль адъюванта для синтеза аллергических антител;
• широкое применение сывороток в лечебных целях, которые сами могут являться аллергенами;
• значительный рост простых и сложных химигеских веществ, потенциальных аллергенов, окружающих человека (лекарства, препараты бытовой химии, пестициды и гербициды в сельском хозяйстве, воздух и вода, загрязненные промышленными отходами).
Считается, что в среднем аллергические заболевания охватывают около 10 % населения земного шара.
Механизмы перехода защитной иммунной реакции в аллергическую реакцию повреждения
Какие же конкретные механизмы реактивности способствуют переводу иммунной реакции защиты в реакцию повреждения? Это следующие:
• повышенная проницаемость кожных или слизистых барьеров, ведущая к поступлению в организм АГ, которые в обычных условиях либо не поступают, либо их поступление ограничено;
Аллергия. Понятие и общая характеристика. Актуальные проблемы аллергии435
• особенности иммунного ответа, которые характеризуются как дисфункцией иммунокомпетентных клеток, так и нарушением количества образующихся антител и соотношения (дисбалансом) разных классов иммуноглобулинов;
• нарушение образования и соотношения различных медиаторов иммунного ответа, способствующее развитию воспаления;
• нарушение фагоцитоза.
Причины (экологические факторы) аллергических реакций и заболеваний
Вещества, вызывающие развитие аллергической реакции (заболевания), получили название аллергены.
Аллергены могут быть АГ, с многочисленными антигенными детерминантами, и биологически активными веществами, представляющими смесь АГ (пыльца трав, частицы эпидермиса).
Аллергены обладают чужеродностью и, часто, макромолекулярностью, хотя гаптены тоже могут обладать аллергенными свойствами, становясь АГ только после соединения с белками тканей организма (метаболиты лекарств, простые химические вещества — йод, бром, хром, никель).
При этом образуются так называемые комплексные АГ, специфичность которых определяется специфичностью гаптена.
По химигеской структуре аллергены являются белками, белково-полисахаридными комплексами (сывороточные, тканевые, бактериальные аллергены) или могут быть полисахаридами или соединениями полисахаридов с липоидами (аллерген домашней пыли, бактериальные аллергены).
Основные экологические факторы, приводящие к развитию аллергии (рис. 28-2):
1. Инфекционные агенты (грибковые, вирусные, паразитарные).
Роль инфекционных агентов в аллергизации населения, развитии аллергических реакций и заболеваний общеизвестна. Наиболее активны из инфекционных аллергенов — грибковые аллергены. За ними идут бактериальные, вирусные, паразитарные. Следует подчеркнуть, что структурные элементы бактерий очень часто могут действовать как адъюванты (при вакцинации), вызывая сенсибилизацию организма.
Однако инфекция, вызывая воспаление, приводит к повышению проницаемости слизистых оболочек, кожи, что, в свою очередь, способствует проникновению в организм других аллергенов и развитию полисенсибилизации.
436
Лекция 28
Инфекционные агенты
(грибковые, бактериальные, вирусные, паразитарные)
Пыльца растений
Пыль
домашняя (шерсть, перхоть животных)
производственная
Бронхиальная астма
Поллинозы (аллергический, ринит, конъюнктивит) Крапивница
Аллергические дерматиты
Укусы кровососущих насекомых (комары, Отек Квинке
мошки, москиты, гнус)
Ужаление перепончатокрылых (пчелы, шмели, осы)
Химические вещества
—Анафилактический шок
Сывороточная болезнь
Лекарства
Пищевые продукты
Ф Поствакцинальные аллергические осложнения (лихорадка, гиперемия, отек, сыпь, феномен Артюса)
^“Ф Аллергические заболевания ЖКТ
Рис. 28-2. Экологические факторы и аллергические заболевания
Сенсибилизация — процесс приобретения организмом повышенной чувствительности к тому или иному аллергену, процесс выработки специфических антител.
Различают сенсибилизацию пассивную и активную.
2. Пыльца растений. Вызываемые пыльцой растений аллергические заболевания — поллинозы. Поллинозы занимают значительное место в общей аллергической заболеваемости. В разных регионах России поллинозами страдают 1-5 % населения. На сенсибилизацию населения к пыльце в значительной мере влияют региональные особенности', распространенность тех или иных растений, степень агрессивности (аллергенности) пыльцы этих растений. Так, наибольшей аллергоопасностью в средней полосе России обладают тимофеевка, мятлик, ежа сборная, овсяница луговая. В Краснодарском и Ставропольском краях основным растительным аллергеном является сорная трава — амброзия.
3. Другой наиболее частой причиной неинфекционной аллергии является сенсибилизация к домашней пыли. 4-15 % населения страдает аллергией к домашней пыли. Состав домашней пыли весьма сложен: это и остатки органических веществ (шерсть, шелк, перхоть, перья, пыльца растений), и отходы пластмасс, синтетических тканей, различные виды грибов, бактерий и др. Однако главным аллергизирующим фактором домашней пыли являются микроскопические клещи семейства Pyroglyphidae, которые и определяют ее аллергенную активность.
Аллергия. Понятие и общая характеристика. Актуальные проблемы аллергии 437
Важными факторами, влияющими на распространение клещей, являются температура воздуха и влажность. Поэтому более высокая сенсибилизация к клещам — в регионах с влажным и теплым (среднегодовым) климатом.
4. Сложной экологической обстановкой характеризуются такие регионы России, как Сибирь и Дальний Восток. Суровая, длительная зима, вечная мерзлота, перепады температур (суточные и сезонные) — все это создает условия, способствующие размножению в колоссальных количествах кровососущих насекомых (гнус, москиты, мошки, комары). Аллергия к кровососущим насекомым вызывает тяжелые аллергические реакции в виде генерализованной экссудативной крапивницы, отека Квинке, лихорадки.
5. Неуклонный рост химического производства, внедрение химии в быт увеличивают вероятность контакта с химическими веществами, обладающими сенсибилизирующими свойствами, и рост профессиональной аллергии, вызванной воздействием химических соединений. Наиболее частые химические аллергены: скипидар, эпоксидные смолы, красители, лаки и другие вещества.
6. Воздействию металлов-аллергенов подвергаются значительные контингенты рабочих горнорудной, металлургической промышленности, жители крупных промышленных регионов.
• Воздействие таких металлов, как хром, никель, кобальт, марганец (электросварка, литейное, горнорудное производство), приводит к развитию аллергигеских дерматозов.
• Одним из эффектов биологического действия бериллия, платины, палладия является сенсибилизация организма и возникновение аллергических заболеваний органов дыхания.
7. Особое значение в последние годы приобретает проблема лекарственной аллергии. Связано это с увеличением производства и внедрением в медицинскую практику высокоактивных, длительно действующих (адъювантных) лекарственных препаратов.
Большинство лекарственных препаратов, сывороток и вакцин обладают способностью сенсибилизировать организм, вызывая при повторном введении аллергические реакции различной степени тяжести.
Механизм развития аллергических реакций
В развитии любой аллергической реакции можно выделить три стадии.
I. Стадия иммунных реакций (иммунологигеская).
Начинается с первого контакта организма с аллергеном и заключается в образовании в организме аллергических антител и их накоплении.
438
Лекция 28
В результате организм становится сенсибилизированным или повышенно чувствительным к специфическому аллергену.
При повторном попадании в организм специфического аллергена происходит образование комплексов АГ + АТ, которые и обусловливают следующую II стадию аллергической реакции.
И. Стадия биохимических реакций (патохимигеская).
Суть ее состоит в выделении готовых и образовании новых биологически активных веществ (медиаторов аллергии) в результате сложных биохимических процессов, запускаемых комплексами АГ + АТ.
III. Стадия клинических проявлений (патофизиологигеская).
Представляет ответную реакцию клеток, органов и тканей организма на образовавшиеся в предыдущей стадии медиаторы.
Кооперация иммунокомпетентных клеток при реализации иммунного ответа в стадию иммунных реакций
После внедрения в организм АГ активирует макрофаги (побуждая их к выделению ИЛ-1), обрабатывается ими и распознается ТЫ (рис. 28-3).
Связывание аллергена поверхностным иммуноглобулином на В-клетке
Интернализация образованного комплекса в В-клетку
Активация Th2
Синтез IgE
Секреция IgE
Первичные клетки-мишени
Процессинг (фрагментация) аллергена
Представление комплекса аллергенный пептид + МНС на В-клетке
Распознавание ТИ2-рецептором комплекса аллергенный пептид + МНС
Секреция ИЛ-4, ИЛ-3, ИЛ-5, ИЛ-6
IgE-связывающие
(IgE-УФ, 1дЕ-ТФ)
(тучная клетка, базофил)
Рис. 28-3. Схема индукции IgE-ответа в иммунную стадию аллергической реакции
Аллергия. Понятие и общая характеристика. Актуальные проблемы аллергии 439
Т-лимфоцит-помощник 1 посредством двух сигналов (специфического от АГ и неспецифического от ИЛ-2 и ИФН-у) включает в антителогенез В-лимфоцит.
После получения обоих сигналов активированный В-лимфоцит образует клон антителообразующих клеток, которые и экспрессируют в кровь антитела (иммуноглобулины разных изотипов: IgM, IgG, IgA).
При повторном поступлении АГ продукция антител осуществляется в более короткие сроки и характеризуется их большим количеством, так как в организме надолго сохраняется «иммунологигеская память», т.е. активированные АГ долгоживущие Т-лимфоциты.
Механизм регуляции продукции 1дЕ
Продукция pearHHOB-IgE-антител (основных в аллергических реакциях) подвержена тем же закономерностям, что и синтез иммуноглобулинов других классов.
Однако синтез реагинов-^Е-антител индуцируется взаимодействием аллерген-представляющих клеток с Т-хелперами 2(Th2), приводящими к преимущественной стимуляции образования ИЛ-4 и одновременно к подавлению секреции ИФН-у, что переключает активированные IgM-IgG-несущие В-клетки на синтез IgE.
Аллерген связывается с поверхностными иммуноглобулиновыми молекулами аллерген-специфических В-клеток, которые в 10 000 раз более эффективны в представлении очень малых количеств растворимых АГ Т-клеткам, чем макрофаги.
Аллерген-иммуноглобулиновый комплекс интернализируется в клетку, где (как и в макрофагах) происходит фрагментация (процессинг) исходной молекулы аллергена на пептидные фрагменты.
Пептидные компоненты аллергена представляются затем на поверхности В-клеток в ассоциации с молекулами II класса главного комплекса гистосовместимости (МНС II класса).
В такой форме комплекс «аллергенный пептид + МНС» связывается Т-клеточным рецептором (TCR). Таким образом устанавливается контакт В-клетки с Т-клеткой.
Распознавание комплекса «аллергенный пептид + МНС» Т-клеточ-ным рецептором активирует Т-клетку (Th2).
Аллерген-специфические Т-клетки (Th2) стимулируют продукцию ИЛ-4, а также других цитокинов (ИЛ-3, ИЛ-5, ИЛ-6).
Сигнал, осуществляемый ИЛ-4, ответствен за выбор изотипа IgE, на который происходит переключение синтеза иммуноглобулинов.
440
Лекция 28
Однако само по себе действие ИЛ-4 на В-клетки еще недостаточно для запуска секреции IgE.
К сигналам, необходимым для специфического IgE-синтеза, относятся также IgE-связывающие факторы с разной физиологической направленностью (IgE-усиливающий фактор — IgE-УФ — усиливает образование IgE, IgE-тормозящий фактор — IgE-ТФ угнетает продукцию IgE), которые поддерживают образование IgE.
Точкой приложения действия этих факторов является несущая поверхностный IgE В-клетка памяти.
IgE-УФ способствует дифференцировке этих клеток в IgE-секретирующие плазматические клетки, IgE-ТФ тормозит продукцию IgE плазматическими клетками.
Набор цитокинов, секретируемых Т-хелперными клетками-2 (ИЛ-3, ИЛ-5, ИЛ-6) наряду с синтезом IgE, способствуют пролиферации, созреванию и активации тучных клеток, базофильных и эозинофильных гранулоцитов с вовлечением их в IgE-опосредованный иммунный ответ.
Тучные клетки и базофилы после антигениндуцированного перекрестного взаимодействия рецепторов для IgE на их клеточной мембране секретируют не только медиаторы аллергии (гистамин, лейкотриены, простагландины), но и цитокины, соответствующие цитокинам Т-хелперных клеток. Тем самым происходит пролонгирование и усиление IgE-опосредованного иммунного ответа, замыкается порочный круг. Возникший синтез IgE поддерживается на определенном уровне и благодаря этому осуществляется механизм IgE-зависимого высвобождения медиаторов аллергического воспаления.
Принципы терапии аллергических реакций
Исходя из современных представлений об этиологии и механизмах развития аллергии, диктуются принципиальные подходы к этиопатоге-нетической терапии аллергических заболеваний (табл. 28-1):
• Специфигеские методы терапии в основном направлены на ликвидацию причины заболевания:
предотвращение реакции АГ + АТ (устранение контакта с аллергеном);
блокада продукции IgE-антител (восстановление супрессивных влияний Thl — иммунокоррекция и переключение синтеза IgE на IgG, IgM — гипосенсибилизация).
• Неспецифигеские методы терапии. Исходя из современных представлений о механизмах развития аллергии можно выделить еле-
Аллергия. Понятие и общая характеристика. Актуальные проблемы аллергии 441
дующие принципиальные подходы к патогенетической терапии аллергических заболеваний:
предотвращение (супрессия) синтеза аллергических антител путем изменения функции иммунорегуляторных лимфоцитов — Tht и Th2 (иммуномодуляторы):
предотвращение (супрессия) синтеза аллергических антител, вмешиваясь в природу цитокинов (лечение рекомбинантными цитокинами: ИЛ-2, ИФН-у);
поскольку местные нарушения связаны с клетками «быстрого реагирования» (тучные клетки, базофилы, эозинофилы) и их медиаторами (в результате взаимодействия фиксированных на поверхности клеток антител с АГ), весьма перспективным является воздействие фармакологическими средствами на:
- количество клеток-мишеней — ТК, базофилы (глюкокортикоиды);
- рецепторы клеток-мишеней. Сегодня придается большое значение дисбалансу функционального состояния симпатического и парасимпатического отделов вегетативной нервной системы в развитии большинства аллергических реакций. В связи с этим весьма перспективен поиск средств, воздействующих на эти рецепторы. В частности, на p-адренорецепторы (селективные p-адреностимуляторы: сальбутамол и его производные),
- синтез медиаторов (противовоспалительные средства):
- высвобождение медиаторов (теофиллин, катехоламины, простагландины, глюкокортикоиды);
- эффект медиаторов на эффекторные структуры — пораженную ткань, орган (антигистаминные препараты: хлоропи-рамин, дифенгидрамин, прометазин, клемастин, кетотифен и др.).
Таблица 28-1. Принципы лечения аллергических болезней
Специфические Неспецифические
1. Элиминация аллергена 1. Воздействие на фазы аллергической реакции: иммунологическую (введение цитокинов); патохимическую (препараты против Б АВ); патофизиологическую (адренохолиномиметики и др-)
2. Специфическая иммунотерапия (гипосенсибилизация)
3. Иммунокоррекция
2. Воздействие на вторичные механизмы аллергической реакции (нервные, эндокринные и др.)
442
Лекция 28
Псевдоаллергические реакции
В аллергологической практике врачу-аллергологу все чаще приходится иметь дело с большой группой реакций, клинически часто неотличимых от аллергических. Эти реакции имеют аналогичные с аллергическими патохимигескую и патофизиологигескую стадии и получили название псевдоаллергигеские (неиммунологические).
Выявить участие иммунных механизмов не удается.
В развитии псевдоаллергических реакций особую роль играют такие медиаторы, как гистамин, лейкотриены, продукты активации комплемента, ККС.
Выделяют три группы псевдоаллергических реакций:
• реакции, связанные с избытогным освобождением медиаторов (гистамина) из тучных клеток или с нарушением их инактивации. Причины: высокая температура, УФО, ионизирующая радиация, антибиотики, полисахариды;
• реакции, в связи с дефицитом первого компонента комплемента (C1-1NA) и неиммунологической активацией комплемента по альтернативному (пропердиновому) пути. Причины: яд кобры, бактериальные липополисахариды, ферменты: трипсин, плазмин, калликреин, активирующиеся при повреждении;
• реакции, связанные с нарушением метаболизма полиненасыщен-ных жирных кислот (в первую очередь арахидоновой). Причины: анальгетики — ацетилсалициловая кислота, производные пиразо-лона, нестероидные противовоспалительные препараты.
Основные проявления псевдоаллергигеских реакций — крапивница, отек Квинке, бронхоспазм, анафилактоидный шок.
Лекция 29
АУТОИММУННЫЕ БОЛЕЗНИ
НЛ. Богуш
Аутоиммунные болезни (АИБ) — хронические рецидивирующие заболевания, при которых повреждение органов и тканей происходит в результате иммунных реакций, направленных на собственные ткани организма.
АИБ поражают 5-7 % населения земного шара. На сегодняшний день насчитывают около 40 нозологических единиц. Женщины заболевают чаще, чем мужчины, при этом характерно возникновение заболевания в молодом возрасте.
Аутоантигены по химической природе — белки, нуклеиновые кислоты, фосфолипиды, сахара, стероиды.
Характерно появление аутоантител, которые подразделяют на:
• органоспецифигеские'. к рецепторам (тиреотропного гормона, ацетилхолина, адренокортикотропного гормона), к специфическим для органов белкам (тиреоглобулину), пептидным гормонам (инсулину):
• клетогно-специфигеские: к белковым компонентам мембран различных клеток (эритроцитов, тромбоцитов, лейкоцитов);
• органонеспецифигеские'. к нуклеиновым кислотам и нуклеопротеидам, цитоплазматическим белкам, белкам цитоскелета, стресс-белкам и др.
В основу данной классификации АИБ положена иммунологическая специфичность аутоантител.
К аутоиммунным болезням относят:
• органоспецифигеские АИБ (тиреоидит Хашимото, первичная микседема, диффузный токсический зоб, или базедова болезнь, первичная надпочечниковая недостаточность, или болезнь Аддисона, сахарный диабет I типа, атрофический гастрит при пернициозной анемии, миастения гравис и др.);
444
Лекция 29
• органонеспецифигеские АИБ (системная красная волчанка, ревматоидный артрит, склеродермия);
• аутоиммунные заболевания крови (аутоиммунная гемолитическая анемия, аутоиммунная тромбоцитопения, аутоиммунный агранулоцитоз).
Этиология АИБ неизвестна. Даже судя по названиям болезней видно, что найти общую причину столь разных заболеваний очень трудно. Патогенез более понятен — агрессия собственной иммунной системы против своих тканей. Что является антигеном (АГ)? Если нормальные АГ клеточных мембран или межклеточного вещества распознаются как «чужие» АГ и подвергаются атаке иммунной системы, то в этом случае развиваются истинные или первичные АИБ, в основе которых лежит срыв иммунологической толерантности. Если нормальные ткани подверглись сначала какому-либо изменению и потом из них образовались измененные АГ (неоантигены), ставшие инициаторами иммунной атаки, в таком случае это будут вторичные АИБ. К сожалению, неизвестность этиологии не позволяет достоверно определить, первичная это или вторичная АИБ.
Иммунологигеская толерантность — отсутствие иммунного ответа на АГ собственных тканей в норме (в эксперименте можно вызвать иммунологическую толерантность в ответ на чужеродный АГ). Т-лимфоциты проходят обучение этой толерантности в тимусе, В-лимфоциты — в костном мозге (центральная толерантность). Известно, что если незрелый лимфоцит-тимоцит обладает средней аффинностью (сродством) к молекулам главного комплекса гистосовместимости (ГКГ), то он продолжит дифференцировку; если не обладает — погибнет в результате апоптоза (позитивная селекция); если обладает очень высокой аффинностью — также погибнет (отрицательная селекция).
Прошедшие селекцию лимфоциты существуют в организме всю жизнь, так как они поддерживаются стимулирующими сигналами от тех же комплексов «молекулы ГКГ + свой пептид» уже на периферии. Без такого сигнала лимфоцит погибает. Сигнал на выживание стимулирует пролиферацию данного клона лимфоцитов и увеличение их количества до нормального уровня, при этом активные цитокины не синтезируются и иммунное воспаление не развивается.
Возникающие иногда аутоагрессивные клоны лимфоцитов не обязательно будут повреждать свои клетки, так как существует еще и периферическая толерантность. Для окончательной и полной активации лимфоцитов необходимы, во-первых, ко-стимулирующие молекулы
Аутоиммунные болезни
445
(CD40-CD40L, B7-CD28 и для Т-, и для В-лимфоцитов), возникающие на АПК только при повреждении и воспалении тканей. Во-вторых, во время контакта Т-лимфоцита со своей АПК активация этого лимфоцита подавляется Т-регулирующими клетками. Наконец, после повторной нежелательной стимуляции инициируется апоптоз такого лимфоцита. При АИБ происходит «обход» или «срыв» иммунологигеской толерантности.
Предполагаются следующие механизмы, ответственные за эту патологию.
• Появление в организме аутоагрессивных клонов лимфоцитов в результате генетической предрасположенности к АИБ.
• Нарушение механизмов апоптоза, с помощью которого из организма удаляются неправильно активированные лимфоциты, или нарушение периферической толерантности в результате дисбаланса провоспалительных (ИЛ-1, ФНО-а, ГМ-КСФ) и антивоспалитель-ных (ИЛ-4, ИЛ-10, растворимый антагонист ИЛ-1, растворимые рецепторы для ФНОа) цитокинов.
• синтез генетически измененных молекул клеток и соединительной ткани (в частности, суставных сумок, при ревматоидном артрите).
• Существование в организме инфекций, способных инициировать аутоиммунные процессы.
АГ микроорганизмов могут иметь антигенные детерминантные группы (эпитопы), сходные (например, по аминокислотной последовательности) с такими же группами АГ хозяина (антигенная мимикрия). (АГ мембраны стрептококка перекрестно реагируют с волокнами сердечной мышцы, АГ Е. coli — с эпителием толстой кишки, АГ р-гемолитического стрептококка — с базальной мембраной почечного клубочка и т.д.). Иммунный ответ на инфекционные АГ может распространиться и на перекрестные АГ тканей.
Микробные суперантигены вызывают неспецифическую поликлональную активацию В-клеток, в результате будут активированы не только необходимые клоны плазмоцитов для синтеза защитных антител, но и аутоагрессивные клоны, начинающие вырабатывать антитела против своих тканей.
Повреждение и воспаление стимулируют посредством цитокинов синтез на АПК ко-стимулирующих молекул (В7). Появившиеся цитокины способствуют привлечению в очаг лимфоцитов, способствуют индукции молекул ГКГ II класса на клетках поврежденных тканей. Такие клетки начинают выполнять не свой-
446
Лекция 29
ственную им роль АПК и презентируют свои пептиды как АГ, на которые развивается иммунный ответ.
Многие патогенные факторы способны вызвать изменение АГ (создание неоантигенов) мембран и межклеточного вещества:
• ультрафиолетовое, ультразвуковое облучение, высокая температура повреждают белки организма, меняя их антигенную характеристику (в частности разрушая водородные связи, меняют третичную структуру):
• химические, лекарственные вещества могут, как гаптены, присоединяться к нормальным АГ и образовывать комплексные АГ, чужеродные для организма: при этом белковая часть отвечает за иммуногенность, а гаптен — за специфичность АГ;
• появление «скрытых» АГ (о чем свидетельствует обнаружение антисердечных антител после операций на сердце) или «забарьерных» АГ, т.е. в норме ограниченных от тесного контакта с лимфоцитами (мозг, передняя камера глаза, тестикулы, коллоид щитовидной железы), после травм этих тканей:
• вирусы (особенно вирус герпеса, цитомегаловирус, вирус Эпштейна-Барр) способны в результате интеграции двух геномов — вирусного и хозяина, приводить к появлению новых промежуточных АГ;
Все вышеизложенные условия помогают обойти естественную толерантность и развить иммунное повреждение тканей. Раз начавшееся иммунное воспаление приводит к повреждению клеток, высвобождая для контакта с лимфоцитами новые внутриклеточные, ранее скрытые АГ, на которые лимфоциты развивают ответ. Лавинообразно начинает расти количество новых АГ и, следовательно, дальнейшее иммунное повреждение тканей. Поскольку антиненами становятся свои собственные органы и ткани, избавиться от которых невозможно, заболевание имеет хронический характер.
Независимо от этиологии, патогенез большинства АИБ включает в себя элементы II, III и IV типов иммунного ответа (по классификации Джелла и Кумбса).
II тип — комплементзависимый цитотоксический тип
Особенности: синтезируются антитела IgM-IgG классов, способные в комплексе (АГ-АТ) активировать комплемент; специфический АГ находится на мембране клеток или внеклеточном матриксе (базальной мембране, коллагене) первично либо вторично (например, в результате адсорбции гаптена).
Аутоиммунные болезни
447
• I стадия патогенеза — стадия иммунныхреакций. После кооперации в лимфатическом узле АПК с АГ + Т-хелпер2 (Тх2) + В-лимфоцит (Вл) происходит размножение Тх2, синтез ими ИЛ (ИЛ-4,5,6,10), способствующих пролиферации, дифференцировке Вл, переключению в Вл синтеза класса иммуноглобулинов с исходного IgM на IgG, превращение Вл в плазмоциты, синтез и секреция ими АТ. Все это занимает не менее 4-7 дней от момента проникновения АГ сквозь барьерные ткани.
• II стадия — стадия патохимигеских реакций. АТ соединяется с АГ Fae-концами, и на поверхности клетки или базальной мембраны образуется комплекс АГ-АТ. После этого IgM-IgG-антитело становится способным активировать комплемент по классическому пути. Появляются медиаторы: С3а, С5а — анафилатоксины; С3а, С5а — хемоаттрактанты, С3в, С4в — опсонины. Эти медиаторы привлекают нейтрофилы, моноциты, лимфоциты, эозинофилы, естественные киллеры (ЕК). Нейтрофилы активируются и выделяют свои медиаторы: ФАТ, лейкотриены, бактерицидные вещества, лизосомальные ферменты, токсические продукты кислорода, цитокины. Активированные тромбоциты высвобождают серотонин и другие медиаторы, а тучные клетки и базофилы крови — гистамин. В плазме может образоваться брадикинин.
• III стадия — патофизиологигеская. В ткани в ответ на медиаторы и действия нейтрофилов развивается воспаление (например, воспаление базальной мембраны при подостром гломерулонефрите с полулуниями). Клетки, нагруженные антителами (в комплексе АГ-АТ), ухудшают свою функцию и подвергаются уничтожению. Происходит их: 1) фагоцитоз (С3в, С4в и ориентированный наружу Fc-фрагмент антитела = опсонины); 2) лизис (С5в6789 — мембраноатакующий комплекс комплемента); 3) гибель в результате лизиса (действие перфоринов) или апоптоза (действие гранзимов) при антителозависимой клеточной цитотоксичности (АЗКЦ). АЗКЦ реализуется естественными киллерами (ЕК), К-клетками — большими гранулярными лимфоцитами-киллерами, моноцитами/ма-крофагами, эозинофилами и нейтрофилами, так как все эти клетки имеют рецепторы для Fc-фрагмента IgG-AT, через который и прикрепляются к клетке-мишени. В отличие от ЕК и К-лимфоцитов, имеющих перфорины и гранзимы, другие клетки убивают клетку-мишень своими токсическими продуктами (ФНО-а, кислородными радикалами).
448
Лекция 29
Таков патогенез аутоиммунных гемолитической анемии, тромбоцитопении, агранулоцитоза, частично сахарного диабета 1-го типа, одной из форм ГН. Вышеперечисленные эффекторные механизмы разрушают эритроциты при гемолитической болезни новорожденных при резус-конфликте и переливании несовместимых групп крови (гетероиммун-ные реакции).
К этому классическому взгляду на патогенез II типа иммунопатологии следует добавить и другие варианты развития АИБ, обнаруженные в последнее время. Они касаются судьбы клеток, ставших через АГ мишенями для антител. Оказалось, что механизмы повреждения клеток-мишеней не ограничиваются иммуноглобулиновой активацией комплемента и последующей деструкцией клетки. Клетка может существовать, но значительно нарушать свою функцию. Например:
• Антитела синтезируются против рецепторов на поверхности клетки. Если эти рецепторы — рецепторы для факторов роста, то ростовой сигнал блокируется, и развивается атрофия (первичная атрофия коры надпочечников; атрофический гастрит при пернициозной анемии). Если мишень — рецепторы для связывания ацетилхолина на мышцах, нарушается проведение сигнала с нерва на мышцу (миастения гравис). Если антитела реагируют с рецептором для тиреотропного гормона на щитовидной железе, то АТ оказывают стимулирующее действие, развивается тиреотоксикоз (базедова болезнь, диффузный токсический зоб). Такой необычный антире-цепторный стимулирующий механизм было предложено выделить в самостоятельный V тип иммунопатологии Ройтом, дополнительно к I—IV вариантам Джелла и Кумбса.
• Антитела против рецепторов приводят к ускоренной интернализации (погружению внутрь клетки) комплекса рецептор-АТ и уменьшению рецепторов на поверхности клетки, дополнительно к аналогичной интернализации, связанной со старением и повреждением рецепторов (еще один механизм в патогенезе миастении гравис).
• Антитела против внутриклеточных и противоядерных АГ способны проникнуть внутрь клетки и вызвать в ней нарушение метаболизма, нарушение ее специфической функции и даже ускоренную гибель (первичная микседема).
Итак, ко II типу иммунопатологии относятся следующие АИБ.
I. Органоспецифигеские АИБ: частично тиреоидит Хашимото, первичная микседема, первичная надпочечниковая недостаточность,
Аутоиммунные болезни
449
атрофический гастрит при пернициозной анемии, миастения гравис.
II. Аутоиммунные заболевания крови: аутоиммунная гемолитическая анемия, аутоиммунная тромбоцитопения, аутоиммунный агранулоцитоз.
Патогенез аутоиммунного агранулоцитоза. Попадание в организм гаптена (возможно, лекарства) привело к адсорбции этого химического вещества на мембране нейтрофилов, вызвало через 5-7 дней появление аутоантител класса IgG, фиксацию АТ Fae-концами к АГ на поверхности нейтрофилов, активацию комплемента по классическому пути и разрушению нейтрофилов через лизис (АЗКЦ или С5в, 6,7,8,9) или фагоцитоз. В итоге развиваются агранулоцитоз — лейкопения (общее количество лейкоцитов становится менее 4х109/л) и выраженная нейтропения (общее количество сегментоядерных нейтрофилов менее 800-1000 клеток в 1 мкл).
Ill тип—иммунокомплексный тип
Особенности: синтезируются антитела IgM-IgG классов, способные в комплексе АГ-АТ активировать комплемент: АГ растворимый; образуются циркулирующие иммунные комплексы — ЦИК.
Для их существования необходим ряд условий.
1. В ЦИК больше молекул АГ, чем АТ.
2. Небольшой молекулярный вес ЦИК — около миллиона дальтон.
3. Недостаточная функция макрофагов, утилизирующих в норме ЦИК.
Уменьшение выведения ЦИК может быть связано также с уменьшением количества комплемента и нарушением адсорбции ЦИК эритроцитами. Последние доставляют ЦИК в селезенку, где ЦИК поглощаются селезеночными макрофагами.
Стадии патогенеза. АГ бывают экзогенные: чужеродный белок при сывороточной болезни, бактерии, например стрептококк при ГН, вирусы. Эндогенные АГ представлены клеточными и тканевыми АГ, циркулирующими в крови.
I и II стадии — аналогичные вышеизложенным для цитотоксического типа. Во время II стадии комплемент активируется антителом, находящимся в составе ЦИК.
III патофизиологическая стадия имеет свои особенности, обусловливающие разнообразие мест поражения. ЦИК могут образовываться в крови и фиксироваться на стенках сосудов, вызывая васкулиты (системное заболевание). Если АГ находится в органе, то ЦИК будут возникать в этих органах — иммунный комплекс in situ, вызывая там иммунное
450
Лекция 29
воспаление (повреждение эндотелия клубочков почек при ГН). Имеет значение отрицательный заряд ЦИК, валентность АГ, аффинность АТ к различным компонентам тканей. Наиболее часто депозиты ЦИК встречаются в суставах, микрососудах кожи, сердце, почках, серозных полостях. Независимо от локализации, механизм повреждения одинаков: ЦИК инициируют патохимическую стадию и посредством медиаторов, цитокинов и привлеченных лейкоцитов вызывают иммунное воспаление. Отмечается важная роль ФНО, привлекающего нейтрофилы. Они дегранулируют (экзоцитоз), выделяются эластаза, коллагеназа, гиалуронидаза, приводящие к повышению проницаемости сосудистой стенки, базальной мембраны клубочка почек и т.п. Подчеркивается роль тромбоцитов, которые не только способствуют тромбозу и ишемическим повреждениям, но и выделяют факторы роста, стимулирующие ангиогенез и синтез соединительной ткани.
Характерно развитие васкулитов, геморрагий, тромбозов и некрозов.
В случае однократного массивного поступления АГ в организм (белки чужеродной сыворотки, стрептококк) заболевание может завершиться после катаболизма ЦИК. Если АГ попадает в организм неоднократно или персистирует долго, что характерно для аутоантигенов, то развивается хроническое заболевание.
По III иммунокомплексному типу развиваются следующие заболевания:
• Органонеспецифигеские АИБ: системная красная волчанка, ревматоидный артрит, склеродермия, болезнь Шенлейна-Геноха. При лечении сыворотками — сывороточная болезнь.
• Органоспецифигеские АИБ: иммунокомплексный ГН, экзогенный аллергический альвеолит и др. ЦИК играют важную роль в патогенезе инфекций, сопровождаемых бактериемией и вирусемией.
При ревматоидном артрите в крови можно обнаружить циркулирующий ревматоидный фактор. Это — AT IgG, М классов против Fc-фрагмента IgG. При болезни Шенлейна-Геноха развивается системный васкулит. Экзогенный аллергический альвеолит вызывает белок кала и перьев птиц, зараженный актиномицетами.
Сывороточная болезнь. Открыта К. Пирке при лечении больных дифтерией лошадиной сывороткой. У больных на 9-10-й день лечения развивались лихорадка, кожная сыпь, артриты, нефриты, перикардиты, увеличение лимфоузлов и селезенки. Белок лошадиной сыворотки являлся АГ, к нему в течение 5-7 дней вырабатывались IgG антитела, которые образовывали со все еще циркулирующим в крови АГ ЦИК. По мере
Аутоиммунные болезни
451
нарастания титра АТ нарастало и количество ЦИК. Они фиксировались в стенке микрососудов кожи, активировали комплемент, привлекали лейкоциты и тромбоциты, активировали тучные клетки и фактор Хагемана, вызывая повышение проницаемости, васкулиты, кожную сыпь. В других органах ЦИК способствовали развитию иммунного воспаления. Развивался ответ острой фазы.
Феномен Артюса. Местная иммунокомплексная реакция. Подопытного кролика сенсибилизировали повторными внутрикожными введениями стерильной чужеродной сыворотки. Через 7-10 дней очередная инъекция приводила к появлению отека в месте введения, тяжелым кровоизлияниям и даже образованию язвы. Ответная реакция, в отличие от немедленного типа, развивалась в течение 4-12 ч. В данном случае ЦИК образовались в месте скопления АГ в коже. Васкулит, тромбоз и некроз формировали характерную клиническую картину.
IV клеточно-опосредованный тип иммунопатологии
Особенности: повреждение реализуется эффекторными клетками — лимфоцитами и макрофагами, а не АТ. Медиаторы лимфокины продуцируются активированными лимфоцитами и макрофагами. Кожные реакции проявляются замедленно — через 24-72 ч (максимально выражены). Возможна пассивная сенсибилизация, но не сывороткой или АТ, а живыми лимфоцитами.
К этому типу иммунопатологии относятся следующие группы болезней:
• контактный дерматит в ответ на гаптены — простые химические вещества;
• реакции Пирке, Манту и другие, как проявление гиперчувствительности при специфической инфекции (туберкулезе, бруцеллезе, сифилисе):
• отторжение трансплантата;
• аутоиммунные болезни: сахарный диабет 1-го типа, тиреоидит Ха-шимото, первичная надпочечниковая недостаточность, миастения гравис, ревматоидный артрит.
Принципиально в реализации этого типа иммунопатологии можно выделить 2 эффекторных механизма:
• первый — гиперчувствительность замедленного типа (ГЗТ);
• второй — прямая клеточно-опосредованная цитотоксичность.
Гиперчувствительность замедленного типа
Главный защитный механизм организма на внутриклеточные патогены. Классическим примером является кожная реакция на туберкулин
452
Лекция 29
(белково-липополисахариновый компонент, полученный из бацилл туберкулеза).
При первичном попадании возбудителя туберкулеза в организм развивается иммунная стадия — I стадия иммунного ответа. АПК презентирует АГ, в присутствии ИЛ-12 CD4’ ТхО превращаются в Тх1, которые пролиферируют и становятся АГ-специфическими (сенсибилизированными). Часть из них превращается в клетки памяти. ИЛ-12 синтезируется дендритными клетками, макрофагами и нейтрофилами в очаге инфицирования.
Раствор туберкулина, введенный внутрикожно, создает повторный контакт организма со специфическим АГ. Через 8-12 ч начинает проявляться реакция, достигающая максимума через 24-72 ч. В месте инъекции кожа краснеет и уплотняется, появляется папула. Морфологически вокруг расширенных сосудов скапливаются мононуклеары (макрофаги и лимфоциты), откладывается фибрин, что дает уплотнение этого участка ткани. Установлено, что эти лимфоциты — в основном CD4’ Thr Можно предположить следующую цепочку событий. Вторичное появление АГ привлекло в этот очаг АГ-специфические Тх1 памяти, которые после контакта с АГ начинают усиленно синтезировать и секретировать ИФ-у, ФНО и другие цитокины (развивается II стадия — патохимиге-ская). ФНО способствует расширению сосудов, повышению их проницаемости, активации эндотелиоцитов с появлением на их поверхности молекул адгезии, что способствует эмиграции моноцитов/макрофагов и лимфоцитов. Синтезируются также ИФ-у, ФНО, ИЛ-1; фактор, активирующий макрофаги = ФАМ; фактор, угнетающий миграцию макрофагов, = ФУМ и вызывающий задержку и скопление макрофагов в очаге с АГ и др. Под влиянием этих цитокинов макрофаги накапливаются, активируются, усиливают свою фагоцитарную способность и выработку токсических продуктов кислорода и бактерицидных веществ.
Главными эффекторными клетками при ГЗТ являются макрофаги. Они мигрируют к месту нахождения АГ, фагоцитируют его и расщепляют. Для усиления киллерных способностей макрофагов необходимы цитокины, которые продуцируются АГ-активированными Thr Синтезируя ИЛ-1, ФНО-а, ИЛ-6, ГМ-КСФ и ИЛ-3, макрофаги «присоединяют» силы всего организма — развивается ответ острой фазы. Кроме того, местно медиаторы макрофагов способствуют развитию воспаления. Все это вместе помогает избавиться от повреждающего агента или ограничить его распространение.
Следовательно, III патофизиологигеская стадия будет заключаться в развитии местного иммунного воспаления. В случае, если не удается
Аутоиммунные болезни
453
справиться с АГ, клетки, содержащие АГ, окружаются макрофагами, частично превращающимися в эпителиоидные клетки, и лимфоцитами. Возникают гранулемы — признак хронического воспаления, характерного для IV типа иммунопатологии.
Прямая клеточно-опосредованная цитотоксичность
С помощью этого механизма организм борется с вирусной инфекцией, убивая собственные вирусинфицированные клетки.
I стадия, иммунная. Прецитотоксический лимфоцит CD8’ распознает АГ с помощью своего специального рецептора и активируется. Это означает, что, во-первых, на его поверхности появляются рецепторы к фактору роста ИЛ-2, и, во-вторых, он сам начинает вырабатывать ИЛ-2. Дополнительно ИЛ-2 синтезируют Тх1, активированные АПК с АГ. Под влиянием фактора роста CD8*-лимфоциты пролиферируют и дифференцируются в антигенспецифические или сенсибилизированные; часть из них превращается в клетки памяти. Постепенно накапливаясь (в течение 5-7 дней), лимфоциты устремляются к клеткам, содержащим АГ. После контакта с АГ через Т-клеточный рецептор лимфоцит вбрасывает в этой области содержимое гранул в инфицированную клетку. Гранулы образуются только в сенсибилизированном лимфоците и только в области АГ-специфического Т-клеточного рецептора. В гранулах содержатся перфорины и гранзимы, которые попадают в клетку-мишень. Перфорины образуют в клетке пору, через которую инъецируются гранзимы. Последние стимулируют апоптоз. Клетка погибает, причем при таком способе гибели разрушается не только собственная ДНК, но и ДНК вируса. Если апоптоз почему-либо не может произойти, то через пору входят NaCl, вода, и клетка осмотически лизируется.
III стадия, патофизиологигеская. Главными клетками в этом механизме являются цитотоксические лимфоциты (ЦТЛ) киллеры CD8*. ЦТЛ, активированные АГ, продуцируют цитокины (II патохимическая стадия): ИФ-у, ФНО-а, ФНО-р (он же лимфотоксин), ФАМ, ФУМ и др. Они помогают в АГ презентации, в дифференцировке Th0 в Thp в привлечении других клеток воспаления, непосредственно воздействуют на вирусы.
В АИБ одновременно могут встречаться и прямая цитотоксичность, и ГЗТ как эффекторные механизмы.
Например, при аутоиммунном тиреоидите Хашимото обнаруживаются ЦТЛ, сенсибилизированные к тиреоглобулину и микросомальному АГ (тиреоидной пероксидазе). Считается, что это ЦТЛ ответственны за апоптоз фолликулярных клеток. К этим же АГ щитовидной железы и к АГ ядер клеток выявляются также AT IgG.
454
Лекция 29
При сахарном диабете 1 -го типа поджелудочная железа инфильтрирована мононуклеарами, где обнаруживаются Thr ЦТЛ и макрофаги. Обсуждается роль AT IgG в механизмах разрушения железы. То же можно сказать о патогенезе первичной надпочечниковой недостаточности, ревматоидном артрите, миастении гравис.
Особенностью контактного дерматита, относящегося к IV типу, является то, что простое химическое вещество (никель, платина, хром, ртуть, некоторые лекарства) при попадании на кожу, соединяясь с ее белками, превращается в полноценный АГ. Иммунный ответ развивается не только на гаптен, но и на носитель, которым являются белки кожи. В I стадию образуются Thj и ЦТЛ, клетки памяти (5-7 дней). При продолжающемся контакте к АГ устремляются Thr ЦТЛ, которые выделяют после контакта с АГ цитокины или лимфокины (II стадия), способствуют эмиграции моноцитов/макрофагов, лимфоцитов и других клеток воспаления. ВIII стадии в области контакта с АГ развиваются повреждение клеток с АГ и иммунное воспаление.
Реакция отторжения трансплантата происходит в основном по механизму IV типа иммунопатологии. Различают сверхострое, острое и отсроченное отторжение. Сверхострое реализуется антителами, которые активируют комплемент, повреждают стенки сосудов, вызывают тромбоз и отторжение трансплантата. Подчас это заметно уже на операционном столе. Такой тип отторжения наблюдается при повторных пересадках, когда на чужеродный АГ уже были выработаны АТ в достаточном количестве.
Острое отторжение — нормальная реакция, первичный иммунный ответ происходит в течение 10-14 дней после пересадки. Эффекторными механизмами (в зависимости от вида трансплантата) могут быть и АТ, и ЦТЛ, и Thj + макрофаги. Если в эксперименте после первой пересадки пересадить этот орган второй раз, то отторжение произойдет быстрее (через 3-5 дней), так как все эффекторные механизмы будут быстро мобилизованы благодаря клеткам памяти.
Отсроченное отторжение трансплантата если произойдет, то по вышеприведенным механизмам. Отложено оно по времени только из-за применения иммунодепрессивной терапии.
Лекция 30
ПАТОФИЗИОЛОГИЯ ИММУНОДЕФИЦИТНЫХ состояний
Ж.М. Салмаси
Все иммунодефицитные состояния (ИДС) можно разделить на 2 группы: первичные (врожденные) иммунодефицитные состояния и вторичные (приобретенные).
Первичные иммунодефицитные состояния
Первигные иммунодефициты — это тяжелые заболевания, в большинстве случаев приводящие к гибели (чаще всего от присоединившихся инфекций) до 20 лет, а при тяжелых комбинированных иммунодефицитах — в пределах первого года жизни.
Эффективная терапия этих заболеваний, которая была бы направлена на устранение их причины, пока отсутствует.
Условно-первичные иммунодефициты можно разделить по преобладающему типу поражений звеньев иммунной системы на 3 типа:
• комбинированные иммунодефициты;
• иммунодефициты с преимущественным поражением клеточного иммунитета;
• преимущественно гуморальные иммунодефициты.
К первым относят заболевания, в основе которых лежат генетические дефекты, затрагивающие ранние этапы развития лимфоцитов, которые еще не разделились на Т- и В-линии. Во вторую группу входят иммунодефициты, при которых нарушается развитие Т-клеток и поражаются опосредуемые ими реакции клеточного иммунитета; к этой же группе относятся дефекты фагоцитирующих клеток. В группу гуморальных иммунодефицитов включают патологию, в основе которой лежат нарушение развития В-клеток и Т-хелперов гуморального ответа, а также патология компонентов комплемента.
Примером комбинированных ИДС могут служить заболевания, основой которых являются мутации ферментов пуринового метаболизма — аденозиндезаминазы и пуриннуклеотидфосфорилазы. Аденозин-
456
Лекция 30
дезаминаза катализирует превращение аденозина и дезоксиаденозина соответственно в инозин и дезоксиинозин, а пуриннуклеотидфосфори-лаза — превращение дезоксигуанозина и гуанозина в гуанин, а также инозина и дезоксиинозина в гипоксантин. При отсутствии аденозинде-заминазы накапливаются дезоксиаденозин, аденозин, дезокси-АТФ и цАМФ (наиболее токсичен первый), что приводит к гибели как Т-, так и В-клеток, причем особенно сильно поражаются развивающиеся тимоциты. При дефиците пуриннуклеотидфосфорилазы накапливаются дезоксигуанозин, ГТФ и дезокси-ГТФ, токсичные только для лимфоцитов Т-ряда.
Еще один характерный пример — синдром «голых» лимфоцитов. При нем вследствие генетически обусловленного нарушения образования транскрипционных факторов отсутствует экспрессия всех генов МНС II класса. В результате не может осуществиться положительная селекция СП4*-клеток, и эта популяция не формируется.
При аутосомном варианте тяжелого комбинированного иммунодефицита наблюдается нокаут или мутации генов рекомбиназ RAG-1 и RAG-2. Повреждение любого из них приводит к тяжелому комбинированному иммунодефициту, вызванному блокадой развития В- и Т-лимфоцитов на стадии, предшествующей перестройке генов антигенраспознающих рецепторов.
Некоторые комбинированные виды иммунодефицита возникают при локализованных дефектах генов мембранных молекул адгезии. Следствием таких мутаций является нарушение миграции клеток, в первую очередь нейтрофилов и моноцитов/макрофагов, а также их взаимодействий с клетками других типов Примером могут служить фенотипически сходные поражения, развивающиеся как результат наследственных дефектов экспрессии интегринов (их общей цепи, CD18) и углеводных детерминант (CD15), распознаваемых селектином L (CD62L). Эти поражения представляют собой два варианта LAD-синдрома — дефицита адгезии лейкоцитов (синдрома ленивых лейкоцитов).
При атаксии-телеангиэктазии наблюдается поражение совершенно различных функций, обусловленное дефектностью репарации ДНК и нестабильностью хромосом, а также дефектами клеточного цикла. Это приводит к неожиданному сочетанию симптомов: комбинированному иммунодефициту (недоразвитие тимуса, дефицит Т-клеток и иммуноглобулинов «поздних» изотипов — IgG2, IgG4, IgE, IgA), неврологическим отклонениям (атаксия), к поражению сосудистой стенки (телеангиэктазии), нарушению пигментации.
Патофизиология иммунодефицитных состояний
457
Следствием комплексного генетического дефекта является синдром Вискотта-Олдрига, при котором отсутствует «привязанность» к эмбриональным закладкам, а нарушаются скорее определенные ферментативные процессы и субклеточные компоненты. Это геморрагический синдром, вызванный тромбоцитопенией, в сочетании с экземой и комбинированным иммунодефицитом. При этом заболевании проявляется дефект экспрессии мембранных сиалогликопротеинов (в частности, наиболее важного из них CD43), аномально функционирует цитоскелет, что отражается на подвижности клеток, их форме, межклеточных взаимодействиях, а это, в свою очередь, влияет на эффективность иммунных и многих других процессов.
Классическим примером Т-клетогного иммунодефицита является синдром Ди Джорджи, развивающийся вследствие генетического дефекта, приводящего к нарушению развития преимущественно производных 3-й и 4-й жаберных щелей, что приводит к дефекту развития тимуса (тимус не заселяется лимфоидными предшественниками и не происходит развития Т-лимфоцитов) и гистогенетически родственных органов (паращитовидной железы, сосудов сердца и т.д.).
Причиной В-клетогных иммунодефицитов может служить как поражение В-лимфоцитов (их развития или экспрессии генов иммуноглобулинов), так и дефектность Т-клеток (снижение хелперной активности). Примером первого варианта может быть агаммаглобулинемия Брутона, сцепленная с полом. Дефектный ген, расположенный в Х-хромосоме, блокирует активность гена тирозинкиназы btk, участвующей в развитии В-лимфоцитов.
При большинстве проявлений общего вариабельного иммунодефицита, напротив, преобладает недостаточность Т-хелперной функции. Еще одним примером такого рода патологии может служить гипер-IgM-синдром. Основой заболевания является нарушение экспрессии на Т-клетках молекулы CD154 (CD40L), являющейся лигандом рецептора CD40 В-клеток. В результате в В-клетку не передается сигнал, ответственный за переключение изотипов иммуноглобулинов, и образуются только IgM-антитела. Синтез всех остальных изотипов антител резко снижен.
Среди селективных дефицитов изотипов иммуноглобулинов наиболее частым является дефицит IgA. При нем присутствуют В-лимфоциты, несущие мембранный IgA (т.е. процесс переключения изотипов осуществляется), однако не образуются плазматические клетки, секретирующие IgA-антитела.
458
Лекция 30
Генетические поломки, затрагивающие сами В-лимфоциты и гены иммуноглобулинов, лежат в основе болезни тяжелых цепей, дефицита к-цепей и других гуморальных иммунодефицитов.
Дефекты компонентов комплемента представлены весьма полно: практически все основные факторы классического и альтернативного путей активации могут быть объектами генетического поражения. При этом проявляется однообразная симптоматика: снижение устойчивости к некоторым инфекциям (в частности, нейссериальной) и иммунокомплексная патология с волчаночным синдромом.
Клинические проявления наследственных иммунодефицитов
Основным симптомокомплексом, отражающим нарушение иммунной защиты при этих заболеваниях, является инфекционный синдром. Он проявляется в частом (6 раз и более в год) развитии простудных респираторных заболеваний. Характерной чертой иммунодефицитов, часто определяющей постановку диагноза, является развитие инфекционных процессов, причиной которых служат оппортунистические агенты, т.е. сапрофиты, внедрению которых организм с нормальным состоянием иммунной системы способен надежно противостоять. К ним относятся Pneumocystis carinii, Candida, цитомегаловирус, энтеровирусы типа Эхо и др. Системное поражение кандидами при иммунодефиците неясной этиологии выделяют даже в отдельный синдром, относящийся к наиболее частым первичным иммунодефицитам.
При инфекционных процессах у больных первичными иммунодефицитами на первом месте среди поражаемых органов стоят легкие с бронхиальным аппаратом, затем следуют ЛОР-органы и ротовая полость, почти в той же степени уязвимы кожа и ее производные (ногти и т.д.). Существенно ниже частота поражения урогенитального тракта. ЖКТ страдает достаточно часто, однако его поражение не всегда можно однозначно связать с инфекцией, поскольку энтериты с упорной диареей могут служить проявлением ряда иммунологических аномалий вплоть до аутоагрессии типа реакция трансплантат против хозяина (РТПХ), сопровождающей иммунодефициты. В общем, поражение различных органов и систем инфекционными агентами наблюдается у 75-100 % больных наиболее распространенными первичными иммунодефицитами. При некоторых иммунодефицитах регистрируется септицемия (30 % при синдроме Вискотта-Олдрича).
Иногда при первичных иммунодефицитах обнаруживаются признаки аутоиммунных поражений. Наиболее часто они проявляются как синдром, сходный с ревматоидным артритом (45 % случаев агаммаглобули
Патофизиология иммунодефицитных состояний
459
немии с поражением В-клеток), аутоиммунное поражение эндокринных органов (до 50 % при кожно-слизистом кандидозе) и системы крови, синдром Шегрена, хронический гепатит и, наконец, в форме системных синдромов типа волчаночного (при дефиците факторов комплемента).
Иммунологические нарушения еще одной группы, которая проявляется лишь при некоторых первичных иммунодефицитах, составляют аллергоподобные расстройства. Эти нарушения, как правило, не связаны с действием аллергенов. Обычно в их основе лежит гиперфункция определенных звеньев систем, участвующих в реализации аллергических реакций. Такие реакции в форме псевдоаллергического дерматита регистрируются, например, у всех больных с rHnep-IgE-синдромом и синдромом Вискотта-Олдрича.
Наконец, наблюдается (особенно в случаях преимущественного нарушения клеточного иммунитета) повышения частоты злокачественных опухолей вследствие ослабления противоопухолевого надзора. К счастью, этот тип последствий иммунной недостаточности проявляется не при всех первичных иммунодефицитах. При атаксии-телеангиэктазии развитие злокачественных процессов регистрируется в 10-20 % случаев, причем почти исключительно в форме лимфо- и миелопролиферативных заболеваний. Резкое увеличение частоты развития опухолей лимфоретикулярного генеза (до 15 %) наблюдается при синдроме Вискотта-Олдрича, сцепленной с полом агаммаглобулинемии (6 %) и ряде других первичных иммунодефицитных состояний.
Помимо нарушений, имеющих в своей основе иммунологическую недостаточность, при первичных иммунодефицитах, обусловленных дисгенезиями тканей и системными пороками, выявляются другие (неиммунологические) нарушения, рассматриваемые в клинических курсах, посвященных этим заболеваниям. Поскольку первичные иммунодефициты — это патология, регистрируемая в основном у детей, важно, что они нередко сопровождаются задержкой развития, которая особенно часто регистрируется при атаксии-телеангиэктазии и синдроме Вискотта-Олдрича.
Вторичные иммунодефицитные состояния
Вторигные, или приобретенные, иммунодефициты определяют как нарушения иммунной защиты организма, развившиеся в постнатальном периоде вследствие действия ненаследственных индукторных факторов (внешних или внутренних). Вторичные иммунодефицитные состояния распространены чрезвычайно широко. В большей или меньшей степени отклонения в иммунной системе сопутствуют всем заболеваниям, они проявляются в экстремальных ситуациях.
460
Лекция 30
Наиболее частые причины развития вторичных ИДС:
• нарушения обмена веществ, в том числе вызванные алиментарными факторами;
• инфекционный процесс;
• эндокринная патология;
• действия лекарственных препаратов;
• стресс;
• действие ионизирующей радиации.
К примеру, недостаток поступления в организм незаменимых аминокислот с белком пищи (строгое вегетарианство, неполное голодание) приводит, кроме прочего, к нарушению синтеза иммуноглобулинов и рецепторов Т-лимфоцитов и к развитию ИДС. Аналогичные нарушения развиваются при недостаточном поступлении в организм витаминов и микроэлементов (в первую очередь железа и цинка).
Особого внимания заслуживают иммунодефициты инфекционной природы, поскольку они непосредственно связаны с иммунной системой, которая вовлекается в инфекционный процесс как основной компонент защиты и в то же время становится объектом поражения. При любом инфекционном заболевании наблюдается развитие воспаления и ответа острой фазы. В свою очередь, развитие этих патологических состояний сопровождается усиленным синтезом медиаторов воспаления (простагландины, гистамин, лейкотриены и др.), цитокинов, белков острой фазы, вторичных пептидов, которые вызывают функциональную блокаду клеток иммунной системы. Роль вторичного мессенджера, реализующего иммунодепрессивный эффект в этих случаях наиболее часто играет цАМФ. Таким образом, любая перенесенная инфекция вызывает развитие транзиторного иммунодефицита. Поэтому наличие инфекционного процесса или нахождение в состоянии реконвалесцен-ции являются противопоказаниями для проведения профилактических прививок.
Особого внимания заслуживают инфекции, вызываемые лимфотропными вирусами. Вирус Эпштейна-Барр, часто присутствующий в организме человека в латентной форме, поражает В-лимфоциты. В условиях умеренного климата этот вирус вызывает развитие инфекционного мононуклеоза. Сущность инфекционного мононуклеоза состоит во временной доброкачественной пролиферации В-лимфоцитов. Причина пролиферации В-клеток при этом заключается в том, что один из продуктов вируса является транскрипционным фактором, способствующим индукции генов, причастных к митогенезу. В определенных регионах
Патофизиология иммунодефицитных состояний
461
Африки, где распространены кофакторы, вызывающие формирование транслокаций в лимфоцитах, у больных развивается злокачественное заболевание — лимфома Беркитта. Тот же вирус при содействии других кофакторов вызывает два других заболевания — рак носоглотки (также имеющий эндемический характер) и лимфогранулематоз.
Наиболее известной патологией, вызываемой действием лимфотропного вируса, является ВИЧ-инфекция.
ВИЧ-инфекция
Патогенез. Проникновение вируса в клетку возможно при наличии на клетке-мишени рецептора для данного вируса. Рецепторами для ВИЧ являются дифференцировочный АГ CD4, а также неспецифические, не зависящие от наличия CD4, компоненты. Фиксация вируса через gpl20 ВИЧ-1 (или gpl05 в случае инфицирования ВИЧ-2) с мембранным рецептором CD4 клетки хозяина блокирует основную функцию этих иммунокомпетентных клеток — восприятие сигналов от АПК. В организме человека имеется целый ряд клеток, имеющих рецепторы для ВИЧ (CD4’-лимфоциты, CDS’-лимфоциты, дендритные клетки, моноциты, эозинофилы, мегакариоциты, нейроны, микроглия, сперматозоиды). Последующая за рецепцией репликация вируса ведет к гибели клеток, выпадению функции, ими выполняемой, развитию иммунодефицита.
Наличие СВ4-рецептора на многих и не только иммунокомпетентных клетках, возможность поражать и клетки, не имеющие этого рецептора, определяют политропность ВИЧ и полиморфизм клинической картины. Степень поражения тех или иных, содержащих СВ4-рецепторы, клеток зависит от плотности этих рецепторов на мембране клеток. Наиболее высока плотность на Т-хелперной субпопуляции лимфоцитов, что и определяет во многом патогенез болезни. Но степень поражения клеток-мишеней вирусом зависит также и от возможности репликации вируса в том или ином виде клеток. Очевидно, осуществляется репликация в основном в лимфоцитах с СО4’-фенотипом и моноцитах/макрофагах. Поэтому главной мишенью ВИЧ являются хелперные лимфоциты человека, при этом угнетение Th] выражено в большей степени, чем обеспечиваются и вирусная патология, и онкогенез.
Механизмы формирования иммунодефицита при СПИД. Основным механизмом гибели клеток, инфицированных ВИЧ, является некроз, т.е. нарушение целостности клеток, вызванное в первую очередь повреждением ее мембраны. Детально механизмы цитонекроза не выяснены, но полагают, что он обусловлен прямым цитопатогенным действием
462
Лекция 30
вируса. При заражении лимфоцитов крови человека in vitro уже в первые часы оболочечные АГ ВИЧ появлялись на 80 % СП4*-клеток; в течение 3-х сут численность СП4*-клеток не изменялась, затем начинала резко уменьшаться параллельно с высвобождением вирионов в среду.
В последние годы стали накапливаться факты о том, что достаточно большая часть Т-лимфоцитов, в том числе неинфицированных, гибнет по механизму апоптоза. Еще один из механизмов, приводящих к гибели ВИЧ-инфицированных клеток, связан с формированием под влиянием ВИЧ синцития, т.е. слияния клеток с формированием многоядерного образования, не способного выполнять обычные функции клеток данного типа и в итоге обреченного на гибель. Образование синцития связано со слиянием не только мембран клетки и вируса, но и соседних клеток, связавших частицу ВИЧ. В основе этого процесса лежит взаимодействие молекул CD4 и gpl20.
Отнюдь не все типы клеток, чувствительных к заражению ВИЧ, подвергаются цитопатогенному действию. Ему подвержены только CD4 -лимфоциты и мегакариоциты. Ни макрофаги, ни эпителиальные, ни многие другие клетки, инфицированные ВИЧ, не подвергаются цитолизу. Однако для большинства из них получены данные, свидетельствующие о снижении функций после инфицирования вирусом СПИД. Это относится и к пораженным вирусом Т-лимфоцитам, не успевшим подвергнуться цитолизу.
Описанные выше цитопатогенные воздействия ВИЧ и его белков на СП4*-лимфоциты составляют основу патогенеза иммунодефицита на уровне ВИЧ-инфицированного организма. На высоте заболевания наиболее ярким лабораторным показателем является резкое снижение содержания CD4’-клеток. Условную границу снижения содержания этих клеток, за которой обычно следуют клинические проявления в виде присоединяющейся инфекции, составляет 200-250 клеток в 1 мкл (20 % нормального содержания). Соотношение CD47CD8* на высоте заболевания снижается до 0,3 и менее.
К основным факторам подавления Т-зависимых реакций иммунной системы при СПИДе относятся:
• снижение числа СП4*-клеток-хелперов вследствие их гибели:
• ослабление их функций под влиянием инфицирования и действия растворимых белков ВИЧ (gpl20 блокирует СП4+-клетки, вызывает потерю CD4 или неэффективную активацию клеток):
• сдвиг баланса Th1/Th2 в сторону Th2, тогда как эффективная противовирусная защита может реализоваться с участием Thp
Патофизиология иммунодефицитных состояний 463
• индукция супрессорных клеток белком gpl20 и ВИЧ-ассоцииро-ванным белком р67.
Обусловленное этими факторами снижение способности организма к иммунному ответу затрагивает как его клеточную, так и гуморальную формы. Результатом этого становится комбинированный иммунодефицит, делающий организм уязвимым к инфекционным агентам, в том числе условно-патогенным (отсюда развитие оппортунистических инфекций). Дефицит клеточного иммунитета играет определенную роль в развитии лимфотропных опухолей, а сочетание иммунодефицита и действия некоторых белков ВИЧ (в частности, р14) — в развитии саркомы Капоши.
Несмотря на то что симптоматика ВИЧ-инфекции крайне разнообразна, могут быть выделены несколько типов течения заболевания.
• Легогный тип. Он проявляется гипоксемией, болями в груди, рассеянными легочными инфильтратами на рентгенограммах легких. Характеризуется развитием пневмоний, наиболее часто вызываемых Pneumocystis carinii. Заболевание отличается высокой летальностью. Нередко при ВИЧ-инфекции пневмонии могут провоцироваться цитомегаловирусом или Legionella pneumophila. Поражение бронхолегочного аппарата — одно из самых постоянных и частых проявлений ВИЧ-инфекции. По данным патологоанатомических вскрытий, у умерших от СПИДа более чем в 60 % случаев отмечено поражение дыхательной системы.
• НейроСПИД — преимущественное поражение центральной нервной системы (встречается в 30 % случаев заболевания). Этот тип заболевания может протекать в следующих формах:
— абсцессы, криптококковый менингит, прогрессирующая многоочаговая лейкоэнцефалопатия, подострый энцефалит;
— опухоли — первичная и вторичная лимфомы головного мозга;
— сосудистые осложнения — небактериальный тромботический эндокардит, церебральная геморрагия, связанная с тромбоцитопенией;
— поражение ЦНС с очаговыми мозговыми повреждениями и асептическим менингитом.
• Желудогно-кишегный тип. У больных этой формой ВИЧ-инфекции отмечают выраженную диарею, нарушения всасывания и стеаторею. У всех больных выявляются гистологические изменения био-птатов тощей и прямой кишки (атрофия ворсинок, гиперплазия крипт) с очаговой регенерацией клеток в области основания крипт.
464
Лекция 30
Как правило, из оппортунистических инфекций выявляют кандидоз пищевода и желудка, криптоспоридиоз. Поражения ЖКТ, как и легких, являются одной из основных причин смерти при ВИЧ-инфекции.
• СПИД, ассоциированный с лихорадкой неясного генеза. Характеризуется уменьшением массы тела, недомоганием, слабостью. Клинически этот тип характеризуется диссеминированной инфекцией, вызванной микобактериями, обнаруживаемыми в крови, моче, мокроте, костном мозге, лимфатических узлах, печени, легких.
• Диссеминированная форма. У части больных с развернутой картиной ВИЧ-инфекции отмечаются нефротический синдром с почечной недостаточностью, поражения органа зрения. Поражения кожи при этом чаще всего проявляются саркомой Капоши, васкулитами, опоясывающим лишаем, микозами. Могут иметь место и другие генерализованные инфекции.
Принципы легения. До недавнего времени среди противовирусных препаратов, непосредственно действующих на ВИЧ, использовался только зидовудин (азидотимидин, ретровир, тимазид). Этот препарат за счет ингибирования обратной транскриптазы тормозит репликацию ВИЧ-1, ВИЧ-2 и других ретровирусов. Применение зидовудина позволило снизить летальность, уменьшить количество оппортунистических инфекций в 5 раз. Недостатком терапии зидовудином является формирование устойчивости вирусов при приеме свыше 6 мес. Побочные эффекты касаются, прежде всего, влияния препарата на гемопоэз. Нейтропения, анемия возникают примерно у 50 % пациентов, у части больных возникает миопатия.
Внедрение аналогов нуклеозидов нового поколения, ингибирующих обратную транскриптазу, синтез ряда ненуклеозидных ингибиторов обратной транскриптазы и ингибиторов протеазы коренным образом изменили терапию ВИЧ-инфекции. Активная противовирусная терапия в настоящее время позволяет перевести процесс в хроническое течение. К наиболее перспективным аналогам нуклеозидов с активностью против обратной транскриптазы относятся диданозин, ставудин, ламиву-дин. Среди ненуклеозидных аналогов с активностью против обратной транскриптазы в клиническую практику внедрен невирапин (вирамун). Среди ингибиторов протеазы в лечении ВИЧ-инфекции используются индинавир, саквинавир, ритонавир, нелфинавир.
Современная концепция применения антивирусных препаратов построена на комплексном применении средств с различными точ
Патофизиология иммунодефицитных состояний
465
ками приложения. Чаще всего прибегают к тритерапии, включающей комбинацию двух ингибиторов обратной транскриптазы с одним из ингибиторов протеазы. За счет применения тритерапии удалось снизить показатель смертности больных в 3 раза по сравнению с монотерапией.
Патогенез иммунодефицита, вызванного действием ионизирующей радиации
Довольно быстро после открытия ионизирующих лучей была установлена их способность снижать резистентность к инфекционным АГ, а также уменьшать содержание лимфоцитов в большей степени, чем других клеток крови.
Иммунодефицит развивается почти немедленно после общего внешнего облучения организма.
Действие радиации на иммунную систему обусловлено прежде всего двумя эффектами:
• повреждением естественных барьеров, отделяющих внутреннюю среду организма от внешней, что приводит к усилению биологической агрессии (повреждение эпителия кишечника, дыхательных путей и других органов);
• избирательным повреждением лимфоцитов, а также всех делящихся клеток, в том числе клеток-предшественников иммуноцитов и клеток, вовлекаемых в иммунный ответ, что делает организм беззащитным перед биологической агрессией.
Радиационная гибель клеток осуществляется по двум механизмам.
Делящиеся клетки гибнут в митозе в связи с нерепарируемым повреждением ДНК и хромосомного аппарата, что проявляется нарушением деления клеток. Митотическая гибель проявляется в форме некроза.
Вторая разновидность радиационной гибели затрагивает покоящиеся клетки. При облучении всех клеток, кроме лимфоидных, интерфазная гибель наступает при очень высоких дозах (десятки грей), и лишь покоящиеся лимфоциты погибают при дозе 0,5-3,0 Гр. В основе интерфазной гибели лежит развитие апоптоза. В-клетки несколько более радиочувствительны, чем Т-клетки.
Таким образом, лимфоциты подвержены как бы двойной атаке радиации: покоящиеся клетки гибнут вследствие индукции апоптоза, а выжившие лимфоциты в случаях вступления в иммунный ответ гибнут в митозе. Это проявляется на уровне иммунной системы опустошением лимфоидных органов и развитием лимфопении вплоть до полного исчезновения лимфоцитов из циркуляции. Дозозависимое опустошение
466
Лекция 30
органов иммунной системы вследствие преимущественно интерфазной гибели лимфоцитов проявляется уже через сутки после облучения в дозах, превышающих 1 Гр.
Иммунодефицит при стрессе
Стресс представляет собой эволюционно выработанную реакцию организма на действие экстремальных факторов. В основе реакции лежат повышенная выработка АКТГ и индуцированная ею гиперпродукция стероидных гормонов надпочечников, а также катехоламинов. В свою очередь, глюкокортикоидные гормоны обладают выраженным лимфотропным действием.
Реакция лимфоцитов зависит от концентрации глюкокортикоидов, а следовательно, от интенсивности стресса.
Воздействия умеренной интенсивности вызывают преимущественно перераспределение лимфоцитов. Незрелые кортикальные тимоциты эмигрируют из тимуса и поступают в основном в костный мозг. Сюда же мигрирует часть зрелых Т-клеток. При слабом стрессе массовая гибель лимфоцитов отсутствует. Функциональная активность лимфоцитов и макрофагов несколько уменьшается, и суммарный уровень ответа, особенно гуморального, снижается. Значимость подобных перестроек в иммунной системе в плане эффективности иммунной защиты и целесообразности для организма оценить пока трудно, но они едва ли могут расцениваться как проявления иммунодефицита.
Иная ситуация возникает при интенсивных стрессорных воздействиях, когда концентрация глюкокортикоидов весьма возрастает, что вызывает апоптоз лимфоцитов. К действию глюкокортикоидов наиболее чувствительны кортикальные CD4*CD8+-тимоциты (это определило их первоначальное обозначение как кортизолчувствительных клеток). Среди периферических лимфоцитов В-клетки более устойчивы, чем Т-клетки. Предшественники Т- и В-лимфоцитов устойчивы к действию глюкокортикоидов.
Результатом всех этих изменений является подавление формирования гуморального иммунного ответа и некоторых форм клеточного ответа. Уже развившийся иммунный ответ при этом не ингибируется.
Возрастные иммунодефициты
Известно два типа физиологических иммунодефицитов, связанных с возрастом, — иммунодефицит раннего постнатального периода и иммунодефицит при старении.
Иммунодефицит раннего возраста связан с тем, что формирование иммунной системы не завершается к моменту рождения. У новорожден
Патофизиология иммунодефицитных состояний
467
ных из тимуса выселяются лишь yS-клетки с ограниченной способностью к распознаванию АГ, и лишь позже на протяжении нескольких недель периферия иммунной системы заселяется оф-клетками. В течение всего периода колонизации лимфоидных органов Т-лимфоцитами функция тимусзависимого звена иммунной системы остается сниженной. Кроме того, у младенцев снижена выработка цитокинов. Недостаточность продукции интерферона-у отрицательно влияет на функцию макрофагов, а низкая секреторная активность ТЬ2-клеток и недостаточность контактных сигналов, опосредованных CD40, — на выработку антител.
Вследствие этого спектр иммуноглобулинов крови у маленьких детей существенно отличается от показателей взрослых. Высокое содержание IgG в крови новорожденных отражает поступление иммуноглобулинов этого класса из организма матери вследствие процесса Fc-зависимого транспорта через плаценту. Иммуноглобулины других классов не преодолевают плацентарный барьер в связи с отсутствием на поверхности клеток трофобласта соответствующих Fc-рецепторов, а также из-за больших размеров молекул IgM и IgA. В связи с тем что время полужизни молекул IgG в циркуляции составляет примерно 20-23 сут, этот белок некоторое время сохраняется в организме в раннем постнатальном периоде. Однако из собственных иммуноглобулинов в организме новорожденных образуются лишь IgM (и то в малых количествах). Их синтез достигает уровня взрослых к концу первого года жизни. Собственный синтез IgG проявляется в возрасте около 6 мес, но полного развития он достигает лишь к 5-6 годам. Еще медленнее формируется способность к образованию IgA и IgE, которая завершается к 10 годам; параллельно формируется способность к развитию аллергических реакций немедленного типа.
В результате сочетания описанных выше процессов оказывается, что у детей первых лет жизни проявляется естественный гуморальный иммунодефицит, затрагивающий все изотопы. Причем в возрасте около 6 мес выраженность этого дефицита достигает максимума, поскольку к этому периоду в организме ребенка исчерпываются запасы материнских IgG, а собственные IgG только начинают синтезироваться. После года спектр гуморального дефицита сужается, и последний полностью ликвидируется к 10 годам. Все сказанное имеет отношение к повышенной восприимчивости детей к простудным заболеваниям и инфекциям.
Развитие иммунодефицита при старении в наибольшей степени связано с возрастной инволюцией тимуса. Снижение секреторной активности эпителия тимуса проявляется в том, что секреция основного гормона
468
Лекция 30
тимуса тимулина неуклонно уменьшается начиная с периода полового созревания. К возрасту 60 лет гормон практически не удается обнаружить функциональными тестами. Уровень других гормонов тимуса с возрастом также снижается, хотя и несколько слабее. Атрофия эпителиального ретикулума, происходит практически в течение всей жизни, однако резкое опустошение тимуса, отражающее утрату одновременно эпителиальных и лимфоидных клеток, проявляется позже 60 лет.
Функциональные эффекты старения, как правило, достаточно долго компенсируются и не приводят к проявлениям иммунодефицита. Снижение численности Т-лимфоцитов на периферии (особенно в циркуляции) регистрируется обычно после 60-70 лет и в большей степени затрагивает Т-хелперы, чем цитотоксические Т-лимфоциты, а среди хелперов — в большей степени Thj-, чем ТЬ2-клетки. При этом численность В-лимфоцитов и NK-клеток существенно не изменяется, а активность фагоцитов даже повышается.
Ослабление иммунной защиты определяется характером описанных выше изменений в иммунной системе старых людей. Прежде всего это ослабление затрагивает реакции, обусловленные Т-клетками (подавлены кожные реакции на динитрохлорбензол, распространенные АГ и митогены), хотя явного роста заболеваемости, обусловленной снижением Т-клеточной защиты, практически не происходит. Полагают, что ослабление Т-клеточного надзора служит одной из причин, способствующих повышению частоты опухолей в старости.
Происходят подавление тимусзависимого гуморального ответа и в то же время повышение концентрации иммуноглобулинов, преимущественно IgG и IgA. При этом особенно важно снижение созревания аффинитета, приводящее к преобладанию низкоаффинных антител. Проявления аллергических процессов с возрастом ослабевают. Наблюдается накопление аутоантител как к распространенным (ДНК, коллаген, IgG), так и органоспецифическим (белки щитовидной железы) АГ. Они определяются в повышенных титрах примерно у 50 % старых людей. Гиперпродукцию аутоантител связывают с ослаблением контроля со стороны Т-супрессоров.
Таким образом, возрастные нарушения в иммунной системе, хотя и редко, могут служить основой развития клинических форм иммунодефицитов.
Лекция 31
ПАТОФИЗИОЛОГИЯ ДЫХАНИЯ I
Г.В. Порядин
Распространенность, этиология, классификация заболеваний органов дыхания
Основные функции легких:
• обмен газов между внешней средой и организмом;
• регуляция КОС через концентрацию СО2 в крови.
Эти процессы необходимы для клеточного метаболизма. Эффективный газообмен возможен при интеграции и координации функций различных органов и систем, а также при тесном сопряжении физиологических процессов, вовлеченных в газообмен. Расстройство функций интегративных систем, а также патологические изменения любого из функциональных компонентов дыхательной системы: паренхимы легких, грудной клетки, альвеолярно-капиллярной мембраны, нервной или гуморальной регуляции дыхания, легочного кровообращения — могут привести к тяжелым расстройствам дыхания, нарушающим гомеостаз, и к развитию болезни.
Заболевания легких занимают существенное место в общей структуре заболеваемости населения. Достаточно сказать, что на долю наиболее трудоспособного возраста приходится около 70 % случаев так называемых неспецифических заболеваний легких (НЗЛ). В последние годы, в связи с возрастанием загрязненности воздушного бассейна, наметилась тенденция к прогрессирующему росту частоты заболеваний легких.
Общая этиология заболеваний системы дыхания
В качестве возбудителей заболеваний органов дыхания могут выступать бактерии (пневмококки, стрептококки, стафилококки), вирусы (аденовирусы, вирусы гриппа, кори, ветряной оспы), риккетсии, про-
470
Лекция 31
стейшие, грибки. Причиной заболеваний системы дыхания могут стать травмы, попадание инородных тел, опухоли, аллергены.
Классификация заболеваний системы дыхания
На сегодня не существует единой классификации болезней органов дыхания. Существующие ныне подходы основаны на международных принципах деления бронхолегочных заболеваний с учетом таких критериев, как:
• особенности клинического течения;
• особенности этиологии и патогенеза;
• морфологические и физиологические (функциональные) изменения;
• преимущественная локализация процесса и ряд других признаков.
Например, воспалительные процессы, возникающие в системе дыхания, могут иметь острое или хроническое течение. В соответствии с этим патологические процессы в легких делятся на острые (острый бронхит, пневмония, бронхоэктатическая болезнь) и хронические (хронический абсцесс, эмфизема).
Другим примером подхода к классификации заболеваний легких является преимущественное поражение: либо воздухопроводящих путей (бронхит, бронхиальная астма, бронхостенозы), либо респираторных отделов легких (альвеолиты, пневмония, пневмосклероз).
Поскольку эффективность газообменной функции системы дыхания определяется взаимодействием трех основных физиологических процессов — вентиляции, диффузии, перфузии, то патогенетический подход как раз и учитывает эти изменения.
Расстройства альвеолярной вентиляции в результате изменения проходимости верхних дыхательных путей — трахеи, бронхов, бронхиол (воспаление, отек, обтурация, спазм), так называемые обструктивные нарушения дыхания (преобладает закрытие просвета дыхательных путей).
Обструктивные расстройства — это такая форма патологии системы внешнего дыхания, при которой увеличено сопротивление току воздуха в дыхательных путях при их закупорке, сужении или отеке.
Обструктивные легочные расстройства очень распространены. В настоящее время известно около 100 заболеваний, сопровождающихся бронхообструктивным синдромом. Бронхообструктивный синдром является основным выражением бронхиальной астмы, обструктивной эмфиземы легких, хронического бронхита, бронхоэктатической болезни,
Патофизиология дыхания I
471
экспираторного стеноза, стенотического ларинготрахеита, муковисцидоза и других заболеваний.
Пригины обструктивных нарушений дыхания:
• обтурация дыхательных путей рвотными массами, инородными телами, бронхостеноз, сдавление трахеи и главного бронха увеличенными лимфатическими узлами, загрудинным зобом, опухолью средостения;
• инфекции (туберкулез легких, сифилис, грибковые поражения, хронический бронхит, пневмония);
• аллергические поражения дыхательных путей (анафилактический шок, анафилаксия, бронхиальная астма);
• отравления (передозировка) медикаментами (холинотропными препаратами, p-блокаторами, вагостимуляторами).
Механизмы сужения воздухоносных путей (бронхоконстрикции).
• Важным механизмом сужения воздухоносных путей является сокращение гладкой мускулатуры воздухоносных путей в ответ на различные стимулы (бронхоспазм), что суживает просвет воздухоносных путей и повышает сопротивление дыхательных путей.
• В основе другого механизма сужения воздухоносных путей лежит утолщение стенок воздухоносных путей (отек слизистой оболочки, гиперплазия и гипертрофия вследствие их хронического воспаления, клеточная инфильтрация).
• Частичная закупорка просвета воздухоносных путей накапливающимся секретом, слизистыми пробками и фрагментами поврежденных клеток с увеличением их сопротивления.
Наиболее характерные изменения функциональных показателей легких при обструктивных процессах
Для оценки вентиляционной способности легких (состояния внешнего дыхания), а также для решения вопроса, по какому типу (обструктивному или рестриктивному) развивается ДН, в клинической практике применяются различные функциональные показатели, определение которых ведется либо с помощью спирометрии (статические — объемные показатели), либо пневмотахометрии (динамические показатели).
Основными показателями спирометрии являются: дыхательный объем (ДО.УТ), представляющий собой инспираторный объем во время спокойного дыхания, ЖЕЛ.УС, представляющая собой максимальный объем воздуха, который можно вдохнуть или выдохнуть, остаточный объем (00, RV), количество воздуха которое остается в легких даже
472
Лекция 31
после максимального выдоха, общая емкость легких (ОЕЛ, TLC), представляющая сумму жизненной емкости и остаточного объема, функциональная остаточная емкость (ФОЕ, FRC), объем воздуха в легких в состоянии покоя по окончании обычного выдоха.
К динамигеским параметрам системы дыхания относятся: дыхательный ритм (ЧД), МОД, представляющий собой произведение ДО и ЧД, максимальная вентиляция легких (МВЛ), составляющая произведение ЖЕЛ и форсированной частоты дыхания, объем форсированного выдоха за 1 с (ОФВР FEVj), выражаемого в процентах к форсированной жизненной емкости легких (ФЖЕЛ, FVC), форсированный экспираторный поток между 25 и 75 % форсированной жизненной емкости легких (ФЭП 25-75 %, FEF 25-75 %), позволяющий оценивать среднюю объемную скорость воздушного потока.
У больных с обструктивными заболеваниями выявляются характерные изменения (табл. 31-1): снижение объемной скорости воздушного потока, МВЛ, ЖЕЛ (VC), ФЭП (FEF) 25-75 %, ФЖЕЛ (FVC), ОФВ, (FEVJ, отношения ОФВуФЖЕЛ (FEVj/FVC %). Происходит увеличение легочных объемов ДО (VT), ФОЕ (FRC), ООЛ (RV) и ОЕЛ (TLC) из-за задержки воздуха в легких.
Таблица 31-1. Данные дыхательных тестов в норме и при патологии
Показатель Данные тестов в норме Данные тестов при обструктивных нарушениях Данные тестов при рестриктивных нарушениях
ДО (VT) 0,5 л т 1(N)
ЧД(О 12-18/мин т ТТ
МОД (VE) 6-10 л/мин ? т
МВЛ 90-120 л/мин i
рд 1,5-2,0 л 1 и
ЖЕЛ (VC) 3,5-5,0 л N(l) 1 (всегда)
ФЖЕЛ (FVC) 1 1
ОЕЛ (TLC) 4,5-6,5 л ? J-(N)
00 (RV) 1,0-1,5 л т 1(N)
ФОЕ (FRC) 2,5-3,0 л ? i(N)
МАВ 4,5-5 л/мин и u
0ФВ1 (FEV1) и 4,1
ИНД. ТИФФНО 70-85 % ^всегда N(T)
вдох/ выдох 1:1,2 1:1,4-1,8 N
Патофизиология дыхания!
473
Наиболее частыми обструктивными заболеваниями легких являются — бронхиальная астма, обструктивная эмфизема, хронический бронхит.
Бронхиальная астма (БА) — одно из тяжелейших заболеваний человека. Наиболее распространенное аллергическое заболевание (3-7 %).
На бронхиальную астму приходится 67-72 % бронхообструктивных состояний. БА характеризуется резко выраженным изменением внешнего дыхания, в связи с бронхиальной обструкцией, нарушением газообмена между внешней средой и организмом.
Обязательным признаком бронхиальной астмы является приступ удушья в течение нескольких часов.
Расстройство дыхания при БА чаще имеет экспираторный характер и сопровождается ощущением сжатия грудной клетки. Грудная клетка находится в положении максимального вдоха (расширяется).
В дыхании принимают участие все мышцы шеи, плечевого пояса, спины, брюшной стенки.
Патогенез приступа бронхиальной астмы
В последнее время все большее значение в формировании обструктивного синдрома придается роли гиперреактивности бронхов (рис. 31-1).
Другим важным патогенетическим механизмом БА являются изменения в иммунокомпетентной системе, что нашло отражение в современной классификации БА (инфекционно-аллергическая и неин-фекционно-аллергическая или атопическая).
Большое значение в патогенезе БА играет вегетативная нервная система. В физиологических условиях в регуляции тонуса гладких мышц бронхов участвуют несколько механизмов. Холинергические (парасимпатические) нейрогуморальные раздражители: ацетилхолин (мощный
Сенсибилизация Врожденные дефекты мембран и рецепторного аппарата клеток-мишеней
Длительные инфекции дыхательных путей
Рис. 31-1. Патогенез приступа бронхиальной астмы
Повышение раздражительности (реактивности) бронхов
Воздействие Обострение аллергена инфекции дыхательных путей
Приступ бронхиальной астмы
Физико- Психическое химические возбуждение раздражители
474
Лекция 31
стимулятор сокращения). Воздействуя на М-рецепторы, ацетилхолин вызывает сужение бронхов. Норадреналин или адреналин (симпатический раздражитель). Раздражение адренергических волокон дает двойной эффект, но как результирующий — расширение бронхов.
Сильное сокращающее или расслабляющее действие на тонус гладких мышц бронхов оказывает неадренергическая, нехолинергическая нервная система (субстанция Р, ВИП).
Бронхоспазм может быть обусловлен: или усилением бронхосуживающих стимулов (повышением холинергической, а-адренергической активности или субстанции Р), или понижением p-адренергической активности, или ВИП-высвобождения.
Обструктивные изменения бронхов при БА можно объяснить также влиянием воспалительных медиаторов, которые освобождаются из тучных клеток, расположенных в дыхательных путях. Особое место среди них занимает гистамин, обусловливающий спазм гладкой мускулатуры, гиперемию с повышением проницаемости капилляров и увеличением секреции.
В последние годы в патогенезе БА большое значение придают простагландинам (PGF2a и PGE2).
В значительной мере обструкции бронхов способствуют отек слизистой оболочки и инфильтрация.
Нарушение вентиляции может быть обусловлено изменением вязко-эластигеских свойств легогной ткани (воспаление, отек, атрофия, склероз, опухоли, ателектаз), так называемые рестриктивные расстройства дыхания (в основе лежит нарушение растяжимости или спадение альвеолярной ткани легких).
Рестриктивные расстройства могут быть связаны с:
• расстройствами центральной регуляции дыхания (интоксикации, травмы черепа, позвоночника, инфаркт головного мозга, воспаление, отек, опухоль мозга, кома и др.);
• патологическими изменениями в дыхательной мускулатуре (нарушение иннервации мышц, сократительной способности мышц — при миастении, полиомиелите, ботулизме, столбняке, эпилепсии ит.д.);
• нарушением анатомической целостности и герметичности плевральной полости (пневмотораксы, травмы, деформации, сдавления грудной клетки).
Все эти механизмы являются экстрапулъмональными (внелегочны-ми), поскольку патологический процесс локализуется вне ткани легко
Патофизиология дыхания I 475
го, а вентиляционная ДН получила название регуляторной дыхательной недостаточности (с нарушением регуляции дыхания).
Нарушение вентиляции легких может наступить также вследствие прямого поражения легких при:
• изменении вязкоэластических свойств легочной ткани или в результате потери эластических волокон (диффузные фиброзы различного происхождения — первичная эмфизема легких, пневмосклероз, пневмофиброз, альвеолиты; очаговые изменения в легких — опухоли, ателектазы; отек легких различного происхождения — воспалительный, застойный) — поражение паренхимы;
• снижении активности сурфактанта (поверхностно-активного вещества, выстилающего внутреннюю стенку альвеол) — ателектазы, отек легких, эмболия легочной артерии, гидроторакс, пневмония.
При рестриктивных расстройствах дыхания главным патофизиологическим механизмом является ограниченное расправление легких, что проявляется снижением легочных объемов (см. табл. 31-1, включая TLC (ОЕЛ), ФОЕ (FRC), RV (00), VC (ЖЕЛ), ФЖЕЛ (FVC), ОФВ, (FEV1).
Наряду с определением легочных объемов в клинической практике для выявления ограничения экспираторного потока в легких проводится измерение отношения объемной скорости воздушного потока и объема легких, что можно оценить по петле поток-объем (рис. 31-2).
Петля поток-объем представляет собой график максимальных объемных скоростей потока воздуха на выдохе и вдохе как функции объема легких. Петля состоит из двух половин — экспираторной (экспираторная жизненная емкость), представляющей максимальное усилие выдоха от уровня общей емкости легких (ОЕЛ, TLC) до уровня остаточного объема (00, RV), и инспираторной (инспираторная жизненная емкость), пред-
Рис. 31-2. Петля поток-объем
—10 /к'Х. Выдох
~5 4 A. RV
/ \1
/______________Объем легких
Т (% ЖЕЛ, VC)
TLC /
\ 7 Вдох
МО \
476
Лекция 31
сгавляющей максимальное усилие вдоха из прежде достигнутого остаточного объема (00, RV) назад к общей емкости легких (ОЕЛ, TLC).
После некоторого периода спокойного дыхания пациент делает максимальный вдох (нижняя кривая). Объем легких в точке максимального вдоха есть TLC (ОЕЛ). Вслед за этим пациент делает форсированный выдох (FVC) (верхняя кривая).
Воздушный поток во время вдоха в определенной степени симметричен: наивысшая его скорость достигается приблизительно в средней точке кривой. Эта точка называется максимальной объемной скоростью вдоха (МОС50%ВД) при 50 % жизненной емкости легких.
Максимальная же объемная скорость экспираторного воздушного потока — пиковый экспираторный поток (ПОС, PEF) достигается по ходу выдоха очень рано.
Из кривой поток-объем может быть установлена скорость воздушного потока между 25 и 75 % форсированной жизненной емкости легких.
При обструктивных расстройствах в результате увеличения сопротивления ВП (бронхиальная астма) FVC может быть сохранена, но воздушный поток снизится и FEVj/FVC % уменьшится. Как видно из рис. 31-3, наклон кривой заметно снижен по сравнению с нормой.
Рестриктивные расстройства характеризуются ограничением наполнения грудной клетки воздухом (легкие жесткие и с трудом расправляются). Функция ВП обычно остается нормальной, и скорость воздушного потока не изменяется (рис. 31-4). Хотя FVC и FEV( снижаются, отношение FEV^FVC % остается нормальным.
Экспираторная часть петли поток-объем также описывает функциональные отклонения, свойственные обструктивным и рестриктивным расстройствам.
У пациентов с рестриктивными болезнями петля поток-объем выглядит как уменьшенный вариант нормальной (все величины снижены) и сдвинута вправо.
Рис. 31-3, Петля поток-объем (инспираторная часть) при обструктивных и рестриктивных расстройствах
Патофизиология дыхания I
477
Рис. 31-4. Петля поток-объем (экспираторная часть) при обструктивных и рестриктивных расстройствах
При болезнях обструкции ВП форма петли поток-объем заметно изменена (объемные скорости потока выдоха уменьшены при всех объемах легких), и кривая сдвинута влево.
Нарушение диффузионной способности легких
Наряду с механическими свойствами ВП и легочной паренхимы, определяющими движение газа в альвеолы и обратно, решающим процессом для обеспечения тканевого метаболизма являются газообмен через альвеолярно-капиллярную мембрану, т.е. диффузионная способность легких, или скорость, с которой газ проходит через альвеолярнокапиллярную мембрану.
Движение газа через альвеолярно-капиллярную мембрану происходит путем диффузии. Согласно закону Фика, скорость диффузии газа через мембрану (VG) определяется площадью поверхности мембраны (А) и ее толщиной (d), молекулярным весом (МВ) и растворимостью (а) газа в мембране, а также разницей парциальных давлений по обе стороны мембраны (Р1вальвеолах - Р2вкрови).
VG = к х (А-е-d) х (а + "VMB) х (?! - Р2).
Из уравнения следует, что скорость диффузии газа через альвеолярнокапиллярную мембрану возрастает с:
• увеличением площади поверхности мембраны (в норме диффузионная поверхность легких равно 50-100 м2), растворимости и градиента давления газа по обе стороны мембраны;
• уменьшением толщины мембраны и молекулярного веса газа;
• увеличением размеров тела (массы, роста, площади поверхности);
478
Лекция 31
• увеличением объема легких;
• во время физической нагрузки;
• увеличением альвеолярного РСО2.
Диффузионная способность легких снижается с:
• увеличением альвеолярного РО2;
• увеличением толщины альвеолярно-капиллярной мембраны (отек легких, воспалительный процесс) и молекулярного веса газа;
• уменьшением диффузионной поверхности (резекция легких, легочная эмфизема, пневмонии, фиброз легких).
Диффузионная способность легких снижается при хронических обструктивных заболеваниях легких. Снижение связано с уменьшением площади поверхности альвеолярно-капиллярного контакта и объема капиллярной крови.
Диффузионная способность легких снижается при заболеваниях, поражающих паренхиму легких и характеризующихся рестриктивными расстройствами дыхания. Снижение связано с интерстициальным отеком, фиброзом и деструкцией капилляров, в результате чего увеличивается диффузионное расстояние, утрачиваются капилляры, снижается объем капиллярной крови и нарушается баланс между альвеолярной и капиллярной перфузией.
Нарушение перфузионной способности легких (патология легочного кровотока)
Движущей силой легочного кровотока (перфузии легких) является различие давления в правом желудочке и левом предсердии, а главным регулирующим механизмом — легочное сосудистое сопротивление.
Факторы, влияющие на легочный кровоток:
• объем циркулирующей крови;
• работа правого желудочка;
• кровенаполнение левого предсердия;
• легочное сосудистое сопротивление, регулируемое баро- и хеморецепторами;
• внутриальвеолярное давление и действие силы тяжести.
Все названные факторы тесно взаимосвязаны, и серьезные изменения любого из них могут стать основой патологии дыхания.
На практике все большее и большее значение имеет соотношение вентиляции и перфузии В/П. Показатель, выражающий соотношение между кровообращением и дыханием. В норме на каждый литр крови, протекающей через легкие в 1 мин, должно приходиться около 0,8 л альвеолярного воздуха, т.е. показатель вент/перфуз (В/П) = 0,8.
Патофизиология дыхания I
479
В/П = мин.альвеолярная вент (МАВ) + мин.кровоток через легкие =
4 л/мин + 5 л/мин = 0,8.
Нарушение соответствия вентиляции/перфузии легких наблюдается как при обструктивных (бронхиты, бронхиальная астма), так и рестриктивных (пневмонии, эмфизема, пневмоторакс, ателектазы) заболеваниях легких.
Патофизиология дыхательной недостаточности
Патологические изменения любого из функциональных компонентов дыхательной системы — паренхимы легких, грудной клетки, альвеолярнокапиллярной мембраны, нервной или гуморальной регуляции дыхания, легочного кровообращения - могут привести к тяжелым расстройствам дыхания: развитию дыхательной недостаточности (ДН).
В общем плане ДН — это состояние системы дыхания, при котором нарушен газовый гомеостаз, или его поддержание обеспечивается благодаря постоянному напряжению системы внешнего дыхания.
ДН подразделяют на несколько типов (рис. 31-5).
А. В зависимости от механизмов, лежащих в основе развития ДН, на:
• вентиляционную (при нарушении вентиляции легких, нарушении центральной регуляции дыхания);
• диффузионную (при диффузионной дыхательной недостаточности удлиняется путь и затрудняется прохождение газов через аэроге-матический барьер. В норме толщина альвеолярно-капиллярной мембраны равна 0,5 мкм);
• перфузионную*,
• смешанную.
Нарушения центральной регуляции дыхани
Нарушения вентиляции легких
перфузии тканевого
легких дыхания
Нарушения Нарушения Нарушения транспорта кислорода через АГБ
Диспноэ
Гипоксия
Гипоксемия
Дыхательная недостаточность
вентиляционная диффузионная перфузионная смешанная
Рис. 31-5. Патофизиологические механизмы развития дыхательной недостаточности
480
Лекция 31
ДСЛ (диффузная способность легких) = количество поглощенного за 1 мин О2 (мл) -ь РО2 в альвеолярном возд - РО2 в лег.капил.
У здоровых ДСЛ = 15-20 мл/1 мм рт.ст./мин (300/100-80) — это количество газа (О2), которое способно пройти через альвеолярную/ка-пиллярную мембрану за 1 мин при разности парциального давления по обе стороны мембраны, равной 1 мм рт.ст.
Б. ДН может развиться при нарушении регуляции дыхания в результате дисфункции:
• респираторных нейронов при заболеваниях центральной нервной системы (ЦНС):
• при нейромышечной патологии:
• при болезнях грудной клетки.
Соответственно выделяют несколько типов дыхательной недостаточности.
Б-1. Центрогенная дыхательная недостаточность — ДН может быть следствием нарушений центральной регуляции дыхания, встречающихся при передозировке наркотических анальгетиков, метаболической энцефалопатии (при гипотиреозе, гипонатриемии и др.), инфекционном энцефалите, внутричерепных объемных образованиях, таких, как опухоли или гематомы, идиопатических гиповентиляционных синдромах и других заболеваниях.
В норме регуляция дыхания осуществляется сложной системой рецепторов, обеспечивающих нейрохимическую (по отклонению газового состава артериальной крови) и нейромеханическую (по возбуждению механорецепторов) стимуляцию дыхательного центра (ДЦ), расположенного в варолиевом мосте и продолговатом мозге.
При нарушениях афферентной импульсации (например, при метаболических нарушениях или застойной сердечной недостаточности), повышении порога возбудимости ДЦ (например, при передозировке наркотических анальгетиков), органических поражениях ДЦ (травма, опухоли и т.д.) происходит искажение центральной респираторной посылки (ЦРП) с развитием гиповентиляции легких, апноэ (остановка дыхания), дискоординации дыхательных мышц, нарушений частоты и ритма дыхания.
Наиболее характерным проявлением центрогенной ДН является альвеолярная гиповентиляция или снижение минутной вентиляции легких без существенного изменения их функции (состояние, сходное с задержкой дыхания).
Б-2. Нейромышечная дыхательная недостаточность — может развиться при нарушении передачи нервного импульса дыхательным
Патофизиология дыхания I
481
мышцам или нарушении их функции. Чаще она возникает при травмах или заболеваниях спинного мозга с поражением передних мотонейронов шейного или грудного отделов, некоторых экзогенных интоксикациях (отравлении курареподобными веществами, мускарином, фосфорорганическими соединениями и др.), а также при нарушении сократимости дыхательным мышц, например, при судорожном синдроме любого происхождения, миастении и т.д. Кроме того, нейромышечная ДН может развиваться при тяжелых водно-электролитных нарушениях, особенно при выраженной гипокалиемии.
При нейромышечной ДН всегда нарушается функция дыхательных мышц, в результате чего в той или иной мере уменьшается их способность выполнять работу по обеспечению дыхания. При этом, несмотря на адекватную посылку из дыхательного центра, мышцы не создают достаточного уровня трансторакального и трансдиафрагмального давления, необходимого для поддержания нормальной альвеолярной вентиляции.
Б-3. «Каркасная» дыхательная недостаточность
Основной механизм развития ДН при заболеваниях этой группы связан с уменьшением податливости грудной клетки («каркаса») и увеличением эластической работы дыхания, ведущей к утомлению дыхательных мышц.
Снижение податливости грудной клетки отмечается при выраженном кифосколиозе и других врожденных или приобретенных аномалиях позвоночного столба или ребер, ожирении (при условии увеличения массы тела более 130 кг), травмах грудной клетки, некоторых хирургических вмешательствах (торакопластика). Заболевания плевры в ряде случаев также приводят к развитию ДН вследствие ограничения дыхательной экскурсии легких, сдавления легочной паренхимы или структур средостения. К наиболее часто встречающимся патологическим процессам относятся выраженный фиброз плевры (шварты), скопления воздуха (пневмоторакс) или жидкости (гидроторакс) в плевральных полостях. Наконец, податливость грудной клетки уменьшается при ограничениях подвижности диафрагмы, обусловленных заболеваниями органов брюшной полости (напряженный асцит, внутрибрюшные опухоли, увеличения печени и селезенки, острый перитонит, выраженные боли в области послеоперационной раны и т.д.).
В. В зависимости от парциального давления газов в артериальной крови дыхательная ДН подразделяется на:
• гипоксемигескую (при которой РаО2 менее 55 мм рт.ст.) (паренхиматозную, легочную) или ДН I типа;
482
Лекция 31
• гиперкапнигескую (при которой РаСО2 превышает верхний предел нормы — 45 мм рт.ст.) (вентиляционную, насосную) или ДН II типа.
Гипоксемическая ДН характеризуется выраженной артериальной гипоксемией (РаО2 менее 60 мм рт.ст.), трудно корригируемой кислоро-дотерапией. Обычно эта форма ДН, как следует из названия, возникает при тяжелых паренхиматозных заболеваниях легких, таких, как пневмония, ателектаз или отек легких, а также при некоторых заболеваниях нижних дыхательных путей (ХОБЛ, бронхиальная астма и др.).
Основные механизмы гипоксемии:
• снижение парциального давления кислорода во вдыхаемом воздухе;
• общая гиповентиляция легких;
• нарушения диффузии газов через альвеолярно-капиллярную мембрану;
• регионарное нарушение вентиляционно-перфузионных отношений;
• шунт (прямой сброс венозной крови в артериальную систему кровообращения);
• снижение парциального напряжения кислорода в смешанной венозной крови.
Гиперкапническая дыхательная недостаточность обусловлена первичным уменьшением эффективной легочной вентиляции (альвеолярная гиповентиляция), что нарушает выведение СО2 и нередко приводит к серьезным нарушениям КОС. При этом уровень гиперкапнии прямо пропорционален степени уменьшения альвеолярной вентиляции.
Основные механизмы развития гиперкапнии:
• повышение продукции углекислоты;
• снижение минутной вентиляции легких (гиповентиляция);
• увеличение объема физиологического «мертвого пространства».
Г. В зависимости от состояния механизмов компенсации ДН подразделяют на:
• компенсированную (если нормальный газовый состав вытекающей из легких крови обеспечивается напряжением компенсаторных механизмов);
• декомпенсированную (если из легких вытекает кровь, не соответствующая по своему составу артериальной).
Д. По времени развития на:
• острую (минуты, часы);
Патофизиология дыхания I
483
• подострую (сутки, недели),
• хроническую (месяцы, годы).
Острая ДН требует проведения интенсивной терапии, так как может представлять непосредственную угрозу для жизни больного. При быстром развитии ДН не успевают включиться компенсаторные механизмы со стороны других органов и систем организма (сердце, почки и др.). Наиболее характерным признаком острой ДН является нарушение КОС, в частности респираторный алкалоз (при ДН I типа — вследствие избыточного выведения углекислоты) или респираторный ацидоз (при ДН II типа — в результате задержки выведения углекислоты). Кроме того, острая ДН практически всегда сопровождается выраженными нарушениями со стороны сердечно-сосудистой системы. Не случайно острейшая форма ДН носит название асфиксии (в переводе с греческого означает «без пульса») и требует оказания экстренной медицинской помощи.
Хроническая ДН развивается в течение нескольких месяцев или лет. Начало ее может быть постепенным, или же она может развиться при неполном восстановлении после острой ДН. При хронической ДН происходит активация компенсаторных механизмов, направленных на улучшение доставки (транспорта) кислорода к тканям: углубление и/или учащение дыхания, нормализация КОС за счет почечных механизмов его регуляции, рост периферического кровотока в результате тахикардии и повышения сердечного выброса, увеличение массы эритроцитов и НЬ крови, вследствие повышенной выработки эритропоэтина почками, изменение диссоциации оксигемоглобина, замедление тканевого метаболизма и др.
Лекция 32
ПАТОФИЗИОЛОГИЯ ДЫХАНИЯ II
Г.В. Порядин
Нарушение регуляции дыхания (патологические формы дыхания)
Все формы нарушения нормального характера дыхания (частоты, глубины, соотношения фаз дыхательного цикла, ритмичности) именуются общим термином — диспноэ (в отличие от нормального дыхания — эупноэ).
Выделяют две большие группы диспноэ:
• ремиттирующие (или волнообразные, равномерные):
• интермиттирующие (или перемежающиеся, неравномерные).
Ремипирующие формы диспноэ
К ремиттирующим формам относят диспноэ различной выраженности — от изменений, стоящих на границе нормы дыхания и являющихся индивидуальной особенностью данного лица, до форм, характеризующих крайние степени расстройства жизнедеятельности, вплоть до терминальных состояний организма.
Гиперпноэ (тахипноэ, полипноэ)
Тахипноэ — повышение частоты дыхания свыше 24 в минуту.
Как правило, тахипноэ сопровождается уменьшением дыхательного объема (ДО). Минутная альвеолярная вентиляция (МАВ) практически не меняется. Тахипноэ возникает при:
• гипоксии;
• гиперкапнии;
• умеренном ацидозе.
Близкой к тахипноэ формой диспноэ является полипноэ (или гипервентиляция) — увеличение частоты и глубины дыхания, при котором возрастает МОД и МАВ. Полипноэ возникает при:
Патофизиология дыхания II
485
• острых нарушениях мозгового кровообращения (мозговой инсульт);
• сухом плеврите;
• межреберной невралгии;
• ателектазе легких;
• крупозной пневмонии.
Тахипноэ и полипноэ свидетельствуют о повышенном возбуждении дыхательного центра, которое может наступать в результате:
• повышения содержания в крови СО2;
• недостатка О2;
• эмоционального перевозбуждения;
• локальных патологических процессов в области дыхательного центра.
Патогенез учащения дыхания при гиперпноэ связан с двумя механизмами:
• облегчение функции дыхательного центра (ДЦ) в продолговатом мозге (вдоха и выдоха) в результате потери торможения со стороны нисходящих кортикальных нейронов (острые нарушения мозгового кровообращения);
• укорочение рефлекса Геринга-Брейера, тормозящего вдох, в результате повышения возбудимости рецепторов в альвеолах и интерстиции (при воспалении: сухой плеврит, пневмонии, межреберная невралгия).
Брадипноэ — редкое, менее 12 дыхательных движений в минуту, дыхание. Замедление дыхательных движений наступает в результате понижения возбудимости ДЦ. Наблюдается при передозировке наркотических препаратов или фармакологических средств, пищевых отравлениях, гипероксии, гипокапнии, длительной гипоксии. Эта форма диспноэ обозначается еще термином олигопноэ, или гиповентиляция.
Одним из тяжелых инфекционных заболеваний, которое сопровождается расстройством дыхания с резким учащением дыхательных движений, является ботулизм (частота дыхания может доходить до 30-40 в минуту). Нарушается ритм дыхания, главным образом затрудняется вдох. Вдох совершается при усиленном напряжении дополнительных дыхательных мышц.
Возникновение одной из форм ремиттирующего диспноэ — большого дыхания Куссмауля (рис. 32-1) типично для больных в состоянии диабетической и уремической комы, при отравлении метиловым спиртом или для спортсменов после чрезвычайно больших физических нагрузок — «дыхание загнанного зверя»).
486
Лекция 32
Рис. 32-1. Дыхание Куссмауля Рис. 32-2. Апнейстическое дыхание
Большое дыхание Куссмауля — признак глубокого ацидоза; оно характеризуется глубокими, шумными вдохами с участием основной и вспомогательной дыхательной мускулатуры. Дыхание Куссмауля возникает в результате нарушения возбудимости дыхательного центра на фоне гипоксии мозга, ацидоза, интоксикации.
При хронических анемиях, гипоксии головного мозга, интоксикации ЦНС наступает угнетение пневмотаксического отдела дыхательного центра с появлением апнейстигеского дыхания (рис. 32-2).
Апнейстическое дыхание характеризуется медленным развитием вдоха, инспираторной задержкой с форсированным коротким выдохом. Длительность вдохов многократно превышает продолжительность выдохов. Данный вид диспноэ может быть результатом снижения тонуса отделов ДЦ, расположенных в области варолиева моста, либо следствием выпадения афферентной сигнализации, поступающей в ДЦ по блуждающим нервам (интоксикации ЦНС: ботулизм, передозировка наркотиками, отравление фосфор-органическими соединениями).
Интермиттирующие формы диспноэ
Характеризуются неравномерностью дыхательных циклов, степень которой может существенно варьировать. Эти формы нарушения внешнего дыхания обозначаются еще как периодическое дыхание.
Дыхание Чейна-Стокса
Дыхание Чейна-Стокса (рис. 32-3) — форма диспноэ, характеризующаяся тем, что периоды волнообразного дыхания перемежаются длительными дыхательными паузами (апноэ). В основе дыхания Чейна-Стокса лежат функциональные изменения (угнетение) возбудимости ДЦ (кровоизлияния в область ствола, передозировка морфина).
Патофизиология дыхания II 487
—^4/\/w
Рис. 32-3. Дыхание Чейна-Стокса
Периодическое дыхание Чейна-Стокса может встречаться и в норме (в частности, во время сна у маленьких детей и пожилых людей), но главным образом оно возникает у больных:
• с заболеваниями головного мозга;
• сердечно-сосудистыми заболеваниями.
Соответственно отличается и механизм его развития.
Дыхание Чейна-Стокса центрально-нервного происхождения
Причины:
• деструкции пирамидного тракта (заболевания промежуточного мозга, эпидемический энцефалит, псевдобульбарный паралич, двусторонние инфаркты в глубоких отделах мозговых полушарий);
• функциональные расстройства ДЦ (снижение возбудимости ДЦ при уремии).
Патогенез
Во всех этих случаях в основе возникновения периодического дыхания лежит снижение возбудимости нейронов бульбарного дыхательного центра. В результате гипоксии, либо прямого поражения опухолью, либо вследствие влияний, приходящих из вышележащих отделов головного мозга.
При уменьшении возбудимости ДЦ обычная его стимуляция оказывается слишком слабой, чтобы поддерживать его постоянную ритмическую активность. Эта активность возникает лишь при накоплении (в период апноэ) избытка СО2 в крови, вызывающего сильное возбуждение хеморецепторов. Постепенное накопление СО2 в крови выше обычного порога возбудимости ДЦ приводит к его возбуждению, возникает первый вдох. В дальнейшем возбудимость ДЦ поддерживается и постепенно усиливается импульсами, поступающими из проприорецепторов дыхательных мышц. Как только избыток СО2, благодаря усилению легочной вентиляции, удаляется, стимуляция ослабевает, и ДЦ тормозится снова. Наступает апноэ.
Дыхание Чейна-Стокса циркуляторного происхождения
Может возникнуть при гипертонической болезни и других сердечнососудистых заболеваниях.
488
Лекция 32
Рис. 32-4. Дыхание Биота
Патогенез
При недостаточности кровообращения сердечный выброс (ударный и минутный объемы) уменьшен и соответственно увеличивается время кровотока. Поэтому главным фактором, вызывающим дыхание Чейна-Стокса у больных с сердечной недостаточностью является длительное увеличение времени кровообращения. В результате происходит задержка передачи информации о состоянии газов крови к дыхательным нейронам.
Периодическое дыхание Биота
Вторая классическая форма периодического дыхания (рис. 32-4).
Причинами данной формы диспноэ могут стать травмы, опухоли мозга, менингоэнцефалит, интоксикации, нарушения кровоснабжения и глубокая гипоксия стволовых структур мозга. Патологическое дыхание характеризуется появлением пауз длительностью до 30 с и более, разделяющих периоды довольно регулярного дыхания.
Этиология и патогенез отдельных патофизиологических синдромов
Бронхоастматигеский статус. Стойкий бронхообструктивный синдром, при котором становятся неэффективными бронхолитики. Синдром является осложнением бронхиальной астмы, хронического обструктивного бронхита, эмфиземы легких, интерстициального пневмофиброза и др. ХОЗЛ. Летальность достигает 5-16 %.
Патогенез
Дыхательная недостаточность при астматическом статусе.
Физиологическим механизмом дыхательной недостаточности при астматическом статусе является экспираторное закрытие дыхательных путей (ЭЗДП).
Суть ЭЗДП заключается в том, что в физиологических условиях (рис. 32-5, а) в ходе выдоха (в самом его конце) в различных легочных зонах задерживается газ, при этом его давление внутри альвеолы распределяется равномерно. Дыхательные пути закрываются постепенно начиная с
Патофизиология дыхания II
489
Рис. 32-5. Экспираторное закрытие дыхательных путей
нижних отделов легких до достижения уровня ООЛ. При этом давление газов распределяется равномерно.
При сужении бронхов (рис. 32-5, б — форсированный выдох) мышцы грудной клетки напряжены, что ведет к увеличению осевого давления в бронхах. Радиально направленного давления внутри бронха становится недостаточно, чтобы предупредить его спадение при выдохе. Кроме того, при форсированном выдохе растет напряжение мышц грудной клетки, что ведет к возрастанию экстрапульмонального давления. ЭЗДП наступает рано, прежде, чем из легких выйдет воздух. Поэтому ЭЗДП в условиях бронхообструкции получило название раннее ЭЗДП.
В патогенезе дыхательной недостаточности при астматическом статусе можно выделить три этапа.
• Основу составляют отек, воспаление, закупорка дыхательных путей вязкой мокротой. Это ведет к развитию первого этапа астматического статуса — обструкции дыхательных путей: затрудняется вдох, делается активным и удлиненным выдох. Больной активно выдыхает задержанный воздух, напрягая мышцы выдоха, повышая тем самым внутриплевральное давление. Это ведет к сдавлению мелких бронхов и развитию ЭЗДП и экспираторного стеноза.
• Неэффективность работы дыхательных мышц (повышенные расходы кислорода) ведет к росту гипоксии. Присоединяется правожелудочковая недостаточность.
• Поражение метаболизма (усугубление гипоксии), развитие респираторного и метаболического ацидоза, гипогидратация, надпочечниковая недостаточность (результат длительного приема больными глюкокортикоидных гормонов).
Легочная артериальная гипертензия
Различают первичную и вторичную легочную артериальную гипертензию (ЛАГ).
490
Лекция 32
Первигная ЛАГ возникает при первичном тромбозе мелких ветвей легочной артерии, при легочной артериопатии, артериитах, коллагено-зах, интоксикации и др.
Вторигная ЛАГ возникает при ХОЗ Л и других хронических заболеваниях легких (силикоз, туберкулез).
В патогенезе дыхательной недостаточности при ЛАГ можно выделить 4 звена:
• нарушение легочного кровотока;
• нарушение вентиляционно-перфузионных соотношений;
• нарушение альвеолярно-капиллярной диффузии;
• альвеолярное шунтирование крови.
Все указанные механизмы приводят к дыхательной недостаточности разной степени тяжести (одышка, гипоксемия, правожелудочковая недостаточность).
Сердегная астма (СА) — неспособность сердца обеспечить необходимый сердечный выброс при достаточном венозном возврате. Возникает СА при инфаркте миоркарда, миокардите, врожденных (приобретенных) пороках сердца, нарушениях сердечного ритма и т.д.
Патогенез дыхательной недостаточности при СА включает два механизма.
• Левожелудочковая недостаточность с задержкой крови в малом круге кровообращения. Вначале развивается легочная венозная, а затем легочная артериальная гипертензия. Кровенаполнение легких увеличивается, повышается легочное капиллярное давление. Возрастает (в 2-3 раза) внесосудистый объем воды в легких, возникает гипоксемия. В связи с отеком бронхиолы теряют свою эластичность, развивается раннее ЭЗДП.
• Рестриктивный. Для раскрытия жестких, отечных, переполненных кровью легких требуется повышенное мышечное усилие. Возрастает работа дыхательных мышц. Это ведет к усилению гипоксии, еще большему увеличению проницаемости альвеолярно-капиллярной мембраны — отеку легких.
Тромбоэмболия легогной артерии (ТЭЛА). Занимает 3-е место среди причин гибели больных (1-е — ИБС, 2-е — злокачественные новообразования). Возникает в связи с тромбозами, тромбофлебитами, ХНЗЛ, катетеризацией сосудов.
По характеру течения различают:
• молниеносную форму (минуты);
• острую форму (часы);
Патофизиология дыхания II
491
• подострую форму (сутки);
• рецидивирующую форму.
По степени поражения легочного сосудистого русла:
• массивную ТЭЛА (эмболия ствола и главных ветвей легочной артерии);
• субмассивную ТЭЛА (эмболия долевых ветвей);
• эмболию мелких ветвей легочной артерии.
Основу дыхательной недостаточности при ТЭЛА составляют:
• нарушение перфузии легких;
• легочная артериальная гипертензия;
• резкое нарушение вентиляционно-перфузионных (В/П) соотношений.
Патогенез дыхательной недостаточности при ТЭЛА сложный и складывается из нескольких этапов'.
• механическое перекрытие легочного кровотока эмболом;
• генерализованный артериолоспазм в малом круге кровообращения;
• выделение биологически активных веществ (серотонина, гистамина, простагландинов);
• бронхоспазм, ведущий к обструктивному типу ДН;
• перегрузка давлением. В результате напряженной работы правого желудочка для преодоления высокого легочно-артериального сопротивления развивается острая правожелудочковая недостаточность.
В результате ограничения объема поступающей из малого круга в левый желудочек крови резко снижается выброс левого желудочка.
Все это снижает коронарный кровоток, ведет к тяжелой ишемии миокарда вплоть до развития фибрилляции сердца. Развиваются коллапс в большом круге кровообращения, острая ДН.
Респираторный дистресс-синдром взрослых
Форма острой ДН, характеризующаяся острым началом, выраженной гипоксемией, не устраняемой оксигенотерапией, интерстициальным отеком и диффузной инфильтрацией легкого. Синонимами являются синдром шокового легкого, синдром дыхательных расстройств и ряд других.
Причинами развития синдрома могут стать множественные травмы, ожоги, массивные трансфузии, сепсис, шок (геморрагический, травматический, анафилактический, кардиогенный), длительная гиповолемия.
492
Лекция 32
Патогенез дыхательной недостаточности
Ведущим в патогенезе являются повышение более чем в 2 раза проницаемости альвеолярно-капиллярной мембраны и увеличение внесосуди-стого объема воды — диссеминированная, двусторонняя инфильтрация легких (отек легких).
В повреждении альвеолярно-капиллярной мембраны участвуют свободные радикалы, альдегиды (при гидролизе арахидоновой кислоты), простагландины, лейкотриены, активированный комплемент, бактериальные токсины.
Другим важным механизмом развития синдрома являются нарушение диффузии газов в результате утолщения альвеолярно-капиллярной мембраны и снижение растяжимости легких как следствие инактивации сурфактантной системы, что происходит в результате образования гиалиновых мембран из фибриногена — плазменного белка. Легкие становятся жесткими, образуются микроателектазы с отеком и нарушением микроциркуляции, возникают гиповентиляция и шунтирование крови, благодаря чему артериальная гипоксемия не поддается оксигенотера-пии.
Респираторный дистресс-синдром новорожденных
Проявляется тем, что после первых вдохов ДО начинает постепенно снижаться, хотя дыхательные мышцы развивают большие усилия, а трахея и бронхи проходимы. Вдох становится более коротким, напоминая глотательные движения («дыхание лягушки»).
Возникновение и развитие респираторного дистресс-синдрома новорожденного (РДСН) связаны с рядом особенностей и спецификой системы дыхания у детей.
• У детей более высокий метаболизм, поэтому потребность в кислороде и уровень газообмена у них выше, чем у взрослых. Однако компенсаторные возможности дыхания у детей меньше.
• Абсолютные величины легочных объемов у грудных детей очень малы (количество альвеол в 10-15 раз меньше, чем у взрослых). Легкие более полнокровны и менее эластичны, а дыхательные пути очень узки. Это заведомо увеличивает вероятность возникновения у младенцев и обструктивных, и рестриктивных форм ДН.
• Грудная клетка у детей по сравнению со взрослыми находится как бы в положении вдоха (ребра расположены перпендикулярно к позвоночнику). По этой причине увеличить объем вентиляции
Патофизиология дыхания II
493
младенцы могут в основном за счет учащения (тахипноэ), а не углубления дыхания, что, с одной стороны, менее эффективно, а с другой — энергетически невыгодно.
Респираторный дистресс-синдром новорожденных может возникнуть с первых же секунд жизни ребенка. Это самая частая причина смерти новорожденных в 1-2 сут.
Во время родов жидкость, заполняющая легкие, выдавливается при сжатии грудной клетки в естественных родовых путях. Поверхностное натяжение в слое жидкости между слипшимися альвеолами очень велико и препятствует расправлению легких. Если новорожденный не способен преодолеть это сопротивление, то легкие не расправляются, и у ребенка развивается дистресс-синдром.
Первый крик новорожденного — важнейшая приспособительная реакция, облегчающая полное раскрытие легких. Чем звонче крик, тем Уже голосовая щель, тем больше давление в альвеолах, тем лучше они раскрываются.
Патогенез дыхательной недостаточности
Этот синдром представляет дефицит сурфактанта в результате ишемии легочной ткани.
• Изменение легочного кровотока нарушает образование сурфактанта, который продуцируется альвеолярными клетками II типа. При нарушении продукции сурфактанта поверхностное натяжение в альвеолах становится очень высоким, и новорожденный должен преодолеть очень большие силы сцепления между молекулами жидкости, покрывающей стенки спавшихся альвеол.
• Ишемизированная альвеолярно-капиллярная мембрана становится более проницаемой, и через нее идет транссудация плазмы внутрь альвеолы, а также некоторых белков, которые инактивируют даже те небольшие количества сурфактанта, которые содержатся в альвеолах и образуют пленку — гиалиновую мембрану.
Врожденная аномалия, спазм легочных артериол при внутриутробной асфиксии, инактивация сурфактанта амниотической жидкостью при внутриутробных вдохах способствуют развитию дистресс-синдрома. Активный синтез сурфактанта происходит в 36-37 нед беременности, поэтому у недоношенных частота данной патологии резко возрастает.
Лекция 33
ПАТОФИЗИОЛОГИЯ КИШЕЧНОГО ПИЩЕВАРЕНИЯ
Л.Ю. Семенова
Деятельность желудочно-кишечного тракта (ЖКТ) направлена на удовлетворение энергетических и пластических потребностей организма.
Болезни кишечника, его тонкого и толстого отделов развиваются часто. Кишечник работает как единое целое, объединяя деятельность желудка, печени и системы желчевыделения, а также поджелудочной железы. При этом кишечник осуществляет важнейшие функции пищеварения: расщепление питательных веществ до малых молекул и транспорт их через стенку кишки. В тонкой кишке заканчиваются процессы переваривания пищи, одновременно идут процессы всасывания, которые заканчиваются в толстой кишке.
Патология одного звена пищеварительной системы сопровождается дисфункцией и патологией другого звена ЖКТ. В целом ЖКТ обладает высокими компенсаторными возможностями.
Патологические процессы, развивающиеся в кишечнике, вызывают расстройства пищеварения и могут проявляться изменением секреции, моторики, всасывания, инкреции, эвакуации и состава микрофлоры.
Выделяются два пика заболеваемости: детство и старость.
В детском возрасте выявляется неспособность адекватно реагировать на изменение характера питания, а также легкость инфицирования ЖКТ, обусловленная недоразвитыми навыками гигиены. Кроме того, именно в детском возрасте дебютирует ряд врожденных энтеропатий, таких как лактазная недостаточность.
В старости сказываются накапливаемые с возрастом атрофические изменения слизистых оболочек, запоры, дивертикулезы, а также опухолевые процессы.
Пригины расстройств ЖКТ достаточно многообразны, что позволяет объединить их в несколько групп:
Патофизиология кишечного пищеварения
495
• нарушения в питании, т.е. недоедание, переедание либо изменения качества пищи (дисбаланс белков, углеводов и жиров);
• нарушения, связанные с патологическим воздействием микробной флоры (бактерии, вирусы, простейшие, гельминты);
• радиационные поражения и действие отравляющих веществ;
• злоупотребление алкоголем и никотином;
• психоэмоциональные травмы и стрессы;
• профессиональные вредности;
• эндокринные расстройства.
Для заболеваний кишечника характерно развитие как острых осложнений (дегидратация, сепсис, кровопотеря, шок), так и хронических (нарушение питания, дефицитные состояния по витаминам, микроэлементам, белкам, жирам или закупорка ЖКТ).
Патогенетические механизмы нарушения кишечного пищеварения
• Нарушение расщепления питательных веществ:
— нарушение полостного пищеварения (дистантного);
— нарушение мембранного пищеварения (пристеночного).
• Нарушение всасывания питательных веществ.
• Нарушение выделения питательных веществ.
Нарушение расщепления питательных веществ
Полостное пищеварение
Полостное пищеварение нарушается при:
• нарушении выделения желчи;
• нарушении секреции сока поджелудочной железы;
• нарушении функции двенадцатиперстной кишки.
Полостное (дистантное) пищеварение представлено на всех участках ЖКТ, осуществляется с помощью ферментов, которые покидают клетки и на расстоянии от места образования в просвете кишечника встречают субстрат.
Нарушение выделения желчи
При заболеваниях ЖКТ может встречаться как недостаточное поступление желчи в просвет кишечника (гипохолия), так и отсутствие поступления желчи (ахолия).
Желчи выделяется приблизительно до 0,5 л/с. Стимулируют секрецию желчи гастрин, секретин, глюкагон, холецистокинин, бомбезин, вазоинтестинальный полипептид. Угнетают секрецию соматостатин, вещество Р.
496
Лекция 33
Функции желчи:
• нормализует pH, так как в составе имеет много бикарбонатов;
• активирует липазу и эмульгирует жиры;
• способствует всасыванию жирорастворимых витаминов;
• стимулирует перистальтику;
• обладает бактерицидным действием;
• образует мицеллы с жирными кислотами.
Гйпохолия и ахолия встречаются при нарушении проходимости желчных путей и расстройствах регуляции образования и выделения желчи.
Причины нарушения желчеотделения:
• закупорка желчных протоков камнем, механическое препятствие паразитами;
• рак головки поджелудочной железы, рак фатерова сосочка;
• воспаление желчного пузыря и протоков (желчнокаменная болезнь — ЖКБ), дуодениты, гепатиты;
• дискинезии желчевыводящих путей.
Последствия гипохолии и ахолии:
• уменьшение pH в двенадцатиперстной кишке;
• отсутствие активности липазы и нарушение эмульгирования жиров приводит к нарушению переваривания и всасывания жиров и жирорастворимых витаминов.
Так, нарушение всасывание витамина К приводит к снижению синтеза факторов свертывания крови, и развивается кровоточивость, а нарушение всасывания витамина А приводит к снижению сумеречного зрения:
• стеаторея (при нарушении расщепления жиров в кале появляются капли нейтрального жира более 10 г/с). Кал становится бесцветным или имеет вид белой глины. Могут наблюдаться понос, запор, метеоризм;
• нарушение переваривания белков и углеводов (жир обволакивает пищевой комок и препятствует поступлению в него протеолитических и амилолитических ферментов);
• креаторея (в кале появляются непереваренные мышечные волокна);
• усиление процессов гниения, брожения, метеоризм и аутоинтоксикация в результате снижения перистальтики и развития дисбактериоза, так как отсутствуют бактерицидные свойства желчи.
Патофизиология кишечного пищеварения
497
Нарушение секреции сока поджелудочной железы
Начинается секреция сока поджелудочной железы в условно-рефлекторную фазу пищеварения, а также в результате возникновения местных изменений в кишечнике — поступление пищи, изменение pH, растяжение кишки химусом, нервные раздражения от вагуса, которые способствуют секреции сока поджелудочной железы (до 1,5-2,0 л/с).
Стимулируют секрецию сока поджелудочной железы гастрин, холецистокинин, бомбезин, вещество Р, секретин и вазоинтестинальный полипептид. Тормозят секрецию соматостатин, панкреатический полипептид.
Синдром панкреатической ахилии связан с нарушением поступления в кишечник ферментов поджелудочной железы.
Нарушение оттока сока поджелудочной железы встречается при следующих изменениях:
• камень (общий проток с желчью), закупорка или сдавление протока поджелудочной железы извне опухолью;
• длительный спазм сфинктера Одди;
• воспаления в панкреатическом протоке (отек);
• опухолевые и дистрофические процессы поджелудочной железы;
• повышение давления в двенадцатиперстной кишке при дуодените (заброс желчи из просвета двенадцатиперстной кишки в просвет панкреатических протоков).
Панкреатит — воспаление поджелудочной железы, вызванное ее поражением собственными ферментами (самопереваривание).
Воспалительные процессы в ткани поджелудочной железы приводят к развитию панкреатита, который может быть острым и хроническим. В этиологии острого панкреатита существенное значение придают злоупотреблению алкоголем, перееданию жирной пищи, желчным камням, механическому повреждению, инфекциям (дуодениты) и интоксикациям. Любая причина вызывает повреждение ацинозных клеток. В результате происходит повышение секреции панкреатического сока и затруднение его оттока из поджелудочной железы. Развивается гипертензия в протоках поджелудочной железы и происходят забрасывание в протоки веществ, активирующих агрессивные ферменты поджелудочной железы (трипсин, химотрипсин, карбоксипептидаза, эластаза, фосфолипаза).
Ключевую роль при этом играет активация фосфолипазы А2, вызывающая разрушение мембраны клеток с освобождением из них ферментов. Следствием этого является аутолиз (самопереваривание) ткани железы, отек и жировой некроз ее отдельных участков, запуск всех плаз
498
Лекция 33
менных систем (фибринолитической, калликреин-кининовой, системы свертывания крови, системы комплемента) с последующим нарушением гемодинамики, дыхания и других систем организма. В тяжелых случаях развивается панкреатический шок, так как трипсин начинает переваривать катехоламиновые рецепторы, которые перестают отвечать на действие катехоламинов. Определенную роль в патогенезе панкреатитов играют нарушения кровоснабжения железы и иммунологические процессы.
Клиническая картина
У больных наблюдаются коллапс и опоясывающая боль, многократная рвота. Для тяжелого острого панкреатита характерно нарушение всех видов обмена: водно-электролитного обмена и КОС — наблюдается гипокалиемия, обусловленная потерей калия с рвотными массами. Снижение кальция и его задержка в очагах жирового некроза. Функциональная недостаточность сердечно-сосудистой системы, печени, почек и легких приводит к сдвигу pH в сторону ацидоза. Углеводный обмен — гипергликемия, глюкозурия. Белковый обмен — гипоальбуминемия, гиперглобулинемия. Липидный обмен — увеличение общих липидов, НЭЖК и холестерина. В крови: увеличение амилазы и липазы, нейтрофильный лейкоцитоз.
Лечение
Холод, голод и дренирование протоков поджелудочной железы. Ингибиторы сериновых протеаз - а2-макроглобулин, а,-антитрипсин, антитромбин-3, а, -антихимотрипсин.
Хронигеский панкреатит (в 70-80 % случаев причина — хронический алкоголизм на фоне избыточного употребления белка в пищу, ЖКБ). Также причинами развития являются ожирение, атеросклероз кровеносных сосудов поджелудочной железы, хронические интоксикации свинцом, ртутью, фосфором, мышьяком. К возникновению хронического панкреатита могут привести спазмы, воспалительный стеноз или опухоль, препятствующие выделению панкреатического сока в двенадцатиперстную кишку, дисфункция сфинктера Одди, органическая патология двенадцатиперстной кишки (стриктуры), врожденные аномалии желчевыводящих путей.
Один из механизмов хронического панкреатита — задержка выделения и внутриорганная активация панкреатических ферментов, постепенно осуществляющих аутолиз паренхимы поджелудочной железы, реактивное разрастание и рубцовое сморщивание соединительной ткани, которое затем приводит к склерозированию органа. Немаловажное значение имеет нарушение кровообращения в поджелудочной железе,
Патофизиология кишечного пищеварения 499
а также большое значение имеет аутоагрессия. Происходит развитие деструктивно-дегенеративных изменений в поджелудочной железе.
При хроническом панкреатите алкоголь стимулирует концентрирование сока поджелудочной железы, густой секрет делает «пробки», в которые впадает бикарбонат кальция (появляются участки обезыст-вления). Повышение концентрации белка приводит к появлению преципитатов белка, которые закупоривают протоки железы и возникает самопереваривание с последующим фиброзным перерождением. Постепенное замещение соединительной тканью приводит к функциональной недостаточности железы.
В норме в поджелудочной железе вырабатывается литостатин, который уменьшает преципитацию белка и агрегацию бикарбоната кальция. Если человек употребляет алкоголь, происходят инактивация литостатина и активация ПОЛ. Появление свободных радикалов, метаболитов алкоголя, приводит к повреждению клеток поджелудочной железы и запускает воспаление, некроз и ее фиброз.
Алкоголь вызывает спазм сфинктера Одди и увеличивает внутрипротоковое давление, что сопровождается забросом энтерокиназы в протоки поджелудочной железы. Выделение энтерокиназы приводит к активации трипсина и вызывает переваривание поджелудочной железы.
При разрушении 95 % ткани поджелудочной железы нарушается расщепление белков (гипоальбуминемия, белковые отеки), липидов и в меньшей степени углеводов.
Проявления хронического панкреатита: опоясывающая боль, отсутствие ферментов кишечника, снижение массы тела, дефицитные состояния по витаминам A, D, К, В12, В6, фолиевой кислоты, кальция, синдром мальабсорбции, проявляющийся диарей, стеатореей, креатореей, в кале — повышение креатина и зерен крахмала.
Нарушение функции двенадцатиперстной кишки
В двенадцатиперстную кишку открываются протоки многих желез, происходит гомеостазирование химуса. С помощью ферментов, входящих в состав панкреатического и кишечного соков, происходит гидролиз белков, жиров и углеводов.
В двенадцатиперстной кишке вырабатываются гормоны ЖКТ: секретин, холецистокинин, мотилин, гастроингибирующий и вазоинтестинальный полипептид.
Дуоденит — воспалительно-дистрофическое заболевание слизистой оболочки двенадцатиперстной кишки. При этом происходят структурная перестройка элементов слизистой оболочки кишки и постепенная
500
Лекция 33
атрофия железистого аппарата двенадцатиперстной кишки. Острый дуоденит встречается редко, как правило, процесс бывает хроническим. Выделяют первичный хронический дуоденит, когда воспалительный процесс в двенадцатиперстной кишке является самостоятельным заболеванием, и вторичный хронический дуоденит. В этом случае дуоденит сопровождает другие заболевания органов пищеварения (гастрит, язвенную болезнь, панкреатит, холецистит и др.).
В патогенезе в качестве главных факторов выделяют кислотнопептический фактор, гиперсекрецию протеолитических ферментов, аутоиммунные процессы, вегетативные расстройства. Характерно развитие дискинезии двенадцатиперстной кишки за счет блуждающего нерва. Пища задерживается в результате стаза, происходит растяжение кишки, что сопровождается рвотой, болью, коллапсом.
Нарушение мембранного пищеварения
Мембранное пищеварение — это особая форма фермент-субстратных взаимоотношений, происходит на стенке тонкой кишки за счет фиксированных ферментов (олигосахаридазы, олигопептидазы, фосфатазы) и осуществляет заключительные этапы пищеварения. Фермент, вырабатываемый клеткой, остается на ее мембране, а субстрат сам подходит к ферменту.
Строение слизистой оболочки тонкой кишки. Крупные ворсины и крипты покрыты энтероцитами, на которых имеются микроворсинки, от них отходят гликокаликсные нити, которые имеют поры. На порах задерживаются ферменты, позволяя им контактировать с субстратом. Гликокаликс не пропускает крупные продукты и микроорганизмы, обеспечивает стерильность пищеварения. Обладает адсорбирующими свойствами — имеются рецепторы для бактерий, АГ, лекарственных средств, витаминов.
Через поры гликокаликса проходят для мембранного пищеварения до 3 л кишечных ферментов для расщепления белков до аминокислот, дисахарид до моносахаров, жиров до моноглицеридов.
Нарушение структуры гликокаликса наблюдается при:
• активации бактериальной флоры (инфекционный агент вызывает сглаживание крипт, снижение числа ворсин);
• повышении моторной функции кишечника — субстрат не успевает встретиться с ферментом: симптом раздраженного кишечника в виде спастического колита либо в результате эмоциональных расстройств, либо нарушение нервной и гуморальной регуляции.
Патофизиология кишечного пищеварения
501
При нарушении мембранного пищеварения затрудняется транспорт и нарушается расщепление питательных веществ, необходимых для всасывания.
Причины нарушения мембранного пищеварения:
• неподготовленность химуса к мембранному пищеварению;
• нарушение структуры и ультраструктуры стенки тонкой кишки, повреждение микроворсин;
• нарушение микрофлоры кишечника;
• нарушение ферментативного слоя кишечника;
• нарушение выделительной, двигательной функции тонкой кишки;
• резекция тощей кишки.
Нарушения мембранного пищеварения бывают первичные (наследственные) и вторичные (приобретенные). Разнообразные процессы в стенке кишечника (воспалительные, дистрофические, опухолевые) приводят к снижению выделения кишечного сока и его ферментов. При недостаточности кишечных ферментов нарушается их перемещение на поверхность микроворсинок кишки.
Ферментопатии могут иметь генетический характер или быть вторичными по отношению к основному заболеванию.
Синдром недостатогности дисахаридаз — дефицит лактазы, выявляется непереносимость молока. Возникают боли в животе спазматического характера, метеоризм и растяжение живота, диарея, а также наблюдается снижение психического и физического развития.
Недостатогность лактазы относится к аутосомно-доминантному типу наследования — не расщепляется и не переваривается лактоза, происходит разложение в толстой кишке лактозы бактериями на органические кислоты: повышение осмотических веществ и образование большого количества газов (СО2, метан, водород). Наблюдаются брожение и секреция жидкости в просвет кишки — диарея. Непереносимость лактозы проявляется диареей уже после первых кормлений грудным молоком. Стул быстро нормализуется после исключения соответствующего дисахарида.
Непереносимостью лактозы страдают и взрослые люди. У них прием молока также вызывает желудочно-кишечные расстройства. Недостаточность лактазы у таких людей развивается в онтогенезе, т.е. не сразу после рождения. Дефекты генов лактазы широко распространены у восточных народов, в Центральной Азии, среди северных народностей, американских негров и индейцев.
Лактазная активность снижается с возрастом и при повышении кислотности желудочного сока.
502
Лекция 33
Недостатогность сахаразы приводит к непереносимости сахарозы. Патогенез связан с развитием мощной флоры кишечника и энтеритом. Клиническая картина та же, но проявляется позже, с момента введения в рацион ребенка сахара (сладкая вода, прикорм), т.е. когда ребенок переходит на смешанное вскармливание и молочный сахар (лактоза) заменяется сахарозой. Развиваются хроническая диарея, гипотрофия.
Также встречается недостатогность трегалозы (трегалоза содержится в водорослях, грибах, насекомых). Проявления аналогичны недостаточности лактазы.
Глютеновая болезнь (целиакия, спру) вызвана непереносимостью глютенсодержащих пищевых продуктов вследствие ферментативной недостаточности кишечного эпителия. При этом не расщепляется белковый компонент клейковины некоторых злаков — пшеницы, ржи, овса, ячменя. Глютен и продукт его неполного переваривания — глиадин являются токсическими фракциями, вызывают атрофию кишечной стенки, сглаживание микроворсинок, инициируют иммунологические реакции. Механизм — врожденное отсутствие ферментов, некоторых пептидаз, может быть аутоиммунной природы: антитела к глиадину (аденовирус 12 имеет общую структуру с глютеном). При этом развивается атрофия кишечных ворсинок, связанная с локальной иммунной реакцией. Глиадин поступает в слизистую оболочку и активирует Т-лимфоциты, продуцирующие цитокины (ФНО, ИЛ-2, IFN), возникают повреждение и атрофия ворсинок, что приводит к снижению абсорбционной поверхности и дефициту ферментов тонкой кишки.
Характеризуется развитием синдрома мальабсорбции с диареей, стеатореей, зловонным пенистым стулом с газами, формируется большой отвислый живот и снижается мышечный тонус. Симптомы ликвидируются после перехода больных на безглютеновую диету (замена хлебобулочных изделий и блюд из злаков на рис и кукурузу).
Эффективность усвоения питательных веществ в ЖКТ человека достигается тем, что полостное пищеварение сопряжено с мембранным пищеварением и всасыванием питательных веществ. Во времени и в пространстве эти процессы являются вероятностными. Их распределения частично перекрывают друг друга. Таким образом, полостное пищеварение завершается мембранным пищеварением, а переваривание сменяется всасыванием. Поэтому недостаточность мембранного и пристеночного пищеварения существенно нарушает процессы всасывания питательных веществ.
Патофизиология кишечного пищеварения
503
Нарушение всасывания питательных веществ
Возникает в результате нарушения мембранного и полостного пищеварения, т.е. нарушаются одновременно расщепление и всасывание питательных веществ. Недостаточное пищеварение в желудке приводит к нарушению всасывания белков. Недостаток панкреатических ферментов может проявиться в нарушении всасывания белков, жиров, углеводов. Недостаток желчи в кишечнике приводит к нарушению эмульгирования и всасывания жиров, жирорастворимых витаминов, кальция.
Причины нарушения всасывания:
• воспалительные процессы слизистой оболочки тонкой кишки — энтериты (инфекционные агенты: холера, брюшной тиф, дизентерия);
• токсическое поражение слизистой оболочки тонкой кишки (эндотоксины при уремии, сахарном диабете; токсические яды — свинец, мышьяк, сулема, грибы);
• иммунные поражения слизистой оболочки тонкой кишки;
• гетические дефекты слизистых оболочек;
• атрофические процессы слизистой оболочки;
• потеря части тонкой кишки, обширная резекция кишки при сосудистых заболеваниях (более 50 % кишки), что снижает компенсаторные возможности и уменьшает поверхность всасывания.
Синдром мальабсорбции, развивающийся при этом, зависит от длины и области удаленного кишечника. Резекция до 50 % тонкой кишки обычно переносится нормально, при сохранении интактности проксимального и дистального отделов кишечника. Если резекция более обширна или удалению подлежат проксимальные или дистальные сегменты, могут развиться выраженная мальабсорбция и тяжелая диарея. Так, при высокой резекции тонкой кишки наблюдаются дефицит железа, фолиевой кислоты, понос, стеаторея, мальабсорбция. Низкая резекция (дистальный отдел) способствует возникновению мальабсорбции, поноса, стеатореи, дефицита витаминов В12, A, D, Е, К и желчных кислот.
В результате снижения всасывания питательных веществ в тонкой кишке развивается синдром мальабсорбции — комплекс нарушений, включающих диарею, признаки гиповитаминоза, другие дефицитные состояния, могут быть отеки и снижение массы тела.
Так, снижение всасывания белков приводит к гипопротеинемии, гипоонкотическим отекам, отрицательному азотистому балансу, повышению аммиака в крови. Невсасывание углеводов приводит к осмо
504
Лекция 33
тической диарее. Снижение всасывания жиров приводит к стеаторее, дефициту жирорастворимых витаминов A, D, Е, К, снижению массы тела (истощение). Снижение всасывания железа, витамина В12, фолатов приводит к развитию анемии, глоссита, хейлоза, стоматита; витамина В2 — к ангулярному стоматиту, витамина Bj — к парестезиям, неврологической симптоматике; витамина D — к болям в костях, тетании, остеомаляции; витаминов К и С — к кровоточивости; витамина А — к нарушению сумеречного зрения (гемералопия), сухости кожи и роговицы, кератомаляции.
Наблюдаются обезвоживание организма и как следствие падение сосудистого тонуса, гипопротеинемия в результате нарушения гидролиза белков и нарушения всасывания аминокислот. Отмечаются признаки гиповитаминоза, стеаторея.
Нарушение выделения питательных веществ
За сутки в ЖКТ поступает 9 л жидкости; 2 л в составе пищи, 1 л слюны, 2,5 л желудочного сока и 0,5 л желчи, 1,5 л панкреатического сока и 1,5 л секрета тонкой кишки. Большая часть жидкости всасывается в тонкой кишке, причем 4-5 л в тощей и 3-4 л в подвздошной. Примерно 1 л жидкости поступает в толстую кишку, где всасывается еще 900 мл. Оставшиеся 100 мл выделяются с калом.
Основная функция толстой кишки — реабсорбция электролитов и воды. Главным фактором изменения количества жидкости и ионов в содержимом толстой кишки является реабсорбция натрия за счет его активного транспорта.
Нарушение всасывания в толстой кишке приводит к потерям воды, натрия и калия.
Регуляция работы кишечника осуществляется парасимпатическими влияниями — вагуса (сокращение и секреция) и симпатическими нервами (снижение оттока и снижение секреции), рефлекторной дугой со стенки самой кишки, биологически активными веществами (амины — изменяют перистальтику).
Работа кишечника монотонная. На вершинах ворсинок происходит всасывание, а в криптах — секреция в просвет кишки.
Основную роль в процессах всасывания и секреции играет уровень кальция в энтероцитах: чем он больше, тем выше секреция жидкости, натрия и хлора и ниже всасывание; чем меньше уровень, тем выше всасывание и ниже секреция.
Патофизиология кишечного пищеварения
505
Существуют следующие проявления нарушения выделения питательных веществ: понос и запор.
Понос (частый или жидкий стул) — результат увеличенной секреции.
Причины:
• при воспалении кишечника происходит затруднение всасывания жидкости:
• при механическом или химическом раздражении слизистой оболочки плохо измельченной, грубой пищей;
• при действии бактериальных токсинов или токсинов, содержащихся в недоброкачественной пище или образующихся в организме, которые увеличивают перистальтику, секрецию слизи, повышают проницаемость;
• нередко при психических переживаниях, чувстве страха, сопровождающихся изменением вегетативной регуляции;
• диарея может встречаться в результате снижения площади контакта химуса со стенкой кишечника (гипертиреоз, резекция);
• на фоне приема некоторых лекарственных средств (при противоопухолевой химиотерапии);
• после бега на марафонскую дистанцию;
• прием пищи после длительного периода голодания;
• смена климата и пищевых условий (диарея путешественников) и др.
В развитии поноса имеют значение затруднение всасывания жидкости из кишечника, усиление перистальтики, что приводит к быстрому продвижению пищи по кишечнику и сопровождается усилением секреции слизи, вызывая ускорение опорожнения кишечника.
Диарея опасна развитием обезвоживания, сгущением крови, общим истощением. Наблюдаются артериальная гипотензия, вплоть до коллапса, гипонатриемия, гипокалиемия, гипохлоремия, ацидоз.
Запор — ослабление перистальтики, происходит задержка опорожнения кишечника.
Причины:
• скудное питание — снижение рефлекторного раздражения стенок толстой кишки минимальным количеством содержимого — гипе-рацидные гастриты (комок в желудке активно обрабатывается желудочным соком);
• бедность пищи клетчаткой;
• недостаток в пище солей калия и кальция;
• повышенная кислотность желудочного сока;
506
Лекция 33
• снижение тонуса вагуса;
• обезвоживание (лихорадка, полиурия) вплоть до появления ко-пролитов, при сахарном диабете, микседеме — отек слизистой оболочки;
• при отравлении тяжелыми металлами (свинец);
• рубцы, спайки;
• при заболеваниях прямой кишки: трещина, геморрой приводят к застою в прямой кишке;
• ослабление тонуса мышц кишечника или их сильное сокращение.
Скопившиеся пищевые массы вызывают растяжение кишечной стенки — боль, вплоть до болевого шока; развиваются упорная рвота, обезвоживание, сгущение крови, потеря хлоридов.
Хронический запор сопровождается болями и вздутием живота, развитием дисбактериоза, нарушениями обменных процессов в организме, токсическим отравлением, нарушениями иммунитета и т.д. Вторичными, психологическими последствиями запора могут быть повышенная раздражительность, нервозность, неуверенность в себе, снижение самооценки и, как результат, частые неудачи в личной и деловой жизни.
При застое кишечного содержимого может быть отравление продуктами белкового обмена. Происходят их гниение и брожение, токсины поступают в кровь и развивается кишечная аутоинтоксикация сероводородом, скатолом, путресцином, кадаверином. Развивается мереотизм.
Дисбактериоз кишечника
Макроорганизм и населяющая его микрофлора, в том числе кишечник, являются сбалансированной экологической системой.
Микрофлора кишечника подразделяется на две части: облигатную (микроорганизмы, постоянно входящие в состав нормальной флоры, которые играют важную роль в метаболических процессах и защите организма хозяина от инфекции) и факультативную (бактерии, часто встречающиеся у здоровых людей, но являющиеся условно-патогенными в случае снижения резистентности макроорганизма).
В случае ослабления иммунной системы происходят нарушения и в микрофлоре кишечника. Представители нормальной микрофлоры (молочнокислые и кишечные палочки, бифидобактерии и др.) сокращаются в количестве, а количество микроорганизмов (стафилококк, синегнойная палочка, кандида и т.д.) увеличивается.
В тонкой кишке начинают доминировать эшерихия коли (Е. coli), клебсиеллы, лактобациллы, энтерококки.
Патофизиология кишечного пищеварения
507
В толстой кишке уменьшаются или исчезают бифидобактерии и относительно возрастает количество Е. coli, клебсиелл, стрептококков, стафилококков, протея, дрожжевых грибов.
Дисбактериоз кишегника — клинико-лабораторный синдром, характеризующийся количественными и качественными изменениями в составе нормальной кишечной микрофлоры и изменением ее мест обитания.
Физиологическая роль кишечной микрофлоры
• Кишечные палочки, энтерококки, бифидобактерии и ацидофильные палочки обладают выраженными антагонистическими свойствами.
• Кишечная микрофлора участвует в формировании иммунитета. Наличие микрофлоры оказывает постоянный антигенный тренирующий эффект.
• Питание и регенерация кишечной стенки.
• Регуляция функций ЖКТ. Продукты жизнедеятельности бифидо-и лактобактерий усиливают всасывание кальция, железа, витаминов D и С.
• Облигатная микрофлора толстой кишки в норме обеспечивает конечный гидролиз белков, омыление жиров, сбраживание углеводов, которые не всосались в тонкой кишке.
• Некоторые поступающие с пищей вещества могут метаболизироваться только кишечной микрофлорой. Так, амилолитическая микрофлора расщепляет целлюлозу.
• Под влиянием ферментов нормальной микрофлоры в подвздошной кишке осуществляется деконъюгация желчных кислот.
• Детоксикационная и антиканцерогенная функция. Способность нейтрализовать многие токсические субстраты и метаболиты (нитраты, ксенобиотики, гистамин, стероиды).
• Синтетическая функция. Синтез витаминов группы В (Вг В2, В6, В12), С, К, РР, фолиевой кислоты. Синтез гормонов и биологически активных веществ, принимающих участие в регуляции функций не только ЖКТ, но и печени, сердечно-сосудистой, нервной, эндокринной систем, кроветворении и др.
• Регуляция обмена холестерина, оксалатов.
Дисбактериоз всегда вторичен, т.е. является не самостоятельным заболеванием, а осложнением других патологических состояний.
Этиология. Развитию дисбактериоза благоприятствуют ферментативная недостаточность, резкое изменение привычного образа жизни,
508
Лекция 33
голодание, авитаминоз, истощение организма в связи со злокачественными образованиями, хирургическими операциями в брюшной полости, ожоговой болезнью, лучевое воздействие, активная терапия гормонами, аллергизация, применение химиопрепаратов, антибиотиков, иммунодефициты (при лейкозах, СПИДе), злоупотребление алкоголем, нарушения питания.
Патогенез. Увеличение количества токсических веществ в кишечнике приводит к нарушению всасывания питательныых веществ, изменяется pH в кислую сторону, угнетается функция пищеварительных ферментов, происходит преждевременная деконъюгация желчных кислот (разрушение). Появляется стеаторея, которая связана с уменьшением в просвете кишки конъюгированных желчных кислот, обеспечивающих эмульгирование жиров и активацию панкреатической липазы. Нарушается всасывание жирорастворимых витаминов A, D, Е и К.
Бактериальные токсины и протеазы могут связывать витамин В)2.
Избыточная микробная флора приводит к повреждению эпителия тонкой кишки, так как метаболиты некоторых микроорганизмов обладают цитотоксическим действием. Наблюдаются уменьшение высоты ворсинок, углубление крипт, дегенерация микроворсинок, митохондрий и эндоплазматической сети.
Конкуренты за питательные вещества выделяют токсины и свои метаболиты, перегружают печень и почки, снижают их функции, ухудшают регенерацию кишечного эпителия, повышают проницаемость кишечной стенки, изменяют перистальтику.
Клинические проявления дисбактериоза
Местные проявления:
• диспептический синдром, который всегда сопровождает изменения кишечного биоценоза. Типичные проявления — нарушение стула (диарея или, реже, — запор), вздутие живота, урчание:
• болевой синдром, который обусловлен поражением тонкой и толстой кишки.
Системные проявления крайне разнообразны и нередко выходят на первый план, скрывая истинную причину заболевания. Практически нет ни одного органа, который не будет вовлечен в патологический процесс. Все они обусловлены мальабсорбцией, мальдигестией, синдромом раздражения толстой кишки, иммунными нарушениями либо транслокацией кишечных микроорганизмов.
Лекция 34
ПАТОФИЗИОЛОГИЯ ЯЗВЕННОЙ БОЛЕЗНИ ЖЕЛУДКА И ДВЕНАДЦАТИПЕРСТНОЙ КИШКИ
Ж.М. Салмаси
На сегодняшний день язвенная болезнь (ЯБ) желудка и двенадцатиперстной кишки является одним из наиболее часто встречающихся заболеваний внутренних органов у человека. По данным мировой статистики, распространенность ЯБ среди взрослых составляет 6-10 %. Только в США ежегодно регистрируется 350 000 свежих случаев заболевания, 100 000 больных с ЯБ подвергается оперативному лечению, а 6000 человек умирают от осложнений ЯБ.
Язвенная болезнь — это хроническое заболевание с циклическим, рецидивирующим течением, характеризующееся возникновением язвенного дефекта в слизистой оболочке желудка или в луковице двенадцатиперстной кишки.
ЯБ следует отличать от возникающих вторично, как осложнение основного заболевания, или от симптоматических гастродуоденальных язв вследствие прямого повреждающего действия на стенку желудка. При излечении основного заболевания или прекращении действия повреждающего фактора происходит спонтанное рубцевание симптоматических язв. Примерами симптоматических гастродуоденальных язв являются:
• стресс-язвы, возникающие после тяжелых операций, травм, ожогов, сепсиса, психической травмы и т.п.;
• лекарственные язвы, развивающиеся на фоне длительного приема глюкокортикостероидных препаратов, противоспалительных средств (ацетилсалициловой кислоты, индометацина и др.), резерпина и некоторых других лекарств;
• язвы, возникающие при попадании в желудок высокоагрессивных веществ, например концентрированных кислот и щелочей;
• язвы, сопутствующие заболеваниям с резким нарушением микроциркуляции в тканях, таким как атеросклероз, ревматизм, сахарный
510
Лекция 34
диабет, лейкозы, хронические заболевания печени, почек, поджелудочной железы и др.
Механизм развития язвенного дефекта при симптоматических гастродуоденальных язвах связан с основным заболеванием.
Этиология и патогенез ЯБ значительно сложнее, патогенез ЯБ включают в себя много звеньев. Расширение представления врачей о патогенезе ЯБ сопровождалось появлением соответствующих теорий ульцерогенеза (ульцерогенез — механизм образования язвы, от лат. Ulcus — язва).
Теории ульцерогенеза
1. Механическая теория Ашоффа объясняла механизм формирования язв травматизацией слизистой оболочки желудка и двенадцатиперстной кишки пищевыми продуктами, обращая внимание на то, что язвы часто локализуются на малой кривизне желудка (пищевой дорожке). Действительно, в эксперименте грубая пища вызывает повреждение слизистой оболочки желудка и может вызвать развитие гастрита, однако не приводит к формированию язвенной болезни.
2. Воспалительная теория, согласно которой язва развивается на месте воспаления и некроза, например, на фоне гастрита. Действительно, хронический гастрит предшествует ЯБ у многих больных, однако далеко не у всех.
3. Сосудистая теория Вирхова. С точки зрения этой теории, причиной развития язвенного дефекта является нарушение периферического кровообращения, сопровождающееся ишемией и некрозом, что хорошо объясняет механизм формирования симптоматических язв (при атеросклерозе, коллагенозах, сахарном диабете и др.). Однако морфологические исследования не подтвердили наличия грубых расстройств микроциркуляции у больных с ЯБ.
4. Согласно пептической теории Квинке язва является результатом самопереваривания слизистой оболочки при желудочной гиперсекреции. Действительно, у большинства больных ЯБ двенадцатиперстной кишки кислотность и переваривающая способность желудочного сока повышены. Однако у больных ЯБ желудка кислотность может быть нормальной или сниженной. Кроме того, у многих людей на фоне гиперсекреции и гиперхлоргидрии язвенная болезнь не развивается.
5. Согласно кортиковисцеральной теории Быкова-Курцина, язвенная болезнь является органным проявлением коркового невроза, развивающегося в результате срыва равновесия между процессами возбуждения и
Патофизиология язвенной болезни желудка и двенадцатиперстной кишки 511
торможения в ЦНС при перенапряжении одного из них (в том числе при стрессах). При этом нарушается центральная регуляция секреторной и моторной функции желудка и кишечника. Однако известно, что ЯБ формируется только у части людей с неврозами, и далеко не у всех больных ЯБ невротизация предшествовала заболеванию.
6. Инфекционная теория ЯБ развивается с 1982 г., когда были открыты микроорганизмы Helycobacter pylori, о роли которых в патогенезе ЯБ и хронического гастрита речь пойдет ниже. Однако сразу следует отметить, что инфицированность населения Н. pylory многократно превышает заболеваемость ЯБ, кроме того, существуют значительные ножницы в региональном распространении хеликобактериоза и ЯБ.
Современная концепция патогенеза ЯБ. По современным представлениям, ЯБ обусловлена нарушением равновесия между факторами, обеспечивающими сохранность слизистой оболочки желудка и двенадцатиперстной кишки (защитными), и факторами, способствующими ее повреждению (агрессивными).
Защитные факторы слизистой оболочки желудка и двенадцатиперстной кишки:
• выработка слизи, покрывающей поверхность желудка;
• способность эпителия слизистой оболочки желудка и двенадцатиперстной кишки к регенерации;
• нейроэндокринная регуляция желудочной секреции;
• секреция бикарбонатов.
Ульцерогенные (агрессивные) факторы:
• внутренние ульцерогенные факторы:
— кислотно-пептический фактор;
— нарушение моторно-эвакуаторной функции (дисмоторика);
— нарушение нейроэндокринной регуляции организма;
• внешние ульцерогенные факторы. К внешним ульцерогенным факторам можно отнести все факторы внешней среды, вызывающие повреждение слизистой оболочки желудка и двенадцатиперстной кишки, однако особое значение имеют следующие:
— алиментарные факторы;
- микроорганизмы;
— лекарственные вещества;
— вредные привычки;
— стрессорные воздействия.
Определенную роль в развитии ульцерогенеза у человека играет наследственная предрасположенность.
512
Лекция 34
Нарушение работы защитных факторов слизистой оболочки
Нарушение образования слизи. Желудочная слизь в норме представляет вязкую, клейкую пленку толщиной 1-1,5 мм с коллоидными тяжами, связывающими ее с эпителиальными клетками. Мукоидные вещества, щелочные компоненты и физические свойства слизи охраняют эпителий слизистой оболочки от действия соляной кислоты и пепсина, от механического и химического повреждения. Слизь, состоящая из гликопротеинов и протеогликанов, в норме не подвержена пептическому перевариванию. Принципиально важно, что слизь обладает селективной (избирательной) проницаемостью для ионов и является барьером для ионов Н\ Бикарбонаты слизи позволяют удерживать pH на поверхности эпителиальных клеток на уровне 7,0-7,4 при pH в просвете желудка 1,4-2,0. Кроме того, водонерастворимый гель слизи действует как фильтр, не пропуская к эпителию крупномолекулярные вещества, в том числе пепсин. Слизь — это первый иммунный барьер. В ней также происходит домембранное пристеночное пищеварение. При нарушении слизеобразования наблюдается снижение содержания в желудочном соке гликопротеинов, в первую очередь N-ацетилнейраминовой кислоты (NANA).
При ЯБ желудка наблюдается как снижение синтеза слизи, так и изменение ее состава. За счет снижения содержания гликопротеинов в желудочной слизи снижаются барьерные свойства слизи, что резко облегчает обратную диффузию Н+. Существует предположение, что у больных ЯБ желудка имеется генетически детерминированное угнетение синтеза гликопротеинов.
Слизеобразование в желудке тормозится под влиянием ингибиторов синтеза простагландинов и глюкортикоидов.
Увеличивают продукцию слизи серотонин, соматостатин, бомбезин, эндогенные опиоиды, простагландины Е и F. Способствуют слизеобра-зованию в желудке адрено- и холиномиметики, механическое растяжение желудка.
Нарушение способности слизистой оболочки желудка к регенерации
В норме слизистая оболочка желудка хорошо регенерирует, полное обновление ее происходит в среднем за 5 сут. После биопсии слизистой оболочки ее дефект заживает в течение 3-4 сут. У животных в эксперименте после удаления 70-80 % площади слизистой оболочки желудка практически полное ее восстановление происходило за 11 нед.
Патофизиология язвенной болезни желудка и двенадцатиперстной кишки 513
Регенерация эпителия слизистой обологки замедляется: при нарушении нейроэндокринной регуляции, в частности под влиянием глюкокортикоидных гормонов и ингибиторов синтеза простагландинов, при дефиците пластических веществ (голодание, гиповитаминозы), действии вредных веществ (алкоголь).
Важнейшую роль в нарушении регенерации желудочного эпителия и образования слизи играет нарушение микроциркуляции в желудочной стенке. В эксперименте нарушение кровоснабжения желудка приводит к формированию язвенных дефектов стенки желудка у экспериментальных животных. У человека развитие язвы желудка преимущественно за счет нарушения микроциркуляции может наблюдаться при развитии ЯБ на фоне сахарного диабета, гипертонической болезни, облитерирующих заболеваний сосудов.
Следует подчеркнуть, что нарушение регенерации слизистой оболочки в сочетании с нарушениями слизеобразования рассматриваются как главные патогенетические факторы развития ЯБ желудка у больных с нормальной или пониженной секрецией, а атрофический гастрит у больных с гиперсекрецией НС1 и пепсина — как предъязвенное состояние.
Нарушения нейроэндокринной регуляции желудочной секреции
У здорового человека важную роль в регуляции работы желудка играет антродуоденальное торможение. В норме, когда pH в антральном отделе желудка снижается до 2,0-2,5, происходит торможение выделения гастрина из G-клеток и, следовательно, по принципу обратной связи прекращается выделение НС1 из обкладочных клеток. Выделение НС1 прекращается также в том случае, когда pH в двенадцатиперстной кишке снижается до 4,0 и ниже. Если же нарушается нервная или гуморальная регуляция, то торможение образования НС1 будет происходить при более низких величинах pH, что способствует повреждению слизистой оболочки.
Значительные изменения секреторной функции желудка могут наблюдаться при нарушениях функции вегетативной нервной системы, которые, в свою очередь, часто сопутствуют неврозам. Возникающая при вегетоневрозе активация парасимпатической нервной системы сопровождается усиленным выделением ацетилхолина из пресинаптиче-ских терминалей и приводит к повышению секреции гастрина и в итоге к гиперпродукции соляной кислоты в желудке. А чрезмерная активация симпатической нервной системы вызывает спазм артериол желудочной стенки и ухудшение кровоснабжения слизистой оболочки желудка, со всеми вытекающими последствиями.
514
Лекция 34
Существенную роль в патогенезе ЯБ у отдельных пациентов может играть снижение образования D-клетками слизистой оболочки антрального отдела желудка гормона соматостатина. У здорового человека соматостатин регулирует выделение желудочного сока, тормозя секрецию соляной кислоты обкладочными клетками.
Важнейшую роль в патогенезе развития гастродуоденальных язв играет нарушение синтеза простагландинов в желудочной стенке. У здоровых людей простагландины Е и F синтезируются поверхностноямочным эпителием желудка. Их противоязвенный эффект обусловлен несколькими механизмами, они:
• способны значительно подавлять секрецию НС1;
• обладают цитопротекторным действием:
• обеспечивают стимуляцию слизеобразования, усиление синтеза гликопротеинов, повышение гидрофобности слизи;
• усиливают кровообращение:
• активируют секрецию бикарбонатов.
Во время секреции НС1 их количество в желудочном соке увеличивается в 2-3 раза. Уменьшают образование простагландинов в желудке ненаркотические анальгетики (ацетилсалициловая кислота, индометацин и др.). В связи с выраженной противоязвеннной активностью препараты простагландинов предложены к применению в клинике.
Эстрогены, секретин и некоторые другие гормоны также усиливают защитные свойства слизистой оболочки и тормозят ульцерогенез.
Секреция бикарбонатов
Помимо соляной кислоты в состав желудочного сока входят и бикарбонаты, вырабатываемые поверхностными эпителиальными клетками слизистой оболочки желудка. Бикарбонаты желудочного сока участвуют в стабилизации слизистого геля, поддерживают градиент pH между эпителием желудка и его просветом, нейтрализуют избыток соляной кислоты.
Стимуляторами образования бикарбонатов являются ацетилхолин, простагландины и некоторые гормоны ЖКТ, а ингибируется синтез бикарбонатов а-адреномиметиками, ингибиторами синтеза ПГ и этанолом.
Действие ульцерогенных факторов
Внешние ульцерогенные факторы
Наиболее важные внешние ульцерогенные факторы:
• микроорганизмы;
• алиментарные факторы;
Патофизиология язвенной болезни желудка и двенадцатиперстной кишки 515
• лекарственные вещества;
• вредные привычки и стрессорные воздействия.
Роль микроорганизмов
В 1982 г. Б. Маршалл и И. Уоррен выделили и описали паразитирующие у человека в пилорическом отделе желудка спиралевидные грамотрицательные микробы Helicobacter pylori. Есть мнение, что Н. pylori инфицировано около 25 % взрослого населения, но у половины из них инфицирование протекает бессимптомно (бактерионосительство). Эта бактерия обнаруживается у 50 % людей, страдающих диспепсией (без язвы), у 84 % больных хроническим гастритом, у 70-100 % больных язвенной болезнью желудка, у 90-93 % больных язвенной болезнью двенадцатиперстной кишки.
Свойства Н. pylori
Эти микроорганизмы живут под слизью, но над слизистой оболочкой. Они обладают уреазной активностью — расщепляют мочевину. При этом ионы NH4* связывают ионы Н‘, микробы окружаются облачком нейтрального аммония, что позволяет им сохранять жизнеспособность в очень кислой среде (даже при pH = 2). Расщепление мочевины в желудке может быть причиной неприятного запаха изо рта у больных. Присутствие уреазы в желудочном соке больного — специфический признак инфицирования этими микробами. Н. pylori обладают высокой фосфатазной и каталазной активностью, разрушают Н2О2, что обеспечивает им защиту от фагоцитов, вырабатывают протеолитические ферменты, разрушающие полимеры слизи. В результате ионы Н’ и пепсин могут проникать в слизистую оболочку. Н. pylori оказывают выраженный цитотоксический эффект против эпителиальных клеток — повреждают их мембраны, вызывают грубую структурную перестройку. Наконец, эти бактерии могут быть причиной развития аутоиммунных процессов в слизистой оболочке желудка. Предполагают, что Н. pylori, благодаря своей способности выделять в больших количествах СО2, могут быть причиной метеоризма, вздутия живота, нарушения моторики желудка, изжоги и т.п. Доказана возможнссть инфицирования Н. pylori. Как правило, в семьях наблюдается инфицированность одним и тем же штаммом всех членов семьи.
На сегодняшний день большинство гастроэнтерологов считают, что хроническое инфицирование Н. pylori в сочетании с анацидным состоянием является фактором риска для развития рака желудка, в сочетании с гипоацидным состоянием — диспептического синдрома без язвы. Если при хроническом инфицировании секреция НС1 восстанав
516
Лекция 34
ливается почти до нормы, то может развиться ЯБ желудка, а на фоне гиперацидного состояния очень велик риск развития ЯБ двенадцатиперстной кишки.
Алиментарные факторы
Возникновению ЯБ могут способствовать: голодная гиперсекреция при нерегулярном питании, избыток в рационе продуктов, вызывающих гиперсекрецию желудочного сока (мясо, кофе, сухое вино, пиво, сильногазированные напитки) или, наоборот, дефицит белка (когда уменьшается нейтрализация НС1 и пепсина); содержание в пище раздражающих веществ в большом количестве (перец, уксус и т.п.). В эксперименте можно получить у крыс эрозии и язвы слизистой оболочки желудка, если их содержать на рационе с большим количеством подобных раздражающих веществ. В Грузии, где в пищу употребляют много острых приправ, ЯБ встречается в 2,5 раза чаще, чем в России. Этим же объясняется большое число больных ЯБ в некоторых регионах Индии, Индонезии, Африки.
Лекарственные препараты
Привести к образованию язв в желудке могут все препараты, которые подавляют синтез простагландинов, такие как индометацин, бутадион и другие противовоспалительные средства. Наиболее выражен ульцероген-ный эффекту салицилатов, сочетающих торможение синтеза простагландина (ПГ) с прямым раздражающим действием на слизистую оболочку желудка. Мощным ульцерогенным действием обладают глюкокортикоиды, которые одновременно уменьшают образование слизи, угнетают регенерацию, вызывают гиперсекрецию НС1 и пепсина, нарушают моторику желудка и двенадцатиперстной кишки. Вызвать образование язв желудка могут катехоламины, вызывающие нарушение микроциркуляции и ишемическое повреждение эпителия желудка, и резерпин, резко усиливающий продукцию гистамина и секрецию НС1. Медикаментозные язвы, как правило, являются вторичными симптоматическими язвами и исчезают после отмены вызвавших их препаратов. Однако, по данным американских авторов, примерно 1/3 часть хронических, длительно не рубцующихся язв желудка и duodenum связаны с систематическим приемом препаратов из группы ненаркотических анальгетиков (аспирин, анальгин, патентованные препараты против головной боли и т.п.).
Вредные привычки
Курение. 30 % больных ЯБ — заядлые курильщики. Под влиянием никотина сужаются сосуды, нарушается микроциркуляция в слизистой оболочке желудка, ухудшается регенерация ее эпителия, повышается
Патофизиология язвенной болезни желудка и двенадцатиперстной кишки 517 секреция НС1 и пепсина, нарушается моторика, угнетается функция поджелудочной железы. Алкоголь вызывает нарушение нервной и гуморальной регуляции секреторной и моторной функции ЖКТ, усиливает выделение гастрина (особенно пиво) и НС1 (белое вино), при постоянном употреблении вызывает атрофию слизистой оболочки желудка, нарушает слизеобразование, выделение желудочных бикарбонатов.
Стресс
Стресс может быть причиной развития вторичной симптоматической язвы, не переходящей в хроническое заболевание. Механизм развития стрессовых язв связан в первую очередь с повышением концентрации глюкокортикоидов в крови, а также с гиперкатехоламинемией и нарушением регуляции системы эндогенных опиоидов. Стресс-язвы воспроизводятся и в эксперименте на животных. Однако многие механизмы стресса могут быть привлечены и к объяснению патогенеза ЯБ (о чем будет сказано ниже). К тому же статистика показывает, что число больных ЯБ резко возрастало в годы войны и послевоенные годы, их значительно больше среди лиц, профессии которых сопряжены с определенными психоэмоциональными перегрузками, умственным перенапряжением, риском и т.п.
Внутренние ульцерогенные факторы
Кислотно-пептический фактор
Язвенной болезни (особенно двенадцатиперстной кишки) часто предшествует и сопутствует гиперпродукция НС1 и пепсина. У этих больных обнаруживается гиперплазия обкладочных (отвечающих за синтез НС1) и главных (синтез пепсина) клеток слизистой оболочки желудка. Естественным стимулятором обкладочных клеток является гистамин, через него также опосредуются эффекты ацетилхолина и гастрина.
Гистамин, который в физиологических условиях выделяется энте-рохромафинноподобными клетками и частично тучными, действует на Н2-гистаминовые рецепторы обкладочных клеток. В условиях патологии количество гистамина может быть резко повышенно (в основном из тучных клеток), в результате чего НС1 выделяется в больших количествах. Кроме того, гистамин может действовать и как медиатор воспаления (повышать проницаемость сосудов и т.п.).
Гастрин в физиологических условиях, с одной стороны, стимулирует желудочную секрецию (НС1 + пепсин), с другой, улучшает регенерацию эпителия и усиливает слизеобразования. Тем не менее избыточная продукция гастрина способствует возникновению язвенных дефектов
518
Лекция 34
слизистой оболочки желудка. Наиболее ярко ульцерогенный эффект гастрина проявляется при синдроме Золлингера-Эллисона. Это заболевание возникает вследствие выраженной гипергастринемии, чаще всего связанной с гастринпродуцирующей опухолью (гастриномой) в желудке или поджелудочной железе. В результате при синдроме Золлингера-Эллисона возникает выраженная гиперпродукция НС1, приводящая к возникновению множественных, постоянно рецидивирующих язв.
Пепсин. Считают, что пепсин лишь закрепляет повреждающее действие НС1 (точнее Н-ионов). Установлено также, что при избытке НС1 возрастает ферментативная активность пепсина. Кроме того, у части больных с ЯБ наблюдается продукция пепсина с очень высокой протеолитической активностью. Так, если у здоровых людей коэффициент агрессивности желудочного сока, отражающий активность пепсина, равен 0,3, то у больных ЯБ он составляет около 1 (еще один фактор наследственной предрасположенности).
Стимулируют желудочную секрецию: повышение тонуса вагуса, глюкокортикоиды, гастрин, гистамин, паратгормон, бомбезин, энкефалины, холецистокинин-панкреозимин и др.
Угнетают желудочную секрецию: минералокортикоиды, секретин, глюкагон, кальцитонин, соматостатин, люлиберин, ГИП (гастроингибирующий пептид), ВИП, бульбогастрон, энтерогастрон и др.
Нарушение моторики желудка и двенадцатиперстной кишки
Развитию язвенной болезни двенадцатиперстной кишки способствуют усиление моторики желудка, сопровождающееся преждевременной эвакуацией пищевой кашицы в двенадцатиперстную кишку, когда НС1 еще недостаточно нейтрализована пищей; ослабление моторики двенадцатиперстной кишки и дуоденостаз (кислое желудочное содержимое долго задерживается в двенадцатиперстной кишке).
Напротив, ослабление моторики желудка, застой пищевых масс в желудке вследствие пилороспазма (длительный спазм привратника может быть связан с медленной нейтрализацией кислой реакции пищевой кашицы в двенадцатиперстной кишке) или стеноза привратника способствуют длительному контакту слизистой оболочки желудка с агрессивным содержимым и формированию язв желудка.
Особое значение в патогенезе ЯБ имеет дуоденогастралъный рефлюкс — антиперистальтика и заброс в желудок дуоденального содержимого, включая желчь и панкреатический сок. Этот рефлюкс наблюдается у 90 % злостных курильщиков.
Патофизиология язвенной болезни желудка и двенадцатиперстной кишки 519
Последствия дуоденогастрального рефлюкса связаны с тем, что ЖК и панкреатические ферменты дуоденального содержимого:
• вызывают дегенерацию желудочной слизи и облегчают агрессивное действие на слизистую оболочку НС1 и пепсина;
• повреждает клеточные мембраны эпителиальных клеток вплоть до их разрушения;
• тормозят регенерацию слизистой оболочки, способствуют возникновению ее атрофии;
• вызывают развитие стойкой воспалительной реакции, в том числе за счет дегрануляции тучных клеток; в результате развивается антральный гастрит с дисплазией и желудочного эпителия (эпителий желудочного типа замещается на кишечный).
Дуоденогастральный рефлюкс представляет серьезный фактор риска для формирования ЯБ желудка, хронического атрофического гастрита, рака желудка.
Нарушения нервной н эндокринной регуляции
У больных ЯБ выявляются изменения психики (различные невротические синдромы), вегетоневроз (у 76 % больных), вегетососудистая дистония (в 15 %). Эти нарушения могут предшествовать развитию ЯБ и участвовать в патогенезе, но могут быть и вторичными по отношению к ней.
Язву желудка и двенадцатиперстной кишки в эксперименте можно воспроизвести у животных путем раздражения ядер гипоталамуса, парасимпатичнских нервов (на любом уровне). При повышении тонуса вагуса усиливается секреция НС1 и пепсина, как в результате непосредственной стимуляции главных и париетальных клеток, так и через усиленную продукцию гастрина и гистамина; также уменьшается образование слизи и нарушается моторика. В то же время ваготомия в эксперименте предупреждает развитие язвы. В настоящее время разрабатываются различные модификации операции ваготомии у больных ЯБ (органосберегающие операции).
Однако язва воспроизводится в эксперименте на животных и другим путем — при повышении тонуса симпатических нервов (раздражением постганглионарных симпатических нервов солнечного сплетения, симпатических центров гипоталамуса, просто введением кетахоламинов в больших дозах). Адреноблокаторы предупреждают развитие язвы. При высокой концентрации катехоламинов в стенке желудка ослабляется кровоток и тормозится регенерация, могут возникнуть ишемия и некроз. Таким образом, срыв нервной регуляции секреторной и моторной
520
Лекция 34
функции желудка в любую сторону (вегетоневроз) может способствовать развитию ЯБ.
Наследственная предрасположенность
Наследственная предрасположенность может играть существенную роль в развитии ЯБ. По данным некоторых авторов, наследственность отягощена у 40 % больных. Факторы наследственной предрасположенности существенно различаются.
Наибольшее значение имеет наличие ЯБ у ближайших родственников. Из фенотипических признаков предрасположенности к ЯБ следует указать наличие I группы крови, что связано с нарушением синтеза гликопротеинов желудочной слизи. У лиц с I группой крови риск заболеть ЯБ на 35 % больше, чем у лиц с другими группами крови.
Среди фенотипических признаков, характерных для больных ЯБ, отмечаются светлые волосы, голубые глаза, промежуточный тип стопы (1 и 2 пальцы равны по длине), радиальный тип кисти (указательный палец почти равен среднему), уменьшение окружности плеча. Что же при этом наследуется? Наследуются морфологические и функциональные особенности ЖКТ, количество обкладочных и других секреторных клеток, их реактивность, качественный состав вырабатываемой защитной слизи, особенности центральной и местной регуляции секреторной и моторной функции ЖКТ.
В настоящее время выделяют 7 морфофункциональных типов желудочной секреции:
• нормальный (с нормальной секрецией НС1 и пепсина);
• гиперхлоргидрический (с гиперсекрецией НС1, пепсин в норме);
• гиперпепсиногеновый (с гиперсекрецией пепсина, НС1 в норме);
• пилорический (гиперхлоргидрическо-пепсиногеновый) — с гиперсекрецией НС1 и пепсина;
• гипохлоргидрический (с гипосекрецией НС1, пепсин в норме);
• гипопепсиногеновый (с гипосекрецией пепсина, НС1 в норме);
• ахилический (с резко сниженной или отсутствием секреции НС1 и пепсина).
Принципы консервативного лечения язвенной болезни
У подавляющего большинства больных с язвенной болезнью пилорической части желудка и двенадцатиперстной кишки наблюдается гиперсекреция соляной кислоты и пепсина, что и определяет тактику консервативного лечения этих больных.
Патофизиология язвенной болезни желудка и двенадцатиперстной кишки 521
Диета
Больным ЯБ важно соблюдать режим питания, желателен частый дробный прием пищи (каждые 3-4 ч).
Из пищевого рациона больных исключаются блюда и продукты, раздражающие слизистую оболочку желудка и двенадцатиперстной кишки и стимулирующие секрецию соляной кислоты (концентрированные бульоны, жареные блюда, грубая растительная клетчатка, специи, копчености, маринады, алкогольные и газированные напитки, крепкие чай и кофе).
Фармакологические средства, применяемые при язвенной болезни
• Препараты, подавляющие кислотно-пептическую активность желудочного сока:
— блокаторы Н2-гистаминовых рецепторов (циметидин, ранитидин, фамотидин);
— блокаторы мускариновых рецепторов (холиноблокаторы — атропин, платифиллин); оптимальное использование селективных блокаторов Mj-холинорецепторов (гастроцепин);
— блокаторы протонного насоса (Н+/К*-АТФазы) (омепразол);
— антацидные препараты, нейтрализующие НС1 (алмагель, маа-локсидр.).
• Антихеликобактерные средства:
— препараты висмута (де-нол);
— метронидазол (трихопол);
— антибиотики (амоксициллин, оксациллин, доксициллин, тетрациклин).
На сегодняшний день для борьбы с инфекцией Н. pylori чаще всего используют схему лечения с использованием одновременно 3-х или в крайнем случае 2-х антибактериальных препаратов.
• Протективные средства:
— препараты простагландина Е (сайтотек);
— препараты коллоидного висмута (де-нол, трибемол);
— сульфатированные полисахариды (сукральфат, вентер).
Препараты ПГ значительно подавляют желудочное кислотообра-зование, ускоряют процессы регенерации эпителия, усиливают синтез желудочной слизи. Препараты висмута и сукральфат, взаимодействуя с белками слизи, образуют защитную пленку, препятствующую обратной диффузии ионов Н*.
522
Лекция 34
Болезни оперированного желудка
После резекции желудка у многих больных развиваются тяжелые осложнения и одно из самых тяжелых — демпинг-синдром (ДС). Через 10-30 мин после приема пищи (особенно молока, легко всасываемых углеводов, горячей пищи или большого объема пищи) у больных, прооперированных по Бильрот-I или Бильрот-П, развивается приступ общей слабости, сопровождающийся сердцебиением, повышением АД, болями в сердце, головокружением, приливом жара к лицу, усиленным потоотделением, сонливостью, урчанием и схваткообразными болями в животе, профузным поносом. Патогенез этого синдрома до конца не ясен. Большинство исследователей считает, что ДС — результат быстрой эвакуации («сброса») гиперосмотического желудочного содержимого из культи желудка в тонкую кишку. Присходит раздражение ее рецепторов, активация симпатоадреналовой и ККС, выброс большого количества серотонина (до 80 % эндогенного серотонина содержится в энтерохромаффинных клетках ЖКТ). Возрастают образование брадикинина и содержание в крови адреналина. Расширенные мезентериальные сосуды переполняются кровью, вследствие повышения их проницаемости резко усиливается транссудация жидкости в просвет кишечника, что ведет к ускорению перистальтики и развитию профузного поноса. ОЦК резко уменьшается, возникает ишемия головного мозга и сердца (головокружение, боли в сердце и т.п.). Адреналин усиливает спазм сосудов (АД повышается) и активирует гликогенолиз (повышается содержание сахара в крови). Любопытно, что при ДС углеводы, находясь еще в ротовой полости, рефлекторно активируют распад гликогена и вызывают гипергликемию.
При длительно существующем демпинг-синдроме у больных развиваются анемия, гиповитаминоз, угнетается функция поджелудочной железы (серотонин ингибирует секретин), гипергликемия вызывает недостаточность инсулярного аппарата, нарушается функция печени. Принципы лечения ДС: ограничение углеводов, питание малыми порциями, седативные средства, симптоматическое лечение.
Лекция 35
ПАТОФИЗИОЛОГИЯ ПЕЧЕНИ
Н.И. Бережнова
Пегень — жизненно важный орган, участвующий в поддержании гомеостаза в организме. Основа структуры печени — долька, формирующаяся из гепатоцитов. Число гепатоцитов составляет более 300 миллиардов! При этом каждый гепатоцит представляет собой химическую лабораторию, в которой происходит около тысячи различных реакций.
Функции печени
Метаболигеская функция. В печени осуществляется синтез аминокислот и важнейших белков: всех альбуминов и некоторых глобулинов крови, почти всех факторов свертывания крови, транспортных белков — трансферрина, транскортина и других, а также тирозина и холинэстеразы. В ней идет синтез белков острой фазы (БОФ): С-реактивного белка, церулоплазмина, гаптоглобина, антипротеаз cq-антитрипсина и антихемотрипсина. Не менее интенсивно происходят в печени и распад белков, а также обезвреживание конечного продукта белкового обмена — аммиака.
Существенную роль играет печень в обмене липидов, который тесно связан с выделением желчи, необходимой для расщепления и всасывания жиров в кишечнике. В печени идет синтез триглицеридов, липопротеидов, фосфолипидов, холестерина, желчных кислот, кетоновых тел.
Печень активно участвует в обмене углеводов. Галактоза и фруктоза в печени превращаются в глюкозу, идут синтез и распад гликогена, окисление глюкозы, образование глюкозы из некоторых аминокислот, синтез молочной, пировиноградной и глюкуроновой кислот. Гепатоциты являются одним из важнейших депо гликогена в организме.
Печень является важным депо витаминов: A, D, К, РР, В12, фолиевой кислоты и других. ЖК, синтезируемые гепатоцитами, необходимы для всасывания в кишечнике жирорастворимых витаминов (A, D, Е, К). Печень играет роль депо для железа, меди, цинка, марганца, молибдена.
524
Лекция 35
Печень участвует в поддержании и коррекции КОС. Она служит поставщиком протеинового буфера крови, поддерживает нормальный уровень аммиака в крови. Гепатоциты используют в глюконеогенезе лактат, пируват и аминокислоты. В печени синтезируются кетоновые тела и гидрокарбонат.
Экскреторная функция — образование и выделение желчи в кишечник. Основными органическими компонентами печеночной желчи являются ЖК, фосфолипиды (лецитин), холестерин и желчные пигменты (билирубин).
ЖК синтезируются из холестерина, на что расходуется около 40 % его содержания в организме. ЖК поступают в тонкую кишку для осуществления мицеллярной фазы переваривания и абсорбции жиров. Основная масса ЖК (более 90 %) в тонкой кишке всасывается и по системе воротной вены возвращается в печень (печеночно-кишечный круговорот), поэтому в крови их содержание незначительное. ЖК являются важнейшим стабилизатором коллоидного состояния желчи. Нарушение состава желчи может способствовать образованию конкрементов в желчевыводящих путях.
Участие печени в пигментном обмене заключается в захвате из крови неконъюгированного (несвязанного, непрямого) билирубина, образовавшегося в ретикулоэндотелиальной системе из НЬ при разрушении эритроцитов, конъюгации его с глюкуроновой кислотой и экскреции в желчь в форме конъюгированного (связанного с глюкуроновой кислотой, прямого) билирубина.
Детоксицирующая функция — обезвреживание ряда токсических продуктов, поступающих в печень по системе воротной вены. В ходе восстановительных реакций обезвреживаются нитросоединения. Путем гидролиза подвергается детоксикации ряд лекарственных веществ, например сердечные гликозиды. В печени метаболизируют многие гормоны: глюкокортикоиды, альдостерон, АДГ, эстрогены, инсулин, тироксин, инактивируются серотонин и гистамин.
Барьерная функция осуществляется за счет защитного действия клеток Купфера, которые путем фагоцитоза удаляют из крови микроорганизмы, их токсины, АГ, иммунные комплексы, жировые капли, отжившие клетки крови и т.д. 95 % веществ с антигенными свойствами обезвреживается купферовскими клетками печени. Специфические (иммунные) защитные реакции осуществляются лимфоцитами печеночных лимфатических узлов.
Патофизиология печени
525
Этиологические факторы поражения печени
Инфекционные агенты: вирусы, простейшие, гельминты, бактерии, грибы.
Токсигеские вещества: полициклические ароматические углеводороды, хлор- и фосфорорганические пестициды, этанол и его суррогаты, фенолы, соединения фосфора, соли тяжелых металлов, четыреххлористый углерод, токсины растительного происхождения (токсины бледной поганки и др.), лекарственные препараты (сульфаниламиды, антибиотики, средства для наркоза, снотворные (хлоралгидрат), психотропные (аминазин), ненаркотические анальгетики (индометацин, парацетамол) ит.д.
Физигеские воздействия: радиация, механические повреждения.
Алиментарные факторы: повышение потребления животных жиров, дефицит липотропных факторов и белков.
Нарушение пегеногного кровотока: локальное (тромбоз печеночной артерии, воротной вены, цирроз печени) и системное (хроническая застойная сердечная недостаточность).
Нарушение оттока желги.
Опухоли (первичные и метастазы).
Врожденные дефекты: аномалии развития печени (каверноматоз воротной вены и др.), нарушение метаболизма (гликогенозы), наследственные ферментопатии.
Аутоиммунные процессы в печени.
Печеночные синдромы и их биохимические маркеры
Цитолитигеский синдром (нарушение целостности гепатоцитов)
Характерны:
• повышение активности индикаторных ферментов АлАТ, АсАТ, ЛДГ и ее изоферментов — ЛДГ-4 и ЛДГ-5;
• гипербилирубинемия, главным образом за счет повышения конъюгированного билирубина (КБ).
Синдром холестаза (внутри- или внепеченочный)
Проявляется:
• повышением активности экскреторных ферментов (маркеров холестаза) — щелочной фосфатазы (ЩФ), ГГТ, лейцинаминопептидазы (ЛАП), 5-нуклеотидазы;
• повышением содержания в крови конъюгированного билирубина, желчных кислот, холестерина, фосфолипидов, р-липопротеидов.
526
Лекция 35
Синдром пегеногно-клетогной недостатогности
Характеризуют:
• понижение активности холинэстеразы:
• уменьшение содержания в сыворотке крови общего белка преимущественно за счет альбумина (снижение альбумино-глобулинового коэффициента); уменьшение количества I, II, V, VII, IX, X, XIII факторов свертывания крови;
• гипохолестеринемия;
• гипербилирубинемия, преимущественно за счет неконъюгирован-ного билирубина (НКБ) (из-за нарушения его обезвреживания);
• повышение содержания аммиака (не превращается в мочевину) и других токсических продуктов.
Иммуновоспалительный синдром, обусловленный сенсибилизацией иммунокомпетентных клеток и активацией фагоцитарной системы.
Проявляется:
• повышением уровня у-глобулинов сыворотки крови;
• положительными белково-осадочными пробами (тимоловая, сулемовая, Вельтмана и др.);
• повышением уровня IgG, IgM, IgA, появлением неспецифических антител, в том числе к ДНК, гладкомышечным волокнам, митохондриям, изменением реакции БТЛ.
Патофизиологические синдромы
Гепатомегалия (увеличение печени) — наиболее частый симптом болезней печени. Он может быть обусловлен дистрофией гепатоцитов, лимфомакрофагальной инфильтрацией при острых и хронических гепатитах, развитием регенераторных узлов и фиброза при циррозах; застоем крови при сердечной недостаточности, внутрипеченочным и внепеченочным холестазом; очаговыми поражениями при опухолях, абсцессах, кистах.
Гепатолиенальный синдром — сочетанное увеличение печени и селезенки, обусловленное общей связью с воротной веной, общим лимфооттоком, общей иннервацией, а также принадлежностью к единой системе мононуклеарных фагоцитов.
Гиперспленизм — синдром, который выражается усилением и извращением функции селезенки, сопровождающимся повышенным разрушением полноценных клеток крови, что проявляется лейкопенией, тромбоцитопенией и анемией.
Астеновегетативный синдром сопровождает большинство заболеваний печени. Этот синдром проявляется слабостью, подавленным
Патофизиология печени
527
настроением, раздражительностью, бессонницей, снижением работоспособности и может указывать на печеночно-клеточную недостаточность.
Диспептический синдром наблюдается при многих заболеваниях печени. Плохой аппетит, тошнота, тяжесть в эпигастрии, отрыжка, вздутие живота, запор могут быть обусловлены печеночно-клеточной недостаточностью, внутри- и внепеченочным холестазом, портальной гипертензией. Возможно похудание (синдром «плохого питания»), доходящее иногда до кахексии.
Геморрагический синдром, который проявляется появлением петехиальной сыпи и кровоизлияний в кожу. Развитие синдрома обусловлено: снижением синтеза в гепатоцитах факторов свертывания крови (I, II, V, VII, IX, X, XIII): уменьшением количества тромбоцитов вследствие гиперспленизма; повышенным потреблением как тромбоцитов, так и факторов свертывания крови, активацией фибринолитической системы.
Портальная гипертензия. Повышение давления в бассейне воротной вены, обусловленное нарушением кровотока либо в портальной, либо в печеночной, либо в нижней полой венах.
Печеночный кровоток характеризуется соединением артериального и венозного потоков в центральной вене печеночных долек. Если в прекапиллярной части артериальной системы давление составляет 110-120 мм рт.ст., то в венулах — всего 5-10 мм рт.ст. Эта огромная разница должна была бы привести к исключительно артериальному потоку крови, что предотвращается системой специальных сфинктеров, выравнивающих этот перепад давления. В результате через воротную вену идет 75 % всего потока крови, а через артерию только 25 %. Таким образом, речь идет не только о выравнивании давления, но и о создании преимущества для венозного потока, идущего с очень низким давлением. Ежеминутно через сосуды печени протекает 1500 мл крови.
Различают три формы портальной гипертензии:
• подпеченочный блок, обусловленный врожденной аномалией портальной вены либо сдавлением портального коллектора, в результате чего повышается давление в портальной вене:
• надпеченочный блок, развивающийся вследствие тромбоза печеночных вен; сдавления печеночных вен (синдром Бадда-Киари), правожелудочковой сердечной недостаточности:
• внутрипеченочный блок, который связан с диффузными заболеваниями печени. В 70 % случаев причиной возникновения этого блока
528
Лекция 35
является цирроз печени. В основе этой формы лежит дезорганизация структуры печени за счет регенерации и образования ложных долек. Нарушается сосудистая архитектоника, гибнут сфинктеры и устраняется препятствие на пути артериального потока. Воспалительные процессы в паренхиме печени, сопровождающиеся набуханием и повышением давления внутри самой печеночной дольки, приводят к сдавлению венозных сосудов.
Типичным следствием портальной гипертензии является формирование феномена шунтирования, в частности между воротной веной и системой нижней полой вены вне печени (портокавальные шунты).
Известны три основные локализации таких шунтов: кожные анастомозы в области пупка, анастомозы в нижней трети пищевода и геморроидальные вены.
Шунтирование крови частично или полностью выключает антитоксическую функцию печени, так как кровь с большим количеством токсических веществ, поступающих из кишечника по воротной вене, минует паренхиму печени и попадает в общий кровоток. Результатом этого является тяжелая интоксикация организма.
Портальную гипертензию сопровождают следующие симптомы и синдромы: гепатомегалия, спленомегалия или гиперспленизм, понижение питания (человек с тонкими, худыми конечностями и большим за счет асцита животом имеет фигуру «паука»); варикозное расширение вен нижней трети пищевода, желудка, геморроидальных вен, вен передней брюшной стенки (симптом «голова медузы* ); геморрагигеский синдром (кровотечение из варикознорасширенных вен является одной из самых частых причин смерти больных), асцит, интоксикация.
Асцит. Скопление жидкости в брюшной полости. Наиболее часто встречается при циррозе печени. Асцитическая жидкость представляет собой как бы ультрафильтрат плазмы, ее компоненты находятся в динамическом равновесии с составляющими плазмы.
Факторы, способствующие выходу жидкости из сосудов в брюшную полость
• Портальная гипертензия, при которой повышается как синусоидальное гидростатическое давление, вызывающее транссудацию плазменного фильтрата через стенки синусоидов в пространство Диссе, так и гидростатическое давление во всех венозных сосудах печени.
• Понижение ОД плазмы вследствие понижения синтеза альбуминов в печени и потери их при удалении асцитической жидкости.
Патофизиология печени
529
• Усиление лимфообразования (до 15-20 л при норме 8-9 л!) вследствие блокады венозного оттока с развитием динамической лимфатической недостаточности и просачиванием лимфы через порозные стенки лимфатических сосудов; характерна лапароскопическая картина «плагущей» печени с многочисленными лимфатическими кистами и выделяющейся по каплям лимфой.
• Гиперальдостеронизм, обусловленный активацией РААС вследствие ухудшения почечного кровотока, а также понижением инактивации альдостерона в печени и снижением его клиренса. Гиперальдостеронизм приводит к накоплению в организме натрия и воды.
• Стимуляция выработки АДГ (вазопрессина), обусловленная (относительно) сниженным эффективным внутрисосудистым объемом крови, и, возможно, в ряде случаев гиперосмоляльностью плазмы за счет задержки натрия (имеются также данные об усиленном выходе натрия в асцитическую жидкость и развитии гипонатриемии разведения). Снижение эффективного внутрисосудистого объема крови связано, в свою очередь, с неадекватной дилатацией артериальных сосудов вазоактивными веществами, недостаточно инактивирующимися в печени, а также уменьшением эффективной онкотической всасывательной силы на фоне дефицита альбуминов.
При большом количестве асцитической жидкости могут наблюдаться следующие дополнительные симптомы: пупочные, паховые и диафрагмальные грыжи; варикозное расширение вен голеней; смещение диафрагмы вверх; пищеводный рефлюкс, приводящий к появлению эрозий слизистой оболочки пищевода и кровотечениям из варикозных вен.
Болевой синдром и чувство тяжести в правом подреберье возникают вследствие растяжения фиброзной оболочки печени и бывают при остром вирусном гепатите, застойной сердечной недостаточности, внепеченочном холестазе. Воспалительное поражение капсулы печени (перигепатит) и спаечный процесс между фиброзной оболочкой и париетальной брюшиной также приводят к болям в правом подреберье. Острая боль в области желчного пузыря характерна для приступа холецистита, в том числе калькулезного.
Холестаз. Синдром, характеризующийся недостаточным выделением в кишечник, накоплением в желчных протоках и возможным попаданием в кровь всех или отдельных компонентов желчи.
Различают:
• холестаз парциальный — в кишечник выделяется мало желчи;
530
Лекция 35
• холестаз диссоциированный — задерживается только КБ или только ЖК;
• холестаз тотальный — в кишечник не поступают все компоненты желчи.
По механизму развития холестаз может быть внутрипегеногный (пер-вигный) — при повреждениях самой печени, например, при гепатитах, хо-лангиолитах, при повышении проницаемости желчных капилляров, при сгущении желчи и тому подобное и внепегеногный (вторигный) — при нарушении проходимости общего желчевыводящего протока (обтурация камнем, сдавление опухолью или воспалительным инфильтратом в области двенадцатиперстной кишки и головки поджелудочной железы).
Ахолия. Синдром, обусловленный отсутствием или уменьшением желчи в кишечнике в результате первичного или вторичного холестаза.
Для него характерны: ахолигный (цвета мела или замазки) кал (из-за отсутствия в нем пигмента стеркобилина), часто с серебристым оттенком из-за содержания непереваренных жиров (стеаторея). При ахолии в кишечник не попадают ЖК, необходимые для активации липазы, эмульгирования и переваривания жиров. Жиры могут обволакивать белки, делая их недоступными пищеварительным ферментам, что приводит к развитию креатореи (в кале обнаруживаются мышечные волокна). Длительное нарушение всасывания жиров влечет за собой уменьшение всасывания жирорастворимых витаминов — A, D, Е и К с симптомами, соответствующими данным гиповитаминозам.
Отсутствие бактерицидного действия желчных кислот в кишечнике приводит к развитию дисбактериоза, усилению процессов гниения и брожения, метеоризму, нарушению моторной функции кишечника (запор или понос).
Холемия
Холемия (дословно — желчь в крови). Синдром, характеризующийся увеличением в крови желчных кислот и их солей вследствие внутрипе-ченочного или внепеченочного холестаза.
Синдром проявляется кожным зудом, брадикардией, ослаблением сократимости миокарда, артериальной гипотензией, общей астенией, депрессией, раздражительностью, головной болью, утомляемостью, ночной бессонницей и сонливостью днем.
Эти изменения обусловлены непосредственным действием желчных кислот на нервные окончания, на вегетативную нервную систему (усиление тонуса блуждающего нерва, особенно на фоне дефицита катехоламинов и тироксина, обусловленного недостатком тирозина), на
Патофизиология печени 531
мышечные клетки (ослабление работы ионных насосов, уменьшение синтеза АТФ). При тяжелой холемии может быть разрушение клеток крови — эритроцитов, лейкоцитов и тромбоцитов.
Увеличение желчных кислот в крови сопровождается повышенным выделением их с мочой. При этом мота сильно вспенивается при встряхивании, что обусловлено уменьшением ее поверхностного натяжения.
Желтуха (icterus). Симптом, характеризующийся появлением желтой окраски кожи, склер и слизистых оболочек в результате отложения желчных пигментов (билирубина) при увеличении их содержания в крови более 30-35 мкмоль/л.
Билирубин образуется в основном при разрушении гема (гемолиз эритроцитов), а также миоглобина и тканевых цитохромов. В клетках ретикулоэндотелиальной системы он превращается в биливердин, который, в свою очередь, переходит в несвязанный, прямой билирубин (НКБ), транспортирующийся кровью с помощью альбумина. НКБ из крови поступает в гепатоциты, где из него образуется конъюгированный (связанный, непрямой) билирубин (КБ). Этот процесс катализируется ферментом УДФ-глюкуронилтрансферазой. КБ (моноглюкуронид) выделяется из гепатоцитов в желчные капилляры, где преобразуется в диглюкуронид. С желчью КБ выводится в двенадцатиперстную кишку, где кишечные бактерии восстанавливают пигмент с образованием вначале мезобилирубина, а затем уробилиногена (УБ). Из тонкой кишки часть образовавшегося УБ всасывается и по воротной вене попадает в печень, где полностью расщепляется. Оставшийся УБ попадает в толстую кишку, где преобразуется в стеркобилиноген (СБ), придающий нормальное окрашивание фекалиям и выделяющийся с ними в окисленной на воздухе форме — стеркобилина. Часть СБ, всасываясь в слизистой оболочке толстой кишки, по геморроидальным венам попадает в общий кровоток, фильтруется в почках и выделяется с мочой. Содержание общего билирубина в крови составляет 6,8-20,5 мкмоль/л. Около 70 % этого количества составляет НКБ, а около 25 % приходится на долю КБ. По уровню общего билирубина оценивают степень тяжести желтухи: при ее легкой степени общий билирубин плазмы повышается до 86 мкмоль/л; при средней — до 171 мкмоль/л; при тяжелой — составляет более 171 мкмоль/л. В мочу может попадать только КБ при повышении его содержания в крови более 34 мкмоль/л (нерастворимый НКБ связан с альбумином и не проходит через почечный фильтр).
Желтуха как синдром предполагает, кроме желтушности кожных покровов и слизистых оболочек, наличие характерного комплекса симптомов, объединенных общим патогенезом.
532
Лекция 35
Виды желтух:
• приобретенные — надпеченочная, подпеченочная, печеночная (табл. 35-1);
• наследственные — Жильбера, Криглера-Найяра и Дабина-Джон-
сона.
Таблица 35-1. Сравнительная характеристика желтух
Кровь Моча Кал
НКБ КБ ЖК Хол. НКБ КБ УБ СБ ЖК СБ
Норма ++ + следы + - - (+) + - +
Надпеченочная желтуха тт т следы + - - +т т - тт
Подпеченочная желтуха т тт тт т - + - - + -(Т)
Печеночная желтуха (легкая) т тт тт 1 - ++ ± ++ ±
Печеночная желтуха (тяжелая) тт т Т(-) и - ± - - + -
Обозначения: НКБ — неконьюгированный билирубин; КБ — конъюгированный билирубин; ЖК — желчные кислоты; Хол. — холестерин: УБ — уробилин; СБ — стеркобилин.
Надпеченочная или гемолитическая желтуха сопровождает все наследственные и приобретенные гемолитические анемии.
В результате повышенного распада эритроцитов из РЭС в общий кровоток поступает больше НКБ, из которого в печени образуется больше КБ, а в кишечнике соответственно — больше УБ и СБ. Кал становится гиперхоличным (очень темным из-за повышенного содержания в нем стеркобилина). Печень не может извлекать из оттекающей от кишечника крови весь УБ (относительная печеночная недостаточность), и УБ из кишечно-печеночного кругооборота попадает в общий кровоток и выделяется с мочой. В геморроидальных венах больше всасывается СБ и поэтому его больше в моче. Уробилинурия и стеркобилинурия являются причиной потемнения цвета мочи.
Билирубинурии нет, так как, несмотря на повышение в крови растворимого КБ, его количество не превышает 34 мкмоль/л и с мочой он не выделяется.
В крови при надпегеногной желтухе повышено содержание общего билирубина, главным образом за счет НКБ, незначительно повышено количество КБ, при этом БП всегда остается меньше 25 %. Гипербилирубинемия сопровождается отложением НКБ в коже, слизистых
Патофизиология печени
533
оболочках, склерах глаз — появляется желтуха с характерным лимонножелтым оттенком. При чрезмерном увеличении НКБ в крови (более 250 мкмоль/л) он, не связавшись с белком, проходит через гематоэнцефалический барьер и повреждает ядра головного мозга («ядерная желтуха») с картиной энцефалопатии. Такое нарушение особенно характерно для тяжелых форм гемолитической болезни новорожденных и наследственных желтух.
Кроме гипербилирубинемии для надпеченочной желтухи характерны увеличение в крови сывороточного железа, а также все признаки гемолитической анемии: снижение НЬ, числа эритроцитов и Ht, рети-кулоцитоз и нейтрофильный лейкоцитоз, ускорение СОЭ, а также клинические проявления гипоксического синдрома. Синдромы холемии и печеночно-клеточной недостаточности отсутствуют!
Подпеченочная, или механическая (обтурационная), желтуха развивается на фоне внепеченочного (вторичного) холестаза.
Желчь не поступает в кишечник — развивается синдром ахолии, переполняет печеночные желчные протоки и капилляры. При повышении в них давления более 270 мм вод.ст. они повреждаются, и путем регургитации печеночная желчь попадает в кровь — развивается синдром холемии.
В крови при этом увелигивается содержание общего билирубина, главным образом за счет КБ (БП больше 50 %). Отложение пигмента в тканях сопровождается появлением желтухи с характерным зеленым оттенком. Содержание КБ в крови обычно превышает 34 мкмоль/л, и он выделяется с мочой (билирубинурия), придавая ей темный цвет (цвет крепкого чая или пива). Моча сильно вспенивается при встряхивании из-за присутствия желчных кислот (результат холемии). Уробилина и стеркобилина в моге нет (результат ахолии).
Наличие у больного холестаза подтверждается соответствующими маркерами — увеличением в крови ЩФ, ГГТП, ЛАП, 5-нуклеотидазы, а также — холестерина, липопротеидов.
Маркеры цитолитигеского синдрома (АЛТ, ACT и др.) обычно не изменены, однако при хроническом внепеченочном холестазе возможно вторичное повреждение паренхимы печени и тогда могут появиться признаки и умеренно выраженного цитолитического синдрома, и печеночно-клеточной недостаточности.
Печеночная, или паренхиматозная, желтуха развивается вследствие повреждения самой печени и может проявляться признаками внутрипеченочного холестаза, ахолии (гипохолии) и холемии —
534
Лекция 35
холестатигеская форма желтухи, или без холестаза и ахолии, но с преобладанием синдрома печеночно-клеточной недостаточности — гепатоцеллюлярная форма. Однако в большинстве случаев развивается смешанная форма — с холестазом, а- или гипохолией, холемией, с признаками цитолиза и печеночно-клеточной недостаточности.
Первым признаком недостаточности функции гепатоцитов (например, в 1-й стадии вирусного гепатита) может быть потемнение моги за сгет увелигения в ней УБ, так как поврежденные гепатоциты не смогут извлекать весь УБ из крови, оттекающей от кишечника.
При легких формах гепатита (или на его первой стадии) процесс захвата НКБ из крови гепатоцитами и процесс его обезвреживания (конъюгация с УДФ ГК) не нарушаются, т.е. печень перерабатывает НКБ в КБ.
Однако нарушается процесс экскреции КБ в желгные капилляры, так как транспортные ферментные системы, обеспечивающие перенос КБ через билиарные полюса гепатоцитов в желчные капилляры, являются энергозависимыми и страдают первыми. Воспалительная или дегенеративная деструкция пегеногных клеток, кровеносных сосудов, желгных протоков и капилляров сопровождается попаданием в кровь пегеногной желги: в крови повышается содержание общего билирубина, главным образом КБ (БП всегда высокий), в меньшей степени — НКБ. КБ превышает уровень 34 мкмоль/л в крови и выделяется с мочой, делая ее темной (билирубинурия). Развивается холемия, но слабее выраженная, чем при подпеченочной желтухе (ослаблен синтез желчных кислот в печени). При наличии внутрипеченочного холестаза (иногда его может не быть) наблюдается а- или гипохолия.
При тяжелых формах гепатита (или в период разгара болезни) начинают нарушаться процесс захвата гепатоцитами НКБ из крови и его конъюгация (ослабляется обезвреживающая функция печени). В крови на фоне высокого уровня общего билирубина начинают снижаться количество КБ (однако БП всегда остается повышенным) и расти содержание НКБ, который не выделяется с мочой — моча больного светлеет (исчезает билирубинурия). В тканях откладывается НКБ и КБ — желтый цвет кожи, склер и слизистых оболочек, имеет рыжевато-коричневый оттенок цвета охры. Чем больше повреждаются гепатоциты, тем меньше в печени синтезируется желчных кислот — уменьшаются проявления холемии (меньше зуд, меньше вспенивается моча и др.), но сильнее проявляются признаки дефицита желчи и желчных кислот в кишечнике (см. ахолия).
Патофизиология печени
535
При всех формах печеночной желтухи в зависимости от ее тяжести в большей или меньшей мере выявляются маркеры синдромов: холестаза (ЩФ, ГГТП, ЛАП и др.), цитолиза (АЛТ, ACT и др.), но главными являются признаки пегеногно-клетогной недостатогности.
Наследственные желтухи
• Синдром Жильбера (тип наследования аутосомно-доминантный) характеризуется уменьшением в печени глюкоронилтрансферазы и ослаблением конъюгации НКБ, количество которого в крови умеренно повышается.
• Синдром Криглера-Найяра (тип наследования смешанный) характеризуется полным отсутствием глюкоронилтрансферазы, в результате чего КБ в печени не образуется совсем (симптом бесцветной желчи), а количество необезвреженного НКБ достигает 260-550 мкмоль/л и вызывает развитие «ядерной желтухи» и тяжелой энцефалопатии.
• Синдром Дабина-Джонсона (тип наследования аутосомно-доминантный) проявляется развитием парциального холестаза без хо-лемии (в крови повышается содержание только КБ) и меланозом печени («болезнь черной печени»).
Печеночно-клеточная недостаточность. Синдром, который характеризуется ослаблением метаболической, детоксикационной и барьерной функций печени.
Нарушения метаболических функций печени проявляются изменениями углеводного, белкового, липидного, водно-электролитного обмена и КОС, обмена витаминов и микроэлементов.
Углеводный обмен характеризуется склонностью больных с патологией печени к гипогликемии, развитие которой объясняется уменьшением запасов гликогена в печени, ослаблением процессов гликогенолиза и глюконеогенеза. Гипогликемия проявляется слабостью, головокружением, вегетативными расстройствами. Дефицит глюкуроновой кислоты влечет за собой ослабление детоксикационной функции печени, в том числе обезвреживание НКБ.
Белковый обмен характеризуется прежде всего уменьшением в крови альбуминов, синтез которых в печени резко падает. Дефицит альбуминов в крови приводит к снижению эффективной онкотической всасывательной силы (ЭОВС) и появлению гипергидратации и отеков, а при наличии портальной гипертензии — участвует в развитии асцита. Синтез а- и р-глобулинов или также уменьшается, или увеличивается (например, при вирусном гепатите А, что обнаруживается положительной
536
Лекция 35
тимоловой пробой). При инфекционных, иммунных формах патологии печени в крови повышается содержание у-глобулинов.
Диспротеинемия приводит к снижению альбумино-глобулинового (А/Г) коэффициента, увеличению СОЭ.
Характерны увеличение в крови аминокислот {гипераминоацидемия) вследствие того, что они не используются в синтезе белка, и выделение аминокислот с мочой {аминоацидурия).
Чем больше снижена функция печени, тем больше в крови токсичного конечного продукта белкового обмена — аммиака, который не превращается в печени в мочевину, поэтому содержание ее в крови уменьшается.
В поврежденной печени нарушается синтез важнейших факторов свертывания крови: I, И, V, VII, IX, X, XIII и других, что приводит к развитию геморрагигеского синдрома. Маркером тяжести гепатоцеллюлярной недостаточности может служить протромбиновый индекс крови.
Чем ниже протромбиновый индекс, тем тяжелее патология печени.
Нарушение синтеза в печени транспортных белков может повлечь за собой развитие железодефицитной анемии {уменьшение синтеза трансферрина), В12-дефицитной анемии {уменьшение транскобаламина в сочетании с уменьшением депонирования витамина В12и фолиевой кислоты в печени). Нарушается в печени синтез и таких витаминов, как витамин А и В6.
Дисбаланс гормонов и гормональные расстройства могут развиться как из-за дефицита соответствующих транспортных белков, так и из-за нарушения метаболизма гормонов в печени, в частности эстрогенов, глюкокортикоидов, альдостерона, тироксина и др.
Жировой обмен при синдроме печеночно-клеточной недостаточности характеризуется нарушением синтеза и окисления триглицеридов, фосфолипидов, липопротеидов, холестерина, желчных кислот и кетоновых тел. В крови снижено содержание холестерина и антиатерогенных форм липопротеидов, в кишечнике — дефицит желчных кислот.
Ослабление антитоксигеской функции пегени выражается в нарушении обезвреживания НКБ и аммиака, токсических продуктов (ароматические углеводороды, нитробензол, индол, скатол, фенол, путресцин, кадаверин), поступающих от кишечника по портальной системе, а также многих лекарств (сульфаниламидов, сердечных гликозидов и др.).
Ослабление функции печеночных макрофагов (клеток Купфера) приводит к ослаблению барьерной функции: в кровь попадают необезвре-женные микробы и их токсины, АГ и иммунные комплексы, что влечет
Патофизиология печени
537
за собой снижение устойчивости к инфекции, развитие аллергических и иммунных заболеваний. В тяжелом случае может развиваться картина токсемии (с лейкоцитозом, лихорадкой, гемолизом эритроцитов, эрозиями кишечника, почечной недостаточностью и т.п.), а при возникновении портокавального шунтирования — тяжелый токсигеский шок.
Печеночная недостаточность (ПН) — состояние печени, которое не обеспечивает постоянство внутренней среды организма — гомеостаз.
Виды печеночной недостаточности:
• абсолютная и относительная (на фоне функциональных перегрузок);
• тотальная (снижены все функции печени) и парциальная (нарушены отдельные функции);
• малая (без энцефалопатии) и большая печеночная недостаточность (с энцефалопатией).
Патогенетигеские формы пегеногной недостатогности:
• экскреторная (холестатическая);
• гепатоцеллюлярная (при воспалении, дистрофии, некрозе печени, энзимопатиях);
• сосудистая (при нарушении местного или общего кровообращения);
• смешанная.
Клинигеские формы: острая и хроническая, с диффузным или очаговым поражением печени.
Экспериментальными моделями пегеногной недостатогности могут быть: полное или частичное удаление печени, формирования фистул (Экка и Экка-Павлова), перевязка желчных протоков, ангиостомия по Лондону, токсическое повреждение печени гепатотропными ядами и др.
Печеночная кома. Синдром, развивающийся на фоне абсолютной, тотальной, большой печеночной недостаточности, главным проявлением которого является развитие печеночной энцефалопатии или гепато-церебрального синдрома.
Печеночная кома проявляется нарушением психики, сознания, кровообращения, дыхания, судорогами, гипоксическим и геморрагическим синдромом, печеночной желтухой, гипогликемией, гипопротеинемией, нарушением водно-электролитного обмена и КОС, а также характерным запахом изо рта (запах сырой печени, заплесневелой соломы), обусловленным накоплением в крови ароматического вещества меркаптана.
Развитие энцефалопатии объясняется появлением в крови большого количества веществ — церебротоксиноъ, к которым относят:
538
Лекция 35
• аммиак, выключающий а-кетоглютаровую кислоту из цикла Кребса, что сопровождается уменьшением образования АТФ;
• белковые метаболиты — фенол, индол, скатол, амины и др.;
• жирные кислоты — масляная, капроновая, валериановая, повреждающие липидные компоненты клеточных мембран, нарушающие передачу возбуждений в ганглиях;
• производные пировиноградной и мологной кислот — ацетоин, бути-ленгликоль;
• аминокислоты — фенилаланин, триптофан, тирозин и метионин;
• ложные нейромедиаторы — октопамин и р-фенилэтиламин, нарушающие синаптическую передачу путем вытеснения из синапсов норадреналина и допамина.
Нарушение КОС может выражаться как в развитии метаболического ацидоза из-за дефицита буферных оснований (белков, бикарбоната) и накопления органических кислот (лактата, пирувата, кетоновых тел и др.), так и в развитии метаболического гипокалиемического алкалоза. Последний развивается на фоне гиперальдостеронизма, сопровождающегося выведением из организма калия.
Кома может развиться на фоне тяжелого вирусного гепатита, токсической дистрофии и циррозе печени, при портальной гипертензии, при остром нарушении печеночного кровообращения и травмах печени.
Внепегеногные симптомы: бледность (вследствие развития анемии); следы расчесов на коже вследствие зуда при холемии; ксантоматоз, обусловленный нарушением холестеринового обмена; телеангиэктазии (сосудистые звездочки); пальмарная эритема (покраснение ладоней); малиновый язык; гинекомастия (увеличение молочных желез у мужчин).
Появление сосудистых звездочек, печеночных ладоней, малинового языка и гинекомастии объясняют избытком в крови эстрогенов вследствие нарушения их метаболизма в печени.
Лекция 36
ПАТОФИЗИОЛОГИЯ ПОЧЕК I
Г.П. Щелкунова
Почечные (ренальные) симптомы и синдромы
Почки являются важнейшими органами, обеспечивающими гомеостаз организма. Они поддерживают нормальный объем жидкости в организме и ОЦК (изоволемию), нормальную концентрацию осмотически активных веществ в жидкостях (изотонию), нормальную осмоляльность и ионный состав (изоионию); участвуют в регуляции pH крови; выводят конечные продукты азотистого обмена; играют важную роль в регуляции артериального давления, эритропоэза и гормонального статуса.
Главным структурным элементом почки является нефрон. В каждой почке содержится около 1 млн нефронов, в которых осуществляются главные процессы: фильтрация первичной мочи в клубочковом аппарате, реабсорбция — обратное всасывание воды и растворенных веществ из первичной мочи в кровь в канальцах и собирательных трубках, секреция эпителиальными клетками канальцев различных веществ в мочу, экскреция — выведение из организма путем фильтрации и секреции токсичных продуктов обмена, гормонов, лекарств и других ненужных организму компонентов; инкреция — синтез и высвобождение биологически активных веществ, например ренина, эритропоэтинов, урокиназы, почечных простагландинов, кининов и др. При патологии почек все эти функции могут нарушаться и приводить к появлению характерных для нефропатий симптомов и синдромов.
Изменения величины суточного диуреза
Здоровый взрослый человек в оптимальных условиях должен выделять за сутки около 1,0-1,5 л воды. Величина суточного диуреза зависит как от процесса клубочковой фильтрации, так и от процесса канальцевой реабсорбции воды. Количество профильтрованной первичной мочи
540
Лекция 36
находится в зависимости от числа функционирующих нефронов, т.е. от площади фильтрации, и от величины эффективного фильтрационного давления (ЭФД) в капиллярах клубочков: ЭФД = ГД - ОД - ДМП (где ГД — гидростатическое давление крови в капиллярах клубочков, ОД — онкотическое давление крови, ДМП — давление в мочевыводящих путях). Гидростатическое давление поддерживается на уровне 70-75 мм рт.ст. даже при условии колебания систолического системного АД в пределах от 90 до 190 мм рт.ст. благодаря автоматическому изменению тонуса приносящей и выносящей артериол клубочков (повышение тонуса выносящих артериол повышает ГД, уменьшение их тонуса или повышение тонуса приносящих артериол понижает ГД в капиллярах клубочков). Так как ОД в норме 25-30 мм рт.ст., а ДМП — около 20 мм рт.ст., то нормальное ЭФД = 20-30 мм рт.ст.
Вся внеклеточная жидкость организма (около 14 л) за стуки проходит почечный фильтр 12 раз. Почечный кровоток составляет 1100 мл/мин, а плазмоток — 600 мл/мин. В результате клубочковой фильтрации за 1 мин образуется 80-120 мл мочи (за сутки — 150-170 л), однако в мочевой пузырь за ту же минуту выделяется только около 1 мл конечной мочи, а нормальный суточный диурез не превышает 1,5 л, 98-99 % воды, содержащейся в первичной моче, реабсорбируется. Около 80 % воды реабсорбируется в проксимальных канальцах и нисходящей части петли Генле по осмотическому градиенту с участием противоточно-множительного механизма, а 18-19 % возвращается в кровь в дистальных канальцах и собирательных трубках почек под влиянием АДГ.
Подробный механизм участия АДГ в регуляции величины диуреза описан в лекции о патофизиологии водно-электролитного обмена.
Изменения величины суточного диуреза могут проявляться в виде полиурии, олигурии и анурии.
Полиурия — увеличение суточного диуреза более 1,5 л в большинстве случаев не связана с нарушением функции почек.
Ее причинами могут быть:
• повышенное поступление воды в организм, приводящее к увеличению ОЦК и почечного кровотока, повышению ГД в клубочковых капиллярах, а также торможению выделения АДГ из задней доли гипофиза и уменьшению реабсорбции воды в целях сохранения водного баланса;
• повышенное содержание в моче осмотически активных веществ, например глюкозы при сахарном диабете или мочевины при хронической почечной недостаточности;
Патофизиология почек I
541
• уменьшение осмоляльности плазмы крови, например при гипонатриемии, сопровождающееся торможением выделения АДГ;
• несахарный диабет при врожденном или приобретенном дефиците АДГ;
• прием диуретиков.
Полиурия может развиться в 1-й стадии лихорадки из-за уменьшения тонуса приносящих артериол клубочков (значит, увеличения ГД и ЭФД), а также торможения выделения АДГ. Увеличение диуреза часто наблюдается в 1-й стадии артериальной гипертензии, при волнении и некоторых стрессах, когда небольшие дозы адреналина в результате активации симпатической нервной системы приводят к повышению тонуса выносящих артериол клубочков (т.е. к повышению ГД и ЭФД), а также торможению секреции АДГ.
Полиурия погегного происхождения может быть обусловлена нарушением функции канальцев, их нечувствительностью к АДГ («нефрогенный несахарный диабет», азотемическая стадия ХПН, калиопеническая нефропатия при синдроме Конна и др.), а также патологическими изменениями мозгового вещества почек, приводящими к нарушению противоточно-множительного механизма (пиелонефрит).
Олигурия — уменьшение суточного диуреза менее 1 л также может быть внепочечного и почечного происхождения. Уменьшение диуреза на фоне нормальной функции почек наблюдается при обезвоживании, снижении артериального давления ниже 80 мм рт.ст. (шок, коллапс, сердечная недостаточность), при увеличении осмоляльности плазмы крови (гипернатриемия), сопровождающейся увеличенной секрецией АДГ (осморегулирующий рефлекс), а также при увеличении в плазме крови белка (переливание белковых кровезаменителей). В этих случаях происходит или уменьшение фильтрации первичной мочи, так как снижается ЭФД (за счет уменьшения ГД и увеличения ОД), или увеличение реабсорбции воды в канальцах в целях компенсации гиповолемии.
Олигурия погегного происхождения развивается при уменьшении площади клубочковой фильтрации, например, при хронической почечной недостаточности, когда постепенно уменьшается число функционирующих нефронов, а также при снижении ЭФД. Причиной уменьшения ЭФД может быть снижение ГД в капиллярах клубочков, обусловленное аномалией, склерозом или тромбозом почечных сосудов, повышением тонуса приносящих артериол (например, под влиянием больших доз адреналина, при избыточной секреции ренина и ангиотензина-П). ЭФД может снизиться по причине увеличения ДМП, обусловленного
542
Лекция 36
затруднением оттока мочи по мочевыводящим путям на уровне канальцев и собирательных трубок (повреждение при острой почечной недостаточности), мочеточников (перегиб, сдавление, обтурация), мочевого пузыря и уретры (аденома предстательной железы). В ряде случаев в капиллярах клубочков может повыситься ОД за счет сгущения крови.
Следует помнить, что даже значительное уменьшение клубочковой фильтрации не всегда сопровождается олигурией, если при этом снижена реабсорбция воды в почечных канальцах и собирательных трубках почек.
Об анурии обычно говорят в том случае, когда суточный диурез больного не превышает 30-50 мл. Анурия может развиться при снижении систолического артериального давления ниже 50 мм рт.ст., когда фильтрация в клубочках практически прекращается (ЭФД = 0), что часто наблюдается на фоне шока, коллапса, сердечной недостаточности и т.п.
С анурии или олигурии начинается острая почечная недостаточность вследствие тотального повреждения нефронов. Анурией завершается хроническая почечная недостаточность при склерозировании 80-90 % нефронов.
Так как по величине суточного диуреза нельзя судить о величине клубочковой фильтрации, то для объективной ее оценки в клинической практике используют показатель — клиренс креатинина. Он рассчитывается по формуле: С = М : К х Д, где С — клиренс (показатель очищения), М — концентрация креатинина в моче, К — концентрация креатинина в крови, Д — величина минутного диуреза. Клиренс показывает, какое количество крови очищается в почках от креатинина за 1 мин, а также какое количество первичной мочи за 1 мин фильтруется в капсулу Шумлянского-Боумена.
Креатинин постоянно присутствует в крови и у людей старше 11 лет выводится из организма только путем фильтрации в клубочках почек (он не реабсорбируется и не секретируется в канальцах). Поэтому по клиренсу креатинина можно оценивать сразу две важнейшие функции почек — экскреторную и фильтрационную. В норме этот показатель равен 80-120 мл/мин. Это означает, что за 1 мин в почках путем фильтрации очищается от креатинина (а значит, и от многих других токсичных веществ) 100-120 мл крови и за эту же минуту фильтруется 80-120 мл первичной мочи. Чем ниже клиренс креатинина, тем больше нарушена фильтрационная и экскреторная функция почек.
Можно определять клиренс и по отношению к другим веществам, например экзогенному инулину или гипосульфиту (норма — 100-120 мл/мин),
Патофизиология почек I
543
фенолроту (400 мл/мин). Клиренс мочевины равен 55-70 мл/мин. Фе-нолрот в почках не только фильтруется, но и секретируется в канальцах, а мочевина после фильтрации в клубочках частично в канальцах реабсорбируется, поэтому их клиренсы столь значительно отличаются от клиренса креатинина. В некоторых нефрологических клиниках определяют клиренсы «средних молекул», мукопротеидов, пепсиногена, альбуминов, иммуноглобулинов и т.д.
Изменения плотности мочи
Первичная моча по плотности равна плотности плазмы крови, но по мере движения по канальцам и собирательным трубкам почек происходит ее концентрирование и разведение. Плотность конечной мочи у здорового человека колеблется в течение суток и зависит от воднопищевого режима. При водной нагрузке она может снижаться до 1002, а при сухоедении — повышаться до 1032 и более.
При нарушении концентрационной функции канальцев почек, например, при пиелонефрите, интерстициальном нефрите, начальных стадиях хронической почечной недостаточности у больных выявляется гипостенурия. При этом во всех порциях мочи, взятых у больного в течение суток через каждые 4 ч (проба Зимницкого), плотность мочи низкая, а после 18-часового сухоедения без воды (проба на концентрационную функцию) плотность мочи ниже 1020 (у здоровых людей должна быть всегда выше). Выявление у больного гипостенурии свидетельствует о наличии у него отека, воспаления или склероза мозгового вещества почек. Состояние, при котором плотность мочи равна 1010-1012 (соответствует плотности плазмы крови) и не меняется в течение суток (монотонная), называется изостенурией. Она свидетельствует о неспособности почек как концентрировать, так и разводить мочу и является признаком более тяжелого поражения нефрона, например, при острой и хронической почечной недостаточности.
Изменения pH мочи
Нормальная реакция мочи слабо кислая, так как за сутки с мочой выделяется 30-60 ммоль Н+, 10-30 ммоль NaH2PO4 (титруемые кислоты), 11-27 г NH4C1. Благодаря фильтрации в клубочках почек сульфатов и фосфатов, а также процессам ацидо- и аммониогенеза в канальцах, их способности реабсорбировать бикарбонат, почки участвуют в регуляции КОС. При респираторном или метаболическом ацидозе (кроме почечного) моча становится более кислой, так как в этом случае с мочой
544
Лекция 36
больше выводится Н' и больше реабсорбируется НСО3~. При респираторном или метаболическом алкалозе с мочой меньше выводится Н4, но больше бикарбоната, поэтому моча становится щелочной. Следует заметить, что в кислой моче легче образуются уратные и оксалатные камни, а в щелочной — фосфатные.
При тубулопатиях или почечной недостаточности процессы ацидо-и аммониогенеза в канальцах почек нарушаются, что ведет к задержке Н4 в организме и развитию почечного метаболического ацидоза. Реакция мочи у таких больных становится щелочной. В некоторых случаях для оценки способности почек выводить Н4 проводят пробу с нагрузкой аммония хлоридом (при нормальной функции канальцев моча закисляется).
«Мочевой синдром»
«Мочевой синдром» — сочетание у больного протеинурии, гематурии и лейкоцитурии (один из наиболее часто встречаемых синдромов, выявляемых у больных с почечной патологией).
Протеинурия
Протеинурия — выделение с мочой белка более 300 мг/сут. Следует вспомнить, что через гломерулярную мембрану легко фильтруются аминокислоты, пептиды и некоторые низкомолекулярные белки (АДГ, инсулин, глюкагон, АКТГ, СТГ, паратгормон, лизоцим, фрагменты легких цепей, p-микроглобулины и др.). Этот процесс зависит как от размера белковых молекул, так и от электрического заряда клубочковой мембраны и фильтруемых белков. Белки высокой молекулярной массы через клубочковую мембрану не проходят, а альбумины фильтруются только частично, так как, имея отрицательный заряд, они отталкиваются от отрицательно заряженной мембраны капилляров клубочков. Почти все содержащиеся в первичной моче белки путем пиноцитоза реабсорбируются в проксимальных и меньше в дистальных канальцах почек. Небольшое количество белков секретируется в мочу из тубулярного эпителия, в том числе в восходящей толстой части петли Генле — белок Тамма-Хорсфалля (гликопротеин ММ 7 000 000). Поэтому у взрослых людей в норме с мочой может выделяться от 100 до 300 мг белка в сутки (не более 0,3 г/л), а у детей — 30-60. Среди выделяемых в норме белков 40 % — альбуминов, 10 % — IgG, 5 % — легких цепей, 3 % — IgA, остальное — белок Тамма-Хорсфалля и др. При этом р- и у-глобулинов в моче быть не должно.
Патофизиологияпочек!
545
Проба на белок считается отрицательной, если с мочой выделяется менее 0,1 г/л белка, положительной (+) — до 0,3 г/л. Протеинурия считается умеренной (++) при содержании в моче белка до 1 г/л и резко выраженной (+++) — более 5 г/л. При pH мочи более 8 протеинурия может быть ложноположительной.
По своему происхождению протеинурия может быть: функциональная, клубочковая, канальцевая, смешанная, протеинурия переполнения, секреторная и гистурия.
Функциональная протеинурия наблюдается у людей после тяжелой физической нагрузки («маршевая» протеинурия), при стрессах, застойной сердечной недостаточности, лордозе, дегидратации, алиментарной гиперпротеинемии и т.п. Она не превышает 1-5 г/сут и носит кратковременный преходящий характер. При этом с мочой главным образом выводятся альбумины (40-80 %), а-глобулины, ферменты, пептиды, иммуноглобулины, тканевые белки.
Клубогковая протеинурия обычно обусловлена повышением проницаемости гломерулярной мембраны или изменением ее заряда или заряда фильтруемых белков. При «нефропатиях с минимальными изменениями» (особенно у детей) может быть нарушена только способность гломерулярного фильтра задерживать отрицательно заряженные молекулы, например альбумин или равный ему по размеру трансферрин. В этом случае индекс селективности — соотношение в моче IgG/ трансферрин <0,1 (селективная протеинурия).
При нарушении барьерных свойств клубочковой мембраны по отношению к размеру фильтруемых частиц этот индекс >0,1. С мочой теряются трансферрин, альбумины, а также IgG и другие высокомолекулярные белки (неселективная протеинурия). Это наблюдается при грубом повреждении гломерул медиаторами воспаления, протеолитическими ферментами, активированными метаболитами кислорода при таких заболеваниях, как ГН, амилоидоз, диабетическая нефропатия, системная красная волчанка, хронические инфекции, опухоли и др.
Канальцевая протеинурия развивается по причине нарушения реабсорбции белка в поврежденных канальцах на фоне его нормальной фильтрации и обычно не превышает 3 г/сут. О повреждении канальцев может свидетельствовать наличие в моче р2-микроглобулинов, которые, легко фильтруясь в клубочках, должны полностью реабсорбироваться в канальцах.
Смешанная протеинурия наблюдается при одновременном повреждении клубочков и канальцев почек. Она может быть умеренной — от 1 до 3 г/л и выраженной — даже до 80 и 100 г/л.
546
Лекция 36
Протеинурия переполнения связана с гиперпротеинемией, сопровождающейся увеличением фильтрации белка и перегрузкой транспортных тубулярных систем.
Секреторная протеинурия обусловлена повышенной секрецией белка эпителием канальцев, в том числе белка Тамма-Хорсфалля, которого в норме за сутки секретируется от 30 до 60 мг.
Гистурия возникает при распаде структур канальцев.
При оценке протеинурии в последние годы используют индекс селективности (ИС). Его рассчитывают по соотношению клиренса иммуноглобулина G и клиренса альбуминов (ИС = С IgG-s-C альб.х100 %). Протеинурия с ИС <15 % называется высокоселективной. В моче обнаруживается белок молекулярной массой менее 70 000, 80 % которого составляют альбумины. Протеинурия с ИС 15-30 % называется селективной (умеренной). В моче содержатся те же белки + гаптоглобин. Если ИС превышает 30 %, то протеинурию называют неселективной. При этом в моче обнаруживаются белки с молекулярной массой более 70 000 (в том числе у-глобулины, а2-макроглобулины, гликопротеиды, трансферрин и т.п.). Неселективная протеинурия свидетельствует о грубом повреждении гломерулярного фильтра и тубулярного эпителия. Она выявляется у больных при ГН, амилоидозе, системной красной волчанке, диабетической нефропатии и др.
При выраженной протеинурии моча может быть мутной и легко вспенивается при встряхивании.
Главным следствием протеинурии являются гипопротеинемия, снижение онкотического давления крови и развитие гипергидратации с отечным синдромом (см. патофизиологию водно-электролитного обмена).
Гематурия
Гематурия — повышенное выделение с мочой эритроцитов. При макрогематурии моча больного мутная и имеет вид мясного смыва. Микрогематурия выявляется при микроскопическом исследовании мочи. В норме в поле зрения обнаруживается 2-3 эритроцита, а в 1 мл мочи их не более 1000 (проба Нечипоренко). Гематурия может быть внепочеч-ная (пузырная, мочеточниковая) и почечная.
Внепогегная гематурия может наблюдаться при циститах, уретритах, опухолях или травмах мочевыводящих путей (например, выходящим камнем).
Погегная гематурия характерна для таких заболеваний, как ГН, туберкулез или опухоль почки и т.п. Долгое время считалось, что нали
Патофизиология почек I 547
чие почечной гематурии при ГН свидетельствует о грубом повреждении гломерулярного фильтра и значительном повышении проницаемости капилляров клубочков. В последние годы появилось утверждение, что эритроциты не могут пройти через трехслойную мембрану клубочковых капилляров (даже при повышении ее проницаемости) и вероятнее всего попадают в мочу из перитубулярных капилляров. Одним из доказательств этого может быть сочетание макрогематурии с селективной слабовыраженной протеинурией у больного ГН.
Для дифференциальной диагностики почечной и внепочечной гематурии в клинике используют «трехстаканную пробу» — забор 3 порций мочи. При почечной гематурии наибольшее число эритроцитов обнаруживается в средней порции, кроме того, при микроскопии они имеют вид «теней» — выщелоченные, измененные. При внепочечной гематурии (у урологических больных) эритроцитов больше в 1-й и 3-й порциях мочи и под микроскопом они имеют свежий неизмененный вид.
Лейкоцитурия
Лейкоцитурия — повышенное выделение с мочой лейкоцитов. Она может быть скрытая, явная и даже пиурия (гной в моче). В норме лейкоцитов в моче 2-5 в поле зрения или не более 2000 в 1 мл (проба Не-чипоренко). Лейкоцитурия свидетельствует о наличии воспалительного процесса или в паренхиме почек, или в мочевыводящих путях.
Кроме белка, эритроцитов и лейкоцитов в моче могут встречаться и другие патологические компоненты.
Цилиндрурия
Цилиндрурия. В моче здоровых людей встречается 0-1 цилиндр в поле зрения или не более 100 в 1 мл мочи, причем только гиалиновые. Основой цилиндров является гель, образующийся из белка Тамма-Хорсфалля в кислой среде с участием электролитов. В зависимости от дополнительных включений цилиндры могут быть: лейкоцитарные, эритроцитарные, эпителиальные, зернистые, восковидные.
Бактериурия
Бактериурия. В норме моча должна содержать не более 10 000 микробов в 1 мл. Патологическая бактериурия характерна для инфекционных нефропатий (пиелонефрит и др.).
Кристаллы солей (ураты, оксалаты, фосфаты) обнаруживаются в большом количестве при мочекаменной болезни.
548
Лекция 36
Аминоацидурия
Аминоацидурия (повышенное выделение с мочой аминокислот) может быть почечная и внепочечная. Аминокислоты легко фильтруются в клубочках почек, но 90 % их должно реабсорбироваться в проксимальных канальцах. В норме с мочой должно выделяться не более 1,1 г аминокислот в сутки. При тубулопатиях (синдром Фанкони и др.), повреждениях эпителия канальцев (дегенерация, некроз) реабсорбция аминокислот нарушается, и они выделяются с мочой.
Внепогегная аминоацидурия является следствием гипераминоацидемии, например, при повышенном распаде тканевых белков (голодании, ожоговой болезни, обширном инфаркте, распаде опухоли), печеночной недостаточности из-за нарушения синтеза белков в печени, наследственных дефектах аминокислотного обмена (фенилкетонурии, оксалу-рии др.). В этом случае развивается функциональная недостаточность транспортных канальцевых систем из-за их перегрузки.
Глюкозурия
Глюкозурия (выделение сахара с мочой) чаще не связана с патологией почек. Глюкоза, легко прошедшая гломерулярный фильтр, полностью реабсорбируется эпителием канальцев с участием ферментов — гексо-киназы, глюкозо-6-фосфатазы. При любой гипергликемии (алиментарной, диабетической), превышающей уровень 8,88 ммоль/л (у некоторых людей «почечный порог для глюкозы» может быть выше), глюкоза полностью не реабсорбируется и выделяется с мочой.
Погегная глюкозурия развивается при наследственных тубулопатиях (например, синдроме Фанкони) и хронических заболеваниях почек; при отравлении свинцом, ртутью, флоридзином, флорэтином.
Глюкоза — осмотически активное вещество, поэтому присутствие ее в моче в больших количествах мешает реабсорбции воды и вызывает осмотический диурез. Глюкозурия, как правило, сопровождается полиурией, гипогидратацией, жаждой и полидипсией.
Липидурия
Липидурия является следствием гиперлипидемий, характерна для нефротического синдрома (особенно первичного) и сопровождается появлением в моче зернистых (содержащих жир) и восковидных цилиндров.
Патофизиология почек I
549
Билирубинурия
Билирубинурия (выделение с мочой конъюгированного билирубина) развивается в том случае, когда количество этого желчного пигмента в крови превышает 34 мкмоль/л. Характерна для печеночной и подпеченочной желтух, сопровождается увеличением в моче желчных кислот. Моча при этом становится темной из-за присутствия пигмента и легко вспенивается при встряхивании, так как ЖК уменьшают поверхностное натяжение жидкости. Неконъюгированный билирубин в моче никогда не обнаруживается, так как является нерастворимым и не проходит клубочковый фильтр. Нормальная моча имеет светло-желтую окраску из-за присутствия в ней небольшого количества стеркобилина.
Внепочечные (экстраренальные) симптомы и синдромы, характерные для нефропатий
Азотемия — повышение в крови остаточного азота более 30 ммоль/л или его главных компонентов (мочевины более 6 ммоль/л, креатинина более 0,1 ммоль/л).
Под остаточным азотом понимают совокупность азотсодержащих веществ плазмы крови (50 % его составляет мочевина, 25 % — азот аминокислот, 8 % — эрготианин, 5 % — креатин, 5 % — азот полипептидов и небелковых соединений, 4 % — мочевая кислота, 2,5 % — креатинин, 0,5 % — аммиак и индикан). У здоровых людей уровень остаточного азота колеблется от 7 до 12 ммоль/л. При нефропатиях, сопровождающихся нарушением фильтрационной и экскреторной функций почек (например, при ГН), он значительно повышается. При острой и хронической почечной недостаточности (при уремии) может достигать 350 ммоль/л. Чем выше уровень остаточного азота в крови (или мочевины и креатинина), тем больше нарушена экскреторная функция почек.
Азотемия всегда сопровождается тяжелой интоксикацией организма и нарушением функций многих систем: нервной, сердечно-сосудистой, пищеварительной, системы крови и других (см. острую и хроническую почечную недостаточность в гл. 37).
Гипергидратация, отеки. Нарушения водного и электролитного баланса характерны для большинства заболеваний почек. Все случаи олигурии (см. выше) сопровождаются задержкой жидкости в организме и выраженным отечным синдромом. При этом у больных увеличивается ОЦК (объем циркулирующей крови) и происходит перегрузка сердца с
550
Лекция 36
его дилатацией или гипертрофией, растет артериальное и внутричерепное давление. На фоне анурии легко развиваются отек мозга, отек легких и другие грозные осложнения.
В патогенезе «почечных» отеков ведущую роль могут играть следующие факторы:
• увеличение эффективного фильтрационного давления (ЭФД) в капиллярах, обусловленное гиперволемией и ростом гидростатического давления (механический фактор);
• снижение эффективной онкотической всасывающей силы (ЭОВС) в капиллярах вследствие протеинурии и гипопротеинемии (онкотический фактор);
• активация РААС и вторичный альдостеронизм, спровоцированный уменьшением почечного кровотока и повышенной секрецией ренина из ЮГА;
• повышение проницаемости сосудов, например, при ГН с генерализованным капилляритом или других аутоиммунных заболеваниях (мембраногенный фактор);
• лимфатическая недостаточность, обусловленная перегрузкой лимфооттока.
Для «почечных» отеков характерна локализация на лице (пастозность лица) и особенно в области глаз. Однако при тяжелой гипергидратации отеки могут быть генерализованными, например, при нефротическом синдроме (см. ниже) и на фоне анурии.
Гипогидратация может развиться у больных на фоне полиурии в тех случаях, когда потеря воды с мочой не компенсируется ее адекватным поступлением в организм. Обезвоживание не менее опасно, чем гипергидратация, так как при этом может снизиться ОЦК, АД, сердечный выброс, повыситься вязкость крови и риск тромбоза. В результате нарушения центрального и периферического кровообращения разовьется гипоксия, ацидоз.
Дисбаланс электролитов характеризуется изменением содержания в крови натрия, калия, кальция, магния, ионов хлора, бикарбоната. Однако эти изменения зависят от конкретной нозологической формы нефропатии (см. ниже).
Нефротигеский синдром может быть первигным (липоидный нефроз) и вторигным — при ГН, пиелонефрите и любых других заболеваниях почек, сопровождающихся протеинурией более 3 г/сут. Его патогенез складывается из сочетания резко выраженной гипергидратации и отечного синдрома с гиперлипидемией, повышенной свертываемостью крови и снижением иммунитета.
Патофизиология почек I
551
Главными в механизме нефротических отеков являются:
• снижение онкотического давления крови и ЭОВС:
• вторичный альдостеронизм;
• лимфатическая недостаточность. При этом может развиваться ана-сарка (отек всего тела, всех органов), асцит и другие водянки.
Гиперлипидемия характеризуется повышением в крови больных холестерина и ЛПНП на фоне снижения ЛПВП, вследствие чего повышается риск развития атеросклероза, артериальной гипертензии и ишемической болезни сердца. У больных появляется липидурия с большим количеством зернистых цилиндров. Механизм этих нарушений имеет несколько объяснений. В целях компенсации дефицита белка печень усиливает его синтез. Параллельно повышается синтез липопротеидов и холестерина (общий метаболический путь). С мочой теряется фактор липолиза и до 1 % ЛПВП, что способствует увеличению ЛПНП, которые легко откладываются в стенках сосудов, в том числе в гломерулах и в мезангии. Повреждение почек усиливается. Снижается активность ЛХАТ (лецитинхолестеринацетилтрансфера-зы) — модулятора обмена триглицеридов в плазме, катализатора эсте-рификации холестерина. Снижается клиренс триглицеридов, так как избыток холестерина подавляет ЛПЛ.
Повышенный риск тромбообразования у больных с нефротическим синдромом обусловлен увеличением в крови прокоагулянтов на фоне дефицита антикоагулянтов и ослабления фибринолитической системы. На фоне неселективной протеинурии с мочой выводятся антитромбин-111 и иммуноглобулины.
Нарушение синтеза и потеря иммуноглобулинов приводят к снижению гуморального иммунитета и повышает риск развития инфекционных осложнений.
Погегная артериальная гипертензия осложняет течение большинства нефропатий. Ее патогенез связывают, прежде всего, с активацией РААС, так как при заболеваниях почек часто уменьшается почечный кровоток, приводящий к повышенной секреции ренина из ЮГА. При этом повышение тонуса резистивных сосудов (их спазм) обусловлен как действием на гладкие мышцы сосудистой стенки ангиотензина-П, так и вазопрессина (АДГ). Секреция АДГ усиливается из-за повышения осмоляльности плазмы крови, обусловленной гипернатриемией (вторичный альдостеронизм). Кроме того, избыток в крови Na* приводит к повышению чувствительности сосудов к катехоламинам, отеку эндотелиоцитов и увеличению ОЦК (задержка воды способствует нор
552
Лекция 36
мализации осмоляльности плазмы, но одновременно повышает АД). Гиперволемия особенно выражена у больных с олигурией и анурией при почечной недостаточности. В ряде случаев поврежденные почки уменьшают выработку важнейших депрессорных факторов — простагландинов и кининов. Артериальную гипертензию, в патогенезе которой этот механизм является ведущим, принято называть «ренопривной». У некоторых больных важную роль в патогенезе почечной артериальной гипертензии играет наследственная предрасположенность. При выраженном склерозе почечной артерии гипертензия может носить злокачественный характер и приводить к необходимости пересадки почки.
Анемия часто развивается у больных с патологией почек. В ее патогенезе могут играть роль несколько факторов: уменьшение продукции эритропоэтинов в ЮГА; потеря с мочой эритроцитов, железа, трансферрина при хронической патологии почек, сопровождающейся гематурией и протеинурий; повреждение костного мозга, усиление неэффективного эритропоэза, нарушение включения железа в гем под влиянием уремических токсинов, ингибиторов эритропоэза (см. хронигескую погегную недостатогность в гл. 37).
Лекция 37
ПАТОФИЗИОЛОГИЯ ПОЧЕК II
Г.П. Щелкунова
Этиология и патогенез заболеваний почек
Все нефропатии принято делить на две группы — иммунные и неиммунные.
К иммунным нефропатиям относятся гломерулонефрит (ГН), нефропатии при системных аутоиммунных заболеваниях (системная красная волчанка, узелковый периартериит, ревматизм), нефропатия беременных (под вопросом). В этих случаях развивается иммуноопосредован-ное воспаление, при котором образующиеся медиаторы повреждают главным образом гломерулярный аппарат, а также другие структуры нефрона (см. гломерулонефрит).
К неиммунным нефропатиям относятся:
• инфекционно-воспалительные заболевания почек (пиелонефрит, абсцесс почки, туберкулезное, сифилитическое поражение почек и т.п.);
• метаболигеские нефропатии (при амилоидозе, сахарном диабете, подагре и др.);
• токсигеские нефропатии (при отравлениях ядами и лекарствами, при радиационном повреждении и синдроме длительного раздавливания тканей (краш-синдром);
• вторигные нефропатии (при нарушениях обмена электролитов, недостаточности кровообращения):
• сосудистые нефропатии (атеросклероз почечной артерии с тяжелой формой злокачественной гипертонии и т.п.);
• опухоли почки;
• врожденные нефропатии (поликисгоз, тубуло- и энзимопатии, например, синдром Фанкони, болезнь Альпорта и др.).
554
Лекция 37
Пиелонефрит
Пиелонефрит (ПН) — генетически обусловленное инфекционновоспалительное заболевание мочевыводящих путей и паренхимы почек.
Составляет 39 % всех нефропатий. О наследственной предрасположенности к этому заболеванию свидетельствуют выявление у этих больных различных аномалий нефронов, повышенная плотность рецепторов к инфекционным агентам в мочевыводящих путях и особый настрой иммунной системы, характеризующийся наличием характерных генетических маркеров в комплексе гистосовместимости. Инфекционные агенты (кишечная палочка, стрептококки, стафилококки, вирусы, грибы, микоплазмы и др.) проникают в мочевыводящие пути чаще по мочеточникам (восходящий путь), реже — гематогенно или лимфогенно. Способствуют развитию заболевания снижение иммунитета и общей резистентности организма, изменения гормонального баланса, а также нарушения уродинамики при мочекаменной болезни, перегибах и стриктурах мочеточников, аденоме предстательной железы и т.п. Воспалительный процесс, как правило, охватывает лоханки, чашечки и все мозговое вещество почек с находящимися в нем канальцами, петлями Генле и собирательными трубками почек. Корковое вещество с расположенными в нем гломерулами в патологический процесс вовлекается редко. Это и определяет клинические особенности ПН.
В типичных случаях развития ПН можно выделить три главных клинических синдрома: «мочевой», поллакиурический и интоксикационный.
В «мотевом» синдроме ведущим является выраженная лейкоцитурия (может быть пиурия). Обычно наблюдается микрогематурия (редко макрогематурия) с неизмененными эритроцитами в моче. Протеинурия умеренная (редко >3 г/л), не клубочковая. Характерны также цилиндрурия и патологическая бактериурия (>10 ООО в 1 мл).
Поллакиуригеский синдром включает полиурию, дизурию (частые и болезненные мочеиспускания), тупые боли в поясничной области и ощущения холода в пояснице. Развитие полиурии объясняется нарушением концентрационной функции канальцев и собирательных трубок из-за воспалительного отека мозгового вещества почек и нарушения противоточно-множительного механизма в петлях Генле, из-за повреждения эпителия канальцев и собирательных трубок почек. Полиурия сочетается с гипосгенурией и полидипсией. Иногда при скрытом течении
Патофизиология почек II
555
ПН нарушение концентрационной функции почек является единственным проявлением болезни, и в этом случае полезным может оказаться проведение пробы с 18-часовым сухоедением.
Интоксикационный синдром является проявлением общих реакций организма на инфекцию («ответ острой фазы») и характеризуется наличием у больных лихорадки с ознобом (иногда озноба без лихорадки), нейтрофильного лейкоцитоза со сдвигом влево и ускорения СОЭ. У разных больных может преобладать любой из этих синдромов. Описаны септические формы острого ПН с резко выраженным интоксикационным синдромом, абдоминальные формы с картиной «острого живота», гематурические формы с макрогематурией.
Течение хронического ПН чаще бывает моносимптомным (например, лейкоцитурия или полиурия), но может быть в форме нефротического синдрома, или проявляться только артериальной гипертензией. Нарушение фильтрационной и экскреторной функции клубочков для ПН не характерно, поэтому снижение клиренса креатинина, появление азотемии у больных может свидетельствовать о развитии у них почечной недостаточности. Для диагностики ПН, кроме традиционных исследований мочи и функциональных почечных проб, дополнительно используют рентгенографическое и ультразвуковое исследование, эхографию и тепловидение. Лечат больных пиелонефритом антибиотиками, иммуностимуляторами и нестероидными противовоспалительными препаратами, проводят восстановление уродинамики и фитотерапию.
Гломерулонефрит
ГН — генетически обусловленное иммуноопосредованное воспаление с преимущественным исходным поражением гломерул и последующим вовлечением в патологический процесс всех почечных структур.
Составляет около 37 % всех нефропатий. В большинстве случаев выявляются признаки наследственной предрасположенности к данному заболеванию, в том числе генетические маркеры в главном комплексе гистосовместимости.
Различают два патогенетических варианта этого заболевания — наиболее часто встречающийся иммунокомплексный ГН и ГН с антительным механизмом, который по классификации иммунопатологий Джейла и Кумбса правильнее называть цитотоксигеским. Последний встречается редко.
556
Лекция 37
Иммунокомплексный ГН по патоморфологическим признакам подразделяют на три вида: мезангиально-пролиферативный, мембранозный и мембранозно-пролиферативный.
ГН с антителъным механизмом (цитотоксигеский) патоморфологически делится на два вида: экстракапиллярный (быстропрогрессирующий) и синдром Гудпасчера (сочетание ГН с поражением легких).
ГН обычно развивается через 10-20 сут после перенесенного инфекционного заболевания, в процессе которого образуются и накапливаются в большом количестве иммунные комплексы (АГ + АТ) или появляются АТ к структурам гломерул. Сенсибилизацию организма могут вызвать стрептококки (особенно гемолитический стрептококк типа А), стафилококки, энтерококки, диплококки, тифозная салмонелла, бледная трепонема, малярийный плазмодий, токсоплазма, грибы (чаще Candida albicans), а также вирусы герпеса, гепатита, Эпштейна-Барра и др. В редких случаях ГН развивается после применения лекарств, введения чужеродных сывороток и попадания ядов.
После образования и накопления комплексов АГ-АТ в различных структурах гломерулярного аппарата происходит активация системы комплемента с появлением хемоаттрактантов, анафилатоксинов и цитолитигеских {мембраноатакующих) комплексов. Корковое вещество почек инфильтрируется лейкоцитами. Активированные нейтрофилы, макрофаги, тромбоциты, эндотелиоциты и другие клетки выделяют медиаторы, под влиянием которых формируется воспаление.
Главную роль в его патогенезе играют: гистамин, простагландины, лейкотриены, активированные метаболиты кислорода, протеазы, вызывающие повышение проницаемости клубочковых и перитубулярных капилляров; фактор активации тромбоцитов (ФАТ), стимулирующий агрегацию тромбоцитов и тромбоз капилляров гломерул; факторы роста (ФР), приводящие к склерозированию сосудов; цитокины (ИЛ), ФНО, усиливающие местные и общие иммунные реакции организма.
Под влиянием этих процессов значительно уменьшаются число функционирующих клубочков и площадь фильтрации с одновременным повышением проницаемости гломерулярной мембраны, что сопровождается нарушением фильтрационной и экскреторной функций почек в сочетании с мочевым синдромом. При благоприятном течении процесса воспаление не распространяется на мозговое вещество почек и не нарушает функцию канальцев и собирательных трубок. При неблагоприятном развитии заболевания патологический процесс распространяется на все отделы нефрона и приводит к развитию острой или хронической почечной недостаточности (рис. 37-1).
Патофизиология почек II
557
Острый гломерулонефрит встречается редко (всего 2 % всех ГН) и чаще у детей. Он может протекать как моносимптомно (только с «мочевым» синдромом), так и в развернутой форме — с ярко выраженными ренальными и экс-траренальными симптомами и синдромами.
Наиболее типичные ренальные симптомы:
• олигурия (чаще умеренная, реже — в тяжелых случаях анурия):
• снижение клубогковой фильтрации и клиренса креатинина',
• «могевой синдром»: макрогематурия (моча в виде мясного смыва), обычно умеренная лейкоцитурия, протеинурия (клубочковая, и в зависимо-
Инфекция
Сенсибилизация
Образование и циркуляция в крови ИК
i
Оседание ИК в клубочках почек
Синтез антител к антигенам БМК
Образование ИК на БМК
Активация комплемента
Привлечение и активация лейкоцитов
Воспаление
I высвобождение медиаторов
Тромбоз капилляров клубочков
4 почечного кровотока
t проницаемости сосудов почек
«Мочевой» синдром
i 4 клубочковый фильтрации Активация РААС .
I диуреза Азотемия
Гипергидратация
f АД отеки
сти от степени проницаемо-сти клубочковой базальной мембраны — селективная или неселективная). У некоторых больных, теряющих с мочой более 3 г белка в сут-
Рис. 37-1. Патогенез гломерулонеф-ритов: I — иммунокомплексного; II — цитотоксического (с антительным механизмом) типов. Обозначения: ИК—иммунныекомплексы; БМК—базальная мембрана клубочков; РААС — ренин-ангиотензин-альдостероновая система; АД — артериальное давление
ки, может развиться нефротический синдром.Гипо- или изостенурии обычно нет. Ее появление прогностически неблаго
приятно, так как свидетельствует о распространении патологического процесса на канальцевые структуры и развитии почечной
недостаточности.
Наиболее характерные экстраренальные синдромы:
• азотемия (от умеренной до значительной с явлениями энцефалопатии и даже эклампсии);
• артериальная гипертензия:
• гипергидратация и отеки.
558
Лекция 37
В крови выявляют высокий титр специфических антител (чаще антистрептолизина О) и снижение фракций комплемента. Лейкоцитоз не характерен. Больные часто жалуются на сильную головную боль, снижение зрения и боли в поясничной области.
Продолжительность острого ГН — около 2 мес. При тяжелом его течении угрозами для жизни больного могут быть: энцефалопатия, острая почечная недостаточность и острая сердечная недостаточность. Главные принципы лечения острого ГН — диета с ограничением поваренной соли и белка, а также применение стероидных противовоспалительных препаратов.
Хронический ГН составляет 98 % всех ГН. Часто в течение нескольких лет протекает скрыто или моносимптомно. У одних больных может быть только артериальная гипертензия, у других — только отечный или только мочевой синдром. Описаны клинические случаи, когда больные впервые обращались к врачу уже во второй и даже в третьей стадии хронической почечной недостаточности. Этот факт должен побуждать врача к тщательному обследованию функций почек при малейшем подозрении на их неблагополучие и особенно после перенесенных инфекционных заболеваний. Следует подчеркнуть, что в последние годы окончательный диагноз острого или хронического ГН ставят только на основании результатов пункционной биопсии почки, позволяющей обнаружить иммунные комплексы (депозиты иммунных комплексов, симптом «горбов» и т.п.) в гломерулярных структурах и установить морфологический вариант ГН. Типичным морфологическим проявлением хронического ГН является обнаружение в микропрепаратах «полулуний». При хроническом ГН в патологический процесс вовлекается как корковое, так и мозговое вещество почек, повреждаются все отделы нефронов, нарушаются все функции почек и заболевание характеризуется как хроническая почечная недостаточность.
Хроническая почечная недостаточность
Хроническая почечная недостаточность (ХПН) — синдром, развивающийся вследствие уменьшения числа и изменения функции оставшихся нефронов, сопровождающийся нарушением всех функций почек и завершающийся развитием уремии.
ХПН в 80 % случаев развивается на фоне хронического ГН, в 18 % — на фоне хронического пиелонефрита и только 2 % приходится на поли-кистоз и другие нефропатии. При хроническом воспалении паренхимы почек (иммуноопосредованном или инфекционном) происходят тром
Патофизиология почек II
559
боз и постепенное склерозирование гломерулярных сосудов. Количество функционирующих клубочков и площадь фильтрации мочи уменьшаются. Оставшиеся неповрежденными нефроны гипертрофируются и выполняют большую нагрузку. По мере склерозирования (сморщивания) почек нарастает картина почечной недостаточности с характерными ренальными и экстраренальными проявлениями.
В развитии ХПН принято выделять 3 стадии: латентную, азотемическую и уремическую. В некоторых клиниках каждую из этих стадий подразделяют на А- и Б-подстадии.
Латентная стадия, несмотря на склерозирование даже половины клубочков, из-за компенсаторной гипертрофии оставшихся нефронов в течение многих месяцев и даже лет может протекать незаметно со слабо выраженными проявлениями в виде мочевого синдрома, или отеков, или артериальной гипертензии. Однако функциональные резервы почек уже снижены, и это выявляется специальными пробами. Снижен клиренс креатинина, указывающий на уменьшение клубочковой фильтрации, но не менее 50 % должной величины (т.е. не <50-60 мл/мин), и на снижение экскреторной функции. Концентрация креатинина в крови начинает расти, но не превышает 0,18 ммоль/л.
Азотемигеская стадия характеризуется склерозированием более 50 % нефронов и выраженными признаками почечной недостаточности. Клиренс креатинина может составлять 50-10 % должной величины, а концентрация креатинина в крови колеблется от 0,19 ммоль/л в начале стадии до 0,71 ммоль/л — в конце. Растет величина остаточного азота, концентрация мочевины и других азотсодержащих веществ, что и определило название данной стадии ХПН. У больных выявляется «мочевой» синдром, гипо- или изостенурия, почти у всех — артериальная гипертензия, анемия, компенсированный почечный ацидоз. Эти изменения свидетельствуют о нарушении всех функций почек.
Следует обратить внимание на то, что в течение нескольких лет, пока развивается эта стадия, несмотря на значительное уменьшение клубочковой фильтрации до 50 и даже 10 мл/мин (вместо 80-120 мл/мин), суточный диурез у больных может оставаться нормальным и даже повышаться. У ряда больных выявляются полиурия и никтурия (преобладание ночного диуреза над дневным), а также полидипсия. Объясняют этот феномен как нарушением концентрационной функции канальцев почек из-за повреждения мозгового вещества и уменьшением чувствительности эпителиальных рецепторов к АДГ, так и развитием так называемого осмотического диуреза. Дело в том, что через оставшиеся
560
Лекция 37
гипертрофированные канальцы на фоне нарастающей азотемии протекает моча, содержащая все больше мочевины. Осмотически активная мочевина препятствует реабсорбции воды в канальцах почек. Абсолютное же количество выводимой из организма мочевины уменьшается. Задержка ее в крови приводит к росту осмоляльности плазмы на фоне уменьшения осмоляльности мочи. Концентрационный индекс — отношение осмоляльности мочи к осмоляльности плазмы — снижается (в норме он равен 3,3), что в сочетании с гипо- или изостенурией характеризует нарушенную функцию канальцев почек. Осмоляльность повышается не только в крови, но и в интерстиции и внутри клеток, так как мочевина легко проникает через клеточные мембраны. Возникающая при этом жажда и большее поступление воды в организм, несмотря на повышенный или нормальный диурез, способствует развитию внутри- и внеклеточной гипергидратации.
Важно помнить, что каждое новое увеличение мочевины и креатинина в крови является признаком прогрессирования заболевания, которое подтверждается также результатами патоморфологического исследования пунктата почечной ткани (число склерозированных нефронов увеличивается, а функционирующих — уменьшается).
Уремигеская стадия начинается, как правило, с появления олигурии и особенно выражена на фоне анурии. Клиренс креатинина при этом снижается до 10 мл/мин и ниже, а концентрация креатинина в крови колеблется в интервале 0,72-1,24 ммоль/л. Уремия дословно означает «мочекровие», т.е. другими словами, задержка в крови тех токсических продуктов метаболизма (прежде всего азотистых), которые должны выводиться из организма с мочой.
Уремия — синдром, характеризующийся отравлением организма уремическими токсинами, тяжелой азотемией, гипергидратацией и нарушениями электролитного баланса, а также некомпенсированным метаболическим ацидозом и нарушением функции всех важнейших систем.
К уремигеским токсинам, которых сегодня описано уже более 200, относят мочевину, аммиак, креатинин, гуанидин, гуанидин-янтарную кислоту, метилгуанидин, ароматические и алифатические амины, ураты, фенолы, гидрокислоты, так называемые средние молекулы (фрагменты пептидов с мол. массой около 500) и др. Уремические токсины вызывают гемолиз эритроцитов, повреждение костного мозга с усилением неэффективного эритропоэза и нарушением включения железа в гем, повреждение слизистых оболочек ЖКТ и дыхательных путей, блокирование многих ферментов и повреждение клеточных мембран,
Патофизиология почек II
561
угнетение иммунитета, неврологические и психические расстройства и другие тяжелые последствия. Например, избыток в крови аммиака приводит к связыванию а-кетоглютаровой кислоты, выключению ее из цикла Кребса и уменьшению образования АТФ. Повышение NH3 также нарушает процесс переаминирования аминокислот и синтеза мочевины, стимулирует синтез холестерина и кетоновых тел.
Мочевина относительно нетоксичный продукт до тех пор, пока концентрация ее в крови не превысит 20 ммоль/л. Превышение этого порога приводит к тому, что мочевина начинает выделяться из организма через кожу и слизистые оболочки, в серозные полости (особенно в полость перикарда и плевральную полость), вызывая их повреждение и воспаление. Она может также вызывать перегруппировку альбуминов, менять структуры белков, делая их гетерогенными, способными стимулировать аутоиммунные процессы.
При уремии нарушаются все виды обмена веществ, и прежде всего водный и электролитный баланс. Пшергидратация в сочетании с гипе-росмоляльным синдромом приводит к гиперволемии, еще большему повышению АД, перегрузке сердца, повышению внутричерепного давления и может повлечь за собой отек мозга, отек легких или РДСВ (респираторный дистресс-синдром взрослых). Концентрация К2+ и Mg в крови обычно повышается из-за массивного повреждения клеток. Концентрация Са2*, Na+ и С1* в большинстве случаев снижается, что можно объяснить нарушением их всасывания в кишечнике, потерей с рвотными массами и поносом, перемещением в клетки и костную ткань, а также гидремией.
Уремия всегда сопровождается тяжелейшим некомпенсированным ацидозом, так как через оставшиеся гломерулы не фильтруются сульфаты и фосфаты, которые, задерживаясь в крови, уменьшают ее щелочные резервы. Но главным в механизме ацидоза является нарушение в канальцах процессов ацидо- и аммониогенеза и реабсорбции бикарбоната. К тому же на фоне развивающейся гипоксии и нарушенного обмена вещества усилено образование Н* (накапливаются лактат, пируват, кетоновые тела и другие кислые продукты).
У 100 % больных ХПН выявляется артериальная гипертензия еще в первых стадиях, но на фоне уремии она достигает критических цифр, так как повышенный тонус артериол в результате активации РААС сочетается с тяжелой гиперволемией.
Изменения со стороны центральной нервной системы можно назвать уремигескойэнцефалопатией. Она проявляется сильной головной болью, нарушениями сна, патологическим рефлексами, менингеальными сим
562
Лекция 37
птомами, а также помрачением и утратой сознания. Ее патогенез связан прежде всего с гипергидратацией, повреждением нервных структур уремическими токсинами, ацидозом, а также с дисбалансом электролитов.
Гипертрофия миокарда, аритмии, боли в сердце, шум трения перикарда (который врачи называют «похоронным звоном уремика») и другие проявления нарушения сердечной деятельности называют уремитеской кардиопатией, механизм которой имеет аналогичные объяснения.
Гиперсаливация, тошнота, рвота, понос, желудочно-кишечные кровотечения и другие нарушения ЖКТ объединяются термином уремиге-ская гастро- и энтеропатия.
Их развитие обусловлено выделением уремических токсинов через слизистые оболочки.
Из-за возникновения легочной гипертензии (гипергидратация + сердечная недостаточность) и повышения проницаемости легочных сосудов у больных развивается уремигеский пневмонит с одышкой, хрипами, кашлем и другими признаками дыхательной недостаточности. Наибольшую угрозу представляет возможное развитие отека легких или РДСВ.
Почти у всех больных с хронической ПН наблюдается ренальная остеодистрофия (остеомаляция, остеопороз, оссалгии), так как под влиянием избытка паратгормона (его также относят к уремическим токсинам) костная ткань теряет кальций. Все усугубляется гипокальциемией.
Если к этому добавить наличие у больных тяжелой анемии и резкого снижения иммунитета, то становится понятным, что продолжительность 3-й стадии ХПН, в отличие от двух предыдущих, может быть не более нескольких недель.
Уремия — это финал необратимого процесса склероза почек, поэтому единственным радикальным способом лечения больных в этой стадии ХПН является трансплантация почки. Гемодиализ показан только на период подбора донора. Для того чтобы замедлить развитие ХПН, необходимо еще в 1-й и во 2-й стадии проводить лечение основного заболевания (ГН, пиелонефрита), специальными диетами и лекарственными препаратами уменьшать азотемию, периодически проводить гемодиализ с целью освобождения крови от токсических продуктов и восстановления водно-электролитного баланса и КОС.
Острая почечная недостаточность
Острая почечная недостаточность (ОПН) — синдром, развивающийся в результате нарушения всех функций почек и характеризующийся азотемией, нарушением водно-электролитного баланса и КОС.
Патофизиология почек II
563
Различают 4 формы ОПН — преренальная, ренальная, постренальная и аренальная.
Преренальная ОПН или острая циркуляторная нефропатия может развиться при шоке, коллапсе, острой сердечной недостаточности, тяжелой гиповолемии, т.е. в тех случаях, когда из-за резкого падения системного артериального давления ниже 80 мм рт.ст. уменьшаются почечный кровоток и ГД в капиллярах клубочков. При этом ЭФД снижается и даже приближается к нулю, фильтрация первичной мочи уменьшается или прекращается совсем. Падение АД ниже 50 мм рт.ст. влечет за собой развитие анурии.
Ренальная ОПН может быть инфекционной, токсической и сосудистой. Она может развиться при сепсисе, холере, краш-синдроме, при отравлении грибами, ртутью, хлороформом и другими ядами, а также при тромбозе или эмболии сосудов почек. У детей ОПН иногда осложняет течение острого ГН. Во всех этих случаях происходят повреждение всех отделов нефрона и нарушение всех функций почек.
Постренальная ОПН является следствием нарушения оттока мочи, например, при обтурации или сдавлении мочевыводящих путей (камни, опухоли, стриктуры, перегибы мочеточника и т.п.).
Аренальная ОПН может развиться в тех редких случаях, когда человек по какой-то причине лишается единственной имевшейся у него почки.
Механизм развития ОПН включает в себя несколько патогенетических факторов:
• нарушение почечного кровотока, приводящее к снижению ЭФД и уменьшению фильтрации мочи в почечных клубочках;
• гипоксическое повреждение канальцев, сопровождающееся отеком и некрозом эпителия (считается главным);
• повышенный выход мочевины и воды через поврежденные канальцы и собирательные трубки в мозговое вещество почек с последующим отеком интерстиция и сдавлением канальцев снаружи;
• обтурация мочевыводящих путей цилиндрами, спущенным эпителием, детритом, нарушающая отток мочи и приводящая к повышению ДМП и снижению ЭФД в клубочках, т.е. к еще большему уменьшению фильтрации первичной мочи.
В патогенезе ОПН важную роль может играть включающийся часто порочный круг (рис. 37-2). Уменьшенный почечный кровоток, приводящий к снижению ЭФД и клубочковой фильтрации, одновременно вызывает гипоксическое повреждение канальцев и нарушение их функ-
564
Лекция 37
| почечного кровотока и
| клубочковой фильтрации
Повреждение канальцев
i
4 реабсорбции Na’
f Na’ в канальцевой моче
Раздражение ЮГА
f ренина
f АТ-11
Спазм приносящих артериол клубочков —►
4 ГД ЭФД-ЦЗФД
» Олигурия Анурия
{ГД-ЦЭФД
Рис. 37-2. Порочный круг в патогенезе ОПН
ции, в том числе уменьшение реабсорбции Na*. Увеличение концентрации натрия в моче, протекающей по канальцам, приводит к стимуляции ЮГА и выбросу ренина. Образующийся ангиотензин-П вызывает спазм приносящих артериол клубочков, т.е. еще большее снижение ГД и ЭФД. Таким образом, данный порочный круг усиливает развитие олигурии и анурии.
В развитии ОПН принято выделять 4 периода: начальный, период олигоанурии, период полиурии (восстановления диуреза) и выздоровления.
Проявления и особенности натального периода зависят от причины ОПН (шок, коллапс, сепсис, отравление, краш-синдром, ДВС-синдром и Т.Д.).
Самым опасным является второй олигоануригеский период, продолжающийся обычно 1-2 нед. Многие врачи состояние больного в этом периоде не без оснований характеризуют как уремию. И хотя 2-я стадия ОПН действительно имеет много сходства с 3-й стадией ХПН, главное отличие их в том, что уремия при ХПН завершает необратимый патологический процесс склерозирования и сморщивания почек, а длительное (годами) течение болезни приводит к серьезным нарушениям со стороны других важнейших систем организма. Во 2-й стадии ОПН величина клубочковой фильтрации (клиренс креатинина) может быть снижен до 10 мл/мин и менее. Больной за сутки выделяет очень мало (50 мл и менее) темной мутной мочи, содержащей цилиндры, белок, эритроциты. Нарушение экскреторной и фильтрационной функции почек влечет за собой развитие тяжелой азотемии и гипергидратации с картиной энце
Патофизиология почек II
565
фалопатии. Остаточный азот растет до 300-350 ммоль/л. Содержание мочевины в крови повышается до 20 ммоль/л и выше, концентрация креатинина — до 0,9-1,3 ммоль/л. Задержка воды приводит к гиперволемии, перегрузке сердца и его дилатации, к повышению внутричерепного давления и появлению менингеальных симптомов, спутанности сознания. На фоне выраженного генерализованного отечного синдрома наибольшую угрозу для жизни больного представляет развитие отека мозга, отека легких и респираторного дистресс-синдрома (РДСВ), так как на фоне легочной гипертензии под влиянием эндогенных токсических веществ более чем в два раза повышается проницаемость легочных сосудов.
Так как при ОПН повреждаются все отделы нефронов, в том числе и канальцы, обеспечивающие регуляцию КОС в организме, выпадение этой важнейшей функции влечет за собой развитие тяжелого некомпенсированного почечного ацидоза. Мочевина и другие токсичные продукты метаболизма, которые не могут удаляться из организма с мочой, начинают выделяться через кожу (может быть дерматит), через слизистые оболочки (возникают язвенно-некротические повреждения слизистой оболочки ЖКТ с тошнотой, рвотой, кровотечениями), в полость перикарда (сухой перикардит с шумом трения перикарда), через легкие (может развиться пневмония, РДСВ).
Дисбаланс электролитов обычно проявляется гиперкалиемией (реже гипокалиемией) и гипермагниемией из-за массивного разрушения клеток. Концентрация натрия, кальция и хлора обычно снижается. Это может быть обусловлено уменьшением их всасывания в кишечнике, потерей с рвотными массами, перемещением в клетки и гидремией. У больных может развиться анемия, геморрагический синдром (иногда и ДВС-синдром). Характерно также резкое снижение иммунитета.
Главными угрозами для жизни больного в этом периоде могут быть: присоединение инфекции, отек мозга или легких, РДСВ, сердечная недостаточность, остановка сердца. При увеличении К+ в крови более 7 ммоль/л, мочевины — до 24 ммоль/л, появлении дыхания Куссмау-ля (признак тяжелого ацидоза) и угрозы вышеназванных осложнений больному показан гемодиализ. Необходимо проводить восстановление водно-электролитного баланса и КОС, выводить токсические метаболиты, уменьшать азотемию, проводить профилактику инфекции и других осложнений.
Патологический процесс при ОПН, в отличие от ХПН, является обратимым, поэтому за то время, пока аппарат «искусственная почка» будет
566
Лекция 37
выполнять функции поврежденных органов, в почках начнется процесс восстановления.
Полиуритеский период (восстановления диуреза) называется так потому, что вначале происходит восстановление процесса фильтрации в клубочках и, хотя клиренс креатинина до конца периода остается сниженным, из-за нарушенной концентрационной функции канальцев развивается полиурия и гипостенурия. Если в предыдущем периоде больному угрожала гипергидратация, то в этом периоде — не менее опасное по своим последствиям — обезвоживание. Сохраняется «мочевой» синдром и резкое снижение иммунитета. Только к концу периода возвращаются к норме клиренс креатинина и величина остаточного азота в крови.
Для того чтобы нормализовались все показатели гомеостаза, необходимо около двух лет (период выздоровления). У некоторых больных после ОПН могут развиться хронический пиелонефрит, хроническая почечная недостаточность или артериальная гипертензия.
Мочекаменная болезнь
Мочекаменная болезнь (уролитиаз) — распространенное заболевание, встречающееся в любом возрасте, даже у новорожденных. Камни локализуются чаще в правой почке, в 15-20 % случаев в обеих почках, у детей и стариков камни чаще образуются в мочевом пузыре.
Камни почек (нефролитиаз) — заболевание полиэтиологическое. Важнейшими причинами образования камней являются прежде всего врожденные патологические изменения в почках и мочевыводящих путях: энзимопатии (тубулопатии) с преимущественным нарушением функции проксимальных и дистальных канальцев почек, пороки ана-томигеского развития могевыводящих путей, наследственные нефрозе- и нефритоподобные синдромы. Наиболее распространенными тубулопатиями, способствующими образованию камней в почках, являются: оксалурия, цистинурия, аминоацидурия, галактоземия, фруктозе-мия, лактоземия и рахитоподобные заболевания. При тубулопатиях в почках скапливаются вещества, идущие на построение камня.
Камни почек и мочевыводящих путей чаще всего состоят из оксалата кальция, фосфата кальция, мочевой кислоты, магний-аммоний-фосфата, цистина.
Факторы, способствующие образованию камней почек на фоне тубулопатий, разделяют на экзо- и эндогенные. Эндогенные — в свою очередь, делятся на общие и местные (почечные).
Патофизиология почек II
567
К экзогенным факторам относятся климатические и географические условия, флора и фауна, особенности питания и т.д. Важную роль играют температура и влажность воздуха, характер почвы, состав питьевой воды и содержание в ней минеральных солей. Например, у жителей жарких стран на фоне тенденции к обезвоживанию выделяется более концентрированная моча, способствующая камнеобразованию. Растительная и молочная пища способствует ощелачиванию мочи, а мясная — ее закислению. Питьевая вода с избытком известковых солей уменьшает кислотность мочи и повышает содержание солей кальция в организме. Высокую частоту нефролитиаза у жителей Заполярья связывают с полигиповитаминозом, дефицитом УФЛ и преобладанием в рационе мясной и рыбной пищи.
Среди эндогенных факторов, способствующих нефролитиазу, особое место занимает гиперфункция околощитовидных желез (гиперпарати-реоидизм), сопровождающийся нарушением фосфорно-кальциевого обмена, увеличением Са2+ в крови и в моче. Гиперкальциемия и каль-цийурия наблюдаются также при остеомиелитах, травмах костей, остеопорозах и т.д.
Нефролитиаз может развиться на фоне заболеваний ЖКТ (хронический гастрит, колит, язвенная болезнь), так как при этих патологиях возможны нарушения КОС и обмена кальция. У детей на фоне длительного обезвоживания (при дизентерии, токсической диспепсии) в почках легко осаждаются соли мочевой кислоты, дающие начало образованию камней.
Наконец, важную роль в образовании камней играют местные по-гегные факторы: застой мочи, нарушение секреции и экскреции ее составных элементов, воспалительные процессы. Нарушению уродинамики способствуют врожденные аномалии почек (удвоение и дистопия, подковообразная почка, клапаны и сужения мочеточника, пузырно-мочеточниковый рефлюкс и т.п.), а также гидронефроз, нефротуберку-лез, воспалительные стриктуры мочеточников, беременность и другие причины. На фоне воспаления в почках нарушается поверхностное натяжение между слизистой оболочкой чашечно-лоханочной системы и мочой, усиливающее явления адсорбции. Некоторые микробы (стафилококк, протей, синегнойная палочка) способны расщеплять мочевину мочи, что приводит к ее защелачиванию и выпадению в осадок фосфатов. Фосфатные камни чаще встречаются у женщин на фоне нарушения уродинамики и инфекции мочевыводящих путей. В образовании оксалатных и уратных камней большую роль играют общие метаболические нарушения, что чаще наблюдается у мужчин.
568
Лекция 37
Согласно коллоидно-кристаллоидной теории в моче содержатся защитные коллоиды, препятствующие кристаллизации солей. При нарушении функции канальцев в моче появляется много полисахаридов и мукопротеидов, которые нарушают коллоидное равновесие. Накопление в перенасыщенном растворе мочи кристаллоидов и выпадение их в осадок с последующей кристаллизацией ведут к образованию камней.
Согласно теории матрицы первоначально образуется белковый остов, на котором вторично откладываются соли. Ядром камня всегда является органическая субстанция, которая может стать материалом образования всего камня (белкового, цистинового), или чаще только матрицей, на которую оседают различные соли.
По химическому составу различают оксалатные, фосфатные, уратные, карбонатные камни. Реже встречаются камни цистиновые, ксантиновые, белковые и холестериновые.
Наиболее характерными проявлениями мочекаменной болезни являются: приступообразные боли в поясничной области (почечная колика), гематурия, пиурия и дизурия. Эти симптомы часто сочетаются с желудочно-кишечными расстройствами и общей интоксикацией, головной болью, лихорадкой, лейкоцитозом и другими проявлениями ответа острой фазы (ООФ). Мелкие камни могут самопроизвольно выходить с мочой, а крупные, находясь длительно в лоханках или чашечках почки или в мочеточнике (уретеролитиаз), часто приводят к серьезным осложнениям: к острому или хроническому калькулезному пиелонефриту, калькулезному пионефрозу (гнойное расплавление почки) или гидронефрозу. Эти осложнения, в свою очередь, могут повлечь за собой развитие нефрогенной артериальной гипертензии, а также острой или хронической почечной недостаточности.
К принципам легения могекаменной болезни относятся лечение воспалительных заболеваний почек, восстановление уродинамики, нормализация обменных процессов, диетотерапия, дробление камней или удаление их хирургическим путем.
Лекция 38
ПАТОФИЗИОЛОГИЯ ШОКОВЫХ состояний
Т.Ю. Ругинская
Шок (по англ. — удар) — остро развивающийся угрожающий жизни патологический процесс, характеризующийся тяжелыми нарушениями системного кровотока, микроциркуляции, клеточного метаболизма и глубокими нарушениями функций многих органов и тканей.
Впервые шок был описан в 1895 г. Больной со сложными множественными переломами и незначительными кровотечениями не реагирует на окружающее, смотрит на врача индифферентно. Черты его лица обострены, холодный пот, пульс почти не прощупывается, температура тела снижена. Мышечного паралича нет, но больной не пытается двигаться, чувствительность сохранена, отмечается апатия.
Шок возникает при неадекватной перфузии тканей кровью, что приводит к недостаточному снабжению тканей кислородом и неадекватному удалению из ткани продуктов обмена. Поскольку нарушение перфузии тканей может быть результатом многих факторов, которые приводят к снижению функции сердца как насоса или снижению объема крови и давления, шок может иметь множество причин и различные клинические проявления.
По этиологии различают следующие виды шока:
• кардиогенный — вызванный сердечной недостаточностью;
• нейрогенный — вызванный нарушением тонуса гладких мышц сосудов;
• септигеский — вызванный инфекцией;
• гиповолемигеский — вызванный недостаточностью внутрисосудистого объема крови;
• аллергигеский — результат аллергической реакции немедленного типа;
• комбинированные виды шока-, травматический — комбинация гиповолемического и септического шока.
570
Лекция 38
В зависимости от тяжести шока выделяют три главные стадии шока: • непрогрессирующая (компенсаторная):
• прогрессирующая;
• стадия необратимых изменений.
Непрогрессирующая стадия шока отличается от прогрессирующей отсутствием порочных кругов, появление которых во второй стадии означает быстрое самоускоряющееся повреждение тканей. О стадии необратимых изменений говорят тогда, когда самые энергичные терапевтические воздействия на больного не способны вывести его из состояния шока.
Гиповолемический шок
Этот вид шока возникает при уменьшении объема циркулирующей крови (ОЦК). ОЦК у здорового человека составляет 7-8 % массы тела. Уменьшение ОЦК приводит к падению давления наполнения циркуляторной системы и к снижению венозного возврата в сердце. Снижение венозного возврата сопровождается уменьшением минутного объема сердца (МОС) и падением артериального давления (АД). Возникают условия для неадекватной перфузии органов и ишемического повреждения тканей.
Гиповолемия может быть обусловлена:
• кровопотерями: при ранениях сосудов, болезнях ЖКТ или легких, разрыве маточных труб и т.д.;
• плазмопотерями: при ожогах, перитонитах:
• потерями жидкости через ЖКТ, кожу, почки: при поносе, рвоте, обильном потении, сахарном и несахарном диабетах, при приеме диуретиков.
Тяжесть гиповолемического шока определяется величиной гиповолемии, скоростью с которой она развивается и состоянием реактивности пострадавшего. Оценка тяжести гиповолемического шока может быть сделана на основании измерений основных показателей системного кровообращения — МОС и АД (табл. 38-1). На практике чаще всего измеряют кровяное давление. При этом надо помнить, что АД само по себе не определяет тяжести шока, поскольку шок — результат неадекватного кровоснабжения. В отдельных (редких) случаях шок может развиться при нормальном кровяном давлении. Напротив, низкое кровяное давление, даже в половину ниже нормы, не обязательно указывает на шок (рис. 38-1). На графике видно, что уменьшение ОЦК на 10 % не сопровождается существенными изменениями МОС и среднего АД. Заметное прогрессивное падение МОС и АД обнаруживается, когда ОЦК снижа-
Патофизиология шоковых состояний
571
Рис. 38-1. Изменения артериального давления и минутного объема сердца (в %) от контроля при острой кровопотери
ется более 10-15 %. К шоку обычно приводит потеря более 30 % крови. Если человек теряет более половины объема крови, смерть наступает через несколько часов.
Таблица 38-1. Корреляция клинических проявлений и дефицита объема крови при гиповолемическом шоке
Тяжесть шока Клинические проявления Снижения ОЦК, %
Отсутствует Отсутствуют До 10 % (500 мл)
Легкая Брадикардия или минимальная тахикардия, АД — норма. Кожа бледная, холодные конечности, снижение температуры тела и диуреза. 15-25 %
Средняя Тахикардия (100-120 в минуту), пульс слабого наполнения, снижение систолического давления 90-100 мм рт.ст., усиление потоотделения, бледность кожи, олигурия 25-35 % (1250-1750)
Тяжелая Тахикардия выше 120 в минуту, АД ниже 60 мм рт.ст., цианоз, ступор, анурия Свыше 50 % (2500)
Понижение объема крови сопровождается немедленной активацией компенсаторных механизмов: нервных и гуморальных. Падение АД улавливается барорецепторами дуги аорты и каротидного синуса, средняя частота разрядов этих рецепторов снижается, активируется симпатическая нервная система. Увеличение симпатического тонуса (срочная компенсация в течение нескольких минут) через активацию а-адренорецепторов приводит к спазму артериол кожи и органов брюшной полости. Сосуды сердца и головного мозга не имеют а-адренорецепторов и поэтому не отвечают на активацию симпатикуса.
572
Лекция 38
Создаются условия, при которых большая часть оставшейся в организме крови направляется в сосуды жизненно важных органов: мозга и сердца. Происходит централизация кровообращения.
Увеличение симпатического тонуса влечет за собой ряд других последствий: увеличение тонуса вен, возрастание частоты и силы сердечных сокращений, стимуляцию синтеза и секреции некоторых гормонов: адреналина — мозговым веществом надпочечников, ренина — юкстагломерулярным аппаратом почек, аргенин-вазопрессина (АДГ) — нейронами гипоталамуса.
Увеличение тонуса вен сопровождается увеличением давления наполнения циркуляторной системы и нормализацией венозного возврата крови в сердце, что нормализует МОС. Увеличение частоты и силы сердечных сокращений препятствует снижению МОС (МОС = УО х ЧСС). Частота сердечных сокращений растет пропорционально тяжести кровопотери и может достигать 200 в минуту. Длительный спазм артериол кожи и органов брюшной полости сопровождается снижением гидростатического давления в капиллярах, что способствует переходу жидкости из внесосудистого пространства внутрь сосудистого русла. Разведение крови (гемодилюция) обнаруживается уже через 10-30 мин после начала кровопотери и достигает максимума через 24-48 ч. Восстановление ОЦК за счет внутренних запасов воды — достаточно эффективный механизм при острых кровопотерях и является важнейшим фактором, определяющим прогноз при шоке. Известно, что удовлетворительный транспорт кислорода может быть обеспечен всего лишь половиной нормального объема эритроцитов, тогда как снижение ОЦК на одну треть опасно для жизни. Гемодилюция — это отставленный механизм компенсации.
Другим отставленным (пролонгированным) механизмом компенсации гиповолемии является ренин-ангиотензин-альдостероновая система (РААС), которая активируется при увеличении тонуса симпатической нервной системы и/или при снижении почечного кровотока. И то, и другое происходит при гиповолемии. Активация РААС приводит к увеличению реабсорбции натрия почками. Таким образом, увеличивается осмоляльность крови, что приводит к переходу жидкости из внесосудистого пространства в кровеносное русло и восстановлению ОЦК. Увеличение осмоляльности крови сопровождается также активацией осморецепторов головного мозга пострадавшего, который начинает испытывать жажду. Активация центральных осморецепторов приводит к рефлекторному усилению продукции АДГ (аргинин-вазопрессин), который резко ограничивает выведение воды почками, увеличивая про
Патофизиология шоковых состояний
573
ницаемость для воды почечных сосочков собирательных трубочек. Продукция вазопрессина может быть увеличена и симпатической активацией нервных волокон, которые иннервируют соответствующие ядра гипоталамуса.
Если гиповолемия невелика, а активация компенсаторных механизмов предупреждает ишемическое повреждение тканей, вызванное гиповолемией, шок не прогрессирует. Если же гиповолемия значительна, а механизмы компенсации недостаточны, то развивается вторая, прогрессирующая стадия шока, которая характеризуется возникновением «порочных кругов». «Порочный круг» — патогенетическая цепочка с положительной обратной связью, появление которой усиливает повреждение. При шоке таких кругов формируется много. Рассмотрим типичные примеры.
Порочный круг, способствующий повреждению миокарда
Снижение ОЦК приводит через снижение давления наполнения к снижению венозного возврата крови в сердце, а следовательно, и к падению МОС и АД. Это ведет к уменьшению коронарного кровотока и снижению сократимости миокарда. Снижение сократимости миокарда — к еще большему падению МОС и дальнейшему снижению АД и т.д.
Порочный круг, способствующий недостаточности вазомоторного центра
Вызванное гиповолемией снижение МОС и падение АД приводят к уменьшению церебрального кровотока и к нарушению деятельности сосудодвигательного центра. Одним из последствий этого может быть падение тонуса симпатической нервной системы. Расстраиваются механизмы централизации кровообращения, падает давление, что сопровождается еще большим уменьшением мозгового кровотока и еще большим угнетением сосудодвигательного центра.
Важнейшее патогенетическое звено при шоке — нарушения микроциркуляции. Снижение МОС и продолжительный спазм артериол сопровождаются ишемией многих тканей. В таких тканях накапливаются продукты ишемического повреждения, ионы водорода — возникает ацидоз, снижается напряжение кислорода. Все эти факторы вызывают расширение микроциркуляторных сосудов и срыв механизмов централизации кровообращения — происходит падение АД и МОС, а следовательно, ишемия тканей нарастает. Накапливающиеся местно продукты ишемического повреждения тканей оказывают прямое повреждающее действие на стенку микроциркуляторных сосудов. Повреждение стенки вызывает активацию механизмов свертывания крови, адгезию тромбоци-
574
Лекция 38
тов и тромбоз сосудов. Синдром диссеминированного внутрисосудистого свертывания (ДВС) крови — одно из тяжелых осложнений при шоке. Кроме адгезии и агрегации тромбоцитов и снижения скорости кровотока, вызванных прямым повреждением стенки, процесс потенциируется усиленным высвобождением фактора Виллебранда из эндотелиальных клеток и адреналина, из активированных тромбоцитов. Сладжирование эритроцитов еще больше расстраивает микроциркуляцию.
Другим последствием повреждения сосудов является увеличение их проницаемости для белка. Это сопровождается понижением онкотического давления крови и вызывает переход части жидкости из сосуда во внесосудистое пространство. ОЦК еще более снижается, а шок все более прогрессирует.
Токсические вещества, накапливающиеся в ишемизированных тканях, оказывают системное действие на организм. Развивается синдром полиорганной недостаточности. Полиорганная недостаточность определяется как синдром прогрессирующей, но потенциально обратимой недостаточности двух органов или систем органов и более, удаленных от места первичного повреждения. Термин полиорганной недостаточности был введен в 1975 г.
К числу наиболее часто повреждаемых органов относятся легкие, печень, почки, ЖКТ, кроветворная система, ЦНС, сердечно-сосудистая система.
Полиорганная недостаточность — главная причина смерти в палатах интенсивной терапии. При установленной полиорганной недостаточности смертность достигает 75 %. Прогноз определяется числом пораженных органов. Наиболее вероятными причинами являются: инфекции, гипоперфузия из-за множественных травм или геморрагического шока либо длительное воспаление. Бактериальные инфекции — одна из наиболее частых причин.
Нарушение микроциркуляции приводит к ишемии и повреждению эндотелия. Активированные эндотелиальные клетки экспрессируют молекулы адгезии для лейкоцитов и секретирует вещества, активирующие лейкоциты: ИЛ-1, ИЛ-8, ФАТ, ФНО. Активированные лейкоциты секретируют цитокины и активные метаболиты кислорода, повреждающие ткани. Поврежденные ткани также высвобождают токсические пептиды — в частности, миокардиальный токсический фактор, снижающий сократимость сердца.
Повреждение кишечной стенки сопровождается нарушением барьерных свойств кишечника. Населяющие кишечник грамотрицательные
Патофизиология шоковых состояний
575
бактерии поступают в кровоток, вызывая отравление организма эндотоксинами. Повреждение гепатоцитов приводит к нарушению детоксикационной функции и к расстройствам обмена веществ. В крови растет содержание молочной кислоты — возникает метаболический ацидоз. Повреждение клеток поджелудочной железы сопровождается активацией содержащихся в ней протеолитических ферментов, которые также поступают в кровоток, воздействуя на белки крови и белки клеточных мембран.
Как правило, при шоке нарушается функция почек. Падение АД сопровождается снижением фильтрационного давления, а снижение почечного кровотока может привести к некрозу почечных канальцев, Эпителий таких канальцев слущивается внутрь их просвета, что прекращает деятельность отдельных нефронов.
Главным критерием необратимости процессов считают метаболические маркеры — развитие генерализованной деполяризации клеточных мембран, истощение выброса катехоламинов, блокады адренорецепторов сердца и сосудов, прогрессирующее падение возбудимости и сократимости миокарда вследствие тяжелого ацидоза. Отрицательную роль в нарушении микроциркуляции играет истощение тонуса прекапиллярных сфинктеров. Формируется polling (карман, ловушка) с депонированием в нем крови. В результате снижаются АД и центральное венозное давление. Момент образования polling является гранью перехода шока в необратимую стадию (сосудистый маркер, предшествующий метаболическому) и его можно уловить клинически.
В основе метаболической декомпенсации лежат серьезные нарушения гомеостаза, которые приводят к тяжелым повреждениям всех органов. Однако ближайший прогноз определяется степенью поражения сердца.
Повреждение мышц приводит к увеличению внеклеточной концентрации ионов калия, фосфатов, миоглобина, креатина и креатинина. Креатинин токсичен для нервных клеток, миоглобин обладает нефротоксичностью, особенно при олигурии и ацидурии.
Поражение почек может быть следствием не только действия токсических веществ, но и снижением давления в почечных сосудах, что является результатом снижения ОЦК и АД. Эффективное фильтрационное давление при этом может упасть до нуля, а снижение почечного кровотока может привести к некрозу почечных канальцев и гибели отдельных нефронов.
Еще в стадию компенсации организм мобилизует и запускает нейроэндокринные механизмы со стимуляцией симпатической нервной
576
Лекция 38
системы, надпочечников, щитовидной железы и гипофиза с ростом выброса АДГ, тироксина, катехоламинов, глюкагона, АКТГ и кортизола. Эти гормоны изменяют обмен веществ: усиливается гликогенолиз, снижаются синтез и периферическое действие инсулина, усиливается распад белка, характерен выраженный отрицательный азотистый баланс.
На определенной стадии прогрессии шока самые энергичные терапевтические воздействия, включая переливание крови и кровезаменителей, введение кардиотропных и вазотропных препаратов и другие, не могут сохранить жизнь больному. В этом случае говорят о необратимом шоке. На этой стадии врачебное вмешательство может вернуть на короткое время АД и МОС к норме, но это не останавливает разрушительных процессов в организме. Через некоторое время возникает новое необратимое падение АД, и больной погибает. Пороговым критерием необратимости считают метаболическую декомпенсацию — развитие генерализованной деполяризации клеточных мембран: или истощение выброса катехоламинов, блокада рецепторов сердца и сосудов, прогрессирующее падение возбудимости и сократимости миокарда. Ближайший прогноз определяется степенью повреждения сердца.
Нарушение микроциркуляции также играет отрицательную роль в переходе шока в необратимую стадию. Это связано с истощением тонуса прекапиллярных сфинктеров, формированием polling (ловушка, карман) и депонированием в нем крови, следовательно, понижением АД и центрального венозного давления. Это можно уловить клинически.
Травматический шок
Этот вид шока возникает при массивных ранениях или размозжениях тканей, открытых и закрытых переломах трубчатых костей и т.д. Одним из обязательных составляющих травматического шока является гиповолемия. В одних случаях она возникает вследствие кровопотери из раненых сосудов, в других — вследствие кровопотери из микроцир-куляторных сосудов с повышенной проницаемостью в травмированных участках тела. Снижение ОЦК — потеря внутрисосудистой и внесосу-дистой жидкости приводит к нарушению макро- и микроциркуляции, в результате возникают гипоперфузия жизненно важных органов и гипоксическое повреждение клеток. При этом происходит активация систем: комплемента, свертывания, фибринолиза, ККС. Активация гуморальных систем активирует клеточные системы. Главными клетками при травматическом шоке являются тромбоциты и полиморфно
Патофизиология шоковых состояний
577
ядерные нейтрофилы, выделение медиаторов из которых и появление свободных кислородных радикалов включают системную воспалительную реакцию.
Нарушения органов — это выражение системного воспалительного ответа, который может быть результатом как неинфекционного, так и инфекционного повреждения тканей. Как правило, травматический шок осложняется инфекциями. До 60 % больных с множественной травмой — инфицированы. Причиной инфекции обычно бывают граммотрица-тельные бактерии. Развитию инфекций способствуют ранения органов брюшной полости, местные и общие нарушения кровообращения, иммуносупрессия. Выделение бактерий и токсинов из кишечника провоцирует типический ответ хозяина, который вызывает массивную активацию клеточных систем: моноциты, макрофаги, эндотелиальные клетки, нейтрофилы, сопровождающийся высвобождением цитокинов, протеаз, эйкозаноидов, свободных радикалов.
Комбинация геморрагических поражений с инфекционными способствует быстрому развитию синдрома ДВС и РДСВ.
Диагноз инфекций обычно затруднен системными реакциями организма на повреждение: лихорадкой, тахикардией, гипервентиляцией, нарушением функций нервной системы, изменениями клеточного состава периферической крови. Между тем инфекция — главная причина смерти больных спустя одну неделю после травмы.
Другим важным компонентом травматического шока является токсемия, которая обусловлена попаданием в кровь продуктов распада поврежденных клеток.
Специальным осложнением травматического шока может быть жировая эмболия, возникающая у больных с переломами длинных трубчатых костей. Самые опасные нарушения при этом связаны с эмболией сосудов легких и головного мозга.
В некоторых случаях травматический шок осложняется рабдомиолизом. Травматический рабдомиолиз или краш-синдром — результат длительного сдавления конечностей, при котором повреждение скелетных мышц приводит к истечению содержимого миоцитов в кровь. Синдром характеризуется: гиповолемическим шоком; гиперкалиемией, которая вызывает кардиотоксикоз, аритмию и возможную остановку сердца; метаболическим ацидозом вследствие накопления молочной кислоты; появлением миоглобина в крови, что приводит к миоглобинурии и острой почечной недостаточности. Все эти проявления угрожают жизни, и краш-синдром характеризуется высокой смертностью.
578
Лекция 38
Существенным фактором патогенеза травматического шока, как и кардиогенного, является боль. Боль может нарушать регуляцию сосудистого тонуса, что приводит к увеличению внутрисосудистой емкости, снижению венозного возврата в сердце и снижению МОС.
Принципы патогенетической терапии гиповолемического и травматического шоков:
• остановка кровотечения;
• восстановление ОЦК и КОС с коррекцией концентрации калия;
• нормализация гемодинамики;
• оксигенотерапия;
• если нет тампонады миокарда — стимуляция сократимости миокарда:
• снижение постнагрузки;
• обезболивание.
Кардиогенный шок
Это синдром, обусловленный острой сердечной недостаточностью, вызванной резким снижением эффективности работы сердца как насоса. Кардиогенный шок характеризуется следующими показателями гемодинамики: низким МОС и низким АД (систолическое давление обычно ниже 80 мм рт.ст.), сердечный индекс ниже 1,81, коронарный резерв — снижен, высоким давлением наполнения левого желудочка, обычно выше 18 мм рт.ст. (в норме — 0-8 мм рт.ст.) и высоким центральным венозным давлением. Таким образом, главная особенность кардиогенного шока — прекардиальный застой крови. Самая частая причина кардиогенного шока — массивные инфаркты миокарда левого желудочка. Шок возникает обычно, когда поражено не менее 45 % всей массы миокарда. Кроме того, кардиогенный шок могут вызвать операции на сердце; острые нарушения внутрисердечного кровотока, связанные с разрывом межжелудочковой перегородки или папиллярной мышцы, с разрывом стенки желудочков и тампонадами сердца; тяжелые нарушения ритма сердца; стенозы аортального или митрального клапанов, которые сопровождаются нарушениями выведения или заполнения сердца кровью.
Клинигеские симптомы кардиогенного шока: низкое АД (особенно систолическое), низкое пульсовое давление, повышение центрального венозного давления, бледность кожи, снижение температуры кожи. Нарушение почечного кровотока приводит к олигурии и анурии. Возможны психические расстройства. В тяжелых случаях — потеря сознания.
Патофизиология шоковых состояний
579
В патогенезе кардиогенного шока, как и в патогенезе гиповолемического шока, существенная роль принадлежит активации симпатической нервной системы, которая возникает немедленно вслед за падением АД. Повышение симпатического тонуса приводит к усилению частоты и силы сердечных сокращений и к повышению АД благодаря централизации кровообращения. Однако, если при гиповолемическом шоке такой ответ целесообразен, при кардиогенном шоке он может привести к нежелательным последствиям. Поскольку у больных кардиогенным шоком резко снижен коронарный резерв, увеличение частоты и силы сердечных сокращений вместе с ростом АД вызовут резкое несоответствие между притекающей к сердцу кровью и его метаболическими потребностями. Подобное несоответствие резко снижает сократимость сердца — шок прогрессирует. Возникает порочный круг, который приводит к еще большему снижению МОС и увеличению давления наполнения левого желудочка. Последнее приводит к возрастанию конечного диастолического давления в левом предсердии и к увеличению давления в легочных капиллярах — возникает угроза отека легких.
Принципы патогенетической терапии кардиогенного шока:
• опиаты для снятия боли;
• оксигенотерапия;
• сердечные гликозиды (осторожно);
• препараты, расширяющие коронары;
• (3-адреноблокаторы;
• антагонисты кальция;
• противоаритмические препараты;
• тромболитики.
Септический шок
Септический шок — остро развивающийся патологический процесс, вызванный инфекционными агентами и характеризующийся рядом системных нарушений, включая гипотензию, гипоперфузию, полиорган-ную недостаточность. Этот вид шока возникает при диссеминированных инфекциях, как осложнение сепсиса. Наиболее часто встречается при маточных перитонитах, перитонитах, связанных с разрывами кишечника, при генерализованных инфекциях, возникающих после банальных стрептококковых и стафилококковых повреждений кожи. Может возникнуть при генерализованной газовой гангрене.
Причины: бактерии, вирусы, простейшие, грибы.
580
Лекция 38
Самая частая причина — грамотрицательные бактерии — Е. coli, протей. Граммположительные бактерии — пневмо-, стафило-, энтерококки, клостридии — реже вызывают сепсис.
К числу вирусов-возбудителей, вызывающих сепсис, относятся арбовирусы и вирусы геморрагической лихорадки, реже — цитомегаловирус и вирус герпеса.
Среди простейших — плазмодиум фальципарум. Грибки редко вызывают сепсис. Причиной могут быть длительное применение антибиотиков или катетерная инфекция, чаще всего — кандидоз.
Клинические симптомы септического шока:
• высокая лихорадка:
• генерализованная вазодилатация, особенно выраженной в инфицированных тканях;
• сладжирование эритроцитов, глубокая гипотензия:
• снижение артериовенозной разницы по кислороду:
• дыхательный алкалоз — в начале септического шока, позже — метаболический ацидоз.
Лихорадка обусловлена высоким содержанием в организме одного из главных медиаторов ответа острой фазы — ИЛ-1, который синтезируется многими клетками (моноцитами, макрофагами, эндотелиальными клетками) под влиянием микроорганизмов.
Вазодилатация в инфицированных тканях есть результат микробной активации ферментных систем крови, в ходе которой образуются вазоактивные кинины, высвобождаются гистамин и другие сосудорасширяющие вещества. Высокий МОС обусловлен расширением сосудов и высоким уровнем метаболизма, который, в свою очередь, является следствием повышения температуры тела и высокого содержания в крови катаболических гормонов.
Сладжирование эритроцитов обусловлено, вероятно, агглютинацией эритроцитов под влиянием бактериальных токсинов.
При септическом шоке первичные нарушения обнаруживаются в расстройствах периферического кровообращения. Различают гипердинамическую и гиподинамическую фазы развития шока.
Гипердинамигеская фаза обусловлена действием патогенных бактериальных токсинов, которые открывают артериовенозные шунты и нарушают использование кислорода тканями. В результате снижается общее периферическое сопротивление сосудов, АД падает и рефлекторно увеличиваются частота сердечных сокращений, ударный объем и МОС. При этом происходят перегрузка сердца повышенным объемом и
Патофизиология шоковых состояний
581
снижение сократимости сердца. Но так как перфузия капилляров снижена, нарушается транспорт кислорода из капилляров в клетки.
Это приводит к гипоксии тканей и повышению сосудистой проницаемости. В результате снижаются ОЦК и венозный возврат в сердце, уменьшается МОС и АД. Наступает гиподинамическая фаза. Застою крови в венах способствует спазм артериол. Вовлечение сердечно-сосудистой системы обусловлено воздействием на сердце и сосуды микробов и их токсинов, а также появлением в крови большого количества вазоактивных веществ и нарушением нервной регуляции.
Одним из важнейших патогенетических механизмов септического шока является поражение легких в виде отека или синдрома респираторного дистресса. При этом виде шока отек легких развивается без повышения давления в капиллярах, но в результате прямого повреждения альвеолярно-капиллярной мембраны. Синдром респираторного дистресса связан с быстрой активацией комплемента и накоплением в легких большого количества нейтрофилов и альвеолярных макрофагов, которые продуцируют различные вещества, в том числе активные метаболиты кислорода, повреждающие альвеолярно-капиллярную мембрану.
Специальной формой септического шока является эндотоксигеский шок. Этот вид шока может возникнуть при завороте кишок и последующей гангрене значительного сегмента толстой кишки, населенного граммотрицательными бактериями. Эти бактерии содержат эндотоксины — высокомолекулярные липополисахариды, обладающие сильным биологическим действием. Нарушение барьерных свойств кишечника приводит к поступлению эндотоксинов в кровоток, а затем в ткани, где они вызывают дегрануляцию тучных клеток и выделение гистамина. Гистамин расширяет периферические сосуды, повышает их проницаемость, вызывает аритмии сердца, т.е. нарушает гемодинамику. В больших концентрациях гистамин может вызвать гистаминовый шок.
В поздней стадии развивается синдром полиорганной недостаточности как системный ответ на неконтролируемую инфекцию. Раньше всего повреждаются легкие в результате образования анатомических и функциональных артериовенозных шунтов. Функциональные шунты возникают из-за перфузии невентилируемых участков легких. Шунтируемый объем крови в раннюю стадию может составлять 20-30 %, а в позднюю — 60-80 % от МОС, что приводит к артериальной гипоксемии. Повышенная проницаемость легочных капилляров ведет к отеку легких.
Почки повреждаются чаще, чем при других формах шока, вследствие двустороннего некроза коркового слоя — развивается острая почечная
582
Лекция 38
недостаточность. Экзотоксины клостридий обладают свойствами не-фротоксинов.
Сердце обнаруживает резкое снижение сократимости, что ведет у примерно 50 % больных к развитию острой сердечной недостаточности.
В печени, как и при гиповолемическом и кардиогенном шоках, возникают центрилобулярные повреждения вплоть до некрозов.
Повреждения других органов в большинстве случаев — результат ДВС-синдрома.
Принципы патогенетической терапии при септическом шоке:
• удаление возбудителей;
• хирургическое удаление септических органов;
• восстановление ОЦК, особенно на ранних стадиях;
• антибиотики после высева культуры и установления чувствительности к антибиотикам;
• коррекция электролитного состава и КОС;
• оксигенотерапия;
• сердечные гликозиды;
• антиагреганты;
• вазоактивные препараты с преобладанием вазопрессоров.
Анафилактический шок
Этот вид шока подробно разбирают в лекциях по иммунопатологии. Принадлежит к числу наиболее быстро развивающихся патологических процессов. В основе его развития лежит аллергическая реакция немедленного типа. Анафилактический шок может развиваться по I или III типу иммунопатологических реакций по классификации Джелла и Кумбса. При анафилактическом шоке кожные проявления часто предшествуют системным гемодинамическим расстройствам — это отличительная черта этого вида шока — первоначальное отсутствие симпатоадреналовой реакции. Если анафилактический шок протекает с потерей сознания, больной может погибнуть за 5-30 мин от удушья или через 24-48 ч и более в связи с вторичными тяжелыми необратимыми изменениями жизненно важных органов (ГН, кишечные кровотечения, миокардит, отек или кровоизлияния в головной мозг).
Нейрогенный шок
Нейрогенный шок характеризуется нарушением сосудистого тонуса и увеличением емкости сосудистого русла. Иногда емкость сосудов воз
Патофизиология шоковых состояний
583
растает столь значительно, что даже нормального ОЦК не хватает для адекватного заполнения сердечно-сосудистой системы. При этом, как и в случае с гиповолемическим шоком, падает давление наполнения циркуляторной системы, уменьшается венозный возврат в сердце, падает МОС. Это происходит при нарушении нервного контроля за тонусом сосудов, а также дисбалансе между симпатическими и парасимпатическими влияниями. Тонус вен падает, большая часть крови аккумулируется в венах и не участвует в циркуляции. Возникает нейрогенный шок. Он может развиться при глубокой анестезии в результате угнетения вазомоторного центра, при спинномозговой анестезии, травмах мозга, которые сопровождаются нарушением вегетативной регуляции.
В противоположность гиповолемическому шоку частота сердечных сокращений в нейрогенном шоке ниже нормы, кожа сухая и теплая.
Этот вид шока является редким и обычно проходящим.
Нейрогенный шок следует отличать от обморока. Обморок — приступ кратковременной утраты сознания, обусловленный преходящим снижением мозгового кровотока. Основную роль в его развитии играет снижение АД, вызванное дилатацией сосудов мышц и внутренних органов. Вазодепрессивный обморок наблюдается, как правило, у молодых людей с повышенной эмоциональной лабильностью и всегда развивается под влиянием определенного фактора, характерного для каждого больного. Психоэмоциональные ситуации (вид крови, волнение, испуг) имеют преимущественное значение у женщин, в то время как болевое воздействие провоцирует обмороки у мужчин. К физическим факторам относятся пребывание на жаре, в душном помещении, длительное стояние, выраженная усталость. Обморок не приводит к существенным нарушениям гемодинамики и повреждениям тканей.
Принципы патогенетической терапии нейрогенного шока:
• корреляция причины шока;
• улучшение перфузии тканей;
• сосудосуживающие препараты;
• симпатомиметики;
• препараты, улучшающие коронарный кровоток.
предметный указатель
А
Абазия 286
Агранулоцитоз 136
Агранулоцитоз аутоиммунный 449
Адаптация 13
Адаптоген 34
Аддисона-Бирмера анемия 119
Адиадохокинез 286
Азотемия 549
Акромегалия 335
Акселерация прогрессирующая 141
Аксонопатия 273
Аксон-рефлекс 52
Алкалемия 208
Алкалоз 220, 231
Алкоголизм 22,27
Аллерген 435
Аллергия 431
Альдостерома 339
Альдостерон 189
Альдостеронизм 190,339 Альтерация 19,50,72,255
Амилаза 159
Аминоацидемия 148 Аминоацидурия 536,547
Аммониогенез 213
Амфифил 46
Аналгезия 299
Анаплазия 238
Анасарка 202
Анафилаксия 59
Анемия 105
Аноксия 260
Антиоксидант 43
Антиплазмин 92
Антипротеиназа 80
Антитрипсин 91
Антитромбин 91
Анулоцит 115,117
Анурия 542
Апоптоз 49
Апудома 318
Апудопатия 318
Апудоцит 317
Аритмия 381
Артюса феномен 432,451
Асинергия 286
Ассоциация эозинофильнобазофильная 142
Астазия 286
Астма бронхиальная 362,473
Астма сердечная 379,490
Асцит 204,528
Атаксия 456
Атипизм 237
Аутоантиген 443
Ахолия 495,530
Ахрезия 117
Ацидемия 208
Ацидогенез 213
Ацидоз 47, 220
Ашоффа теория 510
Б
Базофилия 132
Бактериурия 547
Баланс азотистый 146
Батиста М.Б. 10
Бейлиса эффект 399
Белок 145
Бенс-Джонса белок 429
Беркитта лимфома 238,245,461
Бернара-Сулье тромбоцитопатия 87,94
Бернар К. 351
Билирубинурия 548
Богомолец А.А. 10
Предметныйуказатель
585
Болезнь 15
- Аддисона 133,164,193,320,343
- Аддисона-Бирмера 119
- Альцгеймера 266, 267,318
- базедова 327
- Беркитта 138
- Бернара-Сулье 87,94
- бронзовая 164
- Вакеза 93,138
- Виллебранда 88,94
- гипертоническая 342,358
- Гланцманна 94
- глютеновая 502
-Дауна 267
-Дюшенна 155
- Иценко-Кушинга 137,165,196
- ишемическая сердца 383
- Кули 126
- Минковского-Шоффара 127
- Ниманна-Пика 259
- Олбрайта 331
- Паркинсона 284
- Помпе 259
- Рейно 358,360
- Реклингаузена 332
- Симмондса-Шиена 309
-средиземноморская 126
- сывороточная 427,450
- тромботическая 93
- Хашимото 321
- Ходжкина 137,138
- черной печени 535
- Шенлейна-Геноха 94,450
- язвенная желудка и
двенадцатиперстной кишки 509
Боль 50,289
-фантомная 302
Брадикинин 60,410
Брадипноэ 485
Бронхоконстрикция 471
Брутона агаммаглобулинемия 457
Буфер 205
Быкова-Курцина теория 510
В
Вильмса опухоль 238
Виментин 239
Випома 318
Вирхова теория 510
ВИЧ 461
Воздействие 13
Волчанка системная красная 95
Воспаление 11,19,38,50,75
- хроническое 73
Г
ГаллиотЛ. 10
Галлюцинация 35
Гастрин 517
Гастринома 318
Гастромукопротеин 120
ГеккерА.Ф. 10
Гемартроз 96
Гематокрит 99
Гематурия 546
Гемобластоз 138
Гемоглобиноз 126
Гемоглобинопатия 126,155
Гемоглобинурия маршевая 124
Гемостаз 85
Гемофилия 96
Гендерсона-Хассельбаха уравнение
206, 217
Генле петля 213
Гентингтона хорея 284
Гепарин 91
Гепатоз жировой 183
Гепатомегалия 526
Гигантизм 334
Гидролаза 71
Гипалгезия 299
Гипералгезия 300
Гипервентиляция 484
Гиперволемия 101
Гипергликемия 164,171
Гиперемия 51
586
Предметный указатель
Гиперинсулизм 171 Гиперкальциемия 263 Гиперкатехоламинемия 376 Гиперкортицизм 190 Гиперлактатацидемия 161 Гиперлипемия 176 Гиперлипидемия 550 Гипернатриемия 262 Гиперпаратиреоз 332 Гиперпноэ 484 Гйперспленизм 526 Гипертензия - артериальная почечная 551 - портальная 527 - ренопривная 413 Гипертиреоз 327
Гипертония артериальная 396 Гипертриглицеридемия 176 Гйпертрофия сердца 370 Гиперурикемия 156 Гиповентиляция 485 Гйповолемия 100 Гипогидратация 550 Гипогликемия 163 Гипокальциемия 263 Гипоксия 11 Гипонатриемия 263 Гипопаратиреоз 331 Гипопротеинемия 151 Гйпостенурия 543 Гипотиреоз 323 Гипохолия 495 Гипохромия 117 Гйстамин 56,517 Гистогенез 237 Гистурия 546
Гланцманна тромбастения 94
Гликогеноз 259 Гликогенолиз 160 Гломерулонефрит 555 Глюкозурия 548 Глюкокортикоиды 354 Голодание 149 Гомеостаз 39
Гормон 305
Гранулема 73
Графи лейкоз 245
ГЪмбла ионограмма 214
Д
ДВС-синдром 97
Дерматомиозит 282
Диабет несахарный 338
Диабет сахарный 166
Дисгидрия 192
Дисметрия 286
Диспноэ 484
Диспротеинемия 536
Дистрофия белковая 152
Дисфория 28
Дифиллоботриоз 120
Дрейф генов 15
Дуоденит 499
Дыхание
- апнейстическое 486
- Биота 488
- загнанного зверя 485
- Куссмауля 485
- Чейна-Стокса 486
Дюшенна дистрофия 155,281
Е
Единица моторная 269
Ж
Желтуха 531
Жолли тельца 121
3
Зависимость 28,32
Заеды 115
Запор 505
Здоровье 14
Зиггаарта-Андерсона карты 216
Зимницкого проба 543
Предметный указатель
587
И
Изоволемия 539
Изоиония 539
Изоосмоляльность 215
Изостенурия 543
Изотония 539
Иммунодефицит 455
Иммунодефицит возрастной 466
Иммунопатология 427
Инбридинг 15
Индекс
-Брока 180
- Кетле 180
Инкреция 539
Инсулинома 171
Интегрин 68
Интерлейкин 60,76
Интерферон 421
Инфильтрат 67
Инфильтрат лейкемический 142
Исход 10
К
Канцероген 243
Капоши саркома 245,463
Карбоангидраза 209
Карликовость 335
Карман горячий 110
Кассирского классификация 102
Каузалгия 301
Кахексия 151
- тиреопривная 325
Квашиоркор 152
Квинке отек 200
Квинке теория 510
Кебота кольца 121
Кератин 238
Кетоацидоз 227
Кетоз 167
Кинин 60
Кислота фолиевая 119
Классификация по Фредериксону 177
Клетка - пенистая 37 - пузырчатая 125 - Пуркинье 264 - шванновская 265 Коагуляция 89 Кобаламин 118 Койлонихии 115 Колика почечная 568 Кома 169,170
Кома печеночная 537 Комплемент 48,61,420 Конституция 417
Креаторея 530
Кретинизм 325,330
Криз бластный 141
Круг порочный 9,19
Ксантома бугорчатая 178
Ксеродерма 250
Кумбса тест 125
Л
Лактоферрин 112
Лапа когтистая 300
Лейкемия 138
Лейкин 421
Лейкон 129
Лейкопения 135
Лейкотриен 59 Лейкоцитурия 547 Лейкоцитоз 131 Лептин 181 Лизин 421 Лизоцим 421 Лилипутизм 335 Лимфопения 137 Лимфоцитоз 132 Липидоз 259
Липидурия 548
Липопротеинлипаза 81
Липотиаминпирофосфат 162
Лихорадка 11,79 Локомоция 241
588
Предметный указатель
М
Макроцит 121
Макроцитоз 136
Маразм алиментарный 151
Маргинация 67
Маршалл Б. 515
Мегалобласт 121
Мегалоцит 121
Медиатор 56
Мейнерта ядро 267
Меланоз печени 535
Мечников И.И. 69
Миастения 280
Миастения псевдопаралитическая 160
Мигрень 360
Миелоз фуникулярный 122
Миелолейкоз 141
Микросфероцитоз 127
Микроцит 115
Микседема 323,330
Миопатия 281
Митоген 252
Мицелла 47
Моделирование 20
Молони лейкоз 245
Молочница 115
Мононейропатия 172
Моноцитоз 132
Моноцитопения 137
Мукополисахаридоз 259
Мурамидаза 421
Н
Нанизм 331
Наркомания 22,32
Недостаточность полиорганная 574
Нейропатия периферическая 266
Нейтрофил ез 132
Нейтрофилия 81
Некроз 49
Неоантиген 444
Нефроз липоидный 550
Нефролитиаз 566
Нефропатия 553
Никтурия 340
Новообразование 236
Нозология 13
Номограмма 216
Норма 14
Ноцицептор 51,291
О
Обморок 583
Обструкция 470
Одди сфинктер 497
Одышка 379
Ожирение 180
Окисление липидов перекисное 42
Окраска
- по Райту 102
- по Романовскому-ГЪмзе 102
Олигопноэ 485
Олигурия 541
Онкобелок 249
Онкогенез 245
Опохмеление 29
Опухоль 236
Оротацидурия 157
Остеодистрофия почечная 333,562
Ответ острой фазы 75
Отек 198,379
Отек легких 365
Отторжение трансплантата 454
П
Павлова фистула 537
Пангипопитуитаризм 309,336
Панкреатит 497
Панмиелофтиз 110
Паралич 283
Паралич бульбарный 272
Пастера эффект 239
Патогенез 9,11,17 Патофизиология -клиническая 11 - частная 11
Предметный указатель
589
Пепсин 518
Перигепатит 529
Пермеация 240
Перфузия 478
Пиелонефрит 554
Плазмин 92
Плеоморфизм 237
Повреждение 37,40,49,56, 75
Подагра 155
Показатель цветовой 106
Полидипсия 340
Полиомиелит 271
Полипноэ 484
Полиурия 540
Полихроматофилия 125
Поллиноз 436
Полунин А.И. 10
Понос 505
Постнагрузка 366
Прайс-Джонса кривая 128
Предболезнь 15,27
Преднагрузка 366
Престаз 54
Привыкание 35
Причина 17
Провал лейкемический 139
Прогерия 110
Пропердин 421
Простагландин 58
Простациклин 59,411
Протеин С 91
Протеинурия 544
Протоонкоген 243
Псевдоаллергия 442
Псевдогиперпаратиреоз 317
Пуазейля-Хагена закон 399
Пьянство 27
Р
Рабдомиолиз 577
Радикулоплексопатия 172
Раздражимость 416
Реабсорбция 539 Реактивность 415,427
Реакция
- Манту 451
- Пирке 451
- сосудистая 19
Реакция лейкемоидная 134
Регургитация 366
Резистентность 425
Резорбция 81
Ремнант 175
Ремоделирование миокарда 369
Ретикулоцит 102
Ретинобластома 250
Рефлекс
- Бабинского 273
- Геринга-Брейера 485
Рефлюкс 518
Роллинг 68
С
Саногенез 20
Сдвиг лейкемоидный 142
Сдвиг хлоридный 210, 221
Секреция 539
Селектин 68
Селье Г. 82,348
Сенсибилизация 436
Серотонин 57
Симптом
- ватных ног 122
- голова медузы 528
- желательности алкоголизации 27
- Пламмера-Уинсона 115
- раздраженного кишечника 500
- «синий» костный мозг 122
-Тинеля 301
Сингера-Никольсона мембрана 39
Синдром 16
- LAD 456
- абстинентный 28,31
- астеновегетативный 526
- Бадда-Киари 527
- Блума 250
- болевой 529
- Вискотта-Олдрича 137,457
590
Предметный указатель
- геморрагический 137,527
- гепатолиенальный 526
- гепатоцеребральный 537
- «голых» лейкоцитов 456
- Дабина-Джонсона 535
- Дауна 423
-демпинг 522
- Ди Джорджи 137,457
- диспептический 527
- Дресслера 395
- Жильбера 535
- Золлингера-Эллисона 119,318, 518
- Иммерслунда-Грасбека 119
- иммунновоспалительный 526
- интоксикационный 555
- Итона-Ламберта 240
- Иценко-Кушинга 137,184,321,405
- Конна 340,405
- краш 577
- Криглера-Найяра 535
-Ламберта-Итона 278
- ленивых лейкоцитов 136,423
-Луи-Барр 137
- мальабсорбции 503
- мочевой 544,554
- недостаточности дисахаридаз 501
- нефротический 550
- паранеопластический 239
- Паркинсона 284
- печеночно-клеточной
недостаточности 535
- поллакиурический 554
- респираторный дистресс 491
-сидеропенический 114
- Симмондса-Шиена 336
- тестикулярной феминизации 316
- Тиннеля 276
- тромбогеморрагический 97
- Уотерхауса-Фридериксена 312
- Фанкони 548
- цитолитический 525
- Чедиака-Хигаси 423
- Элерса-Данло 155
Сирингомиелия 299
Склероз амиотрофический боковой 271
Склероз рассеянный 265
Сладж 55
Спазм 51
СПИД 24,461
Спирометрия 471
Спру 502
Стаз 54
Старлинг 190
Статус бронхоастматический 488
Стеатоз печени 183
Стеаторея 530
Стояние краевое 67
Стресс 13,343,466
Стрессор 343
Супрессия 441
Т
Талассемия 126
Тамм-Хорсфалля белок 546
Тахикардия 380
Тахипноэ 484
Телеангиоэктация 456
Тельца
- Боткина-Гумпрехта 144
- Гейнца-Эрлиха 127
- Князькова-Деле 135
Тетания тиреопривная 331
Тиреотоксикоз 327
Токсикомания 22,32
Толерантность 444
Точка установочная 79
Трансферрин 111
Трансцитоз 63
Триада мерзебургская 327
Тромбинемия 97
Тромбоз 92
Тромбоксан 59
Тромбофилия 93
Тромбоцитоз реактивный 142
Тромбоцитопения 95
Тубулярный некроз 365
ТЭЛА 490
Предметный указатель
у
Ульцерогенез 510
Уоллеровское перерождение 274
Уоррен И. 515
Уремия 560
Уролитиаз 566
Ф
Фагосома 71
Фагоцитоз 69,418
Фактор
- Виллебранда 87
- Касла 119
- Хагемана 60,86
ФАТ 59
Феохромоцитома 165,307
Ферментопатия 155,501
Ферритин 111
Фибриногенопатия 155
Фибринолиз 91
Фибринолизин 92
Фильтрация 539
ФНО 60,77
Фолат 119
Франка-Старлинга закон 367
Франка-Старлинга механизм 367
X
Хантера язык 122
Хашимото тиреоидит 321
Хемотаксис 69
Хилоз 176
Хлороз 114
Холдейна эффект 210
Холемия 530
Холестаз 529
Ц
Целиакия 502
Церебротоксин 537
Цилиндрурия 547
591
Цитиноз 149
Цитокин 76
Ш
Шок 83
- спинальный 282
-токсический 537
Э
Эйфория 29,34
Экка фистула 537
Экскреция 539
Экссудат 65
Экссудация 54, 63
Электонейтральность 214
Эмиграция 67
Эндокринопатия 319
Эндотелии 412
Энергодефицит 40
Энзимопатия 127
Энурез 114
Эозинопения 137
Эозинофилия 132
Эпштейна-Барр вирус 245
Эритремия 142
Эритрон 102
Эритропоэтин 105
Эритроцитоз 99,104
Эритроцитопатия 126
Этиология 9,11
Эупноэ 484
Эффект
-зобогенный 311
- соскальзывания 371
Я
Язва 509
АЬс
Myasthenia gravis 160
Pica chlorotica 115
ПРИГЛАШЕНИЕ К СОТРУДНИЧЕСТВУ
Издательская группа «ГЭОТАР-Медиа» приглашает к сотрудничеству авторов и редакторов медицинской литературы.
ИЗДАТЕЛЬСТВО СПЕЦИАЛИЗИРУЕТСЯ НА ВЫПУСКЕ учебной литературы для вузов и колледжей, атласов, руководств для врачей, переводных изданий
По вопросам издания рукописей обращайтесь в отдел по работе с авторами Тел. 3(495) 921-39-07.
I
Угебнос издание
Патофизиология
Курс лекций
Под редакцией
Геннадия Васильевича Порядина
Зав. редакцией
А.В. Андреева Выпускающие редакторы Н.А. Галахова, Д.А. Фомина
Редактор
Е.Д. Руденко
Корректоры
М.Ю. Никитина, Е.И. Макеева Подготовка оригинал-макета
С.И. Евдокимов
Подписано в печать 21.11.2013. Формат 60x90 ’/16. Бумага офсетная. Печать офсетная.
Объем 37 усл. печ. л. Тираж 1500 экз. Заказ 4611.
ООО Издательская группа «ГЭОТАР-Медиа». 115035, Москва, ул. Садовническая, д. 9, стр. 4. Тел.: 8 (495) 921-39-07.
E-mail: info@geotar.ru, http://www.geotar.ru.
Отпечатано в ОАО «Можайский полиграфический комбинат», 143200, г. Можайск, ул. Мира, д. 93.
Учебное пособие соответствует требованиям действующего Федерального государственного образовательного стандарта высшего профессионального образования. Подготовлено сотрудниками кафедры патофизиологии Российского национального исследовательского медицинского университета имени Н.И. Пирогова Минздрава России.
Пособие состоит из 38 лекций, в которых последовательно разбираются вопросы общей и частной патофизиологии. Теоретический материал иллюстрирован таблицами и рисунками.
Предназначено студентам и аспирантам всех факультетов медицинских вузов.
> Молекулярные механизмы повреждения клетки
> Воспаление. Ответ острой фазы
> Патология опухолевого роста. Канцерогенез
> Роль реактивности организма в патологии
> Патофизиология:
- алкоголизма, наркоманий и токсикоманий
- системы гемостаза
- обмена веществ и кислотно-основного состояния
- нервной системы
- эндокринной системы. Эндокринопатии
- сердечно-сосудистой системы
- иммунной системы. Иммунопатология
- внешнего дыхания
- желудочно-кишечного тракта
- почек
- шоковых состояний
www.geotar.ru
www.medknigaservis.ru
Патофизиология