/


































































































































































































































































































































































































































Автор: Мороз Б.Б.
Теги: патологическая физиология формы развития заболеваний патогенез учение о происхождении заболеваний общая патология медицина
ISBN: 5-225-04388-7
Год: 2001




Текст
АКТУАЛЬНЫЕ
ПРОБЛЕМЫ
ПАТОФИЗИОЛОГИИ
(избранные лекции)
Под редакцией
академика РАМН Б.Б.Мороз
Учебная
литература
для студента*
медицинских
вузов
УДК 616-092
ББК 52.5
А43
Федеральная программа книгоиздания России
Рецензент — Г.В.Порядин, профессор, заведующий кафед-
рой патофизиологии Российского государст-
венного медицинского университета.
Актуальные проблемы патофизиологии: Избранные лек-
А43 ции/Под ред. Б.Б.Мороза. — М.: Медицина, 2001. — 424 с.:
ил. ISBN 5-225-04388-7
Излагается современное состояние ряда актуальных проблем па-
тофизиологии. Авторами преследовалась цель всесторонне осветить
изучаемые проблемы для наиболее полного о них представления.
Важное место при этом занимает описание молекулярно-биологичес-
ких механизмов патофизиологии и новейших материалов, касающих-
ся данной темы.
Для студентов и аспирантов.
ББК 52.5
ISBN 5-225-04388-7 © Коллектив авторов, 2001
Все права авторов защищены. Ни одна часть этого издания не может
быть занесена в память компьютера либо воспроизведена любым спосо-
бом без предварительного письменного разрешения издателя.
Авторский коллектив
Гущин Игорь Сергеевич — доктор медицинских наук, член-
корреспондент РАМН, заведующий отделом аллергологии и
клинической иммунологии в Институте иммунологии ГНЦ
М3 РФ.
Игнатьева Галина Алексеевна — доктор медицинских наук,
профессор, ведущий научный сотрудник лаборатории биотех-
нологии в Институте иммунологии ГНЦ М3 РФ.
Кукушкин Михаил Львович — доктор медицинских наук, руко-
водитель лаборатории патофизиологии боли в НИИ общей па-
тологии и патофизиологии РАМН.
Лихтенштейн Анатолий Владимирович — доктор биологичес-
ких наук, руководитель лаборатории биохимии опухолей в
НИИ канцерогенеза Российского онкологического научного
центра РАМН.
Макаров Владимир Александрович — доктор медицинских
наук, профессор, руководитель лаборатории патологии и фар-
макологии гемостаза в Гематологическом научном центре
РАМН.
Пшенникова Мая Григорьевна — доктор биологических наук,
профессор, ведущий научный сотрудник лаборатории молеку-
лярных механизмов адаптации в НИИ общей патологии и па-
тофизиологии РАМН.
Решетняк Виталий Кузьмич — доктор медицинских наук, про-
фессор, заместитель директора Учебно-научного центра при
Медицинском центре управления делами Президента.
]Шапот Владимир Сергеевич! — доктор биологических наук,
профессор, член-корреспондент РАМН.
Ярилин Александр Александрович — доктор медицинских наук,
профессор, заведующий отделом клеточной иммунологии в
Институте иммунологии ГНЦ М3 РФ.
3
Содержание
Предисловие. — Б. Б. Мороз...............................9
Лекция 1. Апоптоз: природа феномена и его роль в норме
и при патологии. — А.А.Ярилин...........................13
Введение................................................13
1.1. Проявления, механизмы развития и регуляция апоптоза
на уровне клетки .......................................15
1.1.1. Феноменология и методы выявления апоптоза....15
1.1.2. Многообразие пусковых механизмов апоптоза....20
1.1.3. Пути передачи внутриклеточных сигналов к развитию
апоптоза (частные события)..........................24
1.1.4. Общий путь индукции апоптоза.................28
1.1.5. Эндогенные регуляторы апоптоза...............30
1.2. Роль апоптоза в многоклеточном организме...........32
1.2.1. Апоптоз, процессы формообразования и клеточного
гомеостаза на уровне организма......................32
1.2.2. Роль апоптоза в иммунных процессах...........36
1.3. Место апоптоза в патологии.........................41
1.3.1. Апоптоз как компонент типовых патологических
процессов...........................................44
1.3.2. Патологические процессы, обусловленные ослабле-
нием апоптоза.......................................45
1.3.3. Патологические процессы, связанные с усилением
апоптоза............................................48
Заключение..............................................51
Список литературы.......................................52
Лекция 2. Иммунная система и патология. — Г.А. Игнатьева 57
Введение................................................57
2.1. Словарь сокращений и терминов .....................58
2.1.1. Субпопуляции клеток иммунной системы.........58
2.1.2. Молекулы клеточных мембран...................58
2.1.3. Цитокины.....................................59
2.1.4. Конкретные иммунологические эффекторные реак-
ции ................................................60
2.2. Определение понятия «иммунитет»....................60
2.3. Главные функции иммунной системы...................63
4
2.4. Анатомия и физиология иммунной системы. Основные зако-
номерности развития иммунного ответа....................67
2.4.1. Органы лимфопоэза.............................68
2.4.2. Характеристика лимфоцитов.....................72
2.5. Гуморальные факторы иммунитета .....................82
2.6. Стадии развития иммунного ответа....................85
2.7. Иммунная подсистема кожи........................... 92
2.8. Иммунная система слизистых оболочек.................93
2.9. Патологические процессы с участием иммунной системы . . 96
2.9.1. Полноценная иммунная система..................96
2.9.2. Генетические дефекты в иммунной системе.......99
2.9.2.1. Патологические процессы с участием иммун-
ной системы — истинные аутоиммунные
заболевания....................................111
2.9.2.2. Патологические процессы с участием иммун-
ной системы при общем тяжелом патологи-
ческом процессе в организме....................112
2.9.3. Иммуностимулирующая терапия, неспецифичная
по антигену.........................................117
Список литературы ......................................118
Лекция 3. Аллергия: аллергены, индукция и регуляция
синтеза иммуноглобулина Е. — И. С. Гущин................121
Введение................................................121
3.1. Аллергены и аллергенность..........................124
3.1.1. Номенклатура аллергенов......................126
3.1.2. Идентификация и очистка аллергенов...........126
3.1.3. Нативные аллергены как гетерогенная и изменчивая
популяция...........................................130
3.2. Иммуноглобулин Е: индукция и регуляция синтеза .... 133
3.2.1. Модель запуска синтеза IgE...................134
3.2.2. Сигнал индукции синтеза IgE, обеспечиваемый
интерлейкином-4 (и интерлейкином-13)................137
3.2.3. Сигнал индукции синтеза IgE, обеспечиваемый
взаимодействием CD40 с CD154........................139
3.2.4. Т-клетки у/5 и регуляция синтеза IgE.........142
3.2.5. Независимая от взаимодействия CD40 с CD154
индукция синтеза IgE‘...............................143
3.2.6. Вспомогательные молекулы, усиливающие
и сдерживающие влияния..............................144
3.2.7. Избирательность включения ТЬ2-клеток в IgE-ответ . 150
3.2.8. Возможные способы оценки опосредуемого ТЬ2-клет-
ками аллергического ответа в клинических условиях . 152
3.3. Некоторые замечания о противоаллергическом лечении . . 153
Список литературы ......................................154
Лекция 4. Опухолевый рост: ткани, клетки, молекулы. —
А.В.Лихтенштейн, | В. С.Шапот\..........................156
Введение...............................................156
Терминологический словарь..............................157
5
4.1. Опухолевый рост: общая характеристика..............159
4.1.1. Классификация................................ 159
4.1.2. Краткие эпидемиологические данные.............160
4.1.3. Этиологические факторы канцерогенеза..........164
4.1.4. Характерные свойства опухолей.................167
4.1.5. Взаимоотношения опухоли и организма...........169
4.1.6. Стадии развития...............................174
4.2. Опухолевые клетки..................................177
4.2.1. Изменения кариотипа и хромосомные аберрации 177
4.2.2. Признаки клеточной трансформации в культуре . . . 178
4.2.3. Иммортализация опухолевых клеток..............178
4.2.4. Межклеточная кооперация.......................179
4.3. Молекулярные механизмы опухолевого роста...........179
4.3.1. Эндокринная, паракринная и аутокринная регуляция 180
4.3.2. Митогенная «рефлекторная дуга» ...............181
4.3.3. Клеточный цикл................................184
4.3.4. Перенос митогенного сигнала...................186
4.3.5. Реализация митогенного сигнала................196
4.3.6. Апоптоз.......................................202
4.3.7. Механизмы опухолевой трансформации............208
Заключение..............................................218
Список литературы ......................................218
Лекция 5. Феномен стресса. Эмоциональный стресс
и его роль в патологии. — М.Г.Пшенникова................220
Введение................................................220
5.1. Феномен стресса....................................222
5.1.1. Стресс-реакция................................223
5.1.2. Стресс-система ...............................225
5.1.3. Стресс-лимитирующие системы...................230
5.1.4. Роль соотношения активностей стресс-системы
и стресс-лимитирующих систем в реакции организма
на стрессоры........................................235
5.1.5. Адаптивные и повреждающие эффекты стресс-реакции 238
5.2. Эмоциональный стресс и связанные с ним патологические
состояния...............................................249
5.2.1. Особенности эмоциональных стрессов и эмоциональной
стресс-реакции .....................................249
5.2.2. Стрессорные патологические состояния и их возмож-
ные механизмы ......................................253
5.2.2.1. Роль стресс-системы в формировании эмоцио-
нального стресса и патогенезе стрессорных
повреждений....................................253
5.2.3. Виды патологических состояний у человека, связанные
с эмоциональным стрессом, и их механизмы............259
5.2.3.1. Сердечно-сосудистая система ..259
5.2.3.1.1. Ишемическая болезнь сердца
и инфаркт миокарда.......................266
5.2.3.1.2. Внезапная сердечная смерть .... 281
5.2.З.1.З. Гипертоническая болезнь.......287
6
5.2.3.2. Желудочно-кишечный тракт..............291
5.2.3.3. Система крови и иммунная.система при
эмоциональном стрессе..........................297
5.2.3.4. Психический статус при эмоциональном стрессе
и посттравматическое стрессовое расстройство 313
5.2.3.4.1. Нарушения психического статуса . . 314
5.2.3.4.2. Посттравматическое стрессовое
расстройство.............................316
5.2.4. Основы предрасположенности и устойчивости
к стрессорным повреждениям..........................322
5.3. Принципы профилактики и коррекции стрессорной
патологии...............................................328
5.3.1. Профилактика и коррекция с помощью защитных
эффектов адаптации к факторам среды ................329
5.3.2. Коррекция с помощью фармакологических средств 334
5.3.3. Использование приемов психотерапии при стрессор-
ных психосоматических расстройствах.................338
Список литературы.......................................339
Лекция 6. Боль: физиологические и патофизиологические
аспекты. — В. К. Решетник, МЛ. Кукушкин..............354
Введение................................................354
6.1. Характеристика боли ...............................355
6.2. Физиология боли....................................357
6.2.1. Анатомо-функциональная организация ноцицептивной
системы...........................................357
6.2.1.1. Ноцицепторы и их афференты..........357
6.2.1.2. Периферические алгогены.............358
6.2.1.3. Первое переключение ноцицептивной инфор-
мации (первичное ноцицептивное реле) ... 359
6.2.1.4. Восходящие спинальные тракты (пути)
ноцицептивных афферентов.....................361
6.2.1.5. Таламические ядра, воспринимающие
ноцицептивную афферентацию ..................363
6.2.1.6. Обработка ноцицептивной информации в коре
больших полушарий............................365
6.2.2. Антиноцицептивная система мозга............368
6.3. Патофизиология боли...............................372
6.3.1. Соматогенные болевые синдромы................373
6.3.1.1. Механизмы возникновения первичной
гипералгезии...................................373
6.3.1.2. Механизмы развития вторичной гипералгезии 375
6.3.2. Патофизиология нейрогенных болевых синдромов . . 378
6.3.2.1. Этиология нейрогенных болевых синдромов . 378
6.3.2.2. Клинические проявления нейрогенных болевых
синдромов......................................378
6.3.2.3. Периферические механизмы нейрогенной боли 379
6.3.2.4. Центральные механизмы нейрогенной боли 380
6.4. Болевые синдромы в клинической практике...........383
Список литературы .....................................388
7
Лекция 7. Патология гемостаза. — В.А. Макаров.................390
Введение......................................................390
7.1. Современные представления о свертываемости крови ... 391
7.1.1. Механизмы свертывания крови........................391
7.1.2. Механизмы ингибирования свертывания крови.
Фибринолиз...........................................398
7.2. Современные представления о природе тромбозов......400
7.2.1. Основные причины развития тромбозов................400
7.2.2. Лабораторная диагностика вероятности развития
тромбозов............................................404
7.3. Геморрагии...............................................407
7.3.1. Виды и основные причины развития геморрагий . . . 407
7.3.2. Лабораторная диагностика геморрагических состояний 413
7.4. Синдром диссеминированного внутрисосудистого
свертывания (ДВС-синдром) ...............................415
7.5. Лекарственная коррекция патологии гемостаза..............416
Заключение....................................................419
Список литературы ............................................419
Предисловие
В биологии и медицине в последнее время происходят со-
бытия, меняющие привычные представления о механизмах
патологических процессов. Суть этих событий — последние
достижения в молекулярной биологии и генетике. Вместе с
тем и в содержании традиционных проблем патофизиологии
произошли большие перемены — появились новые пробле-
мы, представления, идеи, что должно найти отражение и в
преподавании патофизиологии. Придать курсу патофизиоло-
гии современное звучание, изложить механизмы патоло-
гического процесса от изменений на уровне молекул до кли-
нического проявления — сложная задача, тем более что мно-
гие звенья патогенеза ряда заболеваний еще не расшифро-
ваны.
Предлагаемая читателю книга представляет собой в своей
основе лекции, предназначенные для опубликования в журна-
ле «Патологическая физиология и экспериментальная тера-
пия». Авторы лекций являются специалистами, активно участ-
вующими в научных исследованиях. Цель лекций — использо-
вать комплексный, интегральный подход, осветить каждую
проблему с разных сторон в их единстве для наиболее полного
о ней представления. Важное место при этом занимает моле-
кулярно-биологическая интерпретация явлений. Другая осо-
бенность лекций заключается в том, что сущность патофизио-
логического процесса анализируется на основе описания и со-
временного понимания физиологических закономерностей.
Излагая новейшие сведения о механизмах патологических
процессов, которые еще не получили необходимого освеще-
ния в учебниках и руководствах, авторы имели в виду, что лек-
ции должны создать базу для понимания закономерностей,
наблюдаемых у человека. Лекции предназначены в первую
очередь для преподавателей патофизиологии, а также клини-
ческих дисциплин, но в то же время и для читателей с самыми
различными профессиональными интересами, так как в них
речь идет о проблемах, знание современного состояния кото-
рых необходимо каждому врачу. В лекциях отражена попытка
компромисса между строго научным изложением и интереса-
ми широкого круга врачей и биологов.
9
Лекция А.А.Ярилина «Апоптоз: природа феномена и его
роль в норме и при патологии» посвящена программирован-
ной гибели клеток, которая в последние годы привлекла при-
стальное внимание медиков. Апоптоз является важнейшей
физиологической реакцией, определяющей гомеостаз. Гибель
отдельных клеток полезна и необходима на разных стадиях
онтогенеза для поддержания постоянства численности клеток,
для обеспечения правильного соотношения численности кле-
ток различных типов, для «разборки» определенных структур.
Назначение апоптоза состоит также в удалении генетически
дефектных, мутантных клеток. Нарушение механизмов кон-
троля и реализации апоптоза приводит к ослаблению или уси-
лению программированной гибели клеток. Последствиями ос-
лабления апоптоза являются развитие аутоиммунной патоло-
гии и повышение вероятности возникновения опухолей. След-
ствием усиления может быть формирование врожденных де-
фектов развития некоторых органов и тканей. Старение орга-
низма также рассматривается с учетом явлений апоптоза. При
расшифровке механизма действия на организм ряда неблаго-
приятных факторов окружающей среды, химических и физи-
ческих (ионизирующее излучение), апоптоз имеет важное зна-
чение. Ряд лекарственных средств реализует свое действие
через индукцию апоптоза, и его нарушение, например, служит
одной из причин множественной лекарственной резистент-
ности. В лекции рассмотрены феноменология и молекулярные
механизмы апоптоза, но главное внимание уделено его роли в
патологии. Понятие об апоптозе, развившееся в рамках мор-
фологии и биохимии, приобрело в наше время универсальное
общебиологическое значение для понимания течения физио-
логических и патологических процессов, для диагностики за-
болеваний и поиска новых подходов к их терапии.
Лекция Г.А.Игнатьевой «Иммунная система и патология»
привлечет внимание читателя не только потому, что в литера-
туре отсутствуют публикации, в которых в концентрирован-
ном виде сформулированы самые современные представления
об иммунитете. Наряду с объективным описанием самых пос-
ледних данных в лекции весьма выражено собственное отно-
шение автора ко многим вопросам, что может явиться пово-
дом для размышления. Сформулировано определение имму-
нитета, в основе которого лежат представления о молекуляр-
ных механизмах распознавания своих и чужих антигенов, опи-
саны взаимосвязь и сопряжение иммунитета с другими меха-
низмами защиты, обосновывается положение о том, что при
любом завершенном иммунном ответе имеет место деструкция
собственных тканей. Иммунный ответ характеризуется как
физиологическая реакция в норме; обсуждаются условия, при
которых он сопровождается признаками альтерации тканей и
воспаления (иммунопатогенеза), т.е. взаимоотношения нормы
10
и патологии, связанные с количеством патогенного фактора и
невозможностью его полной элиминации из организма. Им-
мунная система может оказаться несостоятельной, если орга-
низм столкнется с новыми факторами окружающей среды в
связи с антропогенной деятельностью или новыми вирусами
на базе природных ретровирусов. Подробно описаны патоло-
гические процессы с участием иммунной системы и выражено
отношение автора к методам иммунокорректирующего ле-
чения.
Лекция И.С.Гущина посвящена аллергии. Учение об ал-
лергии сегодня нельзя себе представить вне принципиально
новых сведений, полученных на молекулярном уровне. Имен-
но эти материалы занимают центральное место в лекции, ко-
торая отражает ключевой механизм аллергии — индукцию и
регуляцию синтеза IgE. Новые материалы приведены практи-
чески во всех разделах, в которых рассматривается цепь после-
довательных событий от момента первичного действия аллер-
гена на иммунную систему до завершения ответа образовани-
ем и поддержанием продукции аллергенспецифического IgE-
ответа. Обсуждается возможность создания способов, позво-
ляющих предсказывать индивидуальную склонность к типу
иммунной реакции, ответственной за образование аллергичес-
ких антител.
В лекции А.В.Лихтенштейна и [В С.Шапота! в обобщенном
виде изложены основные представления об опухолевом росте
на организменном, клеточном и молекулярном уровнях. Впе-
рвые в учебной литературе описаны молекулярные механизмы
опухолевого роста, механизм деления клетки, действующие
начала которого обладают либо промитогенной (протоонкоге-
ны и протоонкобелки), либо антимитогенной (супрессоры)
активностью, механизмы защиты — репарация ДНК и апо-
птоз. Подробно описана роль в канцерогенезе гена-супрессора
р53 и гена-супрессора АРС. Генетические события рассматри-
ваются на фоне освещения других аспектов опухолевого роста
и там, где это возможно, при различных опухолях у человека,
благодаря чему у читателя создается ясное представление о со-
стоянии проблемы в целом. Совершенно очевидно, сколь зна-
чительной трансформации должен подвергнуться данный раз-
дел патофизиологии при преподавании, учитывая содержание
этой лекции.
Современная жизнь, к сожалению, сопряжена с огромны-
ми психологическими перегрузками. Медицинские последст-
вия острого и хронического эмоционального стресса являются
предметом изучения различных дисциплин. В лекции
М.Г.Пшенниковой освещается проблема эмоционального
стресса и его роли в патологии с учетом всех основных аспек-
тов проблемы. Материалы лекции могут быть использованы
для преподавания не только патофизиологии, но и клиничес-
11
ких дисциплин, так как эмоциональный стресс может быть
причиной нарушения функции различных систем организма.
В лекции описана роль эмоционального стресса в патогенезе
ишемической болезни сердца, внезапной сердечной смерти,
гипертонической болезни, язвенной болезни желудка и две-
надцатиперстной кишки, в изменениях системы крови и им-
мунитета, описаны стрессорные нарушения психологического
статуса. Впервые в патофизиологической учебной литературе
освещены посттравматические стрессовые расстройства и ме-
ханизмы предрасположенности и устойчивости к стрессорным
повреждениям, принципы их профилактики и коррекции. Па-
тофизиологические эффекты эмоционального стресса развер-
тываются как следствие неадекватной реакции физиологичес-
ких механизмов, определяющих адаптацию организма. В лек-
ции изложено и современное представление о механизмах
формирования адаптивной реакции на различных уровнях ин-
теграции организма.
В.К.Решетняк и М.Л.Кукушкин рассматривают в своей
лекции принципиальные различия между физиологической и
патологической болью, морфофункциональную организацию
систем, осуществляющих регуляцию болевой чувствительнос-
ти, излагают механизмы развития болевых синдромов, приво-
дят схемы их патогенетической терапии.
В лекции В.А.Макарова «Патология гемостаза» привлека-
ют внимание современная схема свертывания крови, новые
данные о патогенезе тромбозов и лечении геморрагий.
Авторы лекций надеются, что представленные материалы
окажутся полезными для преподавателей патофизиологии и
других медицинских дисциплин, для научных работников,
врачей и студентов.
Академик РАМН Б. Б. Мороз
Лекция 1
Апоптоз: природа феномена и его роль
в норме и при патологии
А.А.Ярилин
Введение
Термин «апоптоз» (греч. — опадание листьев) введен в на-
учный обиход в 1972 г. [Kerr et al., 1972] для обозначения
формы гибели клеток, прототипом которой является гибель
тимоцитов под действием глюкокортикоидов. Эта форма кле-
точной смерти была отождествлена с ранее описанной про-
граммированной гибелью клеток: разница обозначений отра-
жает способы идентификации гибели — морфологический в
первом и биохимический во втором случае. Использование
двух терминов как равнозначных сохраняется до настоящего
времени, хотя и вызывает критику. Поэтому считаем целесо-
образным сформулировать определение обоих понятий.
• Программированная гибель — активная форма гибели
клетки, являющаяся результатом реализации ее генети-
ческой программы или ответом на внешние сигналы и
требующая затрат энергии и синтеза макромолекул de
novo (биохимический способ определения).
• Апоптоз — форма гибели клетки, проявляющаяся в
уменьшении ее размера, конденсации и фрагментации
хроматина, уплотнении наружной и цитоплазматических
мембран без выхода содержимого клетки в окружающую
среду (морфологический способ определения).
Несмотря на то что фактор программированности и актив-
ный характер гибели обычно являются более принципиальны-
ми, чем сопутствующие ей морфологические изменения, чаще
используется термин «апоптоз», вероятно, из-за его краткости.
Апоптоз обычно противопоставляется другой форме гибе-
ли клеток — некрозу, который развивается при воздействии
внешних по отношению к клетке повреждающих агентов и не-
адекватных условий среды (гипоосмия, крайние значения pH,
гипертермия, механические воздействия, влияние агентов, по-
вреждающих мембрану, формирование пор в мембране с учас-
тием факторов комплемента) и проявляется набуханием клет-
ки и разрывом наружной мембраны с выходом содержимого
клетки в среду.
13
Апоптоз универсально распространен в мире многоклеточ-
ных организмов; аналогичные ему проявления отмечены у
дрожжей, трипаносом и некоторых других одноклеточных.
Ему подвержены все виды тканей.
История изучения апоптоза поучительна. После описания
феномена апоптоза, введения термина и достаточно четкого
обозначения биологической значимости данного явления в
1972 г. наступило десятилетие его полузабвения. Однако уже в
1980 г. A.H.Wyllie, в лаборатории которого сформировалось
понятие «апоптоз», связал его с процессами деградации хро-
матина, катализируемыми эндонуклеазами. В 1984 г. была по-
казана значимость макромолекулярных синтезов для реализа-
ции апоптоза, т.е. подтвержден активный характер процесса.
Интересно, что на этом раннем этапе развития учения об
апоптозе большой вклад в его разработку был внесен радио-
биологами, причем в первую очередь российскими. В 1979 г.
К.П.Хансон, основываясь на данных Skalka и соавт. (1976) о
регулярном характере деградации хроматина лимфоцитов при
действии на них ионизирующей радиации, высказал мысль о
том, что радиационная гибель лимфоцитов в интерфазе пред-
ставляет собой проявление программированной гибели, т.е.
апоптоза. Связь радиации с апоптозом была обоснована в на-
чале 80-х годов в ряде российских и зарубежных лабораторий.
С конца 80-х годов внимание к апоптозу резко возросло, что в
значительной степени связано с установлением роли апоптоза
в селекции тимоцитов и в других иммунологических феноме-
нах. С начала 90-х годов интерес к апоптозу становится всеоб-
щим, число посвященных ему публикаций в последние годы
непрерывно растет.
Бурный рост интереса к проблеме апоптоза связан с рядом
обстоятельств. Прежде всего появились методические возмож-
ности регистрации различных проявлений апоптоза и анализа
его молекулярных механизмов, которые оказались тесно свя-
занными с механизмами других «актуальных» феноменов (на-
пример, активации клеток и связанной с ней биологической
сигнализации). Далее изучение апоптоза оказалось очень про-
дуктивным для понимания ряда важнейших явлений и про-
цессов, включая иммунный гомеостаз и онкогенез. Наконец, в
связи с апоптозом возникла необходимость пересмотра ряда
концептуальных основ физиологии и патологии. Вместо преж-
них представлений о гибели клеток многоклеточного организ-
ма как отрицательном по значимости явлении, идентифици-
руемом с некрозом, сформировался новый взгляд, согласно
которому гибель части клеток в пределах организма является
закономерным и необходимым процессом и само существова-
ние многоклеточного организма подразумевает баланс жизни
и смерти на уровне составляющих его клеточных популяций.
Менее очевидной представляется роль апоптоза в развитии па-
14
тологических процессов. Создается впечатление, что эта
форма гибели клеток (в отличие от некроза) не является обя-
зательной составляющей типовых патологических процессов;
скорее можно говорить о нарушениях самого апоптоза как ос-
нове ряда заболеваний.
Данный обзор посвящен рассмотрению и оценке места
апоптоза в патологии, однако для понимания этой проблемы
невозможно обойтись без знаний о природе данного явления
и его механизмах, без участия в реализации нормальных фи-
зиологических процессов. Мы рассмотрим проявления и био-
логическую значимость апоптоза на двух уровнях. Сначала
будут представлены наиболее принципиальные данные о мо-
лекулярных основах осуществления и регуляции апоптоза на
уровне клетки, а затем — сведения о месте апоптоза в целост-
ном организме в условиях нормы и патологии.
Здесь представлены лишь основные сведения об этом про-
цессе; для более глубокого ознакомления с природой и меха-
низмами апоптоза рекомендуем обратиться к многочислен-
ным обзорам, опубликованным в последние годы [Барышни-
ков А.Ю., Шишкин Ю.В., 1996; Ярилин А.А., 1996, 1998;
Cohen J.J., 1992; Green D.R. et al., 1992; Onici Y., 1994; Rus-
sel J.H., 1995; Kroemer G., 1995; McConkey D.J. et al., 1996;
Kroemer G. et al., 1997; Rowan S. et al., 1997; Wong B., Choi Y.,
1997; Komblau S.M., 1998; Krammer P.H., 1999; Rathmell G.C.,
Thompson C.B., 1999].
1.1. Проявления, механизмы развития и регуляция
апоптоза на уровне клетки
1.1.1. Феноменология и методы выявления
апоптоза
Наиболее характерные проявления апоптоза определяются
тем, что первые события, связанные с его осуществлением,
начинаются в ядре. В ядре регистрируются первые морфологи-
ческие признаки апоптоза — конденсация хроматина с фор-
мированием его осмиофильных скоплений, прилежащих к
ядерной мембране. Позже появляются вдавления ядерной
мембраны и происходит фрагментация ядра. Отшнуровавшие-
ся фрагменты ядра, ограниченные мембраной, обнаруживают-
ся вне клетки; их называют апоптотическими тельцами. В ци-
топлазме происходят расширение эндоплазматического рети-
кулума, конденсация и сморщивание гранул. Важнейший при-
знак апоптоза — снижение трансмембранного потенциала ми-
тохондрий. Клеточная мембрана утрачивает ворсинчатость и в
то же время образует пузыревидные вздутия. Клетки округля-
ются и отделяются от субстрата. Проницаемость мембраны
15
Некроз
Апоптоз
Рис. 1.1. Развитие некроза и апоптоза клеток. Характерные морфо-
логические проявления.
повышается лишь в отношении небольших молекул, причем
это происходит позже изменений в ядре: если конденсация
хроматина проявляется в пределах 1 ч, то повышение прони-
цаемости для пропидия йодида — только через 3—5 ч. Как уже
упоминалось, одной из наиболее характерных особенностей
апоптоза является уменьшение объема клетки в противопо-
ложность ее набуханию при некрозе. В схематической форме
морфологические проявления апоптоза и некроза отражены
на рис. 1.1. Следует обратить внимание на увеличение разме-
ров клетки и ранние изменения мембраны, но не ядра при не-
крозе и уменьшение размеров клетки с ранними изменениями
в ядре, но не в цитоплазме при апоптозе. Признаки апоптоза и
некроза сопоставлены в табл. 1.1.
Таблица 1.1. Сравнительная характеристика апоптоза н некроза
клеток
Показатели Апоптоз Некроз
Пусковой фактор Сигнал, воспринимаемый мембранными рецептора- ми, или отсутствие сигна- ла Токсические и мем- бранотропные аген- ты, неадекватные внешние условия
16
Продолжение табл. 1.1
Показатели Апоптоз Некроз
Скорость развития 1-12 ч В пределах 1 ч
Локализация пер- вичного поврежде- ния В ядре В мембране
Причины гибели Деградация ДНК, нару- Нарушение целости
клетки шение энергетики клетки мембраны
Изменения размера Уменьшение (сморщива- Увеличение (набуха-
клетки ние) ние)
Изменения ядра Конденсация хроматина, пикноз, фрагментация Набухание
Изменения в цито- плазме Конденсация цитоплаз- мы, уплотнение гранул Лизис гранул
Изменения клеточ- ной мембраны Потеря микроворсинок, образование вздутий Нарушение целости
Состояние ДНК Разрывы с образованием сначала крупных, затем мелких фрагментов Неупорядоченная деградация
Энергозависимость Зависит Не зависит
Зависимость от синтеза макромоле- кул » » »
Примеры проявле- Метаморфоз, отрицатель- Гибель клеток вслед-
ния Методы выявления: ная селекция лимфоци- тов, гормонозависимая атрофия, интерфазная ра- диационная гибель лим- фоцитов ствие гипоксии, дей- ствия токсинов, ви- русного цитолиза, комплементзависи- мого цитолиза
морфологические Сморщивание клетки Набухание клетки
тинкториальные Ослабление окрашивае- мости ДНК-тропными красителями Окрашиваемость суправитальными красителями
цитофлуоримет- Г иподиплоидность, Окрашиваемость
рические уменьшение размеров клетки, появление фос- фатидилсерина на мем- бране йодидом пропидия
электрофоретиче- Формирование дискрет- Размазанное пятно
с кие ных фракций («лесенки») при электрофорезе
при электрофорезе ДНК ДНК
2—1385
17
Указанные морфологические проявления апоптоза изуче-
ны в модельных опытах in vitro. Апоптотические клетки труд-
но выявить в тканях in situ в связи с тем, что они быстро фаго-
цитируются не только макрофагами и нейтрофилами, но и
другими окружающими клетками, не являющимися «профес-
сиональными» фагоцитами. Быстрота фагоцитирования связа-
на с появлением на их поверхности (вследствие реорганиза-
ции мембраны) некоторых молекул, распознаваемых фагоци-
тами; такими молекулами являются фосфосерин, тромбоспон-
дин, десиалированные мембранные гликоконъюгаты. В ре-
зультате фагоцитоза, которому клетки подвергаются уже в
процессе развития апоптоза, их содержимое не выделяется в
межклеточное пространство, как это бывает при некрозе,
когда вокруг гибнущих клеток скапливаются их активные
внутриклеточные компоненты, включая энзимы, закисляется
среда, что способствует гибели других клеток и развитию очага
воспаления. Апоптоз поражает индивидуальные клетки и
практически не отражается на их окружении.
Главное проявление апоптоза — деградация хроматина. Ее
основой служит ферментативное расщепление ДНК, которое
происходит в несколько этапов с формированием все более мел-
ких фрагментов. Сначала образуются фрагменты, включающие
700; 200—250; 50—70 тыс. пар нуклеотидов (т.п.н.), затем —
фрагменты, содержащие 30—50 т.п.н. [Zhivotovsky В. et al., 1994].
На этой стадии происходят конденсация хроматина и инвагина-
ции ядерной мембраны. После реализации этого этапа процесс
становится необратимым. Затем наступает очередной этап дег-
радации, который изучен наиболее полно и служит широко при-
нятым опознавательным признаком апоптоза, — межнуклео-
сомная дезинтеграция ДНК, т.е. разрывы нитей ДНК, находя-
щихся между нуклеосомами. При этом образуются фрагменты,
кратные по величине 180—200 пар нуклеотидов, что соответст-
вует протяженности нити ДНК в пределах одной нуклеосомы.
Этот тип деградации обычно завершается в течение суток. При
всей типичности для апоптоза этого типа расщепления ДНК из-
вестны примеры, когда процесс ограничивается образованием
более крупных фрагментов.
Деградация хроматина при апоптозе является активным
процессом, зависящим от температуры, источников энергии,
синтеза РНК и белка de novo. Различные этапы деградации
ДНК катализируются разными формами эндонуклеаз, отлича-
ющимися субстратной специфичностью и условиями проявле-
ния активности (см. далее).
Несмотря на безусловно ключевую роль деградации ДНК в
осуществлении апоптоза, между этими процессами нельзя по-
ставить знак равенства. Во-первых, морфологические и био-
химические изменения в цитоплазме и мембранах, характер-
ные для апоптоза (снижение трансмембранного потенциала
18
митохондрий, накопление реактивных форм кислорода и т.д.),
могут быть индуцированы в энуклеированных (безъядерных)
клетках, т.е. в ситуации, когда деградация хроматина невоз-
можна в связи с его отсутствием. Во-вторых, непосредствен-
ной причиной смерти клетки при апоптозе являются не разру-
шение ДНК и прекращение работы генов, поскольку их по-
следствия сказываются несколько позже того срока, когда на-
ступает апоптотическая гибель клетки. Предполагают, что ее
гибель обусловлена энергетическим голодом, вызванным рас-
ходованием АТФ на работу по репарации поврежденной ДНК.
Методы выявления апоптоза достаточно разнообразны.
Ориентировочная идентификация апоптоза возможна на ос-
новании вышеназванных морфологических признаков. Более
доказательные подходы к оценке апоптоза основаны на выяв-
лении формирующихся разрывов ДНК, ее деградации и поте-
ри клеткой части генетического материала. Часто используют
электрофоретическое разделение полидезоксирибонуклеотид-
ных фрагментов, экстрагируемых из клеток; на электрофоре-
грамме фрагменты образуют характерную «лесенку» (серию
прерывистых полос), отражающую дискретный характер меж-
нуклеосомных разрывов [Zhyvotovsky В. et al., 1995]. Другие
методы основаны на синтезе олигонуклеотидов из меченых
предшественников, катализируемом дезоксирибонуклеоти-
дилтрансфердзой (TdT), и их встраивании в ДНК через ее сво-
бодные концы, формирующиеся в участках разрывов. Встра-
ивание меченых олигонуклеотидов затем выявляется с помо-
щью цитофлуорометрических или морфологических методов.
На этом, в частности, основан TUNEL (TdT-mediated dUTR-
biotin nick end-labeling) — метод оценки апоптоза [Gavrieli Y. et
al., 1992].
В настоящее время широко используется скрининговый
цитофлуорометрический метод определения численности апо-
птотических клеток, основанный на утрате ими части хрома-
тина. При этом фиксированные клетки окрашивают йодидом
пропидия, который, проникая в ядро, взаимодействует с ДНК,
что позволяет выявить процент гиподиплоидных клеток с по-
мощью проточной цитофлуорометрии [McCloskey T.W. et al.,
1994]. На регистрации снижения трансмембранного потенци-
ала митохондрий основан метод определения апоптоза по
изменению окрашивания липофильными катионными флуо-
рохромами, например йодидом дигексилоксикарбоцианина
или родамином, тропными к внутренней поверхности мембра-
ны митохондрий. Наконец, широкое распространение полу-
чил метод раннего цитофлуорометрического обнаружения
апоптоза по связыванию меченого аннексина V с фосфатидил-
серином, который, как уже отмечалось, появляется на поверх-
ности клеток, подвергающихся апоптозу. Параллельно клетки
(нефиксированные) окрашивают йодидом пропидия, который
э*
19
способен проникать лишь в клетки, подвергающиеся некрозу.
Окрашивание одним аннексином служит показателем раннего
апоптоза, окрашивание йодидом пропидия — свидетельством
некроза, окрашивание обоими красителями — показателем
позднего апоптоза, к которому присоединился вторичный не-
кроз [Van Engeland М. et al., 1998].
1.1.2. Многообразие пусковых механизмов
апоптоза
В развитии апоптоза выделяют 3 стадии: индукторную, эф-
фекторную и стадию деградации. Если две последние едины
для всех разновидностей апоптоза, то первая стадия проявля-
ется весьма разнообразно в зависимости от типа клеток и ин-
дукторных факторов. Это разнообразие можно объединить в
две группы, в одну из которых войдут ситуации, когда про-
грамма гибели включается внешними факторами, а в дру-
гую — случаи, когда эта программа предсуществует в клетке и
реализуется в отсутствие защитных факторов (табл. 1.2).
Таблица 1.2. Разновидности сигналов, приводящих к развитию
аноптоза
Происхождение сигнала Природа сигнала Примеры
Внеклеточные сигналы Внутриклеточные сигналы Дефицит факто- ров Антиген Гормон Цитокин или его аналог (FasL) Повреждение хро- мосом, другие из- менения хроматина Дефицит цитоки- нов Дефицит антигенов Дефицит корецеп- тора Отрицательная селекция ти- моцитов Действие глюкокортикоидов Fas-зависимый апоптоз, ци- толиз, вызванный ФНОа или ФНО(3 Радиационная гибель лим- фоцитов в интерфазе. Дейст- вие топоизомераз Гибель кроветворных клеток при дефиците цитокинов. Гибель активированных Т-клеток в отсутствие ИЛ-2 Гибель В-клеток в зароды- шевых центрах Апоптоз при активации Т-клеток в отсутствие сигна- ла с CD28
20
Наиболее детально изучен вариант с индукцией апоптоза
внешними воздействиями — экзогенной индукцией. Только в
этом случае можно с определенностью говорить о рецепции
(восприятии) сигнала, вызывающего развитие апоптоза. Наи-
более полно проанализированы условия экзогенной индукции
апоптоза Т-лимфоцитов, особенно их незрелой формы — кор-
тикальных тимоцитов. Классическими индукторами апоптоза
тимоцитов служат кортикостероиды. Как известно, они про-
никают в клетку и проявляют свое действие через рецепторы,
локализующиеся в ядре. Поскольку отнюдь не все клетки гиб-
нут под действием кортикостероидов, возникает вопрос о воз-
можности различной интерпретации клеткой однотипного
сигнала. Примером специфичности действия индукторов
апоптоза на клетки определенных типов могут служить также
эстрогены и андрогены, которые индуцируют апоптоз преиму-
щественно эпителиальных клеток.
Более распространенным вариантом восприятия сигналов
является рецепция сигналов к апоптозу через мембранные ре-
цепторы, при которой проблема интерпретации сигнала также
весьма актуальна. Так, сигнал, поступающий в Т-лимфоциты
через рецептор для антигена (комплекс TCR-CD3), который в
норме приводит к активации пролиферации клеток, в опреде-
ленных условиях вызывает их апоптоз, обозначаемый как ак-
тивационный. Сигнал может подаваться при воздействии на
рецептор антигена, суперантигена, митогенного, лектина,
антител, способных перекрестно сшивать молекулы рецепто-
ра. Индукция апоптоза при этом может происходить при вы-
полнении одного из следующих условий:
• при отсутствии дополнительного сигнала (костимуляции
через молекулу CD28 рецептора);
• при повторном действии лиганда на активированные
клетки;
• в отсутствие ростовых факторов, прежде всего интерлей-
кина 2 (ИЛ-2);
• после предварительного перекрестного сшивания моле-
кул CD4.
Аналогичная ситуация создается при активации В-лимфоци-
тов через связывание рецептора BCR (В cell receptor); условием
индукции апоптоза в этом случае является, в частности, отсутст-
вие костимуляции через мембранную молекулу CD40. Сигнал к
развитию апоптоза может подаваться также через другие мем-
бранные структуры лимфоцитов — CD2, молекулы главного
комплекса гистосовместимости класса I Рг и р2-интегрины и т.д.
Апоптоз могут индуцировать некоторые цитокины, в первую
очередь фактор некроза опухоли а (ФНО). Сигнал гибели пода-
ется через один из двух рецепторов ФНО — р55 (TNFR1). Эти
рецепторы ФНО ответственны за реализацию самых разнооб-
21
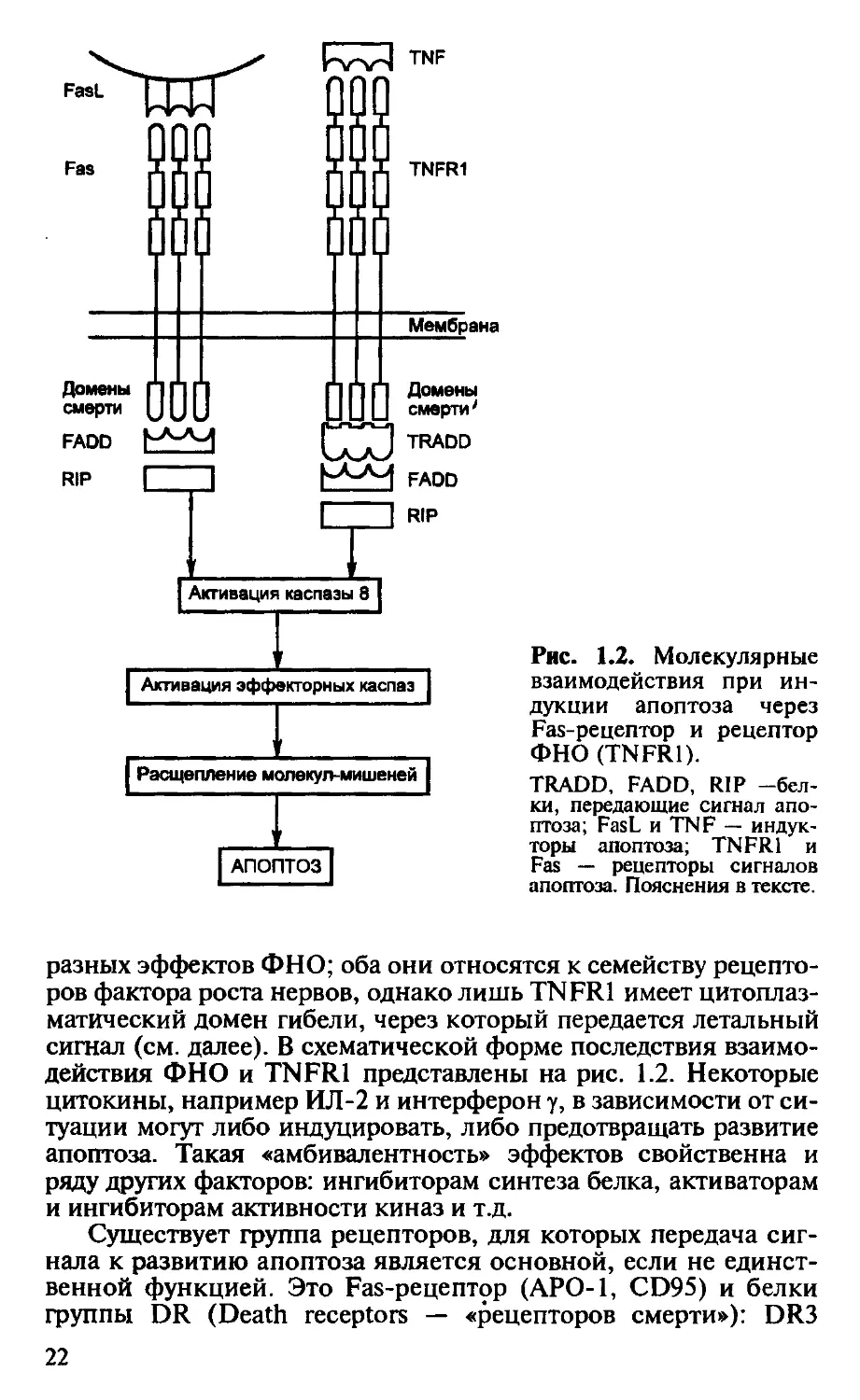
FasL
Мембрана
Домены
смерти
FADD
RIP
Домены
смерти'
TRADD
FADD
RIP
Активация каспазы 8
Активация эффекторных каспаз
Расщепление молекул-мишеней
АПОПТОЗ|
Рис. 1.2. Молекулярные
взаимодействия при ин-
дукции апоптоза через
Fas-рецептор и рецептор
ФНО (TNFR1).
TRADD, FADD, RIP -бел-
ки, передающие сигнал апо-
птоза; FasL и TNF — индук-
торы апоптоза; TNFR1 и
Fas — рецепторы сигналов
апоптоза. Пояснения в тексте.
разных эффектов ФНО; оба они относятся к семейству рецепто-
ров фактора роста нервов, однако лишь TNFR1 имеет цитоплаз-
матический домен гибели, через который передается летальный
сигнал (см. далее). В схематической форме последствия взаимо-
действия ФНО и TNFR1 представлены на рис. 1.2. Некоторые
цитокины, например ИЛ-2 и интерферон у, в зависимости от си-
туации могут либо индуцировать, либо предотвращать развитие
апоптоза. Такая «амбивалентность» эффектов свойственна и
ряду других факторов: ингибиторам синтеза белка, активаторам
и ингибиторам активности киназ и т.д.
Существует группа рецепторов, для которых передача сиг-
нала к развитию апоптоза является основной, если не единст-
венной функцией. Это Fas-рецептор (АРО-1, CD95) и белки
группы DR (Death receptors — «рецепторов смерти»): DR3
22
(APO-3), DR4 (TRAIL-R1), DR5 (TRAIL-R2). Все они отно-
сятся к тому же семейству рецепторов фактора роста нервов, к
которому принадлежат рецепторы ФНО, а также мембранные
молекулы CD30 и CD40, и, подобно TNFR1, имеют цитоплаз-
матический «домен смерти» (см. рис. 1.2). Лучше других изу-
чен Fas-рецептор (36 кДа), который появляется на поверхнос-
ти клеток спонтанно (на гепатоцитах, на части кортикальных
тимоцитов) или под влиянием активации (на зрелых лимфо-
цитах). Естественным лигандом этого рецептора и источником
сигнализации, приводящей к развитию апоптоза, является
Fas-лиганд — молекула с мол. массой 46 кДа (в мембране он
представлен преимущественно тримерной формой), гомоло-
гичная ФНО, CD40L и ряду других молекул, служащих лиган-
дами для семейства рецептора фактора роста нервов. Fas-ли-
ганд присутствует на клетках «иммунологически привилегиро-
ванных органов», о чем далее будет сказано подробнее.
Ситуация с предсуществующей программой гибели клетки
(эндогенной индукцией апоптоза) выяснена в значительно
меньшей степени, чем индукция апоптоза внешними агентами.
Ряд примеров эндогенного формирования программы гибели
может быть почерпнут из развития кроветворных клеток. На не-
которых этапах этого развития клетки автоматически гибнут,
будучи лишены сывороточных факторов и/или определенных
цитокинов. В роли факторов выживания кроветворных клеток
обычно выступают колониестимулирующие факторы и другие
гемопоэтические цитокины. Аналогичная ситуация возникает
при активации лимфоцитов: Т-клетки, стимулированные анти-
геном или митогеном, погибают, если их лишить ИЛ-2. В этом
случае гибель может быть предотвращена добавлением любо-
го из цитокинов, рецепторы которых имеют общую у-цепь, —
ИЛ-2, ИЛ-4, ИЛ-7, ИЛ-9 и ИЛ-15. Очевидно, защитный сигнал
в этом случае подается через эту у-цепь. Наконец, большинство
клеток различных типов при росте в культуре (и, вероятно, in
vivo) подвергаются апоптозу, если их лишить контакта с меж-
клеточным матриксом, например переведя в суспензию. В этом
случае защитный сигнал передается через молекулы р,-интегри-
нов. Приведенные примеры, однако, не проясняют, в чем состо-
ит программа гибели и как она формируется.
Примером эндогенного сигнала к развитию апоптоза явля-
ется также накопление нерепарированных разрывов ДНК, на-
личие которых реализуется при участии ядерного белка р53.
Дополнительным условием гибели при этом является слабый
синтез эндогенных защитных факторов типа Вс1-2 (см. далее).
Это происходит, например, в кортикальных тимоцитах, а
также при радиационном поражении лимфоцитов и т.д. В пос-
леднем случае факторами, способствующими включению про-
граммы апоптоза, могут служить, помимо разрывов ДНК, про-
дукты переокисления липидов, а также экспрессия облучен-
23
ными клетками Fas-лиганда. Эндогенным условием развития
апоптоза может быть повышение внутриклеточного уровня
Са2+, которое можно моделировать действием кальциевых ио-
нофоров. Реализации эндогенной программы гибели клетки
могут препятствовать, помимо экзогенных факторов выжива-
ния, эндогенные факторы типа Bcl-2, Bcl-xL, о которых будет
подробнее сказано далее.
Таким образом, условия индукции апоптоза весьма разно-
образны и могут быть сведены к действию внешних факторов,
воспринимаемому рецепторами (не всегда специализирован-
ными на этой функции), и/или отсутствию защиты от реали-
зации внутренней программы апоптоза. В зависимости от си-
туации (тип клеток, их состояние, сопутствующие воздейст-
вия) один и тот же фактор может как индуцировать, так и по-
давлять развитие апоптоза.
1.1.3. Пути передачи внутриклеточных сигналов
к развитию апоптоза (частные события)
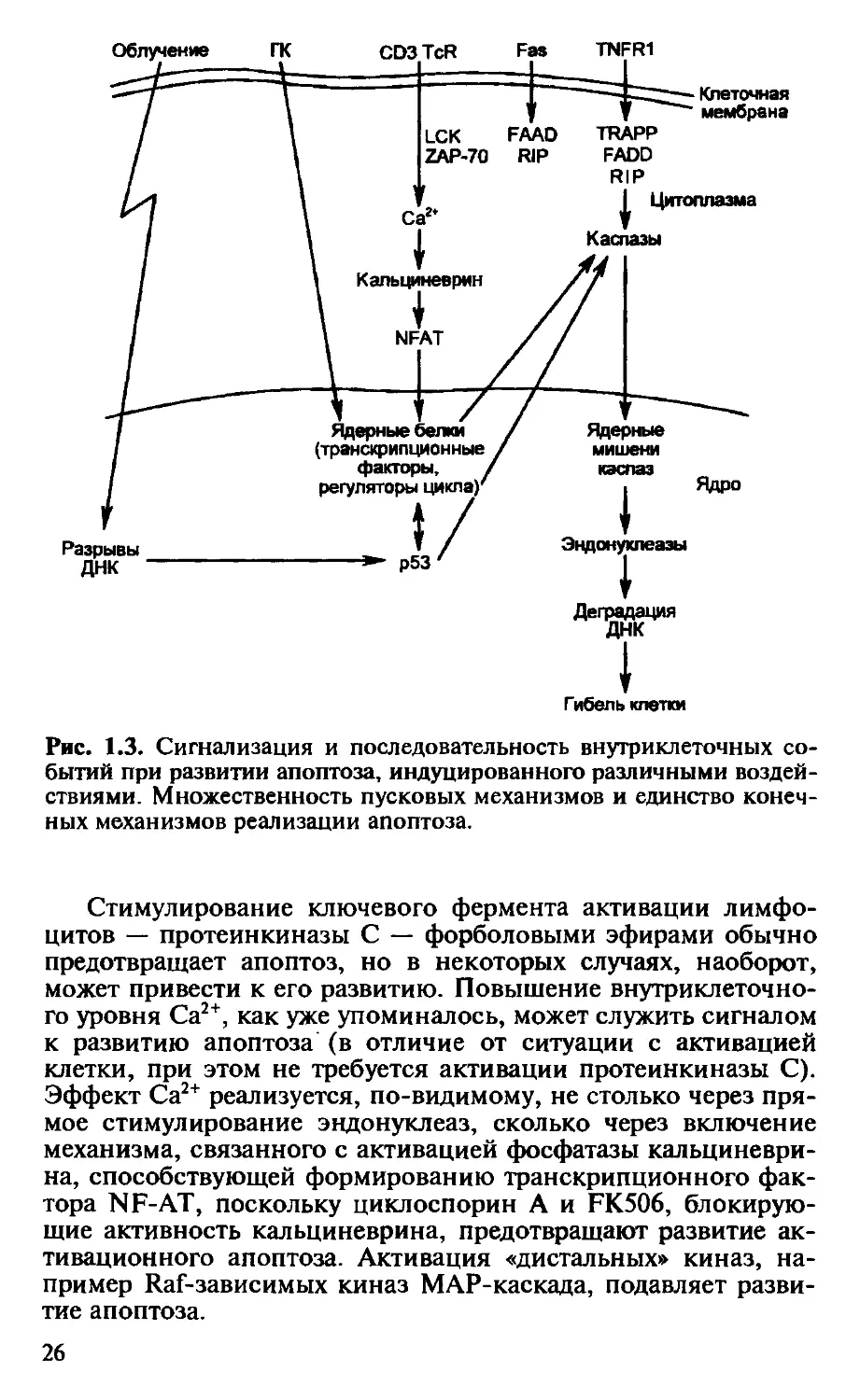
В процессе передачи внутриклеточного сигнала, приводя-
щего к развитию апоптоза, существуют две фазы. Более ран-
няя из них весьма вариабельна, и ее характер зависит от вида
пусковых механизмов. Следующая фаза универсальна для всех
разновидностей апоптоза (рис. 1.3, табл. 1.3).
Как уже указывалось выше, первая фаза передачи апопто-
генного сигнала может быть различной в зависимости от вида
сигнала, что иллюстрируют следующие примеры.
В случае Fas-зависимого апоптоза связывание Fas-лиганда с
тримерным Fas-рецептором приводит к конформационным из-
менениям в цитоплазматическом домене смерти Fas-рецептора.
Это создает возможность его связывания с аналогичным доме-
ном адапторной молекулы FADD (Fas-associated death domain), а
затем — с таким же доменом белка RIP (Receptor interacting pro-
tein) (см. рис. 1.2). Образующийся комплекс активирует фер-
мент каспазу 8, или FLICE (FADD-like IL-ip-converting enzyme),
что означает включение общего пути развития апоптоза (см.
далее). Аналогичные события происходят при действии ФНО
через рецептор TNFR1. Только в этом случае с рецептором взаи-
модействует адапторный белок TRADD (TNFR-associated death
domain), с которым связывают FADD и RIP (см. рис. 1.2).
В случаях передачи сигнала к апоптозу через рецепторы
для антигена на начальных этапах используются факторы, уча-
ствующие в запуске обычных активационных сигналов, преж-
де всего ферменты тирозинкиназы, связанные с мембранными
рецепторами (в случае связывания TCR — р561ск и ZAP-70).
Как и при активации лимфоцитов, передача сигнала регулиру-
ется тирозинфосфатазами — CD45 и 1D.
24
Таблица 1.3. Факторы, причастные к развитию апоптоза
Факторы и структура
Роль в развитии апоптоза
Рецепторы: Fas (Аро-1, CD95), рецептор ФНО (TNFR1), TCR-CD3, BCR Молекулы, связанные с рецепторами: FADD, TRADD, RIP (связаны с Fas, TNFR1); тирозинкиназы р56|с|!, ZAP-70 (связаны с TCR-CD3) Онкосупрессор и связанные с ним факторы: р53, р21WAFI Факторы передачи активационного сигнала: Са2+, кальциневрин Транскрипционные факторы: C-myc, Nur-77, ALG-2, ALG-3, NFAT, NFkB, TDAG51 Факторы, связанные с циклинами: Киназа p34cdc, фосфатаза cdc25A Факторы, контролирующие апоптоз: Ингибиторы: Bcl-2, BcI-Xl, Mell, Bfll, А20. Стимуляторы: Вах, Bcl-xs, Bad, Bak, Bik Каспазы: Каспазы 1—13, Mch-6 Мишени каспаз в ядре: PARP (поли-АДФ-рибозилполиме- раза), Rb, топоизомераза I, ламин, фодрин, UlsnRNP, ДНК-зависимые протеинкиназы Эндонуклеазы: Цитоплазматическая ДНК-полиме- раза I, ядерная I-подобная ДНК- полимераза, ядерная эндонуклеаза CAD, Са2+, Mg2+-3aBHCHMbie эндо- нуклеазы pl8 и р49 Вирусные факторы: р53 бакуловируса, СппА вируса ос- повакцины, Е1В12 К аденовируса, BHR вируса Эпштейна—Барр Воспринимают внешние сиг- налы к развитию апоптоза Обеспечивают передачу апо- птогенных сигналов с ре- цепторов внутрь клетки р53 воспринимает эндоген- ные сигналы к развитию апоптоза, р21 определяет их передачу Внутриклеточная передача апоптогенного сигнала Участвуют в активации генов, необходимой для реализации апоптоза Провоцируют развитие апо- птоза, обеспечивая продви- жение клеток по циклу Ингибиторы подавляют реа- лизацию программы гибели клетки, стимуляторы способ- ствуют ей Конвергенция сигналов, при- водящих к реализации апо- птоза Предположительно обеспечи- вают осуществление эффек- торной фазы (активации эн- донуклеаз, деградации ДНК) Обусловливают деградацию хроматина, в частности меж- нуклеосомные разрывы ДНК Подавляют развитие апоптоза клеток-мишеней вирусов
25
Облучение
Разрывы
ДНК
Рис. 1.3. Сигнализация и последовательность внутриклеточных со-
бытий при развитии апоптоза, индуцированного различными воздей-
ствиями. Множественность пусковых механизмов и единство конеч-
ных механизмов реализации апоптоза.
Стимулирование ключевого фермента активации лимфо-
цитов — протеинкиназы С — форболовыми эфирами обычно
предотвращает апоптоз, но в некоторых случаях, наоборот,
может привести к его развитию. Повышение внутриклеточно-
го уровня Са2+, как уже упоминалось, может служить сигналом
к развитию апоптоза (в отличие от ситуации с активацией
клетки, при этом не требуется активации протеинкиназы С).
Эффект Са2+ реализуется, по-видимому, не столько через пря-
мое стимулирование эндонуклеаз, сколько через включение
механизма, связанного с активацией фосфатазы кальциневри-
на, способствующей формированию транскрипционного фак-
тора NF-АТ, поскольку циклоспорин А и FK506, блокирую-
щие активность кальциневрина, предотвращают развитие ак-
тивационного апоптоза. Активация «дистальных» киназ, на-
пример Raf-зависимых киназ MAP-каскада, подавляет разви-
тие апоптоза.
26
Таким образом, сигнальные пути, приводящие к активации и
апоптозу лимфоцитов, на начальных этапах передачи сигна-
ла имеют общие компоненты.
В реализации апоптоза участвует ряд транскрипционных
факторов, ответственных за активацию клеток (т.е. за их
выход из фазы покоя в цикл) или за продвижение по циклу.
К ним прежде всего относятся факторы Nur-77 и с-тус. Экс-
прессия с-тус в условиях неполноты ростового сигнала приво-
дит к экспрессии гена — активатора циклинзависимых ки-
наз — фосфатазы cdc 25 А; их преждевременная экспрессия в
клетках, не защищенных от реализации программы гибели,
приводит эти клетки к выходу в цикл и автоматическому раз-
витию апоптоза. Этот механизм срабатывает, в частности, при
Fas-опосредованном апоптозе.
К механизмам выхода клеток в цикл также имеет отноше-
ние уже упоминавшийся фактор, участвующий во включении
механизма апоптоза, — р53 [Levine et al., 1994]. Этот белок на-
зывают онкосупрессором, поскольку его присутствие приво-
дит к гибели клеток с нарушениями в геноме, так как при му-
тациях гена р53 и нарушении синтеза нативного белка р53
такие клетки выживают и становятся источником злокачест-
венного роста. Белок р53 участвует также в индукции радиа-
ционного апоптоза. В норме белок р53 не синтезируется, экс-
прессия его гена индуцируется облучением, действием проти-
воопухолевых химиопрепаратов и ряда других агентов. Пока
не вполне ясно, каким образом он воспринимает сигнал о раз-
рывах ДНК и других генетических нарушениях. Его участие в
индукции апоптоза связано, с одной стороны, с активацией
гена WAF, кодирующего ингибитор циклинзависимых киназ
p21WAFi/cipf, и ПрОВОКацией вступления клетки в цикл (с учас-
тием продуктов генов Rb и с-тус), а с другой стороны — с
супрессией синтеза Вс1-2 и активацией фактора Вах, димерная
форма которого активирует каспазу Ich-1, т.е. обусловливает
переход ко второму — универсальному этапу индукции апо-
птоза (см. далее).
К аналогичному результату приводят начальные события
запуска гибели клеток-мишеней при действии на них цитоток-
сических Т-лимфоцитов или естественных киллеров. В этих
случаях в мембране клеток-мишеней в результате полимериза-
ции перфорина образуются поры, и в эти клетки проникают
гранзимы — протеазы, специфически разрывающие пептид-
ные связи около остатков аргинина. Мишенями этих гранзи-
мов служат неактивные предшественники сериновых протеаз,
которые под влиянием гранзимов переходят в активную
форму и участвуют в развитии апоптоза. Гранзимы (прежде
всего гранзим В) благодаря активации киназы p34cdc обуслов-
ливают также несвоевременное вступление клетки в цикл.
27
Таким образом, ранние этапы передачи внутриклеточного
сигнала, приводящего к развитию апоптоза, различны при
действии разных индукторов. Для них характерно использова-
ние факторов, участвующих в передаче других внутриклеточ-
ных сигналов, в частности сигналов, приводящих к активации
клетки. Эти различные пути приводят к одинаковым результа-
там, обязательными компонентами которых являются актива-
ция каспаз, способствующая развитию апоптоза, и неадекват-
ное вступление клеток в цикл.
1.1.4. Общий путь индукции апоптоза
Известно два относительно автономных проявления обще-
го пути индукции апоптоза — активация сериновых протеаз и
снижение трансмембранного потенциала митохондрий.
Анализ генетического контроля апоптоза у нематоды
Caenorhabditis elegans показал, что реализация апоптоза детер-
минируется двумя генами — ced-З и ced-4. Эффекты продукта
ced-4 неизвестны, но продукт ced-З идентифицирован как цис-
теиновая протеаза, и его гомологи обнаружены у млекопитаю-
щих. Они образуют семейство ферментов, называемых каспа-
зами [Alnemri E.S., 1996; Thomberry N.A., Lazebnik Y., 1998].
Именно они являются основными факторами, ответственны-
ми за реализацию апоптоза. Выше было показано, что «част-
ные» этапы развития апоптоза завершаются активацией фер-
ментов каспаз. Каспазы представляют собой подсемейство
цистеиновых протеиназ, специфичных по отношению к участ-
кам полипептидных молекул, содержащих остатки аспараги-
новой кислоты.
В покоящейся клетке присутствуют неактивные молеку-
лы — предшественники каспаз, которые представляют собой
гетеродимеры, состоящие из цепей с мол. массой около 20 и
10 кДа и содержащие ингибиторный домен. При активации
этот домен отщепляется, а гетеродимеры объединяются по
два, формируя тетрамер. Каспазы разделяют на две группы —
инициаторные каспазы (каспазы 8, 9; вероятно, также 2 и 10) и
эффекторные каспазы (каспазы 3, 6, 7); функция других каспаз
и их роль в развитии апоптоза пока точно не установлены.
Разделение каспаз на указанные группы отражает каскадный
характер их функционирования.
Индукторы апоптоза приводят к активации инициаторных
каспаз. Так, активирование каспазы 8 происходит при включе-
нии рецепторного механизма запуска апоптоза с участием мо-
лекулы FADD, а каспазы 9 — при действии цитотоксических
агентов с участием множества факторов — APAF-1, цитохро-
ма-с, дезоксиаденозинфосфата и др. Функция инициаторных
каспаз состоит в активации эффекторных каспаз, которые
28
имеют около 40 различных молекул-мишеней в ядре, мито-
хондриях и цитоплазме. К числу ядерных мишеней каспаз от-
носятся ферменты, связанные с репарацией ДНК, в частности
PARP (poly-ADP-ribosylpolymerase), белок Rb, принимающий
участие в контроле клеточного цикла, ДНК-зависимые проте-
инкиназы, которые участвуют в перестройке и репарации
ДНК, белок ICAD/DFF45, совмещающий функции активатора и
ингибитора ДНКазы CAD, а также топоизомераза I, ламин,
фодрин, малый ядерный рибонуклеопротеин U1 и т.д.
Основным механизмом, с помощью которого каспазы участ-
вуют в реализации апоптоза, является инактивация бел-
ков — ингибиторов ДНКаз.
Так, расщепление ингибиторного белка I0*0 приводит к ак-
тивации ДНКазы CAD (caspase-activated desoxyribonuclease), ко-
торая ответственна за межнуклеосомную фрагментацию ДНК.
Действуя на ядерные молекулы, каспазы подавляют процессы
репарации, репликации и реорганизации ДНК, что делает не-
возможным нормальное функционирование генов. Кроме того,
каспазы реализуют свои эффекты путем ослабления отрицатель-
ной регуляции апоптоза, например, вследствие расщепления
фактора Вс1-2. Еще один механизм их действия обусловлен пря-
мым разрушением клеточных структур, например ядерной плас-
тины, образованной белками — ламинами, или структур цито-
скелета, содержащих мишени каспаз — гельсолин, киназы FAK,
РАК2 и т.д. Это приводит к характерным изменениям формы
клеток, нарушению их подвижности, ослаблению адгезии и вза-
имосвязей с окружающими клетками.
Хотя в настоящее время ясно, что механизмы апоптоза не
сводятся исключительно к деградации ДНК, этому процессу и
осуществляющим его ферментам отводится очень важная роль
в апоптотической гибели клеток. Деградация ДНК при апо-
птозе реализуется в 3 этапа, которым соответствуют форми-
рование однонитевых разрывов, образование крупных (50—
200 т.п.н.) фрагментов и формирование мелких (180—200 пар
нуклеотидов) фрагментов ДНК вследствие двуните вых меж-
нуклеосомных разрывов [Zhivotovsky В. et al., 1994]. За форми-
рование этих разрывов ответственны ферменты эндонуклеазы.
Несмотря на пристальное внимание к данной проблеме, успе-
хи в изучении эндонуклеаз, участвующих в реализации апо-
птоза, до сих пор невелики.
Эндонуклеазы, обусловливающие реализацию межнуклео-
сомной деградации хроматина, должны отвечать ряду крите-
риев:,локализоваться в ядре апоптотических клеток, обладать
специфичностью к межнуклеосомному участку ДНК, способ-
ностью катализировать одно- и двунитевые разрывы, отщеп-
лять фрагменты как с 5’-, так и с З’-концов, активироваться
активаторами апоптоза и подавляться его ингибиторами. В той
29
или иной степени этим критериям соответствует ряд фер-
ментов: резидентная Са2+-, К^2+-зависимая эндонуклеаза
(18 кДа), в неактивной форме входящая в состав высокомоле-
кулярного комплекса (100 кДа) и освобождающаяся из него в
активной форме под влиянием индукторов апоптоза; упоми-
навшаяся выше ядерная каспазозависимая ДНКаза (CAD);
Са2+-, Mg2+-3aBHCHMaa эндонуклеаза, присутствующая в ядре в
формах с мол. массой 45, 47 и 49 кДа; цитоплазматические
ДНКазы 1 и II; ядерная I-подобная ДНКаза. Тем не менее во-
прос о реальном вкладе этих эндонуклеаз в осуществление
апоптоза окончательно не решен.
Общепризнанной является роль в развитии апоптоза изме-
нений, затрагивающих митохондрии. К ним относятся про-
цессы, которые сопровождаются потерей митохондриями
мембранного потенциала с утратой положительного заряда
внутренней поверхности мембраны, повышением проницае-
мости мембраны и ее дезинтеграцией, нарушением цепей
транспорта электронов, снижением содержания восстанов-
ленного глутатиона, NAD(P)H2, усиленным образованием
супероксиданиона, выходом Са2+ и митохондриальных белков
(в том числе причастных к запуску апоптоза) в цитоплазму
[Kroemer G. et aL, 1997; Waterhouse N.G., Green D.R., 1999].
Среди белков, поступающих из митохондрий в цитоплазму,
особенно важен цитохром-с, активирующий каспазу 9. С ми-
тохондриями связаны продукты протоонкогенов семейства
bcl-2, баланс которых изменяется в пользу факторов, стимули-
рующих апоптоз. Нарушения метаболических процессов в ми-
тохондриях приводят к энергетическому голоду клетки —
одной из главных непосредственных причин апоптотической
гибели клеток. Таким образом, в процессе апоптоза происхо-
дит формирование энергетического дефицита за счет значи-
тельных затрат энергии на реализацию метаболических реак-
ций, сопровождающих апоптоз, а также на постоянно идущие
процессы репарации поврежденных структур.
Таким образом, к гибели клетки приводят различные нару-
шения, проявляющиеся при развитии апоптоза. Среди них
основными являются процессы, приводящие к деградации
жизненно важных молекул и структур клетки, а также ис-
черпание ее энергетических ресурсов.
1.1.5. Эндогенные регуляторы апоптоза
В сигнальном пути, приводящем к развитию апоптоза, вы-
деляют контрольные точки, в которых возможен выбор между
реализацией программированной гибели (схема 1.1) и выжи-
ванием клетки [Oltvai Z.N., Korsmeyer S.J., 1994]. Этот выбор
30
Схема 1.1. Контрольные точки при развитии
апоптоза и факторы, определяющие выбор
между индукцией и блокадой апоптоза
Пояснения в тексте.
осуществляется с участием молекул, являющихся продуктами
протоонкогенов семейства Вс1-2 (см. табл. 1.3) [Hockenbe-
ry D.M., 1992; Pellegrini М., Strasser А., 1999]. Ген bcl-2 являет-
ся гомологом гена ctd-9 С. elegans. Белок Вс1-2 имеет мол.
массу 26 кДа и локализуется на наружной поверхности мито-
хондрий; он причастен к регуляции трансмембранного потен-
циала. Гиперпродукция Вс1-2 вследствие транслокации его
гена в локус иммуноглобулина сопряжена с развитием фолли-
кулярной В-клеточной лимфомы и рака носоглотки. Экспрес-
сия гена bcl-2 регулируется в процессе развития лимфоцитов.
По крайней мере в иммунной системе с синтезом Вс1-2 корре-
лирует устойчивость клеток к индукции апоптоза; особенно
ярко это проявляется в зародышевых центрах, где выживае-
мость активированных В-лимфоцитов прямо связана с экс-
прессией гена bcl-2. Гиперэкспрессия трансфецированного
гена bcl-2 приводит к таким же последствиям, как мутация
генов Fas-рецептора или Fas-лиганда. Аналогичная актив-
ность характерна для родственных белков — Bcl-xL, Bill, Mell,
а также гетеродимера белков Вс1-2 и Вах.
Противоположной активностью обладает гомодимер
Вах/Вах: он является одним из факторов, активирующих кас-
пазы. Сходное действие оказывают его гомологи Bad, Bak, Bik,
а также Bcl-xs.
Bcl-xL и Bcl-xs, обладающие противоположным влиянием
на апоптоз, образуются в результате альтернативного сплай-
синга продукта одного гена. Комбинация указанных факторов
и соотношение их уровней в клетке определяют выбор в пер-
вой контрольной точке.
31
На этом же этапе проявляют свое действие вирусные инги-
биторы апоптоза, которые делят на 3 класса — серпины (белок
вируса осповакцины СппА и др.), белок бакуловируса р35 и
белки семейства JAP.
Выбор во второй контрольной точке (см. схему 1.1) опре-
деляется соотношением двух продуктов (длинного и коротко-
го) альтернативного сплайсинга транскриптов гена Ich — IchL
и Ichs. Гомодимер Icht/Icht обусловливает развитие апоптоза,
а гетеродимер Ich^/Ichj предотвращает его. Этот этап является
точкой приложения действия вышеупомянутых ингибиторов
апоптоза вирусного происхождения — белка СппА вируса ос-
повакцины и белка р35 бакуловируса.
Лишь после вовлечения в процесс каспаз развитие апопто-
за становится необратимым, и клетку неминуемо ждет гибель.
1.2. Роль апоптоза в многоклеточном организме
1.2.1. Апоптоз, процессы формообразования
и клеточного гомеостаза на уровне
организма
Если массовая гибель клеток многоклеточного организма
по механизму некроза (например, вследствие гипоксии) часто
ассоциируется с гибелью всего организма, то апоптоз клеток
рассматривается скорее как условие нормального существова-
ния организма. В наиболее общей форме назначение апоптоза
(в сочетании с его альтернативой — пролиферацией) состоит в
определении размеров и «архитектуры» организма, что прояв-
ляется:
• в поддержании постоянства численности клеток;
• в определении формы организма и его частей;
• в обеспечении правильного соотношения численности
клеток различных типов;
• в удалении генетически дефектных клеток.
Эти функции апоптоза реализуются на уровне клеточных по-
пуляций — в процессе дифференцировки клеток и поддержания
постоянства их численности. Роль апоптоза в популяциях неде-
лящихся клеток минимальна; обычно она сводится к реакции на
внешние воздействия (типа ионизирующей радиации). Напро-
тив, в формирующихся и обновляющихся популяциях клеток
апоптозу принадлежит существенная роль фактора, уравнове-
шивающего процессы пролиферации и корригирующего диффе-
ренцировку. Из этого следует, что интенсивность апоптоза
выше в начальные периоды онтогенеза, в частности во время
эмбриогенеза, а во взрослом организме апоптоз продолжает иг-
рать большую роль лишь в быстро обновляющихся тканях.
32
Назначение апоптоза в клеточных популяциях можно
сформулировать таким образом:
• поддержание численности клеток в популяции на задан-
ном уровне;
• определение уровня численности клеток и его изменение
под влиянием внешних (по отношению к клетке) сигна-
лов вплоть до полной элиминации данного типа клеток;
• селекция разновидностей клеток внутри популяции (в
том числе элиминация клеток с генетическими дефекта-
ми).
Хотя общетеоретические представления в данной области
не разработаны, накоплено достаточно много фактов, позво-
ляющих иллюстрировать вышесказанное.
Наиболее простой иллюстрацией значимости апоптоза для
многоклеточного организма являются данные о роли этого
процесса в поддержании постоянной численности клеток не-
матоды Caenorhabditis elegans [Horvitz R., 1999]. Данному виду
свойствен очень жесткий контроль общей численности кле-
ток. Показано, что этот гомеостаз обеспечивается путем апо-
птоза части клеток. Его индукция и осуществление контроли-
руются набором из 14 генов, из которых один (ces-Г) обуслов-
ливает выбор апоптотического пути, два (ced-З и ced-4) — его
реализацию, а остальные или ингибируют апоптоз, или отве-
чают за фагоцитоз и расщепление погибших клеток.
Роль апоптоза в поддержании численности отдельных кле-
точных популяций иллюстрирует увеличение численности эн-
дотелиальных клеток и увеличение размера сосудов у мышей с
прицельной инактивацией («нокаутом») гена braf, контроли-
рующего апоптоз эндотелиальных клеток у мышей. In vitro ре-
гуляция численности клеток в популяции проявляется в ин-
дукции их апоптоза при достижении определенного уровня
плотности.
Роль апоптоза в формообразовании иллюстрируют резуль-
таты исследования локализации апоптотических клеток в про-
цессе морфогенеза внутреннего уха у куриных эмбрионов.
Апоптозу подвергаются клетки тех участков закладки внутрен-
него уха, которые играют роль в формировании полуцирку-
лярных каналов. Подавление апоптоза путем гиперэкспрессии
гена bcl-2 обусловливает задержку или отсутствие формирова-
ния просвета названных каналов. Морфогенетические анома-
лии, связанные с блокадой апоптоза, могут быть вызваны
также ингибированием биохимических процессов, лежащих в
основе апоптоза. Так, ингибиторы каспаз задерживают закры-
тие нервной трубки у куриных эмбрионов.
Роль апоптоза в дифференциации отдельных частей орга-
нов и их формообразовании изучается на примере становле-
ния в эмбриогенезе органов. Ярким примером этой роли явля-
3 — 1385
33
ется процесс формирования коры головного мозга. Так, у че-
ловеческого плода критическим периодом кортикогенеза яв-
ляется срок с 12 до 23 нед беременности, когда происходят ин-
тенсивная пролиферация клеток вентрикулярной зоны и миг-
рация нейронов. Наиболее интенсивный апоптоз регистриру-
ется в постмитотических клетках вентрикулярной зоны и
вдоль путей миграции нейронов в промежуточной зоне; он от-
сутствует в кортикальной пластинке. Роль апоптоза при этом
сводится к селекции клеток, которым предстоит участвовать в
формировании коры. Другим примером селективного апопто-
за определенного типа клеток может служить массовая гибель
(апоптоз) интернейронов (но не мотонейронов) серого веще-
ства спинного мозга крыс вскоре после рождения.
Массовым апоптозом целых клеточных популяций объяс-
няют такие ключевые события эмбриогенеза, как атрофия ги-
похорды у зародышей амфибий. В качестве причины внезап-
ной утраты гемопоэтической функции печенью эмбрионов на-
зывают тотальный апоптоз кроветворных клеток вследствие
снижения восстановительного потенциала в их микроокруже-
нии. Это снижение обусловлено деятельностью гепатоцитов,
которая приводит к ослаблению активности у-глутаматтранс-
феразы и уменьшению концентрации глутатиона.
Наконец, апоптозом клеток в связи со снижением кон-
центрации соответствующих гормонов обусловлена быстрая
атрофия гормонально-зависимых тканей. Процессы такого
рода периодически совершаются в женских половых органах в
течение менструального цикла. Аналогичные процессы в
предстательной железе происходят при снижении концентра-
ции андрогенов.
Роль апоптоза в гомеостазе обновляющихся популяций
клеток во взрослом организме показана также на примере кле-
ток сперматогенного эпителия, кроветворных клеток, энтеро-
цитов ворсинок слизистой оболочки тонкой кишки. Во всех
этих клеточных популяциях апоптозу подвергаются клетки на
определенных стадиях развития. Подверженность клеток
апоптозу чаще всего определяется ослаблением биосинтеза
эндогенных ингибиторов апоптоза — Bcl-2, Bcl-xL и др. Так, в
сперматогенном эпителии эти факторы синтезируются слабее
всего в мейотических и постмейотических клетках и сильно —
в сперматогониях; в соответствии с этим в первом случае
апоптоз наблюдается, а во втором — отсутствует.
Зависимость развития апоптоза от соотношения факторов
с противоположными эффектами наглядно иллюстрирует
судьба кроветворных клеток. Ранние кроветворные предшест-
венники, несущие маркер CD34, не имеют на своей поверх-
ности Fas-рецептора; половина этих клеток содержит Вс1-2.
Под влиянием фактора некроза опухоли или у-интерферона
Fas-рецептор появляется, а содержание Вс1-2 снижается, при-
34
чем клетки становятся более подверженными апоптозу. Даль-
нейшая их судьба определяется соотношением цитокинов,
способствующих развитию апоптоза (трансформирующего
фактора роста р, фактора некроза опухоли, хемокина MIP-1) и
защищающих от него (ИЛ-3 и фактора стволовых клеток для
наиболее юных кроветворных клеток, ГМ-, Г- и М-колоние-
стимулирующих факторов и ИЛ-10 для клеток миелоидного
ряда, ИЛ-7 для клеток лимфоидного ряда, эритропоэтина для
эритроидных клеток, ИЛ-5 и ГМ-КСФ для эозинофилов).
Очень важным фактором в реализации апоптоза развиваю-
щихся клеток, в частности кроветворных, является контакт с
межклеточным матриксом, обусловленный мембранными ин-
тегринами. Межклеточные контакты могут как способство-
вать, так и препятствовать развитию апоптоза. Уже упомина-
лось о контактной индукции апоптоза в плотных культурах
клеток, где контакты были очень интенсивными. В то же
время утрата контактов с межклеточным матриксом также яв-
ляется причиной апоптоза клеток, в частности для клеток,
«теряющих свой дом». Апоптоз неизбежно развивается при от-
делении клеток от субстрата (при утрате способности или воз-
можности прикрепляться к поверхностям и переходе прилипа-
ющих клеток в суспензию). Приобретение устойчивости к та-
кого рода переходам может быть связано с генетическими
перестройками и обычно означает малигнизацию клеток.
Важную роль играют факторы, защищающие клетки от
апоптоза. Помимо упомянутых цитокинов, поддерживающих
выживаемость кроветворных клеток, аналогичные факторы
существуют для других типов клеток. Примерами таких факто-
ров могут служить фактор роста фибробластов, поддерживаю-
щий жизнеспособность фибробластов, коллаген типа II, необ-
ходимый для выживания хондроцитов, фактор роста нервов и
нейротрофины, защищающие от апоптоза нейроны, и т.д.
Целую систему факторов, контролирующих развитие
апоптоза клеток различных типов, образуют морфогенетичес-
кие белки из костной ткани. Они действуют главным обра-
зом на клетки, происходящие из мезодермы, вызывая (реже
ингибируя) развитие апоптоза. Спектр этих факторов, обла-
дающих определенной степенью специфичности в отноше-
нии клеток разных типов, неодинаков в различных органах,
что в определенной степени влияет на судьбу образующих их
клеток. Эти факторы участвуют в формировании архитектуры
определенных участков тела, например суставных полостей,
в которых локализуются два типа белков этой группы —
ВМР-2 и ВМР-7, причем первый содержится на хрящевых
поверхностях, формирующих сустав, а второй — в околохря-
Щевой зоне.
Приведенные примеры лишь выборочно отражают участие
апоптоза в определении размеров и формы тела, а также в
з*
35
процессах морфогенеза и поддержания гомеостаза клеточных
популяций. Факты такого рода быстро накапливаются, и в
ближайшее время можно ожидать новых обобщений в этой
области. Пока же они продолжают оставаться в значительной
степени разрозненными. Есть лишь одна область знаний, в
которой роль апоптоза определилась достаточно точно и раз-
носторонне: это иммунология. К краткому обзору места апо-
птоза в иммунологических процессах мы обратимся в следую-
щей главе.
1.2.2. Роль апоптоза в иммунных процессах
Можно выделить 3 стадии развития лимфоцитов, в которых
роль апоптоза велика и реализуется по-разному (табл. 1.4):
• стадия до формирования антигенраспознающего рецеп-
тора;
• период формирования рецептора, последующей селек-
ции и созревания лимфоцитов;
• период функционирования зрелых лимфоцитов [Яри-
лин А.А., 1996].
Таблица 1.4. Роль апоптоза на различных этапах развития Т-лим-
фоцитов
Этапы развития Экспрессия генов факторов, связанных с апоптозом Проявления апоптоза
Развитие пред- шественников Т-лимфоцитов (до экспрессии TCR) Селекция кло- нов и диффе- ренцировка суб- популяций В костном мозге отсутствует экс- прессия bcl-2, в ви- лочковой железе на стадии CD25+ — сильная экспрессия bcl-2, слабая — fas На стадии CD4+, CD8+ слабая экс- прессия bcl-2, сильная — fas, позже — наоборот До стадии CD25+ апоптоз раз- вивается в отсутствие ИЛ-7 и фактора стволовых клеток, в процессе перестройки генов рецептора — при непродук- тивной перестройке СО4+СВ8+-тимоциты выжи- вают при условии распознава- ния аутологичных молекул ГКГ на мембране эпителиаль- ных клеток (положительная селекция), наконец, тимоциты гибнут при распознавании ау- тологичных комплексов пеп- тид-молекула ГКГ на поверх- ности дендритных клеток (от- рицательная селекция)
36
Продолжение табл. 1.4
Этапы развития Экспрессия генов факторов, связанных с апоптозом Проявления апоптоза
Функциониро- вание зрелых Т-клеток На покоящихся клетках — сильная экспрессия bcl-2, но не fas. Активи- рованные Т-клетки несут Fas и со вре- менем теряют Вс1-2 Покоящиеся клетки устойчи- вы к индукции апоптоза. Ак- тивированные клетки гибнут при дефиците сигнала, по- вторной стимуляции, связыва- нии CD4, в поздние сроки — спонтанно. Цитотоксические клетки индуцируют апоптоз клеток-мишеней
В первой из этих стадий лимфоциты фактически еще не
выделяются из общего семейства кроветворных клеток-пред-
шественников. Ранее мы уже говорили о том, что все эти клет-
ки нуждаются в факторах выживания, предохраняющих их от
самопроизвольного развития апоптоза. Это свидетельствует о
предсуществовании в этих клетках программы гибели и отсут-
ствии внутренней защиты в виде продукции факторов Вс1-2 и
Bcl-xL. Основным экзогенным фактором выживания пред-
шественников лимфоцитов обоих классов служит ИЛ-7. На
стадиях, непосредственно предшествующих началу перестрой-
ки генов антигенраспознающих рецепторов — CD19+CD76+
проВ-клеток и CD44+CD25+ проТ-клеток, синтезируется
Вс1-2, и клетки приобретают устойчивость к индукторам апо-
птоза, что означает их выход из пула малодифференцирован-
ных клеток-предшественников.
Далее следует стадия «драматических» перестроек генов
В-клеточного и Т-клеточного рецепторов (BCR и TCR соот-
ветственно), важной составляющей которых являются разры-
вы и рекомбинации нитей ДНК. Это само по себе может слу-
жить сигналом к развитию апоптоза (с участием фактора р53).
Однако высокий уровень экспрессии Вс1-2 и Bcl-xL препятст-
вует этому. Лишь в случае неудачной реализации перестройки
рецепторных генов клетка подвергается апоптозу. После за-
вершения перестроек и экспрессии генов рецепторов (BCR на
В-лимфоцитах и TCR на Т-лимфоцитах) происходит резкое
снижение экспрессии Вс1-2 и Bcl-xL. Это событие означает на-
чало процесса селекции клонов — выбраковки ненужных и
опасных для организма клонов лимфоцитов.
Процесс селекции достаточно полно изучен для Т-клеток.
Объектом селекции являются кортикальные тимоциты, содер-
жащие на поверхности мало рецепторных комплексов TCR-
37
CD3 и несущие одновременно корецепторы CD4 и CD8. Эти
клетки практически лишены Вс1-2 и Bcl-xL, но содержат на
своей поверхности Fas-рецептор. Такое соотношение факто-
ров, препятствующих и способствующих развитию апоптоза,
обрекает их на гибель в отсутствие специальных факторов за-
щиты. Источником защитных сигналов для них служит взаи-
модействие их рецептора (TCR) с молекулами главного ком-
плекса гистосовместимости (ГКГ) эпителиальных клеткок
микроокружения в глубоких слоях коры вилочковой желе-
зы, — распознавание аутологичных молекул ГКГ. Если клетка
несет рецептор, способный распознавать эти молекулы, она
получает «поддерживающий» сигнал, который приводит к уси-
лению экспрессии генов рецептора TCR, активации и проли-
ферации клетки. Остальные клетки «игнорируются», что озна-
чает для них неизбежное развитие апоптоза. Пока не вполне
ясно, что служит непосредственным сигналом к развитию
апоптоза в данном случае — действие глюкокортикоидов и пу-
риновых нуклеотидов, присутствующих в микроокружении,
или активные воздействия со стороны окружающих эпители-
альных клеток (всех или определенных их субпопуляций).
Положительная селекция сопряжена с диффенцировкой ти-
моцитов на субпопуляции клеток CD4+CD8 и CD4'CD8+, осно-
вой которой является выбор способа распознавания антигенных
пептидов в составе молекулы I класса (в их распознавании уча-
ствует молекула CD8) или молекулы II класса (распознаваемой с
участием CD4). При этом синтез корецептора CD4 в сочетании
с TCR, распознающим комплекс пептида с молекулой ГКГ
I класса (не комплементарных корецептору), приводит к апо-
птозу клетки, так же как экспрессия гена CD8 в сочетании с
TCR, распознающим пептид в составе молекул ГКГ II класса.
Причиной развития апоптоза в этом случае может служить
«неполнота» сигнала от распознавания молекул ГКГ, который
оказывается недостаточным для включения защиты от апоптоза.
После положительной селекции тимоциты подвергаются
второму туру отбора — отрицательной селекции (пока неясно,
как соотносятся во времени этот этап селекции и деление на
субпопуляции). На этой стадии выбраковываются аутореак-
тивные клетки, т.е. тимоциты, чьи рецепторы распознают ау-
тологичные пептиды в составе аутологичных молекул ГКГ.
«Проба» на распознавание осуществляется при контакте тимо-
цитов, прошедших положительную селекцию и содержащих на
своей поверхности больше молекул TCR-CD3, чем в предыду-
щий период развития, с дендритными клетками, богатыми
мембранными молекулами ГКГ обоих классов. Пространст-
венно этот процесс приурочен к кортико-медуллярной и, воз-
можно, мозговой зонам вилочковой железы. В случае распо-
знавания аутологичного комплекса возникает летальный сиг-
нал, приводящий к развитию апоптоза, т.е. к удалению потен-
38
циально аутореактивных клонов Т-клеток. До сих пор нет чет-
кого ответа на вопрос, почему распознавание аутологичных
молекул на поверхности эпителиальных клеток защищает ти-
моциты от апоптоза, а их распознавание на мембране денд-
ритных клеток индуцирует апоптоз тимоцитов. Предполагает-
ся, что исход зависит от интенсивности воздействия, опреде-
ляемой степенью сродства рецептора по отношению к распо-
знаваемому комплексу и плотности молекул TCR и ГКГ на
поверхности клеток.
Поскольку не все антигены представлены в вилочковой
железе, процесс выбраковки аутореактивных клонов и фор-
мирования аутотолерантности продолжается после эмиграции
Т-клеток из вилочковой железы. Однако в этом случае значи-
тельная часть аутоспецифических клеток не погибает, а бло-
кируется вследствие индукции анергии и подавления их ак-
тивности супрессорными клетками.
Процесс селекции развивающихся В-лимфоцитов изучен
слабее. Однако известно, что и они подвергаются двум этапам
селекции. При этом положительную селекцию определяет рас-
познавание неизвестного фактора(ов) микроокружения, тогда
как отрицательная селекция происходит аналогцчно тому, как
она осуществляется в популяции Т-клеток.
Реальность процессов отрицательной селекции ярко проде-
монстрирована в различных системах с трансфекцией генов, де-
терминирующих синтез неких чужеродных для данного вида мо-
лекул и одновременно рецептора, специфичного в отношении
этой молекулы. Поскольку при этом все лимфоциты несут ре-
цептор, специфичный к «аутоантигену», они становятся объек-
том отрицательной селекции и полностью элиминируются.
Зрелые Т- и В-лимфоциты, подвергшиеся селекций, в поко-
ящемся состоянии лишены мембранного Fas-рецептора и экс-
прессируют протоонкогены bcl-2 и bcl-xL, что определяет их ус-
тойчивость к индукторам апоптоза. Положение меняется на об-
ратное при активации лимфоцитов соответствующим антигеном
или митогеном. Моноклональные антитела анти-Fas не действу-
ют на покоящиеся лимфоциты и убивают активированные Т- и
В-клетки. Активированные лимфоциты могут подвергнуться
апоптозу при самых различных воздействиях — повторной сти-
муляции через перекрестное сшивание рецептора, при недостат-
ке факторов роста (для Т-клеток — ИЛ-2, для В-клеток — глав-
ным образом ИЛ-4), при действии глюкокортикоидов и т.д.
Апоптоз может развиться и в процессе активации, например
после предварительного связывания молекул CD4 при отсутст-
вии костимуляции Т-клеток через CD28, а В-клеток — через
CD40 при отсутствии ростовых факторов.
Дифференцировка В-лимфоцитов в клетки, секретирую-
щие IgG-антитела, происходящая в зародышевых центрах, со-
провождается интенсивным мутагенезом (он происходит в ос-
39
новном в темных зонах зародышевых центров лимфоидных
органов на стадии центробластов). Конечное назначение му-
тагенеза в данном случае — появление клонов клеток с более
высоким сродством к антигену, чем у исходных В-лимфоци-
тов, однако при этом не все мутации приводят к повышению
сродства рецептора к антигену, большинство из них даже сни-
жают это сродство. Требуются отбор клонов с высоким срод-
ством рецепторов к антигену и элиминация прочих клонов.
Это происходит в базальных участках светлых зон зародыше-
вых центров при взаимодействии В-лимфоцитов (центроци-
тов) и фолликулярных дендритных клеток, несущих антиген,
на фоне ослабления синтеза Вс1-2 в В-клетках. При сильном
сродстве рецептора В-центроцитов к антигену их взаимодей-
ствие стабилизируется с помощью связывания молекулы CD40
В-клеток с ее лигандом CD 154 (CD40L), которое служит ис-
точником сигнала выживания В-клеток. Лимфоциты, не полу-
чившие этого сигнала, гибнут. После завершения этапа селек-
ции центроциты перемещаются в апикальные участки светлых
зон, а затем мигрируют в мантию зародышевых центров, где
дифференцируются в плазматические клетки—продуценты
антител и В-клетки памяти. Уже в апикальном отделе светлых
зон в В-клетках проявляется экспрессия bcl-2; она возрастает в
мантийной зоне. На этих этапах клетки В-ряда становятся ус-
тойчивыми к индукторам апоптоза.
После дифференцировки Т-лимфоцитов в эффекторные
клетки — цитотоксические Т-лимфоциты, Т-клетки — проду-
центы цитокинов, а также в Т-клетки памяти в них также синте-
зируется Вс1-2, и они временно приобретают устойчивость к ин-
дукторам апоптоза. Однако эффекторные клетки, несущие мар-
кер CD45R0, все-таки легче направить на путь апоптоза, чем
«наивные» Т-лимфоциты, несущие маркер CD45RA. Кроме
того, образование Вс1-2 в эффекторных клетках иммунной сис-
темы как Т-, так и В-типа продолжается недолго; его прекраще-
ние неизбежно приводит к гибели клеток. В клетках памяти экс-
прессия «защитных» протоонкогенов типа bcl-2 продолжается
дольше, что обусловливает длительный срок их жизни. Особен-
но стабильной оказывается продукция Вс1-2 у клеток памяти с
долгим сроком жизни, если они периодически испытывают воз-
действие специфического антигена, локализующегося на по-
верхности дендритных клеток зародышевых центров.
Известно еще по меньшей мере два аспекта функционирова-
ния лимфоцитов, непосредственно связанных с проявлениями
апоптоза. Один из этих аспектов — реализация цитотоксической
активности лимфоцитов. Клетки-мишени естественных килле-
ров и цитотоксических Т-лимфоцитов гибнут при морфологи-
ческих и биохимических проявлениях апоптоза, хотя при этом
примешиваются некоторые черты некротической гибели клеток.
Эта двойственная природа гибели с преобладанием апоптоза
40
может быть проанализирована на примере наиболее универсаль-
ного механизма действия киллеров. Этот механизм состоит из
формирования в мембране клетки-мишени пор (вследствие
полимеризации перфорина) и поступления через эти поры сери-
новых протеаз — гранзимов (главным образом гранзима В), ко-
торые включают механизм апоптоза. Среди других механизмов
цитотоксического действия Т-лимфоцитов (но не естественных
киллеров) могут быть названы Fas-опосредованный апоптоз
(в клетках-мишенях увеличивается присутствие Fas-рецепторов,
а цитотоксические Т-лимфоциты «нарабатывают» Fas-лиганд) и
апоптоз, индуцируемый фактором некроза опухоли или лимфо-
токсином через рецептор 1-го типа (TCRF1).
Другая область функционирования иммунной системы,
для которой оказалась важной реализация апоптоза, — защита
«иммунологически привилегированных» зон в организме от
эффекторных клеток иммунной системы. Недавно выясни-
лось, что по крайней мере две такие зоны — внутренние среды
глаза и семенники — выстланы клетками, синтезирующими
Fas-лиганд. В глазу это клетки эпителия, эндотелия, радужки,
сетчатки, ресничных телец, в семенниках — эпителиальные,
эндотелиальные и клетки Сертоли (сустентоциты). В результа-
те эффекторные Т-лимфоциты, продуцирующие Fas-рецептор,
неизбежно гибнут в процессе миграции через этот «барьер»
вследствие взаимодействия указанного рецептора с Fas-лиган-
дом барьерных клеток, приводящего к апоптозу этих агрессив-
ных Т-лимфоцитов [Griffith T.S., 1997].
Таким образом, апоптоз является одним из ключевых про-
цессов, определяющих формирование антигенспецифичес-
кой составляющей иммунной системы и в значительной сте-
пени — реализацию ее эффекторных функций.
Особенно важной оказывается роль апоптоза в обеспечении
«неотвечаемости» лимфоцитов, т.е. отсутствия воздействия лим-
фоцитов на собственные компоненты организма, что достигает-
ся, с одной стороны, формированием аутотолерантности вслед-
ствие апоптоза аутореактивных клонов лимфоцитов, а с другой
стороны — исключением некоторых областей организма из-под
контроля иммунной системы с помощью «барьера», преодоле-
вая который, активированные клетки подвергаются апоптозу.
1.3. Место апоптоза в патологии
Материал предшествующих глав свидетельствует о том, что в
реализации и регуляции апоптоза участвуют многочисленные
внутриклеточные факторы. Он может быть вызван действием
разнообразных внешних агентов, и от его осуществления зави-
сят многие важные процессы, реализуемые на уровне организ-
41
Схема 1.2. Роль апоптоза в патологии
Апоптоз
Участие апоптоза а
формировании типовых
патологических процессов
Изменение выраженности
апоптоза
Гибель лимфо-
цитов и энте-
роцитов при
стрессе
Ослабление
Усиление
Гибель клеток
при септичес-
ком шоке
Локальная ги-
бель клеток
при реоксиге-
нации после
ишемии
Повышение
вероятности
развития зло-
качественных
опухолей
Врожденные
дефекты тка-
ней, уродства
Повреждение
клеток Т-килле-
рами при
аутоиммунных
процессах
Сочетание
волчаночного
и лимфопро-
лиферативно-
го синдромов
Панцитопении,
первичные им-
мунодефициты
Нейродегене-
ративные про-
цессы
Замедление
гибели клеток-
эффекторов в
позднюю фазу
немедленной
аллергии
Бактериальные
инфекции (эф-
фект суперанти-
генов)
Вирусные ин-
фекции, в том
числе СПИД
Действие неблагоприятных факторов
среды, цитотоксической терапии
ма. Из этого следует неизбежность существования патологии,
связанной с нарушениями в звеньях индукции и регуляции
апоптоза (Onici Y., 1994]. Далее мы рассмотрим роль апоптоза в
качестве обязательного компонента патологических процессов,
а также две группы патологических процессов, связанных с ос-
лаблением и усилением апоптоза (табл. 1.5; схема 1.2).
42
Таблица 1.5. Патология, связанная с нарушениями апонтоза
Группа заболеваний Заболевания Комментарии
Заболевания, связанные с ослаблением апоптоза
Аутоиммунные процессы Семейный аутоим- мунный лимфопро- лиферативный син- дром, связанный с дефицитом Fas Системная красная волчанка, ревмато- идный артрит, син- дром Бехчета Аналоги у мышей — носители мутаций генов Ipr, gid и транс- фектанты по гену bcl-2. СКВ- подобный синдром в сочета- нии с накоплением клеток CD3+CD4CD8B220+ Ослабление апоптоза невыяс- ненного генеза. Образование растворимого Fas-рецептора
Злокачествен- ные опухоли Лимфома Беркита Лейкозы и солид- ные опухоли Транслокация и гиперэкспрес- сия генов bcl-2 и с-тус, ослаб- ление апоптоза Мутации и экспрессия гена р53 приводят к ослаблению апоптоза, что часто коррели- рует с прогрессированием и метастазированием опухолей и их устойчивостью к терапии
Заболевания, связанные с усилением апоптоза
Врожденные аномалии Синдром Дауна и др. Преобладание апоптоза при формировании локальных структур
Болезни крови (цитопении) Миелодисплазии, апластическая, Fe-, фолат-, Вп-дефи- цитные анемии, тромбоцитопения, нейтропения, бо- лезнь Кастлемана Усиление апоптоза клеток от- дельных или всех ростков кро- ветворения в процессе разви- тия
Инфекционные (бактериальные) заболевания Различные инфек- ционные процессы, сепсис Апоптоз клеток иммунной системы вызывают суперанти- гены, токсины; при сепсисе — накапливающиеся в крови ФНОа
Вирусные ин- фекции Различные вирус- ные заболевания, в том числе СПИД Индукторами апоптоза при СПИДе служат вирусные фак- торы, в частности gpl20, взаи- модействующий с CD4
43
Продолжение табл. 1.5
Группа заболеваний Заболевания Комментарии
Дистрофичес- кие заболевания нервной систе- мы Другие заболе- вания Боковой амиотро- фический склероз, болезнь Альцгейме- ра, спинальная мы- шечная атрофия Инфаркт миокарда Токсичные гепати- ты Усиление апоптоза нейронов и других клеток в определенных участках центральной нервной системы Преобладание апоптоза мио- цитов в период «реперфузии» миокарда Апоптоз гепатитов под дейст- вием ядов, в том числе этанола
1.3.1. Апоптоз как компонент типовых
патологических процессов
Выше была показана ключевая роль апоптоза в реализации
ряда физиологических процессов, особенно связанных с под-
держанием клеточного гомеостаза и с развитием организма. В то
же время механизм апоптоза не входит в качестве обязательной
составляющей в реализацию большинства типовых патологи-
ческих процессов. В этом можно убедиться на примере воспали-
тельной реакции. Хотя для воспаления характерно повреждение
тканей, гибель клеток при этом происходит преимущественно
по механизму некроза и сопровождается выходом содержимого
клеток в межклеточное пространство, что может стать причиной
гибели (некроза) соседних клеток и расплавления тканей. Одна-
ко на завершающих этапах воспаления апоптозу принадлежит
более важная роль, поскольку в этот период происходит устра-
нение активированных клеток иммунной системы, выполнив-
ших свои функции. То же относится к аллергическому воспале-
нию, при котором упомянутая элиминация эффекторных кле-
ток к тому же затруднена в связи с их способностью к самопод-
держанию благодаря выработке аутокринных цитокинов (так,
активированные эозинофилы секретируют ГМ-КСФ, защи-
щающий от апоптоза) [Wedi et al., 1997].
Одной из основ опухолевого процесса является нарушение
упоминавшегося выше механизма устранения генетически из-
мененных и чужеродных клеток, основой которого служит
апоптоз. Но развитие опухоли и реализация ее отрицательного
действия на организм (т.е. опухолевая патология как таковая)
происходят без обязательного участия апоптоза в качестве ве-
дущего механизма.
При инфекционных процессах развитие апоптоза зависит
от конкретных факторов, выделяемых микроорганизмами (су-
44
перантигены — экзотоксины часто выступают в качестве ин-
дукторов активационного апоптоза, о чем будет сказано далее)
и не определяется инфекционным патологическим процессом
как таковым. Важное исключение составляет септический
шок, который развивается в ответ на гиперпродукцию ФНОа
макрофагами и другими клетками при действии эндотоксинов
[Leist М. et al., 1995]. Наряду с другими многочисленными
проявлениями при этом шоке значительно усиливается гибель
от апоптоза клеток, несущих рецептор TNFRI.
Лишь при стрессе апоптозу принадлежит роль одного из
ключевых механизмов, определяющих специфику реакции ор-
ганизма на стрессор. Его участие в реализации стрессорной
реакции связано с повышенным выделением стероидных гор-
монов корой надпочечников [Cohen J.J., 1992]. Именно чувст-
вительностью к действию стероидов определяется спектр тка-
ней, поражаемых при стрессе. К таким тканям относятся в
первую очередь лимфоидная ткань (особенно кора вилочковой
железы) и слизистые оболочки, прежде всего кишечника. Ре-
зультатом является, например, такое типичное проявление
стресса, как опустошение лимфоидных органов.
Таким образом, апоптоз, как правило, не является неотъ-
емлемой составляющей и обязательным компонентом патоло-
гических процессов, хотя обычно сопутствует им (например,
как одно из проявлений стресса или орудие эффекторных им-
мунных механизмов). Однако есть смысл подойти к рассмот-
рению роли апоптоза в патологии с другой стороны — путем
анализа результатов нарушения механизмов, участвующих в
реализации или контроле апоптоза.
1.3.2. Патологические процессы,
обусловленные ослаблением апоптоза
На основании знаний о роли апоптоза в осуществлении
физиологических процессов можно предположить, что недо-
статочность проявления апоптоза должна отразиться на про-
цессах морфогенеза, элиминации клеток с генетическими по-
ломками, становления аутотолерантности и выражаться в
форме разного рода дефектов развития, аутоиммунных про-
цессов и злокачественных опухолей. Однако моделирование
ослабления апоптоза путем трансфекции мышам гена bcl-2 в
форме, предусматривающей его спонтанную экспрессию во
всех или в определенных клетках, показало, что спектр прояв-
ляющихся при этом дефектов уже, чем можно было ожидать.
Основное последствие гиперпродукции Вс1-2 состоит в раз-
витии системных аутоиммунных процессов (вплоть до волча-
ночного синдрома), а также в накоплении необычных клеток
Т-ряда с фенотипом клеток CD3-TCRap+CD4’CD8 B220+ (т.е.
45
клеток, лишенных корецепторов и несущих маркер В-клеток
В220), в норме обнаруживаемых в ограниченном количестве
лишь в некоторых органах, например в печени. У таких мы-
шей не наблюдалось повышения частоты развития злокачест-
венных опухолей. Лишь при одновременной гиперэкспрессии
трансфецированного гена с-тус у мышей наряду с аутоиммун-
ными развивались лимфопролиферативные процессы [Stras-
ser A. et al., 1991].
Патология, фенотипически идентичная описанной выше
для мышей с гиперпродукцией Вс1-2, была описана ранее у
мышей линии MRL, несущих мутации Ipr (lymphoproliferation)
и gid (generalized limphoproliferative disease), которые использу-
ются в качестве моделей системных аутоиммунных процессов,
в частности системной красной волчанки. Генетический ана-
лиз показал, что мутация Ipr затрагивает ген Fas-рецептора
[Watanabe-Fukunaga R. et al., 1992], мутация gid — ген Fas-ли-
ганда [Takashi R. et al., 1994]. У этих мышей с возрастом опре-
деляется широкий спектр аутоантител (в том числе к нативной
ДНК), развивается аутоиммунный гломерулонефрит, гипер-
трофируются периферические лимфоидные органы с накопле-
нием в них Т-клеток указанного выше мембранного феноти-
па, что и обозначается как лимфопролиферативный синдром,
хотя он и лишен признаков злокачественности. Существенно,
что селекция клонов Т-лимфоцитов в тимусе этих мышей про-
исходит, хотя и медленнее, чем в норме, а селекция на пери-
ферии отменяется полностью [Sprent J., Webb S., 1995]. Таким
образом, во-первых, нарушение синтеза Fas-рецептора и Fas-
лиганда имеет сходные последствия, проявляющиеся в нару-
шении формирования аутотолерантности и экспансии субпо-
пуляции Т-клеток с неизвестной функцией, в норме имеющей
ограниченное распределение в иммунной системе; во-вторых,
эти же последствия наблюдаются при повышенной продукции
фактора Вс1-2, ограничивающего апоптоз; в-третьих, от экс-
прессии генов fas и bcl-2 сильнее зависит селекция клонов
Т-лимфоцитов на периферии, чем в тимусе.
Поскольку среди последствий генетически обусловленного
ослабления апоптоза в иммунной системе доминируют прояв-
ления системной аутоиммунной патологии, возник вопрос о
состоянии апоптоза и связанных с ним факторов при аутоим-
мунных заболеваниях человека, прежде всего при системной
красной волчанке. При классической системной красной вол-
чанке (см. табл. 1.5) апоптоз Т-клеток обычно нарушен
[Kaneko Н. et al., 1996; Budagyan V.M. et al., 1998], но иногда
регистрируется усиление апоптоза лимфоцитов in vitro, что
коррелирует с усилением их спонтанной активации. Синтез
Fas-рецептора при системной волчанке, как правило, не нару-
шен, а синтез Fas-лиганда даже повышен. В то же время опи-
сан семейный аутоиммунный лимфопролиферативный син-
46
дром, обусловленный дефектом гена fas [Fischer G.H. et al.,
1995], одним из проявлений которого могут быть васкулиты и
гломерулонефрит, свойственные волчаночному синдрому;
значительно реже у человека регистрируется мутация гена fast,
имеющая аналогичные проявления.
При ревматоидном артрите Т-лимфоциты, присутствую-
щие в пораженной суставной полости, имеют признаки, «об-
рекающие» их на развитие апоптоза (высокая «выработка»
Fas-рецептора и низкая — Вс1-2), однако они не подвергаются
апоптозу; предполагается, что в данном случае срабатывает
некий механизм, препятствующий реализации апоптоза
Т-клеток (нейтрофилы в тех же суставных полостях подверга-
ются массовому апоптозу) [Salmon М. et al., 1997]. Следует от-
метить, что при ревматоидном артрите усиливается активаци-
онный апоптоз Т-лимфоцитов периферической крови.
При системной красной волчанке и некоторых других рев-
матических заболеваниях (например, ювенильном ревматоид-
ном артрите) усилена выработка клетками растворимой
формы Fas-рецептора [Cheng J. et al., 1994], накопление кото-
рого в микроокружении лимфоцитов может препятствовать
реализации Fas-зависимого апоптоза, например апоптоза ак-
тивированных Т-лимфоцитов или Т-клеток, подвергающихся
отрицательной селекции на периферии. Нами показано, что у
части больных системной красной волчанкой для включения
активационного апоптоза требуется присутствие клеток-ин-
дукторов в количестве, на два порядка большем, чем требуется
для развития апоптоза активированных Т-клеток здоровых
людей [Никонова М.Ф. и др., 1996]. Образование растворимой
формы Fas-рецептора особенно характерно для острого
Т-лимфобластного лейкоза. Оно обусловлено альтернативным
сплайсингом транскриптов гена fas, что приводит к формиро-
ванию белка, лишенного трансмембранного участка.
Принципиально важная закономерность была установлена
при анализе мишеней аутоантител при системной красной
волчанке. Оказалось, что они одновременно являются мише-
нями действия каспаз [Utz Р.Х. et al., 1997], обусловливающих
включение эффекторного механизма апоптоза (см. раздел 1.4).
Кроме того, аутоантитела при данном и родственных заболе-
ваниях направлены против серин-треониновых киназ, кото-
рые активируются при клеточном стрессе.
Вторую большую группу заболеваний, к генезу которых
имеет отношение подавление апоптоза, образуют злокачест-
венные опухоли, особенно имеющие гематогенное происхож-
дение [Komblau S.M. et al., 1998]. В этом случае ключевым со-
бытием, способствующим развитию патологии, чаще всего
служат соматические мутации, затрагивающие ген р53 (обыч-
но его экзоны 5—8) [Trulson J.A., Millhauser G.L., 1999]. Выше
упоминалось, что фактор р53 трансформирует сигнал о нере-
47
парированных разрывах цепей ДНК в сигнал к развитию
апоптоза. Благодаря этому элиминируются клетки с поврежде-
ниями генетического аппарата, спонтанными или индуциро-
ванными (например, облучением) [Levine A.J. et al., 1994].
В нормальных клетках белок р53 не выявляется, а при опухо-
лях его мутантную форму синтезируют до 70 % трансформиро-
ванных клеток. Однако большой разброс частоты мутаций р53
при различных злокачественных опухолях не позволяет сде-
лать универсального заключения относительно его роли в па-
тогенезе злокачественных процессов.
Аномалии фактора р53, а также других внутриклеточных
факторов, контролирующих апоптоз, в процессе развития опу-
холи имеют отношение к ее прогрессированию. Лишившись
такого контроля, клетки, утрачивающие связи с межклеточ-
ным матриксом и другими факторами нормального микро-
окружения, не гибнут, а благополучно развиваются в чуждой
для них среде, что способствует метастазированию опухолей.
Общеизвестным является факт рекомбинации гена bcl-2
при лимфоме Беркита и некоторых формах фолликулярных
лимфом, когда он транслоцируется из хромосомы 18 в ген Igh
хромосомы 14 [McDonnell T.J. et al., 1989]. При тех же типах
лимфом выявляется аналогичное перемещение гена с-тус из
хромосомы 8 в тот же локус Igh. Не вызывает сомнений связь
патогенеза лимфом с этими транслокациями, которая реализу-
ется через повышение резистентности измененных клеток к
индукции апоптоза.
Мутация гена р53, гиперпродукция Вс1-2 и Bcl-xL, актива-
ция каспаз являются основой формирования устойчивости к
лечебным воздействиям, основанным на индукции апоптоза
опухолевых клеток, — рентгено-, радио- и химиотерапии
[Searle J. et al., 1975]. Наиболее полной формой такой резис-
тентности является множественная лекарственная устойчи-
вость опухолевых клеток.
1.3.3. Патологические процессы,
связанные с усилением апоптоза
Заболеваниям, основой которых является усиление апо-
птоза клеток организма, принадлежит, по-видимому, не мень-
шее место в патологии. Наиболее выраженные формы такого
рода нарушений, при которых в процесс апоптоза тотально
вовлекаются клетки любых типов, наблюдаются при беремен-
ности. Они обычно несовместимы с развитием плода и приво-
дят к внутриутробной гибели. Известны также более ограни-
ченные нарушения такого типа, которые проявляются как де-
фекты развития с формированием «минус-ткани». Другим
примером могут служить некоторые врожденные поражения
48
сердца. Во II триместре развития плода в сердце происходит
перестройка тканевых структур, в которой важная роль при-
надлежит апоптозу. Эти процессы нарушаются при синдро-
ме Дауна, что влечет за собой формирование миопатии [Sa-
phier C.J., Yeh J., 1998]. Другой пример, относящийся к па-
тологии сердца, аритмогенная кардиопатия правого желу-
дочка, при которой в значительной степени вследствие уси-
ленного апоптоза миоцитов происходит их замещение жировой
тканью [Ahmad F. et al., 1998].
Распространенными вариантами патологии, развивающей-
ся в сформировавшемся организме в связи с усилением апо-
птоза, являются разного рода аплазии и дегенеративные про-
цессы. Наиболее разнообразные их формы описаны в области
патологии системы крови. Чаще всего они развиваются вслед-
ствие недостаточности «факторов выживания» костномозго-
вых клеток-предшественников. Так, в эксперименте направ-
ленная инактивация гена ИЛ-7 приводит к тотальной лимфо-
пении, в значительной степени связанной с гибелью предше-
ственников В- и Т-лимфоцитов на стадиях, предшествующих
формированию антигенраспознающих рецепторов [Von Free-
den-Jeffry U. et al., 1997]. Аналогичная патология, связанная
с мутацией гена общей у-цепи рецепторов для ИЛ-7, ИЛ-2,
ИЛ-4, ИЛ-9 и ИЛ-15, описана у человека как тяжелый комби-
нированный иммунодефицит [Puck J.M., 1993]. Сходные при-
чины лежат в основе апластической анемии, анемии при де-
фиците железа, фолатов, витамина В12, талассемии, тромбоци-
топении, нейтропении, панцитопении, хотя конкретные меха-
низмы гибели клеток-предшественников в этих случаях не
всегда установлены. Отключение генов факторов выживания в
ряде случаев не приводит к указанным последствиям в силу
избыточности контроля выживания клеток. Особенно при-
стально изучается роль апоптоза в патогенезе миелодисплас-
тических процессов, приводящих к панцитопении в результате
апоптоза стволовых клеток и ранних кроветворных предшест-
венников [Gersuk G.M. et al., 1998]. Повышенная готовность к
развитию апоптоза Т-лимфоцитов обнаружена при мульти-
центрической болезни Кастлемана.
Большую группу заболеваний, связанных с усилением
апоптоза, образуют инфекционные процессы. Индукторами
апоптоза служат бактериальные эндотоксины (например, ли-
пополисахарид кишечных микроорганизмов) и экзотоксины
(в частности, стафилококков, бордетелл и т.д.). Последние,
как правило, выступают в роли суперантигенов, вызывая мас-
совую пролиферацию Т-лимфоцитов с их последующей гибе-
лью по механизму апоптоза [Kawabe Y., Ochi А., 1991]. Массо-
вый апоптоз, опосредованный фактором некроза опухоли и
его рецепторами 1-го типа, развивается при сепсисе. При ви-
русных инфекциях сосуществуют факторы, индуцирующие и
4—1385
49
ингибирующие апоптоз (вирусам «невыгодна» тотальная ги-
бель клеток-мишеней).
Особая ситуация складывается при СПИДе. Установлено,
что доля инфицированных клеток среди гибнущих Т-лим-
фоцитов невелика. Гибель лимфоцитов происходит по меха-
низму апоптоза, и ее выраженность коррелирует с быстрым
прогрессированием заболевания [Ameisen J.C., Capron А., 1991;
Groux Н. et al., 1992]. Апоптозу подвергаются предварительно
активированные лимфоциты, в основном несущие маркер
клеток памяти CD45R0. Полагают, что одним из механизмов,
повышающих их чувствительность к активационному апопто-
зу, может быть перекрестное связывание молекул CD4 мем-
бранным гликопротеином ВИЧ-1 gpl20 (что моделируется
при действии на Т-клетки моноклональных антител к CD4)
[Groux Н. et al., 1992], а также связывание gpl20 с рецептора-
ми хемокинов [Berndt С. et al., 1998]. Вирусный белок Tat спо-
собен вызывать апоптоз и сенсибилизировать клетки к его ин-
дукторам. Определенную роль в активационном апоптозе CD4+
клеток играет корецептор CD28, поскольку антитела к этой
молекуле предотвращают гибель Т-клеток больных СПИДом.
Более того, клетки CD4+, инфицированные вирусом ВИЧ-1,
менее чувствительны к индукции апоптоза, поскольку один из
вирусных белков, Nef, подавляет его развитие.
Другим примером болезней, связанных с усилением апо-
птоза, являются заболевания нервной системы, вызываемые
атрофией ее определенных участков. Как правило, атрофия
является следствием индукции апоптоза соответствующих
клеток в неположенное время или в чрезмерно выраженном
масштабе. К таким заболеваниям относятся боковой амиотро-
фический склероз, болезнь Альцгеймера, спинальная мышеч-
ная атрофия и другие заболевания нервной системы. Иден-
тифицирован «ген болезни Альцгеймера», продукт которого
оказался транскрипционным фактором ALG-2. При этом за-
болевании идентифицированы факторы, накопление которых
способствует развитию апоптоза нервных клеток, — пептид
Аа [La Ferla F.M. et al., 1955], р-амилоид и др.
Существует ряд заболеваний, при которых в реализации
основного поражения решающая роль принадлежит апоптозу.
Примерами могут служить инфаркт миокарда: апоптоз являет-
ся преобладающей формой гибели миоцитов в период «репер-
фузии» участка миокарда, ранее подвергшегося ишемии
[Saikumar Р. et al., 1998], токсический (в частности, алкоголь-
ный) гепатит и т.д.
Со временем увеличивается доля патологических процес-
сов, основывающихся на усилении апоптоза, которое вызва-
но действием внешних апоптогенных факторов. Первое
место среди них занимает ионизирующая радиация. В связи с
тем что она индуцирует апоптоз преимущественно лимфоид-
50
ных клеток, эта сторона ее действия проявляется в иммунной
недостаточности, хотя вызываемые облучением нарушения
кроветворения, по крайней мере частично, обусловлены ин-
дукцией апоптоза клеток-предшественников. Аналогичным
эффектом обладают многие химиотерапевтические препара-
ты, используемые при лечении опухолей, а также гормоны,
прежде всего глюкокортикоиды, широко применяемые при
лечении различных заболеваний. Источником апоптогенных
факторов служит внешняя среда. Нормальное окружение че-
ловека практически не является источником воздействий,
вызывающих апоптоз, однако такие факторы могут накапли-
ваться во внешней среде при формировании экологического
неблагополучия. Наиболее ярким примером агрессивных
факторов, загрязняющих среду, которые способны вызвать
гибель клеток путем индукции апоптоза, является диоксан,
повреждающий, в частности, эпителиальные клетки вилочко-
вой железы.
Таким образом, в основе достаточно большого числа пато-
логических процессов лежат нарушения процесса апоптоза.
У взрослых могут регистрироваться лишь дефекты с ограни-
ченными фенотипическими проявлениями, поскольку орга-
низмы с обширными дефектами такого рода гибнут на ранних
этапах онтогенеза. Наиболее характерными проявлениями не-
достаточности апоптоза служит развитие аутоиммунных про-
цессов и злокачественных новообразований, проявлениями
усиленного апоптоза — аплазии и дегенеративные процессы,
а также некоторые уродства с дефектами тканей.
Заключение
Из материала, представленного в обзоре, следует, что
апоптоз играет первостепенную роль в нормальных процессах
морфогенеза, а также в поддержании клеточного гомеостаза.
В то же время его значение в формировании типовых патоло-
гических процессов является значительно более скромным (в
отличие от некроза, практически не реализуемого в здоровом
организме, но активно «используемого» при развитии патоло-
гии). Однако проблема «апоптоз и патология» имеет и иной
аспект: важной составляющей или даже основой заболеваний
может быть нарушение выраженности апоптоза — его усиле-
ние или ослабление. При усилении апоптоза развиваются по-
вреждения, затрагивающие обычно определенные типы клеток
и состоящие в уменьшении их численности. При ослаблении
апоптоза решающим для развития болезни становится прояв-
ление активности клеток, в норме подлежащих ограничению
или полной элиминации. Часто заболевания этой группы име-
ют стандартные проявления — в форме аутоиммунного и
4*
51
доброкачественного лимфопролиферативного синдромов; к
этой группе примыкают злокачественные опухоли, формиро-
ванию которых способствует ослабление механизма удаления
генетически измененных клеток, основанного на апоптозе.
Сведения о месте апоптоза в развитии патологии человека в
схематически обобщенной форме представлены на схеме 1.2 и
в табл. 1.5.
Хотя вклад апоптоза в патологию находится еще в про-
цессе осмысления, уже сейчас ясно, что в одних случаях он
может быть ключевым, а в других — определяет тот патоло-
гический фон, на котором развертываются специфические
процессы. И то, и другое не может не учитываться при разра-
ботке стратегии лечения соответствующих заболеваний. Это
побуждает к развитию особой отрасли фармакологии, кото-
рая посвящена разработке путей регулирования и коррекции
апоптоза [Kroemer G., 1995]. «Фармакология апоптоза» в на-
стоящее время лишь формируется; однако стремительный
прогресс в изучении молекулярных основ апоптоза вселяет
надежду на ее быстрое развитие и столь же быстрое внедре-
ние в медицинскую практику разработок, основанных на
учении об апоптозе.
Список литературы
Барышников А.Ю., Шишкин Ю.В. Программированная клеточная ги-
бель (апоптоз)//Рос. онкол. журн. — 1996. — № 1. — С. 58—61.
Никонова М.Ф., Буланова Е.Г., Станислав М.А. и др. Различная готов-
ность к развитию апоптоза активированных Т-лимфоцитов в норме и
при СКВ//Иммунология. — 1997. — № 4. — С. 21—24.
Хансон К.П. Радиобиология. — 1979. — Т. 19. — С. 814—817.
Ярилин А.А. Апоптоз и его место в иммунных процессах//Иммуноло-
гия. - 1996. - № 6. - С. 10-22.
Ярилин А.А. Апоптоз: природа феномена и его место в целостном ор-
ганизме//Пат. физиол. — 1998. — № 2. — С. 38—48.
Ahmad F., Li D., Karibe A. et al. Localization of gene responsible for ar-
rhythmogenic right ventricular dysplasia to chromosome 3p23//Circula-
tion. - 1998. - Vol. 98. - P. 2791-2795.
Alnemri E.S., Livingston D.J., Nickolson D.W. et al. Human ICE/CED-3
protease nomenclature//Cell. — 1996. — Vol. 87. — P. 171—172.
Ameisen J.C., Capron A. Cell dysfunction and depletion in AIDS: the pro-
grammed cell death hupotesis//Immunol. Today. — 1991. — Vol. 12. —
P. 102-105.
52
Berndt C., Mopps В., Angermuller S. et al. CXCR4 and CD4 mediate a
rapid CD95-independent cell death in CD4+T cells//Proc. nat. Acad. Sci.
USA. - 1998. - Vol. 95. - P. 12556-12561.
Budagyan V.M., Bulanova E.G., Sharova N.I. et al. The resistance of acti-
vated T cells from SLE patients to apoptosis induced by human thymic
stromal cells//Immunol. Lett. — 1998. — Vol. 60. — P. 1—5.
Cheng J., Zhou T., Liu C. et al. Protection from Fas-mediated apoptosis by
a soluble form of the Fas molecule//Science. — 1994. —Vol. 263. —
P. 1759-1762.
Cohen J.J. Glucocorticoid-induced apoptosis in the thymus//Sem. Immu-
nol. - 1992. - Vol. 4. - P. 363-369.
Fischer G.H., Rosenberg F.J., Straus S.E. et al. Dominant interfering Fas
gene mutations impair apoptosis in a human autoimmune lymphoprolifera-
tive syndrome//Cell. — 1995. — Vol. 81. — P. 935—946.
Gavrieli Y, Sherman Y, Ben Sasson A. Identification of programmed cell
death in situ via specific labelling of nuclear DNA fragmentation//!. Cell.
Biol. - 1992. - Vol. 119. - P. 493-501.
Gersuk G.M., Beckman C, Loken M.R. et al. A role for TNFa, Fas and
Fas-ligand in marrow failure associated with myelodysplastic syndrome//
Brit. J. Haemat. - 1998. - Vol. 103. - P. 176-188.
Green D.R., Bissonnette R.P., Glynn J.M., Shi Y. Activation-induced apop-
tosis in lymphoid systems//Sem. Immunol. — 1992. — Vol. 4. — P. 379—
388.
Griffith T.S., Ferguson T.A. The role of FasL-induced apoptosis in immune
previlege//Immunol. Today. — 1997. — Vol. 18. — P. 240—244.
Groux H., Torpier G., Monte D. et al. Activation-induced death by apop-
tosis in CD4i cells from human immunodeficiency virus-infected asymp-
tomatic individuals//!, exp. Med. — 1992. — Vol. 175. — P. 331—340.
Hockenbery D.M. The bcl-2 oncogen and apoptosis//Sem. Immunol. —
1992. - Vol. 4. - P. 413-420.
Horvitz R. Genetic control of programmed cell death in the nematode
Caenorhabditis elegans//Cancer Res. — 1999. — Vol. 59, suppl. —
P. 170 Is-1706s.
Kaneko H., Saito K., Hashimoto H. et al. Preferential elimination of CD28+
T cells in SLE and the relation with activation-induced apoptosis//Clin.
exp. Immunol. — 1996. — Vol. 106. — P. 218—229.
Kawabe Y., Ochi A. Programmed cell death and extrathymic reduction of
Vp8+ CD4+ T cells in mice tolerant to st. Aureus enterotoxin B//Natu-
re. - 1991. - Vol. 349. - P. 245-248.
53
KerrJ.F.R., WyllieA.H-, Curie A.R. Apoptosis: a basic biological phenome-
non with wide ranging implications in tissue kinetics//Brit. J. Cancer. —
1972. - Vol. 26. - P- 239-257.
Krammer P.H. CD95 (APO-i/Fas)-mediated apoptosis: live and let die//
Adv. Immunol. — 1999. — Vol. 71. — P. 163—210.
Komblau S.M. The role of apoptosis in the pathogenesis, prognosis, and
therapy of hematological maIignancies//Leukemia. — 1998. — Vol. 12,
suppl. 1. — S. 41-46-
Kroemer G. The pharmacology of T cell apoptosis//Adv. Immunol. —
1995.-Vol. 58. — P-211—296.
Kroemer G., Zamzani N., Susin S.A. Mitochondrial control of apoptosis//
Immunol. Today. — 1997. — Vol. 18. — P. 44—51.
LaFerla F.M., Tinkle B.T., Bieberich C.J. et al. The Alzheymer’s Ap pep-
tide induces neurodegeneration and apoptotic cell death in transgenic
mice//Nature Genetics. — 1995. — Vol. 9. — P. 21—30.
Leist M., Gartner E, JHg S., Wendel A. Activation of the 55 kDa TNF re-
ceptor is necessary artd sufficient for TNF-induced liver failure, hepatocyte
apoptosis, and nitrite release//!. Immunol. — 1995. — Vol. 154. —
P. 1307-1316.
Levine A.J., Perry ME., Chang A. et al. The role of the p53 tumour-sup-
pressor gene in tumorigenesis//Brit. J. Cancer. — 1994. — Vol. 69. —
P. 409-416.
McCloskey T IK, Oytizu N., Coronezi M., Pahwa S. Use of a flow cytomet-
ric assay to quantitate apoptosis in human lymphocytes//Clin. Immunol.
Immunopath. — 1994- — Vol. 71. — P. 14—18.
McConkey D.J., Zhifotovsky Rr Orrenius S. Apoptosis-molecular mecha-
nisms and biomedical implications//Molec. Aspects Med. — 1996. — Vol.
17. _ p. 1-Ц0.
McDonnell T.J., Deane N., P[att F.M. et al. Bcl-2-immunoglobulin trans-
genic mice demonstrate extended В cell survival and follicular lymphopro-
liferation//Cell. - 1989. - Vol. 57. - P. 79-88.
Nagata S., Goldstein P. The Fas death factor//Science. — 1995. — Vol.
267. - P. 1449—1456.
Nakamura S., Sugiti M., Matoba H. et al. Insufficient expression of Fas
antigen on helper T cells in Behcet’s disease//Brit. J. Ophtalmology. —
1996. - Vol. 80. —F 174-176.
Oltvai Z.N., KorsmWer S.J. Checkpoints of dueling dimers foil death
wishes//Cell. - 1991- - Vol. 79. - P. 189-192.
54
Onici К, Kizaki H. Apoptosis and diseases//Hum. Cell. — 1994. — Vol.
7. - P. 27-32.
Puck J.M. X-linked immunodeficiencies//Adv. Hum. Genet. — 1993. —
Vol. 21. - P. 107-135.
Pellegrini M., Strasser A. A portrait of the Bcl-2 protein family: life, death,
and the whole picture//! clin. Immunol.— 1999.— Vol. 19.— P. 365—377.
Rathmell J. C., Thompson C.B. The central effectors of cell death in the im-
mune system//Ann. Rev. Immunol. — 1999. — Vol. 17. — P. 781—828.
Rowan S., Fischer D.E. Mechanisms of apoptotic cell death//Leukemia. —
1997. - Vol. 11. - P. 457-465.
Russel J.H. Activation-induced apoptosis of nature T cells in the regulation
of immune responses//Curr. Opin. Immunol. — 1995. — Vol. 7. —
P. 382-388.
Saikumar P., Dong Z., Weinberg J.M., Venkatachalam M.A. Mechanisms of
cell death in hypoxia/reoxygenation injury//Oncogene. — 1998. — Vol.
17. - P. 3341-3349.
Salmon M., Pilling D., Borthwick NJ., Akbar A.N. Inhibition of T cell
apoptosis — a mechanism for persistence in chronic inflammation//Immu-
nologist. - 1997. - Vol. 5. - P. 87-92.
Saphier C.J., Yeh J. Altered apoptosis levels in hearts of human fetuses with
Down syndrome//Amer. J. Obstet. Gynecol. — 1998. — Vol. 179. —
P. 962-965.
Skalka M., Matyasova J., Cejkova M. DNA in chromatin of irradiated lym-
phoid tissues degrades in vivo into regular fragments//FEBS Lett. —
1976. - Vol. 72. - P. 271-274.
Searle J., Lawson T.A., Abbott P.J. et al. An electromicroscopie study of the
mode of cell death induced by cancer-chemotherapeutic agents in popula-
tion of proliferating normal and neoplastic cells//J. Pathol. — 1975. —
Vol. 115. — P. 129-138.
Sprent J., Webb S.R. Intrathymic and extrathymic clonal deletion of T
cells//Curr. Opin. Immunol. — 1995. — Vol. 7. — P. 196—205.
Steller H. Mechanisms and genes of cellular suicide//Science. — 1995. —
Vol. 267. - P. 1145-1149.
Strasser A., Harris A. W., Cory S. Bcl-2 transgene inhibits T cell death and
perturbs thymus self-censorship//Cell. — 1991. — Vol. 67. — P. 889—899.
Takashi R., Tanaka M., Brannan C.I. et al. Generalized lymphoprolifera-
tive syndrome im mice caused by a point mutation in the Fas ligand//
Cell. - 1994. - Vol. 76. - P. 969-976.
55
Thornberry N.A., Lazebnik Y. Caspases: enemies within//Science. —
1998. - Vol. 281. - P. 1312-1316.
Trulson J.A., Millhauser G.L. The effect of mutations on peptide models of
the DNA binding helix of p53: evidence for a correlation between structure
and tumorigenesis//Biopolimers. — 1999. — Vol. 49. — P. 215—224.
Utz X., Hottelet M., Schur P.H., Anderson P. Proteins phosphorylated dur-
ing stress-induced apoptosis are common targets for autoantibody produc-
tion in patients with SLE//J. exp. Med. — 1997. — Vol. 185. — P. 843—
854.
Изл Engeland M., Nieland L.J.W., Ramaekers F.C.S. et al. Annexin V-af-
finity assay: a review on an apoptosis detection system based on phospha-
tidylserine exposure//Cytometry. — 1998. — Vol. 31. — P. 1—9.
Von Freeden Jeffry U., Solvason N., Howard M., Murray R. The earliest T
lineage-commited cells depend on IL-7 for Bcl-2 expression and normal
cell cycle progression//Immunity. — 1997. — Vol. 7. — P. 147—154.
Watanabe-Fukunaga R., Brannan C.I., Copeland N.G. et al. Lymphoprolif-
eration disorder in mice explained by defects in Fas antigen that mediates
apoptosis//Nature. — 1992. — Vol. 356. — P. 314—317.
Waterhouse N.J., Green D.R. Mitochondria and apoptosis: HQ or high-se-
curity prison?//! Clin. Immunol. — 1999. — Vol. 19. — P. 378—387.
Wedi B., Raap U., Lewrick H., Kapp A. Delayed eosinophil programmed
cell death in vitro: a common feature of inhalant allergy and extrinsic and
intrinsic atopic dermatitis//! Allergy clin. Immunol. — 1997. — Vol.
100. - P. 536-543.
Winoto A. Cell death in the regulation of immune responses//Curr. Opin.
Immunol. - 1997. - Vol. 9. - P. 365-370.
Wong B., Choi Y. Pathways leading to cell death in T cells//Curr. Opin.
Immunol. - 1997. - Vol. 9. - P. 358-364.
WyllieA.H. Glucocorticoid-induced thymocyte apoptosis is associated with
endogenous endonuclease activation. Int. Rev//Cytol. — 1980. — Vol.
68. - P, 251-306.
Zhyvotovsky B., Wade D., Nicotera P., Orrenius S. Role of nucleases in
apoptosis//Int. Arch. Allergy Immunol. — 1994. — Vol. 105. — P. 333—
338.
Zhyvotovsky B., Gahn A., Ankercrone M. et al. Multiple proteases are in-
volved in thymocyte apoptosis//Exp. Cell Res. — 1995. — Vol. 221. —
P. 404—412.
Лекция 2
Иммунная система и патология
Г. А. Игнатьева
Введение
В иммунологии как науке и медицинской специальности
до настоящего времени еще не устоялись до однозначно
понимаемых основные понятия и термины, начиная с терми-
на, обозначающего предмет данной науки, — «иммунитет».
Главные события иммунитета происходят на молекулярном
и клеточном уровнях, которые стали доступными для ис-
следований в последние 25—30 лет, а самые сильные гно-
сеологические методы — генетический нокаут (knockout) и
трансгенные модели на животных — это работы 90-х годов
XX в. Но хотя и очевидно, что в данном предмете мы пре-
бываем в периоде интенсивного накопления новой инфор-
мации, тем не менее медицинская практика нетерпелива и
подчас слишком смело вмешивается в организм больных
людей с иммунокорригирующей терапией и здоровых людей
с вакцинацией, не дожидаясь появления общей теории имму-
нитета.
Теории иммунитета, такой, которая бы имела не предпо-
ложительно-описательный характер (свойственный теориям
иммунитета начала и середины века), а предсказательный,
прогностический, нет.
В наше время реальный спрос есть только на такую
теорию, которая бы умела на основании знаний о предмете
рассчитать последствия иммунотропной терапии как «на
конце иглы», так и через сутки, месяцы, годы и по-
жизненно.
Отсюда вывод: систематизация знаний, несмотря на их
быстрое развитие, необходима хотя бы для того, чтобы ока-
зать проясняющее и как следствие — сдерживающее влияние
на энергичное и массовое внедрение иммунотропных мето-
дов лечения во имя соблюдения первого врачебного принци-
па «не навреди».
Вот для того, чтобы «не навредить», достаточно уже
имеющихся знаний. Довести эти знания до врачей в система-
тизированном и понятном виде — главная задача данной ра-
боты.
57
2.1. Словарь сокращений и терминов
2.1.1. Субпопуляции клеток иммунной системы
В-лимфоцит — лимфоцит, специализирующийся на био-
синтезе иммуноглобулинов.
В-1 — субпопуляция В-лимфоцитов, представляющая
собой отдельный самоподдерживающийся росток, отселяю-
щийся от общей СКК в период эмбрионального развития; ло-
кализуется преимущественно в плевральной и брюшной по-
лостях; эти лимфоциты несут отличительный мембранный
маркер — молекулу CD5.
В-2 — субпопуляция В-лимфоцитов, дифференцирующих-
ся из стволовой кроветворной клетки на территории костного
мозга.
Т-лимфоциты — лимфоциты, имеющие на мембране осо-
бые рецепторы для антигенов (TcR) и особую молекулярную
систему проведения сигналов с мембраны внутрь клетки —
комплекс CD3 и ^-полипептид.
Тар — субпопуляция Т-лимфоцитов, имеющая рецептор
для антигена, состоящий из полипептидных цепей а и Р; диф-
ференцируется из стволовой кроветворной клетки в вилочко-
вой железе.
Туб — субпопуляция Т-лимфоцитов, имеющая рецептор
для антигена, состоящий из полипептидных цепей у и б; диф-
ференцируется из стволовой кроветворной клетки преимуще-
ственно в слизистой оболочке желудочно-кишечного тракта;
в вилочковой железе составляет минорную субпопуляцию —
около 0,5 % всех тимоцитов.
ЦТЛ — цитотоксические Т-лимфоциты.
Th — Т-хелперы.
ТЫ — Т-хелперы 1-го типа.
Th2 — Т-хелперы 2-го типа.
ТЬЗ — Т-хелперы 3-го типа.
ThO — Т-хелперы 0-го типа.
• NK — нормальные киллеры.
DC — дендритные клетки.
АРС — антигенпредставляющие клетки.
СКК — стволовые кроветворные клетки.
IEL — внутриэпителиальные лимфоциты.
2.1.2. Молекулы клеточных мембран
TcR — рецептор Т-лимфоцитов для антигена.
BcR — рецептор В-лимфоцитов для антигена.
МНС — главный комплекс гистосовместимости (гены и
кодируемые ими белки).
58
МНС I класса и МНС П класса — две отдельные состав-
ляющие комплекса МНС; экспрессируются независимо друг
от друга.
Молекулы CD (от Cluster definition) — молекулы клеточной
мембраны различного предназначения.
Номера (CD1, CD2, CD3 и т.д. до CD130) обозначают от-
дельные молекулы.
FcR — рецепторы на мембране клетки, предназначенные
для связывания молекул иммуноглобулинов за их Fc (кон-
стантный) конец. Например, FcTRIII — рецептор 3-го типа
(низкоаффинный) для связывания IgG за у-цепь. Эта молекула
(FcyRIII) по номенклатуре CD называется CD 16.
LFA-1 — 1-й тип молекул клеточной мембраны, ассоции-
рованных с функцией лимфоцита/лейкоцита.
LFA — 3-й тип молекул клеточной мембраны, ассоцииро-
ванных с функцией лимфоцита/лейкоцита.
ICAM — молекулы межклеточной адгезии.
Интегрины — групповое название молекул клеточных мем-
бран, предназначенных для удержания нужных клеток друг
около друга и в нужном месте в тканях.
Селектины (синоним — homing-рецептор) — молекулы
мембран лимфоцитов и лейкоцитов, отвечающие за сродство
данного лимфоцита/лейкоцита к определенной ткани и органу
в организме.
Адрессины — молекулы-партнеры для селектинов на мем-
бране клеток эндотелия сосудов в определенных тканях.
2.1.3. Цитокины
IL — интерлейкины. Номера (IL-1, IL-2, IL-3 и т.д.) обо-
значают отдельные молекулы разных цитокинов.
IFN — интерфероны.
TNF — фактор некроза опухолей.
LT — лимфотоксин.
GM-CSF — фактор, стимулирующий пролиферацию кле-
ток-предшественников гранулоцитов/макрофагов.
G-CSF — фактор, стимулирующий пролиферацию кле-
ток-предшественников гранулоцитов.
M-CSF — фактор, стимулирующий пролиферацию кле-
ток-предшественников макрофагов.
TGFp — трансформирующий фактора роста р.
Хемокины — групповое название цитокинов, обеспечива-
ющих движение лимфоцитов и лейкоцитов из циркуляции в
заданный очаг в тканях.
59
2.1.4. Конкретные иммунологические
эффекторные реакции
ГЗТ — гиперчувствительность замедленного типа.
АЗКЦТ — антителозависимая клеточная цитотоксичность.
2.2. Определение понятия «иммунитет»
Im-munis — по латыни «невредимый, чистый, неприкосно-
венный ...». В медицине издавна понятие «иммунный» относи-
лось к способности того или иного человека не заболевать за-
разной болезнью, когда другие вокруг него болеют.
Биологический феномен некой особой невосприимчивос-
ти к какой-либо заразной болезни был известен людям и в
давние времена. Например, иммунитет к чуме у отдельных
людей описывает врач Thucydides из Афин в V в. до нашей
эры. В античном Китае существовала традиция прививания
детей в целях защиты их от заболевания оспой путем вдыхания
через нос истолченных в порошок корочек с оспенных папул
людей, выздоравливающих от оспы. Для более энергичной и
широкой практики массовой иммунизации общечеловеческий
многовековой опыт не имел, очевидно, предпосылок. Преоб-
разовательная врачебная деятельность по вакцинации в отно-
шении многих инфекционных болезней и на больших популя-
циях людей — это новое явление нашего века, опирающееся
уже не на опыт древних, а на персональные работы E.Jenner
(1878) и L.Pasteur (1881). Вторая причина неоднозначности
терминологии в иммунологии — лингвистическая. Основной
поток научной информации в этой области знаний в XX в.
идет на английском языке, в котором по сравнению с латин-
ским и русским языками меньше языковых возможностей для
выражения однозначного соответствия термина и явления
природы или объекта. Поэтому то, что привычно устраивает
англоязычное мышление, не всегда удовлетворяет русскоязыч-
ной логике. Например, в английском языке используют тер-
мин «врожденный иммунитет» (innate or natural immunity).
Под этим понимают вполне определенный перечень клеток,
молекул и физиологических механизмов (бактерицидные фер-
менты биологических жидкостей, фагоцитоз, систему компле-
мента и т.п.), которые, однако, не лежат в основе практики
античных китайцев, греков, работ Дженнера, Пастера и совре-
менных иммунологов. То, что лежит в основе практики анти-
чных китайцев и остальных перечисленных выше исследовате-
лей, в современном английском называют «приобретенным
иммунитетом» (acquired immunity).
Для удовлетворения потребностям русскоязычной логики,
а также следуя понятиям древних, мы предлагаем называть ту
60
материю, которая в английском языке названа innate immu-
nity, не иммунитетом, а резистентностью с указанием кон-
кретного определения (прилагательного) — системы компле-
мента, лизоцима, фагоцитоза, эозинофильной цитотоксичнос-
ти и т.д., а иммунитетом называть то биологическое свойст-
во, которому соответствуют глагол «иммунизировать», сущест-
вительное «иммунизация», прилагательное «иммунный». За
этим свойством стоит система особых клеток, а именно лим-
фоцитов, развившихся в эволюции позже тех клеток и их про-
дуктов, которые обеспечивают врожденную резистентность
(к инфекциям и инвазиям). Эта система — иммунная система.
Анатомический синоним иммунной системы — лимфоидная
система. И эти особые, эволюционно новые клетки — лимфо-
циты, и только они, обладают новым, уникальным свойст-
вом — молекулярного распознавания. Лимфоциты взаимодейст-
вуют со всеми клетками крови, с эндотелием сосудов, по кото-
рым и через стенку которых лимфоциты попадают в ткани.
Лимфоциты предназначены для контактов со всеми клетками
организма. Лимфоциты через специальные рецепторы воспри-
нимают информацию от нервной системы, от эндокринной
системы, уровень глюкозы в крови и т.д. Но это не значит, что
иммунная (лимфоцитарная) система сливается до неразличи-
мости со всем организмом и в список иммуноцитов надо
включать эндотелий венул, фибробласты рыхлой соединитель-
ной ткани и т.д. Есть вполне конкретные и ясные свойства
у лимфоцитарного иммунитета, по которым он может быть
идентифицирован как особое биологическое свойство много-
клеточных организмов. Эти свойства мы опишем в данной ра-
боте. Лимфоциты функционируют не сами по себе, не вне ор-
ганизма, следовательно, их взаимосвязи с другими клетками,
тканями и органами в целостном организме — единственно
возможная реальность. Более того, лимфоцит, специально
дифференцированный для уникальной функции распознава-
ния, все свои клеточные силы отдает именно этой функции, а
доведение дела защиты организма от того, что распознал лим-
фоцит, он передает другим клеткам (кроме цитотоксических
лимфоцитов — Т и NK — нормальных киллеров). Эти кон-
кретные взаимосвязи мы проследим в той мере, в которой они
известны на сегодня.
Итак, иммунитет — некое защитное свойство многоклеточ-
ных организмов. Лимфоциты и иммунитет существуют всего у
1,4 % биологических видов животных, начиная с челюстных
рыб, тогда как у 98,6 % видов лимфоцитов нет. Эти 1,4 %
видов, имеющих иммунную систему, отличаются от остальных
98,6 % малым количеством потомства вплоть до единичного у
человека. При столь малом числе потомков для выживания
вида необходимо выживание по возможности каждого инди-
видуального организма. Вот для выживания индивидуумов в
61
нестерильной внешней среде обитания и понадобилась особая
защитная система — иммунная. Чтобы понять место иммуни-
тета, рассмотрим, какие способы (механизмы, уровни) защи-
ты, например, от какой-либо инфекционной болезни имеются
у человека и млекопитающих.
1. Ментальный (поведенческий — избегать контактов с за-
раженными, мыть руки, правильно стерилизовать медицин-
ские инструменты, одеваться по погоде и т.п.).
2. Покровные барьерные ткани (кожа, слизистые оболочки).
3. Сосудистые реакция.
4. Микробоцидные экзосекреты (соляная кислота желудка,
бактерицидные компоненты слюны, литические пищевари-
тельные ферменты кишечника, эндогенные пептиды-антибио-
тики, вырабатываемые, например, нейтрофилами, и т.п.).
5. Первичный фагоцитоз микробных тел нейтрофилами и
макрофагами. Этот способ клеточной защиты происходит от
пищеварительной функции одноклеточных организмов. Одна
и та же клетка — фагоцит — способна поглотить с целью пере-
варивания разные предложенные ей объекты.
6. Белки острой фазы — С-реактивный протеин и маннан-
связывающий лектин. Эти белки вырабатывает печень. По
своей природе они способны связывать множество наиболее
распространенных микроорганизмов при их попадании в
кровь. Для комплексов белков острой фазы с патогенами на
фагоцитах существуют специальные рецепторы, и фагоциты
связывают, поглощают и расщепляют такие комплексы, т.е.
белки острой фазы — опсонины.
7. Лимфоцитарный иммунитет: каждый индивидуальный
лимфоцит (и его клональные дочерние лимфоциты-потомки)
несет на своей поверхности индивидуальный антигенсвязываю-
щий рецептор, которым лимфоцит потенциально способен фи-
зически связать (это и есть суть молекулярного распознава-
ния) антигены, которые по своей химической природе могут
образовать с рецептором комплементарные связи (ионные,
ван-дер-ваальсовы, водородные и гидрофобные с надлежащей
аффинностью).
Молекулярный объект, который потенциально может свя-
зать какой-либо лимфоцит своим распознающим рецептором,
называют антигеном. По химической природе антигены быва-
ют белками, полисахаридами, фосфолипидами и производны-
ми названных соединений. С точки зрения внешней среды
антигенами как веществами, способными индуцировать им-
мунный ответ на себя, являются макромолекулы. Низкомоле-
кулярные соединения, попадающие из внешней среды во
внутреннюю, организм выводит через почки и желудочно-ки-
шечный тракт без применения особых механизмов расщепле-
ния. Макромолекулы, тем более микроорганизмы, так просто
не выводятся из организма. Поэтому иммунная система их
62
распознает и направляет на них те или иные механизмы де-
струкции. Во внутренней среде для антигенраспознающих ре-
цепторов лимфоцитов объектом распознавания являются
малые фрагменты молекул внешних антигенов. Эти малые
фрагменты (5—9 аминокислот пептида, отдельные участки мо-
лекул полисахаридов, фосфолипидов и т.п.) называют анти-
генными эпитопами.
Антигенами являются не только чужеродные соединения,
но в той же (если не в большей) мере антигенами для лимфо-
цитов являются молекулы своего собственного организма.
Антигенраспознающий рецептор лимфоцита формируется в
процессе дифференцировки, которую называют иммунопоэзом.
Иммунопоэз большинства лимфоцитов происходит сугубо во
внутренней среде организма, без доступа экзогенных субстан-
ций, на территории лимфопоэтических органов (костного
мозга, вилочковой железы). Таким образом, дифференцировка
и отбор антигенраспознающих рецепторов, в том числе и
предназначенных для связывания потенциальных чужеродных
антигенов, проходят во взаимодействиях исключительно со
своими, эндогенными, антигенами. Итак, для выживания
многоклеточных организмов с малым числом потомков по
сравнению с одноклеточными необходимо решать две эволю-
ционно новые проблемы — морфогенез и защита от инфек-
ций. Решение этих двух проблем и является эволюционно
новой функцией новой системы клеток — иммунной системы.
2.3. Главные функции иммунной системы
1. Морфогенетическая (обеспечение своевременного рас-
познавания и аккуратной элиминации уже ненужных клеток,
тканей и органов в онтогенезе многоклеточного организма).
2. Противоинфекционная, или, шире, — антиинвазивная.
Сюда также относят дополнительный иммунный барьер, по-
могающий пищеварительной системе в работе со значитель-
ным ежедневным потоком пищевых антигенов, и реакции
лимфоцитов на ингаляторные и аппликаторные внешние ве-
щества, проникающие во внутреннюю среду организма.
Другие естественные предназначения системы не очевид-
ны. Но в «списке» функций иммунной системы есть еще по
крайней мере один целиком искусственный, антропогенный,
а именно ятрогенный пункт — иммунитет против так назы-
ваемых аллоантигенов, т.е. антигенов чужеродных тканей,
трансплантированных во внутреннюю среду организма. Не
было бы медицинских пересадок органов и тканей, включая
переливание крови и введение кровепрепаратов, не было бы и
понятия о трансплантационном иммунитете. В природе нет
естественных процессов, при которых уже рожденные особи
63
обменивались бы с другими особями кровью, тканями, орга-
нами, разве только каннибалы-сыроеды с обширными ульце-
ративными колитами. Привнесение во внутреннюю среду
одного организма тканей от другой особи — чисто искусствен-
ное, ятрогенное явление. По этой причине оно требует к себе
особого отношения, ибо если в природе нет процессов такого
рода, то в эволюции млекопитающих, в том числе в иммунной
системе, не мог идти отбор молекулярных механизмов адап-
тации к таким ситуациям и защитного контроля над такими
ситуациями. Есть основания полагать, что именно это лежит в
основе неспособности иммунной системы санировать орга-
низм от кровяных инфекций, т.е. инфекций, распространяю-
щихся в популяции с переливанием крови и введением про-
дуктов медицинского назначения, полученных из крови или
тканей млекопитающих. В настоящее время такими инфек-
циями являются ВИЧ-инфекция (СПИД) и прионные инфек-
ции типа губчатого энцефалита (бешенства) коров, заразных
для человека по пищевому пути. Может быть, стоит хотя бы
временно признать, что бессмысленно рассчитывать на вакци-
ну от СПИДа. Вакцина в классическом понимании (и на се-
годня другого понимания, основываясь на здравом смысле,
пока никто не предложил) рассчитана на мобилизацию им-
мунных механизмов, нредсуществующих в организме, заложен-
ных в процессе лимфопоэза, т.е. генетической дифференцировки
лимфоцитов до встречи с антигеном. А если в генетической
программе лимфоцитов эволюционно и не закладывались ме-
ханизмы защиты, адекватные патогенным свойствам ретрови-
русов, прионов и неизвестных нам новых инфекций, эволю-
ция которых идет темпами, несоизмеримыми с темпами эво-
люции млекопитающих? Тогда вакцинировать от таких ин-
фекций так же полезно, как заставлять спеть красивую мело-
дию человека, от природы лишенного и слуха, и голоса.
Опыт показывает, что антигены, т.е. то, что распознает им-
мунная система, а именно антигенраспознающие рецепторы
лимфоцитов, — это молекулы наружных мембран клеток. По
биохимической природе антигены относятся к белкам и их
производным — гликопротеидам, липопротеидам. Антигенами
бывают также чистые углеводы, липополисахариды, фосфори-
лированные производные органических молекул. Принципи-
ально, что иммунные механизмы работают с внеклеточным
материалом и с поверхностью клеток, но не внутри клеток, не
в ядре, не с геномом, т.е. иммунная система распознает то, что
в классической генетике называют фенотипом. Внешние ве-
щества, микроорганизмы и продукты их деградации становят-
ся объектом, на который организм развивает иммунный ответ,
только в том случае, когда эти внешние вещества прочно свя-
зываются с поверхностью клеток организма и/или с молекула-
ми межклеточного матрикса.
64
Таким образом, определение иммунитета следующее.
Иммунитет — особое биологическое защитное свойство
многоклеточных, осуществляемое специализированными
клетками — лимфоцитами и состоящее в молекулярном рас-
познавании клеточного фенотипа во внутренней среде орга-
низма с целью деструкции и элиминации поврежденных па-
тогеном (инфекционными микроорганизмами, гельминтами,
лекарственными препаратами и т.п.) клеток и молекул меж-
клеточного матрикса.
Иммунитет = распознавание + деструкция. Это определе-
ние отражает пределы действия иммунной защиты — вне и
снаружи клеток собственного тела. Иммунная система не ра-
ботает с геномом клеток. Поэтому вирусные инфекции с кова-
лентным встраиванием вируса в геном, а также малоизучен-
ные внутриклеточные прионные инфекции, видимо, недо-
ступны санирующим механизмам иммунитета. А иммунитет,
похоже, эволюционно самый последний и самый сильный
биологический защитный механизм у многоклеточных. Поэ-
тому, вероятно, в защите от ретровирусных и прионных ин-
фекций человеку надо бы полагаться на ментальные механиз-
мы защиты, а не на бессознательный иммунитет. Распознава-
ние патогена — специальная функция лимфоцитов. На де-
струкцию и элиминацию патогена лимфоциты «нанимают»
все клетки крови:
• эритроциты (сорбируют растворимые иммунные ком-
плексы с IgM и комплементом и уносят их на расщепле-
ние в синусоиды печени и селезенки);
• нейтрофилы (фагоцитируют иммунные комплексы с IgG
и продуцируют активные формы кислорода и гидролити-
ческие ферменты, а также пептиды-антибиотики и, воз-
можно, другое);
• макрофаги (фагоциты и эффекторы воспаления по типу
ГЗТ — гиперчувствительности замедленного типа);
• эозинофилы (активируются иммунными комплексами с
IgE и IL-5 и продуцируют токсичные протеины гранул —
эозинофильный катионный протеин, миелопероксидазу,
которые способны убить гельминта, — это естественный
механизм противогельминтного иммунитета в норме.
При патологии продукты активированных эозинофилов
вызывают деструктивное воспаление в тканях, например
в стенке бронхов при бронхиальной астме;
• тучные клетки (активируются IgE и антигеном, секрети-
руют биологически активные вещества, обеспечивающие
быстрое развитие в норме защитных сосудистых реакций
и реакций миоконстрикции, например, в желудочно-ки-
шечном тракте. При патологии те же медиаторы тучных
5—1385
65
клеток обеспечивают развитие деструктивных аллерги-
ческих воспалительных реакций;
• базофилы (содержат те же медиаторы, что и тучные клет-
ки, только анатомически локализованы в крови, тогда
как тучные клетки — в соединительной ткани).
Иммунологическое распознавание — это физическое свя-
зывание (нековалентное, не катализируемое ферментами,
комплементарное за счет ван-дер-ваальсовых водородных гид-
рофобных и ионных связей) неопределенно большого числа
разнообразных молекул-антигенов (как своих, так и чужерод-
ных) с антигенспецифичными распознающими рецепторами
лимфоцитов. Иммунологическое распознавание — уникальное
свойство лимфоцитов, которого нет ни у каких других клеток
в организме многоклеточных. Оно возникает в результате осо-
бой и молекулярно весьма «трудоемкой» дифференцировки
лимфоцитов в процессе лимфопоэза. В результате этой диф-
ференцировки на наружной мембране лимфоцита экспресси-
руется рецептор для антигена. Кроме этого рецептора, в боль-
шинстве лимфоцитов экспрессируется еще некоторое опреде-
ленное количество молекул на мембране, предназначенных
для определенных межклеточных взаимодействий (молекулы
адгезии клеток и корецепторы).
В распознавших антиген лимфоцитах еще экспрессируют-
ся определенные гены цитокинов, предназначенных для орга-
низации определенных межклеточных коммуникаций. На осу-
ществление второй половины иммунного ответа — деструкции
и элиминации — большинство специализированных субпопу-
ляций лимфоцитов уже себя «не тратят» (исключение — толь-
ко цитотоксические лимфоциты-эффекторы). Распознав анти-
ген и синтезировав определенные цитокины, лимфоцит «нани-
мает» (посредством цитокинов) для деструкции и элимина-
ции антигена лейкоциты крови, известные как классические
клетки-эффекторы общей воспалительной реакции. Лейкоци-
ты имеют каждый свой специализированный тип протеолити-
ческих, окислительных, цитотоксических, вазоактивных био-
логически активных веществ, локализованных внутриклеточ-
но или секретируемых, но имеющих общее предназначение —
отторгнуть, выбросить из организма, расщепить. Деструктив-
ные, окислительные, цитотоксические, вазоактивные меха-
низмы макрофагов, эозинофилов, базофилов, тучных клеток
неспецифичны по антигену, т.е. одни и те же в случае любого
антигена. Но лимфоцит, распознавший антиген, собирает в
нужном месте, активирует и направляет на свой антиген де-
структивные биохимические механизмы общевоспалительных
белых клеток крови.
Отсюда ясно видна роль иммунной системы в развитии па-
тологических процессов. Иммунный ответ по самой своей
66
сути, по цели направлен на деструкцию и элиминацию патоге-
на из организма. Но патогены, попав во внутреннюю среду, не
являются патогенами, если они не проникли внутрь клетки
или не связались с межклеточным матриксом. Поэтому разру-
шить патоген — всегда означает разрушить собственную клет-
ку или молекулы межклеточного матрикса. Целостный орга-
низм не чувствует боли от какого-либо иммунного ответа
только в тех случаях, когда количество разрушаемого и убирае-
мого антигена невелико и работа воспалительных лейкоцитов-
эффекторов остается под порогом чувствительности анализа-
торов нервной системы. Если же количество антигена больше
порогового, особенно если антиген самовоспроизводящий
(микроорганизмы, гельминты), то деструктивная фаза иммун-
ного ответа будет восприниматься с симптомами rubor, tumor,
calor, dolor, т.е. как болезненный процесс. Его локализация,
степень распространенности, продолжительность, качествен-
ные особенности будут определяться качествами и количест-
вом антигена, а также индивидуальными способностями им-
мунной системы особи и связанных с этой системой сосуди-
стой системы и органов детоксикации (печень) и выделения.
В данной лекции мы перечислим конкретные варианты пато-
логических процессов, развивающихся с участием иммунной
системы.
2.4. Анатомия и физиология иммунной системы.
Основные закономерности развития
иммунного ответа*
В тексте мы будем использовать ряд английских терминов
без перевода, местами — транслитерацией по двум причинам:
а) из уважения к лидирующему языку первоисточника факти-
ческой информации (точнее, к работающим в этой области
*В данной главе почти вся фактическая информация относится к
иммунной системе человека. Структура и функции иммунной систе-
мы человека и мыши (главного экспериментального животного в им-
мунологии) в высокой степени сходны. Такое сходство свидетель-
ствует о высокой степени консервативности гомологичных призна-
ков иммунной системы. Этот консерватизм относится не только к
паре человек—мышь. Например, строение и свойства 5 классов им-
муноглобулинов консервативны у всех млекопитающих, что указыва-
ет на то, что молекулы иммуноглобулинов возникли до начала видо-
образования млекопитающих. Такая эволюционная консерватив-
ность в свою очередь указывает на то, что строение и функции этих
молекул эволюционно «выгодны», т.е. приспособительно очень хоро-
ши. Мы не рассматриваем в данной работе немногочисленные из-
вестные несовпадения в устройстве иммунокомпетентных молекул
человека и мыши. Ссылки на эксперименты по генетическому но-
кауту и трансфекции генов относятся только к мышиным моделям.
5
67
людям) и б) для лучшего понимания международных обще-
принятых устойчивых сокращений.
Итак, анатомический синоним иммунной системы — лим-
фоидная система. Однако понять суть функционирования им-
мунной системы можно, только проследив конкретные взаи-
мосвязи лимфоидной системы по крайней мере с системой
клеток крови и кровеносных сосудов. Эти две системы — бли-
жайшие, на которые опирается в своей работе система лимфо-
цитарного иммунитета. В организме взрослого человека при-
мерно 1013 лимфоцитов. Только 0,2—2 % из них в каждый дан-
ный момент времени циркулируют в крови, остальные нахо-
дятся в лимфоидных органах, в неинкапсулированной лимфо-
идной ткани слизистых оболочек и среди клеток иных, нелим-
фоидных тканей организма (например, внутриэпителиальные
лимфоциты слизистых оболочек — IEL — intraepitelial lympho-
cytes).
Иммунная система устроена органно-циркуляторно. Орга-
ны иммунной системы следующие.
Центральные (органы лимфопоэза):
а кроветворный костный мозг (орган гемопоэза и лимфо-
поэза В-2-лимфоцитов);
▲ вилочковая железа (орган лимфопоэза Тоф-лимфоци-
тов);
▲ плевральная полость (В-1-лимфоциты) и слизистая обо-
лочка ЖКТ (Туб-лимфоциты).
Периферические лимфоидные органы (места развития им-
мунного ответа):
▲ лимфатические узлы;
▲ селезенка;
▲ неинкапсулированные лимфоидные образования сли-
зистых оболочек (пейеровы бляшки, аппендикс, минда-
лины и др.);
▲ лимфоидные подсистемы барьерных тканей и органов —
кожи, слизистых оболочек, печени, матки.
2.4.1. Органы лимфопоэза
Кроветворный костный мозг и вилочковая железа являются
органами лимфопоэза, и на этом основании их называют
центральными. В костном мозге поддерживается пул стволо-
вых кроветворных клеток (СКК), из которых дифференциру-
ются все клетки крови, в том числе и все лимфоциты. На
территории костного мозга в условиях микроокружения
именно костномозговых стромальных клеток проходит лим-
фопоэз большей части В-лимфоцитов (В-2-субпопуляции).
68
Аббревиатура «В» — от Bone marrow. Есть еще В-1-субпопу-
ляция В-лимфоцитов. На этих лимфоцитах экспрессируется*
маркерная молекула CD5, поэтому часто в литературе эти
лимфоциты называют CD5 В-клетками. Кроме того, на мем-
бране этих В-лимфоцитов не экспрессируется IgD, хотя экс-
прессируется IgM. Их лимфопоэз отличается от лимфопоэза
общеизвестных В-2-лимфоцитов тем, что клетка-предшест-
венник CD5 В-лимфоцитов еще в эмбриогенезе покидает
костный мозг и физиологическая регенерация CD5 В-лимфо-
цитов поддерживается в течение взрослой жизни в перифе-
рических тканях с преимущественной локализацией в плев-
ральной и брюшной полостях. Функционально В-1-лимфо-
циты отличаются от В-2, о чем будет сказано далее.
На территории вилочковой железы из коммитированной к
тимической дифференцировке клетки — близкому потомку
СКК — проходит дифференцировка более чем половины всех
Т-лимфоцитов («Т» от Thymus-dependent). Раньше полагали,
что дифференцировка всех Т-лимфоцитов происходит в ви-
лочковой железе, пока не открыли Т-лимфоциты с рецепто-
ром уб, большинство которых дифференцируется экстратими-
чески преимущественно в стенке кишечника. В тимусе Туб со-
ставляют менее 0,5 % общего числа тимоцитов.
Лимфоциты — единственные из клеток крови, которые за-
кономерно имеют обязательно двухэтапную дифференциров-
ку: первый этап — лимфопоэз (на территории органов лимфо-
поэза — костного мозга и вилочковой железы), второй этап —
иммуногенез на территории периферических лимфоидных ор-
ганов.
Иммуногенез — развитие иммунного ответа, в результате
которого из зрелого неиммунного лимфоцита образуется клон
иммунных лимфоцитов. Затем этот клон иммунных лимфоци-
тов покидает территорию периферического лимфоидного ор-
гана и через циркуляцию достигает очага повреждения тканей,
где присутствует патоген. Здесь на месте иммунный лимфоцит
продуцирует провоспалительные цитокины, с помощью кото-
рых «нанимает» те или иные лейкоциты общевоспалительного
назначения для деструкции пораженной патогеном ткани. Это
есть эффекторная фаза иммунного ответа.
Существенно, что синтез и экспрессия на мембране анти-
генраспознающего рецептора лимфоцита происходят во время
лимфопоэза, т.е. в отсутствие чужеродных антигенов. В ре-
* Выражение «экспрессия (экспрессируется) в/на клетке» здесь и да-
лее означает выход (выставление) молекулы, рецептора и т.п. из
клетки на наружную мембрану клетки. Применительно к гену (ге-
нам) «экспрессия» означает принятый в молекулярной биологии тер-
мин, выражающий работу гена (транскрипцию и т.д.). — Примеч. ав-
тора.
69
зультате пожизненно идущего лимфопоэза здоровый организм
млекопитающего формирует порядка 1018 клонов Т-лимфоци-
тов и 1016 клонов В-лимфоцитов. Каждый один клон лимфоци-
тов (все клетки клона — дочерние клетки одного лимфоцита,
поделившегося правильным митозом, следовательно, тождест-
венны друг другу) экспрессирует один или два варианта анти-
генсвязывающих рецепторов. По данным последних лет, при-
мерно на 30 % Т-лимфоцитов находят два варианта антиген-
связывающих рецепторов. Этот факт дает возможность понять
в ряде случаев механизмы развития аутодеструктивного им-
мунного воспаления тех или иных тканей в организме.
Уникальное свойство дифференцировки лимфоцитов со-
стоит в неповторимой перестройке ДНК генов антигенраспоз-
нающего рецептора в каждой отдельной дифференцирующейся
клетке. В результате при редкостном внешнем однообразии
малых лимфоцитов разнообразие антиТенсвязывающих рецеп-
торов на их поверхности составляет, по расчетам, Ю16-18 вари-
антов. Таким образом, лимфоциты — «антимикробы» (предна-
значенные для связывания разнообразных микроорганизмов)
внутри консервативно дифференцированных, зримо неэволю-
ционирующих многоклеточных организмов. Такое свойство
не найдено больше ни у одного типа клеток многоклеточных
организмов и даже эукариот вообще.
В органы лимфопоэза заходят недифференцированные
клетки-предшественники. Из этих органов (костного мозга и
вилочковой железы) выходят завершившие лимфопоэз неим-
мунные лимфоциты. В английском языке такие лимфоциты
называют naive or virgin. Эти лимфоциты предназначены для
встречи с антигенами. Один из наиболее стабильных маркеров
клеточной мембраны неиммунных Т-лимфоцитов — молекула
CD45RA (определенная изоформа мембранного фермента ти-
розинфосфатазы). Маркер Т-лимфоцитов памяти (иммунных,
но вышедших из состояния активации) — CD45RO (другая
изоформа трансмембранного фермента тирозинфосфатазы).
В краткой форме лимфопоэз Т-лимфоцитов в вилочковой же-
лезе заключается в следующей последовательности событий.
1. Самая ранняя коммитированная клетка-предшествен-
ник, пришедшая из костного мозга, пролиферирует некоторое
число раз в корковом слое. Особенным свойством лимфоид-
ных клеток-предшественников является сохранение в них
уникального репарирующего хромосомы фермента стволовой
кроветворной клетки — теломеразы. Это указывает на то, что
лимфоциту предназначено еще не раз пройти циклы пролифе-
рации в течение жизни организма. Клетки-предшественники,
коммитированные к другим росткам кроветворения, утрачива-
ют способность синтезировать теломеразу.
2. На мембране экспрессируется одна из полипептидных
цепей молекулы CD3-8.
70
3. Происходит синтез информационной РНК для сурро-
гатной легкой цепи пре-рецептора — рТа. Экспрессируется
много CD25 и CD44 на мембране. Экспрессируется ген конце-
вой дезоксирибонуклеотидилтрансферазы — TdT.
4. Начинается перестройка ДНК генов TcR, в первую оче-
редь гена p-цепи. Эта перестройка идет под контролем опреде-
ленных генов — Rag-1 и Rag-2 (rearrangement-activating gene 1
и 2). После экспрессии p-цепи происходит коэкспрессия
одновременно CD4 и CD8 (тимоциты на этой стадии называ-
ют двойными позитивными; в вилочковой железе молодого
организма таких клеток порядка 80 %).
5. Происходят перестройка a-цепи TcR и экспрессия
цельного гетеродимерного рецептора TcRap.
6. Тимоциты со сформировавшимся TcR вступают в ак-
тивное взаимодействие с молекулами МНС I или II класса,
укомплектованными эндогенными пептидными антигенами на
клеточной мембране дендритных и эпителиальных клеток ви-
лочковой железы, и проходят процессы позитивной и негатив-
ной селекции. Позитивная селекция состоит в гибели (по ме-
ханизму апоптоза) тимоцитов, не связавших своим TcR ника-
кого антигена. Позитивная селекция уносит порядка 80 % ти-
моцитов. Негативная селекция состоит в индукции апоптоза
таких тимоцитов, которые «слишком сильно связали» свой ли-
ганд в вилочковой железе. Негативная селекция уносит около
15—19 % тимоцитов. В результате завершают лимфопоэз и вы-
ходят в периферические лимфоидные органы всего 1 % или
менее тимоцитов.
В зависимости от того, с каким комплексом:пептид-МНС-1
или пептид МНС-II — на антигенпредставляющих клетках
тимуса (эпителиальные, дендритные, макрофаги) связался
TCR тимоцита, на клеточной мембране и в геноме тимоцита
закрепляется экспрессия только одной из двух ключевых коре-
цепторных молекул: либо CD4 (которая комплементарно свя-
зывается с собственными МНС-П), либо CD8 (которая ком-
плементарно связывается с собственными МНС-I). Соответст-
венно из CD4'-тимоцита в будущем в периферических тканях
получится какой-либо из Т4-лимфоцитов (ТЫ, 2 ... или ЦТЛ).
Из СП8+-тимоцитов получится цитотоксический Т-лимфоцит
(ЦТЛ). Только таким тимоцитам в норме суждено мигриро-
вать из тимуса в периферические ткани и там ждать встречи со
своим антигеном, тождественным или подобным тому «учеб-
ному» своему пептиду, с которым этот тимоцит контактировал
в период лимфопоэза в тимусе. После встречи с антигеном в
периферических тканях начнется второй этап дифференци-
ровки Т-лимфоцита — иммуногенез. В лимфоидной системе
выделены специальные места, предназначенные для встречи
лимфоцитов с антигенами. Для экзогенных антигенов, попа-
дающих во внутреннюю среду через покровные барьерные
71
ткани, такие места — регионарные лимфатические узлы и не-
инкапсулированная лимфоидная ткань слизистых оболочек.
Для антигенов, попадающих во внутреннюю среду через
кровь, это селезенка.
Интенсивность рециркуляции лимфоцитов через систем-
ное кровяное русло высока: время одного цикла рециркуля-
ции — около 1 ч; например, через 1 лимфатический узел за
сутки проходит примерно 25-109 лимфоцитов. Чтобы лимфо-
цит попал в определенный лимфоидный орган или в опреде-
ленную нелимфоидную ткань, на клеточной мембране лимфо-
цита экспрессируются определенные молекулы, которые на-
зываются homing-рецепторы (от английского home — дом).
Например, homing-рецептор лимфоцитов, необходимый для
их перемещения в лимфатический узел, — это известная, оха-
рактеризованная мембранная молекула, имеющая название
L-селектин (L-selectin). А на эндотелиальных клетках постка-
пиллярных венул того органа, где нужен этот лимфоцит, экс-
прессируются вполне определенные молекулы, комплементар-
ные определенному homing-рецептору. Эти молекулы на эндо-
телии сосудов называют адрессинами (adressins). Адрессин для
L-селектина — молекула GlyCAM-1 (от glycan-bearing cell
adhesion molecule 1). Миграция лимфоцитов из крови в пери-
ферическую лимфоидную ткань происходит преимущественно
через кубовидный (или высокий) эндотелий посткапиллярных
венул HEV (high endotelial venules). Эти эндотелиальные клет-
ки формируются не в результате конститутивной дифферен-
цировки сосуда, а под воздействием цитокинов, продуцируе-
мых Т-лимфоцитами. Антигены на территорию лимфатичес-
ких узлов приносят в себе дендритные клетки из покровных
тканей по афферентным лимфатическим сосудам. Через эф-
ферентный лимфатический сосуд лимфоциты уходят из лим-
фоидного органа в системную циркуляцию — в кровь, из
крови — снова в ткани (круг замкнулся).
2.4.2. Характеристика лимфоцитов
Клетки иммунной системы, их дифференцировка, меха-
низмы функционирования, взаимодействия, гуморальные
факторы иммунитета и взаимосвязи иммунной системы с
нервной, гормональной, сосудистой и др. в настоящее время
изучены весьма детально. Объем конкретной фактической
информации по многим позициям столь значителен, что за-
ведомо не укладывается в рамки данной лекции. Поэтому мы
приведем краткую «паспортную» справку о составе «иммуно-
логической материи», т.е. о том, какие существуют субпо-
пуляции лимфоцитов и какие гуморальные факторы имму-
нитета.
72
Морфологически единообразные малые лимфоциты отли-
чаются друг от друга следующими свойствами:
1) специфичностью антигенсвязывающего рецептора (это
клон лимфоцитов);
2) функциями в иммунном ответе (это функциональная
субпопуляция лимфоцитов).
Функциональная специализация лимфоцитов коррелирует
с тремя типами клеточных белков:
• с определенными молекулами на наружной клеточной
мембране (в плане корреляции с функцией мембранные
белки называют маркерами, в англоязычной литературе
часто антигенами с тем или иным наименованием); в
последние 20 лет для обозначения функционально зна-
чимых молекул клеточной мембраны в иммунологии
принята международная номенклатура CD (от cluster
definition/differentiation);
• с определенными молекулами продуцируемых цитоки-
нов;
• с определенными молекулами, синтезируемыми лимфо-
цитом для секреции из клетки и не являющимися цито-
кинами (это, например, иммуноглобулины В-лимфоци-
тов и перфорин ЦТЛ и NK).
Строго специфичным элементом дифференцировки лим-
фоцита (любого), отличающим лимфоцит от всех остальных
клеток организма без исключения, являются только антиген-
связывающие рецепторы. Молекулы клеточной мембраны, ха-
рактерные для лимфоцитов, представлены в организме и на
клетках каких-то других направлений дифференцировки, но
в иных количествах, иных сочетаниях и иных взаимосвязях.
Еще более выразительная интерференция дифференцировки
лимфоцитов с нелимфоидными клетками отмечается у ци-
токинов. Всего в настоящее время выделено, охарактеризова-
но и молекулярно клонировано более 80 цитокинов. Термин
cytokine в данном случае точно выражает смысл: любой ци-
токин — это молекула, вырабатываемая какими-то клетками
в организме и предназначенная для оказания определенного
воздействия на другую(ие) клетку(и). На клетках-мишенях
для цитокина имеются специфические рецепторы (с опре-
деленным спектром перекрестных связей). Большинство ци-
токинов (за исключением 4 из них) действуют в микроок-
ружении, т.е. узко локально в тканях в месте продукции. Те
же цитокины, которые синтезируют дифференцированные
лимфоциты, вырабатывают и клетки за пределами иммунной
системы. Цитокины лимфоцитов в свою очередь воздейст-
вуют как на лимфоциты, так и на клетки также за предела-
ми этой системы, чем обеспечивается биохимическая вза-
имосвязь иммунной системы с остальной плотью и кровью
73
организма. Но беспорядка в продукции цитокинов при этом
в здоровом организме нет. Каждый тип дифференцирован-
ных лимфоцитов экспрессирует гены определенных цитоки-
нов, т.е. продуцирует строго определенный набор цитокинов
и в определенной последовательности во времени под влия-
нием тех или иных регуляторных воздействий извне на лим-
фоцит. Внешние регуляторные биохимические воздействия
(цитокины, гормоны эндокринных желез, нейромедиаторы)
на лимфоцит исходят как от других лимфоцитов, так и от не-
лимфоидных клеток — дендритных, макрофагов, клеток эн-
дотелия сосудов, кератиноцитов в коже (на IEL эпидермиса
и дермы), эпителия ЖКТ (на IEL в ЖКТ) со стороны нерв-
ной и эндокринной систем.
У млекопитающих есть два больших типа лимфоцитов,
различающихся по характеру клеточной дифференцировки:
Т (от «тимусзависимые») и В (от «bone marrow dependent»).
В-лимфоциты специализируются на синтезе иммуноглобули-
нов — секреторных белков с антигенсвязывающими свойства-
ми. Рецептор В-лимфоцитов для антигена называется BcR (от
В-cell receptor).
Собственно антигенсвязывающая часть BcR является мо-
лекулой иммуноглобулина. Но в состав рецептора, кроме им-
муноглобулина, входят еще 4 долипептидные цепи — по 2 с
двух сторон от молекулы иммуноглобулина. Эти мембранные
молекулы обозначают Iga и Igp, и их назначение — проведе-
ние сигнала о связывании антигена собственно иммуноглобу-
линовым компонентом рецептора внутрь клетки.
Дефинитивная стадия дифференцировки В-лимфоцита —
плазматическая клетка. Дифференцировка В-2-лимфоцитов
характеризуется экспрессией генов иммуноглобулинов того
или иного из 5 классов, а также некоторого числа мембранных
CD-молекул, характерных для В-лимфоцитов вообще. Под-
черкнем функциональное значение рецепторов к белкам сис-
темы комплемента — CD19, CD21, CD23, CD35. Их экспрес-
сия на В-лимфоцитах строго необходима для организации от-
вета В-лимфоцита на антиген антителопродукцией. Knockout
генов этих рецепторов приводит к неспособности В-лимфоци-
тов продуцировать антитела.
Среди В-лимфоцитов выделяют 2 субпопуляции, которые
отличаются одна от другой местом прохождения лимфопоэза
и функционально. Эти субпопуляции называют В-1 (CD5+) и
В-2 (общеизвестные). Кроме того, В-лимфоциты различаются
по степени зрелости и классу секретируемых иммуноглобули-
нов, а тем самым и по функциональному вкладу в иммунный
ответ, так как каждый класс иммуноглобулинов имеет особые
и определенные функции. Как и всякий лимфоцит, В-лимфо-
циты делят на неиммунные (никогда не контактировавшие со
своим антигеном) и иммунные (после связывания со своим
74
антигеном). CD5 В-лимфоциты, дифференцирующиеся вне
костного мозга, характеризуются тем, что они продуцируют
иммуноглобулины без взаимодействия с Т-лимфоцитами. Их
иммуноглобулины почти всегда класса IgM. Принципиальная
особенность этих иммуноглобулинов — широкая перекрестная
реактивность, причем их антигены — бактериальные поли-
сахариды. Могут ли CD5 В-лимфоциты распознавать бел-
ковые антигены, — еше вопрос. По биологическому смыслу
CD5 В-лимфоциты аналогичны Т-лимфоцитам с рецептором
TcRyS.
Т-лимфоциты. Лимфопоэз в тимусе заканчивается образо-
ванием двух больших субпопуляций Т-лимфоцитов — CD8+ (в
будущем ЦТЛ) и CD4+ (в будущем Thl, Th2, Th ... или ЦТЛ) и
по крайней мере одной минорной — CD4“, CD8-. Все без ис-
ключения Т-лимфоциты имеют на клеточной мембране ре-
цептор Т-лимфоцитов для антигена, который называют TcR
(от T-cell receptor). Он состоит из 8 полипептидных цепей. Две
из них центрально расположены (это гетеродимеры аир или
у и 8, причем именно они вступают в ионные, водородные,
ван-дер-ваальсовы и гидрофобные взаимодействия с антиге-
нами). Кроме антигенсвязывающего гетеродимера, в состав
TCR входит комплекс трансмембранных полипептидов CD3,
состоящий из 3 типов цепей (обозначаемых как е, у и 8). Эти
цепи по две (е + 8 и у + е) примыкают к антигенсвязывающим
цепям аир или у и 8 TCR, связаны с последними внутри мем-
браны ионными связями и служат для проведения сигнала
внутрь клетки. С трансмембранной частью антигенсвязываю-
щих цепей TcR связаны еще две одинаковые £-цепи, также
уходящие внутрь клетки и обеспечивающие проведение сиг-
нала.
Гены, кодирующие молекулы 1g, и гены, кодирующие мо-
лекулы TcR, имеют одно уникальное свойство, не только от-
личающее их от других генов других клеток в пределах орга-
низма, но свойство, уникальное по сравнению со всеми извест-
ными генами всех известных позвоночных. Это свойство — ре-
комбинация ДНК в соматических клетках. Единственный тип
соматических клеток, в которых происходит такое явление, —
лимфоциты. Только два типа молекул белков синтезируются
на перестроенных генах — это TcR и 1g. На всех Т-лимфоцитах
TcR имеют единую четвертичную молекулярную структуру.
Дифференцировка Т-лимфоцитов изучена весьма подробно.
Описание молекулярно-клеточных параметров этой диффе-
ренцировки занимает более 2 печатных листов текста. Поэто-
му в данной работе мы с сожалением опустим это описание и
констатируем как факт характеристики функционально разли-
чающихся субпопуляций Т-лимфоцитов по малому числу оп-
ределяющих параметров: составу TcR, мембранным молеку-
лам CD4, CD8, CD45R и основным Т-клеточным цитокинам.
75
Т-лимфоциты-«хелперы» — CD4+TcRa0 (ThO, ТЫ, Th2,
Th3), а также CD4+ ЦТЛ. Молекула CD4 экспрессирована на
субпопуляции Т-лимфоцитов с рецептором TcRaP, получив-
шей более 25 лет назад название Т-хелперов (Th). К слову ска-
зать, среди нелимфоидных клеток молекула CD4 экспрессиру-
ется, например, на нейронах, макрофагах, эозинофилах.
Гетерогенность CD4+ Т-лимфоцитов в периферических
лимфоидных тканях была показана 10 лет назад (публикации
1989 г.) Mossman и Koffman в работах по клонированию
Т4-лимфоцитов мышей in vitro. Проанализировав свойства
десятков клонов, авторы увидели, что клоны можно сгруппи-
ровать всего в два типа, различающихся между собой по про-
дуцируемым цитокинам. Один тип, названный ТЫ, — это
продуценты гамма-интерферона и IL-2. Второй тип, назван-
ный Th2, — это продуценты IL-4, IL-5, IL-6, IL-10. Была
сформулирована парадигма об «иммунном отклонении», в
которой говорилось, что в процессе развития иммунного от-
вета из исходно одного типа зрелых неиммунных лимфоци-
тов CD4+ ThO получаются либо ТЫ, либо Th2. Их функции
различны в соответствии с так называемым профилем проду-
цируемых цитокинов. ТЫ, продуценты гамма-интерферона,
посредством этого интерферона активируют макрофаги, ко-
торые создают очаг воспаления, известный как ГЗТ. Th2 по-
средством IL-4 переключают биосинтез иммуноглобулинов в
В-лимфоцитах на IgE, что инициирует цепь физиологических
или патологических процессов с вовлечением тучных клеток.
IL-5 как продукт Th2 активирует эозинофилы и соответст-
венно способствует развитию эозинофильных воспалитель-
ных процессов в тканях. Практическим врачам идея о двух
типах Т4-лимфоцитов весьма понравилась своей простотой:
гиперфункция ТЫ — клинические случаи ГЗТ, гиперфунк-
ция Th2 — клинические случаи реакций гиперчувствитель-
ности немедленного типа. Но, как показывают последующие
эксперименты, такой простоты, хоть и красивой, какую на-
блюдали Mossman и Koffman на клонах лимфоцитов мыши in
vitro, не существует у той же мыши in vivo, а у человека и in
vitro этого нет. Приведем известные опубликованные дан-
ные, ставящие под сомнение парадигму Thl/Th2, по крайней
мере в столь простом ее виде.
1. У мышей in vivo нокаут генов y-IFN или IL-4 не приво-
дит к нарушениям функций, приписываемых Th2.
2. Нокаут гена y-IFN у мышей линии NOD не защищает
от развития диабета (ТЫ-патогенез).
3. При экспериментальном аутоиммунном энцефаломие-
лите (ЕАЕ) (Th 1-патогенез) y-IFN предотвращает развитие
клиники, а антитела к y-IFN усиливают патологию.
4. У мышей с дефицитом IL-4 нарушены Th-1 — опосре-
дованные цитотоксические процессы.
76
У человека в терминальных стадиях дифференцировки им-
мунных Т4-лимфоцитов, вероятно, наступает узкая специали-
зация по продукции 1—2 биологически активных веществ
(БАВ), т.е. на стадии эффекторных процессов у человека суб-
популяций Т4-лимфоцитов не две, а много. Кроме того, в ра-
ботах последних 3 лет по клонированию in vitro Т-лимфоцитов
человека среди Т-лимфоцитов из кожи больных с синдромом
Лайела значительный процент среди прочих Т-лимфоцитов
составляют СВ4+-цитотоксические Т-лимфоциты с перфо-
рин-гранзимным механизмом действия. Раньше полагали, что
перфорин-гранзимные ЦТЛ — это всегда CD8+ Т-лимфоциты.
Было время, когда CD8+ Т-лимфоциты называли
ЦТЛ/супрессоры. Однако в последние 10 лет понятие о Т-суп-
рессорах как особой субпопуляции Т-лимфоцитов, развиваю-
щейся в ходе лимфопоэза, перестало существовать. Супрессию
иммунного ответа сейчас понимают как процесс, в который
вовлечено несколько действующих факторов. Во-первых, это
элиминация патогена из организма, т.е. первопричины, ини-
циирующей иммунный ответ. Нет причины — нет ответа.
Ранее возбужденные к иммунному ответу лимфоциты ликви-
дируются несколькими способами. Во-первых, с помощью
глюкокортикоидов, физиологические концентрации которых
индуцируют апоптоз активированных лимфоцитов. Во-вто-
рых, Th3 (стадия развития иммунных Т4-лимфоцитов, харак-
теризующаяся высокой продукцией антипролиферативного в
отношении лимфоцитов цитокина — TGFp) ингибируют про-
лиферацию (следовательно, прогрессию иммунного ответа) и
Т-, и В-лимфоцитов. В-третьих, на активированных в режиме
иммунного ответа лимфоцитах увеличивается экспрессия ре-
цепторов к факторам некроза опухолей (TNF), FasL, CD30 и
других мембранных рецепторов, связывание которых с их ли-
гандами инициирует в лимфоците-носителе рецептора апо-
птоз.
CD4+ Т-лимфоциты любой функциональной специализа-
ции объединяет одно общее свойство, касающееся особеннос-
ти их антигенрас познающего рецептора — TcR. Тс Rap в ком-
плексе с мембранной молекулой CD4 способен связывать (т.е.
распознавать) только пептидные антигены и только в том слу-
чае, если эти пептиды связаны в комплекс с молекулами МНС
II класса (МНС-II) на поверхности клеток своего организ-
ма. Сама молекула CD4 взаимодействует с молекулами МНС-П.
Можно сказать, что на CD4+ Т-лимфоцитах дееспособен ре-
цепторный комплекс такого состава: {CD4+ TcRap + CD3}.
Компоненты по отдельности не распознают антиген и не за-
пускают дифференцировку лимфоцита. Молекулы МНС-П
экспрессируются на мембране только определенных клеток в
организме: дендритных клеток, В-лимфоцитов, макрофагов,
эндотелии сосудов. Эти клетки имеют групповое название
77
антигенпредставляющих клеток (АРС — от antigen presentin
cells). CD4+ Т-лимфоциты способны «увидеть» своим рецепто-
ром пептидный антиген только на клеточной мембране на-
званных АРС и в том случае, если определенная группа АРС
предварительно поглотила антиген из внешней среды и под-
вергла его частичному ферментативному расщеплению и
конъюгации со своими клеточными молекулами МНС-П. До-
лимфоцитарная обработка антигена называется процессингом
антигена. Никто ни разу не описал наблюдения, чтобы CD4+
Т-лимфоцит распознал (связял своим TCRa0) свободный
антиген.
«Профессиональными» антигенпредставляющими клетка-
ми являются клетки, экспрессирующие МНС-П наряду с
МНС-I, а также определенный набор корецепторных молекул,
связывание которых с комплементарными молекулами-лиган-
дами на лимфоците необходимо и достаточно для инициации
развития иммунного ответа. Таких клеток у млекопитающих
всего три типа:
• дендритные — единственные с конститутивной экспрес-
сией МНС-П;
• В-лимфоциты;
• макрофаги.
МНС-П есть еще на клетках эндотелия сосудов, но на них
в норме нет костимуляторных молекул. Самыми эффективны-
ми и исключительно профессиональными (т.е. без других
функций) АРС являются дендритные клетки. Это единствен-
ные клетки, способные представить антиген в первичном им-
мунном ответе. Гистогенетически дендритные клетки проис-
ходят из той же стволовой кроветворной, причем, вероятно, из
общего предшественника лимфоцитов (общего для Т, В и
НК). Дендритные клетки не только обслуживают иммунный
ответ на экзогенные антигены. Например, в вилочковой желе-
зе, вероятно, именно дендритные клетки предоставляют эндо-
генные петиды дифференцирующимся Т-лимфоцитам таким
образом, что формируется тот самый репертуар антигенрас-
познающих рецепторов Т-лимфоцитов, с которым только и
живет конкретный организм в течение своего онтогенеза.
Дендритные клетки — клетки, которые первыми связыва-
ют все вещества, проникающие через барьер кожи и слизис-
тых оболочек. Они не фагоциты. Дендритные клетки способ-
ны прочно фиксировать антигены на своей клеточной мем-
бране, выполнять неглубокий эндоцитоз и частичное фермен-
тативное расщепление антигена. Загрузившись антигеном,
дендритные клетки мигрируют по лимфодренажу. Они тропны
к лимфатическим узлам, куда и несут антиген. В лимфатичес-
ком узле эти дендритные клетки имеют гистологический вид
интердигитальных дендритных клеток. Именно эти клетки
78
предоставляют антиген для распознавания лимфоцитам. На
дендритных клетках за короткое время, пока они мигрируют
из барьерных тканей в лимфоидные органы, экспрессируются
в большом количестве молекулы МНС-П и костимуляторные
мембранные молекулы В7.
В-лимфоциты в роли АРС характеризуются тем, что они
способны «уловить» своим иммуноглобулиновым рецептором
низкие дозы растворимых антигенов (за счет высокой аффин-
ности антигенраспознающих рецепторов по определению).
Очевидно, что В-лимфоциты заметно проявляют себя при по-
вторных попаданиях антигена в организм (надо, чтобы при
первичном ответе размножился соответствующий клон В-лим-
фоцитов).
Макрофаги не представляют антиген в первичном иммун-
ном ответе. Они способны выполнять функции АРС только в
ранее иммунизированном организме. Это объясняется тем,
что функция АРС для макрофага вторична по отношению к
его санитарным обязанностям. К последним относится фаго-
цитоз комплексов антиген—антитело, которые макрофаг эф-
фективно «ловит» своим рецептором к Fc-фрагменту имму-
ноглобулинов с целью поглотить и расщепить, т.е. необходи-
мо, чтобы антитела к антигену уже выработались в предыду-
щем цикле иммунного ответа. Фрагменты расщепленного
антигена из поглощенного комплекса макрофаг экспонирует
на клеточной мембране в комплексе с МНС-П и, таким обра-
зом, работает как АРС для следующего цикла иммунного отве-
та. Экспрессия комплексов МНС-П — пептид на эндотелии
вносит свой вклад в обеспечение регионарной локализации
процессов иммунного воспаления в месте проникновения
антигена в ткани.
За открытие факта, что Т-лимфоциты (с рецептором ар)
способны распознавать только пептидные антигены и только
при условии, что эти пептиды укомплектованы в соединение с
собственными молекулами клеточной мембраны, называемы-
ми алогично, но исторически обусловленно антигенами глав-
ного комплекса гистосовместимости (МНС — major histocom-
patibilyity complex I или II классов), Peter C. Doherty и Rolf M.
Zinkemagel получили в 1996 г. Нобелевскую премию по фи-
зиологии и медицине. Первые публикации этих авторов были
сделаны в 1974 г. Первая экспериментальная работа, в кото-
рой наблюдали этот феномен, но не могли еще интерпретиро-
вать так, как можем мы сейчас, принадлежит Peter A. Gorer и
относится к 1936 г. Теперь мы также знаем, что это явление
обязательного «двойного распознавания» — антигена + МНС —
закономерно только для Т-лимфоцитов с рецептором TcRap,
на которых экспрессирована одна из двух молекул — CD4 или
CD8. Эти Т-лимфоциты предназначены для распознавания
пептидных антигенов: они самые точные, т.е. высокоспеци-
79
фичные по индивидуальным антигенам. Иначе, без «двойного
распознавания», связывают антигены Т-лимфоциты с рецеп-
тором TcRyS, а также субпопуляция Tap/CD4~ CD8~. Эти лим-
фоциты могут работать как с белковыми антигенами (причем
непроцессированными), так и с гликолипидами и полифос-
фатами.
Цитотоксические Т-лимфоциты — CD8+TcRa0. CD8+
Т-лимфоциты с рецептором TcRaP работают с антигеном по
иной программе, а именно: молекула CD8 на клеточной мем-
бране Т-лимфоцита предназначена для взаимодействия (свя-
зывания) с молекулами МНС-I на поверхности клеток, несу-
щих антиген, т.е. CD8+TcRaP Т-лимфоциты также способны
распознать свой антиген, если только антиген уже связан с
МНС, но не II, а I класса на клетках своего организма. МНС-1
экспрессируются на всех клетках, кроме безъядерных эритро-
цитов. Но это не значит, что антигенпредставляющими клет-
ками для CD8+TcRap Т-лимфоцитов может стать любая клет-
ка организма, если ее молекулы МНС-I связали некий анти-
ген, для которого среди CD8+ Т-лимфоцитов нашелся компле-
ментарный TcR. Чтобы быть АРС, мало нести на своей по-
верхности антиген в комплексе с МНС. Облигатно необходи-
мо наличие костимуляторных молекул, которых на любых
клетках нет, а есть они только на профессиональных АРС.
Молекулы МНС-I способны связывать надлежащим образом
пептидные фрагменты (антигена) в цитозоле клетки. В цито-
золе присутствуют преимущественно белки, которые и синте-
зировались внутри данной клетки. Такими белками, которые
должны «интересовать» защитную иммунную систему, явля-
ются белки вирусов, бактерий и простейших, инфицировав-
ших клетки организма (т.е. внутриклеточные инфекции).
CDS^TcRaP Т-лимфоциты предназначены для дифферен-
цировки в цитотоксические лимфоциты-эффекторы (ЦТЛ,
или CTL, — cytotoxic T-lymphocytes). ЦТЛ сами, непосредст-
венно своим клеточным телом, «работают» над деструкцией,
разрушением клетки-мишени, на мембране которой «просту-
пили» инфекционные антигены в комплексах с молекулами
МНС-I. Вот для связывания и выполнения функций иммун-
ных лимфоцитов-эффекторов костимуляторные молекулы уже
не нужны. Механизм деструкции клеток-мишеней состоит в
индукции апоптоза с участием особых ферментов — гранзи-
мов, которые лимфоцит «впрыскивает» в клетку-мишень через
поры, сформированные в мембране мишеней с помощью пер-
форина лимфоцитов.
Как видим, Т-лимфоциты с TcRa(3, CD4+ и CD8+ способ-
ны «работать» с патогеном, только если его «поднесли» им
другие клетки «на блюдечке с нужной каемочкой». Такая при-
хотливость страхует организм от слишком легкой индукции
именно иммунного ответа и вслед за ним иммунного воспале-
80
ния, ибо это — самое жесткое по механизмам воспаление,
легко переходящее грань между поддержанием здоровья с по-
мощью иммунного ответа и патологическими процессами с
компонентой иммунного воспаления.
ТуЗ-лимфоциты. Т-лимфоциты с рецептором TcRyS сущест-
венно отличаются от Т-лимфоцитов с рецептором TcRap сво-
ими антигенраспознавательными способностями. ТуЗ нужны
организму на барьерах (они и локализуются в слизистых обо-
лочках и коже) для быстрого связывания внешних антигенов,
когда нет времени для предварительного процессинга в АРС.
TcRyS так устроен, что он не нуждается в предоставлении
антигена MHC-I/II, способен сам связывать непроцессиро-
ванные антигены, в том числе небелковые. Но зато степень
специфичности TcRyS существенно меньше, чем у TcRap:
один и тот же лимфоцит с одним и тем же TcRyS способен
связывать довольно широкий набор антигенов. ТуЗ специали-
зируются на инфекционных и пищевых антигенах, эволюци-
онно характерных для класса млекопитающих. Функциональ-
ные свойства лимфоцитов ТуЗ изучены меньше, чем других
лимфоцитов (они открыты менее 10 лет назад). По тем дан-
ным, которые есть, одни ТуЗ могут работать как ЦТЛ, дру-
гие — как ТЫ, третьи — как ТЬ2. Замечено, что именно ТуЗ
адоптивно переносят состояние толерантности к перорально
вводимым антигенам на экспериментальных моделях.
Нормальные киллеры (NK). NK дифференцируются из
общей клетки-предшественника всех лимфоцитов, т.е. той же,
из которой развиваются Т- и В-лимфоциты. Функционально
они являются цитотоксическими клетками-киллерами и, как
и все лимфоциты, продуцентами цитокинов, в частности у-ин-
терферона. Молекулярные механизмы собственно цитолиза
клеток-мишеней тождественны у NK и у ЦТЛ. Но на NK не
выявлены антигенраспознающие рецепторы ни TcR, ни 1g-
типов. В настоящее время известны две субпопуляции НК.
Одна из них циркулирует преимущественно в крови и являет-
ся эффектором антителозависимой клеточной цитотоксичнос-
ти (АЗКЦТ). На клетку-мишень этих NK наводят антитела,
специфичные к мембранным антигенам клетки-мишени: сна-
чала антитела связываются с антигеном на клетке, а затем к
этому комплексу антиген+антитело прикрепляется клетка NK
через имеющийся у NK низкоаффинный (низко-, а не высо-
коаффинный, чтобы не связывать свободные иммуноглобули-
ны) рецептор к Fc-фрагменту IgG (этот рецептор обозначают
FcyRIII, а в CD-номенклатуре эта же молекула значится как
CD16 и часто используется как маркер NK).
АЗКЦТ с антителами класса IgE или IgA развивают эози-
нофилы. Вторая субпопуляция NK локализована в печени и в
слизистой оболочке матки и выполняет функции супрессо-
ров — убивает лимфоциты, активированные на пищевые анти-
6—1385
81
гены (в печени) или на аллоантигены плода (в матке). От
Т-лимфоцитов NK отличаются тем, что контакты со своими
клетками с нормальным уровнем экспрессии антигенов МНС
ингибируют активность NK, но при контактах со своими клет-
ками со сниженным уровнем экспрессии МНС их активность
возрастает.
Т-лимфоциты CD4 CD8 TcRa[3+. Такие лимфоциты выявле-
ны в периферических тканях сравнительно недавно. В отличие
от обычных Tap-лимфоцитов, которые либо CD4+, либо CD8+ и
распознают только пептидные антигены, укомплектованные
либо в МНС-П (для CD4+), либо в МНС-I (для CD8+), двойные
негативные периферические Tap-лимфоциты предназначены
для распознавания непептидных антигенов. Они способны свя-
зывать липогликаны и соединения миколевой кислоты (харак-
терные соединения микобактерий), укомплектованные неполи-
морфной МНС-подобной молекулой CD1 на поверхности анти-
генпредставляющих (или инфицированных) клеток.
2.5. Гуморальные факторы иммунитета
Гуморальные факторы в иммунной системе, имеющие
лимфоцитарное происхождение, делятся на два типа: антите-
ла (иммуноглобулины) и цитокины. Главная гуморальная сис-
тема, «нанимаемая» лимфоцитами и их продуктами для реали-
зации эффекторных (деструктивных — цитотоксических и
протеолитических) стадий иммунного ответа, — это система
белков комплемента сыворотки крови. Молекулярная «красо-
та» иммуноглобулинов и белков комплемента, а также труд ис-
следователей, весьма подробно изучивших названные объек-
ты, стоят подробного описания. Но по объему оно превосхо-
дит рамки данной лекции. Мы его опустим. Напомним, что у
человека есть 9 вариантов (изотипов) молекул иммуноглобу-
линов: IgD, IgM, IgGl, IgG2, IgG3, IgG4, IgAl, IgA2, IgE.
Более подробно приведем, однако, перечень цитокинов, по-
скольку 90-е годы стали принципиально новым этапом в
нашем познании цитокинов. В настоящее время цитокин по-
лучает право на самостоятельное название, если описан(ы)
биологический(е) феномен(ы), клонирован ген, получен ре-
комбинантный биологически активный препарат цитокина,
на моделях трансгенных мышей и мышей с нокаутом гена дан-
ного цитокина показана его роль в целостном организме in
vivo.
Посредством цитокинов лимфоциты и сами работают (как
над собой, так и в межклеточных взаимодействиях), и «вплете-
ны» в целостный организм. Это происходит потому, что, во-
первых, клетки других тканей (строма кроветворных и лимфо-
идных органов, кератиноциты, эндотелий сосудов, нелимфо-
82
идные клетки крови и т.д ) вырабатывают лимфотропные ци-
токины и, во-вторых, цитокины вообще не строго специфич-
ны для лимфоцитов, т.е. те же самые цитокиновые гены экс-
прессируются и в нелимфоидных клетках: какие и где — изу-
чено весьма конкретно и детально. Цитокины перекидывают
один из самых больших «мостов» между системой иммунитета
и всем остальным организмом.
По функции цитокины делят на 4 группы.
1. Цитокины доиммунного воспаления. Их выработку стиму-
лируют непосредственно инфекционные микроорганизмы в
моноцитах/макрофагах, кератиноцитах кожи, возможно, в
других клетках барьерных тканей. К этой группе относят ин-
терферон-а, интерферон-p, провоспалительные цитокины —
TNF, IL-1, IL-6, хемокины, IL-12.
2. Лимфоцитарные цитокины (главные клетки-продуцен-
ты — Т-лимфоциты) — регуляторы активации, пролиферации
и дифференцировки лимфоцитов. В норме главным сигналом
для выработки этих цитокинов является связывание TcR со
специфическим антигеном (т.е. иммунологическое распозна-
вание). К этой группе относят IL-2, IL-4, TGFp.
IL-2 — самый эффективный стимулятор митозов Т- и
В-лимфоцитов. IL-4 индуцирует дифференцировку Th2; в
В-лимфоцитах он индуцирует переключение синтеза иммуног-
лобулинов на классы IgE и IgGl.
TGFp (transforming grows factor p — трансформирующий
фактор роста p — самый эффективный антипролиферативный
цитокин для лимфоцитов.
3. Цитокины — регуляторы иммунного воспаления. Их выра-
батывают главным образом иммунные лимфоциты (но не
только). Они предназначены для активации функции клеток
общевоспалительного назначения, которых лимфоциты «на-
нимают» для деструкции и элиминации антигена, т.е. нейтро-
филов, макрофагов, эозинофилов, NK.
К этой группе цитокинов относят интерферон-у (IFNy),
LT, IL-10, IL-5, IL-12.
IFNy — самый эффективный стимулятор активности мак-
рофагов. Кроме того, этот интерферон стимулирует актив-
ность нейтрофилов, NK, эндотелия сосудов. Он является ко-
фактором дифференцировки CD4+ Т-лимфоцитов в ТЫ, в
В-лимфоцитах IFNy индуцирует переключение синтеза имму-
ноглобулинов на классы IgG2 и IgG3 и ингибирует переклю-
чение на IgGl и IgE. Этот цитокин индуцирует дифференци-
ровку CD8+ Т-лимфоцитов в ЦТЛ. При этом он супрессирует
дифференцировку ТЬ2-лимфоцитов и как следствие — эффек-
торные реакции эозинофилов.
LT (lymphotoxin) — эффективный активатор функций ней-
трофилов; активирует эндотелий сосудов, способствуя адгезии
и экстравазации лейкоцитов.
6*
83
IL-10 продуцируется Th2, а также некоторыми активи-
рованными В-лимфоцитами, макрофагами, кератиноцитами.
Его эффекты — ингибиция продукции цитокинов макрофага-
ми (TNF, IL-1, IL-12, хемокинов), ингибиция экспрессии
генов МНС-П и в итоге — ингибиция воспаления, зависящего
от Т-лимфоцитов. IL-10 стимулирует дифференцировку
В-лимфоцитов, индуцирует переключение синтеза иммуног-
лобулинов на класс IgG4.
IL-5 продуцируют ТЬ2 и активированные тучные клетки.
Его главные функции — стимуляция дифференцировки и ак-
тивизации эозинофилов. Кроме того, он стимулирует проли-
ферацию В-лимфоцитов и, вероятно, индуцирует переключе-
ние синтеза иммуноглобулинов на класс IgA.
IL-12 продуцируют активированные макрофаги и В-лим-
фоциты. Этот цитокин является самым сильным стимулято-
ром NK, причем он не только активирует цитолитическую
функцию, но и индуцирует пролиферацию. Кроме того, IL-12
стимулирует дифференцировку CD4+ Т-лимфоцитов в ТЫ и
дифференцировку CD8+ в зрелые ЦТЛ.
4. Цитокины — факторы роста. Цитокины этой группы
стимулируют пролиферацию и дифференцировку клеток-
предшественников лейкоцитов в костном мозге. Продуциру-
ются они локально клетками стромы костного мозга (адипо-
цитами, фибробластами, эндотелием). К этой группе относят
C-kit-лиганд, IL-3, GM-CSF, M-CSF, IL-7, IL-9, IL-11.
C-kit-лиганд (он же называется stem cell factor) — цитокин,
связывающийся с рецептором c-kit на мембране стволовой кро-
ветворной клетки и тем поддерживающий ее пролиферацию и
готовность к восприятию дифференцирующих сигналов от дру-
гих факторов (CSFs). Knockout гена c-kit-лиганда детален.
IL-3 — мультиростковый колониестимулирующий фактор.
Его продуцируют CD4+ Т-лимфоциты обоих типов (ТЫ и
Th2). Он поддерживает пролиферацию ранних клеток-предше-
ственников всех ростков лейкоцитов в костном мозге. C-kit и
IL-3 стимулируют дифференцировку тучных клеток.
GM-CSF (гранулоцит/макрофаг-колониестимулирующий
фактор) продуцируется активированными Т-лимфоцитами,
активированными моноцитами, эндотелием и фибробластами.
У человека этот цитокин стимулирует пролиферацию не толь-
ко предшественников лейкоцитов, но и предшественников
эритроцитов и мегакариоцитов. Кроме того, этот цитокин ак-
тивирует зрелые лейкоциты.
M-CSF (моноцит-колониестимулирующий фактор) проду-
цируется макрофагами, клетками эндотелия и фибробластами.
Он действует на предшественники макрофагов, более зрелые,
чем те, на которые действует GM-CSF.
G-CSF (гранулоцит-колониестимулирующий фактор) дей-
ствует на клетки-предшественники гранулоцитов после GM-
84
CSF, стимулируя их пролиферацию и дифференцировку в
костном мозге. Этот цитокин в отличие от остальных в данной
группе может действовать дистантно из очага воспаления, что
объясняет механизм усиления созревания и выхода из костно-
го мозга нейтрофилов при наличии очага воспаления в пери-
ферических тканях.
IL-7 продуцируется клетками стромы костного мозга и
действует на развивающиеся клетки—потомки общей лимфо-
идной клетки-предшественника, поддерживая их пролифера-
цию и дифференцировку.
IL-9 поддерживает пролиферацию клеток-предшественни-
ков тучных клеток в костном мозге. В опытах in vitro этот ци-
токин поддерживает пролиферацию ряда линий Т-лимфоци-
тов. Его эффекты на Т-лимфоциты in vivo неизвестны.
IL-11 продуцируется клетками стромы костного мозга и
стимулирует пролиферацию и дифференцировку мегакарио-
цитов.
В системной циркуляции можно выявить только 4 цитоки-
на, а именно TNF, IL-1, IL-6 и G-CSF, но не в норме, а при
тяжелых септических процессах. Другие цитокины в норме не
выходят в циркуляцию и действуют лишь локально в микроо-
кружении по месту выработки. Общим свойством цитокинов
является то, что они не вырабатываются клетками «про запас»,
т.е. не депонируются в клетках в виде молекул-предшествен-
ников. Их синтез активизируется импульсно, по сигналу и на-
чинается с транскрипции тРНК цитокинов с соответствую-
щего гена. Такой характер биосинтеза называют brief, self-lim-
ited, т.е. краткосрочный и самоограничивающийся. Информа-
ционные РНК цитокинов нестабильны, короткоживущи. Про-
дукция их клеткой носит транзиторный характер. Вместе взя-
тые, эти свойства объясняют общее предназначение цитоки-
нов: их высочайшая биологическая активность проявляется
оперативно по сигналу в том месте, где нужно (согласно сиг-
налу), и не дольше, чем нужно. Естественно, что цитокин ока-
зывает эффект исключительно на клетки, на мембране кото-
рых экспрессирован рецептор, способный связать данный ци-
токин. Рецепторы к цитокинам делят на молекулярные семей-
ства с известными вариантами и степенью авидности связыва-
ния с цитокином.
2.6. Стадии развития иммунного ответа
Ограниченные размеры статьи не позволяют нам привес-
ти описание разных вариантов развития иммунного ответа в
зависимости от природы антигена, дозы, входных ворот во
внутреннюю среду организма, кратности введения антигена.
Все эти факторы обязательно отражаются на конкретном им-
85
мунном ответе. Мы же приведем ключевые стадии развития
абстрактного иммунного ответа на такой антиген, который
макроорганизм по генетическим причинам может процесси-
ровать, конъюгировать со своими молекулами МНС, на ко-
торый в организме есть специфические TcR и BcR. Этот
антиген попадает в организм впервые через барьерные ткани
(например, через кожу) и по качеству представляет собой
природный микроорганизм. Стадии развития иммунного от-
вета таковы.
Антиген проникает через барьерные ткани. -> Часть анти-
гена фиксируют АРС; антиген стимулирует выработку цитоки-
нов клетками барьерных тканей, которые готовят сосуды
(формируется очаг доиммунного воспаления) -> дендритные
клетки, поглотившие антиген в барьерных тканях, мигрируют
по путям лимфодренажа в тимусзависимые зоны перифери-
ческих лимфоидных органов, где представляют антиген для
распознавания Т-лимфоцитам. Сюда же из крови приходят
В-лимфоциты. -> Лимфоциты распознают антиген TcR и
BcR. -> Т- и В-лимфоциты взаимодействуют. -> Лимфоциты
пролиферируют. -> Додифференцировка лимфоцитов (имму-
ногенез). -> Иммунные лимфоциты мигрируют через кровь в
очаг повреждения в ткани, где организуют деструкцию клеток,
поврежденных антигеном, лейкоцитами общевоспалительного
назначения (макрофагами, эозинофилами, базофилами, туч-
ными клетками, нейтрофилами) или делают это сами (ЦТЛ)
(формируется очаг иммунного воспаления). -> Разрушенный
антиген выводится из организма.
Подробнее процесс первичного иммунного ответа можно
описать следующим образом.
1. Антиген связывают дендритные клетки барьерной ткани
(клетки Лангерганса в коже) и с тканевой жидкостью мигри-
руют в регионарный лимфатический узел. По «дороге» про-
цессируют антиген до пептидных фрагментов и конъюгируют
эти пептиды со своими молекулами МНС-П. Одновременно
антиген непосредственно стимулирует другие клетки кожи, а
именно кератиноциты и макрофаги, и те в минимальных, но
значимых количествах продуцируют хемокины, TNF, которые
активируют эндотелий в месте внедрения антигена, индуциру-
ют на нем экспрессию молекул адгезии для лимфоцитов и
лейкоцитов (очаг доиммунного воспаления).
2. В лимфатическом узле дендритные клетки представляют
антиген для распознавания Т-лимфоцитам. Сюда же через
кровь попадают В-лимфоциты.
3. Клоны Т- и В-лимфоцитов, связавшие антиген, взаимо-
действуют друг с другом, быстро индуцируют синтез собст-
венных факторов роста (IL-2) и вступают в обязательную
пролиферацию.
86
4. Размножившиеся лимфоциты антигенраспознавших
клонов дифференцируются согласно их специализации, полу-
ченной в лимфопоэзе: CD4+ — в тот или иной тип Т-хелперов
(ТЫ, Th2 или ЦТЛ), CD8+ — в ЦТЛ, В — в антителопродуцен-
ты. Дифференцированные антигенспецифичные лимфоциты
мигрируют из лимфатического узла в ткани к месту внедрения
антигена согласно экспрессии на них нужных молекул адге-
зии, распознающих очаг доиммунного воспаления в ткани.
В-лимфоциты секретируют антитела в кровь с целью перехва-
та всосавшегося системно антигена.
5. На месте наибольшей концентрации антигена активи-
рованные антигенспецифичные лимфоциты рекрутируют
(«нанимают») своими цитокинами тот или иной тип клеток
общевоспалительного назначения, способных осуществить де-
струкцию ткани, с которой связан антиген с целью выведения
из организма (нейтрофилы, макрофаги, эозинофилы, тучные
клетки, базофилы).
6. В случае элиминации антигена из организма иммунный
ответ на него затихает до нуля по следующим причинам:
а) исчез антиген как стимулирующий сигнал № 1;
б) из активированных CD4* Т-лимфоцитов развивается
субпопуляция ТЬЗ, продуцирующих в повышенном количест-
ве среди прочих цитокинов супрессорный цитокин TGFp, ко-
торый останавливает пролиферацию лимфоцитов и продук-
цию ими провоспалительных цитокинов;
в) активированные лимфоциты погибают по механизму
апоптоза; индуцируют этот апоптоз физиологические кон-
центрации глюкокортикоидных гормонов.
7. В части прореагировавших на антиген Т-лимфоцитов
формируются молекулярные механизмы защиты от апоптоза.
Эти клетки выходят из состояния активации, в них выключа-
ется биосинтез цитокинов, но при этом они не погибают, а
циркулируют по организму длительное время. Эти лимфоциты
получили название клеток иммунологической памяти. При
повторном попадании одноименного антигена в организм его
встретят уже существенно более многочисленные и дифферен-
цированные представители антигенспецифичного клона. На
этом свойстве лимфоцитов основана медицинская практика
вакцинации.
Обратим внимание на три процесса в иммунном ответе —
пролиферацию лимфоцитов, взаимодействие клеток в иммун-
ном ответе и апоптоз.
Пролиферация лимфоцитов. Пролиферация в периферичес-
ких тканях — уникальное свойство лимфоцитов, отличающее
лимфоцит от всех без исключения других лейкоцитов. Другие
лейкоциты пролиферируют только на территории гемопоэти-
ческой ткани (в костном мозге у взрослых) и в норме не про-
лиферируют на периферии. Антигенспецифичный лимфоцит
87
же принципиально не может выполнить свои функции, не
пройдя значительного числа циклов пролиферации именно на
периферии, где лимфоцит распознает свой антиген. Поэтому
пролиферация — специфичный признак иммунного ответа.
Если при исследовании какого-то физиологического или па-
тологического процесса с участием клеток крови и лимфоид-
ной системы не регистрируется пролиферация, то это какой
угодно процесс, только не иммунный ответ. Обязательность
пролиферации для лимфоцитов после того, как они распозна-
ли антиген, объясняется тем, что всего во взрослом организме
более 109 вариантов разнообразных рецепторов для антигена,
т.е. более 109 вариантов клонов лимфоцитов. Так как общее
число лимфоцитов в организме человека порядка 10'3, частота
встречаемости лимфоцитов одного клона в здоровом организ-
ме порядка 1 из 10 000. Поэтому, чтобы обеспечить количест-
венно адекватный ответ на случайно попавший в организм
антиген, соответствующий клон лимфоцитов должен проли-
ферировать пропорционально и с запасом относительно дозы
антигена.
Взаимодействие клеток в иммунном ответе. Много лет по-
лагали, что в иммунном ответе Т-лимфоциты-хелперы играют
руководящую и управляющую роль, а В-лимфоциты — испол-
нители-антителопродуценты. Современные исследования по-
казывают, что взаимодействия клеток в иммунном ответе
более равноправны. Описывать многообразие всех известных
на сегодня межклеточных взаимодействий в иммунном ответе
в данной статье мы не имеем возможности, поэтому перечис-
лим главные.
1. Взаимодействие Т-лимфоцита с антигеном на антиген-
представляющей клетке. При распознавании антигена Т-лим-
фоцитом должны реализоваться следующие парные взаимо-
действия между молекулами мембраны лимфоцита и молеку-
лами мембраны АРС: TcR с комплексом антигена с МНС,
CD4 с МНС-П или CD8 с МНС-I и так называемые костиму-
ляторные взаимодействия (CD28 с В7, LFA-1 с ICAM, CD2 с
LFA-3 и т.д.). Только после этого могут начаться пролифера-
ция Т-лимфоцитов и секреция ими про пролиферативных ци-
токинов.
2. Взаимодействие В- и Т-лимфоцитов. Чтобы Т-лимфоцит
начал синтезировать цитокины, управляющие дифференци-
ровкой В-лимфоцитов (переключением классов иммуноглобу-
линов), необходимо взаимодействие такой молекулы мембра-
ны Т-лимфоцита, которая называется CD40L (СЕ)40-лиганд),
с молекулой CD40 на мембране В-лимфоцита, распознавшего
антиген. Иными словами, чтобы Т-лимфоцит начал управлять
дифференцировкой В-лимфоцита, сначала должна поступить
«заявка» от В-лимфоцита, которая является для Т-лимфоцита
сигналом на выработку цитокинов.
88
3. Взаимодействие лимфоцитов с эндотелием сосудов и с
клетками периферических тканей. Без знания конкретных вза-
имодействий между молекулами клеточной адгезии в настоя-
щее время не понять ни нормального хода развития иммунно-
го ответа, ни патогенеза той или иной патологии в тканях. По-
чему, например, у одних пациентов аллергия немедленного
типа выражается в виде крапивницы, у других — в виде брон-
хиальной астмы, у.третьих — в виде отека Квинке? Этиологи-
ческих факторов в патогенезе каждого из названных заболева-
ний больше, чем один, но тканевая локализация может зави-
сеть от аномалии именно в молекулах адгезии. Их выделено и
охарактеризовано в настоящее время значительное число: ин-
тегрины (удерживают клетки друг около друга в нужном
месте), хемокины (обеспечивают передвижение клеток в очаг
по молекулам межклеточного матрикса), селектины (homing-
рецепторы на лимфоцитах и лейкоцитах), адрессины (молеку-
лы-лиганды для селектинов на эндотелии в ткани).
Апоптоз. Апоптоз — это запрограммированная гибель клет-
ки (PCD — programmed cell death). Биологическое явление за-
программированной клеточной смерти было открыто более
20 лет назад зоологами при исследовании стадий развития
круглых червей. По мере изучения иммунной системы выяс-
нилось, что у млекопитающих апоптоз наиболее присущ
именно лимфоцитам и, более того, является одним из основ-
ных механизмов работы иммунной системы, альтернативным
пролиферации. Выделяют всего два варианта смерти клеток —
некроз и апоптоз. Некроз — травматическая гибель клетки. На
уровне ткани некроз сопровождается воспалительным процес-
сом. Апоптоз — гибель клетки «изнутри». Сигнал на запуск
апоптоза может прийти как снаружи клетки (для этого есть
специальные рецепторы на мембране, например Fas и др.), так
и из генома, но в обоих случаях собственно механизм гибели
клетки осуществляется определенными ферментами, специ-
ально для этого предназначенными и синтезированными
самой клеткой. Цитологически апоптоз проявляется фрагмен-
тацией ДНК на куски размером 200—300 пар нуклеотидов и
постепенной фрагментацией ядра и цитоплазмы, т.е. всей
клетки. При этом внутриклеточное содержимое даже фрагмен-
тов остается окруженным мембраной, не вытекает в межкле-
точное пространство и на уровне ткани проходит без призна-
ков воспаления.
В норме апоптозу подвергаются следующие лимфоциты:
• на стадии лимфопоэза — лимфоциты, отвергнутые при
позитивной и негативной селекции (плохо реагирующие
со своими МНС);
• в периферических тканях: а) лимфоциты, уже «отрабо-
тавшие» в иммунном ответе, после завершения элимина-
89
ции антигена; б) лимфоциты, получившие часть, но «не
комплект» всех необходимых для правильной работы
сигналов: апоптозу подвергаются лимфоциты, у которых
TcR или BcR связал антиген, а других необходимых сиг-
налов лимфоцит не получил (связь с МНС, CD40 с
CD40L и т.д.); апоптозу подвергаются лимфоциты, полу-
чившие митогенный сигнал, но не получившие специ-
фического антигена на свой TcR или BcR. Последние
две ситуации укладываются в понятие «индуцированной
активацией клеточной смерти» (activation-induced cell
death - AICD).
Клетка, претерпевающая апоптоз, способна посылать
смертельный сигнал соседним клеткам (клеточное братоубий-
ство — fratricide). Мы обращаем на это в данной статье внима-
ние в том плане, что применение иммуностимулирующих пре-
паратов, неспецифичных по определенному заданному анти-
гену, очевидно, небезопасно. Более того, иммуностимулято-
ром можно убить «нечаянно» неведомое число клонов лимфо-
цитов, предназначенных для каких-то будущих или актуаль-
ных антигенов.
Генетические дефекты в клеточном аппарате апоптоза,
приводящие к недостаточности апоптоза лимфоцитов, приво-
дят к развитию лимфопролиферативных процессов, с высокой
частотой переходящих в злокачественные лимфолейкозы.
Эффекторные механизмы иммунного ответа. Теперь перей-
дем к эффекторным механизмам «доведения до конца» им-
мунного ответа, т.е. к механизмам деструкции и элиминации
распознанных лимфоцитами поврежденных патогеном тканей.
Механизмы деструкции делятся на два типа: 1) антителозави-
симые и 2) Т-лимфоцитзависимые, антителонезависимые.
Опыт показывает, что все антигены — те или иные органи-
ческие вещества: преобладают белки, но имеется и достаточ-
ное количество углеводов, липополисахаридов, полифосфа-
тов, т.е. всего того, из чего состоят инфекционные микроорга-
низмы и естественная пища млекопитающих.
Деструктивные биохимические механизмы происходят от
далекой пищеварительной функции одноклеточных. Поэтому
все эффекторные механизмы иммунного ответа многоклеточ-
ных используют тот или иной аппарат протеолиза и свободно-
радикального окисления. Применительно к иммунному ответу
известны следующие эффекторные процессы.
1. Цитолиз клеток-мишеней непосредственно ЦТЛ (в меж-
клеточном контакте работает белок перфорин, а также индук-
торы апоптоза — сериновые протеазы — каспазы, или гран-
зимы).
2. Антителозависимая клеточная цитотоксичность
(АЗКЦТ) — цитолиз клеток-мишеней клетками-эффекторами
90
только при условии, что с мембраной клетки-мишени связано
специфическое антитело; у клетки-эффектора есть рецептор
к Fc-фрагменту иммуноглобулина соответствующего класса.
С антителами класса IgG в АЗКЦТ клетками-эффекторами
«работают» в первую очередь NK. С антителами IgE и IgA
клетками-эффекторами в АЗКЦТ «работают» эозинофилы.
Молекулярный токсический механизм NK тождествен тако-
вому у ЦТЛ (перфорин и индукция апоптоза). У макрофагов
и нейтрофилов для этого служат их протеазы и ферменты
перекисного окисления, у эозинофилов — токсичные белки
(большой основной протеин — major basic protein, катионный
протеин эозинофилов — eosinophil cationic protein и др.).
3. Комплементзависимый лизис клеточных патогенов (бак-
терий), связанных антителами.
4. Фагоцитоз нейтрофилами и макрофагами растворимых
либо корпускулярных комплексов антиген+антитело и рас-
щепление их в лизосомах или перекисным окислением, или
анаэробными протеазами типа катепсинов до низкомолеку-
лярных катаболитов, которые клетка выбрасывает через свою
мембрану, а из циркуляции их выводят почки, печень, желу-
дочно-кишечный тракт (ЖКТ).
5. Гиперчувствительность замедленного типа. От иммун-
ной системы в данной реакции выступают лимфоциты — про-
дуценты ylFN, т.е. ТЫ или CD8+ ЦТЛ, клетками-исполните-
лями являются активированные макрофаги. Главный цито-
кин — активатор макрофагов у-интерферон, продуцируемый
ТЫ или иммунными CD8+ Т-лимфоцитами.
6. Гиперчувствительность немедленного типа — это совокуп-
ность сосудистых и гладкомышечных реакций, обеспеченных
биологически активными веществами (БАВ) тучных клеток и
базофилов (биогенными аминами, липидными медиаторами,
протеинами гранул). От иммунной системы в данном случае ра-
ботают Th2 или CD4- CD8- Т-лимфоциты и В-лимфоциты, ко-
торые под воздействием IL-4 продуцируют IgE. Клетки-эффек-
торы (базофилы и тучные клетки) связаны с системой лимфоци-
тарного иммунитета таким образом, что на них экспрессирова-
ны высокоаффинные рецепторы к IgE и IgG4 (высокоаффин-
ные — т.е. способные связать свободный иммуноглобулин до
связывания его в комплекс с антигеном). БАВ тучных клеток и
базофилов в отличие от цитокинов в значительном количестве
синтезируются этими клетками «впрок», чтобы по сигналу клет-
ка немедленно выбросила эти вещества в межклеточные про-
странства или/и в кровь. Цель защитных физиологических про-
цессов, вызываемых БАВ (расширение сосудов, быстрая экссу-
дация плазмы из сосудов в ткани, энергичный спазм гладкой
мускулатуры бронхов и кишечника), — попытаться быстро вы-
бросить из организма во внешнюю среду или тампонировать
отеком проникающий в организм патоген.
91
2.7. Иммунная подсистема кожи
Иммунная система кожи состоит из следующих клеток:
• клетки Лангерганса (эпидермальные дендритные клет-
ки);
• внутриэпителиальные лимфоциты (IEL — intraepitelial
lymphocytes);
• периваскулярные лимфоциты в соединительнотканном
слое дермы. Это Т-лимфоциты (как CD4+, так и CD8+).
Большинство из них имеют маркеры активации:
CD45RO и высокий уровень a-цепи рецептора для ин-
терлейкина-2 (CD25).
Процесс иммунного ответа на антигены, проникающие
через кожу, представляется как последовательность следующих
событий. Антиген связывают и поглощают эндоцитозом денд-
ритные клетки. Они мигрируют по афферентным лимфатичес-
ким сосудам в парафолликулярные зоны регионарных лимфати-
ческих узлов (ЛУ). Во время миграции DC частично расщепля-
ют антиген, оформляют его в комплексы с молекулами МНС II
или/и МНС I. В ЛУ DC оформляются гистологически как ин-
тердигитальные дендритные клетки и представляют антиген для
распознавания неиммунным Т-лимфоцитам. Активированные
антигеном Т-лимфоциты мигрируют из лимфатических узлов
через кровь. Те из них, на которых экспрессирована специаль-
ная мембранная молекула, обеспечивающая тропизм к коже
CLA-1 (Cutaneous lymphocyte antigen-1, по химической природе
эта молекула — сиаловое производное карбогидрата; лигандом
для этой молекулы на клетках эндотелия является Е-селектин),
возвращаются в дерму и эпидермис. Среди активированных
Т-лимфоцитов кожи преобладает функциональная субпопуля-
ция Т4 — ЦТЛ с перфорин-гранзимным эффекторным механиз-
мом. Есть также клетки Thl. Их собственная функция после ак-
тивации антигеном состоит в интенсивной продукции опреде-
ленных цитокинов, а именно у-интерферона, IL-2 и т.д. Эти ци-
токины, и в первую очередь у-интерфнгрон, являются сильными
активаторами макрофагов. Макрофаги в достаточно большом
количестве присутствуют в периваскулярных пространствах
дермы. И именно макрофаги, активированные Т-хелперами 1-го
типа, распознавшими антиген, являются исполнительными эф-
фекторными клетками, пытающимися подвергнуть поврежден-
ные патогеном клетки и межклеточный матрикс деструкции и
элиминации. В результате «работы» активированных в коже
макрофагов развивается физиологическая, а при большой дозе
антигена (и/или при нарушениях межклеточного равновесия
или регуляции) — патофизиологическая реакция, которая в на-
чале века получила название кожной реакции гиперчувствитель-
ности замедленного типа (ГЗТ).
92
Еще один эффекторный иммунный механизм защиты
кожи от проникновения внешних антигенов — IgA в димерной
форме, который присутствует во всех экзосекретах кожи
(сальном, потовом, ушной сере).
Эффекторный механизм гиперчувствительности замедленно-
го типа. В эксперименте ГЗТ можно адоптивно перенести от
иммунного животного интактному только с сенсибилизиро-
ванными к определенному антигену CD4+ Т-лимфоцитами,
а именно ТЫ. В меньшей степени, но и иммунные CD8+
Т-лимфоциты способны рекрутировать макрофаги для выпол-
нения ГЗТ. Главный цитокин — медиатор ГЗТ у-интерферон.
Внутрикожным введением препарата у-интерферона можно
имитировать ГЗТ на моделях. Кроме того, в организации реак-
ции ГЗТ участвуют такие цитокины, как IL-2, TNF и LT. Ре-
акция ГЗТ развивается до видимых глазом симптомов за 24—
48 ч после попадания антигена в кожу. В очагах ГЗТ можно
наблюдать следующую гистологическую картину. Через 4—6 ч
в зонах вокруг посткапиллярных венул развивается инфильт-
рация нейтрофилами, которая длится недолго и к 12-му часу
сменяется инфильтратом мононуклеаров — Т-лимфоцитов и
моноцитов. Эндотелий в очаге становится сильно проницае-
мым для белков крови, и происходит значительная экссудация
фибриногена, который уже в периваскулярных тканях поли-
меризуется в фибрин. Именно выпадение фибрина создает то,
что клинически воспринимается как симптом индурации
очага ГЗТ. Высокие локальные концентрации цитокинов, в
том числе IL-2, обеспечивают не только аутокринную актива-
цию пролиферации антигенспецифичных лимфоцитов, но и
паракринную активацию пролиферации «соседних» лимфоци-
тов. Это объясняет тот факт, что более 90 % Т-лимфоцитов в
очаге ГЗТ неспецифичны по причинному антигену. Разреше-
ние процесса (излечение) наступает в результате того, что ак-
тивированные макрофаги справляются с задачей санации
очага от причинного антигена, т.е. фагоцитируют его и рас-
щепляют с помощью механизмов клеточного метаболизма.
Внутриэпидермальные лимфоциты кожи (IEL) составляют
примерно 2 % всех лимфоцитов, присутствующих в коже.
Большинство IEL — CD8+ Т-лимфоциты. Значительная часть
IEL (у мышей большая) — это Ту8.
2.8. Иммунная система слизистых оболочек
Иммунная система слизистых оболочек состоит из следую-
щих компонентов:
• внутриэпителиальные лимфоциты (IEL);
• лимфоциты lamina propria;
93
• лимфоидные фолликулы слизистой оболочки (или неин-
капсулированная лимфоидная ткань слизистых оболо-
чек; в тонкой кишке — это пейеровы бляшки).
Большинство IEL — Т-лимфоциты, причем именно CD8+
Т-лимфоциты. У человека примерно 10 % IEL составляют ТуЗ.
У мышей на долю ТуЗ приходится не менее 50 % всех IEL. На
IEL слизистых оболочек обнаружена особая молекула клеточ-
ной мембраны — интегрин, названный HML-1 (от human mu-
cosal lymphocyte antigen-1), являющийся homing-рецептором
лимфоцитов в эпителий слизистых оболочек. Эпителиальные
клетки слизистой кишечника продуцируют IL-7, который дей-
ствует как фактор роста для IEL. На IEL обнаружен рецептор
для IL-7.
Лимфоциты в lamina propria гетерогенны. Большинство из
них, однако, это активированные CD4+ Т-лимфоциты. Эти
лимфоциты мигрируют в lamina propria из регионарных мезен-
териальных лимфатических узлов, где они были активированы
антигеном и пришли в lamina propria для организации эффек-
торных процессов. В качестве исполнительных клеток-эффек-
торов в lamina propria используются присутствующие здесь в
достаточном количестве эозинофилы, тучные клетки, макро-
фаги. В lamina propria присутствует много активированных
В-лимфоцитов (которые мигрируют сюда из мезентериальных
лимфатических узлов и пейеровых бляшек) и их дифференци-
рованных потомков — плазматических клеток, продуцирую-
щих большие количества иммуноглобулинов.
Неинкапсулированная лимфоидная ткань слизистых обо-
лочек (NLT) (в слизистой тонкого кишечника это в первую
очередь пейеровы бляшки; сюда же включают аппендикс,
лимфоидную ткань глоточного кольца Пирогова и все менее
гистологически оформленные лимфоидные скопления во всех
остальных слизистых оболочках (дыхательной системы, моче-
половой системы). У взрослых 50—70 % клеток пейеровых
бляшек составляют В-лимфоциты, 10—30 % — Т-лимфоциты.
Отсюда очевидна главная функциональная нагрузка пейеро-
вых бляшек — обеспечение иммуногенеза В-лимфоцитов и
дифференцировки их в антителопродуцирующие плазматичес-
кие клетки. Эпителиальные клетки слизистой оболочки, по-
крывающие пейеровы бляшки, не имеют характерных «пище-
варительных» ворсинок. Они называются М-клетками (от
membranous). Их отличает высокий уровень пиноцитоза, по-
средством которого они транспортируют макромолекулы с по-
верхности слизистой в субэпителиальные лимфоидные фолли-
кулы (пейеровы бляшки). Сами М-клетки, вероятно, не вы-
полняют функции антигенпредставляющих клеток, а только
транспортируют антигены. Среди Т-лимфоцитов пейеровых
бляшек существенно больше, чем в иных лимфоидных тканях,
94
содержание Th2, продуцирующих среди своих характерных
цитокинов и IL-5. Этот цитокин — один из трех (второй —
TGF-р, третий — IL-10), которые контролируют переклю-
чение синтеза иммуноглобулинов в дифференцирующемся
В-лимфоците на специальный секреторный класс — IgA.
Переключению на IgA способствует присутствие в месте дей-
ствия дендритных клеток. Продукция IgA в слизистой оболоч-
ке ЖКТ весьма интенсивна. Она составляет до 70 % от общей
ежедневной продукции иммуноглобулинов в организме, и у
взрослого человека это в среднем 3 г новообразованного IgA в
день. Но синтез IgA — далеко не единственный эффекторный
иммунный механизм, препятствующий внедрению во внут-
реннюю среду организма внешних антигенов через барьер
ЖКТ. В ЖКТ задействованы и другие механизмы иммунитета.
Например, больше 90 % синтезируемого IgE секретируется в
просвет кишечника.
ТуЗ связывают микробные и пищевые антигены не только
пептидной, но и полисахаридной, липополисахаридной, фос-
фолипидной природы, причем без предварительного процес-
синга и без представления в комплексе с молекулами МНС
или в связи с неклассическими молекулами МНС, например
с молекулой CD1. Связывание антигена рецептором TCRyS
предположительно имеет следствием, во-первых, физический
перехват антигена на барьере и, во-вторых, либо деструкцию
патогена, либо индукцию толерантности к данному антигену.
Во всяком случае, в эксперименте специфическую толерант-
ность можно адоптивно перенести интактному животному от
животного, получавшего антиген перорально, выделенными
ТуЗ-лимфоцитами. Кроме того, у мышей с эксперименталь-
ным нокаутом генов уЗ не удается индуцировать толерант-
ность пероральным введением антигена. Описаны такие за-
щитные реакции на территории ЖКТ, в которых суть про-
цесса определяют ТЬ2-лимфоциты. Например, при энтераль-
ной инфекции Heligmosomoides polygyrus защитный иммуни-
тет зависит от достаточной активации Th2, выработки ими
IL-4, который избирательно переключает синтез иммуногло-
булинов в В-лимфоцитах на класс IgE. IgE фиксируется на
тучных клетках (ТК). Лейкотриен D4, гистамин и простаглан-
дин Е2, выбрасываемые ТК, обеспечивают надлежащую ин-
тенсивность сокращения гладких мышц кишечника, доста-
точную для выброса из организма возбудителя вовне. Учас-
тие именно вышеперечисленных молекул и клеток в меха-
низме защитного иммунитета к данной кишечной инфекции
доказано в изящных молекулярно-генетических эксперимен-
тах на моделях на мышах (мыши SCID; мыши с мутацией
w/wv, дефицитные по тучным клеткам; мыши, обработанные
антителами к c-kit; мыши с генетическим нокаутом по гену
липоксигеназы и т.д.).
95
2.9. Патологические процессы с участием
иммунной системы
Из приведенного нами краткого обзора устройства нор-
мальной иммунной системы видно, что в ней есть чему «ло-
маться» и весьма много мест для поломки — столько, сколь-
ко генов программируют существование и функционирова-
ние системы иммунитета. Проанализировать все возможные
варианты патогенеза нарушений в иммунной системе или с
ее участием под силу разве еще не написанной никем про-
грамме для компьютера. Отдавая себе в этом отчет, мы рас-
смотрим патологические процессы с участием иммунной
системы более крупным планом, не претендуя в каждом кон-
кретном случае на возможность проследить всю цепь собы-
тий — от клинического симптома до дисфункции определен-
ных генов. Из вышеизложенного видно, что один и тот же
клинический симптом может иметь разные молекулярно-кле-
точные механизмы патогенеза. Например, выброс БАВ из
тучных клеток и соответствующую клиническую симптома-
тику может вызвать не только связывание антигена с антите-
лом IgE на поверхности тучной клетки, но и физический
фактор переохлаждения (холодовые аллергии), психический
стресс, фармакологические лабираторы мембраны тучных
клеток и т.д.
Известно 5 вариантов патологических процессов с участи-
ем иммунных реакций крупными блоками.
I. Собственно иммунная система полноценна.
II. В клетках иммунной системы есть генетические дефек-
ты (первичные иммунодефициты).
III. Истинные аутоиммунные заболевания.
IV. Организм в целом подвергается тяжелому системному
патогенному воздействию (шок любой этиологии,
ВИЧ-инфекция, психический стресс и др.), вторич-
ные иммунодефициты.
V. Аллергические заболевания.
2.9.1. Полноценная иммунная система
Под полноценной иммунной системой понимают, что все
субпопуляции лимфоцитов и лейкоцитов присутствуют в до-
статочных количествах и в правильных пропорциях, не нару-
шен биосинтез иммуноглобулинов, в наличии все цитокины.
Никаким лабораторным анализом невозможно выявить откло-
нений, а человек болеет именно по причине мучительного или
несостоятельного иммунного ответа.
Причины такого положения могут быть следующими.
96
1. Деструктивная фаза любого, в том числе вполне нор-
мального, здорового иммунного ответа — это по сути воспале-
ние того или иного рода. Очевидно, что на уровне эффектор-
ных деструктивных процессов, которыми по смыслу заканчи-
вается любой иммунный ответ, переход нормы в патологию
может происходить по двум причинам:
• количество антигена выше того порога, при котором де-
струкция ткани происходит «незаметно» для анализато-
ров центральной нервной системы организма, и мы
видим rubor, tumor, calor и чувствуем dolor. Передози-
ровки антигенов болезненны для организма при самой
сильной и здоровой иммунной системе. Иначе говоря,
нарушение меры есть отклонение от нормы (патология
по сути);
• по мере прогрессирования первичного заболевания
(чаще всего инфекционного или лимфопролиферативно-
го) в иммунной системе развивается и нарастает наруше-
ние баланса между различными субпопуляциями лимфо-
цитов и цитокинами, фиксируются и прогрессируют по-
рочные круги. Нарушение баланса может быть систем-
ным, а может происходить в той или иной ткани, в тка-
невых иммунных подсистемах (кожа или слизистые обо-
лочки). Системные нарушения иногда можно выявить по
анализу крови (ВИЧ-инфекция, первичные иммуноде-
фициты, лейкозы и радиационные поражения). Иммун-
ный дисбаланс в тканях может никак не проявляться в
анализе периферической крови, имея при этом сильно
выраженную клиническую симптоматику в тканях
(кожа, слизистые оболочки, как, например, при очаго-
вых пародонтитах, и др.).
2. Генетический контроль иммунного ответа обеспечивает-
ся не только генами, кодирующими дифференцировку лимфо-
цитов и их продуктов, но и генетическими системами за пре-
делами собственно иммунной системы. Это надо понимать,
чтобы отдавать себе отчет в принципиальных ограничениях —
эволюционных, филогенетических, популяционных — защит-
ных возможностей иммунитета человека по отношению к ин-
фекциям, пищевым и ингаляционным антигенам. Допустим,
что у какого-то определенного человека гены рецепторов для
антигенов, мембранных молекул, всех цитокинов у лимфоци-
тов и их партнеров лейкоцитов в полном порядке. С любой ли
инфекцией может справиться его иммунная система? Заведо-
мо нет. Во-первых, хотя репертуар TcR и BcR велик, но в нем
может не оказаться TcR и BcR, адекватных антигенам именно
данного микроба. Это вполне реальная ситуация, касающаяся
новых и быстро эволюционирующих возбудителей, к которым
относятся, например, ретровирусы (возбудитель СПИДа из их
7—1385 97
числа). Во-вторых, самая главная генетическая система за пре-
делами иммунной системы, определяющая принципиальную
возможность иммунного ответа, — это система генов главного
комплекса гистосовместимости (название неудачное, но усто-
явшееся) — гены МНС. Если белки МНС (которые, как мы
помним, экспрессируются на всех без исключения клетках ор-
ганизма) по каким-то причинам не в состоянии сформировать
правильный комплекс с антигеном-пептидом, то и при нали-
чии TcR, специфичного к данному антигену, и при полной
гармонии всех белков и цитокинов в лимфоцитах и лейкоци-
тах Т-лимфоцитарного иммунного ответа на данный антиген
быть не может. В каких ситуациях можно столкнуться с этим
на практике? Дело в том, что в отличие от TcR/BcR, разнооб-
разие генов которых достигается перестройкой ДНК в сомати-
ческих клетках (в лимфоцитах), гены МНС каждый человек
наследует, как и все остальные гены, инвариантно, т.е. имеет
только то, что унаследовал от матери и отца. Разнообразие
генов МНС, а значит и диапазон способностей белков МНС
связывать пептидные антигены, достигается накоплением му-
таций (аллелей) в популяции людей. На уровне популяции
отбор идет на приспособление к тем инфекционным микроор-
ганизмам, пищевым и ингаляционным антигенам, которые
присутствуют в среде обитания. Поэтому если человек, предки
которого во многих поколениях жили в определенном регио-
не, переезжает в далекие места, где в биосфере много новых
для МНС данного человека антигенов, то существует вероят-
ность, что он начнет болеть как иммунодефицитная особь,
хотя на прежнем месте жительства не болел. Подобные клини-
ческие ситуации регистрируют у многих групп мигрантов из
отдаленных регионов (например, у строителей Байкало-Амур-
ской магистрали, приехавших из южных регионов). Понятно,
что среди причин заболеваний имеют место и отсутствие
активной иммунизации новыми антигенами, и неприспособ-
ленность нейроэндокринной системы к новому климату, но
только этими факторами не удается полностью объяснить кар-
тину заболеваемости и ее динамику.
Непредставляемость антигенов молекулами МНС проявля-
ется и в случае новых, быстро эволюционирующих инфекций, и
в случаях нагрузки на организм слишком искусственных неоан-
тигенов антропогенного происхождения (химия синтетическая,
химия отходов при сгорании топлива, мусора и т.д., неестест-
венная экология жилища, искусственные пищевые добавки и
т.п.). Вот здесь проявляется несоразмерность материи организма
(МНС/TcR в данном случае) веществам из внешней среды,
внедряющимся во внутреннюю среду организма. Отсутствие
меры — патология по сути. Возможно даже, патологию (как
противоположность нормы) можно и определить как несораз-
мерность нормы реакции генома особи факторам конкретной
98
среды обитания. Если не реализуется природная программа раз-
вития иммунного ответа, то возникают, так сказать, «дисфунк-
ции», т.е. такие варианты реакций иммунной системы, в кото-
рых преобладают не защитные процессы, а повреждение тканей.
Современные врачи наблюдают это у весьма значительного
числа пациентов с аллергиями разного рода. В основе патогене-
за аллергий, которые клинически выглядят как гиперреактив-
ность, может лежать как раз недостаточность (т.е. дефицит) того
звена иммунного ответа, который необходим для уравновешива-
ния гиперреактивности и в норме присутствует у неаллергиков.
2.9.2. Генетические дефекты в иммунной системе
Вспомним формулу: иммунитет = распознавание антигена
(лимфоциты) + деструкция ткани, поврежденной патогеном
(лимфоциты, лейкоциты, комплемент). Следовательно, кли-
нические проявления недостаточности иммунитета будут
иметь место как при дефектах в лимфоцитарном распознава-
нии, так и при дефектах в нелимфоцитарных механизмах эли-
минации антигена, которые используют лимфоциты. Напо-
мним суть: без лимфоцитов, однако при полной сохранности
лейкоцитов и комплемента никакого иммунного ответа не бы-
вает; одни (без лимфоцитов) механизмы доиммунной клеточ-
ной и гуморальной резистентности не справляются с реаль-
ным эволюционно возникшим и непрерывно развивающимся
множеством инфекционных микроорганизмов, гельминтов,
искусственных пищевых и лекарственных веществ.
Мы не будем в данной лекции пытаться проследить, какая
иммунодефицитная патология развивается при дефектах в кон-
кретных генах. Конкретных нозологических единиц первичных
иммунодефицитов насчитывают около 30. Генов, которые
можно при этом подозревать «на дефектность», много, и приве-
дение их полного списка сейчас — не цель нашей работы. Но
зато гены, контролирующие иммунный ответ, — как раз цель
фундаментальных экспериментальных исследований в мире по
генетическому нокауту. Эти работы начаты около 5 лет назад,
они чрезвычайно дорогостоящи, но тем не менее прогрессивно
развиваются, и в ближайшие годы будет получена значительная
фактическая информация о том, как реализуется морфогенез
организма млекопитающего при полном физическом отсутствии
того или иного определенного гена, и что, напротив, происходит
при заведомой гиперфункции того или иного определенного гена
(модель трансгенных мышей, особенно с тканеспецифической
транскрипцией экзогена) в организме мышей (модели генети-
ческого нокаута в настоящее время разработаны только для
мышей). Мы же приведем корреляции между вариантами кли-
нической патологии и дефектами в том или ином конкретном
7
99
иммунологическом механизме в паре с эффекторным механиз-
мом (т.е. дефект может быть либо на уровне лимфоцитов, либо в
эффекторном звене — клинические проявления и лабораторные
исследования в ряде случаев позволяют дифференцировать
место повреждения). Понятно, что у реального больного челове-
ка чаще имеют место сочетанные дефекты, и в клинической
практике нельзя не учитывать наличие патологических процес-
сов одновременно в разных органах и системах.
Соответственно названным компонентам иммунной систе-
мы перечислим те клетки и внеклеточные ферментативные
системы (в данном случае это система комплемента), на кото-
рые опирается тот или иной тип лимфоцитов: лимфоцит как
бы «нанимает» их для разрушения и выведения из организма
распознанного лимфоцитом антигена. Иными словами, назо-
вем эффекторные механизмы иммунитета (табл. 2.1).
Таблица 2.1. Соответствие между субнопуляциями лимфоцитов и
эффекторными механизмами разрушения и выведения антигена
Лимфоцит и его продукт Эффекторный механизм
1. В-лимфоцит, продуцирующий
антитела классов:
IgM Сорбция иммунных комплексов на эритроцитах и транспортировка их на деструкцию в печень и селезенку
IgGl 1. Комплексы Ag+IgGl эффективно фагоцитируют макрофаги (опсонизация). 2. Слабо, но фиксирует комплемент (Clq). 3. Опосредует АЗКЦТ с участием NK и макрофагов
IgG2 1. Слабо, но фиксирует комплемент (Clq). 2. Опосредует АЗКЦТ
IgG3 1. Эффективно активирует комплемент. 2. Комплексы Ag+IgG3 эффективно фагоцитируют макрофаги (опсонизация)
IgG Способен вызвать дегрануляцию базофилов и туч- ных клеток
IgE 1. Сорбция антигенов на дендритных клетках. 2. АЗКЦТ в паре с эозинофилами против гельмин- тов. 3. Сосудистые и миоконстрикторные реакции с участием тучных клеток и базофилов
2. ЦТЛ: Непосредственно сами перфорируют мембрану
CD8+TcRa|3 клетки-мишени и дополнительно индуцируют в
CD8+TcRy3 CD4+ ней апоптоз. В итоге — смерть клетки-мишени
100
Продолжение табл. 2.1
Лимфоцит и его продукт Эффекторный механизм
3. Т-хелперы 1-го типа (ТЫ) 1. Активируют макрофаги, которые развивают ре- акцию по типу ГЗТ. 2. Взаимодействуют с В-лимфоцитами, обеспечи- вая их пролиферацию и переключение класса 1g с IgM на IgG. 3. Как продуценты IFNy поддерживают все зави- сящие от IFNy процессы
4. Т-хелперы 2-го типа (Th2) 1. Продуцируют 1L-4, который индуцирует пере- ключение в В-лимфоцитах синтеза Ig с IgM на IgE и секрецию IgE в просвет кишки при кишечных гельминтозах. Обеспечивают защиту от гельминтов по механизму АЗКЦТ с эффекторами эозино- филами. В патологии вызывают аллергию по типу гиперчувствительности немедленного типа. 2. Продуцируют IL-5, который активирует эозино- филы. Переключают синтез Ig на IgA. IgE и IgA опосредуют АЗКЦТ эозинофилов. Это главный деструктивный защитный механизм против гель- минтных инфекций
Корреляция патологических состояний с дефектами в отдель-
ных звеньях иммунитета. При анализе клинического состояния
конкретного больного и попытки дифференциальной диагнос-
тики иммунопатогенетического звена в общем патологическом
процессе следует не упускать из виду ряд ориентиров.
1. Непосредственное вовлечение иммунной системы в па-
тогенез имеет место только при следующих симптомоком-
плексах:
• инфекционный синдром (вне эпидемических очагов —
рекуррентные и оппортунистические инфекции);
• аллергические болезни (в подавляющем большинстве
случаев);
• аутоиммунные болезни;
• лимфопролиферативные заболевания.
2. Патология иммунного ответа может быть антигенспеци-
фична либо распространяться на большинство антигенов или
на все антигены. Если патология антигенспецифична, то кри-
тический фактор патогенеза локализован в клоне лимфоцитов
или на уровне генов/антигенов МНС. Если очевидна неспеци-
фичность патологии по антигену, то «больное место» либо в
лимфопоэзе каких-то (какой-то) функционально-различных
101
субпопуляций лимфоцитов, либо в эффекторных нелимфоци-
тарных механизмах расщепления антигенов. При антигенспе-
цифичном процессе в большинстве случаев никакой пользы
не может принести лабораторное исследование на «иммунный
статус» вообще, за исключением ситуаций, когда болезнь раз-
рушила и иммунную систему (как, например, при СПИДе).
Нужны другие, строго антигенспецифичные лабораторные ис-
следования преимущественно молекулярно-генетическими
методами.
3. Генетические дефекы могут быть наследственными и
приобретенными в результате случайных мутаций. Наследст-
венные дефекты проявляются, как правило, с детства в так на-
зываемых первичных иммунодефицитных заболеваниях. Гене-
тические дефекты, приобретенные от случайных мутаций, ма-
нифестируют в любом возрасте (когда случилась мутация).
Если мутация происходит в дифференцированном лимфоците,
то она элиминируется вместе с апоптозом данного лимфоци-
та, и никто этого никогда не заметит, если мутация не онко-
генная. Клиническая манифестация генетического иммуноде-
фицита у взрослых происходит, очевидно, только в случаях,
когда мутация случилась в стволовой кроветворной клетке.
В тезисной форме отметим, какие патологические процессы
можно наблюдать при тех или иных дефектах в иммунной сис-
теме, неспецифичных по антигену. Начнем с гуморальных
факторов иммунитета (табл. 2.2).
Таблица 2.2. Клинические проявления различных дефектов гумо-
ральных факторов иммунитета
Дефект гуморального звена имму- нитета (антитела, комплемент) Клинические проявления
Агаммаглобулинемия (полная или по отдельным классам) Рекуррентные бактериальные ин- фекции верхних дыхательных пу- тей, кожи, ЖКТ. Терапия: медика- ментозная антибактериальная, за- местительная — препараты 1g или цельной плазмы периодически по- жизненно
Гипериммуноглобулинемия (необходим дифференциальный иммунохимический анализ классов 1g) Недостаточность компонентов комплемента: Cl, С2, СЗ, С4 В-клеточные лейкозы; плазмоцито- мы; прогредиентные инфекции с наличием у возбудителя суперанти- генов (например, СПИДа) Болезни иммунных комплексов: системный васкулит (системная красная волчанка — СКВ), гломе- рулонефрит; пиогенные инфекции
102
Продолжение табл. 2.2
Дефект гуморального звена имму-
нитета (антитела, комплемент)
Клинические проявления
Недостаточность компонентов
комплемента: С5, С6, С7, С8, а
также пропердина и фактора D
Недостаточность ингибитора
С1 (дизрегуляция системы ком-
племента в сторону повышен-
ной активации)
Недостаточность фактора DAF
(ускорителя распада СЗ) -» уси-
ление активности СЗ конверта-
зы; дефект CD59
Инфекции Neisseria вплоть до дис-
семинированных форм; другие пио-
генные инфекции
Наследственный или приобретен-
ный ангионевротический отек.
Возможны также СКВ, пиогенные
инфекции
Внутрисосудистый гемолиз эритро-
цитов -> пароксизмальная ночная
гемоглобинурия
Антитела связывают антиген. Факта связывания бывает до-
статочно для реализации защитного эффекта только в случаях,
когда свободный антиген — сильный яд и в патогенности анти-
гена на первый план выступает его острая токсичность. Связы-
вание с антителом инактивирует яд. В таком случае антитела на-
зывают нейтрализующими. Кстати, не всякое антитело против
данного антигена проявляет свойства нейтрализации. Комплекс
антиген+антитело — более крупное молекулярное образование,
чем свободный антиген, и циркуляция в крови макромолекуляр-
ных агрегатов является патогенным фактором. Комплексы
антиген+антитело могут выпадать в осадок на стенках сосудов,
инициируя воспалительный процесс (васкулиты). Клиренс
крови от комплексов антиген+антитело осуществляют компле-
мент и эритроциты, а также фагоциты (макрофаги, нейтрофи-
лы). Патогенный избыток комплексов антиген+антитело возни-
кает в случаях, когда в организме имеется перманентный источ-
ник антигена (истинные аутоиммунные болезни, хронические
прогредиентные инфекции) и/или дефект комплемента. Так
случается при системных васкулитах (системная красная вол-
чанка, иммунокомплексный гломерулонефрит и т.п.).
При генетических дефектах мембранолитических компо-
нентов комплемента (С6—С8; дефект С9 клинически не про-
является) у пациентов можно наблюдать разве что хроничес-
кие инфекции, вызванные внутриклеточными микроорганиз-
мами рода Neisseria. Другие внутриклеточные инфекции де-
структивный механизм комплемента «уступил» цитотоксичес-
ким Т-лимфоцитам (ЦТЛ) и NK (АЗКЦТ).
При недостаточности литических внутриклеточных систем
у макрофагов (лизосомальных ферментов, ферментов пере-
103
кисного окисления, NO-синтазы и др.) они фагоцитируют,
сколько могут, иммунные комплексы, но не справляются с их
расщеплением и выведением. Клинически это выглядит как
хроническая гранулематозная болезнь.
Во всех случаях генетических дефектов гуморальных факто-
ров иммунитета лечение может быть только заместительным
(препараты человеческой крови), плюс симптоматическая тера-
пия (антибиотики) и общеукрепляющие методы адаптационной
терапии (диета и т.п.). Врачу следует четко отдавать себе отчет,
что назначение пациенту заместительной терапии препаратами
крови или тканей человека, а в настоящее время и животных
(в связи с появлением прионных инфекций типа губчатого эн-
цефалита, или бешенства коров) означает, что пациент навсегда
попадает в группу риска по СПИДу, прионным инфекциям и тем
кровяным инфекциям, о которых мы пока не знаем. Поэтому
назначение любых препаратов из крови и тканей животных и че-
ловека врач (для большей ответственности — консилиум) имеет
право делать только по абсолютно витальным показаниям.
Дефекты в клеточных реакциях иммунитета проявляются в
различных вариантах патологических процессов (табл. 2.3).
В общем виде следует помнить, что в норме CD8+ Тар-лимфо-
циты (преимущественно по функции ЦТЛ) являются ведущим
эффекторным механизмом санации организма от вирусных
инфекций. CD4+ Тар-лимфоциты предназначены для распо-
знавания антигенов, укомплектованных с МНС-П, к которым,
как показывает опыт, относятся такие инфекционные возбу-
дители, как внутриклеточные бактерии и простейшие, а также
всевозможные растворимые антигены, в том числе из разряда
ксенобиотиков (среди которых много аллергенов). Вспомним,
что ТуЗ «работают» на барьерах и специализируются на бакте-
риальных и пищевых антигенах. Вспомним также, что функ-
циональные субпопуляции ТЫ и Th2 находятся в альтерна-
тивных взаимоотношениях, т.е. активное функционирование
одной подддерживает саму себя (аутокринная стимуляция) и
ингибирует вторую субпопуляцию из названных.
Таблица 2.3. Клинические проявления различных дефектов Т-лим-
фоцитов
Вариант дефекта
Клинические проявления
Дефект ТуЗ
Дефект Thl
Абсцессы с геморрагиями в ЖКТ, вызван-
ные Listeria, Eimeria. У мышей с нокаутом
гена 3 — летальный туберкулез
Недостаточность всех реакций, зависящих
от у-интерферона и IL-2 (снижение ГЗТ,
уменьшение продукции IgG и т.д.)
104
Продолжение табл. 2.3
Вариант дефекта Клинические проявления
Дефект Thl/Th3* с гиперфункцией Th2 Дефект Th2 Аллергические болезни по немедленному типу (опосредованные IgE) Недостаточность противогельминтной за- щиты (-> гельминтозы)
Дефект Th2/Th3* с гиперфункцией ТЫ Аллергические болезни по типу ГЗТ с той или иной органной локализацией. Этот ме- ханизм существен в патогенезе аллергичес- кого энцефаломиелита, синдрома воспален- ного кишечника, некоторых дерматозов
*Под дефектом Th3 понимают недостаточность иммуносупрессорных
цитокинов TGFp и IL-10.
Аллергии. Профессиональные аллергологи называют все
эффекторные механизмы иммунных реакций механизмами ал-
лергического повреждения тканей. С точки зрения иммуноло-
га, эффекторные механизмы иммунитета работают не только у
больных-аллергиков, но и у здоровых людей: пока иммунная
система и организм в целом сбалансированы и нет превыше-
ния меры какого-то антигена во внутренней среде организма,
все эффекторные механизмы «работают» постоянно и непре-
рывно, но без rubor, tumor, color, dolor. Аллергия же — всегда
болезнь. С точки зрения иммунолога аллергией можно назы-
вать патологические процессы, вызванные эффекторными ме-
диаторами (тучных клеток и базофилов при немедленных ре-
акциях, макрофагов при ГЗТ), в том числе и без участия соб-
ственно иммунологических механизмов (TcR, IgE, IgG4) при
иных патогенных этиологических факторах (нейромедиаторы,
лабираторы мембран тучных клеток, физические факторы и
т.д.). Однако аллергологи предпочитают антигеннезависимую
клинику аллергий называть псевдоаллергиями, а аллергиями
называть только антигензависимую клинику. В данном случае
антиген называют аллергеном и аллергию — истинной аллер-
гией. Можно с этим согласиться, но все-таки не называть ал-
лергией замедленного типа, например, отторжение трансплан-
тированной почки. Дело в том, что чужую почку отторгает
всякий несингенный организм в силу нормальной природы
вещей, а аллергия на какой бы то ни было антиген развивается
все-таки у меньшинства людей, даже если это меньшинство к
концу XX в. достигает 20 % населения западных стран. Alios
все-таки иной, т.е. не такой, как все. В патогенезе истинных
аллергий следует искать превышение меры антигена персо-
105
нально для данного индивидуума, нарушение у него баланса
субпопуляций лимфоцитов, дисбаланс цитокинов или/и ре-
цепторов к ним, иммуноглобулинов, молекул межклеточной
адгезии и т.п. Следует отдавать себе отчет в том, что при ал-
лергиях, строго специфичных по аллергену, генетические де-
фекты локализованы в клоне (или небольшом числе клонов)
лимфоцитов и их принципиально нельзя зарегистрировать в
антигеннеспецифичных анализах параметров общего иммун-
ного статуса. Поэтому их таким образом и искать не стоит. Те
достоверные сдвиги в субпопуляциях лимфоцитов, которые
может зарегистрировать анализ антигеннеспецифичного им-
мунного статуса, могут отражать либо антигеннезависимую
патологию, либо колоссальную дозу антигена во внутренней
среде (такую, как при ВИЧ-инфекции и ей подобных).
Классические клинические группы аллергических болез-
ней немногочисленны. К ним относят бронхиальную астму,
аллергический ринит, атопический дерматит, аллергические
реакции на пищевые аллергены и аллергические синдромы на
лекарственные препараты. Принято считать, что в патогенезе
истинных аллергий немедленного типа задействованы гомо-
цитотропные антитела, относящиеся к классу иммуноглобули-
нов Е. Давно известно, что на тучных клетках и базофилах
имеется особый высокоаффинный рецептор для IgE — FceRI.
Такой же FceRI экспрессирован на дендритных клетках. Кон-
станта связывания IgE с этим рецептором максимальна из всех
известных связей иммуноглобулиновых молекул и составляет
порядка 10~|0моль/л. Главное, что с этим рецептором IgE свя-
зываются до их связывания с антигеном (что отличает IgE от
антител, относящихся к иммуноглобулинам других классов, и
их связей с их класс-специфичными FcR). При последующем
попадании растворимого аллергена во внутреннюю среду
организма этот аллерген быстро связывается с IgE, заранее
фиксированным на тучных клетках (в тканях) и базофилах
(в крови). Связывание с аллергеном является сигналом для
быстрого выброса из названных клеток готовых медиаторов
(у человека это в первую очередь биогенный амин гистамин,
у грызунов — серотонин), а также для индукции синтеза de
novo липидных медиаторов метаболитов арахидоновой кисло-
ты: простагландина D2, лейкотриенов С4, D4, Е4 (которые в
совокупности называют медленно реагирующей субстанцией
анафилаксии); В4; тромбоцитактивирующего фактора PAF.
Кроме того, в активированных тучных клетках индуцируется
синтез цитокинов — TNF, IL-1, IL-4, IL-5, IL-6, IL-8, IL-3,
GM-CSF. Активированные тучные клетки секретируют также
особые протеазы — триптазу, химазу, карбоксипептидазу и ка-
тепсин G. Именно медиаторы из тучных клеток и базофилов
обеспечивают развитие патогномоничных симптомов и син-
дромов аллергических реакций немедленного типа и повреж-
106
дения тканей системно или местно: вазодилатацию, сокраще-
ние гладкой мускулатуры бронхов и кишечника, гиперсекре-
цию желез слизистых оболочек, локальный протеолиз. Если
этиологический фактор (аллерген) не элиминируется быстро,
то наступает продолжение «аллергической драмы» в виде реак-
ции поздней фазы, которая объясняется инфлюксом в ткани
нейтрофилов, эозинофилов, базофилов, лимфоцитов, созда-
ющих очаги воспаления.
Как мы уже отмечали, аллергические реакции всегда пато-
логичны. Никому не удалось ни раньше, ни теперь усмотреть в
них защитные компоненты. Поэтому неизбежен вопрос: под
давлением каких природных факторов отбор закрепил IgE в
эволюции, каковы защитные функции IgE? Все ли нам ясно в
клеточных механизмах аллергических реакций? В 60-е годы
была обнаружена ассоциация продукции IgE в организме с
гельминтными инфекциями. В последние годы стало извест-
но, что рецепторы для IgE имеются не только на тучных клет-
ках и базофилах, но и на других клетках. Такой же высокоаф-
финный рецептор FccRl найден, например, на дендритных
клетках (клетках Лангерганса). Выделены два других молеку-
лярных типа рецепторов для IgE, менее аффинных. Один из
них — Fc£RII — обнаружен на тромбоцитах, лимфоцитах, эо-
зинофилах и эпителиальных клетках тонкой кишки (энтеро-
цитах), второй — EBP (epsilon binding protein) — на эозинофи-
лах и энтероцитах. Следовательно, у IgE есть какие-то природ-
ные функции вне поля событий, связанных с аллергиями. Что
это за функции — еще предстоит изучить. Пока известно толь-
ко то, что IgE опосредует реакции антителозависимой клеточ-
ной цитотоксичности против гельминтов, в которых клетка-
ми-эффекторами являются эозинофилы, синтезирующие и
секретирующие в процессе АЗКЦТ токсичные белки — major
basic protein (главный основной протеин) и eosinophil cationic
protein (катионный протеин эозинофилов).
Среди недавно полученных фактов есть довольно неожи-
данные — о закономерностях IgE-ответа на кишечные гель-
минтные инфекции. Например, показано, что, хотя концент-
рация IgE в крови на пике ответа составляет всего 30 мкг/мл,
что заметно меньше концентрации самого малочисленного из
IgG — IgG4 (его концентрация 600—700 мкг/мл), в относи-
тельном выражении прирост IgE составляет минимум два по-
рядка над исходным фоном, что существенно значительнее,
чем для любого из субклассов IgG. Но самое интересное со-
стоит в том, что природное место IgE не в крови, где присутст-
вует менее 1 % всего синтезированного IgE, а в кишечнике.
Более 99 % IgE секретируется энтероцитами в просвет кишки,
где IgE и «работает» против гельминтов.
Зоологам известно, что у позвоночных животных гель-
минтные инфекции являются ведущим фактором, контроли-
107
рующим численность и ареалы распространения популяции.
Поэтому можно думать, что гельминтные инфекции являют-
ся сильным фактором отбора защитных структур и функций
в иммунной системе позвоночных. И человек — вряд ли ис-
ключение. Прослеживается ли какая-либо связь гельминто-
зов с аллергиями у людей? Прослеживается и весьма сильная
обратная корреляция: в популяциях, в которых гельминтоза-
ми поражено практически все детское население, почти не
встречаются аллергии. И наоборот, в популяциях, саниро-
ванных в силу образа жизни от гельминтов, аллергиям под-
вержено 20—40 % представителей популяции. К последним
относятся высокие социальные слои населения развитых за-
падных стран. Конкретные цифры поражают. Например, в
Венесуэле на одном экологическом фоне обследовали группу
богатых и образованных людей в сравнении с группой абори-
генов, ведущих антисанитарный, с цивилизованной точки
зрения, образ жизни. Среди аборигенов гельминтозами пора-
жено более 88 % жителей; средний уровень общего IgE в сы-
воротке у них составил 13 088 МЕ/мл, но при этом аллерги-
ческие болезни выявлены менее чем у 2 %. У богатых и об-
разованных сограждан гельминтозы обнаружены менее чем
у 10 %, средний уровень общего IgE в сыворотке составил
370 МЕ/мл, а аллергические болезни наблюдали у 43 % об-
следованных. Исследование проводили специалисты из США
в течение 5 лет, гипо- или гипердиагностику аллергий пред-
полагать нет оснований. Аналогичная картина выявлена при
сравнении рядом живущих цивилизованных жителей запад-
ной Австралии и аборигенов из Папуа-Новая Гвинея. Среди
западных австралийцев бронхиальная астма выявлена у 7 %
детского населения и 28 % — взрослого. Среди аборигенов
Папуа у детей астмы вообще не нашли, а среди взрослых
этим заболеванием страдали только 0,3 % обследованной по-
пуляции. Эти данные позволяют предположить, что такой
природный фактор, как гельминтозы в детстве, обеспечивают
постнатальный онтогенез иммунной системы, предохраняю-
щий организм в дальнейшей жизни от аллергических «болез-
ней цивилизации».
В экспериментах и клинических работах выявлено, что в
процессе естественного иммунного ответа на гельминтные ин-
фекции активно «работают» внутриэпителиальные ТЬ2-лим-
фоциты слизистой оболочки кишечника. Вырабатываемый
ими цитокин IL-4 обеспечивает локальное (в слизистой обо-
лочке кишки) переключение синтеза иммуноглобулинов
В-лимфоцитами на класс IgE, а также активную экскрецию
этого IgE в просвет кишечника. Возможно, что в отсутствие
природных взаимоотношений с гельминтами формируется
«неправильный», аллергогенный баланс Thl/Th2 субпопуля-
ций лимфоцитов. Конечно, такая гипотеза, как и любая моно-
108
гипотеза, не объясняет патогенез аллергий во всех случаях.
В других молекулярно-генетических исследованиях нашли,
например, что у 1/5 обследованных больных аллергией (всего
было обследовано 20 человек) в молекулах иммуноглобулинов
класса Е присутствуют дополнительные участки молекулы (ве-
роятно, в результате дупликации участков генов) и нарушена в
сторону гиперэкспрессии молекулярная регуляция этих генов.
В контрольной группе здоровых людей такой молекулярной
патологии генов IgE не обнаружено.
Несмотря на неполноту и пробелы в теоретических пред-
ставлениях о молекулярных и клеточных механизмах аллер-
гий, диагностика и лечение аллергических болезней — наибо-
лее разработанная область иммунологии с точки зрения кли-
нических успехов. Аллергии лучших других болезней, развива-
ющихся с участием иммунной системы в патогенезе, контро-
лируются медициной. В общем виде алгоритм лечения аллер-
гических болезней может иметь следующий вид.
1. Если в этиологии есть один (или мало) аллерген экзо-
генного происхождения и его удается выявить, то элиминация
аллергена часто может сама по себе стать радикальным мето-
дом излечения.
2. При том же условии наличия этиологического аллерге-
на, но невозможности полностью элиминировать его из среды
обитания больного есть такой метод лечения, который назы-
вают специфической иммунотерапией — СИТ. Он показан при
IgE-зависимых аллергиях и состоит в осторожной курсовой
иммунизации больного малыми дозами причинного антигена.
Как мы писали выше, наблюдения показывают, что иммуни-
зация малыми дозами способна сдвинуть равновесие в пользу
Thl, хотя механизм явления по сути неизвестен.
3. Для улучшения качества жизни больного в ожидании ра-
дикального излечения или при невозможности радикального
излечения больным назначают глюкокортикостероидные пре-
параты, причем в последние годы предпочитают топические
стероиды (местные), а не системные. Глюкокортикостероиды
ингибируют пролиферацию и активацию лейкоцитов, что объ-
ясняет их противовоспалительный эффект.
4. Патогенетической симптоматической терапией является
назначение антагонистов медиаторов тучных клеток и базофи-
лов (в первую очередь антигистаминных препаратов) и инги-
биторов активации тучных клеток и базофилов (кромогликат
натрия).
5. Другие назначения зависят от конкретной нозологичес-
кой формы заболевания, локализации ведущего патологичес-
кого процесса (при бронхиальной астме — бронходилататоры,
при атопическом дерматите — средства, способствующие эпи-
телизации кожи, при пищевых аллергиях — препараты пище-
варительных ферментов и т.д.).
109
6. В случае системной аллергической реакции немедленно-
го типа — анафилактического шока — применяют cito общую
противошоковую терапию — эпинефрин (адреналин) с целью
быстрой вазоконстрикции и реанимационные меры по пока-
заниям (интубация гортани при тотальном отеке и др.). После
выведения из ургентного состояния проводят интенсивную
симптоматическую терапию.
7. При обширных процессах из разряда острых токсико-ал-
лергических реакций (ОТАР) на медикаменты типа синдрома
Стивенсона—Джонсона (Stevens—Johnson syndrome) или син-
дрома Лайелла (Lylle’s disease) — массивного некролиза кожи
и слизистых оболочек — для спасения жизни больного необ-
ходим специализированный стационар, имеющий стерильные
палатки (как для ожоговых больных) и персонал, обученный
методам интенсивной терапии общего назначения — дезин-
токсикационной, терапии для стабилизации гемодинамики,
профилактики синдрома ДВС (диссеминированного внутри-
сосудистого свертывания), системной противоинфекционной
терапии, «цитоусмиряющей» терапии глюкокортикоидами и
новыми препаратами антицитокинового назначения (цикло-
спорин, или сандиммун). Вышеназванные лечебные меры не
имеют иммунологической специфики. Эта специфика должна
проявиться в осознании и попытке выявления этиологическо-
го фактора — аллергена.
Весьма часто аллергенами при синдромах острого некро-
лиза кожи бывают ксенобиотики — лекарственные препара-
ты. Поэтому при лечении таких больных необходимо прояв-
лять суперосторожность при введении лекарственных пре-
паратов. Иммунопатогенез этих клинических состояний
малоизвестен. В очагах поражения вокруг погибающих кера-
тиноцитов иммуногистохимическими методами выявляют
повышенное содержание Т-лимфоцитов и еще больше моно-
цитов. Клонирование Т-лимфоцитов из очагов поражения
кожи при синдроме Лайелла показало, что большая часть
Т-лимфоцитов — это CD4+ Т-лимфоциты с рецептором ар.
Но удивительно то, что по функциональной дифференциров-
ке эти Т4-лимфоциты оказались перфорин-гранзимными
ЦТЛ. CD8+ ЦТЛ среди клонов тоже были (всего охаракте-
ризовано 400 клонов), но немного. Раньше принято было
считать, что перфорин-гранзимные ЦТЛ — это всегда CD8+
Т-лимфоциты. Клиника развивается в острое состояние за
7—21 сут от начала приема причинного лекарственного пре-
парата. Описывают конкретные клинические случаи, когда
синдром Стивенсона—Джонсона развивается вслед за erythe-
ma multiforme, вызванной вирусом ветряной оспы. Клини-
ческое излечение в таких случаях достигается интенсивным
курсом глюкокортикостероидов (преднизон по 30 мг/день в
течение 5—7 дней).
ПО
2.9.2.1. Патологические процессы с участием иммунной
системы — истинные аутоиммунные заболевания
Аутоиммунным заболеванием следует считать только
такое, при котором лимфоцит, запускающий механизмы де-
струкции, распознает истииио собствеиный(ые) антиген(ы).
В таких случаях тактика лечения в первую очередь должна быть
нацелена на прерывание цепи сигналов от лимфоцита на ме-
ханизмы деструкции. Современные методы лечения аутоим-
мунных болезней крайне грубы, и иммунологи честно сравни-
вают их с лечением шизофрении лоботомией. Суть в том, что
аутоиммунные болезни — проявления нарушений той самой
морфогенетической функции иммунитета, которую мы упоми-
нали в начале статьи. Морфогенез вообще — самое трудное
«место» в живом организме для нашего познания, и патогене-
тические механизмы развития истинных аутоиммунных болез-
ней нам известны лишь минимально. Исследования показыва-
ют, например, что никаких необычных рецепторов для антиге-
нов у лимфоцитов не обнаруживают. Вероятно, дело в том, что
в норме аутораспознающие лимфоциты есть, но они не ини-
циируют и не направляют на свои клетки нелимфоцитарные
механизмы деструкции, а при аутоиммунных заболеваниях по
каким-то причинам начинают направлять. Лечат аутоиммун-
ные болезни в наши дни иммуносупрессивными средствами
системного назначения: глюкокортикоидными гормонами,
сандиммуном (синоним — циклоспорин), т.е. препаратом, до-
вольно избирательно ингибирующим биосинтез цитокинов в
Т-лимфоцитах, но во всех клонах без разбору с соответствую-
щими побочными эффектами, иногда иммунодепрессорами из
арсенала трансплантологов и онкологов.
Часто и ошибочно к аутоиммунным относят процессы, при
которых имеет место повреждение тканей иммунными меха-
низмами. Но вспомним, что всякий иммунный ответ предпо-
лагает деструкцию собственных тканей, поврежденных пато-
геном.
Между инфекционным антигеном и антигенами тканей
организма часто, если не всегда, бывают перекрестно реаги-
рующие эпитопы (микроорганизмы эволюционируют в этом
направлении). Причинный антиген — микробный, и в тактике
лечения первичной должна быть не иммуносупрессивная те-
рапия, а противоинфекционная (хирургическая и/или медика-
ментозная санация очага инфекции). До санации очага не по-
казана ни иммуносупрессивная, ни тем более иммуностиму-
лирующая терапия. Хотя иммуносупрессивная терапия обще-
системного действия (сандиммун, глюкокортикоиды) дает
противовоспалительный эффект и, следовательно, клиничес-
кое улучшение, но такое лечение не патогенетично, а улучше-
ние поверхностное и временное.
111
Еще одним внешним источником повышенной деструкции
собственных тканей иммунными механизмами являются хи-
мические вещества антропогенного происхождения (химичес-
кие добавки к пище, медикаменты, косметика, бытовая и
строительная химия и т.д.), которые, проникая во внутрен-
нюю среду организма, в прямом смысле денатурируют наши
родные молекулы, превращая их в раздражающие иммунную
систему антигены. В данном случае патологический процесс
разрушения собственных тканей иммунными механизмами
также некорректно называть аутоиммунным. Здесь наша им-
мунная система борется с химическими загрязнениями на по-
верхности своих клеток, а не с нативными клетками. Патоге-
нетическим будет лечение, направленное на элиминацию хи-
мических агентов, а не иммуносупрессия своей иммунной
системы, чтобы она «не мешала» присутствию чужеродной
синтетической химии в нашей крови и на наших клетках.
2.9.2.Z. Патологические процессы с участием иммунной
системы при общем тяжелом патологическом
процессе в организме
В таких случаях может иметь место выведение иммунной
системы из строя, так сказать, «в том числе», т.е. на фоне об-
щего тяжелого патологического процесса. В таких случаях не-
обходимо лечить основное заболевание в соответствии с его
этиологией. Если этиологический фактор поддается устране-
нию и в иммунной системе не возникают генетические дефек-
ты (как с неизбежностью происходит при СПИДе и с реаль-
ной вероятностью — при радиационном воздействии), то ни-
какой специальной иммунокорригирующей терапии, скорее
всего, не потребуется. Иммунная система вернется к сбалан-
сированному состоянию. Если же на фоне основного заболе-
вания происходит повреждение генов в иммунной системе,
то пациент попадает в группу II из вышеназванных (см. раз-
дел 2.9).
Взаимосвязь иммунной системы с нервной и эндокринной
системами. Кратко коснемся взаимосвязей иммунной системы
с другими общеорганизменными системами адаптации —
нервной и гормональной. В мире издается несколько между-
народных научных журналов, где печатают работы, касающие-
ся исследований этих вопросов (Journal of Neuroimmunology;
Neuroendocrinology; Neuropsychobiology и др.). В западных
странах есть официальная врачебная специальность — психо-
нейроиммунология. Но не обязательно быть врачом — доста-
точно внимательно отнестись к общечеловеческому истори-
ческому жизненному опыту, чтобы заметить взаимосвязь за-
щитных возможностей организма человека по отношению к
112
инфекциям (а это основная функция иммунной системы) с
психическим, неврологическим и эндокринным статусом. На-
пример, если учитывать, что лимфоциты — главные клетки
иммунной системы — общаются со своими целевыми объекта-
ми не иначе, как циркулируя по крови и проникая в ткани
через стенки сосудов, то понятно, что результат функциони-
рования лимфоцитов зависит от состояния сосудов и всех со-
бытий, происходящих с ними. А нейроэндокринная регуляция
кровеносной системы — классика общей физиологии и общей
патологии. Но в данной работе мы не беремся описывать взаи-
мосвязи этих систем, чтобы «не утонуть в море» взаимоотно-
шений разной степени опосредованности. Отметим лишь не-
многие факты конкретных морфологических, клеточных и мо-
лекулярных взаимосвязей иммунной системы с нервной и эн-
докринной. Мы умышленно выделим наблюдения, сделанные
в экспериментах in vitro, за немногими исключениями, так как
в экспериментах in vivo «черный ящик» цепочки опосредован-
ных взаимовлияний всегда имеет неопределенные размеры.
Итак, во-первых, о прямых связях иммунной системы с
нервной. На гистологических препаратах лимфоидных орга-
нов наблюдают нервные окончания (по крайней мере для ад-
ренергических нервных волокон) не только в стенках сосудов,
но и в паренхиме, в межклеточных пространствах, а иногда в
прямой связи с мембраной лимфоцита. Во-вторых, на Т- и
В-лимфоцитах и на макрофагах выявлены и подсчитаны холи-
нергические рецепторы мускаринового типа (блокируемые ат-
ропином). На лимфоцитах таких рецепторов примерно
200 штук на клетку, на макрофагах — 400 штук на клетку. Это
на порядок с лишним меньше, чем принято считать для ней-
ронов, но зато константа связывания с лигандом холинерги-
ческого рецептора на лимфоците (109 моль/л) на порядок
выше, чем оценивается для нервной системы. На макрофагах
найден рецептор для гипоталамического кортикотропин-ри-
лизинг-фактора (CRF). В культуре клеток in vitro, т.е. без по-
средников, CRF индуцирует биосинтез интерлейкина-1 в мак-
рофагах. Иммунная система сопряжена с нервной системой
еще и общим биосинтезом нейропептидов. Функциональное
значение этой общности для организма непонятно, но факты
показывают, например, что IL-1 из макрофагов индуцирует в
В-лимфоцитах (и только в них из лимфоцитов) биосинтез та-
кого нейропептида, как р-эндорфин.
Особое значение в системе наших знаний о взаимоотноше-
ниях иммунной системы с эндокринной имеют представления
о физиологической роли и конкретных механизмах действия
глюкокортикоидных гормонов, поскольку эти гормоны не
один десяток лет применяют в качестве противовоспалитель-
ных медикаментов, причем при заболеваниях с очевидным во-
влечением в патогенез иммунной системы (ревматические,
8—1385
113
аутоиммунные, аллергические болезни). Кортикостероидные
гормоны вовлечены в иммунопоэз и иммуногенез на правах
имманентных факторов, контролирующих названные процес-
сы. Как именно? На этот счет известно на сегодня следующее.
Источником кортикостероидных гормонов, воздействующих
на лимфоциты, являются не только надпочечники и систем-
ная циркуляция. Кортикостероиды синтезируются еще и не-
лимфоидными клетками вилочковой железы, где создается
надлежащая локальная концентрация этих гормонов. Их эф-
фект — индукция апоптоза (т.е. запрограммированной клеточ-
ной смерти) клонов тимоцитов, отсекаемых по правилам се-
лекции (а это 95 % всех внутритимических лимфоцитов).
Главный эффект физиологических концентраций системных
кортикостероидов на лимфоциты в периферических тканях —
также индукция апоптоза, но не в каждом первом встречном
лимфоците, а только в лимфоцитах, подвергшихся активации.
Глюкокортикоиды являются «исполнителями» так называемой
индуцированной активацией клеточной смерти лимфоцитов
(AICD — activation-induced cell death). AICD — вариант апо-
птоза периферических лимфоцитов, закономерной гибели
однажды активированных клеток.
При нормальном иммунном ответе в ранние сроки от на-
чала его развития происходит активация гипоталамо-гипофи-
зарно-надпочечниковой системы (ГГНС), что можно зареги-
стрировать лабораторными анализами на гормоны. Особенно
наглядно эти процессы видны на экспериментальных моделях
иммунизации грызунов суперантигенами (например, мышей
линии Balb/c и С57В1-энтеротоксинами А и В стафилококка —
SEA, SEB), для которых молекулярно охарактеризованы гены
и белки антигенраспознающих TcR. На этих моделях для на-
глядности использованы дозы SEB, сравнимые с теми, что
имеют место при септическом шоке, и строго показано, что
сам по себе факт введения в организм даже значительных доз
SEB не вызывает активации ГГНС, если в организме животно-
го по генетическим причинам нет Т-лимфоцитов с распознаю-
щим именно данный антиген рецептором. Показано, что
именно CD4+ Т-лимфоциты, распознавшие свой антиген (а не
макрофаги или иные клетки), начинают активно продуциро-
вать TNFa. Этот цитокин через циркуляцию достигает гипота-
ламуса, в котором есть специфические рецепторы для него (и
еще, по-видимому, для IL-1). Специфический сигнал с этих
рецепторов активирует продукцию кортикотропин-рилизинг-
фактора, что в свою очередь активирует продукцию АКТГ в
гипофизе и, следовательно, кортикостероидов в надпочеч-
никах.
Строгие эксперименты показывают, что именно кортико-
стероиды вызывают апоптоз, т.е. физически элиминируют из
организма активированные суперантигеном клоны лимфоци-
114
тов, и тем самым останавливают деструктивный компонент
иммунного ответа. Если тот же антиген при тех же условиях
вводят адреналэктомированным животным, то они умирают
при явлениях разлитых воспалительных процессов, индуциро-
ванных цитокинами лимфоцитов, активированных антигеном.
Смертность от SEB также возрастает, если животному перед
антигеном вводят фармакологический антагонист глюкокор-
тикоидных рецепторов — препарат RU-38486 (в этом случае
глюкокортикоиды не имеют молекулярной возможности оста-
новить выработку провоспалительных цитокинов активиро-
ванными антигеном лимфоцитами и привлеченными ими
макрофагами). Тот же вывод следует из наблюдений за крыса-
ми линии Lewis. У крыс этой линии генетически детермини-
рована гипореактивность ГГНС, и они генетически предрас-
положены к хроническим лимфоцитзависимым воспалитель-
ным процессам. В данном случае представляется вероятным,
что «после этого» — значит «вследствие этого». Гиперактива-
ция иммунной системы, особенно системная, как при септи-
ческом шоке, потенциально детальна для организма. Анализ
этих данных обосновывает ту клиническую практику, что при
септическом шоке (или сравнимых состояниях) показано вве-
дение экзогенных глюкокортикоидных препаратов.
По совокупности данных на клеточном уровне за корти-
костероидами «числятся» следующие эффекты в отношении
иммунной системы:
а) индукция апоптоза тимоцитов на территории вилочко-
вой железы в период лимфопоэза;
б) индукция апоптоза периферических лимфоцитов, под-
вергнутых активации (индуцированная активацией клеточная
смерть — AICD);
в) ингибиция активности ТЫ — снижение ими продукции
IL-2 (и, следовательно, ограничение пролиферации лимфоци-
тов) и IFNy.
Снижение уровня IFNy в свою очередь имеет следствием
уменьшение активности макрофагов (в том числе процессинг
и представление антигена); уменьшение активности NK; сдвиг
функциональной дифференцировки Т-хелперов от ТЫ в сто-
рону Th2.
Таким образом, весьма вероятно, что именно глюкокорти-
коидные гормоны являются той основной «материей» в орга-
низме, которая ответственна за физиологическую супрессию
иммунного ответа. Кроме них, на супрессию иммунного отве-
та «работает» такой цитокин, как трансформирующий фактор
роста р (TGFp), и с большей долей компромиссов — цитоки-
ны с частичными противовоспалительными эффектами типа
IL-10. Все вместе они «пришли на смену» в наших знаниях об
иммунитете Т-супрессорам, которые за последние 10 лет как
бы перестали существовать в нашем сознании, поскольку их
8
115
никому не удалось выделить как физически индивидуальную
субпопуляцию лимфоцитов.
В завершение лекции кратко разберем еще два метода им-
мунокорригирующего лечения: вакцинацию и иммуностиму-
ляторы.
Вакцинация. Вакцинация — целенаправленное введение в
организм человека заданного антигена в неагрессивной форме
и в неагрессивных, но иммуногенных дозах с целью индукции
защитного иммунного ответа и формирования иммунологи-
ческой памяти для профилактики реального инфекционного
заболевания в будущем.
Вакцинация — теоретически самый лучший метод имму-
нотерапии и иммунопрофилактики. Вакцинация — антиген-
специфичная стимуляция иммунитета. Но есть проблемы,
наиболее трудные из которых мы обозначим. Самая крупная
из трудных проблем — биологическая опасность самих вакци-
нирующих препаратов вне зависимости от целевого антигена.
Дело в том, что все современные вакцинирующие препараты
получают методами биотехнологии с использованием сыворо-
ток и клеток животных. У животных, как становится известно
нам чем дальше, тем больше, есть чрезвычайно опасные для
человека инфекции типа прионных и ретровирусных. Очис-
тить вакцину от примесей, потенциально содержащих эти ин-
фекции, принципиально невозможно (без потери собственно
вакцинирующего антигена). Такое серьезное сопутствующее
явление заставляет признать, что, вакцинируя население, ме-
дицина несознательно нарушает основной принцип — «не на-
вреди». Выход — в разработке вакцинирующих препаратов без
применения каких бы то ни было методов биотехнологий, воз-
можно, методами чисто химического синтеза. Как знает
любой врач, в настоящее время таких вакцин нет.
Вторая проблема состоит в применении живых аттенуиро-
ванных вирусных вакцин. В действии аттенуации (ослабление
патогенных свойств лабораторными манипуляциями in vitro
или на животных) скрыт еще один врачебный самообман.
Дело в том, что, выпуская ранее аттенуированный живой мик-
роорганизм в живые человеческие тела, мы не можем отме-
нить и даже контролировать дальнейшую его эволюцию в сто-
рону нарастания патогенности, его генетических рекомбина-
ций с другими микроорганизмами (а таких фактов много) с
образованием новых форм жизни. Поэтому, как бы ни хорош
был защитный эффект живых вакцин, а они действительно
лучшие, применяя их, врач также не соблюдает заповедь «не
навреди».
Третья проблема уже обсуждалась нами выше. Она состоит
в том, что вакцина — одна на всех людей в популяции, а анти-
генпредставляющие возможности MHC-I/II у каждого челове-
ка свои согласно семейной наследственности. Поэтому зако-
116
номерным является то, что не каждого человека в принципе
может защитить какая бы то ни была стандартная вакцина.
Делом разработчиков вакцин должны стать разумное реше-
ние проблем и создание вакцинирующих препаратов, удовле-
творяющих следующим необходимым критериям.
1. Нельзя использовать никакие сыворотки и клетки жи-
вотных и человека при производстве препаратов массового на-
значения. Исключение может представить лишь персональное
изготовление для индивидуума вакцинирующего препарата с
использованием in vitro его собственной крови или тканей.
2. Вакцина не должна индуцировать патогенные иммунные
процессы (типа усиливающих инфекцию антител и др.).
3. Вакцина должна эффективно индуцировать протектив-
ный иммунитет.
4. Если вакцинация, напротив, имеет цель подавить какой-
либо нежелательный иммунный процесс в организме, то вак-
цинный препарат должен индуцировать антигенспецифичную
иммунологическую толерантность (т.е. ареактивность или де-
лению клона лимфоцитов).
5. Врач-иммунолог должен уметь контролировать создание
заданного иммунитета у человека лабораторными методами
исследования.
Общая задача всей современной медицины, а вероятно, и
всего общества, но отнюдь не только иммунологии — сосредо-
точиться, собраться с силами и начать настоящие разработки
по созданию биологически безопасных вакцинирующих пре-
паратов.
2.9.3. Иммуностимулирующая терапия,
неспецифичная по антигену
На фармацевтическом рынке, причем на отечественном
гораздо больше, чем во всем остальном мире (на что тоже по-
лезно обратить внимание — а почему?), есть препараты под
рубрикой «иммунокорректоры», «иммуномодуляторы» или
«иммуностимуляторы». Такие препараты не содержат специ-
фических антигенов. Весь мир, и мы вместе с ним, еще очень
мало знаем об иммунной системе — иначе бы у нас уже была
«предсказательная» общая теория иммунитета, а ее нет. Поэ-
тому кто же будет утверждать, включая разработчиков имму-
нокорригирующих препаратов, что они точно знают механиз-
мы действия своих препаратов и предвидят последствия от их
применения во всех компонентах иммунной системы и в орга-
низме в целом, да еще во времени? Идеология применения
иммуностимуляторов гораздо сложнее, чем может показаться
на непрофессиональный взгляд, что «чем больше иммунитета,
тем лучше». Положительные эффекты от применения имму-
117
ностимуляторов можно читать в рекламе на препараты. Мы же
перечислим проблемы и последствия, которые не принято
подвергать процедуре рекламы.
1. Стимуляция лимфоцитов вообще, в том числе тех, для
которых сию минуту нет антигена, приводит к апоптозу — ги-
бели «холостых» клонов. Какой-то антиген вскоре может по-
пасть в организм, а его клон убит предыдущей неспецифичес-
кой стимуляцией. Самое трудное в этой ситуации заключается
в том, что на будущее в принципе нельзя составить прогноза,
если не брать на себя функции Бога: какой антиген попадет в
организм и насколько он будет опасен?
2. У врача должна быть обоснованная уверенность, что на-
значаемая им иммуностимуляция не усилит аутореактивные
или аллергогенные патогенные клоны лимфоцитов.
3. У врача должна быть обоснованная уверенность также в
том, что иммуностимулятор не «сорвет» естественную про-
грамму апоптоза лимфоцитов и не возникнет лейкоза, так как
пролиферация — обязательный процесс в иммунном ответе и
стимуляция иммунного ответа не может не означать стимуля-
цию пролиферации, по крайней мере лимфоцитов.
Среди доступных в аптеках препаратов иммунокорригирую-
щего назначения есть такие, которые получают из животного и
даже человеческого сырья. Следовательно, каждый человек, по-
лучивший такой препарат, с неизбежностью попадает в группу
риска по СПИДу и другим «кровяным» инфекциям (вирусным,
прионным, которые фильтруются через любой микропористый
фильтр и весьма устойчивы к термо- и химиообработкам). Поэ-
тому врач, назначающий препараты из животного и человечес-
кого сырья, должен полностью отдавать себе отчет, что показа-
ния к назначению стоят вышеназванного потенциального риска
по инфекциям. ВОЗ еще в 1990 г. рекомендовала всем практи-
ческим врачам квалифицировать процедуру назначения крове-
препаратов как процедуру но витальным показаниям. В отсутст-
вие витальных показаний следует брать на себя труд по поиску
иных, как жизнь показывает, более трудоемких терапевтических
решений. Иначе ятрогенная трансмиссия медленных, скрытых
инфекций может превзойти количественно всю пользу от совре-
менной медицины.
Список литературы
Петров Р.В. Иммунология. — М.: Медицина, 1987.
Хаитов Р.М., Игнатьева Г.А. СПИД. — М., 1992.
Хаитов Р.М., Игнатьева Г.А., Сидорович И.Г. Иммунология. — М.:
Медицина, 2000.
118
Порошина Ю.А., Гущин И. С., Латышева Т.В. и др. Острые токсико-ал-
лергические реакции на медикаменты (клиника, лечение, профилак-
тика): Методические рекомендации Минздрава СССР. — М., 1988.
Гущин И. С. Немедленная аллергия и противоаллергические лекарст-
венные средства//Ма1епа Medica. — 1993. — Vol. 1. — Р. 7—29.
Лусс Л.В., Ильина Н.И., Прокопенко В.В. и др. Опыт работы аллерго-
логического консультативного центра Института иммунологии М3
СССР//Проблемы региональной аллергологии. — Ташкент, 1989. —
С. 123.
Лусс Л.В., Прокопенко В.Д., Репина Т.Ю. Истинные и ложные аллер-
гические реакции на пищевые продукты. — М.: Крон-пресс, 1996.
Лусс Л.В. Аллергические и псевдоаллергические реакции на медика-
менты//Рус.мед.журн. — 1996. — № 1.
Федосеева В.Н., Порядин Г.В., Ковальчук Л.В. и др. Руководство по
иммунологическим и аллергологическим методам в гигиенических
исследованиях. — М.: Промедэк, 1993.
Guitart J. Immunopathology of Stevens—Johnson syndrome//Allergy
Proc. - 1995. - Vol. 16, N 4. - P. 163-165.
Niessner M., Volk B.A. Altered Thl/Th2 cytokine profiles in the intestinal
mucosa of parients with inflammatory bowel disease as assessed by quanti-
tative reversed transcribed polymerase chain reaction (RT-PCR)//Clin.
exp. Immunol. — 1995. — Vol. 101, N 3. — P. 428—435.
Wallace W.A.H., Ramage E.A., Lamb D., Howie S.E.M. A type 2 (Th2-like)
pattern of immune response predominates in the pulmonary intestitium of
patients with cryptogenic fibrosing alveolitis (CFA)//Clin. exp. Immu-
nol.- 1995. - Vol. 101, N 3. - P. 436-441.
Paliogianni F., Hama N., Balow J.E. et al. Glucocorticoid-mediated regula-
tion of protein phosphorylation in primary human T cells//J. Immunol. —
1995. - Vol. 155. - P. 1809-1817.
Vanhecke D., Leclercq G., Plum J., Vandekerckhove B. Characterization of
distinct stages during the differentiation of human CD69+CD3+thymo-
cytes and identification of thymic emigrants//J. Immunol. — 1995. — Vol.
155. - P. 1862-1872.
Shi Y, Radvanyi L.G., Sharma A. et al. CD28-mediated signaling in vivo
prevents activation-induced apoptosis in the thymus and alters peripheral
lymphocyte homeostasis//J. Immunol.— 1995.—Vol. 155.— P. 1829—1837.
Papavasiliou F, Jankovic M., Suh H. The cytoplasmic domains of immu-
noglobulin (Ig) a and IgP can independently induce the precursor В cell
transition and allelic exclusion//J. exp. Med. — 1995. — Vol. 182, N S.-
Р. 1389-1394.
119
Larsen C.G., Thomsen M.K., Gesser B. et al. The delayed-type hypersensi-
tivity reaction is dependent on IL-8//J. Immunol. — 1995. — Vol. 155. —
P. 2151-2157.
Aalberse R.C. Atopy and ectopic immune response//Immunol. and cell
Biol. - 1996. - Vol. 74, N 2. - P. 201-205.
Thornton B.P., Vetvicka V., Ross G.D. Function of C3 in a humoral re-
sponce: iC3b/C3dg bound to an immune complex generated wiyh natural
antibidy and a primary antigen promotes antigen uptake and the expression
of co-stimulatory molecules by all В cells, but only stimulates Ig synthesis
by antigen-specific В cells//Clin. exp. Immunol. — 1996. — Vol. 104,
N 3.- P. 531-537.
Valenta R., Steinberg P., Duchene M., Kraft D. Immunological and struc-
tural similarities among allergens: prerequisite for a specific and compo-
nent-based therapy of allergy//Immunol. cell Biol. — 1996. — Vol. 74,
N 2. - P. 187-194.
Spronk P.E., Van Der Gun B.T.F. et al. Anti-dsDNA production coincides
with concurrent В and T cell activation during development of active dis-
ease in systemic lupus erythematosus (SLE)//Clin. exp. Immunol. —
1996,- Vol. 104, N 3. - P. 446-453.
Pelletier E. Wain-Hobson S. AIDS is not caused by the extreme genetic
variability of HIV/J. of NIH Research. — 1996. — Vol. 8. — P. 45—50.
Shurin G. et al. Hypothalamic-pituitary-adrenal activation by the bacterial
superantigen staphylococcal enterotoxin B: role of macrophages and T
cells//Neuroendocrinology. — 1997. — Vol. 65, N 1. — P. 18—28.
Лекция 3
Аллергия: аллергены, индукция
и регуляция синтеза иммуноглобулина Е
И. С. Гущин
Введение
Смысл, вкладываемый в настоящее время в термин «аллер-
гия», существенным образом отличается от того, который со-
ответствовал этому термину в начале XX в. С. von Pirquet
(1906) ввел термин «аллергия» для обозначения состояния «из-
мененной реактивности» (к антигенной стимуляции) безотно-
сительно к тому, проявляется ли эта «измененная» реактив-
ность иммунитетом или повышенной чувствительностью к
антигену. Сейчас термин «аллергия» принято использовать
как синоним иммунологически опосредованных реакций по-
вышенной чувствительности вообще и даже в узком смысле —
IgE-зависимых реакций. Термин «гиперчувствительность»
(специфическая) как раньше, так и теперь применяют для
обозначения нежелательных клинических реакций на антиге-
ны или аллергены. Сложилась ситуация, при которой термины
«аллергия» и «гиперчувствительность» взаимозаменяемо ис-
пользуют для описания нежелательной «преувеличенной» от-
ветной реакции «системы иммунитета» на антигенные или ал-
лергенные продукты. В этом легко заметить явное противоре-
чие в дефинициях терминов «иммунитет» и «система иммуни-
тета», так как функцией последней может быть формирование
«иммунитета» («невосприимчивости», «устойчивости», «защи-
ты»), а не повышение чувствительности к антигену. «Аллерги-
ческий энцефаломиелит» никак нельзя определить как «им-
мунный энцефаломиелит». Равным образом состояние гипер-
чувствительности к аутоантигенам куда более оправданно обо-
значать как аутоаллергическое, но не «аутоиммунное», коль
скоро в данном случае речь не идет о формировании иммуни-
тета к собственным антигенам (например, не аутоиммунный
тиреоидит, а аутоаллергический тиреоидит).
Приведенные очевидные терминологические противоре-
чия явились поводом для обоснования точки зрения о необхо-
димости возвратиться к толкованию термина «аллергия» в по-
нимании С. von Pirquet. Справедливости ради следует заме-
тить, что такого рода взгляд и в прежние годы разделяли оте-
чественные исследователи.
121
Упомянутые противоречия предлагают устранить путем ис-
пользования термина «аллергические болезни» вместо термина
«аллергия» (в современном понимании), оставив за «аллергией»
обозначение биологического ответа организма с измененной ре-
активностью на антиген, не реализованного («некоммитирован-
ного» ответа) в то или иное клиническое состояние (в состояние
иммунитета или гиперчувствительности). У конкретного инди-
вида такой некоммитированный ответ может привести либо к
иммунитету, либо к гиперчувствительности (к аллергическому
заболеванию). Таким образом, предлагают под аллергией (ал-
лергическим ответом) понимать состояние, характеризующееся
образованием антител и специфически реагирующих лимфоци-
тов и обеспечивающее «вооруженность» организма либо для ре-
акций иммунитета, либо для гиперчувствительности (аллерги-
ческих болезней) [КауА.В., 1997].
Ограничение смыслового использования термина «аллер-
гия» в рамках «аллергические болезни» нашло отражение в ме-
дицинской практике «клинической аллергии» («клинической
аллергологии»), когда специалисты этой области медицины
(«aiuiepracTbi»/«allergists», по международной терминологии)
заняты диагностикой, лечением и профилактикой только но-
зологически ограниченной группы состояний гиперчувстви-
тельности, а не широкого спектра расстройств системы, обес-
печивающей состояние иммунитета организма. Во многих
странах эти специалисты заняты ведением больных IgE-onoc-
редованными заболеваниями, такими как поллиноз со всеми
его проявлениями, аллергическая бронхиальная астма, аллер-
гический ринит, атопический дерматит, аллергические реак-
ции на пищевые продукты, лекарственные аллергические ре-
акции. Кроме того, в компетенцию этих специалистов входит
и ведение пациентов с так называемыми ложными аллергичес-
кими реакциями, требующими профессионального дифферен-
циального диагноза, лечения и профилактики, сходных с та-
ковыми при ведении пациентов с истинными аллергическими
заболеваниями. Другие болезни гиперчувствительности,
такие, например, как болезни комплексов антиген — антите-
ло, гемолитическая болезнь у новорожденных, контактный
дерматит и некоторые другие, отнесены к компетенции специ-
алистов по болезням соответствующих органов и систем.
Среди классификаций реакций гиперчувствительности, иг-
рающих ту или иную роль при аллергических заболеваниях,
наиболее общепринятой является классификация, предложен-
ная R.R.A.Coombs и P.G.H.Gell более трети века тому назад (в
1963 г.). По мере получения новых сведений конкретные меха-
низмы развития каждого из типов реакций гиперчувствитель-
ности дополнялись и конкретизировались.
По этой классификации IgE-опосредованные реакции, как
известно, отнесены к I типу. Основной механизм этого типа
122
реакций гиперчувствительности состоит в следующем. IgE-
антитела фиксируются на обладающих высокой аффинностью
к этому иммуноглобулину специализированных рецепторах,
представленных в клеточной мембране тучных клеток и базо-
филов (а также на моноцитах, клетках Лангерганса; менее
представлены они и на эозинофилах, но функция рецептора
на этих клетках точно не определена). Антиген/аллерген пере-
крестно связывает IgE-антитела и соответственно клеточные
рецепторы этого иммуноглобулина. Перекрестное связывание
активирует тучные клетки и базофилы, в результате чего из
клеток секретируются медиаторы, которые действуют на дру-
гие клетки и ткани и вызывают тем самым внешние быстро
развивающиеся проявления реакции (ранняя фаза — возникает
в пределах нескольких минут после действия антигена): сокра-
щение гладкой мускулатуры, изменение микроциркуляции,
повышение сосудистой проницаемости, отек ткани, раздраже-
ние периферических нервных рецепторов, гиперсекреция сли-
зистых желез. Наряду с этими эффектами отдельные медиато-
ры вызывают миграцию и хемотаксис других клеток — участ-
ников реакции: эозинофилов, ТЬ2-клеток, базофилов, моно-
цитов, нейтрофилов, которые, будучи активированы накапли-
вающимися медиаторами и, возможно, IgE-опосредованным
механизмом, также секретируют медиаторы, дополняющие
внешние проявления тканевой реакции. Поскольку на при-
влечение этих клеток затрачивается относительно продолжи-
тельное время, то вызываемая ими реакция отсрочена по от-
ношению к моменту действия антигена/аллергена (поздняя
фаза — возникает через 6—8 ч после действия антигена).
Не исключено, что, помимо IgE-антител, I тип реакций ги-
перчувствительности может отчасти опосредоваться и под-
классом IgG-антител, но функциональное и соответственно
клиническое значение взаимодействия с IgG-антителами все
еще остается неопределенным. В реальной клинической си-
туации в той или иной мере могут участвовать разные типы
гиперчувствительности, но особенность классификации со-
стоит в том, что она выделяет один из наиболее существенных
механизмов запуска реакции и не претендует на исчерпываю-
щее описание всех событий, сопутствующих каждому типу ги-
перчувствительности .
В цепи последовательных событий, завершающихся внеш-
ними (клиническими) признаками аллергии, определяющим
является этап индукции поступающим в организм аллергеном
особого типа иммунологического ответа. Этот ответ состоит в
образовании аллергических антител, принадлежащих к IgE,
которые фиксируются на клетках воспаления, «вооружая» их
специфическим механизмом узнавания вновь поступающего в
организм аллергена. Возникновение IgE-ответа тем самым
предопределяет формирование аллергенспецифической сенси-
123
билизации (повышения чувствительности) тканей организма.
Выражением такой сенсибилизации становятся, при условии
повторного (разрешающего) действия аллергена на сенсибили-
зированный организм, аллергенспецифические системные и
тканевые реакции. В зависимости от того, в каких системах,
органах и тканях произойдет встреча аллергена с фиксирован-
ными на клетках воспаления IgE-антителами, возникают ха-
рактерные проявления, создающие клиническую картину со-
ответствующих аллергических заболеваний в виде системной
анафилаксии (анафилактического шока), аллергической брон-
хиальной астмы, аллергического конъюнктивита и ринита,
атопического дерматита, аллергической крапивницы и пр.
К настоящему времени накоплен очень большой объем
знаний о всех этапах аллергического процесса. Поэтому удов-
летворительно рассмотреть эти вопросы в одной лекции не-
возможно. В связи с этим данная лекция ориентирована на
описание лишь тех этапов, которые являются обязательными
для запуска всего аллергического процесса, и посвящена опи-
санию природы и свойств аллергенов, индукции аллергеном
синтеза IgE-антител и его регуляции.
События, следующие за этапом образования IgE-антител и
состоящие в фиксации IgE на специализированных рецепторах
клеток воспаления, в аллергенспецифической активации этих
клеток, вовлечении их в аллергический процесс, в образовании
и секреции из клеток разнообразных посредников (медиаторов)
аллергии, в действии последних на периферические ткани и воз-
никновении основных патофизиологических эффектов в виде
сокращения гладкой мускулатуры, повышения сосудистой про-
ницаемости, отека ткани, раздражения периферических нерв-
ных окончаний, гиперсекреции слизистых желез, подробно опи-
саны в недавно опубликованных обобщающих отечественных
работах [Гущин И.С., 1998; Пыцкий В.И. и др., 1998]. В этих же
работах даны описания давно устоявшихся и ставших общепри-
нятыми терминов и понятий (активная и пассивная сенсибилиза-
ция, прямая и обратная анафилаксия, атопия, системная ана-
филаксия, анафилаксия тканей и органов in vivo и in vitro, гино-
сенсибилизация/десенсибилизация и пр.).
3.1. Аллергены и аллергенность
Несмотря на огромные успехи, достигнутые за последние
20 лет в химии и биохимии белков, в технологии рекомби-
нантной ДНК, в идентификации специфической аллергенной
активности, в установлении перекрестных свойств эпитопной
характеристики антигенных молекул, и сегодня невозможно
дать исчерпывающий ответ на вопрос, почему же все-таки оп-
ределенные молекулы действуют именно как аллергены.
124
Аллергенность — это особенный способ действия антигена,
результатом которого является индукция IgE-ответа. Поэтому
для характеристики этого действия важно, с одной стороны,
установление структурных особенностей аллергенных моле-
кул, а с другой — выяснение сущности превращений этих мо-
лекул в организме в виде их переработки и/или взаимодейст-
вий с эндогенным субстратом.
Большинство естественных аллергенов являются белками
и имеют молекулярную массу в пределах от 10 до 70 кДа.
Антигены с меньшим размером молекулы сами по себе, не бу-
дучи полимеризованы, не образуют эффективного мостика
между фиксированными на клетке молекулами IgE-антител и
не запускают аллергическую реакцию. Аллергены, имеющие
в нативном состоянии массу, значительно превышающую
70 кДа, с трудом проникают или вообще не проникают через
барьерные ткани (слизистые оболочки) и потому не могут в
таком виде достигать IgE-антител, фиксированных на клеточ-
ных рецепторах. Такие крупные аллергены являются высоко-
полярными соединениями, и их парентеральное поступление
в очень малых количествах (нано- и микрограммы) вызывает
выраженную сенсибилизацию. Сенсибилизация в этом случае
(при парентеральном введении) становится возможной пото-
му, что преодолен гистогематический барьер.
Аллергены являются Т-клеточными антигенами. Понятно,
что особый интерес представляет выяснение строения множе-
ственных В- и Т-клеточных эпитопов аллергенов. В-клеточ-
ные эпитопы характеризуются специфическими последова-
тельностями аминокислотных остатков, что определяет трех-
мерную конфигурацию молекулы. Т-клеточные эпитопы пред-
ставляют собой очень короткие пептидные фрагменты в пре-
делах 12—13 аминокислотных остатков. Перекрестная анти-
генная реактивность определяется структурной гомологией
молекул разного происхождения.
Структурные характеристики сами по себе еще недостаточ-
ны для объяснения возможности индукции IgE-ответа. Для
этого необходимо понимание как механизма генетического
предрасположения определенных лиц к формированию IgE-
ответа, так и особенностей способов превращений (переработ-
ки) поступающего в организм (и на поверхностные ткани —
на слизистые оболочки) материала, обладающего аллергенны-
ми свойствами. Самостоятельный интерес при этом представ-
ляет способ процессирования антигена в антигенпредставляю-
щих клетках, что может повлиять на характер последующего
ответа.
Биохимическая активность аллергенного материала также
может быть фактором аллергенности. В последнее время стало
известно, что многие важные аллергены (аллергены пыльцы
растений, бытовые аллергены — аллергены постельного кле-
125
ща, ядов насекомых) обладают ферментативной активностью.
Эта активность может, с одной стороны, повышать аллерген-
ный потенциал всего аллергенного продукта, влияя на харак-
тер его превращений по пути проникновения через поверх-
ностные барьерные ткани или на процессирование молекулы в
аллергенпредставляющих клетках, а с другой — приводить к
образованию новых молекул, которые затем могут быть вовле-
чены в индукцию IgE-ответа.
3.1.1. Номенклатура аллергенов
Номенклатура аллергенов была унифицирована в 1994 г.
Номенклатурным субкомитетом по аллергенам (при ВОЗ). Эта
номенклатура основана на таксономическом названии рода и
вида источника аллергена. Сокращенное название аллергена
приводится следующим образом. Первые три буквы латинско-
го названия рода; отступ; первая буква вида; отступ; арабская
цифра, например Der f 1 — Dermatophagoides farinae. Одна и та
же цифра означает гомологичные аллергены разных видов: на-
пример, антиген 5 из Dactylis glomerulata и Phleum pratense —
Phi p 5 и Dac g 5. Дополнительные буквы в сокращенном обо-
значении рода или вида используют для того, чтобы избежать
путаницы: например, Ves v 5 — Vespula vulgaris, но Ves vi 5 —
Vespula vidua. Изоформы аллергенов и их варианты обознача-
ют дополнительными четырьмя цифрами. Первые две из них
характеризуют изоаллерген, а следующие две — вариант. На-
пример, четыре изоаллергена Amb а 1 (Ambrosia artemisiifolia)
обозначают как Amb а 1.01; Amb а 1.02 и т.д., а варианты —
как Amb а 1.0101; Amb а 1.0102 и т.д.
3.1.2. Идентификация и очистка аллергенов
Лечебные и диагностические аллергены, наиболее часто
используемые в клинической практике, представляют собой
водно-солевые экстракты исходного аллергенного сырья. Та-
ковы водно-солевые экстракты пыльцы растений, домашней
пыли, шерсти животных, экстракты насекомых и пр. Такие
экстракты содержат не один, а несколько аллергенов, т.е. тех
действующих начал, которые способны вызвать сенсибилиза-
цию, переключение синтеза иммуноглобулинов на синтез IgE
и взаимодействовать с аллергенспецифическим IgE, вызывая
аллергическую реакцию. Между разными аллергенами может
существовать структурная гомология в той или иной степени,
что объясняет столь распространенные перекрестные аллер-
генные свойства. Так, например, лица, имеющие повышенную
чувствительность к аллергену березы, одновременно реагиру-
126
ют на пыльцу орешника, ольхи и на некоторые фрукты и
овощи (в частности, на яблоки). Индивидуальные аллергены,
содержащиеся в данном экстракте, классифицируют на глав-
ные (основные), промежуточные и второстепенные. Такое
подразделение основано на частоте реагирования на эти ком-
поненты лиц с повышенной чувствительностью к цельному
экстракту. За главные компоненты принимают те, на которые
реагируют не менее 50 % лиц, имеющих повышенную чувстви-
тельность к цельному экстракту. Для идентификации аллерге-
нов и определения их состава используют методы химическо-
го, иммунохимического и биохимического анализа. Среди них
наиболее часто применяют методы иммуноэлектрофореза,
электрофореза в додецилсульфате натрия — полиакриламид-
ном геле (sodium dodecyl sulphate-polyacrylamide gel electropho-
resis — SDS-PAGE) и изоэлектрофокусирования в комплексе с
иммуноблоттингом. Поскольку каждый из этих методов имеет
серьезные ограничения, окончательное заключение можно
сделать только на основании сопоставления результатов, по-
лученных разными способами оценки.
SDS-PAGE позволяет разделять белки по размеру их моле-
кулы. Додецил натрия представляет собой сильный анионный
детергент, который связывается с белками и маскирует собст-
венный заряд белковых молекул. В таких условиях полиакрил-
амидный гель выполняет роль простого молекулярного сита.
Параллельное использование одновременно с образцами ал-
лергена белковых маркеров с известной величиной молекуляр-
ной массы (мол. массы) позволяет достаточно точно опреде-
лить величину молекулярной массы компонентов, входящих в
состав аллергена. Следует иметь в виду, что на результаты раз-
деления может оказывать влияние гликозилирование белков,
приводящее к значительному увеличению молекулярной
массы белковых молекул. Использование восстанавливающих
агентов (меркаптоэтанола или динитрофенола), разрушающих
дисульфидные связи, позволяет достичь диссоциации димер-
ных и полимерных белков и повысить их растворимость.
Затем разделенные белки наносят на нитроцеллюлозные или
фторполивиниловые мембраны. Нанесенные на мембраны
белковые пятна («блоты») инкубируют в присутствии сыворо-
ток крови больных, имеющих повышенную чувствительность
к данному аллергену и соответственно содержащих IgE-анти-
тела. Связывание IgE-антител с аллергенными белками иден-
тифицируют при помощи использования анти-IgE-антител,
меченных либо |251, либо ферментом соответственно аутора-
диографически или по образованию хемилюминесцентного
продукта. Результаты метода (Western blot technique) зависят от
полноты ренатурации аллергенов в ходе процесса «блоттинга».
Изоэлектрофокусирование позволяет разделять белки в со-
ответствии с их изоэлектрической точкой в градиенте pH, со-
127
здаваемом смесью соответствующих амфотерных молекул.
Иммунохимическую идентификацию аллергенов проводят тем
же приемом, что и при выполнении SDS-PAGE. Толкование
результатов нередко затрудняется тем, что изоформы аллерге-
нов обладают гетерогенностью заряда, разной степенью гли-
козилирования и другими вариабельными характеристиками.
Перекрестный иммуноэлектрофорез представляет собой
комбинацию первичного электрофоретического разделения в
геле агарозы и последующего электрофореза под прямым
углом в геле агарозы, содержащем специфическую антисыво-
ротку. В зоне эквивалентных соотношений концентраций
антигена и антител возникают преципитаты в виде «ракетных»
следов. Выявление в них аллергенов достигается при помощи
использования сыворотки крови больных, чувствительных к
данному аллергену, с последующим добавлением анти-lgE-
антител, меченных 1251 (метод ауторадиографии). Понятно, что
в этом случае результаты будут зависеть от качества антисыво-
ротки, которая должна «распознавать» все антигенные компо-
ненты аллергена, а также от достижения оптимальной кон-
центрации антител для возникновения иммунопреципитации.
Данные, характеризующие молекулярный размер и точку
изоэлектрофокусирования, являются обоснованием выбора
методов очистки конкретного аллергена. Многие аллергены
(ингаляционные и пищевые) представляют собой гликопро-
теины с мол. массой от 5 до 70 кДа и имеют точку изоэлектро-
фокусирования при pH от 4,0 до 7,0. Однако встречаются и
белки со свойствами основания (катионные белки). У разных
изоформ аллергенов могут быть разные величины молекуляр-
ной массы и суммарного заряда, что зависит от соответствую-
щих аминокислотных замещений и различий углеводных ком-
понентов.
Поскольку аллергены являются растворимыми белками и
гликопротеинами, то для их очистки используют принципи-
ально те же методы, что и для очистки белков. Первый этап
очистки состоит в экстракции исходного сырья. При этом
должны быть созданы условия максимального извлечения ал-
лергенов при минимальной степени их разрушения и денату-
рации. Это достигается прежде всего подбором величины pH,
ионной силы раствора, буферного состава, продолжительнос-
ти экстракции в зависимости от особенных свойств данного
аллергена. Использование низкой температуры в период
экстракции снижает степень разрушения экстрагируемых мо-
лекул. В тех случаях, когда в экстрагируемом материале могут
присутствовать протеолитические ферменты, необходимо ис-
пользовать ингибиторы протеаз, как, например, при приготов-
лении аллергенов грибов и постельного клеща. При довольно
длительных процедурах выделения аллергенов необходимо
применять бактериостатические агенты-консерванты (мертио-
128
лат). Однако использовать их допустимо в очень низкой кон-
центрации (0,005 %).
Различные физико-химические методы очистки базируют-
ся на электрофоретическом и хроматографическом разделении
материала, различающегося величиной молекулярной массы,
заряда и гидрофобными свойствами. Для хроматографическо-
го разделения в настоящее время используют варианты высо-
коразрешающей жидкостной хроматографии (HPLC), преиму-
щества которой состоят в большой емкости колонок, высоко-
разрешающих свойствах используемых смол, большой скорос-
ти разделения и высокой степени воспроизводимости.
Высокая степень очистки достигается с помощью методов
аффинной хроматографии. При этом в качестве аффинного
материала применяют либо соответствующие моноклональные
антитела, либо лектины. Последние, разумеется, могут быть
использованы в том случае, если аллергены являются глико-
протеинами. Лектин-аффинная хроматография имеет преиму-
щества при выделении гликопротеинов из большого объема и
как этап последующего выделения из материала, полученного
ионообменной хроматографией, так как связывание с лекти-
ном не зависит от высокой концентрации солей. Избиратель-
ность выделения аллергенов лектиновой хроматографией оп-
ределяется характером углеводных компонентов гликопротеи-
нов. Конканаваллин А распознает структуры, содержащие
связанную с альфа-группами маннозу, а агглютинин пророст-
ков пшеницы — олигосахара, связанные с аспарагином через
сиаловую кислоту, N-ацетилглюкозу и остатки N-ацетилга-
лактозамина. Роль охарактеризованных углеводных остатков в
связывании с IgE может быть установлена сравнительной
оценкой этого свойства гликозилированных и дегликозилиро-
ванных аллергенных белков.
Чрезвычайно важной теоретической и практической про-
блемой является вопрос о перекрестных свойствах разных ал-
лергенов. Работы, посвященные изучению этих свойств, очень
многочисленны и разнообразны. В одной из них в последнее
время получены результаты, позволяющие удачно объяснить
перекрестные свойства аллергенов пыльцы разных видов рас-
тений. Показано, что такие перекрестные свойства могут быть
связаны с растительными профилинами, содержащимися в
пыльце растений. Эти профилины распознаются актином,
входящим в состав оболочки растительной клетки. Выделение
профилинов осуществимо хроматографией на колонках, со-
держащих полипролин. Таким образом может быть получен
продукт, ответственный за общие аллергенные свойства цело-
го ряда аллергенов пыльцы растений.
Ферменты, входящие в состав аллергенов и ответственные
за их аллергенную активность, могут быть выделены аффин-
ной хроматографией с использованием соответствующих суб-
9—1385 129
стратов или ингибиторов этих ферментов. Так, один из инди-
видуальных аллергенов, входящих в состав D. pteronissinus,
имеет высокую гомологию с глутатион-5-трансферазой и
может быть выделен на колонке, содержащей глутатион. Груп-
па аллергенов D. pteronissinus и D. farinae представляет собой
трипсиноподобные серинэстеразы. Соответственно они могут
быть выделены на аффинных колонках, содержащих ингиби-
торы трипсина.
Наконец, молекулярное клонирование аллергенов делает
принципиально возможным синтез неограниченного количе-
ства клонально чистого белка или модифицированных аллер-
генов и пептидов для последующего изучения их функции.
3.1.3. Нативные аллергены как гетерогенная
и изменчивая популяция
Приведенные выше общие сведения о способах выделения
и идентификации аллергенов, входящих в состав разнообраз-
ных продуктов, абсолютно необходимы для понимания про-
блемы природы аллергенов и аллергенности. К сожалению,
эти сведения, даже в столь общей форме, преподносятся, как
правило, лишь в узко специализированной литературе. Наибо-
лее существенные выводы, которые вытекают из результатов
изучения очищенных и идентифицированных аллергенов,
могут быть следующими.
• Аллергенный состав большинства продуктов, являющих-
ся носителями аллергенов, оказывается в той или иной
степени гетерогенным.
• Отсюда следует, во-первых, что для достижения высоко-
го лечебного эффекта необходимо использование макси-
мально полноценных по составу аллергенов, а не только
их главных компонентов, и, во-вторых, главные аллерге-
ны в высокоочищенной (индивидуальной) форме могут
быть претендентами на эффективные препараты для спе-
цифической диагностики.
• Помимо собственно аллергенных молекул, исходные ал-
лергенные продукты содержат, как правило, компонен-
ты, которые влияют на механизм проникновения самих
аллергенов в организм, способы его превращений в орга-
низме и на интенсивность IgE-ответа к аллергену.
• Сами аллергенные молекулы нередко могут обладать до-
полнительной физиологической и биохимической актив-
ностью, что также сказывается на формировании IgE-от-
вета и на последующей аллергической реакции.
• Общие аллергенные свойства являются распространен-
ной и важной характеристикой разнообразных аллерге-
130
нов, объясняющей отчасти наличие гиперчувствитель-
ности к широкому перечню нативных аллергенных про-
дуктов у одного и того же пациента.
Перечень известных в настоящее время аллергенов чрезвы-
чайно разнообразен. Ни одна из существующих классифика-
ций не является исчерпывающей, причем приходится одно-
временно использовать несколько принципов для группирова-
ния разных аллергенов. Группируют аллергены по происхож-
дению (аллергены пыльцы растений, грибов, насекомых, жи-
вотных, лекарственные аллергены и пр.), по способам поступ-
ления в организм (аэроаллергены, пищевые, контактные ал-
лергены), по встречаемости в тех или иных условиях (быто-
вые, профессиональные). Аллерген одной из групп может от-
носиться и к большинству других групп аллергенов. Так, на-
пример, латекс представляет собой и профессиональный, и
бытовой аллерген, он может быть и аэроаллергеном, и кон-
тактным аллергеном. Если латексовый аллерген приготовлен
из сока растения, то он содержит довольно большое число ин-
дивидуальных аллергенов и может выявлять гиперчувствитель-
ность у лиц, профессионально связанных как с начальными
этапами заготовки сырья, его обработки, так и с конечным
этапом производства изделий из латекса. Поскольку в такой
аллерген входят компоненты, имеющие перекрестные свойст-
ва с другими растительными аллергенами, то реакции на этот
аллерген могут возникать у лиц, имеющих гиперчувствитель-
ность к некоторым фруктам. Латексовый аллерген, приготов-
ленный из конечного продукта, содержит меньшее число ал-
лергенных компонентов, выявляет повышенную чувствитель-
ность, чаще формирующуюся при контакте с готовыми латек-
совыми изделиями как в профессиональных, так и в бытовых
условиях.
Наиболее подробно изученной группой аллергенов, имею-
щих важное клиническое значение, являются аллергены пыль-
цы растений. Водно-солевые экстракты пыльцы могут содер-
жать около 40 различных белков, но лишь некоторые из них
имеют клиническое значение. Аллергические заболевания
наиболее часто вызываются пыльцой ветроопыляемых расте-
ний, так как ее концентрация в период цветения намного пре-
вышает концентрацию пыльцы растений, опыляемых насеко-
мыми. Кроме того, морфология пыльцы ветроопыляемых рас-
тений такова, что пыльцевые зерна являются относительно
легкими, легко переносятся на большие расстояния. Размер
пыльцевых зерен относительно небольшой (20—40 мкм в диа-
метре), что обеспечивает их легкое проникновение в воздухо-
носные пути.
Среди растений травы являются семейством, наибольшим
по числу представителей близких родов. Детально охарактери-
9*
131
зованы аллергены относительно небольшого числа видов рас-
тений средней полосы и субтропиков. Однако уже эти сведе-
ния показывают, что имеется существенное физико-химичес-
кое и иммунохимическое сходство между видами в пределах
субсемейств. Пыльца злаковых и луговых трав, сорняков явля-
ется наиболее часто встречающимся причинным фактором по-
вышенной чувствительности к пыльце растений. Среди дере-
вьев наиболее часто аллергические болезни вызывает пыльца
березы, ольхи, орешника, клена, дуба и пр.
Серьезной проблемой является изменчивость аллергенных
свойств. Эти изменения могут возникать, в частности, вслед-
ствие взаимодействия природных аллергенов с некоторыми
продуктами антропогенного происхождения. Такая вероят-
ность допускалась отечественными аллергологами еще в нача-
ле 60-х годов. Тогда же были приведены возможные свиде-
тельства в пользу того, что некоторые химические соедине-
ния, загрязняющие окружающую среду, могут изменять аллер-
генные свойства пыльцы растений и приводить к образованию
новых специфичностей. Сейчас это направление приобретает
актуальность в связи с ухудшающейся экологической ситуа-
цией во многих странах.
Изменчивость свойств аллергенов может зависеть и от осо-
бенностей их основного источника. Так, например, наиболь-
шую значимость среди аллергенов постельных клещей пред-
ставляют пищеварительные ферменты. Разнообразие аллер-
генного «репертуара» в данном случае может отражать преиму-
щественную стимуляцию разных типов ферментов в зависи-
мости от характера питания клещей: профиль ферментов, про-
дуцируемых клещами, питающимися эпидермисом человека и
животных, отличается от профиля ферментов тех же клещей,
выращиваемых на грибах и на зерновых отходах. В связи с
этим полученные из таких источников аллергены могут отли-
чаться друг от друга и иметь разную диагностическую и лечеб-
ную аллергенную активность. Важной проблемой является ге-
терогенность аллергенного состава практически всех естест-
венных источников аллергенов. Это убедительно продемон-
стрировано на примере аллергенов пыльцы растений. Так, ал-
лергенный состав пыльцы амброзии представлен по крайней
мере шестью индивидуальными аллергенами, имеющими кли-
ническое значение. То же самое показано на примере аллерге-
нов животного происхождения и грибов. В культуре Aspergillus
fumigatus определено не менее 5 аллергенов с точкой изоэлек-
трофокусирования от 4,4 до 4,8 и мол. массой 20; 29; 34; 38 и
45 кДа. Выраженная аллергенная активность аллергена
кошки, помимо аллергена Fel d 1, продуцируемого слюнными,
слезными и сальными железами, представлена и альбумином.
Аллерген шерсти собак также гетерогенен (аллерген Com f 1 с
мол. массой около 25 кДа по данным SDS-PAGE и аллерген с
132
мол. массой около 18 кДа). Аллергенная гетерогенность еще
более увеличивается за счет изоформ аллергенов. Например,
имеется не менее 4 изоформ главного аллергена мышей Mus
m 1 (преальбумин, обнаруживаемый в моче и волосяных фол-
ликулах).
Все это лишний раз подтверждает приведенное выше поло-
жение о том, что оптимальным требованием для лечебных ал-
лергенов является получение максимально полного аллерген-
ного состава из аллергенного источника. Применение для ле-
чебных целей высокочистых рекомбинантных аллергенов, не
воспроизводящих такой полноты, не может обеспечить высо-
кой терапевтической эффективности лечебных препаратов.
3.2. Иммуноглобулин Е:
индукция и регуляция синтеза
Иммуноглобулин Е (IgE) — ключевая молекула, опреде-
ляющая особенности формирования и проявления классичес-
ких аллергических состояний, к которым относится атопия.
IgE обладает уникальной способностью связываться своим
Fc-фрагментом с высокоаффинными рецепторами на тучных
клетках и базофилах. Фиксированные на этих рецепторах IgE-
антитела перекрестно связываются аллергеном, что запускает
цепь биохимических событий, приводящих к развитию аллер-
гических реакций в виде таких клинических проявлений, как
аллергический ринит, бронхиальная астма, кожные проявле-
ния, анафилактический шок.
Молекула IgE состоит, как и молекулы других мономерных
иммуноглобулинов, из четырех полипептидных цепей: двух
легких и двух тяжелых. Легкие цепи не отличаются от к- и X-
цепей иммуноглобулинов G, А, М, D, а тяжелые цепи харак-
терны только для данного класса иммуноглобулинов.
Тяжелые е-цепи включают в себя 5 доменов: один вариабель-
ный (VH) и четыре константных (Се1, Се2, С£3 и Се4). Каждая тя-
желая цепь имеет внутрицепьевые дисульфидные мостики. Две
дисульфидные связи соединяют между собой тяжелые цепи.
Каждая легкая цепь присоединена к тяжелой одной дисульфид-
ной связью. Таким образом, в молекуле IgE имеются 4 межце-
пьевые дисульфидные связи. Мол. масса IgE соответствует 180
кДа. 12 % мол. массы приходится на углеводные компоненты.
Характерное свойство термолабильности IgE связано со
структурными изменениями в области доменов С£3 и Се4-
Домен С62 нечувствителен к прогреванию (при 56 °C).
Не исключено, что IgE гетерогенен. Во всяком случае, есть
основания считать, что существует изоформа IgE, которая
преимущественно образуется при аллергенспецифической ин-
дукции IgE-ответа.
133
IgE по сравнению с другими иммуноглобулинами имеет
самый короткий период полужизни — около 2,5 дня и самое
низкое содержание в крови. Содержание IgE в сыворотке
крови новорожденных не превышает 0,5 кЕд/л. В течение пер-
вого года жизни концентрация IgE в крови ступенчато воз-
растает и продолжает увеличиваться до 5-летного возраста.
Пределы концентраций IgE в сыворотке крови практически
здоровых лиц, выбранных среди большого числа представите-
лей человеческой популяции, соответствуют величинам от 1
до 180 кЕд/л (средняя геометрическая — порядка 20 кЕд/л).
Средняя геометрическая величина остается довольно посто-
янной, несколько уменьшаясь в возрастной группе старше
70 лет. Курящие имеют более высокий уровень сывороточного
IgE, чем некурящие.
Различия в содержании IgE у практически здоровых и у
больных атопическими заболеваниями более заметны в ран-
нем периоде жизни. Это объясняют тем, что обе группы по-
разному реагируют на первую аллергенную экспозицию.
Понятно, что открытие IgE и установление преимущест-
венной принадлежности к нему аллергических антител (реаги-
нов) явилось толчком к настойчивому поиску тех специализи-
рованных механизмов, которые индуцируют и регулируют
продукцию именно этого класса (изотипа) иммуноглобулинов.
Эти поиски, осуществлявшиеся более четверти века, привели
к накоплению огромного фактического материала, который
был систематизирован в соответствии с общими положениями
фундаментальной иммунологии, и, таким образом, к началу
90-х годов была сформулирована принципиальная концепция,
удачно объясняющая на настоящий момент механизм индук-
ции и регуляции IgE (и аллергенспецифического IgE) и не
противоречащая большинству ранее полученных эксперимен-
тальных данных. Это не значит, что все прежние сведения уч-
тены в этой концепции. Просто ограниченность наших зна-
ний пока не позволяет найти обоснованное место таким неуч-
тенным сведениям в цепи событий и процессов, запускающих,
поддерживающих и усиливающих или тормозящих синтез и
секрецию IgE.
3.2.1, Модель запуска синтеза IgE
В ходе становления иммунного ответа В-лимфоцит последо-
вательно синтезирует иммуноглобулины разных изотипов. Этот
феномен последовательного переключения с одного изотипа
иммуноглобулина на другой (изотипическое переключение) по-
зволяет одному В-клеточному клону продуцировать антитела
одной и той же специфичности, но с разными эффекторными
функциями, соответствующими строению их тяжелых цепей.
134
Рис. 3.1. Ключевые элементы взаимодействия клеток при индукции
синтеза IgE.
Для того чтобы произошло переключение на определен-
ный изотип, необходимо присутствие двух сигналов. Первый
сигнал доставляется посредником-цитокином, который акти-
вирует транскрипцию на специфическом регионе иммуногло-
булинового локуса (Ig-локус). Этот стимул определяет изоти-
пическую специфичность. Другой сигнал активирует рекомби-
национный процесс, приводящий к переключающей рекомби-
нации ДНК.
Аллергенспецифические В-клетки связывают аллерген
(антиген) своими поверхностными иммуноглобулиновыми
молекулами (антигенспецифические В-клетки приблизитель-
но в 10 ООО раз более эффективны в предоставлении очень
малых количеств растворимых антигенов Т-клеткам, чем
макрофаги). Аллерген-иммуноглобулиновый комплекс ин-
тернализируется в клетку, где (в эндосомах так же, как и в
фагоцитах) происходит фрагментация (процессинг) исходной
молекулы антигена на пептидные фрагменты. Эти пептиды
представляются затем на поверхности В-клетки в ассоциации
с молекулами II класса главного комплекса гистосовмести-
мости (Major Histocompatibility Complex — МНС). В такой
форме комплекс антигенных пептидов с молекулами II клас-
са МНС связывается Т-клеточным рецептором (TCR). Таким
образом, контакт В-клетки с Т-клеткой устанавливается
«родственным» распознаванием. Распознавание комплекса
«антиген — молекулы II класса главного комплекса гистосов-
местимости» Т-клеточным рецептором активирует Т-клет-
ку (Th) и приводит к возникновению двух очень важных со-
бытий. Во-первых, возникает секреция лимфокинов, в част-
ности интерлейкина-4 (IL-4), который обеспечивает один не-
135
Схема 3.1. Индукция IgE-ответа (пояснения в тексте).
Связывание аллергена поверхностным
иммуноглобулином на В-клетке
Интернализация образованного
комплекса в В-клетку
I
Процессинг аллергена
I
Представление аллергенных пептидов на В-клетке
в комплексе с молекулами II класса МНС
I
Распознавание Т-клеточным рецептором комплекса
пептидов молекулами II класса МНС
I
।----Активация Т-клетки----->
Секреция IL-4/IL-13 Экспрессия CD40L (CD154)
на Т-клетке
I
Связывание CD40 на В-клетке с
CD40L (CD154) на Т-клетке
Активация транскрипции на Запуск,переключающий
специфическом регионе lg рекомбинации на синтез
локуса IgE
► Синтез IgE
Секреция IgE
136
обходимый сигнал для индукции синтеза IgE. Во-вторых, на
Т-клетке представляется лиганд для CD40 (CD40L, или, по
новой классификации, CD154). На покоящейся Т-клетке
CD 154 отсутствует. Представление этой молекулы на Т-клет-
ке, сопровождающее активацию клетки, делает Т-клетку пол-
ностью компетентной в индукции образования IgE. Связы-
вание CD40 на В-лимфоците своим лигандом (CD 154) на
Т-лимфоците обеспечивает другой сигнал (помимо секреции
IL-4), который запускает переключающую рекомбинацию на
синтез IgE (рис. 3.1, схема 3.1). В ходе этого процесса возни-
кают разнонаправленные взаимодействия, вовлекающие в ре-
акцию вспомогательные молекулы, которые усиливают про-
дукцию IgE.
3.2.2. Сигнал индукции синтеза IgE,
обеспечиваемый интерлейкином-4
(и интерлейкином-13)
Взаимодействие IL-4 со своим рецептором (IL-4R) на по-
верхности В-клетки — один из двух необходимых сигналов
для переключения на синтез IgE. В пользу того, что IL-4 яв-
ляется важнейшим и обязательным фактором для индук-
ции IgE, получены многочисленные и разнообразные доказа-
тельства в условиях как in vitro, так и in vivo. В условиях in
vitro IL-4 оказался единственным из использованных цито-
кинов (добавленным в рекомбинантной форме), способ-
ным индуцировать синтез IgE. Инактивация IL-4 в услови-
ях in vivo тормозила развитие IgE-ответа. Так, инъекция
мышам моноклональных анти-1Ь-4-антител резко угнетала
образование у них аллергенспецифических IgE в ответ на ал-
лергенную стимуляцию (инфицирование личинками гель-
минта).
Влияние предупреждения взаимодействия IL-4 с IL-4R на
образование IgE было проверено и другими способами. Один
из них состоял в связывании IL-4R. В качестве такого специ-
фического для этого рецептора агента использована мутантная
форма IL-4. Такой мутант (аминокислота тирозин замещена у
него в положении 124 аспартатом) связывается с IL-4R, но
при этом не способен передавать сигнал. Использование
такой мутантной формы IL-4 делало невозможным развитие
IgE-ответа.
Другой способ заключался в том, что достигалось предуп-
реждение действия IL-4 (за счет блокады взаимодействия IL-4
с IL-4R) связыванием его фрагментом IL-4R. Добавление к
системе отвечающих В- и Т-клеток растворимой формы вне-
клеточного домена рецептора блокировало переключение на
синтез IgE после внесения в систему IL-4.
137
Таким образом, очевидно, что блокада сигнала, осущест-
вляемого IL-4, приводит к блокаде переключения на синтез
IgE.
Наконец, опыты с «выбиванием» гена наглядно продемон-
стрировали, что при невозможности продуцировать IL-4 тор-
мозится способность синтезировать и IgE. Мыши, дефицит-
ные по гену для IL-4, утрачивают свойство образовывать ал-
лергенспецифические противопаразитарные IgE. В меньшей
степени, но все же подавлялся и IgGl-ответ. Продукция же
других изотипов у таких мышей не нарушалась.
Генетический анализ сцепления признаков показал, что
ген IL-4 или, по другим данным, участок, близко расположен-
ный к этому гену в регионе 5q 31.1 (для IL-13, IL-5, 1L-9), от-
ветствен за регуляцию продукции общего IgE. Отсюда сделано
допущение, что полиморфизм регуляторного региона этого
гена может привести к предрасположению к аллергии за счет
того, что при антигенной стимуляции будет возникать избы-
точная секреция цитокинов, индуцирующих синтез IgE. Прав-
да, исследования, выполненные в самое последнее время
одной из авторитетных групп, изучающих генетику аллергии,
не подтвердили данных о таком расположении гена, ответст-
венного за регуляцию синтеза IgE.
Надежность функционирования биологических систем
обеспечивается, в частности, существованием альтернатив-
ных механизмов. Поэтому можно было предполагать, что
ключевые звенья регуляции запуска синтеза IgE могут нахо-
диться под контролем альтернативных молекул. Видимо, это
действительно так. Сравнительно недавно показано, что IL-
13 имеет многие свойства, сходные с IL-4, в частности, обла-
дает активностью, запускающей синтез IgE. Гомология IL-4
и IL-13 в целом не очень высокая. Она соответствует всего
лишь 30 %. Однако участки, ответственные за связывание с
рецептором, оказываются сходными. Это находит отражение
в том, что мутантная форма IL-4 конкурентно блокирует свя-
зывание с рецептором как IL-4, так и IL-13. При этом соот-
ветственно блокируется индукция синтеза IgE, вызванная и
IL-4, и IL-13. Вместе с тем идентичности нет ни между сами-
ми интерлейкинами, ни между их рецепторами. Об отсутст-
вии идентичности IL-4 и IL-13 свидетельствует, например,
то, что некоторые лиганды, специфически связывающиеся с
IL-4, не связываются с IL-13. На различие рецепторов для
IL-4 и IL-13 указывает отсутствие действия IL-13 на Т-клет-
ки человека, на которых есть IL-4R (рецептор для IL-4), но,
видимо, нет рецептора для IL-13. Показано также, что IL-13R
(рецептор для IL-13) не имеет субъединицы у-цепи, общей
для рецепторов IL-2, IL-4, IL-7, IL-9 и IL-15. Сравнительная
характеристика биологической активности IL-4 и IL-13 при-
ведена в табл. 3.1.
138
Таблица 3.1. Биологическая активность IL-4 и IL-13
Тип клеток, продукция и секреция продуктов, функция IL-4 IL-13
Моноциты/макрофаги: МНС II класса t т
Fcy-рецепторы г г
CD23 т
провоспалительные цитокины г г
хемокины г г
IL-1 га т ?
антигенпредставляющая функция т ?
В-клетки: CD23, CD71, CD72, МНС II класса ? ?
пролиферация + +
дифференцировка (Ig-переключение) ? т
Т-клетки: пролиферация +
CD8a + —
Th 2 + —
Примечание, t усиление, Ф угнетение, + индукция, — отсутст-
вие действия.
3.2.3. Сигнал индукции синтеза fgE,
обеспечиваемый взаимодействием
CD40c CD154
Связывание CD40 на В-клетках лигандом CD154, пред-
ставленным на активированных Т-клетках, обеспечивает вто-
рой сигнал, необходимый для переключения на синтез IgE.
Действие CD 154 может быть замещено in vitro монокло-
нальными анти-СВ40-антителами. Демонстрация этого фе-
номена явилась первой иллюстрацией возможности актива-
ции В-клетки для синтеза IgE вызванным антителами связы-
ванием поверхностного антигена В-клетки. Эти же материа-
лы позволяют предположить, что соответствующие антитела
(например, аутоантитела) или иные лиганды в растворимой
форме в естественных условиях могут облегчать запуск син-
теза IgE, если присутствует IL-4- (или IL-13-) опосредован-
ный стимул.
CD40 является поверхностным гликопротеином с мол.
массой 50 кДа, представленным на В-лимфоцитах человека,
на активированных цитокинами моноцитах, фолликулярных
дендритных клетках, эпителиальных клетках (включая эпите-
139
лий вилочковой железы) и на клетках некоторых типов карци-
ном и меланом, но не на Т-клетках. CD40 принадлежит к су-
персемейству рецепторов фактора некротизации опухоли
(TNFR), которое включает TNFRI и TNFRII, рецептор факто-
ра роста нервной ткани (NGFR), CD30, CD27 и Fas(CD95).
Члены этого семейства имеют идентичную последователь-
ность аминокислотных остатков в экстраклеточных регионах,
которые содержат множественные повторения, богатые цисте-
ином. Общая структурная основа экстраклеточного домена
находит свое выражение в способности членов суперсемейства
TNFR взаимодействовать с семейством молекул, родственных
TNF. Однако сходство CD40 с указанными рецепторами,
представленными на В-клетке, не означает, что их связывание
с соответствующими лигандами приводит к тому же действию,
что и связывание CD40. Так, NGF и анти-СО40-антитела ока-
зывают противоположное действие на IL-4-опосредованную
индукцию синтеза IgE мононуклеарными клетками перифери-
ческой крови человека. В отличие от стимулирующего дейст-
вия анти-СО40-антител NGF тормозит индукцию синтеза IgE,
вызываемую интерлейкином-4, причем это торможение не
связано с угнетением функции рецептора для IL-4. Кроме
того, добавление анти-СГ)40-антител и IL-4 обеспечивает сиг-
нал, устойчивый к тормозящему действию NGF. Антагонисти-
ческий эффект NGF и IL-4 проявляется и в действии на син-
тез других изотипов иммуноглобулинов: IL-4 предупреждает
усиление продукции IgA и IgM, вызванное NGF. Таким обра-
зом, хотя NGFR и CD40 принадлежат к одному суперсемейст-
ву рецепторов и обладают пролиферативными эффектами на
В-лимфоциты, они по-разному взаимодействуют с IL-4-зави-
симой регуляцией продукции IgE.
CD40 играет важную роль в выживаемости, росте и диффе-
ренцировке В-клеток. Передача сигнала через CD40 спасает
В-клетку от индуцированного апоптоза. Отдельные виды
анти-СГ)40 моноклональных антител запускают выраженную
пролиферацию высокоочищенных покоящихся В-клеток в от-
сутствие других костимулов.
Лиганд для CD40—CD 154 представляет собой мембранный
гликопротеин, который транзиторно появляется на поверх-
ности активированных ТЫ- и ТЬ2-клеток. Клетки, трансфе-
цированные CD 154, вызывают синтезирование IgE и мыши-
ными, и человеческими В-клетками в присутствии IL-4, в то
время как растворимый белок — продукт слияния CD40-Ig —
тормозит IL-4-зависимый синтез IgE клетками периферичес-
кой крови человека. Эти данные показывают, что взаимодей-
ствие CD40 и CD 154 выступает в качестве необходимого сиг-
нала продукции IgE.
Центральная роль взаимодействия CD40 и CD 154 в запус-
ке синтеза IgE (или вообще в изотипическом переключении)
140
подтверждена обнаружением дефекта переключения у боль-
ных с rHnep-IgM-синдромом, связанным с Х-хромосомой
(HIGM), который возникает вследствие мутации CD154.
Такая мутация приводит к нарушению взаимодействия CD40
и CD154. Исключение этого взаимодействия у мышей, у кото-
рых отсутствует ген CD40 или ген CD154, приводит к тому,
что у таких животных не развивается IgG-, IgA- или IgE-ответ
на тимусзависимые антигены и в лимфоидных органах таких
животных (CD40 или CD154 КО мыши) не удается обнару-
жить соответствующие зародышевые центры. Ответ на тимус-
независимые антигены у них сохраняется.
Неспособность В-клеток новорожденных человека вклю-
чаться в синтез иммуноглобулинов также связана с неполно-
ценностью взаимодействия молекул CD40 и CD 154, поскольку
у новорожденных снижены как способность образования
CD 154 на Т-клетках, так и чувствительность В-клеток к дейст-
вию агонистов CD40.
Таким образом, все эти наблюдения, выполненные в усло-
виях in vitro и in vivo, позволили убедиться в том, что взаимо-
действию CD40 и CD154 принадлежит решающая роль в обра-
зовании зародышевых центров, активации В-клеток, изотипи-
ческом переключении и продукции антител.
Взаимодействие CD40 и CD 154 находится под довольно
жестким контролем. Т-клетки приобретают через CD40 ком-
петентность в активации В-клеток только после представле-
ния на них (Т-клетках) CD 154. Это требует в свою очередь
TCR-зависимой активации Т-клеток. Этот процесс, по-види-
мому, может происходить только на ограниченных анатоми-
ческих участках: во вторичных лимфоидных органах в зонах,
где происходят родственные Т- и В-клеточные взаимодей-
ствия.
Субпопуляция CD4+ Т-клеток памяти, находящаяся в за-
родышевых центрах, содержит преформированный CD 154,
который быстро и транзиторно появляется на клеточной по-
верхности после TCR-опосредованной активации. Скорость,
с которой Т-клетки могут представить CD 154 на своей по-
верхности, может быть фактором, определяющим судьбу кле-
ток зародышевых центров. Дело в том, что центроциты либо
покидают светлую зону в течение нескольких ближайших
часов после активации, либо погибают in situ вследствие
апоптоза. Функция CD 154 очень ограничена во времени.
Такое ограничение связано в известной степени с тем, что
взаимодействие этой молекулы с CD40 вызывает быстрый
эндоцитоз поверхностного CD 154, с одной стороны, и вы-
свобождение В-клетками растворимого CD40 — с другой. Эта
растворимая форма ингибирует синтез мРНК лиганда —
CD154.
141
3.2.4. Т-клетки у/S и регуляция синтеза IgE
Т-лимфоциты распознают процессированный антиген
(пептид) своими специфическими поверхностными рецепто-
рами, состоящими из пары полипептидных цепей (а/p или
у/8). В отличие от антител, которые специфически распознают
пространственную структуру (форму) антигена, Т-клеточные
рецепторы специфически распознают последовательность не-
больших пептидных фрагментов (эпитопов), являющихся про-
дуктом процессирования интактной молекулы антигена в
антигенпредставляющей клетке.
CD 154 представляется как на активированных а/рТ-клет-
ках, так и на у/8Т-клетках. Первые клетки представлены в пе-
риферических лимфоидных органах и ТСЯ+-тимоцитами, а
вторые находятся в эпителиальных тканях (в кишечном эпите-
лии, матке, языке, у мышей — в эпидермисе), но не в перифе-
рических лимфоидных органах. Поскольку представление
CD154 менее выражено на у/8Т-клетках, чем на а/рТ-клетках,
то первые менее эффективны в индукции синтеза IgE, однако
они проявляют компетентность в индукции изотипического
переключения на IgE в условиях in vitro в присутствии экзо-
генного IL-4. Мыши, дефицитные по а/рТ-клеткам, продуци-
руют иммуноглобулины всех изотипов с достаточно высоким
уровнем IgE и IgGl, что указывает на вполне ощутимую роль
у/8Т-клеток в изотипическом переключении и в условиях in
vivo. Потенциальная роль у/8Т-клеток в индукции IgE-ответа
подкрепляется и тем, что у/8Т-клетки способны различать на
разных стадиях инфекции ТЫ- и ТЬ2-индуцирующие патоге-
ны и продуцировать наборы цитокинов соответствующего
(ТЫ- или Th2-) профиля (особенности функционального про-
филя ThO-, ТЫ- и ТЬ2-клеток — табл. 3.2). Таким образом,
цитокины, продуцируемые у/8Т-клетками, не только осущест-
вляют элиминацию патогенов, но могут принимать участие в
формировании того цитокинового окружения, которое влияет
на дифференцировку CD4+ Т-клеток в ТЫ- или ТЬ2-клетки.
Таблица 3.2. ThO-, ТЫ- и ТЪ2-фуикциональиый профиль
Функция Thl ThO Th2
Секреция цитокинов IL-2
IFN-y + + + + + + —
TNF-P + + + + —
TNFa + + + + + +
GM - CSF + + +
IL-3 + + + +
142
Продолжение табл. 3.2
Функция ТЫ ThO Th2
IL-4 + + + +
IL-5 — + + + +
IL-6 4- +
IL-9 — + +
IL-10 + +
IL-13 + + + + +
Цитолитическая активность + + + + + ±
Синтез IgE — ± + + +
Синтез IgM, IgA, IgG:
низкое отношение Т- и В-клеток 4- 4- + + 4- 4-
высокое отношение Т- и В-клеток + + + + +
Представляет интерес то, что адоптивный перенос неболь-
шого числа у/8Т-клеток от толерантных к овальбумину мышей
избирательно подавляет ТЬ2-зависимый синтез IgE (без влия-
ния на IgG-ответ). Этот тип клеток принадлежал к СО8+-клет-
кам и продуцировал большие количества интерферона-у
(IFN-y).
Таким образом, эти материалы свидетельствуют о том, что
у/8Т-клетки могут не только принимать участие в регуляции
IgE-ответа, но и оказывать разное по направлению действие
на этот процесс в зависимости от условий.
3.2.5. Независимая от взаимодействия CD40
с CD154 индукция синтеза IgE
На существование способов такой индукции указывает ряд
сведений, которые, естественно, еще потребуют специального
экспериментального анализа. Тем не менее эти сведения за-
служивают упоминания.
Так, неактивированные фиксированные Т-клетки перифе-
рической крови (т.е. без представленного на них CD154) и Т-
клетки больных HIGM (с функционально неактивным CD 154)
выступают в роли клеток-помощников тонзиллярным В-клет-
кам в запуске синтеза IgE в присутствии IL-4. Вспомогатель-
ная функция достигалась стафилококковым суперантигеном,
адсорбированным на Т-клетках до момента их фиксации, при-
чем обработка Т-клеток суперантигеном не приводила к появ-
лению CD 154. Так возникает подозрение о возможности акти-
вации В-клеток независимо от представления CD154 на
Т-клетках. Правда, следует еще убедиться в том, что такие аль-
143
тернативные воздействия не замещают взаимодействие CD40—
CD 154 за счет включения в общий путь, следующий за связы-
ванием CD40 с рецептором.
Известно также, что стимуляция интерлейкином-4 и виру-
сом Эпштейна — Барр (EBV) вызывает независимый от Т-кле-
ток синтез IgE В-лимфоцитами человека. В этом случае эф-
фект может быть объяснен возможностью включения в общий
путь активации, опосредуемый CD40. Для этого привлекают
данные о существовании так называемого фактора 1, ассоции-
рованного с рецептором для CD154 (CRAF-1). CRAF-1 рас-
сматривают как один из компонентов СО40-сигнальной сис-
темы. С-конец CRAF-1 непосредственно и специфически вза-
имодействует с цитоплазматическим участком CD40. CRAF-1
может также взаимодействовать с цитоплазматическим доме-
ном так называемого латентного инфекционного мембранно-
го белка 1 (LIMP-1), который кодируется EBV и может инду-
цировать трансформацию В-лимфоцитов. Таким образом,
способность EBV индуцировать синтез IgE может быть связа-
на с кодируемым этим вирусом белком, активирующим CD40-
сигнальный путь.
Синтез IgE может быть также запущен в высокоочищенной
клеточной взвеси нормальных или лейкозных В-клеток, не не-
сущих поверхностный IgE (запуск синтеза de novo), добавле-
нием IL-4 и гидрокортизона. Эффект специфичен для глюко-
кортикостероида, так как он не воспроизводится стероидными
половыми гормонами. Пока не дано приемлемого объяснения
такому механизму запуска синтеза IgE.
3.2.6. Вспомогательные молекулы,
усиливающие и сдерживающие влияния
Показано, что несколько пар вспомогательных молекул
(CD27 и В7, LFA-1 и ICAM-1, CD2 и CD58) участвуют в Т- и
В- клеточных взаимодействиях, приводящих к синтезу IgE.
Взаимодействия в пределах этих пар лиганд — рецептор спо-
собствуют активации и/или повышают опосредуемую Т-клет-
ками активацию В-клеток, которая следует за связыванием
CD40 с CD 154. Наибольшую вспомогательную роль в этом
процессе выполняет рецепторно-лигандная пара CD28 и В7
(CD80). CD28 КО мыши (лишенные гена CD28) имеют сни-
женный исходный уровень иммуноглобулинов, и у них подав-
лена способность к изотипическому переключению после ин-
фицирования вирусами.
Связывание CD28 на Т-клетках с В7 (CD80) на В-клетках
может быть составной частью механизма, который усиливает
клеточное взаимодействие между Т- и В-клетками, опосредуе-
мое связыванием CD40 и CD 154. Связывание CD40 приводит
144
Схема 3.2. Пример подключения вспомогательных,
усиливающих синтез IgE взаимодействий (CD28 + CD80)
CD40 (В-клетка) + CD154 (Т-клетка)
г~
Экспрессия CD80 (В-кпетка)
+
CD28 (Т-клетка)
!
Усиление
___________________________________L____________________________________
экспрессии CD154 (Т-клетка) секреции IL-4 дифференцировки ТЬ2-клеток
(Т-клетка)
I
Усиление экспрессии CD80 (В-клетка)
к представлению В7 (CD80) на В-клетках. Связывание же
CD28 увеличивает представление CD154 на Т-клетках и, что
особенно важно, усиливает секрецию IL-4 и дифференциров-
ку ТЬ2-клеток. IL-4 в свою очередь увеличивает выраженность
представления В7 (CD80) на В-клетках (схема 3.2).
Таким образом, взаимодействие CD28 и В7 (CD80) может
усиливать и секрецию IL-4, и СО40-опосредованную актива-
цию В-клеток и тем самым потенцировать оба обязательных
сигнала, необходимых для индукции синтеза IgE. В согласии
с этим представлением находятся данные о том, что моно-
клональные анти-СО28-антитела подавляют синтез IgE, осу-
ществляемый в присутствии 1L-4 и преактивированного клона
Т-клеток.
Значительное увеличение представительства В7 (CD80)
возможно также и при перекрестном связывании молекул II
класса МНС на В-клетках. Это усиление особенно выражено
при одновременном связывании ICAM-1.
Слабый IgE-индуцирующий сигнал, обеспечиваемый свя-
зыванием LFA-3 (CD58) с CD2, по-видимому, не зависит от
механизма, опосредуемого CD40.
Из сказанного следует предположение о том, что те клет-
ки, которые, во-первых, способны синтезировать IL-4 (и/или
IL-13) и, во-вторых, могут представлять CD154, при опреде-
ленных обстоятельствах замещают функцию Т-клеток в ин-
дукции синтеза IgE. Наиболее вероятными кандидатами на
исполнение такой роли могут быть тучные клетки и базофилы.
Эти клетки человека способны при активации секретировать
IL-4 (и IL-13) и представлять CD154. Поэтому они в принципе
могли бы обеспечивать оба необходимых сигнала для синтеза
IgE. Действительно, при определенных экспериментальных
10— 1385 145
условиях показана способность этих клеток индуцировать
синтез IgE. Однако наиболее вероятно участие этих клеток не
в первичной индукции IgE-ответа, а в его усилении. Это сле-
дует из того, что интенсивная активация клеток и соответст-
венно секреция IL-4 осуществляются IgE-зависимым механиз-
мом. Иными словами, для вовлечения тучных клеток и базо-
филов в регуляцию синтеза IgE требуется присутствие в орга-
низме аллергенспецифического IgE и соответствующего ал-
лергена. Если же это условие соблюдено, то вероятно, что при
очередной аллергенной стимуляции тучных клеток и базофи-
лов они могут потенцировать IgE-ответ на тот же самый ал-
лерген. Правда, нельзя исключить и того, что в случае поступ-
ления в организм в этот период новых аллергенов тучные
клетки и базофилы могут стать соучастниками индукции IgE-
ответа на новые аллергены. Такая вероятность могла бы объ-
яснить хорошо известный феномен постепенного расширения
спектра повышенной чувствительности к разным аллергенам у
пациентов с аллергическими заболеваниями.
Сравнительно давно было показано, что одним из эффек-
тов IL-4 может быть увеличение выраженности представления
на некоторых клетках низкоаффинного рецептора для IgE.
Этот вид рецептора в отличие от высокоаффинного рецептора
для IgE, присутствующего на тучных клетках, базофилах и не-
которых других клетках, обозначается как рецептор второго
типа — Fee RII (CD23). До последнего времени появляются
сведения, которые заставляют признать участие CD23 в регу-
ляции синтеза IgE.
CD23 является дифференцировочным антигеном В-кле-
ток, несущих поверхностные иммуноглобулины М и D. Этот
антиген представляет собой сиалогликопротеин с мол. массой
порядка 45 кДа, содержит одну N-связанную углеводную цепь
комплексного типа. Его аминокислотная последовательность
выведена из последовательности клонированной ДНК. Рецеп-
тор состоит из 320 аминокислотных остатков. Положение его
в клеточной мембране необычно. Аминоконцевой участок
ориентирован внутрь клетки, а карбоксильный — наружу. Ре-
цептор состоит из небольшого цитоплазматического домена
(23 аминокислотных остатка), трансмембранного домена
(20 аминокислотных остатков) и большого внеклеточного до-
мена (277 аминокислотных остатков). CD23 описан на многих
клетках, включая В-клетки, моноциты, эозинофилы, тромбо-
циты, эпидермальные клетки Лангерганса.
Особенным свойством этого белка является способность
к аутопротеолитическому перевариванию. За счет этого ре-
цептор распадается на растворимые фрагменты с мол. массой
37; 33; 25—27 и 12 кДа. Все эти фрагменты (за исключением
последнего) связывают IgE (IgE-связывающие факторы —
IgE-СФ).
146
Известно два подтипа низкоаффинного рецептора для
IgE — CD23a и CD23b. Они отличаются друг от друга несколь-
кими аминокислотными остатками цитоплазматического до-
мена, а внеклеточные домены являются идентичными. CD23a
выявляется лишь на нестимулированных В-клетках, а
CD23b — на В-клетках, стимулированных IL-4, а также на мо-
ноцитах, эозинофилах, Т-клетках.
Регуляция выраженности представления CD23 и соответ-
ственно регуляция образования IgE-СФ связаны между собой:
чем больше представлен рецептор, тем более вероятно образо-
вание растворимых IgE-СФ.
Представление CD23 индуцируется IL-4 и подавляется
IFN-a. IFN-y тормозит представление CD23 на В-клетках, но
не на моноцитах, тромбоцитах, клетках Лангерганса. Повы-
шенное присутствие CD23 обнаруживается при некоторых па-
тологических состояниях, для которых характерно усиление
образования IgE: при острых аллергических заболеваниях, при
паразитарных инфекциях.
Вполне вероятно, что CD23 выполняет различные функ-
ции и спектр участия этой молекулы в иммунологически опо-
средованных реакциях может быть шире, чем сейчас представ-
ляется. Во всяком случае, связанный с мембраной CD23 опо-
средует IgE-зависимую цитотоксичность, направленную про-
тив паразитов и выполняемую эозинофилами, макрофагами,
тромбоцитами. Кроме функций, обусловленных сродством к
IgE, рецептор может исполнять роль молекулы клеточной
адгезии за счет своих лектиноподобных свойств. У лектиново-
го домена CD23 обнаружена гомология с семейством молекул
клеточной адгезии (ELAM1, GMP 140).
В прямых испытаниях показано, что высокоочищенный
естественный IgE-СФ или рекомбинантный IgE-СФ с мол.
массой 37 кДа усиливает IL-4-опосредо ванный синтез IgE. Ве-
роятнее всего, что IgE-СФ, не индуцируя синтез IgE, действу-
ет на более поздний этап продукции IgE, чем IL-4, — иными
словами, на активированные IL-4 IgE-секретирующие клетки.
CD23 взаимодействует главным образом с CD21 на тех В-
клетках, которые преимущественно осуществляют продукцию
IgE. Антитела к CD23 тормозят in vitro IL-4-опосредованный
синтез IgE и антигенспецифический IgE-ответ изотипспеци-
фическим образом. На секретирующих IgE-клетках CD23 не
представлен. Поэтому тормозящий эффект антител к CD23 на
возникший синтез IgE опосредуется, по-видимому, не пря-
мым действием на секретирующие иммуноглобулин клетки, а
нейтрализацией растворимых IgE-СФ, которые высвобожда-
ются из IgM/IgD-несущих В-клеток, стимулированных IL-4,
или из моноцитов.
Поддержание синтеза IgE на определенном уровне может
зависеть и от действия других лимфокинов. Так, синтез IgE
ю*
147
человека, вызванный IL-4, усиливается IL-5 и IL-6. Для до-
стижения оптимального уровня продукции IgE, видимо, необ-
ходим IL-6, так как антитела именно к IL-6 существенно по-
давляют секрецию IgE.
Возникший синтез IgE поддерживается на определенном
уровне и за счет других факторов, которые могут оказывать
сдерживающее влияние на синтез IgE. IFN-a, как правило,
подавляет продукцию IgE и IgE-СФ клетками, стимулирован-
ными IL-4. В испытаниях на мышах IFN-y как в условиях in
vivo, так и на клетках in vitro тормозит продукцию IgE. Данные
на человеческом материале не столь однозначны. IFN-y как
подавляет, так и усиливает вызванный IL-4 синтез IgE клетка-
ми человека, что может быть связано с невыясненными осо-
бенностями условий испытаний. В отдельных наблюдениях,
выполненных на больных, страдающих аллергией, показано,
что введение таким больным IFN-y не оказывало заметного
влияния на уровень IgE в сыворотке крови. Таким образом,
пока нельзя сделать окончательное заключения о роли IFN-y в
регуляции синтеза IgE у человека.
В модельных же опытах на животных показано, что уро-
вень продукции IgE зависит от соотношения образующихся
IL-4 и IFN-y. Последнее в свою очередь определяется пре-
имущественной активацией клеток, продуцирующих IL-4 или
IFN-y. У мышей эти лимфокины образуются разными клет-
ками: Thl-клетки продуцируют IFN-y, а также IL-2 и лимфо-
токсин, а ТТ12-клетки — IL-4 и IL-5, но не IL-2 и IFN-y.
У человека хелперные Т-клетки не могут быть подразделены
на два аналогичных типа. Большинство клеток, продуцирую-
щих IL-4, образуют одновременно либо IFN-y, либо IL-2,
или оба этих медиатора. И все же некоторые сведения о вза-
имоотношении IFN-y и IL-4 в продукции IgE могут быть по-
лучены на клеточных системах человека in vitro. Стимуляция
аллогенными клетками вызывает продукцию IFN-y в клеточ-
ной культуре. Как и следует ожидать, такая культура клеток
не продуцирует IgE. Если же в нее в самом начале культиви-
рования внести IL-4, то удается вызвать синтез IgE и пода-
вить образование IFN-y.
Высвобождаемый активированными макрофагами IL-12
тормозит синтез IgE, по-видимому, за счет усиления продук-
ции IFN-y и частично — за счет угнетения продукции IL-4, о
чем будет сказано далее.
Известно, что связывание с лигандом молекулы CD 14 на
моноцитах активирует эти клетки и дает отрицательный сиг-
нал, останавливающий пролиферацию Т-клеток. Вместе с тем
есть данные, свидетельствующие о том, что связывание с ли-
гандом CD 14 может реализоваться действием непосредствен-
но на В-клетки. Так, связывание определенных эпитопов
CD14 моноклональными антителами тормозит IL-4-зависи-
148
мый синтез IgE мононуклеарными клетками периферической
крови человека. Такое торможение не связано ни с высвобож-
дением тормозных монокинов (IL-10, TGF-p, простагланди-
нов), ни с угнетением накопления усиливающих IgE-ответ
факторов. Кроме того, торможение продукции IgE, вызванное
связыванием CD 14, воспроизводится на обедненной Т-клет-
ками клеточной взвеси, стимулированной 1L-4 и анти-СО-40
моноклональными антителами или IL-4 и гидрокортизоном.
Взятые вместе, эти данные позволяют допустить, что лигация
(связывание с лигандом) CD 14 оказывает сдерживающее регу-
ляторное действие непосредственно на В-клетки.
Пока точно не установлены ни временные параметры вы-
свобождения усиливающих и тормозных посредников, ни
конкретные точки приложения их действия, ни их взаимная
связь в ходе IgE-ответа, но уже стало очевидным, что выясне-
ние всех этих вопросов может объяснить механизм удержания
продукции IgE на определенном уровне. Примеры потенци-
альных взаимодействий и молекул — участников механизма
индукции, усиления или торможения образования IgE приве-
дены в табл. 3.3.
Т аб л и ц а 3.3. Взаимодействия и молекулы, запускаю-
щие, тормозящие или усиливающие синтез IgE
Взаимодействия/факторы Функция
Антиген (пептид) в комплексе с МНС II класса TCR/CD3 CD40/CD154 IL-4 IL-13 Индукция переключения на синтез IgE в В-клет- ках
IFN-y IFN-a TGF-P IL-8 IL-12 Моновалентный SCD23/CD21 PGE2 PAF Торможение синтеза IgE В-клетками
IL-2 IL-5 IL-6 IL-9 CD23/CD21 CD80/CD28? Усиление синтеза IgE В-клетками
149
3.2.7. Избирательность включения
П2-клеток в IgE-omeem
До сих пор не получено удовлетворительного ответа на во-
прос о том, почему в одних случаях возникает преимуществен-
но IgE-ответ (и соответственно преимущественное накопле-
ние и включение в реакцию ТЬ2-клеток), а в других — IgG-
ответ (и соответственно преимущественное накопление и
включение в реакцию Thl-клеток). Очевидным является лишь
то, что единственного объяснения этого феномена быть не
может. Скорее всего, механизм такой избирательности может
быть понят при учете нескольких факторов.
Во-первых, преимущественное накопление ТЬ2-клеток и
индукция IgE-ответа могут определяться природой самого
антигена, вернее природой полученных процессированием
антигена пептидов, представляемых в ассоциации с молекула-
ми II класса МНС Т-клеточному рецептору (TCR). Принци-
пиально функциональная неоднородность разных участков
(детерминант, эпитопов) одного и того же антигена проиллю-
стрирована на примере наименьшего по размеру (мол. масса
4500 кДа) антигенного компонента аллергена пыльцы амбро-
зии. Показано, что для одного и того же пациента эпитопы ал-
лергена, ответственные за связывание с IgE, отличаются от
эпитопов, ответственных за связывание с IgG. Прямые испы-
тания, выполненные на лицах, инфицированных гельминтами
и не имевших в анамнезе указаний на атопические заболева-
ния, установили, что антигенспецифические Т-клеточные
клоны, полученные от таких лиц, имеют характеристики Th2-
клеток. Эти клетки продуцировали много IL-4 и мало IL-2 и
IFN-y, а в условиях in vitro индуцировали синтез IgE. У боль-
ных атопическими заболеваниями аллергенспецифические
CD4+ Т-клеточные клоны, естественно, имели характеристики
ТЬ2-лимфоцитов и обладали вспомогательным действием на
синтез IgE in vitro. Одновременно у этих же самых пациентов
Т-клеточные клоны, специфичные по отношению к неаллер-
генным антигенам, имели характеристику ТЫ-клеток. Поэто-
му не лишним будет подчеркнуть известный факт, состоящий
в том, что у атопических больных сохранена способность фор-
мировать и IgG-ответ при неаллергенной антигенной стиму-
ляции.
Интересно напомнить, что при специфической иммуноте-
рапии атопических больных происходит образование аллер-
генспецифических IgG и перестройка Т-клеточного предста-
вительства в сторону Thl-клеток. Очевидно, что в этом случае
у того же самого пациента достигается переключение на дру-
гой — ТЫ-зависимый IgG-ответ той же аллергенной специ-
фичности. Иными словами, изменение режима подачи аллер-
генного материала приводит к изменению характера иммунно-
150
го ответа. Поэтому, вероятнее всего, что избирательное воз-
никновение ТЬ2-ответа зависит от природы антигена, с одной
стороны, и от пути и режима его поступления в организм в ес-
тественных условиях — с другой.
Во-вторых, лимфоцитарный Т-клеточный профиль может
зависеть от наследственных факторов. Из многочисленных ар-
гументов, которые обычно приводятся в пользу такой связи,
наиболее убедительными являются следующие два: 1) CD4+ Т-
клеточные клоны атопических лиц способны все же продуци-
ровать заметные количества IL-4 и IL-5 в ответ на стимуляцию
бактериальными антигенами (PPD и стрептокиназа), которые
обычно вызывают у неатопических лиц ответ, характеризую-
щийся Thl-цитокиновым профилем; 2) Т-клеточные клоны,
полученные из лимфоцитов пупочной вены новорожденных
атопических родителей, продуцировали больше IL-4, чем лим-
фоциты новорожденных неатопических лиц.
Кроме того, средовые факторы также могут приобретать ре-
шающее значение в изменении регуляции иммунного ответа в
сторону Th2-THna. В качестве примера можно привести то, что
одновременное воздействие аллергена и содержащих полиаро-
матические углеводороды частиц выхлопных газов вызывает
значительное усиление синтеза мРНК цитокинов (IL-4, IL-5,
IL-6, IL-10, IL-13), являющихся маркерами ТЬ2-ответа, и 16-
кратное увеличение продукции аллергенспецифических IgE. Та-
кого рода воздействия, создающие экзогенно вызванные формы
предрасположенности к аллергии, приобретают сейчас особое
значение как наиболее потенциально опасные.
Надо принимать во внимание и существование комплекс-
ных цитокиновых влияний, которые усиливают как диффе-
ренцировку Th-клеток, так и синтез и секрецию IgE.
Образуемый ТЪ2-клетками IL-4 тормозит продукцию IFN-y
Т-клетками человека и тем самым в еще большей степени сме-
щает дифференцировку предшественников в ТЬ2-клетки,
которые в свою очередь дополнительно секретируют IL-4.
Напротив, IFN-y способствует дифференцировке клеток в
ТЫ-лимфоциты. Возникает ситуация, когда антиген запускает
ответ ТЫ- или ТЬ2-типа, выбор которого зависит от природы
и способа воздействия антигена и от генетических или средо-
вых предпосылок. Характер антигенспецифических Т-клеток
прогрессивно смещается в выбранную сторону, и в процесс
вовлекается дополнительное число Т-клеток того же цито-
кинового профиля. В этом отношении IL-13, лимфокин
ТЬ2-клеток, индуцирующий продукцию IgE, отличается от
других лимфокинов. Он не оказывает действия на Т-клетки и
потому вряд ли участвует в описанном процессе смещения
клеток в сторону ТЬ2-лимфоцитов. Этим можно объяснить то,
что у мышей, у которых отсутствует ген IL-4, но имеется ген
IL-13, ТЬ2-ответ оказывается слабовыраженным.
151
Опосредованное IFN-y смещение дифференцировки Т-кле-
ток в сторону ТЫ-лимфоцитов может индуцироваться дру-
гими монокинами, например IL-12. Некоторые вирусы и
внутриклеточные бактерии обладают стимулирующим дейст-
вием на продукцию IL-12 макрофагами. Этот лимфокин инду-
цирует образование IFN-y Т- и NK-клетками (NK — естест-
венные киллеры). Таким образом, антигенпредставляющая
(макрофаг) клетка может представить антиген Th-клеткам
одновременно с индукцией цитокинов, обеспечивающих диф-
ференцировку клеток в сторону ТЫ-фенотипа. Тормозящее
действие IL-12 на синтез IgE дополняется тем, что он оказы-
вает влияние не только на клетки-продуценты IFN-y, но и на
продуценты IL-4. Последнее проявляется в торможении IL-12
вызванной антигеном продукции IL-4. На В-клетки IL-12, по-
видимому, не действует, так как он не подавляет синтез IgE во
взвеси очищенных В-клеток.
Приведенные выше сведения были привлечены для обо-
снования предположения о том, что повышение заболевае-
мости аллергическими болезнями в развитых странах может
быть связано, хотя бы отчасти, с уменьшением заболеваемости
в детском возрасте (т.е. в период, когда формируется сенсиби-
лизация к аэроаллергенам) инфекционными болезнями, по-
скольку снижается вероятность стимуляции и поддержания
иммунного ответа ТЫ-типа, сдерживающего, по этой гипоте-
зе, Т112-тип. Подтверждение такого предположения получено
в эпидемиологических исследованиях, показавших, что сни-
жение заболеваемости туберкулезом и, возможно, другими ин-
фекциями в детском возрасте совпадает с утяжелением тече-
ния и повышением распространенности атопических заболе-
ваний за последнее десятилетие в развитых странах.
3.2.8. Возможные способы оценки опосредуемого
ТН2-клетками аллергического ответа
в клинических условиях
Вне зависимости от установления конкретных механизмов
избирательной дифференцировки Т-клеток при аллергичес-
ком ответе в сторону 'П12-лимфоцитов идентификация их по
надежным маркерам доступными методами имела бы важное
значение для предсказания формирования предрасположен-
ности к аллергии.
Обоснованным подходом к решению этого вопроса являет-
ся прежде всего определение спектра лимфокинов, продуци-
руемых спонтанно и при соответствующей стимуляции клет-
ками, полученными от пациента. Однако этот прием является
довольно трудоемким, чтобы быть использованным в широ-
кой клинической практике. Более подходящей была бы оцен-
152
ка клеточных маркеров, свидетельствующих об активации
ТЬ2-опосредованного пути иммунного ответа. Кандидатом на
такой маркер может быть CD30. Между способностью Th-кле-
ток продуцировать лимфокины ТЬ2-профиля и представлени-
ем на них CD30 существует прямая связь. Большинство аллер-
генспецифических ТН2-клеток клонов человека, активирован-
ных соответствующим аллергеном, демонстрирует наличие
CD30 и одновременно секретирует IL-4 и IL-5. Это свидетель-
ствует, в частности, о том, что представление CD30 может
быть неким образом связано с дифференцировочным и акти-
вационным этапом Т-клеток человека, продуцирующих цито-
кины, характерные для ТЬ2-клеток.
Оценка выраженности представления CD30 может претен-
довать на место показателя, дающего общую характеристику
клеточного профиля IgE-ответа. Синтез шРНК для CD30 по-
вышается в мононуклеарных клетках периферической крови
атопических больных, но растворимая форма CD30 при этом
не обнаруживается в крови. Между уровнем синтеза шРНК
для CD30 и уровнем IgE существует прямая связь. CD30
может быть представлен как на Т-, так и на В-лимфоцитах. На
В-лимфоцитах он представлен главным образом на CD19+-
клетках (CD19 имеет отношение к активации В-клеток), что
установлено при использовании взвеси чистых В-клеток ато-
пических лиц.
Выяснение предсказательной или диагностической значи-
мости оценки этого и/или других маркеров имеет важное зна-
чение как для выявления предрасположенности к аллергии,
так и для определения эффективности терапевтических воз-
действий.
3.3. Некоторые замечания
о противоаллергическом лечении
Начальные этапы аллергического процесса, заключающие-
ся в образовании IgE-антител, являются объектом воздействия
противоаллергических лечебных приемов. Эти приемы состо-
ят в торможении образования IgE-антител или в переключе-
нии иммунологического ответа на синтез другого класса анти-
тел, которые лишены способности фиксироваться на клетках
воспаления, но конкурируют с IgE-антителами за связывание
с аллергеном и тем самым уменьшают вероятность его дейст-
вия на клетки, вооруженные IgE-антителами. Таким свойст-
вом обладает давно используемый в практической аллерголо-
гии метод аллергенспецифической гипосенсибилизации (им-
мунотерапии), состоящий во введении в сенсибилизирован-
ный организм возрастающих доз аллергена, к которому выяв-
лена повышенная чувствительность. В настоящее время разра-
153
батываются и предложены разные варианты и модификации
этого метода, направленные на повышение его клинической
эффективности и безопасности. Этот метод является важней-
шим, но далеко не единственным способом лечения больных
аллергическими заболеваниями. Другие методы (фармаколо-
гические в том числе) направлены на торможение последую-
щих звеньев аллергического процесса: вовлечения в ответ кле-
ток воспаления, секреции из них медиаторов, действия медиа-
торов на периферические ткани. Исходя из знаний о много-
этапное™ аллергического процесса и взаимосвязи друг с дру-
гом отдельных этапов, сформулировано важнейшее положе-
ние терапии аллергических состояний. Это положение заклю-
чается в том, что противоаллергическое лечение может быть
высокоэффективным только в том случае, если использован
комплексный подход, учитывающий терапевтическое действие
на разные звенья аллергического процесса, а также форму,
стадии, степень тяжести конкретного аллергического заболе-
вания и медицинский, биологический и социальный «портрет*
конкретного пациента. Поэтому понимание комплексного
подхода к лечению аллергических заболеваний и своевремен-
ного использования набора тех или иных лечебных приемов
строится на знании обо всем аллергическом процессе и дано в
специальных обобщающих работах [Гущин И.С., 1998; Пыц-
кий В.И. и др., 1998; КауА.В., 1997].
Список литературы
Адо А.Д. Общая аллергология. — М.: Медицина, 1978.
Гущин И. С. Аллергическое воспаление и его фармакологический кон-
троль. — М.: Фармарус Принт, 1998. — 256 с.
Пыцкий В.И., Адрианова Н.В., Артамасова А.В. Аллергические заболе-
вания. — М.: Триада-Х, 1998. — 454 с.
Abbas К.A., Lichtman А.Н., PoberJ.S., eds. Cellular and Molecular Immu-
nology. 2nd/Ed. W.B.Saunders Co. — Philadelphia, 1994.
de Boer B.A., Kruize Y.C., Rotmans P.J., Yazdanbakhsh M. Interleukin-12
suppresses immunoglobulin E production but enhances immunoglobulin
G4 production by human peripheral blood mononuclear cells//Infect.
Immun. - 1997. - Vol. 65. - P. 1122-1125.
Bonnefby J.Y., Gauchat J.F., Life P. et al. Pairs of surface molecules in-
volved in human IgE regulation: CD23—CD21 and CD40—CD40L//
Europ. Respir. J. — 1996. — Vol. 22, suppl. — P. 63s—66s.
Bonnefby J.Y., Gauchat J.F., Lecoanet-Henchoz S. et al. Regulation of
human IgE synthesis//Ann. NY Acad. Sci.— 1996.— Vol. 796.— P. 59—71.
154
Diaz-Sanchez D. The role of diesel exhaust particles and their associated
polyaromatic hydrocarbons in the induction of allergic airway disease//Al-
lergy. — 1997. — Vol. 52, suppl. 38. — P. 52—56.
Esnault S., Benbemou N., Lavaud F., Guenounou M. Spontaneous CD30
mRNA expression in peripheral blood mononuclear cells from atopic pa-
tients with high IgE serum levels//Clin. exp. Immunol. — 1996. — Vol.
106. - P. 67-72.
Kay A.B. Concepts of allergy and hypersensitivity//Allergy and Allergic
Diseases/Ed. by A.B.Kay. — Oxford: Blackwell Sci., 1997. — Vol. 1. —
P. 23-35.
Keane-Myers A., Gause W.C., Linsley P.S. et al. B7-CD28/CTLA-4
costimulatory pathways are required for the development of T helper cell
2-mediated allergic airway responses to inhaled antigens//J. Immunol. —
1997. - Vol. 158. - P. 2042-2049.
Leung D.Y. Mechanisms controlling the human immunoglobulin E re-
sponse: new directions in the therapy of allergic diseases//Pediatr. Res. —
1993. - Vol. 33. - P. S56-S61.
Levy F., Kristofic C., Heusser C., Brinkmann V. Role of IL-13 and CD4 T
cell-dependent IgE production in atopy//Int. Arch. Allergy Immunol. —
1997. - Vol. 112. - P. 49-58.
Renz H. The central role of T-cells in allergic sensitization and IgE regula-
tion//Exp. Dermatol. — 1995. — Vol. 4. — P. 173—182.
Romagnani S. Development of type 2 T-helper cells in allergy//Curr. Opin.
Immunol. — 1994. — Vol. 6. — P. 838—845.
Romagnani S. The «Th2 hypothesis» in allergy.— In: Progress in Allergy
and Clinical Immunology/Eds. — A.K.Ohling, J.G.Huerta. — Seattle:
Hogrefe and Huber Publisher, 1997. — P. 12—16.
Vercelli D., Geha R.S. Structure, function and synthesis of IgE. — In: Al-
lergy, 2nd ed/Ed by A. Kaplan. — Philadelphia: W.B. Saunders, 1997. —
P. 82-91.
Лекция 4
Опухолевый рост: ткани, клетки, молекулы
А.В.Лихтенштейн,\В.С.Шапот
Опухоль разрушает человека уникальным и ужасаю-
щим способом. Плоть от его собственной плоти, она
в силу каких-то неведомых причин превращается в
нечто неуправляемое, хищное, неистово разножаю-
щееся. Опухоль — самая упорная, страшная и в то
же время наименее познанная из всех болезней челове-
ка, несмотря на 70 лет ее экспериментального изуче-
ния... В чем причина такого превращения?
P.Rous (лауреат Нобелевской премии 1966 г.)
Введение
Прогресс, достигнутый в последние годы в биологии, обу-
словлен в определяющей степени теми чрезвычайными уси-
лиями, которые предпринимаются мировым научным сообще-
ством в борьбе со злокачественными новообразованиями.
Причины канцерогенеза коренятся так глубоко в самой осно-
ве живого и столь тесно переплетены с такими фундаменталь-
ными явлениями, как деление клетки, межклеточные взаимо-
действия, смерть, старение и бессмертие, что проблема рака
вышла за пределы чисто медицинские и приобрела масштабы
интеллектуального вызова человечеству. Исследование канце-
рогенеза, изначально преследовавшее вполне определенную
прикладную цель (лечение рака), приобрело общебиологичес-
кое значение еще и потому, что здесь произошло слияние со-
вершенно, казалось бы, не связанных между собой областей.
Опухолевый рост — та общая платформа, на которой объеди-
нились усилия генетиков, эпидемиологов, медиков, молеку-
лярных биологов, эмбриологов, включая исследователей столь
экзотических объектов, как, например, нематоды (именно
здесь был достигнут решающий прорыв в исследовании апоп-
тоза). Этот факт подчеркивает замечательный консерватизм
фундаментальных биологических процессов, позволяющий
использовать в исследованиях наряду с традиционными экс-
периментальными животными значительно более простые и
доступные анализу организмы.
Только в последней четверти XX в. появились реальные
предпосылки решения проблемы злокачественных новообра-
зований. Главные из них — расшифровка структуры, функции
156
и регуляции гена, развитие генной и клеточной инженерии и
особенно компьютеризация научного поиска (создание до-
ступных любому исследователю мировых банков данных, лег-
кость межлабораторных коммуникаций, возможность деталь-
ного анализа и сопоставления гигантских генетических текс-
тов, выявление гомологии генов, принадлежащих эволюцион-
но далеким видам).
Достигнутый благодаря этому прогресс сопоставим по
своему масштабу и значению для повседневной жизни челове-
ка с научными революциями начала века в физике. Научная
революция в биологии, совершаемая на наших глазах, еще да-
леко не завершена. На начало следующего века отнесено
окончание проекта «Геном человека», который имеет целью
расшифровку всех нуклеотидных последовательностей ДНК
человека для проникновения в суть генетических программ,
управляющих жизнью клетки и организма.
Интенсивность исследований в этой области и их специа-
лизация чрезвычайно велики. Даже первые шаги внутри мик-
рокосмоса клетки дают представление о колоссальной слож-
ности ее структурной и функциональной организации, делаю-
щей задачу сколько-нибудь полного обзора нереальной. Поэ-
тому казалось целесообразным, не вдаваясь в детали, привести
в данном изложении лишь наиболее важные сведения об опу-
холевом росте, имея при этом в виду три уровня проблемы —
тканевый, клеточный и молекулярный.
Терминологический словарь
Аберрации (хромосомные) — крупные нарушения структуры хромо-
сом
Аддукты ДНК — химические соединения, ковалентно связанные с
ДНК и модифицирующие ее структуру
Аллель — одна из альтернативных форм гена, находящихся в одном и
том же локусе на гомологичных хромосомах
Анэуплоидия — отклонения от нормального числа хромосом в клетке
Апоптоз — программируемая клеточная гибель
Аутосома — любая, кроме половых, хромосома (у человека 22 пары
аутосом)
Геном — совокупность всех генов организма (полная последователь-
ность всей клеточной ДНК)
Гетерозигота — организм, обладающий двумя различными аллелями
на гомологичных хромосомах
Гомозигота — организм, обладающий двумя одинаковыми аллелями
на гомологичных хромосомах
157
Дальтон — единица молекулярной массы
Делеция — потеря участка хромосомы
Дисплазия — нарушение структуры ткани
Доминанта — признак, который проявляется (экспрессируется) в ор-
ганизме, гетерозиготном по соответствующему гену
Кариотип — характерные для соматической клетки число, размер и
форма хромосом
Катаплазия (анаплазия) — снижение дифференцировки ткани
кДа — килодальтон
Клон — совокупность клеток, являющихся потомками единственной
клетки и получающихся из нее в процессе последовательных клеточ-
ных делений
Локус — определенный участок на хромосоме
Метаплазия — стойкое изменение морфофизиологических свойств
клеток (ткани), сопровождающееся превращением их в клетки
(ткань) другого типа
Метастазирование (диссеминация) — распространение опухоли по
организму
Митогенный сигнал — внешний по отношению к клетке стимул
(в частности, ростовой фактор), индуцирующий клеточное деление
Мутаген — химический или физический агент, вызывающий мутации
Мутация — изменение нуклеотидной последовательности ДНК в оп-
ределенном участке
Онкогены — гены, вызывающие нерегулируемое клеточное деление
Проканцерогены — канцерогены, требующие для проявления своей
активности метаболической активации
Промотор — последовательность ДНК, узнаваемая РНК-полимераза-
ми и определяющая начало транскрипции гена
Протоонкогены — нормальные клеточные гены, способные при на-
рушении их структуры (трансформации в онкогены) индуцировать
опухолевый рост
Прямые канцерогены — канцерогены, не требующие метаболичес-
кой активации
Рак — злокачественная опухоль, происходящая из эпителиальных
клеток
Саркома — злокачественная опухоль, происходящая из клеток соеди-
нительной ткани
Транскрипция — синтез РНК на матрице ДНК
Транслокация — перенос генетического материала с одной хромосо-
мы на другую
158
Трансляция — синтез белка на матрице мРНК
Трансформация — превращение нормальной клетки в опухолевую
Энхансер — последовательность ДНК, усиливающая транскрипцию
данного гена
4.1. Опухолевый рост: общая характеристика
Клиническая онкология включает множество заболеваний,
существенно различающихся своим течением, прогнозом и
методами лечения в зависимости от происхождения опухоли,
ее гистологической формы, локализации и ряда других факто-
ров.
4.1.1. Классификация
Опухоль (новообразование, бластома, неоплазма) — пато-
логическое разрастание, отличающееся от других патологичес-
ких разрастаний (гиперплазия, гипертрофия, регенерация
после повреждения) наследственно закрепленной способнос-
тью к неограниченному, неконтролируемому росту. Существу-
ет два основных типа опухолей — доброкачественные и злока-
чественные.
Доброкачественные опухоли растут, раздвигая прилежащие
ткани, иногда сдавливая их, но обычно не повреждая; в неко-
торых случаях они инкапсулируются. Доброкачественные опу-
холи, как правило, не оказывают неблагоприятного воздейст-
вия на организм, поэтому их можно рассматривать как мест-
ные разрастания, не препятствующие отправлению жизненно
важных функций. Их клиническое значение невелико. Ис-
ключение составляют лишь те случаи, когда локализация опу-
холи является фактором, угрожающим жизнедеятельности ор-
ганизма, например при ее возникновении в головном мозге и
сдавлении вследствие этого нервных центров.
Злокачественные опухоли — многочисленная группа тяже-
лых, хронических заболеваний, заканчивающихся, как прави-
ло, летальным исходом, если отсутствовала или запоздала ле-
чебная помощь. Злокачественные опухоли характеризуются
инвазивным ростом, они инфильтрируют прилегающие ткани,
образуют перифокальные очаги воспаления, часто метастази-
руют в близлежащие лимфатические узлы и отдаленные ткани,
оказывают генерализованное воздействие на весь организм,
расстраивая его гомеостаз. Все последующее изложение по-
священо описанию именно этого типа опухолей.
Гистологические типы опухолей. Организм человека состо-
ит из клеток примерно 100 разных типов и почти все они
159
могут трансформироваться в опухолевые. В зависимости от
типа трансформированных клеток опухоли подразделяются на
раки (происходят из клеток эпителия) и саркомы (происходят
из клеток соединительной ткани). Поскольку первые возника-
ют примерно в 10 раз чаще, чем вторые, термином «рак» за-
частую пользуются расширительно для обозначения всех зло-
качественных новообразований. Вместе с тем благодаря по-
всеместному присутствию в организме соединительнотканных
элементов саркомы могут появиться практически в любом ор-
гане или ткани.
Локализация и гистологический тип опухоли во многом оп-
ределяют скорость ее роста, чувствительность к тем или иным
терапевтическим воздействиям, способность давать метастазы и
рецидивы и в конечном итоге — клиническое течение и прогноз.
Поэтому гистологический диагноз опухоли имеет первостепен-
ное значение для выбора стратегии лечения.
Существует много форм раков, например аденокарцинома
(рак железистый, возникающий из эпителия желез), рак па-
пиллярный (образует сосочковые структуры), бронхоальвео-
лярный (из эпителия бронхов), раки плоскоклеточные, перст-
невидноклеточные, овсяноклеточные, мелкоклеточные, гиган-
токлеточные (по форме образующих их клеток), рак медулляр-
ный (по внешнему сходству с тканью мозга), скирр («твердый»
рак с преобладанием стромальных элементов), рак эпидермо-
идный (по сходству с многослойным плоским эпителием
кожи) и т.д.
Саркомы подразделяются (по своей локализации) на сар-
комы костей, мягких тканей и органов, а по типу исходных
клеток — на фибросаркомы, липосаркомы, лейомиосаркомы и
рабдосаркомы (происходят из мышечных элементов), а также
лимфосаркомы, хондросаркомы и т.д.
4.1.2. Краткие эпидемиологические данные
Распространение. Опухолевые заболевания поражают
практически всех представителей животного мира и даже не-
которые растения. Они широко распространены, являясь вто-
рой после сердечно-сосудистых заболеваний причиной меди-
цинской смертности. В современном мире примерно каждый
четвертый индивидуум на протяжении жизни непосредствен-
но сталкивается с онкологической патологией и примерно
каждый пятый погибает по этой причине. Для младенца, ро-
дившегося в России в 1992 г., вероятность заболеть злокачест-
венным новообразованием на протяжении предстоящей
жизни составляет 19,6 % (для мальчика) и 16,0 % (для девоч-
ки), а вероятность умереть от этой патологии — 16,5 % (для
мальчика) и 10,8 % (для девочки).
160
Рост числа онкологических заболеваний объясняется, по
крайней мере частично, присущим развитым странам общим
постарением населения и увеличением канцерогенной нагруз-
ки. Частота возникновения злокачественных опухолей растет
с возрастом («болезнь пожилых»), хотя нередки (и в последнее
время становятся все более частыми) случаи их появления в
относительно раннем и даже детском и младенческом возрас-
те. Если учесть то обстоятельство, что клиническим проявле-
ниям опухоли предшествует очень длительный, занимающий
годы и десятилетия латентный период, то вполне вероятно
предположение (M.Sporn), что неопластические «ростки» на
разных стадиях формирования присутствуют в любом организ-
ме: в молодом организме — в меньшем числе и менее продви-
нутые, в пожилом — в большем числе и более «зрелые». С этой
точки зрения, имеет право на существование мнение о неиз-
бежности опухолевого перерождения — просто «не каждый
доживает до своего рака».
Факторы риска. Вероятность возникновения опухоли у от-
дельного человека существенно зависит от условий его прожи-
вания. Многочисленные факторы риска могут быть подразде-
лены соответственно их природе на три основные группы:
дурные привычки, плохие условия труда, загрязнение окру-
жающей среды.
Дурные привычки. К их числу, безусловно, относится таба-
кокурение (около 90 % случаев рака легкого — наиболее час-
той формы рака у мужчин — вызваны этой причиной). Таба-
кокурение значительно повышает риск и многих других форм
опухолей: на его долю приходится примерно 5 % общей меди-
цинской смертности. Чрезмерное употребление алкоголя в
свою очередь увеличивает частоту рака желудка, полости рта,
глотки и печени. Сочетание курения с алкоголизмом дает ку-
мулятивный эффект.
Очень важны факторы питания. Диета, богатая жирами жи-
вотного происхождения и копчеными продуктами, с высоким
содержанием соли и низким содержанием грубых растительных
волокон, с высокой концентрацией нитратов и пестицидов яв-
ляется существенным фактором риска. Напротив, диета, вклю-
чающая овощи, фрукты и витамины (С, А, 0-каротин), оказыва-
ет защитный эффект. По широко распространенному мнению,
рациональное питание населения могло бы снизить заболевае-
мость злокачественными новообразованиями на треть.
Еще один фактор риска — многочисленные случайные поло-
вые связи (промискуитет), повышающие вероятность возник-
новения опухолей с предположительно вирусной этиологией
(например, инфицирование вирусом папилломы человека при
раке шейки матки).
Опасен с онкологической точки зрения чрезмерный загар
(растет риск возникновения меланом, особенно у лиц с пони-
11 — 1385
161
женной способностью к образованию меланина — у голубо-
глазых блондинов).
Условия труда. Классическим примером профессионально-
го рака является описанный в 1897 г. рак мошонки у трубо-
чистов в Лондоне. В последующем список вредных произ-
водств существенно расширился (включая, в частности, ани-
линовое производство, производство асбеста, асфальта и т.д.).
Около 100 веществ, с которыми человек сталкивается в своей
трудовой деятельности, являются предположительно канце-
рогенными. На долю профессионального рака приходится,
по-видимому, 1—4 % всех злокачественных новообразований.
В настоящее время существует особая служба, оценивающая
канцерогенную активность различных химических веществ и
промышленных производств.
Экологические факторы. В эту группу включают факторы
риска, имеющие общегосударственный масштаб и входящие в
сферу компетенции правительственных органов. К их числу
относятся загрязнение водоемов и атмосферы промышленны-
ми отходами, содержащими канцерогены и радиоактивные со-
единения; загрязнение пищевых продуктов пестицидами и
нитратами; строительство промышленных зданий и жилых по-
мещений с использованием материалов, содержащих канцеро-
генные вещества (радон, фенолы и т.п.).
Генетическая предрасположенность. В большинстве случа-
ев опухоли появляются внешне случайно («спорадически»),
т.е. без явно выраженной генетической предрасположенности
к ним. Существуют, однако, так называемые раковые семьи
(5—10 % всех случаев злокачественных новообразований),
когда те или иные виды опухолей с высокой частотой и в от-
носительно раннем возрасте поражают индивидуумов, связан-
ных родственными отношениями. Так, некоторые тяжелые
наследственные заболевания (например, синдромы Ли—Фрау-
мени, Гарднера, Блума, пигментная ксеродерма, анемия Фан-
кони, семейный полипоз кишечника, атаксия-телеангиэкта-
зия и ряд других общим числом свыше 200) характеризуются
чрезвычайно высокой (до 100 %) частотой возникновения зло-
качественных новообразований. В основе предрасположен-
ности к опухолям лежат наследственные генетические дефек-
ты (см. далее).
Психологические факторы. Чрезвычайно труден для анали-
за и далек от окончательного решения вопрос о том, могут ли
быть отнесены к факторам риска различные неблагоприятные
психологические воздействия, в частности психический
стресс. С одной стороны, в клинической практике бытует
убеждение, что тяжелые психические потрясения способству-
ют появлению злокачественных новообразований. Так, еще
Гален отмечал, что женщины-меланхолики чаще болеют
раком, чем женщины-сангвиники. Подобные соображения
162
высказывали и многие современные клиницисты и патологи
[Балицкий К.П., Шмалько Ю.П., 1987]. Результаты обширно-
го статистического исследования, проведенного с помощью
так называемого миннесотского многофазного личностного
теста, показали, что риск возникновения опухоли у пациентов
с признаками глубокой депрессии в 2 раза выше, чем в сред-
нем в исследуемой группе (необходимо отметить, что клини-
ческие проявления болезни обнаруживали спустя 1—17 лет
после психологического обследования). Интересно в то же
время отметить, что заболеваемость раком у больных психи-
ческими заболеваниями (эпилепсия, шизофрения) ниже, чем
в среднем в популяции. Имеются, кроме того, клинические и
экспериментальные данные, свидетельствующие об усиливаю-
щемся на неблагоприятном психологическом фоне процессе
метастазирования опухолей. В экспериментах на животных
также удалось в ряде случаев продемонстрировать стимуляцию
опухолевого роста при различных стрессорных воздействиях
(мигающий свет, интенсивное охлаждение). Предполагается,
что индуцированные стрессом нейроэндокринные сдвиги,
оказывающие, в частности, влияние на иммунный статус орга-
низма, могут опосредованно влиять на процессы возникнове-
ния, прогрессирования и метастазирования опухолей. Дли-
тельный стресс вызывает, в частности, устойчивое повышение
уровня пролактина, инсулина, кортикотропина, кортикосте-
роидов, тиреотропного гормона и ряда других, которые в оп-
ределенных условиях могут стимулировать рост опухолевых
клеток. Кроме того, эти гормоны способствуют подавлению
иммунной реактивности организма, в частности угнетению
системы иммунологического надзора.
Известно также, что надлежащая психологическая под-
держка онкологических больных увеличивает период ремис-
сии после соответствующих лечебных мероприятий и продол-
жительность их жизни.
В то же время в ряде исследований, как клинических, так и
экспериментальных, либо вовсе не обнаружено зависимости
между стрессом и появлением и развитием опухолей, либо пока-
зано прямо противоположное (ингибирующее) влияние стресса
на канцерогенез. Так, торможение роста трансплантированных
и индуцированных канцерогенами опухолей зарегистрировано у
крыс, подвергнутых эмоционально-болевому стрессу. Эти раз-
личия могут быть обусловлены множеством плохо контролируе-
мых факторов, таких как сила и характер стресса, время и про-
должительность воздействия, состояние адаптационно-защит-
ных механизмов организма. По-видимому, в зависимости от
конкретных условий стресс может оказывать как стимулирую-
щее, так и ингибирующее воздействие на иммунный и эндо-
кринный статус организма, а это в свою очередь может опреде-
лять конечный результат [Sklar L., Anisman Н., 1981].
11*
163
4.1.3. Этиологические факторы канцерогенеза
В подавляющем большинстве случаев опухоли у человека
появляются спонтанно, т.е. без видимых причин. Иногда на
фоне полного здоровья этот диагноз ставят пациенту в резуль-
тате случайного обследования, но чаще опухоль дает о себе
знать медленно, но неуклонно нарастающей симптоматикой,
различной для опухолей разного происхождения и локализа-
ции. Невозможно установить в каждом отдельном случае, что
явилось этиологической причиной заболевания. Такая бес-
причинность вносит свой вклад в ту особую психологическую
«ауру», которая существует вокруг этой болезни.
По мере выяснения механизмов канцерогенеза становится
все более очевидным, что, во-первых, возникновение опухо-
ли — процесс многостадийный (причем каждый последующий
«шаг» может быть спровоцирован разными факторами) и, во-
вторых, почти все канцерогены генотоксичны, т.е. вызывают
повреждения ДНК (давно бытует убеждение, что «рак — бо-
лезнь генов»). Канцерогены повреждают ДНК случайно (т.е.
неспецифичны в отношении ее нуклеотидных последователь-
ностей), но при возрастании дозы канцерогенного воздейст-
вия растет, естественно, и вероятность того, что в одной из де-
сятков триллионов клеток организма окажутся «задетыми»
гены, управляющие делением клетки. Поскольку и в окружаю-
щей, и во внутренней среде организма существует великое
множество канцерогенов, то выявить конкретный этиологи-
ческий фактор в большинстве случаев не представляется воз-
можным.
Представление о канцерогенных факторах как генотокси-
ческих агентах способствует унифицированному представле-
нию о механизме канцерогенеза в противоположность еще не-
давно существовавшим, не связанным друг с другом и оспари-
вавшим друг у друга пальму первенства теориям вирусного,
химического и физического канцерогенеза. Ясно, что этиоло-
гических факторов много, тогда как патогенез (в принципи-
альном плане) — един и сводится к нарушению механизма ре-
гуляции клеточного деления на одном из его многочисленных
этапов.
Вирусный канцерогенез. Известно множество вирусов, вы-
зывающих опухоли у животных, — ДНК-содержащие (напри-
мер, вирус обезьян SV40) и РНК-содержащие, или ретровиру-
сы (например, вирус саркомы Рауса).
Хотя в целом роль вирусов в возникновении опухолей че-
ловека, по-видимому, не очень велика, получены свидетельст-
ва вирусной этиологии в отношении лимфомы Беркитта, рака
носоглотки (ДНК-содержащий вирус Эпштейна — Барр), рака
шейки матки (вирус папилломы), а также Т-клеточного лейко-
за взрослых (ретровирус HTLV-1) и некоторых других.
164
Химический канцерогенез. Первые экспериментальные опу-
холи при помощи химического вещества вызвали Yamagiva и
Ishikava (1915) — рак у кроликов, индуцированный каменно-
угольной смолой. В настоящее время известно примерно 20
химических канцерогенов, в том числе производственных, ле-
карственных (гидразид изоникотиновой кислоты), природных,
способных вызвать опухоли у человека. Установлено широкое
распространение в окружающей среде бенз(а)пирена (БП), ос-
новного представителя многочисленной группы полицикли-
ческих ароматических углеводородов (ПАУ), который образу-
ется не только в результате деятельности человека, но и пред-
ставляет собой природный канцероген — содержится в почве,
в выбросах вулканов. К ПАУ относятся метилхолантрен, ди-
метилбенз(а)антрацен и др.
Как отмечалось выше, наиболее опасно в канцерогенном
отношении табакокурение. В 100 сигаретах содержится 1,1 —
1,6 мкг БП, а примесь мышьяка в 15 раз превышает его мак-
симально допустимое количество. Кроме того, в табачном
дыме (состоящем из газовой фазы и твердых частиц) нахо-
дятся дибенз(а)антрацен и никель, канцерогенные для чело-
века. Смертность от рака легкого прямо пропорциональна
числу выкуриваемых в день сигарет: у людей, выкуривающих
16—25 сигарет в день, риск заболеть раком легкого в 30 раз
выше, чем у некурящих. Считается, что у мужчин США в
85—90 % случаев заболевания раком легкого вызваны куре-
нием сигарет.
Большое значение имеют также канцерогенные нитро-
зосоединения, вызывающие опухоли у всех исследован-
ных видов животных. Особое внимание, проявляемое к
этому классу соединений (нитрозометил- и нитрозоэтил-
мочевины, нитрозодиметиламины), обусловлено обнаружен-
ной возможностью их эндогенного синтеза в организме из
нитритов (нитратов) и вторичных аминов, содержащихся
в пище. Кроме того, вторичные амины могут образовы-
ваться в толстой кишке при участии бактериальной флоры,
которая в то же время способна переводить нитраты в ни-
триты.
Химические канцерогены подразделяются на проканцеро-
гены (составляют абсолютное большинство) и прямые канце-
рогены. Проканцерогены превращаются в истинные, конеч-
ные канцерогены только после метаболических превращений,
катализируемых тканевыми ферментами (неспецифическими
оксидазами). Они локализованы главным образом в эндоплаз-
матическом ретикулуме и частично в ядре клетки. ПАУ типа
бенз(а)пирена или диметилбенз(а)антрацена становятся ко-
нечными канцерогенами, превращаясь в соответствующие
эпоксиды. Некоторые проканцерогены становятся конечными
в результате спонтанных реакций.
165
Представители второй группы прямых канцерогенов, на-
пример нитрозамины, 0-пропионлактон, диметилкарбамил-
хлорид, действуют как таковые, не подвергаясь метаболичес-
кой модификации.
Все химические канцерогены реагируют с клеточной ДНК;
ковалентно присоединяясь к ней, образуют многочисленные
аддукты, индуцируют одно- и двунитевые разрывы.
Трансплацентарный канцерогенез обнаружен у эксперимен-
тальных животных и означает появление опухолей у детены-
шей (не у всех и спустя несколько месяцев после рождения) в
результате действия в период их внутриутробного развития
малых (безвредных для матери) доз химических канцерогенов.
Эффект достигается и в том случае, когда канцероген попада-
ет в эмбрион непосредственно, не будучи модифицирован тка-
невыми ферментами матери. У таких детенышей возникают
опухоли различных локализаций, в том числе в головном
мозге. Нервная система зародыша оказалась по крайней мере
в 50 раз более чувствительной к канцерогену, чем у взрослой
крысы.
Человек также должен считаться с этим явлением. Обнару-
жены случаи возникновения светлоклеточного рака влагалища
у девочек и девушек, чьи матери во время беременности при-
нимали диэтилстильбэстрол. В странах Западной Европы по-
вышается частота онкологических заболеваний подростков
(лейкозы, опухоли нервной системы, почек). Умножаются
случаи смерти от этих видов рака у детей моложе 15 лет.
Радиационный канцерогенез. О канцерогенном действии
ионизирующего излучения стало известно уже вскоре после
открытия рентгеновских лучей и естественной радиоактивно-
сти. Так, первый случай лучевого рака кожи описан в 1902 г.
В настоящее время известны индуцированные радиацией
опухоли практически всех органов. Однако чаще всего воз-
никают опухоли кожи и костей, лейкозы, а также эндокрин-
нозависимые опухоли (рак молочной железы и яичников),
причем в коже и костях они появляются преимущественно
после местного облучения, в остальных же органах — после
общего.
Частота и виды злокачественных новообразований, инду-
цированных ионизирующими излучениями, зависят от многих
факторов, в том числе от проникающей способности излуче-
ния, характера воздействия (внешнее или внутреннее), орга-
нотропности радионуклидов, распределения дозы во времени
(облучение острое, хроническое, дробное и т.д.).
Ультрафиолетовый канцерогенез. Длительное воздействие
ультрафиолетового спектра солнечных лучей является основ-
ным индуктором меланом на открытых участках кожи (голова,
шея, руки), при этом наиболее чувствительны блондины со
светлыми кожей и волосами.
166
4.1.4. Характерные свойства опухолей
Злокачественные опухоли характеризуются катаплазией,
метаплазией, дисплазией, инвазивным и деструктивным рос-
том, метастазированием.
Катаплазия (анаплазия) — появление слабодифференциро-
ванных или недифференцированных клеток, похожих на эмбри-
ональные (приставка «ката» обозначает движение вниз, сниже-
ние уровня дифференцировки). Опухоль может утрачивать час-
тично или полностью тканеспецифические признаки. Этот про-
цесс протекает хаотично и нередко приводит к образованию
атипичных клеток, не имеющих аналогов в нормальных тканях.
Метаплазия (от латинского metaplasis — преображение)—
стойкое изменение морфофизиологических свойств клеток
(ткани), сопровождающееся превращением их в клетки
(ткань) другого типа (например, клетки соединительной ткани
начинают образовывать кость в неподходящем месте).
Дисплазия (от латинского dis — нарушение, расстройство и
plasis — форма, образование) — нарушение в опухолевом очаге
характерной для данной ткани структуры, ее атипия (по свое-
му строению, расположению и взаимоотношениям клеточных
элементов опухоль резко отличается от исходной нормальной
ткани).
Инвазивный и деструктивный рост, метастазирование.
Злокачественные опухоли растут, инфильтрируя (прорастая)
окружающие ткани и вызывая их деструкцию. Они часто дают
метастазы (вторичные очаги в отдаленных тканях и органах),
что во многих случаях означает финальную стадию процесса.
Метастазирование — многоэтапный процесс, в ходе кото-
рого отбирается и выживает небольшая субпопуляция метаста-
тических клеток, предсуществующих в «родительской» опухо-
ли. Опухоли обладают весьма гетерогенным метастатическим
потенциалом. Конечный результат определяется как свойства-
ми самих опухолевых клеток, так и условиями внутренней
среды организма — теория «семян» и «почвы» [Paget J., 1889].
Клинически значимые метастазы появляются после много-
этапного отбора опухолевых клеток, причем каждый из этапов
влияет на скорость процесса в целом. Схематически этот про-
цесс можно представить следующим образом.
Первичный очаг — новообразование сосудов — прорастание в
сосуды (лимфатические, капиллярные, венозные) — образова-
ние эмболов (многоклеточных агрегатов, включающих лимфо-
циты и тромбоциты) — транспорт в отдаленные ткани и
органы — задержка в капиллярах — прикрепление — экстра-
вазация (выход из сосудов) — адаптация к микроокруже-
нию — пролиферация — формирование метастаза — мета-
стазирование метастазов.
167
Исключительно важную роль в росте опухоли (как первич-
ного очага, так и его метастазов) играет ангиогенез. Опухоли
диаметром 1—2 мм получают все необходимое посредством
диффузии, однако их дальнейший рост зависит от кровоснабже-
ния и, следовательно, от новообразования сосудов. Опухоль
способна продуцировать стимулирующие ангиогенез факторы,
которые обусловливают врастание сосудов в опухолевый очаг
путем миграции в него эндотелиальных клеток из прилегающей
соединительной ткани и их размножения. Как и в физиологиче-
ских условиях (заживление раны), активность ангиогенеза в опу-
холи определяется балансом регуляторов, позитивных (ангиоге-
нин, фактор роста гепатоцитов, трансформирующие ростовые
факторы аир, фактор некроза опухолей, простагландины Ej и
Е2, интерлейкин-8 и др.) и негативных (ангиостатин, ингибитор
хрящевой ткани, гепариназа, интерфероны аир, тромбоспон-
дин, тканевый ингибитор металлопротеиназ и др.).
Прекращение по тем или иным причинам врастания сосу-
дов в опухоль может на время остановить ее рост и перевести в
«дремлющее» состояние. Более того, поскольку пролиферация
эндотелиальных клеток и связанное с этим новообразование
сосудов, как правило, отстают от роста самой опухоли, в ее
центре часто обнаруживают участки некротического распада.
В изменившихся условиях (при улучшении кровоснабжения)
оставшиеся в живых раковые клетки могут дать начало реци-
диву опухоли даже спустя несколько лет после ее удаления.
В то же время опухоль может продуцировать факторы, бло-
кирующие новообразование сосудов. Именно этим объясня-
ются те нередкие случаи, когда после хирургического удале-
ния основного очага начинается бурный рост «дремавших» до
того метастазов. При этом в организме сдвигается баланс про-
и антиангиогенных факторов в пользу первых; в частности,
падает концентрация циркулирующего в крови ангиостатина
(фрагмента плазминогена размером 38 кДа), продуцируемого
многими неоплазмами.
Формирование метастаза — событие в принципе маловеро-
ятное. В крови онкологических больных, не имеющих мета-
стазов, часто обнаруживают циркулирующие опухолевые клет-
ки. Их подавляющее большинство в кровеносном русле разру-
шается естественными киллерами и макрофагами, тогда как
очень малая часть (менее 0,05 %) выживает благодаря естест-
венному отбору на резистентность к природным «киллерам» и
на способность подавлять их функцию. Сохранившие жизне-
способность клетки задерживаются в узких сосудах того орга-
на, к которому имеют тропность (многие опухоли проявляют
тенденцию к преимущественному метастазированию в опреде-
ленные ткани: например, аденокарцинома молочной железы
метастазирует в кости и головной мозг, а нейробластома — в
печень и надпочечники).
168
4.1.5. Взаимоотношения опухоли и организма
Взаимоотношения злокачественной опухоли и организма
многообразны и противоречивы. С одной стороны, организм,
являющийся для опухоли внешней средой, создает для нее необ-
ходимые условия существования и роста (обеспечивая, напри-
мер, врастание в ее ткань кровеносных сосудов), а с другой — с
большим или меньшим успехом противодействует ее развитию.
Известные сегодня аспекты этой борьбы составляют, по-види-
мому, лишь верхушку айсберга. Так, не выяснены важные во-
просы: является ли психический стресс фактором канцерогенеза
и влияет ли он на течение болезни, «интерферирует» ли опухо-
левый рост с другими патологическими процессами (когда-то,
например, бытовало представление о том, что туберкулез и опу-
холь «не поселяются» в одном легком) и т. д.
Развитие опухоли — интерактивный процесс (акты «агрес-
сии» со стороны опухоли чередуются с ответными «контрме-
рами» организма). Исход этой борьбы предопределен громад-
ным потенциалом «агрессивности» опухоли, с одной стороны,
и ограниченностью защитных ресурсов организма — с другой.
Иммунная защита. Далеко не всякий возникший в орга-
низме клон опухолевых клеток превращается в злокачествен-
ную опухоль. Организм располагает определенными, хотя и
ограниченными, средствами противодействия. На первых эта-
пах действует система так называемой естественной неспеци-
фической резистентности, способная элиминировать неболь-
шое количество (от 1 до 1000) опухолевых клеток. К ней отно-
сятся естественные киллеры — крупные гранулярные лимфо-
циты, составляющие от 1 до 2,5 % всей популяции перифери-
ческих лимфоцитов и независимые от вилочковой железы.
Они могут лизировать не только опухолевые, но и нормальные
поврежденные и эмбриональные клетки, активируются интер-
фероном. К системе естественной резистентности относятся
также макрофаги. Их оружием против небольшого числа опу-
холевых клеток служат кислородные радикалы, образуемые
специфическими оксидазами, и перекись водорода Н2О2. Воз-
можность преодоления опухолевыми клетками первого барье-
ра естественной резистентности определяется их способнос-
тью продуцировать факторы, подавляющие эту систему (про-
стагландины Е), и их устойчивостью к Н2О2.
Что касается специфического противоопухолевого имму-
нитета, то он обычно развивается слишком поздно и не очень
активен. Спонтанные опухоли животных и человека весьма
слабо антигенны и легко преодолевают этот барьер. Однако
зарегистрированы единичные случаи регрессии злокачествен-
ных опухолей (меланомы, нейробластомы у детей), что указы-
вает на принципиальную возможность успешной борьбы орга-
низма с уже сформировавшейся неоплазмой.
169
Системное действие злокачественной опухоли на организм.
Как уже отмечалось, существует большое разнообразие форм
онкологических заболеваний, различающихся происхождени-
ем, локализацией, биологическими свойствами и т.д. Несмот-
ря на различия, при далеко зашедшем процессе все они так
или иначе приводят организм к гибели. В некоторых ситуаци-
ях причины гибели вполне очевидны: при возникающем, на-
пример, на фоне основного заболевания смертельном крово-
течении, при поражении жизненно важного органа (напри-
мер, головного мозга), при опухолевой трансформации кле-
ток, секретирующих биологически активные вещества (напри-
мер, при опухолях эндокринных органов и как следствие —
существенном нарушении гормонального баланса) и т.д.
В большинстве случаев, однако, «местные» проявления
опухолей неясны, смазаны (во всяком случае поначалу, что,
кстати, очень затрудняет их раннюю диагностику) и не затра-
гивают явным образом витальные функции организма. Но по-
скольку губительное и однотипное воздействие на организм
оказывают все злокачественные опухоли независимо от их
формы и локализации, то, по-видимому, существуют и некие
единые механизмы такого воздействия. Полной ясности, од-
нако, в отношении этих механизмов сегодня нет. Далеко не
всегда удается связать те или иные нарушения обмена в орга-
низме больного с биологическими свойствами самой опухоли.
В целом взаимоотношения опухоли и организма сегодня зна-
чительно менее исследованы, чем механизмы собственно кан-
церогенеза.
Опухоль как ловушка питательных веществ. Из-за интен-
сивного роста, требующего пластических и энергетических ре-
сурсов, и особенностей изоферментного спектра, наделяюще-
го опухоль высокой конкурентоспособностью, она становится
«ловушкой» различных питательных веществ, вызывая в орга-
низме состояния дефицита (например, азота, глюкозы, вита-
минов). Известно, что опухоль строит свои структуры, извле-
кая азот не только из пищи, но и из продуктов распада ткане-
вых белков (поначалу преимущественно из мышечной ткани,
а затем и из других). Такое своеобразное паразитирование
ведет иногда к увеличению массы опухоли на фоне уменьше-
ния общей массы тела. Вероятно, основным фактором, обу-
словливающим распад тканевых белков, является гормональ-
ный дисбаланс, в частности возрастание продукции гормонов
коры надпочечников (глюкокортикоидов), стимулирующих
катаболизм тканевых белков.
Опухоль, активно утилизируя глюкозу для синтеза белков и
нуклеиновых кислот и создавая ее дефицит в организме, дей-
ствует как гипогликемический фактор (снижает уровень глю-
козы в крови) и приводит тем самым к напряжению компен-
саторных систем, поддерживающих углеводный гомеостаз.
170
В рамках этой концепции получают свое объяснение многие
проявления системного действия опухоли на организм. Так, в
частности, естественным следствием гипогликемического дав-
ления является отмеченная выше повышенная продукция
глюкокортикоидов (феномен, постоянно сопровождающий
злокачественные новообразования). Глюкокортикоиды же,
как известно, с одной стороны, стимулируют глюконеогенез
(восполнение запасов глюкозы за счет распада тканевых бел-
ков), а с другой — индуцируют распад лимфоидной ткани, что
может вносить свой вклад в ослабление иммунитета. Действие
опухоли как ловушки питательных веществ не ограничивает-
ся, по-видимому, лишь азотистыми соединениями и глюко-
зой, а может распространяться и на другие вещества, напри-
мер витамины. Вместе с тем необходимо подчеркнуть, что
«ловушечная» функция опухоли оказывается значительной и
способной влиять на общий обмен организма, по-видимому,
лишь в относительно далеко зашедших случаях, когда масса
опухоли очень велика.
Опухоль как источник биологически активных соединений.
Опухолевая ткань, создавая, с одной стороны, дефицит опре-
деленных соединений, способна, с другой стороны, продуци-
ровать биологически активные вещества, не свойственные
нормальному организму и в еще большей степени дезоргани-
зующие обмен. Эти вещества могут быть весьма многообразны
по своей природе. Прежде всего это продукты распада самой
опухоли, оказывающие токсическое воздействие. Из-за недо-
статочной васкуляризации почти всегда в центре опухолевого
очага обнаруживаются участки омертвения (некроза). Продук-
ты распада, всасываясь в кровь, разносятся по организму, вы-
зывая повышение температуры и другие проявления общей
интоксикации.
Секретируемые опухолью вещества могут оказывать и спе-
цифическое воздействие. Так, выше уже упоминалось о секре-
ции опухолью различных ростовых и ангиогенных факторов.
Попадая в кровь, они способны оказывать митогенное воздей-
ствие на клетки отдаленных тканей, обладающие соответству-
ющими рецепторами. Кажется вероятным, что именно здесь
кроется причина давно отмеченного феномена — вспышек
митозов в отдаленных от опухоли тканях. К иной форме дис-
тантного действия злокачественных новообразований отно-
сится секреция некоторыми из них так называемых эктопи-
ческих (не свойственных данной ткани) гормонов. Наиболее
известный пример этого — секреция тканью рака легкого или
опухолями дыхательных путей адренокортикотропного гормо-
на или, реже, инсулина и глюкагона.
Особую разновидность составляют опухоли органов эндо-
кринной системы, способные по очевидным причинам оказы-
вать мощное и специфическое воздействие на обмен организма.
171
Паранеопластический синдром — проявление генерализо-
ванного воздействия опухоли на организм. Его формы разно-
образны: состояние иммунодепрессии (повышенная подвер-
женность инфекционным заболеваниям), тенденция к по-
вышению свертываемости крови, сердечно-сосудистая недо-
статочность, мышечная дистрофия, некоторые редкие дерма-
тозы, пониженная толерантность к глюкозе, острая гипогли-
кемия при опухолях больших размеров и ряд других.
Одним из проявлений паранеопластического синдрома яв-
ляется так называемая раковая кахексия (общее истощение
организма), которая возникает в периоде, близком к терми-
нальному, и часто наблюдается при раках желудка, поджелу-
дочной железы и печени. Она характеризуется потерей массы
тела в основном из-за усиленного распада белков скелетных
мышц (частично миокарда), а также истощения жировых депо.
Сопровождается отвращением к пище (анорексией) и измене-
нием вкусовых ощущений. Одна из причин кахексии — повы-
шенный (иногда на 20—50 %) расход энергии, обусловленный,
по-видимому, гормональным дисбалансом.
В сыворотке крови больных, страдающих хроническими
заболеваниями и, в частности, злокачественными новообразо-
ваниями, нередко обнаруживают кахектин — цитотоксичес-
кий полипептидный гормон, известный также как TNF (tumor
necrosis factor — фактор некроза опухолей). Он секретируется
макрофагами и опосредует, воспалительные реакции. Практи-
чески все клетки организма обладают рецепторами к этому
гормону, эффекты которого поэтому могут быть весьма
многообразными: шоковое состояние, падение артериального
давления, расстройства липидного и углеводного обмена, ме-
таболический ацидоз, активация нейтрофилов вплоть до гибе-
ли организма. В экспериментах на животных кахектин инду-
цирует также состояния анорексии и истощения организма,
очень сходные с кахексией при хронических заболеваниях и
злокачественном росте.
Биохимические показатели. Изменения обмена веществ в
организме онкологического больного могут быть настолько
значительными и иногда специфическими, что их выявление
и количественная оценка могут иметь диагностическое и про-
гностическое значение. Так, биохимическое исследование
крови онкологического больного (общий скрининг) позволяет
в ряде случаев судить о степени распространения опухоли, о
функциональном состоянии жизненно важных органов и сис-
тем, о сопутствующих заболеваниях и о локализации опухоли.
Наиболее важными, с этой точки зрения, являются сле-
дующие количественные показатели (имеется в виду содержа-
ние в сыворотке крови): для оценки белоксинтетической
функции печени — общий белок, альбумин, билирубин, ак-
тивность аланиновой трансаминазы; для оценки функции
172
почек и мочевыделительной системы — мочевина; для оценки
инсулярного аппарата поджелудочной железы — глюкоза.
О возможности поражения костей скелета первичной опухо-
лью или метастазами судят по содержанию в сыворотке крови
щелочной фосфатазы.
Опухолевые маркеры — соединения, обнаруживаемые в
биологических жидкостях онкологических больных и синтези-
руемые либо собственно раковыми клетками, либо клетками
нормальных тканей в ответ на инвазию опухоли. Маркерами
опухоли могут быть различные белки (ферменты, гормоны,
антигены — внутриклеточные или ассоциированные с поверх-
ностными мембранами) и метаболиты, концентрация которых
в среде коррелирует с массой опухоли, с ее пролиферативной
активностью и Иногда со степенью злокачественности.
Появление маркеров обусловлено особенностями метабо-
лизма раковой клетки. Так, опухоль может утратить некоторые
изоферменты, присутствующие в гомологичных нормальных
тканях, и, напротив, продуцировать изоформы, характерные
для данной ткани только в период ее эмбрионального разви-
тия. В опухоли может меняться активность лизосомальных и
мембраносвязанных ферментов, могут синтезироваться экто-
пические изоэнзимы и гормоны. Необходимо, однако, под-
черкнуть, что ни в трансформированных клетках, ни в биоло-
гических жидкостях онкологических больных не обнаружены
такие соединения, которые были бы характерны только для
опухоли и не обнаруживались бы и в нормальных тканях на
тех или иных стадиях их развития.
Современные методы позволяют выявить опухолевые марке-
ры в столь малых (фемтамолярных, 10-15 М) концентрациях, что
это дает возможность в ряде случаев следить за ходом заболева-
ния и эффективностью лечения. Опухолевые маркеры можно
подразделить на две основные группы: продуцируемые самой
опухолью и ассоциированные с опухолью (их появление в пос-
леднем случае обусловлено опухолевым процессом как таковым
независимо от того, какой тканью они продуцируются).
Среди маркеров, продуцируемых опухолью, — а-фетопро-
теин (повышенное его содержание в сыворотке крови может
служить указанием на гепатоцеллюлярный рак); раково-эмб-
риональный антиген (отмечен у значительной части больных
раком толстой кишки, поджелудочной железы, молочной же-
лезы и легкого); тканевый полипептидный антиген (обнару-
живается при раке мочевого пузыря, предстательной железы и
почек); хорионический гонадотропин (при опухолях трофо-
бласта содержание маркера в сыворотке крови коррелирует с
массой опухолевых клеток).
Маркеры, ассоциированные с опухолью, — белки острой
фазы воспаления (церулоплазмин, гаптоглобин, а2-глобулины,
С-реактивный белок), некоторые ферменты, иммунные ком-
173
плексы. Белки острой фазы — соединения, синтез которых не-
специфически повышается в ответ на разнообразные патоло-
гические процессы (воспаление, повреждение, злокачествен-
ные новообразования). Их усиленная продукция при опухоле-
вом росте может быть вызвана либо секретируемыми опухо-
лью ростовыми факторами, либо вторичным воспалением.
Лактатдегидрогеназа (ЛДГ) — один из важнейших ферментов
углеводного обмена, представленный 5 изоформами (тетраме-
ры субъединиц Н и М, кодируемых разными генами). Нор-
мальные ткани индивидуальны в отношении изоформ ЛДГ.
При различных онкологических заболеваниях отмечено по-
вышение активности ЛДГ в сыворотке крови, причем при не-
которых формах преобладает М-субъединица, тогда как при
других — Н-субъединица. Креатинкиназа — фермент, поддер-
живающий оптимальные концентрации АТФ и АДФ в клетке,
представлен 4 изоформами. Повышение концентрации креа-
тинкиназы в сыворотке крови отмечается чаще всего у боль-
ных с опухолями желудочно-кишечного тракта.
4.1.6. Стадии развития
Канцерогенез — длительный процесс накопления генети-
ческих повреждений; латентный период (период от начальных
изменений в клетке до первых клинических проявлений)
может составлять 10—20 лет. Возникновение опухоли — про-
цесс многостадийный; выделяют три основные стадии канце-
рогенеза: инициации, промоции и прогрессии.
Инициация — начало в длинной цепи событий, ведущих к
образованию опухолевого очага. Оно заключается в мутации
одного из генов, регулирующих клеточное размножение.
Клетка становится «инициированной», т.е. потенциально спо-
собной к неограниченному делению, но требующей для про-
явления этой способности ряда дополнительных условий.
Инициирующими факторами являются различные канцероге-
ны, вызывающие повреждение ДНК.
Промоция. Существует множество химических веществ, так
называемых промоторов (в частности, форболовые эфиры),
которые не вызывают повреждений ДНК, не являются канце-
рогенами, но хроническое воздействие которых на иницииро-
ванные клетки приводит к возникновению опухоли. Главным
в промоции, по-видимому, является стимуляция клеточного
деления, благодаря которой создается критическая масса ини-
циированных клеток. Это в свою очередь способствует, во-
первых, высвобождению инициированных клеток из-под тка-
невого контроля и, во-вторых, мутационному процессу.
Прогрессия. Существовавшее когда-то представление о том,
что рост опухоли — лишь количественное увеличение числа
174
однородных клеток, чрезвычайно далеко от действительности.
На самом деле наряду с количественным увеличением массы
опухоли она постоянно претерпевает качественные изменения и
приобретает новые свойства — все большую автономность от ре-
гулирующих воздействий организма, деструктивный рост, инва-
зивность, способность к образованию метастазов (обычно от-
сутствующую на ранних этапах) и, наконец, ее поразительную
приспособляемость к меняющимся условиям.
Концепцию прогрессии опухолей сформулировал в 1969 г.
Foulds. Он постулировал, в частности, принцип независимости
возникновения и эволюции признаков злокачественности опу-
холи, который способен объяснить разнообразие опухолевых
фенотипов и путей их эволюции. Именно в этом заключено
принципиальное различие между прогрессией опухоли, которую
никогда нельзя считать завершенной, и нормальной дифферен-
цировкой ткани, которая всегда жестко запрограммирована
вплоть до момента формирования конечной структуры.
Клональное происхождение опухолей. Фундаментальное зна-
чение для онкологии имеет положение о клональном происхож-
дении опухоли, т.е. представление об опухоли как потомстве
(клоне) одной первичной клетки, которая в результате много-
стадийного процесса приобрела способность нерегулируемого
роста. Окружающие опухоль, нормальные клетки в процесс «оз-
локачествления» не вовлечены. Иными словами, первичная
трансформированная клетка передает свои свойства (в том
числе главное — способность неконтролируемого размножения)
только своим потомкам, т.е. «вертикально», но не «горизонталь-
но», как это имеет место при инфекционных заболеваниях.
На поздних стадиях опухолевого процесса нередко проис-
ходит диссеминация (метастазирование) опухоли — ее распро-
странение по всему организму с возникновением вторичных
очагов в отдаленных тканях. Метастазы — не независимо воз-
никшие опухоли, а потомки одной трансформированной клет-
ки, несущие ее родовые черты.
От метастазов следует отличать первично-множественные
опухоли (несколько независимо возникших опухолей у одного
больного). В этих случаях чаще всего имеет место генетическая
предрасположенность к злокачественным новообразованиям.
Клональная гетерогенность опухолей. Трансформирован-
ная клетка в результате многократного деления производит
клон подобных себе клеток — с одинаковыми генотипом и фе-
нотипом. Однако присущая ей нестабильность генома (см.
далее) приводит к появлению новых клонов, различающихся
генотипически и фенотипически. В результате возникает кло-
нальная гетерогенность опухоли; это — ее кардинальное свой-
ство и движущая сила всех последующих метаморфоз.
Популяция опухолевых клеток постоянно находится под
прессом естественного отбора. Селективным преимуществом
175
обладают клоны, наиболее быстро растущие и резистентные к
защитным средствам организма. Благодаря действию отбора
фенотип опухоли постоянно меняется, становясь все более аг-
рессивным и злокачественным. Нестабильность генома и как
следствие клональная гетерогенность опухоли наделяют ее
особой живучестью, приспособляемостью к условиям среды,
сопротивляемостью к терапевтическим воздействиям.
Это положение можно проиллюстрировать ситуацией,
часто возникающей при химиотерапии больных со злокачест-
венными опухолями. Так, в результате применения какого-
либо химиопрепарата гибнет подавляющая доля (предполо-
жим, 99,9 %) всех ее клеток. Однако из-за клональной гете-
рогенности почти наверняка в составе опухоли окажется
клон, резистентный к данному препарату в силу тех или
иных особенностей метаболизма. Оставшиеся 0,1 % клеток
начинают размножаться и постепенно замещают собой массу
погибших. Если продолжительность клеточного цикла при-
нять равной 24 ч, то теоретически для полного восстановле-
ния массы опухоли потребуется всего лишь 10 сут (на самом
деле этот период ремиссии, т.е. временного отступления опу-
холевого процесса, может длиться дольше — месяцы и годы).
Но, поскольку в итоге произошла фенотипическая трансфор-
мация опухоли (один клон заменил собой остальные и опу-
холь в целом приобрела устойчивость к данному химиопре-
парату), следующий цикл лечения окажется значительно
менее эффективным.
Многостадийность канцерогенеза. Еще в ранних эпидемио-
логических исследованиях, в которых сопоставляли такие
признаки опухолей, как время их появления, частота пораже-
ния парных органов и частота семейных случаев, была обо-
снована модель двустадийного канцерогенеза [Knudson, 1972].
Согласно этой модели, первоначального повреждения (мута-
ции) генома половой или соматической клетки недостаточно
для возникновения опухоли, она лишь создает фон для воз-
действия последующих мутагенов. Представления о многоста-
дийности канцерогенеза были подтверждены и детализирова-
ны в последующих исследованиях. Оказалось, в частности, что
развитие рака толстой кишки человека обусловлено несколь-
кими (не менее 7) мутационными событиями, происходящими
в определенной последовательности (см. далее).
Таким образом, непрерывно прогрессируя, претерпевая ка-
чественные изменения и проходя через необратимые стадии,
опухоль «дрейфует» в направлении увеличения злокачествен-
ности. Одной из первых фаз прогрессии являются во многих
случаях доброкачественные новообразования. Известны, од-
нако, факты малигнизации нормальных клеток сразу, минуя
стадию доброкачественной опухоли, как это происходит, на-
пример, при раке толстой кишки.
176
4.2. Опухолевые клетки
Многие свойства трансформированных клеток можно изу-
чать in vitro благодаря присущей им способности легко пере-
ходить в культуру (см. далее), сохраняя опухолевый фенотип.
В этой наиболее простой модельной системе оказываются воз-
можными такие экспериментальные манипуляции, которые
абсолютно исключены in vivo. Именно благодаря широкому
использованию различных клеточных линий оказалось в
принципе возможным выяснить молекулярные механизмы
канцерогенеза.
4.2.1. Изменения кариотипа
и хромосомные аберрации
Задолго до открытий в молекулярной биологии и экспе-
риментальной онкологии, прояснивших генетические меха-
низмы возникновения и эволюции трансформированной
клетки, существовало представление о том, что «рак — бо-
лезнь генов». Об этом свидетельствовало, в частности, одно
из наиболее характерных свойств трансформированных кле-
ток — разнообразные изменения кариотипа (анэуплоидия,
гиперплоидия), хромосомные аберрации (делеции, трансло-
кации, амплификации) и мутации генов, а также присутствие
экстрахромосомных элементов, вовсе не встречающихся в
нормальных клетках. В опухолевом очаге, как правило, обна-
руживаются клетки с существенно различающимся кариоти-
пом. В этом находит свое выражение одно из самых харак-
терных свойств опухолевой клетки — нестабильность ее ге-
нома.
Так, при цитогенетическом анализе зачастую обнаружива-
ется специфическая связь между изменением кариотипа и оп-
ределенным типом опухоли: например, делеция длинного
плеча хромосомы 13 (точнее, участка 13ql4) при ретиноблас-
томе, участка 11р13 — при нефробластоме, длинного плеча
хромосомы 17 — при раке молочной железы; появление так
называемой филадельфийской хромосомы (транслокация
участка длинного плеча хромосомы 9 на длинное плечо хромо-
сомы 22) — при миелоидном лейкозе.
Столь специфическая связь позволила заподозрить локали-
зацию в этих «горячих» точках хромосом неких генов, повреж-
дение (или устранение) которых способствует развитию опу-
холевого процесса. Действительно, дальнейшие исследования
позволили обнаружить во многих из этих локусов так называе-
мые гены-супрессоры, тормозящие клеточное размножение
(см. далее).
12—1385
177
4.2.2. Признаки клеточной трансформации
в культуре
Трансформированные клетки, культивируемые in vitro, от-
личаются от нормальных многими свойствами. Они способны
расти в культуре при низкой концентрации сыворотки и в от-
сутствие ростовых факторов; их размножение ограничено, по-
видимому, лишь питательными ресурсами среды; они посто-
янно пребывают в цикле деления (не переходя в фазу покоя
GO), а если и останавливаются в тех или иных его точках, то
только из-за отсутствия питательных веществ; они могут расти
в суспензии, не испытывая потребности в прикреплении к
внеклеточному матриксу.
Трансформированные клетки способны неограниченно де-
литься вследствие отсутствия у них так называемого контактно-
го торможения, что позволяет им размножаться многослойно
(нормальные клетки, соприкоснувшись своими поверхностями,
прекращают делиться, в результате чего образуют однослойную
культуру). У трансформированных клеток ослаблены адгезив-
ные свойства: они утрачивают способность распластываться на
подложке и прочно прикрепляться к ней. Их деление перестает
зависеть от прикрепления к твердому субстрату; они приобрета-
ют способность размножения в полужидкой среде. Некоторые
опухолевые клетки могут в отличие от нормальных расти в сус-
пензионной культуре — свойство, которое коррелирует с их опу-
холеродностью (способностью образовывать опухоли при имп-
лантации животным). Последняя является главным отличитель-
ным признаком трансформированной клетки.
4.2.3. Иммортализация опухолевых клеток
Нормальные клетки трудно перевести в культуру и практи-
чески невозможно поддерживать ее длительно, поскольку
после определенного числа клеточных делений (пассажей)
скорость роста постепенно замедляется, затем прекращается
полностью и клетки в конце концов погибают (этот феномен
по имени открывшего его ученого называют барьером Хейф-
лика). Число пассажей, которые клетки способны пройти в
культуре, различно у разных видов и зависит от возраста жи-
вотного, от которого эти клетки были исходно получены (чем
моложе организм, тем больше возможное число пассажей).
По-видимому, в клетке существует некий счетчик числа деле-
ний, включающий программу старения и гибели по достиже-
нии какой-то критической величины.
В противоположность этому трансформированные клетки
иммортализованы, т.е. могут делиться неограниченно долго,
не проявляя признаков старения. У них, кроме того, часто об-
178
наруживают различные дефекты механизма апоптоза (про-
граммируемой клеточной гибели), в силу, чего они способны
выживать в условиях, гибельных для клеток нормальных. Бла-
годаря этому свойству культуры опухолевых клеток можно
поддерживать десятилетиями.
4.2.4. Межклеточная кооперация
Установлено, что важную сдерживающую роль по отноше-
нию к трансформированным клеткам могут играть их соседи.
Роль межклеточной кооперации (и межклеточных контактов
как механизма ее реализации) в функционировании нормаль-
ных клеток чрезвычайно велика. «Асоциальность» трансфор-
мированных клеток является, в частности, следствием ослаб-
ления их контактов с другими клетками. В клеточных ассо-
циациях существует сложный баланс митотических стимулов
и тормозов, благодаря которому поддерживается тканевый го-
меостаз. До поры до времени межклеточные взаимодействия
способны держать в «узде» первично трансформированные
клетки. Однако по достижении последними некой критичес-
кой массы они способны ускользнуть из-под тканевого кон-
троля и дать начало опухоли.
В пользу этих представлений свидетельствуют как экспери-
менты in vitro (кокультивирование опухолевых и нормальных
клеток), так и эксперименты in vivo (инокуляция смешанных
культур тех и других экспериментальным животным). В обеих
ситуациях отчетливо проявляется нормализующее опухолевый
фенотип действие нормальных клеток.
4.3. Молекулярные механизмы опухолевого роста
Два типа генов управляют размножением клеток:
протоонкогены, которые играют роль акселераторов
деления, и гены-супрессоры, выполняющие функцию
тормозов. Заклиньте акселератор или уберите тор-
моза — и клетка, будто спущенная с цепи, начнет
безостановочно делиться.
J.M. Bishop (лауреат Нобелевской премии 1989 г.)
Размножение клеток — фундаментальный биологический
процесс, обеспечивающий непрерывную смену клеточных по-
колений, формирование и развитие многоклеточных организ-
мов. Даже у одноклеточных очевидна его колоссальная слож-
ность — за короткий промежуток времени (менее суток) воссо-
здается новая клетка со всем многообразием ее структурных эле-
ментов. Еще сложнее этот процесс у многоклеточных, особенно
12*
179
у млекопитающих в период их эмбрионального развития, когда
абсолютно необходимо строго согласованное во времени и про-
странстве взаимодействие различных клеточных «ансамблей».
Опухолевый рост является следствием нарушения тканево-
го гомеостаза, поддерживаемого балансом клеточной проли-
ферации и клеточной гибели (апоптоза). В самом деле увели-
чение клеточной массы может быть в равной степени обуслов-
лено как усилением первой, так и угнетением второго. Как бы
совершенны сами по себе ни были механизмы поддержания
этого гомеостаза, общее число рождающихся и гибнущих кле-
ток столь велико (у взрослого человека сотни миллиардов тех
и других ежедневно), что вероятность «сбоев» в некоторых из
них вполне реальна, особенно если учесть канцерогенное воз-
действие факторов внешней среды. На молекулярном уровне
наследуемые нарушения механизмов тканевого гомеостаза
обусловлены теми или иными повреждениями структуры
ДНК.
У делящейся клетки с поврежденной ДНК есть альтернати-
ва: либо блок деления до полной репарации повреждений,
либо самоуничтожение (апоптоз). С точки зрения интересов
организма последний вариант (при котором реализуется прин-
цип «нет клетки — нет проблемы»), безусловно, предпочти-
тельнее, поскольку гибель одной клетки не может иметь ника-
ких отрицательных последствий, тогда как ее сохранение таит
для него (организма) смертельную угрозу возникновения
клона дефектных (потенциально опухолевых) клеток. Роль
апоптоза в канцерогенезе поэтому чрезвычайно велика.
Можно полагать, что опухолевый рост в принципе возможен
лишь постольку, поскольку дефектные клетки способны «про-
скальзывать» через защитный барьер апоптоза.
Итак, существует несколько механизмов, повреждение ко-
торых может способствовать опухолевому росту: собственно
механизм деления, действующие начала которого обладают
либо промитогенной (протоонкогены и протоонкобелки),
либо антимитогенной (супрессоры) активностью, и механиз-
мы защиты — репарация ДНК и апоптоз. Далее приведено
краткое описание функционирования этих систем в норме,
после чего рассмотрены те нарушения, которые ведут к злока-
чественной трансформации.
4.3.1. Эндокринная, паракринная
и аутокринная регуляция
В организме существует два типа физиологической регуля-
ции клеточного размножения — эндокринная и паракринная
(рис. 4.1). Эндокринная регуляция осуществляется специали-
зированными органами (железами внутренней секреции), в
180
Эндокринная
регуляция
Паракринная
регуляция
Аутокринная
регуляция
Рис. 4.1. Эндокринная, паракринная и аутокринная регуляции. Регу-
ляторные молекулы (обозначены точками) секретируются клеткой и
действуют вовне (через кровоток, на соседние клетки или на себя
же). Утолщенные полукружия на поверхности клеточной мембра-
ны — рецепторные участки.
числе которых — гипофиз, надпочечники, щитовидная, пара-
щитовидная, поджелудочная и половые железы. Они секрети-
руют продукты своей активности в кровь и оказывают генера-
лизованное воздействие на весь организм.
Паракринная регуляция отличается от эндокринной тем,
что секретируемые клетками активные вещества распростра-
няются диффузией и действуют на соседние клетки-мишени.
Есть множество действующих таким образом митогенных сти-
муляторов (полипептидных ростовых факторов): эпидермаль-
ный фактор роста, фактор роста тромбоцитов, интерлейкин-2
(фактор роста Т-клеток), фактор роста нервов и т.д.
Аутокринная регуляция [Sporn, Todaro, 1980] отличается от
паракринной регуляции тем, что одна и та же клетка является
и источником ростового фактора, и его мишенью. Результат —
непрекращающееся, самоподдерживающееся митогенное
«возбуждение» клетки, приводящее к нерегулируемому раз-
множению. В данной ситуации клетка не нуждается в исходя-
щих из организма митогенных стимулах и становится в этом
отношении полностью автономной. Хотя эта модель была
предложена авторами для объяснения механизма вирусного
канцерогенеза, различные ее модификации применимы, по-
видимому, и для многих других канцерогенных ситуаций.
4.3.2. Митогенная «рефлекторная дуга»
В регуляции сложных систем, как бы различны они ни
были, обнаруживаются общие черты. Так, можно уловить
181
Митоз
Фактор роста
Рис. 4.2. «Рефлекторная дуга» митогенного сигнала. Пояснения в
тексте.
принципиальное сходство между рефлекторной активностью
животного и митотической активностью клетки (рис. 4.2).
В обеих ситуациях на периферии системы существуют раз-
личные специализированные рецепторы (глаз, ухо, тактиль-
ные и обонятельные — в первом случае; рецепторы ростовых
факторов — во втором); в обеих ситуациях воспринимаемые
рецепторами внешние сигналы центростремительным пото-
ком передаются внутрь системы (в виде импульсов по чувстви-
тельным нервам или в виде каскадов реакций фосфорилирова-
ния), и, наконец, в обеих ситуациях после обработки сигнала
в центре (в центральной нервной системе или в клеточном
ядре) соответствующий центробежный поток (в виде импуль-
сов по двигательным нервам или в виде молекул мРНК) по-
ступает к исполнительным органам и индуцирует их актив-
ность (двигательную, секреторную и т.п. — в первом случае и
митотическую — во втором).
Перенос митогенного сигнала от периферии клетки к ее
центру (генетическому аппарату) осуществляется в виде каска-
да реакций фосфорилирования посредством протеинкиназ
(ферментов, фосфорилирующих белки). Существует три типа
протеинкиназ (тирозиновые, сериновые и треониновые) в за-
висимости от их способности фосфорилировать определенные
аминокислоты. Фосфатные группы играют роль молекулярных
переключателей: меняя конформацию определенных белковых
структур (доменов), они могут «включать» или «выключать» их
активность (имеются в виду ферментативная активность,
ДНК-связывающая способность и способность образовывать
белок-белковые комплексы).
182
Центростремительная волна митогенной импульсации в
максимально упрощенном виде сводится к передаче фосфат-
ной группы, наподобие эстафетной палочки, от одной проте-
инкиназы к другой. В конечном итоге она достигает ядерных
регуляторных белков (транскрипционных факторов), активи-
рует их (тоже посредством фосфорилирования) и тем самым
индуцирует перепрограммирование генома. Необходимо отме-
тить, что активность протеинкиназ практически на любом
этапе переноса митогенного сигнала уравновешивается актив-
ностью противодействующих им ферментов — дефосфорили-
рующих белки фосфатаз. Баланс позитивных и негативных
эффектов — фундаментальное свойство регуляции клеточного
деления, проявляемое на любом его уровне.
Противоположно направленный (центробежный, т.е. из ядра
в цитоплазму) поток информации в виде молекул мРНК обу-
словливает специфическую реакцию клетки на митогенный сиг-
нал — синтезируется множество новых белков, выполняющих
структурные, ферментативные и регуляторные функции.
Следует отметить еще ряд характерных особенностей меха-
низма митогенной стимуляции. Во-первых, участвующие в
нем белки, как правило, не уникальны, а образуют семейства,
члены которых объединены сходством структуры и функции,
но различаются в деталях (в частности, субстратной специ-
фичностью, активностью, сайтами фосфорилирования, спо-
собностью связываться со специфическими последователь-
ностями ДНК и т.д.). Это свойство обеспечивает гибкость,
пластичность и надежность всего механизма.
Во-вторых, сигнальные белки во многих случаях имеют
четко выраженное доменное (или модульное) строение. Име-
ется в виду подразделение молекулы белка на отдельные
структурно-функциональные блоки (см., в частности, стро-
ение рецепторов ростовых факторов на рис. 4.4). Другим при-
мером может служить структурная организация регуляторных
белков, в которых можно вычленить ДНК-связывающий и
трансактивирующий домены (см. далее). Функционально
близкие домены обладают структурным сходством даже у
очень отдаленных в эволюционном отношении видов, что де-
лает компьютерный поиск гомологий чрезвычайно эффектив-
ным средством выяснения функций новых белков (именно
таким способом были сделаны многие важные открытия).
В-третьих, в структуре многих сигнальных белков сущест-
вуют своеобразные «стыковочные узлы» (docking sites) разных
типов, предназначенные для белок-белковых взаимодействий.
Поскольку одна молекула может обладать несколькими таки-
ми участками, существует возможность самосборки очень
сложных многокомпонентных конструкций, необходимых и
для переноса сигнала, и для регуляции транскрипции. Присо-
единение к конструкции новых элементов иногда обозначают
183
термином «рекрутирование». Одни и те же структурные блоки
могут формировать существенно разные конструкции, что на-
деляет систему функциональной гибкостью и свойством взаи-
мозаменяемости отдельных ее элементов.
В-четвертых, линейные сигнальные пути не изолированы,
а взаимодействуют друг с другом, что ведет к формированию
сложных функциональных сетей.
В-пятых, нерегулируемое размножение трансформирован-
ной клетки можно представить, если продолжить аналогию с
рефлекторной дугой как следствие возникновения очага «за-
стойного» возбуждения в том или ином звене пути переноса
митогенного сигнала. Повреждение гена и как следствие —
структурный дефект какого-либо из сигнальных белков, спо-
собный зафиксировать его в постоянно активном состоянии
(т.е. сделать независимым от «вышестоящих» регуляторных
инстанций), — один из главных механизмов канцерогенеза.
Нормальные гены, участвующие в переносе митогенного сиг-
нала и потенциально способные на такое превращение, назы-
ваются протоонкогенами.
В-шестых, баланс позитивных и негативных факторов, как
уже отмечалось выше, — фундаментальное свойство любой
сложной регуляторной системы, в том числе и управляющей
клеточным делением. Протоонкогены — элементы позитивной
регуляции; они являются акселераторами клеточного деления и
в случае превращения в онкогены проявляют себя как доми-
нантный признак. Вместе с тем в давних опытах образования ге-
терокарионов (продуктов слияния клеток в культуре) установле-
но, что свойство туморогенности (способности образовывать
опухоли при перевивке животным) ведет себя как признак ре-
цессивный: гетерокарионы, образованные слиянием нормаль-
ных и трансформированных клеток, ведут себя как нормальные.
Это указывает на присутствие в нормальных клетках неких нега-
тивных (тормозящих клеточное деление) факторов, способных
при внесении в опухолевую клетку нормализовать ее. В последу-
ющем такие белковые факторы были идентифицированы; коди-
рующие их гены получили название генов-супрессоров. Полная
трансформация клетки, таким образом, является следствием не-
скольких генетических событий: активации онкогена(ов) и ин-
активации гена(ов), осуществляющих супрессорные функции.
4.3.3. Клеточный цикл
Все главные события канцерогенеза имеют отношение к
циклу деления (рис. 4.3). Клетки организма находятся в одном
из трех возможных состояний: в цикле, в стадии покоя с со-
хранением возможности вернуться в цикл и, наконец, в ста-
дии терминальной дифференцировки, при которой способ-
184
Рис. 4.3. Клеточный цикл (схема не отражает продолжительности от-
дельных фаз цикла). Пояснения в тексте.
ность делиться полностью утрачена (таковы, например, ней-
роны головного мозга). Образовывать опухоли могут, естест-
венно, только клетки, способные делиться.
Цикл удвоения разных клеток человека существенно ва-
рьирует — от 18 ч у клеток костного мозга до 450 ч у клеток
крипт толстой кишки (в среднем 24 ч). Наиболее заметными
его периодами являются митоз (М) и синтез ДНК (фаза S),
между которыми выделяют два промежуточных (gap) перио-
да — G1 и G2. Во время интерфазы (период между двумя деле-
ниями) клетка растет и готовится к митозу.
На протяжении фазы G1 существует ответственный мо-
мент (так называемая точка рестрикции — R), когда решается,
войдет ли клетка в следующий цикл деления или предпочтет
стадию покоя G0, в которой она может находиться неопреде-
ленно долго. Как уже упоминалось, терминально дифферен-
цированные клетки постоянно находятся в стадии покоя,
тогда как сохранившие способность к делению могут вернуть-
ся в цикл при соответствующей стимуляции. Вхождение клет-
ки в цикл деления — процесс вероятностный, определяемый
сочетанием ряда условий (внутренних и внешних); однако
после того как выбор сделан, последующие этапы совершают-
ся автоматически. Хотя клетка может быть блокирована на
той или иной стадии цикла деления, обычно это является
следствием каких-то особых обстоятельств.
В отличие от «асоциальной» опухолевой клетки клетка
нормальная подчиняется исходящим из организма сигналам.
185
Если в момент R у нее имеются все необходимые условия (до-
статочные масса и содержание белков, концентрация кальция,
обеспеченность питательными веществами) и она к тому же
получает митогенный стимул, то ее реакция — вступление в
очередной цикл деления. В отсутствие внешнего сигнала нор-
мальная клетка выходит из цикла, и в этом заключается ее ко-
ренное отличие от клетки опухолевой, которая побуждается к
делению эндогенными стимулами.
Критически важными в цикле являются моменты вхожде-
ния клетки в фазу синтеза ДНК (граница фаз G1/S) и в митоз
(граница G2/M), где действуют своеобразные «контрольно-
пропускные пункты» (checkpoints), которые проверяют це-
лостность (готовность к репликации) ДНК — в первом случае
и завершенность репликации — во втором. Клетки с повреж-
денной ДНК блокируются на границе G1/S, перед началом ее
синтеза, т.е. процесса, способного закрепить повреждения в
виде мутаций, делеций и иных нарушений ее структуры и
передать их потомству. Альтернативой блоку является апоп-
тоз. Конкретный выбор зависит от множества условий, в том
числе от индивидуальных особенностей клетки.
Процесс репликации ДНК сложен и длителен (несколько
часов), весь генетический материал должен быть воспроиз-
веден абсолютно точно, без потерь, излишеств и искажений.
В случае каких-либо отклонений клетки блокируются на под-
ходе к митозу (на границе G2/M) и тоже могут подвергнуться
апоптозу. Значение checkpoints трудно переоценить, посколь-
ку следствием именно их дефектов являются в конечном итоге
и злокачественная трансформация клетки, и прогрессия уже
сформировавшейся опухоли.
4.3.4. Перенос митогенного сигнала
Перенос митогенного сигнала — процесс многоэтапный.
В зависимости от типа клетки и конкретного митогенного сти-
мула реализуется один из множества сигнальных путей. Далее в
качестве «прототипа» описан путь, опосредованный тирозин-
протеинкиназными рецепторами и MAP-киназным каскадом.
Ростовые факторы (регуляторы пролиферации) секретиру-
ются одними клетками и действуют паракринным образом на
других. Это небольшие белки; полипептидная цепь EGF (epi-
dermal growth factor) состоит, например, из 53 аминокислот.
Существует несколько семейств ростовых факторов, члены
каждого из которых объединены структурной гомологией и
функциональным сходством. Одни из них стимулируют про-
лиферацию, например EGF, PDGF (platelet-derived growth fac-
tor, тромбоцитарный фактор роста), а другие (TGF-p, TNF,
интерфероны) — подавляют ее.
186
Рис. 4.4. Прототипическая
структура тирозинкиназ-
ного рецептора, в которой
представлены: аминокон-
цевые (N) и карбоксикон-
цевые (С) участки поли-
пептидной цепи рецепто-
ра, участки связывания
лиганда, АТР и субстрата.
Цистеиновые остатки
(светлые кружочки) фор-
мируют лигандсвязываю-
щий «карман», а тирози-
новые остатки (темные
кружочки) участвуют в вы-
полнении специфической
функции рецептора.
Рецепторы расположены на клеточной поверхности. Каж-
дая клетка обладает своим особым «репертуаром» рецепторов
и соответственно своим особым набором ответных реакций.
Тирозинкиназные рецепторы (ТКР) состоят из нескольких до-
менов: внеклеточного (взаимодействующего с лигандом),
трансмембранного и подмембранного, обладающего тирозин-
протеинкиназной активностью (рис. 4.4).
Структура рецепторов может существенно различаться.
Так, в отличие от рецептора EGF, состоящего из одной поли-
пептидной цепи (1186 аминокислот), другие могут состоять из
нескольких полипептидов. В зависимости от структуры ТКР
подразделяются на три субкласса (рис. 4.5).
Рецепторы I субкласса характеризуются мономерным стро-
ением (состоят из одной полипептидной цепи) и присутствием
во внеклеточном домене двух повторов цистеинбогатых после-
довательностей. Рецепторы субкласса II функционируют как ге-
теротетрамеры и состоят из двух а-субъединиц (формируют ли-
гандсвязывающий домен) и двух р-субъединиц (несущих каждая
по каталитическому домену), соединенных дисульфидными свя-
зями (S-S). Субъединицы аир образуются в результате расщеп-
ления белка-предшественника, кодируемого одним геном. Ре-
цепторы субкласса III отличаются двумя особенностями: отсут-
ствием цистеинбогатых повторов во внеклеточном участке (ли-
гандсвязывающий домен у них организован по-иному) и вклю-
чением длинной последовательности в каталитический домен.
При связывании с ростовыми факторами (например, EGF)
молекулы рецепторов димеризуются, их внутриклеточные до-
187
I П П1
Рис. 4.5. Структурная организация тирозинкиназных рецепторов
разных субклассов.
EGF-R — рецептор эпидермального фактора роста:1-И (рецептор инсулина),
PDGF-R (рецептор фактора роста тромбоцитов). Заштрихованные прямо-
угольники в рецепторах I и II субклассов — цистеиновые повторы в составе
внеклеточных доменов (участвуют в связывании лиганда); светлые прямо-
угольники в подмембранной области — тирозинкиназные домены; светлые
кружочки в рецепторах III субкласса — консервативные участки, обогащенные
цистеином.
мены сближаются и индуцируют межмолекулярное автофос-
форилирование по тирозину. Этот трансмембранный перенос
сигнала — зарождение той волны «возбуждения», которая рас-
пространяется затем в виде каскада реакций фосфорилирова-
ния внутрь клетки и благодаря которой митогенный стимул
достигает в конце концов генетического аппарата ядра. ТК.Р
обладают тирозинкиназной активностью, но по мере продви-
жения сигнала внутрь клетки тип фосфорилирования меняет-
ся на серин/треониновый.
Ras-белки. Одним из наиболее важных является сигнальный
путь с участием Ras-белков (это подсемейство так называемых
G-белков, образующих комплексы с гуаниловыми нуклеотида-
ми; Ras-GTP — активная форма, Ras-GDP — неактивная). Этот
путь — один из основных в регуляции клеточного деления у выс-
ших эукариот, он настолько консервативен, что его компоненты
способны заменить соответствующие гомологи в клетках дрозо-
188
филы, дрожжей и нематод. Он опосредует разнообразные сигна-
лы, исходящие из внешней среды, и функционирует, по всей ве-
роятности, в каждой клетке организма. Ras играет роль своеоб-
разного турникета, через который должен пройти почти любой
из поступающих в клетку сигналов. Критическая роль этого
белка в регуляции клеточного деления известна с середины 80-х
годов, когда активированная форма соответствующего гена (он-
коген Ras*) была обнаружена во многих опухолях человека.
Функционирует этот путь на начальном этапе следующим
образом (рис. 4.6.). Во многих белках, участвующих в переносе
митогенного сигнала, идентифицированы так называемые Src-
гомологичные домены (Src homology domains) SH2 и SH3 —
аминокислотные последовательности, гомологичные таковым
белка Src (одного из первых идентифицированных онкобелков).
Они способны специфически связываться соответственно с
фосфотирозином и с обогащенной пролином аминокислотной
последовательностью (prolin-rich motif). Таким образом, белки,
обладающие SH2- и БНЗ-доменами, способны «стыковаться» с
другими белками, имеющими соответственно либо фосфорили-
рованный тирозин, либо пролиновый «мотив». Важной особен-
ностью SH-доменов является их ограниченная пермиссивность,
т.е. способность обеспечивать взаимодействие не строго с
одним, а с несколькими родственными активаторами.
Выявлена особая группа «адаптерных» белков, обладающих
обоими SH2- и БНЗ-доменами (в частности, Grb2, growth fac-
tor receptor-bound protein 2) и способных поэтому служить свя-
зующим звеном между другими сигнальными элементами.
Благодаря ограниченной пермиссивности белка-адаптера
число возможных, возникающих при его посредничестве
белок-белковых комбинаций весьма велико.
В конкретной ситуации Ras-опосредованного сигнального
пути (например, при взаимодействии EGF с рецептором) ди-
меризация последнего приводит к автофосфорилированию
подмембранного домена. В результате возникает сайт связыва-
ния с адаптерным белком Grb2 (точнее, с его 8Н2-доменом).
В то же время два БНЗ-домена Grb2 обеспечивают его высоко-
аффинное взаимодействие с белком Sosl («рекрутируют» его в
мультибелковый комплекс, локализованный в плазматической
мембране и включающий Ras). Sosl способствует активации
Ras (посредством замены GDP на GTP) и Ras-зависимого ки-
назного каскада (см. далее). Активирующая роль Sosl уравно-
вешивается действием белка GAP (GTPase activating protein),
который повышает ГТФазную активность Ras и способствует
его превращению в неактивный комплекс Ras — GDP.
*Гены обозначаются курсивом в латинской транскрипции; белки —
продукты гена — обычным латинским шрифтом.
189
A
«Б
W Активация рецептора
Рис. 4.6. Распространение митотического сигнала от рецептора к Ras
и далее.
А — рецептор неактивен (ростовые факторы отсутствуют). Б — активация ре-
цепторов ростовыми факторами и начальные этапы передачи митогенного сиг-
нала. Grt>2 связывается с активированным (димеризованным) рецептором по-
средством SH2 домена и с Sos — посредством домена SH3. Ассоциированный с
рецептором Sos инициирует замену GDP на GTP в их комплексе с Ras, запус-
кая тем самым каскад серин-треониновых киназ, которые проводят сигнал
далее к ядру. GAP — GTPase activating protein.
Фактор роста
Рис. 4.7. Обобщенная схема MAP-киназного пути переноса митоген-
ного сигнала. Пояснения в тексте.
MAP-киназный каскад. Ras контролирует МАР-киназный
(mitogen activated protein) каскад, т.е. каскад реакций фосфо-
рилирования, возникающий как следствие митогенной акти-
вации клетки (рис. 4.7).
Как конечное следствие стимуляции Ras повышается
активность двух цитоплазматических серин/треониновых
MAP-киназ (известных также как ERK1 и ERK2 — extracel-
lular signal-regulated protein kinases 1 and 2), которые переме-
щаются в ядро клетки, где фосфорилируют ключевые факто-
ры транскрипции. Активность этих киназ в свою очередь ин-
дуцируется двумя согласованными событиями — фосфори-
лированием входящих в их состав остатков треонина и ти-
розина посредством МАРКК (MAP kinase kinase — киназа
МАР киназы) — фермента, обладающего двойной специфич-
ностью. МАРКК в свою очередь — субстрат фосфорилир-
ования ферментом МАРККК (MAP kinase kinase kinase), об-
ладающей серин/треонинкиназной активностью. Эту по-
следнюю функцию могут выполнять несколько ферментов, в
том числе белок — продукт протоонкогена /?а/, который дав-
но известен тем, что, будучи определенным образом моди-
фицирован, способен вызывать опухолевую трансформацию
клетки.
Интересен путь от активированного Ras до МАР-киназно-
го каскада (точнее до МАРККК). Он в некотором смысле уни-
191
Ras - сенсор Ras - "проводник" Raf - активатор
внеклеточных сигналов белка Raf в мембрану МАР-киназного
Рис. 4.8. Механизм активации МАР-киназного каскада. Внеклеточ-
ные сигналы активируют Ras (см. выше). Комплекс Ras-GTP взаимо-
действует с аминотерминальным регуляторным доменом Raf, фикси-
руя эту МАРКК-киназу в плазматической мембране (знаком вопроса
обозначены некие дополнительные белки мембраны, способствую-
щие фиксации). В цитоплазме Raf находится в комплексе с так назы-
ваемыми «белками теплового шока» (hsp90) и некоторыми другими.
После фиксации в мембране комплекс Ras-Raf диссоциирует и под
воздействием некоторых дополнительных факторов Raf «запускает»
MAP-киназный каскад.
кален, поскольку Ras не является киназой и проявляет только
ГТФазную активность (рис. 4.8).
Оказалось, что активный Ras (т.е. Ras-GTP) может физи-
чески взаимодействовать с N-концевым доменом Raf и рекру-
тировать его в комплекс, локализованный в плазматической
мембране. В этом, по-видимому, и заключается механизм ак-
тивации Raf, где белку Ras отводится лишь роль «проводни-
ка», надобность в котором отпадает сразу же после фиксации
МАРК.К.К в плазматической мембране. Это последнее собы-
тие, по-видимому, запускает «нижележащий» киназный кас-
кад, хотя, возможно, для этого необходим дополнительный
стимул в виде фосфорилирования Raf.
Возникает вопрос: почему в активации Raf принимает
участие не киназа, как на всех остальных этапах, а ГТФаза, ка-
ковой является Ras? Возможно, все дело в кардинальном раз-
личии индуцируемых этими ферментами белковых модифика-
ций. Если киназа действует как молекулярный выключатель
по принципу «да-нет» (on-off switch), то ГТФазу можно уподо-
бить реостату, способному варьировать эффект количествен-
но. Именно количественными различиями в Ras-опосредован-
ной активации МАР-киназного пути можно объяснить тот па-
радоксальный, на первый взгляд, факт, что сигналы, проходя-
192
Схема 4.1. Трехкиназный каскад
МЕКК----► МЕК-----► ERK
МАР-киназный каскад
Rat, Mos, МЕКК1
I
МЕК1.2
I
ERK1.2
JNK/SAPK-киназный каскад
MEKK1
JNKK
I
JNK/SAPK
HOG/рЗВ-киназный каскад
MEKK1
I
JNKK.MKK3
HOG/p38
MAP — киназные модули млекопитающих; ERK — extracellular signal-
regulated kinase; MKK — MAP kinase/ERK kinase; МЕК — то же, что и МАРКИ;
МЕКК — МЕК kinase; JNK/SAPK — Jun-N-terminal kinase/stress-activated
protein kinase; JNKK — JNK kinase. Пояснения в тексте.
щие один и тот же путь, способны вызывать абсолютно разные
клеточные реакции. Так, активация Ras в клетках линии РС12
эпидермальным фактором роста вызывает клеточную проли-
ферацию, а фактором роста нервов — дифференцировку.
Разнообразие сигналов, использующих МАР-киназный
путь, свидетельствует о том, что он «служит» разным целям.
Конкретные же последствия его активации (иногда диамет-
рально противоположные) определяются той конкретной си-
туацией, в которой оказалась данная клетка. Вместе с тем
важно подчеркнуть, что этот сигнальный путь, как и другие,
не «линеен», а входит как элемент в сложную сеть конвергиру-
ющих и ветвящихся информационных потоков.
Наиболее полно изученный и состоящий из 3 ферментов
МАР-киназный каскад является каноническим для ряда дру-
гих 3-киназных каскадов, обнаруженных в клетках млекопита-
ющих и обладающих иными внутриклеточными мишенями
(схема 4.1). Согласно концепции трехкомпонентных МАР-ки-
назных модулей, они, будучи во многих отношениях родствен-
ными, «обслуживают» разные рецепторы и образуют парал-
лельные информационные потоки. Наряду с каноническим
MAP-киназным каскадом (МАРККК -> МАРКИ МАРК)
существуют трехкомпонентные JNK/SAPK (Jun-N-terminal
kinase/stress-activated protein kinase) и HOG/p38 каскады (одни
и те же киназы известны иногда под разными названиями, что
объясняется их независимым обнаружением в разных лабора-
ториях).
Активность ферментов, участвующих в киназных каскадах,
уравновешивается активностью противодействующих им и на-
ходящихся под столь же строгим контролем фосфатаз.
13—1385
193
Схема 4.2. Регуляция транскрипции c-fos и c-jun в ответ
на внеклеточные стимулы (ростовые факторы; цитокины;
повреждение ДНК ультрафиолетовым облучением)
Регуляторные участки (прямоугольники), прилежащие к генам c-fos и
c-jun, взаимодействуют с транскрипционными факторами (на схеме не
представлены), активированными соответствующими протеинкиназами
(последние принадлежат к различным киназным каскадам). SIE (sis-induc-
ible enhancer), SRE (serum response element), TRE (TPA response element) —
регуляторные участки генов; TATA (так называемый TATA-box) — короткая
нуклеотидная последовательность, играющая важную роль в формирова-
нии транскрипционного комплекса; Jak, Jnk, Erk — последние члены раз-
ных киназных каскадов; А — активация c-fos (стрелка показывает начало
считывания РНК); Б — активация c-jun.
Активация транскрипции. Группа генов, определяющих
вхождение клетки в фазу S, активируется транскрипционным
фактором АР-1, представляющим собой комплекс белков —
членов семейств Jun и Fos (гены, их кодирующие, — c-jun и
c-fos, относятся к числу протоонкогенов; с — от cell, обознача-
ет их клеточное происхождение в отличие от вирусных онко-
генов v-jun и v-fos). Эти транскрипционные факторы могут
взаимодействовать между собой с образованием множества
гомо- и гетеродимеров, связывающихся с определенным
участком ДНК и стимулирующих синтез РНК на прилежащих
к этим участкам генов.
MAP-киназы повышают активность АР-1 двояким обра-
зом: а) опосредованным, активируя соответствующие гены и
увеличивая тем самым содержание комплекса в клетке, и
б) прямым, фосфорилируя входящие в их состав сериновые и
треониновые остатки. В первом случае (схема 4.2) МАР-кина-
зы фосфорилируют в строго определенных сайтах соответст-
194
Enhancer
РНК
Рис. 4.9. Транскрипционная триада — типичный эукариотический
транскрипционный комплекс в промоторном участке гена. Белки-ак-
тиваторы (Аг) «садятся» на участок энхансера (регуляторная последо-
вательность ДНК, усиливающая транскрипцию) и стимулируют (++)
синтез РНК посредством белок-белкового взаимодействия своих ак-
тивирующих доменов (волнистая линия) с компонентами базового
аппарата транскрипции. Последний состоит из двух основных ком-
понентов — TBP (TATA-binding protein) и РНК-полимеразы 11 (Pol
II). С ТВР взаимодействует около 10 дополнительных белков — Taf
(TBP-associated factors) и транскрипционный фактор TFIIA (А). В со-
став комплекса с РНК-полимеразой входят транскрипционные фак-
торы TFIIB (В), TFIIE (Е), TFIIF (F), TFIIH (Н) и ряд дополнитель-
ных белков, способствующих правильной посадке комплекса и точ-
ной транскрипции, — Srb, Gall 1, Sin4, Rgrl, а также мультифермент-
ный комплекс Swi/Snf (ДНК-стимулируемая АТФаза), вызывающий
разрушение структуры нуклеосом и движение РНК-полимеразы по
нити хроматина.
вующие транскрипционные факторы (в частности, TCF — ter-
nary complex factor). Последний взаимодействует с так называ-
емым базовым аппаратом транскрипции — мультифермент-
ным комплексом, формирующимся на промоторном участке
гена (в частности, на последовательности так называемого
ТАТА-бокса), в состав которого входит РНК-полимераза, и
активируют его (рис. 4.9).
Активация гена, таким образом, обеспечивается триадой:
а) транскрипционные белки-активаторы, связывающиеся с
энхансерами; б) транскрипционные факторы, взаимодей-
ствующие с ТАТА-боксом; в) РНК-полимераза II в комплек-
се со вспомогательными белками. Гены c-Jun и c-fos отно-
сятся к числу генов немедленного реагирования (immediate-
early genes), чья активность зависит от синтеза новых бел-
13»
195
ков и очень быстро возрастает вслед за клеточной стиму-
ляцией.
Другой путь активации комплекса АР-1 — непосредствен-
ное фосфорилирование Fos (треонина в положении 232) и Jun
(серины в положении 73 и 63). Эти события способствуют, с
одной стороны, формированию активного комплекса Fos/Jun,
а с другой — усилению его активирующего воздействия на ба-
зовый аппарат транскрипции. В фосфорилировании Jun участ-
вует JNK (Jun N-terminal kinase), а в фосфорилировании Fos —
FRK (Fos regulating kinase), входящие в семейство МАР-киназ.
В результате активации генов продуцируются белки, необ-
ходимые для синтеза ДНК и последующего митоза. Некоторые
из новообразованных белков (Fos, Jun, Мус), известные как
белки раннего ответа (immediate-early proteins), выполняют ре-
гуляторные функции; связываясь со специфическими участка-
ми ДНК, они активируют прилежащие гены. Другую группу
составляют ферменты, такие как тимидинкиназа, рибонуклео-
тидредуктаза, дигидрофолатредуктаза, тимидилатсинтаза, ор-
нитиндекарбоксилаза, ДНК-полимеразы, топоизомеразы и
множество других, которые имеют непосредственное отноше-
ние к синтезу ДНК. Усиливается, кроме того, общий белко-
вый синтез, поскольку при каждом цикле удвоения воспроиз-
водятся все клеточные структуры.
Нормальное прохождение клеткой отдельных фаз клеточ-
ного цикла обеспечивается строгой последовательностью и
упорядоченностью во времени многочисленных ферментатив-
ных процессов. Роль «водителя ритма» при этом играют белки
цикл ины, закономерным образом сменяющие друг друга на
протяжении цикла.
4.3.5. Реализация митогенного сигнала
Циклические реакции. Имеется два семейства белков, «дви-
жущих» клеточный цикл: циклин(сус1т)-зависимые се-
рин/треониновые протеинкиназы (Cdk, cyclin-dependent ki-
nases) и сами циклины. Циклины регулируют активность Cdk
и тем самым контролируют способность последних модифи-
цировать клеточные мишени, непосредственно участвующие в
метаморфозах цикла. Cdk активны только в комплексе с
одним из циклинов. Поэтому сборка и активация многочис-
ленных комплексов Cdk-cyclin, а также их диссоциация —
ключевые моменты клеточного цикла (рис. 4.10).
Как следует из их названия, циклины синтезируются и
распадаются в строго определенные моменты цикла, разные
для разных циклинов. Имеется три основных класса: Gl-цик-
лины, необходимые для прохождения G1/S, S-циклины — для
прохождения S-фазы и Сг2-циклины (или митотические) —
196
Фаза G2
Рис. 4.10. Разные циклины комплексируют с Cdk, активируют их в
определенном порядке и «проводят» клетку через критические фазы
цикла.
для вхождения в митоз. В клетках млекопитающих имеется
также несколько семейств Cdk, участвующих в различных ре-
гуляторных событиях. Исчезновение циклина из внутрикле-
точной среды в определенный момент столь же важно, как и
его появление (устранение циклинов из внутриклеточной
среды достигается как их деградацией, так и блоком синтеза).
Например, в митозе (на границе метафазы и анафазы) один из
циклинов быстро деградирует в результате протеолиза; если же
этого не происходит, то митоз не может завершиться и разде-
ления дочерних клеток не происходит.
Первоначально в цитоплазме ооцитов Xenopus был обнару-
жен фактор MPF (maturation promoting factor), который ока-
зался способным индуцировать специфические для митоза
процессы — в частности, дезинтеграцию ядерной мембраны,
конденсацию хроматина, формирование веретена и ряд дру-
гих. После его очистки оказалось, что MPF состоит из Cdk,
названной Cdc2 (она же p34cdc2), в комплексе с митотическим
циклином (cyclin В).
Активация Cdk, необходимой для вхождения клеток в митоз,
требует не только ее ассоциации с циклином, но и двух допол-
нительных модификаций — фосфорилирования треонина в по-
ложении 161 и, напротив, дефосфорилирования тирозина 15.
Эти реакции осуществляются специфическими ферментами
(киназами и фосфатазами), образующими дополнительные
197
Рис. 4.11. Активация циклинзависимой киназы требует не только
формирования ее комплекса с циклином, но и фосфорилирования
треонина 161 (Т161) и дефосфорилирования тирозина 15 (Y15). Ста-
тус тирозина 15 находится под строгим контролем, определяемым
балансом активностей двух ферментов Cdc25 (высокоспецифическая
фосфатаза) и weel (Сбс2-специфическая тирозинкиназа).
уровни контроля. Так, киназа, активирующая Cdk, фосфорили-
рует треонин 161, чего, однако, оказывается недостаточно из-за
присутствия ингибирующего фосфотирозина 15 (степень фос-
форилирования последнего определяется балансом активностей
Cdc2 — специфической тирозинкиназы weel и конкурирующей
с ней фосфатазы Cdc25 — высокоспецифичной и в свою очередь
находящейся под строгим контролем).
После образования cyclin-Cdc2 weel фосфорилирует тиро-
зин 15, инактивируя комплекс. Затем происходит активирующее
фосфорилирование треонина 161 и, наконец, дефосфорилиро-
вание тирозина 15 ферментом Cdc25, что обеспечивает полную,
необходимую для вхождения в митоз активность Cdk (рис. 4.11).
Преодоление G1/S и продвижение в фазе S требует актива-
ции иных киназ — Cdk2, Cdk4 и Cdk6, которые взаимодейст-
вуют уже с G1-фазными циклинами (в частности, с cyclin D).
Комплекс Cdk2 с первым G1-фазным циклином индуцирует
транскрипцию гена следующего цикл ина и т.д., продвигая
клетки все дальше по циклу. Cdk2-cyclin D в самом начале за-
мещается на Cdc2-cyclin Е, а тот в свою очередь на Cdc2-cyclin
А, активирующий аппарат синтеза ДНК (рис. 4.12). Когда
клетка входит в S-фазу, Gl-циклины деградируют и появляют-
ся вновь лишь в G1 следующего цикла.
Контрольно-пропускные пушапы (checkpoints). Любое стрес-
сорное воздействие (например, отсутствие питательных ве-
ществ, гипоксия и в особенности повреждение ДНК) блокиру-
ет движение по циклу в одном из двух упомянутых контроль-
ных пунктов (checkpoints). Во время этих остановок активиру-
ются механизмы надзора, способные: а) обнаружить повреж-
198
Рис. 4.12. Делящиеся клетки человека содержат семейство циклинов.
Точки приложения основных из них представлены на рисунке.
дение ДНК, б) передать сигнал неблагополучия, блокирую-
щий синтез ДНК или митоз, и в) активировать механизмы ре-
парации ДНК. Благодаря этому обеспечивается стабильность
генома. Как упоминалось выше, механизм контроля G1/S бло-
кирует репликацию ДНК и активирует процессы репарации
(или индуцирует апоптоз), тогда как механизм контроля G2/M
запрещает митоз до завершения репликации. Дефекты этих
механизмов могут привести к появлению дочерних клеток с
поврежденным геномом.
В механизме checkpoint участвуют комплексы Cdk-cyclin и
ряд дополнительных белков — Rb, р53 и др. Их совокупность об-
разует систему «тормозов», не позволяющих клетке делиться в
отсутствие адекватных стимулов. Кодирующие эти белки гены
получили название генов-супрессоров. Особое значение этой
системы заключается в том, что раковая трансформация клетки
возможна только после ее инактивации. Важно иметь в виду, что
в соматической клетке существует по два аллеля каждого из
генов, в том числе и генов-супрессоров, и, следовательно, для их
инактивации необходимы два независимых события (например,
делеция одного аллеля и мутация другого). Именно в этом кро-
ется причина того, что «спорадические» опухоли возникают от-
носительно редко (вероятность возникновения в одной клетке
199
A
Cyclin
Активная CdK
Активная CdK
Неактивная Cd К
Рис. 4.13. Структура
активной Cdk и ее
инактивация CKI.
А — активация каталити-
ческой субъединицы Cdk
достигается ее комплекси-
рованием с циклином,
фосфорилированием тре-
онина и дефосфорилиро-
ванием тирозина; Б —
CKI связывается с пол-
ностью активным ком-
плексом Cdk-cyclin и по-
давляет его активность.
нескольких независимых мутаций, причем поражающих один и
тот же локус обеих хромосом, относительно невелика), а «семей-
ные» чрезвычайно часты (в «раковых» семействах один из двух
наследуемых аллелей того или иного гена-супрессора исходно
дефектен). В последнем случае система «тормозов» у всех клеток
данного организма держится лишь на одном нормальном алле-
ле, что резко снижает ее надежность и повышает риск возникно-
вения опухоли. Именно это и происходит при наследственной
ретинобластоме (деления одного аллеля Rb) и других наследст-
венных синдромах (делеция или повреждение одного аллеля р53
или других генов-супрессоров).
В механизме Gl/S checkpoint участвуют, помимо комплек-
са Cdk-cyclin, Cdk-ингибирующие белки (CKI, Cdk inhibitory
proteins). Последние взаимодействуют с комплексом Cdk-cy-
clin и ингибируют его (рис. 4.13).
В семейство CKI входит несколько членов (в частности, р27,
р16, р21), которые продуцируются клеткой в ответ на разные
стимулы и блокируют клеточное деление. Так, антимитогенный
фактор TGFp (transforming growth factor P) индуцирует обрати-
мый блок клеток в поздней Gl-фазе посредством высвобожде-
ния р27 из комплекса с неким ингибитором, после чего р27 свя-
зывается с Cdk2-cyclin Е и подавляет его активность. Напротив,
митоген интерлейкин-2 (IL-2) инактивирует р27 и способствует
тем самым продвижению клеток по циклу. Этот же представи-
тель семейства CKI участвует, по-видимому, в феномене кон-
тактного ингибирования (блоке деления клеток при соприкос-
новении их поверхностей) — характерном свойстве нормальных
клеток, отсутствующем у клеток опухолевых.
200
Митогенный
сигнал
Транскрипция генов,
специфических
для фазы S
Рис. 4.14. Белок р53 координирует различные реакции клетки в ответ
на повреждение ДНК. Увеличение содержания р53 может привести в
конкретных условиях к апоптозу (возможно, через транскрипцион-
ную активацию белка Вах, см. далее), к индукции ферментов репара-
ции (через стимуляцию синтеза белка GADD45) или к блоку клеток в
фазе G1. В последнем случае р53 активирует транскрипцию гена
WAF1/CIP1, в результате чего белок WAF1/CIP1 ингибирует ком-
плекс Cdk2-cyclin. Тем самым предотвращается индуцируемое мито-
генным сигналом фосфорилирование белка-супрессора Rb. Нефос-
форилированный Rb связан с фактором транскрипции E2F и инги-
бирует его. Напротив, в отсутствие тормозящего влияния р53 и
WAF1/CIP1 происходят фосфорилирование Rb, высвобождение E2F
и, как следствие, активация транскрипции «S-фазных» генов. В ре-
зультате этого клетка входит в фазу S клеточного цикла. А — активи-
рующее воздействие; И — ингибирующее воздействие.
Сходную роль выполняет CKI р16, который кодируется
геном-супрессором MTS1 и взаимодействует с Cdk-cyclin D.
CKI pl6 инактивирован во многих линиях опухолевых клеток
и в клетках «семейной» меланомы. Строго упорядоченное во
времени появление CKI обеспечивает их взаимодействие толь-
ко с определенными комплексами Cdk-cyclin.
Особо важную роль в формировании блока G1/S играет CKI
р21 — белок, известный также как WAF1 и CIP1 (рис. 4.14).
201
Синтез р21 индуцируется главным клеточным «хранителем ге-
нома» белком-супрессором р53 (последний в свою очередь акти-
вируется при возникновении различных повреждений ДНК и, в
частности, при у-облучении клеток). WAF1/CIP1 связывается с
комплексом Cdk2-cyclin. Ингибирование последнего предотвра-
щает фосфорилирование супрессора Rb; в дефосфорилирован-
ном состоянии Rb связывается с фактором транскрипции E2F1,
который «запускает» «гены пролиферации», и ингибирует его.
В клетках с интактной ДНК содержание р53 исчезающе
мало и соответственно отсутствует WAF1/CIP1; ничто не пре-
пятствует протеинкиназной активности Cdk2-cyclin и фосфо-
рилированию Rb. Комплекс Rb-E2F1 распадается, и высво-
божденный из-под ингибирующего влияния транскрипцион-
ный фактор E2F1 активирует группу генов, необходимых для
вступления клетки в фазу синтеза ДНК.
У клеток с дефектным или отсутствующим р53 контроль-
ный пункт G1/S неполноценен. Это проявляется в том, что
повреждения ДНК, индуцированные ионизирующей радиа-
цией или каким-либо другим способом, не приводят ни к за-
держке клеток на границе фаз G1/S, ни к апоптозу. В резуль-
тате в популяции накапливаются клетки со множественными
нарушениями структуры ДНК; появляется и со временем на-
растает нестабильность генома, которая ведет к появлению все
новых клеточных клонов. Их естественный отбор лежит в ос-
нове опухолевой прогрессии — постоянного «дрейфа» опухоли
ко все большей автономности и злокачественности. Именно
свойство нестабильности генома наделяет клетки опухоли, с
одной стороны, замечательной приспособляемостью к услови-
ям окружающей среды и «увертливостью» по отношению к ле-
чебным воздействиям, а с другой — все новыми качествами, в
частности способностью метастазирования.
«Выше» р53 в регуляторной цепи расположен белок, коди-
руемый геном ATM (ataxia-telangiectasia mutated). Его функция
состоит в активации р53 в ответ на повреждение ДНК ионизи-
рующей радиацией. В свою очередь р53 активирует группу
генов GADD (Growth Arrest and DNA Damage-inducible), про-
дукты которых, по-видимому, «запускают» механизм репара-
ции (см. рис. 4.14).
4.3.6. Апоптоз
Апоптоз (или программируемая клеточная гибель) — ши-
роко распространенный биологический феномен клеточного
«самоубийства», которое индуцируется либо разнообразными
внешними стимулами, либо неразрешимыми «внутренними»
конфликтами (например, невозможностью репарации повреж-
дений ДНК). Роль апоптоза велика не только в формообразо-
202
вательных процессах во время эмбриогенеза (формирование
органов, замена одних тканей другими, резорбция временных
органов и т.д.), но и в поддержании тканевого гомеостаза во
взрослом организме. В регуляции тканевого гомеостаза кле-
точная гибель выполняет функцию, комплементарную митозу.
У опухолевых клеток программа клеточной гибели во многих
случаях блокирована, что вносит существенный вклад в увели-
чение массы опухоли.
Феноменология. Существует два типа клеточной гибели:
некроз и апоптоз (клеточное «убийство» и «самоубийство» со-
ответственно), различающиеся как индуцирующими эти про-
цессы стимулами, так и механизмом их осуществления.
Некроз — метаболическая катастрофа, вызванная тяжелы-
ми повреждениями клеточных структур. Для него характерно
раннее увеличение объема клетки и митохондрий с последую-
щим их автолизом, тогда как внутриядерные нарушения обна-
руживаются позднее (таким образом, вектор видимых измене-
ний направлен от периферии клетки к ее центру — ядру).
Апоптоз как явление физиологическое распространен го-
раздо шире. С его участием проходят морфообразовательные
процессы в эмбриогенезе, позитивная и негативная селекция
Т- и В-лимфоцитов, индуцированная глюкокортикоидами ги-
бель лимфоцитов, а также гибель клеток при их естественном
старении. Усиленный апоптоз ведет к инволюции органа, что
встречается и в физиологических условиях (например, молоч-
ная железа в постлактационном периоде), и при патологии
(например, при дегенеративных заболеваниях центральной
нервной системы).
Опухолевые клетки имеют селективное преимущество
перед нормальными, поскольку механизм апоптоза у них за-
частую дефектен (в частности, о выживании опухолевых кле-
ток в условиях гипоксии см. далее). Кроме того, у многих из
них развивается резистентность к различным химиопрепара-
там и к облучению, которые индуцируют гибель также по ме-
ханизму апоптоза. Морфологически апоптоз проявляется вна-
чале в конденсации и маргинации хроматина, уменьшении
объема клетки и фрагментации клеточного ядра, в характер-
ной складчатости плазматических мембран (вектор измене-
ний — от центра клетки к периферии). Одним из ключевых
моментов апоптоза, знаменующим необратимость процесса,
является распад ДНК. Процесс заканчивается фрагментацией
клетки с образованием так называемых апоптотических телец,
которые быстро фагоцитируются макрофагами или соседними
клетками без сопутствующей воспалительной реакции.
Механизмы аноптоза. Прорыв в выяснении механизмов
апоптоза у млекопитающих был достигнут в результате изуче-
ния весьма, казалось бы, экзотического объекта — нематоды
Caenorhabditis elegans, на определенной стадии развития кото-
203
рой в строгой очередности погибает 131 соматическая клетка
из 1090 имеющихся [Horvitz, 1986]. Эта оказавшаяся исклю-
чительно ценной модель позволила идентифицировать более
десятка участвующих в апоптозе генов. В частности, ced-З и
ced-4 (cell death abnormal) необходимы для клеточной гибели и
при их дефектах клетки не погибают; напротив, ced-9 кодирует
белок, предотвращающий апоптоз (его мутации сопряжены с
массовой клеточной гибелью). Продукт гена ced-З, как оказа-
лось, является протеазой.
Принципиальное значение имел тот факт, что механизмы
апоптоза чрезвычайно консервативны и сохраняют свои фун-
даментальные черты у весьма далеких в эволюционном отно-
шении организмов. Это обстоятельство позволило идентифи-
цировать у млекопитающих (и, в частности, у человека) гены,
гомологичные генам апоптоза у нематоды.
Гомологи ced-9 — члены семейства bcl-2. У млекопитаю-
щих гомологами ced-9 являются члены семейства bcl-2 [ген
bcl-2 впервые обнаружен в участке хромосомной транслокации
t(14; 18) в В-клеточной фолликулярной лимфоме]. Его роль
антиапоптическая, он предотвращает гибель клеток. Белок,
кодируемый bcl-2 (мол. масса 26 кДа), локализован преимуще-
ственно в мембране митохондрий и не содержит последова-
тельностей с уже известными каталитическими или иными
функциями. Фосфорилирование серина в Вс1-2 инактивирует
его и может, таким образом, играть регуляторную роль.
Механизм апоптоза у млекопитающих неизмеримо слож-
нее, чем у нематод (анализ генов последних был лишь на-
чальным, хотя и очень важным толчком). Так, у bcl-2 млеко-
питающих имеется множество гомологов, образующих семей-
ство и по-разному участвующих в программируемой клеточ-
ной гибели. Все белки этого семейства объединены общнос-
тью структуры — присутствием функционально важных об-
ластей ВН1, ВН2 и ВИЗ (Bcl-2 homology regions 1, 2 и 3), но
могут быть подразделены на две группы соответственно их
антиапоптическому (Bcl-2, BHRF1, MCL-1, Bcl-xL) или про-
апоптическому (Вах, Bcl-xs, Bak) действию. Белок-супрессор
р53 активирует Ьах и ингибирует bcl-2-гены, что, возможно,
является механизмом апоптоза, индуцируемого у клеток с
поврежденной ДНК на границе G1/S (см. выше). Вместе с
тем необходимо заметить, что индукция апоптоза может про-
исходить как опосредованно через р53, так и независимо от
него.
Члены семейства способны к гомо- и гетеродимеризации.
Так, Вах образует гомодимеры и гетеродимеры с Bcl-2, Bcl-xL
и другими белками. Комплексирование проапоптических и
антиапоптических белков ведет, по-видимому, к их взаимному
«погашению», в результате чего конечный результат (индук-
ция апоптоза или, напротив, его отмена) определяется «балан-
204
Схема 4.3. Взаимоотношения семейства Вс1-2
и семейства ICE-подобных протеаз при индукции апоптоза
Bcl-2,Bcl-Xj.
и ДР-
Расщепление
специфических
субстратов --------Апоптоз
(ламины, PARP,
RNP и др.)
Ламины — белки, составляющие основу ядерной мембраны; PARP —
poly (ADP-ribose) polymerase, фермент, участвующий в репарации ДНК;
RNP — рибонуклеопротеидные комплексы. А — активация; И — ингибирова-
ние.
сом сил». Отношение Вс1-2 к Вах определяет чувствительность
данной клетки к апоптическим стимулам (в связи с этим гово-
рят о реостате Bcl-2/Bax). Поскольку гибель опухолевых кле-
ток под воздействием химиопрепаратов и облучения происхо-
дит по механизму апоптоза, то резистентность опухолей к
этим воздействиям может быть обусловлена повышенной экс-
прессией bcl-2.
Гомологи ced-З — члены семейства протеаз. У млекопита-
ющих гомологом гена нематоды ced-З является ген специфи-
ческой цистеиновой протеазы ICE, расщепляющей предшест-
венник интерлейкина-1р. Его искусственная (в условиях
генно-инженерных экспериментов) гиперэкспрессия в куль-
тивируемых клетках вызывает апоптоз. ICE функционирует в
виде тетрамера двух субъединиц размером 10 и 20 кДа и имеет
несколько гомологов с разной субстратной специфичностью.
Хотя участие ICE в апоптозе несомненно, мишени ее протео-
литической активности пока не определены.
Функциональные взаимоотношения между семейством
Вс1-2 и семейством ICE-подобных протеаз в настоящее время
не установлены окончательно, хотя в апоптических сигналь-
ных путях первые явно стоят «выше по течению» (схема 4.3).
Fas/APO-1 (CD95) — рецепторы специфических индукто-
ров апоптоза. Большое функциональное значение имеет меха-
низм апоптоза, индуцируемого через специфические рецеп-
торы — CD95 (трансмембранный белок-рецептор размером
45 кДа, который при связывании со специфическим лигандом
или антителами передает сигнал к апоптозу) и TNF-R (tumor
necrosis factor receptor — рецептор фактора некроза опухолей).
205
Эти рецепторы, объединяемые сходством внеклеточных доме-
нов, входят в состав большого семейства.
Лигандами (молекулами, специфически взаимодействую-
щими с рецепторами TNF-R и CD95), являются соответствен-
но TNF и CD95-L, которые представляют собой трансмем-
бранные белки, но могут функционировать и в растворимой,
«свободной» форме. Особенно интересен с онкологической
точки зрения TNF — цитокин, производимый многими клет-
ками (макрофагами, моноцитами, лимфоидными клетками,
фибробластами) в ответ на воспаление, инфекцию и другие
стрессорные воздействия. Он индуцирует широкий спектр
иногда противоположных по направленности реакций, вклю-
чая лихорадку, шок, некроз опухоли, анорексию; индуцирует
иммунорегуляторные сдвиги, клеточное размножение, диффе-
ренцировку и апоптоз. Эффекты TNF опосредованы через два
рецептора (трансмембранные белки размером 55 и 75 кДа,
TNF-RI и TNF-RII соответственно), по крайней мере один из
которых присутствует во всех клетках.
Взаимодействие трех молекул TNF с TNF-RI «сшивает»
рецепторы с образованием тримера, что запускает каскад пос-
ледующих разветвленных реакций, ответственных за плейо-
тропные эффекты TNF. Их непосредственный итог зависит от
того, какие именно сигнальные белки окажутся рекрутирован-
ными в состав первичного комплекса. Тримеризация рецепто-
ра дает начало самосборке под плазматической мембраной
сложной конструкции, существующей благодаря белок-белко-
вому взаимодействию «стыковочных узлов», получивших на-
звание «доменов смерти» (death domains), — специфической
последовательности, присутствующей во внутриклеточной
части рецептора и абсолютно необходимой для передачи цито-
токсического сигнала (рис. 4.15).
В комплекс рекрутируются белки TRADD (TNFRI — asso-
ciated death domain protein), FADD/MORT1 (Fas — associated
protein with death domain) и RIP (receptor — interacting protein),
С-концевые участки которых содержат «домены смерти» (RIP,
кроме того, является серин/треониновой киназой). В ком-
плексе также участвуют TRAF1 и TRAF2 (TNFR — associated
proteins 1 и 2). В свою очередь N-концевой домен FADD взаи-
модействует с ICE-подобной протеазой MACH (MORT1 — as-
sociating CED homolog), известной и как FLICE (FADD-like
ICE), непосредственно запускающей апоптический протеаз-
ный каскад. В то время как FADD включает механизм апопто-
за, другие участники комплекса индуцируют иные эффекты:
TRAF2 и RIP активируют NF-кВ (транскрипционный фактор
плейотропного действия, оказывающий антиапоптическое
действие) и JNK (см. выше).
Активация комплекса NF-кВ происходит своеобразно:
фосфорилирование ингибирующей субъединицы 1кВ приво-
206
Рис. 4.15. Модель рекрутирования сигнальных молекул при актива-
ции TNF-RI, индуцирующей апоптоз и другие клеточные реакции.
Вызываемая лигандом тримеризация TNF-RI ведет к рекрутирова-
нию TRADD (путем связывания с цитоплазматическим доменом ре-
цептора через «домены смерти» — полностью заштрихованные участ-
ки). TRADD служит платформой для связывания двух дополнитель-
ных белков, обладающих «доменами смерти», — RIP и FADD, а так-
же TRAF2, который не обладает этим доменом; в последнем случае
взаимодействуют N-концевая последовательность TRADD и С-кон-
цевая последовательность TRAF2 (частично заштрихованный учас-
ток). В то время как FADD индуцирует апоптический протеазный
каскад, TRAF2 и RIP активируют транскрипционный фактор широ-
кого спектра действия NF-kZ? (оказывает антиапоптическое дейст-
вие) и киназный каскад. Пояснения в тексте.
дит к ее высвобождению из цитоплазматического комплекса,
включающего также ДНК-связывающие субъединицы р50 и
р65 (последняя известна также как Rel-A). Гетеродимер
p50/Rel-A транслоцируется затем в ядро клетки, где связыва-
ется со специфической последовательностью ДНК.
Таким образом, тримеризация TNF-RI ведет к активации
по крайней мере трех сигнальных путей, что объясняет разно-
образие биологических эффектов TNF.
207
4.3.7. Механизмы опухолевой трансформации
Принципиальное различие между нормальной и опухоле-
вой клеткой заключается в том, что переход от покоя к деле-
нию (GO -> G1) в первом случае инициируется внешними по
отношению к клетке стимулами, а во втором — внутренними.
В молекулярных терминах суть сводится к активации онкоге-
нов и инактивации генов-супрессоров.
Активация онкогенов. В середине 80-х годов было осозна-
но, что некоторые нормальные клеточные гены обладают он-
когенным потенциалом. Так сформировалась концепция он-
когена — «врага внутреннего», возникающего из нормального
гена (протоонкогена) при его повреждении тем или иным спо-
собом. Такая трансформация называется активацией онкоге-
на. Протоонкогены — «акселераторы» клеточного деления, их
функция проявляется как доминантный признак, т.е. стано-
вится явной, при активации даже одного аллеля (напомним,
что выпадение тормозящей функции генов-супрессоров обна-
руживается лишь при утере обоих аллелей).
Возникает вопрос: какие именно гены могут трансфор-
мироваться в онкогены? Из всего изложенного выше следует,
что этим свойством обладают гены, участвующие позитивным
образом в переносе митогенного сигнала независимо от зани-
маемого ими в сигнальном пути места (начиная от ростовых
факторов и кончая транскрипционными факторами), в част-
ности, упоминавшиеся ранее протоонкогены rad, raf, тус,
bcl-2, jun, fos и др. (их число достигает сегодня нескольких де-
сятков и постоянно растет по мере углубления представлений
о механизмах клеточного деления). При повреждении тем или
иным способом одного из этих генов (факторами физически-
ми или химическими, эндогенными или экзогенными) струк-
тура кодируемого им белка может оказаться зафиксированной
в активной конформации. Таким образом, то, что в нормаль-
ной клетке достигается воздействием внешнего стимула и
длится столько же, сколько длится он сам (имеется в виду ак-
тивная конформация сигнального белка), в клетке мутантной
(и в ее потомках) оказывается необратимо закрепленным.
Ущербный сигнальный белок «гонит волну возбуждения» вниз
по течению (downstream) независимо от состояния вышестоя-
щих структур. Возникает состояние «застойного возбуждения»
(см. выше), которое лежит в основе автономного, нерегулиру-
емого деления опухолевой клетки.
Несколько примеров. В разных «спонтанных» опухолях че-
ловека (рак желудка, мочевого пузыря и др.) обнаружена точ-
ковая мутация гена с-На-газ-1 (одного из представителей се-
мейства ras) в кодоне 12 (или в кодоне 61). Установлен, таким
образом, поразительный факт: достаточно замены одного ос-
нования (и соответственно одной аминокислоты в белке),
208
чтобы протоонкоген превратился в онкоген, т.е. приобрел
трансформирующие свойства. При этом кодируемый геном
с-На-газ-1 белок теряет в определенной мере ГТФазную ак-
тивность, в силу чего находится постоянно в «возбужденном»
состоянии, поскольку находящийся с ним в комплексе GTP
не гидролизуется до GDP. Перманентно активный ras «запус-
кает» MAP-киназный каскад независимо от состояния рецеп-
тора и наличия ростовых факторов. В отличие от нормальных
клеток, где митогенный сигнал имеет дискретный, импульс-
ный характер, в клетках с поврежденным ras митогенная сти-
муляция непрерывна.
Клетки в культуре можно трансформировать онкогеном sis,
который, как оказалось, кодирует одну из субъединиц фактора
роста тромбоцитов. В этом случае реализуется в чистом виде
аутокринная регуляция, при которой клетка секретирует фак-
тор роста и стимулирует самое себя. Такое самоподдерживаю-
щееся возбуждение выводит клетку из-под регуляторных влия-
ний организма.
Активация онкогенов может происходить разными путями:
в результате дефекта регуляции их транскрипции или измене-
ния «дозы» гена (при его амплификации), что приводит к не-
адекватному месту и времени избыточному синтезу соответст-
вующего белка (в данном случае нарушение имеет количест-
венный характер), или в результате структурного повреждения
(мутации, делеции), фиксирующего сигнальный белок в ак-
тивной конформации (качественное нарушение).
По первому типу развиваются события, например, в не-
которых случаях мелкоклеточного рака легкого, при нейро-
бластомах и глиобластомах, когда соответственно гены с-мус,
N-myc, c-erb В выходят из-под клеточного контроля и кодируе-
мые ими белковые продукты образуются не вовремя и в из-
бытке (30—50-кратном). К такому же результату могут приво-
дить транслокации содержащего протоонкоген локуса в учас-
ток другой хромосомы, где находятся активные гены. В этом
случае структурная часть гена лишается нормальных регуля-
торных механизмов (промотор, энхансер и т.д.) и ставится под
контроль «чужого» окружения. Таков механизм активации
с-тус при лимфоме Беркитта. Ген транслоцируется из хромо-
сомы 8 в локус генов тяжелых цепей иммуноглобулинов хро-
мосомы 14, где попадает под действие активного промотора и
транскрибируется сверх меры, что и приводит к трансфор-
мации.
По второму типу (т.е. с нарушением структуры кодируемо-
го онкогеном белка) развиваются события при многих формах
опухолей человека (см. приведенный выше пример с онкоге-
ном c-Ha-ras). Действительно, прямое сопоставление нуклео-
тидных последовательностей протоонкогенов и соответствую-
щих им онкогенов, как правило, выявляет в последних ряд от-
14— 1385
209
личий (различные делении и мутации), особых в каждом от-
дельном случае.
Таким образом, активация онкогена обусловлена наруше-
нием его регуляторной или структурной части.
Инактивация генов-супрессоров. Как уже упоминалось, для
полной трансформации клетки в подавляющем большинстве
случаев недостаточно одной лишь активации онкогена. Бес-
контрольному клеточному размножению препятствуют гены-
супрессоры (см. выше). Об их существовании догадывались
давно, исходя из двух групп фактов. Во-первых, в упоминав-
шихся выше опытах слияния клеток и образования гетерока-
рионов было установлено, что в паре «клетка нормальная +
клетка злокачественная» берет «верх» первая (полученный со-
матический гибрид теряет свои туморогенные свойства). Это
свидетельствовало о наличии в нормальной клетке некоего
тормозящего митотическую активность фактора, отсутствую-
щего, по-видимому, в клетке опухолевой, и о том, что соответ-
ствующие гены рецессивны — клетки становятся опухолевыми
при утере обоих аллелей.
Во-вторых, при цитогенетическом исследовании опухоле-
вых клеток постоянно обнаруживаются делеции хромосом, до-
вольно специфические для разных форм рака. Это заставляло
думать (и это предположение полностью оправдалось), что
именно отсутствие соответствующих генов лишает клетку ми-
тотических «тормозов». Например, цитогенетический анализ
больных ретинобластомой (злокачественная опухоль сетчатки
глаза, характеризующаяся зачастую наследственной предрас-
положенностью) выявляет характерную для этой формы деле-
нию в 13-й хромосоме, где при последующем анализе действи-
тельно был идентифицирован ген-супрессор Rb. Наследствен-
ная деления одного аллеля этого гена делает человека предрас-
положенным к заболеванию ретинобластомой или остеосарко-
мами. Цитогенетические наблюдения подобного рода являют-
ся в настоящее время отправной точкой для прицельного по-
иска генов-супрессоров. Гены-супрессоры кодируют белки,
блокирующие каждый своим особым образом разные этапы
митогенной стимуляции (к ним относятся уже упоминавшиеся
р53 — главный «хранитель генома», Rb — формирующий меха-
низм С Gl/S checkpoint, pl6vi р21 — блокирующие активность
комплексов cyclin-Cdk, и др.).
Ген-супрессор р53. Особую роль в канцерогенезе играет ген-
супрессор р53. Продукт этого гена (белок с мол. массой
53 кДа, отсюда его обозначение р53) — регулятор транскрип-
ции, способный активировать одни гены и подавлять другие,
дефектен в большинстве опухолей человека независимо от их
происхождения и локализации (опухоли легких, головного
мозга, толстой кишки, пищевода, молочной железы, шейки
матки, печени, гемопоэтических и ретикулоэндотелиальных
210
тканей и др.). Белок р53 чрезвычайно консервативен (обнару-
жен у многих представителей животного мира), состоит (у че-
ловека) из 393 аминокислот, локализован, как правило, в кле-
точном ядре и может быть подразделен на несколько доменов:
• N-концевой, обогащенный кислыми аминокислотами и
характерный для многих регуляторов транскрипции;
• центральный, обладающий ДНК-связывающими свойст-
вами;
• С-концевой, способствующий олигомеризации (р53
функционирует в виде тетрамера).
Функция р53 в нормальных клетках заключается в связы-
вании со специфическими последовательностями ДНК, нахо-
дящимися в промоторных участках других генов, и в управле-
нии их активностью. Идентифицированы несколько активи-
руемых р53 генов, в том числе ген-ингибитор циклинзависи-
мой киназы p21(WAFl или Cipl), ген GADD45, индуцируемый
повреждениями ДНК; протоонкоген mdm-2, проапоптический
ген Ьах. Экспрессию других генов (например, тус и bcl-2) р53,
напротив, ингибирует. Повреждение ДНК нормальных клеток
приводит к увеличению содержания р53 (за счет главным об-
разом посттранскрипционной регуляции), к индукции р53-за-
висимых генов, задержке клеток на границе G1/S или апоп-
тозу.
Во многих опухолевых клетках (возможно, во всех) гены
р53 делецированы или мутантны. Мутации, которые группиру-
ются в 4 «горячих» участках, по-разному в зависимости от ло-
кализации инактивируют р53:
• missense мутации (замена аминокислоты) в ДНК-связы-
вающем домене лишают его способности связываться с
ДНК и регулировать экспрессию близрасположенных
генов;
• nonsense мутации, укорачивающие белок р53, нарушают
его способность к олигомеризации и комплексообразо-
ванию с другими белками;
• missense мутации в N-концевом домене влияют на его
регуляторные функции.
Возможны и другие причины функциональной инактива-
ции р53. Это происходит, например, в результате инфицирова-
ния клеток теми вирусами, продукты которых способны взаи-
модействовать с р53 (такова, по-видимому, одна из причин
возникновения рака шейки матки у женщин, инфицирован-
ных вирусом HPV папилломатоза человека). К тому же резуль-
тату приводит продукция в избыточном количестве некоторых
внутриклеточных белков, способных связывать р53 и лишать
его функциональной активности, что происходит, в частнос-
ти, при амплификации гена mdm2.
14*
211
OOOOOOOGOQ Нормальные клетки
Онкогенная |
мутация |
@ Начальная опухоль
Последующие I
мутации 1
Большая опухоль: гипоксия
SgStBSm в центре опухоли индуцирует р53;
блок деления, апоптоз
Мутация р53
Клональная
экспансия
Мутентные по р53 клетки
делятся и не претерпевают
апоптоза
Мутантные по р53 клетки
доминируют в опухоли
о Клетки в блоке/апоптоэе
• Мутантные по р53 клетки
Рис. 4.16. Вовлечение р53 в эволюцию опухоли. Мутантные по р53
клетки устойчивы к гипоксии, смертельной для других клеток. Пояс-
нение в тексте.
Клетки с дефектами р53 имеют ряд существенных особен-
ностей — резко возрастающую нестабильность генома, прояв-
ляющуюся, в частности, в амплификации генов, и дефект кон-
трольного пункта G1/S (при внесении в мутантные клетки
копий р53 дикого типа нормальные клеточные реакции вос-
станавливаются). Таким образом, ключевая роль р53 заключа-
ется, по всей вероятности, в поддержании стабильности гено-
ма и в индукции апоптоза в случае нерепарируемых поврежде-
ний ДНК или неразрешимых внутриклеточных конфликтов.
Функциональная инактивация р53 способствует трансформа-
ции нормальных клеток в опухолевые, с одной стороны, и
прогрессии уже возникшей опухоли — с другой.
В опухолевой прогрессии р53 участвует и совершенно осо-
бым образом. Оказалось, что низкое содержание кислорода в
слабо васкуляризованных участках ткани стимулируют синтез
р53 и как следствие этого апоптоз. Мутации р53 отменяют, од-
нако, апоптоз. В результате опухолевые клетки с инактивиро-
ванным р53 получают селективное преимущество над интакт-
ными (рис. 4.16). Таким образом, именно гипоксическая среда
является, возможно, тем плавильным котлом, из которого по-
являются р53 мутанты.
212
«Как в сцене из Дантова «Ада» рождаются эти уродливые
клетки среди распадающихся останков своих предков, неуяз-
вимые и благополучные в условиях, совершенно пагубных для
простых смертных... Глухие к нормальным клеточным регуля-
торам, они бесконтрольно размножаются и постоянно увели-
чивают степень своей ненормальности благодаря амплифика-
ции генов и хромосомным перестройкам. Это только вопрос
времени, когда они выберутся из своего обиталища, пронижут
окружающие нормальные ткани и нападут на своих невинных
соседей...» [Kinzler K.W., Vogelstein В., 1996].
Ген-супрессор АРС (Adenomatous Polyposis Coli) часто де-
фектен при синдромах семейного аденоматозного полипоза и
наследственного неполипозного рака толстой и прямой
кишки. В толстой кишке пациентов с наследственными мута-
циями АРС во второй-третьей декаде жизни возникают тысячи
аденоматозных полипов, причем некоторые из них могут дать
начало опухоли — второй по значимости причины онкологи-
ческой смертности в США. Исследование связанных с этой
патологией наследственных дефектов подтверждает «двуудар-
ную» гипотезу Кнудсона (см. выше). Возникновение множест-
венных опухолей в этом случае обусловлено соматическими
мутациями одного, доставшегося от здорового родителя, нор-
мального аллеля АРС (второй аллель дефектен). «Спорадичес-
кие» раки толстой кишки возникают реже и практически не
бывают множественными, поскольку для их возникновения
необходима инактивация обоих аллелей АРС. Обнаруживае-
мые в опухолевой ткани мутации, инсерции и делеции АРС
приводят к синтезу укороченного и функционально неактив-
ного белка.
Нормальный белок АРС имеет очень большие размеры
(2843 аминокислот), не содержит последовательностей, функ-
ция которых уже была бы известна, и состоит из нескольких
доменов: N-концевого, опосредующего гомоолигомеризацию,
и нескольких дополнительных, обеспечивающих взаимодейст-
вие с другими белками, в том числе с белком-супрессором
DLG и 0-катенинами. Это последнее обстоятельство позволя-
ет предполагать участие АРС по крайней мере в двух процес-
сах: во-первых, в клеточной адгезии, поскольку цитоплазмати-
ческие белки р-катенины совместно с кадгеринами (кальций-
зависимыми белками) участвуют в межклеточных взаимодей-
ствиях, и, во-вторых, в сигнальном пути WNT, приводящем к
семейству регуляторов транскрипции Tcf. Предполагается, что
АРС интегрирует получаемые из разных источников сигналы
и передает их в ядро клетки опосредованно через комплексы
р-катенин/Tcf.
АРС — первый из белков-супрессоров, которых наделяют,
пока сугубо предположительно, совершенно особой функцией
«сторожа» (gatekeeper), обеспечивающего тканевый гомеостаз
213
[Kinzler, Vogelstein, 1996]. Эти белки ответственны за постоян-
ство числа клеток в обновляемых тканях и за адекватность
клеточных реакций в ситуациях, требующих роста ткани (на-
пример, при ее развитии или травме). Повреждение «сторожа»
приводит к нарушению тканевого гомеостаза из-за дисбаланса
клеточной гибели и пролиферации. Ген с подобной функцией
находится в уникальном положении, его нарушение иниции-
рует пролиферацию и, если последуют мутации других генов,
рост опухоли. Напротив, повреждения иных генов при нор-
мальном «стороже» не могут иметь никаких последствий. От-
сюда следует, что определяющим для канцерогенеза может
оказаться не простое накопление мутаций в тех или иных
генах, а порядок появления этих мутаций (схема 4.4). Если уже
первая из них затрагивает АРС, то последующие канцероген-
ные события весьма вероятны, поскольку дефектные клетки
размножаются, увеличивая «базу» для последующих мутацион-
ных событий. Напротив, если первичные события затрагивают
другие гены, то функционально активный «сторож» срабаты-
вает, не позволяя этим клеткам делиться и, более того, инду-
цируя их апоптоз.
Гипотеза «сторожа» возникла как результат анализа мута-
ционного процесса при колоректальном раке человека и жи-
вотных, выявившего повреждения АРС на самых ранних ста-
диях канцерогенеза (уже на стадии микроскопических узел-
ков), тогда как мутации других генов (в частности, ras и р53)
возникают значительно позже (см. схему 4.4). Кроме того, му-
тации р53 обнаруживают в клетках тканей, видимым образом
не измененных, что дает основание предположить, что онко-
генный потенциал этих мутаций может проявиться только на
фоне дефектного АРС. Последний, таким образом, играет оп-
ределяющую роль в инициации колоректального рака, тогда
как дефекты других генных продуктов (онкогенов и супрессо-
ров) — в его прогрессии. Предполагается, что роль «сторожа»
в разных тканях исполняют разные гены: то, что для толстого
кишечника делает АРС, то для сетчатки глаза, возможно, дела-
ет Rb, а для почек — VHL.
Нарушения механизмов репарации ДНК. Инициация и про-
грессия опухоли суть фенотипические проявления поврежде-
ний генотипа (ДНК). Данное положение высвечивает очень
важную защитную функцию механизмов репарации ДНК.
Особенно наглядны в этом отношении некоторые наследст-
венные синдромы (пигментная ксеродерма, синдром Линча и
др.), при которых механизмы репарации неполноценны. У та-
ких больных развиваются множественные опухоли разных
тканей.
Повреждения ДНК могут быть разных типов (многочис-
ленные аддукты ДНК при химическом мутагенезе, тиминовые
димеры — при ультрафиолетовом облучении, одно- и двуните-
214
Схема 4.4. Генетические события (по крайней мере 7),
связанные с возникновением и развитием.рака толстой кишки
Мутации АРС инициируют опухолевый процесс, тогда как прогрессия
опухоли обусловлена последовательно возникающими мутациями других
генов. В криптах кишечника больных с наследственным дефектом АРС по-
являются многочисленные диспластические участки (фокусы дисплазии),
часть из которых прогрессируют при появлении дополнительных мутаций.
Дефекты системы репарации (MMR, mutations in mismatch repair) резко ус-
коряют этот процесс. K-ras (член семейства ras) — онкоген, для активации
которого требуется одно генетическое событие. Другие вовлеченные в
процесс гены относятся к числу супрессоров, для инактивации каждого из
которых требуется по два события (по одному на каждый аллель). В разви-
тии рака толстой кишки участвуют, возможно, и другие гены-супрессоры, в
частности DCC. Их многообразие может явиться причиной вариабельности
клинических проявлений этой формы рака.
вые разрывы — при воздействии ионизирующей радиации).
Соответственно этому существует и несколько типов репара-
ции, в частности эксцизионная репарация, которая выщепля-
ет последовательности ДНК, поврежденные химическими
канцерогенами или ультрафиолетовым облучением (она де-
фектна у больных пигментной ксеродермой, предрасположен-
ных к разным видам кожного рака), и репарация неспаренных
оснований (mismatch repair — MMR). Повреждения последней
215
могут приводить к широкомасштабной нестабильности генома
и различным формам рака, в частности и в особенности к раку
толстой кишки (в этом случае мутации MMR возникают, по-
видимому, вскоре после инактивации АРС; см. схему 4.4).
В геноме широко распространены многократные повторы
2—3 нуклеотидов, образующие так называемые микросател-
литные последовательности. В геноме человека, например,
имеется около 100 000 повторов CA/GT с длиной свыше 24
нуклеотидов. При репликации таких локусов иногда происхо-
дит скольжение (slippage) одной нити ДНК относительно дру-
гой, что приводит к образованию неспаренных (в виде петель)
участков. Удаление таких дефектов структуры — «корректор-
ская» функция MMR, которую выполняют несколько согласо-
ванно действующих белков. Один из них (MSH2) распознает
дефект, связывается с участком неспаренности и рекрутирует
другие белки, в частности MLH1, которые устраняют дефект и
восстанавливают исходную структуру ДНК (схема 4.5).
При дефектах MMR возникает так называемый мутатор-
ный фенотип (частота мутаций возрастает на 2—3 порядка) —
нестабильность микросателлитов охватывает весь геном и от-
ражается на функционировании многих генов, в составе или
вблизи которых расположены микросателлиты. Неполноцен-
ность MMR, резко увеличивающая вероятность новых мута-
ций (в ряду клеточных поколений они накапливаются лавино-
образно), способствует ускоренной прогрессии опухоли. Де-
фекты MLH1 и MSH2 обнаружены при различных семейных и
спорадических формах рака, в частности при раке толстой
кишки. Поскольку гены MMR относятся к числу генов-суп-
рессоров, мутаторный фенотип возникает при инактивации
обоих аллелей соответствующего гена.
Нарушения апоптоза. Апоптоз является эффекторным ме-
ханизмом, «призываемым» к действию самыми разными сти-
мулами. Выше рассмотрены ситуации р53-зависимого апопто-
за. Существуют, однако, и независимые от р53 пути индукции
этого феномена.
Обнаружена связь механизма апоптоза с молекулами кле-
точной адгезии. Давно известно, что пролиферирующие нор-
мальные клетки нуждаются в прикреплении к субстрату — к
внеклеточному матриксу in vivo или к твердой подложке in
vitro. При несоблюдении этого условия клетки останавливают-
ся в фазе G1 из-за подавления синтеза циклина А. Более того,
нормальные клетки, открепленные от субстрата или принуж-
даемые расти в суспензии, претерпевают апоптоз. Этот фено-
мен (получивший название «аноикис» от греческого anoikis —
бездомность) присущ эпителиальным и эндотелиальным клет-
кам, но не фибробластам.
Предполагается, что интегрины — молекулы на поверхнос-
ти клеточных мембран, участвующие в прикреплении клеток к
216
Схема 4.5. Репарация ошибок спаривания,
возникающих при репликации ДНК
А и Б — во время синтеза ДНК могут.происходить ошибки спаривания
из-за ошибочной подстановки нуклеотида ДНК-полимеразой или из-за
скольжения одной нити ДНК относительно другой; В — петля, образован-
ная неспаренными основаниями, распознается ферментами репарации —
MSH2 и GTBP; Г — в комплекс рекрутируются другие ферменты репарации,
выполняющие функции экзонуклеазы, ДНК-полимеразы, лигазы и ряда
других; Д — восстановление нативной структуры ДНК.
твердому субстрату и в их распластывании на нем, индуциру-
ют блокирующий апоптоз каскад реакций с участием FAK
(focal adhesion kinase) и протеинкиназы С, а также Ras и МАР-
киназ. В отсутствие клеточной адгезии программа клеточной
гибели, не встречая противодействия, реализуется, по-види-
мому, с участием bcl-2, Ьах и ICE-подобных протеаз.
Зависимость от субстрата ослаблена или вовсе отсутствует
у опухолевых клеток — многие из них могут расти в суспен-
зии, что объясняется неполноценностью механизма апоптоза.
217
Заключение
В силу многих причин онкологические исследования на-
ходятся на самом переднем крае биологии. Именно в этой
области сделаны фундаментальные открытия, значительно
продвинувшие нас в понимании не только того, как нор-
мальная клетка трансформируется в опухолевую, но и как
она вообще функционирует, как рождается и погибает. Про-
гресс в теоретическом плане, таким образом, очевиден.
Менее существенны, однако, достижения в лечении злокаче-
ственных новообразований. Не удалось пока выявить такие
биохимические особенности опухолевых клеток, которые по-
зволяли бы, с одной стороны, идентифицировать процесс на
самых ранних, еще доступных лечению стадиях его развития
и, с другой — найти ту «магическую пулю», которая избира-
тельно поражала бы злокачественные клетки, не затрагивая
нормальные.
Вместе с тем есть основания для осторожного оптимизма.
Во-первых, становятся все более эффективными профилак-
тические мероприятия, способные резко снизить онкологи-
ческую заболеваемость (профилактика курения, алкоголизма,
других вредных привычек, экологические мероприятия, улуч-
шение условий труда). Наряду с классической триадой (хи-
рургия, химиотерапия, облучение) для лечения онкологичес-
ких больных начинают применяться принципиально новые
методы — иммунотерапия и фотодинамическая терапия, а
также биотехнологические подходы, направленные на акти-
вацию в опухолевых клетках генов-супрессоров и проапопти-
ческих генов. Главной в последнем случае является проблема
избирательной доставки генных конструкций в опухолевые
клетки. Весьма обнадеживающие результаты получены в по-
иске препаратов, блокирующих формирование сосудистого
ложа в опухолевой ткани. Принимая во внимание быстрый
прогресс во всех этих направлениях, можно ожидать ощути-
мых практических результатов уже в ближайшем будущем.
Список литературы
Балицкий К.П., Шмалько Ю.П. Стресс и метастазирование злокачест-
венных опухолей. — Киев: Наукова думка, 1987.
Лихтенштейн А.В., Бассалык Л. С. Введение в проблемы биохимии и
физиологии опухолевого роста//Элементы патологической физиоло-
гии и биохимии/Под ред. И.П.Ашмарина. — М.: Изд-во МГУ, 1992.
Уманский С.Р. Апоптоз: молекулярные и клеточные механизмы//Мо-
лекулярная биология. — 1996. — № 30. — С. 487—502.
218
Шапот В. С. Биохимические аспекты опухолевого роста. — М.: Меди-
цина, 1975.
Албертс Б., БрейД., Льюис Дж. и др. Молекулярная биология клетки.
Т. 1-3. - М.: Мир, 1986.
Ленинджер А. Биохимия. — М.: Мир, 1974.
Льюин Б. Гены. — М.: Мир, 1987.
Хесин Р.Б. Непостоянство генома. — М.: Наука, 1985.
Beutler В., Cerami A. Tumor necrosis, cachexia, shock, and inflammation:
A common mediator//Ann. Rev. Biochem. — 1988. — Vol. 57. — P. 505—
518.
Bishop J.M. Cancer: The rise of the genetic paradigm//Genes and develop-
ment. - 1995. - Vol. 9. - P. 1309-1315.
Fidler I.J., Ellis L.M. The implications of angiogenesis for the biology and
therapy of cancer metastasis//Cell. — 1994. — Vol. 79. — P. 185—188.
Fisher D. Apoptosis in cancer therapy//Cell.— 1994.— Vol. 78,— P. 539—
542.
Fraser A., Evan G. A license to kill//Cell. — 1996. — Vol. 85. — P. 781 —
784.
Hollstein M., Sidransky D., Vogelstein B., Harris. p53 Mutations in human
cancers//Science. — 1991. — Vol. 253. — P. 49—53.
Johnson L., Noble M., Owen D. Active and inactive protein kinases: Struc-
tural basis for regulation//Cell. — 1996. — Vol. 85. — P. 149—158.
Johnson P.F., McKnight S.L. Eukaryotic transcriptional regulatory pro-
teins//Ann. Rev. Biochem. — 1989. — Vol. 58. — P. 799—839.
Marx J. How cells cycle toward cancer//Science. — 1994. — Vol. 263. —
P. 319-321.
Sklar L., Anisman H. Stress and cancer//Psych. Bull.— 1981.— Vol. 89.—
P. 369-406.
Yarden Y., Ullrich A. Growth factor receptor tyrosine kinases//Ann. Rev.
Biochem. - 1988. - Vol. 57. — P. 443-478.
Лекция 5
Феномен стресса.
Эмоциональный стресс
и его роль в патологии
М.Г.Пшенникова
«Стресс есть жизнь и жизнь есть стресс».
Г. Селье
Введение
В настоящее время трудно найти человека, который не
слышал бы о стрессе. Существует мнение, что стресс — это яв-
ление, присущее именно сегодняшней непростой жизни, и что
стресс — явление вредное и с ним надо бороться. В этом вы-
сказывании есть доля истины, но только доля, которая отра-
жает лишь лежащие на поверхности и наиболее заметные не-
гативные последствия стресса.
Термин «стресс» ввел в биологию канадский физиолог
Ганс Селье, когда он в 1936 г. в статье, опубликованной
в журнале «Nature» под заглавием «Синдром, вызываемый
разными повреждающими агентами» [Selye Н., 1936], впер-
вые обозначил это явление. Позже Г.Селье охарактеризовал
это явление как «общий адаптационный синдром», т.е.
«общую неспецифическую нейрогормональную реакцию ор-
ганизма на любое предъявленное ему требование» [Селье Г.,
1960].
Термин «стресс» Г.Селье заимствовал из техники, где он
означает «напряжение, нажим, давление». В общем виде
стресс применительно к человеку можно определить как со-
стояние организма, всегда возникающее при действии на него
различных факторов, или стрессоров, будь это так называемые
физические стрессоры (холод, физическая нагрузка, недоста-
ток кислорода в воздухе и т.п.) или психические (эмоциональ-
ные) стрессоры, которые обычно называют стрессорными си-
туациями (опасность, аварийные и чрезвычайные ситуации,
несчастье, неожиданное радостное известие, конфликтные си-
туации в семье или на работе, цейтнот и др.). В настоящее
время стрессорные ситуации «атакуют» людей все чаще, поэ-
тому столь велики всеобщее внимание к стрессу (эмоциональ-
ному стрессу в особенности) и желание исследователей разо-
браться в его природе и механизмах.
220
В ответ на стрессорное воздействие человек или животное
либо избегает этого воздействия, избавляется от него, либо,
если избавление невозможно, приспосабливается, т.е. адапти-
руется, к новым требованиям среды. При этом при первичном
действии стрессора возникает стресс-реакция, или так назы-
ваемая срочная (аварийная) адаптация’, она дает организму воз-
можность осуществлять жизнедеятельность в новых услови-
ях — в условиях действия стрессора, и в этом ее положитель-
ное значение. Однако эта стресс-реакция весьма расточитель-
на и несовершенна и не обеспечивает организму эффективно-
го устойчивого приспособления к стрессорному воздействию.
Устойчивая долговременная адаптация, характеризующаяся
высокой резистентностью к стрессору, может сформироваться
лишь в результате повторных воздействий этого стрессора. Од-
нако это происходит, если интенсивность и длительность дей-
ствия стрессора умеренны. Если же они чрезмерны, то адапта-
ция не развивается, а возникают повреждения, которые при-
водят к гибели.
Феномены стресса и адаптации, интуитивно предсказан-
ные и в той или иной форме описанные многими поколения-
ми естествоиспытателей и врачей, включая Гиппократа, Де-
карта, Ф.Энгельса, К.Бернара, В.Кэннона, И.М.Сеченова и
А.А.Ухтомского, в настоящее время благодаря успехам биоло-
гических и медицинских наук получают научную базу. В ре-
зультате накоплено большое количество исследований и пуб-
ликаций, посвященных различным аспектам проблемы стрес-
са и стрессорной патологии. Вместе с тем, однако, до сих пор
в литературе еще нет сообщений, содержащих целостное пред-
ставление о стрессе как физиологическом явлении, о механиз-
мах его возникновения и формирования, механизмах, лежа-
щих в основе устойчивости к стрессорным воздействиям, и о
месте стресса в патологии. По-видимому, из-за этого зачастую
путают понятия «стресс» и «стрессорное воздействие»,
«стресс» и «стрессорные повреждения». В научных публикаци-
ях встречаются такие спорные и, по-видимому, ошибочные
выражения, как «защита от стресса», «устойчивость к стрессу»
и др. На самом деле нельзя, да и не нужно защищаться от
«стресса», так как стресс — естественная адаптивная реакция
организма; защищаться следует от стрессорных воздействий
или, если это невозможно, от стрессорных повреждений. То
же относится к понятиям «устойчивость» и «предрасположен-
ность»: организм может быть в разной степени предрасполо-
женным или устойчивым к срессорным нарушениям тех или
иных функций и к стрессорным повреждениям тех или иных
органов, но вовсе не «к стрессу». В связи с изложенным целью
данного сообщения является попытка обобщения и критичес-
кого анализа имеющихся к настоящему времени данных, каса-
ющихся проблемы стресса. При этом последовательно будут
221
рассмотрены основные аспекты этой проблемы: феномен
стресса и связанная с ним терминология, стресс-система, ме-
ханизмы формирования стресс-реакции, особенности эмоцио-
нального стресса и механизмы связанных с ним патологичес-
ких состояний, механизмы устойчивости к стрессорным по-
вреждениям, а также возможности предупреждения и коррек-
ции этих повреждений.
5.1. Феномен стресса
Стресс как биологическая категория — сложное явление,
присущее всему живому от простейших одноклеточных до Ното
sapiens. Стресс неотделим от самого явления жизни. Это объяс-
няется тем, что жизнь — сложное динамическое равновесие ор-
ганизма с окружающей средой, она невозможна без взаимодей-
ствия с этой средой. Солнечный свет, вода, воздух, пища и дру-
гие компоненты среды необходимы для существования живого
организма. При этом непременным условием нормальной жиз-
недеятельности организма и сохранения самой жизни является
поддержание постоянства внутренней среды, или гомеостаз.
Термин «гомеостаз» был введен В. Кэнноном, а сама идея была
впервые высказана в XIX в. Клодом Бернаром, который утверж-
дал, что «постоянство внутренней среды — непременное усло-
вие свободной и независимой жизни».
Гомеостаз проявляется поддержанием физиологических,
биохимических, биоэлектрических и других параметров орга-
низма (артериального давления, температуры тела, количества
форменных элементов крови, парциального давления кисло-
рода в крови и т.п.) на постоянном уровне с минимальными
допустимыми отклонениями. Таким образом, стресс — это со-
стояние нарушенного гомеостаза, а стрессоры — это факторы,
вызывающие нарушение гомеостаза. При этом стрессоры могут
быть как внешние, или экзогенные (факторы окружающей
среды, в том числе информация), так и внутренние, или эндо-
генные, формирующиеся в организме. Они могут возникать
как следствие влияния внешних стрессоров, но самостоятель-
но вызывают нарушение гомеостаза (например, боль, очаг
воспаления и др.). На стрессоры организм отвечает стресс-ре-
акцией, т.е. адаптивным процессом, направленным на восста-
новление гомеостаза и сохранение нормальной жизнедеятель-
ности. Таким образом, сам стресс и стресс-реакция по своей
сути — процессы необходимые и «полезные», а представление
о «вредности» стресса проистекает из тех патологических яв-
лений, которые в действительности возникают при чрезмерно
сильных и/или длительных воздействиях стрессора, приводя-
щих к повреждению, а также при нарушениях работы систем,
осуществляющих стресс-реакцию и формирование адаптации.
222
5.1.1. Стресс-реакция
Содержание стресс-реакции организма зависит от слож-
ности организации живого существа. Так, одноклеточные ор-
ганизмы реализуют свою стресс-реакцию, т.е. адаптируются к
среде обитания, с помощью соответствующих биохимических
изменений в цитоплазме, позволяющих избежать опасности и
сохранить жизнь, например хемо- и фототаксис, цистообразо-
вание (у амебы). Многоклеточные организмы приспосаблива-
ются с помощью координированных нейрогуморальных и
внутриклеточных изменений во многих органах и тканях.
У млекопитающих, включая человека, адаптивная реакция
на стрессор очень сложна. Г.Селье установил и описал явле-
ние стресса как раз применительно к млекопитающим и чело-
веку. При этом Г.Селье на основании экспериментов на кры-
сах, которые подвергались действию различных повреждаю-
щих агентов, описал стресс как «процесс, закономерно проте-
кающий в трех стадиях, последовательно переходящих друг в
друга, проявление которых не зависит от природы повреждаю-
щего агента» [Selye Н., 1936]. Напомним ставшее классичес-
ким описание Г.Селье этих стадий у животных, которое прин-
ципиально может распространяться и на человека.
Первая стадия, или реакция тревоги (в течение 48 ч после
начала воздействия), т.е. в нынешнем понимании — острый
стресс, характеризуется быстрым уменьшением размеров ви-
лочковой железы, селезенки, лимфатических узлов и печени,
исчезновением жировой ткани, отечностью, выходом плев-
рального и перитонеального транссудата, снижением мышеч-
ного тонуса, образованием язв в пищеварительном тракте,
уменьшением липидов и хромаффинных веществ в надпочеч-
никах. Одновременно отмечаются гиперемия кожи, экзоф-
тальм, слезотечение и саливация. При большой силе воздейст-
вия уже эта реакция тревоги может закончиться гибелью орга-
низма, но если организм переносит эту стадию синдрома, то
наступает вторая стадия.
Вторая стадия, или стадия резистентности, начинается
спустя 48 ч после повреждающего действия и характеризуется
увеличением надпочечников, восстановлением их липидных
гранул, вакуолизацией хромаффинных клеток медуллы (моз-
гового слоя) надпочечников, началом исчезновения отечнос-
ти, появлением многочисленных базофилов в гипофизе, тен-
денцией к гиперплазии щитовидной железы, атрофией гонад и
прекращением роста животного. Если повреждающее действие
было не столь сильным, то у животных возрастает резистент-
ность, и в более поздний период второй стадии вид и функция
органов практически возвращаются к норме. Но если действие
повреждающего агента продолжается дольше, то после этой
стадии животные теряют свою резистентность, что приводит к
223
стадии истощения, рассматриваемой автором как третья ста-
дия, которая может привести к гибели.
Определяя стресс-реакцию как «общий адаптационный
синдром», Г.Селье подчеркивал, что это явление характеризу-
ется как неспецифичностью, так и специфичностью. Он
писал, что «... стресс — состояние, проявляющееся специфи-
ческим синдромом, который включает в себя все неспецифи-
чески вызванные изменения в биологической системе».
Иными словами, стресс по своему характеру специфичен, так
как всегда проявляется описанными выше специфическими
симптомами (реакцией вилочковой железы, надпочечников и
т.д.), но одновременно стресс и неспецифичен по своему про-
исхождению, так как возникает при действии на организм
самых разных стрессоров — «механического, физического, хи-
мического, биологического и психического характера». При
этом Г.Селье указывал, и это нам очень важно для дальнейше-
го изложения, что стресс-реакция представляет собой обыч-
ные и закономерные ответы организма не только на прямое по-
вреждающее действие, но и на любые другие «стимулы». К ним
Г.Селье относил мышечные упражнения, температурные из-
менения, яды и другие воздействия, по отношению к которым
«имеет место привыкание или закаливание» [Selye Н., 1936],
т.е., иными словами, к любым факторам окружающей среды.
Развитие исследований феномена стресса показало, что
при действии факторов среды стресс-реакция у высших жи-
вотных характеризуется комплексом поведенческих и физио-
логических изменений в организме. Изменения в поведении
включают возбуждение, настороженность, обострение внима-
ния и познавательных способностей, подавление пищевого и
сексуального поведения, что в целом отражает «ориентировоч-
ную реакцию» и «боевую готовность». Физиологические изме-
нения выражаются в мобилизации функции органов и систем,
ответственных за адаптацию, и переключении энергетических
ресурсов (кислорода и питательных веществ) в эти органы, а
также в изменении активности системы иммунитета. Так, в
ответ на физические стрессоры, механизм действия которых
исследован лучше, чем эмоциональных, адаптивная реакция
состоит в том, что стрессор (схема 5.1) через высшие регуля-
торные центры активирует регуляторную стресс-систему (ха-
рактеристику стресс-системы см. далее), которая объединяет
определенные отделы нервной и эндокринной систем и «не-
специфически» активируется в ответ на любой стрессор, а
также функциональную систему, объединяющую органы и
ткани, «специфически» ответственные за приспособление к
конкретному стрессору (холоду, физической нагрузке, недо-
статку кислорода, т.е. гипоксии, и др.). Например, при физи-
ческой нагрузке функциональная система объединяет соответ-
ствующие моторные центры в головном и спинном мозге и
224
Схема 5.1. Адаптивная стресс-реакция в ответ
на действие физических стрессоров
СТРЕССОР
Пояснения в тексте.
управляемые ими скелетные мышцы, выполняющие необхо-
димую мышечную работу. Активируются также органы дыха-
ния и кровообращения, обеспечивающие увеличенный приток
кислорода и питательных веществ к работающим мышцам и
центрам управления. При этом стресс-система обеспечивает
более полную мобилизацию функциональной системы, специ-
фически ответственной за адаптацию к физической нагрузке,
и координирует адаптационные процессы. Иначе говоря, она
осуществляет «настройку» органов и тканей, вовлеченных в
адаптацию, на функционирование в новых условиях. При
этом влияние стресс-системы в случае достаточно сильного
стрессорного воздействия может оказаться избыточным и
приводить к побочным неблагоприятным эффектам, в част-
ности к стрессорным повреждениям.
5.1.2. Стресс-система
Стресс-система — сложный регуляторный комплекс, ко-
торый помогает координировать гомеостаз в обычных услови-
ях и играет ключевую роль в активации и координации всех
изменений в организме, составляющих адаптивную реакцию
на стрессоры. В соответствии с данными современных иссле-
дований эта система состоит из центрального звена и двух пе-
риферических ветвей, которые осуществляют связь централь-
ного звена со всем организмом (рис. 5.1). Центральное звено
находится в головном мозге: в гипоталамусе и других отделах
ствола мозга. Гипоталамус — «дозорный» центральной нерв-
ной системы, ответственный за нервную регуляцию эндокрин-
15—1385
225
Рис. 5.1. Схематическое изображение стресс-системы, центрального
и периферических ее звеньев и взаимодействия компонентов этих
звеньев между собой, а также со стресс-лимитирующими системами
(см. в тексте раздел 5.1.3).
ГАМК — группы нейронов в гипоталамусе, вырабатывающих гамма-амино-
масляную кислоту (ГАМК); НА — группы нейронов в гипоталамусе и других
отделах ствола мозга («синее пятно»), вырабатывающих норадреналин (НА);
ОП — гоуппы нейронов в гипоталамусе, вырабатывающих опиоидные пептиды
(ОП); КРГ — группы нейронов в гипоталамусе, вырабатывающих кортико-
тропин-рилизинг-гормон (КРГ), т.е. гормон, стимулирующий секрецию АКТГ
226
ных функций, который получает информацию о появлении
стрессора и «запускает» работу стресс-системы.
Центральное звено стресс-системы объединяет три основ-
ные группы нейронов: 1) нейроны паравентрикулярного ядра
гипоталамуса (КРГ-нейроны), которые вырабатывают корти-
котропин-рилизинг-гормон (КРГ), т.е. гормон, стимулирую-
щий секрецию адреналокортикотропного гормона (АКТГ) в
гипофизе (центральной эндокринной железе) и тем самым ак-
тивирующий гипоталамо-гипофизарно-адреналовую систему,
или «ось»; 2) нейроны паравентрикулярного ядра гипоталаму-
са (АВ-нейроны), вырабатывающие гормон аргинин-вазопрес-
син — АВ (чаще всего его называют просто «вазопрессин»);
КРГ-нейроны в ядрах медуллы; 3) группы нейронов (НА-ней-
роны), синтезирующих катехоламины, главным образом нор-
адреналин (НА), в стволе мозга — гипоталамусе и других отде-
лах; ключевую роль играет центр НА-нейронов — «синее
пятно».
Периферические ветви стресс-системы представлены двумя
основными отделами: 1) гипоталамо-гипофизарно-адренало-
вой осью, которая активируется КРГ и конечным продуктом
активации которой являются гормоны глюкокортикоиды,
выделяющиеся из коры надпочечников под влиянием АКТГ;
2) симпатоадреналовой системой, в которую входят симпати-
ческая нервная система (иннервирующая все органы и ткани)
и мозговой слой надпочечников; конечными продуктами ак-
тивации этой системы являются катехоламины норадреналин
и адреналин [Chrousos G.P., Gold P.W., 1992; Harbuz M.S.,
Lightman S.L., 1992; Stratakis C.A., Chrousos G.P., 1995]. Выход
на «периферию» стресс-система имеет также через парасим-
патическую систему путем влияния на центр ее основного
представителя — блуждающего нерва (n. vagus). Однако роль
парасимпатической системы в стресс-реакции изучена недо-
статочно, хотя показано, что возникающая при стимуляции
гипоталамуса гиперинсулинемия реализуется путем влияния
блуждающего нерва на поджелудочную железу.
Существенно, что гормоны и медиаторы, продуцируемые
нейронами центрального звена стресс-системы, обладают
многими функциями, которые определяют весь комплекс про-
цессов, реализующихся при стресс-реакции. Так, например,
(адреналокортикотропного гормона) в гипофизе; АВ — группы нейронов в ги-
поталамусе, вырабатывающих аргинин-вазопрессин (АВ)'НП«У» — нейропеп-
тид «У», вырабатываемый симпатическими нейронами; SP — субстанция «Р»,
вырабатываемая в гипоталамусе и амигдале. ГнРГ — гонадотропин-рилизинг-
гормон, стимулирующий секрецию половых гормонов; РГГР — рилизинг-гор-
мон гормона роста; ТРГ — гормон, стимулирующий секрецию тиреоидных
гормонов; N.V. — центр блуждающих нервов [Chrousos G.P., Gold PAV., 1992;
Stratakis С.А., Chrousos G.P., 1995]. Сплошные стрелки означают активирую-
щее влияние, штриховые стрелки означают тормозное, угнетающее влияние.
15*
227
выяснилось, что КРГ, открытый в 1955 г. M.Saffran и A.V.Scha-
1у как «фактор высвобождения АКТГ из гипофиза», является,
кроме того, самостоятельным нейрогормоном. С одной сторо-
ны, КРГ, выделяясь из КРГ-нейронов гипоталамуса и попадая
в портальную систему гипофиза, «запускает» активацию гипо-
физарно-адреналовой оси (в связи с чем Г.Селье назвал его
«рилизинг-фактором стресса»). С другой стороны, он играет
ключевую роль в реализации поведенческих и гормональных
реакций на эмоциональные стрессоры, участвует в изменении
функций висцеральных органов и иммунной системы при
стрессе. Вазопрессин также обладает многими функциями.
Наряду с КРГ этот гормон является стимулятором секреции
АКТГ [Whitnail М.Н., 1993; Stratakis С.А., Chrousos G.P., 1995].
Но, кроме того, вазопрессин повышает активность симпати-
ческой системы, что усиливает ее действие при стрессе, и на
центральном уровне участвует в реализации «защитного» аг-
рессивного поведения и подавления иммунных реакций орга-
низма при стрессе [Shibasaki Т. et al., 1998] и т.д.
Основные звенья стресс-системы тесно взаимодействуют с
тремя другими отделами ЦНС:
• мезокортикальной и мезолимбической дофаминовыми
системами, которые включают префронтальную кору го-
ловного мозга и nucleus accumbens;
• комплексом амигдала — гиппокамп;
• опиоидергическими нейронами аркуатного ядра гипота-
ламуса, богато иннервируемого НА-содержащими волок-
нами, выходящими из НА-нейронов синего пятна и дру-
гих НА-ергических структур ствола мозга.
Эти отделы имеют особое значение при эмоциональном
стрессе и связанных с ним патологических состояниях, так
как они вовлечены в феномены «оборонительной реакции»,
страха, в изменение болевой чувствительности и влияют на
эмоциональный тонус.
Сигналы о внешних стрессорах поступают в стресс-систе-
му от коры мозга через ассоциативную кору, амигдалу, гиппо-
камп, а также от мезокортико-лимбической системы. Сигналы
о стрессорах от органов чувств, кровеносных сосудов, мышц,
внутренних органов поступают в стресс-систему через крани-
альные и периферические нервы, чувствительные афферент-
ные волокна и кровь. Выход сигналов из центрального звена
стресс-системы осуществляется не только к органам через пе-
риферические ветви системы, как указывалось выше, но также
к другим отделам ЦНС: к коре головного мозга, аркуатному
ядру гипоталамуса, спинному мозгу, амигдале, гиппокампу,
мезокортико-лимбической системе. Таким образом, отделы
ЦНС, из которых сигналы идут к стресс-системе, в свою оче-
редь получают также и сигналы от нее (схема 5.2).
228
Схема 5.2. Формирование стресс-реакции
в ответ на действие внешних и внутренних стрессоров
Пояснение в тексте.
В целом стресс-система получает информацию от окру-
жающей среды и организма через разнообразные сенсорные
системы и кровоток, от «думающего» мозга — через амигдалу
и гиппокамп и от «эмоционального» мозга — через мезокорти-
ко-лимбическую систему.
Основным результатом активации стресс-системы являет-
ся увеличенный «выброс» глюкокортикоидов и катехолами-
нов — главных стресс-гормонов, которые способствуют моби-
лизации функции органов и тканей, ответственных за адапта-
цию, и обеспечивают увеличение их энергообеспечения (см.
рис. 5.1).
Как выяснилось в последние 10 лет, помимо прямых про-
дуктов стресс-системы, в стресс-реакцию вовлечены также ве-
щества, потенцирующие или опосредующие эффекты стресс-
системы, механизм действия которых пока мало изучен: ан-
гиотензин II [Aguilera G. et al., 1995], цитокины, например
интерлейкины [Peristein R.S. et al., 1993], а также тахикины —
нейропептид Y, субстанция Р [Culman J. et al., 1995] и др.
В частности, цитокины опосредуют воздействие стресс-систе-
мы на систему иммунитета, индуцируя активацию, пролифе-
рацию и дифференцировку Т- и В-лимфоцитов и т.д. Нейро-
пептид Y, синтезирующийся в аркуатном ядре и выделяющий-
ся из терминалей аксонов в паравентрикулярных ядрах гипо-
таламуса, содержится также наряду с* катехоламинами в сим-
патических нервных волокнах и надпочечниках, усиливает со-
судосуживающее действие катехоламинов. Вместе с тем ней-
ропептид Y, выделяющийся из аксонов НА-нейронов в гипо-
таламусе, оказывает угнетающее действие на НА-нейроны, но
потенцирующее — на КРГ-нейроны.
229
По концепции Г.Селье, стресс-система активируется в
ответ на любой стрессор неспецифически, т.е. продуцирует
одинаковый «набор» гормонов и медиаторов независимо от
вида стрессора. Однако данные последних 5 лет свидетельст-
вуют, что тип стрессора может определять количественные
различия в реакции центральной НА-системы и центров гипо-
таламо-гипофизарно-адреналовой оси. В частности, показано
[Расак К. et al., 1995], что при действии эмоциональных стрес-
соров наблюдается существенно большая положительная кор-
реляция между подъемом уровня НА в паравентрикулярном
ядре гипоталамуса и опосредованной НА активацией гипота-
ламо-гипофизарно-адреналовой оси, чем при действии физи-
ческих стрессоров.
5.1.3. Стресс-лимитирующие системы
Активность и реактивность стресс-системы регулируются
двумя основными механизмами: механизмом саморегуляции и
механизмом внешней регуляции (см. рис. 5.1).
Механизм саморегуляции реализуется за счет влияния друг
на друга компонентов самой системы. Так, между КРГ-нейро-
нами и НА-нейронами в центральном звене стресс-системы
существуют нервные связи, приводящие к взаимоактивации
этих нейронов: КРГ, выделяемый КРГ-нейронами, активирует
НА-нейроны «синего пятна» и другие НА-нейроны и соответ-
ственно секрецию ими НА, и наоборот: НА-нейроны активи-
руют КРГ-нейроны. По принципу отрицательной обратной
связи гормоны, вырабатываемые в системе, ограничивают
свою собственную продукцию. Кроме того, глюкокортикоиды
ограничивают активность норадреналинового звена стресс-
системы, угнетая синтез, высвобождение и обратный захват
НА в симпатических нейронах.
Механизм внешней регуляции осуществляется модулятор-
ными регуляторными системами, не входящими в стресс-
систему, но тесно с ней связанными. Это так называемые
стресс-лимитирующие системы, которые способны ограни-
чивать активность стресс-системы и чрезмерную стресс-реак-
цию на центральном и периферическом уровнях регуляции
[Меерсон Ф.З., 1993; Меерсон Ф.З., Пшенникова М.Г., 1988,
1989; Юматов, 1997; Малышев И.Ю., Манухина Е.Б., 1998].
К основным центральным стресс-лимитирующим системам
относятся ГАМКергическая система (система нейронов, проду-
цирующих ГАМК, т.е. гамма-аминомасляную кислоту, обла-
дающую тормозным действием на нейроны головного и спинно-
го мозга) и опиоидергическая система, объединяющая нейроны
в гипоталамусе и секреторные клетки в гипофизе, продуцирую-
щие опиоидные пептиды (ОП), также обладающие тормозным
230
Схема 5.3. Взаимодействие стресс-системы
и ГАМК-ергической системы
СТРЕССОР
Активация ГАМК-системы при стрессе приводит к ограничению
стресс-реакции и стрессорных повреждений [Меерсон Ф.З., Пшенникова
М.Г., 1989]. Пояснение в тексте.
действием. КРГ-нейроны, АВ-нейроны и НА-нейроны стресс-
системы взаимосвязаны с ГАМК-нейронами и ОП-нейронами.
Выделяющиеся при активации стресс-системы НА, КРГ и АВ
стимулируют ГАМК-нейроны и ОП-нейроны, они секретируют
ГАМК и ОП, которые в свою очередь ограничивают активность
стресс-системы в целом [Андреев Б.В. и др., 1982; Werling L.L. et
al., 1987; Calogero А.Е., 1995]. Схема 5.3 демонстрирует это поло-
жение на примере ГАМК-системы. Особо следует подчеркнуть
ингибиторное влияние ГАМК и агонистов бензодиазепиновых
рецепторов на функцию КРГ-нейронов, которые, как постули-
руется, координируют эндокринные, метаболические и пове-
денческие реакции организма на стрессоры. Это влияние реали-
зуется главным образом путем угнетения высвобождения КРГ из
терминалей КРГ-нейронов [Calogero А.Е., 1995].
231
Тормозное действие ГАМК и ОП на катехоламиновое
звено стресс-системы осуществляется не только в ЦНС, но и
на периферии: рецепторы для ГАМК и ОП локализованы на
аксонах симпатических нейронов, иннервирующих органы и
ткани, и в самих органах, что обеспечивает ограничение вы-
свобождения катехоламинов и их влияния [Erdo S.L., 1985].
Доказана совместная локализация стресс-гормонов и ОП: ка-
техоламинов и энкефалинов — в хромаффинных пузырьках
медуллы надпочечников [Boarder M.R., McArdle W., 1985], ва-
зопрессина и динорфина — в нейронах гипоталамуса [Whit-
nall М.Н. et al., 1983], глюкокортикоидов и энкефалинов — в
коре надпочечников. В результате «выброс» стресс-гормонов
может сопровождаться выходом ОП, причем интенсивность
этого выхода прямо пропорциональна степени активации
стресс-системы. Доказано также, что в гипофизе АКТГ и ОП
(Р-эндорфины) образуются из общего предшественника (про-
опиомеланокортина) и выделяются при стрессе в эквимоляр-
ных количествах. В регуляции стресс-системы участвуют
также другие нейропептиды. Важная роль отводится субстан-
ции Р. Субстанция Р, образующаяся в гипоталамусе и амигда-
ле, оказывает тормозное действие на КРГ-нейроны и секре-
цию КРГ. Продуцируемая в надпочечниках субстанция Р мо-
дулирует высвобождение из них катехоламинов, уменьшая вы-
брос этих гормонов при стрессе. Показано также, что субстан-
ция Р на центральном уровне угнетает стрессорную гипертен-
зивную реакцию и повышает устойчивость к эмоциональному
стрессу [Юматов А.Е., 1986, 1997]. Определенную роль в анти-
стрессорном противогипертензивном действии субстанции Р
играет способность этого нейропептида угнетать фермент,
превращающий ангиотензин I в ангиотензин II. Названный
«стрессорной молекулой» нейропептид Y обладает угнетаю-
щим действием на НА-нейроны синего пятна (см. рис. 5.1),
но стимулирует активность КРГ-нейронов в гипоталамусе и
потенцирует вазоконстрикторное действие катехоламинов при
стрессе [Zukowska-Grojec Z., 1995].
Действие стресс-системы на уровне органов и тканей огра-
ничивают системы локальной регуляции — локальные стресс-
лимитирующие системы, т.е. системы простагландинов, адено-
зина, ОП и других соединений в самих органах и перифери-
ческих нейроэндокринных структурах [Меерсон Ф.З., 1993;
Пшенникова М.Г., 1987, 1991]. Они угнетают высвобождение
катехоламинов из нервных окончаний и надпочечников и дей-
ствие этих моноаминов на постсинаптическом уровне, умень-
шая тем самым активацию свободнорадикального окисления
(СРО) и ограничивая чрезмерную стресс-реакцию и ее по-
вреждающее действие на органы и ткани. Важную роль в огра-
ничении активации СРО и повреждений при стрессе играют
также относящиеся к локальным стресс-лимитирующим сис-
232
темам антиоксидантные системы в органах и тканях, вклю-
чающие антиоксидантные ферменты (каталазу, супероксид-
дисмутазу и глутатионпероксидазу), а также антиоксиданты —
альфа-токоферол (витамин Е), витамин А, аскорбиновую кис-
лоту и др. [Меерсон Ф.З., 1993]. Таким образом, базальная ак-
тивность стресс-системы и ее активация при стрессе зависят
от активности стресс-лимитирующих систем.
В последнее время появились экспериментальные и теоре-
тические данные, позволяющие причислить к стресс-лимити-
рующим системам также систему генерации окиси азота. Эти
данные свидетельствуют, что оксид азота (NO) является уни-
версальным фактором регуляции физиологических систем (и в
том числе генетического аппарата клеток) и играет важную
роль в механизме стресс-реакции и адаптации организма к
стрессорным воздействиям. Роль NO в стресс-реакции опре-
деляется его свойствами [Kanner J. et al., 1991; Bredt D.S., Sny-
der S.H., 1992; Addicks К. et al., 1994 и др.], а также тем, что
при стрессе происходит резкое изменение его продукции в
разных органах и тканях {Малышев И.Ю., Манухина Е.Б.,
1998]. Показано, что центральное и периферические звенья
стресс-системы снабжены NOepranecKoft иннервацией: ней-
роны стриатума, среднего мозга (superior, interior colliculus,
pontine, tegmentum), гипоталамуса содержат NO-синтазу.
Гипофиз получает от гипоталамуса широко разветвленную
NO-иннервацию [Vanhatolo, Soinila, 1995]. Установлено также,
что NOeprHuecKHe нейроны иннервируют надпочечники, и их
аксоны контактируют с хромаффинными клетками, продуци-
рующими катехоламины [Bredt D.S., Snyder S.H., 1992]. Сим-
патические нейроны наряду с НА содержат в своих термина-
лях и NO, а выделение НА и NO происходит одновременно.
Показано, что в центральном звене стресс-системы NO может
модулировать высвобождение КРГ, вазопрессина, гормона
роста. В частности, высвобождение КРГ из КРГ-нейронов ги-
поталамуса, вызываемое ацетилхолином и холиномиметика-
ми, а также интерлейкинами, опосредовано NO. Под влияни-
ем указанных веществ NO выделяется из вставочных нейро-
нов, поступает в КРГ-нейроны и приводит к секреции из них
КРГ. Установлено, что NO может ограничивать активацию пе-
риферических звеньев стресс-системы. NO ограничивает ак-
тивацию симпатоадреналовой системы за счет угнетения вы-
броса катехоламинов из надпочечников и нервных окончаний
[Addicks A. et al., 1994; Schwarz Р. et al., 1995], угнетение син-
теза NO приводит, напротив, к активации симпатоадренало-
вой системы [Zanchi A. et al., 1995]. Таким образом, NO может
влиять на интенсивность стресс-реакции путем ограничения
активности стресс-системы.
Оказалось также, что NO способен ограничивать повреж-
дающее действие стресс-реакции путем прямого уменьшения
233
Схема 5.4. Защитные эффекты локальных стресс-
лимитирующих систем: системы простагландинов (ПГ)
и окиси азота (NO) при стрессорном поражении сердца.
Пояснение в тексте.
стрессорной активации СРО за счет увеличения активности
антиоксидантных ферментов [Dobashi К. et al., 1997] и экс-
прессии генов, кодирующих эти ферменты. Кроме того, NO
сам обладает антиоксидантными свойствами [Kanner J. et al.,
1991]. Выяснилось также, что NO активирует синтез цитопро-
текторных белков теплового шока семейства HSP70, или
«стресс-белков» [Malyshev LYu et al., 1995, 1996], которые, как
известно, являются важной системой защиты клеток от стрес-
сорных повреждений. Показано, что наряду с простагландина-
ми группы Е и простациклином NO играет ключевую роль в
предупреждении агрегации и адгезии тромбоцитов [Vallance Р.
et al., 1992], что может определять его защитное действие при
стрессорной активации тромбообразования. На схеме 5.4. в
качестве примера представлены противострессорные защит-
ные эффекты NO и ПГ на уровне сердца.
В пользу развиваемого представления о стресс-лимити-
рующем и защитном действии NO говорят также данные о
влиянии снижения продукции NO. Показано, что уменьшение
продукции NO в желудке является важной причиной ише-
мических по своей природе язвенных поражений желудка,
характерных для стресса [Ogino К. et al., 1994]. Показано,
что хроническое ингибирование NO-синтазы и соответст-
234
венно уменьшение продукции NO приводит у крыс к актива-
ции адренергической и ренин-ангиотензиновой систем, что
выражается в подъеме в плазме крови уровня НА, А и ренина
и развитию гипертензии [Zanchi A. et al., 1995].
Важно отметить, что повторные стрессорные воздействия
вызывают не только активацию стресс-лимитирующих систем,
но и приводят к повышению мощности (эффективности) этих
систем. В частности, установлено, что после стрессорных воз-
действий активируется синтез ГАМКА-рецепторов в коре го-
ловного мозга. При этом выяснилось, что активация синтеза
обусловлена воздействием гормона прогестерона, который
увеличивает экспрессию гена ГАМКА-рецепторов и секреция
которого возрастает при стрессе.
Таким образом, стресс-реакция реализуется с помощью из-
менения (главным образом увеличения) продукции медиато-
ров и гормонов компонентами стресс-системы и сопряжен-
ными с ней компонентами стресс-лимитирующих систем ор-
ганизма.
5.1.4. Роль соотношения активностей
стресс-системы и стресс-лимитирующих
систем в реакции организма на стрессоры
Нарушение регуляции стресс-системы, в значительной
мере связанное с недостаточностью функции стресс-лимити-
рующих систем, приводит не только к нарушению реакции
организма на стрессоры, но и к возникновению психических,
эндокринных заболеваний, к болезням системы кровообра-
щения, иммунной системы, обмена веществ или к предрас-
положенности к этим заболеваниям. В то же время введение
медиаторов соответствующих стресс-лимитирующих систем
может корректировать эти нарушения. В качестве примера
можно привести данные о том, что у линейных крыс с на-
следственной гипертонией (линия SHR) наблюдается генети-
чески повышенная активность симпатического звена стресс-
системы [Westfall Т.С., Meldrum M.J., 1985], сочетающаяся с
врожденным уменьшением активности ГАМК-нейронов в
гипоталамусе, иннервирующих центры симпатической регу-
ляции сердечно-сосудистой системы [Hambley J.W. et al.,
1984], а также с врожденным же сниженным содержанием
ОП в тканях [Di Guilio A. et al., 1979]. Показано, что у мута-
генных (lethargic Ihl) мышей, характеризующихся высокой
устойчивостью к судорогам, содержание ГАМКв-рецепторов
в клетках коры головного мозга и таламуса значительно по-
вышено по сравнению с мышами, не устойчивыми к судоро-
гам [Lin F.H. et al., 1995]. В то же время у больных с депрес-
235
сией и некоторыми формами маниакальных расстройств со-
держание ГАМК в плазме крови и мозге значительно ниже
по сравнению со здоровыми людьми, причем это снижение
коррелирует с нарушением настроения [Petty F., 1995]. Важ-
ную роль в генезе депрессивных состояний играет хроничес-
кая активация стресс-системы [Chrousos G.P., Gold P.W.,
1992], которая может приводить к истощению этой систе-
мы. Таким образом, очевидно, что в гиперактивации стресс-
системы и соответственно в страдании этих больных может
быть «виноват» дефицит активности стресс-лимитирующей
ГАМКергической системы.
Установлено также, что генетически детерминированная
и индивидуальная устойчивость животных к повреждениям,
вызванным эмоциональным стрессом, связана с величиной
базального содержания в гипоталамусе и крови нейропепти-
дов: субстанции Р, p-эндорфина, пептида дельта-сна [Юма-
тов Е.А., 1997; Sudakov K.V. et al., 1995], модулирующих ак-
тивность стресс-системы. Так, при сравнении крыс линий
Вистар и Август выявлено, что более устойчивые к наруше-
ниям сердечно-сосудистой системы при длительном тяжелом
эмоциональном стрессорной воздействии крысы Вистар
имеют достоверно большее (в 8 раз) содержание субстанции
Р в гипоталамусе, чем крысы Август, менее устойчивые к
таким нарушениям. Показано также, что у крыс Август суще-
ственно ниже, чем у крыс линии Вистар, содержание в гипо-
таламусе p-эндорфина и пептида дельта-сна, хотя в крови со-
держание p-эндорфина у крыс Август выше, чем у крыс Вис-
тар [Sudakov K.V. et al., 1995]. При этом введение экзогенно-
го пептида дельта-сна крысам Август повышает у них базаль-
ное содержание в гипоталамусе субстанции Р (до уровня,
имеющегося у крыс Вистар) и одновременно увеличивает у
них устойчивость сердечно-сосудистой системы к тяжелому
эмоциональному стрессорному воздействию. Даже однократ-
ное введение субстанции Р вызывает повышение ее содержа-
ния в гипоталамусе и увеличение резистентности животных к
стрессорным повреждениям в течение длительного времени
[Юматов Е.А., 1997; Sudakov K.V. et al., 1995]. Кроме того, у
крыс выявлена корреляция между снижением содержания
субстанции Р в ядрах гипоталамуса с возрастом и возрастным
уменьшением устойчивости к эмоциональному стрессорному
воздействию [Юматов Е.А., 1997]. В связи с этим существен-
ный интерес вызывают данные, показывающие, что у боль-
ных с невротическими нарушениями сна и эссенциальной
гипертензией снижено содержание субстанции Р в крови по
сравнению со здоровыми людьми [Ратсак Р. и др., 1984].
Таким образом, можно полагать, что компенсация этих де-
фектов регуляции стресс-системы может нивелировать ука-
занные нарушения. Действительно, показано [Меерсон Ф.З.
236
и др., 1985], что предварительное введение крысам пептида
дельта-сна или его циклического (более стабильного) произ-
водного предупреждает нарушения сократительной функции
сердечной мышцы при длительном стрессе. Введение суб-
станции Р предупреждает у крыс чрезмерное повышение
уровня катехоламинов в крови при эмоциональном стрессе и
устраняет вызванную стрессом артериальную гипертензию, а
также снижает повышенное АД у крыс линии SHR [Судаков
К.В., Юматов Е.А., 1991]. Предварительное введение р-эн-
дорфина предупреждало нарушение поведения и подавление
секреции половых гормонов у обезьян павианов-гамадрилов
при эмоциональном стрессе [Динзбург А.Л. и др., 1995].
Здесь уместно привести данные, полученные Э.Х.Орловой
совместно с нами еще 13 лет назад [Орлова Э.Х. и др., 1988],
которые показали, что адаптация к физической нагрузке, ха-
рактеризующаяся, как известно, повышением резистентности
организма к стрессорным воздействиям, приводит к повыше-
нию содержания p-эндорфина и других опиоидных пептидов в
надпочечниках, гипоталамусе и других структурах головного
мозга.
Таким образом, очевидно, что устойчивость к стрессорным
повреждениям в значительной мере определяется соотноше-
нием активности стресс-системы и стресс-лимитирующих
систем, которое формируется как на основе генетических
особенностей организма, так и в процессе жизнедеятельнос-
ти под влиянием различных факторов.
Это наше представление по существу уже «витает» в возду-
хе, однако еще не обозначено в литературе. Некоторые иссле-
дователи, занимающиеся проблемой стресса, уже разделяют
такую точку зрения. Е.А.Юматов (1997) пишет: «... животные
проявляют генетически детерминированную индивидуальную
устойчивость к эмоциональному стрессу, которая определяет-
ся особенностями центральной нейрохимической организа-
ции отрицательного эмоционального состояния». При этом он
выдвигает представление о роли пептидергической системы
как фактора, повышающего устойчивость к эмоциональному
стрессу [Юматов Е.А., 1997]. Однако система нейропепти-
дов — это только одна из стресс-лимитирующих систем. Ус-
тойчивость, по-видимому, объясняется результатом сложного
взаимодействия стресс-системы и всех стресс-лимитирующих
систем. Результат этого взаимодействия определяется актив-
ностями этих систем (генетически детерминированными либо
обусловленными действием различных факторов на орга-
низм). К этому ключевому представлению мы еще вернемся в
разделе о механизмах предрасположенности и устойчивости к
стрессорным повреждениям.
237
5.1.5. Адаптивные и повреждающие эффекты
стресс-реакции
Вернемся к событиям, отраженным на схеме 5.1. Как пока-
зано на этой схеме, в результате взаимодействия стресс-систе-
мы и системы, специфически ответственной за адаптацию, до-
стигается восстановление гомеостаза (т.е. адаптация к данно-
му стрессору) и соответственно завершается стресс-реакция.
Однако при прочих равных условиях эта схема реализуется
только в случае, если сила и продолжительность действия
стрессора умеренны, т.е. тогда, когда реализуются положи-
тельные — адаптивные эффекты стресс-реакции. Чрезмерно
сильное стрессорное воздействие вызывает превращение этих
эффектов в повреждающие и приводит к нарушению функций
и повреждениям органов и тканей.
Повреждающие эффекты стресс-реакции возникают как
«издержки» активации стресс-системы в ответ на сильное
стрессорное воздействие и связаны с избыточным выбросом
стресс-гормонов (см. схему 5.1 — «Побочные эффекты стресс-
реакции»).
Для понимания роли стресс-реакции в адаптации организ-
ма к действию стрессоров и возникновении стрессорных по-
вреждений рассмотрим 5 основных, во многом связанных друг
с другом эффектов стресс-реакции (схема 5.5), за счет которых
формируется «срочная» адаптация к факторам среды на уров-
не систем, органов, клеток и которые могут превращаться в
повреждающие эффекты стресс-реакции [Меерсон Ф.З.,
Пшенникова М.Г., 1989].
Первый адаптивный эффект стресс-реакции состоит в мо-
билизации функции органов и тканей путем активации наибо-
лее древнего сигнального механизма стимуляции клетки, а
именно увеличения концентрации в цитоплазме универсаль-
ного мобилизатора функции — кальция, а также путем актива-
ции ключевых регуляторных ферментов — протеинкиназ [Вег-
ridge M.J., 1998]. На рис. 5.2 представлены на примере клетки
сердечной мышцы (кардиомиоцита) 3 основных регуляторных
механизма, за счет которых на уровне плазматической мем-
браны регулируется вхождение кальция (Са2+) в клетку, его со-
держание в саркоплазме, а также уровень функции клетки и ее
генетического аппарата.
При стресс-реакции увеличение концентрации Са2+ в клет-
ке и активация внутриклеточных процессов осуществляются
благодаря двум факторам, сопровождающим стресс-реакцию.
Первый из них — выход Са2+ из костей и повышение его со-
держания в крови под влиянием стрессорного повышения в
крови уровня паратгормона (гормона паращитовидных желез);
это способствует увеличению вхождения указанного катиона в
клетки органов, ответственных за адаптацию. Второй фак-
238
Схема 5.5. Адаптивные эффекты стресс-реакции и превращение их в повреждающие эффекты
СТРЕССОР
5. Развитие "ана-
болической фа-
зы" стресс-реак-
ции: активация
синтеза белков
_______v________
Нерегулируемый
клеточный рост,
который в сочета-
нии со стрессор-
ным иммуноде-
фицитом соста-
вляет основу он-
когенного эффек-
та стресса
Пояснение в тексте.
СТРЕССОР
| Активация стресс-системы |
Высвобождение
стресс-гормонов
шт
АТФ
ДАГ
СТ-рецептор СПК-С
км+Са!*
са-км-пк
Мобилизация функций
и энергообеспечения клетки
РОСТ КЛЕТОЧНЫХ
СТРУКТУР
мРНК Белки
I Глюкокортикоиды |
\ |цАМФ|
Образование
'структурного” следа
адаптации
Рис. 5.2. Регуляция функции и генетического аппарата клетки (на
примере клетки миокарда) с помощью универсального активатора
внутриклеточных процессов кальция (Caz+) и потенциации этой ре-
гуляции при стрессе, осуществляемой стресс-гормонами и медиато-
рами через посредство вторичных мессенджеров.
М — мембрана; СПР — саркоплазматический ретикулум; MX — митохондрия;
КМ — кальмодулин; Са-КМ-ПК — кальмодулинзависимая протеинкиназа;
G — белковая субъединица рецепторного комплекса, осуществляющая сопря-
жение рецептора с ферментом; В-рецептор, а ।-рецептор и аг- соответственно:
р-, ai- и аг-адренорецепторы; Vz — вазопрессиновый рецептор; ФИ — фосфо-
тидилинозитол; Ич>з — инозитолтрифосфат; ДАГ — диацилглицерол; ПК-С —
протеинкиназа С; цАМФ-ПК — цАМФ-зависимая протеинкиназа; СТ-рецеп-
тор — рецептор стероидных гормонов. Ломаной черной стрелкой обозначены
деполяризация мембраны в ответ на импульс возбуждения и потенциал-зави-
симый Са -канал. Сплошные стрелки указывают последовательность процес-
сов. Штриховые стрелки означают активирующее влияние.
240
тор — возросший выброс катехоламинов и других гормонов,
который обеспечивает их увеличенное взаимодействие с соот-
ветствующими рецепторами клеток; в результате происходит
активация механизма вхождения Са2+ в клетку, повышение его
внутриклеточной концентрации, потенциация активации про-
теинкиназ и как следствие — активация внутриклеточных
процессов. Рассмотрим это подробнее. Обратимся к рис. 5.2.
Приходящий к клетке импульс возбуждения (на рисунке обо-
значено черной ломаной стрелкой) вызывает деполяризацию
клеточной мембраны, что приводит к открытию потенциалза-
висимых Са2+-каналов, вхождению внеклеточного Са2+ в клет-
ку, выделению Са2+ из депо, т.е. из саркоплазматического ре-
тикулума (СПР) и митохондрий, и повышению концентрации
этого катиона в саркоплазме. Соединяясь со своим внутрикле-
точным рецептором кальмодулином (КМ), Са2+ активирует
КМ-зависимую протеинкиназу, которая «запускает» внутри-
клеточные процессы, приводящие к мобилизации функции
клетки. Одновременно Са2+ участвует в активации генетичес-
кого аппарата клетки. Гормоны и медиаторы, воздействуя на
соответствующие рецепторы в мембране, потенцируют акти-
вацию этих процессов через посредство вторичных мессенд-
жеров, образующихся в клетке с помощью ферментов, сопря-
женных с рецепторами. Воздействие на а)-адренорецепторы
активирует связанный с ними фермент фосфолипазу С, с по-
мощью которой из фосфолипида мембраны фосфатидилино-
зитола образуются вторичные мессенджеры диацилглицерол
(ДАГ) и инозитолтрифосфат (ИФ3). ДАГ активирует протеин-
киназу С (ПК-С), ИФ3 стимулирует высвобождение Са2+ из
СПР, что потенцирует вызываемые кальцием процессы.
Воздействие на 0-адренорецепторы, а2-адренорецепторы и
вазопрессиновые рецепторы (V2) приводит к активации адени-
латциклазы и образованию вторичного мессенджера цАМФ;
цАМФ активирует цАМФ-зависимую протеинкиназу (цАМФ-
ПК), которая потенцирует клеточные процессы, а также рабо-
ту потенциалзависимых Са2+-каналов, через которые Са2+ вхо-
дит в клетку. Глюкокортикоиды, проникая в клетку, взаимо-
действуют с внутриклеточными рецепторами стероидных гор-
монов и активируют генетический аппарат. Протеинкиназы
играют двоякую роль. Во-первых, они активируют процессы,
ответственные за функцию клетки: в секреторных клетках сти-
мулируется выход соответствующего «секрета», в мышечных
клетках усиливается сокращение и т.п. Одновременно они ак-
тивируют процессы образования энергии в митохондриях —
«фабриках» кислородзависимого синтеза АТФ (носителя энер-
гии), а также в системе бескислородного гликолитического
образования АТФ. Таким образом мобилизуется функция
клетки и органов в целом. Во-вторых, протеинкиназы участву-
ют в активации генетического аппарата клетки, т.е. процессов,
16—1385
241
протекающих в ядре, вызывая экспрессию генов регуляторных
и структурных белков, что приводит к образованию соответст-
вующих мРНК, синтезу указанных белков и обновлению и
росту клеточных структур, ответственных за адаптацию. При
повторных действиях стрессора это обеспечивает формирова-
ние структурной основы устойчивой адаптации к данному
стрессору [ссылки см.: Меерсон Ф.З., Пшенникова М.Г, 1988,
1989].
Однако при чрезмерно сильной и/или затянувшейся стресс-
реакции, когда содержание Са2* и Na+ в клетке чрезмерно увели-
чивается, возрастающий избыток Са2+ может приводить к по-
вреждению клетки. Это может усугубляться при врожденной
неполноценности ферментов и/или других субъединиц катионных
насосов, которые должны обеспечивать своевременное удаление
кальция из цитоплазмы в депо и внеклеточную среду (насос
плазматической мембраны, удаляющий из клетки Са2+,
№+/Са2+-обменный механизм плазматической мембраны, а
также Са2+-насос СПР). Применительно к сердцу такая ситуа-
ция вызывает кардиотоксический эффект [Fleckenstein A. et
al., 1985]: реализуется так называемая кальциевая триада по-
вреждения клеточных структур избытком кальция, которая
складывается из необратимых контрактурных повреждений
миофибрилл, нарушения функции перегруженных кальцием
митохондрий и активации миофибриллярных протеаз и мито-
хондриальных фосфолипаз. Все это может приводить к нару-
шению функции кардиомиоцитов и даже к их гибели и разви-
тию очаговых некрозов миокарда. Этот повреждающий эф-
фект не реализуется как изолированный феномен, а находится
в коренной связи с чрезмерным усилением рассматриваемого
далее второго адаптивного (липотропного) эффекта стресс-ре-
акции.
Второй адаптивный эффект стресс-реакции состоит в том,
что «стрессорные» гормоны — катехоламины, вазопрессин и
др. — прямо или опосредованно через соответствующие ре-
цепторы активируют липазы, фосфолипазы и увеличивают ин-
тенсивность СРО. Это реализуется путем повышения содержа-
ния в клетке кальция и активации зависимых от него кальмо-
дуллин-протеинкиназ, а также за счет повышения активности
зависимых от ДАГ и цАМФ протеинкиназ ПК-С и цАМФ-ПК
(см. рис. 5.2). В результате в клетке повышается содержание
свободных жирных кислот, продуктов СРО, фосфолипидов.
Этот липотропный эффект стресс-реакции меняет структур-
ную организацию, фосфолипидный и жирнокислотный состав
липидного бислоя мембран и тем самым меняет липидное ок-
ружение мембраносвязанных функциональных белков, т.е.
ферментов, рецепторов, каналов ионного транспорта, ионных
насосов, локализованных в мембране. В результате миграции
фосфолипидов и образования лизофосфолипидов, обладаю-
242
щих детергентными свойствами, снижается вязкость и повы-
шается «текучесть» мембраны. Такие изменения умеренной
степени увеличивают «подвижность» полипептидных цепей
функциональных мембраносвязанных белков и повышают их
активность [Архипенко Ю.В. и др., 1983; Меерсон Ф.З., Пшен-
никова М.Г., 1989; Меерсон Ф.З., 1993].
Важным фактором, модифицирующим липидный бислой
мембран, является процесс СРО липидов, который при фи-
зиологических условиях протекает в мембранах клеток всех
без исключения органов. При стресс-реакции или введении
катехоламинов доказана активация СРО в сердце, печени,
скелетных мышцах и других органах [Владимиров Ю.А., Ар-
чаков А.И., 1972; Меерсон Ф.З., 1993]. В частности, на моде-
ли эмоционально-болевого стресса показано, что в зависи-
мости от длительности стресса активация СРО может дости-
гать значительной степени, что выражается увеличением в
2—3 раза содержания гидроперекисей фосфолипидов, шиф-
фовых оснований и других продуктов СРО в миокарде [Ме-
ерсон Ф.З., 1984]. Важно подчеркнуть, что активация СРО
обнаружена также у человека при эмоциональном стрессе —
операторской работе в условиях цейтнота [Прилипко Л.Л. и
др., 1982].
В основе модифицирующего влияния СРО на липидный
бислой мембран лежат реакция радикалов кислорода с нена-
сыщенными жирными кислотами фосфолипидов и образова-
ние таким образом в мембранах полярных гидроперекисей
фосфолипидов, обладающих детергентным действием. В опы-
тах in vitro показано, что при умеренной активации СРО,
когда в процесс вовлечено не более 2—5 % фосфолипидов
мембраны, происходят увеличение «жидкостности» бислоя и
как следствие — нарастание подвижности полипептидных
цепей мембраносвязанных белков и соответственно — по-
вышение их активности. Это явление, в частности, сопровож-
дается повышением активности такого важного фермента сер-
дечной мышцы, как Са2+-АТФаза СПР, которая составляет ос-
нову структуры Са2+-насоса СПР [Архипенко Ю.В. и др.,
1983]. Активация этого фермента играет ключевую роль в ме-
ханизме такого важного адаптивного эффекта, как активация
процесса расслабления миокарда при действии на сердце ка-
техоламинов или при кратковременной умеренной стресс-ре-
акции. Следует отметить также, что повышение содержания
Са2+, свободных жирных кислот и активация СРО стимулиру-
ют фермент синтеза NO NO-синтазу [Малышев И.Ю., Ману-
хина Е.Б., 1998] и, таким образом, сопровождаются увели-
чением продукции NO. Это, несомненно, имеет адаптивное
значение, так как способствует формированию «рабочей ги-
перемии», которая обеспечивает увеличение кровотока в орга-
нах, ответственных за адаптацию (см. далее).
16*
243
Адаптивное значение липотропного эффекта стресс-реак-
ции, очевидно, велико, так как этот эффект может быстро оп-
тимизировать активность всех мембраносвязанных белков, а
следовательно, функцию клеток и органа в целом и способст-
вовать, таким образом, срочной адаптации организма к дейст-
вию факторов среды. Однако при чрезмерно длительной и ин-
тенсивной стресс-реакции усиление именно этого эффекта,
т.е. избыточная активация фосфолипаз, липаз и СРО, может
привести к повреждению мембран и приобретает ключевую
роль в превращении адаптивного эффекта стресс-реакции в
повреждающий.
Повреждающими факторами становятся при этом свобод-
ные жирные кислоты, накапливающиеся в результате избы-
точного гидролиза триглицеридов липазами и при гидролизе
фосфолипидов фосфолипазами, а также лизофосфолипиды,
образующиеся в результате гидролиза фосфолипидов. Высо-
кий уровень жирных кислот повышает содержание их длинно-
цепочечных производных; эти производные и лизофосфоли-
пиды имеют как гидрофильную (полярную), так и гидрофоб-
ную (неполярную) группы, что позволяет им взаимодейство-
вать с фосфолипидами бислоя и внедряться в их молекулы.
В результате меняется структура бислоя мембран. При высо-
ких концентрациях такие соединения образуют мицеллы, ко-
торые «разбивают» мембрану и нарушают ее целостность.
В итоге повышается проницаемость клеточных мембран для
ионов и особенно для Са2+. В частности, показано, что актива-
ция фосфолипазы А2 и образование л изофосфол и пидов при
действии катехоламинов на сердце вызывают повышение про-
ницаемости мембран СПР кардиоцитов и выход из него Са2+,
что приводит к перегрузке этим катионом саркоплазмы и по-
вреждению клеток [Takasu N. et al., 1986].
Повреждающими факторами липотропного эффекта при
интенсивной или затянувшейся стресс-реакции становятся и
продукты активации СРО. При прогрессировании СРО все
большее количество ненасыщенных фосфолипидов окисляет-
ся и в мембранах растет доля насыщенных фосфолипидов в
микроокружении функциональных белков. Это приводит к
уменьшению жидкостности мембраны и подвижности пептид-
ных цепей указанных белков. Возникает явление «вморажива-
ния» этих белков в более «жесткую» липидную матрицу, и как
следствие активность белков снижается или полностью блоки-
руется. Дальнейшее накопление окисленных ненасыщенных
фосфолипидов приводит к образованию так называемых пере-
кисных кластеров, т.е. упорядоченных групп фосфолипидов,
между которыми образуются каналы ионной проводимости,
что ведет опять-таки к повышению проницаемости, а также
разрушению мембран [Архипенко Ю.В. и др., 1983; Меерсон
Ф.З., 1984]. Повреждающим мембраны фактором также стано-
244
вится избыток NO, обладающий токсическим действием [Ма-
лышев И.Ю., Манухина Е.Б., 1998].
Таким образом, чрезмерное усиление липотропного эффекта
стресс-реакции, т.е. ее «липидной триады» (активации липаз и
фосфолипаз, активации СРО и увеличения количества свободных
жирных кислот) может приводить к повреждению биомембран,
что играет ключевую роль в нарушении работы ионных каналов,
рецепторов и ионных насосов. В результате адаптивный липо-
тропный эффект стресс-реакции может превратиться в по-
вреждающий эффект, липидная триада адаптивного эффек-
та — в липидную триаду повреждения клеток.
Третий адаптивный эффект стресс-реакции состоит в моби-
лизации энергетических и структурных ресурсов организма,
что выражается в увеличении в крови концентрации глюкозы,
жирных кислот, аминокислот, а также в мобилизации функ-
ции кровообращения и дыхания. Этот эффект приводит к уве-
личению доступности субстратов окисления, исходных про-
дуктов биосинтеза и кислорода для органов, работа которых
увеличена. Оценивая этот адаптивный эффект стресс-реак-
ции, следует иметь в виду, что главную роль в мобилизации
резерва углеводов и увеличения поступления в кровь глюкозы
играют катехоламины и глюкагон за счет прямой активации
гликогенолиза и гликолиза через аденилатциклазную систему
в печени, скелетных мышцах и сердце. При этом глюкагон вы-
деляется при стрессе несколько позже катехоламинов и как бы
«дублирует» и подкрепляет эффект катехоламинов. Особую
значимость это приобретает в условиях, когда действие ка-
техоламинов реализуется не полностью из-за десенситизации
0-адренорецепторов, вызванной избытком катехоламинов.
В этом случае активация аденилатциклазы осуществляется
через глюкагоновые рецепторы [Ткачук В.А., 1987].
Другим источником глюкозы являются возникающие под
влиянием глюкокортикоидов и в известной степени паратгор-
мона активация гидролиза белков и увеличение фонда свобод-
ных аминокислот, а также активация глюконеогенеза в печени
и скелетных мышцах [Голиков П.П., 1988]. При этом глюко-
кортикоиды, действуя на свои рецепторы на уровне клеточно-
го ядра, стимулируют синтез ключевых ферментов глюконео-
генеза глюкозо-6-фосфатазы, фосфоэтанолпируваткарбокси-
киназы и др. [Голиков П.П., 1988]. Итогом активации глюко-
неогенеза являются трансаминирование аминокислот и обра-
зование из них глюкозы. Важно, что оба гормональных меха-
низма мобилизации глюкозы при стресс-реакции обеспечива-
ют своевременное поступление глюкозы к таким жизненно
важным органам, как мозг и сердце. При стресс-реакции, свя-
занной с острой физической нагрузкой, особое значение при-
обретает возникающая опять-таки под влиянием глюкокорти-
коидов в скелетных мышцах активация глюкозо-аденинового
245
цикла, который обеспечивает образование глюкозы из ами-
нокислот непосредственно в мышечной ткани [Виру А.А., Эй-
лер А. К., 1976].
В мобилизации жировых депо при стрессе главную роль
играют катехоламины и глюкагон, которые опосредованно
через аденилатциклазную систему активируют липазы и липо-
протеинлипазы в жировой ткани, скелетных мышцах, сердце.
В гидролизе триглицеридов крови, по-видимому, играют роль
паратгормон и вазопрессин, секреция которых при стрессе,
как указывалось выше, возрастает. Появившийся таким обра-
зом фонд жирных кислот используется в сердце и скелетных
мышцах.
В целом мобилизация энергетических и структурных ресурсов
выражена- при стресс-реакции достаточно сильно и обеспечива-
ет «срочную» адаптацию организма к стрессорной ситуации,
т.е. является адаптивным фактором. Однако в условиях затя-
нувшейся и/или интенсивной стресс-реакции, когда не происхо-
дит формирования «структурных следов адаптации», иными
словами, не происходит увеличения мощности системы энерго-
обеспечения, интенсивная мобилизация ресурсов перестает быть
адаптивным фактором и приводит к прогрессирующему истоще-
нию организма.
Четвертый адаптивный эффект стресс-реакции может быть
обозначен как «направленная передача энергетических и
структурных ресурсов в функциональную систему, осущест-
вляющую данную адаптационную реакцию». Одним из важ-
ных факторов этого избирательного перераспределения ресур-
сов является хорошо известная, локальная по своей форме
«рабочая гиперемия» в органах системы, ответственной за
адаптацию, которая одновременно сопровождается сужением
сосудов «неактивных» органов. Действительно, при стресс-ре-
акции, вызванной острой физической нагрузкой, доля минут-
ного объема крови, протекающей через скелетные мышцы,
возрастает в 4—5 раз, а в органах пищеварения и почках этот
показатель, напротив, уменьшается в 5—7 раз по сравнению с
состоянием покоя [для ссылок см.: Пшенникова М.Г., 1986].
Известно, что при стрессе развивается увеличение коронарно-
го кровотока, что обеспечивает увеличенную функцию сердца.
Главная роль в реализации этого эффекта стресс-реакции при-
надлежит катехоламинам, вазопрессину и ангиотензину II, а
также субстанции Р, секреция которых, как известно, увеличе-
на при стресс-реакции [Harbuz M.S., Lightman S.L., 1992;
Aguilera G. et al., 1995]. Эти гормоны вызывают сужение сосу-
дов в тех органах и тканях, где этому не препятствуют «рабо-
чая гиперемия» и мобилизация закрытых запасных капилля-
ров. Так, при стрессорных поведенческих реакциях, когда ак-
тивированы локомоции, выделяющаяся в центральном звене
стресс-системы субстанция Р способствует снижению крово-
246
тока в почках, но увеличивает его в скелетных мышцах [Cui-
man J. et al., 1995].
Ключевым локальным фактором рабочей гиперемии, как
уже упоминалось выше, является продуцируемый эндотелием
сосудов оксид азота (NO) — главный вазодилататор, продук-
ция которого возрастает параллельно росту потребления кис-
лорода. «Рабочая гиперемия» обеспечивает увеличенный при-
ток кислорода и субстратов к работающему органу путем вазо-
дилатации в этом органе [Yamabe Т. et al., 1992]. Показано, что
при стресс-реакции умеренной силы происходит рост продук-
ции NO [Малышев И.Ю., Манухина Е.Б., 1998]. Таким обра-
зом, при стрессе увеличенная продукция оксида азота в орга-
нах с повышенной функцией, по-видимому, противостоит
констрикторному действию гормонов, обеспечивая в них ра-
бочую гиперемию и тем самым участвуя в реализации рассмат-
риваемого адаптивного эффекта стресс-реакции.
Очевидно, что перераспределение ресурсов организма при
стрессе, направленное на преимущественное обеспечение органов
и тканей, ответственных за адаптацию, представляет собой
важный адаптивный феномен. Вместе с тем при чрезмерно выра-
женной стресс-реакции он может сопровождаться ишемически-
ми нарушениями функции и даже повреждениями других органов,
не участвующих непосредственно в данной адаптивной реакции,
например ишемическими язвами желудочно-кишечного трак-
та, возникающими у спортсменов при тяжелых длительных
эмоционально-физических нагрузках.
Превращение адаптивного эффекта увеличения коронар-
ного кровотока в повреждающий, на первый взгляд, представ-
ляется маловероятным, так как в сердце преобладает 0-адре-
нергический эффект катехоламинов, что может стимулировать
коронародилатацию и «рабочую гиперемию» миокарда
[Beamish R.E., Dhalla N.S., 1985; Young М.А., Vatner S.F.,
1986]. Однако при большой длительности стресса, особенно
эмоционального, высокие концентрации в крови вазопресси-
на, адреналина и гистамина могут приводить к десенситизи-
ции 0-адренергических рецепторов и на первый план будут
выходить а-адренорецепторы и другие кальциймобилизующие
рецепторы, стимулирующие коронароконстрикцию. В экспе-
риментах такой спазм коронарных сосудов был показан при
действии вазопрессина [Фролькис В.В. и др., 1983] и НА
[Beamish R.E., Dhalla N.S., 1985]. Спазм коронарных сосудов
под влиянием сильного стресса наблюдали у практически здо-
ровых людей [Yasue Н. et al., 1984].
Исследования последних лет позволяют считать, что клю-
чевую роль в реализации данного адаптивного эффекта стресс-
реакции и превращении его в повреждающий играет продукция
NO в сосудистой стенке. При кратковременных и умеренных
стрессорных воздействиях продукция NO возрастает, и это
247
может способствовать рабочей гиперемии, но при сильных и дли-
тельных стресс-реакциях продукция NO снижается, обусловли-
вая возникновение стрессорных спазмов коронарных сосудов, а
также стрессорных гипертензивных состояний.
Пятый адаптивный эффект стресс-реакции состоит в том,
что при однократном достаточно сильном стрессорном воз-
действии вслед за рассмотренной выше хорошо известной «ка-
таболической фазой» стресс-реакции (третий адаптивный эф-
фект) реализуется значительно более длительная «анаболичес-
кая фаза». Она проявляется генерализованной активацией
синтеза нуклеиновых кислот и белков в различных органах
[Меерсон Ф.З., 1993]. Эта активация обеспечивает восстанов-
ление структур, пострадавших в катаболическую фазу, и явля-
ется основой формирования структурных «следов» и развития
устойчивого приспособления к различным факторам среды.
В основе этого адаптавного эффекта лежат процессы, рас-
смотренные при описании первичного эффекта, а именно —
гормональная активация образования вторичных мессендже-
ров ИФ3 и ДАГ, повышения в клетке уровня кальция, а также
действие на клетку глюкокортикоидов. Помимо мобилизации
функции клетки и ее энергообеспечения, этот процесс имеет
«выход» на генетический аппарат клетки (см. рис. 5.2), что
приводит к активации синтеза белков. Кроме того, показано,
что в процессе развертывания стресс-реакции активируется
секреция «приторможенных» в начале реакции соматотропно-
го гормона (гормона роста), инсулина, тироксина, которые
потенцируют синтез белков и могут играть роль в развитии
анаболической фазы стресс-реакции и активации роста кле-
точных структур, на которые приходилась наибольшая нагруз-
ка при стрессорной мобилизации функции клеток. Вместе с
тем следует иметь в виду, что чрезмерная активация этого
адаптивного эффекта, по-видимому, может приводить к нере-
гулируемому клеточному росту. В частности, наряду со стрес-
сорным иммунодефицитом (см. раздел 5.2.3.3.2) это может иг-
рать роль в механизме онкогенного эффекта стресса.
В целом можно заключить, что при затянувшейся по вре-
мени интенсивной стресс-реакции все рассмотренные основ-
ные адаптивные эффекты трансформируются в повреждающие
и именно так могут стать основой стрессорных болезней.
Эффективность адаптивной реакции на стрессоры и вероят-
ность возникновения стрессорных повреждений в значительной
мере определяются, помимо интенсивности и длительности дей-
ствия стрессора, состоянием стресс-системы: ее базальной (ис-
ходной) активностью и реактивностью, т.е. степенью актива-
ции при стрессе, которые обусловлены генетически, но могут ме-
няться в процессе индивидуальной жизни.
Хронически увеличенная базальная активность стресс-сис-
темы и/или избыточная ее активация при стрессе сопровожда-
248
ются повышенным артериальным давлением, нарушением
функции органов пищеварения, подавлением иммунитета.
При этом могут развиваться сердечно-сосудистые и другие за-
болевания. Сниженная базальная активность стресс-системы
и/или неполноценная активация ее при стрессе также небла-
гоприятны. Они приводят к снижению способности организ-
ма адаптироваться к окружающей среде и решать жизненные
задачи, к развитию депрессивных и других патологических со-
стояний.
5.2. Эмоциональный стресс и связанные с ним
патологические состояния
5.2.1. Особенности эмоциональных стрессоров
и эмоциональной стресс-реакции
В «Словаре физиологических терминов»* эмоциональный
стресс применительно к человеку определяется следующим
образом: «Стресс эмоциональный (синоним — стресс психи-
ческий, психологический, психофизиологический; психичес-
кая, нервно-психическая, эмоциональная напряженность) —
понятие, отражающее разновидность общей системной реак-
ции (стресс-синдрома) индивида с характерными объективно
регистрируемыми симптомами на воздействие внутренних или
внешних факторов информационной природы. Определение
«эмоциональный» выделяет особую роль эмоций в генезе
стресса. Термином «эмоциональный стресс» стали именовать
понятие тревоги, конфликта, эмоционального расстройства,
переживания угрозы безопасности, неудачи, досады и т.д. —
такие эмоциональные состояния, которые развиваются у чело-
века, когда он сталкивается с реальными психологически труд-
ными ситуациями либо считает их психологически трудными
или неразрешимыми».
Исследование реакции на эмоциональные стрессоры ука-
зывает как на общность физиологических изменений, возни-
кающих при действии физических и психических стрессоров,
так и на важные различия в механизмах формирования этих
изменений. Цель эмоционального стресса, как и физическо-
го, — адаптировать организм к действию стрессора. Вместе с
тем если при действии физических стрессоров стресс-реакция
возникает в ответ на непосредственное влияние стрессора и
физический стресс можно рассматривать на основе классичес-
кой «рефлекторной» концепции Г.Селье, т.е. как реакцию ор-
ганизма на холод, гипоксию, инфекцию и т.п., то в случае дей-
Словарь физиологических терминов. — М.: Наука, 1987. — С. 364.
249
ствия эмоциональных стрессоров стресс-реакция опосредуется
через сложные психические процессы. Эти процессы включают
оценку воздействующего фактора и сопоставление его с
предыдущим опытом. Таким образом, если физический фак-
тор, как правило, есть «стрессор для всех», то психический
фактор может являться стрессором для одного индивидуума и
быть полностью индифферентным для другого [Соколо-
ва Е.Б., Березин Ф.Б., Барлас Т.В., 1996].
Психический фактор приобретает характер стрессора, если
в результате индивидуальной психологической оценки («перера-
ботки») возникает ощущение угрозы или иная сильная эмоция).
При этом большинство авторов уделяют главное внимание не-
гативным эмоциям, и это оправдано, так как именно отрица-
тельные эмоциональные стрессы чреваты патологическими
последствиями; они способствуют возникновению патологи-
ческих состояний, а также ухудшению уже имеющейся пато-
логии. Однако дело здесь не только в знаке информации или
события, которые воздействуют на человека. Даже положи-
тельные ситуации, такие как успешное поступление в вуз,
вступление в брак, новая желанная работа и т.п., могут стать
тяжелым эмоциональным стрессором, так как они связаны с
разрушением привычных стереотипов, с возможной несостоя-
тельностью, с требованиями новых усилий и т.д. [Lazarus R.S.,
1993]. Иными словами, даже положительные желанные ситуа-
ции могут при определенных условиях вызывать отрицатель-
ные эмоции. Здесь опять-таки решающую роль играет индиви-
дуальное отношение к этим ситуациям.
Вместе с тем К. В.Судаков подчеркивает двоякую роль от-
рицательных эмоций. По его мнению, отрицательная эмоция,
или отрицательный эмоциональный стресс, играет ключевую
роль как в успешной адаптации к условиям жизни, так и в раз-
витии патологических состояний [Судаков К.В., 1998]. Автор
пишет: «Отрицательные эмоции мобилизуют организм на
удовлетворение ведущих биологических и социальных потреб-
ностей, на избегание повреждающих воздействий. В нормаль-
ных условиях жизнедеятельности отрицательные эмоции, как
правило, кратковременны, эпизодичны и при достижении по-
лезных для субъекта результатов завершаются положительны-
ми эмоциями. Отрицательные эмоции усиливаются во всех
случаях, когда субъекты при наличии у них жизненных по-
требностей не имеют возможности достичь социально или
биологически значимых потребных результатов» [Суда-
ков К.В., 1998].
Итак, ощущение угрозы, страха и другие отрицательные
эмоции возникают при действии данного фактора в том слу-
чае, если психологическая оценка обнаруживает очевидное
несоответствие между требованиями действующего фактора и
потребностями данного индивидуума, его физическими и пси-
250
хологическими ресурсами, которые необходимы для удовле-
творения этих потребностей. Иными словами, фактор стано-
вится стрессором, если возникает «конфликт» между требова-
ниями фактора, с одной стороны, и возможностями и потреб-
ностями индивидуума — с другой.
«Ситуации, — пишет К.В.Судаков (1998), — в которых
субъекты при наличии у них выраженных биологических или
социальных потребностей длительно или остро ограничива-
ются (кем-то или чем-то) в их удовлетворении, получили на-
звание конфликтных ситуаций». Согласно хрестоматийной
классификации мотивационно-эмоциональных конфликтов,
предложенной КЛевиным и дополненной Н. Миллером
[Котов А.В., 1999], конфликты бывают эндогенные и экзоген-
ные. Эндогенные возникают как следствие избыточных моти-
ваций, избыточных эмоций, неразрешимых проблем принятия
решения в ситуациях: «приближение — приближение» («бури-
данов осел»), «избегание — избегание» («Сцилла — Харибда»),
«приближение — избегание» (гоголевский персонаж Коробоч-
ка, т.е. «и хочется, и колется») и т.п. Экзогенные конфликты
возникают как проявление фрустрации при наличии непре-
одолимых физических или биологических препятствий на
пути достижения цели, а также при наличии дефицита време-
ни (цейтнот), информации и ресурсов среды для удовлетворе-
ния важных биологических потребностей.
Именно эти конфликтные ситуации приводят к эмоцио-
нальному стрессу. В качестве упрощенного примера можно
привести ситуацию на экзамене. Студенту попался билет,
ответ на вопросы которого он не знает, а результаты экзамена
очень важны для дальнейшей судьбы студента. Налицо вари-
ант экзогенной конфликтной ситуации: «требования» экзаме-
на превышают возможности студента. Результат — острый от-
рицательный эмоциональный стресс.
Важно подчеркнуть, что возникновение конфликтной си-
туации всегда субъективно, так как оценка фактора зависит от
неповторимого индивидуального опыта и психического стату-
са индивидуума. Именно субъективное отношение к фактору
представляет собой важный психологический механизм и опреде-
ляет индивидуальную значимость стрессора [Соколова Е.Б., Бе-
резин Ф.Б., Барлас Т.В., 1996]. Это обстоятельство определяет
тот важный факт, что эмоциональная стресс-реакция может
возникать и развиваться не только в ответ на реальные ситуа-
ции, но и на гипотетические, надуманные ситуации, не свя-
занные, казалось бы, непосредственно с конкретной средой.
Психическими стрессорами могут быть мучительные воспоми-
нания, множество негативно окрашенных сообщений, в част-
ности доставляемых средствами массовой информации. Таким
образом, многие события, информация при определенных ус-
ловиях могут приобретать значение эмоционального стрессо-
251
ра, но вместе с тем ни одна ситуация не вызывает стресс-реак-
цию у всех без исключения индивидуумов. Важность индиви-
дуальной значимости фактора как стрессора уменьшается при
чрезвычайных экстремальных ситуациях, природных или ан-
тропогенных катастрофах, войнах и т.п. Однако даже в этих
условиях, когда ситуация является стрессовой практически
для всех индивидуумов, оказавшихся в «зоне» ее действия,
стресс-реакция не будет одинаковой у всех [Теппеп Н. et al.,
1991].
Таким образом, будет ли данный фактор среды для челове-
ка эмоциональным стрессором или нет, определяется, во-пер-
вых, индивидуальным опытом этого человека (или историей
жизни) и, во-вторых, его психическим статусом (личностными
особенностями). Если первый показатель более понятен и со-
стоит в том, что фактор оценивается на основе индивидуаль-
ного жизненного опыта, то второй — является более сложным
и малоизученным, хотя именно от него главным образом зави-
сят выраженность ответной эмоциональной стресс-реакции, а
также устойчивость организма к стрессорным повреждениям.
Этот второй показатель, т.е. психический статус индивидуума,
в значительной мере определяется генетически обусловлен-
ным исходным состоянием стресс-системы и ее реактивности
при действии эмоционального стрессора. В последующих раз-
делах об этом будет говориться подробнее.
Следует отдельно выделить такой стрессорный фактор,
как действие ионизирующей радиации. Ионизирующая ра-
диация как фактор информационной природы, обозначаю-
щий угрозу безопасности человека, может быть причиной
эмоционального стресса. Из факторов нерадиационной при-
роды именно эмоциональное напряжение наиболее законо-
мерно может сопровождать действие ионизирующего излуче-
ния на организм.
У части населения, пострадавшего от аварии на Черно-
быльской АЭС, и у ликвидаторов этой аварии, облученных в
диапазоне малых доз, были отмечены (по данным психологи-
ческих, психофизиологических и эндокринологических об-
следований) явления хронического эмоционального стресса
и как его следствие — соматические нарушения [Мороз Б.Б.,
Дешевой Ю.Б., 1999]. У этих лиц развитие эмоционального
напряжения было связано со страхом перед последствиями
действия ионизирующей радиации и с изменением социаль-
ных условий жизни. При этом состояние здоровья лиц, кото-
рые испытывали психологический стресс в связи с возмож-
ностью облучения и(или) подвергались действию на орга-
низм ионизирующей радиации в малых дозах, определялось
прежде всего ролью стрессорных повреждений. Так, в период
с 1987 по 1993 г. заболевания органов кровообращения у лик-
видаторов аварии на Чернобыльской АЭС постепенно нарас-
252
тали, и в 1993 г. их показатель превышал общероссийский в
4,3 раза, а показатель заболеваемости органов пищеварения
было в 3,7 раза выше, чем у населения России [Мороз Б.Б.,
Дешевой Ю.Б., 1999].
5.2.2. Стрессорные патологические состояния
и их возможные механизмы
В основе стрессорной патологии лежит нарушение способ-
ности организма отвечать на действие стрессоров адекватной
защитной реакцией. Это происходит в случае возникновения
избыточной или, напротив, недостаточной по силе и/или дли-
тельности стресс-реакции. Адекватная реакция — это состоя-
ние, которое обеспечивает максимальное приспособление ин-
дивидуума к новым условиям жизнедеятельности, к психоген-
ному «давлению» окружающей среды, к информационным
перегрузкам и т.п. При недостаточном развитии стресс-реак-
ции организм не может адекватно реагировать на стрессор и
изменять свое поведение и вегетативные функции в соответст-
вии с «требованиями» ситуации (стрессора). Это состояние ха-
рактерно для людей, которых по классификации И.П.Павлова
относили к «слабому типу нервной деятельности». Именно у
таких людей при действии эмоциональных стрессоров чаще
возникают «стрессорные» болезни, или «психосоматические
расстройства» [Березин Ф.Б., Мирошников М.П., 1996]. Пато-
логические нарушения могут возникать и при чрезмерной
стресс-реакции, которая обычно реализуется при действии
очень сильных стрессоров.
5.2.2.1. Роль стресс-системы в формировании
эмоционального стресса и патогенезе
стрессорных повреждений
На основании оценки состояния компонентов стресс-сис-
темы появилась возможность прогнозирования адекватности
эмоциональной стресс-реакции и возникновения стрессорной
патологии. В зависимости от активации стресс-системы
стресс-реакция будет либо адекватной, либо недостаточной,
либо чрезмерной. Например, по данным G.P.Chrousos,
P.W.Gold (1992), длительная гиперреакция, или гипервозбуж-
дение стресс-системы, может приводить, например, к мелан-
холической депрессии, главными симптомами которой явля-
ются тревога, подавление пищевых и сексуальных реакций,
гипертензия, тахикардия, т.е. то, что обычно характерно для
генерализованной стресс-реакции. При такой патологии отме-
чают хроническую активацию гипоталамо-гипофизарно-адре-
253
наловой оси и симпатической нервной системы [Gold P.W. et
al., 1988; Chrousos G.P., Gold P.W., 1992]. При гипореакции
отмечают сезонные депрессии, например в темное время года.
Эти состояния, наиболее часто встречающиеся у женщин,
особенно в послеродовой период, характеризуются усталос-
тью, синдромом фибромиалгий, ростом аппетита и увеличени-
ем массы тела, сонливостью. Для этих состояний характерно
сниженное содержание КРГ в гипоталамусе. Однако пока нет
четкого представления о физиологических механизмах форми-
рования эмоционального стресса и соответственно формиро-
вания патологических состояний, с ним связанных. Следует
напомнить положение, высказанное П.В.Симоновым (1984) о
том, что эмоциональный стресс как целостное состояние орга-
низма имеет центральное происхождение и первично форми-
руется в эмоциогенных зонах мозга. Исследования П.В.Симо-
нова указывают, что ведущая роль в формировании эмоцио-
нальной стресс-реакции принадлежит четырем структурам го-
ловного мозга: фронтальному отделу неокортекса (новой
коры), гиппокампу, ядрам амигдалы и гипоталамусу. При этом
фронтальные структуры коры необходимы для вероятностного
прогнозирования внешних событий и оценки возможностей
удовлетворения потребностей. С помощью фронтальной коры
определяются события, имеющие высокую степень вероятнос-
ти. Эта функция коры обнаружена, помимо человека, также у
обезьян, собак, кошек и крыс. Функция гиппокампа связана с
выявлением сигналов событий с низкой вероятностью и про-
является в ситуациях, характеризующихся неопределеннос-
тью. При этом «нарастает степень эмоционального напряже-
ния», и это связано с участием гиппокампа в формировании
эмоций и эмоционального поведения. Основная функция
амигдалы проявляется в «оценке» сосуществующих и конку-
рирующих мотиваций и выделении доминирующей потреб-
ности с учетом конкретной ситуации и предшествующего жиз-
ненного опыта.
Главная функция в формировании центрального механизма
эмоционального стресса принадлежит гипоталамусу, как и при
формировании любого вида стресса. Как уже указывалось в раз-
деле 5.1 нашего изложения и будет неоднократно повторяться
далее, гипоталамус является триггером, который «запускает»
деятельность различных структур мозга, необходимых для реа-
лизации доминирующей мотивации, оценки афферентных
сигналов, для определения возможности удовлетворения тре-
бований, предъявляемых организму эмоциональным стрессор-
ным воздействием.
Эмоциональная стресс-реакция формируется на основе
поступления эндогенного (внутреннего) или экзогенного
(внешнего) стрессорного сигнала (см. схему 5.2). При форми-
ровании эмоций эндогенным путем гипоталамус в ответ на гу-
254
моральные и нервные стимулы, поступающие из организма,
вовлекает в возбуждение надгипоталамические лимбические
структуры, и затем возбуждение достигает корковых клеток и
реализуется в поведенческие реакции организма. При форми-
ровании эмоциональной стресс-реакции в ответ на внешний
эмоциональный (информационный) стрессор гипоталамус по-
лучает сигнал от коры мозга, так как первично в ответ на
внешний стимул возбуждение реализуется в коре. Эмоцио-
нальная стресс-реакция включается в результате нисходящих
влияний коры на гипоталамус и лимбические структуры мозга.
При этом доказано, что существует определенная связь между
НА-нейронами «синего» пятна и поведением при эмоциональ-
ном стрессе «тревоги и страха». Нейроны «синего пятна» с
проекцией через кору головного мозга и подкорковые структу-
ры, включая гиппокамп, амигдалу, таламус и гипоталамус, об-
разуют нейроанатомическую структуру, которая реализует бы-
струю модуляцию функции мозга и обеспечивает поведенчес-
кие и вегетативные компоненты эмоциональной стресс-реак-
ции [Bremner J.D. et al., 1996а].
Таким образом, механизмы возникновения стрессорных
патологий нужно «искать» прежде всего в центральных струк-
турах стресс-системы. Действительно, сравнительно недавно
было показано (Brady L.S. et al., 1994], что курс электросудо-
рожной терапии, эффективный в лечении депрессивных со-
стояний у людей, приводит у крыс к стойкой и продолжитель-
ной активации экспрессии генов, кодирующих синтез КРГ и
ключевого фермента синтеза катехоламинов тирозингидрок-
силазы в мозгу — соответственно в паравентрикулярном ядре
гипоталамуса и «синем пятне». При этом выяснилось, что не
имеется какого-либо другого терапевтического воздействия,
курс которого вызывал бы, с одной стороны, такой же хоро-
ший терапевтический эффект и, с другой стороны, столь же
длительную активацию синтеза КРГ и тирозингидроксилазы в
указанных структурах стресс-системы. На основании этих
данных авторы правомерно утверждают, что КРГ является
«главным медиатором» лечебного эффекта электросудорожной
терапии, а депрессивное состояние может быть связано с де-
фицитом КРГ.
Важную роль в возникновении стрессорных патологий, не-
сомненно, играют изменения в центральных ядрах амигдалы и
гиппокампе, тесно связанных со стресс-системой. Показано,
что выключение базолатерального ядра амигдалы вызывает
снижение уровня кортикостерона в крови и предупреждает его
повышение при эмоциональном стрессе. При этом разруше-
ние амигдалы приводило к изменению поведенческих реак-
ций: появлялись вялость, индифферентность. Напротив, акти-
вация (электростимуляция) центральных ядер этой структуры
вызывала реакцию эндокринной, вегетативных систем, а так-
255
же поведенческие реакции, сходные с реакциями при эмоцио-
нальном стрессе, т.е. с реакциями, возникающими при акти-
вации стресс-системы [Gray Y.S., 1990]. В последние 5—6 лет
появились исследования, подтверждающие это положение.
Обнаружилось, что повреждение центральных ядер амигдалы
приводит к нарушению поведенческих реакций, характерному
для посттравматического стрессорного расстройства [Gold-
stein L.E. et al., 1996].
Показано, что сильная эмоциональная стресс-реакция
может приводить к атрофическим изменениям дендритов пи-
рамидных нейронов гиппокампа [Magarinos А.М. et al., 1996].
В исследованиях на самцах Тупай (Tupaia belangeri) установле-
но, что длительно существующий эмоциональный социальный
стресс, вызванный конфликтом «подчиненных» особей с «до-
минирующими», приводит у подчиненных особей к быстрой
потере массы тела, увеличению экскреции с мочой кортизола
и повреждениям пирамидных нейронов гиппокампа (САЗ).
Эти повреждения характеризуются атрофией апикальных
дендритов нейронов: уменьшением их длины и количества
ветвей [Magarinos А.М. et al., 1996]. Авторы показали, что
такую атрофию можно предупредить введением фенитоина —
препарата, ограничивающего возбуждение нейронов. При
этом авторы установили, что подобные повреждения вызыва-
ются также введением глюкокортикоидов. Сходные поврежде-
ния гиппокампа были получены ранее при социальном стрес-
се у обезьян. Можно полагать, что причинами таких повреж-
дений нейронов гиппокампа при стрессе являются избыток
глюкокортикоидов и их катаболический эффект, препятст-
вующий пластическому обеспечению гиперфункции нейро-
нов, а также нарушение продукции нейротропных факторов,
т.е. промоторов роста и дифференцировки нейронов. Так, на
крысах Лонг-Иванс установлено, что социальный стресс или
большие дозы кортикостерона (25—32 мкг/кг), характерные
для такого эмоционального стресса, приводят к снижению си-
наптической пластичности гиппокампа, нарушениям его
функции при неизменном числе нейронов пирамидных полей
СА1 и САЗ, что сопровождается ухудшением обучаемости жи-
вотных. При этом показано, что на фоне адреналэктомии
стресс не вызывал ухудшения памяти и обучаемости. Если
таким адреналэктомированным крысам вводили замещающие
дозы кортикостерона (т.е. поддерживали базальный уровень
гормона), то стресс также не вызывал указанных нарушений.
Если же вводили высокие дозы кортикостерона, приводящие к
повышению его уровня в крови, аналогичному тому, что на-
блюдается при сильном социальном стрессе, то стресс вызы-
вал нарушение обучаемости и памяти [BodnofTSh. et al., 1995].
В связи с приведенными выше данными представляют ин-
терес исследования влияния социального стресса на животных
256
с разным типом высшей нервной деятельности и поведения.
Эти данные свидетельствуют, что более уязвимые для стрес-
сорных повреждений «подчиненные» особи в колониях грызу-
нов (мышей) характеризуются преобладанием активности ги-
пофизарно-адренокортикального звена стресс-системы, а «до-
минирующие» особи — преобладанием симпатического звена
[Ely D.L., 1995]. Существенно, что хроническая гиперактива-
ция гипоталамо-гипофизарно-адреналовой оси в детстве и
юности, в частности, вызванная избыточной физической на-
грузкой, алкоголизмом и т.п., может стать причиной появле-
ния в этот период ряда психоневрологических нарушений,
таких как шизофрения, депрессия, невротическая анорексия
[Stratakis С.A., Chrousos G.P., 1995].
На схеме 5.6 дано гипотетическое представление о формиро-
вании эмоционального стресса и его последствий, построенное
на основании имеющихся к настоящему времени данных. На
схеме 5.6 видно, что фактор среды становится или не становится
для индивидуума эмоциональным стрессором после субъектив-
ной оценки, в основе которой лежат индивидуальный опыт и
психоэмоциональный статус. Если фактор является стрессором,
то эффект его действия, т.е. выраженность стресс-реакции,
будет зависеть от двух обстоятельств: степени устойчивости раз-
личных систем организма к действию фактора и интенсивности
стрессора (силы и продолжительности его действия). При высо-
кой устойчивости в ответ на слабый стрессор (пунктирные
стрелки на схеме 5.6) либо возникает слабая стресс-реакция,
либо не возникает вообще; в ответ на сильный стрессор появля-
ется умеренная стресс-реакция, не вызывающая повреждений.
При низкой устойчивости слабый стрессор вызывает умеренную
стресс-реакцию, не приводящую, как правило, к повреждениям;
в ответ на сильный стрессор возникает стресс-реакция, которая
может привести к повреждениям. При этом в настоящее время
выяснилось, что не бывает универсальной высокой или низкой
устойчивости организма к стрессорам и одинаковой устойчи-
вости к повреждениям всех систем организма. Высокая резис-
тентность одной системы организма к стрессорному поврежде-
нию может сочетаться со сниженной резистентностью другой
системы.
Имеющиеся к настоящему времени данные свидетельству-
ют, что существует три основных обстоятельства, определяю-
щих действие стрессора:
• индивидуальный опыт (история жизни), определяющий
значимость стрессора;
• устойчивость (предрасположенность) к стрессорным по-
вреждениям, обусловленная в значительной мере состоя-
нием стресс-системы;
• сила стрессора.
17 — 1385
257
Схема 5.6. Формирование эмоциональной стресс-
реакции и зависимость последствий стресс-реакции
от силы стрессорного воздействия и резистентности
организма к стрессорным повреждениям
Пунктирные линии означают слабое стрессорное воздействие, сплош-
ные линии — сильное стрессорное воздействие. Пояснение в тексте.
258
5.2.3. Виды патологических состояний у человека,
связанные с эмоциональным стрессом,
и их механизмы
Эмоциональный стресс, как неоднократно уже отмеча-
лось выше, может вызывать так называемые психосоматичес-
кие расстройства, а также потенцировать уже имеющиеся за-
болевания. Однако до сих пор эти расстройства нередко от-
носят к неврологическим и другим болезням и не связывают
с состоянием стресс-системы, стресс-лимитирующих систем
и характером стресс-реакции. Во всяком случае в «Междуна-
родной классификации болезней» (1995) эти виды патологии
распределены по различным разделам в зависимости от орга-
на или системы, в которых развились нарушения. Стрессор-
ные патологии частично внесены в раздел «Невротические,
связанные со стрессом, и соматоформные расстройства»
(F40—F48). В качестве стрессорного повреждения выделено
лишь «посттравматическое стрессовое расстройство» (F43.1).
Такая ситуация тормозит специальное изучение стрессорной
патологии и соответственно разработку методов ее лечения и
профилактики. Целесообразнее было бы выделить подобные
нарушения в особую рубрику, например такую, как «Патоло-
гия стресс-системы и стресс-реакции: стрессорные наруше-
ния психического статуса и функции вегетативных органов и
систем».
5.2.3.1. Сердечно-сосудистая система
Известно, что при эмоциональном стрессе могут возникать
различные нарушения системы кровообращения. Острый эмо-
циональный стресс может провоцировать такие грозные пора-
жения сердечно-сосудистой системы, как внезапная сердечная
смерть, инфаркт миокарда, гипертонический криз, инсульт.
Частые повторные эпизоды эмоциональных стресс-реакций
или хроническое их течение могут играть существенную роль в
патогенезе ишемической болезни сердца (ИБС), гипертони-
ческой болезни, кардиосклероза.
Сердечно-сосудистая стрессорная патология возникает в
результате перехода стресс-реакции из звена адаптации орга-
низма к стрессорному воздействию, когда повышение АД, уве-
личение частоты сердечных сокращений и сократительной
функции сердечной мышцы, «рабочая гиперемия» составляют
необходимый компонент приспособления, в звено патогенеза
заболеваний, когда указанные изменения становятся чрезмер-
но интенсивными и длительными. Этот переход происходит,
как уже было отмечено, при сильных и длительных стресс-ре-
акциях.
17*
259
Очень давно врачи заметили причинно-следственную связь
между сердечными катастрофами и предшествующими эмоци-
ональными потрясениями. В XI в. Авиценна в своем «Каноне
медицины» писал: «... И иногда болезни постигают сердце...
вследствие тех влияний, которые приходят к нему... из мозга,
когда жидкость меланхолии в нем накапливается и... вызывает
трепет и резкий упадок сил, и печаль с предчувствием беды, и
депрессию»*. Эмоциональный стресс как фактор этиологии
заболеваний «тела», и сердца в том числе, вошел в медицину в
середине XVII столетия благодаря работам Томаса Виллиса и
других исследователей (Willis, 1681]. В 1887 г. С.П.Боткин
писал, что «... изменения функции сердца сплошь и рядом нс
идут пропорционально с изменениями в самом сердце, а не-
редко находя тся в зависимости от центральных нервных аппа-
ратов...».
Мы остановимся па рассмотрении роли эмоционального
стресса в провоцировании и развитии наиболее роковых видов
кардиопатологии: ИБС и острого инфаркта миокарда, арит-
мий и внезапной сердечной смерти, артериальной гипертен-
зии. Но перед этим целесообразно рассмотреть явление «пер-
вичного стрессорного повреждения сердца», поскольку важную
роль в кардиопатологии играет не только острое действие
стресс-реакции, т.е. острое влияние избытка гормонов на
сердце («ех tempore»), но и нарушение структуры миокарда,
развивающееся при повторных эпизодах такой реакции.
Причиной изучения явления первичного стрессорного по-
вреждения сердца послужили данные клиники, которые пока-
зали. что у пациентов наблюдали изменения ЭКГ, свидетель-
ствующие о наличии ишемических изменений сердца и даже
инфаркта, которые далее (посмертно) не подтверждались гис-
тологическими исследованиями. Оказалось, что такого рода
изменения ЭКГ дают повреждения миокарда двух видов:
• ишемическое повреждение при инфаркте, которое характе-
ризуется коагуляционным некрозом миофибрилл сердеч-
ной мышцы без выраженных полос сокращения и поли-
морфно-нуклеарным клеточным ответом. При этом
клетки миокарда гибнут в состоянии расслабления;
• повреждение, которое отличается от инфарктного и ха-
рактеризуется гиперсокращением участков миокарда с
выраженными полосами сокращения миофибрилл и по-
явлением гранулярности, т.е. так называемая миофиб-
риллярная дегенерация [Samuels, 1987]. Повреждения
миокарда второго рода ранее были описаны Г.Селье в
эксперименте при эмоциональном стрессе разных моде-
*The Concept of heart failure from Avicenna to Albertini. — Cambridge:
Harvard University Press. 1980. — P. 6.
260
лей. Такие же повреждения наблюдали у животных при
действии больших доз катехоламинов, а также при силь-
ном нейроадренергическом воздействии на сердце, вы-
званном электростимуляцией симпатических звездчатых
узлов и зон гипоталамуса, ответственных за симпатичес-
кую, но не парасимпатическую регуляцию сердца [Samu-
els М.А., 1987]. Сходные повреждения находили у боль-
ных с повреждениями ЦНС и интерцеребральными ге-
моррагиями, не страдающих сердечными заболеваниями.
Таким образом, было сделано заключение, что указанные
контрактурные повреждения сердечной мышцы связаны с мас-
сированным адренергическим действием на сердце, т.е. являют-
ся нейрогенными.
Впервые первичное стрессорное повреждение сердца у
людей, явно перенесших эмоциональный стресс, достаточно
убедительно было показано при ретроспективном обследова-
нии 15 жертв нападения, которые при этом погибли, но не
имели травм внутренних органов [Cebelin М., Hirsch C.S.,
1980]. У 11 из них были обнаружены признаки дегенеративно-
го поражения миофибрилл сердечной мышцы в состоянии ги-
персокращения, которое было охарактеризовано авторами как
«стрессорная кардиомиопатия». Эти люди в анамнезе не
имели каких-либо заболеваний сердца.
Первую попытку количественно оценить повреждение
структур сердца при стрессе в эксперименте осуществили
D.Miller и S. Malov (1977). При электроболевом стрессе они
оценивали повреждение миокарда по выходу из кардиоцитов
ферментов — маркеров повреждения клеточных мембран и по
включению в структуры миокарда меченного технецием ра-
диоактивного пирофосфата. Было показано, что выход фер-
ментов после 4—16 ч стрессорного воздействия был увеличен
в 2—3 раза, а величина включения технеция зависела от
«дозы» стресса: чем длительнее было стрессорное воздействие,
тем больше было включение, а следовательно, и повреждение
миокарда.
Изучение механизма данного явления проводилось весьма
активно в 70—80-е годы XX столетия в комплексных физиоло-
гических, биохимических и цитологических исследованиях на
животных с использованием модели эмоционально-болевого
стресса, сходного с «неврозом тревоги» у человека, и иммоби-
лизационного стресса, т.е. стресса «неволи, лишения свободы»
(Меерсон Ф.З., 1984; Меерсон Ф.З., Пшенникова М.Г., 1988,
1989]. В этих исследованиях было показано, что при увеличе-
нии интенсивности эмоционального стресса адаптивные эф-
фекты стресс-реакции могут превращаться в повреждающие и
в результате может развиваться повреждение сердечной
мышцы.
261
Так, в серии экспериментов, проведенных нами совместно
с Е.Я.Воронцовой и Ф.З.Меерсоном на изолированной сер-
дечной мышце крыс, впервые было установлено явление пост-
стрессорной ригидности миокарда, развивающейся в результате
длительного (6-часового) эмоционально-болевого стрессорно-
го воздействия (ЭБС). Это явление выражалось в резком
уменьшении растяжимости миокарда, нарушении механизма
Франка—Старлинга и депрессии силы сокращения. Стрессор-
ное воздействие приводит также к снижению устойчивости
миокарда к гипоксии и перегрузке кальцием, что выражается в
ускорении развития и увеличении степени гипоксической и
кальциевой контрактур и связано с нарушением энергетичес-
кого обеспечения процессов сокращения и расслабления. Вы-
яснилось, что эти стрессорные нарушения сократительной
функции миокарда эффективно предупреждаются блокатора-
ми адренорецепторов, а также блокаторами фосфолипаз и
липаз; это свидетельствует о том, что они имеют адренерги-
ческую природу и в значительной степени связаны с актива-
цией липотропного адаптивного эффекта стресс-реакции.
Нарушения сократимости и расслабления, вызываемые ли-
потропным эффектом стресс-реакции, связаны с повреждени-
ем клеточных мембран сердечной мышцы и перегрузкой кар-
диоцитов Са2+. Так, в исследованиях В.А.Фролова с примене-
нием электронной микроскопии было показано, что при ин-
тенсивном ЭБС у крыс наблюдаются повреждения сарколем-
мы и внутриклеточных мембран. Повреждения мембран вле-
кут за собой нарушение транспорта катионов в клетке и в том
числе накопление Са2+ в саркоплазме кардиоцитов. Об этом
свидетельствуют результаты исследований сердечной мышцы
методом электронно-микроскопической цитохимии с приме-
нением пироантимоната калия, выявляющего локализацию
Са2+. Увеличение содержания кальция в кардиомиоцитах со-
ставляет, как известно, общее звено патогенеза самых различ-
ных форм повреждения сердечной мышцы — от наследствен-
ных кардиопатий до недостаточности гипертрофированного
сердца. Именно это явление, очевидно, и приводит к разви-
тию стрессорных очаговых контрактурных повреждений мио-
карда, продемонстрированных в морфологических исследова-
ниях и являющихся причиной описанного выше феномена
постстрессорной ригидности сердечной мышцы.
Наблюдаются также стрессорные повреждения проводя-
щей системы сердца [Усынин А.Ф., 1994]. Эти повреждения,
несомненно, играют ключевую роль в возникновении пост-
стрессорной электрической гетерогенности миокарда, пост-
стрессорных нарушений электрической стабильности сердца и
аритмий.
Приведенные выше данные согласуются с результатами
биохимических исследований, в которых было показано, что
262
стрессорное воздействие вызывало нарушение процессов
окисления и фосфорилирования в митохондриях, снижение
содержания креатинфосфата и активности креатинфосфоки-
назы в миокарде. При этом снижались содержание креатин-
фосфата, активность общей креатинфосфокиназы миокарда и
активность этого фермента в митохондриях. Эти изменения
сочетались с резким падением содержания гликогена и ско-
рости его ресинтеза в миокарде. В совокупности эти данные
указывают на постстрессорные нарушения энергетического
обеспечения сократительной функции сердца. Действительно,
было показано, что добавление глюкозы и стимуляция синтеза
гликогена ограничивали стрессорную ригидность и нарушение
сократительной функции миокарда [Меерсон Ф.З., 1993].
Исследования позволили обосновать представление о па-
тогенетической цепи первичного стрессорного повреждения
сердца [Meerson F.Z., 1991]. Схема 5.7 отражает это представ-
ление. На этой схеме указаны семь (I—VII) звеньев патогенеза
первичного стрессорного повреждения сердца; I и II звенья
составляют активация стресс-системы, стресс-реакция, сопро-
вождающий ее выброс стресс-гормонов и их действие на серд-
це. В результате этих событий происходят три группы наибо-
лее существенных изменений на уровне кардиомиоцитов, ко-
торые составляют III звено патогенеза, а именно: увеличение
вхождения Са2+ в кард ио миоциты, мобилизация и уменьшение
резервов гликогена и фосфокреатина и, наконец, реализация
липидной триады. Эти изменения и последующие за ними
(как следствие) процессы в кардиомиоцитах, составляющие
IV—VII звенья патогенеза, подробно рассмотрены выше в раз-
деле 5.1.5 при описании первых трех адаптивных эффектов
стресс-реакции. Центральное звено патогенеза составляет воз-
никающий в результате данных процессов избыток Са2+ в ци-
топлазме кардиомиоцитов (V звено). Это важное явление
имеет два следствия. Во-первых, Са2+ может активировать
процессы, составляющие липидную триаду (штриховая стрел-
ка на схеме 5.7), и, таким образом, замыкается порочный круг,
усугубляющий повреждение миокарда. Во-вторых, избыток
кальция обладает самостоятельным повреждающим действи-
ем, вызывая внутри клетки развитие комплекса изменений
(кальциевую триаду), слагающегося из контрактуры миофиб-
рилл, нарушения функции перегруженных кальцием мито-
хондрий и активации протеаз в миофибриллах и фосфолипаз в
митохондриях, что также усугубляет возникшее повреждение
(VI звено).
Важно отметить, что в большинстве кардиомиоцитов эта
цепочка из первых шести звеньев останавливается на разных
этапах благодаря включению защитных механизмов. В этих
случаях рассмотренные изменения претерпевают «обратное
развитие» и стрессорные нарушения сердечной функции явля-
263
Схема 5.7. Патогенез первичного стрессорного повреждения
сердца [Meerson F., 1991]
СТРЕССОР
Пояснение в тексте.
Факторы защиты
\ Медиаторы цен-
] тральных стресс-
лимитирующих
^систем
ГБлокаторы ре-
J цептороа. Меди-
| агоры локальных
стресс-лимити-
[^рующих систем
f Мембранопро-
текторы: инги-
биторы СРО,
липаз, фосфо-
\ липаз; блока-
л торы вхождения
Са2’, глюкоза,
кофакторы ре-
синтеза глико-
^гена
f
J Ингибиторы про-
теаз и нуклеаз
Ингибиторы
кальмодулина
264
ются транзиторными. Однако в некоторых кардиомиоцитах
все же развиваются необратимые контрактурные изменения
миофибрилл, что приводит к возникновению некробиоза и
мелкоочагового некоронарогенного кардиосклероза (конечное
VII звено процесса на схеме 5.7).
Следует подчеркнуть, что эта схема отражает только прямое
стрессорное повреждение кардиомиоцитов, оставляя в стороне
(за рамками схемы) такие патогенетические механизмы стрес-
сорного повреждения сердца в целом, как возможный стрес-
сорный спазм и тромбоз коронарных сосудов, вызванная
стрессом нагрузка на сердце и коронарный склероз, обуслов-
ленный стрессорным повреждением печени и соответственно
дислипопротеидемией. Во всех этих случаях стресс может вы-
зывать абсолютную или относительную ишемию миокарда,
которая становится непосредственным повреждающим факто-
ром. Этот аспект стрессорного повреждения мы рассмотрим
далее при описании роли стресса в патогенезе ИБС. Здесь же
мы акцентируем внимание на первичном стрессорном, т.е.
главным образом адренергическом, повреждении кардиомио-
цитов.
Какое клиническое значение может иметь это поврежде-
ние? Во-первых, все рассмотренные изменения структуры, ме-
таболизма и функции наблюдались после завершения стрес-
сорного воздействия. Они являются не просто реакцией на
стрессор, а представляют собой относительно стойкие послед-
ствия повреждений, возникающих во время стрессорного воз-
действия. Этот факт в совокупности с клиническими наблю-
дениями дает основание полагать, что именно такие, относи-
тельно стойкие нарушения метаболизма и функции, сохра-
няющиеся после того, как действие стрессора миновало, и на-
капливающиеся от одного стрессорного эпизода к другому,
могут играть роль в развитии первичного некоронарогенного
кардиосклероза и хронической сердечной недостаточности, ко-
торые нередко возникают у людей, ранее не имеющих наруше-
ний системы кровообращения [Меерсон Ф.З., 1984, 1993].
Во-вторых, выраженные стрессорные повреждения струк-
туры проводящей системы сердца могут быть причиной воз-
никновения различных блоков проведения, например блока
правой ножки пучка Гиса у ранее практически здоровых
людей после перенесенного стресса. Возможно также, что не-
которые так называемые идиопатические аритмии некорона-
рогенной природы имеют стрессорное происхождение и связа-
ны с указанными нарушениями проводимости миокарда и по-
вреждениями водителей ритма.
В-третьих (см. правую часть схемы 5.7), возможные средст-
ва защиты от стрессорного повреждения сердца, направлен-
ные на предупреждение или коррекцию определенных звеньев
патогенеза, представляют собой прежде всего медиаторы
265
стресс-лимитирующих систем, ограничивающие интенсив-
ность стресс-реакции, а также средства, блокирующие цепь
патогенеза на разных этапах процесса.
Поиск факторов, повышающих эффективность стресс-ли-
митирующих систем, несомненно, обеспечит прогресс в про-
филактике и лечении стрессорных повреждений. Фактором
такого рода является адаптация к повторным неповреждаю-
щим стрессорным воздействиям, а также к другим факторам
среды, которая повышает мощность природных стресс-лими-
тирующих систем организма и повышает его резистентность к
повреждениям.
5.2.3.1.1. Ишемическая болезнь сердца и инфаркт миокарда
Вопрос о роли эмоционального стресса в патогенезе ИБС
уже давно волновал клиницистов и патофизиологов [Чазов
Е.И., 1975; Lown В., De Silva R., 1978]. Это, безусловно, слож-
ный вопрос, и чтобы определить, как стресс-реакция влияет
на развитие и течение ИБС, несомненно, следовало бы внача-
ле проанализировать патогенетическую цепь ишемического
повреждения как таковую (изолированно от стресса). Однако
эта проблема достаточно подробно обсуждается во всех руко-
водствах по кардиологии и патофизиологии кровообращения,
в монографиях и обзорах, поэтому мы сразу перейдем к рас-
смотрению роли стресса в патогенезе ишемического повреж-
дения миокарда.
Имеющиеся данные позволяют выделить 8 основных пато-
генетических механизмов, посредством которых эмоциональ-
ней стресс способствует развитию ИБС и инфаркта миокарда.
Первым патогенетическим механизмом влияния стресса на
патогенез ИБС являются стрессорная гиперхолестеринемия и
атерогенная дислипопротеидемия.
Наблюдения клиницистов показывают, что трудная психо-
социальная стрессорная ситуация, в которую попадают люди,
вызывает у них подобную дислипопротеидемию, т.е. увеличе-
ние в крови уровня холестерина во фракции липопротеидов
низкой плотности (ЛПНП) и рост индекса атерогенности.
В экспериментах на животных было четко показано, что эмо-
циональный стресс, как интенсивный однократный [Твердо-
хлиб В.П. и др., 1988], так и хронический [Clarkson Т.В. et al.,
1987] приводят к атерогенной дислипопротеидемии, которая
при хроническом стрессе сопровождается развитием коронар-
ного атеросклероза.
Как показано в экспериментах на обезьянах, у «подчи-
ненных» особей, т.е. занимающих в колонии подчиненное по-
ложение, что сопровождается, как известно, хроническим
социальным (эмоциональным) стрессом, отношение в крови
266
содержания общего холестерина крови к содержанию хо-
лестерина во фракции липопротеидов высокой плотности
(Хобш/Хлпвп) в 2 раза больше, чем у «доминирующих» особей,
которые в меньшей степени подвергаются социальному стрес-
су. Это сопровождается у «подчиненных» обезьян увеличени-
ем частоты развития коронарных стенозов по сравнению с
особями соответствующего пола, относящимися к «доминиру-
ющим». Это означает, что эмоциональный стресс приводит к
увеличению в крови доли атерогенных липопротеидов и как
следствие — к развитию коронарного атеросклероза [Clark-
son Т.В. et al., 1987].
Основным механизмом, через который стресс приводит к
атерогенной дислипопротеидемии и способствует развитию
атеросклероза, можно считать стрессорное поражение пече-
ни (главного органа обмена холестерина).
В пользу этого положения свидетельствуют данные о том,
что так называемая семейная гиперхолестеринемия, приводя-
щая к коронарному атеросклерозу, связана с генетическим де-
фектом печени, проявляющимся в нарушении механизма за-
хвата и катаболизма атерогенных ЛПНП [Репин В.С., Су-
хих Г.Т., 1998]. О поражении печени и нарушении ее функции
при стрессе свидетельствуют и экспериментальные данные
[Твердохлиб В.П. и др. 1988], которые показали, что при ЭБС
уменьшение в крови содержания ЛПВП связано с резким па-
дением в ней активности фермента лецитинхолестеринацетил-
трансферазы, который, как известно, катализирует образова-
ние эфиров холестерина из свободного холестерина и тем
самым ускоряет процесс формирования ЛПВП. Как показано,
эстерифицирующая активность этого фермента через 2 ч пос-
ле 6-часового ЭБС уменьшается почти в 3 раза и затем посте-
пенно восстанавливается в течение суток. Данный фермент
является короткоживущим белком, он синтезируется с высо-
кой скоростью в печени и затем переносится в кровь, где осу-
ществляет липопротеиновую конверсию. Поэтому постстрес-
сорное падение активности фермента в крови явно указывает
на нарушение его биосинтеза в печени, что может быть связа-
но со стрессорным повреждением этого органа. На повреж-
дение печени прямо указывает резкое повышение в крови
содержания фермента фруктозо-1,6-дифосфатальдолазы, спе-
цифически отражающее повреждение печеночных клеток.
Клинические данные также свидетельствуют, что стрессорная
ситуация на работе приводит к нарушению функции печени у
людей [Kaneko М. et al., 1996].
Повреждение печени при стрессе, очевидно, обусловлено
чрезмерной активацией СРО в этом органе как результат ли-
потропного эффекта стресс-реакции (см. раздел 5.1.5; схему
5.5). Действительно, показано, что появление в крови фрукто-
267
зо-1,6-дифосфатальдолазы сопутствует значительной актива-
ции СРО в печени (повышению содержания малонового диа-
льдегида на фоне снижения активности антиоксидантного
фермента супероксиддисмутазы). Стрессорное повреждение
печени приводит к нарушению в ней процессов окисления хо-
лестерина, превращения его в желчные кислоты и выведения
из организма, что вызывает в свою очередь гиперхолестери-
немию.
Таким образом, можно заключить, что первый патогенети-
ческий механизм влияния стресса на развитие ИБС, а имен-
но гиперхолестеринемия и атерогенная дислипопротеидемия,
обусловлен глубокими стрессорными нарушениями структу-
ры и функции печени и нарушениями процесса удаления хо-
лестерина из организма.
В результате этого даже при отсутствии избытка холестери-
на в пище стресс может приводить к развитию стенозирующе-
го коронарного склероза и играет важную роль в развитии
ИБС.
Второй и наиболее опасный патогенетический механизм, по-
средством которого стресс может потенцировать тяжелые пос-
ледствия ишемии и инфаркта миокарда, состоит в первичном
стрессорном повреждении миокарда и активации адренергичес-
ких влияний на сердце.
Первичное стрессорное повреждение сердца может иметь
место до эпизода острой ишемии миокарда в результате ранее
перенесенных стресс-реакций. В этом случае очаги повреж-
денного миокарда усугубят возникновение ишемических арит-
мий и нарушение сократительной функции сердца. Первичное
стрессорное повреждение миокарда, т.е. повреждение некоро-
нарного происхождения, может возникать и при ишемии в не-
ишемизированных участках сердечной мышцы в результате
стресс-реакции на боль и страх смерти, сопутствующих ишемии
(см. раздел 5.2.3.1, схему 5.7).
Можно выделить два основных компонента отягчающего
влияния стрессорного повреждения миокарда при ишемии:
аритмогенный эффект стресса (усиление ишемических арит-
мий сердца вплоть до фибрилляции) и нарушение сократи-
тельной функции неишемизированных отделов сердца за счет
стресс-индуцированного повреждения этих отделов.
Аритмогенный эффект стресса. Аритмогенное действие
нейрогенного фактора при ишемии давно хорошо известно.
Ишемия миокарда, вызываемая в эксперименте перевязкой
левой коронарной артерии в условиях глубокого наркоза, при-
водила к аритмиям лишь в 6 % случаев. Стимуляция заднего
гипоталамуса, осуществляющего, как известно, контроль сис-
темы кровообращения, увеличивала при этом число случаев
аритмий в 10 раз [Satinsky J. et al., 1971]. Стрессорные ситуа-
268
ции увеличивают число случаев фибрилляции желудочков при
коронароокклюзии в 3 раза и в 3 раза снижают порог фибрил-
ляции [Verner R.L. et al., 1984]. Интересные данные были по-
лучены В.Lown и сотр. [Lown В., De Silva R., 1978]. Они ис-
пользовали методику И.П.Павлова: помещали собак в станок
и вырабатывали условный рефлекс на электроболевое воздей-
ствие. После этого одно помещение в станок давало стресс-ре-
акцию, оцениваемую по росту уровня катехоламинов в крови.
Такой стресс существенно снижал порог аритмий и фибрилля-
ции сердца, т.е. повышал уязвимость сердца к аритмогенным
факторам.
Аритмогенные эффекты стресса предупреждаются удале-
нием звездчатых симпатических узлов или адреноблокатора-
ми. В то же время стимуляция симпатических сердечных нер-
вов и симпатических узлов репродуцирует аритмогенные эф-
фекты стимуляции среднего мозга даже в условиях стабилизи-
рованных АД и частоты сердцебиений. Клинические исследо-
вания установили наличие возбуждения симпатоадреналового
отдела стресс-системы во время приступов аритмии у людей,
что сопровождалось увеличением экскреции катехоламинов, а
также увеличением продукции цАМФ и уменьшением продук-
ции цГМФ [Дорофеев Г.И. и др., 1985]. Таким образом, не вы-
зывает сомнений, что возбуждение симпатических нервных
центров и адренергическое влияние на сердце играют ключе-
вую роль в патогенезе нейрогенных аритмий.
При одновременной регистрации биоэлектрической актив-
ности сердца и фронтальной коры при аритмиях, вызванных
стрессом или острой ишемией, было установлено, что как дей-
ствие экзогенного стрессора, так и стресс-реакция, спровоци-
рованная ишемической болью (т.е. эндогенным стрессором),
сопровождаются возбуждением определенной зоны фронталь-
ной коры. И в обоих случаях (при стрессе и ишемии) вся цепь
последующих событий, приводящих к фибрилляции сердца,
оказывается кортикально обусловленной [Косицкий Г.И. и
др., 1985; Skinner J.E., 1985]. Путь кора — сердце еще не весь
полностью прослежен. Вместе с тем исследования на живот-
ных и клинические наблюдения показали, что фронтальная
кора по меньшей мере тремя путями может регулировать со-
стояние сердца и системы кровообращения [Косицкий Г.И. и
др., 1985; Skinner J.E., 1985; Blair R.W., 1985, и др.]. Схематич-
но эти пути в общем виде представлены на рис. 5.3.
Первый путь — кортикально-таламическая система, ко-
торая контролирует сенсорные каналы, т.е. выход информа-
ции от сердца и других висцеральных органов на уровень коры
(I на рис. 5.3). Второй путь — фронтальная кора — лимбичес-
кая система, от фронтальной коры к височной доле и ядрам
амигдалы (извилина faciculus). Он обеспечивает эффекторный
выход коры на ядра ствола мозга опосредованно через ядра
269
ХБ
Рис. 5.3. Гипотетическая схема аритмогенного влияния на сердце
стресс-реакции, возникающей при действии внешнего эмоциогенно-
го стрессора, а также в ответ на боль, вызываемую ишемией (внут-
ренний стрессор).
I — кортико-таламический путь; II — кортико-лимбический путь; III — корти-
ко-стволовый путь; ФК — фронтальная зона коры больших полушарий; СП —
синее пятно; NTS — ядра tractus solitarius; ББ — блокада аритмогенного стрес-
сорного влияния введением бета-адреноблокаторов; ХБ — холодовая блокада
аритмогенного стрессорного влияния. Штриховыми линиями обозначены аф-
ферентные пути от сердца в ЦНС [Blair R.W., 1985; Skinner J.E., 1985]. Поясне-
ния в тексте.
амигдалы, которые в свою очередь связаны с гипоталамусом
(II на рис. 5.3). Третий, триггерный, путь (III на рис. 5.3),
наиболее существенный для понимания роли стресс-реакции
в генезе аритмий, связывает фронтальную кору через субтала-
270
мус и дорсальный гипоталамус с ядрами ствола мозга, непо-
средственно регулирующими систему кровообращения, т.е.
кортико-стволовый путь. Данный путь регулирует висцераль-
ные компоненты стресс-реакции, в том числе влияет на уязви-
мость сердца по отношению к аритмогенным факторам. Этот
путь соединен обширными коллатералями с ядрами гипотала-
муса (главным образом паравентрикулярным). Информация с
этих коллатералей передается из гипоталамуса терминалями
нейронов гипоталамуса в области ствола мозга, а именно в
ядра tractus solitarius, моторные кардиоваскулярные ядра, п.
ambiguus, контролирующие сердце и кровообращение. Эти же
отделы гипоталамуса, т.е. получающие информацию из коры
(по третьему пути), получают также терминали аксонов от
амигдалы. Таким образом, гипоталамус, ключевой компонент
центрального звена стресс-системы, является центром путей,
реализующих аритмогенное влияние коры головного мозга
при экзогенно-индуцируемом стрессе и ишемии (эндогенно
вызываемом стрессе в ответ на ишемическую боль). Наиболее
существенные результаты, которые выявили роль этих путей в
механизме аритмий и фибрилляции сердца, были получены в
исследованиях Джеймса Скиннера и его коллег из медицин-
ского колледжа в Хьюстоне в середине 80-х годов [Skinner J.E.,
1985, и др.[. Было установлено, что при окклюзии коронарной
артерии и острой ишемии миокарда у свиней происходит воз-
буждение указанной выше зоны фронтальной коры, законо-
мерно сопровождающееся фибрилляцией сердца. Холодовая
блокада подкорковой зоны, амигдалы, а также блокада рас-
смотренного выше кортико-стволового пути с помощью ин-
терцеребрального введения p-адреноблокатора пропранолола
предупреждают фибрилляцию, остановку сердца и гибель жи-
вотных, несмотря на ишемию миокарда.
Иными словами, было установлено, что ишемия миокарда —
результат окклюзии коронарной артерии, т.е. результат ме-
ханического выключения коронарного кровотока, а реакция
на ишемию в форме фибрилляции и остановки сердца — ре-
зультат сложных межцентральных отношений, реализую-
щихся на уровне головного мозга.
Этот важный вывод убедительно определяет существо вто-
рого патогенетического механизма влияния стресса на разви-
тие ишемического повреждения миокарда. Клинической ил-
люстрацией к этому положению служат данные о том, что у
людей с повреждениями (выключением) лобных долей пол-
ностью отсутствуют вегетативные реакции на все психологи-
чески значимые стимулы (т.е. на эмоциональные стрессоры);
эти люди вообще не подвержены стрессу [Tauber H.L., 1964].
Механизм аритмогенного действия стресс-реакции прибли-
женно можно представить следующим образом. Обратимся
271
вновь к рис. 5.3. Стресс-реакция может возникать в ответ на
внешний стрессор, а также на боль и чувство страха, сопро-
вождающие ишемию миокарда (внутренний стрессор).
Сигнал о внешнем стрессоре воспринимается соответствую-
щими рецепторами (обобщенно изображенными на рисунке в
виде глаза) и по таламо-кортикальной системе (I путь влияния
коры на сердце, см. рис. 5.3) передается в таламус и далее в
воспринимающие нейроны основной коры больших полуша-
рий, отвечающие за вход в кору. Там сигнал «переключается»
и поступает во фронтальную кору. Из фронтальной коры на-
чинается кортико-стволовый путь, как уже было показано
выше (III путь на рис. 5.3), который соединяет фронтальную
кору с таламусом, гипоталамусом и ядрами ствола мозга, не-
посредственно связанными с регуляцией сердца. Основным
звеном этого пути, как указывалось выше, является гипотала-
мус, который «собирает» информацию от вышележащих отде-
лов головного мозга, а также с периферии, в том числе от
сердца. Из гипоталамуса информация при участии стволовых
ядер — «синего пятна», n. ambiguus и др. — поступает в нейро-
ны ядер продолговатого и спинного мозга, осуществляющих
симпатическую и парасимпатическую иннервацию сердца.
При этом именно преобладание симпатического выхода на
сердце создает аритмогенную ситуацию. Электрическая сти-
муляция, или функциональная блокада этого триггерного
пути, вызывает соответственно либо фибрилляцию сердца,
либо ее предотвращение (в случае ишемии). На рис. 5.3 пока-
заны варианты такой блокады, использованные в исследова-
ниях J.E.Skinner и сотр., о которых мы говорили выше [Skin-
ner J.E., Reed J.С., 1981; Skinner J.E., 1985]. Холодовая блокада
подкорковой зоны и амигдалы вызывает предупреждение воз-
никновения аритмий при эмоциональном стрессе, а также
предупреждает возникновение фибрилляции сердца и гибель
животных при острой ишемии сердца. Интерцеребральное
введение p-адреноблокаторов может воздействовать непосред-
ственно на механизм инициации активности в триггерном
пути или предупреждать переключение центральной информа-
ции в триггерном пути на вегетативный выход либо и то, и
другое.
При действии внутреннего стрессора, т.е. при боли, вы-
званной ишемией, болевой сигнал, воспринятый рецепторами
сердца, передается по афферентным волокнам, идущим от
сердца в составе блуждающего нерва, а также по симпатичес-
ким волокнам, в верхние отделы ЦНС по спиноталамическо-
му и спиноретикулярному трактам. Эта информация попадает
в таламус и гипоталамус. Из таламуса по кортико-таламичес-
кой системе, а возможно, также и непосредственно из гипота-
ламуса, сигнал поступает в кору больших полушарий. Далее
«срабатывает» рассмотренная выше схема активации триггер-
272
ного фронтокортико-стволового эфферентного пути к сердцу.
Кроме того, есть основания полагать, что в нейронах заднего
гипоталамуса афферентная информация переключается сразу
на эфферентные симпатические нейроны, аксоны которых по-
ступают в спинальный тракт и являются преганглионарными
волокнами симпатических ганглиев, иннервирующих сердце.
Разумеется, рассмотренный механизм реально намного
сложнее и не все его звенья исследованы. Однако не вызывает
сомнений следующее основное положение, обоснованное
представленными выше исследованиями.
Стресс-реакция независимо от того, вызвана ли она внеш-
ним стрессором или внутренним, является фактором, прово-
цирующим аритмии и фибрилляцию сердца или отягчающим
их протекание при ишемическом поражении миокарда.
Существенно, что этот механизм, первично обусловлен-
ный эмоциональной стрессорной ситуацией окружающего
мира или эндогенным стрессором боли при ишемии, может
«закрепиться» благодаря формированию патологической до-
минанты (по А.А.Ухтомскому) или патологической системы
«застойно» возбужденных центров [Крыжановский Г.Н.,
1997], а также благодаря возникновению при стрессе «дли-
тельного циклического движения процессов возбуждения»
между кортико-лимбическими структурами [Судаков К.В.,
1997а].
Следует подчеркнуть роль адренергического эффекта
стресс-реакции как фактора, приводящего к возникновению или
усугублению сердечных аритмий при ишемии. Во-первых, увели-
чение адренергического влияния на водители ритма сердца
при стрессе создает преобладание адренергического влияния
над холинергическим и тем самым вызывает асинхронизм в
работе водителей ритма. Уже одно это может вызывать арит-
мии сердца вплоть до фибрилляции даже в условиях отсутст-
вия ишемии. В условиях ишемии этот механизм повышает
уязвимость сердца к ишемическим аритмиям и усугубляет тя-
жесть нарушений ритма вплоть до летального исхода. Во-вто-
рых, однократно перенесенная стресс-реакция большой ин-
тенсивности или повторные эпизоды стресса за счет усиления
адренергического влияния на сердце вызывают, как уже было
показано выше, первичное стрессорное повреждение микро-
структур сердечной мышцы и очаговые некротические по-
вреждения. Этот комплекс изменений знаменует собой нару-
шение функционирования мембранного аппарата кардиоци-
тов, который осуществляет генерацию и проведение возбужде-
ния и может играть существенную роль в возникновении эк-
топических очагов, из которых исходят «преждевременные»
импульсы возбуждения, и очагов функционального блока про-
ведения, что при инфаркте и острой ишемии может приводить
18—1385 273
к возникновению хорошо известного феномена reentry — при-
знанную основу желудочковой тахикардии и фибрилляции
сердца.
Нарушение сократительной функции неишемизированных
отделов миокарда. При описании первичного стрессорного
повреждения выше было показано, что это повреждение ха-
рактеризуется постстрессорной ригидностью сердечной
мышцы и снижением ее сократительной функции. Оказалось,
что при экспериментальном инфаркте миокарда левого желу-
дочка у крыс, вызываемого, по Г.Селье, перевязкой левой
нисходящей коронарной артерии, в неишемизированных от-
делах сердца (предсердиях) также развиваются ригидность, на-
рушение сократительной функции и снижение резистентности
к высоким концентрациям кальция и гипоксии. Иными сло-
вами, при ишемическом повреждении сердца в неишемизиро-
ванных отделах возникают те же нарушения функции и струк-
туры сердечной мышцы, что и в интактном сердце после ин-
тенсивного стрессорного воздействия. Предварительное вве-
дение p-блокатора индерала, предупреждающее избыточное
адренергическое влияние на сердце при стрессе, в высокой
степени защищает сократительную функцию неишемизиро-
ванных предсердий при инфаркте левого желудочка. Этот за-
щитный эффект, как и в случае ЭБС, выражался в значитель-
ном уменьшении ригидности и снижения сократительной
функции предсердия, а также в повышении его резистентнос-
ти к гипоксии и перегрузке кальцием (см. раздел 5.2.3.1).
Таким образом, при ишемическом поражении сердца
стресс-реакция, вызываемая внешним стрессором (окружаю-
щей средой) или внутренним стрессором (болью в сердце и
страхом смерти), является патологическим фактором, вызыва-
ющим или потенцирующим нарушения ритма сердца и деп-
рессию сократительной функции неишемизированных отде-
лов миокарда.
Третий патогенетический механизм, через который реализу-
ется роль стресса в патогенезе ИБС и инфаркта миокарда, со-
стоит в том, что сильный адренергический компонент стресс-
реакции, сопутствующей острой ишемии, может приводить к
спазму гладкой мускулатуры анатомически интактных коро-
нарных артерий, и этот достаточно стойкий спазм становится
причиной вторичного ишемического поражения миокарда.
При рассмотрении адаптивных эффектов стресс-реакции
мы отмечали, что вызываемая катехоламинами через р-адре-
норецепторы сосудов «рабочая гиперемия» миокарда (дилата-
ция коронарных сосудов) при увеличении интенсивности и
длительности стресс-реакции может сменяться коронарокон-
стрикцией. Это происходит в связи с десенситизацией р-адре-
норецепторов большими дозами катехоламинов и реализацией
эффекта катехоламинов через констрикторные а,-адреноре-
274
цепторы. В этом смысле очень показательны исследования
[Simons М., Downing S.E., 1985], в которых длительно вводили
животным внутривенно НА и регистрировали коронарный
кровоток. Авторы показали, что вначале в ответ на НА крово-
ток увеличивался и возвращался к исходному уровню, а затем
сопротивление коронарного русла возрастало вдвое, кровоток
снижался, и 48 ч спустя развивалось ишемическое поврежде-
ние сердца. При этом на фоне блокатора а-адренорецепторов
фентоламина коронароспазм и ишемическое поражение не
возникали.
Важное значение в регуляции коронарного кровотока, как
показывают исследования последних 5 лет, имеет NO — мощ-
ный вазодилататор. При умеренной стресс-реакции продукция
NO возрастает [Малышев И.Ю., Манухина Е.Б., 1998], и это
играет ключевую роль в реализации «рабочей гиперемии». При
длительном и интенсивном стрессорном воздействии генера-
ция NO может падать, и этот фактор, очевидно, — одна из ре-
шающих причин возникновения стрессорного коронароспаз-
ма. Поэтому при стенокардии помогают нитроглицерин и дру-
гие нитропрепараты, являющиеся донорами NO.
Установлено, что длительный ЭБС вызывает у крыс суще-
ственное уменьшение отношения содержания в крови корона-
родилататора простациклина (ПГ12), коронароконстриктора и
стимулятора тромбообразования тромбоксана А2 (ТХА2), т.е.
ПГ12/ТХА2, а также отношения коронародилататора ПГЕ, и
коронароконстриктора ПГР2а, т.е. ПГЕ|/ПГЕ2а. Эти измене-
ния могут играть роль в возникновении или потенциации ок-
клюзии коронарных сосудов и усугублении ишемии [Пшенни-
кова М.Г. и др., 1992].
Таким образом, стресс создает благоприятные условия для
возникновения коронароспазма. Врожденная сниженная ак-
тивность стресс-лимитирующих систем предрасполагает к
стрессорным повреждениям, в том числе к нарушению коро-
нарного кровотока при стрессе. Напротив, повышение мощ-
ности этих систем является важным фактором профилактики
стрессорных повреждений и стресс-индуцированного усугуб-
ления ИБС.
Четвертый патогенетический механизм влияния стресса на
патогенез ИБС и инфаркта миокарда состоит в том, что
стресс-индуцированный «выброс» катехоламинов в кровь потен-
цирует свертывание крови и тромбоз коронарных сосудов. След-
ствием возникающей при этом агрегации тромбоцитов являет-
ся выделение из них мощных вазоактивных веществ, особенно
ТХА2, серотонина и гистамина, которые заведомо усиливают
спазм и в сочетании с тромбозом делают его более опасным.
Это имеет место чаще всего в тех зонах коронарного русла, где
изначально имеются атеросклеротические изменения. Дейст-
вительно, клинические наблюдения свидетельствуют о том,
18* 275
что для больных коронарной болезнью интенсивный эмоцио-
нальный («ментальный») стресс очень опасен: он может вызы-
вать ишемию и инфаркт миокарда. Так, холтеровское монито-
рирование ЭКГ у 112 мужчин и 14 женщин (средний возраст
56 лет), страдающих коронарной болезнью, показало, что мен-
тальный стресс вызывал у них резкие ишемические изменения
[Morris J.J. et al., 1996]. При этом важную роль в роковом
стрессорном усугублении коронарной болезни играет стресс-
индуцированный тромбоз. У военных летчиков один ответст-
венный полет на истребителе вызывает повышение активнос-
ти тромбина в 2 раза [Biondi G. et al., 1996].
Как известно, сильным противодействием тромбозу обла-
дают простациклин и ПГЕ. Они ингибируют агрегацию тром-
боцитов, и, таким образом, стресс-лимитирующая система
протекторных ПГ является не только противоконстрикторной,
но и антитромбозной. Указанное выше стресс-индуцирован-
ное уменьшение отношений ПП2/ТХА2 и ПГЕ|/ПГР2а не толь-
ко способствует коронароспазму, но и, несомненно, потенци-
рует тромбообразование или даже обусловливает его возник-
новение при ишемии.
Острая ишемия и инфаркт миокарда могут быть результатом
сочетанного эффекта таких быстродействующих патогене-
тических механизмов, как спазм и тромбоз, и органических
изменений, вызванных медленно развивающимся атероскле-
розом.
На основании изложенного это означает, что при одинако-
вом атеросклеротическом коронаростенозе острая ишемия
может возникать или, напротив, отсутствовать в зависимости
от состояния нейрогуморальной регуляции сосудов, т.е. от ак-
тивности стресс-системы и стресс-лимитирующих систем.
Стресс-реакция достаточной интенсивности на фоне дефици-
та системы протекторных ПГ и снижения генерации NO будет
сильным потенциатором возникновения острой ишемии. На-
против, активация системы протекторных ПГ и увеличение
мощности системы генерации NO могут способствовать
уменьшению угрозы ишемического повреждения сердца при
коронаростенозе.
Таким образом, вопрос о том, возникнет ли стойкий
спазм, тромбоз и в конечном счете инфаркт миокарда, в высо-
кой степени зависит от состояния таких локальных стресс-ли-
митирующих систем, как системы протекторных ПГ и NO.
Пятый патогенетический механизм, через который реализу-
ется роль стресса в патогенезе ишемического повреждения
сердца, формируется на основе такого компонента стресс-ре-
акции, как гипервентиляция. Гипервентиляция закономерно
влечет за собой увеличение напряжения кислорода в крови и
гипокапнический алкалоз. Эти сдвиги в свою очередь умень-
276
шают коронарный кровоток и повышают тонус коронарных
сосудов в связи со снижением в крови содержания Н+, кото-
рое приводит к увеличению связывания Са2+ с тропонином в
миофибриллярном аппарате миоцитов сосудистой стенки с
последующим их констрикторным сокращением. Гипервенти-
ляция и сопутствующий ей гипокапнический алкалоз вызыва-
ют у больных с ИБС уменьшение коронарного кровотока и
появление приступов стенокардии [Groves В. et al., 1977]. Ха-
рактерно, что эти приступы купировались введением Са2+-
блокатора дилтиазема, который не влиял на газовый состав
крови в коронарном русле, т.е. парциальное давление углекис-
лоты оставалось сниженным, а кислорода — увеличенным.
Эти данные не только указывают на роль стрессорной гипер-
вентиляции в обострении ИБС, но и позволяют вновь под-
черкнуть ключевую роль кальция в механизме повреждения.
Шестой патогенетический механизм состоит в том, что дли-
тельная стресс-реакция снижает резистентность миокарда к ги-
поксии и реоксигенации. Это положение было доказано в иссле-
дованиях, где одновременно оценивались сократительная функ-
ция сердца, периферическое сопротивление в сосудах, АД, а
также ударный и минутный объемы сердца и его работа [Белки-
на Л. М. и др., 1983, и др.]. Оценку состояния сердца и кровооб-
ращения осуществляли в условиях нормоксии, 30-минутной
ишемии и последующей реоксигенации сердца у животных,
предварительно перенесших ЭБС (за 2 ч до тестирования). Ока-
залось, что у перенесших ЭБС животных в условиях нормоксии
наблюдалось лишь незначительное статистически недостовер-
ное ухудшение перечисленных параметров функции сердца.
Иное отмечено в условиях ишемии и особенно реоксигенации
сердца: предварительно перенесенный стресс значительно уве-
личивал вызываемое ишемией падение систолического напря-
жения левого желудочка. В еще большей степени перенесенный
стресс потенцировал реоксигенационные (постишемические)
нарушения сократительной функции сердца.
Таким образом, стресс является фактором, отягчающим
повреждение сердца при ишемии и реоксигенации.
Седьмой патогенетический механизм, благодаря которому
стресс усугубляет ишемическое поражение сердца, состоит в
адренергической мобилизации сократительной функции сердеч-
ной мышцы. В сочетании с регуляторно обусловленным по-
вышением сопротивления сосудистого русла это создает зна-
чительную нагрузку на сердце и может существенно потенци-
ровать ишемическое повреждение не только при спазме или
тромбозе, но и при «простом» атеросклеротическом стенози-
ровании коронарных сосудов.
На первый взгляд, это явление, казалось бы, противоречит
тому, что p-адренергический эффект стресса первично приво-
дит к коронарной вазодилатации. G.H.Mudge и соавт. (1976)
277
объясняли это явление тем, что в условиях предшествующего
стеноза крупной коронарной артерии дополнительная нагруз-
ка не может быть компенсирована за счет регуляторного рас-
слабления лежащей ниже стеноза области коронарного русла,
так как достаточный объем кровотока в этой области все
равно не может быть достигнут из-за уменьшенного стенозом
притока. Это объяснение, хотя и неисчерпывающее, согласу-
ется с очень «красивыми» данными, полученными R.L.Verner
и соавт. (1984) при действии эмоционального стресса на собак
с вживленной манжетой на коронарной артерии. Они показа-
ли, что стресс, вызываемый отнятием пищи, приводил у жи-
вотных без стеноза коронарной артерии к р-адренергическим
эффектам, т.е. к увеличению коронарного кровотока и паде-
нию сопротивления в коронарном русле. У животных, кото-
рым создавали стеноз коронарной артерии пережатием манже-
ты, такой стресс, напротив, приводил к увеличению сопротив-
ления в коронарном русле, что свидетельствовало о стресс-ин-
дуцированной коронароконстрикции.
Таким образом, несмотря на всю сложность механизма
рассматриваемых явлений, факт остается фактом — стресс
может усугублять ишемическое поражение сердца, вызывая ад-
ренергическую мобилизацию его функции.
Восьмой патогенетический механизм, посредством которого
стресс-реакция может вызывать нарушение кровообращения и
приводить к ишемии не только сердца, но и мозга, состоит в
выраженном нарушении сократительной функции и тонуса сосу-
дов. В основе этого положения лежат данные, полученные в
последние годы в экспериментах на изолированных венах и
аорте. Во-первых, было показано, что стресс приводит к
уменьшению сократительной функции мускулатуры воротной
вены и снижению ее адренореактивности, причем подобное
нарушение сократительной функции воротной вены возникает
и при инфаркте миокарда. В данном случае решающим факто-
ром, по-видимому, является стресс-реакция, возникающая в
ответ на боль [Манухина Е.Б., 1988]. Таким образом, стресс-
индуцированное снижение сократительной функции порталь-
ной вены может играть роль в избыточном кровенаполнении
портального русла, возникновении артериальной гиповолемии
и коллаптоидных состояний при тяжелом стрессе и инфаркте
миокарда. Во-вторых, было установлено, что при эмоциональ-
ном стрессе и экспериментальном инфаркте миокарда
[Manukhina Е.В. et al., 1996] происходят падение тонуса арте-
риальных сосудов, снижение АД, сопровождающееся умень-
шением чувствительности сосудов к вазоконстрикторным
факторам. Как известно, выраженная гипотензия и гипореак-
тивность сосудов в ответ на констрикторные (адренергичес-
кие) стимулы составляют важное звено в патогенезе кардио-
генного шока в острый период инфаркта миокарда.
278
При изучении механизма падения тонуса сосудов и АД
при стрессе и инфаркте миокарда было обнаружено, что
ключевая роль в этом явлении принадлежит гиперактивации
эндотелия сосудов, которая приводит к увеличению расслаб-
ления гладкой мышцы сосудов и подавлению адренергичес-
ких вазоконстрикторных реакций. Выяснено, что это угне-
тающее действие эндотелия на сократительную функцию со-
судов и их реакцию на вазоконстрикторы связано с продук-
цией эндотелием NO. Действительно, при инфаркте миокар-
да было доказано увеличение синтеза NO в организме [Va-
nin A.F. et al., 1994].
Механизм стимуляции гиперпродукции NO при острой
ишемии и инфаркте миокарда остается во многих аспектах не-
ясным. С современных позиций можно полагать, что важную
роль в этом механизме играют активация липидной триады,
СРО и соответственно увеличение содержания в миоцитах со-
судистой стенки Са2+, свободных радикалов и жирных кислот.
Эти события вызываются самой ишемией миокарда и в еще
большей степени стресс-реакцией, возникающей в ответ на
ишемическую боль и страх смерти и на внешние стрессоры,
т.е. сопутствующую стрессорную ситуацию окружающей сре-
ды. Увеличенная концентрация свободных жирных кислот,
увеличенная концентрация внутриклеточного Са2+ и свобод-
ные радикалы вызывают активацию NO-синтазы, ключевого
фермента продукции NO и рост этой продукции. Данный про-
цесс можно представить в виде гипотетической схемы, приве-
денной на схеме 5.8. Схема показывает, что стресс-реакция,
вызывая мощный липотропный эффект, рассмотренный выше
(см. раздел 5.1.5), приводит к накоплению указанных факто-
ров, которые активируют NO-синтазы: конституитивную NO-
синтазу (cNOS) и индуцибельную, т.е. рецепторзависимую,
NO-синтазу (iNOS). Как следствие возникает гиперпродукция
NO, и в результате всей цепочки процессов могут развиваться
гипотензия, недостаточность кровообращения и кардиоген-
ный шок.
Таким образом, гиперпродукция NO и развивающееся в ре-
зультате уменьшение периферического сопротивления сосу-
дов, а также уменьшение реактивности сосудов к контрак-
тильным факторам, приводящие к острой гипотензии, явля-
ются важным патогенетическим механизмом, посредством
которого стресс может усугублять течение острой ишемии.
В целом рассмотренные патогенетические механизмы,
через которые реализуется роль стресса в патогенезе повреж-
дений сердца при ишемии и инфаркте миокарда, обобщены на
схеме 5.9.
Стресс-реакция является чрезвычайно важным фактором
возникновения, течения и исхода ИБС. Поэтому для рацио-
279
Схема 5.8. Патогенетический механизм, посредством
которого стресс-реакция при острой ишемии миокарда может
вызвать недостаточность кровообращения и кардиогенный шок
[модификация схемы: Manukhina Е.В. et al., 1996]
СЖК — свободные жирные кислоты; cNOS — конститутивная NO-синта-
за; iNOS — индуцибельная NO-синтаза; NF-kB — фактор транскрипции
iNOS; СРО — свободнорадикальное окисление липидов. Пояснение в тексте.
нальной терапии ИБС и ее осложнения — острого инфаркта
миокарда — очень важно оценивать соотношение собственно
ишемических и стрессорных факторов на каждой стадии тече-
ния недуга. Особенно существенно это для понимания меха-
низмов аритмии, внезапной сердечной смерти и для решения
главного вопроса о различиях между первичной ИБС, которая
развивается как прямой результат стенозирующего коронар-
ного атеросклероза, и стрессорной аритмической болезнью
сердца, развивающейся прежде всего как результат первичного
стрессорного повреждения сердца.
280
Схема 5.9. Роль стресса в этиологии и патогенезе
ишемической болезни сердца и инфаркта миокарда
[Меерсон Ф.З., Пшенникова М.Г., 1988]
Ишемия
миокарда
Инфаркт
миокарда
Стресс-реакция
Эндогенные факторы:
боль и страх смерти
Экзогенные факторы:
эмоциональные ситуации
окружающей среды
Стрессорные повреждения пече-
ни, атерогенная дислипидемия
Аритмогенный эффект стресса.
Стрессорная депрессия сократи-
тельной функции неишемизиро-
венных отделов сердца
а-Адренергический и вазопрес-
синовый коронароспазм
Констрикция коронарных сосу-
дов вследствие гипервентиляции
Накопление тромбоксана, умень-
шение отношения простациклин/
тромбоксан, увеличение сверты-
ваемости крови, тромбоз
Уменьшение резистентности ми-
окарда к гипоксии и реоксигеиа -
ции
Общее увеличение сопротивле-
ния сосудов большого круга кро-
вообращения, перегрузка сердца
Уменьшение венозного тонуса,
избыточное депонирование кроаи
Пояснение в тексте.
5.2.3.1.2. Внезапная сердечная смерть
В соответствии с современными представлениями внезапная
сердечная смерть — это смерть внешне здорового человека, поги-
бающего в 50% случаев в течение одного часа после начала сердеч-
ного приступа и в 50 % случаев — в течение первых 10—15 мин.
Если удается записать ЭКГ, то в большинстве случаев регистри-
руется фибрилляция сердца. При этом большой опыт реанима-
тологии показывает, что раннее применение дефибрилляции и
других необходимых мер дает возможность во многих случаях
восстановить ритмическую деятельность сердца и сохранить
жизнь без рецидивов фибрилляции в ближайший отрезок време-
ни [Lown В. et al., 1980]. Это позволяет говорить о том, что в па-
тогенезе внезапной смерти главную роль играют не атеросклеро-
тическое стенозирование сосудов и коронарная болезнь, т.е. вы-
раженные структурные изменения коронарного русла, а другие
факторы. Действительно, значительное число исследований
свидетельствует, что большинство людей, погибших от внезап-
281
ной сердечной смерти, хотя и имели более или менее выражен-
ный коронаросклероз, однако у них не наблюдалось тромбоза
коронарных артерий, некроза миокарда или других органичес-
ких повреждений сердечной мышцы, которые сами по себе
могли бы быть причиной смерти.
Вместе с тем, поскольку коронарный атеросклероз распро-
странен довольно широко и, согласно многим эпидемиологи-
ческим исследованиям, внезапная смерть и коронарная бо-
лезнь имеют общие факторы риска, существует точка зрения,
что внезапная смерть является прямым результатом прогрес-
сирования коронарного атеросклероза. Таким образом, возни-
кает вопрос, в какой степени атеросклероз как таковой может
вызывать фибрилляцию и внезапную смерть и какова доля
участия других отягчающих факторов, например стресса.
Количественный анализ клинических данных показал, что
выраженный коронарный стеноз (75 % в одной или двух артери-
ях) встречался с одинаковой частотой в группе больных ИБС,
погибших от внезапной сердечной смерти, и в группе (контроль)
людей, погибших от других причин. Было показано также, что у
10 % здоровых американских солдат, убитых в Корее, был обна-
ружен выраженный коронаросклероз со стенозом коронарной
артерии, превышающим 75 % просвета сосуда.
Таким образом, даже выраженный атеросклероз едва ли
может считаться главной причиной фибрилляции и внезапной
сердечной смерти. Более того, примерно в половине случаев
внезапной смерти отсутствовали такие общепризнанные пока-
затели ишемии, как тромбоз коронарных сосудов или инфаркт
миокарда. Так, при сравнительном исследовании 868 вскры-
тий, осуществленных в 5 исследовательских центрах США,
было выявлено, что при внезапной сердечной смерти случаи
свежего закрывающего крупную коронарную артерию тромбо-
за составляют от 17 до 46 %. Сходные данные были получены
в гистологических исследованиях А.М.Вихерта и соавт. (1982),
которые установили, что из 100 случаев внезапной сердечной
смерти у мужчин в возрасте 30—60 лет в 98 % случаев был об-
наружен коронарный атеросклероз. Однако лишь в 13 % слу-
чаев наблюдался свежий инфаркт миокарда и в 18 % — свежий
коронарный тромбоз. В 29 % случаев изменения в миокарде
отсутствовали. Авторы пришли к заключению, что в 40 % слу-
чаев острые ишемические изменения в миокарде отсутствова-
ли, хотя степень стенозирующего атеросклероза была такой
же, как в случаях с очагами ишемии миокарда. По данным
многих других авторов, почти у половины погибших от вне-
запной сердечной смерти признаков ишемических поврежде-
ний не было. Таким образом, атеросклероз или даже ишеми-
ческое поражение в виде инфаркта сами по себе не являются
достаточной причиной фибрилляции и внезапной сердечной
смерти. Иными словами, одни люди могут иметь коронарный
282
атеросклероз и жить, а другие — умереть внезапной «сердеч-
ной смертью», не имея атеросклероза.
Что же это за решающий фактор, который является тригге-
ром внезапной сердечной смерти? С большой долей вероят-
ности можно полагать, что таким фактором является стресс.
Так, при изучении обстоятельств, предшествующих внезапной
сердечной смерти у 100 мужчин в возрасте до 70 лет с выяв-
ленным при некропсии коронарным атеросклерозом, было об-
наружено, что 23 % погибших перенесли тяжелое эмоциональ-
ное стрессорное воздействие за 30 мин до смерти, 40 % — в те-
чение последнего дня и 22—23 % испытывали стресс в течение
последних 6 мес и более [Myers A., Dewar Н.А., 1975].
Выяснилось, что существенное значение в этиологии вне-
запной сердечной смерти при скрытой или явной коронарной
болезни имеет биосоциальный статус, который характеризует
наличие эмоционального стресса в жизни человека. Так, изуче-
ние досье 17 000 сотрудников в возрасте 40—64 лет, работающих
в Лондоне и имеющих коронарный атеросклероз, показало, что
в группе сотрудников, занимающих наиболее «низкое» служеб-
ное положение, сердечная смертность возникала почти в 4 раза
чаще, чем среди сотрудников того же возраста, но занимающих
высокое положение [Rose G., Marmot M.G., 1981]. В этом же ис-
следовании было обнаружено, что возраст, АД, курение, масса
тела, уровень холестерина и глюкозы в крови лишь в очень
малой степени ответственны за увеличение сердечной смертнос-
ти. Главным детерминантом смертности у людей с коронарным
атеросклерозом оказался низкий социальный статус и хроничес-
кий прессинг биосоциального стресса.
За счет каких механизмов эмоциональный стресс может
провоцировать аритмии и фибрилляцию и вызывать ишеми-
ческие реакции у больных ИБС и в какой степени он способ-
ствует развитию этих часто фатальных явлений?
На этот вопрос в значительной мере отвечают исследова-
ния, в которых сравнивали реакцию адренергической систе-
мы, ишемическую реакцию сердца, возникновение аритмий
сердца и функцию сердца в ответ на эмоциональную стрессор-
ную нагрузку (решение задач в условиях цейтнота) и физичес-
кую нагрузку (велоэргометр) у больных ИБС и здоровых (кон-
троль) людей. В частности, в работе Э.Ш.Халфина, Н.П.Лями-
ной и Ф.З.Меерсона [Meerson F.Z., 1991] показано, что в
покое уровень экскреции адреналина (А) у больных ИБС су-
щественно выше, чем в контроле. При стрессорной нагрузке
уровень А у них также существенно превышал контроль. Уро-
вень НА в покое у больных ИБС был ниже, чем в контроле, на
22 %, но резко возрастал при стрессорной нагрузке (почти на
НО % против 24 % в контроле). В результате отношение А/НА
у больных в покое оказалось выше почти вдвое, чем у здоро-
вых людей, и имело величину, даже большую, чем у здоровых
283
людей при стрессе. Кроме того, было показано, что у больных
ИБС по сравнению с контролем существенно снижена экскре-
ция предшественника А и НА дофамина как в покое, так и
при стрессе. Это снижение составляло 30—50 %, и чем тяжелее
была форма ИБС, тем более выраженными оказывались раз-
личия с контролем. При этом физиологически значимое отно-
шение дофамин/(А+НА) у больных ИБС как в покое, так и
при стрессе было на 30—40 % меньше, чем в контроле. Пока-
зано, что дофамин ограничивает активность адренергической
системы на центральном уровне [Doda М., Gyorgy L., 1985].
Таким образом, активность адренергической системы — и ба-
зальная, и при стрессорном воздействии у больных ИБС ока-
зывалась выше, чем у здоровых людей, что может быть связа-
но с недостаточностью систем, ограничивающих активность
этой системы, в частности с дефицитом дофамина у этих боль-
ных.
Далее было показано, что при физической нагрузке у боль-
ных ИБС экскреция А возрастала в 1,5—2 раза по сравнению с
покоем, а экскреция НА уменьшалась в той же степени; в ре-
зультате при физической нагрузке отношение А/НА было
меньше, чем при стрессорной нагрузке. При этом изменений
содержания дофамина не наблюдалось.
В целом исследования показали, что ИБС сопровождается
таким мощным аритмогенным изменением адренергической
регуляции, как увеличение отношения А/НА и уменьшение
отношения дофамин/(А+НА). Существенно, что эти измене-
ния обостряются при стрессорных воздействиях значительно
больше, чем при физической нагрузке.
Далее в этих исследованиях было показано, что в ответ на
физическую нагрузку у больных ИБС ишемическая реакция
наблюдалась значительно чаще, чем при стрессорной нагрузке
(в 64 % случаев против 29 % при стрессорной нагрузке). На-
против, появление аритмий наблюдалось при стрессе в 3 раза
чаще, чем при физической нагрузке (в 22 % случаев против
8 % при физической нагрузке). При этом комбинация таких
нагрузок вызывала увеличение числа случаев ишемической ре-
акции, составляющее примерно сумму случаев, наблюдавших-
ся при раздельном действии этих нагрузок (84,8 % случаев).
В отличие от этого аритмогенный эффект комбинированного
действия нагрузок почти не отличался от результата действия
только стрессорной нагрузки (23,9 % случаев).
Эти данные свидетельствуют, что эмоциональный стрессор
является более действенным аритмогенным фактором при
ИБС, чем физическая нагрузка; это связано с более выражен-
ной активацией адренергической регуляции у больных ИБС
при стрессе. Регистрация показателей сократительной функ-
ции сердца показала, что ишемическая реакция на стрессор-
284
ную нагрузку в отличие от реакции на физическую не зависела
от увеличения сократительной функции сердца.
Увеличенная уязвимость сердца у больных ИБС к аритмо-
генному и ишемическому действию стресса является следстви-
ем увеличенного адренергического компонента эмоциональ-
ной стресс-реакции, что может быть связано со снижением
эффективности стресс-лимитирующих систем, а также следст-
вием выраженных изменений в самом сердце. В результате у
больных ИБС даже умеренная эмоциональная стресс-реакция
может вызвать большой аритмогенный эффект, существенно
больший, чем физическая нагрузка, который реализуется не
через увеличенную функцию сердца, а путем прямого дейст-
вия катехоламинов и других стресс-гормонов на миокард.
Таким образом, при ИБС существует три основных меха-
низма, за счет которых главные патогенетические факторы —
ишемия, вызванная стенозирующим атеросклерозом, и чрез-
мерная стресс-реакция, обусловленная недостаточностью
стресс-лимитирующих систем, могут приводить к аритмиям и
фибрилляции сердца'.
• первичное ишемическое повреждение сердца и его арит-
могенное действие;
• первичное стрессорное повреждение сердца и его арит-
могенное действие при отсутствии стенозирующего ко-
ронаросклероза и ишемии;
• комбинированное аритмогенное действие ишемии и
стресс-реакции.
Заключение
Подводя итоги рассмотрения роли стресса в патогенезе
ИБС, аритмической болезни сердца и внезапной сердечной смер-
ти, следует снова сравнить ишемическое и стрессорное повреж-
дения сердца и представить, как их комбинация может привести
к указанным патологиям. Первичное стрессорное повреждение
сердца существенно отличается по этиологии, патогенезу и кли-
ническим проявлениям от того состояния, которое обычно оп-
ределяют термином «ишемическая болезнь». Действительно,
стенозирующий коронаросклероз, вызывающий ишемию мио-
карда, хорошо теперь известен как результат генетического де-
фекта или вызванного алиментарной перегрузкой «полома» ме-
ханизма в печени, ответственного за метаболизм и элиминиро-
вание холестерина. Стрессорное повреждение сердца — это ре-
зультат чрезмерно длительной и сильной стресс-реакции; его
возможно более или менее полно предупредить адаптацией к
коротким, умеренным по силе стрессорным воздействиям, ко-
торые усиливают эффективность стресс-лимитирующих систем
организма, а также медиаторами этих систем.
285
В табл. 5.1 приведены признаки различия между ИБС и
стрессорной аритмической болезнью сердца, при этом на оп-
ределенных стадиях процесса стресс-реакция и ишемия (как
ключевые факторы патогенеза) тесно сопрягаются друг с дру-
гом. Эта таблица, во-первых, демонстрирует несомненные
различия в этиологии и патогенезе этих двух кардиопатологий
и, во-вторых, свидетельствует о необходимости понимания
различий в их прогнозе, профилактике и терапии.
Таблица 5.1. Различия между ИБС и стрессорной аритмической
болезнью сердца
Отличительные признаки ИБС Стрессорная аритмичес- кая болезнь сердца
Этиология Дефекты системы элиминирования хо- лестерина Дефекты стресс-лими- тирующих систем
Ключевое звено Стенозирующий ко- Избыточная стресс-ре-
патогенеза ронарный атероскле- роз -> ишемия мио- карда акция -> стрессорные повреждения сердца
Ишемия в ответ на физическую нагрузку Характерные «ише- мические» изменения ЭКГ и боли в сердце Отсутствует
Аритмии в ответ на Редко на пике на- Отсутствуют или воз-
физическую нагрузку грузки никают после нагрузки
Ишемия в ответ на стрессорную нагрузку Возникает примерно в 30 % случаев Отсутствует
Аритмии в ответ на стрессорную нагрузку Редко (примерно в 10 % случаев) Часто
Аритмии в покое Вторичные; связаны с ишемией, снимают- ся вазодилататорами Первичные; без ише- мии; вазодилататоры не эффективны
Инфаркт миокарда Обширный, ранний Мелкоочаговый, позд- ний
Терапия седативными Частично эффектив- Достаточно эффектив-
препаратами, нейро- на, ограничивает вто- на, ограничивает пер-
лептиками, транкви- ричную стресс-реак- вичную стресс-реак-
лизаторами, адрено- блокаторами цию, вызываемую ишемией цию
Профилактика с по- мощью адаптации к физическим нагруз- кам, мягким стрес- сорным воздействиям Частично эффективна Достаточно эффектив- на
286
Отличаясь по этиологии, патогенезу и клинике, стресс-ре-
акция и ишемия в то же время потенцируют друг друга за счет
трех основных механизмов. Во-первых, стресс-реакция путем
прямого некоронарогенного повреждения проводящей систе-
мы и сократительного миокарда может создавать множествен-
ные зоны деполяризации и нарушения проводимости. Вызван-
ное стрессом увеличение электрической гетерогенности серд-
ца усугубляется нарушением межклеточных контактов избыт-
ком кальция, и все это добавляется к вызванным ишемией
очагам деполяризации и множественным блокам проводимос-
ти. В результате электрическая стабильность сердца, которая
даже у совершенно здоровых людей является относительной,
серьезно нарушается и это приводит к тяжелым аритмиям,
фибрилляции и остановке сердца. Во-вторых, в определенных
условиях стресс может способствовать ускорению развития
коронаросклероза, коронароспазма и тромбоза, усугубляя тем
самым ишемию и приводя к острому инфаркту миокарда, ко-
торый в свою очередь осложняется аритмиями и фибрилля-
цией. В-третьих, ишемический очаг или инфаркт сопровожда-
ется болью и страхом смерти, что вызывает стресс-реакцию,
которая повреждает неишемизированные отделы миокарда,
увеличивает нагрузку на сердце и может осложнять течение и
ухудшать исход первичного ишемического повреждения.
Указанные механизмы, несомненно, должны учитываться
в стратегии и тактике профилактики и терапии ИБС и ин-
фаркта миокарда.
5.2.3.1.3. Гипертоническая болезнь
Со времен исследований Г.Ф.Ланга и В.Н.Черниговского
признается важная роль ЦНС и длительного «эмоционального
перенапряжения» в этиологии и патогенезе гипертонической
болезни. Это представление было подкреплено в эксперимен-
тах на животных. В них было показано, что стойкое повыше-
ние АД удается вызвать, применяя «сшибки» процессов воз-
буждения и торможения в коре мозга, а также путем много-
кратного действия сильных раздражителей, иными словами, в
результате длительного интенсивного действия стрессоров.
Было показано также, что хронически повторяющиеся (на
протяжении нескольких месяцев) отрицательные эмоциональ-
ные ситуации вызывали стойкое повышение АД у взрослых
обезьян бабуинов [Ely D.L., 1995].
Умеренные, но социально значимые стрессоры способны
вызывать постоянные «вспышки» подъема АД, что может вы-
зывать структурные изменения (утолщение) сосудистой стен-
ки и приводить далее к стойкой гипертензии. Клинико-психо-
физиологические исследования больных с начальными ста-
287
днями гипертонической болезни 1Б и ПА стадии, по класси-
фикации А.Л.Мясникова, показали, что у всех этих больных
начало заболевания и его обострения связаны с актуальными
индивидуально значимыми психогенными воздействиями.
Иными словами, причина и усугубление течения гипертони-
ческой болезни в значительной степени связаны с воздействи-
ем стрессогенных жизненных ситуаций [Вейн А.М., 1997].
В этих исследованиях также выяснилось, что в ответ на моде-
лирование эмоционального стресса («дискриминации притя-
заний») у этих больных повышалось АД, увеличивались сер-
дечный выброс и объем циркулирующей крови и повышалось
общее периферическое сопротивление сосудов. При этом у
одной части больных повышение АД в ответ на стресс реали-
зовалось при нарастании общего сердечного выброса, у другой
части повышение АД реализовалось главным образом за счет
возрастания периферического сосудистого сопротивления. Во
втором случае личность больных характеризовалась выражен-
ной тенденцией к длительному переживанию неотреагирован-
ных эмоций, т.е. явлениями «застойных очагов возбуждения»
в эмоциогенных зонах мозга. Развитие хронической гипертен-
зии наблюдали у исходно нормотензивных крыс при психосо-
циальном стрессе [Henry J.P. et al., 1993].
Роль «стойкого» возбуждения отрицательных эмоциоген-
ных центров в генезе артериальной гипертензии, вызываемой
эмоциональным стрессом, была показана в экспериментах на
иммобилизованных бодрствующих животных (кроликах) с
электростимуляцией вентромедиальных ядер гипоталамуса.
Многочасовая стимуляция вызывает повышение АД на 40—
50 мм рт.ст. В экспериментах с адреналэктомией и последую-
щим «заместительным» введением катехоламинов и/или гор-
монов коры надпочечников было выяснено, что для формиро-
вания устойчивой артериальной гипертензии необходимо со-
вместное участие стимуляции эмоциогенных центров и акти-
вации секреции катехоламинов и глюкокортикоидов из надпо-
чечников. Стимуляция эмоциогенных центров обеспечивает
первую транзиторную фазу гипертензии. Включение гормонов
надпочечников обеспечивает вторую фазу развития гипертен-
зии, и это происходит за счет вторичного тонизирующего дей-
ствия гормонов на ретикулярную формацию среднего мозга.
Усиливаются тонические влияния на сосудосуживающие
центры продолговатого мозга, что обусловливает стойкое пре-
обладание прессорных влияний на артериальное русло и раз-
витие устойчивой артериальной гипертензии. Создается свое-
образный порочный круг: первично возникающее под влияни-
ем острого эмоционального стрессорного воздействия возбуж-
дение лимбико-ретикулярных структур мозга вторично устой-
чиво поддерживается «восходящим» действием на него гор-
монов надпочечников. Это в свою очередь порождает непре-
288
рывные тонические нисходящие влияния на сосуды, форми-
руя устойчивую артериальную гипертензию [Судаков К.В.,
19976]. При этом надо учитывать, что под влиянием симпати-
ческой системы активируется ренин-ангиотензиновая система
почек и легких, которая вносит свой существенный вклад в
формирование стрессорной гипертензии. В механизм форми-
рования стрессорной артериальной гипертензии вовлечены
другие регуляторные вазоактивные системы, в том числе кал-
ликреин-кининовая депрессорная система, система ПГ, NO и
др. Именно нарушение согласованного действия всех «заинте-
ресованных» систем регуляции сосудистого тонуса, по-види-
мому, приводит к развитию не только стрессорной гипертен-
зии, но и гипертонической болезни.
Важную роль в патогенезе стрессорной гипертензии игра-
ют нарушения транспорта Са2+ в гладкомышечных клетках со-
судистой стенки артериального русла, возникающие в резуль-
тате стресс-реакции. В исследованиях Ю.В.Постнова и
С.Н.Орлова (1987) показано, что генез первичной гипертензии
непосредственно связан с повреждением мембранных систем
транспорта Са2+ в клетках сосудов, которое приводит к по-
вышению внутриклеточной концентрации этого катиона и как
следствие — к развитию вазоконстрикции. Авторы подчерки-
вают, что эти нарушения мембранных систем катионного
транспорта носят общий характер и проявляются также в по-
вышении содержания Са2+ в клетках крови. Поскольку для ин-
тенсивной стресс-реакции характерны активация СРО и нару-
шение функции клеточных мембран, то очевидно, что в пато-
генезе стрессорной гипертензии это явление играет важную
роль. В пользу данного представления свидетельствуют ре-
зультаты недавних клинико-физиологических исследований
[Gebara О.С. et al., 1996], которые показали, что препарат ве-
рапамил, ограничивающий вхождение Са2+ в клетку, предуп-
реждал повышение АД и агрегацию тромбоцитов, обычно воз-
никающие у людей при ментальном стрессе. При этом у боль-
ных с умеренной гипертензией, длительно (4 нед) получавших
этот препарат, ментальный стресс и холодовой стресс вызыва-
ли значительно меньший подъем АД и не сопровождались
обычной постстрессорной реакцией тромбоцитов, а реакция
на введение адреналина была уменьшена по сравнению с та-
ковой у пациентов, не получавших верапамил.
Важным фактором, определяющим эффект эмоционально-
го стресса как фактора этиологии и патогенеза гипертоничес-
кой болезни, является генетически обусловленная предраспо-
ложенность к стрессорным нарушениям регуляции сосудисто-
го тонуса. Об этом писал еще Г.Ф.Ланг*, который подчерки-
*Ланг Г.Ф. Гипертоническая болезнь. — М.: Медгиз, 1950.
19—1385
289
вал, что гипертензия возникает не у всех людей, а у тех, у ко-
торых есть «некие качества», которые предрасполагают к
этому заболеванию, и среди них важное место занимают «кон-
ституциональные особенности нервной системы».
Механизм данного явления еще недостаточно ясен, однако
имеющиеся к настоящему времени исследования свидетельст-
вуют в пользу этого представления. Вызывает интерес иссле-
дование G.Noll и соавт. (1996), в котором изучалась реакция
на ментальный стресс и гипоксию у людей с нормальным АД,
родители которых также были «нормотензивными», и у людей
с нормальным АД, родители которых страдали эссенциальной
гипертензией. Выяснилось, что в ответ на стресс резко уве-
личивались показатели активности симпатической системы
(АД, уровень в крови НА и эндотелина, импульсация мышеч-
ных симпатических нервов) только у людей, родители которых
страдали гипертензией. Реакция на гипоксию была одинако-
вой у всех испытуемых. Таким образом, у людей, родители
которых страдали гипертензией, выявлена генетически обус-
ловленная повышенная реактивность симпатического звена
нервной системы к стрессу. Это свидетельствует о том, что у
данной группы людей стресс является важным фактором
риска гипертонической болезни, во всяком случае — гиперто-
нической болезни стрессорной этиологии.
Ярким примером генетической обусловленности гипертен-
зивного эффекта стресса является реакция на различного рода
эмоциональные стрессоры у крыс с наследственной спонтан-
ной гипертензией (линия SHR). При сопоставлении реакции
АД на стрессоры у SHR и у крыс линии Вистар показано, что у
крыс Вистар в ответ на стрессоры увеличение АД составляло в
среднем 15 мм рт.ст., а у SHR АД возрастало на 40—45 мм
рт.ст., при этом максимально достижимое АД составляло у
Вистар 165 мм рт.ст., а у SHR — более 200 мм рт.ст. [Ely D.L.,
1995].
Таким образом, очевидно, что существует предрасполо-
женность к стрессорному повышению АД и развитию стойкой
гипертензии, в основе которой лежит увеличенная активация
сосудосуживающих нейрогуморальных систем. Иными слова-
ми, как и при других стрессорных патологических состояниях,
резистентность к стрессорной гипертензии определяется со-
стоянием стресс-системы и стресс-лимитирующих систем.
Показано, что у крыс SHR врожденная увеличенная актив-
ность симпатического звена стресс-системы [Westfall Т.С.,
Meldrum M.J., 1985] связана с врожденным дефицитом актив-
ности стресс-лимитирующей ГАМКергической системы в ги-
поталамусе [Hambley J.W. et al., 1984]. При этом введение аго-
ниста ГАМКА-рецепторов мусцимола в желудочки мозга нор-
мализует увеличенную активность симпатической нервной
системы у этих животных. Эта линия животных характеризует-
290
ся также врожденным низким содержанием ОП в тканях, т.е.
сниженной эффективностью опиоидергической стресс-лими-
тирующей системы [Di Guilio A. et al., 1979].
Показана также роль системы NO в поддержании нормаль-
ного уровня активности прессорных систем — адренергичес-
кой и ренин-ангиотензиновой. Выяснилось, что хроническое
ингибирование NO-синтазы и соответственно уменьшение
продукции NO приводит у крыс к активации этих прессорных
систем, что выражается в повышении в крови уровня НА, А,
ренина и развитии стойкой гипертензии [Zanchi A. et al.,
1995].
Заключение
Хроническое действие интенсивных эмоциональных стрес-
соров является важным фактором этиологии и патогенеза
стойкой артериальной гипертензии и развития гипертоничес-
кой болезни. В основе этого действия лежит «застойная» акти-
вация адренергического звена регуляции тонуса сосудов и на-
рушение Са -транспортирующей системы в клетках гладкой
мышцы сосудов, связанное с адренергической активацией
СРО при стрессе. Важную роль в патологическом действии
эмоционального стресса на систему регуляции сосудистого то-
нуса играет генетически обусловленная или приобретенная
предрасположенность. В основе этой предрасположенности
может лежать врожденный или приобретенный дефект функ-
ционирования отдельных стресс-лимитирующих систем, в том
числе системы продукции NO. Ограничение или исключение
этого дефекта с помощью адаптации к повторным воздействи-
ям факторов среды, а также путем применения медиаторов
или активаторов указанных систем является одним из эффек-
тивных способов предупреждения и коррекции артериальной
гипертензии стрессорной этиологии, а также возможной роли
стресса в усугублении гипертонической болезни иной этио-
логии.
5.2.3.2. Желудочно-кишечный тракт
Язвенные поражения желудочно-кишечного тракта. В пос-
леднее время становится все более очевидной роль эмоцио-
нального стресса в этиологии язвенной болезни желудка и
двенадцатиперстной кишки, а также других патологий желу-
дочно-кишечного тракта (ЖКТ). Во многих случаях выявлена
прямая связь между возникновением или обострением язвен-
ной болезни и наличием в анамнезе больного «острого» или
хронического действия отрицательно окрашенных эмоцио-
19* 291
нальных стрессоров как личного характера (потеря близких,
потеря работы, напряженные ситуации на работе и в семье и
т.п.), так и общего характера (различные катастрофы: земле-
трясения, цунами, война и т.п.). Показано, что первое земле-
трясение в Hanshin-Awaji привело наряду с увеличением числа
респираторных заболеваний (главным образом пневмоний) к
росту числа больных язвенной болезнью желудка: на 39 % уве-
личилось число случаев заболевания с большими язвами и на
34,8 % — с кровоточащими язвами. Результаты эпидемиологи-
ческих опросов, в которых выявлялась роль психосоматичес-
ких факторов в возникновении язвенной болезни, показали,
что острый эмоциональный стресс и хроническое действие от-
рицательных эмоциональных стрессоров, несомненно, явля-
ются важными факторами этиологии болезни. В данных опро-
сах сопоставляли наличие стрессов в «истории жизни» у боль-
ных язвенной болезнью и здоровых людей одинакового воз-
раста, пола и социального статуса. Что касается острого стрес-
са, то эти наблюдения показали, что количество острых стрес-
сорных событий (правда, не таких глобальных, как землетря-
сение) было примерно одинаковым у больных язвенной болез-
нью и у здоровых лиц контрольной группы. Однако хроничес-
ки действующие отрицательно-эмоциональные события, т.е.
«трудности», переживаемые в течение 6 мес и более, встреча-
лись у больных с язвой двенадцатиперстной кишки в 2 раза
чаще, чем у здоровых. При этом наличие язвы двенадцати-
перстной кишки четко ассоциировалось с эмоциональным
стрессом, обусловленным крушением жизненных планов, уг-
розой такого крушения, социальной неустроенностью. Выяв-
лено также, что среди больных было существенно больше
людей с невротическим статусом и депрессиями, чем среди
здоровых лиц контрольной группы. Медико-социологические
исследования, проведенные в Японии, показали, что при про-
чих равных условиях язвенной болезнью желудка чаще страда-
ли люди с неустроенной семейной жизнью, курящие и питаю-
щиеся нерегулярно. Опрос 210 жителей г. Люблина в Польше,
страдающих язвенной болезнью с 16—29 лет, показал, что все
100 % больных язвой желудка считают, что причиной заболе-
вания являются стрессовые ситуации и только половина из
них указали на важную роль плохого питания. Сходные ре-
зультаты получены при опросе жителей Дании. Были обследо-
ваны 59 больных язвой желудка. Из них 75 % считали, что
причиной заболевания являются психические факторы: страх,
горе, тревога. Примерно 80 % врачей высказали такое же мне-
ние. Любопытные данные получены в Западной Германии
[Helmert U., Shea S., 1994]. Было проведено обследование по 7
основным заболеваниям 44 363 пациентов 25—69 лет обоего
пола в течение 1981 — 1991 гг. Сопоставляли количество случа-
ев язвы желудка у людей высокого и низкого социальных
292
классов при одинаковом уровне медицинского обслуживания.
Выяснилось, что «язвенных» пациентов существенно больше
среди мужчин с низким социальным статусом. Статус оцени-
вали по образованию, профессии и годовому доходу семьи.
При этом оказалось, что такие факторы, как курение, туч-
ность или принадлежность к поведенческому типу А, не игра-
ют роли. Главное, как считают исследователи, это — классо-
вое неравенство и психологический стресс, вызванный у
людей низкого социального статуса жизненными трудностя-
ми. Сходные данные получены в США [Levenstein S. et al.,
1995]. Исследователи прослеживали возможные причины по-
явления больных с язвами желудка на протяжении многих лет.
Были выявлены следующие социодемографические характе-
ристики, связанные со случаями язвенной болезни: у жен-
щин — низкое образование, бытовая теснота, проблемы с
детьми, ощущение себя человеком второго сорта; у мужчин —
финансовые трудности, напряженность в семье, принадлеж-
ность к людям не белой расы.
Язвы желудка, как известно, входят в обозначенную
Г.Селье «триаду» стрессорных повреждений у эксперименталь-
ных животных. Эти язвенные поражения желудка являются
ярким и стабильным показателем стрессорного повреждения.
Такой показатель широко используется в экспериментальных
исследованиях как для оценки интенсивности стрессорного
воздействия, так и для изучения резистентности к этому воз-
действию.
Стрессорные язвенные поражения слизистой оболочки же-
лудка являются по своей природе ишемическими «линейными
инфарктами слизистой» и возникают в результате вазокон-
стрикции в желудке, вызванной стрессорной активацией его
адренергической регуляции [Goldman Н., RosoffC., 1968]. При
этом тормозится активность n.vagus и соответственно прекра-
щается выделение желудочного сока (схема 5.10, пунктирные
стрелки). Затем, когда восстанавливается активность n.vagus
после окончания действия стрессора, ишемизированные
участки подвергаются «перевариванию» и изъязвляются
[Desiderate О. et al., 1974].
В эксперименте показано, что большее количество язв воз-
никает при стрессе у животных, принадлежащих к «возбуди-
мому» типу, который характеризуется высокой активацией
(реактивностью) адренергической системы при стрессе [Ано-
хина И.П., 1975; Ведяев Ф.П. и др., 1985]. При этом высокая
активность адренергической системы отмечена как в цент-
ральных звеньях стресс-системы, что выражается в увеличен-
ном «выбросе» НА в структурах среднего мозга и особенно в
гипоталамусе [Анохина, 1975], так и на периферии, что выра-
жается, например, в увеличении содержания НА в желудке. На
фоне блокады адренергической системы с помощью L-ДОФА
293
Схема 5.10. Патогенез стрессорного поражения
желудочно-кишечного тракта
СТРЕССОР
Пояснение в тексте.
и p-адреноблокатора пропранолола стрессорные язвы желудка
не возникали [Анохина И.П., 1975]. Таким образом, очевидно,
что резистентность к стрессорному язвообразованию в значи-
тельной степени определяется реактивностью адренергическо-
го звена стресс-системы к стрессорному воздействию.
Снижение кровотока, ишемия и последующее язвообразо-
вание в желудке при стрессе тем больше, чем меньше продук-
ция кортикостероидов; введение кортикостерона предупреж-
дало этот ишемический эффект стресса [Филаретова Л.П.,
1995]. Данными обстоятельствами, очевидно, определяется и
различная резистентность к стрессорному язвообразованию у
животных разных генетических линий, отличающихся, как те-
перь выяснилось, неодинаковой базальной активностью и ре-
активностью адренергического и кортико-адреналового
[Sudakov К.В. et al., 1995; Пшенникова М.Г. и др., 1996] зве-
ньев стресс-системы. Действительно, показано [Пшенникова
М.Г. и др., 1999, 2000], что эмоциональное стрессорное воз-
действие, вызывающее у крыс линии Вистар четкие язвенные
поражения желудка, вовсе не влияет на крыс линии Август
или вызывает у них ничтожное поражение такого рода. Анализ
этого факта показывает, что у крыс Август базальная концент-
рация катехоламинов (КА) в крови в среднем в 2,6 раза боль-
ше, чем у Вистар. Однако при действии эмоционального
стрессора интенсивность стресс-реакции, оцениваемая по
приросту концентрации КА в крови, у крыс Август примерно
294
в 2 раза меньше, чем у крыс Вистар. Сходные данные получе-
ны для кортикостерона. Оказалось, что базальное содержание
этого гормона в крови у крыс Август больше, чем у Вистар, а
прирост при стресс-реакции у них существенно меньше, чем у
Вистар [Sudakov К.В. et al., 1995; Пшенникова М.Г. и др.,
1996]. У крыс Август стрессорное увеличение содержания в
крови маркера повреждений клеточных мембран креатинфос-
фокиназы существенно меньше, чем у крыс Вистар, при
одном и том же стрессорном воздействии [Пшенникова М.Г. и
др., 1996]. Таким образом, высокая устойчивость ЖКТ у крыс
Август к язвенным поражениям при эмоциональном стрессе
может быть связана с меньшей интенсивностью стресс-реак-
ции, меньшей активацией адренергической системы и соот-
ветственно с меньшей степенью вазоконстрикторного дейст-
вия катехоламинов и меньшей ишемией желудка. Важную
роль в реализации стрессорного язвообразования в желудке,
очевидно, играет состояние стресс-лимитирующих систем.
Так, иммобилизационный стресс, вызывающий язвообразова-
ние, сопровождается снижением содержания ГАМК в гипота-
ламусе, а предварительное введение аналогов ГАМК перед
стрессорным воздействием предупреждало не только сниже-
ние содержания ГАМК в гипоталамусе, но и активацию СРО и
язвообразование в желудке [Бульон В.В., 1995].
Установлено, что у крыс Вистар, более предрасположен-
ных к стрессорным язвенным поражениям желудка, продук-
ция NO, как базальная [Микоян В.Д. и др., 1996; Пшенникова
М.Г. и др., 2000], так и после теплового стресса [Микоян В.Д.
и др., 1996], существенно меньше, чем у крыс линии Август.
Как указывалось выше, было обнаружено, что при стрессе
уменьшается продукция NO в желудке, и это, очевидно, явля-
ется ключевым звеном патогенеза адренергических ишемичес-
ких язвенных поражений желудка. Таким образом, причиной
низкой устойчивости крыс линии Вистар к таким поражениям
по сравнению с крысами линии Август может быть врожден-
ный относительный дефицит продукции NO.
Нарушения функции желудочно-кишечного тракта. Эмоци-
ональный стресс, хронический или сильный однократный,
может вызывать нарушения моторной и секреторной функции
ЖКТ. Среди больных с хроническими болями в ЖКТ обнару-
жено большое число женщин, перенесших тяжелый стресс,
связанный с насилием. Насилие и посттравматические стрес-
сорные расстройства, как будет показано далее, сопровожда-
ются хронической активацией и возможным истощением ги-
поталамо-гипофизарно-адреналовой оси в такой же степени,
как у больных с меланхолической депрессией [De Bellis M.D.
et al., 1994]. Поэтому можно полагать, что причиной такой па-
тологии ЖКТ является хроническая активация определенных
компонентов стресс-системы. Действительно, в эксперименте
295
показано, что при стрессе задерживается освобождение желуд-
ка, снижается его эвакуаторная функция на фоне увеличенной
моторной активности кишечника [Monnikes Н. et al., 1994].
Как показал анализ, это непосредственно связано с актива-
цией стресс-системы: микроинъекции КРГ в паравентрику-
лярное ядро гипоталамуса у крыс вызывает реакцию ЖКТ, по-
добную реакции на эмоциональный стресс: ингибируется эва-
куаторная моторика желудка, стимулируется моторика кишеч-
ника и экскреция фекалий. Этот эффект при стрессе исчезает
после интратекального введения в мозг антагонистов КРГ
[Monnikes Н. et al., 1994]. Такие селективные ответы ЖКТ на
повышение уровня КРГ в гипоталамусе, т.е. стаз желудка и
увеличенная моторика кишечника, реализуются за счет одно-
временного торможения n.vagus и стимуляции сакральной
парасимпатической системы, которые вызываются КРГ и ак-
тивацией НА-нейронов синего пятна соответственно. Пока-
зано, что данное действие КРГ опосредуется интерлейкином
1-Р (ILl-p), выброс которого всегда сопровождает стресс-реак-
цию. Таким образом, стрессорные нарушения моторики ЖКТ
имеют вполне четкую основу в виде увеличенной секреции
КРГ, основного медиатора центрального звена стресс-систе-
мы, и активации синего пятна (см. схему 5.10). По мнению ис-
следователей, КРГ является скрытым звеном связи между
симптомами хронических болей в ЖКТ у людей и наличием у
них в анамнезе перенесенной эмоциональной травмы [Dross-
man D.A., 1994]. КРГ также, возможно, вовлечен в вызывае-
мую стрессом гипермоторику кишечника у больных с синдро-
мом раздражения кишечника.
Хронической активацией гипоталамо-гипофизарно-адре-
наловой оси и/или НА-системы синего пятна можно, по-ви-
димому, объяснить также снижение болевых порогов висце-
ральной чувствительности у больных с функциональными рас-
стройствами ЖКТ, что происходит за счет истощения вызы-
ваемой опиоидными пептидами стресс-аналгезии. Следует
иметь в виду также связанные с хроническими эмоциональны-
ми стрессами синдромы атипических болей в грудной клетке,
характерные для рефлюкса, язвенной болезни ЖКТ, желчно-
каменной болезни. Как выяснилось при тщательном обследо-
вании, у этих людей таких заболеваний не было. Причина ука-
занных синдромов, по-видимому, также имеет центральное
происхождение.
Как было отмечено выше, стресс-система получает инфор-
мацию от «думающего» и «эмоционального» мозга через амиг-
далу, гиппокамп и мезокортико-лимбйческую систему. Как
известно, лимбическая система участвует в регуляции вегета-
тивных функций и в том числе функции ЖКТ. В эксперимен-
тах было установлено, что наблюдаемое при эмоциональном
стрессе угнетение моторики желудка и секреции желудочного
296
сока воспроизводимо путем электростимуляции ядер амигда-
лы, вентрального и дорсального гиппокампа и перегородки.
Электростимуляция амигдалы и дорсального гиппокампа при-
водит как к изменению химического состава желудочного сока
(торможению выделения соляной кислоты), так и к полному
угнетению желудочной секреции, вызываемой условным раз-
дражителем секреции. Таким образом, формирующееся на
уровне центрального звена стресс-системы угнетение функ-
ции желудка при действии эмоциональных стрессов (см. схему
5.10) находится под непосредственным влиянием надгипота-
ламических структур лимбической системы. Иными словами,
стресс-система регулирует функцию ЖКТ под «руководством»
лимбической системы, которая «передает» стресс-системе ин-
формацию о действии эмоционального стрессора (см.
схему 5.2).
Таким образом, при диагностике и коррекции язвенных
поражений ЖКТ и нарушений его функции, несомненно, сле-
дует уточнить наличие в анамнезе стрессорного компонента и
оценить активность звеньев стресс-системы и стресс-лимити-
рующих систем.
5.2.3.3. Система крови и иммунная система
при эмоциональном стрессе
Система крови. Г.Селье [Selye Н., 1936] впервые обнаружил
и описал изменения системы крови, характерные для стресс-
реакции: эозинопению, нейтрофильный лейкоцитоз, лимфо-
пению в периферической крови и инволюцию тимико-лимфа-
тического аппарата, которые регистрировались через 6—48 ч
от начала повреждающего воздействия.
Решающий вклад в изучение изменений системы крови
при стрессе и механизмов этих изменений впервые внесли
фундаментальные экспериментальные исследования П.Д.Го-
ризонтова и его сотрудников [Горизонтов П.Д. и др., 1983].
В этих работах впервые применялись количественные методы
исследования и одновременно изучались реакции разных от-
делов системы крови при воздействиях различных стрессоров.
Было установлено, что при однократном интенсивном эмо-
циональном стрессе (иммобилизация в течение 6 ч) и электро-
болевом воздействии у крыс в периферической крови развивался
выраженный нейтрофилез, при котором содержание нейтро-
филов увеличивалось в 6—7 раз через 6—9 ч после воздейст-
вия. Число лимфоцитов в это же время резко падало. Содер-
жание эозинофилов также резко уменьшалось и через 3—12 ч
после начала воздействия достигало нуля. Через 24 ч после на-
чала воздействия все эти показатели возвращались к норме.
В костном мозге при этом существенно увеличивалось (на 60—
297
70 %) содержание лимфоцитов («лимфоидный пик») и умень-
шалось число зрелых гранулоцитов; после 24 ч эти показатели
возвращались к норме. Содержание бластных клеток грануло-
цитарного ряда (миелобласт — миелоцит) в течение 24 ч после
начала воздействия существенно не менялось, но через 48 ч —
возрастало. Существенных изменений в содержании эритро-
идных клеток костного мозга не обнаружено. Содержание кле-
ток в селезенке и тимусе (лимфоидных органах) после стрес-
сорного воздействия уменьшалось в течение 12 (в селезен-
ке) — 48 (в тимусе) часов. Затем содержание этих клеток по-
степенно нормализовалось.
Наиболее характерными изменениями при однократном и
многократном стрессорном воздействии в стадии мобилиза-
ции общего адаптационного синдрома (по Селье) являются: в
периферической крови — значительный нейтрофилез, лимфо-
пения и эозинопеншг, в костном мозге — лимфоидный пик и
уменьшение содержания зрелых гранулоцитов; в лимфоидных
органах — уменьшение количества (истощение) клеток.
Уменьшение содержания зрелых гранулоцитов в костном
мозге и клеток в лимфоидных органах обусловлено быстрым
выходом этих клеток в периферическую кровь, т.е. стрессор-
ной мобилизацией костномозгового резерва гранулоцитов и
резерва лимфоидных клеток селезенки и тимуса, что главным
образом и обеспечивает наблюдаемое при стрессе пополнение
клеточного состава периферической крови и в том числе пе-
риферический нейтрофилез. Позже в стадии резистентности
развивается гиперплазия костномозгового кроветворения:
число бластных форм гранулоцитарного ряда в костном мозге
увеличивается, т.е. компенсаторно стимулируется гранулоци-
топоэз. При продолжении стрессорного воздействия наступает
стадия истощения.
Анализ возможных механизмов реакции системы крови на
стрессорное воздействие показывает, что важную роль в этой
реакции играет активация адренергического и гипоталамо-ги-
пофизарно-кортико-адреналового звеньев стресс-системы
(Гольдберг Е.Д. и др., 1997]. Показано, что лимфоидные орга-
ны и лимфатические узлы имеют плотную симпатическую ин-
нервацию [Bellinger D.L. et al., 1992; Friedman E.M.,
Irwin M.R., 1995].
Показано, что нейтрофилез в периферической крови, убыль
числа зрелых гранулоцитов и увеличение уровня лимфоидных кле-
ток в костном мозге стимулируются катехоламинами и не за-
висят от гормонов коры надпочечников. При этом увеличение
содержания лимфоцитов в костном мозге (лимфоидный пик)
регулируется через Р-адренорецепторы.
Имеющиеся данные позволяют полагать, что уменьшение
содержания клеток в селезенке при стрессе связано главным
образом с активацией адренергической системы и сокращени-
298
ем гладкой мускулатуры этого органа под влиянием катехол-
аминов, которое реализуется через p-адренорецепторы. При
этом больше «выбрасывается» лимфоцитов меньшего размера,
т.е. В-лимфоцитов, чем более крупных Т-лимфоцитов, поэто-
му в селезенке возникает количественное преобладание
Т-лимфоцитов. Однако Т-лимфоциты также «выбрасываются»
из этого органа. Таким образом, при стрессе, как при экстре-
мальной ситуации, селезенка выполняет функцию срочной
помощи, поставляя в кровоток лимфоидные клетки разных
видов. Эта функция селезенки, по мнению исследователей, не
менее важна, чем функция депонирования клеток красной
крови и «выбрасывания» их в кровоток при кровопотерях.
Уменьшение содержания лимфоидных клеток в тимусе при
стрессе вплоть до опустошения связывают главным образом с
увеличением секреции гормонов гипоталамо-гипофизарно-
кортико-адреналовой системы. В частности, показано, что
при адреналэктомии или гипокортицизме стресс не вызывает
«выброса» тимоцитов; установлено также, что сами Т-клетки
обладают высокой чувствительностью к глюкокортикоидам
[Горизонтов П.Д. и др., 1983]. При этом если селезенка —
орган «скорой помощи», то из тимуса выделение тимоцитов
происходит медленнее и позже, чем из селезенки (через 1—
2 сут).
Предполагается, что данная функция тимуса связана с вос-
полнением пула Т-клеток, израсходованных в начале стресс-
реакции. Однако при дефектах функционирования селезенки
или при ее отсутствии тимус может «ускорить» свою помощь и
становится источником лимфоцитов уже в первые часы дейст-
вия стрессора.
Характерная для стресс-реакции лимфопения, возникаю-
щая вопреки большому «выбросу» в периферическую кровь
лимфоцитов из лимфоидных органов, связана главным обра-
зом с усиленной миграцией лимфоидных клеток из перифери-
ческой крови. Одним из «мест назначения» такой миграции
является костный мозг, и именно этим наряду с другими при-
чинами может быть обусловлено явление лимфоидного пика
в этой структуре. Данный процесс реализуется, как уже упо-
мянуто выше, под влиянием адренергической системы через
Р-адренорецепторы и соответственно тормозится блокаторами
этих рецепторов. Увеличение содержания лимфоидных клеток
в костном мозге в ответ на стрессор имеет большое значение,
так как увеличивает его иммунокомпетентность. Другими
структурами, куда мигрируют лимфоидные клетки из крови,
являются лимфатические узлы, что также создает состояние
их повышенной «готовности» к иммунному ответу. Возможна
миграция таких клеток и в другие органы. При этом установ-
лено, что наряду с обеспечением иммунной функции (см.
далее) лимфатическая система принимает непосредственное
299
участие в регуляции процессов регенерации в различных орга-
нах и тканях. Так, лимфоциты участвуют в регенерации мио-
карда, печени, почек после частичного удаления тканей этих
органов, и при дефиците лимфоидных клеток интенсивность
регенерационных процессов значительно снижается [для ссы-
лок см.: Горизонтов П.Д. и др., 1983; Гольдберг Е.Д. и др.,
1997]. Поэтому мобилизация системы крови при стрессорных
воздействиях выполняет комплексную адаптивную функцию.
Таким образом, изменение системы крови при однократ-
ном стрессорном воздействии, представленное в этом разделе
нашего сообщения, позволяет заключить, что стресс вызывает
срочную мобилизацию всех компонентов системы крови для
реализации адаптивной реакции организма на стресс и прежде
всего для активации иммунной системы.
При хроническом действии стрессоров мобилизация систе-
мы крови и последующая нормализация сменяются угнетени-
ем, что играет ключевую роль в развитии стрессорной имму-
носупрессии и стрессорной иммунопатологии, рассматривае-
мых в последующих разделах. Так, например, в экспериментах
на крысах показано, что при длительном эмоционально-боле-
вом воздействии (3—6 ч в день в течение 14—18 дней) в пери-
ферической крови снижается общее содержание лейкоцитов на
20 %, нейтрофильных гранулоцитов — на 20 %, содержание
эозинофилов — на 70 %, лимфоцитов — на 27 % и моноци-
тов — в 2,5 раза. Содержание эритроцитов существенно не ме-
нялось. Таким образом, происходило уменьшение содержания
в крови лимфоидных клеток. В костном мозге при этом умень-
шалось общее число миелокариоцитов за счет убыли всех
видов клеток, составляющей в среднем от 28 % (для миело-
бластов — нейтрофильных промиелоцитов и миелоцитов) до
68 % (для метамиелоцитов). В лимфоидных органах картина
была неодинаковой: в селезенке существенных отклонений от
нормы не наблюдалось, а в тимусе содержание тимоцитов
резко падало (до 40 % нормы). Таким образом, при длитель-
ном интенсивном действии стрессоров возникает угнетение
системы крови и состояние иммунологической недостаточ-
ности.
Следует отметить, что нарушения системы крови при дли-
тельном эмоциональном стрессорном воздействии усугубля-
ются на фоне ионизирующего облучения в диапазоне даже
малых и низких доз [Мороз Б.Б. и др., 1997].
Иммунная система. Иммунная система как система «бы-
строго реагирования» на чужеродные воздействия тесно связа-
на со стресс-системой и стресс-лимитирующими системами.
Это выражается в том, что структуры центрального аппарата
регуляции иммунной системы локализованы в гипоталамусе,
где находится ключевое звено аппарата нервной регуляции
иммунной системы, гиппокампе, амигдале [Корнева Е.А., Ше-
300
коян В.А., 1982]. Они связаны с холинергическими нейронами
базального ядра Мейнерта и перегородки, с НА-нейронами
синего пятна, ДА-нейронами мезолимбической и нигростиат-
ной системы, наконец, с ГАМК-нейронами хвостатого ядра,
серотонинергическими нейронами ядер шва [Корнева Е.А.,
1990; Крыжановский Г.Н. и др., 1997]. Таким образом, струк-
туры, входящие в состав центрального аппарата регуляции им-
мунной системы, топически совпадают с центральными струк-
турами стресс-системы и стресс-лимитирующих систем.
К настоящему времени выяснилось, что стресс-система и
иммунная система связаны двусторонними связями: влиянием
нейрональных структур на иммунокомпетентные органы и
клетки, с одной стороны, и влиянием иммунной системы на
стресс-систему — с другой [Адо А.Д., 1993; Коляда Т.И. и др.,
1995]. Благодаря этим связям гипоталамус, ключевая структу-
ра центрального аппарата регуляции иммунной системы и
стресс-системы, быстро реагирует на нарушение иммунного
(антигенного) гомеостаза и дает начало сложному эфферент-
ному пути передачи регуляторных влияний на иммунокомпе-
тентные органы и клетки [Коляда Т.И. и др., 1995; Крыжанов-
ский Г.Н. и др., 1997]. Эти влияния осуществляются благодаря
наличию на иммунокомпетентных клетках рецепторов для
нейромедиаторов, гормонов, регуляторных пептидов. Пока-
зано, что иммунокомпетентные клетки имеют рецепторы к
ацетилхолину [Абрамов В.В., 1991], глюкокортикоидам
[Katz А.М. et al., 1985; Голиков П.П., 1988], катехоламинам
[Landmann R. et al., 1984], нейропептидам [Ruff M.R. et al.,
1989], к КРГ [Friedman E.M., Irwin M.R., 1995]. Благодаря
этому обстоятельству возможно прямое воздействие гормонов
и медиаторов на элементы иммунной системы.
Следует подчеркнуть, что существует сходство ответа гипо-
таламуса и других отделов стресс-системы на антиген с ответа-
ми на различные стрессоры. Это понятно, так как антигены по
существу являются стрессорами. В ответ на иммунногенный
антиген сразу возникает увеличение электрической активнос-
ти паравентрикулярных и супраоптического ядер гипоталаму-
са, нейроны которых продуцируют КРГ, аргинин-вазопрессин
и окситоцин. При этом стимулируется секреция этих гормо-
нов, происходит активация оси гипоталамус — гипофиз —
надпочечники [Коляда Т.И. и др., 1995; Stratakis С.А.,
Chrousos G.P., 1995], повышается уровень НА и ДА в заднеги-
поталамической области [Девойно Л.В., 1994], в паравентри-
кулярных и аркуатном ядрах и дорсомедиальном отделе гипо-
таламуса. Затем включаются гиппокамп, амигдала, синее
пятно [Клименко В.М., 1993].
При действии других стрессоров также возникает актива-
ция гипоталамуса и других отделов стресс-системы (как уже
было указано в разделах 5.1.1 и 5.1.2), что соответственно
301
также сопровождается ростом секреции КРГ, аргинин-вазо-
прессина, НА и ДА в центральном звене стресс-системы, ак-
тивацией гипоталамо-гипофизарно-адреналовой оси, а также
активацией нейронов синего пятна и периферической адре-
нергической системы, включая симпатическую систему и моз-
говой слой надпочечников. Таким образом, очевидно, что ак-
тивность различных звеньев стресс-системы и нейрональных
структур, непосредственно с ней связанных (амигдалы, гиппо-
кампа, мезолимбической и др.), а также, по-видимому, стресс-
лимитирующих систем в значительной мере определяет функ-
цию иммунной системы и величину иммунного ответа орга-
низма. В соответствии с этим стресс-реакция, возникающая в
ответ на различные стрессоры, несомненно, включает в себя
изменения иммунологической реактивности организма.
Влияние уровня активности стресс-системы на иммунный
ответ было доказано в многочисленных исследованиях, в ко-
торых осуществляли либо выключение, либо стимуляцию этих
нейрональных структур и оценивали величину иммунного от-
вета на антигены (подробные данные см. в монографии
Г.Н.Крыжановского и соавт., 1997). Так, например, была вы-
явлена зависимость иммунологических реакций от активности
гипоталамуса в исследованиях, где проводили иммунизацию
животных на фоне повреждений (выключения) или электро-
стимуляции (активации) структур гипоталамуса. Показано,
что локальное повреждение заднего гипоталамуса приводит к
снижению активности костномозговых предшественников им-
мунокомпетентных клеток, снижению способности клеток-
предшественников к дифференцировке, к изменению формы
макрофагов и снижению их антиген-активности. При локаль-
ной деструкции заднего поля гипоталамуса снижается способ-
ность к продукции антител в ответ на антигены вплоть до пол-
ного отсутствия признаков синтеза антител и их накопления в
крови. Это было выявлено при иммунизации лабораторных
животных (кроликов, крыс, мышей) различными антигенами:
от вакцины БЦЖ до фракции чумного микроба [Крыжанов-
ский Г.Н. и др., 1997].
Способность к иммунному ответу, митогенная пролифера-
тивная активность Т-лимфоцитов и активность естественных
киллеров снижаются при локальной деструкции переднего и
латерального гипоталамуса [Wrona D. et al., 1991]. При этом
уменьшается отношение Т-хелперов/Т-супрессоров. Предва-
рительная электростимуляция структур гипоталамуса, напро-
тив, приводит к результатам, обратным тем, которые наблюда-
ются при выключении этих структур, а именно увеличивается
синтез антител в ответ на антигены, т.е. активируется иммун-
ный ответ.
Важную роль в регуляции иммунореактивности играет ак-
тивность симпатического звена центрального компонента
302
стресс-системы, представленного нейронами синего пятна.
Показано, что сохранение функциональной и структурной це-
лостности синего пятна необходимо для поддержания имму-
нокомпетентности одного из центральных органов иммунной
системы — костного мозга. Важно подчеркнуть, что гипер-
функция этого норадренергического звена стресс-системы
может приводить к иммунологической недостаточности, по-
скольку системное введение НА оказывает иммунодепрессив-
ное действие. В то же время антагонисты НА в сочетании с ад-
реналэктомией приводят к стимуляции активности иммунной
системы [Besedovsky Н. et al., 1983]. Позже мы приводим рису-
нок (см. рис. 5.5), где показаны активация симпатической
системы и ее «выход» на иммунную систему.
Роль стресс-системы и стресс-лимитирующих систем в фор-
мировании иммунного ответа доказана также при исследовании
влияния на иммунную реакцию предварительного введения гор-
монов и медиаторов этих систем. Как уже указывалось выше,
иммунокомпетентные клетки обладают рецепторами к этим ве-
ществам. Регуляция функций иммунной системы нейрогумо-
ральными факторами осуществляется по антагонистическому
принципу, т.е. вызывая либо стимуляцию, либо угнетение ак-
тивности макрофагов, Т- и В-лимфоцитов в зависимости от
дозы данных веществ, вида антигенов, схемы иммунизации и,
наконец, от фазы иммунного ответа. Тем не менее обобщение
результатов большого числа работ приводит к выводу, что сти-
мулирующее влияние на функции клеток, продуцирующих
антитела, могут оказывать ацетилхолин и дофамин, гормоны ги-
поталамуса (аргинин-вазопрессин и окситоцин), гормоны гипо-
физа (гормон роста, тиреотропный гормон), кортикостероиды
(в физиологической дозе), медиаторы стресс-лимитирующих
систем: ОП (р-эндорфин, Leu-энкефалин), ГАМК, субстанция
Р. Иммуносупрессорными свойствами обладают А и НА, се-
ротонин, а также кортикостероиды (при гиперпродукции).
При этом некоторые из указанных соединений могут индуциро-
вать (окситоцин, аргинин-вазопрессин, субстанция Р), повы-
шать (а- и р-эндорфины, Met-энкефалин) или угнетать (корти-
костероиды и АКТУ) секрецию интерферонов ИФ-а и ИФ-у.
Как видно из изложенного, структуры стресс-системы ока-
зывают регуляторное влияние на иммунную систему. Иммун-
ная система, как было отмечено выше, также влияет на состо-
яние стресс-системы, модулируя ее активность. Механизмы
этого влияния начали проясняться сравнительно недавно бла-
годаря открытию и изучению цитокинов. Эти пептиды проду-
цируются при действии антигенов, а также при действии дру-
гих стрессоров в клетках различных систем, но главным обра-
зом в иммунокомпетентных клетках.
В соответствии с современными представлениями именно
относящиеся к цитокинам интерлейкины — IL-1, IL-2, и IL-6 и
303
Рис. 5.4. Взаимосвязь иммунной системы и стресс-системы: контур
саморегуляции активности иммунной системы.
СП — синее пятно; АВ — аргинин-вазопрессин; Г — гипофиз; КРГ — кортико-
тропин-рилизинг-гормон; АКТГ — аденокортикотропный гормон. Пояснения
в тексте.
фактор некроза опухоли (tumor necrosis factor — TNF) играют
ключевую роль во взаимосвязи между иммунной системой и
центральным звеном стресс-системы. Именно они, образуясь в
ответ на антиген, активируют при иммунных реакциях гипота-
ламус и ось гипоталамус — гипофиз — надпочечники, включая
активацию секреции гипоталамических КРГ и аргинин-вазо-
прессина, гипофизарного АКТГ и глюкокортикоидов [Peristein
R.S. et al., 1993]. Таким образом, интерлейкины являются сигна-
лом нарушения антигенного гомеостаза, который «включает»
стресс-систему (рис. 5.4, стрелка 1). Через активацию гипотала-
мо-гипофизарно-адреналовой оси и соответственно увеличен-
ную секрецию КРГ и глюкокортикоидов иммунная система (по
принципу обратной отрицательной связи) регулирует свою соб-
ственную активность. Действительно, глюкокортикоиды явля-
ются мощным дозозависимым регулятором иммунного ответа и
синтеза интерлейкинов [Sternberg Е.М., Licino J., 1995]. При ги-
перпродукции глюкокортикоиды подавляют, с одной стороны,
стимуляторный эффект цитокинов на гипоталамо-гипофизар-
но-адреналовую ось (см. рис. 5.4, стрелка 2), а с другой — актив-
304
ность иммунокомпетентных клеток (см. рис. 5.4, стрелка 3) и
иммунную/воспалительную реакцию [Bellinger D.L. et al., 1992;
Stratakis C.A., Chrousos G.P., 1995]. Таким образом реализуется
сложный механизм саморегуляции активности иммунной систе-
мы (см. рис. 5.4).
Наличие контура взаимосвязи иммунной системы и
стресс-системы не только обусловливает непосредственное
участие иммунной системы в стресс-реакциях, вызываемых
другими стрессорами, но и определяет влияние стресса на им-
мунореактивность.
Наблюдения на людях и эксперименты на животных убе-
дительно свидетельствуют о влиянии стресса на иммунную сис-
тему и о том, что изменение активности иммунной системы
при эмоциональном стрессе, как и других органов и систем
организма, зависит от трех основных факторов: силы стрессор-
ного воздействия, т.е. от интенсивности стресс-реакции и ее
продолжительности; времени действия стрессора относительно
фазы иммунного ответа; резистентности организма и/или его
иммунной системы к стрессорному повреждению. У здоровых
людей умеренная стресс-реакция может вызывать стимуляцию
активности иммунной системы, усиление неспецифической
противоинфекционной защиты либо незначительное и быстро
проходящее состояние сниженной иммунореактивности. Тя-
желая и длительная стресс-реакция сопряжена с продолжи-
тельным угнетением иммунного ответа вплоть до развития им-
мунодефицитного состояния.
Установлено, что у студентов в период экзаменационной
сессии повышается число В-лимфоцитов и увеличивается уро-
вень иммуноглобулинов в крови при незначительном уменьше-
нии числа Т-лимфоцитов и, в частности, Т-супрессоров. При
этом наиболее существенные изменения возникали у тех студен-
тов, которые в большей мере испытывали чувство тревоги и
страха, что выявлялось при психологическом тестировании.
После каникул параметры иммунологической реактивности, как
правило, нормализовались. У лиц со стабильным психоэмоцио-
нальным статусом, т.е. с незначительной стресс-реакцией или
при ее отсутствии, показатели иммунологической реактивности
оставались в пределах нормы. При умеренной стресс-реакции
на эмоциональные стрессоры у здоровых людей может повы-
шаться активность нормальных киллеров, т.е. клеток, которые
осуществляют контроль антигенного гомеостаза и являются
важным механизмом противоопухолевой защиты.
При действии стрессоров, вызывающих более сильную
стресс-реакцию, возникает угнетение активности иммунной
системы. Так, при сильном однократном эмоциональном
стрессорном воздействии (первый прыжок с парашютом и
т.п.) обнаружено значительное снижение активности нормаль-
ных киллеров. Выраженная депрессия нормальных киллеров
20—1385 305
доказана у космонавтов в ближайшие 2 сут после их спуска с
орбиты. Эти явления ликвидируются через несколько недель,
т.е. являются транзиторными. Однако угнетение механизма
естественной противоопухолевой защиты является фактором
риска онкологических заболеваний, и это следует учитывать,
особенно если столь сильное стрессорное воздействие испы-
тывает человек, имеющий предрасположенность к такого рода
заболеваниям или находящийся в условиях действия онкоген-
ных факторов. Продолжительное и устойчивое снижение ак-
тивности нормальных киллеров отмечено при длительном
психоэмоциональном напряжении. С этим в известной мере
может быть связана стимуляция опухолевого роста у лиц, тя-
жело переживающих горе [Stein М. et al., 1991].
При сильном эмоциональном стрессорном воздействии,
при затяжной стресс-реакции у человека возникают также
снижение содержания иммуноглобулинов в крови и уровня
так называемых нормальных противомикробных антител
[Крыжановский Г.Н. и др., 1997], а также снижение активнос-
ти Т-лимфоцитов и угнетение их реакции на митогены [Stein
М. et al., 1991].
Иммунодепрессивное действие стресса является причиной
того, что вакцинация часто оказывается неэффективной, если
иммунизация проводится на фоне стресса. Это доказано, на-
пример, для прививок против гепатита В [Glaser R. et al.,
1992]. Эмоциональное стрессорное напряжение может приво-
дить также к снижению показателей неспецифической проти-
воинфекционной защиты: снижается число полиморфно-
ядерных лимфоцитов и их способность к фагоцитозу, угнета-
ется вирус-индуцированный синтез интерферона и интерлей-
кинов. Например, после сильного землетрясения в Японии
значительно возросло число случаев острых респираторных за-
болеваний, главным образом пневмоний. Повышение воспри-
имчивости к вирусу Эпштейна—Барр и снижение резистент-
ности к возбудителям респираторных заболеваний отмечено
при длительных тяжелых жизненных ситуациях. Тяжелый
эмоциональный стресс может также провоцировать заболева-
ния, имеющие в основе аллергический или аутоиммунный
компонент, такие как ревматоидный артрит, системная крас-
ная волчанка, множественный склероз [Stratakis С.А.,
Chrousos G.R., 1995].
Таким образом, тяжелый стресс вызывает у человека нару-
шение иммунологического статуса (которое можно определить
как вторичное иммунодефицитное состояние нейрогенной
природы), а также может провоцировать или усиливать прояв-
ление нарушений иммунологического статуса, вызываемых
другими (не нейрогенными) факторами.
Каков механизм стрессорных нарушений иммунологическо-
го статуса? Эта проблема еще далека от полного решения. Вмес-
306
те с тем, определенные успехи в этом направлении достигнуты в
экспериментах на животных. Как показали исследования, у жи-
вотных, как и у людей, умеренное стрессорное воздействие, со-
провождающееся умеренной секрецией гормонов и медиаторов
стресс-системы, приводит к потенциации иммунного ответа.
Напротив, сильное продолжительное стрессорное воздействие,
сопровождающееся значительной активацией стресс-системы,
вызывает иммуносупрессию. В экспериментах на крысах [Меер-
сон Ф.З., Сухих Г.Т., 1985] показано, что непродолжительное
умеренное действие эмоционально-болевого стрессора приво-
дит к увеличению активности нормальных киллеров почти в 2
раза, а затем под влиянием продолжающегося стрессорного воз-
действия развивается выраженная депрессия активности этих
иммунокомпетентных клеток. Умеренная эмоциональная
стресс-реакция сопровождается у крыс повышением иммуно-
компетентности костного мозга в связи с усиливающейся под
влиянием стресса миграцией Т-лимфоцитов из лимфоидных ор-
ганов (см. выше) и увеличением количества клеток-предшест-
венников иммуноцитов [Горизонтов П.Д. и др., 1983]. Повыша-
ется при этом способность Т-лимфоцитов к пролиферации. При
умеренной эмоциональной стресс-реакции у животных обнару-
жены увеличение численности макрофагов и повышение их фа-
гоцитарной активности, увеличение синтеза вирус-индуциро-
ванного интерферона и синтеза интерферона без дополнитель-
ной вирусной стимуляции.
Тяжелый и достаточно длительный стресс подавляет различ-
ные звенья иммунитета: отмечаются стрессорное уменьшение
реакции бластной трансформации Т-лимфоцитов в ответ на ми-
тогены, снижение литической активности Т-лимфоцитов и нор-
мальных киллеров по отношению к опухолевым клеткам-мише-
ням, а также депрессия цитотоксической функции макрофагов.
В основе изменений иммунореактивности при стрессе, по-
видимому, лежит активация стресс-системы и соответственно
действие ее медиаторов, среди которых центральное место зани-
мают КРГ, АКТГ, глюкокортикоиды и катехоламины. Умеренно
увеличенная секреция медиаторов мобилизует систему крови и
оказывает активирующее влияние на иммунный ответ, а значи-
тельно возросшая секреция угнетает иммунореактивность. Так,
еще 50 лет назад ставшие классическими исследования Г.Г.Го-
лодец и Н.В.Пучкова (1948) впервые доказали зависимость фа-
гоцитоза лейкоцитов от активности адренергической системы;
авторы установили на лягушках, что стимуляция симпатической
нервной системы и введение А дозозависимо влияют на фагоци-
тоз. Введение А в физиологических дозах (КГ9—10-8) и слабая
стимуляция симпатической нервной цепочки вызывали актива-
цию фагоцитоза лейкоцитов на введение B.coli, убитых палочек
Коха, частиц кармина и т.п. Введение А в больших дозах (10-7—
10“6) угнетало фагоцитоз.
20*
307
Следует подчеркнуть, что адренергическая система имеет
«надежный выход» на иммунную систему. Как было показано
выше, этот «выход» реализуется благодаря наличию плотной
симпатической иннервации первичных и вторичных лимфо-
идных органов и наличию адренергических рецепторов на им-
мунокомпетентных клетках [Bellinger D.L. et al., 1992; Fried-
man E.M., Irwin M.R., 1995].
В исследованиях Б.А.Фролова и соавт. (1985) на крысах было
показано, что активация первичного иммунного ответа, вызы-
ваемая эмоциональным стрессом, связана с активацией симпа-
тоадреналовой системы и повышением уровня цАМФ в иммун-
нокомпетентных клетках. Авторы убедительно доказали, что им-
мунизация животных эритроцитами барана после перенесенно-
го ЭБС сопровождалась увеличением абсолютного и относи-
тельного числа антителообразующих клеток и титра антител
примерно в 2 раза по сравнению с контролем (без эмоциональ-
но-болевого воздействия). Это явление развивалось на фоне
увеличения продукции катехоламинов в надпочечниках.
Ключевую роль в стрессорной иммуносупрессии играет ак-
тивация гипоталамических КРГ-нейронов и увеличение секре-
ции КРГ. Показано, что введение КРГ в желудочки мозга вы-
зывает дозозависимое снижение активности нормальных кил-
леров и подавление митогениндуцированной пролиферации
лимфоцитов в селезенке и на периферии. Предупреждение по-
вышения уровня КРГ в гипоталамусе при стрессе с помощью
антител к нему предотвращает падение активности естествен-
ных киллеров. Причем удаление надпочечников (т.е. исключе-
ние секреции глюкокортикоидов) не влияло на указанные эф-
фекты К.РГ [Friedman Е.М., Irwin M.R., 1995], что указывает
на самостоятельное (а не через активацию гипоталамо-гипо-
физарно-адреналовой оси) влияние КРГ на иммунную систе-
му. Это влияние реализуется на уровне мозга, т.е. через воз-
действие КРГ на рецепторы мозговых структур, так как пря-
мое введение КРГ в кровь не оказывает влияния на иммунную
систему [Friedman Е.М., Irwin M.R., 1995], хотя КРГ-рецепто-
ры показаны на периферических сайтах иммунной системы,
например на макрофагах.
Имеющиеся к настоящему времени данные позволяют по-
лагать, что иммунодепрессивное действие КРГ реализуется
через активацию центров адренергического звена стресс-сис-
темы, которая приводит к увеличенному влиянию катехолами-
нов симпатической системы и надпочечников на иммунную
систему [Felten D.L. et al., 1987; Sundar S.K. et al., 1990; Fried-
man E.M., Irwin M.R., 1995]. Эту ситуацию демонстрирует ги-
потетическая схема на рис. 5.5.
Увеличение содержания КРГ в паравентрикулярных ядрах
гипоталамуса при стимуляции его секреции под влиянием
стресса (или при введении экзогенного КРГ в желудочки мозга)
308
Рис. 5.5. Гипотетическая схема механизма подавления иммунореак-
тивности при стрессе, связанного с повышением секреции кортико-
тропин-рилизинг-гормона (КРГ) в гипоталамусе и активацией сим-
патической нервной системы.
НА — норадреналин; NPY — нейропептид «Y»; А — адреналин; ГК — глюко-
кортикоиды; СНС — симпатическая нервная система; IL-1 — интерлейкин-1;
АВ — аргинин-вазопрессин. Стрелками со знаком «+» обозначено активирую-
щее влияние, стрелками со знаком «—» обозначено угнетающее влияние [Fel-
ten D.L. et al., 1987; Sundar S.K. et al., 1990; Friedman E.M., Irwin M.R., 1995].
Пояснения в тексте.
приводит к активации симпатической системы. В результате
этой активации НА и колокализованный с ним в симпатических
терминалях нейропептид Y (NPY) воздействуют на иммуноком-
петентные органы и клетки (в частности, на селезенку и лимфа-
тические узлы брыжейки), вследствие чего происходят угнете-
ние процесса образования антител в ответ на антигены, а также
угнетение активности нормальных киллеров и уменьшение ми-
тогениндуцированной пролиферации макрофагов. Под влияни-
ем симпатической регуляции активируется выброс адреналина
из медуллы надпочечников, увеличивается его влияние на им-
мунную систему, что приводит к подавлению иммунного ответа.
Существенно, что при стрессе активация секреции КРГ в гипо-
таламусе происходит не только под влиянием вызываемого
стрессом возбуждения паравентрикулярных ядер, но и под влия-
нием IL-1, продуцируемого макрофагами в ответ на действие
стресс-белков, т.е. белков теплового шока (см. рис. 5.5). Эти
белки синтезируются под влиянием стресса бактериями и вос-
принимаются иммунными клетками как антиген, что усиливает
иммунодепрессивное действие стресса [Акмаев И.Г., 1996; Sun-
dar S.K. et al., 1990]. Естественно, что при действии иммуноген-
ных антигенов на фоне стресса рассмотренная ситуация (т.е. ги-
перактивация КРГ-нейронов гипоталамуса) еще более усугубля-
ется дополнительной продукцией интерлейкинов иммуноком-
петентными клетками.
В целом наличие в центральном регуляторном контуре им-
мунной системы КРГ-звена, по-видимому, определяет депрес-
сивное действие стресса на иммунный ответ: с интенсивной
секрецией КРГ в гипоталамусе при стрессе суммируется сек-
реция КРГ, вызываемая интерлейкинами при ответе на имму-
ногенный антиген, и в результате происходит угнетение им-
мунного ответа от избытка КРГ.
Важную роль в регуляции иммунной системы при стресс-
реакции, несомненно, играет изменение секреции глюкокор-
тикоидов, являющихся, как уже говорилось выше, ключевым
фактором саморегуляции активности иммунной системы в
норме (см. рис. 5.4). В экспериментах показано, что гиперпро-
дукция этих гормонов или введение больших доз извне оказы-
вает депрессивное действие на иммунную систему, введение
же «физиологических доз этих гормонов или умеренное увели-
чение их секреции, напротив, способствует активации иммун-
ного ответа [Dhabhar F.S. et al., 1996, и др.]. Поэтому очевид-
но, что при сильном стрессорном воздействии активация оси
«гипоталамус — гипофиз — надпочечники» вызывает гипер-
продукцию глюкокортикоидов, и это наряду с гиперсекрецией
КРГ приводит к иммунодепрессии (см. рис. 5.5).
Уместно обратить внимание на роль стресс-лимитирующих
систем в регуляции иммунного ответа при стрессе. Показано,
что сильный эмоциональный стресс (иммобилизация в тече-
310
ние ночи) вызывал у мышей резкое уменьшение пролифера-
ции лимфоцитов, стимулированной эритроцитами барана, и
снижение активности нормальных киллеров в селезенке. Вве-
дение Met-энкефалина (медиатора стресс-лимитирующей
опиоидергической системы) снимало указанное подавление
иммунореактивности за счет ограничения стресс-реакции и
предупреждения вызываемого стрессом подъема уровня глю-
кокортикоидов в крови [Marotti Т. et al., 1996].
Таким образом, очевидно, что в реакции иммунной систе-
мы на стресс важную роль играет соотношение активностей
стресс-системы и стресс-лимитирующих систем.
При частых эпизодах сильного стресса, при хроническом
стрессе рассмотренная выше гиперактивация контура КРГ —
симпатическая система — медулла надпочечников может при-
вести к истощению этого контура, дефициту продукции КРГ и
«поломке» регуляции иммунной системы с переходом к «пато-
логическому увеличению» ее функции [Friedman Е.М., Irwin
M.R., 1995]. Это состояние сопровождается нарушением такой
важной функции, как дифференцирование своего и чужого
антигенов. В результате развиваются иммунные «атаки», на-
правленные против собственных клеток или отдельных белков
[Акмаев И.Г., 1996]. В качестве примера можно привести так
называемый стрессорный диабет, когда мишенью аутоиммун-
ных реакций являются В-клетки поджелудочной железы, сек-
ретирующие инсулин. Может быть также, что мишенью им-
мунных атак становится основной белок миелина, оболочки-
«изолятора» нервных волокон. В результате нарушается ин-
нервация мышц и развивается тяжелое заболевание, напоми-
нающее по симптомам заболевание, известное под названием
«рассеянный, или множественный, склероз».
Проблема стрессорных иммунных болезней далека от реше-
ния, однако в пользу приведенных положений свидетельствуют
данные о том, что экспериментальные модели аутоиммунных
болезней человека, в частности ревматоидного артрита, характе-
ризуются сниженной секрецией КРГ и снижением экспрессии
гена КРГ в гипоталамусе [Harbuz M.S. et al., 1995]. Кроме того,
имеются данные о том, что у больных ревматоидным артритом
снижена реакция гипоталамо-гипофизарно-адреналовой оси на
хирургический стресс [Chikanza I.S. et al., 1992 a, b; Stratakis C.A.,
Chrousos G.P., 1995]. Уместно здесь также упомянуть, что одним
из ключевых терапевтических средств, используемых для лече-
ния и профилактики обострения аутоиммунных заболеваний,
являются кортикостероиды и, в частности, гидрокортизон и
другие аналоги кортикостерона. Таким образом, можно пола-
гать, что истощение функции гипоталамо-гипофизарно-корти-
ко-адреналовой системы в результате хронической стрессорной
гиперактивации может стать причиной аутоиммунных состоя-
ний либо провоцировать и усугублять эти состояния, если в ор-
311
ганизме уже имеется дефицит (например, врожденный) функ-
ции этой системы.
Мы подошли к важному аспекту проблемы иммунопатоло-
гии — роли наследственных, т.е. генетически обусловленных,
особенностей стресс-системы в регуляции иммунной системы.
Этот аспект мало изучен, однако наблюдения в клинике и экс-
перименты на животных позволяют полагать, что наследст-
венно обусловленная сниженная активность стресс-системы и
неадекватно слабая стресс-реакция могут быть причиной ауто-
иммунных воспалительных патологий [Stratakis С.А., Chrousos
G.P., 1995; Harbuz M.S. et al., 1995]. Более высокая активность
стресс-системы сопровождается более высокой устойчивостью
иммунной системы к стрессу. Показано, что у крыс линии
Lewis, которые характеризуются низкой реактивностью
стресс-системы и низкой секрецией КРГ в гипоталамусе, на-
блюдается повышенная чувствительность к аутоиммунному
воспалению по сравнению с крысами линии Fisher, которые
характеризуются более высокой активностью системы гипота-
ламус — гипофиз — надпочечники, более высокой секрецией
КРГ и более высокой резистентностью к воспалительным бо-
лезням [Stenberg Е.М., Licino J., 1995]. Далее в экспериментах
на домовых мышах и мышах-полевках установлено, что им-
мунная система у полевок обладает повышенной резистент-
ностью к стрессорному воздействию по сравнению с иммун-
ной системой домовых мышей. У домовых мышей эмоцио-
нальный стресс угнетал индуцируемую введением эритроци-
тов барана продукцию иммуноглобулинов (IgM и IgG), что
было связано с «выбросом» глюкокортикоидов. У полевок
такой же стресс не влиял на иммунореактивность. Выясни-
лось, что полевки обладают более высокой базальной и пост-
стрессорной активностью системы гипоталамус — гипофиз —
надпочечники и для получения у них угнетения иммунного
ответа требуется более высокий уровень в крови глюкокорти-
коидов, чем для домовых мышей [Devries А.С. et al., 1997].
Эти данные согласуются с фактами, полученными ранее на
крысах линий Август и Вистар [Фролов Б.А. и др., 1985].
У крыс линии Август, обладающих, как уже упоминалось
выше, более высоким базальным и постстрессорным уровнем
кортикостерона в крови по сравнению с крысами Вистар, пер-
вичный иммунный ответ на введение эритроцитов барана
после стрессорного воздействия (ЭБС) был существенно боль-
ше, чем у крыс Вистар. Это означает, что у крыс Август устой-
чивость иммунной системы к стрессорному повреждению
выше.
Таким образом, генетические особенности стресс-систе-
мы, несомненно, играют важную роль в резистентности им-
мунной системы к стресс-реакции и механизме стрессорной
иммунопатологии.
312
Заключение
Острый умеренный эмоциональный стресс или повторные
эпизоды умеренного стрессорного воздействия либо не влия-
ют на иммунореактивность организма, либо вызывают транзи-
торную ее стимуляцию. В основе стимуляторного эффекта
стресса лежит активация симпатической системы и действие
физиологических концентраций катехоламинов, вазопрессина
и глюкокортикоидов на иммунную систему. Это явление спо-
собствует повышению резистентности организма к инфек-
циям.
Тяжелый острый эмоциональный стресс вызывает угнете-
ние иммунореактивности. В основе стрессорной иммуноде-
прессии лежит интенсивная секреция КРГ в гипоталамусе с
последующим резким увеличением влияния симпатической
системы и катехоламинов надпочечников на иммунокомпе-
тентные органы и клетки, а также секреция АКТГ и глюко-
кортикоидов (см. рис. 5.5). При эпизодическом характере та-
кого стресса угнетение иммунореактивности является тран-
зиторным и иммунная система нормализуется через несколь-
ко недель после стрессорного эпизода. При более длитель-
ном стрессорном воздействии такого типа развивается стой-
кое иммунодефицитное состояние. Такое состояние приво-
дит к повышению восприимчивости организма к инфекциям
и является важным фактором риска онкологических заболе-
ваний.
Продолжительный (хронический) тяжелый эмоциональ-
ный стресс, вызывающий длительную гиперактивацию конту-
ра «КРГ — катехоламины и глюкокортикоиды — иммунная
система» может приводить к истощению этого контура, дефи-
циту секреции КРГ и глюкокортикоидов и «поломке» меха-
низмов иммунореактивности. Эта ситуация может приводить
к патологическому увеличению иммунореактивности, аутоим-
мунным атакам и как результат — к стрессорным аутоиммун-
ным состояниям или провоцированию аутоиммунных болез-
ней другой этиологии.
5.2.3.4. Психический статус при эмоциональном стрессе
и посттравматическое стрессовое расстройство
Известно, что существенным проявлением стрессорной
патологии человека, которое в значительной мере определяет
вегетативные компоненты стрессорных поражений, является
нарушение психического статуса. В этом разделе мы рассмот-
рим в общем виде стрессорные нарушения психического ста-
туса и более подробно особый и весьма тягостный их вид —
так называемые посттравматические стрессовые расстройства.
313
5.2.З.4.1. Нарушения психического статуса
Остро переживаемые эмоциональные стрессорные ситуа-
ции (психосоциальные, семейные, катастрофы и т.п.) нередко
приводят к психическим нарушениям, большую часть которых
при длительном действии стрессорных ситуаций составляет
развитие невротического статуса. В частности, как показал
ретроспективный анализ, у людей с таким статусом на 50 %
больше, чем у здоровых, выявляется в анамнезе напряженных
и конфликтных стрессорных ситуаций. При этом важную роль
в развитии патологических реакций на эмоциональный стрес-
сор имеет социализация детства, т.е. переживание в детстве
тяжелых стрессорных событий, и базальный психический ста-
тус [Соколова Е.Б. и др., 1996; Судаков К.В., 1998].
Первичное звено в развитии стрессорного нарушения пси-
хического статуса — психическое состояние (фрустрация), ко-
торое возникает при действии отрицательных эмоциональных
стрессоров и проявляется ощущением неудовлетворенности и
психического напряжения, возникающего из-за невозможнос-
ти реализовать те или иные цели. Вероятность развития нару-
шений психического статуса зависит от особенностей личнос-
ти, ресурсов личностной выносливости, т.е. меры способности
активного преодоления трудностей, которая определяется
способностью к построению интегративного поведения. При
хорошей интегративности поведения энергия распределяется
по различным направлениям поведения в соответствии с важ-
ностью реализуемых потребностей. Чем выше способность к
интеграции поведения, тем более успешно преодоление стрес-
согенных ситуаций. Нарушение соответствия между актуаль-
ными потребностями человека и возможностями их удовле-
творения, недостаточность физических и психических ресур-
сов (личностной выносливости), опасения личности, связан-
ные с вероятной неспособностью реализовать значимые уст-
ремления в будущем, а также с возможным ухудшением ситуа-
ции, вызывают тревогу. Тревога — центральный элемент пси-
хического стресса. Она как сигнал неблагополучия и опаснос-
ти активирует механизмы так называемой психической адап-
тации, которая может быть либо в виде пассивного приспо-
собления, либо в виде активного процесса в зависимости от
указанных выше личностных особенностей. Именно активная
психическая адаптация позволяет сохранить физическое и
психическое здоровье, обеспечить адекватное требованиям
среды поведение [Березин Ф.Б., Мирошников М.П., 1996; Со-
колова Е.Б. и др., 1996]. Психическая адаптация направлена
на преодоление (совладание) стрессогенной ситуации. Это пре-
одоление, или преодолевающее поведение, включает сложные
взаимосвязанные когнитивные (мыслительные) и эмоцио-
нальные механизмы, которые обеспечивают планирование це-
314
ленаправленных действий и определяют усилия, ориентиро-
ванные на управление трудно преодолеваемой стрессогенной
ситуацией. Если эмоциональный стресс приводит к стойким
нарушениям психической адаптации, они проявляются кли-
нически выраженными расстройствами психоневрологическо-
го статуса.
В настоящее время психические клинические явления,
обусловленные эмоциональным стрессом, разделяют (весьма
относительно, на наш взгляд) в зависимости от характера
стрессорного воздействия на острые стресс-реакции, которые
возникают после сверхсильного травматического «пережива-
ния» угрожающего характера и завершаются в течение дней, а
иногда и часов, и встречаются сравнительно нечасто; по-
сттравматические стрессовые расстройства, которые пред-
ставляют собой затяжную реакцию на подобного рода пережи-
вания; стресс-реакции в ответ на стрессорные события, влеку-
щие за собой жизненные изменения, т.е. адаптивные реакции;
невротические состояния и личностные декомпенсации, при ко-
торых обычно обнаруживается зависимость от повторного или
хронического воздействия эмоциональных стрессоров (жиз-
ненных ситуаций), трудно разрешимых эмоциональных про-
блем, психических конфликтов, длительного нарастания фру-
страции [Соколова Е.Б. и др., 1996; Тарабрина Н.В. и др.,
1996].
При острых психических стресс-реакциях, классифицируе-
мых в настоящее время как «афективно-шоковые реакции»*,
выраженность тревоги столь велика, что приводит к наруше-
нию ориентировки, сужению сознания, дезорганизации пове-
дения, характеризующегося хаотичностью, ажитацией или, на-
против, — заторможенностью, достигающей степени ступора.
Сюда включаются проявления гнева и депрессии, диссоциа-
тивные, истерические расстройства и т.п. Острые психические
явления обычно сочетаются с вегетативными коррелятами
тревоги, которые весьма многообразны (сердцебиением, чув-
ством удушья, головокружением, потливостью, дискинезией
ЖКТ, тошнотой, вегетососудистыми кризами и т.п.). С ост-
рым стрессом могут ассоциироваться также острые психоти-
ческие расстройства (реактивные психозы).
Более сложное и протяженное во времени стрессорное рас-
стройство психического статуса — это идущее вслед за острым
стрессом катастрофы посттравматическое стрессовое рас-
стройство, в результате которого может развиваться стойкое
расстройство личности. Этот вид стрессорного нарушения
психического статуса будет рассмотрен отдельно далее.
* Руководство по психиатрии/Под ред. А.С.Тиганова. — М.: Медици-
на, 1999. - Т. 2. - С. 492.
315
Адаптивные стресс-реакции в ответ на жизненные изменения,
а также невротические состояния (или личностные декомпенса-
ции) характеризуются нарушением психической адаптации и оп-
ределяются однотипными клиническими проявлениями. Эти
нарушения, как уже упоминалось выше, встречаются чаще
всего. Усиление в настоящее время стрессогенных воздействий,
резкое изменение социальных стереотипов и социальных цен-
ностей, изменение социального статуса большинства населения
не в лучшую сторону увеличивают вероятность нарушений пси-
хической адаптации, так как в таких условиях привычные адап-
тивные формы поведения становятся неадекватными новым об-
стоятельствам, что порождает в конечном итоге тревогу. Поми-
мо тревоги, эти психические нарушения проявляются депрес-
сивными расстройствами, а также личностными декомпенса-
циями, характеризующимися настороженными реакциями аф-
фективно-ригидных личностей. Среди состояний, определяю-
щихся непосредственно выраженной тревогой, выделяются ге-
нерализованное тревожное расстройство, пароксизмальная (эпи-
зодическая) тревога и тревожно-фобические расстройства. Все
эти расстройства сопровождаются вегетативными проявления-
ми тревоги, указанными выше.
Следует еще раз подчеркнуть, что возникновение указан-
ных стрессорных нарушений психической адаптации и психи-
ческого статуса и выраженность этих нарушений зависят от
исходного (базального) психического статуса личности, опре-
деляющего в значительной мере устойчивость организма к
стрессу. Так, например, тревожные расстройства чаще всего
наблюдаются у психастенических, «тревожных» личностей;
депрессивные состояния невротического уровня возникают
чаще всего у психастенических и субдепрессивных личностей
[Березин Ф.Б. и др., 1996; Соколова Е.Б. и др., 1996; Вейн
А.М., 1997]. При этом несомненно, что исходный психичес-
кий статус личности и устойчивость к стрессорному наруше-
нию этого статуса, а также вероятность развития указанных
выше патологий в значительной степени определяются гене-
тически обусловленным и/или измененным в процессе жизне-
деятельности состоянием стресс-системы и ее реактивностью.
5.2.3.4.2. Посттравматическое стрессовое расстройство*
«Посттравматическое стрессовое расстройство (ПТСР)
возникает как отсроченный или затянувшийся ответ на стрес-
совое событие (краткое или продолжительное) исключительно
* Руководство по психиатрии/Под ред. А.С.Тиганова. — М.: Медици-
на, 1999. - Т. 2. - С. 517-526.
316
угрожающего или катастрофического характера, которое
может вызывать глубокий стресс почти у каждого. Предраспо-
лагающие факторы, такие как личностные особенности или
нервное заболевание в анамнезе, могут снизить порог для раз-
вития синдрома или усугубить его течение, но они никогда не
являются необходимыми или достаточными для объяснения
его возникновения...». Такое определение дано ПТСР в пара-
графе F-43.1 «Международной статистической классификации
болезней и проблем, связанных со здоровьем» (1995). В соот-
ветствии с положениями Американской психиатрической ас-
социации F.W. Putnam из национального института психичес-
кого здоровья Бетезды (США) охарактеризовал ПТСР как
психиатрический синдром, определяемый тремя группами
симптомов: 1) симптомы «репереживания», т.е. повторное
воспроизведение в памяти травматических событий, интен-
сивная психологическая и физиологическая реактивность к
стимулам, которые ассоциируются с травмой; 2) симптомы из-
бегания, т.е. попытки избежать напоминаний о травме, ощу-
щение отстраненности и отчуждения; 3) упорные симптомы
гипервозбуждения, т.е. трудности засыпания, нарушение сна,
гипермнительность, болезненно увеличенные реакции на ин-
формацию [Putnam F.W., 1995].
Представления о ПТСР сложились примерно 20 лет назад,
когда после анализа «корейского и вьетнамского психического
синдрома» у прошедших войну американских солдат посттрав-
матическое стрессовое расстройство было включено в класси-
фикационный психиатрический стандарт, подготовленный
Американской психиатрической ассоциацией. Однако только в
1992 г. ПТСР впервые было внесено в «Международную статис-
тическую классификацию болезней и проблем, связанных со
здоровьем». Вместе с тем последствия действия экстремальных
травматических ситуаций изучались уже давно. В результате
были выделены «военный невроз», «невроз истощения, или хро-
нической военной усталости», «неврозы пожаров, землетрясе-
ний» и т.п. [Березин Ф.Б., Мирошников М.П., 1996], которые
практически являются проявлениями эмоционального стресса.
Подобные расстройства наблюдались не только при массовых
катастрофах, но и у узников концлагерей, у солдат, перенесших
плен, а также у других жертв насилия, в частности у изнасило-
ванных. Отечественные исследования последнего времени бази-
ровались в основном на изучении ПТСР у солдат, воевавших в
Афганистане, у лиц, перенесших землетрясение в Армении, ава-
рию на Чернобыльской АЭС [Тарабрина Н.В. и др., 1996].
В частности, уровень психических расстройств у ликвидаторов
аварии на Чернобыльской АЭС постоянно увеличивался с 1987
до 1991 г. и далее стабилизировался на высоком уровне: в 1993 г.
у ликвидаторов этот уровень превосходил общероссийский уро-
вень в 9,6 раза [Мороз Б.Б., Дешевой Ю.Б., 1999].
317
Существенно, что общие закономерности возникновения
и развития ПТСР не зависят от того, какое конкретно травма-
тическое событие явилось этиологическим фактором (стрессо-
ром), если эти события носили экстремальный характер и вы-
зывали интенсивный страх, ужас и ощущение беспомощности,
т.е. безысходности.
Вместе с тем, несмотря на неизбежность возникновения
ПТСР при указанных ситуациях, решающее значение для этого
возникновения, по мнению многих исследователей, имеет субъ-
ективная оценка степени угрозы и выраженность эмоций, обу-
словленных этой угрозой [Тарабрина Н.В. и др., 1996]. Таким
образом, и при ПТСР актуальными остаются индивидуальные
особенности человека, т.е. его история и состояние стресс-сис-
темы. В частности, при обследовании ликвидаторов последст-
вий аварии на Чернобыльской АЭС было установлено, что при-
мерно 20 % из числа обследованных страдали ПТСР, и именно
эти лица субъективно оценивали радиационную опасность как
высокую и считали себя серьезно пострадавшими от облучения,
хотя объективно у них не были выявлены признаки лучевого по-
ражения. Эти лица испытывали во время пребывания в аварий-
ной зоне интенсивные отрицательные эмоции, тревогу и страх
за свое здоровье. Кроме первичных стрессогенных эмоций, у
данных лиц имелись вторичные стрессорные факторы, которые
усиливали эффект первичных, а именно — переоценка любых
недомоганий и рассмотрение их как проявление «ожидаемой»
лучевой болезни, а также страх «отдаленных последствий» облу-
чения [Тарабрина Н.В. и др., 1994].
При ПТСР наблюдаются нейропсихологические наруше-
ния в виде расстройства памяти, познавательных способнос-
тей. Нарушения процессов интеграции информации, памяти и
эмоций, по мнению исследователей, сходны с состоянием,
обозначаемым в неврологии как «диссоциация сознания»
[Carlson Е.В., Putnam F.W., 1993]. Современные представле-
ния о диссоциации свидетельствуют, что диссоциация — до-
вольно распространенный процесс, который присущ в той или
иной степени большинству индивидуумов, но этот процесс
приобретает патологическую значимость только при его экс-
тремальной форме. Однако исследования ПТСР показывают,
что наличие у человека в анамнезе до травмы симптомов дис-
социации является решающим предиктором развития ПТСР.
Конкретный нейрональный механизм формирования
ПТСР еще неясен. Но несомненно, что в основе этого меха-
низма лежат процессы, определяющие формирование самого
эмоционального стресса как такового. При этом, как неодно-
кратно отмечалось в литературе, вопрос о природе отрицатель-
ного эмоционального стресса и, по-видимому, ПТСР «упира-
ется» в расшифровку нейрохимических механизмов, лежащих
в основе «застойных» отрицательных эмоций.
318
Роковой особенностью ПТСР является то обстоятельство,
что для его возникновения достаточно однократного и единст-
венного сильного воздействия, вызывающего потрясение. Это
потрясение закрепляется в памяти и преобразуется затем в
своего рода программы поведения, которым человек неосо-
знанно следует, с одной стороны, и которые способствуют
развитию патологических состояний — с другой. Ключевую
роль в этом процессе, несомненно, играет условнорефлекгор-
ный механизм. Он закрепляет за целым рядом событий и
предметов, которые сопутствовали страшной травматической
ситуации, статус мощного условного сигнала этой ситуации.
В результате «единожды» случившаяся тяжелая ситуация полу-
чает повторы и как бы осуществляется «многожды», т.е. про-
цесс прогрессирует во времени. Не исключена возможность,
что эти условные сигналы обрастают вторично-условными и
т.д. В пользу такого представления свидетельствуют, в част-
ности, давние исследования Ф.П.Ведяева (1975), который изу-
чал роль различных структур лимбической системы мозга в
формировании нейрогуморальных проявлений эмоциональ-
ной стресс-реакции. Он показал, что электростимуляция лим-
бических структур (базальных ядер амигдалы и вентрального
гиппокампа) вызывает у кроликов агрессивно-оборонитель-
ную реакцию, характерную для эмоциональной стресс-реак-
ции в ответ на угрожающий стрессор. Давая одновременно со
стимуляцией ранее индифферентный звуковой сигнал, автор
установил, что этот сигнал дает такую же агрессивно-оборони-
тельную стресс-реакцию, и при этом возникает биоэлектри-
ческая активность в лимбических структурах: амигдале, гип-
покампе, перегородке. Это сопровождается соответствующей
двигательной реакцией, увеличением частоты сердечных со-
кращений, дыхания и т.п. [Ведяев Ф.П., 1975].
Важную роль в предрасположенности к развитию ПТСР и
реализации этого состояния играет активность стресс-системы,
поскольку состояние ПТСР характеризуется определенными из-
менениями в этой системе и стресс-лимитирующих системах.
У больных с ПТСР доказана увеличенная активность симпати-
ческой нервной системы. Это проявляется значительным увели-
чением суточной экскреции с мочой катехоламинов, а также
увеличением частоты сердечных сокращений, АД и увеличенны-
ми эмоциональными реакциями; причем уровень экскреции ка-
техоламинов с мочой позитивно коррелирует с проявлениями
симптомов ПТСР, квалифицируемых как феномен диссоциа-
ции. Как было уже отмечено выше, существует определенная
связь между НА-нейронами мозга, главным образом НА-нейро-
нами синего пятна, и поведением при эмоциональном стрессе
тревоги и страха. Нейроны синего пятна с проекцией через кору
мозга и подкорковые структуры, включая гиппокамп, амигдалу,
таламус и гипоталамус, образуют нейроанатомическую структу-
319
ру, которая позволяет реализовать быструю и глобальную моду-
ляцию функции мозга и реализацию эмоциональной стресс-ре-
акции. Хронический эмоциональный стресс приводит к дли-
тельному застойному возбуждению нейронов синего пятна и
увеличенному «выбросу» НА в иннервируемые структуры
[Bremner J.D. et al., 1996а]. Таким образом, по мнению авторов
этого исследования, НА непосредственно вовлечен в механизм
«обусловленного страха», связанного со стрессом. Это позволяет
понять механизм реализации состояния ПТСР, в развитии кото-
рого важную роль играет этот «обусловленный», или условно-
рефлекторный, страх. Можно думать, что условные сигналы
пережитой страшной ситуации через данный НА-ергический
механизм «подпитывают» состояние посттравматического
стресса, подкрепляя застойные отрицательные эмоции. Дейст-
вительно показано, что у больных с ПТСР и с синдромом пани-
ки увеличена реактивность НА-системы мозга [Bremner J.D. et
al., 1996b].
У пациентов с ПТСР нарушена регуляция активности ги-
поталамо-гипофизарно-адреналовой оси. Обнаружено сниже-
ние уровня секреции АКТГ в гипофизе в ответ на экзогенные
стимуляторы, например введение КРГ [Yehuda R. et al., 1991;
De Bellis M.D. et al., 1994]. При этом доказано, что у людей,
перенесших психическую травму, которая не сопровождается
развитием ПТСР, не наблюдается нарушений активности оси
гипоталамус — гипофиз — надпочечники. У людей, у которых
подобная травма привела к возникновению ПТСР, отмечается
четкая недостаточность функции этой оси. Все эти люди ха-
рактеризуются резко сниженным уровнем кортизола в крови и
моче и соответственно резким повышением числа и чувстви-
тельности глюкокортикоидных рецепторов лимфоцитов. При
этом чем более тягостны проявления ПТСР, тем ниже уровень
кортизола [Yehuda R., 1997].
Существенно, что риск возникновения ПТСР после пере-
несения эмоциональной травмы четко определяется исходным
состоянием стресс-системы. Если до травмы реакция на ост-
рое действие стресса была высокой (т.е. с большим «выбро-
сом» кортизола), то у таких людей ПТСР никогда не развива-
лось; если же реакция была очень слабо выраженной, то
ПТСР развивалось в полной мере. Промежуточное состояние
стресс-реакции характеризовалось возникновением транзи-
торного ПТСР. Также показано, что нанесение повторной
эмоциональной травмы на фоне сниженного (от первой трав-
мы) уровня кортизола приводило к более тяжелому ПТСР.
Выявлена корреляция между ПТСР и активностью стресс-
лимитирующих систем. Показано, что между проявлениями
«диссоциации» и уровнем медиатора опиоидергической
стресс-лимитирующей системы p-эндорфина в цереброспи-
нальной жидкости наблюдается отрицательная корреляция:
320
чем меньше уровень этого пептида, тем более выражены нев-
рологические нарушения. Одновременно обнаружена поло-
жительная корреляция между указанными нарушениями и
уровнем гомованилиновой кислоты (метаболита дофамина) и
5-гидроксииндолуксусной кислоты (метаболита серотонина) в
цереброспинальной жидкости [Demitrack М.А. et al., 1993].
Отмеченный выше уменьшенный базальный уровень р-эндор-
фина в цереброспинальной жидкости у больных с ПТСР
может иметь отношение к снижению болевого порога у таких
больных в покое. Однако в период обострения, возникающего
всегда при действии факторов, связанных с травматическими
событиями (вторичных сигналов), у больных отмечается
яркий анальгетический эффект, эквивалентный в/в ведению
8 мг морфина, причина этого неясна. Возможно, это явление
связано с указанной выше условнорефлекторной активацией
НА-ергической системы, что опосредованно через выброс НА
приводит к активации ОП-нейронов. Однако это предположе-
ние требует специального экспериментального исследования.
Показано, что у крыс «обусловленный страх» (т.е. страх, воз-
никающий, как уже упоминалось выше, при действии факто-
ров, ассоциируемых с перенесенными в прошлом «неприят-
ностями», а в данном случае — с нанесением болевых стиму-
лов) и состояние тревожности характеризуются снижением
ГАМК-передачи в мозгу, выражающимся в уменьшении свя-
зывания меченой ГАМК, и бензодиазепинов с мембранами в
препаратах мозга [Foddi M.S. et al., 1995]. Это означает, что
реализация эмоционального отрицательного стресса и состоя-
ние страха могут зависеть от активности ГАМК-системы и
системы ОП. Иными словами, недостаточность функциониро-
вания данных стресс-лимитирующих систем может быть
одним из эндогенных механизмов, предрасполагающих к раз-
витию ПТСР. В свою очередь активация этих систем или ис-
пользование их медиаторов, по-видимому, могут быть исполь-
зованы для коррекции ПТСР.
Заключение
ПТСР представляет собой стойкую эмоциональную стресс-
реакцию, возникающую у человека в ответ на однократное
действие сверхсильного эмоционального стрессора, вызываю-
щего «потрясение». Эта патологическая застойная реакция ха-
рактеризуется психиатрическим синдромом, сходным с состо-
янием «диссоциации сознания». В основе развития ПТСР
лежит условнорефлекторный механизм: вызванная однажды
эмоциональная стресс-реакция «способна» подкрепляться
вторичными сигналами, т.е. факторами, ситуациями и т.п.,
непосредственно связанными в сознании пациента с первич-
21 — 1385
321
ной причиной потрясения. Важную роль в патогенезе ПТСР
играет состояние стресс-системы и стресс-лимитирующих
систем. ПТСР характеризуется гиперактивацией адренерги-
ческого звена стресс-системы и дефицитом активности ее ги-
поталамо-гипофизарно-кортико-адреналового звена. Исходно
сниженная активность гипоталамо-гипофизарно-кортико-ад-
реналового звена является решающим фактором риска разви-
тия ПТСР. ПТСР характеризуется дефицитом активности
опиоидергической и ГАМКергической, а возможно, и других
стресс-лимитирующих систем.
5.2.4. Основы предрасположенности
и устойчивости к стрессорным
повреждениям
Известно, что разные люди, а также животные одного
вида, но разных генетических линий обладают различной ус-
тойчивостью (резистентностью) к стрессорным воздействиям.
Кроме того, в пределах одной линии степень резистентности у
разных особей также может быть неодинаковой, т.е. существу-
ют индивидуальные различия — различия, приобретенные в
ходе индивидуальной жизни (фенотипические различия). Ин-
дивидуальные различия включают также состояние организма
в момент действия стрессора.
Предрасположенность к стрессорным повреждениям и ус-
тойчивость к ним определяются двумя основными фактора-
ми: генетически обусловленными особенностями организма
и фенотипически обусловленными особенностями, в том
числе состоянием здоровья в момент действия стрессора.
Действительно, накоплено достаточно данных, которые
свидетельствуют о существенном вкладе генотипа в характер
реактивности организма к стрессорам. Ярким примером этому
являются результаты исследований поведения в условиях дей-
ствия эмоционального стрессора потомков полного диаллель-
ного скрещивания инбредных линий мышей. Было установле-
но, что в генофонде линий BALB/C и СЗН/Не сконцентриро-
ваны гены аддитивного действия, ответственные за интенсив-
ную эмоциональную стресс-реакцию, а в генофонде линий
C57BL/6 и AKR/J — гены обратного действия. Иными слова-
ми, генотип мышей первой пары линий определяет большую
чувствительность к стрессорному воздействию и поврежде-
нию, а генотип мышей второй пары линий определяет относи-
тельную резистентность к стрессорному воздействию [Боро-
дин П.М. и др., 1976].
Имеющиеся к настоящему времени данные позволяют по-
лагать, что ключевым звеном генетически обусловленных осо-
322
бенностей организма, определяющих его предрасположен-
ность и устойчивость к стрессу, является уровень активности
различных звеньев стресс-системы и стресс-лимитирующих сис-
тем и степень их активации (реактивности) под влиянием
стрессора. Уровень активности этих систем детерминирован
генетически, т.е. является наследственным, однако может из-
меняться в процессе жизнедеятельности.
В качестве примера можно напомнить приведенные выше
данные, полученные при сопоставлении устойчивости к стрес-
сорным воздействиям у мышей двух популяций: домовых и
полевок. Показано, что при одинаковых по силе и длитель-
ности стрессорных воздействиях у домовых мышей полностью
подавлялся иммунный ответ на введение эритроцитов барана,
а у полевок этот ответ реализовался полностью. Для получе-
ния подавления иммунного ответа у полевок требовалось
более сильное стрессорное воздействие и больший «выброс»
кортикостероидов. Выяснилось, что высокая резистентность к
стрессу у полевок связана с более высокой врожденной актив-
ностью гипофизарно-адреналового звена стресс-системы
[Devries А.С. et al., 1997].
Существенно, что в зависимости от уровня активности и
реактивности разных отделов стресс-системы и стресс-лими-
тирующих систем может оказаться различной резистентность
разных органов и систем к стрессорным повреждениям. Так,
при сравнении устойчивости к стрессорным повреждениям у
крыс двух генетических линий — Август и Вистар — было по-
казано, что крысы Вистар оказались более устойчивыми, чем
крысы Август, к нарушению АД при длительном иммобилиза-
ционном стрессорном воздействии и к тепловому шоку. Одна-
ко к стрессорному язвообразованию в желудке, а также к ост-
рому инфаркту миокарда, важным компонентом которого яв-
ляется стресс-реакция, более устойчивыми оказались крысы
Август. Анализ этих фактов позволяет полагать, что важную
роль в этих межлинейных различиях играют генетически обу-
словленные особенности стресс-лимитирующей NO-системы
и кортико-адреналового звена стресс-системы. Действитель-
но, как уже упоминалось выше, у крыс Август базальная и
постстрессорная концентрация NO в органах существенно
выше, чем у крыс Вистар [Микоян В.Д. и др., 1996]. У крыс
Август выше, чем у Вистар, также и уровень в крови стабиль-
ных метаболитов NO, как в норме, так и при стрессорном воз-
действии, что свидетельствует о более высокой продукции NO
у крыс Август по сравнению с крысами Вистар как в норме,
так и в ответ на действие стрессоров [Пшенникова М.Г. и др.,
2000, 2001]. Именно увеличенная продукция NO, очевидно,
является у крыс Август фактором, предупреждающим или ог-
раничивающим стрессорный спазм сосудов слизистой желудка
и образование в ней «линейных ишемических некрозов», по-
21*
323
скольку, по данным K.Ogino и др. (1994), ключевым звеном
патогенеза ишемических язвенных поражений желудка явля-
ется дефицит в нем продукции NO. Кроме того, NO, как уже
упоминалось выше, ограничивает повреждающее адренерги-
ческое влияние на органы и ткани при стрессорном воздейст-
вии (за счет ингибирования высвобождения НА из терминалей
симпатических волокон и катехоламинов из надпочечников).
Это явление наряду со сниженной адренореактивностью сосу-
дов и миокарда у крыс Август (по сравнению с крысами Вис-
тар) также может определять более высокую устойчивость
крыс этой линии к инфаркту миокарда и стрессорному язво-
образованию в желудке. В то же время важно отметить, что
увеличенная продукция NO у крыс Август по сравнению с
Вистар может быть важной причиной более высокой смерт-
ности у крыс Август, чем у крыс Вистар, при интенсивном
длительном стрессорном воздействии. Действительно, крысы
Август гибнут от тяжелого стресса на фоне глубокого падения
АД, т.е. на фоне коллаптоидной вазодилатации, а именно ги-
перпродукция NO, как было указано выше, ответственна за
резкую гипотензию и кардиогенный шок (см. схему 5.8).
Базальная и постстрессорная активность кортико-адрена-
ловой системы, оцениваемая по содержанию в крови корти-
костерона, у крыс Август также оказалась значительно выше,
чем у крыс Вистар [Sudakov K.V. et al., 1995; Пшенникова М.Г.
и др., 1996]. Этим, по-видимому, также можно объяснить
более высокую резистентность к стрессорному язвообразова-
нию в желудке у крыс Август по сравнению с Вистар, посколь-
ку, по данным Л.П.Филаретовой (1995), кортикостероиды
могут предупреждать стрессорное язвообразование в желудке у
крыс.
Возможно, что определенную роль в механизме более вы-
сокой устойчивости крыс Август по сравнению с крысами
Вистар к стрессорному язвообразованию в желудке и острому
инфаркту миокарда играет также более высокий уровень у них
в крови p-эндорфина [Sudakov K.V. et al., 1995]. Показано, что
p-эндорфин обладает антиадренергическим действием, по-
скольку ограничивает действие катехоламинов на органы и
ткани за счет конкуренции с p-адренорецепторами за влияние
на аденилатциклазный комплекс на уровне постсинаптичес-
кой мембраны. Таким образом, выясняется, что нельзя гово-
рить об абсолютно высокой или низкой устойчивости орга-
низма к стрессорному повреждению. Следует определить в
каждом конкретном случае наиболее и наименее резистентные
к стрессорному поражению органы и системы и учитывать в
этой дифференцированной устойчивости роль активности раз-
личных стресс-лимитирующих систем.
В предыдущих разделах мы неоднократно подчеркивали,
что подверженность стрессорным воздействиям и у живот-
324
ных, и у человека в значительной мере определяется соци-
альным статусом. При этом установлено, что социальный
статус и активность различных звеньев стресс-системы тесно
взаимосвязаны. Показано, что «подчиненные» особи в коло-
ниях грызунов [Ely D.L., 1995] или сообществах обезьян
[Clarkson Т.В. et al., 1987], чаще испытывающие социальный
эмоциональный стресс, характеризуются преобладанием ак-
тивности гипофизарно-адренокортикального звена стресс-
системы, а «доминирующие» особи — преобладанием симпа-
тического звена. Длительное преобладание активности от-
дельных звеньев стресс-системы может приводить к их исто-
щению и дефициту активности. При этом смена социального
статуса сопровождается сменой соотношения активностей
указанных звеньев стресс-системы. Изучение этих обстоя-
тельств может дать ответ на многие вопросы, связанные с
механизмом резистентности организма к стрессу и возмож-
ностью ее повышения.
Известно, что хронический эмоциональный стресс часто
вызывает депрессивные состояния. При этом такие состояния
чаще возникают у женщин [Weissman М.М., Kierman G.L.,
1977; Van Praag H.M., 1982]. Анализ показывает, что причиной
депрессий может быть дефект функционирования серотонин-
ергической системы мозга, так как основной лечебный эф-
фект антидепрессантов заключается в повышении содержания
серотонина в структурах мозга. Эксперименты на животных
[Kennett G.A. et al., 1986] показали, что у самок в отличие от
самцов хронический стресс приводит к снижению содержания
серотонина во фронтальной коре, гиппокампе и гипоталамусе.
Это позволило авторам полагать, что именно этот, больший,
чем у мужчин, дефицит серотонина в мозге во время стресса
является причиной большей предрасположенности женщин к
стресс-индуцированной депрессии. Серотонинергическую
систему, как теперь известно, можно отнести к стресс-лими-
тирующим системам [Меерсон Ф.З., Пшенникова М.Г., 1989;
Меерсон Ф.З., 1993]. Таким образом, резистентность к стрес-
сорной депрессии определяется в значительной мере актив-
ностью данной стресс-лимитирующей системы.
Не вызывает сомнений важность понимания механизмов ре-
зистентности к стрессорному воздействию для разработки мето-
дов ее коррекции и профилактики ее снижения. Особенно акту-
ально это для людей, профессия которых сопряжена с эмоцио-
нальными стрессорными перегрузками. Для этого, несомненно,
важна разработка критериев резистентности и выяснение их ге-
нетических механизмов. Важную роль в развитии этого направ-
ления исследований играет моделирование стрессорной патологии
человека в эксперименте на животных разных генетических линий.
Целесообразность такого моделирования обоснована наличием
общих механизмов эмоционального стресса у человека и живот-
325
ных. Так, А.В.Вальдман (1979)* писал: «Моделирование наруше-
ний психических функций в эксперименте открывает большие
возможности для изучения патологии высшей нервной деятель-
ности человека и для разработки оптимальных способов лече-
ния и профилактики такой патологии» и подчеркивал важность
«моделирования эмоционального стресса с учетом типологичес-
кой характеристики животных».
В экспериментах на животных двух генетических линий
(мыши линии C57BL/6 и BALB/c), обладающих разной устой-
чивостью к эмоциональному стрессору (по поведению в «от-
крытом поле», handling), было показано, что менее резистент-
ными к стрессору являются мыши BALB/c с пассивной стресс-
реакцией замирания и проявлениями «страха», а более устой-
чивыми — мыши C57BL/6 с активной стресс-реакцией [Сере-
денин С.Б., Бадыштов Б.А., 1985]. Показано, что агонисты
бензодиазепиновых рецепторов феназепам и диазепам по-
вышают резистентность к стрессору мышей BALB/c, прирав-
нивая их к C57BL/6 и «убирая» признаки страха, т.е. делая их
поведение более адекватным. Такая коррекция поведения
при стрессе свидетельствует, что у мышей BALB/c, по-види-
мому, имеется генетически обусловленная недостаточность
системы агонистов бензодиазепиновых рецепторов, что позво-
ляет думать о дефиците функционирования у этих животных
ГАМКергической системы, активность которой, как известно,
тесно сопряжена с бензодиазепиновыми рецепторами.
Проведенные исследования создали предпосылки для ис-
пользования агонистов бензодиазепиновых рецепторов для
оценки резистентности поведенческих реакций к эмоциональ-
ному стрессу у человека. Действительно, в последние годы
разработаны критерии резистентности к стрессорным воздей-
ствиям с использованием совокупности психологических,
психофизиологических и биохимических характеристик, реги-
стрируемых в условиях эмоционального стресса с учетом реак-
ции на бензодиазепины. С помощью этих параметров выявле-
ны два основных генетически обусловленных типа стресс-ре-
акции: «активный» и «пассивный» [Бадыштов Б.А., 1998]. «Ак-
тивный» тип характеризуется высокой устойчивостью к эмо-
циональному стрессу по критериям работоспособности и дру-
гим поведенческим показателям в условиях действия эмоцио-
нального (ментального в том числе) стрессора; «пассивный»
тип стресс-реакции характеризуется низкой устойчивостью к
действию стрессора по данным критериям. При этом было ус-
тановлено, что применяемые много лет в клинике для профи-
лактики и коррекции тревожной эмоциональной стресс-реак-
♦ Вальдман А.А. Фармакологическая регуляция эмоционального
стресса. — М.: Медицина, 1979.
326
ции транквилизаторы бензодиазепинового ряда феназепам и
гидазепам в зависимости от типа стресс-реакции могут ока-
заться либо полезными, либо не только бесполезными, но и
ухудшающими работоспособность. Полезными бензодиазепи-
ны оказывались для людей «пассивного» типа стресс-реакции.
В последнем случае они снимали нервозность, повышали ра-
ботоспособность, улучшали самочувствие, т.е. «делали»
стресс-реакцию более адаптивной, адекватной. Людям «актив-
ного» типа стресс-реакции эти препараты, напротив, снижали
работоспособность [Бадыштов Б.А., 1998]. Бензодиазепины,
взаимодействуя с бензодиазепиновыми рецепторами, сопря-
женными на нейронах с ГАМК-рецепторами, усиливают эф-
фект ГАМК и тем самым повышают активность ГАМК-систе-
мы. Поэтому причиной обнаруженного явления, по-видимо-
му, можно считать в первом случае («пассивный» тип) воспол-
нение с помощью бензодиазепинов исходного дефицита ак-
тивности ГАМК-системы и предупреждение гиперактивации
стресс-системы, а во втором случае («активный» тип) — избы-
точную активацию ГАМК-системы под влиянием этих пре-
паратов, приводящую к неоправданному увеличению тормоз-
ного действия ГАМК на стресс-систему и ослабление адаптив-
ной стресс-реакции. В этих исследованиях, как и в экспери-
ментах на животных, вскрылась роль центральной стресс-ли-
митирующей ГАМКергической системы в механизме устойчи-
вости к стрессу.
Следует подчеркнуть, что хорошо известный в настоящее
время метод увеличения резистентности организма к стрес-
сорным повреждениям и заболеваниям путем адаптации к
факторам окружающей среды имеет в своей основе повыше-
ние мощности и эффективности стресс-лимитирующих сис-
тем, ограничивающих или предупреждающих повреждающие
эффекты стресс-реакции и в том числе нарушения поведен-
ческих реакций [Меерсон М.З., Пшенникова М.Г., 1988; Ме-
ерсон Ф.З., 1993].
Как отмечено выше, вторым основным фактором, опреде-
ляющим предрасположенность и устойчивость организма к
стрессорным повреждениям, является его исходное состояние
к моменту действия стрессора. Это обстоятельство не вызыва-
ет сомнений и не нуждается в пространных доказательствах.
Приведем в качестве примера роль исходного состояния же-
лудка в патогенезе стрессорного язвенного повреждения его
слизистой оболочки. Поскольку в механизме формирования
такого повреждения важным звеном является «переваривание»
ишемических очагов в оболочке желудка, образующихся в ре-
зультате вазоконстрикции при стресс-реакции, то при нали-
чии в анамнезе гиперацидного гастрита или просто повышен-
ной кислотности в желудке этот процесс будет развиваться
более активно и стрессорное язвообразование будет более вы-
327
раженным, чем при наличии низкой кислотности. Таким об-
разом, повышенная кислотность является фактором, увеличи-
вающим предрасположенность и снижающим устойчивость
организма к стрессорному поражению желудка. Примеров та-
кого рода немало. Фактором, повышающим риск поврежде-
ний, вызываемых эмоциональным стрессом, является также
неблагоприятная экологическая обстановка, приводящая к на-
рушению функции различных органов и систем организма.
Заключение
Предрасположенность и устойчивость организма к стрес-
сорным повреждениям в значительной мере определяется
двумя основными обстоятельствами: генетически обусловлен-
ными особенностями организма и фенотипически обусловленны-
ми особенностями, в том числе состоянием здоровья к моменту
действия стрессора.
Ключевым звеном генетически обусловленных особеннос-
тей организма, определяющих его предрасположенность и ус-
тойчивость к стрессорным повреждениям, является уровень
активности различных звеньев стресс-системы и стресс-лими-
тирующих систем, а также степень их активации (реактивнос-
ти) под влиянием стрессора. Уровень активности этих систем
детерминирован генетически, т.е. является наследственным,
однако может изменяться в процессе жизнедеятельности.
Нормализация базальной активности стресс-системы и по-
вышение эффективности стресс-лимитирующих систем орга-
низма либо путем его адаптации к факторам среды, либо фар-
макологическими методами (с помощью медиаторов или акти-
ваторов этих систем) является одним из факторов повышения
и коррекции устойчивости организма к стрессорным воздейст-
виям.
В понятие «состояния здоровья» как фактора, определяю-
щего предрасположенность и устойчивость к стрессорным по-
вреждениям, входит наличие заболеваний, принимаемые ме-
дикаментозные средства, возраст, физическая нагрузка, эко-
логическая обстановка, для женщин дополнительно — нали-
чие беременности, лактация, фазы цикла и т.п.
5.3. Принципы профилактики и коррекции
стрессорной патологии
Изложенное в предыдущих разделах свидетельствует, что
важную роль в механизме устойчивости организма к стрессор-
ным повреждениям и соответственно в патогенезе стрессор-
ной патологии играют активность и реактивность стресс-сис-
328
темы и стресс-лимитирующих систем. Показано также, что
естественные медиаторы стресс-лимитирующих систем или
их стабильные химические аналоги повышают устойчивость
организма к стрессорным повреждениям, оказывают профи-
лактическое и терапевтическое действие при стрессорных воз-
действиях главным образом за счет ограничения чрезмерной
или «застойной» стресс-реакции. Поэтому очевидно, что пер-
спективным принципом профилактики и коррекции стрессор-
ных повреждений является применение методов и средств, по-
зволяющих ограничивать чрезмерную активацию стресс-сис-
темы и чрезмерную стресс-реакцию, а также нормализовать
недостаточную активность стресс-системы и соответственно
недостаточную стресс-реакцию (см. раздел 5.2.2). При этом
особое значение имеет применение средств, повышающих
эффективность естественных стресс-лимитирующих систем,
или средств, «подражающих» действию этих стресс-лимитиру-
ющих систем.
Этот принцип в той или иной степени реализуется в при-
меняемых в настоящее время в клинике трех основных мето-
дических приемах, а именно: в адаптационном методе профи-
лактики и коррекции стрессорных повреждений (на базе ис-
пользования защитных эффектов адаптации к факторам окру-
жающей среды); при использовании фармакологических
средств, влияющих на активность стресс-системы и стресс-ли-
митирующих систем; при использовании различных приемов
психотерапии, гипноза и методов традиционной медицины,
направленных главным образом на коррекцию стрессорных
нарушений психического статуса, а также сопровождающих
эти нарушения вегетативных расстройств.
5.3.1. Профилактика и коррекция с помощью
защитных эффектов адаптации
к факторам среды
За последние 15 лет накоплен большой эксперименталь-
ный и клинико-физиологический материал, который свиде-
тельствует о том, что фактором, повышающим эффективность
стресс-лимитирующих систем и одновременно средством ус-
пешной профилактики и коррекции стрессорных поврежде-
ний и заболеваний, имеющих в своей основе стрессорный
компонент, является адаптация к факторам окружающей
среды.
Адаптация к окружающей среде, или фенотипическая адап-
тация, — это процесс, в результате которого организм приоб-
ретает ранее отсутствующую устойчивость к определенному
фактору (или факторам) среды и таким образом получает воз-
можность жить в условиях, ранее не совместимых с жизнью,
329
решать задачи, ранее неразрешимые [Меерсон Ф.З., 1993]. Эта
адаптация базируется на генотипе (совокупности наследуемых
признаков), т.е. генетической программе организма, которая
обеспечивает не заранее готовую адаптацию, а возможности
для ее формирования в соответствии с конкретными требова-
ниями среды. Результаты такой адаптации не передаются по
наследству, поскольку потомство может столкнуться с совсем
другими условиями жизни и должно приспосабливаться к
ним. В основе формирования адаптации лежит переход от
«срочной адаптации», возникающей в ответ на первое дейст-
вие стрессора, к «долговременной адаптации», формирующей-
ся при повторных действиях стрессора и обеспечивающей ус-
тойчивость к действующему стрессору.
Срочная адаптация — немедленный ответ организма на
стрессор. Она реализуется в виде первой стресс-реакции, по-
дробно рассмотренной выше. Она обеспечивает либо поведен-
ческую реакцию избегания, избавления от действия неблаго-
приятного стрессора, либо мобилизацию функциональных
систем, ответственных за приспособление к этому фактору.
Важнейшей чертой этого этапа адаптации является ее расточи-
тельность: деятельность организма протекает почти «на преде-
ле» его физиологических возможностей при почти полной мо-
билизации функционального и энергетического резервов и
тем не менее далеко не в полной мере обеспечивает необходи-
мый адаптивный эффект. Хорошо известно, что бег нетрени-
рованного человека происходит при близких к максимальным
величинах работы сердца, легких, при максимальной мобили-
зации энергетических ресурсов в скелетных мышцах, сердце,
печени. При этом бег не может быть ни достаточно быстрым,
ни достаточно длительным, очень скоро наступает истощение.
Тренированный человек может бежать быстрее и дольше. На
выполнение одной и той же мышечной работы ему потребуют-
ся существенно меньшая мобилизация систем кровообраще-
ния и дыхания и меньшие энергетические затраты, чем нетре-
нированному. При действии эмоциональных стрессоров имен-
но на этом этапе могут возникать стрессорные повреждения.
Это объясняется тем, что срочный этап адаптации реализу-
ется на базе существующего (исходного) уровня функциони-
рования физиологических механизмов, эффективность кото-
рых может оказаться недостаточной для нагрузки, связанной с
действием данного стрессора, и организм вынужден работать
«на износ». Для того чтобы эта срочная, но несовершенная
адаптивная реакция сменилась более совершенной устойчи-
вой адаптацией, необходимы время и повторные действия
стрессора, в результате которых в органах и тканях функцио-
нальной системы, вовлеченной в адаптивную реакцию, фор-
мируется так называемый структурный след адаптации — ма-
териальная база повышения устойчивости организма к дейст-
330
вию стрессора, основа тренированности {Меерсон Ф.З., 1981,
1993; Пшенникова М.Г., 1986, 1994].
Ключевым звеном формирования структурного следа и,
следовательно, устойчивой адаптации является активация
синтеза нуклеиновых кислот и белков, в результате которой
происходит увеличенная «наработка» регуляторных и струк-
турных белков в клетках органов и тканей, ответственных за
адаптацию к данному фактору, причем активируется синтез
белков тех клеточных структур, на которые падает большая
функциональная нагрузка при действии стрессора {Меер-
сон Ф.З., 1981, 1993].
Так, например, при адаптации к недостатку кислорода в
результате повторных сеансов гипоксии активируется синтез
структурных белков в органах дыхания, что приводит к гипер-
трофии (увеличению массы) нейронов дыхательного центра,
дыхательной мускулатуры (диафрагмы и межреберных мышц)
и самих легких, в которых увеличивается количество альвеол.
В результате возрастает мощность аппарата внешнего дыха-
ния, увеличиваются дыхательная поверхность легких и коэф-
фициент утилизации кислорода — возрастает экономичность
функции дыхания. В системе кроветворения активация синте-
за нуклеиновых кислот и белков в костном мозге приводит к
увеличенной продукции эритроцитов, что обеспечивает увели-
чение кислородной емкости крови. В совокупности с увеличе-
нием мощности аппарата внешнего дыхания и ростом функ-
циональных возможностей системы кровообращения это
обеспечивает достаточное поступление кислорода к мозгу,
сердцу и другим органам и тканям, несмотря на гипоксемию,
т.е. недостаток кислорода в крови. Наконец, в клетках самих
органов и тканей активируется синтез белков в митохондриях,
что постепенно приводит к росту числа митохондрий в клет-
ках и активности в них ферментов синтеза АТФ. В итоге эф-
фективность митохондрий возрастает, что обеспечивает увели-
чение способности клеток утилизировать кислород из крови и
образовывать АТФ, несмотря на недостаток кислорода. В ре-
зультате этих процессов организм приобретает устойчивость к
недостатку кислорода и возможность жизни и сохранения
жизненной активности в условиях гипоксии {Пшенникова
М.Г., 1994]. Эти черты характерны и для аборигенов высоко-
горья.
В процессе адаптации к различным стрессорам повышают-
ся также функциональная мощность и эффективность стресс-
системы и стресс-лимитирующих систем. Это обусловлено
формированием структурного следа в клетках этих систем.
В течение последних 4—5 лет показано, что при повторных
действиях стрессоров активируется экспрессия генов, кодиру-
ющих синтез ферментов, ответственных за «наработку» кате-
холаминов в гипоталамусе и надпочечниках и синтез КРГ в
331
КРГ-нейронах в гипоталамусе [Brady L.S. et al., 1994]. Доказа-
ны увеличение мощности стресс-лимитирующих систем и
рост продукции их медиаторов: ГАМК, ОП и ПГ [Орлова Э. и
др., 1988; Пшенникова М.Г., 1991; Пшенникова М.Г. и др.,
1992; Меерсон Ф.З., 1993], NO [Манухина Е.Б. и др., 1997;
Пшенникова М.Г. и др., 2000, и др.]. Это способствует в адап-
тированном организме нормализации регуляции стресс-систе-
мы и предупреждает как развитие чрезмерных стресс-реакций,
так и истощение стресс-системы и стресс-лимитирующих сис-
тем. .
Каков же механизм перехода срочной адаптации в устой-
чивую? Что является стимулом, «запускающим» этот переход?
Окончательный ответ на эти вопросы пока не получен. Они
остаются ключевой проблемой биологии и медицины. Однако
установлено, что все начинается с «выброса» стресс-гормонов,
биологически активных пептидов и других регуляторных фак-
торов во время стресс-реакции.
При стресс-реакции катехоламины, глюкокортикоиды и
другие гормоны воздействуют через определенные рецепторы
на клетки органов и тканей, ответственных за адаптацию. Под
влиянием этих сигналов, как уже было указано выше, в клетке
«запускается» цепь процессов, в результате которой в цито-
плазме активируются протеинкиназы (см. рис. 5.2). Вот на
этом этапе «разыгрываются» ключевые процессы формирова-
ния структурной (материальной) базы устойчивой адаптации.
Наряду с потенцированием функции клетки протеинкиназы
участвуют в механизме активации синтеза нуклеиновых кис-
лот и белков. Именно этот процесс лежит в основе «наработ-
ки» белков клеточных структур, которые обеспечивают увели-
чение мощности и повышение эффективности работы клеток
и органов в целом, вовлеченных в адаптацию к данному стрес-
сору, в том числе увеличение мощности стресс-системы и
стресс-лимитирующих систем. Установлено, что этот процесс
может реализоваться лишь при повторных неповреждающих
воздействиях стрессора, т.е. при повторных кратковременных
увеличениях функции клеток, и требует определенного време-
ни [Меерсон Ф.З., 1981, 1993; Меерсон Ф.З., Пшеннико-
ва М.Г., 1988].
Благодаря указанным процессам устойчивая адаптация об-
ладает так называемыми перекрестными защитными эффекта-
ми. Это означает, что адаптация к какому-либо стрессору по-
вышает устойчивость организма не только к действию данного
стрессора, т.е. имеет прямой защитный эффект, но также
может повышать устойчивость и к действию других стрессо-
ров. Например, у животных, адаптированных к гипоксии в ба-
рокамере, имитирующей условия высокогорья, повышена вы-
носливость к физическим нагрузкам и устойчивость к повреж-
дающему действию эмоциональных стрессоров, в том числе
332
повышена устойчивость иммунной системы к угнетающему
действию эмоциональных стрессоров [Меерсон Ф.З., Сухих
Г.Т., 1985; Сухих Г.Т., 1985], а также повышена устойчивость к
стрессорному язвообразованию в желудке [Пшенникова М.Г.
и др., 1999, 2000]. У животных, адаптированных к физическим
нагрузкам или к эмоциональным стрессорам, повышена ус-
тойчивость к гипоксическому повреждению сердца (инфаркту
миокарда), а также к язвенным поражениям органов пищева-
рения стрессорного происхождения и к нарушениям функции
иммунной системы. В основе этих перекрестных защитных
эффектов адаптации, как и в основе прямых защитных эффек-
тов, лежит наличие структурного следа адаптации, который
повышает функциональные резервы органов и систем. Поэто-
му чем больше органов и систем вовлечено в процесс адапта-
ции к данному стрессору, т.е. чем «богаче» структурный след,
тем большим числом перекрестных защитных эффектов обла-
дает данная адаптация.
Явление защитных эффектов адаптации открывает пер-
спективу использования адаптации к факторам среды как для
повышения устойчивости здорового человека к стрессорным
ситуациям и болезням, так и для лечения некоторых заболева-
ний и коррекции стрессорных повреждений. Достижения в
этой области уже есть, и они составили основу появления
новой медицинской науки — адаптационной медицины и соот-
ветственно новых немедикаментозных способов профилакти-
ки и лечения. На базе этих достижений уже успешно внедрено
в клинику использование адаптации к периодической гипобари-
ческой гипоксии в барокамере в качестве метода профилактики
и лечения патологических состояний, в патогенезе которых
присутствует стрессорный компонент: нарушений функции
иммунной системы (например, аллергических заболеваний),
неврозов, гипертонической болезни, параноидной формы ши-
зофрении и др. [Меерсон Ф.З. и др., 1989; Меерсон Ф.З.,
1993], а также адаптации к нормобарической гипоксии — нор-
мобарической интервальной гипоксической тренировки [Меерсон
Ф.З., 1993; Глазачев О.С., 1997]. При этом следует подчерк-
нуть еще раз, что ключевым механизмом адаптационной про-
филактики и коррекции стрессорной патологии, является,
очевидно, возникающее в процессе адаптации повышение эф-
фективности стресс-лимитирующих систем.
Перспективным в этом плане является также немедика-
ментозный метод традиционной медицины — иглорефлексоте-
рапия и акупунктура, который по существу является методом
адаптации к слабым неповреждающим стрессорным воздейст-
виям [Меерсон Ф.З. и др., 1994]. В частности, в настоящее
время доказано, что, подобно адаптации к физическим нагруз-
кам и стрессорным воздействиям, курс иглорефлексотерапии
или электроакупунктуры активирует опиоидергическую
333
стресс-лимитирующую систему и приводит к повышению в
крови содержания p-эндорфина [Меерсон Ф.З. и др., 1994].
В настоящее время в клинике доказана эффективность этого
вида терапии при вызванных эмоциональным стрессом вегета-
тивных расстройствах [Вейн А.М., 1997].
5.3.2. Коррекция с помощью
фармакологических средств
Для повышения устойчивости к эмоциональным стрессо-
рам и коррекции стрессорных повреждений эффективно ис-
пользуются также психофармакологические средства. Анализ
фармакологических средств, применяемых в настоящее время
для коррекции состояний эмоционального стресса и пост-
стрессорных нарушений, показывает, что центральное место
среди них занимают препараты, направленные на ограничение
активности стресс-системы и соответственно стресс-реакции.
Это — либо прямые блокаторы активности адренергической
системы (блокаторы а- и p-адренорецепторов), либо препара-
ты, повышающие эффективность естественных стресс-лими-
тирующих систем, либо стабильные химические аналоги ме-
диаторов этих систем. Данная ситуация соответствует древ-
нейшему принципу врачевания, которого придерживались ме-
дики со времен Авиценны и Гиппократа, а именно: «не навре-
ди и подражай защитным силам самого организма». В данном
случае речь идет о подражании действию стресс-лимитирую-
щих систем.
Среди таких средств следует назвать прежде всего эффек-
тивные препараты, активирующие функцию ГАМКергической
системы или дублирующие ее. Это агонисты бензодиазепино-
вых рецепторов и стабильные аналоги самой ГАМК [Воронина
Т.А., 1992; Соколова Е.Б. и др., 1996; Петров В.И., 1997; Ба-
дыштов Б.А., 1998]. Препараты бензодиазепинового ряда, свя-
зываясь с бензодиазепиновыми рецепторами, повышают аф-
финность ГАМК-рецепторов и тем самым активируют тормоз-
ную функцию ГАМКергической системы. Поэтому основным
специфическим эффектом данных психотропных средств яв-
ляется анксиолитический, транквилизаторный эффект, кото-
рый облегчает «переживание» сильного эмоционального
стресса, снижая его «цену». Этот эффект выражается в устра-
нении тревоги, страха, беспокойства, волнения. Но вместе с
тем различные бензодиазепины могут давать побочные эффек-
ты: снотворный, седативный, миорелаксантный, которые в
одних условиях желательны, а в других, напротив, нежелатель-
ны. В связи с этим выделяют группы: собственно транквили-
заторы (феназепам, хлордиазепоксид, диазепам, лоразепам и
др.), снотворные (нитрозепам, феназепам, флуразепам и др.),
334
противосудорожные, миорелаксанты (клоназепам, феназепам,
диазепам и др.). Нежелательным побочным эффектом многих
бензодиазепинов, особенно в больших дозах, является способ-
ность нарушать процессы обучения и памяти [Воронина Т.А.,
1992]. Тем самым эти препараты могут подавлять собственные
механизмы защиты, снижая психологическую реактивность на
все эмоционально значимые сигналы, в том числе полезные.
Иными словами, они могут блокировать стресс-реакцию «без
разбора», т.е. вызывают или усиливают «пассивную» резис-
тентность. Поэтому их использование неоценимо при экстре-
мальных ситуациях и сильных стрессорах, но при стрессорах
малой интенсивности длительное их применение нежелатель-
но, так как, формируя толерантность организма к стрессорам
вообще, они предотвращают возможность «активной» адапта-
ции организма [Петров В.И., 1997]. Иными словами, они
могут угашать указанную выше «активную психическую адап-
тацию», необходимую для преодоления стрессогенной си-
туации.
Вместе с тем фармакологи активно продолжают поиски
бензодиазепинов, не обладающих нежелательными побочны-
ми эффектами, не подавляющих «активную психическую
адаптацию». Одним из таких препаратов является сравнитель-
но новый бензодиазепиновый транквилизатор гидазепам. Он
обладает оригинальным спектром фармакологической актив-
ности, сочетая отчетливое анксиолитическое действие со спо-
собностью не нарушать, а напротив, оптимизировать обучение
и оперативную деятельность при малой выраженности побоч-
ных эффектов и низкой токсичности. В связи с этим гидазе-
пам рекомендуют как дневной транквилизатор с выраженной
избирательностью анксиолитического действия [Ворони-
на Т.А., 1992].
Существенный интерес представляют так называемые
ноотропные препараты, являющиеся главным образом произ-
водными ГАМК (фенибут, пикамилон, аминолон, пираце-
там, или ноотропил, и др.). Они поддерживают энергетичес-
кие и пластические процессы в ЦНС на высоком уровне и
позволяют сохранить в условиях стресса адекватный паттерн
внимания, когнитивных и мнестических функций мозга. Они
повышают «активную» резистентность к стрессорам в отли-
чие от бензодиазепинов, повышающих главным образом
«пассивную» устойчивость, и оказывают нормализующее
действие при длительных стрессах и постстрессорных состоя-
ниях. Одним из механизмов действия ноотропных препара-
тов является их влияние на систему естественных возбужда-
ющих медиаторных аминокислот (в том числе глутаминовой
кислоты), что приводит к перестройке пластического метабо-
лизма нейронов и повышению их синтетической активности
в условиях длительных истощающих нагрузок — стрессов
335
[Петров В.И., 1997]. По мнению исследователей, производ-
ные ГАМК и среди них весьма эффективный фенибут соче-
тают транквилизирующий эффект с ноотропным, что позво-
ляет рассматривать эти препараты не только как блокаторы
стресс-реакции, но и как стрессопротекторы [Соколова Е.Б.
и др., 1996; Тарабрина Н.В. и др., 1996]. При стрессорных на-
рушениях психического статуса тревога зачастую сочетается с
депрессивными расстройствами. В этих случаях применяют
препараты, сочетающие анксиолитический и антидепрес-
сантный эффекты. Препараты такого рода тримипрамин,
амитриптилин, пиразидол и др. содержат в механизме своего
действия торможение обратного захвата медиаторов моно-
аминов и обратимую блокаду фермента моноаминоксидазы
(МАО), инактивирующего моноамины. В результате при дей-
ствии таких препаратов снятие тревоги сочетается с возмож-
ностью поддержания активности моноаминовой системы,
прежде всего адренергической, что имеет антидепрессивное
действие [Соколова Е.Б. и др., 1996].
Важную роль в предупреждении и коррекции стрессорных
повреждений играет антиоксидантная стресс-лимитирующая
система организма. Антиоксиданты, как уже неоднократно
упоминалось в данном сообщении, предупреждают чрезмер-
ную активацию СРО и тем самым являются мембранопротек-
торами, ограничивающими стрессорное повреждение клеточ-
ных мембран (см. раздел 5.1.5). В связи с этим в качестве
антистрессорных средств широко используют антиоксиданты
(природные и их синтетические аналоги), а также препараты,
сочетающие антиоксидантную и анксиолитическую актив-
ность, антиоксидантную и антидепрессантную активность и
т.п. Таким образом, эти препараты не только ограничивают
развитие чрезмерной стресс-реакции и нарушения психичес-
кого статуса (т.е. действуют на уровне функции ЦНС), но и
непосредственно ограничивают стрессорные повреждения
клеток, т.е. сочетают ограничение стресс-реакции и ее по-
вреждающих эффектов на клетки. Новым препаратом такого
рода является мексидол (2-этил-6-метил-3-оксипиридин сук-
цинат). Особенностью этого синтетического препарата, раз-
решенного к применению в лечебной практике, является ши-
рокий спектр эффектов на двух уровнях — нейрональном и
сосудистом [Воронина Т.А., Смирнов А.Д., 1999]. Мексидол
является ингибитором СРО, активирует антиоксидантный
фермент супероксиддисмутазу, влияет на физико-химические
свойства клеточных мембран, повышая в них содержание по-
лярных фракций липидов, вязкость липидного бислоя мем-
бран, его текучесть и тем самым увеличивает активность
мембраносвязанных функциональных белков (рецепторов,
ферментов и др.). За счет этих свойств мексидол обладает
антигипоксическим действием, повышает энергетический
336
обмен в клетках, уменьшает тромбообразование, улучшает
мозговое кровообращение и т.д. Иными словами, этот пре-
парат на уровне клеточных мембран повышает резистент-
ность мозга к стрессорному повреждению. Анксиолитическое
действие мексидола связано со способностью препарата мо-
дулировать бензодиазепиновые и ГАМК-рецепторы, повы-
шая функцию ГАМКергической системы, хотя сам мексидол
не обладает свойством связываться с этими рецепторами.
Как показывают результаты исследований, мексидол в дан-
ном случае действует как лиганд, аллостерически потенци-
рующий указанные рецепторы и активирующий ионные ка-
налы мембран нейронов, что приводит к конформационным
изменениям этих мембранных образований. Такая конфор-
мация облегчает эффекты ГАМК и активирует синаптичес-
кие процессы, т.е. в конечном счете активирует эффекты
ГАМК-системы. При этом мексидол не обладает побочными
эффектами традиционных транквилизаторов, в частности не
оказывает миорелаксантного, седативного, эйфоризирующе-
го действия [Воронина Т.А., Смирнов А.Д., 1999]. Высокий
лечебный эффект этого нового препарата при невротических
и неврозоподобных состояниях, острых стрессорных и других
психосоматических нарушениях, доказанный в обширных
клинико-физиологических исследованиях, свидетельствует о
несомненной перспективности создания подобных препара-
тов комплексного действия, основанных на подражании ме-
диаторам естественных защитных стресс-лимитирующих сис-
тем, для коррекции стрессорных повреждений.
Следует далее отметить, что потенциальными перспектив-
ными средствами предупреждения и коррекции стрессорных
повреждений могут быть природные стресс-лимитирующие
нейропептиды или их стабильные химические аналоги.
Е.А. Юматовым и сотр. установлено, что введение животным
пептида субстанции Р и пептида дельта-сна эффективно по-
вышает устойчивость животных к эмоциональным стрессорам
[Юматов Е.А., 1986, 1997]. Показано, что предварительное
введение животным пептида дельта-сна или стабильных его
аналогов защищает от повреждений сердца при эмоциональ-
ном стрессе [Меерсон Ф.З. и др., 1985]. В экспериментах на
животных также показано, что введение стабильного аналога
опиоидного пептида Leu-энкефалина деларгина эффективно
предупреждает и ограничивает поражения сердца при эмоцио-
нальном стрессе и острой ишемии миокарда [Лишманов Ю.Б.
и др., 1987; Меерсон Ф.З., Пшенникова М.Г., 1989]. Таким
образом, изложенное свидетельствует, что использование
принципа «подражания» естественным стресс-лимитирующим
системам является в значительной мере перспективным при
разработке фармакологических средств профилактики и кор-
рекции стрессорных повреждений.
22— 1385
337
5.3.3. Использование приемов психотерапии
при стрессорных психосоматических
расстройствах
Наиболее древним и довольно широко применяемым до
сих пор средством коррекции клинических признаков тяжело-
го эмоционального стресса является психотерапия, хотя точ-
ный механизм ее лечебного действия пока недостаточно ясен.
В настоящее время принято, что в основе психотерапии лежит
«реориентация личности пациента, ее реорганизация», т.е.
восстановление нарушенной интеграции личности [Соколова
Е.Б. и др., 1996]. Иными словами, врач, воздействуя на паци-
ента, зачастую методом гипноза, меняет восприятие больным
стрессогенной ситуации, уменьшая в сознании его эту стрес-
согенность.
Так называемая поведенческая психотерапия основывается
на положении, что различные симптомы (например, фобии) и
невротические формы поведения представляют собой резуль-
тат «неадекватного научения» и могут быть устранены путем
активного «переучивания» [Соколова Е.Б. и др., 1996]. Типич-
ным примером такой терапии может служить систематическая
десенсибилизация, при которой терапевтический эффект до-
стигается рядом последовательных процедур, причем каждая
из них включает в себя постепенное приближение к вызываю-
щим страх ситуациям и последующую релаксацию. При этом
релаксацию начинают в ситуациях (реальных или воображае-
мых), стимулирующих легкую тревогу, и постепенно прибли-
жаются к ситуации, вызывающей выраженный страх.
Когнитивная психотерапия основана на представлении, что
ключевой причиной невротического тревожного или депрес-
сивного реагирования на окружающую среду, или поведения,
обусловленного ригидными концепциями, являются «ошибоч-
ные когнитивные схемы», которые устойчиво сохраняются
(проявления застойного очага возбуждения, возникающего в
эмоциогенных зонах мозга под влиянием стрессора). Терапия
включает в себя оценку процесса возникновения этих схем,
идентификацию когнитивных структур, препятствующих аде-
кватной психической адаптации, и их коррекцию на основе
активного создания более адекватных схем.
Ориентирующая психотерапия направлена на переориента-
цию пациента не столько за счет анализа происхождения не-
адекватных когнитивных схем, сколько за счет введения в эти
схемы новых элементов, которые позволяют изменить эти
схемы, сделав их более адекватными, а стало быть, более адап-
тивными и изменить поведенческие стереотипы.
Релаксационная психотерапия непосредственно направлена
на снижение уровня тревоги и уменьшение выраженности
связанных с ней вегетативных нарушений. Такая терапия
338
включает прогрессирующую миорелаксацию, аутогенную тре-
нировку, различные медитационные техники, позволяющие
«отстраняться» от стрессора, т.е. от стрессогенных ситуаций.
Список литературы
Абрамов В.В. Интеграция иммунной и нервной системы. — Новоси-
бирск, 1991. — 165 с.
Адо А.Д. О взаимодействии нервной и иммунной систем (к механиз-
мам влияния нервной системы на лимфоциты)//Вестн. РАМН. —
1993. № 7. - С. 48-51.
Акмаев И. Г. Современные представления о взаимодействиях регули-
рующих систем: нервной, эндокринной и иммунной//Успехи фи-
зиол. наук. — 1996. — № 1. — С. 3—20.
Андреев Б.В., Ипатов Ю.Д., Никитина З.С., Сытинский И.А. Анти-
стрессорная роль ГАМКергической системы мозга//Журн. высш,
нервн. деят. — 1982. — Т. 32, вып. 3. — С. 511—519.
Анохина И.П. Нейрохимическая характеристика специфических пато-
логических синдромов, возникающих в условиях стрессовых состоя-
ний//Вестн. АМН СССР. - 1975. - № 8. - С. 34-43.
Архипенко Ю.В., Каган В.Е., Козлов Ю.П. Модификация ферментной
системы транспорта Са2+ в саркоплазматическом ретикулуме при
перекисном окислении липидов, молекулярные механизмы изме-
нений активности Са-АТФазы//Биохимия. — 1983. — Т. 48, № 3. —
С. 433-441.
Бадыштов Б.А. Фенотипы реакций здоровых добровольцев на эмоци-
ональный стресс и бензодиазепиновые транквилизаторы: Автореф.
дис. ... докт. мед. наук. — М., 1998. — 47 с.
Белкина Л.М., Мациевский Д.Д., Меерсон Ф.З. Влияние перенесенного
эмоционально-болевого стресса на резистентность к ишемии//Бюлл.
экспер. биол. мед. — 1983. — № 6. — С. 35—38.
Березин Ф.Б., Мирошников М.П. Эмоциональный стресс и психосома-
тические расстройства. Подходы к терапии//Ма!епа Medica. —
1996. - № 1 (9). - С. 29-56.
Бородин П.М., Шюлер Л., Беляев Д.К. Проблемы генетики стресса.
Сообщение 1. Генетический анализ поведения мышей в стрессирую-
щей ситуации//Генетика. — 1976. — Т. XII, № 12. — С. 62—71.
Бульон В.В. Центральные механизмы развития нейрогенного повреж-
дения желудка и его фармакологическая коррекция//Пат. физиол. —
1995. — № 1.-С. 21-23.
22*
339
Ведяев Ф.П. Лимбическая система мозга, эмоциональный стресс и
его эндокринно-вегетативные проявления//Вестн. АМН СССР. —
1975.— № 8.— С. 57-65.
Ведяев Ф.П., Витриченко Е.Е., Мищенко В.П., Тарасенко Л.М. Зависи-
мость ульцирогенного действия эмоционального стресса от индиви-
дуально-типологических особенностей крыс//Пат. физиол.— 1985.—
№ 5. - С. 21-23.
Вейн А.М. Клинические аспекты эмоционального стресса//Эмоцио-
нальный стресс: теоретические и клинические аспекты/Под ред.
К.В.Судакова, В.И.Петрова. — Волгоград, 1997. — С. 138—148, 154—
157.
ВируА.А., Эйлер А.К. Адренокортикальная регуляция белкового обме-
на при длительных физических нагрузках//Бюл. экспер. биол. —
1976. - № 12. - С. 1436-1439.
Вихерт А.М., Галахов И.Е., Матова Е.Е. и др. Гистопатология мио-
карда в случаях внезапной смерти//Внезапная смерть/Под ред.
А.М.Вихерта, Б.Лауна. — М.: Медицина. — 1982. — С. 130—150.
Владимиров Ю.А., Арчаков А.И. Перекисное окисление липидов в
биологических мембранах. — М.: Наука, 1972. — 252 с.
Воронина Т.А. Спектр фармакологической активности гидазепама и
его место среди известных транквилизаторов//Гидазепам/Под ред.
С.А.Андронати, Т.А.Ворониной, Н.Я.Головенко и др. — Киев: Нау-
кова думка, 1992. — С. 63—75.
Воронина Т.А., Смирнов А.Д. Перспективы применения мексидола в
экстремальных ситуациях//Новые технологии, новые лекарственные
средства при чрезвычайных ситуациях природного и техногенного
происхождения. — М., 1999. — С. 3—9.
Глазачев О.С. Клинические аспекты интервальной гипоксической
тренировки при реабилитации стресс-индуцированных психосомати-
ческих нарушений//Эмоциональный стресс: теоретические и клини-
ческие аспекты/Под ред. К.В.Судакова, В.И.Петрова. — Волгоград,
1997. - С. 148-152.
Голиков П.П. Рецепторные механизмы глюкокортикоидного эффек-
та. — М.: Медицина, 1988. — 288 с.
Голодец ЕЕ, Пучков Н.В. О влиянии медиаторов на фагоцитарную де-
ятельность лейкоцитов. Сообщение I и П//Физиол. журн. СССР им.
И.М.Сеченова. - 1948. - Т. 34, №1. - С. 135-150.
Гольдберг Е.Д., Дыгай А.М., Хлусов И.А. Роль вегетативной нервной
системы в регуляции гемопоэза. — Томск: Изд-во Томск. Ун-та,
1997. - 218 с.
340
Горизонтов П.Д., Белоусова О.И., Федотова М.И. Стресс и система
крови. — М: Медицина, 1983. — 293 с.
Девойно Л.В. Центральный механизм допамин-, серотонин-, ГАМК-
и пептидергической иммуномодуляции//Бюлл. СО РАМН. — 1994. —
№ 4. - С. 19-25.
Динзбург А.Л., Чирков А.М., Чиркова С. К. Стресспротективный эф-
фект нейропептидов у обезьян//Пат. физиол.— 1995.— № 1,— С. 19—
21.
Дорофеев Г.И., Шанин Ю.Н., Ефимов Н.В., Ивашкин В.Т. Диагнос-
тическое и прогностическое значение определения циклических
нуклеотидов при нарушениях сердечного ритма//Воен.-мед. журн. —
1985. - № 2. - С. 59-63.
Клименко В.М. Анализ процессов перестройки организации импульс-
ной активности нейронов гипоталамических структур в процессе ре-
акции на антиген//Иммунофизиология/Под ред. Е.А.Корневой. —
Л.: Наука, 1993. - С. 113-129.
Коляда Т.И., Волянский Ю.Л., Васильев Н.В., Мальцев В. И. Адаптаци-
онный синдром и иммунитет. — Харьков: Основа, 1995. — 368 с.
Корнева Е.А. Нарушения нейрогуморальной регуляции функций им-
мунной системы//Вестн. АМН СССР. — 1990. — № 4. — С. 36—42.
Корнева Е.А., Шекоян В.А. Регуляция защитных функций организ-
ма. — Л.: Наука, 1982. — 138 с.
Косицкий Г.И., Михайлова С.Д., Горожанин С.Л., Семушкина Т.М.
Влияние раздражения сенсомоторной зоны коры на развитие ише-
мических аритмий сердца//Вестн. АМН СССР. — 1985. — № 12. —
С. 64-69.
Котов А.В. Мотивационно-эмоциональный конфликт в структуре
поведенческого акта//Психофизиология. — 1999. — Т. 20, № 6. —
С. 62-71.
Крыжановский Г.Н. Общая патофизиология нервной системы. — М.:
Медицина, 1997. — 450 с.
Крыжановский Г.Н., Магаева С.В., Макаров С.В. Нейроиммунопато-
логия. — М., 1997. — 283 с.
Лишманов Ю.Б., Трифонов Ж.В., Цибин А.Н. и др. P-Эндорфин и
стресс-гормоны плазмы крови при состоянии напряжения и адапта-
ции//Бюл. экспер. биол. — 1987. — № 4. — С. 380—383.
Малышев И.Ю., Манухина Е.Б. Стресс, адаптация и оксид азота//
Биохимия. — 1998. — Т. 63, 7. — С. 992—1006.
341
Манухина Е.Б. Портальная вена и ее сократительная функция в
норме и патологии//Успехи физиол. наук. — 1988. — Т. 19, № 3. —
С. 45-66.
Манухина Е.Б., Покидышев Д.А., Малышев И.Ю. Предупреждение ост-
рой гипотензии и гиперактивации эндотелия при тепловом шоке с
помощью адаптации к стрессорным воздействиям//Бюл. экспер.
биол. - 1997. - Т. 124, № 10. — С. 380-383.
Меерсон Ф.З. Адаптация, стресс и профилактика. — М.: Наука,
1981. - 278 с.
Меерсон Ф.З. Патогенез и предупреждение стрессорных и ишемичес-
ких повреждении сердца. — М.: Медицина, 1984. — 272 с.
Меерсон Ф.З. Адаптационная медицина: механизмы и защитные эф-
фекты адаптации. — М.: Hypoxia Medical LTD. — 1993. — 331 с.
Меерсон Ф.З., Заяц В.И., Пшенникова М.Г., Михалева И.И. Пре-
дупреждение нарушений сократительной функции миокарда при
длительном стрессе с помощью пептида дельта-сна и его цикли-
ческого производного//Кардиология. — 1985. — № 8. — С. 104—
106.
Меерсон Ф.З., Пшенникова М.Г. Адаптация к стрессорным ситуациям
и физическим нагрузкам. — М.: Медицина, 1988. — 256 с.
Меерсон Ф.З., Пшенникова М.Г. Стресс-лимитирующие системы орга-
низма и новые принципы профилактической кардиологии. — М.:
НПО Союзмединформ, 1989. — 72 с.
Меерсон Ф.З., Пшенникова М.Г., Кузнецова Б.А. и др. Развитие адапта-
ции к стрессу в результате курса транскраниальной электростимуля-
ции//Бюл. экспер. биол. — 1994. — № 1. — С. 16—18.
Меерсон Ф.З., Сухих ГТ. Стрессорные нарушения в системе противо-
опухолевого иммунитета и их ограничение стресс-лимитирующими
факторами//Вестн. АМН СССР. — 1985. — № 8. — С. 23—29.
Меерсон Ф.З., Твердахлиб В.П., Боев В.М., Фролов Б.А. Адаптация к
периодической гипоксии в терапии и профилактике. — М.: Наука,
1989. - 70 с.
Микоян В.Д., Кубрина Л.Н., Манухина Е.Б. и др. Различия в стимуля-
ции синтеза NO при тепловом шоке у крыс генетически различных
популяций//Бюл. экспер. биол. — 1996. — Т. 121, № 6. — С. 634—
637.
Мороз Б.Б., Дешевой Ю.Б. Роль эмоционального стресса в развитии
соматических нарушений у ликвидаторов аварии на Чернобыльской
атомной станции, облученных в диапазоне малых доз//Радиац. био-
логия, радиоэкология. — 1999. — Т.39, № 1. — С. 97—105.
342
Мороз Б.Б., Дешевой Ю.Б., Лебедев В.Т. и др. Реакция кроветворной
системы при длительном эмоциональном стрессе на фоне действия
у-излучения в низких дозах//Радиацион. биология. Радиоэколо-
гия. - 1997. - Т. 37, вып. 4. - С. 581-589.
Орлова Э.Х., Пшенникова М.Г., Дмитриев АД., Меерсон Ф.З. Увели-
чение содержания иммунореактивных опиоидных пептидов в голов-
ном мозге и надпочечниках крыс под влиянием адаптации к фи-
зической нагрузке//Бюл. экспер. биол. — 1988. — Т. 105, № 2. —
С. 145-148.
Петров В. И. Фармакологическая коррекция эмоционального стрес-
са//Эмоциональный стресс: теоретические и клинические аспекты/
Под ред. К.В.Судакова, В.И.Петрова. — Волгоград, 1997. — С. 127—
134.
Постнов Ю.В., Орлов С.Н. Первичная гипертензия как патология
клеточных мембран. — М., 1987.
Прилипко Л.Л., Орлов О.М., Иванова С.М. и др. Активация перекисно-
го окисления липидов при стрессе у человека, оцениваемая по содер-
жанию пентана в выдыхаемом воздухе//Докл. АН СССР. — 1982. —
Т. 265, №4. - С. 1010-1013.
Пшенникова М.Г. Адаптация к физическим нагрузкам//Руководство
по физиологии. Физиология адаптационных процессов. — М.:
Наука, 1986. - С. 124-221.
Пшенникова М.Г. Роль опиоидных пептидов в реакции организма на
стресс//Пат. физиол. — 1987,— № 3. — С. 85—90.
Пшенникова М.Г. Защитная роль простагландинов при повреждаю-
щих воздействиях//Пат. физиол. — 1991. — № 6. — С. 54—58.
Пшенникова М.Г. Сходство и различия адаптации к гипоксии и физи-
ческим нагрузкам и их защитных эффектов//Нурох!а Med. J. —
1994. - № 3. - Р. 3-10.
Пшенникова М.Г, Бондаренко И.А., Шимкович М.В. и др. Различия в
поведении и устойчивости к язвенному поражению желудка при
стрессе у крыс линии Август и Вистар, адаптированных и неадапти-
рованных к гипоксии//Бюл. экспер. биол. —1999. — Т. 128, № 12. —
С. 638-641.
Пшенникова М.Г, Голубева Л.Ю., Кузнецова Б.А. и др. Различия в
стресс-реакции и формировании адаптации к стрессу у крыс Август и
Вистар//Бюл. экспер. биол. —1996. — Т. 122, № 8. — С. 156—159.
Пшенникова М.Г., Кузнецова Б.А., Копылов Ю.Н. и др. Роль системы
простагландинов в кардиопротекторном действии адаптации к ги-
поксии при стрессе//Кардиология. — 1992. — Т. 32, № 3. - С. 61—
64.
343
Пшенникова М.Г., Попкова Е.В., Бондаренко Н.А. и др. Катехоламины,
оксид азота и устойчивость к стрессорным повреждениям: влияние
адаптации к гипоксии//Рос. физиол. журн. им. И.М.Сеченова. —
2001 (в печати).
Пшенникова М.Г., Смирин Б.В., Бондаренко О.Н. и др. Депонирование
оксида азота у крыс разных генетических линий и его роль в анти-
стрессорном эффекте адаптации к гипоксии//Физиол. журн. им.
И.М.Сеченова. - 2000. - Т. 86, № 2. - С. 174-181.
Ратсак Р., Оеме П., Роске И. и др. Изменения уровня субстанции Р в
организме при различных функциональных состояниях животных и
человека//Экспериментальные и клинические неврозы. — Берлин,
1984. - С. 130.
Репин В.С., Сухих Г.Т. Медицинская клеточная биология. — М.:
БЭБиМ, 1998. - 200 с.
Селье Г. Очерки об адаптационном синдроме. — М.: Медгиз. —
1960. - 254 с.
Середенин С.Б., Бадыштов Б.А. Изучение наследственных различий в
содержании циклических нуклеотидов в плазме крови мышей после
стрессорных воздействий и введения феназепама//Бюл. экспер.
биол. - 1985. - Т. 100, № 11. - С. 586-588.
Симонов П.В. Лимбические структуры мозга и патогенез невро-
зов//Журн. невропатол. и психиатр.— 1984.— Т. 84, № 11.— С. 1665—
1670.
Соколова Е.Б., Березин Ф.Б., Барлас Т.В. Эмоциональный стресс: пси-
хологические механизмы, клинические проявления, терапия//Ма1е-
ria Medica. - 1996. - № 1(9). - С. 5-25.
Судаков К.В. Церебральные механизмы эмоционального стресса//
Эмоциональный стресс: теоретические и клинические аспекты/Под
ред. К.В.Судакова, В.И.Петрова. — Волгоград, 1997а. — С. 59—74.
Судаков К.В. Эмоциональный стресс и артериальная гипертензия:
Эмоциональный стресс: теоретические и клинические аспекты/Под
ред. К.В.Судакова, В.И.Петрова. — Волгоград, 19976. — С. 80—89.
Судаков К.В. Эмоциональный стресс и психосоматическая патоло-
гия//Чтения им. А.Д.Сперанского,— М., 1998,— Вып. X.— С. 11—30.
Судаков К.В., Юматов Е.А. Эмоциональный стресс в современной
жизни. — М.: НПО Союзмединформ, 1991. — 83 с.
Тарабрина Н.В., Соколова Е.Д., Лазебная Е.О., Зеленова М.Е. Пост-
травматическое стрессорное расстройство: психологические и кли-
нические особенности, вопросы терапии//Ма1епа Medica. — 1996. —
№ 1(9). - С. 57-68.
344
Твердохлиб В.П., Озерова И.Н., Творогова М.Г. и др. Влияние эмоцио-
нально-болевого стресса на уровень липидов и эстерификацию хо-
лестерина в крови крыс//Пат. физиол. — 1988. — № 4. — С. 27—29.
Ткачук В.А. Молекулярные механизмы регуляции аденилатциклазной
системы сердца//Регуляция сократительной функции и метаболизма
миокарда/Под ред. Е.И.Чазова, В.Н.Смирнова.— М., 1987.— С. 259—
266.
Усынин А.Ф. Структурно-метаболическое изменение проводящей
системы и миокарда, их коррекция при коронарогенных и стрессор-
ных повреждениях сердца: Автореф. дис. ... д-ра мед. наук. — Ново-
сибирск, 1994.
Филаретова Л.П. Стрессорные язвы желудка: защитная роль гормо-
нов гипоталамо-гипофизарно-адренокортикальной системы//Фи-
зиол. журн. им. И.М.Сеченова. — 1995. — Т. 81, № 3. — С. 50—53.
Фролов Б.А., Афонина С.Н., Меерсон Ф.З. Роль соотношения цАМФ/
цГМФ в постстрессорной активации первичного иммунного ответа//
Пат. физиол. — 1985. — № 5. — С. 23—26.
Фролькис В.В., Головаченко С.Ф., Медведь В.И., Фролькис Р.А. Вазо-
прессин и сердечно-сосудистая система//Успехи физиол. наук. —
1983. - Т. 14, № 2. - С. 56-81.
Чазов Е.И. Эмоциональный стресс и сердечно-сосудистые заболева-
ния//Вестн. АМН СССР. - 1975. - № 8. - С. 3-8.
Юматов Е.А. Центральные нейрохимические механизмы устойчивос-
ти к эмоциональному стрессу: Автореф. дис. ... д-ра мед. наук. — М.,
1986. - 43 с.
Юматов Е.А. Центральные пептидергические механизмы устойчи-
вости к эмоциональному стрессу//Эмоциональный стресс: теорети-
ческие и клинические аспекты/Под ред. К.В.Судакова, В. И. Петро-
ва. — Волгоград, 1997. — С. 134—138.
Addicks К., Bloch W., Feelisch М. Nitric oxide modulates sympathetic neu-
rotransmission ay the prejunctional level//Microscopy Res. Tech. —
1994. - Vol. 29. - P. 161-168.
Aguilera G., Kiss A., Xun Luo, Akbasak B.-S. The renin angiotensin system
and the stress response//Ann. N.Y. Acad. Sci. — 1995. — Vol. 771. —
P. 173-186.
Bateman A., Singh A., Kral R., Solomon S. The immune-hypothalamic-pi-
tuitary-adrenal axis//Endocrine Rev. — 1989. — Vol. 10. — P. 92—112.
Beamish R.E., Dhalla N.S. Involvement of catecholamines in coronary
spasm under stressful conditions//Stress and heart disease/Eds R.E. Bea-
mish, P.K.Singal, N.S.Dhalla. — Martinus Nijhoff Publishing. — Bos-
ton — Dordrecht — Lancaster, 1985. — P. 129—141.
345
Bellinger D.L., Lorton D., Felten S.Y., Felten D.L. Innervation of lymphoid
organs and implications in development, aging, and autoimmunity//Int. J.
Immunopharm. — 1992. — Vol. 14. — P. 329—344.
Berridge M.J., Bootman M.D., Lipp P. Calcium — a life and death signal//
Nature. - 1998. - Vol. 395, N 6703. - P. 645-648.
Besendovsky H., Del Rey A., De Prade H. The immune response evokes
changes in brain noradrenergic neurons//Science. — 1983. — Vol. 217,
N 4610. - P. 564-568.
Biondi G., Farrace S., Mameli G., Marongiu F. Is there a hypercoagulable
state in military fighter pi!ots?//Aviat-Space-Environ. Med. — 1996. —
Vol. 76, N 6. - P. 568-571.
Blair R. W. Noxions cardiac input onto neurons in medullary reticular for-
mation//Bran Res. - 1985. - Vol. 326, N 2.- P. 335-346.
Boarder M.R., McArdle Ж Opioid peptides in human adrenal: partial char-
acterization and presence of adrenal peptide E//J. clin. Endocr. — 1985. —
Vol. 61, N 4. - P. 658-665.
Bodnoff Sh., Humphrey A. G., Lehman J. C. et al. Enduring effects of chronic
corticosterone treatment on spatial learning, synaptic plasticity, and hyp-
pocampal neuropathology in young and middle-aged rats//J. Neurosci. —
1995. - Vol. 15, N. 1, Pt. 1. - P. 61-69.
Brady L.S., Lynn A.B., Glowa J.R. et al. Repeated electroconvulsive shock
produces long-lasting increases in messenger RNA expression of cortico-
tropin-releasing hormone and tyrosine hydroxilase in rat brain-thera-
peutic implications//.!, clin. Invest. — 1994. — Vol. 94, N 3. — P. 1263—
1268.
Bredt D.S., Snyder S.H. Nitric oxide, a novel neuronal messenger//Neu-
ron. - 1992. - Vol. 8. - P. 3-11.
Bremner J.D., Kristal J.H., Southwick. S.M., Charney D.S. Noradrenergic
mechanisms in stress and anxiety: I. Preclinical studies//Synapse. —
1996a. - Vol. 23, N 1. - P. 28-38.
Bremner J.D., Kristal J.H., Southwick S.M., Chamey D.S. Noradrenergic
mechanisms in stress and anxiety: II Clinical studies//Synapse. —
1996b. - Vol. 23, N 1. - P. 39-51.
Calogero A.E. Neurotransmitter regulation of the hypothalamic corticot-
ropin-realising hormone neuron//Ann. N.Y. Acad. Sci. — 1995. — Vol.
771. - P. 31-40.
Carlson E.B., Putnam F.W. An update on the dissociative experiences
scale//Dissociation. — 1993. — Vol. 6. — P. 16—27.
Cebelin M., Hirsch C.S. Human stress cardiomyopathy//Hum. Path. —
1980. - Vol. 11. - P. 123-132.
346
Chikanza I.C., Petrou P., Chrousos G.P., Kingsley G. Defective hypotha-
lamic response to immune/inflammatory stimuli in patients with rheuma-
toid arthritis//Rheum. — 1992a. — Vol. 35. — P. 1281—1288.
Chikanza I.C., Chrousos G.P., Panayi G.S. Abnormal neuroendocrine-im-
mune communication in patients with rheumatoid arthritis//Europ. J. clin.
Invest. - 1992b. - Vol. 22. - P. 635-640.
Chrousos G.P., Gold P. Ж The concepts of stress system disorders: overview
of behavioral and physical homeostasis//J.A.M.A. — 1992. — Vol. 267. —
P. 1244-1252.
Clarkson T.B., Kaplan J.R., Adams M.R., Manuck S.B. Psychosocial influ-
ence on the pathogenesis of atherosclerosis among nonhuman primates//
Circulation. — 1987. — Vol. 76, pt. 2, suppl. — P. 1—29.
Culman J., Jtoi K, Unger Th. Hypothalamic tachykinis. Mediators of
stress responses?//Ann. N.Y. Acad. Sci. — 1995. — Vol. 771. — P. 204—
218.
De Bellis M.D., Chrousos G.P., Dorn L.D. et al. Hypothalamic-pituitary-ad-
renal axis dysregulation in sexually abused girls//J. clin. Endocr. —
1994. - Vol. 78. - P. 249-255.
Demitrack M.A., Putnam F. Ж et al. Relation of dissociative phenomena to
levels of cerebral spinal fluid monoamine metabolites and beta-endorphin
in patients with eating disorders: a pilot study//Psychiatry Res. — 1993. —
Vol. 49. - P. 1-10.
Desiderate 0., Mac Kinnon J.R., Hissom H. Development of gastric ulcers
in rats following termination//.!. Comp. Physiol. — 1974. — Vol. 87. —
P. 208-214.
Devries A.C., Gerber J.M., Richardson H.N. et al. Stress affects corticos-
teroid and immunoglobulin concentrations in male house mice (Mus mus-
culus) and prairie voles (Microtus ochrogaster)//Compar. Biochemistry
and Physiol. - 1997. - Vol. 118. - P. 655-663.
Dhabhar F.S., Miller A.H., Me Ewen B.S., Spencer R.L. Stress-induced
changes in blood leucocyte distrebution. Role of adrenal steroid hor-
mones//!. Immunol. - 1996. - Vol. 157, N 4. - P. 1638-1644.
Di Guilio A., Yang H., Fratta Ж Decreased content of immunoreactive
enkephaline-like peptide in peripheral tissues of spontaneously hyperten-
sive rats//Nature. - 1979. - Vol. 278, N 5705,- P. 646-647.
Dobashi K., Pahan K., Chahal L., Singh I. Modulation of endogenous anti-
oxidant enzymes by nitric oxide in rat C-6 giyal cells//J. Neurochem. —
1997. - Vol. 68. - P. 1806-1903.
Doda M., Gyorgy L. Dopaminergic inhibition of sympathetic activity in the
cat//Pol. !. Pharmacol. — 1985. — Vol. 37. — P. 397— 401.
347
Drossman D.A. Physical and sexual abuse and gastrointestinal illness: what
is the link?//Amer. J. Med. — 1994. — Vol. 97. — P. 105—107.
Ely D.L. Organization of vascular and neurohumoral responses to stress//
Ann. N.Y. Acad. Sci. - 1995. - Vol. 771. - P. 594-608.
Erdo S.L. Peripheral GABAergic mechanisms//Trends Pharm. Sci. —
1985. - Vol. 6. - P. 205-208.
Felten D.L., Felten S.Y., Bellinger D.L. Noradrenergic sympathetic neural
interaction with the immune system: structure and function//lmmunol.
Rev. - 1987. - Vol. 100. - P. 225-260.
Fleckenstein A., Frey M., Fleckenstein-Grun G. Myocardial and vascular
damage by intracellular calcium overload. Preventive actions of calcium
antagonists//Calcium entry blockers and tissue protection. — New York:
Raven Press, 1985. — P. 91 — 105.
Foddi M.S., Cinquanta M., Memmini T. «Conditioned fear», a model for
psychological stress increases [35S] TBPS binding in rat brain//Behav.
Pharmacol. — 1995. — Vol. 6, Suppl. 1. — P. 151 — 152.
Friedman E.M., Irwin M.R. A role for CRH and the sympathetic nervous
system in stress-induced immunosuppression//Ann. N.Y. Acad. Sci. —
1995. - Vol. 771. - P. 396-418.
Gebara O.C., Jimenez A.H., Me Кеппа C. et al. Stress-induced hemody-
namic and hemostatic changes in patients with systemic hypertension: ef-
fect of verapamil//Clin. Cardiol. — 1996. — Vol. 19, N 3. — P. 205—211.
Glaser R., Kiekolt-Glaser J.K., Bonneau R.H. et al. Stress-induced modula-
tion of the immune response to recombinant hepatitis В vaccine//Psy-
chosom. Med. — 1992. — Vol. 54. — P. 22—29.
Gold P. W., Goodwin F, Chrousos G.P. Clinical and biochemical manifesta-
tions of depression: relationship to the neurobiology of stress. Part 2//New
Engl. J. Med. - 1988. - Vol. 319. - P. 413-420.
Goldman H., Rosqff C. Pathogenesis of acute gastric stress ulcers//Amer. J.
Path. - 1968. - Vol. 52. - P. 227-243.
Goldstein L.E., Rasmusson A.M., Bunney B.S. et al. Role of the amygdala in
the coordination of behavioral, neuroendocrine, and prefrontal cortical
monoamine responses to psychological stress in the rat//J. Neurosci. —
1996. - Vol. 16(15). - P. 4787-4798.
Gray Y.S. The organisation and possible function of amygdaloid cortico-
tropin releasing factor pathways// Corticotropin releasing factor: basic and
Clonical studies of neuropeptide. Eds. E.B. De Sonza, C.B.Nemeroff.
CRC Press. Boca Rator. — 1990. — P. 53—68.
Groves B., Marcus E, Ewy G., Phibbs B. Coronary arterial spasm in variant
angina pertoris produced by hyperventilation and valsalva-documented by
348
coronary arteriography//Coronary medicine and surgery/Ed. J.C.Hor-
man. - N.-Y., 1975. - P. 311-315.
Hambley J.W., Johnston G.A.R., Shaw J. Alterations in the sponta-
neously hypertehsive rat//Neurochem. hit. — 1984. — Vol. 6. — P.
813-821.
Harbuz M.S., Jessop D.S., Chowdrey H.S. et al. Evidence for altered control
of hypothalamic CRF in immune-mediated diseases//Ann. N.Y. Acad.
Sci. - 1995. - Vol. 771. - P. 449-458.
Harbuz M.S., Lightman S.L. Stress and the hypothalamo-pituitary-adrenal
axis: acute, chronic and immunological activation//.!. Endocrinol. —
1992. - Vol. 134. - P. 327-339.
Helmert U., Shea S. Social inequalities and health status in western Ger-
many//Public-Health. - 1994. - Vol. 108, N 5. - P. 341-356.
Henry J.P., Liu Y.-Y., Nadra W.E. et al. Psychosocial stress can induce
chronic hypertension in normotensive strains of rats//Hypertension. —
1993. - Vol. 21. - P. 714-723.
Kaneko M., Harada N., Furuya H. et al. The effect of work-related stress
with change of working conditions for workers with alcohol drinking habit
and liver disorder//Nihon — Arukoru — Yakubutsu — Igakkoi — Zasshi. —
1996. — Vol. 31, N I. - P. 81-94.
Kanner J., Harel S., Granit R. Nitric oxide as an antioxidant//Arch. bio-
chem. Biophys. - 1991. - Vol. 289. - P. 130-136.
Kennett G.A., Chaouloff F., Marcou M., Curzon G. Female rats are more
vulnerable than males in an animal model of depression: the possible role
of serotonin//Brain Res. — 1986. — Vol. 382. — P. 416—421.
Landmann R., Burgisser F., Wesp M. Beta-adrenergic receptors are differ-
ent in subpopulations of human circulating lymphocytes//.!. Recept.
Res. - 1984. - Vol. 4, N 1-6. - P. 37-50.
Lazarus R.S. From psychological stress to the emotions: a history of chang-
ing out!ook//Ann. Rev. Psychol. — 1993. — Vol. 44. — P. 1—21.
Levenstein S., Kaplan G.A., Smith M. Sociodemographic characteristics, life
stressors, and peptic ulcer. A prospective study//J. Clin. Gastroenterol. —
1995. - Vol. 21, N 3. - P. 185-192.
Lin F.H., Wang Y, Lin S. et al. GABA(B) receptor-mediated effects in sy-
naptosomes of Lethargic (Ih/In) mice//J. Neurochem. — 1995. — Vol.
65. -P. 2087-2095.
Lown B., De Silva R. Roles of psychologic stress and autonomic nervous
system changes in provocation of ventricular premature complexes//Am. J.
Cardiol. - 1978. - Vol. 41. - P. 979-985.
349
Lown В., De Silva R., Reich P., Mirawski B. Psychophysiologic factor in
sudden cardiac death//Am.J. Psychiatr. — 1980. — Vol. 137. — P. 1325—
1335.
Magarinos A. M., Me Ewen B.S., Flugge G., Fuchs E. Chronic psychosocial
stress causes apical dendritic atrophy of hyppocampal CA3 pyramidal neu-
rons in subordinate tree shrews//! Neurosci. — 1996. — Vol. 16, N 10. —
P. 3534-3540.
Malyshev I. Yu., Malugin A. V., Golubeva L. Yu. et al. Nitric oxide donor in-
duces HSP70 accumulation in the heart and in cultured cells//FEBS
Lett. - 1996. - Vol. 391. - P. 21-23.
Malyshev I. Yu., Manukhina E.B., Mikoyan V.D. et al. Nitric oxide is in-
volved in head-induced HSP70 accumulation//FEBS Lett. — 1995. — Vol.
370. - P. 159-162.
Manukhina E.B., Lapshin A.V., Meerson F.Z. Physical training limits the
fall of blood pressure and the endothelium overactivation in acute myocar-
dial infarction//Physiol. Res. — 1996. — Vol. 45. — P. 261 —266.
Marotti T., Gabrilovac J., Rabatic S. et al. Met-enkephalin modulates
stress-induced alterations of the immune response in mice//Pharmacol.
Biochem. Behav. — 1996. — Vol. 54, N I. — P. 277—284.
Meerson F.Z. Adaptive protection of the heart; protecting against stress and
ischemic damage. — Boca Raton, CRC Press. — 1991. — 340 P.
Meerson F.Z., Pshennikova M.G., Malyshev I. Yu. Adaptive defense of
the organism: architecture of the structural trace and cross protective ef-
fects of adaptation//Ann. N.Y Acad. Sci. — 1996. — Vol. 793. — P. 371 —
385.
Monnikes H., Schmidt B.G., Tebbe J. et al. Microinfusion of corticotropin
releasing factor into the locus coeroleus/subcoeruleus nuclei stimulates
colonic motor function in rats//Brain Res.— 1994.— Vol. 644.— P. 101 —
107.
Morris J.J., O’Connor C.M., Blumenthal J.A. Mental stress-induced myo-
cardial ischemia and cardiac events//!A.M.A. — 1996. — Vol. 275,
N 21. - P. 1651-1656.
Mudge G.H., Grossman W., Mills R.M. et al. Reflex increase in coronary
vascular resistance in patients with ischemic heart disease//N. Engl. J.
Med. - 1976. - Vol. 295. - P. 1333-1340.
Myers A., Dewar H.A. Circumstances attending 100 sudden deaths from
coronary artery disease with coroner’s necropsies//Br. Heart J. — 1975. —
Vol. 37. - P. 1133-1146.
Noll G., Wenzel R.R., Schneider M. et al. Increased activation of sympa-
thetic nervous system and endothelin by mental stress in normotensive
350
offspring of hypertensive parents//Circulation. — 1996. — Vol. 93, N 5. —
P. 866-869.
Ogino K., Takeyama Y., Ischiyama H. et al. Role of nitric ocide in stress
ulcer//Pathophysiology. — 1994. — Vol. 1. — Suppl. — P. 391.
Pacak K., Palkovits M., Kvetnansky R. et al. Effects of various stressors on
in vivo norepinephrine release in the hypothalamic paraventricular nucleus
and on the pituitary-adrenocortical axis//Ann. N-Y Acad. Sci. — 1995. —
Vol. 771. - P. 115-130.
Peristein R.S., Whitnall M.H., Abrams J.S. et al. Sinetgic roles of inter-
leukin-6, interleukin-1 and tumor necrosis necrosis factor in adrenocorti-
cotropin response to bacterial lipopolysacharide in vitro//Endocrino-
logy. - 1993. - Vol. 132. -P. 946-952.
Petty F. GABA and disorders: a brief review and hypothesis//). Affective
Disord. - 1995. - Vol. 34. - P. 275-281.
Putnam F.W. Traumatic stress and pathological dissociation//Ann. N.Y.
Acad. Sci. - 1995. - Vol. 771. - P. 708-715.
Rose G., Marmot M.G. Social class and coronary heart disease//Br. Heart
J. - 1981. - Vol. 45. - P. 13-16.
Ruff M.R., Wahls S.M., Mergenhagen S. et al. Opiate receptor-mediated
chemotaxis of human monocytes//Neuropeptid. — 1989. — Vol. 5. —
P. 363-366.
Samuels M.A. Neurogenic heart disease: a unifying hypothesis//Am. J.
Cardiol. - 1987. - Vol. 60. - P. 15J-19J.
Satinsky J., Kosowsky B., Lown B. Ventricular fibrillation induced by hypo-
thalamic stimulation during coronary occlusion//Circulation. — 1971. —
Vol. 44 — Suppl. 2, Pt. II. - P. 60.
Schwarz P-, Diem R., Dun N.J., Forstermann U. Endogenous and exogenous
nitric oxide inhibits norepinephrine release from rats heart sympathetic
nerves//Circ. Res. — 1995. — Vol. 77. — P. 841—848.
Selye H. Syndrome-produced by diverse nocuous agents//Nature. —
1936. - Vol. 138, N 3479. - P. 32.
Shibasaki T., Hotta M., Sugihara H., Wakabayashi I. Brain vasopressin is
involved in stress-induced supression of immune function in rat//Brain
Res. - 1998. - Vol. 808, N 1. - P. 84-92.
Simons M., Downing S.E. Coronary vasoconstriction and catecholamine
cardiomyopathy//Am. Heart. J. — 1985. — Vol. 109. — P. 297—305.
Skinner J.E. Psychosocial stress and sudden cardiac death: brain mecha-
nisms//Stress and heart disease, (eds. R.E. Beamish, P.K.Singal, N.S.Dhal-
la). — Boston/Dordrecht/Lancaster. — 1985. — P. 44—59.
351
Skinner J.E., Reed J. C. Blocade of a front ocortical-brainstem pathway pre-
vents ventricular fibrillation on the ischemic heart in pigs//Amer. J.
Physiol. - 1981. - Vol. 240. - P. H156-H163.
Stein M., Miller A. H., Trestman R.L. Depression, the immuno system and
health and illness//Arch.Gen.Psychiatry.— 1991.—Vol. 48.— P. 171 — 177.
Sternberg E.M., Chrousos G.P., Wilder R.I., Gold P.W. The stress response
and the regulation of inflammatory disease//Ann. Intern. Med. — 1992. —
Vol. 117, N 10. - P. 854-866.
Sternberg E.M., Licino J. Overview of neuroimmune stress interactions//
Ann. N.Y. Acad. Sci. - 1995. - Vol. 771. - P. 364-371.
Stratakis C.A., Chrousos G.P. Neuroendocrinology and pathophysiology of
the stress system//Ann. N.Y. Acad. Sci. — 1995. — Vol. 771. — P. 1 — 18.
Sudakov K.V., Coghlan J.P., Kotov A. V. et al. Delta-sleep-inducing peptide
sequels in the mechanisms of resistance to emotional stress//Ann. N.Y.
Acad. Sci. - 1995. - Vol. 771. - P. 240-251.
Sundar S.K., Cietpiel M.A., Kilts C. et al. Brain IL-1-induced immunosup-
pression occurs through activation of both pituitary-adrenal axis and sym-
pathetic nervous system by corticotropin-releasing factor//J. Neurosci. —
1990. - Vol. 10, N 11. - P. 3701-3706.
Takasu N., Miyazaki K, Ogawa K, Satake I. Activation of phospholipases
play a role in the development of isoproterenol-induced myocardial dam-
age//! Mol. Cell. Cardiol. - 1986. - Vol. 18. - Suppl. 1. - P. 227-231.
Tauber H.L. Some effects of frontal lobotomy in man//The frontal granu-
lar cortex and behavior (eds. J.M. Warren, K. Akkert) — San Francisco. —
Vc Graw Hill. -1964. - P. 332-333.
Tennen H., Swis J., Affeck G. Personality and daily experience: the promise
and the challenge//! Pers. - 1991. - Vol. 59. - P. 313-337.
Vallance P., Benjamin N., Collier J. The effect of endothelium derived ni-
tric oxide on ex vivo whole blood platelet aggregation in man//Eur. J.
Pharmacol. —1992. — Vol. 42. — P. 37—41.
Vanhatalo S., Soinila S. Nitric oxide synthase in the hypothalamo-pituitary
pathway//! Chem. Neuroanat. — 1995. — Vol. 8. — P. 165—173.
Vanin A.F., Lapshin A. V., Manukhina E.B., Meerson F.Z. Increased nitric
oxide synthesis by rat aorta in experimental myocardial intarction//The bi-
ology of nitric oxide. — Part 3. Clinical and physiological aspects (eds.
S.Moncada, M.Feelish, R.Busse, E.A.Higgs). — Portland Press, Lon-
don. - 1994. - P. 182-185.
Van Praag H.M. Neurotransmitters and CNS disease: depression//Lan-
cet. - 1982. - Vol. 1. - P. 1259-1264.
352
Verrier R.L., Hagestad E.L., Lown B. Behaviorally induced coronary vaso-
constriction in dogs with critical coronary artery stenosis//Fed. Proc. Am.
Soc. Exp. Biol. - 1984. - Vol. 43. - P. 1003.
Weissman M.M., Kierman G.L. Sex differences and the epidemiology of de-
pression//Arch.Gen.Psychiatry. — 1977. — Vol. 34. — P. 98—111.
Werling L.L., Brown S.R., Cox B.M. Opioid receptor regulation of the re-
lease of norepinephrine in brain//Neuropharmacology. — 1987. — Vol.
26. - P. 987-996.
Westfall T.C., Meldrum MJ. Alterations in the release of norepinephrine at
the vascular neuroeffector junction in hypertension//Ann. Rev. Pharmacol.
Toxicol. - 1985. - Vol. 25. - P. 621-641.
Whitnall M.H. Regulation of the hypothalamic corticotropin-releasing hor-
mone neurosecretory system//Progr. Neurobiol. — 1993. — Vol. 40. —
P. 573-629.
Whitnall M.H., Gainer H., Сох B.M., Molineaux C.J. Dynorphin-A-(l-8) is
contained within vasopressin neurosecretory vesicles in rat pituitary//Sci-
ence. - 1983. - Vol. 222. - P. 1137-1140.
Wrona D., Staszewska M., Trojniar W. Lateral hypothalamic lesions de-
crease activity of peripheral blood lymphocytes in rats//J. Neuroimmu-
nol. - 1991. - Suppl. 1. - P. 199.
Yamabe T., Okumura K., Ishizaka T, Yasue H. Role of endothelium-de-
rived nitric oxide in myocardial reactive hyperemia//Am. J. Physiol. —
1992. - Vol. 263. - P. H8-H14.
Yasue H., Takizawa A., Nagao M. et al. Role of coronary spasm in different
anginal syndromes//Acta med. Scand. — 1984. — Vol. 694. —Suppl. —
P. 83-90.
Yehuda R. Stress and glucocorticoids//Science. — 1997. — Vol. 275. —
P. 1662-1663.
Yehuda R., Giller E.L., Southwick S.M. et al. Hypothalamic pituitary adre-
nal axis dysfunction in PTSD//Biol. Psychiatry. — 1991. — Vol. 30. —
P. 1031-1048.
Young M.A., VatnerS.F. Regulation of large coronary arteries//Circ. Res.—
1986. - Vol. 59. - P. 579-596.
Zanchi A., Shaad N. C, Osterheld M. C. et al. Effect of chronic NO synthase
inhibition in rats on renin-angiotensin system and sympathetic nervous
system//Am. J. Physiol. — 1995. — Vol. 37. — P. H2267—H2273.
Zukowska-Grojec Z., Neuropeptide Y. A novel sympathrtic stress hormone
and more//Ann. N.Y. Acad. Sci. — 1995. — Vol. 771. — P. 219—233.
23 — 1385
Лекция 6
Боль: физиологические
и патофизиологические аспекты
В.К.Решетняк, М.Л.Кукушкин
Введение
В своей жизни практически каждый человек испытывал
боль — неприятное ощущение с негативными эмоциональ-
ными реакциями, возникающими вследствие действия на ор-
ганизм экзогенных повреждающих факторов или в результате
эндогенного патологического процесса. И в первом, и во
втором случае боль выполняет сигнальную функцию, предуп-
реждая организм об опасности. Восприятие, проведение и
анализ болевых сигналов обеспечивают специальные нейро-
нальные структуры. Поэтому боль можно рассматривать как
одну из реакций организма, необходимую для нормальной
жизнедеятельности и предупреждающую нас о вредных воз-
действиях.
Вместе с тем врачам известен и другой вид боли, который
имеет патогенное значение для организма. Эта боль делает
людей нетрудоспособными, снижает их активность, вызывает
психоэмоциональные расстройства, приводит к регионарным
и системным нарушениям микроциркуляции, является причи-
ной вторичных иммунных депрессий, нарушения деятельнос-
ти висцеральных систем. Такую боль называют патологи-
ческой.
С античных времен и до настоящего времени борьба с па-
тологической болью была и остается одной из самых актуаль-
ных проблем медицины. Боль не только угнетающе действует
на психику больного и его близких, что само по себе имеет
отрицательное социальное значение, но, кроме того, из-за
огромного числа людей, страдающих хроническими болевы-
ми синдромами, общество несет большой экономический
ущерб. Так, в результате утраты трудоспособности населе-
ния, вызванной болевыми синдромами, экономические поте-
ри в США составляют ежегодно 90 млрд долл. [Bonica J.J.,
1990]. Статистика ВОЗ свидетельствует о том, что в мире
каждый пятый трудоспособный человек ежегодно страдает от
какого-либо острого или хронического болевого синдрома.
В Международной классификации болезней IX пересмотра
перечислено около 40 различных болевых синдромов, кото-
354
рые сопровождают почти 70 % всех хронических заболе-
ваний.
В последние годы число случаев возникновения хроничес-
ких болевых синдромов увеличилось. По данным ВОЗ, наибо-
лее распространены следующие формы хронических болевых
синдромов:
• головная боль, насчитывающая 17 разных видов — от
мигрени до злокачественной цефалгии;
• проявления остеохондроза, дискогенные радикулиты,
люмбалгии и ишиалгии;
• хронические болевые синдромы, связанные с ишемичес-
кой болезнью сердца;
• артралгии в результате воспаления, дистрофических и
аллергических поражений суставов;
• периферические нейроваскулярные болевые синдромы;
• каузалгии и фантомная боль;
• психогенные хронические болевые синдромы, вегетал-
гии, миалгии;
• висцеральная боль при хронических заболеваниях внут-
ренних органов.
Помимо хронических, существует ряд острых болевых син-
дромов, возникающих при внезапных заболеваниях и травмах.
В настоящее время, как свидетельствуют материалы ВОЗ,
только в результате транспортных происшествий ежегодно в
мире получают травмы более 10 млн человек. При этом отме-
чается тенденция к росту числа случаев травматизма.
В данной лекции по понятным причинам не могут быть
рассмотрены все вопросы, связанные с этиологией, патогене-
зом и лечением отдельных болевых синдромов. Основное вни-
мание уделяется главным закономерностям функционирова-
ния ноцицептивной и антиноцицептивной систем, т.е. систе-
мы, воспринимающей и оценивающей повреждающие воздей-
ствия и реализующей ощущение боли (ноцицептивная систе-
ма — от посеге — повреждать и сереге — воспринимать), и
системы, контролирующей ноцицептивную систему и подав-
ляющей боль (антиноцицептивная система), и методам кор-
рекции их нарушений.
6.1. Характеристика боли
Боль всегда субъективна, и ее восприятие зависит от места
и характера повреждения, от природы повреждающего факто-
ра, условий, при которых произошло повреждение, от психо-
логического состояния человека, его индивидуального жиз-
ненного опыта и социального статуса.
23*
355
Боль — понятие многокомпонентное. Она включает в себя:
• перцептуальный компонент (поступление ноцицептив-
ного сигнала в кору головного мозга), позволяющий оп-
ределить место повреждения;
• эмоционально-аффективный компонент, формирующий
неприятное психоэмоциональное переживание;
• вегетативный компонент, отражающий рефлекторные
изменения работы внутренних органов и тонуса симпа-
тоадреналовой системы;
• двигательный компонент, направленный на устранение
действия повреждающих стимулов;
• когнитивный компонент, формирующий субъективное
отношение к испытываемой в данный момент боли на
основе накопленного опыта.
Эксперты Международной ассоциации по изучению боли
дали следующее определение боли.
Боль — это неприятное ощущение и эмоциональное пере-
живание, связанное с реальным или потенциальным повреж-
дением тканей или описываемое в терминах такого повреж-
дения.
Данное определение свидетельствует о том, что ощущение
боли может возникать не только при повреждении ткани или в
условиях риска повреждения ткани, но даже при отсутствии
какого-либо повреждения. В последнем случае определяющим
в механизме возникновения боли является психоэмоциональ-
ное состояние человека (депрессия, истерия или психоз).
Иными словами, интерпретация человеком болевого ощуще-
ния, его эмоциональная реакция и поведение могут не корре-
лировать с тяжестью повреждения.
В зависимости от локализации повреждения боль мо-
жет быть разделена: на соматическую поверхностную (в слу-
чае повреждения кожных покровов), соматическую глубо-
кую (при повреждении костно-мышечной системы) и висце-
ральную.
Боль может быть обусловлена повреждением структур пе-
риферической и/или центральной нервных систем, участвую-
щих в проведении и анализе болевых сигналов. Боль, возни-
кающая при повреждении периферических нервов, называют
нейропатической болью, а при повреждении структур ЦНС —
центральной болью.
Особую группу составляют психогенные боли, которые воз-
никают вне зависимости от соматических, висцеральных или
нейрональных повреждений и в большей степени определяют-
ся психологическими и социальными факторами.
По временным параметрам выделяют острую и хроничес-
кую боль.
356
Острая боль — это новая, недавняя боль, неразрывно свя-
занная с вызвавшим ее повреждением и, как правило, являет-
ся симптомом какого-либо заболевания. Такая боль исчезает
при устранении повреждения. Хроническая боль часто приобре-
тает статус самостоятельной болезни, продолжается длитель-
ный период, и причина, вызвавшая эту боль, в ряде случаев
может не определяться.
6.2. Физиология боли
6.2.1. Анатомо-функциональная организация
ноцицептивной системы
В 1897 г. немецкий невропатолог Фрей опубликовал рабо-
ту, в которой высказал мнение о том, что существуют специ-
альные болевые рецепторы, ответственные за восприятие бо-
левого раздражения (теория специфичности). В следующем
году появилась работа Голдшейдера, который сообщил, что
болевое ощущение возникает не в результате раздражения спе-
циальных болевых рецепторов, а в ответ на сверхсильное раз-
дражение тактильных и температурных рецепторов (теория
интенсивности). На протяжение текущего столетия велись до-
вольно интенсивные дискуссии между сторонниками этих
двух теорий. В итоге, как это часто бывало в медицине, правы
оказались сторонники обеих теорий.
Успехи, достигнутые в этой области за последние два деся-
тилетия [Вальдман А.В., Игнатов Ю.Д., 1976; Решетняк В.К.,
1985; Wall P.D., Melzack R., 1994], позволяют достаточно
полно описать анатомию и физиологию структур ноцицептив-
ной системы, осуществляющей восприятие, проведение и об-
работку информации о различных потенциально или реально
вредных раздражителях, воздействующих на организм.
6.2.1.1. Ноцицепторы и их афференты
Считается, что восприятие повреждающих (ноцигенных)
стимулов осуществляется ноцицепторами — рецепторами,
представляющими собой свободные неинкапсулированные
нервные окончания [Ревенко С.В. и др., 1988]. Ноцицепторы
активируются при сильном механическом (укол, щипок) или
термическом раздражении (нагревание или охлаждение), а
также в результате действия различных химических веществ —
алгогенов (от algos — боль). Поэтому в зависимости от меха-
низма возбуждения ноцицепторы могут быть разделены на ме-
ханоноцицепторы, термоноцицепторы и хемоноцицепторы. Ме-
хано- и термоноцицепторы передают сигналы преимуществен-
357
но через миелинизированные А-дельта волокна (группа III со
скоростью проведения сигнала от 2,5 до 20 м/с), а хемоноци-
цепторы — через немиелинизированные С-волокна (афферен-
ты группы IV со скоростью проведения сигнала менее 2,5 м/с).
Большинство ноцицепторов способны реагировать одновре-
менно на механические, термические и химические стимулы,
иными словами, они полимодальны.
Общим свойством для всех ноцицепторов является более
высокий порог их активации по сравнению с механо- и тер-
морецепторами, реагирующими на тактильные, тепловые
или холодовые раздражения.
Ноцицепторы имеют дискретное распределение по поверх-
ности кожи, что отличает их от механо- и терморецепторов.
Их точечная активация при помощи волосков Фрея или фоку-
сированного ультразвука сразу приводит к возникновению
резкой боли. Представленная характеристика соответствует
описанию специфических ноцицепторов, возбуждающихся
исключительно повреждающими воздействиями.
Вместе с тем наряду со специфическими ноцицепторами
существуют и так называемые неспецифические ноцицепторы
(так же как и первые, они представлены свободными нервны-
ми окончаниями), которые начинают возбуждаться слабыми,
субноцигенными стимулами и при увеличении силы раздраже-
ния прогрессивно усиливают паттерн своего ответа. Их акти-
вация сначала приводит к появлению тактильных или темпе-
ратурных ощущений, и только дальнейшее увеличение интен-
сивности стимула вызывает диффузную боль.
В настоящее время нет сомнений в том, что С- и А-дельта-
афференты, связанные с ноцицепторами, являются основны-
ми проводниками болевой чувствительности. Считается, что
при активации С-волокон возникают ощущения задержанной,
плохо локализованной боли, а при возбуждении А-дельта-аф-
ферентов — быстрая, острая, точно локализованная боль.
Вместе с тем необходимо подчеркнуть, что А-дельта и С-во-
локна не являются проводниками, специфическими для боле-
вой чувствительности, и могут передавать сигналы, не связан-
ные с ноцицепцией.
6.2.1.2. Периферические алгогены
Эндогенные алгогены выделяются при повреждении или
воспалении тканей и не только приводят к выраженной боле-
вой реакции, но и повышают чувствительность ноцицепторов
к последующим раздражениям.
Механизм возбуждающего действия алгогенов на хемоно-
цицепторы недостаточно изучен, однако предполагают, что в
358
его основе лежат механохимические процессы, приводящие к
изменению ионной проницаемости в мембране нервного
окончания. Большое значение в активации хемоноцицепторов
имеет скорость изменения биохимических констант в тканях,
окружающих нервные окончания. Алгогены в минимальных
концентрациях при взаимодействии с хемоноцицепторами вы-
зывают у человека боль.
Различают несколько типов аллогенных веществ:
• тканевые алгогены — гистамин, серотонин, ацетилхо-
лин, простагландины, лейкотриены, цитокины, К+ и Н+;
• плазменные алгогены — брадикинин, каллидин;
• алгогены, выделяющиеся из нервных окончаний, — суб-
станция Р, нейрокинин А, кальцитонин — ген — родст-
венный пептид.
Среди алгогенов, выделяющихся при повреждении тканей,
наибольшее значение в активации и сенситизации ноцицепто-
ров принадлежит брадикинину, простагландинам, цитокинам,
серотонину и гистамину. Брадикинин, высвобождаясь из
плазмы крови в области повреждения, не только самостоя-
тельно может активировать ноцицептивные рецепторы, но и
индуцирует образование из арахидоновой кислоты других ме-
диаторов воспаления — простагландинов, тромбоксанов и
лейкотриенов. В свою очередь выделенные вещества, обладая
выраженным самостоятельным алгогенным действием, потен-
цируют способность гистамина, серотонина и брадикинина
воздействовать на нервные окончания. Вслед за этим из неми-
елинизированных С-афферентов выходят тахикинины (суб-
станция Р и нейрокинин А), которые, увеличивая сосудистую
проницаемость, еще больше повышают локальную концентра-
цию медиаторов воспаления.
6.2.1.3. Первое переключение ноцицептивной информации
(первичное ноцицептивное реле)
Тела нейронов, воспринимающих стимулы от ноцицептив-
ных рецепторов туловища и конечностей, расположены в
спинномозговых ганглиях, а от рецепторов лица — в ганглии
тройничного нерва. От этих клеток большинство центральных
аксонов соответственно входят в спинной мозг через задний
корешок или направляются в продолговатый мозг в составе
тройничного нерва. Некоторая часть болевых афферентов
проникает в спинной мозг через передние корешки. После
входа в спинной мозг А-дельта и С-волокна разделяются на
восходящие и нисходящие ветви и оканчиваются на нейронах
заднего рога, где и происходят первичная переработка ноци-
цептивной информации и передача ее в структуры головного
мозга [Михайлович В.А., Игнатов Ю.Д., 1990].
359
Рис. 6.1. Организация ноцицептивной системы.
С1 — первая соматосенсорная область коры больших полушарий; С2 — вторая
соматосенсорная область коры больших полушарий; ПФК — префронтальная
область коры; ПЦК — передняя цингулярная область коры; ВПЛ — вентропос-
теролатеральное ядро таламуса; ВПМ — вентропостеромедиальное ядро тала-
муса; ВПН — вентропостеронижнее ядро таламуса; ЗГ — задняя группа ядер
таламуса; ЦМ-ПФ центральное медиальное и парафасцикулярное ядра таламу-
са; ЦЛ — центральное латеральное ядро таламуса; МД — медиодорсальное
ядро таламуса; ДС — дорсальные столбы спинного мозга; СТТ — спинотала-
мический тракт; СРТ — спиноретикулоталамический тракт; 1—3, 5—7 — плас-
тины дорсальных рогов спинного мозга.
Выделяют три группы нейронов дорсальных рогов, получа-
ющих ноцицептивные сигналы с периферии (рис. 6.1). Первая
группа нейронов — это специфические ноцицептивные ней-
роны, возбуждающиеся исключительно повреждающими сти-
мулами. Локализованы они преимущественно в 1-й пластине
дорсального рога, где оканчиваются А-дельта и С-афференты,
и имеют небольшие рецептивные поля.
Вторая группа нейронов — это нейроны широкого динами-
ческого диапазона (ШДД), или конвергентные нейроны, кото-
рые возбуждаются как неноцицептивными, так и ноцицептив-
ными стимулами с широких рецептивных полей и располага-
ются преимущественно в 5—7-й пластинах дорсального рога.
Характерной особенностью этих нейронов является то, что
они изменяют паттерн своего ответа при увеличении интен-
сивности наносимых раздражений и долго сохраняют повы-
шенную возбудимость после короткого ноцицептивного сти-
мула. Кроме этого, нейроны ШДД находятся под бблыним
нисходящим тормозным влиянием супраспинальных центров
по сравнению со специфическими ноцицептивными нейрона-
ми. Все это позволяет заключить, что специфические ноци-
360
цептивные нейроны обеспечивают прямую передачу ноцицеп-
тивных сигналов в вышележащие структуры мозга, на основе
которых формируется ощущение острой строго локализован-
ной первичной боли. Нейроны ШДД кодируют интенсивность
ноцицептивного стимула, что придает сигналу соответствую-
щую эмоциональную окраску.
От нейронов первой и второй групп начинаются восходя-
щие афферентные системы, осуществляющие проведение но-
цицептивных сигналов к супраспинальным структурам мозга.
Третья группа нейронов дорсального рога представлена ней-
ронами желатинозной субстанции (2-я пластина), которые вли-
яют на формирование восходящего ноцицептивного потока,
прямо воздействуя на активность клеток первых двух групп.
Именно этим нейронам R.Melzack и P.D.Wall (1965) отвели ве-
дущую роль в теории «воротного контроля боли». Основное по-
ложение этой теории заключается в том, что нейроны желати-
нозной субстанции, активируясь «неболевыми» импульсами,
приходящими с периферии по толстым миелинизированным
волокнам, или нисходящими импульсами из супраспинальных
центров, тормозят нейроны ШДД и тем самым ограничивают
прохождение ноцицептивных сигналов в структуры головного
мозга [Калюжный Л.В., 1984; BonicaJ.J., 1990].
6.2.1.4. Восходящие спинальные тракты (пути)
ноцицептивных афферентов
Основная масса ноцицептивной информации передается в
составе спиноталамического, спиноретикулярного и спиноме-
зенцефалического трактов. Спиноталамический тракт (неос-
пиноталамический тракт) обеспечивает прямое, быстрое по-
ступление ноцицептивных сигналов в ядра таламуса. Спино-
ретикулярный и спиномезэнцефалический тракты представля-
ют собой филогенетически более древний палеоспиноталами-
ческий тракт с полисинаптическим медленным проведением
импульсов.
Как видно на рис. 6.1, спиноталамический тракт (СТТ) об-
разуют аксоны, нейроны которых лежат в 1-й, 5-й и частично
в 6-й пластинах серого вещества заднего рога. После пере-
креста в передней комиссуре волокна СТТ в составе вентрола-
терального канатика поднимаются вверх и оканчиваются в
ядрах таламуса: в специфическом релейном ядре соматосен-
сорной чувствительности — вентро-постеро-латеральном
(ВПЛ), в ассоциативных ядрах медиальной части задней груп-
пы ядер таламуса и неспецифических ядрах интраламинарного
комплекса. Среди нейронов дорсального рога, аксоны кото-
рых образуют СТТ, имеются как специфические ноцицептив-
ные нейроны, так и нейроны широкого динамического диапа-
361
зона. Необходимо отметить, что среди аксонов спиноталами-
ческого тракта, направляющихся в ВПЛ от нейронов 1-й и 2-й
пластин дорсального рога, многие отдают коллатерали к ней-
ронам ретикулярной формации и центрального серого веще-
ства.
Спиноретикулярный тракт берет начало от аксонов, ней-
роны которых лежат в 5-й и 7-й пластинах дорсальных рогов.
Нейроны 1-й пластины не принимают участия в образовании
этого тракта, поэтому большинство волокон спиноретикуляр-
ного тракта составляют аксоны неспецифических полимодаль-
ных нейронов широкого динамического диапазона. Термина-
ли этих нейронов оканчиваются в медиальной ретикулярной
формации продолговатого мозга (ретикулярном гигантокле-
точном ядре, парагигантоклеточном ядре и большом ядре
шва). В свою очередь восходящие проекции от этих ядер на-
правляются к интраламинарным ядрам таламуса, а другая суб-
таламическая ветвь распределяется в структурах лимбический
системы.
Спиномезэнцефалический тракт, так же как и спинотала-
мический, образован аксонами нейронов 1-й и 5—7-й пластин
заднего рога. Основная часть аксонов этого тракта оканчива-
ется в покрышке среднего мозга, центральном сером веществе
и интраламинарных ядрах таламуса. Учитывая тот факт, что
большинство аксонов оканчивается в структурах, контролиру-
ющих проведение болевых сигналов, было высказано предпо-
ложение, что нейроны этого тракта скорее связаны с актива-
цией антиноцицептивных реакций, нежели с формированием
различных ощущений боли. Обширные двусторонние повреж-
дения вентролатеральных канатиков спинного мозга, где про-
ходят волокна спиноталамического, спиноретикулярного и
спиномезэнцефалического трактов, вызывают полную потерю
болевой и температурной чувствительности ниже места по-
вреждения.
В последнее время появились сообщения о том, что клас-
сический неболевой спинальный путь — дорсальные (задние)
столбы, получающий входы от толстых миелинизированных
низкопороговых афферентов, участвует в проведении ноци-
цептивных сигналов. При этом обсуждается наличие как пря-
мых входов от нейронов спинномозговых ганглиев, активиру-
емых ноцицептивными А-дельта-волокнами, так и опосредо-
ванных постсинаптических входов от ноцицептивных клеток
дорсального рога. Полагают, что ноцицептивная информация,
передающаяся по этому тракту, наряду со спиноталамическим
трактом обеспечивает формирование представлений о локали-
зации и качестве болевого стимула. Вместе с тем клинические
наблюдения свидетельствуют о том, что повреждение задних
столбов не только не повышает пороги болевой чувствитель-
ности, а наоборот, снижает их, при этом одновременно нару-
362
шаются все виды дискриминационной чувствительности, ут-
рачивается способность определять положение конечностей,
развивается атаксия. Поэтому в функциональном отношении
задние столбы рассматривают не как систему дублирующих
каналов, обеспечивающих формирование болевого ощущения,
а как афферентное звено, активирующее центральные антино-
цицептивные структуры мозга (на рис. 6.1 это обозначено
пунктирными стрелками).
6.2.1.5. Таламические ядра, воспринимающие
ноцицептивную афферентацию
Большая роль в анализе ноцицептивных сигналов принад-
лежит ядрам таламуса, являющимся основным коллектором,
где собирается и обрабатывается вся сенсорная информация.
Среди большого многообразия ядер таламуса непосредствен-
ное отношение к интеграции боли имеют вентробазальный
комплекс, интраламинарные ядра, задняя группа ядер и ме-
диодорсальное ядро.
Вентробазальный комплекс, включая вентропостеролате-
ральное (ВПЛ) и вентропостеромедиальное (ВПМ) ядра тала-
муса (см. рис. 6.1), является основной передаточной структу-
рой афферентной соматосенсорной системы. Его нейроны ак-
тивируются соматотопически с небольших рецептивных полей
контралатеральной стороны, т.е. ядра, расположенные в пра-
вой части таламуса, воспринимают сигналы от левой стороны
туловища. Ноцицептивные нейроны вентробазального ком-
плекса, так же как и нейроны других соматических модальнос-
тей (т.е. видов чувствительности), имеют упорядоченную то-
пическую организацию и локализованы в наружной области
ядра в виде скорлупы, окружающей центральную зону ядра.
Помимо пространственного расположения ноцицептивных
нейронов отдельно от нейронов других соматических модаль-
ностей в вентробазальном комплексе, существует и топичес-
кая упорядоченность среди нейронов «ноцицептивной зоны».
Так, нейроны дорсальной части ноцицептивной зоны получа-
ют контралатеральные входы от дорсальной поверхности туло-
вища, тогда как нейроны вентральной части активируются
при раздражении вентральной поверхности туловища. Многие
нейроны ноцицептивной зоны вентробазального комплекса
таламуса по своим свойствам относятся к специфическим но-
цицептивным нейронам, что позволяет считать вентробазаль-
ный комплекс частью системы идентификации и локализации
болевых стимулов.
Интраламинарные ядра таламуса: центральное медиальное
(ЦМ), центральное латеральное (ЦЛ) (см. рис. 6.1), парацент-
ральное, срединный центр и парафасцикулярный комплекс
363
относятся к неспецифическим ядрам. Клетки этих ядер яв-
ляются полимодальными и отвечают на многие сенсорные
стимулы (соматические, висцеральные, слуховые, зритель-
ные). Несмотря на то что интраламинарные ядра относятся
к неспецифическим структурам мозга, в них наряду с нейро-
нами ШДД регистрируются специфические ноцицептивные
нейроны, активируемые исключительно болевыми стимулами.
Интраламинарным ядрам таламуса отводится важная роль в
механизмах развития эмоционально-аффективных проявле-
ний протопатической (диффузной) боли. Об этом свидетель-
ствуют и клинические наблюдения. Разрушение интралами-
нарных ядер у людей, страдающих невыносимыми хроничес-
кими болями, приводит к купированию болей за счет устране-
ния тягостного эмоционального компонента, сопровождаю-
щего болевой синдром. При этом не происходит изменений
в оценке прикосновения, укола или температуры, которые
наблюдаются при разрушении вентробазального комплекса
таламуса.
Заднюю группу ядер таламуса (ЗГ), занимающую каудаль-
ную часть дорсального таламуса и включающую в себя подуш-
ку, латеральное заднее ядро, надколенчатое ядро и часть меди-
ального коленчатого тела, относят по своим функциональным
свойствам к ассоциативным ядрам таламуса. Этот комплекс
ядер получает спинальные афферентные проекции, восходя-
щие в вентролатеральных и дорсолатеральных канатиках
спинного мозга. Многие нейроны задней группы ядер активи-
руются болевыми стимулами с широких рецептивных полей,
изменяя характер ответа при увеличении интенсивности раз-
дражения. Особенностью нейронов задней группы ядер явля-
ется их способность одновременно реагировать на возбужде-
ние рецепторов и других сенсорных систем (зрительной и слу-
ховой). Такое свойство является общим для нейронов, входя-
щих в ассоциативные таламопариетальные и таламофронталь-
ные системы мозга, которые возникли в результате дифферен-
цировки неспецифической системы таламуса. Задняя группа
ядер, имеющая тесные связи с теменной ассоциативной
корой, принадлежит к таламопариетальной ассоциативной
системе мозга. Ассоциативные системы мозга имеют особое
значение в определении биологической значимости стимулов
и выработке адекватных ответных реакций на них. Поэтому
значение задней группы ядер в интеграции ноцицептивных
сигналов заключается в координации сложных комплексных
мотивационных, моторных и вегетативных защитных реакций,
возникающих в ответ на повреждающий раздражитель.
Медиодорсальному ядру (МД на рис. 6.1), входящему в та-
ламофронтальную ассоциативную систему мозга, отводится
важная роль в переработке и передаче сигналов во фронталь-
ную кору головного мозга. Нейроны медиодорсального ядра
364
изменяют характер своего ответа при различных ноцицептив-
ных раздражениях с достаточно широких рецептивных полей,
а его разрушение у людей ослабляет аффективный компонент
болевого синдрома.
6.2.1.6. Обработка ноцицептивной информации
в коре больших полушарий
Из таламических ядер ноцицептивные сигналы поступают в
кору больших полушарий, которая является высшим интегра-
тивным звеном, воспринимающим ноцицептивную информа-
цию. Наибольшее значение в анализе ноцицептивной информа-
ции принадлежит соматосенсорной коре (первая и вторая сома-
тосенсорные области — С1 и С2 на рис. 6.1) и фронтальной ас-
социативной области коры больших полушарий. В С1 ноцицеп-
тивные нейроны активируются с небольших рецептивных полей
контралатеральной стороны и располагаются в зоне представи-
тельства соответствующей части тела. Большинство из этих ней-
ронов имеют моносинаптическую связь со специфическими но-
цицептивными нейронами вентробазального комплекса таламу-
са. Однако наряду со специфическими ноцицептивными нейро-
нами в области С1 есть клетки широкого динамического диапа-
зона, активирующиеся с широких рецептивных полей и обла-
дающие конвергентными свойствами. Ноцицептивные нейроны
области С1, так же как и нейроны других соматических модаль-
ностей, структурно организованы в виде колонок, внутри кото-
рых все нейроны обладают сходными свойствами. Так же как и
ноцицептивные нейроны вентробазального комплекса таламуса,
ноцицептивные нейроны области С1 пространственно обособ-
лены от неноцицептивных нейронов и располагаются в более
глубоких слоях коры.
Вторая соматосенсорная область коры больших полушарий
(С2) также имеет нейроны, отвечающие на ноцицептивные
стимулы. В отличие от ноцицептивных нейронов области С1
большинство ноцицептивных нейронов области С2 обладают
конвергентными свойствами, активируются с широких, часто
неустойчивых рецептивных полей и не имеют соматотопичес-
кой организации. Эти отличия обусловлены особенностями
афферентных связей двух областей соматосенсорной коры.
Для области С1 характерно преобладание проекций, идущих
от нейронов вентробазального комплекса таламуса с их высо-
кой пространственной и модальной специфичностью.
Область С2 получает проекции от трех основных талами-
ческих центров: вентробазального комплекса, ЗГ и интрала-
минарных ядер.
Наличие различных афферентных проекций к областям
С1 и С2 определяет не только особенности нейрональной ор-
365
ганизации этих областей, но и их функциональную неравно-
значность в механизмах ноцицепции. При травматическом
повреждении постцентральной извилины коры больших по-
лушарий (область С1) у людей вместе с ухудшением тактиль-
ной, вибрационной, кинестетической и температурной чувст-
вительности наблюдается повышение болевых порогов, а в
некоторых случаях отмечается состояние гемианалгезии на
контралатеральной стороне. Удаление постцентральной изви-
лины у обезьян приводит к нарушению оценки тактильных и
болевых сигналов. Функциональное выключение или разру-
шение области С1 у кошек приводит к снижению кожной
чувствительности и повышению болевых порогов. Удаление
второй соматосенсорной области, наоборот, вызывает сниже-
ние порогов ответных болевых реакций на контралатераль-
ной стороне. При изолированном поражении области С2 у
людей развивается гиперпатия, при которой любые, даже
самые легкие, тактильные стимулы провоцируют развитие
нестерпимой боли.
Таким образом, приведенные факты свидетельствуют о
том, что область С1, участвующая в дискриминационном ана-
лизе всех видов соматической чувствительности, осуществляет
также оценку болевых сигналов, формируя ощущения, связан-
ные с первичной эпикритической болью (т.е. острой, четко
локализуемой болью).
Область С2 как структура, выделяющая биологически
опасные раздражители или ситуации, может претендовать на
роль коркового модулятора, осуществляющего контроль входа
болевых сигналов и координирующего ответные реакции орга-
низма при повреждающих стимулах. Это подтверждается тем,
что ноцицептивные афференты дорсальных и дорсолатераль-
ных канатиков, принимающих участие не столько в формиро-
вании ощущений, связанных с болью, сколько в активации
структур, контролирующих проведение болевых сигналов, в
С2 проецируются в большей степени, нежели в С1. Если при-
нять во внимание также, что область С2 имеет тесные морфо-
функциональные связи с ядрами задней группы таламуса, ко-
торые участвуют в координации сложных комплексных реак-
ций при болевых стимулах, то С2 может считаться конечным
корковым звеном этой быстрой восходящей системы, направ-
ленной на оценку потенциально опасных раздражителей и вы-
работку адекватных защитных реакций.
Фронтальные отделы коры больших полушарий получают
ноцицептивную информацию от трех таламических образова-
ний (вентробазального комплекса, интраламинарных ядер,
медиодорсального ядра) и участвуют в формировании слож-
ных эмоционально-аффективных проявлений боли и связан-
ных с ней психических переживаний. Наглядно об этом свиде-
тельствуют клинические наблюдения за пациентами с удален-
366
ной фронтальной корой или повреждениями путей, ведущих к
ней. Так, у людей, подвергшихся фронтальной лоботомии для
ослабления непереносимых болей, происходило подавление
эмоционального компонента болевого синдрома. Пациенты
отмечали, что перцептуальный компонент боли оставался
таким же, что и до операции, но он не сопровождался нега-
тивными переживаниями.
Основываясь на приведенных данных, всю систему ноци-
цептивной сигнализации можно разделить на три основных
потока, которые и обеспечивают формирование сложного
многокомпонентного болевого ощущения и быстрой ответной
реакции на повреждающий стимул [Решетняк В.К., 1985].
Первый поток специфической ноцицептивной сигнализа-
ции участвует в механизмах возникновения первичной, бы-
строй, четко локализованной боли. К этой системе проведе-
ния относятся высокопороговые специфические ноцицепторы
в различных органах и тканях, связанные с А-дельта и С-во-
локнами, а также специфические ноцицептивные нейроны,
локализованные на различных уровнях ЦНС и в первую оче-
редь в 1-й пластине дорсальных рогов спинного мозга, в ноци-
цептивной зоне вентробазального комплекса таламуса и в об-
ласти С1 коры больших полушарий. Таким образом, ноцицеп-
тивные сигналы, поступающие по этой системе, имеют точно
такую же морфофункциональную организацию, как и любые
другие афферентные системы, обслуживающие ту или иную
модальность (см. выше); это дает право считать, что эпикри-
тическая боль является сенсорной модальностью наравне с
тактильной, температурной и т.д.
Со вторым потоком проведения ноцицептивных сигналов
связывают формирование ощущений вторичной, плохо лока-
лизованной диффузной боли, имеющей выраженные эмоцио-
нально-аффективные и вегетативные проявления. Ноцицеп-
тивная информация в данном случае передается по медлен-
ным немиелинизированным С-афферентам в спинной мозг и
через спиноретикулоталамический и спиномезэнцефаличес-
кий тракты активирует супраспинальные лимбико-ретикуляр-
ные структуры мозга, включая фронтальные отделы коры
больших полушарий.
Третий поток проведения ноцицептивных сигналов на-
правлен на активацию структур антиноцицептивной системы
и формирование немедленной ответной реакции организма,
связанной с устранением действия повреждающего стимула.
Структурной основой данной системы проведения являются, с
одной стороны, задние столбы, вентробазальный комплекс и
задняя группа ядер таламуса, область С2 коры головного
мозга, а с другой стороны — афференты спиномезэнцефали-
ческого тракта, проецирующегося на антиноцицептивные
структуры ствола и среднего мозга.
367
Таким образом, восприятие боли обеспечивается сложноор-
ганизованной ноцицептивной системой, включающей в себя
особую группу периферических рецепторов и центральных
нейронов, расположенных во многих структурах централь-
ной нервной системы и реагирующих на повреждающее воз-
действие.
Иерархическая, многоуровневая организация ноцицептив-
ной системы соответствует нейропсихологическим представ-
лениям о динамической локализации мозговых функций и от-
вергает представления о «болевом центре» как конкретной
морфологической структуре, удаление которой способствова-
ло бы устранению болевого синдрома (см. рис. 6.1).
Данное утверждение также основывается на многочислен-
ных клинических наблюдениях, свидетельствующих о том, что
нейрохирургическое повреждение какой-либо из ноцицептив-
ных структур у больных, страдающих хроническими болевыми
синдромами, приносит только временное облегчение.
6.2.2. Антиноцицептивная система мозга
Как уже упоминалось, болевые ощущения наряду с полез-
ной, сигнальной функцией, предупреждающей организм об
опасности, вызывают и ряд побочных эффектов, таких как тя-
гостное переживание, ограничение подвижности, нарушение
микроциркуляции, снижение иммунной защиты и др. Чрез-
мерная боль может стать причиной шока и смерти, поэтому
организм должен иметь системы, обеспечивающие контроль
за активностью структур, участвующих в восприятии, проведе-
нии и анализе «болевых» сигналов.
Эндогенная система подавления боли получила название
аптиноцицептивпой системы и объединяет структуры спинно-
го и головного мозга [Мелзак Р., 1981; Лиманский Ю.П., 1989;
Игнатов Ю.Д. и др., 1994]. Электрическая стимуляция этих
образований вызывает у человека и животных стойкое сниже-
ние болевой чувствительности. Обезболивающий эффект был
получен при стимуляции центрального серого вещества
(ЦСВ), ядер шва и особенно большого ядра шва, парагиганто-
клеточного и гигантоклеточного ядер ретикулярной форма-
ции, синего пятна, парабрахиальных ядер, черной субстанции,
красного ядра, хвостатого ядра, септальной области, ядер по-
крышки, гипоталамуса, миндалины, дорсальных столбов
спинного мозга, специфических и неспецифических ядер та-
ламуса, внутренней капсулы, фронтальной, моторной и сома-
тосенсорной коры больших полушарий, мозжечка.
Среди перечисленных структур существует тесная взаимо-
связь. Нейроны ЦСВ имеют двусторонние контакты с параги-
368
гантоклеточным и гигантоклеточными ядрами ретикулярной
формации, с ядрами шва, гипоталамуса, синего пятна. В свою
очередь ЦСВ получает нисходящие влияния от фронтальной и
соматосенсорной коры головного мозга, от неспецифических
ядер таламуса.
Аксоны норадренергических нейронов синего пятна дости-
гают интраламинарных ядер таламуса, ЦСВ, входят в гипота-
ламус и миндалевидный комплекс. В нисходящем направле-
нии аксоны нейронов синего пятна направляются к ядрам шва
и в спинной мозг. Афферентные связи синего пятна представ-
лены проекциями из медиальной области фронтальной коры
головного мозга, интраламинарных ядер таламуса, миндале-
видного комплекса, преоптической области, ЦСВ, ядер шва,
латеральной ретикулярной формации продолговатого мозга,
парабрахиальных ядер.
Черная субстанция при помощи дофаминергических путей
связана с фронтальной и сенсомоторной корой головного
мозга, с медиальными, передними и интраламинарными ядра-
ми таламуса, с миндалиной, ЦСВ, синим пятном, ядрами шва
и спинальными нейронами.
Ядра шва образуют мощные восходящие и нисходящие
пути серотонинергической системы, посредством которых они
взаимодействуют с ЦСВ, интраламинарными ядрами таламу-
са, корой больших полушарий, гипоталамусом, гиппокампом,
перегородкой, черной субстанцией, с нейронами дорсальных
рогов спинного мозга.
Обезболивающее действие при активации вышеперечис-
ленных структур мозга достигается посредством торможения
ноцицептивных нейронов на различных уровнях ЦНС. Вместе
с тем наибольший тормозной эффект антиноцицептивных
структур испытывают на себе нейроны дорсальных рогов
спинного мозга (схема 6.1).
Прямые нисходящие проекции к нейронам дорсального
рога имеют кора головного мозга, ретикулярная формация
ствола мозга, ядра шва и синее пятно. Нисходящие волокна от
супраспинальных антиноцицептивных структур проходят в
дорсолатеральных и вентролатеральных канатиках и заканчи-
ваются в различных пластинах дорсальных рогов спинного
мозга.
Показано, что электрическое раздражение ядер дорсаль-
ных канатиков, ядер шва, структур латеральной ретикулярной
формации, ЦСВ, синего пятна, претектального ядра, гипота-
ламуса, таламических ядер и коры головного мозга вызывает
снижение активности ноцицептивных нейронов дорсальных
рогов спинного мозга.
Подавляющее действие, оказываемое системой нисходя-
щих трактов на передачу ноцицептивной афферентации в дор-
сальных рогах спинного мозга, реализуется как за счет прямо-
24—1385
369
Схема 6.1. Антиноцицептивная система мозга
ЛТ — латеральный таламус; МТ — медиальный таламус; ЗГ — задняя
группа ядер таламуса; ЦСВ — центральное серое вещество; ЯШ — ядра
шва; (+) — возбуждающие влияния; (-) — тормозные влияния.
го пре- и постсинаптического торможения ноцицептивных
нейронов, так и путем активации тормозных интернейронов
желатинозной субстанции.
В механизмах развития аналгезии при активации антино-
цицептивных структур наибольшее значение придается серо-
тонинергической, норадренергической и опиоидергической
системам мозга.
Установлено, что серотонинергические нисходящие волок-
на из ядер шва оканчиваются на нейронах спиноталамичес-
кого тракта. Избирательное разрушение нисходящих серо-
тонинергических путей у крыс интратекальным введением
5,6-дигидрокситриптамина усиливает болевую реакцию. И, на-
оборот, интратекальное введение серотонина вызывает у жи-
вотных не только дозозависимую аналгезию, но и угнетение
разрядов нейронов дорсального рога, вызванных болевой тер-
мической стимуляцией. Анальгетический эффект развивается
также при введении в большое ядро шва либо самого серото-
нина, либо ингибиторов его обратного захвата.
370
Участие норадреналина в механизмах обезболивания пока-
зано при его введении животным в синее, пятно, ЦСВ или в
дорсальные рога спинного мозга. От нейронов синего пятна
начинается нисходящий норадренергический путь, разруше-
ние которого значительно ослабляет анальгетические эф-
фекты.
Опиоидергическая система образована нейронами, сома и
отростки которых содержат опиоидные пептиды — бета-эн-
дорфин, мет-энкефалин, лей-энкефалин, динорфин. Высокая
плотность опиоидергических нейронов обнаружена в ядрах та-
ламуса, гипоталамуса, ЦСВ, черной субстанции, ядрах по-
крышки, ядрах шва, желатинозной субстанции дорсальных
рогов спинного мозга. В настоящее время выделяют три ос-
новных подтипа опиоидных рецепторов: мю-, дельта- и каппа-
опиоидные рецепторы, активация которых приводит к стой-
кой аналгезии.
Механизм болеутоляющего действия опиоидных рецепто-
ров связан с пре- и постсинаптическим торможением ноци-
цептивных нейронов на различных уровнях ЦНС [Dicken-
son А.Н., 1994].
В настоящее время считается доказанным, что анальгети-
ческое действие опиатов и опиоидов связано с активацией
как спинальных, так и супраспинальных механизмов. Эпиду-
ральное или интратекальное введение опиатов или опиоидов
у человека и животных приводит к выраженному торможе-
нию ноцицептивных нейронов дорсального рога и к длитель-
ной аналгезии в участках тела, соответствующих зонам вве-
дения.
Использование селективных лигандов позволило устано-
вить, что более 90 % опиоидных рецепторов в дорсальных
рогах спинного мозга представлены мю- и дельта-рецептора-
ми, которые расположены преимущественно на пресинапти-
ческих терминалях С-афферентов, идущих с периферии. В ре-
зультате пресинаптического торможения уменьшается выделе-
ние медиаторов (глутамата и субстанции Р) из центральных
терминалей ноцицептивных афферентов и снижается эффек-
тивность синаптической передачи болевого сигнала.
Постсинаптическое торможение, обусловленное действием
лигандов на опиоидные рецепторы, возникает в результате ги-
перполяризации дендритов или сомы ноцицептивных ней-
ронов.
Возможность развития супраспинальной аналгезии была
впервые продемонстрирована при введении морфина в желу-
дочки мозга. В последующем были представлены убедитель-
ные данные, свидетельствующие о том, что микроинъекции
морфина в различные антиноцицептивные структуры моз-
га — ядра шва, ЦСВ, синее пятно, черную субстанцию, сома-
тосенсорную кору больших полушарий, активируют эти
24*
371
структуры и вызывают у животных аналгезию и торможение
активности спинальных ноцицептивных нейронов. Механизм
супраспинальной аналгезии, реализуемый через опиоидные
рецепторы, до конца не изучен, но существует ряд доказа-
тельств, позволяющих утверждать, что активация опиоидных
рецепторов приводит к усилению нисходящего торможения,
осуществляемого моноаминергическими антиноцицептивны-
ми системами.
Таким образом, антиноцицептивная система мозга представ-
ляет собой в морфологическом и нейрохимическом отноше-
нии гетерогенное образование, структуры которого располо-
жены на всех уровнях ЦНС, начиная от желатинозной суб-
станции спинного мозга и включая кору больших полу-
шарий.
Однако анальгетические структуры функционируют не
изолированно друг от друга, а взаимодействуя между собой,
что обеспечивает избирательное включение нейрохимических
механизмов в зависимости от вида и интенсивности болевых
раздражителей. Активируясь при ноцицептивном воздействии,
структуры антиноцицептивной системы по принципу обрат-
ной связи угнетают передачу болевых сигналов. Постоянно
взаимодействуя, восходящая ноцицептивная и антиноцицеп-
тивная системы мозга осуществляют регуляцию болевой чув-
ствительности и участвуют в адаптации организма к изменяю-
щимся условиям внешней среды.
6.3. Патофизиология боли
В зависимости от патогенеза болевые синдромы подразде-
ляют следующим образом:
• соматогенные болевые синдромы;
• нейрогенные болевые синдромы;
• психогенные болевые синдромы.
Болевые синдромы, возникающие вследствие активации
ноцицептивных рецепторов при травме, воспалении, ишемии,
растяжении тканей, относят к соматогенным болевым синдро-
мам. Клинически среди них выделяют посттравматический й
послеоперационный болевые синдромы, боль при воспалении
суставов, миофасциальные болевые синдромы, боль у онколо-
гических больных, боль при заболеваниях внутренних органов
и многие другие.
Нейрогенные болевые синдромы возникают при повреждении
структур периферической или центральной нервной систем,
участвующих в проведении ноцицептивных сигналов. Приме-
рами таких болевых синдромов являются невралгия тройнич-
372
ного нерва, фантомно-болевой синдром, таламические боли,
каузалгия.
Развитие психогенных болевых синдромов связывают с пси-
хологическими факторами, которые инициируют боль при от-
сутствии каких-либо серьезных соматических расстройств.
Часто боли психологической природы возникают вследствие
мышечного напряжения, которое провоцируется эмоциональ-
ными конфликтами или психосоциальными проблемами.
Психогенная боль может являться составной частью истери-
ческой реакции или возникать как бред или галлюцинация
при шизофрении и исчезать при адекватном лечении основно-
го заболевания. К психогенным болям относят также боли,
связанные с депрессией, которые не предшествуют ей и не
имеют какой-либо другой причины.
6.3.1. Соматогенные болевые синдромы
Клинически соматогенные болевые синдромы проявляют-
ся постоянной болезненностью и/или повышением болевой
чувствительности в зоне повреждения или воспаления. Паци-
енты, как правило, легко локализуют такую боль, четко опре-
деляют ее интенсивность и характер. Со временем зона повы-
шенной болевой чувствительности может расширяться и вы-
ходить за пределы поврежденных тканей. Участки с повышен-
ной болевой чувствительностью к повреждающим стимулам
называют зонами гипералгезии.
Выделяют первичную и вторичную гипералгезию. Первич-
ная гипералгезия охватывает поврежденные ткани, вторичная
гипералгезия локализуется вне зоны повреждения. Области
первичной кожной гипералгезии характеризуются снижением
болевых порогов и болевой толерантности к повреждающим
механическим и термическим стимулам. Зоны вторичной ги-
пералгезии имеют нормальный болевой порог и сниженную
болевую толерантность только к механическим раздражи-
телям.
6.З.1.1. Механизмы возникновения первичной гипералгезии
Патофизиологической основой первичной гипералгезии
является сенситизация (повышение чувствительности) ноци-
цепторов А-дельта и С-афферентов к действию повреждаю-
щих стимулов [Wall P.D., Melzack R., 1994]. Электрофизиоло-
гически сенситизация ноцицепторов проявляется снижением
порога их активации, увеличением частоты и длительности
разрядов в нервных волокнах, что приводит к усилению аффе-
рентного ноцицептивного потока.
373
Сенситизация ноцицепторов происходит в результате вы-
деления в зоне повреждения медиаторов воспаления, включа-
ющих брадикинин, метаболиты арахидоновой кислоты (про-
стагландины и лейкотриены), биогенные амины, пурины и
ряд других веществ, которые, взаимодействуя с соответствую-
щими рецепторами на терминалях ноцицептивных афферен-
тов, повышают чувствительность последних к механическим и
термическим стимулам.
Большое значение в инициации механизмов, обеспечиваю-
щих сенситизацию ноцицепторов, отводится брадикинину,
который может оказывать как прямое, так и непрямое дейст-
вие на чувствительные нервные окончания.
Прямой возбуждающий эффект брадикинина на чувстви-
тельные нервные окончания опосредуется В2-рецепторами и
связан с активацией мембранной фосфолипазы С.
Непрямое возбуждающее действие брадикинина на окон-
чания нервных афферентов обусловлено его воздействием на
различные тканевые элементы (эндотелиальные клетки, фиб-
робласты, тучные клетки, макрофаги и нейтрофилы) и стиму-
лированием образования в них медиаторов воспаления (на-
пример, простагландинов), которые, взаимодействуя с соот-
ветствующими рецепторами на нервных окончаниях, активи-
руют мембранную аденилатциклазу. В свою очередь стимуля-
ция аденилатциклазы и фосфолипазы С через вторичные мес-
сенджеры активирует протеинкиназы, фосфорилирующие
белки ионных каналов. Результатом фосфорилирования бел-
ков ионных каналов является изменение проницаемости мем-
браны для ионов кальция, калия и др., что приводит к по-
вышению возбудимости нервных окончаний и их способности
генерировать нервные импульсы.
Выделение медиаторов воспаления из различных клеток .
происходит не только под влиянием брадикинина; но и в ре-
зультате прямого повреждения тканей или действия нейро-
пептидов С-афферентов, таких как субстанция Р, нейроки-
нин А или кальцитонин-ген-родственный пептид. Эти ней-
ропептиды обладают провоспалительным эффектом, вызывая
расширение сосудов и увеличение их проницаемости. Они
способствуют высвобождению из тучных клеток и лейкоци-
тов простагландина Е2, цитокинов и биогенных аминов, ко-
торые, воздействуя на мембрану нервных окончаний, как
указывалось выше, изменяют возбудимость нервных аффе-
рентов.
Сенситизации ноцицепторов и развитию первичной гипер-
алгезии способствует также воздействие симпатической нерв-
ной системы.
Установлено, что при активации постганглионарных сим-
патических волокон происходит повышение чувствительности
терминалей высокопороговых тонких афферентов, которое ре-
374
ализуется двумя путями: во-первых, за счет повышения сосу-
дистой проницаемости в зоне повреждения и увеличения кон-
центрации медиаторов воспаления (непрямой путь) и, во-вто-
рых, за счет прямого воздействия медиаторов симпатической
нервной системы — норадреналина на альфа2-адренорецепто-
ры, расположенные на мембране ноцицепторов.
6.3.1.2. Механизмы развития вторичной гипералгезии
Клинически область вторичной гипералгезии характеризу-
ется повышением болевой чувствительности к интенсивным
механическим стимулам вне зоны повреждения и может рас-
полагаться на достаточном удалении от места повреждения, в
том числе и на противоположной стороне тела. Этот феномен,
на наш взгляд, может быть объяснен только механизмами
центральной нейропластичности, приводящими к стойкой ги-
первозбудимости ноцицептивных нейронов. Подтверждением
этому служат клинико-экспериментальные данные, свидетель-
ствующие о том, что зона вторичной гипералгезии сохраняет-
ся при введении местных анестетиков в область повреждения
и исчезает в случае блокады активности нейронов дорсального
рога. В электрофизиологических исследованиях было проде-
монстрировано повышение возбудимости и реактивности ней-
ронов спиноталамического тракта к механическим раздраже-
ниям их рецептивных полей, расположенных в зоне вторич-
ной гипералгезии. Сенситизированные нейроны в ответ на
предъявляемые раздражения не только генерировали разряды
с увеличенной частотой, но и сохраняли повышенную актив-
ность продолжительное время.
Такая сенситизация нейронов дорсальных рогов может
быть вызвана различными типами повреждений: термически-
ми, химическими, механическими, возникающими вследствие
гипоксии, острого воспаления или электрической стимуляции
С-афферентов.
В настоящее время большое значение в механизмах сенси-
тизации ноцицептивных нейронов дорсальных рогов спинного
мозга придается возбуждающим аминокислотам и нейропеп-
тидам.
Иммуногистохимическими методами было установлено,
что синаптические терминали многих тонких высокопорого-
вых афферентов содержат в качестве нейромедиатора глута-
мат, аспартат и ряд нейропептидов, таких как субстанция Р,
нейрокинин А, кальцитонин-ген-родственный пептид и др.,
которые высвобождаются из пресинаптических терминалей
под действием ноцицептивных импульсов.
Выделение глутамата из пресинаптических терминалей
происходит при любом ноцицептивном воздействии — корот-
375
ком (укол) или длительном (повреждение тканей или при час-
тотной электростимуляции — более 0,3 Гц). Считается, что ре-
ализация физиологических болевых реакций (например, за-
щитный рефлекс отдергивания) при выделении глутамата
опосредуется через AM РА-рецепторы (alpha-amino-3-hydroxy-
5-methyl-4-isoxazole-propionic acid), в то время как NMDA-ре-
цепторы (N-methyl-D-aspartate) обеспечивают длительную, в
том числе и патологическую гиперактивность ноцицептивных
нейронов.
Активирующее действие глутамата на ноцицептивные ней-
роны потенцируется субстанцией Р, которая как медиатор со-
существует в более 90 % терминалей высокопороговых сенсор-
ных волокон, содержащих глутамат. Субстанция Р, как и дру-
гие нейрокинины, взаимодействуя с N К-1-рецепторами
(neurokinin-1), не только повышает концентрацию внутрикле-
точного Са2+ посредством его мобилизации из внутриклеточ-
ных депо, но и усиливает активность NMDA-рецепторов.
В последнее время важное значение в механизмах сенсити-
зации ноцицептивных нейронов придается оксиду азота (NO),
который в мозге выполняет роль нетипичного внесинаптичес-
кого медиатора. NO образуется в нейронах, содержащих фер-
мент NO-синтетазу из L-аргинина. NO выделяется из клеток
при возбуждении NMDA-рецепторов и взаимодействует с пре-
синаптическими терминалями С-афферентов, усиливая вы-
брос из них глутамата и нейрокининов.
Таким образом, индуцированное ноцицептивной стимуля-
цией высвобождение глутамата и нейропептидов из централь-
ных терминалей С-афферентов вызывает стойкие изменения
возбудимости ноцицептивных нейронов, усиление их спон-
танной активности, увеличение длительности послеразрядов и
расширение рецептивных полей.
Необходимо подчеркнуть, что возникшая вследствие по-
вреждения тканей сенситизация ноцицептивных нейронов
может несколько часов или дней сохраняться и после прекра-
щения поступления ноцицептивных импульсов с периферии.
Иными словами, если уже произошла гиперактивация ноци-
цептивных нейронов, то она не нуждается в дополнительной
«подпитке» импульсами из места повреждения. Долговремен-
ное повышение возбудимости ноцицептивных нейронов свя-
зывают с активацией их генетического аппарата — экспрес-
сией ранних, немедленно реагирующих генов (протоонкоге-
нов), таких как c-fos, c-jun, junB и др. В частности, продемон-
стрирована положительная корреляция между количеством
fos-позитивных нейронов и степенью боли. В механизмах ак-
тивации протоонкогенов важная роль отводится Са2+. При по-
вышении концентрации Са2+ в цитозоле вследствие усиленно-
го его входа через регулируемые NMDA-рецепторами Са-кана-
лы происходит экспрессия c-fos, c-jun, белковые продукты ко-
376
торых участвуют в регуляции долговременной возбудимости
мембраны клетки.
Помимо сенситизации ноцицептивных нейронов дорсаль-
ного рога, повреждение тканей вызывает также повышение
возбудимости и реактивности ноцицептивных нейронов и в
вышележащих центрах, включая ядра таламуса и соматосен-
сорную кору больших полушарий. Таким образом, перифери-
ческое повреждение запускает каскад процессов, затрагиваю-
щих всю ноцицептивную систему от тканевых рецепторов до
корковых нейронов. В целом патогенез соматогенных болевых
синдромов содержит следующие основные звенья:
• раздражение ноцицепторов при повреждении тканей;
• выделение алгогенов и сенситизация ноцицепторов в об-
ласти повреждения;
• усиление ноцицептивного афферентного потока с пери-
ферии;
• сенситизация ноцицептивных нейронов на различных
уровнях ЦНС
При соматогенных болевых синдромах патогенетически
обоснованным считается применение следующих средств:
• подавляющих синтез медиаторов воспаления;
• ограничивающих поступление ноцицептивной импуль-
сации из зоны повреждения в ЦНС;
• средств, активирующих структуры антиноцицептивной
системы.
Для подавления синтеза алгогенов, снижения воспали-
тельных реакций и уменьшения тем самым сенситизации но-
цицепторов используются нестероидные и/или стероидные
противовоспалительные препараты. Ограничение входа но-
цицептивной импульсации в ЦНС достигается при помощи
различного рода блокад местными анестетиками, которые не
только могут предотвратить сенситизацию ноцицептивных
нейронов, но и способствовать нормализации микроцирку-
ляции в зоне повреждения, улучшая восстановление повреж-
денных тканей. Для активации структур антиноцицептивной
системы, осуществляющих контроль за проведением ноци-
цептивной импульсации в ЦНС, может быть использован
целый спектр (в зависимости от клинических показаний) ме-
дикаментозных (наркотические и ненаркотические анальге-
тики, бензодиазепины, агонисты альфа-2-адренорецепторов
и др.) и немедикаментозных (чрескожная электронейрости-
муляция, рефлексотерапия, физиотерапия) средств, снижаю-
щих болевую чувствительность и негативное эмоциональное
переживание.
377
6.3.2. Патофизиология нейрогенных
болевых синдромов
6.3.2.1. Этиология нейрогенных болевых синдромов
Вызванные повреждением периферических нервов или
структур ЦНС нейрогенные болевые синдромы представляют
собой один из клинических парадоксов. Действительно, нару-
шение целости нерва должно приводить к снижению сенсор-
ных ощущений в иннервируемой им области. Однако пациен-
ты с полной денервацией конечности, например при авульсии
плечевого сплетения, часто испытывают в парализованной
руке мучительные боли.
Считается, что нейрогенные болевые синдромы возникают
при повреждении структур, связанных с проведением ноцицеп-
тивных сигналов [Кукушкин М.Л. и др., 1994; Bonica J.J., 1990;
Coderre T.J. et al., 1993]. Так, у пациентов после повреждения пе-
риферических нервов в области постоянной болезненности, по-
мимо парестезии и дизестезии, отмечается повышение порогов
на укол и ноцицептивный электрический стимул. У больных си-
рингомиелией выраженный болевой синдром возникает при
распространении патологического процесса на дорсальные рога
спинного мозга, при этом снижается температурная и болевая
чувствительность. У больных с рассеянным склерозом, страдаю-
щих также приступами болевых пароксизмов, склеротические
бляшки обнаружены в афферентах спиноталамического тракта.
Изолированное поражение вентролатеральных квадрантов
спинного мозга наряду с возникновением спонтанных болей и
дизестезии вызывает снижение болевой и температурной чувст-
вительности. При клиническом обследовании пациентов с тала-
мическими болями, возникающими после цереброваскулярных
нарушений, также отмечается снижение температурной и боле-
вой чувствительности. При этом очаги повреждений, выявлен-
ные методом компьютерной томографии, соответствуют местам
прохождения афферентов соматической чувствительности в
стволе мозга, среднем мозге и таламусе. Спонтанные боли воз-
никают у людей при повреждении соматосенсорной коры, явля-
ющейся конечным корковым пунктом восходящей ноцицептив-
ной системы. Все это свидетельствует о том, что нейрогенный
болевой синдром может возникнуть независимо от места по-
вреждения больпроводящих путей.
6.3.2.2. Клинические проявления нейрогенных
болевых синдромов
Для нейрогенного болевого синдрома характерна следую-
щая клиническая симптоматика: постоянная, спонтанная или
пароксизмальная боль, сенсорный дефицит в зоне болезнен-
378
ности, аллодиния (появление болезненного ощущения при
легком неповреждающем воздействии), гипералгезия и гипер-
патия. Полиморфизм болевых ощущений у разных пациентов
обусловлен характером, степенью и местом повреждения. При
неполном, частичном повреждении ноцицептивных афферен-
тов чаще возникает острая периодическая пароксизмальная
боль, подобная удару электрического тока и длящаяся всего
несколько секунд. В случае полной денервации боль чаще
всего имеет постоянный характер.
Одной из типичных черт нейрогенных болевых синдромов
является аллодиния — болевое ощущение, возникающее при сла-
бом механическом раздражении определенных кожных участ-
ков. В механизме возникновения аллодинии большое значение
придается сенситизации нейронов широкого динамического
диапазона, которые одновременно получают афферентные сиг-
налы от низкопороговых А-бета волокон и высокопороговых С-
волокон. В случае повышения возбудимости ШДД-нейронов
они генерируют усиленный разряд даже в ответ на сигнал, при-
ходящий по низкопороговым А-бета-волокнам.
6.3.2.3. Периферические механизмы нейрогенной боли
Механизмы возникновения нейрогенных болевых синдро-
мов в корне отличаются от болевых синдромов, вызванных
повреждением соматических тканей или внутренних органов.
Развитие нейрогенных болевых синдромов в настоящее
время связывают с морфофункциональными изменениями как
в периферическом травмированном нерве, так и в централь-
ной нервной системе.
При повреждении нерва, как известно, возникают атрофия
и гибель нервных волокон, причем преимущественно гибнут
немиелинизированные С-афференты. Вслед за дегенератив-
ными изменениями начинается регенерация нервных воло-
кон, которая сопровождается образованием невром. Структура
нерва становится неоднородной, что является причиной нару-
шения проведения возбуждения по нерву.
Клинические и экспериментальные работы свидетельству-
ют о наличии в поврежденном нерве эктопической активнос-
ти. Считается, что спонтанные эктопические разряды являют-
ся основой для парестезий и болей у пациентов с поврежден-
ными нервами. Такая электрическая активность зарегистриро-
вана как в невроме, так и в самом нервном волокне. Источни-
ком эктопической активности являются зоны демиелинизации
и регенерации нерва, невромы, а также нервные клетки дор-
сальных ганглиев, связанные с поврежденными аксонами.
Использование специальных методов при проведении
электрофизиологических исследований позволило установить,
379
что генерация невромой спонтанной эктопической активнос-
ти вызвана нестабильностью мембранного потенциала, причи-
ной которой является увеличение на мембране количества на-
триевых каналов.
Эктопическая активность существенным образом отлича-
ется от нормальных разрядов. Если в нормальных условиях
длительность разряда ограничена продолжительностью стиму-
ла, то эктопический разряд имеет не только увеличенную амп-
литуду сигнала, но и большую продолжительность. В результа-
те этот разряд, возникший в одном волокне, может активиро-
вать другие волокна. Подобное перекрестное возбуждение во-
локон, или эфаптическая передача сигнала, наблюдается толь-
ко в условиях патологии и является основой для дизэстезии и
гиперпатии.
Повышение фоновой активности поврежденных нервов в
значительной мере связано с увеличением чувствительности
нервных волокон к механическим и химическим стимулам.
Повышение механо- и хемочувствительности в нервных во-
локнах увеличивает диапазон раздражителей, способных вы-
звать генерацию потенциалов действия.
Изменение возбудимости нервных волокон при поврежде-
нии происходит в течение первых 10 ч и во многом зависит от
аксонального транспорта. Показано, что блокада аксотока за-
держивает повышение механочувствительности в нервных во-
локнах.
6.3.2.4. Центральные механизмы нейрогенной боли
Болевой синдром, вызванный повреждением нервов, наря-
ду с появлением анормальной активности в нервных волокнах
сопровождается также повышением возбудимости и реактив-
ности нейронов дорсальных рогов спинного мозга и вышеле-
жащих структур соматосенсорной системы. Центральная сен-
ситизация при повреждении периферических нервов или дор-
сальных корешков характеризуется увеличением спонтанной
импульсной активности нейронов дорсального рога и появле-
нием у них вспышек высокочастотных разрядов, расширением
рецептивных полей, повышением реактивности нейронов на
периферические раздражения и удлинением времени после-
разрядов.
Одновременно с увеличением нейрональной активности на
уровне дорсальных рогов спинного мозга у животных с экспе-
риментальными моделями нейрогенных болевых синдромов
регистрируется усиление активности нейронов в таламических
ядрах — вентробазальном и парафасцикулярном комплексах, в
соматосенсорной коре больших полушарий. Однако наблю-
даемые изменения активности нейронов в структурах ноци-
380
цептивной системы при нейрогенных болевых синдромах
имеют ряд принципиальных отличий по сравнению с механиз-
мами, приводящими к сенситизации ноцицептивных нейро-
нов у пациентов с соматогенными болевыми синдромами.
Структурной основой нейрогенных болевых синдромов яв-
ляется «агрегат» (группа) взаимодействующих сенситизиро-
ванных (гиперактивных) нейронов с нарушенными тормозны-
ми механизмами и повышенной возбудимостью [Крыжанов-
ский Г.Н., 1980, 1997]. Такие агрегаты способны развивать
длительную самоподдерживающуюся патологическую актив-
ность, для которой необязательна афферентная стимуляция с
периферии. Агрегаты нейронов с патологической активностью
могут возникать вследствие деафферентации структур, осу-
ществляющих проведение и обработку ноцицептивных сигна-
лов на разных уровнях спинного и головного мозга. Напри-
мер, деафферентация спинного мозга у животных путем пере-
резки дорсальных корешков приводит к появлению высоко-
частотных пачечных разрядов в нейронах дорсальных рогов.
Схожая «эпилептиформная» активность в спинном мозге
также была зарегистрирована и у людей, страдающих болевым
синдромом, вызванным травматическим повреждением спин-
номозговых корешков. Формирование таких агрегатов осу-
ществляется синаптическими и несинаптическими механиз-
мами. Одним из условий образования агрегатов при поврежде-
нии нейрональных структур является возникновение устойчи-
вой деполяризации нейронов. Ее возникновению способст-
вуют:
• выделение возбуждающих аминокислот, нейрокининов
и оксида азота из центральных терминалей А-дельта- и
С-волокон;
• дегенерация центральных терминалей А-дельта- и С-во-
локон и транссинаптическая гибель нейронов дорсаль-
ного рога с последующим их замещением глиальными
клетками;
• дефицит опиоидных рецепторов и их лигандов, контро-
лирующих возбуждение ноцицептивных клеток;
• повышение чувствительности тахикининовых рецепто-
ров к своим лигандам — субстанции Р и нейрокинину А.
Воздействия, вызывающие дополнительное растормажива-
ние и гиперактивацию нейронов, также способствуют форми-
рованию и усилению деятельности агрегата гиперактивных
нейронов. Косвенно на это указывают экспериментальные ис-
следования и клинические наблюдения.
Нанесение животным дополнительных болевых раздраже-
ний перед перерезкой нерва не только значительно ускоряет
развитие у них болевого синдрома, но и усиливает его течение.
В противоположность этому ограничение поступления ноци-
381
цептивной импульсации (с помощью местных анестетиков) во
время перерезки нерва задерживает развитие болевого синдро-
ма, а в ряде случаев даже предотвращает его появление.
Важное значение в механизмах образования агрегатов ги-
перактивных нейронов в структурах ЦНС отводится подавле-
нию тормозных реакций, которые опосредуются глицином и
гамма-аминомасляной кислотой (ГАМК). Подтверждением
этому служит возникновение болевого синдрома у крыс при
аппликации на дорсальную поверхность спинного мозга
стрихнина — препарата, блокирующего постсинаптическое
глициновое торможение или веществ, нарушающих ГАМКер-
гическое торможение (например, бикукулина или пикроток-
сина). Внутриспинальное введение стрихнина усиливает раз-
витие болевого синдрома при повреждении седалищного
нерва. Дефицит спинального глицинергического и ГАМКер-
гического торможения возникает и при локальной ишемии
спинного мозга, приводящей к гипервозбудимости нейронов и
развитию выраженной аллодинии. В условиях недостаточнос-
ти тормозных механизмов и повышенной возбудимости ней-
ронов облегчаются синаптические межнейронные взаимодей-
ствия, происходит активация «молчащих» неактивных синап-
сов, что способствует объединению близлежащих сенситизи-
рованных нейронов в единый агрегат.
При формировании нейрогенных болевых синдромов глу-
бокие нейропластические преобразования затрагивают не
только первичное ноцицептивное реле, но и высшие структу-
ры системы болевой чувствительности. Их деятельность изме-
няется настолько, что электростимуляция центрального серо-
го вещества, которая эффективно используется для купирова-
ния болей у онкологических больных, не приносит облегчения
пациентам с нейрогенными болевыми синдромами.
Таким образом, в основе развития нейрогенных болевых
синдромов лежат структурно-функциональные изменения, за-
трагивающие периферические и центральные отделы системы
болевой чувствительности. Под влиянием повреждающих фак-
торов возникает дефицит тормозных реакций, приводящий к
развитию в первичном ноцицептивном реле агрегатов гипе-
рактивных нейронов, продуцирующих мощный афферентный
поток импульсов, который повышает возбудимость супраспи-
нальных ноцицептивных центров, дезинтегрирует их нормаль-
ную работу и вовлекает их в патологические реакции. Проис-
ходящие при этом пластические изменения объединяют ги-
перактивированные ноцицептивные структуры в новую пато-
динамическую организацию — патологическую алгическую
систему, результатом деятельности которой является болевой
синдром.
В целом приведенные клинико-экспериментальные дан-
ные, касающиеся механизмов развития нейрогенных болевых
382
синдромов, позволяют выделить следующие основные этапы
патогенеза нейрогенных болевых синдромов:
• образование невром и участков демиелинизации в по-
врежденном нерве, являющихся периферическими оча-
гами патологического электрогенеза;
• усиление механо- и хемочувствительности в нервных во-
локнах;
• появление перекрестного возбуждения в нейронах дор-
сальных ганглиев;
• формирование агрегатов гиперактивных нейронов с
самоподдерживающейся активностью в ноцицептивных
структурах ЦНС;
• нарушения в работе структур, регулирующих болевую
чувствительность.
Учитывая особенности патогенеза нейрогенных болевых
синдромов, оправданным при лечении данной патологии
будет использование средств, подавляющих патологическую
активность периферических очагов возбуждения и агрегатов
гиперактивных нейронов. Приоритетными в настоящее время
считаются следующие лекарственные средства: антиконвуль-
санты и препараты, усиливающие тормозные реакции в
ЦНС, — бензодиазепины, агонисты ГАМК-а- и ГАМК-Ь-ре-
цепторов, блокаторы кальциевых каналов, антагонисты воз-
буждающих аминокислот, периферические и центральные
блокаторы Na-каналов.
6.4. Болевые синдромы в клинической практике
В принятой в настоящее время международной клиничес-
кой классификации болевых синдромов [Bonica J.J., 1990] от-
ражены следующие проявления боли: локализация боли (голо-
ва, рука, шея и т.д.), система, подвергшаяся повреждению
(нервная, дыхательная, сердечно-сосудистая, мышечно-ске-
летная, кожная и т.д.), характер болевых ощущений (постоян-
ная, возвратная, пароксизмальная и др.), продолжительность
боли (короткая — менее 1 мес, средняя — до 6 мес, длитель-
ная — более 6 мес), интенсивность боли (слабая, средняя,
сильная) и причина боли (травма, инфекционное и неинфекци-
онное воспаление, метаболические нарушения, психоэмоцио-
нальные расстройства и т.д.). В отдельную группу выделены
наиболее известные болевые синдромы: боль при нейропатиях,
фантомные боли, каузалгия, таламическая боль, ревматическая
полимиалгия и др.
Данная классификация позволяет достаточно полно обри-
совать клиническую картину боли и сосредоточить внимание
читателей на этиологии болевого синдрома. Однако схожие по
383
патогенезу болевые синдромы часто оказываются в различных
группах и, наоборот, совершенно различные по патогенезу бо-
левые синдромы представлены в одной группе. Например, в
классификации головных и лицевых болей выделены следую-
щие группы:
• невралгии головы и лица (т.е. нейрогенные боли, возни-
кающие при повреждении соответствующих нервных
структур);
• черепно-лицевые боли мышечно-скелетной природы
(в данной группе наряду с соматогенными болями —
темпоромандибулярным болевым синдромом, также
представлены и головные боли психогенной природы —
головная боль напряжения);
• боли при повреждении ушей, носа и ротовой полости
(в данной группе присутствуют как соматогенные боле-
вые синдромы — зубная боль при воспалении пульпы,
так и психогенная боль — атипичная одонталгия);
• основные нозологические формы головных болей (вмес-
те с нейрогенными болевыми синдромами — мигрень,
кластерная головная боль — присутствует соматогенный
болевой синдром — посттравматическая головная боль);
• боли головы и лица психологической природы (данная
группа представлена психогенными болевыми синдрома-
ми).
На наш взгляд, указанная систематизация болевых синдро-
мов, не учитывающая особенностей их патогенеза, затрудняет
клиническую диагностику болевых синдромов, а следовательно,
и проведение адекватной терапии. Поэтому дальнейшее изложе-
ние клинического материала (на примере головной боли) будет
строиться с учетом этиопатогенетического принципа.
Головные боли — это болезненные или неприятные ощуще-
ния в области головы. Чаще всего причиной данного симпто-
ма является процесс, приводящий к активации ноцицептивных
рецепторов, расположенных в различных тканях головы [Вейн
А.М., Авруцкий М.Я., 1997]. Ноцицептивные рецепторы име-
ются в мягких тканях головы, артериях и венах тканей, покры-
вающих череп, надкостнице черепа, тканях глаза, уха и полос-
ти носа, на отдельных участках твердой мозговой оболочки,
внутричерепных артериях и венах. При раздражении этих
структур боль часто является единственным ощущением. Рас-
пространение сигналов от ноцицептивных рецепторов в ЦНС
обеспечивается тройничным, языкоглоточным и блуждающим
нервами, а также 1—3 шейными корешками спинного мозга.
Раздражение ноцицептивных рецепторов может быть вызвано:
▲ травмой тканей головы (посттравматическая головная
боль);
384
▲ растяжением сосудов головы при артериальных гипо- и
гипертензиях, при аневризмах сосудов мозга, при сни-
жении тонуса вен, при венозных тромбозах и др.;
▲ повышением внутричерепного давления при объемных
внутричерепных процессах, приводящих к натяжению
оболочек, сосудов и нервов внутри черепа;
▲ воспалительным процессом, затрагивающим оболочки
мозга, а также интра- или экстракраниальные сосуды;
▲ токсическим действием лекарственных препаратов или
химических веществ;
▲ длительным напряжением перикраниальных мышц;
▲ заболеванием глаз, ушей, носа.
Нетрудно догадаться, что в патогенетическом отношении
все перечисленные формы головных болей относятся к сома-
тогенным болевым синдромам, и основные принципы тера-
пии будут сводиться к устранению факторов, активирующих
ноцицепторы, и симптоматическому лечению.
Второй причиной, приводящей к возникновению болез-
ненных ощущений в голове, является поражение нервных
структур, осуществляющих передачу в ЦНС болевых сигналов
от тканей головы. Данная группа заболеваний по своему пато-
генезу будет относиться к нейрогенным болевым синдромам.
В качестве примера можно перечислить следующие болевые
синдромы: невралгии (тригеминальная, языкоглоточная,
блуждающего нерва, затылочного нерва и др.), синдромы, вы-
званные поражением соматических и вегетативных ганглиев
(ганглионит тригеминального узла, крылонебного узла, верх-
него шейного симпатического узла и др.), боли, обусловлен-
ные поражением центральных нейронов тригеминальной сис-
темы. Как уже сказано ранее, при поражении больпроводящих
структур в ЦНС формируются агрегаты гиперактивных нейро-
нов, которые настолько изменяют характер афферентного сиг-
нала, что легкое раздражение периферических рецепторов
воспринимается мозгом как повреждающее. Поэтому головная
боль при нейрогенных болевых синдромах имеет, как правило,
пароксизмальный характер и иррадиируют в строго опреде-
ленную область. Приступы болей следуют с определенным ин-
тервалом и запускаются при раздражении триггерных зон. При
лечении нейрогенных головных болей основное внимание
уделяется снижению возбудимости и реактивности централь-
ных ноцицептивных нейронов.
Особое положение в общей структуре головных болей за-
нимают мигрень и головная боль напряжения.
Мигрень характеризуется повторяющимися приступами
пульсирующей односторонней боли, чаще лобно-височно-
глазничной локализации. Среди провоцирующих факторов
мигренозного приступа особо выделяют физическое и эмоци-
25— 1385
385
ональное перенапряжение, употребление алкоголя и продук-
тов, богатых тирамином, нарушение режима сон—бодрствова-
ние. Патогенез мигрени сложен и включает в себя ряд звеньев:
• изменение возбудимости структур лимбико-ретикуляр-
ного комплекса;
• активация системы тройничного нерва;
• изменение тонуса экстра- и интракраниальных сосудов.
На наличие центральной нейрогенной дисфункции у паци-
ентов с мигренью указывают следующие факты: эмоциональ-
ная неустойчивость накануне приступа и появление ауры
(неврологические симптомы в виде зрительных иллюзий, тре-
вожности, вегетативных изменений, повышенной чувстви-
тельности к запахам, свету и шуму); периодически возникаю-
щая в коре больших полушарий распространяющаяся депрес-
сия, приводящая к деполяризации корковых нейронов; повы-
шенная возбудимость в межприступном периоде стволовых
структур мозга.
Участие системы тройничного нерва в формировании миг-
ренозного приступа продемонстрировано в эксперименталь-
ных исследованиях. Так, электрическая стимуляция тригеми-
нального ганглия приводит к развитию нейрогенного воспале-
ния в сосудах твердой мозговой оболочки в результате выделе-
ния алгогенных веществ (см. выше) и активации болевых ре-
цепторов. И, наоборот, болевая стимуляция верхнего сагит-
тального синуса активирует нейроны тригеминального кау-
дального ядра.
Изменение тонуса сосудов головы при мигренозной атаке
происходит в три стадии: вазоспазм, вазодилатация и перива-
скулярный отек. Считается, что пусковым механизмом в изме-
нении тонуса сосудов является повышение активности триге-
миноваскулярной системы, в результате чего выделяются ва-
зоактивные нейрогенные (кальцитонин-ген-родственный пеп-
тид, субстанция Р), плазматические (брадикинин, серотонин)
и тканевые (серотонин, гистамин, простагландины) алгогены,
которые и активируют перивазальные ноцицепторы. Таким
образом, болевое ощущение при мигрени возникает в резуль-
тате активации ноцицептивных рецепторов сосудов и, следо-
вательно, по своему этиопатогенезу мигренозная боль должна
относиться к соматогенным болям. Однако активация перива-
зальных ноцицепторов алгогенами является вторичной реак-
цией, возникающей в результате гиперактивации системы
тройничного нерва вследствие церебральной дисфункции, что
в патогенетическом отношении сближает мигрень с нейроген-
ными болевыми синдромами. Вместе с тем необходимо под-
черкнуть, что центральная дизрегуляция при мигрени в отли-
чие от типичных нейрогенных болевых синдромов (например,
постгерпетическая невралгия) не имеет сколько-нибудь значи-
386
мых внешних причин и обусловлена индивидуальными осо-
бенностями метаболизма различных нейрохимических систем
мозга. При терапии мигрени в межприступный период боль-
шое внимание уделяется контролю возбудимости центральных
структур мозга (бензодиазепины, блокаторы Са2+-каналов,
бета-адреноблокаторы, антидепрессанты). Во время приступа
важное значение приобретают препараты, блокирующие вы-
брос алгогенов (нестероидные противовоспалительные пре-
параты, препараты спорыньи, обладающие вазоконстриктор-
ным действием, агонисты серотониновых рецепторов, восста-
навливающие нормальный тонус сосудов, снижающие прони-
цаемость сосудистой стенки и блокирующие ноцицептивную
импульсацию).
Головная боль напряжения — тупая стягивающая, как пра-
вило, двусторонняя боль. Возникновение данного болевого
синдрома обусловлено рефлекторным напряжением перикра-
ниальных мышц в результате эмоционального дистресса.
Обычно головная боль напряжения возникает у лиц в услови-
ях недостаточной двигательной активности, чья профессио-
нальная деятельность связана с длительной концентрацией
внимания. Тоническое напряжение перикраниальных мышц
приводит к болезненному ощущению в виде «обруча» (или
«каски»), охватывающего голову. Боль при данной патологии
обусловлена возбуждением ноцицепторов мышц вследствие
растяжения тканей и их ишемизации. Однако, как и в случае с
мигренозными болями, возбуждение ноцицептивных рецепто-
ров при головной боли напряжения носит вторичный харак-
тер, а пусковым механизмом являются психоэмоциональные
нарушения. Поэтому при лечении головных болей напряже-
ния патогномоничным будет назначение психотропных пре-
паратов, особенно бензодиазепинового ряда, которые наряду с
подавлением тревожно-депрессивной симптоматики обладают
центральным миорелаксирующим действием. Психотропную
терапию необходимо сочетать с расслаблением перикраниаль-
ных мышц (с помощью постизометрической релаксации, мас-
сажа, иглотерапии и др.), поскольку напряжение этих мышц и
является источником болезненных ощущений. Таким обра-
зом, из приведенных примеров становится ясно, что и миг-
рень, и головные боли напряжения имеют сложный патогене-
тический механизм развития болевого синдрома. При мигрени
нейрогенный механизм (функциональная дизрегуляция цент-
ральных структур болевой чувствительности) сочетается с со-
матогенным (активация алгогенами перивазальных ноцицеп-
торов). В случае головных болей напряжения отмечается ком-
бинация психогенного и соматогенного патогенетических
факторов.
Необходимо подчеркнуть, что для хронических болевых
синдромов характерна комбинация патогенетических механиз-
25*
387
мов, когда к ведущему основному механизму подсоединяется
дополнительный, усугубляющий клиническую картину боли.
Достаточно распространенным явлением считается развитие
депрессивной симптоматики у пациентов с соматогенными
или нейрогенными болевыми синдромами, которая не только
усиливает у пациента ощущение боли, но и посредством нару-
шения вегетативных функций, мышечного тонуса формирует
вторичную психогенную боль. Таким образом, для адекватной
терапии болевых синдромов наряду с определением причины
боли необходимы четкое понимание ведущего патогенетичес-
кого механизма и выделение сопутствующих симптоматичес-
ких проявлений боли.
Список литературы
Вальдман А.В., Игнатов Ю.Д. Центральные механизмы боли. — Л.:
Наука, 1976. — 191 с.
Вейн А.М., Авруцкий М.Я. Боль и обезболивание. М.: Медицина,
1997. - 278 с.
Игнатов Ю.Д., Зайцев А.А., Михайлович В.А., Страшнов В.И. Адренер-
гическая аналгезия. — СПб, 1994. — 213 с.
Калюжный Л. В. Физиологические механизмы регуляции болевой чув-
ствительности. М.: Медицина, 1984. — 215 с.
Кукушкин М.Л., Решетняк В.К., Воробейчик Я.М. Нейрогенные боле-
вые синдромы и их патогенетическая терапия//Анестезиол. и реани-
матол. — 1994. — № 4. — С. 36—41.
Крыжановский Г.Н. Детерминантные структуры в патологии нервной
системы. — М.: Медицина, 1980. — 360 с.
Крыжановский Г.Н. Общая патофизиология нервной системы. — М.:
Медицина, 1997. — 350 с.
Лиманский Ю.П. Основные принципы функциональной организа-
ции ноцицептивных и антиноцицептивных систем мозга//Физиол.
журн. - 1989. - № 2. - С. 110-121.
Мелзак Р. Загадка боли. — М.: Медицина, 1981. — 231 с.
Михайлович В.А., Игнатов Ю.Д. Болевой синдром. — Л.: Медицина,
1990. - 336 с.
Ревенко С.В., Ермишкин В.В., Селектор Л.Я. Периферические меха-
низмы ноцицепции//Сенсорные системы. — 1988. — Т. 2, № 2. —
С. 198-210.
388
Решетняк В.К. Нейрофизиологические основы боли и рефлекторного
обезболивания//Итоги науки и техники. ВИНИТИ. Физиол. челове-
ка и животных. — 1985. —Т.29. — С. 39—103.
Шухов В. С. Боль: механизмы формирования, исследование в клини-
ке//Медицина и здравоохранение. Серия: невропатология и психиат-
рия, вып. 1. — М., 1990. — 62 с.
Bonica J.J. (Ed.) The management of pain. — Philadelphia: Lea and Fe-
biger, 1990.
Coderre T.J., Katz J., Vaccarino A.L., Melzack R. Contribution of central
neuroplasticity to pathological pain: review of clinial and experimental evi-
dence//Pain, 1993. - Vol. 52. - P. 259-285.
Dickenson A.H. Where and how do opioids act.//Proceedings of the 7th
World Congress on Pain, Progress in Pain Research and Management. Vol.
2/Ed G.F.Gebhart, D.L.Hammond, T.S.Jensen. — Seattle: IASP Press,
1994. - P. 525-552.
Wall P.D., Melzack. R. (Ed.) Textbook of pain, 3rd ed. — Churchill Living-
stone, Edinburg, 1994.
Лекция 7
Патология гемостаза
В. А. Макаров
Введение
Свертывание крови (гемостаз) представляет собой сумму
процессов, приводящих к переходу крови из жидкого состоя-
ния в гелеобразное, т.е. к тромбообразованию. В жизнедея-
тельности организма феномен тромбообразования может
иметь различное значение. С одной стороны, благодаря фор-
мированию тромба при нарушении целости кровеносных
сосудов организм защищается от кровопотери. С другой сто-
роны, тромбообразование внутри сосуда ведет к нарушению
кровотока и трофики жизненно важных органов и тканей, что
является важным этапом в патогенезе многих заболеваний.
В частности, образование тромба в коронарных артериях при-
водит к инфаркту миокарда, в сосудах мозга — к тромботичес-
кому инсульту, в артериях поджелудочной железы — к панкре-
атиту. Большую опасность для больного представляют тромбо-
зы мезентериальных сосудов. Часто встречаются тромбозы вен
нижних конечностей. Образование микротромбов является
одной из причин некроза пересаженных органов.
Геморрагии также играют важную роль в патологии, явля-
ясь, в частности, важной причиной смерти матери в период
родов и ребенка в первый месяц жизни (геморрагии новорож-
денных). Геморрагические осложнения могут привести к ле-
тальному исходу не только при геморрагических заболеваниях
(гемофилии, тромбоцитопатии и др.), но и при геморрагичес-
ком инсульте, лучевой болезни, кровотечениях при язвенном
поражении желудочно-кишечного тракта, ранениях в военно-
полевых условиях и в момент массовых катастроф и др.
При сердечно-сосудистых, инфекционных, аутоиммунных,
воспалительных, инфекционных и других заболеваниях часто
развивается ДВС-синдром (синдром диссеминированного
внутрисосудистого свертывания), ведущий к нарушениям
микроциркуляции и геморрагиям. Купирование ДВС-синдро-
ма является важнейшим звеном в лечении указанных заболе-
ваний.
Все эти факты свидетельствуют о необходимости как зна-
ния тонких механизмов функции гемостаза, так и наличия ме-
тодов и средств управления данной функцией организма.
390
В настоящем разделе представлены сведения о современных
взглядах на процесс свертывания крови. Изложены основные
причины возникновения тромбозов и геморрагий, принципы
диагностики патологии гемостаза. Рассмотрены пути лекарст-
венной коррекции тромботических и геморрагических сос-
тояний.
7.1. Современные представления
о свертывании крови
7.1.1. Механизмы свертывания крови
В свертывании крови (гемостазе) различают два звена: кле-
точный и плазменный гемостаз. Под клеточным гемостазом
понимают склеивание форменных элементов крови между
собой (агрегация), их прикрепление к сосудистой стенке или
чужеродной поверхности (адгезия), а также высвобождение из
форменных элементов веществ, активирующих плазменный
гемостаз (реакция высвобождения). Плазменный гемостаз
представляет собой каскад реакций, в которых участвуют фак-
торы свертывания крови, завершающийся процессом фибри-
нообразования. Образовавшийся фибрин подвергается далее
разрушению под влиянием плазмина (фибринолиз).
Артериальные и венозные тромбы содержат фибрин. Кле-
точный состав их различен. В силу высокой скорости кровото-
ка артериальные тромбы содержат в основном тромбоциты с
небольшой примесью эритроцитов и лейкоцитов, оседающих
в сетях фибрина. Венозные тромбы состоят из эритроцитов и
лейкоцитов и небольшого количества тромбоцитов.
Среди клеток крови, участвующих в процессах агрегации и
адгезии, лучше всего изучены изменения, происходящие в
тромбоцитах. Важную роль в образовании первичного тромба
играют и лейкоциты. Тромбоциты и лейкоциты являются «си-
лами быстрого реагирования», образующими агрегаты и адге-
зирующими к поверхности несколько быстрее, чем возникают
отложения фибрина. Следует также учитывать роль эритроци-
тов в образовании тромба и нарушения микроциркуляции.
В силу больших размеров эритроциты, склеиваясь, могут су-
щественно нарушать капиллярный кровоток. Способность
эритроцитов нарушать кровоток в микрососудах связана с на-
рушением деформабильности их мембраны. Повышение жест-
кости эритроцитарной мембраны связано с уменьшением кон-
центрации АТФ в эритроцитах, потерей гиперосмолярности.
Склеивание эритроцитов вызывают глобулины и фибриноген.
Однако механизмы участия лейкоцитов и особенно эритроци-
тов в образовании тромба еще недостаточно изучены. Поэтому
практически во всех современных руководствах авторы при
391
Схема 7.1. Основные индукторы и ингибиторы агрегации
и адгезии тромбоцитов, генерируемые сосудистой стенкой
•------Сосудистая стенка-----.
Ингибиторы агрегации
и адгезии тромбоцитов:
простациклин,
оксид азота,
13-гцдроксиоктадиеновая кислота,
тромбомодулин
Индукторы агрегации
и адгезии тромбоцитов:
коллаген,
фактор Виллебранда,
тромбоспондин,
фибронектин,
фактор агрегации тромбоцитов
Пояснения в тексте.
изложении материалов о клеточном гемостазе вынуждены
ограничиваться описанием изменений, происходящих в тром-
боцитах, составляющих основную массу первичного тромба.
В последние 2 десятилетия знания о биохимических осно-
вах адгезивно-агрегационной реакции тромбоцитов сущест-
венно расширились. Показано, что в физиологических усло-
виях тромбоциты не агрегируют между собой и не приклеива-
ются к сосудистой стенке, так как последняя постоянно гене-
рирует простациклин (простагландин 12) и другие естествен-
ные ингибиторы агрегации и адгезии тромбоцитов (схема 7.1).
Простациклин образуется из арахидоновой кислоты (схема
7.2). Это наиболее мощный физиологический антиагрегант.
Попытки его использования в качестве лекарственного сред-
ства оказались безуспешными, так как он нестоек как хими-
ческое соединение и, кроме того, быстро подвергается био-
Схема 7.2. Участие простагландинов в регуляции
функции тромбоцитов
Тромбоциты
Стенка сосуда
pge2
pgd2
PGHa
Фосфолипиды
Арахидоновая
кислота
PGCj,PGH2
2
------Фосфолипаза Аг
Циклооксигеназа
L_______Тромбоксан-
синтетазе
Тромбоксан А2
Тромбоксан В;
Фосфолипцды
Арахидоновая
кислоте
PGC2, PGH2
Простациклин-______
синтетаза
PGIj
3_______(простациклин)
Агрегация-* — " |
б-кето-РССг
1 — превращение вещества; 2 — активация; 3 — ингибирование.
392
трансформации. Механизм действия простациклина связан с
активацией аденилатциклазы — фермента, превращающего
АТФ в цАМФ, который в свою очередь активирует системы,
приводящие к перемещению ионов кальция из цитоплазмы в
электронно-плотные гранулы, где он прочно связывается с
белками [Макаров В.А., 1982; Струкова С.М., 1995]. Этот эф-
фект очень важен потому, что именно цитоплазматический
кальций, как будет показано далее, является необходимым
участником активации адгезивно-агрегационной способности
тромбоцитов.
Мощным ингибитором агрегации является также оксид
азота [Libby Р., 1987; Moncada S. и др., 1988]. Он активирует
гуанилатциклазу и способствует образованию цГМФ. Послед-
ний уменьшает концентрацию ионов кальция в цитоплазме
тромбоцитов за счет торможения их прохода через плазмати-
ческую мембрану.
Определенное значение в поддержании суспензионной
стабильности тромбоцитов имеют белок тромбомодулин, а
также некоторые другие пептиды. В частности, эндотелии так-
же является естественным антиагрегантом, активирующим
высвобождение простациклина и оксида азота. Тромборезис-
тентности сосудистой стенки способствует также 13-гидрокси-
октадиеновая кислота, тормозящая появление адгезивных (по
отношению к тромбоцитам и нейтрофилам) рецепторов эндо-
телиальных клеток. При повреждении эндотелия обнажается
субэндотелий, появляются адгезивные белки и рецепторы на
поверхности клеток, уменьшаются генерация простациклина,
антиагрегационная активность тромбомодулина и др. К бел-
кам субэндотелия, связывающим клетки крови, относятся
коллаген, фактор Виллебранда, тромбоспондин, фибронектин
(см. схему 7.1). Последний прикрепляет тромбоциты и макро-
фаги к коллагену. Фактор Виллебранда связывается с тромбо-
цитарными рецепторами (интегринами) — белками 1b и
Ilb/IIIa. С рецепторами ПЬ/Ша связываются и другие адгезив-
ные белки: фибриноген, тромбоспондин, фибронектин. Наи-
большая аффинность у этих рецепторов отмечена к фибрино-
гену. Следует отметить, что данные рецепторы появляются на
поверхности тромбоцитов только после активации этих клеток
(высвобождения ионов кальция). Благодаря связыванию с ад-
гезивными белками сосуда тромбоциты распластываются на
субэндотелии. Образующийся в процессе альтернативного ме-
ханизма активации тромбин взаимодействует с рецептором 1b.
Помимо тромбина, активацию тромбоцитов вызывают вы-
деляющиеся из клеток при их повреждении фактор агрегации
тромбоцитов, АДФ, а также повышение содержания в крови
катехоламинов, серотонина и др. Все эти агенты имеют специ-
фические рецепторы на тромбоцитарной плазматической мем-
бране. Связывание этих агонистов с рецепторами приводит к
26— 1385
393
Рис. 7.1. Реакция тромбоцитов на индуктор агрегации.
1 — исходное состояние тромбоцита: кальций в электронно-плотных гранулах;
2 — действие индукторов агрегации, приводящее к переходу ионов кальция в
цитоплазму и секреции содержимого цитоплазмы во внешнее пространство.
освобождению кальция из внутритромбоцитарных депо или
способствует входу ионов кальция в тромбоциты. Свободный
кальций, вышедший в цитоплазму из органелл или проник-
ший из плазмы крови, усиливает появление рецептора ПЬ/Ша
на плазматической мембране, вызывает сокращение контак-
тильных белков и высвобождение из а-гранул тромбоцитов,
фактора Виллебранда, фибриногена, тромбоспондина, факто-
ра V, тромбоцитарного фактора 4, p-тромбоглобулина и др.
(рис. 7.1) (реакция высвобождения). Наряду с кальцием из
электронно-плотных гранул выходят АДФ и серотонин. Каль-
ций активирует фосфолипазу А2, и арахидоновая кислота на-
чинает превращаться в простагландины G2, Н2, а затем в более
мощный естественный индуктор агрегации — тромбоксан Д2
(см. схему 7.2) [Marcus A.J., 1988].
Механизм действия тромбоксана А2 связан с активацией вы-
свобождения кальция из электронно-плотных гранул. Из арахи-
доновой кислоты образуется также 12-гидроксиэйкозатетраено-
вая кислота — аттрактант, включающий в первичный тромб
нейтрофилы. Освобождается также фактор агрегации тромбоци-
тов (ФАТ) — продукт биотрансформации фосфолипидов. АДФ,
серотонин, простагландины G2, Н2, тромбоксан А2, ФАТ, вза-
имодействуя с соответствующими рецепторами, повышают кон-
центрацию ионов кальция, что приводит к появлению рецепто-
ров ПЬ/Ша, активации агрегации и адгезии тромбоцитов.
Таким образом, активация клеточного, в частности тром-
боцитарного, гемостаза имеет характер цеппой реакции. Один
активированный тромбоцит активирует много других. Высво-
394
бождаемые из тромбоцитов белки — фактор Виллебранда,
фибриноген, тромбоспондин — служат клеем, который связы-
вает тромбоциты друг с другом и сосудистой стенкой. Тром-
боспондин связывает тромбоциты с фибрином, коллагеном,
эндотелиоцитами, моноцитами и макрофагами. Моноциты и
макрофаги (как и ряд других клеток) сами также синтезируют
и секретируют тромбоспондин. Именно благодаря тромбос-
пондину агрегация приобретает необратимый характер. Фиб-
ронектин также синтезируется различными типами клеток. Он
связывается не только с рецепторами форменных элементов
крови и эндотелиальных клеток, но и с фибрином, что спо-
собствует упрочению тромба.
Форменные элементы крови играют роль не только в обра-
зовании первичного клеточного тромба, но и в активации
плазменного гемостаза. Они создают поверхность, на которой
происходят реакции активации факторов свертывания крови.
Форменные элементы крови выделяют факторы, активирую-
щие процесс тромбообразования.
Кроме того, в тромбоцитах содержатся митогенный фак-
тор, способствующий росту гладкомышечных элементов и сти-
мулирующий развитие атеросклеротических бляшек, р-тром-
боглобулин, фактор проницаемости сосудов, фактор, уси-
ливающий двигательную и фагоцитарную активность лейко-
цитов и др. В эритроцитах также содержится ряд факторов,
влияющих на различные звенья гемостаза. Подобные актива-
торы содержатся и в лейкоцитах [Кудряшов Б.А., 1975].
Плазменный гемостаз (рис. 7.2) осуществляется в основ-
ном белками, которые называются плазменными факторами
свертывания крови и обозначаются римскими цифрами (в от-
личие от тромбоцитарных факторов, обозначаемых арабскими
цифрами). Буквой «а» отмечают активные факторы свертыва-
ния, ибо в кровотоке циркулируют неактивные комплексы
ферментов, связанные с ингибиторами.
Пути активации белков свертывания крови условно под-
разделяются на внутренний и внешний; они представлены на
рис. 7.2. Инициация фибринообразования на поврежденной
поверхности сосудистой стенки или в участке замедленного
кровотока развивается по внутреннему пути. Появление же в
кровотоке обломков клеточных мембран при травме или
каких-то патологических состояниях запускает внешний меха-
низм свертывания крови.
Свертывание крови по внутреннему пути развивается следу-
ющим образом (см. рис. 7.2). Поверхности коллагена и активи-
рованных тромбоцитов являются высокоаффинными для фак-
тора XII и высокомолекулярного кининогена (ВМК), находяще-
гося в комплексе с прекалликреином и фактором XI. Фактор XII
путем изменения конформации превращается в фактор ХПа.
Фактор ХПа активирует фактор XI. Фактор Х1а превращает фак-
26*
395
ВНУТРЕННИЙ МЕХАНИЗМ
ВНЕШНИЙ МЕХАНИЗМ
Контактная активация
(коллаген, каолин)
XII—L g^xll а] + Калликреин, ВМК
Активация тканевым фактором
Тканевый фактор (ТФ)
УПа+ТФ + С^|
X
VII
TFPI
Фосфоли-
т _ vn
Тромбомодулин
Тромбин
II
Ха
- Антитромбин Ш + гепарин
ФИБРИНОЛИЗ
Протеин С
Протеин S
АРС
Ингибитор
АРС
На
Фибриноген
Пептиды А, В
Фибрин-мономер
V
Са*
ХШ
ХШа
Фибрин-олигомер
| Плазминоген |
1
Плазмин
Калликреин
t-PA
Урокиназа-*
ХШа
АРС
PAI
Сгусток фибрина Фибриноген
х. Фибрин-мономер
><, ПДФ ПДФг
(D-димер) (Фрагмент D)
Рис. 7.2. Механизмы свертывания крови (по З.С.Баркагану и А.П.Момоту, 1999).
АРС — активированный протеин С; ВМК — высокомолекулярный кининоген; TFPI — ингибитор внешнего пути свертывания;
t-PA — тканевый активатор плазминогена; PAI — ингибитор активатора плазминогена; ПДФ — продукты деградации фибрина;
VII — фактор неактивный; Vila — фактор активный. Остальные пояснения в тексте.
тор IX в фактор 1Ха в присутствии ионов кальция. Фактор 1Ха
включается в комплекс, активирующий фактор X. На этом звене
процесса смыкаются внутренний и внешний пути.
Свертывание крови по внешнему пути развивается следую-
щим образом [Ploplis V.A. et al., 1987]. Тканевый фактор
(тромбопластин), содержащийся в эндотелиальных и гладко-
мышечных клетках, в присутствии ионов кальция образует
комплекс с циркулирующим в крови фактором VII, превращая
последний в фактор Vila. Комплекс фактор Vila — тканевый
фактор превращает фактор X в фактор Ха. Фактор Ха в при-
сутствии ионов кальция на фосфолипидной поверхности свя-
зывается с фактором V. Весь этот комплекс действует как про-
тромбиназа, превращая протромбин (II) в тромбин (Па).
При незначительном повреждении сосудистой стенки тка-
невый фактор в комплексе с фактором Vila активирует фактор
IX. Фактор 1Ха комплексируется с фактором VIII в присутст-
вии кальция и фосфолипида. Этот комплекс также активирует
фактор X. Такой путь носит название альтернативный меха-
низм активации. Альтернативный механизм активации ведет к
появлению небольшого количества тромбина, недостаточного
для фибринообразования, но способствующего активации
клеточного гемостаза.
Таким образом, под влиянием протромбиназного комплекса
путем частичного протеолиза образуется тромбин. Субстратом
тромбина является фибриноген. Под действием тромбина от
фибриногена отщепляются 2 фибринопептида А и 2 фибрино-
пептида В. Образуются фибрин-мономеры, которые начинают
полимеризоваться, образуя фибрин-полимер (см. рис. 7.2).
Однако образовавшийся фибрин непрочен и легко расщеп-
ляется протеиназами. Стабилизация фибрина происходит под
влиянием фактора XIII, который под действием тромбина и
ионов кальция превращается в фактор ХШа. Фактор ХШа обра-
зует поперечные сшивки между соседними молекулами фибри-
на. Стабилизация фибрина повышает его резистентность к про-
теазам, в том числе к плазмину, увеличивает эластичность и
прочность сгустка. Фактор ХШа образует также поперечные
сшивки между фибрином и фибронектином, а также другими
адгезивными белками. Все это усиливает взаимодействие фиб-
рина с тромбоцитами и другими форменными элементами
крови, а также с клетками сосудистой стенки. Фактор ХШа во-
влекает в сгусток также а2-антиплазмин, что дополнительно
усиливает резистентность сгустка к фибринолизу. Следует отме-
тить, что упомянутые выше факторы свертывания крови в виде
неактивных предшественников синтезируются в тканях. Факто-
ры II (протромбин), VII, IX и X синтезируются главным образом
в печени. Для их полноценного функционирования в качестве
факторов свертывания необходима их пострибосомальная моди-
фикация под влиянием витамина
397
7.1.2. Механизмы ингибирования свертывания
крови. Фибринолиз
Как известно, локальная инициация процесса тромбообра-
зования не ведет к свертыванию всей крови в кровотоке.
Этому способствует ряд ограничительных механизмов. В кро-
ви циркулирует большое количество ингибиторов факторов
свертывания крови, в частности, в кровотоке циркулирует ин-
гибитор внешнего пути свертывания.
Среди других ингибиторов факторов свертывания важное
место занимает антитромбин III [Струкова С.М., 1995]. Он
блокирует активность тромбина, а также факторов 1Ха, Ха,
Х1а. В присутствии гепарина, вырабатываемого тучными клет-
ками или вводимого извне, эффект антитромбина III резко
возрастает. Ингибиторами тромбина являются также такие
белки, как кофактор гепарина II и а2-макроглобулин. Замед-
ление процесса фибринообразования также связано с тем, что
тромбин захватывается фибриновой сетью.
Важную роль в регуляции фибринообразования играет
также белок тромбомодулин. Он находится на поверхности эн-
дотелия и связывает тромбин. Тромбин, связанный с тромбо-
модулином, не свертывает фибриноген, не активирует агрега-
цию тромбоцитов. Тромбин в комплексе с тромбомодулином в
присутствии ионов кальция активирует циркулирующий в
кровотоке протеин С (схема 7.3). Данный белок синтезируется
в печени, в его построении принимают участие ферментные
системы, зависимые от витамина К(. Активированный проте-
ин С, т.е. протеин Са, ингибирует активность факторов Va и
Villa на фосфолипидной поверхности. Неферментным кофак-
тором активированного протеина С является протеин S, син-
тезирующийся в печени и эндотелии сосудов [Stricklaned D.K.,
Kessler С.М., 1987].
Ингибиторами факторов свертывания являются также
такие антипротеолитические белки, как а(-антитрипсин, а2-
макроглобулин и др.
Разрушение тромба вызывают фибринолитические фермен-
ты. Клетки синтезируют активаторы плазминогена (профиб-
ринолизина), предшественника плазмина (фибринолизина),
двух типов — тканевого и урокиназного. В клетках синтезиру-
ются также ингибиторы указанных активаторов, а также фер-
мент нексин, вызывающий разрушение активаторов.
Тканевый активатор плазминогена связывается с фибри-
ном значительно сильнее, чем с фибриногеном. В силу этого
фибринолитическое действие данного белка более выражено в
зоне тромба, чем в общем кровотоке. Эффективность тромбо-
литического действия тканевого активатора возрастает в при-
сутствии тромбоспондина. Это связано с тем, что последний
связывает плазминоген и ускоряет его гидролиз под влиянием
398
Схема 7.3. Эффекты естественных антикоагулянтов
(-> активация, —> ингибирование)
Антитромбин III
•- Тромбин
Гепарин
Б
А — схема действия протеина С; Б — схема действия антитромбина II.
тканевого активатора. Активатор плазминогена урокиназного
типа содержится в плазме, почках, моче. Под влиянием плаз-
мина или калликреина одноцепочечная молекула активатора
(проурокиназа) превращается в двухцепочечную (урокиназу).
Урокиназа обладает более сильным действием на превращение
плазминогена в плазмин. Однако проурокиназа, подобно тка-
невому активатору плазминогена, обладает большей тропнос-
тью к фибрину, чем к фибриногену, т.е. сильнее проявляет
фибринолитические свойства в зоне тромба. Урокиназа пре-
вращает плазминоген в плазмин вне зависимости от присутст-
вия фибрина.
Активация плазминогена происходит также под влиянием
контактной активации свертывания крови вслед за активацией
фактора XII и образованием калликреина (ХП-зависимый
фибринолиз). Калликреин непосредственно способствует пре-
вращению плазминогена в плазмин. Подобно факторам свер-
тывания крови, активаторы плазминогена имеют специфичес-
кие ингибиторы. Так, выявлены ингибитор активатора плаз-
миногена-1 (PAI-1) и плазмина — а2-антиплазмин. Связанный
со сгустками фибрина и клетками плазмин защищен от дейст-
вия аг-антиплазмина. В плазме же плазмин быстро реагирует с
ингибитором, при этом наступает его необратимая инактива-
ция. а-Антиплазмин способен также связываться с фибрином
в реакции, катализируемой фактором XIII, что повышает ус-
тойчивость сгустка к плазмину. Интересно также, что активи-
399
рованный протеин С связывает РА1-1 и блокирует его дейст-
вие, тем самым активируя фибринолиз.
Наличие в организме естественных антикоагулянтов и
тромболитиков позволило разработать концепцию о противо-
свертывающей системе крови [Кудряшов Б.А., 1975].
7.2. Современные представления
о природе тромбозов
7.2.1. Основные причины развития тромбозов
Главными причинами тромбообразования в артериях явля-
ются повреждение сосудистого эндотелия, локальный ангио-
спазм, активация фибринообразования, снижение фибрино-
литических свойств крови, активация адгезивно-агрегацион-
ной способности тромбоцитов.
Нарушение целости сосудистой стенки и/или изменение
функциональных свойств эндотелиальных клеток могут спо-
собствовать развитию протромботических реакций и индуци-
ровать прогрессирование ряда заболеваний, например атеро-
склероза. Причины, приводящие к травме сосудов, весьма раз-
нообразны. Это экзогенные факторы: механические повреж-
дения, лучевое воздействие, гипер- и гипотермия, токсичные
вещества, лекарственные препараты и т.п. Среди лекарствен-
ных средств наибольшее значение имеют гормональные
контрацептивы, гипертонические растворы с низким pH, ад-
реналин, глюкокортикоиды. К эндогенным факторам относят-
ся биологически активные вещества (тромбин, ряд цитокинов
и т.п.), способные при определенных условиях проявлять мем-
браноагрессивные свойства. Такой механизм поражения сосу-
дистой стенки характерен для многих заболеваний, сопровож-
дающихся склонностью к тромбообразованию [Панченко
Е.П., Добровольский А.Б., 1999].
Общая направленность реакций эндотелиоцитов в ответ на
воздействие протромботических факторов вначале носит за-
щитный характер. Происходит выделение указанных ранее
ингибиторов плазменного и клеточного гемостаза. Но при ис-
тощении ресурса этих веществ наблюдаются уменьшение син-
теза простациклина и оксида азота, усиление синтеза ФАТ,
повышение агонистиндуцированной секреции фактора Вилле-
бранда, повышение экспрессии тканевого фактора и молекул,
участвующих в адгезии лейкоцитов, увеличение секреции PAI-1
и снижение секреции тканевого активатора плазминогена, а
также снижение содержания тромбомодулина [Libby Р., 1987].
Уменьшение содержания тромбомодулина на поверхности
эндотелиоцитов имеет две основные причины. Во-первых, при
400
ряде заболеваний (васкулиты, тромботическая тромбоцитопе-
ническая пурпура, ДВС-синдром и др.) эндотелиальные клет-
ки под действием ряда факторов, в частности интерлейкина-1,
фактора некроза опухолей-аь эндотоксина, гипоксии и т.п.,
теряют со своей поверхности тромбомодулин, обеспечивая тем
самым повышение уровня этого гликопротеина в плазме
крови. Во-вторых, данные факторы также способствуют сни-
жению синтеза тромбомодулина эндотелиальными клетками.
Одной из основных причин приобретения прокоагулянт-
ных свойств интимой сосудов является способность эндотели-
оцитов синтезировать и представлять на своей поверхности
тканевый фактор тромбопластин — основной инициатор про-
цесса свертывания крови. Заболеваниями и патологическими
состояниями, чаще других приводящими к продукции ткане-
вого фактора эндотелиоцитами, являются гипоксия, вирусные
инфекции, аутоиммунные васкулиты, однако-эта реакция аб-
солютно необходима при инициации тромбообразования вне
зависимости от его причин. Количество тромбопластина на
клеточной поверхности является лимитирующим условием
тромбообразования; при достижении определенного уровня
тканевый фактор обеспечивает тромбогенность поврежденной
сосудистой стенки, а степень отложения фибрина на эндоте-
лиоцитах прямо зависит от содержания на них тканевого фак-
тора.
Повышение адгезии лейкоцитов к сосудистой стенке также
является одним из ответов на действие провоцирующих фак-
торов, вызывающих стойкую и длительную активацию эндоте-
лиальных клеток, и способствует выявлению тромбогенных
свойств интимы. Такие факторы, как цитокины или эндоток-
син, обеспечивают увеличение количества адгезивных молекул
на мембране, которые опосредуют связывание эндотелиоци-
тов с клетками белой крови. Механизм повышения коагуляци-
онного потенциала сосудистой стенки под влиянием лейкоци-
тов не совсем ясен, однако не исключено, что в его основе
лежит потенцирование ими продукции фактора агрегации
тромбоцитов в клетках сосудистой стенки, высвобождение ци-
токинов из активированных лимфоцитов и/или прямая акти-
вация эндотелиоцитов адгезированными лимфоцитами и как
следствие этих процессов — повышение содержания тканево-
го фактора на наружной мембране клеток эндотелия. Кроме
того, лейкоциты содержат ряд активаторов различных звеньев
плазменного гемостаза.
Одним из наиболее важных факторов, ограничивающих
прогрессию тромбообразования, инициированного повыше-
нием уровня тромбопластина, является ингибитор внешнего
пути свертывания (см. рис. 7.2). Эта мультивалентная биоло-
гическая субстанция синтезируется главным образом эндоте-
лиальными клетками и может присутствовать в организме как
401
свободно циркулирующее вещество или быть связанной с по-
верхностью эндотелиоцитов. Кроме того, физиологически
значимые количества ингибитора внешнего пути свертывания
(приблизительно 5—10 % от величины плазменного пула) со-
держатся в тромбоцитах. Следует отметить, что объединенные
плазменный и тромбоцитарный пулы этого естественного
антикоагулянта составляют менее чем половину от его общего
количества, присутствующего в сосудистом русле, так как ос-
новная часть ингибитора внешнего пути свертывания связана
с эндотелием. В ответ на действие цитокинов, приводящее к
увеличению экспрессии тканевого фактора, повышается уро-
вень циркулирующего ингибитора внешнего пути свертыва-
ния, однако величина его пула, связанного с сосудистой по-
верхностью, не претерпевает значительных изменений. Инги-
битор внешнего пути свертывания способен предупреждать
прокоагулянтный эффект как фактора Ха, так и комплекса
тканевый фактор — фактор Vila. Механизм его действия за-
ключается в способности связываться с фактором Ха; образо-
вавшееся при этом физиологически активное вещество явля-
ется потенциальным ингибитором комплекса, состоящего из
тканевого фактора (тромбопластина) и фактора VII (см. рис.
7.2).
Чрезвычайно важными результатами нарушения целости
сосудистой стенки являются активация тромбоцитов и индук-
ция процессов их адгезии, реакции высвобождения и агрега-
ции; при этом адгезия предшествует развитию реакции высво-
бождения и агрегации тромбоцитов и является первой ступе-
нью формирования тромба.
Повышенная наклонность крови к свертыванию (тромбофи-
лия) также является важной причиной тромбозов. Тромбо-
филии разделяют на первичные (наследственные) и вторич-
ные (приобретенные).
Особенности тромбофилий подробно классифицированы и
описаны З.С.Баркаганом (1966). Тромбофилии можно разде-
лить на заболевания, связанные с патологией плазменного и
клеточного гемостаза.
Наиболее распространены формы тромбофилий, связан-
ные с патологией плазменного гемостаза. Сравнительно час-
то среди гематогенных тромбофилий встречается недостаточ-
ность антитромбина III. Она может быть как первичной, так
и вторичной. Наследственная форма представляет собой
аутосомно-доминантно наследуемое заболевание. В большин-
стве случаев тормозится синтез этого естественного антикоа-
гулянта. Однако встречаются формы и с аномальной структу-
рой антитромбина III. Вторичный дефицит этого естествен-
ного антикоагулянта может развиваться на фоне ДВС-син-
дрома, приема синтетических прогестинов, используемых в
402
качестве противозачаточных средств. Снижение уровня анти-
тромбина III отмечается при длительном применении гепа-
рина.
Изменения в концентрации, структуре и активности фиб-
риногена могут также носить первичный и вторичный харак-
тер. Описаны различные виды изменений фибриногена, при-
чем некоторые из них ведут к тромбозам. Из вторичных ано-
малий фибриногена следует отметить патологию, связанную с
образованием циркулирующих иммунных комплексов фибри-
ноген—антифибриногеновые антитела.
Часто встречаются дефицит и аномалии протеина С. На-
ряду с врожденными формами этих нарушений дефицит дан-
ного белка может явиться следствием лечения антикоагулян-
тами непрямого действия. Установлено также, что в опреде-
ленных случаях протеин С блокируется ингибиторами этого
белка.
Особенно часто встречается тромбофилия, механизм кото-
рой был расшифрован лишь в 90-х годах. Речь идет о генети-
чески обусловленной аномалии фактора V, который вследст-
вие мутации не инактивируется протеином С (18, 23, 29).
Кроме того, взаимодействие протеина С с фактором V может
носить характер и приобретенной патологии [Stricklaned D.K.,
Kessler С.М., 1987; Dahlback В., 1995; Leroy-Matheron C. et al.,
1996].
Очень часто причиной тромбоза служит появление в крови
антител к фосфолипидно-белковым структурам мембран (вол-
чаночный антикоагулянт), что создает условия для активации
фибринообразования [Brandt J.T. et al., 1995].
Тромбозы отмечаются также при дефиците фактора XII,
при недостаточности каллекреина, высокомолекулярного ки-
ниногена вследствие ослабления фибринолиза.
Фибринолиз замедляется при тромбофилиях, связанных с
дефицитом или аномальной структурой тканевого активатора
плазминогена (t-PA) и активатора плазминогена урокиназного
типа. В ряде случаев нарушены кумуляция и высвобождение
данных активаторов.
Угнетение фибринолиза может развиваться также под
влиянием появления в крови мощных ингибиторов активато-
ров плазминогена и плазмина. Известны как первичные, так
и вторичные формы данных тромбофилий. Нарастание в
плазме таких ингибиторов фибринолиза, как а-антиплазмин,
ингибитор активатора плазминогена-1 (РАТ-1), сц-антитрип-
син, наблюдается при злокачественных новообразованиях,
токсикозах беременных и др. Дефицит плазминогена также
может явиться причиной тромбоза при системных васкули-
тах, ДВС-синдроме, а также при введении больших доз акти-
ваторов плазминогена и препаратов дефибринирующего дей-
ствия.
403
Ряд тромбофилий связан с активацией клеточного гемо-
стаза, в частности с повышением адгезивно-агрегационной
активности тромбоцитов. Это может зависеть от появления в
крови иммунных комплексов, активированных компонентов
системы комплемента, что имеет место при гломерулонефри-
тах, микротромбоваскулитах, гемолитико-уремическом син-
дроме, отторжении трансплантируемых тканей и т.д. При эн-
дотоксемии и эндотоксиновом шоке тромбоциты, как и сосу-
дистая стенка, подвергаются изменениям, ведущим к актива-
ции образования первичных тромбов и внутрисосудистых аг-
регатов [Kubatiev A. et al., 1991]. При ряде острых инфекций,
сепсисе, злокачественных новообразованиях, панкреатите,
шоке и других состояниях отмечается появление в плазме ак-
тивной формы гликопротеина, повышающего агрегацию
тромбоцитов. Повышение агрегационной активности тром-
боцитов наблюдается также при гемолизе, гиперлипидеми-
ях, при введении адреналина и синтетических прогестинов.
Агрегационная активность тромбоцитов возрастает при бо-
лезни Мошковица (тромботическая тромбоцитопеничес-
кая пурпура). Изменение функциональных свойств тромбо-
цитов связано с появлением в крови в высоких концентраци-
ях мощного активатора агрегации белковой природы. В нор-
ме этот белок нейтрализуется другим протеином — ингиби-
тором Бериса—Лайна. Изменение соотношения указанных
белков ведет к активации формирования тромбоцитарных
тромбов.
Показана важная роль в активации тромбоцитов и повреж-
дений сосудистого эндотелия повышения уровня гомоцистеи-
на в крови.
7.2.2. Лабораторная диагностика вероятности
развития тромбозов
При лабораторной диагностике следует учесть, что разви-
тие тромбоза часто провоцируется клиническими факторами
риска. Наиболее важные среди них — оперативное вмешатель-
ство, тяжелая травма, длительная иммобилизация, беремен-
ность с развитием гестоза, роды, прием оральных контрацеп-
тивов, злокачественные новообразования, сердечная и веноз-
ная недостаточность, протезирование сосудов и клапанов
сердца, возраст, инфекции, полицитемии и др.
Существенную роль в развитии венозных и артериальных
тромбозов играют нарушения реологических свойств крови —
повышение ее вязкости, полиглобулии, полицитемии, высо-
кие показатели гематокрита, уровня гемоглобина, эритроци-
тов и тромбоцитов, резкое ускорение СОЭ, повышение уровня
фибриногена и вязкости плазмы.
404
Сдвиги в тромбоцитарно-сосудистом взаимодействии,
проявляющиеся в усилении адгезивности тромбоцитов и чув-
ствительности их к механическому воздействию, опосредован-
ные повышенным уровнем фактора Виллебранда и фибрино-
гена и связанные с наличием крупных гиперактивных тромбо-
цитов, являются компонентами развития преимущественно
артериальных тромбозов. Внутрисосудистая активация тром-
боцитов может быть оценена по маркерам реакции их высво-
бождения — тромбоцитарному фактору 4 и р-тромбоглобули-
ну, уровень которых в плазме измеряется методами иммуно-
ферментного анализа. Предрасположенность к внутрисосуди-
стой активации тромбоцитов можно определить по повыше-
нию уровня тромбоксана В2. При анализе тромбоцитарно-со-
судистого взаимодействия выявление повышенного уровня
антигена фактора Виллебранда и тромбомодулина в крови
свидетельствует о повреждении сосудистой стенки. Опреде-
ленное значение имеет функциональный тест на степень
спонтанной агрегации тромбоцитов, а также при активации
агрегации АДФ, адреналином, арахидоновой кислотой, серо-
тонином, ФАТ и др.
Активация плазменного гемостаза оценивается вначале
грубыми интегральными тестами, такими как АЧТВ (активи-
рованное частичное тромбопластиновое время — провокация
свертывания плазмы фосфолипидами и каолином), тромбино-
вое время (активация свертывания плазмы тромбином), опре-
деление уровня фибриногена. Последний параметр часто по-
вышается при угрозе тромбоза коронарных сосудов [Бокарев
И.Н., 1991]. Самостоятельное значение для риска острой ко-
ронарной смерти имеет сочетание повышенного уровня фак-
тора VII, связанного с активацией внешнего пути свертывания
крови и обменных нарушений, в частности гиперлипидемии.
Высокие уровни фибриногена и фактора VII в сочетании с ги-
перхолестеринемией могут быть отнесены к прогностическим
показателям развития ИБС и использованы для отбора боль-
ных в группу высокого риска с целью проведения первичной
антитромботической профилактики. Высокий риск тромбозов
имеет место при повышении концентрации гомоцистеина в
крови.
К развитию тромбозов может привести недостаточность
основных физиологических ингибиторов коагуляции или их
кофакторов: кофакторов гепарина — антитромбина III и гепа-
ринового кофактора II; протеина С и протеина S; ингибитора
комплекса фактор Vila — тканевый активатор. В связи с этим
данные параметры также необходимо определить при диагнос-
тике вероятности развития тромбоза.
К ряду новых, еще недавно не известных, но широко рас-
пространенных тромбофилий относятся состояния, вызван-
ные резистентностью фактора V к активированному протеи-
405
ну С (РАПС). Среди больных с тромбозами РАПС встречает-
ся, по данным разных авторов, в 20—60 % случаев [Dahl-
back В., 1995]. В связи с этим определение РАПС является
важным показателем в диагностике предрасположенности к
тромбозу.
Присутствие в крови волчаночного антикоагулянта (анти-
тел к фосфолипидно-белковому комплексу мембран) часто со-
провождается повышенной наклонностью к тромбозам [Brandt
et al., 1995]. Около 30 % больных, имеющих волчаночный
антикоагулянт, страдают тромбозами как магистральных, ор-
ганных вен и артерий, так и зон микроциркуляции, вызывая
ишемии и инфаркты органов. Типично развитие нарушений
мозгового кровообращения и даже синдрома эпилепсии, энце-
фалопатии, полинейропатии, нарушений зрения, кожного не-
кроза и прочих нарушений, в основе которых лежит блокада
микроциркуляции. С присутствием волчаночного антикоагу-
лянта в кровотоке связаны также фетоплацентарная недоста-
точность с упорной невынашиваемостью беременности (выки-
дыши или внутриутробная гибель плода), тромбоцитопении
[Brandt et al., 1995].
При появлении в крови антифосфолипидных антител на-
рушается образование или секреция эндотелиального проста-
циклина, усиливаются биологические эффекты тромбоцитар-
ного тромбоксана, стимулируется продукция тканевого факто-
ра эндотелиальными клетками и моноцитами, индуцируя по-
вышенную прокоагулянтную клеточную активность. Кроме
того, in vivo антифосфолипидные антитела ингибируют тром-
бомодулинопосредованную активацию протеина С, конкури-
руют с активированным протеином С и его субстратами за
связывание с фосфолипидной поверхностью, вызывая подав-
ление антикоагулянтной функции протеина С. Угнетается
фибринолиз из-за увеличения активности PAI-1. В связи с из-
ложенным выявление волчаночного антикоагулянта имеет
важнейшее значение в диагностике вероятного развития тром-
бозов.
Наиболее информативным маркером тромбинемии явля-
ются D-димеры — продукты деградации прошитого фактором
ХШа фибрина, которые образуются под действием плазмина
[Bounaneaux Н., 1996; De Moerloose Р. et al., 1994; Estive F. et al.,
1996]. Таким образом, уровень D-димеров отражает интенсив-
ность фибринообразования под действием тромбина и после-
дующий лизис фибрина под влиянием плазмина. Реже в каче-
стве маркера тромбинемии используется определение уровня
фибринопептида А, высвобождающегося из a-цепи фибри-
ногена в процессе образования растворимого фибрина под
действием тромбина. К признакам тромбин- и фибринемии
относится также повышенное содержание в крови фибрин-
мономеров и растворимых фибрин-мономерных комплексов.
406
7.3. Геморрагии
7.3.1. Виды и основные причины
развития геморрагий
Важным видом патологии гемостаза являются геморрагии.
Причиной кровотечений могут быть различные нарушения
плазменного и клеточного гемостаза, а также проницаемости
сосудистой стенки.
Основными причинами кровотечений являются тромбоцито-
пении, тромбоцитопатии, наследственные нарушения плаз-
менного гемостаза, первичный и вторичный гиперфибрино-
лиз, приобретенные коагулопатии, ДВС-синдром, микро-
тромбоваскулиты и др.
Под тромбоцитопениями подразумеваются группы заболе-
ваний и синдромов, при которых кровоточивость обусловлена
снижением количества тромбоцитов. Практически значимо
снижение числа тромбоцитов ниже 100 109/л, кровоточивость
может появляться при уровне тромбоцитов менее 50 109/л, а
угроза серьезных геморрагий — при уровне ниже 30 1 09/л. Это
снижение может быть вызвано либо торможением образова-
ния тромбоцитов, либо их повышенным потреблением или
разрушением. Тромбоцитопении могут быть вызваны иммун-
ной реакцией, связанной с образованием антитромбоцитар-
ных антител, механическим повреждением тромбоцитов (при
гемангиомах, спленомегалии и др.), угнетением пролифера-
ции тромбоцитов в костном мозге (при апластической ане-
мии, лейкозах, гипоплазии кроветворения, болезни Гоше, Ни-
мана—Пика, мукополисахаридозах, лучевой болезни, под вли-
янием цитостатической терапии, при лечении сульфанилами-
дами, антибиотиками и др.), замещением костного мозга опу-
холевой тканью, соединительной тканью (миелофиброз), по-
вышенным потреблением тромбоцитов (при острых формах
ДВС-синдрома), недостатком витамина В12, фолиевой кисло-
ты и др.
Наиболее часто встречаются иммунные формы тромбоци-
топений вследствие появления антитромбоцитарных или
антифосфолипидных (к фосфолипидам мембраны клеток)
антител. При этих заболеваниях антитела могут быть направ-
лены как против антигена самих тромбоцитов, так и против
антигена мегакариоцитов. Практически важно, что часто
тромбоцитопении могут быть обусловлены приемом лекарст-
венных средств. В частности, продукцию тромбоцитов подав-
ляют цитостатические препараты, тиазидные диуретики, эст-
рогены. Образование антитромбоцитарных антител вызывают
сульфаниламидные препараты, алкалоиды хины (хинин, хини-
дин), некоторые антибиотики (новобиоцин и др.), противоси-
407
филитические средства, содержащие мышьяк, некоторые пси-
хотропные средства (карбамазепин и др.), дигитоксин и др.
Подробно вопросы патогенеза, диагностики и лечения тром-
боцитопений рассмотрены в монографии З.С.Баркагана «Ге-
моррагические заболевания и синдромы» (1988).
В клинической практике наблюдается также снижение ад-
гезивно-агрегационной функции тромбоцитов — тромбоцито-
патии. Остановимся на некоторых из них.
В настоящее время показано, что тромбастения Гланцман-
на представляет собой наследственное заболевание, передаю-
щееся по рецессивно-аутосомному типу. Причиной тромбас-
тении является отсутствие на плазматической мембране тром-
боцитов комплекса гликопротеинов ПЬ/Ша. Встречаются
тромбоцитопатии, связанные и с дефектом гликопротеина ПЬ
[Баркаган З.С., 1988].
Под эссенциальной атромбией понимают тромбоцитопатии
без нарушения реакции высвобождения. По-видимому, это
целая группа патологических состояний. От тромбастении
Гланцманна она отличается сохранностью рецепторов ПЬ/Ша.
В этой группе встречаются больные, у которых снижена реак-
ция тромбоцитов на тромбоксан А2, что, возможно, связано с
патологией тромбоксановых рецепторов.
Аномалия Мел-Хеглина — аутосомно-доминантно насле-
дуемое заболевание, характеризующееся нарушением созрева-
ния и аномалией мегакариоцитов, тромбоцитов и нейтрофи-
лов. Предполагается, что заболевание связано с нарушением
фрагментации мегакариоцитов.
При наследственных парциальных дизагрегационных
тромбоцитопатиях могут отсутствовать рецепторы или тром-
бина, или каких-либо других индукторов агрегации. Возмож-
но, нарушается передача сигнала от рецептора к клеточным
органеллам. Патогенез этих состояний мало изучен. К этой же
группе относят афибриногенемию, при которой нарушены
АДФ-агрегация и адгезивность тромбоцитов.
При ряде тромбоцитопатий нарушена реакция высвобожде-
ния индукторов агрегации и адгезивных молекул. Эти тромбо-
цитопатии, по-видимому, имеют различный генез. В данной
группе встречаются формы с активацией аденилатциклазы, сни-
жением активности мембранной фосфолипазы, снижением
концентрации арахидоновой кислоты в мембранных фосфоли-
пидах, с дефицитом или снижением активности циклооксигена-
зы, дефицитом или снижением активности тромбоксансинтета-
зы, снижением содержания в цитоплазме тромбоцитов ионизи-
рованного кальция, фактора активации тромбоцитов и др.
В группу болезней и синдромов недостаточности «пула
накопления и хранения» входят наследственные тромбоцитопа-
тии, при которых нарушено накопление и соответственно вы-
деление АТФ, серотонина, адреналина, фактора 4 или других
408
важных для функции гемостаза веществ. Эти нарушения свя-
заны с отсутствием в тромбоцитах гранул, где накапливается
какое-либо из указанных веществ. К этой же группе могут
быть отнесены больные с недостатком электронно-плотных
телец в тромбоцитах, в которых хранятся кальций, АТФ, серо-
тонин, адреналин, и с дефицитом а-гранул, где накапливают-
ся анти гепариновый фактор 4, p-тромбоглобулин, фибрино-
ген, фибронектин, тромбоцитарный фактор роста, тромбо-
спондин и др. В результате снижения в тромбоцитах количест-
ва а-гранул нарастает концентрация в плазме продуктов, кото-
рые должны храниться в этих органеллах. Это не оказывает
стимулирующего влияния на адгезивно-агрегационные свой-
ства тромбоцитов, так как все эти компоненты должны высво-
бождаться при специфической стимуляции из а-гранул и
иметь плотный контакт с плазматической мембраной тромбо-
цитов. Только в этом случае происходят агрегация и адгезия.
Кроме того, циркулирующие в крови тромбоцитарный фактор
роста и фактор 4 стимулируют рост соединительнотканных
клеток, что способствует развитию миелофиброза. В этой же
группе заболеваний описано нарушение высвобождения кис-
лых гидролаз из тромбоцитов.
Макроцитарная тромбоцитодистрофия Бернара—Сулье
связана с нарушением связывания фактора Виллебранда и
других факторов свертывания крови с рецептором 1b тромбо-
цитов, а также с нарушением строения каналов и микротрубо-
чек тромбоцитов.
В отдельную группу выделяют тромбоцитопатии с дефици-
том и снижением доступности тромбоцитарного фактора 3
тромбоцитов. При этом адгезивно-агрегационные свойства
кровяных пластинок не нарушаются. Вероятно, при данном
состоянии имеет место дефицит фосфолипидов, входящих в
тромбоцитарный фактор 3.
Иногда тромбоцитопатии сочетаются с врожденными ано-
малиями соединительной ткани, нарушениями иммунитета
(гиперэластоз кожи, гипермобильность суставов при синдроме
Элерса—Данло, врожденная грыжа, пролабирование клапа-
нов, синдром Марфана, аномалии скелета, TAP-синдром и
др.), а также с нарушениями пигментации кожи. Это позволи-
ло З.С.Баркагану (1988) выделить особую группу геморраги-
ческих диатезов — геморрагические гематомезенхимальные
дисплазии. При синдроме Вискотта—Олдрича наблюдается
сочетание тромбоцитопатии и тромбоцитопении. При этом на
фоне снижения содержания мегакариоцитов и тромбоцитопе-
нии уменьшено содержание как электронно-плотных телец,
так и а-гранул. Нарушения функции тромбоцитов наблюдают-
ся также при цианотических врожденных пороках сердца, гли-
когенозах, синдроме Дауна. В последнем случае нарушено на-
копление в тромбоцитах серотонина.
27— 1385
409
Приобретенные тромбоцитопатии отмечаются как син-
дром при гемобластозах, миелопролиферативных заболевани-
ях, В12-дефицитной анемии, парапротеинемиях, уремии, гепа-
титах, циррозах печени, различных опухолях. При гемобласто-
зах тромбоцитопатия, по-видимому, связана с нарушением
функции мегакариоцитов и ранней отшнуровкой тромбоци-
тов. При миелопролиферативных заболеваниях, по-видимому,
дисфункция тромбоцитов связана с развитием ДВС-синдрома
и наступающей вследствие этого дегрануляцией тромбоцитов.
При миеломной болезни и макроглобулинемии Вальденстрема
дисфункция тромбоцитов обусловлена обволакиванием их
парапротеинами. Снижение агрегационной способности
тромбоцитов отмечается при уремии, поражениях печени,
ДВС-синдроме, дефиците витамина С, образовании антитром-
боцитарных антител и др.
Особое место в развитии приобретенных дисфункций тром-
боцитов принадлежит лекарственным средствам.
Резкое снижение агрегационной способности тромбоцитов
развивается на фоне приема нестероидных противовоспали-
тельных средств, таких как ацетилсалициловая кислота, ибу-
стрин и др. Механизм действия препаратов этой группы свя-
зан с блокадой циклооксигеназы тромбоцитов. Эффект от
приема ацетилсалициловой кислоты бывает весьма выражен-
ным и продолжительным (до 5—7 дней). Агрегацию тромбо-
цитов тормозят также многие сосудорасширяющие средства
(дипиридамол, трентал и др.). Механизм действия этих соеди-
нений связан с накоплением в тромбоцитах цАМФ. Тормозят
агрегацию также нейролептические средства (аминазин, триф-
тазин, дроперидол и др.), антагонисты кальция, а-адренобло-
каторы, реополиглюкин и др.
Тромбоцитопению и тромбоцитопатию вызывают цитоста-
тические средства. Влиянием на функциональную активность
тромбоцитов обладают и некоторые другие препараты, однако
их эффект не является столь существенным и опасным в
плане развития геморрагического синдрома.
При тромбоцитопениях и тромбоцитопатиях отмечается ка-
пиллярный тип кровотечений, характеризующийся петехи-
ально-пятнистыми кровоизлияниями в кожу и слизистые
оболочки, а также имеют место маточные, десневые и носо-
вые кровотечения.
Одной из наиболее часто распространенных форм гемор-
рагических диатезов является болезнь Виллебранда. Данное
заболевание обусловлено дефицитом белкового комплекса
фактора Виллебранда с фактором VIII. В настоящее время по-
казано, что заболевания этой группы обусловлены либо
уменьшением синтеза, либо качественными аномалиями фак-
410
тора Виллебранда. При болезни Виллебранда отмечается ка-
пиллярно-гематомный тип кровоточивости. Он встречается у
лиц обоего пола обычно уже в детском возрасте [Головина О.Г.,
1996].
Большую группу заболеваний представляют наследствен-
ные и приобретенные нарушения плазменного гемостаза (коагу-
лопатии) [Баркаган З.С., 1988]. Остановимся лишь на наибо-
лее распространенных формах наследственных коагулопатий,
которые могут привести к развитию кровотечений. Одним из
наиболее часто встречающихся наследственных геморрагичес-
ких диатезов является гемофилия А. Данное заболевание обу-
словлено дефицитом или нарушениями структуры фактора
VIII. Ингибиторные формы гемофилии А связаны с выработ-
кой антител к данному фактору свертывания крови. Заболева-
нием в подавляющем большинстве случаев страдают мужчи-
ны. Женская гемофилия встречается крайне редко. Описано
всего 40 хорошо документированных случаев.
Гемофилия В — наследственный геморрагический диатез,
обусловленный дефицитом активности фактора IX. Это свя-
зано в одних случаях с пониженным синтезом фактора, в дру-
гих — с нарушением его структуры. В мировой литературе
описано лишь 14 случаев женской гемофилии В. Крайне ред-
кая патология — одновременный дефицит факторов VIII и IX
(гемофилия АВ). Для больных гемофилией характерен гема-
томный тип кровоточивости, отмечаемый с раннего детского
возраста. Характерно возникновение обильных кровотечений
после любых (даже малых) травм и операций, включая экс-
тракцию зубов. Специфичны также повторяющиеся крово-
течения в суставы, вследствие чего возникают хронические
артрозы. Наблюдаются также межмышечные, внутримышеч-
ные гематомы, поднадкостничные кровоизлияния, а также
желудочно-кишечные и почечные кровотечения.
Достаточно редко встречается наследственный дефицит
фактора XI — гемофилия С. Данная патология в отличие от ге-
мофилий А и В свойственна лицам обоего пола.
Редко наблюдается также дефицит высокомолекулярного
кининогена, витамин К-зависимых факторов (одновременно
всех), фактора V, сочетанный дефицит факторов V и VIII, дефи-
цит и аномалия структуры плазменного прекалликреина, факто-
ра VII, протромбина, фактора XIII. Встречаются также наслед-
ственное снижение количества фибриногена (а-фибриногене-
мия или гипофибриногенемия) и нарушение его структуры.
Среди наследственных коагулопатий следует также отме-
тить наследственный гиперфибринолиз. Описаны формы, свя-
занные с дефицитом а2-антиплазмина, а также повышением
уровня тканевого активатора плазминогена.
Приобретенные нарушения плазменного гемостаза имеют
важное практическое значение. К ним относится изолирован-
27*
411
ный дефицит отдельных факторов свертывания крови. Так,
при системном амилоидозе в отдельных случаях снижается
концентрация фактора X, что связано с быстрой элиминацией
его из кровотока. Изолированный дефицит этого же фактора
был обнаружен при отравлении метилбромидом. Дефицит
факторов VII (или VII+V), IX либо XI описан при невротичес-
ком синдроме. Снижение концентрации фактора VII отмеча-
ется при токсических поражениях печени (например, при ин-
токсикации парами трихлорэтилена).
Значительно больше приобретенных нарушений коагуля-
ционного гемостаза связано с иммунным конфликтом. Часто
встречаются больные с антителами к фактору VIII и фактору
Виллебранда, реже к другим факторам свертывания. Крайне
редко кровоточивость возникает на фоне появления в крово-
токе волчаночного антикоагулянта (за счет развития тромбо-
цитопении, тромбоцитопатии и дефицита протромбина). При
ревматоидном артрите, миеломной болезни, иммунных пан-
цитопениях и др. описано появление у-глобулина — антитром-
бина V.
Приобретенный дефицит витамин К-зависимых факторов
свертывания крови встречается довольно часто. Он может
быть обусловлен разными причинами. Прежде всего — это не-
достаточное образование в кишечнике витамина Кь что имеет
место при геморрагической болезни новорожденных, кишеч-
ном дисбактериозе, энтеропатиях и при приеме лекарств, по-
давляющих рост микроорганизмов — продуцентов витами-
на К.
Причиной дефицита витамин К-зависимых факторов
может явиться также недостаточное всасывание витамина К
из кишечника, что зависит от уменьшения поступления желчи
и отмечается при механической желтухе. Кроме того, синтез
факторов свертывания в печени снижается при острых дис-
трофиях печени, тяжелых формах инфекционного гепатита,
циррозах и др.
При нарушениях синтеза зависимых от витамина К, фак-
торов свертывания крови (II, V, VII и X) наблюдается петехи-
ально-пятнистый и гематомный тип кровоточивости: кровоте-
чения из носа, десен, микро- и макрогематурия. Следует отме-
тить, что при поражениях печени наряду с торможением син-
теза факторов свертывания может развиваться ДВС-синдром,
что, собственно, и является основной причиной кровотече-
ний. Важной причиной нарушения синтеза витамин К-зави-
симых факторов свертывания является антикоагулянтная те-
рапия производными кумарина и феналиндандиона.
Среди других нарушений плазменного гемостаза лекарст-
венного генеза следует отметить геморрагии, связанные с ге-
паринотерапией, а также введением тромболитических
средств. Из лекарственных средств, способных вызвать ге-
412
моррагии, можно выделить нестероидные противовоспали-
тельные средства, прежде всего ацетилсалициловую кислоту.
"Под влиянием этого препарата резко снижается агрегацион-
ная способность тромбоцитов. Геморрагии могут провоциро-
вать некоторые сочетания лекарственных средств. Так, ге-
моррагический эффект антикоагулянтов непрямого действия
возрастает при их сочетании с препаратами гормонов щито-
видной железы, анаболическими стероидами, нестероидными
противовоспалительными средствами, трициклическими
антидепрессантами, ПАСК, амиодароном, клофибратом, хи-
нидином, глюкагоном, антидиабетическими сульфанилами-
дами, химиотерапевтическими средствами (сульфаниламиды
длительного действия, препараты пенициллинового ряда,
тетрациклины).
Геморрагические осложнения также развиваются при
ДВС-синдроме (подробнее рассмотрено далее). Данный син-
дром обусловлен поступлением в кровоток активаторов плаз-
менного и клеточного гемостаза, фибринолиза, что приводит
к истощению указанных систем, развитию тромбозов и гемор-
рагий, гипоксии, ацидозу, дистрофии и дисфункции органов.
Нередко он приводит и к развитию вторичных профузных
кровотечений. Геморрагические осложнения при этом могут
быть связаны либо с тромбоцитопенией, либо с дефицитом
факторов свертывания за счет их потребления, либо с актива-
цией протеолиза, что ведет к разрушению белков сосудистой
стенки и появлению в кровотоке продуктов биотрансформа-
ции фибриногена, лейкоцитарных и тромбоцитарных протеаз
(при этом повышается проницаемость сосудистой стенки),
либо с неправильным применением лекарственных корректо-
ров ДВС-синдрома.
Геморрагии при болезни Рандю—Ослера обусловлены
малой резистентностью, легкой ранимостью сосудов и слабой
сосудистой активацией адгезивно-агрегационных свойств
тромбоцитов. Нередко данное заболевание сочетается с болез-
нью Виллебранда. Имеется и ряд других видов геморрагичес-
ких состояний, связанных не с изменением системы свертыва-
ния крови и фибринолиза, а с поражением соединительной
ткани, эндотелия и др. [Баркаган З.С., 1988].
7.3.2. Лабораторная диагностика
геморрагических состояний
Для ориентировочной диагностики нарушений плазменно-
го гемостаза имеет значение выявление гипокоагуляции в
таких общих тестах, как время свертывания крови, АЧТВ,
протромбиновое время, тромбиновое время [Баркаган З.С.,
МомотА.П., 1999].
413
Вслед за ориентировочными тестами при подозрении на ге-
мофилию проводят количественное определение гемофиличес-
ких факторов (прежде всего VIII и IX). При гемофилии нередко'
возникают специфические иммунные ингибиторы соответст-
венно к фактору VIII или IX, которые необходимо определять.
К одной из наиболее распространенных форм наследст-
венного геморрагического диатеза относится болезнь Вилле-
бранда. Диагностика болезни Виллебранда проводится путем
определения ристомицин-агрегации (агглютинации) в богатой
тромбоцитами исследуемой плазме, что отражает взаимодей-
ствие фактора Виллебранда с собственными тромбоцитами
пациента, так как ристомицин способствует связыванию фак-
тора с тромбоцитарным рецептором, а кроме того, — опреде-
лением активности фактора Виллебранда (ристомицин-кофак-
торной активности) исследуемой плазмы, что основано на ее
способности вызывать в присутствии ристомицина агглютина-
цию нормальных донорских тромбоцитов, внесенных в плазму
больных.
Наиболее тяжелые и разнонаправленные нарушения гемо-
коагуляции развиваются в результате поражения паренхимы
печени при тяжелых и затяжных формах инфекционного гепа-
тита, острой дистрофии печени, хроническом агрессивном ге-
патите, циррозе печени и других ее поражениях. К дефициту
витамин К-зависимых факторов присоединяется нарушение
синтеза других плазменных факторов коагуляции, развивается
печеночная дисфибриногенемия. При тяжелых поражениях
печени может развиться синдром ДВС с потреблением факто-
ров свертывания, тромбоцитов, накоплением продуктов про-
теолиза.
При гемостазиологическом скрининге определяются при
этом признаки тромбин- и фибринемии. При дальнейшем раз-
витии синдрома ДВС кровоточивость связана также с дис-
функцией и внутрисосудистой агрегацией тромбоцитов, гипо-
и дисфибриногенемией, антикоагулянтным действием про-
дуктов протеолиза, дальнейшим потреблением факторов коа-
гуляции, что диктует необходимость выполнения тестов, отра-
жающих динамику этих процессов.
Вторичная недостаточность факторов протромбинового
комплекса развивается также под влиянием антикоагулянтов
непрямого действия, являющихся антагонистами витамина К.
При этом в результате нарушения карбоксилирования факто-
ров II, VII, IX, X образуются их функционально неполноцен-
ные формы. При передозировке таких препаратов развивается
геморрагический синдром, характерный для дефицита факто-
ров протромбинового комплекса. При анализе коагулограмм
определяется снижение уровня соответствующих факторов в
клоттинговых тестах при нормальном содержаний антигена
факторов II, V, IX, X.
414
7.4. Синдром диссеминированного
внутрисосудистого свертывания
(ДВС-синдром)
ДВС-синдром является особой формой патологии гемоста-
за, при которой есть опасность как тромбозов, нарушений
микроциркуляции, так и геморрагий. ДВС-синдром является
наиболее распространенной формой патологии гемостаза.
Возникновение данного синдрома связано с повреждением
тканей и высвобождением в кровоток активаторов плазменно-
го и клеточного гемостаза и фибринолиза. Синдром наблюда-
ется при инфекционных заболеваниях, шоке, травматичных
хирургических вмешательствах, терминальных состояниях, ге-
молизе, акушерской патологии, злокачественных опухолях,
деструктивных процессах в органах и тканях, иммунной и им-
мунокомплексной патологии, острых аллергических реакциях,
обильных кровотечениях, массивных гемотрансфузиях, пере-
дозировке веществ с прокоагулянтной и проагрегантной ак-
тивностью и ингибиторов фибринолиза, гипоксии и др.
[Kubatiev A. et al., 1991; Bick R.L., 1994]. При этих состояниях
происходит активация фибринообразования, агрегации тром-
боцитов и фибринолиза. Однако вслед за активацией развива-
ется истощение активаторов и субстратов этих процессов. По-
степенно отмечаются ингибирование процессов фибринооб-
разования, снижение агрегационной способности тромбоци-
тов в общем кровотоке (наиболее активные склеились и осели
в капиллярном русле), а также угнетение фибринолиза. В кро-
ви накапливаются продукты биотрансформации фибриногена,
пептидов и активированных протеаз.
Различают острый и хронический ДВС-синдромы. Особен-
ность этих синдромов состоит в том, что они могут сопровож-
даться как тромбозами и нарушениями микроциркуляции, так
и геморрагиями. Лабораторное исследование функции гемо-
стаза позволяет выявить наклонность как к тромбообразова-
нию и нарушениям микроциркуляции, так и к геморрагиям.
В частности, определяются повышение уровня фибринопеп-
тида А, фибринмономерных комплексов, продуктов деграда-
ции фибриногена, D-димеров, комплекса тромбин—анти-
тромбин, а также дисфункция тромбоцитов, появление тром-
боцитарных агрегатов, нарастание концентрации тромбоци-
тарного фактора 4 и p-тромбоглобулина. При остром ДВС-
синдроме возможны тромбоцитопения и гипофибриногене-
мия. В связи с быстрым потреблением факторов свертывания
может быть удлинение АЧТВ, тромбинового времени и других
интегральных тестов, заканчивающихся образованием фибри-
на. Возможна и активация фибринолиза, и его истощение
вследствие потребления тканевого активатора плазминогена и
плазминогена.
415
Последние годы характеризуются разработкой новых под-
ходов к диагностике ДВС-синдрома, тромбозов и внедрением
в лабораторную коагулологию иммунохимических, в том
числе иммуноферментных и амидолитических методов с ис-
пользованием хромогенных субстратов [Макаров В.А., 1990].
Совершенствуются клоттинговые (т.е. фиксирующие обра-
зование сгустка) методы, создаются коагулометры, значитель-
но повышающие точность и воспроизводимость исследования.
В России внедряется 10-канальный коагулометр, разработан-
ный лабораторией патологии и фармакологии гемостаза ГНЦ
РАМН и АО «Мелт» [Лабинская Т.А. и др., 1997], снабженный
компьютерной программой для проведения коагулологичес-
ких исследований.
7.5. Лекарственная коррекция патологии гемостаза
Терапевтическая коррекция функции гемостаза осущест-
вляется с помощью различных групп лекарственных препара-
тов. К средствам лечения тромбозов относятся антикоагулян-
ты, антиагреганты, фибринолитические средства и активаторы
противотромботической активности сосудистой стенки. Анти-
коагулянтами называют средства, препятствующие образова-
нию полноценных нитей фибрина. Разделяют антикоагулянты
прямого и непрямого действия.
К антикоагулянтам прямого действия, влияющим непо-
средственно на факторы свертывания крови в сосуде или про-
бирке, относятся сульфатированные мукополисахариды (гепа-
рин, пентозан полисульфат и др.), гирудин и его производные.
Механизм действия гепарина зависит от молекулярной массы
данного сульфополисахарида. Высокомолекулярные полиме-
ры, составляющие основу нефракционированных препаратов,
выпускаемых под названием «гепарин», тормозят активность
тромбина, являются кофактором антитромбина III. Для кон-
троля их активности определяют время свертывания цельной
крови, активированное частичное тромбопластиновое время,
активность антитромбина III и др.
Низкомолекулярные гепарины (фраксипарин и др.) в
меньшей степени нуждаются в наличии антитромбина III. Ме-
ханизм их действия связан с инактивацией фактора Ха. Поэ-
тому именно активность этого фактора служит оптимальным
контролем терапевтической активности этих препаратов. Низ-
комолекулярные гепарины действуют более продолжительно
(до 12 ч), чем нефракционированные (до 4—6 ч), в меньшем
проценте случаев вызывают кровотечения [Haas S., 1996].
В лаборатории патологии и фармакологии гемостаза ГНЦ
РАМН выявлена высокая активность комбинации нефракцио-
нированного гепарина и полисульфата хитозана (хиторин).
416
Полисульфат хитозана малоактивен как антикоагулянт, одна-
ко резко потенцирует активность гепарина. Хиторин обладает
близкой антикоагулянтной активностью к гепарину, но мень-
шей геморрагической активностью и, кроме того, значительно
дешевле [Дрозд Н.Н. и др., 1996].
Гирудин и его производные обладают антитромбиновой ак-
тивностью, не нуждаются в присутствии антитромбина III.
Данные о ряде синтетических ингибиторов тромбина также
опубликованы в последнее время [Струкова С.М., 1995]. Од-
нако для понимания перспективы их внедрения еще требуют-
ся дополнительные исследования.
Антикоагулянты непрямого действия (производные кумари-
на — варфарин, синнумар, а также фенилиндандиона — фени-
лин) тормозят в печени синтез зависимых от присутствия ви-
тамина К факторов свертывания крови II (протромбин), VII,
IX и X. Активность этих препаратов контролируют, определяя
соотношение тромбопластинового времени плазмы больного к
этому параметру плазмы донорского пула.
Способностью тормозить агрегацию тромбоцитов облада-
ют многие препараты (антиагреганты). С этой целью наиболее
часто используют ацетилсалициловую кислоту, дипиридамол,
трентал, тиклопидин. Механизм действия ацетилсалициловой
кислоты связан с торможением активности циклооксигеназы
и торможением синтеза простагландинов G2, Н2 и тромбокса-
на А2. Последние активируют увеличение концентрации иони-
зированного кальция в цитоплазме тромбоцитов. Дипирида-
мол и трентал увеличивают в тромбоцитах содержание цАМФ,
который способствует транспорту кальция из цитоплазмы в
электронно-плотные гранулы. Тиклопидин и клопидогрель
блокируют пуриновые (АДФ) рецепторы. Реопро блокирует
IIIb/IIIa-рецепторы. В настоящее время новые блокаторы этих
рецепторов исследуют во многих лабораториях мира.
В последние годы достигнуты определенные успехи по со-
зданию эффективных и безопасных фибринолитических
средств. Наряду с традиционными препаратами — фибрино-
лизином (плазмином), стрептокиназой, урокиназой внедряют-
ся препараты нового поколения. Механизм действия плазмина
связан с протеолизом фибрина. Стрептокиназа и урокиназа
активируют переход плазминогена в плазмин. Однако эти пре-
параты действуют с одинаковой силой как в зоне тромба, так и
в общем кровотоке. Поэтому эффективные дозы этих средств
создают угрозу геморрагии.
Новые препараты — тканевый активатор плазминогена и
проурокиназа — обладают большей тропностью к фибрину, чем
к фибриногену, поэтому их эффект более выражен в тромбе,
чем в кровотоке. Подобное же характерно для эминазы (ком-
плекса стрептокиназы и лиз-плазминогена). В молекуле эми-
назы имеется анисоильная группировка, на начальном этапе
417
защищающая активный центр комплекса. Благодаря лиз-плаз-
миногену комплекс лучше и быстрее связывается с фибрином,
чем с фибриногеном. На тромбе благодаря эстеразам анисо-
ильная группировка отделяется от комплекса и начинается
переход плазминогена в плазмин и лизис фибрина. Критерием
эффективности тромболизиса на сегодня служит только ан-
гиография.
Важным фактором профилактики тромбозов и нарушений
микроциркуляции является увеличение нротивотромботичес-
кой активности сосудистой стенки.
Показано возрастание антикоагулянтной, антиагрегацион-
ной и фибринолитической активности под действием комби-
нации дипиридамола, фитина, глутаминовой кислоты [Мака-
ров В.А., 1982; Балуда В.П. и др., 1992]. Контроль эффектив-
ности стимуляторов противотромботической активности сосу-
дистой стенки производится путем измерения параметров
свертывания крови, фибринолиза, агрегации тромбоцитов
(или уровня стабильного метаболита простациклина — 6-кето-
простагландина F,) до и после дозированной компрессии на
предплечье [Балуда В.П. и др., 1992J.
Следует подчеркнуть, что в настоящее время для лечения и
профилактики нарушений гемостаза используются и другие
средства. Установлена способность <в-3-полиненасыщенных
жирных кислот накапливать в сосудистой стенке тканевый ак-
тиватор плазминогена, причем под влиянием этих кислот он
медленно выходит в кровоток у интактных животных. Однако
на фоне введения адреналина, иммобилизационного стресса
и др. выход тканевого активатора резко возрастает [Petruchi-
na G.N. et al., 1994J.
Для лечения и профилактики геморрагий используются
различные средства местного и резорбтивного действия. При
гемофилиях вводят препараты дефицитных факторов. При бо-
лезни Виллебранда — криопреципитат, содержащий указан-
ный фактор, связанный с фактором VIII. При геморрагиях на
фоне патологии печени вводят концентрат II, VII, IX, X фак-
торов (PPSB и др.), при гиперфибринолизе — ингибиторы
фибринолиза трасилол, контрикал, аминокапроновую кисло-
ту, амбен и др. Эффект этих средств контролируют путем оп-
ределения степени коррекции недостаточности функции гемо-
стаза в каждом конкретном случае.
Для остановки локальных кровотечений используют тром-
бин, коллаген, целлюлозу. В лаборатории патологии и фарма-
кологии гемостаза ГНЦ РАМН совместно с НИИ текстильных
материалов разработан препарат колетекс-гем, представляю-
щий собой альгинат на текстильном носителе, вызывающий
быструю остановку кровотечений [Белозерская Г.Г. и др.,
1998]. Большой популярностью в мире пользуется фибриновый
418
клей, состоящий из двух флаконов: в первом — тромбин с
кальцием, во втором — фибриноген с ингибитором фибрино-
лиза и фактором XIII (фибринстабилизирующим). На ране эти
компоненты встречаются и образуют тромб независимо от со-
стояния гемостаза у больного. Контроль за эффективностью
этих средств проводят по времени остановки кровотечения и
уровню кровопотери.
Неспецифическим средством коррекции патологии гемо-
стаза при ДВС-синдроме, наклонностях к тромбозу или ге-
моррагии является переливание свежезамороженной донор-
ской плазмы, в которой содержатся как естественные антико-
агулянты, так и прокоагулянты, активаторы и ингибиторы
фибринолиза. При тромбоцитопениях проводят переливание
концентрата тромбоцитов.
Заключение
В последние годы накоплены многочисленные факты о па-
тогенезе нарушений функции гемостаза, разработаны новые
подходы к лечению тромбозов, геморрагий, ДВС-синдрома.
Дальнейший прогресс в гемостазиологии, по-видимому, будет
связан с открытием еще не известных эндогенных регуляторов
плазменного и клеточного гемостаза, уточнением их роли в
возникновении тромбозов и геморрагий. Вероятно, увеличит-
ся количество исследований в области молекулярной природы
мутаций естественных антикоагулянтов, факторов свертыва-
ния и других компонентов, участвующих в плазменном и кле-
точном гемостазе. Несомненно, будут созданы новые подходы
к лекарственной терапии тромбозов и геморрагий. Шире будут
использоваться генно-инженерные препараты естественных
антикоагулянтов, активаторов фибринолиза, факторов свер-
тывания и др. На основе фундаментальных исследований по
структуре рецепторов форменных элементов крови, взаимо-
действующих с эндогенными активаторами их адгезии и агре-
гации, появятся средства, поддерживающие суспензионную
стабильность форменных элементов крови, т.е. предохраняю-
щие организм от тромбозов и нарушений микроциркуляции.
В результате реализации указанных разработок значитель-
но возрастут возможности врача в диагностике и лечении па-
тологии гемостаза.
Список литературы
Балуда В.П., Деянов И.И., Балуда М.В. и др. Профилактика тромбо-
зов. — Саратов, 1992.
419
Баркаган З.С. Геморрагические заболевания и синдромы. М.: Меди-
цина, 1988.
Баркаган З.С. Клинико-генетические варианты, номенклатура и ос-
новы диагностики гематогенных тромбофилий//Пробл. гематол. и
перелив, крови. — 1996. — № 3. — С. 5—15.
Баркаган З.С., Момот А.П. Основы диагностики нарушений гемоста-
за. — М.: Ньюдиамед-АО, 1999.
Белозерская Г.Г., Олтаржевская Н.Д., Лысун Н.В. и др. Гемостатичес-
кий материал «Колетекс-гем»//Хирургия. — 1998. — № 3. — С. SO-
54.
Бокарев И.Н. Тромбофилические состояния и их клинические аспек-
ты//Клин. мед. — 1991. — № 8. — С. 11—17.
Головина О.Г., Папаян Л.П., Белязо О.Е. и др. Особенности диагнос-
тики болезни Виллебранда//Тер. арх. — 1996. — № 4. — С. 58—61.
Дрозд Н.Н., Макаров В.А., Башков Г.В. и др. Влияние совместного
введения гепарина и сернокислого эфира хитозана на функцию гемо-
стаза//Экспер. клин, фармакол. — 1996. — Т. 59, № 1. — С. 30—33.
Кудряшов Б.А. Биологические проблемы регуляции жидкого состоя-
ния крови и ее свертывания. — М.: Медицина, 1975.
Лабинская Т.А., Кострицо П.Р., Пешков А.А. и др. Компьютерная про-
грамма для выполнения коагулологического скрининга на 10-каналь-
ном отечественном коагулометре «АККШ-10-01»//Клин. лабор. диа-
гностика. — 1997. — № 5. — 65 с.
Макаров В.А. Фармакологическая регуляция аггрегационной способ-
ности тромбоцитов: Дис. ... докт. мед. наук. — М., 1982.
Макаров В.А. Актуальные проблемы клинической коагулологии//
Клин. мед. — 1990. — № 6. — С. 3—8.
Панченко Е.П., Добровольский А.Б. Тромбозы в кардиологии: Меха-
низмы развития и возможности терапии. — М.: Спорт и культура,
1999.
Струкова С.М. Гуморальные гемостатические системы при воспале-
нии//Под ред. В.В.Серова, В.С.Паукова. — М.: Медицина, 1995. —
С. 52-80.
Bick R.L. Disseminated Intravascular Coagulation//Semin Thrombosis and
Hemostasis. — 1994. — Vol. 20. — P. 1—132.
Bounameaux H. Plasma Measurement of D-dimer as Diagnostic aid in
Suspected Venous Thromboembolism: an Overview, Abstract, 14 Untem.
Congress on Thrombosis, 1996. — P. 179.
Brandt J.T., Barna L.K., Triplett D.A. Laboratory Identification of Lupus
Coagulation: Results of the Second International Workshop for Identifica-
tion of Lupus Anticoagulant//Thromb. Haemost., 1995. — Vol. 74, N 6 —
P. 1389-1613.
Dahlback B. Resistance of Activated Protein C, the Arg506 to Mutation in
Factor V gene and Venous Thrombosis Functional Tests and DNA-based
Assay//Thromb. Haemost. — 1995. — Vol. 73. — P. 739—742.
De Moerloose P., Minazio P., Reber G. et al. D-dimer Determination to Ex-
lude Pulmonary Embolism//Thromb. Haemost. — 1994. — Vol. 72. —
P. 89-91.
Estive F, Grimaux M., Migand-Fressart M. et al. Individual and Quantita-
tive Rapid Testing of D-dimer Using an Automated System Fibrinolysis//
Fibrinolysis. — 1996. — Vol. 10, suppl. 3. — P. 95.
Haas S. The Role of Low-molecular weight Heparins in the Prevention of
Venous Thrombosis in Surgery with Special Reference to Enoxaparin//
Haemostasis. — 1996. — Vol. 26, suppl. 2. — P. 39—48.
Kubatiev A., Chernousova G., Grachev S. et al. Basic Mechanisms of the
DIC-syndrome Development in Case of System Endotoxemia and Endo-
toxic Shock//Constituent Congress Intern. Society of Pathophysiology. —
Moscow, 1991. — P. 125.
Leroy-Matheron C., Levent M., Pignon J.-M. et al. The 16919-11 A Muta-
tion in the Factor V Gene: Relationship to Activated Protein С (APC) Re-
sistance and Thrombosis in 65 Proteins//Thromb. Haemost. — 1996. —
Vol. 75, N 1. - P. 4-9.
Libby P. The Active Roles of Cell of Blood Vessel Wall in Health and Dis-
ease//Molec. Aspects. Med. — 1987. — Vol. 262. — P. 17356—17361.
Marcus A.J. Eicosanoids: Transcellular Metabolism, Inflammation Basic
principles and clinical correlates/Ed. J.J.Gallin et al. — N.Y.: Raven Press,
1988. - P. 129-137.
Moncada S., Palmer R.M.J., Higgs E.A. The Discovery of Nitric Oxide as
the Endogenous Vasodilator//Hypertension.— 1988.— Vol. 12.— P. 365—
372.
Petruchina G.N., Kalugin S.A., Makarov V.A. et al. Der Einfluss von
Epaden auf einzelne Parameter der Blutgerinnung//Gefassystem und Blut-
geriunnung XXXVII. Hamburger Symposion uber Blutgerinnung, 1994. —
P. 169-171.
Ploplis V.A., Edgington T.S., Fair D.S. et al. Initiation of the entrinsic Path-
way of Coagulation//.!. Biol. Chem.— 1987.— Vol. 262,— P. 9503—9508.
Stricklaned D.K., Kessler C.M. Biochemical and Functional Properties of
PC and PS//Clin.Chem.Acta. - 1987. - Vol. 27. - P. 2885-2890.
Учебное пособие
Актуальные проблемы патофизиологии
(избранные лекции)
Зав. редакцией Т. П. Осокина
Научный редактор М.Г Пшенникова
Редактор издательства Л. В. Левушкина
Художественный редактор С.Л. Андреев
Те^зический редактор В. И. Табенская
Корректор Т.Г Ганина
ЛР № 010215 от 29.04.97. Сдано в набор
30.05.2001. Подписано к печати 03.07.2001.
Формат бумаги 60><90 '/16. Бумага офс. № 1.
Гарнитура Таймс. Печать офсетная. Усл.
печ. л. 26,50. Усл. кр.-отг. 26,50. Уч.-изд. л.
28,01. Тираж 3000 экз. Заказ № 1385.
Ордена Трудового Красного Знамени изда-
тельство «Медицина». 101990, Москва,
Петроверигский пер., 6/8.
Качество печати соответствует качеству
предоставленных диапозитивов.
Отпечатано с готовых диапозитивов в
ФГУП ордена «Знак Почета» Смоленской
областной типографии им. В. И. Смирнова.
214000, г. Смоленск, пр-т им. Ю. Гагарина, 2.
ISBN 5-225-04388-7
ISIIIIIIffll
9 ||785225|,043889|