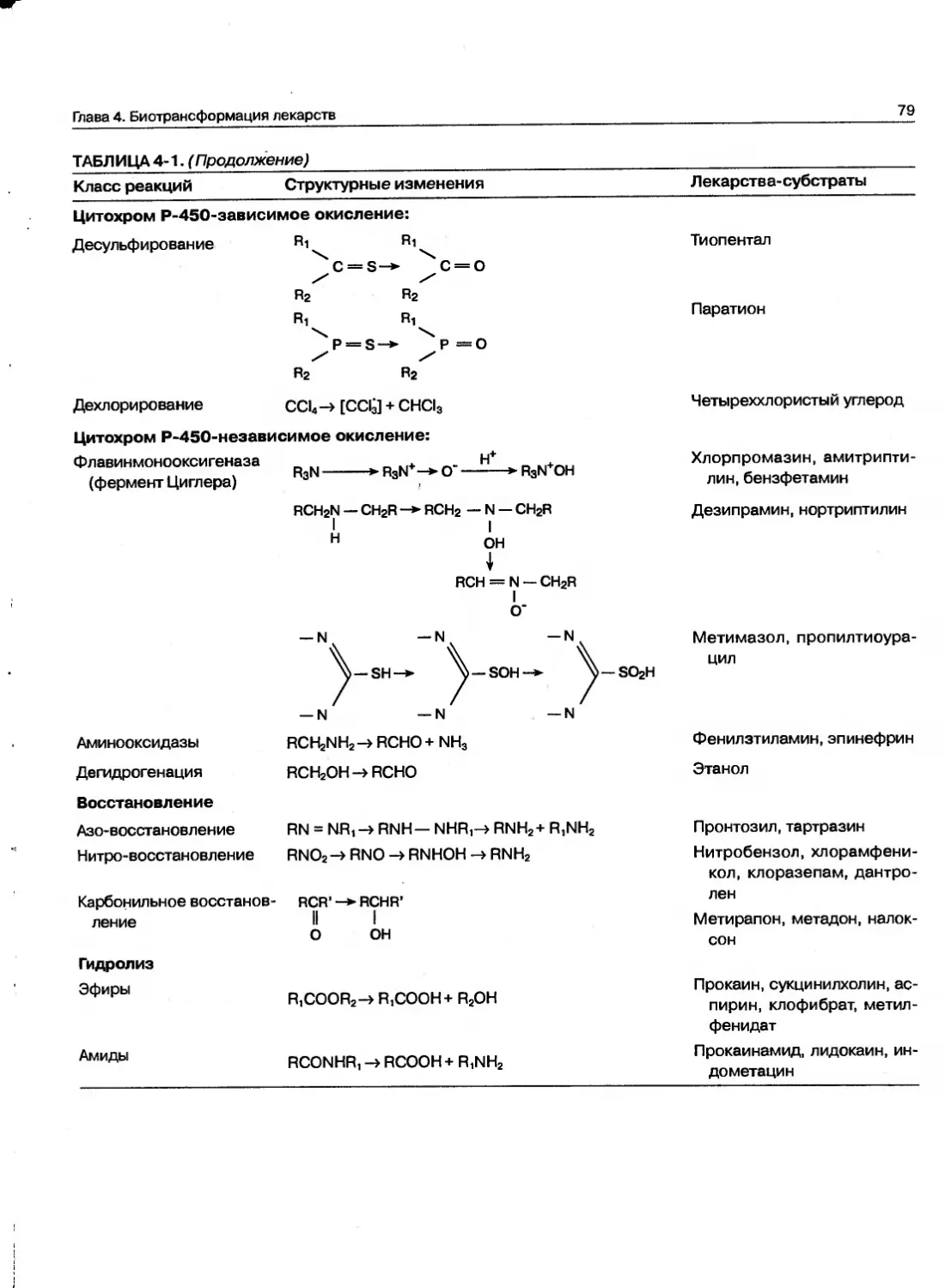
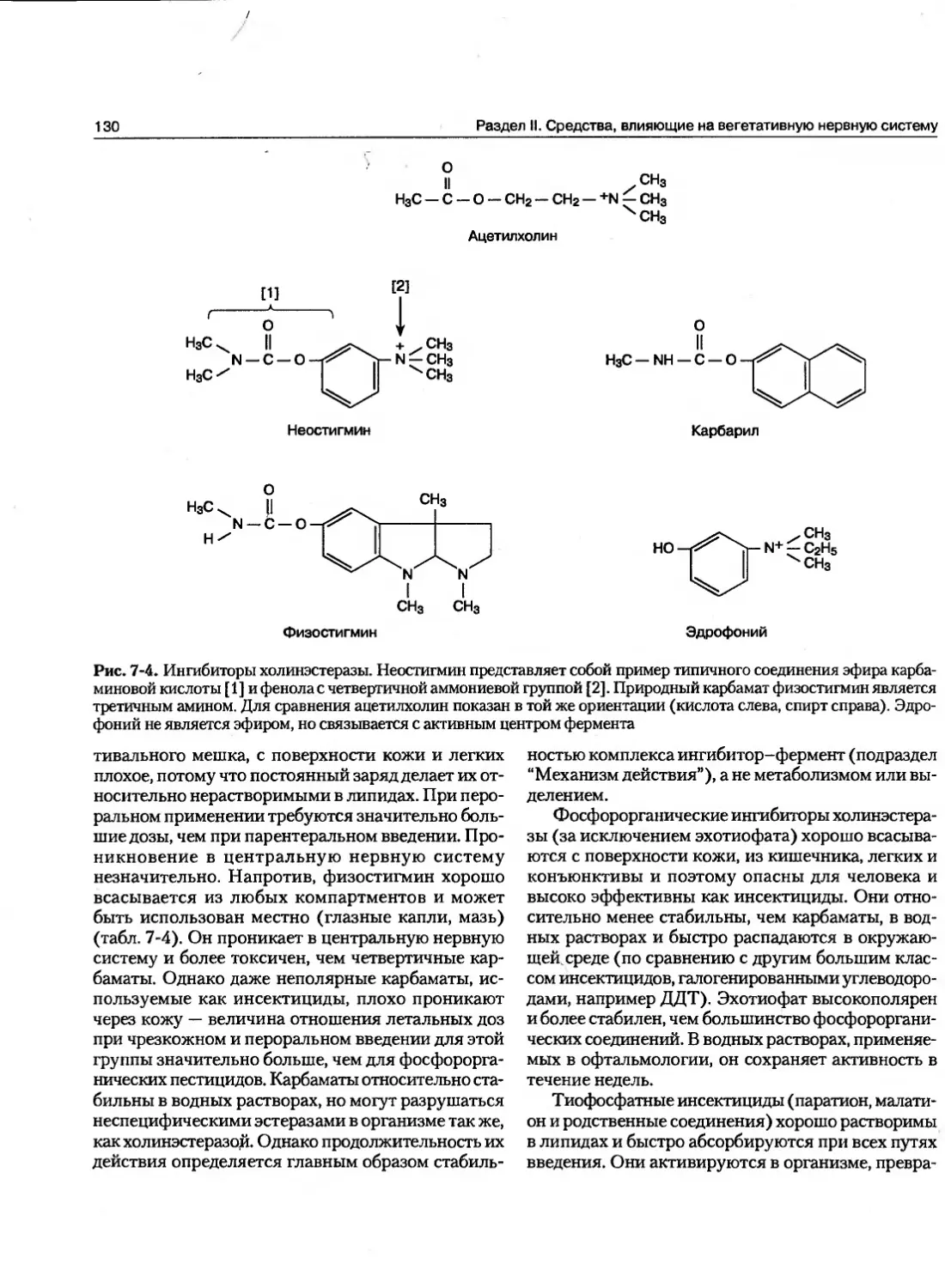
/




































































































































































































































































































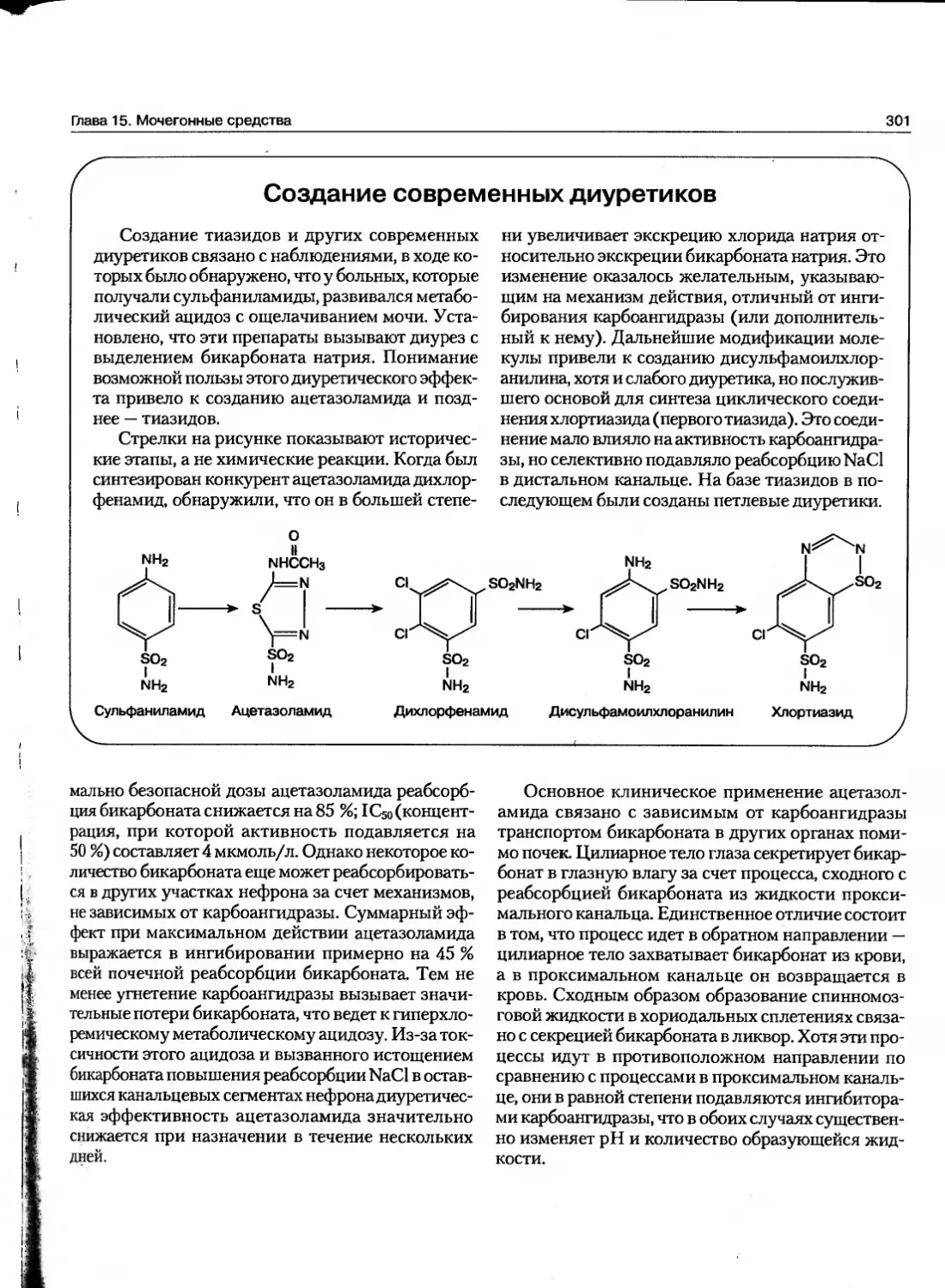

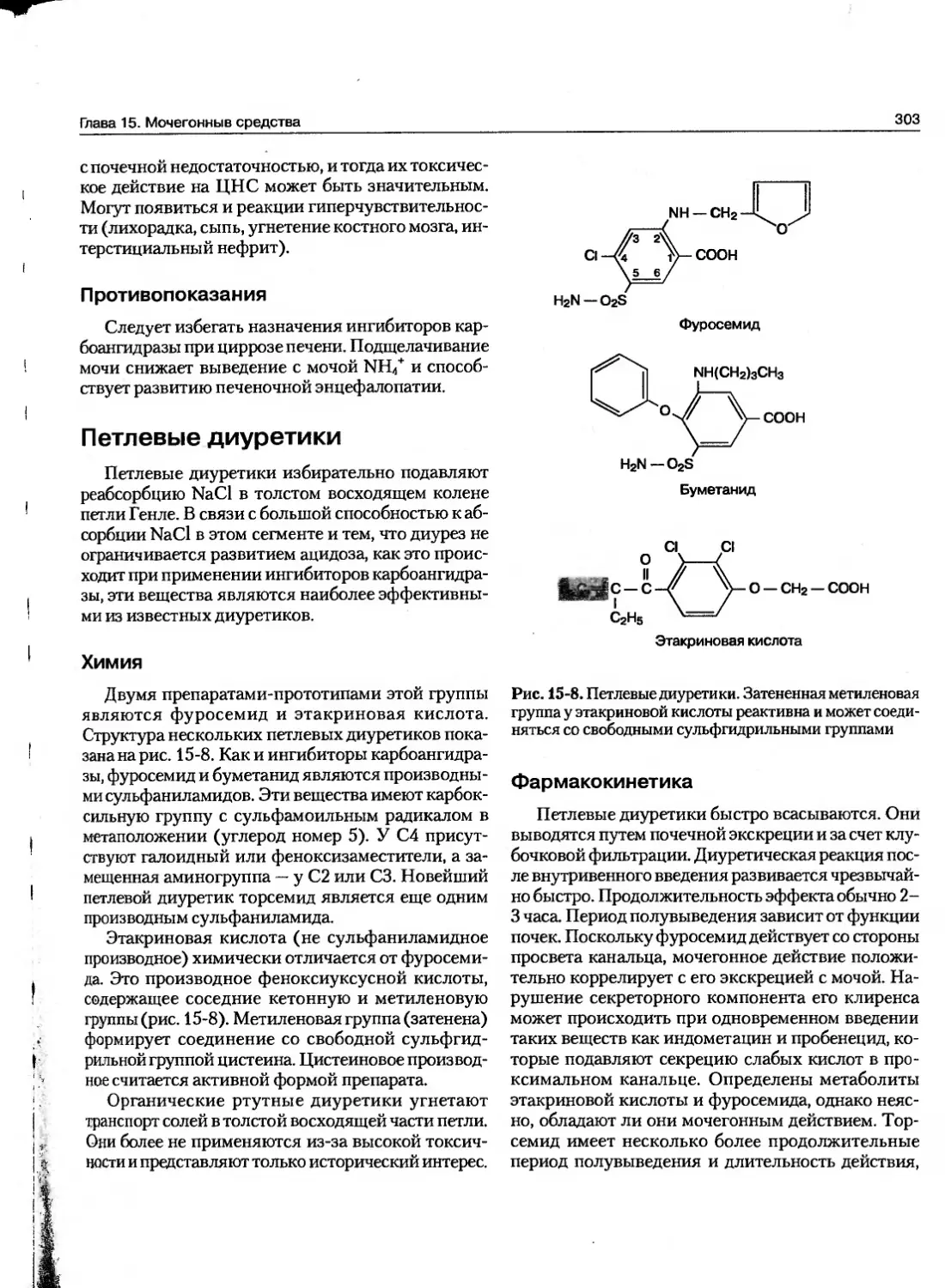



































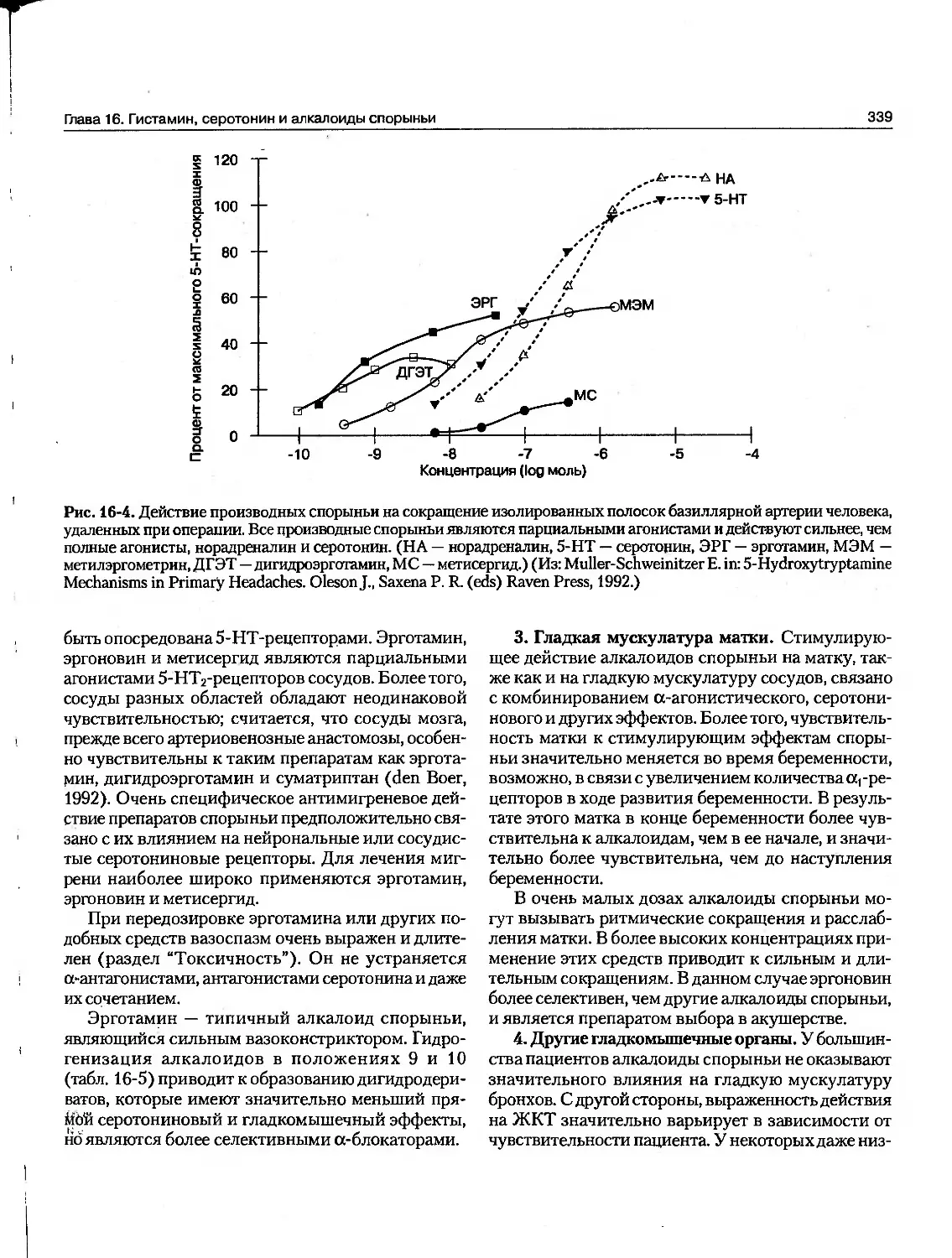

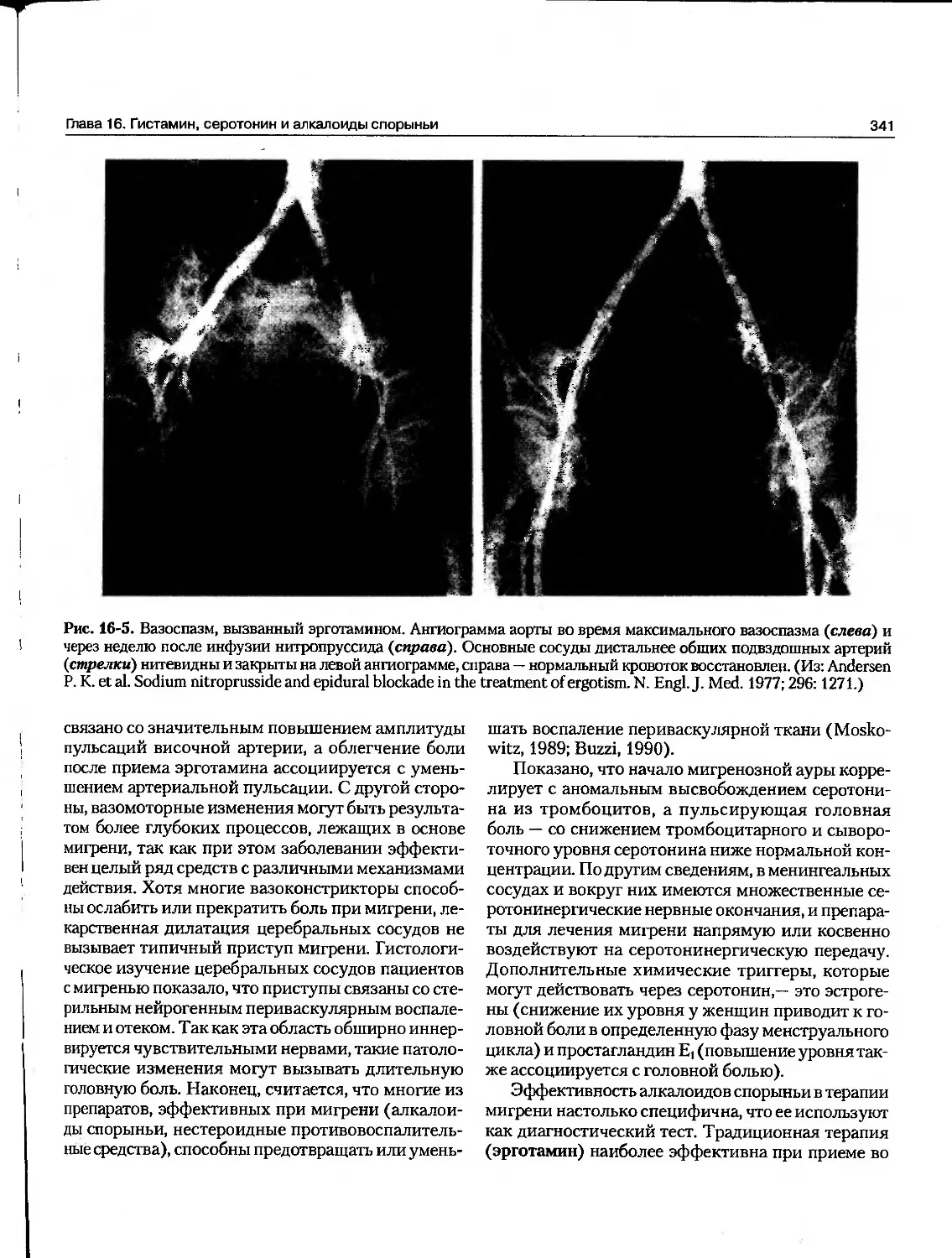































































































































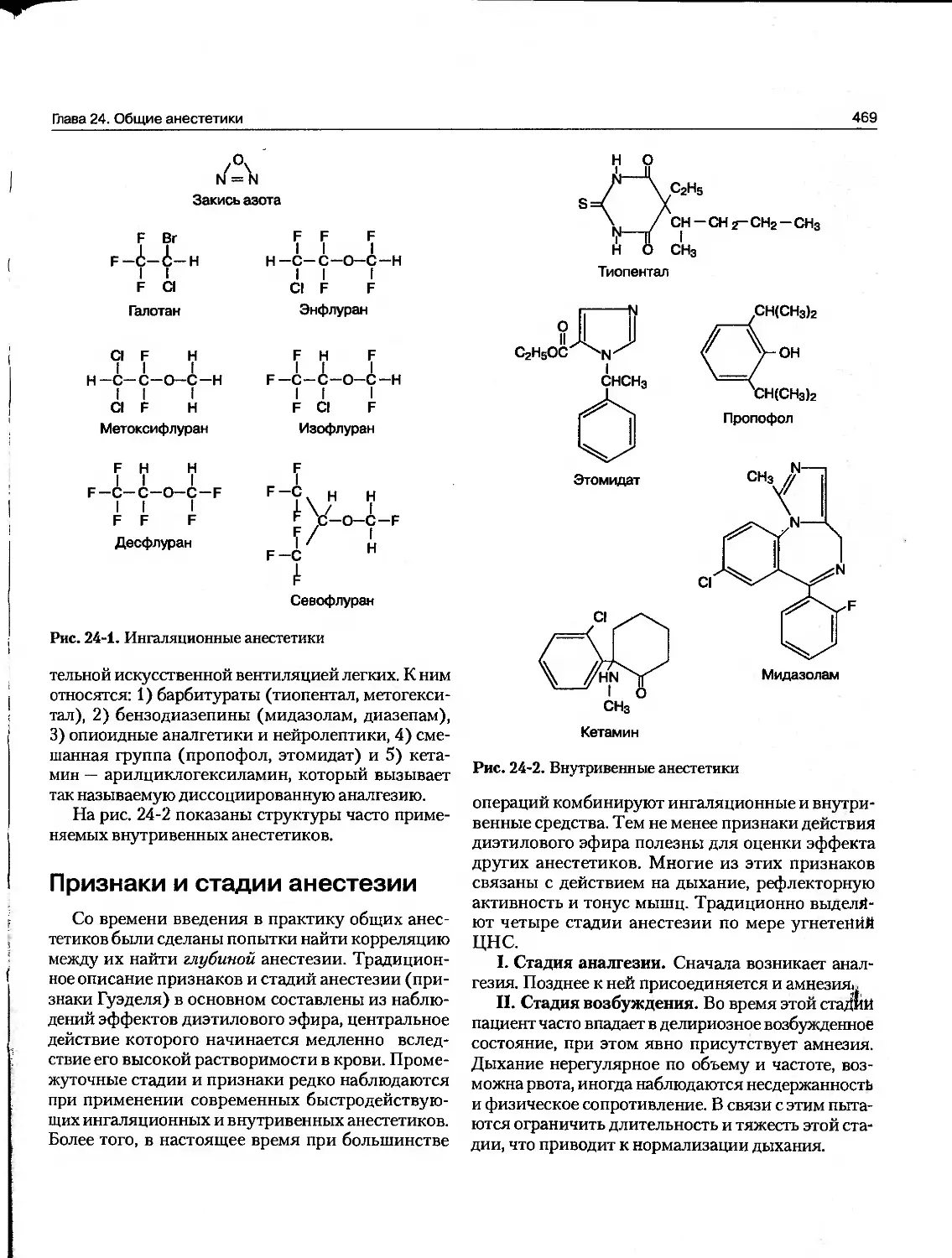































































































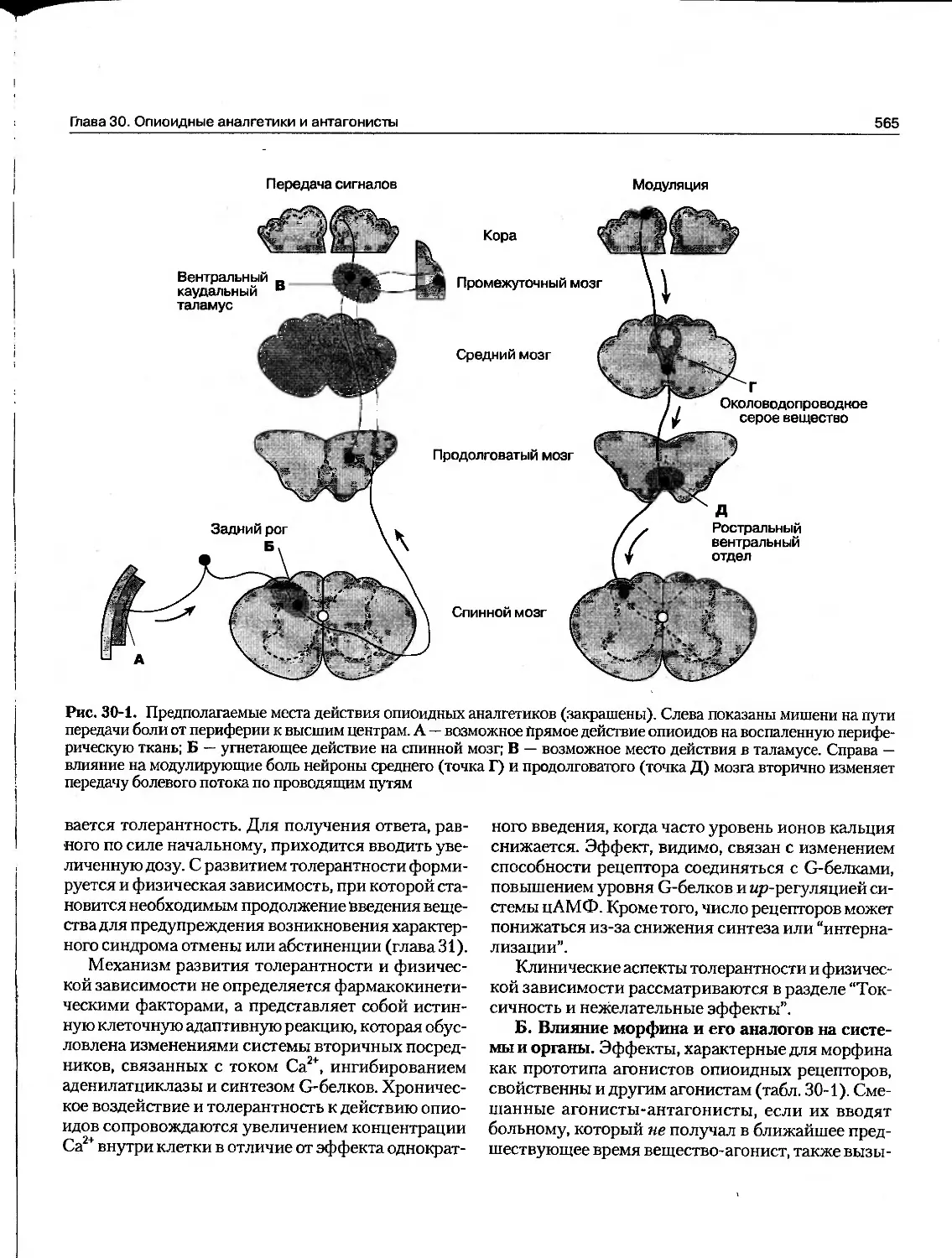
















































Текст
Раздел I
Основные принципы
Введение
Бертрам Г. Катцунг
Фармакологию можно определить как науку, изучающую вещества, которые взаимодействуют с живыми системами посредством химических механизмов, прежде всего путем связывания с регуляторными молекулами и активацией или ингибированием процессов, происходящих в организме. Это могут быть вещества, которые вводят для коррекции патологического процесса у пациента или с целью токсического воздействия на возбудителя заболевания. Терапевтическое применение лекарств может рассматриваться как главная область медицинской фармакологии, которую часто определяют как науку о веществах, используемых для предупреждения, диагностики и лечения заболеваний. Токсикология является разделом фармакологии, изучающим нежелательные эффекты веществ в отношении живых систем от индивидуальной клетки до сложных экосистем.
История
Доисторический человек, несомненно, знал о лечебных и токсических эффектах многих веществ растительного и животного происхождения. Наиболее ранние письменные источники Китая и Египта содержат перечень многих лекарств, включая некоторые малоизвестные в наши дни. Однако большинство из них не приносило пользу или даже вредило больному. Примерно за 2500 лет до н. э. были спорадические попытки ввести рациональные методы в медицину, но все они оказались неэффективными. В те времена еще не понимали необходимости наблюдения и эксперимента для объяснения биологических процессов, в том числе болезней.
Например, считалось, что болезни вызываются избытком желчи или крови в организме, что рану можно лечить, прикладывая бальзам к оружию, которым она нанесена и т. п.
Уверенность в необходимости наблюдения и эксперимента начинает возникать примерно к концу XVII столетия. Как только ценность этих методов в изучении болезней стала очевидной, врачи Великобритании и других стран Европы начали привлекать их для объяснения эффектов традиционных лекарств, используемых в собственной практике. Так начала развиваться предшественница фармакологии — фармакотерапия, т. е. учение о применении лекарств в медицине. Но пониманию механизма действия лекарств еще препятствовало отсутствие методов очистки активных действующих начал из лекарственного сырья и, даже в еще большей степени, отсутствие методов проверки гипотез о действии лекарств. Однако в конце XVIII — начале XIX вв. Франсуа Мажанди и позднее его ученик Клод Бернар положили начало развитию экспериментальной физиологии и фармакологии. В своих опытах они использовали животных. Достижения химии и дальнейшее развитие физиологии в XVIII и XIX столетиях заложили необходимый фундамент для понимания того, как действуют лекарства на уровне органов и тканей. Парадоксально, но реальные достижения базисной фармакологии в XIX в. сопровождались всплеском ненаучного продвижения на рынок под давлением производителей практически бесполезных “патентованных лекарств". Так продолжалось до тех пор, пока около 50 лет назад не были внедрены в медицину концепции рациональной терапии, особенно
концепция контролируемых клинических испытаний, что позволило более полно удовлетворять запросы терапии.
В это время началось бурное развитие исследований во всех областях биологии. По мере внедрения новых идей и новых технических достижений накапливалась информация об активности лекарств и биологическом субстрате их действия — рецепторе. Во второй половине текущего столетия были открыты многие принципиально новые группы лекарств и новые представители известных групп. За последние три десятилетия отмечается более быстрый рост информации о молекулярных основах действия лекарств. В настоящее время изучены механизмы действия многих препаратов, изолированы многочисленные рецепторы, изучена их структура и произведено клонирование. Эти и многие другие достижения обсуждаются в нашей книге.
Продолжается внедрение научных принципов в повседневную лечебную практику. В то же время пациенты, к сожалению, все еще получают большое количество неточной, неполной и ненаучной информации, касающейся фармакологических свойств химических веществ. Это привело к распространению огромного количества дорогих, малоэффективных, а подчас и вредных, средств и росту индустрии “альтернативной медицины”. Часть населения фактически отрицает медицину как науку, не понимая ее основ, и все нежелательные эффекты лекарств считает следствием врачебных ошибок.
Природа лекарств
В самом общем виде лекарство можно определить как любое вещество, которое вызывает изменение биологической функции за счет химического взаимодействия. В громадном большинстве случаев молекула лекарства взаимодействует со специфической молекулой биологической системы, которая выполняет регулирующую роль, т. е. является рецепторной молекулой. Природа рецепторов обсуждается более подробно в главе 2. В очень небольшом числе случаев лекарства, известные как химические антагонисты, могут взаимодействовать непосредственно с другими лекарствами, тогда как некоторые препараты (например, осмотически активные вещества) взаимодействуют почти исключительно с молекулами воды. Лекарства могут быть аналогами веществ, синтезируемых в организме (например, гормоны), или же веществами, которые
не имеют аналогов в организме, т. е. являются ксенобиотиками (от греческого “ксенос” — чужой). Яды — это тоже лекарства. Токсины обычно определяют как яды биологического происхождения, т. е. синтезируемые растениями или животными, в отличие от неорганических ядов типа свинца или мышьяка.
Для того чтобы взаимодействовать химически со своим рецептором, молекула лекарства должна иметь соответствующий размер, электрический заряд, форму и атомное строение. Более того, препарат часто вводят в место, удаленное от точки приложения действия, как например таблетку, принимаемую внутрь при головной боли. Следовательно, лекарство должно иметь необходимые свойства для транспорта от места введения к месту действия. Наконец, лекарственное вещество должно инактивироваться или экскретироваться из организма с определенной скоростью так, чтобы его эффект продолжался соответствующее время.
А. Физическая природа лекарств. Лекарство может быть твердым при комнатной температуре (например, аспирин, атропин), жидким (никотин, этанол) или газообразным (закись азота). Эти факторы часто определяют наилучший путь введения. Некоторые жидкие лекарства легко испаряются и могут использоваться ингаляционно, например галотан, амилнитрит. Наиболее распространенные пути введения рассмотрены в главе 3. В фармакологии представлены различные классы органических соединений — углеводороды, протеины, липиды и их составные части. Многие лекарства являются слабыми кислотами или основаниями. Этот факт важен для их судьбы в организме, поскольку различия pH в разных компартментах организма могут изменять степень ионизации таких лекарств.
Б. Размер молекул. Молекулярные размеры лекарств колеблются от- очень малых (ион лития, мол. м. 7) до очень больших (альтеплаза [t-PA] — белок с мол. м. 59 050). Однако большинство лекарств имеет мол. м. от 100 до 1000. Нижний уровень этого узкого диапазона, возможно, определяется требованиями специфичности действия. Чтобы хорошо соответствовать только одному типу рецепторов, молекула лекарства должна быть уникальна по форме, заряду и т. п., что предупредит связывание с другими рецепторами. Для достижения такого избирательного связывания молекула должна иметь мол. м. не менее 100. Верхняя граница мол. м. определяется, главным образом, необхо-
димостыо перемещения лекарства в организме (например, от места введения к месту действия). Лекарства, значительно превышающие по мол. м. 1000, не смогут легко диффундировать между ком-партментами (глава 3). Следовательно, очень крупные молекулы (обычно белки) должны вводиться непосредственно в компартмент, где они действуют. В случае с альтеплазой (ферментом, растворяющим тромб) лекарство вводят прямо в сосудистое русло путем внутривенной инфузии.
В. Реактивность лекарства и связь лекарства с рецептором. Лекарство взаимодействует с рецептором путем химических связей. Существуют три основных типа связей: ковалентные, электростатические и гидрофобные. Ковалентные связи очень прочны в физиологических условиях. Так, нелегко разорвать ковалентную связь, образованную между активированной формой феноксибензамина и а-рецептором для норадреналина (что ведет к блокаде рецептора). Блокирующий эффект феноксибензамина продолжается еще долго после того, как свободное лекарство покинет кровоток, и прекращается только после синтеза новых «-рецепторов, что занимает приблизительно 48 часов. Другой пример лекарств, образующих высокореактивные ковалентные связи — ДНК-алкилирующие агенты, применяемые для лечения рака, чтобы прервать деление клеток опухоли.
При взаимодействии лекарства и рецептора значительно более распространена электростатическая, а не ковалентная связь. Электростатические связи варьируют от относительно сильного соединения постоянно заряженных ионизированных молекул до более слабых водородных связей и очень слабых дипольных взаимодействий, таких как Вандер-Ваальсовы и подобные им силы. Электростатические связи слабее, чем ковалентные.
Гидрофобные силы обычно очень слабы и, возможно, важны для взаимодействия высоколипофильных лекарств с липидами клеточных мембран, а также для взаимодействия лекарств с внутренними стенками рецепторных “карманов”.
Специфическая природа конкретной связи лекарство-рецептор не так существенна по сравнению с тем фактом, что препараты, слабо связывающиеся со своими рецепторами, обычно более селективны, чем те, которые присоединяются посредством очень сильных связей. Это определяется тем, что слабые связи требуют очень точной “подгонки” лекарства к конфигурации рецептора для того, что
бы осуществилось взаимодействие. Только немногие лекарства могут так точно соответствовать определенной структуре рецепторов. Так, если мы желаем создать селективное короткодействующее лекарство для конкретного рецептора, нужно избегать высокореактивных молекул, которые образуют ковалентные связи, а вместо них выбирать молекулы, образующие слабые связи.
Некоторые вещества, которые почти абсолютно инертны в химическом смысле, тем не менее имеют выраженные фармакологические эффекты. Например, ксенон, относящийся к инертным газам, при повышенном давлении обладает анестезирующим действием.
Г. Форма молекулы лекарства. Форма молекулы лекарства должна быть такой, которая позволит ему связаться с рецептором. Оптимально, когда форма молекулы комплементарна форме рецептора таким же образом, как ключ комплементарен замку. Более того, феномен хиральности (стереоизомерия) столь обычен в биологии, что больше половины всех лекарств являются хиральными молекулами, т. е. имеют пары энантиомеров. Лекарства с двумя асимметричными центрами имеют четыре диастереомера, как например а, p-блокатор ла4 беталол. В большинстве случаев один из этих энантиомеров будет более эффективен, чем его зеркальный энантиомер, что связано с лучшей “подгонкой” к рецепторной молекуле. Например, 5(+) энантиомер парасимпатомиметического лекарства метахолина более чем в 250 раз активнее Л(-) энантиомера. Если представить рецептор в виде перчатки, в которую должна войти молекула лекарства, чтобы вызвать эффект, становится ясно, почему “левосторонние” лекарства будут более эффективны при связывании с рецептором для “левой руки”, чем их “правосторонние” энантиомеры.
Более активный энантиомер для одного типа рецепторов может быть менее активным для другого типа рецепторов, например для рецепторов, ответственных за некоторые нежелательные эффекты. Карведилол — лекарство, взаимодействующее с адренорецепторами, имеет один хиральный центр и, следовательно, два энантиомера (табл. 1-1). Один из этих энантиомеров, 5(-) изомер, является активным р-блокатором. /?(+) изомер в 100 раз слабее действует на p-рецептор. Однако эти изомеры примерно равноактивны как блокаторы «-рецепторов. Кетамин относится к внутривенным анестетикам. Его (+) энантиомер — более активный и менее
ТАБЛИЦА 1 -1. Константы диссоциации (Kd) энантиомеров и рацемата карведилола. Показатель Kd характеризует концентрацию лекарства для 50 % насыщения рецепторов и обратно пропорционален аффинитету (сродству) лекарства к рецептору ’
Форма карведилола Величина, обратная сродству к а-рецепторам (Kd, нмоль/л) Величина, обратная сродству к 0-рецепторам (Kd, нмоль/л)
Л(+) энантиомер 14 45
S(-) энантиомер 16 0.4
Рацемат 11 0.9
’Данные Ruffolo R. R. et al. (Eur. J. Pharmacol., 1990, 38, S82).
токсичный анестетик, чем (-) энантиомер. Тем не менее в качестве лекарства до сих пор используют рацемическую смесь.
Наконец, в связи с тем, что ферменты обычно стереоселективны, один энантиомер лекарственного вещества часто имеет большее сродство к ферменту, метаболизирующему лекарство, чем другой. В результате энантимомеры могут весьма отличаться друг от друга по длительности действия.
К сожалению, большинство исследований клинической эффективности и элиминации лекарств у человека выполнено с применением рацемических смесей лекарств, а не их раздельных энантиомеров. В настоящее время только около 45 % хиральных лекарств, используемых в клинике, доступны как активные энантиомеры — остальные продаются только как рацемические смеси. В результате многие больные получают дозы лекарств, которые на 50 % или более неактивны или даже токсичны. Однако отмечается повышение интереса как на научном, так и на законодательном уровнях, к производству хиральных лекарств в виде их активных энантиомеров.
Д. Рациональный дизайн лекарств. Под целенаправленным синтезом препарата (рациональный дизайн лекарств) понимают создание молекулярной структуры лекарства на основе информации о строении соответствующего ему рецептора. До недавнего времени ни один рецептор не был изучен столь детально, чтобы реализовать подобный подход. Вместо этого лекарства создавались путем случайного тестирования веществ или модификации уже известных лекарств (глава 5). Однако в последние два десятилетия многие рецепторы были выделены и охарактеризованы. Некоторые применяемые сейчас лекарства были созданы путем молекулярного синтеза, основанного на знании трехмерной структуры рецептора. Сейчас доступны компью
терные программы, которые пошагово оптимизируют структуру лекарства, чтобы“подогнать”его к рецептору. Чем больше становится известно о структуре рецептора, тем более осуществимым будет рациональный дизайн лекарств.
Взаимодействие лекарства и организма
Взаимодействия между лекарством и организмом условно делят на два типа. Действие лекарств на организм называют фармакодинамикой. Она детально рассмотрена в главе 2. Фармакодинамические свойства определяют группу, к которой относится лекарство, и часто играют основную роль в решении вопроса о том, является ли эта группа подходящей для лечения больного с данным симптомом или заболеванием. Влияние организма на лекарство называют фармакокинетикой, она рассмотрена в главах 3 и 4. Фармакокинетические процессы определяют абсорбцию, распределение и элиминацию лекарств в организме и имеют огромное практическое значение для выбора и применения конкретного лекарства у определенного пациента, например у больного с нарушенной функцией почек. Следующий раздел дает краткое введение в фармакодинамику и фармакокинетику.
Принципы фармакодинамики
Как уже указывалось, большинство лекарств должны связаться с рецептором, для того чтобы вызвать эффект. Однако на молекулярном уровне связывание лекарства — это только первый шаг из подчас очень сложной последовательности этапов.
А. Типы взаимодействия “лекарство-рецептор”. Лекарство-агонист связывается с рецептором и активирует его, что прямо или косвенно ведет к развитию фармакологического эффекта. Не
которые рецепторы объединены с эффекторным механизмом в одну молекулу и, таким образом, связывание с лекарством вызывает прямой эффект, например открытие ионного канала или активацию фермента. Другие соединены с молекулой эффектора через одну или несколько промежуточных сопряженных молекул. Некоторые типы систем сопряжения лекарство-рецептор—эффектор обсуждаются в главе 2. Лекарство, являющееся фармакологическим антагонистом, путем связывания с рецептором предупреждает его связывание с другими молекулами. Например, блокаторы ацетилхолиновых рецепторов, такие как атропин, являются антагонистами, потому что они закрывают доступ ацетилхолину и сходным с ним лекарствам-агонистам к рецептору. Эти агенты уменьшают эффект ацетилхолина и холиномиметиков в организме. Напротив, лекарства, которые предупреждают связывание ацетилхолина с ацетилхолинэс-теразой (ингибиторы холинэстеразы) замедляют нормальный метаболизм эндогенного ацетилхолина, чем значительно усиливают эффект этого нейротрансмиттера. Таким образом, ингибиторы холинэстеразы обладают в организме больного действием, сходным с эффектом агонистов холи-норецепторов.
Б. Длительность действия лекарства. Прекращение действия лекарства на уровне рецептора обусловлено одной из нескольких причин. В некоторых случаях эффект продолжается, пока лекарство занимает рецептор, и автоматически прекращается при диссоциации. Однако часто действие может сохраняться и после диссоциации лекарства, потому что часть сопряженных молекул еще находится в активированном состоянии. В случае лекарств, которые связываются с рецептором ковалентными связями, эффект может продолжаться до тех пор, пока не синтезируются новые рецепторы, заменяющие инактивированные, как это уже было описано для феноксибензамина. Наконец, некоторые рецепторно-эффекторные системы имеют механизм десенситизации для предупреждения избыточной активации при длительном связывании молекулы препарата с рецептором (более подробно об этом написано в главе 2).
В. Рецепторы и инертные места связывания. Для того чтобы выполнять функцию рецептора, эндогенная молекула должна обладать, во-первых, селективностью во взаимодействии с определенными лигандами (молекулами лекарств) и, во-вторых,
при связывании лекарство должно так повлиять на эндогенную молекулу, чтобы изменить функцию биологической системы (клетки, ткани и т. п.). Первое свойство необходимо, чтобы избежать постоянной активации рецептора путем случайного связывания с многочисленными лигандами. Второе свойство, очевидно, необходимо, чтобы лиганд вызвал фармакологический эффект. В организме содержится множество молекул, способных связывать лекарства, но не все эти эндогенные молекулы являются регуляторными. Связывание лекарства с нерегуляторной молекулой, например альбумином плазмы, не вызывает заметных изменений функции биологических систем, поэтому такой тип связи можно назвать инертным. Однако это связывание не является совершенно безразличным, так как влияет на распределение препарата в организме и может изменять количество свободного лекарства в кровеносном русле. Оба этих фактора важны для фармакокинетики (главы 1 и 3).
Принципы фармакокинетики
При практическом использовании лекарство после введения в организм удобным способом должно достигнуть больного органа. Только в редких случаях его можно прямо доставить к ткани-мишени, например путем местной аппликации противовоспалительного вещества на воспаленную кожу или слизистую оболочку. В других случаях препарат может быть введен внутривенно и попадать с током крови непосредственно в сосуды отдаленного органа-мишени. Обычно лекарство вводят в один из компартментов организма, например в кишечник, откуда оно должно перемещаться к месту действия в другом компартменте, например в мозгу. Для этого необходимо, чтобы лекарство абсорбировалось (всасывалось) в кровь из места введения и доставлялось к месту действия, проникая через различные барьеры, разделяющие эти компартмен-ты. Так, для препарата, который назначается внутрь для оказания действия на ЦНС, эти барьеры включают ткани, образующие стенку кишечника, стенки капилляров кишечника и “гематоэнцефалический барьер” стенок капилляров мозга. Наконец, после того как был вызван эффект, лекарство должно быть элиминировано (выведено) из организма с определенной скоростью путем метаболической инактивации, экскреции или путем сочетания этих процессов.
А. Проникновение. Проникновение лекарств осуществляется с помощью четырех основных механизмов. Наиболее распространена пассивная диффузия в водной или липидной среде. Однако для некоторых лекарств, особенно тех, чьи молекулы слишком велики для простой диффузии, характерна активная диффузия.
1. Диффузия в водной среде. Эта диффузия осуществляется в пределах больших водных ком-партментов организма (интерстициальное пространство, цитозоль и т. п.) через плотные контакты эпителиальных мембран и эндотелий сосудов посредством водных пор, пропускающих молекулы с мол. м. 20 000-30 0001. Водная диффузия молекул лекарств обычно определяется градиентом концентрации вещества и описывается законом Фика (пункт Б). Молекулы лекарств, которые связываются с белками плазмы (чаще всего с альбуминами), не проникают через эти водные поры. Если молекула заряжена, на ее движение также влияют электрические поля (например, мембранный потенциал и в некоторых отделах нефрона — транстубулярный потенциал).
2. Липидная диффузия. Диффузия через липидные среды является наиболее важным лимитирующим фактором проникновения лекарств из-за большого количества липидных барьеров, которые разделяют компартменты организма. Поскольку эти липидные барьеры отделяют водные компартменты, коэффициент распределения липид: вода для лекарства определяет, насколько легко молекула передвигается между водной и липидной средой. В случае слабых кислот и слабых оснований (которые получают или теряют заряженный протон в зависимости от pH среды) способность перемещаться из водной среды в липидную и наоборот зависит от pH среды, поскольку заряженные молекулы связывают воду. Соотношение растворимой в липидах и растворимой в воде форм для слабых кислот или оснований выражается уравнением Гендерсо-на-Хассельбаха.
3. Специальные переносчики. Для некоторых веществ, которые важны для функционирования клеток, но слишком велики или слишком малора
1 Капилляры мозга и тестикул характеризуются отсутствием пор, способствующих водной диффузии многих лекарственных молекул в ткань. Эти ткани, следовательно, “защищены” и являются недоступными для многих лекарств, циркулирующих в крови.
створимы в липидах, чтобы успешно диффундировать через мембраны (пептиды, аминокислоты, глюкоза), существуют специальные молекулы-переносчики. Эти переносчики осуществляют движение по типу активного транспорта или облегченной диффузии. В отличие от пассивной диффузии такие транспортные системы при насыщении могут ингибироваться. Поскольку многие лекарства являются природными пептидами, аминокислотами, сахарами или напоминают их, они могут переноситься указанными транспортными системами через мембраны.
4. Эндоцитоз и экзоцитоз. Некоторые вещества столь велики, что могут попасть в клетку только при помощи эндоцитоза — процесса, при котором вещество обволакивается клеточной мембраной и переносится в клетку путем отщепления образовавшейся везикулы. Вещество может затем высвобождаться внутрь цитозоля посредством разрушения мембраны везикул. Этот процесс участвует в переносе железа и витамина Bt2, которые образуют комплексы с соответствующими протеинами. Обратный процесс (экзоцитоз) ответственен за секрецию целого ряда веществ из клетки. Например, многие нейротрансмиттерные субстанции сохраняются в связанной с мембранами форме в везикулах нервных окончаний для защиты от метаболического разрушения в цитоплазме. Соответствующая активация нервных окончаний вызывает соединение везикул с клеточной мембраной и выброс их содержимого во внеклеточное пространство.
Б. Закон диффузии Фика. Пассивный перенос молекул по концентрационному градиенту описывается законом Фика:
Ток (количество молекул за единицу времени) =
= (Ci-C2)x
Площадь х Коэффициент проницаемости Толщина ’
где Ct — более высокая концентрация, Сг — более низкая концентрация, площадь — площадь, через которую осуществляется диффузия, коэффициент проницаемости — мера подвижности молекул лекарства в среде, где происходит диффузия, и толщина — длина пути, по которому идет диффузия. В случае липидной диффузии коэффициент распределения липид: вода является главным показателем подвижности лекарства, так как он опреде
ляет, насколько легко лекарство проникает через липидные мембраны из водной среды.
В. Ионизация слабых кислот и слабых оснований. Электростатический заряд ионизированных молекул притягивает водные диполи и способствует образованию полярных, относительно водорастворимых и нерастворимых в липидах, комплексов. Поскольку липидная диффузия зависит от высокой растворимости в липидах, ионизация лекарств может существенно уменьшить их способность проникать через мембраны. Очень большое количество применяемых в практике лекарств являются слабыми кислотами или слабыми основаниями (табл. 1-2). Применительно к лекарствам слабую кислоту лучше всего определить как нейтральную молекулу, которая может обратимо диссоциировать на анион (отрицательно заряженную молекулу) и протон (ион водорода). Например, аспирин диссоциирует следующим образом:
С8Н7О2СООН С8Н7О2СОО’ + Н*
Нейтральный Аспирин Протон аспирин анион
Лекарство-слабое основание — можно определить как нейтральную молекулу, которая способна образовать катион (положительно заряженную молекулу) при присоединении протона. Например, ан-тималярийное средство пириметамин участвует в следующих процессах ассоциации-диссоциации: Ci2H11CIN3NH3+^C12Hi1CIN3NH2+ Н+
Пириметамин Нейтральный Протон катион пириметамин
Заметьте, что протонированная форма слабой кислоты нейтральна, следовательно, более липофильна, а для слабого основания нейтральной является непротонированная форма. По закону действия масс равновесие в этих реакциях сдвигается влево в кислой среде (низкое значение pH, избыток протонов) и вправо — в основной среде. Уравнение Гендерсона-Хассельбаха устанавливает связь между соотношением протонированной и непротониро-ванной форм слабых кислот и оснований, рК молекулы и pH среды следующим образом:
Протонированная форма _
У Непротонированная форма а
Это уравнение применимо как к кислым, так и к основным лекарствам. Чем ниже величина pH по отношению к рКа, тем больше будет фракция лекарства в протонированной форме. Так как незаряженная форма более липофильна, в кислой среде большая часть слабых кислот будет находиться в форме, растворимой в липидах, и напротив, для основных лекарств растворимость в липидах будет выше в щелочной среде.
Наиболее важное приложение этого принципа связано с управлением экскрецией лекарства почками. Почти все препараты фильтруются в клубочках. Если лекарство проходит через канальцы в липофильной форме, то его значительная часть будет реабсорбироваться по механизму пассивной диффузии. Когда нужно ускорить выведение вещества, важно предупредить его реабсорбцию из канальцев. Этого можно достигнуть регуляцией pH мочи, чтобы способствовать переходу лекарства в более иони-аированную фсфму, как. показано на \жс. I-1. В результате лекарство будет “улавливаться” в моче. Таким образом, слабые кислоты будут экскретироваться быстрее в щелочной моче, а слабые основания — в кислой. Другими жидкими средами организма, в которых pH отличается от pH крови и может происходить захват или реабсорбция лекарства, являются содержимое желудка и тонкого кишечника, грудное молоко, глазная влага, секреты влагалища и предстательной железы (табл. 1-3).
Как показано в табл. 1 -3, большое число лекарств является слабыми основаниями. Большинство из этих слабых оснований содержит аминогруппы. Азот в нейтральных аминах имеет три атома, связанных с ним, и пару свободных электронов. Три атома могут представлять собой один углерод и два водорода (первичный амин), два углерода и один водород (вторичный амин) или три углеродных атома (третичный амин). Каждая из этих трех форм способна обратимо связывать протон с неподелен-ными электронами, образуя четвертую углеродазотную связь (четвертичный амин). Однако четвертичный амин постоянно заряжен и не имеет свободных электронов, с которыми может обратимо
Первичные Вторичные Третичные
Четвертичные
Н R R R,
R :N: R :М: R :N: R :М: R
Й Й Й Й
ТАБЛИЦА 1 -2. Константы ионизации некоторых распространенных лекарств
Лекарство рка1 Лекарство рКа1 Лекарство рКа’
Слабые кислоты Слабые основания Слабые основания
Ацетаминофен 9.5 Адреналин 8.7 Метадон 8.4
Ацетазоламид 7.2 Амилорид 8.7 Метамфетамин 10.0
Ампициллин 2.5 Амфетамин 9.8 Метилдопа 10.6
Аспирин 3.5 Атропин 9.7 Метисергид 6.6
Хлортиазид 6.8; 9.42 Бупивакаин 8.1 Метопролол 9.8
Хлорпропамид 5.0 Хлордиазепоксид 4.6 Морфин 7.9
Кромолин 2.0 Хлорохин 10.8; 8.42 Никотин 7.9; 3.12
Этакриновая кислота 3.5 Хлорфенирамин 9.2 Норадреналин 8.6
Фуросемид 3.9 Хлорпромазин 9.3 Пентазоцин 9.7
Ибупрофен 4.4; 5.22 Клонидин 8.3 Фенилэфрин 9.8
Леводопа 2.3 Кокаин 8.5 Физостигмин 7.9; 1.82
Метотрексат 4.8 Кодеин 8.2 Пилокарпин 6.9; 1.42
Метилдопа 2.2; 9.22 Циклизин 8.2 Пиндолол 8.8
Пеницилламин 1.8 Дезипрамин 10.2 Прокаинамид 9.2
Пентобарбитал 8.1 Диазепам 3.3 Прокаин 9.0
Фенобарбитал 7.4 Дигидрокодеин 8.8 Промазин 9.4
Фенитоин 8.3 Дифенгидрамин 9.0 Прометазин 9.1
Пропилтиоурацил 8.3 Дифеноксилат 7.1 Пропранолол 9.4
Салициловая кислота 3.0 Эфедрин 9.6 Псевдоэфедрин 9.8
Сульфадиазин 6.5 Эрготамин 6.3 Пириметамин 7.0
Сульфапиридин 8.4 Флуфенаэин 8.0; 3.92 Хинидин 8.5; 4.42
Теофиллин 8.8 Гуанетидин 11.4; 8.32 Скополамин 8.1
Толбутамид 5.3 Гидралазин 7.1 Стрихнин 8.0; 2.32
Варфарин 5.0 Имипрамин 9.5 Тербуталин 10.1
Слабые основания Изопротеренол 8.6 Тиоридазин 9.5
Альбутерол (сальбутамол) 9.3 Канамицин 7.2 Толазолин 10.6
Аллопуринол 9.4; 12.32 Лидокаин 7.9
Алпренолол 9.6 Метараминол 8.6
' рКа соответствуетзначению pH, при котором концентрации ионизированной и неионизированной форм находятся в равновесии.
2 Более одной ионизированной группы.
связываться протон. Следовательно, первичные, вторичные и третичные амины могут подвергаться обратимому протонированию с варьированием их липофильности в зависимости от pH, но четвертичные амины всегда находятся в плохо растворимой в липидах заряженной форме.
Группы лекарств
Рассмотрение каждого существенного факта, касающегося сотен лекарств, которые упоминаются в этой книге, было бы нереально и, к счастью, в большинстве случаев не нужно. Почти все препа
раты, которые применяются в настоящее время, могут быть объединены примерно в 70 групп. Многие из лекарств в пределах каждой группы весьма сходны по фармакодинамическому эффекту, а подчас и по фармакокинетическим свойствам. Для большинства групп можно выделить одно лекарство-прототип, которое имеет типичные для группы наиболее важные характеристики. Это позволяет классифицировать другие лекарства в группе как варианты прототипа, поэтому только лекарство-прототип имеет смысл рассматривать подробно, а для остальных целесообразно обсуждать лишь отличия от прототипа.
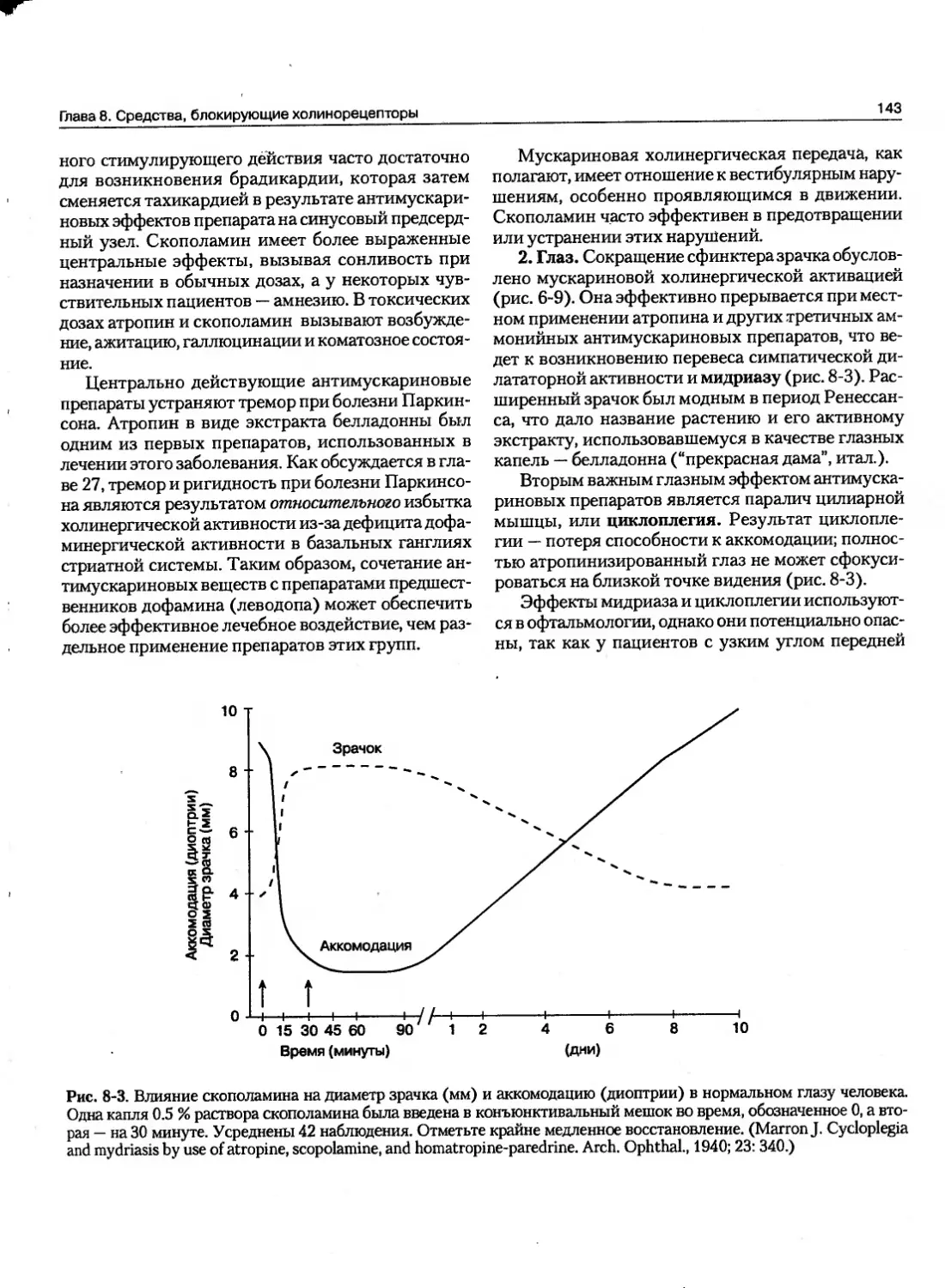
Рис. 1-1. Экскреция слабого основания (пириметамина) с мочой, имеющей более кислую реакцию, чем плазма. В гипотетическом случае, представленном на рисунке, способная к диффузии незаряженная форма лекарства уравновешена с обеих сторон мембраны, но общая концентрация (заряженная и незаряженная формы) в моче почти в восемь раз выше, чем в крови
Источники информации
Студентам, которые хотят ознакомиться с фармакологией с целью подготовки к экзаменам, рекомендуется книга Pharmacology: Examination and Board Review Катцунга и Тревора (Appleton and Lange, 1995). Практическим врачам, которым необходим быстрый доступ к справочному пособию по имеющимся лекарствам и их дозированию, удобнее использовать Drug Therapy под редакцией автора этой главы (Appleton and Lange, 1991).
Ссылки в конце каждой главы этой книги приведены с целью предоставления информации, наиболее важной для усвоения конкретной темы. Три фундаментальных руководства (перечисленные ниже) представляют более полные описания и перечни ссылок:
Bowman W. С., Rand М. J. Textbook of Pharmacology, 2nd ed. Blackwell Scientific, 1980. (A large textbook with a chemical and comparative pharmacology orientation.)
Gilman A. G. et al. (eds) Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 8th ed. Pergamon, 1990. (A large textbook with a medical orientation.)
Patt W. B., Taylor P. Principles of Drug Action. The Basis of Therapeutics, 3rd ed. Churchill Livingstone, 1990. (A specialized text emphasizing gene-
ТАБЛИЦА 1-3. pH-зависимость способности жидких сред организма к "захвату" лекарств
Жидкая среда Диапазон pH Диапазон соотношений концентраций в жидкости и крови для сульфадиазина (кислота, рКа 6.5)' Диапазон соотношений концентраций в жидкости и крови для пириметамина (основание.рКаТ.О)1
Моча 5.0-8.0 0.12-4.65 72.24-0.79
Грудное молоко 6.4-7.62 0.2-1.77 3.56-0.89
Содержимое тощей
и подвздошной кишок 7.5-8.Об3 1.23-3.54 0.94-0.79
Содержимое желудка 1.92-2.592 0.11“ 85.993-18.386
Секрет предстательной железы 6.45-7.42 0.21-1 3.25-1.0
Влагалищный секрет 3.4-4.23 0.114 2.848-452
' Соотношение протонированной и непротонированной форм лекарства рассчитано для каждого крайнего значения pH; pH крови принята за 7.4. Например, соотношение концентраций сульфадиазина в моче составляет 0.12 при pH мочи 5.0 и 4.65 при pH мочи 8.0. Таким образом, сульфадиазин значительно более эффективно захватывается и экскретируется в щелочной моче.
2LentnerC. (ed.) Geigy Scientific Tables, vol.1,8th ed., Ciba Geigy, 1981.
3 Bowman W. C., Rand M. J: Textbook of Pharmacology. 2nded., Blackwell, 1980.
4 Незначительные изменения в соотношениях за пределами физиологических величин pH.
ral principles: receptor concepts, dose-response principles, pharmacokinetics, and biochemical toxicology.)
Ответы на специальные вопросы, связанные с базисными или клиническими исследованиями, можно получить в руководствах по общей фармакологии и в клинических изданиях. Три периодических издания можно рекомендовать как особо полезные источники текущей информации о лекарствах: The New EnglandJournal of Medicine, который публикует много оригинальных исследований, посвященных лекарствам, и обзоры по проблемам фармакологии; The Medical Letter on Drugs and Therapeutics (короткие критические обзоры о новых и уже известных средствах лекарственной терапии) и Drugs, представляющий собой обширные обзоры по лекарствам и группам лекарств.
Следует также упомянуть другие источники информации, доступные в США. “Карманные вкладыши” являются обобщением информации, которую производитель должен представить в документах на продажу лекарств. Physician’s Desk Reference (PDR) является сборником инструкций по приме
нению, публикующимся с дополнениями два раза в год; Facts and Comparisons - наиболее полное информационное издание в формате блокнота с ежемесячным обновлением. Можно рекомендовать такие издания как USPDI (vol. 1. Drug Inf ormation for the Health Care Professional) и AMA Drug Evaluations. Карманный вкладыш включает в себя краткое описание фармакологии продукта, содержит много практически важной информации, а также используется как средство разделения ответственности за нежелательные реакции между производителем и врачом-практиком. Поэтому производитель указывает, как правило, все зарегистрированные токсические эффекты вне зависимости от их частоты.
Полезная и объективная книга, представляющая данные о токсичности лекарств,— это Drug Interactions. Наконец, FDA организовала компьютерный бюллетень, который содержит новости, касающиеся недавно одобренных препаратов, запретов на использование, предупреждений и т. п. Этот бюллетень доступен с помощью персонального компьютера, оборудованного средствами связи и стандартным модемом, по номеру 800-222-0185.
Лекарственные рецепторы и фармакодинамика 2
Генри Р. Бурн, Джеймс М. Робертс
Терапевтические и токсические эффекты лекарств зависят от их превращений в организме пациента. В большинстве случаев лекарственные препараты взаимодействуют со специфическими макромолекулами, при этом изменяются их биохимические или биофизические свойства. Этой идее почти сто лет. В ее основе лежит понятие о рецепторе — компоненте клетки, который взаимодействует с лекарством и инициирует цепочку биохимических превращений, вызывающих в конечном итоге лекарственный эффект.
Первоначально предположение о существовании рецепторов основывалось на наблюдениях химической и физиологической специфичности эффектов лекарств. Так, Эрлих отмечал, что некоторые синтетические органические вещества имеют характерный антипаразитарный эффект, тогда как другие лишены его, несмотря на структурное сходство. Лэнгли обнаружил, что кураре не действует на мышечное сокращение, вызываемое электрической стимуляцией, но блокирует сокращение, вызываемое никотином. Эти элементарные представления послужили отправной точкой для развития фармакодинамики, центральной проблемой которой является изучение эффектов лекарств и их рецепторных механизмов. Концепция рецепторов применительно к эндокринологии, иммунологии и молекулярной биологии стала незаменимой для объяснения многих аспектов биологической регуляции. В настоящее время рецепторы выделяют и изучают их структуру, открывая таким образом путь к точному пониманию молекулярных основ действия лекарств.
Кроме объяснения биологических основ действия, концепция рецепторов имеет чрезвычайно важные практические последствия для создания лекарств и для принятия решений по их применению в клинической практике. Положения, которые
рассматриваются в данной главе, создают основы для понимания механизмов действия и клинического применения лекарств. Они могут быть сформулированы следующим образом.
Рецепторы определяют количественные связи между дозой или концентрацией лекарства и фармакологическими эффектами. Аффинитет рецептора к лекарству определяет концентрацию лекарства, необходимую для образования значительного числа комплексов лекарство-рецептор. Общее количество рецепторов часто лимитирует максимальный эффект, который может вызвать лекарство.
Рецепторы ответственны за избирательность действия лекарств. Размер, форма и электрический заряд молекулы лекарства определяют, будет ли она и с какой легкостью (авидностью) связываться с определенным рецептором среди огромного числа химически разнородных мест связывания, имеющихся в клетке, в организме животного или пациента. Соответственно, модификации химической структуры могут существенно повысить или уменьшить аффинитет нового лекарства к различным классам рецепторов и тем самым изменить его терапевтические и токсические эффекты.
На уровне рецепторов реализуется действие фармакологических антагонистов. Многие лекарства и эндогенные химические вещества с сигнальными свойствами, например гормоны, регулируют функцию макромолекул рецепторов как агонисты, то есть изменение функции рецептора является более или менее прямым результатом связывания с ним. Типичные фармакологические антагонисты связываются с рецепторами, непосредственно не изменяя их функций. Таким образом, эффект типичных антагонистов в клетке или организме больного полностью зависит от их способности предупреждать связывание рецепторов с молекулами аго-
Как открывают рецепторы?
Поскольку сегодняшние представления о рецепторах являются основой создания новых лекарств завтра, важно знать, как происходит открытие новых рецепторов. Этот процесс состоит из нескольких ключевых этапов, приведенных на рис. 2-1. Процесс определения нового рецептора (стадия 1, рис. 2-1) начинается с изучения связи между структурой и активностью группы веществ на общепринятой модели фармакологического эффекта. По характеру связывания радиоактивных лигандов можно предсказать молекулярный состав и аффинность предполагаемого рецептора, что открывает пути для выбора способов его биохимической очистки. Анализ очищенного рецепторного белка позволяет выяснить число субъединиц, их размер и (иногда) дает ключ к пониманию его функционирования (например, стимулирование агонистами рецепторов для инсулина и многих факторов роста вызывает их аутофосфорилирование по тирозиновым остаткам).
Эти “классические” шаги по идентификации рецепторов в настоящее время используются как подготовительный этап в рамках новой мощной экспериментальной стратегии, нацеленной на клонирование сегмента ДНК, кодирующего рецептор (стадии 2-5 на рис. 2-1). Стержнем этой стратегии является способность идентифицировать ДНК-последовательность искомого рецептора в репрезентативном пуле кДНК (комплементарных к иРНК ДНК- последовательностей, полученных с помощью обратной траскрип-тазы и экспрессированных в определенной клетке или ткани). Для выявления соответствующей ДНК исследователь использует как инструмент биохимические и функциональные свойства рецепторного белка. Так, при помощи антител к очищенному рецепторному белку или, используя нуклеотидную последовательность, реконструированную по аминокислотной последовательности рецептора, можно отделить бактериальные колонии, содержащие предполагаемую рецепторную кДНК от колоний, содержащих иные кДНК. В первом случае происходит связывание антител с рецепторным антигеном, эк
спрессированным в бактерии (2А), во втором — гибридизация с рецепторной ДНК. Кроме того, популяцию кДНК можно экспрессировать в ооцитах лягушек или клетках других позвоночных, а затем определить искомую рецепторную кДНК по сигнальной функции белка-рецептора (2В) или по его способности связываться со специфическим лигандом (2Г).
Как только предполагаемая рецепторная кДНК выделена, она “узаконивается” путем тщательного сравнения функции и биохимических свойств рекомбинантного белка с эндогенным рецептором, который исходно дал толчок к поиску (ЗА), После расшифровки последовательности оснований рецепторной ДНК(ЗБ) реконструируется аминокислотная последовательность рецепторного белка. Она сравнивается с аналогичной последовательностью известных рецепторов, после чего заявляют об идентификации нового рецептора (шаг 4).
Молекулярное клонирование кДНК, кодирующей новый рецептор, позволяет получить значительно больше информации, чем при идентификации рецептора “классическим” путем. Выявленная аминокислотная последовательность почти всегда напоминает таковую ранее откры-
еакция
Идентификация кДНК А. Антиген •«-------
Б. ДНК-гибридизация В. Сигнальные функции Г. Связывание с лигандом Д. Полимеразная цепная реакция
Связывание---->Очистка белка)
А.Класс рецептора Б. Общая
< последовательность ДНК.
г Предполагаемая рецепторная кДНК А. Экспрессия, функциональный тест
I__Б. Последовательность____________
Рис. 2-1. Методы, используемые для открытия и описания рецепторов
овый рецептор! )
тых рецепторов. Поэтому часто исследователи могут сразу отнести новый рецептор к уже известному классу. По структуре рецептора можно судить о том, как он работает: является ли он рецепторной тирозинкиназой, семикратно пересекающим мембрану рецептором, сопряженным с G-белком, или др. Расшифровка последовательности ДНК дает инструмент для идентификации клеток и тканей, которые экспрессируют иРНК, кодирующую новый рецептор. Экспрессия кДНК в культуре клеток дает неограниченный источник рекомбинантного рецепторного белка для точного биохимического анализа, тестирования агонистического и антагонистического типов связывания и создания новых лекарств.
Наконец (шаг 5), рецепторная ДНК сама по себе является средством идентификации еще
большего числа рецепторов. Рецепторы определенного класса или подкласса содержат высокоустойчивые области сходных или идентичных аминокислотных последовательностей. Поэтому ДНК-последовательности, соответствующие этим областям, могут использоваться для нахождения последовательностей родственных, но потенциально новых рецепторов путем ДНК-ДНК гибридизации (2Б), или запуском полимеразной цепной реакции, направленной на амплификацию рецепторной ДНК (2Д). С помощью этих “зондов” можно клонировать ДНК, кодирующую рецептор, лиганд которого неизвестен (рецептор-“сирота”). Соответствующий лиганд затем определяют по связыванию с рекомбинантным рецептором и по сигнальной функции.
ниста, что приводит к блокаде биологического ответа. Некоторые весьма полезные в клинической медицине лекарства являются фармакологическими антагонистами.
Макромолекулярная природа лекарственных рецепторов
До недавнего времени о химическом строении рецепторов для большинства лекарств и даже о самом их существовании судили косвенно, на основе анализа структуры лекарств. Однако сейчас многие лекарственные рецепторы выделены, очищены и охарактеризованы биохимическими методами. В дополнении “Как открывают рецепторы?” описаны некоторые методы, с помощью которых обнаруживают и анализируют рецепторы. Большинство из них являются белками: структура полипептидов обеспечивает необходимое разнообразие, требуемую специфичность формы и определенный электрический заряд.
Наиболее изученные лекарственные рецепторы — регуляторные белки, опосредующие действие эндогенных химических сигнальных молекул, таких как нейротрансмиттеры, аутакоиды и гормоны, а также многих лекарств. Молекулярная структура и биохимические механизмы этих рецепторов
рассмотрены в разделе, “Сигнальные механизмы и действие лекарств”.
К другим классам белков-рецепторов лекарств, относятся ферменты, которые могут быть ингибированы (реже активированы) при связывании с лекарствами (например, дигидрофолатредуктаза — рецептор для противоопухолевого лекарства метотрексата); транспортные белки (например, мембранный рецептор для сердечных гликозидов — Na+, К+-АТФаза); структурные белки (например, тубулин — рецептор для противовоспалительного средства колхицина).
В настоящей главе рассматриваются три аспекта функционирования лекарственных рецепторов в порядке возрастания сложности.
1) Детерминация количественной связи между концентрацией лекарства и фармакологическим ответом. С этой точки зрения рецепторы рассматриваются как простые образования, которые характеризуются способностью к связыванию лекарственных лигандов и определенным количеством в клетках или тканях-мишенях.
2) Участие в реализации сигнальных механизмов, являющихся мишенями для важнейших лекарств. В данном аспекте рецепторы рассматривают как сложные молекулы, структура и биохимическая функция которых позволяет объяснить зависимость между концентрацией и эффектом пре
парата, а также избирательность его фармакологического действия.
3) Ключевая роль в развитии терапевтического и токсического действия лекарств в организме больного. Это наиболее сложный вопрос о значении рецепторов в обеспечении избирательности действия лекарств, зависимости между дозой лекарства и его клиническими эффектами (в частности, терапевтической эффективностью в сравнении с токсичностью).
Связь между концентрацией лекарства и эффектом
Зависимость между дозой лекарства и эффектом, наблюдаемым в клинике, может быть весьма сложной. Однако в тщательно контролируемых системах in vitro связь между концентрацией лекарства и его эффектом зачастую проще и может быть описана с математической точностью. В первую очередь мы проанализируем именно эту идеализированную зависимость, потому что она лежит в основе всех более сложных соотношений между дозой и эффектом, которые наблюдаются, когда лекарство дается пациенту.
Кривые “концентрация-эффект” и связывание агонистов с рецепторами
У интактного животного или пациента физиологический ответ на малые дозы лекарства обычно возрастает прямо пропорционально дозе. Однако при увеличении дозы прирост ответной реакции снижается и в конечном счете может быть достигнута доза, при которой не происходит дальнейшего увеличения ответа. В идеальной системе или in vitro связь между концентрацией и эффектом описывается гиперболой (рис. 2-2 А) в соответствии с уравнением:
Emax хС
С + ЕС50
где Е — эффект, наблюдаемый при концентрации С, Е1пах — максимальный эффект, который может вызывать лекарство, и ЕС50 — концентрация лекарства, при которой наблюдается эффект, равный 50 % от максимального.
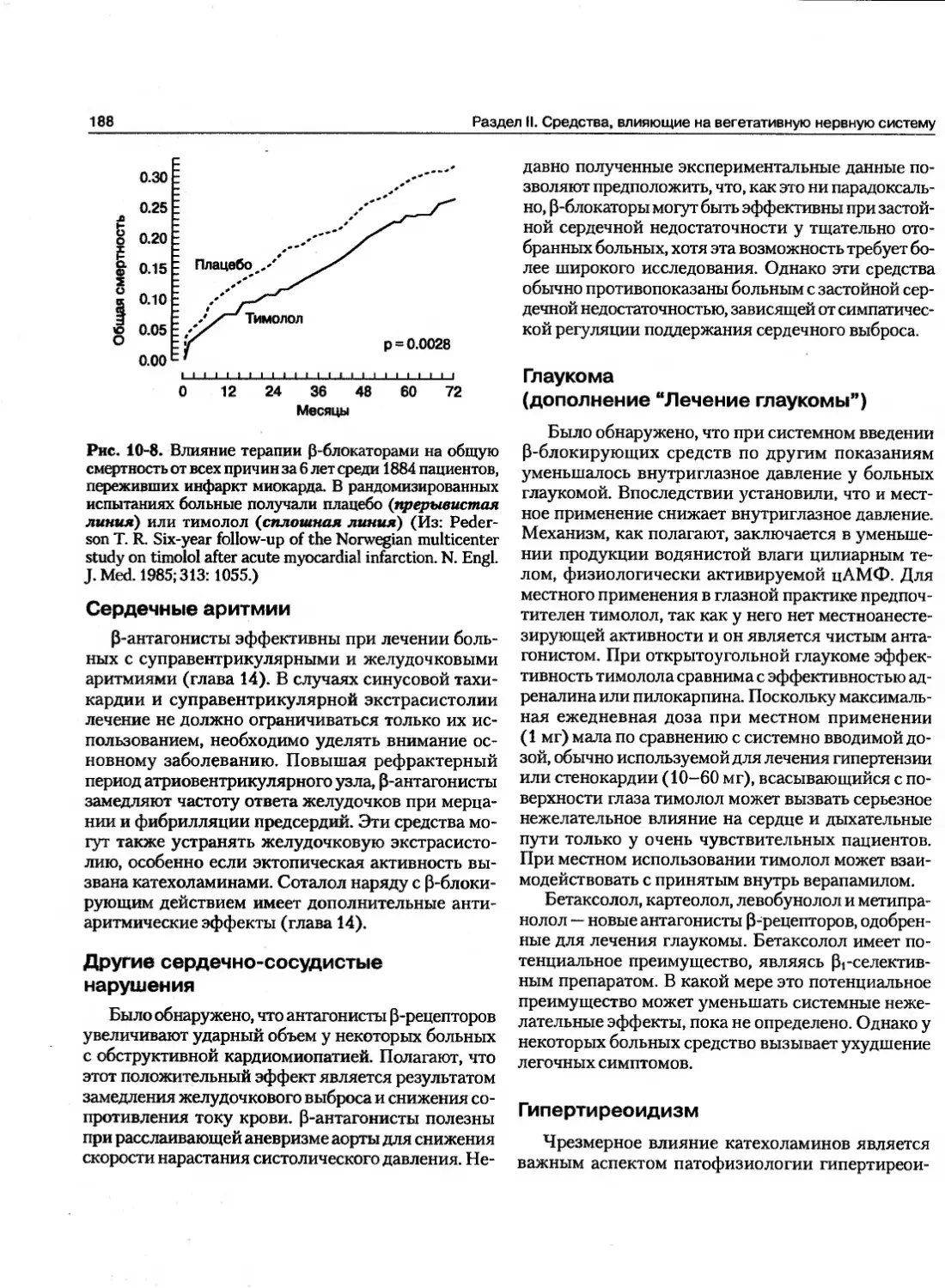
Эта гиперболическая зависимость напоминает закон действия масс, который описывает связывание между двумя молекулами с заданной величиной сродства (аффинитета). Такое сходство наводит на мысль, что лекарства-агонисты действуют именно путем связывания с рецепторами (“оккупации”). Связывание характеризуется определенным сродством лекарства к рецептору. С появлением радиоактивных лигандов (как агонистов, так и ан
Рис. 2-2. Связь между концентрацией лекарства и его эффектом (А) или количеством связанного с рецептором вещества (Б). Концентрации лекарства, при которых его эффект или связывание с рецепторами составляют половину ет максимальных, обозначены ЕС50 и KD соответственно
тагонистов) эти представления были подтверждены на ряде систем “лекарство-рецептор”. В этих системах связь между количеством лекарственного вещества, соединенного с рецептором (В), и концентрацией свободного вещества (С), изображенная на рис. 2-2Б, выражается аналогичным уравнением:
12 Bmax X С C + Kd
где Втах обозначает общую концентрацию рецепторных сайтов (сайтов, связывающихся с лекарственным веществом при бесконечно большой концентрации свободного вещества). KD (равновесная константа диссоциации) обозначает концентрацию сво
бодного вещества, при которой степень связывания составляет половину от максимально возможной. Эта константа обратна показателю аффинитета рецептора: при малой KD степень связывания лекарства с рецептором высока, и наоборот.
Заметим, что ЕС50 может быть равна Ко, но не всегда, что еще будет обсуждаться.
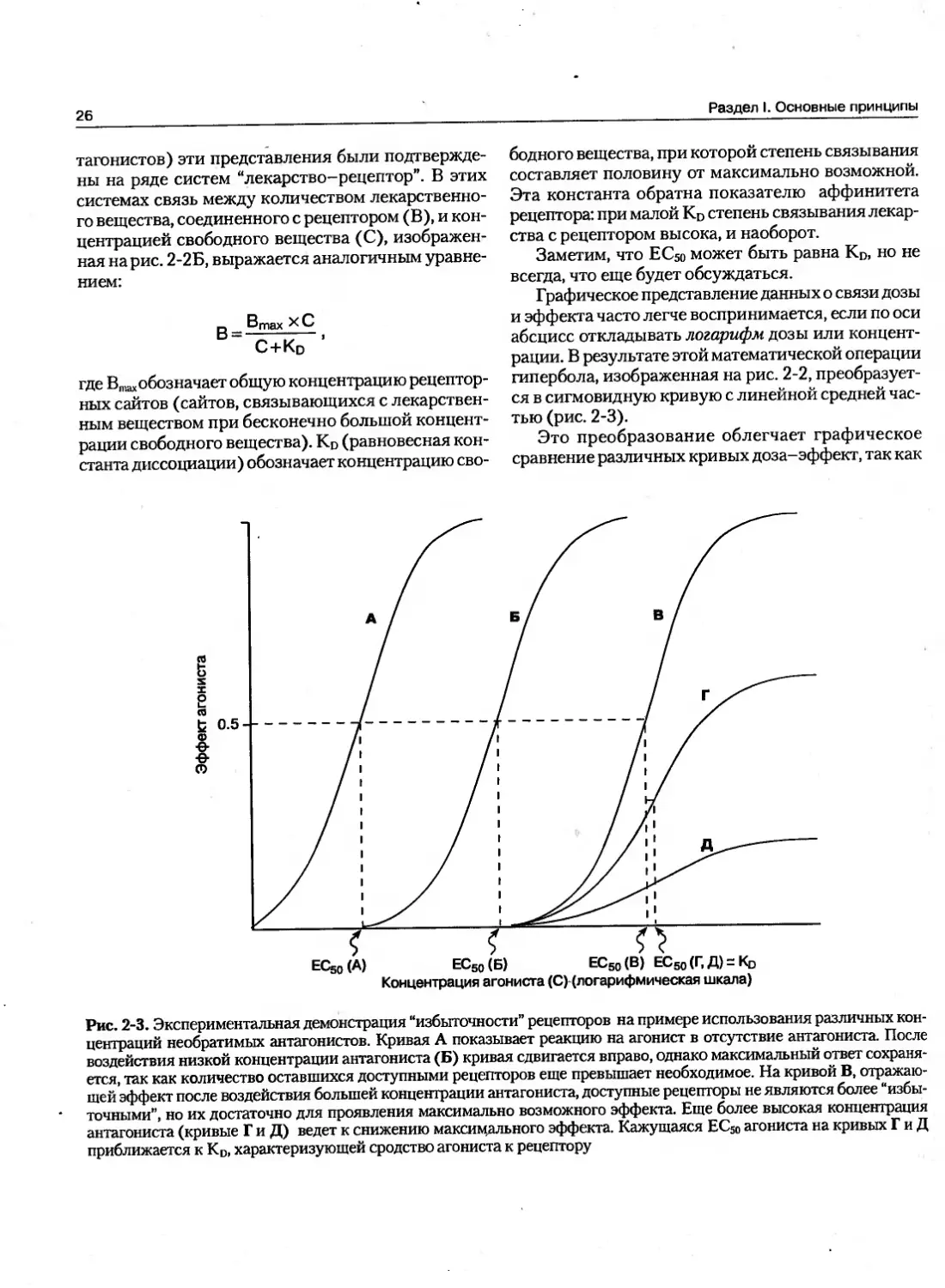
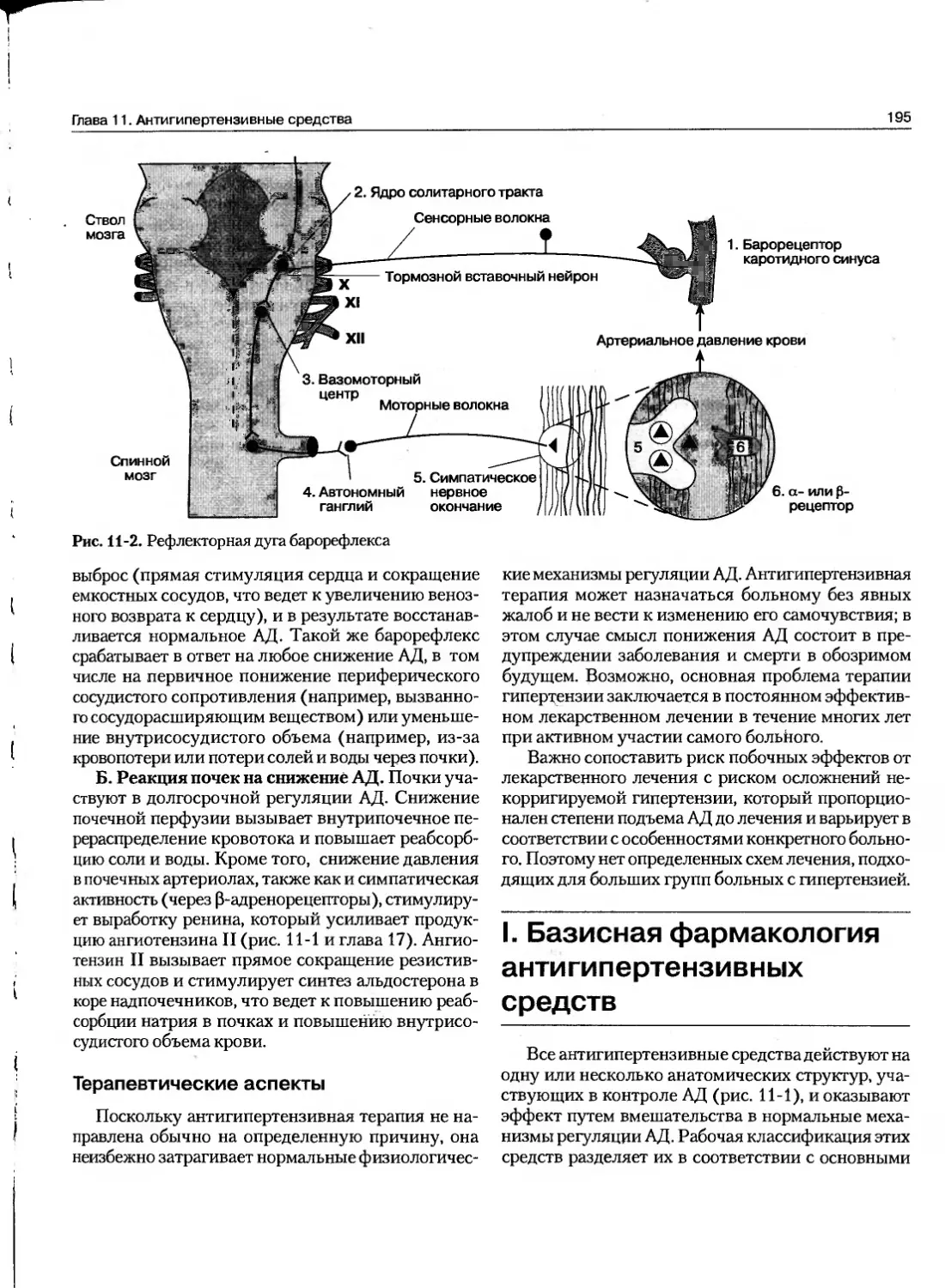
Графическое представление данных о связи дозы и эффекта часто легче воспринимается, если по оси абсцисс откладывать логарифм дозы или концентрации. В результате этой математической операции гипербола, изображенная на рис. 2-2, преобразуется в сигмовидную кривую с линейной средней частью (рис. 2-3).
Это преобразование облегчает графическое сравнение различных кривых доза-эффект, так как
Рис. 2-3. Экспериментальная демонстрация “избыточности” рецепторов на примере использования различных концентраций необратимых антагонистов. Кривая А показывает реакцию на агонист в отсутствие антагониста. После воздействия низкой концентрации антагониста (Б) кривая сдвигается вправо, однако максимальный ответ сохраняется, так как количество оставшихся доступными рецепторов еще превышает необходимое. На кривой В, отражающей эффект после воздействия большей концентрации антагониста, доступные рецепторы не являются более “избыточными”, но их достаточно для проявления максимально возможного эффекта. Еще более высокая концентрация антагониста (кривые Г и Д) ведет к снижению максимального эффекта. Кажущаяся ЕС50 агониста на кривых Г и Д приближается к KD, характеризующей сродство агониста к рецептору
способствует удлинению шкалы концентраций при низких значениях (при которых эффект изменяется быстро) и уплотняет (укорачивает) шкалу при высоких концентрациях, при которых эффект изменяется медленно. Это преобразование не имеет какого-либо специального биологического или фармакологического значения.
Сопряжение рецептора с эффектором и “избыточные” рецепторы
Когда лекарство-агонист связывается с рецептором, происходящие конформационные изменения являются лишь первым из множества последующих событий, обычно требующихся для получения фармакологического эффекта. Промежуточные процессы, происходящие между этапом связывания с рецептором и ответной реакцией на лекарство, часто обозначают термином сопряжение (coupling). Относительная эффективность сопряжения оккупации рецепторов и ответной реакции частично зависит от начального изменения конформации рецептора после связывания. Так, действие полных агонистов в большей степени сопряжено с оккупацией рецепторов, чем эффекты парциальных (неполных) агонистов. Большую роль играют также биохимические события, которые преобразуют связывание рецепторов в ответную клеточную реакцию.
Высокую эффективность взаимодействия рецептор-эффектор можно рассматривать и как результат феномена “избыточности” рецепторов. Об “избыточности” рецепторов для данной фармакологической реакции говорят в том случае, когда максимальный ответ может быть вызван агонистом в концентрации, которая не сопровождается оккупацией всех имеющихся в наличии рецепторов. Избыточные рецепторы качественно не отличаются от неизбыточных. Они не замаскированы и не являются недоступными: в случае их связыванйя возникает фармакологический эффект. В эксперименте избыточность может быть продемонстрирована путем использования необратимых антагонистов для предупреждения связывания агониста с частью имеющихся рецепторов. Даже в этом случае агонист в высоких концентрациях способен вызывать максимально возможный эффект (рис. 2-3). Например, максимальный инотропный ответ сердечной мышцы на катехоламины может быть зарегистрирован при условии, когда даже 90 % Р-рецепторов оккупированы необратимым антаго
нистом. Следовательно, миокард содержит большое количество избыточных рецепторов.
Как можно оценивать феномен “избыточности” рецепторов? В некоторых случаях (если вещество влияет на некоторые регуляторные рецепторы) биохимический механизм более или менее понятен. В этой ситуации эффект активации рецептора, например связывание ГТФ с метаболитом, может быть существенно более продолжительным, чем взаимодействие агониста с рецептором (раздел о G-белках и вторичных посредниках). В данном случае “избыточность” рецепторов является временной в том смысле, что ответ, инициированный связыванием лиганда и рецептора, продолжается дольше, чем процесс связывания сам по себе.
В других случаях, когда биохимические механизмы неизвестны, мы можем представить количественную избыточность рецепторов. Если концентрация или количество нерецепторного клеточного компонента лимитирует сопряжение связывания рецепторов и ответной реакции, максимальный ответ может быть получен без оккупации всех рецепторов. Рис. 2-4 иллюстрирует представление об избыточности рецепторов в этом смысле и помогает понять, как чувствительность клетки или ткани к определенной концентрации агониста может зависеть не только от сродства рецептора и агониста (характеризуемого KD), но также и от общей концентрации рецепторов. Чувствительность может быть выражена величиной ЕСзо, т. е. концентрацией агониста, при которой наблюдается эффект, равный половине от максимального. Кр при взаимодействии агониста с рецептором определяет, какая доля от общего числа рецепторов (В/В1пах) будет связана при данной свободной концентрации (С) агониста независимо от концентрации рецепторов:
В С
Bmax C + Kd
Представим себе клетку с четырьмя рецепторами и четырьмя эффекторами (как на рис. 2-4). В этом случае число эффекторов не лимитирует максимальный ответ и рецепторы не избыточны по количеству. Соответственно агонист в концентрации, равной KD, будет оккупировать 50 % рецепторов и активировать половину эффекторов, что приведет к полумаксимальному эффекту (два рецептора будут стимулировать два эффектора). Теперь вообразим, что число рецепторов возросло в десять раз
Рис. 2-4. Избыточные рецепторы увеличивают чувствительность к лекарству. Слева (А) показан случай, когда свободная концентрация агониста равна концентрации, соответствующей KD: этого достаточно, чтобы связать 50 % из четырех имеющихся рецепторов с образованием двух комплексов рецептор-агонист. (Примечание: когда концентрация агониста равна KD, занята половина рецепторов. Помните, что В/В,„ах- С/( С + Ко).) Оккупация этих двух рецепторов агонистом изменяет их конформацию таким образом, что они активируют две эффекторных молекулы, вызывая ответную реакцию. Поскольку два из четырех эффекторов стимулированы агонист-рецепторными комплексами, величина реакции составляет 50 % от максимальной.
Справа (Б) плотность рецепторов увеличена в 10 раз (показаны не все рецепторы), а KD для связывания агониста с рецептором осталась неизменной. Теперь незначительной концентрации свободного агониста (равной 0.05 от KD) достаточно, чтобы оккупировать два рецептора и, соответственно, активировать две эффекторных молекулы. Таким образом, эффект составит 50 % от максимального, несмотря на то, что концентрация агониста значительно ниже, чем Ко
(до 40), а общее число эффекторов не изменилось. В этом случае большинство рецепторов избыточны. Следовательно, значительно более низкая концентрация агониста будет достаточна для оккупации двух из 40 рецепторов (5 % всех рецепторов) и активации двух из четырех эффекторов (полумакси-мальный ответ). Таким образом, можно изменить чувствительность тканей с избыточными рецепторами путем изменения плотности последних. Заметим, что изменение числа рецепторов, как правило, не влияет на свободную концентрацию вещества, достигаемую при введении определенной дозы. Это связано с тем, что концентрация рецепторов в ткани обычно очень мала по сравнению с эффективными концентрациями лекарств.
Важным биологическим следствием избыточности рецепторов является то, что агонисты с низким сродством к рецепторам могут вызывать полные реакции при низких концентрациях, если ЕС50 ниже, чем KD. Это важно, поскольку лиганды с низким сродством (высокая Ко) быстро диссоциируют, что ведет к прекращению биологического ответа. Высокий аффинитет (низкая KD), напротив, характеризуется медленной диссоциацией агониста и,
соответственно, более медленным прекращением биологического эффекта.
Конкурентные и неконкурентные (необратимые) антагонисты
Антагонисты связываются с рецептором, но не активируют его. Ингибирующий эффект антагонистов основан на предупреждении связывания агонистов (других лекарств или эндогенных регуляторных молекул) с рецепторами и их активации. Антагонисты разделяют на два класса в зависимости от того, обратимо или необратимо они конкурируют с агонистом за оккупацию рецепторов. Два типа рецепторного антагонизма отражены в совершенно различных кривых концентрация-эффект и концентрация-связывание in vitro и имеют важные в практическом отношении отличия.
В присутствии фиксированной концентрации агониста увеличение концентрации конкурентного антагониста прогрессивно подавляет реакцию на агонист вплоть до полного ее ингибирования. Напротив, повышая концентрацию агониста, можно полностью преодолеть эффект конкурентного ан
тагониста, то есть Етах для агониста остается тем же самым для любой фиксированной концентрации антагониста (рис. 2-5А). Поскольку антагонизм является конкурентным, присутствие антагониста требует повышения концентрации агониста, необходимой для данного уровня ответной реакции, и, таким образом, кривая концентрация агониста-эффект сдвигается вправо.
Концентрация агониста (С')> необходимая для получения данного эффекта в присутствии фиксированной концентрации конкурентного антагониста ([I]), выше, чем концентрация агониста (С), требуемая для получения того же эффекта в отсутствие антагониста. Соотношение этих двух концентраций агониста (соотношение доз) связано с константой диссоциации (К;) уравнением Шильда:
Фармакологи часто используют эту зависимость для определения Kj конкурентных антагонистов. Значение К может быть определено достаточно просто и точно даже при отсутствии сведений о связи между оккупацией рецептора агонистом и ответом. На рис. 2-5 изображена кривая концентрация-эффект в присутствии фиксированной концентрации конкурентного антагониста и без него. Сравнение концентраций агониста, требуемых для получения одинакового фармакологического эффекта в этих двух ситуациях, позволяет определить К; антагониста. Если С', например, в два раза превышает С, то [I] = Kj. Значения Kj, полученные в подобных экспериментах, согласуются с теми, которые определяются путем прямого измерения радио-лигандными методами связывания с рецепторами меченых конкурентных антагонистов.
Для клинициста эта математическая зависимость имеет важное прикладное значение в двух аспектах.
Рис. 2-5. Изменения кривых зависимости эффекта от концентрации агониста, вызванные конкурентным антагонистом (А) или антагонистом необратимого действия (Б). В присутствии конкурентного антагониста требуются более высокие концентрации агониста для получения заданного эффекта и, таким образом, концентрация агониста С', требуемая для данного эффекта в присутствии фиксированной концентрации антагониста [I], сдвигается вправо. Повышая концентрацию агониста, можно преодолеть ингибирующее действие конкурентного антагониста. Этого не наблюдается в случае с неконкурентным (необратимым) антагонистом, уменьшающим максимально возможный эффект агониста, хотя при этом может и не измениться ЕС50
1) Степень угнетения, вызванная конкурентным антагонистом, зависит от его концентрации. Таким образом, выраженность и продолжительность действия такого вещества будет зависеть от его концентрации в плазме, и на них будет существенно влиять скорость метаболического клиренса или экскреции. Например, у разных пациентов, получающих одинаковые дозы пропранолола, отмечаются значительные колебания его концентрации в плазме из-за различий в клиренсе лекарства. В результате эффект одной и той же дозы этого конкурентного антагониста норадреналина может варьировать у больных весьма значительно, и дозы должны подбираться индивидуально.
2) Причиной вариабельности клинической реакции на конкурентный антагонист может быть концентрация агониста, конкурирующего за рецептор. Приведем пример с тем же. пропранололом. Когда этот конкурентный антагонист Р-адреноре-цепторов вводят в дозах, достаточных для блокады эффектов базального уровня нейромедиатора норадреналина, частота сердечных сокращений в покое снижается. Однако увеличение высвобождения норадреналина и адреналина, которое происходит при физической нагрузке, изменении позы или при эмоциональном стрессе, может оказаться достаточным для преодоления конкурентного антагонизма пропранолола и учащения пульса. Поэтому врач, определяющий режим дозирования конкурентного антагониста, всегда должен учитывать возможные изменения концентрации эндогенных агонистов, которые могут повлиять на терапевтический эффект.
Некоторые антагонисты рецепторов связываются с рецепторами необратимо или почти необратимо. Сродство антагониста к рецептору может быть столь сильным, что практически рецептор становится недоступным для связывания с агонистом. Другие антагонисты этого класса вызывают необратимые эффекты, потому что после соединения с рецептором они образуют с ним ковалентные связи. При оккупации существенного количества рецепторов таким антагонистом число Оставшихся свободных рецепторов может быть так мало, что высокие концентрации агониста не смогут преодолеть антагонизм, и максимальная ответная реакция на агонист будет недостижима (рис. 2-5Б). Однако если имеется избыточное количество рецепторов, при воздействии антагониста необратимого действия в малых дозах может остаться достаточное ко
личество незанятых рецепторов, что позволяет достичь максимального ответа на агонист, хотя для этого могут потребоваться и более высокие концентрации агониста.
Использование в качестве лекарств антагонистов необратимого действия имеет определенные достоинства и некоторые недостатки. Как только необратимый антагонист занял рецептор, он уже не нужен в несвязанном виде для подавления ответа агониста. Следовательно, продолжительность действия такого необратимого антагониста относительно независима от скорости его элиминации и в большей степени зависит от скорости оборота рецепторных молекул. Необратимый антагонист «-адренорецепторов феноксибензамин используется для контроля гипертензии, вызванной высвобождением катехоламинов при феохромоцитоме (опухоли мозгового слоя надпочечников). Гипотензивный эффект феноксибензамина может сохраняться даже при эпизодическом выделении опухолью больших количеств катехоламинов. В этом случае терапевтически выгодна способность лекарства предупреждать ответы на варьирующие и высокие концентрации агониста. Если происходит передозировка и блокаду «-адренорецепторов нельзя преодолеть, избыточный эффект препарата можно снять “фи-зиологическй”, например с помощью прессорных веществ, действующих помимо «-адренорецепторов.
Парциальные агонисты
В зависимости от максимальной фармакологической ответной реакции, которая возникает, когда оккупированы все рецепторы, агонисты могут быть разделены на два класса. Парциальные (неполные) агонисты вызывают меньшую реакцию при оккупации всех рецепторов, чем полные агонисты. В отличие от полных агонистов для парциальных характерны кривые концентрация-эффект, которые,напоминают аналогичные кривые полных агонистов в присутствии антагонистов, необратимо блокирующих рецепторные сайты (сравните рис. 2-ЗГ и 2-6Б). Тем не менее опыты по радиолигандному связыванию показали, что парциальные агонисты могут занимать все рецепторные сайты (рис. 2-6А) при концентрациях, которые не способны вызвать максимальный ответ по сравнению с ответом после введения полных агонистов (рис 2-4Б). Кроме того, неспособность,
парциальных агонистов вызвать “полный” максимальный ответ не связана со сниженным аффинитетом к рецепторам. Подобные соединения конкурируют, подчас с высоким аффйнитетом,’ за весь пул рецепторов. На способность парциальных агонистов оккупировать всю популяцию рецйтторов указывает тот факт, что они по конкурентному ме
ханизму ингибируют реакции, вызываемые полными агонистами (рис. 2-6В).
Точный молекулярный механизм, объясняющий неполный максимальный ответ на парциальные агонисты, неизвестен. Упрощенно можно представить, что парциальные агонисты оказывают на рецепторы действие, носящее промежуточной
log С [Парциальный агонист или полный агонист]
Рис. 2-6. А: Связывание рецепторов (в %) полным агонистом (в одной концентрации) в присутствии возрастающих концентраций парциального агониста. Поскольку полный агонист (темные квадраты) и парциальный агонист (светлые квадраты) конкурируют за один и тот же рецепторный сайт, при относительном возрастании связывания рецепторов парциальным агонистом связывание полного агониста уменьшается.
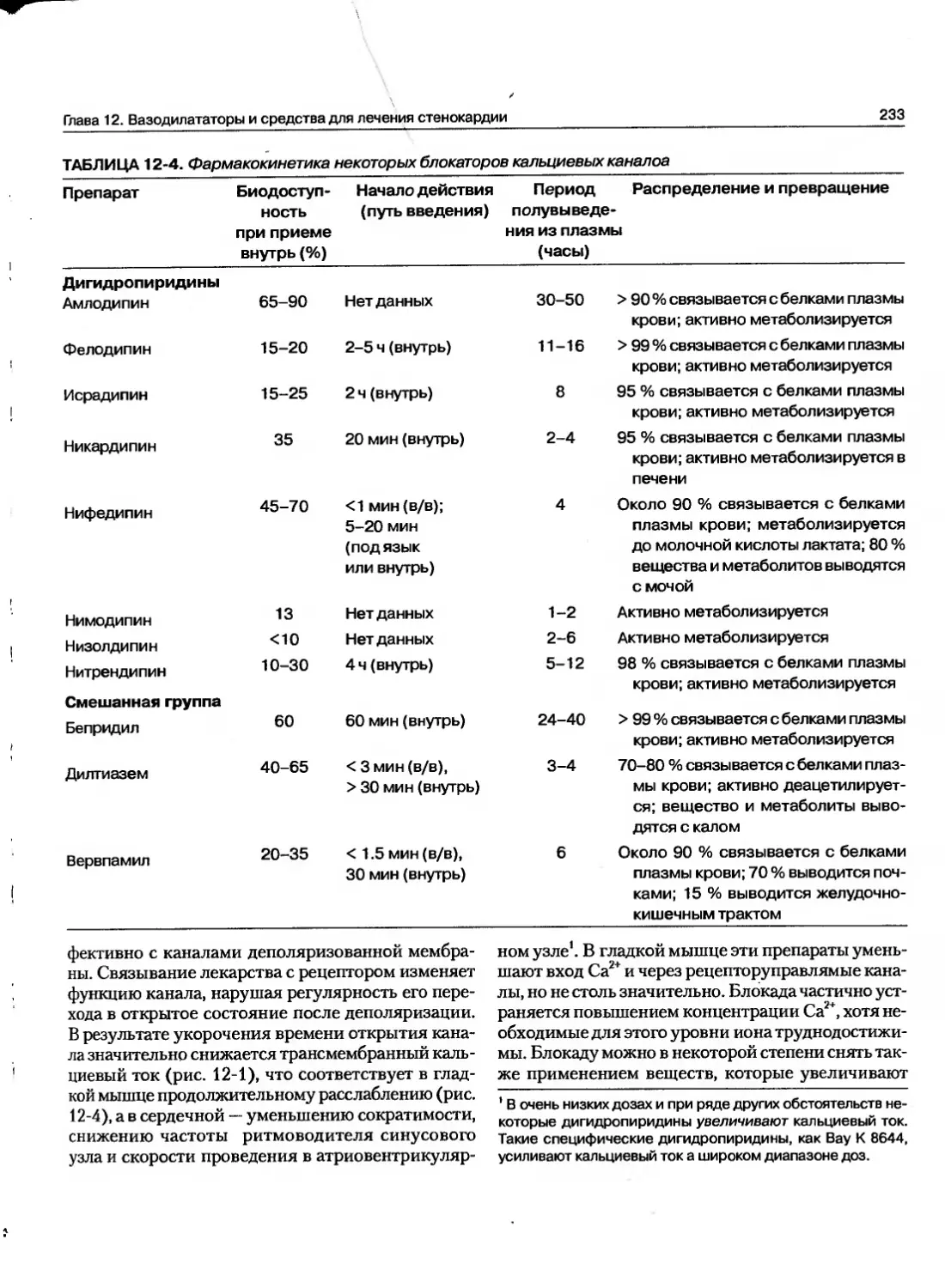
Б: Когда каждое из веществ используется раздельно и измеряется фармакологический эффект, оккупация всех рецепторов парциальным агонистом вызывает меньший максимальный ответ по сравнению с ответом полного агониста при той же степени связывания рецепторов.
В: Одновременное введение полного агониста в определенной концентрации и парциального агониста в возрастающих концентрациях дает ответную реакцию, показанную на нижнем графике. Вклад в общий эффект полного агониста в заданной концентрации (темные квадраты) снижается по мере возрастания концентрации парциального агониста, конкурирующего за рецептор с тем большим успехом, чем выше его концентрация. В то же время вклад парциального агониста (светлые квадраты) возрастает, а общий эффект — сумма реакций на оба вещества (темные треугольники),— постепенно снижается, приближаясь к уровню собственного эффекта парциального агониста (сравните с графиком Б)
характер между эффектом полных агонистов и конкурентных антагонистов. Полный агонист при связывании с рецептором вызывает такое изменение конформации, которое инициирует последующие фармакологические эффекты, тогда как “чистые” конкурентные антагонисты не вызывают подобных изменений конформации рецепторов. С этих позиций парциальные агонисты, хотя и изменяют конформацию рецепторов, но недостаточно для их полной активации. Отражением подобных представлений является термин эффективность лекарства, указывающий на связь между оккупацией рецепторных сайтов и фармакологическим ответом. Лекарство может иметь нулевую эффективность (“чистый” антагонист) или любую степень эффективности выше нуля. Парциальные агонисты можно рассматривать как вещества со столь низкой эффективностью, что даже при полной оккупации всего набора рецепторов они не вызывают максимально возможного ответа, который характерен для более эффективных (полных) агонистов. Читатель сможет убедиться, что многие лекарства, применяемые в качестве конкурентных антагонистов, являются на самом деле слабыми парциальными агонистами.
Другие механизмы антагонизма лекарств
Механизм антагонизма не исчерпывается взаимодействием лекарств или эндогенных лигандов на одном типе рецепторов. Так, химические антагонисты для проявления своего эффекта вовсе не нуждаются в рецепторах. Одно вещество может препятствовать действию другого путем соединения с ним и последующей инактивации. Например, протамин, белок с положительным при физиологическом значении pH зарядом, может использоваться в клинике как антагонист гепарина — антикоагулян-та, имеющего отрицательный заряд. В этом случае протамин просто связывает гепарин и делает его недоступным для взаимодействия с белками, участвующими в образовании кровяного тромба.
Принцип действия целого ряда лекарств основан на использовании физиологического антагонизма между эндогенными регуляторными системами. Многие физиологические функции контролируются реципрокными физиологическими механизмами. Так, катаболическое действие глюкокортикоидных гормонов приводит к увеличению уров
ня сахара в крови. Этому эффекту оказывает физиологическое противодействие инсулин. Хотя глюкокортикоиды и инсулин действуют на разные рецепторно-эффекторные системы, врач иногда вынужден вводить инсулин для противодействия гипергликемическому эффекту глюкокортикоидов независимо от того, является ли причиной гипергликемии эндогенный синтез гормонов (например, опухоль коры надпочечников) или глюкокортикоидная терапия. В целом использование лекарства как физиологического антагониста вызывает менее специфичный и контролируемый эффект по сравнению с эффектом рецепторного антагониста. К примеру, для лечения брадикардии, вызванной повышенным высвобождением ацетилхолина из окончаний блуждающего нерва, при боли, связанной с инфарктом миокарда, врач может использовать изопротеренол. Это агонист Р-адренорецеп-торов, увеличивающий частоту сердечных сокращений, имитируя симпатическую стимуляцию сердца. Однако использование подобного физиологического антагониста может быть менее рациональным и потенциально более опасным, чем применение рецепторспецифичного антагониста типа атропина (конкурентный антагонист ацетилхолиновых рецепторов, замедляющих при возбуждении частоту сердечных сокращений).
Сигнальные механизмы и действие лекарств
До настоящего момента мы обсуждали взаимодействие с рецепторами и эффекты лекарств, анализируя уравнения и кривые концентрация-эффект. Этот абстрактный анализ поясняет некоторые количественные аспекты действия лекарств. Для более полного объяснения мы должны понимать молекулярные механизмы действия лекарств. Это особенно важно для препаратов, которые имитируют или блокируют внутриклеточную сигнальную функцию гормонов и нейромедиаторов.
Исследования последних десяти лет раскрыли важные детали молекулярных процессов превращения внеклеточных сигналов во внутриклеточные, контролирующие функции клетки. Понимание этих важных сигнальных механизмов позволяет нам ставить основные вопросы, имеющие важное значение для клиники. Почему некоторые лекарства вызывают эффекты, которые продолжаются минуты,
часы или даже дни, после того как лекарство уже покинуло организм? Как клеточные механизмы усиления внешних химических сигналов сосуществуют с феноменом “избыточности” рецепторов? Почему химически сходные вещества подчас проявляют чрезвычайную избирательность действия? Могут ли сигнальные механизмы объяснить действие лекарств, не йзаимодействующих с рецепторами? Сегодня уже есть ответы на многие из этих вопросов.
Большинство трансмембранных сигнальных процессов реализуется посредством ограниченного числа молекулярных механизмов. Каждый из них сопряжен с эволюцией определенных семейств белков-переносчиков различных сигналов. К таким белкам относятся рецепторы на клеточной поверхности и внутри клетки, ферменты й другие компоненты, которые генерируют, усиливают, координируют и завершают пострецепторный сигнальный процесс посредством химических вторичных посредников в цитоплазме. В настоящем разделе вначале обсуждаются механизмы переноса химической информации через плазматическую мембрану, затем рассматриваются ключевые свойства вторичных цитоплазматических посредников.
Наиболее подробно изучены четыре основных механизма трансмембранной передачи сигналов (рис. 2-7). В каждом использована различная стратегия для преодоления барьера, созданного двуслойной липидной плазматической мембраной.
Этими стратегиями Являются: 1) проникновение растворимых в липиДйх лйгандов через мембрану и их действие на внутриклеточный рецептор; 2) использование трансмембранного рецепторного белка, внутриклеточная ферментативная активность которого аллд‘стерйчески регулируется лигандом, связывающимся с сайтом на внеклеточном домене белка; 3) закрытие иДи открытие трансмем-браннйх ионных КаЙйЛОй при связывании с лигандом; 4) использование Трансмембранного рецептора для стимуляции ГТФ-связывающего сигнального передающего белка (G-белка), который, в свою очередь, активирует внутриклеточный вторичный посредник.
Для многих внеклеточных лигандов (гормон роста, интерферон, лимфокины) сигнальные механизмы остаются пока не известными. Несмотря на то, что четыре установленных механизма не охватывают все возможности химической передачи сигналов через клеточную мембрану, они объясняют важные сигнальные процессы, используемые в фармакотерапии.
Внутриклеточные рецепторы для агентов, растворимых в липидах
Некоторые биологические сигнальные молекулы достаточно растворимы в липидах, чтобы проникать через плазматическую мембрану и действовать на внутриклеточные рецепторы. К ним отно
Рис. 2-7. Основные трансмембранные сигнальные механизмы. 1. Растворимая в липидах сигнальная молекула проходит через плазматическую мембрану и действует на внутриклеточный рецептор (который может быть ферментом или регулятором транскрипции генов). 2. Сигнальная молекула связывается с внеклеточным доменом трансмембранного белка, активируя таким образом ферментативную активность его цитоплазматического домена. 3. Сигнальная молекула связывается с ионным каналом и регулирует его открытие. 4. Сигнальная молекула связывается с рецептором па поверхности клетки, который сопряжен с эффекторным ферментом посредством G-белка
сится, например, оксид азота (NO), который стимулирует внутриклеточный фермент гуанилатцикла-зу, катализирующую синтез цГМФ. Рецепторы для другого класса жирорастворимых лигандов (кортикостероидов, минералокортикоидов, половых гормонов, витамина D и тиреоидного гормона) стимулируют транскрипцию генов в ядре. Они связываются со специфическими последовательностями ДНК, соседствующими с геном, экспрессия которого подлежит регуляции. Многие из таких последовательностей ДНК (отвечающие элементы — response elements) идентифицированы.
Хорошо изучены детальные молекулярные механизмы, используемые этими “генно-активными" рецепторами. Сходство их структуры указывает на принадлежность к семейству протеинов, происходящих от общего предшественника. “Препарирование” рецепторов с помощью ДНК-рекомбинантной техники позволило понять сущность этих молекулярных механизмов. Например, удаление терминального сегмента глюкокортикоидного рецептора приводит к тому, что образующийся белок связывается с ДНК и стимулирует транскрипцию гена-мишени даже в отсутствие глюкокортикоидов. В физиологических условиях связывание глюкокортикоида с нормальным рецептором также Снимает ингибиторные влияния на транскрипцию гена-мишени. Рис. 2-8 схематически отражает молекулярный механизм действия глюкокортикоидов. В отсутствие гормона рецептор связан с hsp90 — белком, который нарушает “нормальную” упаковку нескольких структурных доменов рецептора. Присоединение гормона к лигандсвязывающему домену способствует высвобождению hsp90. Это позволяет доменам рецептора, связывающим ДНК и активирующим транскрипцию, перейти в их функционально активную конформацию. Активированный рецептор инициирует транскрипцию гена-мишени.
Механизм, характерный для действия гормонов, влияющих на регуляцию экспрессии генов, имеет два терапевтически важных следствия. 1) Все указанные гормоны вызывают эффект после характерного временного интервала от 30 мин до нескольких часов — времени, которое требуется на синтез новых белков. Это означает, что генно-активные гормоны не могут изменить патологическое состояние в течение нескольких минут, например глюкокортикоиды не могут немедленно облегчить симптомы приступа бронхиальной астмы. 2) Эффект этих агентов может продолжаться в течение не
скольких часов или дней, после того как концентрация агониста снизилась до нуля. Существенная длительность эффекта связана прежде всего с относительно медленным биохимическим кругооборотом большинства ферментов и белков, которые могут оставаться активными в клетке в течение часов или дней после синтеза (длительность эффекта может быть также частично связана с высоким аффинитетом рецептора к гормону, результатом чего является замедленная диссоциация гормона). На практике это означает, что полезные (или токсические) эффекты генно-активных гормонов медленно исчезают после прекращения введения гормона и не существует простой временной корреляции между концентрацией гормона в плазме и его эффектами.
Рис. 2-8. Механизм действия глюкокортикоидов. Глюкокортикоидный полипептидный рецептор схематически показан как белок с тремя различными доменами. Белок теплового шока, hsp90, связывается с рецептором в отсутствие гормона и предупреждает его переход в активную конформацию. Присоединение гормонального лиганда вызывает освобождение стабилизирующей молекулы hsp90. Рецептор переходит в активную конфигурацию
Регулируемые лигандами трансмембранные ферменты
Этот класс рецепторных молекул опосредует первый этап передачи сигналов инсулина* эпидермального фактора роста (epidermal growth factor — EGF), тромбоцитарного фактора роста (platelet-derived growth factor — PDGF), предсердного натрийуретического фактора (atrial natriuretic factor — ANF), трансформирующего 0-фактора роста (transforming growth factor-beta — TGF0) и некоторых других трофических гормонов. Рассматриваемые рецепторы являются полипептидами, состоящими из внеклеточного домена, связывающего гормон, и цитоплазматического домена с ферментативной активностью. Это может быть про-теинкиназа, (тирозинкиназа, серинкиназа) или гу-анилатциклаза (рис. 2-9). Во всех случаях два рецепторных домена соединены гидрофобным поли-пептидным сегментом, который пересекает липидный бислой плазматической мембраны.
Тирозинкиназный сигнальный путь начинается со связывания гормона с внеклеточным доменом рецептора. Возникающие изменения конформации рецептора способствуют соединению рецепторных
молекул, что, в .свою очередь, объединяет домены. Тирозиновые остатки в обоих цитоплазматических доменах фосфорилируются (возможно, перекрестно). Это перекрестное фосфорилирование может усиливать аллостерическую регуляцию гормональным лигандом иди увеличивать ее длительность. Например, тирозинкиназная активность аутофосфорилированной молекулы инсулинового рецептора продолжается, после того как инсулин отходит от места связывания.
Различные рецепторы катализируют фосфорилирование тирозиновых остатков различных сигнальных протеинов, но лишь некоторые из этих субстратных белков были идентифицированы. Так, инсулин использует один и тот же класс рецепторов для усиления захвата глюкозы и аминокислот и для регуляции метаболизма гликогена, и триглицеридов в клетке. Сходным образом каждый из факторов роста инициирует в своей клетке-мишени комплексную программу событий от изменения мембранного транспорта протонов, других ионов и метаболитов до характерных изменений экспрессии многих генов. Некоторые из этих реакций связаны с фосфорилированием серин- и треонинкиназами, тогда как другие осуществляются через факторы
+EGF
Рис. 2-9. Механизм активации рецептора EGF — тирозинкипазы. Рецепторный полипептид имеет внеклеточный и цитоплазматический домены, показанные над и под плазматической мембраной. После связывания EGF (кружок) рецептор переходит из неактивного мономерного состояния (слева) в активное димерное состояние (справа), при котором два рецепторных полипептида объединяются с помощью нековалентной связи в плоскости мембраны. Цитоплазматические домены фосфорилируются (Р) в области специфического тирозинового остатка (Y), и стимулируется их ферментативная активность. В результате происходит фосфорилирование субстратного белка (S)
транскрипции, которые могут быть сами по себе субстратами киназ. Рецепторные тирозинкиназы представляют собой интересную мишень для создания лекарств. Пока найдено незначительное число соединений, способных вызывать эффект вследствие ингибирования тирозинкиназной активности. Легко представить терапевтический эффект специфических ингибиторов рецепторов факторов роста, особенно при опухолевых заболеваниях.
Интенсивность и продолжительность действия EGF, PDGF и других веществ, действующих через данный класс рецепторов, ограничены механизмом рецепторной отрицательной обратной связи (down-регуляция). Связывание с лигандом вызывает ускорение эндоцитоза рецепторов, за которым следует их деградация и деградация связанных с ними лигандов. Когда этот процесс происходит со скоростью, превышающей синтез рецептора de novo, общее число рецепторов на клеточной поверхности уменьшается, и реакция клетки на лиганд соответственно снижается.
Многочисленные регуляторы роста и дифференциации, включая TGFP, действуют на рецепторные серинкиназы — другой класс трансмембранных рецепторных ферментов. ANF, важный регулятор объема крови и тонуса сосудов, связывается с трансмембранным рецептором, внутриклеточный домен которого (гуанилатциклаза) способствует образованию цГМФ. Рецепторы обеих групп, подобно ти-розинкиназе, активны в димерной форме.
Регуляция ионных каналов лигандами
Многие лекарства действуют путем имитации или блокирования действия эндогенных лигандов, регулирующих ток ионов через каналы плазматической мембраны. К естественным лигандам такого рода относятся ацетилхолин (АХ), у-аминомас-ляная кислота (ГАМК) и возбуждающие аминокислоты (ВАК — глицин, аспартат, глутамат и др.). Все эти вещества являются синаптическими передатчиками (медиаторами).
Соответствущие рецепторы передают сигнал через плазматическую мембрану посредством увеличения трансмембранной проводимости определенных ионов, что приводит к изменению электрического потенциала мембраны. Например, АХ способствует открытию ионного канала никотинового холинорецептора (АХР), что позволяет Na+ перемещаться по градиенту концентрации в клетку, вызы
вая деполяризацию и соответствующий местный возбуждающий постсинаптический потенциал.
Никотиновый АХР (рис.2-10) — один из наиболее изученных поверхностных рецепторов для гормонов и нейромедиаторов. Этот рецептор представляет собой пентамер, состоящий из пяти полипеп-тидных субъединиц (две a-цепи плюс одна 0-, одна у- и одна 5-цепь с мол. м. от 43 000 до 50 000). Эти полипептиды, каждый из которых пересекает липидный бислой четырежды, образуют цилиндрическую структуру диаметром 8 нм. Когда АХ соединяется с сайтом на а-субъединице, происходят конформационные изменения, ведущие к открытию центрального гидрофильного канала, через который ионы натрия проникают из внеклеточной жидкости в клетку. Интервал времени между связыванием агониста с лигандуправляемым каналом и клеточной реакцией иногда измеряется миллисекундами. Быстрота этого сигнального механизма чрезвычайно важна для моментальной передачи информации через синапсы. Это весьма существенное
Рис. 2-10. Никотиновый ацетилхолиновый рецептор — управляемый лигандом ионный канал. Показана рецепторная молекула, встроенная в прямоугольный участок плазматической мембраны, с внеклеточной жидкостью сверху и цитоплазмой внизу. Рецептор, состоящий из пяти субъединиц (две а-, одна 0-, одна у- и одна 5-цепи), вызывает открытие центрального трансмембранного ионного канала, после того как ацетилхолин (АХ) связывается с сайтом на внеклеточном домене а-субъединицы
отличие от иных молекулярных механизмов передачи сигналов, которые могут требовать секунды, минуты и даже часы, как например в случае генноактивных гормонов.
G-белки и вторичные посредники
Многие внеклеточные лиганды действуют, увеличивая внутриклеточную концентрацию таких вторичных посредников как циклический аденозин-3',5'-монофосфат (цАМФ), ионы кальция или фосфолипиды. В большинстве случаев они используют для передачи сигнала трансмембранную трехкомпонентную систему. Сначала внеклеточный специфический лиганд распознается поверхностным рецептором клетки. Рецептор, в свою очередь, активирует G- белок, расположенный на внутренней поверхности плазматической мембраны. Затем активированный G-белок изменяет функцию эффекторного элемента, обычно фермента или ионного канала. На следующем этапе этот эффекторный элемент изменяет концентрацию внутриклеточного вторичного посредника. Для цАМФ эффекторным ферментом является аденилатциклаза, трансмембранный белок, превращающий внутриклеточ
ный АТФ в цАМФ. Соответствующий G-белок, обозначаемый как Gs, стимулирует аденилатцикла-зу после того, как он активируется гормонами или нейромедиаторами через специфические рецепторы (табл. 2-1).
Gs и другие G-белки связывают и гидролизуют ГТФ (рис. 2-11). Важно, что этот механизм отделяет возбуждение рецептора лигандом от вызванной G-белком активации эффектора, что позволяет усиливать передаваемый сигнал. Например, такой нейромедиатор как норадреналин (НА), может взаимодействовать со своим мембранным рецептором очень непродолжительное время, всего несколько миллисекунд. Однако если при этом произошло связывание ГТФ молекулой Gs, продолжительность активации аденилатциклазы будет зависеть в большей степени от длительности этого связывания, чем от сродства НА к своему рецептору. Действительно, подобно другим G-белкам, ГТФ-свя-занный Gs остается активным в течение десятков секунд, что значительно усиливает исходный сигнал. Этот механизм объясняет, как передача сигналов G-белками способствует рассмотренному феномену “избыточности” рецепторов. Даже если для инициации связывания ГТФ одной молекулой
ТАБЛИЦА 2-1. Сокращенный список эндогенных лигандов и связанных с ними вторичных посредников
Лиганд Вторичный посредник
Адренокортикотропный гормон цАМФ
Ацетилхолин (мускариновые рецепторы) Саг+, фосфоинозитиды
Ангиотензин Са2*, фосфоинозитиды
Катехоламины (оц-адренорецепторы) Са2*, фосфоинозитиды
Катехоламины (p-адренорецепторы) цАМФ
Хорионический гонадотропин цАМФ
Фолликулостимулирующий гормон цАМФ
Глюкагон цАМФ
Гистамин (Н2-рецепторы) цАМФ
Лютеинизирующий гормон цАМФ
Меланоцитстимулирующий гормон цАМФ
Паратиреоидный гормон цАМФ
Тромбоцитарный фактор роста Саг+, фосфоинозитиды
Простациклин, простагландин Е2 цАМФ
Серотонин (5-НТ4-рецепторы) цАМФ
Серотонин (5-HTic- и 5-НТ2-рецепторы) Са2’, фосфоинозитиды
Тиреотропин цАМФ
Тиреотропинрилизинг-гормон Са2*, фосфоинозитиды
Вазопрессин (\А-рецепторы) Са2*, фосфоинозитиды
Вазопрессин (\/2-рецепторы) цАМФ
Рис. 2-11. Зависимый от гуанин-нуклеотида цикл активации-инактивации G-белков. Агонист активирует рецептор (R), что способствует освобождению ГДФ от G-белка (G), позволяя ГТФ связаться с последним. В форме, связанной с ГТФ (G-ГТФ), G-белок регулирует активность эффекторного фермента или ионного канала (Е). Сигнал прекращается после гидролиза ГТФ с последующим возвращением системы к исходному нести-мулированному состоянию. Светлые стрелки отмечают регуляторные моменты
G-белка требуется всего один рецептор, активированный лигандом, медленный гидролиз ГТФ способствует существованию активного G-белка еще длительное время после того, как рецептор диссо
циировал от молекулы агониста. Таким образом, при низких концентрациях агониста доля связанных с ним рецепторов может быть значительно меньше, чем количество G-белков в активном (связанном с ГТФ) состоянии. Если количество активных G-белков коррелирует с фармакологическим ответом, рецепторы будут казаться избыточными, т. е. малая доля рецепторов, занятых агонистом в данный момент, будет вызывать существенно больший эффект.
Семейство G-белков весьма многочисленно (табл. 2-2) — помимо стимулятора аденилатцикла-зы Gs, оно включает в себя другие подсемейства. Члены Gi-подсемейства (i — ингибиторный) вызывают сопряжение рецептора с аденилатциклазой, проявляющееся угнетением функции фермента. В некоторых клетках Gj-белки опосредуют рецепторную стимуляцию системы фосфоинозитидного вторичного посредника и регуляцию К+- и Са++- каналов. Подсемейство Gt включает два G-белка (Gd и Gt2, или “трансдуцины”), которые опосредуют передачу светового сигнала в палочках и колбочках сетчатки.
Неудивительно, что рецепторы, сопряженные с G-белками, структурно похожи друг на друга. Они образуют семейство “серпантинных” рецепторов, называемых так потому, что их полипептидные цепи пересекают плазматическую мембрану семь раз (рис. 2-12). К этому семейству принадлежат
ТАБЛИЦА 2-2. G-белки, их рецепторы и эффекторы
G-белок Рецепторы для Эффектор и сигнальный путь
Gs p-адренергических аминов, глюкагона, гистамина, серотонина и многих других гормонов ТАденилатциклаза —> ТцАМФ
0 0 (5 Ог-адренергических аминов, ацетилхолина (мускариноподобные вещества), опиоидов, серотонина и др. Несколько, в том числе: ТАденилатциклаза -> J-цАМФ Открытие «-каналов сердца -» Т частоты сердечных сокращений
Gok одорантов (обонятельный эпителий) ТАденилатциклаза -> ТцАМФ
Go нейромедиаторов мозга (точно не определены) Пока не выяснены
Go ацетилхолина (например, мускариноподбные вещества), бомбезина, серотонина (5-НТ1С) и др. Тфосфолипаза С -» Тинозитолтрифосфат, диацилглицерол, цитоплазматический Са2*
Gtt, G f2 фотонов (родопсин и цветовые опсины в палочках и колбочках сетчатки) Тфосфодиэстераза цГМФ -> ТцГМФ (фототрансдукция)
рецепторы для адренергических аминов, серотонина, АХ (мускариновые, но не никотиновые), многих пептидных гормонов, одорантов и даже зрительных рецепторов (в колбочках и палочках сетчатки). Амино- и карбокси-концы этих рецепторов расположены соответственно на внешней и внутренней сторонах мембраны. Различные серпантинные рецепторы сходны друг с другом по аминокислотным последовательностям и расположению гидрофобных трансмембранных областей и гидрофильных вне- и внутриклеточных петель, что указывает на их общее происхождение от единого эволюционного предшественника.
Наряду со структурным сходством оказалось, что разные серпантинные рецепторы передают сигналы через плазматическую мембрану сходным образом. Лиганд-агонист, например катехоламин, АХ или фотонактивируемый хромофор фоторецептора сетчатки, может связываться с “карманом”, об
разованным трансмембранными областями рецептора (как на рис. 2-12). Возникшие изменения конформации этих участков передаются цитоплазматическим петлям рецептора, которые в свою очередь активируют соответствующий G-белок, облегчая замещение ГДФ на ГТФ. Биохимические данные указывают на то, что G-белок взаимодействует с аминокислотами в третьей цитоплазматической петле рецепторного полипептида (стрелка на рис. 2-12). Карбоксильные концевые хвосты этих рецепторов, также расположенные в цитоплазме, могут регулировать способность рецептора взаимодействовать с G-белками.
Десенситизация рецепторов
Опосредованные рецепторами реакции на лекарства и гормоны-агонисты часто со временем “де-сенситизируются” (рис. 2-13, сверху). Это означа
Рис. 2-12. Трансмембранное расположение типичного серпантинного рецептора. N (амино)-конец полипептидной цепи рецептора располагается внеклеточно (над плоскостью мембраны), а его С (карбокси)-конец — внутриклеточно. Полипептидная цепь пронизывает плоскость мембраны семь раз. Гидрофобные трансмембранные сегменты обозначены римскими цифрами (I-VII). Агонист (Аг) подходит к рецептору со стороны внеклеточной жидкости и соединяется с сайтом, окруженным трансмембранными участками рецепторного белка. G-белок взаимодействует с внутриклеточными сегментами рецептора, особенно в области третьей цитоплазматической петли между трансмембранными регионами V и VI. Цитоплазматическая терминаль рецептора содержит многочисленные сериновые и треониновые остатки, гидроксильные группы которых (-ОН) могут фосфорилироваться. Это фосфорилирование может ослаблять взаимодействие рецептора и G-белка
ет, что после достижения начального высокого уровня ответ (например, накопление внутриклеточного цАМФ, ток Na+, сокращение мышцы и пр.) постепенно уменьшается в течение секунд или минут, даже несмотря на постоянное присутствие агониста. Десенситизация обычно обратима. Так, через 15 минут после удаления агониста его повторное воздействие ведет к реакции, сравнимой по величине с начальной. (Заметим, что такая обратимость отличает десенситизацию от down-регуляции, т. е. регуляторного уменьшения числа рецепторов, как
было рассмотрено для рецепторной тирозинки-назы.)
Хотя десенситизация характерна для многих видов рецепторов, ее механизм в большинстве случаев остается загадочным (например, вызванная агонистом десенситизация никотинового АХР). Однако некоторые детали молекулярного механизма десенситизации к эффекту агониста были выявлены в отношении [3-адренорецепторов (рис. 2-13, внизу). Связывание агониста приводит к изменению конформации карбоксильного рецептора, что
Рис. 2-13. Возможный механизм десенситизации 0-адренорецептора. Верхняя часть рисунка показывает реакцию агониста 0-адренорецептора (ордината) во времени (абсцисса). Разрыв по оси времени указывает на период отсутствия агониста. Длительность воздействия агониста обозначена диагонально заштрихованными прямоугольниками. Нижняя часть рисунка схематически показывает вызванное агонистом фосфорилирование (Р) киназой 0-адренорецептора (0-АРК) гидроксигруппы (-ОН) карбоксильного участка 0-адренорецептора. Это фосфорилирование вызывает связывание белка 0-аррестина (0-арр), что предупреждает взаимодействие рецептора с Gs. Удаление агониста на короткий период времени позволяет диссоциировать 0-арр, удалить фосфат (PJ от рецептора с помощью фосфатазы и восстановить нормальную реакцию рецептора на агонист
делает его хорошим субстратом для фосфорилирования по остаткам серина (и треонина) с помощью специфической киназы, киназы Р-адренорецептора (Р* АРК). Наличие фосфосеринов повышает аффинитет рецептора к третьему протеину — р-аррести-ну. Связывание Р-аррестина с цитоплазматическими петлями рецептора уменьшает способность рецептора взаимодействовать с Gs, снижая таким образом реакцию на действие агониста (например, стимуляцию аденилатциклазы). После удаления агониста, однако, клеточные фосфатазы отщепляют фосфатные группы от рецептора. Действие Р-АРК прекращается, так что рецептор и, следовательно, реакция на агонист возвращаются в исходное состояние (рис. 2-13, внизу).
Наиболее известные вторичные посредники
А. цАМФ. Действуя как внутриклеточный вторичный посредник, цАМФ участвует в передаче таких гормональных эффектов как мобилизация энергетических запасов (распад углеводов в печени или триглицеридов в жировых клетках, стимулированный катехоламинами (КА), действующими как Р-адреномиметики), задержка воды почками под контролем вазопрессина, поддержание кальциевого гомеостаза (регулируемого гормонами паращитовидных желез) и увеличение частоты и силы сокращений сердечной мышцы (КА с Р-адреноми-метическим действием). Он регулирует также образование стероидов надпочечников и половых желез (в ответ на кортикотропин или фолликулостимулирующий гормон), расслабление гладкой мускулатуры и многие другие эндокринные и нервные процессы.
цАМФ осуществляет большинство своих эффектов, стимулируя цАМФ-зависимыепротеинки-назы (рис. 2-14). Эти тетрамерные киназы состоят из связывающего цАМФ регуляторного (R) димера и двух каталитических (С) цепей. Когда цАМФ связывается с R-димером, высвобождаются активные С-цепи, которые диффундируют через цитоплазму и ядро, где они переносят фосфат от АТФ к соответствующим субстратным белкам, часто имеющим ферментативные свойства.
Специфичность регулирующего действия цАМФ реализуется на определенных белковых субстратах киназы, преобладающих в тех или иных клетках и тканях. Например, в печени содержится
много фосфорилазы и гликогенсинтазы — ферментов, реципрокная регуляция активности которых с помощью цАМФ-зависимого фосфорилирования управляет резервированием и высвобождением углеводов. Адипоциты богаты липазой, цАМФ-зави-симое фосфорилирование которой способствует высвобождению свободных жирных кислот из жировой клетки. Сходным образом фосфорилирование киназы, специфичной в отношении легких цепей миозина (так называемая киназа легких цепей миозина, или КЛЦМ), участвует в расслаблении гладкой мускулатуры при действии р-адреномиме-тических аминов. Другие специфичные для клеток реакции на цАМФ как на вторичный посредник сходным образом зависят от многих ферментов, подлежащих регуляции путем фосфорилирования.
Когда гормональный стимул прекращает действие, внутриклеточные эффекты цАМФ завершаются путем выработки серии ферментов. Происхо-
АТФ АДФ
S S-P
р| ft
Фосфатаза
V Ответ
Рис. 2-14. Схема действия цАМФ в качестве вторичного посредника. Ключевые белковые молекулы: гормональный рецептор, стимулирующий G-белок (G„); аденилат-циклаза(АЦ), фосфодиэстеразы (ФДЭ), гидролизующие цАМФ; цАМФ-зависимые протеинкиназы с регуляторными (R) и каталитическими (С) субъединицами; белковые субстраты (S) киназ и фосфатаз, которые удаляют фосфаты от субстратных белков. Светлые стрелки указывают на регуляторные эффекты
дит дефосфорилирование субстрата различными специфическими и неспецифическими фосфатазами. цАМФ превращается в5'*АМФ под влиянием фосфодиэстераз циклических нуклеотидов (ФДЭ, рис. 2-14). Конкурентное ингибирование распада цАМФ является одним из механизмов действия кофеина, теофиллина и других метилксантинов (глава 19). : !
Б. Кальций и фосфоинозитиды. Еще одна хорошо известная система вторичных посредников связана с гормональной стимуляцией гидролиза фосфоинозитидов (рис. 2-15). Некоторые из гормонов, нейромедиаторов и факторов роста, которые активируют этот путь (табл. 2-1), связываются с рецептором, соединенным с G-белками, тогда как другие связываются с рецепторными тирозинкина-зами. Однако в любом случае важнейший этап процесса — стимуляция мембранного фермента фосфолипазы С (ФЛС), которая специфически гидролизует фосфолипидный компонент плазматической мембраны, обозначаемый как фосфатидилинози-тол-4,5-бифосфат (ФИБФ). ФИБФ распадается на две молекулы, выполняющие функцию вторичного посредника — диацилглицерол (ДАГ) и инози-
Агонист
Рис. 2-15. Са2+-фосфоивозитидвый сигнальный механизм. Ключевые протеины: рецепторы гормонов (R), G-белок (G), специфичная для фосфоинозитида фосфолипаза С (ФЛС), протеинкиназа С (ПК-С), субстрат киназы (S), кальмодулин (КМ) и кальмодулинсвязывающие ферменты (Е), включая киназы, фосфодиэстеразы и др. (ФИБФ — фосфатидилинозитол-4,5,-бифосфат; ДАГ — диацилглицерол. Светлые стрелки обозначают регуляторные влияния)
тол-1,4,5-трифосфат (ИТФ). Первый из этих посредников связан с мембраной, где он активирует фосфолипид- и Са2+-чувствительную протеинкина-зу, обозначаемую протеинкиназа С. Другой посредник (ИТФ) растворим в воде и диффундирует в цитоплазму, где он способствует высвобождению Са2+ из внутриклеточных хранилищ. Повышенная концентрация Са24 в цитоплазме способствует связыванию Са2+ с кальцийсвязывающим белком кальмодулином, регулирующим активность других ферментов, в том числе Са2+-зависимых протеинкиназ.
С учетом вовлечения множества вторичных посредников и протеинкиназ фосфоинозитидный сигнальный путь значительно более сложен по сравнению с системой цАМФ. Например, различные типы клеток могут содержать одну или более специализированных Са2+- и кальмодулинзависимых киназ с ограниченной субстратной специфичностью (например, КЛЦМ) дополнительно к Са2+- и каль-модулинзависимым киназам общего действия, которые могут фосфорилировать широкий круг белковых субстратов. В настоящее время идентифицированы не менее девяти структурно различных типов протеинкиназы С.
Многое из того, что нам известно о биологической роли фосфоинозитидных вторичных посредников, получено при использовании фармакологических агентов, которые активируют или кальциевый, или протеинкиназный пути. Концентрацию Са2+ в цитоплазме можно увеличить путем ионофореза, тогда как протеинкиназу С стимулируют связыванием с форболовыми эфирами или синтетическими диацилглицеролами. Один или оба класса названных агентов могут воспроизвести биологические эффекты, опосредуемые фосфоинозитидным сигнальным механизмом.
Как и в системе цАМФ, существуют способы уменьшения или прекращения сигнальной функции. ИТФ быстро инактивируется путем дефосфорилирования, ДАГ или фосфорилируется с образованием фосфатидной кислоты, которая затем вновь превращается в фосфолипиды или деацетилируется с образованием арахидоновой кислоты. Са2+ активно удаляется из цитоплазмы с помощью Са2+-помп.
Эти и иные нерецепторные элементы Са2+-фос-фоинозитидного сигнального механизма стали в настоящее время мишенями для воздействия новых лекарств. Например, терапевтический эффект иона лития, известного вещества для лечения маниакально-депрессивных состояний, может опосре
доваться влиянием на метаболизм фосфоинозитидов (глава 28).
В. цГМФ. В отличие от цАМФ, универсального передатчика различных сигналов, сигнальная роль цГМФ (циклический гуанозин-3',5'-монофос-фат) доказана лишь для некоторых типов клеток. В слизистой кишечника и гладких мышцах сосудов связанный с цГМФ механизм передачи сигналов функционирует параллельно с цАМФ-системой. Лиганды, узнаваемые рецепторами клеточной поверхности, стимулируют мембраносвязанную гуа-нилатциклазу с образованием цГМФ, а цГМФ действует путем стимуляции цГМФ-зависимой проте-инкиназы. Эффекты цГМФ в этих клетках прекращаются посредством ферментативной деградации циклического нуклеотида и дефосфорилирования субстрата киназы.
Повышенная концентрация цГМФ вызывает расслабление гладкой мускулатуры сосудов за счет опосредованного киназой механизма, итогом работы которого является дефосфорилирование легких цепей миозина. В этих гладкомышечных клетках синтез цГМФ может быть усилен двумя различными трансмембранными сигнальными механизмами с использованием двух разных гуанилатциклаз. Предсердный натрийуретический фактор (ANF) — пептидный гормон, образующийся в крови, стимулирует трасмембранный рецептор, связываясь с внеклеточным доменом. Это событие приводит к активации гуанилатциклазы, располагающейся на внутриклеточном домене рецептора. Другой механизм основан на проницаемости клеточных мембран для лиганда со стимулирующим действием, газообразного оксида азота (NO). Он образуется в клетках эндотелия сосудов в ответ на действие естественных сосудорасширяющих агентов, таких как ацетилхолин и гистамин (NO также называют эндотелиальным релаксирующим фактором). После поступления в клетку NO связывается с цитоплазматической гуанилатциклазой и активирует ее. Ряд важных сосудорасширяющих средств действует за счет образования NO или имитации его эффектов (главы И и 12).
Взаимодействие сигнальных механизмов
Са2+-фосфоинозитидный и цАМФ-сигнальный пути в некоторых клетках реципрокны, тогда как в других — комплементарны. Например, вазопрес
сорные вещества, которые вызывают сокращение гладкой мускулатуры, действуют посредством ИТФ-зависимой мобилизации Са2+, в то время как агенты, вызывающие расслабление гладкой мускулатуры, часто действуют путем повышения уровня цАМФ. С другой стороны, и цАМФ, и фосфоинозитидные вторичные посредники стимулируют высвобождение глюкозы из печени.
Фосфорилирование: общий механизм
Почти все механизмы передачи сигналов с помощью вторичных посредников используют обратимое фосфорилирование. Оно играет ключевую роль на каждом этапе, от рецепторов (аутофосфорилирование тирозинкиназ и десенситизация рецепторов, сопряженных с G-белками) до киназ, регулируемых вторичными посредниками, и, наконец, субстратов этих киназ. Последние сами по себе также могут быть киназами. Фосфатная ковалентная модификация выполняет две главных функции в передаче сигналов — усиление и гибкую регуляцию.
При усилении сигнала, подобно тому как это происходит в процессе связывания ГТФ с G-белком, присоединение фосфорильной группы к остаткам серина, треонина или тирозина молекулы субстрата умножает первоначальный импульс путем своеобразной записи в молекулярной памяти об активации данного пути. Дефосфорилирование “стирает” эту запись, причем медленнее, чем происходит диссоциация комплекса лиганд-рецептор.
Различная субстратная специфичность множества протеинкиназ, регулируемых вторичными посредниками, способствует разветвлению сигнальных путей. Это создает возможности для гибкой регуляции передачи сигнала. Таким образом, цАМФ, Са2+ и другие вторичные посредники используют для реализации различных эффектов в разных клетках имеющиеся в них определенные киназы и их субстраты.
Классы рецепторов и создание лекарств
Как мы могли убедиться, о существовании специфических лекарственных рецепторов судят по результатам изучения связи между строением и активностью группы структурно сходных соединений, которые имитируют эффект лекарства или
противодействуют ему. Таким образом, если группа родственных агонистов проявляет одинаковую относительную активность в отношении двух различных фармакологических эффектов, то вероятно, что эти эффекты опосредуются идентичными или сходными рецепторными молекулами. Кроме того, если оба эффекта реализуются через идентичные рецепторы, конкурентный антагонист будет подавлять оба эффекта при сходных значениях К. Другой конкурентный антагонист будет ингибировать оба эффекта при своем значении К|. Следовательно, изучение связи между строением и действием ряда агонистов и антагонистов может помочь в идентификации рецепторов, опосредующих набор фармакологических эффектов.
С помощью той же экспериментальной процедуры можно доказать, что наблюдаемые эффекты лекарств опосредуются различными рецепторами. В этом случае эффекты будут по-разному активироваться агонистами и иметь неодинаковые значения К; для каждого из конкурентных антагонистов.
Что бы мы ни рассматривали, мы всегда встречаемся с более чем одним классом рецепторов для каждого химического сигнала. Например, изучение зависимости структура-действие у химических аналогов ацетилхолина, гистамина и катехоламинов позволило идентифицировать множество рецепторов для каждого из этих эндогенных лигандов. Методы анализа связывания лигандов и молекулярного клонирования позволили выявить дополнительные рецепторы, например: два класса рецепторов вазопрессина, пять молекулярных видов мускариновых рецепторов (только три из которых выявляются методами лигандного связывания) и множество подтипов рецепторов для дофамина, опиоидных пептидов, серотонина и других веществ.
Почему же существует множество рецепторов для одного лиганда? Ответ может быть однозначным: клетки используют более чем один сигнальный путь для ответной реакции на каждый эндогенный лиганд и, следовательно, им необходим более чем один рецептор для одного и того же лиганда Так, ацетилхолин использует никотиновые АХР для инициации быстрого возбуждающего постсинаптического потенциала (ВПСП) в постганглионарных клетках вегетативных ганглиев и мускариновые рецепторы для медленного ВПСП, который модулирует реактивность к быстрому ВПСП в тех же самых клетках.
Существование множества рецепторов для каждого эндогенного сигнального лиганда создает много разных возможностей для создания лекарств. Хотя каждый эндогенный лиганд вызывает ряд клинических эффектов, с позиций терапии часто выгодно усилить некоторые из них, не затрагивая другие. Незначительные структурные изменения в местах связывания лиганда на двух рецепторах могут привести к тому, что связывание двух родственных лигандов будет происходить с разным аффинитетом. Если эти различия достаточны, может появиться возможность создать лекарство, которое будет действовать селективно, вызывая эффекты только через один из рецепторов. Так, антагонисты Р*ад-ренорецепторов могут блокировать учащение сердечных сокращений, вызванное норадреналином, не предупреждая вазоконстрикторное действие норадреналина через otf-адренорецепторы. Клиническое применение рецепторселективных лекарств рассматривается почти в каждой главе этой книги.
Создание новых лекарств не ограничено агентами, которые действуют на рецепторы внеклеточных химических сигналов. Фармацевтическая химия в наше время способна определить, могут ли элементы сигнальных путей, расположенные дистальнее рецепторов, также служить мишенью селективного и полезного эффекта лекарств. Например, могут быть созданы средства, действующие селективно на G-белки, киназы, фосфатазы или ферменты, разрушающие вторичные мессенджеры.
Связь между дозой лекарства и клиническим эффектом
Мы рассмотрели рецепторы как молекулы и показали, как они могут количественно влиять на связь между дозой или концентрацией лекарства и фармакологическими эффектами, по крайней мере, в идеальной системе. Когда речь идет о больном, врач должен выбрать конкретное лекарство и режим его дозирования, обеспечивающие максимальную терапевтическую эффективность при минимальной токсичности. Поскольку больного нельзя отождествлять с идеальной абстрактной системой, врач не имеет точной информации о физико-химической природе рецепторов данного человека, их плотности и аффинитете к лекарствам. Тем более для принятия рациональных терапевтических решений врач должен понимать, как взаимодей
ствие вещества и рецептора определяет связь между дозой и реакцией больного на лекарства, природу и причины вариабельности фармакологической реактивности, клиническое использование избирательности действия лекарств.
Доза и эффект у пациентов
А. Градуальная связь дозы и эффекта. Для выбора лекарств и определения соответствующей дозы врач должен знать их относительную фармакологическую активность (потентность) и максимальную эффективность. Эти два важных термина, которые часто путают студенты и врачи, могут быть объяснены при рассмотрении рис. 2-16, который отражает градуальные кривые доза-эффект, показывающие связь между дозой четырех различных лекарств и величиной конкретного фармакологического эффекта, например степенью снижения артериального давления у больного с гипертензией или увеличения почечной экскреции натрия у больного с застойной сердечной недостаточностью.
1. Активность. Вещества А и Б можно рассматривать как более активные, чем вещества В и Г из-за относительного положения их кривых доза-эффект вдоль оси абсцисс на рис. 2-16. Активность оценивают по концентрации (ЕС50) или дозе (ED50) лекарства, требуемой для получения эффекта, равного 50 % от максимального. Так, фармакологическая активность вещества А на рис. 2-16 меньше, чем у вещества Б (парциального агониста), поскольку ED5o у А больше, чем ED50 у Б. Отметим, что неко
торые дозы вещества А могут вызывать больший эффект, чем любая доза вещества Б, несмотря на то, что мы определили Б как фармакологически более активное вещество. Причина состоит в том, что вещество А имеет большую максимальную эффективность (пункт 2).
Потентность лекарства зависит от аффинитета рецепторов для связывания (KD) и от эффективности ответной реакции.
Для клинического использования полезно различать активность и эффективность лекарств. Клиническая эффективность лекарства зависит не от его активности (ЕС50), а от максимальной эффективности и его способности достигать соответствующего рецептора. Последняя определяется способом введения, абсорбцией, распределением в организме, удалением из него или от места действия. При принятии решения, какое из двух лекарств вводить больному, врач обычно должен в первую очередь оценить их относительную эффективность, а не относительную активность. Однако фармакологическая активность может в значительной степени определять дозу вводимого препарата. В общем виде низкая активность становится важной лишь тогда, когда ведет к назначению лекарства в несоразмерно больших количествах.
Для терапевтических целей активность лекарства должна быть определена в единицах дозы, обычно в конкретных конечных значениях (например, 50 мг для умеренного седативного эффекта, 1 мкг/кг/мин для увеличения частоты сердечных сокращений на 25 ударов в минуту). Относительная активность — соотношение равноэффективных
Логарифм концентрации
Рис. 2-16. Градуальные кривые доза-эффект для четырех лекарств, иллюстрирующие различную фармакологическую активность и максимальную эффективность
доз (0.2, 10, и т. п.), может быть использована для сравнения веществ друг с другом.
Максимальная эффективность. Этот параметр отражает предел для зависимости доза-эффект по оси ординат. Вещества А, В и Г на рис. 2-16 имеют одинаковую максимальную эффективность, большую, чем у Б. Максимальная эффективность (иногда обозначаемая просто как эффективность) лекарства является решающим фактором для принятия врачом решения в том случае, когда необходим сильный эффект. Она зависит от характера взаимодействия лекарства с рецептором (как в случае с парциальными агонистами)1 или от вовлеченной в фармакологическую реакцию рецептор-эффек-торной системы. Так, диуретики, которые влияют на определенный отдел нефрона, могут вызывать значительно большую экскрецию жидкости и электролитов, чем диуретики, которые действуют в другом отделе. Кроме того, эффективность лекарства в достижении определенного терапевтического действия (например, повышение сократительной способности миокарда) может ограничиваться способностью лекарства вызывать токсическое действие (например, серьезную аритмию сердца).
Б. Форма кривых доза-эффект. Несмотря на то, что кривые А, Б и В (рис. 2-16) приближаются по форме к простой зависимости Михаэлиса-Мен-тен, некоторые клинические эффекты не соответствуют такой зависимости. Очень крутые кривые доза-эффект (например, кривая Г) могут иметь особое клиническое значение, если верхняя часть кривой заходит в область нежелательных эффектов (например, кома, вызываемая седативными и гипнотическими средствами). Крутизна кривой определяется кооперативным взаимодействием нескольких различных эффектов лекарства (например, влияние на мозг, сердце и периферические со
’ Отметим, что “максимальная эффективность” при рассмотрении в терапевтическом контексте не имеет точно того же значения, что и термин, обозначающий более специальное взаимодействие лекарства и рецептора, рассмотренное ранее в этой главе. В идеальной системе in vitro эффективность обозначает относительную максимальную эффективность агонистов и парциальных агонистов, действующих через один и тот же рецептор. В терапии эффективность означает выраженность эффекта, который может быть достигнут у больного. Следовательно, на терапевтическую эффективность могут повлиять особенности взаимодействия лекарства и рецептора, но она твкже зависит и от ряда других факторов, рассматриваемых в тексте.
суды вносят свой вклад в понижение АД). Подобные крутые кривые доза-эффект могут быть связаны также с особенностями рецепторно-эффекторной системы, в которой большинство рецепторов должно быть занято, перед тем как появится какой-либо эффект.
В. Квантовые кривые доза-эффект. Несмотря на важность для характеристики действия лекарств, градуальные кривые доза-эффект имеют ряд ограничений при использовании в клинике. Такие кривые невозможно получить, если фармакологическая реакция имеет дискретный (квантовый) характер, как например предупреждение судорожной реакции, аритмии или смерти. Более того, клиническая значимость количественной связи дозы и эффекта у конкретного больного, вне зависимости от точности их определения, может иметь ограничения при использовании ее у других пациентов в связи с большой вариабельностью между больными по тяжести заболевания и реактивности на лекарственную терапию.
Некоторых сложностей можно избежать путем определения дозы препарата, требуемой для получения эффекта определенной величины у большого числа больных или экспериментальных животных, и графического представления данных о кумулятивном распределении частоты положительных ответов на лекарство в зависимости от логарифма дозы (рис. 2-17). Конкретный квантовый эффект может быть выбран на основании клинической значимости (например, облегчение головной боли) из соображений сохранения безопасности субъектов исследования (например, оценка эффекта при использовании малых доз стимуляторов сердца на уровне прироста частоты сокращений на 20 ударов в минуту) или при использовании альтернативных показателей (например, смерть экспериментального животного). Для большинства лекарств индивидуальные дозы, необходимые для специфического квантового эффекта, распределены логнормально, т. е. распределение частот таких реакций в зависимости от логарифма дозы характеризуется гауссовой кривой нормального распределения (рис. 2-17). Когда эти реакции суммируют, образующаяся кумулятивная кривая частотного распределения представляет собой квантовую кривую доза-эффект (или кривую доза-процент), отображающую процент наблюдений, в которых отмечен фармакологический эффект как функция логарифма дозы (рис. 2-17).
Суммарный Суммарный
« 1 IJ, . . I I 1 « « I |Д 1 1 1. I
1.25 2.5 Т5 10 20 40 80 160 320 640 । Доза (мг) |
ED50
ld50
Рис. 2-17. Квантовые кривые доза-эффект. Затемненные участки (и соответствующие кривые) показывают частоту распределения доз лекарства, которые требуются для получения специфического эффекта, т. е. количество животных (в процентах), которым необходима определенная доза для получения эффекта. Светлые столбики (и соответствующие кривые) указывают на кумулятивную частоту распределения ответных реакций, которая распределена логнормально
Квантовая кривая доза-эффект часто характеризуется средней (медианной) эффективной дозой (ED50), т. е. дозой, после введения которой у 50 % испытуемых наблюдается заданный квантовый эффект. (Заметьте, что обозначение ED50 в этом контексте имеет иной смысл, чем эта же аббревиатура при анализе градуальных кривых доза-эффект.) Сходным образом доза, требуемая для получения определенного токсического действия у 50 % животных, обозначается как средняя токсическая доза (TD50). Если токсический эффект приводит к смерти животного, в эксперименте может быть определена средняя летальная доза (LDS0). Такие измерения позволяют получить удобные показатели для сравнения активности лекарств в эксперименте и в клинике. Так, если ED50 двух веществ для получения определенного квантового эффекта составляют 5 и 500 мг соответственно, то можно утверждать, что первое лекарство в 100 раз более активно, чем второе в отношении определенного фармакологического эффекта. Аналогичным образом можно получить ценный показатель селективности дей
ствия лекарств, сравнивая значения ЕН50для двух различных квантовых, эффектов в популяции (например: подавление кашлевого рефлекса в сравнении с седативным действием для группы опиатных препаратов; увеличение частоты сердечных сокращений в сравнении с вазоконстрикторным действием для симпатомиметических аминов; противовоспалительный эффект в сравнении со способностью задерживать натрий для кортикостероидов и т. п.).
Квантовые кривые доза-эффект могут также использоваться для получения информации о границах безопасности применения конкретного лекарства. Одним из показателей, который используется для сравнения дозы, необходимой для получения желаемого терапевтического эффекта, и дозы, вызывающей нежелательный эффект, является терапевтический индекс. В опытах на животных терапевтический индекс обычно определяют как отношение TD50 к ED5o для заданного эффекта, рассматриваемого как аналог терапевтического. Точность, которая достигается в опытах на животных, позволяет использовать подобный терапевтический индекс для оценки потенциальной эффективности лекарства у человека. Конечно, терапевтический индекс лекарства для человека почти никогда не известен с необходимой точностью. Однако испытания лекарств и накапливающийся клинический опыт, как правило, выявляют диапазоны эффективных и потенциально токсических доз. Иногда эти диапазоны перекрещиваются. Клинически приемлемый риск токсичности зависит прежде всего от тяжести заболевания. Например, диапазон доз, прием которых уменьшает головную боль у подавляющего большинства больных, должен быть значительно ниже, чем диапазон доз, вызывающих серьезные токсические проявления, даже если они отмечаются у минимального числа пациентов. Однако для лечения смертельных болезней, таких как лимфома Ходжкина, допустимое различие между терапевтическими и токсическими дозами может оказаться меньше.
Наконец, отметим, что квантовые и градуальные кривые доза—эффект характеризуют несколько разные виды информации, хотя оба типа кривых имеют сигмовидную форму на полулогарифмическом графике (рис. 2-16 и 2-17). Наиболее важная информация, требуемая для принятия рационального решения, может быть получена при анализе любой из кривых: обе они предоставляют информацию об активности и селективности лекарств.
Градуальная кривая доза-эффект указывает на максимальную эффективность лекарства, квантовая — на потенциальную индивидуальную вариабельность реакции на лекарство.
Вариабельность реакции на лекарства
Реакции на лекарства у больных могут иметь значительные отличия. Конкретный пациент по-разному реагирует на одно и то же лекарство в различное время. В редких случаях у больных проявляются необычные реакции, или идиосинкразия. Она обычно связана с генетическими особенностями метаболизма лекарств или с индивидуальной иммунологической реактивностью, в том числе с аллергическими реакциями.
Количественные вариации в реакции на лекарства в целом встречаются чаще и более важны для клиники. Больной может быть гипореактивен или гиперреактивен по отношению к лекарству, что проявляется в снижении или повышении эффекта данной дозы в сравнении с тем эффектом, который наблюдается у большинства пациентов. {Заметьте: термин гиперчувствительность, как правило, часто означает аллергическую или друхую иммунологическую реакцию на лекарство.) Для некоторых лекарств выраженность реакции на данную дозу может изменяться по ходу курса лечения. При длительном назначении препарата реактивность обычно постепенно снижается, вызывая состояние относительной толерантности к эффекту лекарства. Когда реактивность снижается быстро, говорят о развитии тахифилаксни.
Общее клиническое значение индивидуальной вариабельности реакции на лекарство понятно: врач должен быть готов изменить или дозировку, или выбор лекарства в зависимости от реакции больного. Даже перед введением первой дозы препарата врач обязан оценить факторы, которые могут предсказать направление и выраженность возможных вариаций в реактивности. Это прежде всего способность некоторых лекарств вызывать толерантность или тахифилаксию, а также влияние возраста, пола, размеров тела, сопутствующих заболеваний, одновременного введения других препаратов.
В основе вариабельности реактивности на лекарства у больных или у одного больного в различное время лежат четыре общих механизма. Классификация, представленная ниже, несколько искусственна в том смысле, что большинство вариаций кли
нической реактивности имеют более чем один механизм. Тем не менее она может быть полезной для определения конкретной терапевтической стратегии.
А. Изменение концентрации лекарства в зоне рецептора. Пациенты могут отличаться по скорости всасывания лекарства, его распределению по тканям и выведению препарата из организма (глава 3). Любые из этих фармакокинетических отличий могут изменять концентрацию лекарства, которая создается у соответствующего рецептора и, следовательно, изменять клинический эффект. Некоторые отличия можно предсказать на основе учета возраста, массы тела, пола, характера заболевания, функции печени и почек больного. Подобное предсказание используется для решения о начальном режиме дозирования. Повторные измерения концентрации препарата в крови в ходе лечения полезны для контроля за вариабельностью клинической реакции, обусловленной индивидуальными особенностями фармакокинетики.
Б. Вариации в концентрации эндогенного лиганда рецептора. Этот механизм вносит большой вклад в вариабельность реакции на фармакологические антагонисты. Так, антагонист р-адреноре-цепторов пропранолол заметно замедляет частоту сердечных сокращений у больных с повышенным уровнем эндогенных катехоламинов (как при феохромоцитоме), но не влияет на фоновую частоту сердцебиений у хорошо тренированного бегуна-марафонца. Парциальные агонисты могут вызвать более опасные отличия в реакции, чем антагонисты: саралазин, слабый парциальный агонист рецепторов ангиотензина II, понижает артериальное давление у больных с гипертензией, вызванной повышенной продукцией ангиотензина II, и повышает артериальное давление у больных с низким уровнем выработки ангиотензина.
В. Изменение плотности или функции рецепторов. В эксперименте доказано, что реакция на лекарства может определяться увеличением или уменьшением числа рецепторных сайтов или изменением эффективности сопряжения рецепторов с расположенными дистально эффекторными механизмами. Хотя подобные изменения не были полностью выявлены у человека, вероятно, они ответственны за большинство случаев индивидуальной чувствительности в реакции на некоторые лекарства, особенно действующие на рецепторы гормонов, биогенных аминов и нейромедиаторов. В не
которых случаях изменениё числа рецепторов вызывается другими гормонами, например, гормоны щитовидной железы увеличивают плотность 0-ре-цепторов в миокарде крыс, а также чувствительность сердца к катехоламинам. Сходные изменения, возможно, ответственны за тахикардию при тиреотоксикозе и указывают на целесообразность назначения антагониста p-адренорецепторов пропранолола для облегчения состояния больных.
В других случаях лиганд-агонист способствует снижению плотности (г/огеш-регуляция) или эффективности сопряжения рецептора и эффектора. Специфичные для рецептора механизмы десенситиза-ции, возможно, участвуют в физиологическом процессе адаптации клетки к изменению гормональной или нейромедиаторной стимуляции. Эти механизмы могут играть роль в проявлении двух важных для клиники феноменов: во-первых, тахифи-лаксии или толерантности к эффектам некоторых лекарств (например, биогенным аминам и их аналогам) и, во-вторых, явлении “отдачи", которое сопровождает отмену некоторых лекарств. Эти феномены могут быть связаны как с агонистами, так и с антагонистами. Антагонист может увеличить плотность рецепторов в клетке или ткани-мишени, предупреждая down-регуляцию, вызываемую эндогенным агонистом. Когда введение антагониста прекращают, увеличение плотности рецепторов может способствовать усиленному ответу на физиологическую концентрацию агониста. Потенциально опасные симптомы отмены могут быть связаны с противоположной реакцией после прекращения введения лекарства-агониста. В этом случае число рецепторов, которое уменьшилось в результате действия агониста по механизму отрицательной обратной связи, оказывается слишком малым, для того чтобы эндогенный агонист оказал эффективное собственное стимулирующее действие. Например, отмена клонидина (лекарства, чья агонистическая активность в отношении сс2-адренорецепторов способствует снижению артериального давления) может привести к гипертензивному кризу, возможно, вследствие down-регуляции а2-адренорецепторов (глава 11).
Для влияния на рецептор-специфичные изменения реактивности на лекарство могут быть использованы различные терапевтические стратегии в зависимости от клинической ситуации. Толерантность к действию лекарств требует увеличения дозы или замены препарата. Регуляция рецепторов
по механизму отрицательной или положительной обратной связи (down- или г/р-регуляция) может быть связана с риском последствий прекращения приема некоторых лекарств. В данной ситуации необходимо медленное прекращение лекарственного лечения с тщательным контролем признаков реакции отмены.
Г. Изменение компонентов реакции, расположенных дистальнее рецептора. Хотя лекарстВо инициирует действие путем связывания с рецепторами, наблюдаемая реакция пациента зависит от функциональной интеграции биохимических процессов в клетке-мишени и физиологической рефляции взаимодействия органов и систем. Клинически изменения этих пострецепторных процессов представляют собой наибольший и наиболее важный класс механизмов, ответственных за вариабельность в реактивности на лекарственную терапию.
Перед началом лечения врач должен учесть особенности больного, которые могут изменить реакцию на препарат. Это прежде всего возраст, общее состояние здоровья и, что особенно важно, тяжесть и патофизиологический механизм заболевания. Как только лечение начато, основной причиной отсутствия удовлетворительного результата становится факт неточности или физиологической необоснованности диагноза. Так, при застойной сердечной недостаточности могут оказаться неэффективными препараты, повышающие сократительную способность миокарда, если лежащий в основе патологии механизм связан с нераспознанным стенозом митрального клапана, а не собственно с недостаточностью миокарда. Напротив, лекарственная терапия всегда будет успешна, если она соответствует патофизиологическому механизму заболевания.
Даже при условии, что диагноз точен и лекарство адекватно, лечение все-таки может не дать оптимального результата. Неудовлетворительный терапевтический эффект подчас обусловлен включением компенсаторных механизмов организма больного, которые нивелируют действие препарата. Например, компенсаторное увеличение симпатического тонуса и задержка жидкости почками могут способствовать развитию толерантности к антигипертензивному эффекту вазодилататоров. В этих случаях может потребоваться дополнительное средство для достижения нужного терапевтического эффекта.
Клиническая селективность: сравнение желательного и токсического эффектов лекарств
Хотя мы классифицируем лекарства в соответствии с их принципиальным действием, очевидно, что ни одно лекарство не вызывает только один специфический эффект. В чем причина этого? Маловероятно, что любая молекула лекарства, сможет связаться только с одним типом рецепторных молекул, поскольку число потенциальных рецепторов у каждого пациента имеет астрономическое значение (если учесть, что в геноме человека закодировано более 105 различных пептидных генных .продуктов и что сложность химической структуры каждого из них достаточна для формирования множества различных потенциальных сайтов для связывания).
Соответственно, лекарства скорее селективны, чем специфичны в своем действии, потому что они связываются с одним или несколькими типами рецепторов более прочно, чем с другими. Как мы видели, селективность можно измерить путем сравнения аффинитета лекарств к различным рецепторам или путем сравнения ED50 для различных эффектов препаратов in vivo. В фармакологии и клинической медицине о селективности обычно судят, разделяя виды действия на две категории: желательные (терапевтические) и токсические эффекты. Врачи и фармакологи часто используют термин "побочные эффекты”, рассматривая их как несущественные или развивающиеся побочно по сравнению с главным эффектом, что подчас ошибочно.
Важно понимать, что отнесение определенного эффекта лекарства к терапевтическому или токсическому основано на оценочных суждениях, но не связано с заключением о фармакологическом механизме, лежащем в основе эффекта. Оценка эффекта, в свою очередь, зависит от клинического контекста применения лекарства.
Только благодаря своей селективности, лекарства с успехом применяются в клинической медицине. Для лечения больных и для создания новых препаратов важно анализировать механизмы связи терапевтических и токсических эффектов с целью повышения селективности и эффективности лекарственной терапии. На рис. 2-18 изображены три возможных типа связи между терапевтическими и токсическими эффектами лекарств на основе анализа рецепторно-эффекторных механизмов.
А. Терапевтические и токсические эффекты, опосредуемые одним и тем же рецепторно-эффекторным механизмом. Многие виды токсического действия лекарств являются прямым продолжением их фармакологического терапевтического эффекта. В некоторых случаях (кровоточивость, связанная с применением антикоагулянтов, гипогликемическая кома после введения инсулина) токсического действия можно избежать при правильной дозировке с одновременным мониторингом эффекта (измерение параметров свертывания крови или уровня глюкозы в сыворотке) и предупредительными мероприятиями (профилактика травматических повреждений тканей с кровоизлияниями, регулирование пртребдения углеводов). В иных случаях токсического действия препарата проще избежать, заменяя его на другой, за исключением назначений по жизненным показаниям.
В некоторых ситуациях лекарство определенно показано, но в терапевтических дозах вызывает выраженный токсический эффект. Тогда в схему лечения необходимо добавить другой препарат. Например, празозин (глава 11) понижает артериальное давление при эссенциальной гипертонии, действуя как at-селективный антагонист на рецепторы гладкой мускулатуры сосудов. Вследствие этого действия и при достаточно большой дозе у больного может возникнуть постуральная гипо-
Рецептор Эффектор Ответ
1.D + RvbDR
Токсический эффект
Улучшение
Улучшение
Токсический эффект
3.D +
R^DR,---->Х
R2^DR2--->Y
Токсический эффект
Улучшение
Рис. 2-18. Возможные типы связи между терапевтическим и токсическим эффектами лекарства, опосредуемые различными рецепторно-эффекторными механизмами (D — лекарство; R — рецептор; X, Y — эффекторы)
тензия (заметим, что постуральная гипотензия рассматривается как “побочный эффект” празозина, хотя на самом деле это прямой эффект, связанный с главным терапевтическим действием лекарства). Решение этой проблемы определяется тем, что ар* териальное давление регулируется также изменением объема крови и тонуса гладкой мускулатуры артерий, а не исключительно симпатической иннервацией. Поэтому сочетанное введение диуретиков и вазодилататоров позволяет снизить дозу празозина и избежать постуральной гипотензии, контролируя артериальное давление.
Б. Терапевтические и токсические эффекты, опосредуемые идентичными рецепторами, но различными тканями или различными эффекторными путями. Примерами таких лекарств являются гликозиды наперстянки, которые используют для увеличения сократительной способности миокарда, но которые в то же время способствуют аритмиям сердца, нарушениям функционирования желудочно-кишечного тракта (ЖКТ) и изменениям зрительной функции (все эффекты, вероятно, связаны с ингибированием Ма+,К+-АТФазы в клеточных мембранах). К этой же группе относят метотрексат, применяемый для лечения лейкемии и других новообразований, который также убивает нормальные клетки костного мозга и слизистой ЖКТ (все эффекты опосредуются ингибированием фермента дигидрофолатредуктазы), и производные глюкокортикоидных гормонов, используемые для лечения астмы или воспалительных заболеваний, вызывающие, однако, катаболизм белков, психозы и другие токсические эффекты (все опосредуются сходными или идентичными глюкокортикоидными рецепторами). Таким образом, эти и им подобные нежелательные эффекты многих лекарств опосредуются рецепторами, идентичными тем, которые ответственны за основной терапевтический эффект.
Для предупреждения или устранения данного варианта токсичности используют три стратегии. Во-первых, лекарство всегда следует вводить в наименьшей дозе, которая вызывает приемлемый терапевтический эффект, признавая, что полное устранение симптомов и признаков заболевания может быть недостижимо. Во-вторых, как уже было рассмотрено для празозина, можно снизить дозу (таким образом ограничивая его токсичность), назначая одновременно лекарство, которое оказывает эффект через иные рецепторы и имеет иной профиль
токсичности. Так, при лечении воспалительных заболеваний использование иммуносупрессивных агентов целесообразно сочетать с глюкокортикоидами. В-третьих, селективность действия лекарств может быть увеличена путем управления концентрацией лекарства в районе рецепторов в различных отделах организма. Подобная “анатомическая” селективность может быть достигнута, например, аэрозольной доставкой глюкокортикоидов к бронхам или артериальной инфузией антиметаболита в орган, в котором находятся опухолевые клетки.
В. Терапевтические и токсические эффекты, опосредуемые различными типами рецепторов. Терапевтические преимущества новых химических соединений с улучшенной рецепторной селективностью упоминались ранее в этой главе и детально рассмотрены в последующих главах. Подобные лекарства представлены а- и Р-селективными агонистами адренорецепторов, Нг и Н2-антигистаминны-ми веществами, блокаторами никотиновых и мускариновых рецепторов, рецепторселективными стероидными гормонами. Соответствующие рецепторы группируются в функциональные семейства, каждое из которых реагирует только на ограниченный набор эндогенных агонистов. Эти рецепторы и связанные с ними терапевтические возможности были открыты при анализе эффектов сигнальных веществ — катехоламинов, гистамина, ацетилхолина и кортикостероидов.
Ряд других лекарственных препаратов, не обладая подобной избирательностью, оказывает терапевтические эффекты через одно семейство рецепторов, а токсические — через иные. В качестве примеров можно привести хинидин, производные сульфанилмочевины, тиазидные диуретики, трициклические антидепрессанты, ингибиторы моно-аминоксидазы, фенотиазиновые антипсихотические вещества и многие другие. Вероятно, для некоторых из этих лекарств будет показана возможность действия через рецепторы для эндогенных агонистов, как например в случае мощного аналге-тика морфина. Было обнаружено, что морфин действует на рецепторы, которые в физиологических условиях взаимодействуют с опиоидными пептидами.
Таким образом, способность лекарств связываться с различными классами рецепторов не только важна при лечении больных, но и представляет собой постоянный стимул для создания новых и более эффективных препаратов.
Избранная литература
Becker А. В., Roth R. A. Insulin receptor structure and function in normal and pathological conditions. Annu. Rev. Med. 1990; 41:99.
Berridge M. J. Inositol trisphosphate and calcium signaling. Nature, 1993; 361:315.
Goldstein A., Aronow L., Kalman S. M. Principles of Drug Action: The Basis of Pharmacology. 2nd ed. Wiley, 1974.
Kenakin T. P. Pharmacologic Analysis of Drug-Receptor Interaction. Raven Press, New York, 1987.
Lowenstein C. J., Snyder S. H. Nitric oxide, a novel 'biologic messenger. Cell, 1992; 70:705.
Фармакокинетика
и фармакодинамика: рациональный выбор дозы и времени действия лекарства
Николас X. Г. Холфорд, Лесли 3. Бенет
Целью лекарственной терапии является достижение желаемого лечебного действия при минимальных побочных эффектах. После того как лекарство выбрано для больного, врач должен определить дозу, которая позволит приблизиться к этой цели. Рациональный подход состоит в объединении принципов фармакокинетики и фармакодинамики для выявления связи между дозой и эффектом (рис. 3-1). Фармакодинамика характеризует зависимость концентрация-эффект, тогда как фармакокинетика имеет дело со связью дозы и концентрации (Holford, 1981). Фармакокинетические процессы абсорбции, распределения и элиминации определяют, как быстро и в каких концентрациях лекарство появится в органе-мишени и как долго будет там находиться. Фармакодинамические концепции максимального ответа и чувствительности определяют величину эффекта при определенной концентрации (Ешах и ЕС5о, глава 2).
Рис. 3-1 иллюстрирует основную гипотезу фармакологии. А именно то, что существует связь между полезным или токсическим эффектом лекарства и его концентрацией в легко доступной среде организма (например, в крови). Эта гипотеза получила подтверждения для множества лекарств, что иллюстрируется значениями эффективных и токсических концентраций в табл. 3-1. Очевидное отсутствие такой связи для некоторых лекарств не противоречит основной гипотезе, но указывает на необходимость измерения во времени концентрации лекарства в конкретном месте реализации фармакологического эффекта.
Знание зависимости между концентрацией лекарства и эффектом позволяет врачу учитывать
различные патологические и физиологические характеристики определенного пациента, которые отличают его от усредненного типа ответной реакции на лекарство. Применение принципов фармакокинетики и фармакодинамики дает возможность повышения терапевтической эффективности и снижения токсичности.
Фармакокинетика
“Стандартная” доза лекарства определяется клиническими испытаниями на здоровых добровольцах и пациентах со средней способностью к всасыванию, распределению и элиминации лекарства (глава 5).
Эта доза не подходит для каждого больного. Некоторые физиологические особенности (например, созревание функции органов у детей) и патологические процессы (сердечная или почечная недостаточность) диктуют необходимость поправок в индивидуальные режимы дозирования. Эти процессы изменяют специфические фармакокинетические параметры. Двумя основными параметрами являются клиренс — мера способности организма элиминировать лекарство, и объем распределения — мера кажущегося пространства в органйз-ме, способного вместить лекарство. Эти параметры схематично проиллюстрированы на рис. 3-2, где объем компартментов, в которые диффундирует лекарство, представляет собой объем распределения, а величина внешней “утечки” (рис. 3-2Б й 3-2Г) — клиренс.
Фармакокинетика
Фармакодинамика
Рис. 3-1. Связь между дозой и эффектом может быть разделена на фармакокинетический (доза-концентрация) и фармакодинамический (концентрация-эффект) компоненты. Концентрация является соединительным звеном между фармакокинетикой и фармакодинамикой; достижение необходимой концентрации лежит в основе рационального дозирования лекарств. Три основных процесса фармакокинетики — всасывание, распределение и элиминация
Объем распределения
Объем распределения (VD) связывает количество лекарства в организме с концентрацией лекарства (С) в крови или плазме:
V - Объем лекарства в организме С
Объем распределения можно определить по отношению к крови, плазме или водной части плазмы (несвязанное лекарство) в зависимости от концентрации, которая используется в уравнении [3-1] (С — СКр, С„л или Снеси).
То, что VD, рассчитанный из уравнения [3-1], является кажущимся объемом, можно видеть путем сравнения объемов распределения таких лекарств, как дигоксин и хлорохин (табл. 3-1) с некоторыми физическими объемами в организме (табл. 3-2). VD может значительно превышать любой физический объем, так как VD — это объем, необходимый, для того чтобы равномерно распределить количество лекарства в той концентрации, которая определяется в крови, плазме или в водной части плазмы. Лекарства с очень высокими объемами распределения ха
рактеризуются значительно более высокими концентрациями в тканях вне сосудов, чем внутри сосудистого русла, т. е. они распределены неравномерно. С другой стороны, препараты, которые полностью задерживаются внутри сосудистого русла, имеют минимально возможный показатель VD, равный объему крови, в которой они распределяются, например 0.04 л/кг массы тела (табл. 3-2), или 2.8 л/70 кг для лекарств, которые концентрируются в плазме.
Клиренс
Принципы лекарственного клиренса сходны с концепцией клиренса в физиологии почек, в соответствии с которой клиренс креатинина определяется как скорость элиминации креатинина с мочой в сравнении с его концентрацией в плазме. В простейшем случае клиренс лекарства — это отношение скорости элиминации всеми возможными путями к концентрации лекарства в биологической жидкости:
Скорость элиминации
Количество лекарства в крови
О Время
Рис. 3-2. Модели распределения и элиминации лекарства. Эффект быстрого внутривенного введения представлен как введение известного количества вещества в стеклянный сосуд. Временной ход, отражающий изменение количества вещества в сосуде, показан на графике справа. В первом примере (А) вещество не выходит из сосуда, поэтому трафик показывает только быстрый подъем до максимума с последующим плато. Во втором примере (Б) имеется элиминация, и на графике показано медленное снижение количества вещества в крови после быстрого подъема до максимума. Поскольку уровень жидкости в сосуде снижается, гидростатическое давление, управляющее элиминацией на этой модели, также снижается, и наклон кривой уменьшается, приближаясь асимптотически к постоянному уровню. Это кривая экспоненциального снижения. В третьей модели (В) вещество, введенное в первый компартмент (кровь), находится в равновесии со вторым компарт-ментом (внесосудистый объем), и количество вещества в “крови” экспоненциально снижается до нового стационарного состояния. Четвертая модель (Г) иллюстрирует более реальное сочетание механизмов элиминации и уравновешивания с внесосудис-той средой. Результирующий трафик показывает начальную фазу распределения, за которой следует медленная фаза элиминации. Эти кривые можно преобразовать в линейную форму, отобразив на графике изменение во времени логарифма количества вещества
Клиренс, подобно объему распределения, может определяться относительно крови (CLKp), плазмы (СЬ„Л) или как клиренс несвязанного вещества в водной части плазмы (CLHec„) в зависимости от того, где определяется концентрация лекарства.
Важно отметить аддитивный характер клиренса. Элиминация вещества из организма может происходить с участием процессов, идущих в почках, легких, печени и других органах. Путем деления скорости элиминации в каждом из органов на концент-
ТАБЛИЦА 3-1. Фармакокинетические и фармакодинамические параметры некоторых лекарств
Лекарство Биодоступность при приеме внутрь (F, %) Экскреция с мочой (%) Связывание в плазме (%) Клиренс (л/час/ 70 кг)1 Объем распределения (л/70 кг) Период полувыведения (часы) Эффективные концентрации Токсические концентрации
Ацетаминофен 88 3 0 21 67 2 10-20 мг/л > 300 мг/л
Ацикловир 23 75 15 19.8 48 2.4 — —
Амикацин 98 4 5.46 19 2.3 — —
Амоксициллин 93 86 18 10.8 15 1.7 — —
Амфотерицин 4 90 1.92 53 18 — —
Ампициллин 62 82 18 16.2 20 1.3 — —
Аспирин 68 1 49 39 11 0.25 — —
Атенолол 56 94 5 10.2 67 6.1 1 мг/л —
Атропин 50 57 18 24.6 120 4.3 — —
Каптоприл 65 38 30 50.4 57 2.2 50 нг/мл —
Карбамазепин 70 1 74 5.34 98 15 6.5 мг/л > 9 мг/л
Цефалексин 90 91 14 18 18 0.9 — —
Цефалотин 52 71 28.2 18 0.57 .— —
Хлорамфеникол 80 25 53 10.2 66 2.7 — —
Хлордиазепоксид 100 1 97 2.28 21 10 > 0.7 мг/л —
Хлорохин 89 61 61 45 13000 8.9 15-30 нг/мл 250 нг/мл
Хлорпропамид 90 20 96 0.126 6.8 33 — —
Циметидин 62 62 19 32.4 70 1.9 0.8 мг/л —
Ципрофлоксацин 60 65 40 25.2 130 4.1 — —
Клонидин 95 62 20 12.6 150 12 0.2-2 нг/мл —
Циклоспорин 23 1 93 24.6 85 5.6 100-400 нг/мл > 400 нг/мл
Диазепам 100 1 99 1.62 77 43 300-400 нг/мл —
Дигитоксин 90 32 97 0.234 38 6.7 >10 нг/мл > 35 нг/мл
Дигоксин 70 60 25 7.8 440 39 >0.8 нг/мл > 2 нг/мл
Дилтиазем 44 4 78 50.4 220 3.7 — —
Дизопирамид 83 55 ?2 5.04 41 6 3 мг/л > 8 мг/л
Эналаприл 95 90 55 9 40 3 > 0.5 нг/мл —
Эритромицин 35 12 84 38.4 55 1.6 — —
Этамбутол 77 79 5 36 110 3.1 — > 10 мг/л
Флуоксетин 60 3 94 40.2 2500 53 — —
Фуросемид 61 66 99 8.4 7.7 1.5 — 25 мг/л
Гентамицин 90 10 5.4 18 2.5 — —
Гидралазин 40 10 87 234 105 1 100 нг/мл —
Имипрамин 40 2 90 63 1600 18 100-300 нг/мл > 1 мг/л
Индометацин 98 15 90 8.4 18 2.4 0.3-3 мг/л > 5 мг/л
Лабеталол 18 5 50 105 660 4.9 0.13 мг/л
Лидокаин 35 2 70 38.4 77 1.8 1.5-6 мг/л > 6 мг/л
Литий 100 95 0 1.5 55 22 ' 0.5-1.25 мэкв/л > 2 мэкв/л
Меперидин 52 12 58 72 310 3.2 0.4-0.7 мг/л .—
Метотрексат 70 48 34 9 39 7.2 10 мкмоль/л
Метопролол 38 10 11 63 290 3.2 25 нг/мл .—
Метронидазол 99 10 10 5.4 52 8.5 3-6 мг/л —
ТАБЛИЦА 3-1. (Продолжение)
Лекарство Биодо- Экскре- Связи- Клиренс Объем Период Эффектив- Токси-
ступность ция вание (л/час/ распре- полу- ные ческие
при приеме с в 70 кг)1 деления выве- концент- концент-
внутрь мочой плазме (л/70 кг) дения рации рации
(F, %) (%) (%) (часы)
Мидазолам 44 56 95 27.6 77 1.9 — —
Морфин 24 8 35 60 230 1.9 65 нг/мл —
Нифедипин 50 0 96 29.4 55 1.8 47 нг/мл —
Нортриптилин 51 2 92 30 1300 31 50-140 нг/мл > 500 нг/мл
Фенобарбитал 100 24 51 0.258 38 4.1 10-25 мг/л > 30 мг/л
Фенитоин 90 2 89 В зависи- 45 В зависи- 10 мг/л > 20 мг/л
МОСТИ от мости от
концентрации концентрации4
Празозин 68 1 95 12.6 42 2.9 — —
Прокаинамид 83 67 16 36 130 3 3-14 мг/л > 14 мг/л
Пропранолол 26 1 87 50.4 270 3.9 20 нг/мл —
Пиридостигмин 14 85 36 77 1.9 50-100 нг/мл —
Хинидин 80 18 87 19.8 190 6.2 2-6 мг/л > 8 мг/л
Ранитидин 52 69 15 43.8 91 2.1 100 нг/мл —
Рифампин ? 7 89 14.4 68 3.5 — —
Салициловая кислота 100 15 85 0.84 12 13 150-300 мг/л > 200 мг/л
Сульфаметоксазол 100 14 62 1.32 15 10 — —
Тербуталин 14 56 20 14.4 125 14 2.3 нг/мл —
Тетрациклин 77 58 65 7.2 105 11 — —
Теофиллин 96 18 56 2.88 35 8.1 10-20 мг/л > 20 мг/л
Тобрамицин 90 10 4.62 18 2.2 — —
Токаи нид 89 38 10 10.8 210 14 6-15 мг/л —
Толбутамид 93 0 96 1.02 7 5.9 80-240 мг/л —
Триметоприм 100 69 44 9 130 11 — —
Тубокурарин — 63 50 8.1 27 2 0.6 мг/л —
Вальпроевая кислота 100 2 93 . 0.462 9.1 14 30-100 мг/л >150 мг/л
Ванкомицин — 79 30 5.88 27 5.6 — —
Верапамил 22 3 90 63 350 4 — —
Варфарин 93 3 99 0.192 9.8 37 — —
Зидовудин 63 18 25 61.8 98 1.1 — —
1 Преобразуется в мл/мин путем умножения на 16.6.
2 Варьирует в зависимости от концентрации.
3 Может быть оценен по Ср с помощью уравнения CL = Vmax / (К„, + Ср); Vmax = 415 мг/сут, К„, = 5 мг/л.
4 Варьирует в связи с зависимостью клиренса от концентрации.
5 Прочерк означает отсутствие данных.
ТАБЛИЦА 3.2. Физические объемы (л/кг массы тела) некоторых компартментов, в которых распределяются лекарства
Компартмент и объем Примеры лекарств
Общая жидкость в организме (0.6 л/кг’) Внеклеточная жидкость (0.2 л/кг) Кровь (0.08 л/кг); плазма (0.04 л/кг) Жир (0.2-0.35 л/кг) Кость (0.07 л/кг) Мелкие молекулы, растворимые в воде, например этанол Водорастворимые молекулы большего размера, например ман-нитол Молекулы, прочно связанные с протеинами плазмы, и очень крупные молекулы, например гепарин Молекулы, хорошо растворимые в липидах, например ДДТ Некоторые ионы, например свинец, фтор
1 Усредненные значения. У молодого худощавого человека показатель может быть порядка 0.7 л/кг, а у полной женщины — 0.5 л/кг.
рацию лекарства мы получаем относительный клиренс каждого органа. В совокупности эти отдельные показатели образуют общий системный клиренс:
CL = Скорость почечной элиминации [3-за] почечн с
CL _ Скорость элиминации печенью [З-Зб] С
CL _ Скорость элиминации др. органами [3-3В]
ДР Q
СЬсистемный = СЬПочечн.+ СЬпечени + С1др. [З-Зг]
К “другим” тканям, элиминирующим лекарства, относятся легкие, кровь, мышцы и т. п., где вещества метаболизируются. Пример, приведенный в уравнениях [З-За] — [З-Зг], показывает, что лекарство элиминируется печенью, почками и другими тканями и что эти пути элиминации включают все возможные способы, с помощью которых лекарство выводится из организма.
Два основных органа, в которых осуществляется элиминация лекарств,— это почки и печень. Клиренс неизмененного лекарства, определяемый по выведению с мочой, представляет собой почечный клиренс. В печени элиминация лекарств осуществляется путем биотрансформации исходного вещества в один или несколько метаболитов, путем экскреции неизмененного вещества с желчью или обоими способами одновременно. Пути биотрансформации обсуждаются в главе 4. Для большинства лекарств клиренс является постоянной величиной для диапазонов концентраций в плазме или крови,
обычно наблюдаемых в клинике. Например, если элиминация является ненасыщаемой и ее скорость прямо пропорциональна концентрации, то путем преобразования уравнения [3-2] получаем:
Скорость элиминации = CL х С. [3-4]
Иногда такой процесс обозначают как элиминацию “первого порядка”. Когда клиренс осуществляется таким образом, его можно измерить путем расчета площади под кривой на графике время-концентрация после введения заданной дозы. Клиренс пропорционален дозе, разделенной на величину площади под кривой.
А. Элиминация, ограниченная по объему. Для лекарств, возможности элиминации которых ограничены (например, фенитоин, этанол), клиренс может варьировать в зависимости от концентрации лекарства (табл. 3-1). Подобная ограниченная элиминация обозначается иногда как насыщаемая, зависимая от дозы или концентрации, нелинейная и соответствующая уравнению Михаэлиса-Ментен.
Большинство путей элиминации становятся насыщаемыми, если дозы лекарств достаточно велики. Когда кровоснабжение органа не лимитирует элиминацию, связь между скоростью элиминации и концентрацией математически выражается уравнением [3-5]:
V хС
Скорость элиминации= —^----. [3-51
Кт+С
Это уравнение сходно с уравнением Михаэлиса-Ментен, характеризующим кинетику ферментов. Максимальная способность элиминации соответ
ствует значению V,nax в уравнении Михаэлиса-Мен-тен, а К,„ соответствует концентрации лекарства, при которой скорость элиминации составляет 50 % от максимальной. Важно заметить, что в связи с нелинейностью прирост скорости элиминации становится меньше при увеличении концентрации. При высоких по отношению к К,„ концентрациях скорость элиминации почти не зависит от концентрации, что обозначается как элиминация “псевдо-нулевого порядка”. Если дозировка превышает возможности элиминации, равновесное состояние может не достигаться: концентрация буДет повышаться по мере продолжения введения препарата. Такой характер ограниченной по объему элиминации важен для трех широко используемых веществ — фенитоина, этанола и аспирина. Это необходимо учитывать при судебно-медицинской оценке концентрации этанола в крови.
Б. Элиминация, зависимая от кровотока. В отличие от элиминации, ограниченной по объему, некоторые лекарства элиминируются настолько легко, что при реально возможных в клинике концентрациях кровь, перфузирующая орган, очищается при первом же прохождении лекарства через него. Элиминация подобных препаратов, таким образом, определяется прежде всего кровотоком через орган. Такие лекарства (перечисленные в табл. 4-6) можно обозначить как “легкоэкстрагируемые”, так как они почти полностью захватываются органом из крови.
Период полувыведения
Период полувыведения (П/2) — это время, необходимое для снижения наполовину количества лекарства в организме в процессе элиминации. В простейшем (и наиболее удобном для выбора режима дозирования лекарства случае) организм рассматривается как единое целое (компартмент, камера, что показано на рис. 3-2 Б), имеющее размер, равный VD. Поскольку органы элиминации могут очищать от лекарства только ту кровь или плазму, которая проходит через данный орган, эта кровь или плазма находится в равновесном состоянии с общим объемом распределения. Таким образом, ti/2 будет зависеть как от объема распределения, так и от клиренса:
tf2 =
0.71хУр CL
[3-6]
Однако фармакокинетика многих лекарств может быть описана многокамерными (многочастевы-ми) моделями, как показано на рис. 3-2В и 3-2Г. В этих условиях, когда более чем один показатель периода полувывеДений может быть применен к одному препарату, “истинное”, окончательное значение периода полувыведения, представленное в табл. 3-1, может быть больше, чем рассчитанное по уравнению [3-6]. Тем не менее ti/2 является полезным фармакокинетическим параметром, поскольку характеризует время, необходимое для достижения 50 % стационарной концентрации или для уменьшения концентрации наполовину от стационарной после изменения (например, начала или прекращения) установленной частоты приема лекарства (режима дозирования). Рис. 3-3 показывает временной ход накопления лекарства во время инфузии с постоянной скоростью и временной ход
Рис. 3-3. Временной ход кумуляции и элиминации лекарства. Сплошная линия — концентрации в плазме, отражающие накопление лекарства в процессе инфузии с постоянной скоростью. 50 % от стационарной концентрации достигается через один период полувыведения, 75 % — после двух и более 90 % — после четырех периодов полувыведения. Пунктирная линия — концентрации в плазме, отражающие элиминацию после прекращения инфузии при достижении стационарного уровня. 50 % вещества выводится через один период полувыведения, 75 % — после двух и т. д. В общем случае после начала режима дозирования должно пройти четыре периода полувыведения, пока будет заметен полный эффект, что соответствует приближению кривой кумуляции к отметке выше 90 % по отношению к окончательной стационарной концентрации
1 Постоянная 0.7 в уравнении 3-6 является аппроксимацией величины 0.693, натурального логарифма числа 2. Поскольку элиминация лекарства относится к экспоненциальным процессам, время, необходимое для двукратного снижения концентрации, пропорционально In (2).
элиминации после прекращения инфузии и достижения стационарного состояния.
Период полувыведения сам по себе недостаточен для использования в качестве показателя элиминации лекарства или его распределения. Заболевание может повлиять на физиологические параметры, объем распределения, клиренс, и, таким образом, производный показатель t1/2He обязательно будет отражать ожидаемые изменения элиминации лекарства. Например, у больного с хронической почечной недостаточностью может быть снижен почечный клиренс дигоксина и объем распределения; увеличение периода полувыведения дигоксина при этом не будет столь велико, как можно было бы ожидать по изменению почечной функции. Снижение VD происходит вследствие уменьшения массы скелетной мускулатуры и почек и последующего снижения связывания дигоксина тканями.
Кумуляция лекарств
При повторном введении лекарства оно может накапливаться в организме до тех пор, пока не прекратится введение. Это связано с тем, что теоретически требуется неограниченное время для полной элиминации данной дозы. Практически это означает, что если интервал между дозами короче, чем четыре периода полувыведения, то может возникать кумуляция.
Кумуляция обратно пропорциональна части (фракции) дозы, выведенной за каждый интервал между приемами препарата (выведенная фракция). Эта фракция равна 1 за вычетом оставшейся части лекарства перед введением следующей дозы (оставшаяся фракция). Последнюю можно определить, если известны интервал между дозами и период полувыведения. Общепринятый показатель кумуляции обозначают как фактор кумуляции:
Фактор кумуляции - [3-7]
__________________1__________________
фракция, выведенная за один период " между повторными введениями
=___________1___________
1 - Оставшаяся фракция '
Для лекарства, которое назначается через каждый период полувыведения, фактор кумуляции равен 1/0.5, или 2. Фактор кумуляции определяет отношение стационарной концентрации к концентрации, достигаемой после введения первой дозы. Так, пик концентрации после повторных введений на стационарном уровне будет равен пику концентрации после введения первой дозы, умноженному на фактор кумуляции.
Биодоступность
Биодоступность (биоусвояемость) определяют как часть неизмененного лекарства, достигающую системной циркуляции после любого способа введения (табл. 3-3). Для внутривенного введения лекарств биодоступность равна 1. Данные по биодоступности после энтерального введения лекарств представлены в табл. 3-1 как процент от дозы, попадающий в системную циркуляцию. Для лекарств, вводимых через рот, биодоступность может быть менее 100 % по двум основным причинам: из-за неполного всасывания или элиминации при первом прохождении через печень (first-pass elimination).
А. Степень всасывания. После приема через рот лекарство может абсорбироваться не полностью, например только 70 % дозы дигоксина достигает системной циркуляции. Это связано в основном с недостаточным всасыванием и отчасти с бактериальным метаболизмом дигоксина в кишечнике. Другие лекарства могут быть слишком гидрофильны (например, атенолол) или слишком липофильны (например, ацикловир) для оптимальной абсорбции, и их низкая биодоступность определяется недостаточным всасыванием. Если лекарство слишком гидрофильно, оно не может пройти через содержащие липиды клеточные мембраны, если же лекарство слишком липофильно, то оно недостаточно растворимо в воде, чтобы пройти через водные среды, прилежащие к клетке.
Б. Эффект первого прохождения. После всасывания в кишечнике лекарство доставляется с кровью по портальной венозной системе в печень до того, как оно попадет в системную циркуляцию. Лекарство может метаболизироваться в стенке кишечника или даже в крови воротной вены, но чаще всего именно в печени происходят основные превращения перед попаданием лекарства в общий кровоток. Кроме того, печень может экскретировать лекарства в желчь. Любой из этих способов вносит
ТАБЛИЦА 3-3. Пути введения, их общие характеристики и биодоступность
Путь введения Биодоступность (%) Характеристики
Внутривенный 100 (по определению) Наиболее быстрое начало действия
Внутримышечный <100 Можно вводить большие объемы; инъекции могут быть болезненны
Подкожный <100 Объемы введения меньше, чем при в/м; инъекции могут быть болезненны
Пероральный <100 Наиболее удобен; может быть значительно выражен эффект первого прохождения
Ректальный <100 Меньший эффект первого прохождения, чем при введении через рот
Ингаляционный <100 Часто очень быстрое начало действия
Трансдермальный <100 Обычно очень медленная абсорбция; используется в связи с отсутствием эффекта первого прохождения; большая продолжительность действия
свой вклад в данный тип снижения биодоступности, и весь процесс в целом носит название потери (или элиминации) первого прохождения. Влияние элиминации первого прохождения через печень на биодоступность выражается коэффициентом экстракции (extraction ratio — ER):
ER = Ci-печени/Qi [3~8э]
где Q — кровоток через печень, обычно равный 90 л/час у человека массой 70 кг.
Системная биодоступность лекарства (F) может быть определена при известных величине абсорбции (f) и коэффициенте экстракции (ER):
F = fx(1-ER). [3-86]
Этот процесс показан схематически на рис. 3-4. Лекарство типа морфина почти полностью абсорбируется (f = 1), так что потери в кишечнике ничтожны. Однако коэффициент экстракции в печени для морфина равен 0,67, следовательно, показатель F равен 0,33. Таким образом, биодоступность морфина составляет примерно 33 %, что близко к реальным значениям (табл. 3-1).
В. Скорость всасывания. В дополнение к определению, данному выше, термин биодоступность иногда используется для обозначения как величины или степени абсорбции, так и скорости, с которой введенная доза попадает в общий кровоток.
Различие между величиной и скоростью абсорбции поясняется на рис. 3-5. Скорость всасывания определяется местом введения и особенностями лекарственной формы. Как скорость, так и степень всасывания могут оказывать влияние на клиническую эффективность препарата. У трех различных лекарственных форм, указанных на рис. 3-5, будет
существенное отличие по выраженности клинического эффекта. Лекарственная форма Б потребует введения двойной дозы для достижения концентрации в крови, эквивалентной содержанию формы А. Различия в скорости поступления особенно важны для лекарств, которые даются в виде единичных доз, например для снотворных, назначаемых для облегчения засыпания. В этом случае лекарство в форме А достигнет минимальной эффективной концентрации раньше, чем лекарство в форме В; концентрации действующего вещества в лекарственной форме А также будут достигать более высокого уровня и сохраняться выше минимальной эффективной концентрации более продолжитель-
Рис. 3-4. Печеночный клиренс лекарства отделен от остального системного клиренса с целью иллюстрации влияния кровотока (Q) и коэффициента экстракции ([Cs — С„]/С,) на органный клиренс. Процессы, участвующие в метаболизме при первом прохождении, указаны пунктирными стрелками
всасыввния одинакова
] В — лекарство с полной биодоступностыо, но скорость всасывания наполовину меньше, чем у А
Рис. 3-5. Кривые концентрация-время, поясняющие, как изменения скорости всасывания и степени биодоступности могут влиять на длительность действия и эффективность одинаковой общей дозы лекарства, введенного в трех различных лекарственных формах. Пунктирная линия указывает на минимальную эффективную концентрацию (МЭК) лекарства в крови
ное время. В режиме многократного введения лекарственные формы А и В дадут идентичный средний уровень концентраций в крови, несмотря на то, что форма А будет иметь несколько более высокие максимальные и низкие минимальные значения концентраций.
О механизме всасывания лекарства говорят, что он относится к кинетике всасывания нулевого порядка, когда скорость не зависит от количества лекарства, которое остается в кишечнике, например, когда она определяется скоростью желудочной эвакуации, или когда вводятся лекарственные формы с контролируемым высвобождением. Напротив, когда вся доза растворяется в желудочном и кишечном соке, скорость абсорбции обычно пропорциональна концентрации и о ней говорят как о кинетике всасывания первого порядка.
Коэффициент экстракции и эффект первого прохождения
Системный клиренс, то есть убывание из биологической системы, не изменяется под влиянием биодоступности. Однако он может существенно повлиять на степень доступности, так как опреде
ляет коэффициент экстракции (уравнение [3-8а]). Конечно, терапевтический уровень препарата в крови может быть достигнут при приеме,внутрь, если будет увеличена доза. Однако в этом случае уровень метаболитов лекарства будет значительно возрастать по сравнению с уровнем, который наблюдается после внутривенного введения. Лидокаин и верапамил используются для лечения аритмий сердца и имеют одинаковую биодоступность (20 %), но лидокаин не дают внутрь, потому что его метаболиты, как полагают, обладают токсическим действием наЦНС. В список лекарств, которые активно захватываются печенью, можно включить также изониазид, морфин, пропранолол, верапамил и некоторые трициклические антидепрессанты (табл. 4-6).
Для веществ с высоким ER характерна существенная индивидуальная вариабельность биодоступности из-за отличий в функции печени и печеночном кровотоке. Эти отличия могут объяснить значительные колебания концентраций лекарства у разных людей при введении одинаковых доз. Кроме того, они важны при заболеваниях печени, которые сопровождаются внутрипеченочным или внепеченочным сосудистым шунтированием, а также при хирургических анастомозах между портальной системой и системной венозной циркуляцией. Для лекарств, которые активно экстрагируются печенью, шунтирование кровотока приводит к росту доступности, тогда как для лекарств, которые слабо захватываются печенью (различие входящих и выходящих концентраций несущественно) оно мало влияет на биодоступность. Препараты, перечисленные в табл. 3-1 и имеющие низкий ER,— это хлорпропамид, диазепам, дигитоксин, фенитоин, теофиллин, толбутамид и варфарин.
Альтернативные пути введения и эффект первого прохождения
В клинической медицине используют множество путей введения (табл. 3-3) по следующим причинам: для удобства приема (например, внутрь), возможности увеличить концентрацию в точке приложения и уменьшить ее в других местах (например, местное применение), пролонгировать абсорбцию вещества (например, трансдермальное введение) или избежать эффекта первого прохождения.
Эффекта первого прохождения через печень к!ожно в значительной степени избежать путем ис
пользования сублингвальных таблеток и трансдермальных препаратов и в меньшей степени - при использовании ректальных суппозиториев. Сублингвальное введение дает прямой доступ к системным венам, минуя портальные. Трандермальный путь имеет то же преимущество. Лекарства, которые всасываются из суппозиториев в нижнем отделе прямой кишки, поступают в сосуды, впадающие в нижнюю полую вену, минуя печень. Однако суппозитории имеют тенденцию перемещаться по прямой кишке наверх в область, которая имеет преобладающий венозный отток в печень, например через верхнюю геморроидальную вену. Кроме того, имеются многочисленные анастомозы между верхней и средней геморроидальными венами и, таким образом, лишь 50 % ректальной дозы может миновать печень.
Хотя лекарства, вводимые ингаляционно, не дают печеночный эффект первого прохождения, легкие тоже могут служить местом потерь препарата при первом прохождении за счет экскреции и, возможно, метаболизма лекарств, вводимых парентерально. Легкие выполняют также роль фильтра для частиц, которые могут попадать в организм при внутривенной инъекции.
Временные характеристики действия лекарств
будет наблюдаться через 3 часа. Эналаприл обычно назначают один раз в день, так что пройдет семь периодов полувыведения от момента пика концентрации до окончания интервала между приемами. Концентрация эналаприла после каждого периода полувыведения и соответствующий уровень подавления АПФ показаны на рис. 3-6. Ингибирование АПФ рассчитано на основе модели с Е1пах, где Е,пах — максимальная степень ингибирования, равная 100 %, а ЕСзо составляет примерно 1 нг/мл.
Заметим, что концентрация эналаприла в плазме изменяется за первые 12 часов (четыре периода полувыведения) после достижения пика в 16 раз, тогда как торможение АПФ уменьшается только на 20 %. Поскольку концентрация за этот период времени выше ЕСзо, действие препарата на АПФ почти не меняется. Через 24 часа активность АПФ все еще подавлена на 33 %. Этот пример объясняет, почему лекарство с коротким периодом полувыведения может назначаться один раз в день и поддерживать свой эффект все это время. Ключевой фактор в данном случае — высокая начальная концентрация препарата по отношению к его ЕС50. Даже если концентрация в плазме через 24 часа меньше чем 1 % от максимальной, она составляет половину от ЕСзо. Такая особенность свойственна веществам, влияющим на ферменты, например ингибиторам АПФ, или конкурирующим за рецепторы, например пропранололу.
Принципы фармакокинетики, обсуждаемые в этой главе, и фармакодинамики, рассмотренные в главе 2, создают основу для понимания временных характеристик действия лекарств.
Немедленные эффекты
В простейшем случае действие лекарства прямо связано с его концентрацией в плазме, но это не обязательно означает, что эффекты просто параллельны изменению концентрации во времени. Поскольку связь между концентрацией лекарства и его действием нелинейна (вспомните модель максимального эффекта, рассмотренную в главе 2), эффект не всегда будет прямо пропорционален концентрации.
Рассмотрим действие ингибитора ангиотензин-превращающего фермента (АПФ), например эналаприла. Период его полувыведения составляет около 3 часов. После введения внутрь 10 мг препарата пик концентрации в плазме (около 64 нг/мл)
Время (часы)
г'Т‘, = % ингибирования =концентрация (нг/мл)
Рис. 3-6. Изменение концентрации ингибитора АПФ и его эффекта с течением времени. Темные столбики показывают уровень эналаприла в плазме в нг/мл после однократного приема внутрь. Светлые столбики обозначают степень ингибирования АПФ в %. Отметьте разную конфигурацию кривых концентрация-время (экспоненциальное снижение) и эффект-время (линейное снижение в центральной части кривой)
Когда концентрации находятся в диапазоне между одной четвертой и четырьмя ECS0, временной ход эффекта представляет собой близкую к линейной функцию: в этом диапазоне концентраций в каждый период полувйведения эффект снижается на 13 %. При концентрациях ниже одной четвертой ЕСзоэффект становится почти прямо пропорциональным концентрации и временной ход эффекта соответствует экспоненциальному снижению концентрации. Только тогда, когда концентрация лекарства низка относительно ЕСзо, имеет смысл заключение о “полувыведении эффекта лекарства”.
Отсроченные эффекты
Изменения интенсивности действия лекарства часто запаздывают по отношению к изменениям концентрации в плазме. Такое запоздание может отражать время, требуемое для попадания лекарства из плазмы к месту действия. Это справедливо в отношении почти всех лекарств. Задержка из-за распределения является фармакокинетическим феноменом, который может объяснить задержку эффекта от нескольких минут до часов. Она может наблюдаться и после быстрого внутривенного введения влияющих на ЦНС лекарств, например тиопентала.
Общая причина более отсроченных эффектов препаратов, особенно продолжающихся многие часы или даже дни, состоит в медленном обороте физиологических субстанций, участвующих в проявлении реакции на лекарство. Например, варфа-рин действует как антикоагулянт за счет ингибирования витамин К-эпоксидазы в печени. Это действие варфарина развивается быстро, и ингибирование фермента, вероятно, тесно связано с концентрацией варфарина в плазме. Клинический эффект варфарина, в частности влияние на протромбиновое время, отражает снижение концентрации протромбинового комплекса факторов свертывания (рис. 33-7). Ингибирование витамин К-эпоксидазы снижает синтез этих факторов свертывания, но комплекс имеет длительный период полувыведения (около 14 часов), и именно этот период полувыведения определяет, как долго концентрация факторов свертывания будет достигать нового стационарного уровня и сколько времени потребуется для проявления эффекта лекарства.
Кумулятивные эффекты
Некоторые эффекты лекарств более связаны с кумулятивным, нежели с быстро обратимым действием. Нефротоксичность аминогликозидных антибиотиков (например, гентамицина) выше, когда лекарство вводят в форме постоянной инфузии, чем в виде повторных доз. Полагают, что причиной повреждений почек является накопление аминогликозида в Их коре. Даже если обе схемы введения позволяют получить одинаковые стационарные концентрации, дробное введение дает значительно более высокие пиковые концентрации, которые насыщают механизм захвата лекарства в корковом слое. В результате уменьшается общее накопление аминогликозида. Различие в токсичности определяется характером изменения концентрации препарата и насыщением механизмов захвата.
Эффект многих противораковых лекарств также включает кумулятивное действие (например, степень связывания лекарства с ДНК пропорциональна его концентрации), и оно обычно необратимо. Следовательно, влияние на рост опухоли является результатом кумулятивного воздействия лекарства. Измерение кумулятивного эффекта, в частности по площади под кривой концентрация-время, позволяет предсказать ответную реакцию и индивидуализировать лечение больного.
Приближение к целевой концентрации в схеме рационального режима дозирования
Рациональный режим дозирования основан на предположении, что существует целевая концентрация (target concentration, ТС), которая должна обеспечить желаемый терапевтический эффект. Принимая во внимание фармакокинетические факторы, определяющие связь между дозой и концентрацией, возможно индивидуализировать режим дозирования для приближения к целевой концентрации. В диапазонах эффективных концентраций, указанных в табл. 3-1, у больного должен наблюдаться терапевтический эффект. Начальная целевая концентрация выбирается от нижней границы этого диапазона. В некоторых случаях ТС зависит также от специфических целей лечения, например для контроля фибрилляции предсердий необходи
ма целевая концентрация 2 нг/мл дигоксина, тогда как сердечная недостаточность устраняется при целевой концентрации 1 нг/мл.
Поддерживающая доза
В большинстве клинических ситуаций лекарство вводят так, чтобы поддерживать его постоянный уровень в организме, т. е. с каждой дозой должно вводиться количество препарата, достаточное для восполнения элиминированного после предыдущей дозы. Таким образом, расчет поддерживающей дозы является первостепенной задачей. Клиренс — это наиболее важный фармакокинетический параметр, который должен учитываться при определении рационального режима дозирования для поддержания стационарного состояния. При стационарном состоянии (СС) частота приема препарата (“скорость поступления”) соответствует скорости элиминации (“скорость выведения”). Заменив в уравнении [3-4] С на ТС можно определить необходимую скорость введения для обеспечения поддерживающей дозы лекарства:
Скорость введения лекарстваСс -
= Скорость элиминациисс = CL х ТС. [3-9]
Таким образом, если желаемая ТС известна, клиренс у данного пациента определяет скорость введения лекарства. Если лекарство дается путем, имеющим био доступность ниже 100 %, скорость введения, рассчитываемая по уравнению [3-9], должна быть изменена. Для введения внутрь:
Скорость введения лекарства =
Скорость введения лекарства8НутРь
=--------------F-----------------. [3-10]
внутрь
Если режим назначения лекарства прерывистый, то поддерживающая доза рассчитывается по формуле:
Поддерживающая доза =
= Скорость введения лекарства х
х Интервал между введениями. [3-11]
(Пример расчета поддерживающей дозы приведен в дополнении.)
Заметим, что стационарная концентрация, достигаемая при постоянной инфузии или средняя концентрация после прерывистого введения зависят только от клиренса. Знание объема распределения и периода полувыведения лекарства не нужно для предсказания его средней концентрации в плазме,
Рис. 3-7. Связь между частотой введения теофиллина и его максимальной и минимальной концентрациями в плазме при уровне стационарной концентрации, равном 10 мг/л. Черная восходящая линия показывает концентрацию в плазме, которая достигается при внутривенной инфузии со скоростью 28 мг/ч. Дозы для введения с перерывами в 8 часов (пунктирная линия) составляют 224 мг; для введения через 24 часа (темная линия) — 672 мг. В каждом из трех случаев средняя стационарная концентрация в плазме составляет 10 мг/л
Заказ 3245
Пример: расчет поддерживающей дозы
Для купирования приступа острой бронхиальной астмы необходима целевая концентрация теофиллина 10 мг/л (Holford et al., 1993). Если больной не курит и не имеет других заболеваний, кроме астмы, мы можем использовать для расчета показатель среднего клиренса, указанный в табл. 3-1, т. е. 2.8 л/ч/70 кг. Так как лекарство должно вводиться в виде внутривенной инфузии, показатель F - 1.
Скорость введения лекарства = CL х ТС = = 2.8 л/ч/70 кг х 10 мг/л = = 28 мг/ч/70 кг.
Следовательно, у этого больного расчетная скорость инфузии должна быть 28 мг/ч/70 кг.
После купирования приступа астмы врач может решить поддержать полученный уровень теофиллина в плазме с помощью оральной пролонгированной формы, которую назначают каждые 12 часов, чтобы приблизиться к уровню концентрации при внутривенной инфузии. В соответствии с табл. 3-1 биодоступность препарата
составляет 0.96. Когда интервал между приемами равен 12 часам, величина каждой поддерживающей дозы должна определяться следующим образом:
Поддерживающая доза =
Скорость введения
------------------х
F
х Интервал между приемами =
28мг/ч _
= ' х 12 ч =
0.96
= 350 мг.
Таблетка или капсула, содержащая дозу, близкую к расчетной (350 мг), может быть прописана для приема через 12-часовые интервалы. Если используется 8-часовой интервал между приемами, идеальная доза должна быть 233 мг, а если лекарство дают один раз в день — 700 мг. На практике значение F может быть опущено из расчетов, поскольку оно близко к 1.
которая ожидается при данной скорости введения лекарства или для определения скорости введения для достижения целевой концентрации. Рис. 3-7 показывает, что при различных интервалах между введениями кривые концентрация-время имеют различные максимальные и минимальные значения даже при неизменном среднем уровне 10 мг/л.
Определение скорости введения лекарства и средних стационарных концентраций, рассчитываемых по клиренсу, не зависит от специфики фармакокинетической модели. Напротив, определение максимальных и минимальных стационарных концентраций необходимо проводить с учетом фармакокинетической модели. Расчет фактора кумуляции (уравнение [3-7]) предполагает одночастевую модель распределения и элиминации лекарства (рис. 3-2Б). При определении максимальной концентрации в этом случае допускается, что скорость всасывания значительно выше скорости элиминации. Для расчета оценочных минимальных и максимальных концентраций в клинической ситуации эти допущения обычно обоснованы.
Нагрузочная доза
Когда время до достижения стационарного состояния достаточно существенно, что наблюдается при применении лекарств с продолжительным периодом полувыведения, желательно вводить нагрузочную дозу, которая быстро повысит концентрацию лекарства в плазме до уровня целевой. Теоретически необходимо рассчитать только величину нагрузочной дозы, а не скорость ее введения, и в первом приближении этого достаточно. Объем распределения является фактором пропорциональности, который связывает общее количество лекарства в организме с его концентрацией в плазме. Если нагрузочная доза должна обеспечить целевую концентрацию, то она рассчитывается по уравнению [3-12]:
Нагрузочная доза =
= Количество лекарства в организме сразу после введения нагрузочной дозы =
= VDxTC. [3-12]
В примере с теофиллином, рассмотренном в дополнении, нагрузочная доза равна 350 мг (35 л х 10 мг/л). Для большинства лекарств нагрузочная доза может вводиться тем или иным способом однократно.
До сих пор мы не обращали внимания на то, что некоторые лекарства имеют более сложную многокамерную фармакокинетику, например у них имеется процесс распределения, который иллюстрирует двухкамерная модель на рис. 3-2. В большинстве случаев это упрощение оправдано. Однако иногда фаза распределения не может быть исключена из рассмотрения, особенно при расчете нагрузочной дозы. Если скорость всасывания лекарства высока по сравнению с распределением (это почти всегда верно в случае внутривенного болюсного введения), концентрация лекарства в плазме после соответствующей нагрузочной дозы, рассчитанной с помощью кажущегося VD, может быть существенно выше необходимой. В результате может развиться серьезное, хотя и преходящее, токсическое действие. Это особенно важно учитывать, например при введении противоаритмических средств, таких как лидокаин, когда токсические эффекты могут развиться немедленно. Итак, хотя расчет количества действующего вещества в нагрузочной дозе окажется совершенно верным, скорость введения может иметь подчас решающее значение для предупреждения избыточных концентраций лекарства, поэтому медленное внутривенное введение (в течение минут, а не секунд) почти всегда более предпочтительно. При внутривенном дозировании теофиллина начальные инъекции должны проводиться в течение 20 минут во избежание создания высоких концентраций в плазме в период фазы распределения.
Когда лекарство вводится прерывисто, нагрузочная доза, рассчитанная по уравнению [3-12], позволяет достичь стационарной концентрации, но не определяет пиковую концентрацию в стационарном периоде (рис. 3-7). Для определения пиковой концентрации в стационарном периоде нагрузочную дозу нужно рассчитывать по уравнению [3-13]:
Нагрузочная доза = [3-13]
= Поддерживающая доза х Фактор кумуляции.
Терапевтический лекарственный Мониторинг: связь фармакокинетики и фармакодинамики
Изложенные здесь основные принципы, можно использовать для интерпретации результатов измерения концентрации лекарств в клинике на базе трех основных фармакокинетических переменных: абсорбции, клиренса и объема распределения (и его производной — периода полувыведения), а также двух фармакодинамических переменных: максимального эффекта, который достижим в данном органе (ткани-мишени) и чувствительности ткани к лекарству. Заболевания могут изменять все эти параметры, поэтому важно уметь предсказывать влияние болезни на фармакокинетику для выбора рационального режима дозирования (дополнение “ Стратегия целевой концентрации” ).
Фармакокинетические переменные
А. Абсорбция. Количество лекарства, попадающее в организм, зависит от аккуратности больного при соблюдении режима приема лекарства и от скорости и полноты проникновения лекарства из места введения в кровь.
Передозировка и недостаточная дозировка по сравнению с прописанной дозой, связанные с неаккуратностью приема лекарства больным, часто определяются по грубым отклонениям от ожидаемых значений концентрации лекарства в крови. Если больной достаточно дисциплинирован, то причиной низких концентраций лекарства могут быть нарушения всасывания в тонком кишечнике. Вариации в биодоступности редко связаны с вариабельностью технологии лекарственных форм. Чаще такие различия определяются характером метаболизма в процессе всасывания лекарства.
Б. Клиренс. Изменения клиренса можно предвидеть, когда у больного имеются серьезные нарушения функции почек, печени или сердца. Клиренс креатинина - полезный количественный индикатор почечной функции. С другой стороны, клиренс лекарств может быть ценным показателем функциональных последствий нарушений работы сердца, печени и почек, подчас более точным, чем клинические наблюдения или другие лабораторные тес
ты. Например, когда функция почек изменяется быстро, оценка клиренса аминогликозидных антибиотиков может быть более точным индикатором клубочковой фильтрации, чем определение сывороточного креатинина.
Заболевания печени могут снижать клиренс и удлинять период полувыведения многих лекарств. Однако у некоторых препаратов, которые элиминируются печенью, не происходит изменения этих показателей при нарушениях функции печени. Следовательно, заболевания печени не всегда влияют на собственный печеночный клиренс. В настоящее
Стратегия целевой концентрации
Признание важной роли концентрации как связующего звена фармакокинетики и фармакодинамики способствует созданию стратегии целевой концентрации. Фармакодинамические принципы можно использовать для предсказания концентрации, необходимой для достижения терапевтического эффекта определенной степени выраженности. Эта целевая концентрация затем может быть реализована с помощью фармакокинетических принципов, позволяющих создать подходящий режим дозирования. Стратегия целевой концентрации — это процесс оптимизации дозы у данного больного на основе измерения концентрации лекарства. Она складывается из следующих этапов:
1. Выберите целевую концентрацию.
2. Рассчитайте VD и CL на основе типовых значений (табл. 3-1) и внесения поправок с учетом таких факторов как масса тела и функция почек.
3. Дайте нагрузочную дозу или поддерживающую дозу, рассчитанную с учетом значений ТС, Vdh CL.
4. Зарегистрируйте реакцию больного и определите концентрацию лекарства.
5. Пересмотрите VD и CL на основе результатов измерения концентрации.
6. Повторите шаги 3-6, подбирая необходимую для оптимальной реакции на лекарство поддерживающую дозу.
время нет надежного маркера метаболизирующей функции печени, который можно было бы использовать для предсказания печеночного клиренса подобно тому, как клиренс креатинина является маркером почечного клиренса лекарств.
В. Объем распределения. Кажущийся объем распределения отражает равновесие между связыванием лекарства тканями, что уменьшает его концентрацию в плазме и делает кажущийся объем больше, и связыванием с белками плазмы, которое повышает концентрацию лекарства в плазме и уменьшает показатель кажущегося объема распределения. Изменения связывания как в тканях, так и в плазме, могут изменить кажущийся объем распределения, который рассчитывается по результатам измерения концентрации лекарства в плазме. У пожилых людей наблюдаются относительное уменьшение массы скелетной мускулатуры и тенденция к уменьшению кажущегося объема распределения, например для дигоксина. Объем распределения может быть переоценен у тучных больных, если расчет основан на массе тела, а лекарство плохо проникает в жировую ткань, как в случае с дигоксином. Напротив, теофиллин имеет объем распределения (35 л), близкий по величине к общему объему жидкости в организме. Жировая ткань содержит почти столько же воды, сколько и другие ткани, поэтому кажущийся общий объем распределения теофилллина пропорционален массе тела даже у тучных больных.
Избыточное накопление жидкости (отеки, асцит, плевральный выпот) может заметно увеличить объем распределения лекарств подобных тобрамицину, которые обладают гидрофильными свойствами и характеризуются при отсутствии нарушений водного баланса малым объемом распределения.
Г. Период полувыведения. Различия между клиренсом и периодом полувыведения важны для выяснения механизма влияния болезни па распределение лекарства. Так, период полувыведения диазепама увеличивается с возрастом. Однако выяснилось, что клиренс этого лекарства с возрастом не изменяется (Klotz et al., 1975). Увеличение периода полужизни диазепама в действительности связано с возрастными изменениями объема распределения, тогда как метаболические процессы, ответственные за элиминацию лекарства, оказались относительно постоянными.
Фармакодинамические переменные
А. Максимальный эффект. Все фармакологические ответные реакции имеют максимальные значения (максимальный эффект — Етах). При увеличении концентрации лекарства может быть достигнута точка, за пределами которой не наблюдается дальнейшего прироста реакции.
Если увеличение дозы у данного больного не приводит к усилению клинического эффекта, возможно, что максимум уже достигнут. В этом случае дальнейшее увеличение дозы ведет к возрастанию концентрации лекарства без усиления эффекта. Признание достижения максимума эффекта помогает избежать бесплодного увеличения дозы с сопутствующим риском токсического действия.
Б. Чувствительность. Чувствительность органа-мишени к концентрации лекарства отражается в величине концентрации, требуемой для получения половины от максимального эффекта, т. е. ЕС5|). У больного констатируют снижение чувствительности к лекарству, если при регистрации его концентрации в крови, обычно вызывающей терапевтический эффект, последний не проявляется. Данное явление может быть результатом физиологических нарушений, например гиперкалиемия уменьшает реакцию на дигоксин, или лекарственного антагонизма, например блокаторы кальциевых каналов снижают инотропный эффект дигоксина.
Повышенная чувствительность к лекарству обычно проявляется как преувеличенная ответная реакция на малые или умеренные дозы. Фармакодинамическая природа такой чувствительности может быть подтверждена измерением концентрации лекарства, которая обычно оказывается низкой по сравнению с наблюдающимся эффектом.
Интерпретация результатов измерений концентрации лекарства
Клиренс
Клиренс является наиболее важным фактором, определяющим концентрацию лекарства. Интерпретация результатов измерений концентрации лекарств зависит от ясного понимания трех факторов, которые могут повлиять на клиренс. Это доза,
кровоток и функциональное состояние печени и почек. Каждый из этих факторов должен учитываться при оценке клиренса, рассчитанного на основе измерения концентрации лекарства. Следует также знать, что изменение степени связывания с белками может привести к опрометчивому суждению об изменении клиренса, тогда как на самом деле элиминация лекарства не изменилась (дополнение “Связывание с белками плазмы: насколько это важно?”). Рассмотрим факторы, которые влияют на связывание с протеинами.
А. Концентрация альбуминов. Такие лекарства как фенитоин, салицилаты и дизопирамид интенсивно связываются с альбуминами плазмы. Уровень альбуминов понижается при многих заболеваниях, что ведет к снижению общей концентрации лекарства.
Б. Концентрация агкислого гликопротеина, ос,-кислый гликопротеин является важным белком плазмы, связывающим такие лекарства как хинидин, лидокаин и пропранолол. Его содержание увеличивается при острых воспалительных заболеваниях и вызывает значительные изменения общей концентрации в плазме указанных лекарств, даже если их элиминация не изменена.
В. Ограниченное связывание с белками. Степень связывания лекарств с белками плазмы имеет определенные пределы. В области терапевтических концентраций связывание салицилатов, дизопира-мида, преднизолона с протеинами пропорционально их содержанию в крови. Поскольку концентрация несвязанного лекарства определяется скоростью введения и клиренсом, которые не изменены, при низкой константе связывания с белками увеличение скорости введения вызовет рост концентрации фармакодинамически активного несвязанного вещества. Если степень связывания лекарства с белками плазмы велика, то эффективная концентрация не будет достигнута столь быстро, так как необходимо время для насыщения связывающих белков.
Предыдущее введение лекарств
Для получения максимальной информации от измерения концентрации лекарства необходимо знать, как оно вводилось (кратность, дозу и т. д.). Если же это известно неточно или неизвестно совсем, то измерение лекарственных концентраций теряет свою прогностическую ценность.
Связывание с белками плазмы: насколько это важно?
Связывание с протеинами плазмы часто рассматривают как фактор, играющий важную роль в фармакокинетике, фармакодинамике и при взаимодействии лекарств. Однако нет клинически достоверных примеров изменения распределения или эффективности лекарств, которые можно явно приписать влиянию степени связывания с белками. Представление о том, что если лекарство вытесняется из связи с протеинами, то будет возрастать концентрация несвязанной фракции и усиливаться терапевтический эффект, а возможно, и токсичность, является упрощенным. К сожалению, эта простая теория, оправданная при тестировании в пробирке, не работает в организме, который является открытой системой, способной элиминировать несвязанное лекарство.
Во-первых, существенное, как может показаться, увеличение несвязанной фракции в плазме от 1 % до 10 % способствует высвобождению менее 5 % препарата от общего количества в организме, потому что менее одной трети ле
карства, находящегося в организме, связано с белками плазмы. Примером такого легко связывающегося вещества является варфарин. Лекарство, вытесненное из комплекса с белками плазмы, будет, конечно, попадать в общий объем распределения, так что увеличение на 5 % количества несвязанного вещества в организме вызовет прирост максимум на 5 % фармакологически активной свободной фракции в районе места действия.
Во-вторых, когда количество несвязанного лекарства в плазме возрастает, скорость элиминации будет увеличиваться (если клиренс не изменен) и после четырех периодов полувыведения концентрация несвязанного вещества возвратится к прежнему стационарному уровню. При изучении взаимодействия лекарств на уровне вытеснения из комплекса с протеинами и его клинического значения было показано, что замещающее вещество является также ингибитором клиренса и что именно изменение клиренса несвязанного лекарства является механизмом, объясняющим результаты взаимодействия.
Выбор времени взятия материала для измерения концентрации
Информация о скорости и степени всасывания лекарства у данного больного имеет большое клиническое значение. Обычно абсорбция происходит в течение первых двух часов после введения лекарства. Это время варьирует в зависимости от приема пищи, положения тела больного и его активности. Следовательно, важно избегать взятия крови до завершения всасывания (около двух часов после приема внутрь). Попытки измерить пиковую концентрацию вскоре после приема лекарства внутрь обычно неудачны, так как нет уверенности в завершении процесса всасывания.
Для некоторых лекарств, таких как дигоксин и литий, требуется несколько часов для распределения в тканях. Пробы для определения дигоксина должны забираться по крайней мере через 6 часов после приема последней дозы, а кровь для измерения концентрации лития — перед следующей дозой (обычно через 24 часа после последнего приема).
Аминогликозиды распределяются весьма быстро, но разумно подождать час после приема перед взятием крови.
Клиренс легко определить по скорости введения и стационарной концентрации. Чтобы оценить стационарную концентрацию, образцы крови нужно забирать через определенные интервалы времени. Если стационарное состояние достигнуто (по крайней мере, через три периода полувыведения при постоянном введении), то в образце, полученном примерно посередине интервала между приемами, концентрация препарата будет близка к стационарной.
Первоначальная оценка объема распределения и клиренса
А. Объем распределения. Объем распределения обычно рассчитывают по массе тела (масса 70 кг принята для оценок, приведенных в табл. 3-1). Если больной тучный, то для лекарств, плохо проникающих в жировую ткань (например, тобрамицин и дигоксин), объемы должны рассчитываться, исхо
дя из идеальной для данного роста массы, которую находят следующим образом:
Идеальная масса тела (кг) =
= 52 + 1.9 кг/дюйм роста выше 5 футов
(для мужчин) или 49 + 1.7 кг/дюйм роста выше 5 футов (для женщин). [3-14]
У больных с отеками, асцитом, плевральным выпотом значения объема распределения для ами-ногликозидных антибиотиков (например, тобрамицина) выше, чем можно предсказать по массе тела. У таких пациентов проводят следующую коррекцию массы тела: вычитают из нее оценочное значение массы избытка накопившейся жидкости. Получившуюся “нормальную” массу используют для расчета нормального объема распределения. Затем этот нормальный объем распределения увеличивают на 1 л на каждый расчетный килограмм избыточной жидкости. Эта поправка существенна из-за сравнительно малых объемов распределения водорастворимых лекарств.
Б. Клиренс. Для лекарств, выводимых почками, требуются поправки на клиренс, учитывающие функциональное состояние почек. Поправки удобно делать на основе данных о клиренсе креатинина, определяемого по единичному забору крови (Bjornsson, 1979)2.
Клиренс креатинина =
(л/ч)
160 - Возраст (в годах) 22 х Креатинин в сыворотке (мг%)
Масса тела (кг)
70
[3-15]
(х 0.9 для женщин).
Обратите внимание: если содержание креатинина в сыворотке измеряется в ммоль/л, то знаменатель 22 следует заменить на 250.
У женщин значение клиренса составляет 90 % от расчетного, потому что у них меньше мышечная масса на килограмм массы тела, а именно мышцы определяют продукцию креатинина. В связи со сложностью отбора необходимых проб мочи, кли-
2 Подобное уравнение (уравнение Коккрофта-Гаульта) приведено в главе 62.
рейс креатинина легче рассчитывать указанным образом. Получаемые в данном случае результаты достаточно надежны. У тучных пациентов для расчета клиренса следует использовать идеальную массу тела, а у тяжело больных необходимо вносить поправки на атрофию мышц.
Пересмотр индивидуальных оценочных показателей объема распределения и клиренса
Для эффективного лечения больного необходимо сопоставление предсказанных фармакокинетических параметров и ожидаемых концентраций используемого лекарственного средства с реальными данными измерений. Если эти данные отличаются более чем на 20 % от предсказанных величин, следует пересмотреть оценки VD или CL для данного больного, используя уравнения [3-1] или [3-2]. Если пересчет дает увеличение значений более чем на 100 % или снижение на 50 %, то следует подумать о правильном времени взятия проб и критически пересмотреть “историю” дозирования.
Например, если больной принимает 0.25 мг дигоксина в день, врач может ожидать, что концентрация дигоксина в крови будет около 1 нг/мл. Этот прогноз основан на типовых значениях биодоступности (70 %) и общего клиренса (CLI10.lc,11Mi( + + СЕнсочечный = 4 + 3 = 7 л/ч). Если у больного имеется сердечная недостаточность, непочечный (печеночный) клиренс может быть наполовину меньше из-за печеночного застоя и гипоксии, следовательно, ожидаемый клиренс будет около 5.5 л/ч. Соответственно, ожидаемая концентрация дигоксина в крови будет порядка 1.3 нг/мл. Предположим, что в действительности концентрация составила 2 нг/мл. Сокращение ежедневной дозы наполовину позволит достичь целевой концентрации 1 нг/мл. При пересчете новое значение клиренса составит 3.5 л/ч. Меньшее значение в сравнении с ожидаемой величиной (5.5 л/ч) может отражать наличие дополнительно к печеночному застою нарушения функции почек, связанного с сердечной недостаточностью.
Подобный подход можно использовать, только если достигнуто стационарное состояние. Требуется по крайней мере неделя регулярного приема (три-четыре периода полуэлиминации) для того, чтобы метод дал надежные результаты.
Классическим примером фармакокинетического взаимодействия лекарств являются результаты сочетанного приема хинидина и дигоксина. Влияние хинидина на клиренс дигоксина приводит к удвоению стационарной концентрации дигоксина по сравнению с ожидаемой почти у каждого больного, получающего данное сочетание препаратов.
Избранная литература
Benet L. Z., Mitchell J. R., Sheiner L. B. Pharmacokinetics: The dynamics of drug absorption, distribution, and elimination. In: Goodman and Gilman’s The Pharmacological Basis of Thera
peutics, 8th ed. Gilman A. G. et al. (eds) Perga-mon, 1990.
Benet L. Z., Williams R. L. Design and optimization of dosage regimens: Pharmacokinetics data. In: Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 8th ed. Gilman A. G. et al. (eds) Pergamon, 1990.
Burton M. E., Vasco M. R., Brater D. C. Comparison of drug dosing methods. Clin. Pharmacokinet. 1985; 10:1.
Holford N. H. G., Sheiner L. B. Understanding the dose-effect relationship. Clin. Pharmacokinet. 1981; 6:429.
Биотрансформация лекарств 4
Мария Альмира Коррейя
Человек ежедневно подвергается воздействию множества химических веществ, называемых ксенобиотиками, которые всасываются через легкие, кожу, а чаще попадают внутрь в составе пищи, напитков, лекарств. Воздействие ксенобиотиков среды может оказаться случайным или неизбежным, когда они присутствуют как компоненты воздуха, воды или пищи. Некоторые ксенобиотики безвредны, но многие могут вызывать биологические ответные реакции. Основные токсические эффекты этих веществ обсуждаются в главах 58—60. Они часто зависят от превращения всосавшихся ксенобиотиков в биологически активные метаболиты. Предлагаемое обсуждение проблемы касается ксенобиоти-ков в целом (включая лекарства) и в определенной степени распространяется на эндогенные вещества.
Почему необходима биотрансформация лекарств
Почечная экскреция играет кардинальную роль в прекращении биологического действия очень незначительного числа лекарств, преимущественно представленных малыми молекулами или поляризованных благодаря функциональным группам, полностью ионизированным при физиологических значениях pH. Большинство лекарств лишены этих физико-химических свойств. Фармакологически активные органические молекулы чаще липофильны и остаются неионизированными при физиологических pH. Они обычно сильно связаны с белками плазмы. Такие вещества плохо фильтруются в клубочках. Их реабсорбцию облегчают также липофильные свойства мембран почечных канальцев. Следовательно, большинство лекарств имели бы весьма продолжительное действие, если бы его прекращение зависело только от почечной экскреции. Альтернативный процесс, который может привести к прекращению или изменению биологической активности,— метаболизм лекарства. Как правило,
липофильные ксенобиотики превращаются в более полярные и, следовательно, более легко экскретируемые вещества. Роль, которую метаболизация играет в инактивации жирорастворимых веществ, весьма важна. Например, липофильные барбитураты, такие как тиопентал и пентобарбитал, могли бы иметь чрезвычайно длительный период полувыведения, если бы не превращались в более водорастворимые соединения. С другой стороны, липофильные вещества, которые депонируются в жировой ткани (например, ДДТ), могут сохраняться в течение ряда лет после прекращения воздействия, так как не попадают в основные метаболизирующие органы.
Метаболические продукты часто менее фармакодинамически активны, чем их вещества-прародители, и даже могут быть неактивны. Однако некоторые метаболиты имеют повышенную активность или токсические свойства, в том числе мутагенные, тератогенные, канцерогенные. Заслуживает внимания то обстоятельство, что в синтезе таких эндогенных субстратов как стероидные гормоны, холестерин и желчные кислоты, участвуют многие ферменты, связанные с метаболизмом ксенобиотиков. То же относится к образованию и экскреции эндогенных метаболитов, в частности билирубина — конечного метаболита гема. Наконец, ферменты, метаболизирующие лекарства, используют для создания фармакологически инертных пролекарств, которые превращаются in vivo в активные соединения.
Роль биотрансформации в инактивации лекарств
Большинство метаболических превращений происходит на участке между всасыванием лекарства в системный кровоток и его почечной элиминацией. Незначительная трансформация происходит в просвете или в стенке кишечника. В целом все эти реакции могут быть отнесены к одной из двух
ВСАСЫВАНИЕ
МЕТАБОЛИЗМ ЭЛИМИНАЦИЯ
Лекарство
Лекарство
Лекарство
Липофильное---------------------------► Гидрофильное
Рис. 4-1. Фазы биотрансформации лекарств. Реакции фазы II могут иногда предшествовать реакциям фазы I
категорий реакций, обозначаемых как фаза I и фаза II (рис. 4-1).
В фазе I обычно происходит превращение исходного вещества в более полярный метаболит путем введения или раскрытия функциональной группы (-ОН, -NH2, -SH). Эти метаболиты часто неактивны, хотя в некоторых случаях активность не исчезает, а только изменяется.
Если метаболиты фазы I достаточно полярны, они могут легко экскретироваться. Однако многие из них не элиминируются и подвергаются последующей трансформации, в ходе которой соединяются с эндогенными глюкуроновой, серной, уксусной кислотами или аминокислотами, формируя
высокополярный конъюгат. Подобная конъюгация (реакция синтеза) является отличительным признаком фазы II метаболизма. Огромное количество лекарств подвергаются последовательно реакциям I и II фаз биотрансформации, хотя в некоторых случаях исходное вещество обладает функциональной группой, которая может прямо вступать в конъюгацию. Например, известно, что гидразидный радикал изониазида образует N-ацетил-конъюгат в реакции фазы II. Этот конъюгат является далее субстратом для реакции по типу фазы I, а именно, гидролиза до изоникотиновой кислоты (рис. 4-2). Таким образом, реакции фазы II могут предшествовать реакциям фазы I.
,__„ О Н
/^v II I
N /)— С — N — NH2 (INH)
Рис. 4-2. Фаза II активации изониазида (INH) в гепато-токсичный метаболит
| Фаза II (ацетилирование)
Н Н О
I I II
— N — N — С — СН3 (N-ацетил 1NH)
Фаза I (гидролиз)
О ОН
II II I
С — ОН + СНз —С — N — NH2 (ацетилгидразин)
Изоникотиновая 1
кислота । макромолекул (белков)
К
Гепатотоксичность
Где происходит биотрансформация лекарств?
Хотя многие ткани способны метаболизировать лекарства, главным органом метаболизма лекарств является печень. К другим тканям, отличающимся существенной активностью, относятся желудочно-кишечный тракт, легкие, кожа и почки. После приема внутрь многие лекарства (например, изопротеренол, меперидин, пентазоцин, морфин) всасываются в тонком кишечнике неразрушенными и траспортируются через портальную систему в печень, где они подвергаются активному метаболизму. Этот процесс получил название эффект первого прохождения. Некоторые лекарства, принимаемые перорально (например, клоназепам, хлорпромазин), более интенсивно метаболизируются в кишечнике, чем в печени. Следовательно, кишечный метаболизм может вносить свой вклад в общий эффект первого прохождения. Этот эффект может столь заметно ограничивать биодоступность лекарств, принимаемых внутрь, что приходится использовать альтернативные пути введения для достижения терапевтически эффективных уровней лекарства в крови. В нижних отделах кишечника содержатся микроорганизмы, которые способны осуществлять многие реакции биотрансформации. Кроме того, лекарства могут разрушаться кислотой желудка (например, пенициллин), пищеварительными ферментами (например, инсулин и другие полипептиды) или ферментами в стенке кишечника (например, катехоламины).
Хотя биотрансформация лекарств in vivo может осуществляться путем спонтанных некатализи-руемых химических реакций, громадное большинство лекарств катализируется с помощью специфических клеточных ферментов. На субклеточном уровне эти ферменты могут располагаться в эндоплазматическом ретикулуме, митохондриях, цитозоле, лизосомах или даже в ядре или плазматической мембране.
Микросомальная система оксидаз со смешанной функцией
Многие ферменты, метаболизирующие лекарства, располагаются на липофильных мембранах эндоплазматического ретикулума печени и других
тканей. Когда эти пластинчатые мембраны изолируют путем гомогенизации и фракционирования клетки, они преобразуются в везикулы, называемые микросомами. Микросомы сохраняют большинство морфологических и функциональных характеристик интактных мембран, включая свойство шероховатости или гладкости поверхности, соответственно у шероховатого (рибосомального) и гладкого (нерибосомального) эндоплазматического ретикулума. В то время как шероховатые микросомы в основном связаны с синтезом, гладкие относительно богаты ферментами, ответственными за окислительный метаболизм лекарств. В частности, они содержат важный класс ферментов, известный как оксидазы со смешанной функцией (ОСФ), или монооксигеназы. Активность этих ферментов требует присутствия как восстанавливающего агента (НАДФ-Н), так и молекулярного кислорода. При типичной реакции расходуется (восстанавливается) одна молекула кислорода на молекулу субстрата, и один кислородный атом появляется в продукте реакции, а другой — в форме воды.
В этом окислительно-восстановительном процессе ключевую роль играют два микросомальных фермента. Первый из них флавопротеин НАДФ-Н-цитохром Р-450-редуктаза. Один моль этого фермента содержит по одному молю флавинмононуклеотида (ФМН) и флавинадениндинуклеотида (ФАД). Поскольку цитохром С может служить акцептором электрона, фермент часто обозначают как НАДФ-цитохром С-редуктазу. Второй микросомальный фермент — гемопротеин, называемый цитохромом Р-450, выполняет роль конечной оксидазы. В действительности микросомальная мембрана содержит множество форм этого гемопротеина, и эта множественность возрастает при повторном введении экзогенных химических веществ. Название цитохром Р-450 связано с особыми свойствами этого гемопротеина. В восстановленной форме он связывает моноксид углерода с образованием комплекса с максимальным поглощением света при длине волны 450 нм. Относительное изобилие цитохрома Р-450 по сравнению с редуктазой печени делает процесс восстановления гема цитохрома Р-450 лимитирующей стадией в процессе окисления лекарств в печени.
Процесс микросомального окисления лекарств требует участия цитохрома Р-450, цитохром Р-450-редуктазы, НАДФ-Н и молекулярного кислорода. Упрощенная схема окислительного цикла
представлена на рис. 4-3. Окисленный (Fe3+) цитохром Р-450 соединяется с лекарственным субстратом с образованием бинарного комплекса (этап <—). НАДФ-Н является донором электрона для флаво-протеинредуктазы, которая, в свою очередь, восстанавливает окисленный комплекс цитохром Р-450-лекарство (этап ?). Второй электрон переходит от НАДФ-Н через ту же флавопротеинредуктазу, которая восстанавливает молекулярный кислород и формирует комплекс “активированный кислород"-цитохром Р-450-субстрат (этап —>). Этот комплекс переносит “активированный” кислород на лекарственный субстрат с образованием окисленного продукта (этап X).
Выраженные окисляющие свойства активированного кислорода позволяют окислить большое
количество субстрата. Субстратная специфичность этого комплекса очень низка. Единственным общим свойством разнообразных по структуре лекарств и химических соединений, субстратов этой системы, является высокая растворимость в жирах.
Индукция ферментов
Интересным свойством некоторых структурно различающихся лекарственных субстратов является их способность при повторном введении “индуцировать” цитохром Р-450 путем увеличения скорости его синтеза и/или снижения скорости разрушения. Индукция ведет к ускорению метаболизма и, как правило, к снижению фармакологической активности индуктора и совместно с ним вводимых
Рис. 4-3. Цикл цитохрома Р-450 в окислении лекарств (R-H — исходное вещество, R-OH — окисленный метаболит, е — электрон)
лекарств. Однако в том случае, если лекарство превращается в реактивные метаболиты, такая индукция может усилить опосредованную метаболитами тканевую токсичность.
Различные субстраты способны индуцировать изоформы цитохрома Р-450 с различной мол. м., различной субстратной специфичностью, различными иммунохимическими и спектральными характеристиками. Двумя наиболее исследованными формами являются: 1) цитохром Р-450 2В1 (бывший Р-450b), который индуцируется при введении фенобарбитала, и 2) цитохром Р-450 1А1 (цитохром Pt450, или Р-448), индуцируемый полициклическими ароматическими углеводородами (ПАУ), прототипом которых является 3-метилхолантрен. Кроме того, глюкокортикоиды, макролидные антибиотики, антиконвульсанты и некоторые стероиды способны индуцировать специфические формы, обозначаемые как цитохромы Р-450 ЗА. Изониазид или хроническое потребление этанола индуцируют другую форму — цитохром Р450 2Е1, которая окисляет этанол и активирует канцерогенные нитрозамины. Кло-фибрат, понижающий концентрацию липопротеинов очень низкой плотности, индуцирует еще один изофермент — цитохром Р-450 4А, который ответственен за со-гидроксилирование некоторых жирных кислот, лейкотриенов и простагландинов.
Индуцировать цитохром Р-450 могут также поллютанты окружающей среды. Например, воздействие бензпирена и других полициклических ароматических углеводородов, присутствующих в табачном дыме, жареном на углях мясе и других органических продуктах пиролиза, индуцирует цитохром Р450 1А1 и изменяет скорость метаболизма лекарств как у экспериментальных животных, так и у человека. Другие химические вещества из окружающей среды, способные индуцировать специфические цитохромы Р-450, включают полихлорированные бифенилы, которые широко используют в промышленности как изоляционные материлы и пластификаторы, а также 2,3,7,8,-тетрахлордибен-зо-р-диоксин (диоксин, ТХДД) — следовой побочный продукт при химическом синтезе дефолианта 2,4,5,-Т (глава 58).
Повышенный синтез Р-450 требует усиления процессов транскрипции и трансляции. Выявлен цитоплазматический рецептор для полициклических ароматических углеводородов (например, бензпирена, диоксина), показана транслокация индук-торрецепторного комплекса в ядро с последующей
активацией регуляторных участков генов. Для других классов веществ-индукторов (например, фенобарбитала) аналогичный рецептор еще не найден (Gonzalez, 1989).
Ингибирование ферментов
Некоторые лекарственные субстраты могут ингибировать активность цитохром Р-450-содержа-щих ферментов. Хорошо известным ингибитором является проадифен (SKF 525-А). Это соединение активно связывается с молекулой цитохрома и тем самым конкурентно ингибирует метаболизм потенциальных субстратов. Препараты, содержащие имидазол, такие как циметидин и кетоконазол, связываются с железом гема цитохрома Р-450, инактивируя его, и тем самым эффективно снижают метаболизм как эндогенных субстратов (тестостерона), так и других совместно вводимых лекарств. Макролидные антибиотики, такие как олеандомицин, эритромицин и другие дериваты эритромицина, метаболизируются цитохромом Р-450 ЗА1 до продуктов, которые диссоциируют с железом гема цитохрома и делают его каталитически неактивным. Некоторые субстраты необратимо ингибируют цитохром Р-450 за счет химической модификации апопротеина или гема цитохрома промежуточными продуктами своего метаболизма. Антибиотик хлорамфеникол метаболизируется цитохромом Р-450 до веществ, которые алкилируют его апоцитохром и таким образом инактивирует фермент. Растущий список ингибиторов, которые “атакуют” гем, включает стероиды этинилэстрадиол, норэтин-дрон и спиронолактон, общеанестезирующий агент флюроксен, барбитураты секобарбитал и аллобарбитал, аналгезирующие седативные вещества алли-лизопропилацетилмочевину, диэтилпентенамид и этхлорвинол, растворитель карбондисульфид и пропилтиоурацил.
Реакции фазы II
Исходное вещество или его метаболиты (фаза I), которые содержат подходящие химические группы, часто подвергаются реакциям соединения или конъюгации с эндогенной субстанцией (табл. 4-2). В общем виде конъюгаты являются полярными молекулами, которые легко экскретируются и часто лишены фармакологической активности. Образование конъюгатов связано с проме-
ТАБЛИЦА 4-1. Реакции фазы I
Класс реакций
Структурные изменения
Лекарства-субстраты
Окисление
Цитохром Р-450-зави-симое окисление: Ароматическое гидроксилирование
Ацетанилид, пропранолол, фенобарбитал, фенитоин, фенилбутазон, амфетамин, варфарин, 17а-этинил эстрадиол, нафтален, бензпирен
Алифатическое гидроксилирование
RCH2CH3 — ЯСНгСНгОН
RCH2CH3-»RCHCH3 I ОН
Амобарбитал, пентобарбитал, секобарбитал, хлор-пропамид, ибупрофен, мепробамат, глютетимид, фенилбутазон, дигитоксин
Эпоксидация
\/°\/
RCH=CHR—► R— С — С —R
Алдрин
Окислительное деалкилирование N-деалкилирование
RNHCH3->RNH2+CH2O
О-деалкилирование ROCH3- ->ROH + CH2O
S-деалкилирование RSCH3- •>rsh+ch2o
N-окисление
Первичные амины rnh2-> RNHOH
Вторичные амины R1 Ri
^NH -> N — OH
r2 RZ
Третичные амины Ri Ri
R2—N — ► R2—N = 0
R3 R3
S-окисление Ri Ri
^S- * ^S = O
r2 r2
Дезаминирование OH
RCHCH3 —► R — C — CH3—>- R — CCH3+ NH3
1 1 II
NHZ nh2 0
Морфин, этилморфин, бензфетамин, аминопирин, кофеин, теофиллин
Кодеин, р-нитроанизол
6-метил
Анилин, хлорфентермин
2-Ацетиаминофлюорен, ацетаминофен
Никотин, метаквалон
Тиоридазин, циметидин, хлорпромазин
Амфетамин, диазепам
ТАБЛИЦА 4-1. (Продолжение)_________________
Класс реакций Структурные изменения
Цитохром Р-450-зависимое окисление:
Лекарства-субстраты
Десульфирование
Тиопентал
Дехлорирование
СС14-> [СС!3] + СНС13
Паратион
Цитохром Р-450-независимое окисление:
Флавинмонооксигеназа (фермент Циглера)
* Н* .
R3N------► R3N —► О ----> R3N+OH
RCH2N — CH2R -► RCH2 — N — CH2R
H in
4
RCH = N — CH2R
Четыреххлористый углерод
Хлорпромазин, амитриптилин, бензфетамин
Дезипрамин, нортриптилин
Аминооксидазы
Дегидрогенация
Восстановление
Азо-восстановление
Нитро-восстановление
Карбонильное восстановление
Гидролиз
Эфиры
Амиды
Метимазол, пропилтиоурацил
RCHjNH2->RCHO+NH3 rch2oh->rcho
RN = NRt-> RNH— NHR,-> RNH2+ R,NH2
RNO2-> RNO -» RNHOH -> RNH2
RCR' -► RCHR’
О OH
r,coor2-> r,cooh+ r2oh
RCONHR, -» RCOOH + R,NH2
Фенилзтиламин, эпинефрин
Этанол
Пронтозил,тартразин
Нитробензол, хлорамфеникол, клоразепам, дантро-лен
Метирапон, метадон, налоксон
Прокаин,сукцинилхолин, аспирин, клофибрат, метилфенидат
Прокаинамид, лидокаин, индометацин
ТАБЛИЦА 4-2. Реакций фазы II
Тип конъюгации Эндогенная субстанция Трансфераза (локализация) Типы субстратов Примеры
Глюкуронизация УДФ-глюкуроно-вая кислота УДФ-глюкуронил-трасфераза (микросомы) Фенолы, спирты, кар- Нитрофенол, морфин, боксикислоты, гидро- ацетаминофен,диазепам, ксиламины, сульфона- N-гидроксидапсон суль-миды фатиазол, мепробамат, дигитоксин, дигоксин
Ацетили рование Ацетил-КоА N -ацетилтрансфераза (цитозоль) Амины Сульфонамиды, изониа- зид, клоназепам, дапсон, мескалин
Конъюгация с глутатионом Глутатион Глутатион-SH-S-трансфераза (цитозоль, микросомы) Эпоксиды, ареноксиды, Этакриновая кислота, нитрогруппы, гидро- бромбензол ксиламины
Конъюгаты с гли- Фосфоаденозил Сульфотрансфера- Фенолы, спирты, аро- Эстрон, анилин, фенол,
цином фосфосульфат за (цитозоль) матические амины 3-гидроксикумарин, аце- таминофен, метилдопа
Метилирование S-аденозилме-тионин Трансметилазы (цитозоль) Катехоламины, фено- Допамин, адреналин, пилы, амины, гистамин ридин,гистамин,тиоурацил
Водная конгъю-гация Вода Эпоксидгидролаза (микросомы) (цитозоль) Ареноксиды, цис-дву- Бензпирен-7,8-эпоксид, замещенные и моноза- стирен-1,2-оксид, карба-мещенные оксираны мазепин, эпоксид Алкеноксиды, эпоксиды Лейкотриен А, жирных кислот
жуточными высокоэнергетическими продуктами и специфическими ферментами. Такие ферменты (трансферазы) могут локализоваться в микросомах или в цитозоле. Они катализируют соединение активированного эндогенного вещества (такого как уридин-5'-дифосфат-глюкуронид) с лекарством (или эндогенным веществом) или соединение активированного лекарства (такого как S-KoA-npo-изводного бензойной кислоты) с эндогенным субстратом. Поскольку эндогенные субстраты могут содержаться в продуктах, питание играет важнейшую роль в регуляции процесса конъюгации лекарств.
Конъюгация лекарств, как считают, является конечным событием инактивации и рассматривается как реакция “истинной детоксикации”. Однако эта концепция не абсолютна, так как известно, что некоторые реакции конъюгации (ацилглюкурони-зация нестероидных противовоспалительных веществ, О-сульфатирование N-гидроксиацетилами-нофлуорена и N-ацетилирование изониазида) мо
гут привести к образованию активных продуктов, обладающих гепатотоксичностью.
Метаболизм лекарств с образованием токсичных продуктов
Становится очевидным, что метаболизм лекарств и иных чужеродных химических соединений не всегда является безобидным биохимическим событием, ведущим к детоксикации и элиминации веществ. В действительности, некоторые вещества могут в процессе метаболизма трансформироваться в активные промежуточные продукты, которые токсичны в отношении различных органов. Такие токсические реакции не выражены при незначительном действии исходного вещества, когда механизмы детоксикации не перегружены и количество эндогенных детоксицирующих косубстратов (глутатиона, глюкуроновой кислоты, сульфата) доста
точно. Однако когда эти ресурсы истощены, могут преобладать токсические пути, что ведет к явной органотоксичности или канцерогенезу. Число примеров такой токсичности, вызываемой лекарствами, быстро увеличивается. В качестве примера можно привести гепатотоксичность, вызванную ацетаминофеном (парацетамолом, рис. 4-4).
Это аналгетическое и жаропонижающее средство вполне безопасно в терапевтических дозах. В норме оно соединяется с глюкуроновой и серной кислотами, что в совокупности составляет 95 % Экскретируемых метаболитов. На альтернативный путь цитохром Р-450-зависимой конъюгации с глутатионом (Г-SH) приходятся оставшиеся 5 %. Когда доза ацетаминофена значительно превышает терапевтическую, пути образования глюкуронида и сульфата насыщаются, и возрастает роль цитохром Р-450-за-висимого пути. Гепатотоксичность не выявляется до тех пор, пока количество глутатиона достаточно для конъюгации. Однако постепенно расход глутатиона печени начинает превышать его восстановление и происходит накопление активного и токсичного метаболита. В отсутствие внутриклеточных нуклеофильных веществ типа глутатиона этот активный продукт (как предполагают, N-гидроксилированное производное или N-ацетилбензоиминохинон) реагирует с нуклеофильными группами клеточных макромолекул, например белков, что ведет к гепа-тотоксическому действию (рис. 4-4).
Понимание химической и токсикологической характеристики электрофильной природы активного метаболита ацетаминофена способствовало созданию эффективного антидота — цистеамина и N-ацетилцистеина. Введение более безопасного N-ацетилцистеина в сроки от 8 до 16 часов после передозировки ацетаминофена предупреждает развитие скоротечной гепатотоксичности и летальный исход.
Сходное механистическое объяснение может быть использовано применительно к нефротоксичности фенацетина и гепатотоксичности афлатоксина и бензпирена.
Клиническое значение метаболизма лекарств
Доза и частота приема, необходимые для достижения эффективной концентрации в крови и тканях, могут варьировать у больных из-за индивиду-
Ац-глюкуронид
Ац-сульфат
Цитохром Р-450
Активное
электрофильное соединение (Ац*)
Г-SH
Макромолекулы клетки \ (протеины)
Г-S-Au*
I
Ац-меркагпурат
Ац‘-протеин
Гибель печеночной клетки
Рис. 4-4. Метаболизм ацетаминофена (Ац) с образованием гепатотоксичных метаболитов (Г-SH — глутатион; Г-S — остаток глутатиона; Ац* — реактивный метаболит)
альных различий в распределении лекарств и скорости метаболизма и элиминации. Эти различия определяются генетическими факторами и такими, не связанными с генетикой переменными, как возраст, пол, размер и функция печени, циркадный ритм, температура тела, факторы окружающей среды и питания, в частности воздействие индукторов или ингибиторов метаболизма лекарств. Рассмотрим наиболее важные факторы, влияющие на метаболизм лекарств, которые имеют клиническое значение.
Индивидуальные отличия
Индивидуальные отличия в скорости метаболизма зависят от природы лекарства. Так, в одной и той же популяции при одинаковом уровне в плазме можно наблюдать 30-кратный разброс в скорости метаболизма одного лекарства и лишь двухкратную вариабельность для другого.
Генетические факторы
Генетические факторы, которые влияют на уровень активности ферментов, могут быть ответственны за отличия метаболизма лекарств. Например, сукцинилхолин гидролизуется в два раза медленнее у лиц с генетическим дефектом активности псевдохолинестеразы по сравнению с лицами,
имеющими ее нормальный уровень. Аналогичные фармакогенетические влияния выявлены в отношении ацетилирования изониазида (рис.4-5) и гидроксилирования варфарина. Эффект медленного ацетилирования (изониазида и сходных аминов) вызван скорее синтезом меньшего количества соответствующего фермента, чем его аномальной формой. Врожденный по аутосомному рецессивному типу фенотип медленного ацетилирования наблюдается у 50 % темнокожих и белых жителей США, относительно часто у европейцев, живущих в северных широтах, и значительно реже у жителей Азии и эскимосов. Подобным же образом генетически детерминированные дефекты окислительного метаболизма были выявлены для дебризохина, фенацетина, гуаноксана, спартеина, фенформина и других веществ (табл. 4-3). Эти дефекты наследуются по аутосомно-рецессивному типу и могут проявляться в множественности вариантов метаболической трансформации химических веществ in vivo.
Две генетических вариации полиморфизма биотрансформации лекарств были особенно подробно изучены и внесли некоторую ясность в возможные механизмы. Во-первых, это полиморфизм окисления дебризохина/спартеина, который проявляется у 3-10 % лиц кавказской расы и наследуется по аутосомному рецессивному типу. У этих лиц нарушается цитохром Р-450-зависимое окисление дебризохина, спартеина, фенформина, декстроме-
«25
01 23456789 10 11 12
Концентрация изониазида (мкг/мл)
Рис. 4-5. Генетический полиморфизм метаболизма лекарств. График показывает распределение концентраций изониазида в плазме 267 человек через 6 часов после приема внутрь в дозе 9.8 мг/кг. Это распределение имеет бимодальный характер. Пациенты, с концентрацией в плазме выше 2.5 мг/мл через 6 часов, рассматриваются как медленные ацетиляторы (Evans D. А. Р., Manley К. А., McKusick V. A. Generic control of isoniasid metabolism in man. Br. Med. J., 1960; 2:485.)
торфана, метопролола, буфуралола и некоторых [3-блокаторов и трициклических антидепрессантов. Результаты изучения показывают, что наблюдаемые дефекты окислительного метаболизма лекарств являются врожденными. Точная молекулярная основа дефекта состоит, видимо, в нарушении экспрессии белка цитохрома Р-450, что ведет к нарушению синтеза необходимого изофермента.
Вторым хорошо изученным примером генетического лекарственного полиморфизма является стереоселективное ароматическое ^-гидроксилирование антиконвульсанта мефенитоина. Этот полиморфизм, наследуемый также по аутосомному рецессивному типу, наблюдается у 3-5 % кавказского населения и 18-23 % населения Японии. Он генетически независим от полиморфизма дебризо-хин/спартеинового типа. У нормальных “активных метаболизаторов” S-мефенитоин сначала гидроксилируется в положении 4 фенильного кольца, затем конъюгируется с глюкуроновой кислотой и быстро экскретируется с мочой, тогда как для R-мефени-тоина характерно медленное N-деметилирование до нирванола. “Медленные метаболизаторы”, полностью лишены стереоспецифической S-мефенито-ин-гидроксилазной активности, у них как S-, так и N-энантиомеры, перед экскрецией N-деметили-руются до нирванола. Молекулярная основа этого нарушения, возможно, связана с экспрессией мутантного изофермента Р-450, который напоминает нормальную форму, но имеет достаточную структурную микрогетерогенность для изменения аффинитета к субстрату и стереоселективности, а следовательно,— и функции. Клинически важно учитывать, что безопасность лекарства может быть значительно ниже у лиц, являющихся “медленными метаболизаторами”. Например, в случае недостаточного метаболизма мефенитоина наблюдаются признаки глубокой седации и атаксии после доз препарата, которые хорошо переносятся лицами с нормальным метаболизмом.
Выявляются и другие виды генетического полиморфизма в превращениях лекарств, которые наследуются независимо от уже рассмотренных. Изучение метаболизма теофиллина у монозиготных и дизиготных близнецов, включающее анализ родословной различных семей, показало, что у этих лекарств может наблюдаться отчетливый полиморфизм, наследуемый по рецессивному типу. Генетический полиморфизм лекарств выявлен для окисления аминопурина, толбутамида и карбоцистеина.
ТАБЛИЦА 4-3. Некоторые примеры генетического полиморфизма метаболизма лекарств
Дефект Лекарство и терапевтическое применение Клинические последствия1
Окисление Буфуралол (блокатор p-адренорецепторов) Усиление блокады Р-адренорецепторов, тошнота
Окисление Дебризохин (антигипертензивное средство) Ортостатическая гипотензия
Окисление Этанол Покраснение кожи лица, сердечно-сосудистые симптомы
N-ацетилирование Гидралазин (антигипертензивное средство) Волчаночноподобный синдром
N-ацетилирование Изониазид (антитуберкулезное средство) Периферическая нейропатия
Окисление Спартеин Симптомы окислительной токсичности
Окисление Мефенитоин (антиэпилептическое средство) Токсичность, связанная с передозировкой
Гидролиз эфира Толбутамид (гипогликемическое средство) Кардиотоксичность
Окисление СукцинилхолиН (миорелаксант) Продолжительное апное
’ Наблюдаемые или предсказуемые.
Хотя генетический полиморфизм окисления часто связан со специфическими изоэнзимами цитохрома Р-450, подобные вариации могут иметь отношение и к другим участникам метаболизма. Данные последнего времени о полиморфизме окисления триметиламина, который предположительно метаболизируется флавинмонооксигеназой (фермент Циглера), указывают, что генетические варианты цитохром Р-450-независимых окислительных ферментов могут также участвовать в полиморфизме.
Факторы среды
Факторы среды тоже вносят вклад в индивидуальную вариабельность метаболизма лекарств. Курильщики метаболизируют некоторые лекарства быстрее, чем некурящие, в связи с индукцией ферментов. Промышленные рабочие, подвергающиеся воздействию пестицидов, метаболизируют некоторые препараты быстрее, чем лица, не контактирующие с этими веществами. Такие различия затрудняют определение эффективных и безопасных доз лекарств с небольшим значением терапевтического индекса.
Возраст и пол
Повышенная чувствительность к фармакологическому или токсическому действию лекарств отмечается у очень молодых или пожилых пациентов (глава 62). Хотя это может отражать особенности всасывания, распределения и элиминации, нельзя исключать и различия в скорости метаболизма.
Исследования на других видах млекопитающих показали, что лекарства метаболизируются с пониженной скоростью в препубертатный период и при старении. Замедление метаболизма может быть связано с уменьшением активности ферментов или дефицитом эндогенных кофакторов. Сходные тенденции выявлены у человека, однако достоверные доказательства еще не получены.
Вариации метаболизма, зависящие от пола, показаны у крыс. Молодые крысы-самцы метаболизируют лекарства быстрее, чем взрослые самки или самцы препубертатного возраста. Эти различия в превращении лекарств отчетливо связаны с уровнем андрогенов. Некоторые клинические наблюдения указывают на то, что сходные зависимые от пола отличия метаболизма лекарств существуют и у человека применительно к этанолу, пропранололу, бензодиазепинам, эстрогенам и салицилатам.
Взаимодействия лекарств в процессе метаболизма
Многие субстраты, вследствие относительно высокой липофильности, не только удерживаются около активного центра фермента, но и неспецифически связываются с липидными мембранами саркоплазматического ретикулума. В этом состоянии они могут вызывать индукцию микросомальных ферментов, а также в зависимости от остаточного уровня лекарства в активном центре конкурентно тормозить метаболизм одновременно вводимого лекарства.
В список ферментиндуцирующих лекарств входят различные седативно-гипнотические, противо-
ТАБЛИЦА 4-4. Сокращенный список препаратов, усиливающих метаболизм других лекарств у человека
Индуктор Лекарство, метаболизм которого усиливается
Бензпирен Хлорциклизин Этхлорвинол Глютетимид Гризеофульвин Фенобарбитал и другие барбитураты1 Теофиллин Стероидные гормоны Варфарин Антипирин, глютетимид, варфарин Варфарин Барбитураты, хлорамфеникол, хлорпромазин, кортизол, кумариновые антикоагулянты, деметилимипрамин, дигитоксин, доксорубицин, эстрадиол, фенилбутазон, фенитоин, хинин, тестостерон
Фенилбутазон Фенитоин Рифампин Аминопирин, кортизол, дигитоксин Кортизол, дексаметазон, дигитоксин,теофиллин Кумариновые антикоагулянты, дигитоксин, глюкокортикоиды, метадон, метопролол, оральные контрацептивы, преднизон, пропранолол, хинидин
’ За исключением секобарбитала (см. табл. 4-5 и текст).
судорожные средства, транквилизаторы и инсектициды (табл. 4-4). Пациентам, получающим барбитураты, другие седативные и снотворные средства или транквилизаторы, могут потребоваться значительно более высокие дозы антикоагулянтов варфарина или дикумарола, применяемых перорально. С другой стороны, прекращение приема седативных препаратов может привести к замедленному метаболизму антикоагулянтов и, следовательно, кровоточивости. Сходное взаимодействие наблюдается у больных, получающих такие комбинации лекарств как антипсихотические или седативные с контрацептивными, седативные с противосудорожными, алкоголь с гипогликемическими веществами (толбутамид). Следует также отметить, что
индуктор может усиливать не только метаболизм другого вещества, но и свой собственный. Так, продолжительный прием некоторых лекарств может привести к фармакокинетической толерантности — прогрессивному снижению эффективности из-за ускорения собственного метаболизма.
С другой стороны, одновременное введение двух или более лекарств может привести к нарушению элиминации более медленно метаболизируемого лекарства и удлинению или потенцированию его фармакологического эффекта (табл. 4-5). Как конкурентное субстратное ингибирование, так и необратимая субстратная инактивация фермента, могут увеличить уровень лекарства в плазме и привести к токсическому действию, особенно для препа
ТАБЛИЦА 4-5. Сокращенный список препаратов, угнетающих метаболизм других лекарств у человека
Ингибитор Лекарство, метаболизм которого угнетается
Аллопуринол, хлорамфеникол, изониазид Циметидин Дикумарол Диэтилпентенамид Дисульфирам Этанол Кетоконазол Нортриптилин Оральные контрацептивы Фенилбутазон Секобарбитал Тролеандомицин Антипирин, дикумарол, пробенецид, толбутамид Хлордиазепоксид, диазепам, варфарин, другие вещества Фенитоин Диэтилпентенамид (Новонал) Антипирин, этанол, фенитоин, варфарин Хлордиазепоксид (?), диазепам (?), метанол Циклоспорин, астемизол, терфенадин Антипирин Антипирин Фенитоин, толбутамид Секобарбитал Теофиллин, метилпреднизолон
ратов с небольшим терапевтическим индексом. Так, показано, что эритромицин угнетает метаболизм антигистаминного средства терфенадина и приводит к появлению побочных эффектов в виде сердечных аритмий. Сходным образом аллопуринол удлиняет продолжительность и усиливает химиотерапевтическое действие меркаптопурина путем конкурентного ингибирования ксантиноксидазы. Следовательно, для предупреждения токсического действия на костный мозг у больных, получающих аллопуринол, необходимо снизить дозу меркаптопурина. Циметидин — лекарство, используемое для лечения пептической язвы, потенцирует фармакологическое действие антикоагулянтов и седативных средств. Метаболизм хлордиазепоксида подавляется на 36 % после однократного введения циметидина, эффект устраняется в пределах 48 часов после прекращения приема циметидина.
Нарушение метаболизма может также происходить, если одновременно вводимое вещество необратимо ингибирует общий метаболизирующий фермент, как это наблюдается в случае передозировки секобарбитала и новонала (диэтилпентен-амид). В процессе микросомального окисления этих препаратов инактивируются ферменты, что ведет к нарушению их собственного метаболизма и метаболизма других косубстратов.
Взаимодействие между лекарствами и эндогенными соединениями
Для инактивации ряда лекарств требуется конъюгация с эндогенными субстратами — глутатионом, глюкуроновой кислотой и сульфатом. Соответственно, различные лекарства могут конкурировать за одни и те же эндогенные субстраты, поэтому быстро реагирующие препараты могут эффективно снижать уровень эндогенного субстрата и нарушать метаболизм медленно реагирующего лекарства. Если последнее отличается высокой крутизной кривой доза-эффект или узким диапазоном безопасных доз, может происходить потенцирование его фармакологических и токсических эффектов.
Заболевания, влияющие на метаболизм лекарств
Острые или хронические заболевания, влияющие на структуру или функцию печени, заметно изменяют печеночный метаболизм некоторых ле
карств. К таким заболеваниям относят жировую дистрофию, алкогольный гепатит, активный или неактивный алкогольный цирроз, гемохроматоз, хронический активный гепатит, билиарный цирроз и острый вирусный или лекарственный гепатит. В зависимости от тяжести эти заболевания нарушают функции печеночных ферментов, метаболизирующих лекарства, особенно микросомальных оксидаз, и тем самым значительно влияют на элиминацию лекарств. Например, период полувыведения хлордиазепоксида и диазепама у больных с циррозом печени или острым вирусным гепатитом значительно возрастает. Эти лекарства даже в обычных дозах могут вызвать кому у больных с заболеваниями печени.
При раке печени существенно нарушается метаболизм лекарств у человека. Например, метаболизм аминопирина у больных со злокачественными опухолями печени заметно медленнее, чем у здоровых людей. У этих больных выявляется также значительно уменьшенный клиренс аминопирина. Исследование биоптатов печени пациентов с гепатоцеллюлярной карциномой указывает на нарушение способности к окислительному метаболизму лекарств in vitro. Это сочетается с соответствующим понижением содержания цитохрома Р-450.
Заболевания сердца за счет ограничения печеночного кровотока могут нарушить кинетику лекарств, метаболизм которых лимитируется кровотоком (табл. 4-6). Эта лекарства столь легко метаболизируются печенью, что печеночный клиренс практически равен кровотоку через печень. Заболевания легких также могут влиять на метаболизм лекарств, о чем свидетельствует нарушение метаболизма прокаинамида и прокаина у больных с хронической дыхательной недостаточностью и увеличение периода полувыведения антипирина у больных раком легких. Нарушение активности фермен-
ТАБЛИЦА 4-6. Быстро метаболизируемые лекарства, печеночный клиренс которых лимитируется кровотоком
Алпренолол Лидокаин
Амитриптилин Меперидин
Хлорметиазол Морфин
Дезипрамин Пентазоцин
Имипрамин Пропоксифен
Изониазид Пропранолол
Лабеталол Верапамил
та или его нарушенный синтез фермента при отравлении тяжелыми металлами или порфирии тоже приводят к угнетению печеночного метаболизма лекарств. Так, при отравлении свинцом у человека увеличивается период полувыведения антипирина.
Влияния эндокринных дисфункций на метаболизм лекарств были детально исследованы на экспериментальных моделях у животных, но соответствующие данные для людей с гормональной патологией немногочисленны. Нарушения функции щитовидной железы сочетаются с изменением метаболизма некоторых лекарств и многих эндогенных соединений. Гипотиреоидизм увеличивает период полувыведения антипирина, дигоксина, мети-мазола и практолола, тогда как гипертиреоидизм оказывает противоположное влияние. Немногочисленные наблюдения у больных сахарным диабетом не указывают на очевидные нарушения метаболизма лекарств, судя по периодам полувыведения антипирина, толбутамида и фенилбутазона. В эксперименте у крыс-самцов (аллоксановый или стреп-тозоциновый диабет) выявляются нарушения превращений некоторых препаратов. Эти изменения устраняются при введении инсулина, который не влияет прямо на печеночные ферменты, метаболизирующие лекарства. Нарушение функции гипофиза, коры надпочечников и половых желез значительно изменяет метаболизм лекарств в печени крыс. На основании этих наблюдений можно предполагать, что такие нарушения могут значительно влиять на биотрансформацию препаратов у человека. Пока необходимые доказательства не будут получены в клинических наблюдениях, подобные экстраполяции могут рассматриваться как предположительные.
Избранная литература
Gilmore D. A. et al. Age and gender influence the stereoselective pharmacokinetics of propranolol. J. Pharmacol. Exp. Ther. 1992; 261:1181.
Gonzalez F. The molecular biology of cytochrome P450s. Pharmacol. Rev. 1989; 40: 243.
Gonzalez F. J. et. al. Characterization of the common genetic defect in humans deficient in debrisoquin metabolism. Nature, 1988; 331:442.
Gonzalez F. J. Human cytochromes P450: problems and prospects. Trends Pharmacol. Sci. 1992; 13:346.
Guengerich F. P. (ed.) Mammalian cytochromes P-450: Structure, Mechanism and Biochemistry. Plenum Press, 1986.
Minchin R. F., Boyd M. R. Localization of metabolic activation and deactivation systems in the lung: Significance to the pulmonary toxicity of xeno-biotics. Annu. Rev. Pharmacol. Toxicol. 1983; 23: 217.
Ortiz de Montellano P. R., Correia M. A. Suicidal destruction of cytochrome P-450 during oxidative drug metabolism. Annu. Rev. Pharmacol. Toxicol. 1983; 23:481.
Zhou H-H et al. Racial differences in drug response: Altered sensitivity to and clearance of propranolol in men of Chinese descent as compared with American males. N. Engl. J. Med. 1989; 320:565.
Базисная и клиническая оценка новых лекарств
5
Барри А. Берковиц, Бертрам Г. Катцунг
За последние 60 лет создание новых лекарств произвело революцию в практической медицине, превратив многие ранее фатальные заболевания в почти рядовые. Одной из причин этих достижений является фундаментальный прогресс в средствах создания и проверки новых лекарств, в значительной степени ускорившийся с внедрением новых технологий и в связи с финансовой поддержкой медицинских исследований со стороны правительства. В большинстве стран проверка лекарств в настоящее время регулируется законодательством и тщательно контролируется правительственными учреждениями. В настоящей главе обобщены представления о процессе открытия новых терапевтических средств, их внедрении и контроле. Приведенные примеры отражают опыт США, но пути создания новых лекарств в разных странах в основном сходны.
Первым шагом на пути создания нового лекарства является открытие или синтез новой молекулы потенциального препарата (рис. 5-1). По закону безопасность и эффективность лекарства должны быть тщательно проверены перед тем, как оно попадет на рынок. В дополнение к исследованиям in vitro большинство биологических эффектов молекулы должно быть охарактеризовано у животных до начала клинических испытаний на людях. Их проводят в три этапа (общепринятые фазы), после чего лекарство может быть признано пригодным для медицинского применения. Четвертая фаза (сбора данных) следует за получением разрешения к применению в практике.
На создание и успешное внедрение нового лекарства затрачиваются огромные средства — от 100 до 350 миллионов долларов и более. Эти расходы включают труд, затраченный на поиск новых перспективных веществ (может быть синтезировано 5 000-10 000 соединений перед тем, как будет най
дено нужное), стоимость базисных (доклинических) и клинических исследований и внедрение окончательного вещества-кандидата. Вследствие высокой стоимости работ и элемента риска большинство новых лекарств создаются в лабораториях фармацевтических компаний. В то же время стимулы к успеху в создании лекарств исключительно сильны. Мировой рынок рецептурных лекарств в 1991 г. оценивался в 141 миллиард долларов.
Открытие лекарств
Большинство новых кандидатов в лекарства выявляют с помощью одного из трех подходов: 1) химической модификации известных молекул; 2) скрининга биологической активности большого количества натуральных продуктов, рядов ранее открытых химических структур, больших “библиотек” пептидов или нуклеиновых кислот; 3) направленного синтеза (рациональный дизайн лекарств), основанного на понимании биологических механизмов и химической структуры.
Создание тиазидных диуретиков посредством изменения структуры менее эффективных ингибиторов карбоангидразы (глава 15) является примером первого подхода. Открытие циклоспорина — препарата грибкового происхождения с иммуносупрессивным действием, иллюстрирует второй подход. Пример реализации третьего подхода — создание антагонистов Н2-гистаминоблокаторов (табл. 5-1). На основании представлений о существовании различных типов гистаминовых рецепторов синтезирован циметидин, что более подробно описано в дополнении “Исторический пример: открытие и внедрение антагонистов ^-рецепторов”. Направленный синтез лекарств за последние 10 лет достиг больших успехов, что можно проиллюстрировать примерами создания ингибиторов
| ’ Изучение | in vitro *
Проверка j 4 f Клинические• на животных испытания i
Биологические продукты
Лекарственная субстанция (основное соединение)
(Эффективность^
| Селективность Механизм j действия
Химический синтез
(аффективно лраза е. ли лекарство?)
(эффективно ли лекарство при двойном слепом контроле?)
Метаболизм лекарства, оценка безопасности
Фаза 3?
(безопасно ли лекарство?)
ii-
Появление генериков
Фаза 4
(постмаркетинговое наблюдение)
О
Годы (в среднем)
2
4 IDN Исследование нового лекарства
8-9
NDA Внедрение нового лекарства
Годы после разрешения к применению. Прекращение срока действия патента
Рис. 5-1. Этапы создания и система обязательной проверки лекарств для внедрения на рынок в США. Некоторые требования могут отличаться применительно к препаратам для неотложной терапии
ТАБЛИЦА 5-1. История создания блокаторов Нг-рецепторов
Соединение и характеристики
Структура
Антагонистическая активность (ID50in vivo в мкмоль/кг)’
Гистамин
Исходное вещество
N-гуанилгистамин
Первое активное производное. Слабый парциальный агонист
Буримамид
Производное тиомочевины с удлиненной боковой цепочкой. Малоактивен у человека
Метиамид
Активен у человека, но токсичен
Циметидин
Тиомочевина заменена на N-CN-замес-титель. Обладает высокой активностью и низкой токсичностью. Родоначальник серии лекарств для лечения кислотно-пептических синдромов
' Гистаминстимулируемая секреция кислоты у анестезированных крыс; Ю50 — доза при внутривенном введении, необходимая для 50 % ингибирования. Из: Brimblecombe R. W. et al., 1978; and Black J., 1989.
Исторический пример: открытие и внедрение антагонистов Н2-рецепторов (таблица 5-1)
Идея. Блокаторы Н2-рецепторов сегодня являются лекарствами с наибольшим объемом продаж в мире. Их открытие началось с наблюдения, что гистамин является мощным стимулятором секреции желудка и что классические антигистаминные препараты (известные теперь как блокаторы Н(-рецепторов) не подавляют это действие. Для Джеймса Блэка, создавшего к этому времени первый клинически значимый блокатор p-адренорецепторов, это означало, что могут существовать разные подтипы гистаминовых рецепторов, выполняющие различные функции, и что их можно избирательно возбуждать или блокировать с помощью фармакологических средств.
Клиническая значимость. Пептические язвы очень распространены и могут лечиться путем подавления желудочной секреции. Клиническая потребность в эффективном угнетении выработки кислоты не удовлетворялась существующими лекарствами или возможностями хирургического лечения язвенной болезни.
Биологическая гипотеза. Блэк предположил, что эффект гистамина можно избирательно блокировать на уровне рецептора, опосредующего его влияние на кислотную секрецию. Существовали экспериментальные системы, которые можно было использовать как модели для оценки конечного эффекта гистамина. Предполагали, что некоторые из этих моделей могут быть адекватны язвенной болезни человека.
Химическая гипотеза. Химическая гипотеза была основана на том факте, что существовавшие тогда антигистаминные вещества были неэффективны при заболеваниях желудочно-кишечного тракта, связанных с повышенной кислотностью и не имели структурного сходства с гистамином. Команда исследователей пришла к выводу, что химическая модификация молекулы гистамина может привести к созданию селективного антагониста для гипотетических гистаминовых рецепторов желудка.
Создание. “Охота” началась в 1964 г., когда Блэк приступил к изучению этой проблемы в лаборатории компании “Smith Kline French” в Англии. После преодоления первоначальных трудностей было синтезировано и исследовано большое число соединений, сходных по структуре с молекулой гистамина. Выявленные на доклиническом этапе эффективные и безопасные соединения были переданы на клинические испытания. Первый селективный Н2-антагонист, бури-мамид, оказался не достаточно эффективным. Структуру буримамида несколько изменили и получили более активный метиамид. Клинические испытания этого препарата показали хорошую эффективность, но неожиданно высокую токсичность, проявляющуюся в форме грануло-цитопении. Дальнейшие усилия привели к созданию циметидина. Это вещество успешно прошло клинические испытания и было одобрено в 1974 г. как первый селективный препарат-антагонист Н2-рецепторов. На поиск ушло 12 лет. В 1992 г. объем продаж антагонистов Н2-рецеп-торов составил 4 миллиарда долларов.
Проект “Smith Kline” выполнялся то при возрастающей, то при убывающей финансовой поддержке. Поскольку статистика открытия и внедрения лекарств показывает, что большинство кандидатов в препараты не доходят до медицинского применения, пессимистический прогноз более оправдан. При создании лекарств совершаются два типа ошибок. Лекарства с недостаточной безопасностью и эффективностью могут неадекватно поддерживаться и получить одобрение. С другой стороны, эффективные и безопасные лекарства могут быть преждевременно отвергнуты. В случае с блокаторами Н2-рецеп-торов проект держался на энтузиазме нескольких подвижников идеи и в конце концов оказался успешным.
Послесловие. На решения в области создания лекарств оказывают влияние рыночные факторы. Оценочные данные по рынку лекарств,
применимых при увеличении активности кислотно-пептического фактора, в шестидесятых годах соответствовали объему продаж в 30 миллионов долларов, что, вероятно, слишком мало для того, чтобы оправдать усилия для поиска с негарантированным успехом. Однако оказалось, что исходная рыночная оценка занижена в 100 раз. Новые безопасные и эффективные лекарства могут создавать новые рынки и повышать частоту выявления заболевания. Подобно блокаторам Н2-рецепторов, большинство классов лекарств с высоким потенциалом объема продаж (противотревожные, p-блокаторы, инги
биторы конвертирующего фермента) за последние два десятилетия открыли новые рынки, которые было невозможно предсказать на основе проспективного анализа.
Даже заметные успехи в разработке наиболее продаваемых лекарств, значительно усиливающие компанию и создающие возможности для инвестиций в исследования и развитие, могут оказаться недостаточными для поиска новых препаратов и поддержания необходимых условий роста и выживания компании. В 1989 г. “Smith Kline” слилась с компанией “Beecham Pharmaceuticals”.
ангиотензинпревращающего фермента на основе изучения связи строения и действия блокаторов его активного сайта и компьютерного конструирования гипотетической химической структуры лекарства.
Независимо от источника, проверка молекулы-кандидата состоит из серии экспериментов и оценок, которые обозначают как лекарственный скрининг. Для определения активности и селективности действия лекарств используют множество биологических тестов на молекулярном, клеточном, органном и организменном уровнях. Тип и число начальных скрининговых тестов зависят от фармакологических задач. Антиинфекционные лекарства следует прежде всего тестировать по действию на различные микроорганизмы, гипогликемические — по их способности понижать уровень сахара в крови и т. п. Однако обычно исследуется более широкий спектр активности вещества для установления избирательности его действия. Это дает возможность выявить неожиданные токсические эффекты, а иногда — открыть новые терапевтические возможности. Селекция молекул для дальнейшего изучения наиболее эффективно осуществляется на моделях клинической патологии у лабораторных животных. В случае, когда разработана хорошая экспериментальная модель (например, гипертензия), обычно получаются и адекватные лекарства. Удовлетворительные результаты особенно трудно получить там, где модели еще не разработаны, например при болезни Альцгеймера.
Некоторые исследования, которые выполняются в процессе скрининга, перечислены в табл. 5-2 и определяют фармакологический профиль лекар
ства. Например, широкий набор тестов используется при изучении потенциальных антигипертензивных лекарств, действующих предположительно как антагонисты ос-адренорецепторов сосудов. На молекулярном уровне соединение должно быть проверено на способность связывания с клеточными мембранами, содержащими ос-адренорецепторы, другие рецепторы и ферменты. На начальной стадии изучения должны быть получены данные о влиянии на цитохром Р-450 печени, чтобы определить, может ли интересующая молекула активировать или ингибировать микросомальное окисление.
Для определения эффективности соединения должно быть изучено влияние на клеточные функции. Необходимо получить данные о том, является ли лекарство агонистом, парциальным агонистом или антагонистом сс-рецепторов. Для дальнейшего изучения фармакологической активности и селективности нового соединения в сравнении с эталонными лекарственными средствами должны быть использованы препараты изолированных сосудов. Можно провести и другие исследования in vitro, например на препаратах гладкой мускулатуры бронхов или кишечника. На каждом этапе этого пути соединение должно удовлетворять определенным требованиям, чтобы его дальнейшее изучение имело перспективу.
Исследования на целостном организме животного обычно необходимы для определения влияния лекарства на органы и его действия на моделях заболеваний. Вначале испытания нового средства проводят на интактных животных. Для гипотетического антигипертензивного вещества в последую-
ТАБЛИЦА 5-2. Тестирование фармакологического профиля нового лекарственного средства
Экспериментальный метод или мишень Вид животных/ткань Путь введения Измеряемый параметр
Молекулярный уровень Связывание с рецептором (например, «-адренорецепторы) Фракции клеточных мембран из органов или культуры клеток In vitro Сродство к рецептору и селективность
Активность фермента (на- Симпатические нер- In vitro Угнетение фермента и селективность дей-
пример, тирозингидрок- вы/надпочечники; ствия
силазы, дофамин-Р-гид- очищенные фермен-роксилазы, моноамин- ты оксидазы)
Цитохром Р-450 Печень In vitro Угнетение фермента, влияние на метаболизм лекарств
Клеточный уровень
Клеточная функция Культура клеток In vitro Рецепторная активность: агонизм или антагонизм (например, влияние на циклические нуклеотиды)
Изолированная ткань Кровеносные сосуды: In vitro Влияние на способность сосудов к сокраще-
артерии/вены: серд- нию и расслаблению; избирательность влия-
це, легкие, подвздош- ния на рецепторы сосудов; влияние на дру-
ная кишка (крыса или гую гладкую мускулатуру
морская свинка) Системный уровень, модели заболеваний
Кровяное давление Собаки, кошки (нар- Паренте- Изменения систолических и диастолических
котизированные) рально показателей
Гипертензивные кры- Внутрь Антигипертензивный эффект
сы (бодрствующие)
Влияние на сердце Собаки (ненаркотизи- Внутрь Электрокардиография
рованные)
Собаки (наркотизи- Паренте- Инотропный и хронотропный эффекты, сер-
рованные) рально дечный выброс, общее периферическое сопротивление
Периферическая автоном- Собаки (наркотизи- Паренте- Изменение ответа на известные вещества и
ная нервная система рованные) рально электрическую стимуляцию центральных отделов и периферических автономных нервов
Влияние на дыхание Собаки, морские Паренте- Влияние на частоту и амплитуду дыханий, то-
свинки рально нус бронхов
Диуретическая активность Собаки Внутрь, Натрийурез, калийурез, водный диурез, почеч-
паренте- ный кровоток, скоростьклубочковой фильт-
рально рации
Влияние на ЖКТ Крысы Внутрь Влияние на секреторную функцию и моторику ЖКТ
Уровни гормонов, холесте- Крысы, собаки Паренте- Концентрация в сыворотке
рина, сахара в крови рально, внутрь
Свертывание крови Кролики Внутрь Время свертывания, ретракция сгустка, протромбиновое время
Центральная нервная сис- Мыши, крысы Паренте- Седативный эффект, степень расслабления
тема рально. мускулатуры, локомоторной активности,
внутрь уровень стимуляции
щем необходимо изучать его влияние на животных с гипертензией. Должны быть получены данные о длительности действия и эффективности лекарства после перорального и парентерального введения. Если вещество оказалось фармакологически активным, оно должно быть далее оттестировано на возможность нежелательного действия на основные органы и системы, включая дыхательную, ЖКТ, эндокринную и ЦНС.
В результате этих исследований может возникнуть необходимость дальнейшей химической модификации молекул для достижения более желательных фармакокинетических или фармакодинамических свойств. Так, при пероральном введении лекарство может плохо всасываться или быстро метаболизироваться в печени, что потребует модификации для улучшения его биодоступности. Если лекарство будет приниматься в течение длительного времени, необходимо оценить развитие толерантности. Для лекарств, которые могут вызвать физическую зависимость, надо исследовать возможные последствия злоупотребления. Должны быть расшифрованы механизмы фармакологического действия нового препарата.
В конце концов (может потребоваться несколько модификаций оригинальной молекулы до синтеза перспективного вещества), получают лекарственную субстанцию для создания нового лекарства, т. е. основное соединение. Может быть представлена патентная заявка на новое эффективное соединение или на новое терапевтическое применение известного вещества.
Достижения молекулярной биологии и биотехнологии создали новые подходы к решению проблем синтеза и внедрения лекарств. Детальная информация о структуре рецепторов сделала возможным более рациональное “конструирование” препаратов. Лучшее понимание роли вторичных посредников привело к открытию их рецепторов как нового класса мишеней для воздействия лекарств. Возможность внедрения генов активных пептидов и белков в бактерии, дрожжи или культуру клеток млекопитающих позволяет синтезировать, изолировать и очищать большие количества нужных молекул. В настоящее время доступны для применения полученные по этой технологии человеческие инсулин, гормон роста, интерферон, вакцина против гепатита, тканевой активатор плазминогена, эритропоэтин, антигемофилический фактор и ростовые факторы костного мозга.
Доклиническая оценка безопасности и токсичности
Кандидаты в лекарства, которые прошли первоначальный скрининг и оценку фармакологического профиля, перед клиническими испытаниями должны быть детально исследованы для выявления потенциальных факторов риска. Доклиническая оценка токсичности лекарства зависит от области его предполагаемого использования и включает большинство тестов приведенных в табл. 5-3. Поскольку ни одно химическое соединение не может быть полностью “безопасным” (вещество при определенной дозировке токсично), необходимо оценить риск его применения с помощью специальных тестов.
В ходе доклинических токсикологических исследований должны быть получены следующие основные сведения: 1) острая токсичность — эффекты больших доз и определение порога летальных разовых доз; 2) субхроническая и хроническая токсичность — эффекты многократного (хронического) введения, особенно важные для лекарств, рекомендуемых для длительного применения; 3) влияние на репродуктивные функции, возможная эмбриотоксичность и тератогенность; 4) канцерогенность; 5) мутагенность. В дополнение к исследованиям, перечисленным в табл. 5-3, желательны некоторые количественные оценки. Это определение “неэффективной” (максимально переносимой) дозы — максимальной дозы лекарства, при которой не проявляется токсический эффект; минимальной летальной дозы — наименьшей дозы, которая может вызывать гибель животных и средней летальной дозы (LD50) — дозы, которая вызывает гибель примерно 50 % животных (глава 2). Ранее LD.Wрассчитывали очень точно и использовали для определения соотношения токсических и терапевтических доз соединений. Сейчас считают, что для этих целей высокая точность расчета не обязательна (Malmfors, 1983). Поэтому среднюю летальную дозу оценивают, используя минимум животных. При расчете начальной дозы для человека обычно исходят из того, что она должна составлять 1/100-1/10 от “неэффективной” дозы для животных.
Важно понимать, что возможности доклинических исследований ограничены.
Перичислим основные ограничения.
1) Оценка токсичности отнимает много времени и средств. В последнее десятилетие общую стой-
ТАБЛИЦА 5-3. Тесты оценки безопасности
Тест Подход Комментарий
Острая токсичность Однократная доза, вызывающая гибель при- мерно 50 % животных. Определяется максимально переносимая доза. Обычно используют 2 вида животных, 2 пути введения Сравнивается с терапевтической дозой
Подострая токсичность Три дозы, 2 вида животных. Может быть необходимо исследование продолжительностью около 6 месяцев до клинических испытаний. Чем дольше предполагаемый курс лечения, тем продолжительнее тест на подострую токсичность Биохимические, физиологические, патанатомические исследования; гематологические, гистологические, электронномикроскопические данные. Определение органа, подверженного токсическому действию
Хроническая токсич- От одного до 2 лет. Необходима, если лекарство ность будут использовать у людей п родолжительное время. Обычно исследование производится одновременно с клиническими испытаниями Цели исследования подострой и хронической токсичности — показать, какой орган чувствителен к токсическому действию лекарства. Выполняются те же тесты, что и при оценке подострой токсичности.
Влияние на репродук- Влияние на половое поведение животных, реп-тивные функции родуктивную способность, роды, потомство, частоту врожденных дефектов Оценка фертильности, тератогенного действия, перинатальных и постнатальных эффектов, лактации
Канцерогенный потен- 2 года, 2 вида животных. Требуется в том слу-циал чае, если лекарство будет использоваться в клинике продолжительное время Гематологические, гистологические, патоморфологические исследования
Мутагенный потенциал Влияние на генетическую стабильность бактерий (тест Эймса) или клеток млекопитающих в культуре; тест доминантной летальности у мышей Повышенный интерес к этой проблеме
Исследовательская ток- Определяет последовательность и механизмы сикология токсического действия. Разрабатывает новые методы оценки токсичности Может облегчить рациональное конструирование (направленный синтез) безопасных лекарств
мость доклинических фармакологических и токсикологических испытаний оценивают в среднем в 41 миллион долларов для одного вещества. Для сбора и анализа данных может потребоваться от двух до пяти лет.
2) В доклинических исследованиях используют большое количество животных. В настоящее время достигнут определенный прогресс в уменьшении их числа при той же надежности результатов (Maemfors, 1983; Zbinden, 1981). Все более широко
применяют клеточные и тканевые культуры in vitro, однако прогностическая ценность результатов этих экспериментов еще весьма ограничена. Попытки некоторых общественных групп прекратить все испытания на животных не обоснованны.
3) Экстраполяция экспериментальных данных по токсичности лекарственного средства на человека не вполне надежна (Jelovsek, 1989; Dixon, 1980). Для каждого конкретного вещества общие представления о всех видах токсичности, получен
ные на животных, являются очень важными для оценки прогнозирования побочных эффектов у человека. Однако не всю информацию можно получить на стадии доклинических испытаний.
4) В соответствии с законами статистики редкие побочные эффекты маловероятно определить раньше, чем на этапе клинических испытаний.
Клинические испытания
Только около 10 % соединений, дошедших до клинических испытаний, когда-либо попадут на фармацевтический рынок. Федеральный закон США требует, чтобы испытания новых лекарств на человеке производились в строгом соответствии с детальными руководствами. Однако научную обоснованность результатов нельзя гарантировать путем простого выполнения действующих законов и правил. Планирование и проведение качественных клинических испытаний требует усилий ученого-клинициста или клинического фармаколога, специалиста по статистике и подчас представителей других специальностей. Потребность в тщательном планировании и выполнении основана на трех особенностях терапевтического (как лекарственного, так и нелекарственного) действия на человека.
1) Изменения в ходе естественного течения большинства заболеваний. Многие заболевания имеют тенденцию к волнообразному течению, некоторые со временем самопроизвольно исчезают. Даже при злокачественных опухолях изредка могут наблюдаться спонтанные ремиссии. Правильная схема исследования должна учитывать такую естественную историю изучаемой болезни путем исследования большого количества испытуемых в течение достаточно продолжительного времени. Дополнительная защита от ошибок в оценке, связанных с флуктуацией тяжести проявления болезни, обеспечивается использованием перекрестной схемы испытаний, суть которой состоит в чередовании периодов введения испытуемым исследуемого вещества (опыт), плацебо (контроль) и эталонного препарата (позитивный контроль). Каждая подгруппа больных получает одну из возможных последовательностей лечения. Пример такой схемы приведен в табл. 5-4.
2) Наличие сопутствующих заболеваний и факторов риска. На результаты клинического исследования могут повлиять известные или неизвестные сопутствующие заболевания и факторы риска у ис-
ТАБЛИЦА 5-4. Типичная перекрестная схема клинических испытаний гипотетического нового аналгети-ка "Новент". Каждый период применения продолжается 7 дней с недельным перерывом между ними для выведения предыдущего лекарства
Лекарство, которое получает испытуемый
Группа больных Неделя 1 Неделя 3 Неделя 5
1 Аспирин Плацебо “Новент”
II Плацебо “Новент” Аспирин
III "Новент" Аспирин Плацебо
пытуемого (в том числе образ жизни). Например, некоторые заболевания изменяют фармакокинетику лекарств (главы 3 и 4). Если для оценки эффективности нового лекарства используют мониторинг его концентрации в крови, нужно помнить, что этот показатель может изменяться при наличии сопутствующих заболеваний или одновременном применении другого препарата. Чтобы избежать этой опасности, обычно пользуются перекрестной техникой (когда это выполнимо) и тщательно отбирают пациентов в соответствующие группы. Поэтому должны быть получены точные медицинский и лекарственный анамнезы (включая использование наркотических веществ) и применены статистически корректные методы рандомизации при отборе испытуемых в определенные группы.
3) Предвзятость испытуемого и наблюдателя. Большинство пациентов склонны позитивно реагировать на любое терапевтическое вмешательство, осуществляемое заинтересованным, заботливым персоналом, состоящим из энтузиастов. Проявление этого феномена у испытуемого является плацебо-реакцией и может сопровождаться объективными физиологическими и биохимическими изменениями, так же, как изменениями субъективных жалоб, связанных с болезнью. Плацебо-эффект обычно измеряют путем введения инертного вещества, которое имеет точно такой же внешний вид, запах, консистенцию и т. п., как и активная лекарственная форма. Величина реакции существенно колеблется от больного к больному. Однако частота плацебо-реакций довольно постоянна и наблюдается у 20-40 % больных почти во всех испытаниях. Может отмечаться и плацебо-“токсичность”, сопровождаемая обычно субъективными жалобами на боли в желудке, бессонницу, вялость и т. п.
Предвзятость со стороны больных можно исключить, используя слепой метод испытаний, когда пациенты не знают, какое именно лекарство они получают. При этом можно проводить как перекрестное, так и простое исследование (в последнем случае больные из одной группы в течение всего испытания принимают только один препарат: проверяемый, эталонный или “пустышку”-плацебо). При двойном слепом контроле исключается и предвзятость со стороны медицинского персонала, оценивающего эффективность лечения. В этом случае третий независимый участник контролирует коды, идентифицирующие каждое средство, которые не раскрываются до тех пор, пока не собраны клинические данные.
Комиссия по контролю за пищевыми продуктами и лекарствами
Комиссия по контролю за пищевыми продуктами и лекарствами (Food and Drug Administration — FDA) является административным органом, который наблюдает за процессом оценки лекарств и дает разрешение на их медицинское применение в США. Полномочия FDA регулировать рынок лекарств вытекают из ряда законодательных документов (табл. 5-5). Если путем адекватного тестирования не было доказано, что лекарство “эффективно и безопасно” при применении по определенным показаниям, оно не может быть допущено на рынок1. К несчастью, термин “безопасный” обозначает не одно и то же для больного, врача и для общества в целом. Уже отмечалось, что полное отсутствие риска невозможно (и, вероятно, никогда не будет достигнуто), но этот факт не находит понимания у рядового американца, который полагает, что любое лекарство, продаваемое с одобрения FDA, должно быть лишено серьезных побочных эффектов. Это противоречие является основной причиной судебных тяжб и неудовлетворенности медицинской помощью (Feinstein, 1988; Shulman, 1989).
История регулирования поступления лекарств на рынок отражает развитие медицины и здравоохранения, которое вызвало изменение отношения
' Хотя FDA прямо не контролирует коммерческую деятельность внутри штатов, связанную с лекарствами, ряд законов федерального уровня и уровня штатов контролирует производство и маркетинг лекарств.
общества к лекарственным препаратам (табл. 5-5). Акт о чистой пище и лекарствах (1906 г.) стал законом прежде всего в ответ на разоблачение антисанитарии в мясоперерабатывающей промышленности. Федеральный акт о пищевых продуктах, лекарствах и косметике от 1938 г. был реакцией на серию смертей, связанных с использованием сульфаниламида, который был допущен на рынок до того, как должным образом проверен на безвредность. Талидомид является еще одним примером лекарства, которое изменило методы проверки новых препаратов и стимулировало законодательство в области лекарственного обеспечения. Это вещество было внедрено в Европе в 1957-1958 гг. как “нетоксичный” гипнотик на основании результатов общепринятых тестов на животных. В 1961 г. были опубликованы первые сообщения о том, что прием талидомида беременными женщинами привел к существенному увеличению риска появления редкого врожденного дефекта у их детей — фокомелии, проявляющейся укорочением или полным отсутствием конечностей. Эпидемиологические исследования вскоре представили доказательства связи этого дефекта с использованием талидомида женщинами в первом триместре беременности, после чего лекарство было повсеместно запрещено. Ориентировочно родились 10 000 детей с врожденными дефектами из-за воздействия на мать только одного этого лекарства. Трагедия привела к требованию более серьезной проверки новых лекарств на тератогенность и способствовала утверждению поправки Кефаувера-Харриса в 1962 г„ хотя талидомид не был разрешен к применению в США. К несчастью, этот эпизод способствовал и распространению страха в отношении применения всех лекарств при беременности, вызвал много необоснованных судебных процессов и привел к устранению с рынка лекарств, которые, вероятно, не являлись тератогенными.
Как мы уже говорили, невозможно утверждать, что лекарство абсолютно безопасно, т. е. его применение лишено любого риска. Возможно, однако, прогнозировать большинство опасностей, связанных с применением нового лекарства, и установить некоторые статистические границы частоты их возникновения в популяции. Таким образом, говоря о безопасности лекарства, мы подразумеваем характер и частоту связанных с его приемом побочных эффектов в сравнении с опасностью отказа от лечения данного заболевания.
ТАБЛИЦА 5-5. Основные законодательные документы США, касающиеся лекарств
Законодательный акт Цель и эффект
Акт о чистых пищевых продуктах и лекарствах (1906 г.) Акт о запрете опия (1909 г.) Поправка (1912 г.) к Акту о чистых пищевых продуктах и лекарствах (1906 г.) Акт Харрисона о наркотиках (1914 г.) Запрет на отсутствие этикеток, запрет на фальсификацию лекарств Запрет импорта опия Запрет на ложную или мошенническую рекламу Устанавливает регулирование использования опия, опиатов и кокаина (в 1937 г. добавлена марихуана)
Акт о пищевых продуктах, лекарствах и косметических средствах (1938 г.) Требует, чтобы новые лекарства были как очищенными, так и безопасными (но не требует доказательств эффективности). Принят под давлением FDA
Акт Дурхама-Хамфри (1952 г.) Дает FDA право определять, какое лекарство может продаваться без рецепта
Поправка Кефаувера-Харриса к Акту о пищевых продуктах, лекарствах и косметических средствах (1962 г) Требует доказательств эффективности, равно как и безопасности новых лекарств и лекарств, производимых, начиная с 1938 г.; устанавливает правила предоставления информации о побочных действиях, клиническом тестировании и рекламе новых лекарств.
Акт по всестороннему предупреждению и контролю злоупотребления лекарствами (1970 г.) Поправка по лекарствам-“сиротам” (1983 г.) Устанавливает строгий контроль производства, распределения и прописывания лекарств, вызывающих привыкание; определяет программы предупреждения и лечения наркоманий Дополняет Акт о пищевых продуктах, лекарствах и косметических средствах, стимулирует создание лекарств, которые могут использоваться при лечении болезней, поражающих в США менее 200 000 больных
Акт о конкуренции цен и восстановлении патентов (1984 г.) Упрощает и сокращает процедуру представления заявки на лекарства-генерики. Требует данные по биоэквивалентности. Пролонгирует срок действия патента на время, требуемое для рассмотрения заявки в FDA. Это время не может превышать пяти лет или увеличивать срок на период более четырнадцати-лет после утверждения заявки на новое лекарство (NDA)
Ускорение утверждения лекарств (1992 г.) Допускает ускоренное утверждение FDA лекарств первостепенной важности для здравоохранения. Требует детального постмаркетингового надзора за больными
Фазы клинических испытаний нового лекарства лекарства, 2) информацию о производителе, 3) все данные изучения на животных, 4) клинические планы и разработки протоколов и 5) имена и мандаты
Как только лекарство считается готовым для врачей, которые будут проводить клинические ис-исследования на человеке, в FDA должно быть пытания.
представлено уведомление о праве исследования Подчас требуется от четыре до шести лет клинового лекарства (Notice of Claimed Investigational нических испытаний для накопления всех требуе-Exemption for a New Drug — IND), рис. 5-1. IND мых данных. Тестирование у человека начинают включает: 1) информацию о составе и источниках после завершения необходимых исследований ост-
рой и субхронической токсичности у животных. Хронические исследования безопасности у животных обычно выполняются параллельно с клиническими испытаниями. Клинические испытания проводят в четыре фазы: три до внедрения препарата в медицинскую практику, четвертая — после. Участники испытаний (добровольцы или больные) должны быть информированы об исследовательском статусе лекарства и о возможном риске. Они имеют право соглашаться или не соглашаться участвовать в испытаниях и принимать лекарство. Эти правила основаны на этических принципах, сформулированных в Хельсинкской Декларации (Editor’s Page, 1966). Для проведения испытаний необходимы не только разрешение FDA и спонсорская поддержка, но и одобрение междисциплинарной этической комиссии того учреждения, где они будут проходить.
В фазе 1 оценивают эффекты лекарства в зависимости от дозы на небольшом числе здоровых добровольцев. (Если ожидают, что лекарство может иметь выраженную токсичность, как это бывает в случае лечения рака или СПИДа, в фазе 1 участвуют добровольцы из числа больных, а не обычные волонтеры.) Задача этой фазы — определить различия в реакции на лекарство животных и человека и установить границы безопасного диапазона доз. Эти исследования являются открытыми, а не слепыми, т. е. и испытатель, и испытуемый знают, что за препарат используется в данном случае. На этой фазе испытаний выявляются многие предсказанные виды токсичности. Здесь же часто определяют фармакокинетические параметры всасывания, периода полувыведения и метаболизации. Эта работа выполняется обычно в исследовательских центрах специально обученными клиническими фармакологами.
В фазе 2 лекарство впервые изучают у больных с профильным заболеванием с целью определения его эффективности. Очень детально обследуется небольшое количество пациентов (10-200). Часто используют слепой метод, включая в испытания наряду с тестируемым препаратом плацебо или официальное лекарство (позитивный контроль). И эти испытания обычно проводят в специальных медицинских центрах (например, университетских госпиталях). На этой фазе может быть обнаружен более широкий спектр токсических эффектов.
В фазе 3 лекарство оценивают на значительно большей (иногда это тысячи человек) группе больных с целью дальнейшего определения его эффек
тивности и безопасности. Используя информацию, полученную на предыдущих этапах, испытания проводят таким образом, чтобы минимизировать ошибки, связанные с плацебо-эффектом, изменениями течения болезни и т. п. Поэтому часто применяют двойной слепой метод и перекрестную технику (подобно тому, как показано в табл. 5-4). Испытания фазы 3 обычно проводят в ситуации, приближенной к той, которая ожидается при реальном применении лекарства. Такие исследования довольно трудно спланировать и выполнить: они дороги из-за большого числа больных и массива данных, которые должны быть собраны и проанализированы. Исследователи часто являются специалистами по заболеванию, которое следует лечить с помощью испытуемого лекарства. Именно в фазе 3 могут впервые проявиться некоторые токсические эффекты, особенно связанные с повышенной чувствительностью больного к препарату.
Если результаты, полученные на фазе 3, положительны, следующий этап внедрения — получение разрешения для доступа нового лекарства на рынок. Процедура получения этого разрешения требует представления в FDA заявки на новое лекарственное средство (New Drug Application — ND А). Заявка подчас состоит из сотен томов, полного отчета о всех доклинических и клинических данных, имеющих отношение к рассматриваемому лекарственному средству. Для принятия решения FDA может потребоваться три и более года. В случаях срочной необходимости (например, химиотерапия рака) доклинические и клинические исследования, а также рассмотрение заявки в FDA могут быть ускорены. Если препарат предназначен для лечения серьезных заболеваний, FD А может разрешить обширный, но контролируемый маркетинг до завершения фазы 3; когда речь идет о заболеваниях, представляющих угрозу для жизни, контролируемый маркетинг может быть разрешен даже перед завершением фазы 2 (Young, 1988).
Как только получено разрешение на продажу лекарства, приступают к фазе 4. Она заключается в мониторинге безопасности лекарства в реальных условиях применения у большого числа больных. Окончательное внедрение лекарства в общую практику должно сопровождаться программой неусыпного постмаркетингового надзора. Важность тщательного и полного информирования о токсичности препарата после разрешения FDA на маркетинг понятна, если вспомнить, что многие вызванные
ТАБЛИЦА 5-6. Объем исследования как функция частоты эффекта'
Количество лиц, которые должны получать лекарство, чтобы определить двукратное увеличение возникновения редкого эффекта с вероятностью не обнаружить его в 20 % и сделать ложное заключение о его наличии в 5 % случаев. Необходим один испытуемый, не получающий воздействия, на каждого испытуемого, получающего лекарство. Если прием лекарства увеличивает риск более чем в 2 раза, необходимое количество испытуемых уменьшается.
Частота эффекта в контрольной группе (не получающей лекарство) Пример Необходимое количество испытуемых, получающих лекарство
1/100 Любое врожденное поражение сердца2 1800
1/1000 Незаращения тканей лица 3 18 000
1/10 000 Атрезия трехстворчатого клапана 4 180 000
1/100 000 Инфаркт миокарда5 1 800 000
1 Изменено и воспроизведено с разрешения Finkle W. Report of Joint Commission on Presciprion Drug Use, Appendix V, 1980.
2 Частота всех форм врожденных аномалий развития сердца около 1 на 111 новорожденных.
3 Частота нарушений развития тканей лица около 1 на 700 новорожденных.
4 Частота атрезии трехстворчатого клапана около 1 на 8500 новорожденных.
s Частота инфарктов миокарда у некурящих женщин 30-39 лет около 4 на 100 000.
лекарствами нежелательные эффекты наблюдаются с частотой 1: 10 000 и менее. В табл. 5-6 представлено количество наблюдений, необходимое для выявления связанных с приемом лекарства событий, в зависимости от их частоты в популяции, не получавшей данный препарат (и некоторые примеры таких событий). Вследствие сравнительно небольшого числа испытуемых на первых трех этапах такие маловероятные эффекты лекарств обычно не выявляются до фазы 4 вне зависимости от тщательности проведения испытаний. Фаза 4 не имеет фиксированной продолжительности.
Время, прошедшее от представления патентной заявки до разрешения маркетинга нового лекарства, может составить более 5 лет. Поскольку продолжительность действия патента в США 17 лет, его обладатель (обычно фармацевтическая компания) имеет эксклюзивные права на маркетинг препарата только ограниченное время после утверждения NDA. Из-за длительного рассмотрения документов в FDA время, затраченное на этот этап, иногда добавляют к сроку действия патента. Однако после продления (до 5 лет) патент не может действовать более 14 лет с момента одобрения NDA. После прекращения срока действия патента любая компания может производить и продавать лекарство как генерик без уплаты лицензии обладателю патента. Однако независимо от патентования по закону может быть защищена торговая марка (торговое название лекарства). Поэтому фарма
цевтические компании стремятся дать лекарствам собственные легко запоминающиеся торговые названия. Например, “Либриум” — торговое название противотревожного лекарства “хлордиазепоксид”. По этой же причине рекламные материалы компании акцентируют внимание на торговом названии (глава 66, раздел, рассматривающий прописывание генериков).
“Лекарства-сироты”
Лекарства, предназначенные для терапии редких болезней, так называемые “лекарства-сироты”, могут быть очень сложны для исследования, внедрения и маркетинга. Доказательства их безопасности и эффективности на малых популяциях получить очень сложно. В частности, клиническая проверка лекарств у детей весьма ограничена, даже если речь идет о весьма распространенных болезнях, а ряд редких заболеваний поражает именно детей. Более того, поскольку базисные патофизиологические исследования и изучение механизма развития редких заболеваний обычно не привлекают широкого внимания и финансовой поддержки как в академических учреждениях, так и в промышленности, не всегда известны истинные мишени действия таких лекарств. Кроме того, если пораженная популяция сравнительно невелика на принятие решения о внедрении может повлиять высокая стоимость создания нового лекарства.
Поправка о “лекарствах-сиротах” от 1983 г., которая дополнила Федеральный акт о пищевых продуктах, лекарствах и косметике, дала толчок поиску лекарств для терапии заболеваний, поражающих в США менее чем 200 000 человек. FDA организовала специальный офис по развитию лекарств этой группы, чтобы оказать поддержку ученым, интересующимся данной проблемой. Информация о “лекарствах-сиротах” доступна в ряде агентств, включая Национальную организацию редких заболеваний и Комиссию по лекарствам для редких заболеваний при Ассоциации производителей фармацевтической продукции.
С1993 г. FD А было зарегистрировано описание более 500 биологических или синтетических пре-паратов-“сирот”. FDA одобрила заявки на маркетинг 87 “лекарств-сирот” для лечения более 74 редких заболеваний.
Нежелательные реакции при действии лекарств
Серьезные побочные реакции на лекарства, разрешенные к применению, редки. Однако менее опасные токсические эффекты при использовании некоторых групп лекарств встречаются довольно часто. Угрожающие жизни реакции случаются менее чем у 2 % госпитализированных больных (Adverse drug reactions, 1981). Существуют два основных механизма этих реакций. Первый является следствием развития известного фармакологического эффекта и, следовательно, предсказуем. Токсические реакции такого рода обычно обнаруживаются фармакологами, токсикологами и клиницистами, участвующими в первых трех фазах клинических испытаний лекарства. Реакции второй группы, которые могут иметь иммунологический или другой неизвестный механизм (Rawlins, 1981), обычно неожиданны и могут оставаться нераспознанными до тех пор, пока лекарство не проживет на рынке несколько лет. Эти эффекты обычно выявляются клиницистами. Поэтому важно, чтобы врачи-практики были настороже и следили за различными типами аллергических реакций на лекарства Они включают реакции, опосредованные IgE (анафилаксия, крапивница, сосудистый отек), IgG или IgM (волчаночноподобные реакции), IgG (типа сывороточной болезни с васкулитом) и аллергии клеточного типа, связанные с контактным дерматитом (глава 57).
Оценка изучения лекарства в клинике
Периодическая литература является основным источником клинической информации о новых лекарствах, особенно таких, которые недавно вошли в употребление. Эта информация может касаться новых показаний или появления новых нежелательных эффектов и противопоказаний. Следовательно, врачи-практики должны быть знакомы с источниками такой информации (глава 1) и готовы ее использовать. Некоторые общие принципы оценки клинической информации детально обсуждаются в работе Ригельмана. Они удобно сформулированы в форме вопросов, которые читатель может задать в процессе ознакомления со статьей.
А. Вопросы этики. Были ли предоставлены соответствующие этические и процедурные гарантии пациенту? Было ли получено от него информированное согласие?
Б. Формулировка целей. Каковы цели изучения? Четко ли они определены и сформулированы? Плохо сформулированные цели типа “изучить эффект миноксидила” (антигипертензивного лекарства) со значительно меньшей вероятностью приведут к полезному результату по сравнению с ясно определенными целями, такими как “измерить влияние миноксидила на почечную функцию у мужчин с тяжелой артериальной гипертензией”.
В. Методы исследования. Были ли методы оценки адекватны целям исследования? Указывает ли автор на точность и надежность (воспроизводимость) метода? Достаточно ли чувствителен метод, чтобы выявить незначительные, но биологически важные изменения?
Г. Статистические методы. Как осуществлялся выбор больных? Достаточно ли количество наблюдений? Представляют ли испытуемые популяцию, которая с наибольшей вероятностью будет применять лекарство, или же популяцию, в которой хотелось бы использовать лекарство? Является ли проект продолжительным или амбулаторным исследованием? Были ли больные, которых не включили в группу для оценки результатов лечения? Как их учитывали? Был ли использован плацебо-конт-роль или позитивный контроль? Как пациенты распределялись в различные группы при проведении испытаний? Была ли применима и использована перекрестная схема исследования? Получали ли больные какую-либо другую терапию во время исследования? Как это контролировалось и учитыва
лось? Применялись ли соответствующие статистические тесты?
Д. Заключения. Основаны ли заключения на реальных данных? Имеет ли предлагаемое лекарство существенные преимущества по стоимости, эффективности или безопасности по сравнению с существующими, или оно просто новое?
Экстраполяция результатов проведенных испытаний на другие группы больных должна проводиться с крайней осторожностью.
Хорошо составленный отчет в рецензируемом журнале обычно содержит ясные ответы на все поставленные выше вопросы. Если этого нет, то вполне оправдан скептицизм по отношению к исследованию и заключениям автора.
Избранная литература
Beyer К. Н. Discovery, Development, and Delivery of New Drugs, SP Medical & Scientific Books, 1978.
Black J. Drugs from emasculated hormones: The principle of syntopic antagonism. Science, 1989; 245:486.
Brimblecombe R. W., et al. Charactirezation and development of cimetidine as histamine H2-recep-tor antagonist. Gastroenterology, 1978; 74:339.
Fredd S. The FDA and the physician. Am. J. Gastroenterol. 1988; 83:1088.
Kessler D. A. The regulation of investigation drugs. N. Engl. J. Med. 1989; 320:281.
Riegelman R. K. Studing a Study and Testing a Test: How to Read the Medical Literature. Little, Brown, 1981.
Spilker B. Guide to Clinical Interpretation of Data. Raven Press, 1986.
Zbinden G., Flury-Reversy M. Significance of LD5()-test for the toxicological evaluation of chemical substances. Arch. Toxicol. 1981; 47:77.
Раздел II
Средства, влияющие на вегетативную нервную систему____________________
Введение в фармакологию вегетативной нервной системы
Бертрам Г. Катцунг
Моторная (эфферентная) часть нервной системы может быть разделена на два больших отдела: вегетативный и соматический. Активность автономной (вегетативной) нервной системы (АНС) не находится под прямым контролем сознания. Она охватывает в основном регуляцию висцеральных функций — сердечного выброса, кровотока в разных органах, пищеварения и прочее, т. е. того, что необходимо для жизнедеятельности. Соматический отдел в большей степени неавтономен и касается функций, контролируемых сознанием, таких как движение, дыхание и поддержание позы. Обе системы имеют важные афферентные (чувствительные) входы, которые обеспечивают ощущения и модифицируют моторный выход по рефлекторным дугам различной протяженности и сложности. Нервная система имеет некоторые общие свойства с эндокринной системой организма — другой регуляторной системой, контролирующей функции организма. Это высокий уровень интеграции в мозгу, способность воздействовать на процессы в отдаленных частях тела и широкое использование отрицательной обратной связи. В обеих системах передача информации осуществляется химическими веществами. В нервной системе химическая передача происходит между нервными клетками и между нервными клет
ками и эффекторами. Химическая передача осуществляется посредством выделения малых количеств трансмиттерных субстанций из нервных окончаний в синаптическую щель. Трансмиттер пересекает щель путем диффузии и активирует или ингибирует постсинаптическую клетку, связываясь со специализированной рецепторной молекулой.
Используя препараты, которые имитируют или блокируют действие химических трансмиттеров, мы можем избирательно модифицировать многие вегетативные функции, регулируя деятельность сердечной мышцы, гладких мышц, эндотелия сосудов, желез и пресинаптических нервных окончаний. Препараты, влияющие на вегетативную нервную систему, применяют при многих заболеваниях.
Анатомия автономной (вегетативной) нервной системы
АНС по анатомическим особенностям может быть разделена на две большие части: симпатическую (тораколюмбальную) и парасимпатическую (краниосакральную) нервные системы (рис. 6-1). Оба отдела происходят из ядер центральной нерв-
Парасимпатическая АНС Нервные окончания гладких мышц, сердца, желез
Симпатическая АНС Потовые железы
Симпатическая АНС Нервные окончания гладких мышц, сердца, желез
Симпатическая АНС
Гладкая мускулатура сосудов почек
Соматическая НС
Скелетная мускулатура
Рис. 6-1. Схема анатомических и нейромедиаторных признаков вегетативных и соматических моторных нервов. Показаны только основные трансмиттерные субстанции. Парасимпатические ганглии не показаны, так как они главным образом расположены или около, или в стенках иннервируемых органов. Обратите внимание, что некоторые симпатические постганглионарные волокна выделяют ацетилхолин или дофамин, а не норадреналин. Мозговое вещество надпочечников (модифицированный симпатический ганглий) получает симпатические преганглионарные волокна и выделяет адреналин и норадреналин в кровь. АХ — ацетилхолин; D — дофамин; Ад — адреналин; НА — норадреналин; Н — никотиновые рецепторы; М — мускариновые рецепторы
ной системы и берут начало в преганглионарных нервных волокнах, которые выходят из мозгового ствола или спинного мозга и заканчиваются в моторных ганглиях. Симпатические преганглионарные волокна выходят из центральной нервной системы с грудными и поясничными спинномозговыми нервами, что объясняет альтернативное название этой системы — “тораколюмбальная”. Парасимпатические преганглионарные волокна выходят из центральной нервной системы с черепномозговыми нервами (прежде всего с третьим, седьмым, девятым и десятым) и с третьим и четвертым сакральными спинальными корешками.
Большинство симпатических преганглионарных волокон заканчивается в ганглиях, расположенных
в паравертебральных цепочках, лежащих по обеим сторонам позвоночного столба. Остальные симпатические ганглии являются превертебральными и находятся спереди от позвонков. Из этих ганглиев постганглионарные симпатические волокна направляются к иннервируемым тканям. Большинство парасимпатических преганглионарных нервных волокон заканчивается в клетках ганглиев, распределенных сетью или диффузно в стенках иннервируемых органов. Некоторые же заканчиваются в парасимпатических ганглиях, расположенных снаружи иннервируемых органов; в цилиарном, крылонебном, субмандибулярном, ушном и некоторых брюшных ганглиях. Важно запомнить, что термины “симпатический” и “парасимпатический” являются ана-
Сенсорные волокна
Симпатические постганглионарные
Парасимпатические пре ганглионарные
Слизистая оболочка
Рис. 6-2. Упрощенная диаграмма некоторых цепочек нервной системы кишечника (НСК). НСК получает ветви и из симпатической, и из парасимпатической систем и посылает афферентные импульсы к симпатическим ганглиям и в центральную нервную систему. В НСК идентифицированы многие трансмиттерные или нейромодуляторные субстанции (табл. 6-1). ПМ — слой продольных мышц; МС — мышечное сплетение; КМ — слой круговой мышцы; СМС — субмукозное сплетение; АХ — ацетилхолин; НА — норадреналин; НП — нейропептиды; SP — субстанция Р; 5-НТ — серотонин
томическими и не зависят ни от химического типа медиатора, выделяемого из нервных окончаний, ни от эффекта - возбуждающего или угнетающего.
В дополнение к этим четко определяемым моторным частям вегетативной нервной системы существует большое число афферентных волокон, которые идут с периферии к интегрирующим центрам, включающим нервные сплетения в кишечнике, вегетативные ганглии и центральную нервную систему. Многие чувствительные нейроны, закан-чивающиеся в центральной нервной системе, имеют окончания в интегративных центрах гипоталамуса и продолговатого мозга и вызывают рефлекторную моторную активность, которая поступает
к эффекторным клеткам по эфферентным волокнам, описанным выше. Появляется все больше свидетельств того, что некоторые из этих чувствительных волокон имеют также важную периферическую моторную функцию (неадренергическая, нехолинергическая системы).
Нервная система кишечника (НСК) — это скопление нейронов, расположенных в стенках органов желудочно-кишечного тракта (рис. 6-2). Она иногда рассматривается как третье подразделение АНС. Нервная система кишечника включает мышечное сплетение (сплетение Ауэрбаха) и субмукозное сплетение (сплетение Мейснера) (Furness, 1987). Эти нейрональные сети получают преганглионар-
ные волокна из парасимпатической системы и постганглионарные симпатические аксоны, а также чувствительную импульсацию из стенки кишечника. Волокна из клеток этих сплетений идут к гладким мышцам кишечника и контролируют его моторику. Другие моторные волокна направляются к секреторным клеткам. Чувствительные волокна переносят информацию от слизистой оболочки и рецепторов растяжения к моторным нейронам в сплетениях и к постганглионарным нейронам в симпатических ганглиях. Парасимпатические и симпатические волокна, образующие синапсы с нейронами кишечного сплетения, играют, видимо, модулирующую роль, на что указывает наблюдение, что потеря входа импульсации от обоих отделов АНС не уменьшает активности в сплетениях, а также в гладких мышцах и железах, иннервируемых ими.
Химия нейротрансмиттеров вегетативной нервной системы
Традиционная классификация автономных нервов базируется на природе основного трансмиттера (ацетилхолина или норадреналина), выделяемого из их окончаний и варикозных расширений. Большое число волокон периферической автономной нервной системы синтезируют и выделяют ацетилхолин. Это холинергические волокна. Как видно из рис. 6-1, к ним относятся все преганглионар-ные эфферентные вегетативные волокна и соматические (неавтономные) моторные волокна, идущие к скелетным мышцам. Таким образом, почти все эфферентные волокна, выходящие из центральной нервной системы, холинергические. Кроме того, все парасимпатические постганглионарные и немногие симпатические постганглионарные волокна тоже являются холинергическими. Большинство симпатических постганглионарных волокон выделяют норадреналин. Это норадренергические волокна (часто их называют просто адренергические), т. е. они действуют, выделяя норадреналин. Эти транс-миттерные характеристики представлены схематически на рис. 6-1. Как уже отмечалось, некоторые симпатические волокна выделяют ацетилхолин. Существуют доказательства выделения дофамина некоторыми периферическими симпатическими волокнами. Клетки мозгового вещества надпочечников, которые эмбриологически аналогичны постганглионарным симпатическим нейронам, выделя
ют смесь адреналина и норадреналина. Несколькс лет назад было установлено, что большинство автономных нервов наряду с главными трансмиттерами выделяют несколько дополнительных медиаторных субстанций, или котрансмиттеров. Четыре основных этапа нейротрансмиттерной функции — синтез, хранение, выделение и прекращение действия — представляют потенциальные мишенг для фармакологической терапии. Эти процессы де тально обсуждаются далее.
Холинергическая передача
Окончания холинергических нейронов содержа! большое количество маленьких пузырьков (везикул), окруженных мембранами и сконцентрированных около синаптической части клеточной мембраны (рис. 6-3). Меньшее число везикул болыпегс размера расположено дальше от синаптической мембраны. Везикулы изначально синтезируются в теле нейрона и транспортируются в окончания Там они могут повторно совершать цикл развития несколько раз. Везикулы содержат ацетилхолин в высокой концентрации и другие молекулы (например, пептиды), которые могут действовать как котрансмиттеры. Ацетилхолин синтезируется в цитоплазме из ацетил-КоА и холина при каталитическом действии фермента холинацетилтрансферазы (ХАТ). Ацетил-КоА синтезируется в митохондриях, которых много в нервных окончаниях. Холин транспортируется из внеклеточной жидкости к нервные окончания На+-зависимым мембранным переносчиком (рис. 6-3, переносчик А). Этот переносчик может быть заблокирован группой препаратов, называемых гемихолиниумы. Синтезированный ацетилхолин (АХ) транспортируется из цитоплазмы в везикулы антипортером, перемещающим протоны (рис. 6-3, переносчик Б). Этот переносчик может быть блокирован везамиколом (Anderson, 1983). Синтез ацетилхолина — это очень быстрый процесс, способный поддерживать очень высокую скорость выделения медиатора. Запас ацетилхолина пополняется упаковкой “квантов” молекул ацетилхолина (обычно 1000-50 000 молекул в каждой везикуле).
Выделение трансмиттера зависит от концентрации внеклеточного кальция и происходит, когда потенциал действия достигает окончания и запускав! достаточный приток ионов кальция внутрь клетки. Недавно было продемонстрировано, что повышение
Рис. 6-3. Схема холинергического соединения (без соблюдения масштаба). Холин транспортируется в пресинапти-ческое нервное окончание натрийзависимым мембранным переносчиком (А). Этот транспорт может быть заблокирован препаратами группы гемихолиниума. АХ транспортируется в везикулу на хранение вторым переносчиком (Б), который может быть ингибирован везамиколом. Пептиды, АТФ и протеогликан также Могут запасаться в везикуле. Выделение трансмиттера происходит, когда открываются вольтажчувствительные кальциевые каналы в мембране окончания, обеспечивая приток ионов кальция. Возникающее увеличение внутриклеточной концентрации кальция вызывает слияние везикул с поверхностью мембраны и выброс из клетки ацетилхолина и котрансмиттеров в щель соединения. Этот этап блокируется ботулотоксином. Действие АХ прекращает фермент ацетилхолинэстераза. Рецепторы на пресинаптических нервных окончаниях регулируют выделение трансмиттера
концентрации Са2+ “дестабилизирует” везикулярный запас АХ посредством взаимодействия со специальным белком синаптотагмином, ассоциированным с везикулярной мембраной (Brose et al., 1992). Происходит слияние везикулярной мембраны с мембраной нервного окончания и выброс из клетки (в случае соматических моторных нервов) нескольких сотен квантов ацетилхолина в синаптическую щель. Количество трансмиттера, выделяемого при однократной деполяризации из постганглионарного автономного нервного окончания, вероятно, меньше. В дополнение к ацетилхолину одновременно могут выделяться один или более ко-трансмиттеров (табл. 6-1). Процесс выделения АХ из везикул может быть блокирован ботулиническим токсином, взаимодействующим с другим ассоциированным с везикулами белком, синаптобревином (Schiavo et al., 1992).
После выделения из пресинаптических окончаний молекулы АХ могут связываться с ацетилхолиновым рецептором (холиноцептором) и активировать его. Наконец (и обычно достаточно быстро), весь выделенный АХ распространяется в зоне нахождения молекулы ацетилхолинэстеразы (АХЭ). АХЭ эффективно расщепляет АХ на холин и ацетат, ни один из которых не обладает значительным трансмиттерным эффектом, и тем самым прекращает действие трансмиттера (рис. 6-3). Большинство холинергических синапсов богато АХЭ, поэтому период полувыведения АХ в синапсе очень короткий. АХЭ находится и в других тканях, например в эритроцитах. В плазме крови, печени, глие и многих других тканях найдена другая холинэстераза с меньшей специфичностью для ацетилхолина — бу-тирилхолинэстераза (псевдохолинэстераза).
Адренергическая передача
Адренергические нейроны (рис, 6-4) транспортируют молекулы предшественника в нервные окончания, синтезируют трансмиттер и хранят его в окруженных мембраной везикулах. Однако синтез адренергических трансмиттеров более сложен, чем ацетилхолина (рис. 6-5). В мозговом веществе надпочечников и определенных областях мозга норадреналин превращается в адреналин. Некоторые важные процессы в норадренергических нервных окончаниях являются потенциальными мишенями для действия лекарственных препаратов. Так, превращение тирозина в ДОФА является шагом, ли
митирующим скорость синтеза норадреналина. Этот процесс ингибируется аналогом тирозина — метирозином (рис. 6-4). Высоко аффинный переносчик катехоламинов, расположенный в стенке депонирующих везикул, угнетают резерпиновые алкалоиды (рис. 6-4, переносчик Б). Результатом является снижение запасов медиатора. Другой переносчик транспортирует норадреналин и сходные молекулы в цитоплазму клетки (рис. 6-4, переносчик 1, обычно обозначаемый как захват-1). Он инактивируется кокаином и трициклическими антидепрессантами, в результате чего повышается активность медиатора в синаптической щели.
Выделение везикулярного запаса трансмиттера из норадренергических нервных окончаний сходно с кальцийзависимым процессом, уже описанным для холинергических окончаний. В дополнение к основному трансмиттеру (норадреналин) в синаптическую щель также выделяются АТФ, дофамин-Р-гидроксилаза и некоторые пептидные котранс-миттеры. Симпатомиметики непрямого действия, например тирамин и амфетамин, способны выделять депонированный трансмиттер из норадренергических нервных окончаний. Эти препараты слабо действуют на адренорецепторы (некоторые не действуют вообще), однако влияют на норадренергические нервные окончания, ингибируя обратный захват нейротрансмиттера. Кроме того, в нервных окончаниях они могут вытеснять норадреналин из депонирующих везикул, угнетать моноаминоксида-зу и оказывать другие эффекты, повышающие активность норадреналина в синапсе. Их действие не требует экзоцитоза везикул и не зависит от ионов кальция.
Норадреналин и адреналин могут быть метаболизированы несколькими ферментами (рис. 6-6). Из-за высокой активности моноаминоксидазы митохондрий в нервных окончаниях имеется значительный оборот норадреналина даже в терминалях, находящихся в состоянии покоя. Так как продукты метаболизма экскретируются с мочой, оборот катехоламинов может быть определен при лабораторном анализе всех метаболитов в моче за 24 часа. Однако метаболизм норадреналина, выделяемого норадренергическими нервами, не является главным механизмом его инактивации. Окончание норадренергической передачи является результатом нескольких процессов, в том числе простой диффузии из области рецептора (с окончательным метаболизмом в плазме или печени) и обратным захватом нервным
ТАБЛИЦА 6-1. Некоторые трансмиттерные субстанции, найденные в автономной нервной системе, нервной системе кишечника и в неадренергических, нехолинергических нейронах'
Субстанция Возможная роль
Ацетилхолин (AX) Основной трансмиттер в ганглиях АНС, в соматических нервно-мышечных соединениях и в парасимпатических постганглионарных нервных окончаниях. Главный возбуждающий трансмиттер в гладких мышцах и секреторных клетках в НСК. Возможно, основной межнейронный (“ганглионарный") трансмиттер в НСК
Аденозинтрифосфат (АТФ) Может действовать как котрансмиттер в угнетающих нервно-мышечных соединениях НСК. Угнетает выделение АХ и норадреналина из нервных окончаний АНС
Пептид, связанный с кальцитониновым геном (кокал ьцегенин) Холецистокинин (ХЦК) Обнаружен наряду с субстанцией Р в сердечно-сосудистых чувствительных нервных окончаниях. Присутствует в некоторых секретомоторных нейронах НСК и интернейронах. Стимулятор сердечной деятельности Может действовать как котрансмиттер в некоторых возбуждающих нервно-мышечных нейронах НСК
Дофамин Важный постганглионарный симпатический трансмиттер в кровеносных сосудах почек. Возможно, модуляторный трансмиттер в некоторых ганглиях и НСК
Энкефалин • и родственные опиоидные пептиды Галанин Присутствует в некоторых секретомоторных и промежуточных нейронах в НСК Возможно, ингибирует выделение ацетилхолина и тем самым подавляет перистальтику. Может стимулировать секрецию Присутствует в секретомоторных нейронах, может играть роль в механизмах формирования чувства аппетита-насыщения
у-аминомасляная кислота (ГАМК) Может вызывать пресинаптические эффекты в возбуждающих нервных окончаниях НСК. Оказывает расслабляющее действие на кишечник. Вероятно, не основной трансмиттер в НСК.
Гастринрилизинг-пептид (GRP) НейропептидУ (NPY) Чрезвычайно сильный возбуждающий трансмиттер для гастриновых клеток. Известен как бомбезин у млекопитающих Присутствует в некоторых секретомоторных нейронах в НСК и может угнетать секрецию воды и электролитов в кишечнике. Вызывает долговременную вазоконстрикцию. Является котрансмиттером во многих парасимпатических постганглионарных нейронах и симпатических постганглионарных норадренергических сосудистых нейронах
Оксид азота (NO) Норадреналин (НА) Котрансмиттер ингибиторных нервно-мышечных соединений НСК Основной трансмиттер большинства симпатических постганглионарных нервных окончаний
Серотонин(5-НТ) Субстанция Р и родственные "тахикинины" Котрансмиттер в возбуждающих межнейронных соединениях в НСК Субстанция Р — важный трансмиттер чувствительных нейронов в НСК и в других местах. Тахикинины, вероятно, являются возбуждающими котрансмиттерами АХ в нервно-мышечных соединениях НСК. Обнаружены с ПСКГ в сердечно-сосудистых чувствительных нейронах. Субстанция Р — вазодилататор (вероятно, посредством выделения окиси азота)
Вазоактивный кишечный пептид (VIP) Возбуждающий секретомоторный трансмиттер в НСК, может быть ингибирующим нервно-мышечным котрансмиттером НСК. Вероятный котрансмиттер во многих холинергических нейронах. Вазодилататор (найден во многих периваскулярных нейронах) и сердечный стимулятор
' Из: Furness et. al., 1992; Llewellyn-Smith, 1989; Lundgren, Svanik and Jivegard, 1989; Kromer, 1990; Wharton and Gulbenkian, 1989.
Тирозин
Нервное окончание
Мети розин
Резерпин
Са2+
Бретилиум, гуанетидин
Кальциевый канал
Ауторецептор
Другие рецепторы
Постсинаптическая клетка
Адренорецепторы
Диффузия, метаболизм
НА АТФ, Пептиды
Кокаин, трициклические антидепрессанты
_ Пресинаптические рецепторы
Рис. 6-4. Схема норадренергического соединения (без соблюдения масштаба). Тирозин транспортируется в норадренергическое окончание или варикозное расширение натрийзависимым мембранным переносчиком (А). Тирозин превращается в дофамин (подробности на рис. 6-5), транспортирующийся в везикулу переносчиком (Б), который может быть ингибирован резерпином. Тот же самый переносчик транспортирует норадреналин (НА) и некоторые другие амины в эти гранулы. Дофамин превращается в НА в везикулах с помощью дофамин-Р-гидроксилазы. Выделение трансмиттера происходит, когда потенциал действия открывает вольтажчувствительные кальциевые каналы и увеличивает концентрацию внутриклеточного кальция. Результатом слияния везикул с поверхностью мембраны является выброс норадреналина, котрансмиттеров и дофамин-Р-гидроксилазы. Выделение может быть блокировано гуа-нетидином и бретилиумом. После выделения норадреналин диффундирует из щели или транспортируется в варикозное расширение цитоплазмы (захват 1, блокируется кокаином, трициклическими антидепрессантами) или в постсинаптическую клетку (захват 2). Регуляторные рецепторы находятся на пресинаптическом окончании
НА, АТФ, Пептиды
Гетерорецептор
N-Метилтранс-фераза
Н СН3
С —NH2 I
н
Декарбоксилаза L-аминокислот
Н Н I I
С—-С —1ЧН2
Н Н
Дофамин-р-гидроксилаза
Гидроксилаза (из печени)
Фенилаланин-р-гидроксилаза
Рис. 6-5. Биосинтез катехоламинов. Этап, лимитирующий скорость синтеза (превращение тирозина в ДОФА), может быть ингибирован метирозином (а-метилтирозином). Альтернативные пути, показанные пунктирными стрелками, не имеют физиологического значения у человека. Однако тирамин и октонамин могут накапливаться у пациентов, получающих ингибиторы моноаминоксидазы. (Из: Greenspan F. S., Baxter}. D. (eds): Basic & Clinical Endocrinology, 4th ed. Appleton & Lange, 1994.)
окончанием (захват 1) или перисинаптической глией, или клетками гладких мышц (захват 2) (рис. 6-4).
Котрансмиттеры в холинергических и адренергических нервах
Как уже отмечалось, везикулы холинергических и адренергических нервов, кроме основных трансмиттеров, содержат и другие субстанции. Некото
рые из этих субстанций, идентифицированные к настоящему времени, перечислены в табл. 6-1. Многие из них являются к тому же основными трансмиттерами в неадренергических, нехолинергических нервах, описанных в этой же главе. Роль этих субстанций в функционировании нервов, выделяющих ацетилхолин или норадреналин, не совсем понятна. Они могут обеспечивать медленное, долговременное действие, чтобы дополнять или мо-
АДРЕНАЛИН
НОРАДРЕНАЛИН
ДОФАМИН
З-МЕТОКСИ-4-ГИДРОКСИМИНДАЛЬНАЯ КИСЛОТА (VMA)
Рис. 6-6. Метаболизм катехоламинов катехол-орто-метилтрансферазой (КОМТ) и моноаминоксидазой (МАО). (Из: Greenspan F. S., Baxter J. D. (eds): Basic & Clinical Endocrinology, 4th ed. Appleton & Lange, 1994.)
СН2
С= )
КИСЛОТА
ОН
ГОМОВАНИЛИНОВАЯ КИСЛОТА
СН3О
НО
СН2
СН2
3-МЕТОКСИ-ТИРАМИН
ЫН2
Аудировать более скоротечные эффекты основных трансмиттеров, а также принимать участие в угнетении по механизму обратной связи того же самого или рядом расположенного нервного окончания.
Автономные рецепторы
Очень многое о химической природе автономных рецепторов было выяснено в последнее десятилетие, это направление является одной из наиболее активно развивающихся областей в фармакологии. Сравнительный анализ зависимости актив
ности от структуры серии автономных химических соединений-аналогов привел к обнаружению различных подтипов вегетативных рецепторов, в том числе мускариновых и никотиновых холинорецеп-торов, а-, 0- и дофаминовых адренорецепторов (табл. 6-2). Прогресс молекулярной биологии обеспечил новый подход, сделав возможным идентификацию и экспрессию генов, которые кодируют родственные рецепторы внутри этих групп (глава 2, дополнение “Как открывают рецепторы?”).
Основные подтипы ацетилхолиновых рецепторов были названы по наименованиям естественных
ТАБЛИЦА 6-2. Типы автономных рецепторов и их документированные или вероятные эффекты на периферические автономные эффекторные ткани
Название рецептора Типичная локализация Результат лигандного связывания
Холинорецепторы Нейроны ЦНС, симпатические постганглио- Образование ИТФ и ДАГ, повышение
Мускариновый Mt нарные нейроны, некоторые пресинапти-ческие зоны содержания внутриклеточного кальция
Мускариновый М2 Миокард, гладкие мышцы, некоторые пресинаптические зоны Открытие калиевых каналов, угнетение аденилатциклазы
Мускариновый М3 Экзокринные железы, сосуды (гладкие мышцы и эндотелий) Образование ИТФ и ДАГ, повышение содержания внутриклеточного кальция
Никотиновый Нн Постганглионарные нейроны, некоторые пресинаптические холинергические окончания Открытие Na+, К* каналов, деполяризация
Никотиновый Нм Адренорецепторы Нервно-мышечные конечные пластинки скелетных мышц Открытие Na*, К* каналов, деполяризация
а, Постсинаптические эффекторные клетки, особенно гладких мышц Образование ИТФ и ДАГ, повышение содержания внутриклеточного кальция
а2 Пресинаптические адренергические нервные окончания, тромбоциты, адипоциты, гладкие мышцы Угнетение аденилатциклазы, уменьшение цАМФ
₽. Постсинаптические эффекторные клетки, особенно сердца, адипоциты, мозг, пресинаптические адренергические и холинергические нервные окончания Активация аденилатциклазы и увеличение цАМФ
Постсинаптические эффекторные клетки, особенно гладкие мышцы и сердечная мышца Активация аденилатциклазы и увеличение цАМФ
Рз Постсинаптические эффекторные клетки, особенно адипоциты Дофаминовые рецепторы Активация аденилатциклазы и увеличение цАМФ
D, (DAO.Dg Мозг; эффекторные ткани, особенно гладкие мышцы почечного сосудистого ложа Активация аденилатциклазы и увеличение цАМФ
DZ(DAZ) Мозг; эффекторные ткани, особенно гладкие мышцы; пресинаптические нервные окончания Угнетение аденилатциклазы; увеличение калиевой проводимости
D31 Мозг Угнетение аденилатциклазы
d4' Мозг, сердечно-сосудистая система. Угнетение аденилатциклазы
' Могут быть подтипами О2-рецептора.
алкалоидов, использованных для их идентификации,— мускарина и никотина. Соответственно эти рецепторы называются мускариновыми и никотиновыми. В случае рецепторов, связанных с норадренергическими нервами, не были созданы прилагательные из названий агонистов (норадреналина, фенилэфрина, изопротеренола и т. д.). Поэтому термин адренорецептор стал широко использоваться
для описания рецепторов, которые реагируют на катехоламины, например норадреналин. По аналогии термин холинорецептор обозначает рецепторы (и мускариновые, и никотиновые), которые возбуждаются ацетилхолином. В Северной Америке рецепторы назывались по нервам, которые обычно их иннервируют — адренергические (или норадренергические) рецепторы и холинергические рецепте-
ры. Адренорецепторы разделяются на а- и Р-адре-норецепторы на основании селективности агонистов и антагонистов. Открытие более селективных блокаторов привело к выделению подклассов внутри этих больших типов. Например, внутри класса а-адренорецепторов «г и а2-рецепторы отличаются по селективности и к агонистам, и к антагонистам. Примеры таких селективных веществ приводятся в последующих главах.
Большой прогресс достигнут в выдёлении и очищении рецепторных белков. Во многих случаях были определены их пептидные последовательности и трехмерная конформация. Независимые исследования, основанные на изучении кДНК, подтвердили множественность рецепторов и расширили число подтипов холино- и адренорецепторов. Например, открыты гены для пяти различных мускариновых рецепторов (Buckley, 1989).
Неадренергические, нехолинергические нейроны
Давно известно, что вегетативные эффекторные ткани содержат нервные волокна, которые не обладают гистохимическими характеристиками ни холинергических, ни адренергических волокон. Обнаружены и моторные, и сенсорные нехолинергические, неадренергические волокна. Основными транс-миттерными субстанциями, найденными в этих нервных окончаниях, являются пептиды, но во многих из них присутствуют и другие субстанции, в частности пурины (табл. 6-1). Современные иммунологические методы позволяют точно идентифицировать и количественно определить пептиды, хранящиеся и выделяющиеся из окончаний нервных волокон. Кроме того, обнаружено, что полученный из чилийского перца нейротоксин капсаицин может вызывать выделение трансмиттера из этих нейронов, а в высоких дозах — разрушение нейрона. В настоящее время накоплено большое количество информации, пока полностью не осмысленной.
НСК в стенке кишечника (рис. 6-2) — наиболее хорошо изученная система, содержащая наряду с холинергическими и адренергическими волокнами нехолинергические, неадренергические нейроны. В тонком кишечнике морских свинок, например, эти нейроны дают положительные иммуно-флюоресцентные реакции для одного или нескольких следующих веществ: пептида, связанного с
кальцитониновым геном, холецистокинина, динор-фина, энкефалинов, гастринрилизинг-пептида, 5-гидрокситриптамина (серотонина), нейропептида Y, соматостатина, субстанции Р и вазоактивного кишечного пептида. Некоторые нейроны содержат до пяти различных возможных трансмиттеров. Эта система, очевидно, “интерпретирует” моторный выход АНС и обусловливает необходимую синхронизацию импульсов, которая, например, обеспечивает продвижение вперед, а не назад кишечного содержимого и расслабление сфинктеров при сокращении стенок кишечника.
Чувствительные волокна в нехолинергических, неадренергических системах, возможно, лучше называть “сенсорно-эфферентными” волокнами, так как при активации сенсорного входа они способны выделять трансмиттерные пептиды из самих чувствительных окончаний, из местных ветвей аксона и из коллатералей, заканчивающихся в автономных ганглиях. Эти пептиды — сильные агонисты во многих автономных эффекторных тканях. Пока не известно, модифицируют ли распространенные в клинической практике препараты эти сенсорно-эфферентные процессы, однако ведется активный поиск селективных ингибиторов.
Функциональная организация автономной регуляции
Понимание основ взаимодействия автономных компонентов нервной системы друг с другом и с их эффекторными органами существенно для оценки действия препаратов, влияющих на вегетативную нервную систему, особенно из-за их значительного “рефлекторного” действия.
Центральная интеграция
На высшем уровне два отдела вегетативной нервной системы (средний и продолговатый мозг) и эндокринная система интегрированы друг с другом, с сенсорным входом и с информацией от высших центров центральной нервной системы. Эти взаимодействия таковы, что ранее исследователи называли парасимпатическую систему трофотроп-ной (т. е. приводящей к росту), а симпатическую систему — эрготропной (т. е. приводящей к расходу энергии), активируемой для “борьбы или бегства”. Несмотря на то, что такие термины несколько по
ясняют механизмы регуляции, они дают лишь простое описание, применимое ко многим эффектам этих систем (табл. 6-3). Например, замедление сердечной деятельности и стимуляция пищеварительной активности — это типичные сохраняющие энергию эффекты парасимпатической системы — “покой” и “переваривание пищи”. Напротив, сердечная стимуляция, повышение концентрации сахара в крови и сужение кожных сосудов — ответы, вызываемые симпатической активацией, приспособленные к “битве" или к “борьбе за выживание”.
На более тонком уровне взаимодействий в мозговом стволе, продолговатом и спинном мозгу важными являются кооперативные взаимоотношения между парасимпатической и симпатической системами. Для некоторых органов чувствительные волокна, ассоциированные с парасимпатической системой, оказывают рефлекторный контроль над моторным выходом в симпатической системе. Так, например, чувствительные барорецепторные волокна каротидного синуса в языкоглоточном нерве оказывают большое влияние на симпатический выход из вазомоторного центра. Этот пример еще будет детально описан. Сходным образом парасимпатические чувствительные волокна в стенках мочевого пузыря выражение влияют на симпатическую ингибиторную активность в отношении этого органа. В пределах НСК чувствительные волокна из стенок кишечника образуют синапсы и с преганг-лионарными, и с постганглионарными моторными клетками, которые контролируют гладкие мышцы кишечника и секреторные клетки (рис. 6-2).
Интеграция сердечно-сосудистой функции
Автономные рефлексы особенно важны для понимания сердечно-сосудистых реакций на лекарственные препараты, влияющие на вегетативную нервную систему. Как указано на рис. 6-7, основным контролируемым показателем функции сердечно-сосудистой системы является среднее артериальное давление. Изменения любого фактора, участвующего в регуляции среднего артериального давления (например, периферического сосудистого сопротивления), вызывают сильные гомеостатические вторичные реакции, которые стремятся компенсировать вызванные изменения. Гомеостатические реакции иногда могут быть достаточны для предотвращения любых изменений среднего ар
териального давления идля блокирования действия лекарства на частоту сердечных сокращений. Рассмотрим возможности такой регуляции на примере медленной инфузии норадреналина. Это вещество оказывает прямой эффект и на сердечную мышцу, и на мышцы сосудов; оно является сильным вазоконстриктором и, повышая периферическое сосудистое сопротивление, увеличивает среднее артериальное давление. В отсутствие рефлекторного контроля (например, у пациентов с сердечным трансплантатом) норадреналин повышает частоту и силу сердечных сокращений. Однако у пациентов с интактными рефлексами барорецепторный ответ на повышение среднего артериального давления по механизму отрицательной обратной связи вызывает снижение симпатического влияния на сердце и сильное повышение парасимпатического (вагусного) действия на сердечный пейсмекер. В результате итоговый эффект обычной прессорной дозы норадреналина заключается в заметном увеличении периферического сосудистого сопротивления, умеренном повышении среднего артериального давления и стойком замедлении частоты сердечных сокращений. Брадикардия — рефлекторный компенсаторный ответ, вызываемый этим веществом, противоположен прямому действию препарата. Эта реакция полностью предсказуема при анализе механизмов интеграции сердечно-сосудистой деятельности автономной нервной системой.
Пресинаптическая регуляция
Принцип регуляции по механизму отрицательной обратной связи обнаружен и на пресинаптичес-ком уровне автономной функции. Пока еще не понятые столь полно как барорецепторный рефлекс важные пресинаптические механизмы ингибиторного контроля по принципу обратной связи обнаружены в норадренергических волокнах. Наилучшим образом изученные механизмы включают а2-рецепторы, расположенные на нервных окончаниях. Эти рецепторы активируются норадреналином и сходными молекулами, активация в свою очередь уменьшает дальнейшее выделение норадреналина из этих нервных окончаний (табл. 6-4). Наоборот, пресинаптические p-рецепторы могут облегчать выделение норадреналина. Пресинаптические рецепторы, отвечающие на трансмиттерные субстанции, выделяемые нервными окончаниями, называются ауторецепторами. Ауторецепторы обыч-
ТАБЛИЦА 6-3. Прямой эффект активности автономных нервов и лекарственных препаратов, влияющих на вегетативную нервную систему и некоторые системы органов
Симпатическая система Парасимпатическая система
Органы Действие1 Рецептор2 Действие Рецептор2
Глаз
Радужка /
Радиальная мышца Сокращается а, — —
Циркулярная мышца — — Сокращается М3
Цилиарная мышца (Расслабляется) р Сокращается М3
Сердце
Синоатриальный узел Ускоряется Pl Замедляется М2
Эктопические пейсмекеры Ускоряется ₽1 — ——
Сократимость Повышается ₽1 Замедляется (предсердия) м2
Гладкие мышцы сосудов
Кожа, сосуды внутренних органов Сокращаются а — —
Сосуды скелетных мышц Расслабляются Р2 — —
(Сокращаются) а —
Расслабляются М4 — —
Эндотелий — — Выделяется РФПЭ Ml
Бронхиолярные гладкие мышцы Расслабляются Р2 Сокращаются Мз
Желудочно-кишечный тракт Гладкие мышцы
Стенок Расслабляются Иг5.Рг Сокращаются Мз
Сфинктеров Сокращаются «1 Расслабляются Мз
Секреция — — Повышается Мз
Мышечное сплетение Угнетается а Активируется М,
Гладкие мышцы мочеполовой системы
Стенки мочевого пузыря Расслабляются р2 Сокращаются М3
Сфинктер Матка при беременности Сокращается Расслабляется «1 р2 Расслабляется Мз
Сокращается а Сокращается М3
Пенис, семенные пузырьки Эякуляция а Эрекция М
Кожа
Пиломоторные гладкие мышцы Сокращаются а — —
Потовые железы
Терморегуляторные Активируются М — - -
Апокринные (стресс) Активируются а — —
Мётаболические функции
Печень Глюконеогенез а/р26 — —
Печень Глюкогенолиз а/р2 — —
Жировые клетки Липолиз Рз7 — —
Почки Выделение ренина Pi — —
ТАБЛИЦА 6-3. (Продолжение) Симпатическая система Парасимпатическая система
Органы Действие1 Рецептор2 Действие Рецептор2
Вегетативные нервные окончания Симпатические Снижается М
Парасимпатические Снижается выделение АХ а выделение НА
' Менее важное действие отмечено скобками.
2 Специфический тип рецепторов: а = альфа, р = бета, М = мускариновый. Подтипы мускариновых рецепторов определены, главным образом, в опытах на тканях животных.
3Эндотелий большинства сосудов выделяет РФПЭ (расслабляющий фактор, производимый эндотелием), который вызывает заметную вазодилатацию в ответ на мускариновую стимуляцию. Однако в отличие от рецепторов в кровеносных сосудах скелетных мышц, иннервируемых симпатическими холинергическими волокнами, эти мускариновые рецепторы не имеют иннервации и реагируют только на мускариновые агонисты, циркулирующие в крови.
4 Кровеносные сосуды скелетных мышц имеют симпатические холинергические дилататорные волокна.
5 Вероятно, путем пресинаптического ингибирования парасимпатической активности.
6 Зависит от вида.
7аг угнетают, р, и р3 стимулируют.
но бывают ингибиторными, однако некоторые холинергические волокна, особенно соматические моторные волокна, могут иметь возбуждающие ауторецепторы для ацетилхолина.
Регуляция выделения трансмиттера не ограничена влиянием только самого трансмиттера. Нервные окончания также имеют регуляторные рецепторы, отвечающие на многие другие субстанции (гетерорецепторы). Гетерорецепторы могут активироваться субстанциями, выделяемыми из нервных терминалей, которые образуют синапсы с нервным окончанием. Лиганды для этих рецепторов могут поступать к ним из крови или из близлежащих тканей. Некоторые трансмиттеры и рецепторы, идентифицированные в настоящее время, перечислены в табл. 6-4. Пресинаптическая регуляция разнообразными эндогенными химическими соединениями, вероятно, происходит во всех нервных волокнах.
Постсинаптическая регуляция
Постсинаптическая регуляция может рассматриваться в двух аспектах: модуляция активности на основном рецепторе, которая может уменьшать (увеличивать) число рецепторов или уменьшать‘их восприимчивость — десенситизировать (глава 2); и модуляция другими временными воздействиями.
Первый механизм хорошо продемонстрирован в нескольких рецепторно-эффекторных системах.
Увеличение и уменьшение числа рецепторов, как известно, встречается в ответ на соответствующее снижение или повышение их активации. Крайние формы увеличения числа рецепторов наблюдаются после денервации некоторых тканей, что проявляется в “денервационной суперчувствительности” ткани к активаторам этого типа рецепторов. Например, в скелетных мышцах никотиновые рецепторы в норме ограничены областью концевых пластинок, расположенных на соматических моторных нервных окончаниях. Хирургическая денервация приводит к заметной пролиферации никотиновых хо-линорецепторов по всей поверхности волокна, включая зоны, предварительно не связанные с какими-либо соединениями моторных нервов. Родственный феномен гиперчувствительности, вызванный фармакологической денервацией, наблюдается в автономных эффекторных тканях после введения препаратов, истощающих трансмиттерные запасы и предотвращающих на достаточный период времени активацию постсинаптических рецепторов. Например, введение больших доз резерпина, истощающего запас норадреналина в симпатических волокнах, может вызвать повышение чувствительности гладких мышц и эффекторных клеток сердечной мышцы.
Второй механизм включает модуляцию основных трансмиттер-рецепторных взаимоотношений событиями, вызванными теми же самыми или другими трансмиттерами, действующими на различ-
3 2 Р
СО а ю
>s о
ВАЗОМОТОРНЫЙ ЦЕНТР
Парасимпатическая автономная нервная система '
Барорецепторы
Симпатическая автономная нервная система
Периферическое Частота сосудистое сердечных сопротивление со)фащений
Среднее артериальное давление
Сила Тонус сокращений вен
Сердечный ___Ударный _ Венозный _ Объем
выброс * объем *"” возврат *“ крови
Ангиотензин
Альдостерон
я
ф
s
ф
§ я
Рис. 6-7. Нервный и гормональный контроль сердечно-сосудистой деятельности. Отметьте, что имеются две петли обратной связи: петля вегетативной нервной системы и гормональная петля. Симпатическая нервная система прямо влияет на четыре основных показателя: периферическое сосудистое сопротивление, частоту сердечных сокращений, силу сокращений и венозный тонус. Существует и прямое модулирующее влияние на продукцию ренина (не показано). Эффекты каждой петли обратной связи служат для компенсации изменений артериального давления. Так, например, снижение кровяного давления вследствие потери крови вызовет увеличение симпатической активности и выделение ренина. Наоборот, повышение давления при введении вазоконстрикторных препаратов приведет к уменьшению симпатической активности и снижению выделения ренина, а также повышению парасимпатической (вагусной) активности
ные постсинаптические рецепторы. Ганглионарная передача — типичный пример такой иерархии (рис. 6-8). Постганглионарные клетки активируются (деполяризуются) в результате связывания соответствующего лиганда с никотиновым (нн) ацетилхолиновым рецептором. Возникающий быстрый возбуждающий постсинаптический потенциал (ВПСП) вызывает при достижении порога распространение потенциала действия. За этим событием часто следует малый и медленно развивающийся, но долговременный гиперполяризующий постпотенциал - медленный тормозный постсинаптический потенциал (ТПСП). Гиперполяризация включает открытие калиевых каналов м2-холинорецеп-торами. За ТПСП следует малый медленный возбуждающий постсинаптический потенциал, очевидно, вызывающий закрытие калиевых каналов, связанных с мгхолинорецепторами. Наконец, поздний
очень медленный ВПСП может быть вызван пептидами, выделяющимися из других волокон. Эти медленные потенциалы служат для модуляции реактивности постсинаптических клеток на последующую активность основного возбуждающего пресинаптического нерва, (глава 20).
Фармакологическая модификация функции автономной нервной системы
В передаче нервного импульса участвует много механизмов в разных сегментах автономной нервной системы, поэтому некоторые лекарственные препараты вызывают высокоспецифичные эффекты, в то время как другие являются значительно менее селективными в своем действии. Краткое из-
ТАБЛИЦА 6-4. Ауторецептор, гетерорецептор и модуляторные эффекты в периферических синапсах
Трансмиттер/модулятор Тип рецептора Нервные окончания, в которых обнаружены рецепторы
Угнетающие эффекты Ацетилхолин Норадреналин Дофамин Серотонин(5-НТ) АТФ и аденозин Гистамин Энкефалин НейропептидУ Простагландин ЕЪЕ2 М2 Адренергические, нервная система кишечника а2 Адренергические D2, меньше свидетельств о D, Адренергические 5-НТъ 5-НТ2,5-НТ3 Холинергические преганглионарные Р, Адренергические, автономные пресинаптичес- кие и холинергические нейроны НСК Н3, возможно Н2 Н3 тип идентифицирован в адренергических и се- ротонинергических нейронах ЦНС Д (также ц, %) Адренергические; холинергические НСК * NPY Адренергические, некоторые холинергические ЕР3 Адренергические
Возбуждающие эффекты Адреналин Ацетилхолин Ангиотензин II ₽2 Нм AII-1 Адренергические, соматические моторные холинергические Соматические моторные холинергические Адренергические
ложение этапов передачи импульсов из центральной нервной системы в автономные эффекторные клетки представлено в табл. 6-5. Препараты, блокирующие распространение потенциала действия (местные анестетики), не селективны в своем дей- ствии, так как они влияют на общий для всех нейронов процесс. С другой стороны, препараты, действующие на биохимические процессы, включенные в синтез и хранение трансмиттера, являются более селективными, так как биохимия адренерги-
Преганглионарный Мембранный
Время
Рис. 6-8. Возбуждающие и тормозные постсинаптические потенциалы (ВПСП и ТПСф в клетке вегетативного ганглия. В постганглионарном нейроне может происходить изменение мембранного потенциала, продемонстрированное схематически в записи. Ответ начинается с двух ВПСП на активацию никотиновых (н) рецепторов, первый из которых не достигает порога. За потенциалом действия следует ТПСП, возможно, вызываемый активацией м2-ре-цепторов (с вероятным участием активации дофаминовых рецепторов). За ТПСП, в свою очередь, следует медленный М|-зависимый ВПСП, за ним иногда следует еще один более медленный, индуцированный пептидами, возбуждающий постсинаптический потенциал
ТАБЛИЦА 6-5. Этапы автономной передачи, эффект препаратов
Процесс Препарат Место Эффект
Распространение Местные анестетик^, тет- Аксоны нервов Блокада натриевых каналов;
потенциала действия родотоксин', сакситок- 2 СИН блокада проводимости
Синтез трансмиттера Гемихолиний а-Метилтирозин (метиро-зин) Холинергические нервные Блокирует захват холина и за-окончания: мембрана медляет синтез Адренергические нервные Блокирует синтез окончания и мозговое вещество надпочечников: цитоплазма
Хранение трансмиттера Везамикол Резерпин Холинергические оконча- Препятствует накоплению ния: везикулы Адренергические оконча- Способствует уменьшению ния:везикулы запасов
Выделение трансмиттера Многие3 со-Конотоксин GVIA Ботулотоксин Латротоксин4 Тирамин, амфетамин Мембраны рецепторов Модуляция выделения нервных окончаний Кальциевые каналы Уменьшает выделение транснервных окончаний миттера Холинергические везикулы Препятствует выделению Холинергические и адре- Вызывает взрывное выделе-нергические везикулы ние Адренергические нервные Способствуют выделению окончания трансмиттера
Захват трансмиттера Кокаин, трициклические Адренергические нервные Ингибируют захват, увеличи-
после выделения антидепрессанты 6-гидроксидофамин окончания вают эффект трансмиттера на постсинаптический рецептор Адренергические нервные Разрушает нервные оконча-окончания ния
Активация/блокада рецептора Норадреналин Фентоламин Изопротеренол Пропранолол Никотин Рецепторы на адренерги- Связывается с а-рецептора-ческих соединениях ми, вызывает сокращение Рецепторы на адренерги- Связывается с а-рецептора-ческих соединениях ми, препятствует активации Рецепторы на адренерги- Связывается с р-рецептора-ческихсоединениях ми, активирует аденилат циклазу Рецепторы на адренерги- Связывается с Р-рецептора-ческих соединениях ми, препятствует активации Рецепторы на никотиновом Связывается с никотиновым холинергическом соеди- рецептором, открывает ион-нении (автономные ганг- ные каналы в постсинапти-лии, нервно-мышечные ческой мембране конечные пластинки)
ТАБЛИЦА 6-5. (Продолжение)
Процесс Препарат Место Эффект
Тубокурарин / Нервно-мышечные концевые пластинки Препятствует активации
Бетанехол Рецепторы, парасимпатические эффекторные клетки (гладкие мышцы, железы) Связывается с мускариновыми рецепторами, выделяет трифосфат инозитола и активирует гуанилатциклазу
Атропин Рецепторы, парасимпатические эффекторные клетки Связывается с мускариновыми рецепторами, препятствует активации
Энзиматическая Неостигмин Холинергические синапсы Ингибирует фермент, продле-
инактивация трансмиттера (ацетилхолинестераза) вает и усиливает действие трансмиттера
Транилципромин Адренергические нервные окончания (моноаминооксидаза) Ингибирует фермент, увеличивает хранящийся запас трансмиттера
1 Токсин иглобрюха, калифорнийского тритона.
2Токсин Gonyaulax (микроорганизм красного морского прилива).
3 Норадреналин, дофамин, ацетилхолин, ангиотензин II, различные простагландины и т. д.
“Яд паука “черная едова”, каракурта.
Фармакология глаза
Глаз является хорошим примером органа с множеством функций АНС, контролируемых несколькими различающимися автономными рецепторами. Как показано на рис. 6-9, в передней камере глаза расположено несколько структур, контролируемых АНС. Это три различные мышцы (круговая и радиальная мышцы радужки и цилиарная мышца) и секреторный эпителий цилиарного тела.
Мускариновые холиномиметики вызывают сокращение круговой мышцы радужки и цилиарной мышцы. Сокращение круговой мышцы радужки вызывает миоз, уменьшение размера зрачка. Миоз обычно наблюдается у пациентов, получавших большие системные или малые местные дозы холиномиметиков, особенно фосфорорганических ингибиторов холинэстеразы. Сокращение реснитчатой мышцы вызывает аккомодацию на точку близкого видения. Заметное сокращение цилиарной мышцы, которое часто встречается при интоксикации ингибиторами холинэстеразы, называется цикло
спазмом — спазмом аккомодации. Сокращенная цилиарная мышца также оказывает давление на трабекулярную сеть, открывая ее поры и облегчая отток водянистой влаги в Шлеммов канал. Увеличение оттока уменьшает внутриглазное давление, что очень важно для больных с глаукомой. Все эти эффекты предотвращаются или снимаются препаратами, блокирующими мускариновые рецепторы, например, атропином.
ос-адренорецепторы вызывают сокращение радиально ориентированных расслабляющих мышечных волокон в радужке, в результате чего наблюдается мидриаз. Это обычно отмечается при симпатической активации и при аппликации в конъюнктивальный мешок ос-агонистов, таких как фенилэфрин, p-адренорецепторы на цилиарном эпителии облегчают секрецию водянистой влаги. Блокирование этих рецепторов (Р-блокаторами) уменьшает секреторную активность и снижает внутриглазное давление, что используется при лечении глаукомы.
Рис. 6-9. Структуры передней камеры глаза. На этой схеме представлены ткани со значительными автономными функциями и рецепторы, связанные с АНС. Активация p-адренорецепторов, связанных с цилиарным эпителием, вызывает повышение секреции водянистой влаги (стрелка). Кровеносные сосуды глаза (не показаны) также находятся под вегетативным контролем и влияют на дренажную функцию.
ческой передачи значительно отличается от биохимии холинергической передачи. Активация или блокада рецепторов эффекторных клеток предполагают максимум гибкости и селективности эффекта: адренорецепторы легко отличимы от холиноре-цепторов. Кроме того, некоторые подгруппы рецепторов могут быть избирательно активированы или блокированы внутри каждого большого типа. Некоторые примеры даны в дополнении “Фармакология глаза”. В следующих четырех главах приведены примеры разнообразия процессов автономного контроля.
Избранная литература
Anderson К-Е. Pharmacology of lower urinary tract smooth muscles and penile erectile tissues. Pharmacol. Rev. 1993; 45: 253.
Brose N. et al. Synaptotagmin: A calcium sensor on the synaptic vesicle surface. Science, 1992; 256:1021.
Burnstock G., Hoyle С. H. V. Autonomic Neuroeffector Mechanisms. Harwood Academic Publishers, 1992.
Furchgott R. F. Role of endothelium in responses of vascular smooth muscle to drug. Annu. Rev. Pharmacol. Toxicol. 1984; 24:175.
Furness J. B., Costa M. The Enteric Nervous System. Churchill Livingstone, 1987.
Ruffolo R. R. Jr. et al. Drug receptors and control of the cardiovascular system. Recent advances. Prog. Drug Res. 1991; 36:117.
Starke K., Gothert M., Kilbinger H. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol. Rev. 1989; 69:864 .
1994 receptor nomenclature supplement. Trends Pharmacol. Sci. (Suppl.): 1994.
Средства, активирующие холинорецепторы 7
и ингибирующие холинэстеразу
Ахиллес Дж. Паппано, Август М. Ватанабе
Стимуляторы ацетилхолинового рецептора и ингибиторы холинэстеразы вместе составляют большую группу препаратов, имитирующих эффект ацетилхолина (холиномиметические препараты) (рис. 7-1). В зависимости от типа рецептора (мускариновый или никотиновый) холиномиметики делят по спектру действия. Важным критерием фармакологической классификации является механизм действия. Некоторые препараты прямо активируют хол инорецептор, другие действуют опосредованно, ингибируя гидролиз эндогенного ацетилхолина.
Спектр действия холиномиметических средств
В своих первых исследованиях парасимпатической нервной системы сэр Генри Дэйл обнаружил, что алкалоид мускарин имитирует эффекты активации парасимпатического нерва, т. е. вызывает парасимпатомиметическое действие. При аппликации мускарина действие проявляется на уровне рецепторов автономных эффекторных тканей (гладкие мышцы, сердце, экзокринные железы), а не в ганглиях. Поэтому эффекты самого ацетилхолина и других холиномиметических средств на нейроэффекторные соединения рассматривают как парасимпатомиметические эффекты, вызываемые посредством активации мускариновых рецепторов.
Далее было обнаружено, что алкалоид никотин стимулирует автономные ганглии и нервно-мышечные соединения скелетных мышц, но в малых кон
центрациях может оказывать незначительный эффект и на автономные эффекторные клетки. Рецепторы ганглиев и скелетных мышц были поэтому названы никотиновыми. Установление того факта, что ацетилхолин является единой физиологической трансмиттерной субстанцией как для мускариновых, так и для никотиновых рецепторов, послужило основанием для разделения холинорецепторов на два упомянутых подтипа.
На основании особенностей трансмембранных сигнальных механизмов холинорецепторы относятся или к связанным с G-белками (мускариновые), или к семействам ионных каналов (никотиновые). Мускариновые рецепторы содержат семь трансмембранных сегментов, в которых третья цитоплазматическая петля соединяется с G-белком, функционирующим как внутримембранный передатчик (рис. 2-12). В целом эти рецепторы регулируют синтез внутриклеточных вторичных мессенджеров. Избирательность агонистов определяется подтипами мускаринового рецептора и G-белком, присутствующим в данной клетке (табл. 7-1). Мускариновые рецепторы в основном расположены на плазматических мембранах клеток органов, иннервируемых парасимпатическими нервами, они встречаются также в некоторых тканях, не иннервируемых этими нервами, например на эндотелиальных клетках (табл. 7-1) и втканях, иннервированых холинергическими постганглионарными симпатическими нервами.
Никотиновые рецепторы являются частью трансмембранного полипептида, чьи субъединицы формируют катионселективные ионные каналы (рис. 2-10). Эти рецепторы расположены на плазматических мембранах парасимпатических и сим-
Алкалоиды
ХОЛИНОМИМЕТИЧЕСКИЕ ПРЕПАРАТЫ
_______________ I________________________________________
Нервы Сердце Железы и эндотелий
—► Мускариновые -<—
"Обратимые"
Препараты прямого действия
рецепторы
I ду < Препараты непрямого __
т действия
Холиновые эфиры
—► Никотиновые <—
"Необратимые"
Г
Нервномышечные
“I
Ганглионарные
Рис. 7-1. Основные группы препаратов, активирующих холинорецепторы, рецепторов и тканей-мишеней
патических постганглионарных клеток в автономных ганглиях и также на мембранах соматических мышц, иннервированных моторными волокнами (рис. 6-1).
Вследствие многочисленности точек приложения ацетилхолина и разнонаправленности его действия неселективные стимуляторы холиноре-цепторов в достаточных дозах могут вызывать весь-
ма распространенные и отчетливые нарушения функций систем и органов. Однако желаемые эффекты могут быть достигнуты без или с минимальным побочным действием, поскольку имеются препараты с определенной степенью селективности. Эта селективность действия базируется на нескольких факторах. Некоторые препараты избирательно стимулируют мускариновые рецепторы, в то время
ТАБЛИЦА 7-1. Подтипы и характеристики холинорецепторов
Тип рецептора Другие названия Расположение Структурные особенности Пострецепторный
механизм
м, м„ Нервы 7 трансмембранных сегментов, ИТФ, ДАГ-каскад связанных с G-белком
М2 м2в, сердечные м2 Сердце, нервы, 7 трансмембранных сегментов, Угнетение продук- гладкие мышцы связанных с G-белком ции цАМФ, актива- ция К+-каналов
м3 м2в, железистые м2 Железы, гладкие 7 трансмембранных сегментов, ИТФ, ДАГ-каскад мышцы, эндотелий связанных с G-белком
Мд ? ЦНС 7 трансмембранных сегментов, Угнетение продук- связанных с G-белком ции цАМФ
Ms ? ЦНС 7 трансмембранных сегментов, ИТФ, ДАГ-каскад
Мышечного типа, связанных с G-белком
Нм концевая плас- Нервно-мышечные Пентамер (а2р6у)2 Деполяризация
тинка соединения ске- Na+, IC-каналов
Нейронального летных мышц
Нн типа, ганглионар- Постганглионарные а- и р- субъединицы только как Деполяризация
ный рецептор клеточные тела, а2р2 или а3р3 Na+, К+-каналов дендриты
1 Гены были клонированы, но функционально рецепторы не были неоспоримо идентифицированы.
2 Структура в электрическом органе Torpedo и мышцах плода. В зрелых мышцах субъединица у замещена на субъединицу е. В разных тканях млекопитающих идентифицированы несколько различных а- и р-субъединиц (Lindstrom, 1990).
как другие избирательно активируют никотиновые рецепторы. Отдельные вещества стимулируют предпочтительно никотиновые рецепторы ганглиев и оказывают меньшее действие на никотиновые рецепторы нервно-мышечных соединений. Селективность в отношении органов может быть достигнута и применением соответствующих путей введения (“фармакокинетическая селективность”). Например, мускариновые стимуляторы могут вводиться местно в полость конъюнктивы для воздействия на зрительные функции, минимизируя системные эффекты.
Типы действия
холиномиметических препаратов
Холиномиметики могут быть прямого или непрямого действия (рис. 7-1). Препараты прямого действия непосредственно связываются с мускариновыми или никотиновыми рецепторами и активируют их. Препараты непрямого действия проявляют основной эффект, ингибируя ацетилхолинэсте-разу, которая гидролизует ацетилхолин до холина и уксусной кислоты (рис. 6-3). При инактивации ацетилхолинэстеразы концентрация эндогенного ацетилхолина в синаптических щелях и нейроэф-фекторных соединениях увеличивается, и излишек медиатора в свою очередь стимулирует холинорецепторы, вызывая усиленный ответ. Препараты непрямого действия по эффекту являются усилителями эндогенного ацетилхолина и действуют, главным образом, в местах его физиологического выделения.
Некоторые ингибиторы холинэстеразы даже в низких концентрациях ингибируют также бутирил-холинэстеразу (псевдохолинэстеразу), большинство угнетает этот фермент только в высоких концентрациях. Однако бутирилхолинэстераза не так важна для физиологического завершения действия синаптического ацетилхолина, поэтому ее инактивация играет малую роль в эффектах холиномиме-тиков непрямого действия. Некоторые ингибиторы холинэстеразы имеют и слабое прямое действие. Это особенно свойственно четвертичным карбаматам, например неостигмину, который в дополнение к угнетению холинэстеразы активирует нервно-мышечный никотиновый холинорецептор.
I. Базисная фармакология стимуляторов холинорецепторов прямого действия
Холиномиметики прямого действия по химическому строению разделяются на холиновые эфиры (включая ацетилхолин) и алкалоиды (такие как мускарин и никотин). Немногие из этих препаратов обладают высокой селективностью к мускариновым или к никотиновым рецепторам. Многие влияют на оба типа рецепторов, таким типичным веществом является ацетилхолин.
Химия и фармакокинетика
А. Структура. Четыре хорошо исследованных эфира холина представлены на рис. 7-2. Они имеют постоянный заряд и относительно нерастворимы в липидах, так как содержат четвертичную аммониевую группу. Установлено, что многие естественные и синтетические холиномиметики не являются холиновыми эфирами, некоторые из них показаны на рис. 7-3. Мускариновый рецептор сте-реоселективен: (5)-бетанехол почти в 1000 раз сильнее, чем (й)-бетанехол.
Б. Всасывание, распределение и метаболизм. Холиновые эфиры слабо абсорбируются и из-за гидрофильности плохо проникают в центральную нервную систему. Хотя все они расщепляются в желудочно-кишечном тракте (поэтому не активны при введении внутрь), их чувствительность к гидролизу холинэстеразой в организме заметно различается. Ацетилхолин гидролизуется очень быстро (глава 6), поэтому чтобы достичь достаточного для получения заметных эффектов уровня, внутривенно должны быть введены большие количества препарата. При внутривенном введении даже большая доза вызывает короткий эффект, обычно от.5 до 20 секунд, в то время как внутримышечные и подкожные инъекции приводят только к местным эффектам. Метахолин по меньшей мере в три раза более стоек к гидролизу и вызывает системные эффекты даже при подкожном введении. Эфиры карбаминовой кислоты карбахол и бетанехол чрезвычайно устойчивы к гидролизу холинэстеразой и имеют соответственно значительно большую продолжи-
О ,
II ' 7 у СНз
Н3С — С — О — СН2 — СН2 — N+ — СНз ХСН3
Ацетилхолин
О
II /СН3
Н3С — С — О — CH — СН2 — N+ — СНз
Метахолин (ацетил-р-метилхолин)
О II
H2N — С-О-Н
Карбаминовая кислота
О
II /СНз
H2N —С —О —СН2 —СН2—N+ —СНз
4 СНз
Карбахол (карбамоилхолин)
О
II /СНз
H2N — С-О-СН-СН2 — N+ — СН3
I 4 СНз
СН3
Бетанехол (карбамоил-р-метилхолин)
Рис. 7-2. Молекулярные структуры четырех холиновых эфиров и карбаминовой кислоты. Ацетилхолин и метахолин являются уксусными эфирами холина и 0-метил-холина соответственно. Карбахол и бетанехол — эфиры карбаминовой кислоты тех же спиртов
тельность действия. Присутствие Р-метильной группы (метахолин, бетанехол) уменьшает силу воздействия этих препаратов на никотиновый рецептор (табл. 7-2).
Третичные холиномиметические алкалоиды (пилокарпин, никотин, лобелии; рис. 7-3) хорошо всасываются из большинства мест введения. Никотин в жидком виде достаточно хорошо растворим в липидах и может всасываться через кожу. Мускарин, четвертичный амин, менее полно всасывается из желудочно-кишечного тракта, чем третичные амины, но все же токсичен при приеме внутрь, например с определенными грибами. Выделение этих аминов осуществляется главным образом почками. При кислой реакции мочи клиренс третичных аминов ускоряется. Оксотреморин — чрезвычайно сильный синтетический мускариновый агонист, применяемый в исследовательской работе. Он хорошо проникает в центральную нервную систему. Лобелии, препарат растительного происхождения, имеет меньшую силу, но сходный спектр действия с никотином. Диметилфенилпиперазин (ДМФП) является сильным синтетическим никотиновым стимулятором с малым доступом в центральную нервную систему. Он также используется как средство исследований для избирательного возбуждения ганглионарных никотиновых рецепторов.
Фармакодинамика
А. Механизм действия. Существуют два основных механизма, с помощью которых активация парасимпатической нервной системы влияет на функцию органов. Первый — ацетилхолин может активировать мускариновые рецепторы эффекторных органов, непосредственно изменяя их функцию. Второй — ацетилхолин может взаимодействовать с мускариновыми рецепторами на нервных окончаниях, угнетая выделение из них трансмиттера. С помощью этого механизма выделение ацетилхолина (вероятно и циркулирующих мускариновых агонистов) опосредованно изменяет функции органов, модулируя эффекты парасимпатической и симпатической нервных систем и, возможно, нехолинергических и неадренергических систем.
ТАБЛИЦА 7-2. Свойства холиновых эфиров
Холиновые эфиры Чувствительность Мускариноподобное Никотиноподобное к холинэстеразе действие действие
Ацетилхолина хлорид Метахолина хлорид Карбахо ла хлорид Бетанехола хлорид ++++ +++ +++ + ++++ + незначительная ++ +++ незначительная ++ нет
ГЛАВНЫМ ОБРАЗОМ
МУСКАРИНОПОДОБНОЕ ДЕЙСТВИЕ
Мускарин
Пилокарпин
ГЛАВНЫМ ОБРАЗОМ НИКОТИНОПОДОБНОЕ ДЕЙСТВИЕ
Оксотреморин
Диметилфенилпиперазин (ДМФП)
Рис. 7-3. Структуры некоторых холиномиметических алкалоидов и их синтетических аналогов
Продолжают исследовать субклеточные механизмы, посредством которых мускариновые стимуляторы изменяют клеточные функции. К настоящему времени клонировано множество типов мускариновых рецепторов (глава 6), а некоторые исследованы по характеристикам связывания. При активации мускариновых рецепторов запускается ряд внутриклеточных механизмов, один или боЛее из которых могут выполнять роль вторичных посредников. Все мускариновые рецепторы сопряжены с G-белками и являются рецепторами с семью сегментами (табл. 7-1), пересекающими, как серпантин, мембрану. Важным результатом связывания мускариновых агонистов является активация ИТФ, Д АГ-каскада. Некоторые данные свидетельствуют об участии диацетилглицерола в открытии кальциевых каналов гладких мышц, ИТФ вызывает выделение ионов кальция из эндоплазматического и саркоплазматического ретикулума. Мускариновые агонисты также увеличивают клеточную концентрацию цАМФ. При активации мускариновых рецепторов возрастает и ток ионов калия через клеточные мембраны. Этот эффект опосредуется прямым связыванием активированного G-белка с ка
налом. Наконец, продемонстрировано, что активация мускариновых рецепторов в некоторых тканях (например, в сердце) угнетает активность аде-нилатциклазы, поэтому, мускариновые агонисты могут модулировать повышение уровня цАМФ, вызванное гормонами, например катехоламинами. Такой эффект мускариновых агонистов на образование цАМФ сопровождается ослаблением физиологического ответа органа на стимулирующие гормоны.
Механизм активации никотиновых рецепторов изучен весьма детально, что объясняется преимущественно тремя факторами: 1) рецептор присутствует в чрезвычайно высокой концентрации в мембранах электрических органов некоторых рыб; 2) ос-бунгаротоксин, компонент определенных змеиных ядов, прочно связывается с рецепторами и легко используется в качестве маркера для исследования; и 3) активация рецепторов вызывает легко измеряемые электрические и ионные изменения в вовлеченных клетках. Никотиновый рецептор в мышечных тканях является пентамером четырех типов гликопротеиновых субъединиц (один мономер встречается дважды) с общей мол. м. около 250 000
(рис. 2-10). Никотиновый рецептор в нейронах состоит только из а- и 0-субъединиц (табл. 7-1). Все субъединицы состоят из четырех пересекающих мембрану сегментов. Каждая ос-субъединица имеет рецепторную зону, которая при активации никотиновым агонистом индуцирует конформационные изменения в белке (открытие канала), что позволяет ионам натрия и калия быстро перемещаться по градиенту их концентрации. Если молекула агониста связывается с рецепторным местом одной из двух ос-субъединиц, возможность открытия канала только незначительно увеличивается. Множественное связывание агониста с обеими рецепторными зонами значительно повышает вероятность открытия. Таким образом, основным эффектом активации никотинового рецептора является деполяризация нервной клетки или нервно-мышечной концевой пластинки, индуцированная рецептором.
При стойкой фиксации агониста на никотиновом рецепторе эффекторный ответ прекращается: импульсация в постганглионарном нейроне угасает (ганглионарный эффект), и клетки скелетных мышц расслабляются (эффект нервно-мышечной концевой пластинки). Постоянное присутствие никотиновых агонистов предотвращает электрическое восстановление постсинаптической мембраны. Таким образом, индуцируется состояние “деполяризационной блокады”, которое не устраняется другими агонистами. Этот эффект может быть использован для получения мышечного паралича.
Б. Влияние на системы и органы. Большинство прямых эффектов стимуляторов мускариновых холинорецепторов на системы и органы легко предсказать исходя из проявлений стимуляции парасимпатических нервов (табл. 6-3) и распределения мускариновых рецепторов в организме. Эффекты типичного препарата, такого как ацетилхолин, представлены в табл. 7-3. Эффекты никотиновых агонистов можно также прогнозировать на основе знания физиологии автономных ганглиев и моторной концевой пластинки скелетной мышцы.
1. Глаз. Мускариновые агонисты, введенные в конъюнктивальный мешок, вызывают сокращение гладкой мышцы сфинктера радужки (миоз) и цилиарной мышцы (спазм аккомодации). В результате радужка оттягивается от угла передней камеры и трабекулярная сеть в основании цилиарной мышцы открывается. Оба эффекта облегчают отток водянистой влаги в Шлеммов канал, который дренирует переднюю камеру.
2. Сердечно-сосудистая система. Основными эффектами мускариновых агонистов на сердечнососудистую систему является уменьшение периферического сосудистого сопротивления и изменение частоты сердечных сокращений. Прямые эффекты, приведенные в табл. 7-3, изменяются при участии гомеостатических рефлексов (глава 6, рис. 6-7). Внутривенное введение человеку минимальных эффективных доз ацетилхолина (20-50 мкг/мин) вызывает вазодилатацию, приводящую к уменьшению кровяного давления с рефлекторным увеличением частоты сердечных сокращений. Большие дозы ацетилхолина, помимо гипотензивного эффекта, вызывают брадикардию и снижение проводимости атриовентрикулярного узла.
Прямое действие на сердце мускариновых стимуляторов вызывает следующие эффекты: 1) увеличение тока калия (1к(лх)) в клетках мышцы предсердия, а также в клетках синоатриального и атриовентрикулярного узлов; 2) снижение медленного внутреннего тока ионов кальция в кардиомиоцитах; и 3) уменьшение тока, вызванного гиперполяризацией (If), который лежит в основе диастолической деполяризации. Все эти эффекты участвуют в наблюдаемом замедлении частоты пейсмекера. Эффекты (1) и (2) вызывают гиперполяризацию и уменьшают сократимость предсердных клеток. Прямое замедление синоатриальной частоты и атриовентрикулярного проведения, вызываемое мускариновыми агонистами, часто протекает на фоне противодействия рефлекторной симпатической активации в ответ на снижение кровяного давления. Взаимоуравновешивание парасимпатических и симпатических эффектов является сложным процессом вследствие ранее описанной мускариновой модуляции симпатических влияний, которое выражается в виде угнетения выделения норадреналина и ослабления постсинаптических клеточных эффектов. Таким образом, итоговый эффект на частоту сердечных сокращений зависит от местных концентраций агониста в сердце и сосудах, а также от уровня рефлекторной реактивности.
Парасимпатическая иннервация в желудочках представлена хуже, чем в предсердиях, поэтому активация мускариновых рецепторов приводит к меньшему физиологическому эффекту в желудочках, чем в предсердиях. Однако во время симпатической стимуляции действие мускариновых агонистов на функцию желудочков четко выявляется в связи с мускариновой модуляцией симпатических
ТАБЛИЦА 7-3. Эффекты стимуляторов холинорецепторов прямого действия. Указаны только прямые эффекты; могут быть важны гомеостатические ответы на это прямое действие
Орган Реакция
Глаз Круговая мышца зрачка (сфинктер) Цилиарная мышца Сокращение (миоз) Сокращение (для близкого видения)
Сердце Синоатриальный узел Предсердия Снижение частоты (отрицательный хронотропный эффект) Снижение силы сокращений (отрицательный инотропный эффект). Уменьшение рефрактерного периода
Атриовентрикулярный узел Снижение скорости проведения (отрицательный дромотропный эффект)
Желудочки Увеличение рефрактерного периода. Небольшое снижение силы сокращений
Кровеносные сосуды Артерии Дилатация (посредством РФПЭ) Сокращение (в высоких дозах — прямой эффект)
Вены Дилатация (посредством РФПЭ) Сокращение (в высоких дозах — прямой эффект)
Легкие Бронхиальные мышцы Бронхиальные железы Сокращение (бронхоконстрикция) Стимуляция
Желудочно-кишечный тракт Моторика Сфинктеры Секреция Повышение Расслабление Стимуляция
Мочевой пузырь Детрузор Сфинктер Сокращение Расслабление
Железы Потовые, слюнные, слезные, носоглоточные Секреция
эффектов (“акцентуированный антагонизм”; Levy and Schwartz, 1993).
В организме, в целом, мускариновые агонисты вызывают заметную вазодилатацию. В ранних же исследованиях на препаратах изолированных гладких мышц сосудов часто демонстрировали сократительный ответ на эти вещества. В настоящее время известно (Furchgott et al., 1984), что вызванная ацетилхолином вазодилатация требует присутствия интактного эндотелия. Под действием мускариновых агонистов из эндотелиальных клеток выделяется расслабляющая субстанция (расслабляющий
фактор, продуцируемый эндотелием, или РФПЭ), вызывающая релаксацию гладких мышц. Изолированные сосуды с сохраненным эндотелием стойко воспроизводят вазодилататорный ответ, наблюдаемый в интактном организме. РФПЭ активирует гуанилатциклазу и увеличивает содержание цГМФ в гладких мышцах, что приводит к релаксации (рис. 12-2).
Сердечно-сосудистые эффекты всех холиновых эфиров сходны с эффектами ацетилхолина, главное различие состоит лишь в силе и продолжительности их действия. Из-за резистентности метахолина,
карбахола и бетанехола к действию ацетилхолин -эстеразы меньшие их дозы, введенные внутривенно, достаточны для получения эффектов, сходных с действием ацетилхолина, и продолжительность этих эффектов у синтетических холиновых эфиров больше. Сердечно-сосудистые эффекты большинства естественных холиномиметических алкалоидов и их синтетических аналогов, в основном, сходны с действием ацетилхолина.
Пилокарпин — интересное исключение из ряда холиновых эфиров. Введенный внутривенно (в эксперименте) он может вызывать гипертензию после короткого начального гипотензивного ответа. Долговременный гипертензивный ответ может быть следствием ганглионарной стимуляции, вызванной активацией мгрецепторов на постганглионарной клеточной мембране, что закрывает К+-ка-налы и вызывают медленные возбуждающие (деполяризующие) постсинаптические потенциалы. Этот эффект, подобно гипотензивному эффекту, может быть блокирован антимускариновым препаратом атропином.
3. Дыхательная система. Гладкие мышцы бронхиального дерева сокращаются мускариновыми стимуляторами. К тому же стимулируется секреция желез трахеобронхиальной слизистой. Эта комбинация эффектов может вызывать нежелательные симптомы, особенно у пациентов с астмой.
4. Желудочно-кишечный тракт. Введение мускариновых агонистов, как и стимуляция парасимпатической нервной системы, вызывает повышение секреторной и моторной активности кишечника. Сильно стимулируются слюнные и желудочные железы, менее выражен эффект на поджелудочную железу и малые кишечные железы. Перистальтическая активность повышается во всем кишечнике, и большинство сфинктеров расслабляется. Показано, что стимуляция сокращения в этой системе органов обусловлена деполяризацией клеточной мембраны гладких мышц и увеличенным входом ионов кальция в клетки.
5. Мочеполовая система. Мускариновые агонисты стимулируют мышцу детрузор и расслабляют сфинктер мочевого пузыря, тем самым способствуя отхождению мочи. Матка человека не проявляет заметной чувствительности к мускариновым агонистам.
6. Смешанные секреторные железы. Мускариновые агонисты стимулируют секреторную активность потовых, слезных и носоглоточных желез.
7. Центральная нервная система. Центральная нервная система содержит и мускариновые, и никотиновые рецепторы, причем головной мозг относительно богаче мускариночувствительными зонами, а в спинном мозгу преобладают никотиночувствительные. Физиологическая роль этих рецепторов обсуждается в главе 20. Несмотря на меньшее пропорциональное соотношение в головном мозгу никотиновых рецепторов к мускариновым, никотин и лобелии (рис. 7-3) оказывают выраженное влияние на ствол и кору мозга. Наиболее известный из этих эффектов — умеренное возбуждающее действие никотина, абсорбированного из вдыхаемого табачного дыма. В больших концентрациях никотин вызывает тремор, рвоту и стимуляцию дыхательного центра. В еще более высоких дозах никотин вызывает конвульсии, которые могут закончиться смертельной комой. Летальные эффекты, обусловленные воздействием на центральную нервную систему и факт, что никотин легко всасывается, явились основанием использования его в качестве инсектицида. Синтетический никотиновый стимулятор ди-метилфенилпиперазин (ДМФП) (рис. 7-3) относительно свободен от этих центральных эффектов, поскольку не проникает через гематоэнцефалический барьер.
8. Периферическая нервная система. Автономные ганглии являются важными местами синаптического действия никотина. Все никотиновые препараты, показанные на рис. 7-3, способны вызвать заметную активацию никотиновых рецепторов, проявляющуюся разрядами потенциалов действия в постганглионарных нейронах. Сам никотин отчасти имеет более высокое сродство к нейрональным рецепторам, чем к мышечным рецепторам скелетной мускулатуры. Он оказывает одинаковое действие и на парасимпатические, и на симпатические ганглии. Начальный ответ тем самым часто похож на множественную активацию парасимпатической и симпатической нервных систем. В отношении сердечно-сосудистой системы эффекты никотина, главным образом, симпатомиметические. При парентеральном введении никотина наблюдается злокачественная гипертензия; симпатическая тахикардия может сменяться вызываемой п. vagus брадикардией. В желудочно-кишечном и мочеполовом трактах эффекты, в основном, парасимпатомимети-ческие: обычно наблюдаются тошнота, рвота, диарея и мочеиспускание. Длительное воздействие может привести к деполяризационной блокаде.
Второй класс никотиночувствительных образований состоит из хеморецепторов на окончаниях чувствительных нервов — особенно в коронарных артериях и в каротидных и аортальном телах. Активация этих окончаний вызывает комплекс рефлексов продолговатого мозга, включающих нарушения дыхания и вагусную активацию.
9. Нервно-мышечные соединения. Никотиновые рецепторы на нервно-мышечных концевых пластинках сходны, но не идентичны рецепторам в автономных ганглиях (табл. 7-1). Оба типа отвечают на ацетилхолин и никотин. Однако, как обсуждается в главе 8, рецепторы отличаются по своим структурным требованиям к никотиновым блокирующим препаратам. Когда агонист никотина апплицируют непосредственно на концевые пластинки (ионофоретически или путем внутриартериального введения), возникает немедленная деполяризация, вызывающая увеличение проницаемости для ионов натрия и калия. Зависящий от синхронизации деполяризации концевых пластинок по всей мышце сократительный ответ будет изменяться от дезорганизованных подергиваний отдельных мышечных волокон к сильному сокращению всей мышцы. Деполяризующие никотиноподобные вещества, не подверженные быстрому гидролизу (как и сам никотин), вызывают быстрое развитие деполяризационной блокады. Блокада передачи продолжается, даже когда мембрана реполяризована (главы 8 и 26). В случае скелетных мышц этот блок проявляется как вялый паралич.
II. Базисная фармакология холиномиметиков непрямого действия
Действие ацетилхолина, выделяемого из автономных и соматических моторных нервов, завершается ферментативным разрушением его молекулы. Медиатор гидролизуется ферментом ацетилхоли-нэстеразой, белком с мол. м. около 320 000, который присутствует в высоких концентрациях в холинергических синапсах. Холиномиметики непрямого действия влияют главным образом на активные зоны этого фермента, хотя некоторые из них оказывают и прямое действие на никотиновые рецепторы. Наиболее частым является применение этих соединений в качестве инсектицидов, лишь не
многие имеют терапевтическое значение. Главные различия между веществами этой группы — химические и фармакокинетические, их фармакодинамические свойства почти идентичны.
Химия и фармакокинетика
А. Структура. Обычно ингибиторы холинэстеразы разделяются на три химические группы: 1) простые спирты, несущие четвертичную аммониевую группу, например эдрофоний; 2) эфиры карбаминовой кислоты со спиртами, имеющие четвертичную или третичную аммонийную группу (карбаматы, например неостигмин) и 3) органические производные фосфорной кислоты (фосфорорганические соединения, например изофлурофат). Представители первых двух групп показаны на рис. 7-4. Эдрофоний, неостигмин и амбеноний — это синтетические четвертичные аммонийные препараты, применяемые в медицине. Физостигмин (эзерин) — встречающийся в природе третичный амин с большей растворимостью в липидах, который тоже используется в медицине. Карбарил — типичный представитель большой группы карбаматов-инсектицидов с очень высокой растворимостью в липидах, что позволяет им быстро абсорбироваться в организме насекомых и достигать их центральной нервной системы.
Некоторые из исследованных 50 000 фосфорорганических соединений показаны на рис. 7-5. Многие из этих препаратов (исключение — эхотиофат) хорошо растворимые в липидах жидкости. Изофлурофат (диизопропилфторфосфат, ДФФ) — одно из первых хорошо изученных синтезированных фосфорорганических соединений. Он все еще доступен для применения в клинике, но заменяется препаратами, подобными эхотиофату. Производное тиохолина эхотиофат интересно тем, что сохраняет очень длительную продолжительность действия других фосфорорганических веществ и стабильно в водных растворах. Зоман — чрезвычайно сильный “нервный газ”. Тиофосфатные инсектициды паратион и малатион, как таковые, неактивны, но они способны превращаться в фосфопроизводные в организме животных и в растениях. Эти препараты более стабильны, чем соединения, подобные изо-флюрофату и зоману, что делает их более подходящими для использования в качестве инсектицидов.
Б. Всасывание, распределение и метаболизм. Всасывание четвертичных карбаматов из конъюнк-
О
II /СН3
Н3С — С — О — СН2 — СН2 — +N — сн3
ХСН3
Ацетилхолин
Неостигмин
НзС^
Н-^
Эдрофоний
Рис. 7-4. Ингибиторы холинэстеразы. Неостигмин представляет собой пример типичного соединения эфира карбаминовой кислоты [ 1 ] и фенола с четвертичной аммониевой группой [2]. Природный карбамат физостигмин является третичным амином. Для сравнения ацетилхолин показан в той же ориентации (кислота слева, спирт справа). Эдрофоний не является эфиром, но связывается с активным центром фермента
тивального мешка, с поверхности кожи и легких плохое, потому что постоянный заряд делает их относительно нерастворимыми в липидах. При пероральном применении требуются значительно большие дозы, чем при парентеральном введении. Проникновение в центральную нервную систему незначительно. Напротив, физостигмин хорошо всасывается из любых компартментов и может быть использован местно (глазные капли, мазь) (табл. 7-4). Он проникает в центральную нервную систему и более токсичен, чем четвертичные карбаматы. Однако даже неполярные карбаматы, используемые как инсектициды, плохо проникают через кожу — величина отношения летальных доз при чрезкожном и пероральном введении для этой группы значительно больше, чем для фосфорорганических пестицидов. Карбаматы относительно стабильны в водных растворах, но могут разрушаться неспецифическими эстеразами в организме так же, как холинэстеразой. Однако продолжительность их действия определяется главным образом стабиль
ностью комплекса ингибитор-фермент (подраздел “Механизм действия”), а не метаболизмом или выделением.
Фосфорорганические ингибиторы холинэстеразы (за исключением эхотиофата) хорошо всасываются с поверхности кожи, из кишечника, легких и конъюнктивы и поэтому опасны для человека и высоко эффективны как инсектициды. Они относительно менее стабильны, чем карбаматы, в водных растворах и быстро распадаются в окружающей, среде (по сравнению с другим большим классом инсектицидов, галогенированными углеводородами, например ДДТ). Эхотиофат высокополярен и более стабилен, чем большинство фосфорорганических соединений. В водных растворах, применяемых в офтальмологии, он сохраняет активность в течение недель.
Тиофосфатные инсектициды (паратион, малатион и родственные соединения) хорошо растворимы в липидах и быстро абсорбируются при всех путях введения. Они активируются в организме, превра-
О СН —|| Р
СН —
НзС^ Н3С-^ Н3С^ Н3С-^
Изофлурофат
Диизопропилфторфосфат
СНз СНз
I I О
Н3С —С —СН —О II 1
I p4-f
СНз НзС^ I
Зоман
Рис. 7-5. Структуры некоторых фосфорорганических ингибиторов холинэстеразы. Прерывистые линии указывают на связь, которая гидролизуется при соединении с ферментом. Затененные эфирные связи в структуре малатиона представляют собой участки детоксикации молекулы у млекопитающих и птиц
О
Н5С2- О || ; сн3
P-T-S — СН2 —СН2 —N^CH3
Н5С2- I ХСНз
Эхотиофат
S Н5С2 — О ||
Р —о
Н5С2-С<
NO2
Параоксон
Паратион
S НзС-О^||
Р —S —СН Н3С —I
СгН5
Малаоксон
Малатион
сн2-4м|-с2н5
щаясь в кислородные аналоги (рис. 7-5); этот про- неактивные продукты, у насекомых таких метабо-цесс происходит быстро и у насекомых, и у позво- лических путей нет, поэтому эти препараты можно ночных. Малатион и некоторые другие фосфор- рассматривать как безопасные для продажи насе-органические инсектициды в организме птиц и мле- лению. К сожалению, рыбы не способны метаболи-копитающих метаболизируются другими путями в зировать малатион, и множество рыб погибает от ТАБЛИЦА 7-4. Показания к применению и продолжительность действия ингибиторов холинэстеразы, используемых в терапии
Ингибиторы холинэстеразы Показания к применению Приблизительная продолжительность действия
Спирты Эдрофоний Карбаматы и родственные соединения Неостигмин Пиридостигмин Физостигмин Амбеноний Демекарий Миастения, кишечная непроходимость, аритмии Миастения, кишечная непроходимость Миастения Глаукома Миастения Глаукома 5-15 минут 0,5-2 часа 3-6 часов 0,5-2 часа 4-8 часов 4-6 часов
Фосфорорганические соединения Эхотиофат Глаукома 100 часов
обильного использования этого препарата вблизи водоемов. Паратион не детоксифицируется эффективно у позвоночных, поэтому он является значительно более опасным, чем малатион для человека и домашнего скота.
Все фосфорорганические соединения, за исключением эхотиофата, распределяются по всему организму, включая центральную нервную систему. Поэтому при отравлении этими веществами обнаруживаются признаки поражения центральной нервной системы.
Фармакодинамика
А. Механизм действия. Основная мишень этих препаратов — ацетилхолинэстераза, но ингибируется и бутирилхолинэстераза. Ацетилхолинэстераза — чрезвычайно активный фермент. Сначала ацетилхолин связывается с активным центром фермента и гидролизуется, образуя свободный холин и ацетилированный фермент. Затем гидролизуется аце-тилферментная ковалентная связь. Полный процесс длится приблизительно 150 микросекунд.
Все блокаторы холинэстеразы вызывают свой эффект, ингибируя ацетилхолинэстеразу и тем самым повышая концентрацию эндогенного ацетилхолина в районе холинорецептора. Однако молекулярные детали их взаимодействия с ферментом отличаются соответственно в трех химических подгруппах, упомянутых выше.
Первая группа, главным представителем которой является эдрофоний, содержит четвертичные спирты. Эти вещества обратимо связываются с активным центром, предотвращая таким образом контакт с ацетилхолином. Комплекс фермент-ингибитор не образует ковалентные связи и соответственно является короткоживущим (в пределах 2—10 минут). Вторая группа содержит карбаматные эфиры, например неостигмин и физостигмин. Эти препараты подвергаются двухэтапному гидролизу, аналогично описанному для ацетилхолина. Однако ковалентная связь карбамоилированного фермента, как полагают, более устойчива ко второму этапу, и он соответственно увеличивается (от 30 минут до 6 часов). Третья группа — фосфорорганические соединения. Эти препараты также подвергаются начальному связыванию и гидролизу ферментом, в результате чего фосфорилируется активный центр. Ковалентная фосфатно-ферментная связь чрезвычайно стабильна и гидролизуется в воде очень мед
ленно (сотни часов). После начального этапа связывания-гидролиза фосфорилированный ферментный комплекс может подвергаться процессу, называемому старение. Этот процесс очевидно включает разрушение одной из кислороднофосфатных связей ингибитора, что приводит к дальнейшему укреплению фосфатно-ферментного комплекса. Скорость старения различается у отдельных фосфорорганических соединений. Сильные нуклеофилы, подобные пралидоксиму, способны разрушить фосфорно-ферментную связь, если их использовать до начала процесса старения, и они могут использоваться как “реактиваторы холинэстеразы” при отравлениях фосфорорганическими инсектицидами (глава 8). Как только старение произошло, фер-мент-ингибиторный комплекс становится более стабильным и труднее поддающимся расщеплению даже при использовании оксимных соединений.
Из-за заметных различий в продолжительности действия фосфорорганические ингибиторы иногда называют “необратимыми” ингибиторами холинэстеразы, а эдрофоний и карбаматы рассматривают как “обратимые” ингибиторы. Фактически молекулярные механизмы действия не согласуются с таким упрощенным описанием, однако эти термины часто используются.
Б. Действие на системы и органы. Наиболее известно фармакологическое действие ингибиторов холинэстеразы на сердечно-сосудистую систему, желудочно-кишечный тракт, глаз и нервно-мышечные соединения скелетных мышц. Поскольку основное их действие связано с усилением действия эндогенного ацетилхолина, их эффекты сходны (но не всегда идентичны) с эффектами холиномимети-ков прямого действия.
1. Центральная нервная система. В низких концентрациях растворимые в липидах ингибиторы холинэстеразы вызывают диффузное повышение активности ЭЭГ и субъективно тревожное состояние. В высоких концентрациях они вызывают генерализованные судороги, за которыми могут следовать кома и остановка дыхания.
2. Сердечно-сосудистая система. Ингибиторы холинэстеразы могут увеличивать активность как парасимпатических, так и симпатических ганглиев, и влиять на ацетилхолиновые рецепторы нейроэф-фекторных клеток (сердечной мышцы и гладких мышц сосудов).
В сердце доминируют эффекты парасимпатической системы. Поэтому введение ингибиторов
холинэстеразы, таких какэдрофоний, физостигмин или неостигмин, приводит к эффектам, имитирующим активацию n. vagus. Частота сердечных сокращений уменьшается, атриовентрикулярная проводимость снижается, уменьшается сократимость предсердий и падает сердечный выброс. Понижение сердечного выброса связывают с брадикардией, сниженной сократимостью предсердий и некоторым уменьшением сократимости желудочков. Последний эффект является результатом пресинап-тической модуляции симпатической активности (торможение выделения норадреналина), так же, как и угнетение постсинаптических клеточных симпатических эффектов.
Эффекты ингибиторов холинэстеразы на гладкие мышцы сосудов и кровяное давление менее заметны, чем эффекты мускариновых агонистов прямого действия. Дело в том, что опосредованно действующие препараты могут изменять только тонус сосудов, иннервированных холинергическими нервами, а итоговый эффект на сосудистый тонус отражает активацию как парасимпатической, так и симпатической нервных систем. Поскольку немногие сосуды получают холинергическую иннервацию, холиномиметический эффект ингибиторов холинэстеразы на гладкие мышцы эффекторной ткани минимален. Активация симпатических ганглиев вызывает тенденцию к повышению сосудистого сопротивления.
Таким образом, итоговый сердечно-сосудистый эффект невысоких доз ингибиторов холинэстеразы состоит в умеренной брадикардии, уменьшении сердечного выброса и отсутствии изменения или умеренном снижении кровяного давления. Большие (токсические) дозы этих препаратов вызывают выраженные брадикардию и гипотензию.
4. Нервно-мышечные соединения. Ингибиторы холинэстеразы имеют важные терапевтические и токсические эффекты на нервно-мышечные соединения скелетных мышц. Низкие (терапевтические) концентрации незначительно пролонгируют и усиливают действие физиологически выделяемого ацетилхолина. Это проявляется в увеличении силы сокращения, особенно в мышцах, ослабленных кура-реподобными миорелаксантами, или при миастении. В более высоких концентрациях накопление ацетилхолина может вызвать фибрилляции мышечных волокон. Может также наблюдаться антидромная импульсация моторных нейронов, вызывающая подергивания целых моторных единиц. При отчет
ливом угнетении ацетилхолинэстеразы мембранная деполяризация становится длительной, в результате может произойти деполяризующая нервно-мышечная блокада.
Некоторые четвертичные карбаматные ингибиторы холинэстеразы, например неостигмин, имеют дополнительный прямой никотиновый агонистический эффект на нервно-мышечное соединение. Это может быть частичным объяснением эффективности этих препаратов при лечении миастении.
III. Клиническая фармакология холиномиметиков
Холиномиметики, в основном, применяют для лечения глазных болезней (глаукома), заболеваний желудочно-кишечного тракта и мочевыводящих путей (послеоперационная атония, нейрогенный мочевой пузырь), болезней нервно-мышечных соединений (миастения гравис, нервно-мышечный паралич, вызванный аналогами кураре) и сердца (некоторые типы предсердных аритмий). Ингибиторы холинэстеразы иногда применяются при передозировке атропина. Такрин — новый препарат с антихолинэстеразным и другим холиномиметическим действием применяется для лечения болезни Альцгеймера (глава 62).
Клиническое использование
А. Глаз. Глаукома — это заболевание, характеризующееся повышением внутриглазного давления. Существуют два типа приобретенной глаукомы: первичная, которая может быть подразделена на закрытоугольный и открытоугольный типы, и вторичная, например возникшая в результате травмы, воспаления или хирургических процедур. Мускариновые стимуляторы и ингибиторы холинэстеразы могут понижать внутриглазное давление, облегчая отток водянистой влаги и, возможно, уменьшая скорость ее секреции (рис. 6-9). Из прямых агонистов для лечения глаукомы используются метахолин, карбахол и пилокарпин. Среди ингибиторов холинэстеразы изучалась возможность применения с этой целью физостигмина, демекария, эхотиофа-та и ДФФ. Наиболее часто применяется пилокарпин.
Острая закрытоугольная глаукома — это неотложное состояние, которое обычно сначала купируется лекарственными средствами, но в последующем часто требует хирургического вмешательства для постоянной коррекции. Начальная терапия состоит в назначении прямых мускариновых агонистов и ингибиторов холинэстеразы (например, пилокарпин плюс физостигмин). Как только внутриглазное давление приходит к норме, опасность потери зрения уменьшается, и пациент может быть подготовлен для корригирующей операции (иридэктомии). Открытоугольная глаукома и некоторые случаи вторичной глаукомы являются хроническими заболеваниями, которые не поддаются хирургической коррекции. Терапия поэтому длительная и основывается прежде всего на использовании пара-симпатомиметиков, адреналина, препаратов, блокирующих P-адренорецепторы, и ацетазоламида. Из парасимпатомиметиков в настоящее время наиболее часто применяются пилокарпин (глазные капли или интраконьъюнктивальная пролонгированная форма) и карбахол (капли). Длительнодействующие препараты (демекарий, эхотиофат) используются в тех случаях, когда внутриглазное давление не нормализуется обычными средствами. Другое лечение глаукомы описано в дополнении (“Лечение глаукомы”, глава 10).
Аккомодационная эзотропия (страбизм, вызываемый гиперметропической аккомодационной ошибкой) иногда диагностируется у маленьких детей и лечится холиномиметическими агонистами. Дозирование сходно с таковым при глаукоме, но применяются и более высокие дозы.
Б. Желудочно-кишечный тракт и мочевыводящая система. При заболеваниях, сопровождающихся угнетением активности гладкой мускулатуры без непроходимости, могут использоваться холиномиметики с прямым или непрямым мускариновым эффектом. К этим нарушениям относятся послеоперационный илеус (атония и паралич желудка или кишечника после хирургического вмешательства) и врожденный мегаколон. Задержка мочи может быть послеоперационной, а также вторичной при повреждениях спинного мозга или при заболеваниях (неврогенный мочевой пузырь). Холиномиметики иногда используются для повышения тонуса нижнего пищеводного сфинктера у пациентов с рефлюкс-эзофагитом. Из холиновых эфиров наиболее широко используется при этих нарушениях бетанехол. Для лечения желудочно-кишечных на
рушений его обычно применяют перорально в дозе 10-25 мг 3-4 раза в день. Пациентам с задержкой мочи бетанехол может вводиться подкожно в дозе 5 мг с повторением при необходимости через 30 минут. Из ингибиторов холинэстеразы при этих показаниях наиболее часто используется неостигмин. Для лечения паралитического илеуса или атонии мочевого пузыря неостигмин назначается подкожно в дозе 0.5-1 мг. Если пациенты способны принимать препарат перорально, неостигмин применяется в дозе 15 мг. Во всех этих ситуациях перед назначением холиномиметиков врач должен быть уверен в отсутствии механической непроходимости. В противном случае лекарство может усугубить течение заболевания и даже привести к перфорации в результате повышенного давления.
В. Нервно-мышечные соединения. Миастения гравис — это заболевание, поражающее нервно-мышечные соединения скелетных мышц. Аутоиммунные процессы вызывают выработку антител, снижающих число функциональных никотиновых рецепторов на постсинаптической концевой пластинке. Характерные симптомы слабости и усталости, которые уменьшаются в период покоя и усугубляются при физической нагрузке, могут захватывать любые скелетные мышцы, но наиболее часто вовлекают малые мышцы головы, шеи и конечностей. Частыми проявлениями бывают птоз, диплопия, затруднения речи и глотания, и слабость конечностей. Тяжелое заболевание может поражать все мышцы, включая дыхательную мускулатуру. Эта болезнь имеет сходство с нервно-мышечным параличом, вызываемым d-тубокурарином и похожими недеполяризующими нервно-мышечными блокирующими препаратами (глава 26). Пациенты с миастенией чрезвычайно чувствительны к действию курареподобных средств и других препаратов, вмешивающихся в нервно-мышечную передачу, например аминогликозидных антибиотиков.
Ингибиторы холинэстеразы, но не агонисты ацетилхолиновых рецепторов прямого действия, имеют важное значение для терапии миастении. У некоторых пациентов положительный эффект оказывает иммунодепрессантная терапия, в частности адренокортикостероидами и циклофосфамидом.
Эдрофоний иногда используется для диагностического теста на миастению. После исходного измерения мышечной силы внутривенно вводится 2 мг препарата. Если реакции не наблюдается через 45 секунд, может быть добавочно введено 8 мг
эндрофония. Некоторые врачи разделяют дозу в 8 мг на две дозы 3 и 5 мг, вводимые с 45-секундны-ми интервалами. Если у пациента миастения гравис, обычно наблюдается увеличение мышечной силы, которое сохраняется около 5 минут.
Эдрофоний также применяется для оценки адекватности лечения длительнодействующими ингибиторами холинэстеразы у пациентов с диагносци-рованной миастенией гравис. Если используется большое количество ингибитора холинэстеразы, у больных может развиться парадоксальная слабость вследствие никотиновой деполяризационной блокады моторных концевых пластинок. Подобные состояния сопровождаются симптомами чрезмерной стимуляции мускариновых рецепторов (абдоминальные спазмы, диарея, повышенная саливация, увеличенная бронхиальная секреция, миоз, брадикардия). Малые дозы эдрофония (1-2 мг внутривенно) при передозировке ингибиторов холинэстеразы не вызовут облегчения или даже усилят слабость. С другой стороны, если состояние пациента улучшается после эдрофония, то доза ингибиторов холинэстеразы может быть увеличена. Дифференциальная диагностика выраженной миастении (миастенический кризис) от последствий лекарственной терапии (холинергический кризис), как правило, необходима для тяжелых больных и должна проводиться в условиях стационара с соответствующей аппаратной и медикаментозной поддержкой (например, аппаратами искусственной вентиляции легких).
Длительное лечение миастении гравис обычно включает применение неостигмина, пиридостигми-на или амбенония. Дозы подбирают до оптимального уровня, основываясь на изменении мышечной силы. Это препараты короткого действия и поэтому требуют частого применения (каждые 2-4 часа для неостигмина и каждые 3-6 часов для пиридо-стигмина и амбенония; табл. 7-4). Имеются лекарственные формы пролонгированного действия, но они могут использоваться только перед сном. Длительнодействующие ингибиторы холинэстеразы, такие как фосфорорганические соединения, не применяются, поскольку доза лекарства при этом заболевании может изменяться быстро, что не позволяет регулировать порядок применения этих средств.
Если при использовании ингибиторов холинэстеразы проявляются мускариновые эффекты, их можно нивелировать введением антимускариновых лекарств, таких как атропин. Однако к подобным
эффектам препаратов может развиваться толерантность, так что введение атропина часто и не требуется.
Нередко хирургическую анестезию дополняют нервно-мышечной блокадой с применением недеполяризующих нервно-мышечных релаксантов, таких как кураре, панкурониум и более новых препаратов. После хирургического вмешательства желательно быстро купировать подобный фармакологический паралич. Этого можно легко достигнуть с помощью ингибиторов холинэстеразы: неостигмин и эдрофоний являются препаратами выбора. Для ускорения эффекта их вводят внутривенно или внутримышечно.
Г. Сердце. Короткодействующий ингибитор холинэстеразы эдрофоний в прошлом использовался для лечения суправентрикулярных тахиаритмий, особенно пароксизмальной суправентрикулярной тахикардии. При потенцировании эффектов эндогенного ацетилхолина, выделяемого из нервных окончаний п. vagus в атриовентрикулярном узле, скорость атриовентрикулярной передачи уменьшается, и аномальная суправентрикулярная тахиаритмия превращается в нормальный синусовый ритм. В настоящее время эдрофоний заменен более новыми препаратами (аденозин и блокаторы кальциевых каналов).
Д. Интоксикация антимускариновыми препаратами. Интоксикация атропином потенциально детальна для детей (глава 8) и может вызвать длительные тяжелые поведенческие нарушения у взрослых. Трициклические антидепрессанты при передозировке (часто с суицидальными намерениями) тоже вызывают выраженную мускариновую блокаду (глава 29). Поскольку блокада мускариновых рецепторов, вызываемая этими агентами, является конкурентной по своей природе, она может быть преодолена увеличением количества эндогенного ацетилхолина в нейроэффекторном соединении. Теоретически для снятия блокады могут быть использованы ингибиторы холинэстеразы. По этому показанию используют физостигмин, так как он проникает в центральную нервную систему и уменьшает как центральные, так и периферические симптомы мускариновой блокады. Однако физостигмин сам по себе может быть опасен для центральной нервной системы, и поэтому такая терапия, применяется только у пациентов с опасным повышением температуры тела или с выраженной суправентрикулярной тахикардией.
Токсичность
Токсичность стимуляторов холинорецепторов зависит от степени их всасывания, проникновения в центральную нервную систему и метаболизма.
А. Мускариновые стимуляторы прямого действия. При передозировке пилокарпин и холиновые эфиры вызывают предсказуемые признаки чрезмерного мускаринового возбуждения. К ним относятся тошнота, рвота, диарея, саливация, потливость, кожная вазодилатация и бронхиальная констрик-ция. Все эти эффекты конкурентно блокируются атропином и родственными ему препаратами.
Определенные грибы, особенно из рода Inocybe, содержат мускариновые алкалоиды. Потребление этих грибов в пищу вызывает типичные признаки мускариновой активации длительностью 15-30 минут. Лечат эти состояния атропином, 1-2 мг парентерально. {Amanita muscaria, первый источник получения мускарина, содержит очень малую концентрацию этого алкалоида.)
Б. Никотиновые стимуляторы прямого действия. Никотин сам по себе является основной причиной этого типа отравлений. Проявления острой токсичности алкалоида хорошо известны, но значительно менее важны, чем хронический эффект, ассоциированный с курением. Никотин также применяется во многих инсектицидах.
1. Острая токсичность. Летальная доза никотина для человека приблизительно равна 40 мг, или 1 капле чистой жидкости. Это количество никотина содержится в двух обычных сигаретах. При горении большая часть никотина в сигаретах разрушается или улетучивается с “боковыми потоками” дыма. Попавшие в пищеварительный тракт младенцев или других детей никотиновые инсектициды и табак вызывают рвоту, ограничивающую количество всасывающегося алкалоида.
Токсические эффекты больших доз никотина являются как бы расширением эффектов, описанных ранее. Наиболее опасны: 1) центральное стимулирующее действие, которое вызывает судороги и может привести к коме и остановке дыхания; 2) деполяризация концевых пластинок скелетных мышц, которая может привести к деполяризационной блокаде и параличу дыхания; 3) гипертензия и сердечные аритмии.
Лечение острого отравления никотином является главным образом симптоматическим. Мускариновая активация, возникающая вследствие стиму
ляции парасимпатических ганглиев, может быть купирована атропином. Для лечения центральной стимуляции обычно применяются противосудорожные препараты, такие как диазепам. Нервно-мышечная блокада нечувствительна к фармакологическому лечению, и может потребоваться искусственная вентиляция легких.
Никотин метаболизируется и выделяется относительно быстро. Пациенты, пережившие первые четыре часа после острого отравления, обычно полностью выздоравливают, если не было гипоксии и повреждения мозга.
2. Хроническая токсичность никотина. Цена курения табака для здоровья курильщиков и его социальная значимость для всего населения пока осмыслены не полностью. Однако в сообщении главного хирурга США за 1979 г. о распространении здорового образа жизни и предупреждении заболеваний утверждалось, что “курение сигарет — это определенно важнейшая и единственная причина болезней и преждевременной смерти в США, которую можно предотвратить”. Это положение было подтверждено в докладах Департамента здравоохранения США за 1983 и 1985 гг., которые установили, что до 30 % из более чем 500 000 ежегодных смертей от коронарной болезни сердца и 30 % от более чем 400 000 ежегодных смертей от рака в США были связаны с курением. К сожалению, тот факт, что наиболее важные заболевания, связанные с курением, развиваются медленно, уменьшает здоровое побуждение прекратить курение.
Неизвестно, в какой мере сам никотин участвует в продуцировании хорошо описанных нежелательных эффектов хронического использования табака. Полагают, что высока вероятность повышенного риска сосудистых заболеваний и внезапной коронарной смерти, ассоциированной с курением. Вероятно также, что никотин провоцирует учащение рецидивов у курильщиков с пептической язвенной болезнью.
В. Ингибиторы холинэстеразы. Острые токсические эффекты ингибиторов холинэстеразы, так же как и препаратов прямого действия, являются продолжением их фармакологического действия. Основной источник такой интоксикации — использование пестицидов в сельском хозяйстве и в быту. Приблизительно 100 фосфорорганических соединений и 20 карбаматных ингибиторов холинэстеразы используются в США как пестициды и как ветеринарные антигельминтные средства.
Острая интоксикация у пациентов с тяжелым поражением должна быть быстро распознана и начато своевременное лечение. Доминирующие начальные признаки отравления те же, что и при мускариновой стимуляции: миоз, саливация, потливость, спазм бронхов, рвота и диарея. Обычно быстро следует вовлечение центральной нервной системы, сопровождаемое периферическими никотиновыми эффектами, особенно деполяризующей нервно-мышечной блокадой. Терапия всегда включает: 1) поддержание жизненных функций (особенно дыхания); 2) предотвращение дальнейшего всасывания (промывание кожи и слизистых); 3) применение больших доз атропина парентерально под контролем признаков мускариновой стимуляции. Терапия часто включает также лечение пралидок-симом, как описано в главе 8.
Хроническое воздействие определенных фосфорорганических соединений, включающих некоторые фосфорорганические ингибиторы холинэстеразы, вызывает нейропатию, ассоциированную с демиелинизацией волокон аксонов. Триортокрезилфосфат, который добавляют в машинные масла, является прототипным веществом данного класса соединений. Демиелинизация нервных волокон не является следствием угнетения холинэстеразы.
Препараты
Холиномиметики прямого действия
Ацетилхолин (Миохол)
Глазные капли: 1: 100 (10 мг/мл) раствор
Бетанехол (генерик, Урехолин)
Перорально: таблетки по 5,10,25,50 мг
Парентерально: 5 мг/мл для п/к инъекций
Карбахол
Глазные капли (местно, Изопто Карбахол): 0.75,1.5, 2.25,3 % капли
Глазные капли (внутриглазные, Миостат): раствор 0.01 %
Пилокарпин (генерик, Изопто Карпин) Глазные капли (местно): 0.25,0.5,1, 2,3,4, 6, 8,10 % растворы, 4 % гель
Глазные капли для пролонгированного действия (Окусерт Ппло-20, Окусерт Пило-40): выделение 20 и 40 мкг пилокарпина в час в течение одной недели соответственно
Ингибиторы холинэстеразы
Амбеноний (Мителаза)
Перорально: таблетки по 10 мг
Демекарий (Гуморсол)
Глазные капли: 0.125, 0.25 %
Эхотиофат (Фосфолин)
Глазные капли: порошок для разведения до 0.03,0.06, 0.125,0.25 % растворов
Эдрофоний (генерик, Тензилон)
Парентерально: 10 мг/мл для в/в и в/м инъекций
Изофлурофат (Флороприл)
Глазная мазь: 0.025 %
Неостигмин (генерик, Простигмин)
Перорально: таблетки по 15 мг Парентерально: 1:1000 в 10 мл; 1:2000,1:4000 в 1мл
Физостигмин, эзерин (генерик)
Глазные капли: 0.25 % мазь, 0.25,0.5 % капли Парентерально: 1 мг/мл для в/м или в/в инъекций
Пиридостигмин (Местинон)
Перорально: таблетки по 60 мг; таблетки пролонгированного действия по 180 мг; сироп 15 мг/мл
Парентерально: 5 мг/мл для в/м или медленных в/в инъекций
Избранная литература
Abou-Donia М. В., Lapadula D. М. Mechanisms of organophosphorus ester-induced delayed neurotoxicity: Type I and type II. Annu. Rev. Pharmacol. 1990; 30:405.
Aquilonius S-М., Hartvig P. Clinical pharmacology of cholinesterase inhibitors. Clin. Pharmacokinet. 1986; 11: 236.
Barnes P. J., Minette P., Maclagan J. Muscarinic receptor subtypes in airways. Trends Pharm. Sci. 1988; 9:412.
Berridge M. J., Irvine R. F. Inositol phosphates and cell signalling. Nature, 1989; 341:197.
Bonner T. I. et al. Identification of a family of muscarinic acetylcholine receptor genes. Science, 1987; 237: 527.
Средства, блокирующие холинорецепторы
8
Бертрам Г. Катцунг
Так же, как холинергические агонисты подразделяются на мускариновую и никотиновую подгруппы на основании их аффинитета к специфическим рецепторам, так и антагонисты, действующие на эти рецепторы, делятся на две большие группы: антимускариновые и антиникотиновые средства. К средствам, блокирующим никотиновые рецепторы, относятся ганглиоблокаторы и миорелаксанты. Ганглиоблокирующие препараты имеют очень ограниченное применение и обсуждаются в конце этой главы. Миорелаксанты обсуждаются в главе 26. В данной главе основное внимание уделено лекарственным средствам, блокирующим мускариновые холинорецепторы.
Как обсуждалось в главах 6 и 7, выявлено несколько подтипов мускариновых рецепторов, главным образом на основании результатов экспериментов с использованием лигандного связывания и клонирования кДНК. В настоящее время становится общепринятой стандартная терминология для этих подтипов рецепторов, и имеются свидетельства различий между тремя подтипами, полученные с применением селективных агонистов и антагонистов.
Как отмечалось в главе 1, м,-тип рецепторов расположен в нейронах центральной нервной системы, симпатических постганглионарных телах клеток и во многих пресинаптических участках. м3-рецепто-ры наиболее распространены на мембранах эффекторных клеток, особенно железистых и гладкомышечных. м2-рецепторы расположены в миокарде, гладкомышечных органах и в некоторых нейрональных участках.
I. Базисная фармакология средств, блокирующих мускариновые рецепторы
Эффекты парасимпатической автономной системы могут быть блокированы мускариновыми антагонистами, поэтому их часто называют парасим-патолитическими веществами. Однако они не “лизируют” парасимпатические нервы и имеют некоторые эффекты, которые не объяснить блокадой парасимпатической нервной системы. По этим причинам более предпочтительным является термин “антимускариновые" препараты. Известны природные вещества, обладающие антимускариновым действием и используемые в течение тысячелетий как лекарства, яды и косметические препараты. Прототип таких веществ — атропин. К настоящему времени известно много сходных растительных алкалоидов и изготовлены сотни синтетических анти-мускариновых соединений.
Химия и фармакокинетика
А. Источники получения и химия. Атропин и родственные ему соединения являются третичными аммонийными алкалоидными эфирами тропо-вой кислоты (рис. 8-1).
Атропин (гиосциамин) обнаружен в различных растениях: красавке (Atropa belladonna), или сонной одури, и в разных видах дурмана {Datura stramoni-
Рис. 8-1. Структура атропина (кислород в положении [1] пропущен) или скополамина (кислород присутствует). В гоматропине гидроксиметил в положении [2] замещен гидроксильной группой, кислород в положении [1] отсутствует
ипг), известных под названиями Jamestown weed или Thornapple. Скополамин (гиосцин) содержится в Hyoscyatnus niger, или в белене. Скополамин в растениях встречается в виде /(-)-стереоизомера. Атропин в природе содержится в виде левовращающего изомера гиосциамина, который однако легко рацемизируется, так что коммерческое вещество является рацемическим d,/-гиосциамином. Левовращающие изомеры обоих алкалоидов по меньшей мере в 100 раз сильнее, чем правовращающие.
Полусинтетические третичные аммонийные аналоги могут быть получены этерификацией природного основания, например основания атропина — тропина с разными кислотами. Таким образом, гоматропин — это тропиновый эфир миндальной кислоты. Антимускариновыми эффектами обладают многие полностью синтетические соединения.
Представители третичных аминов (рис. 8-2) часто применяются для воздействия на глаз или на центральную нервную систему. Многие антигистаминные (глава 16), антипсихотические препараты (глава 28) и антидепрессанты (глава 29) имеют сходные структуры и, как можно предсказать, ан-тимускариновые эффекты.
Четвертичные аммонийные антимускариновые вещества проявляют более выраженные периферические эффекты и меньшее влияние на центральную нервную систему. К этим препаратам относятся как полусинтетические, так и синтетические соединения (рис. 8-2).
Б. Абсорбция. Естественные алкалоиды и большинство третичных аммонийных антимускарино-вых препаратов хорошо всасываются в кишечнике
и через конъюнктивальную мембрану. Некоторые препараты, применяемые в соответствующих лекарственных формах, например скополамин, могут всасываться через кожу (трансдермальный путь). В отличие от них только 10-30 % дозы четвертичных аммонийных антимускариновых препаратов всасывается при энтеральном введении, что отражает сниженную липофильность заряженной молекулы.
В. Распределение. Атропин и другие третичные амины широко распределяются после всасывания. Значительный уровень в центральной нервной системе достигается в пределах от 30 минут до часа, и это может потребовать уменьшения дозы препарата, если он используется для действия на периферии. Скополамин особенно быстро и хорошо достигает центральной нервной системы и оказывает более выраженные центральные эффекты, чем большинство других антимускариновых препаратов. В отличие от него производные четвертичных аминов слабо проникают в мозг и поэтому в малых дозах практически не проявляют центральных эффектов.
Г. Метаболизм и экскреция. Атропин после введения быстро исчезает из крови с периодом полувыведения 2 часа. Около 60 % дозы выводится с мочой в неизмененном виде. Оставшаяся часть почти полностью выводится с мочой в виде продуктов гидролиза и конъюгации. Действие препарата на парасимпатические функции быстро снижается во всех органах, исключая глаз. Эффекты на радужку и цилиарную мышцу продолжаются 72 часа или дольше.
Определенные виды животных, например кролики, имеют специфические ферменты (атропин-эстеразы), которые обеспечивают почти полную защиту от токсических эффектов атропина, быстро метаболизируя препарат.
Фармакодинамика
А. Механизм действия. Атропин вызывает обратимую блокаду мускариновых рецепторов, т. е. блокада малыми дозами атропина может быть снята большими концентрациями ацетилхолина или эквивалентных мускариновых агонистов. Это предполагает конкуренцию за общие точки связывания. Блокада мускариновых рецепторов предотвращает возникновение эффектов, описанных в главе 7, таких как образование ИТФ и ингибирование адени-
Четвертичные амины для лечения заболеваний желудочно-кишечного тракта (язвенная болезнь, гипермоторика)
Третичные амины для периферического применения
Тропикамид (мидриатик, циклоплегик)
Третичный амин, используемый при болезни Паркинсона
Четвертичный амин, используемый при астме
Бензтропин
Ипратропиум
Рис. 8-2. Структуры некоторых полусинтетических и синтетических антимускариновых препаратов
латциклазы, которые вызываются ацетилхолином и другими мускариновыми агонистами.
Эффективность антимускариновых препаратов изменяется в зависимости от локализации органа, клетки-мишени и источника агониста. Наиболее чувствительными к атропину являются слюнные, бронхиальные и потовые железы. Значительно менее чувствительны секреторные париетальные клетки желудка. Промежуточной чувствительностью обладают автономные эффекторы гладких мышц и сердца. В большинстве тканей антимускариновые вещества активнее блокируют эффекты экзогенно вводимых агонистов холинорецепторов, чем эндогенного ацетилхолина.
Атропин имеет высокую селективность к мускариновым рецепторам, сила его влияния на никотиновые рецепторы значительно меньше; при клиническом использовании его действие на немускариновые рецепторы, как правило, не обнаруживается.
Атропин одинаково влияет нам,-, м2-> м3-подти-пы мускариновых рецепторов. В отличие от него
некоторые другие антимускариновые вещества обладают умеренной избирательностью к разным подтипам этих рецепторов (табл. 8-1). Большинство синтетических антимускариновых препаратов значительно менее специфичны, чем атропин, в отношении немускариновых рецепторов. Например, некоторые четвертичные аммонийные антимускариновые вещества обладают значительным ганглиоблокирующим действием, а другие — выраженным гистаминоблокирующим эффектом. Антимускариновые эффекты характерны и для некоторых лекарств других групп, например антигистаминных и антидепрессивных препаратов. Их относительная селективность к разным подтипам мускариновых рецепторов не установлена.
Б, Влияние иа системы и органы.
1. Центральная нервная система. В обычно используемых в клинике дозах атропин оказывает умеренное стимулирующее действие на центральную нервную систему, особенно на парасимпатические медуллярные центры, и отсроченный, но длительный седативный эффект. Центрального вагус
ТАБЛ ИЦА 8-1. Подтипы мускариновых рецепторов и их антагонисты (акро'нимами обозначены селективные
антагонисты, использующиеся только в исследовательских работах)
Свойства Подтип м, Подтип м2 Подтип м3
Основная локализация Доминантная эффекторная система Антагонисты Нервы Сердце, нервы, гладкая Железы, гладкая мускула- мускулатура тура, эндотелий Т ИТФ, ДАГ 4- цАМФ, Т тока через ка- Т ИТФ, ДАГ лиевые каналы Пирензепин, телензепин, Галламин’, метоктрамин, 4-DAMP, HHSD дицикломин 2, тригекси- AF-DX 116 фенидил3
Приблизительная константа диссоциации4 Атропин 111 Пирензепин 10 50 200 AF-DX 800 100 3000 HHSD 40 200 2
1 В клинике используется как нервно-мышечный блокатор.
2 В клинике используется как кишечный спазмолитик.
3 В клинике используется для лечения болезни Паркинсона.
4 Относительно атропина. Меньшая величина показателя указывает на более высокий аффинитет.
Обозначения:
AF-DX 116 = 11-({2-[(диэтиламино)метил]-1-пиперидинил) ацетил)-5,11-дигидро-6Н-пиридо-[2,3-Ь](1,4) бензодиазепин-6-он
ДАГ = диацилглицерол
4-DAMP = 4-дифенилацетокси-М-метилпиперидин
HHSD = гексагидросиладифенидол
ного стимулирующего действия часто достаточно для возникновения брадикардии, которая затем сменяется тахикардией в результате антимускариновых эффектов препарата на синусовый предсердный узел. Скополамин имеет более выраженные центральные эффекты, вызывая сонливость при назначении в обычных дозах, а у некоторых чувствительных пациентов — амнезию. В токсических дозах атропин и скополамин вызывают возбуждение, ажитацию, галлюцинации и коматозное состояние.
Центрально действующие антимускариновые препараты устраняют тремор при болезни Паркинсона. Атропин в виде экстракта белладонны был одним из первых препаратов, использованных в лечении этого заболевания. Как обсуждается в главе 27, тремор и ригидность при болезни Паркинсона являются результатом относительного избытка холинергической активности из-за дефицита дофаминергической активности в базальных ганглиях стриатной системы. Таким образом, сочетание антимускариновых веществ с препаратами предшественников дофамина (леводопа) может обеспечить более эффективное лечебное воздействие, чем раздельное применение препаратов этих групп.
Мускариновая холинергическая передача, как полагают, имеет отношение к вестибулярным нарушениям, особенно проявляющимся в движении. Скополамин часто эффективен в предотвращении или устранении этих нарушений.
2. Глаз. Сокращение сфинктера зрачка обусловлено мускариновой холинергической активацией (рис. 6-9). Она эффективно прерывается при местном применении атропина и других третичных аммонийных антимускариновых препаратов, что ведет к возникновению перевеса симпатической ди-лататорной активности и мидриазу (рис. 8-3). Расширенный зрачок был модным в период Ренессанса, что дало название растению и его активному экстракту, использовавшемуся в качестве глазных капель — белладонна (“прекрасная дама”, итал.).
Вторым важным глазным эффектом антимускариновых препаратов является паралич цилиарной мышцы, или циклоплегия. Результат циклопле-гии — потеря способности к аккомодации; полностью атропинизированный глаз не может сфокусироваться на близкой точке видения (рис. 8-3).
Эффекты мидриаза и циклоплегии используются в офтальмологии, однако они потенциально опасны, так как у пациентов с узким углом передней
Рис. 8-3. Влияние скополамина на диаметр зрачка (мм) и аккомодацию (диоптрии) в нормальном глазу человека. Одна капля 0.5 % раствора скополамина была введена в конъюнктивальный мешок во время, обозначенное 0, а вторая — на 30 минуте. Усреднены 42 наблюдения. Отметьте крайне медленное восстановление. (Marron J, Cycloplegia and mydriasis by use of atropine, scopolamine, and homatropine-paredrine. Arch. Ophthal., 1940; 23:340.)
камеры глаза может быть спровоцирована острая глаукома.
Третий глазной эффект антимускариновых препаратов — уменьшение секреции слезной жидкости. Когда пациенты получают большие дозы антимускариновых лекарственных средств, они иногда жалуются на сухость или ощущение “песка” в глазах.
3. Сердечно-сосудистая система. Предсердия богато иннервированы парасимпатическими (вагусными) нервными окончаниями, поэтому синусовый предсердный узел чувствителен к блокаде мускариновых рецепторов. Эффект этих препаратов на изолированном сердце с сохраненной иннервацией проявляется в четкой блокаде вагусного замедления и относительной тахикардии. Выраженная тахикардия наблюдается у пациентов при назначении умеренных и высоких терапевтических доз. Однако, как отмечалось выше, малые дозы вызывают центральную парасимпатическую стимуляцию и часто приводят к начальной брадикардии перед развитием эффектов периферического вагусного блока (рис. 8-4). Те же механизмы участвуют во влиянии на функцию атриовентрикулярного узла. Введение атропина на фоне высокого тонуса п. vagus может значительно уменьшить интервал PR электрокардиограммы, блокируя мускариновые рецепторы сердца. Мускариновые влияния на мышцу предсердия блокируются аналогичным образом и могут вызвать трепетание и фибрилляцию. Желудочки относительно устойчивы к антимускариновым средствам в терапевтических дозах вследствие меньшей мускариновой зависимости. В токсических концентрациях препараты могут вызвать блокаду внутрижелудочкового проведения посредством пока не изученного механизма.
Кровеносные сосуды практически не имеют прямой парасимпатической иннервации. Однако, как отмечалось в главе 6, симпатические холинергические нервы вызывают вазодилатацию сосудов скелетных мышц, которая может быть блокирована атропином. Кроме того, почти все сосуды содержат не связанные с иннервацией мускариновые рецепторы, вызывающие вазодилатацию (глава 7). Эти рецепторы, расположенные на эндотелиальных клетках, в ответ на циркулирующие в крови прямо действующие мускариновые агонисты выделяют расслабляющий фактор (РПФЭ) и легко блокируются антимускарииовыми препаратами. В токсических дозах, а у некоторых пациентов в нормальных дозах, антимускариновые средства вызывают дила-
Рис. 8-4. Кривая зависимости доза-эффект атропина на частоту сердечных сокращений у восьми здоровых людей. Внутривенные дозы вводились последовательно. Меньшие дозы замедляют частоту сердечных сокращений посредством стимуляции центрального ядра вагуса (Porter Т. R. et al. J. Clin. Invest., 1990; 85:1362.)
тацию сосудов кожи, проявляющуюся в виде румянца. Механизм этого явления пока не известен.
Эффекты атропина на сердечно-сосудистую систему у пациентов с нормальной гемодинамикой не приводят к тяжелым последствиям: может отмечаться тахикардия при незначительном изменении кровяного давления. При этом действие введенных мускариновых стимуляторов прямого действия легко предотвращается.
4. Дыхательная система. И гладкая мускулатура, и секреторные железы воздухопроводящих путей получают вагусную иннервацию и содержат мускариновые рецепторы. Даже у здоровых людей после введения атропина могут быть отмечены некоторая бронходилатация и снижение секреции. Значительно более выражен эффект у пациентов с респираторными заболеваниями, хотя при бронхиальной астме антимускариновые препараты менее показаны, чем стимуляторы p-адренорецепторов (глава 19). Тем не менее антимускариновые средства важны для некоторых пациентов с астмой или
хроническими обструктивными легочными заболеваниями. К тому же эти препараты часто используются перед введением ингаляционных анестетиков для уменьшения секреции в трахее и предотвращения ларингоспазма.
5. Желудочно-кишечный тракт. Блокада мускариновых рецепторов изменяет моторику и некоторые секреторные функции кишечника. Однако, поскольку местные гормоны и нехолинергические нейроны тоже модулируют функцию желудочно-кишечного тракта, даже полный мускариновый блок не может полностью подавить активность этой системы органов. Как и в других тканях, более эффективно нейтрализуется действие экзогенно введенных мускариновых стимуляторов, чем последствия парасимпатической (вагусной) нервной активности.
Отмечено влияние антимускариновых препаратов на секрецию слюны. Так, сухость во рту — частый симптом у пациентов, принимающих антимускариновые средства для лечения паркинсонизма или пептических язв (рис. 8-5).
Желудочная секреция угнетается в меньшей степени: объем и количество кислоты, пепсина и муцина уменьшаются, но для этого требуются большие дозы атропина. Базальная секреция блокиру
ется более эффективно, чем вызванная пищей, никотином или алкоголем. Пирензепин и его более сильный аналог телензепин уменьшают желудочную секрецию кислоты с меньшими нежелательными реакциями, чем атропин и ему подобные менее селективные препараты. Это может быть результатом избирательной блокады пресинаптических возбуждающих мускариновых рецепторов на вагусных нервных окончаниях, по-видимому, иЭ-за их большего сродства к мь а не к м3-рецепторам (табл. 8-1). Пирензепин и телензепин испытываются в США. Панкреатическая и кишечная секреции мало чувствительны к атропину, так как эти процессы находятся главным образом не под вагусным, а под гормональным контролем.
Антимускариновые препараты затрагивают моторику гладкой мускулатуры желудочно-кишечного тракта на протяжении от желудка до толстой кишки. В целом стенки внутренних органов расслабляются, уменьшается тонус и пропульсивные движения. Вследствие этого увеличивается время опорожнения желудка и удлиняется время транзита через кишечник. Диарея при передозировке па-расимпатомиметических препаратов легко снимается антагонистами мускариновых рецепторов, которые также могут временно купировать и диарею,
Рис. 8-5. Действие атропина (подкожная инъекция) на саливацию, скорость мочеиспускания (освобождения мочевого пузыря), частоту сердечных сокращений и аккомодацию у здоровых людей. Отметьте, что саливация более чувствительна к действию атропина, а аккомодация менее. (Herxheimer A. Br. J. Pharmacol., 1958; 13:184.)
другой этиологии. Однако кишечный “паралич”, вызываемый антимускариновыми препаратами, длится ограниченное время, так как местные механизмы (глава 6) обычно восстанавливают перистальтику, по крайней мере частично, спустя 1-3 дня после начала терапии антимускариновыми средствами.
Некоторые синтетические антимускариновые препараты, как полагают, имеют ’’спазмолитическую” активность, превышающую их антимускариновые эффекты. Способность этих средств расслаблять кишечник как в отсутствие, так и в присутствии холинергических стимуляторов, является результатом отбора препаратов в скрининговых тестах на изолированных тканях.
6. Мочеполовая система. Гладкие мышцы уретры и стенок мочевого пузыря под влиянием анти-мускаринового действия атропина и его аналогов расслабляются, и опорожнение замедляется (рис. 8-5). Это воздействие полезно при лечении спазма, вызванного умеренным воспалительным процессом, но оно создает риск возникновения задержки мочеиспускания у пожилых пациентов с аденомой простаты. Антимускариновые препараты не оказывают значительного влияния на матку.
7. Потовые железы. Атропин угнетает терморегуляторное выделение пота. Мускариновые рецепторы потовых желез иннервированы симпатическими холинергическими окончаниями и чувствительны к антимускариновым препаратам. У взрослых в этой связи температура повышается только после приема больших доз атропина, а у младенцев и детей даже обычные дозы могут вызвать “атропиновую лихорадку”.
II. Клиническая фармакология средств, блокирующих мускариновые рецепторы
Терапевтическое применение
А. Заболевания центральной нервной системы.
1. Болезнь Паркинсона. Терапия паркинсонизма часто является “упражнением” в комбинировании лекарств (глава 27), так как нет какого-либо одного достаточно эффективного средства для лечения это
го заболевания (обычно продолжительного). Большинство антимускариновых препаратов, предложенных для лечения этой болезни (табл. 27-1), было разработано до появления леводопы. Их использование сопровождалось всеми побочными эффектами, описанными в этой главе, однако эти вещества остаются в арсенале антипаркинсонических препаратов как средства дополнительной терапии.
2. Болезни движения. Антимускариновыми препаратами корригируются определенные вестибулярные расстройства. Скополамин — одно из старейших лекарств против морской болезни, его эф-фективеность не ниже, чем у любого из вновь предложенных препаратов. Его можно назначать в виде инъекций, энтерально илитрансдермально (в виде пластыря). Аппликация пластыря обеспечивает значимое повышение концентрации препарата в крови в течение 24-48 часов. К сожалению, терапевтические дозы препарата, вводимые любым способом, обладают выраженным седативным действием и вызывают сухость во рту.
Б. Офтальмологические заболевания. Точное определение рефракции хрусталика, например у маленьких детей, требует обеспечения цилиарного паралича. К тому же офтальмоскопическое исследование сетчатки облегчается при мидриазе. Поэтому антимускариновые препараты, применяемые местно в виде глазных капель или мазей, необходимы для выполнения полного обследования. Для взрослых и детей старшего возраста предпочтительны короткодействующие препараты (табл. 8-2). Для детей младшего возраста необходим более эффективный атропин, однако при этом повышается возможность антимускаринового отравления. Вытекание препарата из конъюнктивального мешка через носослезный проток в носоглотку можно уменьшить, используя мази.
Антимускариновые препараты не должны использоваться для получения мидриаза, если нетре-
ТАБЛИЦА 8-2. Антимускариновые препараты, применяемые в офтальмологии
Препарат Длительность мидриаза(дни) Длительность циклоплегии (дни)
Атропин 7-10 7-12
Скополамин 3-7 3-7
Гоматропин 1-3 1-3
Циклопентолат 1 1/4-1
Тропикамид 1/4 1/4
буется пролонгированное действие или циклопле-гия. Стимуляторы ос-адренорецепторов, например фенилэфрин, вызывают кратковременный мидриаз, обычно достаточный для офтальмоскопического обследования (глава 9).
Второе офтальмологическое показание для использования антимускариновых препаратов — предотвращение образования синехий (адгезии) при увеитах и иритах. Препараты пролонгированного действия, особенно содержащие гоматропин, являются препаратами выбора в этих случаях.
В. Заболевания желудочно-кишечного тракта. Антимускариновые препараты использовались, главным образом, при пептических язвах, пока не были предложены блокаторы Н2-гистаминовых рецепторов (глава 64, том 2). В настоящее время антимускариновые средства редко принимают по этому показанию, хотя развитие селективных мгблокато-ров, подобных пирензепину, может изменить тенденцию. Из более старых препаратов, применяемых в этом случае, предпочтение можно отдать четвертичным аммонийным антимускариновым средствам, хотя для значительного уменьшения желудочной секреции требуются большие дозы (табл. 8-3).
ТАБЛИЦА 8-3. Антимускариновые препараты, применяемые для лечения желудочно-кишечных и мочеполовых заболеваний
Препараты Обычная доза (мг)
Четвертичные амины
Анизотропии 50 (3 раза вдень)
Клидиниум 2.5 (3-4 раза в день)
Гликопирролат 1 (2-3 раза в день)
Гексоциклиум 25 (4 раза вдень)
Изопропамид 5 (2 раза вдень)
Мепензолат 25-50 (4 раза в день)
Метантелин 50-100(4 раза в день)
Метскополамин 2.5 (4 раза в день)
Оксифенониум 5-10 (4 раза в день)
Пропантелин 15 (4 раза вдень)
Третичные амины
Атропин 0.4 (3-4 раза в день)
Дицикломин 10-20 (4 раза в день)
Оксибутинин 5 (Зраза в день)
Оксифенциклимин 10(2 раза в день)
Скополамин 0.4 (Зраза в день)
Тридигексетил 25-50 (3-4 раза в день)
У пациентов, получающиех такое лечение, может наблюдаться затуманенное зрение, сухость во рту и затрудненное мочеиспускание. В случае пептической язвы желудка антимускариновые препараты могут быть противопоказаны, так как они замедляют опорожнение желудка и продлевают действие кислоты на язвенное ложе.
При обычной диарее путешественников и других умеренно выраженных состояниях гипермоторики антимускариновые препараты могут дать некоторое облегчение. Их комбинация с опиоидными антидиарейными препаратами чрезвычайно эффективна. Однако в этой комбинации очень малые дозы антимускариновых препаратов уменьшают, главным образом, зависимость от опиоидных веществ. Классическая комбинация атропина с дифеноксилатом, неаналгетическим родственником меперидина, существует под разными названиями (например, ломотил) в виде таблеток и жидких форм (глава 64, том 2).
Г. Сердечно-сосудистые заболевания. Отчетливая вагусная гиперактивация иногда сопровождает боль при инфаркте миокарда и может привести к подавлению функции синоатриального или атриовентрикулярного узлов в степени, достаточной для нарушения сердечного выброса. В такой ситуации показано парентеральное введение атропина или сходных антимускариновых препаратов. Некоторые пациенты без явных сердечных заболе-ваний.имеют гиперактивные рефлексы каротидного синуса и испытывают слабость или даже падают в обморок в результате вагусной активации при сдавлении шеи, например тесным воротником. Состояние таких лиц можно улучшить, разумно применяя атропин или родственные антимускариновые лекарства.
Д. Респираторные заболевания. Использование атропина стало частью обычной премедикации перед операциями, когда применяют такие анестетики, как эфир для наркоза, так как их раздражающее действие вызывает заметное повышение секреции в воздухопроводящих путях и сопровождается ла-рингоспазмом. Эти опасные побочные эффекты могут быть предотвращены премедикационной инъекцией атропина или скополамина. Последнее вещество вызывает значительную амнезию событий, ассоциированных с хирургической операцией и родами — эффект, который может считаться желательным. С другой стороны, при использовании антимускариновых препаратов могут значительно
усиливаться послеоперационная задержка мочеиспускания и гипомоторика кишечника. С появлением эффективных и не раздражающих ингаляционных анестетиков, таких как галотан и энфлуран, антимускариновые препараты для премедикации используются все реже.
Вдыхание дыма от сжигаемых листьев дурмана (Datura stramonium) на протяжении веков использовалось как лекарство против бронхиальной астмы. Сигареты “Астмадор”, содержащие Datura stramonium, до недавнего времени использовались с этой целью без рецепта. Как описано в главе 19, повышенный бронхоконстрикторный рефлекс, имеющийся у большинства больных астмой, опосредуется блуждающим нервом, активирующим мускариновые рецепторы бронхиальных гладкомышечных клеток. Ипратропиум (рис. 8-2), синтетический аналог атропина, используется для ингаляций при астме. Ингаляционный путь введения обеспечивает создание максимальной концентрации в тканях-мишенях бронхиального дерева с одновременным уменьшением системных эффектов. Этот способ применения обсуждается более детально в главе 19.
Е. Отравления холинергическими веществами. Тяжелые холинергические отравления — это важные и неотложные медицинские ситуации, особенно в сельской местности, где часто в качестве инсектицидов применяют ингибиторы холинэстеразы, а также среди населения, использующего в пищу дикорастущие грибы. Возможное использование этих соединений в качестве химического оружия (“нервные газы”) требует особого знания методов лечения острых отравлений.
1. Антимускариновая терапия. Как отмечал ось в главе 7, и никотиновые, и мускариновые эффекты ингибиторов холинэстеразы могут угрожать жизни человека. К сожалению, нет надежных методов для прямой блокады никотиновых эффектов ингибиторов холинэстеразы, поскольку и никотиновые агонисты, и блокаторы в результате пролонгированного воздействия вызывают блокаду проведения (глава 26). Для нивелирования мускариновых эффектов должны использоваться третичные (не четвертичные) амины ( предпочтительно атропин), так как необходимо воздействовать как на центральные, так и на периферические эффекты фосфорорганических ингибиторов. Для снижения мускариновых эффектов, таких исключительно сильных средств как паратион и боевые отравляющие вещества, требуются большие дозы атропина:
1 -2 мг атропина могут вводиться внутривенно каждые 5-15 минут до появления признаков действия (сухость во рту, ослабление миоза). Могут потребоваться повторные многократные введения препарата, так как острые эффекты антихолинэстеразных соединений продолжаются в течение 24-48 часов или дольше. Теоретических ограничений для использования атропина в этой угрожающей жизни ситуации нет, и для полного контроля мускаринового возбуждения может потребоваться введение до 1 г атропина в день в период до 1 месяца (LeBlanc, 1986).
2. Реактиваторы холинэстеразы. Для лечения отравлений фосфорорганическими ингибиторами также используется целый класс соединений, способных восстанавливать активный фермент из комплекса фосфорорганического соединения с холинэстеразой. Это оксимы, к которым относятся пралидоксим (РАМ), диацетилмоноксим (DAM) и другие.
Оксимная группа (=NOH) имеет очень высокое сродство к атому фосфора, и оксимы способны гидролизовать фосфорилированный фермент, если комплекс не “постарел” (глава 7). В опытах на животных наиболее эффективен из этих оксимов HL67 — токсогонин (Eyer et al., 1992). У человека наиболее изучено действие пралидоксима; это един-ственный препарат, используемый в клиниках США. Он особенно эффективно восстанавливает активность холинэстеразы, ассоциированной с нервно-мышечными соединениями скелетных мышц. Так как этот препарат имеет положительный заряд, он не проникает в центральную нервную систему и неэффективен против центральных эффектов фосфорорганических ядов. В отличие от пралидоксима диацетилмоноксим проходит через гематоэнцефалический барьер и в эксперименте у животных может частично восстанавливать активность холинэстеразы в центральной нервной системе. Холинэстеразу, связанную карбаматными ингибиторами, оксимы восстанавливают плохо.
Пралидоксим вводится внутривенно в дозе 1-2 г в течение 15-30 минут. Несмотря на вероятность старения фосфатно-ферментного комплекса, недавние исследования показывают, что многократное введение пралидоксима в течение нескольких дней может быть эффективно при тяжелых отравлениях. В чрезмерных дозах пралидоксим может вызывать нервно-мышечную слабость и другие нежелательные явления. Дальнейшие детали лечения от
равлений антихолинэстеразными средствами изложены в главе 60 (том 2).
Еще одна возможность предотвращения чрезмерного угнетения ацетилхолинэстеразы заключается в предварительном применении обратимых ингибиторов фермента для предупреждения образования устойчивых комплексов. Эта профилактика может быть достигнута введением пиридостиг-мина или физостигмина, однако она подходит для чрезвычайных ситуаций, в которых существует угроза летального отравления, например при угрозе применения химического оружия. В опытах на животных показана эффективность лигандов холинэстеразы, производных гемихолиния-3 (Cannon et al., 1991), однако в этих случаях для снижения мускариновой гиперактивации требуется одновременное применение атропина.
Отравления грибами традиционно разделяются на быстро и медленно развивающиеся типы. При быстро развивающемся типе признаки отравления обычно наступают в пределах 15-30 минут после употребления грибов. Отравление проявляется признаками мускаринового возбуждения: тошнотой, рвотой, диареей, вазодилатацией, рефлекторной тахикардией (иногда брадикардией), потливостью, саливацией и иногда спазмом бронхов. В грибе Amanita muscaria кроме мускарина (алкалоид был назван по имени гриба) найдено множество других алкалоидов, включая антимускариновые вещества. Поэтому употребление Amanita muscaria может вызывать симптомы не мускаринового, а атропинового отравления. Другие грибы, особенно из рода
О
II н3с—сх н3с^
С = NOH
Диацетилмоноксим
О = С — NH2 CH —NOH
HL67 (токсогонин)
Inocybe, вызывают быстро развивающееся отравление по типу мускариновой гиперактивации. Эффективное лечение такой интоксикации — парентеральное введение атропина в дозе 1-2 мг.
При медленно развивающемся отравлении, обычно вызываемом Amanita phalloides, Amanita virosa, Galerina autumnalis или Galerina marginata, первые симптомы появляются через 6-12 часов после употребления грибов. Хотя начальными симптомами чаще всего бывают тошнота и рвота, тяжелое отравление сопровождается повреждениями клеток печени и почек аматоксином, который ингибирует РНК-полимеразу. Применение атропина при этой форме отравления грибами неэффективно (глава 60, том 2).
Е. Другие области применения. Атропин и четвертичные аммонийные антимускариновые препараты используются в терапии неотложных состояний при воспалительных заболеваниях мочевого пузыря (табл. 8-3). Их применение обеспечивает некоторое симптоматическое облегчение, хотя не заменяет специфической антимикробной терапии при бактериальных циститах. Оксибутинин часто назначают для снятия спазма мочевого пузыря после урологических операций. Антимускариновые препараты используют и при камнях мочевыводящих путей для уменьшения спазма гладкой мускулатуры мочеточника во время прохождения камня, однако целесообразность их применения в этих ситуациях оспаривается.
Антимускариновые препараты иногда назначают для уменьшения гипергидроза. Хотя эффект не может быть полным из-за большего вовлечения апокринных, а не экзоринных желез.
Нежелательные эффекты
Из-за широкого спектра антимускариновых эффектов лечение атропином или родственными ему соединениями, направленное на одну систему органов, почти всегда вызывает нежелательные (побочные) эффекты в других системах. Например, мидриаз и циклоплегия — “нежелательные” эффекты, когда антимускариновые препараты назначаются для уменьшения секреции или моторики желудочно-кишечного тракта, в то же время они являются “лечебными” при использовании подобных препаратов в офтальмологии.
В более высоких дозах атропин вызывает блокаду всех парасимпатических функций, ее резуль
тат складывается из описанных выше эффектов препарата на системы и органы. Но даже в количествах, измеряемых граммами, атропин является удивительно безопасным веществом для взрослых. Отравления атропином встречаются как результат суицидных попыток, а чаще вследствие попыток вызвать галлюцинации. У отравившегося атропином наблюдаются сухость во рту, мидриаз, тахикардия, горячая и покрасневшая кожа, ажитация и делирий длительностью до недели. Часто поднимается температура тела. Эти эффекты отражены в пословице: “dry as a bone, blind as a bat, red as a beet, mad as a hatter” (“сухой, как кость, слепой, как летучая мышь, красный, как свекла, сумасшедший, как шляпник").
Дети, особенно младенческого возраста, очень чувствительны к гипертермическим эффектам атропина. Хотя иногда случайное введение свыше 400 мг лекарства заканчивалось выздоровлением, в других случаях смерть наступала и от дозы в 2 мг. Таким образом, атропин должен рассматриваться как очень опасный препарат, если передозировка происходит у детей.
Передозировки атропина и препаратов его группы лечат главным образом симптоматически (глава 60, том 2). В прошлом рекомендовали применение физостигмина или других ингибиторов холинэстеразы, однако в настоящее время большинство токсикологов считают физостигмин более опасным и менее эффективным, чем симптоматические средства. Если применение физостигмина необходимо, то внутривенно медленно вводят малые дозы (1-4 мг для взрослых, 0.5-1 мг для детей). Симптоматическое лечение может потребовать коррекции температуры тела с помощью влажных обертываний и борьбы с судорогами с помощью диазепама.
Отравление, вызванное высокими дозами четвертичных аммонийных антимускариновых препаратов, проявляется периферическими признаками парасимпатической блокады без или с небольшими центральными эффектами атропина. Эти более полярные вещества могут привести к значительной блокаде ганглиев с заметной ортостатической гипотензией. При необходимости антимускариновые эффекты можно ослабить с помощью четвертичных ингибиторов холинэстеразы, таких как неостигмин. Для устранения гипотензии применяются симпатомиметические препараты, например фенилэфрин или метоксамин.
Противопоказания
Противопоказания к использованию антимускариновых препаратов не абсолютны. При мускариновой гиперактивации, особенно вследствие передозировки ингибиторов холинэстеразы, всегда можно использовать атропин.
Антимускариновые препараты противопоказаны пациентам с глаукомой, особенно закрытоугольной. У пациентов с мелкой передней камерой системное применение даже умеренных доз может вызвать закрытие угла (и острую глаукому).
Для пожилых людей атропин должен всегда применяться с осторожностью. Его назначения нужно избегать пациентам с аденомой простаты.
Так как антимускариновые препараты замедляют опорожнение желудка, они могут усиливать симптомы у пациентов с язвенной болезнью. При фармакотерапии язвы желудка предпочтение следует отдавать Н2-гистаминоблокаторам и другим препаратам (глава 64, том 2).
III. Базисная и клиническая фармакология ганглиоблокирующих средств
Эти препараты блокируют действие ацетилхолина и сходных агонистов на никотиновые рецепторы как парасимпатических, так и симпатических автономных ганглиев. Некоторые представители группы также (или, возможно, исключительно) блокируют ионные каналы, которые закрываются никотиновыми холинорецепторами. Из-за способности блокировать все вегетативные пути ганглио-блокаторы до сих пор представляют исключительный интерес для фармакологических и физиологических исследований. Однако недостаток селективности ведет к такому широкому спектру побочных эффектов, что эти препараты почти не назначают в клинике. Основным реальным клиническим применением ганглиоблокаторов является их использование для кратковременного снижения кровяного давления.
Химия и фармакокинетика
Все ганглиоблокирующие средства — синтетические амины. Первым препаратом с таким эффектом был тетраэтиламмоний (ТЭА). В связи с очень
короткой продолжительностью действия ТЭА был разработан гексаметоний (С6), который вскоре вошел в клиническую медицину как первый эффективный препарат для коррекции гипертензии. Как показано на рис. 8-6, существует очевидная связь . между структурой естественного агониста ацетил-\ холина и никотиновых антагонистов тетраэтилам-мония и гексаметония. Интересно, что аналог гек-саметония декаметоний (СЮ) является эффективным нервно-мышечным деполяризующим блокатором.
Так как четвертичные аммонийные ганглиоблокирующие соединения плохо и неравномерно всасываются после введения внутрь, был разработан мекамиламин, являющийся вторичным аммонийным соединением. Короткодействующий ганглио-блокатор триметафан неактивен при энтеральном введении и назначается в виде внутривенных вливаний (глава И).
Фармакодинамика
А. Механизм действия. Возможна как деполяризационная, так и недеполяризационная блокада никотиновых рецепторов ганглиев, так же, как и никотиновых рецепторов нервно-мышечных соеди-
СНз СНз
I I
СНз - N+ - СН2 - СН2 - СН2 - СН2 - СН2 - СН2 - +N - СН3
I I
СНз Гексаметоний
Мекамиламин
СН2 —СНз I
СНз — СН2 - N+ - СН2 - СНз I
СН2 —СНз
Тетраэтиламмоний
СНз о
I II
СНз — N+ — СН2 — СН2 — О — С — СНз
I
СНз Ацетилхолин
Рис. 8-6. Некоторые ганглиоблокирующие препараты. Ацетилхолин представлен для сравнения. Структура триметафана приведена в главе 11
нений скелетной мускулатуры (главы 7 и 27). Сам никотин, карбамоилхолин и даже ацетилхолин (при введении с ингибитором холинэстеразы) могут вызывать деполяризационный блок ганглиев.
Все препараты, используемые в настоящее время в качестве ганглиоблокаторов, классифицируют как недеполяризующие конкурентные антагонисты. Однако, результаты многих исследований показывают, что гексаметоний фактически вызывает блокаду, оккупируя места связывания внутри или на поверхности ионного канала, контролируемого холинорецептором, не занимая при этом сам ацетилхолиновый рецептор. Напротив, триметафан, как полагают, блокирует именно никотиновый рецептор, а не канал. В соответствии с данными предыдущих исследований, блокада может быть частично преодолена повышением концентрации естественного агониста, т. е. ацетилхолина.
Как отмечалось в главе 6, другие рецепторы в постганглионарных клетках модулируют процесс ганглионарной передачи. Однако их эффект недостаточен, чтобы преодолеть действие больших доз препаратов группы гексаметония-триметафана.
Б. Влияние на системы и органы.
1. Центральная нервная система. Четвертичные аммонийные соединения и триметафан лишены центральных эффектов, так как не проходят через гематоэнцефалический барьер. Мекамиламин легко попадает в центральную нервную систему и вызывает седативный эффект, тремор, хореоидные движения и психические отклонения.
2. Глаз. Поскольку цилиарная мышца получает главным образом парасимпатическую иннервацию, ганглиоблокаторы вызывают циклоплегию с нарушением аккомодации. Влияние на зрачок не так легко предсказуемо, поскольку радужка получает как симпатическую (вызывающую расширение зрачка), так и парасимпатическую (вызывающую сужение зрачка) иннервацию. Так как в этой ткани преобладает парасимпатический тонус, ганглиоблокаторы обычно приводят к умеренному расширению зрачка.
3. Сердечно-сосудистая система. Кровеносные сосуды получают вазоконстрикторные волокна, главным образом, от симпатической нервной системы, поэтому блокада ганглиев вызывает падение артериолярного и веномоторного тонусов. Кровяное давление может резко снижаться, так как уменьшаются и периферическое сосудистое сопротивление, и венозный возврат (рис. 6-7). Гипотензия осо
бенно заметна в положении стоя (ортостатическая или постуральная гипотензия), поскольку блокируются постуральные рефлексы, которые в норме препятствуют венозному наполнению.
Сердечные эффекты включают в себя снижение / сократимости и умеренную тахикардию, посколь-| ку синусовый предсердный узел управляется парасимпатической нервной системой.
4. Желудочно-кишечный тракт. Препараты уменьшают желудочную секрецию, хотя и недостаточно эффективно, чтобы лечить пептические расстройства. Моторика полностью подавляется, может отмечаться констипация (запор).
5. Другие системы. Нормальная функция гладких мышц мочеполовой системы частично зависит от вегетативной иннервации. В связи с этим ганг-лиоблокаторы вызывают нарушения мочеиспускания, в первую очередь его задержку у мужчин с аденомой простаты. Изменяется сексуальная функция, при этом умеренные дозы препаратов могут нарушить и эрекцию, и эякуляцию.
Ганглиоблокаторы снижают терморегуляторное выделение пота, однако гипертермия, как правило, не выражена (исключая ситуации с высокой температурой окружающей среды), так как кожная вазодилатация обычно достаточна для поддержания нормальной температуры тела.
6. Реакция на вегетотропные препараты. Поскольку эффекторные клеточные рецепторы (мускариновые, а- и Р-) не блокируются ганглиоблока-торами, пациенты, получающие эти средства, полностью чувствительны к действию вегетотропных препаратов, действующих на уровне эффекторов. Фактически эти ответы могут быть усилены, так как отсутствуют гомеостатические рефлексы, которые обычно контролируют вегетативные реакции.
Клиническое применение и токсичность
Наличие более селективных вегетативных блокирующих препаратов ограничивает применение ганглиоблокаторов снижением кровяного давления. Способность триметафана вызывать ортостатический гипотензивный эффект за счет венозного наполнения используется при лечении неотложных гипертензивных состояний, что рассматривается в главе И. Контролируемая гипотензия представляет интерес для нейрохирургии из-за возможности уменьшать кровотечение в операционном поле. Кратковременный управляемый эффект тримета
фана также может использоваться по этому назначению. Триметафан применяют при остром отеке легких для уменьшения давления в легочных сосудах.
Токсичность ганглиоблокаторов ограничивается вегетативными эффектами, которые уже рассмотрены. Для большинства пациентов наличие этих эффектов допустимо только при кратковременном неотложном применении препаратов.
Препараты
Антимускариновые антихолинергические препараты1
Анизотропии (генерик, Валпин)
Перорально: таблетки по 50 мг
Атропин (генерик)
Перорально: таблетки по 0.4,0.6 мг Парентерально: 0.05, 0.1, 0.3, 0.4, 0.5, 0.8, 1 мг/мл для инъекций
Глазные капли (генерик, Изопто Атропин):
0.5, 1, 2, 3 % капли; 0.5, 1 % мазь
Алкалоиды белладонны, экстракт или настойка
(генерик)
Перорально: жидкость 0.27—0.33 мг/мл
Клидиниум (Кварзан)
Перорально: капсулы по 2.5, 5 мг
Циклопентолат (генерик, Циклогил, др.)
Глазные капли: 0.5,1,2%
Дицикломин (генерик, Бентил, др.)
Перорально: капсулы по 10, 20 мг; таблетки по 20 мг; сироп 10 мг/5 мл
Парентерально: 10 мг/мл для инъекций
Флавоксат (Уриспаз)
Перорально: таблетки по 100 мг
Гликопирролат (генерик, Робинул)
Перорально: таблетки по 1, 2 мг
Парентерально: 0.2 мг /мл для инъекций
Гексоциклиум (Трал)
Перорально: таблетки по 25 мг
Гоматропин (генерик, Изопто Гоматропин)
Глазные капли: 2,5 %
1 Антимускариновые вещества для лечения паркинсонизма перечислены в главе 27.
Изопропамид (Дарбид)
Перорально: таблетки по 5 мг
L-Гиосциамин (генерик, Цистоспаз-М,
Левсинекс)
Перорально: таблетки по 0.125,0.15 мг; саморассасывающиеся капсулы по 0.375 мг; эликсиры и растворы по 0.125 мг/5 мл Парентерально: 0.5 мг/мл для инъекций
Ипратропиум (Атровент)
Аэрозоль: 200 стандартных доз в ингаляторе
Мепензолат (Кантил)
Перорально: таблетки по 25 мг
Метскополамин (Памин)
Перорально: таблетки по 2.5 мг
Метантелин (Бантин)
Перорально: таблетки по 50 мг
Оксибутинин (генерик, Дитропан)
Перорально: таблетки по 5 мг, сироп 5 мг/5 мл
Оксифенциклимин (Дарикон)
Перорально: таблетки по 10 мг
Пропантелин (генерик, Про-Бантин)
Перорально: таблетки по 7.5,15 мг
Скополамин (генерик) 1
Перорально: капсулы по 0.25 мг Парентерально: 0.3, 0.4, 0.86, 1 мг/мл для инъекций
Глазные капли (генерик, Изопто Гиосцин): 0.25 % раствор
Трансдермально (Трансдерм-Скоп): пластырь 1.5 мг высвобождает по 0.5 мг в течение 3 дней
Тридигекситил (Патилон)
Перорально: таблетки по 25 мг
Тропикамид (генерик, Мидриацил Офталь-мик, др.)
Глазные капли: 0.5,1 %
Ганглиоблокаторы
Мекамиламин (Инверсии)
Перорально: таблетки по 2.5 мг
Триметафан камзилат (Арфонад)
Парентерально: ампулы 500 мг/10 мл
Реактиваторы холинэстеразы
Пралидоксим (генерик, Протопам)
Парентерально: 1 г во флаконе с 20 мл растворителя для в/в введения;
600 мг в автоинъекторе на 2 мл.
Избранная литература
Amitai Y. et al. Atropine poisoning in children during the Persian Gulf crisis. JAMA, 1992; 268: 630.
Berdie G. E. et al. Angle closure glaucoma precipitated by aerosolized atropine. Arch. Intern. Med. 1991; 151: 1658.
Brown J. H. (ed.) The Muscarinic Receptors, Humana Press, 1989.
DeSilva H. J., Wijewickrema R., Senanayake N. Does pralidqxime affect outcome of management in acute organophosphorus poisoning? Lancet, 1992; 339:1136.
Eglen R. M„ Whiting R. L. Muscarinic receptor subtypes: A critique of the current classification and a proposal for a working nomenclature. J. Auton. Pharmacol. 1986; 6:323.
Mason D. F. J. Ganglion-blocking drugs. In: Physiological Pharmacology: A Comprehensive Treatise. Vol. 3. Root W. S., Hoffman F. G. (eds) Academic Press, 1967.
Salem M. R. Therapeutic uses of ganglionic blocking drugs. Int. Anesthesiol. Clin. 1978; 16:171.
Leadbeater L., Inns R. H., Rylands J. M. Treatment of poisoning by soman. Fundam. Appl. Toxicol. 1985; 5: S225.
Solana R. P. et al. Evaluation of a two-drug combination pretreatment against organophosphorus exposure. Toxicol. Appl. Pharmacol. 1990; 102: 421.
Средства, активирующие адренорецепторы, и другие 9
симпатомиметические средства
Брайен Б. Хоффман
Симпатическая нервная система является важным регулятором активности таких органов как сердце и периферические сосуды, особенно в ответ на стресс (глава 6). Основные эффекты симпатической стимуляции вызываются выделением норадреналина из нервных окончаний, который активирует постсинаптические адренорецепторы. Мозговое вещество надпочечников выделяет при стрессе адреналин, транспортирующийся с кровью к тканям-мишеням. Вещества, имитирующие действие адреналина или норадреналина, так называемые симпатомиметические средства, как и можно ожидать, имеют широкий спектр эффектов. Таким образом, понимание фармакологии этих препаратов является логическим продолжением наших знаний о физиологической роли катехоламинов.
Механизмы и спектр действия симпатомиметических средств
Подобно холиномиметическим средствам сим-патомиметики могут быть сгруппированы по способу действия и по спектру вовлеченных в реализацию эффекта рецепторов. Некоторые из этих веществ (например, норадреналин, адреналин) имеют прямое действие, т. е. непосредственно взаимодействуют с адренорецепторами, активируя их. Другие действуют непрямо, опосредованно: их эффекты зависят от выделения эндогенных катехоламинов. Препараты непрямого действия могут оказывать эффект посредством двух разных механизмов: 1) вытесняя запасы катехоламинов из адренергических нервных окончаний (например, амфетамин и тирамин) или 2) подавляя обратный захват
уже выделенных катехоламинов (например, кокаин и трициклические антидепрессанты). Оба типа симпатомиметиков, прямые и непрямые, в конечном счете вызывают активацию адренорецепторов, ведущую к проявлению некоторых или всех характерных эффектов катехоламинов. Селективность разных симпатомиметиков в отношении различных типов адренорецепторов будет обсуждаться в этой главе.
I. Базисная фармакология симпатомиметических средств
Идентификация адренорецепторов
Понять молекулярные механизмы.действия катехоламинов ученые пытались давно. Крупным концептуальным достижениям в этой области мы обязаны работам Ленгли и Эрлиха, предложившим 75-100 лет назад гипотезу о реализации эффекта веществ посредством их взаимодействия со специфическими “рецептивными” субстанциями. Алк-вист в 1948 г. дал рациональное объяснение большому числу наблюдений, предположив, что катехоламины действуют на два основных типа рецепторов. Он назвал Эти рецепторы а и р. Для «-рецепторов характерен следующий ряд активности адреномиметических средств: адреналин > норадреналин » изопротеренол. Для Р-рецепторов ряд активности выглядит так: изопротеренол > адреналин > норадреналин. Гипотеза Алквиста была под
тверждена разработкой средств, избирательно блокирующих р-, но не а-рецепторы (глава 10).
'А. Подтипы Р-рецепторов. Вскоре после выявления различающихся между собой а- и Р-рецепторов было обнаружено, что имеется по меньшей мере два подтипа p-рецепторов, обозначенных pi и р2. рг и р2-рецепторы определяли по их аффинитету к адреналину и норадреналину: Pt-рецепторы имели приблизительно равное сродство к обоим катехоламинам, в то время как р2-рецепторы имели более высокий аффинитет к адреналину, чем к норадреналину. В настоящее время методом мо-лекулярбго клонирования продемонстрировано существование третьего подтипа Р-рецепторов (Р3) и установлены некоторые свойства каждого из этих подтипов рецепторов (табл. 9-1).
Б. Подтипы а-рецепторов. Вслед за открытием подтипов Р-рецепторов было обнаружено, что имеется также по меньшей мере две подгруппы а-рецепторов: at и а2. Подтипы а-рецепторов были исходно идентифицированы с помощью веществ а-антагонистов, селективных для а(- и аг-рецепторов. Например, а-адренорецепторы были идентифицированы в различных тканях посредством измерения связывания с мечеными радиоактивными соединениями-антагонистами, имеющими, как полагали, высокий аффинитет к этим рецепторам (ди-гидроэргокриптин — к at и а2, празозин — к at и йохимбин — к а2). Эти радиолиганды были использованы для измерения числа рецепторов в тканях и для определения аффинитета других препаратов (посредством вытеснения меченого лиганда), взаимодействующих с данными рецепторами.
Представление о подтипах внутри группы at-рецепторов возникло при анализе результатов фармакологических экспериментов, в которых были получены сложные формы кривых зависимости доза агониста-эффект сокращения гладкой мускулатуры некоторых органов, и из обнаруженных различий аффинитета антагонистов по степени блокирования определенных сократительных реакций. Эти эксперименты продемонстрировали существование двух подтипов at-рецепторов, которые могут быть разделены на основании их обратимого связывания с лигандом WB4101 (atA) или необратимой инактивации хлорэтилклонидином (Ощ, табл. 9-1). Селезенка и печень содержат в основном atB-рецепторы; сердце, неокортекс, почки, каудальная артерия, семенной канатик и гиппокамп, видимо, имеют примерно одинаковое число
а1А- и а1В-рецепторов. Эксперименты с молекулярным клонированием подтвердили существование нескольких подтипов at-рецепторов и установили некоторые их свойства.
Гипотеза о наличии подтипов а2-рецепторов возникла подобным образом на основе фармакологических экспериментов и опытов с радиолигандным связыванием. Человеческие тромбоциты, вероятно, содержат только рецепторы подтипа а2А. Такие, используемые в клинике средства, как празозин, хлорпромазин, йохимбин и оксиметазолин имеют различный аффинитет к а2А- и а2В-рецепторам. Имеются дополнительные фармакологические данные в пользу существования а2С-рецепторов. С помощью молекулярного клонирования идентифицированы по меньшей мере три типа а2-рецепторов, являющихся продуктами различных генов.
В. Дофаминовые рецепторы. Эндогенный катехоламин дофамин, связываясь со специфическими дофаминовыми рецепторами, вызывает разные биологические эффекты (табл. 9-1). Эти рецепторы отличаются от а- и p-рецепторов и особенно важны для функционирования головного мозга (главы 20 и 28), внутренних органов и сосудов почек. Как и в случае с а- и Р-адренорецепторами, существуют убедительные свидетельства наличия множественных форм дофаминовых рецепторов. До настоящего времени были известны два фармакологически различных подтипа дофаминовых рецепторов, называемые Di и D2. В новейших исследованиях с использованием молекулярного клонирования идентифицированы несколько генов для каждого из этих подтипов. Ситуация осложняется присутствием интронов в области, кодирующей О2-подобные рецепторы, что дает возможность вариантного сплайсинга экзонов в этом большом подтипе. К тому же обнаружен полиморфизм длины рестрикции человеческого О2-подобного рецептора (Gingrich, 1993). Обозначения различных подтипов пока не устоялись, и используются альтернативные серии названий: Db D2, D4, D5 или D1A, D2A, D2C, D1B (Receptor Nomenclature, 1994).
Рецепторная избирательность
В табл. 9-2 дана сравнительная характеристика используемых в клинике относительно селективных в отношении а- и Р-адренорецепторов симпа-томиметиков и некоторых неселективных средств. Селективность означает, что вещество предпочти-
ТАБЛИЦА 9-1. Типы и подтипы адренорецепторов
__ Рецептор Агонист Антагонист Эффекты Ген в хромосоме
оь-рецептор «1А Фенилэфрин, мето-ксамин, циразолин Празозин, коринантин WB4101, празозин ? ИТФ, ДАГ обычно для всех подтипов Т ИТФ, ДАГ; Т вход Са2*
«1В СЕС (необратимый) ? ИТФ, ДАГ С5
«>с WB4101.CEC Т ИТФ, ДАГ С8
«10 WB4101 ? Т вход Са2* С20
а2-рецептор Клонидин, ВНТ920 Раувольсцин, йохимбин 4- цАМФ обычно для всех подтипов
«2А Оксиметазолин J- цАМФ; Т К+-каналов; 4- Са2*-каналов СЮ
«2В Празозин 4- цАМФ; -1 Са2*-каналов С2
«2С Празозин 4- цАМФ С4
Р-рецептор Изопротеренол Пропранолол ? цАМФ обычно для всех подтипов
Р, Добутамин Бетаксолол Т цАМФ
Р2 Рз Дофаминовый рецептор Прокатерол, тербуталин BRL37344 Дофамин Бутоксамин Т цАМФ ? цАМФ
D, Фенолдопам Т цАМФ С5
d2 Da d5 Бромокриптин Клозапин 4. цАМФ; ? К*-каналов; Т Са2+-каналов 1 цАМФ ? цАМФ С11 С4
Обозначения:
BRL37344 = натрий-4-(2-[2-гидрокси-{3-хлорфенил}этиламино]пропил)феноксиацетат
ВНТ920 = 6-аллил-2-амино-5,6,7,8-тетрагидро-4Н-тиазоло-[4,5-с/]-азепин
СЕС = хлороэтилклонидин
ДАГ = диацилглицерол
ИТФ = инозитолтрифосфат
WB4101 = Л/-[2-(2.6-диметоксифенокси)этил]-2,3-дигидро-1,4-бензодиоксан-2-метанамин
тельно связывается с одной подгруппой рецепторов в концентрациях, слишком малых, чтобы взаимодействовать с другой подгруппой. Например, норадреналин преимущественно активирует ргрецеп-торы по сравнению с р2-рецепторами. Однако селективность обычно не абсолютна, и при повышении концентрации вещество может также связываться с другими родственными классами рецепторов. В результате “числовая” субклассификация адренорецепторов клинически важна только для средств, имеющих относительно выраженную селективность.
Номенклатура
Номенклатура подтипов адренорецепторов запутана, так как нет общепринятой формальной системы. Разные авторы могут использовать одни и те же названия, говоря о совершенно различных рецепторах. Следовательно, нужно быть внимательным при чтении сообщений о результатах исследований. Ежегодные выпуски номенклатуры рецепторов (1994 Receptor Nomenclature Supplement), публикующиеся в журнале Trends in Pharmacological Sciences, дают полезные сведения о современной но-
ТАБЛИЦА 9-2. Относительная селективность агонистов адренорецепторов
Группа агонистов Относительный
аффинитет к рецепторам
а-агонисты Фенилэфрин, метоксамин Клонидин, метилнорадреналин a1>a2»»>0 a2>a, >»»0
Смешанные а-, 0-агонисты Норадреналин Адреналин a,= a2;0! >>02 ai = a2; 0, = 02
0-агонисты Добутамин Изопротеренол Тербуталин, метапротеренол, альбутерол, ритодрин 0i >02»» a 0i = 02»» a 02» 0i »»a
Дофаминовые агонисты Дофамин D,= D2»0»a
менклатуре, но необходимо знать, что названия и описания приведенных там рецепторов могут быстро изменяться.
Точное число подтипов адренорецепторов, находящихся в тканях человека, не определено, для некоторых из них физиологическое или фармакологическое значение до сих пор не выяснено. По-видимому, возможно создание новых лекарств на основании экспрессии определенных подтипов рецепторов в отдельных тканях-мишенях. Например, определив, какие именно подтипы рецепторов (ocj или а2) находятся в конкретных кровеносных сосудах, можно создать вещества, селективные в отношении разных сосудистых зон, таких как сосуды внутренних органов или коронарные сосуды.
Молекулярные механизмы симпатомиметического действия
Эффекты катехоламинов опосредуются поверхностными рецепторами клеток. Как отмечалось в главе 2, эти рецепторы связаны посредством G-белков с различными эффекторными белками и регулируют их активность. Каждый G-белок является гетеротримером, состоящим из а-, 0- и у-субъеди-ниц. Различные G-белки отличаются а-субъедини-цами; меньшие вариации обнаружены между 0- и у-субъединицами. К наиболее важным для функци
онирования адренорецептора G-белкам относятся: Gs, стимулирующий аденилатциклазу; G, угнетающий этот фермент; Gq, связывающий а-рецептор с фосфолипазой С. Активация катехоламинами рецепторов способствует диссоциации ГДФ из а-субъединицы соответственного G-белка. Затем с G-белком связывается ГТФ, а комплекс распадается на а- и 0-у-субъединицы. Активированная связыванием ГТФ а-субъединица регулирует активность эффектора, например аденилатциклазы, цГМФ-фосфодиэстеразы, фосфолипазы С, ионных каналов. Инактивируется а-субъединица гидролизом связанного ГТФ с образованием ГДФ и Р, и последующей реассоциацией а-субъединицы с 0-у-субъединицей. Комплекс 0-у-субъединиц может иметь дополнительные независимые эффекты, хотя их физиологическое значение пока не выяснено.
Типы рецепторов
А. а-рецепторы. Механизм активации а-рецеп-торов катехоламинами выяснен не так хорошо, как для 0-рецепторов. Было обнаружено, что чувствительный к хлороэтилклонидину а)В-подтип рецепторов стимулирует образование инозитол-1,4,5-трифосфата, в то время как хлороэтилклонидин-резистентный а1Л-подтип может и не активировать фосфолипазу С, но определенно контролирует закрытие мембранных кальциевых каналов.
Наиболее часто наблюдаемым эффектом стимуляции аррецепторов является повышение цитозольной концентрации ионов кальция. За исключением некоторых зон головного мозга этот эффект не опосредован изменением активности аденилатциклазы или концентрации цАМФ в клетках. Механизм, вызывающий это увеличение концентрации Са2+, полностью не выяснен. В определенных гладкомышечных клетках активация а(-рецепторов приводит к входу Са2+ через мембрану, возможно, через открытие рецепторзависимых кальциевых каналов. Активация а(-рецепторов во многих клетках ведет к разрушению полифосфоинозитидов в инозитолтрифосфат и диацилглицерол (рис. 9-1). G-белок, называемый G(|, связывает at -рецептор с фосфолипазой С. Инозитол-1,4,5-трифосфат (ИТФ) способствует выделению депонированного Са2+ из внутриклеточных запасов, что увеличивает цитоплазматическую концентрацию Са2+ и вызывает активацию различных кальцийзависимых протеинкиназ. ИТФ последовательно дефосфорилиру-
Агонист
рецептор
ГДФ
ГТФ
Фосфолипаза С
киназа С
Активированная протеинкиназа С
ИТФ
Са2+- зависимая протеинкиназа
zp, Свободный кальций
Депонированный кальций
Активированная протеинкиназа
Рис. 9-1. Активация реакций, опосредованных а(-рецепторами. Стимуляция ai-рецепторов катехоламинами приводит к активации Сч-белка. а-субъединица этого G-белка активирует эффектор, фосфолипазу С, что приводит к образованию ИТФ (инозитол-1,4,5-трифосфата) и ДАГ (диацилглицерола) из фосфатидилинозитол-4,5-бифосфата(ФДИ 4,5БФ). ИТФ стимулирует выделение кальция (Са2+) из депо, что приводит к увеличению концентрации Са2+ в цитоплазме. Затем Са2+ может активировать Са2+-зависимые протеинкиназы, которые в свою очередь фосфорилируют свои субстраты. ДАГ активирует протеинкиназу С
ется, что в итоге ведет к образованию свободного инозитола. Однако в результате метаболизма ИТФ могут образовываться и накапливаться некоторые другие фосфорилированные изомеры инозитола. Биологическая значимость этих молекул неясна. Диацилглицерол активирует протеинкиназу, называемую протеинкиназой С. Физиологическое значение протеинкиназы С неясно, однако известно, что в некоторых клетках она способствует клеточному делению. Протеинкиназа С может также регулировать долговременные ответы на активацию at-рецепторов.
а2-рецепторы угнетают активность аденилатциклазы и вызывают снижение внутриклеточного уровня цАМФ. Несмотря на то, что этот эффект хорошо документирован, нельзя сказать, является ли это действие единственным или основным следствием активации а2-рецепторов. Так, агонисты а2-рецепторов вызывают агрегацию тромбоцитов и снижение содержания цАМФ в тромбоцитах, однако неизвестно, является ли агрегация результатом снижения уровня цАМФ. а2-угнетение аденилатциклазы регулируется Gj-белком, связанным с
а2-рецептором (рис. 9-2). Как именно активация G, ведет к ингибированию аденилатциклазы — невы-яснено. Существуют два возможных механизма: 1) свободные р-у-комплексы G, комбинируются со свободными а-субъединицами Gs, инактивируя их; 2) а-субъединицы G, прямо ингибируют активность аденилатциклазы. Есть некоторые свидетельства в пользу каждого из этих механизмов. Кроме того, ряд эффектов а2-адренорецепторов не зависит от их способности угнетать аденилатциклазу, а реализуется через активацию калиевых каналов и закрытие кальциевых каналов.
Б. Р-рецепторы. Механизм действия Р-агонис-тов изучен весьма детально. Активация всех трех подтипов рецепторов (Рь р2 и рз) приводит к активации аденилатциклазы и интенсифицирует превращение АТФ в цАМФ (рис. 9-2). Активация аденилатциклазы вызывается стимулирующим Gs-белком. цАМФ является основным вторичным мессенджером Р-рецепторной активации. Например, в печени многих видов животных активация Р-рецепторов усиливает синтез цАМФ, что’приводит к каскаду реакций, кульминацией которых является ак-
АТФ цАМФ
Фермент-Р04 2R
I
Биологический эффект
R2C2
Протеинкиназа
Рис. 9-2. Активация и угнетение аденилатциклазы агонистами, связывающимися с катехоламиновыми рецепторами. Связывание с Р-адренорецепторами стимулирует аденилатпиклазу посредством активации стимулирующего G-белка, G„ что приводит к высвобождению его ос-субъединиц, соединенных с ГТФ. Эта сс,-субъединица прямо активирует аденилатциклазу, в результате чего увеличивается скорость синтеза цАМФ. Лиганды ос2-адренорецепторов угнетают аденилатциклазу, вызывая диссоциацию угнетающего G-белка, Gi( на субъединицы: агсубъединицу, соединенную с ГТФ, и Р-у-единицу. Механизм, посредством которого эти субъединицы ингибируют аденилатциклазу, неизвестен. цАМФ связывается с регуляторной субъединицей (R) цАМФ-зависимой протеинкиназы, что приводит к освобождению активной каталитической субъединицы (С), фосфорилирующей специфические белковые субстраты и модифицирующей их активность
тивация гликогенфосфорилазы. В сердце активация p-рецепторов увеличивает вхождение Са2+ через клеточную мембрану и его секвестрацию внутри клетки. Активация p-рецепторов содействует также расслаблению гладкой мышцы. Хотя механизм этого эффекта не выяснен, он может включать фосфорилирование киназы легких цепей миозина, превращающее ее в неактивную форму (рис. 12-1).
В. Дофаминовые рецепторы. Рецептор D( обычно ассоциируется со стимуляцией аденилатциклазы (табл. 9-1). Например, вызванное Dt-рецептором расслабление гладких мышц, вероятно, происходит вследствие аккумуляции цАМФ в сосудистых зонах, где дофамин является вазодилататором. Было
показано, что О2-рецепторы угнетают активность аденилатциклазы, открывают калиевые каналы и уменьшают ток ионов кальция.
Регуляция рецепторов
Ответы, вызываемые стимуляцией адренорецепторов, не являются неизменными. Количество рецепторов на поверхности клетки и их функциональная активность могут регулироваться катехоламинами, другими гормонами и лекарственными средствами, зависеть от возраста пациента и перенесенных им заболеваний. Эти изменения модифицируют величину физиологического ответа ткани
на катехоламины. Один из наиболее хорошо изученных примеров рецепторной регуляции — это десенситизация адренорецепторов, которая может наблюдаться после применения катехоламинов и других симпатомиметиков. После воздействия агониста на клетку или ткань в течение определенного периода времени эти структуры часто становятся менее реактивными при дальнейшей стимуляции тем же агентом. Синонимами десенситизации являются такие термины как толерантность, рефрак-терность и тахифилаксия. Этот процесс имеет существенную клиническую значимость, поскольку может лимитировать терапевтический ответ на симпатомиметические средства.
Широко исследуются механизмы десенситизации клеток к Р-адреностимуляции. Полагают, что существует по меньшей мере три различных механизма. В некоторых клетках десенситизация вызывается посредством секвестрации рецепторов (быстрое и преходящее состояние, при котором рецепторы делаются временно неспособными активироваться агонистами). Второй процесс типа down-регуляции (т. е. уменьшение числа рецепторов в клетке) может наблюдаться при действии на рецептор ферментов или при ингибировании их синтеза. Третий механизм, фосфорилирование цитоплазматической части рецептора протеинкиназой А или киназой P-адренорецепторов (Р-АРК), может нару
шать связывание p-адренорецепторов с Gs частично путем облегчения ассоциации фосфорилированного Р-адренорецептора с аррестинподобным белком с мол. м. 48 000 (рис. 2-13).
Химия и фармакокинетика симпатомиметических средств
Фенилэтиламин может рассматриваться как исходное соединение — родоначальник симпатомиметических средств (рис. 9-3). Это соединение состоит из бензольного кольца с боковой этиламинной цепью. Заместители могут находиться: 1) в терминальной аминогруппе, 2) в бензольном кольце, 3) у а- или p-углеродных атомов. Включением ОН-групп в третье и четвертое положение бензольного кольца получают вещества, известные как катехоламины.
Модификация фенилэтиламина ведет к изменению аффинитета веществ к а- и p-рецепторам, а также влияет на их внутреннюю способность активировать рецепторы. К тому же химическая структура определяет фармакокинетические свойства этих молекул. Симпатомиметические средства могут активировать одновременно а- и р-рецепторы, однако относительная активность влияния на а-р'ецепторы по сравнению с влиянием на р-рецеп-
Фенилэтиламин
ОН I
СН —СН2 —NH —СНз
Адреналин
Рис. 9-3. Фенилэтиламин и некоторые основные катехоламины. Катехол представлен для сравнения
СН2—СН2—NH2
Дофамин
торы может колебаться от почти чистой «-активации (метоксамин) до преимущественно Р-актива-ции (изопротеренол).
А. Замещения в аминогруппе. Увеличение размера алкильных заместителей в аминогруппе приводит к повышению активности в отношении Р-рецепторов. Например, введение метильной группы в норадреналин, приводящее к образованию адреналина, усиливает активацию р2-рецепторов. Еще более возрастает P-активность при введении изопропильной группы (изопротеренол). Для повышения ргселективности обычно необходимо введение в аминогруппу больших заместителей. Чем больше заместитель в аминогруппе, тем меньше влияние вещества на а-рецепторы, в частности изопротеренол очень слабо действует на а-рецепторы.
Б. Замещения в бензольном кольце. Максимальная а- и P-активность найдена у катехоламинов (веществ, имеющих ОН-группы в третьем и четвертом положениях). Отсутствие одной из этих групп, особенно гидроксила у атома СЗ, без других изменений в кольце может существенно уменьшить биологическую активность вещества. Например, фенилэфрин (рис. 9-4) значительно менее сильное средство, чем адреналин: его a-активность ниже примерно в 100 раз, а P-активность почти отсутствует за исключением случаев применения в очень высоких концентрациях. Однако катехоламины инактивируются катехол-орто-метилтрансферазой (КОМТ) — ферментом, найденным в кишечнике и печени (глава 6). Поэтому отсутствие одной или обеих ОН-групп у фенольного кольца увеличивает биодоступность и продолжительность действия вещества после перорального введения. Более того, отсутствие в кольце ОН-групп обдегчает проник
новение молекулы в центральную нервную систему. Например, эфедрин и амфетамин (рис. 9-4) активны при пероральном введении, имеют большую продолжительность действия и оказывают влияние на центральную нервную систему, что не наблюдается у катехоламинов.
В. Замещения у ато^а углерода в «-положении. Замещения у а-углерода блокируют окисление соединений моноаминооксидазой (МАО) и пролонгируют действие таких средств, особенно некате-холаминов. Примерами а-замещенных соединений являются эфедрин и амфетамин (рис. 9-4). «-метильные соединения называют также фенилизо-пропиламинами. Наряду с устойчивостью к окислению под действием МАО фенилизопропилами-ны имеют повышенную способность вытеснять катехоламины из мест депонирования в норадренергических нервах. Поэтому частично их активность зависит от наличия нормальных запасов норадреналина. Они являются симпатомиметиками непрямого действия.
Г. Замещения у атома углерода в ^-положении. Агонисты прямого действия обычно имеют Р-гидро-ксильную группу за исключением дофамина. Наряду с активирующим действием на адренорецепторы эта гидроксильная группа может быть важна для депонирования симпатомиметических аминов в синаптических везикулах.
Влияние симпатомиметиков на системы и органы
Характерные особенности клеточных эффектов симпатомиметиков показаны в табл. 6-3 и 9-3. Общее действие определенного препарата на интакт-
Фенилэфрин
СН —CH-NH-СНз
ОН СНз
Амфетамин
Эфедрин
Рис. 9-4. Некоторые примеры некатехоламиновых симпатомиметических средств
ТАБЛИЦА 9-3. Распределение типов адренорецепторов
Тип Ткань Эффекты
at Большая часть гладких мышц сосудов (иннервируемых) Дилататор зрачка Пиломоторные гладкие мышцы Печень (крысы) Сердце Сокращение Сокращение (расширение зрачка) Пилоэрекция Гликогено^&з Повышение силы сокращения
а2 Постсинаптические адренорецепторы ЦНС Тромбоциты Адренергические и холинергические нервные терминали Гладкие мышцы некоторых сосудов Жировые клетки Множественные Агрегация Подавление высвобождения медиаторов Сокращение Подавление липолиза
Сердце Повышение силы и частоты сокращений
₽2 Гладкие мышцы дыхательных путей, матки и сосудов Скелетные мышцы Печень(человека) Расслабление гладких мышц Усиление захвата калия Активация гликогенолиза
₽з Жировые клетки Активация липолиза
D, Гладкие мышцы Расширение почечных сосудов
d2 Нервные окончания Модулируют высвобождение медиатора
ный организм зависит от его сравнительной аффинности к рецепторам (а или (3), внутренней активности и от компенсаторных рефлексов, вызванных прямым действием препарата.
Влияние на сердечно-сосудистую систему
А. Кровеносные сосуды. Адренорецепторы регулируют тонус гладкой мускулатуры сосудов, следовательно, катехоламины важны для контроля периферического сосудистого сопротивления и емкости венозного русла, а-рецепторы опосредуют повышение артериального сопротивления, а р2‘Ре-цепторы — расслабление гладких мышц. Типы рецепторов в сосудах разной локализации значительно отличаются (табл. 9-4). Сосуды кожи, так же как и сосуды внутренних органов, в основном содержат а-рецепторы и сужаются под действием адреналина и норадреналина. Сосуды скелетных мышц могут сужаться или расширяться в зависимости от активации а- или 0-рецепторов. Таким образом, общее действие симпатомиметиков на кровеносные сосуды зависит от сравнительной выраженности влияния препарата на а- и 0-рецепторы и от анатомической принадлежности сосудов, на которые он
действует. Кроме того, Di-рецепторы вызывают вазодилатацию сосудов почек, внутренних органов, а также коронарных и церебральных артерий. Активация D ^рецепторов сосудов почек может играть огромную роль в выделении натрия с мочой под действием препаратов дофамина.
Б. Сердце. Прямые эффекты симпатомиметиков на сердце прежде всего определяются ргрецепторами, хотя 02-рецепторы и в меньшей степени а-рецепторы также имеют некоторое значение. Активация Р-рецепторов приводит к повышению тока ионов кальция в клетки сердца. Это вызывает как электрические (рис. 9-5), так и механические последствия. Повышается пейсмекерная активность: нормальная (синоатриальный узел) и аномальная (например, волокна Пуркинье) — положительный хронотропный эффект. Увеличивается скорость проведения по атриовентрикулярному узлу, рефрактерный период уменьшается. Усиливается сократимость сердца (положительный инотропный эффект), ускоряется релаксация. В результате сокращение изолированной сердечной мышцы усиливается, но укорачивается по продолжительности. В интактном сердце внутрижелудочковое давление растет и снижается быстрее, время сердечного выброса уменьшается. Эти прямые эффекты легко продемон-
ТАБЛИЦА 9-4. Реакция сердечно-сосудистой системы на симпатомиметические амины’
Показатель Фенилэфрин Адреналин Изоп ротеренол
Сосудистое сопротивление (тонус)
Кожи, слизистых оболочек (а) ТТ ТТ 0
Скелетных мышц (02. а) т « i или Т и
Почек (а, D,) т > т
Внутренних органов (а) ТТ i или Т2 г
Общее периферическое сопротивление ттт 1 или Т2 и
Тонус вен т т 1
Сердечная деятельность
Сократимость (0,) Оили Т ТТТ ттт
Частота сердечных сокращений (преимущественно 0,) J4- (вагусный рефлекс) Т или i ттт
Ударный объем 0, т, т Т т
Сердечный выброс 1 Т ТТ
Артериальное давление
Среднее ТТ Т 1
Диастолическое ТТ i или Т2 и
Систолическое ТТ ТТ 0 или J-
Пульсовое 0 ТТ ТТ
’ ? — повышение, -I — понижение, 0 — эффект отсутствует.
2 Небольшие дозы снижают, большие дозы повышают.
стрировать в отсутствие рефлексов, вызванных изменениями артериального давления, то есть на изолированных препаратах миокарда и у пациентов с ганглиоблокадой. При нормальной рефлекторной
Рис. 9-5. Действие адреналина на трансмембранный потенциал пейсмекерных клеток (водителей ритма) в сердце лягушки. Запись, помеченная стрелками, была сделана после добавления адреналина. Отметьте увеличение крутизны диастолической деполяризации и снижение интервала между потенциалами действия. Это ускорение импульсации пейсмекерных клеток типично для стимуляторов Pi-рецепторов. (Из: Brown Н., Giles W., Noble S. Membrane currents underlying rhythmic activity in frog sinus venosus. In: The Sinus Node: Structure, Function, and Clinical Relevance. Bonke F. I. M. (ed.), Martinus Nijhoff, 1978.)
активности прямые эффекты на частоту сердечных сокращений могут подавляться рефлекторными реакциями на изменения артериального давления.
С. Артериальное давление. Эффекты симпатомиметиков на артериальное давление можно объяснить на основе их действия на сердце, периферическое сосудистое сопротивление и венозный возврат (рис. 6-7 и табл. 9-4). Сравнительно селективные а-агонисты типа фенилэфрина повышают периферическое артериальное сопротивление и снижают емкость венозного русла. Увеличение сопротивления артериол обычно приводит к значительному повышению артериального давления (рис. 9-6). При сохранении нормальных сердечнососудистых рефлексов повышение артериального давления вызывает опосредованное барорецепторами повышение тонуса вагуса с замедлением частоты сердечных сокращений. Однако сердечный выброс может не снижаться в той же степени, что и частота сердечных сокращений, так как этому противодействует повышение венозного возврата. Более того, прямая стимуляция а-адренорецепторов сердца может оказывать небольшое положительное инотропное действие. Таковы эффекты а-агонис-тов у нормотоников, применение же их у пациентов с гипотензией обычно не приводит к быстрым рефлекторным реакциям, так как в этой ситуации
164
артериальное давление нормализуется, не поднимаясь выше.
Действие 0-агонистов на артериальное давление значительно отличается. Стимуляция [3-рецепторов сердца повышает сердечный выброс. Сравнительно избирательные 0-агонисты типа изопротеренола понижают и периферическое сосудистое сопротивление, расширяя сосуды определенных участков тела (табл. 9-4). Итоговый эффект — небольшое повышение или отсутствие изменения систолического давления при снижении диастолического (рис. 9-6). Действие препаратов, обладающих и а-, и 13-эффектами (например, адреналина и норадреналина), еще будет обсуждаться.
Глаз
Радиальная мышца, расширяющая зрачок, содержит а-рецепторы, их активация веществами типа фенилэфрина вызывает мидриаз (рис. 6-9).
а- и 0-стимуляторы оказывают существенное влияние и на внутриглазное давление. В настоящее время есть сведения о том, что а-агонисты повышают отток внутриглазной жидкости, а 0-аго-нисты усиливают ее продукцию. Эти эффекты важно учитывать при лечении глаукомы. 0-сти-муляторы незначительно расслабляют цилиарную мышцу, вызывая небольшое снижение аккомодации.
Дыхательные пути
Гладкие мышцы бронхов содержат 02-рецепторы, активация которых вызывает расширение бронхов (глава 19 и табл. 9-3). Кровеносные сосуды слизистой оболочки верхних дыхательных путей содержат at-рецепторы; деконгестивное (противоотеч-ное) действие а-стимуляторов имеет клиническое значение (раздел “Клиническая фармакология симпатомиметиков ”).
190/145
АД
ЧСС
145/100
160
170
--- Фенилэфрин
Рис. 9-6. Эффекты а-селективных (фенилэфрин), 0-селективных (изопротеренол) и неселективных (адреналин; симпатомиметиков у собаки после внутривенной болюсной инъекции. (АД — артериальное давление, ЧСС — частотг сердечных сокращений.) В условиях анестезии рефлексы снижены, но не полностью
Желудочно-кишечный тракт
Расслабление гладкой мускулатуры ЖКТ может быть вызвано как а-, так и p-стимуляцией. р-ре-цепторы расположены непосредственно на гладкомышечных клетках и вызывают релаксацию путем гиперполяризации и понижения импульсной активности этих клеток, а-стимуляторы, особенно а2-се-лективные агонисты, снижают активность мышцы непрямым путем — через пресинаптическое подавление высвобождения ацетилхолина и, возможно, других стимуляторов в нервной системе кишечника (глава 6). Опосредованный а-рецепторами эффект, вероятно, имеет большее фармакологическое значение, чем действие Р-стимуляторов. Возбуждение а2-рецепторов приводит к снижению тока ионов натрия и воды в просвет кишечника.
Мочеполовая система
Матка содержит а- и р2-рецепторы. Способность р-агонистов расслаблять матку используют при патологии беременности (раздел “Клиническая фармакология симпатомиметиков”). Мышцы дна мочевого пузыря, сфинктер уретры и простата содержат а-рецепторы, которые опосредуют мышечное сокращение и, следовательно, удержание мочи. Специфический подтип а-рецепторов, участвующий в сокращении простаты и дна мочевого пузыря, неизвестен и в настоящее время активно исследуется. Возбуждение Р2-рецепторов стенки мочевого пузыря вызывает его расслабление. Эякуляция зависит от нормального функционирования а-рецепторов (и, возможно, пуринергических рецепторов), которые активируют семявыводящий проток, семенные пузырьки и простату. Расслабление эректильной ткани, которое обычно следует за эякуляцией, также опосредуется норадреналином, высвобождаемым из симпатических нервов (и, возможно, нейропептидом Y) (deGroat, 1990). а-активация вызывает подобное расслабление эректильной ткани у самок животных.
Экзокринные железы
Слюнные железы содержат адренорецепторы, которые регулируют секрецию амилазы и воды. Однако некоторые симпатомиметики, например клонидин, вызывают ощущение сухости во рту. Механизм этого эффекта не ясен: возможно, он свя
зан с центральным действием, хотя не исключаются и периферические влияния.
Апокринные потовые железы, расположенные на ладонях и вадекоторых других областях, реагируют на а-стимуляторы повышением потоотделения. Эти железы не участвуют в терморегуляции, и их функция обычно отражает состояние психологического стресса. (Диффузно расположенные терморегуляторные эккринные потовые железы регулируются холинергическими симпатическими постганглионарными нервными волокнами, которые активируют мускариновые холинорецепторы — глава 6.)
Метаболические эффекты
Симпатомиметики существенно влияют на промежуточный обмен. Активация Р-адренорецепторов в жировых клетках приводит к липолизу. Р-адре-норецепторы, которые опосредуют этот эффект, относят к рз-адренорецепторам. Человеческие ли-поциты содержат также а2-рецепторы, которые ингибируют липолиз путем снижения уровня внутриклеточного цАМФ. Симпатомиметики усиливают гликогенолиз в печени, что приводит к повышению уровня глюкозы в крови. В печени человека эффекты катехоламинов, вероятно, в основном опосредуются p-рецепторами, хотя роль а-рецепторов полностью отвергать нельзя. В высоких концентрациях катехоламины могут вызывать метаболический ацидоз. Активация р2-рецепторов эндогенным адреналином или симпатомиметиками способствует поступлению ионов калия в клетки с последующим снижением уровня внеклеточного К+. Это может привести к снижению концентрации К+ в плазме во время стресса или защитить от повышения содержания К+ в плазме во время физической нагрузки. Блокада р2-рецепторов оказывает противоположное действие.
Влияние на эндокринную и другие функции
Катехоламины — важные эндогенные регуляторы гормональной секреции некоторых желез. Секреция инсулина усиливается p-рецепторами и подавляется а2-рецепторами. Сходным образом секреция ренина усиливается pt-рецепторами и угнетается а2-рецепторами. Действительно, антагонисты p-рецепторов могут снижать уровень ренина
плазмы хотя бы частично при помощи этого механизма. Адренорецепторы также модулируют секрецию паратиреоидного гормона, кальцитонина, тироксина и гастрина, однако физиологическое значение такого контроля неясно. В высоких концентрациях адреналин и сходные вещества вызывают лейкоцитоз.
Влияние на центральную нервную систему
Действие симпатомиметиков на центральную нервную систему варьирует очень значительно в зависимости от способности лекарственного средства проникать через гематоэнцефалический барьер. Катехоламины почти полностью задерживаются на уровне этого барьера, и их эффекты на ЦНС субъективно отмечают только при массивных инфузиях. Они весьма нежелательны и варьируют от “нервозности” до “ощущения неминуемой катастрофы”. В отличие от этих средств некатехоламино-вые вещества непрямого действия, например амфетамины, которые легко проникают в ЦНС через гематоэнцефалический барьер, вызывают количественно более выраженные эффекты. Они варьируют от незначительной психостимуляции с улучшением внимания при монотонной работе, повышения настроения, возникновения бессонницы, эйфории, анорексии и до психотического поведения на самых высоких уровнях стимуляции. Эти эффекты нельзя однозначно объяснить ни а-, ни р-рецепторными влияниями, они скорее всего вызваны усилением дофаминозависимых процессов в ЦНС.
Специфические симпатомиметики
Катехоламины
Адреналин (эпинефрин) — очень сильный вазоконстриктор и кардиостимулятор. Повышение систолического давления после высвобождения или введения адреналина связано с его положительным хронотропным и инотропным действием на сердце (преимущественно на Pi-рецепторы) и с вазоконстрикцией сосудов различной локализации (а-рецеп-торы). Адреналин также активирует р2-рецепторы некоторых сосудов (например, сосуды скелетных мышц), расширяя их. Вследствие этого общее пе
риферическое сопротивление может падать, что объясняет снижение диастолического давления, наблюдаемое после вве^ния адреналина (рис. 9-6).
Норадреналин (л^вартеренол, норэпинефрин) и адреналин обладают сходными эффектами на Ргрецепторы сердца и сходной выраженностью действия на а-рецепторы. Норадреналин почти не действует на р2-рецепторы. Вследствие этого норадреналин повышает периферическое сосудистое сопротивление и как систолическое, так и диастолическое давление. Компенсаторные вагусные рефлексы обычно нейтрализуют положительный хронотропный эффект норадреналина, но положительное инотропное действие на сердце сохраняется.
Изопротеренол (изопреналин) — очень сильный агонист p-рецепторов, практически не влияющий на а-рецепторы. Он обладает положительными хронотропным и инотропным эффектами. Так как изопротеренол почти исключительно активирует Р-рецепторы, он является сильным сосудорасширяющим средством. Действие препарата приводит к значительному повышению сердечного выброса, связанного со снижением диастолического и среднего артериального давления и с менее выраженным снижением или небольшим повышением систолического давления (табл. 9-4 и рис. 9-6).
Дофамин, непосредственный метаболический предшественник норадреналина, активирует Di-рецепторы разных сосудов, вызывая вазодилатацию. Такое действие на почечные сосуды имеет большое клиническое значение. Вклад активации пресинаптических Ц2-рецепторов, подавляющей высвобождение норадреналина, в реализацию этого эффекта количественно не оценен. Кроме того, дофамин активирует р(-рецепторы сердца. В низких дозах он обладает сравнительно небольшим влиянием на общее периферическое сопротивление. Однако при применении более высоких доз дофамин активирует сосудистые а-рецепторы, вызывая вазоконстрикцию. Таким образом, высокие концентрации дофамина могут имитировать действие адреналина.
Ибопамин — активный при приеме внутрь эфир масляной кислоты метилдофамина (второе название — эпинин). В крови эфирная связь гидролизуется неспецифическими эстеразами с образованием эпинина, который по фармакологическим свойствам сходен с дофамином. Это средство сейчас проходит клинические испытания, его клиническое значение пока не вполне ясно.
Добутамин — относительно селективный синтетический Pi-катехоламин. Как еще будет обсуждаться, добутамин активирует и агрецепторы.
Другие симпатомиметики
Эти лекарственные средства представляют интерес в связи с их фармакокинетическими свойствами (активность при пероральном приеме, проникновение в ЦНС) или в связи с их относительной селективностью действия на определенные подклассы рецепторов.
Фенилэфрин был уже описан как пример относительно селективного а-агониста (табл. 9-2). Он непосредственно активирует эти рецепторы. Не являясь дериватом катехола (рис. 9-4), он не инактивируется КОМТ и действует значительно дольше, чем катехоламины. Фенилэфрин является эффективным мидриатиком, обладает противоотеч-ным действием и может применяться для повышения давления (рис. 9-6).
Метоксамин фармакологически сходен с фенилэфрином, так как действует преимущественно как агонист агрецепторов. Он способен вызывать длительное повышение артериального давления вследствие вазоконстрикции, а также брадикардию из-за активации п. vagus. Метоксамин удобен для парентерального введения, но его клиническое применение ограничено гипотензивными состояниями.
Эфедрин встречается во многих растениях. В Китае его используют уже более 2000 лет. На западе был введен приблизительно 70 лет назад как первый активный при приеме внутрь симпатомиметик. Являясь некатехоловым фенилизопропиламином (рис. 9-4), он обладает высокой биодоступностью и большой продолжительностью действия. Как и многие другие фенилизопропиламины, эфедрин в основном выводится с мочой в неизмененном виде. Он является слабым основанием и его выведение можно ускорить подкислением мочи.
Эфедрин прежде всего действует путем высвобождения катехоламинов из депо. Кроме того, он оказывает незначительный прямой эффект на адренорецепторы. Препарат неселективен, и спектр его эффектов похож на таковой у адреналина. При попадании в ЦНС он вызывает некоторую амфетаминоподобную стимуляцию, которая отсутствует у катехоламинов.
Клинически эфедрин используется, когда необходим пролонгированный эффект, особенно при
пероральном введении. Применяется в основном для уменьшения отека слизистой носа (деконгес-тивное действие) при закапывании препарата в нос и в качестве прессорного агента.
Ксилометазолин и оксиметазолин — это а-аго-нисты прямого действия. Препараты применяют местно с целью деконгестивного действия, так как они вызывают вазоконстрикцию в слизистой носа. В больших дозах оксиметазолин может вызывать гипотензию, предположительно в связи с центральным клонидиноподобным эффектом (глава 11). Как отмечено в табл. 9-1, оксиметазолин обладает значительной аффинностью к а2Л-рецепторам.
Амфетамин является фенилизопропиламином (рис. 9-4) и имеет значение в основном в связи с его употреблением и злоупотреблением в качестве стимулятора ЦНС (глава 31). По фармакокинетическим свойствам он похож на эфедрин, но быстрее проникает в ЦНС. Оказывает более выраженное стимулирующее действие на настроение и уровень бодрствования и подавляет аппетит. Его периферические эффекты в основном связаны с высвобождением катехоламинов. Метамфетамин (N-метиламфетамин) очень похож на амфетамин и обладает еще более выраженными центральными эффектами по сравнению с периферическими. Фенметразин (рис. 31-1) — еще одно производное фенилизопропиламина с амфетаминоподобными эффектами. Его применяют для подавления аппетита. Возможны случаи злоупотребления. Метилфенидат и пемолин — препараты, близкие к амфетамину по спектру действия и потенциалу развития злоупотребления. Аналоги амфетамина эффективны у некоторых детей с гиперактивностью и нарушением внимания (“Клиническая фармакология симпатомиметиков”). Фенилпропа-ноламин обладает слабым по сравнению с амфетамином действием на настроение и присутствует во многих безрецептурных средствах для похудания. Хотя он сравнительно безопасен при приеме в рекомендованных дозах, в больших дозах может вызывать тяжелую гипертензию, повышать риск инсульта и повреждения миокарда. Нет доказательств того, что лечение ожирения этими препаратами приводит к длительному поддержанию сниженного веса.
Рецепторселективные симпатомиметики
oq-селективные препараты не обладают особыми преимуществами перед неселективными препа
ратами. а2-селективные агонисты способны парадоксально снижать давление путем воздействия на ЦНС, хотя их местная аппликация может вызывать вазоконстрикцию. Такие препараты (например, клонидин, метилдопа, гуанфацин, гуанабенз) применяются для лечения гипертензии и обсуждаются в главе 11.
Р-селективные агонисты имеют очень важное значение, так как удалось разделить рг и р2-эффек-ты (табл. 9-2). Хотя это разделение неполное, его достаточно для снижения побочных эффектов в некоторых клинических ситуациях.
К Pi-селективным препаратам относятся добу-тамин и парциальный агонист преналтерол (рис. 9-7). Так как они менее эффективно активируют сосудорасширяющие р2-рецепторы, они повышают сердечный выброс с менее выраженной рефлекторной тахикардией, чем неселективные р-аго-нисты типа изопротеренола. Проявление некоторых важных фармакологических эффектов добутамина существенно зависит от применения смеси разных изомеров препарата. В то время как один изомер является избирательным р(-агонистом, другой стереоизомер действует на а-рецепторы, что уменьшает вазодилатацию и может усиливать положительный инотропный эффект изомера с преимущественно Pf-активностью. Основное ограничение применения этих препаратов (как и других симпатоми
метиков прямого действия) — это развитие толерантности при длительном применении.
р2-селективные препараты занимают значительное место в терапии бронхиальной астмы и обсуждаются в главе 19. Другое показание к применению — необходимость расслабления матки при преждевременных родах (ритодрин). Некоторые примеры препаратов, используемых в настоящее время, приведены на рис. 9-7 и 19-6. Существует еще очень много препаратов этой группы, кроме того, новые средства проходят испытания.
Особые симпатомиметики
Кокаин — местный анестетик, обладающий периферическим симпатомиметическим действием, которое связано с подавлением обратного захвата медиатора в норадренергических синапсах (глава 6). Он хорошо проникает в ЦНС и вызывает более короткий, но более сильный амфетаминоподобный эффект. Эти свойства, а также то, что кокаин можно курить, вдыхать через нос или вводить парентерально для получения более быстрого эффекта, привели к широкому распространению злоупотребления препаратом (глава 31).
Тирамин (рис. 6-5) — это промежуточный продукт метаболизма тирозина в организме, который также находится в высоких концентрациях в фер-
₽1 - СЕЛЕКТИВНЫЕ
О — СН2 — СН - СН2 - NH — СН(СН3)2
ОН
Рис. 9-7. Примеры Р,- и р2-селективных агонистов
₽2 - СЕЛЕКТИВНЫЕ
Преналтерол
СН — СН2 — NH — С(СН3)3
ОН
Тербуталин
ТАБЛИЦА 9-5. Пищевые продукты, содержащие высокие концентрации тирамина или других симпатомиме-тикоа. Если пациент принимает ингибитор МАО, то 20—50 мг тирамина в пище могут значительно повысить артериальное давление (глава 29). Отметим, что сыр, сосиски, маринованная рыба и дрожжевые продукты содержат количества тирамина, которые могут быть опасными. Но возможно, что некоторые другие пищевые продукты также содержат потенциально опасные количества тирамина
Пища Содержание тирамина в средней порции
Пиво Некоторые бобы Сыр зрелый или незрелый Нет данных Незначительное (но содержат дофамин) 0-130 мг (в сырах чеддер, груер и стилтон особенно высокое содержание)
Куриная печень Шоколад Ферментированные колбасные изделия (например, салями, пепперони и т. д.) Копченая или маринованная рыба (например, маринованная сельдь) Улитки Вино (красное) Дрожжи (например, в составе пищевых добавок) 0-9 мг Незначительное (но содержит фенилэтиламин) 0-74 мг 0-198 мг Нет данных 0-3 мг 2-68 мг
монтированных пищевых продуктах типа сыра (табл. 9-5). Он быстро метаболизируется МАО и обычно не активен при пероральном приеме из-за быстрого превращения при первом прохождении. При парентеральном введении тирамин оказывает непрямой симпатомиметический эффект, вызванный высвобождением катехоламинов из депо. Таким образом, спектр его действия похож на таковой у норадреналина. У пациентов, принимающих ингибиторы МАО (особенно ингибиторы изомера МАО-А), действие тирамина может значительно усиливаться, приводя к выраженному повышению артериального давления.
II. Клиническая фармакология симпатомиметиков
Применение симпатомиметиков в терапии базируется на знании физиологических эффектов катехоламинов в тканях. Выбор определенного симпа-томиметика из всей группы этих соединений зависит от того, какой подкласс рецепторов они активируют (ос, Pi или р2), от желательной продолжительности действия и предпочитаемого пути введения. Симпатомиметики — это очень сильные пре
параты, которые действуют на многие органы и системы, особенно на сердце и периферическое кровообращение. Поэтому необходима большая осторожность при парентеральном применении этих препаратов. В большинстве случаев вместо назначения фиксированных доз следует пронаблюдать за фармакологическим ответом на препарат для назначения оптимальной дозы, особенно если его вводят парентерально. Побочные эффекты этих лекарственных средств объяснимы с точки зрения их физиологического действия.
Показания со стороны сердечно-сосудистой системы
А. Состояния, при которых необходимо повысить кровоток или артериальное давление. Гипотензия может возникать вследствие многих причин, например при снижении объема крови, аритмиях, побочных реакциях на антигипертензивные средства и инфекциях. При сохранении нормальной перфузии головного мозга, почек и сердца гипотензия не требует интенсивного лечения. В таких случаях рекомендуется положить больного и обеспечить поддержание достаточного объема циркулирующей крови на время, пока выявляют первичную причину и воздействуют. Использование симпатомиметиков для повышения давления в случаях, не
требующих немедленного вмешательства, может иметь нежелательные последствия (раздел “Токсичность симпатомиметиков”). Симпатомиметики следует использовать для купирования гипотензии при неотложных состояниях, когда декомпенсированы мозговой и коронарный кровотоки. Такие ситуации возникают при массивных кровотечениях, повреждениях спинного мозга, при передозировках антигипертензивных или угнетающих ЦНС лекарственных средств. При этом лечение обычно кратковременно. Оно прекращается с началом внутривенного введения жидкостей или крови. В таких случаях можно применять ос-агонисты прямого действия типа норадреналина, фенилэфрина или ме-токсамина.
Шок — это сложный острый сердечно-сосудистый синдром, приводящий к критическому снижению перфузии жизненно важных тканей и большому спектру системных эффектов. Шок обычно проявляется гипотензией, нарушением сознания, олигурией и метаболическим ацидозом. При отсутствии лечения состояние больного продолжает ухудшаться, становится рефрактерным к лечению и приводит к гибели. Основные механизмы возникновения шока — гиповолемия, сердечная недостаточность и нарушение сосудистого сопротивления. Основные методы терапии шока — восстановление объема циркулирующей крови и устранение причины его возникновения. Хотя симпатомиметики фактически применяются в терапии всех видов шока, их эффективность не всегда определена. При большинстве видов шока вазоконстрикция, вызываемая активацией симпатической нервной системы, уже значительно выражена к началу лечения. Действительно, меры, направленные на снижение, а не на повышение периферического сосудистого сопротивления могут быть более полезны, если улучшается церебральный, почечный и коронарный кровоток. Решение о применении вазоконстрикторов или вазодилататоров лучше всего принимать, основываясь на информации о причине шока, что может потребовать инвазивного мониторирования.
Кардиогенный шок, обычно вызванный обширным инфарктом миокарда, имеет плохой прогноз. В некоторых случаях используются вспомогательная механическая перфузия и экстренная кардиохирургия. Оптимальное возмещение потери жидкости требует постоянного контроля капиллярного легочного давления. В такой ситуации могут помочь препараты с положительным инотропным
эффектом типа дофамина или добутамина. В низких и средних дозах они повышают сердечный выброс и в отличие от норадреналина вызывают незначительную периферическую вазоконстрикцию. Изопротеренол повышает частоту сердечных сокращений и действует сильнее, чем дофамин или добутамин. В главе 13 и в табл. 13-6 рассматривается шок при инфаркте миокарда.
К сожалению, пациент с кардиогенным шоком может не прореагировать на все эти терапевтические меры, и тогда велик соблазн попробовать вазоконстрикторы для поддержания адекватного артериального давления. Хотя коронарный кровоток может при этом улучшиться, положительный эффект нейтрализуется повышением потребность миокарда в кислороде, а также более выраженной вазоконстрикцией сосудов органов брюшной полости. Поэтому цель терапии шока — оптимизировать перфузию тканей, а не артериальное давление.
Б. Состояния, при которых необходимо уменьшить кровоток. Снижение регионарного кровотока необходимо для гемостаза при операциях, при местной анестезии для уменьшения всасывания местных анестетиков и для снижения застоя в слизистых. В каждом из этих случаев желательна активация ос-рецепторов, и выбор препарата зависит от требуемого эффекта, длительности действия и пути введения.
Эффективный фармакологический гемостаз часто необходимый в челюстно-лицевой хирургии требует средств, которые можно применять местнс в высоких концентрациях. Для действия на слизистую носа (при носовых кровотечениях) или на десны (при гингивэктомии) обычно местно используют адреналин. В хирургии носоглотки иногда применяется кокаин, так как он сочетает гемостатический эффект с местноанестезирующим. В ряде случаев кокаин смешивают с адреналином для обеспечения максимального гемостаза при местной анестезии.
Сочетание а-агонистов и местных анестетиког значительно продлевает длительность инфильтрационной анестезии, что позволяет уменьшить общую дозу местного анестетика и снизить его токсичность. В таких случаях наиболее часто применяется адреналин в концентрации 1 : 200 000, используются также норадреналин, фенилэфрин v другие а-агонисты.
Средства, обладающие деконгестивным (проти-воотечным) действием на слизистые (деконгестан-
ты), снижают дискомфорт при сенной лихорадке и в меньшей степени — при обычной простуде. К сожалению, за противоотечным эффектом следует компенсаторная гиперемия, и повторное местное применение этих препаратов в высоких дозах лцэ-жет привести к ишемическим изменениям в слизистых, возможно, вследствие вазоконстрикции питающих артерий. Чаще всего используются такие де-конгестанты как фенилэфрин и фенилпропанола-мин в форме назальных спреев и глазных капель. Большая продолжительность действия при значительно меньшйх местных концентрациях и существенном влиянии на сердце и ЦНС характерна для перорального применения такого средства как эфедрин или одного из его изомеров — псевдоэфедрина. Местные деконгестанты длительного действия — это ксилометазолин и оксиметазолин. Все эти средства продаются без рецепта.
С. Кардиологические показания к применению. Эпизоды пароксизмальной предсердной тахикардии в прошлом купировали а-агонистами типа фенилэфрина или метоксамина. Так как эти препараты вызывают значительную вазоконстрикцию и приводят к повышению давления, рефлекторная активация вагуса может перевести аритмию в синусовый ритм. В таких ситуациях препараты следует назначать в виде медленной внутривенной инфузии; необходимо соблюдать осторожность из-за возможности повышения систолического давления выше 160 мм рт. ст. Этим способом эффективно снимали названные нарушения ритма у гипотензивных пациентов, однако сейчас предпочтение отдают более безопасным альтернативным средствам: верапамилу, аденозину или электроимпульсной терапии (глава 14).
Для временной неотложной терапии полных сердечных блокад и остановки сердца применяют также катехоламины типа изопротеренола и адреналина. Адреналин может быть эффективен при остановке сердца, сердечно-легочной реанимации за счет вызванного им перераспределения кровотока к коронарным и мозговым артериям. Однако более безопасны и эффективны при сердечных блокадах электронные водители ритма. Они должны использоваться в срочном порядке, если есть признаки сердечной блокады высокой степени.
Застойная сердечная недостаточность может быть уменьшена вследствие положительного инотропного действия препаратов типа добутамина и преналтерола. Эти показания обсуждаются в гла
ве 13. Развитие толерантности или десенсибилизации является самым значительным ограничением использования катехоламинов при застойной сердечной недостаточности.
Показания со стороны дыхательной системы
Одно из наиболее весомых показаний к применению симпатомиметиков — это терапия бронхиальной астмы (глава 19). По этому показанию применяют неселективные средства (адреналин), Р-селективные средства (изопротеренол) и [^-селективные средства (метапротеренол, тербуталин, альбутерол). Для большинства пациентов [^-селективные средства оказываются наиболее эффективными и менее токсичными, чем неселективные препараты. С активным использованием Рагонистов при астме связывают увеличение смертности, хотя причина этого не ясна.
Анафилактические реакции
Анафилактический шок и другие реакции немедленного типа (тип I), опосредованные IgE, затрагивают как дыхательную, так и сердечно-сосудистую системы. Синдромы бронхоспазма, застоя в слизистых оболочках, сосудистого отека и сердечно-сосудистого коллапса обычно хорошо снимаются подкожным введением 0.3-0.5 мг адреналина (0.3-0.5 мл 1:1000 раствора адреналина). Адреналин является препаратом выбора вследствие высокой эффективности его действия на а, Рг, р2-рецеп-торы; стимуляция всех типов адренорецепторов помогает обратить этот патологический процесс. При анафилактических реакциях как препараты второй линии могут применяться глюкокортикоиды и антигистаминные препараты (антагонисты Н(-и Н2-рецепторов), однако препаратом первого выбора является адреналин.
Офтальмологические показания к применению
Фенилэфрин является эффективным мидриати-ческим средством и его часто используют при осмотре сетчатки. Он дает также противоотечный эффект при незначительной аллергической гиперемии конъюктивы. Симпатомиметики в форме глазных капель полезны для определения локализации
поражения при синдроме Горнера. (Приложение “Использование базисной фармакологии для решения клинической проблемы”.)
Проявления глаукомы снижаются разными сим-патомиметиками и симпатолитиками (глава 10, раздел “Лечение глаукомы”). Адреналин и р-блокато-ры — наиболее значимые средства. При хроническом применении адреналин (1-2 % раствор, местно) снижает внутриглазное давление в основном за счет повышения оттока внутриглазной жидкости. Эффективность адреналина может быть частично связана с десенситизацией к воздействиям на р-ре-цепторы. Дипивефрин — это аналог адреналина, который образуется этерификацией адреналина с пиваликовой кислотой. Эфир лучше проникает в переднюю камеру глаза, где путем ферментативного гидролиза снова превращается в адреналин. Ап-раклонидин — селективный а2-агонист, который тоже понижает внутриглазное давление и используется после лазеротерапии.
Показания со стороны мочеполовой системы
Как сказано выше, р2-селективные средства расслабляют беременную матку. Ритодрин, тербута-лини подобные препараты используются для предупреждения преждевременных родов. Цель этой терапии — отсрочить роды до максимального созревания плода. Некоторые исследователи показали, что эти препараты могут отсрочить роды на дни и даже недели (Merkatz, 1980). Однако мета-анализ ранее сделанных исследований и недавнего рандомизированного исследования показал, что терапия Р-агонистами не оказывает значительного влияния на перинатальную смертность и даже может привести к повышению материнской заболеваемости (сообщение группы исследователей преждевременных родов Канады, 1992).
Пероральные симпатомиметики иногда применяются для устранения недержания мочи при напряжении (во время кашля или другого усилия).
Использование базисной фармакологии для решения клинической проблемы
Синдром Горнера (Бернара-Горнера) — это состояние (обычно одностороннее), которое возникает вследствие нарушения симпатической иннервации лица. Проявляется он вазодилатацией, птозом, миозом и отсутствием потовыделения на пораженной стороне. Этот синдром может быть следствием либо пре-, либо постганглионарного поражения, например опухоли. Знание его локализации помогает использовать оптимальный метод терапии.
Понимание влияния денервации на метаболизм нейромедиаторов помогает клиницисту использовать определенные препараты для выявления локализации повреждения. В большинстве случаев локальное поражение нерва вызовет дегенерацию дистальной части этого волокна и потерю медиаторов из этого нервного окончания без поражения самого нейрона. Поэтому при преганглионарном нарушении постганглионарный адренергический нейрон остается интактным, в то время как при постганглионарном поражении происходит дегенерация адренергического нервного окончания и потеря всего коли
чества медиаторов (катехоламинов). Так как для эффекта симпатомиметиков непрямого действия необходимы запасы (депо) катехоламинов, эти средства можно использовать для тестирования на наличие неизменных адренергических нервных окончаний. Радужка, являясь хорошо видимой структурой, реагирующей на аппликацию симпатомиметиков,— удобный объект наблюдения.
Если уровень поражения при синдроме Горнера постганглионарный, то симпатомиметики непрямого действия (например, кокаин, гидроксиамфетамин) не приведут к расширению аномально узкого зрачка, так как в нервных окончаниях в радужке нет катехоламинов. Наоборот, зрачок расширится под действием фенилэфрина, который напрямую стимулирует а-рецепто-ры на гладкой мышце радужки. Пациент с пре-ганглионарным поражением, с другой стороны, нормально прореагирует на обе группы препаратов, так как в этом случае постганглионарные волокна и их депо катехоламинов остаются в сохранности.
В таких ситуациях можно попробовать эфедрин или псевдоэфедрин.
Показания со стороны ЦНС
Как уже было сказано, симпатомиметики с амфетаминоподобным действием вызывают повышение настроения (эйфорию); этот эффект является причиной активного злоупотребления препаратами этой группы (глава 31). Амфетамины также стимулируют бодрствование, снижают потребность во сне, что проявляется в повышении внимания при монотонной работе, ускорении и десинхронизации волн ЭЭГ. Терапевтическое приложение этого действия — лечение нарколепсии. Подавление аппетита этими препаратами легко продемонстрировать в экспериментах на животных. При терапии ожирения наблюдается выраженный начальный эффект, но нет сведений о том, что применением одних амфетаминов можно добиться длительного сохранения сниженного веса. И, наконец, последнее показание к применению симпатомиметиков, влияющих на ЦНС,— это гиперкинетический синдром с дефицитом внимания у детей, который пока недостаточно изучен, излишне часто диагностируется и характеризуется непостоянством внимания, гиперкинетическим поведением и проблемами в обучении. Некоторые пациенты с этим синдромом хорошо реагируют на низкие дозы амфетаминоподобных препаратов.
Дополнительные показания к применению
Хотя основное показание к применению а2-аго-ниста клонидина — это лечение гипертензии (глава И), препарат эффективен и при диарее у больных сахарным диабетом с вегетативной нейропатией, возможно, вследствие его способности усиливать абсорбцию соли и воды из кишечника. Кроме того, клонидин эффективен для снижения тяги к наркотикам и алкоголю во время синдрома отмены и может помочь при попытке бросить курить. Клонидин применяют и для уменьшения “приливов" при наступлении климактерического периода, а также используют для коррекции гемодинамической нестабильности при общей анестезии.
Токсичность симпатомиметиков
Побочные эффекты агонистов адренорецепторов прежде всего связаны с распространением их эффектов на сердечно-сосудистую систему и ЦНС.
Побочные эффекты со стороны сердечно-сосудистой системы при применении прессорных агентов — это значительное повышение артериального давления, которое может приводить к кровоизлиянию в мозг или к отеку легких. Усиление работы сердца может вызвать приступ стенокардии и даже инфаркт миокарда. Р-агонисты часто провоцируют синусовую тахикардию и даже серьезные желудочковые аритмии. Симпатомиметики могут привести к повреждению миокарда, особенно после длительной инфузии. Предельная осторожность требуется при лечении пожилых пациентов, больных с гипертензией или стенокардией.
Если побочные эффекты симпатомиметиков требуют принятия срочных мер, необходимо назначение антагонистов специфических адренорецепторов (глава 10). Например, попадание под кожу норадреналина, вводимого внутривенной инфузией, может привести к значительной ишемии; это действие может быть прекращено введением антагонистов а-рецепторов.
ЦНС-токсичность при применении катехоламинов или фенилэфрина встречается редко. Фе-нилизопропиламины часто вызывают двигательное беспокойство, тремор, бессонницу и тревожность. При приеме очень высоких доз может возникнуть параноидное состояние. Кокаин может вызвать ухудшение при судорожных состояниях, инсультах, аритмиях или инфаркте миокарда. Последние три из названных эффектов являются основой токсичности симпатомиметиков. Лечение обсуждается в главе 31.
Препараты
Адреналин (генерик, Адреналина хлорид, др.) Парентерально: 1: 1000 (1мг/мл), 1 :10000 (0.1 мг/мл), 1: 100 000 (0.01 мг/мл) для инъекций
Глазные капли: 0.25, 0.5,1,2% Интраназально: 0.1 % капли и спрэй Аэрозоль для купирования бронхоспазма: 0.16, 0.2, 0.25 мг/спрэй (Приматен Мист, Бронкаид Мист)
Амфетамин, рацемическая смесь (генерик) Перорально: таблетки по 5, 10 мг
Апраклонидин (Иопидин)
Местно: 1 % раствор
Декстроамфетамин (генерик, Декседрин)
Перорально: таблетки по 5,10 мг;
эликсир 5 мг/5 мл
Перорально для пролонгированного действия: капсулы по 5, 10,15 мг
Добутамин (Добутрекс)
Парентерально: 250 мг/20 мл для инъекций
Дофамин (генерик, Интропин)
Парентерально: 40, 80, 160 мг/мл для инъекций; 80,160, 320 мг/100 мл в 5 % растворе глюкозы на воде для инъекций
Изопротеренол (генерик, Изупрел)
Перорально: подъязычные таблетки по 10, 15 мг
Парентерально: 0.2 мг/мл для инъекций
Ксилометазолин (генерик, Отривин, Нео-Синефрин длительного действия, Хлорогист LA)
Интраназально: 0.05, 0.1 % капли и аэрозоль
Мефентермин (Виамин сульфат)
Парентерально: 15, 30 мг/мл для инъекций
Метараминол (генерик, Арамин)
Парентерально: 10 мг/мл для инъекций
Метамфетамин (генерик, Дезоксин) Перорально: таблетки по 5,10 мг Перорально для пролонгированного действия: таблетки по 5,10,15 мг
Метоксамин (Вазоксил)
Парентерально: 20 мг/мл для инъекций
Метилфенидат (генерик, Риталин-SR) Перорально: таблетки по 5,10, 20 мг Перорально для пролонгированного действия: таблетки по 20 мг
Нафазолин (Привин)
Интраназально: 0.05 % капли и спрэй
Норадреналин (Левофед)
Парентерально: 1 мг/мл для инъекций
Оксиметазолин (генерик, Африн, Нео-Синефрин 12-часовой, др.)
Интраназально: 0.025, 0.05 % спрэй
Псевдоэфедрин (генерик, Судафед, др.) Перорально: таблетки по 30, 60 мг; сиропы 15, 30 мг/5 мл; капли 7.5 мг/0.8 мл, 30 мг/мл
Перорально для пролонгированного действия: капсулы по 120 мг
Тетрагидрозолин (генерик, Тизин) Интраназально: 0.05, 0.1 % капли
Фенметразин (Прелудин)
Перорально: таблетки пролонгированного действия по 75 мг
Фенилэфрин (генерик, Нео-Синефрин) Парентерально: 10 мг/мл для инъекций Интраназально: 0.125, 0.16, 0.25, 0.2, 0.5, 1 % капли и спрэй; 0.5 % желе
Фенилпропаноламин (генерик)
Перорально: таблетки по 25, 50 мг Перорально для пролонгированного действия: капсулы по 75 мг
Эфедрин (генерик, капли в нос Ватронол, Эфедрон назад)
Перорально: капсулы по 25, 50 мг; сиропы 11, 20 мг/5 мл
Парентерально: 25,50 мг/мл для инъекций Интраназально: 0.5 % капли, 0.6 % желе
Избранная литература
Caron М. G., Lefkowitz R. J. Catecholamine receptors: Structure, function, and regulation. Recent Prog. Horn. Res. 1993; 48: 277.
DeGroat W. C., Steers W. D. Autonomic regulation of the urinary bladder and sexual organs. In: Central Regulation of Autonomic Functions. Loewy A. D., Spyer К. M. (eds) Oxford, 1990.
Gingrich J. A., Caron M. G. Recent advances in the molecular biology of dopamine receptors. Annu. Rev. Neurosci. 1993; 16: 299.
Ruffolo R. R. Jr. et al. Pharmacologic and therapeutic applications of a2-adrenoreceptor subtypes. Annu. Rev. Pharmacol. Toxicol. 1993; 32: 243.
Sokoloff P. et al. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature, 1990; 347:146.
1994 Receptor Nomenclature Supplement. Trends. Pharmacol. Sci. 1994.
Средства, блокирующие адренорецепторы
10
Брайен Б. Хоффман
Поскольку катехоламины играют важную роль во многих физиологических и патофизиологических процессах, средства, блокирующие адренорецепторы, весьма ценны для клиники. Существенно различаются фармакологические эффекты средств, избирательно влияющих на а- и Р-рецеп-торы. Классификация адренорецепторов на а-, Р* и дофаминовый подтипы, а также последствия их активации обсуждаются в главах 6 и 9. Клиническое значение блокады периферических дофаминовых рецепторов в настоящее время не определено. Напротив, блокада дофаминовых рецепторов центральной нервной системы имеет важное клиническое значение. Средства, влияющие на эти рецепторы, рассматриваются в главах 20 и 28. В данной главе речь идет о средствах, основным эффектом которых является блокада периферических а- или Р рецепторов, и о предотвращении активации этих рецепторов катехоламинами и родственными агонистами.
В базисной фармакологии средства, блокирующие а-рецепторы, очень удобны для экспериментальных исследований функциональной активности автономной нервной системы; в клинике же эти препараты имеют ограниченное применение. Хотя эти средства эффективно блокируют вазоконстрикторные эффекты катехоламинов, их использование при гипертензии (за исключением празозина и его а।-селективных аналогов, а также лабе-талола) дает незначительный результат. Место антагонистов а-рецепторов в терапии заболеваний периферических сосудов неясно, в то время как Р-адреноблокаторы широко используются при многих заболеваниях.
I. Базисная фармакология средств, блокирующих а-рецепторы
Механизм действия
Антагонисты а-рецепторов бывают обратимыми или необратимыми. Антагонисты обратимого действия могут освобождаться из комплексов с а-ре-цепторами, средства необратимого действия — нет. Фентоламин и толазолин являются примерами антагонистов обратимого действия (рис. 10-1). Празозин и лабеталол, используемые главным образом как антигипертензивные средства, являются обратимыми блокаторами а-адренорецепторов так же, как некоторые производные спорыньи (глава 16). Феноксибензамин образует реактивный метаболит этиленимониум (рис. 10-1), который ковалентно связывается с а-рецептором, вызывая необратимую блокаду. Рис. 10-2 иллюстрирует эффекты препаратов обратимого действия в сравнении с необратимыми антагонистами.
Длительность действия обратимых антагонистов в значительной мере зависит от времени полувыведения препарата и от скорости диссоциации комплекса антагонист-рецептор. Чем короче период полувыведения, тем меньше длится эффект. Эффекты необратимых антагонистов могут продолжаться долгое время после полного выведения препарата из плазмы. В случае применения фенокси-бензамина восстановление реактивности ткани после обширной блокады а-рецепторов зависит от синтеза новых рецепторов, на который может потребоваться несколько дней.
Рис. 10-1. Структура некоторых средств, блокирующих а-рецепторы
Фармакологические эффекты
А. Сердечно-сосудистые эффекты. Поскольку артериальный и венозный тонус в большой степени зависят от функционального состояния «-рецепторов гладких мышц сосудов, средства, блокирующие а-рецепторы, вызывают снижение периферического сосудистого сопротивления и кровяного давления (рис. 10-3). Они могут предотвращать прессорные эффекты агонистов а-рецепторов, применяемых в обычных дозах; в случае введения агонистов, влияющих и на а- и на Р2-рецепторы (например, адреналина), а-блокаторы превращают прессорный ответ в депрессорный (рис. 10-3). Это изменение ответа называют извращением эффекта адреналина, а-блокаторы могут вызывать ортостатическую гипотензию и рефлекторную тахикардию. Тахикардия более выражена при введении препаратов, блокирующих а2-пресинаптические рецепторы в сердце (табл. 10-1), так как увеличение вы-
ТАБЛИЦА10-1. Относительная селективность антагонистов адренорецепторов
Тип антагониста Аффинитет к рецепторам
а-антагонисты
Празозин,теразозин, доксазозин а,»» а.
Феноксибензамин а,> а2
Фентоламин а, = а2
Раувольсцин, йохимбин, толазолин а2» а.
Смешанные антагонисты Лабеталол Pl = р2 — «1 > 0.2
Р-антагонисты Метопрололол, ацебутолол, Pi»> Рг
алпренолол, атенолол, бетаксолол, целипролол, эсмолол
Пропранолол,картеолол, Pl= Рг
пенбутолол, пиндолол, тимолол Бутоксамин Р2»> Pl
Рис. 10-2. Кривые зависимости доза-эффект норадреналина в присутствии двух различных средств, блокирующих а-адренорецепторы. Напряжение, вызываемое в изолированных полосках селезенки кошки, богатой а-рецепторами, измерялось в ответ на разные дозы норадреналина. Слева. Обратимый блокатор толазолин в концентрациях 10 и 20 мкмоль/л сдвигает кривую вправо без снижения максимального ответа. Справа. Дибенамин, необратимый блокатор и аналог феноксибензамина, уменьшает максимально достижимую реакцию селезенки на норадреналин в обеих тестируемых концентрациях. (Из: Bickerton R. К. The response of isolated strips of cat spleen to sympathomimetic drugs and their antagonists. J. Pharmacol. Exp. Ther. 1963; 142: 99.)
деления норадреналина вызывает в свою очередь стимуляцию Р-рецепторов в сердце. Хроническое применение а-антагонистов может вызывать компенсаторное увеличение объема циркулирующей крови.
Б. Другие эффекты. К менее значимым эффектам, сигнализирующим о блокаде а-рецепторов в других тканях, относятся миоз, снижение адренергической стимуляции потоотделения, заложенность носа. Блокада а-рецепторов дна мочевого пузыря приводит к снижению сопротивления при мочеиспускании и ретроградной эякуляции. Отдельные а-адреноблокаторы имеют другие важные фармакологические эффекты, что еще будет рассматриваться в этой главе.
Специфические средства
Производное имидазолина фентоламин — сильный конкурентный антагонист а-рецепторов. Фентоламин вызывает уменьшение периферического сопротивления посредством блокады всех подтипов а-рецепторов и дополнительного неадренергического действия на гладкие мышцы сосудов. Стимуляция сердечной деятельности, вызываемая фен-
толамином, может быть частично рефлекторной, не зависящей от барорецепторной активности. В данном случае может играть роль блокада аг-рецепторов. Фентоламин примерно одинаково активен в отношении ар и а2-рецепторов, он угнетает также физиологический ответ на серотонин. Препарат является агонистом мускариновых, Нг и Н2-гиста-миновых рецепторов.
Фентоламин плохо абсорбируется после приема внутрь. Основные нежелательные эффекты препарата связаны с сердечной стимуляцией, которая может вызывать тяжелую тахикардию, аритмии и стенокардию. Активация деятельности желудочно-кишечного тракта может привести к диарее и увеличению продукции кислоты в желудке.
Толазолин сходен с фентоламином. Это менее сильное средство, лучше абсорбирующееся из желудочно-кишечного тракта. Препарат быстро выводится с мочой.
Толазолин имеет очень ограниченное клиническое применение при спастических заболеваниях периферических сосудов и при лечении легочной гипертензии у новорожденных с респираторным дистресс-синдромом. Применяется редко.
Фентоламин
Адреналин до фентоламина
Рис. 10-3. Betray.Влияние блокатора а-рецепторов фентоламина на артериальное давление у анестезированной собаки. Извращение эффекта адреналина продемонстрировано записью ответа на введение адреналина до (в середине) и после (внизу) фентоламина. Все препараты вводились внутривенно. (АД — артериальное давление; ЧСС — частота сердечных сокращений.)
Производные спорыньи (например, эрготамин и дигидроэрготамин) вызывают обратимую блокаду а-рецепторов. Однако большинство клинически значимых эффектов этих средств являются результатом воздействия на другие рецепторы: так, эрготамин при лечении мигрени, вероятно, действует через серотониновые рецепторы (глава 16).
Феноксибензамин ковалентно связывается с а-рецепторами, вызывая необратимую длительную блокаду (14-48 часов). Он отчасти селективен для агрецепторов, но в меньшей степени, чем празозин (табл. 10-1). Кроме того, вещество ингибирует обратный захват выделяемого норадреналина преси-наптическими адренергическими нервными окончаниями, блокирует гистаминовые (Н(), ацетилхолиновые и серотониновые рецепторы. Роль этих вторичных эффектов феноксибензамина у человека не до конца выяснена.
Его фармакологическое действие главным образом связано с блокадой а-рецепторов. Наиболее
важное значение имеет угнетение феноксибензами-ном вазоконстрикции, вызываемой катехоламинами. Хотя у лежащего пациента препарат вызывает относительно небольшое снижение кровяного давления, при высоком симпатическом тонусе, например при переходе пациента в вертикальное положение или при уменьшении объема циркулирующей крови, артериальное давление падает существенно. Из-за рефлекторных эффектов и в связи с частичной блокадой пресинаптических а2-рецепторов в симпатических нервах сердца может увеличиваться сердечный выброс.
Феноксибензамин всасывается после применения внутрь, хотя его биодоступность при этом низка. Препарат обычно назначают перорально, начиная с малых доз (15-20 мг/день) и прогрессивно повышая их до достижения желаемого эффекта. Как правило, для адекватной блокады а-ре-цепторов достаточно менее 100 мг препарата в день.
Нежелательные эффекты феноксибензамина связаны с его блокирующим действием на а-рецепторы; наиболее важны ортостатическая гипотензия и тахикардия. Наблюдаются также заложенность носовых ходов и подавление эякуляции. Так как феноксибензамин проникает в центральную нервную систему, он может вызывать менее специфические эффекты, в том числе слабость, заторможенность и тошноту. Поскольку феноксибензамин является алкилирующим веществом, он может иметь и другие побочные эффекты, которые пока не охарактеризованы. Препарат провоцирует развитие опухолей у животных, однако данных о клинических наблюдениях пока нет.
Празозин эффективен в терапии гипертензии (глава 11). Исходно он рассматривался как прямой вазодилататор, но в настоящее время известно, что основное фармакологическое действие празозина осуществляется за счет чрезвычайно сильного а-рецепторного антагонизма. Он высоко селективен по отношению к at-рецепторам и имеет относительно низкий аффинитет к а2-рецепторам. Это может частично объяснить отсутствие тахикардии при введении празозина в отличие от фентолами-на и феноксибензамина. Празозин приводит к расслаблению и артериальных и венозных гладких мышц.
Празозин активно метаболизируется у человека: при пероральном применении вследствие разрушения в печени доступны только 50 % препарата. Период полувыведения лекарства составляет приблизительно 3 часа.
Теразозин — еще один обратимый а,-селективный антагонист, эффективный при гипертензии (глава 11). Его назначают также пациентам с нарушениями мочеиспускания из-за гипертрофии простаты. Теразозин имеет высокую биодоступность, хотя активно метаболизируется в печени. Только малая часть неизмененного вещества выводится с мочой. Период полувыведения теразозина — около 9-12 часов.
Доксазозин — третий селективный антагонист а,-рецепторов, используемый при гипертензии. Он имеет среднюю биодоступность и активно метаболизируется с выведением очень малой части неизмененного вещества с мочой или калом. Метаболиты доксазозина активны, хотя их вклад в основное действие препарата, вероятно, невелик. Период полувыведения доксазозина приблизительно равен 22 часам.
Другие антагонисты а-адренорецепторов
Индорамин — это еще один (Х|-селективный антагонист, эффективный как антигипертензивное средство. Урапидил — более новый а^антагонист (это его основной эффект), являющийся к тому же слабым а2- и 5-НТ1Л-агонистом и слабым Р,-антагонистом. Он применяется в Европе как антигипертензивное средство. Лабеталол проявляет свойства селективного at-антагониста, а также fJ-блокатора. Такие нейролептические средства как хлорпромазин и галоперидол являются сильными антагонистами а- и дофаминовых рецепторов. Несмотря на то, что они не применяются в клинике для блокады a-рецепторов, этот эффект нейролептиков может приводить к нежелательным осложнениям (например, гипотензии).
Йохимбин — а2-селективное средство, не имеющее определенного клинического значения. Теоретически он может применяться при недостаточности автономной нервной системы, способствуя выделению медиатора путем блокады пресинаптических а2-рецепторов. Йохимбин улучшает состояние некоторых пациентов с болевой формой диабетической нейропатии. Полагают, что препарат активизирует и сексуальную функцию, однако нет достаточных свидетельств в пользу этого эффекта у человека.
Вызывает недоумение тот факт, что а2-антаго-нисты редко используются в клинической практике. Представляет интерес создание высокоселективных антагонистов для угнетения сокрашения гладких мышц при феномене Рейно, для применения при диабете II типа (а2-рецепторы угнетают секрецию инсулина) и при психических депрессиях (Ruffolo et al., 1993). Неизвестно, насколько расширение знаний о множестве подтипов а2-рецепторов приведет к появлению клинически значимых избирательно действующих новых средств.
II. Клиническая фармакология средств, блокирующих а-рецепторы
Феохромоцитома
Основным показанием к применению феноксибензамина и фентоламина является лечение больных с феохромоцитомой. Феохромоцитома — это
опухоль, обычно расположенная-в мозговом веществе надпочечников, которая выделяет смесь адреналина и норадреналина. У пациентов отмечаются многие симптомы катехоламинового возбуждения, включая гипертензию, тахикардию и аритмии.
Диагноз феохромоцитомы обычно ставится на основании результатов химического определения циркулирующих в крови катехоламинов и выделяющихся с мочой их метаболитов, особенно 3-гид-рокси-4-метоксиминд альной кислоты, метанефри-на и норметанефрина. Иногда в качестве диагностического теста при подозрении на феохромоцитому проводится инфузия фентоламина, так как пациенты с опухолью часто демонстрируют в ответ на адреноблокирующие средства большее, чем в среднем, падение кровяного давления. Однако измерение уровня катехоламинов в крови и моче и их метаболитов в моче более безопасно и надежно для диагностики. Инъекция болюсной дозы фентоламина может привести к опасному падению кровяного давления у пациентов с феохромоцитомой. Провокационный тест на феохромоцитому введением гистамина может вызвать значительное повышение кровяного давления, если выполняется неправильно. Во время оперативных манипуляций с феохромоцитомой может наблюдаться выброс катехоламинов из депо; возникающая в результате гипертензия поддается контролю с помощью фентоламина или нитропруссида. Последний имеет ряд преимуществ, поскольку его эффектами легче управлять, а продолжительность его действия меньше.
Антагонисты а-рецепторов наиболее полезны во время предоперационной подготовки пациентов с феохромоцитомой (рис. 10-4). Феноксибензамин, введенный по такой схеме, предотвращает развитие острых гипертензивных эпизодов во время исследований, предпринимаемых для уточнения локализации опухоли, и уменьшает хронические проявления избыточной секреции катехоламинов (например, сокращение объема плазмы), если они наблюдаются. Кроме того, упрощается ход операции. Пероральные дозы от 10 до 20 мг/сутки можно повышать каждые несколько дней до наступления состояния контроля над гипертензией. Некоторые врачи лечат пациентов с феохромоцитомой фенокси-бензамином в течение одной-трех недель до операции, в то время как другие предпочитают более короткую предоперационную подготовку. Феноксибензамин с успехом используют при длительном
Недели
Рис. 10-4. Действие феноксибензамина (Дибензилина) на кровяное давление у пациента с феохромоцитомой. Назначение средства было начато с третьей недели, как показано затененным столбиком. Систолическое и диастолическое давление в положении лежа отмечено кружками, в положении стоя — треугольниками и заштрихованной областью. Обратите внимание, что а-блокатор существенно снижает кровяное давление. Уменьшение ортостатической гипотензии по сравнению с наблюдаемой до лечения, возможно, связано с нормализацией объема крови, который иногда заметно снижен у больных с длительно существующей гипертензией при феохромоцитоме. (Из: Engelman Е., Sjoerdsma A. Chronic medical therapy for pheochromocytoma, Ann. Intern. Med. 1961; 61: 229.)
лечении неоперабельной или метастатической феохромоцитомы. Хотя опыт применения альтернативных средств невелик, известно, что гипертензия у пациентов с феохромоцитомой чувствительна к действию обратимых at-селективных антагонистов или традиционных антагонистов кальция.
В ряде случаев для прекращения действия катехоламинов на сердце после установления блокады a-рецепторов могут потребоваться Р-блокирующие средства. Р-антагонисты не должны применяться до установления эффективной блокады а-рецепторов,
так как не встречающая сопротивления блокада fJ-рецепторов теоретически может вызвать подъем кровяного давления из-за увеличения вазоконстрикции.
Феохромоцитому иногда лечат метирозином, а-метиловым производным тирозина (а-метилти-розин). Это средство является конкурентным ингибитором тирозингидроксилазы и при приеме внутрь в дозах 1-4 г/сутки вмешивается в синтез дофамина (рис. 6-5), снижая вследствие этого количества норадреналина и адреналина, секретируемых опухолью. Метирозин не имеет а-блокирую-щих эффектов, но может действовать как синергист феноксибензамина при лечении больных с феохромоцитомой.
Неотложные гипертензивные состояния
Средства, блокирующие а-адренорецепторы, редко применяются при гипертонических кризах, хотя использование лабеталола в таких ситуациях расширяется. Теоретически а-антагонисты наиболее эффективны, если повышение кровяного давления отражает чрезмерное увеличение концентрации циркулирующих а-агонистов. Следовательно, при феохромоцитоме, передозировке симпатомиметических препаратов или после отмены клонидина для контроля над кровяным давлением может быть полезен фентоламин. Однако на практике чаще используют другие средства (глава 11), так как для безопасного применения фентоламина в этих условиях необходим значительный опыт.
Хроническая гипертензия
Препараты празозиновой группы а1-селективных антагонистов являются эффективными средствами для лечения системной гипертензии легкой и средней тяжести. Они хорошо переносятся большинством пациентов. Их основной нежелательный эффект — ортостатическая гипотензия, особенно выраженная после первой дозы (глава 11). Неселективные а-агонисты не применяются в терапии обычной гипертензии.
Заболевания периферических сосудов
Хотя средства, блокирующие а-рецепторы, пытаются использовать при окклюзионных заболеваниях периферических сосудов, нет доказательств
достоверности их эффекта в случае, если кровоток в сосудах лимитирован морфологическими изменениями. У пациентов с феноменом Рейно и другими нарушениями, включающими чрезмерный обратимый спазм периферических сосудов, Иногда наблюдается улучшение после назначения фентоламина, толазолина или феноксибензамина, тем не менее для многих больных предпочтительнее применение блокаторов кальциевых каналов.
Чрезмерная местная вазоконстрикция
Фентоламин используют и для прекращения выраженной локальной вазоконстрикции, вызванной случайной подкожной инфильтрацией тканей а-агонистами (например, норадреналином) при внутривенном введении, а-антагонист вводится местно в ишемизированные ткани.
Закупорка мочевыводящих путей
В то время как основным лечением гипертрофии простаты является хирургическая операция, обнаружено, что блокада а-рецепторов феноксибенза-мином показана больным с закупоркой мочевыводящих путей при высоком риске оперативного вмешательства. Механизм действия препарата предположительно включает частичное устранение сокращения гладких мышц увеличенной простаты или дна мочевого пузыря. К тому же феноксибензамин может быть эффективен для снятия гипертонуса шейки мочевого пузыря у пациентов со спинальными повреждениями. Однако не установлена безопасность длительного приема феноксибензамина. Некоторым больным с обструктивными симптомами помогают малые дозы а( -селективных антагонистов. В отношении безопасности эти средства более предпочтительны, чем феноксибензамин. При закупорке мочевыводящих путей чаще других а( -блокаторов применяют теразозин.
Сексуальные дисфункции у мужчин
Комбинация антагониста а-адренорецепторов фентоламина с неспецифическим вазодилататором папаверином, инъецируемая прямо в пенис, может вызвать эрекцию у мужчин с сексуальными расстройствами. Долговременна ли эффективность этой терапии — неизвестно. При длительном применении существует риск фиброзных изменений.
Кроме того, резорбтивное действие лекарств может вызвать ортостатическую гипотензию. Возникающий приапизм может потребовать лечения агонистами а-адренорецепторов, такими как фенилэфрин. В качестве альтернативной терапии при нарушениях эрекции были предложены такие вещества как ПГЕ, и вещества, выделяющие оксид азота.
III. Базисная фармакология средств, блокирующих р-рецепторы
Препараты этой группы обладают общим свойством препятствовать действию катехоламинов на Р-адренорецепторы. p-блокирующие средства связываются с fJ-рецепторами и конкурентно снижают их оккупацию катехоламинами или другими Р-агонистами. (Несколько соединений этой группы, используемых только для экспериментальных целей, связываются с Р-рецепторами необратимо.) Большинство применяемых в клинике Р-блокато-ров являются чистыми антагонистами: оккупация этими веществами Р-рецепторов не вызывает активирующего эффекта. Однако некоторые из них относятся к парциальным агонистам. Они вызывают частичную активацию рецептора, хотя и меньшую, чем полные агонисты адреналин и изопротеренол. Средства, блокирующие Р-рецепторы, различаются по их относительному аффинитету к Рг и Р2-рецепторам (табл. 10-1). Некоторые из этих антагонистов имеют большую аффинность к рг, чем к Р2-рецеПторам, и эта селективность имеет важное клиническое значение. Р-антагонисты существенно различаются по фармакокинетическим характеристикам и местноанестезирующим (мембраностабилизирующим) эффектам.
По химическому строению средства, блокирующие Р-рецепторы (рис. 10-5), похожи на сильный Р-агонист изопротеренол (рис. 9-3).
Фармакокинетические свойства антагонистов р-рецепторов
А. Всасывание. Большинство веществ этого класса хорошо абсорбируются после приема внутрь, пик концентрации наблюдается через 1-3 часа. Имеются препараты пропранолола и метопролола пролонгированного действия.
Б. Биодоступность. Пропранолол интенсивно метаболизируется в печени (эффект первого прохождения), его биодоступность относительно низка (табл. 10-2). Пропорция вещества, попадающего в системную циркуляцию, увеличивается при повышении дозы, что позволяет предположить насыщаемость печеночных механизмов экстракции. Основным следствием низкой биодоступности пропранолола является то, что при введении препарата внутрь концентрация вещества в крови оказывается значительно меньшей, чем после внутривенной инъекции той же дозы. Поскольку эффект первого прохождения отличается у разных людей, существует большая индивидуальная вариабельность концентрации пропранолола в плазме, достигаемой после перорального применения. У большинства Р-антагонистов биодоступность ограничена в той или иной степени. Исключение составляют бета-ксолол, пиндолол, пенбутолол и соталол.
В. Распределение и клиренс, р-антагонисты быстро распределяются и имеют большие объемы распределения. Пропранолол и пенбутолол довольно липофильны и легко проходят через гематоэнцефалический барьер (табл. 10-2). Большинство Р-антагонистов имеют период полувыведения в пределах двух-пяти часов. Исключением является эс-молол, который быстро гидролизуется и имеет период полувыведения, приблизительно равный десяти минутам. Пропранолол и метопролол активно метаболизируются в печени и в малом количестве выводятся в неизмененном виде с мочой. Атенолол, целипролол и пиндолол метаболизируются менее полно. Надолол экскретируется в неизмененном виде с мочой и имеет самый длинный период полувыведения из всех применяемых Р-антагонистов (до 24 часов), который еще больше удлиняется при почечной недостаточности. Выведение таких препаратов как пропранолол может замедляться при заболеваниях печени, уменьшающих печеночный кровоток, или при угнетении ферментов печени. Следует заметить, что клинические эффекты этих средств часто более длительное, чем можно ожидать на основании данных о периоде полувыведения.
Фармакодинамика средств, блокирующих Р-рецепторы
Большинство эффектов этих средств развивается вследствие оккупации и блокады Р-рецепторов. Однако некоторые эффекты могут быть связаны
О — СН2 — CH — СН2 — NH — СН(СН3)2 °Н Пропранолол О — СН2 — CH — СН2 — NHC(CH3)3 он Надолол О — СН2 — CH — СН2 — NH — СН(СН3)2 I I он I н Пиндолол ° О —СН2 —СН-СН2 —NH —СН(СН3)2 СН3 — С ОН О Т II NH —С —СН2 —СН2 —СН3 Ацебутолол Рис. 10-5. Структура антагонистов Р-рецепторов с другими механизмами, в том числе с парциальной агонистической активностью в отношении Р-ре-цепторов и местноанестезирующим действием, неодинаково выраженными у разных Р-блокаторов (табл. 10-2). А. Действие на сердечно-сосудистую систему. P-блокирующие средства при длительном приеме уменьшают кровяное давление у пациентов с гипертензией. Реализация этого эффекта может включать действие на сердце и кровеносные сосуды, ренин-ангиотензиновую систему и, возможно, центральную нервную систему. Препараты, блокирующие О — СН2 — CH — СН2 — NH — СН(СН3)2 °н СН2 —СН2 —О —сн3 Метопролол у \ О — СН2 — CH — СН2 — NH — С(СН3)3 Я ZN || | ОН S Тимолол О — СН2 — CH — СН2 — NH — СН(СН3)2 он о Т и сн2 —с—nh2 Атенолол НО —СН —СН2—NH—СН-СН2-СН2 I । Л СН3 С!» о С —NH2 он Лабеталол Р-адренорецепторы, имеют большое значение для лечения больных с гипертензией (глава И). В общепринятых дозах они обычно не вызывают гипотензию у здоровых людей с нормальным кровяным давлением. Антагонисты Р-рецепторов существенно влияют на сердце (рис. 10-6). Их отрицательные инотропный и хронотропный эффекты можно легко предсказать, исходя из роли адренорецепторов в регуляции функций сердца. Результатом блокады адренорецепторов атриовентрикулярного узла является замедление атриовентрикулярной проводимо-
ТАБЛИЦА 10-2. Свойства некоторых средств, блокирующих ^-рецепторы
Препарат Селективность Парциальная агонистическая активность Местноанестезирующее действие Растворимость в липидах Период полувыведения Приблизительная биодоступность
Ацебутолол 0i есть есть низкая 3-4 ч 50
Атенолол 01 нет нет низкая 6-9 ч 40
Бетаксолол 01 нет слабое низкая 14-22ч 90
Бисопролол 01 нет нет низкая 9-12ч 80
Картеолол отсутствует есть нет низкая 6ч 85
Целипролол 01 есть' нет — 4-5 ч 70
Эсмолол 01 нет нет низкая 10 мин —
Лабеталол2 отсутствует есть' есть умеренная 5ч 30
Метопролол 01 нет есть умеренная 3-4 ч 50
Надолол отсутствует нет нет низкая 14-24 ч 33
Пенбутолол отсутствует есть нет высокая 5 ч >90
Пиндолол отсутствует есть есть умеренная 3-4 ч 90
Пропранолол отсутствует нет есть высокая 3.5-6 ч ЗО3
Соталол отсутствует нет нет низкая 12ч 90
Тимолол отсутствует нет нет умеренная 4-5 ч 50
' В отношении рг-рецепторов.
2 Лабеталол вызывает также а,-селективную блокаду.
3 Биодоступность дозозависима.
сти с увеличением интервала PR. Эти эффекты могут быть полезны для некоторых пациентов, но потенциально опасны для других. В сосудистой системе блокада [3-рецепторов противодействует вазодилатации, вызываемой активацией 02-рецепто-ров. Это может сначала привести к увеличению периферического сопротивления из-за не встречающих противодействия эффектов, опосредованных а-рецепторами. Антагонисты 0-рецепторов блокируют выделение ренина под действием симпатической нервной системы. Как отмечается в главе 11, связь между влиянием 0-блокаторов на освобождение ренина и на кровяное давление пока является предметом дискуссий.
Б. Влияние на систему дыхания. Блокада 02-рецепторов в гладких мышцах бронхов может вызвать увеличение сопротивления воздухоносных путей, особенно у пациентов с бронхиальной астмой. Поэтому, если необходима блокада 0t-рецепторов сердца, но нежелательна блокада 02-рецепторов, такие антагонисты 0(-рецепторов как метопролол или атенолол имеют некоторые преимущества перед неселективными 0-антагонистами. Однако пока не существует достаточно специфичных 0(-селективных антагонистов, чтобы полностью исключить взаимодействие с 02-рецепторами. Следо
вательно, нужно избегать назначения этих средств больным астмой.
В. Влияние на глаз. Некоторые 0-блокаторы уменьшают внутриглазное давление, особенно при глаукоме. Считается, что механизм их действия направлен на снижение продукции водянистой влаги (раздел "Клиническая фармакология средств, блокирующих 0-рецепторы” и дополнение "Лечение глаукомы”).
Г. Метаболические и эндокринные эффекты. Такие 0-антагонисты как пропранолол угнетают стимуляцию липолиза симпатической нервной системой. Влияние на метаболизм углеводов не столь очевидно, хотя гликогенолиз в печени, по крайней мере, частично ингибируется блокадой 0-рецепто-ров. Однако основной гормон, повышающий уровень сахара в крови — это глюкагон. Так как неясно, насколько велик вклад 0-антагонистов в поддержание гипогликемии, у пациентов с инсулинзависимым диабетом они должны использоваться с осторожностью. Это особенно важно для диабетиков с неадекватным резервом глюкагона или для пациентов с удаленной поджелудочной железой.
Хроническое применение антагонистов 0-адре-норецепторов приводит к повышению концентрации в плазме холестерина ЛПОНП и снижению
Пропранолол 0.5 мг/кг
1 мкг/кг адреналин
1 мкг/кг адреналин
Сила сердечных сокращений
Рис. 10-6. Эффекты адреналина у наркотизированной собаки до и после введения пропранолола. В присутствии 0-адреноблокатора адреналин не увеличивает ни силу сокращений (измеренную с помощью калиброванного датчика, закрепленного на стенке желудочка), ни частоту сердцебиений. АД повышается на фоне действия адреналина, так как вазоконстрикторная реакция не блокирована. (Из: Shanks R. G. The pharmacology of beta sympathetic blockade. Am. J. Cardiol. 1966; 18:312.)
концентрации холестерина ЛПВП. Эти изменения потенциально неблагоприятны из-за риска кардиоваскулярной патологии. Хотя концентрация ЛПНП обычно не изменяется, часто снижается соотношение холестерин ЛПВП/холестерин ЛПНП, что повышает вероятность поражения коронарных артерий. Эти изменения вызываются и селективными, и неселективными 0-блокаторами, хотя реже встречаются при применении препаратов, обладающих внутренней симпатомиметической активностью (парциальных агонистов). Интересно, что использование антагонистов а-адренорецепторов, таких как празозин, не влияет на липопротеидный спектр плазмы либо повышает концентрацию ЛПВП, что является благоприятным признаком. Механизм, посредством которого антагонисты а- и 0-рецепторов вызывают эти изменения, пока не выяснен.
Д. Эффекты, не связанные с блокадой 0-рецеп-торов. Первый синтезированный 0-блокатор ди-хлоризопротеренол обладал значительной активностью парциального 0-агониста. Предполагается, что сохранение некоторой внутренней симпатомиметической активности желательно для предотвращения таких побочных эффектов, как провокация астмы. Пиндолол и другие парциальные агонисты указаны в табл. 10-2. Однако пока неясно, насколько ценны они для клиники. Эти средства могут быть полезны для пациентов, реагирующих на неселективные 0-антагонисты симптоматической брадикарди
ей или приступом астмы. К тому же продолжение исследования парциальных агонистов оправдано их меньшим нежелательным влиянием на липидный состав плазмы.
Местноанестезирующее действие, известное также как “мембраностабилизирующее”, является важным эффектом некоторых 0-блокаторов (табл. 10-2). Этот эффект — результат типичной местноанестезирующей блокады натриевых каналов и может быть обнаружен в нейронах, мышце сердца и мембране скелетных мышц. Однако вряд ли он проявляется при системном введении препаратов, так как концентрации, достигаемые в этом случае, слишком малы для анестезирующего действия. Соталол — неселективный антагонист 0-ре-цепторов, который не имеет местного анестезирующего действия, но обладает заметными антиарит-мическими эффектами препаратов III класса — блокаторов калиевых каналов (глава 14).
Специфические препараты (табл.10-2)
Пропранолол — типичное 0-блокирующее средство. Он имеет низкую дозозависимую биодоступность в результате активного метаболизма при первом прохождении печени. Имеются пролонгированные формы пропранолола, увеличивающие продолжительность всасывания вещества до 24 часов и
более. Препарат мало влияет на а- и мускариновые рецепторы, однако отличается умеренной блокирующей активностью в отношении центральных серотониновых рецепторов. Не наблюдается заметного парциального антагонистического влияния лекарства на 0-рецепторы.
Метопролол, атенолол и другие препараты (табл. 10-2) являются представителями группы Ргселективных средств. Для больных с бронхокон-стрикторными реакциями на пропранолол применение этих веществ может быть более безопасным. Однако их Pi-селективность довольно умеренна, поэтому они должны применяться у больных с бронхоспазмом в анамнезе с очень большой осторожностью. Pi-селективные антагонисты более предпочтительны, если терапия Р-блокаторами требуется больным диабетом или с заболеваниями периферических сосудов.
Надолол заслуживает внимания в связи с продолжительным действием. Спектр его эффектов сходен со спектром действия тимолола.
Тимолол — неселективное средство, не обладающее местноанестезирующей активностью. Он эффективно понижает внутриглазное давление.
Пиндолол, ацебутолол, картеолол, целипролол и пенбутолол представляют интерес, поскольку являются парциальными агонистами Р-рецепторов. Они эффективны при основных кардиоваскулярных показаниях для группы Р-блокаторов (гипертензии и стенокардии). Хотя для этих парциальных агонистов менее вероятны брадикардия и нарушения липидного состава плазмы, чем для антагонистов, общая клиническая значимость внутренней симпатомиметической активности остается неопределенной.
Целипролол — это новый похожий на пиндолол Pi-селективный антагонист с умеренной способностью активировать 02-рецепторы. Средство эффективно при гипертензии и стенокардии. Имеются ограниченные свидетельства того, что целипролол обладает нежелательным бронхоконстрикторным эффектом при астме и даже может способствовать бронходилатации (van Zyl et al., 1989). Если отсутствие неблагоприятных эффектов у астматиков будет подтверждено более длительными и широкими клиническими испытаниями, целипролол будет иметь значительные преимущества у этой категории больных (раздел “Клиническая фармакология средств, блокирующих 0-рецепторы, подраздел “Гипертензия”).
Лабеталол — обратимый антагонист адренорецепторов, представляющий собой рацемическую смесь двух пар хиральных изомеров (молекула имеет два центра асимметрии). Изомеры SS и RS не активны, SR — сильный а-блокатор, a RR — сильный 0-блокатор. Аффинность лабеталола к «-рецепторам меньше, чем у фентоламина, однако он «гсе-лективен. Его 0-блокирующая активность менее выражена, чем у пропранолола. Гипотензия, вызываемая лабеталолом, сопровождается меньшей тахикардией, чем при введении фентоламина и сходных с ним а-блокаторов.
Эсмолол — 01-селективный антагонист ультракороткого действия. Эфирная связь эсмолола быстро разрушается под действием эстераз эритроцитов с образованием метаболитов, имеющих низкий аффинитет к 0-рецепторам. Вследствие этого эсмолол имеет короткий период полувыведения, около 10 минут. В результате во время постоянной инфузии быстро достигается устойчивая концентрация лекарства, а при ее прекращении быстро заканчивается действие препарата. Эсмолол потенциально более безопасен, чем длительнодействующие антагонисты, для применения у больных в критическом состоянии, которым требуется лечение антагонистами 0-рецепторов. Эсмолол можно использовать для купирования суправентрикулярных аритмий, периоперативной гипертензии и острой ишемии миокарда.
Бутоксамин селективен в отношении 02-рецепторов. Активный поиск таких средств не проводится, так как для них нет очевидных клинических показаний.
IV. Клиническая фармакология средств, блокирующих ^-рецепторы
Гипертензия
Многие средства, блокирующие 0-адренорецеп-торы, оказались эффективны и хорошо переносимы при гипертензии. Хотя у больных с гипертензией наблюдается хороший эффект при лечении только 0-блокаторами, в большинстве случаев эти препараты комбинируют с диуретиками или вазодилататорами. Несмотря на короткий период полувыведения многих 0-антагонистов, эти средства мож
но принимать один или два раза в день, получая при этом адекватный терапевтический эффект. Конкурентный а- и р-антагонист лабеталол также эффективен при гипертензии, хотя его роль окончательно еще не определена. Применение этих средств детально обсуждается в главе И.
В последнее десятилетие значительное внимание уделялось данным о том, что парциальные агонисты (например, пиндолол) повышают безопасность антигипертензивной терапии у больных с заболеваниями дыхательных путей. Теоретически многое говорит в пользу этого, поскольку такие Р-блокаторы являются скорее парциальными агонистами Р2-> чем Pi-рецепторов. Результатом этого должна быть полезная бронходилатация и даже дополнительное гипотензивное действие из-за расслабляющего гладкие мышцы эффекта активации Р2-рецепторов наряду с уменьшением кровяного давления из-за антагонистического влияния на Ргрецепторы. К сожалению, в настоящее время не существует достаточных клинических доказательств этой гипотезы. Одно из потенциальных преимуществ Р-блокаторов, являющихся парциальными агонистами, которое пока не опровергнуто,—
относительно слабое влияние на липидный состав плазмы (Fitzgerald, 1993).
Ишемическая болезнь сердца
Блокаторы Р-адренорецепторов уменьшают частоту приступов стенокардии и улучшают переносимость физической нагрузки у большинства людей, страдающих ишемической болезнью сердца (глава 12). Эти эффекты связаны с блокадой Р-рецепторов сердца, результатом чего является уменьшение работы сердца и снижение потребности в кислороде. Клиническим улучшением может быть замедление и упорядочивание частоты сердечных сокращений (рис. 10-7). Обширные исследования указывают на то, что долговременное применение тимолола, пропранолола, окспренолола или метопролола у больных, перенесших инфаркт миокарда, удлиняет их жизнь (рис. 10-8). Результаты экспериментов на животных наводят на мысль, что использование антагонистов p-рецепторов во время острой фазы инфаркта миокарда может ограничить его размеры. Однако это показание является предметом обсуждения.
Рис. 10-7. Частота сердечных сокращений у пациента с ишемической болезнью сердца, измеряемая телеметрически во время просмотра телевизионных передач. Измерения были начаты через 1 час после приема плацебо (верхний график) или 40 мг окспренолола (нижний график), неселективного р-антагониста с парциальной агонистической активностью. В условиях данного эксперимента под действием препарата не только снижалась частота сердечных сокращений, но и уменьшались ее изменения в ответ на стимулы. (Из: Taylor S.H. Oxprenolol in clinical practice. Am. J. Cardiol. 1983; 52:34D.)
О 12 24 36 48 60 72
Месяцы
Рис. 10-8. Влияние терапии p-блокаторами на общую смертность от всех причин за 6 лет среди 1884 пациентов, переживших инфаркт миокарда. В рандомизированных испытаниях больные получали плацебо (прерывистая линия) или тимолол (сплошная линия) (Из: Pederson Т. R. Six-year follow-up of the Norwegian multicenter study on timolol after acute myocardial infarction. N. Engl. J. Med. 1985; 313:1055.)
Сердечные аритмии
Р-антагонисты эффективны при лечении больных с суправентрикулярными и желудочковыми аритмиями (глава 14). В случаях синусовой тахикардии и суправентрикулярной экстрасистолии лечение не должно ограничиваться только их использованием, необходимо уделять внимание основному заболеванию. Повышая рефрактерный период атриовентрикулярного узла, Р-антагонисты замедляют частоту ответа желудочков при мерцании и фибрилляции предсердий. Эти средства могут также устранять желудочковую экстрасистолию, особенно если эктопическая активность вызвана катехоламинами. Соталол наряду с Р-блоки-рующим действием имеет дополнительные анти-аритмические эффекты (глава 14).
Другие сердечно-сосудистые нарушения
Было обнаружено, что антагонисты Р-рецепторов увеличивают ударный объем у некоторых больных с обструктивной кардиомиопатией. Полагают, что этот положительный эффект является результатом замедления желудочкового выброса и снижения сопротивления току крови. Р-антагонисты полезны при расслаивающей аневризме аорты для снижения скорости нарастания систолического давления. Не
давно полученные экспериментальные данные позволяют предположить, что, как это ни парадоксально, Р-блокаторы могут быть эффективны при застойной сердечной недостаточности у тщательно отобранных больных, хотя эта возможность требует более широкого исследования. Однако эти средства обычно противопоказаны больным с застойной сердечной недостаточностью, зависящей от симпатической регуляции поддержания сердечного выброса.
Глаукома
(дополнение “Лечение глаукомы”)
Было обнаружено, что при системном введении P-блокирующих средств по другим показаниям уменьшалось внутриглазное давление у больных глаукомой. Впоследствии установили, что и местное применение снижает внутриглазное давление. Механизм, как полагают, заключается в уменьшении продукции водянистой влаги цилиарным телом, физиологически активируемой цАМФ. Для местного применения в глазной практике предпочтителен тимолол, так как у него нет местноанестезирующей активности и он является чистым антагонистом. При открытоугольной глаукоме эффективность тимолола сравнима с эффективностью адреналина или пилокарпина. Поскольку максимальная ежедневная доза при местном применении (1 мг) мала по сравнению с системно вводимой дозой, обычно используемой для лечения гипертензии или стенокардии (10-60 мг), всасывающийся с поверхности глаза тимолол может вызвать серьезное нежелательное влияние на сердце и дыхательные пути только у очень чувствительных пациентов. При местном использовании тимолол может взаимодействовать с принятым внутрь верапамилом.
Бетаксолол, картеолол, левобунолол и метипра-нолол — новые антагонисты p-рецепторов, одобренные для лечения глаукомы. Бетаксолол имеет потенциальное преимущество, являясь р(-селектив-ным препаратом. В какой мере это потенциальное преимущество может уменьшать системные нежелательные эффекты, пока не определено. Однако у некоторых больных средство вызывает ухудшение легочных симптомов.
Гипертиреоидизм
Чрезмерное влияние катехоламинов является важным аспектом патофизиологии гипертиреои-
Лечение глаукомы
Глаукома является основной причиной слепоты среди пожилого населения и привлекает внимание фармакологов, поскольку ее хроническая форма часто поддается лекарственной терапии. Основное проявление болезни — бессимптомное вначале повышение внутриглазного давления. Без лечения это вызывает повреждение сетчатки и зрительного нерва с ограничением полей зрения и, наконец, слепоту. Внутриглазное давление легко измеряется при офтальмологическом исследовании.
Выделяют два основных типа глаукомы: открытоугольную и закрытоугольную (или узкоугольную). Закрытоугольная форма наблюдается при неглубокой передней камере, в которой дилатированная радужка может закрывать путь дренажного оттока в углу между роговицей и реснитчатым телом (рис. 6-9). При этой форме ощущается острое и болезненное увеличение внутриглазного давления, которое в неотложном порядке снимают лекарственными средствами или предотвращают хирургическим удалением части радужки (иридэктомия). Открытоугольная форма глаукомы — хроническое заболевание, и лечение его главным образом фармакологическое.
Поскольку внутриглазное давление определяется балансом между образованием жидкости и ее эвакуацией из глазного яблока, существуют две стратегии лечения закрытоугольной глаукомы: уменьшение секреции водянистой влаги и увеличение оттока жидкости. В соответствии с этим для уменьшения внутриглазного давления могут применяться четыре основных группы средств: холиномиметики, а-агонисты, Р-блокаторы и диуретики (табл. 10-3). Из них наиболее популярны Р-блокаторы. Эта популярность — результат удобства (применение один или два раза в день) и относительного отсутствия нежелательных эффектов (исключая осложнения у пациентов с астмой, с искусственным водителем ритма или нарушениями проводимости). Сообщают и о других средствах, уменьшающих внутриглазное давление: a(-aH-тагонистах, простагландине Е2 и марихуане. Применение лекарственных средств при острой закрытоугольной глаукоме ограничено холино-миметиками и осмотическими средствами на дооперационном этапе. Начало действия других препаратов развивается слишком медленно для этой ситуации.
дизма, особенно в отношении сердца (глава 37). Р-антагонисты в этих условиях имеют благотворные эффекты, вероятно, связанные с блокадой адренорецепторов и угнетением периферического превращения тироксина в трийодтиронин. Последнее действие может варьировать у разных Р-блока-торов. Пропранолол особенно эффективен при гипертиреоидном кризе. Его с осторожностью используют в этих условиях для контроля над суправентрикулярной тахикардией, которая часто вызывает застойную сердечную недостаточность.
(глава 27). Малыми дозами пропранолола, особенно при профилактическом приеме, можно снимать соматические проявления тревоги. Пропранолол эффективен также при алкогольной абстиненции. Установлено, что антагонисты Р-рецепторов уменьшают давление в портальной вене у больных циррозом печени. Есть свидетельства, что пропранолол и надолол понижают вероятность начала кровотечения при варикозном расширении вен пищевода и снижают смертность от кровотечений больных циррозом (Poynard et al., 1991).
Неврологические заболевания
По некоторым данным, пропранолол снижает частоту и интенсивность мигренеподобных головных болей. Механизм этого эффекта неизвестен. Так как симпатическая активность может повышать тремор скелетных мышц, неудивительно, что р-ан-тагонисты ослабляют определенные виды тремора
Выбор ^-блокирующего средства
Пропранолол является стандартным препаратом сравнения для новых р-антагонистов, предлагаемых для системного применения. За многие годы очень широкого использования он зарекомендовал
ТАБЛИЦА 10-3. Средства, применяемые при глаукоме
Препараты Механизм действия Способ введения
Холиномиметики Пилокарпин, карбахол, физостиг- Сокращение цилиарной мышцы, Местно: глазные капли или гель, плас-
мин, эхотиофат открытие трабекулярной сети; тиковые пленки с медленным высво-увеличение оттока вождением вещества
а-агонисты Неселективные Адреналин, дипивефрин а2-селективные Увеличение оттока Местно: глазные капли
Апраклонидин Уменьшение секреции жидкости Местно, после лазерной хирургической
операции
р-блокаторы Т имолол,бетаксолол,картеолол, Снижение секреции жидкости Местно: глазные капли
левобунолол, метипранолол цилиарным эпителием
Диуретики Ацетазоламид Снижение секреции из-за недо- Перорально; в клинических испытаниях
статка НСО3_ местнодействующие ингибиторы кар-
боангидразы
Этакриновая кислота Снижение секреции Внутриглазная инъекция с длительными
(исследуется) интервалами, например ежегодно
себя как безопасное и эффективное при многих заболеваниях средство. Некоторые клинически важные сопутствующие эффекты у разных антагонистов Р-рецепторов могут быть опосредованы различными механизмами, поэтому эти препараты нельзя рассматривать как взаимозаменяемые по всем показаниям. Так, при гипертиреоидизме или инфаркте миокарда можно применять лишь определенные Р-блокаторы. Преимущества и недостатки Р-анта-гонистов, являющихся парциальными агонистами, пока недостаточно определены в клинических условиях.
Клиническая токсичность средств, блокирующих Р-рецепторы
Есть сообщения о ряде незначительных токсических эффектов пропранолола. Сыпь, лихорадка и другие проявления лекарственной аллергии редки. Влияние на центральную нервную систему выражается в заторможенности, нарушении сна и депрессии. Редко встречаются психотические реакции. Если у больного развивается депрессия нужно думать о прекращении применения Р-блокаторов, когда это выполнимо клинически. Считается, что ан
тагонисты Р-рецепторов с низкой растворимостью в липидах реже вызывают нежелательное действие на центральную нервную систему, чем соединения с высокой липотропностью (табл. 10-2). Для выработки определенных рекомендаций по применению Р-блокаторов необходимо дальнейшее сравнительное изучение нежелательного воздействия разных препаратов на центральную нервную систему. Однако уже сейчас представляется разумным назначение надолола или атенолола пациентам, нетипично реагирующим на другие Р-антагонисты.
Главные побочные эффекты этих средств связаны с известными последствиями Р-блокады. При применении неселективных препаратов блокада р2-рецепторов часто вызывает ухудшение бронхиальной астмы и других форм обструкции дыхательных путей, но они безопасны в этом отношении для пациентов со здоровой дыхательной системой. Даже незначительная астма может стать очень тяжелой вследствие приема Р-блокаторов. р(-селективные средства меньше влияют на дыхательные пути, чем неселективные препараты, но их надо применять осторожно (если вообще применять) у пациентов с повышенной реактивностью дыхательных путей. Pi-блокаторы обычно хорошо переносятся больными с заболеваниями периферических сосудов легкой и средней тяжести, но необходима осторож
ность, если эти нарушения тяжелые или отмечается синдром вазоспазма.
Блокада Р-рецепторов подавляет сократимость и возбудимость миокарда. У пациентов с нарушениями функции миокарда сердечный выброс может зависеть от симпатической активации. Если этот стимул устраняется Р-блокадой, может наблюдаться декомпенсация. Следует крайне осторожно использовать Р-блокаторы при инфаркте миокарда или компенсированной сердечной недостаточности. Побочный эффект p-блокаторов на сердце нивелируется напрямую действием изопротеренола или с помощью глюкагона (глюкагон оказывает стимулирующее действие на сердце, воздействуя на глюкагоновые рецепторы, которые не блокируются Р-антагонистами), но ни один из этих методов не является полностью безопасным. У чувствительных пациентов даже очень маленькая доза Р-бло-катора (например, 10 мг пропранолола) может вызвать тяжелую сердечную недостаточность. Антагонисты Р-рецепторов способны взаимодействовать с блокатором кальциевых каналов верапамилом. Как побочные эффекты в таких ситуациях были описаны тяжелая гипотензия, брадикардия, сердечная недостаточность и нарушения проводимости. Они могут возникать и у чувствительных пациентов, применяющих местнодействующие (офтальмологические) Р-блокаторы в сочетании с пероральным приемом верапамила.
Может быть опасно внезапное прекращение приема Р-антагонистов после продолжительной терапии. Есть сведения, что у пациентов с ИБС в такой ситуации повышен риск возникновения побочных реакций. Механизм этого эффекта неясен, но может включать “up-регуляцию" Р-рецепторов. До появления более определенных сведений относительно степени риска считается правильным постепенно снижать, а не резко отменять эти препараты.
Частота возникновения гипогликемических эпизодов у больных диабетом, усугубляющимся под действием Р-блокаторов, неизвестна. Тем не менее если возможна альтернативная терапия, не рекомендуется назначать эти средства больным инсулинзависимым диабетом, страдающим гипогликемическими реакциями. В такой ситуации имеют некоторые преимущества Ргселективные препараты, так как восстановление после гипогликемии происходит быстрее, чем при применении неселективных блокаторов.
Р-блокада может маскировать клинические признаки развития гипертиреоидизма. Этот эффект становится все более и более важным, так как постоянно увеличивается количество пациентов, принимающих Р-агонисты в течение длительного времени.
Препараты
а-блокаторы
Доксазозин (Кардура)
Перорально: таблетки по 1, 2,4,8 мг
Феноксибензамин (Дибензилин)
Перорально: капсулы по 10 мг
Фентоламин (Регитин)
Парентерально: 5 мг/мл для инъекций
Празозин (генерик, Минипресс)
Перорально: капсулы по 1, 2, 5 мг
Теразозин (Хитрин)
Перорально: таблетки по 1, 2, 5,10 мг
Толазолин (Присколин)
Парентерально: 25 мг/мл для инъекций
Р-блокаторы
Ацебутолол (Сектрал)
Перорально: капсулы по 200,400 мг
Атенолол (Тенормин)
Перорально: таблетки по 25, 50, 100 мг
Парентерально: 0.5 мг/мл для в/в инъекций
Бетаксолол
Перорально: таблетки по 10, 20 мг (Керлон)
Глазные капли: 0.25 % (Бетоптик)
Бисопролол (Зебета)
Перорально: таблетки по 5, 10 мг
Картеолол
Перорально: таблетки по 2.5, 5 мг (Картрол)
Глазные капли: 1 % (Окупресс)
Эсмолол (Бревиблок)
Парентерально: 10 мг/мл для в/в инъекций;
250 мг/мл для в/в вливания
Лабеталол (Нормодин, Трандат)
Перорально: таблетки по 100, 200, 300 мг
Парентерально: 5 мг/мл для инъекций
Левобунолол (Бетаган Ликвифилм)
Глазные капли: 0.5 %
Метипранолол (Оптипранолол)
Глазные капли: 0.3 %
Метопролол (Лопрессор, Топрол) Перорально: таблетки по 50,100 мг Перорально для пролонгированного действия: таблетки по 50,100, 200 мг Парентерально: 1 мг/мл для инъекций
Надолол (Коргард)
Перорально: таблетки по 20,40, 80, 120, 160 мг
Пенбутолол (Леватол)
Перорально: таблетки по 20 мг
Пиндолол (Вискен)
Перорально: таблетки по 5,10 мг
Пропранолол (генерик, Индерал)
Перорально: таблетки по 10,20,40,60,80, 90 мг; растворы по 4,8, 80 мг/мл
Перорально для пролонгированного действия: капсулы по 60,80,120,160 мг Парентерально: 1 мг/мл для инъекций
Соталол (Бетапейс)
Перорально: таблетки по 80, 160, 240 мг
Тимолол
Перорально: таблетки по 5,10,20 мг (Блокадрен) Глазные капли: 0.25,0.5 % (Тимоптик)
Ингибитор синтеза
Метирозин (Демсер)
Перорально: капсулы по 250 мг
Избранная литература
Babamoto К. Hirokawa W. Т. Doxazozin: A new alpha(-adrenergic antagonist. Clin. Pharm. 1992; 11:415.
Benfield P., Clissold S. P., Brogden R. N. Metoprolol. Drugs, 1986; 31: 376.
Beta blockers and lipophilicity. (Editorial) Lancet, 1987; 1: 900.
Goldberg M. R., Robertson D. Yohimbine: A pharmacological probe of alpha2 adrenoreceptor. Pharmacol. Rev. 1983; 35:143.
Lesar T. S. Comparison of ophthalmic beta-blocking agents. Clin. Pharm. 1987; 6:451.
Limbird L. E. The Alpha-2 Adrenergic Receptors. Humana Press, 1988.
Ruffolo R. R. Jr. et. al. Pharmacologic and therapeutic applications of alpha2-adrenoreceptor subtypes. Annu. Rev. Pharmacol. Toxicol. 1993; 32: 243.
Ruffolo R. R. Jr. The Alpha-1 Adrenergic Receptors. Humana Press, 1987.
Taylor S. H. Intrinsic sympathomimetic activity: Clinical fact or fiction? Am. J. Cardiol. 1983; 52: 16D.
Раздел III
Средства, влияющие на сердечно-сосудистую систему и почки________________
Антигипертензивные средства “| “|
Нил Л. Беновиц
По некоторым оценкам, артериальное давление (АД) крови у 15 % взрослых американцев повышено до степени, требующей медицинского вмешательства. Распространенность этого явления зависит от возраста, расы, образования и многих других факторов. Стойкая артериальная гипертензия приводит к повреждению сосудов почек, сердца и мозга и, как следствие,— к повышению частоты почечной недостаточности, коронарной болезни и инсульта. Эффективное понижение АД с помощью фармакологических средств предупреждает повреждение сосудов и значительно снижает заболеваемость и смертность. Существует достаточно много эффективных лекарств. Знание механизма их антигипертензивного действия и точек приложения позволяет проводить эффективное лечение, а также избегать побочных эффектов.
Гипертензия и регуляция артериального давления
Диагноз
Диагноз гипертензии основывается на повторных и воспроизводимых определениях повышенного АД. Диагноз служит главным образом для предсказания последствий, он редко отражает причины гипертензии.
Эпидемиологические исследования показывают, что риск повреждения почек, сердца и мозга прямо связан со степенью подъема АД. Даже незначительная гипертензия (АД > 140/90 мм рт. ст.) у лиц молодого или среднего возраста повышает риск поражения органов. Риск, а следовательно, и необходимость лечения повышаются пропорционально величине подъема АД. Вероятность поражения органов выше у чернокожих людей при любом уровне АД и в любом возрасте и относительно ниже у женщин в предменопаузный период по сравнению с мужчинами. К другим факторам риска относятся курение, гиперлипидемия, диабет, признаки поражения органов к моменту постановки диагноза и наличие в семейном анамнезе сердечно-сосудистых заболеваний.
Следует заметить, что диагноз гипертензии ставится только на основании измерения АД, а не субъективных ощущений больного, так как гипертензия обычно бессимптомна до тех пор, пока не возникают явные признаки поражения органов.
Этиология гипертензии
Специфическая причина гипертензии может быть установлена лишь у 10-15 % больных. Однако по возможности ее следует выяснить, так как в ряде случаев необходимо хирургическое лечение: при сужении почечной артерии, коарктации аорты,
феохромоцитоме, болезни Кушинга и первичном альдостеронизме.
Если у больного не выявлена конкретная причина гипертензии, говорят об эссенциальной гипертензии.1 В большинстве случаев повышенное АД связано с увеличением общего периферического сопротивления току крови через артериолы, при этом сердечный выброс обычно нормален. Исследования функции автономной нервной системы, барорецепторных рефлексов, ренин-ангиотензин-альдостероновой системы и почек не смогли выявить первичные нарушения, которые можно было бы рассматривать как причину повышения сопротивления периферических сосудов при эссенциальной гипертензии.
Повышенное АД обычно обусловлено несколькими причинами. Данные эпидемиологии указывают на наследственность, психологический стресс, факторы среды и диету (повышенное потребление соли и, возможно, снижение потребления кальция) в развитии гипертензии. Повышение АД с возрастом не происходит в популяции с низким ежедневным потреблением соли. Больные с лабильной гипертензией более склонны по сравнению со здоровыми к повышению АД после солевой нагрузки.
Регуляция кровяного давления в норме
В соответствии с законами гидравлики АД прямо пропорционально произведению кровотока (сердечный выброс, СВ) и сопротивления при прохождении крови через прекапиллярные артериолы (периферическое сосудистое сопротивление, или общее периферическое сопротивление — ОПС):
АД = СВхОПС. [11-1]
Физиологически как у здорового, так и больного человека АД поддерживается путем регуляции СВ и ОПС в трех анатомических областях (рис. 11-1): артериолах, посткапиллярных венулах (емкостные сосуды) и сердце. Четвертая анатомическая контрольная область — почки — вносит весомый вклад в поддержание АД, регулируя объем внутрисосудистой жидкости. Барорефлексы, реализуемые через симпатические нервы, совместно с гуморальными механизмами, включая ренин-ангио-
' Определение первоначально базировалось на том, что подъем АД необходим для адекватной перфузии больного органа (на сегодняшний день зта идея устарела).
Рис. 11-1. Анатомические области, участвующие в регуляции кровяного давления
тензин-альдостероновую систему, координируют функции этих четырех контролирующих зон в поддержании нормального АД.
АД у больного с гипертензией регулируется теми же механизмами, что и у здорового человека с нормальными значениями АД. Регуляция АД при гипертензии отличается от нормы тем, что барорецепторы и почечный контроль давления, сопряженный с объемом крови, имеют “установочную точку” регуляции на более высоком уровне АД. Все антигипертензивные средства воздействуют на эти нормальные механизмы.
А. Постуральный барорефлекс (рис. 11-2). Барорефлексы участвуют в быстрой сиюминутной регуляции АД, например при переходе из горизонтального в вертикальное положение. Центральные симпатические нейроны, берущие начало в вазомоторной зоне продолговатого мозга, тонически активны. Каротидные барорецепторы стимулируются при растяжении стенки сосудов, вызванном давлением изнутри (АД). Активация барорецепторов тормозит центральные симпатические импульсы. Напротив, уменьшение растяжения ведет к снижению барорецепторной активности. Таким образом, при переходе в вертикальное положение давление на барорецепторы снижается вследствие депонирования крови в венах ниже уровня сердца и симпатическая активность растормаживается. Рефлекторно возрастают периферическое сосудистое сопротивление (сокращение артериол) и сердечный
2. Ядро солитарного тракта
Сенсорные волокна
Тормозной вставочный нейрон
XI
XII
Ствол мозга
Спинной мозг
4. Автономный ганглий
Рис. 11-2. Рефлекторная дуга барорефлекса
1. Барорецептор каротидного синуса
Артериальное давление крови
5. Симпатическое нервное окончание
3. Вазомоторный центр Моторные волокна
i. а- или Р-рецептор
выброс (прямая стимуляция сердца и сокращение емкостных сосудов, что ведет к увеличению венозного возврата к сердцу), и в результате восстанавливается нормальное АД. Такой же барорефлекс срабатывает в ответ на любое снижение АД, в том числе на первичное понижение периферического сосудистого сопротивления (например, вызванного сосудорасширяющим веществом) или уменьшение внутрисосудистого объема (например, из-за кровопотери или потери солей и воды через почки).
Б. Реакция почек на снижение АД. Почки участвуют в долгосрочной регуляции АД. Снижение почечной перфузии вызывает внутрипочечное перераспределение кровотока и повышает реабсорбцию соли и воды. Кроме того, снижение давления в почечных артериолах, также как и симпатическая активность (через Р~адренорецепторы), стимулирует выработку ренина, который усиливает продукцию ангиотензина II (рис. 11-1 и глава 17). Ангиотензин II вызывает прямое сокращение резистивных сосудов и стимулирует синтез альдостерона в коре надпочечников, что ведет к повышению реабсорбции натрия в почках и повышению внутрисосудистого объема крови.
Терапевтические аспекты
Поскольку антигипертензивная терапия не направлена обычно на определенную причину, она неизбежно затрагивает нормальные физиологичес
кие механизмы регуляции АД. Антигипертензивная терапия может назначаться больному без явных жалоб и не вести к изменению его самочувствия; в этом случае смысл понижения АД состоит в предупреждении заболевания и смерти в обозримом будущем. Возможно, основная проблема терапии гипертензии заключается в постоянном эффективном лекарственном лечении в течение многих лет при активном участии самого больйого.
Важно сопоставить риск побочных эффектов от лекарственного лечения с риском осложнений не-корригируемой гипертензии, который пропорционален степени подъема АД до лечения и варьирует в соответствии с особенностями конкретного больного. Поэтому нет определенных схем лечения, подходящих для больших групп больных с гипертензией.
I. Базисная фармакология антигипертензивных средств
Все антигипертензивные средства действуют на одну или несколько анатомических структур, участвующих в контроле АД (рис. 11-1), и оказывают эффект путем вмешательства в нормальные механизмы регуляции АД. Рабочая классификация этих средств разделяет их в соответствии с основными
регуляторными зонами или механизмами, на которые они воздействуют (рис. 11-3). Лекарства внутри каждой группы имеют сходный спектр побочных эффектов из-за общего механизма действия. Можно выделить следующие категории антигипертензивных средств.
1) Диуретики, понижающие АД путем выведения из организма натрия и уменьшения объема крови и, возможно, за счет других механизмов.
2) Симпатоплегические (симпатолитические) вещества, которые понижают АД, уменьшая периферическое сосудистое сопротивление, ингибируя функциональную активность сердца и повышая депонирование в венозных емкостных сосудах (последние два эффекта снижают сердечный выброс). Эти вещества далее подразделяют в соответствии с предполагаемыми мишенями на дуге симпатического рефлекса.
p-рецепторы сердца Пропранолол
Другие р-блокаторы
g-рецепторы сосудов
Празозин
и другие Of блокаторы
Гладкая мускулатура сосудов Г идралазин Верапамил
Миноксидил и другие
Нитропруссид блокаторы
Диазоксид кальциевых каналов
Канальцы почек Тиазиды и пр.
р-рецепторы юкстагломерулярных клеток, ______которые высвобождают ренин
Пропранолол
и другие р-блокаторы
Конвертирующий фермент Ангиотензин II -•-??—Ангиотензин I
1
Каптоприл и другие ингибиторы АПФ
Ренин
---------Ангиотензиноген
Рис. 11-3. Точки приложения действия основных классов антигипертензивных средств
3) Прямые вазодилататоры снижают давление, расслабляя гладкую мускулатуру сосудов, тем самым приводя к снижению периферического сосудистого сопротивления и увеличению объема емкостных сосудов.
4) Вещества, которые блокируют выработку или действие ангиотензина, тем самым снижая периферическое сосудистое сопротивление и (потенциально) объем крови.
Перечисленные группы лекарств действуют посредством различных механизмов, что позволяет комбинировать вещества двух или более групп с повышением эффективности и, в некоторых случаях, со снижением токсичности (дополнение “Сравнение моно- и комбинированной терапии при гипертензии”).
Средства, изменяющие баланс натрия и воды
Ограничение потребления натрия с пищей снижает АД у больных с гипертензией. С появлением диуретиков необходимость контроля за количеством натрия в пище стала менее актуальной. Однако контроль над АД с помощью диеты является относительно безвредным лечебным мероприятием и может иметь важное профилактическое значение. Результаты некоторых исследований показали, что даже умеренное ограничение приема натрия с пищей понижает АД (хотя и в различной степени) во многих случаях гипертензии.
Механизм действия и гемодинамические эффекты диуретиков
Диуретики первично понижают АД за счет уменьшения запасов натрия в организме. Вначале они понижают АД, уменьшая объем крови и сердечный выброс; периферическое сосудистое сопротивление может при этом возрастать. Через 6-8 недель после начала приема диуретиков сердечный выброс возвращается к норме, тогда как периферическое сосудистое сопротивление снижается. Считают, что натрий способствует повышению сосудистого сопротивления за счет увеличения жесткости и ней-рореакгивности сосудов. Подобное влияние может быть следствием повышения ионообмена Na+ и Са2+
с итоговым увеличением внутриклеточной концентрации ионов кальция. Эти эффекты устраняются применением диуретиков или ограничением натрия в диете.
Некоторые диуретики дополнительно к мочегонному оказывают прямое сосудорасширяющее действие. Индапамид — нетиазидное сульфониламидное мочегонное, обладающее как диуретическим, так и сосудорасширяющим действием. Влед-ствие вазодилатации сердечный выброс не изменяется или незначительно возрастает. Амилорид ингибирует ответ гладкой мускулатуры на сократительные стимулы, возможно, через влияние на трансмембранный и внутриклеточный транспорт ионов кальция вне зависимости от его влияния на экскрецию ионов натрия.
У большинства пациентов диуретики снижают АД на 10-15 мм рт. ст. и подчас для лечения мягкой и умеренной эссенциальной гипертензии можно ограничиться назначением только этих препаратов. При более тяжелой гипертензии диуретики используют в сочетании с симпатоплегиками и вазодилататорами с целью предупреждения задержки натрия, которую вызывают эти средства. Сосудистая реактивность, т. е. способность к сокращению или расслаблению, снижается под влиянием симпатоплегиков и вазодилататоров, и сосуды становятся похожими на негибкие трубки. Вследствие этого повышается зависимость АД от объема циркулирующей крови. Таким образом, при тяжелой форме гипертензии, когда используют несколько различных лекарств, АД может хорошо контролироваться при объеме крови 95 % от нормальной величины, но не поддаваться коррекции и оставаться высоким, если объем циркулирующей крови составляет 105 % от нормы.
Выбор диуретиков
Место действия в почках и фармакокинетика различных мочегонных средств обсуждаются в главе 15. Тиазидные диуретики подходят для лечения большинства больных с мягкой или умеренной гипертензией при нормальной функции почек и сердца. Более мощные диуретики (например, действующие в области петли Генле) необходимы при тяжелой гипертензии, когда используется ряд лекарств, задерживающих натрий; при почечной недостаточности, когда скорость клубочковой фильтрации менее 30-40 мл/мин; и при сердечной недо
статочности или циррозе печени, когда происходит заметная задержка натрия.
Калийсберегающие диуретики полезны как для предупреждения избыточных потерь калия, особенно у больных, получающих дигиталис, так и для усиления натрийуретического действия остальных диуретиков.
Выбор доз
Хотя фармакокинетика и фармакодинамика различных диуретиков неодинакова, у них имеется общая конечная характеристика терапевтического действия при лечении гипертензии — суточный на-трийурез. Следует признать, что при стабильном состоянии (например, при продолжительной терапии гипертензии) суточная экскреция натрия равна количеству натрия, поступающему с пищей. Диуретики необходимы для того, чтобы предупредить тенденцию задержки натрия у больных с относительно сниженными его запасами. Хотя тиазидные диуретики обладают более выраженным натрийуретическим действием в высоких дозах (до 100— 200 мг гидрохлортиазида) при использовании их в качестве единственного лечебного средства, меньшие дозы (20-25 мг) оказывают такое же антигипертензивное действие, как и высокие. Таким образом, пороговое значение величины снижения запасов натрия может быть достаточным для развития антигипертензивного эффекта. Реакция АД на петлевые диуретики в отличие от реакции на тиазиды продолжает возрастать при многократном превышении обычных терапевтических доз.
Токсичность диуретиков
Наиболее частый нежелательный эффект диуретиков (за исключением калийсберегающих) при лечении гипертензии — дефицит калия. Хотя небольшая гипокалиемия хорошо переносится большинством больных, она может оказаться опасной при приеме дигиталиса, при хронических аритмиях или при инфаркте миокарда. Потери калия сопряжены с реабсорбцией натрия, поэтому ограничение приема натрия с пищей уменьшает потери калия. Диуретики могут также вызывать потери магния, снижать толерантность к глюкозе и увеличивать концентрации в сыворотке липидов и мочевой кислоты. Возможность увеличения риска заболевания коронарных артерий в связи с метаболи
ческими эффектами диуретиков находится в стадии изучения. Получены данные, подтверждающие тот факт, что применение небольших доз диуретиков уменьшает нежелательные метаболические эффекты, не влияя на выраженность антигипертензивного действия.
Вещества, влияющие на симпатическую нервную систему
У больных с умеренной и тяжелой гипертензией наиболее эффективное лекарственное лечение включает применение веществ, угнетающих функции симпатической нервной системы. Вещества этой группы классифицируют в соответствии с местом их действия на симпатическую рефлекторную дугу (рис. 11-2). Этанейроанатомическая классификация объясняет большие различия во влиянии веществ на сердечно-сосудистую систему и позволяет клиницисту предсказывать взаимодействие этих веществ друг с другом и с лекарствами иных классов.
Особенно важно, что подклассы веществ имеют различный характер потенциальной токсичности. Вещества, которые понижают АД, действуя на ЦНС, склонны вызывать седацию и психическую депрессию и могут приводить к нарушениям сна, в том числе к ночным кошмарам. Вещества, нарушающие проведение по автономным ганглиям, в дополнение к глубокой симпатической блокаде нарушают парасимпатическую регуляцию. Вещества, действующие главным образом за счет уменьшения высвобождения норадреналина из симпатических нервных окончаний, вызывают эффекты, сходные с эффектом хирургической симпатэктомии, включая подавление эякуляции и гипотензию, которая появляется при переходе в вертикальное положение и после физической нагрузки. Вещества, блокирующие постсинаптические адренорецепторы, имеют более узкий спектр эффектов в зависимости от класса рецепторов, с которыми они связываются.
Некоторые лекарственные препараты имеют более чем одну точку приложения, но один биохимический механизм действия. Пока нет применяемых в клинике лекарств, которые первично действовали бы на барорецепторы. Алкалоиды вератрума снижают АД, повышая чувствительность барорецепторов, но они представляют лишь теоретичес
кий интерес, потому что вызывают неустойчивые терапевтические эффекты и имеют неприемлемое побочное действие.
Следует отметить, что все вещества, которые понижают АД, изменяя функцию симпатической системы, могут вызывать компенсаторные эффекты, механизм которых не опосредован адренерги-ческойиннервацией. Так, антигипертензивный эффект при применении любого из этих препаратов сопровождается задержкой натрия и увеличением объема циркулирующей крови. По этим причинам симпатоплегические антигипертензивные средства наиболее эффективны, когда их используют совместно с диуретиками.
Симпатолитические средства центрального действия
Механизм и место действия
Метилдопа и клонидин уменьшают симпатические влияния вазопрессорных центров ствола мозга, но сохраняют или даже увеличивают чувствительность этих центров к барорецепторному контролю. Поэтому антигипертензивное и побочное действие этих веществ обычно меньше зависит от положения тела, чем при применении таких веществ, как гуанетидин, который прямо влияет на периферические симпатические нейроны.
Метилдопа (Ь-а-метил-3,4-дигидрофенилала-нин) является аналогом L-ДОФА и превращается в а-метилдофамин и а-метилнорадреналин. Этот путь идет параллельно синтезу норадреналина из ДОФА, как показано на рис. 6-5. а-метилнорадреналин хранится в гранулах окончаний адренергических нервов, из которых он стехиометрически вытесняет норадреналин, высвобождается при возбуждении нерва и взаимодействует с постсинаптическими адренорецепторами. Однако это замещение норадреналина ложным медиатором в периферических нейронах не связано с антигипертензивным действием метилдопы, потому что а-метилнорадреналин является эффективным агонистом а-адренорецепторов, вызывающим периферическую симпатическую реакцию сокращения артериол и венул. Прямая электрическая стимуляция симпатических нервов у животных, получающих ме-тилдопу, вызывает симпатические ответы, сходные с теми, которые наблюдаются у интактных животных.
Антигипертензивный эффект метилдопы связан со стимуляцией центральных а-адренорецепторов а-метилнорадреналином и а-метилдофами-ном, что подтверждается следующим: 1) при центральном введении в желудочки мозга метилдопы животным для понижения АД требуются значительно меньшие дозы препарата, чем при внутривенном введении; 2) антагонисты а-рецепторов, особенно а2-селективные антагонисты, при введении в желудочки мозга блокируют антигипертензивный эффект метилдопы независимо от пути введения (центрального или внутривенного) последнего; 3) активные ингибиторы ДОФА-декар-боксилазы, введенные центрально, устраняют антигипертензивный эффект метилдопы, что указывает на необходимость его метаболизации для оказания эффекта.
Антигипертензивное действие клоиидина, производного 2-имидазолина, было открыто в процессе изучения его как средства местного применения для уменьшения отека слизистой носа. После внутривенного введения клонидин вызывает кратковременный подъем АД, за которым следует более продолжительная гипотензия. Прессорный эффект связан с прямой стимуляцией а-адренорецепторов артериол. Вещество классифицируют как парциальный агонист а-рецепторов, потому что оно также подавляет прессорное действие других а-агонистов.
Существует много доказательств того, что гипотензивный эффект клонидина реализуется на уровне а-адренорецепторов нейронов продолговатого мозга. У животных гипотензивный эффект клонидина предупреждается при центральном введении а-антагонистов. Клонидин уменьшает симпатический и повышает парасимпатический тонус, что ведет к снижению АД и брадикардии. Снижение АД сопровождается падением уровня катехоламинов в циркулирующей крови. Эти наблюдения указывают на то, что кдонидин повышает чувствительность мозговых прессорных центров ствола к барорефлекторному торможению.
Таким образом, исследование механизмов действия клонидина и метилдопы показывает, что нормальная регуляция АД опосредована центральными адренергическими нейронами, которые модулируют барорецепторные рефлексы. Оба вещества лучше связываются с а2-,чем с а(-адренорецепторами. Как указывается в главе 6, а2-рецепторы расположены на пресинаптических адренергических
нейронах, так же как и в некоторых постсинаптических областях. Возможно, что клонидин и а-ме-тилнорадреналин оказывает свое действие в мозгу, уменьшая выделение норадреналина в соответствующих рецепторных зонах. Возможно, что эти вещества действуют на постсинаптические а2-адрено-рецепторы, что ведет к угнетению активности соответствующих нейронов. Клонидин связывается и с неадренергическими зонами, известными как имидазолиновые рецепторы, которые также могут опосредовать антигипертензивные эффекты. В стадии исследования находится ряд новейших соединений, в частности рилменидин, моксонидин, которые имеют значительно более высокую аффинность к ^-имидазолиновому рецептору, чем к а-адрено-рецептору и являются эффективными антигипертензивными средствами у животных и человека (Ernsberger, 1993; McKaigue, 1992).
Гемодинамические эффекты метилдопы и кло-нидина несколько отличаются; клонидин снижает частоту сердечных сокращений и сердечный выброс в большей степени, чем метилдопа. Это указывает на то, что вещества имеют различные точки приложения. Они могут действовать на различные популяции нейронов вазомоторных центров ствола мозга.
Гуанабенз и гуанфацин — новые антигипертензивные средства центрального действия, которые обладают сходным с клонидином центральным а-адреностимулирующим эффектом. Однако данных в пользу их существенных преимуществ по сравнению с клонидином нет.
Метилдопа
Метилдопа применяется для лечения гипертензии незначительной и умеренной степени. Препарат понижает АД в основном за счет уменьшения периферического сосудистого сопротивления с вариабельным уменьшением частоты сердечных сокращений (ЧСС) и сердечного выброса.
Введение метилдопы не затрагивает большинство сердечно-сосудистых рефлексов, и понижение АД существенно не зависит от изменения положения тела. Постуральная (ортостатическая) гипотензия может возникнуть у больных со сниженным объемом циркулирующей крови. Одно из потенциальных преимуществ метилдопы состоит в том, что этот лекарственный препарат вызывает снижение сопротивления сосудов почек.
Фармакокинетика и дозирование
В связи с выраженным эффектом первого прохождения (преимущественно О-сульфатная конъюгация в слизистой оболочке ЖКТ) биодоступность метилдопы низка и составляет в среднем 25 %, причем варьирует у различных пациентов. Около 2/3 вещества, попадающего в системный кровоток, выводится путем почечной экскреции с конечным временем полуэлиминации примерно 2 часа. Нарушение функции почек снижает клиренс препарата.
Метилдопа поступает в мозг посредством механизма активного транспорта ароматических аминокислот. При приеме внутрь метилдопа оказывает максимальное антигипертензивное действие через 4-6 часов, и эффект сохраняется до 24 часов. Поскольку эффект зависит от накопления метаболита (а-метилнорадреналина), действие сохраняется после того, как исходное вещество покидает кровоток.
Метилдопа имеет ограничение максимума эффективности по снижению АД. У большинства больных доза около 2 г вызывает максимальное понижение давления; если результат оказывается неудовлетворительным, повышение дозы, как правило, не дает большего эффекта. Обычная терапевтическая доза составляет 1-2 г/день в несколько приемов. Для многих больных препарат достаточно назначать один раз в день.
Токсичность
Большинство нежелательных эффектов метилдопы имеют центральную природу. Наиболее частым является заторможенность, особенно в начале лечения. При продолжительной терапии больные могут жаловаться на усталость и нарушение способности концентрировать внимание. Сравнительно редко возникают ночные кошмары, психическая депрессия, головокружение и экстрапирамидные нарушения. Не исключено появление лактации, связанной с увеличением секреции пролактина. Она развивается как у мужчин, так и у женщин, получающих метилдопу. Эти побочные эффекты, вероятно, объясняются угнетающим влиянием на дофаминергическую передачу в гипоталамусе.
Другой нежелательный эффект метилдопы — появление положительного теста Кумбса (примерно у 10-20 % больных, получающих препарат бо
лее 12 месяцев), что иногда затрудняет подбор крови для трансфузии и изредка связано с гемолитической анемией, гепатитом и лекарственной лихорадкой. Прекращение приема лекарства обычно приводит к быстрому исчезновению этих нарушений.
Клонидин
Исследование гемодинамики показывает, что понижение АД при приеме клонидина связано с уменьшением сердечного выброса вследствие понижения ЧСС и расслабления емкостных сосудов с уменьшением периферического сосудистого сопротивления, особенно в вертикальном положении (когда в норме симпатический тонус повышается).
Понижение АД клонидином сочетается со снижением сопротивления сосудов почек с сохранением почечного кровотока. Как и метилдопа, клонидин понижает АД у больного в горизональном положении и редко вызывает постуральную гипотензию. Прессорные эффекты клонидина не выявляются после приема терапевтических доз, но при передозировке может возникнуть серьезная гипертензия.
Клонидин
Фармакокинетика и дозирование
У здоровых людей биодоступность клонидина составляет в среднем 75 %, а период полувыведения — 8-12 часов. Около половины препарата элиминируется в неизмененном виде, следовательно, при почечной недостаточности могут быть эффективны меньшие дозы.
Клонидин растворим в жирах и быстро поступает из кровотока в мозг. В связи с относительно коротким периодом полувыведения и тем, что антигипертензивный эффект прямо связан с концентрацией в крови, клонидин можно давать два раза в день с целью “мягкой” коррекции АД. Терапевтические дозы обычно находятся в диапазоне между 0.2 и 1.2 мг в день. Однако в отличие от метилдопы, кривая доза-эффект клонидина такова, что увели
чение дозы ведет к увеличению эффекта, в том числе и токсического. Максимальная рекомендуемая суточная доза — 1.2 мг.
Существует трансдермальная лекарственная форма клонидина, поддерживающая сниженное АД в течение 7 дней при однократной аппликации. Этот препарат вызывает меньшее седативное действие по сравнению с таблетками клонидина, но при таком способе введения возникают местные кожные реакции.
Токсичность
Часто возникают сухость во рту и заторможенность, которые могут быть заметно выражены. Оба вида побочного действия имеют центральную природу, зависят от дозы и совпадают по времени с антигипертензивным эффектом препарата.
Препарат не следует давать больным, склонным к психической депрессии, а при ее появлении лечение следует прекратить. Сочетанное назначение трициклических антидепрессантов может уменьшить антигипертензивный эффект клонидина. Взаимодействие связывают с а-адреноблокирующими свойствами трициклических соединений.
Отмена клонидина после длительного применения, особенно в высоких дозах (более 1 мг в день), может привести к угрожающему жизни гипертензивному кризу, связанному с повышением активности симпатической системы. Через 1-2 дня после отмены препарата у больных появляются беспокойство, тахикардия, головная боль. Хотя вероятность развития серьезного гипертензивного криза точно не определена, она достаточна высока, чтобы помнить о ней всем больным, получающим клонидин. Если препарат требуется отменить, это нужно делать постепенно, заменяя его другим антигипертензивным средством. Лечение больного с гипертензивным кризом состоит в возобновлении приема клонидина или назначении а- и р-адреноблокаторов.
Другие показания к применению клонидина
Клонидин имеет некоторые центральные эффекты, которые можно использовать по другим показаниям. К ним относятся аналгетический эффект, особенно при интратекальном введении (Filos et al., 1992), и ослабление симптомов, возникающих при отмене опиоидов и прекращении курения табака
(Taschner, 1986). Последний эффект, вероятно, отражает способность клонидина снижать симпатический тонус, который обычно повышается при опиатном абстинентном синдроме.
Ганглиоблокаторы
Исторически вещества, которые блокируют способность ацетилхолина стимулировать постганглионарные автономные нейроны, были в числе первых средств, применявшихся для лечения гипертензии. Большинство таких средств в клинике уже не используются из-за плохой переносимости, связанной с их основным действием. Только отдельные препараты этой группы (триметафан) еще применяют в терапии гипертензии.
Ганглиоблокаторы конкурентно блокируют никотиновые холинорецепторы постганглионарных нейронов симпатических и парасимпатических ганглиев. Кроме того, эти вещества могут прямо блокировать никотиновые ацетилхолиновые каналы таким же образом, как и никотиновые нервно-мышечные холинолитики (рис. 26-5).
Триметафан
Триметафана камзилат вводят внутривенно для оказания быстрой помощи при гипертензивном кризе и для создания управляемой гипотензии в нейрохирургии. Зависимость степени ганглионарной блокады от уровня препарата в крови и короткий период его полувыведения позволяют достаточно точно регулировать АД.
Антигипертензивный эффект триметафана в большой степени зависит от задержки крови в емкостных сосудах. Чтобы получить эффективное снижение АД, больной должен находиться в положении полулежа. При избыточном гипотензивном эффекте он может быть уменьшен изменением положения тела (опусканием головного конца кровати).
Триметафан
Побочные эффекты ганглиоблокаторов являются продолжением основного фармакологического действия. Они связаны с симпатоплегией (ортостатическая гипотензия и нарушение половой функции) и парасимпатоплегией (запоры, задержка мочи, провокация приступа глаукомы, затуманенное зрение, сухость слизистой рта и т. д.). Эти побочные эффекты являются основной причиной отказа от использования ганглиоблокаторов для амбулаторного лечения больных с гипертензией.
Вещества, блокирующие адренергические нейроны
Эти вещества понижают АД, предупреждая физиологическое высвобождение норадреналина из постганглионарных симпатических нейронов.
Гуанетидин
В достаточно больших дозах гуанетидин может вызвать глубокую симпатоплегию. Высокая эффективность этого препарата при тяжелой гипертензии лежала в основе его использования в течение многих лет. По этой же причине гуанетидин вызывает все побочные эффекты, которые можно ожидать от “фармакологической симпатэктомии”, включая выраженную постуральную гипотензию, диарею и нарушение эякуляции. Из-за этих побочных эффектов в настоящее время гуанетидин используют редко.
Структура гуанетидина включает основной азот, делающий вещество слишком полярным, чтобы оно проникало в ЦНС. В результате препарат не имеет центральных эффектов, свойственных многим другим препаратам, рассматриваемым в данной главе.
Гуанадрел — гуанетидиноподобное вещество, которое применяется в США. Антигипертензивные агенты бетанидин и дебризохин, не используемые в США, сходны с туанетидином по механизму антигипертензивного действия.
NH
Н II
Гуанетидин
Механизм и место действия
Гуанетидин подавляет высвобождение норадреналина из симпатических нервных окончаний (рис. 11-4). Этот эффект, вероятно, ответственен за большинство проявлений симпатоплегии, развивающейся у больного. Гуанетидин проникает через мембрану симпатического окончания так же, как и норадреналин (механизм захвата 1 типа), что существенно для действия лекарства. После проникновения гуанетидин концентрируется в везикулах, из которых вытесняет норадреналин, в результате чего происходит постепенное уменьшение запасов норадреналина в нервном окончании.
Местноанестезирующие свойства гуанетидина, возможно, связаны с подавлением высвобождения норадреналина на уровне симпатических нервных окончаний. Хотя вещество не нарушает проводимость в аксонах симпатических нервов, может развиваться местная блокада электрической активности мембраны нервных окончаний вследствие их
способности к специфическому захвату и концентрации гуанетидина.
Поскольку для осуществления гипотензивного действия гуанетидина необходим механизм нейронального захвата, вещества, которые препятствуют захвату катехоламинов или вытесняют амины из нервных окончаний (глава 6), блокируют его эффекты. К этим веществам относят кокаин, амфетамин, трициклические антидепрессанты, фенотиазины и феноксибензамин.
Гуанетидин повышает чувствительность к гипертензивным эффектам экзогенных симпатомиметических аминов. Это связано с подавлением нейронального захвата аминов и развивающейся после продолжительной терапии гуанетидином гиперчувствительности эффекторных гладкомышечных клеток, сходной с наблюдаемой после хирургической симпатэктомии (глава 6).
Гипотензивное действие гуанетидина на начальном этапе лечения связано со снижением сердечного выброса вследствие брадикардии и с расслаб
Рис. 11-4. Действие гуанетидина и взаимодействие лекарств в адренергическом нейроне (Г — гуанетидин, НА — норадреналин, ТЦА — трициклические антидепрессанты)
лением емкостных сосудов без выраженных изменений периферического сосудистого сопротивления. При длительном лечении уменьшается периферическое сопротивление сосудов, кроме того, может наблюдаться выраженная задержка натрия и воды.
Фармакокинетика и дозирование
Биодоступность гуанетидина изменчива (3-50 %). Около 50 % вещества выводится почками. Задержка соединения вещества в нервных окончаниях и связывание другими тканями обусловливают очень большой объем распределения и длительный период полувыведения (5 дней). При ежедневном назначении препарата симпатоплегия развивается постепенно (максимум эффекта через 1-2 недели) и сохраняется длительное время после его отмены.
Суточная доза, необходимая для достижения удовлетворительного антигипертензивного эффекта, колеблется у отдельных больных. По этой причине лечение обычно начинают с малых доз, например 10 мг в сутки. Обычно дозу увеличивают через интервалы, не превышающие двух недель. Из-за продолжительного эффекта препарата поддерживающие дозы не следует назначать чаще одного раза в день.
Токсичность
Терапевтическое применение гуанетидина часто сопровождается симптоматической постуральной гипотензией и гипотензией после физической нагрузки, особенно при введении препарата в высоких дозах. Непродуманное применение (слишком большие разовые дозы или слишком быстрое увеличение доз) может привести к опасному снижению сердечного кровотока и кровоснабжения мозга, вплоть до сосудистого шока. Вызванная гуанетиди-ном симпатоплегия у мужчин может привести к задержанной или ретроградной эякуляции. Гуанети-дин нередко вызывает диарею, являющуюся результатом повышенной моторики ЖКТ из-за доминирования парасимпатической системы в регуляции активности гладкой мускулатуры кишечника.
Взаимодействие с другими веществами может осложнить лечение гуанетидином. Симпатомиметические вещества, такие как фенилпропаноламин, в дозах, содержащихся в безрецептурных препара
тах, могут вызвать гипертензию у больных, получающих гуанетидин. Кроме того, гуанетидин может спровоцировать гипертонический криз за счет высвобождения катехоламинов при феохромоцитоме. Когда больным, получающим гуанетидин, вводят трициклические антидепрессанты, антигипертензивный эффект препарата снижается и иногда возникает выраженная гипертензия. Врач, не учитывающий это взаимодействие, может увеличить дозу гуанетидина, если же после этого трициклические антидепрессанты отменить, у больного разовьется гипотензия или сердечно-сосудистый коллапс из-за беспрепятственного действия гуанетидина.
Резерпин
Резерпин — алкалоид, который получают из корней индийского растения Rauwolfia serpentina — был одним из первых широко применявшихся при гипертензии средств. В настоящее время его рассматривают как эффективное и относительно безопасное лекарство для лечения больных с гипертензией легкой и средней тяжести.
Механизм и место действия
Резерпин нарушает способность везикул, содержащих аминергические медиаторы, захватывать и сохранять биогенные амины, возможно, за счет взаимодействия с механизмом захвата, зависимым от Mg2+ и АТФ (рис. 6-4). Этот эффект реализуется во всех тканях, что ведет к истощению запасов норадреналина, дофамина и серотонина в центральных и периферических нейронах. Запасы адреналина в хромаффинных гранулах мозгового вещества надпочечников также снижаются, хотя и в меньшей степени, чем в нейронах. Влияние резерпина на функцию адренергических везикул, видимо, необратимо; следовые количества вещества остаются связанными с мембранами везикул многие дни. Хотя достаточно высокие дозы резерпина могут снизить запасы катехоламинов у животных почти до нуля, меньшие дозы вызывают подавление нейротрансмиссии пропорционально степени истощения запасов аминов.
Истощение периферических аминов, вероятно, является основной причиной антигипертензивного эффекта резерпина, но нельзя исключить и центральный компонент действия. Влияние небольших, но клинически эффективных доз напоминает эф-
фект тех центрально действующих веществ (например, метилдопы), при действии которых симпатические рефлексы остаются в основном неизмененными, АД понижается как в положении лежа, так и стоя, а постуральная гипотензия незначительна. Резерпин легко проникает в мозг, истощение запасов аминов мозга ведет к седации, психической депрессии и симптомам паркинсонизма.
В небольших дозах, применяемых при легкой гипертензии, резерпин понижает АД, уменьшая сердечный выброс и снижая периферическое сосудистое сопротивление.
Фармакокинетика и дозирование
Всасывание, клиренс и метаболизм резерпина детально не исследованы. Вещество быстро исчезает из кровотока, но его эффекты сохраняются долго вследствие необратимой инактивации гранул, депонирующих катехоламины.
Как правило, суточная доза меньше 1 мг (типичная дозировка — 0.25 мг) внутрь однократно. Хотя резерпин выпускают и в инъекционной форме, парентеральное введение применяют редко.
Токсичность
При использовании обычных малых доз резерпина может проявляться незначительная постуральная гипотензия. Большинство нежелательных эффектов препарата связано с действием на мозг или ЖКТ.
Высокие дозы резерпина вызывают заторможенность, усталость, ночные кошмары и тяжелую психическую депрессию. Иногда эти эффекты наблюдаются даже у больных, получающих малые дозы
(0.25 мг в день). Значительно реже малые дозы резерпина вызывают экстрапирамидные нарушения, напоминающие болезнь Паркинсона, что, вероятно, связано с истощением дофамина в структурах полосатого тела. Хотя эти центральные эффекты редки, они могут появиться в любое время, даже спустя месяцы благополучного лечения. Больные с психической депрессией в анамнезе не должны получать резерпин, и если появляются признаки депрессии, лекарство следует отменить.
Сравнительно часто применение резерпина приводит к незначительной диарее и кишечным спазмам, а также повышает желудочную секрецию. Препарат не рекомендуется назначать больным с пептической язвой в анамнезе.
Паргилин
Несмотря на то, что паргилин и другие ингибиторы моноаминоксидазы понижают АД, они не используются в схемах современной терапии гипертензии. Считают, что паргилин понижает АД, увеличивая концентрацию неэффективного ложного трансмиттера в периферических адренергических окончаниях. Препарат угнетает моноаминоксидазу слизистой ЖКТ и печени и тем самым позволяет тирамину, содержащемуся в пище, беспрепятственно поступать в системный кровоток. Тирамин захватывается нервными окончаниями и превращается в октопамин (рис. 6-5), который частично замещает норадреналин в гранулах, депонирующих катехоламины. Поскольку октопамин неактивен как прессорный агонист постсинаптических а-рецепто-ров, симпатоплегия развивается, даже несмотря на повышение общих запасов норадреналина.
Антагонисты адренорецепторов
Фармакология средств-антагонистов а- и Р-ад-ренорецепторов рассмотрена в главе 10. В данной главе внимание концентрируется на двух лекарствах-прототипах — пропранололе и празозине, преимущественно в связи с их использованием в терапии гипертензии.
Пропранолол
Пропранолол — вещество, блокирующее р,-адре-норецепторы,— очень широко используется для понижения АД при легкой и умеренной гипертензии. При тяжелой гипертензии пропранолол особенно полезен для предупреждения рефлекторной тахикардии, которая часто развивается при назначении вазодилататоров прямого действия.
Механизм и место действия
Пропранолол блокирует влияние катехоламинов на Рг и р2-адренорецепторы. Его эффективность при гипертензии так же, как и побочные эффекты, является результатом Р-блокады. Когда пропранолол в первый раз вводят больному, АД понижается вследствие уменьшения сердечного выброса, обусловленного брадикардией. При продолжении лечения сердечный выброс возвращается к норме, но АД остается сниженным из-за уменьшения периферического сосудистого сопротивления.
Блокада p-рецепторов мозга и почек, как предполагают, вносит свой вклад в антигипертензивный эффект данной группы средств. На этот счет существуют противоречивые данные (Pearson, 1989), однако маловероятно, что мозг является первичным местом действия, с которым связана гипотензия, потому что некоторые p-блокаторы, которые плохо проникают через гемато-энцефалический барьер (например, надолол, рассматриваемый ниже), являются эффективными антигипертензивными веществами.
Пропранолол подавляет выработку ренина под влиянием катехоламинов (опосредуется Pt-рецепторами). Эффект пропранолола отчасти связан с угнетением ренин-ангиотензин-альдостероновой системы (Man in’t Veld, 1983). Хотя пропранолол более эффективен у больных с высокой активностью ренина в плазме, он снижает АД и у больных с
гипертензией при нормальном и даже сниженном уровне активности ренина.
При незначительной и умеренной гипертензии пропранолол вызывает значительное снижение АД без выраженной постуральной гипотензии.
Фармакокинетика и дозирование
Как рассматривалось в главе 10, эффективная доза пропранолола при приеме внутрь выше, чем при внутривенном введении, вследствие инактивации при прохождении через печень. Выраженный эффект первого прохождения частично объясняет вариабельность дозировок для получения клинически значимого эффекта. Период полувыведения составляет 3-6 часов.
Лечение больного с гипертензией обычно начинают с суточной дозы 80 мг, разделенной на несколько приемов. Эффективная антигипертензивная дозировка находится в диапазоне 80-480 мг в сутки. Показателями p-блокирующего эффекта пропранолола являются брадикардия в состоянии покоя и снижение ЧСС при нагрузке. Их измерение может служить основой для подбора дозы. Пропранолол можно назначать один или два раза в день.
Токсичность
Основные побочные эффекты пропранолола связаны с блокадой сердечных, сосудистых и бронхиальных p-рецепторов и рассматриваются подробнее в главе 10. Наиболее опасны эти предсказуемые последствия p-адреноблокады для больных со сниженным резервом сократительной функции миокарда, астмой, периферической сосудистой недостаточностью и диабетом.
Когда введение пропранолола после длительного регулярного приема прекращается, у некоторых больных возникает синдром отмены, проявляющийся в форме беспокойства, тахикардии, обострения грудной жабы или увеличения АД. Имеются сообщения о случаях инфаркта миокарда. Хотя частота таких осложнений невелика, пропранолол не следует отменять внезапно. Синдром отмены связывают с “up-регуляцией” или гиперчувствительностью Р-адренорецепторов.
Пропранолол может вызывать эффекты, не связанные отчетливо с р-блокадой, например диарею, запор, тошноту и рвоту. Изредка больные, получа
ющие пропранолол, предъявляют жалобы на центральные эффекты, сходные с теми, которые вызывают метилдопа и клонидин, в том числе ночные кошмары, усталость, депрессию и нарушения сна.
Наконец, пропранолол может повышать уровень триглицеридов плазмы и снижать содержание холестерина в составе липопротеидов высокой плотности, что теоретически способствует атерогенезу.
Другие средства, блокирующие Р-адренорецепторы
Большинство из исследованных 0-блокаторов эффективно снижают АД. Отличия фармакологических свойств от таковых у пропранолола заключаются в том, что они дают им преимущества в некоторых клинических ситуациях.
1. МЕТОПРОЛОЛ
Метопролол примерно одинаково эффективно с пропранололом влияет на 0(-адренорецепторы сердца, но в 50-100 раз менее активен, чем пропранолол, по способности блокировать р2-рецепторы. Хотя метопролол по остальным свойствам сходен с пропранололом, его относительная кардиоселективность может быть использована при коррекции гипертензии у больных астмой, диабетом или периферическими заболеваниями сосудов. Исследования небольшого числа пациентов с астмой показали, что метопролол вызывает меньшую бронхо-констрикцию, чем пропранолол, в дозах, в равной степени подавляющих реакцию на возбуждение Pt-адренорецепторов. Кардиоселективность препарата не абсолютна, и симптомы астмы при введении метопролола могут обостряться. Обычные антигипертензивные дозы метопролола колеблются от 100 до 450 мг в сутки.
2. НАДОЛОЛ, КАРТЕОЛОЛ, АТЕНОЛОЛ И БЕТАКСОЛОЛ
Неселективные антагонисты 0-рецепторов надолол и картеолол, а также атенолол, селективный блокатор Ргадренорецепторов, метаболизируются незначительно и в основном выводятся с мочой. Селективный блокатор Pt-рецепторов бетаксолол преимущественно метаболизируется в печени, но имеет длительный период полувыведения из плазмы. Эти лекарства можно вводить один раз в сутки. Начальные суточные дозы надолола состав
ляют 40 мг, атенолола — 50 мг, картеолола — 2.5 мг, бетаксолола — 10 мг. Коррекция доз для достижения удовлетворительного терапевтического эффекта должна проводиться не чаще 1 раза в 4-5 дней. У больных с пониженной функцией почек следует уменьшать дозы надолола, картеолола и атенолола. Считается, что атенолол вызывает меньше центральных эффектов, чем другие растворимые в жирах р-антагонисты.
3. ПИНДОЛОЛ, АЦЕБУТОЛОЛ И ПЕНБУТОЛОЛ
Пиндолол, ацебутолол и пенбутолол являются парциальными агонистами, т. е. 0-блокаторами с внутренней симпатомиметической активностью. Они проявляют гипотензивное действие, снижая сосудистое сопротивление, но уменьшают сердечный выброс или ЧСС не так выражение, как другие Р-блокаторы, возможно, вследствие значительно большей агонистической, чем антагонистической активности в отношении р2-рецепторов (Aellig, 1987). Это важно учитывать при лечении больных с сердечной недостаточностью, брадиаритмиями или заболеваниями периферических сосудов. Начальные суточные дозы пиндолола составляют 10 мг, ацебутолола — 400 мг и пенбутолола — 20 мг.
4. ЛАБЕТАЛОЛ
Лабеталол применяется в виде рацемической смеси четырех изомеров (он имеет два центра асимметрии). Два изомера (SS и RS) неактивны, третий (SR) является активным а-блокатором, а последний (RR) — активный p-блокатор. Считают, что 0-бло-кирующий изомер обладает селективным Р2-агонистическим и неселективным 0-антагонис-тическим действием. Лабеталол обладает преимущественно 0-блокирующим эффектом: соотношение р- к а-антагонистическому действию после приема внутрь равно 3:1. АД понижается за счет уменьшения сосудистого сопротивления без значительных изменений ЧСС и сердечного выброса. Из-за комбинированной Р* и a-блокирующей активности лабеталол эффективен в коррекции гипертензии при феохромоцитоме и неотложных состояниях. Суточная доза лабеталола для внутреннего применения колеблется от 200 до 2400 мг. При лечении неотложных гипертензивных состояний лабеталол вводят в виде повторных внутривенных болюсных инъекций в дозе 20-80 мг.
Празозин и другие сц-блокаторы
Механизм и место действия
Празозин, теразозин и доксазозин оказывают антигипертензивное действие путем блокады агре-цепторов артериол и венул. Избирательностью действия на а-рецепторы можно объяснить меньшую степень рефлекторной тахикардии после выведения этих веществ, чем после неселективных а-антаго-нистов типа фентоламина. Эта рецепторная селективность позволяет норадреналину избежать тормозного влияния на собственное высвобождение (глава 6) по механизму отрицательной обратной связи, которое опосредуется пресинаптическими а2-рецепторами. В отличие от празозина фентоламин блокирует как пре-, так и постсинаптические а-рецепторы, что ведет к рефлекторной стимуляции симпатических нейронов и высвобождению медиатора, действующего на Р-рецепторы и, соответственно, повышающего ЧСС.
а-блокаторы снижают АД, расширяя как резистивные, так и емкостные сосуды, поэтому АД снижается в большей степени в вертикальном, чем в горизонтальном положении. Если эти вещества принимаются без диуретиков, может произойти задержка солей и жидкости. Препараты проявляют наибольшую клиническую эффективность в сочетании с такими средствами как пропранолол и диуретики.
Фармакокинетика и дозирование
Празозин хорошо всасывается, но подвергается активному метаболизму первого прохождения через печень. Элиминируется почти полностью за счет метаболизма с периодом полувыведения из плазмы 3-4 часа, хотя продолжительность антигипертензивного действия больше. У больных с застойной сердечной недостаточностью концентрации в плазме повышаются в основном за счет снижения эффекта первого прохождения. Теразозин тоже активно метаболизируется (но в меньшей степени) при первом прохождении через печень с периодом полувыведения около 12 часов. Доксазозин имеет промежуточную величину биодоступности и период полувыведения 22 часа.
Лечение празозином должно начинаться с небольшой дозы (1 мг 3 раза в день) для предупреж
дения постуральной гипотензии и обмороков. Дозы могут быть увеличены до 20-30 мг в день. Теразозин часто дают один раз в день в дозах 5-20 мг. Доксазозин обычно назначают однократно, начиная с суточной дозы 1 мг и увеличивая ее до 4 мг или, при необходимости, выше. Хотя длительное лечение этими а-блокаторами вызывает незначительную постуральную гипотензию, у ряда больных в положении стоя после всасывания первой дозы возникает резкое снижение АД. По этой причине первая доза должна быть небольшой и назначаться перед сном. Механизм “феномена первой дозы" неясен, но он развивается чаще у больных с уменьшенным объемом циркулирующей крови и сниженной осмолярностью.
Кроме феномена первой дозы, другие побочные эффекты а.) -блокаторов относительно редки и неопасны. К ним относятся головокружение, сердцебиение, головная боль, утомляемость. У некоторых больных при терапии празозином может проявляться положительный тест на антинуклеарный фактор в сыворотке, но это не связано с ревматическими симптомами. В отличие от диуретиков и 0-блока-торов at-блокаторы могут даже положительно влиять на липидный профиль плазмы.
Другие вещества, блокирующие а-адренорецепторы
В настоящее время исследуется эффект at-селективных блокаторов пинацидила, урапидила и кромокалима. Неселективные вещества фентоламин и феноксибензамин применяются в диагностике феохромоцитомы и при других клинических ситуациях, связанных с повышенным высвобождением катехоламинов (например, фентоламин можно сочетать с пропранололом для лечения больных с синдромом отмены клонидина). Их свойства обсуждаются в главе 10.
Вазодилататоры
Механизм и место действия
К этому классу лекарств относятся пероральные вазодилататоры, гидралазин и миноксидил, применяемые для длительной амбулаторной терапии гипертензии; парентеральные вазодилататоры, нитро
пруссид и диазоксид, используемые при неотложных гипертензивных состояниях, а также блокаторы кальциевых каналов, которые назначают в обеих ситуациях.
Все вазодилататоры, применяемые при гипертензии, расслабляют гладкую мускулатуру артериол, снижая тем самым системное сосудистое сопротивление. Нитропруссид натрия обладает также свойством расслаблять вены. Снижение сопротивления артериол и понижение среднего АД вызывает компенсаторные реакции, опосредованные барорецепторами и симпатической нервной системой (рис. 11-5), а также ренин-ангиотензин-альдостеро-новой системой. Эти компенсаторные реакции препятствуют антигипертензивному действию вазодилататоров. Поскольку симпатические рефлексы не нарушены, лечение вазодилататорами не вызывает ортостатической гипотензии или сексуальных дисфункций.
Вазодилататоры действуют лучше в сочетании с другими антигипертензивными веществами, ко
торые уменьшают компенсаторные сердечно-сосудистые реакции (рис. 11-5 и дополнение “Сравнение моно- и комбинированной терапии при гипертензии”).
Гидралазин
Производное гидразина — гидралазин — расширяет только артериолы. Он используется многие годы, хотя первоначально сомневались в его эффективности из-за быстро развивающейся тахифилак-сии. В настоящее время признаны преимущества
Снижение экскреции riCl)
натрия почками *----Щ----
давления
Повышение высвобождения ренина
Повышение уровня ангиотензина II
Задержка натрия, повышение объема плазмы
Вазодилататоры
I
Снижение системного сосудистого сопротивления
I
Снижение артериального
Повышение активности симпатической нервной
системы
+• системного
сосудистого сопротивления
частоты сократимости емкости сердечных сердца венозного
сокращений русла
Повышение
Повышение
сердечного выброса
> артериального давления
Рис. 11-5. Компенсаторные реакции при введении вазодилататоров — основа для комбинированной терапии с p-блокаторами и диуретиками. 1 — эффект, устраняемый диуретиками; 2 — эффект, устраняемый Р-блокаторами
комбинированного лечения, и гидралазин может применяться более эффективно, особенно при выраженной гипертензии.
Фармакокинетика, метаболизм и дозирование
Гидралазин хорошо всасывается и быстро метаболизируется в печени во время первого прохождения, поэтому биодоступность его низка (в среднем 25 %) и вариабельна. Он метаболизируется частично путем ацетилирования со скоростью, имеющей бимодальное распределение в популяции (глава 4). Как следствие, у людей с высокой скоростью ацетилирования характерны больший эффект первого прохождения, меньшая биодоступность и меньший антигипертензивный эффект данной дозы. Период полувыведения гидралазина колеблется от 2 до 4 часов, но сосудистые эффекты сохраняются дольше, чем концентрация в крови, что соответствует данным о прочном связывании препарата с тканями сосудов.
Обычная суточная доза находится в пределах 40-200 мг. При увеличении доз существует некоторая вероятность развития волчаночноподобного синдрома, рассматриваемого в следующем разделе. Однако высокие дозы вызывают соответствующую вазодилатацию и при необходимости могут назначаться. Прием препарата 2-3 раза в день позволяет достичь мягкого контроля АД.
Токсичность
Наиболее частые побочные эффекты гидралазина — головная боль, тошнота, анорексия, сердцебиение, потливость и сосудистые приливы. У больных с ишемической болезнью сердца рефлекторная тахикардия и симпатическая стимуляция могут провоцировать приступ стенокардии или ишемическую аритмию. При приеме доз 400 мг в день и выше в 10-20 % случаев, особенно при медленном ацетилировании, возникает синдром, характеризующийся артралгией, миалгией, кожными высыпаниями и лихорадкой, напоминающей красную волчанку. Синдром не сопровождается поражением почек и исчезает при прекращении приема гидралазина. Достаточно серьезными, но редкими побочными эффектами препарата являются периферическая нейропатия и лекарственная лихорадка.
Миноксидил
Миноксидил — очень эффективный пероральный вазодилататор. Эффект проявляется посредством открытия калиевых каналов мембран гладкой мускулатуры под влиянием миноксидила сульфата, активного метаболита препарата. Это действие стабилизирует мембрану и ее потенциал покоя, снижая вероятность сокращения. Подобно гидралазину миноксидил расширяет артериолы, но не вены. В связи с выраженным потенциальным антигипертензивным действием можно заменять гидралазин миноксидилом, когда максимальные дозы гидралазина неэффективны. Препарат рекомендуется больным с почечной недостаточностью и тяжелой гипертензией, плохо реагирующим на лечение гидралазином.
Миноксидил
Фармакокинетика, метаболизм и дозирование
Миноксидил хорошо всасывается из ЖКТ и метаболизируется главным образом путем конъюгации в печени. Миноксидил не связывается с белками. Период полувыведения препарата составляет 4 часа. Но гипотензивное действие после однократного приема может сохраняться более 24 часов, что, возможно, связано с присутствием его активного метаболита — миноксидила сульфата.
При гипертензии миноксидил применяют только внутрь. Обычно лечение начинают с суточных доз 5 или 10 мг в два приема, затем суточную дозу постепенно увеличивают до 40 мг. Большие дозировки (до 80 мг) используют для терапии тяжелых форм гипертензии.
Миноксидил даже в большей степени, чем гидралазин, приводит к рефлекторной симпатической стимуляции и задержке натрия и воды. Минокси
дил должен использоваться в сочетании с Р-блока-торами и петлевыми диуретиками.
Токсичность
Тахикардия, сердцебиение, стенокардия и отек наблюдаются, если вводимые больному дозы р-бло-каторов и диуретиков неадекватны. Головная боль, потливость, гипертрихоз, который особенно беспокоит женщин, встречаются относительно часто. Миноксидил является примером того, как побочный эффект у одного человека, может стать терапевтическим у другого. Местное назначение миноксидила в настоящее время используют для стимуляции роста волос при лечении облысения.
Нитропруссид натрия
Нитропруссид натрия — активный вазодилататор для парентерального введения, который используют при неотложных гипертензивных состояниях, а также при тяжелой сердечной недостаточности. Нитропруссид расширяет как артериальные, так и венозные сосуды, что ведет к снижению периферического сосудистого сопротивления и венозного возврата. Эффект связан или с прямой активацией гуанилатциклазы, или с активацией через высвобождение оксида азота. Следствием этого действия является повышение внутриклеточного содержания цГМФ, что способствует расслаблению гладких мышц сосудов.
При отсутствии сердечной недостаточности АД снижается вследствие уменьшения сосудистого сопротивления, тогда как сердечный выброс не изменяется или снижается незначительно. У больных с сердечной недостаточностью и низким сердечным выбросом последний увеличивается из-за снижения постнагрузки.
+NO
CN"
Нитропруссид
Фармакокинетика, метаболизм и дозирование
Нитропруссид является комплексным соединением железа с цианидными группами и нитрозогруппой. Он быстро метаболизируется в эритроцитах с высвобождением цианида. Цианид в свою очередь превращается под влиянием митохондриального фермента роданазы в присутствии донатора серы в тиоцианат. Тиоцианат распределяется во внеклеточной жидкости и медленно выводится почками.
Нитропруссид быстро понижает АД, и его эффект исчезает в течение 1—10 минут после прекращения введения. Препарат вводят путем внутривенной инфузии. Нитропруссид натрия в водном растворе разлагается на свету, поэтому раствор должен готовиться непосредственно перед каждым употреблением и закрываться непрозрачной фольгой. Инфузионные растворы следует готовить заново через несколько часов. Начальная доза составляет 0.5 мкг/кг/мин и может увеличиваться до 10 мкг/кг/мин, если это необходимо для поддержания должного уровня АД. Более высокие скорости инфузии, если она продолжается более часа, могут вызвать появление токсических эффектов. Из-за высокой эффективности и быстрого начала действия препарат следует вводить с помощью инфузионного насоса, а величину АД постоянно контролировать с помощью методов внутриартериальной регистрации.
Токсичность
Наиболее серьезные побочные эффекты, которые могут закончиться летальным исходом, связаны с накоплением цианида, метаболическим ацидозом, аритмиями, избыточной гипотензией. В некоторых случаях появление токсических эффектов после сравнительно небольших доз нитропруссида указывает на нарушение метаболизма цианида. Введение тиосульфата натрия как донатора серы ускоряет метаболизм цианида. Гидроксокобаламин соединяется с цианидом, образуя нетоксичный циан-кобаламин. Оба препарата предлагаются для профилактики и лечения отравления цианидами во время инфузии нитропруссида. Тиоцианат может кумулироваться в процессе длительного введения (неделя и более), особенно у больных с почечной недостаточностью, у которых снижена скорость вы
ведения тиоцианата. Токсическое действие тиоцианата проявляется слабостью, дезориентацией, психозом, мышечными спазмами, судорогами, и диагноз подтверждается, если концентрация в крови превышает 10 мг%. Изредка развивается поздний гипотиреоидизм из-за ингибирования тиоцианатом захвата йода щитовидной железой. Имеются сообщения о метгемоглобинемии во время инфузии нитропруссида.
Диазоксид
Диазоксид представляет собой эффективный и относительно длительно действующий артериолярный дилятатор для парентерального введения, который используют при неотложных гипертензивных состояниях. Инъекция диазоксида приводит к быстрому снижению общего сосудистого сопротивления, сочетающемуся с выраженной тахикардией и повышением сердечного выброса. Изучение механизма действия препарата показало, что он предупреждает сокращение гладкой мускулатуры сосудов, открывая калиевые каналы и стабилизируя мембранный потенциал.
Фармакокинетика и дозирование
Диазоксид сходен по строению с тиазидными диуретиками, но не имеет мочегонного действия. Он прочно связывается с сывороточным альбумином и тканями сосудов. Диазоксид частично метаболизируется, частично экскретируется в неизме-
ненном виде, его метаболические пути недостаточно изучены. Период полувыведения составляет примерно 24 часа, но связь между концентрацией в плазме и гипотензивным действием четко не установлена. Снижение АД после быстрой инъекции развивается в течение 5 минут и продолжается 4-12 часов. Изначально полагали, что быстрые инъекции необходимы, чтобы достичь насыщения препаратом протеинов плазмы и дать возможность свободной форме вещества достичь ткани сосудов, но эта идея не получила убедительных доказательств.
АД снижается и при постоянной инфузии, хотя степень снижения для одной и той же общей дозы больше при быстром введении.
Когда диазоксид появился на фармацевтическом рынке, были рекомендованы быстрые инъекции препарата в дозе 300 мг. Однако избыточного гипотензивного эффекта можно избежать, если начать с меньших доз (75-100 мг). При необходимости дозы по 150 мг могут вводиться повторно каждые 5 минут до требуемой величины снижения АД. Почти все больные реагируют при введении не более 3-4 доз. У больных с хронической почечной недостаточностью гипотензия может наблюдаться от меньших доз препарата в связи со сниженным связыванием с белками. Этим пациентам следует вводить меньшие дозы. Гипотензивное действие диазоксида проявляется в большей степени на фоне введения p-блокаторов для предупреждения рефлекторной тахикардии и связанного с ней увеличения сердечного выброса.
Токсичность
Наиболее важный побочный эффект диазоксида — избыточная гипотензия, связанная с введением рекомендованной фиксированной дозы в 300 мг всем больным. Такая гипотензия может привести к инсульту и инфаркту миокарда. Рефлекторная симпатическая реакция иногда сопровождается стенокардией, признакам ишемии (на ЭКГ) и сердечной недостаточности у больных с ИБС.
Диазоксид угнетает выделение инсулина из поджелудочной железы (возможно, за счет открытия калиевых каналов мембран Р-клеток) и используется для лечения вторичной гипогликемии при инсулиноме. Одним из осложнений применения диазоксида иногда является гипергликемия, особенно у больных с почечной недостаточностью.
В отличие от тиазидных диуретиков диазоксид вызывает задержку натрия и воды почками. Однако в силу того, что препарат используется кратковременно, это нечасто приводит к серьезным последствиям.
Блокаторы кальциевых каналов
В дополнение к антиангинальным (глава 12) и антиаритмическим (глава 14) эффектам блокаторы кальциевых каналов также расширяют перифери
ческие артериолы и понижают АД. Механизм их действия при гипертензии (и отчасти при стенокардии) связан с угнетением входа кальция в гладкомышечные клетки артерий.
Верапамил, дилтиазем и семейство дигидропиридинов (амлодипин, фелодипин, исрадипин, ни-кардипин и нифедипин) примерно равноэффективны по способности снижать АД. В настоящее время в США разрешены к применению различные лекарственные формы этих средств. Некоторые препараты находятся в стадии изучения. На выбор конкретного препарата могут влиять гемодинамические особенности отдельных блокаторов кальциевых каналов. Нифедипин и другие дигидропиридины более селективны как вазодилататоры и имеют менее выраженное угнетающее влияние на сердечные функции по сравнению с верапамилом и дилтиаземом. Рефлекторная симпатическая активация с незначительной тахикардией поддерживает или увеличивает сердечный выброс у большинства больных, принимающих дигидропиридины. Верапамил оказывает наиболее выраженное влияние на сердце и может уменьшать ЧСС и сердечный выброс. Дилтиазем занимает промежуточное положение. Фармакология и токсичность этих средств обсуждается более подробно в главе 12. Дозы блокаторов кальциевых каналов, которые применяют при гипертензии, сходны с теми, которые используют для лечения больных со стенокардией или коронарным спазмом. Нифедипин приобрел популярность как средство оказания неотложной помощи больным с тяжелой гипертензией.
Ингибиторы ангиотензина
Хотя патогенетическая роль ренина, ангиотензина и альдостерона при эссенциальной гипертензии еще однозначно не установлена, тем не менее приданной форме патологии отчетливо выявляются индивидуальные отличия в активности этой системы. Когда у больных с эссенциальной гипертензией измерили потребление натрия (по суточной экскреции с мочой) и концентрацию калия в сыворотке, оказалось, что примерно у 20 % пациентов необычно низкий, а еще у 20 % — необычно высокий уровень активности ренина плазмы. АД больных с гипертензией, сочетающейся с высокой активностью ренина, хорошо поддается коррекции р-ад-реноблокаторами, которые снижают активность
ренина в плазме, и ингибиторами ангиотензина, что подтверждает роль избытка ренина и ангиотензина у этой категории больных.
Механизм и место действия
Высвобождение ренина из коркового слоя почек стимулируется при понижении артериального давления в них, активации симпатических нервов и снижении выведения ионов натрия или повышении их концентрации в дистальных канальцах почек. Ренин действует на свой субстрат, а2-глобулин, с образованием неактивного декапептида ангиотензина I. Ангиотензин I затем конвертируется главным образом в легких в артериальный вазоконстрикторный октапептид ангиотензин II (рис. 11-6), который в свою очередь превращается в надпочечниках в ангиотензин III. Ангиотензин II обладает сосудосуживающим действием и способствует задержке натрия. Ангиотензины II и III стимулируют выделение альдостерона. Ангиотензин может участвовать в поддержании повышенного уровня сопротивления сосудов при гипертензивных состояниях, связанных с высокой активностью ренина плазмы, например при стенозе почечной артерии, некоторых заболеваниях почек и злокачественной гипертензии, а также при эссенциальной гипертензии после лечения ограничением приема натрия, диуретиками и вазодилататорами.
Имеются доказательства существования параллельных систем образования ангиотензина в некоторых других тканях (например, в сердце), которые могут быть ответственны за трофические изменения, такие как гипертрофия сердца. Активность конвертирующего фермента, участвующего в синтезе ангиотензина II в тканях, может быть подавлена с помощью ингибиторов.
Антагонисты ренина и рецепторов ангиотензина II, которые не являются пептидами и, следовательно, активны при приеме внутрь, находятся в стадии изучения (глава 17). Саралазин (см. ниже) все реже применяется в клинике. Лекарства, используемые в настоящее время, действуют на фермент, превращающий ангиотензин I в ангиотензин II.
Саралазин
Саралазин (1-сар-8-ала-ангиотензин II) является аналогом ангиотензина II и конкурентным антагонистом его рецепторов (рис. 11-6). Он блокирует
прессорное действие ангиотензина II, его способность усиливать выделение альдостерона и понижает АД при состояниях, сопряженных с высоким уровнем ренина, например при стенозе почечных артерий. Саралазин в то же время является слабым агонистом, поэтому быстрая инъекция или его введение лицам с низким уровнем ангиотензина II в крови может привести к повышению АД- Использование этого парциального агонистического средства позволило детально исследовать роль ангиотензина II в патогенезе гипертензии.
Ингибиторы ангиотензинпревращающего фермента (АПФ)
Каптоприл (рис. 17-2) и другие средства этой группы угнетают конвертирующий фермент, пеп-тидилдипептидазу, который гидролизует ангиотензин I и превращает его в ангиотензин II, а также (под названием киназы плазмы) инактивирует брадикинин (активный вазодилататор). В отличие от саралазина каптоприл не имеет прессорной активности. Таким образом, гипотензивная активность каптоприла, возможно, связана с ингибирующим влиянием на ренин-ангиотензиновую систему и стимулирующим действием на калликреин-кини-новую систему (рис. 11-6).
Эналаприл (рис. 17-2) является пролекарством, которое путем деэтерификации превращается в ингибитор АПФ эналаприлат с эффектами, сходными с каптоприлом. Эналаприлат применяется только внутривенно при неотложных гипертензивных состояниях. Лизиноприл — это лизиновое производное эналаприлата. Беназеприл, фозиноприл, хинаприл и рамиприл являются недавно внедренными в практику длительнодействующими представителями этой группы. Все они, как и эналаприл, представляют собой пролекарства и превращаются в активные вещества путем гидролиза преимущественно в печени.
Ингибиторы ангиотензина II понижают АД главным образом за счет снижения периферического сосудистого сопротивления. Сердечный выброс и ЧСС существенно не изменяются. В отличие от прямых вазодилататоров эти средства не вызывают рефлекторной симпатической активации и могут безопасно использоваться у людей с ишемической болезнью сердца. Отсутствие рефлекторной
тахикардии может быть связано с перерегулировкой барорецепторов или с повышенной парасимпатической активностью.
Ингибиторы АПФ наиболее эффективны при состояниях, сочетающихся с высоким уровнем активности ренина в плазме, однако четкой корреляции между активностью ренина и выраженностью антигипертензивного эффекта не выявлено. Соответственно, контрольные измерения уровня ренина в плазме становятся необязательными.
Ингибиторы АПФ особенно показаны для лечения больных диабетом, поскольку они уменьшают протеинурию и стабилизируют функцию почек даже в отсутствие снижения АД. Эти преимущества, по-видимому, связаны с улучшением почечной гемодинамики из-за снижения сопротивления эфферентных артериол клубочков и уменьшения внут-риклубочкового капиллярного давления. Ингибиторы АПФ также весьма полезны при застойной сердечной недостаточности (глава 13).
Фармакокинетика и дозирование
Каптоприл быстро всасывается, его биодоступность при приеме натощак около 70 %. Она снижается на 30-40 %, если препарат принимается во время еды, однако антигипертензивное действие каптоприла не изменяется. Он метаболизируется в основном до дисульфидных конъюгатов с другими сульфгидрилсодержащими молекулами. Менее половины дозы каптоприла выводится в неизмененном виде с мочой. Каптоприл распределяется в большинстве тканей организма за исключением ЦНС. Период полувыведения менее 3 часов. Уровень в крови плохо коррелирует с клиническим эффектом.
Каптоприл первоначально вводят в дозах 25 мг 2-3 раза в день за 1-2 часа до еды. Максимальное изменение АД наблюдается через 2-4 часа после приема. Дозы корригируют с 1-2-недельными интервалами до тех пор, пока АД не стабилизируется.
Максимальная концентрация эналаприлата достигается через 3-4 часа после приема эналаприла. Период полувыведения эналаприлата составляет около И часов. Типичные дозы эналаприла 10-20 мг один или два раза в день.
Лизиноприл медленно абсорбируется и достигает максимума концентрации в плазме примерно спустя 7 часов после приема. Его период полувыведения 12 часов. Для большинства больных эффективны дозы 10-80 мг один раз в день.
Ангиотензиноген
Кининоген
Ренин
Калликрвин
Брадикинин
-> Повышение синтеза простагландинов
Ангиотензин I
Конвертирующий фермент (киназа II)
ЕГ......TTiJ О
Неактивный брадикинин
Вазоконстрикция
Секреция альдостерона
Вазодилатация
Повышение периферического сопротивления
т
Задержка натрия и воды
Повышение
кровяного давления
Снижение периферического сосудистого сопротивления
Снижение кровяного давления
Рис. 11-6. Точки приложения действия каптоприла и саралазина. 1 — место действия каптоприла; 2 — место действия саралазина
Все ингибиторы АПФ за исключением фозиноп-рила выводятся главным образом почками, поэтому больным с почечной недостаточностью дозы этих лекарств должны быть снижены.
Токсичность
После приема начальных доз любого ингибитора АПФ может развиться выраженная гипотензия у больных с гиповолемией, связанной с приемом диуретиков, ограничением соли в диете или с потерей жидкости через ЖКТ. Другие побочные эффекты, общие для всех ингибиторов АПФ, включают острую почечную недостаточность (особенно у больных с билатеральным стенозом почечных артерий или стенозом почечной артерии единственной почки), гиперкалиемию, сосудистые отеки и сухой кашель, иногда сопровождаемый хрипами в легких.
Применение ингибиторов АПФ противопоказано во время второго и третьего триместров беремен
ности из-за риска гипотензии плода, анурии и почечной недостаточности, способных привести к нарушениям развития плода и даже его гибели. Каптоприл, особенно при назначении в высоких дозах больным с почечной недостаточностью, может вызвать нейтропению и протеинурию. К менее значимым побочным и более типичным эффектам относятся изменение вкусовых ощущений, кожные аллергические реакции и лекарственная лихорадка, которые могут наблюдаться у 10 % больных. Частота этих побочных эффектов ниже при приеме эналаприла и лизиноприла.
Важные взаимодействия лекарств могут происходить при приеме пищевых добавок с калием или калийсберегающих диуретиков, вызывающих гиперкалиемию. Нестероидные противовоспалительные средства могут нарушать гипотензивное действие ингибиторов АПФ за счет блокады опосредованной брадикинином вазодилатации, механизмы которой, иногда частично, связаны с простагландинами.
II. Клиническая фармакология антигипертензивных средств
Гипертензия является уникальной проблемой для терапии. Обычно это пожизненное заболевание, не вызывающее серьезных симптомов до поздней стадии. Для эффективной терапии этого патологического состояния ежедневно должны применяться лекарства, которые достаточно дороги и часто вызывают побочные эффекты. Таким образом, врач должен установить, что гипертензия постоянна и требует медикаментозного лечения, а также исключить причины гипертензии, которые можно устранить хирургическим путем. Стойкость гипертензии, особенно у лиц с незначительным подъемом АД, должна быть подтверждена измерением АД по меньшей мере при трех посещениях врача. Амбулаторный мониторинг АД может оказаться наилучшим предсказателем риска и, следовательно, необходимости лечения при легкой степени гипертензии. Последние наблюдения показывают, что изолированная систолическая гипертензия и гипертензия у пожилых людей также нивелируются при соответствующем лечении.
После установления диагноза возникает вопрос о том, необходимо ли лечение и какое лекарственное средство нужно применять. Уровень АД, пол, возраст больного, тяжесть поражения органов, обусловленного подъемом АД, и наличие факторов риска со стороны сердечно-сосудистой системы — все должно быть учтено.
Если принято решение о проведении терапии, то нужно выбрать лечебный режим и проинформировать больного о природе гипертензии и необходимости лечения. Выбор лекарственного препарата зависит от уровня АД, степени поражения органов и наличия сопутствующих заболеваний. Значительный подъем АД с угрожающими жизни осложнениями требует неотложного лечения с применением наиболее эффективных средств. Однако большинство больных с эссенциальной гипертензией переносят повышенное АД в течение многих месяцев и даже лет, поэтому лечение лучше начинать постепенно.
Успешное лечение гипертензии требует, чтобы соблюдались рекомендации по диете и приему ле
карств. Больной должен получить сведения о “естественной истории” гипертензии, важности лечения, а также о потенциальных побочных эффектах лекарств. Повторные визиты к врачу должны быть достаточно часты, чтобы больной убедился в том, что врач серьезно относится к его болезни. При каждом посещении необходимо подчеркивать важность лечения и поощрять вопросы, касающиеся дозировки или побочных эффектов лекарств. Для достижения положительных результатов в терапии гипертензии больной должен дисциплинированно выполнять все рекомендации лечащего врача и регулярно самостоятельно измерять АД в домашних условиях.
Амбулаторная терапия гипертензии
Первый шаг в лечении гипертензии может быть нефармакологическим. Как обсуждалось ранее, ограничение приема натрия в диете достаточно эффективно примерно у половины больных с легкой степенью гипертензии. Диета среднего американца содержит около 200 мэкв натрия в день (-10 г). Цель диетотерапии — уровень потребления натрия 70-100 мэкв (3-5 г) в день, что может быть достигнуто, если не солить пищу во время или после приготовления и устранить из диеты продукты с высоким содержанием соли. Эффективность выполнения диетических требований может быть проконтролирована путем измерения экскреции натрия в суточной моче, которая примерно равна суточному потреблению натрия.
Уменьшение массы тела, даже без ограничения натрия в диете, нормализует АД примерно у 75 % тучных больных с легкой и умеренной гипертензией. Понижению АД при гипертензии могут способствовать регулярные физические упражнения.
Фармакотерапия гипертензии легкой степени у большинства больных заключается в приеме одного препарата. Такая “монотерапия” достаточна и для некоторых больных с умеренно выраженной гипертензией. Для начального лечения у таких больных наиболее часто используют тиазидные диуретики или p-блокаторы. Результаты некоторых обширных клинических испытаний показывают, что эти лекарства снижают заболеваемость и смертность, связанные с нелеченной гипертензией. Были опасения, что диуретики и p-блокаторы, неблаго
приятно изменяя сывороточный липидный профиль или нарушая толерантность к глюкозе, могут увеличить риск коронарной болезни, тем самым нивелируя преимущества понижения АД. Альтернативой при выборе начальной терапии могут служить ингибиторы АПФ, блокаторы кальциевых каналов, селективные оц-блокаторы (например, празозин), а-, p-блокаторы (лабеталол) и центральные симпатолитические средства (например, клонидин), хотя пока не доказано, что какой-либо из них изменяет прогноз течения болезни. Наличие сопутствующих заболеваний может повлиять на выбор антигипертензивных средств, поскольку два различных заболевания могут лечиться одним лекарством. Например, Р-блокаторы или блокаторы кальциевых каналов особенно показаны больным, которые страдают стенокардией; диуретики или ингибиторы АПФ — больным с сопутствующей застойной сердечной недостаточностью. Расовые и возрастные факторы тоже могут влиять на выбор лекарств: так, чернокожие больные лучше реагируют на диуретики и блокаторы кальциевых каналов и меньше — на p-блокаторы и ингибиторы АПФ; а пожилые больные лучше поддаются лечению р-бло-каторами и диуретиками.
Если монотерапия не позволяет адекватно корригировать АД, могут быть использованы сочетания лекарств с разным механизмом действия, чтобы эффективно понизить АД и минимизировать токсичность. Если диуретик исходно не использовался, его применяют как второе лекарство. При необходимости назначения трех препаратов часто бывают эффективны сочетания диуретика, прямого вазодилататора (например, гидралазина или блокатора кальциевых каналов) и симпатолитика или ингибитора АПФ.
Оценка АД при посещении врача должна включать измерения в положениях лежа, сидя и стоя. Следует пытаться нормализовать АД (среднее АД < 100 мм рт. ст.) при привычном для больного положении тела или уровне физической активности. Степень ортостатической гипотензии и уменьшение рефлекторной тахикардии при нагрузке являются важными критериями эффективности и соблюдения режима лечения симпатолитиками. Кроме несоблюдения режима, причинами неудачи могут быть чрезмерное потребление натрия и неадекватное лечение мочегонными препаратами с увеличением объема циркулирующей крови (что может быть измерено прямым методом), а также при
ем антидепрессантов, безрецептурных симпатомиметиков и оральных контрацептивов, которые могут взаимодействовать с некоторыми антигипертензивными средствами или непосредственно повышать АД.
Терапия неотложных гипертензивных состояний
Несмотря на большое число больных с хронической гипертензией, неотложные гипертензивные состояния относительно редки. Выраженное или резкое повышение АД может создать серьезную угрозу жизни, что требует экстренного вмешательства. Чаще всего неотложные гипертензивные состояния возникают у больных с тяжелой и мало поддающейся лечению гипертензией, а также у тех, кто резко прекращает антигипертензивную терапию.
Клиническая картина и патофизиология
Неотложные гипертензивные состояния включают гипертензию, связанную с сосудистыми заболеваниями (терминальная злокачественная гипертензия), и гипертензию, связанную с такими гемодинамическими нарушениями как сердечная недостаточность, инсульт или расслаивающая аневризма. Патологический процесс при злокачественной гипертензии проявляется в прогрессирующей арте-риопатии с воспалением и некрозом артериол. Сосудистые повреждения происходят в почках, что способствует выделению ренина, который в свою очередь стимулирует продукцию ангиотензина и альдостерона, что еще больше повышает АД.
Гипертензивная энцефалопатия — классическое проявление злокачественной гипертензии. Для клинической картины характерны сильные головные боли, спутанность сознания, возбуждение. Обычным является затуманивание зрения, тошнота, рвота и местные неврологические нарушения. Если не проводится лечение, синдром может прогрессировать в течение 12-48 часов до развития конвульсий, ступора, комы и даже смерти.
Тактика лечения
Обычно лечение неотложных гипертензивных состояний требует наблюдения за больным в отделении интенсивной терапии с постоянной регист-
Сравнение моно- и комбинированной терапии при гипертензии
Монотерапия при гипертензии (лечение одним лекарством) приобрела большую популярность после внедрения в практику ингибиторов АПФ и селективных а(-блокаторов, поскольку самодисциплина больных при лечении повысилась, а побочные эффекты стали проявляться реже. Однако умеренные и тяжелые гипертензии пока лечат путем сочетания двух и более препаратов. Основой комбинированной терапии (полифармации) является то, что каждое' из лекарств действует на один из множества взаимодействующих компенсаторных регуляторных механизмов поддержания АД (рис. 6-7 и 11-1).
Например, адекватная доза гидралазина вызывает значительное снижение периферического сосудистого сопротивления, вследствие чего происходит падение среднего АД, вызывающее выраженную реакцию в форме компенсаторной тахикардии и задержки натрия и воды. В результате увеличивается сердечный выброс, что может почти полностью устранить эффект гидралазина. Добавление адренергического препарата (например, пропранолола) предупреждает тахикардию; включение в схему лечения диуретика (например, гидрохлортиазида) предупреждает задержку соли и воды. В результате все три вещества повышают чувствительность сердечнососудистой системы к действию каждого из них.
Так, частичное нарушение одного регуляторного механизма (симпатические влияния на сердце) повышает антигипертензивный эффект по другому механизму (периферическое сосудистое сопротивление). В некоторых случаях нормальная компенсаторная реакция рассматривается как побочное действие препарата, которое может быть предупреждено введением второго средства. В случае назначения гидралазина компенсаторная тахикардия и повышенный сердечный выброс могут провоцировать стенокардию у больных с коронарным атеросклерозом. Добавление p-блокатора и диуретика может предупредить этот эффект у многих больных.
На практике, когда гипертензия не снижается адекватно в ответ на монотерапию, добавляют второе лекарство из другого класса с иными механизмами действия и характером побочных эффектов. Если реакция остается неадекватной, несмотря на аккуратный прием лекарства больным, то добавляют третий препарат. Лекарствами, которые с меньшей вероятностью будут эффективными при монотерапии, являются гидралазин и миноксидил. До конца не выяснено, почему такие лекарства как at-блокаторы и блокаторы кальциевых каналов вызывают менее выраженные компенсаторные реакции при той же степени понижения АД.
рацией АД. Экстренное лечение очень важно, хотя следует избегать чрезмерно быстрого понижения АД, так как это может привести к инсульту и инфаркту миокарда. В период лечения следует тщательно учитывать потребление и выведение' Жидкости, ежедневно измерять массу тела как ориентировочный показатель общего объема жидкости.
Для быстрого понижения АД (в течение нескольких часов) используют парентеральные антигипертензивные средства. Как только достигается необходимый уровень АД, следует перейти на пероральный прием антигипертензивных средств, так как это позволяет обеспечить более мягкий и продолжительный лекарственный контроль за состоянием больного. При неотложных гипертензивных
состояниях чаще всего используют вазодилататоры нитропруссид натрия и диазоксид. Другие эффективные парентеральные препараты — это нитроглицерин, лабеталол, триметафан, блокаторы кальциевых каналов, гидралазин, резерпин и метилдопа. Введение внутрь нифедипина, каптоприла, празозина или клонидина также может оказаться полезным при лечении больных с тяжелой гипертензией.
У большинства пациентов отмечается нормальный или сниженный объем циркулирующей крови, хотя у некоторых, например при почечной недостаточности, может наблюдаться гиперволемия. Для предупреждения объемной перегрузки, которая возникает обычно при введении сильнодействую
щих вазодилататоров, применяют диуретики. Поскольку есть вероятность, что у больного нарушена функция почек, следует применять препараты, эффективные при почечной недостаточности, например фуросемид.
Альтернативой петлевым диуретикам может быть диализ, особенно у больных с олигурической почечной недостаточностью. Диализ позволяет удалить избыточную жидкость, откорректировать электролитные нарушения и уменьшить проявления уремии. Последние могут осложнятьобъектив-ную оценку состояния больных при энцефалопатии, связанной с гипертензией.
Препараты
Блокаторы [3-адренорецепторов
Ацебутолол (Сектрал)
Перорально: капсулы по 200,400 мг
Атенолол (генерик, Тенормин)
Перорально: таблетки по 25, 50,100 мг
Парентерально: 0.5 мг/мл для инъекций
Бетаксолол (Керлон)
Перорально: таблетки по 10, 20 мг
Бисопролол (Зебета)
Перорально: таблетки по 5,10 мг
Картеолол (Картрол)
Перорально: таблетки по 2.5, 5 мг
Лабеталол (Нормодин, Трандат)
Перорально: таблетки по 100, 200, 300 мг
Парентерально: 5 мг/мл для инъекций
Метопролол (Лопрессор)
Перорально: таблетки по 50,100 мг
Перорально для пролонгированного действия (Топрол-XL): таблетки по 50, 100, 200 мг Парентерально: 1 мг/мл для инъекций
Надолол (Коргард)
Перорально: таблетки по 20, 40, 80,120,
160 мг
Пенбутолол (Леватол) -1
Перорально: таблетки по 20 мг
Пиндолол (генерик, Вискен)
Перорально: таблетки по 5,10 мг
Пропранолол (генерик, Индерал)
Перорально: таблетки по 10, 20,40, 60, 80, 90 мг; Интенсол, раствор 80 мг/мл Перорально для пролонгированного действия
(Индерал LA): капсулы по 60, 80, 120,160 мг
Парентерально: 1 мг/мл для инъекций
Тимолол (генерик, Блокадрен)
Перорально: таблетки по 5,10, 20 мг
Центральные симпатолитики
Клонидин (генерик, Катапрес)
Перорально: таблетки по 0.1, 0.2, 0.3 мг
Трансдермально (Катапрес-TTS); пластыри, освобождающие 0.1, 0.2, 0.3 мг/сут
Гуанабенз (Витенсин)
Перорально: таблетки по 4,8 мг
Гуанфрцин (Тенекс)
Перорально: таблетки по 1 мг
Метилдопа (генерик, Альдомет)
Перорально: таблетки по 125, 250, 500 мг;
суспензия 250 мг/5 мл
Парентерально: 250 мг/5 мл для инъекций
Блокаторы терминалей постганглионарных симпатических нервов
Гуанадрел (Гилорел)
Перорально: таблетки по 10, 25 мг
Гуанетидин (генерик, Исмелина сульфат)
Перорально: таблетки по 10, 25 мг
Резерпин (генерик, Серпазил)
Перорально: таблетки по 0.1, 0.25, 1 мг
оц-селективные адренолитики
Доксазозин (Кар дура)
Перорально: таблетки по 1, 2,4, 8 мг
Празозин (Минипресс)
Перорально: капсулы по 1, 2, 5 мг
Теразозин(Гитрин)
Перорально: таблетки по 1, 2, 5,10 мг
Ганглиоблокаторы
Мекамиламин (Инверсии)
Перорально: таблетки по 2.5 мг
Триметафан ( Арфонад)
Парентерально: ампулы 500 мг/10 мл
Вазодилататор ы, применяемые при гипертензии Диазоксид (генерик, Гиперстат IV)
Парентерально: ампулы 15 мг/мл, 300 мг/20 мл
Гидралазин (генерик, Апрезолин) Перорально: таблетки по10, 25, 50,100 мг Парентерально: 20 мг/мл для инъекций
Миноксидил (генерик, Лонитен)
Перорально: таблетки по 2.5,10 мг
Нитропруссид (генерик, Ниприд) Парентерально: флаконы по 50 мг
Блокаторы кальциевых каналов
Амлодипин (Норваск)
Перорально: таблетки по 2.5,5,10 мг
Дилтиазем (генерик, Кардизем)
Перорально: таблетки по 30,60, 90,120 мг (не показан при гипертензии)
Перорально для пролонгированного действия (Кардизем SR, Дилакор XL): капсулы по 60, 90,120,180, 300 мг Парентерально: 5 мг/мл для инъекций
Фелодипин (Плендил)
Перорально для пролонгированного действия: таблетки по 5,10 мг
Исрадипин (Динасерк)
Перорально: капсулы по 2.5, 5 мг
Никардипин (Карден)
Перорально: капсулы по 20, 30 мг Перорально для пролонгированного действия (Карден SR): капсулы по 30,45, 60 мг
Нифедипин (генерик, Адалат, Прокардия) Перорально: капсулы по 10, 20 мг (не показан при гипертензии) Перорально для пролонгированного действия (Адалат СС, Прокардия XL): таблетки по 30,60, 90 мг
Верапамил (генерик, Калан, Изоптин) Перорально: таблетки по 40, 80,120 мг Перорально для пролонгированного действия (генерик, Калан SR, Верелан): таблетки по 120, 180, 240 мг;
капсулы по 120, 180, 240 мг
Парентерально: 5 мг/2 мл для инъекций
Ингибиторы
ангиотензинконвертирующего фермента
Беназеприл (Лотензин)
Перорально: таблетки по 5,10, 20,40 мг
Каптоприл (Капотен)
Перорально: таблетки по 12.5, 25, 50,100 мг
Эналаприл (Вазотек)
Перорально: таблетки по 2.5, 5,10, 20 мг Парентерально (Эналаприлат): 1.25 мг/мл для инъекций
Фозиноприл (Моноприл)
Перорально: таблетки по 10, 20 мг
Лизиноприл (Принивил, Зестрил)
Перорально: таблетки по 5,10, 20,40 мг
Хинаприл ( Аккуприл)
Перорально: таблетки по 5,10, 20,40 мг
Периндоприл( Ацеон)
Перорально: таблетки по 2,4, 8 мг
Рамиприл (Алтае)
Перорально: капсулы по 1.25, 2.5, 5,10 мг
Избранная литература
Calhoun D. A., Oparil S. Treatment of hypertensive crisis. N. Eng. J. Med. 1990; 323:1177.
Fletcher A. E., Bullpit C. J. How far should blood pressure be lowered? N. Engl. J. Med. 1992; 326: 251.
Frohlich E. D. Methyldopa: Mechanisms and treatment 25 years later. Arch. Intern. Med. 1980; 140:954.
Gifford R. W. Jr. Management of hypertensive crises. JAMA, 1991; 266: 829.
Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure: The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNCV). Arch. Intern. Med. 1993; 153: 154.
Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure: Nonphar-macological approaches to the control of high blood pressure. Hypertension, 1986; 8:444.
Khoury A. F., Kaplan N. M. Alpha-blocker therapy of hypertension: An unfulfilled promise. JAMA, 1991; 266: 394.
Linas S. L., Nies A. S. Minoxidil. Ann. Intern. Med. 1981; 94: 61.
Luke R. G. Essential hypertension: A renal disease? A review and update of the evidence. Hypertension, 1993; 21:380.
Man in’t Veld A. J., Schalekamp M. A. D. H. Effects of 10 different р-adrenoreceptor antagonists on hemodynamics, plasma renin activity, and plasma norepinephrine in hypertension: The key role of vascular resistance changes in relation to partial agonist activity. J. Cardiovasc. Pharmacol. 1983; 5: S30.
National High Blood Pressure Education Program Working Group: Working Group report on hypertension and chronic renal failure. Arch. Intern. Med. 1991; 151:1280.
National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy: Working Group report on high blood pressure in pregnancy. Am. J. Obstet. Gynecol. 1990; 163: 1689.
Setaro J. F., Black H. R. Refractory hypertension. N. Engl. J. Med. 1992; 327: 543.
SHEP Cooperative Research Group: Prevention of stroke by antihypertensive drug treatment in older
persons with isolated systolic hypertension. JAMA, 1991; 265: 3255.
SHEP Cooperative Research Group: Implications of the Systolic Hypertension in the Eldery Program. Hypertension, 1993; 327: 543.
Stamler J. et. al. INTERSALT study findings: Public health and medical care implications. Hypertension, 1989; 14: 570.
Weber M. A. Clinical pharmacology of centrally acting antihypertensive agents. J. Clin. Pharmacol. 1989; 29: 598.
Weber M. A. Hypertension: Steps forward and steps backward—The Joint National Committee Fifth Report. Arch. Intern. Med. 1993; 153:149.
Working Group on Ambulatory Blood Pressure Monitoring, National High Blood Pressure Education Program Coordinating Committee: National High Blood Pressure Education Program Working Group Report on ambulatory blood pressure monitoring. Arch. Intern. Med. 1990; 150: 2270.
Вазодилататоры и средства для лечения стенокардии
12
Бертрам Г. Катцунг, Кану Чаттерье
Грудная жаба {angina pectoris), или стенокардия,— наиболее распространенное заболевание, связанное с ишемией тканей, при котором используются сосудорасширяющие средства. Боль при этом заболевании обусловлена накоплением метаболитов в миокарде и возникает, когда коронарный кровоток не соответствует потребностям сердца в кислороде. Основой терапии при стенокардии являют-”Ся органические нитраты, например нитроглице-|0Ш. Другая группа вазодилататоров — блокаторы кальциевых каналов, также имеет большое значение, особенно для профилактики приступов. С этой же целью используют p-блокаторы, которые не от-носятЬя к вазодилататорам.
Ишемическая болезнь сердца является наиболее распространенным и серьезно угрожающим здоровью населения заболеванием во многих странах Запада. Самая частая причина стенокардии — атероматозное сужение больших коронарных сосудов (атеросклеротическая, или классическая стенокардия). Однако преходящие спазмы определенных участков этих сосудов, обычно прилегающих к атероматозным бляшкам, также могут вызывать выраженную ишемию миокарда и боль (вариантная, или вазоспастическая стенокардия).
Основная причина грудной жабы заключается в дисбалансе между потребностью сердца в кислороде него доставкой коронарным кровотоком. При классической стенокардии дисбаланс возникает, когда увеличивается потребность миокарда в кислороде, например при физической нагрузке, а коронарный кровоток пропорционально возрасти не может. Появившаяся ишемия обычно сопровождается болью. Следовательно, классическая стенокардия — это “стенокардия напряжения”. В тех случаях, когда ишемия не сопровождается болью, говорят о “молчащей”, или “амбулаторной” ишемии. При вариантной стенокардии доставка кислорода
снижается в результате обратимого коронарного вазоспазма, обычно наслаивающегося на хронический обструктивный процесс.
Теоретически дисбаланс в миокарде между потребностью и доставкой кислорода может устраняться при увеличении его поступления (путем повышения коронарного кровотока) или в результате снижения потребности в кислороде (уменьшение работы сердца). Оба способа используются в клинической практике. При стенокардии, связанной с атеросклерозом, с помощью фармакологических средств легче достичь снижения потребности в кислороде. Традиционная терапия, направленная на достижение этой цели, основана на применении таких активных вазодилататоров как органические нитраты и некоторых других групп лекарств, которые уменьшают работу сердца. Поскольку кровоток ограничен фиксированной атероматозной бляшкой, фармакологически трудно достичь повышения поступления кислорода посредством усиления коронарного кровотока. В этом случае, если снижение потребности в кислороде не устраняет симптомы болезни, могут потребоваться инвазивные методы (коронарное шунтирование или ангиопластика). При вариантной стенокардии спазм коронарных сосудов может быть устранен нитратами или блокаторами кальциевых каналов. Следует подчеркнуть, что не все вазодилататоры эффективны при стенокардии и, напротив, некоторые вещества, эффективные при стенокардии (например, ₽-бло-каторы), не являются вазодилататорами.
Когда происходят изменения характера, частоты, продолжительности приступов и типа провоцирующих их факторов у больного со стабильной стенокардией и когда появляются приступы в состоянии покоя, говорят о нестабильной стенокардии. Это состояние связано с эпизодами повышения тонуса эпикардиальной коронарной артерии или с
образованием небольших тромбоцитарных тромбов в области атеросклеротической бляшки. В большинстве случаев формирование лабильных тромбов в месте расщепившейся бляшки приводит к снижению кровотока. Это указывает на угрожающий инфаркт миокарда и требует энергичного лечения.
История
Впервые стенокардия как самостоятельное заболевание была описана У ильямом Геберденом в конце XVIII в. Во второй половине XIX в. было обнаружено, что амилнитрит способствует временному облегчению состояния. Однако эффективное лечение острых приступов стенокардии стало возможным только после внедрения в 1879 г. нитроглицерина. В дальнейшем для терапии стенокардии были предложены многие другие сосудорасширяющие средства (например, теофиллин, папаверин). Однако клинические испытания с двойным слепым контролем показали, что эффект этих препаратов не отличается от эффекта плацебо. Некоторые из классических исследований плацебо-эффекта были проведены именно у больных со стенокардией. После появления 0-блокаторов стала возможной эффективная профилактика. Позднее было показано, что блокаторы кальциевых каналов также способны предотвращать ангинозные приступы, особенно при вариантной стенокардии.
Патофизиология стенокардии
Факторы, определяющие потребность миокарда в кислороде
Главные и второстепенные факторы, определяющие потребность миокарда в кислороде, перечислены в табл. 12-1. В отличие от скелетной мускулатуры сердечная мышца человека не способна на формирование кислородной задолженности во время нагрузки с последующим ее возмещением. В связи с постоянной активностью потребность сердца в кислороде относительно высока, и при отсутствии нагрузки оно извлекает примерно 75 % поступающего кислорода. Кислородный запрос миокарда повышается, когда возрастают частота сердечных сокращений (ЧСС), сила сокращений, артериальное давление (АД) или объем желудочка. Эти ге-
ТАБЛИЦА 12-1. Факторы, определяющие потребление кислорода миокардом
Главные Второстепенные
Напряжение стенки Энергия активации
Внутрижелудочковое давление Метаболизм Радиус (объем) желудочка в состоянии
Толщина стенки покоя
Частота сердечных сокращений
Сократимость
модинамические изменения часто отмечаются при физической нагрузке и повышении симпатической активности, провоцирующих приступ стенокардии у больных с ишемической болезнью сердца. Влияние базального метаболизма и активации сократительной функции на общее потребление кислорода миокардом незначительно, но в условиях патологии эти второстепенные детерминанты потребления кислорода миокардом могут становиться существенными.
В клинике невозможно измерить все факторы, которые перечислены в табл. 12-1. Поэтому для оценки изменений потребности и расходования кислорода в миокарде часто используют непрямые показатели. Один из них — “двойное произведение” (ЧСС х систолическое АД). Этот показатель тесно связан с индексом “напряжение-время”, который определяет потребность миокарда в кислороде более инвазивным способом.
Детерминанты коронарного кровотока и снабжения миокарда кислородом
Поставка кислорода зависит от поступления кислорода в миокард и его извлечения из крови. Поскольку в покое степень извлечения кислорода в миокарде почти максимальна, резерв для удовлетворения возросших потребностей мал. Более того, содержание кислорода в крови не может значительно возрасти при нормальном атмосферном давлении. Таким образом, повышение потребности миокарда в кислороде в норме достигается за счет увеличения коронарного кровотока.
Коронарный кровоток прямо связан с перфузионным давлением (диастолическим давлением в аорте) и длительностью диастолы. Так как коронарный кровоток во время систолы невелик, продолжительность диастолы становится лимитирующим
фактором перфузии миокарда при тахикардии. Коронарный кровоток обратно пропорционален сопротивлению коронарных сосудов. Последнее определяется внутренними факторами, в частности активностью автономной нервной системы, продуктами метаболизма, различными фармакологическими агентами и внесосудистым механическим давлением на коронарные артерии. Местом ауторегуляции являются резистивные артериолы. Повреждение эндотелия коронарных сосудов изменяет их способность расширяться в ответ на действие нормальных раздражителей.
I. Базисная фармакология средств, применяемых при стенокардии
Действие лекарств при стенокардии
Все три группы лекарств, применяемых при стенокардии, снижают потребность миокарда в кислороде путем уменьшения периферического сосудистого сопротивления, уменьшения сердечного выброса или за счет сочетания указанных эффектов. У некоторых больных доставку кислорода к ишемизированной области может повысить перераспределение коронарного кровотока. При вариантной стенокардии нитраты и блокаторы кальциевых каналов, устраняя спазм коронарных сосудов, повышают снабжение миокарда кислородом. Механизмы сокращения и расслабления гладкой мускулатуры сосудов и места действия блокаторов кальциевых каналов показаны на рис. 12-1. Точки приложения действия нитратов (и оксида азота, высвобождаемого из эндотелиальных клеток нейрогу-моральными факторами) показаны на рис. 12-2.
Нитраты и нитриты
Химия
Эти вещества являются простыми эфирами азотной и азотистой кислот с многоатомными спиртами (рис. 12-3). Они представляют собой чрезвычайно (амилнитрит) или умеренно (нитроглицерин) летучие жидкости либо твердые вещества (изосорбида динитрат). Нитроглицерин является средством-прототипом данной группы. Он использует-
Са2+-каналы
каналов
АТФ
Са2+
Кальмодулин
Комплекс Са2+-кальмодулин
цАМФ
Легкие цепи миозина (ЛЦ).
КЛЦ -*•
(КЛЦ)
* ЛЦ-Р04------► лц
Сокращение Расслабление
Рис. 12-1. Регуляция сокращения гладкой мышцы и место действия блокаторов кальциевых каналов. Сокращение вызывается входом Са2+ (который может ингибироваться блокаторами кальциевых каналов) через трансмембранные кальциевые каналы. Са2+ соединяется с кальмодулином и образует комплекс, который превращает фермент киназу легких цепей миозина в активную форму (КЛЦ*). Последняя фосфорилирует легкие цепи миозина, инициируя тем самым взаимодействие миозина с актином. р-2-агонисты (и другие вещества, повышающие уровень цАМФ) вызывают расслабление гладкой мышцы за счет ускорения инактивации КЛЦ (толстые стрелки) и облегчения удаления Са2+ из клетки (на рис. не показано)
ся и в производстве динамита, но лекарственные формы, используемые в медицине, не взрывоопасны. Обычные таблетки нитроглицерина для сублингвального применения могут терять активность при хранении вследствие испарения и контакта с пластмассовой поверхностью. Следовательно, препарат должен храниться в плотно закупоренной упаковке из стекла. Нитроглицерин не чувствителен к действию света.
Изучение связи между строением препаратов и их действием показало, что все терапевтически активные вещества данной группы способны высвобождать оксид азота (NO) в гладкой мускулатуре сосудов (Harrison and Bates, 1993). К сожалению, все они вызывают перекрестную толерантность при введении в больших дозах. Поэтому при использовании нитратов выбор препарата и способ лечения определяются прежде всего его фармакокинетикой.
Сокращение Расслабление
Рис. 12-2. Механизм действия нитратов, нитритов и других средств, повышающих концентрацию оксида азота (NO) в гладкомышечных клетках. Реакции, ведущие к расслаблению, показаны толстыми стрелками. (КЛЦ* — активированная киназа легких цепей миозина; гуанилат-циклаза* — активированная гуанилатциклаза; ? — неизвестные промежуточные этапы; ЛЦ — легкие цепи.)
Фармакокинетика
На использование органических нитратов существенно влияет наличие высокоэффективной печеночной нитратредуктазы, которая отщепляет нитратную группу от органической молекулы и в ко
нечном счете инактивирует вещество. Следовательно, биодоступность традиционных органических нитратов (нитроглицерина и изосорбида динитрата) при пероральном приеме очень низка (обычно менее 10-20 %). Прием под язык позволяет избежать эффекта первого прохождения, а, значит, предпочтителен для быстрого достижения терапевтической концентрации в крови. При таком пути введения нитроглицерин и изосорбида динитрат быстро всасываются, терапевтический уровень в плазме крови достигается через несколько минут. Однако общая доза в этом случае должна быть ограничена во избежание избыточного эффекта, поэтому продолжительность действия препаратов невелика (15-30 минут). Когда требуется более длительное действие, должны назначаться энтеральные препараты, которые содержат количества вещества, достаточные для получения необходимого уровня в крови действующего начала или активных метаболитов. Другие пути введения нитроглицерина — трансдермальный и трансбуккальный для лекарственных форм с медленным высвобождением.
Амилнитрит и родственные нитриты являются летучими жидкостями. Амилнитрит выпускают в хрупких стеклянных ампулах с защитным тканевым покрытием. Ампулу можно раздавить пальцами, что приводит к быстрому образованию паров, проникающих через защитную ткань, которые следует вдыхать. Ингаляционный путь введения обеспечивает очень быстрое всасывание и, подобно сублингвальному, позволяет избежать эффекта первого прохождения через печень.
н2с—о —no2 I
нс—о —no2 I
н2с—о —no2
Нитроглицерин (глицерилтринитрат)
O2N —о —сн2 сн2 —о —no2
O2N—о —сн2 сн2 —о —no2
Н2С----------------
I нс—о —no2
I о
----------------сн
I
НС----------------
о I
O2N —о —сн ---------------сн2 Изосорбида динитрат
Пентаэритритола тетранитрат
Н3С^
СН — СН2 — СН2 — О — N = О НзС^ Амилнитрит
Рис. 12-3. Химическая структура трех нитратов и амилнитрита
8. Заказ 3245
Период полувыведения неизмененного соединения нитрата после всасывания составляет всего 2-8 минут. Метаболиты с частично удаленными нитрогруппами имеют более продолжительный период полувыведения (до 3 часов). Из метаболитов нитроглицерина (два динитроглицерина и две мононитро-формы) динитропроизводные обладают выраженной сосудорасширяющей способностью; возможно, именно они определяют терапевтический эффект нитроглицерина при приеме внутрь. 5-мо-нонитратный метаболит изосорбида динитрата является активным метаболитом последнего и используется в клинике в виде изосорбида мононитрата. Его биодоступность равна 100 %.
Экскреция, главным образом в форме глюкуронидов денитрированных метаболитов, осуществляется преимущественно почками.
Фармакодинамика
Нитроглицерин и его аналоги являются необычайно селективными средствами: в терапевтических дозах их эффект направлен в основном на клетки гладких мышц. Другое действие, имеющее клиническое значение, связано с влиянием на агрегацию тромбоцитов.
А. Механизм действия на гладкую мускулатуру. Нитроглицерин подвергается денитрированию с высвобождением свободного нитритного иона в гладкомышечной клетке и в других тканях под действием глутатион-S-трансферазы. В результате неизвестных ферментативных реакций из исходной молекулы высвобождается оксид азота (NO) (Harrison and Bates, 1993). Оксид азота является значительно более активным вазодилататором, чем нитрит, который сам по себе может высвобождать оксид азота. Как показано на рис. 12-2, оксид азота вызывает активацию гуанилатциклазы и повышает уровень цГМФ, что является первым этапом в развитии расслабления гладкой мышцы. Может происходить также образование простагландина Е или простациклина (PGI2). Нет сведений о том, что рецепторы автономной системы участвуют в первичной реакции на нитрат, хотя при достижении гипотензивных доз развиваются автономные рефлекторные реакции. Важной особенностью при применении нитратов является развитие толерантности. Толерантность может быть частично связана со снижением содержания сульфгидрильных групп в тканях, однако вещества, восстанавливаю
щие сульфгидрильные группы, не полностью предупреждают или устраняют толерантность. Причина этой клеточной толерантности может быть связана с неизвестной реакцией, ответственной за образование оксида азота из нитрата, так как другие вещества, которые вызывают вазодилатацию через высвобождение оксида азота из эндогенных субстратов, например ацетилхолин, не проявляют перекрестной толерантности с нитратами (дополнение “Феномен коронарного обкрадывания”).
Новый нитрат никорандил, проходящий клинические испытания, сочетает активность по высвобождению NO с открытием калиевых каналов, что является дополнительным способом достижения вазодилатации. Вещества, расширяющие сосуды за счет открытия калиевых каналов, обсуждались в главе И.
Б. Влияние на системы и органы. Нитроглицерин расслабляет все типы гладких мышц независимо от причины мышечного напряжения (рис. 12-4). Он практически не имеет прямого влияния на сердечную или скелетную мускулатуру.
1. Гладкая мускулатура сосудов. Под действием нитроглицерина расслабляются все отделы сосудистой системы от крупных артерий до крупных вен. Вены реагируют при меньшей концентрации, артерии — при несколько более высокой. Артериолы и прекапиллярные сфинктеры расслабляются в меньшей степени, чем крупные артерии и вены, частично из-за рефлекторных реакций (Bassenge, 1986, 1992) и частично потому, что различные сосуды отличаются по способности образовывать оксид азота (см. дополнение). Основное прямое действие нитратов при наличии эффективных концентраций в крови — выраженное расслабление больших вен с увеличением емкости венозного русла и снижением преднагрузки на желудочки. Давление в легочных сосудах и размеры сердца значительно уменьшаются. В связи с увеличением емкости венозного русла может развиваться выраженная гипотензия и даже обморок. Расширение некоторых крупных артерий, включая аорту, может быть весьма значительным из-за их эластичности. Пульсация височных артерий и пульсирующая головная боль, связанная с пульсацией менингеальной артерии, довольно часто сопровождают прием нитроглицерина и амилнитрита. При сердечной недостаточности преднагрузка особенно высока, поэтому нитраты й другие вазодилататоры, снижая ее, могут оказать положительное влияние на сердечный выброс при
К+ Верапамил
Рис. 12-4. Влияние вазодилататоров на сокращение сегментов вен человека in vitro. А. Сокращения, вызванные двумя вазодилататорами,— норадреналином (НА) и калием (К+). Б. Расслабление, вызванное нитроглицерином, 4 мкмоль/л. Расслабление развивается быстро. В. Расслабление, вызванное верапамилом, 2.2 мкмоль/л. Расслабление развивается медленнее, но более продолжительно. (Из: Mikkelsen Е., Andersson К. Е., Bengtsson В. Effects of verapamil and nitroglycerin on contractile responses to potassium and noradrenaline in isolated human peripheral veins. Acta Pharmacol. Toxicol. 1978,41,14.)
этом заболевании (глава 13). Однако и у больных с хронической левожелудочковой недостаточностью, принимающих нитраты, снижение постнагрузки из-за повышения податливости артерий и лучшего сопряжения работы желудочка и аорты также может быть важным элементом улучшения функции желудочков.
Непрямые эффекты нитроглицерина связаны с рефлексами, опосредованными барорецепторами, и с гормональными механизмами регуляции АД (рис. 6-7). Суть этого рефлекса состоит в активации симпатической системы, что приводит к тахикардии и увеличению силы сокращений миокарда. При введении очень быстро действующих веществ (например, при ингаляции амилнитрита) расширение артериол может быть столь велико, что приводит к рефлекторной вазоконстрикции.
В опытах на изолированном по Лангендорфу сердце с перфузией коронарных артерий, а также у здоровых людей без коронарной болезни продемонстрировано, что нитроглицерин вызывает значительное, хотя и преходящее, увеличение коронарного кровотока. Напротив, у больных стенокарди-,ей из-за атеросклеротической обструкции коронарных артерий общий коронарный кровоток не повышается. Некоторые исследования указывают на пе
рераспределение коронарного кровотока из здоровых областей в ишемизированные, но большинство наблюдений свидетельствуют о том, что при стенокардии напряжения нитроглицерин действует в основном за счет снижения потребности миокарда в кислороде вследствие уменьшения преднагрузки и АД (раздел “Клиническая фармакология средств, применяемых при стенокардии”).
2. Другие гладкомышечные органы. В эксперименте показано расслабление гладкой мускулатуры бронхов, желудочно-кишечного тракта (включая билиарную систему), мочеполовой системы. Из-за кратковременности действия эти эффекты нитратов редко находят применение в клинике. В последние годы в некоторых группах населения замечено применение амилнитрита и изобутилнит-рита как средств, усиливающих сексуальные ощущения. Реклама использования изобутилнитрита с этой целью не разрешена, однако препарат продают без рецептов под торговыми названиями Rush, Bolt, Locker Room, Dr. Bananas.
3. Влияние на тромбоциты. Оксид азота, образующийся из нитроглицерина, стимулирует гуани-латциклазу в тромбоцитах так же, как и в гладкой мускулатуре. Происходящее увеличение цГМФ приводит к торможению агрегации тромбоцитов
Феномен коронарного обкрадывания
История создания вазодилататоров для лечения стенокардии отмечена периодами разочарований, когда фармакологи обнаруживали, что новые лекарства, будучи чрезвычайно эффективными сосудорасширяющими средствами у здоровых животных, оказывались неэффективными или даже ухудшали проявления стенокардии у больных. Сейчас известно, что активные артериолярные вазодилататоры (например, гидралазин, дипиридамол) обычно неэффективны при стенокардии и могут снижать перфузию ишемизированных областей. На практике дипиридамол часто используют при коронарографии для выявления зон с пониженной перфузией. С другой стороны, вещества, которые более эффективно расширяют вены, крупные артерии и сравнительно неэффективны в отношении резистивных сосудов (например, нитраты), очень эффективны при стенокардии. Причина этой очевидной аномалии получила название “феномена коронарного обкрадывания”.
Коронарное обкрадывание происходит в том случае, когда две ветви от главной коронарной артерии имеют разную степень обструкции. Например, одна ветвь может быть относительно нормальной и способной к расширению или сужению в ответ на изменение потребности миокарда в кислороде. В другой же — выраженная обструкция, что приводит к расширению артериол ниже места сужения даже при низкой потребности миокарда в кислороде в связи с накоплением метаболитов в ишемизированной
ткани. В состоянии покоя перфузия в этой области может быть адекватной, так как перфузионное давление в основной коронарной артерии стабильно. (Напомним, что перфузия регионарной сосудистой зоны прямо пропорциональна перфузионному давлению в магистральном сосуде и обратно пропорциональна сопротивлению току крови в артериолах.) При введении активного артериолярного вазодилататора артериолы в области без сужения будут расширяться, уменьшая ее сосудистое сопротивление и значительно повышая кровоток, несмотря на то, что ткань исходно получает адекватное кровоснабжение. В результате снижения сопротивления нормального участка сосуда перфузионное давление в магистральном коронарном сосуде падает, и кровоток через суженный участок уменьшается, что усугубляет стенокардию.
Считается, что меньшая сосудорасширяющая активность нитратов в отношении артериол является следствием меньшей способности этих сосудов к образованию оксида азота из молекулы нитрата. Существуют данные о том, что добавление тиолсодержащих соединений, таких как цистеин, может повысить высвобождение оксида азота из нитроглицерина в артериолах (Harrison and Bates, 1993). Если это так, то введение ацетилцистеина с целью предупреждения толерантности к нитроглицерину может привести к тому, что последний будет действовать подобно неэффективным артериолярным вазодилататорам!
(Karlberg, 1992). Возможно, что это действие играет роль в наблюдающемся при внутривенном введении нитроглицерина снижении размеров инфаркта и связанной с инфарктом смертности.
4. Другие эффекты. Нитритный ион реагирует с гемоглобином (который содержит Fe3+) и образует метгемоглобин (который содержит Fe2+). Поскольку у метгемоглобина низкий аффинитет к кислороду, большие дозы нитритов могут привести к псевдоцианозу, тканевой гипоксии и смерти. У взрослого человека уровень нитрита в плазме после приема даже больших доз органических и не
органических нитратов слишком мал, чтобы вызвать значительную метгемоглобинемию. Следует помнить, что нитрит натрия используется как консервирующая добавка к мясным продуктам. У младенцев кишечная флора способна перерабатывать большие количества неорганического нитрата, например из колодезной воды, в нитритный ион. Таким образом, возможно непреднамеренное поступление в организм большого количества нитритного иона, что может вызвать серьезный токсический эффект. Но он находит и терапевтическое применение. Отравление цианидами происходит при со
единении железа цитохромоксидазы с CN”. Железо метгемоглобина имеет очень высокий аффинитет к CN”, поэтому введение нитрита натрия (NaNO2) вскоре после воздействия цианидом восставливает активность цитохромоксидазы. Образующийся ци-анметгемоглобин можно далее подвергнуть детоксикации путем внутривенного введения тиосульфата натрия (Na2S2O3), что приводит к образованию иона тиоцианата (SCN-), менее токсичного и легко экскретируемого. При избыточной метгемоглобинемии внутривенно вводят метиленовый синий.
Токсичность и толерантность
А. Острые побочные эффекты. Основные острые токсические эффекты органических нитратов (ортостатическая гипотензия, тахикардия и пульсирующая головная боль) являются продолжением их терапевтического сосудорасширяющего действия. Течение глаукомы, которая ранее рассматривалась как противопоказание, не ухудшается, нитраты могут безопасно использоваться при повышенном внутриглазном давлении. Однако они противопоказаны при повышении внутричерепного давления.
Б. Толерантность. В эксперименте при постоянном воздействии нитратов на изолированную гладкую мышцу может развиться полная толерантность (тахифилаксия). У человека обычно наблюдается постепенное развитие толерантности к препаратам длительного действия (энтеральные и трансдермальные формы) или при длительной непрерывной внутривенной инфузии в течение нескольких часов (Steering Committee, 1991).
Постоянное воздействие высоких концентраций нитратов может происходить в химической промышленности, особенно при производстве взрывчатых материалов. При высокой степени загрязнения рабочей зоны летучими органическими нитратами рабочие замечают, что в начале недели они страдают от головной боли и головокружения (“болезнь понедельника"). Примерно через день эти симптомы исчезают из-за развития толерантности. В выходные дни, когда воздействие прекращается, толерантность угасает; симптомы возобновляются каждый понедельник. Более серьезный риск промышленного воздействия связан с развитием зависимости. Она может обнаруживаться у рабочих через месяцы и годы и проявляется как вариантная стенокардия, возникающая через 1-2 дня после
прекращения воздействия нитратов. В наиболее серьезных случаях коронарный сосудистый спазм приводит к развитию инфаркта миокарда.
Не доказано, что у больных стенокардией даже при применении больших доз короткодействующих нитратов развивается физическая зависимость.
Механизм развития толерантности не совсем ясен. Как уже отмечалось, определенную роль может играть клеточная толерантность. Установлено, что в гомогенате клеток активация гуанилатцикла-зы снижается при предварительной инкубации с органическими нитратами. Частичное устранение этой ареактивности с помощью тиолсодержащих соединений (например, ацетилцистеина) и отсутствие перекрестной толерантности к другим зависимым от оксида азота вазодилататорам (ацетилхолин, нико-рандил) указывают на то, что оксид азота лишь частично ответственен за толерантность к нитроглицерину. Формирование толерантности в целостном организме, безусловно, опосредовано и системными компенсаторными реакциями. Так, вначале может происходить повышение симпатической импульса-ции, что увеличивает тонус сосудов. После одного или более дней лечения длительнодействующими нитратами задержка воды и солей может устранить благоприятные гемодинамические изменения, вызываемые нитроглицерином (Parker, 1993).
В. Канцерогенное действие нитратов и их производных. Нитрозамины — это небольшие молекулы с общей структурой R2-N-NO, которые образуются при соединении нитратов и нитритов с аминами. Некоторые нитрозамины являются канцерогенами для животных, видимо, после превращения в высокореактивные производные. Хотя нет прямых доказательств того, что эти вещества могут вызывать опухоли у человека, существуют эпидемиологические данные о связи заболеваемости раком пищевода и желудка с содержанием нитратов в пище. Нитрозамины обнаруживаются также в табаке и сигаретном дыме. Не доказано, что малые дозы нитратов, используемые для лечения больных стенокардией, приводят к значительному повышению уровня нитрозаминов в организме.
Механизмы клинического эффекта
Желательные и вредные эффекты вазодилатации, вызванной нитратами, представлены в табл. 12-2.
А. Эффекты нитратов при стенокардии напряжения. Основными гемодинамическими эффекта
ми являются снижение венозного возврата к сердцу и связанное с этим уменьшение внутрисердечного объема. АД падает. Понижение внутрижелудочкового давления и объема левого желудочка ассоциированы с ослаблением напряжения стенки (правило Лапласа) и уменьшением потребности миокарда в кислороде. В редких случаях может происходить парадоксальное повышение потребности миокарда в кислороде вследствие чрезмерной рефлекторной тахикардии и увеличения силы сокращений.
Внутрикоронарное, внутривенное и сублингвальное введение нитратов приводит к увеличению диаметра эпикардиальных коронарных артерий. Наблюдается тенденция к снижению сопротивления артериол, хотя и в меньшей степени. Однако нитраты при обычном системном введении наряду с уменьшением потребности миокарда в кислороде понижают и суммарный коронарный кровоток. Интракоронарное введение малых доз нитроглицерина, которое ведет к увеличению общего коронарного кровотока, но не вызывает системных гемодинамических эффектов, не устраняет симптомов стенокардии при нагрузочной пробе. Однако системное введение нитроглицерина, понижающее АД и объем левого желудочка, облегчает приступ стенокардии, несмотря на сниженный коронарный кровоток. Следовательно, улучшение при стенокардии напряжения после приема нитратов связано главным образом со снижением потребности миокарда в кислороде, а не с повышением коронарного кровотока.
Б. Эффекты нитратов при вариантной стенокардии. При вариантной стенокардии нитраты об
легчают состояние больных за счет расслабления гладких мышц эпикардиальных коронарных артерий и уменьшения коронароспазма. Хотя внутрикоронарное введение нитроглицерина считается наиболее эффективным методом снятия спазма коронарных артерий, этот способ введения по сравнению с другими не имеет большой клинической значимости.
В. Эффекты нитратов при нестабильной стенокардии. Нитраты успешно используют и при лечении нестабильной стенокардии, но точный механизм их терапевтического действия неясен. Стенокардия в состоянии покоя у этих больных может быть связана как с повышением тонуса коронарных сосудов, так и с увеличением потребности миокарда в кислороде, поэтому нитраты могут действовать, расширяя эпикардиальные артерии или снижая одновременно потребность миокарда в кислороде.
Основной механизм продолжительных приступов стенокардии покоя в настоящее время связывают с периодической окклюзией сосуда тромбом (возможно, при участии агрегации тромбоцитов) в месте атеросклеротической бляшки. Как уже обсуждалось, нитроглицерин снижает агрегацию тромбоцитов, и этот эффект может иметь значение при нестабильной стенокардии.
Клиническое применение нитратов
Некоторые формы нитроглицерина и его производных перечислены в табл. 12-3. В связи с быстрым началом действия (1-3 минуты) при приступах стенокардии наиболее часто используют суб-
ТАБЛ ИЦА 12-2. Полезные и вредные эффекты нитратов при лечении стенокардии
Эффекты
Результаты
Потенциально полезные эффекты
Снижение объема желудочков
Снижение артериального давления
Снижение времени изгнания
Расширение эпикардиальных коронарных артерий
Увеличение коллатерального кровотока
Снижение диастолического давления в левом желудочке
Снижение потребности миокарда в кислороде
Уменьшение спазма коронарных артерий
Улучшение перфузии ишемизированного миокарда
Улучшение субэндокардиальной перфузии
Потенциально вредные эффекты
Рефлекторная тахикардия 1
Рефлекторное повышение силы сокращений J Повышение потребности миокарда в кислороде
Снижение времени диастолической перфузии вследствие тахикардии
Снижение перфузии миокарда
лингвальное введение препарата. Так как длительность эффекта невелика (не превышает 2Э-30 минут), он не удобен для поддерживающей терапии. И при внутривенном введении нитроглицерин начинает действовать быстро (минуты), но его гемодинамические эффекты исчезают вскоре после прекращения инфузии. Клиническое значение внутривенных инфузий нитроглицерина, следовательно, ограничивается лечением тяжелых, рецидивирующих форм стенокардии. Медленно всасывающиеся препараты нитроглицерина назначают трансбук-кально, внутрь и в трансдермальных формах. Они обеспечивают нужную концентрацию в крови длительное время, но способствуют развитию толерантности.
Гемодинамические эффекты сублингвальных форм или таблеток для разжевывания изосорбида динитрата, пентаэритритола тетранитрата и эрит-ритила тетранитрата сходны с эффектами нитроглицерина. Рекомендуемые дозы для наиболее распространенных препаратов нитратов длительного действия с указанием продолжительности эффекта представлены в табл. 12-3. Хотя трансдермальное введение поддерживает уровень нитроглицерина в течение 24 часов и более, необходимый гемодинамический эффект обычно не продолжается дольше 8-10 часов. Клиническое применение форм
с медленным высвобождением нитроглицерина для поддерживающей терапии стенокардии ограничено развитием выраженной толерантности. Полагают, что уменьшить или предупредить развитие толерантности можно, соблюдая интервалы между приемами нитратов длительностью не менее 8 часов.
Блокаторы кальциевых каналов
Химия
Первый клинически важный представитель этой группы верапамил был создан в результате попыток синтезировать более активные аналоги алкалоида опийного мака папаверина, являющегося вазодилататором. С тех пор были открыты десятки веществ различной структуры с аналогичным основным фармакологическим действием. На рис. 12-5 показаны четыре химически несходных блокатора кальциевых каналов. Нифедипин является прототипом семейства дигидропиридиновых блокаторов кальциевых каналов. Исследованы десятки представителей этого семейства, шесть из них в настоящее время разрешены к применению в США для терапии стенокардии и по другим показаниям. Наиболее хорошо изучен в этой группе препаратов нифедипин, свойства других дигидропиридинов мож-
ТАБЛ ИЦА12-3. Препараты нитратов и нитритов, используемые при стенокардии
Препараты Дозы Продолжительность действия
Короткого действия
Нитроглицерин, сублингвально 0.15-1.2 мг 10-30 мин
Изосорбид, сублингвально 2.5-5 мг 10-60 мин
Амилнитрит, ингаляционно 0.18-0.3 мл 3-5 мин
Длительного действия
Нитроглицерин, препарат длительного 6.3-13 мг каждые 6-8 ч 6-8 ч
действия для энтерального приема
Нитроглицерин, 2 % мазь 2.5-4 см каждые 4 ч 3-6 ч
Нитроглицерин, трансбуккально 1 -2 мг каждые 4 ч 3-6 ч
с медленным высвобождением
Нитроглицерин, трансдермально 1 0-25 мг каждые 24 ч 8-10 ч
с медленным высвобождением (один пластырь в день)
Изосорбида динитрат, сублингвально 2.5-10 мг каждые 2 ч 1.5-2 ч
Изосорбида динитрат, внутрь 10-60 мг каждые 4-6 ч 4-6 ч
Изосорбида динитрат, для разжевывания 5-10 мг каждые 2-4 ч 2-3 ч
Изосорбида мононитрат 20 мг каждые 12 ч 6-10 ч
Пентаэритритола тетранитрат 40 мг каждые 6-8 ч 6-8 ч
Эритритила тетранитрат 10-40 мг каждые 6-8 ч 6-8 ч
Н3С СНз
СНз
Нифедипин
Дилтиазем
Рис. 12-5. Химическое строение некоторых блокаторов кальциевых каналов
но рассматривать как сходные, если нет специальных оговорок.
Фармакокинетика
Блокаторы кальциевых каналов активны при приеме внутрь. Они характеризуются выраженным эффектом первого прохождения, значительным связыванием с белками плазмы и активной метабо-лизацией (табл. 12-4).
Фармакодинамика
А. Механизм действия. В настоящее время признается существование трех типов потенциалзависимых кальциевых каналов в дополнение к рецеп-торуправляемым (например, через агрецепторы). Потенциалзависимые кальциевые каналы разделяют на L-.T- и N-типы в зависимости от проводимости, длительности открытия и распределения в тка
нях. В некоторых тканях может существовать и четвертый — P-тип. Кальциевые каналы L-типа преобладают в сердце и гладких мышцах и содержат несколько лекарственных рецепторов. Было показано, что нифедипин и другие дигидропиридины имеют одно место связывания, тогда как верапамил и дилтиазем связываются со сходным, но не идентичным рецептором в другой области. Оккупация лекарством рецепторов верапамила или дилтиазе-ма оказывает влияние и на связывание дигидропиридинов. Эти рецепторные области стереоселектив-ны, так как для энантиомеров верапамила, дилтиа-зема и оптически активных производных нифедипина характерны значительные отличия по аффинитету и фармакологической активности.
Блокирующее действие этих веществ напоминает эффект блокады натриевых каналов местными анестетиками: вещества действуют с внутренней стороны мембраны и связываются более эф-
ТАБЛИЦА 12-4. Фармакокинетика некоторых блокаторов кальциевых каналоа
Препарат Биодоступность при приеме внутрь(%) Начало действия (путь введения) Период полувыведе ния из плазм (часы) Распределение и превращение ы
Дигидропиридины
Амлодипин 65-90 Нетданных 30-50 > 90 % связывается с белками плазмы крови; активно метаболизируется
Фелодипин 15-20 2-5 ч (внутрь) 11-16 > 99 % связывается с белками плазмы крови; активно метаболизируется
Исрадипин 15-25 2 ч (внутрь) 8 95 % связывается с белками плазмы крови; активно метаболизируется
Никардипин 35 20 мин (внутрь) 2-4 95 % связывается с белками плазмы крови; активно метаболизируется в печени
Нифедипин 45-70 <1 мин (в/в); 5-20 мин (под язык или внутрь) 4 Около 90 % связывается с белками плазмы крови; метаболизируется до молочной кислоты лактата; 80 % вещества и метаболитов выводятся с мочой
Нимодипин 13 Нетданных 1-2 Активно метаболизируется
Низолдипин <10 Нет данных 2-6 Активно метаболизируется
Нитрендипин Смешанная группа 10-30 4 ч (внутрь) 5-12 98 % связывается с белками плазмы крови; активно метаболизируется
Бепридил 60 60 мин (внутрь) 24-40 > 99 % связывается с белками плазмы крови; активно метаболизируется
Дилтиазем 40-65 < 3 мин (в/в), > 30 мин (внутрь) 3-4 70-80 % связывается с белками плазмы крови; активно деацетилируется; вещество и метаболиты выводятся с калом
Вервпамил 20-35 <1.5 мин (в/в), 30 мин (внутрь) 6 Около 90 % связывается с белками плазмы крови; 70 % выводится почками; 15 % выводится желудочно-кишечным трактом
фективно с каналами деполяризованной мембраны. Связывание лекарства с рецептором изменяет функцию канала, нарушая регулярность его перехода в открытое состояние после деполяризации. В результате укорочения времени открытия канала значительно снижается трансмембранный кальциевый ток (рис. 12-1), что соответствует в гладкой мышце продолжительному расслаблению (рис. 12-4), а в сердечной — уменьшению сократимости, снижению частоты ритмоводителя синусового узла и скорости проведения в атриовентрикуляр
ном узле1. В гладкой мышце эти препараты уменьшают вход Са2+ и через рецепторуправлямые каналы, но не столь значительно. Блокада частично устраняется повышением концентрации Са2+, хотя необходимые для этого уровни иона труднодостижимы. Блокаду можно в некоторой степени снять также применением веществ, которые увеличивают
1 В очень низких дозах и при ряде других обстоятельств некоторые дигидропиридины увеличивают кальциевый ток. Такие специфические дигидропиридины, как Вау К 8644, усиливают кальциевый ток а широком диапазоне доз.
трансмембранный ток ионов кальция, например симпатомиметиками.
Кальциевые каналы Т- и N-типов менее чувствительны к действию блокаторов. Следовательно, ткани, в которых преобладают эти типы (например, нейроны и большинство секреторных желез), менее чувствительны к действию препаратов данной группы, чем сердечная и гладкая мускулатура.
Б. Влияние на системы и органы.
1. Гладкая мускулатура. В большинстве гладких мышц посредством регуляции трансмембранного тока Са2+ поддерживаются нормальный тонус и сократительная реакция. Мышечные клетки расслабляются при введении блокаторов кальциевых каналов (рис. 12-4). Наиболее чувствительны гладкие мышцы сосудов, но сходное расслабление происходит и в гладких мышцах бронхов, желудочно-кишечного тракта, матки. Артериолы проявляют большую чувствительность, чем вены; ортостатическая гипотензия не является частым побочным эффектом. АД может понижаться, особенно после приема нифедипина. Женщины более чувствительны, чем мужчины, к гипотензивному эффекту дил-тиазема. Уменьшение периферического сосудистого сопротивления — один из механизмов, благодаря которому вещества данной группы могут быть эффективны при стенокардии напряжения. У больных с вариантной стенокардией было продемонстрировано снижение тонуса коронарных артерий.
2. Сердечная мышца. Нормальная функция сердечной мышцы в значительной степени зависит от потоков ионов кальция. Генерация импульса в синоатриальном узле и проводимость в атриовентрикулярном узле (так называемый “медленный ответ”, или кальцийзависимый потенциал действия) могут снижаться или подавляться всеми блокаторами кальциевых каналов. Для сопряжения возбуждения и сокращения во всех клетках сердца требуется вход Са2+, поэтому данные вещества дозозависимо снижают сократимость миокарда и сердечный выброс. Ингибирование механической функции — другой механизм действия антагонистов кальция, уменьшающий потребность миокарда в кислороде у больных со стенокардией.
Дополнительная польза от подавления входа ионов кальция была продемонстрирована на модели экспериментального инфаркта миокарда. Поскольку ишемия вызывает деполяризацию мембраны, ток Са2+ в клетку увеличивается. Повышение
уровня внутриклеточного кальция активирует некоторые АТФ-потребляющие ферменты, что способствует истощению ограниченных клеточных энергетических ресурсов и делает сердце еще более чувствительным к ишемическому поражению. Блокаторы кальциевых каналов защищают сердце от повреждающего влияния Са2+, снижая вероятность аритмий и ограничивая размеры инфаркта у экспериментальных животных.
Важные отличия между существующими антагонистами кальция определяются деталями их взаимодействия с ионными каналами сердца и, как указывалось, относительной выраженностью влияния на сердце и гладкие мышцы. Так, бепридил блокирует натриевые каналы сердца, но менее эффективно, чем кальциевые. Блокада натриевых каналов умеренно выражена при действии верапамила и в еще меньшей степени — дилтиазема. Она незначительна после введения нифедипина и других дигидропиридинов. Верапамил и дилтиазем имеют отличную от дигидропиридинов кинетику взаимодействия с рецептором кальциевого канала. В результате они более эффективно купируют тахикардию в Са2+-зависимых клетках, например атриовентрикулярного узла (детально рассматривается в главе 14). С другой стороны, дигидропиридины блокируют кальциевые каналы гладких мышц в концентрациях ниже тех, которые требуются для заметного влияния на сердце; следовательно, они оказывают меньшее депрессивное действие на миокард, чем верапамил и дилтиазем. Бепридил имеет также выраженный блокирующий эффект на калиевые каналы сердца, что ведет к удлинению реполяризации (глава 14) и повышению риска возникновения аритмий.
3. Скелетная мускулатура. Блокаторы кальциевых каналов не угнетают функцию скелетных мышц, потому что эти мышцы используют для сопряжения возбуждения и сокращения внутриклеточный запас Са2+ и не требуют столь значительного трансмембранного кальциевого тока.
4. Мозговой вазоспазм и инфаркт после субарахноидального кровотечения. Представитель группы дигидропиридиновых блокаторов кальциевых каналов нимодипин имеет высокое сродство к мозговым сосудам и уменьшает последствия субарахноидального кровоизлияния, связанного со спазмом сосудов и выходом крови в ткань. Некоторые данные свидетельствуют о том, что блокаторы кальциевых каналов могут также уменьшать пора
жение мозга после тромбоэмболического инсульта. Неясно, с чем связан их положительный эффект (если он действительно существует): с расширением сосудов мозга или с уменьшением потребности нейронов в кислороде.
5. Другие эффекты. Антагонисты кальция мало влияют на сопряжение раздражения и секреции в железах и нервных окончаниях из-за рассмотренной уже тканевой специфичности кальциевых каналов. Верапамил уменьшает выделение инсулина у человека, но требуемые для этого дозы выше, чем те, которые используются при лечении стенокардии.
Существует много данных о том, что блокаторы кальциевых каналов могут влиять на агрегацию тромбоцитов и предупреждать или ослаблять развитие атероматозных повреждений у животных. Клинические исследования пока твердо не установили их роль в свертывании крови и в терапии атеросклероза у человека.
Верапамил может блокировать гликопротеид Р170, ответственный за транспорт многих чужеродных, в том числе лекарственных, веществ из раковых (и не только) клеток. Подобным эффектом обладают и другие препараты этой группы. В отличие от блокады кальциевых каналов это действие не стереоспецифично. Показано, что повышенная экспрессия Р170 сопровождается формированием резистентности онкологических больных к химиотерапии. In vitro было продемонстрировано, что верапамил частично устраняет резистентность раковых клеток ко многим химиотерапевтическим средствам. Первые клинические наблюдения подтвердили это действие (глава 56).
Токсичность
Наиболее важные токсические эффекты блокаторов кальциевых каналов являются прямым продолжением их терапевтического действия. Чрезмерное подавление входящего кальциевого тока может вызывать серьезное угнетение миокарда: брадикардию, атриовентрикулярную блокаду и застойную сердечную недостаточность и даже остановку сердца. Однако эти эффекты редко наблюдаются в клинике. Бепридил удлиняет потенциал действия в сердце и может вызывать опасные аритмии по типу torsade de pointes у чувствительных больных. Он противопоказан больным с серьезными аритмиями в анамнезе или с синдромом удлиненного QT. Па
циенты, получающие p-блокаторы, более чувствительны к кардиодепрессивному действию антагонистов кальция. К менее существенным побочным эффектам (обычно не требующим прекращения лечения) относятся покраснение лица, отеки, головокружение, гиперплазия десен, тошнота и запоры (табл. 12-5).
Механизмы клинических эффектов
Как уже обсуждалось, блокаторы кальциевых каналов понижают силу сокращений миокарда, что в свою очередь уменьшает потребность миокарда в кислороде. Подавление входа Са2+ в гладкой мускулатуре артерий ведет к снижению тонуса артериол и общего сосудистого сопротивления, уменьшению АД и внутрижелудочкового давления. Некоторые из этих веществ (например, верапамил) блокируют также а-адренорецепторы, что может способствовать периферической вазодилатации. В результате всех этих эффектов напряжение стенки левого желудочка уменьшается, что снижает потребность миокарда в кислороде. Урежение сердечных сокращений при назначении верапамила, дил-тиазема или бепридила вызывает дальнейшее снижение потребности миокарда в кислороде. Антагонисты кальция, кроме того, предупреждают местный спазм коронарных артерий, лежащий в основе развития вариантной стенокардии. Применение этих средств оказалось наиболее эффективным способом лечения данной формы стенокардии.
Как указывалось ранее, в клинических условиях различные блокаторы кальциевых каналов отличаются по характеру влияния на сердечнососудистую систему. Синоатриальный и атриовентрикулярный узлы, состоящие в основном из медленно реагирующих клеток, чувствительны к действию верапамила, в меньшей степени — к дилти-азему и значительно меньше реагируют на нифедипин. Таким образом, нифедипин и дилтиазем снижают проведение в атриовентрикулярном узле и эффективны при лечении суправентрикулярных тахикардий, развивающихся по механизму “повторного входа” (reentry), и при назначении с целью снижения реакции желудочков на фибрилляцию или трепетание предсердий. Нифедипин же не влияет на атриовентрикулярную проводимость. Неспецифический антагонизм в отношении симпатической системы наиболее выражен при действии дилтиазема и значительно меньше — вера-
ТАБЛИЦА 12- 5. Сосудистая селективность и клиническое применение некоторых блокаторов кальциевых каналов
Препараты Сосудистая избирательность1 Показания2 Обычные дозы Токсические эффекты
Дигидропиридины
Амлодипин ++ Стенокардия, гипертензия 5-10 мг один раз вдень Головная боль, отек
Фелодипин 5.4 Гипертензия 5-10 мг один раз вдень Головокружение, головная боль
Исрадипин 7.4 Гипертензия 2.5-10 мг каждые 12ч Головная боль, утомляемость
Никардипин 17.0 Стенокардия, гипертензия 20-40 мг каждые 8 ч внутрь Отек, головокружение, головная боль, покраснение лица
Нифедипин 3.1 Стенокардия (гипертензия, 3-10 мкг/кг в/в; мигрень, кардиомиопа- 20-40 мг каждые тия, болезнь Рейно) 8 ч внутрь Гипотензия, головокружение, покраснение лица, тошнота, запор, отеки
Нимодипин ++ Субарахноидальное крово- 60 мг каждые 4 ч течение (мигрень) внутрь Головная боль, понос
Низолдипин ++ Исследуют при стенокар- 20-40 мг один раз дии, гипертензии в день Вероятно, сходные с нифедипином
Нитрендипин Смешанная группа 14.4 Исследуют при стенокар- 20 мг один-два раза дии, гипертензии вдень Вероятно, сходные с нифедипином
Бепридил — Стенокардия 200-400 мг внутрь один раз в день Аритмии, головокружение, тошнота
Дилтиазем 0.3 Стенокардия, гипертензия 75-150 мкг/кг в/в; (болезнь Рейно) 30-80 мг каждые 6 ч внутрь Гипотензия, головокружение, покраснение лица, брадикардия
Верапамил 1.3 Стенокардия, гипертензия, 75-150 мкг/кг в/в; аритмии (мигрень, кар- 80-160 мг каждые диомиопатия) 8 ч внутрь Гипотензия, угнетение миокарда, сердечная недостаточность, отеки
’ Цифровые данные (Spedding, 1990) показывают соотношение активности применительно к сосудам и сердцу; большее число означает большую сосудистую и меньшую сердечную активность: - = миокардиальная депрессия аыражена сильнее, чем вазодилатация; ++ = значительная степень вазодилатации, превышающая эффект депрессии миокарда.
2 В скобках даны показания, не зарегистрированные официально.
памила. Для нифедипина такой эффект не характерен. Следовательно, выраженная рефлекторная тахикардия в ответ на гипотензивное действие возникает чаще после приема нифедипина и реже — после верапамила. Эти отличия в фармакологических эффектах следует учитывать при выборе блокатора кальциевых каналов для лечения конкретного больного со стенокардией.
Клиническое применение блокаторов кальциевых каналов
Помимо применения при стенокардии блокаторы кальциевых каналов эффективны при гипертензии (глава 11) и суправентрикулярных тахиаритмиях (глава 14). Их использование перспективно и при других заболеваниях, в том числе гипер
трофической кардиомиопатии, мигрени, болезни Рейно, постинфарктных состояниях и атеросклерозе (Scriabine, 1987; O’Rourke, 1987; Taira, 1987; Weinstein, 1989).
Утвержденные и еще не зарегистрированные показания к применению препаратов и их дозы обобщены в табл. 12-5. Выбор определенных блокаторов кальциевых каналов должен основываться на знании их фармакологических свойств и потенциальных нежелательных эффектов. Нифедипин, не снижающий атриовентрикулярную проводимость, более безопасен для больных с нарушениями атриовентрикулярного проведения. Верапамил и дилтиазем в сочетании с p-блокаторами могут вызывать атриовентрикулярную блокаду и угнетение функции желудочков. При наличии явной сердечной недостаточности все блокаторы кальциевых каналов могут усугублять ее в результате отрицательного инотропного действия. Контролируемые широкомасштабные исследования выявили более высокую смертность у больных с застойной сердечной недостаточностью, принимавших блокаторы кальциевых каналов; следовательно, они противопоказаны как сосудорасширяющие средства при лечении больных с сердечной недостаточностью. При сравнительно низком АД нифедипин может вызывать его дальнейшее нежелательное понижение. Верапамил и дилтиазем предпочтительнее в подобной ситуации, так как меньше изменяют давление. Больным с предсердной тахикардией, фибрилляцией, трепетанием в анамнезе также следует предпочесть верапамил и дилтиазем, поскольку они имеют антиаритмический эффект. Пациентам, получающим дигиталис, верапамил назначают осторожно, потому что он может повысить уровень дигоксина в плазме вследствие фармакокинетического взаимодействия. Повышение уровня дигоксина отмечено при сочетании препаратов наперстянки с дилтиаземом и нифедипином, но значительно реже, чем при комбинировании с верапамилом.
Средства, блокирующие |3-адренорецепторы
Р-адреноблокаторы, не являющиеся вазодилататорами, тем не менее нашли широкое применение в терапии стенокардии. Их действие связано главным образом с влиянием на гемодинамику и снижением ЧСС, АД и силы сердечных сокращений, что огра
ничивает потребность миокарда в кислороде в покое и при нагрузке. Уменьшение ЧСС способствует также увеличению длительности диастолической перфузии, что улучшает перфузию миокарда. Считают, что p-адреноблокаторы вызывают и перераспределение коронарного кровотока в пользу ишемизированного миокарда из-за неодинакового влияния на сопротивление коронарных сосудов ишемизированных и неишемизированных участков. Однако именно снижение ЧСС и АД с последующим уменьшением потребности миокарда в кислороде рассматривают как наиболее важный механизм лечебного эффекта этих средств при стенокардии и механизм повышения толерантности к нагрузке. p-блокаторы могут быть полезны и при лечении “молчащей” (амбулаторной) ишемии. Поскольку при этом заболевании нет боли, оно обычно диагностируется по типичным признакам ишемии на ЭКГ. Как показано на рис. 12-6, общее “время ишемии” снижается при курсовом введении Р-блокаторов. Этот эффект может частично объяснить уменьшение риска повторного инфаркта и смертности у больных, которые получают лечение P-антагонистами после инфаркта миокарда.
Нежелательные эффекты Р-блокаторов проявляются возрастанием конечного диастолического объема, сопровождающим понижение ЧСС и увеличение периода изгнания. Повышенная потребность миокарда в кислороде, связанная с увеличением диастолического объема левого желудочка, несколько уменьшает терапевтическую ценность Р-блокаторов. Но эти потенциально нежелательные эффекты можно свести до минимума, сочетая Р-ан-тагонисты с нитратами.
II. Клиническая фармакология средств, применяемых при стенокардии
Принципы лечения стенокардии
В дополнение к влиянию на факторы риска коронарного атеросклероза (курение, гипертензия, гиперлипидемия) терапия стенокардии и других проявлений ишемии миокарда основана на снижении потребности миокарда в кислороде и повышении
. . Плацебо
ЧСС (уд/мин)
ЮО и-------------------
r'W'V 60 I...... ' ' г-1".
0 12 24 36 48 60 72
100-гг:----------------
Нормализованное время ишемии
Время (часы мониторной регистрации)
Рис. 12-6. Влияние метопролола на частоту сокращений сердца и нормализованное время ишемии (измеренное при мониторной регистрации ЭКГ) у девяти мужчин с тяжелым заболеванием коронарных артерий. Лекарство уменьшает среднюю частоту сердечных сокращений, размах изменений ЧСС в течение суток и общее время, в течение которого на ЭКГ отмечаются признаки ишемии. (Из: Lambert C.R. et al. Influence of beta-adrenergic blockade defined by time series analysis on circadian variation of heart rate and ambulatory myocardial ischemia. Am. J. Cardiol. 1989, 64,835.)
коронарного кровотока к потенциально ишемизированному миокарду с целью восстановления баланса между снабжением миокарда кислородом и запросом.
Стенокардия напряжения
Во многих исследованиях было показано, что нитраты, блокаторы кальциевых каналов и [3-блокаторы отдаляют начало проявлений стенокардии и депрессии сегмента ST в тесте на тредмилле у больных со стенокардией напряжения (рис. 12-7). Во время нагрузки существенно понижается двойное произведение. Однако, хотя толерантность к нагрузке возрастает, обычно нет изменений в пороге ангинозных проявлений, т. е. в исходной (до лечения) величине произведения частоты и давления, при которой появляется симптоматика.
Для поддерживающего лечения хронической стабильной стенокардии можно применять длительно действующие нитраты, блокаторы кальциевых каналов или [3-блокаторы; выбор определяется индивидуальной реакцией больного. Если пациент
страдает гипертензией, часто адекватна монотерапия блокаторами кальциевых каналов или [3-блока-торами. Нормотензивным больным, как правило, подходят длительно действующие нитраты. Сочетание [3-блокаторов с блокаторами кальциевых каналов (например, пропранолола с нифедипином) или двух различных антагонистов кальция (например, нифедипина и верапамила) может быть более эффективно, чем использование каждого препарата в отдельности. Если реакция на один препарат неадекватна, следует добавить препарат другой группы для повышения эффективности терапии и минимизации нежелательных эффектов (табл. 12-6). Некоторым больным может потребоваться лечение препаратами всех трех групп.
Для больных с постоянной гипертензией, синусовой брадикардией или дисфункцией атриовентрикулярного узла нифедипин или другие более длительно действующие дигидропиридины могут оказаться препаратами выбора.
Нет свидетельств в пользу того, что какой-либо из [3-блокаторов более эффективен при стенокардии напряжения или лучше переносится, чем другие.
Рис. 12-7. Влияние дилтиазема на “двойное произведение” в группе из 20 больных со стенокардией напряжения. В условиях двойного слепого контроля больные выполняли пробу на тредмилле, получая плацебо или одну из трех доз препарата. ЧСС и АД регистрировали через 180 с нагрузки (средняя точка на линиях графика) и во время начала ангинозных симптомов (правая точка). Заметьте, что препарат уменьшает величину двойного произведения во все периоды выполнения теста и увеличивает время появления симптомов. (Из: Lindenberg В. S. et al. Efficacy and safety of incremental doses of diltiazem for the treatment of angina. J. Am. Coll. Cardiol. 1983,2,1129. Использовано с разрешения American College of Cardiology.)
Хирургическая реваскуляризация и ангиопластика являются наиболее действенными методами, восстанавливающими коронарный кровоток и повышающими снабжение миокарда кислородом. Они показаны пациентам, рефрактерным к лекарственной терапии или неспособным адаптироваться к новому стилю жизни.
Вазоспастическая стенокардия
У больных с вариантной стенокардией для лечения и предупреждения ишемических приступов значительно более эффективны, чем р-адренобло-каторы, нитраты и блокаторы кальциевых каналов. Примерно у 70 % пациентов, которые получают нитраты вместе с блокаторами кальциевых каналов, приступы стенокардии полностью устраняются, у 20 % отмечается значительное снижение их частоты. Главным механизмом этого терапевтического эффекта является предупреждение спазма коронарных артерий (в случае присутствия или отсутствия атеросклеротического поражения коронарных артерий). Все доступные в настоящее время антагонисты кальция примерно равноэффективны, выбор конкретного средства зависит от индивидуальных особенностей больного. При вариантной стенокардии хирургическая реваскуляризация и ангиопластика не показаны.
Нестабильная стенокардия
У больных с нестабильной стенокардией с повторяющимися ишемическими приступами в состоянии покоя вследствие расщепления атеросклеротических бляшек и агрегации тромбоцитов может возникать рецидивирующая тромботическая окклюзия пораженной коронарной артерии. Добавление в таких случаях нифедипина к [3-блокаторам и нитратам, как правило, снижает частоту приступов в покое, риск инфаркта миокарда и необходимость неотложной хирургической реваскуляризации миокарда. Однако нифедипин сам по себе не более эффективен в отношении снижения частоты приступов стенокардии в покое, чем нит
ТАБЛ ИЦА 12-6. Действие нитратов и их сочетаний с fi-блокаторами или блокаторами кальциевых каналов
при стенокардии. Нежелательные эффекты обозначены курсивом
Показатель Нитраты 0-блокаторы или блокаторы кальциевых каналов Комбинация нитратов с 0-блокаторами или блокаторами кальциевых каналов
Частота сердечных сокращений Рефлекторное повышение Снижение1 Снижение
Артериальное давление Снижение Снижение Снижение
Конечный диастолический объем Снижение Повышение Нет эффекта или снижение
Сократимость Рефлекторное повышение Снижение1 Нет эффекта
Время изгнания Снижение Повышение Нет эффекта
1 Нифедипин может вызывать рефлекторное повышение частоты и силы сердечных сокращений.
раты или p-блокаторы (Каг et al., 1992). Напротив, верапамил эффективнее контролирует нестабильную стенокардию, чем пропранолол. Уменьшает частоту сердечных приступов у таких больных и аспирин. В этих случаях также показано внутривенное введение гепарина или тромболитических средств.
Препараты
Нитраты и нитриты
Амилнитрит (генерик, Аспиролз, Вапорол) Ингаляционно: капсулы по 0.3 мл
Эритритила тетранитрат (Кардилат)
Перорально: таблетки, подъязычные таблетки по 5, 10 мг
Изосорбида динитрат
(генерик, Изордил, Сорбитрат)
Перорально: таблетки по 5, 10, 20,30,40 мг;
жевательные таблетки по 5, 10 мг Перорально для пролонгированного действия (генерик, Сорбитрат SA, Изо-Вид): таблетки и капсулы по 40 мг Сублингвально: таблетки по 2.5, 5, 10 мг
Изосорбида мононитрат (Измо)
Перорально: таблетки по 20 мг
Нитроглицерин
Сублингвально: таблетки по 0.15, 0.3,0.4,
0.6 мг; 0.4 мг/на дозу в аэрозоле
Перорально для пролонгированного действия (генерик, Нитронг): таблетки по 2.6, 6.5,9 мп капсулы по 2.5, 6.5,9, 13 мг
Защечно (Нитрогард): таблетки по 1,2,3 мг Парентерально (Нитро-Вид IV, Тридил): 0.5, 5 мг/мл для в/в введения Трансдермальные пластыри (Минитран, Нитро-Дур, Трансдерм-Нитро): освобождающие
по 2.5,5,7.5,10,15 мг/сут
Местно (генерик, Нитрол):
мазь 20 мг/мл (25 мм мази содержит около
15 мг нитроглицерина)
Пентаэритритола тетранитрат (генерик, P.E.T.N., Перитрат, др.)
Перорально: таблетки по 10, 20,40,80 мп капсулы пролонгированного действия по 30, 45,80 мг; таблетки пролонгированного действия по 80 мг
Блокаторы кальциевых каналов
Амлодипин (Норваск)
Перорально: таблетки по 2.5, 5, 10 мг
Бепридил(Васкор)
Перорально: таблетки по 200, 300, 400 мг
Дилтиазем (Кардизем)
Перорально: таблетки по 30, 60,90,120 мг Перорально для пролонгированного действия (Кардизем SR, Дилакор XL): капсулы по 60, 90, 120,180, 240, 300 мг
Парентерально: 5 мг/мл для инъекций
Фелодипин (Плендил)
Перорально для пролонгированного действия: таблетки по 5,10 мг
Исрадипин (Динасерк)
Перорально: капсулы по 2.5, 5 мг
Никардипин (Карден)
Перорально: капсулы по 20, 30 мг Перорально для пролонгированного действия (Карден SR): капсулы по 30, 45, 60 мг
Нифедипин (Адалат, Прокардия)
Перорально: капсулы по 10, 20 мг Перорально для пролонгированного действия (Прокардия XL): таблетки по 30, 60, 90 мг
Нимодипин (Нимотоп)
Перорально: капсулы по 30 мг (применяется при субарахноидальных кровотечениях, а не при стенокардии)
Верапамил (генерик, Калан, Изоптин) Перорально: таблетки по 40, 80, 120 мг Перорально для пролонгированного действия: таблетки по 120,180, 240 мг; капсулы по 120,180, 240 мг (одобрен только для применения при гипертензии: Калан SR, Изоптин SR, Верелан) Парентерально: 5 мг/2 мл для инъекций
Р-блокаторы, применяемые при стенокардии
Атенолол (Тенормин)
Перорально: таблетки по 50, 100 мг
Метопролол (Лопрессор)
Перорально: таблетки по 50,100 мг
Пропранолол (генерик, Индерал) Перорально: таблетки по 10, 20, 40, 60, 80, 90 мг; растворы по 4 и 8 мг/мл;
концентрированный раствор 80 мг/мл Перорально для пролонгированного действия (Индерал LA): капсулы по 80, 120, 160 мг
Надолол (Коргард)
Перорально: таблетки по 20,40, 80, 120, 160 мг
Избранная литература
Chatterjee К. Ischemic heart disease. In: Internal medicine. Stein J. H. (ed.) Little, Brown, 1990.
Danish Study Group on Verapamil in Myocardial Infarction: Effect of verapamil on mortality and major events after acute myocardial infarction (The Danish Verapamil Infaction Trial II— DAVIT II). Am. J. Cardiol. 1990; 66: 779.
Dickson R. B„ Gottesmann M. M. Understanding of the molecular basis of drug resistance in cancer reveals new targets for chemotherapy. Trends. Pharmacol. Sci. 1990; 11: 305.
Freedman D. D., Waters D. D. “Second generation” dihydropyridine calcium antagonists. Greater vascular selectivity and some unique applications. Drugs, 1987; 34: 578.
Godfraind T., Miller R., Wibo M. Calcium antagonism and calcium entry blockade. Pharmacol. Rev. 1986; 38: 321.
Harrison D. G., Bates J. N. The nitrqvasodilatators. New ideas about old drugs. Circulation, 1993; 87: 1461.
Jackson C. L., Bush R. C., Bowyer D. E. Mechanism of antiatherogenic action of calcium antagonists. Atherosclerosis, 1989; 80: 17.
Kar S., Waikada Y., Nordlander R. The high-risk unstable angina patient. An approach to treatment. Drugs, 1992; 43: 837.
Moncada S., Palmer R. M. J., Higgs E. A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991; 43: 109.
Multicentre Diltiazem Postinfarction Trial Research Group: The effect of diltiazem on mortality and reinfarction after myocardial infarction. N. Engl. J. Med. 1988; 319:385.
O’Rourke R. A. (ed.) Calcium-entry blockade: Basic concepts and clinical implications. (Symposium.) Circulation, 1987; 75 (Suppl. 6, Part 2): V-l.
Steering Committee, Transdermal Nitroglycerin Cooperative Study: Acute and chronic antianginal efficacy of continuous twenty-four hour application of transdermal nitroglycerin. Am. J. Cardiol. 1991; 68: 1263.
Сердечные гликозиды и другие средства, применяемые при застойной сердечной недостаточности
13
Бертрам Г. Катцунг, Уильям У. Пармлей
Застойная сердечная недостаточность наблюдается, когда сердечный выброс не обеспечивает потребности организма в кислороде. Полагают, что основным дефектом при сердечной недостаточности является нарушение механизма сопряжения возбуждения-сокращения в сердце, однако в патологический процесс вовлекаются многие другие системы и органы, в том числе барорецепторный рефлекс, симпатическая нервная система, почки, ренин-ангиотен-зин-альдостероновая система и вазопрессин.
Сердечные гликозиды (гликозиды наперстянки и подобные им средства), относящиеся к стероидным соединениям, увеличивают сердечный выброс и изменяют электрические характеристики сердца. Они также оказывают влияние на гладкие мышцы и другие ткани. Их основной терапевтический эффект при застойной сердечной недостаточности — увеличение сердечной сократимости (положительный инотропный эффект), что корригирует существующий при недостаточности дисбаланс. Однако у некоторых больных с сердечной недостаточностью эффективность сердечных гликозидов недолговременна. Общепризнано, что обычно используемые гликозиды имеют узкий диапазон безопасных доз и необходимо искать менее токсичные соединения с положительным инотропным действием. При острой недостаточности кандидатами на эту роль могут быть ингибиторы фосфодиэстеразы и некоторые стимуляторы ^-адренорецепторов.
Традиционная терапия застойной сердечной недостаточности в основном направлена на повышение сократительной функции миокарда и на устранение задержки в организме соли и воды. Кли
нические наблюдения показывают, что при некоторых типах сердечной недостаточности существенное облегчение больным дает разгрузка напряженного миокарда путем снижения сосудистого тонуса. В этих случаях можно добиться успеха, используя определенные вазодилататоры. К таким средствам относятся нитропруссид, гидралазин, нитроглицерин, каптоприл, эналаприл, лизиноприл и другие ингибиторы ангиотензинконвертирующе-го фермента.
Контроль сердечной сократимости
Сила сокращения сердечной мышцы определяется несколькими процессами, которые приводят в движение нити актина и миозина в саркомере (рис. 13-1). В конечном счете сокращение происходит при связывании ионов кальция (во время систолы) с актин-тропонин-тропомиозиновой системой, что облегчает взаимодействие актина с миозином. Этот “активирующий" Са2+ выделяется из саркоплазматического ретикулума (СР). Его количество зависит от величины запасов, хранящихся в СР и от количества “триггерного” Са2+, входящего в клетку через кальциевые каналы во время плато потенциала действия. Теоретически каждый из рассмотренных процессов может быть мишенью лекарственной терапии, но практически доказана эффективность только некоторых из этих подходов.
А. Чувствительность сократительных белков к ионам кальция. Факторы, определяющие чувстви-1 тельность к ионам кальция, то есть характер кривой, отражающей укорочение миофибрилл сердца '
Препараты
Рис. 13-1. Схема саркомера сердечной мышцы с обозначением мишеней действия некоторых средств, изменяющих сократимость (нумерованные структуры). Точка 1 — это №+,К+-АТФаза, натриевый насос. Точка 2 — обменник нат-рия/кальция. Точка 3 — потенциалзависимый кальциевый канал. Точка 4 — транспортер Са2+, перекачивающий кальций в саркоплазматический ретикулум (СР). Точка 5 — кальциевый канал в мембране СР, запускающий выделение Са2+ из запасов посредством активации СР “триггера”. Точка 6 — актин-тропонин-тропомиозиновый комплекс; действуя на него Са2+ вызывает взаимодействие актина и миозина, т. е. сокращение
в ответ на изменение цитоплазматической концентрации Са2+, изучены недостаточно, однако некоторые средства влияют на этот процесс in vitro. Например, ряд ингибиторов фосфодиэстеразы (теофиллин и др.) и более новые средства (Solaro, 1993), сдвигают эту кривую в сторону меньших концентраций ионов кальция.
Б. Количество Са2+, выделяемого из саркоплазматического ретикулума. Незначительное увеличение свободного цитоплазматического Са2+, поступающего через открытые потенциалзависимые кальциевые каналы в мембране клетки, вызывает открытие специализированных кальциевых каналов в мембране СР и быстрый выход большого количества ионов в цитоплазму вблизи актин-тропо-нин-тропомиозинового комплекса. Выделяемое ко
личество Са2+ пропорционально его запасам в СР и количеству триггерного иона, входящего в клетку через каналы клеточной мембраны. Рианодин, являющийся веществом с сильным отрицательным инотропным действием, препятствует выделению Са2+ из каналов СР. Средства, усиливающие этот процесс, неизвестны.
В. Количество Са2+, хранящегося в саркоплазматическом ретикулуме. Мембрана саркоплазматического ретикулума содержит эффективный транспортер ионов кальция (кальциевую помпу), который во время диастолы поддерживает очень низкий уровень свободного Са2+ в цитоплазме, закачивая его в ограниченные мембраной цистерны и трубочки СР. Таким образом, количество Са2+, связывающееся СР, отчасти определяется его дос
тупностью для помпы. Это в свою очередь зависит от баланса входа ионов кальция (прежде всего через потенциалзависимые мембранные кальциевые каналы) и их выхода из клетки (главным образом посредством натрий-кальциевого обмена транспортером клеточной мембраны).
Г. Количество триггерного Са2+. Количество триггерного Са2+, который входит в клетку, зависит от доступности кальциевых каналов и продолжительности их открытия. Как было рассмотрено в главах 6 и 9, симпатомиметики усиливают поступление ионов кальция, действуя на эти каналы. Блокаторы кальциевых каналов (глава 12), наоборот, уменьшают вход Са2+ и угнетают сократимость.
Д. Активность натрий-кальциевого обменника. Этот переносчик использует натриевый градиент для транспорта Са2+ против его градиента концентрации из цитоплазмы во внеклеточное пространство. В физиологических условиях внеклеточная концентрация этого иона значительно менее лабильна, чем внутриклеточная. Способность натрийкальциевого обменника осуществлять этот транспорт, следовательно, зависит в большей степени от внутриклеточной концентрации обоих ионов, особенно Na+.
Е. Внутриклеточная концентрация Na+ и активность Ка+,К+-АТФазы. На+,К+-АТФаза, участвуя в транспорте внутриклеточного Na+, является основным определяющим фактором поддержания его концентрации в клетке. С другой стороны, этот процесс обеспечивается поступлением ионов натрия через потенциалзависимые каналы, сопровождающим возникновение потенциалов действия в сердце. №+,К+-АТФаза рассматривается как основная мишень действия сердечных гликозидов. В эксперименте некоторые вещества заметно усиливают вход Na+ во время потенциала действия, препятствуя инактивации натриевых каналов (Hoey et al., 1993). Было продемонстрировано, что они вызывают сильное положительное инотропное действие, однако их роль в клинической практике еще не определена.
Патофизиология сердечной недостаточности
Застойная сердечная недостаточность — это синдром, вызываемый многими причинами, для которого характерно поражение правого желудочка, левого желудочка или обоих одновременно. Сердеч
ный выброс при застойной сердечной недостаточности обычно ниже нормальных значений. Снижение сократимости сердца связано с внутренним биохимическим дефектом и обычно хорошо поддается терапии средствами с положительным инотропным действием. Это типичная сердечная недостаточность: хроническая, возникающая из-за заболеваний коронарных артерий или гипертензии, или острая как следствие инфаркта миокарда. Значительно реже встречается недостаточность с высоким сердечным выбросом. В этих условиях требования организма к снабжению кислородом настолько велики, что даже увеличенный сердечный выброс их не обеспечивает. Сердечная недостаточность с высоким сердечным выбросом может возникнуть в результате гипертиреоидизма, болезни бери-бери, анемии и артериовенозных шунтов. Эта форма недостаточности плохо поддается терапии препаратами с положительным инотропным эффектом.
К основным признакам и симптомам всех типов застойной сердечной недостаточности относятся тахикардия, снижение переносимости физической нагрузки и одышка, периферические и легочные отеки и кардиомегалия. Неадекватный ответ на физическую нагрузку с быстрым возникновением мышечной слабости — это прямое следствие уменьшения сердечного выброса. Другие симптомы являются результатом попыток организма компенсировать внутренний сердечный дефект.
Нейрогуморальная рефлекторная (внешняя) компенсация включает в себя два основных механизма, рассмотренных в главе 6 и показанных на рис. 6-7. Это симпатическая нервная система и ре-нин-ангиотензин-альдостероновый гормональный ответ. У больных с сердечной недостаточностью барорецепторный рефлекс устанавливается на новый уровень регуляции с меньшей чувствительностью к артериальному давлению. В результате барорецепторный сенсорный вход в сосудодвигательный центр снижается даже при нормальном давлении; симпатический выход увеличивается, а парасимпатический — уменьшается (Ferguson, 1992). Усиление симпатической активности вызывает тахикардию, увеличивает сократимость сердца и сосудистый тонус. Повышенный артериальный тонус вызывает увеличение постнагрузки и уменьшает фракцию изгнания, сердечный выброс и почечную перфузию. Возрастание венозного тонуса способствует увеличению давления наполнения желудочков, расширению сердца и увеличению растяжения во
локон. Повышение секреции альдостерона приводит к задержке ионов натрия и воды с увеличением объема крови и в результате — к отеку. Возможно высвобождение и других гормонов.
Наиболее важным внутренним компенсаторным механизмом является гипертрофия миокарда. Увеличение мышечной массы помогает поддерживать сердечную деятельность при таких нежелательных условиях как нагрузка давлением или объемом, частичная потеря функциональных тканей (инфаркт миокарда) или снижение сократимости сердца. Однако после начального улучшения гипертрофия может вызывать ишемию, нарушение диастолического наполнения и изменение геометрии желудочков.
Патофизиология сердечной деятельности
Сердечная деятельность зависит от четырех основных факторов.
А. Преднагрузка. Изменение преднагрузки (давление в предсердиях) весьма существенно сказывается на сердечно-сосудистой деятельности. Если такие показатели деятельности левого желудочка как ударный объем или ударная работа представить как функцию давления наполнения левого желудочка, результирующая кривая будет графиком функции левого желудочка (рис. 13-2). Восходящий участок (давление наполнения < 15 мм рт. ст.) представляет собой классическую зависимость Франка-Старлинга. При давлении приблизительно >15 мм рт. ст. наблюдается плато. Преднагрузка выше чем 20-25 мм рт. ст. вызывает застой в легких. Как отмечалось ранее, преднагрузка при сердечной недостаточности обычно повышена вследствие увеличения объема крови и венозного тонуса. Ограничение потребления соли и терапия мочегонными средствами при застойной сердечной недостаточности направлены на уменьшение давления наполнения левого желудочка. Средства, расширяющие вены (например, нитроглицерин), также снижают преднагрузку, перераспределяя кровь из органов грудной клетки в периферические вены.
Б. Постнагрузка. Постнагрузка — это сопротивление, против которого сердце должно перекачивать кровь. Оно представлено аортальным и системным сосудистым сопротивлением. У больных с застойной сердечной недостаточностью системное
сосудистое сопротивление часто увеличивается. Так как сердечный выброс при хронической недостаточности резко снижается, наблюдается рефлекторное повышение системного сосудистого сопротивления, вызываемое частично увеличением симпатического тонуса и циркулирующих катехоламинов и частично — активацией ренин-ангиотензино-вой системы. Увеличение сосудистого сопротивления в свою очередь может способствовать дальнейшему уменьшению сердечного выброса посредством увеличения сопротивления изгнанию. Это дает основание для применения при застойной сердечной недостаточности средств, понижающих артериолярный тонус.
В. Сократимость. У больных с хронической недостаточностью со сниженным сердечным выбросом основным дефектом является уменьшение собственной сократимости миокарда. При ослаблении сократительной функции замедляется мышеч-
1U ги 30 40
Давление наполнения ЛЖ (мм рт. ст.)
Рис. 13-2. Зависимость работы левого желудочка от давления наполнения у больных с острым инфарктом миокарда. Заштрихованная область — диапазон, характерный для здоровых людей. После инфаркта зависимость может изменяться в широких пределах. У некоторых больных производительность сохраняется на нормальном или повышенном уровне (гиперреактивность). С увеличением размера инфаркта кривая сдвигается вниз и вправо. Сходное подавление наблюдается у больных с хронической застойной сердечной недостаточностью. (Из: Swan Н. J. С., Parmley W. W. Congestive heart failure. In: Pathologic Physiology. Sodeman W. A. Jr., Sodeman T. M. (eds) Saunders, 1979.)
ное укорочение и уменьшается скорость нарастания внутрижелудочкового давления (AP/At). В результате нарушается насосная функция сердца, что сопровождается развитием сердечной недостаточности, например после инфаркта миокарда (рис.13-2). Однако сократимость сердца при этом способна повышаться в ответ на введение средств с положительным инотропным эффектом.
Г. Частота сердечных сокращений. Частота сердечных сокращений — основная детерминанта сердечного выброса. Поскольку при недостаточности ослабляются внутренние функции сердца и уменьшается ударный объем, увеличение частоты сердечных сокращений посредством симпатической активации 3-адренорецепторов является первым компенсаторным механизмом, который включается для поддержания сердечного выброса.
I. Базисная фармакология средств, используемых при застойной сердечной недостаточности
Препараты наперстянки
Лечебные растения, содержащие сердечные гликозиды, были известны древним египтянам 3000 лет назад, однако эти средства использовались непостоянно и с переменным успехом до XVIII в., пока английский врач и ботаник Вильям Визеринг не опубликовал монографию, описывающую клини
ческие эффекты экстракта из наперстянок (в основном из Digitalis purpurea). В его книге “О наперстянке и ее медицинском применении с практическими замечаниями о водянке и других болезнях” (Ап Account of the Foxglove and Some of Its Medical Uses: With Practical Remarks on Dropsy and Other Diseases), опубликованной в 1785 г., детально рассказывалось о показаниях к применению сердечных гликозидов и упоминалось об их токсичности.
Химия
Все используемые обычно сердечные гликозиды, или карденолиды, прототипом которых служит дигоксин (рис. 13-3), состоят из стероидного ядра с ненасыщенным пятичленным лактонным кольцом в семнадцатом положении и сахаров, связанных с агликоном через углерод в третьем положении. Из-за отсутствия легко ионизирующихся групп растворимость сердечных гликозидов не зависит от pH.
Источниками этих веществ являются белая и пурпурная наперстянка (Digitalis lanata and D. purpurea), Strophantus gratus, средиземноморский лук, олеандр, ландыш, молочай и многие другие растения тропической и умеренной зон. Некоторые жабы имеют кожные железы, способные вырабатывать буфадиенолиды, отличающиеся от карденолидов только наличием шестичленного (а не пятичленного) лактонного кольца в семнадцатом положении.
Исследования зависимости между структурой и действием этих средств показали, что лактонное кольцо и стероидное ядро в равной мере необходимы для проявления активности. Другие заместители, особенно сахара в третьем положении, влияют
Дигоксин
Рис. 13-3. Структура типичного сердечного гликозида дигоксина
на фармакокинетические показатели, а именно всасывание, период полувыведения и метаболизм. Отсутствуют клинические доказательства того, что какой-либо из природных карденолидов отличается от других по терапевтическому индексу или максимальной эффективности.
Фармакокинетика
А. Всасывание и распределение. Карденолиды имеют липофильные (стероидное ядро) и гидрофильные (лактонное кольцо, гидроксил, сахар) группы. Как показывает сравнение трех представителей группы — дигоксина, дигитоксина и оубаи-на (табл. 13-1), баланс этих двух факторов имеет важное значение для всасывания, распределения, метаболизма и выведения. Наиболее широко применяется дигоксин.
Дигоксин хорошо всасывается после внутреннего применения. Однако примерно у 10 % пациентов имеются кишечные бактерии, которые инактивируют дигоксин в кишечнике, значительно уменьшая биодоступность и вынуждая принимать поддерживающую дозу выше средней. При лечении таких больных антибиотиками может произойти неожиданное увеличение биодоступности и токсичности дигоксина. Диапазон безопасных доз у сердечных гликозидов очень узок, поэтому даже незначительные изменения биодоступности могут привести к сильному отравлению или к потере эффективности. Показано, что лекарственная форма (из-за особенностей технологии изготовления таблеток) может модифицировать биодоступность дигоксина. В связи с этим препараты дигоксина проходят фармацевтическое тестирование до их продажи в США. Эти ограничения не касаются диги
токсина, так как он хорошо всасывается независимо от условий. Полусинтетическое производное дигоксина — р-метилдигоксин, широко используемый во многих странах,— почти полностью абсорбируется в кишечнике и метаболизируется в организме до дигоксина. Десланозид тоже очень похож на дигоксин (он содержит одну дополнительную группу сахара).
После всасывания в кровь все сердечные гликозиды широко распределяются в тканях, включая центральную нервную систему. Объемы распределения разных средств отличаются и зависят от степени их связывания с белками плазмы. Самая высокая концентрация дигоксина в сердце, почках и печени. В тканях этих органов она в 10-50 раз выше, чем в плазме.
Б. Метаболизм и выделение. Дигоксин мало подвергается метаболизму у человека и в значительной степени выделяется в неизмененном виде почками. Его общий клиренс хорошо коррелирует с клиренсом креатинина и сильно замедляется у больных с заболеваниями почек. Для регуляции дозы дигоксина существуют специальные уравнения и номограммы.
Дигитоксин метаболизируется в печени и выделяется в кишечник с желчью. Его кардиоактивные метаболиты (в том числе дигоксин) так же, как и неизмененный дигитоксин, могут вновь всасываться из кишечника, участвуя в энтеропеченочной циркуляции, которая увеличивает продолжительность периода полувыведения этого средства. Нарушения деятельности почек практически не влияют на период полувыведения дигоксина, поэтому он может назначаться больным с нарушенной или быстро ухудшающейся почечной функцией. С другой стороны, многие препараты, активирующие ферменты
ТАБЛИЦА 13-1. Свойства трех типичных сердечных гликозидов
Оубаин1 Дигоксин Дигитоксин
Растворимость в жирах Низкая Средняя Высокая
(коэффициент масло/вода) Доступность при внутреннем приеме 0 75 >90
(процент всасывания) Период полувыведения(часы) 21 40 168
Связывание с белками плазмы 0 20-40 >90
(процент связывания) Процент метаболизации 0 < 20 >80
Объем распределения (л/кг) 18 6.3 0.6
1 Оубаин как лекарственное средство в США больше не применяется (дополнение “Эндогенные сердечные гликозиды”).
печени (глава 4), могут уменьшать уровень дигоксина в крови, ускоряя его метаболизм.
Оубаин следует вводить парентерально, поэтому его применяют только для неотложной терапии и редко назначают больше нескольких раз. Это средство метаболизируется в малой степени и выводится в основном почками.
Фармакодинамика
Препараты наперстянки обладают множеством прямых и непрямых сердечно-сосудистых эффектов, имеющих как терапевтические, так и токсические (аритмии) последствия. К тому же наблюдается нежелательное влияние на центральную нервную систему и кишечник. Продемонстрировано незначительное прямое действие на почки (диуретический эффект).
На молекулярном уровне все сердечные гликозиды угнетают Ка+,К+-АТФазу — связанный с мембраной транспортер, часто называемый “натриевым насосом”. Идентифицировано несколько изоформ этого фермента. Показано, что они имеют разный аффинитет к сердечным гликозидам. Очень низкие концентрации сердечных гликозидов иногда стимулируют этот фермент. Напротив, в пределах обычных доз доказано их угнетающее действие во всех исследованных тканях. Вероятно, именно этот эффект в основном обусловливает терапевтическое (положительное инотропное) действие на сердце. Поскольку натриевый насос необходим для поддержания нормального потенциала покоя в большинстве возбудимых клеток, токсичность препаратов наперстянки, видимо, также частично связана с их ингибирующим влиянием на фермент. Факт существования рецепторов для сердечных гликозидов на натриевом насосе побудил исследователей предположить наличие эндогенных “диги-талисподобных” веществ (дополнение “Эндогенные сердечные гликозиды”).
А. Действие на сердце. Механизмы влияния препаратов наперстянки на механическую и электрическую функции сердца обсуждаются раздельно, но нужно помнить, что эти процессы происходят в сердце одновременно.
1. Механические эффекты. Терапевтическое действие сердечных гликозидов на механическую функцию сердца заключается в увеличении интенсивности взаимодействия нитей актина и миозина в сердечном саркомере (рис.13-1).Оно происходит
вследствие повышения концентрации свободного кальция вблизи сократительных белков во время систолы. Увеличение концентрации Са2+ является результатом двухэтапного процесса. На первом этапе возрастает внутриклеточный уровень ионов натрия вследствие угнетения На+,К+-АТФазы (1 на рис. 13-1), а на втором ингибируется выведение Са2+ из клетки посредством натрий-кальциевого обмена (2 на рис. 13-1) в связи с увеличением концентрации внутриклеточного Na+. Этот известный механизм, как полагают, могут дополнять два других: облегчение входа Са2+ через потенциалзависимые кальциевые каналы мембраны (3 на рис. 13-1) и воздействие на саркоплазматический ретикулум, результатом которого является активация выделения Са2+ из внутриклеточных депо (5 на рис. 13-1).
Итоговый результат действия сердечных гликозидов в терапевтических концентрациях — отчетливое увеличение сердечной сократимости (рис. 13-4, нижняя запись). В изолированных препаратах миокарда повышается скорость развития как напряжения, так и расслабления с небольшим изменением времени максимального напряжения или с его отсутствием. Продолжительность сократительного ответа не уменьшается (как в случае стимуляции p-адренорецепторов) и не увеличивается (как в случае метилксантинов, таких как теофиллин). Эти эффекты наблюдаются и в нормальном, и в ослабленном миокарде, но в здоровом организме ответы модифицируются сердечно-сосудистыми рефлексами, а у больных или у животных с экспериментальным инфарктом — патофизиологическими механизмами, характерными для застойной сердечной недостаточности.
2. Электрические эффекты. Действие препаратов наперстянки на электрические свойства сердца в интактном организме представляет собой сочетание прямого эффекта и влияния на автономную нервную систему. Прямые эффекты на мембраны клеток сердца развиваются в определенной последовательности — раннее короткое удлинение потенциала действия, за которым следует затяжной период укорочения (особенно фазы плато). Это укорочение длительности потенциала действия, вероятно, является результатом повышения калиевой проводимости, вызываемой увеличением уровня внутриклеточного Са2+. (Влияние ионной проводимости на потенциал действия обсуждается в главе 14.) Эти эффекты могут не сопровождаться проявлениями токсичности. Укорочение потенциала
Эндогенные сердечные гликозиды
Имеют ли все рецепторы регуляторных и транспортных систем эндогенные лиганды наряду с экзогенными, которые мы используем как лекарственные средства? Одни биологи отвечают на этот вопрос утвердительно, доказывая, что эти регуляторы важных процессов сами требуют эндогенных регуляторов. Другие исследователи утверждают, что всегда существует вероятность связывания некоторых совершенно чужеродных лигандных молекул из-за сложной структуры белков. Кроме того, если связывание происходит в критической зоне протеина (активном или аллостерическом центре фермента или др.), то высоко вероятны нарушения функции этого фермента или транспортера, вызывающие фармакологический эффект.
Рецептор для сердечных гликозидов — подходящий случай для обсуждения этого вопроса. Именно высокая аффинность Na+,К?-АТФазы к сердечным гликозидам заставляет предположить возможность наличия эндогенных лигандов похожей структуры. Подобно адренокортикальным гормонам сердечные гликозиды имеют стероидное ядро, но стереохимия кольца существенно отличается от типичных производных холестерина. В то время как многие растения синтезируют эти гликозиды, в животном мире, как полагают, только несколько амфибий (ядовитые жабы) обладают такой способностью.
Результаты большого количества недавно проведенных исследований (рассмотренных в обзоре Blaustein, 1993) с применением чрезвычайно чувствительных иммунохимических методов показали, что оубаин синтезируется надпочечниками и, возможно, мозгом человека и
других млекопитающих. Утверждается, что в определенных обстоятельствах, таких как большое потребление соли и сердечная недостаточность, оубаин выделяется как эндокринное и паракринное вещество. В течение почти 20 лет постулировалось, что некоторые формы гипертензии связаны с вазоконстрикцией, вызываемой эндогенным ингибитором натриевого насоса. Повышенная секреция эндогенного оубаина, обусловленная потреблением соли в больших количествах, может быть важным звеном этой гипотезы. Считают, что при застойной сердечной недостаточности уровень оубаина в плазме положительно коррелирует с тяжестью заболевания.
Однако существуют химические и физиологические причины, подвергающие сомнению эти положения (Kelly, 1992). Даже если спорные наблюдения подтвердятся, многие гипотезы о роли эндогенного оубаина еще потребуют объяснения. Наблюдая повышение уровня оубаина в плазме у больных с сердечной недостаточностью, мы, вероятно, можем заключить, что недостаточность (по крайней мере, у некоторых больных) не вызывается первоначально дефицитом оубаина. Если оубаин действительно усиленно синтезируется у больных с сердечной недостаточностью, то, что это означает: продолжающееся нарастание болезни до стадии, требующей терапии экзогенными гликозидами, неадекватность эндогенного запаса, развитие толерантности или некоторые другие нарушения?
Тем не менее возможно, что еще один рецептор может иметь интересный эндогенный лиганд, демонстрирующий конвергентную эволюцию растений, земноводных и млекопитающих.
действия способствует укорочению предсердной и желудочковой рефрактерности (табл. 13-2).
При использовании более токсических концентраций мембранный потенциал покоя уменьшается (становится менее отрицательным) в результате угнетения натриевого насоса и уменьшения концентрации внутриклеточного К+. При нарастании токсического эффекта за нормально вызванными потенциалами действия следуют колебательные де
поляризующие следовые потенциалы (рис. 13-4, Б). Эти потенциалы (известные как “поздняя следовая деполяризация") ассоциируются с усилением депонирования Са2+ в клетке и колебаниями концентрации свободного внутриклеточного кальция. При подпороговой величине следовые потенциалы могут препятствовать нормальной передаче вследствие дальнейшего уменьшения потенциала покоя. Наконец, постпотенциал может достичь порога,
Сокращение Змг
100 мсек
Рис. 13-4. Влияние сердечного гликозида оубаина на изолированную ткань сердца. Верхняя запись демонстрирует вызванные потенциалы действия во время контрольного периода, в ранней “терапевтической” фазе и позднее, когда наблюдается токсическое действие. Средняя запись показывает величину излучения кальцийопределяющего белка экворина (относительно к максимально возможному, л/л„акс); этот показатель приблизительно пропорционален концентрации свободного внутриклеточного Са2+. На нижней записи представлено напряжение, вызванное потенциалом действия. В ранней фазе действия оубаина (А) наблюдается небольшое укорочение потенциала действия, заметное увеличение концентрации свободного внутриклеточного Са2+ и напряжения сокращения. Токсическая фаза (Б) связана с деполяризацией потенциала покоя, заметным укорочением потенциала действия и появлением осцилятор-ных деполяризаций, нарастания уровня Са2+ и сокращения (стрелки). (Неопубликованные данные, любезно предоставленные Р. Hess и Н. Gil Wier.)
вызывая потенциал действия (преждевременная желудочковая деполяризация, или “эктопическое биение”), соединяющийся с нормальным предшествующим. Если постпотенциалы в проводящей системе Пуркинье регулярно достигают порог таким образом, на ЭКГ будет наблюдаться бигеминия (рис. 13-5). При нарастании токсического эффекта каждый вызванный следовым потенциалом потенциал действия сам по себе вызывает подпороговый постпотенциал, и устанавливается самоподдержи-вающаяся аритмия (желудочковая тахикардия). Если не вмешаться в этот процесс (позволить состоянию ухудшаться), тахикардия может перейти в фибрилляцию желудочков.
Автономные влияния сердечных гликозидов на сердце затрагивают и парасимпатическую, и симпатическую системы и проявляются в терапевтическом и в токсическом диапазонах доз. При введении более низких доз преобладают кардиоселективные парасимпатомиметические эффекты. Фактически эти блокируемые атропином реакции ответственны за значительную часть ранних электрических эффектов препаратов наперстянки (табл. 13-2). Сюда относятся сенситизация барорецепторов, центральная стимуляция вагуса и облегчение передачи в мышечных клетках сердца. Поскольку холи
нергическая иннервация значительно богаче представлена в предсердиях, эти влияния сильнее затрагивают функцию атриального и атриовентрикулярного узлов, чем функционирование системы Пуркинье и желудочков. Некоторые холиномиметические эффекты могут использоваться для терапии определенных аритмий. В токсическом диапа-
НСР ПЖС НСР ПЖС
Vg
st
Рис. 13-5. Запись ЭКГ, демонстрирующая вызванную препаратами наперстянки бигеминию. Комплексы, обозначенные НСР, представляют собой нормальный синусовый ритм: наблюдаются инвертированная волна Т и снижение сегмента ST. Комплексы, обозначенные ПЖС, являются преждевременными желудочковыми сокраще.-ниями (желудочковыми экстрасистолами). Это электрокардиографические проявления деполяризаций, вызван? ных задержанными осцилляторными постпотенциаламй, как показано на рис. 13-4. (Из: Goldman М. J. Principles of Clinical Electrocardiography, 12th ed. Lange, 1986.)
ТАБЛИЦА 13-2. Основные влияния препаратов наперстянки на электрические функции сердца (ПСЭ — парасимпатические эффекты. Прямое — прямые влияния на мембрану)
Переменные Ткани
Мышца предсердия АВ-узел Система Пуркинье, желудочки
Эффективный рефрактерный период Скорость проведения Автоматизм Электрокардиограмма Перед аритмиями Аритмии Г (ПСЭ) Т(ПСЭ) Т (ПСЭ) Г (ПСЭ) Т (Прямое) Т (Прямое) Незначительное Тинтервала PR Предсердная тахикардия, АВ-узловая тахикардия, фибрилляция предсер- АВ-блокада дий X(Прямое) Незначительное ? (Прямое) i интервала QT; инверсия волны Т; снижение сегмента ST Желудочковые экстрасистолы, бигеминия, желудочковая тахикардия, фибрилляция желудочков
зоне доз препараты наперстянки увеличивают симпатический выход. Этот эффект не существенен для типичной карденолидной токсичности, но сенсити-зирует миокард и усиливает все токсические эффекты гликозидов.
Наиболее обычные проявления токсичности гликозидов — атриовентрикулярный узловой ритм, преждевременная деполяризация желудочков, би-геминия и атриовентрикулярная блокада второй степени. Однако считается, что препараты наперстянки могут вызывать фактически все многообразие аритмий. Мембранная кардиотоксичность препаратов наперстянки, рассмотренная ранее, включает деполяризацию покоя (из-за угнетения натриевого насоса) и осцилляторные следовые деполяризации (вызванные перегрузкой внутриклеточным Са2+). Деполяризация потенциала покоя может быть ответственна за часть нарушений проводимости, таких как атриовентрикулярная блокада. Постдеполяризация, вероятно, может провоцировать большинство гликозидных аритмий, в том числе аномальный автоматизм с экстрасистолией, биге-минию и желудочковую тахикардию.
Б. Влияние на другие органы. Сердечные гликозиды влияют на все возбудимые ткани, включая гладкие мышцы и центральную нервную систему. Механизм этих эффектов полностью не исследован, но, вероятно, заключается в угнетении активности №*,К+-АТФазы в этих тканях. Деполяризация, вызываемая ингибированием натриевого насоса, как и можно ожидать, увеличивает спонтанную актив
ность нейронов и гладкомышечных клеток. К тому же повышение уровня внутриклеточного Са2+ может повысить тонус гладких мышц. Это случается при быстром внутривенном введении дигоксина. Однако нужно помнить, что в большинстве случаев застойной сердечной недостаточности, успешно леченной препаратами наперстянки, итоговым эффектом является снижение сосудистого тонуса, возникающее вследствие уменьшения симпатического тонуса.
Наиболее обычной зоной проявления внесердеч-ной токсичности препаратов наперстянки является желудочно-кишечный тракт. Анорексия, тошнота, рвота и диарея могут быть отчасти следствием непосредственного действия гликозидов на желудочно-кишечный тракт, а также результатом их влияния на центральную нервную систему, включая стимуляцию хеморецепторной триггерной зоны.
Воздействие на центральную нервную систему, как уже обсуждалось ранее, обычно заключается в стимуляции вагуса и хеморецепторной зоны. Значительно реже отмечаются дезориентация й галлюцинации (у пожилых) и нарушения зрения. В последнем случае может наблюдаться аберрация цветового восприятия. Сообщалось об ажитации и даже конвульсиях у больных, принимающих препараты наперстянки.
Гинекомастия относится к редким побочным эффектам, отмеченным у мужчин, принимающих препараты наперстянки. Неясно, связана ли она с периферическим эстрогенным действием этих сте
роидных средств или является проявлением гипоталамической стимуляции.
В эксперименте показано, что такие АТФаза-зависимые транспортные процессы как продукция водянистой влаги, цереброспинальной жидкости или реабсорбция натрия в почках угнетаются кар-денолидами. Но эти эффекты не имеют клинического значения.
В. Взаимодействие с калием, кальцием и магнием. Концентрации ионов калия и кальция во внеклеточном пространстве (обычно измеряемые как уровни К+ и Са2+ в сыворотке) имеют важное значение для определения чувствительности пациента к препаратам наперстянки. Калий и препараты наперстянки взаимодействуют двумя путями. Во-первых, они взаимно угнетают связывание с Na+,K+-ATOa-зой. Следовательно, гиперкалиемия ослабляет ингибирование фермента сердечными гликозидами, в то время как гипокалиемия облегчает этот эффект. Во-вторых, гиперкалиемия подавляет аномальный сердечный автоматизм (глава 14), поэтому умеренное увеличение внеклеточного К+ уменьшает эффекты препаратов наперстянки, особенно токсические. Ион кальция облегчает возникновение токсических эффектов сердечных гликозидов, ускоряя переполнение внутриклеточных депо Са2+, что, невидимому, приводит к возникновению ненормального автоматизма. Значит, гиперкальциемия повышает риск аритмий, вызываемых сердечными гликозидами. Эффекты иона магния прямо противоположны действию Са2+. Гипомагниемия, следовательно,— фактор риска развития аритмий. Рассмотренные взаимодействия требуют обязательного определения электролитов сыворотки у больных с аритмиями, вызванными сердечными гликозидами.
Другие средства положительного инотропного действия, применяемые при сердечной недостаточности
Для лечения сердечной недостаточности давно применяются средства, угнетающие активность фосфодиэстераз — семейства ферментов, инакти
вирующих цАМФ и цГМФ. Хотя они имеют положительный инотропный эффект, их эффективность прежде всего связана с вазодилатацией, что еще будет обсуждаться. Хорошо известный метилксантин аминофиллин, способный ингибировать фосфодиэстеразу, часто применялся в терапии острого отека легких. Он не представляет ценности для лечения больных с хронической застойной сердечной недостаточностью и в настоящее время редко применяется в неотложной терапии. Новые ингибиторы фосфодиэстеразы интенсивно изучались в течение нескольких десятилетий, но пока с неопределенными результатами. На сегодняшний день из этой группы наиболее близки к внедрению в практику бипиридины амринон и милринон, но их использование весьма ограничено. Недавно был предложен как кандидат для замены препаратов наперстянки флозехинан, производное фторохинолона со смешанным положительным инотропным и сосудорасширяющим действием, однако в дальнейшем он был изъят из употребления из-за высокой токсичности. Клинические испытания показали, что средство с неизвестным механизмом действия — весна-ринон — может значительно уменьшать риск смерти больных с сердечной недостаточностью (дополнение “Флозехинан и веснаринон”). Для замены препаратов наперстянки применяется также группа стимуляторов p-адренорецепторов.
Бипиридины
Амринон и милринон — бипиридиновые соединения, которые могут назначаться внутрь или парентерально, но выпускаются только в формах для парентерального введения. Период полувыведения 2-3 часа, 10-40 % выводится с мочой.
Фармакодинамика
Бипиридины увеличивают сократимость миокарда без угнетения Ка+,К+-АТФазы или активации адренорецепторов. Они увеличивают входящий кальциевый ток в сердце во время потенциала действия и изменяют внутриклеточные передвижения Са2+, воздействуя на саркоплазматический ретикулум. Они также имеют значительный сосудорасширяющий эффект, который, без сомнения, усиливает лечебное действие при сердечной недостаточности. Биохимические исследования показывают, что большинство их эффектов опосредуется угнетени
ем фосфодиэстеразы (аналогично действию метил-ксантинов). Эти средства и некоторые другие новые ингибиторы фосфодиэстеразы относительно селективны для изофермента III фосфодиэстеразы. Эта форма свойственна сердечной и гладким мышцам. Угнетение активности изофермента вызывает повышение цАМФ, увеличение сократимости и вазодилатацию.
У пациентов с острой сердечной недостаточностью бипиридины увеличивают сердечный выброс и уменьшают давление заклинивания легочных капилляров и периферическое сосудистое сопротивление. Частота сердечных сокращений и артериальное давление меняются мало. Хотя острые эффекты этих препаратов безусловно благоприятны, их токсичность мешает длительному применению.
Токсическое действие амринона проявляется относительно частыми тошнотой и рвотой. Есть сообщения о развитии тромбоцитопении и изменении функции печеночных ферментов у небольшого, но все-таки значительного числа больных. Этот препарат используют только для кратковременного лечения и вводят парентерально. Возникновение аритмий при его применении менее вероятно, чем при лечении препаратами наперстянки. Милринон в меньшей степени, чем амринон, оказывает токсическое действие на костный мозг и на печень, но может вызвать аритмии. В плацебоконтролируемых исследованиях у больных с тяжелой степенью сердечной недостаточности милринон при приеме внутрь увеличивал летальность (Packer et al, 1991). Поэтому, как и амринон, препарат в настоящее время применяют только внутривенно и только при острой сердечной недостаточности.
Стимуляторы 0-адренорецепторов
Общая фармакология этих веществ обсуждается в главе 9. Поиск средств, обладающих положительным инотропным действием с меньшим арит-могенным потенциалом, чем наперстянка, и меньшей способностью учащать сердечные сокращения, чем изопротеренол, привел к созданию р!-селективных препаратов. В то же время успешное применение вазодилататоров при застойной сердечной недостаточности привлекло внимание к воможности .использования селективных р2-агентов в терапии этого заболевания.
Избирательным р(-агонистом, который наиболее широко применяется у больных с сердечной недостаточностью, является добутамин. Этот препарат вызывает увеличение сердечного выброса и одновременно понижает давление наполнения желудочков. Но есть сообщения о развитии незначительной тахикардии и повышении потребности миокарда в кислороде. Таким образом, хотя добутамин и увеличивает сократительную способность миокарда, неясно, какое же преимущество в итоге он может дать больным с ишемической болезнью сердца. Кроме того, необходимо учитывать его способность провоцировать аритмии или стенокардию, также как и тахифилаксию, характерную для всех |3-стимуляторов. Некоторым больным с хронической сердечной недостаточностью могут помогать прерывистые инфузии добутамина.
Был изучен ряд других веществ, влияющих на Р-адренорецепторы и дофаминовые рецепторы. Большинство из них, подобно допамину и добута-мину, оказались неактивны при приеме внутрь, поэтому их относят к препаратам резерва для лечения острой сердечной недостаточности или при рефрактерное™ к средствам, принимаемым внутрь.
Средства без положительного инотропного эффекта, применяемые при сердечной недостаточности
Наиболее часто при хронической сердечной недостаточности применяются сердечные гликозиды, диуретики и ингибиторы ангиотензинпревращаю-щего фермента. При острой недостаточности нашли широкое применение диуретики и вазодилататоры. Таким образом, вещества без положительного инотропного эффекта на сердце играют важную роль в терапии сердечной недостаточности.
Диуретики
Диуретики подробно рассматриваются в главе 15. Механизм их действия при сердечной недостаточности состоит в уменьшении задержки воды
Флозехинан и веснаринон
Сложности на пути создания новых эффективных препаратов для лечения застойной сердечной недостаточности хорошо иллюстрируются историей двух новых лекарств.
Флозехинан — активное при приеме внутрь фторохиноловое соединение длительного действия. Он имеет определенный сосудорасширяющий эффект и, возможно, положительное инотропное действие. Хотя препарат во многих отношениях напоминает бипиридины, механизм его действия неизвестен. Он является слабым и неселективным ингибитором фосфодиэстеразы, не сенсибилизирует миофибриллы сердца к Са2+ и не изменяет активность №+,К+-АТФазы и концентрацию цАМФ. При начальных клинических испытаниях лекарство казалось весьма многообещающим и было одобрено FDA для использовании в США. Рекомендовали начинать лечение с дозы 50 мг в день, постепенно увеличивая ее до 100 мг в день (максимум). Цосле того как лекарство начали применять, обнаружилось, что смертность больных, принимающих 100 мг в день, возросла, поэтому было ойубликовано предупреждение о необходимости избегать высшей дозы, кроме случаев отсутствия реакции на другие лекарства и более низкие дозы. Объем продаж флозехинана, даже для использования в меньших дозах, немедленно и настолько резко снизился, что производители прекратили его поставку на рынок.
Веснаринон — хинолоновое производное, активное при энтеральном приеме. В опытах на изолированной мышце сердца он проявил ряд эффектов, в том числе подавление активности фосфодиэстеразы, увеличение тока ионов каль
ция и удлинение потенциала действия. Каждый из них теоретически должен способствовать инотропному действию (раздел “Контроль сердечной сократимости”). Торможение фосфодиэстеразы может также вызывать вазодилатацию с полезнрй разгрузкой перегруженных желудочков. Несколько клинических испытаний этого препарата (например, Feldman et al, 1993) показали, что он может значительно понижать заболеваемость и смертность на протяжении по меньшей мере 25 недель. Последнее исследование продемонстрировало, однако, что терапевтическая широта препарата мала: если прием 60 мг в день способствует снижению смертности, то прием 120 мг значительно повышает число летальных исходов. Неясно, какой из возможных механизмов действия реализуется при более низкой дозе. Если данные о клинической эффективности веснаринона будут подтверждены при более обширных испытаниях, может возникнуть необходимость поиска иного механизма действия этого препарата при сердечной недостаточности.
Эти данные соответствуют известному положению о том, что при развившейся сердечной недостаточности сердце очень чувствительно к чрезмерной стимуляции. Поэтому лекарства, которые вызывают необходимое увеличение сократительной способности сердца, действуют около абсолютных границ клинической безопасности. Лекарства с положительным инотропным эффектом в этом плане особенно опасны. В настоящее время ни флозехинан, ни веснаринон не применяются в общей клинической практике в США.
и соли и, тем самым, в снижении преднагрузки желудочков. При понижении венозного давления наблюдаются два полезных эффекта: ослабление признаков и симптомов недостаточности (отек) и уменьшение размеров сердца, что приводит к повышению эффективности насосной функции.
Ингибиторы ангиотензин-превращающего фермента
АПФ-ингибиторы рассматриваются в главах 11 и 17. Эти лекарства, обладающие многосторонним действием, снижают уровень ангиотензина II, что уменьшает периферическое сопротивление и, следовательно, постнагрузку. Ингибируя секрецию альдостерона, они устраняют задержку соли и воды
и тем самым снижают преднагрузку. Понижение уровня ангиотензина в тканях способствует также ослаблению симпатических влияний, возможно за счет уменьшения пресинаптического действия ангиотензина на высвобождение норадреналина.
Вазодилататоры
Вазодилататоры эффективны при сердечной недостаточности, так как они уменьшают преднагрузку (за счет венодилатации), снижают постнагрузку (расширяя артериолы) или оказывают оба эффекта одновременно.
II. Клиническая фармакология средств, применяемых
при застойной сердечной недостаточности
Наперстянка при застойной сердечной недостаточности
В связи со своим умеренным, но стабильным положительным инотропным эффектом наперстянка теоретически может ослаблять проявления симптомов и признаков застойной сердечной недостаточности. У таких больных препараты наперстянки повышают ударный объем и сердечный выброс. Повышение сердечного выброса (и, возможно, прямое действие, регулирующее чувствительность барорецепторов) устраняет стимулы, повышающие симпатический тонус, и уменьшает частоту сердечных сокращений и сосудистый тонус. В связи со
снижением конечного диастолического напряжения волокон (в результате повышения систолического выброса и снижения давления наполнения) размеры сердца и потребность миокарда в кислороде снижаются. Наконец, повышение почечного кровотока улучшает клубочковую фильтрацию и понижает контролируемую альдостероном реабсорбцию натрия. Таким образом, отечная жидкость может выводиться, способствуя дальнейшему уменьшению преднагрузки желудочков и риска отека легких.
Введение и дозирование
При длительной терапии препаратами наперстянки необходимо уделять большое внимание фармакокинетике из-за длительного периода полувыведения (табл. 13-3). В соответствии с правилами, изложенными в главе 3, для достижения стационарного состояния при регулярной частоте приема может потребоваться время, равное 3-4 периодам полувыведения, т. е. примерно одной неделе для дигоксина и одному месяцу для дигитоксина. Поскольку очень важно не превысить диапазон терапевтической концентрации дигиталиса в плазме, такой медленный тип “дигитализации” является наиболее безопасным. Если нужен более быстрый эффект, дигитализация может быть быстро достигнута с помощью больших нагрузочных доз (разделенных на 3-4 части и принимаемых в течение 24-36 часов), за которыми следуют поддерживающие дозы. Обычные дозы Для взрослых представлены в табл. 13-3. Когда применяют быстрый (в три дозы) метод дигитализации, важно, чтобы состояние больного оценивали перед каждой очередной дозой. В большинстве медицинских центров применяются чувствительные методы измерения уровня дигоксина в плазме. Измерение проводят в том
ТАБЛИЦА 13-3. Клиническое применение сердечных гликозидов (показатели у взрослых с нормальной функцией печени и почек)
Показатель Дигоксин Дигитоксин
Период полувыведения 40 ч 168 ч
Терапевтическая концентрация в плазме 0.5-2 нг/мл 10-25 нг/мл
Токсическая концентрация в плазме > 2 нг/мл > 35 нг/мл
Дневная доза 0.125-0.5 мг 0.05-0.2 мг
(поддерживающая или при медленной дигитализации)
Доза при быстрой дигитализации 0.5-0.75 мг 0.2-0.4 мг
каждые 8 ч 3 раза каждые 12 ч Зраза
случае, если реакция больного не соответствует ожидаемой при данном режиме дозировки.
Взаимодействия
При назначении гликозидов следует учитывать возможность лекарственных взаимодействий. У всех больных имеется риск развития серьезной аритмии сердца, если возникает гипокалиемия, связанная с приемом диуретиков или с диареей. Более того, у больных, получающих дигоксин, есть риск его передозировки при назначении хинидина, который вытесняет дигоксин из мест связывания в тканях и уменьшает его почечный клиренс. Уровень гликозида в плазме может удвоиться в течение нескольких дней после начала терапии хинидином, и могут появиться симптомы интоксикации. Сходное взаимодействие описано и с другими лекарствами, в частности с нестероидными противовоспалительными средствами и блокаторами кальциевых каналов, но клинически у человека.оно не проявляется. Хинидин не изменяет объем распределения или степень связывания дигоксина с белками. Однако он может увеличивать период полувыведения гликозида. Как уже указывалось, антибиотики, изменяющие желудочно-кишечную флору, повышают биодоступность дигоксина примерно у 10 % больных. Наконец, вещества, которые высвобождают катехоламины, могут сенсибилизировать миокард к диги-талисиндуцируемым аритмиям.
Пониженная реакция на сердечные гликозиды может быть связана с трудноизлечимым и тяжелым заболеванием или с недисциплинированностью больного. У больных, принимающих одновременно холестирамин (глава 34), всасывание дигиталиса снижается. При гипертиреоидизме из-за уменьшения периода полузлиминации дозы препаратов наперстянки, необходимые для достижения терапевтического эффекта, обычно должны увеличиваться.
Другие показания к применению препаратов наперстянки
Дигиталис полезен при лечении предсердных аритмий в связи с кардиоселективным парасимпа-томиметическим эффектом. При трепетании предсердий угнетающее влияние препаратов на атриовентрикулярную проводимость помогает контролировать чрезмерно высокую частоту сокращения же
лудочков. Действие препарата на мускулатуру предсердий может способствовать превращению трепетания в мерцание с последующим снижением частоты сокращений желудочков. При фибрилляции предсердий это же “вагомиметическое” действие позволяет контролировать частоту их сокращения, улучшая наполнение и увеличивая сердечный выброс. Дигиталис используют и для ликвидации пароксизмальной предсердной и атриовентрикулярной узловой тахикардий. Пероральное или, при необходимости, осторожное внутривенное введение дигоксина позволяет быстро купировать такой приступ, возможно, вследствие вагомиметичес-кого действия. Доступность блокаторов кальциевых каналов и аденозина уменьшила популярность применения дигоксина в этих ситуациях.
В настоящее время дигиталис не является препаратом выбора при лечении аритмий, связанных с синдромом Вольфа-Паркинсона-Уайта, потому что он повышает вероятность проведения аритмо-генных импульсов из предсердий через альтернативные быстропроводящие предсердно-желудочковые пути. Он безусловно противопоказан больным с синдромом Вольфа-Паркинсона-Уайта при фибрилляции предсердий (глава 14).
Токсичность
Несмотря на известный риск, препараты наперстянки все еще широко используются. В большом многоцентровом исследовании 17-27 % больных с различными заболеваниями получали препараты наперстянки при поступлении, и у 5-25 % из них выявились токсичные эффекты, потребовавшие, по меньшей мере, прекращения лечения препаратами наперстянки.
Лечение отравлений дигиталисом, проявляющихся как нарушения зрения или желудочно-кишечные расстройства, обычно заключается просто в снижении дозы препарата. Если имеется сердечная аритмия, связанная с действием препаратов наперстянки, необходимо более энергичное вмешательство. При терапии выраженной интоксикации препаратами наперстянки следует осуществлять мониторинг концентрации дигиталиса и калия в сыворотке и ЭКГ. Должен быть откорригирован электролитный статус пациента. У больных, которые не реагируют на препарат достаточно быстро (в пределах 1-2 периодов полувыведения), следует проверить уровни ионов кальция, магния и калия. При
случайных преждевременных деполяризациях желудочков или коротких эпизодах бигеминии может быть достаточно назначения калиевых добавок и отмены сердечных гликозидов. Если аритмия более серьезна, могут потребоваться парентеральное введение калия и противоаритмические средства. Из имеющихся в арсенале противоаритмических препаратов предпочтительнее использовать лидокаин, фенитоин и пропранолол.
При очейь тяжелых интоксикациях препаратами наперстянки (к которым обычно относятся случаи суицидальных передозировок) уровень калия в сыворотке к моменту постановки диагноза, как правило, [повышен (вследствие выхода К+ из внутриклеточных депо). Более того, противоаритмические средства, которые вводят в этой ситуации, могут вызвать остановку сердца. Наилучшие результаты лечения таких больных получены при применении антител к гликозидам наперстянки. Эти антитела вырабатывают путем иммунизации баранов и, хотя ори специфичны для дигоксина, они также связываются с дигитоксином и помогают при тяжелой интоксикации любыми сердечными гликозидами.
Z Аритмии, вызванные препаратами наперстянки, часто ухудшаются при электроимпул ьсной терапии. Такой способ лечения следует держать в резерве на случай индуцированной гликозидами фибрилляции желудочков.
Лечение хронической сердечной недостаточности
Основные этапы лечения больных с хронической сердечной недостаточностью представлены в табл. 13-4. Уменьшение работы сердца — традиционная форма лечения, эффективная в большинстве случаев. Она включает снижение уровня двигательной активности и массы тела, а также, что особенно важно, контроль гипертензии. Следующий важный шаг — ограничение потребления натрия. Несмотря на наличие эффективных мочегонных средств, большую пользу приносит соблюдение бессолевой диеты.
Если имеются отеки, обычно используют мочегонные средства. Разумно начинать с тиазидных диуретиков, переходя по мере необходимости к более сильным препаратам. Фуросемид, буметанид, этакриновая кислота и метозалон являются очень
ТАБЛИЦА 13-4. Этапы лечения хронической сердечной недостаточности
1. Уменьшение нагрузки на сердце
а. Ограничение физической активности
б. Снижение веса
в. Контроль гипертензии
2. Ограничение потребления натрия
3. Ограничение потребления воды (требуется редко) 4. Назначение диуретиков
5. Назначение наперстянки1
6. Назначение вазодилататоров
7. Назначение новых инотропных препаратов ’ Некоторые врачи применяют ингибиторы ангиотензинпре-вращающего фермента перед дигиталисом.
эффективными диуретиками, которые следует держать в резерве для больных с резистентными отеками. Потеря натрия вызывает вторичную потерю калия, которая особенно опасна у больных, получающих препараты наперстянки. Следовательно, у таких пациентов необходимо периодически контролировать уровень электролитов сыворотки. Гипокалиемию можно лечить калиевыми добавками или назначением калийсберегающих диуретиков (глава 15).
При необходимости применять вещества, обладающие положительным инотропным эффектом, обычно выбирают сердечные гликозиды. Следует еще раз подчеркнуть, что препараты наперстянки потенциально токсичны и только примерно у 50 % больных с нормальным синусовым ритмом (как правило, с доказанной систолической дисфункцией) они улучшают состояние при застойной сердечной недостаточности. Лучшие результаты получают у больных с фибрилляцией предсердий. Если принимается решение о назначении гликозидов, в большинстве случаев выбирают дигоксин. Когда симптомы выражены нерезко, медленная дигитализация (табл. 13-3) более безопасна и столь же эффективна, как и быстрая. При умеренной симптоматике можно применять быстрый метод с пероральным приемом, но больного необходимо обследовать перед каждой дозой, обращая особое внимание на сердечный ритм. Если имеются сомнения в характере ритма сердца перед началом терапии или его изменениях в процессе дигитализации, следует регистрировать ЭКГ. Внутривенная дигитализация редко необходима при хронической сердечной недостаточности; она применяется только в стацио
нарных условиях при тщательном мониторном наблюдении.
Определение оптимального уровня дигиталис-ного эффекта может быть затруднительным. У больных с фибрилляцией предсердий снижение частоты сокращений желудочков является наилучшей мерой эффекта гликозида. У пациентов с нормальным, синусовым ритмом симптоматическое улучшение и уменьшение размеров сердца, частоты сердечных сокращений при нагрузке, венозного давления? или отеков указывает на достижение оптимального уровня лекарства в миокарде. К сожалению, ефе раньше могут появляться токсические эффект^. Если используют схему медленной диги-тализац'ии, пропуск одной дозы и/или уменьшение наполовину поддерживающей дозы часто позволяет перейти узкую границу между субоптимальной и токсической концентрациями. Измерение уровней гликозида в плазме полезно у больных, которые необычно рефрактерны или чувствительны к препаратам. Наиболее доступно определение концентрации дигоксина.
При хронической застойной сердечной недостаточности весьма полезны сосудорасширяющие средства. Удобна классификация, разделяющая препараты на селективные артериолярные дилятаторы, венозные дилятаторы и препараты с неизбирательным сосудорасширяющим действием (табл. 13-5). Выбор препарата должен основываться на симптомах, которые наблюдаются у больного, и показателях гемодинамики. Например, больным с высоким давлением наполнения, когда основным симптомом является одышка, для снижения давления наполнения и признаков застоя в легких наиболее показаны венодилататоры. Пациентам со слабостью и усталостью из-за низкого выброса левого желудочка для повышения сердечного выброса назначают артериолярные дилятаторы. У всех больных с тяжелой хронической недостаточностью, рефрактерных к лечению, проблема обычно связана с повышенным давлением наполнения и сниженным сердечным выбросом. При этих обстоятель
ствах требуется расширение как артериол, так и вен. В испытаниях V-HEFT сочетанное лечение гидралазином (расширение артериол) и изосорбида динитратом (расширение вен) способствовало продлению жизни в большей степени, чем при приеме плацебо больными, уже получающими наперстянку и диуретики (Cohn et al., 1986).
В исследовании по сравнению дигоксина и каптоприла как препаратов первой линии для лечения хронической сердечной недостаточности оба вещества вызывали сходные эффекты (Captopril-Digo-xin Multicenter Research group, 1988). В других масштабных испытаниях оценивали эналаприл в сравнении с плацебо или с другими вазодилататорами (CONSENSUS,1987; SOLVD Invesigators, 1991; Cohn et al., 1991). Эти исследования показали, что ингибиторы ангиотезинпревращающего фермента эффективнее, чем плацебо и другие сосудорасширяющие средства, и могут рассматриваться как препараты первой линии для лечения хронической застойной сердечной недостаточности вместе с диуретиками и дигиталисом. Однако больные, состояние которых было стабильным при назначении диуретиков, дигоксина и ингибиторов ангиотезинпревращающего фермента, могут почувствовать себя хуже, прекратив прием дигоксина. Эго означает, что ингибиторы ангиотезинпревращающего фермента в ряде случаев не могут полностью заменить наперстянку (Packer et al., 1993).
Два недавних исследования доказали, что ингибиторы ангиотезинпревращающего фермента также являются ценными средствами для бессимптомных больных с дисфункцией желудочков (Pfeffer et al., 1992; SOLVD Investigators, 1992). Уменьшая пред- и постнагрузку, эти вещества замедляют скорость дилатации желудочков и тем самым отсрочивают начало клинических проявлений застойной сердечной недостаточности. Таким образом, ингибиторы АПФ эффективны при лечении пациентов с разной степенью тяжести заболевания: от бессимптомной до тяжелой сердечной недостаточности.
ТАБЛИЦА 13-5. Вазодилататоры, применяемые при застойной сердечной недостаточности
Артериолярные дилататоры Комбинированные артериолярные и венозные дилататоры Венозные дилататоры
Гидралазин Миноксидил Каптоприл Эналаприл Лизиноприл Нитраты
Лечение острой сердечной недостаточности
Острая сердечная недостаточность часто развивается у больных с хронической сердечной недостаточностью. Приступы нередко связаны с повышенным напряжением, эмоциями, потреблением соли с пищей, неаккуратностью выполнения медицинских назначений или повышенным метаболическим запросом, связанным с лихорадкой, анемией и т. п. Чаще всего причиной острой недостаточности (при наличии или при отсутствии хронической недостаточности) является острый инфаркт миокарда. Многие симптомы острой и хронической сердечной недостаточности идентичны, но их лечение отличается из-за необходимости неотложной помощи и тяжести сосудистого застоя в легких при острой форме.
В связи с необходимостью быстрого распознавания и оценки изменяющегося гемодинамического статуса при острой сердечной недостаточности гораздо более важно, чем при хронической, иметь количественные показатели. Это — давление наполнения левого желудочка (давление заклинивания легочных капилляров) и сердечный выброс (дополнительно к данным о частоте сердечных сокращений и АД). Индекс ударной работы — полезная производная переменная, которая характеризует работу, производимую левым желудочком.
Измерения индекса ударной работы и давления заклинивания легочных капилляров большой группы больных с острым инфарктом миокарда иллюстрирует рис. 13-2. Когда давление наполнения больше 15 мм рт. ст. и индекс ударной работы менее 20 л/мин/м2, смертность высока. Промежуточные значения этих двух показателей позволяют сделать значительно лучший прогноз. Очевидно, что влияние инфаркта миокарда на функцию желудочков разнообразно и что не может быть единой стандартной терапии застойной сердечной недостаточности в этой ситуации.
Группа больных после инфаркта миокарда
Больные с острой застойной сердечной недостаточностью могут быть охарактеризованы на основании трех гемодинамических измерений: АД, давления наполнения левого желудочка и сердечного
индекса. Одна из таких классификаций представлена в табл. 13-6.
1) Гиповолемия. Первая группа включает больных, у которых имеется относительная гиповолемия после инфаркта миокарда в результате приема диуретиков или неадекватного поступления жидкости. Главное гемодинамическое нарушение в этом случае — низкое давление наполнения левого желудочка, что можно исправить назначением жидкости. С увеличением давления наполнения до оптимального уровня 15 мм рт. ст. (рис. 13-6) часто исчезает гипотензия и сердечный выброс увеличивается.
2) Застой в легких. Вторая очень большая группа — больные с выраженным застоем в легких и одышкой. Таким больным полезны диуретики, потому что они уменьшают внутрисосудистый объем. Кроме того, при внутривенном введении мощные
Давление наполнения левого желудочка (мм рт. ст.)
Рис. 13-6. Влияние терапии вазодилататорами на функцию левого желудочка. Подобрана доза, понижающая давление наполнения на 5 мм рт. ст. При введении комбинированного артериального и венозного дилятатора, например нитропруссида, сдвиг функции соответствует верхней кривой, что отражает пониженное общее сопротивление (импеданс) изгнанию крови из желудочков. Однако влияние на ударный объем будет зависеть от начального давления наполнения. У больного с сердечной недостаточностью с начальным давлением наполнения 20 мм рт. ст. ударный объем будет увеличиваться вдоль линии А вверх и влево. Если у больного исходное давление наполнения составляло 10 мм рт. ст., нитропруссид вызовет дальнейший сдвиг кривой вверх, но это приведет к снижению КПД вдоль линии Б. (Из: Chatterrjee К., Parmley W. W. The role of vasodilator therapy in heart failure, Prog. Cardivasc. Dis. 1977; 19:305.)
ТАБЛИЦА 13-6. Терапевтическая классификация вариантов нарушений при остром инфаркте миокарда’
Подгруппа Систолическое артериальное давление | (мм рт. ст.) Давление наполнения левого желудочка (ммрт. ст.) Сердечный индекс
(л/мин/м2) Терапия
1. Гиповолемия < 100 < 10 <2.5 Возмещение объема
2. Застой в легких 100-150 >20 >2.5 Диуретики
3. Периферическая вазодилатация < 100 10-20 >2.5 Не требуется или вазоактивные препараты
4. Недостатокмощности < 100 >20 <2.5 Вазодилататоры; препараты, обладающие инотропным действием
5. Тяжелый шок <90 >20 < 2.0 Вазодилататоры; препараты, обладающие инотропным действием, поддержка циркуляции
6. Инфаркт правого желудочка < 100 RVFP> 10 LVFP<15 <2.5 Возмещение объема при LVFP; препараты, обладающие инотропным действием; избегать диуретиков
7. Митральная недостаточность (регургитация), дефект межжелудочковой перегородки < 100 >20 <2.5 Вазодилататоры; препараты, обладающие инотропным действием; поддержка циркуляции;
хирургическое лечение
' Цифровые показатели являются ориентировочными, а не абсолютными. Приведены значения АД для нормотензивных больных, для гипертоников следует внести поправку. RVFP и LVFP — давление наполнения правого и левого желудочков соответственно.
диуретики, такие как фуросемид, вызывают немедленное увеличение податливости системных вен. Это ведет к периферическому депонированию крови, что снижает центральный объем крови и уменьшает давление наполнения правого и левого желудочков. При лечении больных с острым отеком легких эффективен и морфина сульфат. Морфин уменьшает боль при инфаркте, но механизм, посредством которого он способствует улучшению состояния больных при отеке легких, неясен. При выраженном бронхоспазме может оказаться полезным аминофиллин, так как он сочетает бронходилата-торные, вазодилататорные и позитивные инотропные свойства (глава 19).
3) Периферическая вазодилатация. Третья группа — это больные с гипотензией, у которых в связи с хорошей периферической вазодилатацией конечности остаются теплыми. Эти больные могут не нуждаться в какой-либо терапии, так как давление заклинивания легочных капилляров обычно нормальное и сердечный выброс удовлетворителен.
Однако, если АД очень низкое, следует применять вазоактивные препараты, такие как допамин, для поддержания артериального перфузионного давления на желаемом уровне.
4) “Недостаток мощности”. Четвертая группа представлена больными с пониженным АД, но не до уровня развития шока, в сочетании с повышением давления наполнения левого желудочка и снижением сердечного индекса. В данном случае целесообразно увеличить сердечный выброс и снизить давление наполнения желудочка без дальнейшего понижения АД. Для понижения давления наполнения можно использовать диуретики или нитраты, но они не способствуют повышению сердечного выброса. Чрезвычайно полезны таким больным комбинированные артериолярные и венозные дилятаторы, такие как нитропруссид. Их венодилатирующие эффекты снижают давление в правом и левом предсердиях путем перераспределения крови на периферию. За счет артериолярных сосудорасширяющих свойств препаратов увеличивает
ся сердечный выброс, Поскольку системное сосудистое сопротивление унижается. Главный лимитирующий фактор при Использовании нитропруссида — снижение АД. Однако если доза аккуратно “титруется”, это понижение АД можно свести к минимуму. Существует практическое правило: нитропруссид назначают при системном систолическом давлении 90 мм рт. ст. или выше. Эффекты нитропруссида зависят от уровня давления наполнения левогожелудочка. Это иллюстрируется в форме диаграммы на рис. 13-6. Средняя кривая на рисунке Представляет данные больного с умеренной степёнью сердечной недостаточности. Если у больного высокое давление наполнения левого желудочка (20 мм рт. ст.), введение нитропруссида натрия вызовет сдвиг функции желудочков вверх и влево, как показано линией А. Однако, если больному ввели большую дозу диуретиков и, соответственно, давление наполнения снизилось до 10 мм рт. ст., введение нитропруссида даст иной ре-зультат>Хотя кривая и сдвинется вверх (линия Б), будет отмечаться снижение ударного объема, потому что состояние сердца будет соответствовать крутой восходящей части кривой. Это обострит гипотензию и тахикардию. Если путем нагрузки объемом давление наполнения будет доведено до оптимального значения 15 мм рт. ст., положительный эффект нитропруссида станет очевидным. Этот пример иллюстрирует важность контроля давления наполнения: давление легочного заклинивания должно поддерживаться примерно на уровне 15 мм рт. ст. за счет диуреза или поддержания нужного объема.
5) Тяжелый шок. В пятую группу входят больные с кардиогенным шоком вследствие инфаркта миокарда. У них очень низкое артериальное давление, очень высокое давление наполнения и низкий сердечный индекс. Хотя для уменьшения повышенного давления наполнения можно назначить диуретики, они не повысят сердечный выброс. Более того, для этих больных сомнительна полезность вазодилататоров: они могут вызвать тяжелую гипотензию, которая нарушит перфузию жизненно важных органов. Поскольку смертность в этих условиях очень высока, трудно понять, какая терапия наиболее эффективна. Тем не менее определенные подходы могут быть полезны. Если артериальное давление чрезвычайно низкое, стоит подумать об использовании катехоламинов, например допамина. Тщательно подбирая дозу этого средства, можно
вызвать увеличение артериального давления, обеспечивающее перфузию периферических тканей. В этот момент можно осторожно добавить нитропруссид для уменьшения давления в сосудах легких. Воздействия на сердце этих двух средств имеют тенденцию дополнять друг друга, так как они влияют посредством разных механизмов. Допамин непосредственно повышает сократимость сердца, в то время как нитропруссид вызывает расширение вен и артериол. Добавление лекарств с положительным инотропным действием, таких как амринон или добутамин, также может помочь. Это лечение должно комбинироваться с попытками оптимизации давления наполнения левого желудочка. Эти больные также являются кандидатами для измерения циркуляторного участия (интрааортальный баллон) и проведения сердечной катетеризации и возможной реваскуляризации.
6) Инфаркт правого желудочка. Шестая группа включает больных с инфарктом миокарда нижней части сердца, который в основном затрагивает правый желудочек. Возникающая при этом гипотензия связана с тем, что правый желудочек не способен доставлять достаточное количество крови в левую часть сердца, чтобы обеспечить оптимальное давление наполнения левого желудочка и сердечный выброс. Следовательно, основной терапевтической целью является оптимизация давления наполнения левого желудочка. Это может потребовать объемных инфузий, несмотря на повышенное центральное венозное давление. Нужно избегать применения диуретиков, так как уменьшение объема циркулирующей крови может привести к дальнейшему уменьшению давления наполнения с сопутствующими гипотензией и тахикардией. Можно применять средства инотропного действия.
7) Митральная регургитация и дефект межжелудочковой перегородки. Седьмая группа упоминается для полноты изложения, хотя этих больных можно найти и в других группах, особенно при шоке. Это больные с механическими повреждениями, такими как острая тяжелая митральная регургитация или разрыв межжелудочковой перегородки. В зависимости от тяжести дефекта в таких случаях показана более или менее срочная операция на сердце. Однако для стабилизации состояния больных перед операцией может потребоваться лечение вазодилататорами, в частности нитропруссидом.
Препараты
(Диуретики даны в главе 15)
Препараты наперстянки
Десланозид (Цедиланид-D)
Парентерально: 0.2 Мг/мл для инъекций
Дигитоксин (генерик, Кристодигин)
Перорально: таблетки по 0.05, 0.1, 0.15, 0.2 мг
Дигоксин (генерик, Ланрксикапс, Ланоксин) Перорально: таблетки по 0.125, 0.25, 0.5 мг; капсулы по 0.05,0.1„ 0.2 мг;
эликсир 0.05 мг/мл 1
Парентерально: 0.1, 0.25 мг/мл для инъекций
Антитела к препаратам наперстянки
Дигибайнд (овечий)
Парентерально: флаконы по 40 мг с 75 мг лиофилизированного порошка сорбитола для в/в инъекций. Содержимое флакона связывает примерно 0.6 мг дигоксина или дигитоксина
Симпатомиметики, наиболее часто применяемые при застойной сердечной недостаточ ности
Добутамин (Добутрекс)
Парентерально: 250 мг/20 мл для в/в вливаний
Дофамин (генерик, Интропин, Допастат)
Парентерально: 40, 80,160 мг/мл для в/в инъекций; 80, 160, 320 мг/100 мл для в/в вливаний
Ингибиторы ангиотензинконвертирующего фермента, применяемые при застойной сердечной недостаточности
Каптоприл (Капотен)
Перорально: таблетки по 12.5, 25,50,100 мг
Эналаприл (Вазотек)
Перорально: таблетки по 2.5, 5,10, 20 мг
Лизиноприл (Принивил, Зестрил)
Перорально: таблетки по 5,10, 20,40 мг
Другие препараты, применяемые только при застойной сердечной недостаточности
Амринон (Инокор)
Парентерально: 5 мг/мл для в/в вливаний
Милринон
Парентерально: 5 мг/мл для в/в вливаний
Избранная литература
Braunwald Е. Pathophysiology of heart failure. In: Heart Disease, 3rd ed. Braunwald E. (ed.) Saunders, 1987.
Broddle O-E. Pr and p2-adrenoreceptors in the human heart: Properties, function, and alterations in chronic heart failure. Pharmacol. Rev. 1991; 43: 203.
Chatterjee K. Digitalis, catecholamines, and other positive inotropic agents. In: Cardiology. Parmley W. W., Chatterjee K. (eds) Lippincott, 1988.
CONSENSUS Trial Study Group: Effects of enalapril on mortality in severe congestive heart failure. N. Engl. J. Med. 1987; 316:1429.
Doherty J. E. Clinical use of digitalis glycosides. Cardiology, 1985; 72: 225.
Goto A., Yamada K., Sugimoto T. Endogenous digitalis: Reality or myth? Life Sci. 1991; 48: 2109.
Horn P. T., Murphy M. B. New dopamine receptor agonists in heart failure and hypertension: Implications for therapy. Drugs, 1990; 40:487.
Kelly R. A., Smith T. W. Is ouabain the endogenous digitalis? (Editorial comment.) Circulation, 1992; 86: 694.
Konstam M. A. et al. Effects of the angiotensin converting enzime inhibitor enalapril on the longterm progression of left ventricular disfunction in patients with heart failure. Circulation, 1992; 86: 431.
Parmley W. W. Principles in the management of congestive heart failure. In: Cardiology. Parmley W. W., Chatterjee K. (ed.) Lippincott, 1988.
Pfeffer M. A. et al. On behalf of the SAVE investigators: Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 1992; 327: 669.
Smith T. W. Digitalis: Mechanisms of action and clinical use. N. Engl. J. Med. 1988; 318:358.
Smith T. W., Braunwald E. The management of heart failure. In: Heart Disease, 3rd ed. Braunwald E. (ed.) Saunders, 1987.
Антиаритмические средства “| Д
Люк М. Хон де гем, Дан М. Роден
Одна из серьезных проблем клинической практики — нарушения сердечного ритма. Аритмии возникают примерно у 25 % больных при лечении дигиталисом, у 50 % больных во время общей анестезии и более чем у 80 % пациентов с острым инфарктом миокарда. При аритмиях может потребоваться лечение, потому что слишком частые, слишком редкие или асинхронные сокращения уменьшают сердечный выброс. В ряде случаев наблюдаются более серьезные или даже угрожающие жизни нарушения ритма. Например, преждевременная деполяризация желудочков может привести к их фибрилляции. Таким больным антиаритмические средства могут спасти жизнь. С другой стороны, опасности проти-воаритмической терапии и особенно тот парадоксальный факт, что у некоторых больных она может вызвать смертельные аритмии, заставили переоценить сравнительные преимущества и недостатки данной группы лекарственных средств. В связи с этим считается, что следует избегать лечения бессимптомных аритмий или аритмий с минимальными проявлениями.
Больных с аритмиями можно лечить с помощью лекарственных препаратов, а также такими немедикаментозными средствами как искусственные водители ритма, электроимпульсная терапия, катетеризация, хирургическое лечение. В данной главе рассмотрены фармакология средств, которые устраняют аритмии путем прямого влияния на мембрану клеток миокарда, и иные способы лечения.
Электрофизиология нормального ритма сердца
Электрический импульс, запускающий сокращение сердечной мышцы, возникает с регулярными интервалами в синоатриальном узле (рис. 14-1) обычно с частотой 60-100 в 1 мин. Он быстро распространяется по предсердиям и поступает в атриовентрикулярный узел, который в норме является
единственным путем проведения между предсердиями и желудочками. Проведение через атриовентрикулярный узел медленное и требует примерно 0.15 с (эта задержка обеспечивает сокращение предсердий и проталкивание крови в желудочки). Далее импульс распространяется по системе Гиса-Пуркинье и охватывает все части желудочков. Активация желудочков завершается менее чем через 0.1 с, следовательно, сокращение мускулатуры желудочков происходит синхронно и гемодинамически эффективно.
Аритмии представляют собой процессы деполяризации миокарда, которые отличаются от вышеописанных по одному или более признакам, т. е. они аномальны по месту возникновения импульсов, их частоте, регулярности или характеру проведения.
Ионные основы электрической активности мембраны
Трансмембранный потенциал клеток сердца определяется концентрациями некоторых ионов (главным образом Na+, К+, Са2+) с обеих сторон мембраны и проницаемостью мембраны для каждого из них. Ионные каналы являются основными путями, по которым ионы проникают через мембрану. Эти каналы относительно специфичны для определенного иона, и ионный поток через них контролируется особым затвором (gate), или “воротами” (возможно, гибкими пептидными цепями или энергетическими барьерами). Каждый тип канала имеет свой собственный вид таких ворот (предполагают, что натриевые, калиевые и кальциевые каналы имеют по два типа ворот), и каждый из них открывается и закрывается при особом состоянии мембраны (ионном, метаболическом или в зависимости от трансмембранного потенциала).
Например, известно, что натриевые каналы имеют четыре активационных затвора и один инакти-вационный, расположенный между третьим и четвертым активационными затворами. Эти затворы
Предсердие
Атриовентрикулярный узел
Верхняя полая вена
Синоатриальный узел
.'•'•Г'
мВ
Фаза О
Потенциал покоя
Q S
Трехстворчатый клапан
Митральный
клапан
200 мсек
Рис. 14-1. Схематическое изображение сердца и его электрическая активность в норме (внутриклеточная регистрация в указанных областях и ЭКГ). Синоатриальный узел, атриовентрикулярный узел и клетки Пуркинье имеют пейс-мекерную активность (деполяризация в фазе 4). ЭКГ является отражением на поверхности тела волн деполяризации и реполяризации сердца Зубец Р отражает деполяризацию предсердий, QRS — деполяризацию желудочков, а зубец Т — реполяризацию желудочков. Таким образом, интервал PR позволяет измерить время проведения от предсердий к желудочкам, а длительность комплекса QRS показывает время, необходимое для активации всех клеток желудочка (т. е. время внутрижелудочкового проведения). Интервал QT отражает длительность потенциала действия в желудочках
Реверсия
-100
Желудочек
Фазы потенциала действия
0 - Быстрая деполяризация
1 - Ранняя быстрая реполяризация
2 - Плато
3 - Реполяризация
4 - Диастола
ЭКГ
открываются и закрываются в ответ на изменение потенциала. В большинстве клеток мембранный потенциал покоя отрицательный. Следовательно, Na+, который содержится в большей концентрации вне клетки (140 ммоль/л), чем внутри (10 ммоль/ л), будет быстро входить в клетку по концентрационному и электрическому градиентам, если мемб
рана проницаема для Na+. В покое большинство клеток характеризуются плохой проницаемостью для ионов натрия, но при возникновении потенциала действия они становятся легкопроницаемыми. Сходным образом при каждом потенциале действия Са2+ входит в клетку, а К+ покидает ее. Поэтому клетки должны иметь механизм поддержа
ния стабильного трансмембранного ионного состояния путем создания и поддержания ионных градиентов. Наиболее важным.из активных механизмов является натриевый нарос — На+,К+-АТФаза, рассмотренная в главе 13. Эта помпа и другие активные переносчики ионов вйосят косвенный вклад в создание трансмембранного потенциала за счет поддержания градиентов, необходимых для диффузии через каналы. Кроме того, некоторые насосы и ионообменники участвуют в общем токе ионов (например, путем обмена трех Йонов натрия на два иона калия) и поэтому называются “электроген-ными”.
В каждый момент времени электрический потенциал клеточной мембраны является суммарным отражением рассмотренных процессов. Однако в некоторых случаях полезно сделать упрощения для того, чтобы рассчитать величину трансмембранного потенциала. Допустим, клеточная мембрана высокопроницаема для одного иона (например, в большинстве клеток, находящихся в покое, для К+) и относительно непроницаема для других. Тогда трансмембранный потенциал (Е,„) приближается к потенциалу равновесия ионов (Eion). Потенциал равновесия рассчитывается по уравнению Нернста, которое для ионов натрия и калия выглядит следующим образом (при температуре тела):
Eion = 61 х log (Ce/C|),
где Сс и Cj вне- и внутриклеточные концентрации соответственно, умноженные на их коэффициенты активности. Уравнение Нернста выполнимо на пике реверсии мембранного потенциала при спайке и в периоде покоя в большинстве непейсмекер-ных клеток сердца. Если проницаемость (Р) значительна как для Na+, так и для К+, уравнение Нернста неточно предсказывает мембранный потенциал, и может быть использовано уравнение, которое предложили Гольдман, Ходжкин и Кац:
Emem ~ 61 Xlog
Fk xKt +Fha xNat
<F\xK* +FkaxNaf
Клеточная мембрана во время диастолы
Во время диастолы мембрана непейсмекерных клеток значительно более проницаема для калия, чем для других ионов, так что мембранный потенциал приближается к потенциалу равновесия для
калия (по уравнению Нернста). Для типичных значений Ке (4 ммоль/л) и К (150 ммоль/л) ЕК = = -96 мВ (дополнение “Эффекты калия”).
В пейсмекерных клетках (как нормальных, так и эктопических) во время диастолы развивается спонтанная деполяризация — пейсмекерный потенциал (фаза 4, рис. 14-1). Эта деполяризация связана с постепенным увеличением деполяризующего тока, вызванного повышением проницаемости для натрия или кальция и снижением реполяризующего калиевого тока вследствие уменьшения проницаемости для калия. Эффект изменения внеклеточного калия в пейсмекерных клетках более сложен, чем в непейсмекерных, потому что действие на проницаемость для калия значительно более важно в пейсмекерах (см. дополнение). В ритмоводителе, особенно эктопическом, увеличение концентрации К+ вне клетки в конечном итоге замедляет или прекращает генерацию импульсов. Напротив, гипокалиемия часто активизирует эктопические ритмоводители.
Клеточная мембрана в активном состоянии
В нормальном предсердии, волокнах Пуркинье и клетках желудочков быстрая деполяризация (фаза 0 потенциала действия) зависит от тока Na+. Деполяризация до критического порогового уровня приводит к открытию активационного (ш) затвора натриевого канала (рис. 14-2, средняя часть). Если инактивационный (h) затвор этих каналов еще не закрыт, каналы открываются и проницаемость для Na+ заметно возрастает, значительно превышая проницаемость для любого другого иона. Внеклеточный Na+, следовательно, диффундирует по электрохимическому градиенту внутрь клетки, а мембранный потенциал очень быстро приближается к потенциалу равновесия для натрия, ENa (около +70 мВ, когда Nac = 140 ммоль/л, a Nai = = 10 ммоль/л). Интенсивный натриевый ток длится очень недолго, потому что за открытием т-за-твора при деполяризации быстро следует закрытие h-затвора, то есть инактивация натриевого канала (рис. 14-2, правая часть).
Сходным образом активируется и инактивируется большинство кальциевых каналов, но в случае наиболее распространенного типа кальциевых каналов (L-типа) переход из одного состояния в другое осуществляется более медленно и при более
Эффекты калия
Так же, как лекарственное средство может связываться с двумя или более рецепторами, ионы калия действуют на мембраны двумя различными способами. Как уже отмечалось, градиент концентрации ионов калия (соотношение концентраций снаружи и внутри клетки) определяет потенциал равновесия для калия (Ек) по уравнению Нернста. Это справедливо для всех клеток и искусственных систем, содержащих полупроницаемые мембраны. Следовательно, увеличение внеклеточной концентрации К+ будет понижать (делать менее отрицательным) Ек. Второй важный эффект ионов калия — зависимость проницаемости мембраны для К+ от его внеклеточной концентрации. Последствия возникающих изменений в Рк (проницаемости для К+) лучше всего рассмотреть, используя уравнение Гольдмана-Ходжкина-Каца для потенциала покоя Emem. В соответствии с этим уравнением соотношение проницаемости для ионов калия и натрия определяет, как концентрационные градиенты этих двух ионов будут влиять на мембранный потенциал. Все, что способствует повышению проницаемости для К+, будет усиливать влияние калиевого градиента и приближать мембранный потенциал к Ек. Экспериментально установлено, что увеличение внеклеточной концентрации К+ повышает Рк> поэтому неудивительно, что гиперкалиемия сдвигает мембранный потенциал в сторону Ек.
При комбинации этих двух эффектов калия получается, что гиперкалиемия будет вызывать деполяризацию Ек и одновременно сдвигать Emcm к Ек. В непейсмекерных клетках Рк очень высока (во время диастолы) даже при низкой и
нормальной внеклеточной концентрации К+, и Emem близка к Ек во все периоды времени. С другой стороны, в пейсмекерных клетках Рк относительно низка при нормальных концентрациях калия, и увеличение Рк, вызванное гиперкалиемией, может сдвинуть Етип значительно ближе к Ек. Предлагаемая таблица показывает некоторые гипотетические примеры этих эффектов при Nac, Na, и PNa, принятым за 140, 10 и 1 соответственно.
Внеклеточный калий (Ке) Ki Рк Ек Emem
Непейсмекерная клетка Низкий: 2.5 150 75 -108 -94
Нормальный: 4.0 150 100 -96 -88
Высокий: 10.0 150 300 -72 -71
Пейсмекерная клетка Низкий: 2.5 150 15 -108 -67
Нормальный: 4.0 150 20 -96 -69
Высокий: 10.0 150 60 -72 -66
Заметьте, что в непейсмекерной клетке как потенциал равновесия для калия (Ек), так и потенциал покоя (Emem) становятся менее отрицательными при увеличении концентрации ионов калия. В пейсмекерной клетке происходят те же изменения Ек, но Emem изменяется значительно меньше, поскольку в этом типе клеток изменения проницаемости для калия более важны, чем изменения К,., и оказывают противоположное влияние на потенциал покоя. Следствием повышения уровня внеклеточного К+ будет стабилизация мембраны на уровне, более близком к потенциалу равновесия для калия.
положительном потенциале. Плато потенциала действия (фазы 1 и 2) отражает прекращение натриевого тока, подъем и спад кальциевого тока и медленное развитие реполяризующего калиевого тока.
Окончательная реполяризация (фаза 3) потенциала действия связана с завершением инактивации натриевых и кальциевых каналов и повышением проницаемости для К+. Таким образом, мембранный потенциал вновь приближается к потенциа
лу равновесия для калия. Эти процессы изображены на рис. 14-3.
Влияние потенциала покоя на потенциал действия
Ключевой фактор патофизиологии аритмий и действия противоаритмических средств — связь между потенциалом покоя клетки и потенциалом
Состояние покоя
Наружная сторона
мембраны
Внутренняя сторона мембраны
Активация
Инактивация
Рис. 14-2. Схема натриевого канала в клетка^ сердечной мышцы. Изображение канала и его затворов условно. В состоянии покоя мембрана полностью поляризована (слева), h-затвор открыт, а m-затвор закрыт, что предупреждает ток ионов натрия через канал. При действии йктивируюшего стимула m-затвор открывается, способствуя быстрому входу ионов натрия внутрь клетки (средняя часть рисунка). Примерно через миллисекунду закрывается h-затвор, что ведет к инактивации канала и прекращению натриевого тока (справа). Дополнительные стимулы, воздействующие на инактивированный натриевый канал, не могут открыть его (h-затвор закрыт), следовательно, канал “недоступен”. Область канала, обозначенная R,— предполагаемый рецептор местного анестетика. В состоянии покоя большинство противоаритмических средств имеют низкое сродство к рецептору. Когда канал активируется, вещество достигает рецептор по водной среде внутри клетки (стрелка на средней части рисунка). Когда канал инактивируется, вещество может попадать на рецептор через мембранную фазу (стрелка на правой части рисунка), если оно достаточно хорошо растворимо в липидах мембраны. Большинство противоаритмических средств являются слабыми основаниями, которые находятся частично в нейтральной (растворимой в липидах) и частично в заряженной (растворимой в воде) формах в зависимости от pH и, следовательно, могут достигать предполагаемого места связывания любым из этих способов
действия, который может возникнуть в ней (рис. 14-4, левая часть). В связи с тем, что инакти-вационные затворы натриевых каналов закрыты в диапазоне потенциала от -75 до -55 мВ, значительно меньше натриевых каналов “доступно” для диффузии ионов натрия, если потенциал действия возникает при исходном потенциале покоя -60 мВ по сравнению с исходным потенциалом покоя -80 мВ. Важными следствиями нарушения проницаемости для Na+ являются снижение скорости быстрой деполяризации (обозначаемой для максимальной скорости изменения мембранного потенциала), уменьшение амплитуды потенциала действия, снижение возбудимости и скорости проведения.
Во время плато потенциала действия большинство натриевых каналов инактивировано. В процессе реполяризации происходит восстановление инактивированных каналов (в терминах рис. 14-2 повторное открытие h-затвора), что делает их вновь способными к возбуждению. Другой важный эффект уменьшения величины отрицательного потенциала покоя — удлинение времени восстановления (рис. 14-4 справа). Это означает увеличение эффективного рефрактерного периода, т. е. времени от
начала потенциала действия до момента, когда можно вызвать второй потенциал действия.
Снижение (деполяризация) потенциала покоя1, вызванное гиперкалиемией, блокадой натриевого насоса или ишемическим повреждением, приводит к угнетению натриевых токов во время фазы быстрой деполяризации потенциала действия. Деполяризация потенциала покоя до уровня, превышающего -55 мВ, устраняет натриевый ток, так как все натриевые каналы инактивируются. Однако для таких деполяризованных клеток характерны особые потенциалы действия, возникающие при увеличении проницаемости мембраны для Са2+ или снижении проницаемости для К+. Эти “медленные ответы” с пониженной скоростью фазы деполяризации и медленным проведением зависят от входящих токов Са2+ и являются нормальными для электри-
1 Стойкое снижение потенциала покоя, рассматриваемое в этом разделе, не следует путать с быстрой деполяризацией при действии нормальных раздражителей. Короткие деполяризующие стимулы, вызванные распространяющимся потенциалом действия или раздражением через внешние электроды, приводят к открытию большого числа активационных затворов до того, как закроется большое количество инактивационных затворов.
Рис. 14-3. Схема изменений ионной проницаемости и процессов транспорта, происходящих во время потенциала действия и в последующий период диастолы. Размер и толщина стрелок отображают примерную величину тока через ионные каналы
ческой активности в синоатриальном и атриовентрикулярном узлах, так как их клетки имеют в норме потенциал покоя в пределах от -50 до -70 мВ. Медленные ответы могут играть важную роль при некоторых аритмиях. С помощью современных методов молекулярной биологии и электрофизиологии можно идентифицировать ряд подтипов кальциевых и калиевых каналов. В частности, они различаются по чувствительности к лекарственной терапии, поэтому в будущем могут быть созданы препараты, действующие на определенные подтипы каналов.
Механизмы аритмий
Многие факторы могут вызывать или усугублять нарушения ритма. Это ишемия, гипоксия, ацидоз или алкалоз, электролитный дисбаланс, чрезмерное воздействие катехоламинов, влияние автономной нервной системы, лекарственная токсичность (например, дигиталис или противоаритмичес-кие средства), перерастяжение волокон миокарда и наличие рубцовой или поврежденной ткани. Однако все аритмии возникают из-за: 1) нарушений об
разования импульсов, 2) нарушений проведения импульсов или 3) при сочетании этих факторов.
Нарушения образования импульсов
Период между деполяризациями пейсмекерной клетки является суммой продолжительности потенциала действия и диастолического интервала. Укорочение длительности каждого из них ведет к повышению частоты ритмоводителя. Более важный из этих двух составляющих диастолический интервал зависит в свою очередь от трех факторов: максимального диастолического потенциала, крутизны деполяризации в фазу 4 и порогового потенциала (рис. 14-5). Таким образом, вагусные разряды понижают нормальную частоту водителя ритма, делая максимальный диастолический потенциал более отрицательным и уменьшая скорость нарастания потенциала в фазе 4. Учащение разрядов пейс-мекера часто вызывается увеличением крутизну^, деполяризации фазы 4 в связи с гипокалиемией,^ стимуляцией Р-адренорецепторов, растяжением волокон и частичной деполяризацией при токах повреждения.
Рис. 14-4. Зависимость функционирования натриевого канала от мембранного потенциала, предшествующего раздражителю. Как показано на левом графике, функция натриевых каналов, способных к открытию в ответ на стимул, определяется величиной мембранного потенциала, непосредственно предшествующего стимуляции. Уменьшение этой фракции при деполяризации в отсутствие лекарства (контрольная кривая) происходит в результате закрытия h-затворов канала. Кривая с пометкой “лекарственное вещество” иллюстрирует эффект противоаритмического средства — типичного местного анестетика. Большинство натриевых каналов инактивировано во время фазы плато потенциала действия. Константа времени восстановления от инактивации после реполяризации также зависит от потенциала покоя. График справа показывает, что в отсутствие лекарства при нормальном потенциале покоя (от -85 до -96 мВ) восстановление происходит менее чем за 10 мс. Деполяризованная клетка восстанавливается более медленно (обратите внимание на логарифмическую шкалу). В присутствии вещества, блокирующего натриевые каналы, константа времени восстановления увеличивается, причем значительно больше во время деполяризации, чем при более отрицательном потенциале
Латентные ритмоводители (клетки, имеющие медленную деполяризацию в фазе 4 даже в норме, например некоторые волокна Пуркинье) особенно склонны к ускорению по рассмотренным выше механизмам. Однако все сердечные клетки, включая неактивные в норме предсердные и желудочковые клетки, могут проявлять повторяющуюся пейсме-керную активность, когда деполяризация происходит в определенных условиях, особенно если этому сопутствует гипокалиемия.
Следовая деполяризация (рис. 14-6) — это деполяризация, которая прерывает фазу 3 (ранняя следовая деполяризация, или РСД) или фазу 4 (поздняя следовая деполяризация — ПСД). ПСД, которая обсуждалась в главе 13, часто возникает при увеличении уровня Са2+ внутри клеток. Она усиливается при учащении сердечных сокращений и, как полагают, ответственна за некоторые аритмии, связанные с передозировкой препаратов наперстянки, действием катехоламинов и ишемией миокарда. РСД, как правило, развивается при медленном сердечном ритме и связана с аритмиями, для которых характерно удлинение QT. Такие нарушения рит-
m
5. +20
| 0
| -20 g -40
з -60 -X
« -80 -t™-
Рис. 14-5. Детерминанты частоты ритмоводителя. На рисунке представлен трансмембранный потенциал в пейс-мекерной клетке волокна Пуркинье. Частота ритмоводителя может быть уменьшена четырьмя способами: 1) при более отрицательном диастолическом потенциале, показанном как изменение от -80 до -100 мВ; 2) путем снижения крутизны диастолической деполяризации; 3) при более позитивном пороговом потенциале, что показано как изменение от -65 до -45 мВ; и 4) при увеличении длительности потенциала действия (не показано на рисунке, так как это редкий механизм). Заметьте, что каждое из этих изменений приводит к увеличению интервала между первым и вторым потенциалами действия (сравните изображения второго потенциала сплошной и пунктирной линиями)
Рис. 14-6. Две формы аномальной активности: ранняя (наверху) и поздняя (внизу) следовая деполяризация. В обоих случаях аномальная деполяризация возникает во время или после нормального потенциала действия. Эти виды нарушений относят к “триггерному” автоматизму, т. е. для их появления необходим нормальный потенциал действия
мамогут быть следствием недостаточно изученных врожденных факторов или осложнением терапии некоторыми противоаритмическими и другими лекарственными средствами, влияющими на реполяризацию.
Все ритмоводители, как нормальные, так и аномальные, зависят от диастолической деполяризации в фазе 4. Гиперкалиемия, как уже указывалось, стабилизирует мембранный потенциал и тем самым уменьшает частоту разрядов2. При гипокалиемии мембрана менее проницаема для К+ и, соответственно, мембранный потенциал легче может быть смещен с уровня равновесного потенциала для калия (дополнение “Эффекты калия”), т. е. увеличивается вероятность возникновения спонтанных разрядов.
2 Даже несмотря на то, что К+ деполяризует мембрану и деполяризация может способствовать пейсмекерной активности, эффект стабилизации мембраны более важен , чем деполяризация, поэтому увеличение уровня К* обычно снижает частоту водителя ритма.
Нарушения проведения импульсов
Выраженное подавление проведения импульсов может привести к блокаде, например блокаде атриовентрикулярного узла или блокаде ножки пучка Гиса. Так как предсердно-желудочковое проведение контролирует парасимпатическая система, частичная атриовентрикулярная блокада иногда устраняется атропином. Более распространенным, но более сложным нарушением проведения является “повторный вход” (reentry) (известный также как “круговое движение"), при котором один импульс циркулирует и возбуждает одни и те же участки сердца (рис. 14-7). Путь этого импульса может быть ограничен очень малой областью, например внутри или в непосредственной близости от атриовентрикулярного узла либо может охватывать значительные участки стенки предсердий или желудочков. Более того, циркулирующий импульс часто создает “дочерние импульсы”, которые могут распространяться на остальные области сердца. В зависимости от того, как много кругов совершает импульс перед затуханием, аритмия может проявляться в виде одного или нескольких внеочередных сокращений или же как постоянная тахикардия.
Для возникновения механизма возврата необходимо соблюдение трех условий (рис. 14-7): ^должно быть препятствие (анатомическое или физиологическое) для равномерного проведения, создающее возможность контура, вокруг которого может распространяться фронт волны повторного входа; 2) должен быть односторонний блок в каком-либо месте проведения, т. е. проведение должно затухать в одном направлении, но поддерживаться в противоположном (как показано на рис. 14-7, импульс может постепенно угасать вплоть до полного блока, как только он доходит до более деполяризованной ткани); и 3) время проведения импульса по контуру должно быть достаточно большим, чтобы импульс не застал при обходе препятствия участок ткани в рефрактерном состоянии, т. е. время проведения должно превышать эффективный рефрактерный период. Таким образом, феномен повторного входа зависит от проведения, которое подавлено в определенных критических ситуациях — обычно вследствие ишемии или повреждения миокарда. Если скорость проведения слишком медленная, развивается двусторонний, а не односторонний блок; если повторно входящий импульс слишком слаб, проведение может прекратиться или импульс при-
Импульс, встетивший препятствие и угасший I
В. Нарушение проведения и односторонний блок антероградного импульса
Б. Нормальное проведение
Ретроградный импульс
Г. Ретроградный импульс, проходящий через зону угнетения
Д. Формирование круга повторного хода
Рис. 14-7. Односторонний блок и механизм возврата (reentry). На части А схематически показаны синоатриальный и атриовентрикулярный узлы сердца и проводящая система. Небольшое ответвление в системе Пуркинье обведено кружком в месте входа в стенку желудочка. Часть Б показывает нормальное прохождение и судьбу импульса, который идет по ответвлению. В области бифуркации он расходится на два импульса, которые сталкиваются (и гасят друг друга) после возбуждения мышцы желудочка. Части В и Д показывают последовательность событий, при которых нормальный импульс встречает область одностороннего блока в одной из веточек. Как показано на пути продвижения импульса в зоне угнетения (часть В), этот слабый импульс не способен пройти (“перепрыгнуть”) область блокады. Напротив, волна в неугнетенной веточке способна возбудить весь участок мышечной стенки (часть Г). Поскольку мышечная стенка содержит большую массу клеток, сильная желудочковая деполяризация способна “перепрыгнуть” через участок угнетения, что ведет к прохождению ретроградного импульса (показан черными стрелками на частях Г и Д). Ретроградный импульс может распространяться, если он встретит на своем пути возбудимую ткань, т. е. рефрактерный период будет короче, чем время проведения. Этот импульс повторно возбудит ткань, через которую он ранее проходил, и возникнет аритмия по механизму повторного входа с формированием контура, показанного на рисунке черными стрелками (часть Д)
дет так поздно, что совпадет со следующим регулярным импульсом. С другой стороны, если проведение слишком быстрое, будет возникать двустороннее проведение, а не односторонний блок. Даже при наличии одностороннего блока, если импульс путешествует вокруг препятствия слишком быстро, он может достичь участка ткани, который еще остается рефрактерным.
Замедление проведения может быть связано с подавлением натриевого или кальциевого тока (последнее характерно для атриовентрикулярного узла), а также обоих токов одновременно. Лекарства, которые устраняют повторный вход, вызывают или замедление проводимости в зоне угнетения (путем блокады натриевого или кальциевого токов), или ускорение ее (повышая натриевый или кальциевый токи).
Удлинение (или укорочение) рефрактерного периода также может сделать менее вероятным механизм возврата. Чем длиннее рефрактерный период в ткани около места блока, тем выше шанс, что она будет рефрактерной в момент повторного прихода импульса. (Соответственно, чем короче рефрактерный период в угнетенной области, тем менее вероятно, что разовьется односторонний блок.)
I. Базисная фармакология противоаритмических средств
Механизм действия
Аритмии вызываются аномальной пейсмекер-ной активностью или аномальным распространением импульса. Поэтому целью терапии аритмий является снижение активности эктопического водителя ритма или изменение проведения или рефрак-терности в петлях повторного входа, чтобы прекратить циркуляцию импульса. Основные механизмы достижения этих целей: 1) блокада натриевых каналов, 2) блокада симпатических влияний на сердце, 3) удлинение эффективного рефрактерного периода и 4) блокада кальциевых каналов.
Противоаритмические средства снижают автоматизм эктопических ритмоводителей в большей степени, чем синоатриального узла. Они угнетают также проведение и возбудимость и увеличивают рефрактерный период в деполяризованной ткани
в большей степени, чем в нормально поляризованной. Это достигается прежде всего путем селективной блокады натриевых или кальциевых каналов деполяризованных клеток (рис. 14-8). Терапевтически эффективные средства, блокирующие каналы, имеют высокое сродство к активированным (т. е. во время фазы 0) или инактивированным (т. е. во время фазы 2) каналам, но очень низкий аффинитет к каналам в состоянии покоя. Следовательно, эти вещества блокируют электрическую активность при выраженной тахикардии (много переходов в активированное и инактивированное состояние в единицу времени) или когда имеется значительное уменьшение потенциала покоя (в этом случае будет много инактивированных каналов). Та-
Рис. 14-8. Схема механизма селективного угнетающего действия противоаритмических средств на натриевые каналы. Верхняя часть рисунка показывает популяцию каналов, проходящих цикл активации во время потенциала действия в отсутствие лекарства: R (состояние покоя) -» А (активированный) -» I (инактивированный). Восстановление происходит по пути I -> R. Противоаритмические средства, которые действуют как блокаторы натриевых каналов, могут связываться со своими рецепторами в каналах, как показано вертикальными стрелками, с образованием комплексов лекарство—канал, обозначенных как R-D, А-D и I-D. Сродство лекарств к рецептору зависит от состояния канала, что подчеркнуто различными константами (kul) для этапов R -> R-D, А —> А-D и I —> I-D. Имеющиеся данные для различных блокаторов натриевых каналов показывают, что их сродство к рецептору канала в активированном и инактивированном состоянии значительно выше, чем сродство к каналу в состоянии покоя. Более того, восстановление из состояния I-D в состояние R-D происходит значительно медленнее, чем из I в R. В результате быстрая активность (больше циклов активации и инактивации) и деполяризация потенциала покоя (больше каналов в 1-состоянии) способствуют блокаде каналов и избирательному подавлению аритмических клеток
кой эффект часто описывают как “зависящий от использования” или “зависящий от состояния”, т. е. каналы, которые часто используются1 или находятся в инактивированном состоянии, более чувствительны к блокаторам. Каналы нормальных клеток, которые блокируются лекарственным веществом в период нормальных циклов активации—инактивации, быстро освобождаются от него во время фазы покоя (рис. 14-8). При постоянной деполяризации (мембранный потенциал более положительный, чем -75 мВ) каналы миокарда будут восстанавливаться от блока очень медленно или вообще не восстановятся за это время (правая часть рис. 14-4).
В клетках с аномальным автоматизмом большинство веществ такого типа уменьшают крутизну нарастания фазы 4 (рис. 14-5), блокируя натриевые или кальциевые каналы и тем самым уменьшая отношение натриевой (или кальциевой) проницаемости к проницаемости для калия. В результате мембранный потенциал во время фазы 4 стабилизируется на уровне, более близком к потенциалу равновесия для калия* Кроме того, некоторые вещества могут увеличивать критический уровень порога (делать его более положительным). 0-адреноблокато-ры вызывают непрямое угнетение фазы 4, устраняя положительное хронотропное действие норадреналина в сердце.
При аритмиях типа reentry, связанных с подавлением проводимости до критического уровня, большинство противоаритмических средств еще более замедляют проводимость одним или одновременно двумя следующими способами: 1) устойчивым уменьшением числа доступных незаблокиро-ванныХ каналов, что способствует снижению возбуждающего тока до уровня, ниже требуемого для распространения волны возбуждения (рис. 14-4, слева) и 2) удлинением времени восстановления каналов, которые способны достигнуть исходного состояния покоя и быть готовыми к активации, что увеличивает эффективный рефрактерный период (рис. 14-4, справа)3. В результате ранние экстрасистолы становятся неспособными к распространению, а более поздние импульсы распространяются медленнее и подвергаются двустороннему блоку проведения.
Некоторые вещества удлиняют потенциал действия в сердце, возможно, за счет снижения выходящих реполяризующих токов (например, блокируя калиевые каналы) или усиления входящих токов (например, при активации натриевых или каль
циевых каналов во время фазы 2). Такое удлинение обычно прямо отражается увеличением эффективного рефрактерного периода с уже рассмотренными положительными последствиями. Эти эффекты зависят и от продолжительности сердечного цикла. К сожалению, удлинение обычно мало выражено при продолжительных циклах, т. е. этот эффект проявляет обратную “зависимость от использования”. После длительной компенсаторной паузы удлинение может оказаться чрезмерным и привести к нарушениям реполяризации, ранней следовой деполяризации и к особой форме желудочковой тахикардии, называемой torsade de pointes (трепетание-мерцание желудочков). Предпочтительно “нормальное” удлинение потенциала действия, “зависимое от использования”: при нормальной частоте сердцебиений происходит мало изменений, но во время тахикардии длительность потенциала действия возрастает с каждым сокращением, пока рефрактерный период не становится слишком продолжительным, чтобы поддерживать тахикардию (более детально этот механизм описан при характеристике хинидина, соталола и амиодарона).
С помощью рассмотренных механизмов проти-воаритмические средства могут подавлять эктопический автоматизм и нарушения проведения в деполяризованных клетках, делая их электрически “молчащими”, но мало влияя на электрическую активность нормально поляризованной части сердца. Однако при увеличении доз эти вещества подавляют проведение и в нормальной ткани, иногда провоцируя так называемые лекарственные аритмии. Более того, терапевтическая (антиаритмическая) в нормальных условиях концентрация препарата может стать “проаритмической” (аритмогенной)
3 Существует специфический способ возникновения механизма возврата, связанный с латентными ритмоводителями, особенно в волокнах Пуркинье. Поскольку деполяризация латентных пейсмекеров в фазе 4 дает менее отрицательный исходный уровень для последующего потенциала действия, доступность натриевых каналов для обеспечения быстрой деполяризации будет ниже. На рис. 14-4 показано, что снижение числа доступных каналов с соответствующим уменьшением скорости проведения происходит очень резко в диапазоне от -70 до -60 мВ. Это снижение может оказаться достаточным для одностороннего блока проведения и возникновения повторного входа. Так как противоаритми-ческие средства уменьшают деполяризацию в фазе 4 в латентных ритмоводителях, они (по крайней мере, теоретически) могут повысить скорость проведения в этих элементах проводящей системы сердца.
при частом сердцебиении (больше вероятность блокады), ацидозе (замедление восстановления от блокады при действии многих лекарств) или гиперкалиемии.
Как уже обсуждалось, внеклеточная концентрация К+ значительно влияет на трансмембранный потенциал покоя в непейсмекерных клетках. Следовательно, гиперкалиемия, деполяризуя мембрану и инактивируя больше каналов, усиливает угнетающее действие противоаритмических средств из группы блокаторов натриевых каналов. Снижая концентрацию внеклеточного калия (например, при подъеме pH сыворотки), можно уменьшить токсичность противоаритмических средств за счет гиперполяризации миокарда. Такой подход имеет смысл только при условии нормального или высокого уровня калия в сыворотке, так как гипокалиемия также может вызывать аритмии.
II. Отдельные противоаритмические средства
Противоаритмические средства традиционно разделяют на четыре класса на основании преобладающего механизма их действия. Класс I (самая представительная группа противоаритмических средств) включает блокаторы натриевых каналов, сходные по действию с местными анестетиками (наиболее популярный противоаритмический препарат для парентерального введения лидокаин является одновременно наиболее часто используемым местным анестетиком). Вещества класса I подразделяют на подклассы в соответствии с их влиянием на длительность потенциала действия: средства класса IA увеличивают, а препараты класса IB уменьшают длительность потенциала действия. Вещества класса IC не оказывают заметного действия или в минимальной степени увеличивают длительность потенциала действия. Представители группы IB быстро взаимодействуют (связываются и диссоциируют) с натриевыми каналами. Представители группы IC взаимодействуют с каналами медленно, а группа 1А занимает по этому показателю промежуточное положение. Класс II образуют вещества, снижающие адренергическую активность в сердце. В класс III включают препараты, которые пролонгируют эффективный рефрак-
терный период за счет механизма, отличного от блокады натриевых каналов или дополняющего его. Класс IV — блокаторы кальциевых каналов. В пятый (безымянный) класс входят аденозин, препараты калия и магния.
Важно отметить, что данная классификация не учитывает многие различия между препаратами одной группы и перекрестные фармакологические свойства средств из разных групп. Таким образом, можно выделить определенные эффекты препаратов (блокада натриевых каналов, удлинение потенциала действия и т. п.), но разделение в соответствии с ними лекарственных средств не означает, что препараты одного класса будут оказывать полностью идентичное действие. Это объясняется тем, что данный препарат, например из класса I, может также иметь выраженный эффект, характерный д ля класса III, тогда как другое лекарство класса I может обладать также эффектами класса IV.
В связи с противоречиями традиционной классификации недавно был предложен новый классификационный подход, учитывающий множественность эффектов большинства противоаритмических средств и различные механизмы аритмогенеза (Task Force, 1991). Однако эта система пока не нашла широкого применения.
Средства, блокирующие натриевые каналы (класс I)
Хинидин (класс IA)
Хотя хинин эпизодически применяли как анти-аритмическое средство в XVIII и XIX столетиях, только в начале XX в. диастереомер хинина хинидин начал широко использоваться в практике. Хинидин является одним из наиболее часто применяемых в США противоаритмических средств для приема внутрь.
СН = СН2
Хинидин
Влияние на сердце
Хинидин цодавляет частоту водителей ритма (особенно эктопических), проводимость и возбудимость (прежде всего в деполяризованной ткани) (табл. 14-1). В значительной степени эти эффекты определяются его способностью связываться с активированными натриевыми каналами и блокировать их, хотя вещество блокирует и неактивирова-ванные каналы. Деблокирование происходит медленно и менее полно в деполяризованной клетке по сравнению с полностью поляризованной. Таким образом, хинидин удлиняет рефрактерный период и подавляет возбудимость и проводимость деполяризованной ткани в большей степени, чем нормальной.
Хинидин также удлиняет потенциал действия, что отражается на ЭКГ в виде увеличения интервала QT. Этот эффект обеспечивается блокадой калиевых каналов со снижением реполяризующего выходящего тока (Balser et al., 1991; Snygers et al., 1992). Следует заметить, что любое удлинение потенциала действия уменьшает продолжительность отрицательного диастолического мембранного потенциала и, следовательно, повышает способность хинидина блокировать натриевые каналы. Удлинение потенциала действия и эффективного рефрактерного периода снижает частоту reentry и может уменьшить тахикардию. Этот эффект по типу класса III менее выражен при высокой частоте сердцебиений (когда он нужнее), а в некоторых случаях (при медленном ритме) может быть чрезмерным. В эксперименте показано, что чрезмерное удлинение может вызывать уже описанную раннюю следовую деполяризацию (РСД). У пациентов удлинение потенциала действия и РСД нередко являются причиной torsade de pointes. Эта тахикардия ассоциирована с выраженным удлинением интервала QT и, видимо, провоцирует возникновение хинидиновых синкоп (о них еще пойдет речь). У больных с проявлениями токсичности при приеме хинидина концентрация препарата в плазме часто бывает нормальной.
Внесердечные эффекты
Хинидин обладает а-адреноблокирующим действием, что способствует вазодилатации и рефлекторному увеличению активности синоатриального узла. Эти эффекты наиболее выражены после внутривенного введения.
Токсичность
А. Кардиотоксичность. Хинидин обладает ан-тимускариновым действием на сердце, что ингибирует вагусные эффекты. Это может нивелировать некоторые прямые влияния препарата на мембрану и привести к увеличению частоты синусового ритма и повышению атриовентрикулярной проводимости. При фибрилляции предсердий антимус-кариновое действие хинидина на атриовентрикулярный узел может вызвать чрезмерное повышение частоты сокращений желудочков. При трепетании предсердий трансформация частоты в атриовентрикулярном узле снижается от 4 : 1 или более до 1:1. Однако этот эффект можно предупредить предварительным введением препаратов, замедляющих атриовентрикулярное проведение, например верапамила, Р-блокаторов или дигиталиса.
У незначительного количества больных (1-5 %), получающих хинидин, развивается синдром, называемый “хинидиновая синкопа”, который характеризуется повторяющимися головокружениями и обморочными состояниями. Симптомы являются следствием вызванного препаратом состояния torsade de pointes. Они обычно прекращаются самостоятельно, но могут учащаться и даже становиться непрерывными. (Torsade de pointes и обмороки наблюдаются при назначении многих лекарственных средств, которые удлиняют потенциал действия.)
У больных с синдромом слабости синусового узла хинвдин может подавить пейсмекерную активность синоатриального узла. В токсических концентрациях препарат подавляет электрическую активность в любой части сердца в такой степени, что возникает аритмия или асистолия. Эти токсические эффекты более вероятны, когда концентрация хинидина в сыворотке превышает 5 мкг/мл и при высокой концентрации калия (более 5 ммоль/л). Увеличение длительности QRS на 30 % при введении хинидина обычно считается предвестником серьезных токсических эффектов. Токсические концентрации могут снизить сократимость миокарда и понизить АД.
Б. Внесердечные эффекты. Наиболее частые побочные эффекты связаны с влиянием на желудочно-кишечный тракт и проявляются диареей, тошнотой и рвотой. Эти явления отмечаются у '/зп’/г всех больных. Иногда препарат вызывает явления цинхонизма (головная боль, головокружение,
звон в ушах). Изредка обнаруживаются сыпь, ангионевротический отек, лихорадка, гепатит и тромбоцитопения. Хинидин увеличивает уровень дигоксина в плазме и может способствовать проявлению токсического эффекта дигиталиса у больных, получающих дигоксин или дигитоксин (глава 13).
Фармакокинетика и дозирование
Хинидин обычно назначают внутрь, и он быстро всасывается из ЖКТ. На 80 % связывается с белками плазмы, метаболизируется в печени, но 20 % экскретируется в неизменном виде с мочой. Период полувыведения около 6 часов (табл. 14-2), но может быть более продолжительным у больных с застойной сердечной недостаточностью или заболеваниями печени и почек. Выделение с мочой повышается при ее подкислении. Внутрь препарат вводят обычно в форме солей (сульфата, глюконата или полигалактуроната). Средняя доза 0.2-0.6 г хинидина сульфата назначается 2-4 раза в день. Терапевтическая концентрация в плазме — 3-5 мкг/мл.
Иногда требуется вводить хинидин парентерально. Для внутримышечного введения используют хинидина сульфат в масляном растворе или водорастворимый хинидина глюконат. При соблюдении осторожности хинидин можно ввести внутривенно. При таком способе введения доза не должна превышать 10 мг/кг хинидина глюконата и не должна вводиться со скоростью более 0.5 мг/кг/мин. При этом в результате периферической вазодилатации обычно снижается АД. Поэтому при внутривенном применении хинидина следует тщательно контролировать АД и поддерживать его при необходимости введением плазмозаменяющих растворов.
Терапевтическое применение
Хинидин применяют почти при всех видах аритмий: преждевременных сокращениях предсердий, пароксизмальной фибрилляции и трепетании предсердий, внутрипредсердных и атриовентрикулярных узловых аритмиях по механизму повторного входа, тахикардиях при синдроме Вольфа-Паркинсона-Уайта, преждевременном сокращении желудочков и желудочковых тахикардиях. Для повышения эффективности и ослабления побочных эффектов он может быть назначен вместе с мексилетином. Хинидин также используют в терапии малярии.
Прокаинамид (класс IA)
Влияние на сердце
Электрофизиологические эффекты прокаинамида сходны с наблюдаемыми у хинидина. Препарат несколько менее эффективен в подавлении аномальной эктопической пейсмекерной активности, но лучше блокирует натриевые каналы деполяризованных клеток.
Возможно, наиболее важным отличием между хинидином и прокаинамидом является менее выраженный антимускариновый эффект прокаинамида. Следовательно, его прямое угнетающее влияние на синоатриальный и атриовентрикулярный узлы не столь эффективно тормозится вызванной препаратом блокадой вагусных влияний.
Внесердечные эффекты
Прокаинамид обладает ганглиоблокирующим действием, он может снижать периферическое сосудистое сопротивление и вызвать гипотензию, особенно при внутривенном введении. Однако при терапевтических концентрациях его периферические сосудистые эффекты менее выражены, чем у хинидина. Гипотензия обычно наблюдается только после быстрой внутривенной инфузии прокаинамида.
Токсичность
А. Кардиотоксичность. Кардиотоксический эффект прокаинамида сходен с эффектом хинидина. Может развиваться антимускариновое и кардио-депрессивное действие. Наблюдается и аритмоген-ный эффект.
Б. Внесердечная токсичность. Наиболее неприятный побочный эффект прокаинамида — синдром, напоминающий красную волчанку и обычно проявляющийся артралгией и артритом. У некоторых больных, кроме того, развиваются плеврит, перикардит или паренхиматозное поражение легких. При длительном лечении почти у всех больных находят серологические отклонения (например, повы-
1 ТАБЛИЦА 14-1. Действие противоаритмических средств на мембрану
Препараты Блокада натриевых каналов Рефрактерный период Блокада кальцие-ВЫХ каналов Влияние на пейсмекерную активность Симпато-литичес-кое действие
Нормальные клетки Деполяризованные клетки Нормальные клетки Деполяризованные клетки
Аденозин(Аденокард) 0 0 0 0 0 0 +
Амиодарон(Кордарон) + +++ ТТ ТТТ + XX +
Бретилиум (Бретилол) 0 0 ттт ттт 0 ТХ’ ++
Дизопирамид (Норпейс) + +++ т тт + XX 0
Эсмолол (Бревиблок) 0 + 0 Нет данных 0 XX +++
Флекаинид (Тамбокор) + +++ 0 т 0 XX 0
Имипрамин (исследуется) + ++ Т тт 0 XX 0
Лидокаин(Ксилокаин) 0 +++ X тт 0 XX 0
Мексилетин (Мекситил) 0 +++ 0 тт 0 XX 0
Морицизин(Этмозин) + ++ г X 0 XX 0
Фенитоин (Дилантин) 0 + г т + X +
Прокаинамид (Пронестил и др.) + +++ т ттт 0 X +
Пропафенон (Ригмол) + ++ т тт + XX +
Пропранолол (Индерал) 0 + X тт 0 XX +++
Хинидин + ++ т тт 0 XX +
(много торговых названий)
Соталол(Бетапейс) 0 0 тт ттт 0 XX ++
Токаинид (Тонокард) 0 +++ 0 тт 0 XX 0
Верапамил(Калан, Изоптин) 0 + 0 т +++ XX +
' Бретилиум может временно повышать пейсмекерную частоту, вызывая высвобождение катехоламинов.
шение титра противоядерных антител), но при отсутствии симптоматики нет показаний к прекращению лечения. Волчаночноподобные симптомы развиваются примерно у одной трети больных, длительно получающих прокаинамид.
Другие побочные эффекты — это тошнота и диарея (примерно в 10 % случаев), сыпь, лихорадка, гепатит (менее 5 %) и агранулоцитоз (менее 0.2 %).
Фармакокинетика и дозирование
Прокаинамид можно безопасно вводить внутривенно и внутримышечно. Он хорошо всасывается при приеме внутрь, имея системную биодоступность 75 %. Основной метаболит прокаинамида — N-ацетилпрокаинамид (NAPA, ацекаинид), который является слабым блокатором натриевых каналов и имеет активность, характерную для препаратов класса III. Избыточное накопление NAPA может играть роль в возникновении torsade de pointes при лечении прокаинамидом. Некоторые люди быстро ацетилируют прокаинамид, что приводит к вы
сокой концентрации NAPA. Волчаночноподобный синдром у этих больных возникает реже.
Период полувыведения прокаинамида около 3-4 часов, что требует частого введения. Как прокаинамид, так и NAPA элиминируются главным образом почками. Следовательно, больным с почечной недостаточностью следует уменьшать дозу. Сниженный объем распределения и почечный клиренс при застойной сердечной недостаточности тоже требуют меньших доз. Период полувыведения NAPA значительно дольше, чем у прокаинамида, и, следовательно, способность к кумуляции у них неодинакова. Таким образом, важно измерять уровень в плазме прокаинамида и NAPA, особенно у больных с нарушениями кровообращения или функции почек.
Если требуется быстрый эффект, можно без опасений внутривенно вводить нагрузочную дозу до 12 мг/кг со скоростью 0.3 мг/кг/мин или медленнее. За этой дозой следует ввести поддерживающую дозу 2-5 мг/мин при тщательном наблюдении за уровнем препарата в плазме. Риск токсического
ТАБЛИЦА 14-2. Клинико-фармакологические свойства противоаритмических средств
Препараты Вл1 ляние астоту □атри-□ного зла Влияние на рефрактерный период атриовентрикулярного узла Интервал PR Длительность QRS Интервал QT Эффективность при аритмиях Период полувыведения
на 41
наджелудочковых желудочковых
ani
У
Аденозин (Аденокард) Незначительно ТТТ ТТТ 0 0 ++++ ? <10с
Амиодарон(Кордарон) U’ ТТ ТТ т тттт +++ +++ недели
Бретилиум (Бретилол) Н2 ТТ 0 0 0 0 + 4ч
Дизопирамид (Норпейс) ТТЛ3 П3 ТТ3 ТТ ТТ + +++ 6-8 ч
Эсмолол(Бревиблок) а ТТ ТТ 0 0 + + 10 мин
Флекаинид (Тамбокор) Нет т т ттт 0 +4 ++++ 20 ч
Имипрамин (исследуется) ^1.3 т т т т + Ч-++ 12ч
Лидокаин(Ксилокаин) Нет’ Нет 0 0 0 Нет6 +++ 1 ч
Мексилетин (Мекситил) Нет’ Нет 0 0 0 Нет5 +++ 12ч
Морицизин (Этмозин) Нет Нет т ТТ 0 Нет +++ 2-6 ч5
Фенитоин (Дилантин) Нет Нет 0 т 0 Нет6 + 24 ч
Прокаинамид (Пронестил и др.) i’ ТТ3 ТТ3 ТТ ТТ + +++ 3-4 ч
Пропафенон (Ритмол) 0 т т ттт 0 + +++ 7ч
Пропранолол (Индерал) и ТТ ТТ 0 0 + + 8ч
Хинидин (много торговых названий) ТТ'13 Н3 ТТ3 ТТ ТТ + +++ 6ч
Соталол (Бетапейс) U ТТ ТТ 0 ттт +++ +++ 7ч
Токаинид (Тонокард) Нет’ Нет 0 0 0 Нет6 +++ 12ч
Верапамил (Калан, Изоптин) ТТ ТТ ТТ 0 0 +++ + 7ч
1 Может супрессировать пораженные синусные узлы.
2 Начальная стимуляция высвобождающимся эндогенным норадреналином с последующей депрессией.
3 Антихолинергический эффект и прямое депрессивное действие.
4 Особенно при синдроме Вольфа-Паркинсона-Уайта.
5 Время полувыведения активных метаболитов значительно больше.
6 Может быть эффективным при предсердных аритмиях, вызванных сердечными гликозидами.
воздействия на ЖКТ или сердце возрастает при концентрации прокаинамида в плазме более 8 мкг/мл или концентрации NAPA выше 20 мкг/мл.
Внутрь прокаинамид часто назначают нерационально. Если требуется постоянное противоаритми-ческое действие, в большинстве случаев лучше назначать каждые 6 часов препараты с медленным высвобождением. Для контроля желудочковых аритмий обычно необходима общая доза 2—5 г в день. Больным, у которых накапливается и проявляет активность NAPA, нужно снизить частоту приема. Это необходимо и пациентам с почечными заболеваниями, когда замедлена элиминация прокаинамида.
Терапевтическое применение
Как и хинидин, прокаинамид эффективен в отношении большинства предсердных и желудочковых аритмий. Однако многие клиницисты стараются избежать длительного лечения препаратом из-за высокой частоты приема и вероятности развития волчаночноподобного синдрома. Прокаинамид является препаратом второй линии (после лидокаина) для терапии стойких желудочковых аритмий при остром инфаркте миокарда.
Дизопирамид (класс IA)
Дизопирамида фосфат похож по структуре на изопропамид — вещество, обладающее антимуска-риновыми свойствами.
СН(СН3)2
СН(СН3)2
Влияние на сердце
Эффекты дизопирамида очень сходны с эффектами хинидина. Его антимускариновое действие на сердце даже более выражено, чем у хинидина. Следовательно, при лечении трепетания или мерцания предсердий совместно с дизопирамидом необходимо назначать вещество, которое замедляет атриовентрикулярную проводимость.
Токсичность
А. Кардиотоксичность. Токсические концентрации дизопирамида могут вызывать все электрофизиологические нарушения, характерные для хинидина. Кроме того, у больных с предсуществующей дисфункцией левого желудочка может проявляться отрицательный инотропный эффект. В редких случаях препарат вызывает сердечную недостаточность даже у пациентов без нарушения функции миокарда. Вследствие этого действия дизопирамид не используют в качестве противоаритмического средства первой линии в США. Он должен применяться с большой осторожностью у больных с застойной сердечной недостаточностью.
Б. Внесердечные эффекты. Атропиноподобная активность дизопирамида определяет большинство симптоматических побочных эффектов: задержку мочи (чаще, но не исключительно, у мужчин с гипертрофией простаты), сухость рта, нарушение зрения, запор и ухудшение течения глаукомы. При возникновении этих эффектов может потребоваться отмена препарата.
Фармакокинетика и дозирование
В США дизопирамид применяется только внутрь. Его биодоступность составляет около 50 %. Вещество активно связывается с белками, но места связывания быстро насыщаются при увеличении дозы, что отражается нелинейным увеличением уровня свободной (активной) фракции. Результаты измерения общей концентрации в плазме могут вводить в заблуждение. Вещество экскретируется через почки и имеет период полувыведения примерно 6-8 часов. Обычная доза дизопирамида внутрь составляет 150 мг три раза в день, но может потребоваться ее увеличение до 1 г в день. У больных с почечной недостаточностью следует уменьшать дозы. Из-за опасностей провокации застойной сердечной недостаточности не рекомендуют применять нагрузочные дозы.
Терапевтическое применение
Хотя дизопирамид эффективен при ряде наджелудочных аритмий, в США он разрешен только для лечения желудочковых аритмий. Наиболее часто его применяют, когда хинидин и прокаинамид плохо переносятся больным или неэффективны.
Имипрамин (класс IA)
Имипрамин — трициклический антидепрессант, обладающий противоаритмической активностью. По электрофизиологическим эффектам и клиническому спектру действия он похож на хинидин. Подобно дизопирамиду имипрамин обладает выраженным антимускариновым действием. Период полуэлиминации составляет около 12 часов. Обычная дневная доза — 200 мг/сутки в два приема. Начальная доза должна быть меньше, потому что наиболее сильный побочный эффект препарата (седативный) меньше выражен при медленном увеличении доз. Имипрамин применяется как антидепрессант, его использование в качестве противоаритмического средства не утверждено FDA.
Амиодарон (классы I, II, III и IV)
Амиодарон широко используется как противо-аритмическое и антиангинальное средство в Европе и Южной Америке. В США он разрешен для применения при аритмиях. Препарат очень эффекти
вен при разных нарушениях ритма, но его выраженные побочные эффекты и необычные фармакокинетические свойства ограничили его применение в США.
,1
\ /СгН5
V- О — СН2 — СН2 — N
/ ХС2Н5
I
"О' хсн2 — сн2— сн2— сн3 Амиодарон
Влияние на сердце
Амиодарон имеет широкий спектр влияний на сердце. Препарат очень эффективно блокирует натриевые каналы, но в отличие от хинидина у него относительно низкий аффинитет к активированным каналам по сравнению с инактивированными. Поэтому натриевая блокада под действием амиодарона наиболее выражена в тканях с длительными и частыми потенциалами действия или при менее отрицательном диастолическом потенциале. В терапевтических концентрациях амиодарон также значительно удлиняет потенциал действия, вероятно, за счет блокады калиевых каналов. Это действие препарата выражено и при высокой частоте сокращений. Возможно, в этом причина его эффективности при тахикардиях. Феномен torsade de pointes очень редко наблюдается при использовании препарата. Амиодарон — слабый блокатор кальциевых каналов и, кроме того, неконкурентный ингибитор p-адренорецепторов. Показано, что препарат существенно подавляет аномальный автоматизм.
Амиодарон замедляет синусовый ритм и атриовентрикулярную проводимость, значительно удлиняет интервал QT и пролонгирует QRS. Он повышает рефрактерность предсердий, атриовентрикулярного узла и желудочков. При лечении амиодароном редко возникают новые или ухудшаются существующие желудочковые аритмии. У больных ссиндромом Вольфа-Паркинсона-Уайта препарат замедляет проведение, удлиняет эффективный рефрактерный период и может полностью устранить передачу импульсов через дополнительные пути.
Амиодарон обладает также антиангинальным эффектом. Он может быть связан с неконкурент
ным а- и p-адреноблокирующим действием препарата и со способностью подавлять вход Са2+ в гладкомышечные клетки коронарных сосудов.
Внесердечные эффекты
Амиодарон вызывает расширение периферических сосудов предположительно за счет а-адрено-блокирующего действия и ингибирования кальциевых каналов. Некоторым больным этот эффект полезен, другим из-за него может потребоваться прекращение приема препарата.
Токсичность
А. Кардиотоксичность. У больных с поражением синусного или атриовентрикулярного узлов амиодарон может вызвать симптоматическую брадикардию или блокаду сердца. У чувствительных пациентов он способствует развитию сердечной недостаточности.
Б. Внесердечные эффекты. Амиодарон вызывает ряд внесердечных побочных эффектов, в том числе потенциально опасный легочный фиброз. Количество побочных эффектов возрастает при увеличении кумулятивной дозы и ограничивает применение препарата для длительной терапии.
Амиодарон накапливается в тканях практически всех органов. Наиболее легко обнаруживаются желтовато-коричневые микрокристаллические отложения в роговице, появляющиеся через несколько недель после начала лечения. Эти отложения редко вызывают зрительные симптомы, исключая иногда возникающие ореолы по периферии зрительного поля. Лишь изредка происходит снижение остроты зрения, требующее прекращения лечения или снижения дозы. Примерно у 25 % больных отложения амиодарона в коже приводят к фотодерматитам. Этим людям следует по возможности избегать воздействия прямых солнечных лучей. Менее чем у 5 % пациентов появляются серовато-синие пигментации на коже.
Часто возникают побочные неврологические эффекты: парестезии, тремор, атаксия и головная боль.
Дисфункция щитовидной железы (как гипо-, так и гипертиреоидизм) развивается в среднем у 5 % больных. Функцию щитовидной железы следует контролировать перед началом и в процессе лечения амиодароном.
Препарат, может влиять на желудочно-кишечный тракт (например, запоры у 20 % пациентов),
печень (гепатоцеллюлярный некроз) или легкие (воспаление и фиброз). Легочный фиброз возникает у 5-15 % больных, у части из них он может оказаться фатальным.
Амиодарон взаимодействует с другими лекарствами, уменьшая, в частности, клиренс варфари-на, теофиллина, хинидина, прокаинамида, флекаи-нида и т. п. Хотя амиодарон — высокоэффективное противоаритмическое средство, указанные побочные эффекты ограничивают его клиническое применение в США.
Фармакокинетика и дозирование
Амиодарон имеет очень длительный период полувыведения (13-103 дня). Эффективная концентрация в плазме составляет примерно 1-2 мкг/мл, тогда как концентрация в сердце примерно в 30 раз выше. Даже при больших нагрузочных дозах требуется 15-30 дней для насыщения тканевых депо амиодароном. Используют нагрузочные дозы 0.8-1.2 г в день в течение двух недель, после чего переходят на поддерживающую дозу 0.2-1 г в день. В связи с продолжительным периодом полувыведения достаточно вводить препарат один раз в день. Если возникают побочные эффекты, они долго сохраняются и после отмены препарата.
Терапевтическое применение
Амиодарон очень эффективен как при предсердных, так и при желудочковых аритмиях. Обычно при пароксизмальной фибрилляции предсердий применяют сравнительно низкие дозы (200-400 мг в день). Амиодарон весьма эффективен при наджелудочковых аритмиях у детей (у которых он сравнительно безопасен). Однако опыта долгосрочного применения пока не накоплено. В настоящее время проводится несколько обширных клинических испытаний с целью оценки безопасности амиодарона и его эффективности у больных с прогрессирующими сердечными заболеваниями.
Лидокаин (класс IB)
Лидокаин — противоаритмическое средство, наиболее часто применяемое для внутривенного введения. Он отличается низкой токсичностью и высокой эффективностью при аритмиях, связанных с инфарктом миокарда.
Лидокаин
Влияние на сердце
Лидокаин активно подавляет аномальную активность в сердце и действуёт, как полагают, исключительно на натриевые каналы. Однако это взаимодействие иное, чем у хинидина. В то время как хинидин блокирует главным образом каналы в активированном состоянии, лидокаин блокирует и активированные, и инактивированные натриевые каналы (рис. 14-9). В результате значительная часть (более 50 %) незаблокированных натриевых каналов становится заблокированной во время каждого потенциала действия в волокнах Пуркинье и клетках желудочков, отличающихся продолжительным плато (и соответственно — длительным периодом инактивации). Во время диастолы большинство натриевых каналов нормально поляризованной клетки быстро освобождаются от лекарственного вещества. Так как лидокаин может укорачивать длительность потенциала действия, диастола может удлиняться, тем самым предоставляя дополнительное время для восстановления. В результате лидокаин имеет лишь незначительные электрофизиологические эффекты в нормальной ткани сердца. Напротив, деполяризованные (инактивированные) натриевые каналы остаются в основном заблокированными во время диастолы и еще большее их число может заблокироваться. Таким образом, лидокаин подавляет электрическую активность деполяризованных, аритмогенных участков, но минимально влияет на электрическую активность нормальных тканей. Поэтому лидокаин очень эффективен в отношении аритмий, связанных с деполяризацией (например, при ишемии, токсическом действии дигиталиса), но относительно неэффективен при аритмиях, развивающихся в нормально поляризованных тканях (например, при мерцании и трепетании предсердий).
Токсичность
А. Кардиотоксичность. Лидокаин наименее кар-диотоксичен из применяемых в настоящее время противоаритмических средств. Препарат усилива-
Рис. 14-9. Влияние мембранного потенциала на блокирование и разблокирование натриевых каналов лидокаином. Верхняя запись: потенциалы действия мышечных клеток желудочка; нижняя запись: процент каналов, заблокированных вешеством. Показан сегмент длительностью 800 мс, соответствующий деполяризации мембраны на уровнях -80, -75, -70 и -65 мВ. Перерывы показаны разрывами на записи. Слева: при нормальном потенциале покоя -85 мВ вещество соединяется с открытыми (активированными) и инактивированными каналами во время каждого потенциала действия, но блокада быстро устраняется во время диастолы, потому что аффинитет лекарства к рецептору низок, когда канал возвращается к потенциалу -85 мВ. В середине: произошло метаболическое повреждение, например ишемия из-за закупорки коронарной артерии, вызвавшее постепенную деполяризацию. При последующих потенциалах действия, возникающих на фоне более деполяризованных потенциалов, фракция блокированных каналов повышается, потому что больше каналов остается в инактивированном состоянии при менее отрицательном потенциале (рис. 14-4, слева) и постоянная времени для разблокирования в период диастолы быстро возрастает при менее отрицательных потенциалах покоя (рис. 14-4, справа). В связи с выраженным связыванием препарата возникает блок проведения и утрата возбудимости в ткани, т. е. “больная" (деполяризованная) ткань значительно подавляется
ет желудочковые аритмии менее чем у 10 % больных (хороший показатель). Однако примерно у 1 % больных с острым инфарктом миокарда лидокаин может подавить активность синоатриального узла или ухудшить нарушенную проводимость.
В больших дозах, особенно у больных с сердечной недостаточностью, лидокаин может понизить АД, отчасти за счет снижения сократительной способности миокарда.
В. Внесердечные эффекты. Наиболее частыми побочными эффектами лидокаина, подобно другим
местным анестетикам, являются неврологические: парестезии, тремор, тошнота центрального происхождения, головокружение, нарушения слуха, затрудненная речь и судороги. Последние возникают прежде всего у пожилых или чувствительных больных и дозозависимы, обычно кратковременны и купируются диазепамом, введенным внутривенно. Как правило, если уровень в плазме не превышает 9 мкг/мл, лидокаин хорошо переносится.
Фармакокинетика и дозирование
В связи с выраженным эффектом первого прохождения только 3 % препарата при приеме внутрь доходят до плазмы, поэтому лидокаин должен вводиться парентерально. Лидокаин имеет период полувыведения около двух часов. У взрослых за нагрузочной дозой 150-200 мг, введенной в течение 15 мин, следует поддерживающая инфузия со скоростью 2-4 мг/мин до достижения терапевтического уровня в плазме 2-6 мкг/мл. Определение уровней лидокаина в плазме очень ценно для регулирования скорости инфузии. Отдельным больным с инфарктом миокарда нужны более высокие концентрации, и они их хорошо переносят. Это может быть связано с повышенным уровнем «1-кислого гликопротеина (реактивного белка острой фазы), который связывает лидокаин, уменьшая его свободную фракцию, оказывающую фармакологическое действие.
У больных с застойной сердечной недостаточностью объем распределения лидокаина и общий клиренс могут снижаться. Однако из-за взаимного гашения этих эффектов период полувыведения лидокаина может возрастать не столь значительно, как можно предсказать только по изменению клиренса. У больных с заболеваниями печени плазменный клиренс значительно снижен, а объем распределения часто возрастает. В таких случаях период полуэлиминации может увеличиться в три и более раз. Период полуэлиминации, как правило, определяет время развития стационарного состояния. Так, если стационарные концентрации в норме и у больного с сердечной недостаточностью могут быть достигнуты через 8-10 часов, то при заболевании печени для этого может потребоваться 24-36 часов. Вещества, понижающие печеночный кровоток (например, пропранолол, циметидин), снижают клиренс лидокаина и повышают риск проявления токсичности, если не уменьшается скорость инфузии. При
инфузии, которая продолжается более 14 часов, клиренс снижается и концентрация в плазме возрастает. Заболевания почек не оказывают значительного влияния на распределение лидокаина.
Терапевтическое применение
Лидокаин является препаратом выбора при лечении желудочковой тахикардии и фибрилляции после кардиоверсии. Некоторые данные указывают на то, что лидокаин достоверно понижает частоту фибрилляции желудочков в первые несколько дней после острого инфаркта миокарда. Однако остается предметом споров, следует ли вводить его всем больным, перенесшим инфаркт. Есть сведения, что рутинное профилактическое назначение лидокаина может в действительности повысить риск смерти, вероятно, за счет увеличения частоты возникновения асистолий.
Лидокаин редко оказывается эффективным при наджелудочковых аритмиях, за исключением связанных с синдромом Вольфа-Паркинсона-Уайта или с передозировкой наперстянки..
Токаинид и мексилетин (класс IB)
Токаинид и мексилетин сходны по строению с лидокаином, но не подвергаются метаболизму в печени при первом прохождении. Следовательно, они могут вводиться внутрь. Их электрофизиологическое и противоаритмическое действие похоже на действие лидокаина. Они используются в терапии желудочковых аритмий. Период полувыведения препаратов колеблется между 8 и 20 часами,
их вводят 2-3 раза в день. Обычная суточная доза мексилетина составляет 600-1200 мг, а токаини-да — 800-2400 мг. Оба препарата вызывают дозозависимые побочные эффекты, которые отмечаются уже в терапевтических дозах. Они носят преимущественно неврологический характер (тремор, пелена перед глазами, вялость). Частым побочным эффектом является также тошнота. Высыпания, лихорадка и агранулоцитоз наблюдаются примерно у 0.5 % больных, получающих токаинид.
Мексилетин весьма эффективен для облегчения хронических болей, особенно при диабетической нейропатии и повреждении нервов (Chabal et al., 1992). Обычная доза в этих случаях составляет 450-750 мг в день. Однако это показание не зарегистрировано официально.
Фенитоин (класс IB)
Фенитоин является антиконвульсантом с анти-аритмическими свойствами (глава 23). В связи с ограниченной эффективностью он считается препаратом второй линии при лечении больных с аритмиями. Препарат подавляет эктопическую пейсме-керную активность, блокирует натриевый ток и может регулировать кальциевый ток. Он считается особенно эффективным при аритмиях, вызванных препаратами наперстянки.
Флекаинид (класс 1С)
Флекаинид — активный блокатор натриевых каналов, который применяется в основном в терапии желудочковых аритмий. Препарат также блокирует некоторые калиевые каналы, что позволяет эффективно использовать его при определенных предсердных аритмиях. У флекаинида нет антимуска-ринового действия. Он весьма эффективно подавляет преждевременные сокращения желудочков. Однако может вызывать выраженное ухудшение аритмии даже при введении нормальных доз у больных с желудочковыми тахиаритмиями, а также у больных, перенесших инфаркт миокарда и имеющих эктопические очаги в желудочках (дополнение “Испытания эффективности антиаритмической терапии”). Поэтому флекаинид назначают больным с тяжелыми желудочковыми тахиаритмиями и с более подходящим соотношением пользы и риска от лечения препаратом. Препарат также использу
ют при суправентрикулярных аритмиях, при которых в меньшей степени проявляется склонность к проаритмическому токсическому действию. Он хорошо всасывается и имеет период полувыведения примерно 20 часов. Элиминируется почками. Обычная доза флекаинида составляет 100-200 мг в день.
Пропафенон (класс IC)
Пропафенон имеет некоторое структурное сходство с пропранололом и обладает слабой р-блоки-рующей активностью. Спектр его действия очень сходен со спектром действия хинидина. Активность в качестве блокатора натриевых каналов близка к активности флекаинида. Среднее время полуэлиминации составляет 5.5 часа за исключением лиц с пониженным метаболизмом (10 % населения), у которых эта величина достигает 17 часов. Суточная доза пропафенона 450-900 мг в три приема. Препарат широко используется в Европе. Наиболее частые побочные эффекты — металлический привкус во рту и запоры.
Морицизин (класс IC)
Производное фенотиазина морицизин — анти-аритмическое средство, которое применяют для лечения желудочковых аритмий. Это сравнительно активный блокатор натриевых каналов, не удлиняющий потенциал действия. Он имеет свойства препаратов класса IC. Морицизин образует у человека много метаболитов; некоторые из них, возможно, активны и имеют длительный период полувыведения. Наиболее частые побочные эффекты — головокружение и тошнота. Как и другие активные блокаторы натриевых каналов, он может ухудшать течение аритмий. Обычная доза морицизина внутрь — 200-300 мг в день в три приема.
p-адреноблокаторы (класс II)
Влияние на сердце
Пропранолол и родственные препараты имеют противоаритмические свойства за счет блокады Р-адренорецепторов и прямых мембранных эффектов. Как описано в главе 10, часть из этих веществ селективна в отношении Pi-рецепторов сердца, часть обладает внутренней симпатомиметической
активностью, некоторые — выраженным влиянием на мембрану, а некоторые пролонгируют потенциал действия в сердце. Относительный вклад Р-ад-реноблокирующего и прямого мембранного действия в антиаритмические эффекты этих средств не совсем ясен. Р-блокаторы весьма хорошо переносятся, но их эффективность в подавлении эктопической деполяризации в желудочках ниже, чем у блокаторов натриевых каналов. Однако существуют свидетельства того, что эти препараты могут предупреждать повторные инфаркты и внезапную смерть у больных в периоде восстановления после острого инфаркта миокарда (глава 10).
Короткодействующий Р-блокатор эсмолол преимущественно применяют как противоаритмическое средство во время операций и при других острых аритмиях (более подробно рассмотрено в главе 10). Соталол — неселективное р-блокирующее средство, которое удлиняет потенциал действия (действие по типу препаратов класса III).
Средства, которые удлиняют эффективный рефрактерный период и потенциал действия (класс III)
Эти препараты удлиняют потенциалы действия обычно за счет блокады калиевых каналов в сердечной мышце. В настоящее время наиболее важные вещества этой категории — те, что имеют дополнительные эффекты, характерные для других подгрупп. Хинидин и особенно амиодарон эффективно удлиняют потенциал действия. Для увеличения эффективного рефрактерного периода, как уже отмечалось, имеет значение также их влияние на натриевые каналы. Обе эти характеристики возрастают при приеме бретилиума и соталола. Как известно, удлинение потенциала действия под влиянием таких препаратов характеризуется обратной “зависимостью от использования”, что может спровоцировать феномен torsade de pointes (по всей видимости, амиодарон является исключением). В настоящее время на клинических испытаниях находится большое число новых лекарств с действием по типу класса III (Wong et al., 1992). Предполагается, что
они окажутся менее аритмогенными, чем вещества класса I. Есть данные о том, что некоторые препараты класса III проявляют скорее положительное, чем отрицательное инотропное действие на сердце.
Бретилиум
Бретилиум был первоначально внедрен в практику как антигипертензивное средство. Он участвует в высвобождении катехоламинов нейронами (сначала усиливает, в дальнейшем — подавляет процесс), но имеет и прямые антиаритмические свойства.
Вг
СН3
CH2-+N-C2H5
I сн3
Бретилиум
Влияние на сердце
Бретилиум удлиняет потенциал действия и эффективный рефрактерный период желудочков (но не предсердий). Эффект наиболее выражен в ишемизированных клетках, которые характеризуются укороченным потенциалом действия. В эксперименте показано, что бретилиум значительно увеличивает силу электрического раздражения, необходимого для провокации фибрилляции желудочков (порог фибрилляции желудочков) и задерживает начало фибрилляции после перевязки коронарных сосудов. Антифибрилляторное действие препарата, по всей вероятности, не зависит от его симпатоли-тических свойств.
Так как бретилиум сначала вызывает высвобождение катехоламинов, при первом введении он дает положительный инотропный эффект. Это может служить причиной желудочковых аритмий, следовательно, в начале терапии за пациентом необходимо тщательное наблюдение.
Внесердечные эффекты
Их можно предсказать, исходя из симпатопле-гических свойств препарата. Основной побочный эффект — постуральная гипотензия. Этот эффект можно почти полностью предупредить при совместном введении с бретилиумом трициклических ан
тидепрессантов (таких как протриптилин). После внутривенного болюсного введения препарата могут возникать тошнота и рвота.
Фармакокинетика и дозирование
В США бретилиум используют только внутривенно. Взрослым внутривенную дозу бретилиума тозилата (5 мг/кг) вводят в течение 10 мин. Процедуру можно повторить через 30 мин. Поддерживающая терапия достигается аналогичным болюсным введением каждые 4-6 часов или путем постоянной инфузии со скоростью 0.5-2 мг/мин.
Терапевтическое применение
Бретилиум обычно применяют в неотложных ситуациях, часто при реанимационных мероприятиях, связанных с фибрилляцией желудочков, когда лидокаин и кардиоверсия оказываются неэффективными. В редких случаях бретилиум подавляет преждевременные сокращения желудочков.
Соталол
Соталол — неселективный p-блокатор, который увеличивает продолжительность потенциала действия и является эффективным противоаритмичес-ким агентом. Его D-изомер, исследуемый в настоящее время, не имеет p-блокирующих свойств, но сохраняет влияние на реполяризацию. Соталол можно использовать как при наджелудочковых, так и при желудочковых аритмиях. Обычная эффективная доза составляет 4-6 мг/кг в день в 2-3 приема. Основные побочные эффекты связаны с р-блока-дой (глава 10) и удлинением реполяризации, в том числе torsade de pointes. В отличие от хинидина соталол с большей вероятностью вызывает torsade de pointes после приема больших доз и при высоких концентрациях в плазме.
Соталол
Средства, блокирующие кальциевые каналы (класс IV)
Препараты этой группы, прототипом которых является верапамил, были впервые внедрены в практику в качестве антиангинальных средств; они рассмотрены в главе 12. Верапамил, дилтиазем и бепридил обладают также противоаритмическими свойствами. Нифедипин и другие дигидропиридиновые блокаторы кальциевых каналов проявляют слабый антиаритмический эффект.
Верапамил
Влияние на сердце
Верапамил блокирует как активированные, так и инактивированные кальциевые каналы. Поэтому эффект препарата более выражен в тканях, для которых характерна высокая частота активации, меньшая поляризация в состоянии покоя и в которых активация зависит преимущественно от тока кальция (например, синоатриальный и атриовентрикулярный узлы). При использовании терапевтических доз верапамила проведение в атриовентрикулярном узле и рефрактерный период удлиняются. Как правило, препарат замедляет активность синоатриального узла за счет прямого действия, но его гипотензивный эффект может иногда привести к незначительному рефлекторному повышению частоты импульсов в синоатриальном узле.
Верапамил может подавлять ранние и поздние следовые деполяризации и медленные Са2+-зависи-мые ответы, усиливающиеся в резко деполяризованной ткани.
Внесердечные эффекты
Верапамил вызывает периферическую вазодилатацию, которая может быть полезна при гипертензии и периферических вазоспастических состояниях. Ряд внесердечных эффектов вызывает и его влияние на гладкую мускулатуру (глава 12).
Токсичность
А. Кардиотоксичность. Кардиотоксические эффекты верапамила зависят от дозы, и их можно избежать. Частая ошибка состоит во внутривенном
введении верапамила больному с желудочковой тахикардией, которую неверно признали наджелудочковой. В этом случае может развиться гипотензия и фибрилляция желудочков. Отрицательный инотропный эффект верапамила ограничивает его клиническое применение при поврежденном сердце (глава 12). Верапамил при назначении в высоких дозах, а также у больных с нарушениями предсердно-желудочковой проводимости может вызвать атриовентрикулярную блокаду. Таких пациентов следует лечить атропином, стимуляторами р-адрено-рецепторов или кальцием. У больных с заболеваниями синусного узла верапамил может спровоцировать полное подавление его активности.
Б. Внесердечные эффекты. К второстепенным побочным эффектам относятся запоры, периферические отеки, утомляемость и нервозность.
Фармакокинетика и дозирование
Период полувыведения верапамила примерно 7 часов. Препарат активно метаболизируется в печени: после приема внутрь его биодоступность составляет только около 20 %. Следовательно, верапамил необходимо с осторожностью назначать больным с печеночной дисфункцией. При внутривенном введении требуется значительно уменьшить дозу.
У взрослых без сердечной недостаточности или патологии синоатриального и атриовентрикулярного узлов парентеральная терапия суправентрикулярной тахикардии заключается в болюсном введении 5 мг препарата в течение 2-5 мин, за которым через несколько минут при необходимости можно ввести вторую такую же дозу. Впоследствии 5-10 мг верапамила можно вводить каждые 4-6 часов или производить постоянную инфузию со скоростью 0.4 мкг/кг/мин.
Эффективные дозы для приема внутрь находятся в диапазоне от 120 до 600 мг в день в 3-4 приема.
Терапевтическое применение
Основным показанием к применению верапамила является суправентрикулярная тахикардия по механизму reentry. В этом случае препарат предпочитают более старым средствам (пропранолол, дигоксин, эдрофоний, вазоконстрикторные препараты, кардиоверсия). Верапамил может также уменьшать частоту сокращения желудочков при фибрил-
ляции и трепетании предсердий. Он способствует переходу фибрилляции и трепетания предсердий в синусовый ритм. При желудочковых аритмиях верапамил применяют очень редко.
Дилтиазем и бепридил
Эти препараты сходны по эффективности с верапамилом в терапии наджелудочковых аритмий, в том числе при фибрилляции предсердий. Бепридил может также удлинять потенциал действия и QT, что полезно при некоторых желудочковых аритмиях, но создает риск возникновения torsade de pointes.
Другие противоаритмические средства
Некоторые вещества, используемые для лечения больных с аритмиями, не соответствуют классификационным признакам классов I-IV. К ним относятся препараты наперстянки (глава 13), аденозин, магний и калий.
Аденозин
Механизм действия и клиническое применение
Аденозин — нуклеозид, образующийся в организме. Его период полусуществования в крови менее 10 с. Механизм действия связан с повышением проводимости калия и подавлением вызванного цАМФ входа Са2+ в клетку. Результатом этих действий являются выраженная гиперполяризация и подавление кальцийзависимых потенциалов действия. При болюсном введении аденозин прямо угнетает проводимость в атриовентрикулярном узле и увеличивает его рефрактерность, но незначительно влияет на функцию синоатриального узла. Аденозин является препаратом выбора для неотложной терапии пароксизмальных наджелудочковых тахикардий в связи с высокой эффективностью (90-95 %) и кратковременным действием. Его обычно назначают в виде болюсной дозы 6 мг, после чего при необходимости добавляют еще 12 мг. Препарат иногда помогает при желудочковой тахикардии. Он менее эффективен в присутствии блокаторов
аденозиновых рецепторов, таких как теофиллин г кофеин.
Токсичность
Аденозин вызывает приливы крови примерно у 20 % больных и одышку или ощущение жжения е груди (возможно, в связи с бронхоспазмом) более чем у 10 %. Возможно возникновение очень кратковременного атриовентрикулярного блока. К менее частым побочным эффектам относятся головная боль, гипотензия, тошнота и парестезии.
Магний
Первоначально препараты магния использовали для лечения больных с аритмиями, вызванными передозировкой дигиталиса, у которых отмечалась гипомагниемия. Затем было выявлено их антиарит-мическое действие и у некоторых больных с нормальным уровнем Mg2+ в сыворотке. Механизмь этих эффектов неизвестны, но считается, что препараты магния влияют на натриевые каналы, активность На+,К+-АТФазы, некоторые калиевые и кальциевые каналы. Терапия магнием показана пациентам с дигиталисными аритмиями, если имеется гипомагниемия, а также некоторым больным с torsade de pointes и в ряде случаев острого инфаркта миокарда, в том числе при нормальной концентрациг магния в сыворотке.
Калий
Значение поддержания концентрации ионов калия внутри и вне клетки уже обсуждалось в этой главе. К основным эффектам К+ относятся: 1) деполяризация потенциала покоя и 2) стабилизации мембранного потенциала, связанная с повышением проницаемости для ионов калия. Гипокалиемии приводит к повышению риска ранних и позднил следовых деполяризаций, к эктопической пейсме-керной активности, особенно в присутствии препаратов наперстянки. Гипокалиемия вызывает подавление эктопических водителей ритма (для подавления синоатриального узла требуется выраженная гипокалиемия) и замедление проводимости. Поскольку как недостаток, так и избыток К+ являются потенциально аритмогенными, лечение препаратами калия направлено на нормализацию калиевого градиента и пулов К+ в организме.
III. Принципы
клинического применения противоаритмических средств
Одним из факторов эффективности лечения любым препаратом является соотношение терапевтических и токсических доз. У противоаритмических средств особенно узка граница между эффективностью и токсичностью. Следовательно, врач, прописывающий противоаритмические препараты, должен быть хорошо осведомлен о показаниях, противопоказаниях, факторах риска и клинико-фармакологических характеристиках каждого используемого лекарства.
Оценка до начала лечения
До начала любой противоаритмической терапии следует учесть несколько важных факторов.
1. Должны быть устранены любые аритмоген-ные влияния. Это касается не только отклонений внутреннего гомеостаза, таких как гипоксия или электролитные нарушения (особенно гипокалиемия или гипомагниемия), но и лекарственной терапии. К препаратам, вызывающим аритмию, относятся сердечные гликозиды, все антиаритмические средства и многие вещества, которые не расценивают обычно как влияющие на сердечно-сосудистую систему. Например, высокие дозы эритромицина, пентамидина, трициклических антидепрессантов, антипсихотических средств (особенно тиоридазина), а также “неседативные” антигистаминные средства (терфенадин и астемизол) могут вызвать синдром torsade de pointes. Другие устранимые причины аритмий связаны с сопутствующими заболеваниями. Так, антиаритмическая терапия фибрилляции предсердий малоэффективна при гипертиреоидизме. Большинство больных с аритмиями имеют сопутствующие заболевания сердца. Важно отделить этот органический источник аритмии от триггерных факторов, таких как ишемия миокарда или острая дилатация, которые можно лечить и устранить.
2. Следует точно определить характер аритмии. Например, необоснованное применение верапамила у больных с желудочковой тахикардией, непра
вильно распознанной как наджелудочковая тахикардия, может привести к катастрофической гипотензии и остановке сердца. По мере усовершенствования методов определения механизмов нарушений ритма сердца появится возможность в большей степени индивидуализировать лечение в соответствии с характером аритмии у конкретного больного.
3. Важно установить надежные показатели, по которым можно определять эффективность проводимого лечения. В настоящее время имеется ряд методов для такой оценки. Они включают длительное амбулаторное наблюдение, электрофизиологические исследования, которые воспроизводят определенны# аритмии; воспроизведение аритмии при нагрузке ца тредмилле или применение методов мониторинга для регистрации спорадических, но симптоматических аритмий. Какую бы исходную базу для анализа эффективности терапии ни использовали, следует учитывать феномен смещенной оценки: частота аритмий может колебаться в зависимости от времени года, и больной, вероятнее всего, обратится к врачу, когда имеются симптомы заболевания. Следовательно, даже полностью неэффективные лекарства могут оказать противоарит-мическое действие.
4. Простая констатация аномалии сердечного ритма еще не означает, что необходимо лечение. Четкое обоснование консервативного лечения было представлено в рамках программы “Испытания эффективности антиаритмической терапии” (Cardiac Arrhythmia Suppression Trial — CAST; дополнение).
Польза и риск
Необходимость противоаритмической терапии в действительности определить достаточно трудно. Следует учитывать два вида возможных преимуществ лечения: 1) ослабление симптомов, связанных с аритмией, таких как сердцебиение, обморок или угнетение сердечной деятельности и 2) снижение показателя отсроченной смерти у больных без симптоматики. Среди препаратов, которые обсуждаются в данном разделе, только Р-блокаторы определенно снижают смертность у сравнительно бессимптомных больных. Механизм, лежащий в основе этого эффекта, еще не установлен (глава 10).
Антиаритмическая терапия сопряжена с рядом опасностей. В некоторых случаях риск побочного действия прямо связан с высокими дозами или концентрациями в плазме. В качестве примера можно
Испытания эффективности антиаритмической терапии
Преждевременные сокращения желудочков (ПСЖ) обычно регистрируют у больных, выздоравливающих после инфаркта миокарда. Поскольку эти аритмии сопряжены с повышенным риском внезапной сердечной смерти, многие врачи эмпирически назначали противоаритми-ческую терапию таким больным даже при отсутствии явной симптоматики. В ходе Испытаний эффективности антиаритмической терапии (CAST; Investigators, 1989; Echt et al., 1991) была сделана попытка определить эффективность такого лечения в контролируемых клинических исследованиях. Вначале оценивали влияние некоторых противоаритмических средств на частоту аритмий в открытом испытании. Затем больные, у которых противоаритмическая терапия подавляла ПСЖ, в условиях рандомизации
и двойного слепого контроля получали дальнейшее лечение препаратами или плацебо.
Результаты показали, что смертность среди пациентов, которых лечили флекаинидом и эн-каинидом, была более чем в два раза выше по сравнению с группой, получавшей плацебо. Механизм этого эффекта неясен, хотя вполне возможна корреляция между подавлением проведения при блокаде натриевых каналов и возникновением хронической или острой ишемии миокарда. Каков бы ни был механизм, важный урок, полученный CAST, состоит в том, что решение начать любую форму лекарственной терапии должно основываться на знании (или, по меньшей мере, на разумном предположении), что любой риск перевешивает реальную или потенциальную пользу.
привести вызванный лидокаином тремор или хинидиновый цинхонизм. Иногда развитие побочных реакций не определяется высоким содержанием препарата в плазме (например, агранулоцитоз, вызванный прокаинамидом илитокаинидом). Многие серьезные побочные реакции возникают при сочетании противоаритмической терапии с предшествующими сердечными заболеваниями. Например, у больных с нарушениями проводимости повышен риск полного сердечного блока во время лечения такими препаратами как хинидин или флекаинид, тогда как у больных без нарушений сердечной проводимости этот риск невелик. Схрдным образом развитие застойной сердечной недостаточности чаще возникает во время лечения больных с сократительной дисфункцией левого желудочка. В таких случаях риск сердечной недостаточности особенно велик при использовании 0-блокаторов, верапамила, дизопирамида и флекаинида.
Выявлены некоторые специфические синдромы спровоцированных противоаритмическими средствами аритмий с соответствующими патофизиологическими механизмами развития и факторами риска. Такие вещества как хинидин или соталол, которые удлиняют потенциалы действия в сердце, могут вызвать выраженное удлинение QT и характерные полиморфные желудочковые torsade de
pointes. Терапия torsade de pointes требует распознавания аритмии, отмены любого провоцирующего агента, коррекции гипокалиемии и лечения, направленного на повышение частоты сердечных сокращений (ходьба или введение изопротеренола). Может быть эффективным и внутривенное введение препаратов магния, причем даже у больных с нормальным уровнем Mg2*. Такие, заметно понижающие проводимость вещества, как флекаинид или хинидин, в высоких концентрациях могут увели? чить вероятность возникновения аритмий по типу повторного входа, особенно желудочковую тахикардию с механизмом возврата у больных с инфарктом миокарда в анамнезе, у которых может существовать потенциальный контур для повторной циркуляции импульсов. Лечение в этом случае заключается в своевременном распознавании, отмене провоцирующей терапии и внутривенном введении препаратов натрия. У некоторых больных с такой формой ухудшения аритмии реанимационные мероприятия оказываются неэффективными, и может наступить смерть. Таким образом, врач, выпи? сывающий рецепт, сталкивается со следующим противоречием: у больных, которым лечение антиарц?-? мическими средствами может принести наибрдь? шую пользу (например, при запущенном заболевании коронарных артерий, нарушениях проводимо?
сти, дисфункции левого желудочка и рецидивирующей желудочковой тахикардии), наиболее высок и риск нежелательных реакций на эти средства.
На практике выбор лекарственной терапии часто диктуется не столько ожидаемой эффективностью, сколько профилем токсичности. Наличие конкретных сопутствующих заболеваний может побудить клинициста отвергнуть препараты с определенными побочными эффектами в пользу других лекарственных средств. Помимо упомянутых нарушений функции левого желудочка и проводящей системы, на выбор препарата могут повлиять заболевания других органов. Например, заболевания простаты являются относительными противопоказаниями для назначения дизопирамида, так как этот препарат обычно вызывает задержку мочи даже у здоровых людей. У больных с артритом довольно сложно выявить развитие характерного волчаночноподобного синдрома, вызываемого прокаинамидом; следовательно, этот препарат относительно противопоказан таким пациентам. Сходным образом бывает трудно определить спровоцированные амиодароном нарушения со стороны легких у больных с запущенными легочными заболеваниями. Более того, по некоторым данным, у таких больных повышается риск вызываемых амиодароном нарушений функции легких.
Проведение противоаритмической терапии
Когда решение о начале лекарственной терапии принято, следующий этап состоит в определении того, является ли аритмия непосредственно угрожающей жизни. Если это так, применение нагрузочных доз оправдано. Если же нет, использование нагрузочных доз повышает риск токсического действия, не укорачивает время достижения стационарного состояния и не влияет на окончательное стационарное состояние, которое достижимо. Следовательно, без крайней необходимости не следует прибегать к нагрузочным дозам.
Лекарственная терапия расценивается как эффективная, когда аритмия подавляется (в сравнении с показателями, по которым она оценивалась исходно) и отсутствуют токсические проявления. Напротив, она считается неэффективной, если побочные эффекты возникают раньше, чем устраняется аритмия. Иногда аритмия может рецидивировать, когда уровень препарата в плазме относитель
но высок, а побочные эффекты еще не появились. В этих условиях врач должен решить, может ли разумное повышение дозы подавить аритмию и не вызвать побочного действия.
Мониторный контроль концентраций лекарства в плазме может эффективно дополнять противо-аритмическую терапию. Широкоизвестные терапевтические границы концентраций в действительности получены на основании двух кривых концентрация-эффект. Нижняя граница терапевтического диапазона определяется как концентрация, ниже которой обычно не отмечается клинически значимого действия лекарства. Верхняя граница — это концентрация, выше которой возрастание действия лекарства маловероятно или частота побочных эффектов резко увеличивается. У конкретного больного эффективное или побочное действие лекарства может наблюдаться при концентрациях, ниже обычной минимальной границы терапевтического диапазона, и, напротив, у некоторых пациентов лечебный эффект без признаков побочного действия достигается при концентрациях, больших, чем верхняя граница терапевтического диапазона. Таким образом, применение стратегии мониторного контроля концентраций требует, чтобы врач осознавал потенциальную токсичность, связанную с высокими концентрациями. Измерение концентрации вещества в плазме важно для оценки аккуратности и дисциплинированности больных в выполнении режима при длительном лечении, а также для определения взаимодействия лекарств, которое может привести к очень высоким концентрациям при приеме больших доз. Активные метаболиты некоторых лекарств (они могут оказывать фармакологическое действие, сходное с самим веществом или отличное от него) вносят определенный вклад в действие на больного. Присутствие таких активных метаболитов может затруднить интерпретацию данных измерения уровня лекарства в плазме, а в некоторых случаях делает рутинный мониторинг нецелесообразным.
Препараты
Блокаторы натриевых каналов (класс I)
Амиодарон (Кордарон)
Перорально: таблетки по 200 мг
Дизопирамид (генерик, Норпейс)
Перорально: капсулы поЮО, 150 мг
Перорально с контролируемым высвобождением (генерик, Норпейс CR): капсулы по 100, 150 мг
Флекаинид (Тамбокор)
Перорально: таблетки по 50,100,150 мг
Лидокаин (генерик, Ксилокаин)
Парентерально: 100 мг/мл для в/м инъекций; 10, 20 мг/мл для в/в инъекций;
40, 100, 200 мг/мл для добавления в растворы для в/в введения; готовый раствор
(в 5 % растворе глюкозы) для в/в введения
Мексилетин (Мекситил)
Перорально: капсулы по 150, 200, 250 мг
Морицизин (Этмозин)
Перорально: таблетки по 200, 250,300 мг
Прокаинамид (генерик, Пронестил, др.)
Перорально: таблетки и капсулы по 250,375, 500 мг
Перорально для пролонгированного действия (Прокаин-SR): таблетки по 250, 500,750, 1000 мг
Парентерально: 100, 500 мг/мл для инъекций
Пропафенон (Ритмол)
Перорально: таблетки по 150,300 мг
Хинидина сульфат [83 % хинидина] (генерик)
Перорально: таблетки по 200, 300 мг;
капсулы по 300 мг
Перорально для пролонгированного действия (Хинидекс Экстентабс): таблетки по 300 мг
Хинидина глюконат [62 % хинидина] (генерик)
Перорально для пролонгированного действия (Дураквин): таблетки по 324,330 мг Парентерально: 80 мг/мл для инъекций
Хинидина полигалактуронат [60 % хинидина] (Кардиоквин)
Перорально: таблетки по 275 мг
Токаинид (Тонокард)
Перорально: таблетки по 400, 600 мг
Р-блокаторы, применяемые для лечения аритмий (класс II)
Ацебутолол (Сектрал)
Перорально: капсулы по 200,400 мг
Эсмолол (Бревиблок)
Парентерально: 10 мг/мл, 250 мг/мл для в/в инъекций
Пропранолол (генерик, Индерал)
Перорально: таблетки по 10, 20, 40, 60, 80, 90 мг; растворы 4 мг/мл, 8 мг/мл;
Интенсол, 80 мг/мл
Перорально для пролонгированного действия: капсулы по 80,120,160 мг Парентерально: 1 мг/мл для инъекций
Блокаторы калиевых каналов (класс III)
Амиодарон: см. класс I
Бретилиум (генерик, Бретилол)
Парентерально: 50 мг/мл для инъекций
Соталол (Бетапейс)
Перорально: капсулы по 80, 160, 240 мг
Блокаторы кальциевых каналов (класс IV)
Верапамил (генерик, Калан, Изоптин) Перорально: таблетки по 40,80,120 мг Перорально для пролонгированного действия (Калан SR, Изоптин SR): таблетки и капсулы по 120,180, 240 мг Парентерально: 5 мг/2 мл для инъекций
Дилтиазем (генерик, Кардизем; не рекомендован для лечения аритмий)
Перорально: таблетки по 30, 60,90,120 мг
Перорально для пролонгированного действия: капсулы по 60, 90, 120,180, 240, 300 мг
Парентерально: 5 мг/мл для инъекций
Бепридил (генерик, Васкор; не рекомендован для лечения аритмий)
Перорально: таблетки по 200,300,400 мг
Препараты других групп
Аденозин (Аденокард)
Парентерально: 6 мг/2 мл для инъекций
Магния хлорид
Парентерально: 1г/20 мин;
при необходимости повторить
Мочегонные средства
Харлан Е. Ив, Дэвид Г. Уорнок
15
Изменения объема и нарушения электролитного состава жидкостей организма довольно часты и являются серьезными клиническими проблемами, которые могут приобретать угрожающий жизни больного характер. В настоящее время в терапии подобных нарушений широко применяют лекарственные средства, блокирующие транспортные функции почечных канальцев.
Различные средства, усиливающие мочеотделение, известны с античных времен, но только после 1957 г., когда был синтезирован хлортиазид, появились практичные и мощные диуретические агенты для широкого использования. Таким образом, учение о диуретиках сравнительно молодо. Термин “диурез” обозначает повышение объема мочи, тогда как “натрийурез” — усиленную экскрецию натрия почками. Так как основные натрийуретические средства почти всегда увеличивают и выведение воды, их обычно также называют диуретиками.
Многие мочегонные средства (петлевые диуретики, тиазиды, амилорид и триамтерен) действуют на специфические мембранные транспортные белки на люминальной поверхности эпителиальных клеток почечных канальцев. Другие оказывают осмотическое действие, которое предупреждает реабсорбцию воды в проницаемых для нее участках нефрона (маннитол), угнетают активность ферментов (ацетазоламид) или взаимодействуют с рецепторами гормонов в клетках эпителия почек (спиронолактон).
Каждый мочегонный препарат действует преимущественно на один анатомический сегмент нефрона (рис. 15-1). Поскольку эти сегменты имеют различные транспортные функции, терапевтический эффект действия каждого диуретика станет более понятным, если учитывать его место действия в нефроне и нормальную физиологию этого сегмента. Поэтому первый раздел данной главы посвящен обзору тех особенностей физиологии почечных канальцев, которые имеют отношение к действию диуретиков. Во второй части главы приведена базис
ная фармакология, а в третьей — обсуждается клиническое применение данной группы лекарственных средств.
Транспортные механизмы в канальцах почек
Проксимальные канальцы
Бикарбонат натрия, хлорид натрия, глюкоза, аминокислоты и другие растворенные органические вещества реабсорбируются преимущественно посредством специфического транспорта в начальных отделах проксимальных канальцев. Постоянство осмоляльности жидкости в них поддерживается при помощи пассивной реабсорбции воды. По мере прохождения жидкости вдоль проксимального канальца концентрация растворенных веществ в его просвете понижается относительно концентрации инулина — экспериментального маркера, который не реабсорбируется и не секретируется в почечных канальцах (рис. 15-2). В проксимальной' канальце концентрация инулина повышается вследствие обратного всасывания воды, тогда как здесь реабсорбируются примерно 85 % профильтровавшегося бикарбоната натрия, 40 % хлорида натрия, 60 % воды и практически все профильтровавшиеся органические соединения.
Из различных растворенных в фильтрате веществ, которые всасываются в этом сегменте нефрона, наибольшее отношение к мочегонному действию имеют бикарбонат натрия и хлорид натрия. Из современных диуретиков только один (ацетазоламид) действует преимущественно на проксимальный каналец, блокируя исключительно реабсорбцию бикарбоната натрия. Однако большой теоретический интерес представляет возможность воздействия на проксимальную реабсорбцию хлорида натрия. С учетом большого количества хлорида ййт-
Проксимальный извитой каналец\
NaHCO3 NaCI
NaCI
Са2+ (+ПТГ)
Дистальный извитой каналец
Клубочек
Кора
Проксимальный прямой каналец
Са2+
К+
Н+
Собирательная трубочка
NaCI
(+ альдостерон)
Наружный мозговой слой
Диуретики
(7) Ацетазоламцд
©Осмотические средства (маннитол)
©Петлевые диуретики (например, фуросемид)
(д) Тиазиды
©Антагонисты альдостерона (2,
(б) Антагонисты АДГ
2СГ
Тонкое нисходящее колено
Петля Генле
Толстое восходящее колено
Н20
Тонкое восходящее колено
Собирательный проток
Внутренний мозговой слой
Рис. 15-1. Канальцевая транспортная система и мишени действия диуретиков
рия, который абсорбируется в этой области, вещества, обладающие способностью специфически ингибировать транспорт хлорида натрия в проксимальных канальцах, могут оказаться особенно мощными диуретическими агентами.
Реабсорбция бикарбоната натрия в проксимальных канальцах инициируется действием Ка+,Н+-ионообменника, расположенного на апикальной (люминальной) мембране эпителиальных клеток проксимального канальца (рис. 15-3). Эта транспортная система позволяет Na+ входить в клетку со стороны просвета канальца для обмена в пропорции один к одному с протоном, находящимся внутри клетки. Так как во всех отделах нефрона На+,К+-АТФаза, расположенная на базо-латеральной мембране, “выкачивает” реабсорбированный Na+ из клетки в интерстиций, внутриклеточная концентрация Na+ остается низкой. Протон,
попавший в просвет канальца, взаимодействует с бикарбонатом и образует угольную кислоту Н2СО3. Угольная кислота, подобно бикарбонату, не транспортируется клетками проксимального канальца. Вместо этого она разлагается на СО2 и Н2О, которые легко проникают через мембрану. Эта реакция катализируется ферментом карбоангидразой. Углекислый газ, образующийся при дегидротации угольной кислоты, проникает в клетку проксимального канальца путем простой диффузии и затем вновь регидратируется в угольную кислоту. После диссоциации Н+ вновь пригоден для транспорта с помощью механизма Na+,H+-обмена, и бикарбонат переносится через базолатеральную мембрану и далее в кровь с помощью специфического транспортера (рис. 15-3). Таким образом, реабсорбция бикарбоната в проксимальных канальцах полностью зависит от активности кар-
% длины проксимального канальца
Рис. 15-2. Реабсорбция различных растворов в проксимальном канальце по отношению к длине канальца. (КЖ/П — отношение концентрации в канальцевой жидкости к концентрации в плазме; РП — разность потенциалов по длине канальца.). (Из: Ganong W. F. Review of Medical Physiology, 15th ed. Lange, 1991.)
боангидразы, которая ингибируется диуретиком ацетазоламидом.
В конечном отделе проксимального канальца оставшаяся в просвете жидкость очень напоминает простой раствор NaCl, так как бикарбонат и растворенные органические вещества уже в основном реабсорбировались из канальцевой жидкости. В этих условиях реабсорбция Na+ продолжается, но протон, секретируемый На+,Н+-обменником, далее нейтрализуется бикарбонатом. Это приводит к падению pH в просвете, что активирует еще малоизученный механизм ионообмена СГ на основание (рис. 15-3). Конечным эффектом параллельных Na+/H+ и СГ/основание обменов является реабсорбция NaCl. До настоящего времени нет мочегонного средства, которое влияет на этот процесс.
Рис. 15-3. Ка+,Н+-обменник апикальной мембраны и би-карбонатная реабсорбция в клетке проксимального извитого канальца. Находящаяся в базолатеральной мембране Na+,K+-АТФаза поддерживает внутриклеточные уровни ионов натрия и калия в нормальных пределах. В связи с быстрым уравновешиванием концентрации растворов приблизительно равны в интерстициальной жидкости и крови. Кроме щеточной каймы карбоангидраза (КА) обнаружена и в других участках
Таким образом, действие диуретиков на проксимальный каналец пока ограничивается угнетением реабсорбции бикарбоната натрия ингибиторами карбоангидразы.
В связи с высокой проницаемостью проксимального канальца для воды осмоляльность жидкости в его просвете и концентрация натрия остаются сравнительно постоянными на протяжении всего проксимального канальца (рис. 15-2). Таким образом, реабсорбция воды прямо пропорциональна реабсорбции соли в этом отделе. Следовательно, концентрация веществ типа инулина, которые не проникают через мембрану, будет возрастать по мере реабсорбции воды (рис. 15-2). Если в канальцевой жидкости появляется большое количество плохо проникающих веществ, например глюкозы или маннитола, обратное всасывание воды может привести к росту концентрации этих веществ до точки, когда осмоляльность канальцевой жидкости.пр-вышается и дальнейшая реабсорбция воды блоки
руется. Таков механизм действия осмотических диуретиков.
Секреторные системы для органических кислот расположены в средней трети проксимального канальца (сегмент S2). Эти системы секретируют ряд органических веществ (мочевую кислоту, р-амино-гиппуровую кислоту, диуретики, антибиотики и др.) в жидкость просвета канальца из крови. Секреторные системы для органических оснований (креатинин, прокаинамид, холин и т. д.) расположены в начальном (S,) и среднем (S2) сегментах проксимального канальца. Эти системы важны для поступления диуретиков к месту их действия в просвете сегментов канальца вдоль всего нефрона. Кроме того, в этих местах происходит взаимодействие между диуретиками и мочевой кислотой или другими эндогенными органическими соединениями (например, взаимодействие между диуретиками и пробенецидом).
Петля Генле
Тонкая часть петли Генле не принимает участия в активной реабсорбции соли, но важна для реабсорбции воды. Вода всасывается из тонкого нисходящего участка петли Генле за счет осмотических сил, создаваемых гипертоническим содержимым интерстиция медуллярного слоя. Как и в проксимальных канальцах, плохо проникающие вещества типа маннитола или глюкозы при их присутствии в просвете петли будут препятствовать всасыванию воды и способствовать ее поступлению в более дистальные отделы.
Толстое восходящее колено петли Генле активно реабсорбирует NaCl из просвета (около 35 % профильтровавшегося натрия), но в отличие от проксимального канальца и тонкого колена петли оно очень плохо проницаемо для воды. Реабсорбция соли в толстом восходящем сегменте, следовательно, ведет к разведению канальцевой жидкости, поэтому этот участок называют “разводящий сегмент”. Медуллярная часть толстого восходящего колена вносит свой вклад в гипертоничность мозгового слоя и тем самым играет важную роль в механизме концентрации мочи.
Система транспорта NaCl в люминальной мембране толстого восходящего колена представлена Ка+,К+,2СГ-котранспортером (рис. 15-4). Этот транспортер селективно блокируется диуретиками, известными под названием “петлевые”. Хотя
ПРОСВЕТ Толстое ИНТЕРСТИЦИЙ
входящее Моча Ч колено J Кровь
(+) < потенциал
Мд2+, Са2+
Рис. 15-4. Пути ионного транспорта через люминальную и базолатеральную мембраны клеток толстого восходящего колена. Положительный электрический потенциал в просвете, вызываемый обратной диффузией К+, приводит к реабсорбции двухвалентных катионов по парацел-люлярным путям
На+,К+,2СГ-котранспортер сам по себе электрически нейтрален (два катиона переносятся с двумя анионами), его действие ведет к избыточному накоплению К+ в клетке, так как На+,К+-АТФаза также переносит калий в клетку (с базолатеральной стороны). Это ведет к обратной диффузии К+ в просвет канальца, что формирует положительный электрический потенциал просвета. Этот потенциал создает движущую силу для реабсорбции двухвалентных катионов — Mg2+ и Са2+ — по парацел-люлярным путям (между клетками). Таким образом, подавление транспорта соли в толстом восходящем сегменте петлевыми диуретиками вызывает повышение экскреции с мочой, кроме NaCl, и этих двухвалентных катионов.
Дистальный извитой каналец
В дистальном извитом канальце NaCl всасывается меньше (лишь около 10 % профильтровавшегося NaCl), чем в толстом восходящем участке петли. Дистальный каналец относительно непроницаем для воды и, следовательно, реабсорбция NaCl разводит канальцевую жидкость. Основой механизма транспорта NaCl в дистальном извитом канальце является электрически нейтральный сочетанный транспорт Na+ и СГ (рис. 15-5), который фармако
логически отличен от сочетанного транспорта Na+,K+,2Cr в толстом восходящем колене. NaCl-транспортер блокируется диуретиками группы тиазидов. Поскольку в дистальном извитом канальце не существует повторного цикла переноса К+ через апикальную мембрану, как это происходит в петле Генле, в этом сегменте не создается положительный потенциал в просвете канальца, поэтому Са2+ и Mg2+ не выталкиваются оттуда электрическими силами.
Однако Са2+ активно реабсорбируется эпителиальными клетками дистального извитого канальца через апикальные Са2+-каналы и базолатеральный Ма+,Са2+-ионообменник (рис. 15-5). Этот процесс регулируется гормоном паращитовидных желез. Как будет понятно из дальнейшего изложения, отличия в механизме транспорта Са2+ в дистальном извитом канальце и в петле Генле лежат в основе действия различных групп диуретиков на транспорт Са2+.
Собирательные трубочки
Собирательные трубочки реабсорбируют всего 2-5 % от общего количества всасывающегося в почках NaCl. На первый взгляд это может показаться незначительным. Однако собирательные трубочки играют жизненно важную роль в физиологии по-
Рис. 15-5. Пути ионного транспорта через люминальную и базолатеральную мембраны клетки дистального извитого канальца. Как во всех канальцевых клетках, в базолатеральной мембране находится Ка+,К+-АТФаза (Р — рецептор паратгормона, ПТГ — паратгормон)
чек и действии диуретиков. Как конечное звено реабсорбции NaCl собирательные трубочки ответственны за окончательную концентрацию Na+ в моче. Более того, собирательная трубочка и конечный участок дистального канальца являются областью влияния минералокортикоидов, поэтому данный сегмент канальцевой системы играет важную роль в регуляции объема мочи. Наконец, собирательная трубочка — главное место секреции кадия почками и, следовательно, здесь реализуются фактически все вызванные диуретиками изменения метаболизма калия.
Механизм реабсорбции NaCl в собирательной трубочке отличается от механизмов, свойственных другим участкам канальца. Главные клетки — основное место транспорта Na+ , К+ и Н2О (рис. 15-6), а вставочные клетки — область секреции протонов. В отличие от эпителиальных клеток в начальных
Вставочная клетка
Рис. 15-6. Пути транспорта ионов и воды через люминальную и базолатеральную мембраны клеток собирательной трубочки и собирательного протока. Направленная внутрь диффузия Na+ создает в просвете отрицательный потенциал, приводящий к реабсорбции СГ и выходу К+ (Р — рецептор альдостерона или антидиуретичес-кого гормона) , .ц
сегментах нефрона апикальная мембрана главных клеток собирательных трубочек не содержит систем сочетанного транспорта для Na+ и других ионов. Эти мембраны имеют раздельные ионные каналы для Na+ и К+. Так как они не пропускают анионы, транспорт Na+ или К+ приводит к переносу заряда через мембрану. Поскольку движущая сила для переноса Na+ внутрь главной клетки существенно превышает таковую для выхода К+, преобладает реабсорбция Na+ и создается значительный (10-50 мВ) отрицательный потенциал просвета. Na+, который входит в главную клетку посредством подобного механизма, далее транспортируется в кровь с помощью такой же, как и в других отделах, базолатеральной На+,К+-АТФазы (рис. 15-6). Отрицательный люминальный электрический потенциал способствует обратному транспорту СГ в кровь через па-рацеллюлярные пути и выталкивает К+ из клетки через К+-каналы апикальной мембраны. Таким образом, существует важная связь между поступлением Na+ в собирательные трубочки и секрецией К+. Диуретики, действующие выше собирательных трубочек, повышают поступление Na+ к этому месту и усиливают секрецию К+. Более того, если Na+ поступает с анионом типа бикарбоната, который не может реабсорбироваться так легко, как СГ, отрицательный потенциал просвета увеличивается и еще более возрастает секреция К+. Это является причиной потерь К+, вызываемых многими диуретиками, особенно когда сопутствующее уменьшение объема жидкости усиливает секрецию альдостерона.
Реабсорбция Na+ и сопряженная с ним секреция К+регулируются альдостероном. Этот стероидный гормон, влияя на транскрипцию генов, повышает активность каналов апикальной мембраны и базолатеральной На+,К+-АТФазы, что приводит к возрастанию трансэпителиального электрического потенциала и значительному усилению как реабсорбции Na+, так и секреции К+ (дополнение “Электрический потенциал и секреция калия” и рис. 15-7).
В отсутствие антидиуретического гормона (АДГ) собирательная трубочка (и канал) непроницаемы для воды, что способствует образованию разведенной мочи. Однако это единственное место в нефроне, где регулируется проницаемость мембраны для воды. АДГ вызывает слияние внутриклеточных везикул, содержащих преформированные водные каналы, с апикальными мембранами главных клеток, увеличивая тем самым проницаемость мемб
раны для воды (рис. 15-6). Секреция АДГ регулируется осмоляльностью сыворотки и объемом жидкости. Таким образом, антагонисты АДГ теоретически должны усиливать выведение воды, однако такие вещества пока еще не применяются в клинике.
I. Базисная фармакология мочегонных средств
Ингибиторы карбоангидразы
Карбоангидраза представлена во многих участках нефрона, включая люминальную и базолатеральную мембраны, цитоплазму эпителиальных клеток и эритроциты в сосудах почек. Преобладает же этот фермент в люминальной мембране проксимального канальца (рис. 15-3), где он катализирует дегидратацию Н2СО3 — критическое звено проксимальной реабсорбции бикарбоната. Ингибирование карбоангидразы нарушает этот процесс, вызывая бикарбонатный диурез и уменьшение запасов бикарбоната в организме.
Ингибиторы карбоангидразы были предшественниками современных диуретиков (дополнение “Создание современных диуретиков”). Они относятся к незамещенным производным сульфаниламидов и появились после того, как было замечено, что бактериостатические сульфаниламиды вызывают щелочной диурез и гиперхлоремический метаболический ацидоз. С созданием новых препаратов ингибиторы карбоангидразы стали использоваться редко, но сохранились некоторые специфические области их применения, которые обсуждаются далее.
Химия
Структура ацетазоламида показана в дополнении (стр. 301). Для проявления активности препарата важна сульфаниламидная группа. Алкильные заместители в этой группе полностью блокируют его влияние на активность карбоангидразы.
Фармакокинетика
Все ингибиторы карбоангидразы хорошо всасываются после приема внутрь. Повышение pH мочи при бикарбонатном диурезе возникает в течение 30 минут, достигает максимума через 2 часа и со-
Электрический потенциал и секреция калия
Способность минералокортикоидов увеличивать секрецию К+ за счет влияния на электрический потенциал демонстрируется экспериментом, показанным на рис. 15-7. У адреналэктомирован-ных кроликов трансэпителиальный электрический потенциал (слева) невелик (5-6 мВ) и не изменяется под действием амилорида — диуретика, который блокирует апикальный канал для
Na+. Секреция К+ (справа) в этих условиях очень низка. Однако у животных, получивших дезоксикортикостеронаацетат (ДОКСА) — минералокортикоид, подобный альдостерону,— электрический потенциал падает до -20 мВ, а секреция К+ возрастает в 7 раз. Влияния ДОКСА на электрический потенциал и секрецию К+ устраняются при блокаде Иа+-канала амилоридом.
Электрический потенциал
АЭ кролики
Кролики, леченные ДОКСА
Рис. 15-7. Электрический потенциал и секреция калия. У адреналэктомированных (АЭ) кроликов трансэпителиальный потенциал собирательной трубочки (слева, темный столбик) положителен и секреция К+ (справа, темный столбик) незначительна. При введении кроликам минералокортикоида (кролики, леченные ДОКСА) потенциал изменяется до -20 мВ и секреция К+ увеличивается более чем в 7 раз. Эффекты ДОКСА полностью устраняются (обращаются) амилоридом (светлые столбики). ДОКСА — дезоксикортикостерона ацетат. (Из: Wingo С. S. Cortical collecting tubule potassium secretion: Effect of amiloride, ouabain, and luminal sodium concentration. Kidney Int. 1985; 27:886.)
храняется в течение 12 часов после однократного приема. Экскреция вещества происходит за счет канальцевой секреции в 52-сегменте проксимального канальца. По этой причине при почечной недостаточности дозу надо уменьшать.
Фармакодинамика
Ингибирование активности карбоангидразы существенно подавляет реабсорбцию бикарбонатов в проксимальном канальце. При введении макси-
Создание современных диуретиков
Создание тиазидов и других современных диуретиков связано с наблюдениями, в ходе которых было обнаружено, что у больных, которые получали сульфаниламиды, развивался метаболический ацидоз с ощелачиванием мочи. Установлено, что эти препараты вызывают диурез с выделением бикарбоната натрия. Понимание возможной пользы этого диуретического эффекта привело к созданию ацетазоламида и позднее — тиазидов.
Стрелки на рисунке показывают исторические этапы, а не химические реакции. Когда был синтезирован конкурент ацетазоламида дихлорфенамид, обнаружили, что он в большей степе
ни увеличивает экскрецию хлорида натрия относительно экскреции бикарбоната натрия. Это изменение оказалось желательным, указывающим на механизм действия, отличный от ингибирования карбоангидразы (или дополнительный к нему). Дальнейшие модификации молекулы привели к созданию дисульфамоилхлор-анилина, хотя и слабого диуретика, но послужившего основой для синтеза циклического соединения хлортиазида (первого тиазида). Это соединение мало влияло на активность карбоангидразы, но селективно подавляло реабсорбцию NaCl в дистальном канальце. На базе тиазидов в последующем были созданы петлевые диуретики.
мально безопасной дозы ацетазоламида реабсорбция бикарбоната снижается на 85 %; 1С50 (концентрация, при которой активность подавляется на 50 %) составляет 4 мкмоль/л. Однако некоторое количество бикарбоната еще может реабсорбироваться в других участках нефрона за счет механизмов, не зависимых от карбоангидразы. Суммарный эффект при максимальном действии ацетазоламида выражается в ингибировании примерно на 45 % всей почечной реабсорбции бикарбоната. Тем не менее угнетение карбоангидразы вызывает значительные потери бикарбоната, что ведет к гиперхлоремическому метаболическому ацидозу. Из-за токсичности этого ацидоза и вызванного истощением бикарбоната повышения реабсорбции NaCl в оставшихся канальцевых сегментах нефрона диуретическая эффективность ацетазоламида значительно снижается при назначении в течение нескольких дней.
Основное клиническое применение ацетазоламида связано с зависимым от карбоангидразы транспортом бикарбоната в других органах помимо почек. Цилиарное тело глаза секретирует бикарбонат в глазную влагу за счет процесса, сходного с реабсорбцией бикарбоната из жидкости проксимального канальца. Единственное отличие состоит в том, что процесс идет в обратном направлении — цилиарное тело захватывает бикарбонат из крови, а в проксимальном канальце он возвращается в кровь. Сходным образом образование спинномозговой жидкости в хориодальных сплетениях связано с секрецией бикарбоната в ликвор. Хотя эти процессы идут в противоположном направлении по сравнению с процессами в проксимальном канальце, они в равной степени подавляются ингибиторами карбоангидразы, что в обоих случаях существенно изменяет pH и количество образующейся жидкости.
Клинические показания и дозирование (табл. 15-1)
А. Глаукома. Ингибирование карбоангидразы понижает скорость образования глазной влаги, что ведет к снижению внутриглазного давления. Такой эффект ценен для кратковременного и длительного лечения различных форм глаукомы, поэтому ингибиторы карбоангидразы чаще всего назначают по этому показанию. Предпринимаются попытки создания ингибиторов карбоангидразы для местного применения при лечении глаукомы, но такие препараты пока не внедрены в практику.
Б. Ощелачивание мочи. Мочевая кислота и цистин относительно нерастворимы в кислой моче. Усилить их почечную экскрецию можно, повышая pH мочи при введении ингибиторов карбоангидразы. Таким же образом при назначении ацетазоламида повышается почечная экскреция слабых кислот (например, аспирина). В отсутствие постоянного введения бикарбонатов эти эффекты ацетазоламида относительно кратковременны. Продолжительное лечение требует одновременного назначения бикарбонатов.
В. Метаболический алкалоз. В большинстве случаев метаболический алкалоз является следствием понижения общего уровня К+ в организме и внутрисосудистого объема или высокого уровня минералокортикоидов. Следовательно, его нужно лечить не с помощью ацетазоламида, а корригируя заболевание, лежащее в основе. Когда же алкалоз связан с избыточным назначением диуретиков больным с тяжелой сердечной недостаточностью, а назначение солевых растворов противопоказано из-за повышения давления наполнения сердца, ацетазоламид может быть полезен для коррекции алкалоза и для получения дополнительного диуретического эффекта. Ацетазоламид используют также для быстрой коррекции метаболического алкалоза, который может развиваться в ситуации респираторного ацидоза.
ТАБЛИЦА 15-1. Ингибиторы карбоангидразы, применяемые для лечения глаукомы
Препарат Обычная доза для приема внутрь (1-4 раза в день)
Ацетазоламид 250 мг
Дихлорфенамид 50 мг
Г. Острая горная болезнь. Слабость, головокружение, бессонница, головная боль и тошнота могут наблюдаться у путешественников в горах при быстром подъеме выше 3000 м. Эти симптомы обычно умеренно выражены и длятся несколько дней. В более серьезных случаях быстро прогрессирующий легочный или мозговой отек может угрожать жизни. Ингибируя образование цереброспинальной жидкости и понижая ее pH и pH мозга, ацетазоламид может повышать работоспособность и уменьшать симптомы горной болезни. В целях профилактики его применяют внутрь за 24 часа до восхождения. В одном из исследований (Zell, 1988) выступают против такой профилактики при любительских восхождениях, так как симптомы при этом обычно незначительны.
Д. Другое применение. Ингибиторы карбоангидразы применяются как вспомогательные средства для лечения эпилепсии, некоторых форм гипокалиемического периодического паралича и для повышения экскреции фосфатов с мочой при тяжелой гиперфосфатемии.
Токсичность
А. Гиперхлоремический метаболический ацидоз. Ацидоз связан с хроническим уменьшением запасов бикарбоната в организме под влиянием ингибиторов карбоангидразы. Истощение бикарбонатов ограничивает эффективность диуретического действия двумя или тремя днями.
Б. Почечные камни. Как следствие бикарбонат-урической реакции, вызванной ингибиторами карбоангидразы, развиваются фосфатурия и гипер-кальцийурия. При хроническом применении может также понизиться почечная экскреция растворяющего фактора (например, цитрата). При щелочных значениях pH относительно нерастворимы соли кальция, что означает увеличение риска образования из них почечных камней.
В. Истощение калия почек. Потери калия происходят потому, что NaHCO3, попадающий в собирательные трубочки, вызывает увеличение люминального отрицательного электрического потенциала в этом сегменте и увеличивает экскрецию К1;; Этот эффект можно предупредить одновременным назначением КС1.
Г. Другие токсические эффекты. После приема» больших доз часто наблюдаются сонливость и пгР рестезии. Эти препараты накапливаются у больных)
с почечной недостаточностью, и тогда их токсическое действие на ЦНС может быть значительным. Могут появиться и реакции гиперчувствительности (лихорадка, сыпь, угнетение костного мозга, интерстициальный нефрит).
Противопоказания
Следует избегать назначения ингибиторов карбоангидразы при циррозе печени. Подщелачивание мочи снижает выведение с мочой NH4+ и способствует развитию печеночной энцефалопатии.
Петлевые диуретики
Петлевые диуретики избирательно подавляют реабсорбцию NaCl в толстом восходящем колене петли Генле. В связи с большой способностью к абсорбции NaCl в этом сегменте и тем, что диурез не ограничивается развитием ацидоза, как это происходит при применении ингибиторов карбоангидразы, эти вещества являются наиболее эффективными из известных диуретиков.
Химия
Двумя препаратами-прототипами этой группы являются фуросемид и этакриновая кислота. Структура нескольких петлевых диуретиков показана на рис. 15-8. Как и ингибиторы карбоангидразы, фуросемид и буметанид являются производными сульфаниламидов. Эти вещества имеют карбоксильную группу с сульфамоильным радикалом в метаположении (углерод номер 5). У С4 присутствуют галоидный или феноксизаместители, а замещенная аминогруппа — у С2 или СЗ. Новейший петлевой диуретик торсемид является еще одним производным сульфаниламида.
Этакриновая кислота (не сульфаниламидное производное) химически отличается от фуросемида. Это производное феноксиуксусной кислоты, содержащее соседние кетонную и метиленовую группы (рис. 15-8). Метиленовая группа (затенена) формирует соединение со свободной сульфгидрильной группой цистеина. Цистеиновое производное считается активной формой препарата.
Органические ртутные диуретики угнетают транспорт солей в толстой восходящей части петли. Они более не применяются из-за высокой токсичности и представляют только исторический интерес.
Фуросемид
Буметанид
О — СН2 — СООН
Этакриновая кислота
Рис. 15-8. Петлевые диуретики. Затененная метиленовая группа у этакриновой кислоты реактивна и может соединяться со свободными сульфгидрильными группами
Фармакокинетика
Петлевые диуретики быстро всасываются. Они выводятся путем почечной экскреции и за счет клубочковой фильтрации. Диуретическая реакция после внутривенного введения развивается чрезвычайно быстро. Продолжительность эффекта обычно 2-3 часа. Период полувыведения зависит от функции почек. Поскольку фуросемид действует со стороны просвета канальца, мочегонное действие положительно коррелирует с его экскрецией с мочой. Нарушение секреторного компонента его клиренса может происходить при одновременном введении таких веществ как индометацин и пробенецид, которые подавляют секрецию слабых кислот в проксимальном канальце. Определены метаболиты этакриновой кислоты и фуросемида, однако неясно, обладают ли они мочегонным действием. Торсемид имеет несколько более продолжительные период полувыведения и длительность действия,
чем фуросемид или буметанид. У него есть по меньшей мере один активный метаболит со сравнительно более длительным периодом полувыведения, чем у родительского соединения.
Фармакодинамика
Препараты этой группы подавляют систему сочетанного транспорта Na+, К+, 2СГ в люминальной мембране толстой восходящей части петли Генле. Подавляя этот транспортер, петлевые диуретики снижают реабсорбцию NaCl и снижают нормальный положительный потенциал просвета, связанный с циклическим транспортом К+ (рис. 15-4). Этот электрический потенциал в норме управляет реабсорбцией двухвалентных катионов в петле. Петлевые диуретики, устраняя положительный потенциал в просвете, вызывают увеличение экскреции Са2+ и Mg2+. Продолжительное применение может вызвать значительную гипомагниемию у некоторых больных. Так как Са2+ активно реабсорбируется в дистальном извитом канальце, петлевые диуретики обычно не вызывают гипокальциемию. Однако при расстройствах, которые сопровождаются гиперкальциемией, экскреция Са2+ может быть значительно повышена путем сочетания петлевых диуретиков с инфузией солевого раствора. Этот эффект весьма важен при лечении больных с гиперкальциемией.
Помимо диуретического действия, петлевые диуретики оказывают прямое влияние на кровоток в некоторых сосудистых областях. Механизмы такого эффекта недостаточно изучены. Фуросемид увеличивает почечный кровоток и вызывает перераспределение крови в корковом слое почек. Фуросемид и этакриновая кислота уменьшают проявления застоя в легких и давление наполнения левого желудочка при застойной сердечной недостаточности еще до того, как произойдет измеримое повышение отделения мочи.
Показания к применению и дозирование (табл. 15-2)
Наиболее важными показаниями для применения петлевых диуретиков являются острый отек легких, другие заболевания, сопровождающиеся отеками, и острая гиперкальциемия. Использование препаратов данной группы при указанных состояниях обсуждается далее в разделе клинической фар
макологии. Имеются и другие показания к применению петлевых диуретиков.
А. Гиперкалиемия. При умеренной гиперкалиемии или после неотложной терапии тяжелой гиперкалиемии петлевые диуретики могут значительно повысить экскрецию К+ с мочой, что способствует снижению общего запаса К+ в организме. Этот эффект усиливается при одновременном назначении NaCl и воды.
Б. Острая почечная недостаточность. Петлевые диуретики могут увеличить скорость тока мочи и экскрецию К+ при острой почечной недостаточности. Эти вещества могут способствовать переходу олигурической фазы почечной недостаточности в неолигурическую, что облегчает состояние больных. Однако они не укорачивают продолжительность периода недостаточности функции почек. Если большая нагрузка рентгеноконтрастными веществами провоцирует острую почечную недостаточность или является угрожающей, петлевые диуретики могут помочь промыть канальцы и уменьшить проявления их обструкции. С другой стороны, петлевые диуретики теоретически могут усилить образование цилиндров при миеломе и нефропатии.
В. Передозировка анионов. Бромиды, фториды и иодиды реабсорбируются в толстом восходящем колене петли, следовательно, петлевые диуретики могут быть полезны при лечении отравлений соединениями, содержащими эти ионы. Чтобы заместить потери с мочой Na+, доставить СГ и избежать потери внеклеточной жидкости, одновременно следует вводить солевые растворы.
Токсичность
А. Гипокалиемический метаболический алкалоз. Петлевые диуретики увеличивают поступление соли и воды в собирательные трубки и тем самым способствуют повышению секреции К+ и Н+, что вызывает гипокалиемический метаболический алкалоз.
ТАБЛИЦА 15-2. Дозы петлевых диуретиков’
Препарат Ежедневная доза для приема внутрь
Буметанид 0.5-2 мг
Этакриновая кислота 50-200 мг
Фуросемид 20-80 мг
Торсемид 2.5-20 мг
1 Единичная доза или доза, разделяемая на два приема.
Это токсическое действие является функцией величины диуретического эффекта и может быть устранено замещением К+ и коррекцией гиповолемии.
Б. Ототоксичность. Петлевые диуретики мотуг вызывать связанное с дозой ухудшение слуха, которое обычно носит обратимый характер. Наиболее часто оно бывает у больных с нарушенной функцией почек или у тех, кто получает одновременно другие ототоксичные препараты, например амино-гликозидные антибиотики.
В. Гиперурикемия. Петлевые диуретики могут вызывать гиперурикемию и провоцировать приступы подагры. Это связано с обусловленным гиповолемией повышением реабсорбции мочевой кислоты в проксимальном канальце. Такого эффекта можно избежать, используя более низкие дозы диуретика.
Г. Гипомагниемия. Истощение запасов магния — предсказуемое следствие хронического применения петлевых диуретиков. Оно наблюдается у больных с пищевым дефицитом магния. Это действие можно устранить назначением препаратов магния для приема внутрь.
Д. Аллергические реакции. При лечении фуросемидом иногда наблюдаются кожные высыпания, эозинофилия, реже — интерстициальный нефрит. Они обычно быстро исчезают после отмены препарата. Аллергические реакции, вероятно, связаны с сульфаниламидной частью молекулы; они значительно реже отмечаются при применении этакри-новой кислоты.
Е. Другие токсические эффекты. Петлевые диуретики в большей степени по сравнению с другими могут способствовать дегидратации. Гипонатриемия возникает реже, чем при приеме тиазидов, потому что петлевые диуретики уменьшают максимальную концентрационную способность почек. Тем не менее у больных, которые потребляют больше жидкости в связи с вызванной гиповолемией жаждой, при приеме петлевых диуретиков может развиться выраженная гипонатриемия. Эти препараты известны своим кальцийуретическим действием, но может возникнуть и гиперкальциемия, чаще у больных с выраженной дегидратацией или при наличии других, ранее скрытых причин гиперкальциемии, таких как карцинома легких.
Противопоказания
Фуросемид и буметанид мотуг проявлять перекрестную реактивность у больных, которые чув
ствительны к другим сульфаниламидам. Чрезмерное применение любого диуретика опасно при циррозе печени, пограничной почечной недостаточности или застойной сердечной недостаточности.
Тиазиды
Тиазидные диуретики появились в результате попыток синтеза более сильных ингибиторов карбоангидразы (дополнение “Создание современных диуретиков”). В последующем стало ясно, что тиазиды ингибируют транспорт NaCI независимо от влияния на активность карбоангидразы и что они влияют на транспорт соли в дистальном извитом канальце. Некоторые представители этой группы сохранили выраженную антикарбоангидразную активность, но этот эффект не связан с первичным механизмом их действия.
Химия
Препараты данной группы формально обозначают как бензотиадиазиды, обычно используя сокращенное название тиазиды. Природа гетероциклических колец и заместителей при них может варьировать, но все соединения, как и ингибиторы карбоангидразы, сохраняют незамещенную сульфаниламидную группу (рис. 15-9).
Фармакокинетика
Все тиазиды всасываются при приеме внутрь, но существуют отличия в их метаболизме. Родоначальник группы хлортиазид менее липофилен и должен назначаться в относительно больших дозах. Хлор-талидон медленно всасывается и имеет большую продолжительность действия. Индапамид экскретируется в основном через желчевыводящие пути, но достаточная доля активной формы выводится почками, что обеспечивает мочегонное действие в дистальном извитом канальце.
Все тиазиды выводятся с помощью системы секреции органических кислот и в некоторой степени конкурируют с секрецией мочевой кислоты той же системой. В результате скорость секреции мочевой кислоты может понижаться и соответственно увеличиваться ее содержание в сыворотке. В стационарном состоянии выработка мочевой кислоты и, следовательно, почечная экскреция не изменяются под влиянием тиазидов.
Рис. 15-9. Гидрохлортиазид и родственные средства
Фармакодинамика
Тиазиды подавляют реабсорбцию NaCl со стороны эпителиальной клетки, обращенной в просвет канальца. Считается, что может изменяться и реабсорбция NaCl в дистальном участке проксимального канальца, но в обычных клинических условиях этот эффект незаметен.
О системе транспорта NaCl, которая угнетается тиазидами, известно сравнительно мало. Как уже обсуждалось, этот транспортный механизм является электрически нейтральным NaCl-котранспорте-ром, который отличается от переносчика в области петли Генле. В дистальном извитом канальце существует также активный процесс реабсорбции для Са2+, модулируемый паратиреоидным гормоном (ПТГ) (рис. 15-5). В отличие от петлевых диуретиков, которые угнетают реабсорбцию Са2+ в петле Генле, тиазиды увеличивают ее в дистальном извитом канальце. Механизм такого действия неизвестен, но предполагают, что он связан со снижением концентрации Na+ в клетке из-за блокады вхо
да Na+ под влиянием тиазидов. Понижение уровня внутриклеточного Na+ может усилить Na+, Са2+-об-мен в базолатеральной мембране (рис. 15-5), а это способствует реабсорбции Са2+. Хотя тиазиды редко вызывают гиперкальциемию вследствие повышения реабсорбции, они могут демаскировать гиперкальциемию, связанную с другими причинами • (например, гиперпаратиреоидизмом, карциномой, саркоидозом). Тиазиды оказались полезными в терапии нефролитиаза, связанного с гиперкальций-урией.
Показания к применению и дозирование (табл. 15-3)
Основными показаниями для применения тиазидов являются: 1) гипертензия, 2) застойная сердечная недостаточность, 3) нефролитиаз, связанный с идиопатической гиперкальцийурией, 4) .не-1 фрогенный несахарный диабет. Применение тиа&и-дов при каждом из этих заболеваний рассматривается далее в разделе клинической фармакологиин /у
ТАБЛИЦА 15-3. Дозы тиазидов и родственных диуретиков
Препарат Ежедневная доза для приема внутрь Частота приема
Бендрофторметиазид 2.5-10 мг В один прием
Бензтиазид 25-100 мг В два приема
Хлортиазид 0.5-1 г В два приема
Хлорталидон’ 50-100 мг В один прием
Гидрохлортиазид 25-100 мг В один прием
Гидрофторметиазид 25-100 мг В два приема
Индапамид’ 2.5-10 мг В один прием
Метиклотиазид 2.5-10 мг В один прием
Метолазон* 2.5-10 мг В один прием
Политиазид 1-4 мг В один прием
Хинетазон’ 50-100 мг В один прием
Трихлорметиазид 2-8 мг В один прием
1 Не тиазид, но сульфаниламид, качественно сходный с тиазидами.
Токсичность
А. Гипокалиемический метаболический алкалоз и гиперурикемия. Эти токсические эффекты $ сходны с теми, которые наблюдаются при приеме | j петлевых диуретиков.
г Б. Нарушение толерантности к углеводам. Ги-|| пергликемия может наблюдаться как у больных с ® явно выраженным диабетом, так и у людей с незна-|чительно измененной толерантностью к глюкозе.
Этот эффект связан с нарушением выделения инсулина поджелудочной железой и с уменьшением утилизации глюкозы тканями. Гипергликемия может быть частично обратима при коррекции гипокалиемии.
В. Гиперлипидемия. Тиазиды вызывают увеличение на 5—15% уровня холестерйна в сыворотке и повышение концентрации липопротеидов низкой плотности (ЛПНП). При длительном применении эти показатели обычно понижаются до исходной величины.
Г. Гипонатриемия. Гипонатриемия — важный побочный эффект тиазидных диуретиков. В редких случаях он может представлять угрозу для жизни. Эффект связан с сочетанным гиповолемическим повышением уровня АДГ, снижением способности почек к разведению и повышенной жаждой. Это осложнение можно предупредить путем снижения дозы препарата или ограничения потребления воды.
;Д. Аллергические реакции. Тиазиды являются сульфаниламидами и проявляют перекрестную чувствительность в отношении других представи
телей данной химической группы. Изредка возникают фоточувствительность или генерализованный дерматит. Серьезные аллергические реакции бывают очень редко, но могут проявляться в виде гемолитической анемии, тромбоцитопении и острого некротического панкреатита.
Е. Другие токсические эффекты. Слабость, усталость и парестезии напоминают эффекты ингибиторов карбоангидразы. Имеются сообщения о нарушении потенции, что, вероятно, связано с уменьшением объема жидкости в организме.
Противопоказания
Чрезмерное применение любого диуретика опасно при циррозе печени, пограничной почечной недостаточности или застойной сердечной недостаточности.
Калийсберегающие диуретики
Представители этой группы проявляют антагонизм действию альдостерона в конечной части дистальных канальцев и в собирательных трубочках коркового слоя. Подавление может осуществляться за счет прямого фармакологического антагонизма на уровне рецепторов минералокортикоидов (спиронолактон) или путем угнетения транспорта Na+ через ионные каналы люминальной мембраны (триамтерен, амилорид). Менее выраженный ка-лийсберегающий эффект можно наблюдать у пре-
паратов, подавляющих образование ренина или ангиотензина II (нестероидные противовоспалительные средства, 0-блокаторы, ингибиторы ангиотен-зинпревращающего фермента).
Химия и фармакокинетика
Спиронолактон является синтетическим стероидом, который действует как конкурентный антагонист альдостерона. Начало и длительность его действия, следовательно, определяются кинетикой реакции на альдостерон в ткани-мишени. Инактивация препарата в основном происходит в печени. Для спиронолактона характерно сравнительно медленное начало действия, требующее несколько дней для достижения полного терапевтического эффекта.
Триамтерен метаболизируется в печени, но основным путем элиминации его активной формы и метаболитов является почечная экскреция. О диуретическом эффекте метаболитов известно мало. Амилорид выводится в неизмененном виде с мочой. Поскольку триамтерен активно метаболизируется, он имеет более короткий период полувыведения и должен назначаться чаще, чем амилорид. Структуры спиронолактона, триамтерена и амилорида представлены на рис. 15-10.
Амилорид
Рис. 15-10. Антагонисты альдостерона
Фармакодинамика
Калийсберегающие диуретики уменьшают абсорбцию Na+ в собирательных канальцах и трубочках. Всасывание Na+ (и секреция К+) в этой области регулируются альдостероном. При данной скорости поступления Na+ скорость дистальной секреции К+ положительно коррелирует с уровнем альдостерона. Как уже указывалось, альдостерон увеличивает секрецию К+ путем повышения активности Na+, К+-АТФазы и каналов для Na+ и К+. Всасывание Na+ в собирательных трубочках генерирует отрицательный потенциал просвета, который способствует секреции К+. Антагонисты альдостерона вмешиваются в этот процесс. Сходные эффекты наблюдаются в отношении регуляции Н+ в собирательных трубочках, что отчасти объясняет метаболический ацидоз при назначении антагонистов альдостерона.
Спиронолактон является синтетическим стероидом, который связывается с цитоплазматическими минералокортикоидными рецепторами и пре
дупреждает перемещение рецепторного комплекса в ядро клетки-мишени. Его структура показана на рис. 15-10. Препарат может уменьшать внутриклеточное образование активных метаболитов альдостерона путем ингибирования активности 5а-редуктазы. Триамтерен и амилорид не блокируют рецептор альдостерона, а прямо влияют на вход Na+ через натрийселективные ионные каналы апикальной мембраны собирательных трубочек. Так как секреция К+ в этом отделе нефрона сопряжена со входом Na+, данные вещества тоже являются эффективными калийсберегающими диуретиками.
Другие калийсберегающие агенты
Ингибиторы АПФ (главы Ии 17), хотя и не используются в качестве калийсберегающих диуретиков, проявляют антагонизм эффекту альдостерона, влияя на его секрецию, поэтому их действие часто сопровождается гиперкалиемией. .
Показания к применению и дозирование (табл. 15-4)
Препараты данной группы наиболее эффективны при состояниях, сопряженных с избытком минералокортикоидов: как при первичной гиперсекреции (синдром Конна, эктопическая продукция АКТГ), так и при вторичном альдостеронизме. Последний может возникать при застойной сердечной недостаточности, циррозе печени, нефротическом синдроме и других заболеваниях, ассоциированных с задержкой солей в почках и уменьшением внутрисосудистого объема жидкости. Использование других диуретиков, подобных тиазидам или петлевым агентам, может вызывать или усиливать сокращение объема и тем самым усугублять вторичный альдостеронизм. В случае повышенной секреции минералокортикоидов и продолжающегося поступления Na+ в дистальные отделы нефрона происходит истощение запасов К+. Это связано с секрецией К+ в собирательных трубочках. В этом случае для снижения К+-секреторной реакции и предупреждения истощения запасов внутриклеточного К+ используют калийсберегающие диуретики любого типа.
Токсичность
А. Гиперкалиемия. В отличие от других диуретиков препараты данной группы могут вызывать незначительную, умеренную и даже угрожающую жизни гиперкалиемию. Риск этого осложнения значительно повышается при наличии заболеваний почек или при сочетанном применении с другими лекарствами, угнетающими выработку ренина (Р-бло-каторы, НПВС) или ангиотензина II (ингибиторы АПФ). Поскольку прием большинства других диуретиков ведет к потерям К+, гиперкалиемия может наблюдаться при использовании антагонистов альдостерона в качестве единственного диуретическо
го агента, особенно у больных с почечной недостаточностью. При применении комбинированных препаратов с фиксированными дозами калийсбере-гающих и тиазидных диуретиков можно достигнуть хорошего баланса, при котором гипокалиемия и метаболический алкалоз, вызываемые тиазидами, сглаживаются эффектами антагониста альдостерона. Однако в связи с различиями в биодоступности компонентов некоторых комбинированных лекарственных форм побочные эффекты тиазидов (например, метаболический алкалоз, гипонатриемия) могут преобладать. По этим причинам предпочтительно подбирать дозы двух лекарств раздельно.
Б. Гиперхлоремический метаболический ацидоз. Угнетая секрецию ГГ параллельно с влиянием на секрецию К+, калийсберегающие диуретики могут вызывать ацидоз, сходный с тем, который наблюдается при почечном Канальцевом ацидозе IV типа.
В. Гинекомастия. Синтетические стероиды могут вызывать, эндокринные нарушения, влияя на другие стероидные рецепторы. При назначении спиронолактона описаны гинекомастия и другие побочные эффекты (импотенция, доброкачественная гипертрофия простаты).
Г. Острая почечная недостаточность. Комбинация триамтерена и индометацина иногда вызывает острую почечную недостаточность. Этот эффект не наблюдается у других калийсберегающих диуретиков.
Д. Камни в почках. Триамтерен плохо растворим и может давать осадок в моче, способствуя образованию почечных камней.
Противопоказания
Препараты данной группы могут вызывать тяжелую, даже фатальную, гиперкалиемию у чувствительных больных. Прием К+ внутрь следует прекра
ТАБЛИЦА15-4. Калийсберегающие диуретики и комбинации препаратов
Торговое название Калийсберегающее средство Гидрохлортиазид Частота приема
Альдактазид Спиронолактон 25 мг 25 мг 1-4 раза в день
Альдактон Спиронолактон 25 мг — 1-4 раза в день
Диазид Триамтерен 50 мг 25 мг 1-4 раза в день
Дирениум Триамтерен 100 мг — 1-3 раза в день
Максзид Триамтерен 75 мг 50 мг 1 раз в день
Максзид-25 мг Триамтерен 37.5 мг 25 мг 1 раз в день
Мидамор Амилорид 5 мг — 1 раз в день
Модуретик Амилорид 5 мг 50 мг 1 или 2 раза в день
тить при назначении антагонистов альдостерона. Больные с хронической почечной недостаточностью особо ранимы и им очень редко назначают антагонисты альдостерона. Сочетанное применение других агентов, которые угнетают ренин-ангиотен-зиновую систему (Р-блокаторы или ингибиторы АПФ), повышает вероятность гиперкалиемии. У больных с заболеваниями печени может наблюдаться нарушение метаболизма триамтерена и спиронолактона, что требует корректировки дозы.
Вещества, усиливающие экскрецию воды
Осмотические диуретики
Проксимальный каналец и нисходящая часть петли Генле свободно проницаемы для воды. Осмотические агенты, которые плохо транспортируются, вызывают задержку воды в этих отделах и способствуют водному диурезу. Один из таких препаратов, маннитол, используют главным образом для уменьшения внутричерепного давления, но иногда и для быстрого удаления почечных токсинов, образующихся при остром гемолизе или после введения рентгеноконтрастных веществ.
Фармакокинетика
Маннитол не метаболизируется и его выведение регулируется клубочковой фильтрацией без заметной канальцевой реабсорбции или секреции. Осмотические диуретики плохо всасываются по определению, значит, их следует вводить парентерально. Маннитол экскретируется путем клубочковой фильтрации в течение 30-60 минут. При введении внутрь препарат вызывает осмотическую диарею. Этот эффект можно использовать для потенцирования эффекта препаратов ионообменных смол, связывающих калий, или для элиминации токсических веществ из желудочно-кишечного тракта при сочетанном назначении с активированным углем (глава 60, том 2).
Фармакодинамика
Осмотические диуретики ограничивают реабсорбцию воды прежде всего в тех отделах нефрона, которые свободно проницаемы для воды, т. е.
в проксимальном канальце и нисходящей части петли Генле. Наличие невсасывающегося вещества, такого как маннитол, предупреждает нормальную абсорбцию воды за счет противодействующей осмотической силы. В результате объем мочи увеличивается вместе с экскрецией манни-тола. Соответствующее увеличение скорости тока мочи уменьшает время контакта жидкости с канальцевым эпителием, что уменьшает реабсорбцию Na+. Однако возникающий натрийурез меньше по величине, чем водный, что иногда приводит к гипернатриемии.
Показания к применению и дозирование
А. Увеличение объема мочи. Осмотические диуретики используют для увеличения выведения воды, более выраженного по сравнению с выведением натрия. Этот эффект может быть полезен, когда имеются нарушения почечной гемодинамики или когда значительная задержка Na+ ограничивает реакцию на обычно применяемые диуретики. Осмотические препараты можно использовать для поддержания объема мочи и для предупреждения анурии, которая возникает при большой пигментной нагрузке на почки (например, при гемолизе или рабдомиолизе). Некоторые больные с олигурией не реагируют на осмотические диуретики. В этих случаях перед началом постоянной инфузии следует ввести тест-дозу маннитола (12.5 г внутривенно). Лекарство бесполезно назначать, если в течение трех часов после введения тест-дозы не происходит увеличения мочеотделения до уровня выше 50 мл в час. Если реакция на тест-дозу получена, введение маннитола (12.5-25 г) следует повторять через 1-2 часа для поддержания тока мочи на уровне выше 100 мл в час. Длительное применение маннитола не рекомендуется.
Б. Уменьшение внутричерепного и внутриглазного давления. Осмотические диуретики уменьшают общее содержание воды в организме в большей степени, чем содержание катионов, и тем самым уменьшают внутриклеточный объем. Этот эффект используют для снижения внутричерепного давления при неврологических заболеваниях и внутри^ глазного давления перед офтальмологическими вмешательствами. Маннитол вводят в ДЬзе 1-2 г/кг массы тела. Внутричерепное давлений,' & которым следует установить мониторное наблюдение, должно начать снижаться через 60-90 мин.
Токсичность
А. Увеличение внеклеточного объема. Манни-тол быстро распределяется во внеклеточном секторе и притягивает воду из внутриклеточного. Первоначально это ведет к увеличению объема внеклеточной жидкости и гипонатриемии. Этот эффект может осложнить застойную сердечную недостаточность и способствовать отеку легких. У больных, получающих осмотические диуретики, часто наблюдаются головная боль, тошнота и рвота.
Б. Дегидратация и гипернатриемия. Чрезмерное применение маннитола без адекватного возмещения потерь воды может в конечном счете привести к серьезной дегидратации, потере свободной воды и гипернатриемии. Этого осложнения можно избежать путем тщательного контроля ионного состава сыворотки и водного баланса.
Антагонисты АДГ
к Ряд заболеваний, например опухоли, при кото-с рых секретируются АДГ-подобные пептиды, сопро-
i вождается задержкой воды в результате избыточ-|ного количества АДГ. К сожалению, специфические антагонисты АДГ пока находятся на стадии исследования. Два неселективных агента, Li+ и демек-лоциклин, находят ограниченное применение в некоторых специфических ситуациях.
Фармакокинетика
В настоящее время к антагонистам АДГ можно отнести соли Li+ и производное тетрациклина (де-меклоциклин). Оба препарата активны при приеме внутрь. Литий реабсорбируется в некоторой степени в проксимальных канальцах и далее не абсорбируется и не секретируется. Демеклоциклин метаболизируется в печени.
Фармакодинамика
Антагонисты АДГ угнетают эффекты АДГ на уровне собирательных трубочек. Хотя механизм их действия не доказан достаточно убедительно, считается, что оба агента уменьшают образование цА)ЧФ в ответ на действие АДГ и влияют на эффект цАМФ в клетках собирательных трубочек.
Показания к применению и дозирование
А. Синдром несоответствующей секреции АДГ (СНСАДГ). Антагонисты АДГ используют в терапии СНС АДГ, когда ограничение потребления воды не может исправить нарушение. Обычно это происходит в амбулаторных условиях, когда ограничение нельзя контролировать, или в стационаре, когда большие количества растворов вводят внутривенно вместе с лекарствами. Для лечения используют карбонат лития, но реакция больного не всегда предсказуема. Уровень Li+ в сыворотке следует тщательно контролировать, так как концентрации выше 1 ммоль/л токсичны. Демеклоциклин в дозах 600-1200 мг в день оказывает более адекватное действие и не так токсичен. Уровень лекарства в плазме (порядка 2 мкг/мл) можно определить с помощью стандартных методов измерения тетрациклинов в плазме.
Б. Другие причины увеличения уровня АДГ. Уровень АДГ возрастает также в ответ на уменьшение эффективного объема циркулирующей крови. Когда лечение с помощью возмещения объема невозможно, как при сердечной недостаточности или заболеваниях печени, может возникать гипонатриемия. Как и при СНСАДГ, ограничение воды является средством терапии выбора, но если оно неэффективно, можно назначить демеклоциклин.
Токсичность
А. Нефрогенный несахарный диабет. Если уровень Na+ в сыворотке не контролируется, за СНСАДГ может последовать нефрогенный несахарный диабет с выраженной гипернатриемией. Тогда как Li+ используют преимущественно при аффективных расстройствах, нефрогенный несахарный диабет можно лечить с помощью тиазидных диуретиков или амилорида.
Б. Почечная недостаточность. Как Li+, так и демеклоциклин могут вызвать острую почечную недостаточность. Длительное лечение препаратами Li+ опасно возникновением хронического интерстициального нефрита.
В. Другие токсические эффекты. Побочные эффекты, связанные с лечением препаратам Li+,— это дрожь, кардиотоксическое действие, нарушение функций щитовидной железы и лейкоцитоз (глава 28). Больным с заболеваниями печени не следует назначать демеклоциклин (глава 44, том 2).
Сочетания диуретиков
Петлевые диуретики и тиазиды
У некоторых больных наблюдается рефрактер-ность к обычным дозам петлевых агентов или развивается невосприимчивость после начального периода положительного эффекта. Так как эти средства отличает короткий период полувыведения, рефрактерность может быть связана с чрезмерно большими перерывами между повторными приемами. Задержка Na+ в почках возрастает в тот период времени, когда вещество уже не действует. В этих случаях, помимо укорочения интервала между приемами петлевых диуретиков или увеличения дозы, возможно применение двух диуретиков, действующих на различные отделы нефрона, что обеспечивает синергизм. Петлевые диуретики и тиазиды в сочетании часто вызывают диурез, который недостижим при использовании препаратов каждой из групп в отдельности. Существует несколько причин этого явления. Во-первых, реабсорбция воды и соли как в восходящем толстом участке петли, так и в дистальном извитом канальце может возрастать при блокаде любого из указанных участков. Подавление функции обоих отделов может, следовательно, способствовать более чем аддитивному мочегонному эффекту. Во-вторых, тиазидные диуретики могут вызвать незначительный натрийурез в проксимальном канальце, что обычно маскируется повышением реабсорбции в толстом восходящем колене петли. Поэтому комбинация петлевых диуретиков и тиазидов препятствует реабсорбции натрия, по крайней мере в определенной степени, во всех трех сегментах.
У больных с рефрактерностью к петлевым агентам препаратом выбора из группы тиазидоподобных средств обычно является метолазон. Есть сведения о том, что этот препарат более эффективен, чем другие тиазиды, при почечной недостаточности, но эта точка зрения противоречива. Кроме того, метолазон имеется только в лекарственных формах для приема внутрь, тогда как хлортиазид можно вводить и парентерально.
Сочетание петлевых диуретиков и тиазидов может способствовать мобилизации большого количества жидкости даже у больных, которые проявляют рефрактерность к представителям каждой из этих групп в отдельности. В этих случаях желате
лен тщательный гемодинамический мониторинг. Не следует рекомендовать рутинное применение таких сочетаний в амбулаторных условиях. Тем более, что это часто приводит к истощению запасов калия и может потребовать мониторного наблюдения за состоянием воды и электролитов.
Калийсберегающие диуретики и петлевые агенты или тиазиды
У многих больных, получающих петлевые диуретики или тиазиды, на определенном этапе лечения развивается гипокалиемия. Это состояние можно исправить ограничением приема соли с пищей, что уменьшает поступление натрия в собирательные трубочки. Если гипокалиемия не устраняется таким образом или с помощью приема КС1, добавление калийсберегающих диуретиков поможет значительно снизить экскрецию калия. Такой способ обычно безопасен, но его следует избегать у больных с почечной недостаточностью, когда в ответ на применение калийсберегающих диуретиков может возникать угрожающая гиперкалиемия.
II. Клиническая фармакология диуретиков
В этом разделе обсуждается клиническое применение диуретиков при отечных и иных состояниях. Влияние препаратов данной группы на экскрецию электролитов с мочой продемонстрировано в табл. 15-5.
ТАБЛИЦА 15-5. Изменение содержания электролитов в моче в ответ на мочегонные средства (+- увеличение, - = снижение)
Средства Электролиты мочи
NaCl NaHCO3 к*
Ингибиторы карбоангидразы + +++ +
Петлевые диуретики ++++ - +
Тиазиды ++ + +
Петлевые диуретики + тиазиды +++++ + ++
К*-сберегающие средства +
Отеки
Наиболее частой причиной назначения диуретиков является необходимость уменьшения периферических отеков или отека легких вследствие заболеваний сердца, почек, сосудов или нарушений онкотического давления крови. Задержка соли и воды с образованием отека часто происходит при таком понижении доставки крови к почкам, которое расценивается как недостаточно “эффективный” объем артериальной крови. Применение диуретиков способствует мобилизации отечной жидкости без значительного снижения объема плазмы. Однако чрезмерная терапия мочегонными средствами в этих условиях может привести к дальнейшему понижению эффективного объема артериальной крови со снижением перфузии жизненно важных органов. Следовательно, использование диуретиков для ликвидации отеков требует тщательного мониторного контроля гемодинамического статуса пациента и понимания патофизиологии заболевания.
Застойная сердечная недостаточность
Когда функция сердца нарушается вследствие заболевания, возникающие изменения АД и почечного кровотока воспринимаются организмом как гиповолемия и вызывают задержку в почках соли и воды. Этот первоначально физиологический регуляторный процесс ведет к увеличению внутрисосудистого объема и венозного возврата к сердцу и может частично восстановить сердечный выброс до нормального уровня.
Если имеющееся заболевание способствует нарушению функции сердца, несмотря на возрастание объема плазмы, почки продолжают задерживать соль и воду, которые затем выходят из сосудов и образуют интерстициальную или легочную отечную жидкость. На этом этапе применение диуретиков становится необходимым для ограничения прогрессирования отеков, особенно отека легких. Уменьшение сосудистого застоя в легких при назначении диуретиков улучшает оксигенацию и тем самым функцию миокарда. Отеки, ассоциированные с застойной сердечной недостаточностью, обычно лечат с помощью петлевых диуретиков. В некоторых случаях задержка соли и воды
может стать столь серьезной, что потребуется сочетанное применение петлевых диуретиков и тиазидов.
При лечении диуретиками пациентов с сердечной недостаточностью следует всегда помнить, что сердечный выброс у этих больных частично поддерживается за счет высокого давления наполнения и чрезмерное применение диуретиков может уменьшить венозный возврат и тем самым привести к нежелательному снижению сердечного выброса. Это особенно важно при правожелудочковой недостаточности. Отличительным признаком этого нарушения является системный, а не легочный застой. Если давление наполнения левого желудочка снижается ниже 15 мм рт. ст., вызванное диуретиками сокращение объема может снизить венозный возврат и серьезно нарушить сердечный выброс.
Вызываемый диуретиками метаболический алкалоз является еще одним побочным эффектом, который может нарушать функцию сердца. Обычно его лечат с помощью возмещения калия и восстановления внутрисосудистого объема солевыми растворами, однако при серьезной сердечной недостаточности опасно применение растворов даже для больных, получивших слишком много диуретиков. В этих случаях скорректировать алкалоз поможет ацетазоламид.
Другой серьезный токсический эффект при применении диуретиков, особенно у кардиологических больных,— это развитие гипокалиемии. Она может ухудшить имеющуюся сердечную аритмию и способствовать проявлению токсического действия препаратов наперстянки. Этого осложнения часто удается избежать, ограничивая потребление соли, снижая тем самым доставку натрия в секретирующий К+ участок собирательной трубочки. Пациенты, которые не в состоянии ограничить потребление Na+ с пищей, должны получать внутрь добавки КС1 или К+.-сберегающие диуретики. В противном случае следует прекратить прием тиазидов.
Наконец, следует помнить, что диуретики не могут устранить существующее заболевание сердца. Вещества, которые улучшают сократительную функцию миокарда или уменьшают периферическое сосудистое сопротивление, имеют более прямое отношение к основной проблеме. Диуретики в лучшем случае являются средствами содействующей терапии.
Заболевания почек
При ряде заболеваний почек может нарушаться их важная роль в поддержании объемного гомеостаза. Хотя заболевание почек иногда вызывает солевое истощение, в большинстве случаев почечная патология сопровождается задержкой соли и воды. При тяжелом нарушении функции почек диуретические агенты оказываются малоэффективны из-за нарушения клубочковой фильтрации, которая обеспечивает должный натрийурез. Однако многие больные с умеренной почечной недостаточностью, если у них имеется задержка натрия, могут получать диуретики.
Многие первичные и вторичные заболевания клубочков, например связанные с сахарным диабетом, системной красной волчанкой и другими заболеваниями, сопровождающимися нарушением функции сосудов, проявляются первичной задержкой соли и воды в почках. Причина задержки натрия точно неизвестна, но, возможно, связана с нарушением микроциркуляции в почках и функции канальцев за счет высвобождения вазоконстрикторов, простагландинов, цитокинов и других медиаторов воспаления. Когда у таких больных развивается отек или гипертензия, лечение мочегонными средствами может быть очень эффективно. Если имеется сопутствующая сердечная недостаточность, следует соблюдать рассмотренные выше меры предосторожности.
Некоторые формы заболеваний почек, особенно диабетическая нефропатия, часто сочетаются с развитием гиперкалиемии на сравнительно ранней стадии развития хронической почечной недостаточности. В этих случаях усилить секрецию К+ путем повышения доставки соли в секретирующие К+ собирательные трубочки могут тиазидные или петлевые диуретики.
У больных с заболеваниями почек, ведущими к нефротическому синдрому, часто возникают сложности при попытке регуляции объема. Традиционно считают, что эти пациенты имеют пониженный объем плазмы, связанный с низким онкотическим давлением. Это объяснение, возможно, правильно в ряде случаев нефропатии с “минимальными изменениями”. У этих больных применение диуретиков может вызвать дальнейшее снижение объема плазмы, что нарушает скорость фильтрации и вызывает ортостатическую гипотензию. Однако большинство других случаев нефротического синдрома
ассоциировано с необъяснимой первичной задержкой соли и воды почками, что приводит к увеличению объема плазмы и гипертензии, несмотря на низкое осмотическое давление плазмы. В такой ситуации лечение диуретиками может оказаться полезным для контроля зависимого от объема компонента гипертензии и уменьшения ее участия в развитии заболевания почек.
При выборе диуретика для больного с заболеванием почек нужно помнить о ряде серьезных ограничений. Следует избегать назначения ацетазоламида и калийсберегающих диуретиков из-за их способности усиливать ацидоз и гиперкалиемию соответственно. Тиазидные диуретики обычно неэффективны, когда скорость клубочковой фильтрации падает ниже 30 мл в 1,мин. Таким образом, петлевые диуретики часто являются препаратами выбора при лечении отеков, связанных с недостаточностью почек. В конечном счете чрезмерное применение диуретиков может вызвать нарушение функции почек у всех больных, но у тех, кто страдает почечной недостаточностью, последствия всегда более серьезны.
Цирроз печени
Заболевания печени часто сопровождаются отеками и асцитом в сочетании с повышением гидростатического давления в портальной системе и снижением онкотического давления. Механизмы задержки натрия в этих случаях довольно сложны. Они, вероятно, связаны с комплексом факторов, включающим сниженную перфузию почек из-за системных изменений сосудов, уменьшение объема плазмы вследствие асцита и падение онкотического давления из-за гипоальбуминемии. Уровень альдостерона в плазме обычно повышается в ответ на снижение эффективного объема циркулирующей жидкости.
Когда асцит и отеки становятся выраженными, для инициации и поддержания диуреза может быть полезным лечение диуретиками. Больные с циррозом часто резистентны к петлевым диуретикам: отчасти из-за снижения секреции лекарства в канальцевую жидкость по неизвестному механизму, частично из-за высокого уровня альдостерона, способствующего повышению реабсорбции натрия.В собирательных трубках. С другой стороны, циррОтй-ческий отек необычен по высокой степени реак1гйв-: ности на действие спиронолактона. Некоторая!
больным помогает комбинация петлевых диуретиков. Однако даже в большей степени, чем при сердечной недостаточности, в этих случаях опасна передозировка диуретиков. Она может вызвать значительное уменьшение внутрисосудистого объема, гипокалиемию и метаболический алкалоз. Гепаторенальный синдром и печеночная энцефалопатия являются опасными последствиями чрезмерного применения диуретиков при циррозе печени.
Идиопатический отек
Этот синдром, развивающийся почти исключительно у женщин, выражается в периодических задержках соли и отеках. Несмотря на интенсивное изучение, патофизиология расстройства остается неясной. Некоторые исследования показывают, что прерывистое назначение диуретиков может способствовать развитию этого синдрома. Следовательно, идиопатический отек лучше лечить ограничением потребления соли.
Состояния, не связанные с отеками
Гипертензия
Давно известно, что мочегонное и умеренное сосудорасширяющее действие тиазидов полезно для лечения практически всех больных с эссенциальной гипертензией и почти у двух третей из них оказывается достаточным методом лечения. Умеренное ограничение Na+ в пище (60-100 мэкв в день) потенцирует эффект диуретиков при эссенциальной гипертензии и уменьшает потери К+ в почках.
До недавнего времени тиазидные диуретики были обычно первыми лекарственными средствами, которые назначали больным с легкой степенью эссенциальной гипертензии. По мере выяснения нежелательных эффектов тиазидов и создания новых антигипертензивных средств (глава 11) применение тиазидов как средств выбора несколько сократилось. Диуретики сохранили важную роль в случаях необходимости комплексной лекарственной терапии для контроля АД. Они увеличивают эффективность многих веществ, особенно ингиби-торор ангиотензинконвертирующего фермента. Пациентам, которым назначены сильные вазодила
таторы типа гидралазина и миноксидила, обычно требуется одновременное назначение диуретиков, потому что вазодилататоры часто вызывают заметную задержку жидкости и отек.
Несмотря на побочные эффекты, диуретики продолжают играть большую роль в терапии гипертензии. Это особенно важно из-за их невысокой стоимости и достаточной эффективности.
Нефролитиаз
Примерно две трети всех камней почек содержат фосфат или оксалат кальция. У многих больных с такими камнями выявляется почечная “утечка” кальция, способствующая гиперкальцийурии. Она может лечиться тиазидными диуретиками, которые повышают реабсорбцию кальция в дистальных извитых канальцах и тем самым снижают концентрацию кальция в моче. В этих случаях следует ограничить потребление соли, так как чрезмерное поступление NaCl с пищей подавляет гипокальций-уретический эффект тиазидов. Образование кальциевых камней может быть связано также с повышением всасывания кальция в кишечнике или иметь идиопатическую природу. В этих ситуациях тиазиды будут оказывать эффект при применении в качестве средств дополнительной терапии в сочетании со снижением потребления кальция и другими мероприятиями.
Гиперкальциемия
Иногда гиперкальциемия является причиной неотложных состояний. Так как петля Генле является важным местом реабсорбции кальция, весьма эффективными средствами кальциевого диуреза могут быть петлевые диуретики. Однако сами по себе они могут вызывать заметное уменьшение объема жидкости. Если это происходит, петлевые диуретики становятся неэффективными, потому что реабсорбция кальция в проксимальном канальце возрастает. Следовательно, если требуется достигнуть эффективного кальциевого диуреза, солевые растворы должны вводиться одновременно с петлевыми диуретиками. Обычный метод заключается в инфузии солевого раствора и введении фуросемида (80-120 мг) внутривенно. При появлении диуретического эффекта скорость инфузии солевого раствора следует уравновесить со скоростью мочеотделения для того, чтобы избежать уменьшения
объема. При необходимости к солевому раствору можно добавить калий.
Несахарный диабет
Тиазидные диуретики могут уменьшать полиурию и полидипсию у больных, которые не реагируют на АДГ. Терапевтический эффект связан с уменьшением объема плазмы и сопутствующим снижением скорости клубочковой фильтрации, повышением проксимальной реабсорбции NaCI и воды, снижением поступления жидкости в разводящие сегменты. Таким образом, максимально достижимый объем разведенной мочи снижается. Этот парадоксальный эффект тиазидов может значительно снижать ток мочи у больных с полиурией. Ограничение поступления натрия с пищей усиливает в этом случае эффективность тиазидов. Литий, который используют для лечения маниа-кально’депрессивного психоза, является частой причиной лекарственного несахарного диабета. Тиазидные диуретики полезны при лечении и таких больных. Уровень Li+ в сыворотке в этом случае должен быть объектом мониторного наблюдения, так как диуретики могут понизить почечный клиренс Li+ и повысить его уровень до границ токсического диапазона (глава 28). Литиевая полиурия может быть частично устранена амилоридом, который блокирует вход лития в клетки собирательных трубочек, подобно тому как он блокирует вход Na+.
Препараты
Ацетазоламид (генерик, Диамокс)
Перорально: таблетки по 125,250 мг Перорально для пролонгированного действия: капсулы по 500 мг Парентерально: порошок по 500 мг для инъекций
Амилорид (генерик, Мидамор)
Перорально: таблетки по 5 мг
Бендрофторметиазид (Натуретин)
Перорально: таблетки по 5,10 мг
Бензтиазид (Эксна)
Перорально: таблетки по 50 мг
Буметанид (Бумекс)
Перорально: таблетки по 0.5,1,2 мг Парентерально: ампулы по 0.5 мг/2 мл для в/в и в/м инъекций
Хлортиазид (генерик, Диурил, др.)
Перорально: таблетки по 250, 500 мг; суспензия 250 мг/5 мл
Парентерально: порошок 500 мг для инъекций
Хлорталидон (генерик, Гигротон, др.)
Перорально: таблетки по 15, 25,50,100 мг
Демеклоциклин (Декломицин)
Перорально: таблетки и капсулы по 150 мг;
таблетки по 300 мг
Дихлорфенамид (Даранид)
Перорально: таблетки по 50 мг
Этакриновая кислота (Эдекрин)
Перорально: таблетки по 25, 50 мг Парентерально: порошок 50 мг для в/в инъекций
Фуросемид (генерик, Лазикс, др.)
Перорально: таблетки по 20,40,80 мг; раствор 8,10 мг/мл
Парентерально: 10 мг/мл для в/м и в/в инъекций
Гидрохлортиазид (генерик, Эзидрикс, ГидроДиу-рил, др.)
Перорально: таблетки по 25,50,100 мг;
раствор 10,100 мг/мл
Гидрофторметиазцд (генерик, Диукардин, Салурон) Перорально: таблетки по 50 мг
Индапамид (Лозол)
Перорально: таблетки по 1.25, 2.5 мг
Маннитол (генерик, Осмитрол)
Парентерально: 5,10,15,20,25 % для инъекций
Метиклотиазид (генерик, Эндурон, др.)
Перорально: таблетки по 2.5, 5 мг
Метолазон (Микрокс, Зароксолин) (Внимание, биодоступность Микрокса больше, чем Зароксолина)
Перорально: таблетки по 0.5 мг (Микрокс);
2.5,5,10 мг (Зароксолин)
Политиазид (Ренез)
Перорально: таблетки по 1, 2,4 мг
Хинетазон (Гидромокс )
Перорально: таблетки по 50 мг
Спиронолактон (генерик, Альдактон)
Перорально: таблетки по 25, 50,100 мг
Торсемид (Демадекс)
Перорально: таблетки по 5,10, 20,100 мг
Парентерально: 10 мг/мл для инъекций
Триамтерен (Дирений)
Перорально: таблетки по 50,100 мг
Трихлорметиазид (генерик, Наква) Перорально: таблетки по 2,4 мг
Избранная литература
Brenner В. М., Rector F. С. Jr. (eds) The Kidney, 4th ed. Saunders, 1990.
DeTroyer A. Demeclocycline: Treatment for syndrome of inappropriate antidiuretic hormone secretion. JAMA, 1977; 237: 2723.
Hollifield J. W., Slaton P. E. Thiazide diuretics, hypokalemia, and cardiac arrhythmias. Acta. Med. Scand. 1981; 647 (Suppl.): 67.
Horisberger J.-D., Giebisch G. Potassium-sparing diuretics. Renal. Physiol. 1987; 10:198.
Multicenter Diuretic Cooperative Study Group: Multiclinic comparison of amiloride, hydrochlorothiazide, and hydrochlorothiazide plus amiloride in essential hypertension. Arch. Intern. Med. 1981; 141:482.
Rose B. D. Diuretics. Kidney Int. 1991; 39: 336.
Stanton B. A. Cellular actions of thiazide diuretics in the distal tubule. J. Am. Soc. Nephrol. 1990; 1:832.
Warnock D. G., Eveloff J. NaCl entry mechanisms in the luminal membrane of the renal tubule. Am. J. Physiol. 1982; 242: 561.
Раздел IV
Средства, оказывающие выраженное действие на гладкую мускулатуру Гистамин, серотонин и алкалоиды спорыньи
Алан Буркхалтер, Дэвид Дж. Джулиус, Оскар Л. Фрик
16
Гистамин и серотонин (5-гидрокситрипта-мин) — биологически активные амины, представленные во многих тканях, обладающие комплексом физиологических и патологических эффектов и обычно высвобождающиеся местно. Наряду с эндогенными полипептидами (глава 17), простагландинами и лейкотриенами (глава 18) их иногда называют аутакоидами (в переводе с греческого “са-моизлечивающие”) или местными гормонами в соответствии со свойствами, которыми они обладают.
Роль гистамина и серотонина в нормальной физиологии известна не полностью, эти соединения не применяются в терапии каких-либо заболеваний. Однако препараты, являющиеся селективными антагонистами этих аминов или селективно активирующие определенные подтипы рецепторов, имеют большое клиническое значение. В этой главе прежде всего рассматриваются вопросы базисной фармакологии агонистов аминов и клинической фармакологии селективных агонистов и антагонистов. В конце главы обсуждаются алкалоиды спорыньи — парциальные агонисты серотониновых и ряда других рецепторов.
Гистамин
Гистамин был синтезирован в 1907 г. и позднее изолирован из тканей млекопитающих. Ранние гипотезы о физиологических эффектах гистамина в тканях были основаны на сходстве его действия и симптомов анафилактического шока и повреждения тканей. В эффектах гистамина имеются значительные видовые различия; у людей он является важным медиатором аллергических реакций немедленного типа и реакций воспаления, играет важную роль в секреции желудочного сока, действует как нейромедиатор и нейромодулятор.
I. Базисная фармакология гистамина
Химические свойства и фармакокинетика
Гистамин представляет собой 2-(4-имидазо-ил)этиламин. Он присутствует как в растительных,
так и в животных тканях, а’также является компонентом некоторых ядов и секретов, обладающих раздражающим действием.
Гистамин
Гистамин образуется при декарбоксилировании аминокислоты L-гистидина, катализируемом в тканях млекопитающих ферментом декарбоксилазой (К. Ф. 4.1.1.22). В качестве кофактора выступает пиридоксальфосфат. После образования гистамин либо депонируется, либо быстро инактивируется. Основной путь инактивации включает, во-первых, превращение в метил гистамин, катализируемое имидазол-Ы-метилтрансферазой, и затем окисление в метилимидазолуксусную кислоту с участием ди-аминоксидазы. Второй путь метаболизма — прямое превращение гистамина в имидазолуксусную кислоту под действием диаминоксидазы. Только очень небольшое количество гистамина выделяется в неизмененном виде. Развитие ряда опухолей (системный мастоцитоз, пигментная крапивница, карциноид желудка и иногда миелогенная лейкемия), характеризующихся увеличением числа тучных клеток или базофилов, ведет к повышению секреции гистамина и его метаболитов.
Хотя гистамин присутствует в большинстве тканей, он распределяется очень неравномерно. Основная часть тканевого гистамина существует в связанном виде в гранулах тучных клеток или базофилов: уровень гистамина в некоторых тканях напрямую связан с количеством тучных клеток. Связанная форма гистамина биологически неактивна, но многие стимулы могут вызывать высвобождение гистамина с последующим воздействием на окружающие ткани.
Тучные клетки в большом количестве представлены в местах потенциального повреждения тканей (hoq рот, стопы), на внутренних поверхностях организма, в кровеносных сосудах, особенно в местах сдавления и бифуркаций. Эти клетки в разных тканях отличаются друг от друга, но иногда весьма похожи (например, некоторые тучные клетки слизистой ЖКТ напоминают тучные клетки соединительной ткани). Такие тучные клетки слизистых (атипичные тучные клетки), кроме гистамина, содержат в гранулах хондроитина сульфат вместо ге
парина сульфата и обладают различной чувствительностью к веществам, которые вызывают высвобождение гистамина.
Вне тучных клеток гистамин присутствует в разных тканях,, в том числе в мозгу, где функционирует как нейромедиатор. Тела клеток гистаминерги-ческих нейронов находят в туберомамиллярном ядре задней части гипоталамуса, и они обладают значительными разветвлениями с проекцией в другие участки мозга. В качестве эндогенного нейромедиатора гистамин участвует в реализации многих функций мозга, таких как нейроэндокринный контроль, регуляция деятельности сердечно-сосудистой системы, терморегуляция и возбуждение.
!
Хранение и высвобождение гистамина
В тучных клетках и базофилах человека гранулы содержат гистамин в комплексе с сульфатированными полисахаридами, сульфатом гепарина или хондроитина, а также кислый белок. Связанная форма гистамина может высвобождаться несколькими путями.
А. Иммунологическое высвобождение. Важный патофизиологический механизм высвобождения гистамина из тучных клеток и базофилов связан с иммунологическими реакциями. Эти клетки, сенсибилизированные антителами типа IgE, фиксированными на их поверхностной мембране, де-гранулируются при контакте с соответствующим антигеном (рис. 57-7). Для высвобождения необходима также энергия и ионы кальция. Дегрануляция приводит к одновременному выделению АТФ, гистамина и других медиаторов, которые хранятся вместе в секреторных гранулах. Некоторые из этих соединений, особенно АТФ, в дальнейшем еще больше стимулируют дегрануляцию тучных клеток путем паракринного или аутокринного механизмов. Дегранулированные тучные клетки в течение дней или недель снова накапливают гистамин. Выделившийся таким путем гистамин является медиатором аллергических реакций немедленного типа (тип I). Вещества, выделяющиеся при иммунных реакциях, опосредованных IgG или IgM с последующей активацией каскада комплемента, тоже способствуют высвобождению гистамина из тучных клеток и базофилов.
Кроме того, по механизму отрицательной обратной связи через возбуждение Н2-рецепторов гистамин модулирует свое высвобождение и влияет на
выброс других медиаторов из сенсибилизированных тучных клеток некоторых тканей. У человека такая обратная связь действует в тучных клетках кожи и базофилах, но не в тучных клетках легких, поэтому гистамин может ограничивать интенсивность аллергических реакций в коже и крови.
Эндогенный гистамин модулирует разные иммунные и воспалительные реакции. Возможно, он играет роль в развитии острого воспаления. Освобождающийся при повреждении ткани гистамин вызывает локальную вазодилатацию и выход из сосудов плазмы, которая содержит медиаторы активного воспаления (комплемент, С-реактивныц белок), антитела и клетки-эффекторы воспаления (нейтрофилы, эозинофилы, базофилы, моноциты и лимфоциты). Гистамин подавляет высвобождение содержимого лизосом и некоторые функции Т- и В-лимфоцитов. Большинство из этих эффектов опосредовано Н2-рецепторами через повышение внутриклеточного цАМФ. Выделение пептидов из нервов в ответ на воспаление также, вероятно, модулируется гистамином через пресинаптические Н3-рецепторы.
Б. Химическое и механическое высвобождение. Некоторые амины, в том числе такие препараты как морфин и тубокурарин, могут вытеснять гистамин из комплекса с белками и гепарином в клетках. Этот процесс не требует энергии и не связан с повреждением или дегрануляцией тучных клеток. При потере гранул из тучных клеток также происходит высвобождение гистамина, так как ионы натрия во внеклеточной жидкости быстро вытесняют амин из комплекса. К дегрануляции и выделению гистамина приводит химическое и механичес
кое повреждение клетки. Экспериментальный по лимер диамина — соединение 48/80 — специфичес ки высвобождает гистамин из тучных клеток тка ней путем экзоцитоза, для осуществления которо го необходима энергия и ионы кальция.
Фармакодинамика
А. Механизм действия. Гистамин оказывав' биологическое действие, соединяясь с определен ными рецепторами мембран. Известны три вид; рецепторов, которые называются Нь Н2 и Н3 и опи саны в таблице 16-1. Все три подтипа принадлежа: к большому надсемейству рецепторов, имеющие семь трансмембранных участков и связывающихся с G-белком. Структура Нг и Н2-рецепторов известна. В мозгу они находятся на постсинаптически; мембранах, а Н3-рецепторы в основном пресинап тические. Активация последних приводит к угне тению высвобождения медиаторов, в том числе i самого гистамина, норадреналина, серотонина i ацетилхолина. Активация Н(-рецепторов, которьк имеются в клетках эндотелия и гладких мышц обычно усиливает гидролиз фосфоинозитола и по вышает уровень внутриклеточного кальция. Воз буждение Н2-рецепторов, имеющихся в слизистог желудка, кардиомиоцитах и некоторых иммунньп клетках, увеличивает внутриклеточное содержант цАМФ. Активация Н3-рецепторов, локализованные в некоторых областях ЦНС, угнетает выделенш гистамина из гистаминергических нейронов, чт< скорее всего опосредовано снижением тока Са2+.
Б. Влияние гистамина на органы и ткани. Гистамин обладает выраженным действием на гладкук
ТАБЛИЦА 16-1. Подтипы гистаминовых рецепторов
Подтип рецептора Распределение Пострецепторный механизм Парциальные селективные агонисты Парциальные селективные антагонисты
н, Гладкие мышцы, эндотелий, мозг Т ИТФ, ДАГ 2-(т-фторфенил)-гиста-мин1 Мепирамин, трипролидин
Нг Слизистая желудка, сердечная мышца, тучные клетки, мозг Т цАМФ Димаприт, импромидин Ранитидин, тиотидин
Нз Пресинаптически: мозг, мышечное сплетение кишечника, другие нейроны Связывание с G-белком Я-а-метилгистамин, иметит Тиоперамид, йодофенлро-пит, импромидин
1 Парциальный агонист.
Г истамин
после Hi- и Н2- блокады
Г истамин
Г истамин после Н,- блокады
40 мкг/кг/ч
I-------------1
7120 Г
О
80 г
МЕ
I_I_I_J_I_I_I_I_I_I
-1 01 2345678
Время (мин)
Рис. 16-1. Действие гистамина и его антагонистов на кровяное давление и частоту сердечных сокращений у людей. Гистамин вводили со скоростью 40 мкг/кг/час в течение 5 мин, как отмечено над каждым графиком. В качестве Н|-антагониста использовался хлорфенирамин (10 мг внутривенно). Как Н2-аитагонист применяли циметидин (2 мг внутривенно). (Из: Torsoli A., Lucchelli Р. Е., Brimblecombe R. W. (eds) Н- Antagonists: Н2-Receptor Antagonists in Peptic Ulcer Disease and Progress in Histamine Research. Excerpta Medica, 1980.)
и сердечную мускулатуру, на определенные эндотелиальные и нервные клетки и на секреторные клетки желудка. Чувствительность к гистамину значительно варьирует у разных видов животных. Человек, морские свинки, собаки и кошки очень чувствительны, а мыши и крысы — нет.
1. Сердечно-сосудистая система. У человека инъекция или инфузия гистамина вызывает снижение систолического и диастолического давления и повышение частоты сердечных сокращений (рис. 16-1). Быстрое изменение давления связано с прямой вазодилатацией артериол и расслаблением прекапиллярных сфинктеров, вызванными гистамином; повышение частоты сердечных сокращений объясняется как стимулирующим действием гистамина на сердце, так и рефлекторной тахикардией. При введении гистамина из-за вазодилатации возможны вегетативные “приливы”, ощущение тепла и головная боль. В данном случае вазодилатация опосредована, по крайней мере частично, высвобождением фактора релаксации эндотелия (EDRF; глава 6). Исследование антагонистов гистаминовых рецепторов показало, что в реакциях со стороны сердечно-сосудистой системы на высокие дозы гистамина участвуют Нг и Н2-рецепторы, так как соче
тание блокаторов рецепторов того и другого типа более эффективно предотвращает действие гистамина, чем изолированное применение одного из них (рис. 16-1). Реакция же сердечно-сосудистой системы человека на небольшие дозы гистамина может в достаточной степени подавляться только блокатором Hj-рецепторов.
Действие гистамина на Н(-рецепторы в сосудах микроциркуляторного русла, особенно в посткапиллярах, вызывает отек. Эффект связан с повышением проницаемости сосудистой стенки вследствие раздвижения клеток эндотелия, что приводит к транссудации жидкости и небольших молекул белков в околососудистое пространство. Это действие лежит в основе возникновения крапивницы — проявления высвобождения гистамина в коже. Исследования функции эндотелиальных клеток позволяют предположить, что актин и миозин этих немышечных клеток взаимодействуют при усилении тока Са2+, вызванного веществами типа гистамина. В результате сокращения клеток эндотелия повышается проницаемость сосудов (Curry, 1992).
К прямым влияниям на сердечно-сосудистую систему относятся повышение сократимости и частоты водителя ритма. Эти эффекты прежде всего
11. Заказ 3245
опосредованы Н2-рецепторами. С другой стороны, у человека гистамин может снизить сократимость мышцы предсердий через влияние на Нt-рецепторы. Физиологическое значение этих сердечно-сосудистых эффектов неясно. Они могут быть вызваны высвобождением эндогенного гистамина из тучных клеток. Многие из сердечно-сосудистых симптомов при анафилаксии связаны с гистамином, хотя доказано участие и других медиаторов.
2. Гладкая мускулатура желудочно-кишечного тракта. Гистамин вызывает сокращение гладких мышц кишечника. Индуцированное гистамином сокращение слепой кишки морской свинки является стандартным методом оценки действия гистамина. Кишечник человека обладает меньшей чувствительностью к гистамину, чем кишечник морской свинки, но большие дозы гистамина при участии механизма, опосредованного Нгрецепторами, могут провоцировать диарею.
3. Гладкие мышцы бронхов. У человека и у морских свинок гистамин вызывает бронхоспазм посредством возбуждения Hi-рецепторов. У морских свинок бронхоспазм является причиной смерти при отравлении гистамином, но у здоровых людей после введения им обычных доз гистамина бронхоспазм не выражен. Однако пациенты с астмой очень чувствительны к гистамину. Бронхоспазм у таких больных, видимо, отражает гиперактивный ответ нервной системы, так как они избыточно реагируют и на многие другие стимулы, а их реакция на гистамин может быть предотвращена не только антагонистами Ht-рецепторов, но и ганглиоблокатора-ми (глава 19). Метод провокации со ступенчатым повышением дозы гистамина используют в диагностике гиперреактивности бронхов при подозрении на астму или муковисцидоз. У таких пациентов чувствительность к гистамину может быть повышена в 100-1000 раз.
4. Другие гладкомышечные органы. У человека гистамин обычно не оказывает значительного действия на гладкую мускулатуру глаза и мочеполового тракта, но у беременных женщин при анафилактических реакциях возможен выкидыщ вследствие вызванных гистамином сокращений матки. У некоторых видов животных чувствительность матки к гистамину достаточно высока, и это используется в экспериментальных исследованиях.
5. Нервные окончания. Гистамин является сильным стимулятором чувствительных нервных окончаний, особенно передающих боль и зуд. Эти эф
фекты, опосредованные Hj-рецепторами, являются важной составляющей частью крапивницы, реакции на укусы насекомых и на ожоги крапивой. Установлено, что высокие местные концентрации гистамина могут также деполяризовать эфферентные (аксональные) нервные окончания (пункт 7).
6. Секреторные ткани. Гистамин давно известен как сильный стимулятор желудочной секреции и в меньшей степени — продукции пепсина и внутреннего фактора. Этот эффект вызван активацией Н2-рецепторов на париетальных клетках желудка (Sewing, 1986) или на клетках близлежащих тканей (Mezey, 1992) и связан с повышением активности аденилатциклазы, концентрации цАМФ и внутриклеточного Са2+. Другие стимуляторы секреции кислоты, например ацетилхолин и гастрин, не увеличивают количество цАМФ, хотя их максимальный эффект можно несколько уменьшить антагонистами Н2-рецепторов. Гистамин стимулирует также секрецию в тонком и толстом кишечнике.
В обычных дозах он не оказывает большого влияния на активность других секреторных тканей. Очень высокие концентрации гистамина могут привести к высвобождению гормонов мозгового вещества надпочечников.
7. “Тройной” ответ. Внутрикожная инъекция гистамина вызывает характерные припухлость, потепление и покраснение. Эффект опосредован клетками трех типов: гладкомышечными в микроцирку-’ ляторном русле, эндотелием капилляров или венул и чувствительными нервными окончаниями. В месте инъекции возникает покраснение вследствие расширения мелких сосудов, за которым вскоре следует отек в месте инъекции (волдырь) и неравномерная гиперемия вокруг волдыря. Это вызвано аксональным рефлексом. Как уже отмечалось, гистамин стимулирует нервные окончания. Возникающие импульсы распространяются и по другим ветвям того же аксона, вызывая расширение сосудов за счет высвобождения вазодилататорного нейромедиатора. Эти изменения могут сопровождаться зудом. Волдырь является следствием местного отека.
Сходные местные эффекты возникают прй инъекции либераторов гистамина (вещества 48/80,' морфина и т. д.) внутрикожно или наложением антигенов на кожу сенсибилизированного пациента. Хотя большинство из этих реакций можно предотвратить предварительным введением Ht-блокатора, нельзя отрицать роль Н2-рецепторов в их раз-' витии. '
Другие агонисты гистамина
Замещения в имидазольном кольце значительно влияют на селективность соединений в отношении подгрупп рецепторов гистамина. Например, 2-метилгистамин сравнительно более селективно действует на Нгрецепторы, а 4-метилгистамин является относительно специфичным Н2-агонистом. Бетазол (препарат, использовавшийся для изучения секреторной способности желудка) и импромидин являются Н2-агонистами и Н3-антагонистами. Л-а-метилгистамин — сильный и селективный Н3-агонист, который проникает через гематоэнцефалический барьер. Он также является сильным ингибитором синтеза и высвобождения гистамина.
н н
IX -н
Импромидин
II. Клиническая фармакология гистамина
Клиническое применение
А. Исследование секреторной функции желудка. В прошлом гистамин использовали для определения секреторной функции желудка. Сейчас с этой целью используют пентагастрин, который имеет значительно меньше побочных эффектов.
Б. Диагностика феохромоцитомы. Гистамин может вызывать высвобождение катехоламинов из мозгового вещества надпочечников. Этот эффект не выражен у здоровых людей, но очень существенен при феохромоцитоме. В настоящее время этот опасный тест не применяется, так как разработан химический анализ количества катехоламинов и их метаболитов у пациентов с подозрением на феохромоцитому.
В. Исследование функции легких. В пульмонологических лабораториях аэрозоль гистамина (и других веществ) иногда применяют как провокационный тест на гиперреактивность бронхов.
Токсичность и противопоказания
Выраженность побочных эффектов при применении гистамина, как и эффектов высвобождения гистамина, зависит от дозы. Возможны вегетативные “приливы”, гипотензия, тахикардия, головная боль, крапивница, бронхоспазм и нарушения со стороны ЖКТ. Эти эффекты возникают также после употребления несвежей рыбы типа скумбрии (есть данные о том, что гистамин синтезируется бактериями в организме рыбы).
Гистамин нельзя назначать астматикам (кроме случаев тщательно мониторируемых тестов легочной функции) и пациентам с активной язвой желудка или кровотечением из ЖКТ.
Антагонисты гистамина
Эффекты высвобождаемого в организме гистамина можно ослабить несколькими способами. Физиологические антагонисты, особенно адреналин, действуют на гладкую мускулатуру противоположно гистамину, а также затрагивают другие рецепторные механизмы. Это важно в клинике, так как инъекция адреналина может спасти жизнь при системной анафилаксии и других состояниях, когда происходит массивное выделение гистамина и других медиаторов.
Ингибиторы высвобождения уменьшают дегрануляцию тучных клеток, которая возникает как результат взаимодействия IgE и антигена. Кромо-лин и недокромил, обладающие этим эффектом (глава 19), применяются в терапии астмы, хотя молекулярный механизм их действия в настоящее время неизвестен. Агонисты Р2-адренорецепторов тоже снижают высвобождение гистамина.
Антагонисты гистаминовых рецепторов являются примером третьего подхода к подавлению реакций, опосредованных гистамином. Более чем 45 лет назад появились конкурентные антагонисты действия гистамина на гладкую мышцу. Однако вплоть до открытия антагониста Н2-рецепторов буримамида в 1972 г. было невозможно ингибировать действие гистамина на секрецию кислоты в желудке. Появление селективных антагонистов Н2-ре-цепторов дало возможность не только более точно описать взаимодействие гистамина с рецепторами, но и более эффективно лечить больных с язвенной болезнью. Селективные антагонисты Н3-рецепто-
ров для клинического применения еще не созданы. Однако в эксперименте обнаружено, что сильный и селективный Н3-антагонист тиопирамид в нано-молярных концентрациях конкурирует за места связывания с меченым радиоактивными изотопами агонистом 7?-а-метилгистамином. Оба эти вещества проникают через гематоэнцефалический барьер. Их доступность для экспериментального использования поможет определить физиологическое значение Н3-рецепторов.
Антагонисты Нгрецепторов
Соединения, конкурентно блокирующие связывание гистамина с Нгрецепторами, используют в клинике уже в течение многих лет. В США в настоящее время применяют 18 препаратов. Некоторые из них продаются без рецепта в качестве самостоятельных лекарственных средств или в составе комбинированных препаратов, например противопро-студных и снотворных лекарств (глава 65).
I. Общая фармакология антагонистов Нгрецепторов
Химические свойства и фармакокинетика
Антагонисты Нгрецепторов предыдущих поколений являются стабильными растворимыми в жирах аминами, типичное строение которых представлено на рис. 16-2. Существуют несколько химических подгрупп; структура соединений, относящихся к разным подгруппам, также показана на рисунке. Дозы основных препаратов даны в табл. 16-2. Большинство из них имеют сходные величины абсорбции и распределения. Они быстро всасываются при пероральном введении с пиковыми концентрациями в крови через 1-2 часа, широко распределяются в организме и, кроме новых препаратов (терфенадин, лоратидин, астемизол и меквитазин), легко проникают в ЦНС. Препараты подвергаются значительному метаболизированию в основном микросомальной системой печени. Большинство из них действуют в течение 4-6 часов после приема одной дозы, но меклизин и препараты нового поколения действуют дольше — 12-24 часа. Новые препараты также значительно меньше растворяются в
жирах и практически не попадают в ЦНС. Терфенадин и астемизол имеют активные метаболиты.
Фармакодинамика
А. Блокада гистаминовых рецепторов. Антагонисты Нгрецепторов блокируют действие гистамина по механизму обратимого конкурентного ингибирования. Они почти не действуют на Н2-рецеп-торы и оказывают слабое действие на Н3-рецепто-ры. Например, вызываемый гистамином бронхоспазм или сокращение гладкой мускулатуры ЖКТ полностью блокируются этими препаратами. Но сердечно-сосудистые эффекты гистамина блокируются только частично в связи с тем, что он действует на Н2-рецепторы (рис. 16-1). Эти препараты не влияют на эффекты, опосредованные преимущественно Н2-рецепторами, например повышение синтеза кислоты в желудке и подавление выделения гистамина из тучных клеток.
Б. Эффекты, не связанные с блокадой гистаминовых рецепторов. Блокаторы Нгрецепторов обладают многими эффектами, не связанными с антагонизмом действию гистамина, по-видимому, вследствие структурного сходства этих средств (рис. 16-2) с препаратами, которые действуют на мускариновые холинорецепторы, «-адренорецепторы, серотониновые рецепторы и рецепторы местных анестетиков. Некоторые из этих эффектов имеют терапевтическое значение, другие нежелательны.
1. Седативный эффект. Ht-блокаторы часто проявляют седативный эффект, но его интенсивность варьирует в зависимости от принадлежности к химической подгруппе (табл. 16-2) и неодинакова у разных пациентов. У некоторых из этих препаратов седативное действие настолько выражено, что их применяют в качестве снотворных средств (глава 65) и не используют в дневное время. Однако эффект, проявляемый Н]-антагонистами, больше похож на действие м-холинолитиков, но не седативных и снотворных средств. Иногда при применении Нгблокаторов в обычных дозах у детей (редко у взрослых) вместо седации возникает психомоторное возбуждение. В очень высоких токсических дозах препараты могут вызывать значительную стимуляцию, возбуждение и даже судороги, предшествующие наступлению комы. Считается, что новые Hj-блокаторы почти или совсем не обладают седативным действием. Терфенадин (или его метаболит) селективно действует на Н(-рецепторы и с
X — это: С, О или пропущенная группа
N—X—С—С—N
Общая структура
Эфиры или производные этаноламина
\ /СНз
СН —О —СН2—СН2—N / \
.у СНз
// Дифенгидрамин или дименгидринат
Производные этилендиамина
Т рипеленнамин
Производные пиперазина
Циклизин
Производные алкиламина
Производные фенотиазина
Рис. 16-2. Общая структура Ht-антагонистов и представители главных подгрупп
Терфенадин
трудом проходит через гематоэнцефалический барьер. Астемизол тоже высокоселективен и предположительно не проявляет седативную активность и не действует на вегетативную нервную систему. Лоратадин и цетиризин в этом отношении, по-ви-димому, несколько уступают астемизолу.
2. Уменьшение тошноты и противорвотное действие. Некоторые Н,-блокаторы предупреждают развитие синдрома укачивания (табл. 16-2), правда, они менее эффективны, если пациента уже ука
чало. Определенные Ht-антагонисты, например доксиламин (в составе бендектина), в прошлом широко использовали для лечения тошноты и рвоты при беременности.
3. Противопаркинсонические эффекты. Возможно, некоторые Ht-блокаторы в связи с их анти-холинергическим действием (как бензтропин; глава 27) значительно тормозят развитие симптомов паркинсонизма при приеме ряда антипсихотических препаратов.
ТАБЛИЦА 16-2. Некоторые Нгантигистаминные средства, применяемые в клинике
Лекарственные средства Обычная доза для взрослых Примечания
Этаноламины
Карбиноксамин (Клистин) 4-8 мг Вызывает седацию от легкой до умеренной
Дименгидринат (8-хлоротеофил- 50 мг Заметный седативный эффект; активен при болезнях
линовая соль дифенгидрамина) (Драмамини др.) движения
Дифенгидрамин (Бенадрил и др.) 25-50 мг Заметный седативный эффект; активен при болезнях движения
Доксиламин (Декаприн) Этилендиамины 1.25-25 мг Заметный седативный эффект; в настоящее время применяется только в составе безрецептурных препаратов (“снотворные”)
Антазолин 1-2 капли Компонент офтальмологических растворов
Пириламин (Нео-Антерган) 25-50 мг Умеренная седация; компонент безрецептурных “снотворных” средств
Трипеленнамин (PBZ) Производные пиперазина 25-50 мг Умеренная седация
Циклизин (Марезин) 25-50 мг Легкий седативный эффект; активен при болезнях движения
Меклизин (Бонин и др.) Алкиламины 25-50 мг Легкий седативный эффект; активен при болезнях движения
Бромфенирамин (Диметан и др.) 4-8 мг Легкий седативный эффект
Хлорфенирамин (Хлор-Триметон 4-8 мг Легкий седативный эффект; обычный компонент без-.
и др.) рецептурных средств для лечения простуды
Дексхлорфенирамин (Поларамин) Производные фенотиазина 2-4 мг Легкий седативный эффект; активный изомер хлор-фенирамина
Прометазин (Фенерган и др.) Пиперидины 10-25 мг Заметная седация; противорвотная и антимускарино-вая активность
Астемизол (Гисманал) 10 мг Меньшая частота седации
Терфенадин (Селдан) Разные 60 мг Меньшая частота седации
Ципрогептадин (Периактин и др.) 4 мг Умеренная седация; также имеет антисеротониновую активность
Лоратидин (Кларитин) 10 мг Продолжительное действие, слабый седативный эффект
4. Антихолинергический эффект. Многие Hi-блокаторы, особенно этаноламины и этилендиамины, обладают значительным атропиноподобным действием на периферические холинергические рецепторы. Этим можно объяснить их эффективность при некоторых видах неаллергического ринита. В то же время холинолитический эффект может обернуться задержкой мочи и затуманиванием зрения.
5. Блокада адренорецепторов. Многие из Нрантагонистов, особенно препараты фенотиазиновой подгруппы, обладают способностью блоки
ровать а-адренорецепторы. Это может вызывать ортостатическую гипотензию у чувствительных людей. Блокады 0-рецепторов не возникает.
6. Блокада серотониновых рецепторов. Для некоторых антагонистов Нгрецепторов, прежде* всего ципрогептадина, была продемонстрирована* * способность к выраженной блокаде серотониновых рецепторов. Ципрогептадин внедряли как антисё-ротониновое средство, и в этой книге он описай‘’в'! соответствующей главе. Тем не менее по химичёсЕ!' кой структуре препарат напоминает фенотиазино0"
вне антигистаминные средства и является сильным блокатором Нгрецепторов.
7. Местная анестезия. Большинство антагонистов Нгрецепторов — эффективные местноанестезирующие средства. Они блокируют натриевые каналы возбудимых мембран по тому же механизму, что прокаин и лидокаин. Дифенгидрамин и прометазин на самом деле являются более сильными местными анестетиками, чем прокаин. Их иногда используют в этом качестве у пациентов с аллергией на традиционные анестетики.
II. Клиническая фармакология антагонистов Нгрецепторов
Клиническое применение
А. Аллергические реакции. Блокаторы Нгрецепторов применяются в первую очередь для предотвращения или ослабления аллергических реакций. При аллергическом рините и крапивнице, когда в качестве основного медиатора выступает гистамин, блокаторы Нгрецепторов обычно очень эффективны и являются препаратами выбора. При бронхиальной астме, сопровождающейся выделением множества других медиаторов, эти препараты, как правило, неэффективны.
Высвобождение гистамина может усугублять сосудистый отек, однако его существование в основном поддерживается брадикининами, на которые не действуют антигистаминные препараты. При атопическом дерматите антигистаминные средства типа дифенгидрамина применяются прежде всего с целью седативного действия и некоторого облегчения зуда.
Нгблокаторы используют в терапии аллергических заболеваний типа сенной лихорадки. Обычно предпочтение отдают препаратам с минимальными побочными эффектами: в США это алкиламины и новые пиперидины. Седативный эффект и терапевтическая эффективность разных препаратов варьируют у людей, поэтому обычно в начале лечения пациенту выдают препараты-представители разных групп для определения самого эффективного средства, обладающего минимумом побочных эффектов у этого пациента. Если клиническая эффективность препарата снижается в процессе лечения, переход на препарат другой группы может восстановить ее по, не вполне понятному механизму.
Нгблокаторы нового поколения с незначительными седативными свойствами (терфенадин, лоратадин, астемизол и меквитазин) используются в основном для лечения больных с аллергическим ринитом и хронической крапивницей. В нескольких двойных слепых испытаниях этих средств была показана почти одинаковая их эффективность по сравнению с более старыми препаратами (например, хлорфенирамином). Однако седативный эффект и снижение внимания при выполнении потенциально опасных профессиональных действий (например, работа на станке), которые наблюдаются у 50 % пациентов, использующих традиционные антигистаминные препараты, возникают только у 7 % принимающих астемизол или терфенадин. Препараты нового поколения стоят значительно дороже.
Б. Укачивание и вестибулярные нарушения. Скополамин и определенные Нгблокаторы являются наиболее эффективными из имеющихся средств для предотвращения укачивания. Самые эффективные в этом отношении антигистаминные препараты — дифенгидрамин и прометазин. Пиперазины (циклизин и меклизин) тоже обладают выраженной противоукачивающей активностью, кроме того — меньшим седативным эффектом. Дозировка при укачивании такая же, как при лечении аллергий (табл. 16-2). И скополамин, и Нгблокаторы более эффективно предотвращают укачивание при сочетании с эфедрином или амфетамином.
Считалось, что антигистаминные средства, эффективные для профилактики укачивания, действуют и при болезни Меньера, однако это точно не доказано.
В. Тошнота и рвота при беременности. Было изучено несколько Нгблокаторов с целью их использования при утренней тошноте у беременных. Производные пиперазина исключили из этих исследований, после того как на грызунах были продемонстрированы их тератогенные свойства. Производное этаноламина — доксиламин — предложили для применения с этой целью в составе рецептурного препарата бендектин, содержащего пиридоксин. Вопрос о пороках развития плода в связи с применением бендектина обсуждается далее.
Токсичность
В этой главе уже был описан широкий спектр побочных эффектов Нгантигистаминных препаратов. Некоторые из этих эффектов (седативный, ан-
тимускариновый) имеют терапевтическое применение, особенно в составе безрецептурных средств (глава 65). Тем не менее именно они являются наиболее распространенными нежелательными эффектами при применении антигистаминных средств с единственной целью селективной блокады Нрре-цепторов. Правильный выбор препарата с учетом его терапевтического и побочных эффектов (табл. 16-2), а также применение нескольких лекарств — наиболее эффективные методы минимизации нежелательного действия.
Реже наблюдаются такие системные токсические эффекты как судороги у детей или постуральная гипотензия. Лекарственная аллергия встречается сравнительно часто, особенно после местного применения Ht-блокаторов. Последствия значительной системной передозировки препаратов старого поколения напоминают симптомы передозировки атропина и лечатся таким же образом (главы 8 и 60). Передозировка новых препаратов, особенно астемизола, может вызывать аритмии (Wiley, 1992; Saviuc, 1993). Этот же эффект наблюдается при сочетании с ингибиторами ферментов (подраздел “Лекарственные взаимодействия”).
Возможный тератогенный эффект доксиламина подробно освещался в средствах массовой информации, начиная с 1978 г., после сообщений о пороках развития плода при применении беременными женщинами бендектина. Однако ряд больших проспективных исследований (более 60 000 беременностей), в 3000 случаев из которых матери принимали бендектин, не подтвердил повышение частоты пороков развития у детей. Тем не менее в нескольких контролируемых обследованиях детей с пороками развития констатировали наличие слабой корреляции между использованием бендектина и возникновением определенных пороков. Из-за противоречивости сведений, отрицательного общественного мнения и бесконечных судебных исков изготовитель бендектина снял препарат с производства. Эта ситуация служит прекрасным примером трудностей оценки риска возникновения редкой патологии (глава 5).
Лекарственные взаимодействия
Значительные кардиотоксические эффекты, в том числе удлинение QT и потенциально летальные желудочковые аритмии, возникали у пациентов, принимающих терфенадин или астемизол в сочета
нии с кетоконазолом, итраконазолом или эритромицином. Последние три препарата подавляют метаболизм многих лекарств и, возможно, вызывают значительные повышения концентрации антигистамин ных препаратов в крови. Поэтому терфенадин и ас темизол противопоказаны пациентам, принимающим кетоконазол, итраконазол или эритромицин или пациентам с заболеваниями печени. Такого взаимодействия не отмечается при приеме лоратидина
При использовании Нгблокаторов, обладающих значительным седативным эффектом, одновременное назначение других препаратов, подавляющих ЦНС, вызывает аддитивное усиление эффекта и противопоказано водителям и людям других профессий, требующих постоянного внимания.
Блокаторы Н2-рецепторов (табл.16-3)
Создание блокаторов Н2-рецепторов рассмотрено в главе 5. Совершенствование препаратов этой группы возродило интерес к изучению физиологической роли гистамина и к созданию классификации эффектов агонистов и антагонистов в зависимости от подтипа задействованных гистаминовых рецепторов. Высокая частота язвенной болезни и связанных с ней жалоб на нарушения ЖКТ определили углубленное изучение терапевтического потенциала Н2-блокаторов. Вследствие их способности снижать секрецию кислоты в желудке эти препараты являются одними из наиболее широко применяемых в США средств.
I. Базисная фармакология блокаторов Н2-рецепторов
Химические свойства и фармакокинетика
Первые два Н2-блокатора, буримамид и мети-амид, были дериватами имидазола с длинными боковыми цепями, содержащими тиомочевинную группу. Из-за токсичности эти препараты больше не используют. В настоящее время в США применяют четыре Н2-блокатора: циметидин, ранитидин, фамотидин и низатидин. Их структура и некоторые фармакокинетические свойства приведены в таблице 16-3.
ТАБЛИЦА16-3. Блокаторы ^-рецепторов
Лекарственное средство Биодоступность (при приеме внутрь), % VD, л/кг ti/zp. ч Элиминация
Циметидин 30-80 0.8-1.2 1.5-2.3, повы- Главным образом
ZCH3 . NHCNH CH, 11 CH2 N —C = N шается при почками. Основ-
СНз Сн2 сн2 гл s тяжелой почечной недостаточности ной метаболит — S-оксид
Ранитидин 30-88 1.2-1.9 1.6-2.4, повы- Главным образом
H н шается при почками. Малые
НзС\ CH2 CH2 N N тяжелой по- количества мета-
N —СН2~< ; / А // / X / X / \ / X T S CH2 c CH3 чечной недо- болитов — S-окси-
НзС Ц II СН —no2 статочности да, N-оксида и N-деметила
Фамотидин 37-45 1.1-1.4 2.5-4, повыша- Главным образом
nh2 ется при тя- почками. Основ-
Н2\ H2N 's и 'CH2X/CH\ /2 S CH2“_ -so2nh2 желой почечной недостаточности ной метаболит — S-оксид
Низатидин 75-100 1.2-1.6 1.1-1.6, повы- Главным образом
НзС\ CH2X/CH2X / <1 S CH2 н N \ / \ С СН, II шается при тяжелой почечной недо- почками. Малые количества метаболитов — S-окси-
СН — no2 статочности да, N-оксида и N-деметила
Фармакодинамика
А. Механизм действия. Применяемые в настоящее время Н2-блокаторы обратимо конкурируют с гистамином за Н2-рецепторы. Эти препараты достаточно селективны, то есть практически не оказывают эффектов, опосредованных Нгрецепторами.
Б. Влияние на системы и органы.
1. Секреция кислоты и моторика ЖКТ. Самое главное действие Н2-блокаторов — снижение секреции кислоты в желудке (рис. 16-3). Они ингибируют секрецию кислоты, вызываемую гистамином, гастрином, холиномиметиками и стимуляцией вагу-са. Снижаются объем желудочного секрета и концентрация пепсина. Считается, что способность Н2-блокаторов подавлять все фазы секреции кислоты связана с участием гистамина как конечного общего медиатора этого процесса, однако более логично предположить, что он необходим для проявления
действия гастрина и парасимпатических импульсов. В главе 64 это обсуждается более детально.
Циметидин, ранитидин и фамотидин почти не действуют на гладкую мускулатуру желудка и на нижний сфинктер пищевода. Секреция в других отделах ЖКТ заметно не снижается. Существуют значительные различия между препаратами по силе действия. Низатидин стимулирует сократительную активность желудка, укорачивая время его опорожнения. Возможно, этот эффект связан со способностью низатидина подавлять активность ацетилхо-линэстеразы.
2. Другие эффекты Н2-блокады. В дозах, угнетающих секрецию кислоты в желудке, циметидин и ранитидин почти не влияют на сердце или артериальное давление. Полученные ранее сведения о том, что фамотидин снижает сердечный выброс, в более поздних исследованиях не подтвердились. Отмечено, что низатидин уменьшает частоту сер-
Рис. 16-3. Действие ранитидина на гиперсекрецию пепсина и секрецию кислоты у больного. Желудочное содержимое собиралось желудочным зондом в течение двух 6-часовых периодов. Пепсин измерялся в тысячах единиц (kU), а кислота — в ммоль/30 мин. Во время первого периода с 40-минутными интервалами внутривенно вводили изотонический раствор NaCl (плацебо). Во время второго периода с 40-минутными интервалами внутривенно вводили ранитидин в возрастающих дозах, как показано стрелками. Ранитидин оказывал отчетливое и продолжительное действие на секрецию кислоты и более короткое, но достоверное влияние на продукцию пепсина. (Из: Danilewith М. et al. Ranitidine supression of gastric hypersecretion resistant to cimetidine. N. Engl. J. Med. 1982; 306:20.)
дечных сокращений и сердечный выброс у здоровых людей. Все это соответствует основным представлениям о том, что гистамин оказывает минимальный эффект на регуляцию нормальной функции сердечно-сосудистой системы. Как показано на рис. 16-1, циметидин эффективно подавляет некоторые из воздействий введенного гистамина на сердце и сосуды.
Было предсказано, что Н2-блокаторы могут усиливать некоторые иммунные реакции, блокируя способность гистамина снижать высвобождение медиатора из тучных клеток и базофилов. В эксперименте можно продемонстрировать усиление реакций гиперчувствительности замедленного типа у некоторых больных, получающих циметидин. Однако свидетельств того, что у таких пациентов имеется клинически значимая активация реакций гиперчувствительности, нет.
3. Эффекты, не связанные с блокадой Н2-ре-цепторов. Циметидин и в значительно меньшей степени ранитидин могут подавлять окислительную систему цитохрома Р-450, метаболизирующую лекарственные препараты. Фамотидин и низатидин не обладают этим эффектом. Ранитидин подавляет связывание ацетаминофена с глюкуроновой кислотой у животных. Циметидин и ранитидин могут
снижать почечный клиренс основных препаратов, которые выделяются почечными канальцами. Циметидин связывается с рецепторами андрогенов и оказывает антиандрогенное действие. Ранитидин, фамотидин и низатидин не связываются с андрогенными рецепторами.
II. Клиническая фармакология Н2-блокаторов
Клиническое применение
А. Пептическая язва двенадцатиперстной кишки. Подсчитано, что 10 % взрослых в странах Запада в течение жизни переносят эпизод возникновения пептической язвы. Хотя смертность от этого заболевания низкая, частота возникновения рецидивов и социо-экономическая стоимость болезни высоки. Так как считалось, что “нет кислоты — нет и язвы”, медикаментозная терапия (до появления Н2-блокаторов) фокусировалась на снижении кислотности антимускариновыми препаратами и антацидами. Антимускариновые препараты приходится использовать в высоких дозах, и они оказывают серьезные побочные эффекты. Антациды облегчу-
ют симптомы и в высоких дозах способствуют заживлению язвы. Однако их надо принимать очень часто, а пациенты обычно плохо соблюдают режим приема, кроме как в острой симптоматической фазе заболевания. Высокая эффективность Н2-блокато-ров в отношении снижения кислотности желудка, сочетающаяся с их низкой токсичностью, является большим шагом в прогрессе терапии язвенной болезни. Результаты многих клинических испытаний показали, что эти препараты эффективно снимают симптомы во время острых эпизодов и облегчают заживление язв двенадцатиперстной кишки. Профилактическое применение в сниженных дозах помогает предотвратить рецидивы у многих пациентов. В этих случаях применяют также очень эффективный блокатор секреции кислоты (путем блокирования протонной помпы) омепразол и сукраль-фат, который ускоряет заживление, создавая оболочку вокруг язвы. Как следствие медикаментозной терапии после появления Н2-блокаторов значительно снизилась частота хирургических вмешательств по поводу язвы. В главе 64 представлены более подробные сведения по этому вопросу.
Для облегчения состояния больного с активной дуоденальной язвой достаточно приема Н2-блока-тора один раз в день перед сном. Заживление ускоряется приемом одного из следующих средств: 800 мг циметидина, 300 мг ранитидина, 40 мг фа-мотидина или 300 мг низатидина 1 раз в день курсом до восьми недель. Так как эти препараты выводятся почками, у пациентов с нарушением функции почек дозы необходимо снизить. Поддерживающая терапия после заживления дуоденальной язвы состоит в ежедневном однократном приеме 400 мг циметидина, 150 мг ранитидина, 20мгфамотидина или 150 мг низатидина, что эффективно снижает частоту рецидивов. Большинство пациентов адекватно реагируют на это лечение, но некоторые по непонятным причинам оказываются рефрактерны к нему. Препараты обычно назначают перорально; циметидин, ранитидин и фамотидин можно вводить и внутривенно. При внутривенном введении изредка возникают гипотензия и аритмии.
Б. Язва желудка. У пациентов с активной доброкачественной язвой желудка назначение Н2-бло-каторов облегчает симптомы и ускоряет заживление. Применяют те же дозы, что и при дуоденальной язве.
те. В. Эрозивный эзофагит (гастроэзофагальный рефлюкс). При эрозивном эзофагите обычно тре
буется более частый прием препаратов, чем при язве. Все четыре Н2-блокатора одобрены для применения по этому показанию в таких же или немного более высоких дозах, но разделенных как минимум на два приема.
Г. Гиперсекреторные состояния. Синдром Зол-лингера-Эллисона — это заболевание, как правило, с фатальным исходом, при котором гиперсекреция кислоты в желудке вызывается опухолью, секретирующей гастрин. К гиперсекреции часто приводят системный мастоцитоз и множественные гор-монсекретирующие аденомы. У многих пациентов Н2-блокаторы эффективно контролируют симптомы, связанные с избытком секреции кислоты. Препараты можно применять перед операцией или как основной метод лечения, когда оперативное вмешательство не показано. В таких ситуациях для снижения секреции кислоты обычно требуются намного более высокие дозы Н2-блокаторов, чем при язве. Кроме того, необходимо индивидуально подобрать кратность ежедневного приема препарата.
Д. Другие состояния. У пациентов с грыжей пищеводного отверстия диафрагмы, при стрессовых и ятрогенных язвах польза этих препаратов с очевидностью не доказана.
Токсичность
Н2-блокаторы хорошо переносятся, побочные эффекты возникают только в 1-2 % случаев. Наиболее часты диарея, головокружение, сонливость, головная боль и сыпь. Возможны также запор, рвота и артралгии. Больше всего побочных эффектов вызывает циметидин, меньше — низатидин. Другие более редкие, но серьезные нежелательные эффекты обсуждаются далее.
А. Дисфункция ЦНС. У пожилых пациентов могут проявляться нарушения речи и сознания, делирий. Чаще всего их вызывает циметидин, реже — ранитидин. Частота этого эффекта у фамотидина и низатидина не установлена.
Б. Эндокринные эффекты. Циметидин связывается с андрогенными рецепторами, поэтому встречаются такие антиандрогенные эффекты как гинекомастия у мужчин и галакторея у женщин. Кроме того, у мужчин, которые получают высокие дозы препаратов при гиперсекреторных состояниях, например при синдроме Золлингера-Эллисона, может снижаться количество спермы и возникать обратимая импотенция. Эти эффекты редко наблю
даются при курсе терапии продолжительностью менее 8 недель. Считается, что ранитидин, фамотидин и низатидин не обладают эндокринными эффектами.
В. Гематологические нарушения. Циметидин может вызвать гранулоцитопению, тромбоцитопению, нейтропению и даже в очень редких случаях — апластическую анемию. Есть также отдельные сообщения о подобных эффектах ранитидина. Меньше соответствующей информации имеется относительно фамотидина и низатидина.
Г. Токсическое действие на печень. Циметидин обладает обратимым холестатическим действием. Описаны случаи обратимого гепатита, с желтухой или без нее, при приеме ранитидина. При применении фамотидина и низатидина встречались обратимые нарушения функциональных печеночных тестов.
Д. Беременность и кормление. Исследования на беременных животных не выявили вредных воздействий на плод при приеме Н2-блокаторов. Однако эти препараты проходят через плаценту, то есть их следует назначать только при крайней необходимости. Все четыре препарата секретируются в грудное молоко и поэтому мщуг воздействовать на новорожденного.
Лекарственные взаимодействия
Циметидин подавляет окислительную систему метаболизма лекарств, катализируемую цитохромом Р-450. Препарат уменьшает печеночный кровоток, что может еще больше снизить клиренс других лекарственных средств. Совместное введение циметидина с любым из следующих препаратов может привести к усилению фармакологических эффектов или токсичности. Это варфарин, фенитоин, пропранолол, метапролол, лабеталол, хинидин, кофеин, лидокаин, теофиллин, алпразолам, диазепам, флуразепам, триазолам, хлордиазепок-сид, карбамазепин, этанол, трициклические антидепрессанты, метронидазол, блокаторы кальциевых каналов, препараты сульфонилмочевины. Может потребоваться коррекция дозировки, особенно при сниженной функции почек. Ранитидин в обычных терапевтических дозах не подавляет окислительный метаболизм других препаратов, но может повысить биодоступность этанола более чем на 40 % у здоровых людей. В исследованиях на животных ранитидин снижал связывание ацетаминофена глю
куроновой кислотой. Для фамотидина и низатидина не описаны клинически значимые лекарственные взаимодействия, однако опыт применения этих препаратов пока не велик.
Серотонин (5-гидрокситриптамин)
До идентификации 5-гидрокситриптамина (5-НТ) было известно, что при свертывании крови из сгустка высвобождается вазоконстрикторное вещество. Его назвали серотонином. Независимо в слизистой кишечника был открыт стимулятор гладкой мышцы — энтерамин. Синтез 5-гидрокситриптамина в 1951 г. позволил идентифицировать серотонин и энтерамин как один и тот же метаболит 5-гидрокситриптофана.
Известно, что серотонин играет роль в патогенезе некоторых заболеваний, а, может быть, принимает участие в развитии многих других. Одно из наиболее характерных состояний называется “серотониновый синдром”. Он вызывается взаимодействием серотонинергических средств (особенно блокаторов обратного захвата 5-НТ) и ингибиторов МАО. Это состояние описано в главе 29. Серотонин участвует также в развитии карциноидного синдрома — обычного проявления карциноидной опухоли (новообразования энтерохромаффинных клеток). У пациентов с неоперабельной опухолью антагонисты серотонина могут быть средствами выбора.
I. Базисная фармакология серотонина
Химические свойства и фармакокинетика
Как и гистамин, серотонин широко распространен в природе: находится в растительных и животных тканях, в ядах и секретах, обладающих раздражающим действием. Он представляет собой индо-лэтиламин, образующийся в биологических системах из аминокислоты L-триптофана гидроксилированием индольного кольца, а затем декарбоксилированием аминокислоты. Гидроксилирование по С5 является этапом, ограничивающим скорость про-
Серотонин
цесса, и его можно заблокировать /лхлорфенил-аланином (РСРА, фенклонин) и/>-хлорамфетами-ном. Эти вещества используют в эксперименте для ингибирования синтеза серотонина при карциноидном синдроме.
После синтеза свободная аминокислота хранится или быстро инактивируется, обычно путем окисления, катализируемого моноаминоксидазой. В эпифизе серотонин является предшественником мелатонина, меланоцитстимулирующего гормона. У млекопитающих, в том числе у человека, более 90 % всего серотонина находится в энтерохромаффинных клетках ЖКТ. В крови серотонин обнаруживается в тромбоцитах, которые способны концентрировать амин путем активного переноса, похожего на перенос пузырьками норадренергических или серотонинергических нервных окончаний. Серотонин имеется и в ядрах шва ствола мозга, где содержатся тела триптаминергических (серотонинергических) нейронов, которые синтезируют, хранят и высвобождают серотонин как медиатор. Серотонинергические нейроны мозга участвуют в реализации таких функций как настроение, сон, терморегуляция, ощущение боли и регуляция артериального давления (глава 20). Серотонин может участвовать в развитии таких состояний как депрессия, тревожность и мигрень. Серотонинергические нейроны находятся также в нервной системе кишечника и около кровеносных сосудов. У грызунов (но не у людей) серотонин найден в тучных клетках.
Функция серотонина в энтерохромаффинных клетках неясна. Эти клетки синтезируют серотонин, хранят его в комплексе с АТФ и другими веществами в гранулах и высвобождают в ответ на механические и нейрональные стимулы. Некоторая часть выделенного серотонина захватывается тромбоцитами и хранится в них. Депо серотонина может истощаться резерпином так же, как это происходит с запасами катехоламинов в пузырьках адренергических нервных окончаний (глава 6).
Серотонин метаболизируется моноаминоксидазой, его промежуточный продукт 5-гидроксииндо-лацетальдегид окисляется дальше альдегиддегид
рогеназой. При насыщении этого фермента, например большими количествами ацетальдегида при активации метаболизма этанола, значительная фракция 5-гидроксииндолацетальдегида может быть восстановлена в печени до спирта 5-гидрокси-триптофола. У человека при нормальной диете выделение 5-гидроксииндолуксусной кислоты (5-Ш АА) является мерой синтеза серотонина. Поэтому 24-часовая экскреция 5-Ш АА может быть использована как диагностический тест при опухолях, прежде всего карциноидных, которые синтезируют избыточное количество серотонина. Некоторые пищевые продукты (например, бананы) содержат большие количества серотонина или его предшественников, поэтому их нужно исключить из диеты во время проведения такого теста.
Фармакодинамика
А. Механизм действия. Серотонин, как и гистамин, оказывает множество видоспецифичных эффектов, трудных для обобщения. Действие серотонина опосредовано различными рецепторами клеточных мембран (табл. 16-4). Известно семь основных подтипов 5-НТ-рецепторов, в том числе ионные каналы, регулируемые лигандами, и рецепторы, связанные с G-белком. Среди этих подтипов рецепторов у некоторых до сих пор не определена физиологическая функция. Расшифровка этих функций будет возможна с появлением средств, селективных в отношении определенных подтипов рецепторов, или при использовании генетических методов, вызывающих мутирование или делецию генов, кодирующих эти рецепторы в геноме мыши.
Б. Тканевые и органные системные эффекты.
1. Сердечно-сосудистая система. Серотонин непосредственно вызывает сокращение гладкой мышцы. У человека серотонин является сильным вазоконстриктором всех сосудов, кроме сосудов скелетных мышц и сердца, которые под его действием расширяются. Реализация вазодилатирующего эффекта 5-НТ зависит, по крайней мере частично, от присутствия эндотелиальных клеток сосудов. При повреждении эндотелия возникает коронарный вазоспазм (Golino, 1991). Серотонин может вызывать также рефлекторную брадикардию, активируя хеморецепторы нервных окончаний. После инъекции серотонина часто наблюдается трехфазная реакция артериального давления. Сначала отмечается снижение частоты сердечных сокращений,
ТАБЛИЦА 16-4. Подтипы серотониновых рецепторов
Подтип рецептора Распределение Пострецепторный механизм Парциальные избирательные агонисты Парциальные изби рательн ые антагонисты
5-НТ1Д Ядра шва, гиппокамп 1 цАМФ, К+-каналы 8-OH-DPAT1
5-НТ1В 5-НТ1С Черная субстанция, бледный 1 цАМФ шар, базальные ганглии Хороид, гиппокамп, черная Т ИТФ субстанция СР 931292
5-HT1Daib 5-НТ1Е 5-HT,f Мозг 1 цАМФ Кора, путамен 1 цАМФ Кора, гиппокамп 1 цАМФ Суматриптан
5-НТгд 5-HT2F Тромбоциты, гладкие мышцы, Т ИТФ кора головного мозга Свод желудка ТИТФ а-метил-5-НТ Ритансерин
5-НТз Area postrema, чувствитель- Рецептором является 2-метил-5-НТ, Трописетрон, ондан-
ные и брюшные нервы Na*, «-ионный канал метахлорофе-нил-бигуанид сетрон,гранисетрон
5-НТд ЦНС и нейроны мышечного Т цАМФ 5-метокситрип- Bockaert, 1992,
слоя кишечника, гладкие мышцы тамин,рензап-рид, метоклопрамид (см. с. 345)
б-НТ5аЬ 5-НТ617 Мозг Неизвестен Мозг Т цАМФ Клозапин (5-НТ7)
1 8-OH-DPAT — 8-гидрокси-2-(ДИ-п-пропиламин)тетралин.
2 СР 93129 — 5-гидрокси-3(4-1,2,5,6-тетрагидропиридил)-4-азаиндол.
сердечного выброса и артериального давления, опосредованное хеморецепторами. После этого снижения артериальное давление в результате вазоконстрикции повышается. Во время третьей фазы артериальное давление опять падает из-за вазодилатации сосудов скелетных мышц. К вазоконстрикторному действию серотонина особо чувствительны сосуды легких и почек.
Серотонин сужает вены; веноконстрикция с последующим повышением кровенаполнения капилляров является причиной покраснения кожи после его введения. Он обладает слабым прямым положительным хронотропным и инотропным действием на сердце, не нашедшим применения в клинической практике. Однако длительное повышение уровня серотонина в крови (при карциноидном синдроме) связывают с появлением патологических изменений в эндокарде (субэндокардиальная фиброплазия), которые могут привести к нарушению функции клапанов или электрической проводимости сердца.
Серотонин вызывает агрегацию тромбоцитов, активируя поверхностные 5-НТ2-рецепторы. Этот ответ, в отличие от агрегации во время тромбообг разования, не связан с высвобождением серотонина из тромбоцитов. Физиологическая роль этого эффекта неясна.
2. Желудочно-кишечный тракт. Серотонин индуцирует сокращение гладких мышц ЖКТ, повышая тонус и усиливая перистальтику. Это действие связано с прямым влиянием серотонина на рецепторы гладких мышц и со стимуляцией ганглионарных клеток нервной системы кишечника (глава 6). Серотонин, если и обладает минимальным эффектом в отношении секреции, то обычно ингибиторным. Предполагается, что он играет большую роль как местный гормон, контролирующий моторику ЖКТ. Однако большие дозы антагонистов серотонина не вызывают тяжелых запоров. С другой стороны, гиперпродукция серотонина (и других веществ) карциноидной опухолью приводит к тяжелой диарее.
3. Дыхание. Серотонин имеет слабое прямое стимулирующее влияние на гладкую мускулатуру бронхов у здоровых людей. При карциноидном синдроме в ответ на значительное повышение уровня этого амина возникает бронхоспазм. Серотонин может вызывать и гипервентиляцию в результате хеморецепторного рефлекса или стимуляции сенсорных нервных окончаний бронхов.
4. Нервная система. Как и гистамин, серотонин является сильным стимулятором чувствительных окончаний, в частности ноцицепторов, и, следовательно, причиной возникновения боли и зуда при укусе насекомых и ожоге растениями. Кроме того, он сильный активатор хемочувствительных окончаний в области коронарных сосудов. Активация 5-НТ3-рецепторов на этих афферентных окончаниях блуждающего нерва связана с хеморецепторным рефлексом (рефлекс Бецольда-Яриша). Рефлекторный ответ проявляется значительной брадикардией и гипотензией. Брадикардия вызывается вагусными импульсами и может быть заблокирована атропином. Гипотензия — следствие снижения сердечного выброса из-за брадикардии. Хеморецептор-ный рефлекс могут вызывать многие другие препараты, в том числе Н-холиномиметики и некоторые сердечные гликозиды, например оубаин.
Серотонин присутствует в разных зонах мозга. Его нейромедиаторная роль и связь с эффектами препаратов, действующих на ЦНС, рассматривается в главах 20 и 29.
II. Клиническая фармакология серотонина
Агонисты серотонина
Сам серотонин не применяется в клинической практике. 5-НТ|Л-агонист буспирон привлек широкий интерес как небензодиазепиновый анксиолитик (Глава 21). Новый аналог серотонина суматриптан является 5-НТю-агонистом. Показано, что он эф-
Суматриптан
фективен при приступах мигрени и кластерных головных болях, что свидетельствует о наличии связи между нарушениями обмена серотонина и головными болями такого рода (Moskowitz, 1993).
После подкожного введения 6 мг суматриптана сукцината у 70 % пациентов с приступами мигрени наступает облегчение боли, то есть препарат более эффективен по сравнению с другими использующимися средствами. Побочные эффекты, а именно нарушения чувствительности (покалывание, ощущение тепла и т. д.), головокружение, мышечная слабость, боли в шее и реакции в месте укола, выражены слабо. У 5 % пациентов возникает ощущение дискомфорта в грудной клетке, а иногда даже боль. Суматриптан противопоказан при ишемической болезни сердца и стенокардии Принцметала. Другой недостаток этого препарата заключаются в том, что период его полувыведения меньше двух часов, то есть его эффект короче продолжительности самой головной боли. Следовательно, при продолжительном приступе мигрени может потребоваться прием нескольких доз, хотя препарат одобрен для приема только в количестве двух доз в течение 24 часов. Кроме того, в настоящее время этот препарат стоит очень дорого.
Помимо агонистов и антагонистов, серотонинергическую передачу модулируют такие соединения, как флуоксетин. Эти наиболее широко используемые для лечения больных с депрессией и другими поведенческими нарушениями препараты блокируют обратный захват медиатора. Они рассматриваются в главе 29.
Антагонисты серотонина
Действие серотонина, как и гистамина, можно ингибировать несколькими способами. Такой антагонизм необходим тем редким пациентам, которые страдают от карциноидной опухоли или некоторых других заболеваний.
Как уже отмечалось, синтез серотонина можно подавить р-хлорфенилаланином и р-хлорамфета-мином. Однако эти препараты слишком токсичны. Депонирование серотонина может быть нарушено при помощи резерпина, но симпатолитические эффекты этого средства (глава 11) и высокие уровни высвобождающегося серотонина не допускают его применения при карциноиде. Поэтому главным подходом к терапевтическому ограничению эффектов серотонина остается блокада рецепторов.
Антагонисты рецепторов серотонина
К конкурентным блокаторам серотониновых рецепторов относятся ципрогептадин и несколько экспериментальных средств. Кроме того, парциальными агонистами серотониновых рецепторов являются алкалоиды спорыньи, в последней части этой главы.
Ципрогептадин по химический структуре напоминает фенотиазиновые антигистаминные средства и обладает значительным блокирующим действием на Hj-рецепторы. Эффекты ципрогептадина можно предсказать, зная, Что он действует на гистаминовый и серотониновый рецепторы. Он подавляет действие обоих аминов на гладкую мышцу, в значительной степени проявляет антимускариновые эффекты, вызывает седацию, но не влияет на стимулируемую гистамином желудочную секрецию.
Главное клиническое применение ципрогептадина — терапия гладкомышечных нарушений при карциноидной опухоли и демпинг-синдром после гастрэктомии. Обычная доза для взрослых — 12-16 мг/день в три-четыре приема. Он является также препаратом выбора при крапивнице, вызванной холодом.
Кетансерин блокирует 5-HTiC- и 5-НТ2-рецеп-торы и практически не действует на другие 5-НТ-или на Hi-рецепторы. Он является также ингибитором «,-адренорецепторов сосудов. Кетансерин — антагонист тромбоцитарных 5-НТ2-рецепторов, подавляющий агрегацию тромбоцитов под действием серотонина. Интерес к этому соединению вызван обнаруженной возможностью понижать с его помощью артериальное давление у животных и людей при гипертензии. Однако точные механизмы его гипотензивного действия неизвестны и могут быть опосредованы как аг, так и 5-НТ2-рецеп-торами. Хотя препарат используется в Европе для лечения гипертензии и вазоспазма, в США он не одобрен для применения.
Другой антагонист 5-НТ2-рецепторов ритансе-рин почти не оказывает «-блокирующего действия. Известно, что он может влиять на время кровотечения и уменьшать образование тромбоксана предположительно путем изменения функции тромбоцитов.
Антагонист 5-НТ3-рецепторов ондансетрон одобрен для применения с целью профилактики тошноты и рвоты при химиотерапии рака. Он эф
фективен в дозах 0.1-0.2 мг/кг внутривенно. Сейчас препарат проходит испытания как средство для лечения послеоперационных тошноты и рвоты, тревожности и психоза. Недавно был одобрен грани-сетрон — препарат со сходными свойствами. Другие родственные препараты еще проходят клинические испытания.
Принимая во внимание разнообразие эффектов серотонина и гетерогенность 5-НТ-рецепторов, можно предположить, что и другие селективные 5-НТ-антагонисты могут оказаться полезными в клинике.
Алкалоиды спорыньи
Бертрам Г. Катцунг
Алкалоиды спорыньи вырабатывает грибок Claviceps purpurea (спорынья пурпурная), который инфицирует зерно, особенно пшеницу, при хранении или росте во влажных условиях. Этот грибок, помимо целой группы уникальных алкалоидов, синтезирует гистамин, ацетилхолин, тирамин и другие биологически активные вещества. Алкалоиды спорыньи действуют на «-адренорецепторы, дофаминовые, 5-НТ- и, может быть, другие рецепторы.
Случайный прием этих алкалоидов с загрязненным зерном описывали еще 2000 лет назад как эпидемии отравлений спорыньей (эрготизм). Самые значительные эффекты при отравлении — это деменция и яркие галлюцинации, продолжительный вазоспазм, который может привести к гангрене, и стимуляция гладкой мускулатуры матки, являющаяся причиной невынашивания беременности. Тяжесть каждого эффекта варьировала при разных эпидемиях и зависела скорее всего от состава смеси алкалоидов, синтезированных разными колониями грибка. В средние века отравление алкалоидами спорыньи называли пожаром Св. Антония в честь святого, помощи у которого просили при жгучей боли. Эпидемии таких отравлений наблюдаются до настоящего времени (дополнение “Отравление спорыньей: не только древнее заболевание”), поэтому требуется постоянный контроль пищевого зерна. Часто и во многих областях встречается отравление животных, так как эти же или родственные им грибки могут паразитировать и на пастбищных травах (Porter, 1992).
Кроме названных эффектов, алкалоиды спорыньи оказывают разнообразное действие на цент
ральную и периферическую нервные системы. На основе детального анализа структуры и активности их соответствующих полусинтетических модификаций создано большое количество препаратов с доказанным или потенциальным клиническим действием.
I. Базисная фармакология алкалоидов спорыньи
Химические свойства
и фармакокинетика
Известны два основных семейства соединений, которые содержат тетрациклическое эрголиновое ядро: аминные и пептидные алкалоиды (табл. 16-5).
В состав обеих групп входят препараты, имеющие терапевтическое и токсикологическое значение.
Алкалоиды спорыньи по-разному абсорбируются в ЖКТ. Пероральная доза эрготамина в 10 раз больше, чем внутримышечная, но скорость абсорбции и пиковые уровни в крови после перорального введения можно увеличить, принимая препарат вместе с кофеином. Аминные алкалоиды всасываются также из ротовой полости после ингаляции аэрозоля и из прямой кишки. Абсорбция после внутримышечного введения идет медленно, но обычно достаточно надежна. Бромокриптин хорошо всасывается из ЖКТ.
Алкалоиды спорыньи активно метаболизируются в организме. Первичные метаболиты гидроксилируются по кольцу А, у пептидных алкалоидов модифицируется и пептидная группа. Большая
ТАБЛИЦА 16-5. Главные эрголиновые производные (алкалоиды спорыньи)
Аминные алкалоиды
Ri Re
6-метил-
зрголин -н
Лизергиновая -н кислота
Диэтиламид лизергиновой -н кислоты (ЛСД)
ЭРГОНОВИН _|_|
(эргометрин)
-Н
-СООН
О
II
— С — N(CH2-CH3)2
О СНгОН
II I
— С — NHCHCH3
а-эргокриптин -н -СН(СН3)2 -СН2-СН(СН3)2
Бромокриптин -Вг -СН(СН3)2 -СН2-СН(СН3)2
Метисегрид -СНз
о
II
— С — NH — СН — СН2 — СНз I СНгОН
' У дигидроэрготамина отсутствует двойная связь между С9 и С10.
часть экскреторных продуктов метисергида деметилируется в положении N1.
Фармакодинамика
А. Механизм действия. Алкалоиды спорыньи действуют на несколько рецепторов. Их эффекты являются агонистическими, парциально агонистическими и антагонистическими относительно а-рецепторов и серотониновых рецепторов, и агонистическими в отношении дофаминовых рецепторов ЦНС (табл. 16-6). Кроме того, возможна активация еще не открытых рецепторов. Некоторые члены этой группы имеют сродство к пресинаптическим рецепторам, другие более селективны в отношении постсинаптических. Эти средства оказывают сильное влияние на матку, вероятнее всего, опосредованное агонистическим (или парциально агонистическим) действием на 5-НТ^рецепторы. Структурные различия определяют селективность членов этой группы в отношении конкретных типов рецепторов.
Б. Органные и системные эффекты.
1. ЦНС. Как явствует из традиционных описаний эрготизма, некоторые естественные алкалоиды являются сильными галлюциногенами. Диэтила-мид лизергиновой кислоты (ЛСД, “кислота”), синтезируемый спорыньей, наиболее ярко демонстрирует этот эффект. ЛСД использовали в лабораторных условиях как сильный периферический антагонист 5-НТ2-рецепторов. В настоящее время появились сведения о том, что поведенческие эффекты препарата опосредованы его агонистическим влиянием на пресинаптические (Sadzot, 1989) или постсинаптические (Buckholtz, 1990) 5-НТ2-рецеп-торы ЦНС. Несмотря на активные исследования, ЛСД не нашел клинического применения. Злоупот
ребление этим веществом было очень распространено в течение нескольких десятилетий (глава 31).
Дофаминовые рецепторы ЦНС имеют огромное значение в контроле двигательной активности через экстрапирамидную систему и в регуляции высвобождения пролактина. Влияние бромокриптина на экстрапирамидную систему обсуждается в главе 27. Из всех известных в настоящее время соединений спорыньи бромокриптин обладает самой высокой селективностью в отношении дофаминовых рецепторов гипофиза. Он прямо подавляет секрецию пролактина из клеток гипофиза, активируя регуляторные дофаминовые рецепторы (глава 36). Бромокриптин конкурирует с самим дофамином и агонистами дофамина, например апоморфином, за связывание с соответствующими сайтами.
2. Гладкая мускулатура сосудов. Характер действия алкалоидов спорыньи на гладкую мускулатуру сосудов зависит от препарата, вида животного и от типа сосуда. Эрготамин и родственные соединения вызывают сужение большинства сосудов у человека. Этот частично ингибируемый типичными а-блокаторами эффект существенно выражен, продолжителен и предсказуем (рис. 16-4). Однако он связан также с “извращением действия адреналина” (глава 10) и с блокадой реакции на другие а-агонисты. Это двойное действие является парциально агонистическим (табл. 16-5).
Так как эрготамин медленно диссоциирует из комплекса с а-рецепторами, он вызывает очень длительные агонистические и антагонистические эффекты. Алкалоид практически не действует на Р-рецепторы.
Вазоконстрикция, вызванная алкалоидами спорыньи, в значительной степени связана с парциально агонистическими эффектами в отношении а-адренорецепторов, однако частично она может
ТАБЛИЦА 16-6. Влияние алкалоидов спорыньи на некоторые рецепторы1
Алкалоиды спорыньи а-адреноре-цептор Дофаминовый рецептор Серотониновый рецептор (5-НТ2) Стимуляция гладкой мускулатуры матки
Бромокриптин — +++ — 0
Эргоновин + + -(ПА) +++
Эрготамин --(ПА) 0 +(ПА) +++
Диэтиламид лизергиновой кислоты (ЛСД) 0 +++ -— +
Метисергид +/0 +/0 ---(ПА) +/0
’ Агонистическое влияние обозначено знаком «+», , антагонистическое — «-», отсутствие эффекта — 0. На относительную
аффинность к рецептору указывает количество знаков «+» или «-». ПА означает парциальный агонист (может проявлять и
агонистические, и антагонистические эффекты).
Рис. 16-4. Действие производных спорыньи на сокращение изолированных полосок базиллярной артерии человека, удаленных при операции. Все производные спорыньи являются парциальными агонистами и действуют сильнее, чем полные агонисты, норадреналин и серотонин. (НА — норадреналин, 5-НТ — серотонин, ЭРГ — эрготамин, МЭМ — метилэргометрин, Д ГЭТ—дигидроэрготамин, МС — метисергид.) (Из: Muller-Schweinitzer Е. in: 5-Hydroxytryptamine Mechanisms in Primary Headaches. Oleson J., Saxena P. R. (eds) Raven Press, 1992.)
быть опосредована 5-НТ-рецепторами. Эрготамин, эргоновин и метисергид являются парциальными агонистами 5-НТ2-рецепторов сосудов. Более того, сосуды разных областей обладают неодинаковой чувствительностью; считается, что сосуды мозга, прежде всего артериовенозные анастомозы, особенно чувствительны к таким препаратам как эрготамин, дигидроэрготамин и суматриптан (den Boer, 1992). Очень специфическое антимигреневое действие препаратов спорыньи предположительно связано с их влиянием на нейрональные или сосудистые серотониновые рецепторы. Для лечения мигрени наиболее широко применяются эрготамин, эргоновин и метисергид.
При передозировке эрготамина или других подобных средств вазоспазм очень выражен и длителен (раздел “Токсичность”). Он не устраняется а-антагонистами, антагонистами серотонина и даже их сочетанием.
Эрготамин — типичный алкалоид спорыньи, являющийся сильным вазоконстриктором. Гидрогенизация алкалоидов в положениях 9 и 10 (табл. 16-5) приводит к образованию дигидродериватов, которые имеют значительно меньший пря-Мйй серотониновый и гладкомышечный эффекты, но являются более селективными ос-блокаторами.
3. Гладкая мускулатура матки. Стимулирующее действие алкалоидов спорыньи на матку, также как и на гладкую мускулатуру сосудов, связано с комбинированием ос-агонистического, серотонинового и других эффектов. Более того, чувствительность матки к стимулирующим эффектам спорыньи значительно меняется во время беременности, возможно, в связи с увеличением количества о^-рецепторов в ходе развития беременности. В результате этого матка в конце беременности более чувствительна к алкалоидам, чем в ее начале, и значительно более чувствительна, чем до наступления беременности.
В очень малых дозах алкалоиды спорыньи могут вызывать ритмические сокращения и расслабления матки. В более высоких концентрациях применение этих средств приводит к сильным и длительным сокращениям. В данном случае эргоновин более селективен, чем другие алкалоиды спорыньи, и является препаратом выбора в акушерстве.
4. Другие гладкомышечные органы. У большинства пациентов алкалоиды спорыньи не оказывают значительного влияния на гладкую мускулатуру бронхов. С другой стороны, выраженность действия на ЖКТ значительно варьирует в зависимости от чувствительности пациента. У некоторых даже низ-
Отравление спорыньей: не только древнее заболевание
Как уже отмечалось, эпидемии эрготизма, или отравления загрязненным спорыньей зерном, спорадически встречались, начиная с древних времен. Легко представить социальный хаос, например в средневековом обществе, где люди верили в ведьм, демонов и наказания человека сверхъестественными силами за проступки, если в одно и то же время возникал целый ряд случаев жгучей боли, гангрены, галлюцинаций, судорог и абортов. Такие трактовки причин этих явлений в большинстве современных культур ушли в далекое прошлое. Однако эрготизм в настоящее время не исчез. Наиболее типичные случаи эрготизма были зарегистрированы в 1951 г. в маленькой деревушке Пон-Сент-Эспри во Франции. Эпидемия была описана в Британском медицинском журнале за 1951 г. (Gabbai, 1951) и в дальнейшем в книге (Fuller, 1968). После употребления хлеба из загрязненной муки у нескольких сотен людей возникли галлюцинации, судороги, ишемия, и несколько человек умерли. Сходные случаи описаны и в более позднее время, когда вследствие бедности, голода или неграмотности люди употребляли в пищу загрязненное зерно.
Часто встречается ятрогенный эрготизм, вызванный избыточным применением препаратов
спорыньи. Типичен следующий случай (Andersen et al„ 1977): 58-летняя женщина, применявшая эрготамин в умеренных дозах для лечения мигрени, поступила в больницу после трех дней нарастания симптомов ишемии нижних конечностей (боль, цианоз, болезненность при пальпации и низкая температура кожи). Периферическая пульсация ниже бедренных артерий отсутствовала. Исходная аортограмма показана на рис. 16-5 (слева). Для блокады симпатических вазоконстрикторных импульсов к сосудам таза и нижних конечностей был эпидурально введен местный анестетик, но улучшения не последовало. Через 3 часа начали инфузию нитропруссида натрия. Температура кожи большого пальца ноги резко повысилась с 22 °C до 31 °C. Исчез цианоз, появилась пульсация подколенных и тибиаль-ных сосудов. Попытка прекратить инфузию нитропруссида натрия через 11 часов привела к немедленной вазоконстрикции и цианозу нижних конечностей, поэтому инфузию возобновили и продолжали еще в течение 25 часов с хорошим сохранением циркуляции. Через 36 часов после начала инфузии введение нитропруссида прекратили, вазоконстрикции не возникало. Повторная аортограмма через неделю после лечения была нормальной (рис. 16-5 справа).
кие дозы алкалоидов спорыньи вызывают тошноту, рвоту и диарею; чаще эти симптомы проявляются при употреблении высоких доз. Эти эффекты связаны с действием на рвотный центр ЦНС и на серотониновые рецепторы ЖКТ.
II. Клиническая
фармакология алкалоидов спорыньи
Клиническое применение
А. Мигрень. Классическая головная боль при мигрени характеризуется короткой “аурой”, которая может включать зрительные скотомы или даже ге
мианопсию и нарушения речи, с последующим развитием сильной односторонней пульсирующей головной боли, длящейся в течение нескольких часов вплоть до 1-2 дней. В 60-80 % случаев заболевание носит семейный характер. Более часто встречается у женщин, обычно начинается в юношеском и раннем взрослом возрасте, угасая с течением жизни. Приступы, как правило, провоцируются стрессом, но чаще возникают после, а не во время стрессового эпизода. Частота варьирует от одного в год до двух или более в неделю. Существуют подробнейшие описания приступов пациентами (Creditor, 1982). Хотя характер симптомов варьирует у разных пациентов, тяжесть головной боли в подавляющем большинстве случаев приводит к необходимости лечения.
Патофизиология мигрени включает некий вазо-, моторный механизм, так как начало головной болй
Рис. 16-5. Вазоспазм, вызванный эрготамином. Ангиограмма аорты во время максимального вазоспазма (слева) и через неделю после инфузии нитропруссида (справа). Основные сосуды дистальнее общих подвздошных артерий (стрелки) нитевидны и закрыты на левой ангиограмме, справа — нормальный кровоток восстановлен. (Из: Andersen Р. К. et al. Sodium nitroprusside and epidural blockade in the treatment of ergotism. N. Engl. J. Med. 1977; 296:1271.)
связано co значительным повышением амплитуды пульсаций височной артерии, а облегчение боли после приема эрготамина ассоциируется с уменьшением артериальной пульсации. С другой стороны, вазомоторные изменения могут быть результатом более глубоких процессов, лежащих в основе мигрени, так как при этом заболевании эффективен целый ряд средств с различными механизмами действия. Хотя многие вазоконстрикторы способны ослабить или прекратить боль при мигрени, лекарственная дилатация церебральных сосудов не вызывает типичный приступ мигрени. Гистологическое изучение церебральных сосудов пациентов с мигренью показало, что приступы связаны со стерильным нейрогенным периваскулярным воспалением и отеком. Так как эта область обширно иннервируется чувствительными нервами, такие патологические изменения могут вызывать длительную головную боль. Наконец, считается, что многие из препаратов, эффективных при мигрени (алкалоиды спорыньи, нестероидные противовоспалительные средства), способны предотвращать или умень
шать воспаление периваскулярной ткани (Moskowitz, 1989; Buzzi, 1990).
Показано, что начало мигренозной ауры коррелирует с аномальным высвобождением серотонина из тромбоцитов, а пульсирующая головная боль — со снижением тромбоцитарного и сывороточного уровня серотонина ниже нормальной концентрации. По другим сведениям, в менингеальных сосудах и вокруг них имеются множественные серотонинергические нервные окончания, и препараты для лечения мигрени напрямую или косвенно воздействуют на серотонинергическую передачу. Дополнительные химические триггеры, которые могут действовать через серотонин,— это эстрогены (снижение их уровня у женщин приводит к головной боли в определенную фазу менструального цикла) и простагландин Е( (повышение уровня также ассоциируется с головной болью).
Эффективность алкалоидов спорыньи в терапии мигрени настолько специфична, что ее используют как диагностический тест. Традиционная терапия (эрготамин) наиболее эффективна при приеме во
время продромы, но не при развитии приступа и отсроченном приеме. В аптеках имеется эрготамина тартрат для перорального и сублингвального применения, в виде ректальных свечей и ингаляционной формы. Для улучшения всасывания алкалоида его часто комбинируют с кофеином (100 мг кофеина на 1 мг эрготамина тартрата).
Вызванная эрготамином вазоконстрикция длительна и кумулируется при повторном введении, например в случае сильного приступа. В связи с этим пациентов необходимо предупредить, что для снятия одного приступа нельзя принимать больше 6 мг препарата перорально, а за неделю — более 10 мг. При очень тяжелых приступах можно ввести 0.25-0.5 мг эрготамина тартрата внутривенно или внутримышечно. Некоторые врачи предпочитают лечить пациентов с мигренью, не поддающейся терапии другими препаратами, внутривенным введением 0.5-1 мг дигидроэрготамина. В настоящее время исследуется эффективность интраназального или перорального введения дигидроэрготамина. Этот путь может оказаться перспективным.
В связи с кумулятивными эффектами эрготамина, применяемого для профилактики мигрени искали другие более безопасные препараты. С этой целью используют эргоновин 0.2-0.4 мг 3 раза в день. Препарат аминной подгруппы метисергид (табл. 16-5) оказался эффективным у 60 % пациентов. К сожалению, почти у 40 % больных возникают значительные токсические симптомы. Как показано в табл. 16-6, существуют некоторые различия в проявлении агонистического и антагонистического действия метисергида и эрготамина в отношении серотониновых рецепторов. Метисергид относительно неэффективен при лечении начинающегося или уже активного приступа мигрени вне зависимости от причины. Суточная доза метисергида малеата при использовании для профилактики мигрени равна 4-8 мг. Хотя он практически не имеет быстрой кумулятивной вазоспастической токсичности эрготамина, при хроническом применении может вызывать ретроперитонеальную фиброплазию и субэндокардиальный фиброз, вероятно, вследствие его сосудистых эффектов. Поэтому важно, чтобы пациенты, принимающие метисергид, устраивали “лекарственные каникулы” длительностью 3-4 недели каждые 6 месяцев (раздел “Токсичность и противопоказания”).
Для профилактики мигрени некоторые пациенты с успехом применяют пропранолол и ами
триптилин. Как и метисергид, они не оказывают эффекта при остром приступе мигрени. Исследуемый в настоящий момент блокатор кальциевых каналов флунаризин в предварительных опытах эффективно снижал тяжесть острых приступов и предотвращал их повторное возникновение.
Достойной альтернативой эрготамину в терапии острых приступов мигрени является суматриптан. Однако в настоящее время не существует лекарственной формы этого препарата для перорального применения или хронического использования.
Б. Гиперпролактинемия. Повышение сывороточного уровня гормона передней доли гипофиза пролактина, как правило, вызвано наличием секретирующей опухоли этой железы или применением антагонистов дофамина центрального действия, особенно антипсихотических препаратов. Гиперпролактинемия по механизму отрицательной обратной связи приводит к аменорее и бесплодию у женщин, а также галакторее у пациентов обоих полов.
Бромокриптина мезилат эффективно снижает высокий уровень пролактина у больных с опухолью гипофиза и даже в некоторых случаях приводит к регрессии опухоли. Обычная доза равна 2.5 мг два или три раза в день. Иногда бромокриптин в такой же дозе используют для подавления физиологической лактации. При применении по этому показанию описаны случаи возникновения серьезных сердечно-сосудистых осложнений в послеродовом периоде (Ruch, 1989).
В. Послеродовое кровотечение. Матка в конце беременности крайне чувствительна к стимулирующему действию алкалоидов спорыньи, и даже небольшие их дозы могут вызвать длительный и сильный спазм миометрия, который отличается от его сокращения при естественных родах. На это не обратили должного внимания, когда в XVIII в. алкалоиды спорыньи впервые применили в акушерстве для ускорения родов. В результате значительно повысилась материнская и детская смертность. Следовательно, алкалоиды спорыньи можно использовать только для остановки позднего маточного кровотечения и их никогда не следует назначать перед родами. Введенный внутримышечно эргоновийа малеат в дозе 0.2 мг начинает действовать через 1<-5 минут. Он менее токсичен, чем другие алкалоиды спорыньи, применяемые по этому показанию. Препарат вводят при значительном кровотечения
в момент отхождения плаценты или сразу после этого.
В. Диагностика стенокардии Принцметала. Эргоновин вызывает мгновенную коронарную вазоконстрикцию вследствие спастической реакции (вариантная стенокардия, или стенокардия Принцметала; глава 12). Поэтому этот алкалоид спорыньи можно вводить внутривенно во время коронарной ангиографии для диагностики вариантной стенокардии.
Г. Сенильная церебральная недостаточность. Дигидроэрготоксин, представляющий собой смесь дигидро-а-эргокриптина и трех сходных по структуре дегидрогенизированных пептидных алкалоидов спорыньи (эрголоидные мезилаты), в течение ряда лет предлагался сначала для коррекции сенильных нарушений, а позднее — для терапии деменции при болезни Альцгеймера. Неоспоримых доказательств эффективности такого лечения нет.
Токсичность и противопоказания
Самые частые побочные эффекты алкалоидов спорыньи — это нарушения со стороны ЖКТ, в том числе диарея, тошнота и рвота. В основе этих эффектов лежит активация рвотного центра продолговатого мозга и желудочно-кишечных серотониновых рецепторов. Так как приступы мигрени часто сопровождаются этими симптомами и до терапии, то эти побочные эффекты редко становятся противопоказанием для применения алкалоидов спорыньи. Однако возникновение тошноты, рвоты и диареи при профилактическом применении метисер-гида ограничивают его применение.
Более опасный побочный эффект передозировки таких препаратов как эрготамин и эргоновин — это длительный вазоспазм. Спазмирование гладкой мускулатуры сосудов может привести к гангрене с последующей ампутацией. В большинстве таких случаев нарушается кровообращение в руках и ногах. Однако встречается и инфаркт стенки кишки вследствие вазоспазма мезентериальной артерии. Спазм периферических сосудов под действием алкалоидов спорыньи рефрактерен к действию большинства вазодилататоров, но иногда помогает инфузия больших доз нитропруссида или нитроглицерина (Tfelt-Hansen, 1982) (дополнение ‘‘Отравление спорыньей: не только древнее заболевание”). ; Длительная терапия метисергидом приводит к фцбропластическим изменениям ретроперитоне
ального пространства, плевральной полости и эндокарда. Эти изменения постепенно нарастают в течение нескольких месяцев и могут проявиться как гидронефроз (из-за обструкции мочеточников) или сердечный шум (из-за изменения клапанов сердца). В некоторых случаях патологии клапанов может потребоваться хирургическое вмешательство. К счастью, у большинства пациентов эти токсические проявления регрессируют после отмены препарата. Однако вероятность их возникновения настолько высока, что пациенты, принимающие метисер-гид, должны периодически устраивать “лекарственные каникулы”. При возникновении фиброплазии больные должны находиться под постоянным наблюдением.
Еще один токсический эффект алкалоидов спорыньи — это сонливость, а в случае метисергида — иногда стимуляция ЦНС и галлюцинации. Наркоманы даже использовали метисергид вместо ЛСД.
К противопоказаниям для применения алкалоидов спорыньи относятся обструктивные поражения сосудов и коллагенозы.
Не существует данных о том, что применение эрготамина или метисергида при мигрени опасно во время беременности. Однако большинство клиницистов советуют беременным воздержаться от приема этих препаратов.
Препараты
Антигистаминные препараты (Н^блокаторы)
Астемизол (Гисманал)
Перорально: таблетки по 10 мг
Азатадин (Оптимин)
Перорально: таблетки по 1 мг
Бромфенирамин (генерик, Диметан,
Бромфен, др.)
Перорально: таблетки по 4,8,12 мг;
эликсир 2 мг/5 мл
Перорально для пролонгированного действия: таблетки по 8, 12 мг Парентерально: 10 мг/мл для инъекций
Буклизин (Букладин-С Софтабз) Перорально: таблетки по 50 мг
Карбиноксамин (Клистин)
Перорально: таблетки по 4 мг
Цетиризин (Реактин)
Перорально (пока нет других данных)
Хлорфенирамин (генерик, Хлор-Триметон,
Телдрин, др.)
Перорально: жевательные таблетки по 2 мг; таблетки по 4,12 мг; сироп 2 мг/5 мл Перорально для пролонгированного действия: таблетки и капсулы по 8,12 мг Парентерально: 10,100 мг/мл для инъекций
Клемастин (Тавист)
Перорально: таблетки по 1.34, 2.68 мг;
сироп 0.67/5 мл
Циклизин (Марезин)
Перорально: таблетки по 50 мг
Парентерально: 50 мг/мл для инъекций
Ципрогептадин (генерик, Периактин)
Перорально: таблетки по 4 мг;
сироп 2 мг/5 мл
Дексхлорфенирамин (генрик, Поламин, др.)
Перорально: таблетки по 2 мг;
сироп 2 мг/5 мл
Перорально для пролонгированного действия: таблетки по 4,6 мг
Дименгидринат1 (генерик, Драмамин)
Перорально: таблетки и капсулы по 50 мг; жевательные таблетки по 50 мг;
сироп и эликсир 12.5 мг/4 мл; 15.62 мг/5 мл
Дифенгидрамин (генерик, Бенадрил)
Перорально: таблетки и капсулы по 25, 50 мг; эликсир и сироп 12.5 мг/ 5мл Парентерально: 10, 50 мг/мл для инъекций
Гидроксизин (генерик, Атаракс, Вистарил) Перорально: таблетки по 10, 25, 50,100 мг; капсулы по 25, 50,100 мг; сироп 10 мг/5 мл; суспензия 25 мг/5 мл
Парентерально: 25, 50 мг/мл для инъекций
Лоратидин (Кларитин)
Перорально: таблетки по 10 мг
Меклизин (генерик, Антиверт, др.)
Перорально: таблетки по 12.5, 25, 50 мг; капсулы по 25 мг;
таблетки для жевания по 25 мг
1 Дименгидринат — хлоротеофиллиновая соль дифенгидрамина
Метдилазин (Такарил)
Перорально: таблетки для жевания по 4 мг; таблетки по 8 мг; сироп 4 мг/5 мл
Фениндамин (Нолахист)
Перорально: таблетки по 25 мг
Прометазин (генерик, Фенерган, др.)
Перорально: таблетки по 12.5, 25,50 мг;
сироп 6.25, 25 мг/5 мл
Парентерально: 25 мг/мл для инъекций;
50 мг/мл для в/м инъекций
Ректально: свечи 12.5, 25, 50 мг
Пириламин (генерик)
Перорально: таблетки по 25 мг
Терфенадин (Селдан)
Перорально: таблетки по 60 мг
Тримепразин (Темарил)
Перорально: таблетки по 2.5 мг;
сироп 2.5 мг/5 мл
Перорально для пролонгированного действия: капсулы по 5 мг
Трипеленнамин (генерик, Пеламин, PBZ)
Перорально: таблетки по 25, 50 мг;
эликсир 37.5 мг/5 мл
Перорально для пролонгированного действия: таблетки по 100 мг
Трипролидин (генерик, Актидил, Миидил) Перорально: таблетки по 2.5 мг;
сироп 1.25 мг/5 мл
Н2-блокаторы
Циметидин (Тагамет)
Перорально: таблетки по 200, 300, 400, 800 мг; жидкий 300 мг/5 мл
Парентерально: 300 мг/2 мл, 300 мг/50 мл для инъекций
Фамотидин (Пепсид)
Перорально: таблетки по 20,40 мг; порошок для приготовления суспензии 40 мг/5 мл Парентерально: 10 мг/мл для инъекций
Низатидин ( Аксид)
Перорально: таблетки по 150,300 мг
Ранитидин (Зантак)
Перорально: таблетки по 150,300 мг;
сироп 15 мг/мл
Парентерально: 0.5, 25 мг/мл для инъекций
Агонисты 5-НТ
Суматриптан (Имитрекс)
Парентерально: 6 мг/0.5 мл (автоинъектор) для п/к инъекций
Антагонисты 5-НТ
Гранисетрон (Китрил)
Парентерально: 1 мг/мл для в/в введения
Ондансетрон (Зофран)
Перорально: таблетки по 4,8 мг
Парентерально: 2 мг/мл для в/в введения
Алкалоиды спорыньи
Дигидроэрготамин (D.H.E. 45)
Парентерально: 1 мг/мл для инъекций
Эргоновин (генерик, Эрготрата малеат) Перорально: таблетки по 0.2 мг Парентерально: 0.2 мг/мл для инъекций
Эрготамин (микстура)(генерик, Кафергот, др.) Перорально: таблетки по 1 мг эрготамина/100 мг кофеина Ректально: свечи 2 мг эрготамина/100 мг кофеина
Эрготамина тартрат (Эргомар, Эргостат, др.) Подъязычно: таблетки по 2 мг; дозированный аэрозоль 9 мг/мл (0.36 мг/дозу)
Метилэргоновин (Метэргин)
Перорально: таблетки по 0.2 мг
Парентерально: 0.2 мг/мл для инъекций
Метисергид (Сансерт)
Перорально: таблетки по 2 мг
Избранная литература
Гистамин
Black J. W. et al. Definition and antagonism of histamine H2 receptors. Nature, 1972; 236: 385.
Feldman M., Burton M. E. Drug Therapy: Histamine2 receptor antagonists: Standart therapy for acidpeptic disease. (Two parts.) N. Engl. I. Med. 1990; 323:1672, 1749.
Haaksma E. E. J., Leurs R., Timmerman H. Histamine receptors: Subclasses and specific ligands. Pharmacol. Ther. 1990; 47: 73.
Lin J. H. Pharmacokinetic and pharmacodynamic properties of histamine H2-receptor antagonists. Relationship between intrinsic potency and
effective plasma concentrations. Clin. Pharma-cokinet. 1991; 20: 218.
Schwartz J.-C. et al. Histaminergic transmission in the mammalian brain. Phisiol. Rev. 1991; 71:1.
Torsoli A., Lucchelli P. E., Brimblecombe R. W. (ed.) H2 Antagonists: H2 Receptor Antagonists in Peptic Ulcer Disease and Progress in Histamine Research. (Symposium.) International Congress Series 521. Exerpta Medica, 1980.
Серотонин
Berge B. Ergot compounds: A synopsis. Adv. Biochem. Psychopharmacol. 1980; 23: 3.
Bockaert J. et al. The 5-HTi receptor: A place in the sun. Trends. Pharmacol. Sci. 1992; 13: 141.
Derkach V., Surprenant A., North R. A. 5-HT3 receptors are membrane ion channels. Nature, 1989; 339: 706.
Glennon R. A. Serotonin receptors: Clinical implications. Neurosci. Biobehav. Rev. 1990; 14: 35.
Humphrey P. P. et al. Serotonin and migraine. Ann. N. Y. Acad. Sci. 1990; 600: 587.
Julius D. Molecular biology of serotonin receptors. Annu. Rev. Neurosci. 1991; 14:335.
Moskowitz M. A. et al. Pain mechanisms underlying vasular headaches. Rev. Neurol. 1989; 145:181.
Muller-Schweinitzer E., Weidman H. Basic pharmacological properties. In: Ergot Alcaloids and Related Compounds — Vol. 49 of Handbook of Experimental Pharmacology. Berde B., Schild H. O. (ed.) Springer-Verlag, 1978.
Olesen J., Sazena P. R. (ed.) 5-Hydroxytryptamine Mechanisms in Primary Headaches. Raven Press, 1992.
Peroutka S. J. 5-Hydroxytryptamine-receptor subtypes. Annu. Rev. Neurosci. 1988; 11:45.
Saxena P. R., Ferrari M. D. 5-HTrlike receptor agonists and the pathophysiology of migraine. Trends. Pharmacol. Sci. 1989; 10:200.
Vanhoutte P. M. (ed.) Serotonin and the Cardiovascular System. Raven Press, 1985.
Вазоактивные пептиды
Ян А. Рейд
В большинстве тканей пептиды являются средством сообщения между клетками. Как указывается в главах 6 и 20, они могут играть важную роль в функционировании автономной и центральной нервной систем. Некоторые из пептидов оказывают выраженное прямое действие на гладкие мышцы сосудов и иную гладкую мускулатуру. К ним относятся как вазоконстрикторы (ангиотензин II, вазопрессин, эндотелии и нейропептид Y), так и вазодилататоры (брадикинин и другие кинины, предсердный натрийуретический пептид, вазоактивный интестинальный пептид, субстанция Р, нейротензин и пептид, связанный с геном кальцитонина). В этой главе рассматривается влияние пептидов на гладкую мускулатуру.
Ангиотензин
Биосинтез ангиотензина
Пути образования и метаболизма ангиотензина II суммированы на рис. 17-1. Основные этапы включают ферментативное отщепление ангиотензина I от ангиотензиногена с помощью ренина, превращение ангиотензина I в ангиотензин II с помощью превращающего фермента и расщепление ангиотензина II несколькими пептидазами.
Ренин и факторы, контролирующие секрецию ренина
Ренин представляет собой аспартилпротеазу, которая специфически катализирует гидролитическое отщепление декапептида ангиотензина I от ангиотензиногена. Ренин синтезируется из препрогор-мона, который сначала превращается в неактивный проренин, азатем в активный конечный продукт — гликопротеид, состоящий из 340 аминокислот.
Большая часть ренина (возможно, и весь), находящегося в циркуляции, имеет почечное происхож
дение. Ферменты с рениноподобной активностью присутствуют в некоторых тканях вне почек, в том числе в кровеносных сосудах, матке, слюнных железах и коре надпочечников, но их физиологическая роль неизвестна. В почках ренин синтезируется и хранится в особом отделе нефрона, юкстагломерулярном аппарате, который состоит из афферентной и эфферентной артериол и плотного пятна (macula densa). Афферентная артериола и в меньшей степени эфферентная содержат специализированные гранулярные клетки, называемые юкстагломерулярными клетками, которые являются местом синтеза, хранения и высвобождения ренина. Плотное пятно — специализированный сегмент канальца, тесно связанный с сосудистым компонентом юкстагломерулярного аппарата. Юкстагломерулярный аппарат иннервируется адренергическими нейронами.
Скорость секреции ренина почками является основным фактором, определяющим активность ре-нин-ангиотензиновой системы. Секреция ренина контролируется рядом структур, в том числе сосудистыми рецепторами почек, плотным пятном, симпатической нервной системой и ангиотензином II.
А. Почечный сосудистый рецептор. Сосудистые рецепторы почек функционируют как рецепторы растяжения. Уменьшение растяжения приводит к увеличению высвобождения ренина, и наоборот. Рецепторы расположены в афферентной артериоле, но, возможно, юкстагломерулярные клетки сами по себе чувствительны к изменению растяжения.
Б. Плотное пятно. Macula densa содержит иной тип рецепторов, которые чувствительны к изменениям доставки натрия или хлорида в дистальный каналец. Уменьшение их доставки приводит к стимуляции секреции ренина, и наоборот. Установлен но, что эти реакции опосредуются люминальным механизмом сочетанного транспорта Na+, К+, 20', чувствительного к изменениям концентрации СГв просвете канальца (Lorenz et al., 1991).
1 2 3 4 5 6 7 8 9 10 11 12 13 14
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu-Val-Ile-His-Ans—R
Ангиотензиноген
Ренин
V
Asp-Arg-Val-Tyr-Ile-Hls-Pro-Phe-His-Leu
Ангиотензин I
Конвертирующий фермент
v
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe
Пептидные фрагменты
Рис. 17-1. Химия ренин-ангиотензиновой системы. Показана аминокислотная последовательность N-конца ан-гиотензиногена человека. R обозначает остаток молекулы белка
В. Симпатическая нервная система. В контроле секреции ренина важную роль играет симпатическая иннервация юкстагломерулярного аппарата. При увеличении активности нервного аппарата почек усиливается секреция ренина, тогда как денервация почек приводит к его подавлению. Норадреналин стимулирует секрецию ренина, прямо влияя на юкстагломерулярные клетки. Этот эффект опосредуется p-адренорецепторами и активацией аденилатциклазы с образованием цАМФ. У человека это Ргрецепторы. В некоторых ситуациях норадреналин вызывает непрямое усиление секреции ренина через а-рецепторы. Такая стимуляция связанна с сужением афферентной артериолы, последу-юпщй активацией сосудистого рецептора почек и снижением доставки хлорида натрия к плотному
пятну. В других случаях стимуляция а-адренорецепторов может подавлять секрецию ренина.
На скорость секреции ренина также оказывают влияние адреналин и норадреналин, находящиеся в циркуляции. Эти катехоламины могут действовать посредством тех же механизмов, что и норадреналин, который высвобождается из симпатических нервов почек. Однако есть данные о том, что секреторная реакция ренина на циркулирующие катехоламины опосредуется главным образом вне-почечными Р-рецепторами. Расположение этих рецепторов и способы их влияния на секрецию ренина пока не установлены.
Г. Ангиотензин. Ангиотензин II угнетает секрецию ренина. Этот механизм, связанный с прямым действием пептида на юкстагломерулярные клетки, является основой “короткой петли” отрицательной обратной связи, контролирующей секрецию ренина. Нарушение этого механизма антагонистами ренин-ангиотензиновой системы приводит к стимуляции секреции ренина.
Д. Фармакологические влияния на секрецию ренина. Высвобождение ренина изменяется при действии многих фармакологических агентов. Его усиливают вазодилататоры (гидралазин, миноксидил, нитропруссид), агонисты p-адренорецепторов (изопротеренол), антагонисты а-адренорецепторов и большинство диуретиков и анестетиков. Стимуляция объясняется вовлечением уже рассмотренных механизмов контроля. Вещества, подавляющие выделение ренина, рассматриваются в разделе, касающемся антагонистов ренин-ангиотензиновой системы.
Ангиотензиноген
Ангиотензиноген — пептид, находящийся в циркуляции, от которого ренин отщепляет ангиотензин I. Ангиотензиноген образуется в печени. У человека это гликопротеид с мол. м. примерно 57 000. На рис. 17-1 представлены 14 аминокислот с N-конца молекулы. Концентрация ангиотензиногена в крови человека меньше, чем Кт (концентрация, вызывающая 50 %-ную от максимума реакцию) для взаимодействия ренина и ангиотензиногена. Следовательно, это важный фактор, определяющий скорость образования ангиотензина.
Синтез ангиотензиногена повышается под влиянием кортикостероидов, эстрогенов, гормонов щитовидной железы и ангиотензина II. Концентра
ция ангиотензиногена в плазме увеличивается у больных с синдромом Кушинга и у пациентов, получающих лечение глюкокортикоидами. Она возрастает также при беременности и у женщин, принимающих эстрогенсодержащие оральные контрацептивы.
Ангиотензин I
Хотя ангиотензин I содержит пептидную последовательность, необходимую для проявления всех эффектов ренин-ангиотензиновой системы, он не является биологически активным. Для активации он должен быть превращен в ангиотензин II с помощью конвертирующего фермента (рис. 17-1). На ангиотензин I могут также оказывать действие аминопептидазы плазмы или тканей с образованием [des-Aspl] ангиотензина I, который в свою очередь превращается в [des-Aspl ]ангиотензин II (известный как ангиотензин III) под влиянием конвертирующего фермента.
Конвертирующий фермент (пептидилдипептидаза, PDP, кининаза II)
Конвертирующий (превращающий) фермент является дипептидилкарбоксипептидазой, которая катализирует отщепление дипептидов от С-конца некоторых пептидов. Наиболее важные его субстраты — ангиотензин I, который он превращает в ангиотензин II, и инактивируемый им брадикинин. Кроме того, он расщепляет энкефалины и субстанцию Р, но физиологическое значение этих реакций неясно. Ангиотензин II не гидролизуется конвертирующим ферментом из-за наличия пролинового остатка в предпоследнем положении. Этот фермент широко распространен в организме. В большинстве тканей он располагается на люминальной поверхности клеток эндотелия сосудов и таким образом тесно контактирует с циркулирующей кровью.
Действие ангиотензина II
Ангиотензин II оказывает существенное воздействие на ряд органов, включая гладкую мускулатуру сосудов, кору надпочечников, почки и мозг. Благодаря этим эффектам ренин-ангиотензиновая система играет ключевую роль в регуляции баланса жидкости и электролитов и поддержании АД. Ги
перактивность ренин-ангиотензиновой системы может привести к гипертензии и нарушениям водно-электролитного гомеостаза.
Влияние на артериальное давление
Ангиотензин II является очень активным прессорным веществом: при пересчете на молярные концентрации он примерно в 40 раз активнее, чем норадреналин. Прессорная реакция на ангиотензин II, введенный внутривенно, развивается быстро (через 10-15 с) и поддерживается при постоянной инфузии пептида. Основной компонент прессорной реакции на внутривенное введение ангиотензина II связан с прямым сокращением гладкой мускулатуры сосудов, особенно артериол. Кроме того, ангиотензин II повышает АД за счет влияния на мозг и автономную нервную систему. Прессорная реакция на ангиотензин может сопровождаться незначительной рефлекторной брадикардией или проходить без нее. Очевидно это связано с тем, что ангиотензин, влияя на мозг, настраивает барорефлекторный контроль частоты сердечных сокращений на более высокий уровень АД (Reid, 1992).
Ангиотензин II влияет и на периферическую нервную систему. Он стимулирует автономные ганглии, повышает высвобождение адреналина и норадреналина из мозгового вещества надпочечников и, что наиболее важно, облегчает симпатическую передачу за счет влияния на адренергические нервные окончания. Последний эффект связан как с увеличением высвобождения, так и с уменьшением обратного захвата норадреналина. Ангиотензин II оказывает также прямое положительное инотропное действие на сердце, хотя это имеет меньшее значение.
Влияние на кору надпочечников
Ангиотензин II прямо действует на клубочковую зону коры надпочечников, стимулируя биосинтез и секрецию альдостерона. В больших дозах он усиливает и секрецию глюкокортикоидов.
Влияние на почки
Ангиотензин II непосредственно влияет на почки и вызывает сужение почечных сосудов, повышает проксимальную канальцевую реабсорбцию натрия и, как уже упоминалось, подавляет секрецию ренина.
Влияние на центральную нервную систему
Помимо влияния на ЦНС, которое связано с регуляцией АД, ангиотензин II оказывает центральное действие, проявляющееся усилением жажды (дипсогенный эффект), и увеличивает секрецию вазопрессина и АКТГ. Физиологическое значение влияния ангиотензина II на потребление жидкости и секрецию гормона гипофиза неясно.
Влияние на клеточный рост
Ангиотензин II является митогеном в отношении мышечных клеток сосудов и сердца и может вносить вклад в развитие сердечно-сосудистой гипертрофии (Schelling, 1991). Ряд данных указывает на то, что ингибиторы ангиотензинпревращаю-щего фермента замедляют или предупреждают морфологические изменения после инфаркта миокарда, приводящие к застойной сердечной недостаточности (Konstam et al., 1992).
Рецепторы ангиотензина и механизм действия
Рецепторы ангиотензина II найдены во многих тканях, в том числе в гладкой мускулатуре сосудов, коре надпочечников, почках, матке и мозгу. Подобно рецепторам других полипетидных гормонов, рецепторы ангиотензина II расположены на плазматической мембране клеток-мишеней, что обеспечивает быстрое начало гормональной реакции.
Недавно были выделены два подтипа рецепторов ангиотензина II, названные ATi и АТ2 (Bumpus et al., 1991), которые отличаются по сродству к пептидному (CGP42112A) и непептидным (лозартан [DuP 753], PD 123177) антагонистам и чувствительности к веществам, восстанавливающим сульфгидрильные группы. АТгрецепторы имеют высокое сродство к лозартану и низкое к PD 123177 и CGP 42112 А, тогда как АТ2-рецепторы имеют высокий аффинитет к PD 123177 и CGP 42112А и низкий — к лозартану. Ангиотензин II и саралазин связываются в равной степени с обоими подтипами рецепторов. Связывание ангиотензина II с АТгрецепторами, но не с АТ2-рецепторами, уменьшается при действии таких восстановителей сульфгидрильных групп, как дитиотрейтол. Соотношение подтипов рецепторов варьирует в
разных тканях: ATi преобладают в гладкой мускулатуре сосудов.
Большинство известных эффектов ангиотензина II опосредуются АТ(-подтипом рецепторов. Он принадлежит к надсемейству рецепторов, сопряженных с G-белком. Связывание ангиотензина II с ATi в гладкой мускулатуре сосудов приводит к опосредованному фосфолипазой С образованию инозитолтрифосфата (ИТФ) и диацилглицерола (ДАГ). ИТФ мобилизует Са2+ из саркоплазматического ретикулума, а ДАГ активирует протеинкина-зу С. Эти два эффекта приводят к сокращению гладкой мускулатуры. В других тканях АТгрецепторы сопряжены с другими механизмами передачи сигналов, в том числе с ингибированием аденилатциклазы. Функция и пути передачи сигналов от АТ2-рецепторов пока не изучены.
Метаболизм ангиотензина II
Ангиотензин II быстро исчезает из циркуляции и имеет период полувыведения 15-60 с. Он метаболизируется во время прохождения через большинство сосудистых бассейнов (важным исключением являются легкие) различными пептидазами, которые объединяют под общим названием ангио-тензиназы. Большинство метаболитов ангиотензина II биологически неактивны, но начальный продукт действия аминопептидаз — [des-Aspl] ангиотензин II — сохраняет выраженную биологическую активность.
Антагонисты ренин-ангиотензиновой системы
В настоящее время существует много средств, которые блокируют образование или действие ангиотензина II. Эти вещества могут подавлять секрецию ренина, его ферментативную активность, превращение ангиотензина I в ангиотензин II или влиять на рецепторы ангиотензина II.
Вещества, блокирующие секрецию ренина
Некоторые вещества, влияющие на симпатическую нервную систему, подавляют секрецию ренина. Примерами могут служить клонидин, пропранолол и метилдопа. Клонидин подавляет секрецию
ренина за счет связанного с центральным действием угнетения нервной активности в почках. Он может оказывать и прямое интраренальное действие. Механизм, за счет которого метилдопа подавляет секрецию ренина, не установлен, но он может быть сходным с центральным действием клонидина. Пропранолол и другие Р-адреноблокирующие препараты действуют за счет блокады интра- и экстра-ренальных Р-рецепторов, участвующих в нервном контроле секреции ренина.
Ингибиторы ренина
Действие ренина может быть заблокировано пентапептидом пепстатином, который ингибирует также эффект других протеаз, таких как пепсин и катепсин D. Применение пепстатина in vivo ограничивается его малой растворимостью, но уже синтезирована более растворимая форма — N-ацетил-пепстатин. На основе синтеза аминокислотной последовательности, воспроизводящей область расщепления ангиотензиногена, созданы конкурентные ингибиторы ренина. Основная трудность их использования связана с низкой растворимостью. Однако один из этих пептидов (Pro-His-Pro-Phe-His-Phe-Phe- Val-Tyr-Lys) достаточно растворим и является эффективным ингибитором ренина in vivo. Недавно были созданы ингибиторы ренина, действующие при приеме внутрь. Некоторые из них активны, имеют высокую специфичность в отношении ренина и понижают АД у больных с гипертензией. Тем не менее необходимо дальнейшее повышение эффективности и биодоступности этих ингибиторов при приеме внутрь.
Ингибиторы конвертирующего фермента
Из яда южноамериканской гадюки Bothrops jararaca выделен нонапептидный ингибитор конвертирующего фермента. Высокую активность проявляет и синтетическая форма этого пептида теп-ротид. Однако он активен только при внутривенном введении. В настоящее время широко применяется целый класс ингибиторов ангиотензинпрев-ращающего фермента (АПФ), эффективных при приеме внутрь и действующих на его активный центр. Типичными представителями этого класса являются каптоприл и эналаприл (рис. 17-2). Синтезировано много новых активных ингибиторов АПФ (Salvetti, 1990). Следует заметить, что ингибиторы АПФ не только блокируют превращение ангиотензина I в ангиотензин II, но и ингибируют разрушение других веществ, включая брадикинин, субстанцию Р и энкефалины. Последний эффект участвует в антигипертензивном действии ингибиторов АПФ и объясняет такие побочные реакции как кашель и сосудистый отек. Широкие исследования доказали ценность ингибиторов АПФ при гипертензии и застойной сердечной недостаточности (главы 11 и 13). Не так давно установлено, что эти средства могут также оказывать защитное действие при диабетическом поражении сосудов почек и других заболеваниях ( Bjorck et al., 1992).
Антагонисты ангиотензина
Замена фенилаланина в положении 8 молекулы ангиотензина II алифатическими остатками (глицин, аланин, лейцин, изолейцин или треонин) приводит к образованию активных антагонистов дей
СООСН2СН3
Эналаприл
Рис. 17-2. Ингибиторы ангиотензинпревращающего фермента, активные при пероральном приеме. Эналаприл'* является пролекарством — этиловым эфиром, который гидролизуется в организме ...
СИ, (
HS—СН2—СН—СО—N
Каптоприл
соон
ствия ангиотензина II. Замещение N-концевой аспарагиновой кислоты саркозином (N-метил-гли-цин) удлиняет период полусуществования пептидов и тем самым увеличивает их активность. Наиболее известен из антагонистов [Sar1, Vai5, А1а8]ан-гиотензин-(1-8)октапептид, или саралазин.
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe Ангиотензин II
Sar-Arg-Val-Tyr-Val-His-Pro-Ala Саралазин
Он проявляет некоторую агонистическую активность и может оказывать прессорное действие, особенно при низком уровне ангиотензина II в циркулирующей крови. Саралазин следует вводить внутривенно, что серьезно ограничивает его применение в качестве антигипертензивного средства. Однако он применяется для выявления ренинзависи-мой гипертензии и других гиперренинемических состояний. В целом аналоги ангиотензина менее эффективны по способности понижать АД, чем ингибиторы АПФ. Это различие отражает как парциальную агонистическую активность аналогов ангиотензина, так и потенцирование действия брадикинина ингибиторами АПФ.
Недавно был создан новый класс непептидных антагонистов ангиотензина II. Один из них — DuP 753 (лозартан), является активным и специфичным конкурентным антагонистом AT t-рецепторов ангиотензина (рис. 17-3). Лозартан активен при
приеме внутрь и не проявляет агонистической активности. Он понижает АД у больных с эссенциальной гипертензией и исследуется в качестве средства для терапии гипертензии и застойной сердечной недостаточности. Существуют и антагонисты АТ2-ре-цепторов, например PD 123177 (рис. 17-3), но они пока не применяются в клинике.
Кинины
Биосинтез кининов
Кинины — это группа активных сосудорасширяющих пептидов. Они образуются из белковых субстратов кининогенов под действием ферментов, известных как калликреины, или кининогеназы. С биохимической точки зрения, калликреин-кинино-вая система имеет ряд черт, общих с ренин-ангиотензиновой системой.
Калликреины
Калликреины присутствуют в плазме и некоторых тканях, включая почки, поджелудочную железу, кишечник, потовые и слюнные железы. Это сериновые протеазы, весьма похожие по строению активного центра и каталитическим свойствам на такие ферменты как трипсин, химотрипсин, эластаза, тромбин, плазмин и другие сериновые протеазы. Они являются гликопротеидами. Тканевые каллик-
PD 123177
Ярс. 17-3. Структуры антагониста ангиотензиновых АТ,-рецепторов DuP 753 (лозартан) и антагониста ангиотензиновых АТ2-рецепторов PD 123177
реины имеют мол. м: в диапазоне между 25 000 и 40 000, калликреины плазмы — примерно 100 000.
Плазменный калликреин циркулирует в крови в виде предшественника, прекалликреина, который образуется в печени. Калликреин плазмы может активироваться трипсином, фактором Хагемана и, возможно, самим калликреином. Некоторые калликреины железистой ткани существуют в форме прекалликреинов; другие же находятся в активной форме. В целом, гландулярные калликреины очень отличаются по биохимическим свойствам от плазменных. Было показано, что калликреины могут превращать проренин в активный ренин, но физиологическая роль этого превращения не установлена.
Кининогены
Предшественники кининов кининогены присутствуют в плазме, лимфе и интерстициальной жидкости. По меньшей мере две формы кининогена находятся в плазме: низкомолекулярная форма (LMW-кининоген) и высокомолекулярная форма (HMW-кининоген). Обе являются кислыми гликопротеидами, содержащими одну полипептидную цепь. Около 15-20 % общего количества кининогена плазмы находится в НМ W-форме. Полагают, что LMW-кининоген проходит через стенку капилляров и служит субстратом для тканевых калликреи-нов, тогда как HMW-кининоген содержится в кровотоке и служит субстратом калликреина плазмы.
Образование кининов в плазме и тканях
Путь образования и метаболизма кининов показан на рис. 17-4. У млекопитающих известны три кинина: брадикинин, лизил-брадикинин (известный также как каллидин) и метионил-лизил-бра-дикинин. Они имеют следующие структуры:
Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg
1 23456789
Брадикинин
Lys-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg Лизил-брадикинин (каллидин, Lys-брадикинин)
Met-Lys-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg
Метионил-лизил-брадикинин
(Met-Lys-брадикинин)
Рис. 17-4. Калликреин-кининовая система. Кининаза II идентична конвертирующему ферменту пептидилдипеп-тидазе
Отметим, что все они содержат брадикинин в своей структуре. Каждый кинин образуется из кининогена под действием своего фермента. Брадикинин высвобождается калликреином плазмы, лизил-брадикинин — гландулярным калликреином, а метионил-лизил-брадикинин — пепсином и пепсиноподобными ферментами. Предпочтительным субстратом для плазменного калликреина является HMW-кининоген, а для тканевого калликреина — LMW-кининоген. Часть лизил-брадикинина превращается в брадикинин под влиянием аминопептидазы. Все три кинина обнаруживаются в плазме, но преобладает брадикинин. Все они присутствуют и в моче. Лизил-брадикинин — основной кинин мочи. Возможно, он образуется под влиянием почечного калликреина. Почечная аминопептидаза превращает лизил-брадикинин в брадикинин. Метионил-лизил-брадикинин выявляется в подкисленной моче: кислая среда активирует уропепсиноген, который катализирует образование метионил-лизил-брадикинина из кининогенов мочи.
Действие кининов
Влияние на сердечно-сосудистую систему
Кинины вызывают выраженную вазодилатацию в ряде сосудистых областей, в том числе в сердце, почках, кишечнике, скелетной мускулатуре и печени. В этом отношении кинины примерно в 10 раз активнее, чем гистамин. Вазодилатация может быть
связана с прямым угнетающим действием кининов на гладкую мускулатуру артериол, может опосредоваться высвобождением эндотелиального расслабляющего фактора (endothelium-derived relaxing factor — EDRF), которым является оксид азота, или сосудорасширяющими простагландинами, такими как ПГЕ2и ПП2. В противоположность этому преобладающий эффект простагландинов на вены — суживающий, это может быть связано с прямой стимуляцией гладкой мускулатуры вен или высвобождением веноконстрикторных простагландинов, таких как ПГЕ2а. Кинины также вызывают сокращение большей части висцеральной гладкой мускулатуры.
При внутривенном введении кинины вызывают быстрое падение АД, связанное с их вазодилатирующим эффектом. Гипотензивная реакция на брадикинин очень кратковременна. Внутривенная инфузия пептида не приводит к стойкому понижению АД; длительную гипотензию можно вызвать, прогрессивно наращивая скорость инфузии. Быстрая обратимость гипотензивного действия кининов связана прежде всего с рефлекторным повышением частоты сердечных сокращений, сердечного выброса и сократительной функции миокарда. У некоторых видов животных брадикинин вызывает двухфазные изменения АД: начальную гипотензивную реакцию, за которой следует подъем выше исходного уровня. Увеличение АД, видимо, связано с рефлекторной активацией симпатической нервной системы, хотя следует отметить, что при некоторых условиях брадикинин может непосредственно высвобождать катехоламины из мозгового вещества надпочечников и стимулировать симпатические ганглии. Брадикинин повышает АД и при введении в ЦНС, но физиологическое значение этого эффекта неясно, так как маловероятно, что кинины проходят через гематоэнцефалический барьер. Кинины не оказывают отчетливого влияния на симпатические или парасимпатические нервные окончания.
Расширение артериол, вызванное кининами, приводит к увеличению давления и кровотока через капиллярное русло, тем самым, способствуя току жидкости из крови в ткани. Этот эффект может облегчаться повышением проницаемости капилляров, связанным с сокращением эндотелиальных клеток и расширением межклеточных промежутков, а также вторичным увеличением венозного давления в связи с веноконстрикцией. В результате этих изменений вода и растворенные в ней ве
щества выходят из крови во внеклеточную жидкость, увеличивается ток лимфы, может возникнуть отек.
Влияние на эндокринные и экзокринные железы
Как уже упоминалось, прекалликреины и кал-ликреины обнаружены в некоторых железистых тканях, в том числе в поджелудочной железе, почках, слюнных и потовых железах, и могут выделяться в секреторную жидкость этих органов. Функция, ферментов в этих тканях неизвестна. Ферменты (или активные кинины) могут попадать из органов в кровь и действовать как местные модуляторы кровотока. Так как кинины существенно влияют на гладкую мускулатуру, они могут модулировать тонус выводящих протоков слюнных и поджелудочной желез и участвовать в регуляции моторики желудочно-кишечного тракта. Оказывая действие на трансэпителиальный транспорт воды, электролитов, глюкозы и аминокислот, они регулируют транспорт этих веществ в желудочно-кишечном тракте и почках. Наконец, калликреины участвуют в физиологической активации различных прогормонов, в том числе проинсулина и проренина.
Роль в воспалении
Кинины играют важную роль в воспалительном процессе. Калликреины и кинины способны вызывать все симптомы воспаления. Выработка кининов повышается при воспалительном повреждении, вызванном различными способами.
Влияние на чувствительные нервы
Кинины являются активными веществами, вызывающими болевые ощущения при внутрикожном введении или при аппликации. Они вызывают боль, стимулируя ноцицептивные афференты кожи и внутренних органов.
Рецепторы кининов и механизмы действия
Биологические эффекты кининов опосредуются специфическими рецепторами, расположенными на мембранах тканей-мишеней. Определены два типа рецепторов кининов, которые называют Bt
и В2. (Заметим, что В обозначает брадикинин, а не Р-адренорецептор). Биологическая оценка В,-рецепторных систем показала, что Lys-брадикинин и Met-Lys-брадикинин примерно в 10 и 76 раз соответственно более активны в них, чем брадикинин. Октапептид [<1е8-Аг§9]брадикинин активнее в 10 раз, а гептапептид [<1е8-Р11е8-с1е8-А^9]брадикинин неактивен. К основным В2-рецепторным системам проявляет наибольшее сродство брадикинин, за ним следуют Lys-брадикинин и затем Met-Lys-бра-дикинин. Единственным исключением являются В2-рецепторы, которые участвуют в сокращении гладкой мускулатуры вен: считается, что они более чувствительны к Lys-брадикинину.
Большинство эффектов брадикинина и каллидина опосредуются В2-рецепторами, которые принадлежат к семейству рецепторов, сопряженных с G-белками. Связывание с рецепторами приводит к множественным эффектам передачи сигналов, в том числе мобилизации Са2+, транспорту СГ, образованию NO и активации фосфолипазы С, фосфолипазы А2 и аденилатциклазы.
Метаболизм кининов
Кинины быстро метаболизируются (период полувыведения < 15 с) под действием неспецифических экзо- или эндопептидаз, обычно обозначаемых как кининазы. Достаточно подробно изучены две кининазы плазмы. Кининаза I синтезируется в печени и является карбоксипептидазой, которая отщепляет аргининовый остаток с С-конца. Кининаза II присутствует в плазме и эндотелиальных клетках сосудов. Она идентична АПФ (пептидилдипеп-тидаза). Кининаза II инактивирует кинины путем отщепления С-концевого дипептида фенилаланил-аргинина. Подобно ангиотензину I брадикинин почти полностью гидролизуется во время прохождения через легочное сосудистое русло.
Вещества, влияющие на калликреин-кининовую систему
В настоящее время известны вещества, которые модифицируют активность калликреин-кинино-вой системы, хотя они и не нашли широкого клинического применения. Синтез кининов может быть подавлен ингибитором калликреина апроти-
нином. Обнаружены конкурентные ингибиторы В,-и В2-рецепторов. Примером антагониста В,-рецепторов является [Leu8-des-Arg9J6paflHKHHHH. В2-ре-цепторы блокируются [Thi ,Б-РЬе7]брадики-нином, который в некоторой степени блокирует и Bi-рецепторы. Недавно созданный антагонист В2-рецепторов DArg[Hyp3,Thi5, ОТ1с7,О1с8]бради-кинин (Ное 140) специфично и с высокой активностью ингибирует широкий спектр реакций на брадикинин. Действие кининов, связанное с генерацией простагландинов, может быть неспецифически заблокировано ингибиторами синтеза простагландинов. Наконец, активность кининов можно усилить ингибиторами АПФ, которые блокируют разрушение этих пептидов. Однако сложно определить, связаны ли эффекты ингибиторов АПФ с накоплением кининов или с угнетением синтеза ангиотензина II.
Вазопрессин
Вазопрессин (антидиуретический гормон, АДГ) играет важную роль в долгосрочном контроле АД, который осуществляется благодаря его способности увеличивать реабсорбцию воды в почках. Этот и другие аспекты физиологии вазопрессина рассматриваются в главах 15 и 36.
Существуют свидетельства того, что вазопрессин участвует в краткосрочной регуляции АД за счет сосудосуживающего действия. Пептид повышает общее периферическое сопротивление при введении в дозах, меньших, чем те, которые необходимы для максимальной концентрации мочи. Эти дозы обычно не повышают АД, потому что вазопрессорной активности пептида эффективно противодействует рефлекторное снижение сердечного выброса. Если влияние этого рефлекса устраняется, например при шоке, чувствительность к прессорному действию вазопрессина значительно повышается. Она также возрастает у больных с идиопатической ортостатической гипотензией. Более высокие дозы вазопрессина повышают АД, даже когда барорецепторные рефлексы интактны. Эти дозы обычно выше, чем необходимые для максимальной концентрации мочи, однако прессорная реакция может быть получена, когда концентрация вазопрессина в плазме возрастает до уровня, который достигается во время кровотечения без развития гипотензии.
Рецепторы вазопрессина и антагонисты
Действие вазопрессина опосредуется активацией специфических мембранных рецепторов. Выделяют два подтипа рецепторов вазопрессина — Угрецепторы, ответственные за сосудосуживающее действие пептида, и У2-рецепторы, через которые осуществляется антидиуретическое действие. Vi-эффекты сопряжены с активацией фосфолипазы С, образованием инозитолтрифосфата и повышением внутриклеточной концентрации Са2+. У2-эффекты опосредуются активацией аденилатциклазы.
Синтезированы пептиды, подобные вазопрессину, которые селективны в отношении вазоконстрикторного или антидиуретического действия. Наиболее избирательным современным синтетическим Угсосудосуживающим агонистом является [Phe2, Не3, Огп8]вазотоцин. Селективные У2-антидиурети-ческие аналоги представлены /-деамино [D-Arg8]aprHHHH-Ba3onpeccHHOM (dDVAP) и /-деамино[Уа1\ О-А^8]аргинин-вазопрессином (dVDAVP]. Имеются и специфические антагонисты вазоконстрикторного действия вазопрессина. Одним из наиболее активных V(-антагонистов является [1-(Р-меркапто-0,р-циклопентаметиленпропионовая кислота)-2-(О-метил)тирозин]аргинин-вазопрессин. Это соединение обладает также анти-окситоциновым эффектом, но не ингибирует антидиуретическое действие вазопрессина. Оно не взаимодействует с другими прессорными веществами типа ангиотензина и норадреналина. Недавно получено сообщение об активном конкурентном антагонисте Vi-рецепторов, эффективном при приеме внутрь (Yamamura et al., 1991). Известны и антагонисты антидиуретического действия вазопрессина.
Антагонисты вазопрессорного действия особенно полезны для изучения роли эндогенного вазопрессина в регуляции функции сердечно-сосудистой системы. Антагонисты не оказывают влияния на сердечно-сосудистую систему, когда их вводят животным с уровнем вазопрессина в крови в пределах нормы или ниже. Однако при их введении крысам или собакам в условиях обезвоживания, когда уровень вазопрессина в крови повышен, происходит быстрое снижение АД или общего периферического сопротивления. Блокада вазопрессина понижает также АД у животных с недостаточнос
тью надпочечников и нарушает регуляцию АД при кровотечении. Таким образом, наблюдения показывают, что, благодаря своему вазоконстрикторному действию, вазопрессин оказывает существенное регуляторное влияние на сердечно-сосудистую систему.
Имеются доказательства того, что вазопрессин может быть использован в создании экспериментальной гипертензии у животных и задействован в патогенезе некоторых форм гипертензии у человека. Для исследования этой важной проблемы необходимо применение различных антагонистов вазопрессина.
Предсердный натрийуретический пептид
Предсердия животных содержат пептиды с выраженной натрийуретической активностью, которые обычно обозначают как предсердные натрийуретические пептиды (ANP), кардионатрины, атри-опептины или аурикулицы. Это производные С-концевого участка общего предшественника — npenpoANP, который представляет собой пептид, состоящий из 151 аминокислоты. ANP синтезируется в основном в клетках предсердий, но в небольших количествах и в клетках желудочков. Он также образуется нейронами центральной и периферической нервной систем и в легких. ANP циркулирует в форме пептида из 28 аминокислот, который из 17 остатков формирует кольцо и имеет один дисульфидный мостик. Идентифицированы и другие пептиды, тесно связанные с ANP. К ним относят BNP, который подобно ANP синтезируется главным образом в сердце; CNP, который образуется преимущественно в мозгу. Структуры трех пептидов сходны (рис. 17-5), но существуют отличия в их биологических эффектах.
У здоровых людей концентрация ANP, которую измеряют с помощью радиоиммунных методов, колеблется от 15 до 60 пг/мл. Ряд факторов повышает высвобождение ANP из сердца, но наиболее важным из них является растяжение предсердий. Каким образом растяжение увеличивает выделение ANP — неизвестно, но в этом не участвует нервная система сердца. Кроме того, высвобождение ANP возрастает при увеличении объема жидкости, изменении положения тела из вертикального в горизон
тальное и при физической нагрузке. В каждом случае активация высвобождения ANP связана, вероятно, с растяжением предсердий, но клеточные механизмы этого процесса не установлены. Выработка ANP также усиливается под влиянием глюкокортикоидов, эндотелина, вазопрессина, а-адреноре-цепторов. Наконец, концентрация ANP в плазме повышается при различных патологических состояниях, в том числе при застойной сердечной недостаточности, первичном альдостеронизме, хронической почечной недостаточности и синдроме не-сооответствующей секреции АДГ.
Введение ANP вызывает быстрое и заметное увеличение экскреции натрия и количества мочи. Возрастает скорость клубочковой фильтрации при незначительном изменении почечного кровотока, так что фракция фильтрации увеличивается. Вызванный ANP натрийурез, очевидно, связан как с увеличением клубочковой фильтрации, так и со сниже
нием проксимальной канальцевой реабсорбции. ANP подавляет также секрецию ренина, альдостерона и вазопрессина, и эти изменения способствуют повышению экскреции натрия и воды. Наконец, ANP снижает АД. Гипотензивное действие связано с вазодилатацией, которая обусловлена активированием гуанилатциклазы, повышением уровня цГМФ и снижением концентрации свободного Са2+ в цитозоле. Свой вклад в гипотензивный эффект пептида вносит его способность ингибировать сосудосуживающее действие ангиотензина II и других вазоконстрикторов. Определены три типа рецепторов ANP: ANPa, ANPb и ANPc. Рецепторы ANPa и ANPn связаны с гуанилатциклазой. ANPc-рецепторы участвуют в клиренсе ANP.
ANP разрушается почти во всех тканях и имеет короткий период полувыведения из крови (3-5 минут). Значительная часть клиренса приходится на эндоцитоз, опосредованный ANPc-рецепторами, ко-
Рис. 17-5. Структура предсердных натрийуретических пептидов ANP, BNP и CNP. Последовательности, общие для пептидов, указаны черным цветом
торые широко распределены в тканях организма. ANP разрушается и нейтральной эндопептидазой NEP 24.11, которая метаболизирует несколько вазоактивных пептидов, а также другими ферментами.
Существуют свидетельства того, что ANP участвует в физиологической регуляции экскреции натрия. Так, натрийуретическая реакция на увеличение объема жидкости подавляется, по крайней мере у некоторых видов, при введении антител к ANP или при частичном удалении источника циркулирующего ANP путем резекции ушка предсердия. Тем не менее необходимы дополнительные исследования для того, чтобы полностью прояснить физиологическое значение этого пептида. Благодаря способности уменьшать объем крови и АД, ANP и ингибиторы метаболизирующей его NEP 24.11 исследуются как средства терапии застойной сердечной недостаточности, гипертензии, почечной недостаточности и состояний, сопровождающихся перегрузкой жидкостью.
Эндотелии
Эндотелий является источником ряда веществ с сосудорасширяющей и сосудосуживающей активностью. Один из них — вазоконстрикторный пептид, названный эндотелином, который был впервые
изолирован из культуры эндотелиальных клеток аорты.
Этот пептид состоит из 21 аминокислоты с двумя дисульфидными мостиками. Идентифицированы три изоформы: первоначально выделенный эндотелии (ЕТ-1) и два сходных пептида ЕТ-2 и ЕТ-3. Их структуры показаны на рис. 17-6. Эндотелии широко распределен в организме с наибольшими концентрациями в легких, почках, сердце, гипоталамусе и селезенке. Он присутствует в крови, но в низких концентрациях и, возможно, оказывает местное действие преимущественно по паракринному или аутокринному типу действия, а не как циркулирующий гормон.
Эндотелии вызывает дозозависимое суживающее действие в отношении большинства сосудистых зон. Он в 100 раз более активен, чем норадреналин в отношении мезентериальной артерии человека. Внутривенное введение пептида вызывает временное снижение АД с последующим длительным его повышением. Кроме того, он вызывает сокращение несосудистой гладкой мускулатуры, в том числе трахеи, матки и кишечника. Другие эффекты эндотелина включают положительное инотропное действие, стимуляцию выделения предт сердного натрийуретического гормона и пролиферацию гладкомышечных и других клеток.
Эндотелин-1 (свинья/человек/ крыса/собака)
Lys
Рис. 17-6. Структуры эндотелиновых пептидов эндотелина-1, эндотелина-2 и эндотелина-3. Последовательности, отличающие три пептида, показаны черным цветом
РХIleyHeyTrp) С
Эндотелин-2 (человек)
GluICysYValYTyrTPhdCysYHis
CysjHIs
Lys
Ser
CysySerYCys) N
ру Не у Не XTrp) С
Эндотелин-3 (человек/крыса)
LPPH<ValYTyr
Су
Cys N
Lys
GluJCysifValYTyr
pIHeXHeXTrp) С
Рецепторы эндотелина есть во многих тканях и органах, в том числе в крови, сосудистой стенке, сердечной мышце, ЦНС, легких, почках, надпочечнике, селезенке и кишечнике. Идентифицированы два подтипа рецепторов, которые обозначают ЕТЛ и ЕТВ. ЕТл-рецепторы имеют высокое сродство к ЕТ-1 и низкий аффинитет к ЕТ-3 и расположены в клетках гладкой мускулатуры, где они опосредуют вазоконстрикцию. ЕТв-рецепторы имеют примерно одинаковое сродство к ЕТ-1 и ЕТ-3 и расположены на эндотелиальных клетках сосудов, участвуя в высвобождении EDRF и простаноидов. Оба подтипа рецепторов принадлежат к семейству рецепторов, сопряженных с G-белком. Связываниеэндо-телина с рецепторами приводит к повышению внутриклеточной концентрации ионов кальция, что является следствием входа Са2+ в клетку и стимуляции фосфолипазы С, образования инозитолтрифосфата и выделения Са2+ из эндоплазматического ретикулума.
Эндотелии может играть важную роль в регуляции функций сердечно-сосудистой системы, вносить вклад в патогенез гипертензии и других сердечно-сосудистых заболеваний, но его конкретная роль еще должна быть изучена. Недавно были открыты антагонисты эндотелина, которые имеют перспективы как средства для терапии гипертензии и сосудистых заболеваний.
Вазоактивный интестинальный пептид
Вазоактивный интестинальный пептид (vasoactive interstinal peptide — VIP) содержит 28 аминокислот и сходен по строению с секретином и глюкагоном. Он имеет следующую структуру:
His-Ser-Asp-Ala-Val-Phe-Thr-Asp-Asn-Tyr-Thr-Arg-Leu-Arg-Lys-GIn-Met-Ala-Val-Lys-Lys-Tyr-
Leu-Asn-Ser-Ile-Leu-Asn-NH2
Вазоактивный интестинальный пептид (VIP)
Первоначально VIP был выделен из легких свиньи, а очищенный образец получен из двенадцатиперстной кишки этого вида животных. В настоящее время известно, что VIP широко распределен в ЦНС и в периферической нервной системе человека и нескольких видов животных. Он находится также в циркулирующей крови, но большинство
данных указывает на то, что VIР функционирует скорее как нейротрансмиттер или нейромодулятор, чем как классический гормон.
VIP вызывает выраженную вазодилатацию в большинстве сосудистых областей, в том числе кишечных, легочных, коронарных, почечных и мозговых сосудов. Сосудорасширяющее действие VIP не зависит от влияния на рецепторы норадреналина, ацетилхолина, серотонина и гистамина и, вероятно, связано с прямым влиянием пептида на гладкую мускулатуру сосудов. В результате этого действия АД уменьшается и повышается сердечный выброс. Механизм сосудорасширяющего действия VIP полностью не выяснен. Однако полагают, что он связывается со специфическими рецепторами кровеносных сосудов и стимулирует выработку цАМФ. Этот эффект присущ пептиду, который выделяют из неадренергических, нехолинергических VIP-содержащих нервных волокон, связанных с кровеносными сосудами, или пептиду, содержащемуся в циркулирующей крови.
Кроме расширения сосудов, VIP расслабляет гладкую мускулатуру трахеи и бронхов, желудочно-кишечного тракта, стимулирует секрецию воды и электролитов в кишечнике, способствует гликогенолизу в печени, вызывает возбуждение нейронов и стимулирует выделение нескольких гормонов, в том числе гормона роста, пролактина и ренина. Физиологическое значение этих эффектов еще только изучается.
Субстанция Р
Субстанция Р является ундекапептидом семейства тахикининов. Она имеет следующее строение:
Arg-Pro-Lys-Pro-GIn-GIn-Phe-Phe-Gly-Leu-Met
Субстанция Р
Субстанция Р присутствует в ЦНС в качестве нейротрансмиттера (глава 20) и в желудочно-кишечном тракте, где может играть роль медиатора нервной системы кишечника и местного гормона (глава 6).
Субстанция Р является активным вазодилататором и оказывает заметное гипотензивное действие у человека и некоторых видов животных. Вазодилатация, очевидно, связана с прямым угнетающим действием пептида на гладкую мускулатуру
артериол. Это действие опосредуется специфическими рецепторами, которые отличаются от рецепторов для других вазодилататоров. В отличие от расслабляющего влияния на гладкую мускулатуру артериол субстанция Р стимулирует сокращение гладкой мускулатуры вен, кишечника и бронхов. Она вызывает также секрецию слюнных желез, диурез и натрийурез в почках и ряд эффектов в центральной и периферической нервной системах. Эти разнообразные эффекты могут быть связаны с активацией более чем одного типа рецепторов.
N-концевая область субстанции Р не играет существенной роли в проявлении ее активности. Некоторые фрагменты карбоксильного терминального участка пептида, в том числе окта(4-11)-, гепта(5-11)- и гекса(6-11)-фрагменты могут быть столь же или даже более активны, чем интактный ундекапептид. Синтезированы аналоги субстанции Р, которые являются антагонистами in vivo и in vitro.
Нейротензин
Нейротензин является тридекапептидом, который был впервые изолирован из ЦНС, а в последующем обнаружен в желудочно-кишечном тракте и в крови. Он имеет следующую структуру:
Glu-Leu-Tyr-Glu-Asn-Lys-Pro-Arg-Arg-Pro-Tyr-Ile-Leu
Нейротензин
При введении в периферический кровоток нейротензин вызывает ряд эффектов, в том числе расширение и повышение проницаемости сосудов, гипотензию, увеличение секреции некоторых гормонов передней доли гипофиза, гипергликемию, угнетение секреции соляной кислоты, гастрина и моторики желудка. После введения в спинномозговую жидкость он вызывает гипотермию и аналгезию. Изучение связи структуры и активности пептида показало, что для проявления его биологической активности необходимо наличие пяти или шести С-концевых аминокислотных остатков. В настоящее время физиологическое значение эффектов нейротензина неизвестно.
Пептид, связанный с геном кальцитонина
(кокальцигенин)
Пептид, связанный с геном кальцитонина (calcitonin gene-related peptide — CGRP, кокальци-генин), является одним из трех продуктов гена кальцитонина. Кокальцигенин состоит из 37 аминокислот и примерно на 30 % гомологичен кальцитонину лосося.
Как и кальцитонин, кокальцигенин представлен в больших количествах в С-клетках щитовидной железы. Он широко распределен в центральной и периферической нервной системах, сердечно-сосудистой системе, желудочно-кишечном тракте и мочеполовой системе. В некоторых из этих областей кокальцигенин обнаруживается вместе с субстанцией Р, в других — с ацетилхолином.
Когда кокальцигенин вводят в ЦНС, он вызывает ряд эффектов, в том числе подавление пищевого поведения и гипертензию. При введении пептида в системную циркуляцию развивается гипотензия и тахикардия. Гипотензивное действие ко-кальцигенина связано с его выраженным вазодилатирующим влиянием (кокальцигенин — наиболее активный из всех до сих пор открытых вазодилататоров).
НейропептидУ
Нейропептид Y, впервые изолированный из свиного мозга, состоит из 36 аминокислот. Его структура тесно связана со структурой панкреатического полипептида. Нейропептид Y обнаруживается в высоких концентрациях в мозгу и в периферической нервной системе и является одним из наиболее распространенных нейропептидов. В симпатической нервной системе он часто локализован в норадренергических нейронах и, видимо, функционирует как вазоконстриктор и котрансмиттер норадреналина.
Нейропептид Y оказывает ряд эффектов на ЦНС, в том числе повышает аппетит, вызывает гипотензию, гипотермию и угнетение дыхания. К периферическим эффектам относятся сужение мозговых сосудов, положительное хроно- и инотропное действие на сердце и гипертензия. Эти эффек
ты связаны с пресинаптическим влиянием, угнетением выделения трансмиттера из окончаний симпатических и парасимпатических нервов, а также с постсинаптическим действием, проявляющимся прямой вазоконстрикцией, потенцированием действия вазоконстрикторов и угнетением действия вазодилататоров. Они, очевидно, опосредованы множественными подтипами рецепторов, в том числе постсинаптическими Y,-рецепторами и преси-наптическими У2-рецепторами. Антагонистом вазоконстрикторного эффекта нейропептида Y является производное инозитола D-миоинозитол-1,2,6-трифосфат.
Препараты
Ингибиторы ангиотензинпревращающего фермента: см. главу 11
Вазопрессин: см. главу 15.
Избранная литература
Bathon J. М., Proud D. Bradykinin antagonists. Ann. Rev. Pharmacol. Toxicol. 1991; 31:129.
Bernstein К. E., Alexander R. W. Counterpoint: molecular analysis of the angiotensin II receptor. Endocr. Rev. 1992; 13:381.
Bhoola K. D„ Figueroa C. D., Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992; 44:1.
Bumpus F. M. et al. Nomenclature for angiotensin receptors. Hypertension, 1991; 17: 720.
Cantin M., Genest J. The heart and the atrial natriuretic factor. Endocr. Rev. 1985; 6:107.
Corvol P. et al. Human renin inhibitor peptides. Hypertension, 1990; 16:1.
Dray A., Perkins M. Bradykinin and inflammatory pain. Trends. Neurosci. 1993; 123:61.
Goetz K. L. Physiology and pathophysiology of atrial peptides. Am. J. Physiol. 1988; 254: El.
Greenlee W. J. Renin inhibitors. Med. Res. Rev. 1990; 10: 173.
Griendling К. K., Murphy T. J., Alexander R. W. Molecular biology of the renin-angiotensin system. Circulation, 1993; 87: 1816.
Hedner T. et al. Peptides as targets for antihypertensive drug development. J. Hypertens. 1992; 10: S121.
Iverson L. L. Nonopioid neuropeptides in mammalian CNS. Annu. Rev. Pharmacol. Toxicol. 1983; 23:1.
Kleinert H. D., Baker W. R., Stein H. H. Renin inhibitors. Adv. Pharmacol. 1991; 22: 207.
Laszlo F. A., Laszlo F., De Wied D. Pharmacology and clinical perspectives of vasopressin antagonists. Pharmacol. Rev. 1991; 43: 73.
Peach M. J. Renin-angiotensin system: Biochemistry and mechanisms of action. Physiol. Rev. 1977; 57: 313.
Reid I. A., Morris B. J., Ganong W. F. The reninangiotensin system. Annu. Rev. Physiol. 1978; 40: 377.
Schachter M. Kallikreins (kininogenases): A group of serine proteases with bioregulatory actions. Pharmacol. Rev. 1980; 31:1.
Soffer R. L. (ed.) Biochemical Regulation of Blood Pressure. Wiley, 1981.
Timmermans P. В. M. W. M. et al. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol. Rev. 1993; 45: 205.
Эйкозаноиды: простагландины, тромбоксаны, лейкотриены 1 8 и родственные им соединения
Маркус Хеккер, Мари Л. Фог, ПетерУ. Рамвелл
Простагландины (ПГ) и родственные им производные арахидоновой кислоты являются одними из наиболее сильнодействующих естественных аута-коидов, и в настоящее время их рассматривают как важнейшие регуляторы клеточных процессов. Это подтверждается обилием литературы по эйкозаноидам, множеством клинико-фармакологических испытаний и растущим количеством соединений на фармацевтическом рынке. Несмотря на это, физиологическая роль эйкозаноидов полностью не определена.
Синтез эйкозаноидов
Синтез, реацетилирование и высвобождение арахидоновой кислоты
Простагландины и некоторые другие биологически активные липиды и пептидолипидные кислоты формируются из одного предшественника путем взаимосвязанных цепочек ферментативных реакций. Почти все они являются карбоновыми кислотами. Это тромбоксаны (ТХ), гидроперокси-эйкозатетраеновые кислоты (НРЕТЕ), гидрокси-эйкозатетраеновые кислоты (НЕТЕ), лейкотриены (ЛТ), недавно открытые липоксины (LX) и эпоксиэйкозатетраеновые кислоты (ЕЕТЕ). Термин эйкозаноиды (от греческого eicosa — двадцать) используется для названия этой группы соединений, так как все они могут быть получены из поли-ненасыщенных жирных кислот с С18-, С20- и С22-уг-леродными скелетами.
Среди этих жирных кислот арахидоновая кислота является наиболее важным предшественником биосинтеза эйкозаноидов у человека. Две 18-угле-
Сокращения, используемые в этой главе
АКТГ Адренокортикотропным гормон
АДГ Антидиуретический гормон
ЭФР Эндотелиальный фактор релаксации
ЕЕТЕ Эпоксиэйкозатетраеновая кислота
ФСГ Фолликулостимулирующий гормон
НЕТЕ Гидроксиэйкозатетраеновая кислота
ННТ Г идроксигептадекатриеновая кислота
НРЕТЕ Гидропероксиэйкозатетраено-вая кислота
ЛГ Лютеинизирующий гормон
РФЛГ Рилизинг-фактор лютеинизирующего гормона
ЛТ Лейкотриен
LX Липоксины
НПВС Нестероидные противовоспалительные средства
ФАТ Фактор активации тромбоцитов
ПГ Простагландины
ПМН Полиморфноядерный нейтрофил
ПТГ Паратиреоидный гормон
МРСА Медленнореагирующая субстанция анафилаксии
ТСГ Тиреостимулирующий гормон
ТХ Тромбоксан
родные жирные кислоты (линолевая и «-линоленовая кислоты) и 20-углеродная арахидоновая кислота относятся к незаменимым (эссенциальным) жирным кислотам, которые необходимы для полноценного питания многих видов животных и человека. У большинства млекопитающих арахидоновая кислота образуется из линолевой кислоты путем десатурации и удлинения цепи до дигомо-у-линолено-вой кислоты с последующей десатурацией. Однако линолевая и линоленовая кислоты не синтезируются у человека, то есть должны поступать с пищей. Источником полиненасыщенных эйкозапентаено-вой и докозагексаеновой кислот является жир рыб, обитающих в холодных водах. Информация об эссенциальных жирных кислотах лежит в основе многих систем питания человека.
Эйкозаноиды не депонируются в клетке, их синтез регулируется содержанием предшественников — свободных жирных кислот. Арахидоновая кислота высвобождается из липидов мембраны и из других жиров фосфолипазами, которые активируются специфическими и неспецифическими стимулами. В от
вет на эти стимулы, варьирующие в разных клетках, фосфолипаза А2 или комбинация фосфолипазы С и диглицеридлипазы отщепляют этерифицированную арахидоновую кислоту из положения 2 специфических глицерофосфолипидов, входящих в состав липидного бислоя клеточной мембраны (рис. 18-1).
Количество внутриклеточной свободной арахидоновой кислоты частично контролируется встраиванием арахидоната в липиды клетки. Это реацетилирование регулируется двумя ферментами: ара-хидонил-КоА-синтетазой, которая катализирует образование активной формы арахидоновой кислоты и арахидонил-КоА-трансферазой, присоединяющей арахидонил-КоА ко второму углеродному атому лизофосфолипида. При дальнейшей трансформации фосфолипидного пула может образовываться 1 -алкил-2-ацетил-лизофосфатидилхолин, или фактор активации тромбоцитов (ФАТ) — сильный патофизиологический медиатор астмы и шока (рис. 18-2). Ингибирование любого из этих ферментов ведет к повышению внутриклеточного уровня свободной арахидоновой кислоты.
Арахидоновая кислота (свободная)
ЛТ ТХ
LX
Рис. 18-1. Пути выделения и метаболизма арахидоновой кислоты
О
Рис. 18-2. Реацилирование арахидоновой кислоты в клеточные фосфолипиды (КоА — коэнзим A, GPC — глицеро-фосфатидилхолин, ФАТ — фактор активации тромбоцитов)
Свободная арахидоновая кислота метаболизируется в основном двумя ферментативными путями, в неодинаковой степени выраженными в различных клетках: простагландины и тромбоксаны образуются циклооксигеназным путем, а ЛТ и НРЕТЕ — ли-пооксигеназным. Не в каждой клетке образуются все эти продукты. Например, в сосудистом эндотелии синтезируются прежде всего ПГЕ2 и ПП2, а в тромбоцитах — тромбоксан и 12-НЕТЕ.
Простагландины:
история и номенклатура
Гольдблат и фон Эйлер независимо друг от друга нашли в семенной жидкости человека вазоактивную жирную кислоту, которая снижала артериальное давление у кролика. Фон Эйлер назвал это активное вещество простагландином, так как считал, что оно синтезируется простатой (сейчас известно, что это вещество образуется в семенных пузырьках). Он предположил, что простагландин играет роль в транспорте спермы. В 1962 г. Бергстрём определил структуру первых двух простагландинов и назвал их простагландины Е и F (ПГЕ и ПГЕ) соот
ветственно их способности растворяться в эфире или фосфатном буфере.
Сегодня номенклатура простагландинов включает 10 специфических молекул, обозначаемых буквами от А до J, которые различаются радикалами, присоединенными в положении 9 и 11 циклопентанового кольца. Для биосинтеза простагландинов могут использоваться арахидоновая, дигомо-у-линоленовая и эйкозапентаеновая кислоты. Поэтому каждая группа ПГ состоит из трех серий под номерами 1, 2 и 3. Эти номера определяют степень насыщения (сатурации) алифатической боковой цепи. Свое собственное название имеет только простациклин (ПП2). Для ПГЕ дополнительное обозначение а или Р характеризует пространственную конфигурацию гидроксильной группы при С9.
Биосинтез простагландинов
Первые две реакции биосинтеза простагландинов из полиеновых жирных кислот катализируются про-стагландинэндопероксидсинтетазой. Этот связанный с мембраной гемопротеид, который имеется у большинства животных, обнаружен во всех клетках
организма (кроме эритроцитов и лимфоцитов). Синтез простагландинов из арахидоновой кислоты начинается с оксигенации и циклизации пентанового кольца (циклооксигеназный этап) и приводит к образованию нестабильного С15-гидроперокси-С9,С11-эндопероксида (ПГС2). Пероксидазная реакция, катализируемая тем же ферментом, восстанавливает затем С 15-гидроперокси-группу, образуя эндопероксид ПГН2 (рис. 18-3). Циклооксигеназный путь ингибируется нестероидными противовоспалительными средствами (НПВС), например аспирином и индометацином. Существуют две циклооксигеназы (Сох): Сох I и Сох II. Эти изоферменты гомологичны на 60 % и отличаются функционально. Так, аспирин и индометацин сильнее ингибируют Сох I, чем Сох II, а флурбипрофен лишь несколько сильнее ингибирует Сох I по сравнению с Сох II. Аспирин ацетилирует серин 529 (Сох I) и серин 516 (Сох II), предотвращая синтез простагландинов и ТХА2. Большие количества 15-НЕТЕ возникают из-за ацетилирования Сох II, но не Сох I. В организме доминирующей формой является Сох I.
Эндоперекиси являются промежуточными субстратами для гистоспецифического синтеза разных биологически активных простагландинов. В зависимости от ткани ПГН2 конвертируется в ПГС2 цитозольным ферментом простагландин D-синтета-зой, в то время как ПГЕ2 образуется под действием мембраносвязанной простагландин Е-синтетазы. Оба фермента используют как кофактор восстановленный глутатион. Не выделен фермент, восстанавливающий ПГН2 до ПГЕ2а (пунктирная линия на рис. 18-3). Однако ПГЕ2а может образовываться из ПГЕ при помощи двух других ферментов: простагландин Е-9-кеторедуктазы и 15-гидроксипроста-гландиндегидрогеназы. Присутствие этих двух ферментов (а не их регуляция) определяет, какие эн-доперекисные продукты доминируют в определенной ткани.
Биосинтез простациклина и тромбоксана
Агрегирующие тромбоциты высвобождают тромбоксан А2 (ТХА2), который образуется из того же предшественника (ПГН2), что и простагландины, но отличается тем, что содержит оксан-оксета-новое кольцо. Оксетановое кольцо ТХА2 спонтанно гидролизуется (t)/2=30 с) до биологически неактивного полуацетального тромбоксана В2 (ТХВ2).
ТХА2 в отличие от ТХВ2 вызывает сокращение гладкой мускулатуры и агрегацию тромбоцитов в низких наномолярных концентрациях. Образование ТХА2 катализируется цитохром Р-450-подобным гем-тиолатным белком, тромбоксансинтетазой, которая в основном находится в тромбоцитах и макрофагах. Кроме ТХА2, образуются эквимолярные количества гидроксигептадекатриеновой кислоты (ННТ) и малонового диальдегида, однако их биологические функции неизвестны.
В отличие от ТХА2 простациклин (ПП2), образующийся в основном в эндотелии сосудов и в гладкомышечных клетках, сильно ингибирует агрегацию тромбоцитов и расслабляет гладкую мускулатуру. Он синтезируется связанным с мембраной цитохром Р-450-подобным ферментом — проста-циклинсинтетазой. ПГ12 гидролизуется спонтанно (t)/2 около 3 минут) до биологически неактивного 6-кето-ПГЕ|о. Таким образом, ТХА2и ПГ12 образуются из одного предшественника двумя очень похожими, но гистоспецифическими ферментами. Эта особенность отражает их противоположные (даже антагонистические) биологические свойства. Оба этих соединения быстро гидролизуются до неактивных продуктов, содержание которых легко измерить радиоиммунным методом.
Метаболизм арахидоновой кислоты цитохромом Р-450
Кроме метаболических превращений по циклооксигеназному или липооксигеназному пути, арахидоновая кислота окисляется микросомальными цитохром Р-450-монооксигеназами. В экспериментальных условиях продукты этих реакций действуют на тонус сосудов, транспорт ионов, рост клеток, передачу сигнала, гормональные реакции, гемостаз и гемопоэз. Эта ферментативная система окисляет арахидоновую кислоту до 19-гидрокси- и 19-оксо-эйкозатетраеновых кислот ((0-1-окислением) и до 20-гидроксиэйкозатетраеновой и эйкозатетраен-1,20-диоевой кислот (co-окислением). Микросомальные цитохром Р-450-ферменты печени и почек образуют и другие метаболиты арахидоновой кислоты, в том числе ряд эпоксидов (5,6-оксидо-, 8,9-оксидо-, 11,12-оксидо- и 14,15-оксидоэйкозатетра-еновые кислоты), которые могут далее превращаться в диолы. 11,12-оксидо-продукты являются сильными вазодилататорами и вызывают неоваскуляризацию. Некоторые метаболиты арахидоновой кис-
Арахидоновая кислота
Циклооксигеназа*
ТХВ2
Рис. 18-3. Биосинтез простагландинов и тромбоксанов, Названия соединений указаны в рамках. Звездочки показывают, что циклооксигеназный и пероксидазный пути катализируются одним и тем же ферментом — простагландин-эндопероксидсинтетазой
лоты проявляют свою биологическую активность только после метаболических превращений в клетке. Кроме того, оба типа окисления могут осуществляться одновременно с образованием производных тригидро ксиарахидоновой кислоты (липокси-нов). Эти и другие близкие по структуре соединения исследуются в связи с их действием на гладкую мускулатуру сосудов и эпителий органов пищеварения и почек.
Липооксигеназы, гидропероксиэйкозатетраеновые кислоты и липоксины
Третий, основной путь оксигенации арахидоновой кислоты, осуществляется липооксигеназами, которые встраивают молекулу кислорода в поли-ненасыщенные жирные кислоты (рис. 18-4). Тканевая специфичность липооксигеназ определяется положением углерода, несущего гидропероксигруппу вещества. У человека арахидоновая кислота прежде всего превращается в 5-НРЕТЕ, 12-НРЕ-ТЕ и 15-НРЕТЕ. Эти нестабильные перекиси затем образуют соответствующие гидрокси-дериваты (НЕТЕ). Известны две формы 12-НЕТЕ (Л и Sконфигурации), которые находятся в тромбоцитах и лейкоцитах соответственно.
Ферменты 5-липооксигеназаи 12-липооксигена-за широко распространены в тканях и клетках крови млекопитающих и практически идентичны, за исключением двух аминокислот, определяющих положение субстрата в фермент-субстратном комплексе. Значительная активность 15-липооксигена-зы определяется только в эозинофилах. В отличие от циклооксигеназ липооксигеназы присутствуют и в растениях, например в картофеле и соевых бобах. Это свидетельствует о том, что липооксигеназ-ный путь оксигенации полиеновой жирной кислоты филогенетически более древний. И 5-, и 12-ли-пооксигеназы вносят свой вклад в образование различных тригидроксиэйкозатетраеновых кислот, в том числе липоксинов LXA и LXB (рис. 18-4).
Биосинтез лейкотриенов
Лейкотриены (ЛТ) являются биологически активными веществами, которые синтезируются 5-ли-пооксигеназой в нейтрофилах, моноцитах, макрофагах, тучных клетках и кератиноцитах, а также в легких, селезенке, головном мозгу и сердце
Рис. 18-4. Предполагаемый путь биосинтеза липоксинов. Названия соединений указаны в рамках. 15-НРЕТЕ и 15-НЕТЕ являются субстратами 5-липооксигеназы полиморфноядерных лейкоцитов, которая индуцирует синтез LXA или LXB путем окисления в положении С5 с последующим образованием эпоксида
(рис. 18-5). До того как стала известной их структура, их определяли в перфузатах легкого как мед-леннореагирующую субстанцию анафилаксии (МРСА), которая высвобождается при иммунологической реакции. МРСА является смесью пепти-долейкотриенов ЛТС4, JITD4 и ЛТЕл. Название “лейкотриен” отражает факт их открытия в лейкоцитах и конъюгированную триеновую структуру, определяющую характерный ультрафиолетовый спектр поглощения. Число в названии лейкотриена определяет количество двойных связей. Синтез лейкотриенов идет только 5-липооксигеназным путем, в котором промежуточный 5-НРЕТЕ восстанавливается дегидрогеназой до ЛТА4. В лейкоци-
5-НРЕТЕ
Дегидрогеназа*
ЛТА4
ЛТВ4
CHCONHCH2COOH
лтс4
Рис. 18-5. Биосинтез лейкотриенов. Звездочки показывают, что как липооксигеназные, так и дегидрогеназные реакции контролируются одним и тем же ферментом — 5-липооксигеназой (ГГТП, у-глутамилтранспептидаза)
nh2
— NHCOCH2CH2CHCOOH ггтп
тах человека один фермент отвечает и за синтез 5-НРЕТЕ, и за его дальнейшую трансформацию в ЛТА4. Этот продукт либо гидролизуется до ЛТВ4 или его изомеров, либо превращается в ЛТС4 посредством конъюгации с пептидом глутатионом при помощи глутатион-8-трансферазы. Отщепление глутаминовой кислоты приводит к образованию ЛТО4, а дальнейшее отщепление глицинового остатка дипептидазой — к образованию ЛТЕ4. 5-ли-пооксигеназа нейтрофилов человека стимулируется ионами кальция, которые могут переносить фер
мент из цитозоля к мембране. Этот механизм регулирует участие нейтрофилов в развитии воспаления. В настоящее время активно изучается регуляция циклооксигеназы и липооксигеназы цитокинами, факторами роста и онкогенами.
Метаболизм эйкозаноидов
Определение стабильных продуктов метаболизма эйкозаноидов в плазме представляет значительный интерес для исследователей. Количество этих
веществ можно измерить радиоиммунным методом или ферментной иммуносорбцией (ELISA).
Метаболизм простагландинов по 15-гидроксидегидрогеназному пути
Простагландины быстро метаболизируются в организме. Около 97 % внутривенной дозы ПГЕ2 исчезает из плазмы через 90 секунд. Ферменты деградации простагландинов широко распространены в организме и наиболее активны в легких, поч-
O
0-окисление
©-окисление
Рис. 18-6. Катаболизм простаноидов. Рисунок иллюстрирует ферментативное расщепление ПГЕ2. ПП)2, ПГР2а, 6-кето-ПГГ|„ и ННТ также могут подвергаться этому типу превращений, тогда как ТХВ2 превращается в 2,3-динорпроизводное путем 0-окисления
ках, селезенке, жировой ткани и кишечнике. Особенно интенсивно проходит инактивация циркулирующих простагландинов в сосудах легких (рис. 18-6). Гидроксильная группа при С15, которая необходима для проявления биологической активности всех простагландинов, окисляется до соответствующего кетона ферментом простагландин-15-гидроксидегидрогеназой (ПГДГ). 15-кето-соединение восстанавливается до 13,14-дигидро-деривата простагландин-13-редуктазой. Эти две первые реакции проходят за несколько минут, вторая же фаза катаболизма простагландинов (0- и со-окисление боковых цепей) более длительна. В данном случае образуются полярные дикарбоновые кислоты, которые выделяются с мочой в свободном виде. При метаболизации по другому пути под действием специфической оксидоредуктазы образуются соответствующие 13,14-дигидро-производные. Основные метаболиты ТХВ2 в плазме и моче — это 11-дегид-ро-ТХВ2 и 2,3-динор-ТХВ2 соответственно.
Катаболизм простагландинов цитохромом Р-450
Цитохром Р-450-зависимые монооксигеназы печени и почек катализируют со- и со-1-окисление некоторых простагландинов, в том числе ПГЕ2. Значимость этого метаболического пути в сравнении с дегидрогеназным пока не известна. В полиморфноядерных лейкоцитах человека ЛТВ4 далее подвергается 20-гидроксилированию до 20-гидрокси-ЛТВ4 и превращению при помощи цитохрома Р-450 в дикарбоновую кислоту (20-карбокси-ЛТВ4).
I. Базисная фармакология эйкозаноидов
Механизмы действия эйкозаноидов
Эйкозаноиды — это короткодействующие, высокоактивные местные медиаторы, которые вызывают огромное количество биологических эффектов путем связывания со специфическими поверхностными рецепторами клеток. Некоторые из этих рецепторов выделены и солюбилизированы. Все они связаны с G-белком (глава 2). Однако их фармакологическая специфичность определяется не особы
ми системами внутриклеточных эффекторов или вторичных посредников, а распределением и количеством рецепторов на разных типах клеток.
Например, ПП2 подавляет агрегацию тромбоцитов, связываясь с рецепторами, которые специфически активируют аденилатциклазу. Это приводит к повышению внутриклеточного уровня цАМФ, что в свою очередь активирует специфические проте-инкиназы. Киназы фосфорилируют белки внутриклеточных кальциевых насосов, что повышает концентрацию ионов кальция в клетке. ПГО2 также подавляет агрегацию тромбоцитов, активируя аденилатциклазу, но посредством взаимодействия с другими рецепторами. Связывание же ТХА2 со специфическими рецепторами тромбоцитов активирует метаболизм фосфатидилинозитола, что приводит к образованию инозитол-1,4,5-трифосфата (ИТФ) — другого вторичного посредника. ИТФ мобилизирует внутриклеточный кальций и вызывает агрегацию тромбоцитов. ЛТВ4 посредством вторичного образования ИТФ вызывает активацию, дегрануляцию полиморфноядерных лейкоцитов и образование супероксид-аниона.
Таким образом, сокращение гладкой мускулатуры под действием эйкозаноидов может опосредоваться высвобождением Са2+, а релаксация — образованием цАМФ. Действие эйкозаноидов на ряд систем организма, в том числе иммунную, можно объяснить таким же образом. Многие из сократительных эффектов эйкозаноидов, затрагивающих гладкую мускулатуру, можно предупредить снижением внеклеточной концентрации Са2+ или применением блокаторов кальциевых каналов.
Эффекты простагландинов и тромбоксанов
Основные эффекты простагландинов и тромбоксанов касаются функционирования гладкой мускулатуры и тромбоцитов. Другие важные мишени — это ЦНС, вегетативные постганглионарные нервные терминали, чувствительные нервные окончания, эндокринные органы и жировая ткань.
А. Гладкая мускулатура.
1. Сосуды. Гладкую мускулатуру артерий человека расслабляют ПГЕ2 и ПП2. Эти и другие простагландины вызывают вазодилатацию, активируя аденилатциклазу. Напротив, ТХА2 и ПГР2п являются вазоконстрикторами, особенно в отношении вен. Простациклин, который в основном синтезируется
в эндотелии, первоначально считался циркулирующим гормоном. Однако почти весь ПП2 немедленно разрушается. В микроциркуляции играет также важную роль другой продукт эндотелиального происхождения — ПГЕ2. Вместе с ПГЕ2 и ПП2из эндотелия высвобождается сильный эндотелиальный фактор релаксации (ЭФР) (он идентичен или очень похож на оксид азота). Действие ЭФР в гладкой мускулатуре сосудов (рис. 12-2) опосредуется растворимым ферментом гуанилатциклазой; этот эффект не блокируется НПВС. Следовательно, регуляция микроциркуляции зависит не только от синтеза простагландинов в эндотелии.
2. ЖКТ. Действие простагландинов и лейкотриенов на гладкую мускулатуру желудочно-кишечного тракта различно. Продольная мускулатура сокращается под действием ПГЕ2 и ПГЕ2а, круговая — при участии ПП2 и ПГР2а, а расслабляется под действием ПГЕ2. Введение ПГЕ2 или ПГЕ2а приводит к спастическим болям (раздел “Клиническая фармакология эйкозаноидов”).
3. Дыхательные пути. Гладкая мускулатура дыхательных путей расслабляется ПГЕЬ ПГЕ2 и ПП2, а сокращается под влиянием ТХА2 и ПГР2а.
Б. Тромбоциты. Как уже обсуждалось, агрегация тромбоцитов в значительной степени регулируется эйкозаноидами. ПГЕ( и особенно ПП2 значительно подавляют агрегацию, а ТХА2 усиливает.
В. Репродуктивные органы.
1. Женские репродуктивные органы. Действие простагландинов на матку имеет огромное клиническое значение. Оно рассматривается в разделе “Клиническая фармакология эйкозаноидов”.
2. Мужские репродуктивные органы. Роль простагландинов, обнаруживаемых в сперме, точно не определена. Основной их источник — это семенные пузырьки; предстательная железа и яички синтезируют только малые количества простагландинов. Сперма мужчин, способных к репродукции, содержит около 400 мкг/мл ПГЕ и ПГЕ и их 19-гидро-кси-метаболитов. Причем ПГЕ приблизительно в 20 раз больше, чем ПГЕ, хотя их соотношение варьирует у разных мужчин. Однако у одного человека это соотношение остается постоянным, пока не меняются возрастные характеристики спермы. Факторы, регулирующие количество простагландинов в сперме, неизвестны, но показано, что тестостерон усиливает их синтез. Тромбоксаны и лейкотриены в сперме не найдены. Мужчины со сравнительно низкой концентрацией простагландинов в семенной
жидкости страдают бесплодием. Большие пероральные дозы аспирина снижают содержание простагландинов в сперме.
Г. Центральная и периферическая нервная система.
1. Лихорадка. При введении в желудочки мозга ПГЕ1 и ПГЕ2 повышается температура тела. Пирогены высвобождают интерлейкин-1, который в свою очередь активирует синтез и высвобождение ПГЕ2. Этот процесс блокируется аспирином и другими антипиретиками.
2. Сон. У некоторых видов животных, в том числе у приматов, инфузия в желудочки мозга ПГВ2 вызывает естественный сон (по анализу ЭЭГ).
3. Нейротрансмиссия. ПГЕ подавляет высвобождение норадреналина из симпатических преси-наптических нервных окончаний. Тот факт, что НПВС усиливают высвобождение норадреналина, позволяет предположить, что простагландины играют физиологическую роль в нейротрансмиссии. Следовательно, вазоконстрикция после лечения ингибиторами циклооксигеназы может быть связана с повышенным выбросом норадреналина, а также с ингибированием эндотелиального синтеза вазодилататоров (ПГЕ2 и ПГ11).
Д. Нейроэндокринология. Исследования in vitro и in vivo показали, что некоторые эйкозаноиды вызывают высвобождение гормона роста, пролактина, тиреостимулирующего гормона (ТСГ), адренокортикотропного гормона (АКТГ), фолликулостимулирующего гормона (ФСГ) и лютеинизирующего гормона (ЛГ). Однако соответствующие фармакологические эффекты у пациентов, получающих препараты ПГЕ, не отмечены. ЛТС4 и ЛТЭ4 стимулируют секрецию ЛГ и рилизинг-фактора лютеинизирующего гормона (РФ Л Г).
Эффекты метаболитов липооксигеназного
и цитохром Р-450-путей
Относительно недавно установлено, что 12-НРЕТЕ действует как нейромедиатор в нейронах Aplysia. Кроме того, есть данные, что ЛТС4 в очень высоких разведениях специфически усиливает высвобождение ЛГ и РФЛГ из изолированных клеток передней части гипофиза крыс. Следовательно, продукты липооксигеназного пути могут выполнять нейротрансмиттерную функцию. Эпоксиды арахидоновой кислоты также повышают вы
деление ЛГ из клеток гипофиза. Однако они значительно менее активны, чем ЛТС4.
Биологические функции разных форм гидрокси-и гидропероксиэйкозасновых кислот неизвестны, но их фармакологическая активность очень существенна. Например, 12(5)-НЕТЕ стимулирует миграцию гладкомышечных клеток аорты даже при таких низких концентрациях как 1 фмоль/л. Этот эффект может быть полезен для утолщения внутреннего мышечного слоя после повреждения эндотелия, вызванного, например, ангиопластикой; Стереоизомер 12(Л)-НЕТЕ является сильным ингибитором Ка+,К+-АТФазы роговицы. ЛТВ4 — один из наиболее сильных хемоаттрактантов для нейтрофилов. Он усиливает их адгезию и дегрануляцию, а также образование радикалов кислорода в этих клетках. Пептидолейкотриены, особенно ЛТС4 и JITD4, являются сильными бронхоконстрикторами и вызывают увеличение проницаемости микроцир-куляторного русла, экссудацию плазмы и секрецию слизи — то есть обладают свойствами МРСА. Более того, они снижают сократительную способность миокарда и коронарный кровоток, приводя к сердечной недостаточности. Другие факторы (например, ФАТ) высвобождаются при анафилаксии и еще будут рассмотрены. Считается, что лейкотриены играют роль в развитии воспаления, особенно при хронических заболеваниях.
И LXA, и LXB подавляют цитотоксичность натуральных киллеров. LXA действует на нейтрофилы, как ЛТВ4 и ЛТС4— на сосуды. Таким образом, множество соединений, образующихся в липоокси-геназном пути, регулируют специфические клеточные ответы, важные при воспалении и иммунных реакциях. Метаболиты Цитохромного Р-450-пути влияют на транспортные функции нефрона напрямую или посредством метаболизаций в активные соединения.
Подавление синтеза эйкозаноидов
Кортикостероиды блокируют все известные пути синтеза эйкозаноидов, возможно, через стимуляцию синтеза нескольких ингибиторных белков, которые называются липокортинами. Липокорти-ны уменьшают активность фосфолипазы А2, вероятно, за счет нарушения связывания фосфолипидов и, следовательно, предотвращения высвобож
дения арахидоновой кислоты. Нестероидные противовоспалительные средства (аспирин, индометацин, ибупрофен) блокируют образование и простагландинов, и тромбоксана, ингибируя активность циклооксигеназы. Например, аспирин является длительно действующим ингибитором циклооксигеназы тромбоцитов и, соответственно, биосинтеза ТХА2, так как он необратимо ацетилирует фермент. Тромбоцитарная циклооксигеназа не может быть восстановлена биосинтетическим путем, так как тромбоцит не имеет ядра.
Появление селективных ингибиторов тромбок-сансинтетазы и антагонистов ТХА2-рецепторов потребовало значительных усилий. Полученные соединения весьма полезны для определения эффектов ТХА2 in vitro и in vivo. Сейчас исследуются возможности их использования в терапии легочной гипертензии и преэклампсии. Селективные ингибиторы липооксигеназного пути еще только разрабатываются. За некоторыми исключениями, НПВС не подавляют активность липооксигеназы в концентрациях, которые значительно ингибируют циклооксигеназу. В действительности, предотвратив превращение арахидоновой кислоты циклооксигеназным путем, НПВС могут активизировать метаболизм субстрата через липооксигеназный путь, приводя к повышению уровня в частности флогоген-ных лейкотриенов. Даже внутри циклооксигеназного пути подавление синтеза одного деривата может усилить синтез другого продукта, ферментативно связанного с первым. Поэтому исследователи пытаются разработать препараты, которые ингибируют и циклооксигеназный и липооксигеназный пути.
II. Клиническая фармакология эйкозаноидов
В клиническом использовании: эйкозаноидов существует несколько подходов. Во-первых, созданы стабильные, длительно действующие аналоги ПГЕ( и ПГЕ2, которые повышают внутриклеточные уровни цАМФ и поэтому при определенных обстоятельствах могут считаться “цитопротективными”. Другие сходные соединения, в том числе аналоги простациклина, одобрены для клинического применения в разных странах и сейчас вводятся в клини
ческую практику США. Во-вторых, разработаны ингибиторы фермента и антагонисты рецепторов для снижения экспрессии “патологических” эйкозаноидов, то есть тромбоксанов и лейкотриенов. В-третьих, понимание механизмов синтеза и метаболизма эйкозаноидов привело к созданию новых нестероидных противовоспалительных средств, которые подавляют циклооксигеназу и имеют более выгодные фармакокинетические и фармакодинамические характеристики. Одна цель, как мы уже говорили,— найти двойные ингибиторы, которые блокируют и циклооксигеназный, и липооксигеназный пути. Другая — снизить токсические проявления действия препаратов на ЖКТ и почки. И наконец, в настоящее время активно исследуются способы коррекции диеты для изменения спектра предшественников полиненасыщенных жирных кислот фосфолипидов клеточной мембраны и, следовательно,— регуляции синтеза эйкозаноидов.
Женская репродуктивная система
С момента открытия простагландинов в семенной жидкости приматов и баранов активно изучается физиологическая роль простагландинов в репродукции. Полагают, что простагландины семенной жидкости облегчают имплантацию бластоциста или транспорт яйцеклетки и что секреция простагландинов маткой вызывает лютеолиз. Последнее предположение справедливо для крупного рогатого скота и свиней, но не для человека. Эта находка привела к разработке ветеринарных препаратов ПГЕ2а и его аналогов для синхронизации овуляции самок.
А. Аборт. ПГЕ2 и ПГЕ2а хорошо известны как усилители сокращения матки. Способность простагландинов Е и F и их аналогов прерывать беременность на любом этапе широко используется в клинике. Многие исследования во всем мире показали, что введение простагландина эффективно прерывает беременность. Препараты применяются для абортов во время первого и второго триместра и для подготовки шейки матки перед абортом. Они размягчают шейку, повышая количество протеогликана и изменяя биофизические свойства коллагена.
В ранних исследованиях показано, что внутривенное введение ПГЕ2 и П1Т2а прерывает беременность в 80 % случаев. Успех зависел от дозы, длительности инфузии и количества прошлых беремен
ностей. Ограничивали дозу такие побочные эффекты как рвота, диарея, гипертермия и бронхоспазм.
В настоящее время наиболее часто применяют интраамниотическое, внутримышечное или интра-вагинальное введение. Интраамниотическое введение ПГЕ2а дает почти 100 % результат с меньшим количеством и выраженностью побочных эффектов, чем внутривенное. Препарат динопрост трометамин (дериват ПГЕ2а), который вводили этим путем, был недавно убран с фармацевтического рынка США. Там, где препарат еще применяется, он в основном предпочтителен для прерывания беременности во втором триместре и вводится в виде одной интраамниотической инъекции в дозе 40 мг. Аборт обычно заканчивается в пределах 20 часов. Самый серьезный побочный эффект такого метода введения — это сердечно-сосудистый коллапс. В большинстве таких случаев диагностируют анафилактический шок, но иногда эффект может быть связан с тем, что препарат попадает в циркуляцию и вызывает серьезную легочную гипертензию. У беременных женщин в состоянии наркоза 300 мкг ПГРгц/мин, вводимые внутривенно, повышают сопротивление легочных сосудов на 100 % и усиливают работу правых отделов сердца в 3 раза. Поэтому для развития сердечно-сосудистых осложнений достаточно даже минимальных количеств лекарства, попавших в кровеносное русло при интраам-ниотическом введении 40 мг. Этого можно избежать, вводя препарат под сонографическом контролем.
Для индукции аборта можно применять и внутримышечное введение. Используют карбопрост трометамин (15-метил-ПГЕ2а); метильная группа у С15 продлевает время действия препарата. Карбопрост в отличие от однократной внутриматочной инстилляции динопроста вводится несколько раз до достижения полной дозы 2.6 мг, которой достаточно для индукции аборта.
Синтетический аналог ПГЕ2 динопростон вводится в форме вагинальных свечей. Он напрямую действует на коллагеназу шейки матки и стимулирует сокращения миометрия. Обычно назначают 20 мг, вводя затем повторные дозы каждые 3-5 часов в зависимости от реакции матки. Аборт, как правило, наступает через 90 часов, но почти в 25 % случаев является неполным и требует дополнительного вмешательства.
Активно исследуют возможность использования аналогов ПГЕ для регуляции менструального цик
ла или для очень раннего аборта (в пределах 1-2 недель после последней менструации). В данном случае возникают две проблемы: длительное вагинальное кровотечение и сильные менструальные спастические боли.
Во Франции для раннего аборта применяют ан-типрогестины (например, RU 486) в сочетании с пероральным родостимулирующим простагландином. Доступность и эффективность этой комбинации оспаривается (глава 39). Основные токсические эффекты — спастические боли и диарея.
Б. Стимуляция родов. Многие исследования показали, что ПГЕ2, ПГР2а и их аналоги эффективно инициируют и стимулируют роды. Однако это незарегистрированное показание. При внутривенном введении оба вещества одинаково эффективны, но ПГЕ2<х в 10 раз слабее ПГЕ2. Эти препараты действуют так же успешно, как и окситоцин, и со сходным временным интервалом от индукции родовой деятельности до родов. Побочные эффекты простагландинов умеренные, однако тошнота, рвота и диарея возникают более часто, чем при применении окситоцина. ПГЕ2а более токсичен для ЖКТ, чем ПГЕ2. В рекомендованных дозах ни один из них не вызывает у матери значительных сердечно-сосудистых побочных эффектов. Действительно, для снижения артериального давления и увеличения частоты сердечных сокращений ПГЕ2 надо вливать со скоростью, в 20 раз превышающей используемую при индукции родов. ПГЕ2к является бронхоконстриктором, и у астматиков должен применяться с осторожностью; однако во время индукции родов не возникали ни бронхоспазм, ни приступы астмы. Хотя оба простагландина проходят через фетопла-центарный барьер, токсическое действие на плод встречается редко.
Индукцию родов при пероральном введении ПГЕ2 (0.5-1.5 мг/час) сравнивали с эффектами внутривенной лекарственной формы окситоцина и перорального деривата окситоцина демокситопина. В этих исследованиях ПГЕ2 оказался лучше, чем пероральный дериват окситоцина и практически так же эффективен, как внутривенный окситоцин. Однако в настоящее время существует единственная лекарственная форма ПГЕ2 — динопростон (вагинальные свечи), а при вагинальном введении окситоцин действует несколько сильнее. Вагинальный ПГЕ2 используется и для созревания шейки матки перед индукцией родов. Пероральный ПГР2а обладает слишком большим количеством побочных эф
фектов на ЖКТ, чтобы применяться в такой ситуации.
Теоретически для индукции родов у женщин с преэклампсией или заболеваниями почек или сердца ПГЕ2 и ПГЕ2а должны быть безопаснее окситоцина, так как в отличие от него не оказывают анти-диуретического эффекта. Кроме того, ПГЕ2 обладает натрийуретическим эффектом. Но клиническое значение этих эффектов не документировано. В случаях внутриутробной гибели плода простагландины сами по себе или вместе с окситоцином эффективно вызывают родоразрешение. Иногда при послеродовом кровотечении 15-метил-ПГЕ2а успешно прекращает его в случаях, когда этого не удается добиться окситоцином и метилэргоновином.
В. Дисменорея. Считается, что первичная дисменорея связана с повышением синтеза ПГЕ2 и ПГЕ2а в эндометрии во время менструации с последующим сокращением миометрия и ишемическими болями. НПВС успешно подавляют образование этих простагландинов (глава 35) и в 75-85 % случаев корригируют дисменорею. Некоторые из этих препаратов продаются без рецепта. Аспирин тоже эффективен, но так как он обладает слабым действием и быстро гидролизуется, необходимо частое введение высоких доз.
Сердечно-сосудистая система
У пациентов с гипертензией активно исследовались вазодилататорные эффекты ПГЕ и ПГА. Эти вещества являются также натрийуретиками.
Простациклин снижает периферическое и коронарное сосудистое сопротивление. Его используют в терапии первичной и вторичной легочной гипертензии (вторичная иногда возникает после операций на митральном клапане). Однако его вазодилататорные эффекты не достаточно специфичны для легочных сосудов, поэтому артериальное давление приходится поддерживать прессорными препаратами.
В нескольких исследованиях применяли ПГЕ и ПП2 при феномене Рейно и атеросклерозе периферических сосудов. В последнем случае длительные инфузии использовали для “ремоделирования” сосудистой стенки с целью ускорения регрессии ишемических язв.
А. Тромбоз. Эйкозаноиды участвуют в тромбозе в основном потому, что ТХА2 усиливает агрегацию тромбоцитов, а ПГ12 ее подавляет. Аспирин
ингибирует циклооксигеназу тромбоцитов и вызывает некоторое нарушение свертываемости крови. Незначительная выраженность этого эффекта подтверждается тем, что только небольшие дефекты свертываемости выявляются при заболеваниях с недостаточностью циклоокигеназы и тромбоксансин-тетазы тромбоцитов, то есть у пациентов в анамнезе нет кровотечений или повышения свертываемости. Блокирование любого из этих ферментов подавляет вторичную агрегацию тромбоцитов, вызываемую АДФ, низкими концентрациями тромбина и коллагена или адреналином. Таким образом, эти тромбоцитарные ферменты не являются необходимыми для функции кровяных пластинок, но могут усиливать эффект других агрегирующих стимулов. Очевидно, что основные физиологические факторы, влияющие на процесс свертывания, действуют через иные пути.
Эпидемиологические исследования в США и Великобритании показали, что низкие дозы аспирина снижают риск смерти вследствие инфаркта, но, возможно, повышают общую смертность и частоту развития инсульта (глава 33). Сейчас трудно найти пациентов с риском тромбоэмболии (например, после ортопедических операций или ангиопластики при стенозе коронарных артерий), которые не принимают аспирин. Однако польза аспирина может быть связана и с другими ацетилирующими эффектами, а не только с ацетилированием циклооксигеназы.
Во время развития тромбоза глубоких вен тромбоксан и его метаболиты выделяются с мочой, вероятно потому, что активируется весь пул тромбоцитов. Этот феномен в меньшей степени можно наблюдать при отторжении трансплантата после трансплантации почки. Повышенная экскреция метаболитов тромбоксана возникает также перед инфарктом и во время нестабильной стенокардии.
Простациклин не является циркулирующим гормоном, как считалось раньше; он подавляет агрегацию тромбоцитов, но не их прилипание к эндотелию, распластывание и высвобождение гранул. Аналоги ПГ12 и ПГЕ2 могут ингибировать развитие атеросклероза у животных, получающих холестерин, но при этом в качестве побочного эффекта возникает гипотензия.
Б. Незаращение боталлова протока. Считают, что незаращение фетального боталлова протока связано с местным синтезом ПГЕ2 и ПП2. При определенных врожденных пороках сердца (транспо
зиция магистральных сосудов, атрезия легочного ствола, стеноз легочной артерии) необходимо сохранение открытого боталлова протока до операции. Этого достигают при помощи алпростадила (ПГЕ|). Как и ПГЕ2, ПГЕ| является вазодилататором и ингибитором агрегации тромбоцитов, а также вызывает сокращение гладкой мускулатуры матки и кишечника. Абсолютных противопоказаний нет, но его не рекомендуется применять при респираторном дистресс-синдроме. Побочные эффекты — апноэ, брадикардия, гипотензия и гиперпирек-сия. Из-за быстрого легочного клиренса препарат надо вводить в виде инфузии в начальной дозе 0.05-0.1 мкг/кг/мин (допустимо повышение до 0.4 мкг/кг/мин). Пролонгированное лечение приводит к хрупкости и разрыву протока.
В случае отсроченного заращения боталлова протока для подавления синтеза вазодилататоров ПГЕ2 и ПП2 и закрытия протока можно использовать ингибиторы циклооксигеназы. Недоношенные дети с респираторным дистресс-синдромом вследствие незаращения протока могут успешно лечиться индометацином. Такая терапия снимает необходимость операции.
В. Импотенция или нарушение эрекции. Интракавернозное введение вазодилятатора алпростадила (П ГЕ О становится важным методом лечения импотенции. Однако в США препарат пока не одобрен для применения по этому показанию. Алпрос-тадил можно использовать в виде монотерапии или в сочетании с папаверином или фентоламином.
Дыхательная система
При назначении в аэрозольной форме ПГЕ2 является сильным бронходилататором. К сожалению, он также усиливает кашель, а получить аналог, который обладает только бронхорасширяющим действием, оказалось трудно.
Сильные бронхоконстрикторы ПГР2а и ТХА2 одно время считались основными медиаторами астмы. Однако идентификация лейкотриенов (ЛТС4, JITD4, ЛТЕ4) как МРС А расширила круг эйкозаноидов, являющихся важными медиаторами астмы и других иммунных реакций. Лейкотриены повышают секрецию слизи и усиливают бронхоспазм. Кроме того, они могут участвовать в развитии респираторного дистресс-синдрома у взрослых. Другие патологические медиаторы типа ФАТ не только вызывают бронхоспазм, но и (в зависимости от вида)
могут способствовать высвобождению ПГР2сц тромбоксанов и лейкотриенов. Поэтому много усилий было приложено к поиску антагонистов ФАТ для лечения астмы. К сожалению, этот поиск пока не увенчался успехом. Однако антагонисты ЛТ и ингибиторы липооксигеназы могут оказаться полезными в терапии приступов астмы и реакций гиперчувствительности. Недавние клинические испытания показали, что антагонисты лейкотриенов обладают бронхорасширяющим эффектом у человека. Это доказывает, что ЛТ модулируют тонус стенки дыхательных путей.
Кортикостероиды и кромолин остаются главными связанными с эйкозаноидами препаратами для лечения больных астмой (глава 19). Стероиды подавляют синтез эйкозаноидов и, следовательно, ограничивают количество выделяемого медиатора. Кромолин, возможно, ингибирует высвобождение эйкозаноидов и других медиаторов, например гистамина и ФАТ.
Выделительная система
И в мозговом, и в корковом веществе почек синтезируются простагландины, но синтетические возможности мозгового вещества намного выше. Эти зоны синтезируют несколько гидроксиэйкозатетра-еновых кислот, лейкотриенов, продуктов микросомального цитохром Р-450-окисления и эпоксидов, которые играют важную ауторегуляторную роль в функции почек. Эти соединения влияют и на гемодинамику почки, и на функцию канальцев. Регуляторная роль особенно важна в погранично функционирующих почках, что подтверждается снижением их функции при назначении ингибиторов циклооксигеназы пожилым пациентам или при заболеваниях почек.
Главные продукты эйкозаноидов в корковом веществе почек — ПГЕ2 и ПП2. Оба соединения повышают высвобождение ренина, который обычно находится под контролем [^-адренорецепторов. Клубочки синтезируют также небольшие количества ТХА2. Этот очень сильный вазоконстриктор у здоровых людей не регулирует работу клубочков.
ПГЕ|, ПГЕ2 и ПГ12 повышают гломерулярную фильтрацию вследствие их вазодилататорных эффектов. Эти простагландины усиливают выведение воды и ионов натрия. Дегидратация, видимо, вызвана инактивацией антидиуретического гормона (АДГ) или аденилатциклазы. Непонятно, является
ли натрийуретический эффект следствием прямого подавления реабсорбции Na+ в дистальных канальцах или следствием повышения кровотока в мозговом веществе. Петлевой диуретик фуросемид вызывает некоторые из этих эффектов, стимулируя активность циклооксигеназы. В нормальной почке это приводит к повышению синтеза сосудорасширяющих простагландинов. Поэтому реакция пациента на петлевой диуретик при одновременном введении ингибитора циклооксигеназы будет снижена.
Как и АДГ, ТХА2 усиливает транспорт воды в мочевом пузыре жаб. Это наблюдение позволило предположить, что АДГ частично действует, активируя синтез ТХА2. Нормальная почка синтезирует только малые количества ТХА2. При почечных заболеваниях с клеточной инфильтрацией интер-стиция (например, гломерулонефрит и отторжение трансплантата почки) клетки, участвующие в воспалительной реакции (моноциты-макрофаги), высвобождают значительные количества ТХА2, что вызывает вазоконстрикцию в почке (и возможно, АДГ-подобный эффект) и приводит к снижению ее функции. Введение ингибиторов ТХА2-синтетазы или ее антагонистов (если они доступны) этим пациентам и больным с преэклампсией может улучшить функцию почек.
Гипертензию у животных часто связывают с повышением уровня ТХА2 и снижением концентрации ПГЕ2 и ПГ12. Эти изменения могут быть первопричиной гипертензии либо носить вторичный характер. Активация образования ТХА2 выявлена и при нефротоксических симптомах, вызванных циклоспорином, но причинная связь здесь еще не установлена.
ПГЕ2 может участвовать в экскреции фосфата, так как экзогенный ПГЕ2 является антагонистом подавления реабсорбции фосфата паратиреоидным гормоном (ПТГ) в проксимальных канальцах. Однако физиологическая роль этого эйкозаноида ограничена низким содержанием простагладинов в проксимальных канальцах.
Роль лейкотриенов и продуктов окисления цитохромом Р-450 в почке человека полностью не выяснена. Для определения точных функций этих веществ требуется больше информации. Недавно обнаружено, что -5,-6-эпоксид является сильным вазодилататором у животных. Установлено также, что свободные радикалы атакуют фосфолипиды, содержащие арахидоновую кислоту, с образованием 8-изо-ПГЕ2а, который обладает выраженными тром
боксаноподобными свойствами. Синтез этого вещества не блокируется ингибиторами циклооксигеназы, но может угнетаться антиоксидантами. Этот вазоконстриктор, имеющийся у человека, предположительно является медиатором, опосредующим почечную недостаточность при гепаторенальном синдроме.
ЖКТ
Слово “цитопротекция” употребляется для акцентирования защитного эффекта простагландинов группы Е в отношении язв желудка у животных в дозах, которые не снижают желудочную секрецию. Эти простагландины уменьшают выделение кислоты в желудке. Накоплено много экспериментальных и клинических данных, подтверждающих, что ПГЕ и его аналоги предотвращают появление язв желудка, образующихся при применении стероидов или НПВС. В Европе и США для лечения язв применяют активный при пероральном приеме синтетический аналог ПГЕ! — мизопростол. В США препарат одобрен только для профилактики язв, возникающих при применении НПВС. Этот и другие аналоги ПГЕ (например, энпростил) являются цитопротекторами в низких дозах, а в более высоких — подавляют секрецию соляной кислоты. Характерные побочные эффекты (абдоминальный дискомфорт и иногда диарея) дозозависимы.
При воспалительных заболеваниях кишечника было обнаружено усиление образования лейкотриенов в слизистой толстой кишки. Неизвестно, отражают ли эти изменения патофизиологическую роль лейкотриенов.
Иммунная система
Моноциты-макрофаги — это единственные из главных клеток иммунной системы, которые могут синтезировать все эйкозаноиды. Т- и В-лимфоци-ты являются интересным исключением из общего правила, гласящего, что все ядросодержащие клетки синтезируют эйкозаноиды. Однако в культуре клеток В-клеточной лимфомы наблюдается безре-цепторный захват ЛТВ4 и 5-НЕТЕ. Взаимодействие между лимфоцитами и мононуклеарными фагоцитами может вызывать высвобождение арахидоновой кислоты из клеточных мембран лимфоцитов. Арахидоновая кислота используется затем макрофагами для синтеза эйкозаноидов. Помимо этих
Рис. 18-7. Модуляция взаимодействия макрофага и лимфоцита эйкозаноидами, фактором активации тромбоцитов и кортикостероидами. Кортикостероиды, ПГЕ2и, возможно, ПГ12 подавляют экспрессию интерлейкина-1 (ИЛ-1) и его эффекты в отношении Т-лимфоцитов. Фактор активации тромбоцитов (ФАТ), ЛТВ4 и ЛТО4 усиливают экспрессию ИЛ-1. Сходные угнетающие и стимулирующие влияния оказываются на действие у-интерфе-рона в отношении макрофагов и на эффект интерлейкина-2 (ИЛ-2). Вещества, отмеченные зведочками, как предполагают, оказывают данные эффекты, хотя это еще не подтверждено. (DR — рецептор МНС II класса (major histocompatibility complex — основной комплекс гистосовместимости); Т — Т-лимфоциты.) (Из: Foegh М. L., Ramwell Р. W. PAF and transplant immunology. In: Braquet P. (ed.) The Role of Platelet Activating Factor in Immune Disorders. Karger, 1988.)
клеток, межклеточное взаимодействие с образованием эйкозаноидов характерно для тромбоцитов, эритроцитов, ПМН и эндотелиальных клеток.
Эйкозаноиды модулируют эффекты иммунной системы, что можно продемонстрировать на примере клеточного иммунного ответа. Как показано на рис. 18-7, ПГЕ2 и ПП2, как и кортикостероиды, действуют на пролиферацию Т-клеток. Они угнетают клональную экспансию Т-клеток, инактивируя интерлейкины-1 и -2 и экспрессию антигенов класса II на макрофагах или других антигенпрезентирующих клетках. Лейкотриены, ТХА2 и ФАТ стимулируют клональную экспансию Т-лимфоцитов. Эти веще
ства стимулируют образование интерлейкинов-1 и -2, а также экспрессию рецепторов интерлейкина-2. Лейкотриены усиливают также высвобождение у-интерферона и могут заменять в этом отношении интерлейкин-2. Эти проявляющиеся in vitro эффекты эйкозаноидов и ФАТ соответствуют данным, полученным in vivo на животных с острым отторжением трансплантата. Доказана связь между использованием противовоспалительных стероидов и повышением риска инфекции, но НПВС не влияют существенно на противоинфекционный иммунитет.
А. Отторжение трансплантата. Острое отторжение трансплантата является клеточной реакцией. В некоторых случаях введение ПП2 пациентам после трансплантации прекращает реакции отторжения. Экспериментальные данные (in vitro и in vivo) показали, что ПГЕ2 и ПП2 могут предотвратить пролиферацию Т-клеток и реакцию отторжения. С другой стороны, ингибирование образования ТХА2 и лейкотриенов уменьшает вероятность отторжения органных трансплантатов. У пациентов после пересадки почки при остром отторжении усиливается выделение ТХВ2 с мочой. Пока не доказано, что этот эйкозаноид вызывает развитие данного состояния, однако кортикостероиды все еще являются основным средством при остром отторжении органа.
Б. Воспаление. Аспирин используют для лечения артрита уже почти в течение века, но механизм его действия (подавление активности циклооксигеназы) не был известен до 1971 г. Аспирин и другие НПВС, ингибирующие циклооксигеназу, рассматриваются в главе 35. С клетками, участвующими в воспалении, связан изофермент Сох II. В отличие от простагландинов лейкотриены и некоторые НЕТЕ (например, 12-НЕТЕ) являются сильными хемоаттрактантами. Поэтому исследователи пытаются сейчас разработать препараты, ингибирующие циклооксигеназу II, 5-липооксигеназу и 12-липооксигеназу. ПГЕ2 угнетает вызванную антигенами или митогенами пролиферацию В-лимфо-цитов и их дифференцировку в плазматические клетки, что приводит к подавлению синтеза иммуноглобулина М (рис. 18-8). Одновременное с активацией синтеза ПГЕ2 в моноцитах повышение уровня сывороточного IgE у пациентов после травмы или с болезнью Ходжкина связано с изменением соотношения классов иммуноглобулинов в сторону преобладания IgE под воздействием ПГЕ2.
Рис. 18-8. ПГЕ2 ингибирует активацию и пролиферацию В-лимфоцитов под влиянием Т-лимфоцитов (CD4+). Как митоген-, так и антигениндуцированная пролиферация В-клеток ингибируется ПГЕ2 и стимулируется JITD4. ПГЕ2 подавляет дифференцировку В-лимфоцитов в плазматические клетки и таким образом косвенно ограничивает синтез иммуноглобулина М (IgM)
В. Ревматоидный артрит. При ревматоидном артрите иммунные комплексы откладываются в пораженных суставах, вызывая воспаление, которое усиливается эйкозаноидами и ФАТ. Лимфоциты и макрофаги накапливаются в синовиальных мембранах, а ПМН преимущественно находятся в синовиальной жидкости. Основные эйкозаноиды, синтезируемые ПМН — это лейкотриены, которые усиливают пролиферацию Т-клеток и являются хемоаттрактантами. Макрофаги человека в дополнение к ФАТ и большому количеству лейкотриенов синтезируют циклооксигеназные продукты ПГЕ2 и ТХ А2.
Коррекция метаболизма арахидоновой кислоты при помощи диеты
Так как арахидоновая кислота синтезируется из линолевой и линоленовой кислот пищи, в настоящее время изучаются возможности влияния диеты
на метаболизм арахидоновой кислоты. Используются два подхода. В первом случае в пищу добавляют кукурузное, сафлоровое ( Carthamus tinctorius) и подсолнечное масло, которые содержат линолевую кислоту (С18-полиеновая кислота). Во втором — используют масла, содержащие С20- и С22-полиеновые кислоты (масла из рыб, живущих в холодной воде). Оба типа коррекции диеты изменяют состав фосфолипидов клеточных мембран вследствие вытеснения арахидоновой кислоты жирными кислотами этих масел. Полагают, что в результате снижается синтез ТХА2 и ПГ12, и за этим следуют изменения агрегации тромбоцитов, вазомоторный спазм и метаболизация холестерина. Однако никакая коррекция диеты не изменяет значительно ход развития заболеваний, на которые стремились воздействовать.
Как было сказано ранее, существует множество продуктов окисления разных полиеновых кислот. Наивно пытаться приписывать известные на сегодняшний день эффекты отдельным метаболитам. Для получения удовлетворительного ответа на эти вопросы необходимо проведение тщательно контролируемых клинических испытаний. Тем не менее у пациентов с преобладанием высоконасыщенных жирных кислот в диете повышена агрегация тромбоцитов при сравнении с другими группами населения. Такое питание (например, в Финляндии и США) коррелирует с более высокой частотой инфаркта миокарда, чем при преобладании в пище полиненасыщенных кислот (например, в Италии).
Препараты
Алпростадил (Простин ВР педиатрик)
Парентерально: 500 мкг/мл в ампулах по 1 мл
Карбопрост трометамин (Простин/15М)
Парентерально: 250 мкг карбопроста и 83 мкг трометамина на мл в ампулах по 1 мл
Динопростон (Простин Е2)
Вагинально: свечи 20 мг
Эпопростенол (Простациклин) (Флолан, Циклопростин)
Редкий препарат, применяющийся при необходимости замещения гепарина у некоторых пациентов при гемодиализе
Мизопростол (Цитотек)
Перорально: таблетки по 100, 200 мкг
Бронходилататоры и другие средства, 19
применяемые для лечения астмы
Гомер А. Буше
Астма характеризуется повышенной реактивностью трахеи и бронхов в ответ на различные стимулы и сужением дыхательных путей, проходящим либо спонтанно, либо под действием терапии. Клиническими проявлениями астмы являются повторные эпизодические приступы кашля, одышки, ощущения нехватки воздуха и хрипы. В основе патогенеза астмы лежат следующие изменения дыхательных путей: спазм гладкой мускулатуры, отек слизистой, клеточная инфильтрация и нарушение проходимости пробками из плотной слизи. Из всех этих изменений бронхоспазм является наиболее легко обратимым современными методами терапии, в то время как отек слизистой и клеточная инфильтрация требуют продолжительного лечения противовоспалительными препаратами.
Наиболее часто применяемые бронходилататоры — стимуляторы Р-адренорецепторов (Р-адрено-миметики) (главаЭ) и теофиллин, производное ме-тилксантина. Бронхоспазм купируют и м-холино-литики, блокаторы мускариновых рецепторов (глава 8). Две другие группы препаратов, используемые для лечения астмы, представляют собой противовоспалительные средства: ингибиторы дегрануляции тучных клеток (кромолин и недокромил) и кортикостероиды. Недавние исследования с участием небольшого количества пациентов позволили предположить, что длительное лечение метотрексатом или препаратами золота, которые применяют для лечения ревматоидного артрита, может быть эффективно для лечения тяжелой, стероидзависимой астмы.
В этой главе представлены основные данные по фармакологии кромолина и метилксантинов — препаратов, которые применяются почти исключительно при заболеваниях легких. Другие средства
из вышеупомянутых классов рассматриваются только применительно к терапии бронхиальной астмы.
Патогенез астмы
Рациональный подход к фармакотерапии астмы зависит от понимания основной концепции патогенеза этого заболевания. По классической иммунологической модели, бронхиальная астма — это заболевание, в развитии которого основную роль играет связывание реагиновых антител (IgE) с тучными клетками слизистой оболочки дыхательных путей (рис. 19-1). При повторных контактах с антигеном его взаимодействие с антителом происходит на поверхности тучных клеток и запускает процессы высвобождения уже синтезированных медиаторов, находящихся в гранулах тучных клеток, а также синтез и высвобождение новых медиаторов. В их число входят гистамин, триптаза и другие нейтральные протеазы, лейкотриены С4 и D„, простагландин D2, факторы хемотаксиса эозинофилов (eosinophil chemotactic factor — ECF) и нейтрофилов (neurophil chemotactic factor — NCF) и другие вещества. Путем диффузии медиаторы проникают вглубь стенки дыхательных путей и вызывают спазм гладких мышц, отек, клеточную инфильтрацию, изменяют секрецию слизи либо прямо, либо через механизмы нейрорегуляции.
Однако существуют некоторые особенности астмы, которые не соответствуют этой концепции. У многих взрослых пациентов отсутствуют признаки гиперчувствительности немедленного типа к антигенам и большинство приступов не зависит непосредственно от времени контакта со значительной дозой антигена. Даже в тех случаях, когда в анам-
Рис. 19-1. Иммунологическая модель патогенеза реакции немедленного типа. Контакт с антигеном вызывает синтез IgE, который связывается с тучными клетками в органе-мишени. При повторном контакте с антигеном его взаимодействие с антителом на поверхности тучных клеток запускает высвобождение медиаторов анафилаксии (LTGj и LTD< — лейкотриены С4 и D4; ПГБ2 — простагландин D2; ECF-А — фактор хемотаксиса эозинофилов). (Из: Gold W. W. Cholinergic pharmacology in asthma. In: Asthma Physiology, Immunopharmacology, and Treatment. Austen K. F., Lichtenstein L. M. (eds) Academic Press, 1974.)
незе у пациентов есть астма во время сезона цветения амброзии при положительных кожной пробе и бронхиальных тестах на антиген амброзии, не отмечается корреляции между тяжестью симптомов и количеством антигена в атмосфере. Более того, бронхоспазм может быть вызван неантигенными стимулами, например дистиллированной водой, физической нагрузкой, холодным воздухом, диоксидом серы и гипервентиляцией.
У здоровых людей, не страдающих астмой, ингаляции небольших доз медиаторов анафилаксии (например, гистамина) не вызывают бронхоспазма. У больных астмой ингаляции даже еще меньших доз тех же медиаторов вызывают тяжелый симптоматический бронхоспазм. Эту реакцию можно количественно измерить по снижению форсированного объема выдоха за 1 секунду (ФОВ1). Такую повышенную чувствительность дыхательных путей иногда называют “неспецифической гиперреактивностью бронхов”, чтобы отличить ее от бронхоспазма, вызываемого специфическими антигенами. Считается, что гиперреактивность бронхов — это фундаментальный механизм патогенеза астмы. Она присутствует практически у всех без исключения больных астмой, ее степень коррелирует со степенью тяжести симптомов, а явления, повышающие реактивность бронхов (респираторные вирусные инфекции, контакт с окислителями в загрязненной окружающей среде), часто вызывают обострения болезни.
Механизмы возникновения гиперреактивности бронхов неизвестны, но могут быть связаны с воспалением слизистой оболочки дыхательных путей. Эта гипотеза подтверждается тем, что факторы, повышающие реактивность бронхов (например, ингаляция озона, антигена, респираторные вирусные инфекции) вызывают воспаление слизистой оболочки дыхательных путей. Повышение реактивности бронхов вследствие ингаляции озона коррелирует с возрастанием числа полиморфноядерных лейкоцитов в жидкости бронхоальвеолярного смыва или в биоптатах слизистой оболочки бронхов. Аллергическое повышение реактивности бронхов связано с увеличением числа эозинофилов и полиморфноядерных лейкоцитов в жидкости бронхоальвеолярного лаважа. Продолжительное усиление реактивности бронхов, связанное с поздней астматической реакцией на ингаляцию аллергена (рис. 19-2), по современным представлениям, вызвано воспалительным процессом в дыхательных путях.
Механизмы связи между гиперреактивностью дыхательных путей и воспалением неизвестны, но постепенно накапливающиеся данные свидетельствуют о значительной роли эозинофилов. Концентрация гранулярного эозинофильного белка (основного белка) в откашливаемой мокроте или в бронхоальвеолярной лаважной жидкости коррелирует со степенью реактивности бронхов. Биоптаты слизистых больных астмой и здоровых людей наи-
Рис. 19-2. Сравнение реакции на контрольную ингаляцию и ингаляцию аллергена у двух пациентов. У одного присутствует только ранний ответ (рис.19-2 А), а у другого — как ранний, так и поздний ответ (рис.19-2 Б). Форсированный объем выдоха за первую секунду (ФОВ1) показан слева как процент от контрольного значения. Бронхоспазм приводит к падению значений ФОВ1. Дозы (мг/мл) ингаляционного гистамина, вызывающие снижение ФОВ1 на 20 % (РС20Н) через 8 часов после контрольной ингаляции и ингаляции аллергена, показаны на графике справа. Повышение реактивности проявляется снижением необходимой дозы гистамина. У пациента, данные которого приведены на рис.19-2 А, наблюдается транзиторное снижение ФОВ1 сразу после ингаляции антигена, однако за этим следует быстрое восстановление и нормальные значения ФОВ1 в дальнейшем. Через 8 часов не возникало повышения реактивности на гистамин. В отличие от этого у другого пациента, у которого наблюдался поздний ответ на ингаляцию аллергена (рис.19-2 Б, на левом графике — снижение ФОВ1 через 6 часов), отмечается значительное снижение дозы гистамина, необходимой для провоцирования бронхоспазма (график справа, после ингаляции). (Из: Cockroft D. W. et al. Allergen-induced increase in non-allergic bronchial reactivity. Clin Allergy, 1977; 7:503.)
контрольной ингаляции ингаляции аллергена
более существенно различаются по количеству эозинофилов в и под эпителием дыхательных путей. Иммуногистохимическое исследование показывает повышенные количества другого продукта эозинофилов — эозинофильного катионного белка, который располагается под эпителием и указывает на активацию клеток. Кроме того, показано, что продукты эозинофилов способны вызывать отхождение эпителия и повышение сократительной активности гладкой мускулатуры дыхательных путей. Продукты других клеток, встречающихся в респираторном тракте (макрофагов, тучных клеток, окон
чаний чувствительных нервов и эпителиальных клеток) также способны изменять состояние гладкой мускулатуры дыхательных путей, поэтому вряд ли можно предположить, что антагонист одного медиатора или класса медиаторов будет эффективным и достаточным противоастматическим средством. Однако антагонисты лейкотриенов и ингибиторы липооксигеназного пути дали обнадеживающие результаты на первых стадиях клинических испытаний (Cloud, 1989; Israel, 1993).
Каковы бы ни были механизмы, отвечающие за общее повышение реактивности бронхов при аст
ме, бронхоконстрикция сама по себе является следствием прямого эффекта высвобожденных медиаторов и усиления этого эффекта нервными или гуморальными путями, активированными действием медиаторов. Важность нервных путей показана в основном в экспериментах на лабораторных животных. Так, у собак бронхоспазм, вызванный гистамином, значительно снижался, если предварительно ингалировали местные анестетики, проводили вагусную блокаду, вводили атропин (конкурентный антагонист ацетилхолина) в дозах, влияющих на реакцию гладкой мускулатуры дыхательных путей на гистамин in vitro. Однако результаты исследований на людях неоднозначны. Фармакологическая блокада вагусных эфферентных путей аэрозолем гек-саметония — препарата, который ингибирует проведение через парасимпатические ганглии (глава 8), или сульфатом атропина — постганглионарным антагонистом мускариновых рецепторов, вызывает уменьшение, но не прекращение бронхоспазма в ответ на антигенные и неантигенные стимулы (например, физическую нагрузку, холодный воздух, диоксид серы и дистиллированную воду). Возможно, что нервные импульсы (адренергические, холинергические — глава 6) играют роль в реактивности стенки бронхов, однако тот факт, что возникновение бронхоспазма частично блокируется кромо-лином (ингибитором дегрануляции тучных клеток) позволяет предположить, что и антигенные, и неантигенные стимулы могут вызывать высвобождение медиаторов, ведущее к бронхоспазму как путем прямых воздействий, так и активацией эфферентных парасимпатических путей (рис. 19-3).
Гипотеза, которая выдвигается на основании этих исследований, состоит в том, что астматический бронхоспазм возникает вследствие высвобождения медиаторов и завышенной реакции на их эффекты. Она позволяет предположить, что для эффективного лечения астмы можно использовать препараты с разными механизмами действия (рис. 19-4). Астматический бронхоспазм можно снять или предотвратить, например препаратами, которые блокируют дегрануляцию тучных клеток (кромолин или недокромил, агонисты р-рецепто-ров, блокаторы кальциевых каналов), ингибируют эффекты ацетилхолина, выделяемого из двигательных нервных окончаний вагуса (антагонисты мускариновых рецепторов), или напрямую расслабляют гладкую мускулатуру (симпатомиметики, теофиллин).
Другой подход к лечению астмы направлен не только на профилактику или снятие острого бронхоспазма, но и на уменьшение уровня реактивности бронхов. Так как скорее всего реактивность бронхов связана с воспалением дыхательных путей, а воспаление является составной частью позднего астматического ответа, эту стратегию можно применить, используя продолжительную терапию препаратами, которые предотвращают позднюю реакцию, например кромолином или кортикостероидами. Лекарственные средства, эффективные при других хронических воспалительных заболеваниях, в частности, метотрексат и препараты золота, также могут быть использованы для реализации этого подхода.
I. Базисная фармакология препаратов, используемых для лечения астмы
Кромолин и недокромил
Кромолин натрий (динатрий кромогликат) и недокромил натрий отличаются от большинства противоастматических препаратов тем, что они эффективны только при профилактическом применении. Это стабильные, но крайне нерастворимые соли. При применении в виде аэрозоля (дозированные ингаляторы) они эффективно предотвращают антигенспровоцированную астму и астму физической нагрузки. Их постоянное применение (4 раза в день) может снизить общий уровень реактивности бронхов. Однако эти препараты не оказывают влияния на тонус гладкой мышцы и не снимают бронхоспазм.
Кромолин плохо всасывается в желудочно-кишечном тракте. Его необходимо применять местно, путем ингаляций порошка из микрочастиц или аэрозольного раствора, как упомянуто выше. При ингаляционном или пероральном приеме абсорбируется менее 10 %, а остальная часть выводится из организма в неизмененном виде. У недокромила тоже очень низкая биодоступность, и его применение возможно только в виде дозированного аэрозоля.
Механизм действия
Кромолин более подробно изучен, чем недокромил, но, по-видимому, механизмы действия этих
Рис. 19-3. Механизмы реакции на ингалируемые ирританты. Дыхательные пути представлены поперечным микроскопическим срезом стенки с разветвленными чувствительными вагусными окончаниями, прилегающими к просвету. Афферентные вагусные пути попадают в центральную нервную систему (ЦНС); эфферентные пути от ЦНС доходят до эфферентных ганглиев. Постганглионарные волокна высвобождают ацетилхолин, который связывается с мускариновыми рецепторами на гладкой мышце дыхательных путей. Ингалируемые вещества могут провоцировать бронхоконстрикцию несколькими механизмами. Во-первых, они могут запускать высвобождение химических медиаторов из тучных клеток. Во-вторых, они могут посредством стимуляции афферентных рецепторов вызвать рефлекторную бронхоконстрикцию или высвобождение тахикининов (например, субстанции Р), которые напрямую стимулируют мышечные сокращения
препаратов похожи. Исследования in vitro показали, что оба средства предотвращают, высвобождение гистамина и лейкотриенов из сенсибилизированных тучных клеток бронхолегочного аппарата, очевидно, путем блокирования трансмембранного тока кальция, вызванного взаимодействием IgE и антигена на поверхности тучной клетки (Dahlen, 1989; Bruijnzeel, 1990). Этот ингибиторный эффект специфичен относительно типа клеток и антител,
так как кромолин обладает только незначительным подавляющим эффектом на высвобождение медиаторов из человеческих базофилов или на IgG-ono-средованную дегрануляцию тучных клеток. Действие специфично и относительно разных органов: кромолин подавляет анафилактическую реакцию в легких человека и приматов, но не в коже. В свою очередь это может отражать различия тучных клеток разных локализаций. Например, тучные клет-
Контакт с антигеном (пыль, пыльца и т. д.)
Избегание антигена
Антиген и IgE на тучных клетках
Кромолин Стероиды
Медиаторы (лейкотриены, гистамин и т. д.)
Поздний ответ: Ранний ответ:
воспаление бронхоспазм
Рис. 19-4. Основные стратегии лечения астмы. Терапевтические вмешательства помечены затененными прямоугольниками. (Из: Cockcroft D. W. The bronchial late response in the pathogenesis of asthma and its modulation by therapy. Ann Allergy, 1985; 55; 857.)
ки человеческих легких и кожи отличаются по содержанию нейтральных протеаз. Тучные клетки легких содержат только триптазу, а клетки кожи — и триптазу, и химазу.
Обследования больных астмой подтверждают концепцию о том, что кромолин подавляет высвобождение медиаторов из тучных клеток дыхательных путей. Предварительное лечение кромолином подавляет не только бронхоспазм, вызванный ингаляцией антигена или физической нагрузкой, но и одновременно появление в крови фактора хемотаксиса нейтрофилов (NCF) — предполагаемого мар
кера активации тучных клеток. Этот механизм действия кромолина настолько очевиден, что препарат используют для определения типа бронхиальной реактивности. Если кромолин предотвращает бронхоспазм, то вероятнее всего задействован механизм выделения медиаторов из тучных клеток.
Однако другие данные показывают, что действие кромолина, возможно, не настолько однозначно. Поиск более эффективных препаратов привел к появлению веществ с более выраженным потенциалом подавления дегрануляции тучных клеток in vitro, но большинство из них были неэффективны в отношении ингибирования бронхомоторных реакций in vivo. В ряде экспериментов продемонстрировано, что кромолин ингибирует фосфодиэстеразу, повышая таким образом внутриклеточный уровень цАМФ, и что он влияет на проведение импульса по нервным волокнам, контролирующим гладкомышечный тонус дыхательных путей.
Клиническое применение кромолина и недокромила
Кромолин эффективен в следующих клинических ситуациях. При применении до контакта с аллергеном препарат подавляет немедленную и отсроченную реакции на антиген. Предварительное применение кромолина блокирует также бронхоспазм, вызванный аспирином, физической нагрузкой, различными промышленными загрязнениями (диизо-ционат толуола, древесная пыль, паяльные газы, гидрохлорид пиперазина и некоторые ферменты). Благодаря такому защитному эффекту одной дозы,
Кромолин натрий
Недокромил натрий
кромолин можно вводить непосредственно перед физической нагрузкой или неизбежным контактом с антигеном для профилактики бронхоспазма.
При хронической астме кромолин эффективно снижает тяжесть симптомов и необходимость в бронходилататорах. Но эффект наблюдается не у всех больных. Наиболее вероятен успех при лечении молодых пациентов с экзогенной астмой, однако улучшение наблюдается и у некоторых пожилых пациентов с эндогенной астмой. В настоящее время единственный путь определения эффективности препарата — пробный четырехнедельный курс терапии. В случае улучшения эффективность регулярного применения кромолина приблизительно равна эффективности поддерживающей терапии теофиллином.
Предполагается, что длительное применение кромолина снижает реактивность бронхов, возможно, путем уменьшения воспаления, вызванного химическими медиаторами анафилаксии. В настоящее время считают, что кромолин эффективно предотвращает сезонные приступы у больных аллергической астмой, но менее эффективен, чем ингаляционные стероиды, в снижении реактивности бронхов.
Кромолин плохо всасывается из желудочно-кишечного тракта и поэтому эффективен только при прямом поступлении в дыхательные пути. Сегодня используются два метода введения. Взрослым больным препарат вводят через дозированный ингалятор обычно в дозе 2—4 мг четыре раза в день. Детям, для которых сложно использовать ингалятор, кромолин назначают в виде аэрозоля 1 % раствора.
Раствор кромолина эффективен и при аллергическом рините. Назальный спрей или глазные капли, применяемые несколько раз в день, дают положительные результаты у 75 % больных сенной лихорадкой даже в разгар сезона появления пыльцы.
Вследствие плохой абсорбции побочные эффекты кромолина минимальны и проявляются местно. Это раздражение глотки, кашель, сухость во рту, ощущение давления в грудной клетке и хрипы. Некоторые из этих симптомов можно предотвратить ингаляцией р2*агониста перед применением кромолина. Серьезные побочные эффекты редки. Приблизительно у 2 % больных встречаются обратимые дерматит, миозит или гастроэнтерит. Было отмечено несколько случаев легочной эозинофильной инфильтрации или анафилактической реакции.
Недокромил, обладающий более сильными ингибирующими эффектами на культуре тучных клеток легких у приматов in vitro, показал обещающие результаты в широкомасштабных клинических испытаниях. Он оказался более эффективным, чем кромолин.
Ни кромолин, ни недокромил не могут полностью заменить ингаляционные гормоны, вводимые ингаляционно больным стероидзависимой астмой (Ruffin, 1987).
Метилксантины
Три основных метилксантина — теофиллин, теобромин и кофеин. Они содержатся в чае, какао и кофе. Теофиллин как лекарственный препарат в последнее время уступает свои позиции ингаляционным симпатомиметикам при купировании астматических приступов и ингаляционным противовоспалительным средствам при лечении хронической астмы.
Химия
Теофиллин представляет собой 1,3-диметил-ксантин, теобромин — 3,7-диметилксантин, а кофеин — 1,3,7-триметилксантин. Наиболее часто в клинике применяют теофиллин-этилендиаминовый комплекс аминофиллин. Синтетический аналог теофиллина (дифиллин) менее эффективный и более короткодействующий, чем теофиллин. Фармакокинетика теофиллина рассматривается в разделе “Клиническое применение метил ксантинов”.
I
СН3
Ксантин Теобромин
I I
СНз СН3
Кофеин Теофиллин
Продукты метаболизма препарата — частично деметилированные ксантины (не мочевая кислота), выделяются через почки.
Механизм действия
Предложено несколько механизмов действия метилксантинов, но достоверно не доказана связь ни одного из них с бронхорасширяющим эффектом. В высоких концентрациях in vitro они ингибируют фосфодиэстеразу (рис. 19-5). Так как фосфодиэстераза гидролизует циклические нуклеотиды, ингибирование фермента приводит к повышению концентрации внутриклеточного цАМФ. Этот эффект может объяснить стимулирующее воздействие метилксантинов на сердце и расслабляющее на гладкие мышцы, но не доказано, что in vivo достигаются достаточно высокие концентрации для подавления фосфодиэстеразы. Другой возможный механизм — блокирование аденозиновых рецепторов на поверхности клеток. Рецепторы модулируют активность аденилатциклазы, а аденозин, как показано, вызывает сокращение изолированных гладких мышц дыхательных путей и увеличивает высвобождение гистамина из клеток в легких. Эти эффекты блокируются теофиллином — универсальным антагонистом поверхностных аденозиновых рецепторов. Однако показано, что производные ксантина, не обладающие антагонистическими свойствами по отношению к аденозину (например, энпрофиллин), способны во много раз более эффективно подавлять бронхоспазм у больных астмой.
Бронходилатация Вазодилатация Подавление высвобождения медиаторов
АТФ--------->цАМФ ---------->5'-АМФ
р-стимуляция Теофиллин
Рис. 19-5. Внутриклеточный уровень цАМФ может быть повышен агонистами p-адренорецепторов, которые увеличивают скорость его синтеза, катализируемого адени-латциклазой, или ингибиторами фосфодиэстеразы (теофиллин), которые уменьшают скорость его деградации
Фармакодинамика метилксантинов
Метилксантины действуют на ЦНС, почки, сердечную и скелетные мышцы, а также на гладкую мускулатуру. Из трех соединений теофиллин наиболее селективен в отношении воздействия на гладкую мышцу, а кофеин оказывает самый выраженный эффект на ЦНС.
А. Воздействие на ЦНС. В низких и средних дозах метилксантины (особенно кофеин) вызывают небольшое возбуждение корковых нейронов с повышением уровня бодрствования и снижением чувства усталости. Особо чувствительным людям достаточно чашки кофе (100 мг кофеина), чтобы привести к нервозности и бессоннице. В очень высоких дозах кофеин может стимулировать продолговатый мозг и вызвать судороги. Нервозность и тремор — основные побочные эффекты больших доз аминофиллина у больных астмой.
Б. Действие на сердечно-сосудистую систему. Метилксантины оказывают прямое хронотропное и инотропное действия на сердце. При низких концентрациях препаратов этот эффект связан с повышением высвобождения катехоламинов из-за блокады пресинаптических аденозиновых рецепторов. При более высоких концентрациях препарата (> 10 мкмоль/л) возможно повышение тока кальция в ответ на увеличение содержания цАМФ вследствие ингибирования фосфодиэстеразы. При очень высоких концентрациях (>100 мкмоль/л) нарушается секвестрация кальция в саркоплазматическом ретикулуме. Особенно чувствительных людей несколько чашек кофе могут привести к аритмии, однако у большинства парентеральное введение больших доз метилксантинов вызывает только синусовую тахикардию и повышение сердечного выброса. Иногда метилксантины используют для лечения отека легких при сердечной недостаточности (глава 13). В высоких дозах эти препараты расслабляют также гладкие мышцы сосудов (кроме мозговых сосудов, тонус которых, наоборот, повышается). Однако обычное потребление кофе и других напитков, содержащих метилксантины, в некоторой степени повышает периферическое сосудистое сопротивление и артериальное давление, возможно, через высвобождение катехоламинов.
Метилксантины понижают вязкость крови и в определенных условиях могут улучшать кровоток. Механизм этого действия не вполне ясен, но оно используется при лечении перемежающейся хромо
ты (облитерирующего артериита) производным диметилксантина пентоксифиллином. Но нет сведений, подтверждающих превосходство этого метода над другими.
В. Воздействие на желудочно-кишечный тракт. Метилксантины стимулируют в желудке секрецию соляной кислоты и пищеварительных ферментов. Однако даже декофеинизированный кофе оказывает сильное стимулирующее действие на желудочную секрецию. Следовательно, этот эффект не связан с кофеином.
Г. Действие на почки. Метилксантины (особенно теофиллин) являются слабыми диуретиками. Этот эффект может быть опосредован как увеличением клубочковой фильтрации, так и снижением канальцевой реабсорбции натрия. Но диуретическое действие метилксантинов недостаточно выражено, чтобы его можно было использовать.
Д. Воздействие на гладкую мускулатуру. Расширение бронхов — это главное терапевтическое свойство метилксантинов. Толерантность к ним не развивается. Дозу могут ограничивать побочные эффекты, особенно на ЦНС. В дополнение к прямому действию на гладкую мускулатуру дыхательных путей эти препараты при использовании в достаточных дозах подавляют вызванное антигеном высвобождение гистамина в легочной ткани. Характер действия метилксантинов на мукоцилиарный транспорт неизвестен.
Е. Воздействие на скелетную мускулатуру. Терапевтические эффекты метилксантинов не ограничиваются воздействием на дыхательные пути. Они усиливают сократимость изолированной скелетной мышцы in vitro и способствуют восстановлению сократимости и снятию усталости диафрагмы у больных с хроническими обструктивными заболеваниями легких. Именно действие на диафрагму, а не на дыхательный центр, лежит в основе способности теофиллина улучшать вентиляторную реакцию на гипоксию и уменьшать одышку у пациентов даже с необратимой обструкцией дыхательных путей.
Клиническое применение метилксантинов
Из всех ксантинов теофиллин является наиболее эффективным бронходилататором. Доказана его способность уменьшать обструкцию дыхательных путей при приступах астмы и снижать степень
тяжести и длительность отрыва от работы или учебы при хронической астме. Теофиллин-основание очень слабо растворим в воде, поэтому его применяют в виде солей, содержащих различные количества основного вещества. Две наиболее часто применяемые соли — это аминофиллин, который содержит 86 % теофиллина по массе, и окстрифиллин, содержащий 64 % теофиллина. Чтобы содержание теофиллина было эквивалентно, необходимо корректировать дозу назначаемого препарата. Большинство препаратов хорошо всасываются из желудочно-кишечного тракта, пища не оказывает значительного влияния на их абсорбцию. Всасывание теофиллина из ректальных свечей ненадежно, поэтому суппозитории применяют только в особых ситуациях.
Совершенствование препаратов теофиллина происходит путем изменений физических характеристик препарата, а не его химической структуры. Например, несколько фармацевтических компаний производят сейчас безводный теофиллин в микрокристаллической форме, которая, благодаря увеличению площади поверхности, улучшает растворимость препарата и создает условия для полной и быстрой абсорбции после перорального приема. Кроме того, появились препараты пролонгированного действия (например, Сло-Филлин, Тео-Дур), способные поддерживать терапевтические концентрации теофиллина в крови до 12 часов. Они позволяют уменьшить частоту приема, снизить колебания концентрации в крови и во многих случаях более эффективно лечить ночной бронхоспазм.
Важное преимущество использования теофиллина — возможность мониторирования его уровня в крови для контроля терапии. Терапевтические и токсические эффекты теофиллина связаны с его концентрацией в плазме. Улучшение легочной функции наблюдается при содержании препарата в плазме в пределах 5-20 мг/л. Анорексия, тошнота, рвота, абдоминальный дискомфорт, головные боли, тревожность появляются у некоторых больных при концентрации 15 мг/л и становятся обычными побочными эффектами при уровне теофиллина, превышающем 20 мг/л. Еще более высокие концентрации препарата (> 40 мг/л) могут привести к судорогам или аритмиям, причем их возникновению не всегда предшествуют желудочно-кишечные или неврологические симптомы. Поэтому рациональное применение теофиллина требует знания его фармакокинетики.
Как описано в главе 3, начальная (нагрузочная) доза препарата зависит от объема его распределения (VD), а поддерживающая — от скорости выведения из плазмы. VD теофиллина пропорционален массе тела и практически не зависит от других факторов. В среднем VD = 0.5 л/кг, то есть в плазме можно достигнуть концентрации 10 мг/л нагрузочной дозой теофиллина 5 мг на кг массы тела. Так как аминофиллин содержит 86 % теофиллина, это приблизительно соответствует 6 мг/кг аминофиллина. При внутривенном введении нагрузочная доза дается в течение 30 минут. Более быстрое введение может привести к возникновению временных токсических концентраций препарата в плазме, способных вызвать судороги или аритмии.
Установлено, что фармакокинетика теофиллина очень индивидуальна, особенно — у больных астмой. Так как теофиллин метаболизируется в печени, нарушения ее функции могут изменить период полувыведения препарата. Например, снижение функции печени вследствие цирроза или сердечной недостаточности может замедлить выведение препарата и привести к возникновению токсических концентраций в крови. Индукция ферментов печени при курении или изменениях в диете может повысить клиренс и затруднить достижение нужных концентраций препарата. У здоровых взрослых людей клиренс в среднем равен 0.69 мл/кг/мин (0.041 л/кг/ч). У детей выведение теофиллина идет быстрее (1-1.5 мл/кг/мин; 0.06-0.09 л/кг/час). У новорожденных и детей первых двух лет жизни клиренс самый медленный (глава 61). Даже несмотря на коррекцию поддерживающих доз, концентрация препарата в плазме значительно варьирует.
Внутривенная поддерживающая терапия теофиллином в настоящее время назначается только в особых ситуациях, так как он не усиливает значительно бронходилатацию, достигаемую частой ингаляцией симпатомиметиков. В тех случаях, когда такая терапия все же назначается, у пациентов в стабильном состоянии скорость инфузии должна быть 0.7 мг/кг/ч, а при острых состояниях ее снижают приблизительно до 0.6 мг/кг/ч. В случаях заболевания печени или сердечной недостаточности дозу необходимо снизить еще больше (приблизительно 0.3 мг/кг/ч). Уровень теофиллина в плазме следует измерить через 24 часа после начала лечения. При пероральной терапии препарат назначается для приема каждые 6 часов в начальной дозе, эквивалентной 3-4 мг/кг теофиллина. Изменения
дозы приведут к новой сбалансированной концентрации через 1-2 дня, поэтому дозу можно повышать каждые 2-3 дня до достижения терапевтической концентрации (10-20 мг/л) или до возникновения побочных эффектов.
Симпатомиметики
Агонисты адренорецепторов, в деталях обсуждаемые в главе 9, имеют несколько фармакологических свойств, важных для лечения астмы: они расслабляют гладкую мускулатуру дыхательных путей и ингибируют высвобождение веществ, вызывающих бронхоспазм, из тучных клеток. Адреномиметики могут также повышать мукоцилиарный транспорт, увеличивая цилиарную активность или изменяя состав слизистого секрета. Как и в других тканях, в легких р-агонисты стимулируют адени-латциклазу и образование цАМФ.
Наиболее детально изучено действие агонистов адренорецепторов на дыхательные пути. Они расслабляют гладкую мускулатуру, что приводит к бронходилатации. Несмотря на то, что нет данных о прямой симпатической иннервации гладкой мускулатуры дыхательных путей человека, есть доказательства наличия там адренорецепторов. В общих чертах, стимуляция [}2-рецепторов расслабляет гладкую мускулатуру дыхательных путей, подавляет высвобождение медиаторов и вызывает тремор скелетных мышц (последний эффект — токсический).
В терапии астмы широко используют адреналин, эфедрин, изопротеренол и несколько Р2-селектив-ных препаратов (рис.19-6). Так как адреналин и изопротеренол вызывают значительную кардиостимуляцию (опосредованную активацией Pi-рецепторов), их следует применять только в определенных случаях. Единственное преимущество эфедрина — его низкая стоимость, но этого препарата надо по возможности избегать.
Адреналин (эпинефрин) — это эффективный, быстродействующий при подкожном (0.4 мл 1 : 1000 раствора) или ингаляционном введении (в виде микроаэрозоля в контейнере под давлением, 320 мкг на ингаляцию) бронходилататор. Максимальная бронходилатация достигается через 15 минут после ингаляции и длится 60-90 минут. Так как адреналин стимулирует не только р2-, но и Ргрецепторы, могут возникать такие нежелательные побочные эффекты как тахикардия, аритмии и ухудшение течения стенокардии.
Эфедрин, возможно, имеет самую долгую историю применения из всех противоастматических препаратов: в Китае его использовали в течение 2000 лет до того как ввели в употребление в западной медицине в 1924 г. По сравнению с адреналином эфедрин действует дольше, активен при пероральном приеме, имеет больше центральных эффектов и значительно менее эффективен. Вследствие разработки более эффективных средств, в частности р2-селективных агонистов, эфедрин сейчас редко применяют при астме. Раньше его назначали в стандартных комбинациях вместе с теофиллином и седативным средством в составе коммерческих препаратов. Эти стандартные комбинации делают невозможным изменение концентрации отдельных компонентов, поэтому следует избегать их применения.
Изопротеренол — эффективный бронходилата-тор. При ингаляционном приеме в виде микроаэрозоля из контейнера под давлением в дозе 80-120 мкг он вызывает максимальную бронходилатацию в течение 5 минут. Если для ингаляции используют распылитель, то из-за получения более крупных аэро
зольных частиц препарата требуется большая доза для достижения того же эффекта. Действие изопротеренола длится 60-90 минут. Существует мнение, что причиной повышения смертности больных астмой от сердечных аритмий в Великобритании в 1960-х гг. является применение высоких доз ингаляционного изопротеренола. Однако по поводу этого заключения имеются разногласия.
Р2-селективные препараты
Р2-селективные агонисты адренергических рецепторов — это самые широко применяемые в настоящее время симпатомиметики для лечения астмы (рис. 19-6).
Они эффективны при пероральном приеме, действуют долго и обладают значительной р2-селектив-ностью. Эти препараты структурно отличаются от адреналина по наличию более крупных замещающих группировок в аминогруппе и по положению гидроксильных групп в ароматическом кольце. Метапротеренол, альбутерол, тербуталин и битол-терол выпускаются в виде дозированных ингаля
Изопротеренол
Изоэтарин
Альбутерол (сальбутамол)
НО
Метапротеренол
Рис. 19-6. Структура изопротеренола и некоторых р2-селективных агонистов
торов. Для применения в ручном распылителе разводят 0.3-0.5 мл раствора метапротеренола (5 %) или раствора альбутерола (0.5 %) в 0.3-1.5 мл физиологического раствора. При ингаляционном приеме эти препараты дают бронхорасширяющий эффект, эквивалентный действию изопротеренола. Максимальная бронходилатация наступает через 30 минут и длится 3-4 часа.
Метапротеренол и тербуталин производятся и в таблетированной форме. Обычно назначают по одной таблетке 3 раза в день. Предотвратить основные побочные эффекты — мышечный тремор, нервозность, периодическую слабость — помогает прием половинной дозы препарата в течение первых двух недель терапии.
Только тербуталин выпускается в форме для подкожных инъекций (0.25 мг). Показания к этому способу применения такие же, как для адреналина — тяжелая астма, требующая лечения под наблюдением врача. Однако следует помнить, что большая продолжительность действия тербуталина может привести к кумулятивным эффектам после нескольких инъекций.
К новым р2-селективным агонистам относятся формотерол и сальметерол. Они были разработаны с целью повышения продолжительности действия (12 часов и выше) по сравнению с созданными ранее (4-6 часов). Оба этих препарата - высокоселективные и очень эффективные р2-агонисты; в некоторых тканях сальметерол выступает как парциальный агонист Р-рецепторов. Длительность действия этих препаратов связана с их высокой жирорастворимостью, а не с резистентностью к метаболической деградации. Жирорасгворимость позволяет им присутствовать в мембране гладкомышечных клеток в высоких концентрациях. Считается, что эти растворенные формы являются депо, из которого препарат медленно высвобождается и доставляется к Р рецепторам в течение продолжительного времени (Anderson, 1993).
NHCHO
Формотерол
Хотя агонисты адренорецепторов можно назначать ингаляционно, парентерально или перораль
но, ингаляционное введение приводит к максимальному местному эффекту на гладкую мускулатуру дыхательных путей с наименьшей системной токсичностью. Доставка аэрозоля к месту действия зависит от размера частиц, особенностей дыхания пациента (дыхательного объема и частоты дыхания) и геометрии его дыхательных путей. Даже если частицы оптимальны по размерам (2-5 мкм), 80-90 % от общей дозы аэрозоля остается в ротовой полости или глотке. Частицы меньше 1-2 мкм остаются в выдыхаемом воздухе. Эффективность попадания препарата увеличивается, если задержать дыхание-на вдохе.
Нежелательные эффекты р-агонистов. Ингаляционное применение симпатомиметиков с самого начала вызывало беспокойство по поводу развития тахифилаксии или толерантности к р-агонистам, аритмий из-за стимуляции Pi-рецепторов, а также гипоксии и аритмий как следствия содержания фторированных углеводородов во фреоновом пропелленте.
До сих пор не доказано, что р-агонисты могут вызвать ухудшение клинического течения астмы вследствие развития тахифилаксии. В большинстве исследований обнаружено только незначительное изменение реактивности гладкой мускулатуры дыхательных путей в ответ на p-стимуляцию. Однако результаты одного хорошо спланированного исследования с использованием контрольной группы больных показали, что регулярное применение ингаляционного формотерола приводит к некоторому усилению астматической симптоматики, в том числе реактивности бронхов, по сравнению с состоянием больных, получавших препарат только по необходимости. Другое эпидемиологическое исследование выявило связь между использованием двух или более аэрозольных упаковок фенотерола в месяц и риском смерти или критического состояния вследствие астмы (Spitzer, 1992). Осталось неясным, было ли применение Р-агонистов фактором риска или просто маркером тяжести заболевания. Более поздний сравнительный анализ нескольких публикаций не позволил найти корреляцию между риском смерти от астмы и пероральным или ингаляционным применением р-агонистов (Mullen, 1993).
Было продемонстрировано также, что после введения р-агонистов может снижаться напряжение кислорода в артериальной крови (Ро2), если ухудшается соотношение вентиляция/перфузия в легком. Однако этот эффект незначителен и может
возникнуть при применении любого бронходилата-тора. Кроме того, его выраженность зависит от исходного Ро2 пациента. Может потребоваться назначение кислорода, если Ро2 исходно снижено или значительно понижается за время лечения бронхо-дилататорами.
Вернемся к обсуждению возможности токсического воздействия на миокард фреоновых пропеллентов, содержащихся во всех коммерческих дозированных ингаляторах. Хотя фторуглеродные соединения могут сенсибилизировать сердце к токсическим эффектам катехоламинов, этот эффект наблюдается только при создании в миокарде очень высоких концентраций, которые не достигаются при пользовании дозированными ингаляторами, если соблюдать назначения врача. В целом антагонисты р2-адренорецепторов — это сравнительно безопасные и эффективные бронходилататоры при применении в дозах, позволяющих избежать системных побочных эффектов.
Антагонисты мускариновых рецепторов
В течение сотен лет для лечения астмы использовались листья Datura stramonium. Интерес к антагонистам мускариновых рецепторов повысился после установления роли вагусных влияний на формирование бронхоспазма у лабораторных животных и создания эффективного антагониста мускариновых рецепторов, который плохо всасывается после ингаляционного применения и, следовательно, не имеет системных атропиноподобных эффектов.
Механизм действия
Антагонисты мускариновых рецепторов конкурентно ингибируют эффект ацетилхолина (глава 8). В дыхательных путях ацетилхолин, высвобождаясь из эфферентных вагусных окончаний, вызывает сокращение гладких мышц и повышение секреции слизи. Для ингибирования реакции гладкой мускулатуры на немускариновую стимуляцию требуются очень высокие концентрации препаратов, гораздо более высокие, чем достигаемые при назначении максимальных доз. Такая селективность антагонистов мускариновых рецепторов обусловливает их использование для определения роли парасимпатических механизмов в формировании бронхиального ответа, но ограничивает их применение для про
филактики бронхоспазма. В обычных дозах антагонисты мускариновых рецепторов подавляют только ту часть реакции, которая опосредована мускариновыми рецепторами; роль парасимпатической нервной системы в формировании бронхоспазма существенно варьирует у разных людей.
Клиническое применение антагонистов мускариновых рецепторов
Антагонисты мускариновых рецепторов — эффективные бронходилататоры. При внутривенном введении атропин, прототип этой группы лекарств (глава 8), вызывает бронходилатацию в меньших дозах, чем необходимые для повышения частоты сердечных сокращений. Селективность атропина можно повысить путем ингаляционного введения. Исследования эффективности аэрозоля атропина сульфата показали, что он расширяет исходный диаметр бронхов почти так же, как Р-агонисты, и что это действие длится до 5 часов. У пациентов с эмфиземой эффективность атропина выше, чем р-агонистов. Необходимая доза зависит от размера частиц аэрозоля. При использовании аэрозоля с диаметром частиц 1-1.5 мкм системные эффекты (задержка мочи, тахикардия, нарушение зрительной аккомодации, моторное возбуждение) обычно возникают при вдыхании 2 мг. Таким образом, исходная доза должна быть не больше 1 мг. Оседание части аэрозоля в полости рта нередко приводит к ощущению сухости.
Доза антагонистов мускариновых рецепторов, максимально увеличивающая калибр дыхательных путей в нормальном состоянии, ниже, чем доза, необходимая для подавления бронхоспазма (рис. 19-7), вероятно потому, что в покое выделяется меньшее количество ацетилхолина, чем в ответ на вдыхание ирританта. Это важно знать при лечении больных астмой, так как заболевание характеризуется эпизодическими приступами бронхоспазма, часто в ответ на физическую нагрузку или вдыхание антигенов или ирритантов.
Системные побочные эффекты ограничивают применяемые дозы атропина сульфата. Более селективный препарат ипратропиума бромид — четвертичное аммонийное производное атропина, может использоваться в высоких ингаляционных дозах, так как плохо всасывается и плохо проникает в ЦНС. Изучение этого препарата показало, что степень участия парасимпатических механизмов в развитии
Рис. 19-7. Подавление бронхомоторного ответа на диоксид серы у больного астмой аэрозолями двух дозировок атропина сульфата и аэрозолем метапротеренола. Специфическая резистентность дыхательных путей была измерена через 5 минут эукапнической гипервентиляции (40 л/мин) фильтрованного воздуха с повышенным содержанием SO2. Отметьте, что все препараты вызывают одинаковое изменение исходной специфической резистентности, но разные степени подавления реакции на SO2 бронхоспазма у разных людей варьирует. У некоторых пациентов препарат эффективно подавляет бронхоспазм, у других же — весьма незначительно. То, что введение высоких доз антагонистов мускариновых рецепторов не всегда купирует бронхоспазм, свидетельствует о вовлечении в процесс не только парасимпатической нервной системы. Даже для пациентов, на которых ипратропиума бромид оказывает незначительное действие, степень вызываемой бронходилатации и подавления спровоцированного бронхоспазма могут иметь потенциальное клиническое значение. Препарат может применяться при толерантности к ингаляционным Р-агонис-там. Хотя антагонисты мускариновых рецепторов хуже Р-агонистов подавляют астматический бронхоспазм, они одинаково эффективны (иногда м-хо-линолитики даже эффективнее) у больных с хроническими обструктивными заболеваниями легких, которые имеют частично обратимый компонент.
Кортикостероиды
Несмотря на то, что кортикостероиды применяют в терапии астмы с 1950 г., точный механизм их действия остается неизвестным. Как и кромолин, эти препараты не оказывают прямого расслабляю
щего действия на гладкую мускулатуру дыхательных путей, но они могут значительно увеличивать диаметр дыхательных путей при применении у больного астмой в течение некоторого времени. Результаты ряда исследований позволили предположить, что кортикостероиды уменьшают обструкцию дыхательных путей, потенцируя действие Р-агонистов на Р-рецепторы. Недавние исследования показали, что эти препараты подавляют или модифицируют воспалительный процесс в дыхательных путях. Например, кортикостероиды могут снижать высвобождение арахидоновой кислоты из клеточных мембран, ингибируя тем самым первый этап ее метаболизма — образование эйкозаноидов. Некоторые из этих веществ существенно влияют на функцию дыхательных путей и, может быть, являются причиной нарушения дыхания у больных астмой (глава 18). Действительно, у небольшого процента астматиков острый бронхоспазм может быть вызван приемом аспирина или других нестероидных противовоспалительных препаратов, которые подавляют активность циклооксигеназы, переключая метаболизм арахидоновой кислоты на путь синтеза лейкотриенов.
Клиническое применение кортикостероидов
Хотя эффективность кортикостероидов при лечении астмы очевидна, сохраняются противоречия относительно показаний для гормональной терапии и правил дозирования препаратов. Вследствие серьезных побочных эффектов, возникающих при постоянном приеме, пероральные кортикостероиды назначают только пациентам, у которых отсутствует адекватный ответ на бронходилататоры или при ухудшении состояния, несмотря на поддерживающую терапию бронходилататорами. Лечение обычно начинают с пероральной дозы 30-60 мг преднизолона в день или внутривенной дозы 1 мг/кг метилпреднизолона каждые 6 часов, после облегчения обструкции дыхательных путей общую дневную дозу постепенно снижают. У большинства пациентов терапия кортикостероидами может быть прекращена через неделю или 10 дней, но иногда при снижении дозы наблюдается ухудшение. Так как угнетение надпочечников кортикостероидами зависит от их дозы, а секреция гормонов колеблется в течение дня, ранний утренний прием небольших доз препаратов позволяет уменьшить опасный
эффект. Еше меньшее влияние на надпочечники наблюдается при приеме кортикостероидов через день. К этой схеме лучше всего переходить, постепенно снижая дозы одного дня и одновременно на столько же повышая дозы другого дня.
Самый эффективный метод снижения системных побочных эффектов кортикостероидной терапии — введение препарата в форме аэрозоля. Применение жирорастворимых кортикостероидов, таких как беклометазон, триамцинолон, будесонид и флунизолид, позволяет доставить кортикостероиды в дыхательные пути, минимизируя системную абсорбцию и побочные эффекты. Средняя дневная доза (два вдоха 4 раза в день или четыре вдоха 2 раза в день) так же эффективна для лечения больных астмой легкой и средней степени тяжести, как и пероральные стероиды. Она эквивалентна 10-15 мг/день преднизолона по уровню контроля за состоянием больного и при этом вызывает меньше системных эффектов. Более высокие дозы ингаляционных стероидных препаратов еще эффективнее: для замены ежедневной пероральной терапии кортикостероидами в высоких дозах назначают по 8-12 ингаляционных доз 4 раза в день. Хотя ингаляционные стероиды в таких дозах тоже могут вызывать системные эффекты, они значительно менее выражены, чем при пероральном приеме. Особая проблема, возникающая при частых ингаляциях — это орофарингеальный кандидоз. Риск его развития можно снизить, если рекомендовать пациенту после каждой ингаляции полоскать рот и горло водой. Других осложнений эти препараты, как правило, не дают, но при переходе с перорального приема на ингаляции беклометазона пероральную дозу надо снижать очень медленно, чтобы не вызвать обострения надпочечниковой недостаточности.
Постоянное применение ингаляционных стероидов эффективно уменьшает проявление симптомов заболевания и улучшает легочную функцию у больных астмой легкой степени тяжести. Такая терапия снижает или полностью снимает необходимость в пероральных стероидах у более тяжелых больных. В отличие от Р-агонистов и теофиллина ингаляционные кортикостероиды при их длительном применении снижают реактивность бронхов. Этот важный эффект, который обычно проявляется через 2-4 недели, зависит и от дозы, и от продолжительности лечения. У некоторых пациентов реактивность бронхов снижается очень значительно, но максимум ее снижения иногда достигается только к девятому-
двенадцатому месяцам лечения. Вследствие эффективности и безопасности ингаляционных стероидов в настоящее время их часто назначают пациентам, которым для снятия симптомов недостаточно ингаляций Р-агониста. Некоторые врачи считают глюкокортикоиды препаратами выбора на первом этапе лечения больных легкой формой астмы (при необходимости — в комбинации с Р-агонистом). Лечение проводят в течение десяти-двенадцати недель и затем постепенно отменяют, чтобы определить, нужна ли более продолжительная терапия.
Другие средства, применяемые при астме
Блокаторы кальциевых каналов
Каждая из клеточных функций, которая может нарушаться у больных бронхиальной астмой (сокращение гладкой мускулатуры дыхательных путей, секреция слизи и различных медиаторов, иннервация бронхо-легочного аппарата), зависит в некоторой степени от движения ионов кальция в клетках. Блокаторы кальциевых каналов не влияют на исходный диаметр дыхательных путей, но значительно снижают степень бронхоспазма, который возникает в ответ на различные стимулы. Было показано, что пациентам с бронхоспазмом, возникшим в ответ на физическую нагрузку, гипервентиляцию или ингаляцию аэрозоля гистамина, метахолина или антигена, помогают ингаляции верапамила или нифедипина. Но только в одном исследовании был продемонстрирован полный защитный эффект. В других случаях обнаруживается лишь частичная защита и варьирование этого эффекта у разных людей. Причины недостаточной эффективности блокаторов кальциевых каналов пока не ясны.
Антагонисты лейкотриенов и ингибиторы липооксигеназы
Сведения об участии лейкотриенов в патогенезе многих воспалительных заболеваний (глава 18) и в развитии анафилаксии позволили начать разработку препаратов, которые блокируют синтез этих дериватов арахидоновой кислоты или их рецепторы. Несколько антагонистов лейкотриенов на ранних стадиях клинических испытаний показали определенный положительный эффект у больных астмой (Cloud, 1989; Israel, 1993). В некоторых исследова
ниях доказано, что подобные лекарства блокируют как ранний, так и поздний компоненты астматического ответа на провокацию антигеном (Freidman, 1993). Изучение этих препаратов продолжается.
Доноры оксида азота
Предварительные исследования на животных позволили предположить, что гладкая мускулатура дыхательных путей и сосудов эффективно расслабляется под действием оксида азота. Этот очень липофильный препарат можно назначать при приступах астмы в виде ингаляций газа: он расслабляет не только гладкую мускулатуру дыхательных путей, но и сосуды легких. Несмотря на то, что оксид азота и его доноры могут оказаться эффективными при астме, наиболее вероятно их применение для лечения легочной гипертензии.
Препараты, открывающие калиевые каналы
Кромакалим — это находящийся на испытаниях препарат, имеющий сосудорасширяющее действие, частично предписываемое блокаде а-адренорецепторов, а частично — прямой гиперполяризации мембран гладкомышечных клеток путем-активации калиевых каналов. Полагают, что сходная гиперполяризация возможна и в гладкомышечных клетках дыхательных путей. Хотя in vitro такая релаксация гладкой мышцы легко демонстрируется, результаты исследований на больных астмой противоречивы (Williams, 1990; Kidney, 1993).
II. Клиническая фармакология средств, используемых для лечения астмы
Бронходилататоры
Больным астмой легкой степени с непостоянными симптомами обычно достаточно агониста Р-рецепторов (метапротеренола или альбутерола), принимаемого по мере необходимости. Если симптомы возникают более часто и пациенту требуются частые ингаляции аэрозоля или при необходимости дополнительного лечения ночных приступов,
предпочтение должно отдаваться ингаляционным противовоспалительным препаратам, таким как кромолин или ингаляционный кортикостероид. Теофиллин следует назначать пациентам, у которых симптомы болезни не удается контролировать даже при помощи комбинации ингаляционного кортикостероида и р2-агониста. Если добавление теофиллина не улучшает состояние пациента или возникают серьезные побочные эффекты, очень важно проверить уровень теофиллина в плазме, чтобы выяснить, находится ли он в допустимых терапевтических пределах (10-20 мг/л).
Кортикостероиды
Если обструкция дыхательных путей остается тяжелой, несмотря на терапию бронходилататора-ми, можно назначить пероральные кортикостероиды. После начального курса лечения высокими дозами (например, 30 мг/день преднизолона в течение 3 недель) следует постепенно перейти к лечению минимальной поддерживающей дозой, позволяющей контролировать течение заболевания. По возможности пероральную терапию следует затем заменить на ингаляционную или, если это невозможно, продолжить пероральное введение в минимальных дозах. В последнем случае ежедневный прием следует перевести на прием через день.
Многочисленные недавние исследования показали, что ингаляционные кортикостероиды должны рассматриваться как препараты выбора на первом этапе лечения больных с только что возникшей астмой, так как они также эффективны и менее токсичны, чем р2-агонисты (Juniper, 1990,1991; Haah-tela, 1991; Guidelines, 1991). За 1-2 года наблюдений у пациентов, которые получали местный кортикостероид- будесонид, было меньше приступов астмы и меньше токсических проявлений, чем в группе сравнения, получавшей ингаляционные Р-агонисты.
Кромолин и недокромил
Кромолин и недокромил следует назначать пациентам, которым необходима поддерживающая терапия пероральным теофиллином и ингаляционным препаратом, а также пациентам, у которых астматические симптомы возникают сезонно или после воздействия определенных факторов (физической нагрузки, шерсти животных, других ирритантов). У па
циентов с постоянными или возникающими без очевидной причины симптомами эффективность этих препаратов может быть проверена только назначением четырехнедельного курса ингаляций по 4 раза в день. Если эта терапия эффективна, дозу можно снизить. Поддерживающая терапия кромолином также эффективна, как и поддерживающая терапия теофиллином, и широко применяется у детей.
Антагонисты мускариновых рецепторов
Роль антагонистов мускариновых рецепторов в терапии астмы не определена. При назначении адекватных доз их действие на исходную резистентность дыхательных путей почти равно по силе эффекту симпатомиметиков. Эффекты полных доз антагонистов мускариновых рецепторов и симпатомиметиков не суммируются, то есть у пациента, получавшего антагонист мускариновых рецепторов, не возникает дополнительной бронходилатации при назначении симпатомиметиков. Применение препаратов в обратном порядке тоже не приводит к усилению эффекта: добавление антагониста мускариновых рецепторов только незначительно увеличивает эффект предварительной терапии симпатомиме-тиками. Антагонисты мускариновых рецепторов, возможно, играют большую роль в лечении людей с хроническими обструктивными заболеваниями легких, чем в терапии бронхиальной астмы.
При длительном применении антагонисты мускариновых рецепторов вызывают эффективную бронходилатацию. Хотя предполагалось, что антагонисты мускариновых рецепторов будут “высушивать” секрецию в дыхательных путях, прямые измерения объема секрета одной железы подслизистой оболочки дыхательных путей животных показали, что атропин лишь незначительно снижает секрецию. Однако этот препарат предотвращает избыточную секрецию в ответ на рефлекторную вагусную стимуляцию. Случаев сгущения секрета вследствие введения этих препаратов не описано.
Противовоспалительная терапия
Результаты недавних исследований позволили предположить, что препараты, применяемые для лечения больных ревматоидным артритом, могут
использоваться в терапии хронической стероид-зависимой астмы. Появление альтернативного способа лечения очень важно, так как хроническое лечение кортикостероидами может вызвать остеопороз, катаракту, снижение толерантности к глюкозе, ухудшение течения артериальной гипертензии и изменения внешности по типу синдрома Кушинга. Два двойных слепых проспективных исследования больных преднизолонзависимой астмой показали, что использование метотрексата (15-50 мг/неделю) позволило снизить дозу преднизолона (Mullarkey, 1990; Shiner, 1990). Но получены и противоположные результаты (Erzurum, 1991). В открытом исследовании двадцати больных преднизолонзависимой астмой было обнаружено, что пероральное применение 3 мг золота (ауранофин) дважды в день в течение 24 недель снижало частоту приступов, реактивность бронхов и потребность в преднизолоне. Эти обещающие результаты дают надежду небольшой группе пациентов, у которых астма может контролироваться только высокими дозами перорального преднизолона. Однако до того, как рекомендовать этот способ лечения в медицинскую практику, необходимо провести широкомасштабные клинические испытания.
Лечение
приступов астмы
Лечение пациентов с острым приступом астмы в стационарах требует большего количества объективных исследований легочной функции и большей продолжительности наблюдения, чем при поликлиническом ведении больных хронической бронхиальной астмой. Обычно необходимы повторные измерения напряжения газов в артериальной крови и спирометрия. У пациентов с легкими астматическими приступами ингаляция р-агонистов так же эффективна, как и подкожная инъекция адреналина. Оба метода лечения более эффективны, чем внутривенное введение аминофиллина. Тяжелые приступы требуют ингаляции кислорода, назначения бронходилататоров и кортикостероидов, например комбинации подкожного или аэрозольного р2-агониста, продолжительного внутривенного вливания аминофиллина и внутривенных кортикостероидов.
Препараты
Симпатомиметики для лечения астмы
Альбутерол (Провентил, Вентолин, генерик) Ингаляционно: 90 мкг аэрозоля на ингаляцию; 0.083, 0.5 % раствор для распылителя; капсулы 200 мкг для ингаляций Перорально: таблетки по 2,4 мг; сироп 2 мг/5 мл
Перорально для пролонгированного действия (Провентил Репетабс): таблетки по 4 мг
Битолтерол (Торналат)
Ингаляционно: 0.37 мг аэрозоля на ингаляцию в 300-дозовом контейнере
Эфедрин (генерик)
Перорально: капсулы 25, 50 мг
Парентерально: 25, 50 мг/мл для инъекций
Эпинефрин (генерик, Адреналин, др.) Ингаляционно: 1,1.25, 2.25 % для распылителя; 0.16, 0.2, 0.27 мг аэрозоль эпинефрина в 233- и 350-дозовом контейнере Парентерально: 1: 1000 (1 мг/мл), 1: 200 (5 мг/мл) для инъекций
Этилнорадреналин (Бронкефрин)
Парентерально: 2 мг/мл для инъекций
Изоэтарин (генерик, др.)
Ингаляционно: 0.062, 0.08, 0.1, 0.125, 0.167, 0.17, 0.2, 0.25, 1 % для распылителя;
340 мкг аэрозоля на ингаляцию
Изопротеренол (генерик, Изупрел, др.) Ингаляционно: 0.25, 0.5, 1 % для распылителя; 80,131 мкг аэрозоля на ингаляцию Перорально: таблетки под язык по 10,15 мг Парентерально: 0.2 мг/мл для инъекций
Метапротеренол (Алупент, Метапрел, генерик) Ингаляционно: 0.65 мг аэрозоля на ингаляцию в 75,150 мл контейнерах; 0.6,5 % для распылителя;
Перорально: таблетки по 10, 20 мг; сироп 10 мг/5мл
Пирбутерол (Максаир)
Ингаляционно: 0.2 мг аэрозоля на ингаляцию в 300-дозовых контейнерах
Сальметерол (Серевент)
Ингаляционно: 42 мкг аэрозоля на ингаляцию в 120-дозовых контейнерах
Тербуталин (Бретин, Бриканил) Ингаляционно: 0.2 мг аэрозоля на ингаляцию Перорально: таблетки по 2.5, 5 мг Парентерально: 1 мг/мл для инъекций
Антагонисты мускариновых рецепторов для лечения астмы
Ипратропиум (Атровент)
Аэрозоль: 18 мкг аэрозоля на ингаляцию в 200-дозовых контейнерах
Кромолин натрия и недокромил натрия
Кромолин натрия
Легочный аэрозоль (Интал): 800 мкг аэрозоля на ингаляцию в 200-дозовых контейнерах; капсулы 20 мг для ингаляций;
20 мг/2 мл для распылителя Назальный аэрозоль (Назалкром):
5.2 мг на ингаляцию (при сенной лихорадке) Перорально (Гастрокром): капсулы 100 мг (при пищевой аллергии)
Недокромил натрия (Тайлед)
Легочный аэрозоль: 1.75 мг аэрозоля на ингаляцию в 112-дозовых контейнерах
Аэрозоли кортикостероидов
(см. также главу 38).
Беклометазон (Бекловент, Ванцерил)
Аэрозоль: 42 мкг аэрозоля на ингаляцию в 200-дозовых контейнерах
Дексаметазон (Декадрона фосфат Респихалер) Аэрозоль: 84 мкг аэрозоля на ингаляцию в 170-дозовых контейнерах
Флунизолид (АэроБид)
Аэрозоль: 250 мкг аэрозоля на ингаляцию в 50-дозовых контейнерах
Триамцинолон ацетонид (Азмакорт)
Аэрозоль: 100 мкг аэрозоля на ингаляцию в 240-дозовых контейнерах
Метилксантины:
Теофиллин и производные
Аминофиллин (Теофиллин этилендиамин, 79 % теофиллина) (генерик, др.) Перорально: таблетки по 100, 200 мг; жидкая форма 105 мг/5 мл Перорально для пролонгированного действия: таблетки по 225 мг
Ректально: свечи 250, 500 мг
Парентерально: 250 мг/10 мл для инъекций
Окстрифиллин, теофиллината холин (64 % теофиллина) (генерик, Холедил) Перорально: таблетки по 100, 200 мг; сироп 50 мг/5 мл; эликсир 100 мг/5 мл Перорально для пролонгированного действия: таблетки 400, 600 мг
Теофиллин (генерик, Эликсофиллин, Сло-Филлин, Унифил, Тео-Дур, Тео-24, др.) Перорально: таблетки по 100, 125, 200, 250, 300 мг; капсулы 100, 200 мг; сиропы, эликсиры и растворы 26.7, 50 мг/5 мл Перорально для пролонгированного действия, 8-12 часов: капсулы 50, 60, 65, 75, 100,125, 130, 200, 250, 260, 300 мг Перорально для пролонгированного действия, 8-24 часа: таблетки по 100,200, 300, 450 мг
Перорально для пролонгированного действия, 12 часов: капсулы 50, 75, 125,130, 200, 250, 260 мг
Перорально для пролонгированного действия, 12-24 часа: таблетки по 100, 200, 250, 300, 450, 500 мг
Перорально для пролонгированного действия, 24 часа: таблетки и капсулы по 100, 200, 300, 400 мг
Парентерально: 200, 400,800 мг в контейнере, теофиллин и 5 % глюкоза для инъекций
Другие метилксантины
Дифиллин (генерик, др.)
Перорально: таблетки по 200,400 мг; эликсир 33.3, 53.3 мг/5 мл Парентерально: 250 мг/мл для инъекций
Пентоксифиллин (Трентал)
Перорально: таблетки пролонгированного действия по 400 мг
Избранная литература
Anderson G. Р. Formoterol: Pharmacology, molecular basis of agonism amd mechanism of long duration of a highly potent and selective p2-adrenoreceptor agonist bronchodilator. Life Sci. 1993; 52:2145.
Barnes P. J. A new approach to the treatment of asthma. N. Engl. J. Med. 1989; 321:1517.
Benowitz N. Clinical pharmacology of caffeine. Annu. Rev. Med. 1990; 41: 277.
Bergman К-Ch., Bauer С. P., Overlack A. A placebo-controlled blinded comparison of nedocromil sodium and beclomethasone dipropionate in bronchial asthma. Lung, 1990; 168 (Suppl.): 230.
Bigby T. D., Nadel J. A. Asthma. Chapter 44. In: Inflammation: Basic Principles and Clinical Correlations. Gallin J. L, Goldstein I. M. (eds) Raven, 1992.
Brogden R. N., Sorkin E. M. Nedochromil sodium: An updated review of its pharmacological properties and therapeutic efficacy in asthma. Drugs, 1993; 45: 693.
Check W. A., Kaliner M. A. Pharmacology and pharmacokinetics of topical corticosteroids used for asthma therapy. Am. Rev. Respir. Dis. 1990; 141 (Suppl.): S44.
Dupuy P. M. et al. Bronchodilator action of inhaled nitric oxide in guinea pigs. J. Clin. Invest. 1992; 90: 421.
Garty M. et al. Effect of nifedipine and theophylline in asthma. Clin. Pharmacol. Ther. 1986; 40:195.
Gross N.J. Ipratropium bromide. N. Engl. J. Med. 1988; 319: 486.
Hirsh K. Central nervous system pharmacology of the dietary methylxanthines. In: The Methylxathines Beverages and Foods: Chemistry, Consumption and Health Effects. Spiller G. A. (ed.) Alan Liss, 1984.
Ozenne G. et al. Nifedipine in chronic bronchial asthma: A randomized double-blind crossover trial against placebo. Eur. J. Respir. Dis. 1985; 67: 238.
Persson C. G. A. The pharmacology of anti-asthmatic xanthines and the role of adenosine. Asthma Rev. 1987; 1: 67.
Schleuter D. P. Ipratropium bromide in asthma: A review of the literature. Am. J. Med. 1986; 81 (Suppl. 5A): 55.
Shiner R. L. et al. Randomized, double-blind, placebo-controlled trial of methotrexate in steroiddependent asthma. Lancet, 1990; 336:137.
Spitzer W. O. et al. The use of p2-agonists and the risk of death and near death from astma. N. Engl. J. Med. 1992; 326: 501.
Раздел V
Средства, влияющие
на центральную нервную систему (ЦНС) Введение в фармакологию препаратов, влияющих на ЦНС
20
Роджер А. Николл
Средства, воздействующие на центральную нервную систему (ЦНС), были среди первых, открытых первобытными людьми, и до сих пор являются наиболее широко применяемой группой препаратов. Кроме использования в терапии, препараты, влияющие на ЦНС, применяются в безрецептурных формах для поддержания жизненного тонуса. Кофеин, алкоголь и никотин социально допустимы во многих странах и потребляемы во всем мире. Поскольку некоторые из этих препаратов обладают способностью вызывать привыкание и приводят к тяжелым личным, социальным и экономическим проблемам, то общественность считает необходимым контролировать их использование и доступность.
Механизмы действия различных препаратов на ЦНС не всегда были вполне понятны. Причины многих заболеваний (шизофрения, тревожные состояния и т. д.), при которых используются эти препараты, тоже не совсем ясны, поэтому в прошлом фармакология ЦНС была большей частью описательной. Однако за два последних десятилетия произошли значительные изменения в методологии этого раздела фармакологии. В настоящее время
имеется возможность изучить влияние препарата на индивидуальные клетки и даже на отдельные ионные каналы в пределах синапсов. Информация, полученная с помощью таких исследований, легла в основу нескольких значительных открытий в области изучения ЦНС.
Во-первых, почти все препараты, обладающие центральными эффектами, действуют на специфические рецепторы, модулирующие синаптическую трансмиссию. Только некоторые средства типа общих анестетиков и алкоголя обладают неспецифическим действием на мембраны, но даже такие эффекты, не опосредованные рецепторами, приводят к значительным изменениям синаптической передачи.
Во-вторых, лекарственные препараты являются одним из самых главных инструментов исследования всех аспектов физиологии ЦНС, от механизмов возникновения судорог до долгосрочной памяти. Для таких исследований одинаково важны как агонисты, которые имитируют действие естественных медиаторов (и во многих случаях имеют большую селективность, чем эндогенные вещества), так и антагонисты.
В-третьих, выяснение механизмов действия препаратов с известной клинической эффективностью привело к возникновению самых значительных гипотез о механизмах развития заболеваний. Например, информация о действиях антипсихотических препаратов на дофаминовые рецепторы послужила основой для понимания патофизиологии шизофрении. Исследования эффектов различных агонистов и антагонистов рецепторов у-аминомасляной кислоты (ГАМК) привели к созданию новых концепций патогенеза ряда заболеваний, например тревожных состояний и эпилепсии.
В этой главе излагаются основы функциональной организации ЦНС и ее синаптических медиаторных систем, необходимые для понимания эффектов препаратов, описанных в последующих главах.
Методы изучения фармакологии ЦНС
Несмотря на то, что и ученые, и общественность всегда интересовались действием лекарственных препаратов на ЦНС, детальное описание механизмов синаптической трансмиссии было невозможным до появления стеклянных микроэлектродов, которые позволили осуществлять регистрацию внутриклеточных процессов. Детальные электрофизиологические исследования действия препаратов как на потенциалзависимые, так и на медиатор-зависимые каналы стали намного доступнее благодаря изобретению метода пэтч-кламп, который позволяет регистрировать токи через отдельные каналы. Для картирования локализации разных медиаторов, связанных с ними ферментных систем и их рецепторов широко используются гистохимические, иммунологические и радиоизотопные методы. Наиболее информативным на сегодняшний день является метод молекулярного клонирования. Он дал возможность определить точную молекулярную структуру рецепторов и связанных с ними каналов.
Ионные каналы
Мембраны нервных клеток содержат два типа каналов, классифицируемых в зависимости от механизмов, которые контролируют их открытие и закрытие. Первый механизм действует в потенциалзависимых каналах, второй - в рецепторзависи-мых (химически активируемых). Потенциалзависимые натриевые каналы, рассмотренные на примере каналов миокардиоцитов в главе 14, являются при
мером каналов первого типа и играют в ЦНС важную роль. В нервных клетках эти каналы сконцентрированы в начальном сегменте и аксоне и отвечают за быстрый потенциал действия, который передает сигнал от тела клетки к нервному окончанию. Существует много типов потенциалзависимых кальциевых и калиевых каналов на теле нейрона, дендритах, начальном сегменте, которые действуют значительно медленнее, чем натриевые, и модулируют частоту нейрональных разрядов. Например, некоторые виды калиевых каналов, открывающиеся под действием деполяризации клетки, приводят к замедлению дальнейшей деполяризации и тормозят развитие потенциала действия.
Химически активируемые каналы, которые также называются рецептор- или медиаторзависимы-ми, открываются под действием нейромедиаторов и других химических веществ. Считается, что канал — это интегральная часть рецепторного белка. Такие каналы нечувствительны или только слабо чувствительны к потенциалу мембраны. Рецепторы нейромедиаторов и их ионные каналы концентрируются на постсинаптических мембранах, например никотиновый нервно-мышечный рецептор — на концевой пластинке клеток скелетной мышцы.
Новые данные свидетельствуют о том, что традиционные представления о полной независимости потенциалзависимых и рецепторзависимых каналов требуют пересмотра. В частности, многие рецепторы нейромедиаторов связаны с потенциалзависимыми каналами через систему вторичных посредников.
Синапс и синаптический потенциал
Хорошо известно, что нейроны ЦНС взаимодействуют прежде всего через химические синапсы. (Имеется несколько примеров потенциалзависимых связей между нейронами, играющих роль в синхронизации нейрональных разрядов. Однако маловероятно, что они являются основной точкой приложения действия лекарственных препаратов.) Процесс высвобождения медиатора из пресинапти-ческого окончания был подробно изучен на примере нервно-мышечного синапса позвоночных и гигантского синапса кальмара. Потенциал действия пресинаптических волокон передается на синаптические окончания и активирует потенциалзависи-
мне кальциевые каналы на их мембране (рис. 6-3). Кальциевые каналы, отвечающие за высвобождение медиатора, обычно резистентны к действию блокаторов кальциевых каналов, обсуждаемых в главе 12 (верапамил и другие препараты), но чувствительны к некоторым токсинам морских животных и ионам металлов. Ионы кальция проникают внутрь пресинаптического окончания и инициируют слияние пресинаптических пузырьков с пресинапти-ческой мембраной. Медиатор высвобождается из пузырьков в синаптическую щель и диффундирует к рецепторам постсинаптической мембраны. Связывание медиатора с его рецептором вызывает кратковременное изменение мембранной проводимости (проницаемости для ионов) постсинаптической клетки. Промежуток времени между распространением пресинаптического потенциала действия и началом постсинаптического ответа приблизительно равен 0.5 мс. В основном эта задержка определяется временем высвобождения медиатора, связанного с открытием кальциевых каналов.
В начале 1950-х гг. Экклз с соавторами впервые провели внутриклеточную регистрацию потенциалов спинальных мотонейронов. Когда микроэлектрод попадает в клетку, регистрируется внезапное изменение потенциала, который обычно равен около -70мВ (рис. 20-1). Это потенциал покоя мембраны нейрона. На мотонейрон оказывают влияние два типа путей — возбуждающие и тормозные. Когда стимулируется возбуждающий путь, регистрируется небольшая деполяризация, или возбуждающий постсинаптический потенциал (ВПСП). Воз-
Рис. 20-1. Возбуждающие синаптические потенциалы и образование спайка. На рисунке показан потенциал покоя (-70мВ) постсинаптической клетки. Стимуляция возбуждающих путей (Е) вызывает временную деполяризацию. Повышение силы стимула (второе Е) увеличивает степень деполяризации и позволяет достигнуть порога возникновения пика деполяризации
никновение этого потенциала связано с тем, что возбуждающий медиатор вызывает значительное повышение проницаемости для ионов натрия и калия. Длительность этих потенциалов невелика, обычно менее 20 мс. Изменение интенсивности стимула и, следовательно, числа активированных пресинаптических волокон приводит к разной степени деполяризации. Это означает, что вклад отдельного волокна в ВПСП незначителен. Когда активируется достаточное количество возбуждающих волокон, ВПСП деполяризует постсинаптическую клетку до порогового значения, что вызывает возникновение потенциала действия по типу “все или ничего”.
При стимуляции тормозных путей происходит гиперполяризация постсинаптической мембраны, вызывающая возникновение тормозного постсинаптического потенциала (ТПСП) (рис. 20-2). Для значимого изменения потенциала мембраны необходима одновременная активация нескольких тормозных синапсов. Гиперполяризация в этом случае связана с повышением проницаемости для ионов хлора, проникающих в клетку во время ТПСП. Если в момент ТПСП будет вызван надпороговый ВПСП (рис. 20-2), он не приведет к возникновению потенциала действия, поскольку ТПСП смещает потенциал мембраны в обратную сторону от порога возникновения потенциала действия. Другой тип торможения называется пресинаптическим торможением. В ЦНС он встречается только в чувствительных волокнах, идущих в ствол мозга и спинной
Рис. 20-2. Взаимодействие возбуждающих и тормозных синапсов. Слева: надпороговый стимул воздействует на возбуждающие пути (Е). Справа: такой же стимул действует сразу после стимуляции тормозных путей (I), предотвращающей достижение порога возникновения потенциала действия под влиянием стимуляции возбуждающих путей
мозг. Возбуждающие синаптические терминали этих чувствительных волокон образуют аксоаксо-нальные синапсы (рис. 20-4Б). При активации эти синапсы уменьшают количество медиатора, выделяемого из синапсов чувствительных волокон. Синаптическое торможение при отсутствии анестезии длится десятки миллисекунд.
Места действия лекарственных препаратов
Эффекты почти всех препаратов, влияющих на ЦНС, связаны с воздействием на тот или иной этап химической синаптической трансмиссии; рис. 20-3 иллюстрирует некоторые из них. Эти медиаторза-висимые взаимоотношения можно разделить на пре- и постсинаптические.
Рис. 20-3. Места действия препаратов. Схематическое изображение этапов, воздействие на которые может изменить синаптическую передачу: 1) потенциал действия в пресинаптическом волокне; 2) синтез медиатора; 3) хранение; 4) метаболизм; 5) высвобождение; 6) обратный захват; 7) деградация; 8) рецептор медиатора; 9) рецепторза-висимое повышение или понижение ионной проводимости
Препараты, действующие на синтез, хранение, метаболизм и высвобождение нейромедиаторов, относятся к группе пресинаптических. Подавление синтеза или хранения медиатора снижает синаптическую передачу. Например, р-хлорфенилаланин блокирует синтез серотонина, а резерпин уменьшает количество моноаминов в синапсах, влияя на его внутриклеточное депонирование. У гнетение катаболизма медиатора повышает его концентрацию, и, по некоторым данным, увеличивает количество медиатора, выделяемого при каждом импульсе. Препараты могут влиять и на сам медиатор. Стимулятор амфетамин вызывает высвобождение катехоламинов из адренергических синапсов. Капсаицин высвобождает пептидную субстанцию Р из чувствительных нейронов, а токсин столбняка блокирует высвобождение тормозящих аминокислот-медиаторов. После высвобождения медиатора в синаптическую щель его действие ограничивается либо обратным захватом, либо деградацией. Для большинства нейромедиаторов существуют механизмы захватав синаптическую терминаль и в окружающую нейроглию. Кокаин, например, блокирует захват катехоламинов в адренергических синапсах, потенцируя их действие. Ацетилхолин инактивируется путем ферментативного гидролиза, поэтому ингибиторы холинэстеразы пролонгируют его действие. Пока не определены механизмы захвата многочисленных пептидов ЦНС, и еще предстоит выяснить, разрушаются ли они путем специфической ферментативной деградации.
В постсинаптической области основной мишенью для препаратов является медиатор. Они могут действовать как агонисты нейромедиаторов (например, опиаты, имитирующие эффект энкефалинов) или блокировать функцию рецептора. Рецепторный антагонизм — это механизм действия многих препаратов, влияющих на ЦНС. Примером является блокада стрихнином рецептора тормозного медиатора глицина. Этот эффект, объясняющий индукцию судорог стрихнином, демонстрирует, как блокада тормозных процессов приводит к возбуждению. Обычно рецепторы связаны с одним из двух механизмов изменения проводимости. В большинстве синапсов ЦНС рецепторы связаны с ионными каналами, и активация рецептора приводит к открытию канала на короткое время (от нескольких до десятков миллисекунд). Препараты могут непосредственно действовать на ионные каналы. Например, барбитураты проникают в каналы, связанные с рецепторами многих возбуждающих нейромеди
аторов, и блокируют их. В других случаях рецепторы сопряжены с ферментами и активация последних ведет к метаболическим изменениям в постсинаптической клетке. Такие метаболические изменения могут модифицировать нейрональную функцию, блокируя потенциалзависимые каналы. Эффект может сохраняться долго после окончания взаимодействия медиатора и рецептора, то есть длиться десятки секунд или минуты. Метилксантины — это наиболее хорошо известный пример препаратов, которые изменяют реакцию нейромедиаторов через модуляцию уровня цАМФ. В больших концентрациях метилксантины повышают уровень цАМФ, блокируя его метаболизм, и, следовательно, продлевают его действие на постсинаптическую клетку.
Считается, что общие анестетики и этанол не связываются с какими-либо специфическими рецепторами. Их действие зависит от величины липофильности этих соединений. Они влияют на высвобождение медиатора и на постсинаптическую реактивность посредством взаимодействия с мембранными липидами и белками.
Селективность действия препарата почти полностью обусловлена тем, что разные нейроны используют разные группы медиаторов. Более того, эти различные медиаторы часто бывают сконцентрированы в нейрональных системах, отвечающих за какую-то определенную функцию. Без такой сегрегации было бы невозможно селективно воздействовать на отдельные функции ЦНС, даже если бы один препарат влиял только на одну медиаторную систему. Не вполне понятно, почему в ЦНС имеется столько медиаторов в составе обособленных нейрональных систем, так как первичная функция любого медиатора — это либо возбуждение, либо торможение. Выполнения этой первичной функции можно было бы достигнуть двумя или даже всего одним медиатором. Наличие такой сегрегации открыло большие возможности ученым в плане анализа функций ЦНС и терапии патологических состояний.
Идентификация центральных нейромедиаторов
Селективность действия препарата основана на том, что разные нейрональные пути используют разные медиаторы, поэтому первичной целью нейрофармакологов стало определение медиаторов в путях ЦНС. Для синапсов ЦНС сложнее, чем для
периферических синапсов, доказать, что какое-либо химическое вещество является медиатором. Теоретически для определения медиатора достаточно показать, что стимуляция путей вызывает синтез достаточного количества этого вещества, чтобы спровоцировать постсинаптический ответ. На практике этот эксперимент невозможен по меньшей мере по двум причинам. Во-первых, анатомическая сложность ЦНС не допускает селективной активации определенной группы синаптических терминалей. Во-вторых, имеющиеся методы определение количества высвобождаемого медиатора не совсем точны. Поэтому для определения медиаторов были приняты следующие критерии.
Локализация
Используется несколько подходов для того, чтобы доказать, что предполагаемый медиатор находится в исследуемых путях. В первую очередь — это биохимический анализ локальных концентраций предполагаемых медиаторов, часто комбинируемый с прерыванием этих путей, и микроцитохимические методы. Кроме того, для определения локализации пептидов и ферментов метаболизма небелковых медиаторов весьма информативными являются иммуноцитохимические методы.
Высвобождение
Для того чтобы определить, высвобождается ли медиатор из определенной области, иногда можно выполнить локальный сбор внеклеточной жидкости {in vivo). К тому же, in vitro электрически или химически можно стимулировать срезы ткани мозга, измеряя затем количества исследуемых веществ. Для определения связи между высвобождением медиатора и синаптической передачей надо доказать, что это высвобождение кальцийзависимо. Как уже было отмечено ранее, анатомическая сложность часто затрудняет изучение синаптических терминалей, отвечающих за высвобождение медиатора, а количество медиатора, которое удается собрать в перфузате, является только ничтожной частью истинно высвобождаемого количества.
Синаптическая мимикрия
Наконец, аппликация предполагаемого медиатора должна вызывать реакцию, идентичную действию медиатора, выделяемого при стимуляции не
рва. Микроионофорез, который позволяет чрезвычайно точно локализовать введение препарата, является очень ценным методом оценки действия предполагаемого медиатора. На практике у этого критерия есть две составляющих: физиологическая и фармакологическая идентичность. Для определения физиологической идентичности действия необходимо доказать, что препарат вызывает такие же изменения ионной проводимости постсинаптической клетки, что и медиатор, выделяемый в синапсе. Для этого необходима внутриклеточная регистрация и определение обратимости потенциала и ионной зависимости клеточных ответов. Однако вследствие того, что разные медиаторы могут вызывать одинаковые изменения ионной проводимости, этих данных недостаточно. Поэтому для дальнейшего подтверждения идентичности эффектов изучаемого вещества и естественного медиатора используется селективный фармакологический антагонизм. Вследствие сложности строения ЦНС специфический фармакологический антагонизм синаптических ответов является важным методом идентификации медиатора.
Клеточная организация мозга
Нейрональные системы в ЦНС можно по многим параметрам разделить на два типа: иерархические системы и неспецифические, или диффузные, нейрональные системы.
Иерархические системы
К этим системам относятся все проводящие пути, контролирующие чувствительность и двигательную активность. Обычно эти пути отчетливо прослеживаются, так как состоят из толстых миелинизированных волокон, проводимость по которым иногда превышает 50 м/с. Информация обычно имеет фазный характер, и в системах чувствительности она перерабатывается последовательно путем интеграции на уровне каждого релейного ядра на пути к коре. Повреждение на любом уровне связи приводит к нарушению работы всей системы. В каждом ядре и в коре присутствуют два типа клеток: релейные, или проекционные, нейроны и местные вставочные нейроны (рис. 20-4А). Проекционные нейроны, связываясь друг с другом, передают сигналы на большие расстояния. Тела этих клеток сравнительно крупные, их аксоны имеют коллате
рали, которые дают множество ответвлений в окружении нейрона. Эти нейроны возбуждающие, их влияния на синапсы очень краткосрочные. Медиатором этих клеток в большинстве случаев является глутамат. Местные вставочные нейроны обычно мельче, чем проекционные, и их аксоны ветвятся только в непосредственной близости от тела нейрона. Большинство этих нейронов — тормозные, и они выделяют либо ГАМК, либо глицин. Они образуют синапсы с телами клеток и дендритами проекционных нейронов или друг с другом. Особый класс местных вставочных нейронов в спинном мозгу образует аксоаксональные синапсы с терминалями чувствительных аксонов (рис. 20-4Б). Два основных вида связей этих нейронов (рис. 20-4А) включают возвратные прямые и обратные связи. Во многих чувствительных путях локальные вставочные нейроны могут не иметь аксона и выделять нейромедиатор из дендритных синапсов постепенно (малыми порциями) без возникновения потенциала действия. Некоторые пути, включающие пресинап-тические дендриты местных вставочных нейронов, показаны на рис. 20-4В.
Существует большое разнообразие синаптических связей в иерархических системах, но их нейроны используют только ограниченное количество медиаторов. Следовательно, значительные фармакологические воздействия на эти системы будут иметь существенный эффект на общую возбудимость ЦНС. Например, селективная блокада ГАМК-рецепторов веществами типа пикротоксина приводит к генерализованным судорогам. Таким образом, несмотря на то, что механизм действия пикротоксина достаточно селективен в отношении блокады эффектов ГАМК, общий эффект неспецифичен ввиду широкого распространения системы тормозных ГАМК-синапсов.
Неспецифические, или диффузные, нейрональные системы
Примерами этой категории являются те нейрональные системы, которые содержат один из моноаминов (норадреналин, дофамин или серотонин). К ней относятся также некоторые другие проводящие пути, происходящие из ретикулярной формации, и, возможно, ряд пептидсодержащих структур. Эти системы имеют фундаментальные отличия от иерархических, которые можно проиллюстрировать на примере норадренергических систем.
В ДЕНДРИТ-ДЕНДРИТНОЕ ВЗАИМОДЕЙСТВИЕ
Рис. 20-4. Проводящие пути ЦНС. На рис. А показаны два релейных нейрона и два типа тормозящих связей — обратная и прямая. Тормозные нейроны выделены черным цветом. На рис. Б показаны связи, отвечающие за пресинапти-ческое торможение, при котором аксон тормозного нейрона образует синапс на терминали аксона возбуждающего нейрона. Рис. В: схема реципрокных пре- и постсинаптических синапсов между одной парой дендритов. В триаде аксон образует синапс с двумя дендритами, дендриты образуют синапс друг с другом. В серийных синапсах дендрит может быть постсинаптическим для одного и пресинаптическим для другого дендрита, таким образом соединяя серии дендритов. Дендриты взаимодействуют также посредством электротонических (щелевых) межклеточных связей (две из них показаны на рисунке). Кроме одного аксона, все структуры, показанные на рис. В,— дендриты. (Schmitt F. О, Dev. Р, Smith В. Н. Electrotonic processing of information by brain cells. Science, 1976; 193; 114, Copyright 1976 by the American Association of the Advancement of Science.)
Тела норадренергических клеток в основном находятся в компактном скоплении клеток, именуемом locus ceruleus, расположенном в сером веществе центральной части каудального отдела моста. Число нейронов в этом скоплении незначительно, приблизительно по 1500 на каждой стороне мозга крысы. Аксоны этих клеток очень тонкие и немие-линизированные. И действительно, их долго не уда
валось выявить при помощи классических анатомических методов. Только в середине 1960-х гг., когда для изучения тканей ЦНС применили гистохимический метод формальдегидной флюоресценции, была описана анатомия моноаминсодержащих систем. Так как эти аксоны тонкие и немиелинизиро-ванные, проведение по ним очень медленное (около 0.5 м/с). Аксоны разветвляются несколько раз и
ТАБЛИЦА 20-1. Сводные данные по фармакологии небелковых нейромедиаторов ЦНС
Медиатор Анатомия Подтипы рецепто- Антагонисты Рецепторные механизмы ров и предпочтительные агонисты
Ацетилхолин Тела клеток на всех уровнях; короткие и длинные связи Мотонейроны — синапсы клеток Реншоу Мускариновые (м,): мускарин, McN-A-343 Мускариновые (мг): мускарин, бетанехол Никотиновые: никотин Пирензепин, атропин Атропин Дигидро-0-эритроидин Возбуждающие: снижение проницаемости для К* Тормозящие: повышение проницаемости для К* Возбуждающие: повышение проницаемости для катионов
Дофамин Тела клеток на всехуровнях; короткие, средние и длинные связи D,: SKF 38393 Ds: апоморфин Фенотиазины, SCH23390 Фенотиазины, бутирофеноны Тормозящие: повышают (?) уровень цАМФ Тормозящие: повышение проницаемости для К*
ГАМК Супраспиналь-ные интернейроны; спинальные интернейроны, участвующие в пресинаптичес-ком торможении ГАМКа: мусцимол ГАМКВ: баклофен Бикукуллин, пикротоксин 2-ОН-саклофен, CGP 35348 Тормозящие: повышение проницаемости для СГ Тормозящие (пресинаптические): снижение проницаемости для Са2’; тормозящие (постсинаптические): повышение проницаемости для К*
Глутамат, аспартат Релейные нейроны на всех уровнях N-метил-О-аспартат (NMDA) АМРА, квисквалат, каинат ACPD, квисквалат 2-амино-5-фосфовалерат, СРР CNQX MCPG Возбуждающие: повышение проницаемости для катионов, особенно для Са24 Возбуждающие: повышение проницаемости для катионов, но не для Са24 Возбуждающие: снижение проницаемости для К4
Глицин Спинальные и некоторые стволовые интернейроны Таурин, р-аланин Стрихнин Тормозящие: повышение проницаемости для СГ
Серотонин (5-гидрокси-триптамин) Тела клеток среднего мозга и моста с проекцией на все уровни 5-НТ,А: ЛСД, 8-ОН-DPAT 5-НТгд: ЛСД, DOB 5-НТ3: 2-метил-5-НТ, фенилбигуанид 5-НТ4: BIMU8 Метерголин, спиперон Кетансерин ICS 205930, ондансетрон GR 1138089 Тормозящие: повышение проницаемости для К4 Возбуждающие: снижение проницаемости для К4 Возбуждающие: повышение проницаемости для катионов Возбуждающие: снижение проница-
емости для К4; через цАМФ
ТАБЛИЦА 20-1. (Продолжение)
Медиатор Анатомия Подтипы рецепторов и предпочтительные агонисты Антагонисты Рецепторные механизмы
Норадреналин Тела клеток моста и ствола мозга с проекцией на все уровни ар фенилэфрин а2: клонидин Рр изопротеренол, добутамин рр сальбутамол Празозин Йохимбин Атенолол, практолол Бутоксамин Возбуждающие: снижение проницаемости для К* Тормозящие: повышение проницаемости для К* Возбуждающие: снижение проницаемости для К+; через цАМФ Тормозящие: могут вовлекать элект-рогенный натриевый насос
Сокращения: ACPD - транс-1-амино-циклопентил-1,3-дикарбоксилат; АМРА- О1_-а-амино-3-гидрокси-5-метилизоксол-4-пропионат; BIMU8 - (эндо-Ы-8-метил-8-азабицикло [3.2.1]окт-3-ил)-2,3-дигидро-3-изопропил-2-оксо-1Р-бензими-дазол-1-карбоксамид гидрохлорид; CGP 35348 - 3-аминопропил(диэтоксиметил)фосфиновая кислота; CNQX - 6-циа-но-7-нитрохиноксалин-2,3-дион; СРР - 3-(2-карбоксипиперазин-4-ил)пропил-1-фосфоновая кислота; DOB - 5-бромо-2,5-диметоксиамфетамин; GR113808 - [1 -[2-[(метилсульфонил)амино]этил]-4-пиперидинил]метил-1 -метил-1 Н-индол-3-карбоксилат; 8-OH-DPAT - 8-гидрокси-2(ди-п-пропиламино)тетралин; MCPG - (±)-а-метил-4-карбоксифенилглицин.
постоянно меняют направление хода. Ветви отростков одного нейрона могут иннервировать несколько функционально отличающихся частей ЦНС. В новой коре (неокортексе) у этих волокон имеется тангенциальная организация, и поэтому они могут моносинаптически воздействовать на большие зоны коры. Иннервация коры и ядер иерархических систем диффузна, норадренергические волокна составляют только малую часть всех волокон этих областей. Кроме того, на протяжении аксонов имеются так называемые варикозные расширения, содержащие большое количество пузырьков. В некоторых случаях эти расширения не образуют синаптических контактов, что позволяет предположить возможность диффузного выделения норадреналина, как это происходит в гладкой мышце. Это означает, что клеточные мишени данной системы во многом будут определяться местоположением рецепторов, а не источников медиатора. На этом основании легко сделать вывод о том, что моноаминовые системы не являются передатчиками специфической топографической информации. Более вероятно, что изменение их функций приводит к развитию сходных процессов во многих отделах ЦНС. Поэтому считается, что эта система играет роль в таких глобальных функциях как сон, пробуждение, аппетит и эмоциональное состояние.
Центральные нейромедиаторы
Из мозга было выделено несколько видов мелких молекул (табл. 20-1). Исследования с использованием различных подходов показали, что эти вещества могут быть нейромедиаторами. Сводные данные по некоторым из них представлены в этом разделе.
Аминокислоты
Наиболее интересные для фармакологов аминокислоты можно разделить на две группы: нейтральные аминокислоты: глицин и ГАМК, и кислые аминокислоты: глутамат и аспартат.
Все эти соединения присутствуют в ЦНС в высоких концентрациях и являются значимыми факторами возбудимости нейронов.
А. Нейтральные аминокислоты. Нейтральные аминокислоты относятся к тормозным факторам, повышающим мембранную проницаемость для ионов хлора, имитируя таким образом ТПСП. Концентрация глицина особенно высока в сером веществе спинного мозга. Разрушение нейронов этой области приводит к значительному снижению содержания глицина. Кроме того, стрихнин (конвуль-сант, действующий на спинной мозг и используемый в некоторых крысиных ядах) является селективным антагонистом как эффектов глицина, так и ТПСП в спинном мозгу. Принято считать, что гли
цин высвобождается из ингибиторных местных вставочных нейронов спинного мозга, участвующих в постсинаптическом торможении.
ГАМК-рецепторы делятся на два типа: ГАМКЛ и ГАМКВ. ГАМКЛ открывают хлорные каналы, их действию антагонизируют пикротоксин и бикукуллин, которые вызывают генерализованные судороги. ГАМКв-рецепторы, которые могут быть селективно активированы противоспастическим препаратом баклофеном, связаны с калиевыми каналами на постсинаптической мембране. В большинстве областей мозга ТПСП имеет быстрый и медленный компоненты. Убедительные данные свидетельствуют о том, что ГАМК является тормозным медиатором, отвечающим за оба компонента. Быстрые ТПСП блокируются антагонистами ГАМКЛ, а медленные — антагонистами ГАМКВ. Иммуногистохимические исследования показали, что подавляющее большинство местных вставочных нейронов синтезируют ГАМК. Также синтезирует ГАМК особый класс местных вставочных нейронов в заднем роге спинного мозга. Эти нейроны образуют аксоаксо-нальные синапсы с терминалями чувствительных нервов и отвечают за пресинаптическое торможение (рис. 20-4Б).
Б. Кислые аминокислоты. И глутамат, и аспартат присутствуют в ЦНС в очень высоких концентрациях. Почти все исследованные нейроны сильно возбуждаются под воздействием этих аминокислот. Это возбуждение вызвано активацией рецепторов, которые можно разделить на два класса: ионотропные и метаботропные. Оба класса рецепторов были детально изучены методом молекулярного клонирования. Как видно из их названия, ионотропные рецепторы напрямую регулируют катионселектив-ные каналы. Они подразделяются на 3 подтипа в зависимости от действия селективных агонистов: каината (К), а-амино-5-метилизоксазол-4-пропио-ната (АМРА) и N-метил-О-аспартата (NMDA). АМРА- и К-активируемые каналы проницаемы для ионов калия, натрия и кальция (некоторые подтипы). Они часто группируются вместе и называются HeNMDA-каналы. NMDA-каналы проницаемы для ионов натрия, калия и кальция.
Метаботропные глутаматные рецепторы действуют не на ионные каналы, а на внутриклеточные ферменты. Они селективно активируются транс-1-амино-циклопентил-1,3-дикарбоксилатом (ACPD). Эти рецепторы, связанные с G-белком, активируют фосфолипазу С и ингибируют аденилатцикла-
зу. Биохимические исследования показали, что при перерезке специфических региональных путей падает концентрация глутамата и снижается его каль-цийзависимое высвобождение. Хотя не ясно, участвуют ли ACPD-рецепторы в синаптической трансмиссии, фармакологические исследования показали, что в ней участвуют и NMDA-, и HeNMDA-рецепторы.
Роль NMDA-рецепторов в последнее время привлекает большое внимание. Эти рецепторы определяют величину синаптической пластичности, которая считается основой некоторых типов обучения и памяти. Они селективно блокируются диссоциативным анестетиком кетамином и галлюциногеном фенциклидином, которые оказывают свои эффекты, входя в открытый канал рецептора и блокируя его. Препараты, которые инактивируют этот рецепторный канал, обладают сильной противоэпилеп-тической активностью на экспериментальных моделях, хотя их еще предстоит исследовать в клинике. Особенно интересны данные о том, что блокада NMDA-рецепторов может уменьшать в эксперименте на животных повреждения нейронов, вызванные аноксией. Этот эффект имеет большое потенциальное клиническое значение.
Ацетилхолин
Ацетилхолин был первым веществом, медиаторные свойства которого в ЦНС были определены фармакологически. В начале пятидесятых годов Экклз показал, что возбуждение клеток Реншоу коллатералями двигательных аксонов блокируется антагонистами никотиновых рецепторов. Более того, клетки Реншоу оказались очень чувствительными к антагонистам никотиновых рецепторов. Эти эксперименты были необычными по двум причинам. Во-первых, эти синапсы оставались единственными, медиатор которых был известен, вплоть до конца шестидесятых годов, когда появились сходные данные в отношении нейтральных аминокислот. Во-вторых, по сей день коллатеральный синапс двигательного аксона остается единственным достоверным примером холинергического никотинового синапса в ЦНС млекопитающих, несмотря на достаточно широкое распространение никотиновых рецепторов, показанное методом гибридизации in situ. Большинство реакций на ацетилхолин в ЦНС опосредованы обширной группой мускариновых рецепторов, связанных с G-белком. В некоторых
местах ацетилхолин вызывает медленное торможение нейронов, активируя м2-рецепторы, которые открывают калиевые каналы. Чаще в ответ на распространенное действие ацетилхолина возникает медленное возбуждение, которое опосредуется мрре-цепторами. Эти мускариновые эффекты медленнее, чем никотиновые и чем действие аминокислот на клетки Реншоу. Более того, мускариновое возбуждение необычно тем, что ацетилхолин вызывает его, снижая проницаемость мембраны для калия, то есть действуя отлично от обычного эффекта медиатора.
Ацетилхолин содержат такие системы, как нейроны неостриатума, медиального ядра перегородки и ретикулярной формации. Появление моноклональных антител, которые связываются с ацетил-холинтрансферазой, значительно улучшило методику гистохимического выявления этого медиатора. Холинергические пути играют большую роль в выполнении когнитивных функций, особенно памяти. Пресенильная деменция типа болезни Альцгеймера связана со значительной потерей холинергических нейронов, но специфичность таких нейрональных изменений не доказана, так как отмечается снижение содержания и других предполагаемых медиаторов, например соматостатина.
Моноамины
Моноамины включают катехоламины (дофамин и норадреналин) и серотонин (5-гидрокситрипта-мин). Так как эти соединения присутствуют в ЦНС в очень малых количествах, то их локализацию можно выявить, используя лишь крайне чувствительные гистохимические методы. К ним относят оценку вызванной формальдегидом флюоресценции моноаминов и иммуногистохимический метод определения локализации отдельных ферментов их биосинтеза. Эта система медиаторов является мишенью для действия многих препаратов. Например, стимуляторы ЦНС кокаин и амфетамин действуют прежде всего на катехоламиновые синапсы. Кокаин блокирует обратный захват дофамина и норадреналина, а амфетамины стимулируют высвобождение этих медиаторов из пресинаптических терминалей.
А. Дофамин. Основные системы, содержащие дофамин,— это пути, связывающие черную субстанцию с неостриатумом, и пути, связывающие вентральную тегментальную область с лимбическими структурами, особенно лимбической корой. Клини
ческий эффект противопаркинсонического препарата леводопы связан с действием на первую из названных областей, а эффект антипсихотических препаратов — с последней. Дофаминсодержащие нейроны в туберобазальном гипоталамусе играют важную роль в регуляции гипоталамо-гипофизар-ной функции. Дофамин обычно оказывает медленное тормозное действие на нейроны. Лучше всего это действие можно продемонстрировать на дофаминсодержащих нейронах черной субстанции, в которых Ц2-рецептор открывает калиевые каналы.
Б. Норадреналин (норэпинефрин). Эту систему мы уже рассматривали. Большинство норадренергических нейронов локализуются в locus ceruleus или в латеральной тегментальной области ретикулярной формации. Хотя плотность волокон, иннервирующих разные зоны, варьирует, большинство областей ЦНС имеют диффузный норадренергический компонент. При контакте с нейронами норадреналин может вызвать их гиперполяризацию, повышая проницаемость мембран для ионов калия. Этот эффект опосредуется а2-рецепторами и особенно подробно изучен для locus ceruleus. Во многих участках ЦНС норадреналин усиливает возбуждающие импульсы путем прямых и непрямых механизмов. Непрямые механизмы включают в себя подавление местных вставочных тормозных нейронов. Прямой механизм заключается в блокаде проницаемости для К+, которая снижает частоту нейрональных импульсов. В зависимости от типа нейрона этот эффект опосредуется либо оц-, либо Р~ре-цепторами. Облегчение возбуждающей синаптической трансмиссии связано со многими поведенческими процессами, предположительно включающими норадренергические пути (внимание, бодрствование и т. д.).
В. Серотонин (5-гидрокситриптамин). Большая часть серотониновой (5-гидрокситриптаминовой) системы (5-hydroxytryptamine - 5-НТ) исходит из нейронов ядер шва или средней части моста и передней части ствола мозга. 5-НТ содержится в не-миелинизированных волокнах, которые диффузно иннервируют большинство регионов ЦНС, но их плотность варьирует. В основном 5-НТ оказывает сильное тормозное действие. Это действие опосредовано 5-НТ1Л-рецепторами и связано с гиперполяризацией мембраны, вызванной повышением проницаемости для ионов калия. Установлено, что 5-НТ1Л-рецепторы и ГАМКв-рецепторы действуют на одни и те же калиевые каналы. Эти рецепторы и
калиевые каналы связаны через ГТФ-связывающий белок. Некоторые типы клеток медленно возбуждаются под действием 5-НТ вследствие блокады калиевых каналов, опосредованной 5-НТ2-рецепто-рами. И возбуждение, и торможение могут возникать в одних и тех же нейронах. Предполагают, что 5-НТ-пути играют роль в возникновении галлюцинаций после приема ЛСД, поскольку это вещество может нивелировать периферические эффекты 5-НТ. Однако ЛСД не является антагонистом 5-НТ в ЦНС, и у животных после удаления ядра шва наблюдается типичное поведение, вызванное приемом ЛСД. Другие регуляторные функции 5-НТ-содер-жащих нейронов — это сон, регуляция температуры тела, аппетит и нейроэндокринный контроль.
Пептиды
Выделено значительное количество пептидов ЦНС, которые оказывают большое влияние на поведение животных и на активность отдельных нейронов. Многие из этих пептидов идентифицированы иммуногистохимическими методами. Среди них можно назвать опиоидные пептиды (энкефалины, эндорфины и т. д.), нейротензин, субстанцию Р, соматостатин, холецистокинин, вазоактивный интестинальный полипептид, нейропептид Y и тирео-тропинрилизинг-гормон. Как и в периферической вегетативной нервной системе, в ЦНС пептиды часто сосуществуют в одном нейроне с традиционными непептидными медиаторами. Хорошим примером подходов, которые использовали для определения роли пептидов в ЦНС, является определение роли субстанции Р и ее связей с чувствительными волокнами. Иммуногистохимические исследования показали, что субстанция Р находится в некоторых тонких немиелинизированных волокнах первичных
чувствительных нейронов спинного мозга и ствола мозга. Стимуляция чувствительных нейронов вызывает высвобождение субстанции Р в спинном мозгу. Контакт субстанции Р с нейронами спинного мозга возбуждает те из них, которые активируются болевыми стимулами. Как известно, болевые стимулы селективно активируют немиелинизиро-ванные волокна. Считается, что субстанция Р играет роль возбуждающего медиатора этих волокон — проводников болевых импульсов. Субстанция Р обладает и другими функциями, так как она находится во многих областях ЦНС, не отвечающих за восприятие боли.
Избранная литература
Bloom F. Е. The endorphins: A growing family of pharmacologically pertinent peptides. Annu. Rev. Pharmacol. Toxicol. 1983; 23: 344.
Bowery N. G., Bittiger H., Olpe H-R. GABAd Receptors in Mammalian Function. Wiley, 1990.
Cooper J. R., Bloom F. E., Roth R. H. The Biochemical Basis of Neuropharmacology, 6th ed. Oxford Univ. Press, 1991.
Creese I. Dopamine receptors explained. Trends Neurosci. 1982; 5: 40.
Eccles J. C. The Physiology of Synapses. Academic Press, 1964.
Iversen S. D., Iversen L. L. Behavioral Pharmacology, 2nd ed. Oxford Univ. Press, 1979.
Meltzer H. Y. (ed.) Psychopharmacology: The Third Generation of Progress, Raven Press, 1987.
Shepherd G. M. Neurobiology, 2nd ed. Oxford Univ. Press, 1988.
Седативные и снотворные средства
Энтони Дж. Тревор, Уолтер Л. Вэй
21
Главное клиническое назначение седативных и снотворных средств — вызывать седацию (с одновременным снижением тревожности) или сон. В силу существующих химических различий классификация препаратов этой группы строится на основе клинического применения, а не химической структуры или механизмов действия. Показания к их применению очень широки, седативные и снотворные препараты — одни из наиболее часто применяемых во всем мире.
I. Базисная фармакология седативных и снотворных препаратов
Седативный (анксиолитический) препарат должен снижать ощущение тревоги, обладать успокаивающим эффектом, оказывая при этом минимальное действие на двигательные и мыслительные функции. Степень подавления ЦНС, вызванная седативным средством, должна быть минимальной. Снотворное средство должно вызывать сонливость и облегчать начало и течение сна, максимально идентичного естественному. Снотворный эффект связан с более глубоким подавлением ЦНС, чем седативный. Этого эффекта можно также добиться с помощью большинства седативных препаратов, просто увеличивая их дозу.
Действие седативных и снотворных средств характеризуется градуальным доэозависимым подавлением функции ЦНС. Однако отдельные препараты отличаются по соотношению дозы и степени подавления ЦНС. Два примера таких дозозависимых отношений показаны на рис. 21-1. Линейная кривая препарата А типична для многих седативных и снотворных средств ранних поколений, вклю
чая барбитураты и алкоголь. Для таких препаратов превышение необходимой дозы может привести к наркозу. В еще более высоких дозах они подавляют дыхательный и вазомоторный центры в продолговатом мозгу, приводя к коме и смерти. Отклонения от линейных зависимостей, показанные для препарата Б, потребуют относительно большего увеличения дозы для достижения глубокого подавления ЦНС. Это справедливо для большинства препаратов класса бензодиазепинов вследствие их большей терапевтической широты; они весьма распространены в клинике для лечения тревожных состояний и расстройств сна.
Химическая классификация
Бензодиазепины (рис. 21-2) — это основные седативные и снотворные препараты. Все соединения, представленные на рисунке, относятся к 1,4-бензо-диазепинам. Большинство из них содержит карбоксамидную группу в семичленном гетероциклическом кольце. Для проявления седативного или
Рис. 21-1. Теоретические кривые зависимости доза-эффект для снотворных и седативных препаратов
Хлордиазепоксид
Деметилдиазепам
Оксазепам
Алпразолам
Рис. 21-2. Химические структуры бензодиазепинов
снотворного эффекта требуется замещающее соединение в 7 положении, например галоген или нитрогруппа. Структуры триазолама и алпразолама включают дополнительное триазоловое кольцо в положении 1,2, поэтому их иногда называют триа-золобензодиазепинами.
Химические структуры более ранних и реже используемых седативных и снотворных препаратов
показаны на рис. 21-3. Барбитураты считаются прототипами всего этого класса препаратов из-за активного их использования в прошлом.
Стремление создать новые бензодиазепины и другие седативные и снотворные средства связано с попытками избежать нежелательных эффектов барбитуратов, включая их способность вызывать психологическую и физическую зависимость. К со-
Ri=—СН2—СН=СН2
R2:—СН—СН2—СН2—СН3
СНз
Секобарбитал
Н°ч
СН—СС13 нс/
О С3Н7 О
II I II
Н2Ы—С—О—СН2—С—СНг—О—С—ЫН2
СН3
Мепробамат
Хлоралгидрат
НО—СН2—СОз Трихлорэтанол
Рис. 21-3. Химическая структура барбитуратов и иных седативных и снотворных средств
жалению, эти попытки не всегда успешны. Например, пиперидиндионы типа глютетимида и мети-прилона, которые были введены как “небарбитурат-ные седативы-снотворные”, на самом деле химически родственны и по своему действию неотличимы от барбитуратов. Карбаматы пропавдиола типа мепробамата имеют другую химическую структуру, но идентичны барбитуратам по фармакологическим свойствам, поэтому их клиническое применение быстро сокращается. Класс седативных и снотворных средств включает также вещества простой химической структуры, например спирты (этанол, хлоралгидрат) и циклические эфиры. Хлоралгидрат и родственные соединения типа трихлорэтано-ла, а также паральдегид (не показан), еще используются в настоящее время, особенно в стационарах. В последнее время появились препараты новой химической структуры. Буспирон (азаспиродекавди-он) — это анксиолитик, который по эффектам отличается от традиционных препаратов. Золпидем, имидазопиридиновое снотворное средство, не похожее по структуре на бензодиазепины, имеет сходные с ними фармакологические свойства.
Другие классы препаратов, не показанные на рис. 21-3, также обладают седативным действием. p-блокаторы, например, эффективны при определенных тревожных состояниях и функциональных нарушениях, особенно тех, при которых значительно выражены соматические и вегетативные симп
томы. Парциальный агонист а2-рецепторов (в том числе пресинаптических адренорецепторов мозга) клонидин тоже имеет анксиолитические свойства. Седативные эффекты можно получить, используя антипсихотические транквилизаторы, трициклические антидепрессанты и антигистаминные препараты. Как описано в соответствующих главах, эти препараты отличаются от традиционных седативных и снотворных препаратов и по эффектам, и по клиническим показаниям к применению. Наиболее важным является то, что они не дают общей анестезии и фактически не вызывают привыкания. Так как они часто оказывают значительные эффекты на периферическую вегетативную нервную систему, их иногда называют “вегетато-седативными” препаратами. В некоторых снотворных препаратах, продаваемых без рецепта, присутствуют антигистаминные средства. Однако их влияние на вегетативную нервную систему и большая продолжительность действия могут привести к нежелательным побочным эффектам.
Бензодиазепины и барбитураты
Фармакокинетика
А. Абсорбция. Для лечения тревожных состояний или расстройств сна седативные и снотворные средства обычно назначают перорально. Бензодиа
зепины как слабые основания лучше всего всасываются в двенадцатиперстной кишке (при высоких значениях pH). Их абсорбция зависит от нескольких факторов, включая липофильность. Наибольшую скорость всасывания при приеме внутрь имеет триазолам, далее следуют диазепам и активный метаболит клоразепата, а затем другие распространенные бензодиазепины. Клоразепат конвертируется в активную форму (деметилдиазепам) при гидролизе в желудке. Оксазепам и темазепам всасываются медленнее, чем другие бензодиазепины. Биодоступность некоторых бензодиазепинов, например хлордиазепоксида и диазепама, после внутримышечного введения невелика. Барбитураты и пипе-ридиндионы как слабые кислоты обычно очень быстро всасываются из желудка и тонкой кишки.
Б. Распределение. Транспорт седативных и снотворных средств в кровотоке — это динамический процесс, при котором молекулы препарата поступают и покидают ткани со скоростью, зависимой от величины кровотока, градиентов концентрации и проницаемости биологических барьеров. Растворимость в жирах играет основную роль в определении скорости попадания препарата в ЦНС. Например, диазепам и триазолам более липофильны, чем хлордиазепоксид и лоразепам, поэтому эффекты последних препаратов на ЦНС отсрочены. Тиобарбитураты (например, тиопентал), в которых кислород при С2 замещен серой, легко растворимы в жирах, поэтому их действие на ЦНС начинается очень быстро (глава 24). Мепробамат, напротив, плохо растворим в жирах и медленно проникает в ЦНС даже при внутривенном введении.
Перераспределение препарата из ЦНС в другие ткани является важной характеристикой седативных и снотворных препаратов. Классические исследования тиобарбитуратов показали, что они быстро перераспределяются из мозга сначала в хорошо кровоснабжаемые ткани, в частности скелетные мышцы, а потом в плохо кровоснабжаемую жировую ткань. Эти процессы приводят к прекращению влияния на ЦНС. То же самое справедливо для других седативных и снотворных препаратов, включая бензодиазепины, скорость метаболической трансформации и элиминации которых у человека слишком низкая, чтобы стать причиной сравнительно быстрого прекращения основных фармакологических эффектов.
При назначении бензодиазепинов и других седативных и снотворных средств во время беремен
ности необходимо учитывать, что жирорастворимые препараты могут достигнуть плода через плацентарный барьер. Баланс концентраций веществ между кровыб матери и плода достигается медленнее, чем между центральными сосудами матери и сосудами ЦНС, отчасти в связи с низким кровотоком в плаценте. Однако, если седативные или снотворные средства назначаются незадолго до родов, они могут подавить жизненные функции у новорожденного.
Бензодиазепины и большинство седативных и снотворных средств активно связываются с белками плазмы крови. Например, степень связывания бензодиазепинов альбуминами плазмы крови варьирует между 60 и 95 %. Так как только свободные (несвязанные) молекулы могут проникать в ЦНС, вытеснение седативных и снотворных средств из мест связывания в плазме другими препаратами может изменить их эффект и привести к межгрупповым фармакологическим взаимодействиям. Однако есть только единичные клинически значимые примеры межгрупповых конкурентных взаимодействий седативных и снотворных средств. Таким примером является хлоралгидрат, который повышает антикоагулянтные эффекты варфарина, вытесняя его из мест связывания в плазме.
В. Биотрансформация. Перераспределение препарата в тканях (помимо ЦНС) имеет такое же значение в прекращении действия седативных и снотворных средств на ЦНС, как и его биотрансформация. Почти всем соединениям этого класса необходима метаболическая трансформация в водорастворимые вещества для выведения. Наибольшую значимость для метаболизма имеет система микросомальных ферментов печени. Только единичные препараты этой группы выводятся из организма в неизмененном виде, поэтому величина периода полувыведения зависит в основном от скорости метаболической трансформации.
1. Бензодиазепины. Все бензодиазепины метаболизируются и выводятся печенью. Скорость и особенности метаболизма несколько различаются у разных препаратов. Большинство бензодиазепинов подвергается микросомальному окислению (реакции фазы I), включая N-деалкилирование и алифатическое гидроксилирование. Затем метаболиты подвергаются конъюгации (реакции фазы II) под действием глюкуронозилтрансферазы с образованием глюкуронидов, которые выводятся с мочой. Однако многие метаболиты бензодиазепинов фазы
I сами являются активными веществами с периодом полувыведения, превышающим таковой у исходных препаратов.
Как показано на рис. 21-4, деметилдиазепам, период полувыведения которого составляет 40-140 часов, является активным метаболитом хлордиазе-поксида, диазепама, празепама и клоразепата. Деметилдиазепам затем трансформируется в активное соединение оксазепам. Другие активные метаболиты хлордиазепоксида — это деметилхлордиазепок-сид и демоксепам. Диазепам в основном метаболизируется в деметилдиазепам, а также в темазепам (не показан на рис. 21-4), который далее частично переходит в оксазепам. Флуразепам, применяемый в основном как снотворное средство, окисляется печеночными ферментами до трех активных метаболитов — дезалкилфлуразепама, гидроксиэтилфлу-разепама и альдегида флуразепама (не показан), период полувыведения которых варьирует в пределах 30-100 часов. Такие большие периоды полувыведения могут привести к нежелательному чрезмерному угнетению ЦНС и, как следствие, к сонливости в дневное время. Триазолобензодиазепины (триазолам и алпразолам) метаболизируются «-гидроксилированием с образованием веществ, которые
оказывают очень кратковременный фармакологический эффект, так как быстро конъюгируются в неактивные глюкурониды.
Образование активных метаболитов осложняет исследования фармакокинетики бензодиазепинов, поскольку длительность периода полувыведения исходного препарата может не совпадать с продолжительностью фармакологических эффектов. Те исходные препараты или активные метаболиты, период полувыведения которых продолжителен, вызывают кумулятивные эффекты при приеме нескольких доз. Кумулятивные и следовые эффекты, например дневная сонливость, менее выражены у препаратов типа оксазепама и лоразепама, имеющих короткие периоды полувыведения или метаболизируемых сразу до неактивных глюкуронидов. Некоторые фармакокинетические свойства отдельных бензодиазепинов представлены в табл. 21-1.
2. Барбитураты. За исключением фенобарбитала только незначительное количество барбитуратов выделяется в неизмененном виде. Основные метаболические пути — это окисление печеночными ферментами химических групп при С5, которые различны у всех препаратов. Образуемые спирты,
Хлордиазепоксид Диазепам Празепам Клоразепат (неактивен)
Рис. 21-4. Биотрансформация (метаболизм) бензодиазепинов. (Жирным шрифтом отмечены препараты, применяемые в клинике; * — активные метаболиты.)
ТАБЛИЦА 21 -1. Фармакокинетические свойства бензодиазепинов у человека
Препарат Период полу- выведения (границы, ч) Метаболиты Комментарии
Алпразолам 12-15 Активные: а-алпразолам Быстрое всасывание перорально
Хлордиазепоксид 5-30 Активные: деметил-дериват, демоксепам, оксазепам Низкая биодоступность при внутримышечном введении
Клоразепат 50-100 Активные: деметилдиазепам, Гидролиз до активной формы в же-
(метаболиты) оксазепам лудке
Диазепам 50-150 Активные: деметилдиазепам, темазепам, оксазепам Низкая биодоступность при внутримышечном введении
Флунитразепам 12-24 Активные: деметилфлунитразепам Большой объем распределения
Флуразепам 24-100 (метаболиты) Активные: дезалкил-дериват и др. Длинный период полувыведения активных метаболитов
Лоразепам 10-18 Неактивные: глюкурониды Выведение не зависит от возраста и состояния печени
Нитразепам 24-36 Возможно неактивные. Большой объем распределения
Оксазепам 4-10 Неактивные: глюкурониды Плохое всасывание при пероральном приеме может замедлить начало эффекта
Празепам 30-120 Активные: деметилдиазепам Медленное всасывание при пероральном приеме
Темазепам 5-8 Возможно активные Медленное всасывание при пероральном приеме
Триазолам 3-5 Активные: а-гидрокситриазолам Быстрое всасывание при перораль-
ном приеме
кислоты и кетоны появляются в моче в виде глюкуронидов. За несколькими исключениями производные барбитуратов неактивны. Средняя скорость печеночного метаболизма зависит от особенностей препарата, но обычно она медленная (кроме тиобарбитуратов). Период полувыведения секобарбитала и пентобарбитала колеблется между 18 и 48 часами у разных людей. Для фенобарбитала он составляет 4-5 дней. Многократный прием этих препаратов может привести к кумулятивным эффектам.
Г. Экскреция. Водорастворимые метаболиты барбитуратов и других седативных и снотворных средств выводятся в основном почками. В большинстве случаев нарушения функции почек не оказывают значительного влияния на выделение исходного препарата. Фенобарбитал выводится с мочой в неизмененном виде в определенных пределах (у человека — 20-30 %), при этом скорость выведения можно повысить ощелачиванием мочи. Ощелачивание приводит к повышенной ионизации (фенобарбитал — слабая кислота с рКа = 7.2). В неиз
мененной форме с мочой выводятся только следовые количества бензодиазепинов и 10 % снотворной дозы мепробамата.
Д. Факторы, влияющие на биодиспозицию. На биодиспозицию снотворных и седативных средств могут влиять различные факторы, в первую очередь изменения функции печени вследствие болезней или возраста, а также повышение или снижение активности микросомальных ферментов под действием лекарств.
В целом снижение функции печени приводит к уменьшению скорости трансформации препаратов, метаболизируемых окислительными путями. В группу этих препаратов входят многие бензодиазепины, почти все барбитураты, пиперидиндионы и мепробамат. У пациентов пожилого возраста и у больных с тяжелыми заболеваниями печени период полувыведения этих препаратов обычно значительно длиннее. В таких случаях прием нескольких обычных доз приводит к избыточным эффектам на ЦНС. Поэтому дозу таких препаратов у пожилых пациентов или у больных с нарушениями функций
Гибкость комплекса хлорных каналов и ГА1\Ж-рецепторов
Молекулярная структура хлорного канала, содержащая ГАМК-рецептор,— это одна из наиболее гибких систем организма, реагирующих на лекарственные препараты. Наряду с бензодиазепинами и барбитуратами многие другие препараты, влияющие на ЦНС, связываются с этим важным каналом.
К другим депрессантам ЦНС относят пропофол (важный внутривенный анестетик), алфак-солон (стероидный анестетик), некоторые газообразные анестетики и ивермектин (противо-гельминтное средство). Эти препараты усиливают или имитируют эффекты ГАМК. (Следует заметить, что не доказано, является ли этот механизм единственным или главным для приве
денных средств). Вещества, возбуждающие ЦНС через хлорные каналы, включают пикротоксин и бикукуллин. Эти конвульсанты напрямую блокируют каналы (пикротоксин) или нарушают связывание ГАМК (бикукуллин).
Связывание разных лигандов осуществляется через различные участки макромолекулы канала. Некоторые препараты взаимодействуют аллостерически. Например, бензодиазепины повышают аффинность ГАМК к местам ее связывания, несмотря на то, что они находятся на разных субъединицах. Активация связывания бензодиазепинов, вызываемая барбитуратами, зависит от анестезирующей активности послед-
печени принято снижать. Метаболизм путем конъюгации с глюкуроновой кислотой в меньшей степени зависит от возраста или состояния печени, чем окислительный метаболизм.
Активность микросомальных ферментов печени может быть повышена у пациентов, длительно принимающих препараты из ранних поколений седативных и снотворных средств (индукция ферментов, глава 4). Препараты с длительным периодом полувыведения типа фенобарбитала и мепробамата скорее всего приведут к этому эффекту, который в итоге ускорит их собственный метаболизм и метаболизм других препаратов в печени. Самоиндукция метаболизма — это возможный, но документально не доказанный механизм развития толерантности к седативным и снотворным средствам. Усиление биотрансформации других препаратов под действием барбитуратов — это потенциальный механизм развития межгрупповых взаимодействий препаратов. Длительное применение бензодиазепинов не меняет активности печеночных ферментов.
Фармакодинамика бензодиазепинов и барбитуратов
А. Молекулярная фармакология ГАМКА-ре-цептора. Бензодиазепины, барбитураты и новый седативный и снотворный препарат золпидем связываются в хлорных каналах с молекулами, выпол
няющими функцию ГАМКл-рецепторов, но не с самим местом связывания ГАМК.
Молекулярное клонирование показало, что ГАМКЛ-рецептор — это гетероолигомерный гликопротеин (200-400 кДа), состоящий по меньшей мере из трех субъединиц (а, Р и у), взаиморасположение которых до сих пор неизвестно. Было найдено несколько разных субъединиц одного типа, например шесть разных а, четыре Р и три у. Кроме того, предполагают существование б-, е- и р-субъ-единиц,- Хотя а- или Р-субъединицы сами по себе могут формировать хлорный канал, реагирующий с ГАМК, для нормальной физиологической и фармакологической функции молекулы необходима комбинация по меньшей мере трех типов субъединиц (а, р и у). В различных областях ЦНС рецепторы могут иметь строение разного типа (например (Xi, Pi, у2 или аз, pi, у2), и, таким образом, обладать разными фармакологическими свойствами. Как и у субъединиц н-холинорецептора (с которым ГАМКд-рецепторы гомологичны на 15—20 %), у субъединиц ГАМКд-рецептора есть четыре трансмембранных комплекса.
Разнообразие состава рецептора по комбинации субъединиц говорит о том, что ГАМК может связываться и с а-, и с р-субъединицами, управляя открытием и закрытием хлорного канала. Для чувствительности рецепторного комплекса к бензодиазепинам необходимо наличие у2-субъединицы, что
позволяет предположить, что бензодиазепиновый рецептор расположен на этой структуре или рядом. Разные комбинации субъединиц меняют чувствительность к бензодиазепинам и золпидему. Гипотетическая модель комплекса ГАМК-бензодиазепи-новый рецептор БДП-хлорионный канал показана на рис. 21-5.
Б. Нейрофармакология. ГАМК — это главный тормозный нейромедиатор в ЦНС. Электрофизиологические исследования показали, что бензодиазепины усиливают ГАМК-передачу в ЦНС (включая спинной мозг, гипоталамус, гиппокамп, черную субстанцию, кору мозжечка и мозга). Бензодиазепины повышают эффективность ГАМК-ергическо-го синаптического торможения (через гиперполяризацию мембраны), что приводит к снижению критической нейрональной активности во многих зонах мозга. Бензодиазепины не замещают ГАМК, но
Рис. 21-5. Гипотетическая модель макромолекулярного комплекса ГАМКл-рецептор-хлорионный канал. Это ге-тероолигомерный гликопротеиновый комплекс, который скорее всего состоит из 5 или более трансмембранных субъединиц. ГАМК может связываться с а- и с Р-субъединицами, вызывая открытие хлорионного канала с последующей гиперполяризацией мембраны. Связывание бензодиазепинов с у-субъединицей или с частью а-субъединицы, находящейся под влиянием у-субъедини-цы, облегчает открытие канала, но не оказывает прямого влияния на ток ионов хлора. (Из: Zorumsky С. F., Isenberg К. Е. Insights into the structure and functions of GAB A-benzodiazepine receptors: Ion channels and psychiatry. Ara. J. Psychiatry 1991; 148; 162.)
усиливают эффекты ее действия без прямой активации ГАМКд-рецепторов или связанных с ними хлорионных каналов.
Повышение тока ионов хлора, вызванное взаимодействием бензодиазепинов с ГАМК, связано с повышением частоты открытий каналов (рис. 21-6). Этот эффект может быть ассоциирован в частности с увеличением аффинности к ГАМК.
Барбитураты также облегчают действие ГАМК в разных отделах ЦНС, но в отличие от бензодиазепинов они повышают длительность периода открытия ГАМК-зависимых каналов (рис. 21-6). В высоких концентрациях барбитураты могут быть ГАМК-миметиками, напрямую активирующими хлорионные каналы. Эти эффекты опосредуются через участки рецепторов, отличные от мест связывания бензодиазепинов. Барбитураты менее селективны в своем действии, чем бензодиазепины, так как они параллельно с влиянием на ГАМК-передачу подавляют эффекты возбуждающих аминокислот и оказывают действие на внесинаптические мембраны. Такое множество точек приложения барбитуратов может лежать в основе их способности вызывать хирургическую анестезию (глава 24). Это также объясняет выраженный центральный тормозный эффект, что сужает терапевтический диапазон барбитуратов по сравнению с бензодиазепинами. Действие других седативных и снотворных препаратов типа мепробамата изучено хуже, но скорее всего оно не связано с ГАМК-передачей.
В. Лиганды бензодиазепиновых рецепторов. Найдены три типа взаимодействия лигандов и бензодиазепиновых рецепторов. Агонисты (1) облегчают действие ГАМК. Это в первую очередь — бензодиазепины, применяемые в клинике и обладающие анксиолитическим и антиконвульсивным эффектами. Существуют также и эндогенные агонисты. Так, бензодиазепиноподобные вещества были выделены из мозга животных, никогда не получавших подобные препараты, а бензодиазепиноподобная иммунореактивность определена в тканях мозга человека, фиксированных в парафине за 15 лет до синтеза первого бензодиазепина. Были найдены и эндогенные небензодиазепиновые молекулы, имеющие аффинитет к бензодиазепиновым рецепторам. Такие “эндозепины” облегчают ГАМК-регу-лируемое открытие и закрытие хлорионных каналов в клеточной культуре нейронов. В качестве примера антагонистов (2) можно привести производное бензодиазепинов флумазенил, который блоки-
Контроль
А
<^<1 iJ I / .»«JI. J «а I J ч “,‘rt f- r.^
2пА | 130 мсек
ГАМК 2 мкмоль
Б
VS'.’TM! »'
жштог
ГАМК 2 мкмоль + Диазепам 20 нмоль
ГАМК 2 мкмоль* Фенобарбитал 500 мкмоль
Рис. 21-6. Пэтч-кламп-регистрация одноканальных ГАМК-вызванных токов в нейронах спинного мозга мыши. А. Контроль. Б. Открытие канала (отклонения книзу), вызванное ГАМК. В. Диазепам повышает частоту открытий канала без влияния на его длительность. Г. Фенобарбитал продлевает длительность открытия канала без влияния на частоту. (Twyman R.E. et al. Differential regulation of GABA receptor channels by diazepam and phenobarbital. Ann. Neurol. 1989; 25; 213.)
рует их действие, но не влияет на эффекты барбитуратов,.мепробамата и этанола. Определенные эндогенные соединения, например диазепамсвязыва-ющий ингибитор, также могут блокировать взаимодействие бензодиазепинов со своими рецепторами. Инверсные агонисты (3) вызывают тревогу и судороги — эффекты, которые были найдены у нескольких веществ, особенно у |3-карболинов, например п-бутил-р-карболин-3-карбоксилата. Кроме своего прямого действия, эти молекулы могут блокировать эффекты бензодиазепинов.
Г. Эффекты на органном уровне.
1. Седация. Седация — это подавление реакции на постоянные раздражители со снижением уровня спонтанной активности и мышления. Эти поведенческие изменения происходят уже при приеме минимальных доз седативных и снотворных средств. Еще не известно, являются ли анксиолитические эффекты проявлениями седативного действия. В эксперименте на животных бензодиазепины и седативно-снотворные препараты ранних по
колений вызывали растормаживание поведения, подавленного наказанием, и этот эффект приравнивали к анксиолитическому. Однако это действие более соответствует общему растормаживающему поведенческому эффекту препаратов, ведущему к эйфории, нарушению суждений и потере самоконтроля, который может появиться при приеме доз, достаточных для анксиолитического эффекта. Большинство седативйых и снотворных средств обладают подобным действием у животных, однако бензодиазепины вызывают его в дозах, приводящих лишь к минимальному подавлению ЦНС. Антипсихотические препараты и трициклические антидепрессанты, обладающие седативными свойствами, не эффективны на этой экспериментальной модели. Бензодиазепины в седативных дозах могут вызвать антероградный амнестический эффект (невозможность вспомнить случившееся во время действия препарата).
2. Снотворное действие. Все седативные и снотворные средства вызывают сон при приеме доста
точных доз. Нормальный сон состоит из определенных стадий в соответствии с тремя физиологическими тестами — электроэнцефалографией, электромиографией и электронистагмографией (регистрация латеральных движений глазных яблок). Основываясь на показателях тестов, можно выделить два основных типа сна: сон с медленными движениями глаз (МДГ), составляющий 70-75 % общей продолжительности сна, и сон с быстрыми движениями глаз (БДГ). БДГ- и МДГ-сон возникают циклами с интервалом приблизительно в 90 минут. При БДГ-типе сна регистрируется большинство сновидений. МДГ-сон проходит в четыре стадии (1 -4 ), большую часть (50 %) составляет вторая стадия. За ней следует 8- или медленноволновой сон (стадии 3 и 4), в момент которого могут проявляться сомнамбулизм и возникать ночные кошмары.
Влияние препаратов на стадии сна активно изучалось в основном на здоровых добровольцах, а не на пациентах с нарушениями сна. Эффективность седативных и снотворных средств зависит от нескольких факторов, например от особенностей препарата, дозы, частоты приема. Существуют исключения из правил, но обычно эффекты седативных и снотворных средств на нормальный сон таковы: 1) снижается длительность периода засыпания (латентный период наступления сна); 2) продлевается стадия 2 МДГ-сна; 3) укорачивается стадия медленноволнового сна и 4) уменьшается длительность БДГ-сна.
Более быстрое начало сна и продление второй стадии считаются клинически полезными эффектами. Важность же влияния на БДГ- и медленноволновой сон пока не ясна. Использование седативных и снотворных средств в течение недели и более приводит к развитию толерантности к их влиянию на сон. Синдром отмены после долгого применения препаратов может привести к повышению частоты возникновения и длительности БДГ-сна.
3. Анестезия. Как показано на рис. 21-1, некоторые седативные и снотворные средства в высоких дозах могут подавить ЦНС до стадии общей анестезии (глава 24). Однако достоинства определенного препарата как дополнения к наркозу зависят в основном от химических свойств, определяющих быстроту начала и длительность эффекта. Среди барбитуратов тиопентал и метогекситал очень хорошо растворимы в жирах и быстро проникают в мозг после внутривенного введения. Быстрое перераспределение между тканями приводит к кратко
сти действия этих препаратов, поэтому они удобны для наркоза.
Некоторые бензодиазепины (включая диазепам и мидазолам) используются для наркоза внутривенно (глава 24), но их способность самостоятельно вызывать полноценный хирургический наркоз не доказана. Это подтверждается тем, что применяя бензодиазепины, нельзя снизить до нуля минимальную альвеолярную концентрацию другого анестетика. Интересно, что большие дозы бензодиазепинов, назначенных как дополнение к общей анестезии, могут содействовать возникновению персистирующей постнаркозной респираторной депрессии. Это, видимо, связано с их сравнительно коротким периодом полувыведейия и с образованием активных метаболитов.
4. Противосудорожный эффект. Большинство седативных и снотворных средств способно подавлять развитие и распространение эпилептической активности в ЦНС. Они обладают некоторой селективностью в том смысле, что могут оказывать противосудорожный эффект без значительного подавления ЦНС, то есть при сохраненной умственной и физической активности. Некоторые бензодиазепины, включая клоназепам, нитразепам, лоразепам и диазепам, обладают селективным действием, используемым для профилактики развития судорожных состояний (глава 23). Из барбитуратов для лечения grand mal (больших эпилептических припадков) и джексоновской эпилепсии эффективны фенобарбитал и метарбитал (который в организме превращается в фенобарбитал).
5. Миорелаксация. Некоторые седативные и снотворные средства, особенно относящиеся к карбаматной и бензодиазепиновой группам, оказывают ингибирующий эффект на полисинаптические рефлексы и передачу по вставочным нейронам, а в высоких дозах могут подавлять проведение по скелетному нервно-мышечному соединению. Такие селективные эффекты, приводящие к миорелаксации, легко продемонстрировать на животных, и они могут применяться при мышечных спазмах или заболеваниях суставов (раздел “Клиническая фармакология седативных и снотворных средств”).
6. Действие на дыхание и сердечно-сосудистую систему. В снотворных дозах у здоровых пациентов рассматриваемые препараты практически не нарушают естественного дыхания. Однако у больных с обструктивными заболеваниями легких седативные и снотворные средства даже в терапевти
ческих дозах могут вызвать значительную респираторную депрессию. Эффект зависит от дозы; угнетение дыхательного центра продолговатого мозга — это основная причина смерти после передозировки седативных и снотворных средств.
В дозах, не превышающих снотворные, значительные эффекты на сердечно-сосудистую систему у здоровых людей отсутствуют. При гиповолемии, застойной сердечной недостаточности и других заболеваниях, нарушающих функцию сердечно-сосудистой сисемы, обычные дозы седативных и снотворных средств могут вызвать сердечно-сосудистую депрессию за счет действия на вазомоторные центры продолговатого мозга. Токсические дозы могут угнетать сократимость миокарда и сосудистый тонус за счет центральных и периферических эффектов препаратов и приводить к циркуляторному коллапсу. Дыхательные и сердечно-сосудистые эффекты более выражены, когда седативные и снотворные средства назначаются внутривенно.
Толерантность. Психическая и физическая зависимость
Толерантность — снижение реакции на препарат вследствие его длительного применения — это частое явление при применении снотворных и седативных средств. В некоторых случаях она может привести к необходимости повышать дозу, чтобы продолжить симптоматическое лечение препаратом или чтобы вызвать сон. Важно понимать, что между описываемыми здесь седативными и снотворными средствами, а также между ними и этанолом (глава 22) возникает частичная кросс-толерантность. Механизмы развития толерантности к седативным и снотворным средствам недостаточно известны. Возможно, они связаны с изменением скорости метаболической инактивации при длительном применении (метаболическая толерантность) барбитуратов, однако для большинства седативных и снотворных средств исключительно важную роль играет снижение реактивности нервной системы (фармакодинамическая толерантность).
Полезные свойства (уменьшение тревоги, возникновение эйфории, растормаживание и индукция сна) привели к избыточному применению седативных и снотворных средств (глава 31). Последствия злоупотребления этими препаратами делятся на психологические и физиологические. Психологический компонент может исходно выглядеть как
невротическое поведение, которое трудно отличить от поведения закоренелого кофеиниста или курильщика. Когда пациент начинает ощущать необходимость приема седативных и снотворных средств, то возникают более серьезные осложнения, включающие физическую зависимость и толерантность.
Физическая зависимость может быть охарактеризована как измененное физиологическое состояние, требующее постоянного приема препарата для предотвращения абстинентного синдрома или синдрома отмены. Для седативных и снотворных средств этот синдром характеризуется повышенной тревожностью, бессонйцей, возбудимостью ЦНС, которая может перерасти в судороги. Длительный прием любых седативных и снотворных средств может вызвать физическую зависимость. Тем не менее тяжесть абстинентных симптомов зависит от конкретного препарата и от того, какую дозу получал пациент непосредственно перед прекращением приема. Чем выше была доза, тем выраженнее абстинентные симптомы. Разница по степени их тяжести зависит от периода полувыведения препарата. Так, медленно выводимые препараты достаточно долго остаются в организме. Это смягчает проявления абстинентного синдрома и обеспечивает постепенную отмену лекарств. Использование медикаментов с очень коротким периодом полувыведения для получения снотворного эффекта может привести к возникновению абстинентных симптомов даже между отдельными приемами препарата. Например, триазолам — бензодиазепин с периодом полувыведения около 4 часов, может вызывать дневную тревожность в тех случаях, когда он используется как снотворное средство.
Антагонисты бензодиазепинов: флумазенил
Флумазенил — это один из нескольких производных 1,4-бензодиазепинов с высокой аффинностью к бензодиазепиновым рецепторам, действующий как конкурентный антагонист. Это единственный антагонист бензодиазепиновых рецепторов, используемый в клинике в настоящее время. Он блокирует многие эффекты бензодиазепинов, но не препятствует действию на ЦНС других седативных и снотворных средств, этанола, опиоидов или общих анестетиков. Флумазенил разрешен к применению для устранения депрессии ЦНС при пере
дозировке бензодиазепинов и после их применения для анестезии и в диагностических целях. Препарат снимает седативное действие бензодиазепинов, но его эффект на вызванную бензодиазепинами респираторную депрессию менее предсказуем. Временное улучшение состояния сознания и мышления возникало при приеме флумазенила на фоне печеночной энцефалопатии. При внутривенном введении он действует быстро, но непродолжительно (период полувыведения 0.7-1.3 часа) вследствие быстрого печеночного клиренса. Так как действие всех бензодиазепинов длится дольше, чем действие флумазенила, то седативный эффект восстанавливается, и требуется повторная инъекция флумазецила. Побочные эффекты препарата включают возбуждение, затуманивание сознания, головокружение и тошноту. Флумазенил может вызывать очень тяжелый абстинентный синдром у пациентов с физической зависимостью от бензодиазепинов. У больных, принимающих бензодиазепины вместе с трициклическими антидепрессантами, назначение флумазенила может привести к развитию судорог и аритмий.
Новые седативные
и снотворные средства
Хотя бензодиазепины по-прежнему считаются препаратами выбора для лечения тревожных состояний и бессонницы, к числу их фармакологических эффектов относятся седация и сонливость, синергизм с другими препаратами (особенно алкоголем)
в торможении ЦНС и развитие зависимости при длительном применении. У анксиолитиков, которые действуют не через ГАМК-ергические системы, эти проявления менее выражены. Было исследовано несколько таких новых небензодиазепинов, в том числе буспирон. Другой новый препарат — золпидем действует на бензодиазепиновый рецептор, хотя по структуре отличается от бензодиазепинов.
Буспирон
Буспирон снижает тревожность, не оказывая значительного седативного действия. В отличие от бензодиазепинов он не обладает снотворным, противосудорожным или миорелаксантным свойствами. Буспирон не действует на ГАМК-ергические системы, но оказывает анксиолитический эффект в качестве парциального агониста 5-НТ)А-рецепто-ров. Этот препарат не снимает синдром отмены бензодиазепинов или других седативных или снотворных средств и имеет низкий потенциал зависимости. Буспирон не потенцирует торможение ЦНС, вызванное другими седативными или снотворными средствами, этанолом или трициклическими антидепрессантами. В отличие от бензодиазепинов анксиолитические эффекты буспирона достигают максимума в течение недели, поэтому его применяют только при общих тревожных состояниях. Для лечения панических состояний буспирон неэффективен.
Буспирон быстро всасывается в желудочно-кишечном тракте, но затем подвергается значительным превращениям (эффект первого прохожде
Золпидем
ния). Время его полувыведения 2-4 часа; свободного препарата в моче почти не находят. Печеночные ферменты метаболизируют препарат путем гидроксилирования и N-деалкилирования с образованием нескольких производных, обладающих следовой фармакологической активностью. Дисфункция печени может снизить клиренс препарата.
Буспирон вызывает меньше психомоторных нарушений, чем диазепам, и не препятствует вождению автомобиля, однако тахикардия, сердцебиения, нервозность, желудочно-кишечные нарушения и парестезии возникают чаще, чем при приеме бензодиазепинов.
У больных, принимающих одновременно с бус-пироном ингибиторы моноаминоксидазы, может повышаться артериальное давление.
В настоящее время исследуется несколько разработанные аналогов буспирона (например, ипса-пирон, гепирон, тандоспирон).
Золпидем
Золпидем — это производное имидазопиридина, структурно отличающееся от бензодиазепинов. Препарат связывается с бензодиазепиновыми рецепторами и скорее всего имеет сходные с бензодиазепинами механизмы действия по облегчению ГАМК-передачи. При краткосрочном лечении бессонницы эффективность и побочные эффекты золпидема сходны с таковыми у триазолама. Информация о возникновении толерантности и зависимости при продолжительном применении золпидема отсутствует. Препарат быстро метаболизируется печенью и имеет период полувыведения, равный приблизительно 2-4 часам. При нарушениях функции печени и для пожилых больных рекомендуется снижать дозы.
Седативные и снотворные средства предыдущих поколений
К этой группе препаратов относятся спирты (этхлорвинол, хлоралгидрат), пиперидиндионы (глютетимид, метиприлон), карбаматы (мепробамат) и даже неорганический ион бромида. Несмотря на дешевизну, они редко применяются в клинике. О молекулярных механизмах действия известно немного. Большинство из этих препаратов био-
трансформируются печеночными ферментами до более водорастворимых соединений. Трихлорэта-нол — это фармакологически активный метаболит хлоралгидрата с периодом полувыведения 6-10 часов. Однако его токсическое производное, трихлоруксусная кислота, выводится очень медленно и может накапливаться в организме при регулярном приеме хлоралгидрата. Более того, существует мнение о возможной канцерогенности хлоралгидрата или его метаболитов, поэтому следует воздержаться от использования препарата до выяснения этого вопроса,
II, Клиническая фармакология седативных и снотворных средств
Лечение тревожных состояний
Психологические, поведенческие и физиологические проявления тревожности принимают различные формы. Классическое психическое осознание чувства тревоги сопровождается психическим и моторным напряжением и автономной гиперактивностью. До назначения седативных и снотворных средств необходимо внимательно проанализировать все симптомы. Тревожность часто возникает вторично, на почве органических заболеваний (острого инфаркта миокарда, стенокардии, язвенной болезни желудка и двенадцатиперстной кишки и т. д.), требующих специфической терапии. Другая группа вторичных тревожных состояний (ситуационные тревожные состояния) связана с ситуациями, которые происходят в жизни всего один или несколько раз (пугающая медицинская или стоматологическая процедура, болезнь в семье или другая трагедия). Несмотря на то, что ситуационные тревожные состояния имеют тенденцию к самоограничению, кратковременное применение седативных средств может быть важным для их лечения и для лечения некоторых сопряженных с ними проявлений. Правильным и рациональным является также применение седативных и снотворных средств для премедикации перед операцией или какой-то неприятной медицинской процедурой (табл. 21-2). Если основной жалобой пациента является хроническое тревожное состояние, то для принятия решения о правильности диагноза и не-
ТАБЛИЦА 21 -2. Клиническое применение
седативных и снотворных средств
Снятие тревожности
Снотворный эффект
Седация и амнезия перед медицинскими и хирургическими процедурами
Лечение эпилепсии и других судорожных состояний
Как компонент сбалансированной анестезии при внутривенном введении
Для профилактики синдрома отмены этанола или других седативных и снотворных средств
Для миорелаксации при специфических нервно-мышечных заболеваниях
Для диагностики и для лечения в психиатрии
обходимости медикаментозного лечения следует воспользоваться Диагностическим и статистическим руководством по психическим заболеваниям (Diagnostic and Statistical Manual of Mental Disorders, DSMIII-R). Например, чрезмерная или несоответствующая ситуации тревожность относительно жизненных обстоятельств (генерализованные тревожные состояния), панические расстройства и агорафобия поддаются лекарственной терапии, особенно в сочетании с психотерапией. В некоторых случаях тревожность может быть симптомом других психических проблем, которые потребуют лечения трициклическими антидепрессантами или антипсихотическими препаратами.
Бензодиазепины — это препараты, наиболее часто используемые для лечения тревожных состояний. В 1991 г., например, алпразолам был на пятом месте в числе самых часто назначаемых препаратов в США, а лоразепам и диазепам попали на 18 и 20 места соответственно (среди препаратов-генериков). Многие седативные средства могут оказать анксиолитический эффект, и бывает трудно доказать превосходство одного препарата над другим. Поэтому предпочтение определенного препарата часто зависит от других причин, а не от анксиолитического эффекта. Исключением из общего правила является алпразолам, который особенно эффективен в терапии панических состояний и агорафобий, проявляя большую селективность в этом отношении, чем другие бензодиазепины. Выбор бензодиазепина основывается на нескольких основных фармакологических принципах: 1) сравнительно высокий терапевтический индекс (препарат Б на рис. 21-1) плюс возможность использования флу
мазенил а в случае их передозировки; 2) низкий риск лекарственных взаимодействий, связанный с индукцией печеночных ферментов; 3) низкая скорость выведения, которая может продлевать эффекты на ЦНС; 4) низкий риск физической зависимости с минимальными симптомами отмены.
Недостатки бензодиазепинов — это наличие тенденции к развитию психологической зависимости, образование активных метаболитов, амнестический эффект и высокая стоимость. Бензодиазепины усиливают депрессию ЦНС при совместном применении с другими препаратами седативно-снотворной группы, включая этанол. Исключением является бус-пирон. Пациента необходимо предупреждать о возможности такого осложнения, чтобы избежать проблем с выполнением действий, требующих высокого уровня бодрствования и двигательной координации.
Основное правило применения этих препаратов — использование выбранного препарата с осторожностью, чтобы избежать побочных эффектов. Принимаемые дозы не должны влиять на мыслительные и двигательные функции в рабочее время. Некоторым пациентам удобнее, когда большую часть суточной дозы они принимают перед сном, а днем — только маленькие. Следует стремиться назначать препарат на короткое время, так как долгосрочный прием оправдан только в определенных случаях. Врачу необходимо оценивать эффективность препарата по субъективным ощущениям пациента, поскольку концентрации лекарств в плазме слишком широко варьируют, чтобы быть индикаторным показателем. Следует избегать комбинированной анксиолитической терапии, кроме того, пациентов необходимо предупреждать об опасности приема алкоголя и некоторых препаратов, продающихся без рецепта (глава 65).
Хотя еще используются фенобарбитал, мепробамат и средства, подавляющие вегетативные функции (гидроксизин, дифенгидрамин), но они уже почти полностью и оправданно заменены бензодиазепинами.
При некоторых ситуациях в качестве анксиолитических средств практикуется применение Р-блокаторов (например, пропранолола). Гипер-реактивносгь симпатической нервной системы, связанная с тревожностью, хорошо подавляется р-бло-каторами, кроме того, отмечается некоторое облегчение и несоматического компонента тревоги. Сравнение эффектов Р-блокаторов и бензодиазепинов не
выявило значительной терапевтической разницы, которая дифференцировала бы показания к применению этих препаратов. Побочные эффекты р-бло-каторов на ЦНС включают сонливость, красочные сны и галлюцинации.
Антигипертензивный препарат клонидин тоже использовался в лечении тревожных состояний, в частности панических приступов. Сопутствующее лечение ос-адреноблокаторами (в том числе и трициклическими антидепрессантами) может снизить эффекты клонидина. Синдром отмены клонидина после продолжительного лечения, особенно в высоких дозах, приводит к угрожающим жизни гипертоническим кризам (глава 11).
Лечение нарушений сна
Нарушения сна отличаются большим разнообразием, включающим трудности с засыпанием, частые ночные пробуждения, короткую продолжительность сна и “неосвежающий” сон. Бессонница — это серьезная жалоба, требующая внимательного выявления причин (органических, психологических, ситуационных и т. д.), которые можно лечить без снотворных препаратов. Нефармакологические методы лечения — это правильная диета и физические упражнения, исключение возбуждающих факторов перед сном, комфортная постель, отход ко сну в одно и то же время. Однако иногда пациенту требуется на короткое время седативно-снотворное средство. Следует заметить, что прекращение применения препарата любой группы может вызвать бессонницу по механизму отмены.
Часто появляющиеся сведения о превосходстве какого-либо препарата в связи с его селективным действием на определенные фазы сна не достоверны, так как мало что известно о функциях фаз сна.
Очевидно, что самой важной оценкой эффекта препарата являются клинические критерии. К сожалению, еще не создано идеального снотворного средства, которое бы вызывало сон, не меняя в то же время его естественную структуру. Идеальный препарат должен быстро (короткий латентный период) вызывать сон достаточной продолжительности с минимальной последующей сонливостью, дисфорией и психической или двигательной заторможенностью на следующий день. Еще применяются препараты прошлых поколений типа хлоралгидрата, секобарбитала и пентобарбитала, но обычно предпочтение отдается бензодиазепинам. Следовая седация в течение следующего дня более часто возникает при приеме тех препаратов, у которых более продолжителен период полувыведения и которые трансформируются в активные метаболиты, как например флуразепам. Однако длительность эффекта короткодействующих препаратов типа триазолама, наиболее часто применяемого снотворного препарата в США, может быть слишком мала, и пациенты могут просыпаться очень рано. При ежедневном применении снотворных средств возникает толерантность, требующая повышения дозы для достижения желаемого эффекта. Следует помнить, что при развитии физической зависимости прекращение приема короткодействующих препаратов вызывает более серьезные симптомы отмены, которые могут включать тревожность и бессонницу, двигательное беспокойство, повышение рефлекторной активности и, возможно, судороги. Все бензодиазепины в какой-то степени вызывают антероградную амнезию. Препараты, обычно применяемые для седативного и снотворного эффектов, и их рекомендуемые дозы приведены в таблице 21-3. Внимание! Длительный прием снотворных средств — это необоснованная и опасная практика.
ТАБЛИЦА 21 -3. Дозы препаратов, обычно применяемых для седации и в качестве снотворных
Седативное действие Снотворное действие
Препарат Доза Препарат Доза (перед сном)
Алпразолам (Ксанакс) 0.25-0.5 мг 2-3 раза в день Хлоралгидрат 500-1000 мг
Хлордиазепоксид (Либриум) 10-20 мг 2-3 раза в день Флуразепам (Далман) 15-30 мг
Клоразепат(Транксен) 5-7.5 мг 2 раза в день Лоразепам (Ативан) 2-4 мг
Диазепам (Валиум) 5 мг 2 раза в день Фенобарбитал 100-200 мг
Лоразепам (Ативан) 1 -2 мг 1 -2 раза в день Секобарбитал 100-200 мг
Оксазепам (Сераке) 15-30 мг 3-4 раза в день Темазепам (Ресторил) 10-30 мг
Фенобарбитал 15-30 мг 2-3 раза в день Триазолам (Халцион) 0.125-0.5 мг
Празепам (Центракс) 10-20мг 2-3 раза в день
Другие терапевтические показания
В таблице 21-2 представлены основные показания к применению препаратов седативного и снотворного действия. Описание средств, используемых для лечения судорог и для внутривенного наркоза, приводится в главах 23 и 24.
Для получения седативных и возможных амнестических эффектов во время терапевтических или хирургических процедур, например эндоскопии и бронхоскопии, а также для премедикации перед анестезией предпочтительно используют седативные препараты короткого действия. При тщательном контроле за применением риск случайной или неслучайной передозировки ниже, чем при амбулаторном назначении препарата, поэтому барбитураты используются в этих целях так же часто, как и другие седативные и снотворные средства.
Препараты длительного действия (диазепам и в меньшей степени хлордиазепоксид или фенобарбитал) используют в прогрессивно снижаемых дозах в случае развития синдрома отмены при физической зависимости от этанола или других седативных и снотворных средств.
Мепробамат и бензодиазепины часто применяют как центральные миорелаксанты, несмотря на отсутствие достаточного количества данных, подтверждающих их эффективность без сопутствующей седации. Возможным исключением является диазепам, который хорошо расслабляет скелетные мышцы при спастичности центрального генеза (глава 26).
В психиатрии бензодиазепины, кроме терапии тревожных состояний, назначают для начального лечения мании, а также для лечения больных с депрессивными расстройствами (алпразолам). Седативные й снотворные средства иногда используют-ся для диагностики в неврологии и психиатрии.
Клиническая токсикология седативных и снотворных средств
Прямое токсическое действие
Многие из побочных эффектов препаратов этого класса вызваны дозозависимой депрессией функций ЦНС. У амбулаторных пациентов достаточно
низкие дозы могут привести к сонливости, нарушению суждений, затруднению моторных функций, снижению работоспособности, иногда со значительным влиянием на навыки вождения автомобиля, к изменению личных отношений. Бензодиазепины вызывают значительную дозозависимую антероградную амнезию. Эти препараты влияют на способности к приобретению новых знаний, особенно если это требует напряжения мыслительных процессов, но эффективность использования ранее полученной информации не изменяется. Этим пользуются при проведении неприятных процедур, например эндоскопии, так как правильно подобранная доза позволяет врачу поддерживать контакт с больным во время процедуры, но воспоминаний о процедуре не остается. Нередко наблюдаются остаточные эффекты препаратов, особенно длительнодействующих, принимаемых при нарушениях сна. Пожилым больным, более чувствительным к эффектам седативных и снотворных препаратов, назначают только половинные дозы. Самая частая причина обратимых нарушений сознания у пожилых людей — это передозировка седативных и снотворных препаратов. При приеме высоких доз токсичность может проявляться в виде повышенной сонливости, ощущения утомления, а иногда напоминает интоксикацию этанолом. Разделение лечебных эффектов от побочных затруднено у лекарств, для которых кривая доза-эффект выглядит, как у препарата А на рис. 21-1, например барбитуратов и пи-перидиндионов. Нежелательная депрессия ЦНС часто возникает при слишком длительном лечении, а также при применении препаратов с большим периодом полувыведения или образующих активные метаболиты. Врачи должны учитывать вариабельность доз, вызывающих побочные эффекты. У одного больного сравнительно низкая доза может привести к значительному подавлению ЦНС, у другого подобное состояние может быть вызвано только в 2-3 раза более высокой дозой. Такая вариабельность более выражена у больных с сердечно-сосудистыми, легочными заболеваниями, поражениями печени и у пожилых людей.
Седативные и снотворные препараты из-за своей доступности чаще других используются при умышленных передозировках. В этом отношении бензодиазепины считаются сравнительно безопасными препаратами, так как у них кривая зависимости доза-эффект более пологая. Эпидемиологические исследования смертей, связанных с передози
ровкой препаратов, подтверждают это. Так, в одном исследовании отмечали 0.3 смертей на миллион таблеток диазепама и 11.6 смертей на миллион капсул секобарбитала. Конечно, многие другие факторы тоже оказывают свое влияние, например сопутствующие эффекты этанола. Известно, что самые серьезные случаи передозировок, случайных и намеренных, связаны с применением нескольких препаратов одновременно. В этой ситуации бензодиазепины в комбинациях с другими препаратами могут оказаться далеко не безвредными.
Летальная доза любого седативно-снотворного препарата варьирует в зависимости от пациента и многих других обстоятельств (глава 60, том 2). Если о передозировке стало известно рано и начато своевременное лечение, то летальный исход возникает редко, даже при приеме очень высоких доз. С другой стороны, для большинства седативных и снотворных средств, кроме бензодиазепинов, доза, всего в 10 раз превышающая снотворную, может стать летальной, если вовремя не приняты меры. При тяжелой интоксикации у больного, которому не оказана помощь, центральная респираторная депрессия может осложняться аспирацией желудочного содержимого, что еще более вероятно при сопутствующем приеме этанола. Потеря стволового вазомоторного контроля и прямая депрессия миокарда снижают эффективность реанимационных мероприятий. Таким больным показаны искусственная вентиляция легких, поддержание объема плазмы, диуреза и сердечного выброса и, по-видимому, назначение инотропного препарата типа дофамина, который не влияет на почечный кровоток. Для ускорения выведения некоторых из этих препаратов могут применяться гемодиализ или гемоперфузия (табл. 60-4, том 2). Флумазенил является антагонистом седативного эффекта бензодиазепинов. Однако он действует очень коротко, и его влияние на респираторную депрессию непредсказуемо. Поэтому использование флумазенила при передозировке бензодиазепинов должно сопровождаться постоянным контролем и поддержкой дыхательной функции.
Побочные эффекты седативных и снотворных препаратов, не связанные с их действием на ЦНС, встречаются редко. Реакции гиперчувствительности типа кожной сыпи иногда возникают при приеме любого из препаратов этого класса. Сведения о тератогенности, ведущей к порокам развития плода при приеме пиперидиндионов и некоторых бензо
диазепинов, заставляют проявлять особую осторожность при использовании этих препаратов во время беременности. Так как барбитураты увеличивают синтез порфиринов, они абсолютно противопоказаны при наличии в анамнезе острой периодической порфирии, смешанной порфирии, наследственной копропорфирии или симптоматической порфирии.
Изменения реакции на препарат
В зависимости от дозы и от длительности применения возникают разные степени толерантности к различным эффектам седативных и снотворных препаратов. В этом можно убедиться, анализируя ЭЭГ и измеряя другие характеристики стадий сна у людей, длительно принимающих такие препараты. Конечно же, феномен толерантности распространяется и на иные эффекты. Так, известно, что у людей, злоупотребляющих снотворно-седативными препаратами в дозах, во много раз превышающих обычные, часто не развиваются признаки токсичности. Не следует думать, что степень толерантности одинакова для всех эффектов. Есть данные, что длительное применение седативных и снотворных препаратов не изменяет границы летальных доз. Кросс-толерантность между разными седативными и снотворными средствами, в том числе и этанолом, может привести к неудовлетворительному терапевтическому эффекту при применении стандартных доз у пациентов, ранее злоупотреблявших приемом какого-либо препарата этой группы.
При длительном применении седативных и снотворных препаратов, особенно в возрастающих дозах, может возникнуть физическая зависимость вплоть до степени, превышающей таковую для других препаратов, в том числе и опиоидов. Синдром отмены седативных и снотворных препаратов может иметь угрожающие жизни проявления, от двигательного беспокойства, тревожности, слабости и ортостатической гипотензии до гиперрефлексии и генерализованных судорог. Тяжесть симптомов во многом зависит от дозы, которую получал пациент непосредственно перед прекращением приема, а также от особенностей препарата. Например, барбитураты типа секобарбитала или пентобарбитала (в дозах < 400 мг/день) или диазепама (< 50 мг/день) могут вызвать только легкие симптомы при прекращении приема. Прием более 800 мг/день барбитуратов или 50-60 мг/день диазепама в течение 60-90 дней скорее всего приведет к развитию судо
рог при внезапной отмене препарата. Обычно симптомы отмены более выражены, если препарат имеет короткий период полувыведения, и проявляются в меньшей степени у препарата с длительным периодом полувыведения, за счет смягчения эффекта отмены долгим следовым пребыванием в организме. Кросс-зависимость, то есть способность одного препарата подавлять абстинентный синдром после прекращения приема другого препарата, у седативных и снотворных препаратов выражена значительно. На этом основываются методики коррекции синдрома отмены: используются длительнодействующие препараты типа фенобарбитала и диазепама для снятия синдрома отмены короткодействующих препаратов, в том числе этанола.
Взаимодействия препаратов
Наиболее часто седативные и снотворные препараты взаимодействуют с другими депрессантами ЦНС, приводя к суммации эффекта. Эти взаимодействия используются при подборе адъювантных средств как компонентов анестезии или премедикации. Если их не учитывать, то может возникнуть значительная нежелательная депрессия ЦНС. Очевидна суммация эффекта при одновременном употреблении алкогольных напитков, наркотических аналгетиков, антиконвульсантов, фенотиазинов и других седативных и снотворных препаратов. Менее выраженная, но тем не менее значительная суммация депрессивного влияния на ЦНС, возникает при одновременном приеме антигистаминных, антигипертензивных препаратов и трициклических антидепрессантов.
Взаимодействия, связанные с активностью ферментов печени, могут возникнуть при долгом применении барбитуратов и мепробамата. Например, показано, что у людей барбитураты повышают скорость метаболизма дикумарола, фенитоина, препаратов наперстянки и гризеофульвина, что снижает эффективность этих препаратов. Бензодиазепины при длительном использовании в такие лекарственные взаимодействия не вступают. Циметидин, понижающий печеночный метаболизм многих препаратов, продлевает действие диазепама в два раза, предположительно через подавление его метаболизма. Как уже отмечалось ранее, хлоралгидрат может вытеснять варфарин из связей с белками плазмы, усиливая его антикоагулянтный эффект.
Препараты
Бензодиазепины:
Алпразолам (Ксанакс)
Перорально: таблетки по 0.25, 0.5,1.2 мг
Хлордиазепоксид (генерик, Либриум и др.) Перорально: таблетки и капсулы по 5, 10, 25 мг
Парентерально: 100 мг порошка для инъекций
Клоразепат (Транксен)
Перорально: таблетки и капсулы по 3.75, 7.5, 15 мг
Перорально для пролонгированного действия: таблетки по 11.25, 22.5 мг Парентерально: 100 мг порошка для инъекций
Клоназепам (Клонопин)
Перорально: таблетки по 0.5,1.2 мг
Диазепам (генерик, Валиум и др.) Перорально: таблетки по 2, 5, 10 мг, растворы 5 мг/5 мл, 5 мг/мл Перорально для пролонгированного действия: капсулы по 15 мг Парентерально: 5 мг/мл для инъекций
Эстазолам (ПроСом)
Перорально: таблетки по 1.2 мг
Флуразепам (генерик, Далман) Перорально: капсулы по 15, 30 мг
Галазепам (Паксипам)
Перорально: таблетки по 20,40 мг
Лоразепам (генерик, Ативан, Алзапам) Перорально: таблетки по 0.5,1.2 мг Парентерально: 2.4 мг/мл для инъекций
Мидазолам (Версед)
Парентерально: 1, 5 мг/мл по 1, 2, 5,10 мл в ампулах для инъекций
Оксазепам (генерик, Сераке) Перорально: таблетки по 15 мг, капсулы по 10, 15,30 мг
Празепам (Центракс)
Перорально: таблетки по 10 мг, капсулы по 5,10,20 мг
Квазепам (Дорал)
Перорально: таблетки по 7.5,15 мг
Темазепам (генерик, Ресторил) Перорально: капсулы по 15, 30 мг
Триазолам (Халцион)
Перорально: таблетки по 0.125, 0.25 мг
Антагонист бензодиазепинов
Флумазенил (Ромазикон)
Парентерально: 0.1 мг/мл для в/в инъекции
Барбитураты
Амобарбитал (генерик, Амитал)
Перорально: таблетки по 30, 50, 100 мг (основные), капсулы по 65, 200 мг (натриевая соль)
Парентерально: порошок по 250, 500 мг во флаконах для инъекций
Апробарбитал (Алурат)
Перорально: эликсир 40 мг/5 мл
Бутабарбитал натрия (генерик, Бутизол и др.) Перорально: таблетки по 15, 30, 50,100 мг, капсулы по 15, 30 мг, эликсиры 30, 33.3 мг/5 мл
Мефобарбитал (Мебарал)
Перорально: таблетки по 32, 50, 100 мг
Метарбитал (Гемонил)
Перорально: таблетки по 100 мг
Пентобарбитал (генерик, Нембутал натрия) Перорально: капсулы по 50,100 мг, эликсир 18.2 мг/5 мл Ректально: 30, 60,120, 200 мг в свечах Парентерально: 50 мг/мл для инъекций
Фенобарбитал (генерик, Люминал натрия, др.) Перорально: таблетки по 8, 16, 32, 65, 100 мг, капсулы по 16 мг, эликсиры 15, 20 мг/5 мл Парентерально: 30, 60, 65, 130 мг/мл для инъекций, 120 мг порошка в ампулах для инъекций
Секобарбитал (генерик, Секонал) Перорально: капсулы по 50,100 мг, таблетки по 100 мг Ректально: 50 мг/мл Парентерально: 50 мг/мл для инъекций
Талбутал (Лотузат)
Перорально: таблетки по 120 мг
Разные препараты
Буспирон (Бу Спар)
Перорально: таблетки по 5,10 мг
Хлоралгидрат (генерик, Ноктек, Аквахлорал Суппреттес)
Перорально: капсулы по 250, 500 мг, сиропы 250, 500 мг/5 мл
Ректально: 324, 500, 684 мг в свечах
Этхлорвинол (Плацидил)
Перорально: капсулы по 200, 500, 750 мг
Этинамат (Валмид Пулвулес)
Перорально: капсулы по 500 мг
Глютетимид (генерик, Дориден)
Перорально: таблетки по 250, 500 мг
Гидроксизин (генерик, Атаракс, Вистарил) Перорально: таблетки по 10, 25, 50,100 мг, капсулы по 25, 50,100 мг, сироп 10 мг/5 мл, суспензия 25 мг/5 мл
Парентерально: 25, 50 мг/мл для инъекций
Мепробамат (генерик, Милтаун, Икванил, др.) Перорально: таблетки по 200,400, 600 мг Перорально для пролонгированного действия: капсулы по 200,400 мг
Метиприлон (Нолудар)
Перорально: таблетки по 200 мг, капсулы по 300 мг
Паральдегид (генерик)
Пероральные и ректальные жидкости
Золпидем (Амбиен)
Перорально: таблетки по 5, 10 мг
Избранная литература
Ballenger J. С. Pharmacotherapy of the panic disorders. J. Clin. Psychiatry, 1986; 47 (Suppl.): 27.
Gillin J. C., Byerly W. F. The diagnosis and management of sleep disorders. N. Engl. J. Med. 1990; 322: 239.
Greenblatt D. J., Harmatz J. S., Shader R. I. Clinical pharmacokinetics of anxiolytics and hypnotics in the elderly. Clin. Pharmacokinet. 1991; 21:165.
Langtry H. D., Benfield P. Zolpidem: A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential. Drugs, 1990; 40: 291.
Lin L-H., Whiting P., Harris R. A. Molecular determinants of general anesthetic action: Role of GAB Ал receptor structure.}. Neurochem. 1993; 60:1548.
Спирты
22
Нэнси М. Ли, Чарльз Е. Беккер
Этиловый спирт (этанол) является веществом седативно-гипнотического действия, своеобразным “социальным” лекарством. Злоупотребление алкоголем (алкоголизм) — сложное заболевание, природа и причины которого окончательно не выявлены. Алкоголизм создает серьезные медицинские и общественные проблемы для многих стран.
Этанол и многие другие спирты с потенциально токсическими эффектами используются в промышленности, иногда в огромных количествах. Наряду с интоксикацией этанолом часто встречается отравление метанолом и этиленгликолем, что и является предметом обсуждения в данной главе.
1. Базисная фармакология этанола
Этанол
Фармакокинетика
Этанол (С2Н5ОН), являющийся небольшой растворимой в воде молекулой, быстро и полно всасывается из желудочно-кишечного тракта. Его пары могут легко абсорбироваться в легких. После приема этанола натощак максимальная концентрация в крови достигается в пределах 30 минут. Наличие пищи в кишечнике задерживает всасывание. Распределение происходит быстро, и уровень концентраций в тканях и крови становится приблизительно одинаковым. Объем распределения составляет около 0.7 л/кг.
Более 90 % поступившего этанола окисляется в печени, оставшийся экскретируется через легкие и почки. При приеме обычных доз скорость окисления соответствует кинетике нулевого порядка, т. е. не зависит от времени и концентрации вещества. Количество алкоголя, окисляемое за единицу времени, примерно пропорционально массе тела или печени. При гепатэктомии или печеноч
ной недостаточности скорость исчезновения алкоголя из организма значительно снижается, а иногда элиминация даже полностью прекращается. Взрослый человек может метаболизировать 7-10 г (0.15-0.22 моля) алкоголя в час. Известны два пути метаболизирования спирта до ацетальдегида, который в дальнейшем окисляется (Geokas, 1984).
А. Алкогольдегидрогеназный путь. Главный путь метаболизма алкоголя связан с алкогольдегидрогеназой, 2п2+-содержащим цитозольным ферментом, который катализирует превращение спирта в ацетальдегид в соответствии со следующей реакцией:
Алкогольдегидрогеназа
С2Н5ОН + НАД+—> СН3СНО + НАД-Н + Н+.
Этот фермент находится преимущественно в печени, но имеется и в других органах, например в мозгу и желудке.
У мужчин значительное количество этанола метаболизируется алкогольдегидрогеназой желудка; уровень алкоголя в крови у женщин при приеме внутрь выше, чем у мужчин, тогда как при внутривенном введении половые отличия не выявляются (Frezza et al., 1990).
В приведенной реакции ион водорода переходит от спирта на кофермент никотинадениндинуклео-тид (НАД) с образованием НАД Н. В результате окисление алкоголя создает избыток восстановленных эквивалентов в печени, главным образом в форме НАД-Н. Пока до конца не ясно, может ли хроническое потребление алкоголя влиять на активность печеночной алкогольдегидрогеназы. В действительности алкогольдегидрогеназа сама по себе не является лимитирующим скорость реакции фактором, скорость окисления может зависеть от доступности кофактора НАД. Следовательно, ускорение выведения спирта из крови у больных алкоголизмом может быть не связано с увеличением
активности алкогольдегидрогеназы. Тем не менее 4-метилпиразол (фомепизол) — соединение, применяющееся исключительно как антидот при отравлении метанолом и этиленгликолем, является активным ингибитором алкогольдегидрогеназы (Baud et al., 1986).
Б. Микросомальная этанолокисляющая система (МЭОС). Эта ферментативная система, известная также как оксидазы со смешанной функцией (глава 4), использует в качестве кофактора НАДФ-Н вместо НАД в следующей реакции:
мэос|
С2Н5ОН + НАДФ-Н + Н+ + О2
-у СНзСНО + НАДФ+ + 2Н2О.
Поскольку значения Кт для алкогольдегидрогеназы колеблются от 0.26 до 2 нмоль/л, а для МЭОС от 8 до 10 нмоль/л, полагают, что при концентрациях алкоголя ниже 100 мг% (22 нмоль/л) алкоголь дегидрогеназ а является главной окисляющей системой, тогда как при увеличении концентрации МЭОС начинает играть более значительную роль. При хроническом потреблении алкоголя активность МЭОС значительно возрастает. Как указывается в главе 4, такая индукция ферментативной активности связана с увеличением количества различных компонентов фракции гладкого эндоплазматического ретикулума, участвующих в метаболизме лекарств. В результате хроническое потребление алкоголя может значительно повысить клиренс лекарств, которые метаболизируются микросомальными ферментативными системами печени. Сходным образом другие вещества-индукторы микросомальных ферментов, например барбитураты, могут несколько увеличивать выведение алкоголя из крови. Однако это влияние не столь важно, так как МЭОС не является главным путем превращения этанола.
В. Метаболизм ацетальдегида. Принято считать, что более 90 % ацетальдегида, образовавшегося из спирта, также окисляется в печени. Теоретически несколько ферментативных систем могло бы катализировать эту реакцию. Однако по уровню ацетальдегида в печени после введения этанола (100-350 мкмоль/л) было сделано заключение о преимущественном его окислении митохондриальной НАД-зависимой альдегиддегидрогеназой (кажущаяся Кт для ацетальдегида около 10 мкмоль/л). Продуктом реакции является ацетат,
который может далее метаболизироваться до СО2 и воды. Хроническое потребление алкоголя ведет к понижению скорости окисления ацетальдегида в интактных митохондриях, хотя активность фермента и не изменяется.
Толерантность и физическая зависимость
Потребление алкоголя в больших дозах в течение продолжительного времени ведет к развитию толерантности и физической зависимости. Толерантность к алкогольной интоксикации является сложным процессом, включающим нарушение нормального метаболизма и малоизученные изменения нервной системы. Недавние исследования показали, что толерантность может быть связана с нарушениями транспорта аденозина, ведущими к изменению рецепторзависимого уровня цАМФ (Nagy et al., 1990). Острая толерантность может развиваться через несколько часов после принятия алкоголя, причем как у алкоголиков, так и потребляющих алкоголь в “социальной” обстановке. Хотя у хронических алкоголиков выявлена определенная степень метаболической толерантности, при которой повышается способность метаболизировать этанол, она все-таки недостаточна для объяснения реально существующей клинической толерантности. Наконец, как и в случае с другими седативными и гипнотическими средствами, существует определенный предел тканевой толерантности, так что совсем незначительное превышение летальной дозы сопряжено с возрастающей опасностью смертельного исхода.
У лиц, хронически потребляющих алкоголь, при уменьшении дозы или прекращении его приема развивается синдром отмены, свидетельствующий о физической зависимости. Механизм физической зависимости для веществ седативно-гипнотического действия и алкоголя мало изучен. Предполагают, что интенсивность синдрома отмены определяют доза, частота и продолжительность потребления алкоголя. Когда потребление очень велико, даже простое снижение дозы может привести к развитию синдрома отмены. Толерантность и физическая зависимость, безусловно, являются важными последствиями злоупотребления алкоголем, но спектр заболеваний, сопутствующих алкоголизму, столь широк, что этому феномену трудно дать однозначное определение.
Фармакодинамика этанола при однократном приеме
А. Центральная нервная система. Однократный прием алкоголя существенно влияет на центральную нервную систему. Этанол вызывает седативный эффект, уменьшение тревоги, несвязную речь, атаксию, нарушает способность к суждению, растормаживает поведение — все это обычно называют опьянением. Эти эффекты наиболее заметны при высокой концентрации алкоголя в крови.
Этанол влияет на многие молекулярные процессы. В настоящее время считается, что две наиболее важные мишени его действия — это клеточные мембраны и ферменты мозга. Мелкие органические молекулы, подобные этанолу, могут легко растворяться в липидном бислое клеточных мембран. Исследования показали, что этанол уменьшает вязкость мембран многих типов клеток и даже искусственных систем типа липосом. Общей реакцией на спирт практически всех биологических мембран является увеличение текучести, ответственной за огромное разнообразие его эффектов. К ним относятся изменения структуры и функции рецепторов дофамина, норадреналина, глутамата, опиоидов, таких ферментов как Na+,K+-АТФаза, Са2+-АТФаза, 5'-нуклеотидаза, ацетилхолинэстераза, аденилат-циклаза, ферментов митохондриальной электрон-транспортной цепи, ионных каналов, подобных кальциевым (Lee, 1986). У алкоголиков изменяется активность моноаминоксидазы и аденилатциклазы в тромбоцитах (Tabakoff et al., 1988). Эти изменения могут быть ценными маркерами избыточного потребления алкоголя и, возможно, предрасположенности к алкоголизму.
Этанол может оказывать прямое действие на рецепторзависимые ионные каналы и транспортные молекулы, ассоциированные с клеточными мембранами. Например, острое воздействие этанола увеличивает число ГАМК-рецепторов, что согласуется со способностью ГАМК-миметиков усиливать многие острые эффекты алкоголя. Кроме того, этанол ингибирует NMDA-подтип глутаматных рецепторов в клетках мозга млекопитающих и активирует фосфоинозитидспецифичную фосфолипазу С в гепатоцитах.
Алкоголь оказывает множество разнообразных влияний на клеточном и тканевом уровнях, особенно в нервной системе. Как внеклеточное, так и внутриклеточное введение этанола в тригеминальные
мотонейроны подавляет потенциалы действия, возбуждающие и тормозные постсинаптические потенциалы (ВПСП и ТПСП). Внутриклеточное введение также деполяризует мембрану, а внеклеточное ведет к двуфазным сдвигам потенциала. Установлено, что спирт может как ослаблять, так и усиливать нервно-мышечную передачу.
В клетках Пуркинье мозжечка и в дофаминсодержащих нейронах компактной части черной субстанции и стриатума этанол в малых концентрациях увеличивает, а в больших уменьшает частоту разрядов. В других отделах, включая латеральное коленчатое тело, голубое пятно, ядра шва среднего мозга и нейроны гиппокампа в культуре тканей, отмечены только ингибирующие влияния этанола. Более сложные нейрональные реакции изменяются сходным образом. Например, следовые разряды после высокочастотной стимуляции нейронов гиппокампа и спинальный рефлекс лягушки усиливаются при действии малых и подавляются после введения больших доз этанола (Lee, 1986).
Б. Сердце. Даже при употреблении умеренного количества этанола (концентрация в крови выше 100 мг/100 мл) отмечается значительное угнетение сократительной функции миокарда. При биопсии миокарда у человека до и после инфузии малых количеств алкоголя было обнаружено, что этанол вызывает ультраструктурные изменения, которые MOiyr быть связаны с нарушением миокардиальной функции. Как причину сердечных нарушений рассматривают накопление ацетальдегида, приводящее к изменению запасов катехоламинов в миокарде. Эксперименты на животных подтверждают повреждающее действие этанола на сердечную мышцу. Однако в одном из исследований было показано, что этанол может уменьшить нарушения в миокарде во время аноксии.
В. Гладкая мускулатура. Этанол является вазодилататором, что связано как с центральным действием (угнетение вазомоторного центра), так и с прямым расслабляющим влиянием на гладкую мышцу, вызванным его метаболитом ацетальдегидом. В случае серьезной передозировки при низкой температуре среды может развиваться гипотермия, обусловленная вазодилатацией. Этанол также расслабляет мускулатуру матки, поэтому раньше его использовали внутривенно для прекращения преждевременной родовой деятельности. Однако токсичность спирта для матери и опасность для плода сделали такое применение достоянием истории, тем
более что другие средства (блокаторы кальция, препараты магния, р2-адреностимуляторы и пр.) оказались более эффективны.
Последствия хронического потребления алкоголя
Литература по алкоголизму содержит ограниченные сведения о точной зависимости между дозой хронически потребляемого алкоголя и повреждением жизненно важных систем. Однако некоторые серьезные исследования с адекватным контролем показали, что порог повышения смертности находится в диапазоне 3-5 условных доз (drinks) в день, если за дозу принять среднестатистическое количество регулярно потребляемого алкоголя. Риск резко возрастает на уровне 6 и более доз в день. Смерть, связанная с потреблением алкоголя, происходит из-за заболеваний печени, рака, несчастных случаев и самоубийств.
А. Печень и желудочно-кишечный тракт. Алкоголь в больших дозах вызывает каскад метаболических эффектов, способствующих повреждению печени и ЖКТ. Повышение соотношения НАД-Н/ НАД, рассмотренное выше, зависит от наличия НАД. Предполагают, что измененение этого соотношения приводит к ряду метаболических нарушений в печени. Отмечают ингибирование глюконеогенеза, гипогликемию и кетоацидоз, накопление жиров в паренхиме печени. Такие факторы как наследственность, сопутствующие заболевания, количество алкоголя и продолжительность его применения, вероятно, определяют тяжесть повреждения печени. Вредное влияние на печень оказывает ацетальдегид. Так как его окисление альдегиддегидрогеназой ведет к образованию НАД-Н, некоторые эффекты ацетальдегида могут быть связаны с избытком восстановленного кофермента. Кроме того, ацетальдегид является очень реактивным веществом, которое может иметь и собственное токсическое действие.
В некоторых случаях определяющими для алкогольного повреждения печени оказываются факторы питания. Полагают, что при плохом питании у алкоголиков возникает недостаток глутатиона, т. е. дефицит “уборщиков” токсичных свободных радикалов, повреждающих печень. Могут играть роль и гормональные факторы, поскольку у женщин-алкоголиков риск вызванных алкоголем дисфункций печени выше, чем у мужчин-алкоголиков.
Серьезное заболевание печени может развиваться незаметно и прогрессировать даже на фоне нормального питания. Ожирение печени может переходить в алкогольный гепатит и, наконец, в цирроз. Печеночная недостаточность — одна из причин гибели алкоголиков.
Могут повреждаться и другие отделы ЖКТ. Прием алкоголя активирует желудочную и панкреатическую секрецию и изменяет слизистый барьер, что увеличивает риск гастритов и панкреатитов. Алкогольный гастрит часто вызывает острое желудочно-кишечное кровотечение. Острое действие алкоголя на желудок связано главным образом с токсическим влиянием этанола на мембраны слизистой оболочки и сравнительно меньше — с повышенной кислотной продукцией желудка. Хронические алкоголики склонны к развитию гастритов и к потере протеинов плазмы и крови, что может способствовать анемии и белковой недостаточности. Повреждающее действие алкоголя на тонкий кишечник выражается в диарее, потере веса, дефиците многих витаминов.
Распространенное представление о том, что нарушение пищеварения у алкоголиков связано с пищевым дефицитом, вероятно, не всегда верно. Свой вклад в клиническую картину вносит нарушение всасывания витаминов, особенно водорастворимых. Раньше их дефицит рассматривали как основу большинства патологических эффектов алкоголя, однако заместительные дозы витаминов не защищают полностью от его повреждающего действия. У больных алкоголизмом, несмотря на сравнительно редкий алиментарный дефицит тиамина, могут обнаруживаться его типичные проявления: синдром Вернике-Корсакова, периферическая нейропатия, поражение сердца по типу болезни бери-бери.
Б. Нервная система. Как уже упоминалось, хроническое потребление этанола вызывает толерантность и физическую зависимость. При снижении дозы или отказе от алкоголя развивается синдром отмены, проявляющийся повышенной возбудимостью, а в тяжелой форме — судорогами, токсическим психозом, белой горячкой. Острое и хроническое потребление алкоголя часто ведет к ослаблению краткосрочной памяти. Потребление больших количеств алкоголя длительное время (обычно годами) может вызвать ряд неврологических симптомов. У больного нарушаются интеллектуальные и моторные функции, отмечаются эмоциональная лабильность, искажение восприятия и амнезия. Наиболее
частое неврологическое нарушение при хроническом алкоголизме — генерализованные симметричные поражения периферических нервов, которые начинаются с парестезий дистальных отделов конечностей. Для исключения других причин периферических нейропатий необходим дифференциальный диагноз, однако подобная патология обычно связана с хроническим потреблением алкоголя.
Синдром Вернике-Корсакова является относительно редкой, но тяжелой формой поражений при алкоголизме, которая проявляется параличом наружных глазных мышц, атаксией, изменением мышления с амнезией и нарушением памяти. Он обычно ассоциируется с дефицитом тиамина, но редко выявляется вне связи с алкоголизмом. Болезнь Вернике-Корсакова бывает трудно отличить от острого состояния спутанности, вызванной алкогольной интоксикацией, с последующим присоединением перцептивных и поведенческих нарушений при синдроме отмены. Дифференциальнодиагностическими признаками болезни Корсакова являются большая продолжительность состояния спутанности сознания и относительно слабовыра-женная ажитация, более характерная для синдрома отмены. Учитывая тяжесть этого синдрома, все больные, у которых заподозрена болезнь Корсакова, должны получать лечение тиамином (50 мг внутривенно однократно и 50 мг внутримышечно ежедневно до перехода на полноценное питание).
Алкоголь может также нарушать остроту зрения с двусторонним затуманиванием, которое появляется через несколько недель неумеренного его потребления. При исследовании могут быть выявлены скотомы и снижение остроты видения близких и отдаленных предметов. Изменения обычно двусторонние и симметричные, нередко связанные с дегенерацией зрительного нерва. Тем не менее введение этанола является средством заместительной терапии при отравлении метанолом (раздел IV), который вызывает серьезные нарушения зрительной функции.
В. Кровь. Наиболее частое гематологическое расстройство при хроническом злоупотреблении алкоголем — умеренная анемия из-за дефицита фолиевой кислоты. Железодефицитная анемия может быть связана с кишечным кровотечением. Была описана у таких больных и сидеробластная анемия. Алкоголь рассматривают как одну из причин тяжелых гемолитических синдромов, часть которых связана с гиперлипидемией и серьезными поражения
ми печени. Он прямым воздействием подавляет пролиферацию всех клеточных элементов костного мозга. У алкоголиков описаны аномалии тромбоцитов и лейкоцитов. Такие аномалии могут рассматриваться как.фактрры, способствующие нарушениям гемостаза и повышению частоты инфекций.
Г. Сердечно-сосудистая система. Алкоголь влияет на сердечно-сосудистую систему разными способами. Прямое повреждение миокарда при злоупотреблении алкоголем, как полагают, связано с дефицитом тиамина или с наличием примесей, содержащихся в алкогольных напитках. В настоящее время считают, что алкогольная кардиомиопатия у мужчин связана с эпизодами тяжелых запоев в течение длительного времени независимо от дефицита витаминов или других компонентов пищи. Аритмии возникают при алкогольной абстиненции и, видимо, имеют связь с “социальным” потреблением алкоголя.
Повышение кровяного давления прямо связано с количеством потребленного алкоголя, независимо от ожирения, потребления соли, кофе или курения сигарет. И при подъеме давления, и при потреблении алкоголя регистрируется дефицит магния, но нет убедительных данных о соотношении этих процессов. Пока еще не определена пороговая для нарушения электролитного баланса доза алкоголя.
Любопытно, что заболеваемость коронарной болезнью сердца ниже у лиц, умеренно потребляющих алкоголь (1-3 дозы в день), по сравнению с непьющими (Suh et al., 1992). Имеются данные о том, что алкоголь увеличивает уровень липопротеидов высокой плотности (фракции ЛПВП3) в плазме (Yaskell et al., 1984). Однако по эпидемиологическим наблюдениям, с понижением риска заболеваний сердца связана имеющая меньшую плотность фракция ЛПВП2, а не ЛПВП3. Когда потребление алкоголя сопровождается заболеванием печени, содержание ЛПВП понижено. Клиническое значение этих данных недостаточно ясно. Предполагаемый защитный эффект некоторых напитков, например красного вина, требует дальнейшего изучения.
У больных при синдроме отмены могут развиваться серьезные аритмии, связанные с нарушениями метаболизма калия и магния. Они опасны возникновением припадков, глубоких обмороков и могут вызвать внезапную смерть.
Д. Эндокринная система. Хроническая алкоголизация оказывает выраженное влияние на эндокринную систему, минеральный обмен и водно
электролитный баланс. Клинические наблюдения гинекомастии и атрофии тестикул у алкоголиков с циррозом печени указывают на нарушения в балансе стероидных гормонов. Такая эндокринная патология обнаруживается и у больных с менее серьезными повреждениями печени.
У алкоголиков с хроническими заболеваниями печени нередко нарушен водно-электролитный баланс, что приводит к асцитам, отекам, выпотам. Это может быть связано с угнетением синтеза белков и портальной гипертензией. Изменения содержания калия в организме из-за рвоты и диареи, так же как тяжелый вторичный альдостеронизм, могут приводить к мышечной слабости и усугубляться при применении диуретиков. Изменение обмена таких микроэлементов, как цинк, может проявляться в метаболических нарушениях и бесплодии. У некоторых больных алкоголизмом развивается кетоз, вызванный избытком липолитических факторов, особенно увеличением концентрации кортизола и гормона роста.
Е. Алкогольный синдром плода. Хроническое потребление алкоголя беременной женщиной представляет серьезную опасность для потомства (Abel, 1981; Ernhart et al., 1987). Алкогольный синдром плода может проявляться в виде: 1) задержки роста, 2) микроцефалии (уменьшение размера головы по отношению к размерам тела), 3) нарушения координации, 4) недоразвития средней части лицевого черепа (проявляется уплощением лица), 5) незначительных нарушений развития суставов. В более тяжелых случаях могут наблюдаться врожденные пороки сердца и задержка умственного развития. Возможно, что употребление большого количества алкоголя в первый триместр беременности наиболее опасно для нормального развития плода, тогда как в конце срока оно оказывает большее влияние на питание плода и, следовательно, его массу. Абель (1989) сообщил, что потребление алкоголя крысами-“отцами” уменьшает размеры и уровень активности потомства, снижает содержание тестостерона у крысят-самцов; более того, у родителей-самцов изменяется подвижность сперматозоидов в электрическом поле. Однако эти интересные наблюдения не имеют пока подтверждения у человека. Виндхем (1992) в своей работе по исследованию спонтанных абортов не обнаружил корреляции с потреблением алкоголя отцами.
Ж. Иммунная система. В ряде исследований показано, что хроническое потребление этанола может
изменять функции иммунной системы. Это рассматривается как фактор риска определенных форм рака у алкоголиков. Выявляются нарушение хемотаксиса гранулоцитов, изменения реакции лимфоцитов на митогены, количества Т-клеток, активности естественных киллеров (NK) и уровня фактора некроза опухолей. Даже однократный прием алкоголя может изменять некоторые из этих показателей.
3. Повышение риска заболевания раком. Хроническое потребление алкоголя повышает риск возникновения рака слизистой рта, гортани, глотки, пищевода и печени. Некоторые данные указывают на то, что развитие рака груди не зависит от потребления алкоголя (Schatzkin et al., 1987). Хотя методологические проблемы выявления связи между раком и потреблением алкоголя трудно преодолимы, корреляция во многих случаях очевидна. Значительно больше информации требуется для того, чтобы определить пороговый уровень потребления алкоголя в связи с риском развития рака. В действительности, алкоголь сам по себе, видимо, не является канцерогеном в большинстве тест-систем. Однако алкогольные напитки могут содержать потенциальные канцерогены, образующиеся в процессе ферментации и производства, и могут изменять функцию печени таким образом, что активность потенциальных канцерогенов возрастает.
Взаимодействие алкоголя и лекарств
Взаимодействие алкоголя с другими лекарствами может иметь большое клиническое значение в связи с изменением их фармакокинетики или фармакодинамики.
Чаще всего фармакокинетическое взаимодействие алкоголя и лекарства происходит в результате индуцированной алкоголем пролиферации гладкого эндоплазматического ретикулума печеночных клеток (глава4). Таким образом, продолжительное употребление алкоголя, не вызвавшее гибели гепатоцитов, может ускорить метаболическую биотрансформацию других лекарств. Напротив, однократное применение алкоголя может подавлять метаболизм других лекарств за счет прямого вмешательства в этот процесс или изменения печеночного кровотока. Например, сочетание алкоголя с фенотиазинами, трициклическими антидепрессантами и седативно-гипнотическими средствами замедляет их биотрансформацию и усиливает угнетение психомоторных реакций, что особенно опасно при вождении автомобиля.
Большое клиническое значение имеет также фармакодинамическое взаимодействие. Наиболее важен аддитивный эффект с другими седативногипнотическими препаратами. Алкоголь потенцирует фармакологические эффекты многих неседативных лекарств, включая вазодилататоры и оральные гипогликемические средства, повышает анти-тромбоцитарный эффект аспирина.
Взаимодействие с дисульфирамом обсуждается в этой главе.
II. Клиническая фармакология этанола
Этанол — одно из наименее активных фармакологических средств, хотя наряду с табаком является важнейшим фактором риска заболеваемости и смертности. Однако умеренное потребление этанола сопровождается седативно-гипнотическим эффектом, уменьшает симптомы и гормональные сдвиги, ассоциированные со стрессом, и даже, по данным некоторых исследований, приводит к снижению риска сердечных атак.
Хотя алкоголизму трудно дать точное определение, эпидемиологические данные о потреблении алкоголя предоставляют важную информацию для предсказания факторов, влияющих на здоровье. Примерно 80 % взрослых в США потребляют алкогольные напитки. По этим оценкам, у 5-10% взрослого мужского населения в тот или иной период жизни имеются проблемы, связанные с алкоголем. У женщин, больных алкоголизмом, раньше, чем у мужчин, начинается повреждение мозга и печени. При том же уровне потребления у женщин выше содержание алкоголя в крови, что связано с половыми особенностями фармакокинетики. Наблюдение за почти 90 000 мужчин и женщин в течение десяти лет выявило двукратное увеличение смертности у лиц, которые потребляли 6 и более условных доз алкоголя в день. Среди употреблявших 3-5 доз в день смертность была на 40-50 % выше по сравнению с нормой. Смертность при неумеренном потреблении алкоголя была связана с заболеваемостью раком, циррозом печени и несчастными случаями. Курение, обычное у алкоголиков, тоже являлось потенциальным фактором риска смертности. Результаты исследования показывают, что существует порог повышения риска смертности в диапазоне 2-3 условных доз в день, и
этот риск резко возрастает на уровне 6 и более доз в день. Существуют доказательства того, что малые количества алкоголя (например, одна порция пива в день) снижают риск инфаркта миокарда (по сравнению с лицами, которые никогда не потребляют алкоголь).
Поиск специфических этиологических или предрасполагающих к злоупотреблению алкоголем факторов дал разочаровывающие результаты. Тип личности, серьезные жизненные стрессы, психические расстройства не определяют неизбежность злоупотребления алкоголем. Биотрансформация этанола исключительно вариабельна, и маркеры нарушенного метаболизма алкоголя недостаточно точно предсказывают риск заболевания. Очевидно, что злоупотребление алкоголем не распределено равномерно по всем социальным группам и встречается чаще среди родственников алкоголиков, чем в общей популяции (Devor, 1989). Частота алкоголизма у лиц с биологическими родителями-алкоголиками почти в 4 раза выше, чем в контрольных группах, что подтверждено рядом работ. Трудно различать социальные и генетические факторы риска алкоголизма, но, по современным данным, последние играют ведущую роль. У животных твердо установлен генетический контроль предпочтения алкоголя. У человека выявлена явная связь между аллелем D2 гена дофамина и алкоголизмом (Blum, 1990). Тем не менее детали механизма генетического контроля алкоголизма пока не известны. Изучение изменений ферментов тромбоцитов или трансферрина (Storey et al., 1987) может способствовать разработке лабораторных маркеров неумеренного потребления алкоголя.
Помощь при острой алкогольной интоксикации
У нетолерантных к алкоголю лиц при приеме больших его количеств развиваются типичные эффекты передозировки седативно-гипнотического средства наряду с сердечно-сосудистыми эффектами (вазодилатация, тахикардия) и раздражением желудочно-кишечного тракта. Поскольку толерантность не абсолютна, то даже у хронических алкоголиков может возникать выраженная интоксикация. Степень интоксикации зависит от трех факторов: концентрации этанола в крови, скорости подъема уровня алкоголя и времени, в течение которого со
храняется повышенный уровень этанола в крови. Характер потребления, состояние слизистой желудочно-кишечного тракта и присутствие в организме других лекарственных средств также оказывают влияние на степень интоксикации.
При лечении острого отравления алкоголем наиболее важно предупредить угнетение дыхания и аспирацию рвотных масс. Даже при очень высоком уровне этанола в крови выживание возможно, если поддерживаются функции дыхания и кровообращения. Летальная доза алкоголя широко колеблется в зависимости от степени толерантности. Обычный нетолерантный к алкоголю взрослый может метаболизировать 7—10 г алкоголя в час (количество этанола в 30 мл виски, кружке пива или бокале некрепленого вина). В США юридическое определение интоксикации различно в разных штатах, но наличие алкоголя в крови на уровне 80 мг% (17 ммоль/л) или выше часто рассматривают как свидетельство нарушения психомоторных функций, достаточное для обвинения в управлении автомобилем в нетрезвом виде. Средний уровень концентрации алкоголя в крови в фатальных случаях выше 400 мг%.
Метаболические изменения могут потребовать коррекции гипогликемии и кетоза путем введения глюкозы. Больной алкоголизмом со рвотой и дегидратацией должен также получать растворы электролитов. При сильной рвоте может потребоваться введение большого количества калия до момента нормализации функции почек. Особенно важно распознать снижение запаса фосфатов, которое может усиливаться при введении глюкозы. Низкий резерв фосфатов играет роль в ухудшении заживления ран, неврологическом дефиците и повышении риска инфекции.
Лечение алкогольного абстинентного синдрома
Когда потребление алкоголя резко прерывается, развивается характерный синдром, проявляющийся моторным возбуждением, тревогой, снижением судорожного порога. Тяжесть алкогольного абстинентного синдрома (ААС) обычно пропорциональна дозе и продолжительности злоупотребления алкоголем. Однако она может существенно изменяться при использовании седативных средств и при сопутствующих заболеваниях (например, диа
бет) или травмах. В легкой форме ААС проявляется тремором, тревогой, бессонницей, которые возникают через 6-8 часов после отмены алкоголя. Эти проявления постепенно исчезают в течение 1-2 дней. У некоторых больных развиваются более тяжелые реакции отмены, включая зрительные галлюцинации, общую дезориентацию, значительные нарушения жизненно важных функций. Чем тяжелее ААС, тем более необходима тщательная оценка возможных осложнений. В прошлом переоценивался риск смертности при тяжелом ААС. Прогноз обычно определяется возможными осложнениями.
Главная задача лекарственного лечения в период отмены алкоголя — предупреждение судорог, делирия и аритмий (Romach, 1991). Баланс калия, магния и фосфата можно восстановить быстро, если функция почек не нарушена. Терапия тиамином назначается во всех случаях. Лица в состоянии ААС средней тяжести не нуждаются в какой-либо иной лекарственной помощи.
В тяжелых случаях специфическое лекарственное лечение при детоксикации основано на двух принципах: замена алкоголя длительнодействующим седативно-гипнотическим средством и постепенное уменьшение его дозы. К сожалению, эта наиболее широко применяемая терапия ААС не предупреждает рецидивов алкоголизма. Хотя можно постепенно снижать и дозы самого алкоголя, психологически нежелательно поддерживать прием алкоголя больным. В настоящее время для лечения предпочитают бензодиазепины. Хлордиазепоксид и диазепам имеют фармакологически активные метаболиты, которые могут кумулировать. Оксазепам быстро превращается в неактивный водорастворимый метаболит, который не кумулирует, в связи с чем особенно полезен больным алкоголизмом с тяжелыми заболеваниями печени. Большинство бензодиазепинов плохо всасывается после внутримышечного введения, поэтому для получения быстрого эффекта предпочитают внутривенные инъекции.
Лечение фенотиазинами в период отмены алкоголя потенциально опасно из-за нежелательных эффектов (например, усиления судорог), которые могут свести на нет их достоинства. Иногда используют и антигистаминные препараты, однако без серьезных обоснований.
Некоторые специалисты рассматривают фенитоин как средство предупреждения судорог у алкоголиков, имевших судороги в анамнезе. Так как фенитоин плохо всасывается при внутримышечном
введении, лекарство следует вводить внутривенно. Нагрузочная доза составляет 12-15 мг/кг и затем поддерживается на уровне 300 мг в день с контролем уровня препарата в крови.
После того как больному с ААС оказана неотложная помощь, следует постепенно уменьшать дозы седативно-гипнотических препаратов в течение нескольких недель. За несколько дней трезвости нельзя достичь полной детоксикации организма. Для восстановления нормальной функции нервной системы, особенно сна, может потребоваться несколько месяцев. Вследствие длительного периода, требуемого для полной детоксикации, учитывают другие факторы, которые могут Способствовать этому процессу или помочь предупредить рецидив. В частности, в настоящее время установлено, что тревога и алкоголизм тесно взаимосвязаны (Frances, 1993). Многие алкоголики пьют (по крайней мере отчасти) для того, чтобы уменьшить тревогу и, соответственно, тревога может возрастать в отсутствие алкоголя. Поскольку специалисты по лечению алкоголизма не всегда достаточно осведомлены о тревоге и ее лечении, может быть полезным сотрудничество врачей разных специальностей в этой области. Необходимо также знать и учитывать тот факт, что курение распространено среди алкоголиков и что прекращение курения повышает шанс прекращения злоупотребления алкоголем (Hughes, 1993).
Дисульфирам и другие лекарства для уменьшения потребления этанола
Было обнаружено, что дисульфирам (тётра-этилтиурам) — широко применяемый в резиновой промышленности антиоксидант,— вызывает дискомфорт у пациентов, которые потребляют алкогольные напитки. Дисульфирам сам по себе у непьющего человека не оказывает заметного действия, однако через несколько минут после последующего приема алкоголя развиваются гиперемия лица, пульсирующая головная боль, тошнота, рвота, потоотделение, гипотензия и спутанность сознания. Эффект может продолжаться 30 минут в легких случаях и до нескольких часов — в тяжелых. После исчезновения симптомов больной чувствует себя истощенным и может проспать несколько часов.
Дисульфирам действует путем угнетения альдегиддегидрогеназы. Таким образом, алкоголь метаболизируется как обычно, но происходит накоп
ление ацетальдегида. Симптоматика интоксикации при взаимодейстрии дисульфирама и алкоголя типична для отравления ацетальдегидом и воспроизводится у человека при инфузии ацетальдегида.
Дисульфирам быстро и полностью всасывается из желудочно-кишечного тракта, однако требуется 12 часов для полного проявления его эффекта. Скорость элиминации очень низка, и действие может продолжаться несколько дней после последней дозы. Препарат взаимодействует со многими другими лекарственными средствами.
Дисульфирам рассматривают как фармакологическое вспомогательное средство при лечении алкоголизма, которое должно применяться совместно с терапией, направленной на коррекцию поведения (Chick et al., 1992). При назначении препарата следует информировать больного о содержании алкоголя в обычных безрецептурных лекарствах (табл. 65-3). Лечение дисульфирамом можно начинать только при условии, что больной не принимал алкоголь последние 24 часа. Препарат может вызывать незначительные изменения результатов функциональных печеночных проб. Безопасность дисульфирама при беременности не изучена. Длительность лечения дисульфирамом индивидуальна и определяется реакцией больного и клиническим улучшением. Обычная доза внутрь составляет 250 мг в день перед сном. Использование дисульфирама с целью принудительного лечения по решению суда, вероятно, малоэффективно.
Многие другие лекарства, например метронидазол, некоторые цефалоспорины, гипогликемические средства группы сульфанилмочевины и хлоралгидрат, имеют дисульфирамоподобное действие в отношении метаболизма этанола (Stockey, 1983).
Ведется широкий поиск потенциальных “профилактических” агентов, которые могут ослаблять тягу к этанолу без аверсивного эффекта дисульфирама (bitten, 1991). Последние данные о том, что хроническое злоупотребление алкоголем снижает уровень серотонина и концентрацию ангиотензина, побудило к исследованию терапевтического действия ингибиторов обратного захвата серотонина и ингибиторов ангиотензинпревращающего фермента при хроническом алкоголизме (Naranjo et al., 1990). Уменьшение потребления этанола в этих случаях не было связано с аверсивными эффектами, подобными тем, которые наблюдаются при лечении дисульфирамом. У некоторых групп больных
тягу к алкоголю и его потреблению уменьшают агонисты дофамина (например, бромокриптин) и антагонисты опиоидов (например, налтрексон).
III. Базисная фармакология других спиртов
Другие спирты, родственные этанолу, широко используются в промышленности как растворители и могут вызывать тяжелые отравления (Litovitz, 1986).
Метанол (СН3ОН, метилалкоголь, древесный спирт) является продуктом перегонки древесины. Его используют в домашних отопительных устройствах как добавку к топливу, как промышленный растворитель, как компонент растворов для ксерографии и в питательных смесях для бактериального синтеза белков. В домашних условиях метанол чаще всего хранится в канистрах для использования с целью обогрева и в составе жидкостей для мытья автомобильных стекол. Он может всасываться через кожу, дыхательные пути или желудочно-кишечный тракт. При попадании в ЖКТ метанол быстро всасывается и распределяется в жидкостях организма. Основной механизм элиминации метанола у человека — окисление до формальдегида, муравьиной кислоты и СО2:
о о
II II
СНзОН —► НСН —>НСОН —>СО2 + НгО
Он может удаляться также при вызванной отравлением рвоте, в незначительных количествах — с выдыхаемым воздухом, потом и мочой. Метанол не связывается с активированным углем.
Средние летальные дозы у различных видов животных существенно отличаются. Особенная чувствительность человека к токсическому действию метанола, возможно, связана с фолатзависи-мой продукцией формиата, а не с самим метанолом или промежуточным метаболитом формальдегидом.
Основной фермент, ответственный за метаболизм метанола в печени,— алкогольдегидрогеназа. Этанол имеет более высокое сродство к алкогольдегидрогеназе, чем метанол. Поэтому насыщение фермента этанолом может уменьшать образование формиат-метаболита и часто используется для ле
чения острой интоксикации метанолом. Ингибитор алкогольдегидрогеназы 4-метилпиразол (фомепи-зол) самостоятельно или в сочетании с этанолом оказывает терапевтический эффект при отравлении метанолом и этиленгликолем.
Спирты типа этиленгликоля (СН2ОНСН2ОН) используют в теплообменниках, антифризных составах и как промышленные растворители. Благодаря низкой летучести гликолей, они образуют мало пара при обычной температуре. Однако при использовании в антифризах и теплообменниках они могут присутствовать в парообразной форме, особенно при повышенной температуре. Этиленгликоль более токсичен для человека, чем для многих других видов млекопитающих. Как и другие спирты, этиленгликоль метаболизируется алкогольдегидрогеназой до альдегидов, кислот и оксалата. Оксалат может откладываться в канальцах почек, вызывая острую почечную недостаточность. Своевременное назначение этанола или лечение с помощью 4-ме-тилпиразола может предупредить развитие метаболического ацидоза (Baud et al., 1986).
IV. Клиническая фармакология других спиртов
Лечение интоксикации метанолом
Тяжелые отравления метанолом встречаются обычно у хронических алкоголиков и могут не распознаваться до тех пор, пока не появятся характерные симптомы. Поскольку метанол и его метаболит формиат — гораздо более активные токсины, чем этанол, важно как можно раньше распознать отравление метанолом и начать соответствующую терапию.
Наиболее важный ранний симптом отравления метанолом — нарушение зрения, которое часто описывают как “картину снегопада, метели”. Зрительные расстройства являются универсальной жалобой при эпидемических отравлениях метанолом. Жалобы на затуманенное зрение при нормальных других сенсорных функциях являются серьезным аргументом в пользу диагноза отравления метанолом. В тяжелых случаях запах формальдегида может ощущаться при дыхании больного, этот же запах может иметь и моча. Брадикардия, продолжи
тельная кома, судороги и стойкий ацидоз указывают на неблагоприятный прогноз отравления. Данные физикального обследования при отравлении метанолом обычно неспецифичны. В тяжелых случаях отмечают мидриаз. Атрофия зрительных нервов развивается позднее. Причина смерти при тяжелых отравлениях связана с остановкой дыхания.
При подозрении на отравление важно, чтобы уровень метанола в крови был определен как можно быстрее. Если клинические данные в пользу отравления убедительны, лечебные мероприятия не следует откладывать. Уровень метанола выше 50 мг% является абсолютным показанием для гемодиализа и лечения этанолом, хотя определение уровня формиата в крови является более точным индикатором заболевания. Дополнительные лабораторные данные в пользу отравления включают метаболический ацидоз с повышением содержания анионов и осмоляльности (глава 60, том 2). Снижение содержания бикарбоната сыворотки — характерная черта тяжелого отравления метанолом. Отравление этиленгликолем обычно приводит к возбуждению центральной нервной системы, увеличению уровня мышечных ферментов в крови и гипокалиемии; нарушений зрения не наблюдается. Отравление салицилатами легко определить по их содержанию в крови.
Первая помощь при отравлении метанолом, как и при всех острых отравлениях, состоит в поддержании адекватного дыхания с интубацией трахеи при необходимости. Если пациент в сознании, нет судорог и сохранены рефлексы, следует вызвать рвоту. В случае противопоказаний больного интубируют и производят промывание желудка с помощью толстого зонда после соответствующей защиты дыхательных путей.
Существуют три специфических способа лечения отравлений метанолом: 1) ингибирование алкогольдегидрогеназы для подавления метаболизма метанола с образованием токсичных продуктов, 2) диализ для ускорения выведения метанола и его токсичных метаболитов и 3) введение щелочей с целью коррекции метаболического ацидоза.
Поскольку метанол конкурирует за алкогольдегидрогеназу с менее токсичным этанолом, последний используют для насыщения фермента. Дозозависимые характеристики метаболизма этанола и вариабельность, вызванная его хроническим потреблением, требуют частого контроля уровня этанола в крови для поддержания необходимой кон
центрации. Этанол вводят внутривенно в виде 10 % раствора. После внедрения в клиническую практику ингибитор алкогольдегидрогеназы 4-метилпира-зол может стать эффективным средством при лечении отравлений метанолом.
При диализе этанол также выводится в диализат, что требует коррекции вводимой дозы. Описание гемодиализа приведено в главе 60 (том 2).
В связи с выраженным метаболическим ацидозом при отравлении метанолом необходимо введение бикарбоната. Количество бикарбоната определяют на основе оценки баланса натрия и калия, состояния сердечно-сосудистой системы и изменений pH мочи.
Так как у человека за окисление муравьиной кислоты до СО2 ответственны фолатзависимые системы, при отравлении метанолом может оказаться полезным введение фолиевой кислоты. К настоящему времени этот способ лечения не прошел полной клинической апробации.
Диагностические проблемы возникают, если у больного относительно низкий уровень метанола в крови при наличии зрительной симптоматики. В этом случае нужно повторить лабораторные тесты на метанол и подтвердить диагноз оценкой осмоляльности. Если имеются нарушения зрительной функции, необходимо начать гемодиализ, несмотря на низкий уровень метанола.
Лечение отравлений этиленгликолем
Подробные сообщения об отравлениях этиленгликолем немногочисленны. Как и при отравлении метанолом, может быть задержка в развитии ацидоза и почечной недостаточности. Выделяют три стадии передозировки этиленгликоля. Начальная, когда отмечают транзиторное возбуждение, сменяющееся угнетением ЦНС. Затем развивается выраженный ацидоз и, наконец, развивается отсроченная почечная недостаточность. Результаты офтальмоскопического обследования обычно нормальны, хотя описаны отдельные случаи отека соска зрительного нерва. В моче обычно обнаруживаются кристаллы оксалатов. У больных с отравлением этиленгликолем не определяется запах алкоголя при дыхании. В тяжелых случаях при исследовании плазмы обнаруживаются увеличение активности мышечных ферментов и снижение концентрации кальция. Ключом к диагнозу отравления этиленгликолем является выявление ацидоза, изменений
осмоляльности и наличие оксалатов в моче у больных без симптомов нарушения зрительных функций. Как и при отравлении метанолом, необходимы ранняя инфузия этанола и диализ. Можно будет использовать также лечение 4-метилпиразолом, когда лекарство станет коммерчески доступным.
Препараты
Препараты для лечения острого алкогольного абстинентного синдрома
Диазепам (генерик, Валиум, др.)
Парентерально: 5 мг/мл для инъекций
Тиамин (генерик, Беталин С, Беамин) Парентерально: 100, 200 мг/мл для в/в инъекций
Препараты для предотвращения потребления алкоголя
Дисульфирам (генерик, Антабус)
Перорально: таблетки по 250, 500 мг
Препараты для лечения острого отравления метанолом или этиленгликолем
Этанол (генерик)
Парентерально: 5 % или 10 % этанол и 5 % глюкоза в воде для в/в вливания
Избранная литература
Abel Е. L. (ed.) Fetal Alcohol Syndrome. CRC Press, 1981.
Becker С. E., Roe R. L., Scott R. A. Alcohol as a Drug: A Curriculum on Pharmacology, Neurology and Toxicology. RE Kreiger, 1979.
Brien J. F., Loomis C. W. Pharmacology of acetaldehyde. Can. J. Physiol. Pharmacol. 1983; 61:1.
Charness M. E., Simon R. P., Greenberg D. A. Ethanol and the nervous system. N. Engl. J. Med. 1989; 321: 442.
Chick J. et al. Disulfiram treatment of alcoholism. Br. J. Psychiatry, 1992; 161: 84.
Devor E. J., Cloninger C. R. Genetics of alcoholism. Annu. Rev. Genet. 1989; 23: 19.
Dietrich R. A. et al. Mechanism of action of ethanol: Initial central nervous system actions. Pharmacol. Rev. 1989; 41:489.
Freidman J. D., Klatzky A. R. Is alcohol good for your health? N. Engl. J. Med., 1993; 329:1882.
Holford N. H. Clinical pharmacokinetics of ethanol. Clin. Pharmacokinet. 1987; 13: 273.
Lee N. M., Smith A. S. Ethanol. In: Toxicology of CNS Depressants. Ho I. K. (ed.) CRC Press, 1986.
Romach M. K., Sellers E. M. Management of the alcohol withdrawal syndrome. Annu. Rev. Med. 1991; 42: 323.
Wright C., Moore R. D. Disulfiram treatment of alcoholism. Am. J. Med. 1990; 88: 647.
Противоэпилептические средства1
Роджер Дж. Портер, Брайен С. Мелдрум
23
Приблизительно 1 % населения США страдает эпилепсией, вторым по частоте неврологическим заболеванием после инсульта. Хотя стандартная терапия предотвращает развитие судорог у 80 % больных, 500 000 человек в США страдают от неконтролируемой эпилепсии. Эпилепсия — это гетерогенный симптомокомплекс, хроническое заболевание, характеризующееся периодическим возникновением судорог. Судороги — это состояние временной дисфункции мозга, вызванное аномальными нейрональными разрядами. Причинами судорог может быть большое количество заболеваний, имеющих неврологические проявления, от инфекций до новообразований и травм. В возникновении некоторых видов судорог большую роль играет наследственность.
Противосудорожные препараты, рассмотренные в этой главе, используются также для лечения больных с фебрильными судорогами или с судорогами, являющимися проявлениями другого острого заболевания, например менингита, хотя для таких пациентов термин “эпилепсия” не употребляется, если судорожный синдром не приобретает характер хронического течения. Судороги иногда бывают вызваны токсическими агентами или метаболическими нарушениями, в таких случаях лечение должно быть направлено на устранение соответствующих причин, например на коррекцию гипокальциемии. Однако в большинстве случаев при эпилепсии выбор препарата зависит от эмпирической классификации судорог.
История
До появления противосудорожных средств эпилепсию лечили с помощью трепанации черепа, постановки банок и использования трав и животных экстрактов. В 1857 г. сэр Чарльз Локок для лечения эпилепсии успешно использовал бромид калия. В
1912 г. был впервые применен фенобарбитал, а в течение последующих 25 лет исследованы антикон-вульсантные свойства 35 его аналогов. В 1938 г. на модели судорожных припадков кошек было открыто противосудорожное действие фенитоина.
Между 1935 и 1960 гг. произошли огромные изменения как в испрльзуемых экспериментальных моделях, так и в исследованиях новых противосудорожных препаратов. За этот период появилось и поступило в продажу 13 новых противосудорожных препаратов. После введения определенных требований к качеству лекарственных средств в 1962 г. развитие новых противосудорожных препаратов значительно замедлилось, и за следующие три десятилетия были внедрены только несколько препаратов. Однако в девяностые годы появились новые лекарственные средства.
Современная разработка препаратов для лечения эпилепсии
В течение длительного времени считалось, что можно найти один препарат для лечения всех форм эпилепсии. Сейчас это кажется маловероятным, так как существует много видов судорог, которые развиваются по разным механизмам. Препарат, помогающий при одном типе судорог, может ухудшить ситуацию при другом типе. Фармакология и особенности клинического использования противоэпи-лептических средств значительно различаются. Эти препараты делятся на два фармакологических класса применительно к классификации судорог (табл. 23-1), несмотря на то, что в эксперименте судороги можно вызвать многими способами. Клинические особенности некоторых видов генерализованных судорог, особенно абсанса, коррелируют
1 Авторы благодарят Гарольда Боксенбаума за его помощь в освещении вопросов фармакокинетики в этой главе.
ТАБЛИЦА 23-1. Классификация судорог
Парциальные судороги
Простые парциальные судороги Сложные парциальные судороги Вторично генерализованные парциальные судороги
Генерализованные судороги
Генерализованные тоникоклонические судороги (grand mal)
Абсанс (petit mal)
Тонические судороги
Атонические судороги
Клонические и миоклонические судороги
Инфантильные спазмы* 1___________
1 Инфантильные спазмы — это скорее эпилептический синдром, а не специфический вид судорог; препараты, применяемые при инфантильных спазмах, будут рассмотрены отдельно.
с течением экспериментальных судорог, вызываемых у животных подкожным введением пентилен-тетразола. Парциальные судороги у человека имеют сходство с судорогами, вызванными в эксперименте с использованием максимального электрошока (тест MES). Обычно противосудорожные препараты, эффективные против судорог при электрошоке, изменяют ионный транспорт через возбудимые мембраны. Например, фенитоин эффективен при парциальных и связанных с максимальным электрошоком судорогах, но не влияет на генерализованные (за исключением тоникоклонических) или вызванные инъекцией пентилентетразола судороги. Фенитоин влияет на импульсацию нейронов, изменяя трансмембранный транспорт ионов натрия. С другой стороны, этосукцимид и триметадион, которые эффективны при некоторых генерализованных или моделируемых инъекцией пентилентетразола судорогах, снижают вход Са2+ в клетки через низкопороговые кальциевые каналы Т-типа. Эти два класса противосудорожных препаратов имеют разные механизмы действия, несмотря на структурное сходство многих препаратов обеих групп.
Новые противосудорожные препараты изучают не только на упомянутых экспериментальных моделях, но также используют иные, более рациональные подходы. Известны три основных механизма действия противосудорожных средств: 1) облегчение ГАМК-зависимой (ингибиторной) передачи, 2) подавление возбуждающей (обычно глутаматер-гической) передачи и 3) модификация ионных токов.
I. Базисная фармакология п роти восуд орожн ых препаратов
Химия
Вплоть до 1990 г. в клинике применялись 16 основных противосудорожных препаратов, 13 из них по химическому строению относятся к одной из пяти сходных групп: барбитураты, гидантоины, ок-сазолидиндионы, сукцинимиды и ацетилмочевина. Общей структурой у этих групп является гетероциклическое кольцо со множеством замещений (рис. 23-1). Для препаратов такого строения замещающие группы определяют фармакологический класс: либо анти-MES, либо антипентилентетразол. Малейшие изменения в структуре могут значительно изменить механизм действия и клинические свойства лекарственного средства. Остальные препараты (карбамазепин, вальпроевая кислота и бензодиазепины) схожи по структуре с такими новыми препаратами 1990-х гг., как вигабатрин, окскар-базепин, ламотриджин, габапентин и фелбамат.
Фармакокинетика
Противосудорожные препараты, даже отличающиеся структурно и химически, имеют много сходных фармакокинетических свойств. Хотя многие из этих препаратов плохо растворимы, их абсорбция обычно достаточно хорошая: 80-100 % дозы попадает в кровоток. Биодоступность фенитоина зависит от конкретной формы препарата, определяющей скорость и степень его абсорбции.
За исключением фенитоина, бензодиазепинов и вальпроевой кислоты, противосудорожные препа-
R.
’ Го
О=С----N— Rg
Рис. 23-1. Гетероциклическое кольцо в структуре противосудорожных препаратов. “X” может быть различным: -N- у дериватов гидантоина; -С-N- у барбитуратов; -О- у оксазолидиндионов; -С- у сукцинимидов; -NH2 (N соединяется с С2) у ацетилмочевины. Rt, R2 и R3 варьируют в разных подгруппах
раты лишь незначительно связываются с белками плазмы. Только фенитоин и вальпроевая кислота могут вытеснять другие препараты, в том числе и противосудорожные средства, из комплексов с белками плазмы. Используемые же концентрации бензодиазепина слишком низки для того, чтобы повлиять на связывание других препаратов.
У противосудорожных препаратов низкие показатели экстракции (глава 3). Повышение процента содержания свободного препарата в тканях не влияет на концентрацию свободного препарата, но снижает общую концентрацию. Это заставляет лечащего врача увеличить дозу, что в свою очередь повысит концентрацию свободного препарата и вызовет токсические эффекты. Вальпроат натрия уникален тем, что фракция связанного препарата зависит как от его концентрации, так и от содержания свободных жирных кислот в плазме.
Противосудорожные средства выводятся преимущественно печенью. Препараты типа примидона и бензодиазепинов превращаются в активные метаболиты, которые также выделяются печенью. Однако способность печени метаболизировать противосудорожные препараты достаточно низка и, кроме как для фенитоина, не зависит от их концентрации. Эти лекарства преимущественно распределяются в жидких средах организма. Плазматический клиренс сравнительно низкий, поэтому многие противосудорожные препараты считаются препаратами среднего и длительного действия. Для большинства из них период полувыведения составляет 12 часов. Фенобарбитал и карбамазепин вызывают значительную индукцию активности микросомальных ферментов печени.
Препараты, используемые при парциальных и генерализованных тоникоклонических судорогах
Основные препараты, применяемые для купирования парциальных и генерализованных тоникоклонических судорог,— это фенитоин (и его аналоги), карбамазепин и барбитураты. Однако появление окскарбазепина, вигабатрина и ламотриджи-
на меняет стандарты клинической практики в тех странах, где эти препараты доступны.
Фенитоин
Фенитоин — это самый старый противосудорожный препарат, не обладающий седативным эффектом. Он появился в 1938 г. после систематической оценки влияния препаратов типа фенобарбитала на судороги электрического генеза у лабораторных животных. В течение десятилетий этот препарат был известен под названием дифенилгидантоин (DPH).
Химические свойства
Фенитоин — это дифенилзамещенный гидантоин, обладающий значительно меньшим седативным действием, чем препараты с алкильной группой в положении 5.
Механизм действия
Фенитоин оказывает значительные эффекты на несколько физиологических систем. Он влияет на ток ионов натрия, калия и кальция, на мембранные потенциалы и на концентрации аминокислот и нейромедиаторов норадреналина, ацетилхолина и ГАМК. Фенитоин блокирует посттетаническую потенциацию в препаратах спинного мозга, но роль этого эффекта в подавлении судорог еще не определена.
Исследования клеточной культуры нейронов показали, что фенитоин блокирует продолжительные высокочастотные разряды потенциалов действия (рис. 23-2). В терапевтических концентрациях препарат связывается с инактивированными натриевыми каналами, продлевая это состояние, и изменяет таким образом трансмембранный ток ионов натрия (Macdonald, 1989) (глава 14). Этот
Рис. 23-2. Эффекты трех противосудорожных препаратов на продолжительную высокочастотную импульсацию культуры нейронов. Внутриклеточная регистрация проведена во время импульсов деполяризующего тока продолжительностью 0.75 с (стрелками показаны включения и выключения). В отсутствие препарата серии высокочастотных повторных потенциалов действия заполнили всю длину импульса. Фенитоин, карбамазепин и вальпроевая кислота значительно снизили количество возникающих потенциалов действия. (Macdonald R. L., Meldrum В. S. Principles of antiepileptic drug action. In: Antiepileptic drugs, 3rd ed. Levy R.H. et al. (eds) Raven Press, 1989.)
эффект наблюдается также при лечебных концентрациях карбамазепина и вальпроата и, возможно, обусловливает их противосудорожное действие на моделях электрошока и при парциальных судорогах.
В высоких концентрациях фенитоин подавляет и высвобождение серотонина и норадреналина, усиливает захват дофамина и снижает активность моноаминоксидазы. Препарат взаимодействует с липидами мембраны, стабилизируя ее. Кроме того, в некоторых нейронах фенитоин вызывает парадоксальное возбуждение. Снижение проницаемости клеточной мембраны для ионов кальция может объяснить способность препарата подавлять некоторые кальцийзависимые секреторные процессы, в том числе высвобождение гормонов и нейромедиаторов. Значение этих биохимических реакций и их связь с эффективностью фенитоина пока не ясны.
Механизмы действия фенитоина скорее всего реализуются на нескольких уровнях. Есть данные о том, что в терапевтических концентрациях основной механизм действия фенитоина — это блокада натриевых каналов и подавление образования повторных потенциалов действия.
Клиническое применение
Фенитоин является одним из наиболее эффективных препаратов при парциальных и генерализованных тоникоклонических судорогах. Он эффективен как при первично, так и при вторично генерализованных судорогах.
Фармакокинетика
Абсорбция фенитоина зависит от формы препарата. Размер частиц и добавки влияют на степень и скорость всасывания.
Из желудочно-кишечного тракта фенитоин натрия абсорбируется практически полностью, хотя время достижения максимальной концентрации варьирует между 3 и 12 часами. Предсказать абсорбцию после внутримышечной инъекции невозможно. Поскольку некоторые формы препарата образуют преципитаты в мышцах, этот путь введения не рекомендуется.
Фенитоин легко связывается с белками плазмы крови. Известно, что при уменьшении количества связанного препарата (при уремии и гипоальбуми-немии) снижается общий уровень препарата в плазме, но корреляция между содержанием свободного фенитоина и клиническими проявлениями не установлена. Концентрация лекарства в спинномозговой жидкости пропорциональна уровню свободного препарата в плазме. Он кумулирует в эндоплазматическом ретикулуме клеток мозга, печени, мышц и жировой клетчатки.
Фенитоин в основном метаболизируется парагидроксилированием до 5-(р-гидроксифенил)-5-фенилгидантоина (НРРН), который затем конъюгирует с глюкуроновой кислотой. Метаболиты клинически неактивны и выводятся с мочой. Только малая часть фенитоина выводится в неизмененном виде.
Фармакокинетика выведения фенитоина дозозависима. При очень низких уровнях в крови мета
болизм фенитоина пропорционален скорости попадания в печень, то есть эффекту первого прохождения. Однако при повышении содержания препарата в пределах терапевтических уровней постепенно достигается максимальная способность печени метаболизировать фенитоин (рис. 23-3). Дальнейшие повышения дозы, даже небольшие, могут вызвать значительные изменения концентрации лекарства. В таких случаях период полувыведения препарата значительно повышается, его уровень в плазме начинает нарастать, и у пациента быстро появляются признаки интоксикации.
Период полувыведения фенитоина варьирует от 12 до 36 часов со средним значением 24 часа у большинства пациентов с низким и средним терапевтическими уровнями фенитоина. Более длительные периоды полувыведения характерны для более высоких концентраций. При низком содержании препарата в крови требуется 5-7 дней для достижения стационарного уровня после каждого изменения дозы, при высоком — 4-6 недель до того как уровень стабилизируется.
Рис. 23-3. Нелинейная зависимость концентрации фенитоина в плазме от дозы. Пять разных пациентов (обозначенные соответствующими символами) получали постепенно повышающиеся дозы фенитоина перорально; стабильная концентрация в сыворотке измерялась при каждой дозе. Кривые имеют нелинейный характер, так как с повышением дозы эффект первого прохождения пропорционально снижается. Отметьте также значительную вариабельность уровней в сыворотке крови при приеме у различных пациентов одинаковых доз. (Йз: Jusko W. J. Bioavailability and disposition kinetics of phenytoin in man. In: Quantitative Analytic Studies in Epilepsy. Kellaway P., Peterson I. (eds) Raven Press, 1977.)
Терапевтические концентрации и дозы
Терапевтические концентрации фенитоина в плазме для большинства пациентов составляют 10-20 мкг/мл. Нагрузочную дозу можно давать перорально или внутривенно, последний метод — это метод выбора лечения эпилептического статуса. Пероральную терапию у взрослых начинают с дозы 300 мг/день вне зависимости от массы тела. У некоторых больных это позволяет достичь уровня в плазме около 10 мкг/мл,.что является минимальной терапевтической концентрацией для большинства пациентов. Если судороги продолжаются, необходимо повысить дозу для достижения более высокого, уровня в пределах терапевтических границ. Вследствие своей дозозависимой кинетики фенитоин может вызывать токсические эффекты при минимальном увеличении дозы. Разовое увеличение дозы не должно превышать 25-30 мг у взрослых, после чего требуется время перед дальнейшим повышением дозы для стабилизации уровней в плазме. Частая клиническая ошибка — это увеличение дозы с 300 мг/день до 400 мг/день: через различные периоды времени после этого возникает интоксикация. У детей начальная доза 5 мг/кг/день должна быть в последующем откорректирована в соответствии с уровнями препарата в плазме.
В США производятся два типа перорального фенитоина натрия, различающиеся по скорости растворения. Один абсорбируется быстро, другой — медленнее. Только последний можно использовать для однократного применения. Менять тип назначаемого препарата следует очень осторожно. У некоторых пациентов, длительно получавших фенитоин, возникали низкие уровни препарата в плазме либо из-за плохого всасывания, либо из-за быстрой метаболизации. Самая частая причина низкого уровня — это нарушение режима приема препарата.
Лекарственные взаимодействия и влияние на данные лабораторных исследований
Взаимодействия препаратов с фенитоином прежде всего определяются его связыванием с белками плазмы и особенностями метаболизма. Так как фенитоин образует комплекс с белками плазмы, другие препараты типа фенилбутазона и сульфаниламидов могут вытеснять его из мест связывания. Теоретически такое вытеснение должно вы-
зывать временное увеличение уровня свободного препарата. Снижение связывания с белками (например, при гипоальбуминемии) приводит к уменьшению общей концентрации, а не концентрации свободного препарата. При попытке поддерживать постоянную общую концентрацию в терапевтических пределах повышением дозы препарата может возникнуть интоксикация. Связывание с белками снижается при заболеваниях почек. Препарат обладает аффинностью к тироксинсвязывающему белку, что нарушает достоверность некоторых тестов функции щитовидной железы. Самым надежным методом исследования функции щитовидной железы у пациентов, получающих фенитоин, является измерение тиреотропного гормона.
Было показано, что фенитоин индуцирует активность микросомальных ферментов печени, которые отвечают за метаболизм ряда препаратов. Аутостимуляция своего собственного метаболизма, однако, незначительна. Другие препараты, а именно фенобарбитал и карбамазепин, активируя систему микросомального окисления, снижают стабильные концентрации фенитоина. С другой стороны, изониазид ингибирует метаболизм фенитоина, приводя к повышению стабильных концентраций при совместном приеме этих препаратов.
Токсичность
Дозозависимые побочные эффекты фенитоина, к сожалению, похожи на побочные эффекты других препаратов этой группы, что затрудняет дифференциальный диагноз осложнений у пациентов, принимающих несколько препаратов. Рано возникает нистагм, как и потеря плавности движений глаз, но ни один из этих эффектов не является показанием к снижению дозы. Диплопия и атаксия — это самые частые побочные эффекты, требующие изменения дозы; седативный эффект обычно возникает только при значительных дозировках. У большинства пациентов в той или иной степени наблюдаются гиперплазия десен и избыточное оволосение. Последнее проявление особенно неприятно для женщин. Длительное применение препарата вызывает у некоторых больных огрубение черт лица и умеренную периферическую нейропатию, которая обычно проявляется снижением глубоких рефлексов в нижних конечностях. Продолжительное использование фенитоина может также нарушать метаболизм витамина D, приводя к остеома
ляции. Встречаются также низкий уровень фолиевой кислоты и мегалобластная анемия, но их клиническое значение неясно. Идиосинкразии в отношении фенитоина относительно редки. Более часто встречающаяся типичная кожная сыпь говорит о гиперчувствительности к препарату, который следует в этих случаях отменить. При приеме фенитоина возможна лихорадка, в редких случаях возникают тяжелые эксфолиативные поражения кожи. Лекарственную лимфоаденопатию бывает трудно отличить от злокачественной лимфомы; некоторые исследователи предположили существование причинно-следственной связи между использованием фенитоина и развитием болезни Ходжкина. Но эти данные далеко не окончательны. Гематологические осложнения крайне редки, хотя имеются сообщения о возникновении агранулоцитоза в сочетании с лихорадкой и сыпью.
Мефенитоин, этотоин и фенацемид
Синтезировано много аналогов фенитоина, но только три из них разрешены к ограниченному применению в США. Мефенитоин имеет метильную группу в положении 3 гетероциклического кольца, а одна из фенильных групп в положении 5 замещена этильной группой (рис. 23-1). Этотоин имеет этильную группу в положении 3 гетероциклического кольца и только одну фенильную группу в положении 5. Третий аналог, фенацемид, похож на фенитоин, но в его структуре есть только одно фенильное кольцо в положении 5, а гетероциклическое кольцо разомкнуто с образованием прямой цепи фенилацетилмочевины.
Мефенитоин и этотоин, как и фенитоин, наиболее эффективны при генерализованных тоникоклонических и парциальных судорогах. Однако эти препараты еще не прошли клинических испытаний. Для мефенитоина частота возникновения тяжелых реакций типа агранулоцитоза и дерматита выше, чем для фенитоина. Этотоин можно рекомендовать пациентам с гиперчувствительностыо к фенитоину. Побочные эффекты и токсичность этотоина менее выражены, чем у фенитоина, но этот препарат и менее эффективен, поэтому требуются более высокие дозы.
Оба препарата, мефенитоин и этотоин, в границах терапевтических доз способны, как и фенито
ин, достигать предельной скорости метаболизма. При изменении дозы любого из этих препаратов важен внимательный контроль за пациентом. Ме-фенитоин метаболизируется до 5,5-этил-фенил-ги-дантоина путем деметилирования. Деметилированный метаболит нирванол является основным носителем противосудорожной активности мефенитоина. И мефенитоин, и нирванол далее гидроксилируются и конъюгируются с последующей экскрецией. Терапевтические уровни мефенитоина в среднем составляют 5-16 мкг/мл, содержание в плазме выше 20 мкг/мл считается токсическим. Терапевтические уровни нирванола в плазме равны 25-40 мкг/мл, для этотоина они не установлены.
Линейный аналог фенитоина фенацемид — это токсичный препарат “резерва”, применяемый при рефрактерных парциальных судорогах. Он активен против судорог, вызванных как максимальным электрошоком, так и пентилентетразолом. Механизм его действия неизвестен. Препарат хорошо всасывается и полностью метаболизируется, при этом зависимость эффекта от концентрации пока не установлена. При приеме фенацемида возникают серьезные, а иногда и летальные побочные реакции. К дозозависимым нежелательным эффектам относятся поведенческие изменения типа психозов и депрессивных реакций. Лекарственные осложнения в виде гепатита, нефрита, апластической анемии и т. д. встречаются более часто, чем при приеме других противосудорожных препаратов.
Карбамазепин
Карбамазепин похож на имипрамин и другие антидепрессанты и является трициклическим соединением, эффективным при биполярной депрессии. Он изначально был рекомендован для лечения невралгии тройничного нерва, но оказался полезным и в терапии эпилепсии.
Химия
Хотя это и не заметно на двухмерном изображении (рис. 23-4), карбамазепин имеет сходство с фенитоином. Уреидная группа (-N-CO-NH2), которая присутствует в гетероциклических кольцах других противосудорожных препаратов, есть и в молекуле карбамазепина. Изучение трехмерной структуры показало, что пространственная конформация карбамазепина похожа на наблюдаемую у фенитоина.
Механизм действия
Механизмы действия карбамазепина и фенитоина сходны, карбамазепин также наиболее эффективен при судорогах, вызванных электрошоком. Исследования проницаемости мембран показали, что карбамазепин, как и фенитоин, в терапевтических концентрациях блокирует натриевые каналы и высокочастотные нейрональные разряды в культуре нейронов (рис. 23-2). Он действует пресинапти-чески и затрудняет синаптическую передачу. Эти эффекты, возможно, объясняют противосудорожное действие карбамазепина. Имеются сведения о том, что карбамазепин связывается с аденозиновыми рецепторами, хотя функциональная значимость этих наблюдений пока не известна. Карбамазепин подавляет также захват и высвобождение норадреналина из синаптосом мозга, однако не влияет на захват ГАМК в срезах мозга и на постсинаптическую блокаду, вызываемую ГАМК, что позволяет предположить, что его действие не зависит от ГАМК-ер гической системы.
Клиническое применение
Карбамазепин считается препаратом выбора при парциальных судорогах, многие врачи используют его и в начальной терапии генерализованных тоникоклонических судорог. Он применяется в комбинации с фенитоином у пациентов, состояние которых трудно контролировать. В терапевтических дозах карбамазепин не обладает седативным действием. Он также эффективен у некоторых больных с невралгией тройничного нерва, хотя пожилые пациенты хуже переносят высокие дозы, так как у них возникают атаксия и нестабильность походки.
Фармакокинетика
Скорость абсорбции карбамазепина варьирует у разных пациентов, хотя в конечном итоге препарат всасывается полностью. Максимальная концентрация достигается через 6-8 часов после приема. Замедление всасывания, достигаемое назначением препарата после еды, позволяет пациентам переносить более высокие дозы.
Распределение идет медленно, и его объем приблизительно равен 1 л/кг. Препарат связывается белками плазмы только на 70 %, при этом не вытесняя другие препараты. Карбамазепин обладает очень низким системным клиренсом, который со-
Рис. 23-4. Карбамазепин, окскарбазепин и их метаболиты. Карбамазепин окисляется до эпокси-промежуточных соединений, которые затем образуют дигидроксидериваты. Окскарбазепин не образует эпоксидного метаболита, но быстро конвертируется в 10-гидроксикарбазепин, который затем переходит в дигидрокси-дериваты
ставляет в начале терапии приблизительно 1 л/кг/день. Препарат обладает способностью значительно индуцировать активность микросомальных ферментов печени. В нескольких исследованиях у больных эпилепсией или добровольцев, принимавших препарат более одного месяца, клиренс карбамазепина постепенно увеличился в 2 раза по сравнению с начальным. Обычно период полувыведения, составляющий после начальной дозы препарата 36 часов, снижается при постоянном приеме до 20 часов и менее. Таким образом, в течение первой недели лечения может возникнуть необходимость в существенном пересмотре дозирования препарата. Эти изменения могут в дальнейшем модулировать активность микросомальных ферментов печени. Карбамазепин изменяет также клиренс других препаратов.
Карбамазепин полностью метаболизируется у человека, в частности до 10,11 -дигидрокси-деривата, который затем вступает в конъюгацию. Дигид-рокси-дериват формируется через стадию образо
вания стабильного эпоксида (карбамазепин-10,11-эпоксида), обладающего противосудорожной активностью (рис. 23-4). Вклад этого и других метаболитов в клиническую эффективность карбамазепина неизвестен.
Терапевтические уровни и доза
Карбамазепин считается препаратом выбора при парциальных судорогах. Он применяется только в энтеральной форме. Этот препарат эффективен у детей в дозе 15—25 мг/кг/день. У взрослых применяют дозы от 1 до 2 г/день. Более высокие суточные дозы можно делить на несколько приемов. У пациентов, принимающих препарат 3-4 раза в день, его концентрация в плазме перед первым утренним приемом находится в пределах 4-8 мкг/мл. Хотя некоторые больные жалуются на диплопию при уровнях препарата выше 7 мкг/мл, другие хорошо переносят концентрации выше 10 мкг/мл, особенно при монотерапии. Когда содержание лекарства
контролируется в разное время суток, то оно часто превышает 8 мкг/мл, однако колебания, связанные с абсорбцией, осложняют наблюдение.
Лекарственные взаимодействия
Взаимодействия карбамазепина с другими препаратами почти полностью связаны со способностью препарата индуцировать активность ферментов. Как сказано ранее, повышение метаболической активности ферментов печени может снизить стабильную концентрацию карбамазепина и повысить скорость метаболизма примидона, фенитоина, это-сукцимида, вальпроевой кислоты и клоназепама. Препараты типа пропоксифена, тролеандомйцина и вальпроевой кислоты могут ингибировать клиренс карбамазепина и повышать его стабильные концентрации. Однако другие противосудорожные препараты типа фенитоина, фенобарбитала могут снизить стабильные концентрации карбамазепина путем индукции активности ферментов. О клинически значимых взаимодействиях с другими препаратами на уровне связывания с белками плазмы сообщений не было.
Токсичность
Самые частые дозозависимые побочные эффекты карбамазепина — это диплопия и атаксия. Диплопия часто появляется первой в определенное время дня и длится меньше часа. Перераспределение доз, принимаемых в течение дня, часто снимает эту жалобу. Другие дозозависимые жалобы включают легкое нарушение деятельности желудочно-кишечного тракта, потерю равновесия, а при значительно больших дозах — сонливость. Иногда возникают гипонатриемия и гипергидратация, которые также нередко дозозависимы.
В настоящее время существует настороженность по поводу случаев возникновения гематологических осложнений при приеме карбамазепина, в том числе апластических анемий и агранулоцитоза с летальным исходом. Большинство из них отмечены у пожилых людей с невралгией тройничного нерва и в течение первых четырех месяцев лечения. Легкая персистирующая лейкопения, наблюдаемая у некоторых пациентов, не является основанием для прекращения лечения, но требует тщательного контроля. Наиболее частой идиосинкразической реакцией на препарат является эритематозная кожная
сыпь; другие нарушения типа дисфункции печени встречаются редко.
Окскарбазепин
Окскарбазепин — это новый препарат, по химическому строению похожий на карбамазепин и применяемый по тем же показаниям, но обладающий . меньшей токсичностью. Активность препарата почти полностью базируется на свойствах его 10-гид-рокси-метаболита (рис. 23-4), в который окскарбазепин быстро превращается. У этого вещества период полувыведения такой же, как у карбамазепина, то есть составляет 8-12 часов. Препарат менее эффективен, чем карбамазепин, как на экспериментальных моделях, так и у больных эпилепсией. Для достижения адекватного контроля судорог дозы окскарбазепина должны быть на 50 % выше, чем дозы карбамазепина. В некоторых исследованиях отмечается меньшая частота реакций гиперчувствительности на окскарбазепин. Кроме того, он в меньшей степени, чем карбамазепин, индуцирует активность ферментов печени, ограничивая тем самым влияние на другие препараты. Побочные эффекты, возникающие при приеме окскарбазепина, похожи на эффекты карбамазепина. Препарат продается в некоторых странах Европы, но не в США.
Фенобарбитал
Фенобарбитал — это самый ранний после бромидов используемый в практике противосудорожный препарат. Хотя долгие годы его считали одним из наиболее безопасных средств, в настоящее время рекомендуется преимущественное применение других препаратов, обладающих меньшим седативным эффектом. Многие считают фенобарбитал препаратом выбора только для грудных детей.
Химия
В качестве противосудорожных препаратов применяют четыре производных барбитуровой кислоты — это фенобарбитал, мефобарбитал, метарбитал и примидон. Первые три настолько похожи, что будут рассмотрены вместе. Метарбитал — это метилированный барбитал, а мефобарбитал — это метилированный фенобарбитал. Оба они деметилируются in vivo. рКа этих слабых кислот варьирует между 7.3 и 7.9, поэтому даже небольшие изменения в кислотно-основном балансе могут значитель
но смещать соотношение ионизированной и неио-низированной форм. Это особенно важно для фенобарбитала, используемого наиболее часто, значение рКа которого близко к pH плазмы (7.4).
Изучение трехмерной конформации фенобарбитала и N-метилфенобарбитала показало, что она похожа на структуру молекулы фенитоина. Оба соединения содержат фенольное кольцо и эффективны при парциальных судорогах.
Механизм действия
Точный механизм действия фенобарбитала неизвестен, но большое значение имеют подавление передачи возбуждающего сигнала и усиление тормозящего. Недавние исследования показали, что фенобарбитал может селективно подавлять активность аномальных нейронов, прекращая возникновение и распространение импульсов. Как и фенитоин, фенобарбитал блокирует высокочастотные повторные разряды нейронов, действуя на ток ионов натрия, однако этот эффект проявляется только при высоких концентрациях препарата. В высоких дозах фенобарбитал блокирует также ток ионов кальция через клеточную мембрану (L-и N-типы). Фенобарбитал связывается с аллостерическими регуляторными участками ГАМК-рецептора и усиливает ГАМК-зависимый ток путем продления времени открытия хлорных каналов. Этот препарат также блокирует возбуждающие реакции, особенно опосредованные глутаматным АМРА-рецептором (глава 20). И усиление ГАМК-торможения, и снижение глутамат-опосре-дованного возбуждения отмечаются при терапевтических концентрациях фенобарбитала.
Клиническое применение
Фенобарбитал особенно эффективен при лечении парциальных и генерализованных тоникоклонических судорог, хотя часто его используют при разных типах судорог, особенно при затрудненном их контроле. Мало накоплено сведений о его эффективности при абсансе, атонических атаках и инфантильных спазмах. При этих состояниях препарат может вызвать ухудшение у некоторых больных.
Некоторые врачи предпочитают фенобарбиталу метарбитал либо мефобарбитал (особенно последний) из-за меньшего количества побочных эффектов.
Фармакокинетика
Рассматривается в главе 21.
Терапевтические концентрации и дозы
Терапевтические концентрации фенобарбитала в плазме для большинства пациентов составляют 10-40 мкг/мл. При фебрильных судорогах уровни ниже 15 мкг/мл неэффективны для предотвращения повторного приступа. Значительно труднее определить верхнюю границу терапевтического диапазона, так как многие пациенты переносят длительный прием препарата, при котором концентрация фенобарбитала в плазме превышает 40 мкг/л.
Лекарственные взаимодействия и токсичность
Рассматриваются в главе 21.
Примидон
Примидон, 2-дезоксифенобарбитал (рис. 23-5), появился в начале 1950-х гг. Позже было определено, что примидон метаболизируется до фенобарбитала и фенилэтилмалонамида (РЕМА). Все три соединения являются активными противосудорожными средствами.
Фенилэтилмалонамид (РЕМА)
Рис. 23-5. Примидон и его активные метаболиты
Механизм действия
Хотя примидон превращается в фенобарбитал, по механизму действия он больше похож на фенитоин.
Клиническое применение
Примидон, как и его производные, эффективен при парциальных и генерализованных тоникоклонических судорогах и даже несколько более эффективен, чем фенобарбитал. Раньше он считался препаратом выбора при сложных парциальных судорогах, но последние исследования этого вида судорог у взрослых показали, что карбамазепин и фенитоин обладают более выраженным действием. Были сделаны попытки определить эффективность основного препарата и его двух метаболитов у новорожденных, у которых из-за незрелости ферментативных систем метаболизм протекает очень медленно. Примидон оказался эффективным у этой группы больных, а также у пожилых пациентов, контроль над возникновением судорог у которых наступал до достижения терапевтических концентраций фенобарбитала в плазме. Наконец, исследования судорог, вызванных максимальным электрошоком у животных, показали, что противосудорожное действие примидона не зависит от его превращения в фенобарбитал и РЕМА, и на этой модели оно менее выражено, чем действие фенобарбитала.
Фармакокинетика
Примидон полностью всасывается, достигая максимальных концентраций в среднем через 3 часа после перорального приема, хотя значения этого показателя варьируют. Примидон имеет в жидкостной среде объем распределения 0.6 л/кг. Он плохо связывается с белками плазмы: около 70 % препарата циркулирует в несвязанном виде.
Примидон метаболизируется путем окисления в фенобарбитал, который очень медленно кумулирует, а также расщеплением гетероциклического кольца с образованием РЕМА (рис. 23-5). И примидон, и фенобарбитал также гидроксилируются в пара-положении фенильного кольца, затем конъюгируются и выделяются.
Клиренс примидона более значителен, чем у других противосудорожных препаратов (2 л/кг/день), и соответствует периоду его полу
выведения в 6-8 часов. Клиренс РЕМА в 2 раза меньше клиренса примидона, а клиренс фенобарбитала очень низкий. Появление фенобарбитала по времени совпадает с исчезновением примидона. Поэтому фенобарбитал кумулирует очень медленно, но постепенно достигает терапевтических концентраций у большинства пациентов, получающих терапевтические дозы примидона. Уровни фенобарбитала, образовавшегося из примидона, обычно в 2-3 раза выше, чем уровни самого примидона. РЕМА, который скорее всего оказывает наименьший терапевтический эффект, имеет период полувыведения 8-12 часов и поэтому быстрее, чем фенобарбитал, достигает стабильного уровня в плазме.
Терапевтические концентрации и дозы
Эффективность примидона максимальна при концентрации препарата в плазме 8-12 мкг/мл. Сопутствующие уровни его метаболита фенобарбитала при достижении стабильных значений обычно колеблются в пределах 15-30 мкг/мл. Для достижения таких концентраций необходимы дозы 10-20 мг/кг/день. Однако очень важно начинать прием примидона с низких доз и постепенно повышать их в течение нескольких дней или недель, чтобы избежать седативного действия и нарушений со стороны желудочно-кишечного тракта. При подборе доз важно помнить, что исходный препарат быстро достигает стабильного уровня (30-40 часов), в то время как его метаболитам требуется больше времени: фенобарбиталу — 20 дней и РЕМА — 3-4 дня.
Токсичность
Дозозависимые побочные эффекты примидона сходны с наблюдаемыми у его метаболита фенобарбитала, однако, если исходная доза достаточно высокая, сонливость возникает очень быстро от момента начала лечения. Как для детей, так и для взрослых препарат рекомендуется назначать с медленным повышением дозы.
Вигабатрин
Исследования последних лет, в которых внимание концентрируется на усилении эффектов ГАМК, направлены на поиск ГАМК-агонистов, ингибиторов ГАМК-трансаминазы и ингибиторов захвата
СН2 = СНЧ /СН2ч >СООН СН СН2
I nh2
Вигабатрин
ГАМК. Вигабатрин (у-винил-ГАМК) — наиболее многообещающий из этих новых препаратов, был недавно выпущен для применения в Европе и Южной Америке. Его открытие и использование показали, что усиление ГАМК-трансмиссии является рациональным и обоснованным подходом к лечению эпилепсии. Препарат пока не применяется в США.
Механизм действия
Вигабатрин — это необратимый ингибитор ГАМК-аминотрансферазы (ГАМК-Т) — фермента, отвечающего за деградацию ГАМК. Он действует, повышая концентрацию ГАМК в синапсах, увеличивая этим тормозные эффекты, а также неизвестным способом подавляя повышенную возбудимость нейронов, лежащую в основе возникновения и распространения эпилептических судорог. Этот препарат эффективен на нескольких моделях судорог.
Клиническое применение
Вигабатрин показан прежде всего при парциальных судорогах и при синдроме Веста. Взрослым вигабатрин начинают назначать с дозы 500 мг 2 раза в день; для полной эффективности может потребоваться свыше 1.5 г в сутки. Типичные признаки токсичности — это сонливость, головокружение и прибавка в весе. Менее частые, но более опасные побочные эффекты — психомоторное возбуждение, нарушение сознания и психоз, поэтому наличие психического заболевания является относительным противопоказанием к применению вигабатрина. Длительная отсрочка появления препарата на мировом рынке была связана с обнаружением у крыс и собак обратимого отека миелиновых оболочек при его введении. Тысячи пациентов принимают этот препарат в настоящее время и признаков подобного отека у них не наблюдается.
Фармакокинетика
Абсорбция вигабатрина идет быстро, максимальные концентрации достигаются через 1-3 часа. Период полувыведения препарата приблизительно
равен 6-8 часам, но имеются данные о том, что фармакодинамическое действие вигабатрина более продолжительно и не всегда коррелирует с уровнями в плазме. Препарат подчиняется правилам линейной кинетики, не имеет активных метаболитов, минимально связывается с белками плазмы, выводится преимущественно почками.
Ламотриджин
Ламотриджин был создан на волне представлений о том, что эффективность некоторых противосудорожных препаратов, например фенитоина, связана с их антагонизмом по отношению к фолиевой кислоте. Появились фенилтриазины и, хотя их про-тивофолиевый эффект оказался слабым, некоторые из них эффективно подавляли судороги по механизму, сходному с фенитоином. Конкретный механизм действия ламотриджина неясен, но, как и фенитоин, он предотвращает быструю повторную импуль-сацию нейронов, продлевая стадию инактивации натриевого канала. Период его полувыведения приблизительно равен 24 часам, большая часть препарата выделяется почками в виде глюкуронида. Период полувыведения сокращается назначением препаратов, индуцирующих активность микросомальных ферментов. Ламотриджин эффективен при парциальных судорогах у взрослых при приеме в дозе 100-300 мг/день и при терапевтической концентрации в плазме около 3 мкг/мл. Он также эффективен при абсансе и миоклонических судорогах у детей. Препарат выпускается в некоторых странах Европы, но не в США.
Ламотриджин
Фелбамат и габапентин
Это два других новых интересных препарата, выпуск которых во всем мире планируется на следующие несколько лет. Фелбамат был одобрен и внедрен в США, но затем снят с производства в свя
зи с высоким риском развития апластической анемии. Он эффективен у некоторых пациентов при парциальных судорогах, имеет период полувыведения 20 часов и выводится печенью. Этот препарат повышает уровни фенитоина и вальпроевой кислоты в плазме, но снижает уровень карбамазепина.
Габапентин — это аналог ГАМК, который также эффективен при парциальных судорогах. Его период полувыведения 6 часов, он выводится почками (Rogawski, 1990).
Препараты, применяемые при генерализованных судорогах
Этосукцимид
Этосукцимид был третьим по счету из сукцинимидом, созданным в США в 1960 г. Этосукцимид почти не обладает активностью против судорог, вызванных максимальным электрошоком, но эффективен при пент^лентетразоловых судорогах и потому предложе1| как препарат для лечения исключительно petit mal. Он по-прежнему популярен в связи со своей эффективностью и безопасностью, ему отводится роль препарата первого выбора при абсансе прежде всего из-за гепатотоксичности альтернативного препарата — вальпроевой кислоты.
Химия
Этосукцимид — последний по времени создания препарат из группы веществ, содержащих циклическую уреидную структуру. В США выпускаются три антиэпилептических сукцинимида: этосукцимид, фенсукцимид и метсукцимид. Во всех этих препаратах происходит замещение в положении 2 (сравните с рис. 23-1). Метсукцимид и фенсукцимид имеют фенильные группы, а этосукцимид является 2-этил-2-метилсукцимидом.
Н5С\
Этосукцимид
Механизм действия
Механизм действия сукцинимидов, возможно, связан с кальциевыми каналами. Этосукцимид также подавляет Иа+,К+-АТФазу, снижает церебральный метаболизм и ингибирует ГАМК-аминотранс-феразу. Однако ни один из этих эффектов не проявляется в терапевтических концентрациях. Этосукцимид существенно влияет на токи ионов кальция: он снижает низкопороговые токи (Т-тип). Этот эффект наблюдается в терапевтических концентрациях в таламических нейронах. Считается, что токи ионов кальция Т-типа играют роль водителя ритма в таламических нейронах, генерируя ритмические корковые импульсы абсанса. Подавление этих токов может быть основой эффективности этосукци-мида.
Клиническое применение
Этосукцимид особенно эффективен при абсансе, что можно предсказать и по его действию на различных моделях. Доказательства его эффективности потребовали дополнительных изысканий в определении и диагностике абсанса. Это стало возможным в 1970-е гг., когда обнаружили корреляцию характерных генерализованных волн с частотой 3-1 с на ЭЭТ с нарушением сознания, даже если это нарушение длится всего несколько секунд. Поэтому продолжительная регистрация ЭЭГ обеспечивала необходимый количественный метод определения частоты абсанса и оценки эффективности препаратов. Хотя этосукцимид был выпущен до появления государственного стандарта эффективности, эти методы были использованы при сравнении эффективности вальпроевой кислоты и клоназепама с этосукцимидом.
Фармакокинетика
Препарат полностью абсорбируется после перорального приема. Максимальные уровни достигаются через 3-7 часов после приема капсул. Исследования на животных показали, что длительное пероральное применение раствора может привести к раздражению слизистой желудка.
Этосукцимид равномерно распределяется по перфузируемым тканям, но не проникает в жировую ткань. Объем распределения приближается к общему объему воды в теле, то есть 0.7 л/кг. Препарат не связывается с белками, поэтому концент
рации в спинальной жидкости равны концентрациям в плазме.
Этосукцимид полностью метаболизируется, в основном гидроксилированием. Известны четыре гидроксилированных метаболита этосукцимида. Эти метаболиты затем подвергаются конъюгации и выводятся. Данные о том, что они обладают фармакологической активностью, отсутствуют.
Этосукцимид обладает очень низким общим клиренсом (0.25 л/кг/день), который соответствует периоду полувыведения, приблизительно равному 40 часам, хотя, по некоторым данным, длительность этого периода варьирует от 18 до 72 часов.
Терапевтические концентрации и дозы
Для достижения терапевтических концентраций в 60-100 мкг/мл взрослым требуется прием 750-1500 мг/день, хотя это возможно иногда при меньших или только при более высоких дозах. Считается, что некоторым пациентам для появления эффекта препарата необходимы более высокие (до 125 мкг/мл) концентрации. У таких больных проявлений токсичности не наблюдается. Для этосукцимида характерно линейное соотношение дозы и стабильных уровней в плазме. Если бы не побочные эффекты на желудочно-кишечный тракт, препарат можно было бы назначать для однократного дневного приема. Обычно же его применяют 2 раза в день. Не принципиально, в какое время дня будет контролироваться концентрация препарата, так как он обладает продолжительным периодом полувыведения. Лекарственных форм для парентерального введения нет.
Лекарственные взаимодействия
Назначение этосукцимида совместно с вальпроевой кислотой приводит к снижению клиренса этосукцимида и повышению его стабильной концентрации в связи с угнетением собственного метаболизма. Других важных взаимодействий сукцинимидов с другими препаратами не выявлено.
Токсичность
Самый частый дозозависимый побочный эффект этосукцимида — это раздражение желудочно-кишечного тракта, проявляющееся болью, тошнотой и рвотой. Этого можно избежать, начиная прием с
маленьких доз и постепенно повышая их до терапевтического уровня. При возникновении этого побочного эффекта временное снижение дозы может обеспечить адаптацию к препарату. Этосукцимид — весьма эффективный и безопасный препарат при абсансе, появление сравнительно нетяжелых дозозависимых побочных эффектов не должно являться основанием для прекращения его приема. Другие дозозависимые эффекты — это преходящая сонливость или усталость и значительно реже — головные боли. Изменения поведения обычно наблюдаются в сторону его улучшения.
Недозозависимые побочные эффекты этосукцимида возникают крайне редко. Иногда появляются кожные сыпи. В литературе упоминается по крайней мере один случай синдрома Стивенса-Джонсона. У некоторых пациентов наблюдаются эозинофилия, тромбоцитопения, лейкопения или панцитопения, однако уверенности в том, что их причиной является этосукцимид, нет. Описаны также случаи возникновения системной красной волчанки, но и другие препараты могли сыграть в этом свою роль.
Фенсукцимид и метсукцимид
Фенсукцимид и метсукцимид — это фенилсук-цимиды, которые были созданы и поступили на рынок раньше, чем этосукцимид. Их используют прежде всего при абсансе. Метсукцимид более токсичен, а фенсукцимид — менее эффективен, чем этосукцимид. В отличие от этосукцимида эти препараты эффективны при судорогах, вызванных максимальным электрошоком. Некоторые исследователи использовали метсукцимид при парциальных судорогах. Деметиловый метаболит метсукцимида имеет период полувыведения 25 часов и более, именно он и оказывает основной противосудорожный эффект. Проводились исследования причины низкой эффективности фенсукцимида по сравнению с метсукцимидом. Считается, что это связано с неспособностью деметилового метаболита фенсукцимида к кумуляции.
Вальпроевая кислота и вальпроат натрия
Противосудорожная активность вальпроата натрия и свободной вальпроевой кислоты была выявлена при использовании вальпроата в качестве ра
створителя в процессе поиска других противосудорожных препаратов. Он был выпущен во Франции в 1969 г., но не был разрешен в США до 1978 г. Вальпроевая кислота полностью ионизируется при физиологических значениях pH организма, то есть можно предположить, что именно ион вальпроата является активной частью как вальпроевой кислоты, так и вальпроата натрия.
Химия
Вальпроевая кислота является примером одной из целой серии жирных кислот, обладающих противосудорожной активностью. Эта активность наиболее выражена при наличии пятиатомной углеводородной цепи. Разветвленность и ненасыщенность не оказывают значительного влияния на активность препарата, но могут изменять его липофильность и, соответственно, длительность действия. Амиды и эфиры вальпроевой кислоты также являются активными противосудорожными препаратами.
сн3-сн2-сн2^
сн3-сн2-сн2^
сн-соон
Вальпроевая кислота
Механизм действия
Длительность противосудорожного эффекта плохо коррелирует с уровнем исходного препарата в крови или тканях, что спровоцировало появление разных мнений об активных формах и механизме действия вальпроевой кислоты. Вальпроат эффективен при судорогах, вызванных как максимальным электрошоком, так и пентилентетразолом. Как фенитоин и карбамазепин, вальпроевая кислота в терапевтических концентрациях блокирует высокочастотную импульсацию в культуре нейронов. Ее действие при парциальных судорогах, возможно, является следствием такого влияния на ток ионов натрия. Однако большое внимание уделяется действию вальпроата на обмен ГАМК. В нескольких исследованиях показано увеличение уровня ГАМК в мозгу после введения вальпроата, хотя механизм этого действия остается неясным. Сомнения относительно роли повышения ГАМК в терапевтическом эффекте препарата связаны с тем, что противосудорожное действие развивается до повышения концентрации ГАМК в мозгу. Если эффект вальпроата связан с
ферментами, необходимыми для продукции или разрушения ГАМК, то у него скорее всего более выражен эффект ингибирования ГАМК-аминотранс-феразы (ГАМК-Т, отвечает за разрушение ГАМК), чем потенцирования декарбоксилазы глутаминовой кислоты, катализирующей синтез ГАМК. В очень высоких концентрациях вальпроат подавляет ГАМК-Т в мозгу, блокируя превращение ГАМК в сукцинилсемиальдегид, и этим повышает уровень ГАМК. Однако в сравнительно низких дозах, достаточных для подавления пентилентетразоловых судорог, препарат не меняет содержание ГАМК в мозгу. Вальпроат понижает уровень аспартата в мозгу грызунов, но значение этого явления для развития противосудорожного действия неизвестно.
В высоких концентрациях вальпроат повышает проводимость мембраны для ионов калия. Более того, небольшие его концентрации вызывают гиперполяризацию мембраны. Эти данные привели к предположению, что эффект вальпроата связан с прямым действием на калиевые каналы.
Не исключено, что широкий спектр фармакологических эффектов вальпроата связан с несколькими молекулярными механизмами действия. Причины его эффективности при абсансе еще предстоит выяснить.
Клиническое применение
Вальпроат весьма эффективен при абсансе. Хотя этосукцимид является препаратом выбора при изолированном абсансе, в тех случаях, когда возникает генерализация судорог в тоникоклонические, предпочтение отдается вальпроату. Причина того, что при изолированном абсансе препаратом выбора является этосукцимид, связана с идиосинкразической гепатотоксичностью вальпроата. Вальпроат уникален по своей эффективности при миоклонических судорогах, в некоторых случаях его эффективность поразительна. Этот препарат обладает лечебным действием при генерализованных тоникоклонических судорогах, особенно первично-генерализованных, а также при атонических судорогах. Есть данные об успешном использовании препарата при парциальных судорогах.
Фармакокинетика
Вальпроат хорошо всасывается при пероральном приеме, его биодоступность превышает 80 %. Максимальные концентрации достигаются в пре
делах 2 часов. Пища может замедлить абсорбцию, снижая токсические эффекты препарата при приеме после еды.
рКа вальпроевой кислоты равна 4.7, следовательно, препарат почти полностью ионизируется при физиологических значениях pH крови. Препарат на 90 % связывается с белками плазмы, хотя, если концентрация препарата превышает 150 мкг/мл, связанная фракция препарата несколько уменьшается. Так как препарат высоко ионизирован и связывается с белками, его распределение происходит почти полностью во внеклеточной жидкости с объемом, приблизительно равным 0.15 л/кг.
Около 20 % препарата выделяется в виде прямого конъюгата вальпроата. Остальная часть метаболизируется р- и со-окислением до нескольких соединений, которые затем вступают в реакции конъюгации и выводятся.
Клиренс вальпроата очень низкий, период полувыведения колеблется между 9 и 18 часами. При очень высоких уровнях в крови клиренс вальпроата зависит от дозы. Отмечаются изменения в клиренсе и в связывании с белками при приеме высоких доз препарата.
Натриевая соль вальпроата выпускается в Европе в виде таблеток, защищенных алюминиевой фольгой вследствие их высокой гигроскопичности. В Центральной и Южной Америке используется магниевая соль, которая значительно менее гигроскопична. Свободная вальпроевая кислота впервые была выпущена в США в виде капсул, содержащих кукурузное масло, натриевая соль выпускается в виде сиропа, преимущественно для лечения детей. Сейчас в США продаются таблетки в оболочке devalproex sodium, которая растворяется в кишечнике. Это — улучшенный препарат, представляющий собой комбинацию вальпроата натрия и вальпроевой кислоты в отношении 1:1, имеющий такую же биодоступносгь, как и капсулы, но более медленно растворимый. Его предпочитает большинство пациентов. Максимальные концентрации после приема этих таблеток в оболочке возникают через 3-4 часа.
Терапевтические концентрации и дозы
Некоторым пациентам может быть достаточно 25-30 мг/кг/день, а другим требуется 60 мг/кг или даже больше. Терапевтические концентрации вальпроата колеблются между 50 и 100 мкг/мл. При сомнениях в эффективности этого препарата его не
следует отменять до тех пор, пока уровень утром до приема лекарства не достигнет 80 мкг/мл. Некоторым пациентам необходимы уровни 100 мкг/мл и выше, при этом у них не возникает токсических эффектов.
Лекарственные взаимодействия
Как уже сказано, клиренс вальпроата дозозависим и определяется изменениями эндогенного клиренса и связывания с белками плазмы. В низких дозах вальпроат подавляет свой метаболизм, таким образом снижая эндогенный клиренс. При приеме более высоких доз увеличивается свободная фракция вальпроата, что приводит к неожиданно низким концентрациям препарата. Поэтому в клинике полезно измерять как свободную, так и общую фракции вальпроата. Вальпроат способен вытеснять фенитоин из связей с белками плазмы. Кроме таких взаимодействий, вальпроат подавляет метаболизм некоторых препаратов, в том числе фенобарбитала, фенитоина и карбамазепина, что приводит к повышению их концентраций. Ингибирование метаболизма фенобарбитала вызывает иногда значительное повышение его уровня, что может привести к состоянию ступора или комы.
Токсичность
Наиболее частые дозозависимые побочные эффекты вальпроата — это тошнота, рвота и другие желудочно-кишечные нарушения типа боли в животе или изжоги. Прием этого препарата следует начинать постепенно, чтобы избежать подобных проявлений. Временное снижение дозы обычно облегчает эти симптомы, и затем пациент сможет переносить более высокие дозы. Седативный эффект встречается редко, но при комбинациях с фенобарбиталом может быть очень сильным. При приеме высоких доз часто отмечается легкий тремор. Другие обратимые эффекты, встречающиеся у некоторых больных,— это увеличение веса тела, повышение аппетита и выпадение волос.
Идиосинкразическая токсичность вальпроата проявляется только гепатотоксичностью, но она может быть тяжелой. Известно, что по этой причине в США умерли более 50 человек. Риск особенно велик для детей до 2 лет и для тех, кто принимает несколько препаратов. Начальный уровень активности ACT (аспартатаминотрансферазы) может быть
нормальным, но постепенно он существенно возрастает. Большинство смертей произошло в течение 4 месяцев после начала терапии. Рекомендуется тщательный контроль функции печени с самого начала приема препарата, в некоторых случаях гепатотоксичность обратима после отмены препарата. Другой идиосинкразический эффект вальпроата — это тромбоцитопения, хотя данные об аномальных кровотечениях отсутствуют. Следует отметить, что вальпроат — это эффективный и популярный противосудорожный препарат, и что только у небольшого числа пациентов возникают серьезные токсические эффекты при его применении.
Исследования вальпроата выявили повышенную частоту встречаемости spina bifida у детей, рожденных женщинами, принимавшими вальпроат во время беременности. Кроме того, отмечено повышение частоты возникновения сердечно-сосудистых, орофациальных аномалий и аномалий пальцев у новорожденных. Хотя число этих наблюдений незначительно, их надо учитывать при выборе противосудорожного препарата во время беременности.
Оксазолидиндионы
Первый оксазолидиндион триметадион был предложен как противосудорожный препарат в 1945 г. и оставался препаратом выбора при абсансе до 1950-х гг. Сейчас использование оксазолидин-дионов (триметадиона, параметадиона и диметадиона) очень ограничено.
Механизм действия
Эти соединения эффективны против пентилен-тетразоловых судорог. Триметадион повышает порог судорожной активности после повторной таламической стимуляции. Он оказывает такой же эффект на таламические кальциевые токи, как и этосукцимид (снижает токи Т-типа). Таким образом, можно предположить возможность подавления выполнения таламическими нейронами функции водителя ритма.
Фармакокинетика
Триметадион быстро всасывается, достигая максимальных уровней в течение часа после введения. Он распределяется во все перфузируемые ткани, поэтому объем его распределения приблизительно равен общему объему водной среды. Он не связывается с белками плазмы. Триметадион полностью метаболизируется в печени деметилированием до 5,5-диметил-2,4-оксазолидиндиона (диметадиона), который обладает максимальной противосудорожной активностью среди оксалидиндионов. У препарата сравнительно низкий клиренс (1.6 л/кг/день), который соответствует периоду полувыведения 16 часов. Однако деметилированный метаболит выводится из организма очень медленно и кумулирует в большей степени, чем исходный препарат. Клиренс диметадиона равен 0.08 л/кг/день, у этого метаболита крайне длительный период полураспада (240 часов).
Химия
Оксазолидиндионы содержат гетероциклическое оксазолидиновое кольцо (рис. 23-1) и структурно похожи на другие противосудорожные препараты, появившиеся до 1960-х гг. Их структура включает в себя только короткоцепочечные алкильные замещающие группы в гетероциклическом кольце без фенильной группы.
Н3С
Н3С
5 С=О
----N—СН3
Триметадион
Терапевтические концентрации и дозы
Терапевтические концентрации в плазме для триметадиона пока не установлены, хотя предложено придерживаться уровня триметадиона выше 20 мкг/мл и диметадиона выше 700 мкг/мл. Для достижения этого уровня у взрослых необходима доза 30 мг/кг/день.
Лекарственные взаимодействия
Для оксазолидиндионов известно сравнительно малое количество случаев взаимодействий с другими препаратами, хотя триметадион может конкурентно ингибировать деметилирование других препаратов, например метарбитала.
Токсичность
Самый значительный дозозависимый побочный эффект оксазолидиндионов — это седация. Необычное осложнение — это гемералопия (нарушение зрительной адаптации); она обратима после прекращения приема препарата. Кумуляция диметадиона приводит к легкому метаболическому ацидозу. Триметадион вызывает кожные проявления идиосинкразии, например сыпь или эксфолиативный дерматит, а также токсические гемопоэтические реакции от незначительных изменений клинического анализа крови до злокачественной панцитопении. К другим токсическим реакциям относятся обратимый нефротический синдром, который может быть связан с иммунной реактивностью на препарат, и миастенический синдром. Оксазолидиндионы нельзя использовать во время беременности.
Другие препараты, применяемые для лечения эпилепсии
Далее обсуждаются некоторые препараты, которые нельзя классифицировать по действию на определенный тип судорог.
Бензодиазепины
(подробно рассмотрены в главе 21)
Шесть представителей группы бензодиазепинов играют важную роль в терапии эпилепсии. Хотя многие бензодиазепины похожи химически, некоторые структурные изменения приводят к различиям в их активности. Они обладают двумя механизмами противоэпилептического действия, в разной степени представленными у этих шести препаратов. Так диазепам несколько эффективнее при судорогах, вызванных электрошоком, а клоназепам — против судорог, вызванных пентилентетразолом (последний эффект коррелирует с действием на аллостерический ГАМК-бензодиазепиновый рецептор). Возможные механизмы действия обсуждаются в главе 21.
Диазепам при внутривенном введении очень эффективно прекращает продолжительную судорожную активность, особенно генерализованный тоникоклонический эпилептический статус. Препа
рат также назначается для сочетанной терапии и иногда длительно применяется перорально, хотя этот метод не считается очень эффективным из-за быстрого развития толерантности.
Лоразепам — это новый бензодиазепин, который при внутривенном введении в некоторых исследованиях оказался более эффективным и более длительнодействующим в терапии эпилептического статуса, чем диазепам.
Клоназепам — препарат длительного действия, эффективный при абсансе. Это один из самых лучших противосудорожных препаратов. Иногда он эффективен при миоклонических судорогах, его пробовали применять и при инфантильных спазмах. Клоназепам обладает значительным седативным эффектом, особенно в начале терапии, поэтому начинать лечение надо с малых доз. Максимально переносимые дозы — это обычно 0.1-0.2 мг/кг, но может потребоваться много недель для достижения этого уровня у некоторых пациентов. Терапевтические концентрации в плазме, как правило, меньше 0.1 мкг/мл и их не измеряют в большинстве лабораторий.
Клоразепат дикалий — это бензодиазепин, одобренный в США для сочетанной терапии сложных парциальных судорог у взрослых. Сонливость является частым побочным эффектом, но если дозу препарата повышать постепенно, то ее можно довести вплоть до 45 мг/день.
Нитразепам не производится в США, но применяется во многих других странах, особенно при инфантильных спазмах и миоклонических судорогах. Он менее эффективен, чем клоназепам, и неизвестно, имеет ли какие-либо преимущества перед клоназепамом.
Клобазам не используется в США, но, как и нитразепам,’ применяется во многих других странах. Это 1,5-бензодиазепин (в отличие от других препаратов, являющихся 1,4-бензодиазепинами), дающий меньший седативный эффект, чем другие бензодиазепины. Точно не известно, имеет ли этот препарат значимые преимущества.
Фармакокинетические свойства бензодиазепинов частично определяют их применение. В целом эти препараты хорошо всасываются, широко распределяются и интенсивно метаболизируются с образованием большого числа активных метаболитов. Скорость распределения бензодиазепинов в организме отличается от скорости распределения других противосудорожных препаратов. Диазепам
и лоразепам особенно быстро и широко распределяются в тканях, объем их распределения варьирует между 1 и 3 л/кг. Действие начинается очень быстро, период полувыведения равен 20-40 часам.
Два основных момента ограничивают применение бензодиазепинов. Первый — это значительный седативный эффект, который нежелателен как при лечении эпилептического статуса, так и при поддерживающей терапии. У детей может возникать парадоксальная гиперреактивность, как при применении барбитуратов. Вторая проблема — это толерантность, в связи с которой через несколько месяцев терапии противосудорожный эффект этих препаратов может уменьшаться. В связи с этими ограничениями противосудорожное действие бензодиазепинов не удается использовать в полную силу.
Ацетазоламид
Ацетазоламид — это диуретик, основным действием которого является угнетение карбоангидразы (глава 15). Основной механизм противосудорожной активности препарата связан с кумуляцией диоксида углерода в мозгу; кроме того, препарат, возможно, оказывает прямое действие на нейроны. Ацетазоламид используют при всех видах судорог, но его применение ограничено быстрым развитием толерантности (в течение нескольких недель). Препарат может играть особую роль у женщин, страдающих эпилепсией, обостряющейся во время менструаций, так как периодический прием препарата может предотвратить развитие толерантности и повысить его эффективность. Обычная доза приблизительно равна 10 мг/кг, максимальная — 1000 мг/день.
Эффективность другого ингибитора карбоангидразы султиама не была подтверждена клиническими испытаниями в США. Однако он используется в некоторых других странах.
Бромид
Бромид впервые был использован Лококом в 1857 г. В то время это был первый противосудорожный препарат, обладающий реальной эффективностью. Хотя его применение значительно уменьшилось после появления фенобарбитала в начале XX в., бромид еще иногда назначают, например при судорогах у больных порфирией, которым противопоказаны другие препараты. Период его полувыведения около 12 дней, он назначается взрослым в
дозах 3-6 г/день для получения концентрации в плазме 10-20 мэкв/л. Механизм противоэпилепти-ческого действия бромида неизвестен. Значительные токсические эффекты, к сожалению, часты. К ним относятся кожная сыпь, седация и поведенческие изменения. В настоящее время в США он не применяется.
II. Клиническая фармакология противоэпилептических препаратов
Классификация судорог
Выбор препарата для лечения эпилепсии зависит от характера судорог. Поэтому необходима классификация судорог, чтобы клиницист мог определить их тип и назначить соответствующую терапию. Ошибки в определении типа судорог приводят к назначению несоответствующих препаратов, отсутствию лечебного эффекта, повышению дозы лекарств и возникновению побочных явлений. Как отмечалось ранее, судороги разделяются на две группы: парциальные и генерализованные. Препараты, применяемые при парциальных судорогах, одинаковы для всех типов судорог внутри этой группы, в то время как препараты, применяемые при генерализованных судорогах, различны для каждого их типа. Упрощенная версия международной классификации эпилептических судорог представлена в табл. 23-1.
Парциальные судороги
Парциальными считаются такие судороги, локальное происхождение которых может быть доказано либо клиническими проявлениями, либо регистрацией ЭЭГ, если приступ начинается в каком-либо участке мозга. Существуют три типа парциальных судорог в зависимости от степени вовлечения мозга в аномальную импульсацию.
Наименее осложненный тип парциальных судорог — это простые парциальные судороги, которые характеризуются минимальным распространением аномальных разрядов, что обусловливает сохранение нормального уровня сознания. Например, у пациента может возникать внезапное клоническое подергивание конечности, длящееся 60-90 секунд,
остаточная слабость может длиться 15-30 минут после приступа. Пациент полностью осознает приступ и описывает его в деталях. На ЭЭГ могут выявляться аномальные разряды с четко ограниченной фокальной локализацией.
Сложные парциальные судороги также имеют локализованное начало, но затем нейрональная активность несколько распространяется (обычно в пределах одного полушария мозга) и почти всегда вовлекает лимбическую систему. Большинство (не все) сложных парциальных судорог возникает в височных долях, возможно, вследствие высокой чувствительности этой области мозга к гипоксии или инфекции. Клинически у пациента могут возникать кратковременные предвестники с последующим нарушением сознания, во время которого они могут непрерывно смотреть в одну точку или спотыкаться и даже падать. Однако у большинства наблюдается проявление так называемых моторных автоматизмов, о которых больной впоследствии не помнит. Типичные автоматизмы — это почмокивание губами, глотание, царапанье или даже ходьба. После 30-120 секунд сознание пациента постепенно восстанавливается, но может оставаться чувство усталости в течение нескольких часов после приступа.
Последний тип парциальных судорог — это вторично генерализованные судороги, при которых сразу вслед за парциальными судорогами возникают генерализованные тоникоклонические (grand mat). Этот тип судорог описан в следующем подразделе.
Генерализованные судороги
Генерализованными считаются такие судороги, у которых нет фокального начала. Эта группа достаточно гетерогенна.
Генерализованные тоникоклонические судороги (grand таГ) наиболее тяжелые из всех видов судорог, они характеризуются тонической ригидностью всех конечностей с последующим тремором в течение 15-30 секунд. Этот тремор представляет собой прерывание тонуса периодами релаксации. С удлинением периодов релаксации приступы переходят в клоническую фазу с массивными подергиваниями тела. Клонические подергивания замедляются в течение 60-120 секунд, и пациент обычно остается в ступорозном состоянии. Возможен прикус языка или щеки, обычно возникает недержание
мочи. Первично генерализованные тоникоклонические судороги начинаются без признаков фокального начала, тогда как вторично генерализованным тоникоклоническим судорогам предшествует другой тип судорог, обычно парциальные судороги. Медикаментозное лечение первично и вторично генерализованных тоникоклонических судорог одинаково и основано на использовании препаратов для лечения парциальных судорог.
Абсанс (petit таГ) характеризуется внезапным началом и прекращением. Длится обычно менее 10 секунд и редко дольше 45 секунд. Сознание нарушается, приступы могут также сопровождаться легкими клоническими подергиваниями век или конечностей с изменениями позы, автоматизмами и вегетативными нарушениями. Автоматизмы могут осложнить клиническую дифференцировку со сложными парциальными судорогами у некоторых пациентов. Абсанс (petit таГ) начинается в детстве или юности и может возникать до сотен раз в день. ЭЭГ во время судорог демонстрирует характерные волны частотой 2.5-3.5 Гц. При нетипичном течении наблюдаются приступы с изменением позы, которые начинаются очень внезапно. При таком варианте течения дети часто бывают умственно отсталыми. На ЭЭГ могут возникать медленные волны и зубцы. Такие судороги более рефрактерны к терапии.
Миоклонус встречается в большей или меньшей степени при многих типах судорог, включая генерализованные тоникоклонические судороги, парциальные судороги, абсанс и инфантильные спазмы. Лечение судорог с миоклонусом должно быть ориентировано на тип судорог, а не на миоклонус. Однако у некоторых пациентов миоклонус является главным проявлением судорог, а у ряда больных присутствует частый миоклонус и периодические генерализованные тоникоклонические судороги при отсутствии четких признаков неврологических нарушений. Существует много видов миоклонуса, и попытки его классификации предпринимаются по сей день.
Атонические судороги — это такие судороги, при которых пациент внезапно теряет тонус мышц, поддерживающих тело в определенном положении. При этом пациент неожиданно падает на землю и может получить повреждения. Если пациент сидит, то его голова и тело могут внезапно наклониться вперед. Хотя этот вид судорог чаще всего встречается у детей, но не является редкостью и у взрос
лых. Многие пациенты с атоническими судорогами вынуждены носить шлемы для предотвращения травм головы.
Инфантильные спазмы — это эпилептический синдром, а не тип судорог. Приступы чаще всего двусторонние (хотя могут быть и фрагментарные), и по практическим соображениям они классифицируются как генерализованные судороги. Такие приступы клинически характеризуются короткими повторяющимися миоклоническими подергиваниями с внезапными сгибаниями и разгибаниями тела и конечностей. Однако инфантильные спазмы достаточно гетерогенны. У 90 % пациентов первый приступ возникает в возрасте до 1 года. Большинство пациентов страдают умственной отсталостью, предположительно связанной с той же причиной, что и развитие судорожной патологии. У многих пациентов причина неизвестна, но подозревают различные расстройства, в том числе инфекцию, желтуху новорожденных, tuberous sclerosis и гипогликемию. В некоторых случаях имеются характерные изменения ЭЭГ. Препараты для лечения инфантильных спазмов эффективны только у некоторых больных, терапевтическое действие препаратов в отношении умственной отсталости не доказано, даже если приступы прекращаются.
Терапевтическая стратегия
Для противосудорожных препаратов соотношение уровней препарата в плазме и его эффектов особенно значимо. То же самое справедливо для фармакокинетики этих лекарств. Это соотношение дает значительные преимущества для определения терапевтической стратегии лечения эпилепсии. Терапевтический индекс большинства противосудорожных препаратов низкий, токсические эффекты нередки. Таким образом, эффективное лечение судорог требует знаний терапевтических концентраций и фармакокинетических свойств, а также побочных эффектов каждого препарата. Измерения уровней содержания препаратов в плазме очень полезны при их сопоставлении с клиническими наблюдениями и фармакокинетическими данными (табл. 23-2).
ТАБЛИЦА 23-2. Эффективные уровни шести противосудорожных препаратов в плазме1
Препарат Эффективный Уровень Токсический
уровень (мкг/мл) высокой эффективности2 (мкг/мл) уровень (мкг/мл)
Карбамазепин 4-12 7 >8
Примидон 5-15 10 > 12
Фенитоин 10-20 18 >20
Фенобарбитал 10-40 35 >40
Этосукцимид 50-100 80 > 100
Вальпроат 50-100 80 > 100
1 Из: Porter R. J. Epilepsy: 100 Elementary Principles, 2nd ed. Saunders, 1989.
2 Уровень, к которому при возможности надо стремиться у пациентов с рефрактерными судорогами; такие цифры должны получаться при анвлизе крови утром до первого приема препарата. При монотерапии часто и более высокие уровни не сопровождаются токсическими эффектами.
Лечение эпилепсии
Парциальные и генерализованные тоникоклонические судороги
До недавнего времени выбор препаратов был ограничен фенитоином, карбамазепином или барбитуратами. В последние годы наметилась тенденция ограничивать назначение противосудорожных препаратов с седативным эффектом, в том числе барбитуратов и бензодиазепинов, и назначать их только пациентам, которые не переносят другие лекарства. В 1980-е гг. установилась традиция широко использовать карбамазепин. В настоящее время начало расширяться применение окскарбазепина, а появление вигабатрина и ламотриджина еще в большей степени усложнило выбор препарата.
Генерализованные судороги
Для лечения генерализованных тоникоклонических судорог используют те же препараты, что и против парциальных судорог, кроме того, применяется вальпроат.
При абсансе эффективны три препарата. Два из них не обладают седативным эффектом и поэтому используются чаще: этосукцимид и вальпроат. Клоназепам также очень эффективен, но проявляет дозозависимые побочные эффекты и вызывает развитие толерантности. Препаратом выбора является этосукцимид; у пациентов, резистентных к нему, применяется вальпроат.
Для терапии специфических миоклонических синдромов обычно используют вальпроат. Он не обладает седативным действием и может быть весьма эффективен. Некоторым пациентам помогает клоназепам или другие бензодиазепины, хотя могут потребоваться высокие дозы с сопутствующей седацией и сонливостью. Крайне специфичен миоклонический синдром, или “интенционный миоклонус”, который возникает после тяжелой гипоксии и характеризуется миоклонусом, сопутствующим произвольным движениям. У пациентов с этим необычным нарушением получают хороший лечебный эффект при применении 5-гидрокситрипто-фана.
Атонические судороги часто рефрактерны ко всем известным препаратам, хотя есть сведения, что вальпроат и ламотриджин могут проявлять терапевтическое действие. Бензодиазепины улучшают контроль над судорогами у некоторых таких пациентов, но ухудшают у других. Новый препарат фел-бамат также может оказаться эффективным. Если потеря тонуса мышц тела является следствием судорог другого типа (например, абсанса или сложных парциальных судорог), то все усилия должны быть приложены для их лечения, так как есть основание полагать, что одновременно с судорогами купируется и атонический компонент.
Препараты, используемые при инфантильных спазмах
Терапия инфантильных спазмов, к сожалению, ограничена контролем над развитием судорог и не включает коррекцию других признаков заболевания, например отставания умственного развития. Большинство пациентов получают курс внутримышечного кортикотропина, хотя, по мнению некоторых клиницистов, в той же степени эффективен преднизолон, который можно назначать перорально. К настоящему времени клинические испытания не смогли разрешить эту проблему. В любом слу
чае терапию часто приходится прекращать вследствие развития побочных эффектов. При возобновлении судорог можно назначить повторные курсы кортикотропина или кортикостероидов или попробовать другие препараты. К ним относятся бензодиазепины, в том числе клоназепам и нитразепам, приближающиеся по эффективности при этой патологии к кортикостероидам. Вигабатрин также может применяться для коррекции подобного синдрома. Механизм действия кортикостероидов или кортикотропина при лечении инфантильных спазмов пока не известен.
Несмотря на 30-летний опыт использования кортикотропина и кортикостероидов при инфантильных спазмах, оптимальные дозы и длительность терапии не установлены. В практике применяют различные дозы, но чаще всего назначают 25-40 ед кортикотропина в день. Только некоторые специалисты рекомендуют до 240 ед в день. У 60 % пациентов, реагирующих на лечение, уменьшение количества судорог наблюдается в сроки от 1 до 5 недель. Лечение кортикотропином можно продолжать в течение 3 месяцев или дольше, если развитие побочных эффектов не заставляет его прекратить. При терапии кортикостероидами назначают 2 мг/кг преднизолона или дексаметазон в дозе 0.3 мг/кг.
Токсические эффекты кортикотропина являются характерными для избытка кортикостероидов. К ним относятся гипертензия, кушингоидное ожирение, желудочно-кишечные нарушения, изменения кожи, остеопороз и электролитный дисбаланс.
Эпилептический статус
Существует много форм эпилептического статуса. Наиболее часто встречается генерализованный тоникоклонический эпилептический статус — это угрожающее жизни состояние, требующее неотложных мероприятий по поддержанию сердечнососудистых, дыхательных и метаболических констант и немедленной лекарственной терапии. Фармакотерапия всегда включает внутривенное введение противосудорожных препаратов. Диазепам — наиболее эффективный препарат для большинства пациентов. Для прекращения судорог он вводится внутривенно до достижения максимальной дозы 20-30 мг у взрослых. Внутривенно введенный диазепам может угнетать дыхание (реже функцию сер
дечно-сосудистой системы), поэтому при его использовании должны быть всегда под рукой средства для проведения реанимационных мероприятий. Эффект диазепама длится недолго, но 30-40-минутный перерыв в судорогах позволяет назначить более специфическую терапию. Пациентам, которые в момент осмотра не находятся в состоянии активных судорог, диазепам вводить не обязательно и можно сразу назначить более длительно действующий препарат, например фенитоин. Некоторые врачи предпочитают лоразепам, который эквивалентен диазепаму по эффективности, но действует дольше.
Основным препаратом при лечении эпилептического статуса является фенитоин для внутривенного применения, который достаточно эффективен и не вызывает седации. Его следует давать взрослым в нагрузочной дозе 13-18 мг/кг. Самой частой ошибкой является введение недостаточного количества препарата. Скорость введения не должна превышать 50 мг/мин. Безопаснее всего вводить препарат в виде внутривенной инъекции, но можно и капельно с физраствором. Фенитоин нельзя разводить глюкозой, так как она вызывает его преципитацию. Необходимо тщательное наблюдение за ритмом сердца и давлением, особенно у пожилых людей. По крайней мере частично, кардиотоксичность фенитоина связана с этиленгликолем, в котором он разведен в лекарственной форме.
У пациентов, которые ранее получали лечение по поводу эпилепсии, большая нагрузочная доза фенитоина может вызвать дозозависимые побочные эффекты, например атаксию. Это обычно не является существенным симптомом во время статуса и легко снимается коррекцией уровня препарата в плазме.
Если пациент не реагирует на фенитоин, назначают высокую дозу фенобарбитала: по 100-200 мг внутривенно до общего количества 400-800 мг. Частым осложнением такого лечения является респираторная депрессия, особенно если до этого вводили бензодиазепины. В этом случае не следует колебаться при принятии решения об интубации.
Для лечения генерализованного тоникоклонического эпилептического статуса рекомендованы и другие препараты типа лидокаина; в крайне резистентных случаях требуется общая анестезия.
\
Специальные вопросы токсикологии
п роти восудорожн ых препаратов
Тератогенность
Потенциальная тератогенность противосудорожных препаратов — это важный и неоднозначный вопрос. Он принципиален, потому что тератогенный эффект вследствие хронического употребления препарата миллионами людей во всем мире может иметь огромный резонанс, даже если возникает у небольшого процента больных. Эта область исследований весьма противоречива, так как и эпилепсия, и противосудорожные препараты гетерогейны и для контроля можно найти только очень небольшую группу людей, не получающих лечение. БоЛее того, пациенты с тяжелой эпилепсией, у которых генетические факторы, а не препараты, приводят к порокам развития плода, часто получают много противосудорожных средств в высоких дозах.
Несмотря на эти ограничения, оказывается, 4tb дети, рожденные от матерей, получавших противосудорожные препараты во время беременности, имеют повышенный риск (почти в 2 раза) пороков развития. Один из препаратов, а именно фенитоий, считается причиной развития гидантоинового синдрома плода, хотя не все врачи убеждены в его существовании, а схожий синдром ранее связывали с фенобарбиталом и карбамазепином. Использование вальпроата, как уже говорилось ранее, также ассоциируется с развитием аномалий, в частностй spina bifida. Установлено, что беременная женщина, принимающая вальпроат натрия или вальпроевую кислоту, имеет 1-2 % риск рождения ребенка со spina bifida (Valproate, 1983).
При лечении беременных женщин с эпилепсией большинство эпилептологов считают необходимым снизить до минимума дозы и количество препаратов. Также важно контролировать состояние матери при возникновении судорог.
Синдром отмены
Отмена противосудорожных препаратов, случайная или намеренная, может привести к повыше
нию частоты и тяжести судорог. Необходимо учитывать два фактора: эффект самой отмены и необходимость в подавлении препаратом судорог. В большинстве случаев надлежит принимать во внимание оба эти фактора. Однако важно заметить, что внезапное прекращение приема противосудорожных препаратов обычно не вызывает судорог у людей, не страдающих эпилепсией, если уровни препарата до отмены были в терапевтических пределах.
Синдром отмены имеет особенности для разных препаратов. В целом прекращение приема средств против абсанса проходит легче, чем препаратов против парциальных или генерализованных тоникоклонических судорог. Труднее всего прекратить прием барбитуратов и бензодиазепинов: требуются недели и месяцы с очень медленным снижением дозы до полной отмены, особенно в амбулаторных условиях.
Вследствие гетерогенности эпилепсии полная отмена препаратов — это очень трудное решение. Если у пациента не возникает судорог в течение 3-4 лет, то можно попробовать постепенно отменить препарат.
Передозировка
Противосудорожные препараты — это реальные или потенциальные депрессанты ЦНС, но они редко приводят к летальному исходу. Уровни содержания их в плазме должны подняться очень значительно, чтобы состояние стало угрожающим. Самым опасным следствием передозировки противосудорожных препаратов является респираторная депрессия, которая может быть потенцирована другими препаратами, например алкоголем. Лечение передозировки противосудорожных препаратов симптоматическое, не следует применять стимуляторы. Попытки ускорить выведение противосудорожных препаратов, например ощелачиванием мочи, обычно не дают эффекта. Пытались применить и липидный диализ, но для интерпретации эффективности этого метода пока слишком мало данных.
Препараты
Карбамазепин (генерик, Тегретол)
Перорально: таблетки по 200 мг; таблетки для жевания по 100 мг;
суспензия 100 мг/5 мл
Клоназепам (Клонопин)
Перорально: таблетки по 0.5,1.2 мг
Клоразепат дикалий (генерик, Транксен) Перорально: таблетки и капсулы по 3.75,7.5, 15 мг
Перорально для пролонгированного действия (Транксен-SD): таблетки по 11, 25, 22.5 мг
Диазепам (генерик, Валиум, др.)
Перорально: таблетки по 2, 5, 10 мг;
растворы 5 мг/мл, 5 мг/5 мл
Перорально для пролонгированного действия: капсулы по 15 мг
Парентерально: 5 мг/мл для инъекций
Этосукцимид (Заронтин)
Перорально: капсулы по 250 мг;
сироп 250 мг/5 мл
Этотоин (Пеганон)
Перорально: таблетки по 250, 500 мг
Габапентин (Нейронтин)
Перорально: капсулы по 100, 300, 400 мг
Лоразепам (генерик, Ативан)
Перорально: таблетки по 0.5,1.2 мг
Парентерально: 2.4 мг/мл для инъекций
Мефенитоин (Мезантоин)
Перорально: таблетки по 100 мг
Мефобарбитал (Мебарал)
Перорально: таблетки по 32, 50, 100 мг
Метарбитал (Гемонил)
Перорально: таблетки по 100 мг
Метсукцимид (Гелонтин Капселз)
Перорально: капсулы по 150,300 мг
Параметадион (Парадион)
Перорально: таблетки по 150, 300 мг
Пентобарбитал натрия (генерик, Нембутал)
Парентерально: 50 мг/мл для инъекций
Фенацемид (Фенурон)
Перорально: таблетки по 500 мг
Фенобарбитал (генерик, Люминал натрия, др.)
Перорально: таблетки по 8, 16, 32, 65,100 мг; капсулы по 16 мг; эликсиры 20 мг/5 мл Парентерально: 30, 60, 65, 130 мг для инъекций
Фенсукцимид (Милонтин Капсэльз)
Перорально: капсулы по 500 мг
Фенитоин (генерик, Дилантин, др.)
Перорально: капсулы по 30,100 мг;
таблетки для жевания по 50 мг;
суспензии 30, 125 мг/5 мл
Перорально для пролонгированного действия: капсулы по 30, 100 мг
Парентерально: 50 мг/мл для в/в инъекций
Примидон (генерик, Мизолин)
Перорально: таблетки по 50, 250 мг;
суспензия 250 мг/5 мл
Триметадион (Тридион)
Перорально: таблетки для жевания по 150 мг; капсулы по 300 мг; раствор 40 мг/мл
Вальпроевая кислота (генерик, Депакен, Мипроевая кислота)
Перорально: капсулы по 250 мг; сироп (вальпроат натрия) 250 мг/5 мл
Перорально для пролонгированного действия (Депакот): таблетки (дивальпроат натрия) по 125,250, 500 мг
Избранная литература
Bialer М. Comparative pharmacokinetics of the newer antiepileptic drugs. Clin. Pharmacokinet. 1993; 24: 441.
Brodie M. J. Lamotrigine. Lancet, 1992; 339: 1397.
Dam M., Gram L. Comprehensive Epileptology. Raven Press, 1991.
Meldrum B. S. GABAergic mechanisms in the phato-genesis and treatment of epilepsy. Br. J. Pharmacol. 1989; 27:3S.
Porter R. J. Epilepsy: 100 Elementary Principles, 2nd ed., Saunders, 1989.
Porter R. J. New antiepileptic agents: Strategies for drug development. Lancet, 1990; 336:423.
Treiman D. M. Gamma vinyl GABA: Current role in the management of drug-resistant epilepsy. Epilepsia, 1989; 30 (Suppl. 1): S31.
Общие анестетики
24
Энтони Дж. Тревор, Рональд Д. Миллер
Еще в древние времена предпринимались попытки подавить боль при хирургических процедурах, например введением внутрь этанола и опиатов. В 1846 г. в Бостоне Уильям Мортон впервые продемонстрировал возможность использования диэтилового эфира для анестезии. В течение следующего года Джеймс Симпсон из Шотландии ввел в употребление хлороформ. Через 20 лет после этого были успешно продемонстрированы анестезирующие свойства закиси азота, впервые открытые Дэви еще в 1790-х гг. История современной анестезии началась в 1930-е гг. с внутривенного применения барбитурата тиопентала. Десять лет спустя для миорелаксации начали использовать кураре. Первый современный галогенсодержащий углеводород, галотан, был введен для ингаляционной анестезии в 1956 г. и скоро стал эталоном для новых ингаляционных анестетиков.
В понятие “общей анестезии” обычно включают аналгезию, амнезию, потерю сознания, подавление сенсорных и автономных рефлексов и во многих случаях — релаксацию скелетных мышц. Степень выраженности этих эффектов зависит от препарата, дозы и клинической ситуации.
Идеальный анестетик должен обеспечивать плавное введение в наркоз и достаточно быстрое выведение из него. Он также должен обладать широким терапевтическим диапазоном и не иметь побочных эффектов. Ни один отдельно применяемый анестетик не обладает всеми этими качествами. В современной анестезии чаще всего комбинируют различные препараты, пользуясь их преимуществами и минимизируя побочные эффекты.
Схема анестезии зависит от характера хирургического вмешательства. Для малых операций применяют обезболивание при сохранении сознания, создаваемое бензодиазепинами в комбинации с местными анестетиками. При более серьезных процедурах применяется сбалансированная анестезия с использованием короткодействующих барбитура
тов, закиси азота и внутривенных опиатов. Анестезия, применяемая в “большой” хирургии, включает введение препаратов до операции с целью седативного эффекта и аналгезии, индукцию анестезии тиопенталом или другим быстродействующим внутривенным препаратом и получение глубокой анестезии ингаляционными препаратами или их комбинацией с внутривенными препаратами. Во многих случаях эта схема включает и скелетные миорелаксанты. Для амбулаторной хирургии и для неглубокой анестезии у пациентов с искусственной вентиляцией легких в отделении интенсивной терапии часто используют бензодиазепины и пропофол.
Типы общих анестетиков
Общие анестетики обычно вводятся ингаляци-онно или внутривенно.
Ингаляционные препараты
Химическая структура ингаляционных препаратов показана на рис. 24-1. Закись азота, газ при обычных температуре и давлении, по-прежнему является важным компонентом многих схем анестезии. Анестетики галотан, энфлуран, изофлуран, дес-флуран и метоксифлуран — летучие жидкости. Новый ингаляционный препарат севофлуран пока не одобрен для применения в США. Препараты типа эфира, циклопропана и хлороформа уже не используются в развитых странах вследствие своей потенциальной горючести (эфир, циклопропан) и токсичности (хлороформ).
Внутривенные препараты
Ряд препаратов применяется внутривенно самостоятельно или в комбинациях с другими как компонент сбалансированной анестезии, а также для получения седативного действия у пациентов с дли-
/ \ N =N
Закись азота
I I F CI
Галотан
I I с-о-с-н
I I
F F
Энфлуран
Cl F Н
I I I
н-с-с-о-с-н
I I I
Cl F Н
Метоксифлуран
F Н Н
I I I
F-C-C-O-C-F
I I I
F F F
Десфлуран
F-C-i-0-C-H I I I
F Cl F
Изофлуран
Севофлуран
Рис. 24-1. Ингаляционные анестетики
тельной искусственной вентиляцией легких. К ним относятся: 1) барбитураты (тиопентал, метогекси-тал), 2) бензодиазепины (мидазолам, диазепам), 3) опиоидные аналгетики и нейролептики, 4) смешанная группа (пропофол, этомидат) и 5) кетамин — арилциклогексиламин, который вызывает так называемую диссоциированную аналгезию.
На рис. 24-2 показаны структуры часто применяемых внутривенных анестетиков.
Признаки и стадии анестезии
Со времени введения в практику общих анестетиков были сделаны попытки найти корреляцию между их найти глубиной анестезии. Традиционное описание признаков и стадий анестезии (признаки Гуэделя) в основном составлены из наблюдений эффектов диэтилового эфира, центральное действие которого начинается медленно вследствие его высокой растворимости в крови. Промежуточные стадии и признаки редко наблюдаются при применении современных быстродействующих ингаляционных и внутривенных анестетиков. Более того, в настоящее время при большинстве
Тиопентал
Этомидат
.СН(СН3)2
у- ОН
'СН(СН3)2 Пропофол
Рис. 24-2. Внутривенные анестетики
операций комбинируют ингаляционные и внутривенные средства. Тем не менее признаки действия диэтилового эфира полезны для оценки эффекта других анестетиков. Многие из этих признаков связаны с действием на дыхание, рефлекторную активность и тонус мышц. Традиционно выделяют четыре стадии анестезии по мере угнетений ЦНС.
I. Стадия аналгезии. Сначала возникает аналгезия. Позднее к ней присоединяется и амнезия,.
II. Стадия возбуждения. Во время этой стадаИ пациент часто впадает в делириозное возбужденное состояние, при этом явно присутствует амнезия. Дыхание нерегулярное по объему и частоте, возможна рвота, иногда наблюдаются несдержанность и физическое сопротивление. В связи с этим пытаются ограничить длительность и тяжесть этой стадии, что приводит к нормализации дыхания.
III. Стадия хирургической анестезии. Она начинается с восстановления нормального дыхания и длится до полного прекращения спонтанного дыхания. Описано четыре степени этой стадии в зависимости от движений глаз, размеров и реакции зрачка, которые при определенных условиях могут являться признаками увеличения глубины анестезии.
IV. Стадия угнетения продолговатого мозга. Начинается с момента прекращения спонтанного дыхания. Эта стадия характеризуется значительным подавлением дыхательного и сердечно-сосудистого центров продолговатого мозга. Без полной дыхательной и циркуляторной поддержки быстро наступает смерть.
При современной анестезии стадии трудно разграничить. Это связано с быстрым началом действия многих ингаляционных анестетиков по сравнению с эфиром, а также с преимущественным использованием аппаратного дыхания. Кроме того, действие других препаратов, введенных до или во время операции, может влиять на признаки анестезии. Атропин, используемый для уменьшения секреции, расширяет зрачки; препараты типа тубоку-рарина и сукцинилхолина снижают тонус мышц; опиоидные аналгетики подавляют дыхание. Самый надежный признак наступления стадии III (хирургической анестезии) — потеря корнеального рефлекса и равномерное по частоте и глубине дыхание. Адекватность глубины анестезии для конкретных процедур определяют по реакции сердечно-сосудистой и дыхательной систем на стимуляцию.
Ингаляционные анестетики
Фармакокинетика
Глубина анестезии определяется концентрацией анестетика в ЦНС. Скорость достижения эффективной мозговой концентрации (скорость индукции анестезии) зависит от многих фармакокинетических факторов, влияющих на захват и распределение анестетика. Эти факторы определяют разные скорости поступления анестетиков из легких в кровь и из крови в мозг и другие ткани, а также влияют на скорость восстановления после анестезии.
Захват и распределение
Концентрация индивидуального газа в смеси пропорциональна парциальному давлению, или
напряжению. Эти термины взаимозаменяемы и используются при обсуждении процессов переноса газов в организме. Для достижения требуемой мозговой концентрации анестетика необходим его перенос из альвеолярного газа в кровь и затем в мозг. Скорость этого процесса зависит от растворимости анестетика, его концентрации во вдыхаемом воздухе, показателей вентиляции и от концентрационного градиента анестетика (парциального давления) между артериальной и смешанной венозной кровью.
А. Растворимость. Это один из наиболее важных факторов, влияющих на перенос анестетика из воздуха в артериальную кровь. Коэффициент распределения кровь : газ — важный показатель растворимости, который определяет относительную аффинность анестетика к крови по сравнению с дыхательной смесью. Коэффициент может быть низким, например 0.5 для таких плохорастворимых в крови анестетиков, как закись азота или циклопропан. С другой стороны, у препаратов типа метоксифлурана коэффициент распределения достигает 10. Они очень хорошо растворимы в крови (табл. 24-1). Когда плохо растворимый в крови анестетик диффундирует из легких в кровь, требуется всего несколько молекул и, соответственно, немного времени для повышения его парциального давления (рис. 24-3, вверху). Наоборот, в случае хорошо или средне растворимых анестетиков для значительного повышения парциального давления нужно больше молекул и, следовательно, больше времени (рис. 24-3, внизу). Это обратное соотношение между растворимостью анестетика в крови и скоростью повышения его напряжения в артериальной крови иллюстрируется на рис. 24-4.
Коэффициенты распределения кровь : газ у закиси азота, галотана и метоксифлурана соответственно равны 0.47, 2.3 и 12. Закись азота плохо растворима в крови, быстро достигает высокого напряжения в артериальной крови, что в свою очередь приводит к более быстрому установлению равновесных концентраций в мозгу и крови и к ускорению индукции анестезии.
Метоксифлурановая анестезия развивается еще медленнее: даже через 40 минут после начала ингаляции уровень препарата достигает только 20 % от равновесной концентрации.
Б. Концентрация анестетика во вдыхаемом воздухе. Этот показатель имеет прямое влияние как на максимальное напряжение газа в альвеолах,
ТАБЛИЦА 24-1. Свойства ингаляционных анестетиков
Анестетик Коэффициент Коэффициент Минимальная
распределения кровь: газ1 распределения мозг: кровь1 альвеолярная концентрация2 (%)
Закись азота 0.47 1.1 >100
Десфлуран 0.42 1.3 6-7
Севофлуран 0.69 1.7 2.0
Изофлуран 1.40 2.6 1.40
Энфлуран 1.80 1.4 1.68
Галотан 2.30 2.9 0.75
Метоксифлуран 12.00 2.0 0.16
1 Коэффициенты распределения (при 37 °C) по данным литературы.
2 Минимальная альвеолярная концентрация (МАК) — это концентрация анестетика, при которой у 50 % пациентов нет двигательной реакции при воздействии повреждающего стимула.
так и на скорость повышения напряжения анестетика в артериальной крови. Повышение концентрации вдыхаемого препарата ускоряет анестезию за счет увеличения скорости переноса газа в кровь в соответствии с законом Фика (глава 1). Этот эффект используется в практике применения ингаляционных анестетиков средней растворимости в крови типа энфлурана, изофлурана и галотана, которые дают сравнительно медленное начало анестезии. Например, для ускорения наркоза использует
ся начальная концентрация галотана 3-4 %, затем ее снижают до 1-2 % для поддержания адекватной анестезии.
В. Показатели вентиляции. Скорость повышения напряжения газообразного анестетика в крови напрямую связана со скоростью и глубиной вентиляции и зависит от коэффициента распределения кровь : газ. При повышении легочной вентиляции увеличение артериального напряжения плохо растворимого (обладающего низким коэффициентом)
Рис. 24-3. Зависимость скорости анестезии от растворимости газообразного анестетика. На диаграмме растворимость в крови обозначена как относительный размер, соответствующего компартмента (чем больше растворимость, тем больше компартмент). Относительное парциальное давление препаратов в компартментах обозначено степенью их наполнения. Для достижения той же концентрации или парциального давления, что и в альвеолах, более растворимому анестетику (галотан) потребуется больше времени, чем менее растворимому (при равных концентрациях во вдыхаемом воздухе). Так как концентрация в мозгу повышается с той же скоростью, что и в крови, начало анестезии будет более медленное при применении галотана, чем при использовании закиси азота
Рис. 24-4. Напряжение трех газообразных анестетиков в артериальной крови как функция времени после начала ингаляции. Закись азота относительно нерастворима (коэффициент распределения кровь: газ равен 0.47); мето-ксифлуран гораздо более растворим (коэффициент =12)
анестетика существенно менее выражено, чем для хорошо или средне растворимых препаратов (рис. 24-5). Например, увеличение вентиляции в 4 раза в первые 10 минут анестезии повышает напряжение галотана в артериальной крови почти в 2 раза, а закиси азота — всего на 15 %. Механическая гипервентиляция повышает скорость индукции анестезии при использовании медленных ингаляционных анестетиков. Угнетение дыхания, вызванное другими фармакологическими препаратами, в том числе опиоидными аналгетиками, замедляет анестезию некоторыми ингаляционными анестетиками, что особенно проявляется при отсутствии искусственной вентиляции.
Г. Легочный кровоток. Изменения легочного кровотока влияют на процессы переноса газообразных анестетиков. Повышение кровотока при увеличении сердечного выброса замедляет нарастание напряжения газообразных анестетиков (особенно хорошо и средне растворимых) в артериальной крови. Это связано с тем, что повышенный кровоток подает большее количество крови для контакта с анестетиком, емкость крови увеличивается, и напряжение растет медленно. Снижение легочного кровотока дает обратный эффект, ускоряя повышение напряжения газообразных анестетиков в артериальной
Рис. 24-5. Вентиляция и напряжение газообразного анестетика в артериальной крови. Повышение вентиляции (с 2 до 8 л/мин) оказывает более значительный эффект на уравновешивание концентраций галотана, чем закиси азота
крови. У пациента с циркуляторным шоком сочетание сниженного сердечного выброса (и легочного кровотока) и повышенной вентиляции может ускорять индукцию анестезии некоторыми анестетиками. Менее вероятен такой эффект при использовании закиси азота из-за ее низкой растворимости.
Д. Артериовенозный градиент концентрации. Градиент концентрации анестетика между артериальной и смешанной венозной кровью в основном зависит от его захвата тканями. В зависимости от скорости и объема захвата анестетика тканями в венозной крови, которая возвращается к сердцу, может содержаться значительно меньшее количество анестетика, чем в артериальной. Чем больше разница между этими уровнями, тем большее время требуется для достижения равновесия. Поступление анестетика в ткани регулируется факторами, сходными с теми, которые регулируют поступление анестетика из легких в кровь, а именно коэффициентом распределения кровь: газ, скоростью кровотока в тканях и градиентами концентрации.
Во время индукции анестезии лучше перфузируемые ткани больше влияют на артериовенозный градиент концентрации анестетика. К ним относятся мозг, сердце, печень, почки и другие внутренние
органы, которые в состоянии покоя в сумме потребляют 75 % сердечного выброса. Если анестетик обладает высокой растворимостью в этих тканях, то его концентрация в венозной крови будет исходно очень низкой, и равновесие с артериальной кровью будет достигаться крайне медленно. В стадии поддержания анестезии может продолжаться перераспределение ингаляционных препаратов между тканями со скоростью, которая зависит от их растворимости и от кровотока. Мышцы и кожа, составляющие 50 % от массы тела, снабжаются кровью в среднем в 5 раз хуже, чем другие ткани, и поэтому медленнее кумулируют анестетики. Хотя большинство газообразных препаратов хорошо растворяется в жировой ткани, ее низкая перфузия замедляет кумуляцию, и за время хирургической анестезии не достигаются равновесные концентрации галотана или, например, энфлурана.
Выведение
Время восстановления после ингаляционного наркоза зависит от скорости выведения анестетика из мозга после снижения его концентрации во вдыхаемом воздухе. Многие процессы, которые происходят во время выведения из анестезии, подобны процессам ее индукции. Факторы, контролирующие скорость восстановления после анестезии (выхода из анестезии), включают легочный кровоток и параметры вентиляции, а также растворимость анестетика в тканях, крови и в газообразной форме в легких. Однако существуют два отличия между выходом из анестезии и ее индукцией. Во-первых, перенос анестетика из легких в кровь можно увеличить повышением его концентрации во вдыхаемом воздухе, а обратный процесс ускорить нельзя, так как концентрацию в легких нельзя снизить ниже нуля. Во-вторых, в начале выхода из анестезии напряжение анестетика в разных тканях различно в зависимости от особенностей препарата и длительности анестезии. При введении в анестезию исходное напряжение анестетика во всех тканях равно нулю.
Ингаляционные анестетики, которые сравнительно нерастворимы в крови и мозгу, выводятся быстрее, чем более растворимые. Например, “вымывание” закиси азота и десфлурана происходит быстро, и это обусловливает быстрый выход из анестезии. Галотан приблизительно в пять раз более растворим в крови и в два раза более растворим в моз
гу, чем закись азота, поэтому его выведение идет медленнее, что обусловливает медленный выход из анестезии. На скорость выхода из наркоза может влиять также длительность введения анестетика, особенно при использовании хорошо растворимых анестетиков типа метоксифлурана. Кумуляция препарата в тканях, в том числе в мышцах, коже и жировой ткани, усиливается при длительной ингаляции анестетика. При выходе из анестезии препарат медленно покидает ткани, и его напряжение в артериальной крови также снижается медленно. Таким образом, если анестезия была кратковременной, выход из анестезии будет достаточно быстрым. Выход из длительной анестезии может быть долгим даже при применении не очень хорошо растворимых анестетиков типа галотана.
Ингаляционные анестетики в основном выводятся легкими через выдыхаемый воздух. Однако в этом процессе участвуют также ферменты печени и других тканей. Например, “вымывание” галотана при выходе из анестезии идет быстрее, чем “вымывание” энфлурана, чего нельзя предположить по их растворимости. Это происходит, потому что за время средней по продолжительности операционной анестезии метаболизируются 15-20 % галотана и только 2-3 % энфлурана. Окисление галотана приводит к образованию трифторуксусной кислоты и высвобождению ионов брома и хлора. При низком напряжении кислорода галотан метаболизируется до свободного радикала хлортрифторэтила, который способен реагировать с компонентами мембраны гепатоцитов. Из всех фтористых анестетиков изофлуран и десфлуран метаболизируются наиболее медленно, в моче определяются только следы трифторуксусной кислоты. Медленный метаболизм энфлурана приводит к образованию дифторметоксифторуксусной кислоты и иона фтора, не достигающих токсических уровней. Метоксифлуран метаболизируется печенью быстрее, чем любой другой ингаляционный анестетик, и высвобождает ион фтора в нефротоксических количествах. Закись азота скорее всего не метаболизируется в тканях человека.
Фармакодинамика
Механизм действия
Важный нейрофизиологический эффект всех общих анестетиков — повышение порога импуль-сации клеток (Aloia, 1991). С повышением порога
снижается нейрональная активность. Ингаляционные анестетики (так же, как и внутривенные: барбитураты и бензодиазепины) подавляют спонтанную и стимулируемую активность нейронов многих участков мозга. Они действуют на аксональную и особенно на синаптическую передачу. Ионные механизмы повышения порога импульсации различны. Ингаляционные анестетики вызывают гиперполяризацию нейронов через активацию токов К+, которая приводит к снижению способности инициировать потенциалы действия, то есть к повышению порога возбудимости. Электрофизиологические исследования культуры клеток с использованием пэтч-кламп анализа показали, что изофлуран уменьшает длительность периода открытия катионных каналов, активируемых через никотиновый рецептор, что, предположительно, снижает синаптическую трансмиссию в холинергических синапсах. Бензодиазепины и барбитураты, влияя на открытие ГАМКл-рецептор-регулируемых хлорных каналов, вызывают гиперполяризацию мембраны и снижение частоты импульсации (глава 21). Считается также, что пропофол и некоторые ингаляционные препараты обладают способностью усиливать ингибиторные эффекты ГАМК.
Молекулярные механизмы изменения ионных мембранных токов ингаляционными анестетиками не совсем понятны. Это может быть прямое взаимодействие между молекулами анестетика и гидрофобными участками специфических мембранных белковых каналов. Такой механизм был предложен на основании исследования взаимодействия газообразных анестетиков с никотиновыми холинорецепторными каналами. Взаимодействие вызывает стабилизацию каналов в их закрытом состоянии. Другие теории, которые учитывают значительные структурные отличия газообразных анестетиков, предполагают менее специфические взаимодействия этих препаратов с липидным матриксом мембраны, что приводит к вторичным изменениям функций каналов. Такие теории поддерживаются давними наблюдениями тесной корреляции эффективности (“силы”) анестетика и его растворимости в липидах (принцип Мейера-Овертона). Концепция непрямых взаимодействий через мембранные липиды подтверждена исследованиями поведения анестетиков на искусственных липидных мембранах. Эти исследования показали, что при растворении газа в липидах повышается текучесть мембраны, что предполагает снижение упорядоченнос
ти структуры ее липидов. Такие эффекты могут приводить к изменениям функции мембранных каналов.
В эксперименте общая анестезия обратима при помещении животного в камеру высокого давления. Очень высокое давление (50-100 атм) может вывести его из состояния анестезии. Известно, что высокое давление повышает упорядоченность липидных мембран. Такие эксперименты позволили предположить, что растворение молекул анестетика в липидах мембраны с последующим нарушением упорядоченности вызывает изменения ионных каналов. Однако гипотезы, основанные на изменении вязкости мембран, не могут объяснить, почему небольшие повышения температуры, увеличивающие текучесть мембран в той же степени, что и клинические концентрации анестетиков, на самом деле снижают “силу” действия последних. Кроме того, эти гипотезы не соответствуют клиническим наблюдениям за пожилыми пациентами, у которых обычно снижена потребность в анестетике, несмотря на то, что вязкость мембран повышается с возрастом.
Нейрофармакологические основы анестезии предполагают различную чувствительность разных нейронов и проводящих путей к анестетику. Клетки желатинозной субстанции в дорсальных рогах спинного мозга очень чувствительны к самым низким концентрациям анестетика в ЦНС. Снижение активности этих нейронов прерывает сенсорную трансмиссию по спиноталамическому тракту, в том числе и передачу болевых ощущений (ноцицептивную). Это стадия I анестезии — аналгезия. Растормаживающий эффект анестетиков (стадия II) возникает при более высоких концентрациях анестетика вследствие комплексных нейрональных процессов, в частности подавления множества мелких ингибиторных нейронов, например клеток Гольджи второго типа, а также вследствие парадоксального облегчения процессов, опосредованных возбуждающими медиаторами. Прогрессивное торможение восходящих путей в ретикулярной активирующей системе происходит во время стадии III (хирургической анестезии) совместно с подавлением спинальной рефлекторной активности, которое приводит к расслаблению мышц. Нейроны респираторного и вазомоторного центров продолговатого мозга сравнительно устойчивы к действию общих анестетиков, однако высокие концентрации анестетиков могут вызвать их подавление, что приводит к кардиореспираторному коллапсу (стадия IV).
Характеристики зависимости доза-эффект: минимальная альвеолярная концентрация анестетика
Ингаляционные анестетики поступают в легкие в виде газовой смеси, скорость введения и состав которой легко контролировать. Однако характеристики зависимости доза-эффект газообразных препаратов измерять очень трудно. Во-первых, достижение состояния наркоза зависит от концентрации анестетика в мозгу, а ее в клинике измерить невозможно. Во-вторых, не существует этически правильных методов определения нижних и верхних пределов градуальной кривой зависимости доза-эффект, так как при очень низких концентрациях анестетика больной может испытывать сильную боль, а при высоких возникает значительный риск фатальной сердечно-сосудистой и респираторной депрессии. Несмотря на это, можно определить эффективность анестетика, используя квантовые принципы зависимости доза-оффект.
Во время общей анестезии парциальное давление ингаляционного анестетика в мозгу равно таковому в легких при достижении стабильного состояния. При определенном уровне анестезии в стабильном состоянии измерение альвеолярных концентраций разных препаратов позволит провести сравнение их активности (“силы”). Минимальная альвеолярная концентрация анестетика (МАК) — это концентрация (процент в альвеолярной газовой смеси, или парциальное давление анестетика в процентах от 760 мм рт. ст.), при которой у 50 % пациентов нет реакции на воздействие повреждающего фактора (операционный разрез и др.) Таким образом, МАК представляет собой точку (ED5o) на традиционной квантовой кривой доза-эффект (рис. 2-17). В табл. 24-1 показаны МАК для часто используемых ингаляционных анестетиков. МАК закиси азота более 100 % означает, что она самая слабая в этой группе, так как даже при нормальном барометрическом давлении 760 мм рт. ст. 100 % концентрация закиси азота во вдыхаемой смеси не равна 1 МАК. Доза в 1 МАК предотвращает движения в ответ на операционный разрез у 50 % больных, но разным пациентам может быть необходимо от 0.5 до 1.5 МАК. (Заметим, что МАК не дает информации о характере наклона кривой доза-эффект.) В целом, однако, кривая доза-эффект для ингаляционных анестетиков имеет крутой характер: более 95 % пациентов при
1.1 МАК уже не реагируют на повреждающие стимулы. Измерение МАК в контролируемых условиях позволило количественно оценить действие некоторых факторов на анестезию. Например, МАК снижается у пожилых пациентов, но не зависит от пола, роста и веса. Особое значение имеют эффекты других совместно вводимых препаратов, которые могут значительно изменить потребность в анестезии. Так, при комбинировании с опиоидными аналгетиками или седативными и снотворными средствами МАК снижается, следовательно, должна быть снижена и концентрация вдыхаемого анестетика.
С целью определения зависимости доза-эффект для ингаляционных анестетиков у человека был испробован ряд методов, например измерение ЭЭГ. Однако в связи с невозможностью вычленить влияние других параметров, не связанных с анестезией, эти попытки пока не привели к успеху.
Действие ингаляционных анестетиков на органы и системы
А. Действие на сердечно-сосудистую систему. Галотан, десфлуран, энфлуран и изофлуран снижают среднее артериальное давление прямо пропорционально их альвеолярной концентрации. При применении галотана и энфлурана снижение давления вызвано уменьшением сердечного выброса, так как общее сосудистое сопротивление изменяется незначительно, несмотря на существенные изменения в отдельных сосудистых бассейнах (например, увеличение мозгового кровотока). В отличие от них изофлуран и десфлуран понижают артериальное давление путем значительного снижения общего сосудистого сопротивления; они почти не действуют на сердечный выброс.
Ингаляционные анестетики изменяют частоту сердечных сокращений либо посредством прямого воздействия на деполяризацию синусового узла, либо через изменение активности вегетативной нервной системы. При применении галотана часто наблюдается брадикардия в связи с прямым подавлением частоты сокращений предсердий. Действие метоксифлурана и энфлурана менее выражено, а изофлуран вызывает тахикардию. Большинство этих изменений зарегистрировано у здоровых добровольцев, а не у хирургических больных. Возбужденное состояние пациента перед операцией и стимулирующий эффект операции часто модифициру-
jot реакцию со стороны сердечного ритма на ингаляционные анестетики.
Все эти препараты повышают давление в правом предсердии по дозозависимому механизму, который отражает подавление функциональной активности миокарда. Галотан и энфлуран (в меньшей степени изофлуран) вызывают значительное угнетение миокардиальной функции. Ингаляционные анестетики снижают потребление кислорода миокардом, в основном изменяя параметры, определяющие его потребность в кислороде, а именно артериальное давление и силу сокращений миокарда. Хотя и менее выраженный, чем у других, этот эффект присутствует также у закиси азота. Однако закись азота (одна или в комбинациях с другими ингаляционными анестетиками) вызывает симпатическую стимуляцию, которая может маскировать кардиодепрессивное действие ингаляционных анестетиков. Сочетание закиси азота и галотана или энфлурана, например, вызывает меньше проявлений миокардиальной депрессии, чем некоторые более сильные анестетики, применяемые поодиночке.
Влияние ингаляционных анестетиков на сердечно-сосудистую систему модифицирует ряд факторов. Стимулирующее действие операции, гиперкапния и повышенная длительность анестезии уменьшают подавляющий эффект этих препаратов. Гиперкапния вызывает высвобождение катехоламинов, которые противодействуют понижению артериального давления. Степень снижения артериального давления после 5 часов анестезии меньше, чем после 1 часа; пропранолол блокирует этот адаптивный эффект. Галотан сенсибилизирует миокард к действию катехоламинов и, возможно, к возникновению желудочковых аритмий у пациентов с заболеваниями сердца, которым назначают симпато-миметик прямого или непрямого действия или у которых высоки уровни циркулирующих эндогенных катехоламинов (например, при феохромоцитоме). Другие современные ингаляционные анестетики реже дают аритмогенный эффект.
Б. Действие на дыхательную систему. Все применяемые в настоящее время ингаляционные анестетики, кроме закиси азота, вызывают снижение дыхательного объема и повышение частоты дыхания. Однако учащение дыхания не компенсирует снижение объема, что приводит к уменьшению вентиляции. Все эти препараты являются респираторными депрессантами, о чем судят по снижению реакции на возрастание уровня углекислого газа. Сте
пень подавления вентиляции отличается у разных препаратов, она наиболее выражена у изофлурана и энфлурана. Ингаляционная анестезия повышает уровень Рсо2 в покое (парциальное давление двуокиси углерода в артериальной крови), поднимает порог апноэ (уровень Рсо2, ниже которого возникает апноэ из-за утраты возможности стимуляции дыхания углекислым газом) и снижает вентиляторный ответ на гипоксию. Последний эффект особенно важен, так как субанестетические концентрации (регистрируемые при выходе из наркоза) подавляют нормальное компенсаторное увеличение вентиляции, которое возникает при гипоксии. Все проявления респираторной депрессии анестетиков можно преодолеть, используя во время операции вспомогательную вентиляцию с помощью аппарата искусственной вентиляции легких. Более того, этот эффект анестетиков смягчается самой процедурой оперативного вмешательства, а также увеличением длительности анестезии.
Ингаляционные анестетики подавляют и мукоцилиарный транспорт в дыхательных путях. Следовательно, длительная анестезия может привести к накапливанию слизи и впоследствии — к ателектазам и инфекциям дыхательных путей. Однако эти препараты обладают и бронхорасширяющим эффектом, который используют при лечении астматического статуса. Их применение редко осложняется раздражением дыхательных путей, вызывающим кашель или задержку дыхания. Исключение составляет десфлуран, поэтому при его применении могут возникать проблемы индукции наркоза, несмотря на низкий коэффициент распределения кровь : газ. Раздражающие свойства энфлурана также могут вызвать задержку дыхания, которая замедляет скорость индукции.
В. Действие на мозг. Ингаляционные анестетики снижают скорость метаболизма в мозгу. Несмотря на это, большинство из них повышает мозговой кровоток за счет понижения периферического сопротивления сосудов мозга. Усиление мозгового кровотока часто клинически невыгодно. Например, у пациентов с повышенным внутричерепным давлением вследствие опухоли мозга или травмы ингаляционная анестезия повысит мозговой кровоток и, следовательно, объем крови, что в свою очередь еще больше увеличит внутричерепное давление.
Из всех ингаляционных анестетиков закись азота меньше других увеличивает мозговой кровоток, хотя при добавлении 60 % закиси азота к галотано
вой анестезии мозговой кровоток увеличивается больше, чем при использовании только галотана. В низких дозах все галогенизированные препараты вызывают примерно одинаковое повышение мозгового кровотока. В более высоких — энфлуран и изофлуран обладают менее выраженным эффектом, чем галотан. При гипервентиляции пациента перед анестезией (для снижения Рсо2) повышение внутричерепного давления сводится до минимума.
В дозах до 1—1.5 МАК галотан, изофлуран и энфлуран вызывают сходные эффекты (например, подавление импульсации), обнаруживающиеся на ЭЭГ. При увеличении дозы энфлурана может появиться характерная ЭЭГ-картина зубцов и волн, при которой слуховая стимуляция может вызвать генерализованное подергивание мышц, усиливающееся под действием гипервентиляции. Неблагоприятных клинических последствий такая судорожная активность не имеет. Этот эффект отсутствует у других ингаляционных анестетиков.
Закись азота — слабый анестетик, но она обладает значительной аналгетической и амнезической активностью, что клинически очень важно при использовании этого препарата в комбинациях с другими ингаляционными анестетиками.
Г. Действие на почки. Ингаляционные анестетики в разной степени снижают скорость гломерулярной фильтрации, эффективный почечный кровоток и увеличивают фильтрационную фракцию. Все они повышают периферическое сопротивление сосудов почек. Так как почечный кровоток понижается во время общей анестезии, несмотря на хорошее или даже повышенное перфузионное давление, то скорее всего это связано с нарушением ауторегуляции.
Д. Действие на печень. Все ингаляционные анестетики снижают печеночный кровоток до 15-45 % от уровня до анестезии. Несмотря на временные изменения значений функциональных тестов печени в ходе операции, эти препараты редко повреждают печень. Возможная гепатотоксичность галотана обсуждается отдельно.
Е. Действие на гладкую мускулатуру матки. Закись азота практически не действует на гладкую мускулатуру матки. Однако изофлуран, галотан и энфлуран вызывают значительное ее расслабление. Этот фармакологический эффект можно использовать при необходимости расслабления мускулатуры матки для совершения внутриматочных манипуляций и воздействия на плод во время родов.
Наоборот, при выскабливании матки с целью терапевтического аборта эти препараты могут провоцировать маточное кровотечение.
Токсичность
А. Гепатотоксичность (галотан). Послеоперационный гепатит обычно связан с такими факторами как переливание крови, гиповолемический шок и с другими хирургическими стрессами, а не с токсичностью ингаляционных анестетиков. Однако галоидсодержащие препараты типа галотана мотут вызывать повреждение печени. Еще в первом десятилетии этого века была выявлена гепатотоксичность хлороформа. Галотан начали применять в клинической практике в 1956 г.; к 1963 г. было зарегистрировано несколько случаев послеоперационной желтухи и некроза печени, связанных с использованием галотана. Однако несколько ретроспективных исследований, в которых галотан сравнивали с другими анестетиками, не показали учащения поражений печени при его использовании. В 1966 г. в рамках программы “Национальное исследование галотана в Соединенных Штатах” провели ретроспективный анализ возникновения смертельного массивного некроза печени приблизительно у 850 000 хирургических пациентов. Результаты не привели к каким-либо заключениям и не доказали, что галотан является гепатотоксином. Частота возникновения массивного некроза при применении галотана составляла 7 на 250 000 случаев галотановой анестезии, или приблизительно 1 на 35 000 (не 1 на 10 000, как иногда пишут). Так как галотан является хорошим анестетиком, важно точно определить, является ли он гепатотоксином и ограничивать ли его применение. В отличие от хлороформа и флуроксена, которые могут вызывать жировую инфильтрацию, центрилобулярные некрозы печени и повышение уровней трансаминаз, длительное применение галотана у животных вызывает минимальные признаки повреждения печени. Для выявления возможной гепатотоксичности галотана было использовано несколько экспериментальных моделей. Оказалось, что у крыс, которым сначала давали фенобарбитал, а потом ввели в состояние гипоксии путем снижения кислорода во вдыхаемом воздухе до 7-14 %, галотан дает гепа-тотоксический эффект (так же, как и другие анестетики). Механизм такой гепатотоксичности у животных неясен, но есть предположения, что она мо
жет зависеть от продукции реактивных метаболитов (например, свободных радикалов), которые вызывают либо прямое, либо опосредованное иммунной реакцией повреждение гепатоцитов. Хотя эти результаты воспроизводимы и модель хорошо известна, ее клиническое значение непонятно, так как для моделирования повреждения печени необходима гипоксия.
Недавно было выявлено, что у некоторых людей есть дефект мембраны гепатоцитов, что делает их более чувствительными к повреждению галотаном. Эти люди составляют группу риска возникновения некрозов печени под действием галотана. Сейчас разрабатывается тест для дооперационного определения таких дефектов.
Б. Нефротоксичность (метоксифлуран). В 1966 г. была впервые описана вазопрессин-резис-тентная полиурическая почечная недостаточность у 13 из 41 пациента после метоксифлуранового наркоза при абдоминальных операциях. Впоследствии было показано, что причина почечной недостаточности — неорганический фтор, конечный продукт биотрансформации метоксифлурана.
В. Злокачественная гипертермия. Хотя такие случаи редки, но у генетически предрасположенных пациентов может развиться этот потенциально угрожающий жизни синдром, который включает в себя тахикардию и гипертензию, прогрессирующие до возникновения ацидоза, гиперкалиемии, мышечной ригидности и гипертермии. Начало этого синдрома может быть более острым при применении сукцинилхолина для миорелаксации. Лечение заключается во внутривенном введении дантролена и мероприятиях по снижению температуры тела и восстановлению электролитного и кислотно-основного балансов.
Г. Хроническая токсичность.
1. Мутагенность. В нормальных условиях большинство современных и многие более ранние ингаляционные анестетики не являются мутагенами и, скорее всего, канцерогенами. Однако препараты более старых поколений, содержащие винильную группу (флуроксен и дивиниловый эфир), возможно, мутагенны. Сейчас они уже почти не используются.
2. Канцерогенность. В нескольких эпидемиологических исследованиях обнаружили увеличение частоты возникновения злокачественных опухолей у работников операционных, которые имеют контакт с ингаляционными анестетиками. Однако ни
одно исследование не продемонстрировало существование причинно-следственных связей между анестетиками и раком. Такое увеличение частоты могут вызывать многие другие факторы. Тем более, что в большинстве операционных вентиляторы выводят остатки ингаляционных анестетиков в атмосферу.
3. Действие на репродуктивную функцию. Результаты наиболее регулярных исследований, проводимых для определения репродуктивного здоровья женщин, работающих в операционных, показывают, что у них повышена частота невынашиваний плода. В трактовке этих результатов есть несколько проблем, но в целом можно сказать, что полученные данные не очень убедительны.
Связь же акушерских проблем с операциями и анестезией у беременных не подвергается сомнениям. В США каждый год по меньшей мере 50 000 беременных женщин получают анестезию при операциях по показаниям, не связанным с беременностью. Риск аборта очевидно выше после таких вмешательств. Непонятно, что является причиной невынашивания: сопутствующее заболевание, операция, анестезия или сочетание этих факторов. Другой повод для беспокойства, не нашедший пока подтверждения,— это возможность повышения частоты врожденных аномалий после введения анестетика. Если анестетики и обладают тератогенным действием, то риск, видимо, очень мал.
4. Действие на кровь. Длительное воздействие закиси азота снижает активность метионинсинте-тазы и вызывает мегалобластическую анемию. Это потенциальная профессиональная вредность у работников плохо вентилируемых стоматологических операционных.
Клиническое применение ингаляционных анестетиков
Из всех используемых ингаляционных анестетиков наиболее часто применяются в США закись азота, десфлуран и изофлуран. Галотан нашел широкое применение в педиатрической анестезии. Как было сказано ранее, закись азота — недостаточно сильный препарат, чтобы обеспечить хирургическую анестезию. Поэтому для создания полноценной анестезии ее обычно комбинируют с другими ингаляционными или внутривенными анестетиками. Иногда применяют метоксифлуран (особенно в акушерстве), но не для длительных процедур вслед
ствие его нефротоксичности. До 1960 г. чаще других использовали циклопропан и диэтиловый эфир, сейчас их не применяют из-за горючести и взрывоопасности.
Внутривенные анестетики
Барбитураты ультракороткого действия
Хотя существует несколько барбитуратов ультракороткого действия, для индукции анестезии преимущественно используют тиопентал, часто в сочетании с ингаляционными анестетиками. Общая фармакология барбитуратов обсуждается в главе 21.
После внутривенной инъекции тиопентал быстро проходит через гематоэнцефалический барьер и при введении достаточных доз вызывает сон через период времени, который ему требуется для совершения одного круга по организму. Сходными эффектами обладают и другие барбитураты ультракороткого действия, например тиамилал и ме-тогекситал. Для всех них равновесие концентраций в плазме и в мозгу достигается очень быстро (приблизительно за 1 минуту) в связи с их высокой растворимостью в липидах. Тиопентал быстро диффундирует из мозга и из других хорошо снабжаемых кровью областей и перераспределяется в мышцы, жировую ткань и, в конце концов,— во все ткани (рис. 24-6). Именно из-за такого быстрого выведения препарата из мозга единичная доза тиопентала действует так коротко.
Метаболизм тиопентала идет значительно медленнее, чем его перераспределение, и происходит в основном в печени. После однократной инъекции он метаболизируется со скоростью 12-16 % в час. Менее 1 % вводимой дозы тиопентала выводится в неизмененном виде через почки.
При введении высоких доз тиопентал вызывает дозозависимое снижение артериального давления, ударного объема и сердечного выброса. Это прежде всего связано с угнетением сократимости миокарда и с повышением емкости венозного русла. Действие на общее периферическое сосудистое сопротивление минимально.
Тиопентал, как и другие барбитураты, вызывает респираторную депрессию, снижая чувствительность дыхательного центра продолговатого мозга к двуокиси углерода.
После введения тиопентала происходит снижение уровня церебрального метаболизма и утилизации кислорода пропорционально степени центрального торможения. Мозговой кровоток также снижается, но в значительно меньшей степени, чем потребление кислорода. Это делает тиопентал более адекватным препаратом, чем ингаляционные анестетики, для применения у больных с отеком мозга, так как он не повышает внутричерепное давление и объем мозгового кровотока.
Тиопентал может уменьшать печеночный кровоток и гломерулярную фильтрацию, но не вызывает поражения печени и почек. Барбитураты могут отягощать течение острой интермиттирующей порфирии (глава 21).
Бензодиазепины
В анестезии применяют некоторые препараты группы седативных и снотворных средств, в том числе диазепам, лоразепам и мидазолам (базисная фармакология бензодиазепинов обсуждается в главе 21). Диазепам и лоразепам не растворимы в воде, для их внутривенного введения необходимы растворители, которые могут вызывать местное раздражение. Мидазолам растворим в воде и поэтому вызывает меньшее раздражение, но он становится жирорастворимым при физиологических значениях pH и быстро проникает через гематоэнцефали-
Рис. 24-6. Перераспределение тиопентала после внутривенного болюсного введения. Заметьте, что ось времени нелинейна
ческий барьер. По сравнению с внутривенными барбитуратами бензодиазепины характеризуются более медленным началом действия на ЦНС и вызывают плато центрального торможения, которое не достигает глубины настоящей анестезии. При внутривенном введении бензодиазепины, с одной стороны, продлевают время выхода из анестезии (нежелательный эффект), с другой стороны, вызывают антероградную амнезию (амнезия на события, происходившие после введения препарата), которая полезна в клинике. Так как мидазолам во многих случаях вызывает антероградную амнезию (> 50 %), его часто вводят внутривенно за 15-60 минут до общей анестезии. Мидазолам начинает действовать быстрее, обладает более коротким периодом полувыведения (2-4 часа) и более крутым наклоном кривой доза-эффект, чем другие бензодиазепины, используемые для анестезии. Бензодиазепины применяют для премедикации, для поддержания седативного эффекта в ходе операции и как элемент сбалансированной анестезии в сочетании с другими препаратами. Для ускорения восстановления после седативного действия внутривенных бензодиазепинов сейчас используется их антагонист флумазенил, однако его влияние на респираторную депрессию не всегда предсказуемо. Это препарат короткого действия, поэтому может потребоваться неоднократное его введение.
Анестезия опиоидными аналгетиками
Для общей анестезии, особенно в кардиохирургии, или при других крупных операциях, когда циркуляторный резерв минимален, используют большие дозы опиоидных аналгетиков. В таких ситуациях при внутривенном введении морфина (1-3 мг/кг) или очень сильнодействующего препарата фентанила (50-100 мг/кг) наблюдали лишь минимальные признаки ухудшения циркуляции. В последнее время начали использовать производное фентанила — суфентанил. Несмотря на такие высокие дозы (в табл. 30-2 приведены традиционные дозы аналгетиков), у некоторых больных во время операции сохранялось сознание и оставались неприятные воспоминания. Внутривенные опиоиды могут повышать ригидность грудной стенки, что нарушает вентиляцию, провоцируя послеоперационную респираторную депрессию, которая требует
использования вспомогательной вентиляции и антагонистов опиоидов, например налоксона. Послеоперационные респираторные нарушения можно минимизировать снижением дозы и одновременным введением короткодействующих барбитуратов или бензодиазепинов, обычно вместе с закисью азота. При более коротких операциях внутривенно вводится алфентанил, короткодействующее производное фентанила. Фентанил и дропери-дол (производное бутирофенона) вместе вызывают аналгезию и амнезию и иногда используются с закисью азота при нейролептаналгезии.
Используют также интратекальный и эпидуральный способы введения наркотиков. Этим не достигается обезболивание при операционном разрезе, но обеспечивается полноценная послеоперационная аналгезия.
Пропофол
2,6-Диизопропилфенол (пропофол, дизопро-фол) — это крайне важный внутривенный анестетик. Он вызывает анестезию со скоростью, близкой к скорости внутривенных барбитуратов, а выход из анестезии пропофолом проходит быстрее. В частности, больные раньше начинают ходить после такого наркоза. Более того, пациенты лучше себя чувствуют в ранний послеоперационный период, чем после применения других внутривенных анестетиков. Послеоперационная рвота возникает редко, напротив, считается, что пропофол обладает про-тиворвотным действием. Препарат не вызывает кумулятивных эффектов или отсрочки восстановления сознания после продолжительного введения. Эти полезные свойства привели к частому использованию пропофола как компонента сбалансированной анестезии, а также в качестве анестетика в амбулаторной хирургии. Препарат эффективен и для индукции длительного седативного действия у больных в критическом состоянии. Однако резистентность к его эффектам возникает через несколько дней, то есть по этому последнему показанию пропофол можно применять только в течение ограниченного времени.
После внутривенного введения происходит быстрое распределение препарата (t1/2a - 2-8 мин). Время полувыведения (t1/2p) равно 1-3 часам. Лекарство быстро метаболизируется печенью посредством конъюгации с образованием глюкуронида и сульфата и удаляется с мочой. Менее 1 % пропофо
ла выводится в неизмененном виде. Общий клиренс пропофола превышает печеночный кровоток, что позволяет предположить, что существуют и другие механизмы выведения этого препарата, кроме превращения ферментами печени. Это свойство используется у пациентов, которые не способны метаболизировать другие седативные и снотворные препараты, например мидазолам.
Действие на дыхание сходно с эффектами тиопентала в обычных анестезирующих дозах. Однако пропофол вызывает значительное снижение системного артериального давления во время индукции анестезии, в основном понижая периферическое сосудистое сопротивление. Возможно возникновение апноэ и болевых ощущений в месте инъекции. При применении пропофола были описаны также сокращения мышц, гипотония и (редко) тремор. Реакции гиперчувствительности (гипотензия, сосудистые приливы и бронхоспазм) возникали при введении пропофола в растворителе кремофо-ре и больше не встречались после замены его ин-тралипидом (внутривенная жировая эмульсия). Важный недостаток длительного использования пропофола — его высокая стоимость, превышающая стоимость как бензодиазепинов, так и барбитуратов.
Этомидат
Этомидат — это карбоксилированный имидазол, используемый для индукции анестезии и в схемах сбалансированной анестезии, не требующих его длительного введения. Главное преимущество этого препарата заключается в минимальной депрессии дыхательной и сердечно-сосудистой систем. Этомидат вызывает потерю сознания в течение секунд на фоне небольшой гипотензии, отсутствия действия на частоту сердечных сокращений и низкой частоты возникновения апноэ. Препарат не обладает аналгетическими свойствами, и может потребоваться премедикация опиоидными аналге-тиками для снижения реакции сердечно-сосудистой системы при интубации и для уменьшения количества спонтанных движений. После введения индукционной дозы восстановление наступает в пределах 3 минут.
Распределение этомидата проходит быстро, по двухфазной кривой концентрации в плазме, показывающей периоды полураспределения продолжительностью 3 и 29 минут. Перераспределение пре
парата из мозга в другие области объясняет короткую продолжительность его действия. Этомидат гидролизуется в печени и плазме до неактивных метаболитов. Исследования у человека показали, что почти 90 % препарата в конце концов появляются в моче, из них только 2 % — в неизмененном виде. К сожалению, этомидат вызывает рвоту, боли при инъекциях и миоклонические подергивания, не связанные, однако, с эпилептиформными изменениями ЭЭ Г. Этомидат также может подавлять стероидогенез в коре надпочечников. Длительное введение может привести к гипотензии, дисбалансу электролитов и олигурии.
Кетамин
Кетамин (рис. 24-2) вызывает диссоциированную анестезию, которая характеризуется кататонией, амнезией и аналгезией. Механизм его действия включает блокаду мембранных эффектов возбуждающего нейромедиатора глутамата, опосредованных NMDA-рецепторами. Хотя во многих отношениях кетамин является прекрасным анестетиком, после его использования возникали нарушения ориентации, сенсорные и осязательные иллюзии, красочные сны. Введение диазепама в дозе 0.2-0.3 мг/кг внутривенно за 5 минут до кетамина снижает риск возникновения этого феномена.
Кетамин как липофильный препарат быстро распределяется в хорошо снабжаемые кровью органы, в том числе в мозг, а затем перераспределяется в ткани с пониженной перфузией. Препарат метаболизируется в печени, выводится печенью и почками.
Кетамин — не только сильный аналгетик, но и единственный внутривенный анестетик, который вызывает сердечно-сосудистую стимуляцию. Частота сердечных сокращений, артериальное давление и сердечный выброс обычно значительно увеличиваются. Максимум стимуляции возникает на 2-4 минутах после внутривенного введения и затем медленно уменьшается до нормы за следующие 10-20 минут. Эта стимуляция обусловлена возбуждением симпатической нервной системы и, возможно, подавлением обратного захвата норадреналина в симпатических нервных окончаниях. Повышение уровня адреналина и норадреналина в плазме возникает через 2 минуты после внутривенного введения и возвращается к норме 15 минут спустя.
Кетамин значительно повышает мозговой кровоток, потребность в кислороде и внутричерепное дав
ление. Следовательно, этот препарат, каки большинство ингаляционных анестетиков, потенциально опасен при повышении внутричерепного давления.
У большинства пациентов кетамин слегка снижает частоту дыхания в течение 2-3 минут. Мышечный тонус верхних дыхательных путей сохраняется, их рефлексы высокие (но не всегда). Кетамин почти не вызывает изменений в других органах и системах.
В США кетамин не так часто используется в общей хирургии из-за высокой частоты послеоперационных психических нарушений, связанных с его применением. Его дают пожилым пациентам из группы высокого риска и пациентам в состоянии шока вследствие стимулирующего действия на сердечнососудистую систему. Кетамин используется также в амбулаторной хирургии и у детей при болезненных процедурах (смена повязки при ожоге и др.).
Препараты
В главе 30 приведены лекарственные формы опиоидных препаратов, применяемых для анестезии
Десфлуран ( Супран)
Жидкость: 240 мл для ингаляций
Диазепам (генерик, Валиум, др.)
Перорально: таблетки по 2, 5,10 мг;
растворы 5 мг/мл, 5 мг/5 мл
Перорально для пролонгированного действия: капсулы по 15 мг
Парентерально: 5 мг/мл для инъекций
Энфлуран (Этран)
Жидкость: 125, 250 мл для ингаляций
Этомидат (Амидат)
Парентерально: 2 мг/мл для инъекций
Галотан (генерик, Флуотан)
Жидкость: 125, 250 мл для ингаляций
Изофлуран (Форан)
Жидкость: 100 мл для ингаляций
Кетамин (Кеталар)
Парентерально: 10, 50, 100 мг/мл
для инъекций
Лоразепам (генерик, Ативан, Алзапам) Перорально: таблетки по 0.5,1.2 мг Парентерально: 2.4 мг/мл для инъекций
Метогекситал (Бревитал натрия) Парентерально: порошки по 0.5, 2.5, 5 г для инъекций
Метоксифлуран (Пентран)
Жидкость: 15,125 мл для ингаляций
Мидазолам (Версед)
Парентерально: 1.5 мг/мл для инъекций во флаконах по 1, 2,5, 10 мл
Закись азота (газ в голубых баллонах)
Пропофол (Диприван)
Парентерально: 10 мг/мл для инъекций во флаконах по 20 мл
Тиамилал (Суритал)
Парентерально: растворы для инъекций во флаконах по 1,5,10 г
Тиопентал (Пентотал)
Парентерально: 250, 400, 500 мг в готовых шприцах; растворы по 500 мг, 1 г с растворителем; наборы по 1, 2.5, 5 г Ректально: 400 мг раствора в готовых шприцах
Избранная литература
Aloia R. С., Curtain С. С., Gordon L. М. (eds) Drug and Anesthetic Effects on Membrane Structure and Function. Wiley-Liss, 1991.
Langley M. S., Heel R. C. Propofol: Review of its pharmacodynamic and pharmacokinetic properties and use as an intravenous anesthetic. Drugs, 1988; 35: 334.
Prys-Roberts C., Hug С. C. Jr. Pharmacokinetics of Anesthesia. Blackwell, 1984.
Roth S. H., Miller K. W. Molecular and Cellular Mechanisms of Anesthetics. Plenum Press, 1986.
Swerdlow B. N., Holley F. O. Intravenous anesthetic agents. Pharmacokinetic-pharmacodynamic relationships. Clin. Pharmacokinet, 1987; 12:79.
Ysuda N. et al Desflurane, isoflurane and halothane pharmacokinetics in humans. Anesth. Analg. 1990; 70: S444.
Местные анестетики
25
Рональд Д. Миллер, Люк М. Хондегем
Местные анестетики обратимо блокируют проведение импульса по мембранам аксонов и другим возбудимым мембранам, которые используют натриевые каналы как главный генератор потенциалов действия. Этот эффект может быть применен в клинике для локальной блокады болевых ощущений в определенных частях тела или для прерывания поступления к ним симпатических вазоконстрикторных импульсов. В 1860 г. Ниманн выделил первый препарат, обладающий подобным действием,— кокаин. В 1884 г. Коллер впервые использовал его для анестезии в офтальмологии. Скоро стало известно о его сильных аддиктивных свойствах, но, несмотря на это, его широко использовали в течение последующих 30 лет, так как это был единственный пригодный для применения местный анестетик. В попытке улучшить свойства кокаина Эйнхорн в 1905 г. синтезировал прокаин, ставший основным местным анестетиком на следующие 50 лет. В эти годы создано множество местных обезболивающих средств. Целью поиска новых препаратов было снижение местного раздражающего и повреждающего действия на ткани, уменьшение общей токсичности, более быстрое начало и большая продолжительность анестезии. Самый популярный в настоящее время препарат этой группы лидокаин синтезирован Лефгреном в 1943 г. и считается эталоном местных анестетиков.
Ни один из применяемых сегодня местных анестетиков не идеален, поиск новых препаратов продолжается. Однако в то время как синтезировать химическое соединение со свойствами местного анестетика достаточно легко, крайне трудно добиться значительного снижения токсичности по сравнению с уже существующими препаратами. Основная причина этой сложности состоит в том, что самое серьезное токсическое действие местных анестетиков связано с распространением их локальных терапевтических эффектов на мозг и сосудистую систему.
I. Базисная фармакология местных анестетиков
Химические свойства
Большая часть местных анестетиков состоит из липофильной группы (часто в виде ароматического кольца), соединенной через промежуточную цепь (обычно включающую эфирную или амидную группу) с ионизируемой группой (как правило, третичным амином — табл. 25-1). Для оптимальной активности требуется баланс между гидрофильными и липофильными группами. Кроме общих физических свойств молекул, большое значение имеет их специфическая стереохимическая конфигурация, что проявляется в различной степени выраженности эффекта у стереоизомеров некоторых соединений. Так как эфирные связи (как в прокаине) легче гидролизуются, чем амидные, эфиры обычно имеют более короткое действие.
Местные анестетики являются слабыми основаниями. Для клинического применения они обычно выпускаются в виде солей, так как это улучшает растворимость и повышает стабильность растворов. В средах организма они существуют либо в виде не-ионизированного основания, либо в виде катиона. Соотношение этих двух форм зависит от их рКа и pH жидкостей организма в соответствии с уравнением Гендерсона-Хассельбаха:
, Катионная форма
log----------------—----= рКа - pH.
Неионизированная форма
Так как рКа большинства местных анестетиков находится в пределах 8.0-9.0, большая их часть в жидких средах организма находится в ионизированном катионном состоянии. Рецепторы для местных анестетиков, с которыми наиболее активно связывается катионная форма, расположены на внут
ренней стороне поверхности клеточной мембраны. Связавшись с рецептором, катионы с трудом покидают закрытые каналы. Однако для трансмембранного переноса препаратов необходима неионизиро-ванная форма (рис. 14-2). Это частично объясняет значительно меньшую активность местных анестетиков, которую и стоматологи, и хирурги отмечают в воспаленных тканях, поскольку в них снижен pH и очень мала доля неионизированного местного анестетика, диффундирующего в клетку.
Фармакокинетика
Местные анестетики обычно вводят инъекцион-но в область нервных волокон, проведение по которым необходимо заблокировать. При этом пути введения, абсорбция и распределение не имеют такого значения для времени начала эффекта, какое они имеют для срока окончания анестезии и развития токсического действия на ЦНС и сердечно-сосудистую систему. В случае же локальной аппликации местных анестетиков как начало, так и окончание анестезии определяется способностью препарата к диффузии.
А. Абсорбция. Системная постинъекционная абсорбция местных анестетиков из места введения зависит от нескольких факторов, в том числе от дозы, места введения, связывания препарата с тканями, присутствия вазоконстрикторов и от физико-химических свойств препарата. Аппликации местных анестетиков на области с богатым кровоснабжением, например на слизистую трахеи, приводят к очень быстрой абсорбции и более высокому уровню препарата в крови, чем после инъекции местного анестетика в зоны с менее активным кровоснабжением, например в сухожилие. При региональной анестезии (проводниковой блокаде крупных нервов) максимальные уровни местного анестетика в крови понижаются в зависимости от места введения в следующем порядке: межреберное > корешковое > эпидуральное > в плечевое сплетение > в область седалищного нерва.
Вещества, обладающие вазоконстрикторными свойствами, например адреналин, уменьшают системную абсорбцию местных анестетиков из депо (места инъекции), снижая в нем кровоток. Это особенно выражено для препаратов среднего и короткого действия, например прокаина, лидокаина и ме-пивакаина (но не прилокаина). Снижение локального кровотока увеличивает местную концентра
цию препарата, а значит, и захват его нервом. Кроме того, на фоне вазоконстрикторов общие токсические эффекты уменьшаются вследствие понижения концентрации анестетика в периферической крови практически в 3 раза. Сочетание снижения системной абсорбции и повышения захвата препарата нервом приводят к продлению действия местного анестетика на 50 %. Такой эффект вазоконстрикторов наблюдается в меньшей степени при применении жирорастворимых местных анестетиков длительного действия (бупивакаин, этидокаин), видимо потому, что их молекулы прочно связываются с тканями. К тому же катехоламины могут таким образом изменять функцию нейронов, что аналгезия продлевается, особенно при воздействии на спинной мозг. Исключением является кокаин в связи с его симпатомиметической активностью (табл. 6-5).
Б. Распределение. Амидные местные анестетики широко распределяются в организме после внутривенного болюсного введения. Есть сведения, что в некоторых тканях, например жировой, происходит их секвестрация. После начальной фазы быстрого распределения, во время которой препарат скорее всего накапливается в хорошо перфузируемых тканях, прежде всего в мозгу, печени, почках и сердце, наступает медленная фаза распределения в ткани с меньшим кровоснабжением: мышцы и жировую ткань. Вследствие крайне малого периода полувыведения эфирных препаратов в плазме их распределение в тканях не изучено.
В. Метаболизм и выведение. В печени и в плазме местные анестетики приобретают большую гидрофильность и выводятся с мочой. Так как в неизмененной форме они хорошо проникают через липидные слои, то в этом виде они почти не попадают в мочу. Подкисление мочи вызывает ионизацию третичных оснований до форм, более растворимых в воде. Последние легче выводятся с мочой, так как их реабсорбция затруднена.
Эфирные местные анестетики очень быстро гидролизуются в крови бутирилхолинэстеразой (псев-дохолинестеразой). Поэтому их период полувыведения в плазме обычно очень короткий, в частности для прокаина и хлорпрокаина — менее минуты.
Амидные связи местных анестетиков гидролизуются микросомальными ферментами печени. Скорость метаболизма разных препаратов весьма вариабельна: прилокаин (самый быстрый) > этидокаин > лидокаин > мепивакаин > бупивакаин (са-
ТАБЛИЦА 25-1. Структура и свойства некоторых эфирных и амидных местных анестетиков'
’ Существуют и другие химические типы препаратов, в том числе эфиры (прамоксин), кетоны (диклонин) и дериваты фенетидина (фенакаин).
мый медленный). В результате этого амидные местные анестетики чаще дают токсические эффекты у больных с нарушениями функции печени. Например, средний период полувыведения лидокаина может быть увеличен с 1.8 часов у обычных пациентов до 6 часов и более у пациентов с тяжелыми повреждениями печени. У больных с уменьшенным печеночным кровотоком также следует ожидать замедления инактивации местных анестетиков. Выведение лидокаина у животных после галотановой анестезии идет медленнее, чем у животных, получивших закись азота или кураре. Это может быть связано со снижением печеночного кровотока и вызванной галотаном депрессией микросомального окисления в печени. Пропранолол может продлевать период полувыведения амидных местных анестетиков.
Фармакодинамика
А. Механизм действия. Возбудимые мембраны аксонов, как и мембраны миокарда (глава 14) и тела нейронов (глава 20), поддерживают трансмембранный потенциал на уровне от -90 до -60 мВ. Во время возбуждения натриевый канал открывается, и ток натрия внутрь быстро деполяризует мембрану до потенциала равновесия натрия (+40 мВ). Вследствие этой деполяризации натриевые каналы закрываются (инактивируются) и открываются калиевые каналы. Ток калия наружу реполяризует мембрану до потенциала равновесия калия (около -95 мВ); реполяризация возвращает натриевые каналы в состояние покоя. Трансмембранные ионные градиенты поддерживаются натриевым насосом. Эти процессы похожи на процессы в миокарде, поэтому местные анестетики оказывают сходное действие на все эти ткани.
Функция натриевых каналов может быть нарушена несколькими путями. Биологические токсины типа батрахотоксина, аконитина, вератридина и яда некоторых скорпионов связываются с рецепторами каналов и предотвращают инактивацию. Это приводит к пролонгированию тока натрия через канал, а не к блоку проведения, и некоторые исследователи считают эти вещества агонистами по их действию на натриевые каналы. Токсины морских животных, тетродотоксин и сакситоксин, блокируют эти каналы, связываясь с рецепторами вблизи наружной поверхности клетки. Их эффекты несколько напоминают действие местных анес
тетиков, хотя они и связываются с разными структурами. Местные анестетики связываются с рецептором,. расположенным вблизи внутриклеточного участка канала (рис. 14-2), и вызывают время- и потенциалзависимый блок канала.
При прогрессивном повышении концентрации местных анестетиков на поверхности нервного волокна порог возбуждения повышается, проведение импульсов и скорость возникновения потенциалов действия замедляются, амплитуда потенциалов действия снижается и, наконец, способность генерировать потенциал действия исчезает. Такой нарастающий эффект связан с последовательной блокадой анестетиком все большего числа каналов; в каждом канале связывание приводит к блоку натриевого тока. Если ток ионов натрия заблокирован на определенном отрезке длины нерва, проведение по нерву невозможно. При применении препаратов в минимальных дозах, достаточных для блокады проведения, потенциал покоя значительно не изменяется.
Блокада натриевых каналов большинством местных анестетиков зависит от времени и потенциала'. каналы в состоянии покоя (доминирующее состояние при более отрицательных потенциалах мембраны) имеют значительно меньшую аффинность к местным анестетикам, чем активные (открытые) или инактивированные каналы (доминирующее состояние при более положительных потенциалах мембраны — рис. 14-8). Таким образом, эффект препарата в какой-либо концентрации более выражен в активных действующих аксонах, чем в находящихся в состоянии покоя (рис. 25-1).
Между деполяризациями аксона часть натриевых каналов восстанавливается от блокады местными анестетиками (рис. 14-8). Это восстановление идет в 10-1000 раз медленнее, чем восстановление каналов от нормальной физиологической инактивации, как показано на примере мембраны кардиомиоцита на рис. 14-4. В результате удлиняется рефрактерный период, и нерв может проводить меньшее количество импульсов.
Повышение уровня внеклеточного кальция частично блокирует действие местных анестетиков за счет увеличения поверхностного потенциала мембраны, которое переводит ее в состояние покоя. Наоборот, повышение концентрации внеклеточного калия деполяризует мембранный потенциал и стимулирует инактивацию (рис. 14-9). Это усиливает эффект местных анестетиков.
Хотя было показано, что местные анестетики блокируют много других каналов, в том числе и
химически регулируемые синаптические каналы, не доказана значительная роль этих эффектов в клиническом действии препаратов. С другой стороны, экспериментальные исследования нервных волокон и миокардиоцитов показали, что препараты, которые продлевают потенциал действия, могут значительно повышать чувствительность натриевых каналов к блокаде местными анестетиками (Drach-man, 1991). Это можно объяснить тем, что аффинность активированных и инактивированных каналов к местным анестетикам выше, чем аффинность каналов в состоянии покоя.
Характеристики структуры и активности местных анестетиков. Чем меньше и липофильнее молекулы, тем быстрее они взаимодействуют с рецептором натриевого канала. Эффект также находится в прямой связи с растворимостью в жирах, если при этом гидрофильность препарата достаточна, чтобы диффундировать в место действия. Лидокаин, прокаин и мепивакаин более водорастворимы, чем тетракаин, этидокаин и бупивакаин. Последние препараты действуют сильнее и дольше.
Б. Действие на нервные волокна. Местные анестетики могут блокировать любые нервные волокна, но их действие не ограничивается только потерей чувствительности. Паралич двигательных волокон иногда полезен, однако он сужает возможности пациента выполнять указания врача, например при родах. Во время спинальной анестезии двигательный паралич может вызвать нарушения дыхания, а блокада вегетативных нервов — гипотензию. Разные нервные волокна отличаются по чувствительности к действию местных анестетиков, это зависит от их миелинизации и размера (табл. 25-2). При воздействии местного анестетика на корешок
нерва тонкие волокна В и С блокируются первыми, затем волокна А6. Таким образом, проведение боли исчезает первым, затем подавляются другие виды чувствительности, а далее и двигательные функции.
1. Значение диаметра волокон. Местные анестетики действуют предпочтительно на тонкие волокна, пассивно проводящие электрические импульсы на короткие расстояния. В ходе анестезии такие волокна блокируются первыми, прерывая проведение по коротким участкам нерва. Для прекращения проведения по миелинизированным волокнам необходимо, чтобы блокада распространилась на 3 последовательных узла. Чем толще нерв, тем больше расстояние между узлами, и это частично объясняет большую резистентность толстых волокон к местным анестетикам. Миелинизированные волокна блокируются раньше, чем безмиели-новые того же диаметра. По этой причине преганглионарные В-волокна блокируются раньше, чем безмиелиновые С-волокна.
2. Роль частоты импульсации. Другая причина предпочтительной блокады чувствительных волокон связана с зависимостью действия местных анестетиков от характеристик деполяризации. Вызванная ими блокада более выражена при большей частоте и длительности деполяризации. Сенсорные, особенно болевые, волокна имеют высокую частоту импульсации и сравнительно длительный потенциал действия (до 5 мс). Двигательные волокна посылают импульсы с меньшей частотой и более коротким потенциалом действия (< 0.5 мс). А6- и С-волокна имеют маленький диаметр и участвуют в передаче высокочастотных болевых импульсов. По-
Рис. 25-1. Эффект повторной активации при блокаде тока натрия, вызванной местным анестетиком в миелинизированном аксоне. Было произведено 25 импульсных стимуляций, и полученные токи натрия наложены друг на друга. Отметьте, что токи, вызванные импульсами, постепенно снижались с первого по 25. Длительный период отдыха привел к восстановлению после блокады, которая вновь возникала после возобновления импульсации (нА — наноамперы). (Из: Courtney К. R. Mechanism of frequency-dependent inhibition of sodium currents in frog myelinated nerve by the lidocaine derivative GEA. J. Pharmacol. Exp. Ther. 1975; 195: 225.)
ТАБЛИЦА 25-2. Диаметр и. чувствительность к блокаде нервных волокон разного типа
Тип волокон Вид чувствительности (функция) Диаметр (мкм) Миелинизация Скорость проведения (м/с) Чувствительность к блокаде
ТипА а Проприоцепция, двигатель- 12-20 полная 70-120 +
₽ ная Тактильная, давление 5-12 полная 30-70 ++
Y Контрактильная (мышечные 3-6 полная 15-30 ++
5 веретена) Боль, температура 2-5 полная 12-30 +++
Тип В Преганглионарная вегета- <3 слабая 3-15 ++++
ТипС Дорзальные тивная Боль 0.4-1.2 отсутствует 0.5-2.3 ++++
корешки Боль Постганглионарная 0.3-1.3 отсутствует 0.7-2.3 ++++
этому они блокируются раньше и меньшими концентрациями местных анестетиков, чем Ад-волокна.
3. Эффект положения волокон в пучке. Анатомические особенности, которые иногда изменяют названные правила дифференцированной блокады нервов,— это положение волокна в пучке. В крупных нервных стволах двигательные волокна часто расположены по наружной поверхности и поэтому первыми контактируют с препаратом, который введен в окружающие ткани. Следовательно, в крупных нервных стволах двигательная блокада наступает раньше, чем чувствительная. В конечностях проксимальные чувствительные волокна расположены на наружной поверхности нерва, а дистальные — в его центре. Поэтому во время инфильтрационной блокады крупного нерва анестезия сначала развивается проксимально, а затем распространяется дистально, когда анестетик начинает проникать в центр нерва.
4. Влияния на другие возбудимые мембраны. Местные анестетики вызывают слабую нервно-мышечную блокаду, которая не имеет клинического значения. Однако их влияние на мембраны кардиомиоцитов имеет огромное значение. Некоторые из них в концентрациях, ниже требуемых для блокады нервного проведения, являются отличными противоаритмическими препаратами (глава 14), но все эти препараты в высоких концентрациях могут вызывать аритмии.
II. Клиническая фармакология местных анестетиков
Местные анестетики могут обеспечивать временную, но полную аналгезию некоторых частей тела. Обычно используемые методы введения — это аппликационный, инъекции в область вокруг периферических нервных окончаний и крупных нервных стволов, инстилляция в эпидуральное или субарахноидальное пространство вокруг спинного мозга (рис. 25-2). Кроме того, блокаду вегетативных симпатических волокон можно использовать для изучения роли тонуса симпатической нервной системы у пациентов с периферическим вазоспазмом.
Выбор местного анестетика основывается на необходимой длительности анестезии. Прокаин и хлорпрокаин действуют коротко; лидокаин, мепи-вакаин и прилокаин имеют среднюю продолжительность действия; тетракаин, бупивакаин и эти-докаин действуют долго (табл. 25-1).
Анестезирующий эффект препаратов короткой и средней продолжительности действия можно пролонгировать, повышая их дозу или добавляя вазоконстриктор, например адреналин или фенилэфрин. Вазоконстриктор замедляет удаление препарата из места инъекции. К тому же он снижает вса-
Субарахноидальное пространство спинального канала Спинальная блокада 1гй1 Основание спинного мозга// —jwH. . 1 Паравертебральная блокада
Твердая мозговая оболочка '6' Эпидуральное пространство / у/ Эпидуральная или у' ) перидуральная блокада 1 Эпидуральное пространство каудальногсу^/^ спинномозгового канала J Г wk 11 IB II 'хчК Filum terminale ^11 III Wm/И|| Cauda equina Ж/h &4 vl ж 1 1 111 l/ 11 j ' 1BXs2 NXjA Каудальная блокада
Рис. 25-2. Схема введения местных анестетиков в область спинального канала
сываемость в кровь и, следовательно, общую токсичность.
Начало местной анестезии иногда можно ускорить использованием растворов, насыщенных двуокисью углерода. Высокий тканевой уровень СО2 приводит к развитию внутриклеточного ацидоза (СО2 хорошо проходит через мембраны), который в
свою очередь создает условия для внутриклеточной аккумуляции катионных форм местных анестетиков.
При повторных инъекциях местного анестетика во время эпидуральной анестезии теряется его эффективность (тахифилаксия). Это скорее всего является последствием локального внеклеточного ацидоза. Местные анестетики часто производятся
в виде гидрохлоридов (pH 4.0-6.0). После инъекции препаратов pH повышается до физиологического уровня из-за буферного действия тканей, вследствие чего выделяется достаточное количество свободных оснований, диффундирующих через аксональные мембраны. Однако повторные инъекции используют всю местную буферную емкость. Последующий ацидоз увеличивает количество внеклеточных катионных форм, которые плохо диффундируют в аксон. Клинические последствия этого — очевидная тахифилаксии, особенно в областях ограниченного буферного резерва, например в спинномозговой жидкости.
Токсичность
В конечном итоге местные анестетики абсорбируются из места введения. Если их уровень в крови значительно поднимается, то развиваются эффекты на некоторые органы и системы.
А. ЦНС. Еще с доисторических времен аборигены в Перу жевали листья растения Erythroxylon coca, источника кокаина, для повышения тонуса и снятия усталости. Значительные эффекты, затрагивающие ЦНС, можно получить вдыханием порошкообразного кокаина в нос и курением кокаина в форме основания. Кокаин стал наиболее часто употребляемым наркотиком (глава 31). Считалось, что другие местные анестетики не обладают таким эйфоригенным эффектом кокаина. Однако некоторые исследования показали, что привыкшие к кокаину наркоманы не могут отличить интраназальный кокаин от лидокаина, используемого таким же путем.
Другие центральные эффекты включают сонливость, головокружение, зрительные и слуховые нарушения и двигательное беспокойство. В более высоких концентрациях любые местные анестетики (в том числе и кокаин) могут вызывать нистагм, дрожь и, наконец, тоникоклонические судороги с последующей депрессией ЦНС и смертью. Очевидно, что местные анестетики подавляют корковые тормозящие пути, результатом чего является бесконтрольная активность возбуждающего компонента. Эта стадия несбалансированного возбуждения может затем перейти в генерализованное торможение ЦНС, если и дальше повышать уровень местных анестетиков.
Наиболее серьезные общие токсические реакции этих препаратов связаны с развитием судорог вслед
ствие их избыточных концентраций в крови. Лучший метод предотвращения судорог — это использование минимальной дозы анестетика, необходимой для анестезии. При неизбежности применения больших доз для профилактики судорог целесообразна премедикация бензодиазепинами, например диазепамом в дозе 0.1-0.2 мг/кг парентерально. При возникновении судорог необходимо предотвратить гипоксемию и ацидоз. Хотя оксигенотерапия не предотвращает развитие судорог, гипероксемия после их начала считается благоприятным фактором. Наоборот, гиперкапния и ацидоз повышают риск развития судорог. Следовательно, для терапии судорог рекомендуется гипервентиляция. Гипервентиляция повышает pH крови, что понижает содержание внеклеточного калия. Это гиперполяри-зует трансмембранный потенциал аксонов, что стимулирует развитие низкоаффинного состояния покоя натриевых каналов, приводящее к снижению системной токсичности местных анестетиков.
Судороги, вызванные местными анестетиками, можно лечить и малыми дозами короткодействующих барбитуратов, например тиопенталом в дозе 1 -2 мг/кг или диазепамом — 0.1 мг/кг внутривенно. Двигательный компонент судорог можно подавить короткодействующим миорелаксантом, в частности сукцинилхолином (0.5-1 мг/кг внутривенно). Следует подчеркнуть, что сукцинилхолин не влияет на ЭЭГ — картину центральных корковых процессов развития судорог. В особо тяжелых случаях судорог интубация совместно с введением сукцинилхо-лина и механической вентиляцией легких помогает предотвратить аспирацию желудочного содержимого и облегчить проведение гипервентиляции.
Б. Периферическая нервная система (нейротоксичность). При аппликации слишком больших доз все местные анестетики могут оказывать токсическое действие на ткань нерва. Описано несколько случаев остаточных чувствительных и двигательных нарушений после спинальной анестезии с введением слишком больших объемов хлорпрокаина. Неизвестно однако, обладает ли хлорпрокаин большей нейротоксичностью, чем другие местные анестетики.
В. Сердечно-сосудистая система. Действие местных анестетиков на сердечно-сосудистую систему частично связано с прямым эффектом на мембраны гладких мышц и миокарда и непрямым эффектом на вегетативные нервные волокна. Как указано в главе 14, местные анестетики блокируют нат
риевые каналы в миокарде и этим подавляют аномальную пейсмекерную активность, возбудимость и проводимость. Все эти препараты, кроме кокаина, также уменьшают силу сердечных сокращений и вызывают расширение артерий, что приводит к гипотензии. Сердечно-сосудистый коллапс и смерть возникают только при использовании больших доз, но иногда они случаются и прй применении сравнительно малых доз для инфильтрационной анестезии.
Как уже говорилось, кокаин отличается от других местных анестетиков по своим эффектам на сердечно-сосудистую систему. Блокада обратного захвата норадреналина приводит к вазоконстрикции и гипертензии. Это может ухудшить общее состояние больного при аритмиях. Вызванная кокаином вазоконстрикция может привести к ишемии слизистой носа, а при хроническом использовании — к изъязвлению слизистой и даже к повреждению носовой перегородки. Эти вазоконстрикторные свойства кокаина можно использовать в клинике для уменьшения кровоточивости слизистой оболочки носоглотки при ее повреждении.
Бупивакаин более кардиотоксичен, чем другие местные анестетики. Описано несколько случаев развития не только судорог, но и сердечно-сосудистого коллапса после случайной внутривенной инъекции бупивакаина. Реанимация при этих состояниях была особо трудной или безуспешной. Экспериментальные исследования на животных подтвердили, что бупивакаин действительно токсичнее других местных анестетиков при внутривенном введении. Это связано с тем, что блокада натриевых каналов бупивакаином усилена большей длительностью потенциала действия миокардиоцитов по сравнению с нервными волокнами. В отличие от лидокаина блокирующий эффект бупивакаина проявляется при нормальной частоте сердечных сокращений. Исследования показали, что самое частое изменение ЭКГ при интоксикации бупивакаином — это медленный идиовентрикулярный ритм с широкими комплексами QRS и электромеханической диссоциацией. Реанимация проводится обычными поддерживающими методами (своевременной коррекцией ацидоза гипервентиляцией и введением бикарбоната), а также активным использованием адреналина, атропина и бретилиума. Ропивакаин — это новый исследуемый амидный местный анестетик, похожий по местным эффектам на бупивакаин. Предварительные данные показывают, что он
обладает меньшей кардиотоксичностью, чем бупивакаин.
В. Кровь. Введение больших доз (> 10 мг/кг) прилокаина для региональной анестезии может привести к кумуляции метаболита 0-толуидина — окислителя, который способен переводить гемоглобин в метгемоглобин. При наличии достаточного количества метгемоглобина (30-50 мг/л) у пациента развивается цианоз. Такие уровни метгемоглобинемии переносимы для здоровых людей, но могут вызвать декомпенсацию у пациента с заболеванием сердца или легких, что требует немедленного лечения. В этих случаях применяют восстановители типа метиленового синего или аскорбиновой кислоты для быстрого перевода метгемоглобина в гемоглобин.
Е. Аллергические реакции. Эфирные местные анестетики метаболизируются до дериватов р-ами-нобензойной кислоты. Эти продукты вызывают аллергические реакции у некоторых больных. Амиды не метаболизируются до р-аминобензойной кислоты, и аллергические реакции на препараты этой группы крайне редки.
Препараты
Бензокаин (генерик, др.)
Местно: 5.6 % кремы; 6.20 % гели; 5 % мази;
0.5 % лосьон; 20 % аэрозоль
Бупивакаин (генерик, Маркаин, Сенсоркаин) Парентерально: 0.25, 0.5, 0.75 % растворы для инъекций; 0.25, 0.5, 0.75 % растворы с 1: 200 000 адреналина
Бутамбен пикрат (Бутезин пикрат)
Местно: 1 % мазь
Хлорпрокаин (Незакаин)
Парентерально: 1, 2, 3 % растворы для инъекций
Кокаин ( генерик)
Местно: растворы 40,100 мг/мл; порошок
5, 25 г; растворимые таблетки по 135 мг
Дибукаин (генерик, Нуперкаинал)
Местно: 0.5 % крем; 1 % мазь
Диклонин (Диклон)
Местно: 0.5, 1 % растворы
Этидокаин (Дуранест)
Парентерально: 1 % раствор для инъекций;
1,1.5 % растворы с 1 : 200 000 адреналина для инъекций
Лидокаин (генерик, Ксилокаин, др.) Парентерально: 0.5,1,1.5, 2, 4, 10, 20 % растворы для инъекций;
0.5,1,1.5,2 % растворы с 1: 200 000 адреналина;
1.2 % раствор с 1: 100 000 адреналина;
2 % раствор с 1: 50 000 адреналина Местно: 2.5, 5 % мази; 0.5 % крем;
2 % желе; 2, 4,10 % растворы
Мепивакаин (генерик, Карбокаин, др.) Парентерально: 1, 1.5, 2,3 % растворы для инъекций; 2 % раствор с 1: 20 000 левонордефрина
Прамоксин (Тронотан, Праке)
Местно: 0.5,1 % кремы; 1 % гель и лосьон
Прилокаин (Цитанест)
Парентерально: 4 % растворы для инъекций;
4 % раствор с 1 : 200 000 адреналина
Прокаин (генерик, Новокаин)
Парентерально: 1, 2,10 % растворы для инъекций
Пропоксикаин и прокаин (Равокаин и новокаин) Парентерально: 7.2 мг пропоксикаина с 36 мг прокаина и норадреналина или кобефрина на одну стоматологическую инъекцию объемом 1.8 мл
Тетракаин (Понтокаин)
Парентерально: 1 % растворы для инъекций; 0.2, 0.3 % раствор с 6 % декстрозой для спинальной анестезии
Местно: 0.5 % мазь; 0.5 % глазные капли раствор; 1 % крем; 2 % раствор для полоскания горла
Избранная литература
Butterworth J. F., Strichartz G. R. Molecular mechanisms of local anesthesia: A review. Anesthesiology, 1990; 72: 711.
Catterall W. A. Structure and function of voltagesensitive ion channels. Science, 1988; 242: 50.
Covino B. G. Pharmacology of local anaesthetic agents. Br. J. Anaesth. 1986; 58: 701.
Fleming J. A., Byck R., Barash P. G. Pharmacology and therapeutic applications of cocaine. Anesthesiology, 1990; 73: 518.
Hille B. Local anesthetics: Hydrophilic and hydro-phobic pathways for the drug-receptor reactions. J. Gen. Physiol. 1977; 69:497.
Strichartz G. R. Pharmacology of local anesthetics. In: Anesthesia, 4th ed. Miller R. D. (ed.) Churchill Livingstone, 1994.
Tucker G. T. Pharmacokinetics of local anaesthetics. Br. J. Anaesth. 1986; 58: 717.
Средства, расслабляющие скелетную мускулатуру 26
(миорелаксанты)
Рональд Д. Миллер
Средства, влияющие на скелетную мускулатуру, делятся на две большие терапевтические группы: препараты, используемые во время хирургических процедур и в интенсивной терапии для получения паралича, то есть блокаторы нервно-мышечной передачи, и применяемые для снижения спастичности при ряде неврологических заболеваний, то есть спазмолитики. Члены первой группы в соответствии со своим названием вмешиваются в нервно-мышечную передачу и не действуют на ЦНС. Однако, так как их чаще всего используют как дополнение к общей анестезии, традиционно они изучаются вместе с препаратами, действующими на ЦНС. Препараты второй группы принято называть миорелаксантами “центрального действия”. Однако, по меньшей мере, один из этих препаратов (дантролен) не имеет центрального действия, то есть традиционное название утратило свой смысл.
Блокаторы нервно-мышечной передачи
История
В XVI в. европейские путешественники-исследователи открыли, что жители бассейна Амазонки в Южной Америке используют отравленные стрелы, которые вызывают смерть из-за паралича дыхательных мышц. В дальнейшем этот яд стали активно исследовать. Изучение кураре, компонента этого яда, легло в основу ранних активных исследований в фармакологии. Открытие действующего начала кураре, тубокурарина (d-тубокурари-
на), и синтез его производных оказали большое влияние на анестезиологию и хирургию, а также помогли определить и изучить механизмы нервно-мышечной передачи.
Механизм нервно-мышечной передачи
Теоретически расслабление мышц и паралич могут возникнуть из-за нарушения функции на нескольких уровнях, включая ЦНС, миелинизированные соматические нервы, терминали безмиелино-вых нервов, холинорецепторы, концевые пластин-
Рис. 26-1. Схематическое изображение нервно-мышечного соединения. (П — пузырьки с медиатором; М — митохондрии; АХ — ацетилхолин; АХЭ — ацетилхолинэс-тераза; СМ — складки постсинаптической мембраны). (Drachman D. В. Myasthenia gravis. N. Engl. J. Med. 1978; 298:135.)
ки, мембраны мышечного волокна и сократительный аппарат (рис. 26-1). Механизм нервно-мышечной передачи на уровне концевой пластинки похож на передачу, рассмотренную в главе 6, и состоит из поступления импульса к терминали двигательного нерва, инфильтрации ионами кальция и высвобождения ацетилхолина. Ацетилхолин затем проникает через синаптическую щель к никотиновому рецептору на двигательной концевой пластинке. При взаимодействии ацетилхолина и рецептора повышается проницаемость мембраны в области концевой пластинки в основном для Na+, но также и для К+. Ионы натрия поступают в клетку, деполяризуя мембрану, что обозначают как потенциал концевой пластинки. Величина этого потенциала напрямую связана с количеством высвобождаемого ацетилхолина. Если потенциал небольшой, то проницаемость в зоне концевой пластинки быстро возвращается к норме, и импульс из этой зоны не проводится на остальную часть мембраны. Однако если потенциал концевой пластинки достаточно велик, то мышечная мембрана деполяризуется до порогового значения и потенциал действия проводится по всему мышечному волокну. Сокращение мышцы затем вызывается путем сопряжения возбуждения и сокращения мышечных волокон. Высвободившийся ацетилхолин удаляется из зоны концевой пластинки путем диффузии и быстрой ферментативной деструкции ацетилхолинэстеразой.
Блокада нормального функционирования концевой пластинки может осуществляться двумя путями. Фармакологическая блокада ацетилхолина вызывается препаратами-антагонистами. Они предотвращают вступление этого медиатора в контакт с его рецептором, поэтому деполяризации не возникает. Прототипом этой недеполяризующей подгруппы является тубокурарин. Блокада проведения также может быть вызвана деполяризующим агонистом. (Этот парадоксальный эффект возникает и на ганглионарных никотиновых холиноре-цепторах.) Прототип деполяризующих блокаторов — сукцинилхолин. Сходную деполяризующую блокаду может вызвать и сам ацетилхолин в очень высоких местных концентрациях (например, при интоксикации ингибиторами холинэстеразы), а также никотин и его агонисты. Однако блокаду, вызванную этими препаратами, нельзя адекватно контролировать, следовательно, эти препараты не могут иметь клинического значения при анестезии и операциях.
I. Базисная фармакология блокаторов нервно-мышечной передачи
Химические свойства
Все блокаторы нервно-мышечной передачи имеют структурное сходство с ацетилхолином. Например, сукцинилхолин фактически состоит из двух молекул ацетилхолина (рис. 26-2). В отличие от линейной структуры сукцинилхолина и других деполяризующих препаратов недеполяризующие скрывают свою “двойную ацетилхолиновую” структуру в громоздкой, сравнительно ригидной кольцевой системе двух типов (например, панкуроний — рис. 26-2). Две основные семьи недеполяризующих средств (дериваты изохинолина и стероиды) показаны на рис. 26-3 и 26-4. Еще одно сходство всех средств этого класса—наличие одного или двух четвертичных атомов азота, которые делают эти препараты плохо растворимыми в липидах и, следовательно, предотвращают их попадание в ЦНС.
Фармакокинетика блокаторов нервно-мышечной передачи
Все блокаторы нервно-мышечной передачи вы-сокополярны и неактивны при пероральном приеме. Их всегда вводят внутривенно.
А. Недеполяризующие препараты. Скорость элиминации недеполяризующих блокаторов нервно-мышечной передачи из крови характеризуется быстрой начальной фазой распределения, а затем медленной фазой выведения. Так как блокаторы нервно-мышечной передачи сильно ионизированы, они плохо проходят через мембраны и имеют ограниченный объем распределения — 80-140 мл/кг, который ненамного превышает объем крови.
Тубокурарин (рис. 26-3), метокурин (диметил-тубокурарин) и галламин (не показан) не метаболизируются. Галламин полностью выводится почками (табл. 26-1). В отличие от него только около 50-60 % введенной дозы тубокурарина или метокурина выделяется с мочой за 24 часа. Точный путь выведения остальных препаратов у человека неизвестен, хотя считается, что они выводятся с желчью. Все стероидные миорелаксанты метаболизируются до их 3-гидрокси-, 17-гидрокси- и 1,17-дигидрок-
О СН3
II I
СН3-С — О—СН2—СН2—+N—СН2
I
СНз Ацетилхолин
О СНз
II I
сн2—с -o-ch2-ch2-+n-ch2
I СНз
Панкуроний
Рис. 26-2. Структурная связь сукцинилхолина, деполяризующего препарата, и панкурония, недеполяризующего препарата, с ацетилхолином, медиатором нервно-мышечной передачи. Сукцинилхолин, который исходно называли диацетилхолин, состоит из двух молекул ацетилхолина, связанных через метильные группы уксусной кислоты. Панкуроний можно описать как два ацетилхолиноподобных фрагмента (выделено темным цветом), включенных в стероидную структуру
сипроизводных в основном в печени. 3-гидроксиметаболиты обычно обладают 40-80 % эффективности исходного препарата. Эти продукты не образуются в достаточных количествах, чтобы вызвать значительную дополнительную нервно-мышечную блокаду во время хирургической анестезии. Однако если исходный препарат дается в течение не- ' скольких дней, например при интенсивной терапии, то 3-гидроксипроизводные могут кумулировать и вызывать длительный паралич (Segredo, 1992). Хотя другие метаболиты также обладают способностью к нервно-мышечной блокаде, их эффект незначителен.
Как и тубокурарин, другие длительнодействующие миорелаксанты (доксакурий, метокурин, панкуроний и пипекуроний) в основном выводятся почками (60-90 % дозы, табл. 26-1). В отличие от них миорелаксанты средней продолжительности действия (векуроний и рокуроний) больше выводятся печенью, и их элиминация зависит от печеночного метаболизма. Эти препараты чаще применяются в клинике, чем препараты длительного действия. Векуроний, как и панкуроний, в своем составе имеет стероидное ядро (рис. 26-4), которое отличается только тем, что один из атомов азота третичный, а не четвертичный. Несмотря на такое незначительное структурное отличие, векуроний имеет иные фармакологические свойства. Он действует короче (приблизительно 20-35 мин по сравнению с 60 мин у панкурония), его эффекты на сердечно-сосудистую систему минимальны, и он почти не выводится почками. Только 15 % введенной дозы векурония элиминируется почками, остальные 85 % — печенью в виде либо неизмененного препарата, либо его 3-гидрокси-метаболита. Рокуроний — это новейший дериват стероидов, по фармакокинетическим свойства похожий на векуроний. Рокуроний начинает действовать быстрее, чем любые другие недеполяризующие блокаторы нервно-мышечной передачи. Поэтому он предпочтителен для быстрого начала анестезии с эндотрахеальной интубацией.
Атракурий (рис. 26-3) — это изохинолиновый недеполяризующий миорелаксант, сходный с векуронием по многим характеристикам. Атракурий скорее всего инактивируется путем спонтанного разрушения (элиминация Гоффмана), а не почечными или печеночными механизмами. Основные продукты его распада — это лауданозин и связанная с ним четвертичная кислота, которые не-имеют свойств блокаторов нервно-мышечной передачи. Однако лауданозин очень медленно метаболизируется печенью и имеет длительный период полувыведения (150 мин) по сравнению с исходным соединением, атракурием. Он хорошо проникает через гематоэнцефалический барьер, высокие его концентрации могут вызвать судороги. У собак и кроликов для возникновения судорог необходимы концентрации лауданозина в крови 17 мкг/мл и 3.9 мкг/мл соответственно. Во время операций у человека содержание этого метаболита в крови колеблется от 0.2 до 1 мкг/мл. Однако были сообщения, что при длительном введении атракурия концентрация лауданозина достигала 5.5 мкг/мл. При более низких уровнях
Тубокурарин
Рис. 26-3. Структуры некоторых изохинолиновых блокаторов нервно-мышечной передачи. Эти препараты — недеполяризующие по своему действию
Рис. 26-4. Структуры некоторых стероидных блокаторов нервно-мышечной передачи. Эти препараты — недеполяризующие по своему действию
(0.2-0.8 мкг/мл) лауданозин вызывает повышение потребности в анестетике на 30 %.
Мивакурий имеет самое короткое действие из всех недеполяризующих блокаторов нервно-мышечной передачи (табл. 26-1). Он полностью метаболизируется холинэстеразой плазмы, его фармакокинетика не зависит от печеночного или почечного выведения. Однако у пациентов с почечной недостаточностью уровень холинэстеразы понижен, поэтому длительность действия мивакурия может увеличиваться.
Б. Деполяризующие препараты. Очень короткое действие сукцинилхолина (5-10 мин) в основном связано с его быстрым гидролизом холинэстеразой плазмы (бутирилхолинэстеразой, или псев-дохолиэстеразой) — ферментом, который присутствует в печени и плазме. Очевидно, холинэстераза плазмы метаболизирует сукцинилхолин быстрее, чем мивакурий, так как последний действует дольше (табл. 26-1). Первичный метаболит сукцинилхолина, сукцинилмонохолин, обладает более слабыми свойствами блокатора нервно-мышечной передачи. Затем он метаболизируется до сукцината и холина. Холинэстераза плазмы гидролизует сукцинилхолин с огромной скоростью; в результате этого только очень малая часть исходной внутривенной дозы попадает к нервно-мышечным соедине
ниям. Так как у концевой пластинки холинэстераза плазмы практически отсутствует, действие сукцинилхолина прекращается путем его диффузии во внеклеточную жидкость. Таким образом, холинэстераза плазмы влияет на длительность действия сукцинилхолина, определяя его количество, которое попадает к концевой пластинке.
Нервно-мышечная блокада сукцинилхолином или мивакурием может быть более длительной у пациентов с генетически аномальной холинэстеразой плазмы. У таких больных можно использовать “дибукаиновый тест” на способность метаболизировать сукцинилхолин. При стандартных условиях теста дибукаин подавляет нормальный фермент на 80 %, а аномальный — только на 20 %. Известно много генетических вариантов холинэстеразы плазмы, из которых дибукаинзависимые представляют наибольший интерес для клиники.
Механизм действия
Взаимодействие препарата, холинорецептора и канала концевой пластинки описано на молекулярном уровне. Несколько возможных моделей действия препаратов приведены на рис. 26-5.
А. Недеполяризующие препараты. Все блокаторы нервно-мышечной передачи, используемые в
ТАБЛИЦА 26-1. Некоторые свойства блокаторов нерано-мышечной передачи
Препарат Элиминация Приблизительная длительность действия (мин) Относительная сила действия, по сравнению с тубокурарином
Дериваты изохинолина
Атракурий Спонтанная1 20-35 1.5
Доксакурий Почки >35 6
Метокурин Почки (40 %) >35 4
Мивакурий ХЭ2 плазмы 10-20 4
Тубокурарин Почки(40 %) >35 1
Дериваты стероидов
Панкуроний Почки и печень >35 6
Пипекуроний Почки (60 %) и печень >35 6
Рокуроний Печень (75-90 %) и почки 20-35 0.8
Векуроний Печень (75-90 %) и почки 20-35 6
Другие препараты
Галламин Почки (100 %) >35 0.2
Сукцинилхолин ХЭ2 плазмы (100 %) <8 Другой механизм3
1 Неферментативный и ферментативный гидролиз эфирных связей.
2 Бутирилхолинэстераза (лсевдохолинэстераза).
3 Деполяризующий агонист холинорецепторов.
США, кроме сукцинилхолина, классифицируются как недеполяризующие. Их прототипом является тубокурарин. Они вызывают обратимую блокаду передачи. В низких дозах и при низкой частоте стимуляции они действуют в основном на никотиновые рецепторы и конкурируют с ацетилхолином. В более высоких дозах некоторые из этих препаратов проникают в поры каналов и вызывают блокаду. Это еще больше ослабляет нервно-мышечную передачу и снижает способность ингибиторов холинэстеразы (например, неостигмина) угнетать их действие. Недеполяризующие блокаторы нервно-мышечной передачи могут также блокировать пресинаптические натриевые (но не кальциевые) каналы. В результате эти миорелаксанты вмешиваются в процессы транспорта ацетилхолина из нервных окончаний.
Одним из следствий конкурентной постсинаптической блокады этими препаратами является то, что тетаническая стимуляция, при которой выделяется большое количество ацетилхолина, приводит к транзиторному снятию блокады. Важное клиническое приложение этого принципа — способность ингибиторов холинэстеразы уменьшать блокаду.
Характеристики недеполяризующих блокаторов нервно-мышечной передачи представлены в табл. 26-2 и на рис. 26-6.
Б. Деполяризующие препараты.
1. Фаза I блокады (деполяризующая). Сукци-нилхолин — это единственный деполяризующий блокатор нервно-мышечного проведения, исполь
зуемый в США. Его эффекты почти идентичны эффектам ацетилхолина, только сукцинилхолин действует несколько дольше. Сукцинилхолин взаимодействует с никотиновыми рецепторами и вызывает деполяризацию концевой пластинки, которая распространяется на прилежащие мембраны, вызывая генерализованное дезорганизованное сокращение моторных единиц мышцы. Результаты регистрации токов единичных каналов показали, что деполяризующие блокаторы могут проникать в них, вызывая длительное “мерцание” ионной проводимости (рис. 26-7). Так как ацетилхолин не метаболизируется в синапсе с достаточной эффективностью, мембраны остаются деполяризованными и не реагируют на дополнительные импульсы. Более того, поскольку для поддержания напряжения мышцы требуется поступление повторных импульсов, сопряженное с реполяризацией концевой пластинки, возникает вялый паралич. Фаза I блокады усиливается ингибиторами холинэстеразы. Характеристики деполяризующих блокаторов представлены в табл. 26-2 и на рис. 26-6.
2. Фаза II блокады (десенсибилизирующая). При продолжении контакта с сукцинилхолином начальная деполяризация концевой пластинки снижается и начинается реполяризация мембраны. Несмотря на реполяризацию, мембрана не способна к новой деполяризации под действием ацетилхолина, если сукцинилхолин по-прежнему присутствует. Механизм развития фазы II неясен, но недавние данные (Marshall, 1990) показали, что в этой
Рис. 26-5.. Схема взаимодействия препаратов с холинорецептором на концевой пластинке. Вверху: действие обычных агонистов ацетилхолина — открытие канала. Внизу слева: недеполяризующий блокатор, например тубокурарин, предотвращает открытие канала, связываясь с рецептором. Внизу справа; деполяризующий блокатор, например сукцинилхолин, связывается с рецептором и блокирует канал. Нормального закрытия канала не происходит. Деполяризующие блокаторы могут приводить к “десенсибилизации” концевой пластинки, блокируя ее рецептор и вызывая стойкую деполяризацию. Дополнительное действие препаратов на каналы концевой пластинки может быть вызвано изменениями липидного окружения канала (не показано). Общие анестетики и алкоголь могут нарушать нервно-мышечную передачу по этому механизму
Закрытый канал, норма
Открытый канал, норма
А Недеполяризующий блокатор
Закрытый блокированный канал
Деполяризующий блокатор
Открытый блокированный канал
ТАБЛИЦА 26-2. Сравнение типичных недеполяризующего (тубокурарина)
и деполяризующего
(сукцинилхолина) миорелаксантов
Тубокурарин Сукцинилхолин
Фаза! Фаза!!
Введениетубо- Аддитивное курарина Антагонист Усиливает1
Введение сук- Антагонист цинилхолина Аддитивное Усиливает1
Эффект Антагонист неостигмина Усиливает1 Антагонист
Исходное воз- Нет буждающее действие на скелетную мышцу Фасци куля ции Нет
Ответ на тета- Непродол- Продолжи- Непродол-
нический сти- жительный мул тельный2 жительный
Посттетани- Да ческое расслабление Нет Да
Скорость вое- 30-60 мин2 становления 4-8 мин > 20 мин2
1 Неизвестно, является это взаимодействие аддитивным или синергическим (супераддитивным).
2 Амплитуда снижена, но реакция продолжительная.
фазе блокады блокирование канала может иметь большее значение, чем агонистическое действие сукцинилхолина. Другая гипотеза заключается в том, что на мембране, непосредственно вокруг концевой пластинки, появляется невозбудимая зона. Эта зона предположительно препятствует центробежному распространению импульсов, вызванных действием ацетилхолина на рецептор. Так как конечная пластинка частично реполяризована и еще не реагирует на ацетилхолин, говорят, что мембрана “нечувствительна” к эффектам ацетилхолина, поэтому такую блокаду назвали “десенситизирую-щей”. Какой бы механизм ни был задействован, каналы ведут себя так, как будто они находятся в длительном закрытом состоянии (рис. 26-5).
Позднее, в фазе II, блокада становится очень похожей на блокаду недеполяризующими препаратами, то есть дает непродолжительный ответ на тетаническую стимуляцию (рис. 26-6) и обратима ингибиторами холинэстеразы.
II. Клиническая фармакология блокаторов нервно-мышечной передачи
Паралич скелетных мышц
До появления блокаторов нервно-мышечной передачи адекватной релаксации скелетных мышц можно было достичь только глубокой анестезией, которая часто была связана с опасными угнетающими эффектами на различные системы и органы, особенно на сердечно-сосудистую и дыхательную. Блокаторы нервно-мышечной передачи дали возможность получить адекватную миорелаксацию для хирургического вмешательства без угнетающего действия общей анестезии.
Мониторирование эффекта миорелаксантов во время операции можно проводить, используя чрез-кожную стимуляцию одного из нервов руки с записью вызванных подергиваний (рис. 26-6). Чаще других используется стимуляция по типу блока из четырех раздражений.
А. Недеполяризующие препараты. Во время анестезии внутривенное введение 0.12-0.4 мг/кг тубокурарина сначала вызовет мышечную слабость, затем мышцы полностью парализуются и не будут реагировать на стимуляцию (рис. 26-8). Мышцы, способные на быстрые движения, например мышцы челюстей и глаз, парализуются раньше крупных мышц тела и конечностей. Позже всего парализуется диафрагма и прекращается дыхание. Если поддерживается вентиляция, то проблем не возникает. При выведении препарата восстановление мышечной силы идет в обратном порядке, и диафрагма первой восстанавливает свою функцию. Значительные эффекты названной дозы тубокурарина обычно длятся 30-60 мин, менее очевидные признаки паралича могут сохраняться еще на час. Сила и продолжительность действия других неполяризующих препаратов показаны в табл. 26-1. Кроме длительности действия, другое важное свойство, отличающее недеполяризующие миорелаксанты,— это время до начала эффекта, которое определяет, как скоро можно начинать интубацию. Из используемых или изучаемых в настоящее время недеполяризующих препаратов са-
Без препарата Деполяризующие блокаторы Недеполяризующие блокаторы
Фаза! Фаза II
Четырехи возде мпульсное йствие Постоянная, но сниженная Снижа ющаяся || Снижа ющаяся ||
Ter анус Постоянная, но сниженная Снижа ющаяся Снижа ющаяся
Посттета потенции ническая 1ЦИЯ Отсут ствует Прису гствует Прису тствует
Рис. 26-6. Реакции мышц на разные схемы стимуляции нерва при мониторировании релаксации скелетных мышц. Нарушения, вызванные деполяризующими (деполяризующая и десенсибилизирующая блокада сукцинилхолином) и недеполяризующими миорелаксантами (* — посттетаническое сокращение). (Morgan G. Е., Mikhail М. S. Clinical Anesthesiology. Appleton & Lange, 1992.)
*l~l—
| 4 nA
25 мс
Рис. 26-7. Действие сукцинилхолина на токи одиночного канала концевой пластинки в мышце лягушки. Верхняя запись была сделана в присутствии низких концентраций сукцинилхолина; отклонения книзу соответствуют двум открытиям канала и деполяризующему току внутрь. Нижняя запись была сделана в присутствии значительно больших концентраций сукцинилхолина и показывает длительное “мерцание” канала, когда он постоянно открываетсй или закрывается или когда он “закупорен” препаратом. (Marshall С. G., Ogden D. С., Colquhoun D. The actions of suxamethonium (succinylcholine) as an agonist and channel blocker at the nicotinic receptor of frog muscle. J. Physiol, (bond) 1990; 428:155.)
Галотан (1.25 МАК)
Изофлуран (1.25 МАК)
1 мин
Тубокурарин (3 мг/м2)
I 1 мин Тубокурарин (6 мг/м2)
________ и 1111 щ 111111111 III III
1
23 мин
Рис. 26-8. Нервно-мышечная блокада, вызванная тубокурарином во время анестезии изофлураном и галотаном. Отметьте, что при одинаковых уровнях анестезии изофлуран в большей степени усиливает блокаду, чем галотан
мое быстрое начало действия имеет рокуроний (1-2 мин).
Б. Деполяризующие препараты. После внутривенного введения 0.5-1 мг/кг сукцинилхолина возникают транзиторные фасцикуляции мышц, особенно груди и живота, хотя общая анестезия обычно маскирует этот эффект. В процессе релаксации мышцы рук, шеи и ног расслабляются значительно раньше мышц лица и глотки. За этим следует паралич дыхательной мускулатуры. Начало нервно-мышечной блокады под действием сукцинилхолина очень быстрое, обычно в пределах одной минуты (рис. 26-9). Вследствие быстрого гидролиза холинэстеразой печени и плазмы длительность нервно-мышечной блокады под действием сукцинилхолина обычно не превышает 5-10 минут (табл. 26-1).
Действие на сердечно-сосудистую систему
Векуроний, пипекуроний, доксакуроний и рокуроний почти не действуют на сердечно-сосудистую систему. Все другие используемые в настоящее время недеполяризующие миорелаксанты этими эффектами обладают. Многие из таких эффектов опосредованы гистаминовыми и вегетативными рецепторами (табл. 26-3). Тубокурарин и реже метакурин, мивакурий и атракурий вызывают гипотензию. Эта гипотензия скорее всего опосредована высвобождением гистамина, а при использовании больших доз — блокадой ганглиев. Премедикация антигистаминными препаратами уменьшает гипотензию, вызываемую тубокурарином. Введение панкурония приводит к умеренному учащению сер-
Сукцинилхолин (1 мг/кг)
1 мин 80 % восстановление
Рис. 26-9. Нервно-мышечная блокада под действием сукцинилхолина во время анестезии. Фасцикуляций не видно, так как они маскируются введением анестетика
ТАБЛИЦА 26-3. Действие блокаторов нервно-мышечного проведения на другие ткани
Препарат Действие на вегетативные ганглии Действие на мускариновые рецепторы сердца Тенденция к высвобождению гистамина
Производные изохинолина
Атракурий нет нет слабая
Доксакурий нет нет нет
Метокурин слабая блокада нет слабая
Мивакурий нет • нет слабая
Тубокурарин слабая блокада нет умеренная
Производные стероидов
Панкуроний нет умеренная блокада нет
Пипекуроний нет нет нет
Рокуроний нет нет нет
Векуроний нет нет нет
Другие препараты
Галламин нет сильная блокада нет
Сукцинилхолин стимуляция стимуляция слабая
дечных сокращений и в меньшей степени — к увеличению сердечного выброса почти без изменения периферического сосудистого сопротивления.
Хотя тахикардия прежде всего является следствием вагусной блокады, высвобождение норадреналина из адренергических окончаний и снижение его нейронального захвата также играют свою роль. Галламин повышает частоту сердечных сокращений как за счет ваголитического (антимускаринового) действия, так и вследствие симпатической стимуляции. Симпатическая стимуляция заключается в высвобождении норадреналина из адренергических окончаний в сердце посредством не известных пока механизмов.
Сукцинилхолин может вызывать различные аритмии. Он стимулирует вегетативные холиноре-цепторы: никотиновые в симпатических и парасимпатических ганглиях и мускариновые в синусовом узле сердца. При использовании низких доз возникают отрицательные хроно- и инотропные эффекты, которые снимаются введением атропина. Высокие дозы могут вызывать положительные хроно- и инотропные эффекты. При введении повторной дозы приблизительно через 5 мин после первой часто наблюдается брадикардия. Ее можно предотвратить применением тиопентала, атропина, ганглио-блокаторов и недеполяризующих миорелаксантов. Прямое действие на миокард, повышенная стимуляция мускариновых рецепторов и ганглионарная стимуляция могут приводить к развитию брадикардии.
Другие эффекты
Эти эффекты возникают при применении исключительно деполяризующих миорелаксантов.
А. Гиперкалиемия. Некоторые пациенты реагируют на сукцинилхолин повышенным высвобождением калия в кровь, иногда до такой степени, что возникает остановка сердца. Особенно чувствительны пациенты с ожогами, повреждением нервов или нервно-мышечным заболеванием, закрытой травмой черепа или другой травмой, перитонеальными инфекциями и почечной недостаточностью. Механизм гиперкалиемии неизвестен, но может быть связан с повышенным количеством никотиновых рецепторов, которые расположены вне синапсов на денервированной или неиспользуемой мышце. Эти внесинаптические рецепторы более чувствительны к сукцинилхолину и поэтому более склонны к высвобождению калия, чем синаптические.
Б. Внутриглазное давление. Введение сукцинилхолина приводит к повышению внутриглазного давления, которое проявляется через 1 мин после инъекции, с максимумом на второй-четвертой минуте, и проходит через 5 минут. Механизм этого эффекта в точности не описан, но он может включать сокращение тонических миофибрилл или временное расширение сосудов хороидального сплетения. Несмотря на повышение внутриглазного давления, использование сукцинилхолина при офтальмологических операциях не противопоказано, если не открывать переднюю камеру глаза.
В. Внутрижелудочное давление. У некоторых пациентов, особенно со значительным объемом мышечной ткани, фасцикуляции приведут к повышению внутрижелудочного давления от 5 до 40 см водного столба. Это может вызвать рвоту и опасность аспирации желудочного содержимого.
Г. Мышечные боли. Это основная послеоперационная жалоба у пациентов после применения сукцинилхолина. Из-за субъективных факторов и разных схем исследований реальная частота возникновения этого симптома неизвестна. По разным данным, он возникает у 0.2-20 % пациентов, причем более часто у ходячих больных. Считается, что боль связана с повреждением мышцы, вызванным фасцикуляциями непосредственно перед развитием паралича. Повреждение мышц было подтверждено регистрацией миоглобинурии после применения сукцинилхолина.
Взаимодействие с другими препаратами
А. Анестетики. Ингаляционные анестетики усиливают нервно-мышечную блокаду, вызванную недеполяризующими миорелаксантами, по дозозависимому механизму. Они потенцируют эффект миорелаксантов в таком порядке: изофлуран (рис. 26-8), десфлуран и энфлуран действуют сильнее, чем галотан, который в свою очередь действует сильнее анестезии закисью азота-барбитуратами-безодиазе-пинами-опиоидами. Самые главные факторы этого взаимодействия: 1) торможение структур проксимальнее нервно-мышечного соединения, то есть ЦНС; 2) повышение кровотока в мышце, которое позволяет большей части миорелаксанта попасть в нервно-мышечный синапс (видимо, это справедливо только дли изофлурана); 3) снижение чувствительности постсинаптической мембраны к деполяризации.
Б. Антибиотики. В литературе появилось более 140 сообщений об усилении нервно-мышечной блокады антибиотиками, особенно аминогликозидами. Многие из них, так же как и магнезия, подавляют высвобождение ацетилхолина. Кроме того, они имеют и постсинаптическое действие.
В. Местные анестетики и противоаритмичес-кие препараты. В высоких дозах большинство местных анестетиков блокирует нервно-мышечное проведение, в более низких — усиливает нервно-мышечную блокаду, вызванную как деполяризую
щими, так и недеполяризующими миорелаксантами.
В малых дозах местные анестетики подавляют посттетаническую потенциацию. Считается, что это связано с пресинаптическим эффектом. В более высоких дозах они блокируют сокращения мышц, вызванные ацетилхолином. Этот эффект вызван инактивацией никотиновых рецепторов ионных каналов.
В эксперименте сходным действием обладают противоаритмические препараты, например хинидин. Однако это почти не имеет клинического значения.
Г. Другие блокаторы нервно-мышечного проведения. Действию деполяризующих миорелаксантов препятствует действие недеполяризующих. Для предотвращения фасцикуляций, связанных с применением сукцинилхолина, предварительно вводится малая непарализующая доза тубокурарина. Это уменьшает фасцикуляции и послеоперационные боли, но повышает потребность в сукцинилхо-лине на 50-90 %.
Влияние сопутствующих заболеваний и возраста
Некоторые заболевания могут изменить действие недеполяризующих миорелаксантов. Миастения значительно усиливает нервно-мышечную блокаду, вызванную этими препаратами. В пожилом возрасте наблюдается более длительное их действие, возможно, вследствие замедленного клиренса печенью и почками. В результате доза блокаторов нервно-мышечной передачи должна быть снижена.
Наоборот, пациенты с тяжелыми ожогами и боковым амиотрофическим склерозом резистентны к действию недеполяризующих миорелаксантов. Это, видимо, связано с пролиферацией внесинаптичес-ких рецепторов, что требует дополнительного количества препарата.
Обратимость недеполяризующей блокады нервно-мышечного проведения
Недеполяризующим препаратам эффективно противодействуют ингибиторы холинэстеразы. Их общая фармакология обсуждается в главе 7. Нео-стигмин и пиридостигмин препятствуют недеполяризующей нервно-мышечной блокаде, повышая концентрации ацетилхолина на концевой пластинке в основном за счет ингибирования холинэстера
зы. В меньшей степени эти препараты увеличивают высвобождение медиатора из двигательных нервных окончаний. В отличие от этого эдрофоний противодействует недеполяризующей блокаде только за счет ингибирования холинэстеразы.
Неостигмин также понижает активность холинэстеразы плазмы. Так как мивакурий метаболизируется ею, взаимодействие этих препаратов непредсказуемо. С одной стороны, ослабление нервно-мышечной блокады происходит вследствие увеличения концентрации ацетилхолина в синапсе. С другой — концентрации мивакурия могут быть повышены из-за снижения активности холинэстеразы плазмы. Первый эффект обычно клинически доминирует.
Другие показания к применению блокаторов нервно-мышечного проведения
А. Контроль вентиляции. У пациентов с нарушениями вентиляции в связи с серьезными причинами, например при обструктивных заболеваниях легких, желательно контролировать ее объем для расправления легких. Иногда введением блокаторов нервно-мышечного проведения индуцируется паралич мышц грудной клетки для снятия их сопротивления и прекращения неэффективной спонтанной вентиляции.
Б. Лечение судорог. Блокаторы нервно-мышечного проведения можно использовать для снятия двигательного компонента периферических проявлений судорог, возникших вследствие эпилепсии или токсичности местных анестетиков. Хотя этот подход эффективен в коррекции периферического компонента, он не действует на центральные процессы, так как блокаторы нервно-мышечного проведения в терапевтических дозах почти не проникают через гематоэнцефалический барьер.
Спазмолитики
Бертрам Дж. Катцунг
Спастичность характеризуется усилением тонических рефлексов на растяжение и спазмами мышц сгибателей в сочетании с мышечной слабостью. Она часто связана с центральным параличом, рассеянным склерозом и инсультом. В сочетании с этими
проявлениями могут возникать нарушения функции тазовых органов. В этом разделе обсуждается только спастичность скелетных мышц. Механизмы развития спастичности включают не саму дугу рефлекса на растяжение, а вышерасположенные центры (верхние мотонейроны) с повреждением нисходящих путей, приводящим к гипервозбудимости ос-мотонейронов спинного мозга. Несмотря на это, медикаментозная терапия может облегчить некоторые из симптомов спастичности, воздействуя на рефлекторную дугу рефлекса растяжения или непосредственно на механизмы сопряжения возбуждения и сокращения в скелетной мышце. Компоненты этих процессов показаны на рис. 26-10.
Препараты, воздействующие на рефлекторную дугу, могут изменять работу возбуждающих или тормозных синапсов (глава 20). Таким образом, чтобы снизить гиперактивность рефлекса на растяжение, необходимо сократить активность 1а-волокон, которые возбуждают мотонейрон, или повысить активность тормозных вставочных нейронов. Эти структуры детально изображены на рис. 26-11.
Ряд препаратов, использовавшихся в прошлом, можно назвать депрессантами спинальных полиси-наптических рефлексов (барбитураты: фенобарбитал, эфиры глицерина: мефенезин).
Препараты сходного типа внедряют и сегодня (препараты, применяемые при острых мышечных спазмах). Однако из рис. 26-11 понятно, что неспецифическая депрессия синапсов в дуге рефлекса на растяжение может снизить не только возбуждающие импульсы, но и тормозящие. За два последних десятилетия появилась более специфическая терапия. К сожалению, нет методов, точно и количественно определяющих реакцию как у пациентов, так и на экспериментальной модели, и это не позволяет сравнить эффективность этих препаратов при лечении такой гетерогенной группы состояний.
Диазепам
Как рассмотрено в главе 21, бензодиазепины облегчают действие ГАМК на ЦНС. Диазепам обладает полезным антиспастическим действием. Он, очевидно, действует на ГАМК-синапсы, но место его действия, которое, по крайней мере частично, отвечает за его противоспастическое действие, находится в спинном мозгу, так как он эффективен у пациентов с пересечением спинного мозга. Диазепам можно применять при мышечных спазмах почти любого
Кожные
афферен
ожа
Спинной ^юзг
ентральные г-корешки экстрафузальные эфференты
Веретено
Фузимоторные эфференты
1а-афференты Дорзальный корешок
Миозин 1 Актин
Рис. 26-10. Схема структур, составляющих дугу рефлекса на растяжение. I — тормозный вставочный нейрон; Е — возбуждающая пресинаптическая терминаль; 1а — первичное интрафузальное афферентное волокно; Са2+ обозначает активацию кальция, хранящегося в саркоплазматическом ретикулуме скелетной мышцы. (Young R. R., Delwaide Р. J. Drug therapy: Spasticity. N. Engl. J. Med. 1981; 304:28.)
Мышца
Рис. 26-11. Предполагаемые места спазмолитического действия диазепама и баклофена в спинном мозгу. (Young R. R., Delwaide Р. J. Drug therapy: Spasticity. N. Engl. J. Med. 1981; 304:28.)
генеза, в том числе после местной мышечной травмы. Однако он вызывает седативный эффект у большинства пациентов при применении в дозах, необходимых для снижения мышечного тонуса. Обычно начинают прием с 4 мг/день и постепенно повышают до максимальной дозы — 60 мг/день.
Баклофен
Баклофен (р-хлорфенилГАМК) был разработан как активный при пероральном приеме ГАМК-миметик. Он является эффективным спазмолитиком и действует как агонист ГАМКв-рецепторов. Их активация в ЦНС приводит к гиперполяризации, вероятно, за счет повышения проводимости для К+. Предполагают, что эта гиперполяризация (как в головном, так и в спинном мозгу) вызывает пресинаптическое торможение (рис. 26-11) через снижение тока ионов кальция с последующим уменьшением высвобождения возбуждающих нейромедиаторов. Баклофен также может снижать болевые ощущения у пациентов со спастичностью,
СН- СН2- NH2 СНг-СООН
Баклофен
видимо, за счет подавления высвобождения субстанции Р в спинном мозгу. В этом отношении препарат не менее эффективен, чем диазепам, но вызывает значительно меньший седативный эффект. Кроме того, он не уменьшает мышечную силу, как дантролен. Баклофен быстро и полностью всасывается при пероральном приеме и имеет период полувыведения 3-4 часа. Прием начинают с 15 мг 2 раза в день и повышают (при хорошей переносимости) до 100 мг в день. К побочным эффектам баклофена относится сонливость, к которой постепенно развивается толерантность. У больных эпилепсией была выявлена повышенная судорожная активность.
Двойные слепые краткосрочные и открытые длительные исследования с использованием имплантированных программированных инфузионных помп показали, что интратекалъное введение баклофена может контролировать тяжелую спастичность и облегчать боль, которые не поддаются терапии при пероральном или других парентеральных способах введения препарата. Вследствие плохого выхода баклофена из спинного мозга в этом случае периферических эффектов почти не возникает и большие дозы могут переноситься без осложнений. Несмотря на прекрасный эффект такого введения баклофена, известно несколько случаев сом-нолентных состояний, респираторной депрессии и даже комы. Использование физостигмина, который считается эффективным средством для выведения больного из подобных состояний, само по себе может быть более опасным и менее действенным, чем консервативное лечение.
Другие препараты, действующие на спинной мозг
Предварительные исследования показали, что прогабид и глицин снижают спастичность у некоторых пациентов. Прогабид — это агонист ГАМКЛ-и ГАМКв-рецепторов, одним из его активных метаболитов является сама ГАМК. Глицин — это другой тормозный медиатор (глава 20). Он действует при пероральном приеме и хорошо проходит через гематоэнцефалический барьер. Эффективность этих препаратов сейчас исследуется. Идроциламид и тизанидин — самые новые препараты, возможность использования которых также изучается в настоящий момент.
Дантролен
Дантролен — это дериват гидантоина (как фенитоин) с уникальным механизмом противоспасти-ческого действия вне ЦНС. Его структура показана ниже.
Дантролен снижает силу скелетных мышц, воздействуя на сопряжение возбуждения и сокращения в мышечном волокне. Нормальный сократительный ответ предполагает высвобождение ионов кальция из саркоплазматического ретикулума саркомера. Са2+ вызывает взаимодействие актина и миозина. Дантролен нарушает высвобождение Са2+. Таким образом, он действует не на центральные синапсы, а на внутриклеточные структуры эффекторных органов. Двигательные единицы, которые быстро сокращаются, более чувствительны к действию препарата, чем медленные. Сердечная и гладкие мышцы очень слабо поддаются этому эффекту, возможно потому, что высвобождение в них ионов кальция происходит по другому механизму.
Только около пероральной дозы дантролена абсорбируется, период его полувыведения равен 8 часам. Лечение начинают с 25 мг в день за один прием, постепенно повышая до 100 мг 4 раза в день в зависимости от необходимости и переносимости. Основными побочными эффектами являются генерализованная мышечная слабость, седация и иногда гепатит.
Особое показание к применению дантролена — злокачественная гипертермия, редкое нарушение, которое может быть спровоцировано рядом причин, в том числе общей анестезией и блокаторами нервно-мышечной передачи. У таких больных существует наследственное нарушение способности саркоплазматического ретикулума секвестрировать Са2+. Триггерные стимулы приводят к внезапному и длительному высвобождению Са2+, сопровождающемуся массивными сокращениями мышц, образованием молочной кислоты и повышением температуры тела. Лучшее лечение — это контроль ацидоза и температуры тела, а также ограничение высвобождения ионов кальция. Последнего можно добиться
внутривенным введением дантролена, начиная с 1 мг/кг, при необходимости повторяя инъекции до достижения максимума (10 мг/кг).
Препараты, применяемые при острых мышечных спазмах
Большое количество препаратов предлагается для снятия спазма мышц, вызванного напряжением или местной травмой. Большинство из них действуют как седативные средства на уровне спинного мозга или ствола мозга. Циклобензаприн — это прототип последней группы, который по структуре похож на трициклические антидепрессанты и имеет некоторые схожие с ними свойства, например антимускариновые эффекты. Считается, что он действует на уровне ствола мозга. Циклобензаприн эффективен при мышечных спазмах, развивающихся на фоне церебрального паралича или травмы спинного мозга. Препарат имеет сильный антимускари-новый эффект, и у большинства пациентов вызывает значительную седацию, нарушения сознания, а у некоторых — преходящие зрительные галлюцинации. Доза препарата при остром спазме вследствие травмы равна 20-40 мг/день в три приема.
Препараты
Препараты, блокирующие нервно-мышечную передачу Атракурий (Тракриум)
Парентерально: 10 мг/мл для инъекций
Доксакурий (Нуромакс)
Парентерально: 1 мг/мл для в/в инъекций
Галламин (Флакседил)
Парентерально: 20 мг/мл для инъекций
Метокурин (Метубина иод ид, генерик)
Парентерально: 2 мг/мл для инъекций
Мивакурий (Мивакрон)
Парентерально: 0.5, 2 мг/мл для инъекций
Панкуроний (Павулон)
Парентерально: 1, 2 мг/мл для инъекций
Пипекуроний (Ардуан)
Парентерально: 1 мг/мл для в/в инъекций
Рокуроний(Земурон)
Парентерально: 10 мг/мл для в/в инъекций
Сукцинилхолин (Анектин, др.)
Парентерально: 20, 50, 100 мг/мл для инъекций; 100,500 мг порошок во флаконах для инъекций
Тубокукарин (генерик)
Парентерально: 3 мг (20 единиц)/мл для инъекций
Векуроний (Норкурон)
Парентерально: 10 мг/мл порошок (для инъекций растворить)
Мышечные релаксанты (спазмолитики)
Баклофен (Лиорезал, генерик)
Перорально: таблетки по 10, 20 мг Интратекально: ампулы по 10 мг/20 мл, 10 мг/5 мл
Циклобензаприн (Флексерил, генерик)
Перорально: таблетки по 10 мг
Дантролен (Дантриум)
Перорально: капсулы по 25, 50, 100 мг
Парентерально: порошок по 20 мг во флаконах для инъекций
Диазепам (генерик, Валиум, др.)
Перорально: таблетки по 2,5,10 мг; капсулы пролонгированного действия по 15 мг;
растворы 5 мг/5 мл, 5 мг/мл
Парентерально: 5 мг/мл для инъекций
Избранная литература
Agoston S. et al. Clinical pharmacokinetics of neuromuscular blocking drugs. Clin. Pharmacokinet. 1992; 22: 94.
Davidoff R. A. Antispasticity drugs: Mechanisms of action. Ann. Neurol. 1985; 17:107.
Miller R. D., Savarese J. J. Pharmacology of muscle relaxants and their antagonists. In: Anesthesia, 4th ed. Miller R. D. (ed.) Churchill Livingstone, 1994.
Ochs G. et al. Intrathecal baclofen for long-term treatment of spasticity. A multi-centre study. J. Neurol. Neurosurg. Psychiat. 1989; 52:933.
Penn R. D. et al. Intrathecal baclofen for severe spinal spasticity. N. Engl. J. Med. 1989; 320:1517.
Young R. R„ Weigner A. W. Spasticity. Clin. Orthop. 1987; 219:50.
Фармакотерапия паркинсонизма и других двигательных нарушений
27
Майкл Дж. Аминофф
Существует несколько типов аномальных движений. Тремор представляет собой ритмические колебательные движения вокруг сустава и лучше всего характеризуется по его соотношению с активными движениями. Тремор покоя характерен для паркинсонизма и часто сопровождается ригидностью и нарушением произвольных движений. Тремор может возникать и при поддержании определенной позы (постуральный, позиционный тремор), и при движении (интенционный тремор). Сильный позиционный тремор — это основной признак так называемого доброкачественного эссенциального, или семейного, тремора. Интенционный тремор возникает у пациентов с поражениями ствола мозга или мозжечка, особенно затрагивающими верхнюю ножку мозжечка, а также может быть проявлением алкогольной или лекарственной интоксикации.
Хорея представляет собой нерегулярные, непредсказуемые, непроизвольные движения, которые возникают в разных частях тела и нарушают произвольную двигательную активность. В некоторых случаях в процесс вовлечены проксимальные мышцы конечностей и, так как непроизвольные движения очень размашисты, для их описания используют термин баллизм. Хорея может быть наследственной болезнью, осложнением некоторых других заболеваний, следствием применения ряда препаратов.
Аномальные движения бывают медленными и червеобразными (атетоз), а иногда настолько продолжительными, что их правильнее называть аномальными позами (дистонии). Атетоз или дистония возникают вследствие перинатальной патологии мозга, локальных или генерализованных поражений ЦНС, острых осложнений после приема некоторых препаратов. Кроме того, они могут быть проявлениями сложных неврологических заболеваний
или выступать в качестве изолированного феномена неизвестного генеза, например идиопатической торсионной дистонии, или dystonia musculorum deformans, не поддающейся лечению.
Тик — это внезапные координированные аномальные движения, которые возникают сериями, особенно на лице и голове, чаще у детей, и которые можно подавить усилием воли на короткие промежутки времени. Часто встречаются, например, “по-морщивания” носа или подергивания плечами. Тики могут быть единичными или множественными, преходящими или хроническими. Синдром Жиля де ла Туретта характеризуется хроническими множественными тиками, его фармакотерапия обсуждается в конце главы.
Многие виды двигательных нарушений связывают с поражениями базальных ганглиев, но точная функция этих анатомических структур еще не полностью выяснена, и невозможно связать конкретные симптомы с какой-либо точной локализацией поражения.
Паркинсонизм (Paralysis agitans)
Паркинсонизм характеризуется комбинацией ригидности, брадикинезии, тремора и нестабильности позы, возникающих по многим причинам, но чаще — идиопатически. Патофизиологические основы идиопатического паркинсонизма могут быть связаны с контактом с каким-либо неизвестным нейротоксином или с образованием свободных радикалов в реакциях окисления. Болезнь Паркинсона при отсутствии эффективного лечения обычно быстро прогрессирует. Высокая в норме концентрация дофамина в базальных ганглиях мозга при паркинсонизме снижена. Фармакологические по
пытки восстановить дофаминергическую активность препаратами типа леводопы и агонистов дофамина успешно ослабляют многие клинические проявления этого заболевания. Альтернативный подход к лечению — использование м-холинолити-ков для восстановления нормального баланса холинергических и дофаминергических воздействий на базальные ганглии. Патофизиологические основы этого метода заключаются в том, что при идиопатическом паркинсонизме дофаминергические нейроны черной субстанции, которые в норме подавляют действие ГАМК-ергических клеток в полосатом теле, утрачены (рис. 27-1). (В отличие от этого при хорее Гентингтона исчезают некоторые холинергические нейроны и в еще большей степени — ГАМК-ергические клетки полосатого тела.) Препараты, которые вызывают паркинсонические синдромы, либо являются антагонистами дофаминовых рецепторов (например, антипсихотические препараты; глава 28), либо ведут к деструкции до-
Норма
Черная Полосатое
субстанция тело
।| ।Ацетил-| ГАМК холин
Паркинсонизм
Болезнь Гентингтона
Рис. 27-1. Схематическое изображение последовательности нейронов, нарушающейся при паркинсонизме и хорее Гентингтона. Вверху: Дофаминергические нейроны черной субстанции в норме подавляют ГАМК-ергические импульсы из полосатого тела, в то время как холинергические нейроны оказывают возбуждающий эффект. В середине: При паркинсонизме возникает селективная потеря дофаминергических нейронов. Внизу: При хорее Гентингтона могут быть утеряны некоторые холинергические нейроны, хотя доминирует дегенерация ГАМК-ергических нейронов
фаминергических нигростриатных нейронов (например, МРТР).
Леводопа
Дофамин не проходит через гематоэнцефалический барьер, поэтому при попадании в периферическую циркуляцию не оказывает никакого эффекта при паркинсонизме. Однако в мозг проникает непосредственный метаболический предшественник дофамина — (-)-3-(3,4-дигидроксифенил)-Ь-ала-нин (леводопа), который декарбоксилируется там до дофамина. Синтезировано несколько агонистов дофамина, которые также могут давать хороший эффект, что еще будет обсуждаться в этой главе.
Дофаминовые рецепторы классифицируются в зависимости от их биохимических и фармакологических свойств. Например, некоторые из них способны связываться с аденилатциклазой. При активации этого фермента образуется цАМФ, возможно, являющийся медиатором в реализации ряда (но не всех) специфических эффектов дофамина. Рецепторы, активирующие аденилатциклазу, предложили называть Db а не стимулирующие — D2 (табл. 6-2).
Di-дофаминовые рецепторы расположены в компактной зоне черной субстанции и пресинапти-чески на стриатных аксонах, идущих от кортикальных нейронов и от дофаминергических клеток черной субстанции. Da-рецепторы расположены постсинаптически на стриатных нейронах и пресинап-тически на аксонах черной субстанции, которые принадлежат нейронам базальных ганглиев. Эффективность антипаркинсонических дофаминергических препаратов зависит от стимуляции ими Da-рецепторов, однако для развития максимального эффекта может потребоваться и стимуляция D,-рецепторов. Так, антипаркинсонические свойства имеют агонисты дофамина и парциальные агонисты дериваты алкалоидов спорыньи, например лерготрил и бромокриптин, которые являются сильными стимуляторами Da-рецепторов. Напротив, некоторые блокаторы дофаминовых рецепторов, являющиеся селективными Da-антагонистами, могут вызывать паркинсонизм.
Химические свойства
Напомним, что ДОФА — предшественник дофамина и норадреналина (глава 6). Его структура по
казана на рис. 27-2. Леводопа — это левовращающий стереоизомер ДОФА.
Фармакокинетика
Леводопа быстро всасывается из тонкого кишечника, но его абсорбция зависит от скорости освобождения желудка и от pH в нем. Наличие пищи в желудке может отсрочить появление леводопы в плазме. Более того, некоторые аминокислоты пищи могут конкурировать с препаратом за абсорбцию из кишечника и за транспорт из крови в мозг. Пик концентраций в плазме обычно возникает через 1-2 часа после перорального приема лекарства, период полувыведения чаще всего находится между 1 и 3 часами с некоторыми вариациями у разных людей. Около 2/з дозы появляется в моче как метаболиты в течение восьми часов после приема. Основные метаболиты леводопы — З-метокси-4-гидроксифенил-уксусная кислота (гомованилиновая кислота) и дигидроксифенилуксусная кислота. К сожалению, только около 1-3 % принятого препарата попадает в мозг, а остальная часть метаболизируется экстра-церебрально, в основном декарбоксилированием до дофамина, который не проходит через гематоэнцефалический барьер. Это означает, что для получе-
Дигидроксифенилаланин (ДОФА)
Карбидопа
СН2 I
СН2— СН — N — СНг- С ж СН
СН3
Селегилин (депренил)
Рис. 27-2. Некоторые препараты, применяемые при лечении паркинсонизма
ния эффекта леводопу надо давать в больших количествах. Однако при сочетании леводопы с ингибитором ДОФА-декарбоксилазы, который не проходит через гематоэнцефалический барьер, периферический метаболизм ДОФА замедляется, уровень леводопы в плазме повышается, период полувыведения увеличивается, большее количество ДОФА попадает в мозг (рис. 27-3). Сочетание леводопы и ингибитора периферической ДОФА-декарбоксилазы снижает потребность в леводопе приблизительно на 75 %.
Клиническое применение
При назначении леводопы больным паркинсонизмом необходимо знать, что препарат наиболее эффективен в первые несколько лет приема. Иногда со временем приходится снижать дозу из-за побочных эффектов, развивающихся при приеме таких доз, которые ранее переносились хорошо. Причины этого явления не вполне понятны, но возможна селективная денервация или вызванная препаратом гиперчувствительность. У некоторых пациентов снижается чувствительность к леводопе, и эффективные ранее дозы уже не оказывают желаемого действия. Непонятно, связано ли это с прогрессированием заболевания или с длительностью лечения, хотя накапливаются данные о том, что первая причина наиболее вероятна. Чувствительность к леводопе может быть в конце концов утеряна полностью, возможно, из-за потери дофаминергических нигростриатных нервных терминалей или из-за другого патологического процесса, непосредственно затрагивающего дофаминовые рецепторы полосатого тела. Вследствие этих причин эффективность леводопы часто снижается после 3-4 лет терапии вне зависимости от исходной. Хотя лечение леводопой не тормозит прогрессирования паркинсонизма, полученные недавно данные доказывают, что раннее начало терапии снижает смертность из-за болезни Паркинсона. Однако длительная терапия может привести к ряду проблем, например развитию “on-off” феномена, который рассматривается в подразделе “Побочные эффекты”. Следовательно, время начала терапии леводопой надо определять индивидуально.
По ранее приведенным причинам леводопу обычно назначают вместе с карбид опой (рис. 27-2), ингибитором периферической ДОФА-декарбоксилазы. Синемет — это препарат ДОФА, содержащий
Только леводопа
Леводопа с карбидопой
Рис. 27-3. Судьба леводопы при приеме внутрь и влияние на нее карбидопы (исследования на животных). Ширина каждой стрелки соответствует абсолютному количеству препарата в компартменте, а проценты показывают часть принятой дозы. Эффект добавления карбидопы заключается в сокращении количества лево допы, оставшейся в периферических тканях, и в увеличении фракции, попадающей в мозг (данные Nutt и Fellman, 1984.)
100% 60% 10%
Метаболизм Периферические в ЖКТ ткани (токсичность)
карбидопу и леводопу в фиксированном соотношении (1 : 10 или 1 : 4). Терапию начинают с маленькой дозы, назначая синемет-25/100 (карбидопы 25 мг, леводопы 100 мг) 3 раза в день. В зависимости от терапевтической эффективности и проявления побочных эффектов постепенно повышают дозу. Большинство пациентов в конце концов переходят на синемет-25/250 (карбидопы 25 мг, леводопы 250 мг) 3 или 4 раза в день. Возможно, предпочтительнее синемет-25/100 принимать 3 раза в день, повышая дофаминергическую активность назначением при необходимости агониста дофамина, что помогает снизить колебания эффекта препарата, сейчас с этой целью используются пролонгированные препараты синемета, которые позволяют также снизить кратность приема.
Леводопа может помогать при всех симптомах паркинсонизма, но только частично уменьшает бра-дикинезию и ее последствия. При первичном назна
чении препарат высоко эффективен у */3 пациентов и менее эффективен еще у '/3. Остальные либо не переносят препарат, либо не испытывают никакого эффекта.
Побочные эффекты
А. Желудочно-кишечный тракт. При приеме леводопы без ингибитора ДОФА-декарбоксилазы у 80 % пациентов возникают анорексия, тошнота и рвота. Эти побочные эффекты можно свести до минимума, принимая препарат часто, но маленькими дозами, во время или сразу после еды и повышая дозу препарата очень медленно. Может помочь также прием антацидов за 30-60 мин до приема леводопы. Рвоту объясняют стимуляцией дофамином и ингибиторами периферической декарбоксилазы рвотного центра ствола мозга, находящегося снаружи от гематоэнцефалического барьера. К сча
стью, через несколько месяцев к рвотному эффекту развивается толерантность. Иногда назначаются противорвотные препараты типа фенотиазинов, но они могут снизить и противопаркинсонический эффект леводопы.
При приеме леводопы в комбинации с карбидо-пой нежелательное воздействие на желудочно-кишечный тракт встречается реже (только у 20 % пациентов) и менее выражено.
Б. Влияние на сердечно-сосудистую систему. У пациентов, принимающих леводопу, описаны аритмии, в том числе тахикардии, желудочковые экстрасистолы и редко — фибрилляции. Это объясняют усилением периферического образования катехоламинов. Однако такие аритмии возникают редко даже при наличии заболеваний сердца, риск их возникновения можно снизить использованием ингибитора периферической декарбоксилазы. У пациентов с паркинсонизмом и с заболеваниями сердца польза леводопы значительно перевешивает небольшой риск развития аритмии.
Часто встречается ортостатическая гипотензия, которая протекает в основном бессимптомно и нивелируется в ходе терапии. Возможна также гипертензия, особенно при приеме очень высоких доз леводопы или при одновременном приеме леводопы и неселективных ингибиторов моноаминокси-дазы или симпатомиметиков.
В. Дискинезии. Дискинезии возникают почти у 80 % пациентов, принимающих леводопу в течение длительного времени. Развитие дискинезий дозозависимо, но существуют значительные индивидуальные особенности, определяющие пороговую дозу их возникновения. Чаще страдают пациенты, которые получают леводопу в сочетании с ингибитором периферической декарбоксилазы. Дискинезии могут возникать в процессе лечения при приеме таких доз, с которыми ранее проблем не было. Проблему можно решить, снижая дозу леводопы, но часто при этом усиливаются симптомы паркинсонизма; многие пациенты предпочитают переносить даже выраженные дискинезии, если препарат помогает восстановлению нормальных движений. В некоторых случаях дискинезии менее выражены, если леводопа принимается без карбидопы, в других помогают справиться с этой проблемой “каникулы” (то есть перерыв в приеме препарата). Фармакологические попытки воздействовать на дискинезии при помощи препаратов, используемых для лечения хореи,
не дают желаемых результатов или даже усугубляют паркинсонизм.
Тип вызванных леводопой дискинезий варьирует у разных пациентов, но имеет характерные постоянные особенности у каждого отдельного больного. Хорея, баллизм, атетоз, дистония, миоклонус, тики и тремор могут возникать поодиночке или в любых комбинациях на лице, теле или конечностях. Чаще всего встречается хореоатетоз лица и дистальных отделов конечностей.
Г. Действие на поведение. Есть сообщения о разнообразных нарушениях мышления и психики при применении леводопы. Это депрессии, тревожность, психическое возбуждение, нарушение сознания, бред, галлюцинации, ночные кошмары, эйфория и другие изменения настроения или личности. Такие побочные эффекты чаще встречаются у пациентов, принимающих леводопу вместе с ингибитором ДОФА-декарбоксилазы скорее всего потому, что при таком приеме достигаются более высокие уровни препарата в мозгу. Эти эффекты могут усугубляться сопутствующим заболеванием или операцией. Единственное лечение — это снижение дозы или даже отмена препарата. “Каникулы” в приеме лекарства могут ослабить неблагоприятное воздействие на психику.
Д. Колебания реакции на препарат. Определенные колебания в клиническом эффекте препарата могут учащаться в ходе лечения. У некоторых пациентов они связаны с длительностью приема леводопы, и их называют реакциями снижения (истощения), или акинезиями конца дозы. У других больных колебания эффекта не связаны с длительностью приема препарата (феномен “on-off’)- В последнем случае “off" периоды акинезии чередуются в пределах нескольких часов с “on” периодами хорошей подвижности, но часто с дискинезиями. Этот феномен чаще возникает у пациентов, которые первоначально хорошо поддавались лечению. Точный механизм его развития неизвестен, но было показано, что “off” периоды иногда возникают во время падения уровня леводопы в плазме. Чтобы избежать этого, можно попробовать принимать препарат более часто маленькими дозами в течение дня или добавить к схеме лечения агонист дофамина (например, бромокриптин). Может помочь и снижение потребления пищевого белка до необходимого минимального уровня, а также составление режима питания, при котором белковые продукты употребляются только вечером. Хороший эффект оказы
вает пролонгированный синемет. Короткий отдых от леводопы (“каникулы”) иногда помогает повысить эффективность лечения, но гарантий этот метод не дает, так как наступает только временное улучшение.
Е. Смешанные побочные эффекты. При приеме леводопы возможен мидриаз, вызывающий у некоторых пациентов острый приступ глаукомы. К другим редким побочным эффектам относятся различные гематологические нарушения, положительная реакция Кумбса с признаками гемолиза, приливы, обострение подагры, нарушения вкуса или обоняния, коричневатый цвет слюны, мочи или вагинального секрета, приапизм, легкое, обычно временное, повышение уровня азота мочевины, билирубина, активности щелочной фосфатазы и трансаминаз крови.
Лекарственные “каникулы”
Лекарственные “каникулы” могут ослабить некоторые поведенческие и неврологические побочные эффекты леводопы, но почти не влияют на “on-off” феномен. Пациенты в это время должны быть под наблюдением врача. Прием прекращают постепенно, так как внезапный отказ от препарата может вызвать тяжелую акинезию. По разным наблюдениям, оптимальная длительность перерыва в лечении варьирует от 3 до 21 дня. Почти 2/3 пациентов отмечают увеличение эффекта леводопы при последующем возобновлении лечения. Так как в этом случае назначают меньшие дозы леводопы, побочные эффекты препарата менее выражены. После перерыва леводопу начинают принимать либо с половинной дозы, либо с еще меньшей с постепенным подъемом дозы до половинной. Колебания реакции на препарат (“on-off” феномен) во многих случаях уменьшаются, но только временно. К сожалению, невозможно заранее предсказать реакцию пациента на такие “каникулы”. Более того, перерыв в терапии связан с повышенным риском аспирационной пневмонии, венозного тромбоза, легочной эмболии и депрессии в связи с плохой подвижностью при тяжелом паркинсонизме.
Лекарственные взаимодействия
Фармакологические дозы пиридоксина (витамина В6) усиливают мозговой метаболизм леводопы и поэтому могут ограничить терапевтические эф
фекты препарата, если в его состав не входит ингибитор ДОФ А-декарбоксилазы. Леводопу не следует назначать во время приема ингибиторов моно-аминоксидазы А или в течение двух недель после его прекращения, так как такое сочетание может привести к гипертоническому кризу.
Противопоказания
Препарат противопоказан больным в состоянии психоза, так как может ухудшить их психическое состояние. Его также нельзя назначать пациентам с закрытоугольной глаукомой, но можно — при хронической открытоугольной глаукоме, если внутриглазное давление находится под постоянным контролем. Пациентам с заболеваниями сердца, несмотря на небольшой риск аритмий, леводопу следует давать только в сочетании с карбидопой. Необходимо наблюдать и за больными с активной язвенной болезнью, так как известны случаи желудочно-кишечных кровотечений при приеме леводопы. Поскольку леводопа является предшественником меланина кожи и предположительно может активизировать меланому, не следует выписывать этот препарат при наличии меланомы или подозрительных недиагносцированных поражениях кожи в анамнезе.
Агонисты дофамина
В мозгу больных паркинсонизмом истощены ферменты, отвечающие за синтез дофамина, поэтому препараты, напрямую действующие на дофаминовые рецепторы, могут эффективно дополнять действие леводопы. В отличие от леводопы эти препараты не требуют ферментативной конверсии до активного метаболита, не имеют потенциально токсических метаболитов и не конкурируют с другими веществами за транспорт в крови и за проникновение через гематоэнцефалический барьер. Более того, препараты, специфически действующие на определенные (но не все) дофаминовые рецепторы, имеют меньше побочных эффектов, чем леводопа. Существует несколько агонистов дофамина с антипаркинсонической активностью. Некоторые, например апоморфин, пирибедил и лерготрил, обладают настолько серьезными побочными эффектами, что их нельзя использовать в клинике, но применение других агонистов дофамина вполне возможно.
Бромокриптин
Для лечения паркинсонизма используются некоторые дериваты алкалоидов спорыньи, являющиеся агонистами дофамина (глава 16). В отличие от других дофаминоподобных препаратов они являются парциальными агонистами пресинаптичес-ких дофаминовых В2-рецепторов. Чаще всего в США применяется бромокриптин, его структура показана в табл. 16-5. Его используют также при определенных эндокринологических заболеваниях, прежде всего гиперпролактинемии (главы 16 и 36), но в меньших дозах, чем при лечении больных паркинсонизмом. Препарат в разной степени всасывается в желудочно-кишечном тракте, достигает пикового уровня в плазме в пределах 1-2 часов после перорального приема, выделяется с желчью и калом.
По некоторым данным, бромокриптин может быть препаратом первой очереди при лечении паркинсонизма, так как его использование приводит к меньшему количеству колебаний эффективности и дискинезий, чем при длительном применении леводопы. Терапию можно начать с низкой дозы кар-бидопы и леводопы (например, синеметом 25/100 три раза в день), а затем добавить бромокриптин. Однако некоторые врачи назначают бромокриптин только тем пациентам, у которых в связи с применением леводопы возникли акинезии конца дозы, феномен “on-off” или нарастает рефрактерность к лечению. Может быть эффективным частичное замещение леводопы бромокриптином. Оптимальные результаты получают при сочетании леводопы и бромокриптина в субмаксимальных для каждого препарата дозах. Применение бромокриптина у пациентов, резистентных к леводопе, обычно не дает результатов, а назначение бромокриптина пациентам, получающим оптимальную терапию леводопой, может вызвать серьезные побочные эффекты. При совместном применении с м-холинолитиками или амантадином бромокриптин не вызывает осложнений; эти препараты можно продолжать принимать в прежних дозах.
Клиническое применение
Обычная ежедневная доза бромокриптина при паркинсонизме составляет от 7.5 до 30 мг, но у некоторых пациентов она не вызывает длительного эффекта или плохо переносится. Для минимизации побочных эффектов рекомендуется медленно повы
шать дозу препарата в течение 2-3 месяцев от начального уровня (1.25 мг два раза в день после еды) на 2.5 мг каждые две недели в зависимости от эффективности и переносимости. Доза одновременно принимаемой леводопы должна быть при этом постепенно снижена до половинного уровня по сравнению с дозой до начала приема бромокриптина. Так как некоторые пациенты очень чувствительны к гипотензивному эффекту бромокриптина и могут даже потерять сознание в пределах часа после приема первой дозы, до начала лечения должна назначаться проверочная доза (1 мг), которую следует принимать после еды, лежа в постели. После возникновения сосудистого коллапса пациенты обычно приходят в себя очень быстро и часто хорошо переносят последующие дозы. Лечение бромокриптином следует прекратить, если он приводит к психическим нарушениям, аритмиям, эритромелалгиям, эрготизму или другим неприемлемым побочным эффектам.
Побочные эффекты
А. Желудочно-кишечный тракт. При применении бромокриптина особенно часто возникают анорексия, тошнота и рвота; их можно уменьшить при приеме препарата с едой. Могут появляться запоры, диспепсия, симптомы рефлюкс-эзофагита. Известны случаи кровотечения из язв.
Б. Сердечно-сосудистая система. Достаточно часто, особенно в начале терапии, возникает ортостатическая гипотензия. Безболезненный вазоспазм пальцев — это осложнение длительного лечения, оно обратимо при снижении дозы препарата. Возможные аритмии являются показанием к прекращению приема препарата.
В. Дискинезии. Аномальные движения, сходные с таковыми при приеме леводопы, могут вызываться и бромокриптином. Лечение состоит в снижении общей дозы принимаемых дофаминергических препаратов.
Г. Психические нарушения. К осложнениям дофаминергической терапии паркинсонизма относятся нарушение сознания, галлюцинации, бред и другие психические реакции. Они чаще возникают и тяжелее протекают при приеме бромокриптина, чем леводопы. Эти эффекты исчезают после прекращения приема препарата.
Д. Другие побочные эффекты. Побочными эффектами бромокриптина являются головная боль,
насморк, бессонница, легочные инфильтраты и эритромелалгия. Эритромелалгия представляет собой покраснение, болезненность и припухлость ног (и нередко рук), иногда с артралгией; эти симптомы и признаки исчезают через несколько дней после прекращения приема бромокриптина.
Противопоказания
Бромокриптин противопоказан пациентам с психическими заболеваниями или после недавнего инфаркта миокарда. Его лучше не назначать пациентам с заболеваниями периферических сосудов или язвенной болезнью.
Перголид
Перголид — это агонист дофамина, который напрямую стимулирует Dr и В2-рецепторы. Препарат помогает пациентам, не получающим леводопу, а также продлевает ответ на леводопу у пациентов с колебаниями клинической эффективности. Перголид хорошо переносится больными, но в начале лечения нередки побочные эффекты, похожие на эффекты бромокриптина. Со временем перголид теряет свою эффективность скорее всего в связи с подавлением дофаминовых рецепторов. Неизвестно, дает ли использование перголида на ранних этапах болезни (как при монотерапии, так и в сочетании с низкими дозами леводопы) отсрочку развития вызванных ДОФА побочных эффектов.
Средняя терапевтическая доза перголида — 3 мг в день, начальная доза — обычно 0.05 мг в день; дозу повышают постепенно в зависимости от эффективности и переносимости. Дозу одновременно принимаемой леводопы, возможно, придется снизить.
Ингибиторы моноаминоксидазы
Выделяют два типа моноаминоксидазы. Тип А метаболизирует норадреналин и серотонин, тип В — дофамин. Селегилин (депренил) (рис. 27-2), селективный ингибитор моноаминоксидазы В, тормозит разрушение дофамина. Следовательно, он усиливает и продлевает антипаркинсонический эффект леводопы (можно снизить дозу леводопы) и может уменьшить феномен “on-off”. Поэтому
селегилин используют как дополнение к терапии у пациентов с ухудшающимся ответом на леводопу. Стандартная доза — 5 мг с завтраком и 5 мг — с ланчем. Селегилин может усугублять побочные эффекты леводопы. При монотерапии препарат имеет лишь минимальный антипаркинсонический эффект, но исследования на животных показали, что он может замедлять прогрессирование заболевания. Такой эффект антиоксидантной терапии можно ожидать, если в основе болезни Паркинсона лежит образование свободных радикалов. Однако результаты изучения способности селегилина замедлять прогрессирование заболевания у людей сомнительны. По данным последнего большого многоцентрового исследования, обнаружено некоторое замедление патологического процесса, но это может быть связано чисто с симптоматическим улучшением.
Не следует одновременно назначать леводопу и ингибитор обоих типов моноаминоксидазы, так как это может привести к гипертоническим кризам, вероятно, вследствие периферической кумуляции норадреналина.
Ингибиторы катехол-орто-метилтрансферазы
Подавление ДОФА-декарбоксилазы приводит к компенсаторной активации других путей метаболизма леводопы, особенно к повышению уровня 3-метилдофа в плазме за счет стимуляции катехол-орто-метилтрансферазы (КОМТ). Это в свою очередь снижает эффект леводопы, может быть, из-за конкуренции за активные переносчики через слизистую кишечника и гематоэнцефалический барьер. По этой причине сейчас исследуется возможность применения селективных ингибиторов КОМТ в качестве дополнительной терапии у больных паркинсонизмом, получающих леводопу.
Амантадин
Случайно обнаружено, что противовирусный препарат амантадин обладает противопаркинсони-ческими свойствами. Механизм его действия неясен, но он может усиливать дофаминергическую функцию, влияя на синтез, высвобождение или обратный захват дофамина. Было выявлено высвобождение
катехоламинов из периферических источников при приеме амантадина.
Фармакокинетика
Пиковые концентрации амантадина в плазме достигаются через 1-4 часа после перорального приема. Период полувыведения из плазмы равен 2-4 часам, большая часть препарата выводится в неизмененной форме с мочой.
Клиническое применение
Амантадин менее эффективен, чем леводопа, его эффект короче по продолжительности и часто исчезает всего через несколько недель лечения. Тем не менее во время этого периода он может быть эффективен для снятия брадикинезии, ригидности и паркинсонического тремора. Стандартная доза амантадина — 100 мг перорально два раза в день.
Побочные эффекты
У амантадина есть несколько неприятных побочных эффектов на уровне ЦНС: депрессия, двигательное беспокойство, раздражительность, бессони-ца, психическое и двигательное возбуждение, галлюцинации и нарушение сознания. Все они обратимы при прекращении приема препарата. Передозировка может привести к острому токсическому психозу. При приеме доз, в несколько раз превышающих рекомендованные, возникают судороги.
У пациентов, принимающих амантадин, иногда возникает livedo reticularis, обычно проходящее через месяц после прекращения приема. Были описаны и другие дерматологические реакции. Возникающие нередко периферические отеки не связаны с поражением сердца, почек или печени и проходят под действием диуретиков. Другие побочные эффекты амантадина — это головные боли, застойная сердечная недостаточность, ортостатическая гипотензия, задержка мочи и нарушения желудочно-кишечного тракта (например, анорексия, тошнота, запоры и сухость во рту).
Противопоказания
Препарат следует использовать с осторожностью при наличии в анамнезе судорог или застойной сердечной недостаточности.
Холинолитики
Имеется несколько м-холинолитиков центрального действия, отличающихся по своей эффективности у разных пациентов.
Клиническое применение
Лечение начинают с низкой дозы одного из этих препаратов. Затем дозу постепенно повышают до получения максимального лечебного действия или до развития побочных эффектов, м-холинолитики могут облегчать паркинсонические тремор и ригидность, но почти не действуют на брадикинезию. Если один препарат не оказывает эффекта, то рекомендуется применение других препаратов этой группы, так как они могут быть эффективны. Некоторые из наиболее часто используемых м-холинолитиков приведены в табл. 27-1.
Побочные эффекты
м-холинолитики вызывают неблагоприятное воздействие на ЦНС, в том числе сонливость, замедление мышления, невнимательность, двигательное беспокойство, раздражительность, бред, галлюцинации и нарушения настроения. Эти эффекты иногда провоцируются сопутствующими инфекциями. Они обычно проходят через несколько дней после прекращения приема препарата. Часто возникают сухость во рту, затуманивание зрения, мидриаз, задержка мочи, тошнота и рвота, запор, тахикардия, тахипноэ, повышение внутриглазного давления, сердцебиения и аритмии. Дискинезии редки. Сухость во рту иногда приводит к острому гнойному паротиту.
Если прием препарата прекращают, то это следует делать постепенно для предотвращения обострения паркинсонизма.
Противопоказания
Холинолитики не следует назначать при гипертрофии простаты, обструктивных заболеваниях желудочно-кишечного тракта (например, стенозе привратника или паралитической кишечной непроходимости) и закрытоугольной глаукоме. У больных паркинсонизмом, получающих холинолитики, одновременный прием других препаратов, обладающих холинолитическим действием (например,
ТАБЛИЦА 27-1. Некоторые лекарства
с антимускариновыми
свойствами, используемые
при паркинсонизме
Лекарство Обычная суточная доза (мг)
Бензтропина мезилат 1-6
Бипериден 2-12
Хлорфеноксамин 150-400
Орфенадрин 150-400
Проциклидин 7.5-30
Тригексифенидил 6-20
трициклических антидепрессантов или антигистаминных препаратов), может спровоцировать некоторые из названных побочных эффектов.
О,Ьтрео-3,4-Дигидрокси-фенилсерин (DOPS)
В мозгу больных паркинсонизмом обнаружена региональная недостаточность норадреналина (так же, как и дофамина). Предполагают, что недостаток норадреналина вызывает клинический феномен внезапного транзиторного “замирания”, “замораживания”, прекращения движения у пациентов с тяжелым паркинсонизмом, реакция которых на леводопу непредсказуема. Было показано, что прием В,Ь-тпрео-3,4-дигидроксифенилсерина (DOPS), предшественника норадреналина, вызывает улучшение при таком состоянии, однако не все исследования подтвердили это.
Другие экспериментальные подходы к лечению болезни Паркинсона
Лечебные свойства при болезни Паркинсона приписывали витамину Е, “уборщику” свободных радикалов. Однако последние крупные мультицентровые исследования не обнаружили преимуществ ежедневного приема токоферола (2000 ME) перед традиционной терапией.
У приматов с экспериментальным паркинсонизмом лечение ганглиозидом GMI повышало уровень дофамина в стриатуме и усиливало дофаминергическую иннервацию стриатума. Этот феномен мо
жет стать потенциально новым направлением стратегии лечения болезни Паркинсона у человека.
В настоящее время изучают возможность использования трансплантации дофаминергической ткани (аутотрансплантатов ткани мозгового слоя надпочечников или ткани черной субстанции плода). Результаты противоречивы, а механизм возможного лечебного эффекта неясен.
Общие комментарии к фармакотерапии паркинсонизма
Болезнь Паркинсона имеет обычно прогрессирующее течение. Эффективность лечения леводопой часто со временем уменьшается, возникают нежелательные эффекты, осложняющие длительный курс лечения. Тем не менее накоплены доказательства того, что дофаминергическая терапия на сравнительно ранней стадии может быть наиболее эффективна в ослаблении симптоматики паркинсонизма и может снижать смертность при этом заболевании. Симптоматического лечения умеренно выраженного паркинсонизма, возможно, лучше избегать до тех пор, пока симптомы заболевания не начнут оказывать существенное влияние на образ жизни пациента. Когда лечение становится необходимым, обычно прописывают леводопу в комбинации с карбидопой (синемет). Для оптимального действия может быть необходим амантадин или анти-мускариновый препарат (или оба одновременно). Некоторые нежелательные эффекты длительного лечения леводопой (колебания в реакции на препарат, дискинезии) можно, вероятно, избежать или уменьшить, если сохранять невысокой общую суточную дозу синемета и усиливать дофаминергическую терапию добавлением бромокриптина или перголида. Для улучшения двигательных функций полезна лечебная физкультура. Для больных с тяжелым паркинсонизмом и осложнениями от длительного лечения леводопой (“on-off” феномен) может быть полезной попытка лечения бромокриптином или перголидом. Регуляция потребления белков с пищей также может ослабить проявления “on-off’ феномена.
Лечение селегилином молодых больных или пациентов с умеренно выраженным паркинсонизмом может замедлить прогрессирование болезни и заслуживает серьезного внимания.
Паркинсонизм, вызванный лекарствами
Резерпин и родственный ему тетрабеназин опустошают запасы биогенных аминов, а галоперидол и фенотиазины блокируют дофаминовые рецепторы. Следовательно, эти лекарства могут вызывать синдром Паркинсона. При применении этих препаратов в высоких дозах он обычно возникает в пределах трех месяцев после начала лечения. Симптомы исчезают в течение нескольких недель или месяцев после отмены препаратов. При необходимости лечения предпочитают назначение антимускариновых препаратов. Леводопа не оказывает действия, если продолжается прием нейролептиков, и может даже усилить психические нарушения, по поводу которых исходно назначали антипсихотические препараты.
В 1983 г. была открыта лекарственная форма паркинсонизма у лиц, которые пытались синтезировать и применять наркотик типа меперидина, но в действительности синтезировали и вводили себе Ь-метил-4-фенил-1,2,5,6-тетрагидропиридин. Это соединение (МРТР) вызывает селективное разрушение дофаминергических нейронов черной субстанции и приводит к тяжелой форме паркинсонизма как у животных, так и у людей. Использование этого вещества позволило создать экспериментальную модель для скрининга возможных антипаркин-сонических средств. Токсичность МРТР связана с его окислением моноаминоксидазой В до 1-метил-4-фенилпиридиниума (МРР+).
Другие расстройства движений
Тремор
Тремор — это ритмические колебательные движения. Физиологический постуральный тремор, являющийся нормальным, увеличивается по амплитуде при тревоге, усталости, тиреотоксикозе и после внутривенного введения адреналина или изопротеренола. Пропранолол уменьшает его амплитуду, а при внутриартериальном введении предупреждает реакцию на изопротеренол в перфузируемой конечности преимущественно за счет периферического действия. Некоторые лекарства, особенно бронходилататоры, трициклические антидеп
рессанты и литий, могут вызывать дозозависимое усиление нормального физиологического тремора, которое быстро исчезает при прекращении введения препарата. Тремор, спровоцированный симпа-томиметиками, такими как тербуталин (бронходи-лататор), блокируется пропранололом, антагонистом рг и р2-рецепторов, но не снимается метопрололом, селективным ргантагонистом. Следовательно, такой тремор опосредован в основном р2-рецепторами.
Эссенциальный тремор — это постуральный, иногда семейный тремор, который клинически сходен с физиологическим. В некоторых случаях такого тремора наблюдается дисфункция ргрецепто-ров, так как обнаруживается положительная реакция не только на пропанолол, но и на стандартную дозу метопролола. И все же более эффективно применение пропранолола, но остается неясным, связан ли эффект с его центральным или периферическим действием. Фармакокинетика, фармакологические эффекты и нежелательные реакции пропранолола рассматриваются в главе 10. Необходимая ежедневная доза пропранолола составляет порядка 120 мг (разброс 60-240 мг). Есть сообщения о его побочных эффектах. Препарат должен осторожно использоваться у больных с застойной сердечной недостаточностью, сердечными блокадами, астмой и гипогликемией. Пациента необходимо обучить контролю за пульсом и рекомендовать вызвать врача, если развивается значительная брадикардия. Применение метопролола полезно при лечении тремора у пациентов с сопутствующей легочной патологией, являющейся противопоказанием к назначению пропранолола. В ряде случаев для симптоматического контроля эффективен примидон (антиэпилептический препарат, глава 23) в постепенно возрастающих дозах: до 250 мг три раза в день. Больные с тремором очень чувствительны к примидону и подчас не переносят дозы, применяемые при судорожном синдроме, поэтому следует начинать с дозы 50 мг один раз в сутки и повышать дозу на 50 мг каждые две недели в зависимости от реакции на препарат. Эссенциальный тремор могут подавлять малые дозы алкоголя, но лишь на короткое время. Механизм этого действия неизвестен. В прошлом имели своих приверженцев диазепам, хлордиазепоксид, мефенезин и антипаркинсоничес-кие вещества, но в настоящее время они считаются бесполезными. Некоторым больным помогает изониазид (1200 мг в сутки с 300 мг пиридоксина).
Интенционный тремор — тремор, возникающий при движении, но не в покое. Иногда это проявление токсического действия алкоголя или таких лекарств, как фенитоин. Отмена или уменьшение дозы приводит к облегчению состояния. Удовлетворительного фармакологического лечения интенционного тремора не существует.
Тремор в состоянии покоя обычно связан с паркинсонизмом.
Болезнь Гентингтона
Это преимущественно наследственное заболевание характеризуется прогрессирующей хореей и деменцией, которые обычно начинаются у людей пожилого возраста. Развитие хореи связано с нарушением баланса дофамина, ацетилхолина, ГАМК и, возможно, других нейротрансмиттеров в базальных ганглиях (рис. 27-1). Фармакологические исследования показали, что хорея ассоциируется с функциональной гиперактивностью дофаминергических нигростриатных путей, видимо, из-за повышенной реактивности постсинаптических дофаминовых рецепторов или дефицита нейротрансмиттера, который в норме является антагонистом дофамина. Вещества, нарушающие дофаминергическую передачу путем истощения запасов центральных моноаминов (например, резерпин, тетрабеназин), или путем блокады дофаминовых рецепторов (фенотиазины, бутирофеноны), часто смягчают проявления хореи, тогда как дофаминомиметические препараты, такие как леводопа, скорее обостряют ее проявления.
Уровень ГАМК и фермента ее синтеза (глутаматдекарбоксилазы) в базальных ганглиях больных хореей Гентингтона значительно понижены. Там же отмечается существенное снижение концентрации холинацетилтрансферазы — фермента, отвечающего за синтез ацетилхолина. Эти наблюдения могут иметь патофизиологическое значение и стать основой лечения хореи путем усиления активности центральных ГАМК- и холинергических систем. К сожалению, пока попытки реализации таких направлений фармакологического воздействия принесли разочарование. Поэтому для контроля дискинезии у больных с болезнью Гентингтона по-прежнему чаще всего применяются средства с дофаминергической активностью. Однако все лекарства последней группы могут наряду с уменьшением патологической моторики привести к ятрогенному паркинсонизму.
Резерпин истощает церебральные запасы дофамина, предупреждая его отложение внутри нейронов. Препарат начинают вводить в малых дозах (например, 0.25 мг в день) с постепенным повышением (примерно на 0.25 мг каждую неделю) до тех пор, пока не проявится терапевтическое действие или не возникнет нежелательный эффект. Ежедневная доза 2-5 мг часто подавляет ненормальные движения, но это может сопровождаться нежелательными эффектами: гипотензией, депрессией, седацией, диареей, заложенностью носа. Тетрабеназин напоминает резерпин по способности истощать запасы мозгового дофамина и имеет менее отчетливые нежелательные эффекты, но он не разрешен к применению в США. Могут быть использованы постсинаптические блокаторы дофаминовых рецепторов, таких как фенотиазины и бутирофеноны. Галоперидол начинают вводить в малой дозе (1 мг два раза в день), увеличивая ее каждые 4 дня в зависимости от реакции больного. Если галоперидол не дает эффекта, иногда может помочь лечение перфеназином в возрастающих до 20 мг в сутки дозах. Фармакокинетика и клиническое применение этих лекарств рассмотрены более детально в соответствующем разделе книги.
Другие формы хореи
Если хорея является осложнением тиреотоксикоза, полицитемии, системного волчаночного эри-тематоза, гипокальциемии или цирроза печени, необходимо прежде всего лечить основное заболевание. Лекарственная хорея обычно исчезает после отмены провоцирующих ее препаратов, например леводопы, антимускариновых препаратов, амфетамина, лития, фенитоина или оральных контрацептивов. Острую или медленно прогрессирующую дискинезию могут вызывать также нейролептики. Хорея Сиденхема обычно преходяща и столь умеренно выражена, что редко требуется фармакологическая коррекция. Если же она необходима, как правило, оказываются эффективными лекарства, блокирующие действие дофамина.
Баллизм
Биохимические основы баллизма неизвестны, но фармакологический подход к лечению такой же, как при хорее. Может быть эффективным лечение галоперидолом, перфеназином и другими дофаминоблокаторами.
Атетоз и дистония
Патофизиологические основы этих заболеваний не разработаны и, следовательно, не существует удовлетворительной фармакотерапии. Некоторые больные с дистонией могут положительно реагировать на лечение диазепамом, амантадином, антимускариновыми лекарствами (в высоких дозах), леводопой, карбамазепином, баклофеном, галоперидолом или перфеназином. Попробовать лечение этими препаратами целесообразно, даже несмотря на частые неудачи. Больным с такими местными дистониями, как блефароспазм или тортиколиз, может помочь инъекция ботулотоксина в гиперактивные мышцы.
Тики
Патофизиологический механизм тиков неизвестен. Хронические множественные тики (синдром Жиля де ла Туретта) могут потребовать лечения, если заболевание протекает в тяжелой форме или оказывает значительное влияние на жизнь пациента. Наиболее эффективный фармакологический метод лечения — применение галоперидола. Пациенты лучше переносят это лекарство, если лечение начинают с малых доз (например, 0.25 или 0.5 мг в день) и затем постепенно повышают их в течение нескольких недель. Большинству больных необходимы окончательные суточные дозы от 3 до 8 мг. Если галоперидол неэффективен, могут быть апробированы флуфеназин, клоназепам, клонидин или карбамазепин. Фармакологические свойства этих лекарств обсуждаются в другом разделе книги. Больным, которые плохо переносят галоперидол или не реагируют на него положительно, можно назначить оральный блокатор дофамина пимо-зид, но безопасность этого препарата при долгосрочном лечении не изучена.
Дискинезии, вызванные лекарствами
Фармакологические основы острой дискинезии или дистонии, которые иногда возникают после первых нескольких доз фенотиазинов, не совсем ясны. В большинстве таких случаев оказывается полезным парентеральное введение антимускариновых веществ (2 мг бензтропина внутривенно или 2-5 мг биперидена внутривенно или внутримышечно). Иногда уменьшает проявление ненормальных движений диазепам (10 мг внутривенно).
Тардивная дискинезия — это расстройство, характеризующееся рядом ненормальных движений, которые развиваются после продолжительной терапии нейролептиками. Уменьшение дозы вызывающего это осложнение препарата, блокатора дофаминовых рецепторов, обычно усугубляет дискинезию, тогда как увеличение дозы может подавить ее. Эти клинические наблюдения и ухудшение двигательных расстройств под действием леводопы, указывают на то, что тардивная дискинезия связана с гиперфункцией дофаминергической системы, но ее точные фармакологические механизмы неизвестны. Возможно, что при коротких курсах фенотиазины вызывают обратимую блокаду дофаминовых рецепторов, а при длительных — усиливают дофаминергическую функцию. Зависит ли этот эффект от формирования денервационной гиперчувствительности, увеличения синтеза дофамина или снижения его обратного захвата в центральных синапсах пока не известно.
Известно, что ГАМК-зависимые структуры находятся в стрионигралыюй возвратной петле обратной связи, модулирующей активность дофаминергических клеток в черной субстанции. С другой стороны, ГАМК-эфферентные пути также проходят через стриатум к бледным шарам и таламусу. Опосредованная ГАМК активность может снизить функцию нигростриатных дофаминергических клеток и таким образом уменьшить выраженность непроизвольных движений. Мусцимол, один из структурных аналогов ГАМК, при суточных дозах 5-9 мг внутрь может ослаблять непроизвольную моторику больных с тардивной дискинезией. Однако мусцимол имеет нежелательные поведенческие эффекты, которые ограничивают его применение при этом заболевании. Другие попытки повысить активность ГАМК (например, назначая бензодиазепины или вальпроат натрия) не привели к стабильному положительному эффекту. В ряде случаев удавалось восстановить баланс между дофаминергической и холинергической активностью путем увеличения последней за счет введения внутрь холина или лецитина. Аналогичные наблюдения сделаны при применении деанолацетамидобензоата — вещества, родственного ацетилхолину и в настоящее время не применямого. К лекарствам, дающим с наибольшей вероятностью немедленный симптоматический эффект, относятся те, которые влияют на дофаминергическую функцию, либо истощая ее (резерпин, тетрабеназин), либо блокируя рецепторы (феноти
азины, бутирофеноны). Парадоксально, что именно лекарства, блокирующие рецепторы, сами вызывают дискинезию. Их лучше не применять для предупреждения развития феномена “спирали”, при котором постепенное усиление дискинезии в ответ на введение вещества, назначенного с лечебной целью, требует постоянного увеличения его же дозы для подавления дискинезии.
Поскольку тардивная дискинезия, развивающаяся у взрослых, обычно необратима и нет способов ее эффективного лечения, необходимо сделать все возможное для снижения вероятности ее возникновения. Антипсихотические лекарства должны назначаться только тогда, когда это необходимо, и их надо периодически отменять для оценки эффективности лечения и демаскировки зарождающейся дискинезии. Отметим, что тиоридазин (фенотиазин с пиперидиновым боковым радикалом) — эффективное антипсихотическое средство, которое с меньшей вероятностью вызывает экстрапирамид-ные реакции, возможно потому, что меньше влияет на дофаминовые рецепторы в стриатной системе. Наконец, антимускариновые вещества не должны бесконтрольно назначаться больным, получающим нейролептики, так как эта комбинация может повысить вероятность дискинезии.
Болезнь Вильсона
Болезнь Вильсона — нарушение метаболизма меди, наследуемое по рецессивному типу, биохимически характеризуется снижением в сыворотке крови концентраций меди и церулоплазмина, патофизиологически — увеличением уровня меди в мозгу и внутренних органах и клинически — признаками печеночной и неврологической дисфункций. Лечение требует удаления из тканей избытка меди с последующим поддержанием ее баланса. Наиболее эффективным и доступным в настоящее время является пеницилламин (диметилцистеин) — хелатирующий агент, который образует с медью кольцевидную структуру. Он легко всасывается в желудочно-кишечном тракте и быстро экскретируется с мочой. Обычно начальная доза у взрослых составляет 500 мг 3-4 раза в день. После достижения ремиссии можно снизить дозу до поддерживающей на уровне не менее 1 г в сутки. Побочные эффекты включают тошноту и рвоту, нефротический синдром, волчаночноподобный синдром, пузырчатку, миастению, артропатию, зрительную нейропатию и
различные нарушения со стороны крови. Лечение необходимо контролировать частыми анализами мочи и крови. Медь с продуктами питания должна поступать на уровне ниже 2 мг в сутки путем исключения из рациона шоколада, орехов, устриц, печени, грибов, круп. Может потребоваться дистиллированная или деминерализованная вода для питья, если водопроводная содержит более 0.1 мг/л меди. Целесообразно назначать дисульфид калия (20 мг 3 раза в день с едой), уменьшая всасывание меди в кишечнике. Для больных, которые не переносят пеницилламин, может быть использован новый хелатообразователь триентин в ежедневной дозе 1-1.5 г. У него мало нежелательных эффектов, за исключением незначительной анемии из-за возможного дефицита железа.
Препараты
Амантадин (Симметрел)
Перорально: капсулы по 100 мг; сироп 10 мг/мл
Бензтропин (Когентин, др.)
Перорально: таблетки по 0.5, 1, 2 мг Парентерально: 5 мг/мл для инъекций
Бипериден (Акинетон)
Перорально: таблетки по 2 мг
Парентерально: 5 мг/мл для инъекций
Бромокриптин (Парлодел)
Перорально: таблетки по 2.5 мг;
капсулы по 5 мг
Карбидопа (Лодозин)
Перорально: таблетки по 25 мг
Карбидопа/Леводопа (Синемет)
Перорально: таблетки по 10 мг карбидопы и 100 мг леводопы, 25 мг карбидопы и 100 мг леводопы, 25 мг карбидопы и 250 мг леводопы
Перорально для пролонгированного действия (Синемет CR): 50 мг карбидопы и 200 мг леводопы
Леводопа (Допар, Лародопа)
Перорально: таблетки и капсулы по 100, 250, 500 мг
Орфенадрин (Дизипал)
Перорально: таблетки по 100 мг Перорально для пролонгированного
действия: таблетки по 100 мг
Парентерально: 30 мг/мл для инъекций
Пеницилламин (Купримин, Депен) Перорально: капсулы по 125, 250 мг; таблетки по 250 мг
Перголид (Пермакс)
Перорально: таблетки по 0.05, 0.25,1 мг
Проциклидин (Кемадрин)
Перорально: таблетки по 5 мг
Селегилин (депренил) (Элдеприл)
Перорально: таблетки по 5 мг
Триентин (Куприд)
Перорально: капсулы по 250 мг
Тригексифенидил (Артан, др.)
Перорально: таблетки по 2, 5 мг; эликсир 2 мг/5 мл
Перорально для пролонгированного действия (Артан Секуэлз): капсулы по 5 мг
Избранная литература
Campanella G. et al. Drugs affecting movement disorders. Annu. Rev. Pharmacol. Toxicol. 1987; 27: 113.
Kopin I. J. The pharmacology of Parkinson’s disease therapy: An update. Annu. Rev. Pharmacol. Toxicol. 1993; 32:467.
Lieberman A. N., Goldstein M. Bromocriptine in Parkinson disease. Pharmacol. Rev. 1985; 37:217.
Mannisto O., Kaakola S. New selective COMT inhibitors: Useful adjuncts for Parkinson’s disease? Trends Pharmacol. Sci. 1989; 10:54.
Антипсихотические средства и литий
28
Лео Е. Холлистер
I. Антипсихотические средства
Термины антипсихотические средства и нейролептики взаимозаменяемы и определяют группу лекарственных средств, которые используются в основном для лечения больных шизофренией, но эффективны также при других психозах и ажити-рованных состояниях.
История
Антипсихотические средства используются в клинике уже 40 лет. Резерпин и хлорпромазин были первыми препаратами, применяемыми при шизофрении. Сегодня они практически полностью заменены новыми лекарствами, хотя хлорпромазин до сих пор иногда назначают при психозах. Влияние первых препаратов на развитие психиатрии, особенно в области терапии шизофрении, было очень значительным: существенно снизилось число пациентов, которым требовалась госпитализация в специализированные психиатрические учреждения, психиатрическое мышление приобрело биологическую основу. К сожалению, эти новые веяния не смогли полностью оправдать надежд ни с медицинской, ни с экономической или общечеловеческой точки зрения.
Природа психоза и шизофрении
Термин “психоз” объединяет группу психических заболеваний. Шизофрения — это особый вид психоза, характеризующийся сохранностью восприятия со значительными нарушениями мышления. Патогенез заболевания неизвестен. В резуль
тате серьезных научных исследований, начавшихся после появления первых антипсихотических средств, было установлено, что генетическая предрасположенность является необходимым, но не единственным фактором развития психоза. В некоторой степени это заключение подтверждается семейными случаями шизофрении. Однако определение характера наследования шизофрении на хромосомном уровне при помощи методов молекулярной генетики до сих пор затруднительно вследствие высокой вариабельности фенотипа страдающих этим заболеванием. Молекулярные основы шизофрении еще только предстоит определить полностью, но огромная работа по изучению роли аномалий функций аминных нейромедиаторов, особенно дофамина, уже проделана (дополнение “Дофаминовая гипотеза”). Но и у дофаминовой гипотезы есть значительные недостатки. Можно предположить, что это заболевание значительно сложнее, чем считалось ранее.
Дополнительный фактор, определяющий гетерогенность больных шизофренией,— это наличие анатомических изменений у одних пациентов и отсутствие у других. Ряд исследований с использованием компьютерной томографии и ядерно-магнитного резонанса показал наличие атрофии самых разных структур мозга у некоторых больных шизофренией по сравнению со здоровыми добровольцами той же возрастной группы. У таких пациентов преобладают “негативные” симптомы (эмоциональное притупление, нарушение социальных контактов, когнитивные расстройства), которые плохо поддаются терапии. Более того, у некоторых больных с помощью позитронно-эмиссионной томографии (ПЭТ) показано наличие зон сниженного метаболизма в разных частях мозга.
Базисная фармакология антипсихотических средств
Типы химической структуры
Целый ряд веществ разной химической структуры обладает антипсихотическими свойствами. Их можно объединить в группы (рис. 28-1).
А. Производные фенотиазина. В настоящее время применяют три подтипа семейства фенотиази
нов, отличающиеся по структуре боковой цепи. Алифатические дериваты (например, хлорпромазин) и пиперидиновые дериваты (например, тиоридазин) менее активны, чем производные пиперазина, эффект которых появляется при приеме меньших доз. Последние также более селективны по фармакологическим эффектам (табл. 28-1).
Б. Производные тиоксантена. Типичным представителем группы является тиотиксен. В целом эти соединения несколько менее активны, чем их фенотиазиновые аналоги.
В. Производные бутирофенона. Препараты этой группы в корне отличаются по структуре от
Дофаминовая гипотеза
Дофаминовая гипотеза патогенеза шизофрении — одна из наиболее обоснованных. Она лежит в основе принципов терапии этого заболевания. Существует несколько важных доказательств этой гипотезы: 1) большинство нейролептиков блокирует постсинаптические D2-pe-цепторы в ЦНС, особенно в мезолимбическо-фронтальной системе; 2) препараты, которые повышают дофаминергическую активность, например леводопа (предшественник дофамина), амфетамины (высвобождающие дофамин) или апоморфин (агонист дофаминовых рецепторов прямого действия), либо усугубляют течение шизофрении, либо провоцируют ее начало de novo; 3) по результатам аутопсий установлено, что в мозгу больных шизофренией, которые не получали лечения нейролептиками, повышена плотность дофаминовых рецепторов; 4) ПЭТ продемонстрировала повышение плотности дофаминовых рецепторов у больных, получавших и не получавших лечение, по сравнению со здоровыми людьми; 5) показано, что успешное лечение шизофрении сопровождалось изменением количества метаболита дофамина гомованилиновой кислоты в цереброспинальной жидкости, плазме и моче.
Однако дофаминовая гипотеза разработана отнюдь не полностью. Если бы дефекты обмена дофамина полностью объясняли патогенез шизофрении, то антипсихотические средства были
бы гораздо более эффективны. На самом же деле они только частично помогают одним больным и совсем не эффективны у других. Исследования по клонированию и идентификации множества типов дофаминовых рецепторов помогут развитию и подтверждению этой гипотезы, если будут найдены препараты, селективно действующие на рецептор каждого типа. Традиционные нейролептики связываются с Э2-рецепторами в 50 раз активнее, чем e Dr или В3-рецепторами. До недавнего времени главной целью всех исследований было найти наиболее сильные и наиболее 1)2-селективные препараты. Тот факт, что несколько атипичных нейролептиков действует на D2 в значительно меньшей степени, помогая в то же время при шизофрении, переключил всеобщее внимание на другие типы дофаминовых рецепторов и на недофаминовые рецепторы, особенно 5-НТ2-рецептор. Показано также, что даже среди традиционных фенотиазинов корреляция клинической эффективности с блокадой дофаминовых рецепторов значительно менее выражена, чем с блокадой «-адренорецепторов. В результате направление исследований изменилось и теперь сосредоточено на препаратах, которые действуют на несколько рецепторно-медиаторных систем. Основная цель и надежда фармакологов — создать более эффективные средства с меньшим количеством побочных эффектов, особенно в отношении экстрапирамидной системы.
ПРОИЗВОДНЫЕ ФЕНОТИАЗИНА
ПРОИЗВОДНЫЕ ТИОКСАНТЕНА
Фенотиазиновов ядро
Алифатическая боковая цепь
Тиотиксен (2) — SO2N(CH2)2
Хлорпромазин (2) — CI (10) — СН2—СН2— СН2— N — (СН3)2
Пиперидиновая боковая цепь
Тиоридазин (2) — SCH3 (10) —СН2 —СН2
I
СН3
Трифлуоперазин (2) — CF2
Перфеназин (2) — CI
Пиперазиновая боковая цепь
(10) —СН2 —СН2—СН2 —N N — СН3
(10) —СН2 —СН2—СН2—N
N —СН2 —СН2 —ОН
БУТИРОФЕНОНЫ
N —СН2—СН2 —ОН
Флуфеназин (2) — CF3
(10) —СН2 —СН2 —СН2 —N
Молиндон
Локсапин
БЕНЗАМИД
Рис. 28-1. Структурные формулы фенотиазинов, тиоксантенов, бутирофенонов и представителей смешанной группы нейролептиков. Показаны наиболее типичные представители каждой группы
лекарств из двух предыдущих групп. Наиболее часто используется галоперидол. Дифенилбутилпипе-ридины — родственные соединения. Они более эффективны и дают меньше вегетативных побочных эффектов.
Г. Смешанная группа. Это сравнительно новые препараты, в том числе: дифенилбутилпиперидины (пимозид), дигидроиндолоны (молиндон), дибенз-оксазепины (локсапин), дибензодиазепины (клозапин) и бензамиды (ремоксиприд).
ТАБЛИЦА 28-1. Антипсихотические средства: связь химической структуры с эффективностью и токсичностью
Химический класс Средство Эффективность Экстрапирамид-ная токсичность Седативный эффект Гипотензивный эффект
Фенотиазины:
Алифатические Хлорпромазин Низкая Средняя Сильный Сильный
Пиперазин Флуфеназин Высокая Высокая Слабый Очень слабый
Тиоксантен Тиотиксен Высокая Средняя Средний Средний
Бутирофенон Галоперидол Высокая Очень высокая Слабый Очень слабый
Дибензодиазепин Клозапин Средняя Очень низкая Слабый Очень слабый
Фармакокинетика
А. Абсорбция и распределение. Большинство антипсихотических средств быстро, но не полностью абсорбируются. Более того, многие из них подвергаются значительному пресистемному метаболизму. В частности, хлорпромазин и тиоридазин при пероральном приеме имеют системную доступность 25-35 %, а галоперидол, который метаболизируется в меньшей степени,— около 65 %.
Большинство нейролептиков хорошо растворимы в жирах и связаны с белками на 92-99 %. У них значительные объемы распределения (обычно > 7 л/кг). Возможно, вследствие секвестрации в жидких средах организма они обычно обладают большей продолжительностью действия, чем можно было бы предположить по их периоду полувыведения. При хроническом применении метаболиты хлорпромазина могут определяться в моче в течение нескольких недель после приема последней дозы.
Б. Метаболизм. Большинство нейролептиков почти полностью метаболизируются разными путями. Хотя некоторые из метаболитов сохраняют активность (например, 7-гидроксихлорпромазин и восстановленный галоперидол), считается, что метаболиты не играют большой роли в реализации действия этих препаратов. Единственным исключением является мезоридазин, основной метаболит тиоридазина, который обладает большей активностью, чем исходное соединение. Именно с ним связан лечебный эффект лекарства. Это вещество существует и в виде отдельного препарата.
В. Выведение. Очень малая часть этих препаратов выводится в неизмененном виде, так как они почти полностью метаболизируются до более полярных соединений. Периоды полувыведения (определяемые по метаболическому клиренсу) варьируют между 10 и 24 часами.
Фармакологические эффекты
Первые фенотиазиновые нейролептики (прототип — хлорпромазин) обладали широким спектром действия на ЦНС, вегетативную нервную систему и эндокринные органы. Эти эффекты связывали с блокадой большого количества рецепторов, включая дофаминовые, «-адренорецепторы, мускариновые, Н(-гистаминовые и серотониновые (5-НТ2-рецепто-ры). Из перечисленных рецепторов именно дофаминовые быстро стали объектом всеобщего интереса.
А. Дофаминергическая система. До 1959 г. дофамин не был известен как нейромедиатор ЦНС, а просто считался предшественником норадреналина. Сейчас описаны пять основных дофаминергических систем (путей) в ЦНС. Первый, наиболее тесно связанный с поведением,— это мезолимби-ческо-мезокортикальныйпуть, который проецируется из тел нейронов вблизи черной субстанции в лимбическую систему и неокортекс.
Вторая система (нигростриатныйпуть) состоит из нейронов, которые проецируются из черной субстанции в хвостатое ядро и бледный шар. Эта система участвует в координации произвольных движений.
Третья система (тубероинфундибулярная) соединяет аркуатные ядра и перивентрикулярные нейроны с гипоталамусом и задней долей гипофиза. Дофамин, высвобождаемый этими нейронами, физиологически подавляет секрецию пролактина. Четвертая дофаминергическая система (медулляр-но-перивентрикулярный путь) состоит из нейронов моторного ядра блуждающего нерва, проекция которых плохо известна. Эта система, возможно, участвует в контроле пищевого поведения. Пятая система (инцертогипоталамическая) образует связи внутри гипоталамуса и с латеральными ядрами перегородки. Ее роль еще не определена.
После того как дофамин стал известен как нейромедиатор, в различных экспериментах было показано, что его действие на электрическую активность центральных синапсов и на продукцию цАМФ аденилатциклазой можно заблокировать большинством нейролептиков. В 1960-е гг. пришли к заключению о том, что эти препараты следует считать антагонистами дофамина. Сейчас полагают, что их антипсихотическое действие связано со способностью блокировать эффекты дофамина в мезо-лимбической и мезофронтальной системах. Более того, антагонизм по отношению к дофамину в ниг-ростриатной системе объясняет возникновение паркинсонизма как побочного эффекта при приеме этих средств. Гиперпролактинемия при лечении нейролептиками вызвана блокированием тонического ингибиторного рефлекса на выделение пролактина из гипофиза, который осуществляется дофамином. И наконец, изменение пищевого поведения у многих пациентов может быть связано с действием препаратов на медуллярно-перивентри-кулярную систему. Таким образом, один и тот же фармакодинамический эффект может иметь разные психические, неврологические и эндокринологические последствия.
Б. Дофаминовые рецепторы и их эффекты. В настоящее время описаны пять разных дофаминовых рецепторов, которые делятся на два семейства: Dt- и Б2-подобные рецепторные группы. Dr рецептор является продуктом гена пятой хромосомы. При возбуждении он повышает количество цАМФ посредством активации аденилатциклазы. Локализуется в основном в бледном шаре, nucleus accumbens и обонятельном бугорке. Второй член этого семейства, Ds-рецептор, закодирован в четвертой хромосоме, он также увеличивает уровень цАМФ и находится в гиппокампе и гипоталамусе. Терапевтическая эффективность нейролептиков не коррелирует с их аффинностью к Вгрецептору (рис. 28-2, вверху). Г)2-рецептор, закодированный в одиннадцатой хромосоме, снижает количество цАМФ (подавляя аденилатциклазу) и блокирует кальциевые каналы, открывая при этом калиевые. Он расположен как пре-, так и постсинаптически на нейронах каудопутамен, nucleus accumbens и обонятельного бугорка. И второй член этого семейства рецепторов — Г)3-рецептор, который также кодируется одним из генов одиннадцатой хромосомы, предположительно уменьшает концентрацию цАМФ. Место его локализации — кора лобных до-
10’5
10’6
10’7
10’6
т-ггпт--Г I II 11>1|---r -т I I I Ш|-1 I -1-11 III|
-9-
Сульпирид
Di Клебоприд Молиндон
_ "* Хлорпромазин
Спиперон
г Клозапин
-4>-
Галоперидол _ -*•
: Тиоридазин
Флуфеназйн" Трифлуперазин
Г
Флупентиксол
g 0.1 1 10 100 1000
У Терапевтический диапазон и средние дозы средств для лечения шизофрении (мг/сутки)
Рис. 28-2. Корреляция между терапевтической эффективностью нейролептиков и их аффинностью к Dr (евер-ху) или D2- (внизу) рецепторам дофамина. Эффективность показана по оси абсцисс; она снижается слева направо. Связывающая способность с Di-рецепторами измерена по вытеснению селективного Е^-лиганда SCH 23390; связывающая способность с D2-рецепторами — по вытеснению селективного Э2-лиганда галоперидола. Связывающая способность снижается снизу вверх. (Из: Seeman Р. Synapse, 1987; 1; 133.)
лей, продолговатый и средний мозг. ^-рецепторы, самые молодые из О2-подобных рецепторов, тоже снижают уровень цАМФ. Все дофаминовые рецепторы состоят из семи трансмембранных доменов и связаны с G-белком.
Активация Б2-рецепторов прямыми или непрямыми агонистами (например, амфетаминами, леводопой, апоморфином) приводит к повышению двигательной активности и стереотипии у крыс на мо
дели, которая используется для скрининга антипсихотических средств. При их назначении больным шизофренией течение заболевания ухудшается. Нейролептики стереоселективно блокируют В2-рецепторы, а их связывающая способность в значительной степени коррелирует с выраженностью антипсихотического действия и экстрапирамидных эффектов (рис. 28-1, внизу). Это наблюдение подтолкнуло целый ряд исследований в области определения рецепторсвязывающей способности лекарств.
Длительное лечение нейролептиками в некоторых (но не во всех) случаях приводит к временному повышению уровней метаболита дофамина гомованилиновой кислоты (HVA) в цереброспинальной жидкости, плазме и моче. Через 1-3 недели приема концентрация HVA снижается ниже нормы и остается на этом уровне. Такие изменения можно объяснить следующим образом. Начальный период блокады рецепторов вызывает компенсаторное увеличение оборота медиатора, что приводит к возрастанию уровня HVA. При продолжении терапии повышенный уровень дофамина в синапсе приводит к снижению его высвобождения и оборота.
Как уже отмечалось, эти данные стали интегральной частью дофаминовой гипотезы шизофрении (дополнение “Дофаминовая гипотеза”). Однако пока нет удовлетворительного ответа на многие вопросы, и гипотеза еще полностью не подтверждена. Например, существуют высоко- и низкоаффинные формы дофаминовых рецепторов, и еще не известно, влияет шизофрения или нейролептики на соотношение этих двух форм. Интересно, что вызываемые нейролептиками прогрессивные изменения экстрапирамидной системы (от подавления, напоминающего паркинсонизм, до повышенной активности, проявляющейся дискинезиями) часто возникают через несколько месяцев или даже лет. Этот период значительно дольше, чем сроки обнаружения других рецепторных изменений, связанных с приемом лекарств.
Пока нет возможности определить, играет ли блокада других рецепторов, кроме D2, какую-либо роль в антипсихотическом действии. Селективных В3-антагонистов нет. Существующие селективные Di-антагонисты еще не прошли клинические испытания. Они влияют на действие В2-агонистов, что позволяет предположить взаимодействие между этими двумя типами рецепторов. Обсуждается участие глутаматных, ГАМК- и других рецепторов в
патофизиологии шизофрении и действии нейролептиков. В ближайшие годы должны быть получены ответы на эти вопросы.
В. Отличия между нейролептиками. Хотя все эффективные нейролептики блокируют В2-рецеп-торы, степень этого воздействия по сравнению с влиянием на другие рецепторы варьирует у разных препаратов. Было проведено большое количество экспериментов по исследованию лигандного и рецепторного связывания в попытке выяснить, воздействие на какие рецепторы определяет оптимальную нейролептическую активность. Так, исследования in vitro показали, что хлорпромазин и тиоридазин блокируют а-рецепторы в большей степени, чем.В2-рецепторы. Они также достаточно сильно блокируют серотониновые 5-НТ2-рецепторы. Одйа-ко их аффинность к D,-рецепторам, измеряемая по способности вытеснять селективный В,-лиганд SCH 23390, довольно низка (рис. 28-2). Препараты типа перфеназина и галоперидола в основном действуют на В2-рецепторы; они также оказывают йе-который эффект на 5-НТ2- и а-рецепторы, но почти не действуют на В-рецепторы. Пимозид и ре-моксиприд действуют практически исключительно на В2-рецепторы. Атипичный нейролептик клозапин, который клинически очень отличается от других средств, лучше связывается с В-, 5-НТ2-, а- и гистаминовыми Н-рецепторами, чем с Bt или В2. Рисперидон с одинаковой активностью блокирует 5-НТ2- и В2-рецепторы. Оценка трех различных антипсихотических средств по относительной способности связываться с разными рецепторами показывает, насколько сложно сделать какие-либо однозначные выводы из этих экспериментальных работ:
Хлорпромазин: aj= 5-НТ2 > В2 > В, Галоперидол: В2 > В, = В4 > а( > 5-НТ2 Клозапин: В4 = at > 5-НТ2 > В2 = В(
Очевидно, что связывание с В-рецепторами менее всего сопряжено с клинической эффективйф-стью, но роль других рецепторов оценить трудЙЙ. В настоящее время исследователи ищут атипичные нейролептики, которые либо селективно воздействуют на мезолимбическую систему (для снижения экстрапирамидных эффектов), либо обладают широким спектром действия на рецепторы центральных нейромедиаторов.
Различия антипсихотических средств по побочным эффектам легче связать с определенными рецепторами, чем различия по эффективности
(табл. 28-1 и 28-2). Действие на экстрапирамидную систему связано с влиянием на Э2-рецепторы. Есть надежда, что создание оптимальной комбинации воздействий на разные рецепторы поможет нивелировать эти нежелательные эффекты и повысить клиническую эффективность.
Г. Психические эффекты. Большинство нейролептиков вызывают неприятные субъективные ощущения у здоровых людей. Сочетание сонливости, двигательного беспокойства и вегетативных изменений отличает их от действия традиционных седативных и снотворных средств. У здоровых людей ухудшаются результаты психомоторных и психометрических тестов. Однако психически нездоровые пациенты, наоборот, выполняют эти тесты лучше, после того как лекарственные средства снимают психоз.
Д. Нейрофизиологические эффекты. Антипсихотические средства изменяют частоту и ритм ЭЭГ: обычно наблюдается замедление и повышение синхронизации импульсов. Замедление (гиперсинхрония) иногда бывает фокальной или односторонней, что может привести к ошибочным диагностическим заключениям. На современном оборудовании можно зарегистрировать и количественно охарактеризовать частоту и амплитуду этих изменений.
Изменения ЭЭГ при приеме нейролептиков вначале регистрируются на подкорковых электродах, следовательно, эти средства действуют прежде всего на подкорковом уровне. Вызываемая ими гиперсинхрония может быть причиной активирующего эффекта на ЭЭГ больных эпилепсией, а также причиной возникновения у некоторых людей первичных судорог.
Е. Эндокринные эффекты. Нейролептики оказывают значительное побочное действие на репродуктивную систему. У женщин могут возникать аме-норея-галакторея, ложно-положительные тесты на беременность и повышение либидо, а у мужчин есть опасность снижения либидо и возникновения гинекомастии. Некоторые из этих эффектов опосредованы блокадой тонического подавления дофамином секреции пролактина; другие — повышенной периферической конверсией андрогенов в эстрогены.
Ж. Действие на сердечно-сосудистую систему. Ортостатическая гипотензия и тахикардия в покое часто возникают при применении так называемых “высокодозных” (низкоактивных) фенотиазинов. Среднее артериальное давление, периферическое сопротивление и ударный объем снижаются, часто
та пульса повышается. Эти эффекты можно предсказать, зная действие фенотиазинов на вегетативную нервную систему (табл. 28-2). Могут возникнуть аномалии ЭКГ, особенно при приеме тиоридазина. Возможно удлинение интервала QT и аномальные конфигурации сегмента ST и зубца Т (округлый, плоский, зазубренный). Эти изменения проходят после прекращения приема препарата.
3. Исследования на лабораторных животных. Угнетение условного (но не безусловного) рефлекса избегания — один из наиболее точных тестов для оценки эффективности нейролептиков. Другой тест — подавление вызванного амфетамином или апоморфином стереотипного поведения, что всегда связано с блокадой В2-рецепторов. Антипсихотическое действие предсказывают также по снижению исследовательского поведения без седативного эффекта, возникновению каталептического состояния, угнетению интракраниальной стимуляции зон “награды” и подавлению вызванной апоморфином рвоты. Большинство из этих тестов трудно соотнести с какой-либо моделью клинического психоза.
ТАБЛИЦА 28-2. Побочные эффекты нейролептиков
Тип Проявления Механизм
Вегетативная нервная система Потеря аккомодации, сухость во рту, затруднение мочеиспускания, запор Блокада м-холи-норецепторов'
Ортостатическая гипотензия, импотенция, нарушение эякуляции Блокада а-адренорецепторов
Центральная нервная система Паркинсонический синдром, акатизия, дистонии Блокада дофаминовых рецепторов
Тардивная дискинезия Гиперчувствительность дофаминовых рецепторов
Эндокринная система Токсическое нарушение сознания Аменорея-галакто-рея, бесплодие, импотенция Блокада м-холи-норецепторов Блокада дофаминовых рецепторов, приводящая к гиперпролактинемии
Клиническая фармакология антипсихотических средств
Показания
А. Психиатрические показания. Шизофрения — главное показание для приема антипсихотических средств, являющихся основным методом ее терапии. К сожалению, не всегда больные реагируют на нейролептики, а полного эффекта нельзя достичь ни у одного из них. Так как у некоторых больных шизофренией прогноз благополучный и за первым приступом следует длительная ремиссия, большинство врачей рекомендуют прекращать прием нейролептиков в этой ситуации после достижения ремиссии.
Шизоаффективные состояния более сходны с аффективными расстройствами, чем с шизофренией. Однако психотические проявления этих состояний требуют лечения нейролептиками, которые можно комбинировать с другими средствами, например с антидепрессантами или литием. Маниакальные эпизоды маниакально-депрессивного психоза наиболее эффективно лечатся нейролептиками, хотя в менее тяжелых случаях бывает достаточно лития. После выхода из маниакального состояния прием нейролептиков прекращают. Неманиакальные ажитированные состояния тоже можно лечить нейролептиками (часто в комбинации с бензодиазепинами), но при этом не следует прекращать попытки определить их причину.
Другие психиатрические показания к применению нейролептиков — это синдром Туретта и необходимость контролировать нарушения поведения у больных с сенильной деменцией типа болезни Альцгеймера. Препараты можно применять также вместе с антидепрессантами для коррекции ажитации или психоза у больных с депрессией. Правда, у них выше риск развития тардивной дискинезии, поэтому им следует назначать минимальные дозы. Нейролептики не используют в терапии различных синдромов отмены, например опиоидного.
Одно время нейролептики в малых дозах ошибочно рекомендовали для снятия тревожности при минимальных эмоциональных расстройствах. В таких случаях седативные препараты с противотре-вожным действием (глава 21) лучше со всех точек зрения, в том числе по безопасности и доступности для пациента.
Б. Непсихиатрические показания. Большинство нейролептиков, кроме тиоридазина, обладают сильным противорвотным действием. Оно связано с блокадой как центральных дофаминовых рецепторов (в хеморецепторной триггерной зоне продолговатого мозга), так и периферических (рецепторы желудка). Средства типа прохлорперазина и бенз-хинамида назначают только как противорвотные. Фенотиазины с короткой боковой цепью обладают значительным блокирующим действием на Н(-ре-цепторы и поэтому используются для снятия зуда или, как например прометазин, в качестве предоперационных седативных средств. Бутирофенон дроперидол в сочетании с меперидиноподобным препаратом фентанилом применяют для нейролеп-тоаналгезии. Использование этих средств для анестезии описано в главе 24.
Выбор лекарственного средства
Рациональный выбор нейролептика должен быть основан на различиях химической структуры и соответственно фармакологических свойств препаратов из разных групп, так как они более существенны, чем внутри групп. Например, можно выбрать один препарат из каждой подгруппы фенотиазинов, по одному препарату из тиоксантеновой и бутирофеноновой групп и, возможно, два члена смешанной группы. Примерный набор препаратов показан в табл. 28-3.
Выбор препаратов для узкого применения при определенных симптомах не обоснован, так как они недостаточно селективны. Хотя разрабатываемые сейчас средства считаются более эффективными для коррекции негативных симптомов (эмоционального притупления, снижения социабельности и мотивации), подтверждений этому еще не получено. Практическому врачу не нужно разбираться в мельчайших особенностях каждого нейролептика, но ему необходимо знать эффекты, в том числе и побочные, одного или двух препаратов каждого класса. При выборе лучше всего руководствоваться тем, как больной реагировал на разные антипсихотические средства в прошлом. В последние годы отошли от использования слабых препаратов типа хлорпромазина и тиоридазина и предпочитают применять препараты, обладающие сравнительно высокой силой действия, например тиотиксен, галоперидол и флуфеназин. В настоящее время клозапин назначают только тем пациентам, которые не
ТАБЛИЦА 28-3. Некоторые представители класса нейролептиков
Класс Препарат препаратов Преимущества Недостатки
Фенотиазины
Алифатические Хлорпромазин1 (Торазин) Генерик Много побочных эффектов
Пиперидин Тиоридазин2 (Мелларил) Незначительные экстрапира- Максимальная доза — мидные нарушения; генерик 800 мг/день; нет парентеральной формы; кардио-токсичен
Пиперазин Флуфеназин3 (Пермитил, Проликсин) Есть препарат депо (энантат, (?) Усиливает тардивную деканоат) дискинезию
Тиоксантен Тиотиксен4 (Наван) Есть парентеральная форма; Точно не известны (?) уменьшает тардивную дискинезию
Бутирофенон Галоперидол (Галдол) Есть парентеральная форма; Тяжелые экстрапирамидные генерик нарушения
Дибензоксазепин Локсапин (Локситан) (?) Нет прибавки в весе Точно не известны
Дигидроиндолон Молиндон (Мобан) (?) Нет прибавки в весе Точно не известны
Дибензодиазепин Клозапин(Клозарил) Может действовать на больных, Вызывает агранулоцитоз резистентных к другим пре- почти у 3 % пациентов паратам; незначительная экс-
трапирамидная токсичность
’ Другие алифатические фенотиазины: промазин, трифлупромазин.
2 Другие пиперидиновые фенотиазины: пиперацетазин, мезоридазин.
’Другие пиперазиновые фенотиазины: ацетофеназин, перфеназин, карфеназин, прохлорперазин, трифлуоперазин.
4 Другие тиоксантены: хлорпротиксен.
прореагировали на значительные дозы традиционно применяемых нейролептиков, или тем, у кого значительно выражена тардивная дискинезия. Агранулоцитоз и судороги ограничивают более широкое применение этого препарата.
Дозирование
Диапазон эффективных доз у разных препаратов обычно достаточно широк. В эквивалентных дозах большинство нейролептиков, кроме клозапина, обладают сходной эффективностью. Однако некоторые пациенты, не реагирующие на один препарат, могут поддаваться лечению другим. По этой причине необходимо попробовать несколько средств для того, чтобы найти наиболее эффективное для конкретного пациента. Вполне вероятно, что этот феномен связан с различиями между препаратами по их действию на рецепторы. Больные, которые оказались рефрактерны к терапии несколь
кими нейролептиками в значительных дозах, являются кандидатами для лечения клозапином. Этот препарат в дозах до 900 мг/сутки действует на 30-50 % пациентов, оказавшихся нечувствительными к галоперидолу в дозе 60 мг/сутки. В таких случаях риск, который сопровождает прием клозапина, оправдан.
Соотношения доз некоторых нейролептиков показаны в табл. 28-4.
Концентрация в плазме и клинические эффекты
Попытки определить терапевтический диапазон концентраций нейролептиков в плазме затруднены по многим причинам. Для галоперидола был предложен уровень 2-20 нг/мл, хотя такой диапазон слишком широк. Мониторирование уровня лекарства в плазме, возможное технически, не всегда целесообразно.
ТАБЛИЦА 28-4. Соотношения доз нейролептиков
Препарат Минимальная эффективная терапевтическая доза (мг) Обычный диапазон суточных доз (мг)
Хлорпромазин (Торазин) 100 100-1000
Тиоридазин (Мелларил) 100 100-800
Мезоридазин (Лиданар, Серентил) 50 50-400
Пиперацетазин (Квид) 10 20-160
Трифлуоперазин (Стелазин) 5 5-60
Перфеназин (Трилафон) 10 8-64
Флуфеназин (Пермитил, Проликсин) 2 2-60
Тиотиксен (Наван) 2 2-120
Галоперидол (Галдол) 2 2-60
Локсапин (Локситан) 10 20-160
Молиндон (Лидон, Мобан) 10 20-200
Клозапин(Клозарил) 50 25-600
Рисперидон(Риспердал) 4 4-16
Формы препаратов
Хорошо переносимые парентеральные формы эффективных нейролептиков используют для быстрого начала лечения и для поддерживающей терапии у больных, которые сами не могут следить за приемом лекарств. Так как парентеральное введение повышает биодоступность препарата, то применяемые дозы составляют очень малую часть от пероральных. Флуфеназина деканоат и галоперидола деканоат удобны для длительного парентерального лечения у пациентов, которые не могут принимать препарат перорально.
Режим приема препаратов
Нейролептики можно назначать в несколько приемов в течение времени, необходимого для подбора оптимальной дозы. Но в дальнейшем, даже при пероральном применении, достаточно однократного приема, обычно на ночь. Чем проще схема, тем больше вероятность того, что больной будет ей следовать. Максимальные дозы в лекарственных формах повышаются производителями для возможности одноразового назначения в клинике.
Поддерживающее лечение
У незначительной части больных шизофренией обострение переходит в ремиссию и затем в течение длительного периода лечение не требуется. В большинстве случаев приходится выбирать между лечением “по необходимости” при повторных обострениях и постоянным лечением минимальными дозами. Выбор зависит от социальных факторов, например от наличия семьи и друзей, которые знают симптомы обострения, и от доступности медицинской помощи.
Комбинации препаратов
Сочетание нейролептиков с другими препаратами требует оценки их эффективности. Можно применять трициклические антидепрессанты, но только при четких симптомах депрессии, которая осложняет шизофрению. Они не дают эффекта при эмоциональном притуплении и снижении социабель-ности у шизофреников. Иногда с антипсихотическими препаратами комбинируют литий. Это сочетание эффективно для части больных, у которых нейролептики сами по себе не дают эффекта. Неизвестно, являются ли такие случаи на самом деле нераспознанными маниакальными состояниями. Для снятия бессонницы или тревожности, на которые не действуют нейролептики, могут быть дополнительно назначены седативные средства. Чаще всего с нейролептиками комбинируют противопар-кинсонические препараты.
Побочные эффекты
Большинство побочных эффектов антипсихоти-. ческих средств — продолжение их фармакологических эффектов (табл. 28-1 и 28-2), но некоторые представляют собой проявления аллергии иЛи идиосинкразии.
А. Поведенческие эффекты. Нейролептики являются довольно неприятными препаратами: чем меньше выражен психоз, тем более они неприяТЙы. Многие пациенты прекращают их прием из-за Побочных эффектов, которые можно уменьшить путем снижения дневных и повышения вечерних доз. “Псевдодепрессия”, возможно, связанная с индуцированной нейролептиком акинезией, обычно поддается терапии противопаркинсоНическими средствами. Другая причина “псевдодепрессии” — прием слишком высоких доз, например при частичной
ремиссии. В этом случае снижение дозы должно уменьшить симптомы. Токсические нарушения сознания могут возникать при приеме очень высоких доз препаратов с выраженными м-холинолитичес-кими эффектами.
Б. Неврологические эффекты. Экстрапира-мидные расстройства, которые возникают на ранних этапах лечения — это синдром Паркинсона, акатизия (бесконтрольное моторное беспокойство) и острые дистонии (спастическая кривошея или ретроколлис). Паркинсонический синдром при необходимости можно лечить противопаркинсоничес-кими м-холинолитиками, иногда — амантадином. Со временем он проходит сам по себе, поэтому каждые 3-4 месяца следует делать попытки снижать дозы противопаркинсонических средств. Акатизия и дистонии также могут реагировать на это лечение, но многие врачи предпочитают использовать антигистаминные препараты с седативными и ан-тихолинергическими свойствами, например дифенгидрамин, который назначают парентерально или перорально в виде капсул или эликсира.
Поздняя (тардивная) дискинезия, как понятно из самого названия, возникает на более поздних этапах лечения и проявляется аномальными хорео-атетоидными движениями. Это основной нежелательный эффект нейролептиков. Считалось, что он может быть вызван относительной холинергической недостаточностью вследствие гиперчувствительности дофаминовых рецепторов в системе хвостатое ядро-путамен. У длительно лечившихся пожилых женщин это осложнение развивается чаще, хотя оно может возникнуть у людей любого возраста и пола. Частота тардивной дискинезии варьирует, но в среднем она обнаруживается у 20-40 % больных, проходящих продолжительное лечение. Важна ранняя диагностика, так как далеко зашедшие случаи трудно лечить. Было предложено много методов терапии, но их оценка затруднена тем, что течение этого осложнения крайне вариабельно и иногда оно проходит без лечения. Большинство врачей считают, что начинать лечение необходимо со снижения чувствительности дофаминовых рецепторов, прекратив прием или уменьшив дозу нейролептика. Следующий логический шаг — отмена всех препаратов с центральным антихолинергичес-ким действием, особенно противопаркинсонических средств и трициклических антидепрессантов. Этих двух мер часто бывает достаточно для улучшения. Если они не помогают, то может быть эф
фективно добавление диазепама в дозах до 30-40 мг/день за счет повышения ГАМК-ергической активности. Возможно использование резерпина, хотя при этом существует риск повышения чувствительности рецепторов или усугубления депрессии.
Судороги, хотя и известны как осложнение терапии хлорпромазином, при приеме более сильных средств встречаются крайне редко. Однако у 2-5 % пациентов, принимающих клозапин, возникают первичные судороги.
В. Влияние на вегетативную нервную систему. У большинства пациентов развивается толерантность к м-холинолитическим эффектам нейролептиков. Больным, у которых эти эффекты субъективно очень неприятны или приводят к объективным нарушениям, например к задержке мочи, назначают холиномиметик периферического действия, бетане-хол. При возникновении ортостатической гипотензии или нарушении эякуляции, частых осложнениях терапии хлорпромазином или мезоридазином, следует переходить на прием препаратов с менее выраженными адреноблокирующими свойствами.
Г. Метаболические и эндокринные эффекты. Часто встречается прибавка в весе, которая корректируется диетой. Гиперпролактинемия у женщин приводит к синдрому аменореи-галактореи и бесплодию, у мужчин — к снижению либидо, импотенции и бесплодию.
Д. Токсические или аллергические реакции. При приеме современных эффективных препаратов агранулоцитоз, холестатическая желтуха и кожные поражения возникают редко. В отличие от других средств клозапин вызывает агранулоцитоз у небольшого, но статистически значимого процента пациентов (1-2 %). Этот прогностически неблагоприятный эффект может развиваться быстро, обычно между шестой и восемнадцатой неделями лечения. Неизвестно, имеет ли он иммунную природу, но он обратим при прекращении приема препарата. Из-за риска развития агранулоцитоза пациентам, принимающим клозапин, следует проводить еженедельный клинический анализ крови.
Е. Осложнения со стороны глаз. Частое осложнение терапии хлорпромазином — его отложения в передних структурах глаза (роговице и хрусталике). Они могут ускорять процессы нормального старения хрусталика. Тиоридазин — это единственный нейролептик, вызывающий образование отложений в сетчатке, которые в далеко зашедших случаях могут напоминать пигментную ретинопатию. Об
разование отложений обычно сопровождается появлением коричневатого оттенка у всех предметов в поле зрения. Для снижения риска возникновения этого осложнения максимальную суточную дозу тиоридазина ограничивают 800 мг/день.
Ж. Кардиотоксичность. Тиоридазин в дозах выше 300 мг/сутки почти всегда приводит к незначительным изменениям зубца Т, которые легко обратимы. Передозировка тиоридазина ведет к тяжелым желудочковым аритмиям, нарушениям проводимости и внезапной смерти. Неизвестно, может ли тиоридазин вызывать эти осложнения в терапевтических дозах. В связи с вероятностью усиления ан-тимускаринового и хинидиноподобного эффектов при приеме совместно с трициклическими антидепрессантами такие сочетания препаратов должны применяться с особой осторожностью.
3. Использование при беременности. Хотя нейролептики сравнительно безопасны, при использовании во время беременности возможен некоторый риск. Вопрос о том, применять ли эти средства во время вынашивания ребенка и прерывать ли беременность, если в ее начале использовались нейролептики, решается индивидуально.
И. Злокачественный нейролептический синдром. Это угрожающее жизни осложнение наблюдается у пациентов, которые крайне чувствительны к экстрапирамидным эффектам нейролептиков. Начальный симптом — выраженная мышечная ригидность. При нарушении потоотделения, что нередко бывает при приеме антихолинергических средств, возникает лихорадка, часто достигающая критических значений. Лихорадка и сопровождающий ее стрессовый лейкоцитоз могут заставить ошибочно заподозрить воспаление. Вегетативная лабильность с нарушением пульса и давления — другое проявление изменений среднего мозга. Обычно повышается уровень креатинкиназы, что отражает поражение мышц. Считается, что этот синдром возникает вследствие слишком быстрой блокады постсинаптических дофаминовых рецепторов. Вслед за этим развивается тяжелая форма экстрапирамидного расстройства. В начале его развития пациента следует активно лечить антипар-кинсоническими средствами. Используют миорелаксанты типа дантролена или агонисты дофамина типа бромокриптина, а также диазепам. При наличии лихорадки нужно проводить физическое охлаждение. Сейчас известны так называемые малые формы этого синдрома.
Межлекарственные взаимодействия
Вследствие множественности эффектов антипсихотических средств они вызывают больше фармакодинамических взаимодействий с другими лекарствами, чем фармакокинетических. Аддитивные эффекты могут обнаруживаться при совместном приеме с препаратами, обладающими седативными, а-адреноблокирующими, атихолинергичсекими и хинидиноподобными (для тиоридазина) свойствами.
Описан также ряд фармакокинетических взаимодействий, но они не имеют большого практического значения.
Передозировка
Передозировка нейролептиков редко бывает смертельной, за исключением передозировки мезоридазина и тиоридазина. Обычно сонливость переходит в кому через период возбуждения. Нервно-мышечная возбудимость может повышаться и трансформироваться в судороги. Отмечается миоз, снижаются глубокие рефлексы. Как правило, возникают гипотензия и гипотермия, хотя в дальнейшем может появиться лихорадка. Летальные эффекты мезоридазина и тиоридазина связаны с возникновением желудочковых тахиаритмий.
Пациентов необходимо наблюдать в отделении интенсивной терапии и следить за показателями основных жизненных функций: венозным давлением, газами артериальной крови, электролитами и др. Обоснованы попытки промывания желудка, даже если прошло несколько часов после приема препарата, так как из-за снижения моторики ЖКТ таблетки могут все еще находиться в желудке. Активированный уголь хорошо связывает эти препараты, после него можно дать солевое слабительное. Гипотензия успешно контролируется инфузией жидкостей. Если необходимо применение прессорного препарата, то выбирают дофамин или норадреналин, а не адреналин, p-стимулирующее действие которого при а-блокаде может вызвать вазодилатацию. Судороги можно лечить диазепамом или фенитоином, причем последний назначают сначала в нагрузочной дозе. Терапия передозировок тиоридазина и мезоридазина, которые осложняются аритмиями, сходна с лечением больных после передозировки трициклических антидепрессантов (глава 29).
Новые подходы
Ремоксиприд и раклоприд являются антагонистами D2- и Ц3-рецепторов. Они эффективны и вызывают меньшую экстрапирамидную токсичность. Однако в клинических испытаниях ремоксиприда отмечалось несколько случаев агранулоцитоза. Рисперидон блокирует не только D2-, но и 5-НТ2-ре-цепторы. Зотепин обладает сходным фармакологическим профилем. Таким образом, это новые антипсихотические препараты со смешанным (дофаминовым и недофаминовым) действием. Их место в терапии еще предстоит уточнить.
Психоз, вызванный фенциклидином,— хорошая модель шизофрении. Так как это вещество является антагонистом NMDA-глутаматных рецепторов, были сделаны попытки создания нейролептиков-агонистов NMD А. К настоящему времени в этой области значительного успеха не достигнуто. Однако множественность подходов к синтезу новых нейролептиков рождает надежды. Некоторые препараты, исследуемые сегодня, могут стать прорывом в будущее.
Нетрадиционные лекарственные средства
Эффективность пропранолола в терапии психотических состояний остается неопределенной даже после 30 лет исследований. При добавлении пропранолола к традиционным препаратам может улучшиться состояние пациентов со значительными ажитацией и враждебностью. Высокие дозы бензодиазепинов могут сгладить нарушения поведения так же, как это делали при помощи барбитуратов 35 лет назад. Леводопа, тиреотропинрилизинг-гор-мон, эндорфины и апоморфин оказались неэффективными. Необоснованность мегавитаминной терапии доказана научно, но до сих пор существуют ее сторонники.
Преимущества и ограничения использования нейролептиков
Как отмечено в начале этой главы, нейролептики оказали особое влияние на развитие психиатрии. Во-первых, они переориентировали задачу ухода за больными с психиатрических лечебниц на общество. Для многих пациентов это изменение привело к улучшению жизни в более гуманных условиях и в некоторых случаях сделало возможной жизнь
без постоянных физических ограничений. Для других, к сожалению, трагедия бесцельного существования разыгрывается “на улице”, а не в специализированных заведениях.
Во-вторых, эти препараты перевели психиатрическое мышление на биологический уровень. Отчасти именно благодаря исследованиям, которые подтолкнули поиск эффективных при шизофрении препаратов, мы обязаны нашим сегодняшним знаниям по физиологии ЦНС и фармакологии. Однако, несмотря на интенсивное исследование, шизофрения остается тайной для науки и трагедией для людей. Хотя большинству больных шизофренией нейролептики приносят пользу, никто полностью не выздоравливает.
II. Литий и другие препараты, стабилизирующие настроение
Карбонат лития часто определяют как препарат для лечения маний, но во многих странах его считают стабилизатором настроения, потому что он обладает способностью предотвращать перемены настроения у пациентов с маниакально-депрессивным психозом. Его открытие в 1949 г. было основано на неправильной гипотезе, но стало большой удачей, так как случайно была выбрана правильная доза. Карбамазепин также был известен как стабилизатор настроения, несмотря на то, что не был официально одобрен для применения по этому показанию. Его начали использовать в 70-е годы, основываясь на идее о том, что поскольку маниакальные приступы протекают подобно эпилепсии эпизодически, то и в этой ситуации могут помочь антиконвульсанты. Вальпроат и клоназепам, противосудорожные ГАМК-миметики, также эффективны при этом состоянии.
Причины биполярных аффективных расстройств (маниакально-депрессивного психоза)
Биполярные аффективные (маниакально-депрессивные) расстройства встречаются часто и являются очень серьезным эмоциональным наруше
нием. У пациентов с циклическими приступами мании возникает много симптомов параноидной шизофрении (мания величия, конфликтность, параноидальные мысли и гиперактивность). Для получения хорошего эффекта лития в таких случаях требуется четкая дифференциальная диагностика.
Эпизоды перемены настроения, свойственные этому заболеванию, обычно не связаны с событиями жизни. Его точная биологическая база еще не определена, но считается, что в основе лежат изменения катехоламиновой активности. Препараты, которые повышают эту активность, обостряют маниакальное состояние, а те, которые снижают активность дофамина или норадреналина,— улучшают. Причина внезапного перехода мании в депрессию у некоторых пациентов неясна. Биполярные расстройства имеют четкий семейный характер наследования. Генетические исследования определили по меньшей мере три хромосомы, возможно, связанные с этим заболеванием.
Общая фармакология лития
Фармакокинетика
Литий — это моновалентный катион небольшого размера. Его фармакокинетика отражена в табл. 28-5.
Фармакодинамика
Несмотря на многочисленные исследования, механизм действия лития по-прежнему неясен. Сейчас эти исследования в основном сконцентрированы в трех направлениях. Изучается влияние лития на: 1) электролитный обмен и ионный транспорт; 2) нейромедиаторы и их высвобождение; 3) вторичные мессенджеры, опосредующие действие нейромедиаторов. Последний из этих трех подходов наиболее обещающий.
А. Действие на электролиты и ионный транспорт. По своим свойствам литий очень похож на натрий. Он может замещать натрий при генерации потенциалов действия и в Na+/Na+-o6MeHe через мембрану. Литий подавляет последний процесс, то есть обмен Li+/Na+ постепенно замедляется после начала приема лития. В терапевтических концентрациях (около 1 ммоль/л) литий не оказывает значительного влияния на обмен Na+/Ca2+ или на На+,К+-АТФазу натриевого насоса.
ТАБЛИЦА 28-5. Фармакокинетика лития
Всасывание Почти полное в течение 6-8 часов; пиковый уровень в плазме через 0.5-2 часа
Распределение В основном в жидких средах; медленно проникает в клетки. Начальный объем распределения — 0.5 л/кг, который затем повышается до 0.7-0.9 л/кг; отмечается некоторая секвестрация в костях. Не связывается с белками
Метаболизм Не метаболизируется
Выведение Почти полностью с мочой. Клиренс — приблизительно 20 % от клиренса креатинина. Период полувыведения из плазмы около 20 часов
Б. Действие на нейромедиаторы. Литий усиливает некоторые эффекты серотонина, хотя данные, подтверждающие это, противоречивы. Его действие на норадреналин неоднозначно. В частности, литий может снижать оборот дофамина и норадреналина. Если эти эффекты будут доказаны, то они объяснят механизм его терапевтического действия при маниях. Литий также блокирует развитие гиперчувствительности дофаминовых рецепторов при длительной терапии нейролептиками. Наконец, он может усйливать синтез ацетилхолина, возможно, за счет повышения захвата холина нервными окончаниями. В ряде клинических исследований показано, что повышение холинергической активности может облегчать манию. Однако, как еще будет отмечено, последствия усиления высвобождения ацетилхолина могут быть нейтрализованы действием лития на вторичные посредники.
В. Действие на вторичные посредники. В настоящее время лучше всего изучен характер действия лития на инозитолфосфат. Еще в ранних исследованиях было обнаружено, что в ответ на введение лития изменяется уровень инозитолфосфата в мозгу, но значимость этого эффекта не была понятна, пока не стало известно о функционировании инозитол-1,4,5-трифосфата (ИТФ) и диацилглицеро-ла (ДАГ) в качестве вторичных мессенджеров ос-адренергической и мускариновой передачи (глава 2). Литий ингибирует активность некоторых ферментов нормального метаболизма мембранных фосфоинозитидов, в том числе подавляет превращение инозитолбифосфата (ИБФ) в инозитолмо
нофосфат (ИМФ) и ИМФ — в инозитол (рис. 28-3). Эти блоки приводят к уменьшению количества фос-фатидилинозитол-4,5-бифосфата (ФИБФ), мембранного предшественника ИТФ и ДАГ. С течением времени действие медиаторов на клетку будет снижаться в той же степени, что и активность ФИБФ-зависимых путей. Так как такая активность может быть значительно повышена при мании, то в результате ингибирующего влияния на нее лития происходит селективное подавление гиперактивных нервных путей. Например, недавно было показано, что длительная активация мускариновых рецепторов предотвращает нормальное тормозное действие аденозина на определенные нейроны гиппокампа. Это холинергическое возбуждение можно снять при помощи терапевтических концентраций лития по механизму селективного подавления.
При исследованиях норадренергических эффектов в изолированной ткани мозга обнаружили, что литий может снижать активность норадреналинчувствительной аденилатциклазы. Вероятно, этот механизм имеет отношение и к антидепрессив-ному действию, и к подавлению мании. Его связь с действием лития на ИТФ-механизмы в настоящее время неизвестна.
Снаружи рецептор
Рис. 28-3. Действие лития на ИТФ- и ДАГ-систему вторичных посредников. На схеме показана синаптическая мембрана нейрона. (ФИБФ — фосфатидилинозитол-4,5-бифосфат; ФИ — фосфатидилинозитол; ФИМФ — фос-фатидилинозитолмонофосфат; ФЛС — фосфолипаза С; G — G-белок; Эффекты: активация протеинкиназы С, мобилизация внутриклеточного кальция и т. д.) Литий, ингибируя рециркуляцию субстратов инозитола, вызывает истощение депо вторичных посредников (ФИБФ), тем самым уменьшая их высвобождение. Литий может также участвовать и в других регуляторных механизмах
Клиническая фармакология лития
Биполярные аффективные расстройства (маниакально-депрессивный психоз)
Общепринято считать, что карбонат лития является средством выбора при биполярном расстройстве, особенно в фазе мании. Так как начало его действия медленное, при выраженной мании может потребоваться одновременное применение нейролептиков. Лечебный эффект (инициация ремиссии) наблюдается у 60-80 % пациентов. Однако у тех больных, которым требуется госпитализация, эффект лития регистрируется значительно реже. Такая же зависимость обнаруживается и при длительном лечении. В целом эффект наблюдается в 60 % случаев, но он менее выражен у более тяжелых больных. Это привело к использованию в тяжелых случаях комбинированного лечения.
При не очень выраженных симптомах мании литий сам по себе достаточно эффективен. В более тяжелых случаях почти всегда требуется добавление нейролептиков. После достижения ремиссии нейролептик отменяют, а литий продолжают давать как средство поддерживающей терапии.
В депрессивной фазе маниакально-депрессивного психоза необходимо назначение антидепрессанта (глава 29). По некоторым данным, трициклические антидепрессанты могут усугублять манию и приводить к более быстрой смене циклов заболевания. Несмотря на это, они остаются препаратами выбора, так как данный эффект наблюдается только в единичных случаях. Трудно выбрать конкретный антидепрессант нового поколения, потому что они тоже могут активировать манию. В таких случаях, когда лития недостаточно, чтобы остановить спровоцированные эпизоды мании, эффективным может оказаться карбамазепин.
В отличие от нейролептиков или антидепрессантов, которые действуют на центральную или вегетативную нервные системы, литий оказывает лишь незначительный седативный эффект и не влияет на вегетативную нервную систему. Так как это неорганический ион небольшого размера, то он распределяется в жидких средах организма и не метаболизируется. Его уровни легко измерить в плазме, моче или слюне. Таким образом, кинетику лития легко изучить, а его концентрации в жидких средах
в достаточной степени коррелируют с выраженностью терапевтических эффектов.
Другая важная область применения лития — это профилактика как мании, так и депрессии. Интересно, что так называемый функциональный психоз успешно лечится таким простым веществом как карбонат лития.
Другие показания к применению
Острая эндогенная депрессия обычно не считается показанием к применению лития. Однако повторные обострения, имеющие циклическое течение, поддаются лечению литием или имипрамином, эффекты которых достоверно отличаются от плацебо.
Шизоаффективные расстройства характеризуются сочетанием шизофренических симптомов и нарушением эмоциональных реакций по типу депрессии или возбуждения. В случае возбуждения применяются нейролептики сами по себе или в комбинации с литием, при депрессии — трициклические антидепрессанты.
Признается связь алкоголизма с биполярными расстройствами. При сочетании обоих заболеваний литий может способствовать уменьшению алкогольной зависимости. Однако при отсутствии у алкоголика аффективных расстройств препарат не обладает этим действием.
Монотерапия литием при шизофрении не дает эффекта, но добавление его к нейролептикам может помочь пациентам, которые исходно не реагировали на нейролептики. В такой же ситуации эффективной может оказаться комбинация антипсихотических препаратов с карбамазепином.
Интересное показание для применения лития, которое сейчас исследуется,— это коррекция агрессивного и конфликтного поведения у тюремных заключенных. Хотя этот эффект может оказаться очень полезным, научного обоснования он пока не получил.
Дозирование
До начала лечения литием следует сделать клинический и биохимический анализы крови и анализ мочи, а у пожилых людей и у пациентов с заболеваниями сердца в анамнезе — ЭКГ. Эти тесты послужат оценкой исходного состояния больного для прогнозирования возможных побочных эффектов.
Для определения оптимальных доз карбоната лития следует учитывать вес, возраст пациента и функцию почек. Начальный объем распределения будет равен объему жидких сред организма (около 0.5-0.6 л/кг у женщин и около 0.55-0.7 л/кг у мужчин). Так как литий почти полностью выводится почками, дозу следует снижать при нарушении клиренса креатинина, например у пожилых людей.
Суточная доза лития 0.5 мэкв/кг приведет к созданию желаемой концентрации в пределах 0.6-1.4 мэкв/л после недели лечения при нормальной почечной функции. Каждые 30 мг препарата содержат приблизительно 8 мэкв лития. Суточные дозы могут варьировать у разных людей между 600 и 3600 мг. Большинству требуется 1500-1800 мг/день. Чтобы избежать нарушений со стороны ЖКТ, почти всегда необходимо дробить дозу лития на несколько приемов. Лучше всего принимать литий сразу после еды. Можно попробовать и пролонгированные формы.
Мониторирование лечения
Клиницисты обычно полагаются на измерение концентрации лития для оценки дозы, необходимой при терапии эпизода мании или адекватном длительном лечении. Измерения, как правило, делают через 10-12 часов после последнего приема препарата, поэтому все данные, имеющиеся в литературе, относятся к этому периоду времени.
Через пять дней после начала лечения следует определить концентрацию лития, так как в этот момент она становится стабильной. Если для получения клинического эффекта требуется повышение дозы препарата, то нужно сделать простые арифметические расчеты (новая доза равна произведению предыдущей дозы на желательную концентрацию лития в крови, разделенному на имеющуюся концентрацию лития). Уровень лития при приеме новой дозы необходимо проверить еще через пять дней. После достижения требуемого уровня лития в крови измерения проводят реже, а также дополнительно во время сопутствующих заболеваний и при назначении других препаратов.
Решение о приеме лития с профилактической целью зависит от многих факторов: частоты и тяжести эпизодов, нарастания тяжести каждого последующего эпизода, желания пациента получать пожизненную терапию. Лечение литием следует прекратить после завершения обострения, если оно
первое или если больной не очень ответственно относится к лечению. Пациенты, страдающие несколькими обострениями за год, являются кандидатами для поддерживающего лечения. Некоторым пациентам достаточно уровня 0.6 мэкв/л, но наилучшие результаты были получены при более высоком уровне — 0.9 мэкв/л.
Лекарственные взаимодействия
Почечный клиренс лития снижается на 25 % диуретиками (например, тиазидами), поэтому при совместном применении этих препаратов дозу лития следует уменьшить в той же степени. Сходное снижение клиренса лития было отмечено при приеме некоторых нестероидных противовоспалительных средств, которые блокируют синтез простагландинов. Такого взаимодействия не наблюдали при сочетании лития с аспирином или ацетаминофеном. Все нейролептики могут вызывать более тяжелый экстрапирамидный синдром при комбинированной терапии с литием.
Побочные эффекты и осложнения
На разных этапах терапии могут возникнуть различные побочные эффекты лития. Некоторые достаточно безопасны, но тем не менее их появление может быть сигналом приближающихся серьезных токсических реакций.
А. Неврологические и психические побочные эффекты. Чаще других осложнений терапии литием (даже при приеме терапевтических доз) возникает тремор. Пропранолол, который хорошо помогает при эссенциальном треморе, подавляет и тремор, вызванный литием. При приеме лития возникают и другие неврологические нарушения: хорео-атетоз, двигательная гиперактивность, атаксия, дизартрия и афазия. Психические нарушения — это нарушения мышления, синдром отмены или гиперкинезы. Появление любых новых неврологических илц психических симптомов — это сигнал ко временному прекращению терапии литием и контролю уровня препарата в сыворотке крови.
Б- Действие на функцию щитовидной железы. У большинства пациентов литий снижает функцию щитовидной железы, но этот эффект не прогрессирует и обратим. У небольшого числа больных возникает истинная гипертрофия щитовидной железы, у еще меньшего — гипотиреоидизм. Хотя были
предложения проводить оценку исходного состояния щитовидной железы и мониторирование ее функции, эта процедура экономически не оправдалась.
В. Действие на почки. Полидипсия и полиурия часто возникают даже при приеме терапевтических концентраций, но они обратимы. Основной физиологический механизм этого эффекта — нарушение способности собирательных трубочек задерживать воду под действием антидиуретического гормона, что приводит к повышенному выведению воды (нефрогенному несахарному диабету). Вызванный литием несахарный диабет резистентен к вазопрессину, но проходит при назначении амилорида. Имеется обширная литература о других нефротоксических эффектах при длительной терапии литием, в том числе о хроническом интерстициальном нефрите и гломерулопатии минимальных изменений с нефротическим синдромом. Встречались случаи снижения гломерулярной фильтрации, но значительной азотемии или почечной недостаточности не возникало. Пациентам, принимающим литий, следует избегать дегидратации и связанного с нею повышения концентрации лития в моче, а также периодически проверять концентрационную способность почек.
Г. Отеки. При приеме лития часто возникают отеки, которые могут быть связаны со способностью лития усиливать задержку натрия. У некоторых пациентов можно наблюдать при этом прибавку в весе, хотя здесь не исключаются и другие механизмы.
Д. Побочные эффекты на сердце. Брадикардия-тахикардия (синдром слабости синусового узла) является противопоказанием для назначения лития, так как этот ион еще больше тормозит синусовый узел. На ЭКГ часто возникает уплощение зубца Т, но диагностическое значение этого признака не до конца ясно.
Е. Применение при беременности. Почечный клиренс лития повышается во время беременности и снижается сразу после родов. Даже если уровень лития во время беременности находился в нормальных пределах, после родов могут появиться признаки токсичности. В таких случаях требуется особая осторожность при мониторировании концентрации лития. Литий попадает к новорожденным через грудное молоко, где его концентрация составляет ’/з-’/г от сывороточной. Токсичность лития у новорожденных проявляется летаргией, цианозом, сни
жением сосательного рефлекса и рефлекса Моро, возможна также гепатомегалия.
По вопросу дисморфогенеза нет единого мнения. Опубликован отчет, свидетельствующий о значительном повышении частоты аномалий развития сердца, особенно аномалии Эбштейна, у детей, матери которых принимали литий во время беременности.
Ж. Другие побочные эффекты. На ранних этапах лечения литием были замечены преходящие угреподобные кожные высыпания. Иногда они проходят при временном прекращении приема и больше не возникают при возобновлении лечения. Фолликулит — менее неприятное, но более частое осложнение. Всегда сопровождающий прием лития лейкоцитоз является скорее всего отражением стимуляции лейкопоэза, а не мобилизации имеющегося пула лейкоцитов. Этот “побочный эффект” сейчас используют при лейкопении как терапевтический. У мужчин, принимающих литий, может возникнуть нарушение половой функции.
Передозировки
Ятрогенные передозировки встречаются чаще, чем умышленные или связанные с непреднамеренным случайным приемом лития. Они обычно связаны с кумуляцией лития вследствие изменения состояния пациента, например снижения уровня натрия в крови, применения мочегонных, колебаний почечной функции или при беременности. Так как происходит уравновешивание концентраций лития в тканях и в крови, концентрации лития в крови по своей высоте могут не соответствовать токсичности; любой уровень выше 2 мэкв/л должен считаться потенциально опасным. Литий — это ион небольшого размера, поэтому его легко удалить диализом. Эффективны как перитонеальный, так и гемодиализ, хотя предпочтение отдается последнему. Диализ следует продолжать, пока концентрация в плазме не упадет ниже терапевтической.
Карбамазепин
Карбамазепин появился как альтернатива литию в случаях, когда последний противопоказан больному. Карбамазепин применяется как при острой мании, так и с профилактической целью. Побочные эффекты карбамазепина иногда даже менее выражены, чем побочные эффекты лития. Карба
мазепин применяют отдельно или, в рефрактерных случаях, в комбинации с литием. Механизм действия карбамазепина не вполне ясен, но препарат может снижать сенсибилизацию мозга к повторяющимся колебаниям настроения. Такой механизм действия может быть подобен противосудорожному действию карбамазепина.
Применение карбамазепина для стабилизации настроения идентично его использованию в качестве противосудорожного препарата (глава 23). Начинают прием обычно с 200 мг два раза в день, повышая дозу по необходимости. Поддерживающая доза эквивалентна поддерживающей дозе при эпилепсии (800-1200 мг/сутки). Концентрация в плазме 3-14 мг/л считается оптимальной, хотя терапевтический диапазон пока не определен. Известным побочным эффектом карбамазепина являются гематологические нарушения, но они не доставляют особых проблем при его приеме в качестве стабилизатора настроения. Передозировки карбамазепина требуют неотложной помощи по принципам терапии при отравлении трициклическими антидепрессантами.
Вальпроевая кислота и клоназепам
Действие этих средств при маниакально-депрессивном психозе не так хорошо изучено, как применение лития и карбамазепина. Предварительные исследования показали, что вальпроат натрия помогает предотвратить обострения. Как и карбамазепин, его применяют аналогично использованию при эпилепсии, начиная со 100 мг три или четыре раза в день, постепенно повышая дозу до уровня противоэпилептической. Клоназепам считается неэффективным при монотерапии, но иногда усиливает эффект лития при комбинированном приеме. Он используется в традиционной противосудорожной дозе 0.5-20 мг/день.
Препараты
Антипсихотические средства (нейролептики)
Ацетофеназин (Тиндал)
Перорально: таблетки по 20 мг
Хлорпромазин (генерик, Торазин, др.) Перорально: таблетки по 10, 25, 50,
100, 200 мг; сироп 10 мг/5 мл; концентрат 30, 100 мг/мл
Перорально для пролонгированного действия: капсулы по 30, 75,150, 200, 300 мг Ректально: свечи по 25,100 мг
Парентерально: 25 мг/мл для в/м инъекций
Хлорпротиксен (Тарактан)
Перорально: таблетки по 10, 25, 50,100 мг; концентрат 100 мг/5 мл
Парентерально: 12.5 мг/мл для в/м инъекций
Клозапин (Клозарил)
Перорально: таблетки по 25,100 мг
Флуфеназин (генерик, Пермитил, Проликсин) Перорально: таблетки по 1, 2.5, 5,10 мг; эликсир 2.5 мг/5 мл; концентрат 5 мг/мл Парентерально: 2.5 мг/мл для в/м инъекций
Флуфеназина эфиры (деканоат — генерик, Проликсина энантат, Проликсина деканоат) Парентерально: 25 мг/мл
Галоперидол (генерик, Галдол)
Перорально: таблетки по 0.5, 1, 2, 5,10, 20 мг; концентрат 2 мг/мл
Парентерально: 5 мг/мл для в/м инъекций
Галоперидола эфир (Галдела деканоат) Парентерально: 50,100 мг/мл для в/м инъекций
Локсапин (Локситан)
Перорально: капсулы по 5,10, 25, 50 мг; концентрат 25 мг/мл
Парентерально: 50 мг/мл для в/м инъекций
Мезоридазин (Серентил)
Перорально: таблетки по 10, 25, 50, 100 мг; концентрат 25 мг/мл
Парентерально: 25 мг/мл для в/м инъекций
Молиндон (Мобан)
Перорально: таблетки по 5, 10, 25, 50, 100 мг; концентрат 20 мг/мл
Перфеназин (генерик, Трилафон)
Перорально: таблетки по 2,4, 8,16 мг; концентрат 16 мг/5 мл
Парентерально: 5 мг/мл для в/м или в/в инъекций
Прохлорперазин (генерик, Компазин) Перорально: таблетки по 5, 10, 25 мг; сироп 5 мг/5 мл
Перорально для пролонгированного действия: капсулы по 10,15, 30 мг Ректально: свечи по 2.5, 5, 25 мг Парентерально: 5 мг/мл для в/м инъекций
Пимозид (Орап)
Перорально: таблетки по 2 мг
Промазин (генерик, Спарин)
Перорально: таблетки по 25, 50,100 мг Парентерально: 25, 50 мг/мл для в/м инъекций
Рисперидон (Риспердал)
Перорально: таблетки по 1, 2, 3,4 мг
Тиоридазин (генерик, Мелларил, др.) Перорально: таблетки по 10, 15, 25, 50,100, 150, 200 мг; концентрат 30, 100 мг/мл; суспензия 25,100 мг/5 мл
Тиотиксен (генерик, Наван)
Перорально: капсулы по 1, 2, 5,10, 20 мг;
концентрат 5 мг/мл
Парентерально: 2 мг/мл для в/м инъекций;
порошок растворить до 5 мг/мл для инъекций
Трифлуоперазин (генерик, Стелазин, Супразин) Перорально: таблетки по 1, 2, 5, 10 мг; концентрат 10 мг/мл
Парентерально: 2 мг/мл для в/м инъекций
Трифлупромазин (Весприн)
Парентерально: 10, 20 мг/мл для в/м инъекций
Стабилизаторы настроения
Карбамазепин (генерик, Тегретол)
Перорально: таблетки по 200 мг, таблетки для жевания по 100 мг;
суспензия 100 мг/5 мл
Клоназепам (Клонопин)
Перорально: таблетки по 0.5,1, 2 мг
Карбонат лития (генерик, Эскалит) (Примечание:
300 мг карбоната лития = 8.12 мэкв Li+.) Перорально: капсулы по 150, 300, 600 мг; таблетки по 300 мг; сироп 8 мэкв/5 мл Перорально для пролонгированного действия: таблетки по 300,450 мг
Вальпроевая кислота (генерик, Депакен) Перорально: капсулы по 250 мг;
сироп 250 мг/5 мл
Антидепрессанты
Лео Е. Холлистер
29
Депрессия — одно из наиболее частых психических заболеваний. В каждый момент времени около 5-6 % людей находится в состоянии депрессии, а у 10 % депрессия возникает в какой-либо период их жизни. Симптомы депрессии могут быть мало заметны, и их пропускают как пациенты, так и врачи. Больные с неясными жалобами, которые трудно объяснить соматическими нарушениями, а также люди, которых в жизни называют “невротиками”, могут на самом деле быть в состоянии депрессии.
Депрессия — это гетерогенное заболевание, имеющее различные проявления. Американская ассоциация психиатров пересмотрела третье издание (1987) Диагностического и статистического руководства по психическим заболеваниям (DSM-HI-R). В соответствии с ним дифференцируют несколько диагнозов аффективных расстройств. Чисто депрессивными синдромами являются большая депрессия и дистимия (малая депрессия), а биполярные расстройства и циклотимия состоят из депрессии в сочетании с манией. С учетом этого упрощенная классификация выглядит следующим образом: 1) “реактивная”, или вторичная депрессия (наиболее частая, свыше 60 %), которая возникает в ответ на реальные факторы, например горе, болезнь и т. д.; 2) “эндогенная” депрессия — генетически предопределенное биохимическое нарушение, которое проявляется неспособностью справляться с обычным стрессом (около 25 %); 3) депрессия, связанная с биполярными аффективными расстройствами (маниакально-депрессивным психозом), составляющая около 10-15 %. Препараты, обсуждаемые в этой главе, применяются в основном для лечения депрессии второго типа. Табл. 29-1 показывает методы дифференцировки трех типов депрессии.
Больных с психической депрессией до появления антидепрессантов лечили только электросудорожной терапией. Антидепрессанты не являются стимуляторами ЦНС и противопоказаны при орга
ническом или вызванным лекарственными препаратами угнетений ЦНС. Исследования механизмов действия антидепрессантов в основном касаются их влияния на различные аминные нейромедиаторы.
Патогенез большой депрессии: аминная гипотеза
Вскоре после появления резерпина в начале 1950-х гг. стало ясно, что этот препарат способен вызывать депрессию у пациентов, которые лечились от гипертензии и шизофрении, а также у здоровых людей. В течение нескольких последующих лет фармакологические исследования показали, что основной способ действия резерпина — это нарушение хранения аминных нейромедиаторов типа серотонина и норадреналина в пресинаптических пузырьках нервных окончаний. Резерпин вызывал депрессию и истощал депо аминных нейромедиаторов; поэтому был сделан вывод, что депрессия связана со снижением функции аминных нейромедиаторов. Такое простое заключение привело к появлению так называемой аминной гипотезы депрессии. Главное противоречие этой гипотезы состоит в том, что, несмотря на выраженность фармакологических эффектов трициклических антидепрессантов и ингибиторов МАО, их клинический эффект появляется не раньше, чем через несколько недель или даже месяцев. Были сделаны попытки объяснить это наблюдение развитием медленной компенсации в ответ на начальную блокаду обратного захвата медиатора или ингибирование МАО.
Несмотря на несовершенство аминной гипотезы, она позволила создать экспериментальную модель для поиска новых антидепрессантов. В результате все существующие сейчас антидепрессанты, кроме бупропиона, оказывают основное влияние на хранение, метаболизм или обратный захват серотонина или норадреналина.
ТАБЛИЦА 29-1. Типы депрессии »
Тип Диагностические признаки Комментарии
Реактивная В связи с реальными событиями жизни: Более 60 % всех депрессий. Депрессивный
депрессия утраты; болезнь (инфаркт миокарда, синдром: депрессия, тревожность, сома-
рак); лекарственные препараты (антиги- тические жалобы, напряжение, чувство
пертензивные средства, алкоголь, гор- вины. Может изменяться спонтанно или в
моны); другие психические нарушения (старческие изменения) ответ на попытки лечения
Большая депрессия Провоцирующие реальные события, не- Около 25 % всех депрессий. Депрессивный
(эндогенная) адекватены степени депрессии. Авто- синдром + субъективные признаки: нару-
номная депрессия (не связанная с ре- шение сна, двигательной активности, ли-
альными событиями). Любой возраст (с бидо, аппетита. Обычно поддается дей-
детства до старости). Биологически пре- ствию антидепрессантов или электросу-
допределенная (семейный анамнез) дорожной терапии. Характерны повторные обострения
Биполярное аффек- Есть эпизоды мании. Протекает цикличес- Около 10-15 % всех депрессий. Может
тивное расстрой- ки. Редко изолированная мания. Иногда быть диагностирована как эндогенная,
ство (маниакально- изолированная депрессия. Обычно ма- если гипоманические эпизоды прошли
депрессивный пси- ния + депрессия незамеченными. Карбонат лития стаби-
хоз) лизирует настроение. Мания может потребовать лечения нейролептиками; депрессию лечат антидепрессантами
I. Базисная фармакология антидепрессантов
Химические свойства
Антидепрессантной активностью обладают вещества разной структуры. Их количество постоянно растет, но ни одна из групп не имеет неоспоримых преимуществ.
А. Трициклические антидепрессанты (ТЦА, рис. 29-1). Трициклические антидепрессанты, называемые так из-за характерного трехкольцевого ядра, применяются уже три десятилетия. Они похожи на фенотиазины химически и в меньшей степени — фармакологически. Как и фенотиазины, ТЦА сначала применялись как антигистаминные средства, а потом — как антипсихотические препа-раты. Открытие их антидепрессантивного действия произошло в результате случайного наблюдения. Прототипами этой группы являются имипрамин и амитриптилин.
Б. Гетероциклические соединения, препараты второго поколения. С 1980 г. в практику было введено несколько гетероциклических антидепрессан
тов (антидепрессанты второго поколения). Пять из них применяются в США. На рис. 29-2 представлены четыре препарата. Амоксапин и мапротилин напоминают по структуре трициклические антидепрессанты, а тразодон и бупропион не похожи на них. Эти гетероциклические препараты значительно не отличаются по эффективности от более старых антидепрессантов. Венлафаксин — это более новый и оригинальный по химической структуре препарат второго поколения.
В. Селективные блокаторы обратного захвата серотонина (СБОЗС). Один из недостатков многих антидепрессантов — это множественность фармакологического действия — свойство, унаследованное ими от препаратов фенотиазинового ряда. Насколько известно, м-холинолитические, антигистаминные и ос-адренолитические влияния ТЦА связаны только с их побочными эффектами. С тех пор как стало известно, что антидепрессант с минимальной вегетативной токсичностью — флуоксетин — является высокоселективным блокатором обратного захвата серотонина, все усилия были приложены к поиску средств сходного действия. В настоящее время имеется три селективных блокатора обратного захвата серотонина, еще несколько
^-(CHzhNfCHsh
R2:—Н
Имипрамин
Ri
R1:=CH(CH2)2N(CH3)2
Амитриптилин
R,:—(Ch2)3NCh3
R2:—Н
Дезипрамин
Ri=-(CH2hN(CH3)2
R2:—Cl
Кломипрамин
R1:=CH(CH2)2NHCH3
Нортриптилин
R,:—(Ch2)3NHCH3 Проптриптилин
R!СН2СН(СНэ)СН2М(СНэ)2
R2:—H
Тримипрамин
Рис. 29-1. Структура трициклических антидепрессантов (ТЦА)
проходят клинические испытания. Все они совсем не похожи по структуре на трициклические препараты (рис. 29-3).
Г. Ингибиторы моноаминоксидазы (МАО). Ингибиторы МАО можно разделить на гидразиды, имеющие последовательность C-N-N, например фенелзин и изокарбоксазид, и негидразиды, у которых нет такой группы, например транилципро-мин (рис. 29-4). Транилципромин очень напоминает декстроамфетамин, который и сам является слабым ингибитором МАО. Транилципромин сохраняет некоторые из симпатомиметических эффектов, свойственных амфетаминам. Гидразиды необратимо связываются с МАО, транилципромин тоже имеет пролонгированное действие, хотя оно и обратимо. Эти более старые препараты неселективно блокируют и МАО-А, и МАО-В. Моклобемид — новый селективный МАО-А-ингибитор короткого действия. Множество похожих обратимых ингибиторов МАО-А находится в стадии изучения.
Д. Симпатомиметические стимуляторы. Иногда используются в качестве антидепрессантов декстроамфетамин, другие амфетамины и аналоги амфетамина, например, метилфенидат. Хотя блокада МАО, вызванная амфетаминами, слишком слаба, чтобы оказать значительное антидепрессантное
действие, у некоторых людей она может дополнить эффект других препаратов.
Фармакокинетика
А. Трициклические антидепрессанты. Большинство ТЦА неполностью всасываются и подвергаются активному пресистемному метаболизму. В результате значительного связывания с белками и сравнительно высокой растворимости в жирах объем их распределения очень большой. ТЦА метаболизируются по двум основным путям: трансформацией трициклического ядра и изменением алифатической боковой цепи. Первый путь включает процессы гидроксилирования и конъюгации до глюкуронидов; второй — в основном деметилирование. Монодеметилирование третичных аминов приводит к образованию активных метаболитов, например дезипрамина и нортриптилина (которые существуют и как отдельные лекарственные средства; рис. 29-1). Пропорция монодеметилированных метаболитов может отличаться у разных пациентов. Отношение амитриптилина к его метаболиту нортриптилину смещено в сторону исходного препарата. Наоборот, метаболит имипрамина дезипрамин превышает по количеству исходный препарат. Фар-
Мапротилин
Рис. 29-2. Некоторые гетероциклические антидепрессанты второго поколения
макокинетические параметры разных антидепрессантов представлены в табл. 29-2.
Б. Гетероциклические соединения. Их фармакокинетика сходна с фармакокинетикой трициклических препаратов. Как следует из табл. 29-2, отнюдь не все известно про эти средства. Тем не менее обнаружено, что у них неодинаковая биодоступность, высокая степень связывания с белками, они имеют значительные объемы распределения и, возможно, активные метаболиты. Тразодон и венла
факсин обладают самым коротким периодом полувыведения, что требует введения нескольких доз в течение дня.
В. Селективные блокаторы обратного захвата серотонина. Фармакокинетические параметры этих препаратов обобщены в табл. 29-2. Флуоксетин хорошо всасывается, пиковые концентрации в плазме достигаются в течение 4-8 часов. Его деметилированный активный метаболит, норфлуоксетин, имеет период полувыведения 7-9 дней в ста-
Сертралин
Рис. 29-3. Селективные блокаторы обратного захвата серотонина (СБОЗС)
СН2 — СН2 — NH — NH2
Фенелзин
Рис. 29-4. Некоторые ингибиторы моноаминоксидазы. Фенелзин — это гидразид фенилэтиламина (рис. 9-3), транилпипромин имеет циклопропиловую аминную боковую цепь и напоминает декстроамфетамины (рис. 9-4). Эти средства неселективны и блокируют фермент на очень длительное время. Моклобемид — новый обратимый ингибитор МАО-А короткого действия, который структурно похож на симпатомиметики
ционарный период; период полувыведения исходного средства несколько короче. Флуоксетин подавляет некоторые ферменты, метаболизирующие лекарственные препараты, что приводит к лекарственным взаимодействиям с другими антидепрессантами, а также с другими лекарствами. Сертралин и пароксетин по фармакокинетическим параметрам похожи на ТЦА.
Г. Ингибиторы МАО. Ингибиторы МАО хорошо всасываются из ЖКТ. Гидразидные препараты (фенелзин и изокарбоксазид) ацетилируются в печени и различаются по скорости выведения в зависимости от фенотипа ацетилирования у пациента (глава 4). Однако ингибирование МАО продолжается даже тогда, когда следов этих препаратов (старого поколения) уже не остается в плазме. Поэтому фармакокинетические параметры не играют роли в определении дозы. Рекомендуется определять активность МАО для оценки эффекта, хотя этот метод в клинике не применяется. На практике считается, что эффект будет сохраняться от 7 дней (транилципромин) до 2-3 недель (фенелзин, изокарбоксазид) после прекращения приема лекарства. Моклобемид быстро всасывается и выводится, более 90 % препарата появляется в моче в виде метаболитов уже в течение 12 часов.
ТАБЛИЦА 29-2. Фармакокинетические параметры различных антидепрессантов
Средство Биодоступность (%) Связывание с белквми плазмы (%) Период полувыведения (час) Активные метаболиты Объем распределения (л/кг) Терапевтические концентрации в плазме (нг/мл)
Амитриптилин 31-61 82-96 31-46 Нортриптилин 5-10 80-200
Амоксапин ? ? 8 7-,8-гидрокси ? ?
Бупропион 60-80 85 11-14 ? 20-30 25-100
Кломипрамин ? ? 22-84 Деметил 7-20 240-700
Дезипрамин 60-70 73-90 14-62 ? 22-59 145’
Доксепин 13-45 ? 8-24 Деметил 9-33 30-150
Флюоксетин 70 94 24-96 Норфлуоксетин 12-97 ?
Имипрамин 29-77 76-95 9-24 Дезипрвмин 15-30 > 180
Мапротилин 66-75 88 21-52 Деметил 15-28 200-300
Нортриптилин 32-79 93-95 18-93 10-гидрокси 21-57 50-150
Пароксетин 50 95 24 нет 28-31 ?
Протриптилин 77-93 90-95 54-198 ? 19-57 70-170
Сертралин ? 98 22-35 Деметил 20 ?
Тразодон ? ? 4-9 т-хлорфенилпиперазин ? ?
’ Могут быть эффективны более низкие уровни, но точных данных по этому поводу нет.
Фармакодинамика
А. Действие антидепрессантов на аминные нейромедиаторы. Аминная гипотеза была поддержана исследованиями механизма действия разных групп антидепрессантов (рис. 29-5). ТЦА блокируют обратный захват аминов (норадреналина или серотонина), то есть “выключают” процесс, который прекращает функцию этих медиаторов в синапсе (табл. 29-2, глава 6). Такое действие удлиняет контакт нейромедиатора с рецептором. Ингибиторы МАО блокируют главный путь разрушения медиаторов, что позволяет большему количеству аминов находиться в пресинаптических депо и высвобождаться. Амфетаминоподобные симпатомиметики тоже блокируют аминную помпу, но считается, что они действуют в основном за счет повышения высвобождения катехоламиновых медиаторов. Таким образом, эти три класса средств могут восстановить дефект аминной нейропередачи, хотя и разными механизмами. Некоторые новые препараты второго поколения действуют таким же образом, у других эти эффекты минимальны. Способность тразодона и селективных блокаторов захвата серотонина не влиять на захват нервными окончаниями норадреналина отличает их от способности большинства ТЦА блокировать захват норадреналина в дозах, которые мало влияют на захват серотонина. Так как обе эти группы препаратов являются антидепрессантами, предложен двойной механизм, который включает обе медиаторные системы. Считается, что норадреналиновая система — это основное общее конечное звено механизма антидепрессивного действия и что подавление захвата серотонина связано с норадренергической системой и вызывает сходный результат.
Б. Рецепторные и пострецепторные эффекты. Последнее время особое внимание стали уделять влиянию повышения количества нейромедиатора в синапсе на постсинаптические рецепторы. В исследованиях постсинаптической активности показано, что концентрация цАМФ чаще понижается, а не повышается. Кроме того, наблюдается понижение количества некоторых Р-адренорецепторов, что происходит параллельно клиническому улучшению. Таким образом, начальное повышение количества медиатора с течением времени вызывает компенсаторное понижение рецепторной активности, то есть «/о® «-регуляцию рецепторов. Снижение количества стимулированного норадреналином
Рис. 29-5. Схема, показывающая некоторые возможные точкй приложения действия антидепрессантов. При длительном приеме этих лекарств уменьшается обратный захват норадреналина или серотонина (или обоих одновременно), снижается количество Р-рецепторов и ингибируется образование цАМФ. Ингибиторы МАО действуют на МАО в нервных окончаниях и вызывают те же эффекты в отношении p-рецепторов и синтеза цАМФ
цАМФ и Р-адренорецепторов показано для селективных блокаторов захвата норадреналина, для препаратов смешанного действия (на норадреналин и серотонин), для ингибиторов МАО и для электросудорожной терапии. В меньшей степени это справедливо для селективных блокаторов захвата серотонина, а2-антагонистов и Р-агонистов. Снижение активности серотониновых рецепторов продемонстрировано в основном для препаратов, которые блокируют захват серотонина.
Оказалось достаточно трудным обнаружить изменения серотониновых или Р-адренорецепторов у больных с депрессией. Таким образом, связь вызванных лекарственными средствами изменений рецепторов и эффективности в лечении депрессии неясна. В поисках объяснения действия антидепрессантов внимание постепенно сместилось с изменений медиаторов на изменения рецепторной функции.
В. Эффекты специфических антидепрессантов.
1. Трициклические средства. Препараты первого поколения антидепрессантов обладают разной селективностью в отношении блокады обратного захвата норадреналина или серотонина (табл. 29-3). Характерные электрофизиологические эффекты — это снижение импульсации норадренергических нейронов. В дальнейшем (к моменту появления клинического эффекта) импульсация переходит на
нормальный уровень или выше нормального. Эти средства обладают также множеством побочных эффектов, связанных с их действием на вегететив-ную нервную систему.
2. Гетероциклические средства. Амоксапин — метаболит нейролептика локсапина, сохраняющий некоторые из его свойств. Сочетание антидепрес-сивных и антипсихотических эффектов делает этот препарат очень удобным для терапии депрессии у психотических больных. Однако это антипсихотическое действие может вызвать акатизию, паркинсонизм, синдром аменореи-галактореи и, возможно, отдаленную дискинезию.
Мапротилин (“тетрациклический” препарат) больше всего похож на дезипрамин, в том числе и структурно. Возможно, что он так же, как и более новые антидепрессанты, имеет менее выраженные седативный и м-холинолитический эффекты, чем трициклические средства.
Опыт клинического применения тразодона показал, что эффективность этого препарата непредсказуема: у некоторых больных развивается максимальный эффект, а у других — почти никакого.
Бупропион по структуре похож на амфетамин и стимулятор ЦНС катинон, который применяют на Среднем Востоке в виде ката. В высоких дозах бупропион подавляет обратный захват дофамина в центральных нейронах у животных. Его действие может быть основано преимущественно на дофаминергических эффектах.
3. Селективные блокаторы обратного захвата серотонина. Флуоксетин стал первым применяемым в клинике СБОЗС. Пароксетин и сертралин отличаются в основном более коротким периодом полувыведения. Их эффективность не превышает эффективности препаратов первого поколения, но зато пароксетин и сертралин значительно менее токсичны. Поэтому применение этих средств нашло хороший отклик у пациентов, несмотря на новые побочные эффекты, например головную боль, тошноту и двигательное беспокойство.
Может произойти опасное фармакодинамическое взаимодействие, когда флуоксетин или один из новых селективных блокаторов обратного захвата серотонина применяется вместе с ингибитором мо-ноаминоксидазы. Повышение содержания моноамина и подавление его обратного захвата приводят к значительному повышению количества серотонина в синапсе »- “серотониновому синдрому”. Он включает в себя гипертермию, мышечную ри
гидность, миоклонус и быстрые изменения в состоянии психики и физиологических функций.
4. Ингибиторы МАО. МАО-А — аминооксидаза, которая в первую очередь метаболизирует норадреналин, серотонин и тирамин. МАО-В более селективна по отношению к дофамину. Если с депрессией связаны только норадренергические и серотонинергические синапсы, но не дофаминергические, то селективная блокада МАО-А должна быть эффективным способом лечения. Необратимая блокада МАО ингибиторами первого поколения вызывает кумуляцию тирамина и нарушение пресистемного метаболизма тирамина, содержащегося в пищевых продуктах. В результате этого возрастает риск гипертензивных реакций на тирамин пищи. По существующим на данный момент сведениям, обратимый ингибитор МАО короткого действия моклобемид не дает такого эффекта. Селективный ингибитор МАО-В селегилин теряет свою селективность при приеме в антидепрессивных дозах. Так как он действует на фермент, который метаболизирует дофамин, то он более полезен в терапии болезни Паркинсона (глава 27).
II. Клиническая фармакология антидепрессантов
Показания к применению
Основное показание к применению антидепрессантов — лечение депрессии, но клинический опыт дает обоснование их использования и по некоторым другим показаниям.
А. Депрессия. Это показание специально формулируют расширенно, хотя данные клинических испытаний показали, что антидепрессанты избирательно помогают только при приступах большой депрессии. Большая депрессия диагностируется не столько по степени тяжести, сколько по своим качественным особенностям. Раньше ее называли “эндогенной”, “витальной” или “вегетативной” — по характерным нарушениям ритмов сна, голода, насыщения, полового влечения и двигательной активности. Большая депрессия иногда плохо распознается, поэтому ее нередко не диагностируют и больных не лечат.
Б. Панические состояния. В 1962 г. впервые было обнаружено свойство имипрамина оказывать положительный эффект при острых эпизодах тре
вожности, которые позже стали называться паническими приступами. Недавние исследования показали, что он столь же эффективен, как ингибиторы МАО и бензодиазепины. Однако предпочтение отдается последней группе лекарств, так как больные их лучше переносят, а эффекты развиваются раньше.
В. Обсессивно-компульсивные нарушения. Для лечения этих состояний применяются блокаторы обратного захвата серотонина. Последние исследования концентрируются на флуоксетине и других селективных средствах.
Г. Энурез. Энурез — одно из показаний для применения ТЦА. Существует много доказательств их эффективности при этом состоянии, однако фармакотерапия не является в данном случае методом выбора. Эффект лекарственной терапии длится только в течение периода приема.
Д. Хронические болевые синдромы. Специалисты в области лечения боли считают, что ТЦА играют большую роль в терапии болевых синдромов, причина которых неизвестна. Пока не ясно, являются ли эти состояния болевыми эквивалентами депрессии или депрессия возникает вторично вследствие наличия боли. Не исключается, что ТЦА оказывают прямое действие на пути проведения болевой чувствительности. Иногда в сочетании с ними применяют фенотиазины.
Е. Другие показания. К менее обоснованным показаниям к применению антидепрессантов следует отнести нарушения аппетита (булимия и нервная анорексия), катаплексию при нарколепсии, школьную фобию и гиперкинетический синдром с нарушениями внимания.
Выбор препарата
Сравнения разных антидепрессантов неизменно приводят к заключению, что все они приблизительно одинаково эффективны. Хотя в целом это справедливо, конкретные больные по неизвестной причине лучше реагируют на определенный препарат. Таким образом, выбор средства осуществляется эмпирически. Самый главный ориентир, если больной ранее принимал препараты этой группы — анамнез. Иногда по анамнестическим данным исключают ТЦА, например, если больной раньше хорошо реагировал на ингибиторы МАО.
Антидепрессанты дают более выраженный эффект у пациентов со значительным вегетативным
компонентом: нарушением сна, аппетита, либидо, потерей веса тела и психомоторными нарушениями.
Трициклические и гетероциклические средства в основном отличаются по степени седативного эффекта (амитриптилин, доксепин и тразодон вызывают самый выраженный седативный эффект, а протриптилин — самый незначительный) и м-хо-линолитического эффекта (самый большой — у амитриптилина и доксепина, самый слабый — у тразодона) (табл. 29-3). Хотя существует мнение, что для ажитированных и тревожных больных предпочтительны препараты с более сильным седативным эффектом, а средства с меньшим седативным действием следует назначать пациентам с психомоторной депрессией, эта гипотеза не имеет фактического подтверждения.
Не доказано преимущество ни одного гетероциклического средства по эффективности ни перед одним трициклическим. Исходно основными положительными характеристиками гетероциклических средств называли: 1) более быстрое начало действия; 2) минимизацию побочных седативных и вегетативных эффектов; 3) меньшую токсичность при передозировках. Оказалось трудным найти доказательства более быстрого начала действия гетероциклических средств. Амоксапин и мапротилин дают такое же количество вегетативных побочных эффектов и такое же седативное действие, как и ТЦА; некоторые другие препараты типа тразодона, бупропиона и флуоксетина имеют меньше таких эффектов. Амоксапин и мапротилин примерно так же опасны при передозировках, как и ТЦА; другие новые препараты несколько менее токсичны.
Не существует особых показаний при депрессии для селективных блокаторов обратного захвата серотонина. Дело в том, что пока не определено, с функцией какой системы (норадренергической или серотонинергической) связана депрессия. Несмотря на высокую стоимость, эти средства популярны среди пациентов вследствие хорошей переносимости. Ранее существовавшие подозрения о том, что флуоксетин усиливает суицидные и агрессивные мысли и поведение, еще предстоит доказать; эти симптомы могут являться частью депрессивного синдрома.
Ингибиторы МАО помогают пациентам с так называемой “атипичной” депрессией (это название не совсем правильное и едва ли помогает в определении этого типа расстройств). На эти препараты
ТАБЛИЦА 29-3. Фармакологические различия между антидепрессантами’
Средство м-холинолитическое действие Седативное действие Блокирует механизм обратного захвата:
Серотонина Норадреналина Дофамина
Амитриптилин +++ +++ +++ + 0
Амоксапин ++ ++ + ++ +
Бупропион 0 0 +, 0 +.0 ?
Дезипрамин + + 0 +++ 0
Доксепин +++ +++ ++ + 0
Флуоксетин + + +++ 0, + 0, +
Имипрамин ++ ++ +++ ++ 0
Мапротилин ++ ++ 0 +++ 0
Нортриптилин ++ ++ +++ ++ 0
Пароксетин + 0 +++ 0 0
Протриптилин 0 ++ ? +++ ?
Сертралин + 0 +++ 0 0
Тразодон +++ 0 ++ 0 0
' 0 = нет эффекта, «+» = небольшой, «++» = умеренный, «+++» = высокий,«?» = точно не известно.
лучше всего реагируют депрессивные больные с проявлениями тревожности, фобий, ипохондрии.
При депрессии лишь немногие врачи назначают литий — препарат для терапии маний. Однако иногда комбинация лития и антидепрессанта приводит к усилению эффекта последнего. Другое возможное обоснование применения лития — профилактика повторных эпизодов депрессии.
Дозы
Обычные суточные дозы антидепрессантов представлены в табл. 29-4. Они почти всегда определяются эмпирически; лимитирующим фактором являются побочные эффекты. К некоторым эффектам может развиться толерантность, поэтому обычно лечение начинают с малых доз, постепенно повышая дозу, либо до определенного суточного уровня, либо до уровня, который снимает депрессию, либо до максимально переносимой дозы. Эффективная доза зависит от многих факторов.
Дозы препаратов для лечения острого эпизода депрессии
При амбулаторном лечении начальные дозы ТЦА варьируют от 10 до 75 мг в первый день лечения в зависимости от веса и роста пациента и от переносимости им препарата. Такой консерватив-
ТАБЛИЦА 29-4. Обычные суточные дозы
антидепрессантов
Препарат Доза (мг)
Трициклические антидепрессанты
Амитриптилин 75-200
Кломипрамин 75-300
Дезипрамин 75-200
Доксепин 75-300
Имипрамин 75-200
Нортриптилин 75-150
Протриптилин 20-40
Тримипрамин 75-200
Препараты второго поколения
Амоксапин 150-300
Бупропион 200-400
Мапротилин 75-300
Тразодон 50-600
Венлафаксин 75-225
Ингибиторы МАО
Изокарбоксазид 20-50
Фенелзин 45-75
Транилципромин 10-30
Селективные блокаторы обратного
захвата серотонина
Флуоксетин 10-60
Пароксетин 20-50
Сертралин 50-200
ный подход не обязателен при госпитальном лечении, когда начальные дозы должны быть около 100 мг. После 2-3 дней приема в таких дозах можно начать постепенное повышение: по 25 мг каждый второй или третий день до максимальной дневной дозы — 150 мг. Она поможет достигнуть терапевтического диапазона концентраций у большинства пациентов. Однако если эффекта не отмечается, дозу можно повышать и далее — до 300 мг/сут.
Такая же схема применяется и при назначении гетероциклических средств и селективных блокаторов обратного захвата серотонина. Тразодон, имеющий короткий период полувыведения, принимают 2-3 раза в день. Флуоксетин обычно назначают, начиная с 20 мг/день, и поддерживают этот уровень в течение нескольких недель до того, как становится возможным оценить эффект. Более высокие дозы применяют только в очень редких случаях, а пожилым пациентам назначают 20 мг через день.
Фенелзин изучен лучше всех других ингибиторов МАО. Желательного подавления активности фермента (приблизительно на 80 %) можно достигнуть при приеме дозы 1 мг/кг/сутки. Дозы других ингибиторов МАО надо аккуратно подбирать до получения концентраций препарата, при которых у больного отмечается минимальная ортостатическая гипотензия.
Ингибиторы МАО, бупропион, флуоксетин, сертралин и пароксетин обычно назначают с утра, так как они оказывают стимулирующий эффект и приводят к бессоннице, если их принимать вечером. Почти все другие антидепрессанты обладают седативным эффектом, и их следует принимать на ночь. В этом случае менее заметно и действие на вегетативную нервную систему.
Поддерживающая терапия
Решение о необходимости поддерживающей терапии больного с депрессией принимается в зависимости от течения заболевания в прошлом. Если этот эпизод депрессии первый в жизни больного и хорошо поддавался лечению, то рациональным решением будет постепенно снижать дозу и отменить препарат через несколько месяцев. Если обострение не наступает, то прием препарата можно прекратить до возникновения следующего обострения, предсказать которое нельзя, но известно, что повторные обострения возникают практически всегда.
Однако если данный эпизод у пациента не первый и тяжесть депрессии возрастает с каждым новым эпизодом, то такой пациент — кандидат для поддерживающей терапии. Обычно поддерживающая терапия более эффективна, если используется полная доза, а не частичная. Длительность лечения варьирует, хотя некоторым пациентам требуется лечение в течение всей жизни.
Мониторирование концентраций в плазме
Рутинное мониторирование концентраций антидепрессантов в плазме, хотя и возможно технически, но теоретически не обосновано. Измерение концентраций ТЦА показало, что на определенном этапе лечения около 20 % пациентов неаккуратны в соблюдении режима приема препарата. В таком случае “плохой эффект” препарата может быть объяснен неправильным его применением, что могут подтвердить измерения уровней в плазме. Кровь для такого анализа надо брать по завершении абсорбции, то есть через 10-12 часов после приема последней дозы. Даже при приеме постоянной дозы и взятии крови в одно и то же время наблюдаются колебания общей концентрации препарата и соотношения его метаболитов. Ни при каких обстоятельствах результат такого теста не может ставиться выше клинического суждения врача.
Неэффективность терапии
Почти у трети пациентов, принимающих ТЦА, не возникает лечебного эффекта. При оценке резистентности к лечению следует помнить правило пяти “П”: правильный диагноз, препарат, правильная доза, продолжительность лечения, прочие методы терапии.
Диагноз следует пересмотреть, если больной не реагирует на терапию в течение 2-3 недель при достижении необходимых концентраций в плазме. Если у пациента активное биполярное расстройство (маниакально-депрессивный психоз), то можно добавить литий; если выявляются психотические симптомы — нейролептик. Некоторые клиницисты считают, что лечение следует продолжать в течение минимум нескольких недель или месяцев. Но нужно помнить, что депрессия приводит к тяжелым последствиям, если вовремя не достигнут антидепрес-сивный эффект.
Кроме добавления препаратов для лечения дополнительных недепрессивных симптомов, следует рассмотреть возможность назначения других антидепрессантов. Считается логичным начинать лечение с ТЦА. Если они неэффективны, то следующей группой препаратов в настоящее время считаются селективные блокаторы обратного захвата серотонина, а не ингибиторы МАО и не гетероциклические средства. Большинство врачей в попытках найти эффективный препарат предпочитают пробовать лекарства разных классов, а не разные препараты одного класса.
Необходимо учитывать также дозу и длительность лечения. Чаще всего лечение оказывается неэффективным вследствие неправильного дозирования, в рефрактерных случаях дозу следует повышать до максимально переносимой. Продолжительность лечения одним препаратом до попытки попробовать новый определяется самим врачом.
И, наконец, некоторым больным могут потребоваться совсем другие методы лечения, например электросудорожная терапия. Ее считают терапией отчаяния при эндогенных депрессиях, но она может помочь пациентам, которым не помогла фармакотерапия.
Несоблюдение режима приема препарата — еще одна важная причина неэффективности лечения. Пациентов нужно предупредить, что эффект проявляется медленно, иногда не ранее чем через 3 недели. Неспособность переносить побочные эффекты и разочарование в лечении — основные причины прекращения приема ТЦА и, соответственно, невозможности снять депрессию.
Рекомендована для применения комбинированная терапия ТЦА и ингибиторами МАО, хотя недавние клинические испытания не показали особых преимуществ этого сочетания. Для лечения больных с психотической депрессией или депрессивной фазой маниакально-депрессивного психоза следует назначать антидепрессанты в сочетании с нейролептиками или литием, соответственно. '
Побочные эффекты
Побочные эффекты различных антидепрессантов приведены в табл. 29-5. Большинство из них неопасно, но может значительно повлиять на дисциплинированность пациента в отношении режима приема препарата. Чем тяжелее депрессия, тем больше вероятность того, что больной сможет пе
ренести побочные эффекты. Здоровые же люди отмечают, что даже умеренные дозы этих средств вызывают неприятные ощущения.
Лекарственные взаимодействия
А. Фармакодинамические взаимодействия. Многие из фармакодинамических взаимодействий антидепрессантов с другими препаратами уже рассматривались. Седативный эффект может усиливаться (суммироваться) при применении других седативных средств, особенно алкоголя. Пациентов, принимающих ТЦА, следует предупредить, что это может плохо отразиться на их способности вести машину. Ингибиторы МАО, повышая количество катехоламинов, сенсибилизируют пациентов к действию прямых симпатомиметиков типа тирамина, присутствующего в некоторых пищевых продуктах и напитках, а также типа диэтилпропиона и фенил-пропаноламина, которые являются симпатомиме-тиками, применяемыми в качестве лекарств. Такая сенсибилизация может вызвать гипертонический криз. Исследования моклобемида показали, что этот селективный и обратимый ингибитор МАО-А не вызывает сенсибилизации к тирамину. Опасность взаимодействия ингибиторов МАО и селективных блокаторов обратного захвата серотонина уже упоминалась: серотониновый синдром может привести к смертельному исходу.
Б. Фармакокинетические взаимодействия. ТЦА нейтрализуют антигипертензивное действие гуанетидина (глава 11). Артериальное давление не только возвращается к норме, но может даже повыситься до опасных цифр. Гуанетидин концентрируется в симпатических нервных окончаниях тем же насосом, который блокируется ТЦА. Следовательно, он не достигает места своего действия. Описано такое же влияние ТЦА и на другие антигипертензивные средства типа метилдопы и клонидина. Доксепин может вызывать такой эффект только при применении в высоких дозах, так как он в меньшей степени блокирует этот насос. Ингибиторы МАО увеличивают период полувыведения тех препаратов, которые подвергаются окислительному дезаминированию. Флуоксетин, блокируя метаболизм некоторых средств, может повысить их концентрации в плазме до токсических уровней. Пароксетин и сертралин не обладают таким эффектом.
ТАБЛИЦА 29-5. Побочные эффекты антидепрессантов
Трициклические антидепрессанты Седативные эффекты Симпатомиметические м-холинолитические Сердечно-сосудистые Психиатрические Неврологические Метаболическо- эндокринные Ингибиторы МАО Сонливость, суммация эффектов с другими седативными средствами Тремор, бессонница Затуманивание зрения, запор, задержка мочеиспускания, нарушения мышления Ортостатическая гипотензия, блокады проведения, аритмии Обострение психоза, синдром отмены Судороги Прибавка в весе, нарушения половой функции Головная боль, сонливость, сухость во рту, прибавка в весе, ортостатическая гипотензия, нарушения половой функции
Амоксапин Мапротилин Тразодон, венлафаксин Бупропион Побочные эффекты ТЦА + некоторые побочные эффекты нейролептиков (глава 28) Побочные эффекты ТЦА; дозозависимые судороги Сонливость, головокружение, бессонница, головные боли, похудание Головокружение, сухость во рту, потливость, тремор, ухудшение психозов, в высоких дозах возможны судороги
Флуоксетин и другие блокаторы обратного захвата серотонина Тревожность, бессонница, астения, тремор, потливость, симптомы нарушений ЖКТ, сыпь
Передозировки
А. ТЦА. Передозировки ТЦА очень опасны, тем более что пациенты с депрессией часто склонны к суицидальному поведению. Поэтому не следует выдавать на руки более 1.25 г препарата (50 разовых доз по 25 мг). Когда суицид вероятен, таблетки лучше отдать члену семьи пациента. Лекарства необходимо прятать от детей. И случайная, и преднамеренная передозировка встречаются часто и требуют неотложной помощи. Основные симптомы передозировки включают: 1) кому, шоки иногда метаболический ацидоз; 2) подавление дыхания с тенденцией к внезапному апноэ; 3) возбуждение или делирий как до, так и после нарушения сознания; 4) повышенную нервно-мышечную возбудимость и судороги; 5) гиперпирексию; 6) недержание мочи и кала; 7) множество нарушений со стороны сердечно-сосудистой системы, в том числе блокады проведения и аритмии.
Лечить поражения сердца очень трудно. Следует использовать противоаритмические средства с минимальным тормозным эффектом на сердце. Успешно применяют лидокаин, пропранолол и фенитоин, а хинидин и прокаинамид противопоказаны. Внутривенные инъекции физостигмина (0.5 мг)
до общей дозы 3-4 мг могут привести пациента в сознание и прекратить наджелудочковую тахикардию. При желудочковых аритмиях такой его эффект маловероятен. Однако в отделениях неотложной помощи при отравлениях сейчас уже не применяют физостигмин в подобных ситуацях. Прежде всего необходимо иметь оборудование для реанимации, позволяющее осуществлять продолжительное мониторирование сердца. Следует часто измерять газы артериальной крови и pH, так как и гипоксия, и метаболический ацидоз могут привести к аритмиям. Для восстановления кислотно-основного баланса и для коррекции гипокалиемии может стать необходимым введение бикарбоната натрия и внутривенно хлорида калия. В рефрактерных случаях устанавливают искусственный водитель ритма. В остальном лечение только поддерживающее (глава 60).
Б. Гетероциклические средства. Передозировка амоксапина приводит к тяжелым нейротоксическим последствиям с судорогами, которые плохо поддаются терапии. Отравление мапротилином также вызывает судороги, кроме того, препарат кар-диотоксичен в высокйх дозах. Передозировки других гетероциклических средств не так опасны, и их следует лечить симптоматически.
В. Ингибиторы МАО. Интоксикация ингибиторами МАО встречается редко. Возбуждение, делирий, повышенная нервно-мышечная возбудимость сменяются ступором, судорогами, шоком, гипертермией. Обычно достаточно поддерживающей терапии, хотя могут быть полезны седативные фенотиазины с а-адренолитическими свойствами типа хлорпромазина.
Г. Селективные блокаторы обратного захвата серотонина. Известно несколько случаев смертельного исхода при передозировке флуоксетина, хотя обычно при этом пострадавшие принимали и другие препараты. Можно предложить только симптоматическое лечение, так как вследствие большого объема распределения эти препараты, как и другие антидепрессанты, не выводятся при диализе. Для передозировки характерны судороги, которые можно купировать диазепамом. Известны случаи, когда пациенты выживали после приема 2.6 г сертралина. Отравление пароксетином протекает доброкачественно: при приеме препарата в дозах до 850 мг не наблюдалось проявлений кардиотоксичности.
Препараты
Трициклические антидепрессанты
Амитриптилин (генерик, Элавил, др.)
Перорально: таблетки по 10, 25, 50, 75,100, 150 мг
Парентерально: 10 мг/мл для в/м инъекций
Кломипрамин (Анафранил, показан только при обсессивно-компульсивных расстройствах) Перорально: капсулы по 25, 50, 75 мг
Дезипрамин (генерик, Норпрамин, Пертофран) Перорально: таблетки по 10, 25, 50, 75, 100, 150 мг; капсулы по 25, 50 мг
Доксепин (генерик, Синекван, др.)
Перорально: капсулы по 10, 25, 50, 75, 100, 150 мг; концентрат 10 мг/мл
Имипрамин (генерик, Тофранил, др.) Перорально: таблетки по 10, 25, 50 мг (Имипрамина гидрохлорид);
капсулы (Имипрамина памоат) Парентерально: 25 мг/2 мл для в/м инъекций
Нортриптилин (Авентил, Памелор)
Перорально: капсулы по 10, 25, 50, 75 мг;
раствор 10 мг/5 мл
Протриптилин (Вивактил)
Перорально: таблетки по 5,10 мг
Тримипрамин (Сурмонтил)
Перорально: капсулы по 25, 50, 100 мг
Гетероциклические соединения
Амоксапин (генерик, Асендин)
Перорально: таблетки по 25, 50, 100, 150 мг
Бупропион (Веллбутрин)
Перорально: таблетки по 75,100 мг
Мапротилин (генерик, Лудиомил)
Перорально: таблетки 25, 50, 75 мг
Тразодон (генерик, Дезирел)
Перорально: таблетки по 50, 100,150, 300 мг
Венлафаксин (Эффексор)
Перорально: таблетки по 25, 37.5, 50, 75, 100 мг
Ингибиторы обратного захвата серотонина
Флуоксетин (Прозак)
Перорально: порошок 20 мг;
жидкость 20 мг/5 мл
Пароксетин (Паксил)
Перорально: таблетки по 20, 30 мг
Сертралин (Золофт)
Перорально: таблетки по 50,100 мг
Ингибиторы МАО
Изокарбоксазид (Марплан)
Перорально: таблетки по 10 мг
Фенелзин (Нардил)
Перорально: таблетки по 15 мг
Транилципромин (Парнат)
Перорально: таблетки по 10 мг
Избранная литература
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders (DSM-III-R), 3rd ed. revised. American Psychiatric Association, 1987.
Baldessarini R. J. Current status of antidepressants: Clinical pharmacology and therapy. J. Clin. Psychiatry, 1989; 50:117.
Cesura A. M., Pletcher A. The new generation of monoamine oxidase inhibitors. Prog. Drug Res. 1992; 38: 171.
Опиоидные аналгетики и антагонисты
30
Уолтер Л. Вэй, Е. Леонг Вэй, Говард Л. Филдс
Сто лет назад не существовало антибиотиков, гормональных или антипсихотических средств. В распоряжении врачей было мало истинно полезных лекарств, среди них выделялся морфин, эффективно облегчавший сильную боль различного происхождения. С его помощью также уменьшали диарею, кашель, тревогу и бессонницу. По этим причинам Уильям Ослер назвал морфин “божественным лекарством”.
Термин “наркотик”, часто применяемый к веществам данной группы, неточен, поскольку “наркоз” означает сонливое или оцепенелое (ступорозное) состояние. Термины “опиат” и “опиоидный аналге-тик" более точны: они указывают на аналгетичес-кое действие без погружения в сон или утраты сознания. Под опиоидными аналгетиками обычно понимают все естественные и полусинтетические производные алкалоидов опия так же, как синтетические препараты, имитирующие действие морфина. Опиаты — вещества, которые получают из алкалоидов опийного мака. В последние годы в ситуациях, когда в клинике традиционно использовался морфин, применяют аналгетики, обладающие антагонистическим действием по отношению к морфину (смешанные агонгисты-антагонисты). Более того, существование эндогенных пептидов с анал-гетическими свойствами предполагает, что в будущем в группу опоидных аналгетиков могут войти синтетические пептиды со свойствами опиоидов. Следовательно, в число опиоидов мы должны включить опиаты (производные алкалоидов опия), синтетические опиоиды (агонисты, смешанные агонисты-антагонисты и антагонисты) и опиопептины (такие как Р-эндорфин и энкефалины). Морфин рассматривают как прототип агониста.
История
Источником опия (неочищенная субстанция) и одного из его очищенных извлечений, морфина, является опийный мак Papaver somniferum. Это растение использовалось в течение более 6000 лет, имеются свидетельства его применения в древнем Египте, Греции и Риме. Интересно, что до XVIII в. способность опия вызывать пристрастие не являлась предметом озабоченности.
Современные основы этого раздела фармакологии были заложены немецким фармацевтом Сер-тюрнером, который впервые выделил из опия чистое активное вещество, имеющее щелочную реакцию. Это важнейшее событие позволило стандартизовать активность естественных продуктов. После испытаний вещества на себе и друзьях Сертюр-нер предложил назвать вещество “морфином” в честь греческого бога сна Морфея.
I. Базисная фармакология опиоидных аналгетиков
Источники
Опиум получают из опийного мака, надрезая головки, после того как опали лепестки цветков. Вытекающее из надреза белое смолистое вещество постепенно твердеет и становится коричневым. В таком виде оно называется опием и содержит около 20 алкалоидов, в том числе морфин, кодеин, тебаин и папаверин. Тебаин и папаверин не являются аналгетиками, но тебаин служит предшественником некоторых полусинтетических опиатных
агонистов (например, эторфина, который используется в ветеринарии и в 500-1000 раз активнее морфина) и антагонистов (налоксон). Папаверин — вазодилататор, имеющий в настоящее время ограниченное применение в клинике, однако на его основе создан препарат верапамил, блокирующий кальциевые каналы (глава 12). Основной алкалоид опия — морфин, содержащийся в концентрации около 10 %. Кодеин содержится в концентрации менее 0.5 % и для использования в качестве лекарства его синтезируют из морфина. Культивирование опийного мака в наше время ограничено международными соглашениями, но нелегальное производство опия очень распространено и с трудом поддается контролю.
Химия
В таблице 30-1 перечислены соединения в соответствии с их свойствами агонистов, смешанных агонистов-антагонистов или антагонистов. Некоторые их фармакологические свойства представлены в таблице 30-2. Незначительные различия в химическом строении могут существенно изменить действие этих соединений, превращая агонист в антагонист или в соединение с обоими эффектами, т. е. в “смешанный агонист-антагонист”.
Антагонистические свойства связывают с замещением метильного радикала у атома азота на более крупную группу — аллильную в случае налор-фина и налоксона, метилциклопропановую или метилциклобутановую для некоторых других веществ.
Значительно изменяет фармакокинетические свойства морфина замена гидроксильных групп в положениях СЗ и С6. Метильное замещение у фенольного гидроксила в положении СЗ уменьшает подверженность печеночному метаболизму (эффект первого прохождения) путем образования глюкуроновых конъюгатов в этом положении. Следовательно, такие лекарства как кодеин и оксикодон отличает более высокое соотношение активности в зависимости от вида введения (внутрь/ парентерально) в сравнении с другими препаратами. При ацетилировании обеих гидроксильных групп морфина образуется героин, который значительно быстрее морфина проникает через гематоэнцефалический барьер. Затем он быстро гидролизуется в мозгу до моноацетилморфина и морфина.
Фармакокинетика
А. Всасывание. Большинство опиоидных анал-гетиков хорошо всасывается из мест подкожного и внутримышечного введения, а также со слизистой носа, полости рта и желудочно-кишечного тракта. Кроме того, возможность проникновения фентанила через кожу сделала трансдермальный путь одним из важных путей введения этого препарата. Однако, хотя всасывание через слизистую ЖКТ достаточно быстрое, биодоступность некоторых соединений при пероральном приеме может быть значительно снижена вследствие выраженного эффекта первого прохождения (в данном случае происходит образование глюкуронидов в печени). Следовательно, доза для приема внутрь должна быть увеличена по сравнению с дозой для парентерального введения. Так как количество фермента, ответственного за данную реакцию, весьма вариабельно у разных людей, эффективную дозу для приема внутрь трудно предсказать. Кодеин и оксикодон имеют высокий коэффициент соотношения энте-ральной/парентеральной активности, так как их конъюгация снижена из-за наличия в структуре метильной группы в качестве заместителя у ароматического гидроксила.
Б. Распределение. Поступление опиоидов в органы и ткани зависит от ряда физиологических и химических факторов. Несмотря на то, что опиоиды связываются с протеинами плазмы с разным аффинитетом, они быстро покидают кровь и накапливаются в наибольших концентрациях в тканях с высоким уровнем перфузии, таких как легкие, печень, почки и селезенка. Скелетная мускулатура фиксирует эти препараты в меньшей степени, однако она служит основным резервуаром веществ из-за своей большой массы. Накопление в жировой ткани тоже имеет большое значение, особенно после частого введения больших доз высоколипофильных опиоидов, которые медленно метаболизируются, например фентанила. Концентрации опиоидов в мозгу обычно относительно невысоки по сравнению с другими органами в связи с наличием гематоэнцефалического барьера. Однако через последний могут легко проникать вещества, в которых замещен гидроксил в положении СЗ, например героин и кодеин. Амфотерные соединения (т. е. вещества, имеющие как кислые, так и основные свойства), такие как морфин, поступают в мозг хуже других веществ. Гематоэнцефалический барьер от-
ТАБЛИЦА 30-1. Химические структуры опиоидных аналгетиков и антагонистов
Основная структура Сильные агонисты Слабые и умеренные агонисты Смешанные агонисты-антагонисты Антагонисты
Фенантрены N — СН3 / X СН2 \ / \ Нз / НО XX он Морфин N-СНз / I / Х™2 \ \ СНг / но ^сг о Гидроморфон N —СНз /1 /\сн2 ,—/ но XI , \ /—\ снз / но'^0^0 Оксиморфон N-СНз . X I / Х™2 \ /—\ 012 / Н3С—0 ХУ он Кодеин N — СНз / 1 / \сн2 , / но \1 , \ /—\ снз / н3с-о ту 0 Оксикодон n-СНз /\сн2 \ /—X СН2 / Н3С-0 ХУ 0 Гидрокодон сн2 N—СН2—m СНг .—/1 \ / / Х™2 СН2 / Н04|—1 \ /—\ снг / НО ХУ он Налбуфин сн2 ,n-ch2-ch НгС| \| / \ СН2 /-ЧнзбА-^ (' V—X —1Х-С—с—снз >=4 3—К он снз НО ^0^ О-СНз Бупренорфин n-ch2-ch=ch2 ,—(1 / \сн2 \ /—\ СН2 / но он Налорфин1 n-ch2-ch=ch2 / \сн2 -—/ но У \ \ /—\ СН2 / НО ^0^ 0 Налоксон сн2 N — СН2 — СН /1 \ / \9Н2 сн2 г—/ HoXl—x \ /—\ 012 / НО ^0^ о Налтрексон
Фенил гептил-амины ?Т /"• СНз-СНг-С-С-СНг—CH —N 6 4 Метадон ? । г* Л СНз —СН2-С—0-С- CH—CH2 — Пропоксифен
560 Раздел V. Средства, влияющие на центральную нервную систему (ЦНС)
Заказ 3245
ТАБЛИЦА 30-1. (Продолжение)
Основная структура Сильные агонисты Слабые и умеренные агонисты Смешанные агонисты-антагонисты Антагонисты
Фенилпипери-дины f^ii ° 11 - О - CHj— СН2 1 сн3 Меперидин гл о С “ СНз— СНз 1 СНг-СНг-CeHs Фентанил Н5Сг—О—С— 0 N—СНг—СНг—С—CN Дифеноксилат
Морфинаны N-СНз / \сн2 \ \ снг / но Леворфанол А N-СНз-СН СНг Z1 \ / / \СНг СН2 / НОА|—. \ / \ 012 / НО Буторфанол Н-СНг-СН=СНг / \сн2 \ 7" \СНг / НО Левая лорфан1
Бензоморфаны ^^-N-СНг—СН=С(СН3)2 / \ сн2 //—С / Нз1 \ 7 V сн2 \ - / СНз но Пентазоцин
1 Не является чистым антагонистом. Объяснения в тексте.
30. Опиоидные аналгетики и антагонисты
ТАБЛИЦА 30-2. Основные опиоидные аналгетики
Родовое название Торговое название Примерная доза (мг) ( Активность при энтеральном введении в сравнении < с парентеральным Продолжительность аналгезии (часы) Максимальная эффективность и Способность вызывать пристрастие iзлоупотребление
Морфин 10 Низкая 4-5 Высокая Высокая
Гидроморфон Дилаудид 1.5 Низкая 4-5 Высокая Высокая
Оксиморфон Нуморфан 1.5 Низкая 3-4 Высокая Высокая
Метадон Долофин 10 Высокая 4-6 Высокая Высокая
Меперидин Демерол 60-100 Средняя 2-4 Высокая Высокая
Фентанил Сублимейз 0.1 Применяется только парентерально 1-1.5 Высокая Высокая
Суфентанил Суфента 0.02 Применяется только парентерально 1-1.5 Высокая Высокая
Ал фентанил Алфента Титруется Применяется только парентерально 0.25-0.75 Высокая Высокая
Леворфанол Лево-Дроморан 2-3 Высокая 4-5 Высокая Высокая
Кодеин 30-602 Высокая 3-4 Низкая Средняя
Оксикодон' Перкодан 4.52 Средняя 3-4 Средняя Средняя
Дигидрокодеин’ Дрокод 162 Средняя 3-4 Средняя Средняя
Пропоксифен Дарвон 60-12О2 Применяется только энтерально 4-5 Очень низкая Низкая
Пентазоцин Талвин 30-502 Средняя 3-4 Средняя Низкая
Налбуфин Нубаин 10 Применяется только парентерально 3-6 Высокая Низкая
Бупренорфин Бупренекс 0.3 Применяется только парентерально 4-8 Высокая Низкая
Буторфанол Стадол 2 Применяется только парентерально 3-4 Высокая Низкая
' Выпускается в таблетках, содержащих аспирин.
2 Аналгетическая эффективность данной дозы не эквивалентна 10 мг морфина. Объяснения в тексте.
сутствует у новорожденных. Так как опиоидные аналгетики проникают через плаценту, их применение для обезболивания в акушерстве может привести к рождению ребенка с угнетением дыхания.
В. Метаболизм. Значительная часть опиоидов превращается в полярные метаболиты, которые затем быстро экскретируются почками. Соединения, имеющие свободные гидроксильные группы (морфин и леворфанол), легко конъюгируют с глюк
уроновой кислотой. Эфиры (например меперидин, героин) быстро гидролизуются тканевыми эстеразами. Героин (диацетилморфин) гидролизуется до моноацетилморфина и, наконец, до морфина, который затем конъюгирует с глюкуроновой кислотой. Ранее предполагали, что эти полярные метаболиты, соединенные с глюкуроновой кислотой, неактивны, но последние исследования показали, что морфин-6-глюкуронид обладает даже более выра
женными аналгетическими свойствами, чем сам морфин. У больных с почечной недостаточностью может происходить накопление этих активных метаболитов, что ведет к более продолжительной и глубокой аналгезии, даже несмотря на ограниченное проникновение исходных препаратов в ЦНС. Опиоиды подвергаются в печени и N-деметилиро-ванию, но это неосновной путь метаболизма. У больных с нарушенной функцией почек или при многократном введении высоких доз мепиридина может происходить накопление метаболита нормеперидина. В достаточно высоких концентрациях этот метаболит может вызывать судороги, особенно у детей.
Г. Экскреция. Полярные метаболиты опиоидов экскретируются главным образом с мочой. Через почки выводится и небольшое количество неизмененного вещества. Глюкурониды экскретируются в желчь, но этим путем выводится только малая часть опиоидов.
Фармакодинамика
А. Механизм действия. Морфин и его аналоги селективно связываются с многочисленными рецепторами, посредством чего и осуществляется фармакологический эффект. Области мозга, участвующие в передаче боли и изменении реактивности на ноцицептивные (болевые) стимулы, являются первичными, но не единственными местами действия опиоидов. Области, которые имеют высокое сродство к экзогенным опиоидным лигандам, таким как морфин, содержат высокие концентрации некоторых эндогенных пептидов с опиатоподобными свойствами. Химия и фармакология этих пептидов имеют много общих черт, но существуют биохимические и локализационные отличия. Родовое имя этих веществ — эндорфины (эндогенные морфины). Однако этот термин вызывает сложности из-за сходства с названием одного из главных прототипов опиоидных пептидов — (3-эндорфина. Из всех природных пептидов фармакологический профиль (3-эндорфина наиболее близок к морфину. Вследствие этого предложен термин "опиопептины” как родовой для природных опиоидных пептидов, а термин “эндорфин" — для пептидов, тесно связанных с (3-эндорфином.
Самые короткие пептиды, обладающие прямой опиоидной активностью,— это два пентапептида: метионил-энкефалин (мет-энкефалин) и лейцил-
энкефалин (лец-энкефалин). Их аминокислотная последовательность (тирозин-глицин-глицин-фе-нилаланин) идентична за исключением метиониновой или лейциновой концевых групп. Один или оба пептида включены в три основные белка-предшественника, содержащих 257-265 аминокислот с различными повторяющимися пептидными последовательностями.
Тремя основными пептидами-предшественника-ми являются проопиомеланокортин (ПОМК), про-энкефалин (проэнкефалин А) и продинорфин (про-энкефалин В). Проопиомеланокортин содержит мет-энкефалин, (3-эндорфин и несколько неопиоидных пептидов, включая АКТГ, (3-липотро-пин и меланоцитстимулирующий гормон. Проэнкефалин состоит из шести структурных единиц мет-энкефалина и одной — лей-энкефалина. Продинорфин содержит несколько активных опиоидных пептидов, в которые как фрагмент включен лей-энке-фалин. Это динорфин А, динорфин В (риморфин), а также а- и (3-неоэндорфины. Каждая из этих эндогенных молекул пептида-предшественника присутствует в тех отделах мозга, которые имеют отношение к модуляции боли. Доказано, что они могут активироваться при стрессе, связанном с болью или ожиданием боли. Важно заметить, что эти пеп-тиды-предшественники обнаружены не только в ЦНС, но и во многих других тканях. Проведенные исследования показали, что как эндогенные, так и экзогенные опиоиды могут вызывать аналгезию, опосредованную опиоидными рецепторами вне ЦНС. Это антиноцицептивное действие развивается потому, что происходит активация ц-, к- и 8-рецепторов, расположенных на первичных афферентных нейронах. Другой интересный аспект этих наблюдений состоит в том, что боль, связанная с воспалением, особенно чувствительна к этому периферическому опиоидному действию (Barber, 1993). Периферические механизмы, не опосредованные, по-видимому, опиоидными рецепторами, также важны для аналгезии, которую вызывают активные нестероидные противовоспалительные средства (НПВС) типа кеторолака (глава 35). Недавние исследования показали, что некоторые фенантреновые алкалоиды (морфин и кодеин) могут быть обнаружены как эндогенные вещества в тканях млекопитающих.
1. Типы рецепторов. В различных отделах нервной системы и других тканях идентифицировано несколько типов опиоидных рецепторов. Экзо
генные и эндогенные лиганды связываются с ними в различной степени, и характер связывания между конкретным веществом и специфическим рецептором определяет характерный фармакологический профиль.
Аналгезия на супраспинальном уровне, также как эйфорический эффект, угнетение дыхания и способность вызывать физическую зависимость от морфина (типичные агонистические эффекты), является в основном результатом взаимодействия с ц- и 6-рецепторами, которые также ответственны и за спинальную опиоидную аналгезию. Кроме того, свой вклад в аналгезию на спинальном уровне вносят к-рецепторы. Эти три типа рецепторов были изолированы и клонированы. Сведения о четвертом рецепторе, о-рецепторе, противоречивы, но он может быть связан с дисфорическим, галлюциногенным и кардиостимулирующим эффектами опиоидов. Недавние исследования показали, что каждый тип рецепторов может иметь подтипы (щ, ц2 и т. д.). Так как опиоидное соединение может иметь разную активность как агонист, парциальный агонист или антагонист по отношению к каждому из этих множественных типов рецепторов, неудивительно, что эти вещества могут обладать столь разными эффектами.
Несмотря на гетерогенность, все три типа первичных рецепторов оказывают свое действие посредством механизма, тесно связанного с системой цАМФ и изменением токов ионов кальция и калия.
2. Распределение рецепторов. Места связывания или распознавания опиоидов были выявлены с помощью радиолигандных методов, ауторадиографической и иммуногистохимической техники. Высокая плотность мест связывания отмечена в заднем роге спинного мозга (рис. 30-1, Б) и в некоторых подкорковых областях мозга (таламусе, околоводопроводном веществе среднего мозга и ростральной вентральной части продолговатого мозга (рис. 30-1, В, Г, Д соответственно).
Места связывания опиоидов найдены как на передающих болевые сигналы нейронах спинного мозга, так и на первичных афферентах, передающих информацию в спинной мозг (рис. 30-1, слева). Доказано, что опиоиды и опиопептины ингибируют высвобождение возбуждающих медиаторов из первичных афферентных нейронов. Места связывания опиоидов, участвующие в модуляции боли через нисходящие пути (рис.30-1, справа), включают nucleus raphe magnus в ростральном вентральном
отделе продолговатого мозга и locus coeruleus ствола мозга, околоводопроводное серое вещество и некоторые ядра гипоталамуса и таламуса. Под действием опиоидов на уровне околоводопроводного серого вещества и в ростральном вентральном отделе продолговатого мозга активируются нейроны, которые посылают аксоны к спинному мозгу и ингибируют нейроны, участвующие в передаче боли. Таким образом, связывание опиоидов в этих суп-распинальных отделах значительно усиливает эффект угнетения ноцицептивной передачи на спинальном уровне и способствует повышению болевого порога.
Модулирующие области мозга, связанные с изменением реактивности на болевые раздражители, определены менее точно, чем структуры, связанные с передачей боли. Полагают, что в этих процессах участвует лимбическая система, которая включает переднюю височную и орбитальную лобную кору, а также отделы гипоталамуса и миндалевидного тела, поскольку опиоидные аналгетики уменьшают страх и тревогу и могут вызывать эйфорию. Подтверждает это положение тот факт, что некоторые лимбические структуры отличаются высокой плотностью мест связывания опиоидов.
3. Клеточные эффекты. Опиоиды оказывают свое действие или путем гиперполяризации и торможения постсинаптических нейронов (возможно, за счет увеличения выхода ионов калия), или уменьшая вход ионов кальция в пресинаптические нервные окончания, тем самым снижая высвобождение медиатора. Рис. 30-2 иллюстрирует постсинаптический тормозный эффект опиоидов, действующих на ц-рецептор. Пресинаптическое действие, торможение выделения медиатора, было показано для большого числа нейротрансмиттеров, включая ацетилхолин, норадреналин, дофамин, серотонин и субстанцию Р. Возможно, некоторые из этих передатчиков участвуют в реализации эффекта опиоидов, так как невозможно объяснить все эффекты опиоидов участием только одной трансмиттерной системы. На молекулярном уровне опиоидные рецепторы сопряжены с G-белками и, следовательно, могут влиять на транспорт ионов, внутриклеточное распределение кальция и фосфорилирование белков (глава 20).
4. Толерантность и физическая зависимость. При часто повторяющемся введении в терапевтических дозах морфина или его аналогов происходит постепенная потеря эффективности, т. е. разви-
Рис. 30-1. Предполагаемые места действия опиоидных аналгетиков (закрашены). Слева показаны мишени на пути передачи боли от периферии к высшим центрам. А — возможное Прямое действие опиоидов на воспаленную периферическую ткань; Б — угнетающее действие на спинной мозг; В — возможное место действия в таламусе. Справа — влияние на модулирующие боль нейроны среднего (точка Г) и продолговатого (точка Д) мозга вторично изменяет передачу болевого потока по проводящим путям
вается толерантность. Для получения ответа, равного по силе начальному, приходится вводить увеличенную дозу. С развитием толерантности формируется и физическая зависимость, при которой становится необходимым продолжение введения вещества для предупреждения возникновения характерного синдрома отмены или абстиненции (глава 31).
Механизм развития толерантности и физической зависимости не определяется фармакокинетическими факторами, а представляет собой истинную клеточную адаптивную реакцию, которая обусловлена изменениями системы вторичных посредников, связанных с током Са2+, ингибированием аденилатциклазы и синтезом G-белков. Хроническое воздействие и толерантность к действию опиоидов сопровождаются увеличением концентрации Са2+ внутри клетки в отличие от эффекта однократ
ного введения, когда часто уровень ионов кальция снижается. Эффект, видимо, связан с изменением способности рецептора соединяться с G-белками, повышением уровня G-белков и нр-регуляцией системы цАМФ. Кроме того, число рецепторов может понижаться из-за снижения синтеза или “интернализации”.
Клинические аспекты толерантности и физической зависимости рассматриваются в разделе “Токсичность и нежелательные эффекты”.
Б. Влияние морфина и его аналогов на системы и органы. Эффекты, характерные для морфина как прототипа агонистов опиоидных рецепторов, свойственны и другим агонистам (табл. 30-1). Смешанные агонисты-антагонисты, если их вводят больному, который не получал в ближайшее предшествующее время вещество-агонист, также вызы-
Рис. 30-2. Энкефалин взаимодействует с налоксончувствительным рецептором нейронов locus coeruleus. На рисунке показано внутриклеточное отведение потенциалов in vitro от спонтанноактивного нейрона locus coeruleus в тканевом срезе из области моста. Отклонение наверх — потенциал действия, отклонение вниз — следовая гиперполяризация. Момент введения DADLE ([D-Ala2, D-Leu5 ] энкефалина) показан стрелками с указанием различного количества пульсовых толчков (55 кП, 24 мс, число указано) в микропипетке, содержащей пептид и расположенной над срезом. А — контрольная запись. Б — выборочные записи, сделанные в присутствии различных концентраций налоксона (Williams J. Т., Egan Т. М., North R. A. Enkephalin opens potassium channels on mammalian central neurons. Nature, 1982; 299:74.)
вают аналгезию, но с некоторыми вариациями в эффекте.
Чистые антагонисты и смешанные агонисты-антагонисты, если их вводят пациенту, которому был предварительно введен агонист, проявляют совершенно иные свойства.
1. Влияние на центральную нервную систему. Основные эффекты опиоидных аналгетиков, имеющих сродство к р-рецепторам, связаны с влиянием на ЦНС. Наиболее важными из них являются аналгезия, эйфория, заторможенность и угнетение дыхания. При повторном применении развивается выраженная толерантность ко всем этим эффектам (табл. 30-3).
Аналгезия. Ощущение боли, независимо от ее природы, определяется восприятием ноцицептив
ного раздражителя и реакцией организма на него. Экспериментальные и клинические наблюдения показывают, что опиоидные аналгетики могут увеличивать порог болевого восприятия, однако изменение реакции на боль может быть оценено только по субъективным ощущениям больного. При наличии эффективной аналгезии боль еще может восприниматься больным, но даже при сильной боли устраняется ее всепоглощающий и деструктивный характер.
Эйфория. Обычно после введения морфина больной, ощущающий боль, или наркоман испытывают приятные ощущения и у них исчезают чувства тревоги и дискомфорта. Однако у некоторых больных и здоровых людей, не испытывающих боли, скорее развивается дисфория, а не приятные ощу-
ТАБЛИЦА 30-3. Степень толерантности к некоторым эффектам опиоидов
Высокая степень толерантности Умеренная степень толерантност Минимальная толерантность и или толерантность отсутствует
Аналгезия Брадикардия Миоз
Эйфория, Запор
дисфория Судороги
Заторможенность Антагонистические
Угнетение дыхания эффекты
Уменьшение диуреза
Тошнота и рвота
Подавление кашле-
вого рефлекса
щения после опиоидного аналгетика. Дисфория сопровождается беспокойством и разбитостью. В целом при наличии показаний к применению опиоидного аналгетика наиболее частый аффективный ответ — эйфория.
Седативный эффект. Сонливость и затуманивание сознания являются частыми спутниками действия опиоидов; может наблюдаться нарушение способности к рассуждению. Амнезии не отмечается или она незначительна. У молодых здоровых людей сон вызывается чаще, чем у пожилых. Как правило, сон неглубокий, однако сочетание морфина с другими веществами, угнетающими ЦНС, например седативно-гипнотическими средствами, может потенцировать эффект последних. Более выраженное седативное действие характерно для производных фенантрена и в меньшей степени свойственно синтетическим веществам, таким как меперидин и фентанил. Морфин (фенантрен) в стандартной аналгетической дозе нарушает нормальное соотношение БДГ- и МДГ-фаз сна. Этот эффект, вероятно, характерен для всех опиоидов. В отличие рт человека у некоторых видов животных (кошки, лошади, коровы, свиньи) наблюдается скорее возбуждающий, чем седативный эффект при введении опиоидов. Эти парадоксальные эффекты по крайней мере частично дозозависимы.
Угнетение дыхания. Все опиоидные аналгетики могут вызывать значительное угнетение дыхания за счет торможения стволовых механизмов его регуляции. Рсо2 альвеолярного воздуха может возрастать, но наиболее важная сторона этого угнетающего эффекта — подавление реактивности на дву
окись углерода. Степень угнетения дыхания зависит от дозы опиоидов, ина него влияет характер других сенсорных потоков. Вызванное опиоидами угнетение дыхания можно частично преодолеть различными раздражителями. Поэтому, если на фоне аналгезии действие сильного болевого раздражителя прекращается, угнетение дыхания может неожиданно стать очень выраженным. Небольшое и умеренное снижение дыхательной функции, определяемое по увеличению Рсо2, как правило, легко переносится пациентами без сопутствующей респираторной патологии. Однако значительно хуже переносят этот эффект лица с повышенным внутричерепным давлением, астмой, хроническими обструктивными заболеваниями легких, легочным сердцем.
Подавление кашлевого рефлекса. Подавление кашлевого рефлекса — хорошо известный эффект опиоидов. При патологическом кашле и для поддержки вентиляции через эндотрахеальную трубку вводится кодеин. Однако подавление кашля опиоидами может привести к накоплению секрета с последующей обструкцией дыхательных путей и ателектазом. К данному эффекту опиоидов может развиваться толерантность(табл. 30-3).
Миоз. Сужение зрачка наблюдается при действии почти всех опиоидных агонистов. Миоз относится к эффектам, к которым толерантность не развивается или развивается в малой степени. Этот признак чрезвычайно важен для диагностики передозировки, так как даже у наркоманов, толерантных к большим дозам опиоидов, наблюдается миоз. Эффект можно устранить атропином и опиоидными антагонистами.
Ригидность мышц туловища. После введения ряда опиоидов происходит повышение тонуса скелетной мускулатуры. Считают, что это действие опиоидов реализуется на спинальном, но не супрас-пинальном уровне. Ригидность уменьшает эффективность работы грудных мышц, что может ухудшать легочную вентиляцию. Эффект наиболее выражен при быстром внутривенном введении больших доз липофильных или неполярных опиоидов (фентанил, суфентанил, алфентанил) и может быть устранен опиоидными антагонистами.
Тошнота и рвота. Опиоидные аналгетики активируют триггерную хеморецепторную зону мозгового ствола и могут вызывать тошноту и рвоту. Они оказывают определенное влияние и на вестибулярный аппарат, так как при движении возникновение тошноты и рвоты учащается.
2. Периферические эффекты.
Влияние на сердечно-сосудистую систему. Большинство опиоидов не оказывает заметного прямого действия на сердце, сердечный ритм (за исключением брадикардии) и артериальное давление. Исключения из этого правила рассмотрены при характеристике отдельных препаратов. АД обычно остается стабильным у лиц, получающих опиоиды, до тех пор, пока не возникает нагрузки, при которой может возникнуть гипотензия. Этот гипотензивный эффект, вероятно, связан с расширением периферических артерий и вен, возникающим по разным причинам, включая высвобождение гистамина и подавление центральных механизмов вазомоторной стабилизации. Заметного влияния на сердечный выброс и ЭКГ, как правило, не отмечается. Однако следует соблюдать осторожность у больных с гиповолемией, так как эти больные чувствительны к снижению АД. Если не происходит увеличения Рсо2, то опиоидные аналгетики мало изменяют мозговое кровообращение. Накопление двуокиси углерода приводит к расширению и, соответственно, уменьшению сопротивления мозговых сосудов, увеличивает мозговой кровоток и повышает внутричерепное давление.
Желудочно-кишечный тракт. Запор — хорошо известный эффект опиоидов. Опиоидные рецепторы имеют высокую плотность в ЖКТ, и обстипаци-онный эффект опиоидов опосредован действием как на местную нервную регуляцию (глава 6), так и на ЦНС (Кгошег, 1988). Моторика желудка может понижаться, тогда как тонус, особенно тела желудка, возрастать; базальная секреция понижается. Тонус мускулатуры тонкого кишечника повышается, появляются периодические спазмы, но амплитуда непропульсивных сокращений заметно снижается. В толстом кишечнике пропульсивные перистальтические волны ослабевают, но тонус возрастает, что ведет к задержке прохождения каловых масс и повышает абсорбцию воды — возникает запор. Влияние на толстый кишечник определяет показания для применения опиоидов при диарее. Считают, что бензоморфаны (например, пентазоцин) вызывают меньший обстипационный эффект, чем другие опиоиды.
Желчевыводящие пути. Опиоиды вызывают сокращение гладкой мускулатуры билиарного тракта, в результате чего могут возникать колики. Может сокращаться сфинктер Одди, что приводит к рефлюксу билиарного и панкреатического секретов и повышению уровней амилазы и липазы в плазме.
Мочеполовая система. Опиоиды подавляют функцию почек. Полагают, что у человека это является следствием снижения почечного кровотока. В опытах на животных показано, что наряду с понижением почечной перфузии, уменьшение мочеотделения является следствием повышения секреции антидиуретического гормона. После терапевтических доз опиоидных аналгетиков повышается тонус мочеточника и мочевого пузыря. Увеличение тонуса сфинктера уретры может способствовать задержке мочи, особенно в послеоперационном периоде. При почечно-каменной болезни вследствие повышения тонуса мочеточника возможно возникновение почечной колики.
Матка. Опиоидные аналгетики могут удлинять родовой акт. Механизм этого действия неясен, но снижение тонуса матки может быть связано как с центральным, так и с периферическим эффектами препаратов.
Нейроэндокринные эффекты. Опиоидные аналгетики стимулируют высвобождение антидиуретического гормона, пролактина и соматотропина, но тормозят выделение лютеинизирующего гормона. Эти эффекты могут отражать регулирующую роль эндогенных опиоидных пептидов в деятельности соответствующих нейроэндокринных систем и, вероятно, опосредуются влиянием на гипоталамус.
Другие эффекты. В терапевтических дозах опиоидные аналгетики вызывают покраснение кожи и ощущение тепла, иногда сочетающиеся с усилением потоотделения и зудом. Эти реакции могут быть связаны с центральными эффектами и выделением гистамина.
В. Эффекты смешанных агонистов-антагонистов. Пентазоцин и другие опиоиды, обладающие агонистическими эффектами на одни опиоидные рецепторы и антагонистическими на другие, обычно вызывают седативное действие в дополнение к аналгезии, когда назначаются в терапевтических дозах. В больших дозах наблюдаются потливость, головокружение, тошнота, но выраженное угнетение дыхания менее характерно по сравнению с чистыми агонистами. Если происходит угнетение дыхания, его можно устранить налоксоном, но не такими агонистами-антагонистами, как налорфин. После применения смешанных агонистов-антагонистов иногда отмечают возникновение психотоми-метических эффектов с галлюцинациями, ночными кошмарами и тревогой.
II. Клиническая фармакология опиоидных аналгетиков
Облегчение боли является существенным элементом медицинской практики и требует тщательного выбора типа лекарства, подбора необходимой дозы и постоянной объективной оценки основного заболевания. Существует много ситуаций, при которых аналгезия должна быть обеспечена до того, как будет установлен окончательный диагноз. В острых ситуациях устранение боли облегчает сбор анамнеза, физикальное обследование и, таким образом, ускоряет постановку диагноза.
Использование опиоидов в острых ситуациях отличается от их применения при лечении хронической боли, при которой необходимо учитывать такие факторы как толерантность и физическая зависимость. Во всех случаях факторы, которые учитываются при принятии решения о выборе лекарств, обязывают получить ответ на ряд вопросов: 1) необходима ли аналгезия? 2) не могут ли опиоидные аналгетики замаскировать или изменить симптомы и признаки основного заболевания? 3) не могут ли фармакологические эффекты опиоидов ухудшить течение заболевания, по поводу которого они используются, например, вызывая увеличение внутричерепного давления или подавляя дыхательный центр? 4) могут ли побочные эффекты опиоидов (табл. 30-4) представлять значимую опасность? 5) существуют ли значительные взаимодействия между опиоидным веществом и другими лекарствами, которые получает больной? 6) реально ли развитие толерантности и лекарственной зависимости?
ТАБЛИЦА 30-4. Побочные эффекты опиоидныханалгетиков
Беспокойство, дрожание, гиперактивность (при дисфории)
Угнетение дыхания
Тошнота и рвота
Повышение внутричерепного давления Постуральная гипотензия,усиленная при гиповолемии
Запор
Задержка мочи
Зуд в области крыльев носа, крапивница (чаще при парентеральном введении)
ТАБЛИЦА 30-5. Взаимодействие опиоидов с другими лекарственными средствами
Седативно-гипнотические средства: усиление угнетающего влияния на ЦНС, особенно угнетение дыхания.
Антипсихотические средства: усиление седативного действия. Вариабельное влияние на угнетение дыхания. Усиление сердечно-сосудистых эффектов (антимускариновое и а-адреноблокирующее действие).
Ингибиторы МАО: относительное противопоказание для всех опиоидных аналгетиков из-за высокой частоты гиперпирексической комы; известны также случаи гипертензии.
Табл. 30-2 указывает на диапазон агонистической эффективности различных опиоидных средств. Следовательно, необходимо выбирать лекарство, соответствующее типу и тяжести боли, для того чтобы получить адекватную аналгезию. Выраженность боли неодинакова при разных заболеваниях, например, послеоперационная боль при переломе руки может быть устранена приемом кодеина, тогда как интенсивная боль при почечной колике им не устраняется. Каждое вещество имеет терапевтический “потолок”, и попытка поднять этот потолок увеличением дозы сопряжена с риском значительных побочных эффектов.
Применение опиоидных аналгетиков в клинике
А. Аналгезия. Тяжелая, постоянная боль обычно утихает при назначении более эффективных опиоидов, тогда как острая, кратковременная боль подавляется ими не столь хорошо. Следует попытаться количественно оценить боль, и эта информация должна использоваться для выбора подходящего лекарства и контроля его действия. В процессе выбора особое значение имеют обоснование пути введения (внутрь или парентерально), длительность лечения, информация о применении опиоидов в прошлом.
Боль, связанная с раком и другими терминальными заболеваниями, может и должна быть адекватно устранена, и опасения относительно толерантности и зависимости следует оставить в стороне во имя достижения цели максимального облегче
ния состояния больного. Исследования сотрудников хосписов показали, что использование опиоидов через фиксированный интервал (определенная доза через определенйое время) более эффективно, чем введение по требованию. В настоящее время созданы новые лекарственные' формы морфина, которые обеспечивают более медленное высвобождение алкалоида (например, MS-Contin). Их преимущество заключается в более продолжительном и стабильном уровне аналгезии. Кроме того, стимулирующие лекарства типа амфетамина могут усиливать действие опиоидов и оказаться полезными содействующими (адъювантными) средствами у больных с хронической болью. В экспериментальных и клинических исследованиях показано, что агонисты а2-адренорецепторов (например, клонидин) вызывают аналгезию при разных путях введения, но роль этих веществ в лечении боли еще не выяснена.
Опиоидные аналгетики часто используют при родах. Поскольку опиоиды проникают через плацентарный барьер и попадают в плод, необходимо принимать меры для минимизации опиоидной депрессии дыхания у новорожденного. Если это происходит, то немедленная инъекция антагониста налоксона может устранить угнетение. Препараты фенилпиперидиновой группы (например, меперидин) вызывают меньшее угнетение у новорожденных по сравнению с морфином, что может оправдать их применение в акушерстве.
Острая тяжелая боль при печеночных или почечных коликах часто требует применения сильного опиоидного агониста для адекватной аналгезии. Однако вызванное лекарством повышение тонуса гладкой мускулатуры может способствовать парадоксальному вторичному усилению боли из-за спазма. Увеличение дозы опиоидов обычно обеспечивает достаточную аналгезию.
Б. Острый отек легких. Уменьшение одышки, которое достигается внутривенным введением морфина при отеке легких вследствие левожелудочковой недостаточности, весьма значительно. Механизм действия неясен, но, возможно, связан с уменьшением восприятия недостатка воздуха и связанной с ним тревогой, как и с уменьшением предна-грузки (уменьшение венозного тонуса) и постнагрузки (снижение периферического сопротивления) сердца.
В. Кашель. Подавление кашля достигается при введении субаналгетических доз. Однако в послед
ние годы применение опиоидов для уменьшения кашля снизилось в связи с появлением ряда синтетических соединений, которые не обладают ни анал-гетическим, ни аддиктивным действием. Эти вещества рассматриваются в разделе противокашлевых средств.
Г. Диарея. Диарея почти любого происхождения может купироваться с помощью опиоидных аналгетиков, но, если диарея имеет инфекционную природу, лечение опиоидами не заменяет соответствующую этиологическую терапию. Препараты опия (например, парегорик) долгое время применяли для лечения диареи, но в последние годы появились синтетические заменители с более избирательным влиянием на ЖКТ и незначительным действием на ЦНС или без центрального эффекта (например, дифеноксилат).
Д. Применение для анестезии. Опиоиды часто используют в качестве средств премедикации перед анестезией и операцией из-за свойственных им седативного, анксиолитического и аналгетического действия. Препараты этого класса могут быть использованы во время оперативного вмешательства как дополнение к другим анестезирующим средствам, а в высоких дозах (например, 1 -3 мг/кг морфина или 0.02-0.075 мг/кг фентанила) — как основные анестезирующие агенты (глава 24) в сердечно-сосудистой хирургии и других областях хирургии при высоком риске угнетения функции сердечно-сосудистой системы. В таких ситуациях должна быть обеспечена вспомогательная вентиляция.
Благодаря прямому влиянию на спинной мозг, опиоиды могут применяться для региональной аналгезии путем введения в эпидуральное или субарахноидальное пространство позвоночного столба. Ряд исследований показал, что длительная аналгезия с минимальными побочными эффектами может быть достигнута при эпидуральном введении 3-5 мг морфина с последующей медленной инфузией через катетер в эпидуральное пространство. Вначале предполагали, что эпидуральное введение опиоидов может способствовать избирательной аналгезии без нарушения моторных, вегетативных или сенсорных функций. Однако при инъекции в эту зону может угнетаться дыхание, что потребует применения налоксона. После эпидурального и субарахноидального введения опиоидов часто возникают такие эффекты как зуд, тошнота и рвота, которые также могут при необходимости устраняться налоксоном. В настоящее время предпочитают
эпидуральный путь введения из-за более редких побочных эффектов. Чаще всего используют морфин, но ведется поиск соединений, вызывающих хорошую аналгезию при меньшем числе нежелательных эффектов.
Е. Альтернативные пути введения. В случае нежелательности орального или парентерального введения используются ректальные суппозитории морфина и гидроморфона. В связи с быстрым прогрессом в разработке систем высвобождения лекарств, неудивительно, что опиоиды были адаптированы к этой технологии. Эпидуральный путь для оказания эффекта на спинальном уровне был рассмотрен выше. Другим примером является трансдермальный путь в виде аппликации пластыря для оказания системного действия, который позволяет получить стабильную концентрацию вещества в крови и лучший контроль над болью без повторных парентеральных инъекций. Фентанил оказался наиболее подходящим веществом для такого пути введения и нашел применение для болеутоления у онкологических больных с постоянными болями.
Интраназальный путь используется у больных, непереносящих прием внутрь, и в случае невозможности повторного парентерального введения.
Все чаще используется другой тип контроля над болью. При этом больной сам контролирует устройство для парентеральных (обычно внутривенных) инъекций для получения желаемой степени обезболивания. Сообщения о лучшем контроле над болью при меньшем расходе опиоидов подтверждены хорошо организованными клиническими испытаниями, что делает этот метод полезным для послеоперационного обезболивания. Некоторые проблемы могут возникать из-за технических неисправностей и риска вторичной передозировки вследствие несовершенного программирования устройства.
Токсичность и нежелательные эффекты
Прямые токсические эффекты опиоидных аналгетиков являются продолжением их фармакологического эффекта и проявляются угнетением дыхания, тошнотой, рвотой и запором (табл. 30-4). Кроме того, следует учитывать еще целый ряд особенностей.
А. Толерантность и зависимость. Лекарственная зависимость опиоидного типа характеризуется толерантностью, относительно специфичным синдромом отмены, или абстинентным синдромом, от
ражающим физическую зависимость и выраженное влечение, или психологическую зависимость. Подобно тому, как существуют фармакологические различия между различными опиоидами, отличаются друг от друга потенциал злоупотребления и тяжесть синдрома отмены. Например, абстиненция после отмены сильного агониста характеризуется более выраженными проявлениями, чем после отмены слабого агониста или препарата с умеренной эффективностью. Введение антагониста опиоидов пациенту с опиатной зависимостью приводит к развитию тяжелого синдрома отмены. Слабый опиоидный агонист пропоксифен может вызывать менее выраженную зависимость, но синдром отмены качественно сходен с таковым у других опиоидов. Считается, что опиоиды группы смешанных агонистов-антагонистов имеют меньший потенциал пристрастия, чем у веществ-агонистов.
1. Толерантность. Хотя развитие толерантности начинается после первой же дозы опиоида, она обычно не проявляется клинически ранее чем через 2-3 недели частого введения обычных терапевтических доз. Толерантность развивается в большей степени при введении больших доз через короткие интервалы и менее выражена при введении малых доз с длительными интервалами между введениями.
В зависимости от вещества и регистрируемого эффекта толерантность может приводить к 35-кратному увеличению действующей дозы. Выраженная толерантность обычно развивается к аналгетичес-кому, эйфоризирующему эффектам и угнетению дыхания. Остановка дыхания у обычного пациента при отсутствии толерантности может наблюдаться после введения морфина в дозе 60 мг, тогда как у наркомана, толерантного к опиоидам, введение морфина в дозе 2000 мг с интервалами в 2-3 часа может не вызвать заметного угнетения дыхания. Толерантность развивается также к антидиуретичес-кому, рвотному и гипотензивному, но не к мистическому, судорожному и обстипационному эффектам (табл. 30-3).
Толерантность к эйфоризирующему и респираторному эффектам опиоидов уменьшается в течение нескольких дней после прекращения введения препарата. Привыкание к рвотному эффекту может сохраняться в течение нескольких месяцев. Скорость, с которой толерантность появляется и исчезает, также как и степень толерантности к разным опиоидным аналгетикам, может различаться. Например, толерантность к действию метадона разви
вается медленнее и в меньшей степени, чем к морфину.
Кросс-толерантность (перекрестная толерантность) является очень важной характеристикой опиоидов и подразумевает, что пациент, толерантный к морфину, будет толерантен и к другим опиоидным агонистам. Морфин, меперидин, метадон и их аналоги проявляют кросс-толерантность не только в отношении аналгетического действия, но и в отношении эйфоризирующего, седативного и респираторного эффектов.
Толерантность развивается также к действию аналгетиков группы смешанных агонистов-антагонистов, но в меньшей степени, чем к действию агонистов. Такие эффекты как галлюцинации, заторможенность, гипотермия и угнетение дыхания снижаются после повторного введения смешанных агонистов-антагонистов. Однако толерантность к агонистам-антагонистам обычно не сочетается с толерантностью к опиоидам-агонистам. Важно отметить, что толерантность не развивается к антагонистическим эффектам ни смешанных агонистов-антагонистов, ни чистых антагонистов.
2. Физическая зависимость. Развитие физической зависимости — неизменный спутник толерантности к опиоидам p-типа после их повторного введения. К характерному синдрому отмены — абстинентному синдрому, который отражает преувеличенные эффекты отдачи основных фармакологических свойств опиоидов, ведет невозможность продолжать введение препарата. Симптомы и признаки отмены включают насморк, слезотечение, зевание, озноб, пилоэрекцию (гусиная кожа), гипервентиляцию, гипертермию, мидриаз, мышечные боли, рвоту, диарею, тревогу и враждебность (глава 31). Число и интенсивность признаков и симптомов определяются степенью физической зависимости. Введение опиоида в этот период подавляет проявления абстиненции почти немедленно. Начало, продолжительность и интенсивность абстиненции зависят от препарата и могут быть связаны с его биологическим периодом полу выведения. В случае морфина и героина признаки синдрома отмены обычно появляются через 6-10 часов после последней дозы. Максимум достигается через 36-48 часов, после чего выраженность большинства признаков и симптомов постепенно снижается. К пятому дню большинство эффектов исчезает, но некоторые могут сохраняться в течение месяцев. В случае меперидина синдром отмены в основном
ослабляется в течение 24 часов, тогда как при зависимости от метадона требуется несколько дней для достижения пика абстинентного синдрома. Продолжительность синдрома достигает двух недель. Замедленное исчезновение эффектов метадона определяет менее интенсивный синдром отмены, что является основой его использования для детоксикации героиновых наркоманов. После исчезновения признаков абстиненции исчезает также толерантность, что проявляется восстановлением чувствительности к опиоидным агонистам. Однако, несмотря на утрату физической зависимости от опиоида, влечение к нему может сохраняться многие месяцы.
Преходящий и интенсивный абстинентный синдром, “преципитированный (стимулированный) антагонистом”, может быть вызван у субъекта с физической зависимостью от опиоидов после введения налоксона или другого антагониста. В течение 3 минут после инъекции антагониста появляются признаки и симптомы, сходные с теми, которые наблюдаются после прекращения приема наркотика, максимум достигается через 10-20 минут, синдром постепенно ослабевает в течение часа. Даже в случае метадона, отмена которого приводит к сравнительно мягкому абстинентному синдрому, преципитированный антагонистом абстинентный синдром может быть очень тяжелым.
В случае смешанных агонистов-антагонистов признаки и симптомы абстинентного синдрома могут появляться при резком прекращении повторных введений пентазоцина, циклазоцина или налорфи-на, но этот синдром отличается от того, который вызывают морфин и другие агонисты. Для этого состояния характерны тревога, отсутствие аппетита, потеря веса, тахикардия, озноб, повышение температуры и схваткообразные боли в животе.
3. Психическая зависимость. Эйфория, безразличие к окружающему и заторможенность, вызываемые опиоидными аналгетиками, особенно при внутривенном введении, способствуют формированию компульсивного потребления (неконтролируемого) наркотика. Кроме того, наркоманы испытывают своеобразные ощущения в области живота, которые напоминают интенсивный сексуальный оргазм. Эти факторы являются главной причиной способности опиоидов вызывать злоупотребление и значительно усиливаются при развитии физической зависимости, так как наркотик начинает использоваться на постоянной основе как средство
предупреждения абстинентных симптомов, т. е. для сохранения “нормального" состояния.
Очевидно, что опасность вызвать состояние зависимости является важным фактором при терапевтическом применении опиоидов. Однако ни при каких обстоятельствах адекватное болеутоление не должно ограничиваться из-за способности опиоидов вызывать злоупотребление или из-за законодательных сложностей прописывания наркотиков. Тем не менее клиницист должен соблюдать некоторые принципы для того, чтобы избежать проблем, связанных с толерантностью и зависимостью при применении опиоидных аналгетиков:
а. Определить цели лечения перед его началом. Это поможет уменьшить вероятность развития физической зависимости. В этот процесс следует вовлечь больного.
б. После того как определена терапевтическая доза, следует попытаться ограничить дозировку на этом уровне.
в. Вместо опиоидных аналгетиков, особенно при лечении хронических болевых синдромов, следует попытаться использовать другие группы аналгетиков или соединения с менее выраженным синдромом отмены после прекращения лечения.
г. Производить постоянную оценку продолжающейся терапии аналгетиком и потребности больного в получении опиоидов.
Б. Диагноз и лечение передозировки опиоидами. Диагноз передозировки опиоидами может быть, с одной стороны, простым (наркотизация в анамнезе, следы инъекций, доставка в больницу друзьями, которые употребляют наркотики), с другой — очень трудным, как при любом коматозном состоянии неизвестной природы. Внутривенное введение налоксона в дозе 0.2-0.4 мг может быстро вывести больного из комы, связанной с передозировкой опиоидов, но не других депрессантов ЦНС. Лечение проводится тем же препаратом в дозе 0.4-0.8 внутривенно, при необходимости повторно. При использовании налоксона у новорожденных важно начать с доз 5-10 мкг/кг, в случае отсутствия эффекта повторить дозу до суммарной 25 мкг/кг. Применение антагониста, естественно, не должно заменять другие мероприятия, особенно вспомогательное дыхание.
В. Противопоказания и меры предосторожности при лечении.
1. Применение чистых агонистов вместе со смешанными агонистами-антагонистами. Когда сме
шанный агонист-антагонист типа пентазоцина вводят больному, получающему также агонист (например, морфин), возможно уменьшение степени аналгезии или возникновение состояния абстиненции, поэтому совместное применение агониста со смешанными агонистами-антагонистами должно быть очень осторожным, если вообще допустимым.
2. Использование у больных с черепно-мозговой травмой. Задержка двуокиси углерода, вызванная угнетением дыхания, приводит к церебральной вазодилатации. У больных с повышенным внутричерепным давлением это может вести к тяжелым, даже смертельным нарушениям функции мозга.
3. Применение при беременности. У беременных женщин, которые постоянно применяют опиоиды, может развиться физическая зависимость плода in utero, что проявится синдромом отмены в ранний послеродовый период. Ежедневная доза героина в 6 мг (или эквивалентная доза других опиоидов), принимаемая матерью, может вызывать синдром отмены у новорожденного в легкой форме, а двойная доза — в тяжелой форме, проявляющейся раздражительностью, пронзительным криком, диареей и даже судорожными припадками. Распознавание облегчается тщательным сбором анамнеза и физикальным обследованием. Если симптомы абстиненции относительно слабые, лечение состоит в симптоматическом лечении такими лекарствами как диазепам, при более тяжелой абстиненции применяют камфорную настойку опия (парегорик; 0.4 мг мор-фина/мл) внутрь в дозе 0.12—0.5 мг/кг. Может быть использован внутрь метадон (0.1-0.5 мг/кг).
4. Применение у больных с нарушениями функции легких. У больных со сниженным дыхательным резервом угнетающий дыхание эффект опиоидов может привести к острой респираторной недостаточности.
5. Применение у больных с нарушением функции печени и почек. Морфин и его аналоги метаболизируются главным образом путем конъюгации с глюкуроновой кислотой в печени, поэтому решение об их применении у больных с поражением печени в предкоматозном периоде должно быть сугубо индивидуальным. У больных с нарушением функции почек время полувыведения препаратов увеличивается; поскольку и морфин, и его активный метаболит морфин-6-глюкуронид могут накапливаться, таким больным следует снижать дозу.
6. Применение у больных с эндокринными заболеваниями. Эффект опиоидов может удлинять
ся и усиливаться у больных л недостаточностью надпочечников (Аддисонова болезнь) и гипотиреозом (микседема).
Взаимодействия лекарств
Поскольку тяжелые ИЛи стационарные больные, как правило, нуждаются в большом количестве препаратов, всегда существует возможность лекарственного взаимодействия при введении опиоидных аналгетиков. В табл. 30-5 приведен список некоторых таких взаимодействий и указаны причины нецелесообразности сочетания названных веществ с опиоидами.
Характеристика препаратов
Далее рассматриваются наиболее важные и широко применяемые опиоидные аналгетики и особенности отдельных соединений. Данные о дозировках, примерно эквивалентных 10 мг морфина при внутримышечном введении, эффективность при введении внутрь в сравнении с парентеральным путем, продолжительность аналгезии, максимальная эффективность и потенциал пристрастия и злоупотребления представлены в табл. 30-2.
СИЛЬНЫЕ АГОНИСТЫ
1. ФЕНАНТРЕНЫ
Морфин, гидроморфон и оксиморфои являются сильными агонистами, эффективными при лечении тяжелого болевого синдрома. Эти вещества-прототипы детально рассмотрены выше. Героин — активное и быстро действующее соединение, но его применение запрещено законодательством США. В последние годы проводились попытки возобновить его применение. Однако исследования в условиях двойного слепого контроля не подтвердили мнения о том, что героин более эффективен, чем морфин при лечении тяжелой хронической боли.
2. ФЕНИЛГЕПТИЛАМИНЫ
Метадон имеет фармакодинамический профиль, очень сходный с морфином, но действует более продолжительно. При однократном введении его анал-гетическая активность и эффективность по меньшей мере равны аналогичным показателям морфи
на. Метадон оказывает эффект при введении внутрь. Толерантность и физическая зависимость развиваются более медленно, чем при введении морфина. Признаки и симптомы абстиненции после прекращения приема метадона менее выражены, хотя и более продолжительны, чем при отмене морфина. Эти свойства делают метадон полезным веществом для детоксикации и поддерживающего лечения героиновых наркоманов с частыми рецидивами. Для детоксикации больных, зависимых от героина, малые дозы метадона (5-10 мг внутрь) дают 2-3 раза в день в течение 2-3 дней. После прекращения лечения наркоман испытывает мягкий, но продолжительный синдром отмены. Для поддерживающего лечения при рецидиве опиомании намеренно вырабатывают толерантность к метадону в дозах 50-100 мг в день; в этом состоянии у наркомана развивается перекрестная толерантность к героину, которая предупреждает основные подкрепляющие пристрастие эффекты героина. Родственное соединение левометадила ацетат (1-ос-ацетил-метадол), которое имеет даже более продолжительный период полувыведения, чем метадон, было одобрено FDA для применения в клиниках, проводящих детоксикацию. Этот препарат можно назначать один раз в 2-3 дня.
Смысл поддерживающих программ состоит в том, что блокирование подкрепляющего эффекта нелегальных опиоидов устраняет стремление к их получению и тем самым снижает криминальную активность, повышает возможности психатричес-кого лечения и реабилитации наркоманов. Фармакологические предпосылки применения метадона в поддерживающих программах не вызывают сомнения, резонны и социологические основы, но некоторые метадоновые программы оказались неудачными из-за неправильной организации лечения.
3. ФЕНИЛПИПЕРИДИНЫ
Меперидин и фентанил — наиболее широко применяемые представители этой группы синтетических опиоидов. Меперидин имеет выраженные антимускариновые свойства, которые могут оказаться противопоказанием для применения препарата при тахикардии. Кроме того, он оказывает отрицательное иноторопное действие на сердце. Следует учитывать, что накопление нормеперидина у больных с почечной недостаточностью или получающих
высокие дозы меперидина, может привести к вторичным судорожным реакциям. Подгруппа фентанила в настоящее время включает еще суфентанил и алфентанил. Эти опиоиды отличаются в основном своей активностью и биодоступностыо. Суфентанил в 5-7 раз активнее фентанила. Алфентанил существенно менее активен, чем фентанил, действует быстрее и имеет более короткую продолжительность эффекта.
4. МОРФИНАНЫ
Леворфанол — синтетический опиоидный анал-гетик, напоминающий морфин и не имеющий по сравнению с ним преимуществ.
АГОНИСТЫ С НИЗКОЙ И УМЕРЕННОЙ АКТИВНОСТЬЮ
1. ФЕНАНТРЕНЫ
Кодеин, оксикодон, дигидрокодеин и гидрокодон — все эти соединения менее эффективны, чем морфин, или имеют побочные эффекты, которые ограничивают максимальную переносимую дозу при попытке достичь аналгезию, сравнимую с таковой для морфина. Эти вещества редко используют самостоятельно, чаще в комбинации в сочетанных лекарственных формах с аспирином, ацетаминофеном и другими лекарствами.
2. ФЕНИЛГЕПТИЛАМИНЫ
Пропоксифен химически сходен с метадоном, но имеет низкую эффективность как аналгетик. В различных исследованиях показана его активность в диапазоне от уровня плацебо до уровня половины активности кодеина, т. е. 120 мг пропоксифена соответствуют 60 мг кодеина. Его истинная активность, вероятно, лежит между этими крайними значениями, а аналгетический эффект соответствует действию аспирина в оптимальной дозе. Однако низкая эффективность делает его непригодным для лечения сильной боли даже в сочетании с аспирином. Хотя пропоксифену свойственен низкий потенциал злоупотребления, увеличивающееся количество летальных исходов, связанных с его применением, побудило включить препарат в список контролируемых лекарств.
3. ФЕНИЛПИПЕРИДИНЫ
Дифеноксилат и его метаболит дифеноксин не применяют для обезболивания и назначают при диарее. Они включены в списки минимально контролируемых веществ, так как вероятность злоупотребления ими невысока. Плохая растворимость соединений ограничивает их парентеральное применение. Как антидиарейные средства их используют в сочетании с атропином. Атропин добавляют в малых количествах для уменьшения вероятности злоупотребления.
Лоперамид — другой дериват фенилпипериди-на, применяемый при поносе. Его потенциал злоупотребления низок из-за слабой способности проникновения в мозг.
Обычная начальная доза этих антидиарейных агентов составляет две таблетки, затем препарат принимают по одной таблетке после каждой дефекации при продолжающейся диарее.
СМЕШАННЫЕ
АГОНИСТЫ-АНТАГОНИСТЫ И ПАРЦИАЛЬНЫЕ АГОНИСТЫ
1. ФЕНАНТРЕНЫ
Налбуфин — сильный агонист к-рецепторов и антагонист р-рецепторов, его назначают парентерально. При увеличении дозы отмечается определенный “потолок” угнетающего влияния на дыхание, не характерный для морфина. К сожалению, если угнетение дыхания все-таки происходит, оно относительно резистентно к устранению налоксоном.
Бупренофрин — активный и длительнодействующий дериват фенантрена, который является парциальным агонистом ц-рецепторов. Его длительное действие связано с медленной диссоциацией от р-рецепторов, что делает его эффект устойчивым к устранению налоксоном. Клиническое применение сходно с употреблением налбуфина. Кроме того, некоторые исследования показывают, что бупренофрин может быть столь же эффективен, как и метадон, при детоксикации и поддерживающем лечении лиц, злоупотребляющих героином (Bickel, 1988).
2. МОРФИНАНЫ
Буторфанол вызывает аналгезию, эквивалентную налбуфину и бупренорфину, но оказывает более выраженное седативное действие в эквианалге-тических дозах. Буторфанол относят к к-агонистам.
3. БЕНЗОМОРФАНЫ
Пентазоцин также относят к к-агонистам со слабыми ц-антагонистическими свойствами. Это старейший из применяемых смешанных агонистов-антагонистов. Он может использоваться внутрь и парентерально. Однако в связи с раздражающим действием не рекомендуют вводить пентазоцин подкожно. Как и при применении всех агонистов-антагонистов, необходимо следить, чтобы не ввести препарат больному, получившему чистый агонист, из-за непредсказуемости совместного действия лекарств.
Дезоцин — соединение, структурно сходное с пентазоцином. Он имеет больший аффинитет к ц-рецепторам и меньше взаимодействует с к-рецеп-торами. Хотя указывают, что он эквивалентен по аналгетическому действию морфину, его применение связано с теми же проблемами, которые характерны для всех агонистов-антагонистов.
ПРОТИВОКАШЛЕВЫЕ
СРЕДСТВА
Как уже указывалось, опиоидные аналгетики входят в список средств, наиболее эффективно подавляющих кашель. Этот эффект часто достигается в дозах ниже аналгетических. Рецепторы, опосредующие противокашлевой эффект, вероятно, отличаются от тех, которые связаны с другими эффектами опиоидов. Например, противокашлевой эффект вызывается стереоизомерами опиоидных молекул, которые лишены аналгетического действия и способности вызывать пристрастие.
Физиологический механизм кашля сложен, и мало известно о специфических механизмах действия опиоидных противокашлевых средств. Вероятно, играют роль как центральные, так и периферические эффекты.
Чаще используют такие производные опиоидов как декстрометорфан, кодеин, левопропоксифен и носкапин. Хотя эти вещества (кроме кодеина) в основном лишены побочных эффектов опиоидов, их следует с осторожностью использовать для больных, получающих ингибиторы МАО (табл. 30-5).
Декстрометорфан является правовращающим стереоизомером метилированного производного леворфанола. Он в значительной мере лишен аналгетических и аддиктивных свойств и вызывает меньший обстипационный эффект по сравнению с
кодеином. Обычная противокашлевая доза составляет 15-30 мг 3-4 раза в день. Декстрометорфан включен во многие безрецептурные препараты.
Кодеин, как отмечалось, оказывает противо-кашлевое действие в дозах ниже, чем те, которые требуются для аналгезии. Так, обычно достаточно 15 мг для облегчения состояния больного с кашлем.
Левопропоксифен — стереоизомер слабого опиоидного агониста декстропропоксифена. Он лишен опиоидных эффектов, хотя как побочный эффект отмечают наличие седативного действия. Обычная противокашлевая доза — 50-100 мг каждые 4 часа.
ОПИОИДНЫЕ АНТАГОНИСТЫ
Чистые опиоидные антагонисты, налоксон и налтрексон, являются производными морфина с громоздкими заместителями у атома азота. Эти вещества имеют сравнительно высокое сродство к местам связывания опиоидов типа р-рецепторов и меньший аффинитет к другим рецепторам.
Фармакокинетика
Налоксон мало эффективен при приеме внутрь и имеет короткую продолжительность действия (1 -2 часа) при инъекции. Метаболизируется он путем образования конъюгатов с глюкуроновой кислотой, подобно опиоидам-агонистам со свободными гидроксильными группами. Налтрексон хорошо всасывается после приема внутрь, но может подвергаться быстрому пресистемному метаболизму. Он имеет период полувыведения 10 часов, и однократная доза (100 мг внутрь) блокирует эффект инъекции героина до 48 часов.
Фармакодинамика
Если антагонисты опиоидов вводят в дозах, устраняющих эффект агонистов, но не на их фоне, то заметных проявлений действия антагонистов не наблюдается.
Если антагонист вводят человеку, получившему морфин, антагонист полностью устраняет эффекты опиоида в течение 1-3 минут. При передозировке опиоида антагонист эффективно нормализует дыхание, сознание, величину зрачков, активность кишечника и т. д. У пациентов, имеющих зависимость, которые выглядят нормально при приеме опиоидов, налоксон и налтрексон, как уже опи
сывалось, почти мгновенно вызывают абстинентный синдром.
К антагонистическому действию этих веществ не возникает толерантности, после хронического введения не наблюдается синдрома отмены.
Применение в клинике
Налоксон — чистый антагонист и имеет преимущество перед ранее используемыми слабыми агонистами-антагонистами, которые применяли в основном как антагонисты, например налорфином и леворфанолом.
Налоксон используют главным образом для лечения острой передозировки опиоидов (глава 60). Очень важно учитывать, что налоксон имеет относительно непродолжительный эффект. Это связано с тем, что при выраженном угнетении функций состояние больного может улучшиться после однократного введения налоксона, но кома может рецидивировать через 1-2 часа. Обычная доза налоксона составляет 0.1-0.4 мг внутривенно, при необходимости введение повторяют.
Благодаря своему более продолжительному действию, налтрексон предложен как “поддерживающее” средство для лечения наркоманов. Однократные дозы, вводимые через день, блокируют все эффекты дозы героина. Можно предвидеть, что такой подход к реабилитации не будет популярен у значительной части наркоманов, если у них нет мотивации к прекращению приема наркотиков. Сравнительно недавно большой интерес вызвали сообщения о том, что налтрексон снижает тягу к алкоголю у хронических алкоголиков (Volpicelli, 1992).
Опиоидные антагонисты действуют на некоторых моделях повреждения спинного мозга. Имеются сообщения, что антагонисты можно использовать при сердечно-сосудистых заболеваниях, например при инсульте, когда они могут уменьшать проявления региональной ишемии. К сожалению, клинические исследования не подтвердили положительный эффект антагонистов в этих случаях.
Препараты
(Антидиарейные опиоидные препараты приведены в главе 64)
Опиоидные аналгетики
Алфентанил (Алфента)
Парентерально: 500 мкг/мл для инъекций
Бупренорфин (Бупренекс)
Парентерально: 0.3 мг/мл для инъекций
Буторфанол (Стадол)
Парентерально: 1, 2 мг/мл для инъекций
Интраназально (Стадол NS): аэрозоль 10 мг/мл
Кодеина сульфат или фосфат (генерик)
Перорально: таблетки и быстрорастворимые таблетки по 15, 30, 60 мг
Парентерально: 30, 60 мг/мл для в/м, п/к, в/в инъекций
Дезоцин (Далган)
Парентерально: 5,10,15 мг/мл для в/м и в/в инъекций
Фентанил (Сублимаз)
Парентерально: 50 мкг/мл для инъекций Фентаниловая трансдермальная система (Дурагезик): 25,50, 75,100 мкг/в час
Гидроморфон (генерик, Дилаудид)
Перорально: таблетки по 1, 2,3, 4 мг Парентерально: 1, 2, 3, 4, 10 мг/мл для инъекций
Ректально: свечи по 3 мг
Левометадила ацетат (Орлаам)
Перорально: раствор 10 мг/мл
Внимание: показан только для лечения наркотической зависимости
Леворфанол (Лево-Дроморан)
Перорально: таблетки по 2 мг
Парентерально: 2 мг/мл для инъекций
Меперидин (генерик, Демерол)
Перорально: таблетки по 50, 100 мг;
сироп 50 мг/5 мл
Парентерально: 25, 50,75,100 мгдля п/к, в/м инъекций; 10 мг/мл для в/в вливаний;
50 мг/ в 30 мл, 100 мг/мл в 20 мл
Метадон (генерик, Долофин)
Перорально: таблетки по 5, 10 мг;
растворимые таблетки по 40 мг;
растворы 1, 2, 10 мг/мл
Парентерально: 10 мг/мл для инъекций
Морфина сульфат (генерик, др.)
Перорально: таблетки по 10,15,30 мг;
растворы 10, 20 мг/мл;
растворы 20,100 мг/5 мл
Перорально для пролонгированного действия
(MS-Контин): таблетки по 30, 60 мг Парентерально: 0.5,1, 2,3, 4, 5, 8,10, 15 мг/мл для инъекций
Ректально: свечи по 5,10,20, 30 мг
Налбуфин (генерик, Нубаин)
Парентерально: 10,20 мг/мл для инъекций
Оксикодон (генерик)
Перорально: таблетки по 5 мг;
растворы 1, 20 мг/мл
Оксиморфон (Нуморфан)
Парентерально: 1,1.5 мг/мл для инъекций Ректально: свечи по 5 мг
Пентазоцин (Талвин)
Перорально: таблетки по 50 мг
Парентерально: 30 мг/мл для инъекций
Пропоксифен (генерик, Дарвон Пульвулес, др.) Перорально: капсулы по 32, 65 мг
Внимание: этот препарат не рекомендован
Суфентанил (Суфента)
Парентерально: 50 мкг/мл для инъекций
Комбинации аналгетиков
Кодеин/ацетаминофен
(генерик, Тайленол, w/Кодеин, др.) Перорально: таблетки по 7.5,15, 30 мг кодеина плюс 300 мг ацетаминофена; / таблетки по 12 мг кодеина плюс 120 мг/ ацетаминофена
Кодеин/аспирин
(генерик, Эмпирии компаунд, др.) Перорально: таблетки по 15, 30, 60 мг кодеина плюс 325 мг аспирина
Гидрокодон/ацетаминофен
(генерик, Норсет, Викодин, Лортаб, др.) Перорально: таблетки по 5,7.5 мг гидрокодона плюс 500 мг ацетаминофена
Оксикодон/ацетаминофен
(генерик, Перкоцет, Перкодан, Тилокс, др.) Перорально: таблетки по 5 мг оксикодона плюс 325 или 500 мг ацетаминофена
Оксикодон/аспирин (генерик, Перкодан) Перорально: 4.9 мг оксикодона плюс 325 мг аспирина
Прорпоксифен/ аспирин
(Дарвон компаунд-65, др.)
Перорально: 65 мг пропопксифена плюс 389 мг аспирина плюс 32.4 мг кофеина Внимание: этот препарат не рекомендован
Противокашлевые препараты
Кодеин (генерик, др.)
Перорально: таблетки по 15, 30, 60 мг;
составная часть многих сиропов
Декстрометорфан
(генерик, Бенилин DM, Делзим, др.) Перорально: таблетки по 5 мг; сироп 5,7.5,10, 15 мг/5 мл ; раствор для пролонгированного действия 30 мг, жевательные подушечки по 15 мг; составная часть многих сиропов
Избранная литература
Akil Н. et al. Endogenous opioids: Biology and function. Annu. Rev. Neurosci. 1984; 7: 233.
Benedetti C., Bonica J. J. (eds): Recent advances in intraspinal pain therapy. Acta AnaesthesioL Scand. 1987; 31 (Suppl 85): 1.
Braga P. C. Centrally acting opioid drugs. In: Cough. Braga P. C., Allegra L. (eds) Raven Press, 1989.
Duggan A. W., North R. A. Electrophysiology of opioids. Pharmacol. Rev. 1983; 35: 219.
Goldstein A., Naidu A. Multiple opioid receptors: Ligand selectivity profiles and binding site signatures. Molec. Pharmacol. 1989; 36: 265.
Millan M. J. к-Opioid receptors and analgesia. Trends Pharmacol. Sci. 1990; 11: 70.
Neil A. Tolerance and dependence. In: Advances in Pain Research and Therapy. Benedetti C. et al. (eds) Raven Press, 1990.
Simonds W. F. The molecular basis of opioid receptor function. Endocrine Rev. 1988; 9: 200.
Way E. L., Adler T. K. The pharmacologic implications of the fate of morphine and its surrogates. Pharmacol. Rev. 1968; 12: 383.
Yaksh T. L. CNS mechanisms of pain and analgesia. Cancer Surv. 1988; 7: 55.
Вещества, вызывающие злоупотребление
31
Лео Е. Холлистер
Термин “злоупотребление лекарствами” (drug abuse) нельзя признать удачным, поскольку он определяется общественными нормами морали и может иметь неодинаковое значение для разных людей. Необходимо различать злоупотребление (abuse) и неправильное употребление (misuse). Злоупотребление можно толковать как любое использование вещества с немедицинскими целями. Наиболее типичными иллюстрациями злоупотреблений являются применение лекарств для воздействия на собственное сознание или при занятиях культуризмом и т. п. О неправильном применении можно говорить, когда лекарство используют по несоответствующему показанию, в неадекватной дозе или слишком длительное время. В контексте злоупотребления само вещество имеет меньшее значение, чем обоснование его использования. Например, прием 50 мг диазепама для усиления эффекта дозы метадона является злоупотреблением диазепамом. С другой стороны, прием такой же большой дозы препарата, но с целью анксиолитического действия, является примером неправильного применения.
Зависимость — биологический феномен, часто связанный со “злоупотреблением”. Психическая зависимость проявляется непреодолимым (компульсивным) влечением, при котором человек повторно использует вещество для личного удовлетворения, часто несмотря на известный ему риск для здоровья. Примером могут служить заядлые курильщики. О физической зависимости говорят, когда лишение вещества ведет к развитию клинических симптомов и признаков, часто противоположных тем, ради которых оно принимается. Полагают, что организм приспосабливается в период применения вещества к новому уровню гомеостаза и реагирует противоположным образом, когда вновь созданное равновесие нарушается. Наиболее известным примером является алкогольный абсти
нентный синдром, в меньшей степени синдром отмены может наблюдаться у людей, которые ежедневно выпивают большое количество кофе. Психическая зависимость почти всегда предшествует физической, но не неизбежно ведет к ней. Для обозначения состояния физической и психической зависимости обычно используют термин пристрастие (addiction), но это слово слишком неточно, чтобы достаточно полно отражать существо проблемы.
Толерантность обозначает снижение реакции на введение вещества, что требует увеличения доз для достижения желаемого эффекта. Толерантность тесно связана с феноменом физической зависимости. Она развивается вследствие компенсаторных реакций, которые смягчают фармакодинамическое действие вещества. Метаболическая толерантность связана с повышенным выведением вещества после хронического применения. Поведенческая толерантность, т. е. способность нивелировать эффект вещества,— другой возможный механизм толерантности. Функциональная толерантность, которая может быть наиболее частым вариантом, связана с компенсаторными изменениями рецепторов, эффекторных ферментов или действия веществ на мембраны.
Для предсказания способности веществ вызывать зависимость и оценки вероятности злоупотребления был создан ряд экспериментальных методов. Большинство из этих методов использует технику самовведения веществ животными. Частота подкреплений может изменяться таким образом, чтобы заставить животное выполнять более трудное задание для получения дозы вещества, что создает возможность полуколичественного измерения. Сравнения производят по эффекту стандартного вещества данного класса, например морфина в группе опиоиодов. Лишение зависимого животного привычного вещества используют как для оценки при
роды синдрома отмены, так и для тестирования лекарств, которые могут проявлять перекрестные замещающие свойства со стандартным веществом. Большинство агентов, имеющих значительный потенциал психической или физической зависимости, могут с легкостью выявляться с помощью указанных методов. Однако предсказать истинную способность вызывать злоупотребление достаточно трудно, так как слишком много переменных участвуют в формировании злоупотребления фармакологическими средствами.
Роль фактора культуры
Каждое общество расценивает некоторые вещества как допустимые, приемлемые и отвергает другие как неприемлемые. В США и большинстве стран Западной Европы “национальными лекарствами” являются кофеин, никотин и алкоголь. На Среднем Востоке в перечень приемлемых веществ можно включить каннабис, тогда как алкоголь находится под запретом. Среди некоторых коренных племен Америки галлюциноген пейотль может законно использоваться в религиозных целях. В южноамериканских Андах для уменьшения чувства голода и повышения работоспособности при тяжелой работе в высокогорных условиях используют кокаин. Таким образом, приемлемо или неприемлемо вещество, или, иначе говоря, “употребляется” оно или “злоупотребляется” определяется отношением общества. Большая социальная цена включения какого-либо известного вещества в категорию неприемлемых — часто возникающая криминальная активность. Это связано с тем, что, с одной стороны, поставщики вещества (наркотика) вовлекаются в его нелегальный транспорт, привлеченные возможностью получения сверхприбыли, а с другой — потребители, зависимые от наркотика, для удовлетворения непреодолимой потребности в препарате занимаются грабежами, проституцией и проявляют другие виды антисоциального поведения.
Современное отношение в США к веществам данной группы отражено в Перечне контролируемых веществ (табл. 65-2, том 2), сходным с тем, который публикуют международные контролирующие органы. К сожалению, этих списков придерживаются только законопослушные производители лекарств, но они своеобразно влияют на деятельность незаконных производителей и поставщиков. Узаконенные списки обходят путем синтеза “дизай
нерских” веществ, которые имеют незначительные модификации химической структуры с небольшими изменениями фармакодинамической характеристики или с идентичным эффектом. Перечни должны постоянно пересматриваться, чтобы включать в них вновь созданные вещества.
В связи с большой социальной ценой злоупотребления лекарствами многие страны делают попытки запрета ввоза некоторых препаратов. Хотя установлено, что применение таких веществ как кокаин и марихуана снижается, это трудно связать с успехом деятельности правоохранительных органов. Незначителен прогресс и в снижении спроса на нелегальные вещества. Существует мнение, что единственное резонное решение проблемы состоит в легализации наркотиков. Очевидно, что такие предложения крайне противоречивы.
Любое использование веществ, изменяющих психические функции, основано на сложном взаимодействии трех факторов: человек, применяющий вещество; обстановка, в которой оно используется; и само вещество. Так, личность потребителя и обстановка могут оказывать сильное влияние на то, что субъект будет испытывать после приема вещества. Тем не менее обычно есть возможность определить “стержень” лекарственного эффекта, который присутствует у каждого вне зависимости от обстоятельств, если доза вещества адекватна.
Опиоиды
История
Непенте (греч. “свободный от печали”), упоминаемый в “Одиссее”, возможно, содержал опий. Курение опия до недавних пор было широко распространено в Китае и на ближнем Востоке. Выделение активных алкалоидов опия, так же как и внедрение игл для подкожных инъекций, сделавшее возможным вводить морфин парентерально, увеличили использование опия на Западе. Первая из “эпидемий” применения опиоидов в США последовала за Гражданской войной. Это связано с тем, что облегчение боли опиатами было одним из основных методов лечения, и те, кто выживал после тяжелых ранений, подчас становились физически зависимыми от опиатов. В послевоенный период около 4 % взрослых в США регулярно использовали опиаты. К 1900-м гг. это число снизилось до 1 на 400 человек населения США, но проблема ос
тавалась достаточно серьезной, чтобы оправдать принятие Акта Харрисона о наркотиках перед первой мировой войной. Новая эпидемия использования опиоидов началась примерно в 1964 г. и осталась непобежденной до настоящего времени. По современным оценкам, число зависимых от опиоидов в США недавно стабилизировалось на уровне 400 000 человек.
Химия и фармакология
Наиболее часто злоупотребляемыми веществами группы опиоидов являются героин, морфин, оксикодон и (в профессиональной среде) меперидин. Химия и общие фармакологические свойства этих веществ рассмотрены в главе 30.
При хроническом применении опиоидов развивается толерантность к их психотропному эффекту. Необходимость в увеличении дозы веществ для поддержания желаемого эйфоризирующего эффекта, также как для предупреждения синдрома лишения, приводит к развитию выраженной зависимости. Роль эндогенных опиоидов (эндорфинов) в опиоидной зависимости остается невыясненной.
Клинические аспекты
Наиболее сильными факторами инициации применения опиоидов являются любопытство и социальное давление. Внутривенное введение предпочитают не только потому, что оно наиболее эффективно, но и потому, что оно позволяет создать одномоментную высокую концентрацию наркотика, который проникает в мозг и вызывает “приход” (rush), за которым следуют эйфория, чувство покоя и сонливость (“the nod”). Большинство дозировок героина, которые предлагаются “на улице”, содержат менее 25 мг вещества и вызывают эффект длительностью 3-5 часов. Таким образом, для предупреждения проявлений синдрома отмены у зависимого субъекта может требоваться несколько доз в день. Озабоченность этими требованиями побуждает наркомана постоянно быть в поисках либо денег для приобретения наркотика, либо того, что можно продать для их добычи. Поскольку образцы героина, которые можно купить, имеют очень разную активность, всегда имеется риск передозировки. Рано или поздно у героинистов заканчиваются либо деньги, либо наркотик, и развивается синдром отмены.
Симптомы опиоидной абстиненции появляются через 8-10 часов после употребления последней дозы. Многие из этих симптомов напоминают признаки повышенной активности автономной нервной системы. Вначале появляются слезотечение, ринорея, зевота, потоотделение. Затем развивается беспокойный сон, за которым следуют слабость, озноб, “гусиная кожа”, тошнота и рвота, мышечные боли и непроизвольные движения ("kicking the habit”), а также одышка, гипертермия и гипертензия. Острый период синдрома лишения может продолжаться 7-10 дней. Вторая фаза затянутой (protracted) абстиненции продолжается 26-30 недель и характеризуется гипотензий, брадикардией, гипотермией, мидриазом и пониженной реактивностью дыхательного центра на двуокись углерода.
Потребители героина особенно склонны к полинаркомании и активно применяют алкоголь, седативные средства и стимуляторы. Эти вещества используются не в качестве заменителей опиоидов, а как средства, имеющие желаемый аддитивный эффект. Необходимо быть уверенным, что у больного с синдромом лишения нет одновременно алкогольного абстинентного синдрома или синдрома отмены седативных средств, что может быть более опасным и трудным для лечения.
Наряду с риском смертельной передозировки при опиоидной зависимости может наблюдаться ряд других серьезных осложнений. Гепатит В и СПИД — наиболее вероятные следствия использования загрязненных шприцев. Бактериальные инфекции ведут к таким септическим осложнениям как менингит, остеомиелит и абсцессы в различных органах. Новым осложнением, возникшим на фоне попытки незаконного производства меперидина, стал паркинсонизм у пользователей этого наркотика, причиной которого является специфический нейротоксин МРТР (глава 27). Убийства, самоубийства и несчастные случаи среди героинистов происходят чаще, чем в общей популяции.
Лечение
Своевременное лечение острой передозировки опиоидов может спасти жизнь (главы 30 и 60).
При длительном лечении лиц, зависящих от опиоидов, могут использоваться раздельно или совместно фармакологический и психологический подходы. Существуют различные мнения о предпочтительных способах лечения. Поскольку при
каждом методе лечения используются разные группы больных, результаты сравнивать трудно. Лица, хронически потребляющие опиоиды, предпочитают фармакологические методы, те же, у кого заболевание не столь длительно, более склонны к психотерапевтическому лечению/
Фармакологическое лечение чаще всего используется для детоксикации. Общие принципы детоксикации состоят в том, чтобы заменить наркотик длительнодействующим, принимаемым внутрь и фармакологически эквивалентным веществом, стабилизировать больного на этом веществе и затем постепенно отменить замещающее лекарство. Наилучшим образом для этой цели подходит метадон. Сравнительно недавно для детоксикации стали применять клонидин — центрально действующий симпатолитический агент. Снижая центральный симпатический тонус, клонидин устраняет многие признаки симпатической гиперактивности. Преимущество клонидина в том, что он не является наркотиком.
Хотя провести детоксикацию пациента сравнительно несложно, частота рецидивов (повторение злоупотребления наркотиком) чрезвычайно высока. В некоторых случаях эффективной оказывается поддерживающая терапия метадоном, длительнодействующим и активным при приеме внутрь опиоидом, который замещает героин. Препарат можно давать один раз в день, причем возможна более редкая частота дозировки для больных, получающих длительное лечение. Метадон насыщает опиатные рецепторы и предупреждает быстрое развитие центрального эффекта опиатов при внутривенном введении. Разрешен к применению и имеет некоторые практические преимущества более длительнодействующий гомолог метадона — L-a-ацетилметадол.
Рациональным способом лечения является применение антагонистов наркотиков, поскольку блокада действия опиатов может привести к утрате привыкания. Детально изучен налтрексон, длительнодействующий, активный при приеме внутрь полный антагонист наркотиков. Препарат назначают 3 раза в неделю, дозы достигают 100-150 мг в день. Основной недостаток лечения состоит в том, что лишь немногие наркоманы готовы принимать налтрексон как средство для продолжительного лечения. В отличие от метадона, от которого пациенты становятся зависимыми, налтрексон не обладает такой “удерживающей” способностью.
Психологические методы включают формирование свободных от наркотиков сообществ, основанных на идее о том, что прием наркотика является признаком эмоционального расстройства или неспособности справиться с жизненными стрессами. Наиболее распространенные методы используют психологическое давление групп сверстников с акцентом на конфронтацию. Другие методы основаны на вариантах групповой или индивидуальной психотерапии, дидактическом подходе, формировании альтернативного стиля жизни посредством работы или жизни в коммуне, а также на различных вариантах медитации. Лечение может продолжаться месяцы и годы, и его стоимость определяется степенью участия в нем профессионального штата. Многие наркоманы подчас “устают” от своего пристрастия или “перегорают” даже без всякого лечения.
Барбитураты и другие седативные средства
История
Барбитураты были внедрены в практику в 1903 г. Препараты этой группы с короткой продолжительностью действия по настоящее время являются объектом злоупотребления. Мепробамат вошел в практику в 1954 г. как седативное средство небарбитуратной структуры, лишенное, как предполагали, недостатков старых лекарств. Однако оказалось, что с барбитуратами у него больше общего, чем отличий. В действительности, некоторые небарбитуратные седативно-гипнотические средства, например глютетимид и метаквалон, были во многих отношениях, включая потенциал злоупотребления, хуже барбитуратов. Современная эра бензодиазепинов началась лишь в 1960 г. По мнению многих специалистов, эти препараты являются седативно-гипнотическими средствами выбора. Злоупотребление и физическая зависимость применительно к бензодиазепинам известны, но встречаются реже, чем в случае барбитуратов.
Химия и фармакология
Химические особенности этого класса лекарств рассмотрены в главе 21.
По мере повышения дозы эти препараты вызывают седативный эффект, сон, анестезию, кому и
смерть. Точный механизм развития психической и физической зависимости к ним неизвестен.
Как барбитураты, так и бензодиазепины в соответствии с особенностями их фармакокинетики, могут быть разделены на препараты короткого и длительного действия. Опыт применения барбитуратов показал, что в основном с немедицинскими целями используют короткодействующие препараты, например секобарбитал или пентобарбитал натрия, но не такие длительнодействующие препараты, как фенобарбитал. До некоторой степени такой же подход воспроизводится и для бензодиазепинов. Стало очевидным, что внезапность начала синдрома отмены, так же как и его тяжесть, напрямую зависит от периода полувыведения препарата. Лекарства с длительностью полувыведения в диапазоне 8-24 часа вызывают быстро развивающийся тяжелый абстинентный синдром, тогда как препараты с более продолжительным периодом полувыведения (48-96 часов) вызывают синдром отмены, который начинается позднее и протекает не столь тяжело, хотя и более длительно. Лекарства с периодом полувыведения больше 96 часов обычно отличаются постепенным и мягким снижением эффекта, что уменьшает вероятность развития реакций на отмену препарата. Вещества с очень коротким периодом полувыведения (менее 4 часов) обычно не принимают достаточно часто для поддержания высоких концентраций, поэтому реакция на их отмену возникает редко.
Клинические аспекты
Сегодня никому неизвестно, сколько людей имеют зависимость от седативных средств. Большинство предполагаемых случаев являются вариантами психологической зависимости, возможно, основанной на необходимости в этих лекарствах как форме “заместительной терапии". Физическая зависимость классического типа от бензодиазепинов встречается относительно редко. Обычно это происходит после длительного лечения препаратами в суточных дозах 40 мг диазепама и более или эквивалентных дозах других представителей группы. В течение последних лет большой интерес проявляют к феномену “зависимости от терапевтической дозы”. Многие пациенты с этим синдромом никогда не принимали препараты в дозах выше обычных лечебных доз (15-30 мг в день в пересчете на диазепам). Резкое прекращение приема может сопро
вождаться некоторыми симптомами, которые наблюдаются при классическом синдроме отмены и характеризуются потерей веса, изменениями восприятия, парестезиями и головными болями. Такой синдром обычно связан с постоянным применением того или иного препарата в течение месяцев или лет. Он может быть связан с некоторыми адаптивными изменениями бензодиазепиновых рецепторов при длительном воздействии препаратов. Все больные, получающие седативные препараты продолжительное время, должны прекращать прием постепенно и длительно.
Существует несколько характерных предпосылок злоупотребления седативными средствами. Люди с серьезными эмоциональными расстройствами могут использовать эти препараты в поисках выхода из сложившихся ситуаций в забвении. Однако большинство лиц, злоупотребляющих седативными средствами, используют их для изменения психического состояния и растормаживания, подобно тому, как другие или они же сами используют алкоголь. Наблюдаются варианты чередования применения седативных средств и стимуляторов, когда один тип препаратов используют для противодействия эффектам другого. Увеличивается тенденция применения седативных средств в структуре полинаркоманий.
Эффекты, которых, как правило, добивается потребитель, сходны с теми, которые вызывает алкоголь,— начальное растормаживание с последующей сонливостью. Речь при этом может быть невнятна, очевидно нарушение координации. Так как целью является достаточно частое повторение подобной интоксикации, предпочитают вещества, которые быстро выводятся.
Поскольку седативные средства обычно принимают внутрь, а таблетки или капсулы содержат постоянное количество действующего начала, смертельные передозировки этих препаратов редки. Может развиваться толерантность к седативному эффекту, но не к угнетению дыхания. Следовательно, если препараты потребляют вместе с большим количеством алкоголя, тоже угнетающим дыхание, возможны фатальные отравления.
Синдром отмены седативных средств настолько напоминает алкогольный, что эти типы зависимости входят в общую классификацию (алкоголь-барбитуратный тип). Симптомы и признаки хронической интоксикации короткодействующими препаратами могут уменьшаться в первые 8 часов пос
ле отмены, но затем их сменяют тревога, тремор, подергивания, тошнота и рвота в течение следующих 16 часов. Пропранолол или клонидин могут оказать положительный эффект в уменьшении выраженности этих проявлений. Через 16-48 часов после отмены могут возникать судороги. В тяжелых случаях наблюдаются делирий, галлюцинации и другие психозоподобные проявления.
В случае приема длительнодействующих средств синдром отмены ослаблен. В течение 2-3 дней может не отмечаться никаких признаков, и начальные симптомы могут напоминать рецидив проявлений, для ликвидации которых препарат принимался. Только к четвертому-пятому дням можно быть уверенным, что развивается реакция отмены. Судороги являются поздним проявлением: если они возникают, то не ранее восьмого-девятого дней. После этого синдром затихает. Столь медленная эволюция может позволить некоторым больным разобраться в том, что происходит, и оборвать развитие синдрома, возобновив прием лекарства. Так как синдром отмены обычно слабо выражен, некоторые больные могут пройти его, чувствуя, что что-то не в порядке, но не понимая отчетливо, что же происходит.
Лечение
При лечении абстинентного синдрома, связанного с седативными средствами, принимается во внимание принцип учета времени. Если применялись короткодействующие вещества, назначают фенобарбитал как фармакологически эквивалентный заместительный препарат. Если использовалось длительнодействующее средство, оно же может применяться далее. Больного стабилизируют, назначая дозу, достаточную для устранения признаков и симптомов отмены, и затем вещество длительно и постепенно отменяют. Скорость снижения может составлять 15-25 % от ежедневной дозы в начале лечения и 5-10 % в последующем. Полная детоксикация достигается обычно менее чем за 2 недели.
Для лиц, злоупотребляющих седативными средствами, не создано специальных лечебных программ. Проблема подчас настолько осложнена злоупотреблением другими веществами, что может быть более целесообразно вовлечь больного в программу лечения, разработанную для алкоголиков или зависимых от опиатов. Больные с психически
ми расстройствами, особенно с депрессией, должны одновременно получать специфическое лекарственное лечение соответствующего заболевания.
Стимуляторы
История
Кофеин является, вероятно, наиболее широко применяемым в мире “социальным лекарством”. Большинство людей не считают его фармакологически активным, хотя многие, особенно с возрастом, испытывают его неблагоприятное влияние на сон и сердечный ритм после излишнего количества выпитого кофе. Синдром отмены, характеризующийся летаргией, раздражительностью и головной болью, проявляется у лиц, потребляющих более 600 мг в день (примерно 6 чашек кофе).
Никотин — это одно из наиболее широко применяемых “законных” фармакологических средств. В настоящее время он считается представителем самых коварных аддиктивных веществ, потому что большинство потребителей не испытывают сильных подкрепляющих ощущений при применении никотина, однако при попытках прекратить курение возникает продолжительная тяга к его употреблению. Никотин приводит к центральному возбуждению электрической активности головного мозга и эйфоризирующему эффекту и вызывает высвобождение катехоламинов из периферических адренергических нервов. Быстро развивается толерантность. Несмотря на убедительные свидетельства серьезной опасности для здоровья с вовлечением множества систем и органов (MMVR, 1993), около 28 % взрослых в США еще курят сигареты, так как являются зависимыми от никотина. Для того чтобы отлучить курильщиков от сигареты, используют различные методы, включая замену табака никотином в форме никотинсодержащих жевательных резинок и медленно высвобождающих никотин трансдермальных лекарственных форм. Эти препараты созданы для применения под медицинским наблюдением в постепенно снижающихся дозировках. Однако похоже, что количество вновь появляющихся курильщиков равно числу тех, кто прекращает курение. За последние годы возросло использование бездымных табачных изделий (понюшки, жевательный табак), особенно среди подростков, очевидно, как реакция на новые рекламные кампании, ориентированные на молодежные группы. Эти про
дукты имеют тот же риск возникновения раковых заболеваний и зависимости, что и курительный табак. Краткое описание фармакологии никотина дано в главе 7.
Кокаин используется по меньшей мере в течение 1200 лет в форме привычного жевания листьев коки аборигенами южноамериканских Андов. Впервые кока была импортирована в Европу из' Западного полушария в 1850 г. Кокаин, активное действующее начало коки, был выделен в 1860 г. Его анестезирующие свойства, особенно местноанестезирующее действие, были открыты в 1870-80 гг. Зигмунд Фрейд интересовался этим веществом, полагая, что оно даже может быть панацеей, но энтузиазм знаменитого психиатра угас после несчастного случая с его другом, который пристрастился к кокаину. Коллега Фрейда Карл Келер считается пионером использования кокаина для местной анестезии в глазной хирургии. Это показание к применению сохранилось до настоящего времени.
Амфетамин был синтезирован в конце 1920-х гг. и внедрен в медицинскую практику в 1936 г. Декстроамфетамин является основным представителем этого класса, хотя в последующем появилось много других амфетаминов и их производных, таких как метамфетамин (метедрин, “speed”), фенметразин (прелудин) и метилфенидат (риталин). Число психоактивных гомологов амфетамина продолжает увеличиваться. Первым из них был 2,5-диметокси-4-метиламфетамин (DOM, “STP”). Позднее появились метилендиоксиамфетамин (MDA) и метилендиоксиметамфетамин (MDMA, “экстази”). Последнее вещество имеет в большей степени амфетаминоподобное, нежели галлюциногенное действие.
Было показано, что эти вещества проявляют токсическое действие в отношении серотонинергической системы мозга животных; возможные последствия их действия для человека пока точно не определены.
Близким по действию является естественный алкалоид катинон, который содержится в листьях и стеблях Catha edulis, или ката,— растения, культивируемого на Среднем Востоке и в Африке. Жевание ката приводит к эффектам, неотличимым от эффектов амфетаминов.
Химия и фармакология
Химическая структура различных стимуляторов представлена на рис. 31-1. Несмотря на сходство действия, кофеин, кокаин и амфетамин имеют различную структуру.
Соединение группы метилксантинов, кофеин, оказывает центральное действие (и, возможно, периферическое) путем блокады аденозиновых рецепторов. Другие метилксантины, например теофиллин, имеют такой же механизм действия. Аденозин модулирует активность аденилатциклазы, вызывая в качестве одного из важных периферических эффектов сокращение изолированной гладкой мускулатуры дыхательных путей. Аналогичный механизм, возможно, ответственен за центральный стимулирующий эффект метилксантинов. В высоких концентрациях метилксантины ингибируют фосфодиэстеразу, тем самым подавляя распад цАМФ и увеличивая его внутриклеточную концентрацию. Однако нет уверенности в том, что этот эффект имеет значение для обычных доз ксанти
Кокаин
Рис. 31-1. Химическая структура наиболее известных стимуляторов
Амфетамин
О-СН2-СН2
СН—СН—NH I СН3
Фенметразин
нов. Кокаин связывается с переносчиком дофамина в клетки ЦНС, вызывая блок обратного захвата дофамина, а также норадреналина. Считают, что дофамин важен для функции “системы награды” мозга и увеличение его содержания может иметь отношение к высокому потенциалу зависимости от кокаина. Амфетамины могут действовать разными способами, но в основном путем увеличения высвобождения катехоламинергйческих медиаторов. Они также являются слабыми ингибиторами МАО и, благодаря структурному сходству, возможно, являются прямыми катехоламинергическими агонистами.
Несмотря на сильную психическую зависимость от многих из указанных веществ (кокаин является одним из самых сильных подкрепляющих веществ на модели самовведения у животных), полагают, что физическая зависимость развивается редко. Однако сравнительно новые примеры использования этих наркотиков, особенно амфетаминоподобных веществ, выявили типичный синдром отмены, проявляющийся признаками и симптомами, противоположными тем, которые вызывает само вещество. Лишение наркотика ведет к развитию сонливости, повышению аппетита, чувству опустошенности и депрессии. Этот синдром может длиться несколько дней после прекращения приема вещества. Толерантность развивается быстро, и наркоман может принимать большие дозы в сравнении с принимаемыми по терапевтическим показаниям, например для получения анорексигенно-го эффекта.
Клинические аспекты
Злоупотребление амфетамином появилось в 1940-х гг. Вещество включали в больших количествах в ингаляторы, предназначенные для оказания противоотечного действия при лечении насморка. Во время второй мировой войны амфетамины широко применяли в армии. В послевоенный период молодежь в Японии получила свободный доступ к амфетаминам, что способствовало развитию эпидемии злоупотребления и привело в конечном счете к преследованиям и принятию жестких мер по отношению к пользователям этих препаратов. Тем временем в США вещество продолжали использовать в относительно невысоких дозах для приема внутрь, часто чередуя с барбитуратами. В 1960-х гг. характер использования изменился. Предпочти
тельным наркотиком стал метамфетамин (возможно потому, что его легче синтезировать нелегально), а предпочтительным путем введения — внутривенные инъекции.
Один из способов злоупотребления амфетамином называется “run”. Повторные внутривенные инъекции производятся для получения “rush” — реакции, напоминающей оргазм, за которой следуют чувство психического подъема и выраженная эйфория. Суточные дозы могут достигать 4000 мг. Через несколько дней такой интоксикации у больного может развиться состояние, напоминающее параноидную шизофрению. Типичными проявлениями являются ощущения ползания мурашек под кожей, что приводит к характерным местным расчесам. Наконец, “запой” завершается истощением в связи с отсутствием сна и приема пищи с последующим синдромом отмены, рассмотренным ранее.
Появилась также новая форма злоупотребления амфетамином — аналог кокаинового “crack”, при которой выкуриваются кристаллы метамфетамина-основания (“ice”). При курении в мозг быстро поступают болюсные дозы наркотика, что несколько напоминает внутривенное введение. Поскольку длительность эффекта метамфетамина значительно дольше, чем кокаина, интоксикация может продолжаться несколько часов после одного сеанса курения. Будущее покажет, заменит ли “ice” ранее распространенный “crack” или просто станет еще одной из форм наркотизации. Весь парадокс в том, что попытки законодательного регулирования подчас ведут к появлению более опасных наркотиков. После исключения амфетаминов из официальной медицинской практики (основанного на побочных эффектах при использовании доз на 2-3 порядка более высоких, чем терапевтические) на смену им пришел кокаин. Кокаин можно рассматривать как высокоактивный амфетамин (“superspeed”) со всеми, но усиленными, эффектами последнего. Он значительно сильнее вызывает зависимость, чем амфетамины, и при его применении возможны передозировки, приводящие к смерти.
В настоящее время известны два способа применения кокаина. Первый заключается во вдыхании дозы (“line” — предварительно взвешенная доза препарата, завернутая в бумагу и подготовленная для вдыхания). Второй состоит в выкуривании кристаллов кокаина-основания. Кокаин поставляется в виде гидрохлорида и свободного основания
(“crack”), полученного путем обработки соли щелочью и экстрагирования неполярным растворителем. При выкуривании кокаина в виде свободного основания действующее начало поступает в кровь почти столь же быстро, как и при внутривенном введении, поэтому эффекты наркотика сильнее, чем при вдыхании через нос. Внутривенное введение используется редко в связи с опасностью передозировки. Чистота препаратов и их активность варьируют в очень широких пределах.
Период полусуществования кокаина в плазме очень короткий, и эффект одной дозы продолжается около часа. Соответственно введения с целью достижения эйфорических переживаний могут повторяться много раз в течение дня и ночи.
Кокаин может использоваться спорадически, например во время вечеринок, или регулярно в некоторых группах населения, например в среде рок-музыкантов. Применение кокаина может быть также элементом “репертуара” полинаркоманов. “Speedball” — комбинация кокаина и героина для внутривенного введения, примерно удваивает “rush”, но ослабляет другие эффекты. Пациенты, находящиеся на поддерживающей метадоновой программе, которые не могут получить искомый эффект при введении героина, как правило, переключаются на кокаин, эйфоризирующее действие которого не снимается метадоном. Эпидемия появления “кокаиновых детей”, рожденных матерями-наркоманками, явилась новым вызовом для службы здравоохранения в городах (Volple, 1992). Существует явное патологическое влияние на беременность с повышением внутриутробной заболеваемости и смертности. Будут ли у выживших детей наблюдаться какие-то долгосрочные нарушения, покажут будущие исследования.
Помимо параноидного психоза, связанного с хроническим применением амфетамина, внутривенные инъекции с использованием загрязненных шприцев приводят к тем же инфекционным осложнениям, которые характерны для героина. К специфическим поражениям, связанным с хроническим потреблением амфетамина, относится некротический артериит, который захватывает ряд мелких и средних артерий и ведет к обширным кровоизлияниям в мозг и к почечной недостаточности. Передозировка амфетаминов редко заканчивается летальным исходом и обычно поддается лечению галоперидолом. Выраженное сосудосуживающее действие кокаина приводит к увеличению числа
больных с тяжелым острым гипертензивным синдромом, приводящим к инфарктам миокарда и инсультам. Местноанестезирующее действие кокаина во многом обусловливает его судорожный эффект. Передозировки кокаина обычно приводят к смерти, и жертвы погибают в течение минут из-за аритмий, угнетения дыхания или судорог. Состояние тех, кто выжил в течение 3 часов, обычно полностью нормализуется. Внутривенное введение диазепама, пропранолола или блокаторов кальциевых каналов считается наилучшим способом лечения.
Длительное лечение
Лицам с остаточными эмоциональными расстройствами, шизофреноподобным психозом или депрессией может потребоватся лечение антипсихотическими или антидепрессивными препаратами перед отменой или во время отмены стимуляторов. Был изучен ряд лекарств для ослабления проявлений синдрома отмены кокаина, уменьшения тяги в период абстиненции и облегчения этого состояния. В настоящее время исследуются дезипрамин, карбамазепин, бромокриптин, амантадин, клонидин, бупренорфин, бупропион и другие. Каких-либо специфических программ для лиц, злоупотребляющих стимуляторами, не разработано. Те, кто применяют эти наркотики внутривенно, имеют много общего с героиновыми наркоманами и могут участвовать в программах реабилитации, созданных для больных, зависимых от опиоидов.
Галлюциногены
История
Почти в каждом обществе и в каждой культуре можно найти некоторые растительные продукты (кора, кожура, листья, ягоды и т. п.), которые содержат “галлюциногенные” вещества. Хотя открытие свойств диэтиламина лизергиновой кислоты (ЛСД) произошло в химической лаборатории в 1940-х гг., это тот случай, когда наука просто смоделировала природу. Такие вещества как мескалин и псилоцибин, содержащиеся в бутонах кактуса и “магических” грибах, издавна использовались индейцами Северной и Центральной Америки. Делирианты алкалоидов Atropa belladonna и Datura stramonium также были известны в древних культурах.
Терминология
Хотя мы используем термин “галлюциноген”, он не совсем точен, так как галлюцинации могут быть необязательным эффектом или только одним из эффектов этих веществ. Чтобы подчеркнуть их способность имитировать психозы, часто используют термин “психотомиметики”. Однако состояние, вызываемое этими агентами, не полностью воспроизводит клинику шизофрении. Термин “психоделики” был предложен для того, чтобы оттенить предполагаемое специфическое психотропное действие (“откровение разума”). Однако достигаемый при их помощи эффект не слишком отличается от того, который можно получить при приеме других растормаживающих веществ, включая наиболее распространенный из них — алкоголь.
Вещества, которые мы здесь обозначаем как галлюциногены, используют по разным причинам. Одна из наиболее распространенных связана с тем, что галлюциногены дают новый взгляд на мир и личные проблемы. Первое достигается выраженными в разной степени нарушениями восприятия, тогда как второе — изменением настроения и повышенной склонностью к интроспекции. ЛСД стал галлюциногеном-прототипом, благодаря своей распространенности и потому, что он представляет целое семейство наркотиков со сходными свойствами, и наиболее изучен.
Химия и фармакология
К группе ЛСД-подобных галлюциногенов, кроме ЛСД, относятся мескалин, псилоцибин и родственные соединения. Хотя эти вещества отличаются химически, они имеют некоторые общие клинические свойства и еще более сходные фармакологические характеристики (рис. 31-2). ЛСД — по-лусинтетическое соединение, неизвестное в природе. Он имеет сходство с алкалоидами спорыньи (глава 16). В природных растительных источниках выявлен прежде всего моноэтиламид, но не диэтил-амид. В природе встречаются производное фенил-этиламина, мескалин, и производное индолэтила-мина, псилоцибин. Эти вещества имеют сходство с тремя основными медиаторами — норадреналином, дофамином и серотонином.
Несмотря на многочисленные исследования, механизм действия галлюциногенов типа ЛСД неизвестен. На электроэнцефалограмме их эффект
проявляется признаками возбуждения ЦНС. ЛСД взаимодействует с несколькими подтипами серотониновых 5-НТ-рецепторов мозга. До недавнего времени ЛСД рассматривали как активный антагонист 5-НТ2-подтипа рецепторов. Он изменяет обмен серотонина, что проявляется увеличением в мозгу концентрации его метаболита 5-оксииндолуксус-ной кислоты. Кроме того, ЛСД проявляет агонистическую активность в отношении 5-НТ1Л- и 5-НТ|С-рецепторов. Эти свойства могут быть более тесно связаны с галлюциногенным действием ЛСД, так как ряд других лекарств, обладающих антагонистическим эффектом в отношении 5-НТ2-подти-па рецепторов, не является галлюциногенами.
Галлюциногены-делирианты, примером которых являются скополамин (глава 8) и некоторые синтетические центральные холинолитики, химически и фармакологически отличаются от группы ЛСД. Их эффекты практически полностью объясняются блокадой центральных мускариновых рецепторов. Сходные психотропные эффекты можно наблюдать при терапевтической или преднамеренной передозировке широко применяемых антимускариновых средств, таких как антихолинергические лекарства для лечения паркинсонизма, трициклические антидепрессанты, спазмолитики. Известны случаи злоупотребления этими лекарственными средствами.
Фенциклидин (РСР, “ангельская пыль” и много других жаргонных названий) является синтетическим производным фенилциклогексиламина. Фенциклидин трудно отнести к какой-либо группе, поскольку он во многом отличается от других галлюциногенов, но главная область его применения — получение галлюциногенного эффекта. Вещество было внедрено в практику в качестве “диссоциативного анестетика" в 1957 г. Предполагали, что такие анестетики могут сделать пациента нечувствительным к боли путем “отделения его телесных функций от психических” без выключения сознания. Галлюциногенный эффект был обнаружен у больных, выходящих из состояния общей анестезии. Препарат прекратили применять у человека, но он сохранился в ветеринарной практике под названием сернилан. Кетамин, гомолог фенциклидина, заменил последний в качестве анестетика для применения у человека (глава 24). Он тоже вызывает галлюциногенный эффект.
Начало нелегального использования фенциклидина датируется 1967 г., когда он быстро завоевал
Мескалин
Псилоцибин
3,4-метилендиоксиметамфетамин (MDMA)
Рис. 31-2. Химические формулы диэтиламида лизергиновой кислоты, мескалина, фенциклидина, псилоцибина и производного амфетамина MDMA
репутацию “плохого” наркотика. В течение нескольких последующих лет он продавался под видом ЛСД, тетрагидроканнабинола (ТНС) и других галлюциногенов. С 1970-х гг. фенциклидин получил широкое признание и в настоящее время является одним из наиболее распространенных галлюциногенных веществ. Фенциклидин курят, подмешивая порошок в табак, нюхают, принимают внутрь или вводят внутривенно. К счастью, в последнее время наметилась тенденция к снижению частоты применения этого наркотика.
Первые исследователи фенциклидина полагали, что с его помощью можно получить более адекватную модель шизофрении, чем при использовании ЛСД, потому что он вызывает некое подобие социальной изоляции. В мозгу были идентифицированы рецепторы для фенциклидина, которые тесно взаимосвязаны с опиоидными и-рецепторами, если не идентичны им. Вещество взаимодействует и с NMDA-подтипом глутаматных рецепторов как их антагонист. В ранних исследованиях было также
показано, что фенциклидин блокирует захват дофамина. Наркотик уникален при сравнении с другими галлюциногенами тем, что поддерживает само-введение у экспериментальных животных.
Клинические эффекты
ЛСД вызывает ряд взаимосвязанных соматических, перцептивных и психических эффектов. Головокружение, слабость, тремор, тошнота, парестезии являются наиболее частыми соматическими симптомами. Основные виды нарушения восприятия — затуманенность зрения, искажение перспективы, оформленные зрительные и более аморфные слуховые галлюцинации, изменение восприятия времени. Изменение психики проявляется нарушением памяти, затрудненностью мышления, лабильностью настроения. Физиологические изменения, вызванные ЛСД, напоминают гиперактивность симпатической системы и центральную стимуляцию, что проявляется мидриазом, повышением ча
стоты сердечных сокращений, незначительным повышением АД, тремором и беспокойством. Сходные эффекты вызываются мескалином и псилоцибином, когда их вводят в эквивалентных дозах. Эффекты развиваются быстро, но продолжительность их зависит от дозы и обычно исчисляется в часах. Проявления могут варьировать у разных людей в зависимости от особенностей личности, ожидаемого действия и обстановки, но вышеперечисленные эффекты отмечаются почти у всех. Типично волнообразное течение эффектов.
Обычные дозы ЛСД у человека составляют примерно 1-2 мкг/кг, что характеризует его как один из самых активных фармакологических агентов. Эффективность при парентеральном введении и приеме внутрь примерно одинакова, поэтому почти всегда препарат вводят энтерально. Псилоцибин обычно вводят в дозах 250 мкг/кг, а мескалин в дозах 5-6 мг/кг. Несмотря на отличия в активности, эффекты препаратов почти идентичны.
Скополамин и другие антимускариновые вещества вызывают делирий с дезориентацией, затруднением мышления, значительным нарушением памяти и бредовыми идеями. Если дозы достаточно велики, указанные нарушения могут продолжаться более суток; некоторые утверждают, что требуется несколько дней для восстановления нормальной памяти. Большинство людей, по крайней мере в условиях эксперимента, оценивают эти ощущения как неприятные и не желают их повторить. Некоторые испытывают страх во время действия препаратов, другие преднамеренно их ищут и повторно используют.
Фенциклидин вызывает отрешенность, дезориентацию, нарушение схемы тела, нарушение проприоцепции. Соматические симптомы и признаки включают оцепенение, нистагм, потливость, тахикардию, гипертензию. Эффекты зависят от дозы. Передозировка может привести к летальному исходу, что отличает данный наркотик от наркотиков группы ЛСД, для которых нехарактерен прямой летальный эффект.
Токсичность
Вредные психологические последствия приема галлюциногенов общеизвестны. Панические реакции (“плохое путешествие”) могут быть связаны с передозировкой, они менее характерны при превышении дозы “уличных наркотиков”. Эти реакции
лучше поддаются лечению барбитуратами или бензодиазепинами, чем фенотиазинами. Острые психотические или депрессивные реакции вызываются галлюциногенами обычно у лиц с выраженной предрасположенностью. Они более характерны для фенциклидина, чем для других галлюциногенов. Нарушения мышления могут вести к опрометчивым поступкам, угрожающим жизни, поэтому любой человек, находящийся под влиянием одного из наркотиков данной группы, должен быть под наблюдением до тех пор, пока эффект не прекратится.
При передозировке антимускариновых веществ проводят инфузию физостигмина, но обычно предпочитают симптоматическую поддерживающую терапию.
Опасна передозировка фенциклидина, но при своевременной постановке диагноза она поддается лечению. Фенциклидин экскретируется в желудок, поэтому удаление наркотика из организма может быть ускорено промыванием желудка. Выведение можно интенсифицировать подкислением мочи до pH 5.5, поскольку препарат обладает основными свойствами (глава 60, том 2). Для предупреждения судорог или уменьшения возбуждения назначают диазепам; антипсихотические средства в данном случае могут усиливать эти эффекты. Если за острой интоксикацией следует продолжительное ши-зофреноподобное состояние, можно применить антипсихотические средства по курсовой схеме.
Длительное лечение
Определенных программ лечения для лиц, злоупотребляющих наркотиками этого класса, не существует. Более вероятно, что потребители галлюциногенов сохранят привычку к их применению и у них не разовьется полинаркомания. Наилучшая мера — отделение наркомана от микросоциальной среды, практикующей прием галлюциногенов, но оно должно быть добровольным.
Терапевтическое применение
Признанных терапевтических показаний для применения галлюциногенов не существует. ЛСД предлагали для лечения алкоголизма, но объективные доказательства целесообразности его назначения отсутствуют. Усиление эффектов психотерапии — другая область, предложенная для применения, но и здесь ЛСД не имеет преимуществ по
сравнению с другими лекарствами. Предпринимались попытки применять ЛСД при шизофрении и депрессии, но выше вероятность того, что он усиливает, а не ослабляет эти нарушения. По данным некоторых исследователей, использование препаратов у онкологических больных позволяет снизить дозы опиоидов, но это наблюдение не подтверждено.
Марихуана
История
Каннабис (конопля) применялся в течение тысячелетий, особенно в Китае и Индии. Он был известен в Греции в период ее расцвета, а несколько позднее и в арабских странах. Считается, что около 200-300 миллионов людей применяют каннабис в той или иной форме. Таким образом, это не только старейшее, но и одно из наиболее широко используемых средств, изменяющих психику. С 1960-х гг. в США отмечается значительный подъем потребления марихуаны. Хотя наркотик и был известен в этой стране ранее, количество потребителей было значительно меньше, и они в основном включали представителей национальных меньшинств. В настоящее время в США около 30—40 миллионов человек периодически используют наркотик и значительное число — регулярно. Последние исследования показали, что количество молодых людей, начинающих применять этот наркотик, снизилось после периода резкого подъема, но снизился и возраст лиц, попробовавших наркотик впервые.
Существует предположение, что конопля представлена несколькими видами, однако ботаники не согласны с тем, что существует более одного вида растения: скорее имеется множество морфологических вариантов одного вида. Некоторые растения отличаются превосходными волокнами, являясь сырьем для пеньки, но считаются плохим сырьем для извлечения фармакологически активного начала, и наоборот. Эти свойства связаны с генетическими различиями. Наибольшее количество вещества содержат цветы и мелкие листья Cannabis sativa. Марихуана — это смесь надземных частей растения, которая по внешнему виду напоминает стрижку травяного газона,— отсюда уличное название наркотика — “травка”. При экстракции смолы растения получают более активный продукт — гашиш.
Химия и фармакокинетика каннабиноидов
Каннабис содержит три основных каннабиноида: каннабидиол(CBJD), р9-тетрагидроканнабинол (ТНС) и каннабинол (CBN). Биосинтез начинается с CBD, продолжается образованием ТНС и завершается синтезом CBN (рис. 31-3). Таким образом, по соотношению каннабиноидов в растении можно высчитать его возраст. В растении найдено много других вариаций данной структуры, но за исключением ТНС и его гомологов, другие каннабиноиды не имеют психотропной активности. Содержание ТНС в разных растениях значительно различается, специально выведенные генетические линии могут содержать до 4-6 % ТНС. Большинство растений содержит ТНС в количестве 1-2 %.
Предпочитаемая форма использования наркотика в Западных странах — курение. Высокая растворимость ТНС в липидах обеспечивает быстрый его захват сурфактом легких. Фармакокинетические исследования демонстрируют, что по своим показателям курение почти эквивалентно внутривенному введению за исключением более низкой пиковой концентрации ТНС в плазме. В некоторых странах Востока каннабис принимают внутрь в форме различных сладостей. Скорость всасывания при таком пути введения замедлена и весьма вариабельна, хотя продолжительность действия больше.
ТНС активно метаболизируется, и все еще открывают его новые производные. Один из метаболитов (11-гидрокси-ТНС) более активен, чем родительское соединение. Однако это соединение не столь богато представлено, и можно предположить, что основная активность каннабиса определяется ТНС. Высокая липофильность вещества ведет к активной его секвестрации в липидных компарт-ментах, при этом метаболиты могут экскретироваться в течение недели после однократного применения. Возможно ли накопление неизмененного ТНС, остается невыясненным.
Фармакодинамика каннабиноидов
Механизм действия ТНС является предметом интенсивных исследований. Высокая степень селективности энантиомеров как природных, так и новых каннабиноидов, указывает на существование высокоизбирательного рецептора. Сообщалось о предполагаемом эндогенном лиганде (Devane,
Рис. 31-3. Структуры трех основных каннабиноидов в марихуане. Только ТНС обладает психотропным действием. С5Н|( — л-пентил. Стрелки указывают последовательность биосинтеза
СН3
Каннабинол
1992). Синтетические каннабиноиды-агонисты с высокой активностью и стереоселективностью в поведенческих тестах были использованы для характеристики мест связывания каннабиноидов. Оказалось, что аффинитет при связывании коррелирует с относительной активностью в биологических тестах, указывая на то, что рецептор опосредует и фармакологические эффекты. Места связывания наиболее обильно представлены в эфферентных ядрах базальных ганглиев, ретикулярной части черной субстанции, бледных шарах, гиппокампе и в стволе мозга. Рецептор был клонирован и выявлено, что он сопряжен с G-белком. Исследования с применением позитронной эмиссионной томографии показали, что после введения ТНС его метаболизм усиливается в тех же областях, в которых локализованы рецепторы, что указывает на участие рецепторов в клиническом эффекте вещества. Медиаторную роль в активности ТНС могут играть простагландины, но конкретный механизм остается невыясненным. Высокая липофильность ТНС позволила предположить неспецифический мембранотропный эффект, но это не может объяснить стереоселективность действия.
ТНС оказывает ряд фармакологических эффектов, имеющих определенное сходство с амфетаминами, ЛСД, алкоголем, седативными средствами, атропином и морфином. Таким образом, вещество
не соответствует каким-либо традиционным фармакологическим классификациям и может рассматриваться как представитель отдельного класса.
Малые дозы ТНС могут оказывать эффект, не отличимый от плацебо-эффекта, однако большие дозы оказывают несомненное фармакологическое действие. Опытные потребители определяют действие после двух-трех затяжек. При продолжении курения эффекты возрастают, достигая максимума через 20 минут после его окончания. Большинство эффектов наркотика исчезает через 3 часа, к этому времени его концентрация в плазме становится низкой. Пиковый эффект после энтерального введения может задержаться до 3-4 часов после начала приема, однако он продолжается в течение 6-8 часов.
Ранняя стадия эффекта (“подъем”, “high”) характеризуется эйфорией, бесконтрольным смехом, изменением восприятия времени, деперсонализацией и повышением остроты зрения. Позднее субъект расслабляется, погружается в интроспекцию, испытывает дремотное состояние или даже погружается в сон. Мышление и концентрация внимания затрудняются, хотя усилием воли человек может сосредоточиться.
Двумя характерными признаками интоксикации каннабисом являются повышение частоты сердечных сокращений и покраснение конъюнктивы. Последнее хорошо коррелирует с концентрацией дей
ствующего начала в плазме. Величина зрачков не изменяется. АД может понизиться, особенно в вертикальном положении. Может отмечаться противо-рвотный эффект. Также могут наблюдаться мышечная слабость, тремор, неустойчивость, усиление глубоких сухожильных рефлексов. Практически любой сложный психологический тест выявляет нарушения, если доза достаточно велика. Специфических биохимических изменений у человека при применении наркотика не обнаруживается.
Толерантность наблюдается практически у всех видов животных, которые были исследованы. У человека она очевидна только после длительного интенсивного применения. Различные степени толерантности характерны для различных эффектов вещества, в частности сравнительно быстро развивается привыкание к вызванной наркотиком тахикардии. После хронического потребления очень высоких доз отмечается мягкий абстинентный синдром.
Опасность для здоровья
Использование марихуаны вызывает озабоченность из-за возможного неблагоприятного влияния на здоровье, особенно в связи с тем, что ее применяет в основном молодежь. Сведения о негативных последствиях весьма противоречивы и существует много проблем, касающихся корректности самих исследований. Во-первых, довольно трудно доказать или отвергнуть наличие вредных эффектов у человека на основании экспериментов на животных. Во-вторых, потребителями являются в основном молодые и здоровые люди. В-третьих, каннабис часто используется в сочетании с алкоголем и табаком. И, в-четвертых, многие исследователи исходно настроены против применения каннабиса.
Три эпидемиологических исследования в развивающихся странах не смогли выявить определенных доказательств нарушений здоровья среди активных потребителей каннабиса, но существует вероятность того, что эти исследования малочувствительны. Экспериментальные исследования, при которых испытуемые активно курили каннабис разные по продолжительности периоды, показали снижение уровня тестостерона в сыворотке у мужчин и сужение воздухоносных путей. Данные о влиянии на иммунные механизмы, хромосомы и клеточный метаболизм часто противоречивы. Влияние на плод еще точно не определено.
У активных курильщиков марихуаны могут быть те же проблемы хронического бронхита, бронхообструктивных состояний и метаплазии клеток слизистой, как и у курильщиков табака. Может ухудшиться течение стенокардии в связи с тахикардией, ортостатической гипотензией и повышением содержания карбоксигемоглобина. Водительские навыки, вероятно, тоже должны ухудшаться, но это непросто доказать с помощью обычных тестов. “Амотиваци-онный синдром”, при котором многообещающие молодые люди с очевидными достижениями теряют интерес к школе и карьере и погружаются в наркотическую субкультуру — реальный факт, но нельзя быть уверенным в том, является ли употребление наркотика причиной проблемы или это просто персональный выбор. Острые панические реакции, токсический делирий, параноидные состояния и психозы редки. Повреждения мозга не подтверждены у человека, хотя на животных получены некоторые данные об его ультраструктурных изменениях.
Терапевтическое применение
Одно время каннабис числился в лекарственных формулярах, но не использовался по медицинским показаниям. Сейчас интерес к его применению с терапевтическими целями оживился. Многократно подтверждены наблюдения о снижении внутриглазного давления после приема внутрь TH С. Остается убедиться в том, имеют ли препараты каннабиса какие-либо преимущества по сравнению с другими средствами для лечения глаукомы. Изучалась также возможность уменьшения с их помощью тошноты и рвоты при химиотерапии в онкологии. На фармацевтическом рынке для применения по этому показанию имеется ТНС под названием дро-набинол (Маринол). Оказалось возможным предотвратить тошноту и рвотный рефлекс в дозах, которые лишь незначительно влияют на психику. Другой гомолог левонантрадол, вероятно, может использоваться в медицине как аналгетик, как вещество для уменьшения мышечных спазмов или даже как антиконвульсант. Однако необходимо доказать, что любое из этих средств имеет преимущества перед стандартными лекарствами.
Лечение
Лишь небольшое число лиц, потребляющих каннабис, обращается за медицинской помощью. Тем не менее многие, кто прекратил потребление нар
котика, были приятно удивлены повышением ясности своего мышления. Марихуана рассматривается как заменитель алкоголя, но чаще она потребляется вместе с ним, хотя алкоголизм как осложнение употребления марихуаны, встречается редко. Марихуана иногда используется в структуре полинаркоманий, и в этом случае может потребоваться лечение, которое направлено на избавление от пристрастия к более опасному наркотику.
Вещества, используемые для ингаляции
История
Открытие опьяняющих эффектов ингаляционных препаратов связывают с именем Хемфри Дейви, который в 1799 г. исследовал на себе и других действие закиси азота. По мере внедрения других ингаляционных анестетиков, таких как хлороформ и эфир, они также использовались не по медицинским показаниям. Такое применение сохранилось, к нему добавились случаи ингаляции галотана. В настоящее время преобладают три типа веществ, используемых для вдыхания: 1) промышленные растворители, включая ряд углеводородов (например, толуол); 2) аэрозольные пропелленты (различные фторуглероды); и 3) органические нитриты, такие как амил- или бутилнитрит.
Химия и фармакология
Структура некоторых веществ, используемых для ингаляции, показана на рис. 31-4. Механизм действия ингаляционных анестетиков обсуждается в главе 24.
Клинические аспекты
Закись азота, ранее труднодоступная, в настоящее время продается свободно. При применении она вызывает нарушение концентрации внимания, эйфорию, оцепенение, звон в ушах, неустойчивость походки, зрительные и слуховые расстройства. Она обычно используется в виде смеси 35 % закиси азота с кислородом, поскольку введение 100 % закиси азота может вызвать асфиксию и смерть.
Эфир и хлороформ доступны через магазины химических реактивов. Для ингаляции паров используется ткань, смоченная летучей жидкостью.
Толуол
СН3СОСН2СН3
Метилэтилкетон
CI2C=CHCI (CH3)2CHCH2CH2ONO
Трихлорэтилен Амилнитрит
Рис. 31-4. Структура некоторых часто применяемых для ингаляции веществ
После начального периода возбуждения может происходить потеря сознания. Эфир очень горюч. Галотан обычно доступен только медицинскому персоналу.
Ряд промышленных растворителей, которые могут использоваться для ингаляции, легко доступен, и использование этих веществ не поддается контролю. К ним относятся бензин, растворители для красок, клеи, резиновый цемент, распылители акриловых красителей, крем для обуви, растворители жира. Токсичными ингредиентами могут быть толуол, гептан, гексан, бензол, трихлорэтилен, метилэтилкетон и другие. Вследствие своей доступности эти вещества широко используются. Аэрозольные пропелленты также были легко доступны, хотя в последнее время производители переходят на замену фторуглеродных пропеллентов менее опасными материалами.
Мотивами употребления вдыхаемых веществ являются влияние сверстников, низкая стоимость, легкая доступность, удобная упаковка, быстрая и кратковременная интоксикация и повышение настроения. Основные потребители — мальчики-подростки из низших социально-экономических слоев общества. Как правило, у них имеются существенные проблемы в школе и в семье. При применении упомянутых веществ развивается психологическая зависимость.
Клинические эффекты промышленных растворителей кратковременны и продолжаются только 5-15 минут. Обычно какую-либо ткань смачивают растворителем и вдыхают его пары. Аэрозольные пропелленты вдыхают с помощью пластиковых мешков. Эйфория и чувство опьянения сменяются дезориентацией, ощущением замедления хода времени и, возможно, галлюцинациями.
Органические нитриты заслужили репутацию сексуальных усилителей. Амилнитрит используется в медицине для лечения грудной жабы в форме хрупких стеклянных ампул, покрытых материей. Когда ампулу разбивают, она издает хлопающий звук (отсюда прозвище “хлопушка”). “Уличные” препараты (изобутилнитрит в бутылках) в некоторых регионах легко доступны. Ингаляция вызывает головокружение, учащение сердцебиений, понижение АД, покраснение кожи. Главное влияние, которое может оказать препарат на сексуальную функцию, видимо, снятие торможения. За исключением единичных примеров метгемоглобинемии, связанной с избытком нитритов, имеется мало сообщений о серьезных вредных эффектах.
Токсичность
Хлороформ оказывает выраженное токсическое действие на печень и почки. Ряд вредных эффектов вызывают промышленные растворители: повреждение печени и почек, поражение периферических нервов и, возможно, повреждение мозга, угнетение костного мозга, заболевания легких. Ингаляция фторуглеродов может привести к неожиданной смерти из-за желудочковой аритмии или асфиксии. Нитриты сравнительно безопасны, но могут оказаться опасными для лиц с сердечнососудистыми заболеваниями.
Избранная литература
Bock G. R., Whelan J. (eds) Cocaine: Scientific and Social Dimensions. (Ciba Foundations Symposium 166.) Wiley, 1992.
Collier H. O. J. Physiological basis of opiate dependence. Drug Alcohol Depend. 1983; 11:15.
Devane W. A. et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science, 1992; 258: 1946.
Grabowski J., Dworkin S. I. Cocaine: An overview of current issues. Int. J. Addict. 1985; 20:1065.
Hollister L. E. Chemical Psychoses: LSD and Related Drugs. Thomas, 1968.
Hollister L. E. Health aspects of cannabis. Pharmacol. Rev. 1986; 38: 1.
Johanson C.-E., Fischman M. W. The pharmacology of cocaine related to its abuse. Pharmacol. Rev. 1989; 41: 3.
Kalix P., Braenden O. Pharmacological aspects of the chewing of khat leaves. Pharmacol. Rev. 1985; 37: 149.
Koob G. F., Bloom F. E. Cellular and molecular mechanisms of drug dependence. Science, 1988; 242: 715.
Nestler E. J. Molecular mechanisms of drug addiction. J. Neurosci. 1992; 12: 2439.
Smith D. E. et al. (eds) Amphetamine Use, Misuse and Abuse. G. K. Hall, 1979.
WHO: Report of the advisory group meeting on adverse health consequences of volatile solvents/ inhalants. Mexican Institute of Psychiatry, Mexico City, Mexico. 1986; April: 21.
Указатель
Большинство препаратов указано под их родовыми названиями. Там, где возможно, если указано торговое название лекарства, ссылка на страницу относится к имеющимся препаратам. Для более детального описания следует смотреть ссылку на родовое название.
А
Абсанс, 462
Абстинентный синдром, 572
Авентил, 458.
См. также Нортриптилин, 556
Автономная нервная система (АНС), 101-120
Агонисты, 15-16, 22
а-агонисты и глаукома, 190
Адалат, 168,185.
См. также Нифедипин, 220,240
АДГ. См. Антидиуретический гормон
Аддикция (пристрастие), 579
Аденилатциклаза, 159
Аденозинтрифосфат (АТФ), 107
Аденозин, 277,278,288,293
Адреналин, 166,173-174,389,395
Адренергическая передача, рецепторы 106-109,154-160
Адренергические нейроны, 106-107, 203
Адренергических нейронов блокаторы, 202
Адреноблокаторы, 175-192 а-адреполитики селективные, 219 р-адрепорецептор(ы), 40,155-156, 158-159,242,253,386-391
Азатадин, препараты, 343
Азмакорт, 396.
См. также Триамцинолон ацетонид
Азо-восстановление, 79
Азота оксид, 226,394
Акатизия, 534
Аквахлорал суппретте, 347.
См. также Хлоралгидрат, 429
Акинетон, 430.
См. также Бипериден, 522 Аккомодационная эзотропия, 133 Аккуприл, 168.
См. также Квинаприл, 202
Аконитин, 486
Аксид, 344.
См. также Низатидин
Актидил, 344
Алзапам, 347.
См. также Лоразепам, 428 Алкалоз метаболический, 302-305 Алкалоиды спорыньи, 336-345 Алкалоиды, 152,336-345 Алкогольдегидрогеназный путь, 431 Аллергеп(ы), 381-383 Аллопуринол, 19,84 Алпренолол, 85,176 Алтае, 168.
См. также Рамиприл, 220
Алупепт, 318.
См. также Метапротеренол, 396
Алурат, 429
Алфента, 475.
См. также Алфентанил, 577
Алфентанил, 462,577
Альбумин и измерение концентраций лекарств, 70
Альбутерол, 389,396
Альдактоп, 247-248.
См. также Спиронолактон, 316
Альдомет, 168.
См. также Метилдопа, 229
Альдостерон, 213,308
Альпразолам 412,425,428
Альпростадил, препараты, 377 Амантадин, 516-517
Amanita phalloides, 149
Amanita virosa, 149
Амбеноний, 131,137
Амбиен, 348.
См. также Золпидем, 429 Амидат, 393.
См. также Этомидат, 482
Амиды, 79,484
Амикацин, 56
Амилнитрит, 225,231,240,594-595
Амилорид, 308-309,316
Амин(ы), 80,141,546
Аминные алкалоиды, структура, 337 Аминокислоты,407-408
Аминооксидазы, 79
Аминофиллин, 385
Амиодарон, 280-282
Амитал, 429
Амитриптилин, 342,552,555
Амлодипин, 213,220,240
Амобарбитал, 429
Амоксапин, 545-557
Амринон, 252-257
Амфетамин, 161,173,584-587
Аналгезирующие агенты, 558 Аналгезия, 558,566,567,540 Анафранил, 458.
См. также Кломипрамин, 556 Ангиотензин, 197,213-214,346-351 Ангиотензиноген, 347
Ангиотепзинпревращающий фермент (АПФ), ингибиторы, 214-215
Анектин, 417.
См. также Сукцинилхолин, 508
Анестезия аналгетиками, опиоиды, 391
Анестезия, 420,468-470,480-482, 483-492
Анизотропии, 147
Анионная передозировка и петлевые диуретики, 303-305
Антабус, 359.
См. также Дисульфирам, 439,442 Антагонист(ы), 16,22 Антагонисты Н,-рецепторов, 324-
328
Антагонисты Н2-рецепторов, 88-90, 328-332
Антазолин, 326
Антиаритмические агенты, 273-293, 504
Антиверт, 272.
См. также Меклизин, 344 Антигипертензивные агенты, 193-221
Антигистаминные средства, 323-332 Антидепрессанты, 544-557 Антимускариновые средства, 139-
150
Антипсихотические средства, 524-543
Антихолинергические агенты, 139-
153
Апраклонидин, 190
Апресолин, 168.
См. также Гидралазин, 220
Апробарбитал, 429
Апротинин, и калликреин-кинино-вая система, 354
Арамин, 130.
См. также Метараминол, 174 Арахидоновая кислота, 362-368,377 Ардуан, 417.
См. также Пипекуроний, 508 Аритмии, 265-294.
См. также Сердечные аритмии Ароматическое гидроксилирование, 79
Артан, 430.
См. также Тригексилфени-дил, 523
Артериовенозный концентрационный градиент и ингаляционная анестезия, 472
Арфонад, ИЗ, 168.
См. также Триметафан; триметафана камзилат 153,219
Асендин, 458.
См. также Амоксапин, 556
Аспартат, 406
Астемизол, 326,343
Астма, 379-398
Астокорт, 3.
См. также Триамцинолон;
Триамцинолона ацетонид,
Триамцинолона ацетат, 396
Атаракс, 272,348.
См. также Гидроксизин, 344
Атенолол, 183-185,207
Атетоз, 509,521
Ативан, 347,379,393.
См. также Лоразепам, 428,466,
482
Атонические судорога, 462-463
Атракурий, 498,503
Атровент, 113,319.
См. также Ипратропиум, 153,396
Атропин, 139-146
Атропина сульфат и моторика
бронхов, 391-392
АТФ. См. Аденозин трифосфат
Ауторецепторы, 117
Африн, 174.
См. также Оксиметазолин
Ацебутолол, 186,207
Ацеон, 220
Ацетазоламид, 19,190,301,316,461
Ацетальдегида метаболизм, 432
Ацетаминофен, 19,56,80
Ацетил-р-метилхолин, 124
Ацетилирование, 80,81
Ацетилхолин (АЦХ), 36,104-105,
124,408-409,493
Ацетилхолина хлорид, 124
Ацетилхолииэстераза, 104,129
Ацетофеназин, 542
Ацидоз 303,309-310
АэроБид, 396
Б
Баклофен, 506-507
Баллизм, 520
Бантин, 113.
См. также Метантелин, 153
Барбитураты, 415-421,582-584
Барорефлекс, постуральный, 194—
195
Батрахотоксин и натриевые каналы, 486
Беамин, 359.
См. также Тиамин, 442
Беклометазон при астме, 396 G-белок, 37-39
Белладонны алкалоиды, препараты, 152
Бенадрил, 272.
См. также Дифенгидрамин, 344
Беназеприл, 214,220
Бендрофторметиазид, 307,316 Бензо[а]пирен, и метаболизм лекарств, 84
Бензодиазепины, 413-422,582-584
Бензокаин, 485,491
Бензоморфаны, 576 Бензтиазид, 307,316 Бензтропин, 141,518,522 Бенилин DM, 476.
См. также Декстрометорфан, 578
Бентил, 112.
См. также Дицикломин, 152
Бепридил, 288
Бетазол, 323
Бетаксолол, 207
Беталин S, 359.
См. также Тиамин, 442
Бетанидин, 202
Бетапейс, 145.
См. также Соталол, 192
Бетоптик, 191.
См. также Бетаксолол, 219
Бикарбонат, 296
Биодоступность, 60-62 Биотрансформация, 73-86 Бипериден, 518,522 Бипиридины, 252-253 Биполярные аффективные расстройства, 536-537
Бисопролол, 184,191,219
Битолтерол, 389-390
Блокадрен, 192,219.
См. также Тимолол
Блокаторы кальциевых каналов, 212-213,231-237,287-288,393 р-блокаторы, 182-191,207,285 Болезни движения (укачивание, кинетозы), 146,327
Ботулизма токсин, 118 Бревиблок, 145,227.
См. также Эсмолол, 191,292
Бревитал натрия, 482
Бретилиум, 279,280,292 Бретилол, 227.
См. также Бретилиум, 292 Бретин, 319.
См. также Тербуталин, 396 Бриканил, 319.
См. также Тербуталин, 396 Брокефрин, 395 Бромид, и эпилепсия, 461 Бромокриптин, 515-516 Бромокриптина мезилат и гиперпролактинемия, 342
Бромфен, 272.
См. также Бромфенирамин, 343 Бромфенирамин, 326, 343 Бронхиолярная гладкая мускулатура и гистамин, 322
Бронходилататоры, 379-398 Букладин-S Софттабс, 343 Буклизин, препараты, 343 Бумекс, 247.
См. также Буметанид, 316 Буметанид, 304,316 Бупивакаин, 491 Бупренекс, 475.
См. также Бупренорфин, 577 Бупренорфин, 560,562,577 Бупропион, 547-548,552 Буримамид, 88 БуСпар, 347.
См. также Буспирон, 422-423 Буспирон, 335,422-423 Бутабарбитал, препараты, 429 Бутамбен, препараты, 491 Бутезин Пикрат, 491 Бутизол, 429 Бутирилхолинэстераза, и ингибиторы холинэстеразы, 130-131 Бутирофенона производные, 525-
526
Бутоксамин, 176,186 Буторфанол, 562,562,577 Буфадиенолиды, 246-247
В
Вазоактивные пептиды, 346-360
Вазоактивный интестинальный пептид, (VIP) 107,358
Вазодилататоры, 197,208-213,222-241,255
Вазоксил, 174.
См. также Метоксамин Вазопрессин, 354-355.
См. также Антидиуретический гормон
Вазотек, 220,262.
См. также Эналаприл
Валиум, 428,442,466, 508,417.
См. также Диазепам
Вапорол, 240.
См. также Амилнитрит Васкор, 240,292.
См. также Бепридил Векуроний, 497,498,503,508 Веллбутрин, 556.
См. также Бупропион Венлафаксин, 556 Вентолин, 396.
См. также Альбутерол Верапамил, 213,220,232-287,240, 287-288,292
Вератридин, 484
Вератрума алкалоиды, 198-199
Версед, 428,482.
См. также Мидазолам
Веснаринон, 254
Весприн, 542
Вивактил, 556.
См. также Протриптилин Вигабатрин, 453-454
Вискен, 192,219.
См. также Пиндолол
Вистарил, 344,429.
См. также Гидроксизин Витензин, 219.
См. также Гуанабенц Внутривенные анестетики, 479-482
Г
Габапентин, 454-457,466
Галанин, роли, 107
Галдол деканоат, 542
Галдол, 446.
См. также Галоперидол 542
Галламин, 494,508
Галлюциногены, 587-591
Галоперидол, 526,532-533,542
Галотан, 482
Гамма-аминомасляная кислота
(ГАМК), 407-410
Ганглиоблокирующие средства, 150-153,202
Ганглионарный никотиновый ацетилхолиновый рецептор, 493
Гастрин-рилизинг пептид, роли, 107
Гастрокром,319.
См. также Кромолин натрия 396
Гексаметоний, 151
Гелонтин Капселе, 379.
См. также Метсуксимид 466
Гемихолиний и автономная передача, 118
Гемонил, 429,466
Генерализованные судороги, 444,
463-464
Героин, 574,581
Гетероциклические антидепрессанты, 545,547,550
Гигротон, 247.
См. также Хлорталидон 316
Гидрокодон, 575
Гидрокодон/ацетаминофен, препараты, 578
10- Г идроксикарбазепин, структура, 450
15-Гидроксидегидрогеназный путь, 368
6-Гидроксидофамин и автономная передача, 118-119
Гидролиз, 79
Гидромокс, 247.
См. также Хинетазон, 316
Гидроморфон, 574
Гидропероксиэйкозатетраеновые
кислоты,361
Гидрохлортиазид, 306-307,316
Гилорел, 168.
См. также Гуанадрел 219
Гиосциамин, 112,958.
См. также Атропин 153
Гиосцин, ИЗ.
См. также Скополамин 153
Гиперкалиемия, 304,309
Гипернатриемия и осмотические диуретики, 311
Гиперреактивность, 48
Гиперстат IV, 168.
См. также Диазоксид, 219
Гипертензия, 193-221
Гипертермия, злокачественная, 478,
507-508
Гипертонический криз, 181
Гиперурикемия, 305,307
Гиперхлоремический метаболический ацидоз, 302,309
Гиперчувствительность немедленного типа, 380
Гиперчувствительность, 48
Гипокалиемический метаболичес-
кий алкалоз, 304-305
Гипомагниемия и петлевые диуретики, 305
Гипонатриемия и диуретики, 307
Гипореактивность, 48
Гипотензия и симпатомиметические средства, 169-170
Гисманал, 272.
См. также Астемизол, 343
Гистамин, 318-323
Гитрин, 145,168.
См. также Теразозин 219
Глаукома, 133-134,171-172,189,190
Гликопирролат, 141,147,152,958
Глицин, 407-408,416
Глутамат, 407-408
Глюкуронидация, 80
Глютетимид, 429
Гоматропин, 146,152
Гранизетрон, препараты, 272
Гуанабенц, 200
Гуанадрел, 202
Гуанетидин, 202-204
Гуанфацин, 200
Гуморсол, 100.
См. также Демекарий, 137
д
Далган, 475.
См. также Дезоцин, 577
Далман, 347.
См. также Флуразепам, 428
Дантриум, 417.
См. также Дантролен, 508
Дантролен, 508
Даранид, 247.
См. также Дихлорфенамид, 316
Дарбид, 112,959.
См. также Изопропамид, 153
Дарикон, 113,959.
См. также Оксифенциклимин, 153
Деалкилирование, окислительное, 78
N-деакилирование, 78
О-деакилирование, 78 S-деалкилирование, 78 Дебризохин, 202
Дезаминирование, 78 Дезипрамин, 85,546,556 Дезирел, 458.
См. также Тразодон, 556
Дезоцин, 576
Декадрон, 319,605.
См. также Дексаметазон, 396
Декломицин, 316
Декседрин, 130.
См. также Декстроамфетамин, 173
Дексозин, 130.
См. также Метамфетамин, 174 Декстроамфетамин, 583 Декстрометорфан, 576
Делзим, 476.
См. также Декстрометорфан, 578 Демадекс, 248.
См. также Торсемид, 304,317 Демерол, 475.
См. также Меперидин, 577 Демсер, 145.
См. также Метирозин, 192 Депакен, 379,446.
См. также Вальпроевая кислота, 467,542
Депакот, 379,446.
См. также Вальпроевая кислота, 467,542
Депен, 430.
См. также Пеницилламин, 523 Деполяризующие блокирующие средства, 499-501
Деполяризующие средства, 493 Депренил, 430.
См. также Селегилин, 523 Десенситизация, 16,39-41 Десланозид, препараты, 262 Десфлуран, структура, 469 Диазепам, 428,460-461,505 Диазоксид, 212
Диамокс, 247.
См. также Ацетазоламид, 316 Диацетилхолин, 417.
См. также Сукцинилхолин, 508 Дибензилин, 145.
См. также Феноксибензамин, 191 Дибензодиазепин, 526,527,532 Дибукаин, препараты, 491 Дигибайнд, 262 Дигидроиндолон, 532 Дигидрокодеин, 575
Дигидроксифенилаланйн, структу-
ра, 511
Дигидропиридины,213
Дигидроэрготамин, 341,345
Дигиталис, 246-252
Дигитоксин, 247
Дигоксин, 247
Дигоксиновая иммунная сыворотка, препараты, 262
Дизипал, 430.
См. также Орфенадрин, 522
Дизопирамид, 278-279
Диклон, 491
Диклонин, препараты, 491
Дилантин, 379.
См. также Фенитоин, 467
Дилтиазем, 213,220
Дименгидринат, 326,344
Диметилтубокурарин, 417.
См. также Метокурин, 508
Динасерк, 168,185.
См. также Исрадипин, 220
Динопростон, 377
Дипивефрин, 172
Диприван, 393.
См. также Пропофол, 482
Дирений, 248.
См. также Триамтерен, 317
Дисменорея и эйкозаноиды, 373
Диссоциативная анестезия, 481
Дистония, 509,521
Дисульфирам, 440-442
Дисульфитрам, 439
Дитропан, ИЗ.
См. также Оксибутинин, 153
Диукардин, 247.
См. также Гидрофторметиазид, 316
Диуретики, 197-198,253-254,294-
317
Диурил, 247.
См. также Хлортиазид, 316
Дифенгидрамин, 325-326
Дифеноксилат, 575
Дифеноксин, 575
Дифиллин, препараты, 397
Дихлорфенамид, 316
Дицикломин, 147,152
Диэтиламид лизергиновой кислоты (ЛСД), 337,587-591
Добутамин, 167,173,253
Доза
нагрузочная, 66-67
неэффективная, 93
поддерживающая, 65-66 срединная летальная, 93
Доза-эффект — кривая зависимости, 45,47
Доксазозин, 176
Доксакурий, 508
Доксепин, 522,556
Доксиламин, 326
Долофин, 475.
См. также Метадон, 577
Допар, 430.
См. также Леводопа, 522 Допастат, 202.
См. также Дофамин, 262
Дорал, 428
Дориден, 348.
См. также Глютетимид, 429 Дофамин, 166,510,514,525,527 Дофаминергическая система и антипсихотические средства, 527-529
Драмамин, 272.
См. также Дименгидринат, 344
Дураквин, 227,801.
См. также Хинидин, 292
Дуранест, 402.
См. также Этидокаин, 491
3
Зантак, 344.
См. также Ранитидин Зароксолин, 316.
См. также Метолазон, 306 Заронтин, 455-456,466.
См. также Этосукцимид Застойная сердечная недостаточность, 242-263
Зебета, 191,219.
См. также Бисопролол Земурон, 508.
См. также Рокурониум Зестрил, 220,262.
См. также Лизиноприл Злоупотребление ингалянтами, 594-595
Золофт, 556.
См. также Сертралин
Золпидем, 423,429
Зоман, 131
Зофран, 342.
См. также Ондансетрон
И
Ибопамин, 166
Идиосинкразия лекарственная, 48 Идроциламид, 507
Измо, 240.
См. также Изосорбид мононитрат Изо-Бид, 240.
См. также Изосорбид динитрат Изокарбоксазид, 552,556 Изопропамид, 147,153 Изопротеренол, 160,174,396 Изоптин, 220,240,292.
См. также Верапамил Изопто Гоматропин, 152.
См. также Гоматропин Изопто Карбахол, 137.
См. также Карбахол Изопто Карпин, 137.
См. также Пилокарпин Изордил, 240.
См. также Изосорбида динитрат Изосорбида мононитрат, 240 Изофлуран, 469,482 Изофлурофат, 137 Изоэтарин, 389,396
Изупрел, 174,396.
См. также Изопротеренол Имидазолиновый рецептор, 200 Имипрамин, 278,280,546,548-552 Имитрекс, 345.
См. также Суматриптан Импромидин, 323 Инверсии, 153,219.
См. также Мекамиламин Ингаляционные анестетики, 470-479
Ингибиторы холинэстеразы, 133-135
Индапамид, 306,307,316
Индерал, 192,219,240,292.
См. также Пропранолол Индорамин, 179 Инозитол-1,4,5-трифосфат (ИТФ), 42-43,122,156,369
Инокор, 262.
См. также Амринон Интал, 396.
См. также Кромолин натрия Интропин, 262.
См. также Дофамин Инфантильные спазмы, 444
Инфаркт миокарда, 222-223,259-261
Ионизация, константы 18-19 Иопидин, 173.
См. также Апраклонидин Йохимбин, 176,179 Ипратропиум, 141,153,396 Исмалин сульфат, 219.
См. также Гуанетидин Исрадипин, 220,240 Ишемическая болезнь сердца, 141-
142
К
Калан, 168,185,227-228.
См. также Верапамил 220,240,292 Калиевые каналы, модуляторы, 292, 394
Калийсберегающие диуретики, 307-310
Калликреин(ы), 351-352 Калликреин-кининовая система, 351-354
Кальцитониновый ген-связанный пептид (ССКР.ко-кальцигенин), 107,359
Каннабидиол, 592
Каннабиноиды,591-594
Каннабинол, 592
Кантил, 113.
См. также Мепензолат, 153 Капотен, 168,203.
См. также Каптоприл, 220 Каптоприл, 214-216,220,356 Карбамазепин, 449-451,463,466,540 Карбаминовая кислота, структура
124
Карбамоил-р-метилхолин, структура, 124
Карбамоилхолин, структура, 124
Карбахол, 124,157,190
Карбахола хлорид, свойства, 125
Карбидопа, 511,522
Карбидопа/леводопа, 511,522
Карбиноксамин, 326,343
Карбоангидразы ингибиторы, 299-
303
Карбонильное восстановление, 79
Карбопрост, препараты, 377 Карден, 168,185.
См. также Никардипин, 220,240
Кардизем, 168,185,228.
См. также Дилтиазем, 220,240, 292
Кардилат, 184.
См. Также Эритритила тетранитрат, 240
Кардиогенный шок, 170,261
Кардиоквин, 227.
См. также Хинидин, 292
Кардионатрин, 355
Кардура, 145,168.
См. также Доксазозин, 191, 219
Картеолол, 190,191,207
Картрол, 145,167.
См. также Картеолол, 190,191,207
Катапрес, 168.
См. также Клонидин, 219
Катехол, структура, 160
Катехоламины, 160,166-167
Катехол- О-метилтрансфераза (КОМТ),516
Катинон, 585
Кафергот, 272-273.
См. также Эрготамин, 345
Квазепам, 428
Квантовая кривая “Доза-эффект”, 47
Кварзан, 152.
См. также Клидиниум
Кемадрин, 523.
См. также Проциклидин
Керлон, 191,212.
См. также Бетаксолол
Кеталар, 481-482.
См. также Кетамин
Кетамин, 481,588
Кетансерин, 336
Кининогены, 351-352
Кинины, 351-353
Китрил, 345.
См. также Гранизетрон
Кларитин,272.
См. также Лоратадин, 344
Клемастин, препараты, 344
Клиренс, 54-59
Клистин, 272.
См. также Карбиноксамин 343
Клобазам, 460-461
Клозапин, 526,527,532,542
Клозарил, 446.
См. также Клозапин
Кломипрамин, 552,556
Клоназепам, 460,542
Клонидин, 199,201-202,219
Клонопин, 428,466,542.
См. также Клоназепам Клоразепат, 416,428,460 Когентин, 430.
См. также Бензтропин, 522 Кодеин, 576,577
Кодеин/аспирин, препараты, 578 Кодеин/ацетаминофен, препараты, 578
Кокаин, 168,585-587
Компазин, 446.
См. также Прохлорперазин, 542 Конвертирующий фермент, 348 Конкурентные антагонисты, 28-30 Конотоксин и автономная передача, 118-119
Концентрация в крови-время, кривая, 62
Конъюгация, 77-80
Коргард, 145,168,185.
См. также Надолол, 192,219,241 Кордарон, 227.
См. также Амиодарон, 292 Кортикостероиды, 392-393,396 Кортикостероиды, препараты аэрозольные, 396
Котрансмиттеры, 103,107 Кофеин, 385,584 Кристодигин, 202.
См. также Дигитоксин, 262 Кромакалин, 382-385,396 Кромолин, 382-385,396 Кросс-толерантность, 572 Ксанакс, 428.
См. также Алпразолам Ксенобиотики, 13,73 Ксйлокаин, 292,492.
См. также Лидокаин Ксилометазолин, 167,174 Куприд, 523.
См. также Триентин
Куприд, 430.
См. также Триентин, 522,523 Купримин, 430.
См. также Пеницилламин, 523 Кураре, 493
Л
Лабеталол, 176,179,191,207,219 Лазикс, 316.
См. также Фуросемид
Ламотридджин, 454 Ланоксикапс, 262.
См. также Дигоксин Ланоксин, 262;
См. также Дигоксин Лародопа, 522.
См. также Леводопа Леваллорфан, структура, 561 Леватол, 192,219.
См. также Пенбутолол Левобунолол, 192 Леводопа, 510-514 Лево-Дроморан, 577.
См. также Леворфанол Левометадил ацетат, препараты, 577 Левонантрадол, 593 Левопропоксифен, 576 Леворфанол, 575,577
Левофед, 174.
См. также Норадреналин Левсинекс, 153
Лейкотриены (ЛТ), 366-367,393-
394
Лейцин-энкефалин, 563 Лекарства основания, 18-19
Лекарства-кислоты, 18-19,328-330 Лекарственные рецепторы, 22-25, 27-32
Либрий, 428.
См. также Хлордиазепоксид Лиганд(ы) 36-37
Лидокаин, 282-209,292,485,492
Лизиноприл,214
Лиорезал, 508. См. также Баклофен Липоксины, 361-362
Липооксигеназы, 366,393-394
Литий, 311,536-542
Лобелии, 91
Лодозин, 430.
См. также Карбидопа
Лозартан, 280
Лозол, 247.
См. также Индапамид Локсапин, 434,438,446 Локситан, 446.
См. также Локсапин
Лонитен, 168.
См. также Миноксидил Лоперамид, 474,956 Лопрессор, 145,168,185.
См. также Метопролол Лоразепам, 337,347,374,379,393 Лоратидин, 257,272
Лортаб, 476
Лотензин, 168.
См. также Беназеприл
Лотузат, 347
Лудиомил, 458.
См. также Мапротилин Люминал натрия, 347,379.
См. также Фенобарбитал
м
Магний, 195,224-225
Магния хлорид, препараты, 288,292 Максаир, 396
Максимальная эффективность, 46 Малаоксон, 131
Маннитол, 310-311,316
МАО ингибиторы, 516,548,555,556 Мапротилин, 547,548,552,556 Марезин, 344.
См. также Циклизин Марихуана, 591-594 Маркаин, 492.
См. также Бупивакаин Марплан, 556.
См. также Изокарбоксазид Мебарал, 429,442 Медицинская фармакология, определение, 12 Мезантоин, 466.
См. также Мефинитоин Мезоридазин, 533,542 Мекамиламин,151 Меклизин, 326,344 Мексилетин, 278-279,284,292 Мекситил, 292.
См. также Мексилетин Мелларил, 542.
См. также Тиоридазин Мепензолат, 153 Меперидин, 562,574-575,577 Мепивакаин, 492 Мепробамат, 413,429 Местинон, 137.
См. также Пиридостигминн Местные анестетики, 84,395-402, 414,966
Метадон, 562,574,577 Метамфетамин, 174,585 Метанол, 431,440-441 Метантелин, 147,153 Метапролол, аффинитет
к рецептору, 176,192 Метапротеренол, 389-390,396 Метараминол, 174
Метарбитал, препараты, 429,466 Метахолин, структура, 124 Метдилазин, препараты, 344 Метиамид, 88
Метиклотиазид, 307,316 Метилдопа, 200-201,219 Мети ленгликоль, 431-441
3,4-Метилендиоксиметамфетамин (MDMA), 589
Метилксантины, 385-388,396-397 4-Метилпиразол, 431 а-Метилтирозин, 118
Метилприлон, 429 Метилфенидат, 167,174 1-Метил-4-фенил-1,2,5,6-тетрагид-
ропиридин (МРТР), 519 Метилэргоновин, 345 Метилэтилкетон, 594 Метилпранолол, 191
Метирозин, 118,179-182,192 Метисергид, 342,345
Метогекситал, препараты, 482 Метоксамин, 167,174
Метоксифлюран, 469,471,482 Метокурин, 498,503,508 Метолазон, 306,316
Метопролол, 183,186,192,207,219 Метскополамин, 153
Метсуксимид, 456,466 Метубина иодид, 507.
См. также Метокурин Метэргин, 345 Мефенитоин, 448-449 Мефентермин, препараты, 174 Мефобарбитал, препараты, 429,466 Мивакрон, 508.
См. также Мивакурий Мивакурий, 496,498,503,508 Мигрень, 340-342 Мидазолам, 428,469,482 Мидамор,316.
См. также Амилорид Мидриаз, 143 Мидриацил, 153.
См. также Тропикамид Мизолин, 467.
См. также Примидон Мизопростол, 377 Миидил, 344
Микрокс, 316.
См. также Метолазон Милонтин, 466.
См. также Фенсукцимид Милринон, 252,262 Милтаут, 429.
См. также Мепробамат Минипресс, 191,219.
См. также Празозин Минитран, 240.
См. также Нитроглицерин Миноксидил, 210-211,220 Миоклонические судороги, 462 Миостат, 137.
См. также Карбахол
Миохол, 137.
См. также Ацетилхолин Мипроевая кислота, 467,541-542.
См. также Вальпроевая кислота Мителаза, 137.
См. также Амбеноний
Мобан, 542.
См. также Молиндон Моклобемид, 548 Молиндон, 526,532-533,542 Моноприл, 220.
См. также Фозиноприл Мороицизин, 278,279,285,292 Морфин, 559-577 Морфинаны, 561,575
Мускарин, 121
Мускариновых рецепторов блокаторы, 139-152,391-392
Мусцимол, 521
н
Наван, 542.
См. также Тиотиксен Нагрузочная доза, 43-44 Надолол, 183-184,186,192,207,219, 241
Назалкром, 396.
См. также Кромолин натрия Налбуфин, 560,562,575,578 Налоксон, 560,562 Налорфин, 560,575 Налтрексон, 560,575,582 Нардил, 556.
См. также Фенелзин Натрий-кальций обменник, 243-244 Натрия вальпроат, 446,456-459,463 Натрия нитропруссид, 211-212
Натуретин, 316.
См. также Бендрофторметиазид Нафазолин, 174 Неадренергические, нехолинергические нейроны,! 12 Недеполяризующие блокаторы, 494-499,500-502
Недокромил, 382-385,394-395,396 Незакаин, 491 Нейролептики, 524 Нейролептический злокачественный синдром, 535
Нейролептоаналгезия, 480 Нейронтин, 466.
См. также Габапентин Нейропептид Y, 359-360 Нейротензин, 359 Нембутал, 429,466.
См. также Пентобарбитал Необратимые антагонисты, 28-30 Нео-Синефрин, 174.
См. также Оксиметазолин,
Фенилэфрин, Ксилометазолин Неостигмин, 119,130-131,137 Нервно-мышечная блокада, 493-
505
Нернста уравнение, 266 Нефролитиаз, 315 Низатидин, 329,344 Низолдипин, 233-236 Никардипин, 213,220,233,236,240 Никотин, 136,584-585 Никотиновый рецептор, 36,498-499 Нимодипин, 233-236,240 Нимотоп, 240.
См. также Нимодипин Нитразепам, 412,416,460 Нитраты, 224-231,240 Нитрендипин, 233-236 Нитриты, 224-231 Нитро-Бид IY, 240.
См. также Нитроглицерин Нитрогард, 240.
См. также Нитроглицерин Нитроглицерин, 224-231,240 Нитро-Дур, 240.
См. также Нитроглицерин Нитрол, 240.
См. также Нитроглицерин Нитронг, 240.
См. также Нитроглицерин Нитропруссид, 220 Нифедипин, 213,220,233-236,240
Новокаин, 492.
См. также Прокаин Ноктек, 429.
См. также Хлоралгидрат Нолахист, 344.
См. также Фениндамин
Нолудар, 429
Норадреналин, 106-107,118,160, 166,174,177,407,409
Норваск, 213,220
См. также Амлодипин Нормодин, 191,219.
См. также Лабеталол
Норпейс, 292.
См. также Дизопирамид Норпрамин, 556.
См. также Дезипрамин Нортриптилин, 546,548,556 Нубаин, 578.
См. также Налбуфин Нуморфан, 578.
См. также Оксиморфон Нуперкаинал, 491 Нуромакс, 508.
См. также Доксакурий
О
Общие анестетики, 381-393 Объем распределения, 54 Ограниченное связывание с белками и концентрация лекарства, 69 Окисление, 78 Оксаазолидиндионы, 459-460 Оксазепам, 412,416,428 Оксибутинин, 153 Оксикодон, 560,562,579,581 Оксиметазолин, 174 Оксиморфон, 560,562,579 Оксимы, 149 Оксифенциклимин, 153 Окскарбазепин, 450 Оксотреморин, 125 Окстрифиллин, 397 Окупресс, 191,219.
См. также Картеолол Ондансетрон, 336,345 Опиаты, 558 Опиоиды, 558-578 Оптимин, 343 Оптипранолол, 191.
См. также Метипранолол
Орап, 542.
См. также Пимозид
Органические нитриты, 595
Орлаам, 577
Орфенадрин, 518,522
Осмитрол, 316.
См. также Маннитол
Осмотические диуретики, 310-311
Отравление холинергическими средствами, 148-149
Отривин, 174.
См. также Ксилометазолин
Оубаин,247-249
п
Павулон, 508.
См. также Панкуроний Паксил, 556.
См. также Пароксетин Паксипам, 428 Памелор, 556.
См. также Нортриптилин Памин, 153.
См. также Метскополамин Панкуроний, 495,498,508 Парадион, 466 Паральдегид, 429 Параметадион, 466 Параоксон, 131
Паратион, 131 Паргилин, 205 Парлодел, 522.
См. также Бромокриптин Парнат, 556.
См. также Транилципромин Пароксетин, 547,548,552,556 Пароксизмальная тахикардия и симпатомиметические агенты 169-173
Парциальные агонисты, 30-32 Пеганон, 379.
См. также Этотоин Пеламин, 344.
См. также Трипеленнамин Пемолин, 167
Пенбутолол, 184,186,192,219 Пеницилламин, 522-523
Пентазоцин, 576,578
Пентаэритрола тетранитрат, 231,240 Пентобарбитал, 413,429,466 Пентоксифиллин, 397
Пентотал, 482.
См. также Тиопентал
Пентран, 482.
См. также Метоксифлюран Перголид, 516,523
Периактин, 344.
См. также Ципрогептадин Периндоприл, 220
Период полувыведения, 59-60 Перитрат, 240.
См. также Пентаэритрола тетранитрат
Перкодан, 578
Перкоцент, 578
Пермакс, 516,523
См. также Перголид
Перметил, 542.
См. также Флуфеназин Пертофран, 556.
См. также Дезипрамин Перфеназин, 526,533,542 Петлевые диуретики, 237-239,244 Петля Генле, 232-233 Пилокарпин, 131,190
Пимозид, 526,542
Пиндолол, 176,183,192 Пипекуроний, 497,498,503,508
Пиперацетазин, 533
Пирбутерол, 396
Пирензепин, 141,142
Пиридостигмин, 131,137
Пириламин, 326,344
Плазма, связывание лекарств, 69
Плацебо-реакция, 95 Плацидил, 429.
См. также Этхлорвинол Плендил, 220,240.
См. также Фелодипин Поддерживающая доза, 65-66 Поламин, 344.
См. также Дексхлорфенирамин Политиазид, 307,316
Понтокаин, 492.
См. также Тетракаин Постнагрузка, 245 Постуральный барорефлекс, 194-
195
Потенциалы действия, 268-270, 285-286
Празепам, 425,428
Празозин, 175-176,179,191,219
Праке, 492
Пралидоксим (РАМ), 148-149,153
Прамоксин, 492
Предсердные аритмии и дигиталис, 255-258
Предсердный натриуретический пептид (ANP), 355-357
Предсердный натриуретический фактор (ANF) и трансмембранные энзимы, 35-37
Прелудин, 174.
См. также Фенметразин Пренальтерол, 168 Привин, 174 Прилокаин, 485,492 Примидон, 452-453,467 Принивил, 262.
См. также Лизиноприл Приодерм. См. Малатион Присколин, 191.
См. также Толазолин Про-Бантин, 153.
См. также Пропантелин Провентил, 396.
См. также Альбутерол Прогабид, 507 Прозак, 556.
См. также Флюоксетин Прокаин, 485,492 Прокаинамид, 277-279,292 Прокардиа, 240.
См. также Нифедипин Проликсин, 542.
См. также Флуфеназин Промазин, 542 Прометазин, 325,344 Промышленные растворители, 594 Пронестил, 292.
См. также Прокаинамид Пропантелин, 141,147,153 Пропафенон, 277-279,292 Пропоксифен, 560,562,575,578 Пропофол, 469,480-482 Пропранолол, 118,176,183-186, 192,206-207,219,240,278-279, 292,342
ПроСом, 428
Простагландины, 361-378 Простациклин, 364-367 Простигмин, 137.
См. также Неостигмин Простин, 377
Противокашлевые средства, 576 Противоэпилептические агенты, 443-467
Протопам, 153.
См. также Пралидоксим Протриптилин, 546,548,556 Прохлорперазин, 542 Проциклидин,523 Псевдоэфедрин, 174 Псилоцибин,588-589 Психическая зависимость, 421,531, 572-571
Р
Рамиприл, 214,220
Ранитидин, 344
Раувольсцин, 176
Реактиваторы холинэстеразы, 148-
149
Регитин, 191.
См. также Фентоламин Резерпин, 204-205,219,519 Ремоксиприд, 526
Ренез, 316.
См. также Политиазид
Ренин, 213,346-351
Ренин-ангиотензиновая система, 346-351
Ресторил, 428.
См. также Темазепам Рецепторы, 22-23.
См. также Лекарственные рецепторы а-рецептор(ы), 154-156 Риспердал, 542 Риталин-SR, 174.
См. также Метилфенидат Ритансерин, 336 Ритмол, 292.
См. также Пропафенон Ритодрин, 167,172 Робинул, 152.
См. также Гликопирролат Рокуроний, 497,498,503,508 Ромазикон, 428.
См. также Флумазенил
С
Сальбутамол (альбутерол), 389 Салурон, 316.
См. также Гидрофторметиазид Сальметерол, 396
Сансерт, 345.
См. также Метисергид Саралазин, 213-214 Связывание, лекарство-рецептор,
14
Севофлуран, 469
Седативно-снотворные средства, 4'11-430,582-584
Секобарбитал, 413,429 Секонал, 429.
См. также Секобарбитал Сектрал, 197,219,292.
См. также Ацебутолол Селдан, 344.
См. также Терфенадин Селегилин, 511,516,523 Селективные ингибиторы обратного захвата серотонина, 544-556
Сенильная церебральная недостаточность, и алкалоиды спорыньи, 343
Сенсоркаин, 492.
См. также Бупивакаин Сераке, 428.
См. также Оксазепам Сердечная мышца и блокаторы кальциевых каналов, 232
Сердечные гликозиды, 242-263 Сердца остановка, и симпатомиметические агенты, 128
Сердце
блокада и симпатомиметические средства, 128
замена клапана,
и антипсихотические средства, 440 и гистамин, 253
и литий, 445
и метопролол, 183
и острое потребление алкоголя, 352
и сердечные гликозиды, 192-195 и симпатомиметики, 122
и холиномиметики, 99 недостаточность.
См. также Застойная сердечная недостаточность однонаправленный блок и reentry, 212 острая, лечение, 200-202 хроническая, лечение, 199-200 частота, 191
Сердечные аритмии, 188,264 Серентил, 542.
См. также Мезоридазин Серотонин, 332-336,406,409-410 Серпазил, 219,
См. также Резерпин Сертралин, 547,548,552,556 Симметрел, 522.
См. также Амантадин Симпатомиметики, 154-174,388-
391
Симпатомиметические агенты, анафилаксия, 171
Синдром отмены, вызванный антагонистом опиатов, 572
Синекван, 556.
См. также Доксепин Синемет, 430.
См. также Карбидопа/леводопа 522
Скополамин, 140-146,153 Следовая деполяризация, 269-273 Сложные парциальные судороги, 461-462
Сло-Филлин, 397.
См. также Теофиллин Снотворные, 333.
См. также Седативные-снотвор-ные средства. 411-430
Сократимость сердца, контроль, 242-244
Сорбитрат, 240.
См. также Изосорбита динитрат Соталол, 192,278-279,292 Спазмолитики, 493,505-508 Спарин, 542.
См. также Промазин Спиронолактон, 307-308,316 Спирты,350-359.
См. также Этанол, 431-442 Средняя летальная доза, 93 Средняя токсическая доза, 46 Средняя эффективная доза, 46 Стадол, 577.
См. также Буторфанол Стелазин, 542.
См. также Трифлуоперазин Стенокардия (грудная жаба), 222 Стенокардия напряжения, 237-240 Стереоизомеризм, 14 Стероидные нервно-мышечные блокаторы, 497,498,503
Сублимаз, 577.
См. также Фентанил
Судороги, 420,444-466,505
Сукцинилхолин, 495,498,503,508
Сульфаниламид, 301 Суматриптан, 335,345 Супразин, 542.
См. также Трифлуоперазин Суритал, 482 Сурмонтил, 556.
См. также Тримипрамин Суфента, 578.
См. также Суфентанил Суфентанил, 562,578 Субстанция Р, 107,358-359
т
Тагамет, 344.
См. также Циметидин Такарил, 344 Талбутал, 429 Талвин, 578.
См. также Пентазоцин Тамбокор, 292.
См. также Флекаинид Тарактан, 542 Тегретол, 466,542.
См. также Карбамазепин Телдрин, 344.
См. также Хлорфенирамин Темазепам, 416,429 Темарил, 344 Тенекс,219.
См. также Гуанфацин Тензилон, 137.
См. также Эндрофоний Тенормин, 191,219,240.
См. также Атенолол Теобромин, 385,397 Тео-Дур, 319.
См. также Теофиллин Теофиллин, 385,397 Теразозин, 176,179,191 Терапевтический индекс, 47 Тербуталин, 172,389,396 Тетрагидрозолин, 174 Тетрагидроканнабинол, 592 Тетракаин, 485,492 Тетраэтиламмоний, 151 Тетраэтилтиурам, 442.
См. также Дисульфирам Тетродотоксин, 118,486 Тиазидные диуретики, 305-307 Тиамилал, 482
Тиамин, 442
Тизин, 174
Тимолол, 183,184,186,190,192 Тиндал, 542
Тиопентал, 479,482
Тиоридазин, 526,528,539,542 Тиотиксен, 526,528,539,542 Тирамин, 118,168-169 Тирозинкиназы,35-36 Токаинид, 278-279,284,292 Толазолин, 175-176,191 Толерантность, 48,579 Тонокард, 292.
См. также Токаинид Торазин, 541.
См. также Хлорпромазин Торналат, 396.
См. также Битолтерол Торсемид, 304,317 Тофранил, 556.
См. также Имипрамин Тразодон, 547-548,552,556 Тракрий, 507.
См. также Атракурий Трал, 152.
Трандат, 191,219.
См. также Лабеталол Транилципромин, 548,552,556 Транксен, 428,466.
См. также Клоразепат Тремор интенционный, 520 Триазолам, 412,416 Триамтерен, 308,309,317 Тридил, 240.
См. также Нитроглицерин Тридион, 467.
См. также Триметадион Триентин, 523 Трилафон, 542.
См. также Перфеназин Тримепразин, 344 Триметадион, 459,467 Триметафан, 151,202 Тримипрамин, 552,556 Трифлуоперазин, 526,542 Трифлупромазин, 542 Трихлорметиазид, 317 Трихлорэтанол, 413 Трициклические антидепрессанты, 545-547,549-550,552
Тромбоксаны, 361-362 Тропикамид, 141,146,153 Тубокурарин, 493,496,498, 503,508
У
Углеводная толерантность и тиазиды, 307
Унифил, 397.
См. также Теофиллин
Урапидил, 179
Урехолин, 137.
См. также Бетанехол
ф
Фактор кумуляции, 60 Фармакодинамика, 22-52 Фармакокинетика, 53-72 Фелбамат, 454-555 Фелодипин, 213,220 Фенантрены, 560,575 Фенацемид, 448-449,466 Фенелзин, 552,556 Фенерган, 344.
См. также Прометазин Фенилпропаноламин, 167,174 Фенилэтиламин, 156 Фенилэфрин, 156-161,174 Фениндамин, 344 ' Фенитоин, 278-279,284,467 Фенметразин, 167,174,585 Фенобарбитал, 413-415,425,429 Феноксибензамин, 176-182,191 Фенотерол, 389 Фенотиазины, 325,326,525,527,532 Фенсукцимид, 456,466 Фентанил, 562,577 Фентоламин, 176-182,191,208 Фенурон, 466.
См. также Фенацемид Фенциклидин, 588-589 Физическая зависимость, 421,432, 564-565,579
Физологический антагонизм, 32 Физостигмин, 130-131,137,190 Флавинмонооксигеназа, 79 Флавоксат, препараты, 152 Флакседил, 417.
См. также Галламин, 580 Флекаинид, 284-289,292 Флексерил, 508 Флозехинан, 254 Флолан, 377 Флороприл, 101.
См. также Изофлурофат 137 Флумазенил, 418-419,429 Флунаризин, 342
Флунитразепам, фармакокинетика, 416
Флуоксетин, 548,556 Флуотан, 393.
См. также Галотан, 482 Флуразепам, 412,416,428,924 Флуфеназин, 526,532,533,542 Фозиноприл, 214 Фомепизол, 432
Форан, 393.
См. также Изофлуран, 482 Формотерол, 390
Фосфодиэстеразы ингибиторы, 242, 252
Фосфолин, 136-137.
См. также Экотиофат Фосфорорганические ингибиторы холинэстеразы, 129-133,148
Функциональная толерантность, 579 Фуросемид, 303-304,316
X
Халцион, 347. См. также Триазолам 429
Хинаприл, 214,220
Хинетазон, 307,316
Хинидин, 275-277,292
Хлоралгидрат 413,429
Хлордиазепоксид, 84,412,416,425,
428
Хлорпрокаин,491
Хлороформ, 594
Хлорпромазин, 526,527,532,541
Хлорпротиксен, 542
Хлорталидон, 306,316
Хлортиазид, 301,307,316 Хлор-Триметон, 272.
См. также Хлорфенирамин, 344 Хлорфенирамин, 326,344 Хлорциклизин и лекарственный метаболизм, 84
Холедил, 397
Холецистокинин, 107
Холина эфиры, 124
Холинергическая передача, 74,75
Холинолитики, 517
Холиномиметические средства, 123-138
Хорея, 419,428.
См. также Хорея Гентингтона, 509,520
ц
Цедиланид-D, 262
Целипролол, 176,184,186 Центракс.
См. также Празепам, 428
CNP, структура, 356
Цетиризин, препараты, 344
Циклизин, 325,326,344
Циклобензаприн, 508
Циклогил, 112.
См. также Циклопентолат, 152
Циклопентолат, 146,152
Циклоплегия, 143
Циклопростин, 377 Ципрогептадин, 326,344
Цистоспаз-М, 153
Цитанест, 402.
См. также Прилокаин, 492
Цитотек, 303.
См. также Мизопростол, 377 Цитохром Р-450,75-86, метаболизм простагландинов, 364-368
ч
Четвертичные амины, 141,147
Эдекрин, 247.
См. также Этакриновая кислота,
316
Эдрофоний, 134,137
Эзидрикс, 247.
См. также Гидрохлортиазид, 316 Эйкозаноиды, 361-378 Экванил, 348.
См. также Мепробамат, 429
Эксна, 247.
См. также Бензтиазид, 316
Элавил, 458.
См. также Амитриптилин, 556 Элдеприл, 430.
См. также Селегилин, 523
Электрический потенциал и секреция калия, 300 Эликсофиллин, 319.
См. также Теофиллин, 397 Элиминация, ограниченная
по объему, 58-59
Эмпирии, 578
Эналаприл, 214,220,350 Эндогенная депрессия, 538-539 Эндорфины, 563
Эндотелии, 357-358
Эндурон, 247.
См. также Метиклотиазид, 316 Энкефалины, 563 Энфлуран, 482 Эпилепсия, 443-467 Эпинин, 166 Эпоксидация, 78 Эпоксиэйкозатетраеноевая кислота, 361
Эпопростенол, препараты, 377 Эргомар, 273.
См. также Эрготамин, 345 Эргостат, 273.
См. также Эрготамин, 345 Эрготамин, 345 Эрготрата малеат, 272.
См. также Эргоновин, 345 Эскалит, 542 Эсмолол, 184,186
Эссенциальная гипертензия, 194 Эссенциальный тремор, 519 Эстазолам, препараты, 428 Этакриновая кислота, 190,304,316 Этанол, 350-357,747.
См. также Спирты, 431-442 Этидокаин, 485,491 Этиленгликоль, 441-442 Этилнорадреналин, препараты, 396 Этинамат, препараты, 429 Этмозин, 227.
См. также Морицизин, 292 Этомидат, 482 Этосуксимид, 455-456,466 Этотоин, 448-449 Этран, 392-393,
См. также Энфлуран, 482 Этхлорвинол, 429
Эфедрин, 161,167,389,396
Эфедрон интраназальный, 130,318.
См. также Эфедрин, 174,396 Эфир, 594
Эфиры, метаболизм, 79
Эффексор, 556
Эффект первого прохождения, 75
Эхотиофат, 133,137
Содержание
Том 1
Предисловие научного редактора перевода..........................................5
Предисловие......................................................................7
Авторы...........................................................................9
Раздел I. Основные принципы....................................................12
Глава 1. Введение.............................................................12
Глава 2. Лекарственные рецепторы и фармакодинамика............................22
Глава 3. Фармакокинетика и фармакодинамика: рациональный выбор дозы и времени действия лекарства.................................................53
Глава 4. Биотрансформация лекарств............................................73
Глава 5. Базисная и клиническая оценка новых лекарств.........................87
Раздел II. Средства, влияющие на вегетативную нервную систему..................101
Глава 6. Введение в фармакологию вегетативной нервной системы................101
Глава 7. Средства, активирующие холинорецепторы и ингибирующие холинэстеразу.121
Глава 8. Средства, блокирующие холинорецепторы...............................139
Глава 9. Средства, активирующие адренорецепторы, и другие симпатомиметические средства...............................154
Раздел III. Средства, влияющие на сердечно-сосудистую систему и почки...........193
Глава 10. Антигипертензивные средства.........................................193
Глава 11. Вазодилататоры и средства для лечения стенокардии...................222
Глава 12. Сердечные гликозиды и другие средства, применяемые при застойной сердечной недостаточности.....................................243
Глава 13. Антиаритмические средства..........................................265
Глава 14. Мочегонные средства................................................295
Раздел IV. Средства, оказывающие выраженное действие на гладкую мускулатуру.........................................................318
Глава 15. Гистамин, серотонин и алкалоиды спорыньи...........................318
Глава 16. Вазоактивные пептиды...............................................347
Глава 17. Эйкозаноиды: простагландины, тромбоксаны, лейкотриены и родственные им соединения........................................ 360
Глава 18. Бронходилататоры и другие средства, применяемые для лечения астмы.... 378
Раздел V. Средства, влияющие на центральную нервную систему (ЦНС)..................399
Глава 19. Введение в фармакологию препаратов, влияющих на ЦНС.................. 399
Глава 20. Седативные и снотворные средства.......................................410
Глава 21. Спирты............................................................... 4Й0
Глава 22. Противоэпилептические средства______________________________________ 442
Глава 23. Общие анестетики..................................................... 469
Глава 24. Местные анестетики.....................................................482
Глава 25. Средства, расслабляющие скелетную мускулатуру (миорелаксанты).......................................... 492
Глава 26. Фармакотерапия паркинсонизма и других двигательных нарушений.........................................508
Глава 27. Антипсихотические средства и литий................................... 525
Глава 28. Антидепрессанты.........................................................545
Глава 29. Опиоидные аналгетики и антагонисты................................... 559
Глава 30. Вещества, вызывающие злоупотребление....................................578
Указатель...............................................................*...........596